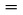|
31.5.2008
|
FR
|
Journal officiel de l'Union européenne
|
L 142/1
|
RÈGLEMENT (CE) no 440/2008 DE LA COMMISSION
du 30 mai 2008
établissant des méthodes d'essai conformément au règlement (CE) no 1907/2006 du Parlement européen et du Conseil concernant l'enregistrement, l'évaluation et l'autorisation des substances chimiques, ainsi que les restrictions applicables à ces substances (REACH)
(Texte présentant de l'intérêt pour l'EEE)
LA COMMISSION DES COMMUNAUTÉS EUROPÉENNES,
vu le traité instituant la Communauté européenne,
vu le règlement (CE) no 1907/2006 du Parlement européen et du Conseil du 18 décembre 2006 concernant l'enregistrement, l'évaluation et l'autorisation des substances chimiques, ainsi que les restrictions applicables à ces substances (REACH), instituant une agence européenne des produits chimiques, modifiant la directive 1999/45/CE et abrogeant le règlement (CEE) no 793/93 du Conseil et le règlement (CE) no 1488/94 de la Commission ainsi que la directive 76/769/CEE du Conseil et les directives 91/155/CEE, 93/67/CEE, 93/105/CE et 2000/21/CE de la Commission (1), et notamment son article 13, paragraphe 3,
considérant ce qui suit:
|
(1)
|
Conformément au règlement (CE) no 1907/2006, il convient que des méthodes d'essai soient adoptées au niveau communautaire aux fins de la réalisation d'essais sur des substances lorsque de tels essais sont nécessaires pour produire des informations sur les propriétés intrinsèques desdites substances.
|
|
(2)
|
La directive 67/548/CEE du Conseil du 27 juin 1967 concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives à la classification, l'emballage et l'étiquetage des substances dangereuses (2) définissait, à l'annexe V, les méthodes permettant de déterminer les propriétés physico-chimiques, la toxicité et l'écotoxicité des substances et préparations. L'annexe V de la directive 67/548/CEE a été supprimée par la directive 2006/121/CE avec effet au 1er juin 2008.
|
|
(3)
|
Il y a lieu que les méthodes d'essai de l'annexe V de la directive 67/548/CEE soient intégrées dans le présent règlement.
|
|
(4)
|
Le présent règlement n'exclut pas l'utilisation d'autres méthodes d'essai à condition que ladite utilisation soit conforme aux dispositions de l'article 13, paragraphe 3, du règlement (CE) no 1907/2006.
|
|
(5)
|
Il importe que les principes de remplacement, de réduction et d'affinement de l'utilisation des animaux dans les procédures soient pleinement pris en compte lors de l'élaboration des méthodes d'essai, notamment lorsque des méthodes appropriées et validées permettant de remplacer, réduire ou affiner les essais sur les animaux deviennent disponibles.
|
|
(6)
|
Les dispositions du présent règlement sont conformes à l'avis du comité institué par l'article 133 du règlement (CE) no 1907/2006,
|
A ARRÊTÉ LE PRÉSENT RÈGLEMENT:
Article premier
Les méthodes d'essai à appliquer aux fins du règlement (CE) no 1907/2006 sont définies à l'annexe du présent règlement.
Article 2
La Commission procède, si nécessaire, à une révision des méthodes d'essai établies dans le présent règlement en vue de remplacer, réduire ou affiner les essais menés sur les animaux vertébrés.
Article 3
Toutes les références à l'annexe V de la directive 67/548/CEE s'entendent comme des références au présent règlement.
Article 4
Le présent règlement entre en vigueur le jour suivant celui de sa publication au Journal officiel de l'Union européenne.
Il s'applique à compter du 1er juin 2008.
Fait à Bruxelles, le 30 mai 2008.
Par la Commission
Stavros DIMAS
Membre de la Commission
(1) JO L 396 du 30.12.2006, p. 1; rectifié au JO L 136 du 29.5.2007, p. 3.
(2) JO 196 du 16.8.1967, p. 1. Directive modifiée en dernier lieu par la directive 2006/121/CE du Parlement européen et du Conseil (JO L 396 du 30.12.2006, p. 850); rectifiée au JO L 136 du 29.5.2007, p. 281.
ANNEXE
PARTIE A: MÉTHODES DE DÉTERMINATION DES PROPRIÉTÉS PHYSICO-CHIMIQUES
TABLE DES MATIÈRES
|
A.1.
|
TEMPÉRATURE DE FUSION/DE CONGÉLATION |
|
A.2.
|
TEMPÉRATURE D'ÉBULLITION |
|
A.5.
|
TENSION SUPERFICIELLE |
|
A.8.
|
COEFFICIENT DE PARTAGE |
|
A.10.
|
INFLAMMABILITÉ (SOLIDES) |
|
A.11.
|
INFLAMMABILITÉ (GAZ) |
|
A.12.
|
INFLAMMABILITÉ (AU CONTACT DE L'EAU) |
|
A.13.
|
PROPRIÉTÉS PYROPHORIQUES DES SOLIDES ET DES LIQUIDES |
|
A.15.
|
TEMPÉRATURE D'INFLAMMATION SPONTANÉE DES LIQUIDES ET DES GAZ |
|
A.16.
|
TEMPÉRATURE RELATIVE D'INFLAMMATION SPONTANÉE POUR LES SOLIDES |
|
A.17.
|
PROPRIÉTÉS COMBURANTES (SOLIDES) |
|
A.18.
|
DÉTERMINATION DE LA MASSE MOLÉCULAIRE MOYENNE EN NOMBRE ET DE LA DISTRIBUTION DES MASSES MOLÉCULAIRES DES POLYMÈRES |
|
A.19.
|
DÉTERMINATION DE LA TENEUR EN POLYMÈRES DE FAIBLE MASSE MOLÉCULAIRE |
|
A.20.
|
COMPORTEMENT DE DISSOLUTION EXTRACTION DES POLYMÈRES DANS L'EAU |
|
A.21.
|
PROPRIÉTÉS COMBURANTES (LIQUIDES) |
A.l. TEMPÉRATURE DE FUSION/DE CONGÉLATION
1. MÉTHODE
La plupart des méthodes décrites se basent sur les lignes directrices de l'OCDE (1). Leurs principes fondamentaux sont décrits dans les références (2) et (3).
1.1. INTRODUCTION
Les méthodes et les appareils décrits ci-après permettent de déterminer la température de fusion des produits chimiques, quel que soit leur degré de pureté.
La sélection de la méthode dépend de la nature de la substance à tester. Par conséquent, le facteur limitant dépend du fait que la substance peut être facilement ou difficilement pulvérisable ou peut ne pas l'être du tout.
Pour certaines substances, il est préférable de déterminer la température de congélation ou de solidification. C'est pourquoi les normes de ces déterminations figurent également dans cette méthode.
Lorsqu'il est difficile, du fait de certaines propriétés de la substance, de mesurer les paramètres cités ci-dessus, il peut être approprié de rechercher un point d'écoulement.
1.2. DÉFINITIONS ET UNITÉS
La température de fusion est définie comme étant la température à laquelle se produit la transition de phase de l'état solide à l'état liquide, à la pression atmosphérique. Idéalement, cette température correspond à la température de congélation.
Étant donné que la transition de phase de nombreuses substances a lieu dans un certain intervalle de température, celle-ci est souvent décrite comme un intervalle de fusion.
Conversion des unités (K en oC)
t = T – 273,15
|
t
|
:
|
température Celsius en degrés Celsius ( oC)
|
|
T
|
:
|
température thermodynamique en Kelvin (K)
|
1.3. SUBSTANCES DE RÉFÉRENCE
Il n'est pas nécessaire d'employer des substances de référence dans tous les cas où l'on étudie une nouvelle substance. Elles devraient servir en premier lieu à vérifier de temps à autre la fiabilité de la méthode et à permettre les comparaisons avec les résultats obtenus avec d'autres méthodes.
Certaines substances d'étalonnage sont énumérées dans la référence (4).
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La température (intervalle de température) de la phase de transition de l'état solide, à l'état liquide, ou de l'état liquide à l'état solide, est déterminée. En pratique, les températures du début et de la fin de la fusion/congélation sont déterminées au cours du chauffage/refroidissement, à pression atmosphérique, de la substance à étudier. Cinq types de méthodes sont décrites; la méthode en tube capillaire, la méthode par bloc chauffant, la mesure de la température de congélation, les méthodes d'analyse thermique et la détermination du point d'écoulement (mise au point pour les huiles de pétroles).
Dans certains cas, il peut être commode de mesurer la température de congélation au lieu de la température de fusion.
1.4.1. Méthode du tube capillaire
1.4.1.1. Dispositif de mesure de la température de fusion comportant un bain liquide
Une petite quantité de substance finement broyée est placée dans un tube capillaire et tassée avec soin. Le tube est chauffé en même temps qu'un thermomètre dans le bain liquide et l'accroissement de température est ajusté à un peu moins d'1 K/mn pendant la fusion réelle. Les températures correspondant au début et à la fin de la fusion sont relevées.
1.4.1.2. Dispositif de mesure de la température de fusion comportant un bloc métallique
Le protocole est le même que celui décrit au paragraphe 1.4.1.1, si ce n'est que le tube capillaire et le thermomètre sont placés dans un bloc de métal chauffé et observés à travers des ouvertures aménagées dans ce dernier.
1.4.1.3. Détection photo-électrique
L'échantillon contenu dans le tube capillaire est chauffé automatiquement dans un cylindre métallique. Par une ouverture aménagée dans celui-ci, un faisceau de lumière est envoyé à travers la substance à tester vers une cellule photo-électrique soigneusement étalonnée. Au moment de la fusion, les propriétés optiques de la plupart des substances sont modifiées en ce sens que l'opacité fait place à la transparence. De ce fait, l'intensité de la lumière qui atteint la cellule photo-électrique augmente et envoie un signal d'arrêt à l'indicateur digital qui affiche la température d'un thermomètre à résistance de platine placé dans l'enceinte chauffante. Cette méthode n'est pas applicable à certaines substances fortement colorées.
1.4.2. Méthodes avec bloc chauffant
1.4.2.1. Méthode au banc chauffant de Kofler
Le banc chauffant de Kofler est composé de deux pièces de métal de conductivité thermique différente, qui sont chauffées électriquement. Il est construit de façon à ce que le gradient de température soit quasi linéaire sur toute sa longueur. La température de ce banc chauffant peut varier de 283 à 573 K grâce à un dispositif de lecture de la température comprenant un curseur avec un index et une réglette graduée, spécialement conçus pour le banc en question. Pour déterminer une température de fusion, une fine couche de substance est directement déposée sur la surface du banc. En quelques secondes, il se forme une ligne fine de séparation entre la phase fluide et la phase solide. La température à la hauteur de cette ligne est lue en plaçant l'index face à cette dernière.
1.4.2.2. Microscope à fusion
Différents microscopes à platine chauffante sont utilisés pour déterminer des températures de fusion avec de très pentes quantités de substance. La température est généralement mesurée à l'aide d'un thermocouple sensible, mais parfois aussi à l'aide d'un thermomètre à mercure. Le dispositif type de mesure de la température de fusion grâce à un microscope à platine chauffante comporte une enceinte chauffante qui contient une platine métallique sur laquelle est posée une lame de verre destinée à recevoir l'échantillon. Le centre de la platine métallique est percé d'un trou permettant le passage de la lumière provenant du miroir éclairant le microscope. Lors de l'utilisation, l'enceinte est fermée à l'aide d'une plaque de verre pour empêcher la circulation d'air dans la région où se trouve l'échantillon.
Le chauffage de l'échantillon est réglé au moyen d'un rhéostat. Pour effectuer des mesures très précises sur des substances optiquement anisotropes, il est possible d'utiliser de la lumière polarisée.
1.4.2.3. Méthode du ménisque
Cette méthode s'applique spécialement aux polyamides.
La température à laquelle se déplace un ménisque d'huile de silicone, emprisonné entre une surface chauffée et une lamelle couvre-objet placée au-dessus de l'échantillon de polyamide à étudier, est déterminée visuellement.
1.4.3. Méthode de détermination de la température de congélation
L'échantillon est introduit dans un tube à essai spécial et placé dans un appareil permettant la détermination de la température de congélation. L'échantillon est agité doucement durant tout le refroidissement et la température est relevée à intervalles déterminés. Dès que quelques relevés indiquent une température constante, cette dernière est considérée comme la température de congélation (après correction de l'erreur relative au thermomètre).
Il convient d'éviter un refroidissement trop fort en maintenant un équilibre entre la phase liquide et la phase solide.
1.4.4. Analyse thermique
1.4.4.1. Analyse thermique différentielle (ATD)
Cette méthode consiste à enregistrer la différence de température entre la substance et la substance de référence en fonction de la température pendant que la substance et la substance de référence sont soumises à un programme contrôlé de variation de température. Lorsque l'échantillon subit une transition impliquant un changement d'enthalpie, ce changement est indiqué par un mouvement endothermique (fusion) ou exothermique (congélation) sur la ligne de base de l'enregistrement de la température.
1.4.4.2. Analyse calorimétrique différentielle (ACD)
Cette technique consiste à soumettre une substance et un produit de référence à un même programme de variation contrôlée de la température et à enregistrer la différence d'énergie absorbée par la substance et par le produit de référence, en fonction de la température. Cette énergie correspond à l'énergie nécessaire pour annuler la différence de température entre la substance et le matériel de référence, Lorsque l'échantillon subit une transition impliquant un changement d'enthalpie, celui-ci est indiqué par un mouvement endothermique (fusion) ou exothermique (congélation) sur la ligne de base de l'enregistrement du flux de chaleur.
1.4.5. Point d'écoulement
Cette méthode, mise au point pour les huiles de pétrole, s'applique aux substances huileuses dont la température de fusion est basse.
Après un chauffage préliminaire, l'échantillon est refroidi à une vitesse spécifique et son écoulement est observé à des intervalles de 3 K. La température la plus basse à laquelle on observe un mouvement de la substance est considérée comme le point d'écoulement.
1.5. CRITÈRES DE QUALITÉ
L'applicabilité et la précision des différentes méthodes utilisées pour déterminer la température de fusion/intervalle de fusion sont énumérées dans le tableau suivant.
TABLEAU: APPLICABILITÉ DES MÉTHODES
A. Méthodes du tube capillaire
|
Méthode de mesure
|
Substances pulvérisables
|
Substances difficilement pulvérisables
|
Gamme de températures
|
Précision approximative (1)
|
Norme existante
|
|
Dispositifs de mesure de la température de fusion comportant un bain de liquide
|
oui
|
seulement quelques unes
|
de 273 à 573 K
|
±0,3 K
|
JIS K 0064
|
|
Dispositifs de mesure de la température de fusion comportant un bloc métallique
|
oui
|
seulement quelques unes
|
de 293 à 573 K
|
±0,5 K
|
ISO 1218 (E)
|
|
Détection photoélectrique
|
oui
|
plusieurs avec des dispositifs d'application
|
de 253 à 573 K
|
±0,5 K
|
|
B. Méthodes utilisant une surface chauffée et méthode de mesure de la température de congélation
|
Méthode de mesure
|
Substances pulvérisables
|
Substances difficilement pulvérisables
|
Gamme de températures
|
Précision approximative (2)
|
Norme existante
|
|
Banc chauffant de Kofler
|
oui
|
non
|
de 283 à > 573 K
|
±1,0 K
|
ANSI/ASTM D 345176
|
|
Microscope a fusion
|
oui
|
quelques-unes seulement
|
de 273 à > 573 K
|
±0,5 K
|
DIN 53736
|
|
Méthode du ménisque
|
non
|
spécifique aux polyamides
|
de 293 à > 573 K
|
±0,5 K
|
ISO 1218 (E)
|
|
Méthodes de mesure de la température de congélation
|
oui
|
oui
|
de 223 à 573 K
|
±0,5 K
|
par ex. BS 4695
|
C. Analyses thermiques
|
Méthode de mesure
|
Substances pulvérisables
|
Substances difficilement pulvérisables
|
Gamme de températures
|
Précision approximative (3)
|
Norme existante
|
|
Analyse thermique différentielle
|
oui
|
oui
|
de 173 à 1 273 K
|
jusqu'à 600 K ±0,5 K jusqu'à 1 273 K ±2,0 K
|
ASTM E 537-76
|
|
Analyse calorimétrique différentielle
|
oui
|
oui
|
de 173 à 1 273 K
|
jusqu'à 600 K ±0,5 K jusqu'à 1 273 K ±2,0 K
|
ASTM E 537-76
|
D. Point d'écoulement
|
Méthode de mesure
|
Substances pulvérisables
|
Substances difficilement pulvérisables
|
Gamme de températures
|
Précision approximative (4)
|
Norme existante
|
|
Point d'écoulement
|
pour les huiles de pétrole et les substances huileuses
|
pour les huiles de pétrole etles substances huileuses
|
De 223 à 323 K
|
±3,0 K
|
ASTM D 9766
|
1.6. DESCRIPTION DES MÉTHODES
Les procédures de presque toutes les méthodes d'essai ont été décrites dans des normes internationales et nationales (voir annexe 1).
1.6.1. Méthodes en tube capillaire
Lorsqu'elles sont soumises à une élévation lente de la température, les substances finement pulvérisées montrent habituellement les stades de fusion représentés à la figure 1.
Figure 1

Durant la détermination de la température de fusion, il y a lieu de relever la température au début et à la fin de la fusion.
1.6.1.1. Dispositif de mesure de la température de fusion comportant un bain liquide
La figure 2 décrit un type d'appareil de mesure de la température de fusion standardisé (JIS K 0064); l'appareil est en verre et toutes les spécifications sont exprimées en millimètres.
Figure 2

Bain liquide:
Il convient de choisir un liquide approprié en fonction de la température de fusion à déterminer; on utilisera, par exemple de la paraffine liquide pour des températures de fusion ne dépassant pas 473 K et de l'huile de silicone pour les températures de fusion ne dépassant pas 573 K.
Pour les températures de fusion dépassant 523 K, un mélange composé de trois parties d'acide sulfurique et de deux parties de sulfate de potassium (en rapport de masse) peut être utilisé. Il convient de prendre des précautions appropriées pour utiliser un mélange de ce type.
Thermomètre:
Seuls peuvent être utilisés les thermomètres qui répondent aux exigences des normes suivantes ou de leurs équivalents:
ASTM E 1-71, DIN 12770, JIS K 8001.
Procédé:
La substance sèche est finement broyée dans un mortier et introduite dans un tube capillaire scellé à une extrémité, la hauteur du remplissage étant fixée à environ 3 mm après tassement. Pour obtenir un échantillon uniformément tassé, il faut laisser tomber le tube capillaire d'une hauteur d'environ 700 mm à l'intérieur d'un tube de verre posé verticalement sur un verre de montre.
Le tube capillaire rempli est placé dans le bain de telle sorte que la partie centrale du réservoir à mercure du thermomètre soit en contact avec la partie du tube capillaire où se trouve l'échantillon. En général, le tube capillaire est introduit dans l'appareil au moment où le bain est à environ 10 K en dessous de la température de fusion.
Le chauffage du bain est réglé pour que l'accroissement de température soit d'environ 3 K par minute. Le liquide doit être agité. À environ 10 K en dessous de la température de fusion attendue, l'accroissement de température est réglé à un maximum de 1 K par minute.
Calcul:
Le calcul de la température de fusion s'effectue au moyen de la formule suivante:
T = TD+0,00016(TD – TE)n
où
|
T
|
=
|
température de fusion corrigée, exprimée en K
|
|
TD
|
=
|
température lue sur le thermomètre D, exprimée en K
|
|
TE
|
=
|
température lue sur le thermomètre E, exprimée en K
|
|
n
|
=
|
nombre de graduations de la colonne de mercure sur la tige émergente du thermomètre D.
|
1.6.1.2. Dispositif de mesure de la température de fusion comportant un bloc métallique
Appareil:
L'appareil comprend:
|
—
|
un bloc métallique cylindrique, dont la partie supérieure est creuse et forme une enceinte (voir figure 3),
|
|
—
|
un bouchon métallique, percé de deux ou plusieurs trous, permettant l'introduction des tubes dans le bloc métallique,
|
|
—
|
un système de chauffage du bloc métallique, qui peut être constitué d'une résistance électrique dans le bloc,
|
|
—
|
un rhéostat pour régler la puissance dans le cas d'un chauffage électrique,
|
|
—
|
quatre fenêtres en verre résistant à la chaleur, percées dans les parois latérales de l'enceinte, en position diamétralement opposée. Un oculaire permettant d'observer le tube capillaire est installé en face d'une de ces fenêtres. Les trois autres fenêtres permettent d'éclairer l'intérieur de l'enceinte à l'aide de lampes,
|
|
—
|
un tube capillaire, en verre résistant à la chaleur, scellé à une extrémité (voir point 1.6.1.1).
|
Thermomètre:
Voir nonnes mentionnées au point 1.6.1.1. Des instruments de mesure thermoélectriques de précision équivalente sont aussi utilisables.
Figure 3

1.6.1.3. Détection photo-électrique
Appareil et procédé:
L'appareil consiste en une enceinte métallique pourvue d'un système de chauffage automatisé. Trois tubes capillaires sont remplis comme prévu au point 1.6.1.1 et placés dans le four.
Plusieurs programmes d'accroissement linéaire de la température permettant d'étalonner l'appareil. L'accroissement de température approprié est électriquement réglé à un taux constant et linéaire présélectionné. Des enregistreurs indiquent la température réelle du four et la température de la substance dans les tubes capillaires.
1.6.2 Méthode de la surface chauffée
1.6.2.1 Banc chauffant de Kofler
Voir annexe.
1.6.2.2 Microscope à fusion
Voir annexe.
1.6.2.3 Méthode du ménisque (polyamides)
Voir annexe.
Aux alentours de la température de fusion, l'accroissement de la température doit être inférieur à 1 K/mn.
1.6.3. Méthodes de détermination de la température de congélation.
Voir annexe.
1.6.4. Analyses thermiques
1.6.4.1. Analyse thermique différentielle
Voir annexe
1.6.4.2. Analyse calorimétrique différentielle
Voir annexe.
1.6.5. Détermination du point d'écoulement
Voir annexe.
2. DONNÉES
La correction du thermomètre s'impose dans certains cas.
3. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés) et, éventuellement, l'étape de purification préliminaire,
|
|
—
|
une estimation de la précision.
|
La température de fusion indiquée dans le rapport est donnée par la moyenne de deux mesures au moins qui ne dépassent pas les limites de la précision estimée (voir tableau).
Si la différence de température entre le début et la fin de la fusion se trouve dans les limites de précision de la méthode, la température relevée au stade final de la fusion est considérée comme la température de fusion; sinon les deux températures sont indiquées.
Si la substance se décompose ou se sublime avant que la température de fusion soit atteinte, la température à laquelle cela se produit doit être mentionnée.
Toutes les informations et observations pertinentes pour l'interprétation des résultats doivent être mentionnées, en particulier les impuretés et l'état physique de la substance.
4. RÉFÉRENCES
|
1)
|
OCDE, Paris, 1981, Ligne directrice no 102, décision du Conseil C(81) 30 final.
|
|
2)
|
UIPAC, B. Le Neindre, B. Vodar, eds. Experimental thermodynamics, Butterworths, London, 1975, vol. II, 803-834.
|
|
3)
|
R. Weissberger ed., Technique of organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed, Interscience Publ., New York, 1959, vol. I, Part I, Chapter VII.
|
|
4)
|
UICPA, Mesures physico-chimiques: Catalogue de substances de référence pour les laboratoires nationaux, Chimie pure et appliquée, 1976, vol. 48, 505-515.
|
Annexe
Pour plus de détails, on peut consulter par exemple les normes suivantes.
1. Méthodes du tube capillaire
1.1. Dispositif de mesure de la température de fusion comportant un bain liquide
|
ASTM E 324-69
|
Standard test method for relative initial and final melting points and melting range of organic chemicals
|
|
BS 4634
|
Method for the determination of melting point and/or melting range
|
|
DIN 53181
|
Bestimmung der Schmelzintervalles von Harzen nach Kapillarverfahren.
|
|
JIS K 00-64
|
Testing methods for melting point of chemical products.
|
1.2. Dispositif de mesure de la température de fusion comportant un bloc métallique
|
DIN 53736
|
Visuelle Bestimmung der Schmelztemperatur von teilkristallinen Kunststoffen
|
|
ISO 1218 (E)
|
Plastics — polyamides — determination of «melting point»
|
2. Méthodes du bloc chauffant
2.1. Banc chauffant de Kofler
|
ANSI/ASTM D 3451-76
|
Standard recommanded practices for testing polymeric powder coatings
|
2.2. Microscope à fusion
|
DIN 53736
|
Visuelle Bestimmung der Schmelztemperatur von reilkristallinen Kunsrstoffen.
|
2.3. Méthode du ménisque (polyamides)
|
ISO 1218 (E)
|
Plastics — polyamides — determination of «melting point»
|
|
ANSI/ASTM D 2133-66
|
Standard specification for acetal resine injection moulding and extrusion materials
|
|
NF T 51-050
|
Résines de polyamides. Détermination du «point de fusion». Méthode du ménisque.
|
3. Méthodes de détermination de la température de congélation
|
BS 4633
|
Method for the determination of crystallizing point
|
|
BS 4695
|
Method for Determination of Melting Point of Petroleum Wax (cooling curve)
|
|
DIN 51421
|
Bestimmung des Gefrierpunktes von Flugkraftstoffen, Ottokraftstoffen und Motorenbenzolen
|
|
ISO 2207
|
Cires de pétrole: détermination de la température de figeage.
|
|
DIN 53175
|
Bestimmung des Erstarrungspunktes von Fettsäuren
|
|
NF T 60-114
|
Point de fusion des paraffines
|
|
NF T 20-051
|
Méthode de détermination du point de cristallisation (point de congélation)
|
|
ISO 1392
|
Method for the determination of the freezing point.
|
4. Analyse thermique
4.1. Analyse thermique différentielle
|
ASTM E 537-76
|
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis
|
|
ASTM E 473-85
|
Standard definitions of terms relating to thermal analysis
|
|
ASTM E 472-86
|
Standard practice for reporting thermoanalytical data
|
|
DIN 51005
|
Thermische Analyse, Begriffe
|
4.2. Analyse calorimétrique différentielle
|
ASTM E 537-76
|
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis
|
|
ASTM E 473-85
|
Standard definitions of terms relating to thermal analysis
|
|
ASTM E 472-86
|
Standard practice for reporting thermoanalytical data
|
|
DIN 51005
|
Thermische Analyse, Begriffe
|
5. Détermination du point d'écoulement
|
NBN 52014
|
Échantillonnage et analyse des produits du pétrole: point de trouble et point d'écoulement limite — Monsterneming en ontleding van aardolieproducten: Troebelingspunt en vloeipunt
|
|
ASTM D 97-66
|
Standard test method for pour point of petroieum oils
|
|
ISO 3016
|
Petroleum oils — Determination of pour point.
|
A.2. TEMPÉRATURE D'ÉBULLITION
1. MÉTHODES
La plupart des méthodes décrites se fondent sur les lignes directrices de l'OCDE (1). Les principes fondamentaux sont décrits dans les références (2) et (3).
1.1. INTRODUCTION
Les méthodes et dispositifs décrits ici peuvent être appliqués aux substances liquides et à température de fusion faible, à condition qu'elles ne subissent pas de réaction chimique au-dessous de la température d'ébullition (par exemple, auto-oxydation, réarrangement, dégradation, etc.). Les méthodes s'appliquent aux substances liquides pures et impures.
L'importance donnée aux méthodes recourant à la détection photoélectrique et à l'analyse thermique est due au fait que ces méthodes permettent de déterminer non seulement la température de fusion, mais également la température d'ébullition. De surcroît, les mesures peuvent être effectuées de manière automatique.
La «méthode dynamique» a l'avantage de pouvoir être également utilisée pour la détermination de la pression de vapeur et il n'est pas nécessaire de ramener la température d'ébullition aux conditions normales de pression (101,325 kPa), puisque la pression normale peut être maintenue au cours de la mesure à l'aide d'un manostat.
Observations:
L'influence des impuretés sur la détermination de la température d'ébullition dépend beaucoup de la nature de l'impureté. Si l'échantillon contient des impuretés volatiles, susceptibles d'avoir une incidence sur les résultats, il convient de purifier la substance.
1.2. DÉFINITIONS ET UNITÉS
La température d'ébullition normale est définie comme la température à laquelle la pression de vapeur d'un liquide s'élève à 101,325 kPa.
Si la température d'ébullition n'est pas mesurée à la pression atmosphérique normale, la dépendance de la pression de vapeur vis-à-vis de la température peut être décrite à l'aide de l'équation de Clausius-Clapeyron:

où:
|
p
|
=
|
pression de vapeur de la substance exprimée en pascal
|
|
Δ Hv
|
=
|
chaleur de vaporisation en J mol – 1
|
|
R
|
=
|
constante molaire des gaz parfaits = 8,314 J mol – 1 K – 1
|
|
T
|
=
|
température thermodynamique exprimée en K
|
La température d'ébullition est établie en tenant compte de la pression ambiante au moment de la mesure.
Conversions
Pression (unité: kPa)
|
100 kPa
|
=
|
1 bar = 0,1 MPa
(l'utilisation du «bar» est toujours permise mais non recommandée);
|
|
133 Pa
|
=
|
1 mm Hg = 1 Torr
(l'utilisation des unités «mm Hg» et «Torr» n'est pas permise);
|
|
1 atm
|
=
|
atmosphère standard = 101,325 Pa
(l'utilisation de l'unité «atm» n'est pas permise).
|
Température (unité: K)
t = T - 273,15
|
t
|
:
|
température Celsius, exprimée en degré Celsius ( oC)
|
|
T
|
:
|
température thermodynamique, exprimée en kelvin (K)
|
1.3. SUBSTANCES DE RÉFÉRENCE
II n'est pas nécessaire d'utiliser des substances de référence dans tous les cas où l'on étudie une nouvelle substance. Ces substances de référence doivent servir en premier lieu à vérifier la fiabilité de la méthode de temps à autre et à permettre la comparaison avec les résultats obtenus à l'aide d'autres méthodes.
Certaines substances d'étalonnage figurent dans les méthodes énumérées dans l'annexe.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Cinq méthodes de détermination de la température d'ébullition (intervalle d'ébullition) sont fondées sur la mesure de la température d'ébullition, deux autres sont fondées sur une analyse thermique.
1.4.1. Méthode de l'ébulliomètre
Bien que les ébulliomètres aient été à l'origine mis au point pour la détermination du poids moléculaire par élévation de la température d'ébullition, ils se prêtent également aux mesures exactes de la température d'ébullition. Un appareil très simple est décrit dans la norme ASTM D 1120-72 (voir annexe). Dans cet appareil, le liquide est chauffé jusqu'à ébullition à la pression atmosphérique (conditions d'équilibre).
1.4.2. Méthode dynamique
Cette méthode comporte la mesure de la température de recondensation de la vapeur à l'aide d'un thermomètre approprié placé dans le reflux pendant l'ébullition. La pression peut être modifiée dans cette méthode.
1.4.3. Méthode par distillation à la température d'ébullition
Cette méthode comporte la distillation du liquide, la mesure de la température de recondensation de la vapeur et la détermination de la quantité de distillat.
1.4.4. Méthode selon Siwoloboff
Un échantillon est chauffé dans un tube à essai, qui est immergé dans le liquide contenu dans un bain chauffant. Un capillaire scellé, contenant une bulle d'air dans sa partie inférieure, est plongé dans le tube à essai.
1.4.5. Détection photoélectrique
Suivant le principe de Siwoloboff, l'ascension des bulles permet une mesure photoélectrique automatique.
1.4.6. Analyse thermique différentielle
Cette technique permet d'enregistrer la différence de température entre la substance et une substance de référence, en fonction de la température, pendant que la substance et la substance de référence sont soumises au même programme de variation contrôlée de la température. Lorsque l'échantillon subit une transition impliquant un changement d'enthalpie, celui-ci est indiqué par un mouvement endothermique (ébullition) sur la ligne de base de l'enregistrement de la température.
1.4.7. Analyse calorimétrique différentielle
Cette technique permet d'enregistrer la différence entre l'énergie absorbée par une substance et par un produit de référence, en fonction de la température, alors qu'ils sont soumis au même programme de variation contrôlée de la température. Cette énergie correspond à l'énergie nécessaire pour annuler la différence de température entre la substance et le matériel de référence. Lorsque l'échantillon subit une transition impliquant un changement d'enthalpie, celui-ci est indiqué par un mouvement endothermique (ébullition) sur la ligne de base de l'enregistrement du flux de chaleur.
1.5. CRITÈRES DE QUALITÉ
L'applicabilité et la précision des différentes méthodes utilisées pour déterminer la température d'ébullition/intervalle d'ébullition sont indiquées dans le tableau 1.
Tableau 1
Comparaison des méthodes
|
Méthode de mesure
|
Précision estimée
|
Norme existante
|
|
Ébulliomètre
|
±1,4 K (jusqu'à 373 K) (5)
(6)
±2,5 K (jusqu'à 600 K) (5)
(6)
|
ASTM D 1120-72 (5)
|
|
Méthode dynamique
|
±0,5 K (jusqu'à 600 K) (6)
|
|
|
Méthode de distillation (intervalle d'ébullition)
|
±0,5 K (jusqu'à 600 K)
|
ISO/R918, DIN 53171, BS 4591/71
|
|
Méthode selon Siwoloboff
|
± 2 K (jusqu'à 600 K) (6)
|
|
|
Détection photoélectrique
|
±0,3 K (à 373 K) (6)
|
|
|
Calorimétrie
|
±0,5 K (jusqu'à 600 K)
±2,0 K (jusqu'à 1 273 K)
|
ASTM E 537-76
|
|
Analyse calorimétrique différentielle
|
±0,5 K (jusqu'à 600 K)
±2,0 K (jusqu'à 1 273 K)
|
ASTM E 537-76
|
1.6. DESCRIPTION DES MÉTHODES
Les procédures de certaines méthodes d'essai ont été décrites dans des normes internationales et nationales (voir annexe).
1.6.1. Ébulliomètre
Voir annexe.
1.6.2. Méthode dynamique
Voir méthode d'essai A.4 pour la détermination de la pression de vapeur.
La température d'ébullition est enregistrée à une pression de 101,325 kPa.
1.6.3. Méthode de distillation (intervalle d'ébullition)
Voir annexe.
1.6.4. Méthode selon Siwoloboff
L'échantillon est introduit dans un tube à essai ayant environ 5 mm de diamètre et chauffé dans un appareil servant à la détermination de la température de fusion (figure 1).
La figure 1 donne un exemple d'appareil normalisé servant à déterminer la température de fusion et la température d'ébullition (JIS K 0064) (l'appareil est en verre et toutes les spécifications sont données en millimètres).
Figure 1
Un tube capillaire (capillaire d'ébullition) scellé à environ 1 cm au-dessus de son extrémité inférieure est plongé dans le tube à essai contenant la substance à étudier. La partie scellée du capillaire doit se trouver au-dessous de la surface du liquide. Le tube à essai contenant le capillaire doit être attaché au thermomètre avec un élastique ou fixé au moyen d'un support latéral (voir figure 2).
|
Figure 2
Méthode selon Siwoloboff
|
Figure 3
Méthode modifiée
|
|

|

|
Le liquide du bain est choisi en fonction de la température d'ébullition. Pour des températures allant jusqu'à 573 K, on peut utiliser de l'huile de silicone. La paraffine liquide ne convient que pour des températures inférieures à 473 K. Au départ, le chauffage du bain doit être réglé de façon à obtenir un accroissement de température de 3 K/min. Le liquide du bain doit être agité. À environ 10 K au-dessous de la température d'ébullition présumée, le chauffage est réduit de telle sorte que l'accroissement de température ne dépasse pas 1 K par minute. À l'approche de la température d'ébullition, des bulles commencent à s'échapper rapidement du capillaire à l'ébullition.
La température d'ébullition est déterminée par la température à laquelle, lors d'un refroidissement momentané, le chapelet de bulles s'interrompt et le liquide s'élève soudain dans le capillaire. La température relevée à ce moment précis correspond à la température d'ébullition de la substance.
Dans la méthode modifiée (figure 3), la température d'ébullition est déterminée dans un capillaire à température de fusion. L'extrémité de ce dernier est étirée en une fine pointe d'environ 2 centimètres de longueur (a) et une petite quantité de la substance à tester est aspirée à l'intérieur. La pointe est alors scellée en emprisonnant une petite bulle d'air. Lors du chauffage dans l'appareil servant à déterminer la température de fusion (b), la bulle d'air se dilate. La température d'ébullition correspond à la température à laquelle l'échantillon de la substance atteint le niveau de la surface du bain de liquide (c).
1.6.5. Détection photoélectrique
Un échantillon de la substance à tester est chauffé dans un tube capillaire placé à l'intérieur d'un bloc métallique chauffant.
Un faisceau de lumière est envoyé, par les ouvertures aménagées dans le bloc, à travers la substance vers une cellule photoélectrique étalonnée de façon précise.
Durant l'élévation de la température de l'échantillon, quelques bulles d'air s'échappent du capillaire d'ébullition. Lorsque la température d'ébullition est atteinte, le nombre de bulles augmente très fortement. La modification de l'intensité lumineuse qui s'ensuit est enregistrée par la cellule, qui envoie un signal d'arrêt à l'indicateur de température (thermomètre à résistance de platine placé dans le bloc).
Cette méthode est particulièrement utile parce qu'elle permet d'effectuer des déterminations au-dessous de la température ambiante jusqu'à 253,15 K (– 20 oC) sans aucune modification de l'appareil. L'instrument doit simplement être placé dans un bain réfrigérant.
1.6.6. Analyse thermique
1.6.6.1. Analyse thermique différentielle
Voir annexe.
1.6.6.2. Analyse calorimétrique différentielle
Voir annexe.
2. DONNÉES
Pour de faibles écarts par rapport à la pression normale (maximum ±5 kPa), les températures d'ébullition peuvent être ramenées à Tn par l'équation de Sidney Young:
Tn = T + (fT × Δ p)
où:
|
Δ p
|
=
|
(101,325 - p) [attention au signe]
|
|
p
|
=
|
pression barométrique en kPa
|
|
fT
|
=
|
vitesse de variation de la température d'ébullition avec la pression, en K/kPa
|
|
T
|
=
|
valeur mesurée de la température d'ébullition, en K
|
|
Tn
|
=
|
valeur corrigée de la température d'ébullition, en K, à la pression normale.
|
Les facteurs de correction de la température (fT) et les équations de calcul des approximations figurent dans les normes internationales et nationales citées dans le texte (valable pour de nombreuses substances).
Par exemple, la méthode DIN 53171 donne les corrections pour les solvants contenus dans les peintures:
Tableau 2
Facteurs de correction de la température (fT)
|
Température T (K)
|
Facteur de correction fT (K/kPa)
|
|
323,15
|
0,26
|
|
348,15
|
0,28
|
|
373,15
|
0,31
|
|
398,15
|
0,33
|
|
423,15
|
0,35
|
|
448,15
|
0,37
|
|
473,15
|
0,39
|
|
498,15
|
0,41
|
|
523,15
|
0,44
|
|
548,15
|
0,45
|
|
573,15
|
0,47
|
3. RÉSULTATS
Le procès-verbal d'essais contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés) et, éventuellement, l'étape de purification préliminaire,
|
|
—
|
une estimation de la précision.
|
La température d'ébullition indiquée dans le procès-verbal est la moyenne d'au moins deux mesures qui se situent à l'intérieur des limites de précision estimées (tableau 1).
Les valeurs mesurées des températures d'ébullition ainsi que leurs moyennes doivent être indiquées et la (les) pression(s) à laquelle (auxquelles) ont été effectuées les mesures doit (doivent) être notée(s) en kPa. La pression doit de préférence être proche de la pression atmosphérique normale.
Toutes les informations et observations pertinentes pour l'interprétation des résultats doivent être fournies, notamment en ce qui concerne les impuretés et l'état physique de la substance.
4. RÉFÉRENCES
|
(1)
|
OCDE, Paris, 1981, Ligne directrice no 103, décision du Conseil C(81) 30 final.
|
|
(2)
|
UICPA, B. Le Neindre, B. Vodar, eds. Expérimenta! thermodynamics, Buttenworths, London, 1975, vol. II.
|
|
(3)
|
R. Weissberger éd.: Technique of organic chemistry, Physical Methods of Organic Chemistry, 3rd ed., Interscience Publ., New York, 1959, vol. 1, Part I, Chapter VIII.
|
Annexe
Pour plus de détails techniques, on peut, par exemple, consulter les normes suivantes:
1. Ébulliomètre
|
ASTM D 1120-72
|
Standard test method for boiling point of engine anti-freezes
|
2. Méthode de distillation (intervalle d'ébullition)
|
ISO/R 918
|
Test Method for Distillation (Distillation Yield and Distillation Range)
|
|
BS 4349/68
|
Method for determination of distillation of petroleum products
|
|
BS 4591/71
|
Method for the determination of distillation characteristics
|
|
DIN 53171
|
Losungsmittel fur Anstrichstoffe, Bestimmung des Siedeverlaufes
|
|
NF T 20-608
|
Distillation: détermination du rendement et de l'intervalle de distillation
|
3. Analyse thermique différentielle et analyse calorimétrique différentielle
|
ASTM E 537-76
|
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis
|
|
ASTM E 473-85
|
Standard definitions of terms relating to thermal analysis
|
|
ASTM E 472-86
|
Standard practice for reporting thermoanalytical data
|
|
DIN 51005
|
Thermische Analyse, Begriffe
|
A.3. DENSITÉ RELATIVE
1. MÉTHODE
Les méthodes décrites se fondent sur les lignes directrices de l'OCDE (1). Les principes fondamentaux sont décrits dans la référence (2).
1.1. INTRODUCTION
Les méthodes de détermination de la densité relative décrites s'appliquent aux substances solides et liquides, quel que soit leur degré de pureté. Les diverses méthodes à appliquer sont énumérées dans le tableau 1.
1.2. DÉFINITIONS ET UNITÉS
La densité relative D20
4 des solides ou des liquides est le rapport entre la masse d'un volume de substance à tester, déterminée à 20 oC, et la masse du même volume d'eau, déterminée à 4 oC. La densité relative est un nombre sans dimension.
La masse volumique, P, d'une substance est le quotient de la masse, m, par son volume, v.
En unités SI, la masse volumique, P, est exprimée en kg/m3.
1.3. SUBSTANCES DE RÉFÉRENCE (1) (3)
Il n'est pas nécessaire d'employer des substances de référence dans tous les cas où l'on étudie une nouvelle substance. Elles devraient servir en premier lieu à vérifier de temps à autre la fiabilité de la méthode et à permettre les comparaisons avec les résultats obtenus avec d'autres méthodes.
1.4. PRINCIPE DES MÉTHODES
Quatre types de méthodes sont utilisés.
1.4.1. Méthodes de flottabilité
1.4.1.1 Aréomètre (pour les liquides)
On peut obtenir des mesures de densité suffisamment précises et rapides à l'aide d'aréomètres flottants qui permettent de déduire la densité d'un liquide à partir de la profondeur d'immersion repérée sur une échelle graduée.
1.4.1.2 Balance hydrostatique (pour les liquides et les solides)
La différence entre le poids d'un échantillon mesuré dans l'air et dans un liquide approprié (par exemple l'eau) peut servir à déterminer sa densité.
Dans le cas des solides, la densité mesurée n'est représentative que de l'échantillon utilisé. Pour déterminer la densité d'un liquide, on pèse un corps d'un volume v tout d'abord dans l'air, puis dans le liquide.
1.4.1.3 Méthode du corps immergé (pour les liquides) (4)
Dans cette méthode, la densité d'un liquide est déterminée à partir de la différence entre les résultats de la pesée du liquide avant et après immersion d'un corps de volume connu dans le liquide à étudier.
1.4.2. Méthodes pycnométriques
Pour les solides ou les liquides, on peut utiliser des pycnomètres de forme variée dont les volumes sont connus. La densité est calculée à partir de la différence de poids entre le pycnomètre plein et le pycnomètre vide, d'une part, et de son volume connu, d'autre part.
1.4.3. Pycnomètre de comparaison à air (pour les solides)
La densité d'un solide de forme quelconque peut être mesurée, à la température ambiante, à l'aide d'un pycnomètre de comparaison à gaz. Le volume d'une substance dans l'air ou dans un gaz inerte est mesuré dans une éprouvette graduée de volume variable. Pour le calcul de la densité, une mesure de masse est effectuée après la mesure du volume.
1.4.4. Densimètre oscillant (5) (6) (7)
La densité d'un liquide peut être mesurée à l'aide d'un densimètre oscillant. Un oscillateur mécanique, ayant la forme d'un U, vibre à une fréquence de résonance spécifique qui dépend de sa masse. L'introduction d'un échantillon modifie la fréquence de résonance de l'oscillateur. L'appareil doit être étalonné à l'aide de deux substances liquides de densités connues, choisies de préférence de façon à ce que leurs densités couvrent l'intervalle de mesure.
1.5. CRITÈRES DE QUALITÉ
L'applicabilité des différentes méthodes utilisées pour la détermination de la densité relative est représentée au tableau.
1.6. DESCRIPTION DES MÉTHODES
Les références des normes citées en exemple, qui peuvent être consultées pour obtenir des détails techniques supplémentaires, sont jointes à l'annexe.
Les mesures doivent être réalisées à 20 oC, et au moins en double.
2. DONNÉES
Voir normes.
3. RÉSULTATS
Le procès-verbal contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés) et, éventuellement, l'étape de purification préliminaire.
|
La densité relative 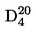 doit être indiquée selon la définition figurant au point 1.2 en même temps que l'état physique de la substance examinée.
doit être indiquée selon la définition figurant au point 1.2 en même temps que l'état physique de la substance examinée.
Toutes les informations et observations pertinentes pour l'interprétation des résultats doivent être mentionnées, en particulier en ce qui concerne les impuretés et l'état physique de la substance.
Tableau
Applicabilité des méthodes
|
Méthode de mesure
|
Densité
|
Viscosité dynamique maximale possible
|
Normes existantes
|
|
solide
|
liquide
|
|
|
|
oui
|
5 Pa s
|
ISO 387,
ISO 649-2,
NF T 20-050
|
|
1.4.1.2.
|
Balance hydrostatique
|
|
|
|
|
|
|
|
oui
|
|
|
ISO 1183 (A)
|
|
|
|
oui
|
5 Pa s
|
ISO 901 et 758
|
|
1.4.1.3.
|
Méthode du corps immergé
|
|
|
oui
|
20 Pa s
|
DIN 53217
|
|
|
|
|
|
ISO 3507
|
|
|
oui
|
|
|
ISO 1183 (B),
NF T 20-053
|
|
|
|
oui
|
500 Pa s
|
ISO 758
|
|
1.4.3.
|
Pycnomètre de comparaison à air
|
|
oui
|
|
|
DIN 55990 partie 3,
DIN 53243
|
|
1.4.4.
|
Densimètre oscillant
|
|
|
oui
|
5 Pa s
|
|
4. RÉFÉRENCES
|
(1)
|
OCDE, Paris, 1981, Ligne directrice no 109, décision du Conseil C(81) 30 final.
|
|
(2)
|
R. Weissberger éd., Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed, Chapter IV, Interscience Publ., New York, 1959, vol. I, Part 1.
|
|
(3)
|
IUPAC, Recommended reference materials for realisation of physico-chemical properties, Pure and applied chemistry, 1976, vol. 48, 508.
|
|
(4)
|
Wagenbreth, H., Die Tauchkugel zur Bestimmung der Dichte von Flüssigkeiten, Technisches Messen tm, 1979, vol. 11, 427-430.
|
|
(5)
|
Leopold, H., Die digitale Messung von Flüssigkeiten, Elektronik, 1970, vol. 19, 297-302.
|
|
(6)
|
Baumgarten, D., Füllmengenkontrolle bei vorgepackten Erzeugnissen — Verfahren zur Dichtebestimmung bei flüssigen Produkten und ihre praktische Anwendung — Die Pharmazeutische Industrie, 1975, vol. 37, 717-726.
|
|
(7)
|
Riemann, J., Der Einsatz der digitalen Dichtemessung im Brauereilaboratorium, Brauwissenschaft, 1976, vol. 9, 253-255.
|
Annexe
Pour plus de détails techniques, on peut consulter, par exemple, les normes suivantes.
1. Méthodes de flottabilité
1.1. Aréomètre
|
DIN 12790, ISO 387
|
Hydrometer; general instructions
|
|
DIN 12791
|
Part I: Density hydrometers; construction, adjustment and use
Part II: Density hydrometers; standardized sizes, designation
Part III: Use and test
|
|
ISO 649-2
|
Laboratory glassware: Density hydrometers for general purpose
|
|
NF T 20-050
|
Produits chimiques à usage industriel — détermination de la densité des liquides — méthode aréométrique
|
|
DIN 12793
|
Laboratory glassware: range find hydrometers
|
1.2. Balance hydrostatique
Pour les substances solides
|
ISO 1183
|
Method A: Methods for determining the density and relative density of plastics excluding cellular plastics
|
|
NF T 20-049
|
Produits chimiques à usage industriel — détermination de la densité des solides autres que les poudres et les produits alvéolaires — méthode de la balance hydrostatique
|
|
ASTM-D-792
|
Specific gravity and density of plastics by displacement
|
|
DIN 53479
|
Testing of plastics and elastomers; determination of density
|
Pour les substances liquides
|
ISO 901
|
ISO 758
|
|
DIN 51757
|
Testing of mineral oil and related materials; determination of density
|
|
ASTM D 941-55, ASTM D 1296-67 et ASTM D 1481-62
|
|
ASTM D 1298
|
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method
|
|
BS 4714
|
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method
|
1.3. Méthode du corps immergé
|
DIN 53217
|
Testing of paints, varnishes and similar coating materials; determination of density; immersed body method
|
2. Méthodes pycnométriques
2.1. Pour les substances liquides
|
ISO 3507
|
Pycnometers
|
|
ISO 758
|
Liquid chemical products; determination of density at 20 oC
|
|
DIN 12797
|
Gay-Lussac pycnometer (for non-volatile liquids which are not too viscous)
|
|
DIN 12798
|
Lipkin pycnometer (for liquids with a kinematic viscosity of less than l00,10 – 6 m2 s – 1 at 15 °C
|
|
DIN 12800
|
Sprengel pycnometer (for liquids as DIN 12798)
|
|
DIN 12801
|
Reischauer pycnometer (for liquids with a kinematic viscosity of less than 100,10 – 6 m2 s – 1 at 20 °C, applicable in particular also to hydrocarbons and aqueous solutions as well as to liquids with higher vapour pressure, approxi-mately 1 bar at 90 oC)
|
|
DIN 12806
|
Hubbard pycnoter (for viscous liquids of all types which do not have a too high vapour pressure, in particular also for paints, varnishes and bitumen)
|
|
DIN 12807
|
Bingham pycnometer (for liquids, as in DIN 12801)
|
|
DIN 12808
|
Jaulmes pycnometer (in particular for ethanol-water mixture)
|
|
DIN 12809
|
Pycnometer with ground-in thermometer and capillary side tube (for liquids which are not too viscous)
|
|
DIN 53217
|
Testing of paints, varnishes and similar products; détermination of density by pycnometer
|
|
DIN 51757
|
Point 7: testing of mineral oils and related materials; determination of density
|
|
ASTM D 297
|
Section 15: Rubber products -chemical analysis
|
|
ASTM D 2111
|
(Method C: Halogenated organic compounds)
|
|
BS 4699
|
Method for determination of specific gravity and density of petroleum products (graduated bicapillary pycnometer method)
|
|
BS 5903
|
Method for determination of relative density and density of petroleum products by the capillary-stoppered pycnometer method
|
|
NF T 20-053
|
Produits chimiques à usage industriel — détermination de la densité des solides en poudre et des liquides — méthode pycnométrique
|
2.2. Pour les substances solides
|
ISO 1183
|
Method B: Methods for determining the density and relative density of plastics excluding cellular plastics
|
|
NF T 20-053
|
Produits chimiques à usage industriel — détermination de la densité des solides en poudre et des liquides — méthode pycnométrique
|
|
DIN 19683
|
Determination of the density of soils
|
3. Pycnomètre de comparaison à air
|
DIN 55990
|
Part 3: Prüfung von Anstrichstoffen und ähnlichen Beschichtungsstoffen; Pulverlack; Bestimmung der Dichte
|
|
DIN 53243
|
Anstrichstoffe; Chlorhaltige polymere; Prüfung
|
A.4. PRESSION DE VAPEUR
1. MÉTHODES
La plupart des méthodes décrites se fondent sur les lignes directrices de l'OCDE (1). Les principes fondamentaux sont énoncés dans les références (2) et (3).
1.1. INTRODUCTION
Il est souhaitable de disposer d'informations préliminaires concernant la structure, la température de fusion et la température d'ébullition de la substance avant de procéder à cet essai.
Il n'existe aucune méthode de mesure qui soit applicable à toute la gamme des pressions de vapeur. C'est pourquoi plusieurs méthodes sont recommandées pour la mesure des pressions de vapeur allant de < 10 – 4 Pa à 105 Pa.
D'une manière générale, les impuretés ont sur la pression de vapeur une incidence dont l'importance dépend en grande partie de la nature de l'impureté.
Si l'échantillon contient des impuretés volatiles qui peuvent avoir une influence sur le résultat, il convient de purifier la substance. Il peut également être nécessaire d'indiquer la pression de vapeur pour le produit technique.
Certaines méthodes décrites ici utilisent des appareils contenant des parties métalliques; ceci doit être pris en considération pour les essais concernant des substances corrosives.
1.2. DÉFINITIONS ET UNITÉS
La pression de vapeur d'une substance est la pression de saturation au-dessus d'une substance solide ou liquide. À l'équilibre thermodynamique, la pression de vapeur d'une substance pure est uniquement fonction de la température.
L'unité SI de pression à utiliser est le pascal (Pa).
Ci-après les unités couramment employées autrefois et leurs facteurs de conversion:
|
1 torr (– 1 mm Hg)
|
= 1,333 × l02 Pa
|
|
1 atmosphère
|
= 1,013 × 105 Pa
|
|
1 bar
|
= 105 Pa
|
L'unité SI de température est le kelvin (K).
La constante molaire des gaz parfaits R est égale à 8,314 J mol – 1 K – 1
La dépendance de la pression de vapeur vis-à-vis de la température est décrite par l'équation de Clausius-Clapeyron:

où:
|
p
|
=
|
pression de vapeur de la substance, exprimée en pascal
|
|
Δ Hv
|
=
|
chaleur de vaporisation, exprimée en J mol – 1
|
|
R
|
=
|
constante molaire des gaz parfaits en J mol – 1K – 1
|
|
T
|
=
|
température thermodynamique, exprimée en kelvin.
|
1.3. SUBSTANCES DE RÉFÉRENCE
Il n'est pas nécessaire d'employer des substances de référence dans tous les cas où l'on étudie une nouvelle substance. Elles doivent servir en premier lieu à vérifier de temps à autre la fiabilité de la méthode et à permettre la comparaison avec les résultats obtenus avec d'autres méthodes.
1.4. PRINCIPE DES MÉTHODES
Le présent paragraphe propose sept méthodes de détermination de la pression de vapeur qui sont applicables à différentes gammes de pression de vapeur. Dans le cadre de chaque méthode, la pression de vapeur est déterminée à différentes températures. Dans une gamme de températures limitée, le logarithme de la pression de vapeur d'une substance pure est une fonction linéaire de l'inverse de la température.
1.4.1. Méthode dynamique
La méthode dynamique est fondée sur la mesure de la température d'ébullition qui correspond à une pression spécifiée.
Gamme recommandée:
de 103 à 105 Pa.
Cette méthode a aussi été recommandée pour la mesure de la température d'ébullition normale lorsque celle-ci ne dépasse pas 600 K.
1.4.2. Méthode statique
Dans le présent procédé, la pression de vapeur s'établissant dans un système fermé à l'équilibre thermodynamique est déterminée à une température spécifiée. Cette méthode est applicable aux solides et aux liquides qui contiennent un ou plusieurs composants.
Gamme recommandée:
de 10 à 105 Pa.
Cette méthode peut aussi être utilisée dans une gamme allant de 1 à 10 Pa, à condition de prendre des précautions.
1.4.3. Isoténiscope
Il s'agit d'une méthode normalisée qui est aussi un procédé statique. Toutefois, elle n'est pas applicable aux systèmes à plusieurs composants. La norme ASTM D-2879-86 contient des informations supplémentaires.
Gamme recommandée:
de 100 à 105 Pa.
1.4.4. Méthode d'effusion: balance de pression de vapeur
La quantité de substance quittant une cellule par unité de temps, par une ouverture de taille connue, est déterminée dans des conditions de vide telles que le retour de la substance dans la cellule soit négligeable (par exemple, par mesure de l'impulsion imprimée à une balance sensible par un jet de vapeur ou encore par la mesure de la perte de poids de la cellule).
Gamme recommandée:
10 – 3 à 1 Pa.
1.4.5. Méthode d'effusion: par perte de poids ou par collecte du vaporisat
Cette méthode repose sur l'estimation de la masse de la substance à tester qui s'écoule, par unité de temps, d'une cellule de Knudsen (4) sous forme de vapeur, au travers d'un capillaire et dans des conditions de vide poussé. La masse de la vapeur formée peut être mesurée, soit en déterminant la perte de masse de la cellule, soit par condensation de la vapeur à basse température; la quantité de substance volatilisée peut alors être déterminée à l'aide d'une analyse chromatographique. La pression de vapeur est calculée en appliquant l'équation d'Hertz-Knudsen.
Gamme recommandée:
de 10 – 3 à 1 Pa.
1.4.6. Méthode de saturation des gaz
Un courant de gaz porteur inerte est envoyé à travers la substance de telle façon qu'il en ressorte saturé de vapeur. La quantité de matière transportée par un volume connu de gaz porteur peut être mesurée après avoir été recueillie dans un collecteur approprié ou estimée à l'aide d'une technique analytique directe. Le résultat obtenu permet de calculer la pression de vapeur à une température donnée.
Gamme recommandée :
de 10 – 4 à 1 Pa.
Cette méthode est aussi applicable à une gamme de 1 à 10 Pa, à condition de prendre des précautions.
1.4.7. Méthode du rotor
Dans la jauge à rotor, l'élément de mesure réel est une petite bille d'acier en suspension dans un champ magnétique qui est soumise à une rotation à grande vitesse. La pression du gaz est déduite du ralentissement de la bille d'acier dû à la pression.
Gamme recommandée:
de 10 – 4 à 0,5 Pa.
1.5. CRITÈRES DE QUALITÉ
Le tableau suivant présente une comparaison de différentes méthodes de détermination de la pression de vapeur, pour ce qui est du domaine d'application, de la répétabilité, de la reproductibilité, de la gamme de mesure et des possibilités de normalisation.
Tableau
Critère de qualité
|
Méthode de mesure
|
Substance
|
Estimation de la répétabilité (7)
|
Estimation de la reproductibilité (7)
|
Gamme recommandée
|
Norme existante
|
|
solide
|
liquide
|
|
|
fusion basse
|
X
|
jusqu'à 25 %
|
jusqu'à 25 %
|
de 103 Pa à 2 × 103 Pa
|
—
|
|
|
|
|
de 1 à 5 %
|
de 1 à 5 %
|
de 2 × 103 Pa à 105 Pa
|
—
|
|
|
X
|
X
|
de 5 à 10 %
|
de 5 à 10 %
|
de 10 Pa à 105 Pa (8)
|
NFT 20-048 (5)
|
|
|
X
|
X
|
de 5 à 10 %
|
de 5 à 10 %
|
de 102 Pa à 105 Pa
|
ASTM-D
2879-86
|
|
1.4.4.
|
Méthode d'effusion, balance de pression
|
|
X
|
X
|
de 5 à 20 %
|
jusqu'à 50 %
|
de 10 – 3 Pa à 1 Pa
|
NFT 20-047 (6)
|
|
1.4.5.
|
Méthode d'effusion, perte de poids
|
|
X
|
X
|
de 10 à 30 %
|
—
|
de 10 – 3 Pa à 1 Pa
|
—
|
|
1.4.6.
|
Méthode de saturation des gaz
|
|
X
|
X
|
10 à 30 %
|
jusqu'à 50 %
|
de 10 – 4 Pa à 1 Pa (8)
|
—
|
|
1.4.7.
|
Méthode par rotation
|
|
X
|
X
|
de 10 à 20 %
|
—
|
de 10 – 4 Pa à 0,5 Pa
|
—
|
1.6. DESCRIPTION DES MÉTHODES
1.6.1. Mesure dynamique
1.6.1.1. Appareil
L'appareil de mesure comporte un appareil à reflux avec réfrigérant en verre ou en métal (figure 1), un dispositif de mesure de la température et un système de régulation et de mesure de la pression. L'appareil de mesure type représenté sur le schéma est en verre et se compose de cinq parties:
Un tube large partiellement à double paroi, comprenant un joint rodé, un réfrigérant, un récipient de refroidissement et un orifice d'admission.
Un cylindre de verre relié à une pompe Cottrel, qui est monté sur la section du tube où s'effectue l'ébullition et qui possède une surface rugueuse pour éviter les soubresauts lors de l'ébullition.
La température est mesurée à l'aide d'un détecteur approprié (par exemple, un thermomètre à résistance ou un thermocouple à enveloppe) immergé dans l'appareil au point de mesure (no 5, figure 1) par un orifice d'admission approprié (par exemple un joint mâle rodé).
Cet appareil est relié à un dispositif de régulation et de mesure de la pression.
L'ampoule, qui sert de volume tampon, est reliée à l'appareil de mesure par l'intermédiaire d'un tube capillaire.
L'appareil à reflux est chauffé à l'aide d'un élément chauffant (par exemple, une cartouche chauffante) inséré dans le bas de l'appareil en verre. Le courant nécessaire pour le chauffage est établi et contrôlé au moyen d'un thermocouple.
Une pompe à vide permet d'obtenir le vide désiré, entre 102 Pa et 105 Pa environ.
Une soupape appropriée est utilisée pour contrôler l'arrivée d'air ou d'azote afin d'établir la pression souhaitée (entre 102 et 105 Pa environ) et d'assurer la ventilation.
La pression est mesurée à l'aide d'un manomètre.
1.6.1.2. Mesure
On mesure la pression de vapeur en déterminant la température d'ébullition de l'échantillon à plusieurs pressions spécifiées comprises entre 103 et 105 Pa environ. La température d'ébullition est atteinte lorsque la température reste constante à une pression donnée. Cette méthode n'est pas applicable aux substances moussantes.
La substance est placée dans une colonne propre et sèche. Quand les solides ne sont pas sous forme de poudre, le remplissage peut poser des problèmes. Toutefois, il est parfois possible d'y remédier en chauffant la gaine de refroidissement. Après remplissage, l'appareil est scellé au collet et la substance est dégazée. La plus basse des pressions désirées est alors établie et le système de chauffage est mis en marche en même temps que le détecteur de température est connecté à un enregistreur.
L'équilibre est atteint lorsque la température d'ébullition enregistrée est constante pour une pression donnée. Il faut prendre particulièrement soin d'éviter les soubresauts pendant l'ébullition. En outre, il peut se produire une condensation complète sur le réfrigérant. Lors de la détermination de la pression de vapeur de solides dont la température de fusion est basse, il convient de prendre soin d'éviter le bouchage du condenseur.
Après enregistrement de ce point d'équilibre, une pression plus élevée est établie. Le processus se poursuit de la même manière jusqu'à ce qu'une pression de 105 Pa soit atteinte (environ 5 à 10 mesures en tout, qui seront répétées pour vérification à des pressions décroissantes).
1.6.2. Mesure statique
1.6.2.1. Appareil
L'appareil comporte un récipient où placer l'échantillon ainsi qu'un système de chauffage et de refroidissement permettant de contrôler et de mesurer la température de l'échantillon. Il comporte également un dispositif de réglage et de mesure de la pression. Les figures 2a et 2b illustrent les principes fondamentaux de cette méthode.
La cellule où est placé l'échantillon (figure 2a) est reliée d'un côté à un robinet à vide poussé et, de l'autre, à un tube en U rempli d'un liquide manométrique approprié. L'autre extrémité du tube en U se termine par une tubulure dont les branches mènent à la pompe à vide, à un récipient à azote ou à la soupape de ventilation et à un manomètre.
Le tube en U peut être remplacé par une jauge de pression munie d'un indicateur de pression (figure 2b).
Pour amener l'échantillon à la température choisie, le récipient contenant l'échantillon ainsi que le robinet et le tube en U ou la jauge de pression sont plongés dans un bain maintenu à une température constante de ±0,2 K. La température est mesurée au niveau de la paroi extérieure du récipient contenant l'échantillon, ou à l'intérieur de celui-ci.
Une pompe à vide munie d'un piège réfrigérant en amont est utilisée pour faire le vide dans l'appareil.
Dans la méthode 2a, la pression de vapeur de la substance est mesurée indirectement à l'aide d'un indicateur de zéro. Cette méthode tient compte du fait que la densité du fluide dans le tube en U est modifiée par un changement important de la température.
Différents liquides, huiles de silicone et phtalates, peuvent être utilisés comme indicateur de zéro dans le tube en U, selon la gamme de pressions et le comportement chimique de la substance. La substance à tester ne doit pas se dissoudre ou réagir d'une manière notable avec le liquide du tube en U.
Pour le manomètre, le mercure peut être utilisé pour une gamme de pression d'air normale jusqu'à 102 Pa, alors que les huiles de silicone et les phtalates conviennent au-dessous de 102 Pa et jusqu'à 10 Pa. Quant aux manomètres à membrane chauffables, ils peuvent même être utilisés pour des pressions inférieures à 10 – 1 Pa. Il existe d'autres jauges de pression qui peuvent aussi être utilisées au-dessous de 102 Pa.
1.6.2.2. Mesure
Avant la mesure, toutes les parties de l'appareil schématisé sur la figure 2 sont soigneusement nettoyées et séchées.
Pour la méthode 2a, le tube en U est rempli avec le liquide choisi, qui doit être dégazé à une température élevée avant d'effectuer les lectures.
La substance à tester est placée dans l'appareil, qui est alors fermé, et la température est suffisamment abaissée pour permettre le dégazage. La température doit être assez basse pour que l'air soit aspiré, mais sans toutefois modifier la composition du produit dans le cas de systèmes constitués de plusieurs composants. L'équilibre peut, si nécessaire, être établi plus rapidement par agitation.
L'échantillon peut être refroidi plus efficacement, par exemple à l'aide d'azote liquide (attention à la condensation de l'air et du liquide de la pompe) ou d'un mélange d'éthanol et de glace carbonique. Pour les mesures à basse température, il convient d'utiliser un bain thermostaté relié à un cryostat.
Pour faire le vide dans l'appareil, l'air est aspiré pendant plusieurs minutes, le robinet qui se trouve au-dessus du récipient contenant l'échantillon étant ouvert. Le robinet est ensuite fermé et l'échantillon est amené à la plus basse des températures voulues. L'opération de dégazage doit, si nécessaire, être répétée plusieurs fois.
Lorsque l'échantillon est chauffé, la pression de vapeur augmente, ce qui modifie l'équilibre du liquide dans le tube en U. Pour compenser ce changement, on fait entrer de l'azote ou de l'air dans l'appareil en ouvrant le robinet jusqu'à ce que le liquide indicateur de pression soit revenu au zéro. La pression nécessaire peut être lue sur un manomètre de précision à température ambiante. Cette pression correspond à la pression de vapeur de la substance à une température donnée.
La méthode 2b est similaire, mais la pression de vapeur est lue directement.
La dépendance de la pression de vapeur vis-à-vis de la température est déterminée à des intervalles appropriés (environ 5 à 10 mesures en tout à intervalles courts) jusqu'au maximum désiré. Les mesures à basse température doivent être répétées pour vérification.
Si les valeurs obtenues en effectuant des mesures répétées ne coïncident pas avec la courbe obtenue pour des températures croissantes, cela peut être dû à l'une des raisons suivantes:
|
1.
|
L'échantillon contient encore de l'air (par exemple, dans le cas de produits d'une viscosité élevée) ou des substances dont la température d'ébullition est basse, qui ont été libérés pendant le chauffage et peuvent être éliminés par aspiration après une étape de surréfrigération supplémentaire.
|
|
2.
|
La température de refroidissement n'est pas assez basse. Il faut alors utiliser de l'azote liquide comme réfrigérant.
S'il s'avère que le problème est dû à l'une de ces deux raisons, il convient de répéter les mesures.
|
|
3.
|
La substance subit une réaction chimique dans la gamme de température utilisée (par exemple: décomposition, polymérisation).
|
1.6.3. Isoténiscope
Pour une description complète de cette méthode, se référer à la référence 7. Le principe de l'appareil de mesure est décrit dans la figure 3. À l'instar de la méthode statique décrite au point 1.6.2, la méthode à l'isoténiscope est applicable aux solides et aux liquides.
Dans le cas des liquides, la substance elle-même sert de liquide de remplissage dans le manomètre auxiliaire. Une quantité de liquide suffisante pour remplir l'ampoule et la partie courte du manomètre est versée dans l'isoténiscope. L'isoténiscope est ensuite relié à une pompe à vide; une fois le vide fait dans l'appareil, celui-ci est rempli d'azote. L'évacuation et la purge de système sont répétées deux fois afin d'éliminer l'oxygène résiduel. L'isoténiscope rempli est placé en position horizontale de telle sorte que l'échantillon forme une couche fine dans l'ampoule et dans le manomètre (partie en U). La pression du système est réduite à 133 Pa et l'échantillon est chauffé doucement jusqu'au début de l'ébullition afin d'éliminer les gaz dissous fixés. L'isoténiscope est ensuite placé de telle manière que l'échantillon revienne remplir entièrement l'ampoule et la branche courte du manomètre. La pression est maintenue comme pour le dégazage; la pointe de l'ampoule est chauffée à l'aide d'une petite flamme jusqu'à ce que la vapeur provenant de l'échantillon soit suffisante pour déplacer une partie de l'échantillon de la partie supérieure de l'ampoule et de la branche du manomètre vers la partie manomètre de l'isoténiscope, créant ainsi un espace dépourvu d'azote et rempli de vapeur.
L'isoténiscope est ensuite placé dans un bain thermostaté, et la pression d'azote est réglée de manière à être la même que celle de l'échantillon. La balance de pressions est indiquée par la partie manomètre de l'isoténiscope. À l'équilibre, la pression de vapeur de l'azote est égale à celle de la substance.
Dans le cas des solides, le manomètre est rempli avec l'un des liquides énumérés au point 1.6.2.1, choisi en fonction de la gamme de pression et de température. L'ampoule du côté de la partie la plus longue de l'isoténiscope est remplie avec le liquide dégazé du manomètre. Le solide à examiner est ensuite introduit dans l'ampoule et dégazé à haute température. L'isoténiscope est alors incliné de telle sorte que le liquide du manomètre puisse couler dans le tube en U. La pression de vapeur est mesurée en fonction de la température conformément à la description présentée au point 1.6.2.
1.6.4. Méthode d'effusion: balance de pression de vapeur
1.6.4.1. Appareil
Plusieurs modèles d'appareil sont décrits dans la littérature (1). L'appareil représenté à la figure 4 illustre le principe général de la méthode. Il se compose principalement d'une cuve à vide poussé en acier inoxydable ou en verre, d'une pompe avec dispositif de mesure du vide et d'un équipement incorporé permettant de mesurer la pression de vapeur sur une balance. Les équipements suivants sont incorporés à l'appareil:
|
—
|
Un four d'évaporation avec bride et canal d'alimentation rotatif. Il s'agit d'un récipient cylindrique, fabriqué par exemple en cuivre ou en un alliage chimiquement résistant d'une bonne conductivité thermique. Un récipient de verre doublé d'une paroi de cuivre peut également être utilisé. Le four a un diamètre de 3 à 5 cm et une hauteur de 2 à 5 cm, environ; il possède d'un à trois orifices d'évaporation de sections différentes. Le chauffage du four est produit, soit par une plaque chauffante située au-dessous, soit par une spirale chauffante qui l'entoure à l'extérieur. Afin d'éviter une dissipation de la chaleur vers le plateau de base, le système de chauffage est fixé sur ce dernier à l'aide d'un métal de faible conductivité thermique (acier au nickel-argent ou au chrome-nickel); par exemple, si le four utilisé est muni de plusieurs orifices, un tuyau d'acier au nickel-argent est attaché à un canal d'alimentation rotatif. Ce dispositif présente l'avantage de permettre l'introduction d'une barre de cuivre, ce qui permet d'assurer le refroidissement par l'extérieur à l'aide d'un bain réfrigérant.
|
|
—
|
Le couvercle du four en cuivre possédant trois orifices de sections différentes, situés à 90 degrés l'un de l'autre, permet de couvrir plusieurs gammes de pression de vapeur au sein de la gamme de mesures globales (orifices d'un diamètre de 0,30 à 4,50 mm environ). Des ouvertures larges sont utilisées pour les pressions de vapeur basses et vice versa. L'orifice choisi ou une position intermédiaire dans le flux de vapeur (orifice du four — bouclier — plateau de la balance) est sélectionné par rotation du four, ce qui permet de diriger le flux moléculaire sur le plateau de la balance au travers de l'orifice du four, ou de l'en dévier. La température de la substance est mesurée à l'aide d'un thermocouple ou d'un thermomètre à résistance placé à un endroit approprié.
|
|
—
|
Le plateau d'une microbalance ultrasensible est placé sous le bouclier (voir ci-après). Le plateau de la balance a un diamètre de 30 mm environ. L'aluminium, revêtu d'une couche d'or, est un matériau approprié.
|
|
—
|
Le plateau de la balance est entouré par un cylindre en laiton ou une enceinte de réfrigération en cuivre. Certains types de balances disposant d'un orifice pour le fléau de la balance et d'un orifice dans le bouclier permettant le passage du flux moléculaire assurent une condensation complète de la vapeur sur le plateau de la balance. La dissipation de la chaleur vers l'extérieur est assurée par une barre de cuivre connectée à l'enceinte de réfrigération. Cette barre thermiquement isolée au moyen d'un tube d'acier au chrome-nickel, par exemple, traverse le plateau de base. Sous le plateau de base, la barre plonge dans un vase de Dewar contenant de l'azote liquide, ou de l'azote liquide circule au travers de cette barre. L'enceinte de réfrigération est ainsi maintenue à une température d'environ - 120 oC. Le plateau de la balance n'est refroidi que par rayonnement, ce qui suffit pour la gamme de pressions soumises à l'étude (refroidir une heure environ avant le début des mesures).
|
|
—
|
La balance est placée au-dessus de l'enceinte de réfrigération. On peut citer, parmi les balances qui conviennent pour cette méthode, la microbalance électronique ultrasensible à deux bras (8) ou la balance ultrasensible à bobine mobile (voir la ligne directrice 104 de l'OCDE, édition 12 mai 1981).
|
|
—
|
Le plateau de base comporte aussi des connexions électriques pour thermocouples (ou pour thermomètres à résistance) et bobines de chauffage.
|
|
—
|
Le vide est produit dans le récipient à l'aide d'une pompe à vide partiel ou d'une pompe à vide poussé (le vide requis, correspondant à une pression d'environ 1 à 2 × 10 – 3 Pa, est obtenu après deux heures de pompage). La pression est contrôlée à l'aide d'un manomètre à ionisation approprié.
|
1.6.4.2. Mesure
Le récipient est rempli avec la substance à tester et le couvercle est fermé. Le bouclier et l'enceinte de réfrigération sont glissés de l'autre côté du four. L'appareil est fermé et les pompes à vide sont branchées. La pression à obtenir avant le commencement de la mesure est d'environ 10 – 4 Pa. Le refroidissement de l'enceinte de réfrigération commence à 10 – 2 Pa.
Une série d'étalonnage à la plus basse des températures choisies commence dès que le vide nécessaire est atteint. L'orifice approprié dans le couvercle est ouvert, et le jet de vapeur qui traverse le bouclier juste au-dessus de l'orifice vient frapper le plateau réfrigéré de la balance. Ce dernier doit être d'une taille suffisante pour garantir que le jet qui traverse le bouclier l'atteint tout entier. L'impulsion du jet de vapeur exerce une force sur le plateau de la balance, et les molécules se condensent au contact de sa surface froide.
Cette impulsion et la condensation simultanée produisent un signal sur l'enregistreur. L'évaluation des signaux fournit deux types d'informations:
|
1.
|
L'appareil décrit ici permet de déterminer la pression de vapeur directement à partir de la force exercée sur le plateau de la balance [il n'est pas nécessaire pour cela de connaître le poids moléculaire (2)]. Des facteurs géométriques, tels que la section de l'orifice du four et l'angle du flux moléculaire, doivent être pris en considération pour l'évaluation des lectures.
|
|
2.
|
La masse du condensat peut être mesurée en même temps et on peut en déduire la vitesse d'évaporation. La pression de vapeur peut également être calculée à partir de la vitesse d'évaporation et du poids moléculaire à l'aide de l'équation de Hertz (2).
|

où:
|
G
|
=
|
vitesse d'évaporation (kg s – 1 m – 2)
|
|
M
|
=
|
masse molaire (g mol – 1)
|
|
T
|
=
|
température (K)
|
|
R
|
=
|
constante molaire des gaz parfaits (J mol – 1 K – 1)
|
|
p
|
=
|
pression de vapeur (Pa)
|
Lorsque le vide nécessaire est atteint, une série de mesures est entreprise à la plus basse des températures choisies.
Pour les mesures suivantes, la température est augmentée par petits intervalles jusqu'au moment où la plus haute valeur choisie est atteinte. L'échantillon est alors refroidi une nouvelle fois, et une deuxième courbe de pression de vapeur peut être enregistrée. Si les résultats du second enregistrement ne confirment pas ceux du premier, il est possible que la substance se décompose aux températures choisies pour la détermination.
1.6.5. Méthode d'effusion — par perte de poids
1.6.5.1. Appareil
L'appareil utilisé dans la présente méthode comporte principalement les éléments suivants:
|
—
|
une cuve dont la température et la pression peuvent être contrôlées, dans lequel sont placées les cellules à effusion,
|
|
—
|
une pompe à vide poussé (par exemple, une pompe à diffusion ou une pompe turbomoléculaire) accompagnée d'une jauge à vide,
|
|
—
|
un piège contenant de l'azote liquide ou de la glace carbonique.
|
La figure 5 présente un exemple de cuve à vide en aluminium, munie d'un système de chauffage électrique, contenant quatre cellules à effusion en acier inoxydable. La feuille d'acier inoxydable, d'une épaisseur de 0,3 mm et percée d'un orifice à effusion d'un diamètre de 0,2 à 1,0 mm, est fixée à la cellule à effusion à l'aide d'un couvercle fileté.
1.6.5.2. Mesure
La substance de référence et la substance à tester sont versées dans la cellule à effusion. Le diaphragme métallique percé d'un orifice est fixé à l'aide du couvercle fileté, et la cellule est pesée avec une précision de 0,1 mg. La cellule est placée dans l'appareil thermostaté où la pression est alors réglée à une valeur correspondant au dixième de la pression attendue. On laisse entrer de l'air dans l'appareil à des intervalles de temps définis entre 5 et 30 heures, et la perte de masse de la cellule à effusion est déterminée grâce à une nouvelle pesée.
Pour s'assurer que les résultats ne sont pas perturbés par la présence d'impuretés volatiles, la cellule est pesée à intervalles réguliers, ce qui permet de vérifier que le taux d'évaporation est constant, au moins pendant deux intervalles de temps.
La pression de vapeur p dans la cellule d'effusion est calculée au moyen de l'équation suivante:
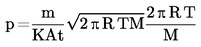
ou:
|
p
|
=
|
pression de vapeur (Pa)
|
|
m
|
=
|
masse de la substance qui quitte la cellule pendant le temps t (kg)
|
|
t
|
=
|
temps (s)
|
|
A
|
=
|
surface de l'orifice (m2)
|
|
K
|
=
|
facteur de correction
|
|
R
|
=
|
constante molaire des gaz parfaits (J mol – 1 K – 1)
|
|
T
|
=
|
température (K)
|
|
M
|
=
|
masse moléculaire (kg mol – 1)
|
Le facteur de correction K dépend du rapport de la longueur sur le rayon de l'orifice cylindrique:
|
rapport
|
0,1
|
0,2
|
0,6
|
1,0
|
2,0
|
|
K
|
0,952
|
0,909
|
0,771
|
0,672
|
0,514
|
On peut écrire l'équation suivante:

où  représente la constante de la cellule à effusion.
représente la constante de la cellule à effusion.
Cette constante E peut être déterminée à l'aide de substances de référence (2,9), en appliquant l'équation suivante:

où
|
p(r)
|
=
|
pression de vapeur de la substance de référence (Pa)
|
|
M(r)
|
=
|
masse moléculaire de la substance de référence (kg × mol – 1)
|
1.6.6. Méthode de saturation des gaz
1.6.6.1. Appareil
L'appareil utilisé dans la présente méthode comporte un certain nombre d'éléments qui sont schématisés sur la figure 6 a et décrits ci-après (1).
Gaz inerte:
Le gaz porteur ne doit pas réagir avec la substance à tester. L'azote convient dans la plupart des cas, mais d'autres gaz peuvent parfois être employés (10). Le gaz choisi doit être sec (voir figure 6 a, numéro 4: sonde d'humidité relative).
Contrôle du flux gazeux:
Un système adéquat de contrôle des gaz est indispensable pour assurer un flux constant et suffisant à travers la colonne de saturation.
Collecteurs de vapeur:
Leur choix dépend des caractéristiques de l'échantillon, ainsi que de la méthode d'analyse utilisée. La vapeur doit être recueillie quantitativement et sous une forme qui permette l'analyse ultérieure. Pour certaines substances, on utilisera des collecteurs contenant des liquides tels que l'hexane ou l'éthylène glycol. Pour d'autres, on recourra à des adsorbants solides.
Au lieu de recueillir la vapeur pour l'analyser ultérieurement, il est possible d'utiliser des techniques analytiques directes, comme la chromatographie, pour déterminer la quantité de matériel transporté par un volume connu de gaz porteur. De surcroît, la perte de masse de l'échantillon peut être mesurée.
Enceinte calorifugée:
Pour les mesures à différentes températures, il peut être nécessaire de prévoir une enceinte calorifugée.
Colonne de saturation:
La substance à tester en solution est déposée sur un support inerte qui est ensuite tassé dans la colonne de saturation. Les dimensions de celle-ci et la vitesse d'écoulement doivent être choisies de façon à assurer la saturation complète du gaz porteur. La colonne doit être thermostatée. Pour les déterminations s'effectuant à des températures supérieures à la température ambiante, l'espace entre la colonne et les collecteurs doit être chauffé pour empêcher la condensation de la substance.
Afin de diminuer la diffusion de la matière, un tube capillaire peut être placé après la colonne de saturation (figure 6 b).
1.6.6.2. Mesure
Préparation de la colonne de saturation:
Après mise en solution dans un solvant très volatil, une partie de la substance d'essai est ajoutée à une quantité donnée du matériau de support. La quantité de substance ajoutée à une quantité adaptée du support doit être suffisante pour maintenir la saturation pendant toute la durée de l'essai. Le solvant est complètement évaporé à l'air ou dans un évaporateur rotatif, et le matériau bien homogénéisé est introduit dans la colonne. Une fois l'échantillon thermostaté, un courant d'azote sec est envoyé à travers l'appareil.
Mesure:
Les collecteurs ou le détecteur direct sont reliés aux tubes de sortie de la colonne et le temps est enregistré. Le débit est vérifié au début de l'expérience et à intervalles réguliers en cours d'expérience, grâce à un débitmètre à bulles (ou encore, en continu, à l'aide d'un débitmètre massique).
Il faut mesurer la pression à la sortie de la colonne de saturation, soit:
|
a)
|
en intercalant une jauge à pression entre le saturateur et les pièges (cette méthode peut se révéler peu satisfaisante en raison de l'accroissement du volume mort et de la surface d'adsorption); soit
|
|
b)
|
en déterminant la perte de charge du système de pièges utilisé, en fonction du débit dans une expérience séparée (cette méthode peut se révéler peu satisfaisante pour les collecteurs à liquide).
|
Le temps qu'il faut pour recueillir la quantité de substance nécessaire aux différentes méthodes d'analyse est déterminé au cours d'essais préliminaires ou par estimation. Au lieu de recueillir la substance pour l'analyser ultérieurement, il est possible d'avoir recours à des techniques analytiques quantitatives directes (comme la chromatographie). Avant de calculer la pression de vapeur à une température donnée, il y a lieu d'effectuer des essais préliminaires pour déterminer le débit maximal qui saturera complètement le gaz porteur avec la vapeur de la substance. Il faut pour cela que le gaz porteur passe dans le saturateur suffisamment lentement pour qu'aucune vitesse inférieure ne fournisse de pression de vapeur calculée plus importante.
Le choix de la méthode d'analyse sera fonction de la nature de la substance à tester (par exemple, chromatographie en phase gazeuse ou gravimétrie).
La quantité de substance transportée par un volume connu de gaz porteur est déterminée.
1.6.6.3. Calcul de la pression de vapeur
La pression de vapeur est calculée à partir de la densité de vapeur (M/V) au moyen de l'équation suivante:

où:
|
p
|
=
|
pression de vapeur (Pa)
|
|
W
|
=
|
masse de la substance évaporée (g)
|
|
V
|
=
|
volume de gaz saturé (m3)
|
|
R
|
=
|
constante molaire des gaz parfaits (J mol – 1 K – 1)
|
|
T
|
=
|
température (K)
|
|
M
|
=
|
masse molaire de la substance à tester (g mol – 1)
|
Les volumes mesurés doivent être corrigés pour tenir compte des différences de pression et de température entre le débitmètre et le saturateur thermostaté. Si le débitmètre est placé en aval du collecteur de vapeur, des corrections peuvent être nécessaires pour tenir compte de l'évaporation éventuelle des produits contenus dans les collecteurs (1).
1.6.7. Méthode du rotor (8, 11, 13)
1.6.7.1. Appareil
La présente méthode peut être réalisée à l'aide d'une jauge à viscosité à rotor représentée à la figure 8. Le montage expérimental est schématisé à la figure 7.
L'appareil de mesure type comporte une tête de mesure à rotor placée dans une enceinte thermostatée (réglée avec une précision de 0,1 oC). Le récipient qui contient l'échantillon est placé dans une enceinte thermostatée (réglée avec une précision de 0,01 oC); tous les autres éléments du montage sont maintenus à une température supérieure afin d'éviter la condensation. Un dispositif de pompe à vide poussé est connecté au système au moyen de vannes appropriées.
La tête de mesure à rotor est constituée d'une bille d'acier (d'un diamètre de 4 à 5 mm) placée dans un tube. La bille est mise en suspension et stabilisée dans un champ magnétique, généralement au moyen d'aimants permanents et de bobines de contrôle.
La bille est mise en mouvement par les champs rotatifs produits par les bobines. La vitesse de rotation de la bille est mesurée grâce aux bobines de lecture qui déterminent sa faible, mais toujours présente, magnétisation latérale.
1.6.7.2. Mesure
Lorsque la bille a atteint une vitesse de rotation donnée v(o), (généralement de 400 révolutions par seconde environ), on coupe l'alimentation, et la décélération, due à la friction par le gaz, commence.
La chute de la vitesse de rotation est mesurée en fonction du temps. La friction due à la suspension magnétique étant négligeable par rapport à la friction du gaz, la pression du gaz est donnée par l'équation suivante:

où:
|

|
=
|
vitesse moyenne des molécules de gaz
|
|
r
|
=
|
rayon de la bille
|
|
ρ
|
=
|
densité massique de la bille
|
|
σ
|
=
|
coefficient de transfert tangentiel du moment (σ = 1 si la bille a une surface sphérique idéale)
|
|
t
|
=
|
temps
|
|
v(t)
|
=
|
vitesse de rotation après un temps t
|
|
v(o)
|
=
|
vitesse de rotation initiale
|
Cette équation peut également s'écrire de la manière suivante:

où tn, et tn - 1, sont les temps requis pour effectuer un nombre donné N de révolutions. Ces intervalles de temps tn et tn - 1 se succèdent et tn > tn - 1.
La vitesse moyenne de la molécule de gaz  est donnée par l'équation suivante:
est donnée par l'équation suivante:

où:
|
T
|
=
|
température
|
|
R
|
=
|
constante molaire des gaz parfaits
|
|
M
|
=
|
masse molaire
|
2. DONNÉES
Quelle que soit la méthode choisie, la pression de vapeur doit être déterminée à deux niveaux de température au moins. Trois niveaux ou plus sont préférables, entre 0 oC et 50 oC, pour vérifier la linéarité de la courbe de la pression de vapeur.
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés) et, s'il y a lieu, l'étape de purification préliminaire,
|
|
—
|
au moins deux valeurs pour la pression de vapeur et la température, de préférence comprise entre 0 et 50 oC,
|
|
—
|
toutes les données brutes,
|
|
—
|
une courbe de log p en fonction de 1/T,
|
|
—
|
une estimation de la pression de vapeur à 20 oC ou à 25 oC.
|
En cas de modification (changement d'état, décomposition):
|
—
|
nature de la modification,
|
|
—
|
température à laquelle la modification survient à la pression atmosphérique,
|
|
—
|
pression de vapeur à 10 oC et à 20 oC au-dessous et au-dessus de la température de transition (sauf en cas de passage de l'état solide à l'état gazeux).
|
Toutes les informations et remarques pertinentes pour l'interprétation des résultats doivent être signalées, en particulier les impuretés et l'état physique de la substance.
4. RÉFÉRENCES
|
(1)
|
OCDE, Paris, 1981, Ligne directrice no 104, décision du Conseil C(81) 30 final.
|
|
(2)
|
Ambrose, D. in B. Le Neindre; B. Vodar, (Eds.): Experimental Thermodynamics, Butterworths, London, 1975, Vol. II.
|
|
(3)
|
R. Weissberger ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed, Chapter IX, Interscience Publ. New York, 1959, Vol. I, Part I.
|
|
(4)
|
Knudsen, M. Ann Phys. Lpz., 1909, vol. 29, 1979; 1911, vol. 34, 593.
|
|
(5)
|
NF T 20-048 AFNOR (septembre 1985). Produits chimiques à usage industriel — Détermination de la pression de vapeur des solides et des liquides dans le domaine 10 – 1 à 105 Pa. Méthode statique.
|
|
(6)
|
NF T 20-047 AFNOR (septembre 1985). Produits chimiques à usage industriel — Détermination de la pression de vapeur des solides et des liquides dans le domaine 10 – 3 à 1 Pa. Méthode de la balance de pression de vapeur.
|
|
(7)
|
ASTM D 2879-86 Standard test method for vapour pressure-temperature relationship and initial decomposition temperature of liquids by isoteniscope.
|
|
(8)
|
G. Messer, P. Röhl, G. Grosse and W. Jitschin. J. Vac Sci.Technol.(A), 1987, vol. 5 (4), 2440.
|
|
(9)
|
Ambrose, D.; Lawrenson, I.J.; Sprake, C.H.S. J. Chem Thermodynamics 1975, vol. 7, 1173.
|
|
(10)
|
B.F. Rordorf. Thermochimica Acta, 1985, vol. 85, 435.
|
|
(11)
|
G. Comsa, J.K. Fremerey anb B. Lindenau. J. Vac. Sci. Technol., 1980, vol. 17 (2), 642.
|
|
(12)
|
G. Reich. J. Vac. Sci. Technol., 1982, vol. 20 (4), 1148.
|
|
(13)
|
J.K. Fremerey. J. Vac Sci. Technol.(A), 1985, vol. 3 (3), 1715.
|
Annexe 1
Méthode d'estimation
INTRODUCTION
Des valeurs calculées de la pression de vapeur peuvent être utilisées pour:
|
—
|
choisir la méthode expérimentale,
|
|
—
|
fournir une estimation ou une valeur limite au cas où la méthode expérimentale ne peut pas être appliquée pour des raisons techniques (notamment lorsque la pression de vapeur est très faible),
|
|
—
|
contribuer à déceler les cas où il est justifié de ne pas effectuer de mesures expérimentales car la pression de vapeur serait < 10 – 5 Pa à la température ambiante.
|
MÉTHODE D'ESTIMATION
La pression de vapeur des liquides et des solides peut être estimée à l'aide de la corrélation de Watson modifiée (a). La seule donnée expérimentale requise est le point d'ébullition normal. Cette méthode est applicable à des pressions comprises entre 105 et 10 – 5 Pa.
Cette méthode est décrite en détail dans «Handbook of Chemical Properry Estimation Methods» (b).
CALCUL
D'après la méthode décrite dans la référence (b), la pression de vapeur est calculée de la manière suivante:
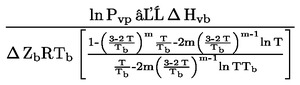
où:
|
T
|
= température choisie
|
|
Tb
|
= point d'ébullition normal
|
|
Pvp
|
= pression de vapeur à la température T
|
|
ΔHvb
|
= température de vaporisation
|
|
ΔZb
|
= facteur de compressibilité (estimé à 0,97)
|
|
m
|
= facteur empirique dépendant de l'état physique à la température choisie.
|
De plus:
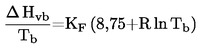
où KF est un facteur empirique qui tient compte de la polarité de la substance. Les facteurs KF pour plusieurs types de composés sont énumérés à la référence (b).
Il est fréquent que l'on dispose de données mentionnant un point d'ébullition à une pression réduite. Dans ce cas, la pression de vapeur est calculée comme suit [conformément à (b)]:

où T1 est le point d'ébullition à la pression réduite P1.
PROCÈS-VERBAL
Lorsque la méthode d'estimation est utilisée, le procès-verbal doit comporter une documentation détaillée concernant le calcul.
RÉFÉRENCES
|
(a)
|
K.M. Watson, Ind. Eng. Chem., 1943, vol. 35, 398.
|
|
(b)
|
W.J. Lyman, W.F. Reehl, D.H. Rosenblatt. Handbook of Chemical Property Estimation Methods, Mc Graw-Hill, 1982.
|
Annexe 2
Figure 1
Appareil de détermination de la courbe de pression de vapeur, suivant la méthode dynamique

Figure 2a
Appareil de détermination de la courbe de pression de vapeur, selon la méthode statique (en utilisant un manomètre à tube en U)

Figure 2b
Appareil de détermination de la courbe de pression de vapeur, suivant la méthode statique (en utilisant un indicateur de pression)
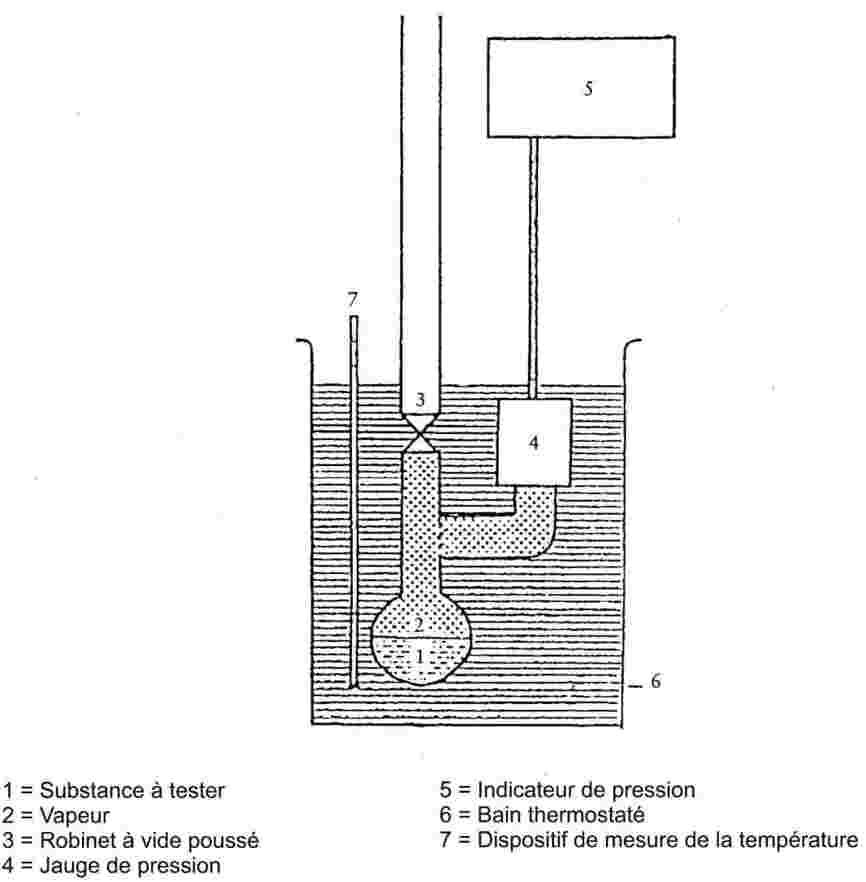
Figure 3
Isoténiscope (voir référence 2)

Figure 4
Appareil de détermination de la courbe de pression de vapeur, suivant la méthode de la balance de pression de vapeur

Figure 5
Modèle d'appareil pour l'évaporation à basse pression grâce à la méthode d'effusion, avec une cellule d'effusion d'un volume de 8 cm3

Figure 6a
Exemple de système de débit permettant de déterminer la pression de vapeur par la méthode de saturation des gaz
Figure 6b
Modèle de système de détermination de la pression de vapeur par la méthode de saturation des gaz avec un capillaire placé après la chambre de saturation
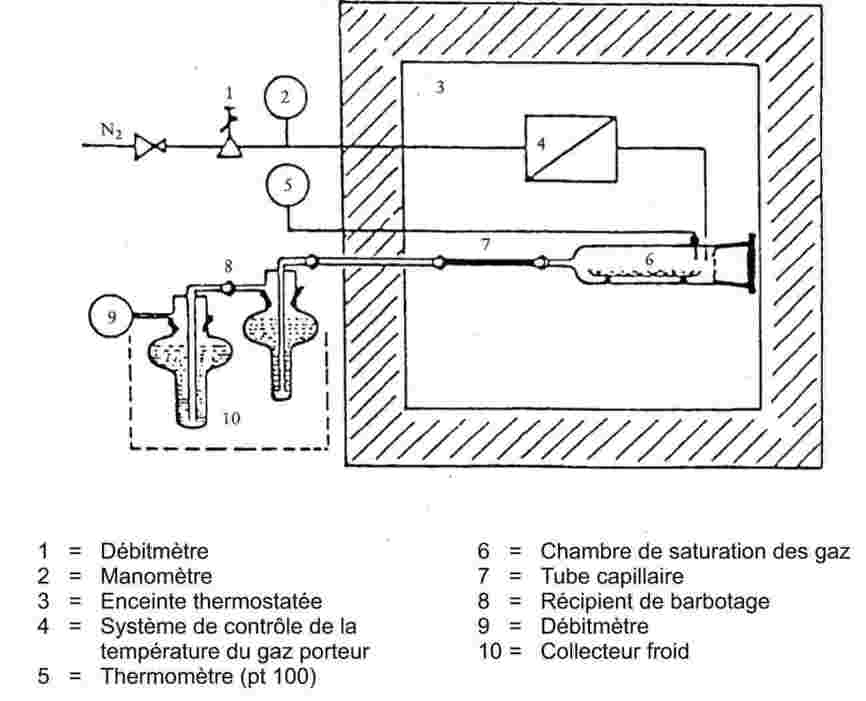
Figure 7
Exemple de dispositif expérimental pour la méthode du rotor
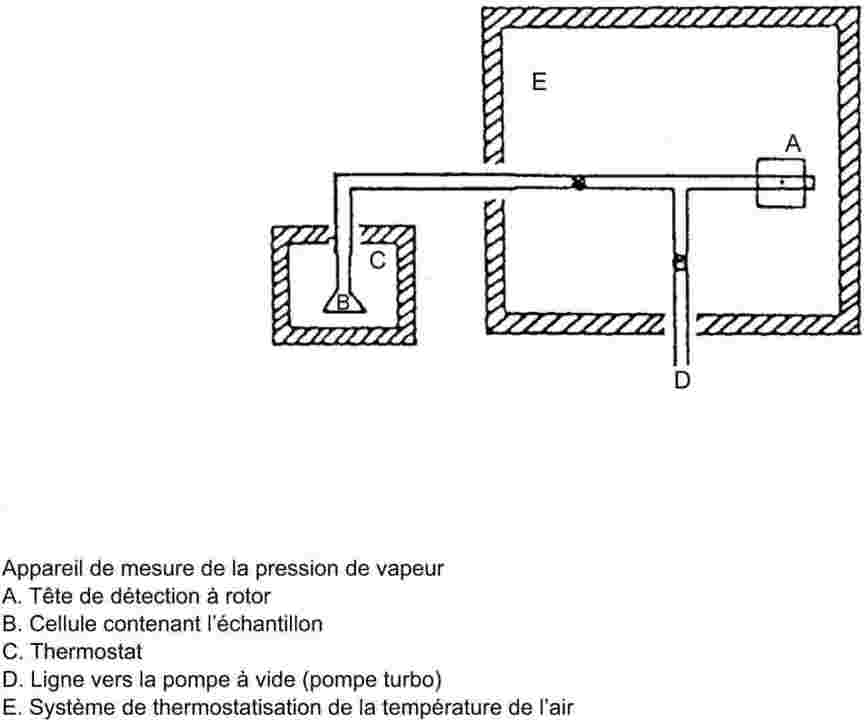
Figure 8
Modèle de tête de mesure à rotor

A.5. TENSION SUPERFICIELLE
1. MÉTHODE
Les méthodes décrites se fondent sur les lignes directrices de l'OCDE (1). Leurs principes fondamentaux sont décrits dans la référence (2).
1.1. INTRODUCTION
Les méthodes décrites s'appliquent à la mesure de la tension superficielle des solutions aqueuses.
Avant d'effectuer ces essais, il sera bon d'avoir des informations préliminaires sur l'hydrosolubilité, la structure, les propriétés de la substance en matière d'hydrolyse et la concentration critique pour la formation de micelles.
Les méthodes suivantes s'appliquent à la plupart des substances chimiques, quel que soit leur degré de pureté.
La mesure de la tension superficielle par la méthode du tensiomètre à anneau est limitée aux solutions aqueuses ayant une viscosité dynamique inférieure à 200 mPa s environ.
1.2. DÉFINITIONS ET UNITÉS
L'enthalpie libre de surface par unité de surface constitue la tension superficielle.
La tension surperficielle s'exprime en:
N/m (en unités SI) ou
mN/M (en sous-unités SI)
1 N/m = 103 dynes/cm
I mN/m = 1 dyne/cm dans le système cgs qui n'est plus utilisé.
1.3. SUBSTANCES DE RÉFÉFENCE
Il n'est pas nécessaire d'employer des substances de référence dans tous les cas où l'on étudie une nouvelle substance. Elles doivent servir en premier lieu à vérifier de temps à autre la fiabilité de la méthode et à permettre de comparer les résultats obtenus avec différentes méthodes.
Des substances de référence couvrant une vaste gamme de tensions superficielles sont données dans les références (1) et (3).
1.4. PRINCIPE DES MÉTHODES
Les méthodes sont fondées sur la mesure de la force maximale qu'il faut exercer verticalement sur un étrier ou un anneau en contact avec la surface du liquide étudié, placé dans un récipient approprié, afin de le séparer de cette surface, ou sur une plaque, dont un des bords est en contact avec la surface, afin d'assurer l'arrachement du film formé.
Les substances hydrosolubles à une concentration supérieure à 1 mg/ml sont testées en solution aqueuse à une seule concentration.
1.5. CRITÈRES DE QUALITÉ
Ces méthodes fournissent une plus grande précision que ce qui est probablement requis pour le contrôle de la protection de l'environnement.
1.6. DESCRIPTION DES MÉTHODES
La substance est dissoute dans de l'eau distillée à une concentration correspondant à 90 % de la saturation de l'hydrosolubilité de cette substance; si cette concentration excède 1 g/l, l'essai est effectué avec une concentration de 1 g/l. Les substances dont l'hydrosolubilité est inférieure à 1 mg/l n'ont pas besoin d'être testées.
1.6.1. Méthode de la plaque
Voir ISO 304 et NF T 73-060 (substances tensioactives — détermination de la tension superficielle par arrachement de films liquides).
1.6.2. Méthode de l'étrier
Voir ISO 304 et NF T 73-060 (substances tensioactives — détermination de la tension superficielle par arrachement de films liquides).
1.6.3. Méthode de l'anneau
Voir ISO 304 et NF T 73-060 (substances tensioactives — détermination de la tension superficielle par arrachement de films liquides).
1.6.4. Méthode de l'anneau harmonisée OCDE
1.6.4.1. Appareil
Des tensiomètres commerciaux se prêtent à cette mesure. Ils comportent les éléments suivants:
|
—
|
un porte-échantillon mobile,
|
|
—
|
un corps de mesure (anneau),
|
|
—
|
un récipient de mesure.
|
1.6.4.1.1. Porte-échantillon mobile
Le porte-échantillon mobile sert de support au récipient de mesure thermostaté, qui contient le liquide à étudier. Il est monté sur le même socle que le dynamomètre.
1.6.4.1.2. Dynamomètre
Le dynamomètre (voir figure) est placé au-dessus du porte-échantillon. L'erreur dans la mesure de la force ne doit pas dépasser ± 10 – 6 N, ce qui équivaut à une erreur limite de ±0,1 mg dans une mesure de masse. Dans la plupart des cas, l'échelle de mesure des tensiomètres vendus dans le commerce est étalonnée en mN/m, si bien que l'on peut lire directement la tension superficielle en mN/m avec une précision de 0,1 mN/m.
1.6.4.1.3. Corps de mesure (anneau)
L'anneau est habituellement constitué par un fil de platine ou de platine-iridium ayant une épaisseur de 0,4 millimètres environ et une circonférence moyenne de 60 millimètres. Cet anneau est suspendu horizontalement à l'étrier de montage qui est relié par une tige métallique au dynamomètre (voir figure).
Figure
Corps de mesure
(toutes les dimensions sont exprimées en millimètres)
1.6.4.1.4. Récipient de mesure
Le récipient de mesure qui contient la solution à tester doit être un récipient en verre thermostaté. Il doit être conçu de telle sorte que, au cours de la mesure, la température du liquide et de la phase gazeuse au-dessus de la surface reste constante et qu'il n'y ait pas d'évaporation. Des récipients cylindriques en verre d'un diamètre intérieur d'au moins 45 millimètres répondent à ces exigences.
1.6.4.2. Préparation de l'appareil
1.6.4.2.1. Nettoyage
La verrerie doit être nettoyée avec soin. Le cas échéant, elle sera lavée au mélange sulfochromique chaud, puis à l'acide phosphorique sirupeux (83 à 98 % en poids de H3PO4), rincée abondamment à l'eau courante et ensuite à l'eau bidistillée jusqu'au moment où l'on obtient une réaction neutre, et, enfin, séchée ou bien rincée avec une partie du liquide à mesurer.
L'anneau sera tout d'abord rincé abondamment à l'eau pour éliminer toutes les traces de substances hydrosolubles, plongé quelques secondes dans le mélange sulfochromique, rincé à l'eau bidistillée jusqu'à ce que l'on obtienne une réaction neutre, et, enfin, rapidement séché au-dessus d'une flamme de méthanol.
Note:
Les traces de substances qui ne sont pas dissoutes ou détruites par l'acide chromosulfurique ou l'acide phosphorique, telles que les silicones, doivent être éliminées à l'aide d'un solvant organique approprié.
1.6.4.2.2. Étalonnage de l'appareil
La validation de l'appareil consiste à vérifier le point zéro et à le régler de telle sorte que l'indication donnée par l'appareil permette une détermination fiable en mN/m.
Montage:
L'appareil sera mis à niveau, par exemple à l'aide d'un niveau à bulle placé sur le socle du tensiomètre, en ajustant des vis de réglage.
Réglage du point zéro:
Après montage de l'anneau sur l'appareil et avant son immersion dans le liquide, l'indicateur du tensiomètre doit être réglé à zéro; on vérifiera le parallélisme de l'anneau en utilisant la surface du liquide comme miroir.
Étalonnage:
L'étalonnage de l'appareil peut être effectué par l'une des deux méthodes suivantes:
|
a)
|
à l'aide d'une masse: ce procédé utilise des cavaliers de masse connue (entre 0,1 et 1,0 g), qui sont placés successivement sur l'anneau. Le facteur d'étalonnage Φa, par lequel il y a lieu de multiplier toutes les lectures de l'instrument, sera déterminé au moyen de l'équation (1):
|
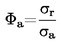
|
(1)
|
où
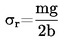 (mN/m) (mN/m)
|
m
|
=
|
masse du cavalier (g)
|
|
g
|
=
|
accélération de la pesanteur (981 cm s – 2 au niveau de la mer)
|
|
b
|
=
|
circonférence moyenne de l'anneau (cm)
|
|
σa
|
=
|
lecture du tensiomètre après placement du cavalier sur l'anneau (mN/m);
|
|
|
b)
|
à l'aide d'eau: ce procédé utilise de l'eau pure dont la tension superficielle, par exemple à 23 oC, est égale à 72,3 mN/m. Il est plus rapide que l'étalonnage à l'aide de cavaliers, mais il comporte toujours le risque que la tension superficielle de l'eau soit modifiée par des traces de substance tensioactive.
Le facteur d'étalonnage Φb, par lequel il faut multiplier toutes les lectures de l'appareil, sera déterminé à l'aide de l'équation ci-après (2):
|

|
(2)
|
où:
|
σo
|
=
|
valeur donnée dans la littérature pour la tension superficielle de l'eau (mN/m)
|
|
σg
|
=
|
valeur mesurée de la tension superficielle de l'eau (mN/m) à cette même température.
|
|
1.6.4.3. Préparation des échantillons
Les solutions aqueuses de la substance à tester seront préparées compte tenu des concentrations requises. Il est impératif que la dissolution de la substance soit complète.
La solution ainsi préparée doit être maintenue à une température constante (±0,5 oC). Étant donné que la tension superficielle d'une solution placée dans le récipient de mesure se modifie après un certain temps, il faut effectuer plusieurs mesures à des moments différents et tracer une courbe donnant la tension superficielle en fonction du temps. Lorsqu'il n'y a plus de modification, on a atteint un état d'équilibre.
La poussière ou les vapeurs d'autres substances faussent la mesure. Le travail doit donc s'effectuer sous une cloche de protection.
1.6.5. Conditions de l'essai
Les mesures doivent être effectuées à une température de 20 oC environ, sans variation supérieure à ±0,5 oC.
1.6.6. Déroulement de l'essai
Les solutions à mesurer seront transférées dans le récipient de mesure soigneusement nettoyé, en prenant soin d'éviter la formation de mousse; ensuite, le récipient de mesure sera placé sur le porte-échantillon qui sera élevé jusqu'à ce que l'anneau soit immergé au-dessous de la surface de la solution. Le porte-échantillon est alors abaissé graduellement et uniformément (à une vitesse d'environ 0,5 cm/mn) pour retirer l'anneau de la surface du liquide et ce, jusqu'à ce que la force maximale soit atteinte. La couche du liquide accroché à l'anneau ne doit pas s'en détacher. Une fois les mesures achevées, l'anneau sera immergé à nouveau sous la surface, et les mesures répétées jusqu'à ce que l'on parvienne à une tension superficielle constante. À chaque détermination, le temps écoulé depuis le transfert de la solution dans le récipient de mesure sera enregistré. Des lectures seront effectuées à la valeur maximale de la force nécessaire pour retirer l'anneau de la surface du liquide.
2. DONNÉES
Pour calculer la tension superficielle, on multipliera tout d'abord la valeur lue sur l'appareil en mN/m par le facteur d'étalonnage Φa ou Φb (selon la méthode d'étalonnage utilisée). On obtiendra alors une valeur qui n'est qu'une approximation et doit ensuite être corrigée.
Harkins et Jordan (4) ont établi de manière empirique des facteurs de correction pour les valeurs de tension superficielle obtenues par la méthode de l'anneau, facteurs qui dépendent des dimensions de l'anneau, de la densité du liquide et de sa tension superficielle.
Étant donné le travail qu'exige la détermination du facteur de correction propre à chaque mesure à partir des tables de Harkins & Jordan, l'utilisation d'une méthode simplifiée de lecture directe de la tension superficielle corrigée à partir du tableau ci-après est autorisée pour les solutions aqueuses (on recourra à l'interpolation pour les lectures qui se situent entre deux valeurs du tableau.)
Tableau
Correction des valeurs mesurées de la tension superficielle
Valable seulement pour les solutions aqueuses, p≈1 g/cm3
|
r
|
= 9,55 mm (rayon moyen de l'anneau)
|
|
r
|
= 0,185 mm (rayon du fil constituant l'anneau)
|
|
Valeur expérimentale (mN/nr)
|
Valeur corrigée (mN/m)
|
|
Étalonnage à l'aide de cavaliers (voir 1.6.4.2.2. (a)]
|
Étalonnage à l'aide d'eau [voir 1.6.4.2.2. (b)]
|
|
20
|
16,9
|
18,1
|
|
22
|
18,7
|
20,1
|
|
24
|
20,6
|
22,1
|
|
26
|
22,4
|
24,1
|
|
28
|
24,3
|
26,1
|
|
30
|
26,2
|
28,1
|
|
32
|
28,1
|
30,1
|
|
34
|
19,9
|
32,1
|
|
36
|
31,3
|
34,1
|
|
38
|
33,7
|
36,1
|
|
40
|
35,6
|
38,2
|
|
42
|
37,6
|
40,3
|
|
44
|
39,5
|
42,3
|
|
46
|
41,4
|
44,4
|
|
48
|
43,4
|
46,5
|
|
50
|
45,3
|
48,6
|
|
52
|
47,3
|
50,7
|
|
54
|
49,3
|
52,8
|
|
56
|
51,2
|
54,9
|
|
58
|
53,2
|
57
|
|
60
|
55,2
|
59,1
|
|
62
|
57,2
|
61,3
|
|
64
|
59,2
|
63,4
|
|
66
|
61,2
|
65,5
|
|
68
|
63,2
|
67,7
|
|
70
|
65,2
|
69,9
|
|
72
|
67,2
|
72
|
|
74
|
69,2
|
—
|
|
76
|
71,2
|
—
|
|
78
|
73,2
|
—
|
Ce tableau a été établi sur la base de la correction de Harkins-Jordan et correspond à celle de la norme DIN (DIN 53914) concernant l'eau et les solutions aqueuses (densité ρ = 1 g/cm3), et pour un anneau vendu dans le commerce dont les dimensions sont R = 9,55 mm (rayon moyen de l'anneau) et r = 0,185 mm (rayon du fil constituant l'anneau). Le tableau donne les valeurs corrigées des mesures de tension superficielle effectuées après un étalonnage au moyen de cavaliers ou d'eau pure.
Une autre solution consiste à calculer la tension superficielle, sans étalonnage préalable, selon la formule suivante:

où
|
F
|
=
|
la force mesurée sur le dynamomètre au point de rupture du film
|
|
R
|
=
|
le rayon de l'anneau
|
|
f
|
=
|
le facteur de correction (1)
|
3. RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
le type d'eau ou de solution utilisé,
|
|
—
|
les spécifications précises de la substance étudiée (identité et impuretés),
|
|
—
|
le résultat des mesures: tension superficielle (lue) en indiquant à la fois les différentes lectures et leur moyenne arithmétique, ainsi que la valeur moyenne corrigée (compte-tenu du facteur dû au matériel et du tableau de correction),
|
|
—
|
la concentration de la solution,
|
|
—
|
la température de l'essai,
|
|
—
|
l'âge de la solution utilisée, en particulier le temps écoulé entre la préparation de la solution et sa mesure,
|
|
—
|
l'évolution de la tension superficielle en fonction du temps à partir du moment où la solution a été transférée dans le récipient de mesure,
|
|
—
|
toutes les informations et observations pertinentes pour l'interprétation des résultats doivent être signalées, notamment les impuretés et l'état physique de la substance.
|
3.2. INTERPRÉTATION DES RÉSULTATS
Étant donné que la tension superficielle de l'eau est de 72,75 mN/m à 20 oC, les substances dont la tension superficielle est inférieure à 60 mN/m, dans les conditions propres à cette méthode, doivent être considérées comme des produits tensioactifs.
4. RÉFÉFENCES
|
(1)
|
OCDE, Paris, 1981, Ligne directrice 115, décision du Conseil C(81) 30 Final.
|
|
(2)
|
R. Weissberger ed., Technique of organic chemistry, Chapter XIV, Physical Methods of Organic Chemistry, 3rd ed, Interscience Publ. New York, 1959, Vol. I, Part I.
|
|
(3)
|
Pure Appl. Chem., 1976, vol. 48, 511.
|
|
(4)
|
Harkins, W.D., Jordan, H.F., J. Amer. Chem. Soc, 1930, vol. 52, 1751.
|
A.6. HYDROSOLUBILITÉ
1. MÉTHODE
Les méthodes décrites sont basées sur les lignes directrices de l'OCDE (1).
1.1. INTRODUCTION
Il est utile de disposer d'informations sur la formule développée, la pression de vapeur, la constante de dissociation et l'hydrolyse (en fonction du pH) de la substance pour réaliser cet essai.
Une seule méthode ne suffit pas à couvrir toute la gamme des solubilités dans l'eau.
Les deux méthodes d'essai décrites ci-après permettent de couvrir toute la gamme des solubilités, mais elles ne sont pas applicables aux substances volatiles:
|
—
|
la première, ci-après dénommée «méthode par élution sur colonne», s'applique aux substances essentiellement pures à faible solubilité (< 10-2 g/l) et stables dans l'eau,
|
|
—
|
la deuxième, ci-après dénommée «méthode du flacon», s'applique aux substances essentiellement pures à solubilité élevée (> 10-2 g/1) et stables dans l'eau.
|
L'hydrosolubilité de la substance étudiée peut être considérablement affectée par la présence d'impuretés.
1.2. DÉFINITIONS ET UNITÉS
L'hydrosolubilité d'une substance est la concentration massique de saturation de la substance dans l'eau à une température donnée. Elle s'exprime en unités de masse par volume de solution. L'unité SI est le kg/m3 (g/l peut également être utilisé).
1.3. SUBSTANCES DE RÉFÉRENCE
Il n'est pas nécessaire d'employer des substances de référence dans tous les cas où l'on étudie une nouvelle substance. Elles doivent servir en premier lieu à vérifier de temps à autre la fiabilité de la méthode et à permettre de comparer les résultats obtenus avec différentes méthodes.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La quantité approximative d'échantillon et le temps nécessaire pour obtenir la concentration massique de saturation doivent être déterminés par un essai préliminaire simple.
1.4.1. Méthode par élution sur colonne
Cette méthode repose sur l'élution d'une substance à étudier avec de l'eau a partir d'une microcolonne remplie avec un support inerte, tel que des billes de verre ou du sable, chargé avec un excès de substance à étudier. L'hydrosolubilité est déterminée lorsque la concentration massique de l'éluant est constante. Elle est indiquée par un plateau de concentration en fonction du temps.
1.4.2. Méthode du flacon
La substance (les solides doivent être pulvérisés) est dissoute dans l'eau à une température quelque peu supérieure à la température d'essai. Lorsque la saturation est atteinte, le mélange est refroidi, maintenu à la température d'essai et agité jusqu'à ce que l'équilibre soit atteint. Une autre possibilité consiste à effectuer la mesure directement à la température d'essai, si un échantillonnage approprié assure que l'équilibre de saturation est atteint. Après quoi, la concentration massique de la substance dans la solution aqueuse, qui ne doit contenir aucune particule non dissoute, est déterminée suivant une méthode analytique appropriée.
1.5. CRITÈRES DE QUALITÉ
1.5.1. Rentabilité
La méthode par élution sur colonne permet d'obtenir une répétabilité < 30 %, et la méthode du flacon, une répétabilité < 15 %.
1.5.2. Sensibilité
La sensibilité dépend de la méthode d'analyse, mais la concentration massique peut être déterminée jusqu'à 10-6 grammes par litre.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Conditions de l'essai.
L'essai doit de préférence être effectué à 20 oC ±0,5 oC. Si l'on suspecte une incidence de la température sur la solubilité (> 3 %/ oC), on utilisera également deux autres températures, supérieure et inférieure d'au moins 10 oC à la température initiale choisie. Dans ce cas, la température doit être ajustée à ±0,1 oC. La température choisie sera maintenue constante dans toutes les parties de l'appareillage où elle peut avoir une influence.
1.6.2. Essai préliminaire
Dans une éprouvette graduée et bouchée de 10 ml, ajouter à 0,1 g environ d'échantillon (les substances solides doivent être réduites en poudre) des volumes croissants d'eau distillée à température ambiante, suivant la progression indiquée dans le tableau suivant:
|
0,1 g soluble dans «x» ml d'eau
|
0,1
|
0,5
|
1
|
2
|
10
|
100
|
> 100
|
|
Solubilité approximative (grammes par litre)
|
> 1 000
|
1 000 à 200
|
200 à 100
|
100 à 50
|
50 à 10
|
10 à 1
|
< 1
|
Après chaque addition de la quantité d'eau indiquée, agiter vigoureusement le mélange pendant dix minutes, puis vérifier visuellement s'il contient des parties d'échantillon non dissoutes. Si, après addition de 10 ml d'eau, l'échantillon, ou des parties de celui-ci, n'est pas dissous, répéter l'expérience dans une éprouvette de 100 ml avec de plus grands volumes d'eau. Si la solubilité est faible, le temps nécessaire pour dissoudre la substance peut être considérablement plus long (24 heures au moins sont à prévoir). La solubilité approximative est indiquée dans le tableau sous le volume d'eau ajoutée dans lequel s'effectue la dissolution complète de l'échantillon. Si la substance reste, selon toute apparence, insoluble, il faut augmenter le temps au-delà de 24 heures (96 heures au maximum), ou procéder à une nouvelle dilution afin de déterminer s'il y a lieu d'utiliser la méthode par élution sur colonne ou la méthode par solubilité en flacon.
1.6.3. Méthode par élution sur colonne
1.6.3.1. Support, solvant et éluant
Dans la méthode par élution sur colonne, le matériel support doit être inerte. On peut employer des billes de verre et du sable. Pour appliquer la substance à tester au support, il faut utiliser un solvant volatile approprié de qualité analytique. L'eau bidistillée dans un appareil de verre ou de quartz peut être utilisée comme éluant.
Remarque:
Ne pas utiliser l'eau provenant directement d'un échangeur d'ions organique.
1.6.3.2. Charge du support
Peser et transférer 600 mg environ de matériel support dans un ballon à fond rond de 50 ml.
Peser une quantité appropriée de substance à étudier et la dissoudre dans le solvant choisi. Une quantité déterminée de cette solution est ajoutée au support. Le solvant doit être complètement évaporé, par exemple dans un évaporateur rotatif, afin d'assurer la saturation en eau du support qui, sinon, ne se ferait pas en raison de l'effet de répartition à la surface.
La charge du support peut poser des problèmes (résultats erronés) si la substance à étudier est déposée sous forme d'huile ou d'une phase cristalline différente. Le problème devrait être examiné expérimentalement et décrit en détail dans le procès-verbal.
Laisser tremper le support chargé pendant au moins 2 heures dans environ 5 ml d'eau, puis transférer la solution dans la microcolonne. Une autre possibilité consiste à verser le support chargé sec dans une microcolonne préalablement remplie d'eau et à équilibrer pendant 2 heures environ.
Mode opératoire:
L'élution de la substance à partir du support peut s'effectuer selon deux systèmes différents:
|
—
|
pompe de recirculation (voir figure 1),
|
|
—
|
réservoir d'eau (voir figure 4).
|
1.6.3.3. Méthode de colonne d'élution avec pompe de recirculation
Appareil
Le schéma d'un système couramment utilisé est indiqué sur la figure 1. La figure 2 montre une microcolonne appropriée, bien que toute autre dimension soit acceptable, à condition de satisfaire aux critères de reproductibilité et de sensibilité. La colonne doit avoir un volume tampon correspondant à cinq fois au moins le volume d'eau contenu dans le lit de la colonne et pouvoir contenir au moins cinq échantillons. On peut cependant réduire la dimension si l'on utilise un appoint de solvant pour remplacer les cinq volumes précités, éliminés avec les impuretés.
La colonne doit être reliée à une pompe de recirculation, capable de donner un débit approximatif de 25 ml/heure environ, au moyen de raccords en polytétrafluoroéthylène (PTFE) et/ou en verre. La colonne et la pompe, lorsqu'elles sont couplées, doivent pouvoir permettre l'échantillonnage de l'effluent et l'équilibrage à pression atmosphérique du réservoir. Le matériau dans la colonne est soutenu par un petit tampon de laine de verre (5 mm), qui sert également à filtrer les particules. La pompe de recirculation peut être, par exemple, une pompe péristaltique ou une pompe à membrane (veiller à ce que la matière du tube ne cause aucune contamination et/ou adsorption).
Mesure:
La circulation dans la colonne est amorcée. Le débit recommandé est d'environ 25 ml/h (ce qui correspond à dix volumes de lit par heure pour la colonne décrite ici). Les cinq premiers volumes (minimum) sont écartés afin d'éliminer les impuretés solubles dans l'eau. Après quoi, faire fonctionner la pompe de recirculation jusqu'à atteindre l'état d'équilibre défini par cinq échantillons successifs dont les concentrations ne diffèrent pas de façon aléatoire de plus de ± 30 %. Ces échantillons doivent être séparés l'un de l'autre par un intervalle de temps correspondant au passage d'un volume d'éluant équivalent à dix fois au moins le volume du lit de la colonne.
1.6.3.4. Méthode par élution sur colonne avec réservoir tampon
Appareillage (voir figures 3 et 4)
Réservoir: le raccordement au réservoir est assuré par un joint de verre rodé, lui-même raccordé à un tube en polytétrafluoroéthylène. Débit recommandé: environ 25 ml/h. Les fractions successives de l'éluant seront recueillies et analysées suivant la méthode choisie.
Mesure:
Les fractions provenant du milieu de l'intervalle d'élution où les concentrations sont constantes (± 30 %) dans au moins cinq fractions consécutives seront utilisées pour déterminer l'hydrosolubilité.
Dans les deux cas (pompe de recirculation ou réservoir d'eau) on répétera l'opération en réduisant le débit de moitié. Si les résultats des deux opérations concordent, l'essai est satisfaisant; si l'on constate une solubilité apparemment plus élevée au débit inférieur, on réduira une nouvelle fois celui-ci de moitié jusqu'à ce que deux opérations successives donnent la même solubilité.
Dans les deux cas (pompe de recirculation ou réservoir d'eau), rechercher dans les fractions la présence éventuelle de matière colloïdale par détection de l'effet Tyndall (diffusion de la lumière). La présence de telles particules fausse les résultats et l'essai doit être répété en améliorant l'action de filtration de la colonne.
Noter le pH de chaque échantillon. Effectuer une deuxième opération à la même température.
1.6.4. Méthode par solubilité en flacon
1.6.4.1. Appareil
Cette méthode requiert le matériel suivant:
|
—
|
verrerie et instrumentation normale de laboratoire,
|
|
—
|
dispositif approprié pour agiter les solutions à des températures constantes contrôlées,
|
|
—
|
si nécessaire en présence d'émulsions, centrifugeuse (de préférence thermostatée),
|
|
—
|
équipement pour la détermination analytique.
|
1.6.4.2. Mesure
Évaluer, à partir de l'essai préliminaire, la quantité de produit nécessaire pour saturer le volume d'eau choisi. Celui-ci dépend de la méthode analytique et de l'intervalle de solubilité. Peser environ cinq fois la quantité de matière déterminée ci-avant et l'introduire dans trois récipients en verre pourvus d'un bouchon, également en verre (par exemple, flacons ou tubes à centrifuge). Ajouter le volume d'eau choisi dans chaque récipient, que l'on bouchera hermétiquement. Agiter ces récipients bouchés à 30 oC (utiliser un dispositif agitateur ou mélangeur susceptible d'opérer à température constante, par exemple, un agitateur magnétique en bain-marie contrôlé par thermostat). Après une journée, retirer l'un des récipients et rééquilibrer pendant 24 heures à température d'essai; agiter de temps à autre. Le contenu du récipient est ensuite centrifugé à température d'essai et la concentration du composé dans la phase aqueuse limpide est déterminée selon une méthode analytique appropriée. Les deux autres ballons sont traités de la même façon après un premier équilibrage à 30 oC pendant deux et trois jours respectivement. Si les concentrations des deux derniers récipients au moins concordent avec la reproductibilité requise, l'essai est satisfaisant. Répéter l'ensemble de l'essai en utilisant des temps d'équilibrage plus longs si les résultats des récipients 1, 2 et 3 accusent une tendance à la progression.
La mesure peut également être effectuée sans préincubation à 30 oC. Pour estimer la vitesse à laquelle s'établit l'équilibre de saturation, prélever des échantillons jusqu'à ce que le temps d'agitation n'ait plus d'effet sur la concentration de la solution à étudier.
Le pH de chaque échantillon doit être noté.
1.6.5. Analyse
Une méthode d'analyse spécifique de la substance est préférable pour ces déterminations, de petites quantités d'impuretés solubles pouvant entraîner des erreurs sensibles dans la solubilité mesurée. Exemples de méthodes: chromatographie en phase gazeuse ou liquide, méthodes titrimétriques, méthodes photométriques, méthodes voltamétriques.
2. DONNÉES
2.1. MÉTHODE PAR ÉLUTION SUR COLONNE
La valeur moyenne déterminée sur au moins cinq échantillons consécutifs prélevés durant le plateau de saturation doit être calculée pour chaque opération, de même que la déviation standard. Les résultats doivent être exprimés en unités de masse par volume de solution.
La comparaison des moyennes calculées sur deux essais effectués avec des débits différents doit donner une répétabilité inférieure à 30 %.
2.2. MÉTHODE DU FLACON
Les résultats individuels doivent être indiqués pour chacun des trois flacons; on fera la moyenne, exprimée en unités de masse par volume de solution, des résultats considérés comme constants (répétabilité inférieure à 15 %). Cette opération peut exiger de convertir des unités de masse en unités de volume, en utilisant la densité lorsque la solubilité est très élevée (> 100 grammes par litre).
3. RÉSULTATS
3.1. MÉTHODE PAR ÉLUTION SUR COLONNE
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
résultats de l'essai préliminaire,
|
|
—
|
spécification précise de la substance (identité et impuretés),
|
|
—
|
concentrations individuelles, débits et pH de chaque échantillon,
|
|
—
|
moyenne et déviation standard d'au moins cinq échantillons provenant du plateau de saturation pour chaque opération,
|
|
—
|
moyenne de deux opérations acceptables consécutives,
|
|
—
|
température de l'eau pendant le processus de saturation,
|
|
—
|
méthode d'analyse utilisée,
|
|
—
|
nature du matériel employé comme support,
|
|
—
|
signe d'instabilité chimique éventuelle de la substance pendant l'essai et la méthode utilisée,
|
|
—
|
toute information pertinente pour l'interprétation des résultats, notamment les impuretés et l'état physique de la substance.
|
3.2. MÉTHODE DU FLACON
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
résultats de l'essai préliminaire,
|
|
—
|
spécification précise de la substance (identité et impuretés),
|
|
—
|
résultats analytiques individuels et moyenne lorsque plus d'une valeur est déterminée pour un même flacon,
|
|
—
|
pH de chaque échantillon,
|
|
—
|
moyenne des valeurs pour les différents flacons en concordance,
|
|
—
|
méthode analytique utilisée,
|
|
—
|
signe d'instabilité chimique éventuelle de la substance pendant l'essai et la méthode utilisée,
|
|
—
|
toute information pertinente pour l'interprétation des résultats, notamment les impuretés et l'état physique de la substance.
|
4. RÉFÉRENCES
|
(1)
|
OCDE, Paris, 1981, Ligne directrice no 105, décision du Conseil C(81) 30 Final.
|
|
(2)
|
NF T 20-045 AFNOR (septembre 1985). Produits chimiques à usage industriel — Détermination de la solubilité dans l'eau des solides et liquides à faible solubilité — Méthode de l'élution sur colonne
|
|
(3)
|
NF T 20-046 AFNOR (septembre 1985). Produits chimiques à usage industriel — Détermination de la solubilité dans l'eau des solides et liquides à faible solubilité — Méthode du flacon
|
ANNEXE
Figure 1
Méthode par élution sur colonne avec pompe de recirculation

Figure 2
Microcolonne type
(dimensions en millimètres)
Figure 3
Microcolonne type
(dimensions en millimètres)

Figure 4
Colonne d'élution avec réservoir d'eau
A.8. COEFFICIENT DE PARTAGE
1. MÉTHODE
La méthode par «agitation en flacon» décrite est basée sur les lignes directrices de l'OCDE (1).
1.1. INTRODUCTION
Il est utile de disposer d'informations préliminaires sur la formule développée, la constante de dissociation, l'hydrosolubilité, l'hydrolyse, la solubilité dans le n-Octanol et la tension superficielle de la substance pour exécuter cet essai.
Les mesures concernant les substances ionisables ne doivent être réalisées qu'avec leur forme non ionisée (acide libre ou base libre) obtenue à l'aide d'un tampon approprié dont le pH est inférieur (acide libre) ou supérieur (base libre) au pK, au moins d'une unité pH.
La présente méthode d'essai comporte deux techniques distinctes: la méthode par agitation en flacon et la chromatographie en phase liquide haute performance (HPLC). La première est applicable lorsque la valeur de log Pow (voir définition ci-après) tombe dans l'intervalle compris entre 2 et 4, et la seconde dans l'intervalle compris entre 0 et 6. Avant d'effectuer l'une de ces techniques expérimentales, il y a lieu de procéder à une estimation préliminaire du coefficient de partage.
La méthode par agitation en flacon ne s'applique qu'aux substances essentiellement pures, solubles dans l'eau et dans le n-Octanol. Elle n'est pas applicable aux substances tensioactives (pour lesquelles il convient de fournir une valeur calculée ou une estimation fondée sur les solubilités individuelles dans l'eau et le n-Octanol).
La méthode de HPLC ne s'applique pas aux acides et aux bases forts, aux complexes métalliques, aux substances tensioactives et aux substances qui réagissent avec l'éluant. Pour ces produits, il convient de fournir une valeur calculée ou une estimation fondée sur les solubilités individuelles dans l'eau et le n-Octanol.
La méthode de HPLC est moins sensible à la présence d'impuretés dans le composé à tester que la méthode par agitation en flacon. Les impuretés peuvent toutefois, dans certains cas, compliquer l'interprétation des résultats en rendant incertaine l'identification des pics. Pour des mélanges qui donnent des pics non résolus, il faut noter la limite inférieure et la limite supérieure de log P.
1.2. DÉFINITIONS ET UNITÉS
Le coefficient de partage (P) est défini comme étant le rapport des concentrations à l'équilibre (ci) d'une substance dissoute dans un système biphasique consistant en deux solvants quasiment non miscibles. Dans le cas du n-Octanol et de l'eau:

Le coefficient de partage (P) est donc le quotient de deux concentrations; il est généralement indiqué sous la forme de son logarithme, base 10 (log P).
1.3. SUBSTANCES DE RÉFÉRENCE
Méthode par agitation en flacon
Il n'est pas nécessaire d'employer des substances de référence dans tous les cas où l'on étudie une nouvelle substance. Elles doivent servir en premier lieu à vérifier de temps à autre la fiabilité de la méthode et à permettre de comparer les résultats obtenus avec différentes méthodes.
Méthode de HPLC
Pour établir une corrélation entre les données mesurées par HPLC et le coefficient de partage d'un composé, il faut établir une courbe d'étalonnage de log P par rapport aux données chromatographiques, à l'aide de six points de référence au moins. Le choix des substances de référence appropriées est laissé à l'utilisateur. Au moins un des composés de référence doit, si possible, avoir un Pow supérieur, et un autre un Pow inférieur à celui de la substance à étudier. Pour des valeurs de log P inférieures à 4, l'étalonnage peut reposer sur les données obtenues avec la méthode par agitation en flacon. Pour des valeurs de log P supérieures à 4, l'étalonnage peut être effectué à partir de valeurs validées citées dans la littérature, si celles-ci concordent avec des valeurs calculées. 11 est préférable, pour une plus grande précision, de choisir des composés de référence dont la structure est apparentée à celle de la substance à étudier.
Des listes de valeurs de log P ont été établies pour un grand nombre de produits chimiques (2) (3). Si l'on ne dispose pas de données relatives au coefficient de partage de composés de structures voisines, il faut utiliser un étalonnage plus général, établi avec d'autres composés de référence.
L'annexe II présente une liste de substances de référence recommandées et la valeur de leur Pow.
1.4. PRINCIPE DE LA MÉTHODE
1.4.1. Méthode par agitation en flacon
Pour déterminer un coefficient de partage, il faut parvenir à un équilibre entre l'ensemble des éléments constitutifs du système s'influençant mutuellement et déterminer les concentrations des substances dissoutes dans les deux phases. Une étude bibliographique sur ce sujet fait apparaître que plusieurs techniques différentes permettent de résoudre ce problème, à savoir le mélange complet de deux phases suivi de leur séparation dans le but de déterminer la concentration à l'équilibre de la substance étudiée.
1.4.2. Méthode de HPLC
La HPLC est effectuée sur des colonnes analytiques remplies d'une phase solide contenant de longues chaînes d'hydrocarbures (par exemple C8, C18) liées chimiquement à de la silice, vendue dans le commerce. Les produits chimiques déposés sur une telle colonne se déplacent sur toute sa longueur à des vitesses différentes en raison de leur différence de taux de partage entre la phase mobile et la phase fixe hydrocarbonée. Les mélanges de produits chimiques sont élués par ordre d'hydrophobicité: les produits hydrosolubles sont élués les premiers et les produits liposolubles, les derniers, proportionnellement à leur coefficient de partage entre l'eau et les hydrocarbures. Ainsi peut on établir une relation entre le temps de rétention sur une telle colonne (à phase inversée) et le coefficient de partage n-Octanol/eau. Le coefficient de partage est déduit du facteur de capacité k, défini par l'expression suivante:

où tR = temps de rétention de la substance à étudier et t0 = temps moyen nécessaire à une molécule de solvant pour traverser la colonne (temps mort).
Il n'est pas nécessaire de disposer d'une méthode analytique quantitative; il suffit de déterminer les temps d'élution.
1.5. CRITÈRES DE QUALITÉ
1.5.1. Répétabilité
Méthode par agitation en flacon
Pour garantir la précision du coefficient de partage, on doit procéder à des déterminations en double, dans trois conditions d'essai différentes, la quantité de substance spécifiée ainsi que le rapport des volumes de solvant pouvant être modifiés. Les valeurs déterminées de ce coefficient, exprimées sous la forme de leurs logarithmes communs, doivent se situer dans un intervalle de ±0,3 unité log.
Méthode de HPLC
Afin d'augmenter la fiabilité des mesures, on procédera à des déterminations en double. Les valeurs de log P issues de mesures individuelles doivent se situer dans un intervalle de ±0,1 unité log.
1.5.2. Sensibilité
Méthode par agitation en flacon
L'intervalle de mesure de la méthode est déterminé par la limite de détection du procédé analytique. Celle-ci doit permettre de déterminer les valeurs de log Pow dans l'intervalle de - 2 à 4 (cet intervalle peut occasionnellement, lorsque les conditions le permettent, être étendu à des valeurs de log Pow allant jusqu'à 5) lorsque la concentration du soluté n'excède pas 0,01 mol par litre dans l'une ou l'autre phase.
Méthode de HPLC
La méthode de HPLC permet d'estimer des coefficients de partage dans une gamme de valeurs de log Pow comprises entre 0 et 6.
Normalement, le coefficient de partage d'un composé peut être estimé avec une précision de ± 1 unité log de la valeur obtenue avec la méthode par agitation en flacon. On peut trouver des exemples de corrélations dans la littérature (4) (5) (6) (7) (8). Une plus grande précision peut généralement être obtenue lorsque la courbe de corrélation est établie avec des composés de référence de structure voisine (9).
1.5.3. Spécificité
Méthode par agitation en flacon
La loi de partage de Nernst ne s'applique qu'à température, pression et pH constants pour les solutions diluées. Rigoureusement, elle ne s'applique qu'à une substance dispersée entre deux solvants purs. Les résultats peuvent être affectés par l'apparition simultanée, dans l'une ou dans les deux phases, de plusieurs solutés différents.
La dissociation ou l'association de molécules dissoutes se traduit par des déviations par rapport à la loi de partage citée. Ces déviations s'expliquent par le fait que le coefficient de partage dépend dès lors de la concentration de la solution.
En raison des équilibres multiples en présence, cette méthode d'essai ne doit pas être appliquée sans correction aux composés ionisables. Dans ce cas, il faut envisager de remplacer l'eau par des solutions tampons dont le pH doit être distant d'au moins une unité pH du pKa de la substance; il faut tenir compte de l'importance de ce pH pour l'environnement.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Estimation préliminaire du coefficient de partage
La valeur du coefficient de partage est estimée de préférence à l'aide d'une méthode de calcul (voir annexe 1), ou bien, s'il y a lieu, à partir du rapport des valeurs de solubilité de la substance à étudier dans des solvants purs (10).
1.6.2. Méthode par agitation en flacon
1.6.2.1. Préparation
n-Octanol: la détermination du coefficient de partage doit être effectuée en recourant à un réactif de qualité analytique.
Eau: utiliser de l'eau distillée ou bidistillée dans un appareil de verre ou de quartz. Pour les composés ionisables, remplacer, si c'est justifié, l'eau par des solutions tampons.
Note:
Ne pas utiliser l'eau provenant directement d'un échangeur d'ions.
1.6.2.1.1. Présaturation des solvants
Avant de déterminer un coefficient de partage, les phases du système de solvants sont saturées réciproquement par agitation à température d'essai. Pour ce faire, il est pratique d'agiter pendant vingt-quatre heures, dans un agitateur mécanique, deux grands flacons contenant du n-Octanol pur de qualité analytique ou de l'eau, avec une quantité suffisante de l'autre solvant, puis de les laisser reposer jusqu'à ce que les phases se séparent et que l'on parvienne à un état de saturation.
1.6.2.1.2. Préparation de l'essai
Le volume liquide doit remplir presque complètement le récipient d'essai afin d'éviter toute perte de matière due à la volatilisation. Le rapport de volume et les quantités de substance à utiliser sont fixés comme suit:
|
—
|
estimation préliminaire du coefficient de partage (voir ci-avant),
|
|
—
|
quantité minimale de substance à tester requise par le procédé analytique utilisé,
|
|
—
|
limite de concentration maximale dans chaque phase de 0,01 mol par litre.
|
Trois essais sont effectués. Pour le premier, on utilise le rapport n-Octanol/eau calculé, pour le second, ce rapport est réduit de moitié et pour le troisième, il est doublé (par exemple 1:1, 1:2, 2:1).
1.6.2.1.3. Substance à étudier
Une solution de réserve est préparée dans du n-Octanol présaturé d'eau. La concentration de cette solution mère doit être déterminée avec précision avant de l'utiliser pour déterminer le coefficient de partage. Cette solution doit être stockée dans des conditions qui garantissent sa stabilité.
1.6.2.2. Conditions de l'essai
La température d'essai doit être maintenue constante (± 1 oC) et se situer dans l'intervalle compris entre 20 oC et 25 oC.
1.6.2.3. Mesure
1.6.2.3.1. Établissement de l'équilibre de partage
Pour chaque série de conditions d'essai, préparer en double des récipients d'essai contenant les quantités requises, mesurées avec précision, des deux solvants ainsi que la quantité nécessaire de solution de réserve.
Mesurer le volume des phases de n-Octanol. Placer les récipients d'essai dans un agitateur approprié ou les agiter à la main. Lorsqu'on utilise un tube à centrifuger, une méthode recommandée consiste à retourner rapidement le tube à 180o autour de son axe transversal de telle sorte que l'air éventuellement retenu traverse les deux phases. L'expérience a montré que cinquante rotations de ce type suffisent généralement pour établir l'équilibre de partage. Pour plus de certitude, il est recommandé d'effectuer cent rotations en cinq minutes.
1.6.2.3.2. Séparation des phases
On peut, si nécessaire, centrifuger le mélange pour séparer les phases. Cette opération est effectuée à l'aide d'une centrifugeuse de laboratoire maintenue à température ambiante, ou, si l'on utilise une centrifugeuse sans contrôle de température, les tubes a centrifuger doivent être gardés à température d'essai pendant au moins une heure avant l'essai.
1.6.2.4. Analyse
Pour déterminer le coefficient de partage, il est nécessaire de déterminer la concentration de la substance à étudier dans les deux phases. Pour ce faire, prélever une portion de chacune des deux phases de chaque tube pour chaque série de conditions d'essai et les analyser suivant le procédé choisi. La quantité totale de substance présente dans les deux phases doit être calculée et comparée avec la quantité de substance initialement introduite.
La phase aqueuse doit être échantillonnée selon un procédé réduisant au minimum le risque d'inclusion de traces de n-Octanol: on peut utiliser à cet effet une seringue en verre à aiguille interchangeable. Tout d'abord remplir partiellement la seringue d'air, lequel est ensuite expulsé doucement, tout en insérant l'aiguille dans la couche de n-Octanol. Prélever un volume adéquat de phase aqueuse. Retirer rapidement la seringue de la solution et enlever l'aiguille. Le contenu de la seringue pourra alors être utilisé comme échantillon aqueux. La concentration doit être déterminée dans les deux phases distinctes de préférence par un procédé spécifique à la substance. Exemples de méthodes analytiques susceptibles de convenir:
|
—
|
méthodes photométriques,
|
|
—
|
chromatographie en phase gazeuse,
|
|
—
|
chromatographie en phase liquide haute performance.
|
1.6.3. Méthode HPLC
1.6.3.1. Préparation
Appareil
Il est nécessaire de disposer d'un chromatographe à phase liquide, muni d'une pompe à débit régulier et d'un dispositif de détection approprié. Il est recommandé d'utiliser une valve à injection portant des boucles à injection. La présence de groupes polaires dans la phase stationnaire peut entraver considérablement le fonctionnement de la colonne d'HPLC. C'est pourquoi les phases stationnaires doivent contenir un minimum de groupes polaires (11). On peut utiliser des phases inverses à microparticules ou des colonnes prêtes à l'emploi vendues dans le commerce. Une colonne de sécurité peut être placée entre le système d'injection et la colonne analytique.
Phase mobile
Le solvant d'élution est préparé avec du méthanol et de l'eau de qualité HPLC et dégazé avant utilisation. Il convient d'employer une élution isocratique et d'utiliser un rapport méthanol/eau contenant un minimum de 25 % d'eau. Normalement, un mélange méthanol-eau 3:1 (v/v) convient pour éluer des composés de log P 6 en moins d'une heure, à un débit de 1 millilitre/minute. Pour les composés dont le log P est élevé, il peut être nécessaire de réduire le temps d'élution et celui des composés de référence en diminuant la polarité de la phase mobile ou la longueur de la colonne.
Les substances très peu solubles dans le n-Octanol ont tendance à donner des valeurs de log Pow anormalement basses avec la méthode HPLC; les pics formés par ces composés font parfois partie du front du solvant. Ce phénomène est probablement dû au fait que le processus de partage est trop lent pour atteindre l'équilibre dans un laps de temps normal pour une séparation par HPLC. Pour parvenir à une valeur fiable, il peut être utile de diminuer le débit ou le rapport méthanol/eau.
Les substances à tester et les composés de référence doivent être solubles, dans la phase mobile, à des concentrations suffisantes pour permettre leur détection. On ne peut employer d'additifs avec le mélange méthanol-eau que dans des cas exceptionnels, car ils modifient les propriétés de la colonne. Les chromatogrammes avec additifs doivent obligatoirement être effectués avec une autre colonne du même type. Si le mélange méthanol-eau n'est pas approprié, on peut utiliser d'autres mélanges solvant organique-eau, par exemple éthanol-eau ou acétonitrile-eau.
Le pH de l'éluant est d'une importance capitale pour les composés ionisables. Il doit se situer dans l'intervalle de pH auquel fonctionne la colonne, qui est habituellement compris entre 2 et 8. Il est recommandé de tamponner. Il faut prendre soin d'éviter la précipitation saline et la détérioration de la colonne qui se produisent avec certains mélanges phase organique/tampon. Il n'est pas recommandé d'effectuer des mesures de HPLC avec des phases stationnaires à base de silice à un pH supérieur à 8 parce qu'une phase mobile alcaline peut altérer rapidement le fonctionnement de la colonne.
Solutés
Les composés de référence doivent être les plus purs possibles. Les composés utilisés à des fins d'essai ou d'étalonnage sont, si possible, dissous dans la phase mobile.
Conditions de l'essai
La température ne doit pas varier de plus de ± 2 K pendant les mesures.
1.6.3.2. Mesure
Calcul du temps mort t0
Le temps mort t0 peut être déterminé en utilisant soit une série d'homologues (par exemple n-alkyl-méthyl-cétones) soit des composés organiques non retenus (par exemple thiourée ou formamide). Pour calculer le temps mort (t0) à l'aide d'une série homologue, il faut injecter un jeu d'au moins 7 membres d'une série homologue et déterminer leurs temps de rétention respectifs. Les temps de rétention bruts tr(nc + 1) sont tracés en fonction de tr(nc). L'intersection a et la pente b de l'équation de régression:
tr(nc + 1) = a + b tr(nc)
sont déterminés (nc = nombre d'atomes de carbone). Le temps mort est défini par l'équation suivante
t0 = a/(1 - b)
Courbe d'étalonnage
L'étape suivante consiste à construire une courbe de corrélation entre log k et log P pour des composés de référence appropriés. En pratique, il s'agit d'injecter simultanément un jeu de cinq à dix composés de référence standard dont le log P est proche de la gamme prévue et de déterminer les temps de rétention, de préférence sur un intégrateur enregistreur lié au système de détection. Les logarithmes des facteurs de capacité, log k, correspondants sont calculés et tracés en fonction du log P déterminé avec la méthode par agitation en flacon. L'étalonnage est effectué à intervalles réguliers, au moins une fois par jour, afin de pouvoir tenir compte de changements éventuels du fonctionnement de la colonne.
Détermination du facteur de capacité de la substance à tester
Injecter la substance à tester dans une quantité aussi petite que possible de phase mobile. Déterminer (en double) le temps de rétention qui permet de calculer le facteur de capacité k. Le coefficient de partage de la substance à tester peut être déduit par interpolation à partir de la courbe de corrélation des produits de référence. Une extrapolation s'impose pour les coefficients de partage très faibles ou très élevés. Il faut alors accorder une attention particulière aux limites de confiance de la courbe de régression.
2. DONNÉES
Méthode par agitation en flacon
La fiabilité des valeurs de P ainsi déterminées peut être vérifiée en procédant à une comparaison de la moyenne des déterminations effectuées en double avec la moyenne générale.
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés) et, s'il y a lieu, l'étape de purification préliminaire,
|
|
—
|
si les méthodes ne sont pas applicables (par exemple substance tensioactive), il convient de fournir une valeur calculée ou estimée à partir des solubilités individuelles dans l'eau ou le n-Octanol,
|
|
—
|
toutes les informations et remarques pertinentes pour l'interprétation des résultats doivent être signalées, en particulier les impuretés et l'état physique de la substance.
|
Pour la méthode par agitation en flacon:
|
—
|
les résultats de l'estimation préliminaire, s'il y a lieu,
|
|
—
|
la température à laquelle est effectuée la détermination,
|
|
—
|
les données relatives aux procédés analytiques utilisés pour déterminer les concentrations,
|
|
—
|
le temps et la vitesse de centrifugation, s'il y a lieu,
|
|
—
|
les concentrations mesurées dans les deux phases pour chaque détermination (cela signifie qu'un total de douze concentrations doit être consigné),
|
|
—
|
le poids de la substance à étudier, le volume de chaque phase utilisée dans chaque récipient à essai et la quantité totale calculée de substance à étudier présente dans chaque phase une fois l'équilibre atteint,
|
|
—
|
les valeurs calculées du coefficient de partage (P) et la moyenne doivent être mentionnées pour chaque série de conditions d'essai ainsi que la moyenne de l'ensemble des déterminations. Toute indication de dépendance du coefficient de partage vis-à-vis de la concentration doit être mentionnée,
|
|
—
|
la déviation standard des valeurs individuelles de P par rapport à leur moyenne,
|
|
—
|
la moyenne P de l'ensemble des déterminations exprimée par son logarithme (base 10),
|
|
—
|
la valeur théorique calculée de Pow, si elle a été déterminée, ou si la valeur mesurée est > 104,
|
|
—
|
le pH de l'eau utilisée et de la phase aqueuse pendant l'expérience,
|
|
—
|
si l'eau est remplacée par un tampon, justifier son utilisation, et donner sa composition sa concentration et son pH ainsi que le pH de la phase aqueuse avant et après l'expérience.
|
Pour la méthode par HPLC:
|
—
|
le résultat de l'estimation préliminaire, s'il y a lieu,
|
|
—
|
les substances à étudier et les substances de référence ainsi que leur degré de pureté,
|
|
—
|
la gamme de température à laquelle sont effectuées les déterminations,
|
|
—
|
le pH auquel sont effectuées les déterminations,
|
|
—
|
une description détaillée de la colonne analytique et de la colonne de sécurité ainsi que de la phase mobile et des moyens de détection,
|
|
—
|
des données relatives à la rétention et les valeurs de log P citées dans la littérature pour les composés de référence utilisés pour l'étalonnage,
|
|
—
|
une description détaillée de la ligne de régression ajustée (log k en fonction de log P),
|
|
—
|
des données relatives à la rétention moyenne et à la valeur interpolée de log P pour le composé à étudier,
|
|
—
|
une description du matériel et des conditions expérimentales,
|
|
—
|
les quantités de produit à étudier et de substances de référence introduites dans la colonne,
|
|
—
|
le temps mort et la méthode utilisée pour le calculer.
|
4. RÉFÉRENCES
|
(1)
|
OCDE, Paris, 1981, Ligne directrice no 107, décision du Conseil C(81) 30 final.
|
|
(2)
|
C. Hansch and A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York, 1979.
|
|
(3)
|
Log P and Parameter Database, A tool for quantitative prediction of bioactivity (C. Hansch, chairman; A.J. Leo. dir.) — Available from Pomona College Medical Chemistry Project 1982, Pomona College, Claremont, California 91711.
|
|
(4)
|
L. Renberg, G. Sundström and K. Dundh-Nygärd, Chemosphere, 1980, vol. 80, 683.
|
|
(5)
|
H. Ellgehausen. C. D'Hondt and R. Fuerer, Pestic. Sci., 1981, vol. 12, 219.
|
|
(6)
|
B. McDuffie, Chemosphere, 1981, vol. 10, 73.
|
|
(7)
|
W.E. Hammers et al., J. Chromatogr., 1982, vol. 247, 1.
|
|
(8)
|
J.E. Haky and A.M. Young, J. Liq. Chromat., 1984, vol. 7, 675.
|
|
(9)
|
S. Fujisawa and E. Masuhara, J. Biomed Mat. Res., 1981, vol. 15, 787.
|
|
(10)
|
O. Jubermann, Verteilen und Extrahieren, in Methoden der Organischen Chemie (Houben Weyl), Allgemeine Laboratoriumpraxis (édité par E. Muller), Georg Thieme Verlag, Stuttgart, 1958, Band I/1, 223-339.
|
|
(11)
|
R.F. Rekker and H.M. de Kort, Euro. J. Med. Chem., 1979, vol. 14, 479.
|
|
(12)
|
A. Leo, C. Hansch and D. Elkins, Partition coefficients and their uses, Chem Rev., 1971, vol. 71, 525.
|
|
(7) (13)
|
R.F. Rekker, The Hydrophobic Fragmentai Constant, Elsevier, Amsterdam, 1977.
|
|
(14)
|
NF T 20-043 AFNOR (1985). Produits chimiques à usage industriel — Détermination du coefficient de partage — Méthode par agitation en flacon.
|
|
(15)
|
C.V. Eadsforth and P. Moser, Chemosphere, 1983, vol. 12, 1459.
|
|
(16)
|
A. Leo, C.Hansch and D. Elkins, Chem. Rev, 1971, vol. 71, 525.
|
|
(17)
|
C. Hansch, A. Leo, S.H. Unger, K. H. Kim, D. Nikaitam and E. J. Lien, J. Med. Chem., 1973, vol. 16, 1207.
|
|
(18)
|
W.B. Neely, D.R. Branson and G.E. Blau, Environ. Sci. Technol., 1974, vol. 8, 1113.
|
|
(19)
|
D.S. Brown and E.W. Flagg, J. Environ. Qual., 1981, vol. 10, 382.
|
|
(20)
|
J.K. Seydel and K.J. Schaper, Chemische Struktur und biologische Aktivität von Wirkstoffen, Verlag Chemie, Weinheim, New York, 1979.
|
|
(21)
|
R. Franke, Theoretical Drug Design Methods, Elsevier, Amsterdam, 1984.
|
|
(22)
|
Y.C. Martin, Quantitative Drug Design, Marcel Dekker, New York, Basel, 1978.
|
|
(23)
|
N.S. Nirrlees, S.J. Noulton, C.T. Murphy, P.J. Taylor; J. Med. Chem., 1976, vol. 19, 615.
|
Annexe
METHODES DE CALCUL/ESTIMATION
INTRODUCTION
Une introduction générale aux méthodes de calcul des données et des exemples figurent dans le «Handbook of Chemical Property Estimation Methods» (a).
Les valeurs calculées du Pow peuvent servir:
|
—
|
à choisir la méthode expérimentale appropriée (gamme pour la méthode par agitation du flacon: log Pow: de - 2 à 4 T; gamme pour l'HPLC: log Pow: de 0 à 6),
|
|
—
|
à déterminer les conditions d'essai appropriées (par exemple, les substances de référence pour l'HPLC, le rapport de volume n-Octanol/eau pour la méthode par agitation du flacon),
|
|
—
|
de contrôle propre au laboratoire relatif aux erreurs expérimentales éventuelles,
|
|
—
|
à fournir une estimation de Pow au cas où les méthodes expérimentales ne peuvent être appliquées pour des raisons techniques.
|
MÉTHODES D'ESTIMATION
Estimation préliminaire du coefficient de partage.
La valeur du coefficient de partage peut être estimée à l'aide de la solubilité de la substance à tester dans les solvants purs:

MÉTHODES DE CALCUL
Principe des méthodes de calcul
Toutes les méthodes de calcul reposent sur la fragmentation formelle de la molécule en sous-structures appropriées pour lesquelles on dispose de données précises concernant le log Pow. Le log Pow de la molécule entière est alors calculé en ajoutant la somme des valeurs des fragments correspondantes et la somme des termes de correction pour les interactions moléculaires.
Il existe des listes de constantes de fragments et de termes de correction (b) (c) (d) (e) dont certaines sont régulièrement mises à jour (b).
Critères de qualité
En général, la fiabilité de la méthode de calcul est inversement proportionnelle à la complexité du composé étudié. Dans le cas de molécules simples, dont le poids moléculaire est faible et qui contiennent un ou deux groupes fonctionnels, on peut prévoir une déviation de 0,1 à 0,3 unité de log P entre les résultats obtenus à l'aide de différentes méthodes de fragmentation et la valeur mesurée. La marge d'erreur peut être plus importante dans le cas de molécules plus complexes. Cette marge d'erreur dépend de l'existence des constantes de fragments et de leur fiabilité ainsi que de la possibilité d'identifier les interactions intramoléculaires (par exemple les liaisons hydrogènes) et de l'utilisation correcte des termes de correction (ce qui ne pose aucune difficulté avec un logiciel informatique CLOGP-3) (b). Dans le cas des composés ionisables, il est important de prendre en considération la charge ou le degré d'ionisation.
Calcul
Méthode π de Hansch
La constante du substituant hydrophobe d'origine, π, établie par Fujita et al. (f) est définie comme suit:
πx = log Pow (PhX) - log Pow (PhH)
où Pow (PhX) est le coefficient de partage d'un dérivé aromatique et Pow (PhH), celui du composé parent;
(par exemple πCl = log Pow (C6H5Cl) - log Pow (C6H6) = 2,84 - 2,13 = 0,71).
Conformément à sa définition la méthode π s'applique surtout à la substitution aromatique. Les valeurs de π d'un grand nombre de substituants ont été rassemblées (b) (c) (d). Elles sont utilisées pour calculer le log P de molécules aromatiques ou de sous-structures.
Méthode de Rekker
Selon Rekker (g), la valeur de log Pow est calculée de la manière suivante:

où fi représente les constantes des divers fragments moléculaires et ai leur fréquence dans la molécule à l'étude. Les termes de corrections peuvent être exprimés sous la forme du multiple entier d'une constante unique Cm (appelée «constante magique»). Les constantes de fragments fi et Cm ont été déterminées à partir d'une liste de 1 054 valeurs de Pow obtenues expérimentalement (825 composés) à l'aide d'une analyse de régression multiple (c) (h). La détermination des termes d'interaction est effectuée conformément aux règles établies décrites dans la littérature (e) (h) (i).
Méthode de Hansch-Leo
Selon Hansch et Léo (c), la valeur de log Pow est calculée à l'aide de l'équation suivante:

où fi représente les constantes des divers fragments moléculaires, Fj les termes de correction et ai et bj les fréquences correspondantes. Une liste de valeurs de fragments (atomes ou groupes) a été déterminée par essai et erreur à partir de valeurs expérimentales de Pow, ainsi qu'une liste de termes de correction Fj (appelés «facteurs»). Les termes de correction ont été classés en plusieurs classes (a) (c). Il est relativement long et compliqué de tenir compte de toutes les règles et de tous les termes de correction mais des logiciels ont été mis au point (b).
Méthode mixte
Le calcul de log Pow pour des molécules complexes peut être considérablement amélioré si l'on découpe la molécule en infrastructures de plus grande dimension, pour lesquelles on dispose de valeurs de log Pow fiables, provenant soit de tableaux (b) (c) soit de mesures. De tels fragments (par exemple des hétérocycles, anthraquinone, azobenzène) peuvent être associés avec les valeurs de π définies selon Hansch ou avec les constantes de fragments définies par Rekker ou par Leo.
Observations
|
i)
|
Les méthodes de calcul ne s'appliquent qu'à des composés partiellement ou totalement ionisés lorsqu'il est possible de tenir compte des facteurs de correction nécessaires.
|
|
ii)
|
Si les liaisons hydrogène intramoléculaires peuvent être déterminées, les termes de correction correspondants (de +0,6 à +1,0 unité log P) doivent être ajoutés (a). Les modèles stériques ou les données spectroscopiques relatives à la molécule peuvent indiquer la présence de telles liaisons.
|
|
iii)
|
Si plusieurs formes tautomériques sont possibles, les calculs doivent être basés sur la forme la plus probable.
|
|
iv)
|
Les révisions des listes de constantes de fragments doivent être soigneusement suivies.
|
Procès-verbal
Lors de l'utilisation de méthodes de calcul/estimation. Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
description de la substance (mélange, impuretés, etc.),
|
|
—
|
indication concernant toute liaison hydrogène intramoléculaire possible, charge, ou tout effet inhabituel (par exemple tautomérisme),
|
|
—
|
description de la méthode de calcul,
|
|
—
|
identification ou base de données,
|
|
—
|
particularités du choix des fragments,
|
|
—
|
documentation détaillée du calcul.
|
RÉFÉRENCES
|
(a)
|
W.J. Lyman, W.F. Reehl and D.H. Rosenblatt (ed.), Handbook of Chemical Property Estimation Methods, McGraw-Hill, New York, 1983.
|
|
(b)
|
Pomona College, Medicinal Chemistry Project, Claremont, California 91711, USA, Log P Database and Med. Chem. Software (Program CLOGP-3).
|
|
(c)
|
C. Hansch, A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York, 1979.
|
|
(d)
|
A. Leo, C. Hansch, D. Elkins, Chem. Rev., 1971, vol. 71, 525.
|
|
(e)
|
R.F. Rekker, H.M. de Kort, Eur. J. Med. Chem. — Chim Ther., 1979, vol. 14, 479.
|
|
(f)
|
T. Fujita, J. Iwasa and C. Hansch, J. Amer. Chem. Soc, 1964, vol. 86, 5175
|
|
(g)
|
R.F. Rekker, The Hydrophobic Fragmental Constant, Pharmacochemistry Library, Elsevier, New York, 1977.
|
|
(h)
|
C.V. Eadsforth, P. Moser, Chemosphere, 1983, vol. 12, 1459.
|
|
(i)
|
R.A. Scherrer, ACS — American Chemical Society, Washington, D.C. 1984, Symposium Series 255, p. 225.
|
Annexe II
Substances de référence recommandées pour la méthode HPLC
|
No
|
Substance de référence
|
Log Pow
|
pKa
|
|
1
|
2-butanone
|
0,3
|
|
|
2
|
4-acétylpyridine
|
0,5
|
|
|
3
|
aniline
|
0,9
|
|
|
4
|
acétaniline
|
1,0
|
|
|
5
|
alcool benzylique
|
1,1
|
|
|
6
|
p-méthoxyphénol
|
1,3
|
pKa = 10,26
|
|
7
|
acide phénoxy-acétique
|
1,4
|
pKa = 3,12
|
|
8
|
phénol
|
1,5
|
pKa = 9,92
|
|
9
|
dinitrophénol
|
1,5
|
pKa = 3,96
|
|
10
|
benzonitrile
|
1,6
|
|
|
11
|
phénylacétonitrile
|
1,6
|
|
|
12
|
alcool 4-méthylbenzylique
|
1,6
|
|
|
13
|
acécophénone
|
1,7
|
|
|
14
|
2-nitrophénol
|
1,8
|
pKa = 7,17
|
|
15
|
acide 3-nitrobenzoïque
|
1,8
|
pKa = 3,47
|
|
16
|
4-chloraniline
|
1,8
|
pKa = 4,15
|
|
17
|
nitrobenzène
|
1,9
|
|
|
18
|
alcool cinnamique
|
1,9
|
|
|
19
|
acide benzoïque
|
1,9
|
pKa = 4,19
|
|
20
|
p-crésol
|
1,9
|
pKa = 10,17
|
|
21
|
acide cinnamique
|
2,1
|
pKa = 3,89 cis pKa = 4,44 trans
|
|
22
|
anisole
|
2,1
|
|
|
23
|
méthylbenzoate
|
2,1
|
|
|
24
|
benzène
|
2,1
|
|
|
25
|
acide méthylbenzoïque
|
2,4
|
pKa = 4,27
|
|
26
|
chlorophénol
|
2,4
|
pKa = 9,1
|
|
27
|
trichloréthylène
|
2,4
|
|
|
28
|
atrazine
|
2,6
|
|
|
29
|
éthylbenzoate
|
2,6
|
|
|
30
|
2,6-dichlorobenzonitrile
|
2,6
|
|
|
31
|
acide chlorobenzoïque
|
2,7
|
pKa = 3,82
|
|
32
|
toluène
|
2,7
|
|
|
33
|
naphtol
|
2,7
|
pKa = 9,34
|
|
34
|
2,3 dichloroaniiine
|
2,8
|
|
|
35
|
chlorobenzène
|
2,8
|
|
|
36
|
allyl-phényléther
|
2,9
|
|
|
37
|
bromobenzène
|
3,0
|
|
|
38
|
éthylbenzène
|
3,2
|
|
|
39
|
benzophénone
|
3,2
|
|
|
40
|
4-phénylphénone
|
3,2
|
pKa = 9,54
|
|
41
|
thymol
|
3,3
|
|
|
42
|
1,4-dichlorobenzène
|
3,4
|
|
|
43
|
diphénylamine
|
3,4
|
pKa = 0,79
|
|
44
|
naphtalène
|
3,6
|
|
|
45
|
phénylbenzoate
|
3,6
|
|
|
46
|
isopropylbenzène
|
3,7
|
|
|
47
|
2,4,6-trichlorophénol
|
3,7
|
pKa = 6
|
|
48
|
biphényl
|
4,0
|
|
|
49
|
benzylbenzoate
|
4,0
|
|
|
50
|
2,4-dinitro-6sec.butylphénol
|
4,1
|
|
|
51
|
1,2,4-trichlorobenzène
|
4,2
|
|
|
52
|
acide dodécanoïque
|
4,2
|
|
|
53
|
diphényléther
|
4,2
|
|
|
54
|
n-butylbenzène
|
4,5
|
|
|
55
|
phénanthrène
|
4,5
|
|
|
56
|
fluoranthène
|
4,7
|
|
|
57
|
dibenzyl
|
4,8
|
|
|
58
|
2,6-diphénylpyridine
|
4,9
|
|
|
59
|
triphénylamine
|
5,7
|
|
|
60
|
DDT
|
6,2
|
|
|
Autre substance de référence dont le log Pow est faible
|
|
1
|
acide nicotinique
|
-0,07
|
|
A.9. POINT D'ÉCLAIR
1. MÉTHODE
1.1. INTRODUCTION
Pour réaliser cet essai, il est utile de disposer d'informations préliminaires sur l'inflammabilité de la substance. Le mode opératoire est applicable aux substances liquides dont les vapeurs peuvent être enflammées par des sources d'inflammation. Les méthodes d'essai énumérées dans le présent document ne sont valables que pour les intervalles de point d'éclair spécifiés dans les méthodes individuelles.
Il convient de tenir compte dans le choix de la méthode des réactions chimiques éventuelles entre la substance et le support de l'échantillon.
1.2. DÉFINITIONS ET UNITÉS
Le point d'éclair est la température la plus basse, corrigée pour une pression de 101,325 kPa, à laquelle le liquide d'essai dégage des vapeurs, dans les conditions définies dans la méthode d'essai, en quantité telle qu'il en résulte dans le récipient d'essai un mélange vapeur/air inflammable.
Unités: oC
t = T - 273,15
(t est exprimé en oC et T en K)
1.3. SUBSTANCES DE RÉFÉRENCE
Il n'est pas nécessaire d'employer des substances de référence dans tous les cas où l'on étudie une nouvelle substance. Elles devraient servir en premier lieu à vérifier de temps à autre la fiabilité de la méthode et à permettre de comparer les résultats obtenus avec différentes méthodes.
1.4. PRINCIPE DE LA MÉTHODE
La substance est placée dans un récipient d'essai que l'on chauffe ou que l'on refroidit à la température d'essai, suivant le mode opératoire décrit dans la méthode d'essai individuel. Des essais d'inflammation doivent être effectués afin de s'assurer que la substance dégage ou non des vapeurs inflammables à la température d'essai,
1.5. CRITÈRES DE QUALITÉ
1.5.1. Répétabilité
La répétabilité dépend de l'intervalle de point d'éclair et de la méthode d'essai utilisée; maximum 2 oC.
1.5.2. Sensibilité
La sensibilité dépend de la méthode d'essai utilisée.
1.5.3. Spécificité
La spécificité de certaines méthodes d'essai est limitée à certains intervalles de point d'éclair et dépend de données relatives à la substance (par exemple haute viscosité).
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Préparation
Un échantillon de la substance à tester est placé dans un appareil d'essai conforme aux points 1.6.3.1 ou 1.6.3.2.
Par mesure de sécurité, il est conseillé d'utiliser pour les substances énergétiques ou toxiques, une méthode n'exigeant qu'un échantillon de petite taille (2 cm3 environ).
1.6.2. Conditions d'essai
L'appareil doit, dans la limite des règles de sécurité, être placé à l'abri des courants d'air.
1.6.3. Mode opératoire
1.6.3.1. Méthode de l'équilibre
Voir normes ISO 1516, ISO 3680, ISO 1523, ISO 3679.
1.6.3.2. Méthode du non-équilibre
Appareil d'Abel:
Voir normes BS 2000 partie 170, NF M07-011, NF T66-009.
Appareil d'Abel-Pensky:
Voir normes EN 57, DIN 51755 partie 1 (pour des températures de 5 à 65 oC), DIN 51755 partie 2 (pour des températures inférieures à 5 oC), NF M07-036.
Appareil Tag:
Voir norme ASTM D 56.
Appareil de Pensky-Martens:
Voir normes ISO 2719, EN 11, DIN 51758, ASTM D 93, BS 2000-34, NF M07-019.
Observations:
Lorsque le point d'éclair, déterminé par une méthode basée sur le non-équilibre (point 1.6.3.2.), a une valeur de 0 ± 2 oC, 21 ± 2 oC ou 55 ± 2 oC, il importe de confirmer cette valeur par une méthode basée sur l'équilibre en utilisant le même appareil.
Seules les méthodes susceptibles de donner la température du point d'éclair peuvent être utilisées pour une notification.
Pour déterminer le point d'éclair des liquides visqueux (peintures, gommes et substances similaires) contenant des solvants, il ne faut utiliser que des appareils et des méthodes d'essai permettant de déterminer le point d'éclair des liquides visqueux.
Voir normes ISO 3679, ISO 3680, ISO 1523, DIN 53213 partie 1.
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés),
|
|
—
|
la méthode utilisée ainsi que toute variante éventuelle,
|
|
—
|
les résultats et toute observation supplémentaire pertinente pour l'interprétation des résultats.
|
4. RÉFÉRENCES
Aucune.
A.10. INFLAMMABILITÉ (SOLIDES)
1. MÉTHODE
1.1. INTRODUCTION
Avant de procéder à cet essai, il est utile de disposer d'informations préliminaires sur les propriétés explosives potentielles de la substance.
Cet essai ne doit être appliqué qu'aux substances poudreuses, granuleuses ou pâteuses.
De façon à ne pas englober toutes les substances susceptibles d'être enflammées, mais uniquement celles qui brûlent rapidement ou celles dont la combustion est particulièrement dangereuse d'une façon ou d'une autre, ne sont considérées comme très inflammables que les substances dont la vitesse de combustion dépasse une certaine limite.
La propagation de l'incandescence dans une poudre métallique peut être particulièrement dangereuse en raison des difficultés que présente l'extinction du feu. Les poudres métalliques sont considérées comme très inflammables si l'incandescence se propage dans tout l'échantillon en un temps déterminé.
1.2. DÉFINITIONS ET UNITÉS
Le temps de combustion est exprimé en secondes.
1.3. SUBSTANCES DE RÉFÉRENCE
Non spécifiées.
1.4. PRINCIPE DE LA MÉTHODE
La substance est disposée de manière à former une bande ininterrompue ou une traînée de poudre d'environ 250 mm de longueur. Un essai préliminaire est effectué afin de déterminer si, par allumage à la flamme d'un brûleur à gaz, il se produit une propagation de la combustion avec ou sans flamme. Si la combustion se propage sur une distance de plus de 200 mm en un temps déterminé, un essai complet est effectué pour déterminer la vitesse de combustion.
1.5. CRITÈRES DE QUALITÉ
Non établis.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Essai préliminaire
La substance est disposée en bande ininterrompue ou en traînée de poudre d'environ 250 mm de longueur par 20 mm de largeur et 10 mm de hauteur sur une plaque non combustible, non poreuse et peu thermoconductrice. La flamme chaude d'un brûleur à gaz (diamètre minimum 5 mm) est appliquée à l'une des extrémités de la traînée de poudre jusqu'à ce que celle-ci s'enflamme, ou pendant un maximum de 2 minutes (5 minutes pour les poudres métalliques ou d'alliages métalliques). Noter si la combustion se propage le long de la traînée sur une distance de 200 mm pendant une durée d'essai de 4 minutes (40 minutes pour les poudres métalliques). Si la substance ne s'enflamme pas et ne propage pas la combustion, avec ou sans flamme, sur une distance de 200 mm pendant une durée d'essai de 4 minutes (ou 40 minutes), la substance n'est pas considérée comme très inflammable, et il n'est pas nécessaire de poursuivre l'essai. Si la combustion de la substance se propage sur une distance de 200 mm sur la traînée de poudre en moins de 4 minutes (ou de 40 minutes pour les poudres métalliques), le mode opératoire décrit ci-dessous (aux points 1.6.2 et suivants) doit être suivi.
1.6.2. Essai de vitesse de combustion.
1.6.2.1. Préparation
Les substances poudreuses ou granuleuses sont versées en vrac dans un moule de 250 mm de longueur et de section transversale triangulaire dont la hauteur et la largeur intérieure sont respectivement de 10 et 20 millimètres. Disposer de part et d'autre du moule, dans le sens de la longueur, deux plaques métalliques destinées à jouer le rôle de supports latéraux. Ces plaques dépassent de 2 mm le bord supérieur de la section triangulaire transversale (voir figure). Ensuite, laisser tomber trois fois le moule d'une hauteur de 2 cm sur une surface dure. Rajouter de la substance s'il y a lieu. Enlever ensuite les plaques latérales et racler le trop-plein. Placer une plaque non combustible, non poreuse et peu thermoconductrice sur le moule, retourner l'ensemble et démouler.
Les substances pâteuses sont étalées sur une plaque non combustible, non poreuse et peu thermoconductrice sous la forme d'un cordon de 250 mm de longueur et d'environ 1 cm2 de section transversale.
1.6.2.2. Conditions d'essai
Dans le cas de substances sensibles à l'humidité, effectuer l'essai le plus vite possible après avoir retiré la substance de son récipient.
1.6.2.3. Mode opératoire
Disposer le tas dans une hotte, perpendiculairement au sens du courant d'air.
La vitesse de l'air doit suffire à empêcher la fumée de se répandre dans le laboratoire et ne doit pas être modifiée au cours de l'essai. Le flux d'air doit former un écran qui entoure l'appareil.
L'une des extrémités du tas est enflammée à l'aide de la flamme chaude d'un brûleur à gaz (diamètre minimal 5 mm). Lorsque le tas a brûlé sur une distance de 80 mm, mesurer la vitesse de combustion sur les 100 mm suivants.
Répéter l'essai six fois, en utilisant chaque fois une plaque propre et froide, à moins d'observer un résultat positif avant.
2. DONNÉES
Le temps de combustion observé lors de l'essai préliminaire (1.6.1) et le temps de combustion le plus court observé au cours de l'essai (1.6.2.3) sont pertinents pour l'évaluation.
3. RÉSULTATS
3.1. PROCÈS-VERBAL
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés),
|
|
—
|
une description de la substance à essayer, son état physique, y compris son taux d'humidité,
|
|
—
|
les résultats des essais préliminaires et des essais de vitesse de combustion, si ces derniers ont été effectués,
|
|
—
|
toute observation supplémentaire pertinente pour l'interprétation des résultats.
|
3.2. INTERPRÉTATION DES RÉSULTATS
Les substances poudreuses, granuleuses ou pâteuses doivent être considérées comme très inflammables lorsque le temps de combustion observé au cours de l'un des essais effectués conformément au mode opératoire décrit au point 1.6.2 est inférieur à 45 secondes. Les poudres métalliques ou d'alliages métalliques doivent être considérées comme très inflammables lorsqu'elles peuvent être enflammées et que la flamme ou la zone de réaction s'étend à tout l'échantillon en 10 minutes ou moins.
4. RÉFÉRENCES
|
(1)
|
NF T 20-042 (septembre 1985). Produits chimiques à usage industriel — Détermination de l'inflammabilité des solides.
|
Annexe
Figure
Moule et accessoires nécessaires à la confection des tas
(toutes les dimensions sont exprimées en millimètres)

A.11. INFLAMMABILITÉ (GAZ)
1. MÉTHODE
1.1. INTRODUCTION
La présente méthode permet de déterminer si des gaz mélangés à l'air à température ambiante (20 oC environ) et à la pression atmosphérique sont inflammables et, s'ils le sont, dans quel intervalle de concentration. Des mélanges contenant des concentrations croissantes du gaz à tester dans de l'air sont exposés à une étincelle électrique et on observe si l'inflammation se produit.
1.2. DÉFINITIONS ET UNITÉS
L'intervalle d'inflammabilité est l'intervalle de concentration entre les limites d'explosion supérieure et inférieure. Les limites d'explosion supérieure et inférieure sont les concentrations dans l'air du gaz inflammable auxquelles l'inflammation ne se propage pas.
1.3. SUBSTANCE DE RÉFÉRENCE
Non spécifiée.
1.4. PRINCIPE DE LA MÉTHODE
La concentration du gaz dans l'air est augmentée graduellement et le mélange est exposé à une étincelle électrique à chaque étape.
1.5. CRITÈRES DE QUALITÉ
Non fixés.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Appareil
Le récipient d'essai est un cylindre en verre d'un diamètre intérieur de 50 mm au moins et d'une hauteur de 300 mm au moins, disposé verticalement. Les électrodes d'inflammation sont distantes l'une de l'autre de 3 à 5 mm et placées à 60 mm du fond du cylindre. Le cylindre est équipé d'une soupape. L'appareil doit être protégé par un blindage pour limiter les dégâts d'une explosion éventuelle.
La source d'inflammation est une étincelle inductive entretenue d'une durée de 0,5 seconde, produite par un transformateur à haute tension avec une tension de sortie de 10 à 15 kV (la puissance maximale est de 300 W). Un modèle d'appareil approprié est décrit dans la référence (2).
1.6.2. Conditions d'essai
L'essai doit avoir lieu à température ambiante (20 oC environ).
1.6.3. Mode opératoire
Remplir le cylindre en verre d'un mélange air-gaz de concentration connue à l'aide de pompes doseuses. Faire jaillir une étincelle au travers de ce mélange et observer si une flamme se détache de la source d'inflammation et se propage de manière indépendante. La concentration du gaz est modifiée par étapes de 1 % en volume jusqu'à ce que l'inflammation décrite ci-dessus se produise.
Si la structure chimique du gaz laisse supposer qu'il doit être ininflammable et s'il est possible de calculer la composition du mélange stœchiométrique avec l'air, ne soumettre à essai, par étapes de 1 %, que des mélanges dans une gamme comprise entre 10 % de moins que la composition stœchiométrique et 10 % de plus.
2. DONNÉES
La propagation de la flamme constitue la seule donnée d'information valable pour la détermination de cette propriété.
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés),
|
|
—
|
une description, incluant les dimensions, de l'appareil utilisé,
|
|
—
|
la température à laquelle l'essai a été réalisé,
|
|
—
|
les concentrations d'essai ainsi que les résultats obtenus,
|
|
—
|
le résultat de l'essai: gaz ininflammable ou très inflammable,
|
|
—
|
lorsque l'on conclut à l'ininflammabilité, il convient de préciser l'intervalle de concentration sur lequel a porté l'essai, par étapes de 1 %,
|
|
—
|
toute information et observation pertinente pour l'interprétation des résultats.
|
4. RÉFÉRENCES
|
(1)
|
NF T 20-041 (septembre 1985). Produits chimiques à usage industriel — Détermination de l'inflammabilité des gaz.
|
|
(2)
|
W.Berthold, D.Conrad, T.Grewer, H.Grosse-Wortmann, T.Redeker und H.Schacke. «Entwicklung einer Standard-Apparatur zur Messung von Explosionsgrenzen». Chem.-Ing.-Tech., 1984, vol. 56, 2, 126-127.
|
A.12. INFLAMMABILITÉ (AU CONTACT DE L'EAU)
1. MÉTHODE
1.1. INTRODUCTION
Cette méthode d'essai peut être utilisée pour déterminer si la réaction d'une substance avec l'eau ou l'air humide entraîne le dégagement d'une quantité dangereuse d'un gaz ou de plusieurs gaz, susceptibles d'être très inflammables.
Elle peut être appliquée à la fois aux substances solides et liquides mais non aux substances qui s'enflamment spontanément au contact de l'air.
1.2. DÉFINITIONS ET UNITÉS
Très inflammables: substances qui, au contact de l'eau ou de l'air humide, dégagent une quantité dangereuse de gaz très inflammables à raison d'un débit minimal de 1 litre/kg par heure.
1.3. PRINCIPE DE LA MÉTHODE
L'essai de la substance comporte plusieurs phases décrites ci-après; si l'inflammation intervient à une quelconque de ces phases, il n'est pas nécessaire de poursuivre l'essai. Si on sait que la substance ne réagit pas violemment avec l'eau, procéder à la phase 4 (voir 1.3.4).
1.3.1. Phase 1
Placer la substance à tester dans un bac contenant de l'eau distillée à 20 oC et noter si le gaz s'enflamme ou non.
1.3.2. Phase 2
Placer la substance à tester sur un papier filtrant flottant sur de l'eau distillée à 20 oC contenue dans une capsule et noter si le gaz qui se dégage s'enflamme ou non. Le papier-filtre ne sert qu'à maintenir la substance en place, ce qui accroît les probabilités d'inflammation.
1.3.3. Phase 3
Mettre la substance à tester en tas sur une hauteur de 2 cm et un diamètre de 3 cm environ. Ajouter quelques gouttes d'eau au tas ainsi constitué et noter si le gaz qui se dégage s'enflamme ou non.
1.3.4. Phase 4
Mélanger la substance d'essai avec de l'eau distillée a 20 oC et mesura le débit du gaz pendant 7 heures, à intervalles d'une heure. Si ce débit est variable ou s'il s'accroît après 7 heures, le temps de mesure doit être prolongé jusqu'à 5 jours au maximum. L'essai peut être arrêté si le débit excède 1 l/kg par heure à un moment donné.
1.4. SUBSTANCE DE RÉFÉRENCE
Non spécifiée.
1.5. CRITÈRES DE QUALITÉ
Non indiqués.
1.4. 1.6. DESCRIPTION DES MÉTHODES
1.6.1. Phase 1
1.6.1.1. Conditions de l'essai
L'essai est effectué à température ambiante (20 oC, environ).
1.6.1.2. Mode opératoire
Placer une petite quantité (approximativement 2 mm de diamètre) de la substance à tester dans un bac contenant de l'eau distillée. Noter si: i) il y a dégagement de gaz, ii) le gaz s'enflamme. Si le gaz s'enflamme, il est inutile de poursuivre l'essai de la substance, celle-ci étant dès lors considérée comme substance dangereuse.
1.6.2. Phase 2
1.6.2.1. Appareil
Papier-filtre flottant sur la surface de l'eau distillée dans un récipient approprié, par exemple une capsule de 100 mm de diamètre.
1.6.2.2. Conditions de l'essai
L'essai est effectué à température ambiante (20 oC, environ).
1.6.2.3. Mode opératoire
Placer une petite quantité (approximativement 2 mm de diamètre) de la substance à tester au centre du papier-filtre. Noter si: i) il y a dégagement de gaz, ii) le gaz s'enflamme. Si le gaz s'enflamme, il est inutile de poursuivre l'essai de la substance, celle-ci étant dès lors considérée comme substance dangereuse.
1.6.3. Phase 3
1.6.3.1. Conditions de l'essai
L'essai est effectué à température ambiante (20 oC, environ).
1.6.3.2. Mode opératoire
Mettre la substance à tester en tas sur une hauteur de 2 cm et un diamètre de 3 cm environ en aménageant au sommet un petit cratère. Ajouter quelques gouttes d'eau dans le creux. Noter si: i) il y a dégagement de gaz, ii) le gaz s'enflamme. Si le gaz s'enflamme, il est inutile de poursuivre l'essai de la substance, celle-ci étant dès lors considérée comme substance dangereuse.
1.6.4. Phase 4
1.6.4.1. Appareil
L'appareil est monté comme indiqué sur la figure.
1.6.4.2. Conditions de l'essai
S'assurer que le récipient contenant la substance à tester est exempt de particules pulvérulentes (< 500 5m). Si celles-ci représentent plus de 1 % en poids du total, ou si l'échantillon est friable, réduire la substance en poudre avant de procéder à l'essai, afin de réduire la dimension des particules pendant le stockage et la manutention; sinon, la substance est utilisée telle qu'elle est reçue. L'essai doit être réalisé à température ambiante (20 oC environ) et à pression atmosphérique.
1.6.4.3. Mode opératoire
Verser de 10 à 20 ml d'eau dans l'entonnoir à robinet de l'appareillage et placer 10 g de substance dans la fiole conique. Le volume de gaz qui se dégage peut être mesuré par tout moyen approprié. Ouvrir le robinet de l'entonnoir pour laisser pénétrer l'eau dans la fiole conique et déclencher un chronomètre. Le dégagement gazeux est mesuré toutes les heures pendant sept heures. Si, pendant cette période, le dégagement gazeux devient irrégulier, ou si, à la fin de cette période, son débit augmente, il convient de poursuivre les mesures pendant cinq jours. Si, à un moment quelconque des mesures, le débit du dégagement gazeux excède 1 litre/kg par heure, l'essai peut être interrompu. L'essai doit être réalisé en triple.
Si l'identité chimique du gaz est inconnue, celui-ci doit être analysé. Si le gaz contient des constituants très inflammables, ou si l'on ignore si l'ensemble du mélange est très inflammable, préparer et essayer un mélange de même composition conformément à la méthode d'essai (A.11).
2. DONNÉES
La substance est considérée comme dangereuse si:
|
—
|
une inflammation spontanée intervient lors d'une phase quelconque du déroulement de l'essai,
|
|
—
|
un gaz inflammable se dégage à un débit supérieur à 1 litre/kg de substance par heure.
|
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés),
|
|
—
|
une description détaillée de toute préparation préalable de la substance à essayer,
|
|
—
|
les résultats de l'essai (phases 1, 2, 3 et 4),
|
|
—
|
l'identité chimique du gaz qui se dégage,
|
|
—
|
le débit du dégagement gazeux, si la phase 4 (1.6.4) est effectuée,
|
|
—
|
le résultat de l'essai: gaz ininflammable ou très inflammable,
|
|
—
|
toute observation supplémentaire pertinente pour l'interprétation des résultats.
|
4. RÉFÉRENCES
|
(1)
|
Recommandations relatives au transport des marchandises dangereuses. Épreuves et critères, 1990, Nations unies, New York.
|
|
(2)
|
NF T 20-040 (septembre 1985), Produits chimiques à usage industriel. Détermination de l'inflammabilité des gaz engendrés par hydrolyse des produits solides et liquides.
|
Annexe
Figure
Appareillage

A.13. PROPRIÉTÉS PYROPHORIQUES DES SOLIDES ET DES LIQUIDES
1. MÉTHODE
1.1. INTRODUCTION
Cet essai est applicable aux substances solides et liquides, qui, en petite quantité, s'enflamment spontanément, peu de temps après être entrées en contact avec l'air à température ambiante (20 oC environ).
Les substances dont l'inflammation spontanée n'intervient qu'après une exposition de plusieurs heures ou de plusieurs jours à température ambiante, ou à des températures élevées, ne sont pas couvertes par cette méthode d'essai.
1.2. DÉFINITIONS ET UNITÉS
Les substances sont considérées comme ayant des propriétés pyrophores si elles s'enflamment ou causent la carbonisation dans les conditions décrites au point 1.6.
L'auto-inflammabilité des liquides peut également exiger d'être évaluée selon la méthode A.15 (température d'inflammation spontanée des liquides et des gaz).
1.3. SUBSTANCES DE RÉFÉRENCE
Non spécifiées.
1.4. PRINCIPE DE LA MÉTHODE
La substance, solide ou liquide, est ajoutée à un porteur inerte et mise en contact avec l'air à température ambiante pendant une période de cinq minutes. Si les substances liquides ne s'enflamment pas, elles sont absorbées sur un papier-filtre qui est exposé à l'air, à température ambiante (20 oC environ), pendant cinq minutes. Si la substance, solide ou liquide, enflamme ou carbonise un papier-filtre, elle est considérée comme pyrophorique.
1.5. CRITÈRES DE QUALITÉ
Répétabilité: en raison de l'importance que revêt l'aspect «sécurité», un seul résultat positif est suffisant pour conclure que la substance est très inflammable.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Appareillage
Remplir une capsule de porcelaine de 10 cm de diamètre environ de terre d'infusoires, sur une épaisseur de 5 mm environ, à température ambiante (20 oC environ).
Remarque:
La terre d'infusoires, ou toute autre substance inerte comparable généralement disponible, sera prise comme représentative du sol sur lequel la substance pourra être accidentellement répandue.
Les essais concernant les liquides qui ne s'enflamment pas au contact de l'air lorsqu'ils sont en contact avec un porteur inerte seront effectués avec un papier-filtre sec.
1.6.2. Réalisation de l'essai
a) Solides pulvérulents
Verser 1 ou 2 cm3 de substance pulvérulente à tester, d'une hauteur d'environ 1 m sur une surface non combustible et observer si la substance s'enflamme pendant la chute ou pendant les cinq premières minutes de tassement.
L'essai est répété six fois, à moins qu'une inflammation ne survienne.
b) Liquides
Verser environ 5 cm3 de liquide à essayer dans une capsule de porcelaine préparée et observer si la substance s'enflamme dans les cinq minutes.
S'il ne survient pas d'inflammation au cours de six essais, effectuer l'essai suivant:
Déposer à l'aide d'une seringue 0,5 ml de l'échantillon à essayer sur un papier-filtre à cet usage et observer si le papier-filtre s'enflamme ou se carbonise dans les cinq minutes qui suivent le dépôt du liquide. L'essai est effectué trois fois, à moins qu'une inflammation ou une carbonisation ne survienne.
2. DONNÉES
2.1. TRAITEMENT DES RÉSULTATS
L'essai peut être interrompu dès que l'un des essais donne un résultat positif.
2.2. ÉVALUATION
Si la substance s'enflamme dans les cinq minutes après avoir été ajoutée à un porteur inerte et exposée à l'air, ou si un liquide carbonise ou enflamme un papier-filtre dans les cinq minutes après avoir été déposé sur ce dernier et exposé à l'air, il est considéré comme pyrophore.
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés),
|
|
—
|
les résultats de l'essai,
|
|
—
|
toute observation supplémentaire pertinente pour l'interprétation des résultats.
|
4. RÉFÉRENCES
|
(1)
|
NF T 20-039 (septembre 1985), Produits chimiques à usage industriel — Détermination de l'inflammabilité spontanée des solides et liquides.
|
|
(2)
|
Recommandations relatives au transport des marchandises dangereuses — Épreuves et critères, 1990, Nations unies, New York.
|
A.14. DANGER D'EXPLOSION
1. MÉTHODE
1.1. INTRODUCTION
Cette méthode permet de déterminer si une substance solide ou pâteuse présente un danger d'explosion lorsqu'elle est soumise à l'effet d'une flamme (sensibilité thermique), à un choc ou à une friction (sensibilité aux stimulations mécaniques), et si une substance liquide présente un danger d'explosion lorsqu'il est soumis à l'effet d'une flamme ou d'un choc.
La méthode comprend trois parties:
|
a)
|
un essai de sensibilité thermique (1),
|
|
b)
|
un essai de sensibilité mécanique (choc) (1),
|
|
c)
|
un essai de sensibilité mécanique (friction) (1).
|
La méthode fournit des données permettant d'évaluer la probabilité d'amorcer une explosion par certaines stimulations ordinaires. Elle n'a pas pour objet d'affirmer qu'une substance n'est pas susceptible d'exploser dans certaines conditions.
La méthode est apte à déterminer si une substance présentera un danger d'explosion (sensibilité thermique et mécanique) dans les conditions particulières définies par la directive. Elle repose sur un certain nombre de types d'appareils qui sont largement utilisés sur le plan international (1) et qui donnent en règle générale des résultats probants. Il est cependant admis qu'elle n'a pas une valeur définitive. D'autres appareils que ceux qui sont mentionnés peuvent être utilisés, à condition qu'ils soient reconnus à l'échelle internationale et que les résultats puissent être mis en corrélation avec les résultats obtenus avec les appareils spécifiés.
Il n'est pas nécessaire d'effectuer les essais lorsque les informations disponibles, d'ordre thermodynamique (par exemple: chaleur de formation, chaleur de décomposition) ou structural (absence de certains groupes réactifs (2) dans la formule développée), montrent que la substance n'est, sans aucun doute, pas susceptible de se décomposer rapidement en libérant des gaz ou de la chaleur (à savoir, que ce matériau ne présente aucun risque d'explosion). Il est inutile de réaliser un essai de sensibilité mécanique à la friction pour les liquides.
1.2. DÉFINITIONS ET UNITÉS
Substances explosibles:
substances qui sont susceptibles d'exploser sous l'effet d'une flamme, ou qui sont sensibles au choc ou à la friction dans l'appareil spécifié (ou dont la sensibilité mécanique est supérieure à celle du 1,3-dinitrobenzène dans un autre appareil).
1.3. SUBSTANCES DE RÉFÉRENCE
1,3-dinitrobenzène, produit technique cristallisé, tamisé (qui passe au travers d'une maille de 0,5 mm) pour la méthode d'essai par friction et par choc.
Perhydro-l,3,5,-trinitro-l,3,5-triazine (RDX, hexogène, cyclonite — CAS 121-82-4), recristallisé à partir d'une solution aqueuse de cyclohexanone, tamisé mouillé au travers d'une maille de 250 μm et retenu par une maille de 150 μm) puis séché à 103 ± 2 oC (pendant 4 heures) pour la seconde série d'essais par friction et par choc.
1.4. PRINCIPE DE LA MÉTHODE
Des essais préliminaires sont nécessaires pour établir les conditions de sécurité devant présider à l'exécution des trois essais de sensibilité.
1.4.1. Essais de sécurité de manipulation (3)
Pour des raisons de sécurité, avant d'effectuer les essais proprement dits, des échantillons très réduits (10 mg environ) de substance sont soumis à échauffement sans confinement dans la flamme d'un brûleur à gaz, à un choc dans toute forme appropriée d'appareil, et à friction en utilisant un maillet et une enclume ou tout autre type de dispositif à friction. Ces essais ont pour objectif de déterminer si la substance est sensible et explosible au point de devoir s'entourer de précautions particulières pour réaliser les essais de sensibilité prescrits, notamment les essais de sensibilité thermique, afin d'éviter tout dommage corporel pour l'expérimentateur.
1.4.2. Sensibilité thermique
Cette méthode consiste à chauffer la substance dans un tube d'acier, fermé par des plaques à orifice dont le trou peut avoir différents diamètres, pour déterminer si la substance ou la préparation est susceptible d'exploser dans des conditions de température très élevée et de confinement défini.
1.4.3. Sensibilité mécanique (choc)
Cette méthode consiste à soumettre la substance au choc d'une masse spécifiée tombant d'une hauteur définie.
1.4.4. Sensibilité mécanique (friction)
La méthode consiste à soumettre des substances solides ou pâteuses à une friction entre des surfaces standard dans des conditions spécifiées de charge et de mouvement relatif.
1.5. CRITÈRES DE QUALITÉ
Non indiqués.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Sensibilité thermique (effet d'une flamme)
1.6.1.1. Appareil
L'appareil consiste en un tube d'acier non réutilisable et de son système de fermeture réutilisable (figure 1), installé dans un dispositif de chauffage et de protection. Chaque tube, d'un diamètre interne de 24 mm, d'une longueur de 75 mm et d'une épaisseur de paroi de 0,5 mm, est réalisé par un processus d'emboutissage profond à partir d'une tôle (voir appendice). Les tubes sont pourvus d'une bride de fermeture à leur extrémité ouverte afin de pouvoir les fermer par une plaque à orifice. Il s'agit d'une plaque solidement assujettie au tube par un joint fileté en deux parties (écrou et écrou borgne), résistante à la pression et munie d'un orifice central. L'écrou et l'écrou borgne sont en acier au chrome-manganèse (voir annexe) ne produisant aucune étincelle jusqu'à 800 oC. Les plaques ont une épaisseur de 6 mm et sont fabriquées avec un acier résistant à la chaleur (voir annexe). L'expérimentateur dispose d'une série de plaques dont l'orifice présente différents diamètres.
1.6.1.2. Conditions de l'essai
L'essai est normalement réalisé avec la substance telle qu'elle est reçue; toutefois, dans certains cas, par exemple si elle est comprimée, coulée ou condensée d'une manière quelconque, il peut être nécessaire de la désagréger avant de procéder à l'essai.
Pour les solides, la masse de matériel à utiliser pour chaque essai est déterminée par une opération préliminaire à deux étapes. Placer un volume de substance de 9 cm3 dans un tube taré, puis tasser en appliquant une force de 80 N sur toute la surface de la section du tube. D'autres méthodes de remplissage doivent parfois être utilisées pour des raisons de sécurité ou lorsque l'état physique de l'échantillon peut être modifié par la compression; par exemple, si la substance est très sensible à la friction, il ne faut pas tasser. Si le matériel est compressible, en rajouter et tasser jusqu'à ce que le tube soit rempli jusqu'à 55 mm du bord. Déterminer la masse totale utilisée pour remplir le tube jusqu'à ce niveau, puis ajouter de la substance encore deux fois, en tassant à chaque fois avec une force de 80 N. Ensuite, rajouter du matériel et tasser, ou en enlever, de manière à ce que le tube soit rempli jusqu'à 15 mm du bord. Effectuer une seconde opération préliminaire en commençant avec une quantité tassée correspondant au tiers de la masse totale déterminée lors de l'opération précédente. Ajouter deux fois de la substance en tassant avec une force de 80 N et ajuster le niveau à 15 mm du bord du tube en ajoutant du matériel ou en enlevant. La quantité de produit solide déterminée lors de cette seconde opération est utilisée pour chaque essai; le remplissage est effectué en trois fois avec des quantités égales de substance comprimées afin que leur volume soit de 9 cm3, quelle que soit la force nécessaire pour y parvenir. (Cette opération peut être facilitée par l'emploi d'anneaux d'espacement.)
Les liquides et les gels sont versés dans le tube jusqu'à une hauteur de 60 mm, en prenant soin d'éviter la formation de bulles d'air avec les gels. L'écrou borgne est glissé sur le tube par le bas, la plaque à orifice appropriée est insérée et l'écrou est serré après qu'a été appliqué un lubrifiant à base de bisulfite de molybdène. Il est important de vérifier qu'aucune substance n'est emprisonnée entre la bride et la plaque, ni dans les filetages.
Le chauffage est assuré par du propane venant d'une bouteille industrielle, équipée d'un régulateur de pression (60 à 70 mbar), grâce à un débitmètre, et distribué régulièrement (ce que l'on vérifie en observant les flammes des brûleurs) par un collecteur à quatre brûleurs. Les brûleurs sont situés autour de la chambre d'essai, comme indiqué à la figure 1. Les quatre brûleurs consomment ensemble 3,2 litres de propane par minute, environ. Il est possible d'utiliser d'autres gaz de chauffage et d'autres brûleurs, mais la vitesse de chauffage doit correspondre à celle qui est spécifiée à la figure 3. Pour tous les appareils, la vitesse de chauffage doit être vérifiée régulièrement en utilisant des tubes remplis de phtalate de dibutyle, comme indiqué à la figure 3.
1.6.1.3. Déroulement des essais
Chaque essai est poursuivi jusqu'à ce que le tube se soit fragmenté ou que le tube ait été chauffé pendant cinq minutes. Un essai aboutissant à la fragmentation du tube en trois parties, ou plus, qui peuvent parfois être reliées l'une à l'autre par d'étroites bandes de métal, comme l'illustre la figure 2, est considéré comme produisant une explosion. Un essai aboutissant à un plus petit nombre de fragments ou à une absence de fragmentation, est considéré comme ne produisant pas d'explosion.
Une série de trois essais est d'abord effectuée avec une plaque dont l'orifice a un diamètre de 6,0 mm et, si aucune explosion ne se produit, une seconde série de trois essais est réalisée avec une plaque dont l'orifice a un diamètre de 2,0 mm. S'il se produit une explosion au cours de l'une des séries d'essais, aucun essai supplémentaire n'est requis.
1.6.1.4. Évaluation
Le résultat de l'essai est considéré comme positif s'il se produit une explosion au cours de l'une des séries d'essais mentionnées ci-avant.
1.6.2. Sensibilité mécanique (choc)
1.6.2.1. Appareil (figure 4)
Le mouton de choc classique comprend essentiellement: un bloc de fonte avec une embase, une enclume, une colonne, des guides, des masses tombantes, un mécanisme de libération et un support pour l'échantillon. L'enclume d'acier (100 mm de diamètre × 70 mm de hauteur) est vissée sur un bloc d'acier (230 mm de longueur × 250 mm de largeur × 200 mm de hauteur) avec une embase coulée (450 mm de longueur × 450 mm de largeur × 60 mm de hauteur). Une colonne, qui consiste en un tube d'acier étiré sans soudure, est fixée dans un support vissé au dos du bloc d'acier. Quatre vis ancrent l'appareil dans un bloc de béton (60 × 60 × 60 cm) de telle sorte que les rails soient absolument verticaux et que la chute de la masse tombante ne soit pas entravée. L'expérimentateur dispose de masses de 5 et 10 kg, en acier trempé et dont la surface d'impact, d'un diamètre minimal de 25 mm, est en acier traité HRC 60 à 63.
L'échantillon à essayer est enfermé dans une matrice de choc constituée de deux cylindres coaxiaux en acier trempé, placés l'un au dessus de l'autre dans un cylindre d'acier creux faisant office de bague de guidage. Les cylindres d'acier trempé doivent avoir un diamètre de 10 (-0,003, -0,005) mm et une hauteur de 10 mm, des surfaces polies, des arêtes arrondies (rayon de courbure 0,5 mm) et une dureté de HRC 58 à 65. Le cylindre creux doit avoir un diamètre extérieur de 16 mm, un alésage de 10 (+0,005, +0,010) mm et une hauteur de 13 mm. La matrice de choc est placée sur une enclume intermédiaire (26 mm de diamètre et 26 mm de hauteur) en acier, centrée par une bague munie de perforations permettant aux vapeurs de s'échapper.
1.6.2.2. Conditions de l'essai
L'échantillon doit avoir un volume de 40 mm3, ou un volume compatible avec l'appareil utilisé. Les substances solides doivent être essayées à l'état sec et préparées comme suit:
|
a)
|
les substances pulvérulentes sont tamisées (maille de 0,5 mm); la fraction séparée par tamisage est entièrement utilisée pour l'essai;
|
|
b)
|
les substances comprimées, coulées ou condensées sont désagrégées et tamisées; la fraction de 0,5 à 1 mm de diamètre séparée par tamisage est utilisée pour l'essai et doit être représentative de la substance originale.
|
Les substances sous forme de pâte doivent être essayées à l'état sec si c'est possible, ou, en tout cas, après avoir enlevé la plus grande quantité possible de diluant. Pour ce qui est des substances liquides, l'essai est réalisé avec un intervalle de 1 mm entre le cylindre d'acier du haut et celui du bas.
1.6.2.3. Déroulement des essais
On effectue une série de six essais en faisant tomber une masse de 10 kg d'une hauteur de 0,40 m (40 J). S'il se produit une explosion au cours des six essais effectués à 40 J, une autre série de six essais doit être effectuée en faisant tomber une masse de 5 kg d'une hauteur de 0,15 m (7,5 J). Dans d'autres appareils, l'échantillon est comparé avec la substance de référence choisie selon la procédure établie (technique du «up-and-down», etc.).
1.6.2.4. Évaluation
Le résultat de l'essai est considéré comme positif si une explosion (l'inflammation ou un bruit intense équivaut à une explosion) se produit au moins une fois au cours des six essais réalisés avec l'appareil de choc indiqué ou si l'échantillon est plus sensible que le 1,3-dinitrobenzène ou le RDX lors d'un autre essai par choc.
1.6.3. Sensibilité mécanique (friction)
1.6.3.1. Appareil (figure 5)
L'appareil consiste en une plaque de base en fonte sur laquelle est monté le dispositif de friction proprement dit comprenant un crayon fixe en porcelaine et une plaquette mobile en porcelaine. La plaquette est fixée dans un coulisseau se déplaçant entre deux rails. Le coulisseau est relié par une barre d'entraînement et un engrenage de transmission excentrique à un moteur électrique, de telle sorte que la plaquette se déplace une fois seulement sur une distance de 10 mm, en arrière et en avant sous le crayon. Le crayon peut être chargé, par exemple, à 120 ou 360 newtons.
Les plaquettes de porcelaine plates sont en porcelaine blanche technique (rugosité de 9 à 32 μm) et ont les dimensions suivantes: 25 mm de longueur, 25 mm de largeur et 5 mm de hauteur. Le crayon cylindrique qui est également en porcelaine blanche technique (longueur 15 mm, diamètre 10 mm), présente des extrémités sphériques rugueuses dont le rayon de courbure est de 10 mm.
1.6.3.2. Conditions de l'essai
L'échantillon doit avoir un volume de 10 mm3, ou un volume compatible avec l'appareil utilisé.
Les substances solides sont essayées à l'état sec et préparées comme suit:
|
a)
|
les substances pulvérulentes sont tamisées (maille de 0,5 mm); la fraction séparée par tamisage est entièrement utilisée pour l'essai;
|
|
b)
|
les substances comprimées, coulées ou condensées sont désagrégées et tamisées; la fraction tamisée d'un diamètre inférieur à 0,5 mm est utilisée pour l'essai.
|
Les substances sous forme de pâte doivent être essayées à l'état sec si possible. Si la substance ne peut pas être préparée à l'état sec, la pâte (après avoir enlevé la plus grande quantité possible de diluent) est essayée sous la forme d'un film de 0,5 mm d'épaisseur, 2 mm de largeur et 10 mm de longueur préparé à l'aide d'un gabarit.
1.6.3.3. Déroulement des essais
Placer le crayon de porcelaine sur l'échantillon soumis à essai et accrocher le poids. Lors de la réalisation de l'essai, les marques laissées par l'éponge sur la plaquette de porcelaine doivent être transversales par rapport à la direction du mouvement. Veiller à ce que le crayon repose sur l'échantillon, à ce que la quantité de substance essayée soit suffisante et à ce que la plaquette se déplace correctement sous le crayon. Les substances pâteuses seront appliquées sur le plateau à l'aide d'une gabarit de 0,5 mm d'épaisseur avec une rainure de 2 × 10 mm. La plaquette de porcelaine accomplit sous le crayon un mouvement de va-et-vient sur une distance de 10 mm dans chaque direction en 0,44 secondes. N'utiliser chaque partie de la surface de la plaquette et du crayon que pour un seul essai; les deux extrémités de chaque crayon serviront pour deux essais et les deux surfaces de la plaquette pour trois essais.
Une série de six essais est effectuée avec une charge de 360 N. Si un résultat positif survient au cours de ces six essais, il convient d'effectuer une autre série de six essais avec une charge de 120 N. Dans d'autres appareils, l'échantillon est comparé avec la substance de référence choisie en suivant un mode opératoire établi (technique «up-and-down», etc.).
1.6.3.4. Évaluation
Le résultat de l'essai est considéré comme positif s'il se produit au moins une explosion (un crépitement, un bruit intense ou une inflammation sont équivalents à une explosion) au cours de l'un des essais réalisés avec l'appareil de friction indiqué ou s'il satisfait aux critères équivalents pour un autre essai de friction.
2. DONNÉES
En principe, une substance est considérée comme présentant un risque d'explosion dans le sens de la directive dés qu'un résultat est positif lors des essais de sensibilité thermique, au choc ou à la friction.
3. RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
l'identité, la composition, la pureté, le contenu en humidité, etc., de la substance soumise à essai,
|
|
—
|
la forme physique de l'échantillon; préciser s'il a été désagrégé, cassé ou tamisé,
|
|
—
|
les observations relevées au cours des essais de sensibilité thermique (par exemple masse de l'échantillon, nombre de fragments, etc.),
|
|
—
|
les observations relevées au cours des essais de sensibilité mécanique (par exemple formation d'une quantité considérable de fumée ou décomposition complète sans bruit intense, flammes, étincelles, crépitation, etc.),
|
|
—
|
les résultats de chaque type d'essai,
|
|
—
|
si un autre appareil a été utilisé, fournir une justification scientifique ainsi que la preuve de la corrélation entre les résultats obtenus avec l'appareil spécifié et les résultats obtenus avec l'appareil utilisé,
|
|
—
|
tout commentaire utile, se référant notamment aux essais pratiqués avec des produits similaires, susceptible d'être pertinent pour une interprétation correcte des résultats,
|
|
—
|
toutes observations supplémentaires pertinentes pour l'interprétation des résultats.
|
3.2. INTERPRÉTATION ET ÉVALUATION DES RÉSULTATS
Le rapport d'essai doit mentionner tous les résultats considérés comme erronés, anormaux, ou non représentatifs. Lorsqu'un des résultats est rejeté, fournir une explication et indiquer les résultats de tout essai substitutif ou supplémentaire. À moins de pouvoir expliquer un résultat anormal, celui-ci doit être accepté tel quel et utilisé pour classer la substance en conséquence.
4. RÉFÉRENCES
|
(1)
|
Recommandations relatives au transport des marchandises dangereuses, Épreuves et critères, 1990, Nations unies, New York.
|
|
(2)
|
Bretherick, L., Handbook of Reactive Chemical Hazards, 4th edition, Butterworths, London, ISBN 0-750-60103-5, 1990.
|
|
(3)
|
Koenen, H., Ide, K.H. und Swart, K.H. Explosive Stoffe, 1961, vol.3, 6-13 and 30-42.
|
|
(4)
|
NFT 20-038 (septembre 1985). Produits chimiques à usage industriel — Détermination du danger d'explosion.
|
Annexe
Exemple de spécification de matériel pour les essais de sensibilité thermique (voir norme DIN 1623)
|
(1)
|
Tube: Spécification de matériel No 1.0336.505 g
|
|
(2)
|
Plaque à orifice: Spécification de matériel No 1.4873
|
|
(3)
|
Écrou et écrou borgne: Spécification de matériel No 1.3817
|
Figure 1
Appareil d'essai de sensibilité thermique
(dimensions en mm)

Figure 2
Essai de sensibilité thermique
Exemples de fragmentation

Figure 3
Étalonnage de la vitesse d'échauffement pour essai de sensibilité thermique TEMPS (s)

Courbe de température en fonction du temps obtenue en chauffant du phtalate de dibutyle (27 cm3) dans un tube fermé (plaque à orifice de 1,5 mm) au moyen d'un débit de propane de 3,2 litres/minute. La température est mesurée à l'aide d'un thermocouple chromel/alumel gainé d'acier inoxydable de 1 mm de diamètre, placé en position centrale, 43 mm sous le bord du tube. Entre 135 oC et 285 oC, la vitesse d'échauffement doit se situer entre 185 et 215 K/minute.
Figure 4
Appareil d'essai de sensibilité au choc
(dimensions en mm)

Figure 4
suite
Figure 5
Appareil d'essai de sensibilité à la friction

A.15. TEMPÉRATURE D'INFLAMMATION SPONTANÉE DES LIQUIDES ET DES GAZ
1. MÉTHODE
1.1. INTRODUCTION
Les substances explosives qui s'enflamment spontanément au contact de l'air à température ambiante ne doivent pas être soumises à cet essai. Le mode opératoire est applicable aux gaz, aux liquides et aux vapeurs qui peuvent être enflammés par une surface chaude, en présence d'air.
La température d'inflammation spontanée peut être réduite considérablement par la présence d'impuretés catalytiques, par la matière de la surface ou par une augmentation du volume du récipient d'essai.
1.2. DÉFINITIONS ET UNITÉS
Le degré d'inflammabilité spontanée est exprimé en termes de température d'inflammation spontanée. La température d'inflammation spontanée est la température la plus basse à laquelle s'enflamme la substance à essayer mélangée avec de l'air dans les conditions définies dans la méthode d'essai.
1.3. SUBSTANCES DE RÉFÉRENCE
Les substances de références sont citées dans les normes (voir 1.6.3). Elles devraient servir en premier lieu à vérifier de temps à autre la fiabilité de la méthode et à permettre de comparer les résultats obtenus avec différentes méthodes.
1.4. PRINCIPE DE LA MÉTHODE
La méthode permet de déterminer la température minimale de la surface interne d'une enceinte qui provoquera l'inflammation d'un gaz, d'une vapeur ou d'un liquide injecté dans l'enceinte.
1.5. CRITÈRES DE QUALITÉ
La répétabilité varie en fonction de l'intervalle de température d'inflammation spontanée et de la méthode d'essai utilisée.
La sensibilité et la spécificité dépendent de la méthode d'essai utilisée.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Appareil
L'appareil est décrit dans la méthode évoquée au point 1.6.3.
1.6.2. Conditions de l'essai
Un échantillon de la substance à essayer est essayé selon le point 1.6.3.
1.6.3. Mode opératoire
Voir normes IEC 79-4, DIN 51794, ASTM-E 659-78, BS 4056, NF T 20-037.
2. DONNÉES
Noter la température d'essai, la pression atmosphérique, la quantité d'échantillon utilisée et le laps de temps qui s'écoule avant que l'inflammation se produise.
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
les spécifications précises de la substance (identité et impuretés),
|
|
—
|
la quantité d'échantillon utilisée, la pression atmosphérique,
|
|
—
|
les résultats des mesures (température d'essai, résultats concernant l'inflammation, laps de temps correspondants),
|
|
—
|
toute observation supplémentaire pertinente pour l'interprétation des résultats.
|
4. RÉFÉRENCES
Aucune.
A.16. TEMPÉRATURE RELATIVE D'INFLAMMATION SPONTANÉE POUR LES SOLIDES
1. MÉTHODE
1.1. INTRODUCTION
Les substances explosibles et les substances qui s'enflamment spontanément au contact de l'air à température ambiante ne doivent pas être soumises à cet essai.
L'objectif en est de fournir des données préliminaires sur l'inflammabilité spontanée des substances solides soumises à de hautes températures.
Si la chaleur engendrée, soit par une réaction de la substance avec l'oxygène, soit par décomposition exothermique, ne se dissipe pas assez rapidement dans l'environnement, l'auto-échauffement entraîne l'inflammation spontanée. L'inflammation spontanée se produit par conséquent lorsque la vitesse de production de chaleur excède la vitesse de perte de chaleur.
Le procédé d'essai est utile en tant que test préliminaire de sélection des substances solides. Compte tenu de la nature complexe de l'inflammation et de la combustion des solides, la température d'inflammation spontanée déterminée selon cette méthode d'essai ne doit servir qu'à des fins de comparaison.
1.2. DÉFINITIONS ET UNITÉS
La température d'inflammation spontanée telle qu'elle est déterminée par cette méthode est la température ambiante minimale exprimée en degrés Celsius ( oC), à laquelle un certain volume d'une substance s'enflamme spontanément dans des conditions définies.
1.3. SUBSTANCES DE RÉFÉRENCE
Aucune.
1.4. PRINCIPE DE LA MÉTHODE
Placer un volume défini de la substance soumise à essai dans un four à température ambiante; enregistrer la courbe température/temps au centre de l'échantillon, la température du four étant portée à 400 oC, ou à la température du point de fusion si celle-ci est inférieure, à la vitesse de 0,5 oC/mn. Pour les besoins de cet essai, la température du four à laquelle l'échantillon atteint une température de 400 oC par auto-échauffement est appelée température d'inflammation spontanée.
1.5. CRITÈRES DE QUALITÉ
Aucun.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Appareil
1.6.1.1. Four
Un four de laboratoire à température programmable (d'un volume de 2 litres environ) équipé d'une circulation d'air naturel et d'un clapet d'explosion. Pour éviter tout risque potentiel d'explosion, il convient d'empêcher que les gaz de décomposition puissent entrer en contact avec les éléments de chauffage électrique.
1.6.1.2. Cube en treillis de fil métallique
Couper un morceau de treillis en fil d'acier inoxydable d'une maille d'une taille de 0,045 mm, selon le modèle indiqué à la figure 1. Plier le treillis et l'attacher avec du fil métallique en forme de cube ouvert.
1.6.1.3. Thermocouples
Thermocouples appropriés.
1.6.1.4. Enregistreur
Tout enregistreur à deux canaux, étalonné à l'intervalle 0 — 600 oC ou à une tension correspondante.
1.6.2. Conditions de l'essai
Les substances sont testées telles qu'elles sont reçues.
1.6.3. Mode opératoire
Remplir le cube de la substance soumise à essai. Tasser doucement en ajoutant de la substance jusqu'à le remplir complètement. Suspendre ensuite le cube au centre du four à température ambiante. Placer un thermocouple au centre du cube et l'autre entre le cube et la paroi du four afin d'enregistrer la température du four.
Les températures du four et de l'échantillon sont enregistrées en continu pendant que la température du four est portée à 400 oC, ou au point de fusion si cette valeur est plus basse, à une vitesse de 0,5 oC/min.
Lorsque la substance s'enflamme, le thermocouple de l'échantillon indiquera une très forte montée de température par rapport à la température du four.
2. DONNÉES
La température du four à laquelle la température de l'échantillon atteint 400 oC par auto-échauffement est pertinente pour l'évaluation (voir figure 2).
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
une description de la substance à essayer,
|
|
—
|
les résultats des mesures incluant la courbe température/temps,
|
|
—
|
toute observation supplémentaire pertinente pour l'interprétation des résultats.
|
4. RÉFÉRENCES
|
(1)
|
NF T 20-036 (septembre 1985). Produits chimiques à usage industriel — Détermination de la température relative d'inflammation spontanée des solides.
|
Figure 1
Modèle de cube d'essai de 20 mm

Figure 2
Courbe type température/temps

A.17. PROPRIÉTÉS COMBURANTES (SOLIDES)
1. MÉTHODE
1.1. INTRODUCTION
Il est utile de disposer d'informations préliminaires sur les propriétés explosives éventuelles de la substance avant d'effectuer cet essai.
Cet essai n'est pas applicable aux liquides, aux gaz, aux substances explosives ou très inflammables ou aux peroxydes organiques.
Cet essai est inadapté quand l'examen de la structure chimique montre que la substance ne peut sans aucun doute avoir de réaction de type exothermique avec un combustible.
Un essai préliminaire doit être effectué pour s'assurer que cet essai ne nécessite pas de précautions particulières.
1.2. DÉFINITIONS ET UNITÉS
Temps de combustion: temps de réaction, exprimé en secondes, pris par la zone de réaction pour se propager à travers un tas, selon la procédure décrite au point 1.6.
Vitesse de combustion: exprimée en millimètres par seconde.
Vitesse maximale de combustion: valeur la plus élevée parmi les vitesses de combustion obtenues avec des mélanges contenant 10 à 90 % en poids de comburant.
1.3. SUBSTANCE DE RÉFÉRENCE
Le nitrate de barium (de pureté analytique) est utilisé comme substance de référence pour l'essai et l'essai préliminaire.
Le mélange de référence est composé de nitrate de barium et de cellulose en poudre, préparé conformément au point 1.6 et possédant la vitesse de combustion maximale (il s'agit généralement d'un mélange contenant 60 % de nitrate de barium en poids).
1.4. PRINCIPE DE LA MÉTHODE
Il convient, pour des raisons de sécurité, d'effectuer un essai préliminaire. Il est inutile de continuer l'essai si l'essai préliminaire montre clairement que la substance a des propriétés oxydantes. Lorsque ce n'est pas le cas, la substance est soumise à l'essai complet.
Dans l'essai complet, la substance à essayer et une substance combustible définie sont mélangées dans des proportions variables. Chacun de ces mélanges est alors disposé en tas, que l'on enflamme à une extrémité. La vitesse maximale de combustion est comparée à la vitesse maximale de combustion du mélange de référence.
1.5. CRITÈRES DE QUALITÉ
Lorsqu'elle est requise, toute méthode de broyage et de mélange est valable pour autant que l'écart entre la vitesse maximale de combustion et la moyenne arithmétique, dans les six essais, ne dépasse pas 10 %.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Préparation
1.6.1.1. Substance à essayer
L'échantillon à essayer est traité comme suit afin d'obtenir une granulométrie inférieure à 0,125 mm: tamiser la substance à essayer et broyer la fraction restante; répéter cette opération jusqu'à ce que tout l'échantillon soit passé au travers du tamis.
Toutes méthodes de broyage et de tamisage respectant les critères de qualité requis peuvent être utilisées.
Avant de préparer le mélange, sécher la substance à 105 oC jusqu'à obtention d'un poids constant. Si la température de décomposition de la substance est inférieure à 105 oC, la substance doit être séchée à une température moins élevée.
1.6.1.2. Substance combustible
On utilise de la cellulose en poudre comme substance combustible. Elle doit être du type utilisé en chromatographie en couche mince ou en colonne. Un type de cellulose dont la longueur de plus de 85 % des fibres est comprise entre 0,020 mm et 0,075 mm s'est avérée adéquate. La poudre de cellulose est tamisée au moyen d'un tamis à mailles de 0,125 mm. Il faut utiliser le même lot de cellulose pour tout l'essai.
Avant de préparer le mélange, sécher la poudre de cellulose à 105 oC jusqu'à ce que son poids soit constant.
Si de la sciure de bois est utilisée lors de l'essai préliminaire, préparer une sciure de bois tendre qui passe au travers d'un tamis d'une maille de 1,6 mm, mélanger, puis sécher à 105 oC pendant 4 heures en couche de moins de 25 mm d'épaisseur. Refroidir la sciure et la conserver jusqu'à utilisation dans un récipient hermétique aussi rempli que possible. Elle sera de préférence utilisée dans les 24 heures après séchage.
1.6.1.3. Source d'allumage
Utiliser la flamme d'un brûleur à gaz (diamètre; 5 mm minimum). Si une autre source d'allumage est utilisée (par exemple lorsque l'essai est effectué dans une atmosphère inerte), fournir la description et la justification.
1.6.2. Mode opératoire
Note:
Les mélanges de comburants avec de la cellulose ou de la sciure de bois doivent être considérés comme des explosifs potentiels et manipulés avec prudence.
1.6.2.1. Essai préliminaire
La substance séchée est grossièrement mélangée à de la cellulose ou à de la sciure de bois sèche dans des proportions de 2 (substance à essayer) pour 1 (cellulose ou sciure de bois) en poids et le mélange est disposé en petit tas conique de 3,5 cm (diamètre de base) × 2,5 cm (hauteur) par remplissage sans tassement d'une forme conique (par exemple un entonnoir de laboratoire en verre dont le tube est bouché).
Le tas est placé sur une plaque froide, non-combustible, non poreuse, et peu thermo-conductrice. L'essai doit être conduit dans une hotte aspirante (voir point 1.6.2.2).
La source d'allumage est mise en contact avec le cône. L'intensité et la durée de la réaction sont observées et enregistrées.
La substance doit être considérée comme comburante si la réaction est intense.
Lorsqu'il y a doute quant au résultat, il est nécessaire de pratiquer l'essai complet décrit ci-après.
1.6.2.2. Essai complet
Préparer des mélanges oxydant/cellulose contenant de 10 à 90 % de comburant, en poids, par incréments de 10 %. Pour les cas limites utiliser des mélanges comburant/cellulose intermédiaires pour déterminer la vitesse maximale de combustion avec plus de précision.
On réalise un tas au moyen d'un moule métallique d'une longueur de 250 mm et dont la section triangulaire transversale a une hauteur intérieure de 10 mm et une largeur intérieure de 20 mm. Placer comme cadres latéraux des deux côtés du moule, dans le sens de la longueur, deux plaques métalliques dépassant de 2 mm le bord supérieur de la section triangulaire (figure). Remplir ce dispositif avec un excès de mélange, sans tasser. Après avoir laissé tomber le moule une fois d'une hauteur de 2 cm sur une surface dure, racler l'excès avec une raclette tenue obliquement. Enlever alors les cadres latéraux et égaliser la surface de la poudre à l'aide d'un rouleau. Disposer une plaque non-combustible non-poreuse et peu thermo-conductrice sur le moule, retourner et démouler.
Disposer le tas dans une hotte perpendiculairement au sens de courant d'air.
Le débit d'air doit être suffisant pour empêcher les vapeurs de se répandre dans le laboratoire et il ne doit pas subir de modification au cours de l'essai. Le courant d'air doit former un écran entourant l'appareil.
Vu les propriétés hygroscopiques de la cellulose et de certaines substances à essayer, l'essai doit être effectué aussi rapidement que possible.
Enflammer une extrémité du tas par contact avec la flamme.
Mesurer le temps de réaction sur une distance de 200 mm après que la zone de réaction a parcouru une distance initiale de 30 mm.
L'essai est effectué avec la substance de référence et au moins une fois avec chacun des mélanges de substance à essayer et de cellulose.
Si l'on trouve une vitesse maximale de combustion significativement plus grande que celle de la substance de référence, l'essai peut être arrêté; autrement, les essais doivent être répétés cinq fois avec chacun des trois mélanges qui donnent les vitesses de combustion les plus élevées.
Si l'on soupçonne que le résultat est un «faux positif», répéter l'essai en remplaçant la cellulose par une substance inerte dont la taille des particules est la même, comme le kieselguhr. Une autre possibilité consiste à renouveler l'essai avec le mélange substance à essayer/cellulose dont la vitesse de combustion est la plus élevée, dans une atmosphère inerte (contenu d'oxygène < 2 % v/v).
2. DONNÉES
Pour des raisons de sécurité, la vitesse maximale de combustion — et non la valeur moyenne — sera retenue pour caractériser les propriétés comburantes de la substance examinée.
Pour l'évaluation d'un mélange donné, retenir la vitesse de combustion la plus élevée mesurée au cours des six essais.
Porter sur un graphique la vitesse de combustion la plus élevée de chaque mélange en fonction de la concentration de comburant. Déduire de ce graphique la vitesse de combustion maximale.
Les six vitesses de combustion mesurées au cours d'une même opération pour le mélange qui a donné la vitesse de combustion maximale ne doit pas s'écarter de plus de 10 % de la moyenne arithmétique; dans le cas contraire, il importe d'améliorer les méthodes de broyage et de mélange.
Comparer la vitesse maximale de combustion obtenue avec la vitesse maximale de combustion du mélange de référence (voir point 1.3).
Si les essais sont réalisés dans une atmosphère inerte, comparer la vitesse maximale avec la vitesse de réaction du mélange de référence dans une atmosphère inerte.
3. RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
l'identité, la composition, la pureté, le contenu en humidité etc., de la substance à essayer,
|
|
—
|
tout traitement de l'échantillon à essayer (par exemple, broyage, séchage...),
|
|
—
|
la source d'allumage utilisée dans les essais,
|
|
—
|
les résultats des mesures,
|
|
—
|
le type de réaction (par exemple combustion rapide superficielle, combustion à travers toute la masse, toute observation concernant les produits de combustion,...),
|
|
—
|
toute observation supplémentaire, y compris une description de l'intensité (flamme, étincelle, fumée, lente incandescence, etc.) et la durée approximative de la réaction observée au cours de l'essai préliminaire de sécurité/dépistage pour la substance à essayer comme pour la substance de référence,
|
|
—
|
les résultats des essais effectués avec un matériel inerte, s'il y a lieu,
|
|
—
|
les résultats des essais effectués dans une atmosphère inerte, s'il y a lieu.
|
3.2. INTERPRÉTATION DES RÉSULTATS
Une substance est à considérer comme comburante si:
|
a)
|
lors de l'essai préliminaire, il y a réaction intense;
|
|
b)
|
lors de l'essai, la vitesse maximale de combustion de la substance soumise à essai est supérieure ou égale à celle du mélange de référence composé de cellulose et de nitrate de barium.
|
Pour éviter un «faux» résultat positif, il faut aussi tenir compte des résultats obtenus en essayant la substance mélangée avec un matériel inerte et/ou dans une atmosphère inerte, lors de l'interprétation des résultats.
4. RÉFÉRENCES
NF T 20-035 (septembre 1985). Produits chimiques à usage industriel — Détermination des propriétés comburantes des solides.
Annexe
Figure
Moule et accessoires nécessaires à la confection de tas
(toutes les dimensions sont en millimètres)

A.18. DÉTERMINATION DE LA MASSE MOLÉCULAIRE MOYENNE EN NOMBRE ET DE LA DISTRIBUTION DES MASSES MOLÉCULAIRES DES POLYMÈRES
1. MÉTHODE
Cette méthode de chromatographie sur gel perméable reprend la ligne directrice OCDE TG 118 (1996). Les principes fondamentaux et tous autres renseignements techniques sont donnés en référence (1).
1.1. INTRODUCTION
Les propriétés des polymères sont tellement diversifiées qu'il est impossible de décrire une méthode unique indiquant avec précision des conditions de séparation et d'évaluation qui recouvrent toutes les éventualités et particularités rencontrées dans la séparation des polymères. Les systèmes complexes de polymères, notamment, se prêtent rarement à la chromatographie sur gel perméable. Lorsque la chromatographie sur gel perméable n'est pas applicable, on peut déterminer la masse moléculaire moyenne à l'aide d'autres méthodes (voir annexe). Dans de tels cas, il convient de fournir tous les détails de la méthode utilisée et de justifier son choix.
La méthode décrite ci-après est basée sur la norme D1N 55672 (1). Celle-ci fournit des indications détaillées sur la manière de réaliser les expériences et d'évaluer les données. S'il est nécessaire de modifier les conditions expérimentales, ces changements doivent être justifiés. D'autres normes peuvent être utilisées, à condition d'en mentionner toutes les références. La méthode décrite fait appel à des échantillons de polystyrène de polydispersité connue pour l'étalonnage et il y aura peut-être lieu de la modifier pour l'adapter aux cas de polymères particuliers, par exemple, les polymères solubles dans l'eau et les polymères ramifiés à longue chaîne.
1.2. DÉFINITIONS ET UNITÉS
La masse moléculaire moyenne en nombre Mn et la masse moléculaire moyenne en poids Mw sont déterminées à l'aide des équations suivantes:
où
Hi est l'amplitude du signal du détecteur, par rapport au niveau de référence, correspondant au volume de rétention Vi;
Mi est la masse moléculaire de la fraction de polymère correspondant au volume de rétention Vi et
n est le nombre de points expérimentaux.
La largeur de la distribution des masses moléculaires, qui représente une mesure de la polydispersité du système, est donnée par le rapport Mw/Mn.
1.3. SUBSTANCES DE RÉFÉRENCE
La méthode par chromatographie sur gel perméable étant une méthode relative, il est nécessaire de procéder à un étalonnage. Celui-ci est généralement effectué au moyen de polystyrènes étalons à chaîne linéaire et à distribution étroite, dont les masses moléculaires moyennes Mn et Mv ainsi que la distribution des masses moléculaires sont connues. La courbe d'étalonnage ne peut être utilisée pour déterminer la masse moléculaire d'un échantillon inconnu que si les conditions de séparation des échantillons et des étalons ont été sélectionnées de manière identique.
Une relation déterminée entre la masse moléculaire et le volume d'élution n'est valable que dans les conditions particulières d'une expérience donnée. Parmi ces conditions figurent, avant tout, la température, le solvant (ou le mélange de solvants), les conditions de chromatographie, ainsi que la colonne de séparation ou le système de colonnes.
Les masses moléculaires de l'échantillon déterminées de cette manière constituent des valeurs relatives et sont désignées par le terme «masses moléculaires en équivalent polystyrène». Cela signifie qu'en fonction des différences chimiques et structurelles entre l'échantillon à tester et les étalons, les masses moléculaires peuvent dévier des valeurs absolues de manière plus ou moins importante. Si d'autres étalons sont utilisés, par exemple le polyéthylène glycol, le poly(éthylène oxyde), le poly(méthacrylate de méthyle) ou l'acide polyacrylique, il importe d'en indiquer la raison.
1.4. PRINCIPE DE LA MÉTHODE
La chromatographie sur gel perméable permet de déterminer la distribution des masses moléculaires ainsi que les masses moléculaires moyennes (Mn, Mw). La chromatographie sur gel perméable constitue un type particulier de chromatographie liquide dans lequel l'échantillon à tester est séparé en fonction des volumes hydrodynamiques de chaque constituant (2).
La séparation s'effectue à mesure que l'échantillon progresse dans une colonne remplie d'un matériau poreux, généralement un gel organique. Les petites molécules peuvent pénétrer dans les pores tandis que les grosses molécules en sont exclues. Le parcours des grosses molécules est, de ce fait, plus court; elles sont éluées en premier lieu. Les molécules de taille moyenne pénètrent dans certains des pores; elles sont éluées plus tard. Les molécules les plus petites, dont le rayon hydrodynamique moyen est plus petit que les pores du gel, peuvent pénétrer dans tous les pores. Elles sont éluées en dernier lieu.
Dans la situation idéale, c'est la taille des espèces moléculaires qui régit entièrement la séparation, mais, en pratique, il est difficile d'éviter tout au moins l'interférence de certains effets d'absorption. Un remplissage irrégulier de la colonne, de même que des volumes morts, peuvent aggraver la situation (2).
La détection s'effectue, par exemple, par mesure de l'indice de réfraction ou de l'absorption dans l'UV pour donner une courbe de distribution simple. Cependant, pour pouvoir associer à la courbe des valeurs de masses moléculaires réelles, il est nécessaire d'étalonner la colonne par passage de polymères de masse moléculaire connue et, idéalement, d'une structure aussi proche que possible, par exemple divers polystyrènes étalons. Une courbe de Gauss en résulte généralement, parfois avec une dissymétrie marquée du côté des faibles masses moléculaires, l'axe vertical indiquant la quantité, en poids, des espèces de différentes masses moléculaires éluées, tandis que sur l'axe horizontal figure le logarithme de la masse moléculaire.
1.5. CRITÈRES DE QUALITÉ
La reproductibilité (écart-type relatif) du volume d'élution doit être au plus égale à 0,3 pour cent. Il y a lieu d'assurer la reproductibilité de l'analyse grâce à une correction mettant en jeu un étalon interne, si un chromatogramme est évalué en fonction du temps et ne satisfait pas aux critères mentionnés plus haut (1). Les polydispersités dépendent des masses moléculaires des étalons. Dans le cas des polystyrènes étalons, les valeurs typiques sont:
|
Mp < 2 000
|
Mw/Mn < 1,20
|
|
2 000 ≤ Mp ≤ 106
|
Mw/Mn < 1,05
|
|
Mp > 106
|
Mw/Mn < 1,20
|
(Mp est la masse moléculaire de l'étalon correspondant au maximum du pic)
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Préparation des solutions de polystyrène étalon
On dissout les polystyrènes étalons en les mélangeant soigneusement à l'éluant choisi. On préparera les solutions en observant les recommandations du fabricant.
Les concentrations des étalons choisis dépendent de différents facteurs, comme le volume d'injection, la viscosité de la solution et la sensibilité du détecteur. Le volume d'injection maximum doit être adapté à la longueur de la colonne, en veillant à ne pas surcharger celle-ci. Les volumes d'injection courants pour des séparations analytiques par chromatographie sur gel perméable, au moyen d'une colonne de 30 cm × 7,8 mm, varient normalement entre 40 et 100 ml. Il est possible d'injecter des volumes plus importants, mais ceux-ci ne peuvent dépasser 250 μl. On doit déterminer le rapport optimal entre le volume d'injection et la concentration avant l'étalonnage de la colonne.
1.6.2. Préparation de la solution de l'échantillon
En principe, la préparation des solutions de l'échantillon s'effectue dans les mêmes conditions. L'échantillon est dissous dans un solvant approprié, par exemple le tétrahydrofurane (THF), en agitant soigneusement. Il ne doit, en aucun cas, être dissous dans un bain à ultrasons. Si nécessaire, la solution de l'échantillon est purifiée au moyen d'un filtre à membrane d'une dimension de pores comprise entre 0,2 et 2 μm.
La présence de particules non dissoutes doit être mentionnée dans le rapport final, dans la mesure où elle peut résulter d'espèces de masse moléculaire élevée. Il faut recourir à une méthode appropriée pour déterminer le pourcentage en poids des particules non dissoutes. Les solutions doivent être analysées dans un délai de 24 heures.
1.6.3. Appareillage
|
—
|
un réservoir à solvant,
|
|
—
|
un système de dégazage (si nécessaire),
|
|
—
|
un amortisseur d'impulsions (si nécessaire),
|
|
—
|
un système d'injection,
|
|
—
|
des colonnes de chromatographie,
|
|
—
|
un débitmètre (si nécessaire),
|
|
—
|
un système d'enregistrement et de traitement des données,
|
|
—
|
un récipient pour recueillir les solutions usées.
|
On doit vérifier que le système de chromatographie sur gel perméable est inerte vis-à-vis des solvants utilisés (par exemple, utilisation de capillaires en acier lorsque le solvant est le THF).
1.6.4. Injection et système de distribution du solvant
Un volume défini de la solution de l'échantillon est chargé sur la colonne dans une zone étroitement définie, à l'aide d'un échantillonneur automatique ou manuellement. Si on opère manuellement, le fait de retirer ou d'enfoncer le piston de la seringue trop rapidement peut provoquer des modifications dans la distribution des masses moléculaires observées. Le système de distribution du solvant doit, dans la mesure du possible, être exempt de pulsations; il est souhaitable qu'il comporte un amortisseur d'impulsions. Le débit est de l'ordre de 1 ml/min.
1.6.5. Colonne
Selon la nature de l'échantillon a tester, le polymère est caractérisé en ayant recours à une seule colonne ou à plusieurs colonnes connectées en série. Un certain nombre de matériaux poreux pour colonne de propriétés définies (par exemple taille des pores, limites d'exclusion) sont disponibles dans le commerce. Le choix du gel de séparation ou celui de la longueur de la colonne dépendent, d'une part, des propriétés de l'échantillon à tester (volumes hydrodynamiques, distribution des masses moléculaires), d'autre part, des conditions particulières de la séparation, telles que la nature du solvant, la température et le débit (1) (2) (3).
1.6.6. Plateaux théoriques
La colonne ou la combinaison de colonnes utilisée pour la séparation doit être caractérisée par le nombre de plateaux théoriques. Pour ce faire, dans le cas où le solvant d'élution est le THF, il y a lieu de faire passer une solution d'ethylbenzène ou d'un autre soluté non polaire approprié sur une colonne de longueur connue. Le nombre de plateaux théoriques est donné par l'équation suivante:
|

|
ou
|
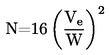
|
où:
|
N
|
est le nombre de plateaux théoriques
|
|
Ve
|
est le volume d'élution correspondant au maximum du pic
|
|
W
|
est la largeur du pic au niveau de la ligne de base
|
|
W1/2
|
est la largeur du pic à mi-hauteur.
|
1.6.7. Efficacité de la séparation
Outre le nombre de plateaux théoriques, paramètre déterminant la largeur de bande, l'efficacité de séparation est une autre grandeur caractéristique; c'est la pente de la courbe de calibrage qui la détermine. L'efficacité de séparation d'une colonne se détermine au moyen de la relation suivante:
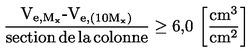
où
|
Ve, Mx
|
est le volume d'élution du polystyrène de masse moléculaire Mx
|
|
Ve,(10.Mx)
|
est le volume d'élution du polystyrène de masse moléculaire 10 fois supérieure
|
La résolution du système est généralement définie comme suit:

où,
|
Ve1, Ve2
|
sont les volumes d'élution des deux polystyrènes étalons au maximum du pic
|
|
W1, W2
|
sont les largeurs des pics au niveau de la ligne de base
|
|
Mp M2
|
sont les masses moléculaires correspondant au maximum des pics (elles devraient différer d'un facteur 10).
|
La valeur de R pour le système de colonnes doit être supérieure à 1,7 (4).
1.6.8. Solvants
Tous les solvants doivent être d'une grande pureté (on emploie du THF à 99,5 % de pureté). Le réservoir de solvant (mis, au besoin, sous atmosphère de gaz inerte) doit être assez grand pour permettre l'étalonnage de la colonne et plusieurs analyses d'échantillons. Le solvant sera dégazé avant d'être pompé vers la colonne.
1.6.9. Régulation de la température
La température des composants internes critiques (boucle d'injection, colonne(s), détecteur et tuyaux) doit être constante et compatible avec le choix du solvant.
1.6.10. Détecteur
Le détecteur sert à enregistrer de manière quantitative la concentration de l'échantillon qui est élue de la colonne. En vue d'éviter un élargissement indésirable des pics, le volume de la cuvette du détecteur sera choisi aussi petit que possible. Il ne doit pas excéder 10 μl, sauf pour les détecteurs par diffusion de la lumière et les viscosimètres. La détection s'effectue généralement par réfractométrie différentielle. Si les propriétés spécifiques de l'échantillon ou du solvant d'élution l'exigent, on peut toutefois utiliser d'autres types de détecteurs tels que les détecteurs UV/VIS ou IR, les viscosimètres, etc.
2. RÉSULTATS ET PROCÈS-VERBAL D'ESSAI
2.1. RÉSULTATS
On se référera à la norme DIN (1) pour ce qui concerne les critères d'évaluation détaillés, de même que pour les spécifications relatives à la collecte et au traitement des données.
Pour chaque échantillon analysé, il est nécessaire de réaliser deux expériences indépendantes. Leurs résultats seront analysés séparément.
Les paramètres Mn, Mw, Mw/Mn, Mp doivent être déterminés pour chaque mesure. Il est nécessaire d'indiquer de façon explicite que les valeurs mesurées sont des valeurs relatives correspondant a des équivalents de masse moléculaire de l'étalon utilisé.
Après avoir déterminé les volumes et les temps de rétention (éventuellement corrigés au moyen d'un étalon interne), on porte en graphique les valeurs de log Mp (Mp représentant les maxima des pics des polymères d'étalonnage) en fonction de l'une de ces quantités. Deux points d'étalonnage au moins sont nécessaires pour chaque facteur 10 de masse moléculaire; cinq points de mesure au moins sont requis pour la courbe dans son ensemble, qui doit couvrir la masse moléculaire estimée de l'échantillon. Le point extrême de la courbe d'étalonnage du côté des faibles masses moléculaires peut être défini grâce au n-hexylbenzène ou à un autre soluté non polaire approprié. Les masses moléculaires moyennes en nombre et en poids sont généralement déterminées par un traitement électronique des données fondé sur les formules décrites dans la section 1.2. Si on a recours à un traitement manuel, il est opportun de consulter l'ASTM D 3536-91 (3).
La courbe de distribution doit être présentée sous la forme d'un tableau ou d'un graphique (fréquence différentielle ou pourcentages cumulatifs en fonction du log M). Dans la représentation graphique, une puissance de dix de masse moléculaire doit normalement correspondre à une largeur de 4 cm environ tandis que pour le maximum du pic une hauteur de 8 cm environ est adéquate. Dans le cas de courbes de distribution cumulatives, la différence entre 0 et 100 pour cent sur l'axe des ordonnées doit être d'environ 10 cm.
2.2. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai doit comporter les renseignements suivants:
2.2.1. Substance à tester
|
—
|
renseignements disponibles sur la substance à tester (nature, additifs, impuretés),
|
|
—
|
description du traitement de l'échantillon, observations, problèmes.
|
2.2.2. Dispositif expérimental
|
—
|
réservoir d'éluant, gaz inerte, dégazage de l'éluant, composition de l'éluant, impuretés,
|
|
—
|
pompe, amortisseur d'impulsions, système d'injection,
|
|
—
|
colonnes de séparation (fabricant, toutes précisions sur les caractéristiques des colonnes, telles que taille des pores, nature du matériel de séparation etc., nombre, longueur et ordre des colonnes utilisées),
|
|
—
|
nombre de plateaux théoriques de la colonne (ou de la combinaison de colonnes), efficacité de la séparation (résolution du système),
|
|
—
|
informations sur la symétrie des pics,
|
|
—
|
température de la colonne, mode de régulation de la température,
|
|
—
|
détecteur (principe de mesure, type, volume de la cuvette),
|
|
—
|
débitmètre, s'il y en a un (fabricant, principe de mesure),
|
|
—
|
système utilisé pour rassembler et pour traiter les données (matériel et logiciel).
|
2.2.3. Étalonnage du système
|
—
|
description détaillée de la méthode utilisée pour établir la courbe d'étalonnage,
|
|
—
|
précisions sur les critères de qualité propres à cette méthode (par exemple, coefficient de corrélation, erreur quadratique moyenne, etc.),
|
|
—
|
informations sur toutes les extrapolations, hypothèses et approximations effectuées au cours de la procédure expérimentale, ainsi que sur l'évaluation et le traitement des données,
|
|
—
|
toutes les mesures effectuées pour établir la courbe d'étalonnage doivent être présentées dans un tableau qui comporte, pour chaque point d'étalonnage, les informations suivantes:
|
—
|
fabricant de l'échantillon,
|
|
—
|
valeurs caractéristiques Mp, Mn, Mw, Mw/Mn des étalons fournis par le fabricant ou déduites de mesures ultérieures, complétées d'indications relatives à la méthode de détermination,
|
|
—
|
volume d'injection et concentration d'injection,
|
|
—
|
valeur de Mp utilisée pour l'étalonnage,
|
|
—
|
volume d'élution ou temps de rétention corrigé mesuré aux maxima des pics,
|
|
—
|
Mp calculée au maximum du pic,
|
|
—
|
pourcentage d'erreur sur la Mp calculée et sur la valeur d'étalonnage.
|
|
2.2.4. Évaluation
|
—
|
évaluation en fonction du temps: toutes méthodes visant à améliorer la reproductibilité requise (méthode de correction, étalon interne, etc.),
|
|
—
|
préciser si l'évaluation a été effectuée à partir du volume d'élution ou à partir du temps de rétention,
|
|
—
|
indiquer les limites de l'évaluation si un pic n'est pas complètement analysé,
|
|
—
|
décrire les méthodes de lissage lorsqu'on y a recours,
|
|
—
|
décrire les procédés de préparation et de prétraitement de l'échantillon,
|
|
—
|
indiquer la présence de particules non dissoutes, s'il y en a,
|
|
—
|
indiquer le volume d'injection (μl) et la concentration d'injection (mg/ml),
|
|
—
|
mentionner les observations témoignant d'effets qui engendrent des déviations par rapport au profil idéal de chromatographie sur gel perméable,
|
|
—
|
décrire en détail toutes les modifications aux procédures d'essais,
|
|
—
|
préciser les intervalles d'erreur,
|
|
—
|
consigner toutes autres informations et observations utiles pour interpréter les résultats.
|
3. RÉFÉRENCES
|
(1)
|
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1.
|
|
(2)
|
Yau, W.W., Kirkland, J.J. and Bly, D.D. (eds) (1979). Modern Size Exclusion Liquid Chromatography, J. Wiley & Sons.
|
|
(3)
|
ASTM D 3536-91 (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography-GPC), American Society for Testing and Materials, Philadelphie, Pennsylvanie.
|
|
(4)
|
ASTM D 5296-92 (1992). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution of Polystyrène by High Performance Size-Exclusion Chromatography, American Society for Testing and Materials, Philadelphie, Pennsylvanie.
|
Annexe
Exemples d'autres méthodes de détermination de la masse moléculaire moyenne en nombre (Mn) des polymères
La chromatographie sur gel perméable constitue la méthode de choix pour la détermination de Mn, en particulier lorsqu'on dispose d'un ensemble de substances étalons de structure comparable à la structure du polymère. Toutefois, lorsque le recours à la chromatographie sur gel perméable présente des difficultés pratiques ou que l'on peut s'attendre à ce que la substance ne satisfasse pas à un critère réglementaire pour la Mn (ce qui demande confirmation), il existe des méthodes de remplacement, par exemple:
1. Utilisation des propriétés colligatives
|
1.1.
|
Ébullioscopie et cryoscopie:
consistent à mesurer l'élévation du point d'ébullition (ébullioscopie) ou l'abaissement du point de congélation (cryoscopie) d'un solvant lorsque le polymère est ajouté. La méthode est basée sur le fait que la dissolution d'un polymère dans un liquide exerce sur les points d'ébullition et de congélation de celui-ci un effet qui dépend de la masse moléculaire du polymère (1) (2).
Le domaine d'application correspond à Mn < 20 000.
|
|
1.2.
|
Abaissement de la pression de vapeur:
consiste à mesurer la pression de vapeur d'un liquide de référence avant et après addition de quantités connues de polymères (1) (2).
Le domaine d'application correspond à Mn < 20 000 (théoriquement; en pratique, ne présente toutefois qu'une valeur limitée).
|
|
1.3.
|
Osmométrie à membrane:
repose sur le principe de l'osmose, à savoir la tendance naturelle des molécules de solvant à traverser une membrane semi-perméable à partir d'une solution diluée vers une solution concentrée de manière à réaliser l'équilibre. Dans l'essai, la concentration de la solution diluée est nulle tandis que la solution concentrée contient le polymère. Le passage de solvant à travers la membrane induit une différence de pression qui dépend de la concentration et de la masse moléculaire du polymère (1) (3) (4).
Le domaine d'application correspond à des valeurs de Mn comprises entre 20 000 et 200 000.
|
|
1.4.
|
Osmométrie en phase vapeur:
consiste à comparer la vitesse d'évaporation d'un aérosol de solvant pur à celle de trois aérosols au moins contenant le polymère à différentes concentrations (1) (5) (6).
Le domaine d'application correspond à Mn < 20 000.
|
2. Analyse des groupes terminaux
Pour utiliser cette méthode, il est nécessaire de connaître à la fois la structure globale du polymère et la nature des groupes terminaux situés en bout de chaîne (ceux-ci doivent pouvoir être distingués de ta chaîne principale par exemple, par leur spectre RMN ou par titrage et formation de dérivés). La détermination de la concentration moléculaire des groupes terminaux présents sur le polymère peut, ensuite, fournir une valeur de la masse moléculaire (7) (8) (9).
Le domaine d'application correspond a des valeurs de Mn pouvant atteindre 50 000 (avec une fiabilité décroissante).
3. Références de l'annexe
|
(1)
|
Billmeyer, F.W. Jr. (1984). Textbook of Polymer Science, 3rd Edn., John Wiley, New York.
|
|
(2)
|
Glover, C.A. (1975). Absolute Colligative Property Methods, Chapter 4, in Polymer Molecular Weights, Part I (ed. P.E. Slade, Jr.), Marcel Dekker, New York.
|
|
(3)
|
ASTM D 375-79 (1979). Standard Practice for Détermination of Number-Average Molecular Weight of Polymers by Membrane osmometry, American Society for Testing and Materials, Philadelphie, Pennsylvanie.
|
|
(4)
|
Coll, H. (1989). Membrane osmometry, in: Détermination of Molecular Weight (ed. A.R. Cooper), J. Wiley and Sons, pp. 25-52.
|
|
(5)
|
ASTM D 3592-77 (1977). Standard recommended practice for Determination of Molecular Weight by Vapour Pressure, American Society for Testing and Materials, Philadelphie, Pennsylvanie.
|
|
(6)
|
Morris, C.E.M. (1989). Vapour Pressure Osmometry in Determination of Molecular Weight (ed. A.R. Cooper), J. Wiley and Sons.
|
|
(7)
|
Schröder, E. Müller, G. & Arndt, K.-F. (1989). Polymer Characterisadon, Carl Hanser Verlag, Munich.
|
|
(8)
|
Garmon, R.G. (1975). End-Group Determinations, Chapter 3 in Polymer Molecular Weights, Part I, (ed. P.E. Slade, Jr.) Marcel Dekker, New York.
|
|
(9)
|
Amiya, S. et al. (1990). Pure and Applied Chemistry, 62, 2139-2146.
|
A.19. DÉTERMINATION DE LA TENEUR EN POLYMÈRES DE FAIBLE MASSE MOLÉCULAIRE
1. MÉTHODE
Cette méthode de chromatographie sur gel perméable reprend la ligne directrice OCDE TG 118 (1996). On trouvera dans les références les principes fondamentaux et autres renseignements techniques.
1.1. INTRODUCTION
Les propriétés des polymères sont tellement diversifiées qu’il est impossible de décrire une méthode unique indiquant avec précision des conditions de séparation et d’évaluation qui recouvrent toutes les éventualités et particularités rencontrées dans la séparation des polymères. Les systèmes complexes de polymères, notamment, se prêtent rarement à la chromatographie sur gel perméable. Lorsque la chromatographie sur gel perméable n’est pas applicable, on peut déterminer la teneur en polymères de faible masse moléculaire à l’aide d’autres méthodes. Dans de tels cas, il convient de fournir tous les détails de la méthode utilisée et de justifier son choix.
La méthode décrite ci-après est basée sur la norme DIN 55672 (1). Celle-ci fournit des indications détaillées sur la manière de réaliser les expériences et d’évaluer les données. S’il est nécessaire de modifier les conditions expérimentales, ces changements doivent être justifiés. D’autres normes peuvent être utilisées, à condition d’en mentionner toutes les références. La méthode décrite fait appel à des échantillons de polystyrène de polydispersité connue pour l’étalonnage et il y aura peut-être lieu de la modifier pour l’adapter aux cas de polymères particuliers, par exemple, les polymères solubles dans l’eau et les polymères ramifiés à longue chaîne.
1.2. DÉFINITIONS ET UNITÉS
On définit arbitrairement une masse moléculaire faible comme étant une masse moléculaire inférieure à 1 000 daltons.
La masse moléculaire moyenne en nombre Mn et la masse moléculaire moyenne en poids Mw sont déterminées à l’aide des équations suivantes:
où
|
Hi
|
est l’amplitude du signal du détecteur, par rapport au niveau de référence, correspondant au volume de rétention Vi, par rapport au niveau de référence.
|
|
Mi
|
est la masse moléculaire de la fraction de polymère correspondant au volume de rétention, Vi et n est le nombre de points expérimentaux.
|
La largeur de la distribution des masses moléculaires, qui représente une mesure de la polydispersité du système, est donnée par le rapport MW/Mn.
1.3. SUBSTANCES DE RÉFÉRENCE
La méthode par chromatographie sur gel perméable étant une méthode relative, il est nécessaire de procéder à un étalonnage. Celui-ci est généralement effectué au moyen de polystyrènes étalons à chaîne linéaire et à distribution étroite, dont les masses moléculaires moyennes Mn et Mw, ainsi que la distribution des masses moléculaires sont connues. La courbe d’étalonnage ne peut être utilisée pour déterminer la masse moléculaire d’un échantillon inconnu que si les conditions de séparation des échantillons et des étalons ont été sélectionnées de manière identique.
Une relation déterminée entre la masse moléculaire et le volume d'élution n'est valable que dans les conditions particulières d'une expérience donnée. Parmi ces conditions figurent, avant tout, la température, le solvant (ou le mélange de solvants), les conditions de chromatographie, ainsi que la colonne de séparation ou le système de colonnes.
Les masses moléculaires de l'échantillon déterminées de cette manière constituent des valeurs relatives; on les désigne par le terme «masses moléculaires en équivalent-polystyrène». Cela signifie qu'en fonction des différences chimiques et structurelles entre l'échantillon à tester et les étalons, les masses moléculaires peuvent dévier des valeurs absolues de manière plus ou moins importante. Si d'autres étalons sont utilisés, par exemple le polyéthylèneglycol, le poly(éthylène oxyde), le poly(méthacrylate de méthyle) ou l'acide polyacrylique, il importe d'en indiquer la raison.
1.4. PRINCIPE DE LA MÉTHODE
La chromatographie sur gel perméable permet de déterminer la distribution des masses moléculaires ainsi que les masses moléculaires moyennes (Mn, Mw). La chromatographie sur gel perméable constitue un type particulier de chromatographie liquide dans lequel l'échantillon à tester est séparé en fonction des volumes hydrodynamiques de chaque constituant (2).
La séparation s'effectue à mesure que l'échantillon progresse dans une colonne remplie d'un matériau poreux, généralement un gel organique. Les petites molécules peuvent pénétrer dans les pores tandis que les grosses molécules en sont exclues. Le parcours des grosses molécules est, de ce fait, plus court; elles sont éluées en premier lieu. Les molécules de taille moyenne pénètrent dans certains des pores; elles sont éluées plus tard. Les molécules les plus petites, dont le rayon hydrodynamique moyen est plus petit que les pores du gel, peuvent pénétrer dans tous les pores. Elles sont éluées en dernier lieu.
Dans la situation idéale, c'est la taille des espèces moléculaires qui régit entièrement la séparation, mais, en pratique, il est difficile d'éviter l'interférence de certains effets d'absorption au moins. Un remplissage irrégulier de la colonne, de même que des volumes morts, peuvent aggraver la situation (2).
La détection s'effectue, par exemple, par mesure de l'indice de réfraction ou de l'absorption dans l'UV pour donner une courbe de distribution simple. Cependant, pour pouvoir associer à la courbe des valeurs de masses moléculaires réelles, il est nécessaire d'étalonner la colonne par passage de polymères de masse moléculaire connue et, idéalement, d'une structure aussi proche que possible. Une courbe de Gauss en résulte généralement; elle présente parfois une dissymétrie marquée du coté des masses moléculaires faibles, l'axe vertical indiquant la quantité, en poids, des espèces de différentes masses moléculaires éluées, tandis que sur l'axe horizontal figure le logarithme de la masse moléculaire.
La teneur en polymères de faible masse moléculaire est déduite de cette courbe. Le calcul ne peut être exact que si les espèces à faible masse moléculaire se comportent de la même manière, par unité de masse, que l'ensemble du polymère.
1.5. CRITÈRES DE QUALITÉ
La reproductibilité (écart-type relatif) du volume d'élution doit être au plus égale à 0,3 pour cent. Il y a lieu d'améliorer la reproductibilité de l'analyse grâce à une correction mettant en jeu un étalon interne, si un chromatogramme est évalué en fonction du temps et ne satisfait pas aux critères mentionnés plus haut [voir informations complémentaires en (1)]. Les polydispersités dépendent des masses moléculaires des étalons; dans le cas des polystyrènes étalons:
|
Mp < 2 000
|
Mw/Mn < 1,20
|
|
2 000 ≤ Mp ≤ 106
|
Mw/Mn < 1,05
|
|
Mp > 106
|
Mw/Mn < 1,20
|
(Mp est la masse moléculaire de l'étalon correspondant au maximum du pic)
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Préparation des solutions de polystyrène étalon
On dissout les polystyrènes étalons en les mélangeant soigneusement à l'éluant choisi. On préparera les solutions en observant les recommandations du fabricant.
Les concentrations des étalons choisis dépendent de différents facteurs, comme le volume d'injection, la viscosité de la solution et la sensibilité du détecteur. Le volume d'injection maximum doit être adapté à la longueur de la colonne, en veillant à ne pas surcharger celle-ci. Les volumes d'injection courants pour des séparations analytiques par chromatographie sur gel perméable, au moyen d'une colonne de 30 cm × 7,8 mm, varient normalement entre 40 et 100 μl. Il est possible d'injecter des volumes plus importants, mais ceux-ci ne peuvent dépasser 250 μl. On doit déterminer le rapport optimal entre le volume d'injection et la concentration avant l'étalonnage de la colonne.
1.6.2. Préparation de la solution de l'échantillon
En principe, la préparation des solutions de l'échantillon s'effectue selon les mêmes critères. L'échantillon est dissous dans un solvant approprié, par exemple le THF, en agitant soigneusement. Il ne doit, en aucun cas, être dissous dans un bain à ultrasons. Si nécessaire, la solution de l'échantillon est purifiée au moyen d'un filtre à membrane d'une dimension de pores comprise entre 0,2 et 2 μm.
La présence de particules non dissoutes doit être mentionnée dans le rapport final, dans la mesure où elle peut résulter d'espèces de masse moléculaire élevée. Il faut recourir à une méthode appropriée pour déterminer le pourcentage en poids des particules non dissoutes. Les solutions doivent être analysées dans un délai de 24 heures.
1.6.3. Correction liée à la présence d'impuretés et d'additifs
S'agissant de la teneur en espèces de M < 1 000, il est généralement nécessaire d'apporter une correction pour tenir compte de la présence de composés particuliers non polymériques (par exemple des impuretés ou des additifs), sauf si la teneur mesurée n'excède pas un pour cent. Cette correction peut être réalisée par analyse directe de la solution de polymère ou de l'éluat de la chromatographie sur gel perméable.
Au cas où l'éluat est trop dilué pour permettre une analyse après passage à travers la colonne, il convient de le concentrer. Il est éventuellement nécessaire d'évaporer l'éluat à sec puis de le redissoudre. La concentration de l'éluat sera effectuée dans des conditions telles qu'aucune modification de sa composition n'intervienne. Le traitement de l'éluat après la phase de chromatographie sur gel perméable est fonction de la méthode d'analyse utilisée pour la détermination quantitative.
1.6.4. Appareillage
Un appareil de chromatographie sur gel perméable se compose des éléments suivants:
|
—
|
un réservoir à solvant,
|
|
—
|
un système de dégazage (si nécessaire),
|
|
—
|
un amortisseur d'impulsions (si nécessaire),
|
|
—
|
un système d'injection,
|
|
—
|
des colonnes de chromatographie,
|
|
—
|
un débimètre (si nécessaire),
|
|
—
|
un système d'enregistrement et de traitement des données,
|
|
—
|
un récipient pour recueillir les solutions usées.
|
On doit vérifier que le système de chromatographie sur gel perméable est inerte vis-à-vis du solvant utilisé (par exemple, utilisation de capillaires en acier lorsque le solvant est le THF).
1.6.5. Injection et système de pompage du solvant
Un volume défini de la solution de l'échantillon est chargé sur la colonne dans une zone étroitement définie, à l'aide d'un échantillonneur automatique ou manuellement. Si on opère manuellement, le fait de retirer ou d'enfoncer le piston de la seringue trop rapidement peut provoquer des modifications dans la distribution des masses moléculaires observées. Le système de pompage du solvant doit, dans toute la mesure du possible, être exempt de pulsations; il est souhaitable qu'il comporte un amortisseur d'impulsions. Le débit est de l'ordre de 1 ml/min.
1.6.6. Colonne
Selon la nature de l'échantillon à tester, le polymère est caractérisé en ayant recours à une seule colonne ou à plusieurs colonnes connectées en série. Un certain nombre de matériaux poreux pour colonne de propriétés définies (par exemple taille des pores, limites d'exclusion) sont disponibles dans le commerce. Le choix du gel de séparation ou celui de la longueur de la colonne dépendent, d'une part, des propriétés de l'échantillon à tester (volumes hydrodynamiques, distribution des masses moléculaires), d'autre part, des conditions particulières de la séparation, telles que la nature du solvant, la température et le débit (1) (2) (3).
1.6.7. Plateaux théoriques
La colonne ou la combinaison de colonnes utilisée pour la séparation doit être caractérisée par le nombre de plateaux théoriques. Pour ce faire, dans le cas où le solvant d'élution est le THF, il y a lieu de faire passer une solution d'éthylbenzène ou d'un autre soluté non polaire approprié sur une colonne de longueur connue. Le nombre de plateaux théoriques est donné par l'équation suivante:
|
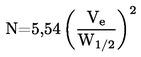
|
ou
|
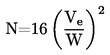
|
où
|
N
|
est le nombre de plateaux théoriques
|
|
Ve
|
est le volume d'élution correspondant au maximum du pic
|
|
W
|
est la largeur du pic au niveau de la ligne de base
|
|
W1/2
|
est la largeur du pic à mi-hauteur.
|
1.6.8. Efficacité de la séparation
Outre le nombre de plateaux théoriques, paramètre déterminant la largeur de bande, l'efficacité de séparation est une autre grandeur caractéristique; c'est la pente de la courbe de calibrage qui la détermine. L'efficacité de séparation d'une colonne se détermine au moyen de la relation suivante:

où
|
VeMx
|
est le volume d'élution du polystyrène de masse moléculaire Mx
|
|
Ve,(10.Mx)
|
est le volume d'élution du polystyrène de masse moléculaire 10 fois supérieure.
|
La résolution du système est généralement définie comme suit:
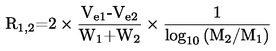
où.
|
Ve1, Ve2
|
sont les volumes d'élution des deux polystyrènes étalons au maximum du pic.
|
|
W1, W2
|
sont les largeurs des pics au niveau de la ligne de base.
|
|
M1, M2
|
sont les masses moléculaires correspondant au maximum des pics (elles doivent différer d'un facteur 10).
|
La valeur de R pour le système de colonnes doit être supérieure à 1,7 (4).
1.6.9. Solvants
Tous les solvants doivent être d'une grande pureté (on emploie du THF à 99,5 pour cent de pureté}. Le réservoir de solvant (mis, au besoin, sous atmosphère de gaz inerte) doit être assez grand pour permettre l'étalonnage de la colonne et plusieurs analyses d'échantillons. Le solvant sera dégazé avant d'être pompe vers la colonne.
1.6.10. Régulation de la température
La température des composants internes critiques [boucle d'injection, colonne(s), détecteur et tuyaux] doit être constante et compatible avec le choix du solvant.
1.6.11. Détecteur
Le détecteur sert à enregistrer de manière quantitative la concentration de l'échantillon qui est élué de la colonne. En vue d'éviter un élargissement indésirable des pics, le volume de la cuvette du détecteur sera choisi aussi petit que possible. Il ne doit pas excéder 10 μl, sauf pour les détecteurs par diffusion de la lumière et les viscosimètres. La détection s'effectue généralement par réfractométrie différentielle. Toutefois, si les propriétés spécifiques de l'échantillon ou du solvant d'élution l'exigent, d'autres types de détecteurs peuvent être utilisés tels que les détecteurs UV/VIS ou IR, les viscosimètres, etc.
2. RÉSULTATS ET PROCÈS-VERBAL D'ESSAI
2.1. RÉSULTATS
On se référera à la norme DIN (1) pour ce qui concerne les critères d'évaluation détaillés, de même que pour les spécifications relatives à la collecte et au traitement des données.
Pour chaque échantillon analysé, on réalisera deux expériences indépendantes. Leurs résultats seront analysés séparément.
Il est nécessaire d'indiquer explicitement que les valeurs mesurées sont des valeurs relatives correspondant a des équivalents de masse moléculaire de l'étalon utilisé.
Dans tous les cas, il est essentiel de déterminer aussi les données relatives aux blancs, ceux-ci étant traités de la même manière que les échantillons.
Après avoir déterminé les volumes et les temps de rétention (éventuellement corrigés au moyen d'un étalon interne), on porte en graphique les valeurs de log Mp (Mp représentant les maxima des pics des polymères d'étalonnage) en fonction de l'une de ces quantités. Deux points d'étalonnage au moins sont nécessaires pour chaque facteur 10 de masse moléculaire; cinq points de mesure au moins sont requis, pour la courbe dans son ensemble qui doit couvrir la masse moléculaire estimée de l'échantillon. Le point extrême de la courbe d'étalonnage du côté des faibles masses moléculaires peut être défini grâce au n-hexylbenzène ou à un autre soluté non polaire approprié. On détermine la portion de la courbe correspondant aux masses moléculaires inférieures à 1 000 et on la corrige, si nécessaire, pour tenir compte des impuretés et des additifs. Les courbes d'élution sont généralement évaluées au moyen d'un système électronique de traitement des données. Si on a recours à un traitement manuel, il est opportun de consulter l'ASTM D 3536-91 (3).
Si un polymère insoluble est retenu sur la colonne, sa masse moléculaire est vraisemblablement supérieure à celle de la fraction soluble et il faut en tenir compte pour éviter de surestimer la teneur en polymères de faible masse moléculaire. On trouvera en annexe des indications permettant de corriger la teneur en polymères de faible masse moléculaire pour tenir compte des polymères insolubles.
La courbe de distribution doit être présentée sous la forme d'un tableau ou d'un graphique (fréquence-différentielle ou pourcentages cumulatifs en fonction du log M). Dans la représentation graphique, une puissance de dix de masse moléculaire doit normalement correspondre à une largeur de 4 cm environ, tandis que pour le maximum du pic une hauteur de 8 cm environ est adéquate. Dans le cas de courbes de distribution cumulatives, la différence entre 0 et 100 pour cent sur l'axe des ordonnées doit être d'environ 10 cm.
2.2. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai doit comporter les renseignements suivants:
2.2.1. Substance à tester
|
—
|
renseignements disponibles sur la substance à tester (nature, additifs, impuretés),
|
|
—
|
description du traitement de l'échantillon, observations, problèmes.
|
2.2.2. Dispositif expérimental
|
—
|
réservoir d'éluant, gaz inerte, dégazage de l'éluant, composition de l'éluant, impuretés,
|
|
—
|
pompe, amortisseur d'impulsions, système d'injection,
|
|
—
|
colonnes de séparation (fabricant, toutes précisions sur les caractéristiques de la colonne, telles que taille des pores, nature du matériel de séparation, etc., nombre, longueur et ordre des colonnes utilisées),
|
|
—
|
nombre de plateaux théoriques de la colonne (ou de la combinaison de colonnes), efficacité de la séparation (résolution du système),
|
|
—
|
informations sur la symétrie des pics,
|
|
—
|
température de la colonne, mode de régulation de la température,
|
|
—
|
détecteur (principe de mesure, type, volume de la cuvette),
|
|
—
|
débitmètre s'il y en a un (fabricant, principe de mesure),
|
|
—
|
système utilisé pour rassembler et pour traiter les données (matériel et logiciel).
|
2.2.3. Étalonnage du système
|
—
|
description détaillée de la méthode utilisée pour établir la courbe d'étalonnage,
|
|
—
|
précisions sur les critères de qualité propres à cette méthode (par exemple, coefficient de corrélation, erreur quadratique moyenne, etc.),
|
|
—
|
informations sur toutes les extrapolations, hypothèses et approximations effectuées au cours des processus expérimentaux, ainsi que sur l'évaluation et le traitement des données,
|
|
—
|
toutes les mesures effectuées pour établir la courbe d'étalonnage doivent être présentées dans un tableau qui comporte, pour chaque point d'étalonnage, les informations suivantes:
|
—
|
fabricant de l'échantillon,
|
|
—
|
valeurs caractéristiques Mp, Mo, Mw, Mw/Mn des étalons fournies par le fabricant déduites de mesures ultérieures, complétées d'indications relatives à la méthode de détermination,
|
|
—
|
volume d'injection et concentration d'injection,
|
|
—
|
valeur de Mp utilisée pour l'étalonnage,
|
|
—
|
volume d'élution ou temps de rétention corrigé mesuré aux maxima des pics,
|
|
—
|
Mp calculée au maximum du pic,
|
|
—
|
pourcentage d'erreur sur la Mp calculée et sur la valeur d'étalonnage.
|
|
2.2.4. Renseignements sur la teneur en polymères de faible masse moléculaire
|
—
|
description des méthodes d'analyse utilisées et de la manière dont les expériences ont été menées,
|
|
—
|
informations sur le pourcentage que représente la teneur en espèces de faible masse moléculaire (poids/poids) par rapport à l'ensemble de l'échantillon,
|
|
—
|
informations sur les impuretés, les additifs et les autres substances non polymériques en pourcentage pondéral de l'ensemble de l'échantillon.
|
2.2.5. Évaluation
|
—
|
évaluation en fonction du temps: toutes méthodes visant à améliorer la reproductibilité requise (méthode de corrections, étalon interne, etc.),
|
|
—
|
préciser si l'évaluation a été effectuée à partir du volume d'élution ou à partir du temps de rétention,
|
|
—
|
indiquer les limites de l'évaluation si un pic n'est pas complètement analyse,
|
|
—
|
décrire les méthodes de lissage lorsqu'on y a recours.
|
|
—
|
décrire les procédés de préparation et de prétraitement de l'échantillon,
|
|
—
|
indiquer la présence de particules non dissoutes, s'il y en a,
|
|
—
|
indiquer le volume d'injection (μl) et la concentration d'injection (mg/ml),
|
|
—
|
mentionner les observations témoignant d'effets qui engendrent des déviations par rapport au profil idéal de chromatographie sur gel perméable,
|
|
—
|
décrire en détail toutes les modifications aux procédures d'essais,
|
|
—
|
préciser les intervalles d'erreur,
|
|
—
|
consigner toutes autres informations et observations utiles pour interpréter les résultats.
|
3. RÉFÉRENCES
|
(1)
|
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1.
|
|
(2)
|
Yau, W.W., Kirkland, J.J. et Bly, D.D. (eds.) (1979). Modem Size Exclusion Liquid Chromatography, J.Wiley & Sons.
|
|
(3)
|
ASTM D 3536-91 (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography-GPC), American Society for Testing and Materials, Philadelphie, Pennsylvanie.
|
|
(4)
|
ASTM D 5296-92, (1992). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution of Polystyrene by High Performance Size-Exclusion Chromatography, American Society for Testing and Materials, Philadelphie, Pennsylvanie.
|
Annexe
Indications permettant de corriger la teneur en polymères de faible masse moléculaire pour tenir compte des polymères insolubles
La présence d'un polymère insoluble dans un échantillon entraîne une perte de masse au cours de l'analyse par chromatographie sur gel perméable. Le polymère insoluble est irréversiblement retenu sur la colonne ou sur le filtre à échantillon lorsque la partie soluble de l'échantillon traverse la colonne. Si l'indice de réfraction différentiel (dn/dc) du polymère peut être estimé ou mesuré, il est possible d'estimer la masse que l'échantillon a perdue sur la colonne. Dans ce cas, on effectue une correction à l'aide d'un étalonnage externe du réfractomètre, avec des étalons dont on connaît la concentration et le dn/dc. Dans l'exemple qui suit, on utilise un étalon de poly(méthacrylate de méthyle).
L'étalonnage externe pratiqué lors de l'analyse des polymères acryliques consiste à analyser par chromatographie sur gel perméable une solution étalon de concentration connue de poly(méthacrylate de méthyle) dans du tétrahydrofuranne; les résultats servent à calculer la constante du réfractomètre selon l'équation suivante:
K = R/(C × V × dn/dc)
où
|
K
|
=
|
est la constante du réfractomètre (microvolts secondes/ml)
|
|
R
|
=
|
est la réponse de l'étalon de poly(méthacrylate de méthyle) (microvolts secondes)
|
|
C
|
=
|
est la concentration de l'étalon de poly(méthacrylate de méthyle) (mg/ml)
|
|
V
|
=
|
est le volume d'injection (ml) et
|
|
dn/dc
|
=
|
est l'indice de réfraction différentiel d'une solution de poly(méthacrylate de méthyle) dans du tétrahydrofuranne (ml/mg).
|
Les données suivantes caractérisent généralement un étalon de poly(méthacrylate de méthyle):
|
R
|
=
|
2 937 891;
|
|
C
|
=
|
1,07 mg/ml;
|
|
V
|
=
|
0,1 ml;
|
|
dn/dc
|
=
|
9 x 10 – 5 ml/mg.
|
La valeur de K résultante, 3,05 x 1011, est ensuite utilisée pour calculer la réponse théorique du détecteur, c'est-à-dire celle qu'on obtiendrait si 100 pour cent du polymère injecté était élué à travers le détecteur.
A.20. COMPORTEMENT DE DISSOLUTION — EXTRACTION DES POLYMÈRES DANS L'EAU
1. MÉTHODE
La méthode décrite reprend la version révisée de la ligne directrice OCDE TG 120 (1997). On trouvera davantage de renseignements techniques en référence (1).
1.1. INTRODUCTION
Dans le cas de certains polymères, tels que les polymères en émulsion, un travail préparatoire initial peut être nécessaire avant utilisation de la méthode exposée. La méthode n'est pas applicable aux polymères liquides ni aux polymères qui réagissent avec l'eau dans les conditions d'essai.
Lorsque la méthode est difficile ou impossible à mettre en pratique, le comportement de dissolution-extraction des polymères peut être étudié par d'autres méthodes. Dans ce cas, la méthode utilisée devra être entièrement détaillée et justifiée.
1.2. SUBSTANCES DE RÉFÉRENCE
Néant
1.3. PRINCIPE DE LA MÉTHODE
Le comportement de dissolution — extraction des polymères en milieu aqueux est déterminé par la méthode du flacon (voir A.6, Solubilité dans l'eau, méthode du flacon), en y apportant les modifications décrites ci-après.
1.4. CRITÈRES DE QUALITÉ
Néant
1.5. DESCRIPTION DE LA MÉTHODE
1.5.1. Appareillage
L'appareillage nécessaire à l'application de la méthode est le suivant:
|
—
|
un dispositif permettant de réduire l'échantillon en poudre, tel qu'un broyeur produisant des particules de taille déterminée
|
|
—
|
un système d'agitation avec possibilité de régler la température
|
|
—
|
un système de filtration sur membrane
|
|
—
|
un dispositif d'analyse
|
1.5.2. Préparation de la solution de l'échantillon
Il convient de réduire d'abord un échantillon représentatif à l'état de particules d'une taille comprise entre 0,125 et 0,25 mm en utilisant des tamis appropriés. Il peut être nécessaire de refroidir pour garantir la stabilité de l'échantillon ou pour procéder au broyage. Les matériaux du type du caoutchouc peuvent être pulvérisés à la température de l'azote liquide (1).
S'il n'est pas possible d'obtenir des particules de la dimension requise, on doit s'efforcer de réduire autant que possible la taille des particules et consigner le résultat. Dans le rapport, il est nécessaire d'indiquer comment l'échantillon pulvérisé a été conservé avant l'analyse.
1.5.3. Procédure
On pèse trois échantillons de la substance à tester, de 10 g chacun, dans trois récipients pourvus de bouchons en verre et on ajoute 1 000 ml d'eau dans chacun des récipients. Si la manipulation de 10 g de polymère s'avère impossible, il convient d'utiliser la quantité la plus grande qui puisse être manipulée, le volume d'eau étant ajusté en proportion.
Les récipients sont soigneusement bouchés, puis agités à 20 oC. On doit employer un dispositif d'agitation qui puisse fonctionner à température constante. Après une période de 24 heures, le contenu de chaque récipient est centrifugé ou filtré et la concentration du polymère dans la phase aqueuse limpide est déterminée à l'aide d'une méthode d'analyse appropriée. S'il n'existe pas de méthode permettant une analyse adéquate de la phase aqueuse, on peut obtenir une estimation de la solubilité-extractibilité totale en pesant, après séchage, le résidu filtré ou le précipité centrifugé.
Il est également nécessaire de différencier au plan quantitatif les impuretés et les additifs, d'une part, et les espèces de faible masse moléculaire, d'autre part. Dans le cas d'une détermination par gravimétrie, il est important de réaliser un essai à blanc, c'est-à-dire sans utiliser la substance à tester, de manière à pouvoir tenir compte des résidus générés par la méthode expérimentale.
On peut déterminer de la même manière le comportement de dissolution — extraction des polymères dans l'eau à 37 oC à pH 2 et pH 9 tel que décrit pour l'expérience réalisée à 20 oC. Les valeurs de pH peuvent être obtenues par addition de tampons adéquats ou d'acides et de bases appropriés tels que l'acide chlorhydrique, l'acide acétique, les hydroxydes de sodium et de potassium de qualité pour analyse ou l'ammoniac.
Il convient de mener un ou deux essais, suivant la méthode d'analyse utilisée. Lorsqu'on dispose de méthodes suffisamment spécifiques pour l'analyse directe du polymère en phase aqueuse, un essai tel que décrit plus haut devrait suffire. Mais quand ces méthodes n'existent pas et que la détermination du comportement de dissolution-extraction du polymère ne peut se faire que par une analyse indirecte, c'est-à-dire uniquement par la détermination de la teneur en carbone organique total (COT) de l'extrait aqueux, il est nécessaire de réaliser un essai supplémentaire. Celui-ci doit aussi être effectué en trois exemplaires, avec des échantillons de polymère dix fois plus petits et les mêmes quantités d'eau que celles qui ont été utilisées dans le premier essai.
1.5.4. Analyse
1.5.4.1. Essais réalisés sur des échantillons d'une seule taille
Il est possible que l'on dispose de méthodes permettant une analyse directe des polymères en phase aqueuse. Sinon, l'analyse indirecte des polymères dissous ou extraits peut aussi être envisagée. Pour ce faire, on détermine la teneur totale en parties solubles et on la corrige pour tenir compte des substances non polymériques.
L'analyse de l'extrait aqueux pour déterminer la teneur totale en espèces polymériques peut être réalisée;
soit par une méthode suffisamment sensible, par exemple.
|
—
|
la détermination du COT par la digestion au persulfate ou au dichromate pour donner du CO2, suivie d'une estimation par IR ou d'une analyse chimique,
|
|
—
|
la spectrométrie d'absorption atomique (SAA) ou son équivalent en émission d'un plasma couplé par induction (PCI), dans le cas des polymères contenant du silicium ou un métal,
|
|
—
|
l'absorption UV ou la spectrofluorimétrie pour les polymères arylés,
|
|
—
|
la chromatographie en phase liquide couplée à la spectrométrie de masse pour les échantillons de faible masse moléculaire,
|
soit par évaporation à sec, sous vide, de l'extrait aqueux, suivie d'une analyse du résidu par spectroscopie (IR, UV, etc.) ou par SAA-PCI.
Si une telle analyse de la phase aqueuse est impossible, l'extrait aqueux sera obtenu au moyen d'un solvant organique non miscible à l'eau (un hydrocarbure chlore, par exemple). Le solvant est alors évaporé et le résidu analysé, par ex. par IR, UV ou SAA-PCI, pour déterminer sa teneur en polymère notifié. Tous les constituants de ce résidu qui s'avèrent être des impuretés ou des additifs doivent être soustraits dans le but de déterminer le degré de dissolution — extraction du polymère lui-même.
Lorsque de telles substances sont présentes en quantités relativement importantes, il peut être nécessaire de soumettre le résidu à une analyse par chromatographie en phase liquide à haute performance ou en phase gazeuse, par exemple, afin de différencier ces impuretés des monomères et des dérivés des monomères présents, de telle sorte que la teneur réelle en ces dernières espèces puisse être déterminée.
Dans certains cas, une simple évaporation à sec du solvant organique, suivie de la pesée du résidu sec, peut suffire.
1.5.4.2. Essais réalisés sur des échantillons de deux tailles différentes
La teneur en carbone organique total doit être déterminée pour tous les extraits aqueux.
Une détermination par gravimétrie est réalisée sur la partie non dissoute ou non extraite de l'échantillon. Si, après la centrifugation ou la filtration du contenu de chaque récipient, des résidus de polymère adhèrent encore à la paroi d'un récipient, il faut rincer ce dernier avec le filtrat jusqu'à ce qu'il ne comporte plus aucune trace visible de résidus. Après quoi, le filtrat est à nouveau filtré ou centrifugé. Les résidus déposés sur le filtre ou dans le tube de centrifugation sont séchés à 40 oC sous vide et pesés. On poursuit le séchage jusqu'à obtention d'une masse constante.
2. RÉSULTATS
2.1. ESSAIS RÉALISÉS SUR DES ÉCHANTILLONS D'UNE SEULE TAILLE
Les résultats obtenus pour chacun des trois flacons, ainsi que les valeurs moyennes, doivent être consignés et exprimés en unités de masse par volume de solution (généralement en mg/l) ou en unités de masse par masse d'échantillon de polymère (habituellement en mg/g). En outre, la perte de masse de l'échantillon (calculée en divisant la masse du soluté par la masse de l'échantillon initial) sera également mentionnée. Les écarts-types relatifs doivent être calculés. Les diverses valeurs seront consignées à la fois pour le produit total (polymère + principaux additifs, etc.) et pour le polymère seul (à savoir, après soustraction de la contribution relative à de tels additifs).
2.2. ESSAIS RÉALISÉS SUR DES ÉCHANTILLONS DE DEUX TAILLES DIFFÉRENTES
Les différentes concentrations du carbone organique total dans les extraits aqueux provenant des deux séries de trois expériences, ainsi que la valeur moyenne pour chaque série, doivent être consignées et exprimées en unités de masse par volume de solution (généralement en mgC/l), ainsi qu'en unités de masse par masse de l'échantillon initial (généralement en mgC/g).
S'il n'y a pas de différence entre les résultats correspondant aux rapports taille de l'échantillon/volume d'eau élevés et bas, cela peut indiquer que tous les constituants susceptibles d'être extraits l'ont effectivement été. Si tel est le cas, une analyse directe n'est, en principe, pas nécessaire.
Les différentes masses des résidus doivent être consignées et exprimées en pourcentage des masses initiales des échantillons. On calculera les valeurs moyennes pour chaque expérience. La différence entre 100 et le pourcentage obtenu représente le pourcentage de matières solubles et extractibles dans les échantillons de départ.
3. PROCÈS-VERBAL D'ESSAI
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai doit comporter les renseignements suivants:
3.1.1. Substance à tester
|
—
|
renseignements disponibles sur la substance à tester (nature, additifs, impuretés, proportion d'espèces de faible masse moléculaire).
|
3.1.2. Conditions expérimentales
|
—
|
description des méthodes utilisées et des conditions expérimentales,
|
|
—
|
description des méthodes d'analyse et de détection.
|
3.1.3. Résultats
|
—
|
résultats de solubilité — extractibilité en mg/l: toutes les valeurs et la valeur moyenne obtenues pour les essais d'extraction dans les différentes solutions, ventilées en polymères et impuretés, additifs, etc.,
|
|
—
|
résultats de solubilité — extractibilité en mg/g de polymère,
|
|
—
|
concentrations du carbone organique total dans les extraits aqueux, masse du soluté et pourcentages calculés, si on en fait la mesure,
|
|
—
|
pH de chaque échantillon,
|
|
—
|
informations sur les valeurs obtenues dans les essais à blanc,
|
|
—
|
description des méthodes d'analyse et de détection,
|
|
—
|
si nécessaire, mention de l'instabilité chimique de la substance à tester durant la procédure d'essai et durant la procédure d'analyse,
|
|
—
|
toutes les informations importantes pour interpréter les résultats.
|
4. RÉFÉRENCES
|
(1)
|
DIN 53733 (1976). Zerkleinerung von Kunststofferzeugnissen fur Prüfzwecke.
|
A.21. PROPRIÉTÉS COMBURANTES (LIQUIDES)
1. MÉTHODE
1.1. INTRODUCTION
La présente méthode vise à déterminer l'aptitude d'un liquide à accroître la vitesse de combustion ou l'intensité de la combustion d'une substance combustible, ou de provoquer l'inflammation spontanée d'une substance combustible avec laquelle il est mélangé de manière homogène. La méthode repose sur l'épreuve des Nations unies pour les liquides comburants (1) et en est l'équivalent. Toutefois, comme cette méthode A.21 est avant tout conçue pour satisfaire aux exigences du règlement 1907/2006, une seule substance de référence est nécessaire pour effectuer la comparaison. Il peut être nécessaire d'effectuer des tests et des comparaisons avec d'autres substances de référence lorsque les résultats du test doivent être utilisés à d'autres fins (9).
Il n'est pas nécessaire d'effectuer ce test lorsque l'examen de la formule structurale établit indubitablement que la substance est incapable de présenter une réaction exothermique avec un combustible.
Il est utile de disposer d'informations préliminaires sur les éventuelles propriétés explosives de la substance avant de pratiquer ce test.
Ce test ne s'applique pas aux solides, gaz, substances explosives ou hautement inflammables, ni aux peroxydes organiques.
Il n'est pas nécessaire d'effectuer ce test lorsque l'on dispose déjà, pour la substance concernée, des résultats de l'épreuve des Nations unies pour les liquides comburants (1).
1.2. DÉFINITIONS ET UNITÉS
Le temps moyen de montée en pression est la moyenne des temps mesurés pour que le mélange testé produise une élévation de pression de 690 kPa à 2 070 kPa au-dessus de la pression atmosphérique.
1.3. SUBSTANCE DE RÉFÉRENCE
La substance de référence est une solution aqueuse d'acide nitrique (qualité pour analyse) à 65 % (poids/poids) (10).
Le cas échéant, si l'expérimentateur prévoit que les résultats de ce test seront peut-être utilisés à d'autres fins (9), il peut également être indiqué de tester d'autres substances de référence (11).
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Le liquide à tester est mélangé avec de la cellulose fibreuse dans un rapport massique de 1 pour 1 et placé dans une bombe. Si le mélange s'enflamme spontanément lors du mélange ou du remplissage, il n'est pas nécessaire de poursuivre l'essai.
Si le mélange ne s'enflamme pas spontanément, il y lieu d'effectuer l'essai complet. Le mélange est chauffé dans la bombe et le temps moyen que met la pression pour s'élever de 690 kPa à 2 070 kPa au-dessus de la pression atmosphérique est déterminé. Ce résultat est comparé au temps moyen de montée en pression pour le mélange 1:1 de la (des) substance(s) de référence et de cellulose.
1.5 CRITÈRES DE QUALITÉ
Dans une série de cinq essais sur une substance, aucun résultat ne devrait s'écarter de plus de 30 % de la moyenne arithmétique. Les résultats qui diffèrent de plus de 30 % de la moyenne devraient être rejetés, la procédure de mélange et de remplissage améliorée et les essais répétés.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Préparation
1.6.1.1. Substance combustible
Comme matériau combustible on utilise de la cellulose fibreuse séchée ayant une longueur de fibre comprise entre 50 et 250 μm pour un diamètre de 25 μm (12). On la fait sécher en couche de moins de 25 mm d'épaisseur à 105 oC pendant 4 heures jusqu'à obtention d'un poids constant, puis on la maintient dans un dessiccateur en présence de dessiccant jusqu'à refroidissement et utilisation. La teneur en eau doit être inférieure à 0,5 % en masse (rapportée au poids sec) (13). Si cela est nécessaire pour satisfaire à cette condition, la durée de séchage doit être prolongée (14). Le même lot de cellulose doit être utilisé tout au long des essais.
1.6.1.2. Appareillage
1.6.1.2.1. Bombe
On doit disposer d'une bombe. Le dispositif d'essai est constitué par une bombe cylindrique en acier de 89 mm de longueur et de 60 mm de diamètre extérieur (voir figure 1). La bombe comporte deux plats usinés en des points diamétralement opposés (réduisant sa largeur à cet endroit à 50 mm), ce qui permet de l'immobiliser pour le serrage du bouchon de mise à feu et du bouchon à évent. Elle est alésée intérieurement à 20 mm et comporte aux deux extrémités un chambrage de 19 mm de profondeur taraudé au pas de 1 en tube normalisé BSP (British Standard Pipe) ou son équivalent métrique. Une prise de pression est vissée latéralement dans le corps de la bombe à 35 mm d'une extrémité, et à un angle de 90o par rapport aux plats. Elle se visse dans un chambrage de 12 mm de profondeur taraudé au pas de 1/2 en tube normalisé BSP (ou l'équivalent métrique). Si nécessaire, un joint en un matériau inerte est utilisé pour assurer l'étanchéité au gaz. La prise de pression fait saillie latéralement de 55 mm par rapport au corps de la bombe et elle est percée d'un trou axial de 6 mm. Elle comporte à son extrémité extérieure un chambrage taraudé pour recevoir un capteur de pression du type à diaphragme. Tout autre type de dispositif de mesure de la pression peut être utilisé, à condition qu'il résiste aux gaz chauds et produits de décomposition et qu'il puisse réagir à des accroissements de pression de 690 à 2 070 kPa en moins de 5 ms.
L'extrémité de la bombe la plus éloignée du raccord est fermée par un bouchon fileté qui porte deux électrodes, dont l'une est isolée du corps du bouchon et l'autre mise à la masse. L'autre extrémité est fermée par un disque de rupture en aluminium de 0,2 mm d'épaisseur (taré à une pression d'environ 2 200 kPa), maintenu en place par un bouchon comportant un évent de 20 mm. Si nécessaire, un joint inerte est utilisé pour assurer la bonne étanchéité entre le bouchon et la bombe. Un porte-bombe spécial (figure 2) permet de maintenir la bombe dans la position voulue pendant les essais. Il est généralement constitué par une embase en acier doux de 235 mm × 184 mm × 6 mm, sur laquelle est soudé obliquement un tube de section carrée (de 70 mm × 70 mm × 4 mm) de 185 mm de longueur.
À une extrémité du tube carré, on a enlevé une certaine longueur de métal sur deux faces opposées, ce qui laisse une longueur de 86 mm de tube carré prolongée par deux côtés plats. Les extrémités de ces plats sont coupées à 60o par rapport à l'axe du tube et soudées à la plaque d'embase. Une encoche de 22 mm de large et de 46 mm de profondeur est découpée sur un côté en haut du tube carré, de telle manière que lorsque la bombe repose sur le support, bouchon de mise à feu vers la base, le raccord de prise de pression vienne s'y loger. Une entretoise en acier de 30 mm de largeur et 6 mm d'épaisseur est soudée sur la paroi intérieure du tube du côté orienté vers le bas. Deux trous taraudés dans le côté opposé reçoivent des vis à molettes de 7 mm, qui servent à fixer la bombe. Deux rebords en acier de 12 mm de largeur et de 6 mm d'épaisseur, soudés sur les flancs du support à la base de la section carrée soutiennent la cuve par le fond.
1.6.1.2.2. Système d'allumage
Le système d'allumage est constitué par un enroulement de fil au Ni/Cr de 25 cm de longueur, de 0,6 mm de section, et d'une résistance électrique de 3,85 ohm/m. Le fil a été enroulé en forme de bobine sur un mandrin de 5 mm de diamètre; ses extrémités sont reliées aux électrodes du bouchon de mise à feu. La bobine doit avoir l'une des configurations présentées à la figure 3. La distance entre la face supérieure du bouchon et le point le plus bas de l'enroulement de chauffage doit être de 20 mm. Si les électrodes ne sont pas réglables en longueur, les deux sections de fil chauffant situées entre la bobine et la face supérieure du bouchon doivent être isolées par une gaine de céramique. Le fil doit être alimenté par une source électrique stable pouvant fournir une intensité d'au moins 10 A.
1.6.2. Réalisation de l'essai
(15)
La bombe montée, avec son capteur de pression, mais non fermée par son disque de rupture, est posée bouchon d'allumage vers le bas dans son support. On mélange 2,5 g du liquide à essayer avec 2,5 g de cellulose séchée dans un bécher en verre à l'aide d'un agitateur en verre (16). Pour des raisons de sécurité, lors de cette opération, le manipulateur devrait s'abriter derrière un écran de protection. Si le mélange s'enflamme spontanément au cours du mélange ou du remplissage, il n'est pas nécessaire de poursuivre l'essai. On remplit la bombe en plusieurs fois en tassant par petits chocs contre un objet dur; on s'assure qu'il n'y a pas de vide autour de l'enroulement de chauffage et que le mélange est directement en contact avec celui-ci. On doit cependant veiller à ne pas déformer l'enroulement en tassant le mélange car cela pourrait entraîner des résultats erronés (17). On met en place le disque de rupture et on visse le bouchon à évent en le bloquant. La bombe chargée est alors posée sur le porte-bombe, disque de rupture vers le haut, l'ensemble étant placé dans une hotte blindée ou dans une chambre de tir. On raccorde les bornes extérieures du bouchon de mise à feu à une source électrique et on applique au dispositif de chauffage un courant de 10 A. Il ne devrait pas s'écouler plus de 10 minutes entre le début de la préparation du mélange et le moment de la mise sous tension.
Le signal émis par le capteur de pression est enregistré sur un système permettant d'effectuer un enregistrement permanent de la courbe pression/temps et une analyse de cette courbe (enregistreur de signaux transitoires couplé à un enregistreur à bande de papier par exemple). Le mélange est chauffé jusqu'à rupture du disque ou pendant une durée d'au moins 60 s. S'il n'y a pas rupture, on doit attendre que le mélange ait refroidi avant d'ouvrir la bombe prudemment au cas où celle-ci serait encore sous pression. Cinq essais sont exécutés avec le mélange et avec chacune des substances de référence. On note le temps nécessaire pour monter de 690 kPa à 2 070 kPa (pression manométrique). Le temps moyen de montée en pression est calculé.
Dans certains cas, des substances peuvent engendrer une augmentation de pression (trop élevée ou trop faible), due à des réactions chimiques non caractéristiques des propriétés comburantes de ces substances. Il peut alors se révéler nécessaire de répéter l'épreuve en remplaçant la cellulose par une substance inerte, par exemple la diatomite (kieselguhr), afin de s'assurer de la nature de la réaction.
2. DONNÉES
Temps de montée en pression à la fois pour la substance à essayer et pour la (les) substance(s) de référence. Temps de montée en pression pour les essais avec une substance inerte, le cas échéant.
2.1. TRAITEMENT DES RÉSULTATS
Le temps moyen de montée en pression est calculé à la fois pour la substance à essayer et pour la (les) substances(s) de référence.
Le temps moyen de montée en pression est calculé pour les essais réalisés, avec une substance inerte (le cas échéant).
Le tableau 1 présente quelques exemples.
Tableau 1
Exemples de résultats
(18)
|
Substance (19)
|
Temps moyen de montée en pression pour un mélange 1:1 avec de la cellulose
(ms)
|
|
Dichromate d'ammonium, solution aqueuse saturée
|
20 800
|
|
Nitrate de calcium, solution aqueuse saturée
|
6 700
|
|
Nitrate ferrique, solution aqueuse saturée
|
4 133
|
|
Perchlorate de lithium, solution aqueuse saturée
|
1 686
|
|
Perchlorate de magnésium, solution aqueuse saturée
|
777
|
|
Nitrate de nickel, solution aqueuse saturée
|
6 250
|
|
Acide nitrique, 65 %
|
4 767 (20)
|
|
Acide perchlorique, 50 %
|
121 (20)
|
|
Acide perchlorique, 55 %
|
59
|
|
Nitrate de potassium, 30 % solution aqueuse
|
26 690
|
|
Nitrate d'argent, solution aqueuse saturée
|
(21)
|
|
Chlorate de sodium, 40 % solution aqueuse
|
2 555 (20)
|
|
Nitrate de sodium, 45 % solution aqueuse
|
4 133
|
|
Substance inerte
|
|
|
Eau: cellulose
|
(21)
|
3. COMPTE RENDU
3.1. COMPTE RENDU DE L'ESSAI
Le compte rendu de l'essai doit comprendre les informations suivantes:
|
—
|
l'identité, la composition, la pureté, etc. de la substance essayée,
|
|
—
|
la concentration de la substance,
|
|
—
|
la méthode de séchage de la cellulose employé,
|
|
—
|
la teneur en eau de la cellulose utilisé,
|
|
—
|
le résultat des mesures,
|
|
—
|
le résultat des essais réalisés avec une substance inerte, le cas échéant,
|
|
—
|
les temps moyens de montée en pression calculés,
|
|
—
|
tout écart par rapport à cette méthode et les raisons de celui-ci,
|
|
—
|
toute information ou remarque complémentaire présentant un intérêt pour l'interprétation des résultats.
|
3.2. INTERPRÉTATION DES RÉSULTATS (22)
Pour l'évaluation des résultats, on se fonde:
|
a)
|
sur le fait que le mélange substance à essayer/cellulose s'enflamme spontanément ou non;
|
|
b)
|
sur la comparaison du temps moyen de montée de 690 kPa à 2 070 kPa (pression manométrique) avec le temps moyen obtenu pour la (les) substance(s) de référence.
|
Une substance liquide doit être considérée comme comburante lorsque:
|
a)
|
un mélange 1:1, en poids, de la substance et de cellulose s'enflamme spontanément; ou
|
|
b)
|
un mélange 1:1, en poids, de la substance et de cellulose présente un temps moyen de montée en pression inférieur ou égal au temps moyen de montée en pression d'un mélange 1:1, en poids, d'une solution aqueuse d'acide nitrique à 65 % (poids/poids) et de cellulose.
|
Afin d'éviter les faux positifs, il y a lieu, si nécessaire, de tenir également compte, lors de l'interprétation des résultats, des résultats obtenus dans les essais de la substance avec un matériel inerte.
4. RÉFÉRENCES
|
(1)
|
Recommandations on the Transport of Dangerous Goods, Manual of Tests and Criteria. 3rd revised edition. UN Publication No: ST/SG/AC.lO/11/Rev. 3, 1999, page 342. Test O.2: Test for oxidizing liquids.
|
Figure 1
Bombe
Figure 2
Porte-bombe
Figure 3
Système d'allumage
(1) Valeur dépendante du type d'instrument et du degré de pureté de la substance.
(2) Valeur dépendante du type d'instrument et du degré de pureté de la substance.
(3) Valeur dépendante du type d'instrument et du degré de pureté de la substance.
(4) Valeur dépendante du type d'instrument et du degré de pureté de la substance.
(5) Cette précision n'est valable que pour les appareils simples comme celui décrit dans la norme ASTM D 1120-72; il est possible de l'améliorer en utilisant des ébulliomètres plus perfectionnés.
(6) Valable seulement pour les substances pures. Dans le cas contraire, son utilisation doit être justifiée.
(7) Dépendant du degré de pureté.
(8) Ces méthodes peuvent aussi être utilisées dans une gamme de 1 à 10 Pa, à condition de prendre des précautions.
(9) Comme par exemple dans le cadre du règlement des Nations unies sur les transports.
(10) L'acide doit être titré avant l'essai, afin de confirmer sa concentration.
(11) Par exemple: acide perchlorique à 50 % (poids/poids) et chlorate de sodium à 40 % (poids/poids) sont utilisés dans la référence 1.
(12) Par exemple Cellulose en poudre Whatman pour chromatographie sur colonne CF 11, no de catalogue 4021 050.
(13) Confirmé par titrage Karl-Fisher par exemple.
(14) On peut également obtenir cette teneur en eau, par exemple, en chauffant à 105 oC pendant 24 heures sous vide.
(15) Les mélanges de substances comburantes avec de la cellulose doivent être traités comme potentiellement explosifs et manipulés avec le plus grand soin.
(16) Dans la pratique, cela peut être réalisé en préparant un mélange 1:1 du liquide à essayer et de cellulose en quantité plus importante que ce qui est nécessaire pour l'essai et en transférant 5 ±0,1 g dans la bombe. Un nouveau mélange doit être préparé pour chaque essai.
(17) Il convient notamment d'éviter tout contact entre les spires adjacentes de la bobine.
(18) Voir référence (1) pour le classement au transport selon le système des Nations unies.
(19) Les solutions saturées doivent être préparées à 20 oC.
(20) Valeur moyenne d'après des essais interlaboratoires de comparaison.
(21) Pression maximale de 2 070 kPa non atteinte.
(22) Voir la référence 1 pour l'interprétation des résultats des essais effectués selon le règlement des Nations unies sur le transport, qui utilisent plusieurs substances de référence.
PARTIE B: MÉTHODES DE DÉTERMINATION DE LA TOXICITÉ ET DES AUTRES EFFETS SUR LA SANTÉ
TABLE DES MATIÈRES
INTRODUCTION GÉNÉRALE
|
B.1 bis.
|
TOXICITÉ ORALE AIGUË — MÉTHODE DE LA DOSE PRÉDÉTERMINÉE |
|
B.1 ter.
|
TOXICITÉ ORALE AIGUË — MÉTHODE DE LA CLASSE DE TOXICITÉ AIGUË |
|
B.2.
|
TOXICITÉ AIGUË (INHALATION) |
|
B.3.
|
TOXICITÉ AIGUË (CUTANÉE) |
|
B.4.
|
TOXICITÉ AIGUË: IRRITATION/CORROSION CUTANÉE |
|
B.5.
|
TOXICITÉ AIGUË: IRRITATION/CORROSION OCULAIRE |
|
B.6.
|
SENSIBILISATION CUTANÉE |
|
B.7.
|
TOXICITÉ (ORALE) PAR ADMINISTRATION RÉPÉTÉE (28 JOURS) |
|
B.8.
|
TOXICITÉ À DOSES RÉPÉTÉES (28 JOURS) (INHALATION) |
|
B.9.
|
TOXICITÉ À DOSES RÉPÉTÉES (28 JOURS) (ADMINISTRATION CUTANÉE) |
|
B.10.
|
MUTAGÉNICITÉ — ESSAI IN VITRO D'ABERRATION CHROMOSOMIQUE SUR CELLULES DE MAMMIFÈRE |
|
B.11.
|
MUTAGÉNICITÉ — ESSAI IN VIVO D'ABERRATION CHROMOSOMIQUE SUR MOELLE OSSEUSE DE MAMMIFÈRE |
|
B.12.
|
MUTAGÉNICITÉ — ESSAI IN VIVO DU MICRONOYAU SUR ÉRYTHROCYTES DE MAMMIFÈRE |
|
B.13/14.
|
MUTAGÉNICITÉ — ESSAI DE MUTATION RÉVERSE SUR BACTÉRIES |
|
B.15.
|
TESTS DE MUTAGENÈSE ET DE DÉPISTAGE DE CANCÉROGENÈSE MUTATION GÉNIQUE — SACCHAROMYCES CEREVISIAE
|
|
B. 16.
|
RECOMBINAISON MITOTIQUE — SACCHAROMYCES CEREVISIAE
|
|
B.17.
|
MUTAGÉNICITÉ — ESSAI IN VITRO DE MUTATION GÉNIQUE SUR CELLULES DE MAMMIFÈRE |
|
B.18.
|
LÉSION ET RÉPARATION D'ADN — SYNTHÈSE NON PROGRAMMÉE DE L'ADN (UDS) — CELLULES DE MAMMIFÈRE IN VITRO |
|
B.19.
|
ESSAI IN VITRO D'ÉCHANGE DE CHROMATIDES-SŒURS |
|
B.20.
|
TEST DE LÉTALITÉ RÉCESSIVE LIÉE AU SEXE SUR DROSOPHILA MELANOGASTER
|
|
B.21.
|
TESTS DE TRANSFORMATION SUR CELLULES DE MAMMIFÈRE IN VITRO |
|
B.22.
|
TEST DE LÉTALITÉ DOMINANTE CHEZ LE RONGEUR |
|
B.23.
|
ESSAI D'ABERRATION CHROMOSOMIQUE SUR SPERMATOGONIES DE MAMMIFÈRE |
|
B.24.
|
SPOT TEST CHEZ LA SOURIS |
|
B.25.
|
TRANSLOCATION HÉRÉDITAIRE CHEZ LA SOURIS |
|
B.26.
|
ESSAI DE TOXICITÉ SUBCHRONIQUE PAR VOIE ORALE — TOXICITÉ ORALE À DOSES RÉPÉTÉES — RONGEURS: 90 JOURS |
|
B.27.
|
ESSAI DE TOXICITÉ SUBCHRONIQUE PAR VOIE ORALE — TOXICITÉ ORALE À DOSES RÉPÉTÉES — NON-RONGEURS: 90 JOURS |
|
B.28.
|
TOXICITÉ DERMIQUE SUBCHRONIQUE — ÉPREUVE DE 90 JOURS SUR DES RONGEURS |
|
B.29.
|
TOXICITÉ SUBCHRONIQUE PAR INHALATION — EXPÉRIENCE DE 90 JOURS SUR DES RONGEURS |
|
B.30.
|
ÉTUDE DE LA TOXICITÉ CHRONIQUE |
|
B.31.
|
ÉTUDE DE LA TOXICITÉ POUR LE DÉVELOPPEMENT PRÉNATAL |
|
B.32.
|
ÉTUDE DE CANCÉROGÉNÈSE |
|
B.33.
|
ÉTUDE COMBINÉE DE CANCÉROGÉNÈSE ET DE TOXICITÉ CHRONIQUE |
|
B.34.
|
TEST DE REPRODUCTION SUR UNE GÉNÉRATION |
|
B.35.
|
ÉTUDE DE TOXICITÉ POUR LA REPRODUCTION SUR DEUX GÉNÉRATIONS |
|
B.37.
|
NEUROTOXICITÉ RETARDÉE DES SUBSTANCES ORGANOPHOSPORÉES APRÈS EXPOSITION AIGUË |
|
B.38.
|
NEUROTOXICITÉ RETARDÉE DES SUBSTANCES ORGANOPHOSPHORÉES — ÉTUDE PAR ADMINISTRATION RÉITÉRÉE SUR 28 JOURS |
|
B.39.
|
ESSAI IN VIVO DE SYNTHÈSE NON PROGRAMMÉE DE L'ADN (UDS) SUR CELLULES HÉPATIQUES DE MAMMIFÈRE |
|
B.40.
|
CORROSION CUTANÉE IN VITRO: ESSAI DE RÉSISTANCE ÉLECTRIQUE TRANSCUTANÉE (RET) |
|
B.40 bis.
|
CORROSION CUTANÉE IN VITRO: ESSAI SUR MODÈLE DE PEAU HUMAINE |
|
B.41.
|
ESSAI DE PHOTOTOXICITÉ IN VITRO 3T3 NRU |
|
B.42.
|
SENSIBILISATION CUTANÉE: ESSAI DE STIMULATION LOCALE DES GANGLIONS LYMPHATIQUES |
|
B.43.
|
ÉTUDE DE NEUROTOXICITÉ CHEZ LES RONGEURS |
|
B.44.
|
ABSORPTION CUTANÉE: MÉTHODE IN VIVO |
|
B.45.
|
ABSORPTION CUTANÉE: MÉTHODE IN VITRO |
INTRODUCTION GÉNÉRALE
A. CARACTÉRISATION DE LA SUBSTANCE À TESTER
Avant d'entreprendre toute étude de toxicité, il faut connaître la composition de la substance à tester, y compris les principales impuretés, ainsi que ses propriétés physico-chimiques dont sa stabilité.
Les propriétés physico-chimiques de la substance fournissent des informations importantes pour le choix de la voie d'administration, pour la conception des différentes études, ainsi que pour la manipulation et le stockage de la substance.
La mise au point d'une méthode d'analyse permettant une évaluation qualitative et quantitative de la substance à tester (y compris, si possible, de ses principales impuretés) dans le véhicule d'administration et dans le matériel biologique doit précéder la mise en œuvre de l'étude.
Toutes les informations concernant l'identification, les propriétés physico-chimiques, la pureté et le comportement de la substance à tester doivent être consignées dans le procès-verbal d'essai.
B. SOIN DES ANIMAUX
Lors des essais de toxicité, il est essentiel de procéder à des contrôles stricts des conditions ambiantes et d'utiliser des techniques appropriées de soin des animaux.
i) Conditions d'hébergement
Les conditions ambiantes dans les locaux ou enceintes réservés aux animaux d'expérience doivent être adaptées à l'espèce utilisée pour l'essai. Pour les rats, les souris et les cobayes, la température ambiante doit être de 22 oC ± 3 oC et l'humidité relative de 30 à 70 %; pour les lapins, la température doit être de 20 oC ± 3 oC et l'humidité relative de 30 à 70 %.
Certaines techniques expérimentales sont particulièrement sensibles aux effets thermiques et, dans de tels cas, des indications précises concernant les conditions adéquates figurent dans la description de la méthode d'essai. Dans tous les essais de toxicité, la température et l'humidité doivent être contrôlées et consignées, et doivent figurer dans le rapport final d'étude.
Un éclairage artificiel doit garantir l'alternance de 12 heures de lumière et 12 heures d'obscurité. Les détails concernant les conditions d'éclairage doivent être consignés dans le rapport final d'étude.
Sauf si la méthode prévoit d'autres conditions, les animaux doivent être hébergés individuellement ou mis en cage par petits groupes de même sexe. Les cages collectives ne doivent pas contenir plus de cinq animaux.
Il est important que les comptes rendus d'expérimentation animale précisent le type de cage utilisé et le nombre d'animaux par cage lors de l'exposition à la substance chimique et pendant toute période d'observation qui suit.
ii) Conditions d'alimentation
Le régime alimentaire doit répondre à tous les besoins nutritionnels de l'espèce soumise à l'expérience. Lorsque les substances à tester sont administrées dans la nourriture, il se peut que la valeur nutritionnelle de cette dernière soit amoindrie par une interaction entre la substance et un ingrédient de l'alimentation. Cette éventualité doit être prise en compte lors de l'interprétation des résultats de l'essai. Les régimes alimentaires classiquement utilisés en laboratoire sont acceptables, l'eau de boisson étant fournie à satiété. Le choix du régime alimentaire peut être guidé par la nécessité de garantir une proportion appropriée de substance en cas d'administration par cette méthode.
Les contaminants alimentaires qui agissent sur la toxicité ne doivent pas être présents à des concentrations susceptibles d'influencer les résultats.
C. MÉTHODES ALTERNATIVES
L'UE attache une grande importance à la mise au point et à la validation de méthodes alternatives qui fournissent autant d'informations que les expérimentations animales actuelles, mais qui nécessitent moins d'animaux, minimisent leurs souffrances et permettent d'éviter leur sacrifice.
Dès que de telles méthodes sont disponibles, elles doivent être envisagées, chaque fois que cela est possible, pour la caractérisation des dangers et pour la classification et l'étiquetage des substances en fonction des dangers intrinsèques.
D. ÉVALUATION ET INTERPRÉTATION
L'extrapolation directe à l'homme des résultats des expériences sur les animaux et des essais in vitro n'est possible que dans certaines limites; il faut en tenir compte lors de l'évaluation et de l'interprétation des essais; aussi, lorsque des effets indésirables ont été mis en évidence chez l'homme, ces données peuvent être utilisées pour confirmer les résultats expérimentaux.
E. RÉFÉRENCES BIBLIOGRAPHIQUES
La plupart des méthodes présentées ici sont élaborées dans le cadre du programme de lignes directrices de l'OCDE en matière d'essais. Ces méthodes doivent être mises en œuvre conformément aux bonnes pratiques de laboratoire, afin de garantir l'acceptation mutuelle des données.
De plus amples informations peuvent être obtenues dans les références citées dans les lignes directrices de l'OCDE et dans d'autres publications pertinentes.
B.1 bis. TOXICITÉ ORALE AIGUË — MÉTHODE DE LA DOSE PRÉDÉTERMINÉE
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 420 (2001) de l'OCDE.
1.1. INTRODUCTION
Les méthodes traditionnelles pour évaluer la toxicité aiguë utilisent comme effet observé la mort des animaux. En 1984, la British Toxicology Society (BTS) a proposé une nouvelle approche pour les essais de toxicité aiguë, utilisant des doses prédéterminées (1). Cette méthode évitait d'utiliser la mort des animaux comme effet observé et s'appuyait plutôt sur l'observation de signes manifestes de toxicité apparaissant après traitement à une dose prédéterminée. À la suite des études de validation in vivo au Royaume-Uni (2) et au niveau international (3), cette procédure a été adoptée comme ligne directrice en 1992. Après cela, les propriétés statistiques de la méthode de la dose prédéterminée ont été évaluées dans une série d'études utilisant des modèles mathématiques (4) (5) (6). Ensemble, les études in vivo et celles fondées sur des modèles mathématiques ont démontré que la procédure était reproductible et utilisait moins d'animaux auxquels elle occasionnait moins de souffrance que les méthodes traditionnelles. Elle permet de classer les substances par ordre de toxicité, de la même manière que les autres méthodes d'essai de toxicité aiguë.
Des indications permettant de choisir la méthode d'essai la plus appropriée pour un but donné sont présentées dans le document d'orientation sur les essais de toxicité orale aiguë (7). Ce document contient également de plus amples informations sur la conduite et l'interprétation de la méthode d'essai B.1 bis.
La méthode de la dose prédéterminée a comme principe de n'utiliser, pour l'étude principale, que des doses modérément toxiques et d'éviter d'administrer des doses qui peuvent s'avérer létales. De même, il n'est pas nécessaire d'administrer des doses dont on sait qu'elles provoquent des douleurs et une détresse importante du fait de propriétés corrosives ou sévèrement irritantes. Au cours de l'essai, les animaux moribonds et les animaux souffrant de façon manifeste ou montrant des signes de détresse grave devront être euthanasiés. Ces animaux devront être pris en compte dans l'interprétation des résultats au même titre que les animaux morts au cours de l'essai. Les critères pour décider d'euthanasier les animaux moribonds et ceux qui souffrent de façon manifeste, ainsi que les orientations pour reconnaître une mort prévisible ou imminente, font l'objet d'un autre document d'orientation (8).
La méthode fournit des informations qui permettent à la fois l'évaluation des dangers et le classement des substances selon le système général harmonisé de classification (SGH) des substances ayant une toxicité aiguë (9).
Le laboratoire doit rassembler toutes les informations disponibles sur la substance d'essai avant de procéder à l'essai. Ces informations comprennent l'identité et la structure chimique de la substance, ses propriétés physico-chimiques, les résultats obtenus dans tous les autres essais de toxicité in vitro et in vivo, les données toxicologiques concernant des substances structurellement apparentées, ainsi que le ou les usages escomptés de la substance. Ces informations sont nécessaires pour rassurer les personnes concernées quant à la pertinence de l'essai pour la protection de la santé humaine et elles seront utiles dans le choix de la dose initiale appropriée.
1.2. DÉFINITIONS ET UNITÉS
Toxicité orale aiguë: effets défavorables apparaissant après l'administration par voie orale d'une dose unique de substance ou de plusieurs doses données sur une période de 24 heures.
Mort différée: l'animal ne meurt ni n'apparaît moribond en l'espace de 48 heures, mais meurt ultérieurement au cours de la période d'observation de 14 jours.
Dose: la quantité de substance d'essai administrée. La dose s'exprime en poids de substance d'essai par unité de poids de l'animal d'expérience (par exemple mg/kg).
Toxicité manifeste: terme général désignant des signes évidents de toxicité qui surviennent à la suite de l'administration d'une substance d'essai [voir (3)]. Ces signes doivent être tels que l'on puisse s'attendre à ce qu'une augmentation de la dose administrée entraîne l'apparition de douleurs importantes et des signes persistants de détresse profonde, un état moribond [pour les critères, voir (8)] et probablement de la mortalité pour la plupart des animaux.
SGH: système général harmonisé de classification et d'étiquetage. Une activité conjointe de l'OCDE (santé humaine et environnement), du comité d'experts sur le transport des matières dangereuses (propriétés physico-chimiques) et du BIT (communication des dangers) et coordonnée par l'IOMC (Interorganisation Programme for the Sound Management of Chemicals).
Mort imminente: il y a mort imminente lorsqu'on s'attend à ce qu'un état moribond ou la mort interviennent avant le prochain moment d'observation prévu. Les signes indicatifs de cet état chez les rongeurs comprennent les convulsions, la position latérale, la position couchée et les tremblements [voir (8) pour plus de détails].
DL
50
(dose létale 50 %): dose unique d'une substance d'essai, obtenue par calcul statistique, susceptible d'entraîner la mort de 50 % des animaux lorsqu'elle est administrée par voie orale. La valeur de la DL
50
est exprimée en poids de la substance par unité de poids corporel de l'animal d'expérience (mg/kg).
Dose limite: désigne une dose qui est la limite supérieure pour l'essai (2 000 ou 5 000 mg/kg).
État moribond: désigne l'état précédant la mort ou l'incapacité à survivre, même si un traitement est donné [pour de plus amples détails, voir (8)].
Mort prévisible: présence de signes cliniques indiquant que la mort va intervenir à un moment déterminé dans le futur, avant la fin projetée de l'expérience, par exemple incapacité à atteindre l'eau ou la nourriture [pour plus de détails, voir (8)].
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
Des groupes d'animaux du même sexe reçoivent des doses prédéterminées de 5, 50, 300 et 2 000 mg/kg selon une procédure séquentielle. Exceptionnellement, une dose additionnelle de 5 000 mg/kg peut être envisagée (voir section 1.6.2). La dose initiale, choisie sur la base d'une étude d'orientation, est celle qui est susceptible de faire apparaître certains signes de toxicité, sans toutefois provoquer des effets toxiques graves ou la mort. Les manifestations cliniques et les effets associés à la douleur, à la souffrance et à une mort imminente sont décrits dans un document d'orientation de l'OCDE (8). D'autres groupes d'animaux reçoivent des doses plus fortes ou moins fortes en fonction de l'absence ou de la présence d'effets toxiques ou de mortalité. On continue la procédure jusqu'à ce que l'on identifie la dose qui occasionne un effet toxique évident ou la mort d'un seul animal. La procédure est également interrompue lorsque la dose la plus forte ne donne lieu à aucun effet observé ou lorsque la mort survient à la dose la plus faible.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Choix de l'espèce animale
Le rat est l'espèce préférée, mais d'autres espèces peuvent être utilisées. Normalement, on utilise des femelles (7). L'étude de la littérature sur les essais traditionnels de DL50 permet de conclure qu'il y a peu de différences de sensibilité entre sexes et que, lorsqu'il y a une différence, les femelles sont en général légèrement plus sensibles (10). Si, toutefois, compte tenu des propriétés toxicologiques et toxicocinétiques de composés structurellement voisins, il apparaît que les mâles sont probablement plus sensibles, il convient d'employer des mâles. Dans ce cas, il faut fournir une justification adéquate.
On utilise de jeunes animaux adultes sains issus de souches couramment utilisées en laboratoire. Les femelles doivent être nullipares et non gravides. Au début de l'essai, chaque animal doit être âgé de 8 à 12 semaines et son poids doit se situer dans un intervalle de ± 20 % du poids moyen des animaux précédemment exposés.
1.4.2. Conditions d'hébergement et d'alimentation
La température du local des animaux d'expérience doit être maintenue à 22 oC (± 3 oC). L'humidité relative doit atteindre au moins 30 % et rester de préférence inférieure à 70 %, mais en dehors des heures de nettoyage du local, on s'efforcera de maintenir le taux d'humidité autour de 50 à 60 %. On appliquera un éclairage artificiel alternant 12 heures de lumière et 12 heures d'obscurité. Pour l'alimentation des animaux, on peut utiliser la nourriture classique de laboratoire avec de l'eau potable à satiété. Les animaux peuvent être groupés par dose. Toutefois, le nombre d'animaux par cage ne doit pas faire obstacle à une observation précise de chaque animal.
1.4.3. Préparation des animaux
Les animaux sont choisis au hasard, marqués pour permettre une identification individuelle et gardés dans leurs cages pour les acclimater aux conditions de laboratoire pendant au moins cinq jours avant l'expérience.
1.4.4. Préparation des doses
En général, il convient d'administrer la substance d'essai à volume constant quelle que soit la dose testée, en faisant varier la concentration de la préparation. Lorsqu'un produit liquide ou un mélange font l'objet de l'essai, l'utilisation du produit non dilué, donc à concentration constante, peut être plus appropriée pour l'évaluation du risque de cette substance. Certaines autorités réglementaires l'exigent. Dans tous les cas, il ne faut pas dépasser le volume de dose maximal. Le volume maximal de liquide qui peut être administré en une seule fois dépend de la taille de l'animal d'essai. Chez les rongeurs, ce volume ne doit pas dépasser 1 ml/100 g de poids corporel, sauf dans le cas de solutions aqueuses pour lesquelles on peut utiliser 2 ml/100 g de poids corporel. Il est recommandé d'utiliser une solution aqueuse chaque fois que cela est possible, sinon on peut utiliser une solution dans de l'huile (par exemple de l'huile de maïs) et éventuellement une solution dans d'autres véhicules. En ce qui concerne les véhicules non aqueux, leur toxicité doit être connue. Les doses doivent être préparées juste avant l'administration, sauf si la stabilité de la préparation pendant la durée de la période d'utilisation est connue et jugée acceptable.
1.5. MODE OPÉRATOIRE
1.5.1. Administration des doses
La substance d'essai est administrée en une seule dose par gavage à l'aide d'une sonde gastrique ou de toute autre canule pour intubation appropriée. Lorsqu'il n'est pas possible d'administrer la dose en une seule fois, celle-ci peut être fractionnée sur une période n'excédant pas 24 heures.
Les animaux doivent être à jeun avant l'administration de la substance (on supprimera la nourriture, mais pas l'eau, pendant toute une nuit pour les rats et pendant 3 à 4 heures pour les souris). Après la période de jeûne, les animaux doivent être pesés; la substance d'essai leur est ensuite administrée. Après l'administration de la substance, les animaux peuvent être à nouveau privés de nourriture, pendant 3 à 4 heures pour les rats et pendant 1 à 2 heures pour les souris. Si la dose est administrée par fractions sur un certain laps de temps, il peut s'avérer nécessaire, en fonction de la durée du traitement, d'alimenter et de faire boire les animaux.
1.5.2. Étude d'orientation
Le but de l'étude d'orientation est de permettre la sélection d'une dose initiale appropriée pour l'étude principale. L'administration des doses se fait de façon séquentielle aux animaux individuellement selon les schémas de l'annexe 1. L'étude d'orientation prend fin lorsqu'une décision au sujet de la dose initiale de l'étude principale peut être prise (ou lorsqu'une mortalité est observée à la dose prédéterminée la plus faible).
Pour la dose initiale de l'étude d'orientation, on choisit un niveau parmi les suivants: 5, 50, 300 et 2 000 mg/kg. Le niveau choisi est celui pour lequel on peut s'attendre à observer des signes de toxicité évidente, si possible sur la base d'indications obtenues à partir de données in vivo et in vitro sur la même substance et sur des substances structurellement voisines. En l'absence de telles informations, la dose initiale sera de 300 mg/kg.
Les animaux sont traités à 24 heures d'intervalle au moins. Tous les animaux sont observés pendant au moins quatorze jours.
Exceptionnellement, et uniquement lorsque cela est nécessaire pour satisfaire à une exigence réglementaire particulière, l'utilisation d'une dose maximale prédéterminée supplémentaire de 5 000 mg/kg peut être envisagée (voir annexe 3). Par souci de protection des animaux, l'essai de substances en catégorie 5 du SGH (2 000 - 5 000 mg/kg) est déconseillé et ne doit être envisagé que lorsqu'il est très probable qu'il donnera des résultats qui présenteront un intérêt direct pour la protection de la santé des hommes et des animaux ou de l'environnement.
Dans le cas où un animal ayant reçu la dose prédéterminée la plus faible (5 mg/kg) mourrait au cours de l'étude d'orientation, la procédure normale est de terminer l'étude et de classer la substance en catégorie 1 du SGH (voir l'annexe 1). Cependant, il peut s'avérer nécessaire de confirmer la classification, et la procédure facultative supplémentaire ci-après est alors proposée. Un deuxième animal reçoit une dose de 5 mg/kg. Si ce deuxième animal meurt, la classification en catégorie 1 du SGH est confirmée et l'étude prend fin immédiatement. Si le deuxième animal survit, un maximum de trois animaux supplémentaires reçoivent chacun 5 mg/kg. Étant donné le risque de mortalité élevé, il convient de procéder de manière séquentielle par souci de protection des animaux. L'intervalle de temps entre chaque administration doit être suffisant afin de pouvoir démontrer que l'animal précédent a des chances de survivre. Si un deuxième animal meurt, l'administration séquentielle sera immédiatement arrêtée et aucun autre animal ne recevra de dose. Avec la mort d'un deuxième animal (indépendamment du nombre d'animaux soumis à l'essai au moment où celui-ci est arrêté), le résultat est A (2 morts ou plus), et la règle de classification présentée à l'annexe 2 pour la dose prédéterminée de 5 rng/kg s'applique: catégorie 1 s'il y a 2 morts ou plus, et catégorie 2 s'il y a seulement 1 mort. En outre, l'annexe 4 propose une méthode de classification selon le système communautaire en attendant la mise en place du nouveau SGH.
1.5.3. Étude principale
1.5.3.1. Nombre d'animaux et niveaux des doses
Les schémas de l'annexe 2 indiquent la marche à suivre après administration de la dose initiale. Il y a trois possibilités: arrêter l'essai et attribuer la classification appropriée, administrer la dose prédéterminée supérieure ou administrer la dose inférieure. Cependant, par souci de protection des animaux, les doses ayant entraîné la mort au cours de l'étude d'orientation ne sont pas réappliquées lors de l'étude principale (voir annexe 2). L'expérience démontre que le résultat le plus probable à la dose initiale est que la substance pourra être classée et qu'il sera inutile de prolonger l'essai.
En général, on utilise au total cinq animaux du même sexe à chaque niveau de dose étudié. Ce groupe de cinq animaux est constitué de l'animal utilisé dans l'étude d'orientation, auquel est administrée la dose sélectionnée, et de quatre animaux supplémentaires (sauf dans les rares cas où une dose utilisée dans l'étude principale n'a pas été utilisée dans l'étude d'orientation).
L'intervalle de temps à respecter entre l'administration de chaque niveau de dose dépend du moment où se manifestent les effets toxiques, de leur durée et de leur gravité. L'administration de la dose suivante doit être retardée jusqu'à ce que l'on ait obtenu la certitude que les animaux précédemment soumis au traitement vont survivre. Un laps de temps de 3 ou 4 jours entre les administrations à chaque niveau de dose est recommandé, si nécessaire, pour permettre l'observation de signes de toxicité différée. Cet intervalle peut être ajusté selon qu'il convient, par exemple, en cas de réponse peu concluante.
Lorsque l'on envisage d'utiliser une dose prédéterminée maximale de 5 000 mg/kg, il y a lieu de suivre la procédure présentée à l'annexe 3 (voir également section 1.6.2).
1.5.3.2. Essai limite
L'essai limite est utilisé principalement lorsque l'expérimentateur dispose d'informations indiquant que la substance d'essai n'est probablement pas toxique, c'est-à-dire que sa toxicité ne se manifeste qu'au-delà des doses limites réglementaires. Des informations concernant la toxicité de la substance d'essai peuvent être déduites des connaissances acquises sur des substances, produits ou mélanges similaires déjà testés, compte tenu de l'identité et du pourcentage des composants importants sur le plan toxicologique. Quand on ne dispose pas d'informations concernant la toxicité de la substance, ou lorsqu'on s'attend à une substance toxique, il faut effectuer l'essai principal.
Selon la procédure normale, une étude d'orientation à la dose initiale de 2 000 mg/kg (exceptionnellement 5 000 mg/kg), suivie du traitement de quatre animaux supplémentaires à cette même dose, sert d'essai limite dans cette ligne directrice.
1.6. OBSERVATIONS
Les animaux doivent être observés individuellement au moins une fois pendant les 30 premières minutes suivant l'administration de la substance et régulièrement pendant les premières 24 heures, avec une attention particulière au cours des 4 premières heures; l'observation doit ensuite être quotidienne, pendant 14 jours en tout, sauf dans le cas des animaux qui meurent en cours d'étude ou qui doivent être retirés de l'étude et euthanasiés pour leur épargner des souffrances excessives. Toutefois, la durée d'observation ne doit pas être fixée de manière rigide. Elle doit être fonction des réactions de toxicité, du moment où elles apparaissent et de la durée de la période de récupération. Elle peut donc être prolongée si nécessaire. Les moments où les signes de toxicité apparaissent et disparaissent sont importants, surtout si les effets toxiques ont tendance à être différés (11). Toutes les observations sont enregistrées de façon systématique, une fiche individuelle étant établie pour chaque animal.
D'autres observations peuvent s'avérer nécessaires lorsque les animaux continuent à manifester des signes de toxicité. Les observations doivent porter sur les modifications de la peau, des poils, des yeux et des muqueuses, ainsi que de l'appareil respiratoire, du système circulatoire, du système nerveux central et autonome, de l'activité somatomotrice et du comportement. L'attention portera en particulier sur l'observation des diverses manifestations de tremblement, convulsions, salivation, diarrhée, léthargie, sommeil et coma. Les principes et critères résumés dans le document d'orientation sur les effets sur l'homme doivent être pris en considération (8). Les animaux moribonds et les animaux souffrant manifestement ou présentant des signes graves de détresse doivent être euthanasiés. Qu'il s'agisse d'animaux euthanasiés pour raisons éthiques ou d'animaux retrouvés morts, le moment de la mort doit être enregistré de façon aussi précise que possible.
1.6.1. Poids corporel
Le poids de chaque animal doit être déterminé peu de temps avant l'administration de la substance d'essai et au moins une fois par semaine ensuite. Les variations de poids doivent être calculées et enregistrées. À la fin de l'essai, les animaux survivants sont pesés puis euthanasiés.
1.6.2. Pathologie
Tous les animaux d'essai (y compris ceux qui sont morts au cours de l'essai ou ceux qui ont été retirés de l'étude pour des raisons éthiques) doivent être soumis à une autopsie à l'échelle macroscopique. Pour chaque animal, toutes les altérations pathologiques macroscopiques doivent être enregistrées. Chez les animaux qui survivent 24 heures ou plus après administration de la dose initiale, l'examen microscopique des organes présentant des signes évidents de pathologie doit également être envisagé, car il peut fournir des renseignements utiles.
2. RÉSULTATS
Les résultats obtenus pour chaque animal doivent être présentés. Toutes les données doivent être résumées dans un tableau indiquant, pour chaque groupe d'essai, le nombre d'animaux utilisés, le nombre d'animaux présentant des signes de toxicité, le nombre d'animaux retrouvés morts pendant l'essai ou sacrifiés pour des raisons éthiques, et pour chaque animal, le moment de la mort, la description des effets toxiques et leur évolution dans le temps ainsi que leur réversibilité éventuelle et les résultats de l'autopsie.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit contenir, s'il y a lieu, les renseignements suivants:
|
|
Substance d'essai:
|
—
|
état physique, pureté et, s'il y a lieu, propriétés physico-chimiques (compris isomérisation),
|
|
—
|
données relatives à l'identification, y compris numéro CAS.
|
|
|
|
Véhicule (le cas échéant):
|
—
|
justification du choix du véhicule, s'il ne s'agit pas d'eau.
|
|
|
|
Animaux d'essai:
|
—
|
espèce/souche utilisée,
|
|
—
|
état microbiologique des animaux, s'il est connu,
|
|
—
|
nombre, âge et sexe des animaux (le cas échéant, justification du choix de mâles à la place de femelles),
|
|
—
|
origine, conditions d'hébergement, régime alimentaire, etc.
|
|
|
|
Conditions de l'essai:
|
—
|
détails sur la formulation de la substance d'essai, notamment état physique du produit administré,
|
|
—
|
détails sur le mode d'administration, notamment volume des doses et moment de l'administration,
|
|
—
|
détails sur la qualité de la nourriture et de l'eau (y compris le type de régime et sa provenance ainsi que celle de l'eau),
|
|
—
|
justification du choix de la dose initiale.
|
|
|
|
Résultats:
|
—
|
tableau des réactions aux différentes doses pour chaque animal (c'est-à-dire nombre d'animaux montrant des signes de toxicité, y compris mortalité, nature, gravité et durée des effets),
|
|
—
|
tableau des poids corporels et des variations du poids,
|
|
—
|
poids individuel des animaux le jour du traitement, et ensuite à intervalles d'une semaine, et au moment de la mort ou du sacrifice,
|
|
—
|
date et heure de la mort si celle-ci intervient avant le moment prévu du sacrifice,
|
|
—
|
pour chaque animal, moment d'apparition et évolution des signes de toxicité et réversibilité éventuelle,
|
|
—
|
pour chaque animal, résultats de l'autopsie et observations histopathologiques.
|
|
|
|
Discussion et interprétation des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
British Toxicology Society Working Party on Toxicity (1984). Special report: a new approach to the classification of substances and preparations on the basis of their acute toxicity. Human Toxicol., 3, p. 85-92.
|
|
(2)
|
Van den Heuvel, M.J., Dayan, A.D. and Shillaker, R.O. (1987). Evaluation of the BTS approach to the testing of substances and preparations for their acute toxicity. Human Toxicol., 6, p. 279-291.
|
|
(3)
|
Van den Heuvel, M.J., Clark, D.G., Fielder, R.J., Koundakjian, P.P., Oliver, G.J.A., Pelling, D., Tomlinson, N.J. and Walker, A.P. (1990). The international validation of a fixed-dose procedure as an alternative to the classical LD50 test. Fd. Chem. Toxicol. 28, p. 469-482.
|
|
(4)
|
Whitehead, A. and Curnow, R.N. (1992). Statistical evaluation of the fixed-dose procedure. Fd. Chem. Toxicol., 30, p. 313-324.
|
|
(5)
|
Stallard, N. and Whitehead, A. (1995). Reducing numbers in the fixed-dose procedure. Human Exptl. Toxicol. 14, 315-323. Human Exptl. Toxicol.
|
|
(6)
|
Stallard, N., Whitehead, A. and Ridgeway, P. (2002). Statistical evaluation of the revised fixed dose procedure. Hum. Exp. Toxicol., 21, p. 183-196.
|
|
(7)
|
OECD (2001). Guidance Document on Acute Oral Toxicity. Environmental Health and Safety Monograph Series on Testing and Assessment No 24. Paris
|
|
(8)
|
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment No 19.
|
|
(9)
|
OECD (1998). Harmonised Integrated Hazard Classification for Human Health and Environmental Effects of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p. 11 [http://webnetl.oecd.org/oecd/pages/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-0,FF.html].
|
|
(10)
|
Lipnick, R.L., Cotruvo, J.A., Hill, R.N., Bruce, R.D., Stitzel, K.A., Walker, A.P., Chu, I., Goddard, M., Segal, L, Springer, J.A. and Myers, R.C. (1995). Comparison of the Up-and-Down, Conventional LD50, and Fixed-Dose Acute Toxicity Procedures. Fd. Chem. Toxicol. 33, p. 223-231.
|
|
(11)
|
Chan, P.K and A.W. Hayes (1994). Chapter 16 Acute Toxicity and Eye Irritation in: Principles and Methods of Toxicology. 3rd Edition. A.W. Hayes, Editor. Raven Press, Ltd. New York, USA.
|
ANNEXE 1
SCHÉMA POUR L'ÉTUDE D'ORIENTATION
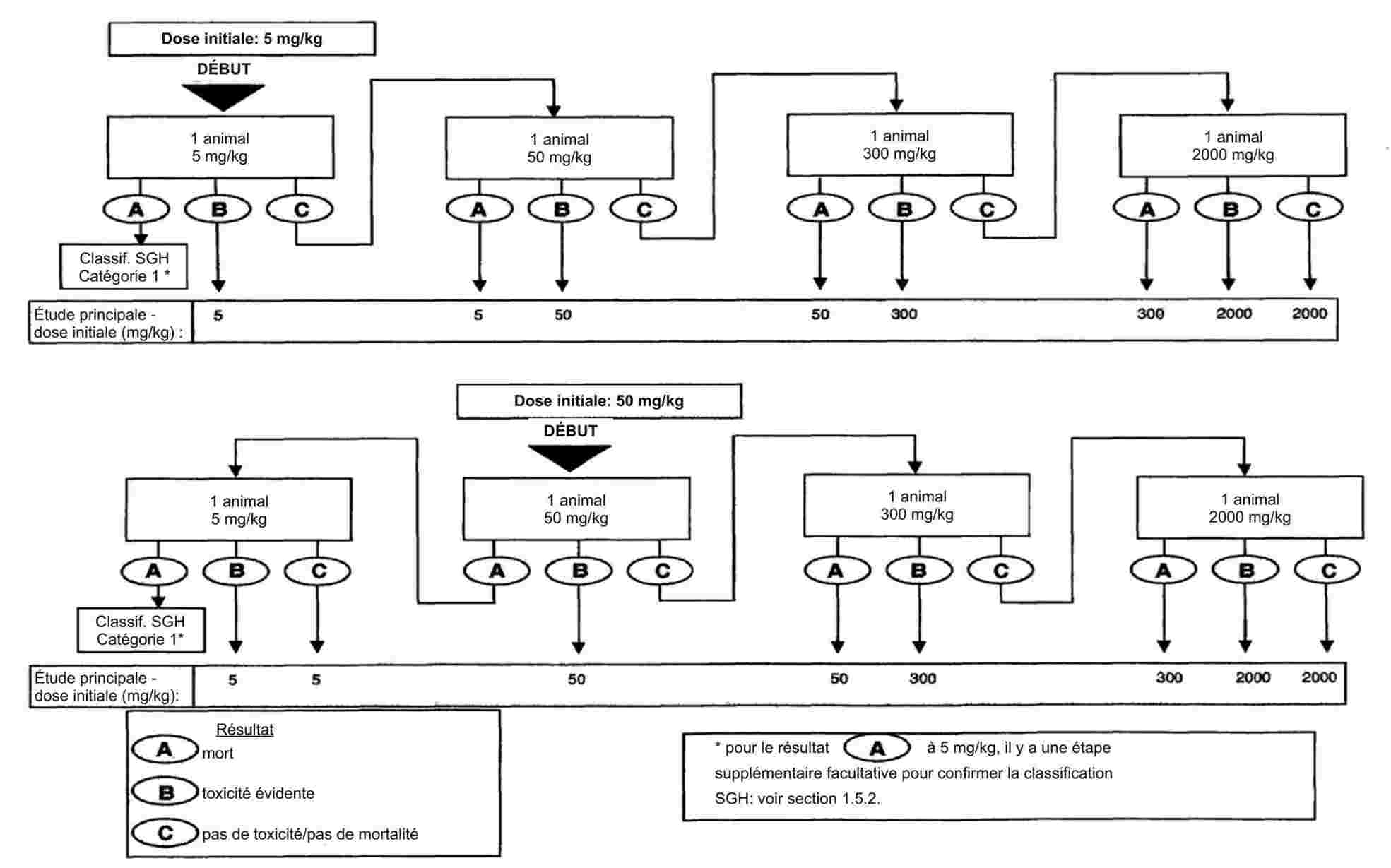

ANNEXE 2
SCHÉMA POUR L'ÉTUDE PRINCIPALE

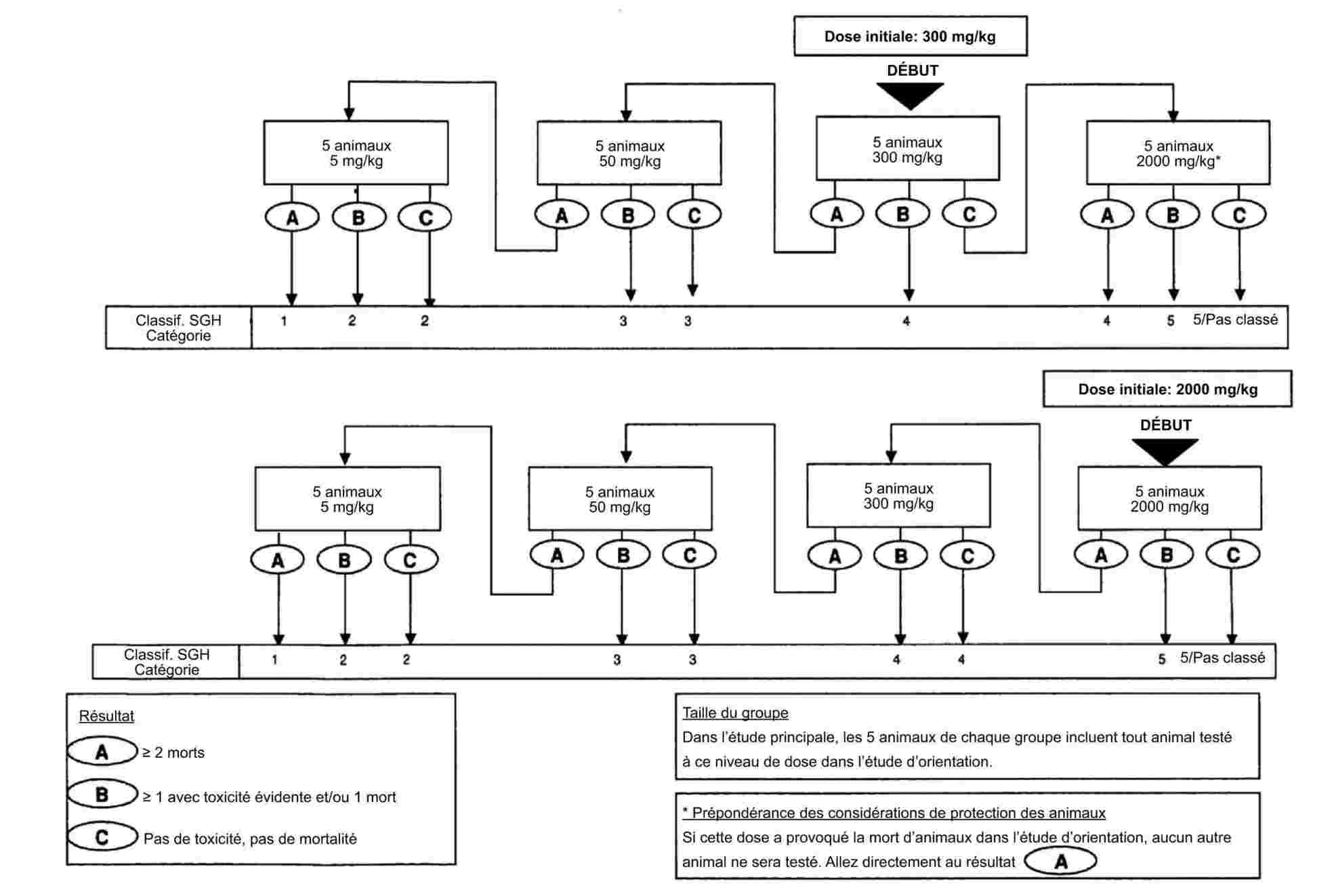
ANNEXE 3
CRITÈRES POUR LA CLASSIFICATION DE SUBSTANCES D'ESSAI DONT ON PRÉSUME QUE LA DL50 EST SUPÉRIEURE À 2 000 mg/kg, SANS RECOURIR À L'ESSAI
Les critères pour la catégorie de danger 5 sont destinés à permettre l'identification de substances qui ont une toxicité aiguë relativement faible mais qui, dans certaines conditions, peuvent se révéler dangereuses pour des populations vulnérables. Ces substances sont présumées avoir une DL50 orale ou cutanée comprise dans la plage 2 000 - 5 000 mg/kg ou correspondant à des doses équivalentes par d'autres voies. Des substances d'essai peuvent être classées dans la catégorie SGH de danger 5, définie par 2 000 mg/kg < DL50 < 5 000 mg/kg, dans les cas suivants:
|
a)
|
lorsque, en fonction de la mortalité, l'un des schémas de l'annexe 2 oriente la substance vers cette catégorie;
|
|
b)
|
lorsque l'on est déjà en possession de données fiables indiquant que la DL50 se situera dans la plage des valeurs de la catégorie 5; ou lorsque d'autres études sur des animaux ou l'observation d'effets toxiques chez l'homme suscitent de sérieuses inquiétudes pour la santé humaine;
|
|
c)
|
par extrapolation, évaluation ou mesure de données, lorsque la classification dans une catégorie de plus grand danger n'est pas justifiée, et
|
—
|
il existe des informations fiables faisant état d'effets toxiques significatifs chez l'homme, ou
|
|
—
|
une certaine mortalité est observée en testant par voie orale jusqu'aux valeurs de la catégorie 4, ou
|
|
—
|
lorsqu'un jugement d'expert confirme l'existence de signes cliniques significatifs de toxicité lors d'un essai mené jusqu'aux valeurs de la catégorie 4, hormis diarrhée, hérissement des poils ou aspect mal soigné, ou
|
|
—
|
quand un jugement d'expert confirme l'existence d'informations fiables indiquant la possibilité d'effets aigus significatifs d'après les résultats des autres études sur animaux.
|
|
ESSAIS À DOSES SUPÉRIEURES À 2 000 mg/kg
Exceptionnellement, et uniquement lorsque cela est nécessaire pour satisfaire à une exigence réglementaire particulière, l'utilisation d'une dose maximale supplémentaire de 5 000 mg/kg peut être envisagée. Par souci de protection des animaux, l'essai de substances à la dose de 5 000 mg/kg est déconseillé et ne devrait être envisagé que lorsqu'il est fort probable que les résultats d'un tel essai présenteront un intérêt direct pour la protection de la santé des hommes et des animaux (9).
Étude d'orientation
Les critères de décision de la procédure séquentielle présentée à l'annexe 1 sont étendus de manière à s'appliquer également à une dose de 5 000 mg/kg. Ainsi, lorsqu'une dose initiale de 5 000 mg/kg est utilisée dans l'étude d'orientation et que le résultat A (mortalité) est obtenu, il faut tester un deuxième animal à 2 000 mg/kg; si le premier résultat est B ou C (toxicité évidente ou pas de toxicité), on pourra choisir 5 000 mg/kg comme dose initiale dans l'étude principale. De même, si on choisit une dose initiale différente de 5 000 mg/kg, l'essai se poursuivra à la dose de 5 000 mg/kg en cas d'obtention d'un résultat B ou C à 2 000 mg/kg. L'obtention d'un nouveau résultat A à 5 000 mg/kg imposera une dose initiale de 2 000 mg/kg pour l'étude principale, alors qu'un résultat B ou C imposera 5 000 mg/kg comme dose initiale pour cette étude.
Étude principale
Les critères de décision de la procédure séquentielle présentée à l'annexe 2 sont étendus de manière à s'appliquer également à une dose de 5 000 mg/kg. Ainsi, lorsqu'une dose initiale de 5 000 mg/kg est utilisée dans l'étude principale et que le résultat A (> 2 morts) est obtenu, il faut tester un second groupe à 2 000 mg/kg. Si le premier résultat est B (toxicité évidente et/ou ≤ 1 mort) ou C (pas de toxicité), la substance ne sera pas classée dans le cadre du SGH. De façon similaire, si une dose initiale différente de 5 000 mg/kg est choisie, l'essai se poursuivra à la dose de 5 000 mg/kg en cas d'obtention d'un résultat C à 2 000 mg/kg. L'obtention d'un nouveau résultat A à 5 000 mg/kg entraînera la classification de la substance dans la catégorie 5 du SGH; si le résultat obtenu est B ou C, la substance ne sera pas classée dans le cadre du SGH.
ANNEXE 4
MÉTHODE D'ESSAI B.1 bis
Conseils pour une classification selon le système de l'UE en attendant la mise en place effective du système général harmonisé de classification (SGH) [voir référence (8)]
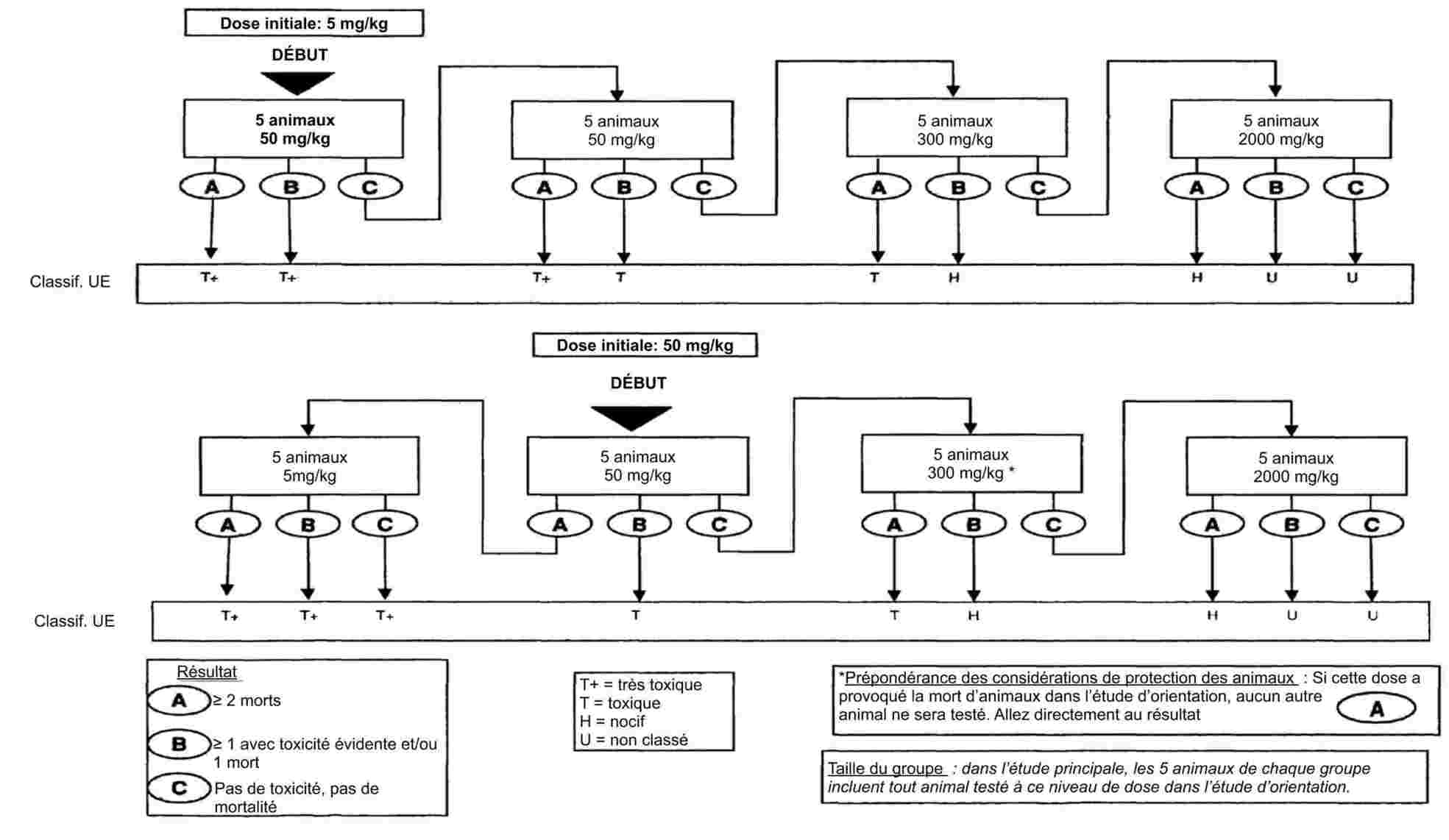
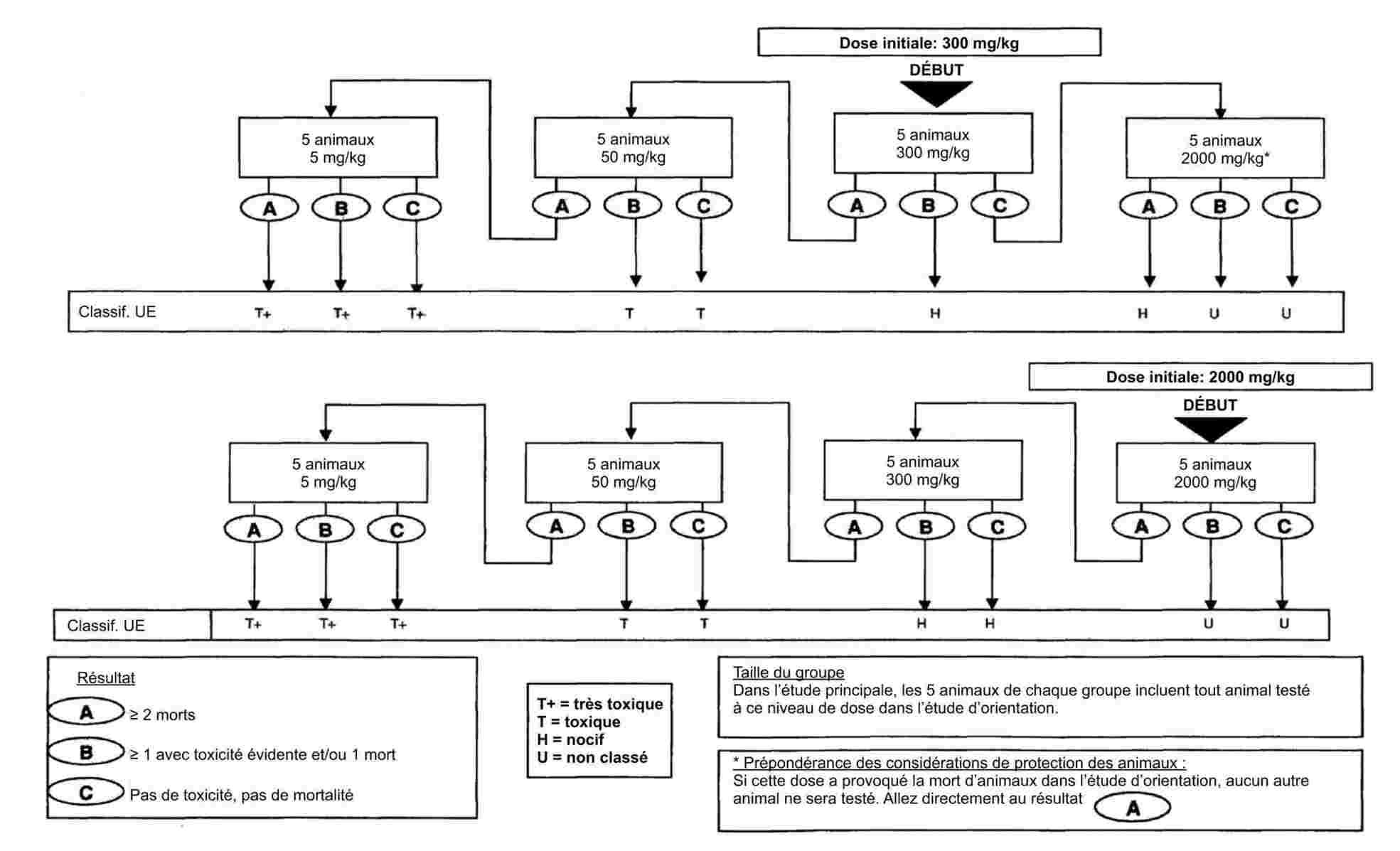
B.1 ter. TOXICITÉ ORALE AIGUË — MÉTHODE DE LA CLASSE DE TOXICITÉ AIGUË
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 423 (2001) de l'OCDE.
1.1. INTRODUCTION
La méthode de la classe de toxicité aiguë (1) décrite dans cet essai est une procédure séquentielle qui utilise trois animaux de même sexe à chaque étape. Suivant la mortalité et/ou l'état moribond des animaux, deux à quatre étapes sont en moyenne nécessaires pour évaluer la toxicité aiguë de la substance d'essai. Cette procédure est reproductible, utilise très peu d'animaux et permet de classer des substances par ordre de toxicité de la même manière que les autres méthodes de toxicité aiguë. La méthode par classe de toxicité aiguë est fondée sur des évaluations biométriques (2) (3) (4) (5) avec des doses prédéterminées, convenablement espacées de manière à permettre le classement des substances les unes par rapport aux autres aux fins de l'évaluation des dangers. La méthode, telle qu'elle a été adoptée en 1996, a été largement validée in vivo par rapport aux données de DL50 issues de la littérature, tant sur le plan national (6) que sur le plan international (7).
Des indications permettant de choisir la méthode d'essai la plus appropriée pour un but donné sont présentées dans le document d'orientation sur les essais de toxicité orale aiguë (8). Ce document contient également de plus amples informations sur la conduite et l'interprétation de la méthode d'essai B.l ter.
Il n'est pas nécessaire d'administrer des substances d'essai à des niveaux de dose dont on sait qu'elles provoquent des douleurs et une détresse importante du fait des propriétés corrosives ou sévèrement irritantes. Au cours de l'essai, les animaux moribonds et les animaux souffrant de façon manifeste ou montrant des signes de détresse grave devront être euthanasiés. Ces animaux devront être pris en compte dans l'interprétation des résultats, au même titre que les animaux morts pendant l'essai. Les critères pour décider de tuer les animaux moribonds et ceux qui souffrent de façon manifeste ainsi que des orientations pour reconnaître une mort prévisible ou imminente font l'objet d'un autre document d'orientation (9).
La méthode utilise des doses prédéterminées et donne des résultats qui permettent le classement des substances selon le système général harmonisé de classification (SGH) des substances ayant une toxicité aiguë (10).
En principe, la méthode ne vise pas à calculer une valeur précise de DL50, mais elle permet de déterminer dans quelle gamme de doses la substance doit être considérée comme létale, puisque la mort d'une partie des animaux reste le principal effet observé dans cet essai. La méthode permet de déterminer une DL50 uniquement dans le cas où au moins deux doses donnent une mortalité supérieure à 0 % et inférieure à 100 %. Grâce à l'utilisation d'un choix de doses prédéterminées, indépendamment de la substance d'essai, et au lien explicite entre classification et nombre d'animaux dans différents états observés, la cohérence entre laboratoires est favorisée.
Le laboratoire doit rassembler toutes les informations disponibles sur la substance d'essai avant de procéder à l'essai. Ces informations comprennent l'identité et la structure chimique de la substance, ses propriétés physico-chimiques, le résultat de tout autre essai de toxicité réalisé in vivo ou in vitro sur la substance, les données toxicologiques concernant des substances structurellement apparentées, ainsi que le ou les usages escomptés de la substance. Ces informations sont nécessaires pour rassurer les personnes concernées quant à la pertinence de l'essai pour la protection de la santé humaine et elles seront utiles dans le choix de la dose initiale appropriée.
1.2. DÉFINITIONS
Toxicité orale aiguë: effets défavorables apparaissant après l'administration par voie orale d'une dose unique de substance ou de plusieurs doses données sur une période de 24 heures.
Mort différée: l'animal ne meurt ni n'apparaît moribond en l'espace de 48 heures, mais meurt ultérieurement au cours de la période d'observation de 14 jours.
Dose: la quantité de substance d'essai administrée. La dose s'exprime en poids de substance d'essai par unité de poids de l'animal d'expérience (par exemple mg/kg).
SGH: système général harmonisé de classification et d'étiquetage. Une activité conjointe de l'OCDE (santé humaine et environnement), du comité d'experts sur le transport des matières dangereuses (propriétés physico-chimiques) et du BIT (communication des dangers) et coordonnée par l'IOMC (Interorganisation Program for the Sound Management of Chemicals).
Mort imminente: il y a mort imminente lorsqu'on s'attend à ce qu'un état moribond ou la mort interviennent avant le prochain moment d'observation prévu. Parmi les signes qui sont indicatifs de cet état chez les rongeurs, il y a les convulsions, la position latérale, la position couchée et les tremblements [pour de plus amples détails, voir (9)].
DL50 (dose orale létale 50 %): dose unique d'une substance d'essai, obtenue par calcul statistique, susceptible d'entraîner la mort de 50 % des animaux lorsqu'elle est administrée par voie orale. La valeur de la DL50 est exprimée en poids de la substance d'essai par unité de poids corporel de l'animal d'expérience (mg/kg).
Dose limite: désigne une dose qui est la limite supérieure pour l'essai (2 000 ou 5 000 mg/kg).
État moribond: l'état avant la mort ou l'incapacité à survivre, même si un traitement est donné [pour de plus amples détails, voir (9)].
Mort prévisible: présence de signes cliniques indiquant que la mort va intervenir à un moment déterminé dans le futur, avant la fin projetée de l'expérience, par exemple incapacité à atteindre l'eau ou la nourriture [pour plus de détails, voir (9)].
1.3. PRINCIPE DE L'ESSAI
Le principe de cet essai est que, avec un processus séquentiel et un nombre minimal d'animaux par étape, il est possible d'obtenir des informations sur la toxicité aiguë de la substance d'essai qui sont suffisantes aux fins de sa classification. Une dose déterminée de la substance est administrée par voie orale à un groupe d'animaux. La substance est testée selon une procédure séquentielle utilisant trois animaux de même sexe (généralement des femelles) à chaque étape. L'absence ou la manifestation de mortalité liée à la substance dans un groupe ayant reçu une dose à une étape donnée détermine l'étape suivante, c'est-à-dire:
|
—
|
administration de la même dose à trois animaux supplémentaires,
|
|
—
|
administration de la dose immédiatement supérieure ou immédiatement inférieure à trois animaux supplémentaires.
|
Des détails concernant le mode opératoire sont décrits à l'annexe 1. La méthode permet de se prononcer sur la classification de la substance d'essai dans une classe de toxicité délimitée par des valeurs préalablement fixées de DL50.
1.4. DESCRIPTION DE LA MÉTHODE
1.4.1. Choix de l'espèce animale
Le rat est l'espèce préférée mais d'autres espèces de rongeurs peuvent être utilisées. Normalement, on utilise des femelles (9). L'étude de la littérature sur les essais traditionnels de DL50 montre en effet qu'il y a peu de différence de sensibilité entre sexes et que, lorsqu'il y a des différences, les femelles sont en général légèrement plus sensibles (11). Si, toutefois, compte tenu des propriétés toxicologiques et toxicocinétiques de composés structurellement voisins, il apparaît que les mâles sont probablement plus sensibles, il convient d'employer des mâles. Dans ce cas, il faut fournir une justification adéquate.
On utilise de jeunes animaux adultes sains, issus de souches couramment utilisées en laboratoire. Les femelles doivent être nullipares et non gravides. Au début de l'essai, chaque animal doit être âgé de 8 à 12 semaines et son poids doit se situer dans un intervalle de ± 20 % du poids moyen des animaux précédemment exposés.
1.4.2. Conditions d'hébergement et d'alimentation
La température du local des animaux d'expérience doit être de 22 oC (± 3 oC). L'humidité relative doit atteindre au moins 30 % et rester de préférence inférieure à 70 %, mais en dehors des heures de nettoyage du local, on s'efforcera de maintenir le taux d'humidité autour de 50 à 60 %. On appliquera un éclairage artificiel alternant 12 heures de lumière et 12 heures d'obscurité. Pour l'alimentation des animaux, on peut utiliser la nourriture classique de laboratoire avec de l'eau potable à satiété. Les animaux peuvent être groupés par dose. Toutefois, le nombre d'animaux par cage ne doit pas faire obstacle à une observation précise de chaque animal.
1.4.3. Préparation des animaux
Les animaux sont choisis au hasard, marqués pour permettre une identification individuelle et gardés dans leurs cages pour les acclimater aux conditions de laboratoire pendant au moins cinq jours avant l'expérience.
1.4.4. Préparation des doses
En général, il convient d'administrer la substance d'essai à volume constant quelle que soit la dose testée, en faisant varier la concentration de la préparation. Lorsqu'un produit liquide ou un mélange font l'objet de l'essai, l'utilisation du produit non dilué, donc à concentration constante, peut être plus appropriée pour l'évaluation du risque de cette substance. Certaines autorités réglementaires l'exigent. Dans tous les cas, il ne faut pas dépasser le volume de dose maximal. Le volume maximal de liquide pouvant être administré en une seule fois dépend de la taille de l'animal d'essai. Chez les rongeurs, ce volume ne doit pas dépasser 1 ml/100 g de poids corporel, sauf dans le cas de solutions aqueuses pour lesquelles on peut utiliser 2 ml/100 g de poids corporel. Il est recommandé d'utiliser une solution aqueuse chaque fois que cela est possible, sinon on peut utiliser une solution dans de l'huile (par exemple de l'huile de mais) et éventuellement une solution dans d'autres véhicules. En ce qui concerne les véhicules non aqueux, leur toxicité doit être connue. Les doses doivent être préparées juste avant l'administration, sauf si la stabilité de la préparation pendant la durée de la période d'utilisation est connue et jugée acceptable.
1.5. MODE OPÉRATOIRE
1.5.1. Administration des doses
La substance d'essai est administrée en une seule dose en utilisant une sonde gastrique ou toute autre canule pour intubation appropriée. Lorsqu'il n'est pas possible d'administrer la dose en une seule fois, celle-ci peut être fractionnée sur une période n'excédant pas 24 heures.
Les animaux doivent être à jeun avant l'administration de la substance (on supprimera la nourriture, mais pas l'eau, pendant toute une nuit pour les rats et pendant 3 à 4 heures pour les souris). Après la période de jeûne, les animaux doivent être pesés; la substance d'essai leur est ensuite administrée. Après l'administration de la substance, les animaux peuvent être à nouveau privés de nourriture, pendant 3 à 4 heures pour les rats et pendant 1 à 2 heures pour les souris. Si la dose est administrée par fractions sur un certain laps de temps, il peut s'avérer nécessaire, en fonction de la durée du traitement, d'alimenter et de faire boire les animaux.
1.5.2. Nombre d'animaux et niveaux des doses
Trois animaux sont utilisés à chaque étape. Pour la dose initiale, on choisit un niveau parmi les quatre suivants: 5, 50, 300 et 2 000 mg/kg de poids corporel. Le niveau choisi est celui pour lequel on peut s'attendre à observer une mortalité chez les animaux traités. Les schémas de l'annexe 1 décrivent la marche à suivre pour chacune des doses initiales. En outre, l'annexe 4 propose une méthode de classification selon le système communautaire en attendant la mise en place du nouveau SGH.
Lorsque certaines informations donnent à penser qu'il est peu probable que la dose initiale la plus élevée (2 000 mg/kg de poids corporel) provoque une mortalité, il convient de procéder à un essai limite. En l'absence de telles informations sur la substance d'essai, la dose initiale qui est recommandée par souci de protection des animaux est 300 mg/kg de poids corporel.
L'intervalle de temps à respecter entre l'administration de chaque niveau de dose dépend du moment où se manifestent les effets toxiques, de leur durée et de leur gravité. L'administration de la dose suivante doit être retardée jusqu'à ce que l'on ait obtenu la certitude que les animaux précédemment soumis au traitement vont survivre.
Exceptionnellement, et uniquement lorsque cela est nécessaire pour satisfaire une exigence réglementaire particulière, l'utilisation d'une dose maximale prédéterminée supplémentaire de 5 000 mg/kg peut être envisagée (voir annexe 2). Par souci de protection des animaux, l'essai de substances en catégorie 5 du SGH (2 000 - 5 000 mg/kg) est déconseillé et ne doit être envisagé que lorsqu'il est très probable qu'il donnera des résultats qui présenteront un intérêt direct pour la protection de la santé des hommes et des animaux ou de l'environnement.
1.5.3. Essai limite
L'essai limite est utilisé principalement lorsque l'expérimentateur dispose d'informations indiquant que la substance d'essai n'est probablement pas toxique, c'est-à-dire que sa toxicité ne se manifeste qu'au-delà des doses limites réglementaires. Des informations concernant la toxicité de la substance d'essai peuvent être déduites des connaissances acquises sur des substances, produits ou mélanges similaires déjà testés, compte tenu de l'identité et du pourcentage des composants importants sur le plan toxicologique. Quand on ne dispose pas d'informations concernant la toxicité de la substance, ou lorsqu'on s'attend à une substance toxique, il faut effectuer l'essai principal.
Un essai limite à la dose de 2 000 mg/kg peut être effectué sur six animaux (trois animaux par étape). Exceptionnellement, un essai limite à la dose de 5 000 mg/kg peut être réalisé sur trois animaux (voir annexe 2). Si une mortalité liée à la substance est observée, il peut s'avérer nécessaire de poursuivre l'essai à la dose immédiatement inférieure.
1.6. OBSERVATIONS
Les animaux doivent être observés individuellement au moins une fois pendant les 30 premières minutes suivant l'administration de la substance et régulièrement pendant les premières 24 heures, avec une attention particulière au cours des 4 premières heures; l'observation doit ensuite être quotidienne, pendant 14 jours en tout, sauf dans le cas des animaux qui meurent en cours d'étude ou qui doivent être retirés de l'étude et euthanasiés pour leur épargner des souffrances excessives. Toutefois, la durée d'observation ne doit pas être fixée de manière rigide. Elle doit être fonction des réactions de toxicité, du moment où elles apparaissent et de la durée de la période de récupération. Elle peut donc être prolongée, si nécessaire. Les moments où apparaissent et disparaissent les signes de toxicité sont importants, particulièrement quand on constate un certain retard dans l'apparition de ces signes (12). Toutes les observations sont enregistrées de façon systématique, une fiche individuelle étant établie pour chaque animal.
D'autres observations peuvent s'avérer nécessaires lorsque les animaux continuent à manifester des signes de toxicité. Les observations doivent porter sur les modifications de la peau, des poils, des yeux et des muqueuses, ainsi que de l'appareil respiratoire, du système circulatoire, du système nerveux central et autonome, de l'activité somato-motrice et du comportement. L'attention portera en particulier sur l'observation des diverses manifestations de tremblement, convulsion, salivation, diarrhée, léthargie, sommeil et coma. Les principes et critères résumés dans le document d'orientation sur les effets sur l'homme doivent être pris en considération (9). Les animaux moribonds et les animaux souffrant manifestement ou présentant des signes graves de détresse doivent être euthanasiés. Qu'il s'agisse d'animaux euthanasiés pour raisons éthiques ou d'animaux retrouvés morts, le moment de la mort doit être enregistré de façon aussi précise que possible.
1.6.1. Poids corporel
Le poids de chaque animal doit être déterminé peu de temps avant l'administration de la substance d'essai, et au moins une fois par semaine ensuite. Les variations de poids doivent être calculées et enregistrées. À la fin de l'essai, les animaux survivants sont pesés puis euthanasiés.
1.6.2. Pathologie
Tous les animaux (y compris ceux qui sont morts au cours de l'essai ou ceux qui ont été retirés de l'étude pour des raisons éthiques) doivent être soumis à une autopsie à l'échelle macroscopique. Pour chaque animal, toutes les altérations pathologiques macroscopiques doivent être enregistrées. Chez les animaux qui survivent 24 heures ou plus à l'administration de la dose initiale, l'examen microscopique des organes présentant des signes évidents de pathologie doit également être envisagé, car il peut fournir des renseignements utiles.
2. RÉSULTATS
Les résultats obtenus pour chaque animal doivent être présentés. Toutes les données doivent être résumées dans un tableau indiquant, pour chaque groupe d'essai, le nombre d'animaux utilisés, le nombre d'animaux présentant des signes de toxicité, le nombre d'animaux retrouvés morts pendant l'essai ou sacrifiés pour des raisons éthiques, et, pour chaque animal, le moment de la mort, la description des effets toxiques et leur évolution dans le temps ainsi que leur réversibilité éventuelle et les résultats de l'autopsie.
3. RAPPORT
3.1. Rapport d'essai
Le rapport d'essai doit contenir, s'il y a lieu, les renseignements suivants:
|
|
Substance d'essai:
|
—
|
état physique, pureté et, s'il y a lieu, propriétés physico-chimiques (compris isomérisation),
|
|
—
|
données relatives à l'identification, y compris numéro CAS.
|
|
|
|
Véhicule (le cas échéant):
|
—
|
justification du choix du véhicule, s'il ne s'agit pas d'eau.
|
|
|
|
Animaux d'essai:
|
—
|
espèce/souche utilisée,
|
|
—
|
état microbiologique des animaux, s'il est connu,
|
|
—
|
nombre, âge et sexe des animaux (le cas échéant, justification du choix de mâles à la place de femelles),
|
|
—
|
origine, conditions d'hébergement, régime alimentaire, etc.
|
|
|
|
Conditions de l'essai:
|
—
|
détails sur la formulation de la substance d'essai, notamment état physique du produit administré,
|
|
—
|
détails sur le mode d'administration, notamment volume des doses et moment de l'administration,
|
|
—
|
détails sur la qualité de la nourriture et de l'eau (y compris le type de régime et sa provenance ainsi que celle de l'eau),
|
|
—
|
justification du choix de la dose initiale.
|
|
|
|
Résultats:
|
—
|
tableau des réactions aux différentes doses pour chaque animal (c'est-à-dire nombre d'animaux montrant des signes de toxicité, y compris mortalité; nature, gravité et durée des effets),
|
|
—
|
tableau des poids corporels et des variations de poids,
|
|
—
|
poids individuels des animaux le jour du traitement, et ensuite à intervalles d'une semaine, ainsi qu'au moment de la mort ou du sacrifice,
|
|
—
|
date et heure de la mort si celle-ci intervient avant le moment prévu du sacrifice,
|
|
—
|
pour chaque animal, moment d'apparition et évolution des signes de toxicité et réversibilité éventuelle,
|
|
—
|
pour chaque animal, résultats de l'autopsie et observations histopathologiques le cas échéant.
|
|
|
|
Discussion et interprétation des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
Roll, R., Höfer-Bosse, Th. And Kayser, D. (1986). New Perspectives in Acute Toxicity Testing of Chemicals. Toxicol. Lett., Suppl. 31, p. 86
|
|
(2)
|
Roll, R., Riebschläger, M., Mischke, U. and Kayser, D. (1989). Neue Wege zur Bestimmung der akuten Toxizität von Chemikalien. Bundesgesundheitsblatt 32, p. 336-341.
|
|
(3)
|
Diener, W., Sichha, L., Mischke, U., Kayser, D. and Schlede, E. (1994). The Biometric Evaluation of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 68, p. 559-610
|
|
(4)
|
Diener, W., Mischke, U., Kayser, D. and Schlede, E. (1995). The Biometric Evaluation of the OECD Modified Version of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 69, p. 729-734.
|
|
(5)
|
Diener, W., and Schlede, E. (1999). Acute Toxicity Class Methods: Alterations to LD/LC50 Tests. ALTEX 16, p. 129-134
|
|
(6)
|
Schlede, E., Mischke, U., Roll, R. and Kayser, D. (1992). A National Validation Study of the Acute-Toxic-Class Method — An Alternative to the LD50 Test. Arch. Toxicol. 66, p. 455-470.
|
|
(7)
|
Schlede, E., Mischke, U., Diener, W. and Kayser, D. (1994). The International Validation Study of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 69, p. 659-670.
|
|
(8)
|
OECD (2001). Guidance Document on Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment No 24. Paris.
|
|
(9)
|
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment No 19.
|
|
(10)
|
OECD (1998). Harmonized Integrated Hazard Classification System For Human Health And Environmental Effects Of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p. 11 [http://webnetl.oecd.org/oecd/pages/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-0,FF.html].
|
|
(11)
|
Lipnick, R.L., Cotruvo, J.A., Hill, R.N., Bruce, R.D., Stitzel, K.A., Walker, A.P., Chu, I., Goddard, M., Segal, L., Springer, J.A. and Myers, R.C. (1995). Comparison of the Up-and Down, Conventional LD50, and Fixed Dose Acute Toxicity Procedures. Fd. Chem. Toxicol 33, p. 223-231.
|
|
(12)
|
Chan, P.K. and Hayes, A.W. (1994). Chap. 16. Acute Toxicity and Eye Irritancy. Principles and Methods of Toxicology. Third Edition. A.W. Hayes, Editor. Raven Press, Ltd, New York, USA.
|
ANNEXE 1
MARCHE À SUIVRE POUR CHACUNE DES DOSES INITIALES
REMARQUES GÉNÉRALES
Les divers schémas d'essai présentés dans la présente annexe indiquent la marche à suivre pour chaque dose initiale.
|
—
|
Annexe 1 A: dose initiale de 5 mg/kg de poids corporel
|
|
—
|
Annexe 1 B: dose initiale de 50 mg/kg de poids corporel
|
|
—
|
Annexe 1 C: dose initiale de 300 mg/kg de poids corporel
|
|
—
|
Annexe 1 D: dose initiale de 2 000 mg/kg de poids corporel
|
En fonction du nombre d'animaux trouvés morts ou euthanasiés, la marche à suivre est indiquée par les flèches.
ANNEXE 1 A
MODE OPÉRATOIRE POUR UNE DOSE INITIALE DE 5 MG/KG DE POIDS CORPOREL
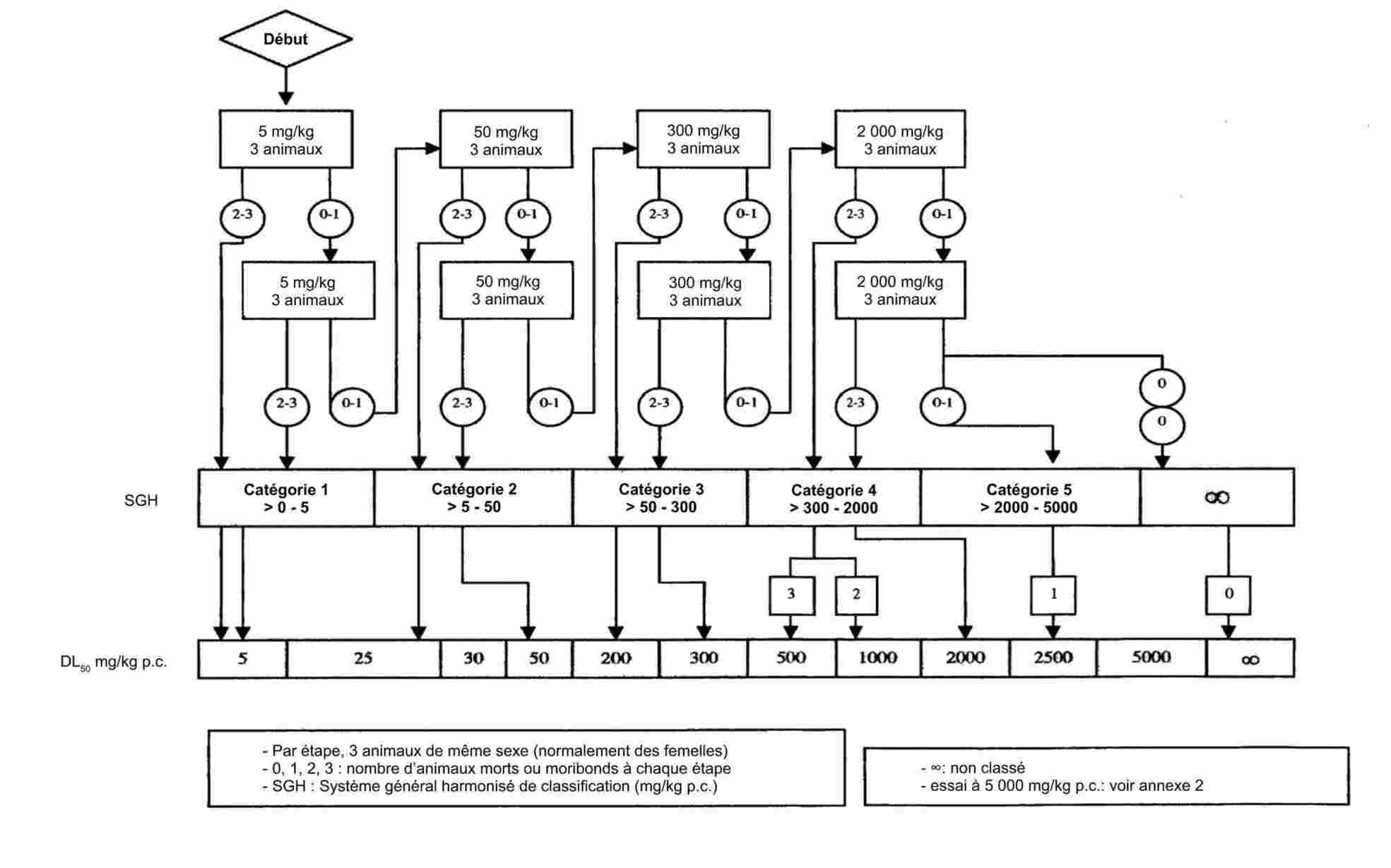
ANNEXE 1 B
MODE OPÉRATOIRE POUR UNE DOSE INITIALE DE 50 MG/KG DE POIDS CORPOREL

ANNEXE 1 C
MODE OPÉRATOIRE POUR UNE DOSE INITIALE DE 300 MG/KG DE POIDS CORPOREL

ANNEXE 1 D
MODE OPÉRATOIRE POUR UNE DOSE INITIALE DE 2 000 MG/KG DE POIDS CORPOREL
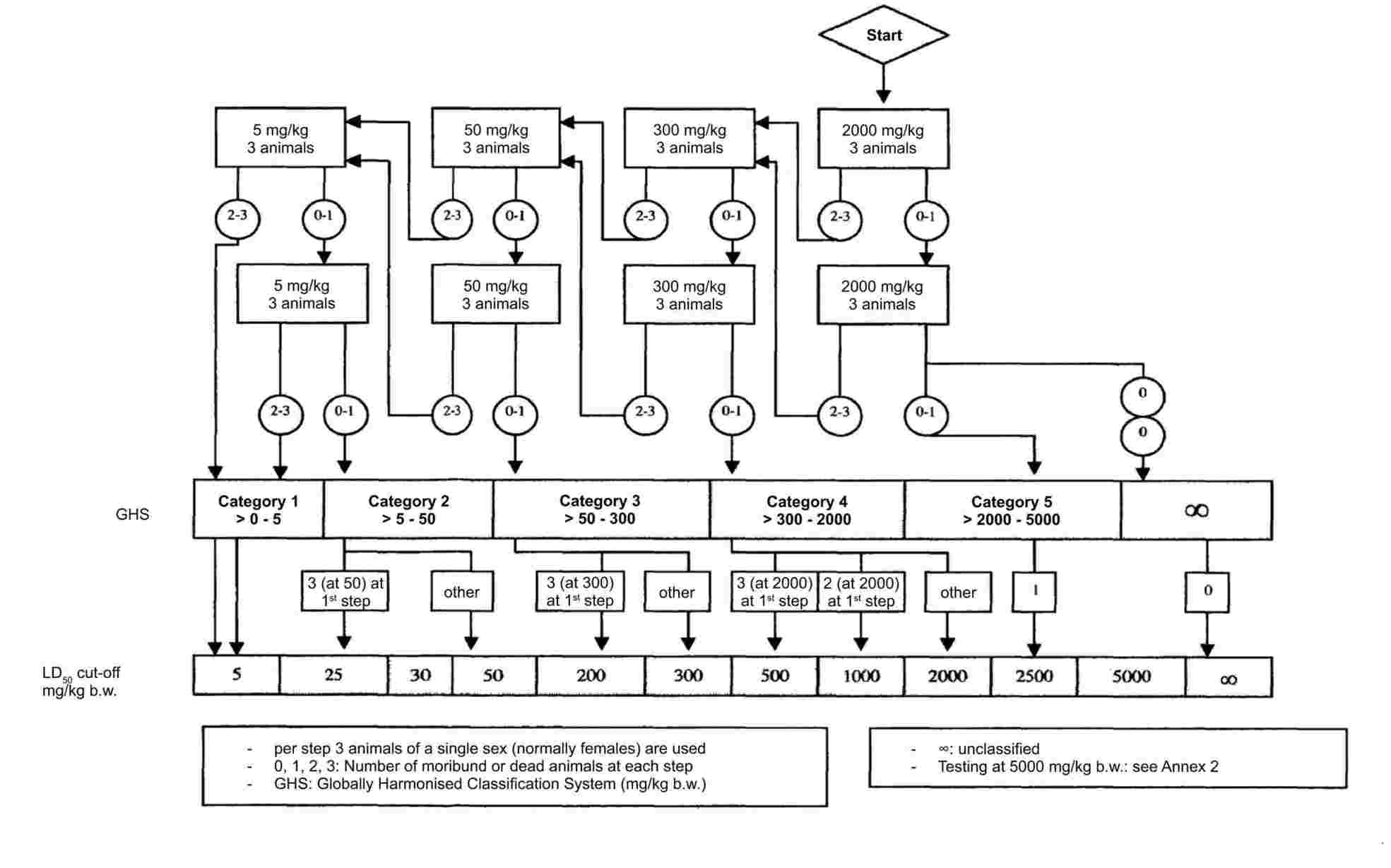
ANNEXE 2
CRITÈRES POUR LA CLASSIFICATION DE SUBSTANCES D'ESSAI DONT ON PRÉSUME QUE LA DL50 EST SUPÉRIEURE À 2 000 MG/KG, SANS RECOURIR À L'ESSAI
Les critères pour la catégorie de danger 5 sont destinés à permettre l'identification de substances qui ont une toxicité aiguë relativement faible mais qui, dans certaines conditions, peuvent se révéler dangereuses pour des populations vulnérables. Ces substances sont présumées avoir une DL50 orale ou cutanée comprise dans la plage 2 000 - 5 000 mg/kg ou correspondant à des doses équivalentes par d'autres voies. Les substances d'essai devraient être classées dans la catégorie SGH de danger 5, définie par 2 000 mg/kg < DL50 < 5 000 mg/kg, dans les cas suivants:
|
a)
|
lorsque, en fonction de la mortalité, l'un des schémas des annexes 1A à 1 D oriente la substance vers cette catégorie;
|
|
b)
|
lorsque l'on est déjà en possession de données fiables indiquant que la DL50 se situera dans la plage des valeurs de la catégorie 5; ou lorsque d'autres études sur des animaux ou l'observation d'effets toxiques chez l'homme suscitent de sérieuses inquiétudes pour la santé humaine;
|
|
c)
|
par extrapolation, évaluation ou mesure de données, lorsque la classification dans une catégorie de plus grand danger n'est pas justifiée, et
|
—
|
il existe des informations fiables faisant état d'effets toxiques significatifs chez l'homme, ou
|
|
—
|
une certaine mortalité est observée en testant par voie orale jusqu'aux valeurs de la catégorie 4, ou
|
|
—
|
lorsqu'un jugement d'expert confirme l'existence de signes cliniques significatifs de toxicité lors d'un essai mené jusqu'aux valeurs de la catégorie 4, hormis diarrhée, hérissement des poils ou aspect mal soigné, ou
|
|
—
|
quand un jugement d'expert confirme l'existence d'informations fiables indiquant la possibilité d'effets aigus significatifs d'après les résultats des autres études sur animaux.
|
|
ESSAIS À DOSES SUPÉRIEURES À 2 000 MG/KG
Par souci de protection des animaux, l'essai de substances à la dose de 5 000 mg/kg est déconseillé et ne devrait être envisagé que lorsqu'il est fort probable que les résultats d'un tel essai présenteront un intérêt direct pour la protection de la santé des hommes et des animaux (10). Aucun essai ne doit être effectué à des doses plus élevées.
Lorsqu'un essai à 5 000 mg/kg est nécessaire, une seule étape suffit (c'est-à-dire trois animaux). Si le premier animal traité meurt, l'essai se poursuit à la dose de 2 000 mg/kg comme indiqué dans les schémas de l'annexe 1. Si le premier animal traité survit, deux autres animaux sont traités. Si un seul des trois animaux meurt, la DL50 est présumée supérieure à 5 000 mg/kg. Si les deux animaux meurent, l'essai se poursuit à la dose de 2 000 mg/kg.
ANNEXE 3
MÉTHODE D'ESSAI B.1 tris Conseils pour une classification selon le système de l'UE en attendant la mise en place effective du système général harmonisé de classification (SGH) [voir référence (8)]


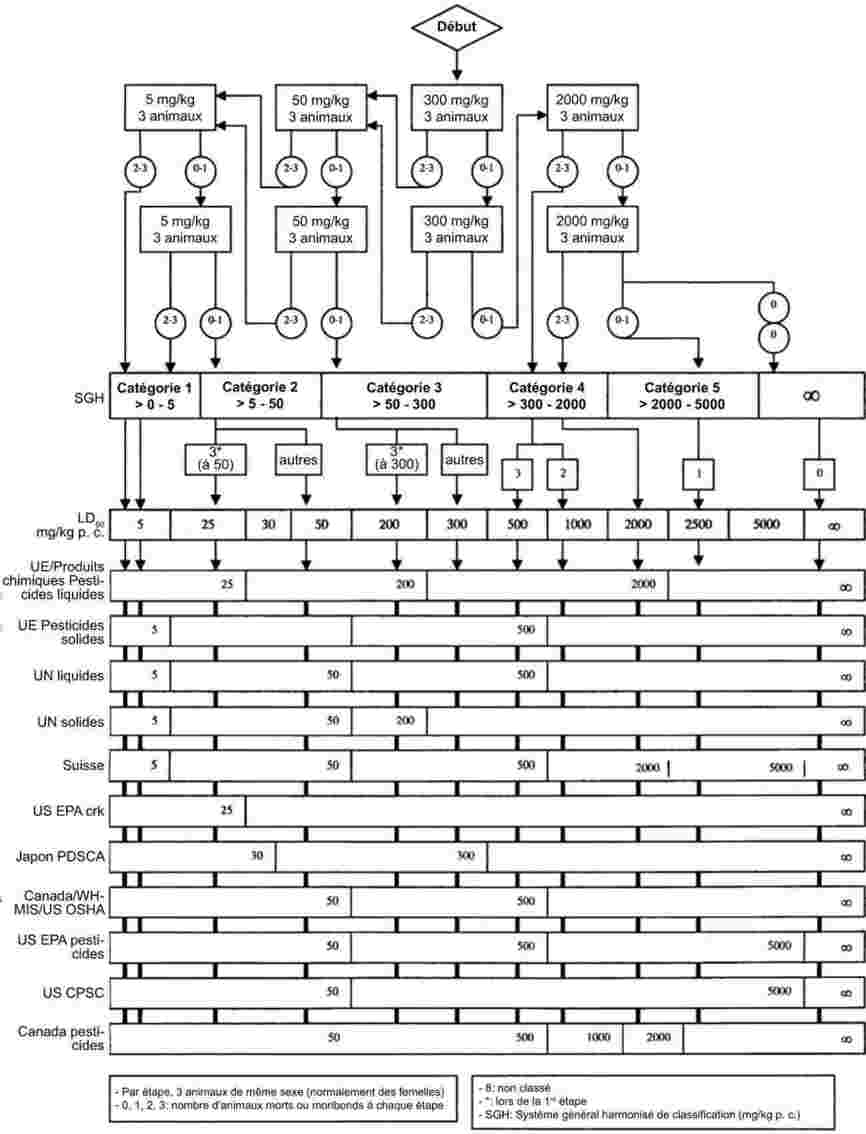

B.2. TOXICITÉ AIGUË (INHALATION)
1. MÉTHODE
1.1. INTRODUCTION
Il est utile de disposer d'informations préliminaires sur la distribution de la taille des particules, la pression de vapeur, le point de fusion, le point d'ébullition, le point d'éclair et l'explosivité (le cas échéant) de la substance.
Voir également l'introduction générale, partie B (point A).
1.2. DÉFINITIONS
Voir introduction générale, partie B (point B).
1.3. SUBSTANCES DE RÉFÉRENCE
Aucune.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Plusieurs lots d'animaux d'expérience sont exposés à des concentrations croissantes de la substance à tester pendant une période déterminée, une seule concentration étant utilisée par lot. On observe ensuite les effets et la mortalité dus à la substance. Les animaux qui meurent pendant l'essai sont autopsiés ainsi que ceux qui survivent à la fin de l'expérience.
Si des animaux montrent des signes d'inconfort et de douleur intenses et durables, il peut être nécessaire de les euthanasier; il n'y a pas lieu d'effectuer l'essai lorsque l'on sait que l'administration de la substance à étudier peut entraîner une détresse ou des douleurs intenses du fait des propriétés corrosives ou irritantes.
1.5. CRITÈRE DE QUALITÉ
Néant.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Préparation
Les animaux sont maintenus dans des conditions d'hébergement et d'alimentation propres à l'expérience au minimum pendant au moins les 5 jours qui la précèdent. Avant l'expérience, des animaux adultes jeunes et sains sont distribués selon les règles du hasard dans les différents lots d'expérience. Il n'est pas nécessaire de soumettre les animaux témoins à une exposition simulée, à moins que le dispositif d'exposition utilisé ne l'exige.
Il peut être nécessaire de microniser les substances solides à tester afin de les réduire en particules d'une taille appropriée.
On peut, au besoin, ajouter à la substance à tester un véhicule approprié en vue d'obtenir la concentration adéquate de celle-ci dans l'atmosphère; il faut alors prévoir un groupe témoin pour ce véhicule. Si un véhicule ou d'autres additifs sont utilisés pour faciliter l'administration, ils doivent être non toxiques et il est possible de s'appuyer pour cela sur des données bibliographiques appropriées.
1.6.2. Conditions de l'essai
1.6.2.1. Animaux d'expérience
Sauf contre-indication, le rat est l'espèce préférée. Il faut utiliser des souches courantes d'animaux de laboratoire. Pour chaque sexe, au début de l'essai, l'intervalle de variation du poids des animaux utilisés ne doit pas excéder ± 20 % de la valeur moyenne requise.
1.6.2.2. Nombre et sexe
Dix rongeurs au moins (cinq femelles et cinq mâles) sont utilisés pour chaque niveau de concentration. Les femelles doivent être nullipares et non gravides.
Note: Il faut essayer d'utiliser un nombre plus restreint d'animaux pour les tests de toxicité aiguë sur des animaux appartenant à un ordre plus élevé que celui des rongeurs. Les concentrations doivent être soigneusement choisies, en multipliant les efforts visant à ne pas dépasser des doses modérément toxiques. Il faut éviter dans ce type d'essais d'administrer des doses létales de la substance à tester.
1.6.2.3. Concentrations d'exposition
Les concentrations doivent être en nombre suffisant, au moins trois, et espacées correctement pour produire des lots présentant une gamme d'effets toxiques et de taux de mortalité. Les données doivent être suffisantes pour permettre de tracer une courbe doses/réponses et, si possible, permettre une détermination acceptable de la CL50.
1.6.2.4. Essai de limite
Si une exposition de 5 mâles et de 5 femelles à une concentration de 20 milligrammes par litre d'un gaz ou de 5 milligrammes par litre d'un aérosol ou de particules pendant 4 heures (ou lorsque cela n'est pas possible en raison des propriétés chimiques ou physiques, voire explosives, de la substance d'essai, une exposition à la concentration maximale possible) ne cause la mort d'aucun animal en 14 jours, on peut juger inutile de poursuivre l'expérience.
1.6.2.5. Temps d'exposition
La durée de l'exposition doit être de quatre heures.
1.6.2.6. Dispositif expérimental
Les animaux doivent être exposés à la substance au moyen d'un dispositif d'exposition dynamique capable de maintenir un courant d'air permettant au moins 12 renouvellements de l'atmosphère de l'enceinte d'exposition par heure, et assurant une teneur suffisante en oxygène ainsi qu'une répartition uniforme du produit à tester dans l'atmosphère de l'enceinte d'exposition. Si l'on utilise une chambre d'exposition, celle-ci doit être conçue de manière à éviter autant que possible l'entassement des animaux et à optimiser l'exposition par inhalation à la substance d'essai. En règle générale, pour assurer la stabilité de la composition de l'atmosphère d'une chambre, le volume total des animaux d'expérience ne doit pas dépasser 5 % du volume de la chambre d'essai. On peut aussi avoir recours à un système d'exposition soit oro-nasal, soit de la tête seule, soit du corps entier en chambre individuelle; les deux premiers types d'exposition présentent l'avantage de limiter la pénétration de la substance d'essai par d'autres voies.
1.6.2.7. Période d'observation
La période d'observation doit s'étendre au moins sur 14 jours. Cependant, cette durée ne doit pas être limitée de façon rigide. Elle doit être déterminée en fonction des réactions de toxicité, de leur vitesse d'apparition et de la durée de la période de récupération; elle peut donc être prolongée si nécessaire. Le moment d'apparition ou de disparition des symptômes de toxicité ainsi que le moment de la mort sont importants, surtout en cas de mortalité retardée.
1.6.3. Mode opératoire
Peu de temps avant l'exposition, les animaux sont pesés, puis exposés à la concentration d'essai dans l'appareillage décrit pendant une durée de quatre heures après stabilisation de la concentration dans la chambre. La stabilisation doit être rapide. La température à laquelle s'effectue l'essai doit être maintenue à 22 oC ± 3 oC. L'idéal est de maintenir l'humidité relative entre 30 et 70 %, mais, dans certains cas (par exemple certains essais d'aérosols), cela peut être impossible. Le maintien d'une pression légèrement négative à l'intérieur de la chambre (≥ 5 mm d'eau) empêchera la substance d'essai de s'échapper dans le laboratoire. La nourriture et l'eau doivent être retirées pendant l'exposition. Il convient d'utiliser un dispositif permettant de produire et de contrôler l'atmosphère d'essai. Le système doit permettre de créer des conditions d'exposition stables aussi rapidement que possible. La chambre doit être conçue et fonctionner de telle façon qu'il y soit maintenue une distribution homogène de la substance dans l'atmosphère.
Il convient de mesurer ou de surveiller:
|
a)
|
le débit d'air (en permanence);
|
|
b)
|
la concentration réelle de la substance d'essai. Elle doit être mesurée dans la zone de respiration au moins trois fois au cours de l'exposition (certaines atmosphères, comme les aérosols à forte concentration, peuvent nécessiter un contrôle plus fréquent). Pendant la période d'exposition, la concentration ne doit pas varier de plus de ± 15 % par rapport à la valeur moyenne. Toutefois, dans le cas de certains aérosols, ce degré de précision peut ne pas être obtenu et un écart plus grand est alors acceptable. En ce qui concerne les aérosols, la taille des particules doit être analysée aussi souvent que nécessaire (au moins une fois par groupe d'exposition);
|
|
c)
|
la température et l'humidité, en permanence si possible.
|
Les observations ont lieu pendant et après l'exposition et sont enregistrées systématiquement; une fiche individuelle doit être établie pour chaque animal. Les observations doivent être fréquentes le premier jour. Un examen clinique attentif doit être pratiqué au moins une fois par jour ouvrable, d'autres observations doivent être faites quotidiennement, en agissant de façon à réduire le nombre d'animaux perdus pour l'étude, par exemple grâce à l'autopsie ou à la conservation au froid des animaux trouvés morts ainsi qu'en isolant et en sacrifiant des animaux faibles ou moribonds.
Les observations doivent concerner les modifications de la peau et des poils, des yeux, dés muqueuses, de l'appareil respiratoire, du système circulatoire ainsi que des systèmes nerveux autonome et central, de l'activité somato-motrice et du comportement. Une attention particulière sera apportée à la respiration, aux tremblements, convulsions, salivation, diarrhées, léthargie, sommeil et coma. Le moment de la mort doit être noté avec autant de précision que possible. Le poids de chaque animal doit être déterminé chaque semaine après l'exposition ainsi qu'au moment de la mort.
Les animaux qui meurent pendant l'expérience et ceux qui survivent à la fin de l'expérience sont autopsiés en recherchant en particulier toutes les modifications des voies respiratoires supérieures et inférieures. Toutes les modifications doivent être consignées. Si nécessaire, des tissus doivent être prélevés pour un examen histopathologique.
2. DONNÉES
Les données doivent être récapitulées dans un tableau indiquant, pour chaque lot d'expérience, le nombre d'animaux au début de l'essai, le moment de la mort de chaque animal, le nombre d'animaux présentant d'autres symptômes de toxicité, la description des effets toxiques et les résultats de l'autopsie. Le poids de chaque animal doit être déterminé et enregistré lorsque la survie dépasse une journée. Les animaux qui sont euthanasiés à cause de l'inconfort et des souffrances dus à la substance sont enregistrés en tant que morts dues à la substance. La CL50 doit être déterminée selon une méthode reconnue. L'évaluation des données doit inclure la relation, si elle existe, entre l'exposition des animaux à la substance à tester et l'apparition ainsi que la gravité de toutes les anomalies, y compris les anomalies du comportement et les anomalies cliniques, les lésions macroscopiques, les changements de poids corporel, la mortalité et tout autre effet toxique éventuel.
3. RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
espèce, souche, origine, conditions ambiantes, régime alimentaire, etc.,
|
|
—
|
conditions de l'essai: description de l'appareillage d'exposition, y compris conception, type, dimensions, source d'air, système de génération produisant l'aérosol, méthode de conditionnement d'air et, le cas échéant, stabulation des animaux en chambre d'essai. Le dispositif de mesure de la température, de l'humidité, de la concentration des aérosols et de la distribution de la taille des particules doit être décrit.
|
Données relatives à l'exposition:
Elles doivent être présentées sous forme d'un tableau indiquant les valeurs moyennes ainsi qu'une mesure de la variabilité (par exemple écart type); elles devront, si possible, comprendre:
|
a)
|
débit d'air dans le dispositif d'inhalation;
|
|
b)
|
température et humidité de l'air;
|
|
c)
|
concentrations nominales (quantité totale de substance d'essai introduite dans le dispositif d'inhalation, divisée par le volume d'air);
|
|
d)
|
le cas échéant, nature du véhicule;
|
|
e)
|
concentrations réelles dans la zone de respiration;
|
|
f)
|
diamètre aérodynamique médian en masse (DAMM) et écart type géométrique (ETG);
|
|
g)
|
durée de stabilisation;
|
|
h)
|
durée d'exposition;
|
—
|
tableau de données de réponses par sexe et par dose (à savoir nombre d'animaux morts ou sacrifiés au cours de l'essai, nombre d'animaux présentant des symptômes d'intoxication, nombre d'animaux exposés),
|
|
—
|
moment de la mort pendant ou après exposition, raisons et critères pour lesquels les animaux ont été euthanasiés,
|
|
—
|
toutes les observations,
|
|
—
|
valeur de la CL50 pour les animaux de chaque sexe, déterminée à la fin de la période d'observation (en précisant la méthode de calcul),
|
|
—
|
intervalle de confiance de 95 % pour la CL50 (lorsque le calcul est possible),
|
|
—
|
courbe dose/mortalité et pente de cette courbe (lorsque la méthode de calcul le permet),
|
|
—
|
tous les résultats d'examens histopathologiques,
|
|
—
|
discussion des résultats (en tenant compte, en particulier, de l'effet que peuvent avoir sur la valeur calculée de la CL50 les animaux euthanasiés au cours de l'essai),
|
|
—
|
interprétation des résultats.
|
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B (point D).
4. RÉFÉRENCES
Voir introduction générale, partie B (point E).
B.3. TOXICITÉ AIGUË (CUTANÉE)
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B (point A).
1.2. DÉFINITIONS
Voir introduction générale, partie B (point B).
1.3. SUBSTANCES DE RÉFÉRENCE
Aucune.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Des doses croissantes de la substance à tester sont appliquées sur la peau à plusieurs lots d'animaux d'expérience, une seule dose étant utilisée par lot. On observe ensuite les effets et la mortalité. Les animaux qui meurent pendant l'essai sont autopsiés ainsi que ceux qui survivent à la fin de l'expérience.
Si des animaux montrent des signes de détresse et de douleur intenses et durables, il peut être nécessaire de les euthanasier; il n'y a pas lieu d'effectuer l'essai lorsque l'on sait que l'administration de la substance à étudier entraîne une détresse ou des douleurs intenses du fait de ses propriétés corrosives ou irritantes.
1.5. CRITÈRE DE QUALITÉ
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Préparation
Les animaux sont maintenus dans des conditions d'hébergement et d'alimentation propres à l'expérience au minimum pendant les 5 jours qui la précèdent. Avant l'expérience, des animaux adultes jeunes et sains sont distribués selon les règles du hasard dans les différents lots d'expérience. Vingt-quatre heures environ avant l'épreuve, on tond ou l'on rase les poils de la région dorsale du tronc des animaux en évitant toute lésion de la peau susceptible de modifier sa perméabilité. La surface à préparer pour l'application de la substance ne doit pas être inférieure à 10 % de la surface corporelle. Lorsque le test concerne des substances solides qui, le cas échéant, peuvent être pulvérisées, la substance à tester doit être suffisamment humectée au moyen d'eau ou, au besoin, d'un véhicule approprié, de manière à assurer un bon contact avec la peau. Si l'on utilise un véhicule, son influence sur la pénétration de la substance dans la peau doit être prise en considération. Les substances liquides sont généralement appliquées non diluées.
1.6.2. Conditions de l'essai
1.6.2.1. Animaux d'expérience
On peut utiliser des rats ou des lapins adultes. On peut également utiliser d'autres espèces, mais il faut dans ce cas en justifier l'utilisation. Il faut utiliser des souches courantes d'animaux de laboratoire. Pour chaque sexe, au début de l'essai, l'intervalle de variation du poids des animaux utilisés ne doit pas excéder ± 20 % de la valeur moyenne requise.
1.6.2.2. Nombre et sexe
Cinq animaux au moins sont utilisés pour chaque dose. Ils doivent être de même sexe. Si l'on utilise des femelles, elles doivent être nullipares et non gravides. Si l'on dispose d'informations qui montrent qu'un sexe est nettement plus sensible, les essais seront pratiqués sur des animaux de ce sexe.
Note: Il faut essayer d'utiliser un nombre plus restreint d'animaux lorsque les tests de toxicité aiguë sont effectués sur des animaux appartenant à un ordre plus élevé que celui des rongeurs. Les doses doivent être soigneusement choisies, en s'efforçant au maximum de ne pas dépasser des doses modérément toxiques. Il faut éviter dans ce type d'essais d'administrer des doses létales de la substance à tester.
1.6.2.3. Doses
Les doses doivent être en nombre suffisant, au moins trois, et espacées de manière à produire des lots présentant une gamme d'effets toxiques et de taux de mortalité. Tout effet irritant ou corrosif doit être pris en considération lors du choix des doses. Les données doivent être suffisantes pour permettre de tracer une courbe doses/réponses et, si possible, permettre une détermination acceptable de la DL50.
1.6.2.4. Essai de limite
Un essai de limite peut être effectué en administrant une dose au moins égale à 2 000 mg/kg de poids corporel à un lot de 5 mâles et 5 femelles, en suivant le mode opératoire décrit ci-après. Si l'on observe une mortalité due à la substance, il peut être nécessaire de réaliser une étude complète.
1.6.2.5. Période d'observation
La période d'observation doit s'étendre au moins sur 14 jours. Cependant, sa durée ne doit pas être limitée de façon rigide. Elle doit être déterminée en fonction des réactions de toxicité, de leur vitesse d'apparition et de la durée de la période de récupération; elle peut donc être prolongée en cas de besoin. Le moment où les signes de toxicité apparaissent et disparaissent, leur durée ainsi que le moment de la mort sont importants, surtout en cas de tendance à une mortalité retardée.
1.6.3. Mode opératoire
Les animaux doivent être placés dans des cages individuelles. La substance d'essai doit être appliquée uniformément sur une surface à peu près égale à 10 % de la surface totale du corps. Dans le cas de substances hautement toxiques, la surface traitée peut être moindre, mais la substance d'essai doit être appliquée de manière à former un film aussi mince et uniforme que possible.
Les substances d'essai doivent être maintenues en contact avec la peau au moyen d'un pansement de gaze poreux et d'un sparadrap non irritant pendant une période de 24 heures. La partie traitée doit, en outre, être convenablement couverte, de manière à maintenir en place le pansement de gaze et la substance d'essai et à éviter que les animaux puissent ingérer cette dernière. On peut utiliser des appareils de contention pour empêcher les animaux d'ingérer la substance d'essai mais une immobilisation complète n'est pas recommandée.
À la fin de la période d'application, la substance d'essai résiduelle doit être éliminée, si possible, avec de l'eau, ou au moyen d'un autre procédé de nettoyage de la peau.
Les observations doivent être consignées systématiquement au fur et à mesure qu'elles sont effectuées, en établissant une fiche individuelle pour chaque animal. Les animaux doivent être observés fréquemment le premier jour. Un examen clinique attentif doit être pratiqué au moins une fois par jour ouvrable, d'autres observations doivent être faites quotidiennement, en prenant des mesures de façon à réduire le nombre d'animaux perdus pour l'étude, par exemple autopsie ou conservation au froid des animaux trouvés morts, ainsi qu'en isolant et sacrifiant les animaux faibles ou moribonds.
Les observations doivent concerner les modifications des poils, de la peau traitée, des yeux, des muqueuses ainsi que de l'appareil respiratoire, du système circulatoire ainsi que des systèmes nerveux autonome et central, et enfin de l'activité somato-motrice et du comportement. Une attention particulière sera apportée aux tremblements, convulsions, salivation, diarrhées, léthargie, sommeil et coma. Le moment de la mort doit être enregistré avec autant de précision que possible. Les animaux qui meurent pendant l'expérience et ceux qui survivent à la fin de l'expérience sont autopsiés. Toutes les modifications pathologiques macroscopiques doivent être enregistrées. Si nécessaire, des tissus doivent être prélevés pour un examen histopathologique.
Estimation de la toxicité pour l'autre sexe
Après avoir mené à terme l'étude portant sur des animaux appartenant à un sexe, la substance est administrée au moins à un lot de 5 animaux de l'autre sexe, afin de déterminer si les animaux de ce sexe ne sont pas nettement plus sensibles à la substance à tester. L'utilisation d'un plus petit nombre d'animaux peut être justifiée dans des circonstances particulières. Si l'on dispose d'informations appropriées qui montrent que les animaux du sexe testé sont considérablement plus sensibles, on peut se dispenser d'effectuer des essais sur les animaux de l'autre sexe.
2. DONNÉES
Les données doivent être récapitulées sous forme de tableaux indiquant, pour chaque lot d'expérience, le nombre d'animaux au début de l'essai, le moment de la mort de chaque animal, le nombre d'animaux présentant d'autres signes d'intoxication, la description des effets toxiques et les résultats de l'autopsie. Le poids de chaque animal doit être déterminé et noté peu de temps avant l'application de la substance d'essai, puis une fois par semaine et au moment de la mort; les modifications du poids doivent être calculées et enregistrées lorsque la survie dépasse une journée. Les animaux qui sont euthanasiés à cause des souffrances et de l'inconfort dus à la substance sont enregistrés comme s'ils étaient morts du fait du composé étudié. La DL50 doit être déterminée par application d'une méthode reconnue.
L'évaluation des données doit inclure une évaluation de la relation, si elle existe, entre l'exposition des animaux à la substance à tester et l'apparition ainsi que la sévérité de toutes les anomalies, y compris les anomalies de comportement et les anomalies cliniques, les lésions macroscopiques, les modifications de poids corporel, la mortalité et tout autre effet toxique éventuel.
3. RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
espèce, souche, origine, conditions ambiantes, régime alimentaire, etc.,
|
|
—
|
conditions expérimentales (y compris le procédé de nettoyage de la peau et le type de pansement: occlusif ou non),
|
|
—
|
doses (avec indication du véhicule le cas échéant, et des concentrations),
|
|
—
|
sexe des animaux sur lesquels l'essai a été réalisé,
|
|
—
|
tableau de données de réponses par sexe et par dose (à savoir nombre d'animaux morts ou sacrifiés au cours de l'essai, nombre d'animaux présentant des symptômes d'intoxication, nombre d'animaux exposés),
|
|
—
|
moment de la mort après administration, raisons et critères justifiant l'euthanasie des animaux,
|
|
—
|
toutes les observations,
|
|
—
|
valeur de la DL50 pour le sexe soumis à une étude complète, déterminée à 14 jours (en précisant la méthode de calcul),
|
|
—
|
intervalle de confiance de 95 % pour la DL50, lorsque le calcul est possible,
|
|
—
|
courbe dose/mortalité et pente de cette courbe (lorsque la méthode de calcul le permet),
|
|
—
|
toutes les constatations histopathologiques,
|
|
—
|
résultats de tout essai réalisé sur l'autre sexe,
|
|
—
|
discussion des résultats (en tenant compte, en particulier, de l'effet que peuvent avoir sur la valeur de la DL50 calculée les animaux euthanasiés au cours de l'essai),
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B (point D).
4. RÉFÉRENCES
Voir introduction générale, partie B (point E).
B.4. TOXICITÉ AIGUË: IRRITATION/CORROSION CUTANÉE
1. MÉTHODE
Cette méthode est équivalente à la ligne directrice TG 404 (2002) de l'OCDE.
1.1. INTRODUCTION
La mise à jour de cette méthode a porté plus particulièrement sur les possibilités d'améliorer le traitement des animaux de laboratoire et sur l'évaluation de toutes les informations existantes se rapportant aux substances d'essai en vue d'éviter les tests inutiles sur animaux. La présente méthode recommande d'analyser la valeur probante des résultats pertinents déjà disponibles avant de procéder à l'essai in vivo de corrosion/irritation décrit ci-après. Les données manquantes pourront être comblées par des essais séquentiels (1). La démarche expérimentale, décrite en annexe, inclut la réalisation d'essais in vitro validés et acceptés. Il est également recommandé d'opter, le cas échéant, pour une application successive plutôt que simultanée des trois timbres sur l'animal dans l'essai in vivo initial.
Pour assurer à la fois la fiabilité des résultats scientifiques et le bien-être animal, on ne procédera pas aux essais in vivo tant que la valeur probante de toutes les données relatives au caractère éventuellement corrosif ou irritant pour la peau de la substance n'aura pas été analysée. Ces données comprendront les résultats d'études menées sur des humains et/ou des animaux, des données relatives à l'effet corrosif ou irritant d'une ou plusieurs substances structurellement proches de la substance d'essai ou de mélanges de ces substances, des données démontrant la forte acidité ou alcalinité de la substance (2) (3), et des résultats d'essais in vitro ou ex vivo validés et acceptés (4) (5) (5a). Cette analyse devrait réduire la nécessité de tester in vivo l'effet corrosif ou irritant sur la peau des substances pour lesquelles des études antérieures ont déjà livré suffisamment d'informations quant à ces deux aspects.
La démarche expérimentale préconisée, de type séquentiel, comporte des essais de corrosion et d'irritation in vitro ou ex vivo validés et acceptés, et est décrite dans l'annexe de la présente méthode. Cette démarche, élaborée au cours d'un atelier de l'OCDE (6) et recommandée à l'unanimité par les participants à cet atelier, a été adoptée à titre de recommandation dans le cadre du Globally Harmonized System for the Classification of Chemical Substances (GHS) (système de classification des substances chimiques harmonisé à l'échelon mondial) (7). Bien que cette stratégie séquentielle ne fasse pas partie intégrante de la méthode d'essai B.4, elle constitue cependant la procédure recommandée avant d'entreprendre les essais in vivo. S'agissant de nouvelles substances, on préconise de suivre une démarche expérimentale par étapes pour obtenir des résultats scientifiques fiables sur l'effet corrosif ou irritant de la substance. En ce qui concerne les substances existantes dont les données sur l'effet corrosif ou irritant sont insuffisantes, on suppléera à ces dernières en appliquant cette stratégie. Le choix d'une autre démarche expérimentale ou la décision de ne pas procéder par étapes doivent être justifiés.
Si l'analyse de la valeur probante des résultats ne permet pas de déterminer l'effet corrosif ou irritant, on envisagera un essai in vivo compatible avec la démarche séquentielle (voir annexe).
1.2. DÉFINITIONS
L'irritation cutanée désigne l'apparition de lésions cutanées réversibles consécutives à l'application d'une substance d'essai durant une période de quatre heures au maximum.
La corrosion cutanée désigne la survenue de lésions cutanées irréversibles, et plus précisément d'une nécrose visible à travers l'épiderme et dans le derme, à la suite de l'application d'une substance d'essai durant une période de quatre heures au maximum. La corrosion cutanée se manifeste par des ulcères, des saignements, des croûtes saignantes et, au terme de la période d'observation de 14 jours, par une décoloration due au pâlissement de la peau, des zones d'alopécie totale et des escarres. Un examen histopathologique sera envisagé en cas de lésions douteuses.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
Une seule dose de la substance d'essai est appliquée sur la peau de l'animal choisi pour l'expérience, les zones non traitées de la peau de l'animal servant de témoin. L'expérimentateur observe et note selon une échelle de valeurs le degré d'irritation ou de corrosion à intervalles déterminés, et le décrit de façon plus détaillée afin de fournir une évaluation complète des effets. La durée de l'étude doit être suffisante pour permettre d'évaluer la réversibilité des effets observés.
Les animaux qui manifestent des signes persistants de détresse et/ou de douleur aiguës à n'importe quel stade de l'essai doivent être euthanasiés, et ces symptômes seront pris en compte dans l'évaluation de la substance. Les critères régissant la décision d'euthanasier les animaux moribonds et souffrant fortement sont définis dans un document cité en référence (8).
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparation de l'essai in vivo
1.4.1.1. Sélection de l'espèce animale
On choisira de préférence de jeunes adultes sains parmi les lapins albinos. L'utilisation d'une autre espèce sera justifiée, le cas échéant.
1.4.1.2. Préparation des animaux
Environ 24 heures avant l'essai, la région dorsale du tronc des animaux sera tondue à ras. On prendra soin de ne pas égratigner leur peau et seuls des animaux présentant une peau saine et intacte seront utilisés.
La fourrure de certaines souches de lapins est plus touffue par endroits et ce phénomène est plus marqué à certaines périodes de l'année. Ces plages à forte pilosité ne doivent pas recevoir la substance d'essai.
1.4.1.3. Conditions d'hébergement et d'alimentation
Les animaux sont placés dans des cages individuelles. La température du local expérimental est réglée à 20 oC (± 3 oC) pour les lapins. L'humidité relative doit atteindre au moins 30 % et rester de préférence inférieure à 70 %, mais en dehors des heures de nettoyage du local, on s'efforcera de maintenir le taux d'humidité autour de 50 à 60 %. On appliquera un éclairage artificiel, alternant 12 heures de lumière et 12 heures d'obscurité. Les lapins seront nourris avec un mélange classique pour animaux de laboratoire et boiront de l'eau potable à volonté.
1.4.2. Mode opératoire
1.4.2.1. Application de la substance d'essai
La substance d'essai est appliquée sur une petite zone (environ 6 cm2) de la peau et recouverte par une compresse de gaze, maintenue en place à l'aide d'un sparadrap non irritant. Si l'application directe est impossible (dans le cas de liquides ou de certaines pâtes, par exemple), la substance d'essai est d'abord appliquée sur la compresse de gaze, laquelle est ensuite placée sur la peau. La compresse doit être maintenue en contact souple avec la peau à l'aide d'un pansement semi-occlusif durant la période d'exposition. Si la substance d'essai est déposée sur la compresse, celle-ci doit être fixée sur la peau de façon que la substance y soit répartie uniformément et entre bien en contact avec celle-ci. On fera en sorte que l'animal n'ait pas accès à la compresse et ne puisse ingérer ou inhaler la substance d'essai.
Les substances liquides sont généralement testées à l'état non dilué. Si la substance d'essai est solide (elle peut être pulvérisée si nécessaire), il y a lieu de l'humidifier avec la plus petite quantité d'eau (ou au besoin avec un autre véhicule approprié) nécessaire à assurer un bon contact avec la peau. Lorsqu'on utilise un véhicule autre que l'eau, l'influence éventuelle du véhicule sur l'irritation de la peau par la substance d'essai doit être minimale.
À la fin de la période d'exposition, qui dure normalement 4 heures, on enlève ce qui peut l'être de la substance d'essai restante, avec de l'eau ou un solvant approprié sans interférer avec la réaction ni altérer l'intégrité de l'épiderme.
1.4.2.2. Dose
Une dose de 0,5 ml de liquide ou de 0,5 g de solide ou de pâte est appliquée sur la plage à tester.
1.4.2.3. Essai initial (essai d'irritation/corrosion cutanée in vivo sur un seul animal)
Il est fortement recommandé de commencer par pratiquer l'essai in vivo sur un seul animal, surtout lorsqu'on pense que la substance risque d'être corrosive. Cette précaution obéit à la démarche expérimentale séquentielle (voir annexe 1).
Dès lors qu'une substance est jugée corrosive d'après l'analyse de la valeur probante des résultats, tout essai sur animal s'avère superflu. Pour la plupart des substances risquant d'être corrosives, il n'est généralement pas nécessaire de procéder à un essai in vivo. Toutefois, si l'on estime que les données disponibles ne sont pas assez convaincantes, on peut réaliser un essai limité sur un animal en suivant la procédure décrite ci-après. Jusqu'à trois timbres d'essai sont appliqués successivement sur l'animal. Le premier timbre est enlevé après trois minutes. Si aucune réaction cutanée grave n'est constatée, un deuxième timbre est appliqué et retiré après une heure. Si les observations effectuées à ce stade indiquent que l'exposition peut être étendue à quatre heures sans que cela fasse trop souffrir l'animal, l'expérimentateur appliquera un troisième timbre durant quatre heures et attribuera une cote à la réaction.
Si un effet corrosif est détecté à l'issue d'une des trois expositions séquentielles, l'essai s'achève immédiatement. Si aucun effet corrosif n'est relevé après l'enlèvement du troisième timbre, l'animal est gardé en observation durant 14 jours, à moins qu'un effet corrosif se déclare avant.
Dans les cas où l'on s'attend à ce que la substance d'essai soit peut-être irritante, mais pas corrosive, un seul timbre sera appliqué sur un animal durant quatre heures.
1.4.2.4. Essai confirmatoire (essai d'irritation cutanée in vivo sur des animaux supplémentaires)
Si l'essai initial ne révèle aucun effet corrosif, il convient de confirmer la réaction irritante ou négative sur deux animaux supplémentaires, traités chacun avec un timbre maintenu durant quatre heures. Si l'essai initial produit un effet irritant, l'essai confirmatoire peut être conduit en mode séquentiel ou par l'exposition simultanée de deux animaux supplémentaires. Au cas exceptionnel où l'essai initial ne serait pas pratiqué, deux ou trois animaux peuvent être traités au moyen d'un seul timbre appliqué durant quatre heures. Si l'on utilise deux animaux et qu'ils expriment la même réaction, il n'est pas nécessaire de poursuivre l'essai. Dans le cas contraire, le troisième animal est également testé. L'utilisation d'animaux supplémentaires pourra être requise si les réactions sont équivoques.
1.4.2.5. Période d'observation
La durée de la période d'observation devrait être suffisante pour permettre d'évaluer complètement la réversibilité des effets observés. Il faudra cependant mettre fin à l'expérience dès que l'animal montre des signes persistants de douleur ou de détresse aiguës. La réversibilité des effets est déterminée par l'observation des animaux sur une période s'étendant jusqu'à 14 jours après l'enlèvement des timbres. Si la réaction s'avère réversible avant le quatorzième jour, l'expérience s'achève à ce moment-là.
1.4.2.6. Observations cliniques et cotation des réactions cutanées
L'observation des signes d'érythème et d'œdème chez tous les animaux et la cotation des réactions s'effectuent au bout de 60 minutes et ensuite 24, 48 et 72 heures après l'enlèvement du timbre. S'agissant de l'animal du test initial, la plage soumise à l'épreuve est aussi examinée immédiatement après l'enlèvement du timbre. Les réactions cutanées sont cotées et consignées conformément à l'échelle figurant dans le tableau ci-après. Si la peau présente des lésions qui ne caractérisent pas une irritation ou une corrosion après 72 heures, il pourra être nécessaire d'observer l'animal jusqu'au quatorzième jour afin de déterminer la réversibilité des effets. En plus de l'observation de l'irritation, tous les effets toxiques locaux, tels qu'un dessèchement de la peau, et tout effet systémique nocif (par exemple des effets se manifestant par des signes cliniques de toxicité et sur le poids corporel) doivent être relevés et décrits en détail. L'examen histopathologiques est à envisager en cas de réactions équivoques.
La cotation des réactions cutanées est forcément subjective. L'harmonisation de la cotation des réactions cutanées et l'appui aux laboratoires d'essai ainsi qu'au personnel chargé d'effectuer et d'interpréter les observations passent par une formation adéquate des expérimentateurs au système de cotation utilisé (voir tableau ci-après). Un manuel illustré sur la cotation de l'irritation cutanée et d'autres lésions pourrait être utile (9). La cotation des réactions cutanées devrait faire l'objet d'une évaluation à l'aveugle.
2. RÉSULTATS
2.1. PRÉSENTATION DES RÉSULTATS
Les résultats de l'étude devraient être récapitulés dans un tableau joint au rapport d'essai final et couvrir tous les aspects énumérés au paragraphe 3.1.
2.2. ÉVALUATION DES RÉSULTATS
Le degré d'irritation cutanée devrait être évalué en considération de la gravité des lésions et de leur caractère réversible ou non. Les cotes individuelles ne fournissent pas une valeur absolue des propriétés irritantes d'une substance, celles-ci étant évaluées parallèlement à d'autres effets de la substance. Ces cotes individuelles doivent plutôt être considérées comme des valeurs de référence, à évaluer en association avec toutes les autres observations effectuées au cours de l'étude.
L'évaluation des réactions d'irritation doit tenir compte de la réversibilité des lésions cutanées. Si des réactions, telles qu'une alopécie (sur une aire limitée), une hyperkératose, une hyperplasie et une desquamation, persistent jusqu'à la fin de la période d'observation de 14 jours, il y a lieu de considérer la substance d'essai comme irritante.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Justification de l'essai in vivo: analyse de la valeur probante des résultats disponibles avant l'essai, notamment des résultats de la démarche expérimentale séquentielle:
|
—
|
description des données pertinentes livrées par des essais précédents,
|
|
—
|
données obtenues à chaque étape de la démarche expérimentale,
|
|
—
|
description des essais in vitro effectués, exposant le détail des procédures et les résultats obtenus avec les substances d'essai et de référence,
|
|
—
|
justification de l'étude in vivo après analyse de la valeur probante des résultats disponibles.
|
|
|
|
Substance d'essai:
|
—
|
données d'identification (par exemple, numéro CAS, source, pureté, impuretés connues, numéro de lot),
|
|
—
|
état physique et propriétés physico-chimiques (par exemple, pH, volatilité, solubilité, stabilité),
|
|
—
|
s'il s'agit d'un mélange: composition et pourcentages relatifs des constituants.
|
|
|
|
Véhicule:
|
—
|
identification, concentration (s'il y a lieu), volume utilisé,
|
|
—
|
justification du choix du véhicule.
|
|
|
|
Animaux d'expérience:
|
—
|
espèce/souche utilisée, justification de l'utilisation éventuelle d'un animal autre que le lapin albinos,
|
|
—
|
nombre d'animaux de chaque sexe,
|
|
—
|
poids de chaque animal au début et à la fin de l'essai,
|
|
—
|
âge des animaux au début de l'essai,
|
|
—
|
source des animaux, conditions d'encagement, régime alimentaire, etc.
|
|
|
|
Conditions expérimentales:
|
—
|
technique de préparation du site d'application,
|
|
—
|
détails concernant la composition du timbre et la technique d'application de ce dernier,
|
|
—
|
détails sur la préparation, l'application et l'enlèvement de la substance d'essai.
|
|
|
|
Résultats:
|
—
|
tableau faisant apparaître, pour chaque animal et à chaque relevé, les cotes attribuées aux réactions d'irritation/corrosion observées,
|
|
—
|
description de toutes les lésions observées,
|
|
—
|
description circonstanciée de la nature et du degré d'irritation ou de corrosion observé, et de tout effet histopathologique,
|
|
—
|
description de tout autre effet local néfaste (par exemple dessèchement de la peau) et des effets systémiques.
|
|
4. BIBLIOGRAPHIE
|
(1)
|
Barratt, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, I., Giuliani, A., Gray, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995) The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23, p. 410-429.
|
|
(2)
|
Young, J.R., How, M.J., Walker, A.P., Worth, W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, p. 19-26.
|
|
(3)
|
Worth, A.P., Fentem, J.H., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Liebsch, M. (1998) Evaluation of the proposed OECD Testing Strategy for skin corrosion. ATLA 26, p. 709-720.
|
|
(4)
|
ECETOC (1990) Monograph No 15, «Skin Irritation», European Chemical Industry, Ecology and Toxicology Centre, Brussels.
|
|
(5)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, p. 483-524.
|
|
(5a)
|
Méthode d'essai B.40. Corrosion cutanée.
|
|
(6)
|
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Held in Solna, Sweden, 22 - 24 January 1996 (http://wwwl.oecd.org/ehs/test/background.htm).
|
|
(7)
|
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://wwwl.oecd.org/ehs/Class/HCL6.htm).
|
|
(8)
|
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications. Series on Testing and Assessment No 19 (http://wwwl.oecd.org/ehs/test/monos.htm).
|
|
(9)
|
EPA (1990). Atlas of Dermal Lesions, (20T-2004). United States Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, DC, August 1990.
[peut être obtenu sur demande auprès du secrétariat de l'OCDE].
|
Tableau I
COTATION DES RÉACTIONS CUTANÉES
Formation d'érythème et d'escarres
|
Pas d'érythème …
|
0
|
|
Érythème très léger (à peine perceptible) …
|
1
|
|
Érythème bien défini …
|
2
|
|
Érythème modéré à grave …
|
3
|
|
Érythème grave (rouge violacé) à formation d'escarres empêchant la cotation de l'érythème …
|
4
|
Maximum possible: 4
Formation d'œdème
|
Pas d'œdème …
|
0
|
|
Œdème très léger (à peine perceptible) …
|
1
|
|
Œdème léger (pourtour de la zone œdémateuse bien délimité par une enflure nette)…
|
2
|
|
Œdème modéré (enflure d'environ 1 mm) …
|
3
|
|
Œdème grave (enflure de plus de 1 mm s'étendant au-delà de l'aire exposée) …
|
4
|
Maximum possible: 4
Un examen histopathologique pourra être réalisé en cas de réaction équivoque.
ANNEXE
Démarche expérimentale séquentielle pour les essais d'irritation et de corrosion cutanées
CONSIDÉRATIONS GÉNÉRALES
Pour concilier la fiabilité des résultats scientifiques et le bien-être animal, il importe d'éviter l'utilisation inutile d'animaux d'expérience et de réduire au minimum toute procédure expérimentale susceptible de déclencher des effets graves chez les animaux. Avant d'envisager un essai in vivo, on évaluera toutes les informations relatives à l'éventuel pouvoir corrosif/irritant d'une substance sur la peau. Il se peut que les données disponibles suffisent à classer une substance d'essai quant à son pouvoir irritant ou corrosif, sans qu'il soit nécessaire d'effectuer des essais sur animaux. Ainsi, le recours à une analyse de la valeur probante des résultats et l'adoption d'une démarche expérimentale séquentielle limiteront au maximum la nécessité de pratiquer des essais in vivo, surtout lorsqu'il est probable que la substance va engendrer de graves effets.
Il est recommandé d'évaluer les informations existantes concernant le pouvoir irritant ou corrosif des substances à l'aide d'une analyse de la valeur probante des résultats, afin d'établir la nécessité de réaliser des études supplémentaires, autres que des études cutanées in vivo, pour contribuer à caractériser ce pouvoir. Si cette nécessité se confirme, une démarche expérimentale séquentielle est préconisée pour produire les données expérimentales pertinentes. S'agissant des substances qui n'ont pas encore fait l'objet d'essais, il convient d'obtenir l'ensemble des données permettant d'évaluer le pouvoir corrosif ou irritant de la substance par une démarche séquentielle. La démarche expérimentale exposée dans cette annexe a été élaborée au cours d'un atelier de l'OCDE (1) avant d'être entérinée et complétée dans le cadre du système harmonisé de classification intégrée des risques pour la santé humaine et l'environnement liés aux substances chimiques (Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances), adopté par la vingt-huitième réunion conjointe du comité sur les produits chimiques et du groupe de travail sur les produits chimiques, les pesticides et la biotechnologie, en novembre 1998 (2).
Si cette démarche expérimentale séquentielle ne fait pas partie intégrante de la méthode d'essai B.4, elle constitue cependant la procédure recommandée pour déterminer l'effet irritant ou corrosif sur la peau. Cette procédure représente à la fois la meilleure pratique et une référence éthique dans ce domaine. La méthode d'essai décrit le mode opératoire de l'essai in vivo et récapitule les facteurs à prendre en considération avant d'entamer l'essai. La démarche indique comment évaluer les données existantes relatives aux propriétés irritantes ou corrosives des substances et présente les différentes étapes permettant d'obtenir des données pertinentes sur les substances qui réclament d'autres études ou qui n'ont pas encore été étudiées. Elle recommande également, dans certaines circonstances, la réalisation d'essais in vitro ou ex vivo d'irritation et de corrosion cutanées, validés et acceptés.
DESCRIPTION DE LA STRATÉGIE D'ÉVALUATION ET D'ESSAI
Avant d'entreprendre les essais inscrits dans la démarche expérimentale séquentielle (figure), il faut évaluer toutes les informations disponibles afin d'établir la nécessité d'effectuer des essais cutanés in vivo. Bien que l'évaluation de certains paramètres particuliers puisse livrer des informations significatives (par exemple un pH extrême), la totalité des informations existantes doit être prise en considération. L'analyse de la valeur probante des résultats portera sur toutes les données pertinentes concernant les effets de la substance en question, ou de ses analogues; elle débouchera sur une décision qui devra être justifiée. Il conviendra d'accorder une place prépondérante aux données se rapportant aux humains et aux animaux, et d'examiner ensuite les résultats des essais in vitro ou ex vivo sur la substance. Les essais in vivo de substances corrosives devront être évités chaque fois que cela est possible. Les facteurs pris en compte dans la démarche expérimentale sont repris ci-après:
Évaluation des données existantes sur les humains et les animaux (étape 1). Les données existantes sur l'être humain, par exemple des études cliniques ou sur les conditions de travail et des rapports sur des cas relevant de ces disciplines, et/ou les résultats d'essais sur animaux, par exemple des essais de toxicité par exposition cutanée unique ou répétée, sont à examiner en premier, car ils livrent des informations directement liées aux effets sur la peau. Les substances dont le pouvoir irritant ou corrosif est avéré et celles dont le caractère non corrosif et non irritant a été clairement démontré ne doivent pas faire l'objet d'études in vivo.
Analyse des relations structure-activité (RSA) (étape 2). Le cas échéant, les résultats d'essais sur des substances structurellement proches doivent être pris en considération. S'il existe suffisamment de données relatives aux humains et/ou aux animaux sur des substances structurellement proches ou sur des mélanges de ces substances pour attester leur pouvoir corrosif ou irritant, on peut supposer que la substance étudiée provoquera les mêmes réactions. Auquel cas, il n'est plus forcément nécessaire de tester la substance. Des résultats négatifs lors d'études de substances structurellement proches ou de mélanges de ces substances ne constituent pas une preuve suffisante de l'effet non corrosif ou non irritant d'une substance dans le cadre de la démarche expérimentale séquentielle. On déterminera le pouvoir corrosif ou irritant pour la peau en appliquant une méthode d'analyse RSA validée et acceptée.
Propriétés physico-chimiques et réactivité chimique (étape 3). Les substances présentant des valeurs de pH extrêmes, par exemple < 2,0 ou > 11,5, sont susceptibles d'induire de fortes réactions locales. Si une valeur de pH extrême est un indice décisif quant au caractère corrosif d'une substance, son pouvoir tampon doit également entrer en ligne de compte (3) (4). Si le pouvoir tampon donne à penser que la substance peut ne pas être corrosive pour la peau, cette hypothèse demande à être confirmée par un autre essai, de préférence un essai in vitro ou ex vivo validé et accepté (voir étapes 5 et 6).
Toxicité cutanée (étape 4). S'il est prouvé qu'une substance chimique est très toxique par voie cutanée, une étude d'irritation ou de corrosion cutanée risque d'être impossible à réaliser in vivo, la quantité de substance d'essai normalement appliquée pouvant dépasser la dose très toxique et entraîner ainsi la mort des animaux ou les faire terriblement souffrir. En outre, lorsque des études de toxicité cutanée ont déjà été menées sur des lapins albinos jusqu'à la dose limite de 2 000 mg/kg de poids corporel, ou au-delà, et qu'aucune irritation ou corrosion cutanées n'ont été constatées, il n'est plus forcément nécessaire d'effectuer un essai supplémentaire d'irritation ou de corrosion cutanée. Il convient de faire attention à plusieurs aspects lorsqu'on évalue la toxicité cutanée aiguë à partir d'études antérieures. À titre d'exemple, les informations rapportées sur les lésions cutanées peuvent être incomplètes. Il se peut que les essais et les observations aient été effectués sur une autre espèce que le lapin, or, la sensibilité aux substances est très variable d'une espèce à l'autre. De même, la forme sous laquelle la substance d'essai a été appliquée sur les animaux peut ne pas convenir à l'évaluation de l'irritation et de la corrosion cutanées [par exemple la dilution des substances (5)]. Néanmoins, lorsque des études bien conçues de toxicité cutanée ont été correctement menées sur le lapin, leurs résultats négatifs peuvent être considérés comme une démonstration suffisante du caractère non irritant ni corrosif de la substance.
Résultats des essais in vitro ou ex vivo (étapes 5 et 6). Les substances dont le pouvoir fortement irritant ou corrosif a été mis en évidence par un essai in vitro ou ex vivo validé et accepté (6) (7), conçu pour évaluer ces effets particuliers, ne doivent pas être testées sur des animaux. Il est en effet très vraisemblable que ces substances produiront des effets semblables in vivo.
Essai in vivo sur des lapins (étapes 7 et 8). Si la décision d'effectuer une étude in vivo a été prise sur la base d'une analyse de la valeur des résultats, celle-ci doit débuter par un essai initial réalisé sur un seul animal. Si les résultats de cet essai indiquent que la substance est corrosive pour la peau, il ne faut pas poursuivre les essais. Si l'essai initial ne montre aucun effet corrosif, il y a lieu de confirmer l'absence de réaction ou une réaction d'irritation sur un ou deux animaux supplémentaires exposés à la substance durant quatre heures. Lorsque l'essai initial fait apparaître un effet irritant, l'essai confirmatoire doit être conduit sur un mode séquentiel, ou par l'exposition simultanée des deux animaux supplémentaires.
BIBLIOGRAPHIE
|
(1)
|
OECD (1996). Test Guidelines Programme: Final Report on the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Held on Solna, Sweden, 22 - 24 January 1996 (http://wwwl.oecd.org/ehs/test/background.htm).
|
|
(2)
|
OECD (1998). Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://wwwl.oecd.org/ehs/Class/HCL6.htm).
|
|
(3)
|
Worth, A.P., Fentem, J.H., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdail, D.J., Liebsch, M. (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26, p. 709-720.
|
|
(4)
|
Young, J.R., How, M.J., Walker, A.P., Worth, W.M.H. (1988). Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substances, Without Testing on Animals. Toxic In Vitro, 2 (1), p. 19-26.
|
|
(5)
|
Patil, S.M., Patrick, E., Maibach, H.I. (1996) Animal, Human, and In Vitro Test Methods for Predicting Skin Irritation, in: Francis N. Marzulli and Howard I. Maibach (editors): Dermatotoxicology. Fifth Edition ISBN 1-56032-356-6, Chapter 31, p. 411-436.
|
|
(6)
|
Méthode d'essai B.40.
|
|
(7)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsail, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, p. 483-524.
|
Figure
PROCÉDURE D'ESSAI ET D'ÉVALUATION DE L'IRRITATION ET DE LA CORROSION CUTANÉES

B.5. TOXICITÉ AIGUË: IRRITATION/CORROSION OCULAIRE
1. MÉTHODE
Cette méthode est équivalente à la ligne directrice TG 405 (2002) de l'OCDE.
1.1. INTRODUCTION
La révision de la présente méthode a porté plus particulièrement sur l'évaluation préalable de toutes les informations existantes se rapportant aux substances d'essai, afin d'éviter les essais inutiles sur les animaux de laboratoire et contribuer ainsi à préserver le bien-être animal. Cette méthode préconise d'analyser la valeur probante des résultats pertinents déjà disponibles (1) avant de procéder à l'essai in vivo de corrosion/irritation oculaire décrit ci-après. Il est conseillé de combler le manque de données par des essais séquentiels (2) (3). La démarche expérimentale décrite à l'annexe de la présente méthode inclut la réalisation d'essais in vitro validés et acceptés. Il est en outre recommandé d'effectuer un essai d'irritation/corrosion cutanée in vivo, afin de prédire le pouvoir corrosif pour les yeux avant d'envisager un essai oculaire in vivo.
Afin de concilier la fiabilité des résultats scientifiques et le bien-être animal, on ne procédera pas aux essais in vivo avant d'avoir analysé la valeur probante de toutes les données relatives au caractère éventuellement corrosif ou irritant de la substance pour les yeux. Ces données comprendront les résultats d'études menées sur des humains et/ou des animaux de laboratoire, des données relatives à l'effet corrosif ou irritant d'une ou plusieurs substances structurellement proches de la substance d'essai ou de mélanges de ces substances, des données démontrant la forte acidité ou alcalinité de la substance (4) (5), et des résultats d'essais in vitro et ex vivo d'irritation et de corrosion cutanées validés et acceptés (6) (6a). Ces études pourront être effectuées avant ou après l'analyse de la valeur probante des résultats.
Pour certaines substances, une telle analyse peut faire ressortir la nécessité de mener des études in vivo afin de déterminer le pouvoir corrosif/irritant de la substance pour les yeux. Dans de tels cas, il est préférable, avant d'envisager un essai oculaire in vivo, de commencer par tester in vivo les effets cutanés de la substance et de les évaluer conformément à la méthode d'essai B.4 (7). L'analyse de la valeur probante des résultats, couplée à la stratégie d'essai séquentielle, devrait réduire la nécessité de tester in vivo l'effet corrosif ou irritant sur les yeux des substances pour lesquelles des études antérieures ont déjà livré suffisamment d'informations quant à ces deux aspects. Si le pouvoir corrosif ou irritant pour les yeux est impossible à déterminer par la démarche séquentielle, même après réalisation d'un essai in vivo d'irritation/corrosion cutanée, on peut tester in vivo l'effet corrosif ou irritant sur les yeux.
La démarche expérimentale préconisée, de type séquentiel, comporte des essais de corrosion et d'irritation in vitro ou ex vivo validés, et est décrite dans l'annexe de la présente méthode. Cette démarche, élaborée au cours d'un atelier de l'OCDE (8) et recommandée à l'unanimité par les participants à cet atelier, a été adoptée à titre de recommandation dans le cadre du système de classification des substances chimiques harmonisé à l'échelon mondial (Globally Harmonized System for the Classification of Chemical Substances — GHS) (9). Bien que cette stratégie séquentielle ne fasse pas partie intégrante de la méthode d'essai B.4, elle constitue cependant la procédure recommandée avant d'entreprendre les essais in vivo. S'agissant de nouvelles substances, cette démarche expérimentale par étapes est préconisée pour obtenir des résultats scientifiques fiables sur l'effet corrosif ou irritant de la substance. En ce qui concerne les substances existantes dont les données sur l'effet corrosif ou irritant sur la peau et les yeux sont insuffisantes, on suppléera à ces dernières en appliquant cette stratégie. Le choix d'une autre démarche expérimentale ou la décision de ne pas procéder par étapes doivent être justifiés.
1.2. DÉFINITIONS
L'irritation oculaire désigne des altérations oculaires qui surviennent à la suite de l'application d'une substance d'essai sur la surface antérieure de l'œil, et qui sont totalement réversibles dans les 21 jours suivant le traitement.
La corrosion oculaire désigne la survenue de lésions tissulaires de l'œil ou une grave détérioration de la vision, qui sont provoqués par l'application d'une substance d'essai sur la surface antérieure de l'œil, et qui ne sont pas totalement réversibles dans les 21 jours suivant le traitement.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance d'essai est appliquée en une seule dose sur l'un des yeux de l'animal d'expérience, l'œil non traité servant de témoin. On évalue le degré de l'irritation ou de la corrosion oculaires en cotant la gravité des lésions affectant la conjonctive, la cornée et l'iris, à intervalles déterminés. Les autres réactions de l'œil et les troubles systémiques sont également décrits de manière à fournir une évaluation complète des effets. La durée de l'étude doit être suffisante pour permettre d'évaluer la réversibilité des effets.
Les animaux qui manifestent des signes persistants de détresse et/ou de douleur aiguës à n'importe quel stade de l'essai doivent être euthanasiés, et ces symptômes seront pris en compte dans l'évaluation de la substance. Les critères régissant la décision d'euthanasier les animaux moribonds et souffrant fortement sont définis dans un document cité en référence (10).
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparation de l'essai in vivo
1.4.1.1. Sélection de l'espèce animale
On choisira de préférence de jeunes adultes sains parmi les lapins albinos. L'utilisation d'une autre espèce ou souche sera justifiée, le cas échéant.
1.4.1.2. Préparation des animaux
Les deux yeux de chaque animal susceptible de participer à l'essai doivent être examinés dans les 24 heures précédant le début de l'essai. Les animaux qui présentent des signes d'irritation oculaire, des défauts oculaires ou une lésion de la cornée seront écartés.
1.4.1.3. Conditions d'hébergement et d'alimentation
Les animaux sont placés dans des cages individuelles. La température du local expérimental est réglée à 20 oC (± 3 oC) pour les lapins. L'humidité relative doit atteindre au moins 30 % et rester de préférence inférieure à 70 %, mais en dehors des heures de nettoyage du local, on s'efforcera de maintenir le taux d'humidité autour de 50 à 60 %. On appliquera un éclairage artificiel, alternant 12 heures de lumière et 12 heures d'obscurité. Les lapins seront nourris avec un mélange classique pour animaux de laboratoire et boiront de l'eau potable à volonté.
1.4.2. Mode opératoire
1.4.2.1. Application de la substance d'essai
L'expérimentateur introduit la substance d'essai dans le sac conjonctival de l'un des yeux de chaque animal, après avoir délicatement écarté la paupière inférieure du globe oculaire. Il ramène ensuite doucement les deux paupières l'une contre l'autre et les maintient dans cette position pendant environ une seconde afin d'éviter toute perte de substance. L'autre œil, qui ne subit pas de traitement, sert de témoin.
1.4.2.2. Irrigation
Il ne faut pas laver les yeux des animaux traités pendant au moins 24 heures après l'instillation de la substance d'essai, à moins que celle-ci soit à l'état solide (voir paragraphe 1.4.2.3.2) ou déclenche immédiatement des effets corrosifs ou irritants. Au besoin, un lavage pourra être effectué à l'issue de ce délai.
L'utilisation d'un groupe d'animaux satellite pour étudier l'influence du lavage n'est pas indiquée, à moins qu'elle ne se justifie d'un point de vue scientifique. Le cas échéant, on utilisera deux lapins. Les conditions du lavage doivent être décrites minutieusement en indiquant, par exemple, le moment du lavage, la composition et la température de la solution ophtalmique, la durée, le volume et la vitesse d'application.
1.4.2.3. Dose
1.4.2.3.1. Essai de liquides
Pour les liquides, on utilise une dose de 0,1 ml. Il faut éviter d'instiller la substance directement dans l'œil avec un vaporisateur à pression; il est préférable d'en expulser d'abord le contenu, de prélever 0,1 ml de celui-ci dans une fiole et de l'instiller dans l'œil.
1.4.2.3.2. Essai de solides
Dans le cas des solides, pâtes ou substances particulaires, la quantité utilisée doit avoir un volume de 0,1 ml ou un poids ne dépassant pas 100 mg. La substance d'essai sera broyée finement. Il convient de mesurer le volume de la substance solide après l'avoir légèrement tassée, par exemple en tapotant le récipient de mesure. Si la substance d'essai solide n'a pas encore été évacuée de l'œil de l'animal par des mécanismes physiologiques au premier moment d'observation, à savoir une heure après le traitement, l'œil peut être rincé à l'aide d'une solution saline ou à l'eau distillée.
1.4.2.3.3. Essai des aérosols
Il est recommandé de prélever une dose du contenu de tous les vaporisateurs à pression et aérosols avant de l'instiller dans l'œil. La seule exception concerne les substances conditionnées en bombes aérosol sous pression, qui sont impossibles à recueillir préalablement à l'instillation car elles se vaporisent. Dans ce cas, l'expérimentateur maintient l'œil de l'animal ouvert et administre la substance à tester en un seul jet d'environ une seconde, émis à 10 cm et directement en face de l'œil. Cette distance peut être modulée en fonction de la pression du jet et de sa composition. Il faut veiller à ce que la pression du jet n'endommage pas l'œil. Dans certains cas, il pourra être nécessaire d'évaluer l'ampleur des dégâts «mécaniques» risquant d'être causés à l'œil par la force du jet.
La dose d'aérosol peut être estimée grâce à une simulation de l'application menée comme suit: la substance est projetée à travers une ouverture de la taille d'un œil de lapin placée directement devant un papier. L'augmentation de poids du papier donne une idée approximative de la quantité administrée dans l'œil du lapin. Pour une substance volatile, la dose peut être estimée à partir du poids du récipient (dans lequel elle est recueillie) avant et après utilisation.
1.4.2.4. Essai initial (essai in vivo de l'effet irritant/corrosif sur les yeux mené sur un seul animal)
Conformément à la démarche expérimentale séquentielle (voir annexe 1), il est fortement recommandé de commencer par pratiquer l'essai in vivo sur un seul animal.
Si les résultats de cet essai, mené selon la procédure décrite, indiquent que la substance est corrosive ou fortement irritante pour l'œil, il n'y a pas lieu de mener d'autres essais d'irritation oculaire.
1.4.2.5. Anesthésie locale
L'anesthésie locale se pratique cas par cas. Si l'analyse de la valeur probante des résultats montre que la substance risque de provoquer une douleur, ou si l'essai initial révèle que l'animal souffrira, un anesthésique local peut être administré avant l'instillation de la substance d'essai. Le type, la concentration et la dose d'anesthésique doivent être choisis avec soin pour éviter que son utilisation n'influe sur la réaction à la substance d'essai. L'œil témoin subira une anesthésie identique.
1.4.2.6. Essai confirmatoire (essai d'irritation oculaire in vivo sur des animaux supplémentaires)
Si l'essai initial ne fait apparaître aucun effet corrosif, la réaction négative ou d'irritation demande à être confirmée sur un ou deux animaux supplémentaires. Si l'essai initial provoque une forte irritation, indiquant que l'essai confirmatoire pourrait donner lieu à un effet très marqué (irréversible), on recommande de conduire l'essai confirmatoire sur un mode séquentiel en n'utilisant qu'un seul animal à la fois, plutôt que d'exposer les deux animaux simultanément. Si le deuxième animal manifeste des signes de corrosion ou d'irritation grave, l'essai s'arrête là. On pourra utiliser d'autres animaux s'il y a lieu de confirmer des réactions d'irritation faibles à modérées.
1.4.2.7. Période d'observation
La durée de la période d'observation doit être suffisante pour permettre d'évaluer à fond l'ampleur et la réversibilité des effets observés. Il faut cependant mettre un terme à l'expérience dès qu'un animal manifeste des signes persistants de détresse ou de douleur aiguës (9). Pour déterminer la réversibilité des effets, les animaux doivent normalement être observés durant 21 jours après l'administration de la substance d'essai. Si la réversibilité est constatée avant ce délai, l'expérience prend fin à ce moment-là.
1.4.2.7.1. Observations cliniques et cotation de la gravité des réactions oculaires
On examine les yeux 1, 24, 48 et 72 heures après l'application de la substance d'essai. Les animaux ne seront maintenus à l'épreuve que le temps nécessaire pour obtenir un résultat concluant. Les animaux manifestant des signes persistants de douleur ou de détresse aiguës doivent être euthanasiés rapidement, et ces symptômes sont à prendre en compte dans l'évaluation de la substance d'essai. Il faudra également euthanasier les animaux qui présentent les lésions oculaires suivantes à la suite de l'instillation: perforation de la cornée ou ulcération profonde de la cornée associée à un staphylome; présence de sang dans la chambre antérieure de l'œil; opacité cornéenne de niveau 4 persistant durant 48 heures; absence de réflexe protomoteur (réaction indienne de niveau 2) durant 72 heures; ulcération de la membrane conjonctivale; nécrose des conjonctives ou de la membrane nictitante, ou décollement du tissu nécrosé. L'euthanasie s'impose parce que ces lésions sont généralement irréversibles.
Les animaux qui ne présentent pas de lésions oculaires peuvent être écartés, mais seulement à partir du quatrième jour suivant l'instillation. Les animaux affectés de lésions légères à modérées doivent être gardés en observation jusqu'à ce que ces lésions disparaissent, ou durant 21 jours, soit jusqu'au terme de l'étude. L'expérimentateur déterminera la nature et la gravité des lésions ainsi que leur réversibilité en observant les animaux aux septième, quatorzième et vingt et unième jours.
L'intensité des réactions oculaires (conjonctives, cornée et iris) doit être consignée à chaque examen (tableau I). Toute autre lésion oculaire (par exemple un pannus, une coloration) et les troubles systémiques doivent également être signalés.
Pour examiner les réactions, on peut s'aider d'une loupe binoculaire, d'une lampe à fente portative, d'un biomicroscope ou d'un autre appareil approprié. Après l'enregistrement des observations effectuées à la vingt-quatrième heure, l'examen des yeux peut se poursuivre à la fluorescéine.
La cotation des réactions oculaires est forcément subjective. L'harmonisation de la cotation des réactions oculaires et l'appui aux laboratoires d'essai ainsi qu'au personnel chargé d'effectuer et d'interpréter les observations passent par une formation adéquate des expérimentateurs au système de cotation utilisé. La cotation des réactions cutanées devrait faire l'objet d'une évaluation à l'aveugle.
2. RÉSULTATS
2.2. ÉVALUATION DES RÉSULTATS
Le degré d'irritation oculaire devrait être évalué en considération de la gravité des lésions et de leur caractère réversible ou non. Les cotes individuelles ne fournissent pas une valeur absolue des propriétés irritantes d'une substance, celles-ci étant évaluées parallèlement à d'autres effets de la substance. En revanche, les cotes individuelles ont une valeur de référence et ne sont significatives que lorsqu'elles sont appuyées par une description et une évaluation complètes de toutes les observations.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Justification de l'essai in vivo: analyse de la valeur probante des résultats d'essais préexistants, notamment ceux qui procèdent de la démarche expérimentale séquentielle:
|
—
|
description des données pertinentes livrées par des essais précédents,
|
|
—
|
données obtenues à chaque étape de la démarche expérimentale,
|
|
—
|
description des essais effectués in vitro, détaillant les procédures utilisées et mentionnant les résultats obtenus avec les substances d'essai et avec les substances de référence,
|
|
—
|
description de l'étude d'irritation/corrosion cutanée menée in vivo, mentionnant les résultats obtenus,
|
|
—
|
justification de l'étude in vivo après analyse de la valeur probante des résultats disponibles.
|
|
|
|
Substance d'essai:
|
—
|
données d'identification (par exemple, numéro CAS, source, pureté, impuretés connues, numéro de lot),
|
|
—
|
état physique et propriétés physico-chimiques (par exemple, pH, volatilité, solubilité, stabilité, réactivité avec l'eau),
|
|
—
|
s'il s'agit d'un mélange: composition et pourcentages relatifs des constituants,
|
|
—
|
si on a eu recours à une anesthésie locale: identification, pureté, type, dose et interaction potentielle avec la substance d'essai.
|
|
|
|
Véhicule:
|
—
|
identification, concentration (s'il y a lieu), volume utilisé,
|
|
—
|
justification du choix du véhicule.
|
|
|
|
Animaux, d'expérience:
|
—
|
espèce/souche utilisée, justification de l'utilisation éventuelle d'un animal autre que le lapin albinos,
|
|
—
|
âge de chaque animal au début de l'essai,
|
|
—
|
nombre d'animaux de chaque sexe dans les groupes d'essai et témoin (le cas échéant),
|
|
—
|
poids de chaque animal au début et à la fin de l'essai,
|
|
—
|
source des animaux, conditions d'encagement, régime alimentaire, etc.
|
|
|
|
Résultats:
|
—
|
description de la méthode utilisée pour coter l'irritation à chaque moment d'observation (par exemple lampe à fente portative, biomicroscope, fluorescéine),
|
|
—
|
présentation sous forme de tableaux des réactions d'irritation ou de corrosion relevées chez chaque animal et à chaque moment d'observation jusqu'à la fin de la participation de chaque animal à l'essai,
|
|
—
|
description circonstanciée du degré et de la nature de l'irritation ou de la corrosion observées,
|
|
—
|
description de toute autre lésion oculaire observée (par exemple vascularisation, formation d'un pannus, adhérences, coloration),
|
|
—
|
description des effets non oculaires locaux et des troubles systémiques, ainsi que des observations histopathologiques, le cas échéant.
|
|
|
|
Discussion des résultats.
|
3.2. INTERPRÉTATION DES RÉSULTATS
L'extrapolation à l'humain des résultats d'études d'irritation oculaire menées sur des animaux de laboratoire n'est valable que dans une certaine mesure. Dans bien des cas, le lapin albinos est plus sensible que l'espèce humaine aux substances irritantes ou corrosives pour l'œil.
Lors de l'interprétation des résultats, il faut savoir reconnaître une irritation consécutive à une infection secondaire, laquelle ne doit pas être prise en compte.
4. BIBLIOGRAPHIE
|
(1)
|
Barratt, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, I., Giuliani, A., Gray, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995) The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23, p. 410- 429.
|
|
(2)
|
De Silva, O., Cottin, M., Dami, N., Roguet, R., Catroux, P., Toufic, A., Sicard, C., Dossou, K.G., Cerner, I., Schlede, E., Spielmann, H., Gupta, K.C., Hill, R.N. (1997) Evaluation of Eye Irritation Potential: Statistical Analysis and Tier Testing Strategies. Food Chem. Toxicol 35, p. 159-164.
|
|
(3)
|
Worth, A.P. and Fentem J.H. (1999) A general approach for evaluating stepwise testing strategies ATLA 27, p. 161-177.
|
|
(4)
|
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, p. 19-26.
|
|
(5)
|
Neun, D.J. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, p. 227-231.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, p. 483-524.
|
|
(6a)
|
Méthode d'essai B.40. Corrosion cutanée.
|
|
(7)
|
Méthode d'essai B.4. Toxicité aiguë: irritation/corrosion cutanée.
|
|
(8)
|
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Held in Solna, Sweden, 22 - 24 January 1996 (http://www.oecd.org/ehs/test/background.htm).
|
|
(9)
|
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://www.oecd.org/ehs/Class/HCL6.htm).
|
|
(10)
|
Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications. Series on Testing and Assessment No 19 (http://www.oecd.org/ehs/test/monos.htm).
|
Tableau I
Cotation des réactions oculaires
Cornée
Opacité: degré de densité (les observations porteront sur les zones les plus denses) (1)
|
Pas d'ulcération ni d'opacité …
|
0
|
|
Zones d'opacité (autres qu'un léger ternissement de l'éclat normal) dispersées ou diffuses; détails de l'iris nettement visibles …
|
1
|
|
Zone translucide aisément discernable; détails de l'iris légèrement masqués …
|
2
|
|
Zone nacrée; détails de l'iris complètement invisibles; dimension de la pupille à peine discernable …
|
3
|
|
Cornée opaque; iris non discernable à travers l'opacité …
|
4
|
Maximum possible: 4
NOTES
Iris
|
Normal …
|
0
|
|
Plis nettement plus profonds, congestion, tuméfaction, hyperhémie péricornéenne modérée ou conjonctives injectées; iris réactif à la lumière (une réaction lente est positive) …
|
1
|
|
Hémorragie, destruction marquée, ou absence de réaction à la lumière …
|
2
|
Maximum possible: 2
Conjonctives
Rougeur (s'applique aux conjonctives palpébrale et bulbaire, mais pas à la cornée ni à l'iris)
|
Normal …
|
0
|
|
Hyperhémie de certains vaisseaux sanguins (yeux injectés) …
|
1
|
|
Coloration pourpre diffuse, vaisseaux sanguins difficilement discernables les uns des autres …
|
2
|
|
Coloration rouge soutenu diffuse …
|
3
|
Maximum possible: 3
Chémosis
Tuméfaction (s'applique aux paupières et/ou aux membranes nictitantes)
|
Normal …
|
0
|
|
Tuméfaction légèrement supérieure à la normale …
|
1
|
|
Tuméfaction patente avec éversion partielle des paupières …
|
2
|
|
Tuméfaction avec paupières à demi closes …
|
3
|
|
Tuméfaction avec paupières plus qu'à demi closes …
|
4
|
Maximum possible: 4
ANNEXE
Démarche expérimentale séquentielle pour les essais d'irritation et de corrosion oculaires
CONSIDÉRATIONS GÉNÉRALES
Pour concilier la fiabilité des résultats scientifiques et le bien-être animal, il importe d'éviter l'utilisation inutile d'animaux d'expérience et de réduire au minimum toute procédure expérimentale susceptible de déclencher des effets graves chez les animaux. Avant d'envisager un essai in vivo, on évaluera toutes les informations disponibles concernant l'éventuel pouvoir corrosif/irritant d'une substance sur les yeux. Il se peut que les données disponibles suffisent à classer une substance d'essai quant à son pouvoir d'irritation ou de corrosion oculaire, sans qu'il soit nécessaire d'effectuer des essais sur animaux. Ainsi, le recours à une analyse de la valeur probante des résultats et l'adoption d'une démarche expérimentale séquentielle limiteront au maximum la nécessité de pratiquer des essais in vivo, surtout si la substance risque d'engendrer des réactions violentes.
Il est recommandé d'évaluer les informations existantes concernant le pouvoir irritant ou corrosif des substances sur les yeux, à l'aide d'une analyse de la valeur probante des résultats, afin d'établir la nécessité de réaliser des études supplémentaires, autres que des études in vivo sur l'œil, pour contribuer à caractériser ce pouvoir. Si cette nécessité se confirme, une démarche expérimentale séquentielle est préconisée pour produire les données expérimentales nécessaires. S'agissant des substances qui n'ont pas encore fait l'objet d'essais, il convient d'obtenir l'ensemble des données permettant d'évaluer le pouvoir corrosif ou irritant de la substance par une démarche séquentielle. La démarche expérimentale exposée dans la présente annexe a été élaborée au cours d'un atelier de l'OCDE (1). Elle a par la suite été entérinée et complétée dans le cadre du système harmonisé de classification intégrée des risques pour la santé humaine et l'environnement liés aux substances chimiques (Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances), adopté par la vingt-huitième réunion conjointe du comité sur les produits chimiques et du groupe de travail sur les produits chimiques, les pesticides et la biotechnologie, en novembre 1998 (2).
Si cette démarche expérimentale séquentielle ne fait pas partie intégrante de la méthode d'essai B.5, elle constitue cependant la procédure recommandée pour déterminer l'effet irritant ou corrosif sur les yeux. Cette procédure représente à la fois la meilleure pratique et une référence éthique dans ce domaine. La méthode d'essai décrit le mode opératoire de l'essai in vivo et récapitule les facteurs à prendre en considération avant d'envisager un tel essai. La démarche séquentielle préconise une analyse de la valeur probante des données existantes concernant les propriétés d'irritation ou de corrosion oculaires des substances et présente les différentes étapes permettant d'obtenir des données pertinentes sur les substances qui nécessitent d'autres études ou qui n'ont pas encore été étudiées. Elle préconise, dans un premier temps, la réalisation des essais in vivo ou ex vivo validés et acceptés, et ensuite, dans certaines circonstances, la mise en œuvre des études d'irritation/corrosion cutanée de la méthode d'essai B.4 (3) (4).
DESCRIPTION DE LA DÉMARCHE EXPÉRIMENTALE SÉQUENTIELLE
Avant d'entreprendre les essais inscrits dans la démarche expérimentale séquentielle (figure), il faut évaluer toutes les informations disponibles afin d'établir la nécessité d'effectuer des essais oculaires in vivo. Bien que l'évaluation de certains paramètres particuliers puisse livrer des informations significatives (par exemple un pH extrême), la totalité des informations existantes doit être prise en considération. L'analyse de la valeur probante des résultats portera sur toutes les données pertinentes concernant les effets de la substance en question et de ses analogues; elle débouchera sur une décision qui devra être justifiée. Il conviendra d'accorder une place prépondérante aux données se rapportant aux humains et aux animaux, et d'examiner ensuite les résultats des essais in vitro ou ex vivo sur la substance. Les essais in vivo de substances corrosives devront être évités chaque fois que cela est possible. Les facteurs pris en compte dans la démarche expérimentale sont repris ci-après:
Évaluation des données existantes sur les humains et les animaux (étape 1). Les données existantes sur l'être humain, notamment études cliniques, études sur les conditions de travail et rapports se rapportant à ces études, et/ou les résultats d'essais réalisés sur des animaux dans le cadre d'études oculaires sont à examiner en premier, car ils livrent des informations directement liées aux effets sur les yeux. Il convient ensuite d'évaluer les résultats des études d'irritation/corrosion cutanée menées sur l'homme et/ou l'animal. Les substances ayant un pouvoir avéré d'irritation ou de corrosion oculaires, de même que les substances qui provoquent une irritation ou corrosion cutanées ne doivent pas être instillées dans les yeux des animaux; ces dernières substances sont à considérer comme étant également corrosives et/ou irritantes pour les yeux. Les substances dont le caractère non corrosif et non irritant a été démontré de manière suffisante par des études oculaires antérieures ne doivent pas non plus faire l'objet d'essais in vivo sur l'œil.
Analyse des relations structure-activité (RSA) (étape 2). Le cas échéant, les résultats d'essais sur des substances structurellement proches doivent être pris en considération. S'il existe suffisamment de données relatives aux humains et/ou aux animaux sur des substances structurellement proches ou sur des mélanges de ces substances pour attester leur pouvoir corrosif ou irritant sur les yeux, on peut supposer que la substance étudiée provoquera les mêmes réactions. Auquel cas, il n'est plus forcément nécessaire de tester la substance. Des résultats négatifs lors d'études de substances structurellement proches ou de mélanges de ces substances ne constituent pas une preuve suffisante de l'effet non corrosif ou non irritant d'une substance dans le cadre de la démarche expérimentale séquentielle. On déterminera le pouvoir corrosif ou irritant pour la peau et pour l'œil en appliquant une méthode d'analyse RSA validée et acceptée.
Propriétés physico-chimiques et réactivité chimique (étape 3). Les substances présentant des valeurs de pH extrêmes, par exemple < 2,0 ou ≥ 11,5, sont susceptibles d'induire de fortes réactions locales. Si une valeur de pH extrême est un indice décisif quant au caractère corrosif ou irritant d'une substance pour l'œil, son pouvoir tampon doit également entrer en ligne de compte (3) (4). Si le pouvoir tampon donne à penser que la substance peut ne pas être corrosive pour l'œil, cette hypothèse demande à être confirmée par un autre essai, de préférence un essai in vitro ou ex vivo validé et accepté (voir étapes 5 et 6).
Prise en compte des autres informations existantes (étape 4). Toutes les informations disponibles concernant la toxicité systémique par voie cutanée doivent être évaluées à ce stade. Il convient également d'évaluer la toxicité cutanée aiguë de la substance d'essai. Si la substance d'essai s'est révélée très toxique par voie cutanée, il n'est pas nécessaire de la tester sur les yeux. Même s'il n'y a pas nécessairement de lien entre la toxicité cutanée aiguë et l'irritation/corrosion oculaire, on peut considérer que, si un agent est très toxique par voie cutanée, il entraînera également une toxicité élevée en cas d'instillation dans l'œil. Ces données peuvent aussi être prises en considération entre les étapes 2 et 3.
Résultats des essais in vitro ou ex vivo (étapes 5 et 6). Il est inutile de tester sur des animaux les substances dont le pouvoir fortement irritant ou corrosif a été mis en évidence par un essai in vitro ou ex vivo validé et accepté (7) (8) pour l'évaluation spécifique de l'irritation/corrosion oculaire ou cutanée. Il est en effet très vraisemblable que ces substances produiront des effets semblables in vivo. S'il n'existe pas d'essais in vitro/ex vivo validés et acceptés, il convient de passer directement à l'étape 7 en ignorant les étapes 5 et 6.
Évaluation in vivo du pouvoir irritant ou corrosif de la substance sur la peau (étape 7). S'il n'existe pas suffisamment d'éléments pour déterminer de façon concluante le pouvoir d'irritation/corrosion oculaire d'une substance à partir des résultats des études susmentionnées, ce pouvoir doit être évalué en premier lieu in vivo, suivant la méthode d'essai B.4 (4) et son annexe (9). S'il apparaît que la substance provoque une corrosion ou une forte irritation cutanée, il y a lieu de la considérer comme corrosive ou irritante pour l'œil, à moins que d'autres éléments ne tendent à prouver le contraire. Auquel cas, un essai oculaire in vivo ne serait pas nécessaire. Si la substance n'est pas corrosive ni très irritante pour la peau, il y a lieu de pratiquer un essai oculaire in vivo.
Essai in vivo sur des lapins (étapes 8 et 9): l'étude oculaire in vivo doit débuter par un essai initial réalisé sur un seul animal. Si les résultats de cet essai indiquent que la substance est très irritante ou corrosive pour l'œil, il ne faut pas poursuivre les essais. Si cet essai ne fait apparaître aucun effet de corrosion ni d'irritation marquée, un essai confirmatoire est pratiqué sur deux animaux supplémentaires.
BIBLIOGRAPHIE
|
(1)
|
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Held in Solna, Sweden, 22 - 24 January 1996 (http://wwwl.oecd.org/ehs/test/background.htm).
|
|
(2)
|
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://wwwl.oecd.org/ehs/Class/HCL6.htm).
|
|
(3)
|
Worth, A.P. and Fentem J.H. (1999). A General Approach for Evaluating Stepwise Testing Strategies. ATLA 27, p. 161- 177.
|
|
(4)
|
Méthode d'essai B.4. Toxicité aiguë: irritation/corrosion cutanée.
|
|
(5)
|
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, p. 19-26.
|
|
(6)
|
Neun, D.J. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, p. 227-231.
|
|
(7)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, PA, Curren, R.D., Earl, L.K., Edsail, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, p. 483-524.
|
|
(8)
|
Méthode d'essai B.40. Corrosion cutanée.
|
|
(9)
|
Annexe à la méthode d'essai B.4. Démarche expérimentale séquentielle pour les essais d'irritation et de corrosion cutanées.
|
Figure
PROCÉDURE D'ESSAI ET D'ÉVALUATION DE L'IRRITATION ET DE LA CORROSION OCULAIRES
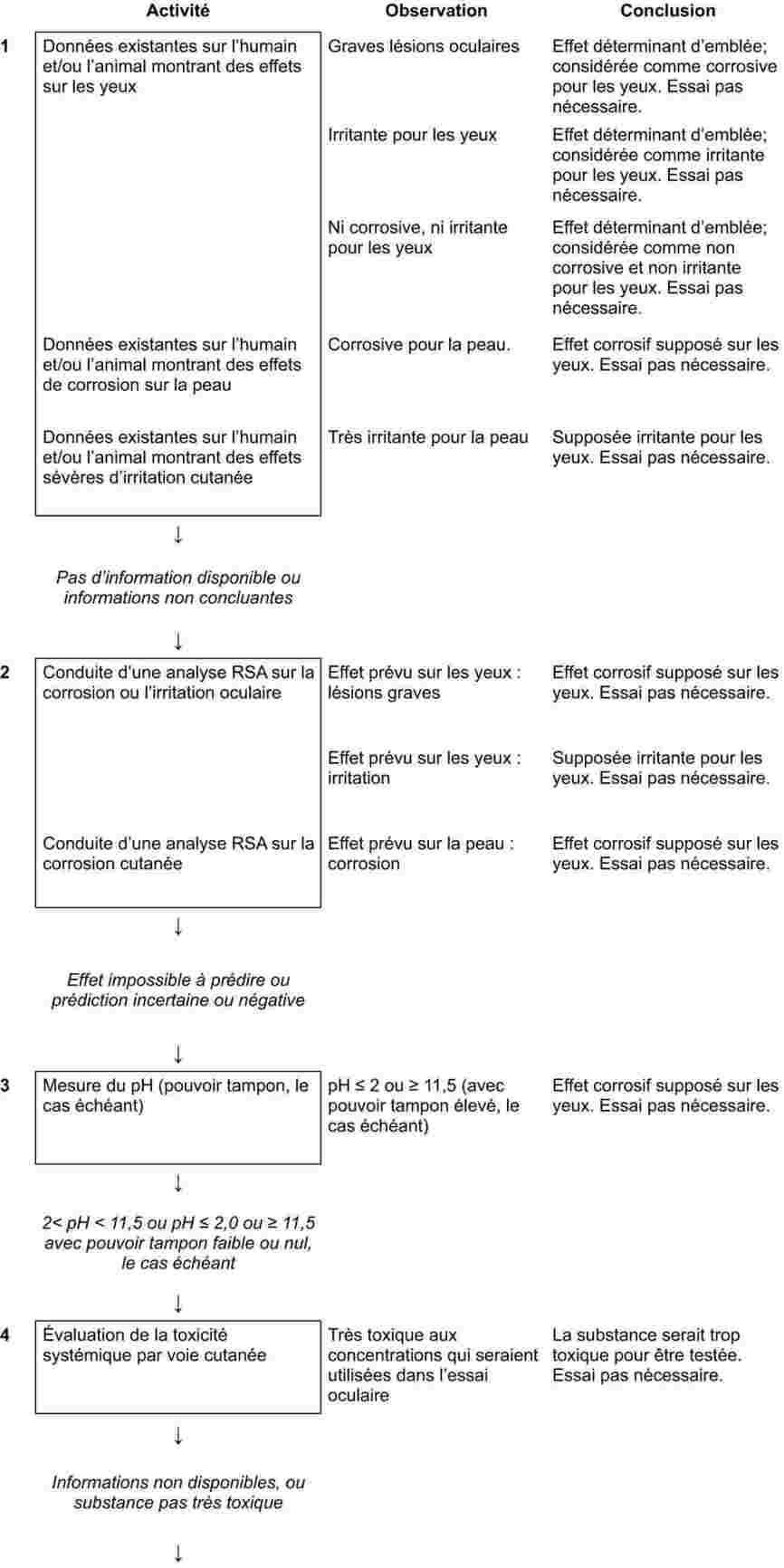

B.6. SENSIBILISATION CUTANÉE
1. MÉTHODE
1.1. INTRODUCTION
Remarques
La sensibilité des essais et leur capacité à détecter les substances susceptibles d'entraîner une sensibilisation cutanée chez l'homme sont des critères importants dans un système de classification de la toxicité établi à des fins de santé publique.
Il n'existe pas de méthode d'essai unique qui permette d'identifier de manière adéquate toutes les substances ayant un potentiel de sensibilisation cutanée pour l'homme et qui puisse être appliquée à toutes les substances.
Divers facteurs tels que les caractéristiques physiques d'une substance, y compris son pouvoir de pénétration cutanée, doivent être pris en considération lors du choix de l'essai.
Deux types d'essai utilisant des cobayes ont été développés: d'une part, les essais avec adjuvant dans lesquels l'état est potentialisé par la dissolution ou la mise en suspension dans de l'adjuvant complet de Freund (ACF) de la substance à tester, et d'autre part les essais sans adjuvant.
Les essais avec adjuvant ont généralement un meilleur pouvoir prédictif du potentiel sensibilisant cutanée de la substance testée avec plus de précision que les méthodes qui n'utilisent pas l'adjuvant complet de Freund. C'est la raison pour laquelle leur utilisation est préférable.
L'essai de maximalisation chez le cobaye (GPMT: Guinea Pig Maximisation Test) est un essai avec adjuvant très répandu. Bien qu'il existe plusieurs autres méthodes pour détecter le potentiel de sensibilisation cutanée, le GPMT est l'essai avec adjuvant de prédilection.
Les essais sans adjuvant (l'essai de Buehler étant préférable) sont considérés comme moins sensibles vis-à-vis de nombreuses classes de produits chimiques.
Dans certains cas, l'essai de Buehler qui consiste en une application topique, peut s'avérer préférable à l'essai de maximalisation qui nécessite une injection intradermique. L'utilisation de l'essai de Buehler doit être scientifiquement justifiée.
L'essai de maximalisation chez le cobaye (GMPT) et l'essai de Buehler sont décrits dans cette méthode. D'autres méthodes peuvent être utilisées, à condition qu'elles soient correctement validées et que leur utilisation soit scientifiquement justifiée.
Si un résultat positif est obtenu dans un test de dépistage reconnu, une substance peut être considérée comme un sensibilisant potentiel et il peut ne pas être nécessaire de réaliser un essai complémentaire sur le cobaye. Cependant si un résultat négatif est obtenu dans un tel test de dépistage, un essai sur le cobaye doit être effectué en utilisant la procédure décrite dans la présente méthode d'essai.
Voir également introduction générale, partie B.
1.2. DÉFINITIONS
La sensibilisation cutanée (dermite allergique de contact) est une réaction immunologique cutanée à une substance. Chez l'homme, la réaction peut se caractériser par un prurit, un érythème, un œdème, des vésicules, des bulles ou une association de ces manifestations. Dans les autres espèces, la réaction peut différer et prendre uniquement la forme d'un érythème ou d'un œdème.
Exposition d'induction: exposition expérimentale d'un sujet à une substance dans le but d'induire une hypersensibilité.
Période d'induction: période d'au moins une semaine consécutive à l'exposition d'induction, au cours de laquelle une hypersensibilité peut s'installer.
Exposition de déclenchement: exposition expérimentale, après période d'induction, d'un sujet préalablement exposé à la substance, dans le but de vérifier si le sujet présente une réaction d'hypersensibilité.
1.3. SUBSTANCES DE RÉFÉRENCE
La sensibilité et la fiabilité de la technique expérimentale utilisée doivent être vérifiées tous les six mois à l'aide de substances ayant des propriétés connues de sensibilisation cutanée faible à modérée.
Pour un essai conduit selon les règles, les substances faiblement ou moyennement sensibilisantes provoquent normalement une réaction d'au moins 30 % dans les méthodes avec adjuvant et 15 % dans les méthodes sans adjuvant.
Les substances suivantes seront de préférence utilisées:
|
Numéro CAS
|
Numéro EINECS
|
Dénomination EINECS
|
Dénomination usuelle
|
|
101-86-0
|
202-983-3
|
α-hexylcinnamaldéhyde
|
α-hexylcinnamaldéhyde
|
|
149-30-4
|
205-736-8
|
benzothiazole-2-thiol (mercaptobenzothiazole)
|
kaptax
|
|
94-09-7
|
202-303-5
|
benzocaïne
|
nordcaïne
|
Dans certaines circonstances dûment justifiées, il est possible d'utiliser d'autres substances répondant aux critères ci-dessus.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Les animaux d'expérience sont dans un premier temps exposés à la substance à tester par une injection intradermique et/ou une application épidermique (exposition d'induction). Après une période de repos de 10 à 14 jours (période d'induction) au cours de laquelle une réaction immunitaire peut se produire, les animaux sont exposés à une dose de déclenchement. L'étendue et le degré de la réaction cutanée des animaux sont alors comparées à celles de la réaction observée chez les animaux témoins qui ont reçu un placebo lors de 1 induction et qui ont été soumis à l'exposition de déclenchement.
1.5. DESCRIPTION DES MÉTHODES D'ESSAI
Si l'élimination de la substance à tester s'avère nécessaire, ceci doit être fait en utilisant de l'eau ou un solvant approprié afin de ne pas modifier la réaction existante ou l'intégrité de l'épiderme.
1.5.1. Essai de maximalisation chez le cobaye (GMPT)
1.5.1.1. Préparation
De jeunes cobayes albinos, sains, sont acclimatés aux conditions du laboratoire pendant au moins cinq jours avant le début de l'essai. Avant l'essai, les animaux sont répartis au hasard en lots à traiter et lots témoins. Les poils sont tondus, rasés ou épilés chimiquement selon la méthode utilisée. Il faut veiller à ne pas érafler la peau. Les animaux sont pesés avant le début et à la fin de l'essai.
1.5.1.2. Conditions de l'essai
1.5.1.2.1. Animaux d'expérience
Souches de cobayes albinos couramment utilisées en laboratoire.
1.5.1.2.2. Nombre et sexe
On peut utiliser des animaux de l'un ou l'autre sexe. Si l'on utilise des femelles, elles doivent être nullipares et non gravides.
Le lot traité comprend au minimum 10 animaux et le lot témoin, au moins 5. Si le lot traité comprend moins de 20 animaux et le lot témoin moins de 10, et qu'il n'est pas possible de conclure que la substance à tester est un agent sensibilisant. Dans ce cas, il est fortement recommandé d'utiliser d'autres animaux afin d'arriver à un total d'au moins 20 animaux d'essai et 10 animaux témoins.
1.5.1.2.3. Doses
La concentration de substance utilisée pour chaque exposition d'induction doit être bien tolérée par l'organisme des animaux et doit correspondre à la concentration maximale entraînant une irritation cutanée légère à modérée. La concentration utilisée pour l'exposition de déclenchement doit correspondre à la concentration maximale n'entraînant pas d'irritation. Si nécessaire, les concentrations appropriées peuvent être déterminées à partir d'un essai pilote impliquant deux ou trois animaux. À cet effet, il convient d'utiliser des animaux traités par l'adjuvant complet de Freund.
1.5.1.3. Mode opératoire
1.5.1.3.1. Induction
Jour 0 — lot traité
Trois séries de deux injections intradermiques d'un volume de 0,1 ml chacune sont pratiquées dans la région scapulaire préalablement débarrassée de ses poils, de part et d'autre de la ligne médiane.
Injection 1: mélange 1:1 (v/v) adjuvant complet de Freund/eau ou sérum physiologique
Injection 2: la substance à tester dans un véhicule adéquat, à la concentration sélectionnée
Injection 3: la substance à tester à la concentration voulue, mélangée dans un rapport 1:1 (v/v) à de l'adjuvant complet de Freund ou du sérum physiologique.
Pour l'injection 3, les substances hydrosolubles sont dissoutes dans la phase aqueuse avant d'être mélangées avec l'adjuvant complet de Freund. Les substances liposolubles ou insolubles sont d'abord mises en suspension dans 1 adjuvant complet de Freund, puis mises en phase aqueuse. La concentration finale de la substance à tester doit être égale à celle utilisée pour l'injection 2.
Les injections 1 et 2 sont pratiquées à proximité l'une de l'autre et le plus près possible de la tête, tandis que l'injection 3 est effectuée vers la partie caudale de la zone d'essai.
Jour 0 — lot témoin
Trois séries de deux injections intradermiques d'un volume de 0,1 ml chacune sont réalisées aux mêmes endroits que chez les animaux traités.
Injection 1: mélange 1:1 (v/v) adjuvant complet de Freund/eau ou sérum physiologique
Injection 2: le véhicule non dilué
Injection 3: formulation à 50 % du véhicule dans un mélange 1:1 d'adjuvant complet de Freund/eau ou sérum physiologique.
5e — 7e jour — lot traité et lot témoin
Environ 24 heures avant l'application topique d'induction et si la substance ne provoque pas d'irritation cutanée, la zone d'essai rasée et/ou tondue est badigeonnée avec 0,5 ml de lauryl sulfate de sodium à 10 % dans de la vaseline, afin de créer une irritation locale.
6e — 8e jour — lot traité
La zone d'essai est à nouveau débarrassée de ses poils. Un papier filtre (2 x 4 cm) imprégné de la substance dans le véhicule approprié est appliqué sur la zone d'essai et maintenu en contact avec la peau à l'aide d'un pansement occlusif pendant 48 heures. Le choix du véhicule doit être justifié. Les substances solides sont réduites en poudre fine et incorporées dans un véhicule approprié; les substances liquides peuvent éventuellement être appliquées directement.
6e — 8e jour — lot témoin
La zone d'essai est à nouveau débarrassée de ses poils. Le véhicule seul est appliqué comme indiqué précédemment sur la zone d'essai et maintenu en contact avec la peau pendant 48 heures à l'aide d'un pansement occlusif.
1.5.1.3.2. Déclenchement
20e — 22ème jour — lot traité et lot témoin
Les flancs des animaux traités et des animaux témoins sont débarrassés de leurs poils. Un patch ou une cupule chargés de la substance à tester est appliquée sur un des flancs des animaux et, s'il y a lieu, un patch ou une cupule imprégnés uniquement du véhicule est appliquée sur l'autre flanc. Les pastilles sont maintenues en contact avec la peau pendant 24 heures à l'aide d'un pansement occlusif.
1.5.1.3.3. Observation et cotation: lot traité et lot témoin
|
—
|
Environ 21 heures après le retrait de la pastille, la zone étudiée est nettoyée et rasée et/ou tondue et épilée si nécessaire;
|
|
—
|
3 heures plus tard environ (à peu près 48 heures après le début de l'application de déclenchement), la réaction cutanée est observée et cotée d'après l'échelle indiquée en annexe;
|
|
—
|
environ 24 heures après cette observation, on procède à une seconde observation (72 heures) et à une nouvelle cotation.
|
Il est recommandé de procéder à une lecture en aveugle chez les animaux traités et les animaux témoins.
S'il est nécessaire de préciser les résultats obtenus lors de la première exposition de déclenchement, une seconde exposition de déclenchement (c'est-à-dire redéclenchement), si nécessaire avec un nouveau lot témoin, sera envisagée environ une semaine après la première exposition. La nouvelle exposition de déclenchement peut également être effectuée sur le lot témoin initial.
Toutes les réactions cutanées et toutes les observations inhabituelles, y compris les réactions systémiques, résultant d'une exposition d'induction ou de déclenchement, doivent être observées et cotées d'après l'échelle de Magnusson/Kligman (voir annexe). D'autres techniques telles qu'un examen histopathologique ou la mesure de l'épaisseur du pli cutané peuvent être utilisées pour préciser les réactions douteuses.
1.5.2. Essai de Buehler
1.5.2.1. Préparation
De jeunes cobayes albinos, sains, sont acclimatés aux conditions du laboratoire pendant au moins cinq jours avant le début de l'essai. Avant l'essai, les animaux sont répartis au hasard en lots à traiter et lots témoins. Les poils sont coupés, rasés ou éventuellement épilés chimiquement selon la méthode utilisée. Il faut veiller à ne pas érafler la peau. Les animaux sont pesés avant le début et à la fin de l'essai.
1.5.2.2. Conditions de l'essai
1.5.2.2.1. Animaux d'expérience
Souches de cobayes albinos couramment utilisées en laboratoire.
1.5.2.2.2. Nombre et sexe
On peut utiliser des animaux de l'un ou l'autre sexe. Si l'on utilise des femelles, elles doivent être nullipares et non gravides.
Le lot traité comprend au minimum 20 animaux et le lot témoin, au moins 10.
1.5.2.2.3. Doses
Pour chaque exposition d'induction, la concentration de substance à utiliser est la concentration maximale entraînant une irritation modérée mais non excessive. Pour l'exposition de déclenchement, la concentration à utiliser est la concentration maximale n'entraînant pas d'irritation. Si nécessaire, les concentrations appropriées peuvent être déterminées à partir d'un essai pilote impliquant deux ou trois animaux.
Pour les substances hydrosolubles, il convient d'utiliser comme véhicule de l'eau ou une solution diluée non irritante de surfactant. Pour les autres substances, il est préférable d'utiliser un mélange d'éthanol à 80 % dans de l'eau pour l'induction, et de l'acétone pour le déclenchement.
1.5.2.3. Mode opératoire
1.5.2.3.1. Induction
Jour 0 — lot traité
Un des flancs des animaux est rasé. La compresse servant à réaliser le test est imprégnée de la substance à tester incorporée dans un véhicule approprié (le choix du véhicule doit être justifié; les substances liquides peuvent au besoin être appliquées directement). La compresse est appliquée sur la zone d'essai et maintenue en contact avec la peau pendant 6 heures à l'aide d'un pansement occlusif ou d'une cupule et d'un pansement approprié.
Le système doit être occlusif. Un tampon de ouate rond ou carré de 4 à 6 cm2 convient. Il est préférable d'utiliser un dispositif de contention approprié pour garantir l'occlusion. Si l'on utilise des bandages, des expositions supplémentaires peuvent s'avérer nécessaires.
Jour 0 — lot témoin
Un des flancs des animaux est rasé. Le véhicule seul est appliqué de la façon décrite pour les animaux traités. La compresse servant à réaliser le test est maintenue en contact avec la peau pendant 6 heures à l'aide d'un pansement occlusif ou d'une cupule et d'un pansement approprié. S'il peut être prouvé qu'il n'est pas nécessaire de simuler le traitement sur le lot témoin, cette étape peut être omise, un contrôle ne recevant rien peut être utilisé.
6e — 8e jour et 13e — 15e jour — lot traité et lot témoin
La même application que celle décrite au jour 0 est réalisée sur la même zone (débarrassée de ses poils si nécessaire) du même flanc au 6e — 8e jour, puis à nouveau au 13e — 15e jour.
1.5.2.3.2. Déclenchement
27e — 29e jour — lot traité et lot témoin
Le flanc non traité des animaux du lot traité et du lot témoin est rasé. Une compresse occlusive ou une cupule contenant la quantité appropriée de la substance à tester, à la concentration maximale n'entraînant pas d'irritation, est appliquée sur la partie postérieure du flanc non traité des animaux du lot traité et du lot témoin.
S'il y a lieu, on appliquera également une compresse occlusive ou une cupule ne contenant que le véhicule sur la partie antérieure du flanc non traité des animaux du lot traité et du lot témoin. Les compresses ou cupules sont maintenues en contact avec la peau à l'aide d'un pansement approprié pendant 6 heures.
1.5.2.3.3. Observations et cotation
|
—
|
Environ 21 heures après le retrait de la compresse, la zone d'essai est débarrassée de ses poils;
|
|
—
|
environ trois heures plus tard (à peu près 30 heures après le début de l'exposition de déclenchement), la réaction cutanée est observée et cotée d'après l'échelle indiquée en annexe;
|
|
—
|
environ 24 heures après cette observation (soit environ 54 heures après l'exposition de déclenchement), la réaction cutanée est à nouveau observée et cotée.
|
Il est recommandé d'effectuer une lecture en aveugle de la réaction dans le lot traité et le lot témoin.
S'il est nécessaire de préciser les résultats obtenus lors de la première exposition de déclenchement, une seconde exposition de déclenchement, si nécessaire avec un nouveau lot témoin, sera envisagée environ une semaine après la première exposition. La nouvelle exposition de déclenchement peut également être effectuée sur le lot témoin initial.
Toutes les réactions cutanées et toutes les observations inhabituelles, y compris les réactions systémiques, résultant d'une exposition d'induction ou de déclenchement (c'est-à-dire de redéclenchement), doivent être consignées et cotées d'après l'échelle de Magnusson/Kligman (voir annexe). D'autres techniques telles qu'un examen histopathologique ou la mesure de l'épaisseur du pli cutané peuvent être utilisées pour préciser les réactions douteuses.
2. RÉSULTATS (GMPT ET ESSAI DE BUEHLER)
Les résultats sont récapitulés sous forme de tableaux indiquant, pour chaque animal, la réaction cutanée lors de chaque observation.
3. COMPTE RENDU (GMPT ET ESSAI DE BUEHLER)
Lorsqu'un test de dépistage [par exemple essai local sur ganglions lymphatiques (LLNA), essai de gonflement de l'oreille de souris (MEST)], est effectué préalablement à l'essai sur le cobaye, il convient d'en fournir la description ou la référence, ainsi que les détails du mode opératoire et les résultats obtenus avec la substance à tester et les substances de référence.
Procès-verbal d'essai (GMPT et essai de Buehler)
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
|
Animaux d'expérience:
|
—
|
souche de cobayes utilisée,
|
|
—
|
nombre, âge et sexe des animaux,
|
|
—
|
origine, conditions d'hébergement, régime alimentaire, etc.,
|
|
—
|
poids de chaque animal au début de l'essai.
|
|
|
|
Conditions expérimentales:
|
—
|
technique de préparation du site d'application,
|
|
—
|
détails sur les matériaux utilisés pour l'application et la technique d'application,
|
|
—
|
résultats de l'étude pilote et conclusions concernant les concentrations d'induction et de déclenchement à utiliser dans l'essai,
|
|
—
|
détails concernant la préparation, l'application et l'élimination de la substance à tester,
|
|
—
|
justification du choix du véhicule,
|
|
—
|
concentrations du véhicule et de la substance à tester utilisées pour les expositions d'induction et de déclenchement, et quantité totale de substance appliquée pour l'induction et le déclenchement.
|
|
|
|
Résultats:
|
—
|
résumé des résultats du dernier contrôle de sensibilité et de fiabilité (voir 1.3), y compris informations sur la substance, la concentration et le véhicule utilisés,
|
|
—
|
toute observation effectuée sur chaque animal, cotations comprises,
|
|
—
|
description de la nature et de la gravité des effets observés,
|
|
—
|
toute observation histopathologique.
|
|
4. RÉFÉRENCES
Cette méthode reprend la ligne directrice 406 de l'OCDE.
Appendice
TABLEAU
échelle de Magnusson et Kligman pour la cotation des réactions cutanées à une exposition de déclenchement
0 = pas de modification apparente
1 = érythème discret ou localisé
2 = érythème modéré et confluent
3 = érythème intense avec gonflement
B.7. TOXICITÉ (ORALE) PAR ADMINISTRATION RÉPÉTÉE (28 JOURS)
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance à tester est administrée quotidiennement, par voie orale, à doses croissantes, à plusieurs lots d'animaux d'expérience, à raison d'une dose par lot pendant 28 jours. Pendant la période d'administration, les animaux sont observés attentivement chaque jour pour déceler les éventuels signes de toxicité. Les animaux qui meurent ou qui sont sacrifiés en cours d'expérience ainsi que ceux qui survivent jusqu'à! a fin de l'essai sont autopsiés.
Cette méthode insiste davantage sur les effets neurologiques; les observations cliniques doivent être effectuées avec grand soin pour obtenir le maximum d'informations possible. La méthode vise à identifier les produits chimiques ayant un potentiel neurotoxique qui pourrait nécessiter des études plus approfondies. En outre, cette méthode peut fournir des indications sur les effets immunologiques et sur la toxicité de la substance pour les organes de la reproduction.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparation
De jeunes animaux adultes, sains, sont répartis au hasard en lots traités et lots témoins. Les cages seront disposées de façon à minimiser les éventuels effets dus à l'emplacement des cages. Les animaux sont identifiés individuellement et maintenus dans leur cage pendant au moins cinq jours avant le début de l'étude, pour leur permettre de s'acclimater aux conditions régnant dans le laboratoire.
La substance à tester est administrée par gavage ou dans l'alimentation ou l'eau de boisson. La méthode d'administration orale dépend du but de l'étude et des propriétés physico-chimiques de la substance.
Si nécessaire, la substance à tester est dissoute ou mise en suspension dans un véhicule approprié. Chaque fois que possible, on utilisera plutôt une solution/suspension aqueuse, ou sinon une solution/émulsion huileuse (par exemple huile de maïs) et en dernier lieu d'autres véhicules. Si le véhicule utilisé n'est pas de l'eau, ses caractéristiques toxiques doivent être connues. Il convient de déterminer la stabilité de la substance à tester dans le véhicule.
1.4.2. Conditions de l'essai
1.4.2.1. Animaux d'expérience
Les expériences sont de préférence effectuées sur le rat, mais d'autres espèces de rongeurs peuvent être utilisées. Les animaux adultes jeunes et sains sont issus de souches couramment utilisées en laboratoires. Les femelles doivent être nullipares et non gravides. L'administration de la substance doit débuter le plus tôt possible après le sevrage, et dans tous les cas, avant que les animaux aient atteint l'âge de neuf semaines.
Au début de l'étude, l'écart de poids entre les animaux doit être minime et ne doit pas dépasser — 20 % du poids moyen de chaque sexe.
Lorsqu'une étude par administration orale répétée est effectuée préalablement à une étude à long terme, il est préférable d'utiliser des animaux de même souche et de même origine pour les deux études.
1.4.2.2. Nombre et sexe
Dix animaux au moins (5 femelles et 5 mâles) sont utilisés pour chaque dose. Si le protocole expérimental prévoit des sacrifices en cours d'étude, le nombre d'animaux doit être augmenté du nombre de sacrifices prévus.
En outre, un lot satellite de 10 animaux (cinq de chaque sexe) peut être traité à la dose la plus élevée pendant 28 jours et placé en observation pendant 14 jours après l'arrêt du traitement pour étudier la réversibilité, la persistance ou l'apparition tardive des effets toxiques. Un lot satellite de 10 animaux témoins est également utilisé.
1.4.2.3. Niveau des doses
On utilise en général au moins trois lots d'essai et un lot témoin. A l'exception de la substance à tester, les animaux du lot témoin reçoivent le même traitement que les animaux des lots traités. Si un véhicule est utilisé pour administrer la substance à tester, le volume maximal de ce véhicule sera administré au lot témoin.
S'il ressort de l'évaluation d'autres données que l'administration d'une dose de 1 000 mg/kg/j ne devrait pas entraîner d'effets, il est possible d'effectuer un essai de limite. En l'absence de données pertinentes, on peut procéder à une étude exploratoire pour aider à déterminer les doses à utiliser.
Les doses doivent être choisies en tenant compte de toutes les données disponibles concernant la toxicité et les aspects toxicocinétiques de la substance à tester ou des substances de structure analogue. La dose la plus élevée doit produire des effets toxiques sans entraîner la mort ni des souffrances intenses. Une série de doses décroissantes est ensuite choisie, dans le but de mettre en évidence une relation entre la réaction et la dose administrée et d'objectiver une absence d'effets adverses à la dose la plus faible (dose sans effet adverse observé). L'écart optimal entre deux doses est souvent un facteur 2 à 4; il est préférable de prévoir une quatrième dose plutôt que d'avoir un écart excessif entre deux doses (plus qu'un facteur 10).
Pour les substances qui sont administrées dans l'alimentation ou dans l'eau de boisson, il est important de vérifier que les quantités de substance d'essai administrées n'interfèrent pas sur la nutrition normale et l'équilibre hydrique. Si la substance est administrée dans l'alimentation, on peut utiliser une concentration alimentaire constante (ppm) ou une dose constante par rapport au poids de l'animal; la solution choisie doit être précisée. Si la substance est administrée par gavage, la dose doit être administrée chaque jour à heure fixe et si nécessaire ajustée pour rester constante par rapport au poids de l'animal.
Lorsqu'une étude par administration orale répétée est effectuée préalablement à une étude à long terme, le régime alimentaire doit être identique dans les deux études.
1.4.2.4. Essai limite
Lorsqu'un essai réalisé selon les méthodes décrites dans la présente étude, à une dose d'au moins 1 000 mg/kg poids corporel/jour, ou en cas d'administration dans l'alimentation ou l'eau de boisson, à une concentration équivalente (en fonction du poids corporel), ne produit pas d'effets toxiques observables et que les données relatives à des substances de structure apparentée ne laissent pas présumer une toxicité, il peut s'avérer inutile d'effectuer une étude complète sur trois doses. Dans ce cas, un essai limite se justifie, sauf si l'exposition humaine fait apparaître la nécessité d'utiliser une dose plus élevée.
1.4.2.5. Période d'observation
La période d'observation dure 28 jours. Les animaux des lots satellites prévus pour des observations complémentaires seront maintenus en observation, sans aucun traitement, pendant 14 jours supplémentaires au moins, afin de détecter l'apparition retardée, la persistance ou la réversibilité des effets toxiques.
1.4.3. Mode opératoire
La substance à tester est administrée aux animaux sept jours sur sept pendant 28 jours. Si la substance n'est administrée qu'à raison de cinq jours par semaine, ce choix doit être justifié. En cas d'administration par gavage, la dose doit être administrée en une fois à l'aide d'une sonde œsophagienne ou d'une canule à intubation appropriée. Le volume maximal de liquide pouvant être administré en une fois dépend de la taille de l'animal. Ce volume ne doit pas excéder 1 ml/100 g de poids corporel, mais peut atteindre 2 ml/100 g de poids corporel lorsqu'il s'agit de solutions aqueuses. Sauf en ce qui concerne les substances irritantes ou corrosives pour lesquelles une augmentation de la concentration entraînerait des effets exacerbés, la variabilité des volumes d'essai doit être minimisée par un ajustement des concentrations, afin que le volume reste constant quelle que soit la dose.
1.4.3.1. Observation générale
L'observation clinique générale doit être effectuée au moins une fois par jour, de préférence au(x) même(s) moment(s) de la journée, en fonction de la période post-administration où les effets sont les plus marqués. L'état de santé des animaux doit être consigné. Au moins deux fois par jour, tous les animaux sont examinés pour déterminer la morbidité et la mortalité. Les animaux moribonds ou présentant des signes de détresse ou de souffrance intenses sont immédiatement retirés, euthanasiés et autopsiés.
Tous les animaux sont soumis à une observation clinique détaillée une fois avant la première exposition (pour permettre des comparaisons sur un même sujet) et au moins une fois par semaine par la suite. Ces observations doivent être effectuées sur les animaux sortis de leur cage et placés dans une enceinte standard, et doivent de préférence avoir lieu toujours au même moment. Les observations doivent être soigneusement consignées, de préférence à l'aide de systèmes de notation explicitement définis par le laboratoire d'essai. Il convient de veiller à ce que les conditions expérimentales varient le moins possible, il est souhaitable que les personnes qui effectuent les observations ne soient pas informées du traitement administré. Les observations doivent porter, entre autres, sur les modifications de la peau, du poil, des yeux, des muqueuses, des sécrétions et excrétions et de l'activité autonome (par exemple larmoiement, hérissement du poil, respiration inhabituelle). Il convient également de noter tout changement de comportement, de posture ou de réaction à la manipulation, ainsi que la présence de mouvements cloniques ou toniques, comportements stéréotypés (par exemple, toilettage excessif, animaux qui tournent en rond de façon répétitive) ainsi que tout comportement bizarre (automutilation, marche à reculons).
Au cours de la quatrième semaine d'exposition, la réactivité aux différents stimuli sensoriels (auditifs, visuels et proprioceptifs), ainsi que la force de préhension et l'activité motrice sont évaluées. Les méthodes utilisables à cet effet sont détaillées dans les publications de référence (voir introduction générale, partie B).
Les observations fonctionnelles de la quatrième semaine d'exposition peuvent être omises si l'étude est effectuée préalablement à une étude subchronique (sur 90 jours). Dans ce cas, les observations fonctionnelles doivent faire partie de l'étude complémentaire. En revanche, l'existence de données résultant des observations fonctionnelles effectuées lors de l'étude préliminaire par administration réitérée peut faciliter le choix des doses pour l'étude subchronique complémentaire.
Exceptionnellement, les observations fonctionnelles peuvent également être omises pour les lots qui montrent des signes de toxicité tels que les examens fonctionnels perdraient de leur valeur.
1.4.3.2. Poids corporel et consommation de nourriture et d'eau
Tous les animaux doivent être pesés au moins une fois par semaine. La consommation d'aliments et d'eau doit être mesurée au minimum une fois par semaine. Si la substance est administrée dans l'eau de boisson, la consommation d'eau doit également être mesurée une fois par semaine au minimum.
1.4.3.3. Hématologie
Les examens hématologiques suivants doivent être réalisés à la fin de la période d'essai: hématocrite, concentration en hémoglobine, numération des hématies et des leucocytes et formule leucocytaire, numération des plaquettes et mesure du temps/potentiel de coagulation.
Les échantillons de sang doivent être prélevés en un point déterminé, juste avant ou pendant le sacrifice des animaux, et conservés dans des conditions appropriées.
1.4.3.4. Biochimie clinique
Des analyses biochimiques cliniques visant à étudier les principaux effets toxiques sur les tissus et en particulier sur le foie et les reins doivent être effectuées sur les échantillons de sang prélevés sur tous les animaux juste avant ou pendant le sacrifice (à l'exception des animaux retrouvés moribonds ou sacrifiés en cours d'essai). Il est préférable que les animaux soient à jeun depuis la veille (2). Ces déterminations effectuées dans le sérum ou le plasma concernent notamment le sodium, le potassium, le glucose, le cholestérol total, l'urée, le créatinine, les protéines totales et l'albumine, et au moins deux enzymes révélatrices des effets hépatocellulaires (telles que l'alanine, aminotransférase, l'aspartate aminotransférase, la phosphatase alcaline, la gamma-glutamyl transpeptidase et la sorbitol-deshydrogénase). La détermination d'autres enzymes (d'origine hépatique ou autre) ainsi que des acides biliaires peut dans certains cas fournir des informations utiles.
Éventuellement, des analyses d'urine peuvent être effectuées au cours de la dernière semaine de l'étude, comprenant un recueil du volume urinaire en un temps donné: aspect, volume, osmolalité ou densité, pH, protéines, glucose, de sang ou de cellules sanguines.
En outre, des études portant sur les marqueurs sériques des lésions tissulaires doivent être considérées. Les autres déterminations éventuellement nécessaires lorsque les propriétés de la substance à tester sont susceptibles de modifier les profils métaboliques concernant le calcium, le phosphate, les triglycérides à jeun, les hormones spécifiques, le méthémoglobine et la cholinestérase. Ces analyses devront être effectuées systématiquement pour les substances appartenant à certaines classes et au cas par cas pour les autres.
Dans l'ensemble, la démarche adoptée doit être souple et pouvoir être adaptée en fonction de l'espèce utilisée et des effets observés et/ou prévisibles de la substance considérée.
Si les données historiques de base recueillies antérieurement sont insuffisantes, les paramètres hématologiques et biochimiques devront être déterminés avant le début de l'expérience.
1.4.3.5. Autopsie
Tous les animaux de l'étude doivent faire l'objet d'une autopsie détaillée comportant un examen approfondi de la surface externe du corps, de tous les orifices ainsi que des cavités crânienne, thoracique et abdominale et de leur contenu. Le foie, les reins, les glandes surrénales, les testicules, les épididymes, le thymus, la rate, le cerveau et le cœur de tous les animaux seront débarrassés de tous les tissus adhérents et pesés le plus rapidement possible pour éviter le dessèchement.
Les tissus ci-après seront conservés dans le milieu de fixation le plus approprié en fonction du type de tissu et des examens histopathologiques prévus: tout tissu présentant des lésions macroscopiques, l'encéphale (régions représentatives: cerveau, cervelet et protubérance annulaire), la moelle épinière, l'estomac, le colon et l'intestin grêle (y compris plaques de Peyer), le foie, les reins, les surrénales, la rate, le cœur, le thymus, la thyroïde, la trachée et les poumons (conservés par insufflation d'un fixateur et immersion), les gonades, les organes génitaux annexes (par exemple: utérus, prostate), la vessie, les ganglions lymphatiques (de préférence un ganglion sur le trajet de la voie d'administration et un autre à distance pour couvrir les effets systématiques), nerfs périphériques (sciatique ou tibial) de préférence très près du muscle, et une coupe de moelle osseuse (ou lame fraîchement montée de moelle recueillie par aspiration). En fonction des observations cliniques et des autres résultats, il peut s'avérer nécessaire d'examiner d'autres tissus. Tous les organes considérés comme organes cibles potentiels du fait des propriétés de la substance doivent également être conservés.
1.4.3.6. Examen histopathologique
Pour tous les animaux du lot témoin et du lot traité à la dose la plus élevée, il est pratiqué un examen histopathologique complet des organes et tissus conservés. Cet examen devra être pratiqué pour tous les lots exposés à des doses inférieures si des modifications induites par la substance à tester sont observées chez les animaux du lot traité à la dose la plus élevée.
Toutes les lésions macroscopiques doivent être examinées.
Lorsqu'un lot satellite est utilisé, un examen histopathologique doit être pratiqué sur les organes et tissus sur lesquels des effets ont été observés dans les lots traités.
2. RÉSULTATS
Les résultats doivent être indiqués pour chaque animal, individuellement. En outre, tous les résultats doivent être récapitulés sous forme de tableaux indiquant, pour chaque lot d'expérience, le nombre d'animaux utilisé, le nombre d'animaux retrouvés morts au cours de l'essai ou sacrifiés pour des raisons humanitaires, le moment de la mort ou du sacrifice, le nombre d'animaux présentant des signes de toxicité ainsi qu'une description de ces signes, le moment d'apparition, leur durée et leur gravité, le nombre d'animaux présentant des lésions, le type de lésion et le pourcentage d'animaux présentant chaque type de lésion.
Si possible, les résultats numériques seront évalués au moyen d'une méthode statistique appropriée et reconnue. La méthode statistique doit être choisie lors de la conception de l'étude.
3. COMPTE RENDU
PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les informations suivantes:
|
|
Animaux d'expérience:
|
—
|
espèce/souche utilisée,
|
|
—
|
nombre d'animaux, âge et sexe,
|
|
—
|
origine, conditions d'hébergement, régime alimentaire, etc.,
|
|
—
|
poids de chaque animal au début de l'essai, à intervalles d'une semaine par la suite et à la fin de l'essai.
|
|
|
|
Conditions expérimentales:
|
—
|
justification du choix du véhicule si autre que de l'eau,
|
|
—
|
justification du choix de la dose,
|
|
—
|
détails concernant la formulation/préparation de la substance, la concentration obtenue, la stabilité et l'homogénéité de la préparation,
|
|
—
|
détails concernant l'administration de la substance,
|
|
—
|
le cas échéant, conversion de la concentration de la substance dans la ration alimentaire/l'eau de boisson (ppm) en dose effective (mg/kg p.c./jour),
|
|
—
|
détails sur la qualité de la nourriture et de l'eau.
|
|
|
|
Résultats:
|
—
|
poids corporel/modifications du poids corporel,
|
|
—
|
consommation d'aliments et, le cas échéant, consommation d'eau,
|
|
—
|
réponse toxique par sexe et par dose, signes de toxicité compris,
|
|
—
|
nature, gravité et durée des effets cliniques observés (réversibilité éventuelle),
|
|
—
|
évaluation de l'activité sensorielle, de la force de préhension et de l'activité motrice,
|
|
—
|
tests hématologiques et valeurs de référence applicables,
|
|
—
|
tests de biochimie clinique et valeurs de référence applicables,
|
|
—
|
poids corporel au moment du sacrifice et poids des organes,
|
|
—
|
résultats de l'autopsie,
|
|
—
|
description détaillée de tous les résultats de l'examen histopathologique,
|
|
—
|
données relatives à l'absorption si disponibles,
|
|
—
|
le cas échéant, traitement statistique des résultats.
|
|
4. RÉFÉRENCES
Cette méthode reprend la ligne directrice 407 de l'OCDE.
B.8. TOXICITÉ À DOSES RÉPÉTÉES (28 JOURS) (INHALATION)
1. MÉTHODE
1.1. INTRODUCTION
Il est utile de disposer d'informations préliminaires sur la distribution de la taille des particules, la pression de vapeur, le point de fusion, le point d'ébullition, le point d'éclair et l'explosivité (si applicables) de la substance.
Voir également l'introduction générale, partie B (point A).
1.2. DÉFINITIONS
Voir introduction générale partie B (point B).
1.3. SUBSTANCES DE RÉFÉRENCE
Aucune.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Plusieurs lots d'animaux d'expérience sont exposés quotidiennement, pendant une période déterminée, à des concentrations croissantes de la substance à tester, une seule concentration étant utilisée par lot et ce, pendant une durée de 28 jours. Lorsqu'on utilise un véhicule en vue d'obtenir une concentration adéquate de la substance d'essai dans l'atmosphère, il y a lieu de prévoir un lot témoin pour le véhicule. Durant la période d'administration, les animaux sont observés quotidiennement afin de déceler les symptômes de toxicité. Les animaux qui meurent en cours d'expérience sont autopsiés de même que ceux qui sont encore en vie à l'issue de l'expérience.
1.5. CRITÈRE DE QUALITÉ
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Préparation
Les animaux sont maintenus dans des conditions d'hébergement et d'alimentation propres à l'expérience au moins pendant les 5 jours qui la précèdent. Avant l'expérience, des animaux jeunes et sains sont répartis selon les règles du hasard entre les différents lots requis. Au besoin un véhicule approprié peut être utilisé pour obtenir une concentration adéquate de substance dans l'atmosphère. Si l'on utilise un véhicule ou un autre additif pour faciliter l'administration, celui-ci doit être réputé non toxique et cela peut être étayé par des données publiées, si nécessaire.
1.6.2. Conditions de l'essai
1.6.2.1. Animaux d'expérience
Sauf contre-indication, le rat est l'espèce utilisée de préférence. Il faut utiliser des animaux jeunes et sains d'une souche courante d'animaux de laboratoire.
Au début de l'expérience, l'intervalle de variation de poids entre les animaux utilisés ne doit pas excéder ± 20 % de la valeur moyenne requise.
1.6.2.2. Nombre et sexe
Dix animaux au moins (cinq femelles et cinq mâles) sont utilisés pour chaque lot. Les femelles doivent être nullipares et non-gravides. S'il est prévu dans le protocole expérimental des sacrifices intermédiaires en cours d'expérience, il y a lieu d'ajouter la quantité d'animaux nécessaire pour ces sacrifices. De plus, un lot satellite de 10 animaux (5 par sexe) peut être traité à la dose la plus élevée pendant 28 jours et faire l'objet d'une observation relative à la réversibilité, la persistance ou l'apparition tardive d'effets toxiques durant les 14 jours qui suivent l'arrêt du traitement. On utilise aussi un lot satellite témoin de 10 animaux (cinq animaux par sexe).
1.6.2.3. Concentration d'exposition
On utilise au moins trois concentrations ainsi qu'un témoin ou un lot témoin pour le véhicule (correspondant à la concentration de véhicule présente dans l'atmosphère inhalé par le groupe de traitement le plus élevé). Excepté l'exposition à la substance d'essai, les animaux du lot témoin doivent être traités de la même manière que les sujets des lots d'expérience. La concentration la plus élevée doit être suffisamment forte pour produire des effets toxiques mais sans toutefois entraîner la mort (ou rarement). La concentration la plus faible ne doit faire apparaître aucun effet toxique. Lorsque l'on dispose d'une estimation concernant l'exposition de l'homme, la concentration la plus faible doit être supérieure à cette valeur. Dans les conditions idéales, la concentration intermédiaire doit produire des effets toxiques minimaux observables. Si l'on utilise plusieurs concentrations intermédiaires, celles-ci doivent être espacées de façon à entraîner une gradation des effets toxiques. En cas de mortalité dans les lots à concentrations faibles et intermédiaires, ainsi que dans les lots témoins, celle-ci devra être minime pour que l'évaluation des résultats soit valable.
1.6.2.4. Durée d'exposition
L'exposition quotidienne doit être de 6 heures, mais d'autres durées peuvent se révéler nécessaires pour répondre à certaines exigences particulières.
1.6.2.5. Dispositif expérimental
Les animaux doivent être exposés à la substance d'essai au moyen d'un dispositif d'exposition dynamique capable de maintenir un flux d'air continu permettant au moins 12 renouvellements de l'atmosphère de l'enceinte d'exposition par heure, et assurant une teneur en oxygène suffisante et une répartition uniforme du produit à tester dans l'air. Si l'on utilise une chambre d'exposition, celle-ci doit être conçue de manière à éviter autant que possible l'entassement des animaux et à optimiser l'exposition par inhalation à la substance d'essai. En règle générale, pour assurer la stabilité de l'atmosphère d'une chambre, le «volume» total des animaux d'expérience ne doit pas dépasser 5 % du volume de la chambre d'essai. On peut aussi avoir recours à un système d'exposition soit oro-nasal, soit de la tète seule, soit du corps entier en chambre individuelle; les deux premiers types d'exposition présentent l'avantage de limiter la pénétration de la substance d'essai par d'autres voies.
1.6.2.6. Période d'observation
Les animaux d'expérience devront être observés quotidiennement, en vue de déceler les symptômes de toxicité, durant toute la période de traitement et de récupération. Le moment de la mort ainsi que celui auquel les symptômes de toxicité apparaissent et disparaissent doivent être notées.
1.6.3. Mode opératoire
Les animaux sont quotidiennement exposés a la substance à tester à raison de 5 à 7 jours par semaine pendant une période de 28 jours. Les animaux de tout groupe satellite destiné à des observations de réversibilité d'effet doivent être gardés en vie pendant 14 jours encore, sans traitement, afin de déceler la régression ou la persistance des effets toxiques. La température à laquelle s'effectue l'essai doit être maintenue à 22 oC ± 3 oC.
Dans les conditions idéales, l'humidité relative doit être maintenue entre 30 et 70 % mais, dans certains cas, cela peut se révéler impossible (par exemple, essai de certains aérosols). Le maintien d'une pression légèrement négative à l'intérieur de la chambre (≤ 5 mm d'eau) empêchera la fuite de substance d'essai vers l'extérieur. La nourriture et l'eau doivent être retirées pendant l'exposition.
Il y a lieu d'utiliser un système d'inhalation qui fonctionne dans des conditions dynamiques comportant un dispositif approprié de contrôle analytique de la concentration. Pour établir les concentrations d'exposition appropriées, il est recommandé de procéder à un essai préliminaire. Le débit devra être réglé ajusté pour assurer des concentrations homogènes dans toute la chambre. Le système doit permettre d'obtenir des conditions d'exposition stables aussi rapidement que possible.
II y a lieu de mesurer ou de contrôler:
|
a)
|
le débit d'air (en permanence).
|
|
b)
|
la concentration réelle de la substance d'essai. Elle doit être mesurée dans la zone de respiration; pendant la période d'exposition quotidienne, la concentration ne doit pas varier de plus de ± 15 % par rapport à la valeur moyenne. Toutefois, dans le cas de certains aérosols, ce degré de contrôle peut ne pas être obtenu et un écart plus grand est alors acceptable. Durant toute la durée de l'étude, il faut garder les concentrations aussi constantes que possible d'un jour à l'autre. En ce qui concerne les aérosols, la taille des particules doit être analysée au moins une fois par semaine pour chaque groupe d'exposition.
|
|
c)
|
la température et l'humidité, en permanence si possible.
|
Les observations ont lieu pendant et après l'exposition et sont notées systématiquement; une fiche individuelle doit être établie pour chaque animal. Tous les animaux doivent être observés quotidiennement et les signes de toxicité, y compris leur moment d'apparition, leur intensité et leur durée doivent être enregistrés. Les observations doivent concerner les modifications de la peau et des poils, des yeux, des muqueuses, de l'appareil respiratoire, du système circulatoire ainsi que des systèmes nerveux autonome et central, de l'activité somatomotrice et du comportement. Les animaux sont pesés toutes les semaines. Il est aussi recommandé d'effectuer des mesures hebdomadaires sur la consommation alimentaire. Il est nécessaire d'observer régulièrement les animaux afin de veiller à ce que, dans la mesure du possible, ceux-ci ne soient pas perdus pour l'expérience pour des raisons telles que cannibalisme, autolyse des tissus ou erreur au cours des remises en cage. A l'issue de l'expérience, tous les animaux survivants, appartenant aux lots traités, non satellites, sont autopsiés. Les animaux trouvés moribonds ou dans un état de détresse ou de douleur intenses au cours de l'essai doivent être immédiatement retirés, euthanasiés et autopsiés.
Les examens figurant ci-après seront effectués à l'issue de la période d'essai sur tous les animaux (y compris les témoins):
|
1.
|
examen hématologique comprenant au moins l'hématocrite, la concentration en hémoglobine, le dénombrement des érythrocytes et des leucocytes, la formule leucocytaire ainsi qu'une étude de la coagulation;
|
|
2.
|
examen biochimique clinique du sang comprenant au moins un paramètre des fonctions hépatiques et rénales: alanine aminotransférase sérique (anciennement connue sous le nom de transaminase glutamopyruvique) aspartate aminotransférase sérique (anciennement connue sous le nom de transaminase glutamo-oxalo-acétique), urée, albumine, créatinine, bilirubine totale et protéines sériques totales.
|
Les autres déterminations éventuellement nécessaires à une évaluation toxicologique adéquate concernent le calcium, le phosphore, les chlorures, le sodium, le potassium, la glycémie à jeun, l'analyse des lipides, les hormones, l'équilibre acido-basique, la méthémoglobine et l'activité cholinestérasique.
D'autres analyses biochimiques cliniques peuvent, si nécessaire, être effectuées pour approfondir l'étude des effets observés.
1.6.3.1. Autopsie
Tous les animaux soumis à l'essai doivent faire l'objet d'une autopsie macroscopique. Au moins le foie, les reins, les glandes surrénales, les poumons et les testicules doivent être pesés à l'état humide le plus rapidement possible après la dissection afin d'éviter le dessèchement. Organes et tissus (système respiratoire, foie, reins, rate, testicules, glandes surrénales, cœur et tout autre organe présentant des lésions macroscopiques ou des modifications de leur volume) doivent être conservés dans un milieu approprié en vue éventuels d'examens histopathologiques ultérieurs. Les poumons doivent être prélevés entiers, pesés et traités au moyen d'un fixateur approprié permettant de conserver la structure pulmonaire intacte.
1.6.3.2. Examen histopathologique
Pour le lot exposé à la dose la plus élevée et le(s) lot(s) témoin(s), il faut pratiquer un examen histologique systématique de tous les organes et des tissus conservés. Les organes et tissus qui auront présenté des lésions induites par la substance à tester au niveau a la dose la plus élevée, devront être examinés dans tous les lots exposés à toutes les doses inférieures. Les animaux de tout lot satellite devront faire l'objet d'un examen histologique particulièrement axé sur les organes et les tissus pour lesquels les lésions ont été constatées dans les autres lots traités.
2. DONNÉES
Les données doivent être récapitulées sous forme de tableaux indiquant, pour chaque lot d'expérience, le nombre d'animaux au début de l'essai et le nombre d'animaux présentant chaque type de lésion.
Tous les résultats observés doivent être évalués au moyen d'une méthode statistique appropriée. Toute méthode statistique reconnue peut être utilisée.
3. RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
espèce, souche, origine, conditions ambiantes, régime alimentaire, etc.,
|
|
—
|
conditions de l'essai:
Description du dispositif d'inhalation, y compris conception, type, dimensions, source d'air, système de génération d'aérosols, méthode de conditionnement d'air, traitement de l'atmosphère rejetée hors de l'enceinte et, le cas échéant, modalités d'hébergement des animaux en chambre d'essai. L'équipement de mesure de température, d'humidité et, s'il y a lieu, de la stabilité des concentrations des aérosols ou de la distribution de taille des particules doit être décrit.
Données relatives à l'exposition:
elles doivent être représentées sous la forme de tableaux indiquant des valeurs moyennes ainsi qu'une mesure de la variabilité (par exemple, écart type); elles doivent, si possible, inclure:
|
a)
|
les débits d'air dans le dispositif d'inhalation,
|
|
b)
|
la température et l'humidité de l'air,
|
|
c)
|
les concentrations nominales (quantité totale de substance d'essai introduite dans le dispositif d'inhalation, divisée par le volume d'air),
|
|
d)
|
le cas échéant, nature du véhicule,
|
|
e)
|
concentrations réelles dans la zone de respiration,
|
|
f)
|
le diamètre aérodynamique médian en masse (DAMM) et l'écart-type géométrique (ETG),
|
|
|
—
|
données concernant la réponse toxique par sexe et par concentration,
|
|
—
|
moment de la mort en cours d'expérience ou indication que les animaux ont survécu à l'expérience,
|
|
—
|
description des effets toxiques ou autres; concentration sans effet,
|
|
—
|
moment de l'observation de tout symptôme anormal et évolution de celui-ci,
|
|
—
|
données relatives à la prise de nourriture et à l'évolution du poids corporel,
|
|
—
|
examens hématologiques pratiqués et résultats complets,
|
|
—
|
examens biochimiques cliniques pratiqués et résultats complets,
|
|
—
|
description détaillée de tous les résultats histopathologiques,
|
|
—
|
traitement statistique des résultats, si possible,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale partie B (point D).
4. RÉFÉRENCES
Voir introduction générale partie B (point E).
B.9. TOXICITÉ À DOSES RÉPÉTÉES (28 JOURS) (ADMINISTRATION CUTANÉE)
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B (point A).
1.2. DÉFINITIONS
Voir introduction générale, partie B (point B).
1.3. SUBSTANCES DE RÉFÉRENCE
Aucune.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance à tester est appliquée quotidiennement, à doses croissantes, sur la peau de plusieurs lots d'animaux d'expérience pendant une période déterminée, à raison d'une seule dose par lot, pendant une durée de 28 jours. Durant la période d'application, les animaux sont observés quotidiennement afin de déceler les signes de toxicité. Les animaux qui meurent en cours d'expérience ainsi que ceux qui sont encore en vie à l'issue de l'expérience sont autopsiés.
1.5. CRITÈRE DE QUALITÉ
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Préparation
Les animaux sont maintenus dans des conditions d'hébergement et d'alimentation propres à l'expérience au minimum pendant les 5 jours qui la précèdent. Avant l'expérience, des animaux jeunes et sains sont répartis selon les règles du hasard entre les différents lots témoins et traités. Peu de temps avant l'essai, on tond la région dorsale du tronc des animaux. On peut avoir recours au rasage mais, dans ce cas, l'opération doit être effectuée 24 heures environ avant l'essai. Il est en général nécessaire de répéter les opérations de tonte ou de rasage à des intervalles d'une semaine environ, en évitant toute lésion de la peau. La surface à préparer pour l'application de la substance ne doit pas être inférieure à 10 % de la surface corporelle. Le poids de l'animal doit être pris en considération pour décider de la taille de la zone à épiler, et de la dimension de la surface à traiter. Lorsque le test concerne des substances solides qui, le cas échéant, peuvent être pulvérisées, la substance à tester doit être humectée au moyen d'eau ou, au besoin, d'un véhicule approprié, de manière à garantir un bon contact avec la peau. Les substances liquides sont généralement appliquées non diluées. On procède à une application quotidienne à raison de 5 à 7 jours par semaine.
1.6.2. Conditions de l'essai
1.6.2.1. Animaux d'expérience
On peut utiliser des rats, des lapins ou des cobayes adultes. On peut également utiliser d'autres espèces, mais il faut dans ce cas en justifier l'utilisation.
Au début de l'essai, l'intervalle de variation du poids des animaux utilisés ne doit pas excéder ± 20 % de la valeur moyenne requise.
1.6.2.2. Nombre et sexe
Dix animaux au moins (cinq femelles et cinq mâles), à la peau saine, sont utilisés pour chaque dose. Les femelles doivent être nullipares et non-gravides. S'il est prévu dans le protocole expérimental des sacrifices intermédiaires en cours d'expérience, il y a lieu d'ajouter la quantité d'animaux nécessaire pour ces sacrifices. De plus, un lot satellite de 10 animaux (5 par sexe) peut être traité à la dose la plus élevée pendant 28 jours et faire l'objet d'une observation relative à la réversibilité, la persistance ou l'apparition tardive d'effets toxiques durant les 14 jours qui suivent l'arrêt du traitement. On utilise aussi un lot satellite témoin de 10 animaux (cinq animaux par sexe).
1.6.2.3. Doses
On utilise au moins trois doses ainsi qu'un lot témoin ou, le cas échéant, un lot témoin traité avec le véhicule. La période d'exposition devra être d'au moins 6 heures par jour. La substance à tester doit être appliquée chaque jour au même moment et les quantités à administrer doivent faire l'objet d'une adaptation régulière (hebdomadaire ou bihebdomadaire) afin de conserver un niveau de dose constant par rapport au poids corporel des animaux. Excepté l'administration de substance d'essai, les animaux du lot témoin doivent être traités de la même manière que les animaux des lots d'expérience. Lorsqu'un véhicule est utilisé pour faciliter l'administration, celui-ci sera administré au lot témoin dans les mêmes conditions qu'aux lots traités et la quantité de véhicule correspondra à celle reçue par le groupe traité avec la dose de substance d'essai la plus élevée. La dose la plus élevée doit être suffisamment forte pour produire des effets toxiques mais sans toutefois entraîner la mort (ou rarement). La dose la plus faible ne doit faire apparaître aucun effet toxique. Lorsque l'on dispose d'une estimation concernant l'exposition de l'homme, la dose la plus faible doit être supérieure à cette valeur. Dans les conditions idéales, la concentration intermédiaire doit produire des effets toxiques minimaux observables. Si l'on utilise plusieurs concentrations intermédiaires, celles-ci doivent être espacées de façon à entraîner une gradation des effets toxiques. En cas de mortalité dans les lots recevant les doses faibles et intermédiaires, ainsi que dans les lots témoins, celle-ci devra être minime pour que l'évaluation des résultats soit valable.
Si l'application de la substance d'essai provoque une irritation cutanée grave, les concentrations doivent être réduites, ce qui peut entraîner une diminution, voire une disparition, des autres effets toxiques à la dose la plus élevée. De plus, si les lésions cutanées sont très graves, il peut se révéler nécessaire d'arrêter l'expérience et de la recommencer avec des concentrations plus faibles.
1.6.2.4. Essai «limite»
Si une expérience préliminaire réalisée avec une dose de 1 000 mg/kg de poids corporel ou avec une dose plus élevée en fonction de l'exposition possible pour l'homme, n'a provoqué aucun effet toxique, il peut s'avérer inutile de poursuivre l'expérience.
1.6.2.5. Période d'observation
Les animaux d'expérience doivent faire l'objet d'une observation quotidienne afin de déceler les symptômes d'intoxication. Le moment où les symptômes d'intoxication apparaissent et disparaissent ainsi que le moment de la mort doivent être consignés.
1.6.3. Mode opératoire
Les animaux doivent être placés dans des cages individuelles. Dans les conditions idéales, la substance d'essai est administrée aux animaux 7 jours sur 7, pendant une période de 28 jours. Les animaux de tous les lots satellites destinés à des observations complémentaires doivent être gardés en vie pendant 14 jours encore, sans traitement, afin de constater la régression ou la persistance des effets toxiques. La durée de l'exposition doit être au moins de 6 heures par jour.
La substance d'essai doit être appliquée uniformément sur une surface représentant environ 10 % de la surface corporelle totale mais lorsqu'il s'agit de substances hautement toxiques, la surface couverte peut être réduite. La couche de substance doit être aussi mince et uniforme que possible.
Durant l'exposition, la substance d'essai est maintenue en contact avec la peau au moyen d'une compresse de gaze poreuse et d'un sparadrap non irritant. La surface traitée doit, en outre, être convenablement couverte de manière à maintenir en place le pansement de gaze et la substance d'essai et de manière à éviter que les animaux puissent ingérer la substance à tester. Des appareils de contention peuvent être utilisés pour empêcher l'ingestion de la substance mais une immobilisation complète n'est pas recommandée. Il est également possible d'utiliser la technique du «collier de protection».
À l'issue de la période d'exposition, il faut, si possible, éliminer la substance résiduelle avec de l'eau ou par tout autre procédé adéquat de nettoyage de la peau.
Tous les animaux doivent être observés quotidiennement et les signes de toxicité ainsi que le moment de leur apparition, leur intensité et leur durée doivent être notés. Les observations doivent concerner les modifications de la peau et des poils, des yeux, des muqueuses, de l'appareil respiratoire, du système circulatoire ainsi que des systèmes nerveux autonome et central, de l'activité somatomotrice et du comportement. Les animaux doivent être pesés chaque semaine. Il est également recommandé d'effectuer des mesures hebdomadaires sur la consommation alimentaire des animaux. Il est nécessaire d'observer régulièrement les animaux afin de veiller à ce que, dans la mesure du possible, ceux-ci ne soient pas perdus pour l'expérience pour des raisons telles que cannibalisme, autolyse des tissus au cours des remises en cages. A l'issue de l'expérience, tous les animaux survivants appartenant aux lots traités, non satellites, sont autopsiés. Les animaux moribonds et les animaux dans un état de détresse ou de douleur intenses doivent être immédiatement retirés, euthanasiés et autopsiés.
Les examens figurant ci-après seront effectués à l'issue de la période d'essai sur tous les animaux (y compris les témoins):
|
1.
|
examen hématologique comprenant au moins l'hématocrite, la concentration en hémoglobine, le dénombre ment des érythrocytes et des leucocytes, la formule leucocytaire ainsi qu'une étude de la coagulation;
|
|
2.
|
examen biochimique clinique du sang comprenant au moins un paramètre des fonctions hépatiques et rénales: alanine aminotransférase sérique (anciennement connue sous le nom de transaminase glutamopyru-vique), aspartate aminotransférase sérique (anciennement connue sous le nom de transaminase glutano-oxalo-acétique), urée, albumine, créatinine, bilirubine totale et protéines sériques totales.
|
D'autres déterminations éventuellement nécessaires à une évaluation toxicologique adéquate concernent le calcium, le phosphore, les chlorures, le sodium, le potassium, la glycémie à jeun, l'analyse des lipides, les hormones, l'équilibre acido-basique, la méthémoglobine, l'activité cholinestérasique.
D'autres analyses biochimiques cliniques peuvent, si nécessaire, être effectuées pour approfondir l'étude des effets observés.
1.6.4. Autopsie
Tous les animaux soumis à l'essai doivent faire l'objet d'une autopsie macroscopique. Au moins le foie, les reins, les glandes surrénales et les testicules doivent être pesés à l'état humide le plus rapidement possible après la dissection afin d'éviter le dessèchement. Organes et tissus, à savoir, peau normale et traitée, foie, reins, rate, testicules, glandes surrénales, cœur et les organes-cibles (c'est-à-dire ceux qui présentent des lésions importantes ou des modifications de leur volume) doivent être conservés dans un milieu approprié en vue d'éventuels examens histopathologiques ultérieurs.
1.6.5. Examen histopathologique
Pour le lot exposé à la dose la plus élevée et le lot témoin, il faut pratiquer un examen histologique systématique de tous les organes et des tissus conservés. Les organes et tissus qui auront présenté des lésions attribuables à la substance à tester à la dose la plus élevée devront être examinés dans tous les lots ayant été exposés à des doses plus faibles. Les animaux de tout lot satellite devront faire l'objet d'un examen histologique particulièrement axé sur les organes et les tissus pour lesquels les lésions ont été constatées dans les lots traités.
2. DONNÉES
Les données doivent être récapitulées sous forme de tableaux indiquant, pour chaque lot d'expérience, nombre d'animaux au début de l'essai et le nombre d'animaux présentant chaque type de lésion.
Tous les résultats observés doivent être évalués au moyen d'une méthode statistique appropriée. Toute méthode statistique reconnue peut être utilisée.
3. RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
données concernant les animaux (espèce, souche, origine, conditions ambiantes, régime alimentaire, etc.),
|
|
—
|
conditions de l'essai, (y compris le type de pansement: occlusif ou non-occlusif),
|
|
—
|
doses (avec, le cas échéant, le véhicule) et concentrations,
|
|
—
|
dose sans effet, lorsque c'est possible,
|
|
—
|
données relatives à la réponse toxique par sexe et par dose,
|
|
—
|
indication du moment de la mort en cours d'expérience ou des survies au terme de l'expérience,
|
|
—
|
effets toxiques ou autres,
|
|
—
|
moment de l'observation de tout symptôme anormal et évolution de celui-ci,
|
|
—
|
données relatives à la prise de nourriture et à l'évolution du poids corporel,
|
|
—
|
examens hématologiques pratiqués et résultats complets,
|
|
—
|
examens biochimiques cliniques pratiqués et résultats complets,
|
|
—
|
description détaillée de tous les résultats histopathologiques,
|
|
—
|
traitement statistique des résultats, si possible,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B (point D).
4. RÉFÉRENCES
Voir introduction générale, partie B (point E).
B.10. MUTAGÉNICITÉ — ESSAI IN VITRO D'ABERRATION CHROMOSOMIQUE SUR CELLULES DE MAMMIFÈRE
1. MÉTHODE
Cette méthode est une adaptation de la ligne directrice OCDE TG 473, Essai d'aberration chromosomique in vitro chez les mammifères (1997).
1.1. INTRODUCTION
L'essai d'aberration chromosomique in vitro est destiné à détecter les agents qui provoquent des aberrations chromosomiques de structure dans des cellules de mammifère en culture (1) (2) (3). Les aberrations de structure peuvent être de deux types: chromosomiques ou chromatidiennes. La majorité des mutagènes chimiques induisent des aberrations de type chromatidien, mais parfois de type chromosomique. Une augmentation de la fréquence de polyploïdie peut indiquer qu'une substance chimique possède la faculté de provoquer des aberrations du nombre des chromosomes. Cet essai n'est cependant pas conçu pour évaluer les aberrations de nombre et il n'est généralement pas utilisé à cette fin. Les mutations chromosomiques et les événements liés sont à l'origine de beaucoup de maladies génétiques humaines et on a de bonnes raisons de penser que les mutations chromosomiques et événements liés, touchant les oncogènes et les gènes suppresseurs de tumeurs des cellules somatiques, jouent un rôle dans l'induction de cancers chez l'homme et chez les animaux de laboratoire.
L'essai d'aberration chromosomique in vitro peut être pratiqué sur des cultures de lignées cellulaires établies, des souches cellulaires ou des cultures de cellules primaires. Les cellules utilisées sont choisies en fonction de leur potentiel de croissance en culture, de la stabilité de leur caryotype, du nombre et de la diversité des chromosomes et de la fréquence des aberrations chromosomiques spontanées.
Les essais in vitro requièrent généralement l'utilisation d'une source exogène d'activation métabolique. Ce système d'activation métabolique est incapable de reproduire parfaitement les conditions présentes in vitro chez les mammifères. Il faut éviter absolument d'introduire des conditions qui conduiraient à des résultats positifs ne reflétant pas une mutagénicité intrinsèque et pouvant provenir de modification du pli, de l'osmolalité, ou d'une forte cytotoxicité (4) (5).
Cet essai est utilisé pour le dépistage de substances pouvant être mutagènes et cancérigènes pour les mammifères. Bien que de nombreux composés donnant un résultat positif lors de cet essai soient cancérigènes pour les mammifères, il n'y a pas de corrélation parfaite entre cet essai et la cancérogénicité. La corrélation dépend de la classe chimique mais d'autre part, de plus en plus d'arguments laissent penser qu'il existe des agents cancérigènes que cet essai ne permet pas de détecter parce qu'ils agissent par des mécanismes qui n'impliquent pas des lésions directes de l'ADN.
Voir aussi l'introduction générale de la partie B.
1.2. DÉFINITIONS
Aberration de type chromatidien: lésion chromosomique de structure se traduisant par une cassure d'une seule chromatide ou par une cassure et une réunion entre chromatides.
Aberration de type chromosomique: lésion chromosomique de structure se traduisant par une cassure, ou par une cassure et une réunion, des deux chromatides sur le même site.
Endoreduplication: processus dans lequel le noyau, après une phase S de implication, n'entre pas en mitose mais commence une autre phase S. Il en résulte des chromosomes avec 4, 8, 16, ... chromatides.
Lacune («gap»): lésion achromatique plus petite que la largeur d'une chromatide, avec un défaut d'alignement minimal des chromatides.
Index mitotique: nombre de cellules en métaphase divisé par le nombre total de cellules dans une population une indication de la vitesse de prolifération de cette population.
Aberration de nombre: modification du nombre de chromosomes par rapport au nombre normal caractéristique des cellules employées.
Polyploïde: état multiple du nombre de chromosomes haploïdes (n), autre que la diploïdie (c'est-à-dire 3n, 4n, etc.).
Aberration de structure: modification de la structure des chromosomes, détectable par un examen au microscope au stade de la métaphase et se présentant sous la forme de délétions, de cassures, de modifications intrachromosomiques ou interchromosomiques.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
Des cultures de cellules sont exposées à la substance d'essai avec et sans activation métabolique. Après avoir été exposées à la substance d'essai, les cultures de cellules sont, à intervalles préétablis, traitées par une substance qui bloque les cellules en métaphase (par exemple de la clochicine ou du Colcemid®), récoltées et teintées. Les cellules en métaphase sont soumises à un examen microscopique afin de déceler les aberrations chromosomiques.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparations
1.4.1.1. Cellules
Diverses lignées cellulaires, souches cellulaires ou cultures de cellules primaires, y compris des cellules humaines, peuvent être utilisées (par exemple les fibroblastes de hamster chinois, les lymphocytes du sang périphérique de l'homme ou d'autres mammifères).
1.4.1.2. Milieu et conditions de culture
Il convient d'utiliser un milieu de croissance et des conditions d'incubation (récipients de culture, concentration de CO2, température et degré d'humidité) appropriés pour la croissance cellulaire. La stabilité du nombre modal de chromosomes et l'absence de contamination par des mycoplasmes doivent être vérifiées régulièrement dans les lignées cellulaires établies et les souches cellulaires. En cas de contamination, elles ne doivent pas être utilisées. La durée normale du cycle cellulaire de la lignée cellulaire et les conditions de culture utilisées doivent être connues.
1.4.1.3. Préparation des cultures
Lignées et souches cellulaires établies: les cellules sont multipliées à partir de stocks cellulaires, ensemencées dans un milieu de culture à une densité telle que les cultures ne confluent pas avant la récolte, et incubées à 37 oC.
Lymphocytes: du sang complet traité avec un anticoagulant (héparine, par exemple) ou des lymphocytes isolés issus de donneurs sains sont ajoutés à un milieu de culture contenant un mitogène (par exemple la phytohémagglutinine) et sont incubés à 37 oC.
1.4.1.4. Activation métabolique
Les cellules doivent être exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique approprié. Le système le plus couramment utilisé est une fraction postmitochondriale enrichie en cofacteurs (S9), préparée à partir de foie de rongeur traité avec des inducteurs enzymatiques, tels que de l'Aroclor 1254 (6) (7) (8) (9) ou un mélange de phénobarbitone et de ß-naphthoflavone (10) (11) (12).
La fraction postmitochondriale est généralement utilisée à des concentrations allant de 1 a 10 % v/v dans le milieu de culture final. Le choix du système d'activation métabolique peut dépendre de la classe chimique de la substance d'essai. Dans certains cas il peut être préférable d'utiliser plus d'une concentration de la fraction postmitochondriale.
Plusieurs procédés nouveaux, comme la création par génie génétique de lignées cellulaires exprimant des enzymes activantes spécifiques, sont susceptibles de fournir une activation endogène. Le choix des lignées cellulaires utilisées doit être justifié scientifiquement (par exemple par l'importance de l'isoenzyme du cytochrome P450 pour le métabolisme de la substance d'essai).
1.4.1.5. Substances d'essai/préparation
Les substances d'essai solides sont préparées par dissolution ou suspension dans un véhicule ou un solvant approprié, puis éventuellement diluées avant le traitement des cellules. Les substances d'essai liquides peuvent être ajoutées directement au système d'essai et/ou diluées avant traitement. Il convient d'utiliser des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage.
1.4.2. Conditions expérimentales
1.4.2.1. Solvant/véhicule
Le solvant/véhicule choisi ne doit pas réagir chimiquement avec la substance d'essai ni altérer la survie des cellules et l'activité du mélange S9. L'emploi d'un solvant/véhicule inhabituel doit être justifié par des données faisant état de sa compatibilité. On préconise d'envisager d'abord l'utilisation d'un véhicule/solvant aqueux, chaque fois que cela est possible. Dans le cas d'essais conduits avec des substances instables dans l'eau, on utilisera des solvants organiques exempts d'eau. L'eau peut être enlevée par addition d'un tamis moléculaire.
1.4.2.2. Concentrations d'exposition
La cytotoxicité, la solubilité dans le système d'essai et les modifications de pH et d'osmolalité sont des critères à prendre en considération pour déterminer la dose la plus élevée à utiliser.
La cytotoxicité est mesurée, avec et sans système d'activation métabolique, dans l'expérience principale par un indicateur approprié de l'intégrité et de la croissance cellulaires, tel que le degré de confluence des colonies cellulaires, le nombre de cellules viables ou l'index mitotique. Il peut être utile de déterminer la cytotoxicité et la solubilité lors d'un essai préliminaire.
Il convient d'utiliser au moins trois concentrations analysables. Dans le cas d'une substance cytotoxique, on utilisera des concentrations qui couvrent un domaine allant d'une toxicité nulle ou légère à la toxicité maximale, ce qui signifie en général des concentrations séparées par un facteur compris entre 2 et √10 au maximum. La concentration la plus élevée doit entraîner au moment de la récolte, une réduction significative (au moins 50 %) du degré de confluence, du nombre de cellules ou de l'index mitotique. Ce dernier varie dans le temps après le traitement et fournit seulement une mesure indirecte des effets cytotoxiques et cytostatiques. Cependant, l'index mitotique est une mesure acceptable pour des cultures en suspension pour lesquelles d'autres mesures de toxicité peuvent être incommodes ou irréalisables. Des données sur la cinétique du cycle cellulaire, par exemple le temps de génération moyen (TGM), peuvent fournir des compléments d'information. Toutefois, le TGM est une moyenne générale qui ne révèle pas toujours l'existence de «sous-populations en retard»; même de faibles augmentations du temps de génération moyen peuvent conduire à un retard très important du moment où le nombre d'aberrations est optimal.
En ce qui concerne les substances relativement non cytotoxiques, la concentration maximale doit être la plus basse des trois concentrations suivantes: 5 μl/ml, 5 mg/ml ou 0,01 M.
Pour les substances relativement insolubles qui ne sont pas toxiques aux concentrations solubles, la dose la plus élevée à tester doit être une concentration supérieure à la limite de solubilité dans le milieu de culture à la fin de la période de traitement. Dans certains cas (par exemple si la toxicité n'apparaît qu'à des concentrations supérieures à la plus faible concentration insoluble), il est recommandé de tester plusieurs concentrations auxquelles une précipitation est visible. Il peut être utile d'évaluer la solubilité au début et à la fin du traitement, celle-ci pouvant varier au cours de l'exposition dans le système d'essai en raison de la présence de cellules, de S9, de sérum, etc. On conclut à l'insolubilité lorsqu'une précipitation est observée à l'œil nu. Le précipité ne doit pas interférer avec l'évaluation des résultats.
1.4.2.3. Témoins négatifs et positifs
Des témoins positifs et négatifs (solvant ou véhicule), avec et sans activation métabolique, doivent être inclus simultanément dans chaque expérience. En présence d'un système d'activation métabolique, la substance utilisée comme témoin positif doit être celle qui exige l'activation pour donner une réponse mutagène.
On utilise comme témoin positif un clastogène connu à des niveaux d'exposition qui conduisent à un accroissement détectable et reproductible par rapport au bruit de fond, démontrant la sensibilité du système d'essai.
Les doses des témoins positifs doivent être choisies de manière que les effets soient nets et que l'identité des lames codées ne soit pas évidente. Quelques exemples de témoins positifs figurent dans le tableau ci-dessous.
|
Condition d'activation métabolique
|
Substances
|
Numéro CAS
|
Numéro EINECS
|
|
En l'absence d'activation métabolique exogène
|
Méthanesulfonate de méthyle
|
66-27-3
|
200-625-0
|
|
Méthanesulfonate d'éthyle
|
62-50-0
|
200-536-7
|
|
Éthylnitrosourée
|
759-73-9
|
212-072-2
|
|
Mitomycine C
|
50-07-7
|
200-008-6
|
|
N-oxyde de 4-nitroquinoléine
|
56-57-5
|
200-281-1
|
|
En présence d'activation métabolique exogène
|
Benzo[a]pyrène
|
50-32-8
|
200-028-5
|
|
Cyclophosphamide
Monohydrate de cyclophosphamidée
|
50-18-0
6055-19-2
|
200-015-4
|
D'autres témoins positifs appropriés peuvent être utilisés. De plus, l'emploi de témoins positifs appartenant à la même classe chimique sera envisagé, s'ils sont disponibles.
Le solvant ou le véhicule doit être utilisé seul en tant que témoin négatif dans le même milieu de culture, dans les mêmes conditions que la substance d'essai et pour chaque temps de prélèvement. On utilisera également des témoins non traités, à moins que des essais antérieurs n'aient démontré que le solvant/véhicule choisi n'a pas d'effet délétère ou mutagène.
1.4.3. Mode opératoire
1.4.3.1. Traitement avec la substance d'essai
Les cellules en prolifération sont traitées avec la substance d'essai en présence et en l'absence de système d'activation métabolique. Le traitement des lymphocytes doit débuter environ 48 heures après la simulation mitogénique.
|
1.4.3.2.
|
On réalise normalement des cultures dupliquées à chaque niveau de dose. Des cultures dupliquées sont aussi vivement recommandées pour les témoins négatifs (solvant). Si des essais antérieurs ont démontré que la variation entre cultures dupliquées est minime (13) (14), l'utilisation d'une seule culture pour chaque concentration peut être envisagée.
Les substances gazeuses ou volatiles doivent être testées en utilisant des méthodes appropriées, par exemple dans des récipients de culture hermétiquement fermés (15) (16).
|
1.4.3.3. Récolte des cultures
Pour la première expérience, les cellules sont exposées à la substance d'essai pendant 3 à 6 heures, en présence et en l'absence d'activation métabolique. On prélève ensuite des échantillons après un temps correspondant à environ 1,5 fois la durée du cycle cellulaire normal après le début du traitement (12). Si l'essai s'avère négatif, que ce soit avec ou sans activation métabolique, on effectue un essai supplémentaire sans activation. le traitement se poursuivant jusqu'au prélèvement d'échantillons après une période équivalente à environ 1,5 fois la durée du cycle cellulaire normal. Certaines substances peuvent être détectées plus facilement en prolongeant le temps de traitement et de prélèvement au-delà de 1,5 fois la durée du cycle cellulaire. Les résultats négatifs obtenus avec activation métabolique demandent à être confirmés cas par cas. Si la confirmation des résultats négatifs n'est pas jugée nécessaire, une justification doit être fournie.
1.4.3.4. Préparation des chromosomes
Les cultures de cellules sont traitées avec du Colcemid® ou de la colchicine pendant 1 à 3 heures avant la récolte. Chaque culture est récoltée et traitée séparément en vue de la préparation des chromosomes. Celle-ci consiste en un traitement hypotonique, une fixation et une coloration des cellules.
1.4.3.5. Analyse
Toutes les lames, y compris celles des témoins positifs et négatifs, doivent être codées individuellement avant l'analyse au microscope. Le procédé de fixation provoque souvent la rupture d'une certaine fraction des cellules en métaphase, ce qui entraîne une perte des chromosomes. Pour cette raison, toutes les cellules examinées doivent contenir un nombre de centromères égal au nombre modal ± 2. Pour chaque dose et chaque témoin, il y a lieu d'examiner au moins 200 cellules en métaphase bien étalées et, le cas échéant, réparties de façon égale entre les cultures en double exemplaire. Ce nombre peut être réduit lorsqu'un grand nombre d'aberrations sont observées.
Bien que l'essai soit destiné à détecter les aberrations chromosomiques de structure, il est important de signaler d'éventuels cas de polyploidie et d'endoreduplication.
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
L'unité expérimentale étant la cellule, on détermine le pourcentage de cellules présentant une ou plusieurs aberrations de structure. Une liste des différents types d'aberrations chromosomiques de structure doit être établie avec leur nombre et leur fréquence dans les cultures traitées et les cultures témoins. Les lacunes sont enregistrées séparément et rapportées mais ne sont généralement pas incluses dans la fréquence totale des aberrations.
On indiquera aussi les résultats des mesures concomitantes de cytoxicité pour toutes les cultures traitées et les témoins négatifs dans les principaux essais d'aberration chromosomique.
Les données seront présentées séparément pour chaque culture. En outre, toutes les données doivent être résumées sous tonne de tableaux.
Il n'est pas obligatoire de vérifier une réponse nettement positive. Par contre, les résultats équivoques doivent être clarifiés par un ou des essais supplémentaires, de préférence dans des conditions expérimentales modifiées. La nécessité de confirmer les résultats négatifs est discutée au point 1.4.3.3. Lors des expériences complémentaires, il faut envisager la modification des paramètres de l'étude afin d'élargir l'éventail des conditions évaluées. Les paramètres susceptibles d'être modifiés comprennent l'intervalle entre les niveaux de dose et les conditions d'activation métabolique.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Plusieurs critères permettent de déterminer si un résultat est positif, notamment un accroissement lié à la dose ou un accroissement reproductible du nombre de cellules présentant des aberrations. En premier lieu, il s'agir de prendre en considération la pertinence biologique des résultats. L'évaluation des résultats peut s'appuyer sur des méthodes statistiques (3) (13) mais la signification statistique ne doit pas être le seul facteur décisif pour conclure à une réponse positive.
Une augmentation du nombre de cellules polyploïdes peut signifier que la substance d'essai est capable d'inhiber les processus mitotiques et d'induire des aberrations de nombre. Une augmentation du nombre de cellules ayant des chromosomes endoredupliqués peut indiquer que la substance d'essai est capable d'inhiber la progression du cycle cellulaire (17) (18).
Si les résultats obtenus avec une substance d'essai ne répondent pas aux critères indiqués ci-dessus, on peut conclure que la substance en question n'est pas mutagène dans cet essai.
Bien que la plupart des essais donneront des résultats nettement positifs ou négatifs, dans de rares cas l'ensemble des résultats d'un essai n'autorise pas de conclusion catégorique sur l'activité de la substance étudiée. Les résultats peuvent rester équivoques ou discutables indépendamment du nombre de répétitions de l'essai.
Des résultats positifs à l'essai d'aberration chromosomique in vitro indiquent que la substance d'essai induit des aberrations chromosomiques de structure dans des cellules somatiques de mammifère en culture. Des résultats négatifs signifient que, dans les conditions expérimentales, la substance d'essai n'induit pas d'aberration chromosomique dans des cellules somatiques de mammifère en culture.
3. RAPPORT
RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Solvant/véhicule:
|
—
|
justification du choix du véhicule,
|
|
—
|
solubilité et stabilité de la substance d'essai dans le solvant/véhicule, si elles sont connues.
|
|
|
|
Cellules:
|
—
|
type et source des cellules,
|
|
—
|
données sur le caryotype et raisons du choix du type cellulaire utilisé,
|
|
—
|
absence de mycoplasme, le cas échéant,
|
|
—
|
données concernant le cycle cellulaire,
|
|
—
|
sexe des donneurs de sang, sang complet ou lymphocytes isolés, mitogène utilisé,
|
|
—
|
nombre de repiquages, le cas échéant,
|
|
—
|
méthodes d'entretien des cultures cellulaires, le cas échéant,
|
|
—
|
nombre modal de chromosomes.
|
|
|
|
Conditions de l'essai:
|
—
|
identité de l'inhibiteur de 1a métaphase, sa concentration et la durée d'exposition des cellules,
|
|
—
|
justification du choix des concentrations et du nombre de cultures, y compris des données relatives à la cytotoxicité et aux limites de solubilité, si elles sont disponibles,
|
|
—
|
composition du milieu et, le cas échéant, la concentration de CO2,
|
|
—
|
concentration de la substance d'essai,
|
|
—
|
volume de véhicule et de substances d'essai ajouté,
|
|
—
|
température d'incubation,
|
|
—
|
densité cellulaire au moment de l'ensemencement, si nécessaire,
|
|
—
|
type et composition du système d'activation métabolique, y compris les critères d'acceptabilité,
|
|
—
|
témoins positifs et négatifs,
|
|
—
|
méthodes de préparation des lames,
|
|
—
|
critères de dénombrement des aberrations,
|
|
—
|
nombre de métaphases analysées,
|
|
—
|
méthodes de mesure de la toxicité,
|
|
—
|
critères pour conclure que l'étude est positive, négative ou équivoque.
|
|
|
|
Résultats:
|
—
|
signes de toxicité (degré de confluence, données sur le cycle cellulaire, comptage des cellules, index mitotique),
|
|
—
|
signes de précipitation,
|
|
—
|
données sur le pH et l'osmolalité du milieu de traitement, si elles ont été déterminées,
|
|
—
|
définition appliquée aux aberrations, y compris les lacunes,
|
|
—
|
nombre de cellules présentant des aberrations chromosomiques et type d'aberration, séparément pour chaque culture traitée et culture témoin,
|
|
—
|
changements de la ploïdie, le cas échéant,
|
|
—
|
relation dose-réponse, si possible.
|
|
—
|
analyses statistiques, le cas échéant,
|
|
—
|
données concernant les témoins négatifs (solvant/véhicule) et positifs concomitants,
|
|
—
|
données historiques concernant les témoins négatifs (solvant/véhicule) et positifs (étendues, moyennes, écarts-types).
|
|
|
|
Discussion des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
Evans, H. J. (1976), Cytological Methods for Detecting Chemical Mutagens, in: Chemical mutagens. Princi-ples and Methods for their Detection. Vol. 4, Hollaender, A. (ed) Plenum Press, New York and London. pp. 1-29.
|
|
(2)
|
Ishidate. M. jr. and Sofuni, T. (1985), The In Vitro Chromosomal Aberration Test Using Chinese Hams ter Lung (CHL) Fibroblast Cells in Culture, in: Progress in Mutation Research, Vol. 5. Ashby, J. et al., (eds) Elsevier Science Publishers, Amsterdam-New York-Oxford, pp. 427-432.
|
|
(3)
|
Galloway, S. M.. Armstrong, M. J., Reuben, C, Colman, S., Brown, 13., Cannon, C, Bloom, A. D., Nakamura, F., Ahmed, M., Duk, S., Kimpo, j., iviargolin, c. H., Resnick, M. A., Andersen. G. and Zeiger, E. (1978), Chromosome aberration and sister chromatic exchanges in Chinese hamster ovary cells: Evaluation of 108 chemicals, Environs. Molec. Mulogen 10 (suppl. 10), pp. 1-175.
|
|
(4)
|
Scott, D., Galloway, S. M., Marshall, R. R., Ishidate, M. Jr., Brusick, D., Ashby, J. and Myhr, B. C. (1991), Genotoxicity under Extreme Culture Conditions. A report from 1CPEMC Task Group 9, Mutation Res., 257, pp. 147-204.
|
|
(5)
|
Monta, T., Nagaki, T., Fukuda. I. and Okumura, K. (1992), Clastogenicity of low pH to Various Cultured Mammalian Cells, Mutation Res., 268. pp. 297-305
|
|
(6)
|
Ames. B. N., McCann. J. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test, Mutation Res., 31, pp. 347-364.
|
|
(7)
|
Maron. D. M. and Ames, B. N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113. pp. 17 3-215.
|
|
(8)
|
Natarajan, A. T., Tates, A. D., van Buul, P. P. W., Meijers, M. and de Vogel, N. (1976), Cytogenetic Effects of Mutagen/Carcinogens after Activation in a Microsomal System In Vitro, J. Induction of Chromosome Aberrations and Sister Chromatid Exchange by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes, Mutation Res., 37, pp. 83-90.
|
|
(9)
|
Matsuoka, A., Hayashi, M. and Ishidate, M. Jr. (1979), Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro, Mutation Res., 66, pp. 277-290.
|
|
(10)
|
Elliot, B. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Report of UK environmental Mutagen Society Working Party. Alternatives to Aroclar 1254-induced S9 in In Vitro Genotoxicity Assays, Mutagenesis, 7, pp. 175-177.
|
|
(11)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, in: de Serres, F. J., Fonts, J. R., Berid, J. R. and Philpot, R. M. (eds), In Vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, pp. 85-88.
|
|
(12)
|
Galloway, S. M., Aardema. M. J., Ishidate, M. Jr., Iven, J. L., Kirkland, D. J., Morita, T., Mosesso. P., Sofuni, T. (1994), Report from Working Group on in In Vitro Tests for Chromosomal Aberrations, Mutation Res., 312. pp. 241-261.
|
|
(13)
|
Richardson, C, Williams, D. A., Allen, J. A., Amphlett, G., Chanter, D. O. and Phillips, B. (1989). Analysis of Data from In Vitro Cytogenetic Assays, in: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D. J., (ed) Cambridge University Press, Cambridge, pp. 141-154.
|
|
(14)
|
Soper, K. A. and Galloway, S. M. (1994), Replicate Flasks are not Necessary for In Vitro Chromosome Aberration Assays in CHO Cells. Mutation Res., 312, pp. 139-149.
|
|
(15)
|
Krahn, D. F., Barsky, F. C. and McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, in: Tice, R. R., Costa, D. L, Schaich, K. M. (eds), Genotoxic Effects of Airborne Agents, New York, Plenum, pp. 91-103.
|
|
(16)
|
Zamora, P. O., Benson, J. M., Li, A. P. and Brooks, A. L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental Mutagenesis, 5, pp. 795-801.
|
|
(17)
|
Locke-Huhlc, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation induced G2 arrest, Mutation Res., 119. pp. 403-413
|
|
(18)
|
Huang, Y., Change, C. and Trosko, J. E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells, Cancer Res., 43, pp. 1362-1364.
|
B.11. MUTAGÉNICITÉ — ESSAI IN VIVO D'ABERRATION CHROMOSOMIQUE SUR MOELLE OSSEUSE DE MAMMIFÈRE
1. MÉTHODE
Cette méthode est une adaptation de la ligne directrice OCDE TG 475, Essai d'aberration chromosomique sur moelle osseuse de mammifères (1997).
1.1. INTRODUCTION
L'essai in vivo d'aberration chromosomique est destiné à identifier les agents qui provoquent des aberrations chromosomiques de structure dans des cellules de moelle osseuse d'animaux, généralement des rongeurs (1, 2, 3, 4). Les aberrations de structure peuvent être de deux types: chromosomiques ou chromatidiennes. Une augmentation de la fréquence de polyploïdie peut indiquer qu'une substance chimique possède la faculté de provoquer des aberrations du nombre des chromosomes. La majorité des mutagènes chimiques induisent des aberrations de type chromatidien, mais parfois de type chromosomique. Les mutations chromosomiques et les événements connexes sont à l'origine de beaucoup de maladies génétiques humaines et on a de bonnes raisons de penser que les mutations chromosomiques et événements connexes, touchant les oncogènes et les gènes suppresseurs de tumeurs des cellules somatiques, jouent un rôle dans l'induction de cancers chez l'homme et chez les animaux de laboratoire.
Les rongeurs sont couramment utilisés dans cet essai. Cet essai se pratique sur la moelle osseuse parce que c'est un tissu très vascularisé, formé d'une population de cellules au cycle rapide et faciles à prélever et traiter. D'autres espèces et tissus cibles ne font pas l'objet de cette méthode.
Cet essai d'aberration chromosomique se prête particulièrement bien à l'évaluation du risque mutagène, en ce sens qu'il permet de prendre en compte des facteurs de métabolisme in vivo, de pharmacocinétique et de processus de réparation de l'ADN. Ces facteurs peuvent cependant varier d'un animal à un autre et d'un tissu à un autre. Un essai in vivo est aussi utile pour vérifier un effet mutagène détecté par un essai in vitro.
On n'aura pas recours à cet essai s'il est manifeste que la substance d'essai ou un métabolite réactif n'atteindra pas le tissu cible.
Voir aussi l'introduction générale de la partie B.
1.2. DÉFINITIONS
Aberration de type chromatinien: lésion chromosomique de structure se traduisant par une cassure d'une seule chromatide ou par une cassure et une réunion entre chromatides.
Aberration de type chromosomique: lésion chromosomique de structure se traduisant par une cassure, ou par une cassure et une réunion, des deux chromatides sur le même site.
Endoreduplication: processus dans lequel le noyau, après une phase S de réplication, n'entre pas en mitose mais commence une autre phase S. Il en résulte des chromosomes avec 4, 8, 16, ... chromatides.
Lacune: lésion achromatique inférieur à la largeur d'une chromatide avec un défaut d'alignement minimal des chromatides.
Aberration de nombre: modification du nombre de chromosomes par rapport au nombre normal caractéristique des cellules employées.
Polyploïdie: état multiple du nombre de chromosomes haploïdes (n), autre que la diploïdie (c'est-à-dire 3n, 4n, etc.).
Aberration de structure: modification de la structure des chromosomes, détectable par un examen au microscope au stade de la métastase et se présentant sous la forma de délétions, de cassure, de modifications intrachromosomiques ou interchromosomiques.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
Les animaux sont exposés à la substance d'essai par une voie d'exposition appropriée et sont sacrifiés à des moments appropriés après le traitement. Avant le sacrifice, les animaux sont traités avec une substance qui bloque les cellules en métaphase (par exemple de la colchicine ou du Colcemid®). Ensuite, les préparations chromosomiques réalisées à partir des cellules de moelle osseuse sont colorées, et les cellules en métaphase sont examinées en vue de la mise en évidence d'aberrations chromosomiques.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparations
1.4.1.1. Sélection des espèces, animales
Bien que le rat, la souris et le hamster chinois soient habituellement utilisés, n'importe quelle espèce de mammifère appropriée peut être utilisée. Il convient d'utiliser des animaux adultes, jeunes et sains issus de souches couramment utilisées en laboratoire. Au début de l'étude, la différence pondérale entre les animaux doit être minimale et ne pas dépasser ± 20 % du poids moyen de chaque sexe.
1.4.1.2. Conditions d'encagement et régime alimentaire
Les conditions générales décrites dans l'introduction générale de la partie B sont appliquées mais le taux d'humidité devrait se situer aux environs de 50-60 %.
1.4.1.3. Préparation des animaux
Les animaux sont répartis par randomisation entre les groupes témoins et les groupes traités. Les cages doivent être disposées de telle manière qu'il n'y ait pas d'effet dû à leur emplacement. Les animaux sont identifiés individuellement. Ils sont acclimatés aux conditions du laboratoire pendant au moins cinq jours.
1.4.1.4. Préparation des doses
Les substances d'essai solides sont dissoutes ou mises en suspension dans un véhicule ou un solvant approprié, puis si nécessaire diluées avant d'être administrées aux animaux. Les substances d'essai liquides peuvent être administrées directement ou après dilution. Il convient d'utiliser des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage.
1.4.2. Conditions expérimentales
1.4.2.1. Solvant/véhicule
Le solvant/véhicule choisi ne doit pas avoir d'effet toxique aux doses utilisées ni présenter de réaction chimique avec la substance d'essai. Le recours à des solvants/véhicules inhabituels doit être justifié par des données sur leur compatibilité. On préconise d'envisager d'abord l'utilisation d'un véhicule/solvant aqueux, chaque fois que cela est possible.
1.4.2.2. Témoins
Des témoins positifs et négatifs (solvant ou véhicule) doivent être inclus pour chaque sexe et dans chaque essai. Les animaux des groupes témoins doivent être manipulés exactement comme ceux des groupes traités, mise à part l'administration de la substance d'essai.
Les témoins positifs doivent produire des aberrations chromosomiques de structure in vivo à des niveaux d'exposition supposés provoquer un accroissement détectable par rapport au bruit de fond. Les doses de témoin positif doivent être choisies afin que les effets soient évidents mais que l'identité des lames codées ne soit pas révélée immédiatement à l'examinateur. Le témoin positif peut être administré par une voie différente de celle utilisée pour la substance d'essai et un seul prélèvement d'échantillon peut suffire. L'emploi de témoins positifs appartenant à la même classe chimique peut être envisagé, s'ils sont disponibles. Les substances indiquées dans le tableau ci-dessous sont des exemples de témoins positifs.
|
Substance
|
Numéro CAS
|
Numéro Einecs
|
|
Méthanesulfonate d'éthyle
|
62-50-0
|
200-536-7
|
|
Éthylnitrosourée
|
759-73-9
|
212-072-2
|
|
Mitomycine C
|
50-07-7
|
200-008-6
|
|
Cyclophosphamide
Monohydrate de cyclophosphamide
|
50-18-0
6055-19-2
|
200-015-4
|
|
Triéthylènemélamine
|
51-18-3
|
200-083-5
|
Lors de chaque prélèvement, il faut inclure un lot d'animaux témoins négatifs traités seulement avec le solvant ou le véhicule et par ailleurs manipulés de la même façon que les groupes traités. Ceci peut être évité si l'on dispose de données acceptables sur la variabilité interindividuelle et la fréquence spontanée de cellules comportant des aberrations chromosomiques provenant de témoins historiques. Dans le cas où un seul prélèvement est effectué pour les témoins négatifs, le premier temps de prélèvement est le plus approprié. En outre, des témoins non traités doivent aussi être inclus, à moins que des données historiques ne démontrent que le solvant/véhicule choisi n'induit aucun effet délétère ou mutagène.
1.5. MODE OPÉRATOIRE
1.5.1. Nombre et sexe des animaux
Chaque groupe traité et groupe témoin doit comprendre au moins cinq animaux analysables par sexe. Si, au moment de l'étude, on dispose de données historiques sur la même espèce et avec la même voie d'administration démontrant l'absence de différence notable de toxicité entre les sexes, la réalisation de l'essai sur un seul sexe est acceptable. Si l'exposition humaine est spécifique du sexe, par exemple dans le cas de certains produits pharmaceutiques, des animaux du sexe approprié doivent être utilisés pour l'essai.
1.5.2. Modalité de traitement
Il est préférable d'administrer la substance d'essai en une seule fois. Elle peut aussi être administrée en doses fractionnées (deux doses le même jour avec un intervalle de quelques heures au maximum) afin que l'administration d'un grand volume soit plus aisée. D'autres modalités de traitement sont acceptables s'ils sont scientifiquement justifiés.
Les échantillons sont prélevés après le traitement à deux moments différents de la même journée. Pour les rongeurs, le premier prélèvement après le traitement est effectué après une période de 1,5 fois la durée normale du cycle cellulaire (généralement de 12 à 18 heures). Étant donné que le temps requis par l'absorption et le métabolisme de la substance d'essai ainsi que l'effet de celle-ci sur la cinétique du cycle cellulaire peuvent influer sur le moment optimal de détection des aberrations, on recommande d'effectuer un autre prélèvement 24 heures après le premier. Si le traitement s'étend sur plus d'une journée, le prélèvement doit avoir lieu après une période de 1,5 fois la durée normale du cycle cellulaire après la dernière administration.
Avant d'être sacrifiés, les animaux reçoivent une dose appropriée d'une substance qui bloque les cellules en métaphase (par exemple du Colcemid® ou de la colchicine) par injection intrapéritonéale. Les prélèvements sont effectués après une période appropriée, qui est de 3 à 5 heures pour la souris et de 4 à 5 heures pour le hamster chinois. Les cellules sont prélevées de la moelle osseuse et analysées en vue de la détection d'aberrations chromosomiques.
1.5.3. Niveaux de dose
Si une étude préliminaire est réalisée afin d'évaluer les niveaux de dose requis, cette étude doit être effectuée dans le même laboratoire en utilisant la même espèce, la même souche, des animaux du même sexe et la même modalité de traitement que ceux de l'étude principale (5). En cas de toxicité, on utilise trois niveaux de dose pour le premier prélèvement. Ces niveaux de dose doivent couvrir un domaine allant d'une toxicité nulle ou légère à la toxicité maximale. Pour les prélèvements suivants, seule la dose la plus élevée est utilisée. Celle-ci est définie comme la dose qui produit des signes de toxicité tels qu'ils font supposer que des doses plus fortes, administrées selon les mêmes modalités, seraient létales. Les substances possédant une activité biologique spécifique à des doses faibles et non toxiques (comme les hormones et les mitogènes) peuvent faire exception aux critères d'établissement des doses et doivent être évaluées cas par cas. La dose la plus élevée peut aussi être définie comme étant celle qui produit certains indices de toxicité dans la moelle osseuse (par exemple une diminution de plus de 50 % de l'index mitotique).
1.5.4. Test limite
Si un essai réalisé avec au moins 2 000 mg/kg de poids corporel, en une seule dose ou en deux doses administrées le même jour, n'engendre aucun effet toxique observable et si une génotoxicité est improbable d'après les données relatives à des substances de structure voisine, on peut considérer qu'une étude complète avec trois niveaux de doses n'est pas nécessaire. Pour les études de plus longue durée, la dose limite est de 2 000 mg/kg de poids corporel par jour dans le cas d'un traitement d'une durée allant jusqu'à 14 jours et de 1 000 mg/kg de poids corporel par jour pour les études plus longues. Suivant l'importance de l'exposition humaine probable, on peut envisager d'utiliser des doses plus fortes dans l'essai de dose limite.
1.5.5. Voies d'administration
La substance d'essai est généralement administrée par gavage à l'aide d'une sonde gastrique ou d'une canule d'intubation adaptée, ou par injection intrapéritonéale. D'autres voies d'exposition sont acceptables lorsqu'elles peuvent être justifiées. Le volume maximal de liquide administré en une fois par gavage ou par injection dépend de la taille de l'animal. Ce volume ne devrait pas dépasser 2 ml/100g de poids corporel. Une justification doit être fournie si des volumes plus importants sont administrés. Il convient de réduire au minimum la variation du volume administré en ajustant la concentration à tous les niveaux de doses, sauf pour les produits irritants ou corrosifs dont les effets sont généralement exacerbés lorsqu'on augmente la concentration.
1.5.6. Préparation des chromosomes
La moelle osseuse est extraite immédiatement après le sacrifice, exposée à une solution hypotonique et fixée. Les cellules sont alors étalées sur des lames et colorées.
1.5.7. Analyse
L'index mitotique sert de mesure de la cytotoxicité et doit être déterminé sur un minimum de 1 000 cellules pour chaque animal traité (y compris les témoins positifs) et pour chaque témoin négatif non traité.
L'analyse des aberrations chromosomiques doit porter sur au moins 100 cellules de chaque animal. Ce nombre peut être réduit lorsque les aberrations observées sont nombreuses. Toutes les lames, y compris celles des témoins négatifs et positifs, doivent être codées indépendamment avant l'analyse au microscope. Comme le procédé de fixation provoque souvent la rupture d'une certaine fraction des cellules en métaphase, entraînant une perte de chromosomes, toutes les cellules examinées doivent contenir un nombre de centromères égal à 2n ± 2.
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
Les résultats individuels pour chaque animal doivent être présentés sous forme de tableaux. L'unité expérimentale est l'animal. Pour chaque animal, on indiquera le nombre de cellules examinées et on évaluera le nombre d'aberrations par cellule ainsi que le pourcentage de cellules présentant une ou plusieurs aberrations de structure. Les différents types d'aberrations chromosomiques de structure doivent être consignés avec leur nombre et leur fréquence pour les groupes traités et les groupes témoins. Les lacunes sont enregistrées séparément et rapportées mais ne sont généralement pas incluses dans la fréquence totale des aberrations. S'il n'y a pas de différence de réponse entre sexes, les données des deux sexes peuvent être combinées dans l'analyse statistique.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Plusieurs critères permettent de déterminer si un résultat est positif, notamment un accroissement dose-dépendant du nombre relatif de cellules présentant des aberrations ou une augmentation nette du nombre de cellules présentant des aberrations dans un seul groupe traité à un seul temps de prélèvement. En premier lieu, il s'agit de déterminer la pertinence biologique des résultats. L'évaluation des résultats peut s'appuyer sur des méthodes statistiques (6) mais la signification statistique ne doit pas être le seul facteur décisif pour conclure à une réponse positive. Les résultats équivoques doivent être clarifiés par des essais supplémentaires réalisés de préférence dans des conditions expérimentales modifiées.
Une augmentation du nombre de cellules polyploïdes peut signifier que la substance d'essai est capable d'induire des aberrations de nombre. Une augmentation du nombre de cellules présentant des chromosomes endoredupliqués peut indiquer que la substance d'essai est capable d'inhiber la progression du cycle cellulaire (7) (8).
Si les résultats obtenus avec une substance d'essai ne répondent pas aux critères indiqués ci-dessus, on peut conclure que la substance en question n'est pas mutagène dans cet essai.
Bien que la plupart des essais donneront des résultats manifestement positifs ou négatifs, dans de rares cas l'ensemble des résultats d'un essai n'autorise pas de conclusion catégorique sur l'activité de la substance étudiée. Les résultats peuvent rester équivoques ou discutables indépendamment du nombre de répétitions de l'essai.
Des résultats positifs obtenus dans cet essai in vivo d'aberration chromosomique indiquent qu'une substance induit des aberrations chromosomiques dans la moelle osseuse de l'espèce testée. Des résultats négatifs signifient que, dans les conditions de l'essai, la substance testée n'induit pas d'aberrations chromosomiques dans la moelle osseuse de l'espèce testée.
Le fait que la substance d'essai ou ses métabolites atteignent la circulation générale ou spécifiquement le tissu cible (toxicité systémique) doit être discuté.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Solvant/véhicule:
|
—
|
justification du choix du véhicule,
|
|
—
|
solubilité et stabilité de la substance d'essai dans le solvant/véhicule, si elles sont connues.
|
|
|
|
Animaux d'expérience:
|
—
|
espèce/souche utilisée,
|
|
—
|
nombre, âge et sexe des animaux,
|
|
—
|
source, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
poids individuel des animaux au début de l'essai, y compris l'intervalle des poids, la moyenne et l'écart-type dans chaque groupe.
|
|
|
|
Conditions expérimentales:
|
—
|
données relatives aux témoins positifs et négatifs (solvant/véhicule),
|
|
—
|
résultats de l'étude de détermination des doses utiles, si elle a été réalisée,
|
|
—
|
justification du choix des doses employées,
|
|
—
|
détails concernant la préparation de la substance d'essai,
|
|
—
|
détails concernant l'administration de la substance d'essai,
|
|
—
|
justification du choix de la voie d'administration,
|
|
—
|
méthodes pour vérifier que la substance a atteint la grande circulation ou le tissu cible, le cas échéant,
|
|
—
|
conversion de la concentration (ppm) de la substance d'essai dans l'eau de boisson ou la nourriture en dose réelle (mg/kg de poids corporel/jour), s'il y a lieu,
|
|
—
|
détails sur la qualité de la nourriture et de l'eau,
|
|
—
|
description détaillée des modalités de traitement et de prélèvement,
|
|
—
|
méthodes de mesure de la toxicité,
|
|
—
|
nature et concentration de l'inhibiteur du fuseau mitotique et durée du traitement,
|
|
—
|
méthodes de préparation des lames,
|
|
—
|
critères de dénombrement des aberrations,
|
|
—
|
nombre de cellules analysées par animal,
|
|
—
|
critères de décision concernant les résultats positifs, négatifs et équivoques.
|
|
|
|
Résultats:
|
—
|
type et nombre d'aberrations, séparément pour chaque animal,
|
|
—
|
nombre total d'aberrations par groupe, moyennes et écarts-types,
|
|
—
|
nombre de cellules présentant des aberrations par groupe, moyennes et écarts-types,
|
|
—
|
changements de la ploïdie, le cas échéant,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
analyses statistiques, le cas échéant,
|
|
—
|
données concernant les témoins négatifs concomitants,
|
|
—
|
données concernant les témoins négatifs antérieurs (étendues, moyennes et écarts-types),
|
|
—
|
données concernant les témoins positifs concomitants.
|
|
|
|
Discussion des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
Adler, J. D. (1984), Cytogenctic Tests in Mammals, in: Mutagenidty Testing: a Practical Approach. S. Vcnitt and J. M. Parry (eds.). IRL Press, Oxford, Washington D. C, pp. 275-306.
|
|
(2)
|
Preston, R. J., Dean, B. J., Galloway, S., Holden, H., McFee, A. F. and Shelby, M. (1987), Mammalian In Vivo Cytogenetic Assays: Analysis of Chromosome Aberrations in Bone Marrow Cells, Mutation Res., 189, 157-165.
|
|
(3)
|
Richold, M.. Chandly, A., Ashby, J., Gatehouse D. G., Bootman, J. and Henderson, L. (1990), In Vivo Cytogenetic Assays, in: D. J. Kirkland, (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing Report. Part I revised, Cambridge University Press, Cambridge, New Cork, Port Chester, Melbourne, Sydney, pp. 115-141.
|
|
(4)
|
Tice, R. R. Hayashi, M., MacGregor, J. T., Anderson, D., Blakey, D. H., Holden, H. E., Kirch-Volders, M, Oleson Jr., F. B., Paccierotti, F., Preston R. J., Romagna F., Shimada, H., Sutou, S. and Vannier, B. (1994), Report from the Working Group on the In Vivo Mammalian Bone Marrow Chromosomal Aberration Test, Mutation Res., 312, pp. 305-312.
|
|
(5)
|
Fielder, R. J., Alleen, J. A., Boobis, A. R., Botham, P. A.. Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson- Walker, G., Morton, D. B., Kirkland, D. J. and Richold, M. (1992), Report of British Toxicology Society/UK, Environmental Mutagen Society Working Group: Dose setting in In Vivo Mutagenicity Assays, Mutagenesis, 7, pp. 313-319.
|
|
(6)
|
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Pap- worth, D. G. and Savage, J. R. K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays, in: UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report Part III. Statistical Evaluation of Mutagenicity Test Data, D. J. Kirkland, (ed.) Cambridge University Press, Cambridge, pp. 184-232.
|
|
(7)
|
Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation-induced G2 arrest, Mutation Res., 119, pp. 403-413.
|
|
(8)
|
Huang, Y., Change, C. and Trosko, J. E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells, Cancer Res., 43, pp. 1363-1364.
|
B.12. MUTAGÉNICITÉ — ESSAI IN VIVO DU MICRONOYAU SUR ÉRYTHROCYTES DE MAMMIFÈRE
1. MÉTHODE
Cette méthode est une adaptation de la ligne directrice OCDE TG 474, Test du micronoyau sur les érythrocytes de mammifère (1997).
1.1. INTRODUCTION
L'essai du micronoyau in vivo sur des cellules de mammifère est pratiqué en vue de détecter des lésions des chromosomes ou de l'appareil mitotique d'érythroblastes résultant de l'action d'une substance d'essai. Il repose sur l'analyse d'érythrocytes prélevés dans la moelle osseuse et/ou dans le sang périphérique d'animaux, généralement des rongeurs.
L'essai du micronoyau est destiné à identifier les substances qui provoquent les lésions cytogénétiques se traduisant par la formation de micronoyaux contenant des fragments de chromosomes ou des chromosomes entiers.
Lorsqu'un érythroblaste de moelle osseuse se transforme en érythrocyte polychromatique, le noyau principal est expulsé et chaque micronoyau qui s'est formé peut subsister dans le cytoplasme anucléé. La visualisation des micronoyaux est facilitée par l'absence du noyau principal. Un accroissement de la fréquence des micronoyaux dans les érythrocytes polychromatiques des animaux traités est le signe d'une lésion chromosomique induite.
La moelle osseuse des rongeurs est couramment utilisée dans cet essai, les érythrocytes polychromatiques étant produits dans ce tissu. Le comptage des érythrocytes (polychromatiques) immatures micronucléés dans le sang périphérique est également acceptable chez toute espèce où l'on a démontré l'incapacité de la rate à éliminer les érythrocytes micronucléés ou qui présente une sensibilité suffisante pour permettre la détection des agents provoquant des aberrations chromosomiques de structure ou de nombre. Plusieurs critères permettent de distinguer les micronoyaux. Un de ces critères est la détection de la présence ou de l'absence dans les micronoyaux d'un centromère ou d'ADN centromérique. La fréquence des érythrocytes immatures (polychromatiques) micronucléés est le principal critère. Quand les animaux sont traités en continu pendant quatre semaines ou plus, on peut également rechercher dans le sang périphérique la proportion des érythrocytes matures (normochromatiques) contenant des micronoyaux par rapport à un nombre donné d'érythrocytes matures.
Cet essai in vivo du micronoyau pratiqué sur des érythrocytes de mammifère se prête particulièrement bien à l'évaluation des risques mutagènes, en ce sens qu'il permet de prendre en compte des facteurs du métabolisme in vivo, de pharmacocinétique et des processus de réparation de l'ADN, malgré la variabilité de ces facteurs en fonction de l'espèce, du tissu et de l'effet génétique recherché. Un essai in vivo sert aussi à vérifier un effet mutagène détecté dans un système in vitro.
On n'aura pas recours à cet essai s'il est manifeste que la substance d'essai ou un métabolite réactif n'atteindra pas le tissu cible.
Voir aussi l'introduction générale de la partie B.
1.2. DÉFINITIONS
Centromère: portion d'un chromosome qui au cours de la division cellulaire est lié aux fibres du fuseau mitotique permettant un mouvement ordonné des chromosomes filles vers les pôles des cellules filles.
Micronoyau: petit noyau, présent en plus du noyau principal des cellules et séparé de celui-ci, produit pendant la télophase de la mitose (méiose) par des chromosomes ou des fragments de chromosomes retardataires.
Érythrocyte normochromatique: érythrocyte mature dépourvu de ribosomes et pouvant être distingué des érythrocytes immatures polychromatiques à l'aide de substances colorant sélectivement les ribosomes.
Érythrocyte polychromatique: érythrocyte immature se trouvant à un stade de développement intermédiaire, qui renferme encore des ribosomes et peut ainsi être distingué des érythrocytes matures normochromatiques à l'aide de substances colorant sélectivement les ribosomes.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
Les animaux sont exposés à la substance d'essai par une voie d'exposition appropriée. Si on utilise la moelle osseuse, les animaux sont sacrifiés à des moments appropriés après le traitement, la moelle est extraite, préparée sur des lames et colorée (1) (2) (3) (4) (5) (6) (7). Lorsqu'on utilise le sang périphérique, ce dernier est prélevé à des moments appropriés après le traitement, les cellules sanguines sont étalées sur des lames et colorées (4) (8) (9) (10). En cas d'essai sur cellules du sang périphérique, il faut recueillir les cellules aussi rapidement que possible après la dernière exposition. Les lames préparées sont soumises à une analyse en vue de la détection de micronoyaux.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparations
1.4.1.1. Sélection des espèces animales
L'utilisation de souris ou de rats est recommandée en cas d'essai sur moelle osseuse, bien que n'importe quelle espèce de mammifère appropriée puisse être employée. Si l'essai porte sur le sang périphérique, on recommande d'utiliser des souris. Il est toutefois possible d'utiliser n'importe quelle espèce de mammifère appropriée, à condition que sa rate n'élimine pas les érythrocytes micronucléés ou que sa sensibilité soit suffisante pour permettre la détection d'agents qui provoquent des aberrations chromosomiques de structure ou de nombre. Il convient d'utiliser des animaux adultes, jeunes et sains issus de souches couramment utilisées en laboratoire. Au début de l'étude, la différence pondérale entre les animaux ne doit pas dépasser ± 20 % du poids moyen de chaque sexe.
1.4.1.2. Conditions d'encagement et régime alimentaire
Les conditions générales décrites dans l'introduction générale de la partie B sont appliquées mais le taux d'humidité devrait se situer aux environs de 50-60 %.
1.4.1.3. Préparations des animaux
Les animaux sont répartis par randomisation entre les groupes témoins et les groupes traités. Les animaux sont identifiés individuellement. Ils sont gardés dans leur cage pendant au moins cinq jours avant le début de l'étude, de façon à les acclimater aux conditions du laboratoire. Les cages doivent être disposées de telle manière qu'il n'y ait pas d'effet dû à leur emplacement.
1.4.1.4. Préparation des doses
Les substances d'essai solides sont dissoutes ou mises en suspension dans un véhicule ou un solvant approprié, puis éventuellement diluées avant d'être administrées aux animaux. Les substances d'essai liquides peuvent être administrées directement ou après dilution. Il convient d'utiliser des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage.
1.4.2. Conditions expérimentales
1.4.2.1. Solvant/véhicule
Le solvant/véhicule choisi ne doit pas avoir d'effet toxique aux doses utilisées ni présenter de réaction chimique avec la substance d'essai. Le recours à des solvants/véhicules inhabituels doit être justifié par des données faisant état de leur compatibilité. On préconise d'envisager d'abord l'utilisation d'un véhicule/solvant aqueux, chaque fois que cela est possible.
1.4.2.2. Témoins
Des témoins positifs et négatifs (solvant ou véhicule) doivent être inclus pour chaque sexe et chaque essai. Les animaux des groupes témoins sont manipulés exactement comme ceux des groupes traités, mise à part l'administration de la substance d'essai.
Les témoins positifs doivent produire des micronoyaux in vivo à des niveaux d'exposition supposés conduire à un accroissement détectable par rapport au bruit de fond. Les doses de témoin positif doivent être choisies de façon que les effets soient nets et que l'identité des lames codées ne soit pas révélée immédiatement à l'examinateur. Le témoin positif peut être administré par une voie différente de celle utilisée pour la substance d'essai et un seul prélèvement d'échantillon peut suffire. En outre, l'emploi de témoins positifs appartenant à la même classe chimique peut être envisagé, s'ils sont disponibles. Les substances indiquées dans le tableau ci-dessous sont des exemples de témoins positifs.
|
Substance
|
Numéro CAS
|
Numéro Einecs
|
|
Méthanesulfonate d'éthyle
|
62-50-0
|
200-536-7
|
|
N-éthyl N-nitrosourée
|
759-73-9
|
212-072-2
|
|
Mitomycine C
|
50-07-7
|
200-008-6
|
|
Cyclophosphamide
Monohydrate de cyclophosphamide
|
50-18-0
6055-19-2
|
200-015-4
|
|
Triéthylènemélamine
|
51-18-3
|
200-083-5
|
Lors de chaque prélèvement, il faut inclure un lot d'animaux témoins négatifs traités seulement avec le solvant ou le véhicule et par ailleurs manipulés de la même façon que les groupes traités. Ceci peut être évité si l'on dispose de données acceptables sur la variabilité interindividuelle et la fréquence spontanée de cellules comportant des micronoyaux provenant de témoins historiques. Dans le cas où un seul prélèvement est effectué pour les témoins négatifs, le premier temps de prélèvement est le plus approprié. En outre, des témoins non traités doivent aussi être inclus, à moins que des données historiques ne démontrent que le solvant/véhicule choisi n'induit aucun effet délétère ou mutagène.
Si le sang périphérique est utilisé, un échantillon prélevé avant le traitement peut aussi tenir lieu de témoin négatif, mais uniquement dans les études de courte durée sur sang périphérique (par exemple 1 à 3 traitements) lorsque les résultats se situent dans l'intervalle auquel on peut s'attendre sur la base de données historiques.
1.5. MODE OPÉRATOIRE
1.5.1. Nombre et sexe des animaux
Chaque groupe traité et groupe témoin doit comprendre au moins cinq animaux analysables par sexe (11). Si, au moment de l'étude, on dispose de données d'études antérieures effectuées avec la même espèce et ayant utilisé la même voie d'administration, qui démontrent l'absence de différence importante de toxicité entre les sexes, il suffit d'effectuer l'essai avec des animaux d'un seul sexe. Si l'exposition humaine est spécifique du sexe, par exemple dans le cas de certains produits pharmaceutiques, des animaux du sexe approprié doivent être utilisés pour l'essai.
1.5.2. Modalité de traitement
Aucune modalité particulière de traitement (un, deux ou davantage de traitements à intervalles de 24 heures) ne peut être recommandée. Les prélèvements qui proviennent d'une étude avec des procédures d'administration prolongées sont acceptables si l'essai est positif ou, pour les essais négatifs, si un effet toxique est observé ou que la dose limite a été employée et que la substance a été administrée jusqu'au moment du prélèvement. La substance d'essai peut aussi être administrée en doses fractionnées (deux doses le même jour avec un intervalle de quelques heures au maximum) afin que l'administration d'un grand volume soit plus aisée.
L'essai peut être effectué de deux façons:
|
a)
|
La substance d'essai est administrée en une seule fois aux animaux. Des échantillons de moelle osseuse sont prélevés au moins deux fois, jamais avant 24 heures et pas au-delà de 48 heures après le traitement et à un intervalle approprié. Des prélèvements effectués moins de 24 heures après le traitement doivent avoir une justification. Les échantillons de sang périphérique sont prélevés au moins deux fois jamais avant 36 heures et pas au-delà de 72 heures après le traitement et à un intervalle approprié. Si une réponse positive est observée lors d'un prélèvement, il n'est pas nécessaire de procéder à un autre prélèvement.
|
|
b)
|
Dans le cas où deux doses ou plus sont administrées (par exemple deux doses ou plus à intervalle de 24 heures), les échantillons de moelle osseuse doivent être prélevés une seule fois entre 18 et 24 heures après le dernier traitement, et ceux de sang périphérique une seule fois entre 36 et 48 heures après le dernier traitement (12).
|
Des temps de prélèvements supplémentaires peuvent être envisagés si nécessaire.
1.3.3. Niveaux de dose
Si une étude préliminaire est réalisée afin d'évaluer les niveaux de dose requis, cette étude doit être effectuée dans le même laboratoire en utilisant la même espèce, la même souche, des animaux du même sexe et la même modalité de traitement que ceux de l'étude principale (13). En cas de toxicité, on utilise trois niveaux de dose pour le premier prélèvement. Ces niveaux de doses doivent couvrir un domaine allant d'une toxicité nulle ou légère à la toxicité maximale. Pour les prélèvements suivants, seule la dose la plus élevée est utilisée. Celle-ci est définie comme la dose qui produit des signes de toxicité tels qu'ils font supposer que des doses plus fortes, administrées selon les mêmes modalités, seraient létales. Les substances possédant une activité biologique spécifique à des doses faibles et non toxiques (comme les hormones et les mitogènes) peuvent faire exception aux critères d'établissement des doses et doivent être évaluées cas par cas. La dose la plus élevée peut aussi être définie comme étant celle qui produit certains indices de toxicité dans la moelle osseuse (par exemple une diminution de la proportion d'érythrocytes immatures par rapport au nombre total d'érythrocytes dans la moelle osseuse ou le sang périphérique).
1.5.4. Test limite
Si un essai réalisé avec au moins 2 000 mg/kg de poids corporel, en une seule dose ou en deux doses administrées le même jour, n'engendre aucun effet toxique observable et si une génotoxicité est improbable d'après les données relatives à des substances de structure voisine, on peut considérer qu'une étude complète avec trois niveaux de doses n'est pas nécessaire. Pour les études de plus longue durée, la dose limite est de 2 000 mg/kg de poids corporel par jour dans le cas d'un traitement d'une durée allant jusqu'à 14 jours et de 1 000 mg/kg de poids corporel par jour pour les études plus longues. Suivant l'importance de l'exposition humaine probable, on peut envisager d'utiliser des doses plus fortes dans l'essai de dose limite.
1.5.5. Voie d'administration
La substance d'essai est généralement administrée par gavage à l'aide dune sonde gastrique ou d'une canule d'intubation adaptée, ou par injection intrapéritonéale. D'autres voies d'exposition sont acceptables lorsqu'elles peuvent être justifiées. Le volume maximal de liquide administré en une fois par gavage ou par injection dépend de la taille de l'animal. Ce volume ne devrait pas dépasser 2 ml/100 g de poids corporel. Une justification doit être fournie si des volumes plus importants sont administrés. Il convient de réduire au minimum la variation du volume administré en ajustant la concentration à tous les niveaux de doses, sauf pour les produits irritants ou corrosifs dont les effets sont généralement exacerbés lorsqu'on augmente la concentration.
1.5.6. Préparation de la moelle osseuse et du sang
Les cellules de moelle osseuse sont généralement prélevées dans le fémur ou le tibia immédiatement après le sacrifice. Les cellules sont extraites, préparées et colorées suivant des méthodes bien établies. Le sang périphérique est prélevé dans la veine caudale ou un autre vaisseau sanguin approprié. On effectue immédiatement une coloration supravitale des cellules sanguines (8) (9) (10) ou des frottis qui sont ensuite colorés. L'emploi d'un colorant spécifique de l'ADN [par exemple, l'organe d'acridine (14) ou Hoechst 33258 plus pyronine-Y (15)] peut éliminer une partie des artefacts dus à l'utilisation d'un colorant non spécifique de l'ADN. Cet avantage n'exclut pas l'emploi de colorants classiques (Giemsa, par exemple). D'autres méthodes, telles que celle basée sur l'emploi de colonnes de cellulose pour enlever les cellules nucléées (16), peuvent être utilisées à condition que leur efficacité pour la préparation de micronoyaux dans le laboratoire ait été prouvée.
1.5.7. Analyse
Le pourcentage d'érythrocytes immatures par rapport au nombre total d'érythrocytes (immatures + matures) est déterminé pour chaque animal en comptant au moins 200 érythrocytes dans le cas de la moelle osseuse et 1 000 érythrocytes dans le cas du sang périphérique (17). Toutes les lames, y compris celles des témoins positifs et négatifs, doivent êtres codées indépendamment avant l'analyse au microscope. L'incidence des érythrocytes immatures micronucléés est déterminée sur la base de l'examen d'au moins 2 000 érythrocytes immatures par animal. On peut obtenir des informations supplémentaires en recherchant la présence de micronoyaux dans les érythrocytes matures. A l'examen des lames, le pourcentage d'érythrocytes immatures par rapport au nombre total d'érythrocytes ne doit pas être inférieur à 20 % de la valeur des témoins. Lorsque les animaux sont traités en continu pendant 4 semaines ou davantage, on peut aussi examiner au moins 2 000 érythrocytes matures par animal pour déterminer l'incidence des érythrocytes matures micronucléés. Des systèmes d'analyse automatisée (analyse d'images et cytométrie de flux pour cellules en suspension) peuvent remplacer des techniques manuelles s'ils sont justifiés et validés.
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
Les résultats individuels pour chaque animal doivent être présentés sous forme de tableaux. L'unité expérimentale est l'animal. Le nombre d'érythrocytes immatures examinés, le nombre d'érythrocytes immatures micronucléés et le nombre d'érythrocytes immatures par rapport au nombre total d'érythrocytes doivent être présentés séparément pour chaque animal analysé. Lorsque les animaux sont traités en continu pendant quatre semaines ou davantage, il convient de fournir également les données relatives aux érythrocytes matures, si elles ont été recueillies. Le pourcentage d'érythrocytes immatures par rapport au nombre total d'érythrocytes et, si cela est jugé pertinent, le pourcentage d'érythrocytes micronucléés seront indiqués pour chaque animal. S'il n'y a pas de différence de réponse entre sexes, les données des deux sexes peuvent être combinées dans l'analyse statistique.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Plusieurs critères permettent de déterminer si un résultat est positif, notamment une augmentation, dose-dépendante, du nombre de cellules micronucléées ou une augmentation nette du nombre de cellules micronucléées dans un seul groupe traité à un seul temps de prélèvement. En premier lieu, il s'agit de déterminer la pertinence biologique des résultats. L'évaluation des résultats peut s'appuyer sur des méthodes statistiques (18) (19) mais la signification statistique ne doit pas être le seul facteur décisif pour conclure à une réponse positive. Les résultats équivoques doivent être clarifiés par des essais supplémentaires réalisés de préférence dans des conditions expérimentales modifiées.
Si les résultats obtenus avec une substance d'essai ne répondent pas aux critères indiqués ci-dessus, on peut conclure que la substance en question n'est pas mutagène dans cet essai.
Bien que la plupart des essais donneront des résultats nettement positifs ou négatifs, dans certains cas rares l'ensemble des résultats d'un essai n'autorise pas un jugement catégorique sur l'activité de la substance étudiée. Les résultats peuvent rester équivoques ou discutables indépendamment du nombre de répétitions de l'essai.
Des résultats positifs obtenus dans cet essai in vivo du micronoyau indiquent que la substance induit la formation de micronoyaux qui résultent de lésions des chromosomes ou de l'appareil mitotique dans les érythroblastes de l'espèce testée. Des résultats négatifs signifient que, dans les conditions de l'essai la substance testée n'induit pas la formation de micronoyaux dans les érythrocytes immatures de l'espèce testée.
Le fait que la substance d'essai ou ses métabolites atteignent la circulation générale ou spécifiquement le tissu cible (toxicité systématique) doit être discute.
3. RAPPORT
RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Solvant/véhicule:
|
—
|
justification du choix du véhicule,
|
|
—
|
solubilité et stabilité de la substance d'essai dans le solvant/véhicule, si elles sont connues.
|
|
|
|
Animaux d'expérience:
|
—
|
espèce/souche utilisée,
|
|
—
|
nombre, âge et sexe des animaux,
|
|
—
|
source, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
poids individuel des animaux au début de l'essai, y compris l'intervalle des poids, la moyenne et l'écart-type dans chaque groupe.
|
|
|
|
Conditions expérimentales:
|
—
|
données relatives aux témoins positifs et négatifs (solvant/véhicule),
|
|
—
|
résultats de l'étude de détermination des doses utiles, si elle a été réalisée,
|
|
—
|
justification du choix des doses employées,
|
|
—
|
détails concernant la préparation de la substance d'essai,
|
|
—
|
détails concernant l'administration de la substance d'essai,
|
|
—
|
justification du choix de la voie d'administration,
|
|
—
|
méthodes pour vérifier que la substance a atteint la circulation générale ou le tissu cible, le cas échéant,
|
|
—
|
conversion de la concentration (ppm) de la substance d'essai dans l'eau de boisson ou la nourriture en dose réelle (mg/kg de poids corporel/jour), s'il y a lieu,
|
|
—
|
détails sur la qualité de la nourriture et de l'eau,
|
|
—
|
description détaillée des modalités de traitement et de prélèvement,
|
|
—
|
méthodes de préparation des lames,
|
|
—
|
méthodes de mesure de la toxicité,
|
|
—
|
critères de dénombrement des érythrocytes immatures micronuclées,
|
|
—
|
nombre de cellules analysées par animal,
|
|
—
|
critères de décision concernant les résultats positifs, négatifs et équivoques.
|
|
|
|
Résultats:
|
—
|
proportion d'érythrocytes immatures par rapport au nombre total d'érythrocytes,
|
|
—
|
nombre d'érythrocytes immatures micronuclées, donné séparément pour chaque animal,
|
|
—
|
moyenne ± écart-type des érythrocytes micronuclées immatures par groupe,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
analyses et méthodes statistiques appliquées,
|
|
—
|
données concernant les témoins négatifs concomitants et antérieurs,
|
|
—
|
données concernant les témoins positifs concomitants.
|
|
|
|
Discussion des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
Heddle, J. A. (1973), A Rapid In Vivo Test for Chromosomal Damage, Mutation Res., 18, pp. 187-190.
|
|
(2)
|
Schmid, W. (1975), The Micronucleus Test, Mutation Res., 31, pp. 9-15.
|
|
(3)
|
Heddle, J. A., Salamone, M. F., Hite, M., Kirkhart, B., Mavournin. K., MacGregor, J. G. and Newell, G. W. (1983), The Induction of Micronuclei as a Measure of Genotoxicity, Mutation Res., 123, pp. 61-118.
|
|
(4)
|
Mavournin, K. H., Blakey, D. H., Cimino, M. C, Salamone, M. F. and Heddle, J. A. (1990), The In Vivo Micronucleus Assay in Mammalian Bone Marrow and Peripheral Blood. A report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 239, pp. 29-80.
|
|
(5)
|
MacGregor, J. T., Schlegel, R., Choy, W. N., and Wehr, C. M. (1983), Micronuclei in Circulating Erythrocytcs: A Rapid Screen for Chromosomal Damage During Routine Toxicity Testing in Mice, in: Developments in Science and Practice of Toxicology, ed. A. W. Hayes, R. C. Schnell and T. S. Miya, Elsevier, Amsterdam, pp. , 555-558.
|
|
(6)
|
MacGregor, J. T., Heddle, J. A., Hite, M., Margolin, G. H., Ramel, C, Salamone, M. F., Tice, R. R. and Wild, D. (1987), Guidelines for the Conduct of Micronucleus Assays in Mammalian Bone Marrow Erythrocytes, Mutation Res., 189, pp. 103-112.
|
|
(7)
|
MacGregor, J. T., Wehr, C. M., Henika, P. R., and Shelby, M. E. (1990), The in vivo Erythrocyte Micronucleus Test: Measurement at Steady State Increases Assay Efficiency and Permits Integration with Toxicity Studies, Fund. Appl. Toxicol. 14, pp. 513-522.
|
|
(8)
|
Hayashi, M., Morita, T., Kodama, Y., Sofuni, T. and Ishidate, M. Jr. (1990), The Micronucleus Assay with Mouse Peripheral Blood Reticulocytes Using Acridine Orange-Coated Slides, Mutation Res., 245, pp. 245-249.
|
|
(9)
|
The Collaborative Study Group for the Micronucleus Test (1992). Micronucleus Test with Mouse Peripheral Blood Erythrocytes by Acridine Orange Supravital Staining: The Summary Report of the 5th Collaborative Study by CSGMT/JEMMS, MMS, Mutation Res., 278, pp. 83-98.
|
|
(10)
|
The Collaborative Study Group for the Micronucleus Test (CSGMT/JEMMS, MMS: The Mammalian Mutagenesis Study Group of the Environmental Mutagen Society of Japan) (1995). Protocol recommended for the short-term mouse peripheral blood micronucleus test, Mutagenesis, 10, pp. 153-159.
|
|
(11)
|
Hayashi, M., Tice, R. R., MacGregor, J. T., Anderson, D., Blackey, D. H., Kirsch-Volders, M., Oleson, Jr. F. B., Pacchierotti, F., Romagna, F., Shimada, H., Sutou, S. and Vannier, B. (1994), in vivo, Rodent Erythrocyte Micronucleus Assay, Mutation Res., 312, pp. 293-304.
|
|
(12)
|
Higashikuni, N. and Sutou, S. (1995), An optimal, generalised sampling time of 30 16 h after double dosing in the mouse peripheral blood micronucleus test, Mutagenesis. 10, pp. 313-319.
|
|
(13)
|
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson-Walker, G., Morton, D. B., Kirkland, D. J. and Rochold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose Setting in in vivo Mutagenicity Assays, Mutagenesis, 7, pp. 313-319.
|
|
(14)
|
Hayashi, M., Sofuni, T. and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, pp. 241-247.
|
|
(15)
|
MacGregor, J. T., Wehr, C. M. and Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 3 3258 and Pyronin Y, Mutation Res., 120, pp. 269-275.
|
|
(16)
|
Romagna, F. and Staniforth, C. D. (1989), The automated bone marrow micronucleus test, Mutation Res., 211, pp. 91-104.
|
|
(17)
|
Gollapudi. B. and McFadden. L. G. (1995), Sample size for the estimation of polychromatic to normochromatic erythrocyte ratio in the bone marrow micronucleus test. Mutation Res., 347, pp. 97-99.
|
|
(18)
|
Richold, M., Ashby, J., Bootman, J., Chandley, A., Gatehouse, D. G. and Henderson, L. (1990), In vivo Cytogenetics Assay, in: D. J. Kirkland (ed.), Basic Mutagenicity tests. L/KEMS Recommended Procedures, UKEMS Sub-Committee on Guidelines for Mutagenicity Testing Report, Part 1. revised. Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 115-141.
|
|
(19)
|
Lovell. D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D. G. and Savage, J. R. K. (1989), Statistical Analysis of In vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Statistical Evaluation of Mutagenicity Test Data. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report, Part III, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 184-232.
|
B.13/14. MUTAGÉNICITÉ — ESSAI DE MUTATION RÉVERSE SUR BACTÉRIES
1. MÉTHODE
Cette méthode est une adaptation de la ligne directrice OCDE TG 471, Essai de mutation réverse sur des bactéries (1997).
1.1. INTRODUCTION
L'essai bactérien de mutation réverse est pratiqué sur des souches de Salmonella typhimurium et d'Escherichia coït auxotrophes à l'égard d'un acide aminé. Il sert à détecter des mutations ponctuelles résultant de la substitution, de l'addition ou de la délétion d'une ou de plusieurs paires de base de l'ADN (1) (2) (3). Le principe de cet essai repose sur la détection de mutations qui inversent des mutations présentes dans la souche d'essai et rétablissent ainsi la capacité fonctionnelle des bactéries de synthétiser un acide aminé essentiel. Les bactéries révertantes sont détectées d'après leur capacité de se développer en l'absence de l'acide aminé requis par la souche d'essai parentale.
Les mutations ponctuelles sont à l'origine de beaucoup de maladies génétiques humaines et de nombreuses données tendent à démontrer que des mutations ponctuelles frappant les oncogènes et les gènes suppresseurs de tumeurs dans des cellules somatiques jouent un rôle dans le développement de tumeurs tant chez l'homme que chez les animaux de laboratoire. L'essai bactérien de mutation réverse est rapide, peu coûteux et relativement facile à effectuer. Beaucoup de souches utilisées possèdent entre autres particularités, celles de les rendre plus sensibles aux mutations: séquences d'ADN plus sensibles aux mutations au niveau des sites de réversion, augmentation de perméabilité cellulaire pour les grosses molécules, élimination des systèmes de réparation de l'ADN ou favorisation (augmentation) des processus induisant des erreurs lors de la réparation de l'ADN. La spécificité des souches d'essai peut fournir des informations utiles sur les types de mutations induites par les agents génotoxiques. On dispose pour les essais bactériens de mutation réverse de très nombreux résultats pour une grande variété de structures et d'une méthodologie d'essai qui est bien au point et qui peut être appliquée à des substances ayant des propriétés physico-chimiques très diverses, y compris les composés volatils.
Voir aussi l'introduction générale de la partie B.
1.2. DÉFINITIONS
Un essai de mutation réverse chez Salmonella typhimurium ou Escherichia coli détecte, chez une souche dont la croissance requiert la présence d'un acide aminé (respectivement histidine ou trytophane), des mutations qui transforment cette souche en une souche dont la croissance s'effectue indépendamment d'un rapport extérieur de cet acide aminé.
Les mutagènes provoquant la substitution de paires de base sont des agents qui entraînent le remplacement d'une base de l'ADN. Lors d'un essai de réversion, cette modification peut avoir lieu sur le site de la mutation initiale ou sur un autre site du génome bactérien.
Les mutagènes décalant le cadre de lecture sont des agents qui provoquent l'addition ou la délétion d'une ou de plusieurs paires de base dans l'ADN, modifiant ainsi le cadre de lecture dans l'ARN.
1.3. CONSIDÉRATIONS PRÉLIMINAIRES
L'essai bactérien de mutation réverse porte sur des cellules procaryotes qui différent des cellules de mammifère notamment du point de vue de l'absorption, du métabolisme, de la structure des chromosomes et des processus de répartition de l'ADN. Les essais in vitro nécessitent généralement une activation métabolique exogène. Comme les systèmes d'activation métabolique in vitro ne peuvent pas reproduire parfaitement le métabolisme de cellules de mammifère in vivo, les essais ne fournissent pas directement des informations sur le pouvoir mutagène et carcinogène d'une substance chez les mammifères.
L'essai de mutation réverse sur bactéries est couramment employé comme première étape pour la détection de l'activité génotoxique en général et des mutations ponctuelles en particulier. De nombreuses données démontrent que beaucoup de substances chimiques qui se révèlent positives dans cet essai présentent aussi une activité mutagène dans d'autres essais. Certains agents mutagènes ne sont pas décelés par ces systèmes d'essai. Ces défaillances peuvent tenir à la nature particulière de l'effet détecté, à des différences sur le plan de l'activation métabolique ou à des différences de biodisponibilité. D'autre part, les facteurs qui amplifient la sensibilité de l'essai de mutation réverse sur bactéries peuvent conduire à une surestimation de l'activité mutagène.
L'essai de mutation réverse sur bactéries peut ne pas convenir pour l'évaluation de certaines classes de substances chimiques, par exemple les composés fortement bactéricides (certains antibiotiques, par exemple) et ceux dont on soupçonne (ou on sait) qu'ils interfèrent spécifiquement avec le système de réplication des cellules de mammifère (par exemple les inhibiteurs de la topoisomérase ou certains analogues de nucléosides). Dans de tels cas, des essais de mutation sur des cellules de mammifère peuvent s'avérer plus appropriés.
Bien que de nombreux composés qui donnent un résultat positif à cet essai soient cancérogènes pour les mammifères, la corrélation n'est pas parfaite. Celle-ci dépend de la classe chimique et il existe des substances cancérogènes qui ne sont pas détectées par cet essai parce qu'elles agissent par des mécanismes non génotoxiques ou par des mécanismes qui sont absents des cellules bactériennes.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Des bactéries en suspension sont exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique exogène. Dans le cas de la méthode d'incorporation directe (méthode par étalement), ces suspensions sont mélangées avec la gélose de surface et déposées immédiatement sur le milieu minimal. Dans le cas de la méthode avec préincubation, le mélange de bactéries en suspension-substance testée est incubé et ensuite mélangé avec un couche de gélose de surface avant d'être étalé sur le milieu minimal. Dans les deux techniques, après deux ou trois jours d'incubation, les colonies révertantes sont comptées et leur nombre est comparé à celui des revenants spontanés dans les boîtes témoins traitées avec le solvant.
Plusieurs méthodes permettant de réaliser l'essai de mutation réverse sur bactéries ont été décrites. Parmi celles qui sont communément utilisées figure la méthode par incorporation directe (1) (2) (3) (4), la méthode de la préincubation (2) (3) (5) (6) (7) (8), la méthode de la fluctuation (9) (10) et la méthode par suspension (11). Des méthodes modifiées pour les essais avec des gaz ou des vapeurs ont été décrites (12).
Les modes opératoires décrits se réfèrent principalement aux méthodes d'incorporation directe et de préincubation. Chacune de ces méthodes peut être utilisée avec et sans activation métabolique. L'utilisation de la méthode avec préincubation permet de détecter plus efficacement l'effet mutagène de certaines substances. Ces substances appartiennent à des classes chimiques qui comprennent les nitrosamines aliphatiques à chaînes courtes, les métaux bivalents, les aldéhydes, les colorants azoïques et les composés diazoïques, les alcaloïdes pyrollizidiniques, les composés allyliques et nitrés (3). Il est également admis que certaines classes de mutagènes ne sont pas toujours détectées par les méthodes conventionnelles telles que l'incorporation directe ou la préincubation. Ces substances doivent être considérées comme des cas particuliers et il est fortement recommandé d'utiliser d'autres méthodes de détection. On a pu mettre en évidence les cas particuliers suivants (ainsi que des exemples de méthodes qui pourraient servir à leur détection): colorants azoïques et composés diazoïques (3) (5) (6) (13), gaz et substances chimiques volatiles (12) (14) (15) (16), glycosides (17) (18). Tout écart par rapport au mode opératoire standard demande à être scientifiquement justifié.
1.5. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.5.1. Préparations
1.5.1.1. Bactéries
On laisse des cultures fraîches de bactéries se développer jusqu'à la fin de la phase exponentielle ou jusqu'au début de la phase stationnaire de croissance (environ 109 cellules par ml). Des cultures en fin de phase stationnaire ne doivent pas être utilisées. Il est essentiel que les cultures utilisées pour cet essai présentent une concentration élevée de bactéries viables. La concentration peut être déterminée à partir des données historiques sur les courbes de croissance des souches bactériennes utilisées ou, lors de chaque essai, par la détermination du nombre de cellules viables à l'aide de la méthode par étalement.
La température d'incubation recommandée est de 37 oC.
Au moins cinq souches de bactéries doivent être employées. Celles-ci doivent comprendre quatre souches de S. typhimurium (TA1535, TA1537 ou TA97a ou TA97, TA98 et TA100) pour lesquelles des comparaisons entres laboratoires ont démontré la fiabilité et la reproductibilité de la réponse. Ces souches, qui comportent des paires de bases GC sur les sites primaires de réversion, risquent toutefois de ne pas déceler certains mutagènes oxydants, des agents de réticulation et des hydrazines. Ces substances mutagènes peuvent être détectées à l'aide de souches d'E. coli WP2 ou de S. typhimurium TA102 (19) qui ont une paire de base AT sur le site primaire de réversion. Par conséquent, il est recommandé de mettre en œuvre la combinaison de souches suivante:
|
—
|
S. typhimurium TA1535, et
|
|
—
|
S. typhimurium TA1537 ou TA97 ou TA97a, et
|
|
—
|
S. typhimurium TA98, et
|
|
—
|
S. typhimurium TA100, et
|
|
—
|
E. coli WP2 uvrA ou E. coli WP2 uvrA (pKM101) ou S. typhimurium TA102.
|
Pour les mutagènes à pouvoir réticulant, il peut être préférable d'inclure la souche TA102 ou d'ajouter une souche d'E. coli pourvue d'un mécanisme de réparation de l'ADN [E. coli WP2 ou E. coli WP2 (pKM101), par exemple].
La préparation des cultures souches, la vérification des marqueurs et le stockage doivent s'effectuer suivant des méthodes reconnues. Pour chaque culture souche congelée, il faut démontrer que la croissance exige la présence d'un acide aminé (histidine pour S. typhimurium et tryptophane pour E. coli). D'autres caractéristiques phénotypiques demandent également à être vérifiées, à savoir la présence ou l'absence de plasmides de résistance, s'il y a lieu [résistance à l'ampicilline des souches TA98, TA100 et TA97a ou TA97, WP2 uvrA et WP2 uvrA (pKM101), et résistance à l'association ampicilline/tétracycline de la souche TA102], ainsi que la présence de mutations caractéristiques (à savoir la mutation rfa chez S. typhimurium d'après la sensibilité au cristal violet et la mutation uvrA chez E. coli ou uvrB chez S. typhimurium d'après la sensibilité aux UV) (2) (3). D'autre part, les souches doivent produire un nombre de colonies de révertants spontanés par boîte qui se situe dans la gamme de fréquence prévue sur la base des données historiques du laboratoire et de préférence être comparable aux valeurs citées dans la littérature.
1.5.1.2. Milieu
On utilise une gélose minimale appropriée (composée, par exemple, de milieu minimal E de Vogel-Bonner et de glucose) et une gélose de recouvrement contenant de l'histidine et de la biotine ou du tryptophane pour permettre quelques divisions cellulaires (1) (2) (9).
1.5.1.3. Activation métabolique
Les bactéries sont exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique approprié. Le système le plus communément utilisé est une fraction post-mitochondriale enrichie en cofacteur (S9), préparée à partir de foie de rongeur traité avec des indicateurs enzymatiques comme Aroclor 1254 (1) (2) ou une combinaison de phénobarbital et de β-naphthoflavone (18) (20) (21). La fraction post-mitochondriale est généralement utilisée à une concentration comprise entre 5 et 30 % v/v dans le mélange S9. Le choix et la composition du système d'activation métabolique peuvent varier suivant la classe chimique de la substance d'essai. Dans certains cas, il peut être intéressant d'utiliser plusieurs concentrations de la fraction post-mitochondriale. Dans le cas de composés azoïques et diazoïques, un système d'activation métabolique réducteur peut s'avérer plus approprié (6) (13).
1.5.1.4. Substance d'essai/préparation
Les substances d'essai solides sont préparées par dissolution ou suspension dans un véhicule ou un solvant approprié, plus éventuellement diluées avant le traitement des bactéries. Les substances d'essai liquides peuvent être ajoutées au système d'essai directement ou après dilution. Il convient d'utiliser des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage.
Le solvant/véhicule choisi ne doit pas réagir chimiquement avec la substance d'essai ni altérer la survie des bactéries et l'activité du mélange S9 (22). L'emploi d'un solvant/véhicule inhabituel doit être justifié par des données faisant état de sa compatibilité. On préconise d'envisager d'abord l'utilisation d'un véhicule/solvant aqueux, chaque fois que cela est possible. Dans les essais de substances qui sont instables dans l'eau, on utilisera des solvants organiques exempts d'eau.
1.5.2. Conditions expérimentales
1.5.2.1. Souches utilisées (voir point 1.5.1.1)
1.5.2.2. Concentration d'exposition
La cytotoxicité et la solubilité dans le mélange d'essai final sont des critères à prendre en considération pour déterminer la dose la plus élevée à mettre en œuvre.
Un essai préliminaire destiné à déterminer la toxicité et les caractéristiques de solubilité peut être utile. La cytotoxicité peut être détectée par une réduction du nombre de révertants, un éclaircissement ou une diminution du tapis bactérien ou une diminution du taux de survie des cultures traitées. Les systèmes d'activation métabolique peuvent avoir une influence sur la cytotoxicité d'une substance. On conclut à l'insolubilité lorsqu'une précipitation dans le mélange final est observée à l'œil nu dans les conditions réelles de l'essai.
Pour les substances d'essai solubles et non cytotoxiques, on recommande une concentration maximale de 5 mg ou 5 μl par boîte. Pour les substances d'essai non cytotoxiques qui sont insolubles à 5 mg ou 5 μl par boîte, l'essai doit comprendre une ou plusieurs concentrations auxquelles on observe une insolubilité. Pour les substances qui sont déjà cytotoxiques à une concentration inférieure à 5 mg ou 5μl/boîte, l'essai doit être effectué jusqu'à une concentration cytotoxique. Le précipité ne doit pas interférer avec l'évaluation des résultats.
Pour un premier essai, il convient de tester au moins cinq concentrations analysables différentes de la substance d'essai, avec des intervalles d'environ une demi-unité logarithmique, c'est-à-dire √10. Pour établir une relation dose-réponse, il peut s'avérer nécessaire de réduire ces intervalles. Dans le cas de substances d'essai contenant des quantités substantielles d'impuretés potentiellement mutagènes, il peut être utile d'effectuer des essais avec des concentrations supérieures à 5 mg ou 5 μl/boîte.
1.5.2.3. Témoins négatifs et positifs
Des témoins positifs spécifiques de la souche, et des témoins négatifs (solvant ou véhicule), doivent être inclus dans chaque essai, avec ou sans activation métabolique. Dans le cas des témoins positifs, on utilisera une concentration permettant de démontrer l'efficacité de chaque essai.
Pour les essais avec système d'activation métabolique, le ou les témoins positifs de référence doivent être choisis en fonction du type de souche bactérienne utilisé.
Les substances suivantes sont des exemples de témoins positifs pour les essais avec activation métabolique:
|
Substance
|
Numéro CAS
|
Numéro Einecs
|
|
Diméthyl-9,10 anthracène
|
781-43-1
|
212-308-4
|
|
Diméthyl-7,12 benz[a]anthracène
|
57-97-6
|
200-359-5
|
|
3enzo[a]pyrène
|
50-32-8
|
200-028-5
|
|
Amino-2 anthracène
|
613-13-8
|
210-330-9
|
|
Cyclophosphamide
|
50-18-0
|
200-015-4
|
|
Monohydrate de cyclophosphamide
|
6055-19-2
|
|
La substance suivante convient comme témoin positif pour la méthode d'activation métabolique réductrice:
|
Substance
|
Numéro CAS
|
Numéro Einecs
|
|
Rouge Congo
|
573-58-0
|
209-358-4
|
L'amino-2 anthracène ne peut être utilisé comme indicateur unique de l'efficacité du mélange S9. S'il est utilisé, il faut également caractériser chaque lot de S9 avec un mutagène qui requiert une activation métabolique par des enzymes microsomalcs, benzo[a]pyrène et diméthylbenzanthracène par exemple.
Les substances suivantes sont des exemples de témoins positifs spécifiques de souches pour les essais sans activation métabolique exogène:
|
Substance
|
Numéro CAS
|
Numéro Ëinecs
|
Souche
|
|
Azoture de sodium
|
26628-22-8
|
247-852-1
|
TA 1535 et TA 100
|
|
Nitro-2 fluorène
|
607-57-8
|
210-138-5
|
TA 98
|
|
Amino-9 acridine
|
90-45-9
|
201-995-6
|
TA 15 37, TA 97 et TA 97a
|
|
ICR 191
|
17070-45-0
|
241-129-4
|
TA 1537, TA 97 et TA 97a
|
|
Hydropéroxyde de cumène
|
80-15-9
|
201-254-7
|
TA 102
|
|
Mitomycine C
|
50-07-7
|
200-008-6
|
WP2 uvrA et TA 102
|
|
N-éthyl-N-nitro-N-nitrosoguanidine
|
70-25-7
|
200-730-1
|
WP2, WP2 uvrA et WP2 uvrA (pKM101)
|
|
1-oxyde de 4-nitroquinoléine
|
56-57-5
|
200-281-1
|
WP2, WP2 uvrA et WP2 uvrA (pKM101)
|
|
Furylfuramide (AF2)
|
3688-53-7
|
|
Souches contenant des plasmides
|
D'autres témoins positifs appropriés peuvent être utilisés comme référence. L'utilisation de témoins positifs appartenant à la même classe chimique doit être envisagée, s'ils sont disponibles.
L'essai doit comprendre des témoins négatifs constitués de solvant ou véhicule sans substances d'essai, auxquels sont appliquées les mêmes conditions expérimentales qu'à la substance d'essai. Il faut également inclure des témoins non traités, à moins que des essais antérieurs n'aient démontré que le solvant choisi n'a pas d'effet délétère ou mutagène.
1.5.3. Mode opératoire
Pour la méthode par étalement (1) (2) (3) (4) sans activation métabolique, on ajoute généralement 0,05 ml ou 0,1 ml de la solution à tester, 0,1 ml de culture bactérienne fraîche (contenant environ 108 cellules viables) et 0,5 ml de tampon stérile à 2,0 ml de gélose de recouvrement. Pour l'essai avec activation métabolique, on mélange habituellement 0,5 ml du mélange d'activation métabolique contenant une quantité adéquate de fraction post-mitochondriaie (entre 5 et 30 % v/v), les bactéries et la substance d'essai, ou une solution de celle-ci, avec 2,0 ml de gélose de recouvrement. Le contenu de chaque tube est mélangé et versé sur la surface d'un milieu minimal gélosé. On laisse la gélose de recouvrement se solidifier avant incubation.
Dans la méthode de la préincubation (2) (3) (5) (6), la substance d'essai, ou une solution de celle-ci, est préincubée avec la souche d'essai (environ 108 cellules viables) et un tampon stérile ou 0,5 ml du système d'activation métabolique généralement pendant 20 minutes ou plus à 30-37 oC. On ajoute ensuite la gélose de recouvrement et on verse le tout sur la surface d'un milieu minimal gélosé. On mélange généralement 0,05 ou 0,1 ml de la substance d'essai, ou d'une solution de celle-ci, 0,1 ml de bactéries et 0,5 ml de mélange S9 ou de tampon stérile avec 2,0 ml de gélose de recouvrement. Pendant la préincubation, les tubes doivent être aérés par agitation.
Pour une bonne estimation de la variation, chaque concentration doit être testée dans trois boîtes de Petri. Le nombre de boîtes peut être limité à deux si cela se justifie scientifiquement. La perte accidentelle d'une boîte n'invalide pas nécessairement l'essai.
Les substances gazeuses ou volatiles doivent être testées en utilisant des méthodes appropriées, par exemple dans des récipients hermétiquement fermés (12) (14) (15) (16).
1.5.4. Incubation
Toutes les boîtes d'un essai donné doivent être incubées à 37 oC pendant 48-72 heures. À la fin de la période d'incubation, on compte dans chaque boîte le nombre de colonies révertantes.
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
Les données doivent être présentées sous la forme du nombre de colonies révertantes par boîte. Le nombre de colonies révertantes dans les boîtes de témoins négatifs (témoins traités avec le solvant et, le cas échéant, témoins non traités) et positifs doit également être mentionné. Le dénombrement par boîte, le nombre moyen de colonies révertantes par boîte et l'écart-type doivent être indiqués pour la substance d'essai ainsi que pour les témoins positifs et négatifs (non traités et/ou traités au solvant).
Il n'est pas nécessaire de vérifier une réponse nettement positive. Les résultats équivoques doivent être clarifiés par des essais supplémentaires réalisés de préférence dans des conditions expérimentales modifiées. Les résultats négatifs demandent à être confirmés cas par cas. Si la confirmation de résultats négatifs n'est pas jugée nécessaire, une justification doit être fournie. Pour les essais de confirmation, il convient d'envisager une modification des conditions expérimentales, afin d'élargir la gamme des conditions évaluées. Les paramètres susceptibles d'être modifiés comprennent l'écart entre les doses, la méthode de traitement (étalement ou préincubation en milieu liquide) et les conditions d'activation métabolique.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Plusieurs critères permettent de déterminer si un résultat est positif, notamment un accroissement, dose-dépendant, du nombre de révertants ou une augmentation reproductible du nombre de colonies de revenants par boîte à une ou plusieurs concentrations pour au moins une souche, avec ou sans activation métabolique (23). En premier lieu, il s'agit de prendre en considération la signification biologique des résultats. L'évaluation des résultats peut s'appuyer sur des méthodes statistiques (24) mais la signification statistique ne doit pas être le seul facteur décisif pour conclure à une réponse positive.
Si les résultats obtenus avec une substance d'essai ne répondent pas aux critères indiqués ci-dessus, on peut conclure que la substance en question n'est pas mutagène dans cet essai.
Bien que la plupart des essais donneront des résultats manifestement positifs ou négatifs, dans certains cas rares l'ensemble des résultats d'un essai n'autorise pas de conclusion catégorique sur l'activité de la substance étudiée. Les résultats peuvent rester équivoques ou discutables indépendamment du nombre de répétitions de l'essai.
Des résultats positifs obtenus à l'essai de mutation réverse sur bactéries indiquent que la substance induit des mutations ponctuelles par substitution de bases ou décalage du cadre de lecture dans le génome de Salmonella typhimurium et/ou Escherichia coli. Un résultat négatif signifie que, dans les conditions de l'essai, la substance n'est pas mutagène pour l'espèce utilisée dans l'essai.
3. RAPPORT
RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Solvant/véhicule:
|
—
|
justification du choix du solvant/véhicule,
|
|
—
|
solubilité et stabilité de la substance d'essai dans le solvant/véhicule, si elles sont connues.
|
|
|
|
Souches:
|
—
|
nombre de cellules par culture,
|
|
—
|
caractéristiques des souches.
|
|
|
|
Conditions expérimentales:
|
—
|
quantité de substance d'essai par boite (mg/boîte ou μl/boîte) et justification du choix des doses et du nombre de boîtes par concentration,
|
|
—
|
type et composition du système d'activation métabolique, y compris les critères d'acceptabilité,
|
|
—
|
procédés de traitement.
|
|
|
|
Résultats:
|
—
|
signes de précipitation,
|
|
—
|
dénombrement par boîte,
|
|
—
|
nombre moyen de colonies révertantes par boîte et écart-type,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
analyses statistiques, le cas échéant,
|
|
—
|
données concernant les témoins négatifs (solvant/véhicule) et positifs concomitants (étendues, moyennes et écarts-types),
|
|
—
|
données concernant les témoins négatifs (solvant/véhicule) et positifs antérieurs (étendues, moyennes et écarts-types).
|
|
|
|
Discussion des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
Ames, B. N., McCann, J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res., 31, pp. 347-364.
|
|
(2)
|
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res., 113, pp. 173-215.
|
|
(3)
|
Gatehouse, D., Haworth, S., Cebula, T., Gocke, E., Kier, L., Matsushima, T., Melcion, C, Nohmi, T., Venitt, S. and Zeiger, E. (1994), Recommendations for the Performance of Bacterial Mutation Assays, Mutation Res., 312, pp. 217-233.
|
|
(4)
|
Kier, L. D., Brusick, D. J., Auletta, A. E., Von Halle, E. S., Brown, M. M., Simmon, V. F., Dunkel, V., McCann., Mortelmans, K., Prival, M., Rao, T. K. and Ray V. (1986), The Salmonella typhimurium/Mammalian Microsomal Assay: A Report of the U.S. Environmental Protection Agency Gene-lox Program, Mutation Res., 168, pp. 69-240.
|
|
(5)
|
Yahagi, T., Degawa, M., Seino, Y.Y., Matsushima, T., Nagao, M., Sugimura. T. and Hashimoto, Y. (1975), Mutagenicity of Carcinogen Azo Dyes and their Derivatives. Cancer Letters, 1, pp. 91-96.
|
|
(6)
|
Matsushima, M., Sugimura, T., Nagao, M., Yahagi, T., Shirai, A. and Sawamura, M. (1980), Factors Modulating Mutagenicity Microbial Tests, in: Short-term Test Systems for Detecting Carcinogens. ed. Nor- poth K. H. and Garner, R. C, Springer, Berlin-Heidelberg-New York, pp. 273-285.
|
|
(7)
|
Gatehouse. D. G.. Rowland, I. R.. Wilcox, P., Callender, R. D. and Foster R. (1980), Bacterial Mutation Assays, in: Basic Mutagenecity Tests: UKEMS Part 1 Revised, ed. D. J. Kirkland, Cambridge University Press, pp. 13-61.
|
|
(8)
|
Aeschacher. H. U.. Wolleb, U. and Porchet. L. (1987). Liquid Preincubation Mutagenicity Test for Foods. J. Food Safety, 8, pp. 167-177.
|
|
(9)
|
Green, M. H. L, Muriel, W. J. and Bridges, B. A. (1976), Use of a simplified fluctuation test to detect low levels of mutagens, Mutations Res., 38, pp. 33-42.
|
|
(10)
|
Hubbard, S. A., Green, M. H. L, Gatehouse, D. and J. W. Bridges (1984), The Fluctuation Test in Bacteria, in: Handbook of Mutagenicitv Test Procedures, 2nd Edition, ed. Kilbey, B. J., Legator, M., Nichols, W. and Ramel, C, Elsevier, Amsterdam-New York-Oxford, pp. 141-161.
|
|
(11)
|
Thompson, E. D. and Melampy, P. J. (1981), An Examination of the Quantitative Suspension Assay for Mutagenesis with Strains of Salmonella typhimurium. Environmental Mutagenesis, 3, pp. 453-465.
|
|
(12)
|
Araki, A., Noguchi, T., Kato, F. and T. Matsushima (1994), Improved Method for Mutagenicity Testing of Gaseous Compounds by Using a Gas Sampling Bag, Mutation Res., 307, pp. 335-344.
|
|
(13)
|
Privai, M. J., Bell, S. J., Mitchell, V. D., Reipert, M D. and Vaughan, V. L. (1984), Mutagenicity of Benzidine and Benzidine-Congener Dyes and Selected Monoazo Dyes in a Modified Salmonella Assay, Mutation Res., 136, pp. 33-47.
|
|
(14)
|
Zeiger, E., Anderson B. E., Haworth, S., Lawlor, T. and Mortelmans, K. (1992), Salmonella Mutagenicity Tests. V. Results from the Testing of 311 Chemicals, Environ. Mol. Mutagen., 19, pp. 2-141.
|
|
(15)
|
Simmon, V., Kauhanen K. and Tardiff, R. G. (1977), Mutagenic Activity of Chemicals Identified in Drinking Water, in Progress in Genetic Toxicology, D. Scott, B. Bridges and F. Sobels (eds.) Elsevier, Amsterdam, pp. 249-258.
|
|
(16)
|
Hughes, T. J., Simmons, D. M., Monteith, 1. G. and Claxton, L. D. (1987), Vaporisation Technique to Measure Mutagenic Activity of Volatile Organic Chemicals in the Ames/Salmonella Assay, Environmental Mutagenesis, 9, pp. 421-441.
|
|
(17)
|
Matsushima, T., Matsumoto, A., Shirai, M., Sawamura, M. and Sugimura T. (1979), Mutagenicity of the Naturally Occurring Carcinogen Cycasin and Synthetic Methylazoxy Methane Conjugates in Salmonella typhimurium, Cancer Res., 39, pp. 3780-3782.
|
|
(18)
|
Tamura, G., Gold, C, Ferro-Luzzi, A. and Ames, B. N. (1980), Fecalase: A Model for Activation of Dietary Glycosides to Mutagens by Intestinal Flora, Proc. Nail. Acad. Sri, USA, 77, pp. 4961-4965.
|
|
(19)
|
Wilcox, P., Naidoo, A., Wedd, D. J. and Gatehouse, D. G. (1990), Comparison of Salmonella typhimurium TA 102 with Escherichia coli WP2 Tester strains, Mutagenesis, 5, pp. 285-291.
|
|
(20)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer or Metabolic Activation Systems, in: In Vitro Metabolic Activation in Mutagenesis Testing, eds. F. J. de Serres et al. Elsevier, North Holland, pp. 85-88.
|
|
(21)
|
Elliot, B. M., Combes, R. D., Elcombe, C. R., Tatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-induced S9 in in vitro Genotoxicity Assays, Mutagenesis, 7, pp. 175-177.
|
|
(22)
|
Maron, D., Katzenellenbogcn, J. and Ames, B. N. (1981), Compatibility of Organic Solvents with the Salmonella/Microsome Test, Mutation Res., 88, pp. 343-350.
|
|
(23)
|
Claxton, L. D., Allen J., Auletta, A., Mortelmans, K., Nestmann, E. and Zeiger, E. (1987), Guide for the Salmonella typhimurium/Mammalian Microsomc Tests for Bacterial Mutagenicity, Mutation Res., 189, pp. 83-91.
|
|
(24)
|
Mahon, G. A. T., Green, M. H. L., Middleton, B., Mitchell, I., Robinson, W. D. and Tweats, D. J. (1989), Analysis of Data from Microbial Colony Assays, in: UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Part II. Statistical Evaluation of Mutagenicity Test Data, ed. Kirkland, D. J., Cambridge University Press, pp. 28-65.
|
B.15. TESTS DE MUTAGENÈSE ET DE DÉPISTAGE DE CANCÉROGENÈSE MUTATION GÉNIQUE — SACCHAROMYCES CEREVISIAE
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITION
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Diverses souches haploïdes et diploïdes de la levure Saccharomyces cerevisiae peuvent être utilisées pour mesurer la production de mutations géniques induites par des agents chimiques en présence et en l'absence d'activation métabolique.
La mesure de mutation en avant dans les souches haploïdes peut être réalisée notamment par la mesure du passage des mutants rouges exigeant l'adénine (ade-1, ade-2) à une forme blanche doublement exigeante pour l'adénine ainsi que des systèmes sélectifs tels que l'induction de la résistance à la canavanine et la cydoheximide.
La méthode de mutation reverse la plus largement utilisée implique l'utilisation de la souche haploïde XV 185-14C qui porte les mutations non-sens ochre ade 2-1, arg 4-17, lys 1-1 et trp 5-48, lesquelles sont réversibles par des mutagènes provoquant des substitutions de bases qui induisent des mutations à un site spécifique ou des mutations suppressives ochre. La souche XV 185-14C porte également le marqueur his 1-7, une mutation contresens principalement révertée par des mutations de second site et le marqueur hom 3-10 qui est réverté par des mutagènes conduisant au décalage du cadre de lecture du code génétique (frameshift mutagens).
La seule souche diploïde de S. cerevisiae largement validée est la souche D7, homozygote pour ilv 1-92.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Les solutions des substances d'essai ainsi que celles des substances témoins seront préparées, immédiatement avant l'essai, à l'aide d'un véhicule approprié. Lorsqu'il s'agit de substances organiques non solubles dans l'eau, les solvants organiques tels que l'éthanol, l'acétone ou le diméthylsulfoxyde (DMSO) devraient être utilisés à une concentration ne dépassant pas 2 % v/v. La concentration finale du véhicule ne doit pas affecter de manière significative la viabilité des cellules ni les caractéristiques de croissance.
Activation métabolique
Les cellules devraient être exposées aux substances d'essai en présence et en l'absence d'un système d'activation métabolique exogène approprié.
La méthode la plus fréquemment utilisée consiste à ajouter la fraction post-mitochondriale préparée à partir de foies de rongeurs prétraités par des inducteurs enzymatiques additionnée de co-facteurs. L'usage d'autres espèces, tissus, fractions post-mitochondriales, ou méthodes peut aussi convenir pour l'activation métabolique.
Conditions d'essai
Souches d'expérience
La souche haploïde XV 185-14C et la souche diploïde D7 sont les plus fréquemment utilisées dans les études de mutation génique. D'autres souches peuvent également convenir.
Milieux
Des milieux de culture appropriés sont utilisés pour déterminer la survie cellulaire et le nombre de mutants.
Utilisation de témoins négatifs et positifs
Des témoins positifs, des témoins non traités et des témoins en présence de solvant devraient être simultanément réalisés. Des substances chimiques témoins positives appropriées devraient être utilisées pour concrétiser chaque effet mutagène spécifique.
Concentration d'exposition
Au moins 55 concentrations convenablement espacées devraient être utilisées. Dans le cas de substances toxiques, la concentration maximale testée ne fera pas descendre le taux de survie en dessous de 5 à 10 %. Les substances relativement insolubles dans l'eau seront testées jusqu'à leur limite de solubilité par des méthodes appropriées. En ce qui concerne les substances non toxiques très solubles dans l'eau, la concentration la plus élevée sera déterminée cas par cas.
Conditions d'incubation
Les boîtes sont incubées dans l'obscurité à une température de 28 à 30 oC pendant 4 à 7 jours.
Fréquences de mutation spontanée
On utilisera des subcultures dont les fréquences de mutations spontanées se situent dans les limites normales admises.
Nombre de répétitions
Trois boîtes au moins seront utilisées par concentration, en vue de déterminer les prototrophes produits par mutation génique et d'observer la viabilité des cellules. Dans le cas d'expériences utilisant des marqueurs tels que hom 3-10 à faible taux de mutation, le nombre des boîtes utilisées peut être augmenté afin de fournir des données statistiquement pertinentes.
Conduite de l'essai
Les souches de S. cerevisiae sont habituellement traitées au cours d'un essai en milieu liquide impliquant l'utilisation de cellules en phase stationnaire ou de croissance. Les expériences initiales devraient être faites sur des cellules en croissance. 1 — 5 × 107 cellules/ml sont exposées à la substance d'essai pendant une période allant jusqu'à 18 heures, à une température de 28 à 37 oC, le mélange étant agité; une quantité adéquate d'un système d'activation métabolique est ajoutée pendant le traitement. Si la première expérience fournit des résultats négatifs, une deuxième expérience devrait être réalisée en utilisant des cellules en phase stationnaire. Si la première expérience fournit des résultats positifs, ceux-ci sont confirmés par une expérience indépendante. À l'issue du traitement, les cellules sont centrifugées, lavées et ensemencées sur un milieu de culture approprié. Après incubation, les boîtes sont examinées afin de déterminer le taux de survie et l'induction de mutations géniques.
2. RÉSULTATS
Les résultats devraient être présentés sous forme de tableaux indiquant le nombre de colonies comptées, le nombre de mutants, le taux de survie et la fréquence de mutants. Tous les résultats devraient être confirmés au cours d'une expérience indépendante. Les résultats devraient être évalués par des méthodes statistiques adéquates.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
conditions d'essai: cellules en phases stationnaire ou de croissance; composition des milieux; température d'incubation et durée de celle-ci; système d'activation métabolique,
|
|
—
|
conditions de traitement: concentration d'exposition; méthode et durée de traitement; température de traitement; témoins positifs et négatifs,
|
|
—
|
nombre de colonies comptées; nombre de mutants; taux de survie et fréquence de mutants; relation dose-réponse, si possible; évaluation statistique des résultats,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.16. RECOMBINAISON MITOTIQUE — SACCHAROMYCES CEREVISIAE
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITION
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE
Chez Saccharomyces cerevisiae, on peut déceler la recombinaison mitotique intergénique (ou plus généralement, entre un gène et son centromère) et intragénique. Dans le premier cas, on parle de croissing-over mitotique lequel donne naissance à des échanges réciproques, tandis que, dans le second cas, les échanges sont le plus souvent non réciproques et l'on parle de conversion génique. Le crossing-over est généralement déterminé par la production de colonies ou de secteurs récessifs homozygotes à partir d'une souche hétérozygote, tandis que la conversion génique est déterminée par la production de revertants prototrophes dans une souche auxotrophe hétéroallélique portant deux allèles défectueux différents du même gène. Les souches les plus couramment utilisées pour déceler une conversion génique mitotique sont D4 (hétéroallélique pour ade 2 et trp 5), D7 (hétéroallélique pour trp 5), BZ34 (hétéroallélique pour arg 4) et JD1 (hétéroallélique pour his 4 et trp S). croissing-over mitotique produisant des secteurs homozygotes de couleur rouge et rose peut être déterminé dans D5 ou dans D7 (qui mesure aussi la conversion génique mitotique et la mutation reverse pour ilv 1-92), ces deux souches étant hétéroalléliques complémentaires pour les allèles ade 2.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Les solutions des substances d'essai ainsi que celles des substances témoins ou de référence devraient être préparées, immédiatement avant l'essai, à l'aide d'un véhicule approprié. Dans le cas de substances organiques insolubles dans l'eau, on utilisera des solvants organiques, tels que l'éthanol, l'acétone et le Diméthylsulfoxyde (DMSO), a une concentration ne dépassant pas 2 % v/v. La concentration finale du véhicule ne devrait pas affecter de manière significative la viabilité des cellules ni les caractéristiques de croissance.
Activation métabolique
Les cellules seront exposées aux substances d'essai en présence et en l'absence d'un système d'activation métabolique exogène. La méthode la plus fréquemment utilisée consiste à ajouter la fraction post-mitochondriale préparée à partir de foies de rongeurs prétraités par des inducteurs enzymatiques additionnée de co-facteurs. L'usage d'autres espèces, tissus, fractions post-mitochondriales, ou méthodes peut aussi convenir pour l'activation métabolique.
Conditions d'essai
Souches d'expérience
Les souches les plus fréquemment utilisées sont les diploïdes D4, D5, D7 et JD1. D'autres souches peuvent convenir.
Milieux
Des milieux de culture adéquats sont utilisés en vue de déterminer le taux de survie et la fréquence de recombinaisons mitotiques.
Utilisation de témoins négatifs et positifs
Des témoins positifs, des témoins non traités et des témoins en présence de solvant seront simultanément réalisés. Des substances chimiques témoins positives appropriées seront utilisées pour chaque type spécifique de recombinaison.
Concentration d'exposition
Au moins cinq concentrations convenablement espacées de substance d'essai seront utilisées. La cytotoxicité et la solubilité sont au nombre des facteurs à prendre en considération. La concentration la plus faible ne doit avoir aucun effet sur la viabilité des cellules. Dans le cas de produits chimiques toxiques, la concentration maximale testée ne diminuera pas le taux de survie en dessous de 5 à 10 %. Les produits chimiques relativement insolubles dans l'eau seront testés jusqu'à leur limite de solubilité par des méthodes appropriées. Dans le cas de produits chimiques non toxiques franchement solubles dans l'eau, la concentration d'essai maximale sera déterminée cas par cas.
Les cellules peuvent être exposées aux substances d'essai en phase stationnaire ou en phase de croissance pendant des périodes allant jusqu'à 18 heures. Toutefois, dans le cas d'un traitement de longue durée, les cultures seront examinées au microscope afin de déceler la formation de spores; la présence de celles-ci rendant l'essai nul.
Conditions d'incubation
Les boîtes sont incubées, dans l'obscurité, à une température de 28 à 30 oC, pendant 4 à 7 jours. Les boîtes utilisées pour la détermination des secteurs homozygotes rouge et rose produits par croissing-over mitotique seront conservées dans un réfrigérateur à ± 4 oC pendant 1 à 2 jours encore avant le dénombrement de manière à ce que la pigmentation des colonies concernées puisse s'intensifier.
Fréquences de recombinaisons mitotiques spontanées
On utilisera des subcultures dont les fréquences de recombinaisons mitotiques spontanées se situeront dans la gamme normalement admise.
Nombre de répétitions
Trois boîtes au moins seront ensemencées par concentration afin de déterminer la viabilité ainsi que le nombre de prototrophes produits par conversion génique mitotique. Dans le cas de la détermination d'homozygotie récessive produite par croissing-over mitotique, le nombre des boîtes sera augmenté pour obtenir un nombre adéquat de colonies.
Conduite de l'essai
Les souches de S. cerevisiae sont habituellement traitées au cours d'un essai en milieu liquide impliquant l'utilisation de cellules en phase stationnaire ou de croissance. Les expériences initiales devraient être faites sur des cellules en croissance. 1 — 5 × 107 cellules/ml sont exposées à la substance d'essai pendant une période allant jusqu'à 18 heures à des températures de 28 à 37 oC, le mélange étant agité; une quantité adéquate d'un système d'activation métabolique est ajoutée pendant le traitement. Si la première expérience fournit des résultats négatifs, une deuxième expérience devrait être réalisée en utilisant des cellules en phase stationnaire. Si la première expérience fournit des résultats positifs, ceux-ci sont confirmés par une expérience indépendante appropriée. À l'issue du traitement, les cellules sont centrifugées, lavées et ensemencées sur un milieu de culture adéquat. Après incubation, les boîtes sont examinées afin de déterminer le taux de survie et l'induction de recombinaison mitotique.
2. RÉSULTATS
Les résultats devraient être présentés sous forme de tableaux indiquant le nombre de colonies comptées, le nombre de recombinants, le taux de survie et la fréquence de recombinants.
Tous les résultats devraient être confirmés au cours d'une expérience indépendante.
Les résultats devraient être évalués par des méthodes statistiques adéquates.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
conditions d'essai: cellules en phase stationnaire ou en phase de croissance; composition des milieux; température d'incubation et durée d'incubation; système d'activation métabolique,
|
|
—
|
conditions de traitement: concentration d'exposition; méthode et durée de traitement; température de traitement; témoins positifs et négatifs,
|
|
—
|
nombre de colonies comptées; nombre de recombinants; taux de survie et fréquence de recombinaison: relation dose-réponse, si possible; évaluation statistique des résultats,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B. 17. MUTAGÉNICITÉ — ESSAI IN VITRO DE MUTATION GÉNIQUE SUR CELLULES DE MAMMIFÈRE
1. MÉTHODE
Cette méthode est une adaptation de la ligne directrice OCDE TG 476, Essai in vitro de mutation génique sur cellules de mammifères (1997).
1.1. INTRODUCTION
L'essai in vitro de mutation génique sur cellules de mammifères permet de détecter des mutations géniques induites par des substances chimiques. Les lignées cellulaires appropriées comprennent des cellules de lymphome de souris L5178Y, les lignées CHO-AS52 et V79 de cellules de hamster chinois et les cellules lymphoblastoïdes humaines TK6 (1). Dans ces lignées cellulaires, les critères génétiques les plus couramment utilisés sont une mutation sur les loci de la thymidine kinase (TK) et de l'hypoxanthine-guanine phosphoribosyl transférase (HPRT), ainsi qu'un transgène de la xanthine-guanine phosphoribosyl transférasc (XPRT). Les essais de mutation TK, HPRT et XPRT détectent des gammes différentes d'effet génétiques. L'emplacement autosomique de TK et XPRT peut permettre de détecter des effets génétiques (par exemple des délétions importantes) qui ne sont pas détectés sur le locus HPRT de chromosomes X (2, 3, 4, 5, 6).
L'essai in vitro de mutation génique sur cellules de mammifère peut être pratiqué sur des cultures de lignées cellulaires établies ou de souches cellulaires. Les cellules employées sont choisies en fonction de leur potentiel de croissance en culture et de la stabilité de la fréquence de mutation spontanée.
Les essais in vitro requièrent généralement une source exogène d'activation métabolique qui ne peut pas reproduire parfaitement les conditions présentes in vivo chez les mammifères. Il faut éviter absolument d'introduire des conditions qui conduiraient à des résultats positifs ne reflétant pas une mutagénicité intrinsèque et pouvant provenir de modifications du pH ou de l'osmolalité ou d'une forte cytotoxicité (7).
Cet essai est utilisé pour le dépistage de substances pouvant être mutagènes et cancérigènes pour les mammifères. Bien que de nombreux composés donnant un résultat positif à cet essai soient cancérigènes pour les mammifères, il n'y a pas de corrélation parfaite entre cet essai et la cancérogénicité. La corrélation dépend de la classe chimique et de plus en plus d'éléments laissent penser qu'il existe des agents cancérigènes que cet essai ne permet pas de détecter parce qu'ils agissent par des mécanismes non génotoxiques ou par des mécanismes qui sont absents des cellules de mammifères (6).
Voir aussi l'introduction générale de la partie B.
1.2. DÉFINITIONS
Mutation directe: mutation génique de la forme parentale en une forme mutante, qui engendre une modification ou une perte de l'activité enzymatique de la fonction de la protéine codée.
Mutagènes provoquant la substitution de paires de bases: substances qui entraînent le remplacement d'une ou de plusieurs bases de l'ADN.
Mutagènes décalant le cadre de lecture: substances qui entraînent l'addition ou la délétion d'une ou de plusieurs paires de bases dans la molécule d'ADN.
Temps d'expression du phénotype: période pendant laquelle des produits géniques non modifiés disparaissent des cellules qui viennent de subir une mutation.
Fréquence de mutants: rapport cellules mutantes observées/cellules viables.
Croissance totale relative: augmentation du nombre de cellules dans le temps par rapport à une population témoin; elle est calculée comme le produit de la croissance en suspension par rapport au témoin négatif et de l'efficacité de clonage par rapport au témoin négatif.
Croissance relative en suspension: augmentation du nombre de cellules pendant la période d'expression par rapport au témoin négatif.
Viabilité: efficacité de clonage des cellules traitées au moment de l'étalement dans des conditions sélectives après la période d'expression.
Taux de survie: efficacité de clonage des cellules traitées au moment de l'étalement à la fin de la période de traitement; le taux de survie est généralement exprimé par rapport au taux de survie de la population cellulaire témoin.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
Les cellules déficientes en thymidine kinase (TK) par suite de la mutation directe TK-/- —> TK-/- sont résistantes aux effets cytotoxiques de la trifluorothymidinc (TFT), un analogue de la pyrimidine. Les cellules non déficientes en TK sont sensibles à la TFT, ce qui entraîne une inhibition du métabolisme cellulaire et l'arrêt de la division cellulaire. Ainsi, les cellules mutantes peuvent proliférer en présence de TFT, tandis que les cellules normales, qui contiennent de la TK, ne le peuvent pas. De la même façon, des cellules déficientes en HPRT ou XPRT sont sélectionnées par leur résistance à la thio-6 guanine (TG) ou à l'aza-8 guanine (AG). Lorsqu'un analogue d'une base ou une substance apparentée à l'agent sélectif est soumis à un essai de mutation génique sur des cellules de mammifère, il convient d'en étudier soigneusement les propriétés. Il faut, par exemple, examiner toute toxicité sélective soupçonnée de la substance d'essai vis-à-vis de cellules mutantes et non mutantes. Par conséquent, la performance du système ou de l'agent de sélection doit être confirmée lors de l'essai de substances ayant une similitude de structure avec l'agent de sélection (8).
Des cellules en suspension ou en culture monocouche sont exposées à la substance d'essai, avec et sans activation métabolique, pendant une période appropriée. Les cellules sont repiquées afin de déterminer la cytotoxicité et de laisser le phénotype s'exprimer avant la sélection des mutants (9, 10, 11, 12, 13). On détermine la cytotoxicitc en mesurant l'efficacité de clonage relative (taux de survie) ou la croissance totale relative des cultures après la période de traitement. Les cultures traitées sont maintenues dans un milieu de croissance pendant une période de temps suffisante, caractéristique de chaque locus et du type de cellule choisi, afin de permettre une expression phénotypique presque optimale des mutations induites. On détermine la proportion de mutants en mettant en culture un nombre connu de cellules dans un milieu contenant l'agent sélectif pour détecter les cellules mutantes et dans un milieu exempt d'agent sélectif pour déterminer l'efficacité de clonage (viabilité). Les colonies sont comptées après une durée d'incubation appropriée. On compare le nombre de colonies mutantes en milieu sélectif au nombre de colonies en milieu non sélectif pour obtenir la proportion de mutants.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparations
1.4.1.1. Cellules
Divers types de cellules conviennent pour ce test, entre autres: sous-clones de L5178Y, CHO, AS52, V79 ou TK6. Les cellules utilisées doivent avoir une sensibilité prouvée aux mutagènes chimiques, une efficacité de clonage élevée et une fréquence de mutation spontanée stable. On doit veiller à ce que les cellules ne soient pas contaminées par des mycoplasmes. Des cellules contaminées ne doivent pas être utilisées.
L'essai doit être conçu de façon à avoir une sensibilité et une puissance prédéterminées. Le nombre de cellules, de cultures et de concentrations de la substance d'essai utilisées doit refléter ces paramètres définis à l'avance (14). À chaque stade de l'essai, le nombre minimal de cellules viables survivant au traitement doit être basé sur la fréquence de mutation spontanée. Une règle générale consiste à utiliser un nombre de cellules au moins égal à dix fois l'inverse de cette fréquence. Cependant, il est recommandé d'utiliser au moins 106 cellules. Pour s'assurer de la performance constante de l'essai, il faut disposer de données antérieures adéquates relatives au système cellulaire utilisé.
1.4.1.2. Milieux et conditions de culture
On doit utiliser des milieux de culture et des conditions d'incubation appropriés (récipients de culture, température, concentration de CO2 et humidité). Les milieux doivent être choisis en fonction des systèmes sélectifs et du type de cellules utilisés dans l'essai. Il est particulièrement important de choisir des conditions de culture qui assurent une croissance optimale des cellules pendant la période d'expression, ainsi que la capacité des cellules, tant mutantes que non mutantes, à former des colonies.
1.4.1.3. Préparation des cultures
Les cellules sont multipliées à partir de cultures souches, ensemencées dans le milieu de culture et incubées à 37 oC. Avant l'emploi des cultures pour l'essai, il peut être nécessaire de les débarrasser des cellules mutantes qu'elles contiennent.
1.4.1.4. Activation métabolique
Les cellules doivent être exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique approprié. Le système le plus couramment utilisé est une fraction post-mitochondriale enrichie en cofacteur (S9), préparée à partir de foie de rongeur traité avec des inducteurs enzymatiques, par exemple Aroclor 1254 (15, 16, 17, 18) ou un mélange de phénobarbital et de β-naphthoflavone (19, 20).
La fraction post-mitochondriale est généralement utilisée à une concentration comprise entre 1 et 10 % v/v dans le milieu final. Le choix du système d'activation métabolique peut dépendre de la classe chimique de la substance d'essai. Dans certains cas il peut y avoir avantage à utiliser plus d'une concentration de la fraction post-mitochondriale.
Plusieurs procédés nouveaux, comme la création de lignées cellulaires génétiquement modifiées exprimant des enzymes activantes spécifiques, sont susceptibles de fournir une activation endogène. Le choix des lignées cellulaires utilisées doit être justifié scientifiquement (par exemple par la pertinence de l'isoenzyme du cytochrome P4 50 pour le métabolisme de la substance d'essai).
1.4.1.5. Substance d'essai/préparation
Les substances d'essai solides sont dissoutes ou mises en suspension dans un solvant ou un véhicule approprié, puis éventuellement diluées avant le traitement des cellules. Les substances d'essai liquides peuvent être ajoutées directement ou après dilution au système d'essai. Il convient d'utiliser des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage.
1.4.2. Conditions expérimentales
1.4.2.1. Solvant/véhicule
Le solvant/véhicule choisi ne doit pas réagir chimiquement avec la substance d'essai ni altérer la survie des cellules et l'activité du mélange S9. L'emploi d'un solvant/véhicule inhabituel doit être justifié par des données faisant état de sa compatibilité. On préconise d'envisager d'abord l'utilisation d'un véhicule/solvant aqueux, chaque fois que cela est possible. Dans les essais de substances qui sont instables dans l'eau, on utilisera des solvants organiques exempts d'eau. L'eau peut être enlevée à l'aide d'un tamis moléculaire.
1.4.2.2. Concentration d'exposition
La cytotoxicité, la solubilité dans le mélange d'essai final et les modifications de pH et d'osmolalité sont des critères à prendre en considération pour déterminer la dose la plus élevée à mettre en œuvre.
La cytotoxicité doit être mesurée, avec et sans système d'activation métabolique, dans l'expérience principale par un indicateur pertinent de l'intégrité et de la croissance cellulaire, tel que l'efficacité de clonage relative (taux de survie) ou la croissance totale relative. Il peut être utile de déterminer la cytotoxicité et la solubilité dans un essai préliminaire.
Il convient d'utiliser au moins quatre concentrations analysables. Dans le cas d'une substance cytotoxique, ces concentrations doivent couvrir un domaine allant d'une toxicité nulle ou légère à la toxicité maximale, ce qui signifie généralement des concentrations séparées par un facteur compris entre 2 et √10 au maximum. Si la concentration la plus forte est basée sur la cytotoxicité, elle devrait donner un taux de survie relatif (efficacité de clonage relative) ou une croissance totale relative d'environ 10 à 20 % (mais non inférieure à 10 %). En ce qui concerne les substances relativement non cytotoxiques, la concentration maximale doit être la plus basse des trois concentrations suivantes: 5 μl/ml, 5 mg/ml ou 0,01 M.
Les substances relativement insolubles doivent être testées jusqu'à des concentrations égales ou supérieures à la limite de solubilité dans les conditions de culture. L'insolubilité doit être mise en évidence dans le milieu de traitement final auquel les cellules sont exposées. Il peut être utile d'évaluer la solubilité au début et à la fin du traitement, celle-ci pouvant varier au cours de l'exposition dans le système d'essai en raison de la présence de cellules, de S9, de sérum, etc. On conclut à l'insolubilité lorsqu'une précipitation est observée à l'exil nu. Le précipité ne doit pas interférer avec l'évaluation des résultats.
1.4.2.3. Témoins
Des témoins positifs et négatifs (solvant ou véhicule) doivent être inclus dans chaque expérience avec et sans activation métabolique. Quand l'essai est effectué avec activation métabolique, la substance utilisée comme témoin doit être celle qui exige l'activation pour donner une réponse mutagène.
Des exemples de substances pouvant servir comme témoins positifs figurent dans le tableau ci-dessous.
|
Condition d'activation métabolique
|
Locus
|
Substance
|
No CAS
|
No Einecs
|
|
Absence d'activation métabolique exogène
|
HPRT
|
Méthanesulfonate d'éthyle
|
62-50-0
|
200-536-7
|
|
Éthylnitrosourée
|
759-73-9
|
212-072-2
|
|
TK (petites et grandes colonies)
|
Méthanesulfonate de méthyle
|
66-27-3
|
200-625-0
|
|
XPRT
|
Méthanesulfonate d'éthyle
|
62-50-0
|
200-536-7
|
|
Éthylnitrosourée
|
759-73-9
|
212-072-2
|
|
Présence d'activation métabolique exogène
|
HPRT
|
Méthyl-3 cholanthrène
|
56-49-5
|
200-276-4
|
|
N-Nitrosodiméthylaminc
|
62-75-9
|
200-549-8
|
|
Diméthyl-7,12 benzanthracène
|
57-97-6
|
200-359-5
|
|
TK (petites et grandes colonies)
|
Cvclophosphamide
|
50-18-0
|
200-015-4
|
|
Monohydrate de cyclophosphamide
|
6055-19-2
|
|
|
Benzo[a]pyrène
|
50-32-8
|
200-028-5
|
|
Méthyl-3 cholanthrène
|
56-49-5
|
200-276-5
|
|
XPRT
|
N-Nitrosodiméthylamine (pour des niveaux élevés de S9)
|
62-75-9
|
200-549-8
|
|
Benzo[a]pyrène
|
50-32-8
|
200-028-5
|
D'autres substances de référence appropriées peuvent être utilisées comme témoins positifs, par exemple la 5-bromo 2'-désoxyuridine (no CAS 59-14-3, no Einecs 200-415-9) si le laboratoire possède des contrôles historiques relatifs à cette substance. L'emploi de témoins positifs appartenant à la même classe chimique doit être envisagé, s'ils sont disponibles.
Des témoins négatifs, constitués uniquement du solvant ou du véhicule dans le milieu de traitement et traités de la même façon que la substance d'essai, doivent être inclus. Il faut également inclure des témoins non traités, à moins que des essais antérieurs n'aient démontré que le solvant choisi n'a pas d'action délétère ou mutagène.
1.4.3. Mode opératoire
1.4.3.1. Traitement avec la substance d'essai
Les cellules en prolifération sont traitées avec la substance d'essai en présence et en l'absence d'activation métabolique. La durée d'exposition doit être suffisante (une durée de trois à six heures est généralement efficace) et peut s'étendre sur un ou plusieurs cycles cellulaires.
On peut réaliser les cultures en un seul ou en double exemplaire pour chaque concentration testée. Dans le cas des cultures en un seul exemplaire, il faut augmenter le nombre des concentrations afin d'assurer un nombre adéquat de cultures à analyser (par exemple, au moins huit concentrations analysables). Les cultures servant de témoins négatifs (solvant) sont faites en double.
Les substances gazeuses ou volatiles doivent être testées en utilisant des méthodes appropriées, par exemple dans des récipients de culture hermétiquement fermés (21, 22).
1.4.3.2. Détermination du taux de survie, de la viabilité et de la proportion de mutants
À la fin de la période d'exposition, les cellules sont lavées et mises en culture en vue de la détermination de leur taux de survie et de l'expression du phénotype mutant. La détermination de la cytotoxicité commence généralement après la période de traitement par la mesure de l'efficacité de clonage relative (taux de survie) ou de la croissance totale relative des cultures.
À chaque locus correspond un temps bien défini qui est nécessaire à l'expression presque optimale du phénotype des mutants nouvellement induits (HPRT et XPRT ont besoin d'au moins 6 à 8 jours et TK d'au moins 2 jours). Pour déterminer le nombre de mutants et l'efficacité de clonage, les cellules sont placées dans un milieu en présence et en l'absence d'agent(s) sélectif(s). On commence à mesurer la viabilité, qui permet de calculer la proportion de mutants, à la fin de la période d'expression en déposant les cultures dans un milieu non sélectif.
Si la substance d'essai est positive à l'essai sur L5178Y TK-/-, la taille des colonies doit être déterminée dans au moins une des cultures traitées (à la concentration positive la plus élevée) et dans les témoins négatifs et positifs. Si la substance d'essai est négative à l'essai sur L5178Y TK+/-, la taille des colonies doit être déterminée dans les témoins négatifs et positifs. Dans le cas des essais sur TK6TK+/-, on peut également déterminer la taille des colonies.
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
Les données doivent comprendre la détermination de la cytotoxicité et de la viabilité, le nombre de colonies et la proportion de mutants dans les cultures traitées et les cultures témoins. Dans le cas d'une réponse positive à l'essai sur L5178Y TK+/-, on dénombre les colonies en distinguant les petites des grandes colonies pour au moins une concentration de la substance d'essai (concentration positive la plus élevée) et pour les témoins négatifs et positifs. La nature moléculaire et cytogénétique des deux sortes de colonies de mutants (petites et grandes) a été étudiée en détail (23, 24). Dans l'essai TK+/-, les colonies sont recensées sur la base du critère de croissance normale (grandes colonies) et de croissance faible (petites colonies) (25). Les cellules mutantes qui ont subi les altérations génétiques les plus importantes ont un temps de duplication plus long et forment donc de petites colonies. Ces altérations vont de la perte du gène entier à des aberrations chromosomiques se manifestant dans le caryotype. L'induction de petites colonies de mutants a été mise en rapport avec des substances chimiques qui engendrent des aberrations chromosomiques importantes (26). Les cellules mutantes moins affectées croissent à des vitesses semblables à celles des cellules parentales et forment de grandes colonies.
Il convient d'indiquer le taux de survie (efficacité de clonage relative) ou la croissance totale relative. La proportion de mutants doit être exprimée comme le nombre de cellules mutantes divisé par le nombre de cellules survivantes.
Les données individuelles concernant chaque culture doivent être indiquées. En outre, toutes les données doivent être résumées sous forme de tableaux.
Il n'est pas nécessaire de vérifier une réponse manifestement positive. Les résultats équivoques doivent être clarifiés par des essais supplémentaires réalisés de préférence dans des conditions expérimentales modifiées. Les résultats négatifs demandent à être confirmés cas par cas. Si la confirmation de résultats négatifs n'est pas jugée nécessaire, une justification doit être fournie. Pour les essais de confirmation effectués en cas de résultats négatifs ou équivoques, il convient d'envisager une modification des paramètres de l'étude, afin d'élargir la gamme des conditions évaluées. Les paramètres susceptibles d'être modifiés comprennent l'écart entre les doses et les conditions d'activation métabolique.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Plusieurs critères permettent de déterminer si un résultat est positif, notamment un accroissement dose-dépendant ou un accroissement reproductible de la proportion de mutants. En premier lieu, il s'agit de prendre en considération la pertinence biologique des résultats. L'évaluation des résultats peut s'appuyer sur des méthodes statistiques mais la signification statistique ne doit pas être le seul facteur décisif pour conclure à une réponse positive.
Si les résultats obtenus avec une substance d'essai ne répondent pas aux critères indiqués ci-dessus, on peut conclure que la substance en question n'est pas mutagène dans cet essai.
Bien que la plupart des essais donneront des résultats nettement positifs ou négatifs, dans certains cas rares l'ensemble des résultats d'un essai n'autorise pas une conclusion catégorique sur l'activité de la substance étudiée. Les résultats peuvent rester équivoques ou discutables indépendamment du nombre de répétitions de l'essai.
Des résultats positifs d'un essai in vitro de mutation génique sur cellules de mammifère indiquent que la substance d'essai induit des mutations géniques dans les cellules de mammifère des cultures utilisées. Une relation dose-réponse positive, si elle est reproductible, est des plus significatives. Des résultats négatifs signifient que, dans les conditions de l'essai, la substance testée n'induit pas de mutation génique dans les cellules de mammifère des cultures utilisées.
3. RAPPORT
RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Solvant/véhicule:
|
—
|
justification du choix du véhicule/solvant,
|
|
—
|
solubilité et stabilité de la substance d'essai dans le solvant/véhicule, si elles sont connues.
|
|
|
|
Cellules:
|
—
|
type et source des cellules,
|
|
—
|
données concernant le cycle cellulaire,
|
|
—
|
nombre de repiquages, le cas échéant,
|
|
—
|
méthodes d'entretien des cultures cellulaires, le cas échéant,
|
|
—
|
absence de mycoplasme, le cas échéant.
|
|
|
|
Conditions de l'essai:
|
—
|
justification du choix des concentrations et du nombre de cultures, y compris des données relatives à la cytotoxicité et aux limites de solubilité, si elles sont disponibles,
|
|
—
|
composition du milieu, concentration de CO2,
|
|
—
|
concentration de la substance d'essai,
|
|
—
|
volume de véhicule et de substance d'essai ajouté,
|
|
—
|
température d'incubation,
|
|
—
|
densité cellulaire pendant le traitement,
|
|
—
|
type et composition du système d'activation métabolique, y compris les critères d'acceptabilité,
|
|
—
|
témoins positifs et négatifs,
|
|
—
|
durée de la période d'expression,
|
|
—
|
critères pour conclure que les résultats des essais sont positifs, négatifs ou équivoques,
|
|
—
|
méthodes utilisées pour compter le nombre de cellules viables et de cellules mutantes,
|
|
—
|
définition de colonies qui sont prises en compte pour le type et pour la taille, y compris les critères pour les «petites» et «grandes» colonies.
|
|
|
|
Résultats:
|
—
|
signes de précipitation,
|
|
—
|
données sur le pH et l'osmolalité pendant l'exposition à la substance d'essai, si elles ont été déterminées,
|
|
—
|
taille des colonies si elle a été déterminée pour au moins les témoins négatifs et positifs,
|
|
—
|
aptitude du laboratoire à déceler des mutants en petites colonies avec le système L5178Y TK+/-, le cas échéant,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
analyses statistiques, le cas échéant,
|
|
—
|
données concernant les témoins négatifs (solvant ou véhicule) et positifs concomitants,
|
|
—
|
données concernant les témoins négatifs (solvant ou véhicule) et positifs antérieurs (étendues, moyennes, écarts-types).
|
|
|
|
Discussion des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
Moore, M.M., DeMarini, D. M., DeSerres, F. J. and Tindall, K. R. (eds.) (1987), Ranbury Report 28: Mammalian Cell Mutaginesis, Cold Spring Harbor Laboratory, New York.
|
|
(2)
|
Chu, E. H. Y. and Mailing, H. V. (1968), Mammalian Cell Genetics. II. Chemical Induction of Specific Locus Mutations in Chinese Hamster Cells in Vitro. Proc. Natl. Acad. Sci. USA. 61, pp. 1306-1312.
|
|
(3)
|
Liber, H. L. and Thilly, W. G. (1982), Mutation Assay at the Thymiuine Kinase Locus in Diploid Human Lymphoblasts, Mutation Res., 94, pp. 467-48 5.
|
|
(4)
|
Moore, M. M., Harington-Brock. K., Doerr, C. L. and Dearfield, K. L. (1989), Differential Mutant Quantitation at the Mouse Lymphoma TK and CHO HGPRT Loci, Mutagenesis. 4, pp. 394-403.
|
|
(5)
|
Aaron, C. S. and Stankowski, Jr. L. F. (1989), Comparison of the AS52/XPRT and the CHO/HPRT Assays: Evaluation of Six Drug Candidates, Mutation Res, 223, pp. 121-128.
|
|
(6)
|
Aaron, C. S., Bolcsfoldi, G., Glatt, H. R., Moore, M., Nishi, Y., Stankowski, Jr. L. F., Theiss, J. and Thompson, E. (1994), Mammalian Cell Gene Mutation Assays Working Group Report. Report of the International Workshop on Standardisation of Genotoxicity Test Procedures. Mutation Res, 312, pp. 235-239.
|
|
(7)
|
Scott, D., Galloway, S. M., Marshall, R. R., lshidate, M., Brusick, D., Ashby, J. and Myhr, B. C. (1991), Gcnotoxicity Under Extreme Culture Conditions. A report from ICPEMC Task Group 9, Mutation Res, 257, pp. 147-204.
|
|
(8)
|
Clive, D., McCuen, R., Spector, J. F. S., Piper, C. and Mavournin, K. H. (1983), Specific Gene Mutations in L5178Y Cells in Culture. A Report of the U. S. Environmental Protection Agency Gene-Tox Program, Mutation Res, US, pp. 225-251.
|
|
(9)
|
Li, A. P., Gupta, R. S., Heflich, R. H. and Wasson. J. S. (1988), A Review and Analysis of the Chinese Hamster Ovary/Hvpoxanthine Guanine Phosphoribosyl Transferase System to Determine the Mutagenicity of Chemical Agents: A Report of Phase III of the U. S. Environment Protection Agency Gene-Tox Program. Mutation Res, 196, pp. 17-36.
|
|
(10)
|
Li, A. P., Carver. J. H., Choy, W. N., Hsie, A. W., Gupta, R. S., Loveday, K. S., O'Neill, J. P., Riddle, J. C, Stankowski, L. F. Jr. and Yang, L. L. (1987), A Guide for the Performance of the Chinese Hamster Ovary Cell/Hypoxanthine-Cuanine Phosphoribosyl Transferase Gene Mutation Assay, Mutation Res, 189, pp. 135-141.
|
|
(11)
|
Liber, H. L, Yandell, D. W. and Little, J. B. (1989), A Comparison of Mutation Induction at the TK and HPRT Loci in Human Lymphoblastoid Cells: Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK Locus, Mutation Res, 216, pp. 9-17.
|
|
(12)
|
Stankowski, L. F. Jr., Tindall, K. R. and Hsie, A. W. (1986), Quantitative and Molecular Analyses of Ethyl Methanosulphonate — and ICR 191-Induced Molecular Analyses of Ethyl Methanosulphonate — and ICR 191-Induced Mutation in AS 52 Cells, Munition Res, 160, pp. 133-147.
|
|
(13)
|
Turner, N. T., Batson, A. G. and Clive, D. (1984), Procedures for the L5178Y/TK-/- — TK-/- Mouse Lymphoma Cell Mutagenicity Assay, in: Kilbey, B. J. et al (eds.) Handbook of Mutagenicity Test Procedures, Elsevier Science Publishers, New York, pp. 239-268.
|
|
(14)
|
Arlett, C. F., Smith, D. M., Clarke, G. M., Green, M. H. L, Cole, J., McGregor, D. B. and Asquith, J. C. (1989), Mammalian Cell Gene Mutation Assays Based upon Colony Formation, in: Statistical Evaluation of Mutagenicity Test Data, Kirkland D. J., ed., Cambridge University Press, pp. 66-101.
|
|
(15)
|
Abbondandolo, A., Bonatti, S., Corti, G., Fiorio, R., Loprieno, N. and Mazzaccaro, A. (1977), Induction of 6-Thioguanine-Resistant Mutants in V79 Chinese Hamster Cells by Mouse-Liver Microsome-Activated Dimethylnitrosamine, Mutation Res, 46, pp. 365-373.
|
|
(16)
|
Ames, B. N., McCann, J. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res, 31, pp. 347-364.
|
|
(17)
|
Clive, D., Johnson, K. O., Spector, J. F. S., Batson, A. G. and Brown M. M. M. (1979), Validation and Characterisation of the L5178Y/-/- Mouse Lymphoma Mutagen Assay System, Mutation Res, 59, pp. 61-108.
|
|
(18)
|
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res, 113, pp. 173-215.
|
|
(19)
|
Elliott, B. M., Combes, R. D., Elcomc, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-lnduced S9 in In Vitro Genotoxicity Assays, Mutagenesis. 7, pp. 175-177.
|
|
(20)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polychlorinatcd Biphenyls as an Inducer of Metabolic Activation Systems, in In Vitro Metabolic Activation in Mutagenesis Testing, de Serres. F. J.. Fonts, I. R.. Bend, J. R. and Philpot. R. M. (eds.) Elsevier, North-Holland, pp. 85-88.
|
|
(21)
|
Krahn, D. F., Barsky. F. C. and McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, In: Tice, R. R., Costa, D. L., Schaich, K. M. (eds.), Genoloxic Effects of Airborne Agents, New York, Plenum, pp. 91-103.
|
|
(22)
|
Zamora, P. O., Benson, J. M., Li, A. P. and Brooks, A. L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental Mutagenesis. 5, pp. 795-801.
|
|
(23)
|
Applegate, M. L., Moore, M. M., Broder, C. B., Burreli, A. and Hozier, J. C. (1990), Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells, Proc. Natl. Acad. Sci. USA, 87, pp. 51- 55.
|
|
(24)
|
Moore. M. M., Clive, D., Hozier, J. C. Howard, B. E., Batson, A. G., Turner, N. T. and Sawyer, J. (1985), Analysis of Trifluorothymidine-Resistant (TFT1) and Mutants of L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 151, pp. 161-174.
|
|
(25)
|
Yandell, D. W., Dryja, T. P. and Little, J. B. (1990), Molecular Genetic Analysis of Recessive Mutations at a Heterozygous Autosomal Locus in Human Cells, Mutation Res., 229, pp. 89-102.
|
|
(26)
|
Moore, M. M. and Doerr, C. L. (1990), Comparison of Chromosome Aberration Frequency and Small- Colony TK-Deficient Mutant Frequency in L5178Y/TK+/- — 3.7.2C Mouse Lymphoma Cells, Mutagenesis 5, pp. 609-614.
|
B.18. LÉSION ET RÉPARATION D'ADN — SYNTHÈSE NON PROGRAMMÉE DE L'ADN (UDS) — CELLULES DE MAMMIFÈRE — IN VITRO
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITION
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
L'essai de synthèse non programmée de l'acide désoxyribonucléique (ADN) (UDS) mesure la synthèse de réparation de l'ADN après excision et élimination d'un fragment d'ADN contenant la région lésée par des agents chimiques et physiques. L'essai est basé sur l'incorporation de thymidine tritiée (3H-TdR) dans l'ADN de cellules de mammifère ne se trouvant pas en phase S du cycle cellulaire. On peut déterminer l'incorporation de 3H-TdR en examinant par autoradiographie ou par comptage en scintillation liquide (LSC) l'ADN provenant de cellules traitées. Les cellules de mammifère en culture, à moins que des hépatocytes primaires de rat ne soient utilisés, sont traitées avec la substance d'essai en présence et en l'absence d'un système d'activation métabolique exogène. L'UDS peut également être mesurée par des méthodes in vivo.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Les substances d'essai ainsi que les substances témoins ou de référence seront, soit préparées en milieu de culture, soit dissoutes ou mises en suspension dans des véhicules appropriés, puis diluées en milieu de culture avant d'être utilisées dans l'essai. La concentration finale du véhicule ne devrait pas affecter la viabilité cellulaire.
Des cultures primaires d'hépatocytes de rats, de lymphocytes humains ou des lignées cellulaires établies (par exemple fibroblastes humains diploïdes) peuvent être utilisées pour cet essai.
Les cellules seront exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique approprié.
Conditions d'essai
Nombre de cultures
Au moins deux cultures cellulaires pour l'autoradiographie et six cultures cellulaires (ou moins si justifié scientifiquement) pour le comptage en scintillation liquide sont nécessaires pour chaque pont expérimental.
Utilisation de témoins négatifs et positifs
Des témoins simultanés positifs et négatifs (non traités et/ou véhicule), avec et sans activation métabolique, devraient être inclus dans chaque expérience. Pour l'essai sur hépatocytes de rat, on peut, par exemple, utiliser le 7,12-DMBA (7,12-diméthylbenzanthracène) ou le 2-AAF (2-acétylaminofluorène). Dans le cas de lignées cellulaires établies, on peut, pour les essais par autoradiographie et LSC réalisés sans activation métabolique, utiliser comme témoin positif par exemple le 4-NQO (4-nitroquinoline-N-oxyde); lorsqu'on a recours à des systèmes d'activation métaboliques, la N-diméthylnitrosamine est un des composés témoins positifs utilisables.
Concentration d'exposition
Une gamme de concentrations de la substance d'essai devrait être utilisée afin de permettre de déterminer au mieux la réponse. La concentration maximale devrait produire certains effets cytotoxiques. Les substances relativement insolubles dans l'eau seront testées jusqu'à leur limite de solubilité. En ce qui concerne les substances non toxiques franchement solubles dans l'eau, la concentration maximale à tester devrait être déterminée cas par cas.
Cellules
Pour l'entretien des cultures on aura recours à des milieux de culture, une concentration en CO2, une température et une humidité appropriées. Les lignées cellulaires établies devraient être périodiquement contrôlées du point de vue de la contamination par Mycoplasme.
Activation métabolique
Aucun système d'activation métabolique n'est utilisé pour des cultures primaires d'hépatocytes. Les lignées cellulaires établies et les lymphocytes sont exposés à la substance d'essai en présence et en l'absence d'un système d'activation métabolique approprié.
Conduite de l'essai
Préparation des cultures
Des lignées cellulaires établies sont obtenues à partir de cultures souches (par exemple par trypsinisation ou par agitation), ensemencées dans des récipients de culture à une densité adéquate et incubées à 37 oC.
Des cultures à court terme d'hépatocytes de rat sont établies en permettant aux hépatocytes récemment dissociés dans un milieu approprié de s'attacher à la surface de croissance.
Les cultures de lymphocytes humains sont réalisées par des techniques appropriées.
Traitement des cultures avec la substance d'essai
Hépatocytes primaires de rat:
Des hépatocytes primaires de rat récemment isolés sont traités au moyen de la substance d'essai dans un milieu contenant 3H-TdR, pendant un laps de temps adéquat. À l'issue de la période de traitement, le milieu sera éliminé, les cellules seront rincées, fixées et séchées. Les lames sont plongées dans une émulsion autoradiographique [alternativement une pellicule photographique (stripping film) peut être utilisée], exposées, développées, colorées et comptées.
Lignées cellulaires établies et lymphocytes:
Techniques autoradiograpbiques: Les cultures cellulaires sont exposées à la substance d'essai pendant des périodes de temps adéquates, puis traitées à la 3H-TdR. La durée de l'exposition sera fonction de la nature de la substance, de l'activité du système métabolique et du type de cellules. Pour déceler l'acmé de «UDS», la 3H-TdR sera ajoutée soit en même temps que la substance d'essai, soit dans les quelques minutes qui suivent l'exposition à la substance d'essai. Le choix de l'une ou l'autre de ces techniques sera déterminé par d'éventuelles interactions entre la substance d'essai et la 3H-TdR. Afin de pouvoir faire la distinction entre l'UDS et la réplication semi-conservatrice de l'ADN, on peut inhiber cette dernière en utilisant, par exemple, un milieu déficient en arginine, une faible teneur en sérum ou en ajoutant de l'hydroxyurée dans le milieu de culture.
Mesures l'USC de l'UDS: Avant de procéder au traitement avec la substance d'essai, l'entrée des cellules en phase S sera bloquée de la manière décrite ci-dessus; les cellules sont ensuite exposées à la substance d'essai comme décrit pour l'autoradiographie. À l'issue de la période d'incubation, l'ADN est extrait des cellules et la teneur totale en ADN ainsi que la quantité de 3H-TdR incorporée sont déterminées.
Il faut noter que lorsque des lymphocytes humains sont utilisés, il n'est pas nécessaire de supprimer la réplication semi-conservative de l'ADN dans des cultures non stimulées.
Analyse
Déterminations autoradiographiques
Pour déterminer l'UDS dans des cellules en culture, on ne compte pas les noyaux en phase S. Au moins 50 cellules par concentration seront comptées. Les James seront codées avant comptage. Plusieurs champs choisis au hasard suffisamment éloignés les uns des autres seront comptés sur chaque lame. On déterminera la quantité de 3H-TdR incorporée dans le cytoplasme en comptant trois surfaces de la taille du noyau dans le cytoplasme de chaque cellule comptée.
Détermination LSC
On devrait utiliser un nombre adéquat de cultures pour chaque concentration et pour les témoins lors des déterminations LSC-UDS.
Tous les résultats sont confirmés au cours d'une expérience indépendante.
2. RÉSULTATS
Les résultats sont présentés sous la forme de tableaux.
2.1. DÉTERMINATIONS AUTORADIOGRAPHIQUES
La quantité de 3H-TdR incorporée dans le cytoplasme ainsi que le nombre de grains comptés par noyau cellulaire seront enregistrés séparément.
La moyenne, la médiane et le mode peuvent être utilisés pour décrire la distribution de la quantité de 3H-TdR incorporée dans le cytoplasme ainsi que le nombre de grains par noyau.
2.2. DÉTERMINATIONS LSC
Pour les déterminations LSC, l'incorporation de 3H-TdR sera indiquée sous la forme de dpm/μg d'ADN. La moyenne des dpm/μg d'ADN moyen avec son écart type peut être utilisé pour décrire la distribution de l'incorporation.
Les données devraient être évaluées par des méthodes statistiques appropriées.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
cellules utilisées, densité et nombre de passages au moment du traitement, nombre de cultures cellulaires,
|
|
—
|
méthodes utilisées pour l'entretien des cultures cellulaires, y compris milieu, température et concentration en CO2,
|
|
—
|
substance d'essai, véhicule, concentration et justification du choix des concentrations utilisées dans l'essai,
|
|
—
|
détails relatifs aux systèmes d'activation métabolique,
|
|
—
|
témoins positifs et négatifs,
|
|
—
|
technique autoradiographique utilisée,
|
|
—
|
méthodes utilisées pour bloquer l'entrée des cellules en phase S,
|
|
—
|
techniques utilisées pour extraire l'ADN et déterminer la quantité totale d'ADN dans les déterminations LSC,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
évaluation statistique,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.19. ESSAI IN VITRO D'ÉCHANGE DE CHROMATIDES-SŒURS
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITION
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
L'essai d'échange des chromatides-sœurs (SCE) constitue un test à court terme destiné à déceler les échanges réciproques d'ADN entre deux chromatides-sœurs d'un chromosome se dédoublant. Les SCE-s représentent l'échange réciproque de produits de réplication de l'ADN à des loci apparemment homologues. Le processus d'échange implique probablement une cassure et une réunion de l'ADN, bien que l'on sache peu de choses de sa base moléculaire. Pour déceler des SCE-s, il est nécessaire de pouvoir marquer différemment les chromatides-sœurs; ceci est rendu possible par l'incorporation de bromodésoxyuridine (BrdU) dans l'ADN chromosomique pendant deux cycles cellulaires.
Les cellules de mammifère in vitro sont exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique exogène de mammifère, le cas échéant, et mises en culture pendant deux cycles de réplication dans un milieu contenant de la BrdU. Après traitement avec un inhibiteur du fuseau (par exemple colchicine) afin d'accumuler les cellules en phase de mitose de type métaphasique (c-métaphase), les cellules sont récoltées et des préparations de chromosomes sont réalisées.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
|
—
|
Des cultures primaires (lymphocytes humains) ou des lignées cellulaires établies (par exemple cellules ovariennes de hamsters chinois) peuvent être utilisées pour l'essai. Les 'lignées cellulaires seront contrôlées du point de vue de la contamination par des Mycoplasmes.
|
|
—
|
Des milieux de culture et des conditions d'incubation appropriés (par exemple température, récipients de culture, concentration en CO2 et humidité) seront utilisés.
|
|
—
|
Les substances d'essai peuvent être soit préparées dans des milieux de culture soit dissoutes ou mises en suspension dans des véhicules appropriés avant le traitement des cellules. La concentration finale d'un véhicule dans le milieu de culture ne devrait pas affecter significativement la viabilité des cellules ou le taux de croissance et les effets sur la fréquence de SCE seront contrôlés à l'aide d'un solvant témoin.
|
|
—
|
Les cellules seront exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique exogène de mammifère. Lorsqu'on utilise des types de cellules ayant une activité métabolique intrinsèque, le taux et la nature de l'activité devraient être appropriés à la classe chimique testée.
|
Conditions d'essai
Nombre de cultures
Deux cultures séparées au moins seront utilisées pour chaque point expérimental.
Utilisation de témoins positifs et négatifs
Des témoins positifs utilisant une substance à action directe et une substance nécessitant une activation métabolique devraient être inclus dans chaque expérience: un témoin devrait être également utilisé pour le véhicule.
Les substances ci-après peuvent par exemple, être utilisées comme témoins positifs:
|
—
|
substance à action directe:
|
—
|
méthanesulfonaté d'éthyle,
|
|
|
—
|
substance à action indirecte:
|
Le cas échant, un témoin positif supplémentaire appartenant à la même classe chimique que la substance d'essai peut être inclus.
Concentration d'exposition
Au moins trois concentrations de la substance d'essai, convenablement espacées, devraient être utilisées. La concentration maximale produira un effet toxique significatif mais permettra encore une réplication cellulaire adéquate. Des substances relativement insolubles dans l'eau seront testées jusqu'à leur limite de solubilité par des méthodes appropriées. Dans le cas de substances non toxiques franchement solubles dans l'eau, la concentration maximale de la substance d'essai devrait être déterminée cas par cas.
Conduite de l'essai
Préparation des cultures
Des lignées cellulaires établies sont obtenues à partir de cultures souches (par exemple par trypsinisation ou par agitation), ensemencées dans des récipients de culture à une densité adéquate et incubées à 37 oC. Dans le cas de cultures en couche monocellulaire, le nombre de cellules par récipient de culture devrait être ajusté de manière à ce que les cultures ne soient pas confluentes à plus de 50 % au moment de la récolte. Les cellules peuvent également être utilisées sous une forme de culture en suspension. Les cultures de lymphocytes humains sont préparées à partir de sang hépariné, en utilisant des techniques appropriées, et incubées à 37 oC.
Traitement
Des cellules en phase exponentielle de croissance sont exposées à la substance d'essai pendant un laps de temps adéquat, dans la plupart des cas une à deux heures peuvent suffire, mais, dans certains cas, le traitement peut être prolongé pour couvrir jusqu'à deux cycles cellulaires complets. Les cellules n'ayant pas une activité métabolique intrinsèque suffisante devraient être exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique approprié. À l'issue de la période d'exposition, les cellules sont lavées de manière à éliminer la substance d'essai et mises en culture, en présence de BrdU pendant deux cycles de réplication. Une autre méthode consiste à exposer simultanément les cellules à la substance d'essai et à la BrdU pendant deux cycles cellulaires complets.
Les cultures de lymphocytes humains sont traitées pendant qu'elles se trouvent dans un état semi-synchrone.
Les cellules sont analysées au cours de leur seconde division après traitement, garantissant ainsi l'exposition des phases les plus sensibles du cycle cellulaire à la substance d'essai. Toutes les cultures additionnées de BrdU seront manipulées dans l'obscurité ou sous un faible éclairage provenant de lampes incandescentes jusqu'au moment de la récolte des cellules, afin de réduire la photolyse de l'ADN contenant de la BrdU.
Récolte des cellules
Les cultures cellulaires sont traitées au moyen d'un inhibiteur du fuseau (par exemple colchicine) une à quatre heures avant leur récolte. Chaque culture est récoltée et traitée séparément pour la préparation des chromosomes.
Préparation des chromosomes et coloration
Les préparations de chromosomes sont effectuées par des méthodes cytogénétiques standard. Plusieurs techniques peuvent être utilisées pour colorer les lames de manière à faire apparaître les SCE (par exemple la méthode par fluorescence plus Giemsa).
Analyse
Le nombre de cellules analysées sera fonction de la fréquence spontanée témoin de SCE. Habituellement, on analyse au moins 25 métaphases bien étalées par culture afin de compter les SCE. Les lames sont codées avant l'analyse. Pour les lymphocytes humains, seules les métaphases contenant 46 centromères sont analysées. Pour les lignées cellulaires établies, seules les métaphases contenant ± 2 centromères du nombre modal sont analysées. Il sera précisé si une inversion de coloration au niveau de centromère est considérée comme un SCE ou non. Les résultats sont confirmés au cours d'une expérience indépendante.
2. RÉSULTATS
Les résultats devraient être présentés sous la forme de tableaux. Le nombre de SCE par métaphase et le nombre de SCE par chromosome sont donnés séparément pour toutes les cultures traitées et témoins.
Les résultats seront analysés par des méthodes statistiques adéquates.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
cellules utilisées, méthodes d'entretien de la culture cellulaire,
|
|
—
|
conditions d'essai: composition des milieux, concentration en CO2, concentration de la substance d'essai, véhicule utilisé, température d'incubation, temps de traitement, inhibiteur du fuseau utilisé, concentration et durée du traitement avec celui-ci, type de système d'activation de mammifère utilisé, témoins positifs et négatifs,
|
|
—
|
nombre de cultures cellulaires par point expérimental,
|
|
—
|
détails relatifs à la technique utilisée pour la préparation des lames,
|
|
—
|
nombre de métaphases analysées (données indiquées séparément pour chaque culture),
|
|
—
|
nombre de SCE par cellule et par chromosome (données indiquées séparément pour chaque culture),
|
|
—
|
critère de comptage des SCE,
|
|
—
|
justification du choix des doses,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
évaluation statistique,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.20. TEST DE LÉTALITÉ RÉCESSIVE LIÉE AU SEXE SUR DROSOPHILA MELANOGASTER
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Le test de létalité récessive liée au sexe (Sex-linked recessive lethal test: SLRL) utilisant Drosophila melanogaster décèle l'apparition de mutations (mutations ponctuelles ainsi que petites délétions), dans la lignée germinale de l'insecte. Cette épreuve constitue un essai de mutation en avant permettant de déceler des mutations à environ 800 loci du chromosome X, soit environ 80 % de tous les loci du chromosome X. Le chromosome X représente approximativement un cinquième du génome haploïde complet.
Les mutations du chromosome X de Drosophila melanogaster sont phénotypiquement exprimées chez les mâles porteurs du gène mutant. Lorsque la mutation est létale à l'état hémizygote, sa présence est déduite de l'absence d'un des groupes de descendance mâle sur les deux normalement produits par une femelle hétérozygote. L'épreuve SLRL exploite cette caractéristique en utilisant des chromosomes spécifiquement marqués et possédant des réarrangements de structure.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Stocks
Des mâles d'une lignée de type sauvage bien définie ainsi que des femelles contenant le chromosome Muller-5 peuvent être utilisés. D'autres lignées de femelles adéquatement marquées et porteuses de chromosomes X inversés de façon multiple peuvent également être utilisées.
Substance d'essai
Les substances d'essai sont dissoutes dans l'eau. Les substances insolubles dans l'eau peuvent être dissoutes ou mises en suspension dans des véhicules appropriés (par exemple un mélange d'éthanol et de Tween-60 ou 80), puis être diluées dans de l'eau ou dans une solution saline avant d'être administrées. L'utilisation de diméthylsulfoxyde comme véhicule devrait être évitée.
Nombre d'animaux
La sensibilité et la signification de l'essai seront prédéterminées. La fréquence de mutants spontanés observée dans le témoin approprié influencera fortement le nombre de chromosomes traités à analyser.
Voie d'administration
L'administration peut être orale, par injection ou par exposition à des gaz ou des vapeurs. L'ingestion de la substance d'essai peut se faire par l'intermédiaire d'une solution sucrée. Si nécessaire, les substances peuvent être dissoutes dans une solution à 0,7 % de NaCl et injectées dans le thorax ou l'abdomen.
Utilisation de témoins négatifs et positifs
Des témoins négatifs (véhicule) et positifs seront inclus. Toutefois, si le laboratoire dispose de données de contrôles historiques adéquats, des témoins simultanés ne sont pas nécessaires.
Niveau d'exposition
Trois niveaux d'exposition seront utilisés. Pour une première évaluation, on peut utiliser un seul niveau d'exposition de la substance d'essai, à savoir, soit la concentration maximale tolérée soit la concentration produisant certains signes de toxicité. Dans le cas de substances non toxiques, on devrait avoir recours à l'exposition à la concentration maximale praticable.
Conduite de l'essai
Des mâles de phénotype sauvage (âgés de 3 à 5 jours) sont traités avec la substance d'essai et individuellement accouplés à plusieurs femelles vierges de la lignée Muller-5 ou d'une autre porteuse d'un marqueur adéquat (à chromosomes X inversés de façon multiple). Tous les deux à trois jours, ces femelles sont remplacées par d'autres femelles vierges afin de couvrir le cycle germinal complet. La descendance de ces femelles est examinée afin de relever les effets létaux correspondant aux effets sur le sperme mature, les spermatides à des stades précoces, moyens ou tardifs, les spermatocytes et les spermatogonies au moment du traitement.
Les femelles F1 hétérozygotes provenant des croisements susmentionnés sont accouplées individuellement (c'est-à-dire une femelle par flacon) avec leurs frères. À la génération F2, chaque culture est examinée afin de déceler l'absence de mâles du type sauvage. Si une culture s'avère provenir d'une femelle F1 porteuse d'un gène létal sur le chromosome X parental (c'est-à-dire qu'aucun mâle porteur du chromosome traité n'est observé), les filles de cette femelle présentant le même génotype seront testées afin de vérifier si la létalité se répète dans la génération suivante.
2. RÉSULTATS
Les résultats devraient être présentés sous forme de tableaux afin de montrer le nombre de chromosomes testés, le nombre de mâles infertiles et le nombre de chromosomes létaux à chaque concentration d'exposition et à chaque période d'accouplement pour chaque mâle traité. Le nombre de groupes de tailles différentes par mâle est indiqué. Les résultats de l'essai seront confirmés au cours d'une expérience séparée.
Des méthodes statistiques appropriées seront utilisées pour évaluer le test de létalité récessive liée au sexe. Le regroupement de gènes létaux récessifs provenant d'un seul mâle sera pris en considération et évalué par une méthode statistique adéquate.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
lignée: lignées ou souches de drosophiles utilisées, âge des insectes, nombre de mâles traités, nombre de mâles stériles, nombre de cultures F2 établies, nombre de cultures F2 sans progéniture, nombre de chromosomes porteurs d'un gène létal décelé à chaque stade de la cellule germinale
|
|
—
|
critères retenus pour déterminer la taille des groupes traité,
|
|
—
|
conditions d'essai: description détaillée du programme de traitement et de prélèvement, niveau d'exposition, données relatives à la toxicité, témoins négatifs (solvant) et positifs, si nécessaire,
|
|
—
|
critères de comptage des mutations létales,
|
|
—
|
relation exposition/effet, si possible,
|
|
—
|
évaluation des données,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.21. TESTS DE TRANSFORMATION SUR CELLULES DE MAMMIFÈRE IN VITRO
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Des systèmes de culture de cellules de mammifère peuvent être utilisés pour déceler des modifications phénotypiques in vitro induites par des substances chimiques associées à une transformation maligne in vivo. Parmi les cellules les plus fréquemment utilisées figurent C3H10T'1/2, 3T3, SHE, rat Fisher; les tests sont fondés sur des modifications de la morphologie cellulaire, la formation des foyers ou des modifications liées à la croissance ou non dans un agar semi-solide. Il existe des systèmes moins fréquemment utilisés qui décèlent d'autres modifications physiologiques ou morphologiques dans les cellules a l'issue d'une exposition à des cancérogènes chimiques. Aucun des critères analysés dans ces tests in vitro n'a de relation mécanistique établie avec le cancer. Certains systèmes de tests sont capables de déceler des promoteurs de tumeur. On peut déterminer la cytotoxicité en mesurant l'effet de la substance d'essai sur les aptitudes à former une colonie (efficacité de clonage) ou sur les taux de croissance des cultures. La mesure de la cytotoxicité a pour but de déterminer si l'exposition à la substance d'essai a été toxicologiquement significative mais ne peut servir à calculer la fréquence des transformations dans toutes les épreuves, étant donné que certaines de celles-ci peuvent nécessiter une incubation prolongée et/ou un réensemencement.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Cellules
Suivant l'épreuve de transformation effectuée, diverses lignées cellulaires ou cellules primaires peuvent être utilisées. L'investigateur veillera à ce que les cellules utilisées pour l'épreuve présentent la modification phénotypique adéquate après exposition à des cancérogènes connus et à ce que la preuve de la validité et de la fiabilité de l'essai effectué dans le laboratoire de l'investigateur soient étayées par des documents.
Milieu
On utilisera des milieux et des conditions d'essai convenant à la méthode de transformation utilisée.
Substance d'essai
Les substances d'essai peuvent être soit préparées dans des milieux de culture, soit dissoutes ou mises en suspension dans des véhicules appropriés avant le traitement des cellules. La concentration finale du véhicule dans le milieu de culture n'affectera ni la viabilité des cellules, ni le taux de croissance, ni l'incidence de transformation.
Activation métabolique
Les cellules sont exposées à la substance d'essai en présence et en l'absence d'un système approprié d'activation métabolique de mammifère. Si l'on utilise des types de cellules ayant une activité métabolique intrinsèque, la nature de l'activité sera reconnue comme appropriée à la classe chimique testée.
Conditions d'essai
Utilisation de témoins positifs et négatifs
Des témoins positifs utilisant une substance à action directe et une substance nécessitant une activation métabolique seront inclus dans chaque expérience: un témoin négatif (véhicule) sera également utilisé.
Les substances ci-après peuvent, par exemple, être utilisées comme témoins positifs:
|
—
|
substances chimiques à action directe:
|
—
|
méthanesulfonate d'éthyle,
|
|
|
—
|
substances nécessitant une activation métabolique:
|
—
|
4-diméthylaminoazobenzène,
|
|
—
|
7,12-diméthylbenzo(a)anthracène.
|
|
Si cela se justifie, un témoin positif supplémentaire appartenant à la même classe chimique que la substance testée devrait être inclus.
Concentrations d'exposition
Plusieurs concentrations de la substance d'essai devraient être utilisées. Ces concentrations devraient produire un effet toxique en rapport avec la concentration, la concentration maximale produira un faible taux de survie, la concentration minimale un taux de survie voisin de celui observé pour le témoin négatif. Les substances relativement insolubles dans l'eau seront testées jusqu'à leur limite de solubilité par des méthodes appropriées. Dans le cas de substances non toxiques très solubles dans l'eau, la concentration maximale devrait être déterminée cas par cas.
Conduite de l'essai
Les cellules sont exposées pendant un laps de temps adéquat dépendant du système cellulaire utilisé, et ceci peut impliquer un nouveau traitement assorti d'un changement de milieu (et si nécessaire, un nouveau mélange d'activation métabolique) si l'exposition est prolongée. Les cellules n'ayant pas une activité métabolique intrinsèque suffisante devraient être exposées à la substance d'essai en présence et en l'absence d'un système d'activation métabolique approprié. À l'issue de la période d'exposition, les cellules sont lavées de manière à éliminer la substance testée et mises en culture dans des conditions permettant l'apparition du phénotype transformé étudié et l'incidence de cette transformation est déterminée. Tous les résultats sont confirmés au cours d'une expérience indépendante.
2. RÉSULTATS
Les résultats devraient être présentés sous la forme de tableaux et peuvent être combinés différemment suivant la méthode utilisée, par exemple comptage par boîte, boîtes positives ou nombre de cellules transformées. Le cas échéant, le taux de survie sera exprimé en pourcentage des taux témoins; la fréquence de transformation correspondra au nombre de cellules transformées par nombre de survivantes. Les résultats devraient être évalués par des méthodes statistiques appropriées.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
type de cellule utilisée, nombre de cultures cellulaires, méthodes d'entretien des cultures cellulaires,
|
|
—
|
conditions d'essai: concentration de la substance d'essai, véhicule utilisé, temps d'incubation, durée et fréquence du traitement, densité cellulaire durant le traitement, type de système d'activation métabolique exogène utilisé, témoins positifs et négatifs, spécification du phénotype étudié, système sélectif utilisé (le cas échéant), justification du choix des doses,
|
|
—
|
méthode utilisée pouf compter les cellules viables et transformées,
|
|
—
|
évaluation statistique,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.22. TEST DE LÉTALITÉ DOMINANTE CHEZ LE RONGEUR
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La létalité dominante provoque la mort de l'embryon ou du fœtus. L'induction de létalité dominante par exposition à une substance chimique indique que la substance a altéré le tissu germinal de l'espèce étudiée. Il est généralement admis que la létalité dominante est imputable à une lésion chromosomique (anomalies structurelles et numériques). Si les animaux traités sont des femelles, la mort de l'embryon peut également être due à des effets toxiques.
En général, les mâles sont exposés à la substance d'essai et accouplés avec des femelles vierges non traitées. Les différents stades des cellules germinales peuvent être testés séparément grâce à l'utilisation de périodes d'accouplement séquentielles. L'accroissement du nombre d'implants morts par femelle dans le groupe traité par rapport au nombre d'implants morts par femelle dans le groupe témoin reflète les pertes après implantation. Les pertes avant implantation peuvent être estimées par comptage des corps jaunes ou en comparant le nombre total d'implants par femelle dans le groupe traité et dans le groupe témoin. L'effet total de létalité dominante est égal à la somme des pertes avant et après implantation. Le calcul de l'effet total de la létalité dominante est basé sur la comparaison entre le nombre d'implants vivants par femelle dans le groupe testé et celui enregistré pour le groupe témoin. Une réduction du nombre des implants à certaines périodes peut être due à la destruction de cellules (c'est-à-dire de spermatocytes et/ou de spermatogonies).
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Lorsque c'est possible, les substances d'essai sont dissoutes ou mises en suspension dans une solution saline isotonique. Les substances chimiques insolubles dans l'eau peuvent être dissoutes ou mises en suspension dans des véhicules appropriés. Le véhicule utilisé ne devrait pas interférer avec la substance d'essai et ne devrait pas produire d'effets toxiques. Des préparations fraîchement réalisées de la substance d'essai sont utilisées.
Conditions d'essai
Voie d'administration
La substance d'essai ne devrait être généralement administrée qu'une seule fois. En fonction des informations toxicologiques disponibles, un programme de traitement répété peut être utilisé. Les voies d'administration habituelles sont l'intubation orale ou l'injection intrapéritonéale. D'autres voies d'administration peuvent être appropriées.
Animaux d'expérience
Il est recommandé d'utiliser des rats ou des souris. De jeunes animaux ayant atteint leur pleine maturité sexuelle sont répartis au hasard entre les groupes traités et témoins.
Nombre et sexe
Un nombre adéquat de mâles traités est utilisé compte tenu de la variabilité spontanée du caractère biologique étudié. Le nombre retenu devrait être fondé sur la sensibilité de détection et le degré de signification qui ont été prédéterminés. Par exemple, dans une expérience type, le nombre de mâles retenus pour chaque groupe de dose devrait être suffisant pour obtenir environ 30 à 50 femelles gravides par période d'accouplement.
Utilisation de témoins négatifs et positifs
Généralement des témoins positifs et négatifs (véhicule) seront inclus simultanément dans chaque expérience. Si des expériences récentes effectuées dans le même laboratoire ont donné des résultats acceptables pour des témoins positifs, ces résultats peuvent être utilisés à la place d'un témoin positif simultané. En ce qui concerne les substances témoins positives, on utilisera une dose faible appropriée (par exemple MMS, i.p., 10 mg/kg), afin de démontrer la sensibilité du test.
Doses
Normalement trois niveaux de dose sont utilisés. La dose élevée produira des signes de toxicité ou réduira la fécondité des animaux traités. Dans certains cas, un seul niveau de dose élevé peut suffire.
Épreuve limite
Les substances non toxiques sont testées à raison de 5 g/kg en cas d'administration unique et de 1 g/kg/jour en cas d'administration multiple.
Conduite de l'essai
Plusieurs schémas de traitement sont possibles. La méthode la plus fréquente est celle de l'administration unique. D'autres schémas de traitement peuvent être utilisés.
Chaque mâle est accouplé séquentiellement à 1 ou 2 femelles vierges non traitées à des intervalles de temps appropriés après le traitement. Les femelles devraient être laissées avec les mâles pendant au moins un cycle œstral complet ou jusqu'à ce que l'accouplement soit constaté par la présence de sperme dans le vagin ou la présence d'un bouchon vaginal.
Le nombre d'accouplements après traitement est déterminé par le programme de traitement et sera tel que tous les stades des cellules germinates traitées soient impliqués.
Les femelles sont sacrifiées durant la seconde moitié de leur gestation et le contenu de leur utérus est examiné, afin de déterminer le nombre d'implants vivants et morts. Les ovaires peuvent être examinés afin de déterminer le nombre de corps jaunes.
2. RÉSULTATS
Les résultats devraient être présentés sous la forme de tableaux indiquant le nombre de mâles, le nombre de femelles gravides ainsi que le nombre de femelles non gravides. Les résultats de chaque accouplement, y compris l'identité de chaque mâle et de chaque femelle, sont présentés séparément. Pour chaque femelle, la semaine d'accouplement et la dose reçue par les mâles ainsi que les fréquences d'implants vivants et morts sont rapportés.
Le calcul de l'effet total de la létalité dominante est basé sur une comparaison du nombre d'implants vivants par femelle dans le groupe testé avec le nombre d'implants vivants par femelle dans le groupe témoin. Le rapport entre implants vivants et implants morts dans le groupe traité comparé avec le rapport correspondant dans le groupe témoin est analysé en vue d'indiquer la perte après implantation.
Si les résultats sont enregistrés sous la forme de mortalité précoce et de mortalité tardive, les tableaux devraient faire clairement ressortir cette différence. Si la perte avant implantation est évaluée, il y a lieu d'en présenter les résultats. Cette perte peut être exprimée par la différence entre le nombre de corps jaunes et le nombre d'implants ou par la réduction du nombre moyen d'implants par femelle par rapport aux accouplements témoins.
Les résultats sont évalués par des méthodes statistiques appropriées.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
espèces, souche, âge et poids des animaux utilisés, nombre d'animaux de chaque sexe dans les groupes traités et témoins,
|
|
—
|
substance d'essai, véhicule, niveaux de dose testés et justification du choix des doses, témoins négatifs et positifs, données relatives à la toxicité,
|
|
—
|
voie et programme de traitement,
|
|
—
|
programme d'accouplement,
|
|
—
|
méthode utilisée pour constater l'accouplement,
|
|
—
|
critères de comptage des effets de la létalité dominante,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
évaluation statistique,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.23. ESSAI D'ABERRATION CHROMOSOMIQUE SUR SPERMATOGONIES DE MAMMIFÈRE
1. MÉTHODE
Cette méthode est une adaptation de la ligne directrice OCDE TG 483, Essai d'aberration chromosomique sur des spermatogonies de mammifères (1997).
1.1. INTRODUCTION
L'essai in vivo d'aberration chromosomique sur spermatogonies de mammifère est destiné à détecter les substances qui causent des aberrations de structure dans les spermatogonies de mammifère (1, 2, 3, 4, 5). Les aberrations de structure peuvent être de deux types: chromosomiques ou chromatidiennes. La majorité des mutagènes chimiques induisent des mutations chromatidiennes, mais des aberrations chromosomiques se produisent également. Cet essai n'est pas conçu pour évaluer les aberrations de nombre et n'est pas utilisé systématiquement dans ce but. Les mutations chromosomiques et les événements liés sont à l'origine de nombreuses maladies génétiques humaines.
Cet essai évalue des atteintes chromosomiques qui surviennent dans tes spermatogonies et devrait par conséquent permettre de prévoir l'introduction de mutations transmissibles à la descendance dans les cellules germinales.
Pour cet essai, on utilise généralement des rongeurs. Cet essai cytogénétique in vivo détecte des aberrations chromosomiques dans les spermatogonies en mitose. D'autres cellules cibles ne font pas l'objet de cette méthode.
Afin de détecter des aberrations chromatidiennes dans les spermatogonies, il faut examiner la première division cellulaire mitotique après le traitement, avant que ces aberrations ne disparaissent lors des divisions cellulaires ultérieures. L'analyse de chromosomes méiotiques peut fournir des informations complémentaires utiles; elle est destinée à mettre en évidence des aberrations de type chromosomique aux stades de la diacinèse-métaphase I, lorsque les cellules traitées se transforment en spermatocytes.
Cet essai in vivo est conçu pour vérifier si les mutagènes actifs sur les cellules somatiques le sont également sur les cellules germinales. L'essai sur spermatogonies se prête particulièrement bien à l'évaluation du risque mutagène, puisqu'il permet de tenir compte de facteurs de métabolisme in vivo, de pharmacocinétique et de processus de réparation de l'ADN.
Les testicules contiennent plusieurs générations de spermatogonies présentant des sensibilités diverses au traitement chimique. De ce fait, les aberrations détectées représentent une réponse globale des populations de spermatogonies traitées, dans lesquelles les cellules différenciées, plus nombreuses, prédominent. En fonction de leur position dans le testicule, les différentes générations de spermatogonies sont ou non exposées aux substances présentes dans la circulation générale en raison de la barrière physique et physiologique des cellules de Sertoli et de la barrière sang-testicule.
L'utilisation de cet essai ne convient pas s'il est manifeste que la substance d'essai ou un métabolite réactif n'atteindra pas le tissu cible.
Voir aussi l'introduction générale de la partie B.
1.2. DÉFINITIONS
Aberration de type chromatidien: lésion chromosomique de structure se traduisant par une cassure d'une seule chromatide ou par une cassure et une réunion entre chromatides.
Aberration de type chromosomique: lésion chromosomique de structure se traduisant par une cassure, ou par une cassure et une réunion, des deux chromatides sur le même site.
Lacune: une lésion achromatique inférieure à la largeur d'une chromatide, avec un défaut d'alignement minimal des chromatides.
Aberration de nombre: une modification du nombre de chromosomes par rapport au nombre normal caractéristique des cellules employées.
Polyploïdie: état multiple du nombre de chromosomes haploïdes (n), autre que la diploïdie (c'est-à-dire 3n, 4n, etc.)
Aberration de structure: modification de la structure des chromosomes, détectable par un examen au microscope au stade de la métaphase et se présentant sous la forme de délétions, de cassures, de modifications intrachromosomiques ou interchromosomiques.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
Les animaux sont exposés à la substance d'essai par une voie d'exposition appropriée et sont sacrifiés à des moments appropriés après le traitement. Avant le sacrifice, les animaux sont traités avec une substance qui bloque les cellules en métaphase (par exemple la colchicine ou Colcemid®). Ensuite, les préparations chromosomiques réalisées à partir des cellules germinales sont colorées et les cellules en métaphase sont examinées pour mettre en évidence les aberrations chromosomiques.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparations
1.4.1.1. Sélection des espèces animales
On utilise communément des hamsters chinois et des souris. Des mâles d'autres espèces de mammifères appropriées peuvent cependant être employés. Il convient d'utiliser des animaux adultes jeunes et sains, issus de souches de laboratoire courantes. Au début de l'étude, la variation pondérale des animaux doit être minimale et ne doit pas excéder ± 20 % du poids moyen.
1.4.1.2. Conditions d'encagement et régime alimentaire
Les conditions générales décrites dans l'introduction générale de la partie B sont appliquées mais le taux d'humidité devrait se situer aux environs de 50-60 %.
1.4.1.3. Préparation des animaux
De jeunes mâles sains sont répartis par randomisation entre les groupes témoins et les groupes traités. Les cages doivent être disposées de telle manière qu'il n'y ait pas d'effet dû à leur emplacement. Les animaux sont identifiés individuellement. Ils sont acclimatés aux conditions du laboratoire pendant au moins cinq jours.
1.4.1.4. Préparation des doses
Les substances d'essai solides sont dissoutes ou mises en suspension dans un véhicule ou un solvant approprié, puis éventuellement diluées avant l'administration aux animaux. Les substances d'essai liquides peuvent être administrées directement ou après dilution. Il convient d'utiliser des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage.
1.4.2. Conditions expérimentales
1.4.2.1. Solvant/véhicule
Le solvant/véhicule choisi ne doit pas avoir d'effet toxique aux doses utilisées ni présenter de réaction chimique avec la substance d'essai. Le recours à des solvants/véhicules inhabituels doit être justifié par des données sur leur compatibilité. On préconise d'envisager d'abord l'utilisation d'un véhicule/solvant aqueux, chaque fois que cela et possible.
1.4.2.2. Témoins
Des témoins positifs et négatifs (solvant ou véhicule) doivent accompagner chaque essai. Les animaux des groupes témoins doivent être manipulés exactement comme ceux des groupes traités, mise à part l'administration de la substance d'essai.
Les témoins positifs doivent produire des aberrations chromosomiques de structure in vivo dans les spermatogonies lorsqu'ils sont administrés à des niveaux d'exposition supposés produire un accroissement détectable par rapport au bruit de fond.
Les doses de témoin positif doivent être choisies afin que les effets soient nets mais que l'identité des lames codées ne soit pas révélée immédiatement à l'examinateur. Le témoin positif peut être administré par une voie différente de celle utilisée pour la substance d'essai et un seul prélèvement d'échantillon peut suffire. L'emploi de témoins positifs appartenant à la même classe chimique peut être envisagé, s'ils sont disponibles. Les substances indiquées dans le tableau ci-dessous sont des exemples de témoins positifs.
|
Substance
|
No CAS
|
No Einecs
|
|
Cyclophosphamide
Monohydrate de cyclophosphamide
|
50-18-0
6055-19-2
|
200-015-4
|
|
Cyclohexylamine
|
108-91-8
|
203-629-0
|
|
Mitomycine C
|
50-07-7
|
200-008-6
|
|
Acrylamide monomère
|
79-06-1
|
201-173-7
|
|
Triéthylènemélamine
|
51-18-3
|
200-083-5
|
Lors de chaque prélèvement, il faut inclure un lot d'animaux témoins négatifs traités seulement avec le solvant ou le véhicule et par ailleurs manipulés de la même façon que les groupes traités. Ceci peut être évité si l'on dispose de données acceptables sur la variabilité interindividuelle et la fréquence spontanée de cellules comportant des aberrations chromosomiques provenant de témoins historiques. En outre, des témoins non traités doivent aussi être inclus, à moins que des données historiques ne démontrent que le solvant/véhicule choisi n'induit aucun effet délétère ou mutagène.
1.5. MODE OPÉRATOIRE
1.5.1. Nombre d'animaux
Chaque groupe traité et groupe témoin doit comporter au moins cinq mâles analysables.
1.5.2. Modalités de traitement
La substance d'essai est de préférence administrée en une seule fois ou en deux fois (c'est-à-dire en un seul ou deux traitements). Elle peut aussi être administrée en doses fractionnées (deux doses le même jour avec un intervalle de quelques heures au maximum) afin que l'administration d'un grand volume soit plus aisée. D'autres modalités de traitement doivent être scientifiquement justifiées.
Dans le groupe qui reçoit la dose la plus forte, deux prélèvements d'échantillons sont effectués après le traitement. La cinétique cellulaire pouvant être influencée par la substance d'essai, le premier et le deuxième prélèvement ont lieu respectivement environ 24 heures et 48 heures après le traitement. Pour les autres doses, le prélèvement est effectué après 24 heures ou 1,5 cycle cellulaire après le traitement, à moins que l'on sache qu'il existe un autre délai de prélèvement plus propice à la détection des effets (6).
D'autres temps de prélèvement peuvent être employés. Par exemple, dans le cas de substances chimiques qui pourraient induire des chromosomes retardataires ou d'avoir des effets indépendants de la phase S, il peut être judicieux d'effectuer des prélèvements plus précoces (1).
La pertinence d'un traitement répété doit être évaluée au cas par cas. Après un traitement répété, les animaux doivent être sacrifiés 24 heures (1,5 cycle cellulaire) après le dernier traitement. De plus, d'autres temps de prélèvements peuvent être employés si nécessaire.
Avant d'être sacrifiés, les animaux reçoivent une dose appropriée d'un agent bloquant la métaphase (par exemple du Colcemid® ou de la colchicine) par injection intrapéritonéale. Les échantillons sont prélevés après des délais appropriés. Ce délai est d'environ 3-5 heures pour la souris et de 4-5 heures environ pour le hamster chinois.
1.5.3. Niveaux de dose
Si une étude préliminaire est réalisée afin d'évaluer les niveaux de dose requis, cette étude doit être effectuée dans le même laboratoire en utilisant la même espèce, la même souche, des animaux du même sexe et les mêmes modalités de traitement que ceux de l'étude principale (7). En cas de toxicité, on utilise trois niveaux de dose pour le premier prélèvement. Ces niveaux de dose doivent couvrir un domaine allant d'une toxicité nulle ou légère à la toxicité maximale. Pour les prélèvements suivants, seule la dose la plus élevée est utilisée. Celle-ci est définie comme la dose qui produit des signes de toxicité tels qu'ils font supposer que des doses plus fortes, administrées suivant la même modalité de traitement, seraient létales.
Les substances possédant une activité biologique spécifique à des doses faibles et non toxiques (comme les hormones et les mitogènes) peuvent faire exception aux critères d'établissement des doses et doivent être évaluées cas par cas. La dose la plus élevée peut aussi être définie comme étant celle qui produit certains indices de toxicité dans les spermatogonies (par exemple une diminution des mitoses des spermatogonies par rapport à la première et la deuxième métaphase méiotique; cette diminution ne devrait pas dépasser 50 %).
1.5.4. Test limite
Si un essai réalisé avec au moins 2 000 mg/kg de poids corporel, en une seule dose ou en deux doses administrées le même jour, n'engendre aucun effet toxique observable et si une génotoxicité est improbable d'après les données relatives à des substances de structure voisine, on peut considérer qu'une étude complète avec trois niveaux de doses n'est pas nécessaire. Suivant l'importance de l'exposition humaine probable, on peut envisager d'utiliser des doses plus fortes dans le test limite.
1.5.5. Voies d'administration
La substance d'essai est généralement administrée par gavage à l'aide d'une sonde gastrique ou d'une canule d'intubation adaptée, ou par injection intrapéritonéale. D'autres voies d'exposition sont acceptables lorsqu'elles peuvent être justifiées. Le volume maximal de liquide administré en une fois par gavage ou par injection dépend de la taille de l'animal. Ce volume ne devrait pas dépasser 2 ml/100 g de poids corporel. Une justification doit être fournie si des volumes plus importants sont administrés. Il convient de réduire au minimum la variation du volume administré en ajustant la concentration à tous les niveaux de doses, sauf pour les produits irritants ou corrosifs dont les effets sont généralement exacerbés lorsqu'on augmente la concentration.
1.5.6. Préparation des chromosomes
Des suspensions cellulaires obtenues à partir d'un ou des deux testicules immédiatement après le sacrifice sont exposées à une solution hypotonique et fixées. Les cellules sont ensuite étalées sur des lames et colorées.
1.5.7. Analyse
Au moins 100 cellules en métaphase bien étalées sont analysées par animal (soit un minimum de 500 métaphases par groupe d'essai). Ce nombre peut être réduit lorsque les aberrations observées sont nombreuses. Toutes les lames, y compris celles des témoins positifs et négatifs, doivent être codées indépendamment avant l'analyse au microscope. Le procédé de fixation provoquant souvent la rupture d'une certaine fraction des cellules en métaphase, ce qui entraîne une perte de chromosomes, les cellules examinées doivent contenir un nombre de centromères égal à 2n ± 2.
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
Les résultats individuels pour chaque animal doivent être présentés sous forme de tableaux. L'unité expérimentale est l'animal. Le nombre de cellules présentant une ou plusieurs aberrations de structure et le nombre d'aberrations par cellule doivent être évalués pour chaque animal. Les différents types d'aberrations chromosomiques de structure doivent être consignés avec leur nombre et leur fréquence pour les groupes traités et témoins. Les lacunes sont enregistrées séparément et rapportées mais ne sont généralement pas incluses dans la fréquence totale des aberrations.
Pour les cellules en méiose, il conviendra de prendre comme élément de cytotoxicité le rapport du nombre de spermatogonies en métaphase de première mitose/nombre de spermatogonies en métaphase de seconde mitose. Cette analyse sera réalisée à la fois chez les animaux traités et les contrôles négatifs sur un échantillon de 100 cellules en division pour chaque animal. Dans le cas où seules des mitoses sont observées, l'index mitotique doit être déterminé dans au moins 1 000 cellules de chaque animal.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Plusieurs critères permettent de déterminer si un résultat est positif, notamment un accroissement, dose-dépendant, du nombre relatif de cellules présentant des aberrations ou une augmentation nette du nombre de cellules présentant des aberrations dans un seul groupe traité à un seul moment de prélèvement. En premier lieu, il s'agit de prendre en considération la pertinence biologique des résultats. L'évaluation des résultats peut s'appuyer sur des méthodes statistiques (8) mais la signification statistique ne doit pas être le seul facteur décisif pour conclure à une réponse positive. Les résultats équivoques doivent être clarifiés par des essais supplémentaires réalisés de préférence dans des conditions expérimentales modifiées.
Si les résultats obtenus avec une substance d'essai ne répondent pas aux critères indiqués ci-dessus, on peut conclure que la substance en question n'est pas mutagène dans cet essai.
Bien que la plupart des essais donneront des résultats nettement positifs ou négatifs, dans certains cas rares l'ensemble des résultats d'un essai n'autorise pas un jugement catégorique sur l'activité de la substance étudiée. Les résultats peuvent rester équivoques ou discutables indépendamment du nombre de répétitions de l'essai.
Des résultats positifs obtenus dans cet essai in vivo d'aberration chromosomique sur spermatogonies indiquent que la substance induit des aberrations chromosomiques dans les cellules germinales de l'espèce testée. Des résultats négatifs signifient que, dans les conditions de l'essai, la substance testée n'induit pas d'aberrations chromosomiques dans les cellules germinales de l'espèce testée.
Le fait que la substance d'essai ou ses métabolites n'atteignent pas le tissu cible devrait être discuté.
3. RAPPORT
RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Solvant/véhicule:
|
—
|
justification du choix du véhicule,
|
|
—
|
solubilité et stabilité de la substance d'essai dans le solvant/véhicule, si elles sont connues.
|
|
|
|
Animaux d'expérience:
|
—
|
espèce/souche utilisée,
|
|
—
|
nombre et âge des animaux,
|
|
—
|
source, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
poids individuels des animaux au début de l'essai, y compris l'intervalle des poids, la moyenne et l'écart-type dans chaque groupe.
|
|
|
|
Conditions expérimentales:
|
—
|
résultats de l'étude de détermination des doses utiles, si elle a été réalisée,
|
|
—
|
justification du choix des doses employées,
|
|
—
|
justification du choix de la voie d'administration,
|
|
—
|
détails concernant la préparation de la substance d'essai,
|
|
—
|
détails concernant l'administration de la substance d'essai,
|
|
—
|
justification du choix des moments de sacrifice,
|
|
—
|
conversion de la concentration (ppm) de la substance d'essai dans l'eau de boisson ou la nourriture en dose réelle (mg/kg de poids corporel/jour), s'il y a lieu,
|
|
—
|
détails sur la qualité de la nourriture et de l'eau,
|
|
—
|
description détaillée des modalités de traitement et de prélèvement,
|
|
—
|
méthodes de mesure de la toxicité,
|
|
—
|
nature et concentration de l'inhibiteur du fuseau mitotique et durée du traitement,
|
|
—
|
méthodes de préparation des lames,
|
|
—
|
critères de dénombrement des aberrations,
|
|
—
|
nombre de cellules analysées par animal,
|
|
—
|
critères de décision concernant les résultats positifs, négatifs et équivoques.
|
|
|
|
Résultats:
|
—
|
taux de mitoses des spermatogonies par rapport à la première et la deuxième métaphase méiotique,
|
|
—
|
type et nombre d'aberrations, séparément pour chaque animal,
|
|
—
|
nombre total d'aberrations par groupe,
|
|
—
|
nombre de cellules présentant des aberrations par groupe,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
analyses statistiques, le cas échéant,
|
|
—
|
données sur les témoins négatifs concomitants,
|
|
—
|
données sur les témoins négatifs antérieurs (étendues, moyennes et écarts-types),
|
|
—
|
données sur les témoins positifs concomitants,
|
|
—
|
changements de la ploïdie, le cas échéant.
|
|
|
|
Discussion des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
Adler, I. D. (1986), Clastogenic Potential in Mouse Spermatogonia of Chemical Mutagens Related to their Cell-Cycle Specifications, in: Genetic Toxicology of Environmental Chemicals. Part B: Genetic Effects and Applied Mutagenesis, Ramel, C, Lambert B., and Magnusson, J. (eds.) Liss, New York. pp. 477-484.
|
|
(2)
|
Adler, I. D. (1984), Cytogenic tests in Mammals, in: Mutagenicity Testing: a Practical Approach, ed. S. Venitt and J. M. Parry. IRL Press, Oxford, Washington DC, pp. 275-306.
|
|
(3)
|
Evans, E. P., Breckon, G. and Ford. C. E. (1964), An Air-Drying Method for Meiotic Preparations from Mammalian Testes, Cytogenetics and Cell Genetics. 3, pp. 289-294.
|
|
(4)
|
Richold, M., Ashby, J., Chandley, A., Gatehouse, D. G. and Henderson L. (1990), In Vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenidty Testing. Report. Part I revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 115-141.
|
|
(5)
|
Yamamoto, K. and Kikuchi, Y. (1978), A New Method for Preparation of Mammalian Spermatogonial Chromosomes, Mutation Res., 52. pp. 207-209.
|
|
(6)
|
Adler, I. D., Shelby, M. D., Bootman,]., Favor, j., Generoso, W., Pacchierotti, F., Shibuya, T. and Tanaka, N. (1994), International Workshop on Standardisation of Genotoxicity Test Procedures. Summary Report of the Working Group on Mammalian Germ Cell Tests, Mutation Res., 312, pp. 313-318.
|
|
(7)
|
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson- Walker, G., Morton, D. B., Kirkland D. J. and Richold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working group: Dose setting in In Vivo Mutagenicity Assays, Mutagenesis, 7, pp. 313-319.
|
|
(8)
|
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D. G. and Savage, J. R. K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Statistical Evaluation of Mutagenicity Test Data. UKEMS Sub-Committee on Guidelines for Mulagenicity Testing, report. Part III, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 184-232.
|
B.24. SPOT TEST CHEZ LA SOURIS
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITION
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Il s'agit d'une épreuve in vivo effectuée sur souris dont les embryons en développement sont exposés à la substance d'essai. Les cellules visées des embryons en développement sont des mélanoblastes et les gènes cibles sont ceux responsables de la pigmentation des poils. Les embryons en développement sont hétérozygotes pour un certain nombre de ces gènes. Une mutation au niveau de l'allèle dominant ou la perte de l'allèle dominant (suite à divers événements génétiques) d'un tel gène dans un mélanoblaste se traduit par l'expression du phénotype récessif dans les cellules qui en sont issues, ce qui entraîne l'apparition d'une tache d'une autre couleur dans le pelage de la souris nouveau-née. Le nombre de descendants porteurs de taches (mutations) est compté et la fréquence de celles-ci est comparée à celle observée pour la descendance issue des embryons traités uniquement avec le solvant. Le spot test chez la souris décèle des mutations présumées somatiques au niveau des cellules fœtales.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Lorsque c'est possible, les substances d'essai sont dissoutes ou mises en suspension dans une solution saline isotonique. Les substances insolubles dans l'eau sont dissoutes ou mises en suspension dans des véhicules appropriés. Le véhicule utilisé ne devrait pas interférer avec la substance d'essai et ne devrait pas produire d'effets toxiques. Des préparations de la substance d'essai fraîchement réalisées seront utilisées.
Animaux d'expérience
Des souris de la souche T (nonagouti, a/a; chinchilla, pink eye, cchp/cchp; brown, b/b; dilute, short ear, d se/d; piebald spotting, s/s) sont accouplées soit avec la souche HT (pallid, nonagouti, brachypodie, pa a bp/pa a bp; leaden fuzzy, In fz/ln fz; pearl pe/pe) ou avec C57 BL (nonagouti, a/a). D'autres croisements appropriés tels que NMRI (nonagouti, a/a; albino, c/c) et DBA (nonagouti, a/a; brown, b/b; dilute d/d) peuvent être utilisés à condition de donner une descendance nonagouti.
Nombre et sexe
On traite un nombre suffisant de femelles gravides de manière à obtenir un nombre adéquat de descendants vivants pour chaque dose utilisée. La taille de l'échantillon dépend du nombre de taches observées chez les souris traitées ainsi que l'importance du nombre de données témoins. Un résultat négatif ne peut être accepté que si au moins 300 descendants de femelles traitées avec la dose maximale ont été examinés.
Témoins négatifs et positifs
Simultanément, on devrait disposer de résultats de souris traitées uniquement avec le véhicule (témoins négatifs). Des données témoins historiques provenant du même laboratoire peuvent être rassemblées afin d'améliorer la sensibilité de l'essai, à condition que ces données soient homogènes, à l'issue d'un traitement avec un produit chimique réputé mutagène dans cet essai. Si la substance d'essai ne se révèle pas mutagène, des données obtenues récemment par le même laboratoire pour un contrôle positif connu pour être mutagène dans cette épreuve devraient être disponibles.
Voie d'administration
Les voies d'administration habituelles sont l'intubation orale et l'injection péritonéale des femelles gravides. Le traitement par inhalation ou d'autres voies d'administration est utilisé, le cas échéant.
Doses
On utilise au moins deux doses dont une faisant apparaître des signes de toxicité ou réduisant le taille des portées. Dans le cas de substances non toxiques, on aura recours à un traitement à la dose maximale utilisable.
Conduite de l'épreuve
Un traitement unique est normalement administré aux huitième, neuvième et dixième jours de gestation, le jour 1 étant le jour où l'on observe pour la première fois la présence d'un bouchon vaginal. Ces jours correspondent à 7,25, 8,25 et 9,25 jours après la conception. Des traitements successifs peuvent être effectués pendant ces jours.
Analyse
Trois à quatre semaines après la naissance, la descendance est codée et examinée pour la présence de taches. On distingue trois classes de taches:
|
a)
|
les taches blanches situées à moins de 5 mm de la ligne médio-ventrale présumées résulter de mort cellulaire (WMVS);
|
|
b)
|
les taches jaunes, de type agouti, associées aux mamelles, aux organes sexuels, à la gorge, aux régions axillaire et inguinale ainsi qu'au milieu du front, présumées être dues à un manque de différenciation (MDS)
|
|
c)
|
les taches pigmentées et blanches, réparties au hasard sur le pelage, présumées être dues à des mutations somatiques (RS).
|
Les trois classes sont comptées mais seule la dernière c'est-à-dire RS, a une signification génétique. Les problèmes que pose la distinction entre les types MDS et RS peuvent être résolus en examinant des échantillons de poils au microscope à fluorescence.
Les anomalies morphologiques macroscopiques évidentes observées sur la descendance seront notées.
2. RÉSULTATS
Les résultats sont présentés en donnant le nombre total de jeunes examinés et le nombre de jeunes présentant une ou plusieurs taches présumées être dues à une mutation somatique. Les résultats sont également présentés en se référant à la portée comme unité. Les résultats relatifs aux animaux traités et aux témoins négatifs sont comparés par des méthodes statistiques appropriées.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
souches utilisées dans le croisement,
|
|
—
|
nombre de femelles gravides dans les groupes traités et témoins,
|
|
—
|
taille moyenne des portées dans les groupes traités et témoins à la naissance et au sevrage,
|
|
—
|
doses de la substance d'essai,
|
|
—
|
jour de la gestation auquel le traitement est administré,
|
|
—
|
nombre total de jeunes examinés ainsi que le nombre de ceux présentant une WMVS, MDS et RS dans les groupes traités et témoins,
|
|
—
|
anomalies morphologiques macroscopiques,
|
|
—
|
relation dose/effet de RS si possible,
|
|
—
|
évaluation statistique,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.25. TRANSLOCATION HÉRÉDITAIRE CHEZ LA SOURIS
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITION
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPES DE LA MÉTHODE D'ESSAI
L'épreuve de translocation héréditaire chez la souris décèle des changements chromosomiques structurels et numériques dans les cellules germinales de mammifères tels qu'ils sont mis en évidence dans la descendance de première génération. Les types de changements chromosomiques détectés sont des translocations réciproques et, si la descendance femelle est incluse, la perte du chromosome X. Les porteurs de translocations et les femelles XO présentent une fertilité réduite permettant la sélection d'une descendance F1 en vue d'une analyse cytogénétique. Une stérilité totale est due à certains types de translocations (autosome X et type c-t). Les translocations sont observées cytogénétiquement dans les cellules méiotiques en diacinèse-métaphase I des individus mâles, qu'il s'agisse de mâles F, ou de fils de femelles F1. Les femelles XO sont identifiées cytogénétiquement par la présence de 39 chromosomes seulement dans des mitoses de la moelle osseuse.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Les substances d'essai sont dissoutes dans une solution saline isotonique. Si elles sont insolubles dans l'eau, elles sont dissoutes ou mises en suspension dans des véhicules appropriés. On utilise des solutions fraîchement préparées de la substance d'essai. Si un véhicule est utilisé pour faciliter le traitement, il ne doit pas interférer avec la substance d'essai et ne pas produire d'effets toxiques.
Voie d'administration
Les voies d'administration sont habituellement l'intubation orale ou l'injection intrapéritonéale. D'autres voies d'administration peuvent être appropriées.
Animaux d'expérience
Pour faciliter l'élevage et la vérification cytologique, ces expériences sont effectuées avec des souris. Aucune souche spécifique de souris n'est requise. Toutefois, la taille moyenne d'une portée de la souche utilisée devra être supérieure à 8 et être relativement constante.
Des animaux sains ayant atteint leur maturité sexuelle seront utilisés.
Nombre d'animaux
Le nombre d'animaux nécessaires dépend de la fréquence de translocations spontanées ainsi que du taux minimal d'induction requis pour un résultat positif.
L'essai consiste habituellement en une analyse de la descendance mâle F1. Au moins 500 descendants mâles F1 sont testés par groupe de dose. Si la descendance femelle F1 est incluse, 300 mâles et 300 femelles sont nécessaires.
Contrôles
Des résultats de contrôles adéquats provenant d'épreuves réalisées simultanément et de contrôles historiques doivent exister. S'il existe des résultats acceptables de contrôles positifs provenant d'expériences récemment effectuées dans le même laboratoire, ces résultats peuvent être utilisés à la place de contrôles positifs simultanés.
Doses
Une dose est testée, il s'agit habituellement de la dose maximale associée à la production d'effets toxiques minimaux mais n'affectant pas le comportement reproducteur ou la survie. Pour établir une relation dose/réponse, deux autres doses plus faibles sont nécessaires. Dans le cas de substances non toxiques, on devrait avoir recours à une exposition à la dose maximale possible.
Conduite de l'essai
Traitement et accouplement
Deux programmes de traitement existent. L'administration unique de la substance d'essai est la méthode la plus fréquente. La substance d'essai peut également être administrée 7 jours sur 7 pendant 35 jours. Le nombre d'accouplements après traitement est déterminé par le programme de traitement et sera tel que tous les stades des cellules germinales traitées soient impliqués. À l'issue de la période d'accouplement, les femelles sont placées dans des cages individuelles. Lorsqu'elles ont mis bas, la date, la taille de la portée et le sexe des jeunes sont enregistrés. Toute la descendance mâle est sevrée et toute la descendance femelle est écartée à moins d'être incluse dans l'expérience.
Recherche des hétérozygotes de translocation
On utilise une des deux méthodes disponibles:
|
—
|
analyse de la fécondité de la descendance F1 et vérification ultérieure d'éventuels porteurs de translocations par analyse cytogénétique,
|
|
—
|
analyse cytogénétique de tous les mâles F1 sans sélection préalable par vérification de la fécondité.
|
|
a)
|
Analyse de la fécondité:
La fécondité réduite d'un individu F1 peut être déterminée en observant la taille de la portée et/ou en analysant le contenu utérin des partenaires femelles.
Il y a lieu de fixer les critères de détermination de la fécondité normale et réduite de la souche de souris utilisée.
Observation de la taille de la portée: Les mâles F1 à tester sont placés dans des cages individuelles avec des femelles provenant soit de la même expérience soit de la colonie. Les cages sont inspectées quotidiennement à partir du dix-huitième jour qui suit l'accouplement. La taille de la portée et te sexe de la descendance F2 sont enregistrés au moment de la naissance et les jeunes sont ensuite éliminés. Si la descendance femelle F1 est testée, les descendants F2 provenant de petites portées sont conservés en vue d'une analyse plus approfondie. Les femelles porteuses de translocation font l'objet d'une vérification par analyse cytogénétique d'une translocation dans n'importe lequel de leurs descendants mâles. Les femelles XO sont identifiées par la modification du rapport des sexes dans leur descendance, rapport qui passe de 1:1 à 1:2 mâles/femelles. Dans une méthode séquentielle, les animaux F1 normaux ne font pas l'objet d'une autre vérification si la première portée F2 atteint ou dépasse une valeur normale prédéterminée, sinon une deuxième ou une troisième portée F2 sont observées. Les animaux F1 qui ne peuvent pas être classés comme normaux après observation de jusqu'à trois portées F2 maximum sont soit soumis à un nouveau contrôle par le biais d'une analyse du contenu utérin de leurs partenaires femelles soit directement soumis à une analyse cytogénétique.
Analyse du contenu utérin: La réduction de la taille des portées chez les porteurs de translocation est due à la mort des embryons, de sorte qu'un grand nombre d'implants morts indique la présence d'une translocation chez l'animal soumis au test. Chaque mâle F1 à tester est accouplé à 2 ou 3 femelles. On constate la conception en examinant les femelles tous les matins afin de déceler la présence de bouchons vaginaux. Les femelles sont sacrifiées 14 à 16 jours plus tard et le nombre d'implants vivants et morts présents dans leur utérus est enregistré.
|
|
b)
|
Analyse cytogénétique:
Les préparations de testicules sont effectuées par la méthode de séchage à l'air. Les porteurs de translocations sont identifiés par la présence de configurations multivalentes en diacinèse métaphase I dans les spermatocytes primaires. L'observation d'au moins 2 cellules présentant une association multivalente constitue la preuve nécessaire que l'animal testé est porteur d'une translocation.
Si aucune sélection par analyse de fécondité n'a été effectuée, tous les mâles F1 sont soumis à un examen cytogénétique. Un minimum de 25 diacinèses métaphases I par mâle doivent être analysées au microscope. L'examen des métaphases mitotiques, des spermatogonies ou de la moelle osseuse est requise pour les mâles F1 ayant de petits testicules et présentant un arrêt méiotique avant diacinèse ou pour les femelles F1 suspectées d'être XO. La présence inhabituelle d'un chromosome long et/ou court dans 10 cellules est la preuve d'une translocation particulière entraînant la stérilité du mâle (type c-t). Quelques translocations X autosomes provoquant la stérilité du mâle peuvent uniquement être identifiées par une analyse des bandes de chromosomes mitotiques. La présence de 39 chromosomes dans 10 mitoses sur 10 est la preuve d'un état XO chez une femelle.
|
2. RÉSULTATS
Les résultats sont présentés sous forme de tableaux.
La taille moyenne des portées et le rapport entre les sexes sont enregistrés à la naissance et au sevrage pour chaque période d'accouplement.
Lors de l'évaluation de la fécondité des animaux de F1, la taille moyenne des portées issues de tous les accouplements normaux ainsi que la taille individuelle des portées issues d'animaux F1 porteurs de translocation sont présentées. En ce qui concerne l'analyse du contenu utérin, le nombre moyen d'implants vivants et morts issus d'accouplements normaux et le nombre d'implants vivants et morts pour chaque accouplement de porteurs de translocation F1 sont notés.
Lors de l'analyse cytogénétique de la diacinèse-métaphase I, le nombre des différents types de configurations multivalentes et le nombre total de cellules sont relevés pour chaque porteur de translocation.
Pour les individus stériles F1, le nombre total d'accouplements et la durée de la période d'accouplement sont indiqués. Le poids des testicules et les détails de l'analyse cytogénétique sont indiqués.
Pour les femelles XO, on indique la taille moyenne de la portée, le rapport entre les sexes dans la descendance F2 ainsi que les résultats de l'analyse cytogénétique.
Si d'éventuels porteurs de translocation F1 sont présélectionnés par des tests de fécondité, les tableaux mentionneront combien parmi eux ont été confirmés comme étant des hétérozygotes de translocation.
Les données relatives aux témoins négatifs ainsi que les expériences témoins positives sont présentées.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
souche de souris, âge des animaux, poids des animaux traités,
|
|
—
|
nombre d'animaux parents de chaque sexe dans les groupes traités et témoins,
|
|
—
|
conditions d'essai, description détaillée du traitement, doses, solvants, programme d'accouplement,
|
|
—
|
nombre et sexe des jeunes par femelle, nombre et sexe des jeunes élevés en vue d'une analyse de translocation,
|
|
—
|
moment et critères de l'analyse de translocation,
|
|
—
|
nombre et description détaillée des porteurs de translocation, y compris données relatives à l'élevage et au contenu utérin, si possible,
|
|
—
|
méthodes cytogénétiques et détails de l'analyse microscopique, de préférence avec clichés,
|
|
—
|
évaluation statistique,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.26. ESSAI DE TOXICITÉ SUBCHRONIQUE PAR VOIE ORALE TOXICITÉ ORALE À DOSES RÉPÉTÉES — RONGEURS: 90 JOURS
1. MÉTHODE
La méthode décrite pour cet essai de toxicité orale subchronique reprend la ligne directrice no 408 de l'OCDE (1998).
1.1. INTRODUCTION
Lors de l'appréciation et de l'évaluation des caractéristiques toxiques d'un produit chimique, la détermination de la toxicité subchronique par voie orale à doses répétées peut s'effectuer après avoir obtenu des informations préliminaires sur la toxicité à partir d'essais de toxicité aigué ou à doses répétées sur 28 jours. L'étude sur 90 jours fournit des informations sur les dangers que peut entraîner pour la santé une exposition réitérée durant une période prolongée, période qui s'étend du sevrage jusqu'à l'état adulte. L'étude fournira des informations sur les principaux effets toxiques, indiquera les organes cibles et les possibilités d'accumulation et pourra donner une estimation de la dose sans effet toxique (NOAEL) qui pourra être utilisée pour sélectionner des doses en vue d'études chroniques et pour établir les critères de sécurité concernant l'exposition humaine.
La méthode accorde davantage d'importance aux effets neurologiques et donne des indications relatives aux effets sur le système immunitaire et sur la reproduction. Elle insiste également sur la nécessité d'observer très attentivement les animaux sur le plan clinique, en vue d'obtenir le plus d'informations possible. Cette étude devrait permettre de repérer les produits chimiques susceptibles d'avoir une action neurotoxique ou des effets sur le système immunitaire ou les organes reproducteurs, pouvant justifier des études plus approfondies.
Voir également introduction générale, partie B.
1.2. DÉFINITIONS
Dose: quantité de substance d'essai administrée. La dose est exprimée en poids (g, mg), en poids de substance d'essai par unité de poids corporel de l'animal d'expérience (par exemple, mg/kg) ou en concentration constante dans la nourriture (ppm).
Dosage: terme général recouvrant la dose, sa fréquence et la durée de l'administration.
DSET: abréviation de dose sans effet toxique (NOAEL: No Observable Adverse Effect Level}, c'est-à-dire la dose la plus élevée à laquelle aucun effet nocif lié au traitement n'est observé.
1.3. PRINCIPE DE LA MÉTHODE
Différentes doses de la substance d'essai sont administrées quotidiennement par voie orale à plusieurs groupes d'animaux de laboratoire, à raison d'un niveau de dose par groupe et ce pendant une période de 90 jours. Les animaux sont observés attentivement pendant la période d'administration afin de déceler d'éventuels signes de toxicité. Les animaux qui meurent ou qui sont sacrifiés au cours de l'essai sont autopsiés: au terme de l'essai, les animaux suivants sont également sacrifiés et autopsiés.
1.4. DESCRIPTION DE LA MÉTHODE
1.4.1. Préparation des animaux
Il convient d'utiliser des animaux en bonne santé, ayant été acclimatés aux conditions du laboratoire pendant au moins 5 jours et qui n'ont pas encore été sujets d'expériences. L'espèce, la race, la source, le sexe, le poids et/ou l'âge des animaux d'expérience doivent être précisés. Les animaux seront répartis au hasard entre les groupes traités et les groupes témoins. Les cages doivent être placées de façon telle que l'influence éventuelle de leur disposition sur les résultats de l'étude soit réduite au minimum. Un numéro d'identification unique est attribué à chaque animal.
1.4.2. Préparation des doses
La substance d'essai est administrée par gavage ou dans les aliments ou dans l'eau de boisson. Le mode d'administration par voie orale dépend de l'objectif de l'étude et des propriétés physico-chimiques de la substance d'essai.
S'il y a lieu, la substance d'essai est dissoute ou mise en suspension dans un véhicule approprié. On recommande, chaque fois que les circonstances le permettent, d'envisager d'abord l'utilisation d'une solution ou d'une suspension aqueuse, ensuite une solution ou une émulsion dans une huile (par exemple l'huile de maïs) et en dernier lieu une solution dans d'autres véhicules. La toxicité des véhicules autres que l'eau doit être connue. La stabilité de la substance d'essai dans les conditions d'administration doit être déterminée.
1.4.3. Conditions d'essai
1.4.3.1. Animaux d'expérience
L'espèce préférée est le rat, bien que d'autres espèces de rongeurs, notamment la souris, puissent être utilisées. Il convient d'employer de jeunes animaux adultes, sains, issus de souches de laboratoire courantes. Les femelles doivent être nullipares et non gravides. L'administration doit commencer dès que possible après le sevrage et en aucun cas au-delà de l'âge de 9 semaines. Au début de l'étude, les différences de poids entre les animaux utilisés doivent être minimales et ne pas excéder ± 20 % de la moyenne du poids de chaque sexe. Lorsque l'essai est conduit à titre d'étude préliminaire à une étude de toxicité chronique à long terme, il faut utiliser des animaux issus de la même souche et de la même source dans les deux études.
1.4.3.2. Nombre et sexe
Au moins 20 animaux (10 femelles et 10 mâles) doivent être utilisés pour chaque dose. Si on a prévu de sacrifier des animaux en cours d'essai, il convient d'accroître ce nombre du nombre d'animaux qui doivent être sacrifiés en cours d'essai. En fonction des connaissances dont on dispose au préalable sur le produit chimique ou sur un produit de structure très proche, on pourra envisager d'inclure un groupe satellite supplémentaire de 10 animaux (5 par sexe) dans le groupe témoin et dans le groupe traité à la plus forte dose, afin d'observer, une fois la période de traitement terminée, la persistance ou la réversibilité de tout effet toxique. La durée de cette période post-traitement devrait être fixée en fonction des effets observés.
1.4.3.3. Niveaux de dose
Il faut utiliser au minimum trois doses et un groupe témoin, sauf si un essai limite est pratiqué (voir paragraphe 1.4.3.4). Les niveaux de dose peuvent être établis en fonction des résultats obtenus lors d'études à doses répétées ou d'études préliminaires et doivent tenir compte de toutes les données toxicologiques et toxicocinétiques disponibles sur la substance d'essai ou de substances apparentées. Sauf contraintes dues aux propriétés physico-chimiques de la substance ou à ses effets biologiques, la dose la plus élevée doit être choisie en vue de provoquer un effet toxique, mais ni la mort ni d'intenses souffrances. Une série de doses décroissantes doit être sélectionnée en vue de mettre en évidence tout effet lié à la dose ainsi qu'une dose sans effet toxique (DSET) à la dose la plus faible. Des intervalles correspondant à un facteur 2 ou 4 sont souvent les plus appropriés entre les doses décroissantes et l'inclusion d'un quatrième groupe d'essai est souvent préférable à la fixation de très grands intervalles (par exemple séparés par un facteur supérieur à 6-10) entre les doses.
Le groupe témoin sera un groupe non traité ou un groupe recevant le véhicule si la substance est administrée dans un véhicule. Exception faite de l'administration de la substance à tester, les animaux du groupe témoin doivent être traités de la même manière que ceux des groupes d'essai. Si un véhicule est employé, on administrera au groupe témoin le plus grand volume de véhicule utilisé. Si la substance d'essai est incorporée aux aliments et qu'elle entraîne une diminution de la prise de nourriture, il peut être utile d'utiliser un groupe témoin nourri en parallèle afin de déterminer si la diminution est due aux caractéristiques organoleptiques ou à des altérations toxicologiques du modèle d'essai.
Il convient également d'être attentif aux caractéristiques suivantes du véhicule ou d'autres additifs, selon le cas: effets sur l'absorption, la distribution, le métabolisme ou la rétention de la substance d'essai; effets sur les propriétés chimiques de la substance d'essai susceptibles de modifier sa toxicité; et effets sur la consommation alimentaire et hydrique ou sur l'état nutritionnel des animaux.
1.4.3.4. Essai limite
Si un essai pratiqué avec une seule dose équivalant à au moins 1 000 mg/kg de poids corporel/jour, en suivant le mode opératoire décrit pour cette étude, ne produit aucun effet nocif observé et s'il n'y a pas de raison de penser que la substance soit toxique compte tenu des données dont on dispose au sujet de substances ayant une structure analogue, on peut considérer qu'il n'est pas nécessaire de réaliser une étude complète avec trois niveaux de dose. L'essai limite est valable sauf dans le cas où l'exposition humaine puisse advenir à des doses plus élevées.
1.5. MODE OPÉRATOIRE
1.5.1. Administration des doses
Les animaux reçoivent une administration quotidienne de la substance d'essai, sept jours sur sept, durant 90 jours. Tout autre régime d'administration, par exemple cinq jours par semaine, doit être justifié. Lorsque la substance d'essai est administrée par gavage, une dose unique devrait être dispensée aux animaux par une sonde gastrique ou une canule d'intubation appropriée. Le volume maximal de liquide administrable en une fois dépend de la taille de l'animal. Le volume ne doit pas dépasser 1 ml/100 g de poids corporel, sauf dans le cas des solutions aqueuses où il peut aller jusqu'à 2 ml/100 g de poids corporel. Sauf pour les substances irritantes ou corrosives, qui provoqueront normalement des effets exacerbés aux concentrations plus élevées, il convient de réduire au minimum la variabilité du volume d'essai en ajustant la concentration pour obtenir un volume constant à toutes les doses.
Si la substance d'essai est administrée dans les aliments ou l'eau de boisson, il importe de s'assurer que sa quantité n'interfère pas avec la nutrition normale ni avec l'équilibre hydrique. Lorsque la substance d'essai est incorporée aux aliments, on peut soit appliquer une concentration constante dans les aliments (ppm), soit une dose constante par rapport au poids corporel de l'animal; il y a lieu de spécifier la méthode utilisée. Si la substance est administrée par gavage, la dose doit être administrée à la même heure chaque jour et ajustée si nécessaire pour rester constante par rapport au poids corporel de l'animal. Lorsqu'une étude sur 90 jours sert de préliminaire à une étude de toxicité chronique à long terme, un régime alimentaire semblable doit être utilisé dans les deux études.
1.5.2. Observations
La période d'observation doit durer au moins 90 jours. Les animaux d'un groupe satellite destinés à des observations ultérieures ne doivent recevoir aucun traitement pendant une période appropriée pour que l'on puisse constater la persistance ou la disparition des effets toxiques.
Il faudra effectuer un examen clinique général au moins une fois par jour, de préférence aux mêmes heures, en tenant compte de la période après l'administration pour laquelle des effets observables les plus marqués sont prévisibles. L'état clinique des animaux doit être noté. Deux fois par jour au minimum, généralement au début et à la fin de chaque journée, tous les animaux doivent être examinés afin de déceler des symptômes de morbidité et de mortalité.
Un examen clinique détaillé doit être pratiqué sur tous les animaux au moins une fois avant la première exposition (pour pouvoir effectuer des comparaisons sur un même individu), et ensuite une fois par semaine. Ces examens doivent être effectués hors de la cage, de préférence dans une enceinte normalisée, et à heure fixe. Ils doivent être soigneusement consignés, de préférence en utilisant un système de notation explicitement défini par le laboratoire qui réalise l'essai. Les conditions d'observation doivent demeurer aussi constantes que possible. Les symptômes relevés devraient inclure, de façon non limitative, les changements affectant la peau, la fourrure, les yeux, les muqueuses, la fréquence des sécrétions et des excrétions ainsi que l'activité autonome (sécrétion de larmes, horripilation, diamètre de la pupille, respiration anormale). Il convient également de noter les changements dans la démarche, le maintien et les réactions à la manipulation ainsi que la présence de mouvements cloniques ou toniques, de stéréotypes (par exemple, soins corporels excessifs, animaux tournant en rond de façon répétitive) et de comportements bizarres (par exemple, automutilation, marche à reculons) (1).
À l'aide d'un ophtalmoscope ou d'un appareillage approprié équivalent, il y a lieu d'effectuer, avant l'administration de la substance à tester et au terme de l'étude, un examen ophtalmologique, de préférence sur tous les animaux mais au minimum sur le groupe d'animaux exposés à la dose la plus élevée et sur le groupe témoin. Si on décèle des changements dans les yeux de ces animaux, tous les autres animaux doivent être examinés.
Vers la fin de la période d'exposition, mais en aucun cas avant la onzième semaine, il faut vérifier la réactivité sensorielle à différents types de stimuli (1) (auditifs, visuels et proprioceptifs, par exemple) (2) (3) (4), et évaluer la force de préhension (5) et l'activité motrice (6). Des détails supplémentaires sur les méthodes utilisables figurent dans les références respectives. Des méthodes non décrites dans les références peuvent aussi être appliquées.
Les observations fonctionnelles préconisées vers la fin de l'étude ne sont pas indispensables si on dispose d'observations fonctionnelles provenant d'autres études et que les examens cliniques quotidiens n'ont pas révélé de troubles fonctionnels.
Exceptionnellement, les observations fonctionnelles peuvent aussi être omises pour des groupes qui par ailleurs présentent des symptômes de toxicité à un degré tel qu'ils interféreraient sensiblement avec le déroulement de l'essai fonctionnel.
1.5.2.1. Poids corporel et consommation de nourriture et d'eau
Tous les animaux doivent être pesés au moins une fois par semaine. On mesurera la consommation de nourriture au moins une fois par semaine. Si la substance d'essai est administrée dans l'eau de boisson, la consommations d'eau doit aussi être mesurée au moins une fois par semaine. Il est également recommandé de mesurer la consommation d'eau, dans le cas d'étude où la substance d'essai est administrée dans les aliments ou par gavage, si celle-ci risque d'être modifiée.
1.5.2.2. Hématologie et biochimie clinique
Des prélèvements de sang doivent être pratiqués à des sites déterminés et les échantillons, stockés, s'il y a lieu, dans des conditions appropriées. À la fin de la période d'essai, des prélèvements sont réalisés juste avant le sacrifice des animaux ou au cours de celui-ci.
On procédera aux examens hématologiques suivants au terme de la période d'essai et sur les prises de sang effectuées en cours d'essai, le cas échéant: hématocrite, concentration d'hémoglobine, numération des érythrocytes et des leucocytes, formule leucocytaire, numération des plaquettes et mesure du temps de coagulation.
Les analyses de biochimie clinique destinées à étudier les principaux effets toxiques sur les tissus, et en particulier sur les reins et le foie, devraient être pratiquées sur des échantillons de sang prélevés sur chaque animal juste avant son sacrifice ou au cours de celui-ci (à l'exception des animaux trouvés moribonds et/ou sacrifiés avant la fin de l'essai). A l'instar des examens hématologiques, les analyses de biochimie clinique peuvent être conduites sur des échantillons de sang prélevés en cours d'essai. Il est recommandé de faire jeûner les animaux durant la nuit qui précède la prise de sang (3). Les analyses effectuées sur le plasma ou le sérum devraient comprendre le sodium, le potassium, le glucose, le cholestérol total, l'urée, l'azote uréique du sang, la créatinine, les concentrations totales de protéines et d'albumine et plus de deux enzymes indicatrices des effets hépatocellulaires (telles que l'alanine aminotransférase, l'aspartate aminotransférase, la phosphatage alcaline, la gamma glutarmyle transpeptidase et la sorbitol déshydrogenase). Des mesures d'activités enzymatiques supplémentaires (d'origine hépatique ou autre) et des acides biliaires, susceptibles de fournir des informations utiles dans certaines circonstances, peuvent également être incluses.
Les analyses d'urine suivantes peuvent être réalisées, à titre facultatif, au cours de la dernière semaine de l'étude sur des échantillons d'urine prélevés à des moments déterminés: apparence, volume, osmolalité ou densité, pH, protéines, glucose, sang et cellules sanguines.
Il faut envisager par ailleurs de rechercher les indicateurs sériques de lésions générales des tissus. Si les propriétés connues de la substance d'essai risquent ou sont soupçonnées d'affecter des courbes métaboliques connexes, d'autres analyses devraient être pratiquées, notamment celles du calcium, du phosphore, des triglycérides à jeun, d'hormones spécifiques, de la méthémoglobine et de la cholinestérase. Ces analyses doivent être définies pour certaines classes de produits chimiques ou au cas par cas.
Il convient d'une façon générale de suivre une démarche souple, adaptée à l'espèce et aux effets observés et/ou escomptés d'une substance donnée.
Si les données historiques sont insuffisantes, il y a lieu d'envisager la détermination de paramètres d'hématologie et de biochimie clinique avant de commencer l'étude; il est généralement déconseillé d'obtenir ces données avant le traitement (7).
1.5.2.3. Autopsie
Tous les animaux de l'étude feront l'objet d'une autopsie complète et détaillée, comportant l'examen attentif de la surface externe du corps, de tous les orifices ainsi que des cavités crânienne, thoracique et abdominale et de leur contenu. Le foie, les reins, les glandes surrénales, les testicules, les épididymes, l'utérus, les ovaires, le thymus, la rate, le cerveau et le cœur de tous les animaux (à l'exception des animaux moribonds et/ou sacrifiés avant la fin de l'essai) doivent être disséqués et pesés à l'état frais dès que possible afin d'éviter leur dessiccation.
Les tissus suivants doivent être conservés dans le milieu de fixation le plus approprié, à la fois pour le type de tissu et pour l'examen histopathologique prévu: tous les organes présentant des lésions macroscopiques, l'encéphale (les régions représentatives du cerveau, du cervelet, de la moelle et du pont), la moelle épinière (à trois niveaux: cervical, médio-thoracique et lombaire), l'hypophyse, la thyroïde, les glandes parathyroïdes, le thymus, l'œsophage, les glandes salivaires, l'estomac, l'intestin grêle et le gros intestin (y compris les plaques de Peyer), le foie, le pancréas, les reins, les glandes surrénales, la rate, le cœur, la trachée et les poumons (traités par gonflement avec un fixateur, puis immergés), l'aorte, les gonades, l'utérus, les organes génitaux auxiliaires, les glandes mammaires des femelles, la prostate, la vessie, la vésicule biliaire (souris), les ganglions lymphatiques (de préférence un ganglion lymphatique faisant partie du territoire de drainage dont dépend la voie d'administration et un autre faisant partie d'un autre territoire de drainage afin de détecter les éventuels effets systémiques), les nerfs périphériques (sciatique ou poplité interne) de préférence proche du muscle, un prélèvement de moelle osseuse (et/ou ponction de moelle osseuse examinée directement), la peau et les yeux (si des changements ont été relevés au cours des examens ophtalmologiques). Les observations cliniques et les observations macroscopiques peuvent conduire à l'examen de tissus supplémentaires. Tous les organes considérés comme des cibles vraisemblables de la substance d'essai, compte tenu de ses propriétés, devraient également être conservés.
1.5.2.4. Histopathologie
Un examen histopathologique complet doit être pratiqué sur les tissus et organes conservés de tous les animaux appartenant au groupe témoin et au groupe traité à la dose la plus élevée. Si des changements liés au traitement sont constatés dans le groupe traité à la dose la plus élevée, il faudra étendre cet examen aux animaux de tous les groupes traités.
Toutes les lésions macroscopiques doivent être examinées.
Si un groupe satellite est inclus, les tissus et organes sur lesquels des effets ont été observés dans les groupes traités doivent faire l'objet d'un examen histopathologique.
2. RÉSULTATS ET RAPPORT
2.1. RÉSULTATS
Les résultats correspondant à chaque animal doivent être fournis. En outre, tous les résultats doivent être résumés sous forme de tableaux faisant apparaître, pour chaque groupe d'essai, le nombre d'animaux au début de l'essai, le nombre d'animaux trouvés morts au cours de l'essai ou euthanasiés, le moment de la mort ou du sacrifice, le nombre d'animaux présentant des signes de toxicité et une description des symptômes de toxicité observés, notamment le moment de leur apparition, leur durée et leur gravité, le nombre d'animaux présentant des lésions, les types de lésions et le pourcentage d'animaux affecté par chaque type de lésion.
S'il y a lieu, les résultats numériques doivent être évalués par une méthode statistique appropriée et largement reconnue. Le choix des méthodes statistiques et des résultats à analyser doit intervenir au stade de rédaction du protocole d'étude.
2.2. RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
2.2.1. Substance d'essai
|
—
|
État physique, pureté et propriétés physico-chimiques,
|
|
—
|
données permettant l'identification chimique,
|
|
—
|
véhicule (le cas échéant) justification du choix du véhicule, s'il est autre que l'eau.
|
2.2.2. Animaux d'expérience
|
—
|
Espèce et souche utilisée,
|
|
—
|
nombre, âge et sexe des animaux.
|
|
—
|
source, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
poids de chaque animal au début de l'essai.
|
2.2.3. Conditions d'essai
|
—
|
Justification du choix des doses,
|
|
—
|
détails concernant la préparation contenant la substance d'essai, son incorporation dans la nourriture, la concentration obtenue, la stabilité et l'homogénéité de la préparation,
|
|
—
|
détails relatifs à l'administration de la substance d'essai,
|
|
—
|
doses réelles (mg/kg de poids corporel/jour) et facteur de conversion de la concentration de la substance d'essai dans la nourriture ou l'eau de boisson (en ppm) en dose réelle, s'il y a lieu,
|
|
—
|
détails concernant la qualité de la nourriture et de l'eau.
|
2.2.4. Résultats
|
—
|
Poids corporel et variation de poids corporel,
|
|
—
|
consommation de nourriture et d'eau, le cas échéant,
|
|
—
|
données sur la réponse toxique par sexe et par dose et description des signes de toxicité,
|
|
—
|
nature, gravité et durée des observations cliniques (réversibles ou non),
|
|
—
|
résultats de l'examen ophtalmologique,
|
|
—
|
évaluations de l'activité sensorielle, la force de préhension et l'activité motrice (le cas échéant),
|
|
—
|
paramètres hématologiques et valeurs normales de référence,
|
|
—
|
paramètres de biochimie clinique et valeurs normales de référence,
|
|
—
|
poids du corps et des organes des animaux à leur mort et rapports poids de l'organe/poids corporel,
|
|
—
|
description détaillée de tous les résultats histopathologiques,
|
|
—
|
données relatives à l'absorption, le cas échéant,
|
|
—
|
traitement statistique des résultats, s'il y a lieu.
|
Discussion des résultats.
Conclusions.
3. BIBLIOGRAPHIE
|
(1)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document No 60.
|
|
(2)
|
Tupper, D. E., Wallace, R.B. (1980). Utility of the Neurologic Examination in Rats. Acta Neurobiol. Exp., 40, pp. 999-1003.
|
|
(3)
|
Gad, S. C. (1982). A Neuromuscular Screen for Use m Industrial Toxicology. J. Toxicol. Environ. Health, 9, pp. 691-704.
|
|
(4)
|
Moser, V. C, Mc Daniel, K. M., Phillips, P. M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol, 10S, pp. 267-283.
|
|
(5)
|
Meyer O. A, Tilson H. A., Byrd W. C, Riley M. T. (1979). A Method for the Routine Assesment of Fore- and Hindlimb grip Strength of Rats and Mice. Neurobehav. Toxivol., 1, pp. 233-236.
|
|
(6)
|
Crofton K. M., Howard J. L, Moser V. C, Gill M. W., Reiter L. W., Tilson H. A., MacPhail R. C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, pp. 599-609.
|
|
(7)
|
Weingand K., Brown G., Hall R. et al. (1996). ‘Harmonisation of Animal Clinic Pathology Testing in Toxicity and Safety Studies’, Fundam. & Appl. Toxicol, 29, pp. 198-201.
|
B.27. ESSAI DE TOXICITÉ SUBCHRONIQUE PAR VOIE ORALE TOXICITÉ ORALE À DOSES RÉPÉTÉES NON-RONGEURS: 90 JOURS
1. MÉTHODE
La méthode décrite pour cet essai de toxicité orale subchronique reprend la ligne directrice no 409 de l'OCDE (1998).
1.1. INTRODUCTION
Lors de l'appréciation et de l'évaluation des caractéristiques toxiques d'un produit chimique, la détermination de la toxicité subchronique par voie orale à doses répétées peut s'effectuer après avoir obtenu des informations préliminaires sur la toxicité à partir d'essais de toxicité aiguë ou à doses répétées sur 28 jours. L'étude sur 90 jours fournit des informations sur les dangers que peut entraîner pour la santé une exposition réitérée durant une période de croissance rapide jusqu'au début de l'âge adulte. L'étude fournira des informations sur les principaux effets toxiques, indiquera les organes cibles et les possibilités d'accumulation et pourra donner une estimation de la dose sans effet toxique (NOAEL) qui pourra être utilisée pour sélectionner des doses en vue d'études chroniques et pour établir les critères de sécurité concernant l'exposition humaine.
La méthode d'essai permet de mettre en évidence les effets nocifs de l'exposition aux produits chimiques chez des non-rongeurs; elle ne doit être utilisée que dans les cas suivants:
|
—
|
lorsque les effets observés dans d'autres études font ressortir la nécessité d'éclaircir et de préciser certains points chez une deuxième espèce non-rongeurs, ou
|
|
—
|
lorsque les études toxicocinétiques montrent que l'utilisation d'une espèce particulière de non-rongeur constitue le choix le plus pertinent en tant qu'animal d'expérience, ou
|
|
—
|
lorsque d'autres raisons précises justifient l'utilisation d'une espèce de non-rongeur.
|
Voir également introduction générale, partie B.
1.2. DÉFINITIONS
Dose: quantité de substance d'essai administrée. La dose est exprimée en poids (g, mg), en poids de substance d'essai par unité de poids corporel de l'animal d'expérience (par exemple, mg/kg) ou en concentration constante dans la nourriture (ppm).
Dosage: terme général recouvrant la dose, sa fréquence et la durée de l'administration.
DSET: abréviation de dose sans effet toxique (NOAEL: No Observable Adverse Effect Level), c'est-à-dire la dose la plus élevée à laquelle aucun effet nocif lié au traitement n'est observé.
1.3. PRINCIPE DE LA MÉTHODE
Différentes doses de la substance d'essai sont administrées quotidiennement par voie orale à plusieurs groupes d'animaux de laboratoire, à raison d'une valeur de dose par groupe et ce pendant une période de 90 jours. Pendant la période d'administration, les animaux sont observés attentivement afin de déceler d'éventuels symptômes de toxicité. Les animaux qui meurent ou qui sont sacrifiés en cours d'essai sont autopsiés et, au terme de l'essai, les animaux survivants sont également sacrifiés et autopsiés.
1.4. DESCRIPTION DE LA MÉTHODE
1.4.1. Sélection de l'espèce animale
L'espèce non-rongeur couramment utilisée est le chien, dont la race doit être définie; on se sert fréquemment du beagle. D'autres espèces, par exemple le porc ou le porc nain (mini porc), peuvent aussi être utilisées. Les primates ne sont pas recommandés et leur utilisation doit être justifiée. Il convient d'employer de jeunes animaux en bonne santé et, dans le cas du chien, l'administration de la substance à tester devrait commencer de préférence à l'âge de 4-6 mois, et jamais au delà de 9 mois. Lorsque l'étude est préliminaire à une étude de toxicité chronique à long terme, il faut utiliser la même espèce et la même race dans les deux études.
1.4.2. Préparation des animaux
Il convient d'utiliser de jeunes animaux, en bonne santé, acclimatés aux conditions du laboratoire et n'ayant pas encore été sujets d'expérience. La durée de l'acclimatation dépendra de l'espèce d'essai sélectionnée et de sa provenance. Il est recommandé de compter au moins 5 jours pour les chiens ou les porcs élevés à cette fin dans une animalerie résidente et au moins deux semaines pour ces mêmes animaux s'ils proviennent de sources extérieures. L'espèce, la race, la source, le sexe, le poids et/ou l'âge des animaux d'expérience doivent être précisés. Les animaux seront répartis au hasard entre les groupes traités et le groupe témoin. Les cages doivent être placées de façon telle que l'influence éventuelle de leur disposition sur les résultats soit réduite au minimum. Un numéro d'identification distinct doit être attribué à chaque animal.
1.4.3. Préparation des doses
La substance à tester peut être administrée dans la nourriture ou dans l'eau de boisson, par gavage ou dans des capsules. Le mode d'administration par voie orale dépend de l'objectif de l'étude et des propriétés physicochimiques de la substance d'essai.
S'il y a lieu, la substance d'essai est dissoute ou mise en suspension dans un véhicule approprié. On recommande, chaque fois que les circonstances le permettent, d'envisager d'abord l'utilisation d'une solution ou d'une suspension aqueuse, ensuite une solution ou une émulsion dans une huile (par exemple l'huile de maïs) et en dernier lieu une solution dans d'autres véhicules. La toxicité des véhicules autres que l'eau doit être connue. La stabilité de la substance d'essai dans les conditions d'administration doit être déterminée.
1.5. MODE OPÉRATOIRE
1.5.1. Nombre et sexe des animaux
Il faut employer au moins 8 animaux (4 femelles et 4 mâles) à chaque dose. Si on a prévu de sacrifier des animaux en cours d'essai, il convient d'accroître ce nombre du nombre d'animaux qui doivent être sacrifiés en cours d'essai. Le nombre d'animaux survivants au terme de l'étude doit être suffisant pour permettre une évaluation significative des effets toxiques. En fonction des connaissances dont on dispose au préalable sur le produit chimique ou sur une substance de structure très proche, on envisagera d'inclure un groupe satellite supplémentaire de 8 animaux (4 par sexe) dans le groupe témoin et dans le groupe traité à la concentration la plus élevée, en vue d'observer, une fois la période de traitement écoulée, la réversibilité ou la persistance de tout effet toxique. La durée de cette période post-traitement doit être fixée en fonction des effets observés.
1.5.2. Dosage
Il faut utiliser au minimum 3 doses et un groupe témoin, sauf si un essai limite est pratiqué (voir paragraphe 1.5.3). Les niveaux de dose peuvent être établis en fonction des résultats obtenus lors d'études à doses répétées d'études préliminaires et doivent tenir compte de toutes les données toxicologiques et toxicocinétiques disponibles sur la substance d'essai ou de substances apparentées. Sauf contraintes dues aux propriétés physico-chimiques de la substance d'essai ou par ses effets biologiques, la dose la plus élevée doit être choisie en vue de provoquer un effet toxique, mais ni la mort ni d'intenses souffrances. Une série de doses décroissantes doit être sélectionnée en vue de mettre en évidence tout effet lié à la dose ainsi qu'une dose sans effet toxique (DSET) à la dose la plus faible. Des intervalles correspondant à un facteur 2 ou 4 sont souvent les plus appropriés entre les doses décroissantes et l'inclusion d'un quatrième groupe d'essai est souvent préférable à la fixation de très grands intervalles (par exemple séparés par un facteur supérieur à 6-10) entre les doses.
Le groupe témoin sera un groupe non traité ou un groupe recevant le véhicule si la substance est administrée dans un véhicule. Exception faite de l'administration de la substance à tester, les animaux du groupe témoin doivent être traités de la même manière que ceux des groupes d'essai. Si un véhicule est employé, on administrera au groupe témoin le plus grand volume de véhicule utilisé. Si la substance d'essai est incorporée aux aliments et qu'elle entraîne une diminution de la prise de nourriture, il peut être utile d'utiliser un groupe témoin nourri en parallèle afin de déterminer si la diminution est duc aux caractéristiques organoleptiques ou à des altérations lexicologiques du modèle d'essai.
Il convient également d'être attentif aux caractéristiques suivantes du véhicule ou d'autres additifs, selon le cas effets sur l'absorption, la distribution, le métabolisme ou la rétention de la substance d'essai, effets sur les propriétés chimiques de la substance d'essai susceptibles de modifier sa toxicité, et effets sur la consommation alimentaire et hydrique ou sur l'état nutritionnel des animaux.
1.5.3. Essai limite
Si un essai pratiqué avec une seule dose équivalant à au moins 1 000 mg/kg de poids corporel/jour, en suivant le mode opératoire décrit pour cette étude, ne produit aucun effet nocif observé et s'il n'y a pas de raison de penser que la substance soit toxique compte tenu des données dont on dispose au sujet de substances ayant une structure analogue, on peut considérer qu'il n'est pas nécessaire de réaliser une étude complète avec trois niveaux de dose. L'essai limite est valable sauf dans le cas où l'exposition humaine puisse advenir à des doses plus élevées.
1.5.4. Administration des doses
Les animaux reçoivent une administration quotidienne de la substance d'essai, sept jours sur sept, durant 90 jours. Tout autre régime d'administration, par exemple 5 jours par semaine, doit être justifié. Lorsque la substance d'essai est administrée par gavage, une dose unique devrait être dispensée aux animaux par une sonde gastrique ou une canule d'intubation appropriée. Le volume maximal de liquide administrable en une fois dépend de la taille de l'animal. Le volume devrait normalement être le plus petit possible. Sauf pour les substances irritantes ou corrosives, qui provoqueront normalement des effets exacerbés aux concentrations plus élevées, il convient de réduire au minimum la variabilité du volume d'essai en ajustant la concentration pour obtenir un volume constant à toutes les doses.
Si la substance d'essai est administrée dans les aliments ou l'eau de boisson, il importe de s'assurer que sa quantité n'interfère pas avec la nutrition normale ni avec l'équilibre hydrique. Lorsque la substance d'essai est incorporée aux aliments, on peut soit appliquer une concentration constante dans les aliments (ppm), soit une dose constante par rapport au poids corporel de l'animal; il y a lieu de spécifier la méthode utilisée. Si la substance est administrée par gavage ou dans une capsule, la dose doit être administrée aux mêmes heures chaque jour et ajustée si nécessaire pour rester constante par rapport au poids corporel de l'animal. Lorsqu'une étude sur 90 jours sert de préliminaire à une étude de toxicité chronique à long terme, un régime alimentaire semblable doit être utilisé dans les deux études.
1.5.5. Observations
La période d'observation doit durer au moins 90 jours. Les animaux d'un groupe satellite destinés à des observations ultérieures ne doivent recevoir aucun traitement pendant une période appropriée pour que l'on puisse constater la persistance ou la disparition des effets toxiques.
Il faudra effectuer un examen clinique général au moins une fois par jour, de préférence aux mêmes heures, en tenant compte de la période après l'administration pour laquelle des effets observables les plus marqués sont prévisibles. L'état clinique des animaux doit être noté. Au moins deux fois par jour, généralement au début et à la fin de chaque journée, tous les animaux doivent être examinés afin de déceler des symptômes de morbidité et de mortalité.
Un examen clinique détaillé doit être pratiqué sur tous les animaux au moins une fois avant la première exposition (pour pouvoir effectuer des comparaisons sur un même individu), et ensuite une fois par semaine. Ces examens doivent être effectués, si possible, hors de la cage, de préférence dans une enceinte normalisée, et à heure fixe. Les conditions d'observation doivent demeurer aussi constantes que possible. Les symptômes de toxicité doivent être soigneusement notés, notamment le moment de leur apparition, leur durée et leur gravité. Les observations devraient inclure, de façon non limitative, les changements affectant la peau, la fourrure, les yeux, les muqueuses, la fréquence des sécrétions et des excrétions ainsi que l'activité autonome (par exemple, sécrétion de larmes, horripilation, diamètre de la pupille, respiration anormale). Il convient également de noter les changements dans la démarche, le maintien et les réactions à la manipulation ainsi que la présence de mouvements cloniques ou toniques, de stéréotypes (par exemple, soins corporels excessifs, animaux tournant en rond de façon répétitive) et de comportements bizarres.
À l'aide d'un ophtalmoscope ou d'un appareillage approprié équivalent, il y a lieu d'effectuer, avant l'administration de la substance à tester et au terme de l'étude, un examen ophtalmologique, de préférence sur tous les animaux mais au minimum sur le groupe d'animaux exposés à la dose la plus élevée et sur le groupe témoin. Si on décèle des changements liés au traitement dans les yeux de ces animaux, tous les autres animaux doivent être examinés.
1.5.5.1. Poids corporel et consommation de nourriture et d'eau
Tous les animaux doivent être pesés au moins une fois par semaine. On mesurera la consommation de nourriture au moins une fois par semaine. Si la substance d'essai est administrée dans l'eau de boisson, la consommation d'eau doit aussi être mesurée au moins une fois par semaine. Il est également recommandé de mesurer la consommation d'eau, dans le cas d'étude où la substance d'essai est administrée dans les aliments ou par gavage, si celle-ci risque d'être modifiée.
1.5.5.2. Hématologie et biochimie clinique
Des prélèvements de sang doivent être pratiqués à des sites déterminés et les échantillons, stockés, s'il y a lieu, dans des conditions appropriées. À la fin de la période d'essai, des échantillons sont prélevés juste avant le sacrifice des animaux ou au cours de celui-ci.
Au début de l'essai et, par la suite, soit tous les mois, soit à partir du milieu de la période d'essai et à la fin de celle-ci, il y a lieu d'effectuer un examen hématologique en mesurant l'hématocrite, la concentration d'hémoglobine, la numération des érythrocytes et des leucocytes, la formule leucocytaire, la numération des plaquettes et l'étude de paramètres de la coagulation tels que le temps de coagulation, le temps de prothrombine ou le temps de thromboplastine.
Des analyses de biochimie clinique destinées à l'étude des principaux effets toxiques sur les tissus, et en particulier sur les reins et le foie, devraient être pratiquées sur des échantillons de sang prélevés sur tous les animaux, au début de l'essai et ensuite soit tous les mois, soit au milieu et à la fin de l'essai. Les paramètres qui doivent être analysés comprennent l'équilibre électrolytique, le métabolisme des glucides et les fonctions hépatiques et rénales. Le choix de certaines analyses dépendra des observations sur le mode d'action de la substance d'essai. Les animaux doivent être mis à jeun durant une période dépendant de l'espèce la prise de sang. Les analyses proposées comprennent le calcium, le phosphore, le chlore, le sodium, le potassium, le glucose à jeun, l'alanine aminotransférase, l'aspartate aminotransférase, l'ornithine décarboxylase, la gamma glutamyle transpeptidase, l'azote uréique, l'albumine, la créatinine sanguine et les concentrations totales de bilirubine et de protéines sériques.
Les analyses d'urine doivent être pratiquées au moins au début, au milieu et en fin d'essai, sur des échantillons d'urine prélevés à des moments déterminés. Les paramètres à relever sont l'apparence, le volume, l'osmolalité ou la densité, le pH, les protéines, le glucose, le sang et les cellules sanguines. Des paramètres supplémentaires peuvent être étudiés, s'il y a lieu, afin d'élargir le champ des effets observés.
Il faudrait envisager par ailleurs de rechercher des indicateurs de lésions générales des tissus. D'autres déterminations pourraient être nécessaires à une évaluation toxicologique adéquate: l'analyse des lipides, des hormones, de l'équilibre acido-basique, de la méthémoglobine et de l'inhibition de la cholinestérase. Des analyses de biochimie clinique supplémentaires peuvent être pratiquées, s'il y a lieu, afin d'élargir le champ des effets observés. Celles-ci doivent être définies pour certaines classes de produits chimiques ou cas par cas.
Il convient d'une façon générale de suivre une démarche souple, adaptée à l'espèce et aux effets observés et/ou escomptés d'une substance donnée.
1.5.5.3. Autopsie
Tous les animaux de l'étude feront l'objet d'une autopsie complète et détaillée, comportant l'examen attentif de la surface externe du corps, de tous les orifices ainsi que des cavités crânienne, thoracique et abdominale et de leur contenu. Le foie et la vésicule biliaire, les reins, les glandes surrénales, les testicules, les épididymes, les ovaires, l'utérus, la thyroïde (et les glandes parathyroïdes) le thymus, la rate, le cerveau et le cœur de tous les animaux (à l'exception des animaux moribonds et/ou sacrifiés avant la fin de l'essai) doivent être disséqués et pesés à l'état frais dès que possible afin d'éviter leur dessiccation.
Les tissus suivants doivent être conservés dans le milieu de fixation le plus approprié, à la fois pour le type de tissu et pour l'examen histopathologique prévu: tous les organes présentant des lésions macroscopiques l'encéphale (les régions représentatives du cerveau, du cervelet, de la moelle et du pont), la moelle épinière (à trois niveaux: cervical, médiothoracique et lombaire), l'hypophyse, les yeux, la thyroïde, les glandes parathyroïdes, le thymus, l'œsophage, les glandes salivaires, l'estomac, l'intestin grêle et le gros intestin (y compris les plaques de Peyer), le foie, la vésicule biliaire, le pancréas, les reins, les glandes surrénales, la rate, le cœur, la trachée et les poumons, l'aorte, les gonades, l'utérus, les organes génitaux auxiliaires, les glandes mammaires des femelles, la prostate, la vessie, les ganglions lymphatiques (de préférence un ganglion lymphatique faisant partie du territoire de drainage dont dépend la voie d'administration et un autre faisant partie d'un autre territoire de drainage afin de détecter les éventuels effets systémiques), les nerfs périphériques (sciatiques ou tibiaux), de préférence proches du muscle, un prélèvement de moelle osseuse (et/ou une ponction de moelle osseuse examinée directement) et la peau. Les observations cliniques et les observations macroscopiques peuvent conduire à l'examen de tissus supplémentaires. Tous les organes considérés comme des organes cibles vraisemblables de la substance d'essai, compte tenu de ses propriétés, devraient également être conservés.
1.5.5.4. Histopathologie
Un examen histopathologique complet doit être pratiqué sur les tissus et organes conservés de tous les animaux appartenant au moins au groupe témoin et au groupe traité à la dose la plus élevée. Si des changements liés au traitement sont constatés dans le groupe traité à la dose la plus élevée, il faudra étendre cet examen aux animaux de tous les groupes traités.
Toutes les lésions macroscopiques doivent être examinées.
Si un groupe satellite est inclus, les tissus et organes sur lesquels des effets ont été observés dans les groupes traités doivent faire l'objet d'un examen histopathologique.
2. RÉSULTATS ET RAPPORT
2.1. RÉSULTATS
Les résultats correspondant à chaque animal doivent être fournis. En outre, tous les résultats doivent être résumés sous forme de tableaux faisant apparaître pour chaque groupe d'essai, le nombre d'animaux au début de l'essai, le nombre d'animaux trouvés morts au cours de l'essai ou euthanasiés, le moment de la mort ou du sacrifice, le nombre d'animaux présentant des symptômes de toxicité et une description des signes de toxicité observés, notamment le moment de leur apparition, leur durée et leur gravité, le nombre d'animaux présentant des lésions, les types de lésions et le pourcentage d'animaux affectés par chaque type de lésion.
S'il y a lieu, les résultats numériques devraient être évalues par une méthode statistique appropriée et largement reconnue. Le choix des méthodes statistiques et des résultats à analyser devrait intervenir au stade de la rédaction du protocole d'étude.
2.2. RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
2.2.1. Substance d'essai
|
—
|
État physique, pureté et propriétés physico-chimiques,
|
|
—
|
données permettant l'identification chimique,
|
|
—
|
véhicule (le cas échéant), justification du choix si autre que l'eau.
|
2.2.2. Animaux d'expérience
|
—
|
Espèce et souche utilisée,
|
|
—
|
nombre, âge et sexe des animaux,
|
|
—
|
source, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
poids de chaque animal au début de l'essai.
|
2.2.3. Conditions d'essai
|
—
|
Justification du choix des doses,
|
|
—
|
détails concernant la préparation contenant la substance d'essai, son incorporation dans la nourriture, la concentration obtenue, la stabilité et l'homogénéité de la préparation.
|
|
—
|
détails relatifs à l'administration de la substance d'essai,
|
|
—
|
doses réelles (mg/kg de poids corporel/jour) et facteur de conversion de la concentration de la substance d'essai dans la nourriture ou l'eau de boisson (en ppm) en dose réelle, s'il y a lieu,
|
|
—
|
détails concernant la qualité de la nourriture et de l'eau.
|
2.2.4. Résultats
|
—
|
Poids corporel et variation de poids corporel,
|
|
—
|
consommation de nourriture et d'eau, le cas échéant,
|
|
—
|
données sur la réponse toxique par sexe et par dose et description des signes de toxicité,
|
|
—
|
nature, gravité et durée des observations cliniques (réversibles ou non),
|
|
—
|
résultats de l'examen ophtalmologique,
|
|
—
|
paramètres hématologiques et valeurs normales de référence,
|
|
—
|
paramètres de biochimie clinique et valeurs normales de référence,
|
|
—
|
poids du corps et des organes des animaux à leur mort et rapports poids de l'organe/poids corporel,
|
|
—
|
description détaillée de tous les résultats histopathologiques,
|
|
—
|
données relatives à l'absorption, le cas échéant,
|
|
—
|
traitement statistique des résultats, s'il y a lieu.
|
Discussion des résultats.
Conclusions.
B.28. TOXICITÉ DERMIQUE SUBCHRONIQUE ÉPREUVE DE 90 JOURS SUR DES RONGEURS
1. MÉTHODE
1.1 INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Les doses croissantes de la substance d'essai sont appliquées quotidiennement, sur la peau des animaux de plusieurs lots d'expérience, à raison d'une dose par lot durant une période de 90 jours. Durant la période d'application, les animaux sont observés quotidiennement afin de déceler les manifestations de toxicité. Les animaux qui meurent en cours d'expérience de même que ceux encore en vie à l'issue de celle-ci sont autopsiés.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Préparations
Les animaux sont maintenus dans les conditions d'hébergement et d'alimentation propres à l'expérience pendant au moins les 5 jours qui la précèdent. Avant de commencer l'essai, les jeunes animaux sains sont répartis au hasard en lots traités et lots témoins. Peu de temps avant l'essai, on tond la fourrure de la région dorsale des animaux. On peut avoir recours au rasage, mais l'opération doit alors être effectuée 24 heures environ avant l'essai. Il est habituellement nécessaire de répéter ces opérations toutes les semaines environ et il faut prendre grand soin d'éviter toute lésion de la peau pendant ces opérations. La surface à dégager pour l'application de la substance à tester ne sera pas inférieure à 10 % de la surface corporelle. Le poids de l'animal sera pris en compte pour décider de la zone à dégager et des dimensions de la surface à traiter. Lorsque l'essai porte sur des substances solides qui, le cas échéant, peuvent être pulvérisées, la substance à tester doit être humectée au moyen d'eau ou, au besoin, d'un véhicule approprié de manière à garantir un bon contact avec la peau. Les substances liquides sont généralement testées sans dilution préalable. On procède à une application quotidienne à raison de 5 à 7 jours par semaine.
1.6.2. Conditions expérimentales
1.6.2.1. Animaux d'expérience
Le rat, le lapin ou le cochon d'Inde adultes peuvent être utilisés; d'autres espèces aussi, mais il faut alors en justifier l'emploi. Au commencement de l'expérience, la différence de poids entre les animaux ne dépassera pas ± 20 % de la valeur moyenne. Si une étude dermique subchronique constitue la phase préparatoire d'une étude à long terme, la même espèce et la même souche seront utilisées pour les deux études.
1.6.2.2. Nombre et sexe
20 animaux au moins (10 femelles et 10 mâles) à la peau saine seront utilisés pour chaque niveau de dose. Les femelles seront nullipares et non gravides. Si l'on prévoit de sacrifier certains animaux en cours d'expérience, il y a lieu d'ajouter à ce nombre celui des animaux qu'il est prévu de sacrifier avant la fin de l'expérience. De plus, un groupe satellite de 20 animaux (10 par sexe) peut être traité avec la dose la plus élevée pendant 90 jours et faire l'objet d'une observation quant à la réversibilité, la persistance ou l'apparition tardive des effets toxiques durant les 28 jours qui suivent le traitement.
1.6.2.3. Doses
On utilisera au moins trois doses ainsi qu'un lot témoin ou, le cas échéant, un lot témoin pour le véhicule. La période d'exposition sera d'au moins six heures par jour. La substance à tester sera appliquée chaque jour à la même heure et sa quantité fera l'objet d'un ajustement hebdomadaire ou bi-hebdomadaire afin de conserver une dose constante en rapport avec le poids corporel de l'animal. À l'exception de l'application de la substance à tester, les animaux du lot témoin seront traités de la même façon que les sujets des lots traités. Si un véhicule est utilisé pour faciliter l'application, il sera administré au lot témoin dans les mêmes conditions que pour les lots traités et la dose reçue correspondra à celle du lot traité par la dose la plus élevée. La dose la plus élevée doit être déterminée de manière à produire des effets toxiques mais pas, ou rarement, la mort de l'animal; la dose la plus faible ne doit faire apparaître aucun effet toxique. Si l'on dispose d'informations sur le niveau de l'exposition humaine, la dose la plus faible dépassera cette valeur. L'idéal serait que la dose intermédiaire produise l'effet toxique minimal observable. Si l'on utilise plusieurs doses intermédiaires, l'écart entre les doses sera calculé de manière à entraîner une gradation des effets toxiques. Dans les lots correspondant aux doses faible et intermédiaire ainsi que chez les témoins, le nombre de décès sera faible, afin de permettre une évaluation significative des résultats.
Si l'application de la substance à tester provoque une grave irritation cutanée, les concentrations seront réduites; ceci peut entraîner une diminution, voire une disparition, des autres effets toxiques à la dose la plus élevée. Si les lésions cutanées sont très graves, il peut s'avérer nécessaire d'arrêter l'expérience et de la recommencer avec des concentrations plus faibles.
1.6.3. Essai de limite
Si une expérience préliminaire réalisée avec une dose de 1 000 mg/kg ou avec une dose plus élevée en fonction de la possibilité d'une exposition humaine — pour autant que celle-ci soit connue — n'a provoqué aucun effet toxique, on peut juger inutile de poursuivre l'expérience.
1.6.4. Période d'observation
Les animaux d'expérience feront l'objet d'observations quotidiennes afin de déceler les manifestations de toxicité. Le moment de la mort ainsi que le moment auquel les manifestations de toxicité apparaissent et disparaissent seront notés.
1.6.5. Mode opératoire
Les animaux seront placés dans des cages individuelles. Dans les conditions idéales, la substance d'essai est administrée aux animaux 7 jours sur 7 pendant une période de 90 jours.
Les animaux de tous les groupes satellites devant faire l'objet d'observations complémentaires seront gardés en vie pendant 28 jours encore, sans traitement, afin de constater la guérison ou la persistance des effets toxiques. La durée d'exposition sera de 6 heures par jour.
La substance d'essai sera appliquée uniformément sur une surface à peu près égale à 10 % de la surface totale du corps. Dans le cas de substances hautement toxiques, la surface couverte peut être moins importante mais la couche doit être aussi mince et aussi uniforme que possible.
Durant l'exposition, la substance d'essai est maintenue en contact avec la peau au moyen d'un carré de gaze poreux et d'un sparadrap non irritant. La surface traitée sera, en outre, convenablement couverte de manière à maintenir en place le pansement de gaze et la substance d'essai et à éviter que les animaux puissent ingérer ladite substance. Des appareils de contention peuvent être utilisés pour empêcher l'ingestion de la substance mais une immobilisation complète n'est pas recommandée.
À l'issue de la période d'exposition, il faut, si possible, éliminer tout résidu de substance avec de l'eau ou par tout autre procédé adéquat de nettoyage de la peau.
Tous les animaux seront observés quotidiennement et les manifestations de toxicité ainsi que le moment de leur apparition, leur intensité et leur durée seront enregistrés. Pendant la période d'encagement, on observera notamment les modifications du poil et de la peau, des yeux et des muqueuses, de l'appareil respiratoire, du système circulatoire, du système nerveux autonome et central ainsi que de l'activité somatomotrice et du comportement. La consommation alimentaire et le poids des animaux seront déterminés chaque semaine. Il est nécessaire d'observer régulièrement les animaux afin d'éviter toute perte par cannibalisme, autolyse des tissus ou erreur de mise en place. À l'issue de l'expérience, tous les animaux survivants des lots traités non satellites seront autopsiés. Les animaux moribonds seront immédiatement retirés et autopsiés.
Les examens ci-après sont habituellement effectués pour tous les animaux, y compris ceux du lot témoin:
|
a)
|
Un examen ophtalmologique sera pratiqué à l'aide d'un ophtalmoscope ou d'un appareil équivalent avant d'administrer la substance à tester ainsi qu'au terme de l'expérience, de préférence pour tous les animaux mais au moins pour le lot traité par la dose la plus élevée ainsi que pour le lot témoin. Si des modifications oculaires sont observées tous les animaux seront examinés.
|
|
b)
|
Un examen hématologique comprenant l'hématocrite, la concentration en hémoglobine, la numération des hématies et des leucocytes, la formule leucocytaire ainsi qu'une étude de la coagulation par des épreuves telles que temps de coagulation, temps de Quick, temps de thromboplastine ou numération des plaquettes sanguines sera effectué au terme de l'expérience.
|
|
c)
|
Des déterminations biochimiques cliniques dans le sang seront effectuées à l'issue de l'expérience. L'équilibre électrolytique, le métabolisme glucidique, les fonctions hépatique et rénale présentent un intérêt général pour toutes les études. Le choix d'épreuves spécifiques sera influencé par les observations relatives au mode d'action de la substance. Il est suggéré de doser les substances suivantes: le calcium, le phosphore, les chlorures, le sodium, le potassium, le glucose à jeun (la période de jeûne étant adaptée à l'espèce), les transaminases glutamo-pyruvique (4) et glutamo-oxaloacétique (5), l'ornithine-décarboxylase, la gamma-glutamyl transpeptidase, l'azote uréique, l'albumine, la créatinine plasmatique, la bilirubine totale et les protéines sériques totales. Les autres analyses éventuellement nécessaires à une évaluation toxicologique adéquate concernent les lipides, les hormones, l'équilibre acido-basique, la méthémoglobine, l'activité cholinestérasique. D'autres analyses biochimiques cliniques peuvent, si nécessaire, être effectuées pour approfondir l'étude des effets observés.
|
|
d)
|
Un examen régulier des urines n'est pas nécessaire; un tel examen n'est indiqué qu'en cas de toxicité probable ou manifeste.
|
Si les informations recueillies dans le passé ne sont pas appropriées, les paramètres hématologiques et biochimiques cliniques seront déterminés avant le commencement de l'expérience.
1.6.6. Autopsie
Tous les animaux seront soumis à une autopsie complète comprenant l'examen de la surface externe du corps, de tous les orifices, ainsi que des cavités crânienne, thoracique et abdominale et de leur contenu. Le foie, les reins, les glandes surrénales et les testicules seront pesés, à l'état humide, le plus rapidement possible après la dissection afin d'éviter le dessèchement. Les organes et tissus ci-après seront conservés dans un milieu approprié dans l'éventualité d'examens histopathologiques ultérieurs: toute lésion macroscopique, encéphale — y compris section de la moelle/du pont de Varole, cortex cérébelleux et cortex cérébral, hypophyse, thyroïde/parathyroïde, tout tissu thymique, (trachée), poumons, cœur, aorte, glandes salivaires, foie, rate, reins, glandes surrénales, pancréas, gonades, organes génitaux annexes, vésicule biliaire (si présente), œsophage, estomac, duodénum, jéjunum, iléon, cæcum, colon, rectum, vessie urinaire, ganglion lymphatique représentatif, (femelle: glande mammaire), (musculature de la cuisse), nerf périphérique, (yeux), (sternum avec moelle osseuse), (fémur — y compris surface articulaire), (moelle épinière à trois niveaux — cervical, médiothoracique et lombaire) et (glandes lacrymales). Les tissus mentionnés entre parenthèses ne seront examinés que sur indication des signes de toxicité ou en cas d'atteinte de l'organe.
1.6.7. Examen histopathologique
|
a)
|
La peau normale et la peau traitée ainsi que les organes et tissus de tous les animaux du lot témoin et du lot exposé à la dose la plus élevée doivent faire l'objet d'un examen histopathologique complet.
|
|
b)
|
Toutes les lésions macroscopiques seront examinées.
|
|
c)
|
Les organes-cibles des animaux appartenant aux autres lots traités seront examinés.
|
|
d)
|
Si l'on utilise des rats, les poumons des animaux appartenant aux lots exposés aux doses faible et intermédiaire seront soumis à un examen histopathologique afin de déceler tout signe d'infection qui permette de juger facilement de l'état de santé des animaux. D'autres examens histopathologiques systématiques peuvent ne pas être nécessaires pour les animaux de ces lots mais doivent toujours être effectués pour les organes présentant des lésions dans le groupe traité par la dose la plus élevée.
|
|
e)
|
Lorsqu'un lot satellite est utilisé, un examen histopathologique sera pratiqué sur les tissus et organes présentant des signes de toxicité dans les lots traités.
|
2. RÉSULTATS
Les résultats seront résumés sous la forme de tableaux indiquant, pour chaque expérience, le nombre d'animaux au début de l'essai, le nombre d'animaux atteints de lésions, le type de lésions et le pourcentage d'animaux présentant chaque type de lésion. Les résultats seront évalués au moyen d'une méthode statistique appropriée. Toute méthode statistique reconnue peut être utilisée.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
espèce, souche, origine, milieu ambiant, régime alimentaire, etc.,
|
|
—
|
conditions expérimentales,
|
|
—
|
doses (y compris, le cas échéant, véhicule) et concentrations,
|
|
—
|
dose dépourvue d'effet, si possible,
|
|
—
|
réponse toxique par sexe et par dose,
|
|
—
|
moment de la mort en cours d'expérience ou indication que l'animal était encore en vie au terme de l'expérience,
|
|
—
|
description des effets toxiques ou autres,
|
|
—
|
moment de l'observation de tout symptôme et évolution de celui-ci,
|
|
—
|
quantité de nourriture et poids corporel,
|
|
—
|
observations ophtalmologiques,
|
|
—
|
examens hématologiques pratiqués et résultats,
|
|
—
|
épreuves biochimiques cliniques pratiquées et résultats (y compris, résultats de l'examen des urines),
|
|
—
|
description détaillée de toutes les observations histopathologiques,
|
|
—
|
traitement statistique des résultats, si possible,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.29 TOXICITÉ SUBCHRONIQUE PAR INHALATION EXPÉRIENCE DE 90 JOURS SUR DES RONGEURS
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITION
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Plusieurs lots d'animaux d'expérience sont exposés quotidiennement, pendant une période déterminée, à différentes concentrations de la substance d'essai, à raison d'une concentration par lot, pendant une période de 90 jours. Lorsqu'on utilise un véhicule en vue d'obtenir une concentration appropriée de la substance à tester dans l'atmosphère, il y a lieu de prévoir un lot témoin pour le véhicule. Durant la période d'exposition, les animaux sont observés quotidiennement afin de déceler les manifestations de toxicité. Les animaux qui meurent en cours d'expérience de même que ceux encore en vie au terme de celle-ci sont autopsiés.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Préparations
Les animaux sont maintenus dans les conditions d'hébergement et d'alimentation propres à l'expérience pendant au moins les 5 jours qui la précèdent. Avant de commencer l'essai, les jeunes animaux sains sont répartis au hasard entre le nombre de groupes requis. Au besoin, un véhicule approprié peut être ajouté à la substance à tester en vue d'obtenir une concentration appropriée de celle-ci dans l'atmosphère; si un véhicule ou d'autres additifs sont utilisés pour faciliter l'administration, ils seront réputés non toxiques. Des données publiées peuvent être utilisées le cas échéant.
1.6.2. Conditions d'essai
1.6.2.1. Animaux d'expérience
Sauf contre-indication, le rat est l'espèce préférée. Il faut utiliser de jeunes animaux sains d'une souche de laboratoire courante. Au commencement de l'expérience, la différence de poids entre les animaux utilisés n'excédera pas ±20 % de la valeur moyenne correspondante. Si une étude subchronique par inhalation sert de phase préparatoire à une étude à long terme, la même espèce et la même souche seront utilisées pour les deux études.
1.6.2.2. Nombre et sexe
20 animaux au moins (10 femelles et 10 mâles) seront utilisés pour chaque groupe d'expérience. Les femelles seront nullipares et non gravides. Si l'on prévoit de sacrifier certains animaux en cours d'expérience, il faudra augmenter les effectifs en fonction du nombre d'animaux qu'il est prévu de sacrifier. De plus, un groupe satellite de 20 animaux (10 par sexe) peut être exposé au niveau de concentration le plus élevé pendant 90 jours et faire l'objet d'une observation quant à la réversibilité, la persistance ou l'apparition tardive des effets toxiques pendant les 28 jours qui suivent le traitement.
1.6.2.3. Concentration d'exposition
Il faut prévoir au moins trois concentrations ainsi qu'un lot témoin ou, le cas échéant, un lot témoin pour le véhicule (correspondant à la concentration du véhicule au niveau d'exposition le plus élevé). À l'exception de l'inhalation de la substance d'essai, les animaux du lot témoin seront traités de la même manière que les sujets du groupe d'expérience. On attend de la concentration la plus élevée qu'elle produise des effets toxiques mais pas ou peu de décès; de la concentration la plus faible, qu'elle ne donne aucun effet toxique. Lorsqu'on dispose d'informations sur le niveau de l'exposition humaine, la concentration la plus faible sera supérieure à cette valeur. L'idéal serait que la concentration intermédiaire produise l'effet toxique minimal observable. Si plusieurs concentrations intermédiaires sont utilisées, l'écart entre les niveaux d'exposition sera calculé de manière à entraîner une gradation des effets toxiques. Dans les groupes à concentrations faible et intermédiaire ainsi que dans les groupes témoins, le nombre de morts sera faible afin de permettre une évaluation significative des résultats.
1.6.2.4. Temps d'exposition
L'exposition quotidienne sera de 6 heures après obtention des concentrations dans la chambre d'exposition. D'autres périodes peuvent être utilisées pour répondre à certaines exigences spécifiques.
1.6.2.5. Équipement
Les animaux seront exposés à la substance à tester au moyen d'un dispositif d'inhalation conçu pour maintenir un flux d'air continu assurant au moins 12 renouvellements de l'air par heure, et garantir une teneur en oxygène appropriée et une répartition uniforme du produit à tester dans l'air. Si l'on utilise une chambre, celle-ci sera conçue de manière à obtenir un entassement minimal des animaux d'expérience et une exposition maximale à la substance à tester. En règle générale, pour assurer la stabilité de l'atmosphère de la chambre, le «volume» total des animaux d'expérience ne doit pas dépasser 5 % du volume de la chambre d'essai. On peut aussi avoir recours à un système d'exposition oro-nasal, de la tête seule ou du corps entier en chambre individuelle; les deux premiers types d'exposition permettent de réduire la pénétration par d'autres voies.
1.6.2.6. Périodes d'observation
Les animaux d'expérience seront observés quotidiennement afin de déceler les manifestations de toxicité tout au long de la période de traitement et de récupération. Le moment de la mort ainsi que celui auquel les manifestations de toxicité apparaissent et disparaissent seront notés.
1.6.3. Mode opératoire
Les animaux sont quotidiennement exposés à la substance à tester, à raison de 5 à 7 jours par semaine, pendant une période de 90 jours. Les animaux des groupes satellites destinés à des observations complémentaires seront gardés en vie pendant 28 jours encore, sans traitement, afin de déterminer s'il y a disparition ou persistance des effets toxiques. La température à laquelle s'effectue l'épreuve sera maintenue à 22 oC ± 3 oC. Dans les conditions optimales, l'humidité relative devrait être maintenue entre 30 et 70 % mais, dans certains cas, cela peut s'avérer impossible (par exemple, essais avec aérosols). Pendant l'exposition, les animaux ne recevront ni nourriture ni eau.
On utilisera un système d'inhalation qui fonctionne dans des conditions dynamiques comportant un dispositif approprié de contrôle analytique de la concentration. Il est recommandé de procéder à un essai préliminaire afin de déterminer les concentrations d'exposition appropriées. Le débit d'air devra assurer des concentrations homogènes dans toute la chambre d'exposition. Le système permettra d'obtenir des conditions d'exposition stables aussi rapidement que possible.
Il y a lieu de mesurer et de contrôler:
|
a)
|
le débit d'air (en permanence);
|
|
b)
|
la concentration réelle de la substance d'essai mesurée dans la zone de respiration. Durant la période d'exposition quotidienne, la concentration ne variera pas de plus de ± 15 % de la valeur moyenne. Cependant, dans le cas de poussières et d'aérosols, cette précision peut ne pas être possible et un écart plus grand pourra alors être accepté. Durant toute la durée de l'expérience, les concentrations journalières seront maintenues aussi constantes que possible. Lors de la mise au point du système générateur, il sera procédé à une analyse granulométrique des particules afin de déterminer la stabilité des concentrations d'aérosol. Durant l'exposition, il sera procédé à des analyses aussi fréquentes que nécessaire pour déterminer la stabilité de la répartition granulométrique;
|
|
c)
|
la température et l'humidité;
|
|
d)
|
pendant et après l'exposition, des observations ont lieu et sont systématiquement notées; des fiches individuelles sont établies pour chaque animal. Tous les animaux seront observés quotidiennement et les manifestations de toxicité ainsi que le moment de leur apparition, leur intensité et leur durée seront notés. Pendant la période d'encagement, il convient notamment d'observer les modifications de la peau et du poil, des yeux, des muqueuses, de l'appareil respiratoire, du système circulatoire, du système nerveux autonome et central, de l'activité somatomotrice et du comportement. La consommation alimentaire et le poids des animaux seront déterminés chaque semaine. Il est nécessaire d'observer régulièrement les animaux afin de s'assurer qu'il n'y a pas de perte par cannibalisme, autolyse des tissus ou erreur de mise en place. À l'issue de l'expérience, tous les animaux survivants des lots traités non satellites sont autopsiés, En cours d'expérience, tout animal moribond sera immédiatement retiré et autopsié.
|
Les examens figurant ci-après sont habituellement effectués sur tous les animaux, y compris les témoins:
|
a)
|
Un examen ophtalmologique sera pratiqué à l'aide d'un ophtalmoscope ou d'un appareil équivalent avant l'exposition à la substance à tester ainsi qu'au terme de l'expérience, de préférence sur tous les animaux mais au moins sur le groupe traité au niveau de dose le plus élevé ainsi que sur le groupe témoin. Si des modifications oculaires sont observées, tous les animaux seront examinés.
|
|
b)
|
Un examen hématologique comprenant l'hématocrite, la concentration en hémoglobine, la numération des hématies et des leucocytes, la formule leucocytaire ainsi que l'étude de la coagulation par des épreuves telles que temps de coagulation, temps de Quick, temps de thromboplastine ou numération des plaquettes sanguines sera effectué au terme de l'expérience.
|
|
c)
|
Des déterminations biochimiques cliniques dans le sang seront effectuées à l'issue de l'expérience. L'équilibre électrolytique, le métabolisme glucidique, les fonctions rénale et hépatique présentent un intérêt général pour toutes les études. Le choix d'épreuves spécifiques sera influencé par les observations relatives au mode d'action de la substance. Il est suggéré de doser les substances suivantes: le calcium, le phosphore, les chlorures, le sodium, le potassium, le glucose à jeun (la période de jeûne étant adaptée à l'espèce), les transaminases glutamo-pyruvique (4) et glutamo-oxaloacétique sériques (5), l'ornithine-décarboxylase, la gamma-glutamyl transpeptidase, l'azote uréique, l'albumine, la créatinine plasmatique, la bilirubine totale et les protéines sériques totales. Les autres analyses éventuellement nécessaires pour une évaluation toxicologique adéquate concernent les lipides, les hormones, l'équilibre acido-basique, la méthémoglobine, l'activité cholinestérasique. D'autres analyses biochimiques cliniques peuvent, si nécessaire, être effectuées pour approfondir l'étude des effets observés.
|
|
d)
|
Un examen systématique des urines n'est pas nécessaire; un te! examen n'est indiqué qu'en cas de toxicité probable ou manifeste.
|
Si les informations recueillies dans le passé ne sont pas appropriées, les paramètres hématologiques et biochimiques cliniques seront déterminés avant le commencement de l'expérience.
1.6.4. Autopsie
Tous les animaux seront soumis à une autopsie générale comprenant l'examen de la surface externe du corps, de tous les orifices ainsi que des cavités crânienne, thoracique et abdominale et de leur contenu. Le foie, les reins, les glandes surrénales et les testicules seront pesés, à l'état humide, le plus rapidement possible après la dissection afin d'éviter le dessèchement. Les organes et tissus ci-après seront conservés dans un milieu approprié dans l'éventualité d'examens histopathologiques ultérieurs: toute lésion macroscopique, poumons — ceux-ci doivent être prélevés entiers, pesés et traités au moyen d'un fixateur approprié permettant de conserver la structure pulmonaire (une perfusion avec le fixateur est considérée comme une méthode efficace), tissus du rhino-pharynx, encéphale — y compris section de la moelle/du pont de Varole, cortex cérébelleux et cortex cérébral, hypophyse, thyroïde/parathyroïde, tout tissu thymique, trachée, cœur, aorte, glandes salivaires, foie, rate, reins, glandes surrénales, pancréas, gonades, utérus, (organes génitaux annexes), (peau), vésicule biliaire (si présente), œsophage, estomac, duodénum, jéjunum, iléon, caecum, colon, rectum, vessie, ganglion lymphatique représentatif, (femelle: glande mammaire), (musculature de la cuisse), nerf périphérique, (yeux), sternum avec moelle osseuse, (fémur — y compris surface articulaire), (moelle épinière à trois niveaux — cervical, médiothoracique et lombaire) et (glandes lacrymales). Les tissus mentionnés entre parenthèses ne doivent être examinés que sur indication des signes de toxicité ou en cas d'atteinte de l'organe.
1.6.5. Examen histopathologique
|
a)
|
Les voies respiratoires ainsi que les organes et tissus de tous les animaux du lot témoin et de ceux du lot exposé au niveau le plus élevé feront l'objet d'un examen histopathologique complet.
|
|
b)
|
Toutes les lésions marquées seront examinées.
|
|
c)
|
Les organes-cibles des animaux appartenant à d'autres lots traités seront examinés.
|
|
d)
|
Les poumons des animaux appartenant aux groupes exposés à des doses faible et intermédiaire seront également soumis à un examen histopathologique, les poumons étant un indicateur commode de l'état de santé des animaux. D'autres examens histopathologiques réguliers peuvent ne pas être nécessaires pour les animaux de ces groupes mais doivent toujours être effectués pour les organes présentant des lésions dans le lot traité au niveau d'exposition le plus élevé.
|
|
e)
|
Lorsqu'un groupe satellite est utilisé, un examen histopathologique sera pratiqué sur les tissus et les organes présentant des signes de toxicité dans les autres lots traités.
|
2. RÉSULTATS
Les données seront résumées sous la forme de tableaux, indiquant pour chaque lot d'expérience le nombre d'animaux au début de l'essai, le nombre d'animaux atteints de lésions, le type de lésions et le pourcentage d'animaux présentant chaque type de lésion. Les résultats seront évalués au moyen d'une méthode statistique appropriée. Toute méthode statistique reconnue peut être utilisée.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
espèce, souche, origine, milieu ambiant, régime alimentaire, etc.,
|
|
—
|
conditions expérimentales:
Description de l'appareil d'exposition y compris conception, type, dimensions, source d'air, système générateur de particules et d'aérosols, méthode de conditionnement d'air, traitement de l'air évacué et, le cas échéant, méthode de stabulation des animaux dans la chambre d'essai. L'équipement utilisé pour mesurer la température, l'humidité et, si nécessaire, la stabilité des concentrations d'aérosol ou la granulométrie des particules sera décrit.
Données relatives à l'exposition
Ces données seront présentées sous la forme de tableaux indiquant des valeurs moyennes ainsi qu'une mesure de la variabilité (par exemple écart type) et porteront sur:
|
a)
|
les débits d'air à travers le dispositif d'inhalation;
|
|
b)
|
la température et l'humidité de l'air;
|
|
c)
|
les concentrations nominales (quantité totale de substance à tester introduite dans le dispositif d'inhalation, divisée par le volume d'air);
|
|
d)
|
le cas échéant, la nature du véhicule;
|
|
e)
|
les concentrations réelles dans la zone de respiration;
|
|
f)
|
les dimensions médianes des particules (si nécessaire),
|
|
|
—
|
réponse toxique par sexe et par concentration,
|
|
—
|
moment de la mort en cours d'expérience ou indication que l'animal était encore en vie à la fin de l'expérience,
|
|
—
|
description des effets toxiques ou autres; niveau d'exposition dépourvu d'effet,
|
|
—
|
moment de l'observation de tout signe anormal et évolution de celui-ci,
|
|
—
|
quantité de nourriture et poids corporel,
|
|
—
|
observations ophtalmologiques,
|
|
—
|
examens hématologiques pratiqués et résultats,
|
|
—
|
épreuves biochimiques cliniques pratiquées et résultats (y compris résultats de l'examen des urines),
|
|
—
|
description détaillée de toutes les observations histopathologiques,
|
|
—
|
traitement statistique des résultats, si possible,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.30 ÉTUDE DE LA TOXICITÉ CHRONIQUE
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance à tester est administrée normalement sept jours sur sept, par une voie appropriée, à plusieurs lots d'animaux d'expérience, à raison d'une dose par groupe, pendant une grande partie de leur vie. Pendant et après l'exposition à la substance à tester, les animaux d'expérience sont observés quotidiennement afin de déceler les manifestations de toxicité.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Préparations
Les animaux sont maintenus dans les conditions d'encagement et d'alimentation propres à l'expérience pendant au moins les cinq jours qui la précèdent. Avant de commencer l'essai, les jeunes animaux sains sont répartis au hasard entre le nombre de lots requis.
1.6.2 Conditions expérimentales
1.6.2.1. Animaux d'expérience
Le rat est l'espèce la mieux appropriée.
Compte tenu des résultats d'études antérieures, d'autres espèces (rongeurs ou autres) peuvent être utilisées. De jeunes animaux sains de souches de laboratoire courantes seront utilisés et le traitement commencera dès que possible après le sevrage.
Au début de l'essai, la différence de poids entre les animaux utilisés ne dépassera pas ±20 % de la valeur moyenne. Si une étude orale subchronique a servi de phase préparatoire à une étude à long terme, la même espèce et la même souche seront utilisées pour les deux études.
1.6.2.2. Nombre et sexe
Dans le cas de rongeurs, 40 animaux au moins (20 femelles et 20 mâles) seront utilisés pour chaque dose et chaque lot témoin correspondant. Les femelles seront nullipares et non gravides. Si l'on prévoit de sacrifier certains animaux en cours d'expérience, il y a lieu d'augmenter en conséquence les effectifs des groupes.
Pour les non-rongeurs, un plus petit nombre d'animaux au moins quatre par sexe et par groupe, peut être accepté.
1.6.2.3. Niveaux de dose et fréquence d'exposition
Trois doses au moins seront utilisées en plus des groupes témoins correspondants. La dose la plus élevée sera suffisante pour produire des manifestations nettes de toxicité sans entraîner une mortalité excessive.
La dose la plus faible ne produira aucun effet toxique.
La dose (les doses) intermédiaire(s) aura (auront) une valeur proche de la moyenne entre la dose forte et la dose faible.
Les doses seront choisies en fonction de données obtenues lors d'essais préliminaires et d'études de toxicité effectuées antérieurement.
Il est prévu une exposition quotidienne. Si le produit chimique est administré dans l'eau de boisson ou incorporé à l'alimentation, il devra être constamment disponible.
1.6.2.4. Témoins
On utilisera un groupe témoin en tout point identique au groupe exposé, exception faite toutefois de l'exposition à la substance à tester.
Dans des conditions particulières telles que des études d'inhalation impliquant l'emploi d'aérosols ou l'utilisation d'un émulsifiant à activité biologique non caractérisée dans des études par voie orale, on utilisera un groupe témoin négatif concurrent. Le groupe témoin négatif est traité de la même façon que tous les autres animaux d'expérience, exception faite de l'exposition à la substance à tester ou à un véhicule quelconque.
1.6.2.5. Voie d'administration
Les deux principales voies d'administration sont la voie orale et la voie respiratoire (inhalation). Le choix de la voie d'administration dépend des caractéristiques physiques et chimiques de la substance à tester ainsi que du mode d'exposition probable pour l'homme.
L'utilisation de la voie dermique pose des problèmes pratiques considérables. La toxicité chronique générale due à l'absorption percutanée peut normalement être déduite des résultats de l'essai par voie orale et de la quantité de substance absorbée par voie percutanée lors des essais de toxicité par voie percutanée antérieurs.
1.6.2.6. Études par voie orale
En cas d'absorption gastro-intestinale de la substance à tester, et si l'ingestion est une voie d'exposition humaine, sauf contre-indications, on utilise de préférence la voie orale. Les animaux recevront la substance à tester dans leur nourriture, dissoute dans l'eau de boisson, ou sous la forme de capsule. Dans les conditions idéales, la dose quotidienne sera administrée 7 jours sur 7 car l'administration pendant 5 jours sur 7 peut permettre à l'animal de récupérer ou à la toxicité de décroître durant la période où le traitement est interrompu et affecter, par conséquent, les résultats et les évaluations ultérieures. Cependant, pour des raisons essentiellement pratiques, on peut accepter que la substance soit administrée 5 jours sur 7.
1.6.2.7. Études d'inhalation
Les études d'inhalation posant des problèmes techniques beaucoup plus complexes que pour les autres voies d'administration, des recommandations plus détaillées sont données ici. Il convient également de noter que l'instillation intratrachéale constitue une méthode valable dans certains cas spécifiques.
Les expositions prolongées sont habituellement établies en fonction de l'exposition humaine possible: les animaux sont soit exposés 5 jours sur 7 (exposition intermittente) à raison de 6 heures par jour après équilibrage des concentrations dans la chambre d'essai, soit soumis 7 jours sur 7 (exposition permanente) à une exposition journalière de 22 à 24 heures, une heure par jour environ étant consacrée à l'alimentation des animaux (horaire régulier) et à l'entretien de la chambre d'essai.
Dans les deux cas, les animaux sont habituellement exposés à une concentration fixe du produit à tester. Une différence essentielle entre l'exposition intermittente et l'exposition permanente réside dans le fait que, dans le premier cas, les animaux disposent d'une période de 17 à 18 heures pour se remettre des effets de l'exposition quotidienne et même d'une période plus longue durant les week-ends.
L'exposition intermittente ou permanente sera choisie en fonction des objectifs de l'étude et de l'exposition humaine que l'on se propose de simuler. Il convient, toutefois, de tenir compte de certaines difficultés techniques: par exemple, les avantages qu'offre l'exposition permanente pour simuler les conditions ambiantes peuvent être contrebalancés par la nécessité d'alimenter et de faire boire les animaux pendant l'exposition ainsi que par les contraintes inhérentes à la complexité (et fiabilité) de la génération des aérosols et des vapeurs et des moyens de contrôle.
1.6.2.8. Chambre d'exposition
Les animaux seront exposés à la substance à tester dans des chambres d'inhalation conçues pour assurer un flux d'air dans des conditions dynamiques, avec au moins 12 renouvellements de l'air par heure, garantir une teneur en oxygène appropriée et une répartition uniforme du produit à tester dans l'air. Les chambres d'exposition et les chambres destinées aux animaux témoins seront identiques du point de vue de la construction et de la conception afin de garantir des conditions d'exposition en tout point comparable, à la seule exception de l'exposition à la substance à tester. Une pression légèrement négative est généralement maintenue à l'intérieur de la chambre afin d'empêcher les fuites de la substance à tester dans la zone environnante. Les chambres seront conçues de manière à réduire l'entassement des animaux d'expérience. En règle générale, pour assurer la stabilité de l'atmosphère de la chambre, le volume total des animaux d'expérience ne doit pas dépasser 5 % du volume de la chambre d'essai.
|
1.6.2.9.
|
Mesures ou contrôles à effectuer:
|
(i)
|
le débit d'air: le débit d'air dans la chambre fera, de préférence, l'objet d'un contrôle continu;
|
|
(ii)
|
durant la période d'exposition quotidienne, la concentration ne variera pas de ± 15 % de la valeur moyenne.
Durant toute la durée de l'expérience, les concentrations journalières seront maintenues aussi constantes que possible;
|
|
(iii)
|
température et humidité: pour les rongeurs, la température sera maintenue à 22 oC (± 2 oC) et l'humidité à l'intérieur de la chambre sera de 30 à 70 %, sauf lorsqu'on utilise de l'eau pour mettre la substance à tester en suspension dans l'air dans la chambre d'essai. Ces deux paramètres seront, de préférence, contrôlés en permanence;
|
|
(iv)
|
analyse granulométrique des particules: une répartition granulométrique des particules sera effectuée pour les atmosphères de la chambre d'essai impliquant l'utilisation d'aérosols liquides ou solides. Les particules de l'aérosol seront d'une taille respirable pour l'animal d'expérience utilisé. Des échantillons des atmosphères des chambres d'essai seront prélevés dans la zone respirable par les animaux. L'échantillon d'air prélevé sera représentatif de la répartition des particules auxquelles les animaux sont exposés et représentera, sur une base gravimétrique, la totalité de l'aérosol en suspension, même si une grande partie de celui-ci n'est pas respirable. Les analyses granulométriques seront effectuées fréquemment durant la mise au point du système générateur afin de veiller à la stabilité de l'aérosol puis, par la suite, uniquement lorsqu'il sera nécessaire de déterminer de façon adéquate la stabilité de la répartition des particules auxquelles les animaux ont été exposés.
|
|
1.6.2.10. Durée de l'étude
La durée du traitement sera d'au moins 12 mois.
1.6.3. Mode opératoire
1.6.3.1. Observations
Il sera procédé au moins une fois par jour à un examen clinique attentif. Des observations complémentaires seront faites quotidiennement et des mesures appropriées seront prises afin de réduire la perte d'animaux pour l'étude, par exemple autopsie ou cryobiologie pour les animaux trouvés morts ainsi qu'isolement ou sacrifice des animaux en mauvaise santé. Les animaux seront soigneusement observés afin de déceler l'apparition et l'évolution de toutes les manifestations de toxicité ainsi que pour réduire la perte d'animaux pour cause de maladie, d'autolyse ou de cannibalisme.
Les signes cliniques, y compris les modifications neurologiques et oculaires, ainsi que la mortalité seront relevés pour tous les animaux. Le moment de l'apparition des effets toxiques et leur évolution ainsi que les tumeurs suspectes seront notés.
Le poids de chaque animal sera déterminé et noté une fois par semaine durant les 13 premières semaines de la période d'essai, puis au moins une fois toutes les 4 semaines. La consommation alimentaire sera déterminée chaque semaine durant les 13 premières semaines de l'étude, puis tous les 3 mois environ, à moins que l'état de santé ou les modifications de poids corporel nécessitent une autre fréquence.
1.6.3.1. Examen hématologique
Un examen hématologique (par exemple, teneur en hémoglobine, hématocrite, numération des érythrocytes, des leucocytes, des thrombocytes ou étude de la coagulation) sera effectué au troisième et au sixième mois, puis tous les 6 mois environ ainsi qu'au terme de l'essai, sur des échantillons de sang prélevés chez tous les non-rongeurs ainsi que chez 10 rats/sexe de tous les groupes. Ces échantillons seront, si possible, prélevés chaque fois sur les mêmes rats. De plus, un échantillon sera prélevé chez tous les animaux n'appartenant pas à l'ordre des rongeurs avant de commencer l'essai.
Si les observations cliniques indiquent une détérioration de l'état de santé des animaux durant l'étude, il sera procédé à une numération avec formule leucocytaire chez les animaux atteints.
La formule leucocytaire est établie sur les échantillons des animaux traités par la dose la plus élevée ainsi que sur ceux des animaux du groupe témoin. Cette formule n'est établie pour les groupes exposés à des doses plus faibles que si l'on constate un écart important entre le groupe exposé à la dose la plus élevée et le groupe témoin, ou si l'examen pathologique le justifie.
Examen des urines
Des échantillons d'urine seront collectés pour analyse chez tous les non-rongeurs ainsi que chez 10 rats/sexe de chaque groupe, si possible chez les mêmes rats et aux mêmes moments que pour l'examen hématologique. Qu'il s'agisse des non-rongeurs ou des rongeurs, il sera procédé aux déterminations suivantes:
|
—
|
apparence; volume et densité pour chaque animal,
|
|
—
|
protéines, glucose, cétones, signes discrets d'hémorragie (semi-quantitativement), et
|
|
—
|
microscopie des sédiments urinaires (semi-quantitativement).
|
Chimie clinique
Tous les six mois environ ainsi qu'au terme de l'essai, des échantillons de sang sont prélevés, afin d'effectuer des déterminations chimiques cliniques, chez tous les animaux n'appartenant pas à l'ordre des rongeurs ainsi que chez 10 rats/sexe de chaque groupe, en prenant si possible chaque fois les mêmes rats. De plus, un échantillon sera prélevé sur tous les animaux n'appartenant pas à l'ordre des rongeurs avant de commencer l'essai. Le plasma est préparé à partir de ces échantillons et l'on procède aux déterminations suivantes:
|
—
|
concentrations des protéines totales,
|
|
—
|
concentration de l'albumine,
|
|
—
|
analyse de la fonction hépatique [telle que activité phosphatase alcaline, activité SGOT (5) et activité SGPT (5) ], gamma-glutamyl transpeptidase, ornithine-décarboxylase,
|
|
—
|
métabolisme glucidique tel que glycémie à jeun,
|
|
—
|
exploration fonctionnelle rénale telle qu'azote uréique du sang.
|
Autopsie
Un examen complet sera effectué sur tous les animaux, y compris ceux qui sont morts en cours d'expérience ou qui ont été sacrifiés à l'état moribond. Avant de sacrifier les animaux, des échantillons de sang seront prélevés afin d'établir la formule leucocytaire. Tous les organes ou tissus présentant des lésions macroscopiques visibles, des tumeurs ou des lésions soupçonnées d'être des tumeurs seront conservés. On tentera de mettre en corrélation les observations macroscopiques et microscopiques.
Tous les organes et tissus seront conservés en vue d'un examen au microscope. Il s'agit habituellement des organes et tissus ci-après: encéphale (6) (moelle/pont de Varole, cortex cérébelleux, cortex cérébral), hypophyse, thyroïde (y compris parathyroïdes), thymus, poumons (y compris trachée), cœur, aorte, glandes salivaires, foie (6), rate, reins (6), glandes surrénales (6), œsophage, estomac, duodénum, jéjunum, iléon, caecum, colon, rectum, utérus, vessie urinaire, ganglions lymphatiques, pancréas, gonades (6), organes génitaux annexes, femelle: glande mammaire, peau, musculature, nerf périphérique, moelle épinière (cervicale, thoracique, lombaire), sternum avec moelle osseuse et fémur (y compris articulation), yeux. Bien que l'inflation des poumons et de la vessie urinaire au moyen d'un fixateur constitue la meilleure manière de conserver ces tissus, l'inflation des poumons est essentielle dans les études d'inhalation si l'on veut effectuer les examens histopathologiques appropriés. Dans des études particulières telles que des études d'inhalation, tout le tractus respiratoire sera étudié y compris le nez, le pharynx et le larynx.
Si d'autres examens cliniques sont effectués, les informations obtenues seront communiquées avant de commencer les examens au microscope car elles peuvent fournir des indications précieuses au pathologiste.
Examen histopathologique
Toutes les modifications visibles, en particulier les tumeurs et autres lésions, observées sur un organe feront l'objet d'un examen au microscope. De plus, il est recommandé d'effectuer les examens suivants:
|
a)
|
Examen au microscope de tous les organes et tissus conservés et description complète de toutes les lésions observées
|
1)
|
sur tous les animaux morts ou sacrifiés au cours de l'étude et
|
|
2)
|
sur tous les animaux du (des) groupe(s) exposé(s) à la dose la plus élevée et sur tous les témoins.
|
|
|
b)
|
Les organes ou tissus présentant des anomalies spontanées ou éventuellement induites par la substance à tester seront également examinés dans les groupes exposés aux doses plus faibles.
|
|
c)
|
Si l'expérience met en évidence une altération substantielle de la longévité normale des animaux ou l'induction d'effets susceptibles d'affecter la réponse toxique, les animaux exposés à la dose immédiatement inférieure seront soumis à l'examen mentionné ci-dessus.
|
|
d)
|
L'incidence des lésions atteignant normalement la souche d'animaux utilisée (dans les mêmes conditions d'expérience, c'est-à-dire références antérieures) est indispensable pour évaluer correctement la signification des modifications observées sur les animaux exposés.
|
2. RÉSULTATS
Les données seront résumées sous forme de tableaux indiquant, pour chaque groupe d'expérience, le nombre d'animaux au début de l'essai, le nombre d'animaux atteints de lésions et le pourcentage d'animaux présentant chaque type de lésion. Les résultats seront évalués au moyen d'une méthode statistique appropriée. Toute méthode statistique reconnue peut être utilisée.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
espèce, souche, origine, milieu ambiant, régime alimentaire, etc.,
|
|
—
|
conditions expérimentales:
description de l'appareil d'exposition:
y compris conception, type, dimensions, source d'air, système générateur de particules et d'aérosols, méthode de conditionnement d'air, traitement de l'air évacué et, le cas échéant, mode de stabulation des animaux en chambre d'essai. L'équipement utilisé pour mesurer la température et, si nécessaire, l'humidité, la stabilité de la concentration d'aérosol ou la granulométrie, seront décrits;
données relatives à l'exposition:
elles seront présentées sous forme de tableaux indiquant des valeurs moyennes ainsi qu'une mesure de la variabilité (par exemple écart type) et porteront sur:
|
a)
|
les débits d'air à travers le dispositif d'inhalation;
|
|
b)
|
la température et l'humidité de l'air;
|
|
c)
|
les concentrations nominales (quantité totale de substance à tester introduite dans le dispositif d'inhalation, divisée par le volume d'air);
|
|
d)
|
le cas échéant, la nature du véhicule;
|
|
e)
|
les concentrations réelles dans la zone de respiration;
|
|
f)
|
les dimensions médianes des particules (si nécessaire),
|
|
|
—
|
doses (y compris, le cas échéant, véhicule) et concentrations,
|
|
—
|
réponse toxique par sexe et dose,
|
|
—
|
dose ou concentration dépourvues d'effet,
|
|
—
|
moment de la mort en cours d'expérience ou indication que l'animal était encore en vie à la fin de l'expérience,
|
|
—
|
description des effets toxiques ou autres,
|
|
—
|
moment de l'observation de tout signe anormal et évolution de celui-ci,
|
|
—
|
quantité de nourriture et poids corporel,
|
|
—
|
observations ophtalmologiques,
|
|
—
|
épreuves hématologiques pratiquées et résultats complets,
|
|
—
|
épreuves biochimiques cliniques pratiquées et résultats complets (y compris résultats de l'examen des urines),
|
|
—
|
description détaillée de toutes les observations histopathologiques,
|
|
—
|
traitement statistique des résultats,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.31. ÉTUDE DE LA TOXICITÉ POUR LE DÉVELOPPEMENT PRÉNATAL
1. MÉTHODE
Cette méthode est calquée sur la ligne directrice d'essai no 414 (2001) de l'OCDE.
1.1. INTRODUCTION
La présente méthode d'essai relative à la toxicité pour le développement est destinée à livrer des informations générales concernant les effets d'une exposition prénatale sur la femelle gravide et sur l'organisme en développement qu'elle porte en elle; cela recouvre notamment l'évaluation des effets sur la mère, ainsi que la mortalité fœtale, les anomalies structurelles ou les altérations de croissance du fœtus. Les déficits fonctionnels, qui représentent pourtant un aspect important du développement, ne sont pas étudiés dans le cadre de la présente méthode d'essai. Ces déficits peuvent être étudiés séparément ou en complément à la présente méthode dans le cadre de la méthode d'essai relative à la neurotoxicité pour le développement. Cette dernière méthode ainsi que la méthode relative à la toxicité pour la reproduction sur deux générations indiquent comment dépister les déficits fonctionnels et d'autres effets postnataux.
Il est possible que la présente méthode d'essai nécessite certaines adaptations dans des cas particuliers, compte tenu, par exemple, des propriétés physico-chimiques ou toxicologiques spécifiques de la substance d'essai. Ces adaptations sont acceptables lorsque des données scientifiques convaincantes donnent à penser qu'elles rendront l'essai plus informant. Le cas échéant, ces données scientifiques devront être soigneusement consignées dans le rapport d'essai.
1.2 DÉFINITIONS
Toxicologie du développement: l'étude des effets nocifs sur un organisme en développement, qui peuvent résulter d'une exposition antérieure à la conception, contemporaine au développement prénatal ou postnatale, jusqu'à la maturation sexuelle. La toxicité pour le développement se manifeste principalement par 1) la mort de l'organisme; 2) une anomalie structurelle; 3) une altération de croissance; 4) un déficit fonctionnel. Autrefois, la toxicologie du développement était souvent dénommée “tératologie”.
Effet nocif: toute altération liée au traitement par rapport à une situation de référence, qui diminue la capacité d'un organisme à survivre, se reproduire ou s'adapter à l'environnement. La toxicologie du développement, prise dans son sens le plus large, inclut tous les effets qui interfèrent avec le développement normal du produit de conception, avant et après la naissance.
Altération de croissance: une altération qui touche les organes ou le poids corporel ou la taille de la progéniture.
Altérations (anomalies): altérations structurelles du développement qui comprennent les malformations et les variations (28).
Malformation/Anomalie majeure: changement structurel considéré comme préjudiciable à l'animal (peut aussi être létal) et généralement rare.
Variation/Anomalie mineure: changement structurel considéré comme peu ou pas préjudiciable à pour l'animal; peut être transitoire et peut survenir fréquemment dans la population témoin.
Produit de conception: l'ensemble des produits de la fécondation d'un œuf, à n'importe quel stade du développement entre la fécondation et la naissance, comprenant les membranes extra-embryonnaires et l'embryon ou le fœtus.
Implantation (nidation): la fixation du blastocyste à la muqueuse épithéliale de l'utérus, y compris la pénétration du blastocyste dans l'épithélium utérin et la nidation dans l'endomètre.
Embryon: le premier stade de développement d'un organisme, plus précisément la phase de développement d'un œuf fécondé qui commence après l'apparition du grand axe et s'achève quand toutes les structures principales sont présentes.
Embryotoxicité: nocivité pour la structure, le développement, la croissance et/ou la viabilité d'un embryon.
Fœtus: produit de conception, durant la période post-embryonnaire.
Fœtotoxicité: nocivité pour la structure, le développement, la croissance et/ou la viabilité d'un fœtus.
Avortement: expulsion prématurée hors de l'utérus des produits de conception: embryon ou fœtus non viable.
Résorption: phénomène par lequel un produit de conception qui meurt après l'implantation se résorbe ou a été résorbé.
Résorption précoce: trace d'implantation non accompagnée d'un embryon ou d'un fœtus reconnaissable.
Résorption tardive: embryon ou fœtus mort qui présente des changements dégénératifs externes.
DSET: dose sans effet toxique (correspond à la NOAEL: No Observed Adverse Effect Level)
1.3. SUBSTANCE DE RÉFÉRENCE
Aucune
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Normalement, la substance d'essai est administrée aux femelles gravides, au moins à partir de la nidation et jusqu'à la veille du sacrifice, lequel devrait être programmé à une date aussi proche que possible du jour probable de mise bas, sans être trop tardif pour éviter la perte de données en cas de mise bas prématurée. La méthode d'essai ne porte pas uniquement sur la période de l'organogenèse (comprise, par exemple entre le 5ème et le 15e jour chez les rongeurs et entre le 6e et le 18e jour chez le lapin), mais étudie aussi les effets survenus tout au long de la gestation, depuis le stade pré-implantatoire, selon les besoins, jusqu'à la veille de la césarienne. Les femelles sont sacrifiées peu avant la césarienne, le contenu utérin est examiné et les fœtus étudiés pour détecter les anomalies externes visibles ainsi que les modifications des tissus mous et du squelette.
1.5. DESCRIPTION DE LÀ MÉTHODE D'ESSAI
1.5.1. Choix de l'espèce animale
Il est recommandé de pratiquer l'essai sur l'espèce la plus appropriée et d'employer les espèces et les souches de laboratoire couramment utilisées dans les essais de toxicité prénatale pour le développement. L'espèce de rongeur préféré est le rat et l'espèce non-rongeur préféré est le lapin. Le cas échéant, l'emploi d'une autre espèce doit être justifié.
1.5.2. Conditions d'encagement et d'alimentation
L'animalerie doit être à 22 oC (± 3o) pour les rongeurs et à 18 oC (± 3o) pour les lapins, avec un taux d'humidité relative d'au moins 30 % et de préférence inférieur à 70 % sauf durant le nettoyage de la salle, l'idéal se situant entre 50 et 60 %. On appliquera un éclairage artificiel, avec un cycle de 12 heures de clarté et 12 heures d'obscurité. Un régime alimentaire classique pour animaux de laboratoire, avec eau potable à satiété, conviendra.
L'accouplement doit se dérouler dans des cages appropriées. Bien qu'il soit préférable de placer les animaux s'étant accouplés dans des cages individuelles, l'encagement par petits groupes est aussi acceptable.
1.5.3 Préparation des animaux
Il convient d'utiliser des animaux sains, acclimatés aux conditions du laboratoire pendant au moins 5 jours et n'ayant pas été soumis à d'autres essais. On indiquera l'espèce, la souche, la source, le sexe, le poids et/ou l'âge des animaux d'expérience. Les animaux de tous les groupes d'essai devraient, dans toute la mesure du possible, être du même âge et du même poids. On emploiera de jeunes femelles adultes et nullipares pour chaque dose. On accouplera les femelles avec des mâles de la même espèce et de la même souche, sans accoupler les membres d'une même fratrie. Dans le cas des rongeurs, le jour 0 de la gestation est celui où l'on observe un bouchon vaginal et/ou la présence de sperme; s'agissant des lapins, le jour 0 est ordinairement celui du coït ou de l'insémination artificielle si cette technique est utilisée. Les femelles accouplées seront réparties au hasard entre groupes traités et groupes témoins. On installera les cages de façon à réduire au minimum les éventuels effets liés à leur emplacement. Chaque animal sera désigné par un numéro d'identification propre. En cas d'accouplement par lots, les animaux d'un même lot seront répartis uniformément entre les groupes. De la même façon, les femelles inséminées par un même mâle seront réparties uniformément entre les groupes.
1.6 MODE OPÉRATOIRE
1.6.1 Nombre et sexe des animaux
Chaque groupe traité et témoin doit contenir un nombre de femelles suffisant pour qu'une autopsie puisse être pratiquée sur environ 20 femelles présentant un point d'implantation. Des groupes comptant moins de 16 femelles présentant un point d'implantation risquent d'être inadéquats. La mortalité maternelle n'invalide pas nécessairement l'étude, tant qu'elle demeure inférieure à 10 pour cent environ.
1.6.2 Préparation des doses
Si on emploie un véhicule ou un autre additif pour faciliter l'administration des doses, il convient d'être attentif aux effets sur l'absorption, la distribution, le métabolisme et la rétention ou l'excrétion de la substance d'essai, aux effets sur les propriétés chimiques de la substance d'essai, lesquels sont susceptibles de modifier sa toxicité ainsi qu'aux effets sur la consommation de nourriture ou d'eau et sur l'état nutritionnel des animaux. Le véhicule ne doit pas être toxique pour le développement ni influer sur la reproduction.
1.6.3 Dosage
Normalement, la substance d'essai est administrée quotidiennement depuis la nidation (par exemple 5 jours après l'accouplement) jusqu'à la veille du jour où la césarienne est prévue. Si des études préliminaires, le cas échéant, ne font pas état d'un risque élevé de pertes pré-implantatoires, il est possible d'administrer le traitement pendant toute la durée de la gestation, depuis l'accouplement jusqu'à la veille de la césarienne. Nul n'ignore que le stress ou des erreurs de manipulation pendant la gestation peuvent engendrer des pertes prénatales. Afin de prévenir des pertes fœtales non liées au traitement, on évitera de manipuler inutilement les femelles gravides ou de les soumettre à des facteurs de stress externes comme le bruit.
On utilise au moins trois doses différentes et un groupe témoin en parallèle. Des animaux sains sont répartis au hasard entre les groupes traités et témoins. Les doses doivent être espacées de façon à produire une gradation des effets toxiques. Sauf limitations imposées par les propriétés physiques, chimiques ou biologiques de la substance d'essai, la dose la plus élevée devrait avoir une certaine toxicité pour le développement et/ou la mère (signes cliniques ou diminution du poids corporel), sans entraîner la mort ni provoquer des souffrances importantes. Au moins une dose intermédiaire devrait donner lieu à la plus faible manifestation observable de toxicité. La dose la plus faible ne devrait produire aucun signe de toxicité pour la mère ou pour le développement. Il convient de choisir une série décroissante doses afin de mettre en évidence une éventuelle relation dose-effet et une concentration maximale sans effet nocif observé (DSET). L'écart optimal entre les doses d'une série décroissante est généralement d'un facteur deux ou quatre et l'adjonction d'un quatrième groupe d'essai est souvent préférable à la fixation d'écarts trop importants (par exemple, supérieurs à un facteur dix). Bien que le but soit d'établir une DSET maternelle, les études qui n'aboutissent pas à la détermination de cette valeur sont également acceptables (1).
Les doses doivent être choisies en tenant compte de toutes les données de toxicité disponibles ainsi que des informations complémentaires sur le métabolisme et la toxicocinétique de la substance d'essai ou de substances apparentées. Ces informations seront aussi utiles pour justifier l'échelle des doses.
On incorporera un groupe témoin en parallèle. La substance d'essai n'est pas administrée à ce groupe qui n'est traité que par le véhicule, le cas échéant. Tous les groupes doivent recevoir le même volume de substance d'essai ou de véhicule. Les animaux du ou des groupes témoins seront manipulés de la même façon que les animaux du groupe d'essai. Les groupes témoins auxquels on administre le véhicule doivent recevoir la plus grande quantité utilisée de ce dernier (celle que reçoit le groupe traité à la dose la plus faible).
1.6.4 Essai limite
Si un essai réalisé à une dose d'au moins 1 000 mg/kg de poids corporel/jour, administrée par voie orale, selon la méthode décrite dans cette étude, n'entraîne aucune toxicité apparente chez les femelles gravides ou dans leur progéniture, et qu'aucun effet n'est escompté au vu des données disponibles (concernant, par exemple, des composés présentant une structure et/ou une action métabolique analogues) il n'est pas indispensable de mener une étude complète sur trois doses différentes. Suivant le niveau d'exposition humaine prévu, il peut être nécessaire d'appliquer une dose orale plus élevée dans l'essai limite. Pour d'autres modes d'administration, tels que l'inhalation ou l'application cutanée, ce sont souvent les propriétés physicochimiques de la substance d'essai qui définissent et limitent le niveau maximal d'exposition pouvant être atteint (par exemple, l'application cutanée ne doit faire apparaître aucune toxicité locale grave).
1.6.5 Administration des doses
La substance d'essai ou le véhicule sont habituellement administrés oralement par intubation. Si l'expérimentateur opte pour un autre mode d'administration, il devra motiver sa décision et justifier son choix, et procéder aux modifications nécessaires (2) (3) (4). La substance d'essai doit être administrée à peu près à la même heure chaque jour.
Normalement, on calcule la dose destinée à chaque animal d'après la pesée la plus récente de ce dernier. Il convient toutefois d'être prudent lorsqu'on adapte la dose au cours du dernier tiers de la gestation. On s'appuiera sur les données existantes pour sélectionner la dose de façon à prévenir un excès de toxicité pour la mère. Néanmoins, si on constate une toxicité excessive chez les mères traitées, il faudra les euthanasier. Si plusieurs femelles gravides manifestent des signes de toxicité excessive, on envisagera de sacrifier le groupe traité à cette dose. Si on recourt au gavage, il faudrait de préférence administrer la substance en une seule fois à l'aide d'une sonde gastrique ou d'une canule appropriée. Le volume maximal de liquide administrable en une seule fois dépend de la taille de l'animal. Ce volume ne devrait pas dépasser 1 ml/100 g de poids corporel, sauf dans le cas des solutions aqueuses où l'on peut aller jusqu'à 2 ml/100 g de poids corporel. Lorsqu'on utilise de l'huile de maïs comme véhicule, le volume ne devrait pas excéder 0,4 ml/100 g de poids corporel. On réduira au minimum la variabilité du volume d'essai en adaptant les concentrations de façon à obtenir un volume constant à toutes les doses.
1.6.6 Observation des mères
Les observations cliniques sont effectuées et notées au moins une fois par jour, de préférence à la ou aux mêmes heures, en tenant compte de la période durant laquelle on prévoit que les effets atteindront leur intensité maximale. On consignera l'état des animaux, notamment, les animaux morts ou moribonds, les changements comportementaux pertinents et tous les signes de toxicité apparents.
1.6.7 Poids corporel et consommation de nourriture
Les animaux sont pesés au jour 0 de gestation ou au plus tard au jour 3 si des animaux déjà accouplés sont fournis par un éleveur extérieur, puis au premier jour de traitement et au moins tous les trois jours durant la période d'administration et enfin le jour du sacrifice.
Le relevé de la consommation d'aliments doit être effectué tous les trois jours et coïncider avec les jours de pesée des animaux.
1.6.8 Autopsie
On sacrifiera les femelles un jour avant la date prévue de mise bas. Les femelles qui présentent des signes d'avortement ou de mise bas prématurée avant la date prévue du sacrifice doivent être sacrifiées et faire l'objet d'un examen macroscopique complet.
Juste après le sacrifice ou la mort en cours de l'étude, les mères sont soumises à un examen macroscopique destiné à mettre en évidence d'éventuels changements pathologiques ou anomalies structurelles. Pour demeurer objectif, il est préférable que l'expérimentateur ignore la dose administrée au groupe lors de l'observation des mères au cours de la césarienne et de l'examen subséquent des fœtus.
1.6.9 Examen du contenu utérin
Il y a lieu d'enlever l'utérus juste après le sacrifice ou dès que possible après la mort pour s'assurer de l'état de gravidité des femelles. On examinera aussi les utérus apparemment non gravides (par exemple, par coloration au sulfure d'ammonium chez les rongeurs et par la coloration de Salewski ou par une méthode équivalente appropriée chez les lapins) pour confirmer la non-gravidité (5).
Les utérus gravides sont pesés, col inclus. Les poids des utérus gravides de femelles mortes en cours d'essai ne sont pas déterminés.
On compte le nombre de corps jaunes chez les femelles gravides.
Le contenu utérin est examiné afin de relever le nombre d'embryons ou de fœtus morts et le nombre de fœtus viables. Il est nécessaire de décrire le degré de résorption pour évaluer la date approximative de la mort du produit de conception (voir paragraphe 1.2).
1.6.10 Examen des fœtus
Il convient de déterminer le sexe et le poids corporel de chaque fœtus.
On recherche la présence d'altérations externes sur chaque fœtus.
On examine les fœtus pour voir s'ils présentent des altérations du squelette ou des tissus mous (par exemple des variations et des malformations ou des anomalies) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17) (18) (19) (20) (21) (22) (23) (24). La catégorisation des altérations fœtales est préférable, mais facultative. Si on procède à une catégorisation, il faut stipuler clairement les critères qui définissent chaque catégorie. On vérifiera avec une attention particulière si le développement du tractus génital n'a pas été altéré.
Pour les rongeurs, on prépare environ la moitié de chaque portée en vue de l'examen des altérations squelettiques. Le restant sera préparé en vue de l'examen des altérations des tissus mous, lequel sera conduit au moyen de coupes sériées réalisées suivant des méthodes reconnues ou appropriées ou de techniques soignées de dissection macroscopique.
S'agissant des non-rongeurs, par exemple les lapins, les éventuelles altérations des tissus mous et du squelette doivent être recherchées sur tous les fœtus. On examine les corps de ces fœtus en les disséquant soigneusement pour repérer des altérations des tissus mous; cette opération peut faire appel à une technique permettant d'effectuer un examen plus approfondi de la structure cardiaque interne (25). Les tètes de la moitié des fœtus examinés de cette manière doivent être prélevées et préparées en vue de l'évaluation des altérations des tissus mous (notamment les yeux, le cerveau, les conduits nasaux et la langue), selon des techniques classiques de coupes sériées (26) ou une méthode ayant la même sensibilité. Les corps de ces fœtus et ceux des fœtus intacts restants sont préparés puis examinés pour établir s'ils présentent des altérations squelettiques, à l'aide des mêmes méthodes que pour les rongeurs.
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
Les résultats sont consignés séparément pour chaque femelle et sa progéniture, et résumés sous forme de tableaux, reprenant pour chaque groupe d'essai et chaque génération, le nombre d'animaux présents au début de l'essai, le nombre d'animaux morts durant l'essai ou euthanasiés, la date de chaque mort ou euthanasie, le nombre de femelles gravides, le nombre d'animaux présentant des signes de toxicité, la description des signes de toxicité observés, notamment le moment où ils ont débuté, leur durée et leur gravité, les types d'observations pratiquées sur les embryons ou fœtus et toutes les données pertinentes sur la portée.
On évaluera les résultats numériques par une méthode statistique appropriée, en prenant la portée comme unité pour l'analyse des résultats. Il convient d'utiliser une méthode statistique reconnue; le choix de la méthode doit intervenir au stade de la conception de l'étude et doit être justifié. Les résultats concernant les animaux qui n'ont pas survécu jusqu'à la date programmée du sacrifice doivent aussi être consignés. Ces résultats peuvent être inclus dans les moyennes des groupes, s'il y a lieu. La pertinence des résultats obtenus pour ces animaux et partant, la décision de les inclure ou non dans une moyenne de groupe, doit être appréciée au cas par cas.
2.2. ÉVALUATION DES RÉSULTATS
Les résultats de l'étude de toxicité pour le développement prénatal doivent être évalués du point de vue des effets observés. L'évaluation doit comprendre les informations suivantes:
|
—
|
les résultats des essais maternels et fœtaux, incluant une évaluation de la relation, ou de l'absence de relation, entre l'exposition des animaux à la substance d'essai et la fréquence et la gravité de tous les effets observés,
|
|
—
|
les critères appliqués pour catégoriser, le cas échéant, les altérations externes des fœtus, et les altérations de leurs tissus mous ou de leur squelette,
|
|
—
|
selon les besoins, les données antérieures relatives aux témoins pour affiner l'interprétation des résultats de l'étude,
|
|
—
|
les nombres utilisés pour calculer tous les pourcentages ou indices,
|
|
—
|
l'analyse statistique appropriée des résultats de l'étude; il convient de fournir suffisamment d'informations sur la méthode d'analyse, pour qu'un réviseur ou un statisticien indépendant puisse réévaluer et reconstruire l'analyse.
|
Pour toute étude ne révélant aucun effet toxique, il y a lieu d'envisager des expériences complémentaires destinées à déterminer l'absorption et la biodisponibilité de la substance d'essai.
2.3. INTERPRÉTATION DES RÉSULTATS
Une étude de la toxicité pour le développement prénatal fournit des informations sur les effets de l'exposition répétée à une substance administrée durant la gestation sur les mères et sur le développement intra-utérin de leur progéniture. Les résultats de cette étude doivent être interprétés à la lumière de ceux des études de toxicité subchronique, de reproduction, des études toxicocinétiques et autres. Comme l'étude est centrée à la fois sur la toxicité générale du point de vue de la mère et sur la toxicité pour le développement, ses résultats permettront dans une certaine mesure de distinguer les effets sur le développement qui surviennent en l'absence d'une toxicité générale, des effets qui n'apparaissent qu'à des doses qui sont également toxiques pour la mère (27).
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit inclure les informations particulières énumérées ci-dessous :
|
|
Substance d'essai:
|
—
|
état physique et, s'il y a lieu, propriétés physico-chimiques,
|
|
—
|
identification, y compris le numéro du CAS s'il est connu,
|
|
|
|
Véhicule (le cas échéant):
|
—
|
justification du choix du véhicule s'il diffère de l'eau.
|
|
|
|
Animaux d'expérience:
|
—
|
espèce et souche utilisée,
|
|
—
|
nombre et sexe des animaux,
|
|
—
|
origine, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
poids de chaque animal au début de l'essai.
|
|
|
|
Conditions d'essai:
|
—
|
justification du choix des doses appliquées,
|
|
—
|
détails concernant la préparation de la substance d'essai ou son incorporation aux aliments, la concentration obtenue, la stabilité et l'homogénéité de la préparation,
|
|
—
|
détails sur l'administration de la substance d'essai,
|
|
—
|
conversion de la concentration de la substance d'essai (ppm) dans la nourriture ou l'eau de boisson en dose administrée (mg/kg de poids corporel/jour), s'il y a lieu,
|
|
—
|
détails concernant la qualité de l'alimentation et de l'eau de boisson.
|
|
|
|
Données relatives à la toxicité pour la mère en fonction des doses, précisant entre autres:
|
—
|
le nombre d'animaux au début de l'essai, le nombre de survivants, le nombre de femelles gravides, le nombre d'avortements et de mises bas prématurées,
|
|
—
|
le jour de la mort si celle-ci est intervenue en cours d'essai et le nombre des animaux ayant survécu jusqu'au jour du sacrifice,
|
|
—
|
les résultats concernant les animaux qui n'ont pas survécu jusqu'à la date du sacrifice doivent être consignés, mais non inclus dans les comparaisons statistiques entre les groupes,
|
|
—
|
le jour de l'observation de chaque signe clinique anormal et son évolution ultérieure,
|
|
—
|
le poids corporel, sa variation et le poids de l'utérus gravide, y compris, si on le souhaite, la variation du poids corporel corrigée en fonction du poids de l'utérus gravide,
|
|
—
|
la consommation de nourriture, et d'eau si elle a été mesurée,
|
|
—
|
les résultats de l'autopsie, y compris le poids de l'utérus,
|
|
—
|
les valeurs de la DSET se rapportant aux effets sur la mère et sur le développement.
|
|
|
|
Données concernant les effets sur le développement en fonction des doses, pour les portées attestées par des implantations, notamment:
|
—
|
nombre de corps jaunes,
|
|
—
|
nombre d'implantations, nombre et pourcentage de fœtus vivants et morts et nombre de résorptions,
|
|
—
|
nombre et pourcentage de pertes pré- et postimplantatoires.
|
|
|
|
Données concernant les effets sur le développement en fonction des doses, pour les portées comportant des fœtus vivants, notamment:
|
—
|
nombre et pourcentage de descendants vivants,
|
|
—
|
proportion de mâles et de femelles,
|
|
—
|
poids corporel des fœtus, de préférence par sexe et pour les deux sexes confondus;
|
|
—
|
malformations externes, des tissus mous et du squelette et autres altérations pertinentes,
|
|
—
|
critères de catégorisation, le cas échéant,
|
|
—
|
nombre total et pourcentage de fœtus et de portées présentant une quelconque altération externe, des tissus mous ou du squelette; types et fréquence des différentes anomalies et autres altérations pertinentes.
|
|
4. BIBLIOGRAPHIE
|
(1)
|
Kavlock R.J. et al. (1996) A Simulation Study of the Influence of Study Design on the Estimation of Benchmark Doses for Developmental Toxicity. Risk Analysis 16; 399-410.
|
|
(2)
|
Kimmel, CA. and Francis, E.Z. (1990) Proceedings of the Workshop on the Acceptability and Interpretation of Dermal Developmental Toxicity Studies. Fundamental and Applied Toxicology 14; 386-398.
|
|
(3)
|
Wong, B.A., et al. (1997) Developing Specialized Inhalation Exposure Systems to Address Toxicological Problems. CUT Activities 17; 1-8.
|
|
(4)
|
US Environmental Protection Agency (1985) Subpart E-Specific Organ/Tissue Toxicity, 40 CFR 798.4350: Inhalation Developmental Toxicity Study.
|
|
(5)
|
Salewski, E. (1964) Faerbermethode zum Makroskopischen Nachweis von Implantations Stellen am Uterusder Ratte. Naunyn-Schmeidebergs Archiv fur Pharmakologie und Experimentelle Pathologie 247:367.
|
|
(6)
|
Edwards, J.A. (1968) The external Development of the Rabbit and Rat Embryo. In Advances in Teratology. D.H.M. Woolam (ed.) Vol. 3. Academic Press, NY.
|
|
(7)
|
Inouye, M. (1976) Differential Staining of Cartilage and Bone in Fetal Mouse Skeleton by Alcian Blue and Alizarin Red S. Congenital Anomalies 16; 171-173.
|
|
(8)
|
Igarashi, E. et al. (1992) Frequency Of Spontaneous Axial Skeletal Variations Detected by the Double Staining Techniquefor Ossified and Cartilaginous Skeleton in Rat Foetuses. Congenital Anomalies 32: 381-391.
|
|
(9)
|
Kimmel, C.A. et al. (1993) Skeletal Development Following Heat Exposure in the Rat. Teratology 47:229-242..
|
|
(10)
|
Marr, M.C. et al. (1988) Comparison of Single and Double Staining for Evaluation of Skeletal Development: The Effects of Ethylene Glycol (EG) in CD Rats. Teratology 37; 476.
|
|
(11)
|
Barrow, M.V. and Taylor, W.J. (1969) A Rapid Method for Detecting Malformations in Rat Foetuses. Journal of Morphology 127:291-306.
|
|
(12)
|
Fritz, H. (1974) Prenatal Ossification in Rabbits ss Indicative of Foetal Maturity. Teratology 11; 313-320.
|
|
(13)
|
Gibson, J.P. et al. (1966) Use of the Rabbit in Teratogenicity Studies. Toxicology and Applied Pharmacology 9; :398-408.
|
|
(14)
|
Kimmel, CA. and Wilson, J.G. (1973) Skeletal Deviation in Rats: Malformations or Variations? Teratology 8; 309-316.
|
|
(15)
|
Marr, M.C. et al. (1992) Developmental Stages of the CD (Sprague-Dawley) Rat Skeleton after Maternal Exposure to Ethylene Glycol. Teratology 46; 169-181.
|
|
(16)
|
Monie, I.W. et al. (1965) Dissection Procedures for Rat Foetuses Permitting Alizarin Red Staining of Skeleton and Histological Study of Viscera. Supplement to Teratology
Workshop Manual, pp. 163-173.
|
|
(17)
|
Spark, C. and Dawson, A.B. (1928) The Order and Time of appearance of Centers of Ossification in the Fore and Hind Limbs of the Albino Rat, with Special Reference to the Possible Influence of the Sex Factor. American Journal of Anatomy 41; 411-445.
|
|
(18)
|
Staples, R.E. and Schnell, V.L. (1964) Refinements in Rapid Clearing Technique in the KOH-Alizarin Red S Method for Fetal Bone. Stain Technology 39; 61-63.
|
|
(19)
|
Strong, R.M. (1928) The Order Time and Rate of Ossification of the Albino Rat (Mus Norvegicus Albinus) Skeleton. American Journal of Anatomy 36; 313-355.
|
|
(20)
|
Stuckhardt, J.L. and Poppe, S.M. (1984) Fresh Visceral Examination of Rat and Rabbit Foetuses Used in Teratogenicity Testing. Teratogenesis, Carcinogenesis, and Mutagenesis 4; 181-188.
|
|
(21)
|
Walker, D.G. and Wirtschafter, Z.T. (1957) The Genesis of the Rat Skeleton. Thomas, Springfield, IL.
|
|
(22)
|
Wilson, J.G. (1965) Embryological Considerations in Teratology. In Teratology: Principles and Techniques, Wilson J.G. and Warkany J. (eds). University of Chicago, Chicago, IL, pp 251-277.
|
|
(23)
|
Wilson, J.G. and Fraser, F.C. (eds). (1977) Handbook of Teratology, Vol. 4. Plenum, NY.
|
|
(24)
|
Varnagy, L. (1980) Use of Recent Fetal Bone Staining Techniques in the Evaluation of Pesticide Teratogenicity. Acta Vet. Acad. Sci. Hung. 28; 233-239.
|
|
(25)
|
Staples, R.E. (1974) Detection of visceral Alterations in Mammalian Foetuses. Teratology 9; 37-38.
|
|
(26)
|
Van Julsingha, E.B. and C.G. Bennett (1977) A Dissecting Procedure for the Detection of Anomalies in the Rabbit Foetal Head. In: Methods in Prenatal Toxicology Neubert, D., Merker, H.J. and Kwasigroch, T.E. (eds.). University of Chicago, Chicago, IL, pp. 126-144.
|
|
(27)
|
US Environmental Protection Agency (1991) Guidelines for Developmental Toxicity Risk Assessment. Federal Register 56; 63798-63826.
|
|
(28)
|
Wise, D.L. et al. (1997) Terminology of Developmental Abnormalities in Common Laboratory Mammals (Version 1) Teratology 55; 249-292.
|
B.32 ÉTUDE DE CANCÉROGÉNÈSE
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance à tester est administrée normalement 7 jours sur 7, par une voie appropriée, a plusieurs groupes d'animaux d'expérience, à raison d'une dose par groupe, pendant la majeure partie de leur vie. Durant l'exposition à la substance à tester et après celle-ci, les animaux d'expérience sont observés quotidiennement afin de déceler les signes de toxicité, en particulier la formation de tumeurs.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Les animaux sont maintenus dans les conditions expérimentales d'encagement et d'alimentation pendant au moins 5 jours avant le début de l'étude. Avant de commencer l'étude, les animaux jeunes et en bonne santé sont randomisés et répartis entre le nombre de groupes requis.
Animaux d'expérience
L'espèce préférée est le rat.
Compte tenu des résultats d'études antérieures, d'autres espèces (rongeurs ou autres) peuvent être utilisées. De jeunes animaux sains de souches courantes de laboratoire doivent être utilisés et le traitement doit commencer dès que possible après sevrage.
Au début de l'étude, la différence de poids entre les animaux utilisés ne doit pas dépasser ±20 % de la valeur moyenne. Si un essai de toxicité subchronique par voie orale a été réalisé antérieurement à l'essai à long terme, la même espèce, race/souche doit être utilisée pour les deux essais.
Nombre et sexe
S'agissant de rongeurs, au moins 100 animaux (50 femelles et 50 mâles) doivent être utilisés pour chaque niveau de dose et chaque groupe témoin correspondant. Les femelles doivent être nullipares et non gravides. Si l'on prévoit des sacrifices d'animaux en cours d'expérience, les effectifs doivent être augmentés en conséquence du nombre d'animaux qu'il est prévu de sacrifier avant la fin de l'expérience.
Doses et fréquence d'exposition
Outre les groupes témoins correspondants, trois niveaux de dose au moins devraient être utilisés. La dose supérieure devrait être suffisamment élevée pour montrer un effet de toxicité minimale tel qu'un faible ralentissement de la croissance du poids corporel (moins de 10 %), sans altération notable de la durée de vie normale due à des effets autres que les tumeurs.
La dose la plus faible n'aura aucun effet sur la croissance, le développement et la longévité normale de l'animal et ne produira aucun effet toxique quelconque. En général, cette dose ne sera pas inférieure à 10 % de la dose élevée.
La dose (les doses) intermédiaire(s) aura (auront) une valeur proche de la moyenne entre la dose forte et la dose faible.
Les niveaux de dose prendront en compte les données fournies par les essais et les études de toxicité effectués antérieurement.
L'exposition est de façon générale quotidienne.
Si le produit chimique est administré dans l'eau de boisson ou incorporé à la nourriture, il devra être constamment disponible.
Témoins
Les groupes témoins devront être en tout point identiques aux groupes traités, exception faite de l'exposition à la substance à tester.
Dans des conditions particulières, comme au cours des études par inhalation impliquant l'emploi d'aérosols ou l'utilisation d'un émulsifiant à activité biologique non étudiée au cours d'études par voie orale, on utilisera un groupe témoin supplémentaire non exposé au véhicule.
Voie d'administration
Les trois principales voies d'administration sont la voie orale, la voie cutanée (percutanée) et la voie respiratoire (inhalation). Le choix de la voie d'administration dépend des caractéristiques physiques et chimiques de la substance à tester ainsi que de la voie probable d'exposition humaine.
Études par voie orale:
La voie orale d'administration est préférée, sauf contre-indications, si la substance est absorbée par voie gastro-intestinale, et si l'ingestion est une voie d'exposition humaine. Les animaux devront recevoir la substance à tester dans leur nourriture, dissoute dans l'eau de boisson ou sous la forme de capsule.
Dans les conditions idéales, la dose quotidienne sera administrée 7 jours sur 7 car l'administration 5 jours sur 7 peut permettre à l'animal de récupérer ou à la toxicité de décroître durant la période où le traitement est interrompu et affecter, par conséquent, les résultats et l'évaluation ultérieure. Cependant, pour des raisons essentiellement pratiques, on peut accepter que la substance soit administrée 5 jours sur 7.
Études par voie cutanée:
L'exposition cutanée par badigeonnage de la peau peut être retenue pour simuler une voie d'exposition importante pour l'homme et comme une façon d'induire des lésions cutanées.
Études d'inhalation:
Les études d'inhalation posant des problèmes techniques beaucoup plus complexes que les autres voies d'administration, des recommandations plus détaillées sont données ici. Il convient également de noter que l'instillation intratrachéale peut constituer une méthode valable dans certains cas spécifiques.
Les expositions prolongées sont habituellement établies en fonction de l'exposition humaine possible: les animaux sont soit exposés 5 jours sur 7 (exposition intermittente) à raison de 6 heures par jour après équilibrage des concentrations dans la cage d'expérimentation soit, si une exposition environnementale est possible chez l'homme, exposés 7 jours sur 7 (exposition continue) à raison de 22 à 24 heures par jour, une heure par jour environ étant consacrée à l'alimentation des animaux, selon un horaire régulier, et à l'entretien des cages d'expérimentation. Dans les deux cas, les animaux sont habituellement exposés à une concentration fixe de produits à tester. Une différence essentielle entre l'exposition intermittente et l'exposition continue réside dans le fait que dans le premier cas les animaux disposent d'une période de 17 à 18 heures pour récupérer des effets de l'exposition quotidienne et même d'une période plus longue durant les week-ends.
L'exposition intermittente ou permanente sera choisie en fonction des objectifs de l'étude et de l'exposition de l'être humain qui doit être simulée. Il convient toutefois de tenir compte de certaines difficultés techniques: par exemple, les avantages qu'offre l'exposition continue pour simuler les conditions ambiantes peuvent être contrebalancés par la nécessité d'alimenter et de faire boire les animaux durant l'exposition ainsi que par les contraintes inhérentes à la complexité (et fiabilité) de la génération des aérosols ou des vapeurs et des moyens de contrôle.
Enceintes d'exposition
Les animaux seront exposés à la substance à tester dans des cages d'inhalation conçues pour assurer un flux d'air dynamique d'au moins 12 renouvellements de l'air par heure, une teneur en oxygène appropriée et une répartition uniforme du produit à tester dans l'air. Les enceintes d'exposition et les enceintes témoins seront identiques du point de vue de la construction et de la conception afin de garantir des conditions d'exposition en tous points comparables, exception faite de l'exposition aux substances à tester. Une pression légèrement négative est généralement maintenue à l'intérieur de la chambre afin d'empêcher les fuites de la substance à tester dans la zone environnante. Les enceintes seront conçues de manière à réduire l'entassement des animaux d'expérience. En règle générale, pour assurer la stabilité de l'atmosphère de l'enceinte, le volume total des animaux d'expérience ne doit pas dépasser 5 % du volume de l'enceinte d'exposition.
Mesures et contrôles à effectuer:
|
(i)
|
le débit d'air: le débit d'air dans la chambre fera, de préférence, l'objet d'un contrôle continu;
|
|
(ii)
|
concentration: durant la période d'exposition quotidienne, la concentration de la substance à tester ne variera pas de ± 15 % de la valeur moyenne. Durant toute la durée de l'étude, les concentrations quotidiennes seront maintenues aussi constantes que possible;
|
|
(iii)
|
température et humidité: pour les rongeurs, la température sera maintenue à 22 oC (± 2 oC) et l'humidité à l'intérieur de l'enceinte sera de 30 à 70 %, sauf lorsqu'on utilise de l'eau pour mettre la substance à tester en suspension dans l'air de la cage d'exposition. Ces deux paramètres seront, de préférence, contrôlés en permanence;
|
|
(iv)
|
analyse granulométrique des particules: une répartition granulométrique des particules sera effectuée pour les atmosphères des chambres où des aérosols liquides ou solides sont utilisés. Les particules de l'aérosol seront d'une taille inhalable pour l'animal d'expérience utilisé. Des échantillons de l'air des cages d'exposition seront prélevés à la hauteur des voies respiratoires des animaux. L'échantillon d'air prélevé sera représentatif de la répartition des particules auxquelles les animaux sont exposés et représentera, sur une base gravimétrique, l'ensemble de l'aérosol en suspension, même si une grande partie de celui-ci n'est pas inhalable. Les analyses granulométriques seront effectuées fréquemment durant la mise au point du système de génération afin de veiller à la stabilité de l'aérosol, et ensuite aussi souvent que nécessaire pendant les expositions pour déterminer de façon adéquate la stabilité de la répartition des particules auxquelles les animaux ont été exposés.
|
Durée de l'étude
Un essai de cancérogénicité couvre la majeure partie de la durée de vie normale des animaux d'expérience: 18 mois pour la souris et le hamster et 24 mois pour le rat; cependant pour certaines souches d'animaux ayant une plus grande longévité et/ou un faible taux de tumeur spontanée, l'essai sera de 24 mois pour la souris et le hamster et de 30 mois pour le rat. Il peut également être mis fin à une telle étude lorsque le nombre de survivants dans le groupe exposé à la dose la plus faible ou dans le groupe témoin atteint 25 %. S'il y a manifestement une réponse différente par sexe, chaque sexe sera considéré comme faisant l'objet d'une étude distincte. Lorsque seuls les sujets du groupe exposé à la dose élevée meurent prématurément pour des raisons de toxicité évidentes, il n'est pas nécessaire de mettre fin à l'essai à condition que les manifestations toxiques ne posent pas de problèmes dans les autres groupes. Pour qu'un résultat d'essai négatif soit acceptable, il faut que, par lot, il n'y ait pas plus de 10 % d'animaux perdus pour l'étude en raison d'autolyse, de cannibalisme ou de problèmes organisationnels et que le taux de survie dans tous les lots ne soit pas inférieur à 50 % après 18 mois en ce qui concerne la souris et le hamster et après 24 mois en ce qui concerne le rat.
Mode opératoire
Observations
Les observations quotidiennes sur les animaux en cage porteront notamment sur les modifications de la peau et du poil, des yeux et des muqueuses, de l'appareil respiratoire, du système circulatoire, du système nerveux autonome et central, de l'activité somatomotrice ainsi que du comportement.
Il convient de surveiller régulièrement les animaux afin d'éviter, autant que possible, toute perte pour des raisons telles que cannibalisme, autolyse des tissus ou erreur d'encagement. Les animaux moribonds seront immédiatement mis à part et autopsiés.
Les signes cliniques ainsi que la mortalité seront notés pour tous les animaux. On attachera une attention spéciale à la formation de tumeur; le moment d'apparition, la localisation, les dimensions, l'apparence et l'évolution de toute tumeur nettement visible ou palpable seront notées.
La consommation alimentaire (ainsi que la consommation d'eau lorsque la substance à tester est administrée dans l'eau de boisson) sera déterminée chaque semaine durant les 13 premières semaines de l'étude, puis tous les trois mois environ, à moins que l'état de santé ou les modifications de poids corporel nécessitent une autre fréquence.
Les poids corporels seront relevés individuellement pour tous les animaux une fois par semaine durant les 13 premières semaines de l'étude, puis une fois toutes les 4 semaines au moins.
Examen clinique
Hématologie
Si les observations effectuées pendant la période d'encagement indiquent une détérioration de l'état de santé de certains animaux au cours de l'étude, on établira la formule sanguine des animaux affectés.
Un frottis sanguin est effectué pour tous les animaux au douzième et au dix-huitième mois ainsi qu'avant de les sacrifier. La formule sanguine est établie à partir des échantillons prélevés sur les témoins ainsi que sur les animaux du groupe exposé à la dose la plus élevée. Si ces données, en particulier celles obtenues avant de sacrifier les animaux, ou les données provenant de l'examen histopathologique en font apparaître le besoin, les formules sanguines sont également établies pour les animaux appartenant aux groupes exposés aux doses immédiatement inférieures.
Autopsie
Une autopsie complète sera effectuée pour tous les animaux, y compris ceux morts en cours d'expérience ou sacrifiés à l'état moribond. Tous les organes ou tissus présentant des tumeurs ou des lésions nettement visibles ou des lésions soupçonnées d'être des tumeurs seront conservés.
Les organes et tissus ci-après seront conservés dans un milieu approprié permettant d'éventuels examens histopathologiques ultérieurs: l'encéphale (y compris sections de la moelle/du pont de Varole, cortex cérébelleux et cortex cérébral), hypophyse, tyroïde/parathyroïde, tout tissu thymique, trachée et poumons, cœur, aorte, glandes salivaires, foie, rate, reins, glandes surrénales, pancréas, gonades, utérus, organes génitaux annexes, peau, œsophage, estomac, duodénum, jéjunum, iléon, caecum, colon, rectum, vessie, ganglions lymphatiques représentatifs, glande mammaire femelle, musculature de la cuisse, nerf périphérique, sternum avec moelle osseuse, fémur (y compris les articulations), Mlle épinière à trois niveaux (cervical, médiothoracique et lombaire), yeux.
L'insufflation d'un fixateur dans les poumons et la vessie constitue la meilleure façon de conserver ces tissus; l'insufflation des poumons est absolument nécessaire dans les études d'inhalation si l'on veut procéder à des examens histopathologiques appropriés. Dans les études par inhalation, tout le tractus respiratoire sera conservé, y compris la cavité nasale, le pharynx et le larynx.
Examen histopathologique
|
a)
|
Les organes et tissus de tous les animaux morts ou sacrifiés en cours d'expérience ainsi que de tous les animaux du groupe témoin et du groupe exposé à la dose élevée seront soumis à un examen histopathologique complet.
|
|
b)
|
Toutes les tumeurs nettement visibles ou les lésions soupçonnées d'être des tumeurs dans tous les groupes feront l'objet d'un examen microscopique.
|
|
c)
|
Si l'on observe une différence significative dans l'incidence des lésions néoplasiques entre le groupe exposé à la dose la plus élevée et le groupe témoin, un examen histopathologique sera effectué pour l'organe ou le tissu concerné dans les autres groupes.
|
|
d)
|
Si le taux de survie dans le groupe exposé à la dose élevée est nettement inférieur à celui observé pour le groupe témoin, les animaux du groupe exposé à la dose immédiatement inférieure seront soumis à un examen complet.
|
|
e)
|
Si, dans le groupe exposé à la dose élevée, on observe une induction d'effets toxiques ou autres susceptibles d'affecter la réponse néoplasique, les animaux exposés à la dose immédiatement inférieure feront l'objet d'un examen complet.
|
2. RÉSULTATS
Les données seront résumées sous forme de tableaux indiquant, pour chaque lot d'expérience, le nombre d'animaux au début de l'essai, le nombre d'animaux atteints de tumeurs décelées en cours d'expérience, le moment où celles-ci sont décelées et le nombre d'animaux sur lesquels des tumeurs sont observées à l'autopsie. Les résultats seront évalués au moyen d'une méthode statistique appropriée. Toute méthode statistique validée peut être utilisée.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
espèces souche, origine, milieu ambiant, régime alimentaire, etc.,
|
|
—
|
conditions d'essai:
description du système d'exposition:
y compris conception, type, dimensions, source d'air, système générateur de particules et d'aérosols, méthode de conditionnement d'air, traitement de l'air expiré et, le cas échéant, mode de stabulation des animaux en chambre d'essai. L'équipement utilisé pour mesurer la température, l'humidité et, si nécessaire, la stabilité des concentrations d'aérosol ou la granulométrie seront décrits;
données relatives à l'exposition:
elles seront présentées sous forme de tableaux indiquant des valeurs moyennes ainsi qu'une mesure de la variabilité (par exemple écart type) et porteront sur:
|
a)
|
les débits d'air à travers le dispositif d'inhalation;
|
|
b)
|
la température et l'humidité de l'air;
|
|
c)
|
les concentrations nominales (quantité totale de substance à tester introduite dans le dispositif d'inhalation, divisée par le volume d'air);
|
|
d)
|
le cas échéant, la nature du véhicule;
|
|
e)
|
les concentrations réelles dans la zone de respiration;
|
|
f)
|
les dimensions médianes des particules (si nécessaire),
|
|
|
—
|
niveaux de dose (y compris, le cas échéant, véhicule) et concentrations,
|
|
—
|
incidence des tumeurs par sexe, dose et type de tumeur,
|
|
—
|
moment de la mort en cours d'expérience ou indication que l'animal était encore en vie à la fin de l'expérience,
|
|
—
|
réponse toxique par sexe et dose,
|
|
—
|
description des effets toxiques ou autres,
|
|
—
|
le moment de l'observation de tout signe anormal et évolution de celui-ci,
|
|
—
|
consommation alimentaire et poids corporel,
|
|
—
|
résultats de l'examen hématologique,
|
|
—
|
description détaillée de toutes les observations histopathologiques,
|
|
—
|
traitement statistique des résultats et description des méthodes utilisées,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.33 ÉTUDE COMBINÉE DE CANCÉROGÉNÈSE ET DE TOXICITÉ CHRONIQUE
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Le but d'une étude combinée de la toxicité chronique et de la cancérogénèse est de déterminer les effets chroniques et cancérogènes d'une substance sur une espèce mammifère à l'issue d'une exposition prolongée (à long terme).
C'est pourquoi une étude de cancérogénèse est complétée par au moins un groupe satellite traité et un groupe satellite témoin. La dose utilisée pour le groupe satellite soumis à la dose élevée peut être supérieure à celle utilisée pour le groupe soumis à la dose élevée dans l'étude de cancérogénèse. Dans cette dernière étude, les animaux sont examinés du point de vue de la toxicité générale ainsi que de la réponse cancérogène. Les animaux du groupe satellite traité sont examinés du point de vue de la toxicité générale.
La substance à tester est normalement administrée 7 jours sur 7 par une voie appropriée, à plusieurs groupes d'animaux d'expérience, à raison d'une dose par groupe, pendant la majeure partie de leur vie. Au cours et après l'exposition à la substance, les animaux d'expérience sont observés quotidiennement afin de déceler les signes de toxicité et le développement de tumeurs.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE
Les animaux sont maintenus dans les conditions expérimentales de stabulation et d'alimentation pendant au moins 5 jours avant l'expérience. Avant de commencer l'essai, les jeunes animaux sains sont randomisés et répartis entre les groupes requis (traités et témoins).
Animaux d'expérience
L'espèce préférée est le rat. Compte tenu des résultats d'études antérieures, d'autres espèces (rongeurs ou autres) peuvent être utilisées. De jeunes animaux sains de souches courantes de laboratoire doivent être utilises et le traitement doit commencer dès que possible après sevrage.
Au début de l'étude, la différence de poids entre les animaux utilisés ne doit pas dépasser ± 20 % de la valeur moyenne. Si un essai de toxicité subchronique par voie orale a été réalisé à titre préliminaire à l'essai à long terme, la même espèce, race/souche doit être utilisée pour les deux essais.
Nombre et sexe
S'agissant de rongeurs, au moins 100 animaux (50 femelles et 50 mâles) seront utilisés pour chaque niveau de dose et chaque groupe témoin correspondant. Les femelles seront nullipares et non gravides. Si l'on prévoit des sacrifices d'animaux en cours d'expérience, il y a lieu d'augmenter les effectifs en conséquence.
Le(s) groupe(s) satellite(s) traité(s) pour évaluer les effets pathologiques autres que les tumeurs comprendra (ont) 20 animaux de chaque sexe, tandis que le groupe satellite témoin comprendra 10 animaux de chaque sexe.
Doses et fréquence d'exposition
Pour l'étude de la cancérogénèse, trois doses au moins seront utilisées, outre les groupes témoins correspondants. La dose supérieure devrait être suffisamment élevée pour montrer un effet de toxicité minimale tel qu'un faible ralentissement de la croissance du poids corporel (moins de 10 %), sans altération notable de la durée de vie normale due à des effets autres que les tumeurs.
La dose la plus faible n'aura aucun effet sur la croissance, le développement et la longévité normale de l'animal et ne produira aucun effet toxique. En général, cette dose ne sera pas inférieure à 10 % de la dose élevée.
La (les) dose(s) intermédiaire(s) aura (auront) une valeur proche de la moyenne entre la dose forte et la dose faible.
Les niveaux de dose prendront en compte les données fournies par les essais et les études de toxicité effectués antérieurement.
Pour l'étude de la toxicité chronique, on inclut dans l'étude d'autres groupes traités ainsi qu'un groupe satellite témoin correspondant. Pour les animaux du groupe satellite traité, la dose élevée sera choisie de manière à produire un effet toxique certain.
L'exposition est de façon générale quotidienne.
Si le produit chimique est administré dans l'eau de boisson ou incorporé à la nourriture, il devra être constamment disponible.
Témoins
Le groupe témoin sera en tous points identique au groupe traité, exception faite de l'exposition à la substance à tester.
Dans des conditions particulières, comme au cours des études par inhalation impliquant l'emploi d'aérosols ou l'utilisation d'un émulsifiant à activité biologique non étudiée au cours d'études par voie orale, on utilisera un groupe témoin supplémentaire non exposé au véhicule.
Voie d'administration
Les trois principales voies d'administration sont la voie orale, la voie dermique et la voie respiratoire (inhalation). Le choix de la voie d'administration dépend des caractéristiques physiques et chimiques de la substance à tester ainsi que du mode probable de l'exposition humaine.
Études par voie orale:
La voie orale d'administration est préférée, sauf contre-indications, si la substance est absorbée par voie gastro-intestinale, et si l'ingestion est une voie d'exposition humaine. La substance à traiter sera administrée aux animaux dans leur nourriture, dissoute dans l'eau de boisson ou sous la forme de capsules.
Dans les conditions idéales, la dose quotidienne sera administrée 7 jours sur 7, car l'administration 5 jours sur 7 peut permettre à l'animal de récupérer ou à la toxicité de décroître durant la période où le traitement est interrompu et affecte, par conséquent, les résultats et l'évaluation ultérieure. Toutefois, pour des raisons essentiellement pratiques, on peut accepter que la substance soit administrée 5 jours sur 7.
Études par voie dermique:
L'exposition cutanée par badigeonnage de la peau peut être retenue pour simuler une voie d'exposition importante pour l'homme et comme une façon d'induire des lésions cutanées.
Études d'inhalation:
Les études d'inhalation posant des problèmes techniques beaucoup plus complexes que les autres voies d'administration, des recommandations plus détaillées sont formulées ici. Il convient également de noter que l'instillation intratrachéale peut constituer une autre méthode valable dans certains cas spécifiques.
Les expositions prolongées sont habituellement établies en fonction de l'exposition humaine possible: les animaux sont soit exposés 5 jours sur 7 (exposition intermittente) à raison de 6 heures par jour après équilibrage des concentrations dans la cage d'expérimentation soit, si une exposition environnementale est possible chez l'homme, exposés 7 jours sur 7 (exposition continue) à raison de 22 à 24 heures par jour, une heure par jour environ étant consacrée à l'alimentation des animaux, selon un horaire régulier, et à l'entretien des cages d'expérimentation. Dans les deux cas, les animaux sont habituellement exposés à une concentration fixe de produits à tester. Une différence essentielle entre l'exposition intermittente et l'exposition continue réside dans le fait que dans le premier cas les animaux disposent d'une période de 17 à 18 heures pour récupérer des effets de l'exposition quotidienne et même d'une période plus longue durant les week-ends.
L'exposition intermittente ou permanente sera choisie en fonction des objectifs de l'étude et de l'exposition de l'être humain qui doit être simulée. Il convient toutefois de tenir compte de certaines difficultés techniques: par exemple, les avantages qu'offre l'exposition continue pour simuler les conditions ambiantes peuvent être contrebalancées par la nécessité d'alimenter et de faire boire les animaux durant l'exposition ainsi que par les contraintes inhérentes à la complexité (et fiabilité) de la génération des aérosols ou des vapeurs et des moyens de contrôle.
Enceintes d'exposition
Les animaux seront exposés à la substance à tester dans des cages d'inhalation conçues pour assurer un flux d'air dynamique d'au moins 12 renouvellements de l'air par heure, une teneur en oxygène appropriée et une répartition uniforme du produit à tester dans l'air. Les enceintes d'exposition et les enceintes témoins seront identiques du point de vue de la construction et de la conception afin de garantir des conditions d'exposition en tous points comparables, exception faite de l'exposition aux substances à tester. Une pression légèrement négative est généralement maintenue à l'intérieur de la chambre afin d'empêcher des fuites de la substance à tester dans la zone environnante. Les enceintes seront conçues de manière à réduire l'entassement des animaux d'expérience. En règle générale, pour assurer la stabilité de l'atmosphère de l'enceinte, le volume total des animaux d'expérience ne doit pas dépasser 5 % du volume de l'enceinte d'exposition.
Mesures et contrôles à effectuer:
|
(i)
|
le débit d'air: le débit d'air dans la chambre fera, de préférence, l'objet d'un contrôle continu;
|
|
(ii)
|
concentration: durant la période d'exposition quotidienne, la concentration de la substance à tester ne variera pas de ± 15 % de la valeur moyenne. Durant toute la durée de l'étude, les concentrations quotidiennes seront maintenues aussi constantes que possible;
|
|
(iii)
|
température et humidité: pour les rongeurs, la température sera maintenue à 22 oC (± 2 oC) et l'humidité à l'intérieur de l'enceinte sera de 30 à 70 %, sauf lorsqu'on utilise de l'eau pour mettre la substance à tester en suspension dans l'air de la cage d'exposition. Ces deux paramètres seront, de préférence, contrôlés en permanence;
|
|
(iv)
|
analyse granulométrique des particules: une répartition granulométrique des particules sera effectuée pour les atmosphères des chambres où des aérosols liquides ou solides sont utilisés. Les particules de l'aérosol seront d'une taille inhalable pour l'animal d'expérience utilisé. Des échantillons de l'air des cages d'exposition seront prélevés à la hauteur des voies respiratoires des animaux. L'échantillon d'air prélevé sera représentatif de la répartition des particules auxquelles les animaux sont exposés et représentera, sur une base gravimétrique, l'ensemble de l'aérosol en suspension, même si une grande partie de celui-ci n'est pas inhalable. Les analyses granulométriques seront effectuées fréquemment durant la mise au point du système générateur afin de veiller à la stabilité de l'aérosol, et ensuite aussi souvent que nécessaire pendant les expositions pour déterminer de façon adéquate la stabilité de la répartition des particules auxquelles les animaux ont été exposés.
|
Durée de l'étude
La partie de l'essai portant sur la cancérogénèse couvre la majeure partie de la durée de vie normale des animaux d'expérience. Elle durera 18 mois pour la souris et le hamster et 24 mois pour le rat; toutefois, pour certaines souches d'animaux ayant une plus grande longévité et/ou un faible taux de tumeurs spontanées, la durée sera de 24 mois pour la souris et le hamster et de 30 mois pour le rat. Il peut également être mis fin à une telle étude prolongée lorsque dans le groupe exposé à la dose la plus élevée ou dans le groupe témoin le nombre de survivants atteint 25 %. Lorsqu'il y a manifestement une réponse différente par sexe, chaque sexe sera considéré comme faisant l'objet d'une étude distincte. Lorsque seuls les sujets du groupe exposé à la dose la plus élevée meurent prématurément pour des raisons évidentes de toxicité, il n'est pas nécessaire de mettre fin à l'essai si les manifestations toxiques ne posent pas de problèmes dans les autres groupes. Pour qu'un résultat d'essai négatif soit acceptable, il faut que, par lot, il n'y ait pas plus de 10 % d'animaux perdus pour l'étude, pour cause d'autolyse, de cannibalisme ou de problèmes organisationnels et que le taux de survie dans tous les lots ne soit pas inférieur à 50 % après 18 mois en ce qui concerne la souris et le hamster et après 24 mois en ce qui concerne le rat.
Les groupes satellites de 20 animaux par sexe traités ainsi que les 10 animaux par sexe témoins qui y sont associés, utilisés pour l'essai de toxicité chronique seront conservés dans l'étude pendant au moins 12 mois. Ces animaux sont destinés à être sacrifiés afin d'évaluer la pathologie liée à la substance à tester en dehors de toute modification imputable au vieillissement.
Mode opératoire
Observations
Les observations quotidiennes sur les animaux en cage porteront notamment sur les modifications de la peau et du poil, des yeux et des muqueuses, de l'appareil respiratoire, du système circulatoire, du système nerveux autonome et central, de l'activité somatomotrice et du comportement.
Les animaux du (des) groupe(s) satellite(s) traité(s) seront soumis à un examen clinique à intervalles appropriés.
Il est nécessaire d'observer régulièrement les animaux pour éviter, autant que possible, toute perte pour des raisons telles que cannibalisme, autolyse des tissus ou erreur d'encagement. Les animaux moribonds seront immédiatement mis à part et autopsiés.
Les signes cliniques, y compris les modifications neurologiques et oculaires, ainsi que la mortalité seront notés pour tous les animaux. On accordera une attention spéciale à la formation de tumeurs; la date de formation et l'évolution des effets toxiques seront enregistrées; le moment de formation, la localisation, les dimensions, l'apparence et l'évolution de toute tumeur nettement visible ou palpable seront notés.
La consommation alimentaire (ainsi que la consommation d'eau lorsque la substance à tester est administrée dans l'eau de boisson) sera déterminée chaque semaine durant les 13 premières semaines de l'étude, puis tous les 3 mois environ, à moins que l'état de santé ou les modifications de poids corporel ne nécessitent une autre fréquence.
Les poids corporels seront relevés individuellement pour tous les animaux une fois par semaine durant les 13 premières semaines de l'étude, puis une fois toutes les 4 semaines au moins.
Examen clinique
Examen hématologique
Un examen hématologique (par exemple, teneur en hémoglobine, hématocrite, nombre total d'érythrocytes, nombre total de leucocytes, plaquettes sanguines, ou autres mesures de la coagulation) sera effectué sur des échantillons de sang prélevés sur 10 rats/sexe de tous les groupes au troisième et au sixième mois, puis tous les six mois environ et enfin à l'issue de l'essai. Ces échantillons seront, si possible, prélevés chaque fois sur les mêmes rats.
Si les observations effectuées pendant la période d'encagement indiquent une détérioration de l'état de santé des animaux au cours de l'étude, la formule sanguine est établie sur les animaux affectés. La formule sanguine est réalisée avec des échantillons du lot à la plus forte dose et des témoins. Pour le (les) groupe(s) exposé(s) à la (aux) dose(s) immédiatement inférieure(s), une telle analyse n'est pratiquée que si l'on constate un écart important entre le groupe exposé à la dose la plus élevée et les témoins, ou si l'examen histopathologique le justifie.
Analyse des urines
Des échantillons d'urine seront collectés pour analyse sur 10 rats/sexe de tous les lots; ces analyses seront faites si possible en même temps que l'examen hématologique. Les déterminations suivantes seront faites soit sur les animaux individuellement soit, pour les rongeurs, sur un pool des urines du même lot et du même sexe:
|
—
|
apparence: volume et densité pour les animaux pris individuellement,
|
|
—
|
protéines, glucose, corps cétoniques, hémorragie occulte (semi-quantitativement),
|
|
—
|
microscopie du sédiment urinaire (semi-quantitativement).
|
Chimie clinique
Tous les 6 mois environ ainsi qu'à l'issue de l'essai, des échantillons de sang sont prélevés, afin d'effectuer des déterminations biologiques, chez tous les animaux n'appartenant pas à l'ordre des rongeurs ainsi que chez 10 rats/sexe de tous les lots, si possible, chaque fois sur les mêmes rats. De plus, un échantillon sera prélevé sur tous les animaux n'appartenant pas à l'ordre des rongeurs avant de commencer l'étude. Le plasma, préparé à partir de ces échantillons, est utilisé pour les déterminations suivantes:
|
—
|
concentration en protéines totales,
|
|
—
|
concentration en albumine,
|
|
—
|
tests de la fonction hépatique [tels qu'activité de la phosphatase alcaline, activités SGPT (4) et SGOT (5) ], gamma-glutamyl transpeptidase, ornithine-décarboxylase,
|
|
—
|
métabolisme glucidique tel que glycémie à jeun,
|
|
—
|
tests de la fonction rénale, tels que l'urée sanguine.
|
Autopsie
Un examen complet sera effectué sur tous les animaux, y compris ceux morts en cours d'expérience ou sacrifiés à l'état moribond. Avant de sacrifier les animaux, des échantillons de sang seront prélevés sur tous les animaux afin d'établir la formule sanguine. Tous les organes ou tissus présentant des lésions macroscopiques visibles, des tumeurs ou des lésions soupçonnées d'être des tumeurs seront conservés. On tentera de mettre en corrélation les observations macroscopiques et microscopiques.
Les organes et tissus ci-après seront conservés dans un milieu approprié permettant d'éventuels examens histopathologiques ultérieurs: l'encéphale (7) (y compris sections de la moelle/du pont de Varole, cortex cérébelleux et cortex cérébral), hypophyse, tyroïde/parathyroïde, tout tissu thymique, trachée et poumons, cœur, aorte, glandes salivaires, foie (7), rate, reins (7), glandes surrénales (7), pancréas, gonades (7), utérus, organes génitaux annexes, peau, œsophage, estomac, duodénum, jéjunum, iléon, caecum, colon, rectum, vessie, ganglions lymphatiques représentatifs, glande mammaire femelle, musculature de la cuisse, nerf périphérique, sternum avec moelle osseuse, fémur (y compris les articulations), moelle épinière à trois niveaux (cervical, médiothoracique et lombaire), yeux.
L'insufflation d'un fixateur dans les poumons et la vessie constitue la meilleure façon de conserver ces tissus, l'insufflation des poumons est absolument nécessaire dans les études d'inhalation si l'on veut procéder à des examens histopathologiques appropriés. Dans les études par inhalation, tout le tractus respiratoire sera conservé, y compris la cavité nasale, le pharynx et le larynx.
Si d'autres examens cliniques sont effectués, les informations obtenues seront communiquées avant de commencer les examens microscopiques, car elles peuvent fournir des indications précieuses au pathologiste.
Histopathologie
Pour la partie portant sur l'essai de toxicité chronique:
Tous les organes prélevés chez les animaux du groupe satellite exposé à la dose élevée ainsi que du groupe témoin seront soumis à un examen détaillé. Lorsqu'une pathologie liée à la substance à tester est observée sur le groupe satellite exposé à la dose élevée, les organes cibles de tous les autres animaux de tout autre groupe satellite traité seront soumis à un examen histologique complet et détaillé de même que ceux appartenant aux groupes traités dans le cadre de l'essai de cancérogénèse, à l'issue de cette partie de l'étude.
Pour la partie portant sur l'essai de carcinogénicité:
|
a)
|
Les organes et tissus de tous les animaux morts ou sacrifiés au cours de l'essai ainsi que de tous les animaux du groupe témoin et du groupe exposé à la dose élevée seront soumis à un examen histopathologique complet.
|
|
b)
|
Toutes les tumeurs nettement visibles ou les lésions soupçonnées d'être des tumeurs observées sur tout organe dans tous les groupes feront l'objet d'un examen microscopique.
|
|
c)
|
Si l'on observe une différence significative dans l'incidence des lésions néoplasiques dans le groupe exposé à la dose la plus élevée et le groupe témoin, un examen histopathologique sera effectué pour cet organe ou ce tissu particulier chez les animaux des autres groupes.
|
|
d)
|
Si le taux de survie dans le groupe exposé à la dose élevée est nettement inférieur à celui observé pour le groupe témoin, les animaux du groupe exposé à la dose immédiatement inférieure seront soumis à un examen complet.
|
|
e)
|
Si dans le groupe exposé à la dose élevée, on observe des effets toxiques ou autres susceptibles d'affecter la réponse néoplasique, les animaux exposés à la dose immédiatement inférieure seront soumis à un examen complet.
|
2. RÉSULTATS
Les données seront résumées sous forme de tableaux indiquant, pour chaque lot d'expérience, le nombre d'animaux au début de fessai, le nombre d'animaux présentant des tumeurs ou des manifestations toxiques décelées durant l'essai, le moment de cette détection et le nombre d'animaux présentant des tumeurs à l'autopsie. Les résultats seront évalués au moyen d'une méthode statistique appropriée. Toute méthode statistique validée peut être utilisée.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
espèce, souche, origine, milieu ambiant, régime alimentaire, etc.
|
|
—
|
conditions d'essais:
description du système d'exposition:
conception, type, dimensions, source d'air, système générateur de particules et d'aérosols, méthode de conditionnement d'air, traitement de l'air expiré et, le cas échéant, mode de stabulation des animaux en chambres d'essai. L'équipement utilisé pour mesurer la température, l'humidité et, si nécessaire, la stabilité de la concentration d'aérosols ou la granulométrie des particules seront décrits;
données relatives à l'exposition:
elles seront présentées sous forme de tableaux indiquant des valeurs moyennes ainsi qu'une mesure de la variabilité (par exemple écart type) et porteront sur:
|
a)
|
les débits d'air à travers le dispositif d'inhalation;
|
|
b)
|
la température et l'humidité de l'air;
|
|
c)
|
les concentrations nominales (quantité totale de substance à tester introduite dans le dispositif d'inhalation, divisée par le volume d'air);
|
|
d)
|
le cas échéant, la nature du véhicule;
|
|
e)
|
les concentrations réelles dans la zone de respiration;
|
|
f)
|
les dimensions médianes des particules (si nécessaire),
|
|
|
—
|
niveaux de doses (y compris, le cas échéant, véhicule) et concentrations,
|
|
—
|
incidence des tumeurs par sexe, dose et type de tumeur,
|
|
—
|
moment de la mort en cours d'expérience ou indication que l'animal était encore vivant à la fin de l'expérience, y compris animaux du groupe satellite,
|
|
—
|
réponse toxique par sexe et dose,
|
|
—
|
description des effets toxiques ou autres,
|
|
—
|
moment de l'observation de tout signe anormal et évolution de celui-ci,
|
|
—
|
observations ophtalmologiques,
|
|
—
|
consommation alimentaire et poids corporel,
|
|
—
|
résultats de l'examen hématologique,
|
|
—
|
résultats des épreuves de biochimie clinique (y compris analyse des urines),
|
|
—
|
description détaillée de toutes les observations histopathologiques,
|
|
—
|
traitement statistique des résultats et description des méthodes utilisées,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.34 TEST DE REPRODUCTION SUR UNE GÉNÉRATION
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPES DE LA MÉTHODE D'ESSAI
La substance à tester est administrée à des doses croissantes à plusieurs groupes d'animaux mâles et femelles. Les mâles devraient être traités durant leur croissance et pendant au moins un cycle complet de spermatogenèse (environ 56 jours chez la souris et 70 jours chez le rat) afin de permettre à la substance de produire quelques effets nocifs sur la spermatogenèse.
Les femelles de la génération P seront traitées pendant au moins deux cycles œstraux complets afin de permettre a la substance testée de produire quelques effets nocifs sur l'œstrus. Les animaux sont ensuite accouplés. La substance testée est administrée aux animaux des deux sexes pendant la période d'accouplement, puis uniquement aux femelles durant la période de gestion et d'allaitement. Si l'on veut administrer la substance par voie respiratoire (inhalation), la méthode nécessite des adaptations.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
Avant l'essai, de jeunes animaux adultes et sains sont randomisés et répartis par groupes traités et témoins. Les animaux sont maintenus dans les conditions expérimentales de stabulation et d'alimentation pendant au moins 5 jours avant l'expérience. Il est recommandé d'administrer la substance à tester dans la nourriture ou dans l'eau de boisson. D'autres voies d'administration sont également acceptables. La même méthode d'administration sera utilisée pour tous les animaux durant la période expérimentale appropriée. Si un véhicule ou d'autres additifs sont utilisés pour faciliter le traitement, ils devraient être reconnus sans effet toxique. Le traitement devrait être effectué pendant les 7 jours de la semaine.
Animaux d'expérience
Choix de l'espèce:
L'espèce convenant le mieux est celle du rat ou de la souris. Des souches à faible taux de fécondité ne devraient pas être utilisées. Des animaux sains n'ayant pas été déjà utilisés dans une expérimentation devraient être utilisés. L'espèce, la souche, le sexe, le poids et/ou l'âge des animaux d'expérience seront spécifiés.
Pour évaluer de façon adéquate la fécondité, mâles et femelles devraient tous deux être étudiés. Tous les animaux traités et témoins devraient être sevrés avant le début du traitement.
Nombre et sexe:
Chaque groupe traité et chaque groupe témoin devrait comporter un nombre d'animaux suffisant pour obtenir environ 20 femelles gravides arrivées à terme ou proches de celui-ci.
L'objectif est d'obtenir un nombre de gestations et de portées suffisant pour permettre une évaluation valable de l'influence de la substance sur la fertilité, la gestation, le comportement maternel des animaux de la génération P ainsi que l'allaitement, la croissance et le développement de la génération F1, de la conception au sevrage.
Conditions d'essai
La nourriture et l'eau devraient être fournies à satiété. Lorsqu'elles seront proches du terme, les femelles gravides devraient être placées dans des cages individuelles de mise bas ou de maternité et il peut leur être fourni les matériaux de nidification nécessaires.
Doses
Au moins trois groupes traités et un groupe témoin seront utilisés. Si un véhicule est utilisé pour administrer la substance testée, le groupe témoin recevra le volume maximal de véhicule ayant été utilisé. Si une substance à tester entraîne une réduction de la consommation alimentaire ou de l'assimilation, il peut être nécessaire d'utiliser un groupe témoin apparié. Dans les conditions idéales et à moins que la nature physique/chimique ou les effets biologiques de la substance testée ne s'y opposent dans une certaine mesure, le niveau de dose le plus élevé produira un effet toxique sans toutefois entraîner la mort des animaux parentaux (P). La(les) dose(s) intermédiaire(s) devrait(ent) produire des effets toxiques minima attribuables à la substance testée, et la dose faible ne devrait produire aucun effet nocif observable sur les parents ou leur progéniture. Lorsque la substance est administrée par gavage ou en capsule, la dose administrée à chaque animal devrait être établie en fonction du poids corporel de chaque animal et fera l'objet d'un ajustement hebdomadaire afin de tenir compte des modifications de poids corporel. Pour les femelles pendant la gestation, le traitement peut, si on le souhaite, être établi en fonction du poids corporel au jour 0 ou au jour 6 de la gestation.
Essai de limite
Dans le cas de substances faiblement toxiques, si un niveau de dose d'au moins 1 000 mg/kg n'entraîne aucune interférence sur les capacités de reproduction, des études à d'autres niveaux de doses peuvent être considérées non nécessaires. Si une étude préliminaire effectuée avec le niveau de dose élevé, avec une toxicité évidente chez la mère, n'a aucun effet nocif sur la fertilité, on peut juger inutile d'effectuer des études avec d'autres niveaux de doses.
Mode opératoire
Plan d'expérience
On devrait commencer à administrer quotidiennement la substance aux mâles géniteurs (P) lorsqu'ils auront atteints l'âge de 5 à 9 semaines, après qu'ils auront été sevrés et acclimatés pendant au moins 5 jours. Chez les rats, le traitement est poursuivi pendant 10 semaines, avant la période d'accouplement (pour les souris, 8 semaines). Les mâles devraient être sacrifiés et examinés soit à la fin de la période d'accouplement soit alternativement maintenus en vie avec poursuite de l'administration de la substance dans la nourriture, en vue de la production éventuelle d'une seconde portée, et devraient être sacrifiés et examinés à un moment avant la fin de l'étude. Dans le cas des femelles (P), le traitement devra commencer après au moins 5 jours d'acclimatation et se poursuivre pendant au moins 2 semaines avant l'accouplement. Les femelles P devraient continuer à recevoir leur traitement quotidien durant les 3 semaines de la période d'accouplement, durant la gestation et jusqu'au sevrage des petits F1. Des modifications du schéma de traitement peuvent être envisagées au cas où l'on disposerait d'autres informations sur la substance étudiée, comme l'induction du métabolisme ou la bio-accumulation.
Procédure d'accouplement
Dans les études de toxicité sur la fonction de reproduction, les accouplements peuvent se faire soit 1:1(1 mâle, 1 femelle) soit 1:2 (1 mâle, 2 femelles).
Dans le cas d'un accouplement 1:1, une femelle sera placée avec le même mâle jusqu'à ce qu'elle soit gravide ou que les 3 semaines se soient écoulées. On examinera les femelles chaque matin afin de relever la présence de sperme ou de bouchon vaginal. Le jour 0 de la gestation est défini comme le jour où l'on constate la présence de bouchon vaginal ou de sperme.
Tous les couples ayant failli à l'accouplement devraient être examinés afin de déterminer la cause de l'apparente stérilité.
Ceci peut impliquer des procédures qui permettent des conditions d'accouplement avec des mâles ou des femelles ayant déjà procréé, de procéder à un examen microscopique des organes de reproduction ou à un examen du cycle œstral ou de la spermatogenèse.
Taille de la portée
On laisse les animaux traités durant l'étude de fertilité mettre bas naturellement et élever librement leurs petits jusqu'au sevrage.
Quand une méthode d'homogénéisation des portées est effectuée, il est suggéré d'appliquer le programme suivant: entre le jour 1 et le jour 4 après la naissance, la taille de chaque portée peut être adaptée en éliminant, par sélection, des petits afin d'obtenir, dans toute la mesure du possible, 4 mâles et 4 femelles par portée.
Si le nombre de mâles ou de femelles ne permet pas d'obtenir 4 petits de chaque sexe par portée, un ajustement partiel peut être accepté (par exemple 5 mâles et 3 femelles). Des ajustements ne sont pas possibles pour les portées de moins de 8 petits.
Observations
Pendant toute la période d'essai, chaque animal devrait être observé au moins une fois par jour. Des modifications comportementales significatives, des signes de parturition difficile ou prolongée ainsi que tous les signes de toxicité, y compris la mortalité, devraient être enregistrés. Pendant les périodes de pré-accouplement et d'accouplement, la consommation alimentaire sera déterminée quotidiennement. Après la parturition et durant la lactation, la consommation alimentaire ainsi que la consommation d'eau lorsque la substance à tester est administrée dans l'eau de boisson devraient être déterminées le jour de la pesée des petits. Les mâles et les femelles P devraient être pesés le premier jour du traitement, puis toutes les semaines. Ces observations devraient être notées individuellement pour chaque animal adulte.
La durée de la gestation devrait être calculée à partir du jour 0 de la gravidité. Chaque portée devrait être examinée dès que possible après la naissance afin de déterminer le nombre et le sexe des petits, les mort-nés, les nouveau-nés vivants et la présence d'anomalies macroscopiques.
Les petits morts et les petits sacrifiés le quatrième jour devraient être conservés et examinés en vue de déceler d'éventuelles anomalies. Les petits vivants devraient être comptés et les portées pesées le matin qui suit la naissance ainsi que le quatrième et le septième jour, puis chaque semaine jusqu'au terme de l'étude, où les animaux devraient être alors pesés individuellement.
Les anomalies physiques ou comportementales observées chez les mères ou les petits seront enregistrées.
Pathologie
Autopsie
Au moment du sacrifice ou de la mort en cours d'étude, les animaux de la génération P devraient être soumis à un examen macroscopique afin de déceler toute anomalie structurelle ou toute modification pathologique; une attention particulière étant accordée aux organes de l'appareil de reproduction. Les petits, morts ou moribonds, seront examinés du point de vue des malformations.
Histopathologie
Les ovaires, l'utérus, le col utérin, le vagin, les testicules, l'épididyme, les vésicules séminales, la prostate, la glande coagulante, l'hypophyse et l'(les) organe(s) cible(s) de tous les animaux P seront conservés en vue d'un examen microscopique. Au cas où ces organes n'ont pas été examinés au cours d'autres études à dose multiple, ils devraient être soumis à l'examen histologique pour tous les animaux traités à la dose élevée, tous les animaux témoins ainsi que ceux morts en cours d'expérience, lorsque c'est faisable.
Les organes présentant des anomalies chez ces animaux devraient alors être examinés chez tous les autres animaux P. Dans ce cas, l'examen microscopique sera effectué pour tous les tissus présentant des modifications pathologiques macroscopiques. Comme suggéré dans les procédures d'accouplement, les organes reproducteurs des animaux soupçonnés de stérilité seront soumis à un examen microscopique.
2. RÉSULTATS
Les résultats peuvent être résumés sous forme de tableaux indiquant, pour chaque groupe d'expérience, le nombre d'animaux au début de l'essai, le nombre de mâles féconds, le nombre de femelles gravides, le type de modifications et le pourcentage d'animaux présentant chaque type de modification.
Lorsque c'est possible, les résultats numériques devraient être évalués au moyen d'une méthode statistique appropriée. Toute méthode statistique reconnue peut être utilisée.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
espèce/souche utilisée,
|
|
—
|
réponse toxique par sexe et par dose, y compris fécondité, gestation et viabilité,
|
|
—
|
moment de la mort en cours d'expérience ou indication si l'animal était encore en vie au moment prévu pour le sacrifier à l'issue de l'étude,
|
|
—
|
tableau présentant les poids de chaque portée, les poids moyens des petits et les poids individuels des petits à l'issue de l'étude,
|
|
—
|
effet toxique ou autre sur la reproduction, la descendance, la croissance post-natale,
|
|
—
|
jour de l'observation de tout signe anormal et évolution subséquente,
|
|
—
|
poids corporel des animaux P,
|
|
—
|
description détaillée des observations microscopiques,
|
|
—
|
traitement statistique des résultats, quand cela est nécessaire,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCES
Voir introduction générale, partie B.
B.35 ÉTUDE DE TOXICITÉ POUR LA REPRODUCTION SUR DEUX GÉNÉRATIONS
1. MÉTHODE
Cette méthode est calquée sur la ligne directrice d'essai no 416 (2001) de l'OCDE.
1.1. INTRODUCTION
La présente méthode d'essai relative à la toxicité pour la reproduction sur deux générations est destinée à livrer des informations générales concernant les effets d'une substance d'essai sur l'intégrité et le fonctionnement des appareils reproducteurs mâle et femelle, notamment la fonction gonadique, le cycle œstral, le comportement à l'égard de l'accouplement, la conception, la gestation, la mise-bas, la lactation, le sevrage ainsi que la croissance et le développement de la descendance. L'étude peut aussi montrer les effets de la substance d'essai sur la morbidité et la mortalité néonatales, fournir des données préliminaires sur la toxicité prénatale et postnatale pour le développement et orienter des essais ultérieurs. Cette méthode étudie non seulement la croissance et le développement de la génération F1, mais évalue aussi l'intégrité et le fonctionnement des appareils reproducteurs mâle et femelle, ainsi que la croissance et le développement de la génération F2. Il est possible d'obtenir des informations supplémentaires sur la toxicité pour le développement et les déficits fonctionnels en complétant le présent protocole par des études décrites dans les méthodes d'essai relatives à la toxicité pour le développement et/ou à la neurotoxicité pour le développement, ou en procédant à des études séparées à l'aide de méthodes d'essai appropriées.
1.2 PRINCIPE DE LA MÉTHODE D'ESSAI
La substance d'essai est administrée à différentes doses échelonnées suivant une gradation à plusieurs groupes de mâles et de femelles. On administre la substance aux mâles de la génération P (génération parentale) durant leur croissance et pendant au moins un cycle spermatogène complet (environ 56 jours chez la souris et 70 jours chez le rat) afin de mettre en évidence tous ses effets nocifs sur la spermatogenèse. Les effets sur le sperme sont déterminés d'après plusieurs paramètres, comme la morphologie et la motilité des spermatozoïdes, sur des préparations de tissus et au cours d'un examen histopathologique détaillé. Si l'on dispose de données sur la spermatogenèse provenant d'une précédente étude à doses répétées de durée suffisante, par exemple 90 jours, il n'est pas nécessaire d'inclure les mâles de la génération P dans l'évaluation. Il est toutefois recommandé de conserver des échantillons ou des enregistrements numériques du sperme de la génération P, en vue d'une évaluation ultérieure. Les femelles de la génération P doivent être traitées durant la croissance et pendant plusieurs cycles œstraux complets de manière à pouvoir détecter tous les effets nocifs de la substance d'essai sur le cycle œstral. La substance d'essai est administrée aux animaux de la génération P durant la période d'accouplement, pendant les gestations qui en résultent et jusqu'au sevrage de la descendance F1. Après le sevrage, la descendance F1 continue de recevoir la substance durant toute sa croissance et l'administration se poursuit au cours de l'accouplement et de la production de la génération F2, jusqu'au sevrage de cette dernière.
Tous les animaux subissent un examen clinique et pathologique destiné à mettre en évidence des signes de toxicité; cet examen s'attache en particulier à l'évaluation des effets sur l'intégrité et le fonctionnement des appareils reproducteurs mâle et femelle, ainsi que sur la croissance et le développement de la descendance.
1.3 DESCRIPTION DE LA MÉTHODE D'ESSAI
1.3.1 Choix des espèces
Le rat est l'espèce qui convient le mieux à cet essai. Si on utilise une autre espèce, il faut justifier ce choix et effectuer les adaptations qui s'imposent. Les souches à faible taux de fécondité ou chez lesquelles l'incidence des anomalies du développement est notoirement élevée sont à proscrire. Au début de l'étude, la variation pondérale des animaux utilisés doit être minimale et ne doit pas dépasser 20 % du poids moyen des représentants de chaque sexe.
1.3.2 Conditions d'encagement et d'alimentation
L'animalerie doit être à 22 oC (+ 3 oC), avec un taux d'humidité relative d'au moins 30 % et de préférence inférieur à 70 %, sauf pendant le nettoyage de la salle, l'idéal se situant entre 50 et 60 %. On appliquera un éclairage artificiel, avec un cycle de 12 heures de clarté et 12 heures d'obscurité. Un régime alimentaire classique pour animaux de laboratoire, avec eau potable à satiété, conviendra. Le choix des aliments sera guidé par la nécessité d'y incorporer la substance d'essai, si elle est administrée par cette voie.
Les animaux peuvent être encagés individuellement ou par petits groupes du même sexe. L'accouplement doit se dérouler dans des cages appropriées. Après constatation de la copulation, les femelles s'étant accouplées seront placées dans des cages individuelles aménagées pour la mise-bas ou la maternité. Les rates s'étant accouplées peuvent aussi être encagées par petits groupes et séparées un ou deux jours avant la mise-bas. À l'approche de la mise-bas, on leur fournira des matériaux de nidification appropriés et déterminés.
1.3.3 Préparation des animaux
Il convient d'utiliser de jeunes animaux sains, acclimatés aux conditions du laboratoire pendant au moins 5 jours et n'ayant pas été soumis à d'autres essais. On indiquera l'espèce, la souche, la source, le sexe, le poids et/ou l'âge des animaux d'expérience. Il importe de connaître les liens collatéraux entre les animaux, afin de ne pas accoupler les membres d'une même fratrie. Les animaux sont répartis au hasard entre les groupes témoins et les groupes traités (il est recommandé de les regrouper par tranche de poids corporel). Les cages sont placées de manière à réduire au minimum les éventuels effets liés à leur disposition. Chaque animal est désigné par un numéro d'identification propre. Les animaux de la génération P doivent être identifiés avant le début du traitement; ceux de la génération F1 qui sont sélectionnés pour l'accouplement seront identifiés au moment du sevrage. On notera la portée d'origine de tous les animaux de la génération F1 sélectionnés. Il est également recommandé d'identifier chaque petit dès que possible après la naissance, si on envisage de les peser individuellement ou d'effectuer des essais fonctionnels.
Les animaux de la génération P seront âgés de 5 à 9 semaines au début du traitement. Tous les groupes d'essai doivent être aussi homogènes que possible quant au poids et à l'âge des animaux.
1.4. MODE OPÉRATOIRE
1.4.1. Nombre et sexe des animaux
Chaque groupe d'essai ou témoin doit comporter un nombre d'animaux suffisant pour que l'on puisse disposer en fin d'essai d'au moins 20 femelles gravides à terme ou presque. Pour les substances qui occasionnent des effets indésirables (stérilité, toxicité excessive à la dose élevée, par exemple), cela risque d'être impossible. Le but est d'obtenir suffisamment de femelles gravides pour pouvoir procéder à une évaluation significative de l'action de la substance étudiée sur la fertilité, la gestation, le comportement maternel et la période d'allaitement, la croissance et le développement de la descendance F1, depuis la conception jusqu'à la maturité, et sur le développement de leur descendance F2 jusqu'au sevrage. En conséquence, le fait de ne pas avoir obtenu le nombre souhaité de femelles gravides (c'est-à-dire 20) n'invalide pas nécessairement l'étude, et la situation doit être évaluée au cas par cas.
1.4.2. Préparation des doses
Il est recommandé d'administrer la substance d'essai par voie orale (dans la nourriture, l'eau de boisson ou par gavage) à moins qu'une autre voie (cutanée ou par inhalation) ne soit jugée plus adéquate.
S'il y a lieu, la substance d'essai est dissoute ou mise en suspension dans un véhicule approprié. On recommande, si possible, d'envisager d'abord l'utilisation d'une solution ou d'une suspension aqueuse, puis d'une solution ou d'une émulsion dans l'huile (par exemple l'huile de maïs) et en dernier lieu d'une solution dans d'autres véhicules. Si le véhicule n'est pas aqueux, sa toxicité doit être connue. Il faut déterminer la stabilité de la substance d'essai dans le véhicule.
1.4.3. Dosage
On utilise au moins trois doses différentes et un témoin en parallèle. Sauf limitations imposées par les propriétés physiques, chimiques ou biologiques de la substance d'essai, la dose la plus élevée doit faire apparaître une certaine toxicité, sans entraîner la mort ni provoquer de souffrances aiguës. En cas de mortalité inattendue, si le taux de mortalité de la génération parentale est inférieur à environ 10 pour cent, l'étude reste en générale acceptable. Il convient de choisir une série décroissante de doses afin de mettre en évidence une éventuelle relation dose-effet ainsi qu'une dose sans effets toxiques (DSET). L'écart optimal entre chaque dose de la série décroissante est généralement d'un facteur deux ou quatre, et l'adjonction d'un quatrième groupe d'essai est souvent préférable à la fixation d'écarts trop importants (par exemple, supérieurs à un facteur dix). Si la substance d'essai est incorporée à la nourriture, l'écart entre les doses ne doit pas être supérieur à un facteur trois. Le choix des doses sera orienté par toutes les données existantes sur la toxicité, en particulier les résultats des études à doses répétées. Toute information sur le métabolisme et la cinétique de la substance d'essai ou d'un composé apparenté doit également être prise en considération. Ces informations serviront aussi à justifier l'échelle des doses.
Le groupe témoin sera un groupe non traité ou un groupe recevant le véhicule si la substance est administrée dans un véhicule. Exception faite de l'administration de la substance d'essai, les animaux du groupe témoin seront traités de la même manière que les animaux des groupes d'essai. Si un véhicule est employé, on administrera au groupe témoin le plus grand volume de véhicule utilisé. Lorsque la substance d'essai est administrée mélangée aux aliments et qu'il en résulte une diminution de la prise ou la consommation de nourriture, il peut s'avérer nécessaire d'adjoindre un groupe témoin nourri en parallèle. Les résultats d'études contrôlées destinées à évaluer les effets d'une diminution de la consommation de nourriture sur les paramètres de la reproduction peuvent remplacer l'utilisation d'un groupe témoin nourri en parallèle.
Il convient d'être attentif, le cas échéant, aux effets du véhicule ou des autres additifs sur l'absorption, la distribution, le métabolisme ou la rétention de la substance d'essai, aux effets sur les propriétés chimiques de la substance d'essai, susceptibles de modifier sa toxicité, ainsi qu'aux effets sur la consommation de nourriture ou d'eau et sur l'état nutritionnel des animaux.
1.4.4. Essai limite
Si un essai réalisé à une dose d'au moins 1 000 mg/kg de poids corporel/jour administrée par voie orale ou incorporée dans la nourriture ou l'eau de boisson selon un pourcentage équivalent, conformément à la méthode décrite dans cette étude, n'entraîne pas de signes de toxicité observables chez les parents ou dans leur progéniture et qu'aucun effet toxique n'est escompté au vu des données disponibles concernant des composés présentant une structure et/ou une action métabolique analogues, il n'est pas indispensable de mener une étude complète sur plusieurs doses différentes. L'essai limite n'a pas de raison d'être lorsque le niveau d'exposition humaine implique l'utilisation d'une dose orale plus élevée. Pour d'autres modes d'administration, tels que l'inhalation ou l'application cutanée, les propriétés physico-chimiques de la substance d'essai, comme sa solubilité, limitent souvent la concentration maximale applicable.
1.4.5. Administration des doses
Les animaux reçoivent la substance d'essai quotidiennement, sept jours sur sept. L'administration par voie orale (dans la nourriture, l'eau de boisson ou par gavage) est préférable. Si l'on opte pour un autre mode d'administration, il convient de justifier ce choix et d'apporter les modifications nécessaires, le cas échéant. On appliquera le même mode d'administration à tous les animaux, durant la période expérimentale adéquate. Si la substance d'essai est administrée par gavage, on procédera à l'aide d'une sonde gastrique. Le volume maximal de liquide administré en une seule fois ne doit pas dépasser 1 ml/100 g de poids corporel (0,4 ml/100 g de poids pour l'huile de maïs), sauf dans le cas des solutions aqueuses où l'on peut aller jusqu'à 2 ml/100 g de poids corporel. Sauf dans le cas de substances irritantes ou corrosives dont les effets s'intensifient généralement aux concentrations supérieures, il y a lieu de réduire au minimum la variabilité du volume d'essai en adaptant les concentrations de façon à obtenir un volume constant à toutes les doses. Dans les études par gavage, les petits ne reçoivent normalement la substance d'essai qu'indirectement, par le lait maternel, jusqu'à ce que l'administration directe débute pour eux, à partir du sevrage. Lorsque la substance d'essai est mélangée à la nourriture ou à l'eau de boisson, les petits reçoivent aussi la substance d'essai directement dès qu'ils commencent à s'alimenter seuls, lors de la dernière semaine d'allaitement.
Il est important de s'assurer que les quantités de substances administrées dans la nourriture ou l'eau de boisson n'interfèrent pas avec la nutrition ou l'équilibre hydrique. Si la substance d'essai est ajoutée à la nourriture, elle peut être administrée à concentration constante dans cette dernière (ppm) ou à dose constante par rapport au poids de l'animal; il y a lieu de préciser l'option retenue. Si l'on recourt au gavage, il faut toujours administrer la substance d'essai à peu près à la même heure chaque jour et ajuster la dose au moins une fois par semaine pour qu'elle demeure constante par rapport au poids de l'animal. Cet ajustement devra tenir compte de la diffusion placentaire.
1.4.6. Programmes expérimentaux
L'administration quotidienne de la substance d'essai aux mâles et aux femelles de la génération P doit débuter quand ils sont âgés de 5 à 9 semaines. Pour les mâles et les femelles de la génération F1, elle débutera au sevrage; il faut tenir compte du fait que, lorsque la substance d'essai est administrée dans la nourriture ou l'eau de boisson, il est possible que les petits de la génération F1 soient déjà exposés directement à la substance durant la période d'allaitement. Les deux sexes des générations P et F1 continueront à recevoir la substance pendant au moins 10 semaines avant la période d'accouplement. Le traitement sera poursuivi pour les deux sexes durant les deux semaines de la période d'accouplement. Les mâles qui ne serviront pas à l'évaluation des effets sur la reproduction seront euthanasiés et examinés. Les femelles de la génération P continueront à recevoir la substance d'essai tout au long de la gestation et jusqu'au sevrage de leur descendance F1. Il sera peut-être nécessaire de modifier le programme d'administration des doses à la lumière des informations disponibles sur la substance d'essai, notamment en ce qui concerne la toxicité, l'induction métabolique ou la bioaccumulation. La dose administrée à chaque animal est normalement calculée d'après les résultats de la dernière pesée de l'animal. Toutefois, la prudence s'impose lors de l'ajustement de la dose au cours du dernier tiers de la gestation.
Le traitement des mâles et des femelles P et F1 se poursuit jusqu'à leur sacrifice. Tous les adultes mâles et femelles P et F1 qui ne sont plus nécessaires pour l'évaluation des effets sur la reproduction doivent être euthanasiés. Les descendants F1 qui n'ont pas été sélectionnés pour l'accouplement et tous les descendants F2 seront euthanasiés après le sevrage.
1.4.7 Accouplement
1.4.7.1 Accouplement de la génération parentale (P)
Pour chaque accouplement, on réunira une femelle et un mâle traités à la même dose (accouplement 1:1) jusqu'à ce qu'ils aient copulé ou durant deux semaines. Les femelles subiront un examen quotidien destiné à détecter la présence de sperme ou d'un bouchon vaginal. Le jour 0 de la gestation est défini comme étant celui où l'on observe du sperme ou un bouchon vaginal. Si l'accouplement n'a pas lieu, on peut envisager de faire une deuxième tentative en réunissant les femelles avec des mâles du même groupe ayant fait leurs preuves. Les couples doivent être clairement identifiés dans les résultats. On évitera d'accoupler des membres d'une même fratrie.
1.4.7.2 Accouplement de la génération F1
S'agissant de l'accouplement de la génération F1, on sélectionne au moins un mâle et une femelle de chaque portée, au moment du sevrage, en vue de les accoupler avec d'autres descendants traités à la même dose, mais issus d'une autre portée, afin d'obtenir la génération F2. La sélection des petits au sein d'une même portée doit se faire au hasard s'ils ne diffèrent pas de façon significative quant au poids corporel ou à l'apparence. Si l'on constate de telles différences, on sélectionnera les meilleurs représentants de chaque portée. D'un point de vue pragmatique, il est plus facile d'opérer cette sélection en fonction du poids corporel, mais il pourrait être plus pertinent de se fonder sur l'apparence. Les descendants F1 ne doivent pas être accouplés avant d'avoir atteint leur pleine maturité sexuelle.
On évaluera les couples sans descendance afin de déterminer la cause apparente de leur stérilité. À cet effet, on pourra donner aux animaux d'autres occasions de s'accoupler avec d'autres mâles ou femelles ayant fait leurs preuves, effectuer un examen microscopique des organes reproducteurs, ou analyser les cycles œstraux ou la spermatogenèse.
1.4.7.3. Deuxième accouplement
Dans certains cas, notamment lorsque la taille de la portée est modifiée par le traitement ou lorsqu'un effet équivoque est observé dans le premier accouplement, on préconise d'accoupler une deuxième fois les adultes P ou F1, afin d'obtenir une deuxième portée. Il est recommandé d'accoupler une deuxième fois les mâles ou les femelles qui n'ont pas engendré de portée avec un reproducteur ou une reproductrice ayant fait ses preuves. Si la production d'une deuxième portée est jugée nécessaire, le deuxième accouplement doit avoir lieu environ une semaine après le sevrage de la dernière portée.
1.4.7.4. Taille de la portée
On laisse les animaux mettre bas normalement et élever leur progéniture jusqu'au sevrage. La normalisation de la taille des portées est facultative; si l'on y recourt, il faut décrire en détail la méthode utilisée.
1.5. OBSERVATIONS
1.5.1. Observations cliniques
Une observation clinique générale est réalisée chaque jour; en cas d'administration par gavage, l'heure à laquelle cette observation est réalisée doit tenir compte du moment où l'on prévoit que les effets atteindront leur intensité maximale après administration. On notera les changements de comportement, les signes de mise-bas prolongée ou difficile et tous les symptômes de toxicité. En outre, au moins une fois par semaine, chaque animal devra faire l'objet d'un examen plus détaillé qui pourrait être aisément réalisé à l'occasion d'une pesée de l'animal. Deux fois par jour et une fois par jour pendant le week-end, selon les besoins, on recherchera des signes de morbidité et de mortalité sur tous les animaux.
1.5.2. Poids corporel et consommation de nourriture et d'eau des animaux parents
Les animaux parents (P et F1) sont pesés le jour de la première administration et ensuite au moins une fois par semaine. Les mères (P et F1) sont pesées au minimum aux jours 0, 7, 14, et 20 ou 21 de la gestation, puis aux mêmes jours que leurs petits durant l'allaitement, et enfin le jour de leur sacrifice. Les résultats sont consignés individuellement pour chaque animal adulte. Durant la période qui précède l'accouplement et pendant la gestation, la consommation de nourriture est mesurée au moins une fois par semaine. La consommation d'eau est relevée au moins une fois par semaine si la substance d'essai est administrée dans l'eau.
1.5.3. Cycle œstral
La durée et la normalité du cycle œstral sont évaluées chez les femelles P et Fl à l'aide de frottis vaginaux, avant l'accouplement, et facultativement pendant la période d'accouplement, jusqu'à constatation de preuves d'accouplement. Les cellules vaginales ou cervicales seront prélevées avec soin pour éviter d'agresser la muqueuse et d'induire ainsi une pseudogestation (1).
1.5.4. Paramètres d'évaluation du sperme
On note le poids des testicules et des épididymes de tous les mâles P ou F1 sacrifiés, en conservant un exemplaire de chaque organe pour l'examen histopathologique (voir paragraphes 1.5.7, 1.5.8.1). On réservera les testicules et les épididymes d'un sous-lot composé d'au moins dix mâles de chaque génération (P et F1) pour la numération des spermatides résistantes à l'homogénéisation et des spermatozoïdes stockés dans la queue de l'épididyme. Dans ce même sous-lot de mâles, on recueillera le contenu de la queue de l'épididyme ou du canal déférent en vue d'évaluer la motilité et la morphologie des spermatozoïdes. Si des effets liés au traitement sont observés ou si d'autres études révèlent que la substance peut avoir des effets sur la spermatogénèse, l'évaluation du sperme sera réalisée sur tous les mâles traités aux différentes doses; dans le cas contraire, on pourra limiter la numération aux mâles P et F1 du groupe témoin et du groupe traité à la dose la plus élevée.
Il convient de dénombrer la totalité des spermatides testiculaires résistantes à l'homogénéisation et des spermatozoïdes de la queue de l'épididyme (2) (3). Le nombre de spermatozoïdes mis en réserve dans la queue de l'épididyme peut être déduit de la concentration et du volume des spermatozoïdes dans la suspension utilisée pour compléter les évaluations qualitatives, et du nombre de spermatozoïdes récupérés après le broyage et/ou l'homogénéisation ultérieurs du tissu caudal restant. La numération doit être effectuée au sein du sous-lot de mâles sélectionnés dans les groupes traités aux différentes doses, immédiatement après le sacrifice, à moins qu'on ne réalise des enregistrements vidéo ou numériques ou qu'on ne congèle des échantillons en vue d'une analyse ultérieure. Dans ces circonstances, le groupe témoin et le groupe traité à la dose la plus élevée peuvent être analysés en premier. Si l'on n'observe pas d'effets liés au traitement (révélés par exemple par la numération, la morphologie ou la motilité des spermatozoïdes), il n'est pas nécessaire d'analyser les groupes traités aux autres doses. Si des effets liés au traitement apparaissent dans le groupe traité à la dose la plus élevée, on évaluera aussi les groupes traités aux doses plus faibles.
La motilité des spermatozoïdes dans l'épididyme ou le canal déférent est évaluée ou enregistrée sur support vidéo immédiatement après le sacrifice. Le sperme, prélevé de façon à éviter autant que possible de léser les organes, est dilué en vue de l'analyse de la motilité selon une méthode acceptable (4). Il convient d'évaluer subjectivement ou objectivement le pourcentage de spermatozoïdes devenant progressivement mobiles. Si l'on pratique une analyse du mouvement assistée par ordinateur (5) (6) (7) (8) (9) (10), la motilité progressive est évaluée d'après les seuils définis par l'utilisateur pour la vitesse moyenne de trajectoire, la rectilignité ou l'index linéaire. Si les échantillons sont enregistrés en vidéo (11) ou si les images sont enregistrées d'une autre manière au moment de l'autopsie, l'analyse subséquente pourra se limiter aux mâles P et F1 du groupe témoin et du groupe traité à la dose la plus élevée, à moins que des effets liés au traitement n'aient été observés, auquel cas il faudra aussi évaluer les groupes traités aux doses plus faibles. En l'absence d'une image vidéo ou numérique, tous les échantillons de tous les groupes traités devront être analysés lors de l'autopsie.
On procédera à une évaluation morphologique d'un échantillon de sperme de l'épididyme ou du canal déférent. Les spermatozoïdes (au moins 200 par échantillon) doivent être examinés dans des préparations fixées et en milieu liquide (12), puis classés en tant que spécimens normaux ou anormaux. Les anomalies morphologiques des spermatozoïdes comprennent, par exemple, la fusion, des têtes isolées et des têtes et/ou des queues déformées. L'évaluation doit être réalisée au sein du sous-lot de mâles sélectionnés dans les groupes traités aux différentes doses, immédiatement après le sacrifice ou ultérieurement à partir des enregistrements vidéo ou numériques. Les frottis, une fois fixés, peuvent aussi être analysés à une date ultérieure. Dans ces circonstances, le groupe témoin et le groupe traité à la dose la plus élevée peuvent être analysés en premier. Si l'on n'observe pas d'effets liés au traitement (sur la morphologie des spermatozoïdes, par exemple), l'analyse des groupes traités aux autres doses devient superflue. Si des effets liés au traitement apparaissent dans le groupe traité à la plus dose élevée, il convient d'évaluer également les groupes traités aux doses plus faibles.
Si l'un des paramètres d'évaluation du sperme mentionnés plus haut a déjà été examiné dans le cadre d'une étude de toxicité systémique d'au moins 90 jours, il ne doit pas nécessairement être réévalué au cours de l'étude de la toxicité sur deux générations. Il est cependant recommandé de conserver des échantillons ou des enregistrements vidéo du sperme de la génération P, pour permettre une évaluation ultérieure si nécessaire.
1.5.5. Descendance
On examine chaque portée dès que possible après la mise-bas (jour 0 de l'allaitement), afin d'établir le nombre et le sexe des petits, la mortinatalité, le nombre de naissances vivantes et la présence d'anomalies macroscopiques. Les petits trouvés morts au jour 0 seront de préférence examinés, s'ils ne sont pas macérés, afin de mettre en évidence d'éventuelles anomalies et de déterminer la cause de la mort, et conservés. Les petits vivants doivent être comptés et pesés individuellement à la naissance (jour 0 de l'allaitement) ou le jour 1, et pesés régulièrement par la suite, par exemple les jours 4, 7, 14 et 21 de l'allaitement. On notera les anomalies physiques et comportementales observées chez les mères ou les petits.
Le développement physique de la descendance est consigné essentiellement sous la forme du gain de poids corporel. D'autres paramètres physiques (ouverture des oreilles et des yeux, sortie des dents, développement de la pilosité, par exemple) peuvent fournir des informations supplémentaires, mais ces observations sont de préférence réservées à l'évaluation de la maturité sexuelle (âge et poids corporel au moment de l'ouverture vaginale ou de la séparation balano-préputiale, par exemple) (13). Il est recommandé de réaliser des explorations fonctionnelles (l'activité motrice, les fonctions sensorielles, l'ontogenèse des réflexes, par exemple) de la descendance F1 avant et/ou après le sevrage, notamment celles qui se rapportent à la maturation sexuelle, si elles ne sont pas explorées dans d'autres études. On détermine l'âge auquel sont survenus l'ouverture vaginale ou la séparation balano-préputiale chez les descendants F1 tout juste sevrés qui ont été sélectionnés en vue de l'accouplement. Il convient de mesurer la distance ano-génitale chez les petits F2, le jour de leur naissance, si l'on a constaté une modification de la proportion de mâles et de femelles ou de l'âge de maturation sexuelle dans la génération F1.
Les observations fonctionnelles ne s'imposent plus pour les groupes présentant des signes évidents de toxicité (diminution sensible de la prise de poids, etc., par exemple). Les explorations fonctionnelles, le cas échéant, seront réalisées sur les petits non sélectionnés pour l'accouplement.
1.5.6. Examen macroscopique
Juste après le sacrifice ou la mort si celle-ci survient en cours d'étude, tous les animaux parents (P et F1), tous les petits qui présentent des anomalies externes ou des signes cliniques, ainsi qu'un petit/un sexe/une portée choisi(e) au hasard dans chaque génération (F1 et F2) sont soumis à un examen macroscopique destiné à révéler d'éventuels changements pathologiques ou anomalies structurelles. L'examen des organes reproducteurs doit faire l'objet d'une attention particulière. Il y a lieu d'examiner les petits moribonds qui ont été euthanasiés ainsi que les petits morts, s'ils ne sont pas macérés, afin de détecter d'éventuelles anomalies et/ou établir la cause de la mort, et de les conserver.
On examinera les utérus de toutes les femelles primipares, sans compromettre l'évaluation histopathologique, pour voir s'ils présentent des points d'implantation et les dénombrer.
1.5.7. Pesée des organes
Juste après le sacrifice, on détermine le poids corporel de tous les parents P et F1 ainsi que le poids des organes suivants (les deux exemplaire des organes qui vont par paires doivent être pesés séparément):
|
—
|
testicules, épididymes (entiers et queues),
|
|
—
|
vésicules séminales avec glandes coagulantes et leurs liquides, et prostate (ensemble),
|
|
—
|
cerveau, foie, reins, rate, hypophyse, glande thyroïde, glandes surrénales et organes cibles connus.
|
Les poids corporels des petits F1 et F2 retenus pour l'autopsie sont déterminés après le sacrifice, de même que le poids du cerveau, de la rate et du thymus d'un petit/un sexe/une portée choisi(e) au hasard (voir paragraphe 1.5.6).
Si possible, les résultats de l'autopsie et de la pesée des organes doivent être interprétés à la lumière des observations effectuées dans d'autres études à doses répétées.
1.5.8. Histopathologie
1.5.8.1. Animaux parents
Les organes et tissus ci-après des animaux parents (P et F1), ou des échantillons représentatifs de ces derniers, sont fixés et conservés dans un milieu approprié en vue de l'examen histopathologique.
|
—
|
vagin, utérus avec col, et ovaires (conservés dans un fixateur approprié),
|
|
—
|
un testicule (conservé dans un fixateur approprié), un épididyme, les vésicules séminales, la prostate et une glande coagulante,
|
|
—
|
organe(s) cible(s) précédemment identifié(s) de tous les animaux P et F1 sélectionnés pour l'accouplement.
|
Tous les animaux P et F1 du groupe témoin et du groupe traité à la dose la plus élevée, qui ont été sélectionnés pour l'accouplement, doivent faire l'objet d'un examen histopathologique complet des organes et tissus susmentionnés. L'examen des ovaires des femelles P est facultatif. Il convient également d'examiner les organes présentant des altérations liées au traitement dans les groupes traités à la dose la plus faible et à la dose intermédiaire, afin de faciliter la détermination de la DSET. De plus, une évaluation histopathologique sera pratiquée sur les organes reproducteurs des animaux traités aux doses faibles et moyennes, chez lesquels on soupçonne une diminution de la fertilité, par exemple, ceux qui ne se sont pas accouplés, qui n'ont pas conçu, n'ont pas engendré ou n'ont pas donné naissance à des descendants sains, ou ceux chez qui on a constaté des modifications du cycle œstral ou du nombre, de la motilité ou de la morphologie des spermatozoïdes. Toutes les lésions macroscopiques telles qu'atrophies ou tumeurs doivent être examinées.
Un examen histopathologique détaillé des testicules doit être effectué (par exemple, fixation dans du liquide de Bouin, inclusion dans la paraffine et coupes transversales de 4-5 μm d'épaisseur) afin de mettre en évidence des effets liés au traitement, notamment une rétention de spermatides, l'absence de certaines couches ou types de cellules germinales, la présence de cellules géantes multinucléées ou le décollement des cellules spermatogènes dans la lumière des tubules séminifères (14). L'examen d'un épididyme intact doit porter sur la tête, le corps et la queue, ce qui peut être effectué sur une section longitudinale. On observera l'épididyme pour voir s'il ne s'y produit pas d'infiltration de leucocytes, de changement dans la fréquence des types cellulaires et de phagocytose des spermatozoïdes, et s'il ne renferme pas de cellules aberrantes. L'examen des organes reproducteurs mâles peut être effectué à l'aide d'une coloration à l'acide périodique-Schiff ou à l'hématoxyline.
Après la lactation, l'ovaire devrait contenir des follicules primordiaux et des follicules en croissance ainsi que les grands corps jaunes de la lactation. L'examen histopathologique doit révéler une déplétion qualitative de la population de follicules primordiaux. Les follicules primordiaux des femelles F1 feront l'objet d'une évaluation quantitative; le nombre d'animaux, le choix de la section ovarienne et la taille des échantillons de sections doivent être statistiquement valables pour la méthode d'évaluation employée. L'examen comprend la numération des follicules primordiaux, lesquels peuvent être combinés à de petits follicules en croissance, aux fins de la comparaison entre les ovaires des femelles traitées et témoins (15) (16) (17) (18) (19).
1.5.8.2. Petits sevrés
Les tissus présentant des anomalies macroscopiques et les organes cibles de tous les petits qui présentent des anomalies externes ou des signes cliniques ainsi que ceux du petit/du sexe/de la portée choisi(e) au hasard dans chaque génération (F1 et F2) et non retenu(e) pour l'accouplement seront fixés et conservés dans un milieu approprié en vue de l'examen histopathologique. La description histopathologique complète des tissus conservés sera plus particulièrement axée sur les organes reproducteurs.
2. RÉSULTATS
2.1 TRAITEMENT DES RÉSULTATS
Les résultats obtenus sur chaque animal seront consignés et résumés sous forme de tableaux, reprenant pour chaque groupe d'essai et chaque génération, le nombre d'animaux présents au début de l'essai, le nombre d'animaux morts durant l'essai ou euthanasiés, la date de la mort ou de l'euthanasie, le nombre d'animaux fertiles, le nombre de femelles gravides, le nombre d'animaux manifestant des signes de toxicité, la description des signes de toxicité observés, notamment le moment où ils ont débuté, leur durée et leur gravité, les types d'altérations histopathologiques et toutes les données pertinentes sur la portée.
Les résultats numériques seront évalués à l'aide d'une méthode statistique appropriée et reconnue; le choix des méthodes statistiques doit intervenir lors de la conception de l'étude et doit être justifié. Les modèles statistiques s'appliquant aux relations “dose-effet” peuvent être utiles à l'analyse des résultats. Le rapport doit fournir suffisamment d'informations sur la méthode d'analyse et le logiciel employés, pour qu'un réviseur ou un statisticien indépendant puisse réévaluer et reconstruire l'analyse.
2.2 ÉVALUATION DES RÉSULTATS
Les résultats de cette étude de toxicité pour la reproduction sur deux générations doivent être évalués en fonction des effets observés, notamment à l'autopsie et lors des examens microscopiques. L'évaluation portera sur la relation, ou l'absence de relation, entre la dose administrée de substance d'essai et la présence, la fréquence et la gravité des anomalies, notamment lésions macroscopiques, organes cibles identifiés, altération de la fertilité, anomalies cliniques, altération de la capacité de reproduction et des performances de la portée, modifications du poids corporel, effets sur la mortalité et tout autre effet toxique. Les propriétés physicochimiques de la substance d'essai et, le cas échéant, les données toxicocinétiques doivent être prises en considération lors de l'évaluation des résultats.
Une étude de toxicité pour la reproduction correctement menée doit fournir une estimation satisfaisante de la dose sans effet et permettre de comprendre les effets nocifs de la substance étudiée sur la reproduction, là parturition, l'allaitement et le développement postnatal, notamment la croissance et la maturation sexuelle.
2.3 INTERPRÉTATION DES RÉSULTATS
Une étude de toxicité pour la reproduction sur deux générations fournit des informations sur les effets d'une l'exposition répétée à une substance durant toutes les phases du cycle de reproduction. L'étude livre notamment des informations sur les paramètres de la reproduction ainsi que sur le développement, la croissance et la survie des descendants. Les résultats de l'étude doivent être interprétés à la lumière des résultats des études subchroniques, prénatales, de développement, toxicocinétiques et autres disponibles. Les résultats de la présente étude peuvent servir à apprécier la nécessité de réaliser d'autres essais sur une substance chimique. L'extrapolation des résultats de l'étude à l'homme présente un intérêt limité. Ces résultats se prêtent davantage à la détermination des concentrations sans effet et des niveaux d'exposition humaine acceptables (20) (21) (22) (23).
3. RAPPORT
3.1 RAPPORT D'ESSAI
Le rapport doit contenir les informations suivantes:
|
|
Substance d'essai:
|
—
|
nature physique et, s'il y a lieu, propriétés physico-chimiques
|
|
|
|
Véhicule, le cas échéant:
|
—
|
justification du choix du véhicule s'il diffère de l'eau
|
|
|
|
Animaux d'expérience:
|
—
|
nombre, âge et sexe des animaux
|
|
—
|
origine, conditions d'encagement, régime alimentaire, matériaux de nidification, etc.
|
|
—
|
poids de chaque animal au début de l'essai
|
|
|
|
Conditions d'essai:
|
—
|
justification du choix des doses
|
|
—
|
détails concernant la formulation de la substance d'essai/son incorporation aux aliments, la concentration obtenue
|
|
—
|
stabilité et homogénéité de la préparation;
|
|
—
|
détails concernant l'administration de la substance d'essai;
|
|
—
|
conversion de la concentration de la substance d'essai (ppm) dans la nourriture ou l'eau de boisson en dose administrée (mg/kg de poids corporel/jour), s'il y a lieu;
|
|
—
|
détails concernant la qualité de l'alimentation et de l'eau de boisson.
|
|
|
|
Résultats:
|
—
|
consommation de nourriture, et d'eau si elle a été enregistrée, efficacité alimentaire (gain de poids corporel par gramme d'aliments consommés), et consommation de la substance d'essai par les animaux P et F1, sauf pendant la cohabitation et durant au moins le dernier tiers de la période d'allaitement;
|
|
—
|
données sur l'absorption, le cas échéant;
|
|
—
|
poids corporel des animaux P et F1 sélectionnés pour l'accouplement;
|
|
—
|
données relatives aux portées et au poids des petits;
|
|
—
|
poids corporel au moment du sacrifice et poids absolu et relatif des organes des animaux parents
|
|
—
|
nature, gravité et durée des signes cliniques (qu'ils soient réversibles ou non);
|
|
—
|
date de mort en cours d'étude ou animaux ayant survécu jusqu'au sacrifice;
|
|
—
|
manifestation de la toxicité par sexe et par dose, y compris indices d'accouplement, de fécondité, de gestation, de natalité, de viabilité et d'allaitement; le rapport doit mentionner les chiffres ayant servi au calcul de ces indices;
|
|
—
|
effets toxiques ou autres sur la reproduction, la descendance, la croissance postnatale, etc.;
|
|
—
|
observations à l'autopsie;
|
|
—
|
description détaillée de toutes les observations histopathologiques;
|
|
—
|
nombre de femelles P et F1 ayant un cycle normal et durée du cycle;
|
|
—
|
nombre total de spermatozoïdes dans la queue de l'épididyme, pourcentage de spermatozoïdes progressivement mobiles, pourcentage de spermatozoïdes à morphologie normale et pourcentage de spermatozoïdes par anomalie identifiée;
|
|
—
|
temps écoulé jusqu'à l'accouplement, exprimé en nombre de jours;
|
|
—
|
nombre d'implantations, de corps jaunes, taille de la portée;
|
|
—
|
nombre de naissances vivantes et de pertes postimplantatoires; .
|
|
—
|
nombre de petits affectés d'anomalies macroscopiques; nombre de rabougris s'il a été déterminé;
|
|
—
|
paramètres physiques évalués sur les petits et autres données sur le développement postnatal; il y a lieu de justifier les paramètres physiques évalués;
|
|
—
|
observations fonctionnelles réalisées sur les petits et sur les adultes, suivant le cas;
|
|
—
|
traitement statistique des résultats, le cas échéant.
|
|
|
|
Conclusions, y compris valeurs de la DSET pour les effets sur la mère et sur les petits.
|
4. BIBLIOGRAPHIE
|
(1)
|
Sadleir, R.M.F.S. (1979). Cycles and Seasons, In: Reproduction in Mammals: I. Germ Cells and Fertilization, C.R. Auston and R.V. Short (eds.), Cambridge, New York.
|
|
(2)
|
Gray, L.E. et al., (1989). A Dose-Response Analysis of Methoxychlor-Induced Alterations of Reproductive Development and Function in the Rat. Fundamental and Applied Toxicology 12:92-108.
|
|
(3)
|
Robb, G.W. et al., (1978). Daily Sperm Production and Epididymal Sperm Reserves of Pubertal and Adult Rats. Journal of Reproduction and Fertility 54:103-107.
|
|
(4)
|
Klinefelter, G.R. et al., (1991). The Method of Sperm Collection Significantly Influences Sperm Motion Parameters Following Ethane Dimethanesulfonate Administration in the Rat. Reproductive Toxicology 5:39 44
|
|
(5)
|
Seed, J. et al. (1996). Methods for Assessing Sperm Motility, Morphology, and Counts in the Rat, Rabbit, and Dog:a Consensus Report. Reproductive Toxicology 10(3):237- 244.
|
|
(6)
|
Chapin, R.E. et al, (1992). Methods for Assessing Rat Sperm Motility. Reproductive Toxicology 6:267-273
|
|
(7)
|
Klinefelter, G.R. et al., (1992). Direct Effects of Ethane Dimethanesulphonate on Epididymal Function in Adult Rats: an In Vitro Demonstration. Journal of Andrology 13:409-421.
|
|
(8)
|
Slott, V.L. et al., (1991). Rat Sperm Motility Analysis: Methodologie Considerations. Reproductive Toxicology 5:449-458.
|
|
(9)
|
Slott, V.L. and Perreault, S.D., (1993). Computer-Assisted Sperm Analysis of Rodent Epididymal Sperm Motility Using the Hamilton-Thorn Motility Analyzer. In: Methods in Toxicology, Part A., Academie, Orlando, Florida. pp. 319-333.
|
|
(10)
|
Toth, G.P. et al. (1989). The Automated Analysis of Rat Sperm Motility Following Subchronic Epichlorhydrin Administration: Methodologie and Statistical Considerations. Journal of Andrology 10: 401-415.
|
|
(11)
|
Working, P.K. and M. Hurtt, (1987). Computerized Videomicrographic Analysis of Rat Sperm Motility, Journal of Andrology 8:330-337.
|
|
(12)
|
Linder, R.E. et al., (1992). Endpoints pf Spermatoxicity in the Rat After Short Duration Exposures to Fourteen Reproductive Toxicants. Reproductive Toxicology 6:491-505.
|
|
(13)
|
Korenbrot, C.C. et al., (1977). Preputial Separation as an External Sign of Pubertal Development in the Male Rat. Biological Reproduction 17:298303.
|
|
(14)
|
Russell, L.D. et al., (1990). Histological and Histopathological Evaluation of the Testis, Cache River Press, Clearwater, Florida.
|
|
(15)
|
Heindel, J.J. and R.E. Chapin, (eds.) (1993). Part B. Female Reproductive Systems, Methods in Toxicology, Academic, Orlando, Florida.
|
|
(16)
|
Heindel, J.J. et al., (1989) Histological Assessment of Ovarian Follicle Number in Mice As a Screen of Ovarian Toxicity. In: Growth Factors and the Ovary, A.N. Hirshfield (ed.), Plenum, New York, pp. 421-426.
|
|
(17)
|
Manson, J.M. and Y.J. Kang, (1989). Test Methods for Assessing Female Reproductive and Developmental Toxicology. In: Principles and Methods of Toxicology, A.W. Hayes (ed.), Raven, New York.
|
|
(18)
|
Smith, B.J. et al,. (1991). Comparison of Random and Serial Sections in Assessment of Ovarian Toxicity. Reproductive Toxicology 5:379-383.
|
|
(19)
|
Heindel, J.J. (1999). Oocyte Quantitation and Ovarian Histology. In: An Evaluation and Interpretation of Reproductive Endpoints for Human Health Risk Assessment, G. Daston,. and C.A. Kimmel, (eds.), ILSI Press, Washington, DC.
|
|
(20)
|
Thomas, J. A. (1991). Toxic Responses of the Reproductive System. In: Casarett and Doull's Toxicology, M.O. Amdur, J. Doull, and C.D. Klaassen (eds.), Pergamon, New York.
|
|
(21)
|
Zenick, H. and E.D. Clegg, (1989). Assessment of Male Reproductive Toxicity: A Risk Assessment Approach. In: Principles and Methods of Toxicology, A.W. Hayes (ed.), Raven Press, New York.
|
|
(22)
|
Palmer, A.K. (1981). In: Developmental Toxicology, Kimmel, C.A. and J. Buelke-Sam (eds.), Raven Press, New York.
|
|
(23)
|
Palmer, A.K. (1978). In Handbook of Teratology, Vol. 4, J.G. Wilson and F.C. Fraser (eds.), Plenum Press, New York.
|
B.36 TOXICOCINÉTIQUE
1. MÉTHODE
1.1. INTRODUCTION
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Voir introduction générale, partie B.
1.3. SUBSTANCES DE RÉFÉRENCE
Néant.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance à tester est administrée par une voie appropriée. Suivant l'objectif poursuivi dans l'étude, la substance peut être administrée de façon unique ou réitérée, pendant des périodes de temps déterminées, à un ou plusieurs groupes d'animaux d'expérience. La substance et/ou ses métabolites sont ensuite étudiés dans les liquides de l'organisme, les tissus et/ou les excréta.
Les études peuvent être effectuées avec des formes «marquées» ou «non marquées» de la substance à tester. En cas d'utilisation d'un marquage, la position de celui-ci dans la substance doit fournir un maximum d'informations sur le devenir du composé.
1.5. CRITÈRE QUALITATIF
Néant.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparations
De jeunes animaux adultes et en bonne santé sont acclimatés aux conditions du laboratoire pendant au moins cinq jours avant l'expérience. Avant de commencer l'essai, les animaux sont randomisés et répartis entre les groupes soumis au traitement. Dans des conditions particulières, des animaux très jeunes, gravides ou prétraités peuvent être utilisés.
Conditions d'essai
Animaux d'expérience
Les études toxicocinétiques peuvent être effectuées sur une ou plusieurs espèces animales appropriées et il sera tenu compte des espèces utilisées ou qu'il est prévu d'utiliser dans d'autres études toxicologiques portant sur la même substance à tester. Lorsque des rongeurs sont utilisés pour un essai, la différence de poids entre les animaux ne dépassera pas ± 20 % de la valeur moyenne.
Nombre et sexe
Pour les études d'absorption et d'excrétion, on utilisera au départ un lot de 4 animaux pour chaque dose. Le choix d'un sexe déterminé n'est pas obligatoire mais, dans certains cas, il peut s'avérer nécessaire d'étudier des animaux des deux sexes. En cas de réponses différentes selon le sexe, 4 animaux de chaque sexe seront testés. S'il s'agit de non-rongeurs, un plus petit nombre d'animaux peut être utilisé. Lors de l'étude de la distribution dans les tissus, il sera tenu compte, pour fixer la taille initiale de l'échantillon traité, du nombre d'animaux à sacrifier à chaque date d'examen fixé, ainsi que du nombre d'examens à envisager.
Pour l'étude du métabolisme, la taille de l'échantillon traité sera adaptée aux besoins de cette étude. Pour les études à doses multiples et à plusieurs examens intermédiaires, il sera tenu compte, pour la taille de l'échantillon traité, du nombre d'examens et de sacrifices prévus; le groupe doit, toutefois, se composer d'au moins deux animaux. La taille de l'échantillon traité sera suffisante pour permettre une évaluation acceptable de l'augmentation du taux, du plateau et de la diminution du taux (le cas échéant) de la substance à tester et/ou de ses métabolites.
Doses
Dans le cas d'une administration unique, on utilisera au moins deux niveaux de dose, à savoir une dose faible pour laquelle on n'observe aucun effet toxique et une dose élevée susceptible de modifier les paramètres toxicocinétiques ou pour laquelle on observe des effets toxiques.
Dans le cas d'une administration réitérée, la dose faible est habituellement suffisante mais, dans certaines circonstances, une dose élevée peut aussi s'avérer nécessaire.
Voie d'administration
Les études toxicocinétiques seront effectuées par la même voie et, le cas échéant, avec le même véhicule que celui utilisé ou envisagé pour les autres études de toxicité. La substance à tester est habituellement administrée par voie orale (gavage ou incorporation dans l'alimentation) par application sur la peau ou par inhalation, pendant des périodes de temps déterminées, à plusieurs lots d'animaux d'expérience. L'administration de la substance à tester par voie intraveineuse peut être utile pour déterminer l'absorption relative par les autres voies. De plus, des informations utiles sur le mode de distribution peuvent être obtenues peu après l'administration intraveineuse d'une substance.
La possibilité d'une interférence du véhicule avec la substance à tester sera prise en considération. Une attention particulière sera portée aux différences d'absorption selon que la substance à tester est administrée par gavage ou dans l'alimentation et l'on veillera à déterminer la dose avec précision, en particulier lorsque la substance à tester est incorporée à l'alimentation.
Période d'observation
Tous les animaux feront l'objet d'une observation journalière; les signes de toxicité et autres manifestations cliniques notables ainsi que le moment de leur apparition, leur gravité et leur durée, seront signalés.
Conduite de l'essai
Après avoir pesé les animaux d'expérience, on administre la substance à tester par une voie appropriée.
Absorption
Le taux et le degré d'absorption de la substance administrée peuvent être évalués par différentes méthodes, en présence et en l'absence de groupes de référence (8), par exemple:
|
—
|
détermination de la quantité de substance à tester et/ou de ses métabolites dans les excreta, tels que urine, bile, fèces, air expiré et dans le contenu de la carcasse,
|
|
—
|
comparaison de la réponse biologique (par exemple, étude de toxicité aiguë) obtenue pour les groupes d'expérience, les groupes témoins et/ou les groupes de référence,
|
|
—
|
comparaison de la quantité de substance et/ou de métabolites excrétés par les reins dans les groupes d'expérience et de référence,
|
|
—
|
détermination de l'aire sous la courbe taux plasmatique/temps, de la substance à tester et/ou de ses métabolites et comparaison avec les données obtenues pour un groupe de référence.
|
Distribution
Il existe actuellement deux méthodes pour analyser les modes de distribution, l'une des deux ou toutes les deux pouvant être utilisées:
|
—
|
les techniques d'autoradiographie du corps entier fournissent des informations qualitatives utiles,
|
|
—
|
le sacrifice des animaux, à des périodes différentes après leur exposition et la détermination de la concentration et de la quantité de substance à tester et/ou de ses métabolites dans les tissus et les organes fournissent des informations quantitatives.
|
Excrétion
Dans les études d'excrétion, on collecte l'urine, les fèces, l'air expiré et, dans certains cas, la bile. La quantité de substance à tester et/ou de métabolites présents dans ces excreta sera mesurée à plusieurs reprises après l'exposition, jusqu'à ce que 95 % environ de la dose administrée aient été éliminés, ou pendant sept jours consécutifs, si ce pourcentage n'est pas atteint avant.
Dans certains cas, il peut être nécessaire de tenir compte de la substance à tester éliminée dans le lait des animaux d'expérience qui allaitent.
Métabolisme
Des échantillons biologiques seront analysés par des méthodes adéquates afin de déterminer la voie métabolique et son intensité. Les structures des métabolites seront étudiées et des voies métaboliques appropriées seront proposées lorsqu'il y a lieu de répondre à des questions soulevées par des études toxicologiques antérieures. Il peut s'avérer utile d'effectuer des études in vitro en vue d'obtenir des informations sur les voies métaboliques.
Des informations complémentaires sur la relation entre le métabolisme et la toxicité peuvent être obtenues grâce à des études biochimiques telles que la détermination des effets sur des systèmes d'enzymes métabolisants, de la déplétion en composés endogènes à groupements sulfhydriles non protéiques, de la liaison avec les macromolécules.
2. RÉSULTATS
Selon le type d'étude effectuée, les données seront résumées sous la forme de tableaux étayés, le cas échéant, par des graphiques. Pour chaque groupe d'expérience, les variations moyennes et statistiques des mesures en fonction du temps, des doses, des tissus et des organes seront, le cas échéant, indiquées. Le degré d'absorption ainsi que les quantités excrétées et le rythme de l'excrétion seront déterminés par des méthodes appropriées. Lorsque des études du métabolisme sont effectuées, la structure des métabolites identifiés sera indiquée et les voies métaboliques possibles seront présentées.
3. RAPPORT
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
espèces, souche, source, milieu ambiant, régime alimentaire, etc.,
|
|
—
|
caractéristiques des produits marqués, si ceux-ci sont utilisés,
|
|
—
|
niveaux de dose et intervalles utilisés,
|
|
—
|
voie(s) d'administration et tout véhicule utilisé,
|
|
—
|
effets toxiques et autres observés,
|
|
—
|
méthodes permettant de déterminer la substance d'essai et/ou ses métabolites dans les échantillons biologiques, y compris l'air expiré,
|
|
—
|
mise en tableaux des mesures effectuées par sexe, dose, régime, temps, tissus et organes,
|
|
—
|
indication du degré d'absorption et d'excrétion en fonction du temps,
|
|
—
|
méthodes de caractérisation et d'identification des métabolites dans les échantillons biologiques,
|
|
—
|
méthodes utilisées pour les mesures biochimiques en rapport avec le métabolisme,
|
|
—
|
voies proposées pour le métabolisme,
|
|
—
|
discussion des résultats,
|
|
—
|
interprétation des résultats.
|
3.2. ÉVALUATION ET INTERPRÉTATION
Voir introduction générale, partie B.
4. RÉFÉRENCE
Voir introduction générale, partie B.
B.37 NEUROTOXICITÉ RETARDÉE DES SUBSTANCES ORGANOPHOSPORÉES APRÈS EXPOSITION AIGUË
1. MÉTHODE
1.1. INTRODUCTION
Lorsqu'on évalue les effets toxiques des substances, il est important d'étudier la capacité qu'ont certaines substances à entraîner certains types de neurotoxicité que les autres études de toxicité pourraient ne pas mettre en évidence. Certains composés organophosphorés ont une neurotoxicité différée et doivent faire l'objet de telles études.
Des essais de dépistage in vitro peuvent être utilisés pour identifier les substances susceptibles d'entraîner une polyneuropathie différée; toutefois, des résultats négatifs à l'issue d'essais in vitro ne suffisent pas à prouver que la substance testée n'est pas neurotoxique.
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Les substances organophosphorées comprennent les esters, thioesters organophosphorés non chargés ou anhydrides des acides organophosphoriques, organophosphoniques ou organophos-phoramidiques ou des acides phosphorothioïques, phosphonothioïques ou phosphorothioamidiques apparentés ou d'autres substances pouvant entraîner la neurotoxicité différée parfois observée pour les substances de cette classe.
La neurotoxicité retardée est un syndrome associé à l'installation prolongée et d'ataxie retardée, associée à des axonopathies distales au niveau de la moelle épinière et des nerfs périphériques, et à une inhibition et un vieillissement de l'estérase caractéristique des neuropathies (neuropathy taret esterase — NTE) dans les tissus nerveux.
1.3. SUBSTANCES DE RÉFÉRENCE
Une substance de référence peut être testée sur un lot témoin positif, afin de démontrer que, dans les conditions de l'essai, la réaction de l'espèce testée n'est pas modifiée de façon significative.
Le tri-o-tolyi phosphate est un exemple de produit neurotoxique couramment utilisé [no CAS 78-30-8, no EINECS 201-103-5, nomenclature CAS: acide phosphorique, tris(2-méthylphényl)es-ter]; il est également connu sous le nom de tns-o-crésylphosphate.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance à tester est administrée oralement, en une prise unique, à des poules domestiques qui ont été protégées, le cas échéant, contre des effets cholinergiques aigus. Les animaux sont observés pendant 21 jours pour détecter des troubles du comportement, une ataxie et une paralysie. Des analyses biochimiques sont réalisées, en particulier pour mettre en évidence l'inhibition de l'estérase NTE, sur des poules sélectionnées au hasard dans chaque lot, en général 24 et 48 heures après administration. 21 jours après l'exposition, les poules restantes sont sacrifiées et un examen histopathologique est effectué sur des tissus nerveux choisis.
1.5. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.5.1. Préparation
De jeunes poules adultes exemptes d'affections virales, ne faisant l'objet d'aucune médication susceptible d'interférer et ne présentant pas de troubles de la démarche sont réparties au hasard en lots d'expérience et lots témoins, et sont acclimatées aux conditions du laboratoire pendant au moins 5 jours avant le début de l'étude.
Les cages ou enclos utilisés doivent être suffisamment grands pour que les poules puissent se mouvoir librement et pour que leur démarche puisse être aisément observée.
La substance à tester est généralement administrée oralement, par gavage ou dans des capsules de gélatine, ou par une méthode comparable. Les substances liquides peuvent être administrées directement ou dissoutes dans un véhicule approprié tel que l'huile de maïs; les substances solides doivent si possible être dissoutes car il se peut que d'importantes quantités de solides administrées dans des capsules de gélatine ne soient pas bien absorbées. Si le véhicule est autre que de l'eau, ses caractéristiques toxiques doivent être connues et, dans le cas contraire, déterminées avant le début de l'essai.
1.5.2. Conditions de l'essai
1.5.2.1. Animaux d'expérience
L'espèce recommandée est la poule pondeuse domestique adulte jeune âgée de 8-12 mois (Gallus gallus domesticus). Il convient d'utiliser des races et souches de taille standard; les poules doivent normalement avoir été élevées dans des conditions leur permettant de se mouvoir librement.
1.5.2.2. Nombre et sexe
Outre le lot d'expérience, il faut prévoir un lot témoin recevant le véhicule seul et un lot témoin positif. Le lot recevant le véhicule seul doit être traité de la même façon que le lot d'expérience, exception faite de l'administration de la substance à tester.
Il faut prévoir suffisamment d'animaux dans chaque lot pour que l'on puisse en sacrifier au moins six pour effectuer les déterminations biochimiques (trois à chacun des deux temps de prélèvement) et qu'il en survive six après la période d'observation de 21 jours.
Le lot témoin positif peut être réalisé en parallèle ou provenir d'expériences antérieures récentes. Il doit se composer d'au moins six poules traitées par un produit neurotoxique connu à effet retardé; trois poules seront réservées aux examens biochimiques et trois à la recherche de signes pathologiques. La mise à jour périodique des données historiques recueillies antérieurement est recommandée. De nouveaux lots positifs devront être réalisés si une des conditions essentielles de l'expérience (par exemple souches, alimentation, hébergement) a été modifiée par le laboratoire chargé de l'essai.
1.5.2.3. Niveau de doses
Il convient d'effectuer une étude préliminaire sur un nombre approprié de poules réparties dans des lots traités à différentes doses, afin de déterminer la dose à utiliser pour l'étude principale. Un certain taux de mortalité est inévitable dans cette étude préliminaire. Toutefois, pour éviter la mort consécutive à des effets cholinergiques aigus, il est possible d'utiliser de l'atropine ou un autre agent protecteur ne perturbant pas la réaction neurotoxique différée. Diverses méthodes d'essai peuvent être utilisées pour évaluer la dose non létale maximale d'une substance (voir méthode B.1 bis). Les données recueillies antérieurement sur les poules ou d'autres informations toxicologiques peuvent également être utiles pour déterminer la dose.
La dose utilisée dans l'étude principale doit être aussi élevée que possible, compte tenu des résultats de l'étude préliminaire et de la limite supérieure de 2 000 mg/kg poids corporel. Quel que soit le taux de mortalité, il doit subsister un nombre suffisant d'animaux pour effectuer les analyses biochimiques (six) et l'examen histologique (six) au 21e jour. Il convient d'utiliser de l'atropine ou un autre agent qui ne perturbe pas les réactions neurotoxiques différées, afin d'éviter la mort par effets cholinergiques aigus.
1 .5.2.4. Essai limite
Lorsqu'un essai réalisé selon le mode opératoire décrit ci-après, à une dose d'au moins 2 000 mg/kg p.c/jour, n'entraîne pas d'effets toxiques observables et que les données relatives aux substances de structure voisine ne laissent pas supposer une toxicité, il n'est pas nécessaire d'effectuer une étude à une dose plus élevée. L'essai limite est applicable, sauf lorsqu'une exposition humaine fait apparaître la nécessité d'utiliser une dose plus élevée.
1.5.2.5. Période d'observation
La période d'observation doit s'étendre sur 21 jours.
1.5.3. Mode opératoire
Après administration d'un agent protecteur pour éviter que des effets cholinergiques aigus n'entraînent la mort, la substance à tester est administrée en une prise unique.
1.5.3.1. Observation générale
L'observation doit débuter immédiatement après l'exposition. Toutes les poules sont observées soigneusement plusieurs fois au cours des deux premiers jours, puis au moins une fois par jour, pendant 21 jours ou jusqu'à la date prévue du sacrifice. Tous les signes de toxicité doivent être notés, ainsi que la date d'apparition des troubles du comportement, leur type, leur gravité et leur durée. L'ataxie doit être mesurée à l'aide d'une échelle d'évaluation ordinale comportant au moins quatre niveaux; la paralysie doit être notée. Au moins deux fois par semaine, les poules destinées à la recherche des signes pathologiques doivent être sorties de leurs cages et soumises à une épreuve d'activité motrice forcée, d'escalade d'échelles par exemple, afin de faciliter l'observation de faibles manifestations de toxicité. Les animaux moribonds ou présentant des signes de détresse ou de souffrance intense doivent être immédiatement retirés de l'étude, euthanasiés et autopsiés.
1.5.3.2. Poids corporel
Toutes les poules sont pesées juste avant l'administration de la substance, et au moins une fois par semaine par la suite.
1.5.3.3. Biochimie
Six poules choisies au hasard dans chacun des lots d'expérience et des lots témoins négatifs, ainsi que trois poules prélevées dans le lot témoin positif (si l'essai est réalisé en parallèle sur ce dernier) sont sacrifiées quelques jours après administration; le cerveau et la moelle épinière lombaire sont préparés et analysés pour détecter l'inhibition de l'estérase caractéristique des neuropathies. Il peut en outre s'avérer utile d'analyser l'inhibition de cette estérase sur du nerf sciatique. En règle générale, trois poules du lot témoin et de chaque lot d'expérience sont sacrifiées après 24 heures et trois autres après 48 heures, tandis que les trois poules du lot témoin positif sont sacrifiées après 24 heures. Si l'observation des signes cliniques d'intoxication (souvent estimés par le moment d'apparition des effets cholinergiques) indique que la substance toxique est éliminée très lentement, il peut s'avérer préférable de prélever les tissus sur trois poules à deux autres temps compris entre 24 heures et au plus tard 72 heures après administration.
S'il y a lieu, il est également possible d'effectuer des déterminations de l'acétylcholinestérase (AChE) sur des échantillons. Toutefois, il peut y avoir réactivation spontanée de l'acétylcholinestérase in vivo, ce qui pourrait faire sous-estimer l'activité de la substance en tant qu'inhibiteur de l'AChE.
1.5.3.4. Autopsie
L'autopsie de tous les animaux (sacrifices prévus ou exigés par l'état de l'animal) doit comporter un examen de l'aspect du cerveau et de la moelle épinière.
1.5.3.5. Examen histopathologique
Les tissus nerveux des animaux survivants après la période d'observation et non utilisés pour les études biochimiques doivent faire l'objet d'un examen microscopique. Les tissus doivent être fixés in situ par des techniques de perfusion. Les coupes doivent être pratiquées au niveau du cervelet (plan longitudinal moyen), du bulbe rachidien, de la moelle épinière et des nerfs périphériques. Les coupes de la moelle épinière seront pratiquées dans la partie cervicale supérieure, dans la région thoracique moyenne et dans la région lombo-sacrée. Des coupes devront également être réalisées dans la partie distale du nerf tibial et au niveau de ses ramifications vers les muscles gastroenémiens, ainsi qu'au niveau du nerf sciatique. Les coupes seront colorées à l'aide de colorants appropriés, spécifiques de la myéline et des axones.
2. RÉSULTATS
En règle générale, si les résultats obtenus pour les différents points d'évaluation retenus (biochimie, histopathologie et observation du comportement) sont négatifs, il n'est normalement pas nécessaire de poursuivre les essais de neurotoxicité différée. Les résultats équivoques ou non concluants peuvent nécessiter une poursuite de l'étude.
Les résultats doivent être indiqués pour chaque animal, individuellement. En outre, tous les résultats doivent être présentés sous forme de tableaux indiquant, pour chaque lot d'expérience, le nombre d'animaux présents au début de l'essai, le nombre d'animaux présentant des lésions, des troubles du comportement ou des modifications biochimiques, le type et la gravité de ces lésions ou troubles, ainsi que le pourcentage d'animaux présentant chaque type de lésion ou trouble aux différents niveaux de gravité.
Les résultats de cette étude doivent être évalués du point de vue de l'incidence, de la gravité et de la corrélation entre les effets comportementaux, biochimiques et histopathologiques et tout autre effet observé dans les lots traités et les lots témoins.
Les résultats numériques seront évalués par des méthodes statistiques appropriées et reconnues. Ces méthodes statistiques doivent être choisies lors de la conception de l'étude.
3. COMPTE RENDU
PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
|
Animaux d'expérience:
|
—
|
nombre d'animaux et âge,
|
|
—
|
origine, conditions d'hébergement, etc.,
|
|
—
|
poids de chaque animal au début de l'expérience.
|
|
|
|
Conditions expérimentales:
|
—
|
détails concernant la préparation de la substance à tester, sa stabilité et son homogénéité, s'il y a lieu,
|
|
—
|
justification du choix du véhicule,
|
|
—
|
détails concernant l'administration de la substance à tester,
|
|
—
|
détails concernant la qualité de la nourriture et de l'eau,
|
|
—
|
justification du choix de la dose,
|
|
—
|
spécification des doses administrées, avec indications détaillées concernant le véhicule, le volume et la forme physique de la substance administrée,
|
|
—
|
identité de l'agent protecteur éventuel et détails concernant son administration.
|
|
|
|
Résultats:
|
—
|
données relatives au poids corporel,
|
|
—
|
données concernant la réaction toxique par groupe, y compris la mortalité,
|
|
—
|
nature, gravité et durée des effets cliniques observés (réversibilité éventuelle),
|
|
—
|
description détaillée des méthodes biochimiques et de leurs résultats,
|
|
—
|
résultats de l'autopsie,
|
|
—
|
description détaillée de tous les résultats histopathologiques,
|
|
—
|
le cas échéant, traitement statistique des résultats.
|
|
4. RÉFÉRENCES
Cette méthode reprend la ligne directrice 418 de l'OCDE.
B.38 NEUROTOXICITÉ RETARDÉE DES SUBSTANCES ORGANOPHOSPHORÉES ÉTUDE PAR ADMINISTRATION RÉITÉRÉE SUR 28 JOURS
1. MÉTHODE
1.1. INTRODUCTION
Lorsqu'on évalue les effets toxiques des substances, il est important d'étudier la capacité qu'ont certaines substances à entraîner certains types de neurotoxicité que les autres études de toxicité pourraient ne pas mettre en évidence. Certains composés organophosphorés ont une neurotoxicité différée et doivent faire l'objet de telles études.
Des essais de dépistage in vitro peuvent être utilisés pour identifier les substances susceptibles d'entraîner une polyneuropathie différée; toutefois, des résultats négatifs à l'issue d'études in vitro ne suffisent pas à prouver que la substance testée n'est pas neurotoxique.
Cet essai de neurotoxicité retardée sur 28 jours fournit des informations sur d'éventuels dangers pour la santé pouvant découler d'expositions répétées au cours d'une période déterminée. Il donnera des informations sur la relation dose-effet et pourra fournir une estimation du niveau de dose sans effet néfaste observé, pouvant être utile à l'établissement des critères de sécurité pour l'exposition.
Voir introduction générale, partie B.
1.2. DÉFINITIONS
Les substances organophosphorées comprennent les esters, thioesters organophosphorés non chargés ou anhydrides des acides organophosphoriques, organophosphoniques ou organophos-phoramidiques ou des acides phosphorothioïques, phosphonothioïques ou phosphorothioamidiques apparentés ou d'autres substances pouvant entraîner la neurotoxicité différée parfois observée pour les substances de cette classe.
La neurotoxicité retardée est un syndrome associé à l'installation prolongée et d'ataxie retardée, associée à des axonopathies distales au niveau de la moelle épinière et des nerfs périphériques, et à une inhibition et un vieillissement de l'estérase caractéristique des neuropathies (neuropathy target esterase — NTE) dans les tissus nerveux.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance à tester est administrée quotidiennement par voie orale à des poules domestiques pendant 28 jours. Les animaux sont observés au moins une fois par jour pour rechercher des troubles du comportement, une ataxie et une paralysie, et ce jusqu'au 14e jour après administration de la dernière dose. Des analyses biochimiques sont réalisées, en particulier pour mettre en évidence l'inhibition de l'estérase NTE, sur des poules sélectionnées au hasard dans chaque lot, en général 24 et 48 heures après la dernière administration. Deux semaines après la dernière administration, les poules restantes sont sacrifiées et un examen histopathologique est effectué sur certains tissus nerveux.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Préparation
De jeunes poules adultes exemptes d'affections virales, ne faisant l'objet d'aucune médication susceptible d'interférer et ne présentant pas de troubles de la démarche sont réparties au hasard entre les lots d'expérience et les lots témoins, et sont acclimatées aux conditions du laboratoire pendant au moins 5 jours avant le début de l'étude.
Les cages ou enclos utilisés doivent être suffisamment grands pour que les poules puissent se déplacer librement et pour que leur démarche puisse être aisément observée.
La substance à tester doit être administrée par voie orale chaque jour, sept jours sur sept, de préférence par gavage ou dans des capsules de gélatine. Les substances liquides peuvent être administrées directement ou dissoutes dans un véhicule approprié tel que l'huile de maïs; les substances solides doivent si possible être dissoutes car il se peut que d'importantes quantités de solides administrées dans des capsules de gélatine ne soient pas bien absorbées. Si le véhicule est autre que de l'eau, ses caractéristiques toxiques doivent être connues et, dans le cas contraire, déterminées avant le début de l'essai.
1.4.2. Conditions de l'essai
1.4.2.1. Animaux d'expérience
Il est recommandé d'utiliser de jeunes poules pondeuses adultes (Gallus quitus domesticus) âgées de 8 à 12 mois. Il convient d'utiliser des races et souches de taille standard; les poules doivent normalement avoir été élevées dans des conditions leur permettant de se déplacer librement.
1.4.2.2. Nombre et sexe
En règle générale, il faut prévoir au moins trois lots d'expérience et un lot témoin recevant uniquement le véhicule. Le lot recevant le véhicule seul doit être traité de la même façon que le lot d'expérience, exception faite de l'administration de la substance à tester.
Il faut prévoir suffisamment d'animaux dans chaque lot pour que l'on puisse en sacrifier au moins six pour effectuer les analyses biochimiques (trois lors de chacun des deux temps de prélèvement) et qu'il en survive six après la période d'observation post-traitement de 14 jours.
1.4.2.3. Doses
Les doses doivent être sélectionnées en tenant compte des résultats d'un essai de neurotoxicité retardée après exposition aigué et de toute autre information disponible concernant la toxicité ou la cinétique de la substance à tester. La dose la plus élevée doit être choisie dans le but de produire des effets toxiques et de préférence une neurotoxicité différée, sans entraîner la mort ni des souffrances manifestes. Il convient ensuite de définir une série décroissante de doses afin de mettre en évidence une éventuelle relation dose-effet et une dose minimale n'entraînant aucun effet observable.
1.4.2.4. Essai limite
Lorsqu'un essai réalisé selon le mode opératoire décrit ci-après, à une dose d'au moins 1 000 mg/kg p.c./jour, n'entraîne pas d'effets toxiques observables et que les données relatives aux substances de structure apparentée ne laissent pas supposer une toxicité, il n'est pas nécessaire d'effectuer une étude à une dose plus élevée. L'essai limite est applicable, sauf lorsqu'une exposition humaine fait apparaître la nécessité d'utiliser une dose plus élevée.
1.4.2.5. Période d'observation
Tous les animaux doivent être observés au moins une fois par jour pendant la période d'exposition et pendant les 14 jours suivants, sauf si une autopsie est prévue.
1.4.3. Mode opératoire
La substance à tester est administrée tous les jours, sept jours sur sept, pendant 28 jours.
1.4.3.1. Observation générale
L'observation doit débuter immédiatement après l'exposition. Toutes les poules sont observées attentivement au moins une fois par jour pendant les 28 jours de traitement et pendant les 14 jours suivants ou jusqu'à la date prévue de leur sacrifice. Tous les signes de toxicité doivent être consignés, ainsi que leur date d'apparition, leur type, leur gravité et leur durée. L'observation doit porter sur les troubles du comportement, sans toutefois s'y limiter. L'ataxie doit être mesurée à l'aide d'une échelle d'évaluation ordinale comportant au moins quatre niveaux; la paralysie doit être consignée. Au moins deux fois par semaine, les poules doivent être sorties de leurs cages et soumises à une épreuve d'activité motrice forcée, d'escalade d'échelles, par exemple, afin de faciliter l'observation des effets toxiques minimaux. Les animaux moribonds ou présentant des signes de détresse ou de souffrance intense doivent être immédiatement retirés de l'étude, euthanasiés et autopsiés.
1.4.3.2. Poids corporel
Toutes les poules sont pesées juste avant la première administration de la substance, et au moins une fois par semaine par la suite.
1.4.3.3. Biochimie
Six poules prélevées au hasard dans chacun des lots d'expérience et des lots témoins recevant le véhicule seul sont sacrifiées quelques jours après la dernière administration; le cerveau et la moelle épinière lombaire sont préparés et analysés pour détecter l'inhibition de l'estérase caractéristique des neuropathies (NTE). Il peut en outre s'avérer utile de rechercher l'inhibition de cette estérase sur le nerf sciatique. En règle générale, trois poules du lot témoin et de chaque lot d'expérience sont sacrifiées 24 heures après la dernière administration et trois autres 24 heures plus tard, soit 48 heures après la dernière administration; si les résultats de l'étude par exposition aigué ou d'autres études (étude toxicocinétique, par exemple) indiquent qu'il est préférable de choisir d'autres temps de sacrifice, les temps de sacrifices seront modifiés en conséquence et les documents justificatifs seront fournis.
S'il y a lieu, il est également possible d'effectuer des déterminations de l'acétylcholinestérase (AChE) sur ces échantillons. Toutefois, il peut y avoir réactivation spontanée de l'acétylcholinestérase in vivo, ce qui pourrait faire sous-estimer l'activité de la substance en tant qu'inhibiteur de l'AChE.
1.4.3.4. Autopsie
L autopsie de tous les animaux (sacrifice programmé ou imposé par l'état des animaux) doit comporter un examen de l'aspect du cerveau et de la moelle épinière.
1.4.3.5. Examen histopathologique
Les tissus nerveux des animaux survivants après la période d'observation et non utilisés pour les études biochimiques doivent faire l'objet d'un examen microscopique. Les tissus doivent être fixés in situ par des techniques de perfusion. Les coupes doivent être pratiquées au niveau du cervelet (plan longitudinal moyen), du bulbe rachidien, de la moelle épinière et des nerfs périphériques. Les coupes de la moelle épinière seront pratiquées dans la partie cervicale supérieure, dans la région thoracique moyenne et dans la région sacro-lombaire. Des coupes devront également être réalisées dans la région distale du nerf tibial et au niveau de ses ramifications vers les muscles jumeaux du triceps, ainsi qu'au niveau du nerf sciatique. Les coupes seront colorées à l'aide de colorants appropriés, spécifiques de la myéline et des axones. Dans un premier temps, l'examen microscopique sera effectué sur les tissus conservés de tous les animaux du lot traité à la dose la plus élevée et du lot témoin. Si des effets sont mis en évidence dans le lot traité à la dose la plus élevée, l'examen microscopique devra également porter sur les tissus des animaux des lots traités à la dose intermédiaire et à la dose faible.
2. RÉSULTATS
En règle générale, si les résultats obtenus pour les différents critères d'évaluation retenus dans cette méthode (biochimie, histopathologie et observation du comportement) sont négatifs, il n'est pas nécessaire de poursuivre les essais de neurotoxicité différée. Les résultats équivoques ou non concluants peuvent nécessiter une poursuite de l'étude.
Les résultats doivent être indiqués pour chaque animal, individuellement. En outre, tous les résultats doivent être récapitulés dans un tableau indiquant, pour chaque lot d'expérience, le nombre d'animaux présents au début de l'essai, le nombre d'animaux présentant des lésions, des troubles du comportement ou des modifications biochimiques, le type et la gravité de ces lésions ou symptômes, ainsi que le pourcentage d'animaux présentant chaque type de lésion ou symptôme aux différents niveaux de gravité.
Les résultats de cette étude doivent être évalués du point de vue de l'incidence, de la gravité et de la corrélation entre les effets comportementaux, biochimiques et histopathologiques et tout autre effet observé dans les lots traités et les lots témoins.
Les résultats numériques seront évalués par des méthodes appropriées et reconnues. Ces méthodes statistiques doivent être choisies lors de la conception de l'étude.
3. COMPTE RENDU
PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
|
Animaux d'expérience:
|
—
|
nombre d'animaux et âge,
|
|
—
|
origine, conditions d'hébergement, etc.,
|
|
—
|
poids de chaque animal au début de l'expérience.
|
|
|
|
Conditions expérimentales:
|
—
|
détails concernant la préparation de la substance à tester, sa stabilité et son homogénéité, s'il y a lieu,
|
|
—
|
justification du choix du véhicule,
|
|
—
|
détails concernant l'administration de la substance à tester,
|
|
—
|
détails concernant la qualité de la nourriture et de l'eau,
|
|
—
|
justification du choix de la dose,
|
|
—
|
spécification des doses administrées, avec indications détaillées concernant le véhicule, le volume et la forme physique de la substance administrée,
|
|
—
|
si les déterminations biochimiques ne sont pas pratiquées 24 et 48 heures après la dernière administration, justification du choix d'autres temps.
|
|
|
|
Résultats:
|
—
|
données relatives au poids corporel,
|
|
—
|
données concernant la réaction toxique par groupe, y compris la mortalité,
|
|
—
|
niveau de dose sans effet néfaste observé,
|
|
—
|
nature, gravité et durée des effets cliniques observés (réversibilité éventuelle),
|
|
—
|
description détaillée des méthodes biochimiques et de leurs résultats,
|
|
—
|
résultats de l'autopsie,
|
|
—
|
description détaillée de tous les résultats histopathologiques,
|
|
—
|
le cas échéant, traitement statistique des résultats.
|
|
4. RÉFÉRENCES
Cette méthode reprend la ligne directive 419 de l'OCDE.
B.39. ESSAI IN VIVO DE SYNTHÈSE NON PROGRAMMÉE DE L'ADN (UDS) SUR CELLULES HÉPATIQUES DE MAMMIFÈRE
1. MÉTHODE
Cette méthode est une adaptation de la ligne directrice OCDE TG 486, Essai in vivo de synthèse non programmée de l'ADN (UDS) sur cellules hépatiques de mammifères (1997).
1.1. INTRODUCTION
L'essai in vivo de synthèse non programmée de l'ADN (unscheduled DNA synthesis, UDS) sur cellules hépatiques de mammifère est destiné à identifier les substances qui induisent la réparation de l'ADN dans les cellules hépatiques des animaux traités (1, 2, 3, 4).
Cet essai in vivo constitue une méthode d'étude des effets génotoxiques des substances chimiques dans le foie. L'effet mesuré témoigne de l'altération de l'ADN et de la réparation qui en résulte dans les cellules hépatiques. Comme le foie est généralement le principal site du métabolisme des substances absorbées, il constitue un site approprié pour la détermination in vivo des altérations de l'ADN.
Il ne faut pas avoir recours à cet essai s'il est manifeste que la substance d'essai ou un métabolite réactif n'atteindra pas le tissu cible.
La synthèse non programmée de l'ADN (UDS) est mesurée en déterminant l'incorporation de nucléosides marqués dans les cellules qui ne subissent pas une synthèse programmée (phase S) de l'ADN. La technique la plus couramment utilisée est la détermination par autoradiographie de l'incorporation de thymidine marquée au tritium (3H-TdR). On utilise de préférence le foie de rat pour les essais UDS in vivo. D'autres tissus peuvent être utilisés mais ne font pas l'objet de cette méthode.
La détection d'une UDS dépend du nombre de bases d'ADN excisées et remplacées au site de l'altération. Ainsi, l'essai UDS est particulièrement utile pour détecter la réparation de longues séquences (20-30 bases) induite par la substance. Par contre, la réparation de courtes séquences (1-3 bases) est détectée avec une sensibilité beaucoup plus faible. En outre, des événements mutagènes peuvent résulter d'une non-réparation, d'une réparation erronée d'altérations de l'ADN ou d'une réplication erronée. L'ampleur de l'UDS ne fournit aucune indication quant à la fidélité du processus de réparation. D'autre part, il est possible qu'un mutagène réagisse avec l'ADN sans que l'altération de celui-ci soit réparée par un processus de réparation par excision. Le fait que l'essai UDS ne fournit pas d'informations spécifiques sur l'activité mutagène est compensé par la sensibilité potentielle de ce critère parce qu'il est mesuré dans la totalité du génome.
Voir aussi l'introduction générale de la partie B.
1.2. DÉFINITIONS
Cellules en réparation: nombre net de grains (NNG) supérieur à une valeur préétablie à justifier par le laboratoire effectuant l'essai.
Nombre net de grains (NNG): mesure quantitative de l'activité UDS de cellules lors d'essais UDS autoradiographiques; calculé en soustrayant du nombre de grains nucléaires (GN) le nombre moyen de grains cytoplasmiques dans des zones cytoplasmiques équivalentes au noyau (GC): NNG = GN -GG. Les NNG sont calculés pour les cellules individuelles, puis regroupés pour les cellules d'une culture, de cultures parallèles, etc.
Synthèse réparatrice de l'ADN (UDS): synthèse de réparation de l'ADN après excision et élimination d'un fragment d'ADN contenant une région lésée par des substances chimiques ou physiques.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
L'essai UDS in vivo sur cellules hépatiques de mammifère indique une synthèse de réparation de l'ADN après excision et élimination d'un fragment d'ADN contenant une région altérée par des substances chimiques ou physiques. L'essai repose généralement sur l'incorporation de 3H-TdR dans l'ADN de cellules hépatiques qui présentent une faible fréquence de cellules dans la phase S du cycle cellulaire. L'incorporation de 3H-TdR est généralement déterminée par autoradiographie dans la mesure où cette technique n'est pas sensible aux interférences dues aux cellules en phase S comme c'est le cas du comptage en scintillation liquide, par exemple.
1.4. DESCRIPTION DE LA MÉTHODE
1.4.1. Préparations
1.4.1.1. Sélection de l'espèce animale
Le rat est communément utilisé, bien que n'importe quelle espèce de mammifère appropriée puisse être employée. Il convient d'utiliser des animaux adultes, jeunes et sains issus de souches couramment utilisées en laboratoire. Au début de l'étude, la différence pondérale des animaux doit être minimale et ne pas dépasser ± 20 % du poids moyen de chaque sexe.
1.4.1.2. Conditions d'encagement et régime alimentaire
Les conditions générales décrites dans l'introduction générale de la partie B sont appliquées mais le taux d'humidité devrait se situer aux environs de 50-60 %.
1.4.1.3. Préparation des animaux
De jeunes adultes sains sont répartis par randomisation entre les groupes des animaux témoins et traités. Les cages doivent être disposées de telle manière qu'il n'y ait pas d'effet dû à leur emplacement. Ils sont identifiés individuellement. Ils sont gardés dans leur cage pendant au moins cinq jours avant le début de l'étude, de façon à les acclimater aux conditions du laboratoire.
1.4.1.4. Substance d'essai/préparation
Les substances d'essai solides sont préparées par dissolution ou suspension dans un véhicule ou un solvant approprié, puis éventuellement diluées avant l'administration aux animaux. Les substances d'essai liquides peuvent être administrées directement ou après dilution. Il convient d'utiliser des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les domaines du stockage.
1.4.2. Conditions expérimentales
1.4.2.1. Solvant/véhicule
Le solvant/véhicule choisi ne doit pas avoir d'effet toxique aux doses utilisées ni présenter de réaction chimique avec la substance d'essai. Le recours à des solvants/véhicules inhabituels doit être justifié par des données faisant état de leur compatibilité. On préconise d'envisager d'abord l'utilisation d'un solvant/véhicule aqueux, chaque fois que cela est possible.
1.4.2.2. Témoins
Des témoins positifs et négatifs (solvant/véhicule) doivent être inclus dans chaque partie indépendante de l'expérience. Les animaux des groupes témoins sont traités exactement comme ceux des groupes traités, mise à part l'administration de la substance d'essai.
Les témoins positifs doivent être des substances dont on sait qu'elles produisent une UDS à des niveaux d'exposition supposés conduire à un accroissement détectable par rapport au bruit de fond. Les témoins positifs pour lesquels une activation métabolique est nécessaire doivent être utilisés à des doses qui donnent lieu à une réponse modérée (4). Les doses peuvent être choisies de façon que les effets soient évidents et que l'identité des lames codées ne soit pas révélée immédiatement à l'examinateur. Les substances indiquées dans le tableau ci-dessous sont des exemples de témoins positifs.
|
Moment de prélèvement
|
Substance
|
№ CAS
|
No Einecs
|
|
Précoce (2-4 heures)
|
N-nitrosodiméthylamine
|
62-75-9
|
200-249-8
|
|
Tardif (12-16 heures)
|
N-2-fluorénylacétamide (2-AAF)
|
53-96-3
|
200-188-6
|
D'autres témoins positifs appropriés peuvent être utilisés. Le témoin positif doit être administré par une voie différente de celle utilisée pour la substance d'essai.
1.5. MODE OPÉRATOIRE
1.5.1. Nombre et sexe des animaux
Il convient d'utiliser un nombre adéquat d'animaux afin de tenir compte de variations interindividuelles de la réponse à l'essai. Il doit y avoir au moins 3 animaux analysables par groupe. Lorsqu'on dispose d'une importante quantité de données historiques, les groupes témoins négatifs et positifs peuvent ne comprendre que 1 ou 2 animaux.
Si, au moment de l'étude, on dispose de données historiques effectuées avec la même espèce et ayant utilisé la même voie d'administration, qui démontrent l'absence de différence importante de toxicité entre sexes, on peut effectuer l'essai avec des animaux d'un seul sexe, de préférence des mâles. Si l'exposition humaine est spécifique du sexe, par exemple dans le cas de certains produits pharmaceutiques, des animaux du sexe approprié doivent être utilisés pour l'essai.
1.5.2. Modalité de traitement
Les substances d'essai sont généralement administrées en une seule fois.
1.5.3. Niveaux de dose
Normalement, on utilise au moins deux niveaux de dose. La dose la plus élevée est définie comme la dose qui produit des signes de toxicité tels qu'ils font supposer que des doses plus fortes, administrées suivant les mêmes modalités, seraient létales. En général, la dose la plus faible est comprise entre la moitié et le quart de la dose la plus forte.
Les substances possédant une activité biologique spécifique à des doses faibles et non toxiques (comme les hormones et les mitogènes) peuvent faire exception à ces critères d'établissement de doses et doivent être évaluées cas par cas. Si une étude préliminaire est réalisée afin d'évaluer les niveaux de dose, cette étude doit être effectuée dans le même laboratoire en utilisant la même espèce, la même souche, des animaux du même sexe et la même modalité de traitement que ceux de l'étude principale.
La dose la plus élevée peut aussi être définie comme étant celle qui produit certains indices de toxicité dans le foie (noyaux pyenotiques, par exemple).
1.5.4. Test limite
Si un essai réalisé avec au moins 2 000 mg/kg de poids corporel, en une seule dose ou en deux doses administrées le même jour, n'engendre aucun effet toxique observable et si une génotoxicité est improbable d'après les données relatives à des substances de structure voisine, on peut considérer qu'une étude complète n'est pas nécessaire. Suivant l'importance de l'exposition humaine probable, on peut envisager d'utiliser des doses plus fortes dans l'essai de dose limite.
1.5.5. Voies d'administration
La substance d'essai est généralement administrée par gavage à l'aide d'une sonde gastrique ou d'une canule d'intubation adaptée. D'autres voies d'exposition sont acceptables lorsqu'elles peuvent être justifiées. La voie péritonéale n'est toutefois pas recommandée, étant donné qu'elle peut conduire à une exposition directe du foie à la substance d'essai plutôt que par l'intermédiaire du système circulatoire. Le volume maximal de liquide administre en une fois par gavage ou par injection dépend de la taille de l'animal. Ce volume ne devrait pas dépasser 2 ml/100 g de poids corporel. Une justification doit être fournie si des volumes plus importants sont administrés. Il convient de réduire au minimum la variation du volume administré en ajustant la concentration à tous les niveaux de doses, sauf pour les produits irritants ou corrosifs dont les effets sont généralement exacerbés lorsqu'on augmente la concentration.
1.5.6. Préparation des cellules hépatiques
Les cellules hépatiques des animaux traités sont normalement préparées 12-16 heures après le traitement. Un prélèvement supplémentaire effectué plus tôt (normalement 2-4 heures après le traitement) est en général nécessaire, à moins qu'il y ait une réponse manifestement positive après 12-16 heures. Toutefois, d'autres délais de prélèvement peuvent être utilisés s'ils sont justifiés sur la base de données toxicocinétiques.
Les cultures à court terme de cellules hépatiques de mammifère sont habituellement établies en perfusant le foie in situ avec la collagénase et en laissant les cellules fraîchement dissociées se fixer sur une surface appropriée. Les cellules hépatiques des témoins négatifs doivent présenter une viabilité (5) d'au moins 50 pour cent.
1.5.7. Détermination de l'UDS
Les cellules hépatiques de mammifère fraîchement isolées sont généralement incubées dans un milieu contenant du 3H-TdR pendant une durée appropriée (3-8 heures, par exemple). A la fin de la période d'incubation, le milieu est enlevé des cellules, qui peuvent ensuite être incubées avec un milieu contenant un excès de thymidine non marquée, afin de réduire la radioactivité non incorporée (cold chase). Les cellules sont ensuite rincées, fixées et séchées. Pour les périodes d'incubation plus longues, la cold chase n'est pas absolument nécessaire. Les lames sont plongées dans une émulsion autoradiographique, maintenues dans l'obscurité (réfrigérées pendant 7-14 jours, par exemple), développées et colorées. On procède ensuite au comptage des grains d'argent. On prépare deux à trois lames pour chaque animal.
1.5.8. Analyse
Les préparations sur lame doivent contenir un nombre suffisant de cellules de morphologie normale pour qu'une évaluation probante de l'UDS soit possible. Les préparations sont examinées au microscope en vue de la recherche de signes de cytotoxirité manifeste (pyenose, diminution du niveau de marquage radioactif, par exemple).
Les lames doivent être codées avant le comptage des grains. On analyse normalement 100 cellules de chaque animal sur au moins deux lames; l'analyse d'un nombre inférieur à 100 cellules/animal doit être justifié. Le nombre de grains n'est pas déterminé pour les noyaux en phase S, mais la proportion de cellules en phase S peut être enregistrée.
Le niveau d'incorporation de 3H-TdR dans le noyau et le cytoplasme des cellules morphologiquement normales, tel qu'il est mis en évidence par le dépôt de grains d'argent, doit être déterminé à l'aide de méthodes appropriées.
Le nombre de grains est déterminé au niveau du noyau (grains nucléaires, GN) et des zones du cytoplasme équivalentes au noyau (grains cytoplasmiques, GC). Le nombre de GC est déterminé sur la base soit de la région du cytoplasme la plus fortement marquée, soit de la moyenne de deux ou trois régions proches du noyau choisies au hasard. D'autres méthodes de comptage (cellules entières, par exemple) peuvent être utilisées si on peut les justifier (6).
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
Les données doivent être indiquées individuellement pour chaque culture. En outre, toutes les données doivent être résumées sous forme de tableaux. Le nombre net de grains (NNG) doit être déterminé pour chaque cellule, chaque animal, chaque dose et chaque temps de prélèvement en soustrayant le nombre de GC du nombre de GN. Si des «cellules en réparation» sont comptées, les critères pour définir les «cellules en réparation» doivent être justifiés et se fonder sur des données historiques ou sur des témoins négatifs concomitants. Les résultats numériques peuvent être évalués à l'aide de méthodes statistiques. Dans ce cas, il faut choisir et justifier les tests statistiques avant la réalisation de l'étude.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Exemples de critères permettant de conclure à une réponse positive ou négative:
|
positive
|
i)
|
valeur NNG supérieure à un seuil préétabli justifié sur la base des données historiques du laboratoire, ou
|
|
|
ii)
|
valeur NNG significativement supérieure à celle du témoin concomitant,
|
|
négative
|
i)
|
valeur NNG inférieure ou égale au seuil des contrôles historiques, ou
|
|
|
ii)
|
valeur NNG non significativement supérieure à celle du témoin concomitant.
|
Il convient de prendre en considération la pertinence biologique des données (paramètres tels que la variation interindividuelle, la relation dose-réponse et la cytotoxicité). L'évaluation des résultats peut s'appuyer sur des méthodes statistiques mais la signification statistique ne doit pas être le seul facteur décisif pour conclure à une réponse positive.
Bien que la plupart des essais donneront des résultats nettement positifs ou négatifs, dans certains cas rares l'ensemble des résultats d'un essai n'autorise pas une conclusion catégorique sur l'activité de la substance étudiée. Les résultats peuvent rester équivoques ou discutables indépendamment du nombre de répétitions de l'essai.
Un résultat positif à l'essai UDS in vivo sur cellules hépatiques de mammifère indique que la substance d'essai induit des altérations de l'ADN dans les cellules hépatiques de mammifère in vivo qui peuvent être réparées par la synthèse non programmée de l'ADN in vitro. Un résultat négatif signifie que, dans les conditions de l'essai, la substance d'essai n'induit pas d'altération de l'ADN détectable par cet essai.
Le fait que la substance d'essai atteint la circulation générale ou spécifiquement le tissu cible (toxicité systémique) devrait être discutée.
3. RAPPORT
RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
|
|
Solvant/véhicule:
|
—
|
justification du choix du véhicule,
|
|
—
|
solubilité et stabilité de la substance d'essai dans le solvant/véhicule, si elles sont connues.
|
|
|
|
Animaux d'expérience:
|
—
|
espèce/souche utilisée,
|
|
—
|
nombre, âge et sexe des animaux,
|
|
—
|
source, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
poids individuels des animaux au début de l'essai, y compris l'intervalle des poids, la moyenne et l'écart-type dans chaque groupe.
|
|
|
|
Conditions expérimentales:
|
—
|
données relatives aux témoins positifs et négatifs (solvant/véhicule),
|
|
—
|
résultats de l'étude de détermination des doses utiles, si elle a été réalisée,
|
|
—
|
justification du choix des doses employées,
|
|
—
|
détails concernant la préparation de la substance d'essai,
|
|
—
|
détails concernant l'administration de la substance d'essai,
|
|
—
|
justification du choix de la voie d'administration,
|
|
—
|
méthodes pour vérifier que la substance a atteint ta circulation générale ou le tissu cible, le cas échéant,
|
|
—
|
conversion de la concentration (ppm) de la substance d'essai dans l'eau de boisson ou la nourriture en dose réelle (mg/kg de poids corporel/jour), s'il y a lieu,
|
|
—
|
détails sur la qualité de la nourriture et de l'eau,
|
|
—
|
description détaillée des modalités de traitement et de prélèvement,
|
|
—
|
méthodes de mesure de la toxicité,
|
|
—
|
méthode de préparation et de culture des cellules hépatiques,
|
|
—
|
technique autoradiographique utilisée,
|
|
—
|
nombre de lames préparées et nombre de cellules analysées,
|
|
—
|
critères de décision concernant les résultats positifs, négatifs et équivoques.
|
|
|
|
Résultats:
|
—
|
valeurs moyennes du nombre de grains nucléaires, du nombre de grains cytoplasmiques et du nombre net de grains pour chaque lame, animal et groupe,
|
|
—
|
relation dose-réponse, si possible,
|
|
—
|
évaluation statistique, le cas échéant,
|
|
—
|
données sur les témoins négatifs (solvant/véhicule) et positifs concomitants,
|
|
—
|
données sur les témoins négatifs (solvant/véhicule) et positifs antérieurs (étendues, moyennes et écarts-types),
|
|
—
|
nombre de «cellules en réparation», s'il a été déterminé,
|
|
—
|
nombre de cellules en phase S, s'il a été déterminé,
|
|
—
|
viabilité des cellules.
|
|
|
|
Discussion des résultats.
|
4. BIBLIOGRAPHIE
|
(1)
|
Ashby, J., Lefevre, P. A., Burlinson, B. and Penman, M. G. (1985), An Assessment of the In Vivo Rat Hepatocyte DNA Repair Assay, Mutation Res., 156, pp. 1 — 18.
|
|
(2)
|
Butterworth, B. E., Ashby, J., Bermudez, E., Casciano, D., Mirsalis, J., Probst G. and Williams, G. (1987), A Protocol and Guide for the In Vivo Rat Hepatocyte DNA-Repair Assay, Mutation Res., 189, pp. 123 — 133.
|
|
(3)
|
Kennelly, J. C, Waters, R., Ashby. J., Lefevre, P. A., Burlinson B., Benford, D. J., Dean, S. W. and Mitchell I. de G. (1993), In Vivo Rat Liver UDS Assay, in: Kirkland D. J. and Fox M, (eds.), Supplementary Mutagenicity Tests: UKEM Recommended Procedures. UKEMS Subcommittee on Guidelines for Mulagenicity Testing Report. Part II revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 52 — 77.
|
|
(4)
|
Madle. S., Dean. S. W., Andrae, U., Brambilla, G., Burlinson, B., Doolittle, D. J., Furihata, C, Hertner, T., McQueen, C. A. and Mori, H. (1993), Recommendations for the Performance of UDS Tests In Vitro and In Vivo, Mutations Res., 312, pp. 263 — 285.
|
|
(5)
|
Fautz, R., Hussain, B., Efstathiou, E. and Hechenberger-Freudl, C. (1993), Assessment of the Relation Between the Initital Viability and the Attachment of Freshly Isolated Rat Hepatocytes Used for the In Vivo/In Vitro DNA Repair Assay (UDS), Mutation Res. 291. pp. 21 — 27.
|
|
(6)
|
Mirsalis, J. C, Tyson, C. K. and Butterworth, B. E. (1982), Detection of Genotoxic Carcinogens in the In Vivo/In Vitro Hepafocyte HNA Repair Assay, Environ Mutagen, 4, pp. 553 — 562.
|
B.40. CORROSION CUTANÉE IN VITRO: ESSAI DE RESISTANCE ÉLECTRIQUE TRANSCUTANÉE (RET)
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 430 (2004) de l'OCDE.
1.1. INTRODUCTION
La corrosion cutanée désigne la survenue de lésions irréversibles de la peau après l'application d'un matériel d'essai [selon la définition du système général harmonisé de classification et d'étiquetage des produits chimiques (SGH)] (1). La présente méthode propose une évaluation de la corrosivité qui n'est pas réalisée sur des animaux vivants.
Traditionnellement, l'évaluation de la corrosivité cutanée impliquait le recours à des animaux de laboratoire (2). Dans le souci d'éviter les douleurs et les souffrances ainsi infligées aux animaux, la méthode d'essai B.4 a été révisée afin de permettre la détermination de la corrosion cutanée par la mise en œuvre de méthodes de substitution in vitro.
La conduite d'études de pré-validation a néanmoins été une première étape dans la définition d'essais de substitution susceptibles d'être utilisés pour des essais de corrosivité cutanée menés à des fins réglementaires (3). Par la suite, une étude formelle de validation des méthodes in vitro d'évaluation de la corrosion cutanée (4) (5) a été conduite (6) (7) (8). Sur la base des résultats de ces études et d'autres publications, il est maintenant recommandé d'évaluer la corrosivité cutanée in vivo à l'aide des essais suivants (9) (10) (11) : l'essai utilisant un modèle de peau humaine (voir méthode d'essai B.40 bis) et l'essai de résistance électrique transcutanée (la présente méthode).
Selon une étude de validation et d'autres études publiées, l'essai de résistance électrique transcutanée (RET) sur peau de rat (12) (13) permet de faire la distinction de manière fiable entre substances corrosives et non corrosives pour la peau (5) (9).
L'essai décrit dans cette méthode permet d'identifier les substances et mélanges chimiques corrosifs. Appuyé par d'autres éléments de détermination fondés sur d'autres informations existantes (pH, relations structure-activité, données humaines et/ou animales, par exemple), il permet également d'identifier les substances et mélanges non corrosifs (1) (2) (11) (14). En revanche, il ne fournit pas d'information sur l'irritation cutanée, et contrairement au système général harmonisé de classification (SGH) (1), il ne permet pas non plus la sous-catégorisation des substances corrosives.
Pour une évaluation complète des effets cutanés locaux après une exposition unique, il est recommandé de suivre la démarche expérimentale séquentielle présentée en supplément à la méthode d'essai B.4(2) et proposée dans le système général harmonisé (1). Cette démarche expérimentale prévoit la conduite d'essais in vitro de corrosion cutanée (tel que celui décrit dans la présente méthode) et d'irritation cutanées avant d'envisager des essais sur des animaux vivants.
1.2. DÉFINITIONS
Corrosion cutanée in vivo: survenue de lésions irréversibles de la peau. En l'occurrence, il s'agit de nécrose visible, à travers l'épiderme et jusque dans le derme, suite à l'application d'une substance d'essai pendant une durée allant jusqu'à quatre heures. Les réactions corrosives se traduisent généralement par des ulcères, des saignements, des croûtes sanguinolentes et, à la fin de la période d'observation, soit à 14 jours, par une décoloration due au blanchissement de la peau, des zones complètes d'alopécie, et des cicatrices. Un examen histopathologique peut être envisagé pour évaluer les lésions douteuses.
Résistance électrique transcutanée (RET): mesure de l'impédance électrique de la peau, exprimée par une valeur de résistance en kilo-Ohms. Il s'agit d'une méthode simple et fiable permettant d'évaluer la fonction de barrière de la peau, par l'enregistrement du passage des ions à travers la peau à l'aide d'un pont de mesure de Wheatstone.
1.3. SUBSTANCES DE RÉFÉRENCE
Tableau 1
Substances chimiques de référence
|
Nom
|
No EINECS
|
No CAS
|
|
|
Diamino-1,2 propane
|
201-155-9
|
78-90-0
|
Gravement corrosif
|
|
Acide acrylique
|
201-177-9
|
79-10-7
|
Gravement corrosif
|
|
O-tert. Butylphénol
|
201-807-2
|
88-18-6
|
Corrosif
|
|
Hydroxyde de potassium (10 %)
|
215-181-3
|
1310-58-3
|
Corrosif
|
|
Acide sulfurique (10 %)
|
231-639-5
|
7664-93-9
|
Corrosif
|
|
Acide octanoïque (acide caprylique)
|
204-677-5
|
124-07-02
|
Corrosif
|
|
4-amino-1,2,4-triazole
|
209-533-5
|
584-13-4
|
Non corrosif
|
|
Eugenol
|
202-589-1
|
97-53-0
|
Non corrosif
|
|
Bromure de phénétyle
|
203-130-8
|
103-63-9
|
Non corrosif
|
|
Perchloroéthylène
|
204-825-9
|
27-18-4
|
Non corrosif
|
|
Acide isostéarique
|
250-178-0
|
30399-84-9
|
Non corrosif
|
|
4-(méthylthio)-benzaldéhyde
|
222-365-7
|
3446-89-7
|
Non corrosif
|
La plupart des substances chimiques données ici proviennent de la liste des substances chimiques sélectionnées par le CEVMA dans son étude de validation internationale (4). Les choix du CEVMA sont fondés sur les critères suivants:
|
i)
|
nombre égal de substances corrosives et non corrosives;
|
|
ii)
|
substances disponibles dans le commerce et couvrant la plupart des classes chimiques pertinentes;
|
|
iii)
|
insertion de substances extrêmement corrosives et moins corrosives, de façon à permettre une distinction basée sur le pouvoir de corrosion;
|
|
iv)
|
substances chimiques manipulables en laboratoire sans présenter d'autres risques majeurs que la corrosivité.
|
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Le matériel d'essai est appliqué pour une durée n'excédant pas 24 heures sur la surface épidermique de disques de peau, placés dans un système d'essai à deux compartiments pour lequel les disques de peau font office de séparation entre les compartiments. Ces disques cutanés sont prélevés sur la peau de jeunes rats âgés de 28 à 30 jours et humainement euthanasiés. Les matières corrosives sont identifiées sur la base de leur capacité à provoquer la perte de l'intégrité du stratum corneum normal et de la fonction de barrière, cette perte étant mesurée comme une diminution de la RET en deçà d'une valeur seuil (12). Dans le cas des rats, une valeur de seuil de la RET de 5 kΩ. a été retenue, sur la base de nombreuses données relatives à un large éventail de substances chimiques, desquelles il ressortait que l'immense majorité des valeurs étaient soit très supérieures (souvent > 10 kΩ), soit très inférieures (souvent < 3 kΩ,) à cette valeur de seuil (12). De manière générale, les matériels non corrosifs sur les animaux, mais irritants ou non irritants, ne font pas baisser la RET en dessous de ce seuil. Par ailleurs, l'utilisation d'autres préparations de peau ou d'autres équipements est susceptible de modifier la valeur de seuil, ce qui impose de procéder à une validation supplémentaire.
Une étape de fixation d'un colorant est incorporée dans la procédure d'essai pour confirmer les résultats positifs de la RET présentant des valeurs autour de 5 kΩ. Cette étape de fixation d'un colorant détermine en effet si l'accroissement de la perméabilité ionique est imputable à la destruction physique du stratum corneum. Dans la pratique, la méthode de l'essai RET sur peau de rat prédit très bien la corrosivité in vivo chez le lapin évaluée par la méthode d'essai B.4 (2). A cet égard, il convient de noter que l'essai in vivo sur des lapins est extrêmement prudent concernant la corrosivité et l'irritation cutanées par rapport à l'essai sur modèle de peau humaine (15).
1.5. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.5.1. Animaux
Les rats sont l'espèce la mieux adaptée pour cet essai dans la mesure où la sensibilité de leur peau aux substances chimiques employées a déjà été démontrée (10). L'âge (au moment du prélèvement de la peau) et la souche utilisée sont des critères déterminants, puisqu'il est essentiel que les follicules pileux soient en phase dormante avant le début de la pousse de la fourrure adulte.
Les poils du dos et des flancs des jeunes rats (Wistar ou souche comparable) mâles ou femelles, âgés approximativement de 22 jours, sont soigneusement coupés à l'aide d'une petite tondeuse. Les animaux sont ensuite nettoyés soigneusement avec un linge humide, la zone tondue étant plongée dans une solution antibiotique (contenant, par exemple, de la streptomycine, de la pénicilline, du chloramphénicol et de l'amphotéricine à des concentrations efficaces pour inhiber la croissance bactérienne). Les animaux sont lavés une nouvelle fois avec des antibiotiques le troisième ou le quatrième jours après le premier lavage, et doivent ensuite être utilisés dans un délai de 3 jours, lorsque le stratum corneum a récupéré de la tonte.
1.5.2. Préparation des disques cutanés
Les animaux, âgés de 28 à 30 jours (cet âge est particulièrement important) sont euthanasiés de façon humaine. On prélève la peau dorso-latérale de chaque animal et on la débarrasse soigneusement du tissu adipeux en excès. Des disques cutanés, d'un diamètre approximatif de 20 mm chacun sont prélevés. La peau peut être stockée avant d'utiliser les disques lorsqu'il est établi que les données de contrôle positives et négatives sont équivalentes à celles obtenues avec de la peau fraîche.
Chaque disque cutané est placé sur l'extrémité d'un tube PTFE (polytétrafluoroéthylène) en veillant à ce que la surface épidermique soit en contact avec le tube. Après avoir ajusté de force un joint torique en caoutchouc sur l'extrémité du tube pour maintenir la peau en place, on coupe le tissu excédentaire. Les dimensions du tube et du joint torique sont indiquées à la Figure 2. Le joint torique en caoutchouc est ensuite soigneusement fixé à l'extrémité du tube PTFE de manière étanche à l'aide de vaseline. Le tube est introduit dans une chambre réceptrice contenant une solution de sulfate de magnésium (154 mM) dans lequel il est maintenu par une pince à ressort (Figure 1). Le disque cutané doit être complètement immergé dans la solution de sulfate de magnésium (MgSO4). Entre 10 et 15 disques cutanés peuvent être prélevés de la peau d'un rat.
Avant le début de l'essai, on mesure, pour chaque peau de rat, la résistance électrique de deux disques cutanés, à titre de contrôle de la qualité. Pour que les autres disques d'une même peau soit valables pour l'essai, il faut que les deux disques donnent des valeurs de résistance supérieures à 10 kΩ. Si la résistance est inférieure à 10 kΩ, les disques restants provenant de la même peau doivent être éliminés.
1.5.3. Application des substances d'essai et de contrôle
Pour chaque étude, des substances de contrôle positif et négatif doivent être utilisées parallèlement à la substance d'essai, de façon à garantir la qualité des résultats du modèle expérimental. Dans ce contexte, les disques cutanés employés doivent provenir d'un même animal. Les substances de contrôle positif et négatif recommandées sont respectivement l'acide hydrochlorique 10 M et l'eau distillée.
Les substances d'essai liquides (150 μl) sont appliquées sur la surface épidermique à l'intérieur du tube. Pour l'essai de substances solides, une quantité suffisante du solide est appliquée uniformément sur le disque de peau, de façon à ce que la totalité de la surface de l'épiderme soit couverte. Après avoir ajouté de l'eau désionisée (150 μl) sur les solides, on agite le tube doucement. Afin d'optimiser le contact avec la peau, certains solides doivent être chauffés à 30 oC pour dissoudre la substance d'essai ou être broyés pour obtenir des grains ou une poudre.
On utilise trois disques cutanés pour chaque substance d'essai. Ces substances sont appliquées pendant 24 heures à une température de 20 à 23 oC, puis éliminées complètement par lavage sous l'eau du robinet à 30 oC maximum.
1.5.4. Mesures de la RET
L'impédance de la peau, c'est-à-dire la RET, est mesurée à l'aide d'un pont de mesure de Wheatstone en courant alternatif basse tension (13), présentant les caractéristiques suivantes: tension de 1 à 3 Volts, courant alternatif de forme sinusoïdale ou rectangulaire compris entre 50 et 1000 Hz, et une plage de mesure au moins comprise entre 0,1 et 30 kΩ Le pont de mesure utilisé dans l'étude de validation permettait de mesurer l'inductance, la capacitance et la résistance jusqu'à des valeurs de 2 000 H, 2 000 μF et 2 M respectivement, à des fréquences de 100 Hz ou 1 kHz, en utilisant des valeurs en parallèle ou en série. Pour les besoins de l'essai RET de corrosivité, les mesures sont enregistrées en résistance, à une fréquence de 100 Hz et à l'aide de valeurs en série. Avant la mesure de la résistance électrique, on réduit la tension de surface de la peau en ajoutant un volume suffisant d'éthanol à 70 % pour couvrir l'épiderme. Après quelques secondes, on enlève l'éthanol du tube, puis on hydrate le tissu par l'addition de 3 ml d'une solution de sulfate de magnésium (154 mM). Les électrodes du pont de mesure sont placées de part et d'autre du disque cutané pour prendre la mesure de la résistance en kΩ/disque cutané (Figure 1). Les dimensions des électrodes et la longueur d'électrode en dessous des pinces crocodiles sont indiquées dans la Figure 2. La pince maintenant l'électrode intérieure repose sur la partie supérieure du tube PTFE pendant la mesure de la résistance, afin que la longueur d'électrode immergée dans la solution de sulfate de magnésium reste constante. L'électrode extérieure est introduite dans le compartiment receveur de telle manière qu'elle repose sur le fond de celui-ci. La distance entre la pince à ressort et la partie inférieure du tube PTFE est maintenue constante (Figure 2), cette distance influençant la valeur de résistance obtenue. Par conséquent, la distance entre l'électrode intérieure et le disque cutané doit être constante et minimale (1 à 2 mm).
Il convient de noter que si la valeur de résistance mesurée est supérieure à 20 kΩ, ceci peut être dû au fait que des restes de la substance d'essai couvrent la surface épidermique du disque de peau. On peut essayer de retirer davantage de substance d'essai, par exemple en fermant de façon étanche le tube PTFE avec le pouce revêtu d'un gant et en l'agitant pendant 10 secondes environ. On élimine ensuite la solution de sulfate de magnésium et on répète la mesure de la résistance avec une nouvelle solution de sulfate de magnésium.
Les caractéristiques et dimensions de l'appareil d'essai et de la procédure expérimentale peuvent avoir une incidence sur les valeurs de RET obtenues. Le seuil de corrosivité de 5 kΩ. a été défini sur la base des données obtenues à partir de l'appareil et de la procédure spécifiques décrits dans cette méthode. Avec un autre équipement ou dans des conditions d'essai différentes, les valeurs de seuil et de contrôle peuvent être différentes. Par conséquent, il est nécessaire d'étalonner la méthodologie et la valeur de seuil de résistance en testant différentes substances de référence étalons, choisies parmi celles utilisées dans l'étude de validation (4) (5), ou dans des classes chimiques équivalentes à celles des substances étudiées. Le Tableau 1 présente un ensemble de substances de référence qui conviennent.
1.5.5. Méthodes avec fixation d'un colorant
L'exposition à certains matériels non corrosifs peut entraîner une réduction de la résistance sous la valeur de seuil de 5 kΩ, permettant ainsi le passage d'ions à travers le stratum corneum, ce qui réduit ainsi la résistance électrique (5). Par exemple, les substances organiques neutres et les substances tensio-actives (détergents, émulsifiants et autres agents de surface) peuvent évacuer les lipides de la peau et accroître la perméabilité aux ions de la barrière cutanée. Par conséquent, si les valeurs de RET des substances d'essai sont inférieures à, ou proches de, 5 kΩ, en l'absence de toute lésion visible, une étude de pénétration d'un colorant doit être menée sur les tissus traités et de contrôle, afin de déterminer si ces valeurs de RET résultent d'une perméabilité accrue ou d'une corrosion de la peau (3) (5). S'il s'agit d'une corrosion, autrement dit si le stratum corneum est rompu, le colorant sulforhodamine B appliqué à la surface de la peau pénètre rapidement et teinte les tissus sous-jacents. Ce colorant reste stable au contact d'un large éventail de substances chimiques et n'est pas affecté par la procédure d'extraction décrite ci-après.
1.5.5.1. Application et élimination du colorant Sulforhodamine B
Après l'essai de RET, le sulfate de magnésium est évacué du tube et la peau est soigneusement examinée. Si aucune lésion majeure n'est visible, 150 μl d'une dilution à 10 pour cent (p/v) de sulforhodamine B (Acid Red 52; CI. 45100; numéro EINECS 222-529-8; numéro CAS 3520-42-1) dans de l'eau distillée sont appliqués sur la surface épidermique de chaque disque cutané pendant 2 heures. Les disques cutanés sont lavés ensuite sous un jet d'eau courante à la température ambiante pendant environ 10 secondes pour éliminer le colorant excédentaire ou non fixé. Chaque disque cutané est soigneusement enlevé du tube PTFE et placé dans un flacon (par exemple un flacon à scintillation en verre de 20 ml) contenant de l'eau désionisée (8 ml). Les flacons sont agités doucement pendant 5 minutes pour éliminer complètement tout colorant excédentaire ou non fixé. Après avoir répété l'opération de rinçage, on enlève les disques cutané et on les place dans des flacons contenant 5 ml de dodécylsulfate de sodium (SDS) à 30 pour cent (p/v) dans de l'eau distillée, puis on les incube pendant une nuit à 60 oC.
Après incubation, chaque disque de peau est enlevé et jeté et la solution restante est centrifugée pendant 8 minutes à 21 oC (force centrifuge relative ~175 x g). Un échantillon de 1 ml de surnageant est ensuite dilué dans un rapport de 1 à 5 (v/v) [soit 1 ml + 4 ml] avec du SDS à 30 pour cent (p/v) dans de l'eau distillée. La densité optique (DO) de la solution est mesurée à environ 565 nm.
1.5.5.2. Calcul de la teneur en colorant
La teneur de chaque disque en colorant sulforhodamine B est calculée sur la base des valeurs de DO (5) (coefficient d'extinction molaire de la sulforhodamine B à 565 nm = 8,7 × 104; poids moléculaire = 580). La teneur en sulforhodamine B est déterminée pour chaque disque de peau à l'aide d'une courbe étalon appropriée et on calcule ensuite une teneur moyenne en colorant pour les essais répétés.
2. RÉSULTATS
Les valeurs de résistance (kΩ) et de teneur moyenne en colorant (μg/disque), le cas échéant, pour le matériel d'essai et pour les contrôles positifs et négatifs doivent être présentées sous forme de tableau (données individuelles et moyennes ± écart-type), y compris les données des essais répétés/répliqués, les valeurs moyennes et individuelles.
2. INTERPRÉTATION DES RÉSULTATS
Les valeurs moyennes de la RET sont acceptées à condition que les valeurs des contrôles positifs et négatifs effectués parallèlement se situent dans les fourchettes acceptables pour la méthode en laboratoire d'essai. Les fourchettes acceptables de valeur de résistance pour la méthodologie et l'appareillage décrits ci-avant sont les suivantes:
|
Contrôle
|
Substance
|
Plage de résistance (kΩ)
|
|
Positif
|
Acide hydrochlorique 10 M
|
0,5 – 1,0
|
|
Négatif
|
Eau distillée
|
10 – 25
|
Les valeurs moyennes de la fixation du colorant sont acceptées à condition que les valeurs des contrôles effectués parallèlement se situent dans la fourchette acceptable pour la méthode. Les fourchettes acceptables de teneur en colorant pour les substances de contrôle proposées pour la méthodologie et l'appareillage décrits ci-avant sont les suivantes:
|
Contrôle
|
Substance
|
Plage de teneur en colorant (μg/disque)
|
|
Positif
|
Acide hydrochlorique 10 M
|
40 – 100
|
|
Négatif
|
Eau distillée
|
15 – 35
|
On considère que la substance d'essai est non corrosive pour la peau :
|
i)
|
si la valeur moyenne de la RET pour la substance d'essai est supérieure à 5 kΩ, ou
|
|
ii)
|
si la valeur moyenne de la RET est inférieure ou égale à 5 kΩ, et
|
—
|
que le disque cutané ne présente aucune lésion visible, et
|
|
—
|
que la teneur moyenne en colorant du disque est bien inférieure à la teneur moyenne en colorant du disque de contrôle positif (acide hydrochlorique 10 M) obtenue parallèlement.
|
|
On considère que la substance d'essai est corrosive pour la peau:
|
i)
|
si la valeur moyenne de la RET est inférieure ou égale à 5 kΩ et si le disque cutané est visiblement lésé, ou
|
|
ii)
|
si la valeur moyenne de la RET est inférieure ou égale à 5 kΩ, et
|
—
|
que le disque cutané ne présente aucune lésion visible, et
|
|
—
|
que la teneur moyenne en colorant du disque est bien inférieure à la teneur moyenne en colorant du disque de contrôle positif (acide hydrochlorique 10 M) obtenue parallèlement.
|
|
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit contenir au moins les informations suivantes:
|
|
Substances d'essai et de contrôle:
|
—
|
nom(s) chimique(s), tels que le nom IUPAC ou CAS, et le cas échéant le numéro CAS;
|
|
—
|
pureté et composition de la substance ou de la préparation [en pourcentage(s) en poids] et état physique;
|
|
—
|
propriétés physico-chimiques (telles que l'état physique, le pH, la stabilité, l'hydrosolubilité) utiles pour la conduite de l'étude;
|
|
—
|
le cas échéant, traitement des substances d'essai et de contrôle avant l'essai (par exemple, chauffage, réduction en poudre) ;
|
|
|
|
Animaux d'essai:
|
—
|
âge au moment du prélèvement de peau
|
|
—
|
source, conditions d'hébergement, alimentation, etc.
|
|
—
|
détails de la préparation de la peau
|
|
|
|
Conditions d'essai:
|
—
|
courbes d'étalonnage de l'appareil d'essai;
|
|
—
|
courbes d'étalonnage des performances de l'essai de fixation d'un colorant;
|
|
—
|
détails de la procédure d'essai utilisée pour les mesures de la RET;
|
|
—
|
détails de la procédure d'essai utilisée pour l'évaluation de la fixation d'un colorant, le cas échéant;
|
|
—
|
description de toute modification de la procédure d'essai description des critères d'évaluation utilisés.
|
|
|
|
Résultats:
|
—
|
présentation sous forme de tableau des valeurs de RET et des résultats de l'essai de fixation d'un colorant (le cas échéant), pour chaque animal et chaque échantillon de peau
|
|
—
|
description de tous les effets observés
|
|
4. BIBLIOGRAPHIE
|
(1)
|
OECD (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. Série de l'OCDE sur les essais et évaluations, numéro 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6.
|
|
(2)
|
Méthode d'essai B.4. Toxicité aiguë: irritation/corrosion cutanée.
|
|
(3)
|
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A prevalidation study on in vitro skin corrosivity testing. The report and recommendations of ECVAM Workshop 6. ATLA 23, 219-255.
|
|
(4)
|
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic, in Vitro 12, 471-482.
|
|
(5)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhütter, H.-G., and Liebsch, M. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxic, in Vitro 12, 483- 524.
|
|
(6)
|
OCDE (1996). Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62pp.
|
|
(7)
|
Balls, M., Blaauboer, B.J., Fentem. J.H., Bruner. L., Combes, R.D., Ekwall, B., Fielder. R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski. D., Spielmann, H., and Zucco, F. (1995). Practical aspects of the validation of toxicity test procedures. The report and recommendations of ECVAM workshops. ATLA 23, 129-147.
|
|
(8)
|
CEVMA (Comité de coordination interagences sur la validation des méthodes alternatives). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf
|
|
(9)
|
CEVMA (1998). ECVAM News & Views. ATLA 26, 275-280.
|
|
(10)
|
CEVMA (Comité de coordination interagences sur la validation des méthodes alternatives). (2002). ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/episbrd.pdf.
|
|
(11)
|
OCDE (2002) Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st - 2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat
|
|
(12)
|
Oliver, G.J.A., Pemberton, M.A., and Rhodes, C. (1986). An in vitro skin corrosivity test — modificationsand validation. Fd. Chem. Toxicol. 24, 507-512.
|
|
(13)
|
Botham, P.A., Hall, T.J., Dennett, R., McCall, J.C., Basketter, D.A., Whittle, E., Cheeseman, M., Esdaile, D.J., and Gardner, J. (1992). The skin corrosivity test in vitro: results of an interlaboratory trial. Toxic, in Vitro 6, 191-194.
|
|
(14)
|
Worth AP, Fentem JH, Balls M, Botham PA, Curren RD, Earl LK, Esdaile DJ, Liebsch M (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709-720.
|
|
(15)
|
Basketter, D.A., Chamberlain, M., Griffiths, H.A., Rowson, M., Whittle, E., York, M. (1997). The classification of skin irritants by human patch test. Fd. Chem. Toxicol. 35, 845-852.
|
|
(16)
|
Oliver G.J.A, Pemberton M.A and Rhodes C. (1988). An In Vitro model for identifying skin-corrosive chemicals. I. Initial Validation. Toxic, in Vitro. 2, 7-17.
|
Figure 1
Appareil pour l'essai de RET sur peau de rat

Figure 2
Dimensions du tube PFTE (polytétrafluoroéthylène), des tubes receveurs et des électrodes utilisés

Facteurs importants pour l'appareil ci-dessus:
|
—
|
diamètre intérieur du tube PTFE,
|
|
—
|
longueur des électrodes par rapport au tube PTFE et au tube receveur: le disque cutané ne doit pas être en contact avec les électrodes, et une longueur standard d'électrode doit être en contact avec la solution de sulfate de magnésium,
|
|
—
|
quantité de solution de sulfate de magnésium dans le tube receveur: la profondeur de liquide, par rapport au niveau dans le tube PFTE, doit être telle qu'indiquée dans la figure 1,
|
|
—
|
fixation du disque cutané sur le tube PFTE: la résistance électrique doit être véritablement une mesure des propriétés de la peau.
|
B.40 bis. CORROSION CUTANÉE IN VITRO: ESSAI SUR MODÈLE DE PEAU HUMAINE
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 431 (2004) de l'OCDE.
1.1. INTRODUCTION
La corrosion cutanée désigne la survenue de lésions irréversibles de la peau après application d'un matériel d'essai [selon la définition du système général harmonisé de classification et d'étiquetage des produits chimiques (SGH)] (1). La présente méthode d'essai ne nécessite pas l'utilisation d'animaux vivants ou de tissus animaux pour l'évaluation la corrosivité cutanée.
Traditionnellement, l'évaluation de la corrosivité cutanée impliquait le recours à des animaux de laboratoire (2). Dans le souci d'éviter les douleurs et les souffrances ainsi infligées aux animaux, la méthode d'essai B.4 a été révisée afin de permettre la détermination de la corrosion cutanée par la mise en œuvre de méthodes de substitution in vitro.
La conduite d'études de pré-validation a néanmoins été une première étape dans la définition d'essais de substitution susceptibles d'être utilisés pour des essais de corrosivité cutanée menés à des fins réglementaires (3). Par la suite, une étude formelle de validation des méthodes in vitro d'évaluation de la corrosion cutanée (4)(5) a été conduite (6) (7) (8). Sur la base des résultats de ces études et d'autres publications (9), il est maintenant recommandé d'évaluer la corrosivité cutanée in vivo à l'aide des essais suivants (10) (11) (12) (13): l'essai sur modèle de peau humaine (la présente méthode) et l'essai de résistance électrique transcutanée (voir méthode d'essai B.40).
Selon les études de validation, les essais sur modèles de peau humaine (3)(4)(5)(9) permettent de faire la distinction de manière fiable entre substances corrosives et non corrosives pour la peau. Le protocole d'essai peut aussi fournir des indications permettant de distinguer les substances extrêmement corrosives pour la peau de celles qui le sont moins.
L'essai décrit dans cette méthode permet d'identifier les substances et mélanges chimiques corrosifs. Appuyé par d'autres éléments de détermination fondés sur d'autres informations existantes (pH, relations structure-activité, données humaines et/ou animales, par exemple), il permet également d'identifier les substances et mélanges non corrosifs (1) (2) (13) (14). En revanche, il ne fournit pas d'information sur l'irritation cutanée, et contrairement au système général harmonisé de classification (SGH) (1), il ne permet pas non plus la sous-catégorisation des substances corrosives.
Pour une évaluation complète des effets cutanés locaux après une exposition unique, il est recommandé de suivre la démarche expérimentale séquentielle présentée en supplément à la méthode d'essai B.4 (2) et proposée dans le système général harmonisé (SGH) (1). Cette démarche expérimentale prévoit la conduite d'essais in vitro de corrosion cutanée (tel que celui décrit dans la présente méthode) et d'irritation cutanées avant d'envisager des essais sur des animaux vivants.
1.2. DÉFINITIONS
Corrosion cutanée in vivo: survenue de lésions irréversibles de la peau. En l'occurrence, il s'agit de nécrose visible, à travers l'épiderme et jusque dans le derme, suite à l'application d'une substance d'essai pendant une durée allant jusqu'à quatre heures. Les réactions corrosives se traduisent généralement par des ulcères, des saignements, des croûtes sanguinolentes et, à la fin de la période d'observation, soit à 14 jours, par une décoloration due au blanchissement de la peau, des zones complètes d'alopécie, et des cicatrices. Un examen histopathologique peut être envisagé pour évaluer les lésions douteuses.
Viabilité cellulaire: le paramètre mesurant l'activité totale d'une population cellulaire (par exemple, capacité des déshydrogénases mitochondriales cellulaires à réduire le colorant vital MTT), qui, selon l'effet mesuré et le protocole utilisé pour l'essai, est en corrélation avec le nombre total et/ou la vitalité des cellules.
1.3. SUBSTANCES DE RÉFÉRENCE
Tableau 1
Substances chimiques de référence
|
Nom
|
No EINECS
|
No CAS
|
|
|
Diamino-1,2 propane
|
201-155-9
|
78-90-0
|
Gravement corrosif
|
|
Acide acrylique
|
201-177-9
|
79-10-7
|
Gravement corrosif
|
|
O-tert. butylphénol
|
201-807-2
|
88-18-6
|
Corrosif
|
|
Hydroxyde de potassium (10 %)
|
215-181-3
|
1310-58-3
|
Corrosif
|
|
Acide sulfurique (10 %)
|
231-639-5
|
7664-93-9
|
Corrosif
|
|
Acide octanoïque (acide caprylique)
|
204-677-5
|
124-07-02
|
Corrosif
|
|
4-amino-1,2,4-triazole
|
209-533-5
|
584-13-4
|
Non corrosif
|
|
Eugenol
|
202-589-1
|
97-53-0
|
Non corrosif
|
|
Bromure de phénétyle
|
203-130-8
|
103-63-9
|
Non corrosif
|
|
Perchloroéthylène
|
204-825-9
|
27-18-4
|
Non corrosif
|
|
Acide isostéarique
|
250-178-0
|
30399-84-9
|
Non corrosif
|
|
4-(méthylthio)-benzaldéhyde
|
222-365-7
|
3446-89-7
|
Non corrosif
|
La plupart des substances chimiques données ici proviennent de la liste des substances chimiques sélectionnées par le CEVMA dans son étude de validation internationale (4). Les choix du CEVMA sont fondés sur les critères suivants :
|
(i)
|
nombre égal de substances corrosives et non corrosives;
|
|
(ii)
|
substances disponibles dans le commerce et couvrant la plupart des classes chimiques pertinentes;
|
|
(iii)
|
insertion de substances extrêmement corrosives et moins corrosives, de façon à permettre une distinction sur la base du pouvoir de corrosion;
|
|
(iv)
|
substances chimiques manipulables en laboratoire sans présenter d'autres risques majeurs que la corrosivité.
|
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance d'essai est généralement appliquée sur un modèle tridimensionnel de peau humaine, comprenant au moins un épiderme reconstitué avec un stratum corneum fonctionnel. Les matières corrosives sont identifiées sur la base de leur aptitude à réduire la viabilité cellulaire [déterminée, par exemple, à l'aide de l'essai de réduction du MTT (15)] en dessous de valeurs seuils définies pour des périodes d'exposition déterminées. Le principe de l'essai sur modèle de peau humaine s'appuie sur l'hypothèse selon laquelle les substances chimiques corrosives sont capables de pénétrer dans le stratum corneum par diffusion ou érosion, et sont suffisamment cytotoxiques pour les couches cellulaires sous-jacentes.
1.4.1. Procédure
1.4.1.1. Modèles de peau humaine
Les modèles de peau humaine peuvent être constitués pour l'étude, obtenus dans le commerce (par exemple, les modèles EpiDerm™ et EPISKIN™) (16) (17) (18) (19), ou encore mis au point et constitués dans le laboratoire d'essai (20) (21). Il est entendu que l'utilisation de la peau humaine est soumise à des conditions et considérations d'éthique admises d'ordre national et international. Tout nouveau modèle doit être validé (au moins dans la mesure indiquée au point 1.4.1.1.2). Les modèles de peau humaine utilisés pour cet essai doivent répondre aux conditions et critères suivants:
1.4.1.1.1 Conditions générales applicables au modèle:
L'épithélium doit être constitué à partir de kératinocytes humains. Le modèle doit avoir un stratum corneum fonctionnel avec plusieurs couches sous-jacente de cellules épithéliales vivantes, et comporter le cas échéant une couche stromale. Le stratum corneum doit comporter plusieurs couches, avec un profil lipidique suffisant pour assurer une fonction de barrière offrant une résistance à la pénétration rapide des marqueurs cytotoxiques. Par ailleurs, il est essentiel que le modèle présente des propriétés d'isolement afin que la substance d'essai ne puisse pas contourner le stratum corneum pour pénétrer dans les tissus vivants. En effet, une telle possibilité de contournement diminue la qualité de la modélisation de l'exposition de la peau. Enfin, le modèle de peau ne doit présenter aucune forme de contamination bactérienne (y compris par le mycoplasme) ou mycosique.
1.4.1.1.2 Conditions applicables au modèle fonctionnel:
L'amplitude de la viabilité cellulaire est généralement mesurée par le niveau MTT, ou autres colorants vitaux métaboliquement transformés. Dans ces cas, la densité optique (DO) du colorant (solubilisé) extrait du tissu de contrôle négatif doit être au moins 20 fois supérieure à la DO du solvant d'extraction seul [pour une présentation, voir la référence (22)]. Le tissu de contrôle négatif doit être stable en culture (c'est-à-dire qu'il doit présenter des mesures de viabilité comparables) pendant la durée de la période d'exposition. Le stratum corneum doit être suffisamment solide pour résister à la pénétration rapide de certains produits chimiques marqueurs cytotoxiques (par exemple, Triton X-100 à 1 %). Cette propriété peut être évaluée en fonction du temps d'exposition nécessaire pour réduire de moitié la viabilité cellulaire (TE50) (par exemple, pour les modèles EpiDerm™ et EPISKIN™, cette période est > 2 heures). Par ailleurs, le tissu doit pouvoir être reproduit dans le temps et, de préférence, par différents laboratoires. Enfin, utilisé dans le protocole d'essai choisi, il doit permettre de prévoir le potentiel corrosif des substance chimiques de référence (voir le Tableau 1).
1.4.1.2. Application des substances d'essai et de contrôle
Deux tissus (répliques) sont utilisées pour chaque traitement (c'est-à-dire chaque durée d'exposition), y compris les contrôles. Pour les substances liquides, il faut appliquer une quantité suffisante de substance d'essai pour couvrir uniformément la surface cutanée (25 μl/cm2 au minimum). Pour les substances solides, il faut appliquer une quantité suffisante de substance d'essai pour couvrir régulièrement la peau et ensuite l'humidifier avec de l'eau désionisée ou distillée pour assurer un bon contact avec la peau; si nécessaire, les substances solides doivent être réduites en poudre avant leur application. La méthode d'application doit convenir à la substance chimique mise à l'essai (5). À la fin de la période d'exposition, la surface cutanée doit être nettoyée avec soin afin d'éliminer la substance d'essai à l'aide d'une une solution tampon appropriée ou de NaCl à 0,9 pour cent.
Pour chaque étude, des contrôles positifs et négatifs doivent être utilisés parallèlement à la substance d'essai, de façon à garantir la qualité des résultats du modèle expérimental. Pour cet essai, les substances suggérées pour les contrôles positifs sont l'acide acétique glacial ou l'hydroxyde de potassium 8 N, et pour les contrôles négatifs, l'eau ou une solution NaCl à 0,9 pour cent.
1.4.1.3. Mesures de la viabilité cellulaire
Seules les méthodes quantitatives, validées peuvent être utilisées pour mesurer la viabilité cellulaire. En outre, la mesure de la viabilité doit être compatible avec une application sur un tissu tridimensionnel. La fixation de colorant non spécifique ne doit pas interférer avec la mesure de la viabilité. Par conséquent, les colorants dont la fixation utilise une protéine et ceux qui ne passent pas par une conversion métabolique (par exemple, le rouge neutre) sont à proscrire. L'essai le plus fréquemment employé est l'essai de réduction du MTT [Bromure de 3-(4,5-diméthylthiazole-2-yl)-2,5-diphényltétrazolium, bleu de thiazole; numéro EINECS: 206-069-5, numéro CAS 298-93-1)], qui donne des résultats précis et reproductibles (5), mais d'autres peuvent aussi être utilisés. L'échantillon de peau est placé dans une solution MTT à la concentration appropriée (par exemple, 0,3 - 1 mg/ml) à la température d'incubation voulue et pour une durée de 3 heures. Le précipité bleu de formazan est ensuite extrait à l'aide d'un solvant (isopropanol), puis on mesure la concentration du formazan en déterminant sa DO à une longueur d'onde comprise entre 540 et 595 nm.
L'action chimique de la substance d'essai sur le colorant vital est susceptible d'imiter celle du métabolisme cellulaire et, partant, de conduire à estimation erronée de la viabilité. Il a été montré que cela survient lorsque la substance d'essai n'est pas complètement éliminée par rinçage (9). Si la substance d'essai agit directement sur colorant vital, des contrôles supplémentaires doivent être pratiqués pour détecter et corriger les interférences de la substance avec la mesure de viabilité (9) (23).
2. RÉSULTATS
Pour chaque tissu, les valeurs de DO et les pourcentages calculés de viabilité cellulaire obtenus pour chaque substance d'essai, ainsi que pour les contrôles positifs et négatifs, sont présentés sous forme de tableau, y compris les données des essais répétés, le cas échéant les valeurs moyennes et individuelles.
2.1. INTERPRÉTATION DES RÉSULTATS
La valeur de la DO du contrôle négatif représente une viabilité cellulaire arbitrairement fixée à 100 pour cent. Par conséquent, les valeurs de la DO obtenues pour chaque échantillon d'essai peuvent être utilisées pour calculer un pourcentage de viabilité par rapport au contrôle négatif. La valeur seuil du pourcentage de viabilité cellulaire qui établit la distinction entre les matières corrosives et les matières non corrosives (ou entre différentes classes de substances corrosives), ou encore la ou les procédures statistiques utilisées pour évaluer les résultats et identifier les substances corrosives, doivent être clairement définies et documentées. De même, il doit être démontré qu'elles sont appropriées. De manière générale, ces valeurs de seuil sont établies au cours de la phase d'optimisation de l'essai, testées dans une phase de pré-validation, puis confirmées ensuite dans une étude de validation. A titre d'exemple, l'évaluation de la corrosivité associée au modèle EpiDerm™ est la suivante (9):
La substance d'essai est considérée comme étant corrosive pour la peau:
|
(i)
|
si la viabilité après 3 minutes d'exposition est inférieure à 50 pour cent, ou
|
|
(ii)
|
si la viabilité après 3 minutes d'exposition est supérieure ou égale à 50 pour cent, et la viabilité après une heure d'exposition inférieure à 15 pour cent.
|
La substance d'essai est considérée comme étant non corrosive pour la peau:
|
(i)
|
si la viabilité après 3 minutes d'exposition est supérieure ou égale à 50 pour cent, et la viabilité après une heure d'exposition supérieure ou égale à 15 pour cent.
|
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit contenir au moins les informations suivantes:
|
|
Substances d'essai et de contrôle:
|
—
|
nom(s) chimique(s), tels que le nom IUPAC ou CAS, et le cas échéant le numéro CAS;
|
|
—
|
pureté et composition de la substance ou la préparation (en pourcentage(s) en poids);
|
|
—
|
propriétés physico-chimiques (telles que l'état physique, le pH, la stabilité, l'hydrosolubilité) utiles pour la conduite de l'étude:
|
|
—
|
le cas échéant, traitement des substances d'essai et de contrôle avant l'essai (par exemple, chauffage, réduction en poudre);
|
|
|
|
Justification du choix du modèle de peau et du protocole utilisé.
|
|
|
Conditions d'essai:
|
—
|
système cellulaire utilisé
|
|
—
|
information sur l'étalonnage du dispositif utilisé pour la mesure de la viabilité cellulaire (un spectrophotomètre, par exemple)
|
|
—
|
information complète à l'appui du modèle spécifique de peau humaine, et notamment sa validité
|
|
—
|
détails de la procédure d'essai utilisée
|
|
—
|
doses d'essai utilisées
|
|
—
|
description de toute modification éventuelle de la procédure d'essai
|
|
—
|
référence aux données historiques du modèle
|
|
—
|
description des critères d'évaluation utilisés
|
|
|
|
Résultats:
|
—
|
présentation sous forme de tableau des résultats de chaque échantillon d'essai
|
|
—
|
description des autres effets observés.
|
|
4. BIBLIOGRAPHIE
|
(1)
|
OCDE (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. Série de l'OCDE sur les essais et évaluations, numéro 33, ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001 doc.nsf/LinkTo/env-jm-mono(2001)6.
|
|
(2)
|
Méthode d'essai B.4. Toxicité aiguë: irritation/corrosion cutanée.
|
|
(3)
|
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A prevalidation study on in vitro skin corrosivity testing. The report recommendations of ECVAM Workshop 6 ATLA 23, 219-255.
|
|
(4)
|
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic. In Vitro 12, 471-482.
|
|
(5)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxic. In Vitro 12, 483-524.
|
|
(6)
|
OCDE (1996). Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62pp.
|
|
(7)
|
Balls, M., Blaauboer, B.J., Fentem, J.H., Bruner, L., Combes, R.D., Ekwall, B., Fielder, R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski, D., Spielmann, H., and Zucco, F. (1995). Practical aspects of the validation of toxicity test procedures. Test report and recommendations of ECVAM workshops. ATLA 23, 129-147.
|
|
(8)
|
CEVMA (Comité de coordination interagences sur la validation des méthodes alternatives). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf.
|
|
(9)
|
Liebsch, M., Traue, D., Barrabas, C, Spielmann, H., Uphill, P., Wilkins, S., McPherson, J.P., Wiemann, C, Kaufmann, T., Remmele, M. and Holzhutter, H.G. (2000). The ECVAM prevalidation study on the use of EpiDerm for skin Corrosivity testing. ATLA 28, pp. 371-401.
|
|
(10)
|
CEVMA (1998). ECVAM News & Views. ATLA 26, 275-280.
|
|
(11)
|
CEVMA (2000). ECVAM News & Views. ATLA 28, 365-67.
|
|
(12)
|
CEVMA (Comité de coordination interagences sur la validation des méthodes alternatives) (2002). ICCVAM evaluation of EpiDerm™, EPISKIN™ (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/episjird.pdf.
|
|
(13)
|
OCDE (2002) Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st - 2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat
|
|
(14)
|
Worth AP, Fentem JH, Balls M, Botham PA, Curren RD, Earl LK, Esdaile DJ, Liebsch M (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709-720.
|
|
(15)
|
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity asssays. J.Immunol. Meth. 65, 55-63.
|
|
(16)
|
Cannon, C.L., Neal, P.J., Southee, J.A., Kubilus, J., and Klausner, M., 1994. New epidermal model for dermal irritancy testing. Toxic. In Vitro 8, 889-891.
|
|
(17)
|
Ponec, M., Boelsma, E., Weerheim, A., Mulder, A., Boutwstra, J., and Mommaas, M., 2000. Lipid and ultrastructural characterization of reconstructed skin models. International Journal of Pharmaceutics. 203, 211 -225.
|
|
(18)
|
Tinois E, Gaetani Q, Gayraud B, Dupont D, Rougier A, Pouradier DX (1994). The Episkin model: Successful reconstruction of human epidermis in vitro. In In vitro Skin Toxicology. Edited by A Rougier, AM Goldberg and HI Maibach: 133-140
|
|
(19)
|
Tinois E, Tiollier J, Gaucherand M, Dumas H, Tardy M, Thivolet J (1991). In vitro and post — transplantation differentiation of human keratinocytes grown on the human type IV collagen film of a bilayered dermal substitute. Experimental Cell Research 193: 310-319
|
|
(20)
|
Parentau, NX., Bilbo, P., Molte, C J., Mason, V.S., and Rosenberg, H. (1992). The organotypic culture of human skin keratinocytes and fibroblasts to achieve form and function. Cytotechnology 9, 163-171.
|
|
(21)
|
Wilkins, L.M., Watson, S.R., Prosky, S.J., Meunier, S.F., Parentau, NX. (1994). Development of a bilayered living skin construct for clinical applications. Biotechnology and Bioengineering 43/8. 747-756.
|
|
(22)
|
Marshall, N.J., Goodwin, C.J., Holt, SJ. (1995). A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regulation 5, 69- 84.
|
|
(23)
|
Fentem, J.H., Briggs, D., Chesne', C, Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Rouget, R., and van de Sandt, J.J.M., and Botham, P.A. (2001). A prevalidation study on in vitro tests for acute skin irritation: results and evaluation by the Management Team. Toxic. In Vitro 15, 57-93.
|
B.41. ESSAI DE PHOTOTOXICITE IN VITRO 3T3 NRU
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 432 (2004) de l'OCDE.
1.1. INTRODUCTION
La phototoxicité est définie comme un effet toxique dû à l'application d'une substance sur le corps, qui est soit déclenché ou accentué (et visible à des niveaux de dose faibles) après une exposition ultérieure à la lumière, soit provoqué par l'irradiation de la peau après administration d'une substance par voie systémique.
Le test de phototoxicité in vitro 3T3 NRU permet de déterminer le potentiel phototoxique d'une substance d'essai induit par l'excitation du produit chimique après exposition à la lumière. Cet essai évalue la réduction relative de la viabilité des cellules exposées au produit chimique, en présence ou en l'absence de lumière. Les substances identifiées par cet essai sont susceptibles d'être phototoxiques in vivo après administration systémique et diffusion dans la peau, ou après application topique.
Des effets phototoxiques ont été signalés pour de nombreux types de produits chimiques (1) (2) (3) (4). Le point commun entre ces derniers est leur capacité à absorber l'énergie lumineuse dans la plage de la lumière solaire. D'après la première loi de photochimie (loi de Grotthaus-Draper), la photoréaction nécessite l'absorption d'un nombre suffisant de quanta de lumière. Aussi convient-il, avant d'envisager un essai biologique, de déterminer le spectre d'absorption UV/lumière visible du produit chimique à tester suivant la ligne directrice 101 de l'OCDE. Si le coefficient d'extinction/d'absorption molaire est inférieur à 10 litres × mol-1 × cm-1, le produit chimique n'a pas de potentiel photoréactif et il est inutile de le soumettre à l'essai de phototoxicité in vitro 3T3 NRU ou à tout autre test biologique visant à détecter des effets photochimiques indésirables (1) (5). Voir également l'annexe 1.
Les résultats de l'essai de phototoxicité in vitro 3T3 NRU récemment évalué (6) (7) (8) (9) ont été comparés aux effets de phototoxicité aiguë in vivo chez les animaux et chez les humains, et l'essai s'est révélé fiable pour la prévision de ces effets. L'essai n'est pas conçu pour prévoir d'autres effets nocifs susceptibles de résulter de l'action combinée d'un produit chimique et de la lumière, tels que la photogénotoxicité, la photo-allergie, et la photocancérogénicité, et il ne permet pas non plus d'évaluer le pouvoir phototoxique. Par ailleurs, cet essai n'est pas conçu pour prendre en compte les mécanismes indirects de la phototoxicité, les effets des métabolites de la substance d'essai, ou les effets des mélanges.
Alors que l'utilisation de systèmes d'activation métabolique est nécessaire en règle générale pour tous les tests in vitro de prédiction du potentiel génotoxique ou cancérogène, dans le cas de la phototoxicologie, on ne dispose jusqu'à présent que de rares exemples de produits chimiques qui nécessitent une transformation métabolique pour agir comme une phototoxine in vivo ou in vitro. Par conséquent, il ne paraît pas utile ni scientifiquement fondé de réaliser le présent essai en présence d'un système d'activation métabolique.
1.2. DÉFINITIONS
Irradiance: intensité du rayonnement ultraviolet (UV) ou visible incident sur une surface, mesurée en W/m2 ou en mW/cm2.
Dose de lumière: quantité (= intensité × temps) de rayonnement ultraviolet (UV) ou visible incident sur une surface, exprimée en Joules (= W × s) par unité de surface, par exemple, J/m2 ou J/cm2.
Bandes de longueurs d'ondes de la lumière UV: les désignations recommandées par la CIE (Commission internationale de l'éclairage) sont: UVA (315-400 nm), UVB (280-315 nm) et UVC (100-280 nm). D'autres désignations sont également utilisées; la limite entre UVB et UVA est souvent placée à 320 nm, et les UVA peuvent être subdivisés en UV-A1 et UV-A2 avec une limite à environ 340 nm.
Viabilité cellulaire: paramètre mesurant l'activité totale d'une population cellulaire (par exemple, absorption du colorant vital rouge neutre dans les lysosomes cellulaires), qui, en fonction de l'effet mesuré et du type de test utilisé, correspond au nombre total et/ou à la vitalité des cellules.
Viabilité cellulaire relative: viabilité cellulaire exprimée par rapport aux témoins négatifs (solvant) qui ont été prélevés tout ou long de la procédure (soit +Irr soit -Irr), mais non traités par un produit chimique.
PIF (Photo Irritation Factor — Facteur de photo-irritation): facteur obtenu en comparant deux concentrations cytotoxiques efficaces (CE50) de la substance chimique d'essai, obtenues en absence (-Irr) ou en présence (+Irr) d'une irradiation non cytotoxique par un rayonnement UVA/visible.
CE
50
: concentration de la substance chimique d'essai à laquelle la viabilité cellulaire est réduite de 50 pour cent.
MPE (Mean Photo Effect — Photo-effet moyen): mesure dérivée d'une analyse mathématique du tracé de deux courbes concentration-effet obtenues avec (+Irr) ou sans (-Irr) irradiation non cytotoxique par un rayonnement UVA/visible.
Phototoxicité: réaction toxique aiguë apparaissant après une première exposition de la peau à certains produits chimiques et exposition subséquente à la lumière, ou déclenchée par l'irradiation de la peau après administration systémique d'un produit chimique.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
Le test de phototoxicité in vitro 3T3 NRU repose sur la comparaison de la cytotoxicité d'une substance chimique avec et sans exposition à une dose non cytotoxique de lumière solaire simulée. Dans cet essai, la cytotoxicité est exprimée comme la diminution, en fonction de la concentration, de la fixation du colorant vital rouge neutre [Neutral Red — NR] 24 heures après traitement par le produit chimique mis à l'essai et irradiation (10). Le rouge neutre est un colorant cationique faible qui pénètre facilement dans les membranes cellulaires par non-diffusion, et s'accumule au niveau intracellulaire dans les lysosomes. L'altération de la surface cellulaire de la membrane lysosomale sensible entraîne une fragilité lysosomale et d'autres modifications qui deviennent graduellement irréversibles. Ces modifications induites par l'action de xénobiotiques entraînent une diminution de la fixation du rouge neutre, sur la base de laquelle on peut distinguer les cellules vivantes, des cellules mortes ou abîmées.
Des cellules Balb/c 3T3 sont maintenues en culture pendant 24 heures jusqu'à formation de monocouches. Pour chaque substance chimique à tester, deux plaques à 96 puits sont alors préincubées pendant 1 heure avec 8 concentrations distinctes de la substance chimique. Ensuite, l'une des deux plaques est exposée à la dose d'irradiation non cytotoxique la plus élevée, tandis que l'autre plaque est maintenue à l'obscurité. Dans les deux plaques, le milieu de traitement est ensuite remplacé par un milieu de culture et, après une nouvelle période d'incubation de 24 heures, la viabilité cellulaire est déterminée par le test de fixation du rouge neutre (Neutral Red Uptake — NRU). La viabilité cellulaire relative, exprimée sous forme d'un pourcentage des témoins de solvant non traités, est calculée pour chaque concentration d'essai. Afin de prévoir le potentiel phototoxique, les réponses aux concentrations obtenues en présence et en absence d'irradiation sont comparées, généralement au niveau CE50, c'est-à-dire la concentration inhibant la viabilité cellulaire de 50 pour cent par rapport aux témoins non traités.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1 Préparations
1.4.1.1 Cellules
Une lignée permanente de cellules de fibroblaste de souris, Balb/c 3T3, clone 31, provenant de l'ATCC (American Type Culture Collection, Manassas, Virginie, Etats-Unis), ou de l'ECACC (European Collection of Cell Cultures, Salisbury, Wiltshire, Royaume-Uni) a été utilisée dans l'étude de validation et il est donc recommandé que les cellules proviennent d'un dépositaire de cellules bien qualifié. D'autres cellules ou lignées cellulaires peuvent aussi donner de bons résultats avec le même protocole d'essai si les conditions de culture sont adaptées aux besoins spécifiques des cellules. Il convient toutefois d'en démontrer l'équivalence.
Les cellules doivent être régulièrement contrôlées pour vérifier l'absence de contamination par des mycoplasmes, et ne doivent être utilisées que si aucun n'est trouvé (11).
Il importe de contrôler régulièrement la sensibilité aux UV des cellules Balb/c 3T3 selon la procédure de contrôle qualité décrite dans la présente méthode. Dans la mesure où la sensibilité aux UVA des cellules peut augmenter avec le nombre de passages, il convient d'utiliser des cellules Balb/c 3T3 ayant subi le moins de passages possibles, de préférence moins de 100 (voir point 1.4.2.2.2 et annexe 2).
1.4.1.2. Milieux et conditions de culture
Des milieux de culture et des conditions d'incubation appropriés doivent être utilisés pour le passage systématique des cellules et pendant la procédure d'essai. Par exemple, pour les cellules Balb/c 3T3, le milieu de culture est du DMEM (Dulbecco's modified Eagle's medium) enrichi avec 10 pour cent de sérum de veau nouveau-né, de glutamine (4mM), de pénicilline (100 UI) et de streptomycine (100 μg/ml), et incubé à 37 oC en atmosphère humidifiée, avec une teneur en CO2 de 5 à 7,5 pour cent selon le tampon (voir point 1.4.1.4, second paragraphe). Il est particulièrement important que les conditions de culture cellulaire permettent de maintenir la durée du cycle cellulaire dans les limites normales historiques des cellules ou de la lignée cellulaire utilisées.
1.4.1.3 Préparation des cultures
Des cellules provenant de cultures mères congelées sont mises en culture à une densité appropriée dans le milieu de culture, et sont repiquées au moins une fois avant d'être utilisées pour le test de phototoxicité in vitro 3T3 NRU.
Pour le test de phototoxicité, le milieu de culture est ensemencé avec une densité de cellules telle que les cultures n'arrivent pas à confluence avant la fin de l'essai, c'est-à-dire au moment où la viabilité cellulaire est déterminée, 48 heures après la mise en culture des cellules. Pour les cellules Balb/c 3T3 cultivées sur des plaques à 96 puits, la densité cellulaire recommandée est de 1 x 104 cellules par puits.
Pour chaque substance chimique testée, les cellules sont mises en culture de manière identique sur deux plaques à 96 puits distinctes, qui sont utilisées parallèlement pendant toute la procédure d'essai, dans les mêmes conditions de culture, sauf pendant la période où l'une des plaques est irradiée (+ Irr) et l'autre maintenue à l'obscurité (- Irr).
1.4.1.4 Préparation de la substance d'essai
Les substances chimiques de l'essai doivent être fraîchement préparées, juste avant utilisation, à moins que les données de stabilité ne démontrent qu'elles sont conservables. Il est recommandé de procéder à la manipulation des substances chimiques et au traitement initial des cellules dans des conditions d'éclairage permettant d'éviter la photoactivation ou la dégradation de la substance d'essai avant irradiation.
Les substances chimiques de l'essai doivent être dissoutes dans des solutions salines tamponnées, par exemple, solution saline équilibrée de Earl (Earl's Balanced Salt Solution — EBSS) ou d'autres solutions tamponnées physiologiquement équilibrées, exemptes de constituants protéiques qui absorbent la lumière (par exemple, indicateurs pH colorés et vitamines), afin d'éviter les interférences lors de l'irradiation. Sachant que, pendant l'irradiation, les cellules sont maintenues pendant 50 minutes environ en dehors de l'incubateur CO2, des précautions doivent être prises pour éviter l'alcalinisation. Avec des solutions faiblement tamponnées, telles que l'EBSS, il convient alors d'incuber les cellules à 7,5 pour cent de CO2. Si les cellules sont incubées à 5 pour de CO2 seulement, un tampon plus fort doit être utilisé.
Les substances chimiques ayant une solubilité limitée dans l'eau doivent être dissoutes dans des solvants appropriés. Si un solvant est utilisé, il doit être présent à un volume constant dans toutes les cultures, c'est-à-dire dans les témoins négatifs (solvant) et dans toutes les concentrations de la substance chimique d'essai, et être en outre non cytotoxique à la concentration utilisée. Les concentrations de la substance chimique doivent être sélectionnées de façon à éviter les précipités ou les solutions troubles.
Les solvants recommandés sont le diméthylsulfoxyde (DMSO) et l'éthanol (EtOH). D'autres solvants de faible cytotoxicité peuvent convenir, mais leurs propriétés spécifiques doivent être évaluées avant utilisation, notamment les risques de réaction avec la substance chimique d'essai, l'atténuation de l'effet phototoxique, les propriétés de fixation des radicaux, et/ou la stabilité du produit chimique dans le solvant.
Si nécessaire, on peut recourir à un mélangeur Vortex et/ou à la sonication et/ou à un chauffage à des températures appropriées pour faciliter la solubilisation, à moins qu'une telle manipulation n'affecte la stabilité de la substance chimique.
1.4.1.5 Conditions d'irradiation
1.4.1.5.1 Source de lumière
Le choix d'une source de lumière et d'un filtrage appropriés est le facteur déterminant dans les essais de phototoxicité. Les domaines des UVA et de la lumière visible sont généralement associés à des réactions phototoxiques in vivo (3) (12), tandis que les UVB sont moins pertinents, mais très cytotoxiques, avec une cytotoxicité qui augmente d'un facteur 1 000 entre 313 et 280 nm (13). Parmi les critères présidant au choix de la source de lumière appropriée, il est essentiel que la source de lumière émette des longueurs d'onde absorbées par la substance chimique d'essai et que la dose de lumière (pouvant être obtenue dans un délai d'exposition raisonnable) soit suffisante pour détecter les substances photocytotoxiques connues. En outre, les longueurs d'onde et les doses employées ne doivent pas trop endommager le système d'essai, par exemple par l'émission de chaleur (domaine infrarouge).
La lumière solaire simulée par des simulateurs solaires est considérée comme la source optimale de lumière artificielle. La distribution du pouvoir d'irradiation du simulateur solaire filtré doit être proche de celle de la lumière du jour extérieure donnée dans le document (14). Des arcs au xénon ou des arcs aux halogénures de métal-mercure (dopé) peuvent être utilisés comme simulateurs solaires (15). Ces derniers ont l'avantage d'émettre moins de chaleur et d'être moins chers, mais ils reproduisent moins bien la lumière solaire que les simulateurs utilisant des arcs au xénon. Dans la mesure où tous les simulateurs solaires émettent d'importantes quantités d'UVB, ils doivent être équipés de filtres adéquats pour atténuer les longueurs d'onde UVB hautement cytotoxiques. Dans la mesure où les matériaux plastiques utilisés pour les cultures cellulaires contiennent des stabilisateurs UV, le spectre doit être mesuré à travers le même type de plaques à 96 puits que celui employé dans l'essai. Abstraction faite des mesures prises pour atténuer une part du spectre par filtration, ou des effets de filtre inévitables liés aux appareils, le spectre enregistré sous ces filtres ne doit pas dévier de la norme de lumière du jour extérieure (14). Les documents (8) (16) donnent un exemple de distribution de l'irradiance spectrale du simulateur solaire équipé de filtres qui a été utilisé dans l'étude de validation du test in vitro 3T3 NRU. Voir également la Figure 1 de l'annexe 2.
1.4.1.5.2. Dosimétrie
L'intensité de lumière (irradiance) doit être régulièrement contrôlée avant chaque essai de phototoxicité, à l'aide d'un radiomètre UV à large bande approprié. L'intensité doit être mesurée à travers le même type de plaques à 96 puits que celui employé dans l'essai. Le radiomètre UV doit avoir été étalonné par rapport à la source. Les performances du radiomètre UV doivent être contrôlées. À cet effet, on recommande l'utilisation d'un second radiomètre UV de référence, du même type et étalonné de la même façon. Dans l'idéal, à intervalles plus grands, il conviendrait d'utiliser un spectroradiomètre pour mesurer l'irradiance spectrale de la source de lumière filtrée et pour vérifier l'étalonnage du radiomètre UV à large bande.
Une dose de 5 J/cm2 (dans la plage des UVA) est non cytotoxique pour les cellules Balb/c 3T3 et suffisamment puissante pour exciter les substances chimiques et déclencher des réactions phototoxiques (6) (17). Par exemple, pour obtenir 5 J/cm2 dans un délai de 50 minutes, l'irradiance a été réglée à 1,7 mW/cm2. Voir la Figure 2 de l'annexe 2. En cas d'utilisation d'une autre lignée cellulaire ou d'une source de lumière différente, il peut s'avérer nécessaire d'ajuster la dose d'irradiation en choisissant une dose qui ne soit pas nocive pour les cellules tout en étant suffisante pour exciter les phototoxines standard. La durée de l'exposition à la lumière se calcule de la façon suivante:
|

|
(1 J = 1 Wsec)
|
1.4.2 Conditions d'essai
1.4.2.1 Concentrations de la substance d'essai
Les plages de concentrations du produit chimique testé en présence (+Irr) et en absence (-Irr) de lumière doivent être déterminées de manière adéquate lors d'expériences préalables. Il peut être utile d'évaluer la solubilité au début puis après un délai de 60 minutes (ou à la durée employée dans l'essai), dans la mesure où celle-ci peut varier avec le temps ou au cours de l'exposition. Pour éviter toute toxicité induite par des conditions de culture inappropriées ou des substances chimiques hautement acides ou alcalines, il faut que le pH des cultures cellulaires, auxquelles la substance d'essai a été ajoutée, soit dans une plage comprise entre 6,5 et 7,8.
La concentration la plus élevée de la substance d'essai doit être dans les conditions d'essai physiologiques, par exemple le stress pH ou osmotique doit être évité. Selon la substance d'essai, il peut s'avérer nécessaire d'envisager d'autres propriétés physico-chimiques comme facteurs limitant la concentration d'essai la plus élevée. Pour les substances relativement insolubles, mais non toxiques à des concentrations allant jusqu'au point de saturation, il y a lieu de procéder à des essais pour déterminer la concentration la plus élevée qui puisse être atteinte. De manière générale, il y a lieu d'éviter les effets de précipités de la substance chimique à toutes les concentrations d'essai. La concentration maximale d'une substance d'essai ne doit pas dépasser 1 000 μg/ml, et l'osmolarité ne doit pas dépasser 10 milliosmoles. Il convient d'utiliser une série de dilutions en progression géométrique comprenant 8 concentrations de la substance d'essai avec un facteur de dilution constant (voir point 2.1, second paragraphe).
Si les informations provenant d'expériences préalables établissent qu'une substance chimique n'est pas cytotoxique jusqu'à la concentration limite dans l'obscurité (-Irr), mais se révèle hautement cytotoxique exposée à la lumière (+Irr), les plages de concentrations à choisir pour l'expérience (+Irr) peuvent être différentes de celles utilisées pour l'expérience (-Irr), de façon à garantir une qualité adéquate des résultats.
1.4.2.2 Contrôles/Témoins
1.4.2.2.1. Sensibilité des cellules aux radiations, données historiques:
la sensibilité des cellules à la source lumineuse doit être régulièrement contrôlée (tous les cinq passages environ), par évaluation de leur viabilité après une exposition à des doses croissantes d'irradiation. Pour cette évaluation, plusieurs doses d'irradiation doivent être utilisées, y compris des niveaux significativement supérieurs à ceux de l'essai de phototoxicité 3T3 NRU. La méthode la plus simple pour quantifier ces doses consiste à mesurer les UV à la source. Les cellules sont mises en culture à la densité utilisée dans l'essai de phototoxicité in vitro 3T3 NRU. Elles sont irradiées le jour suivant, et la viabilité cellulaire est déterminée un jour plus tard à l'aide du test NRU. Il faut alors que la dose non cytotoxique la plus élevée (par exemple, 5 J/cm2 [UVA] dans l'étude de validation) soit suffisante pour classer correctement les substances de référence (Tableau 1).
1.4.2.2.2. Sensibilité aux radiations, contrôle de l'essai en cours:
l'essai répond aux critères de qualité si les témoins de contrôle négatifs (solvant) irradiés ont une viabilité supérieure ou égale à 80 pour cent de celle des témoins de contrôle négatifs non irradiés.
1.4.2.2.3. Viabilité des témoins de solvant
la densité optique absolue (OD540 NRU), mesurée dans l'extrait NR des témoins de solvant indique si les 1 × 104 cellules mises en culture par puits se sont développées avec un temps de doublement normal pendant les deux jours de l'essai. Un essai répond aux critères d'acceptation si la densité optique moyenne OD540 NRU des témoins non traités est > 0,4 (c'est-à-dire approximativement vingt fois l'absorbance de fond du solvant).
1.4.2.2.4 Témoin positif:
Pour chaque essai de phototoxicité in vitro 3T3 NRU réalisé, un produit chimique phototoxique connu doit être testé concurremment. La chlorpromazine (CPZ) est recommandée. Dans le cas de la CPZ testée selon le protocole standard dans le test de phototoxicité in vitro 3T3 NRU, les critères d'acceptation suivants ont été définis: CPZ irradiée (+Irr) : CE50 = 0,1 à 2,0 μg/ml; CPZ non irradiée (-Irr) : CE50 = 7,0 à 90,0 μg/ml. Le facteur de photo-irritation (PIF) doit être > 6. Les données historiques du témoin positif doivent être suivies.
À la place de la CPZ, d'autres produits chimiques phototoxiques convenant à la classe chimique ou aux caractéristiques de solubilité de la substance chimique à tester, peuvent aussi être utilisés en tant que témoins positifs parallèles.
1.4.3 Mode opératoire (6) (7) (8) (16) (17) :
1.4.3.1 Premier jour:
Verser 100 μl de milieu de culture dans les puits périphériques d'une plaque de microtitrage de culture tissulaire à 96 puits (= essais à blanc). Dans les puits restants, verser 100 μl d'une suspension cellulaire de 1 × 105 cellules/ml en milieu de culture, (= 1 × 104 cellules/puits). Préparer deux plaques pour chaque série des concentrations de la substance à tester, ainsi que pour les témoins négatifs (de solvant) et positifs.
Incuber les cellules pendant 24 heures (voir point 1.4.1.2) jusqu'à formation d'une monocouche semi-confluente. Cette période d'incubation permet la récupération et l'adhérence des cellules, et leur croissance exponentielle.
1.4.3.2 Deuxième jour:
Après incubation, décanter le milieu de culture pour le séparer des cellules et laver délicatement avec 150μl de la solution tamponnée utilisée pour l'incubation. Ajouter 100 μl de la solution tamponnée contenant la concentration appropriée de la substance chimique à tester, ou uniquement du solvant (témoin négatif). Appliquer 8 concentrations distinctes de la substance chimique à tester. Incuber les cellules avec la substance chimique à tester dans l'obscurité pendant 60 minutes (voir points 1.4.1.2 et 1.4.1.4, second paragraphe).
Sur les deux plaques préparées pour chaque série de concentrations, ainsi que pour les témoins, une plaque est sélectionnée, généralement au hasard, pour la détermination de la cytotoxicité (-Irr) (par exemple, la plaque témoin), et l'autre (la plaque de traitement) pour la détermination de la photocytotoxicité (+Irr).
Pour réaliser la partie (+Irr) de l'essai, irradier les cellules à température ambiante pendant 50 minutes environ à travers le couvercle de la plaque à 96 puits, à la dose maximale de radiations non cytotoxique (voir également annexe 2). Conserver les plaques identiques non irradiées (-Irr) à température ambiante dans une boîte obscure pendant 50 minutes (= durée de l'exposition à la lumière).
Décanter la solution d'essai et laver deux fois avec 150 μl de la solution tamponnée utilisée pour l'incubation, mais ne contenant pas la substance d'essai. Remplacer le tampon par le milieu de culture et incuber (voir le point 1.4.1.2) jusqu'au lendemain (18-22 heures).
1.4.3.3 Troisième jour:
1.4.3.3.1 Évaluation microscopique
Examiner les cellules au microscope à contraste de phase. Noter la croissance, la morphologie et l'intégrité de la monocouche. Les changements morphologiques et les effets sur la croissance cellulaire doivent être enregistrés.
1.4.3.3.2 Test de fixation du rouge neutre
Laver les cellules avec 150 μl de la solution tamponnée préchauffée. Éliminer la solution de lavage en tapotant légèrement. Ajouter 100 μl d'un rouge neutre (3-amino-7-diméthylamino-2-méthylphénazine chlorhydrate, numéro EINECS 209-035-8; numéro CAS 553-24-2; CI. 50040) à 50 μg/ml dans un milieu sans sérum (16) et incuber comme décrit au point 1.4.1.2., pendant 3 heures. Après incubation, éliminer le milieu NR et laver les cellules avec 150 μl du tampon. Décanter et évacuer complètement le tampon en excès par absorption ou centrifugation.
Ajouter exactement 150 μl de solution de désorption NR (solution fraîchement préparée de 49 parts d'eau + 50 parts d'éthanol + 1 part d'acide acétique).
Passer rapidement la plaque de microtitrage à l'agitateur pendant 10 minutes, jusqu'à ce que le NR soit extrait des cellules et forme une solution homogène.
Mesurer la densité optique de l'extrait de NR à 540 nm dans un spectrophotomètre en utilisant les essais à blanc comme référence. Sauvegarder les données dans un format de fichier électronique approprié en vue d'une analyse ultérieure.
2. RÉSULTATS
2.1. QUALITÉ ET QUANTITÉ DES DONNÉES
Les données d'essai doivent permettre une analyse significative des courbes concentration-effet obtenues avec et sans irradiation, et si possible de la concentration de la substance d'essai à laquelle la viabilité cellulaire est réduite de moitié (CE50). Si on constate une cytotoxicité, il y a lieu d'ajuster à la fois la gamme des concentrations et l'intervalle entre chaque concentration, afin qu'il y ait concordance entre la courbe et les données expérimentales.
Pour les résultats clairement positifs ou clairement négatifs (voir point 2.3), l'expérience principale, étayée par une ou plusieurs expériences préliminaires de détermination des gammes de concentrations, est généralement suffisante.
Les tests qui donnent des résultats équivoques, limites ou incertains doivent être vérifiés par un essai supplémentaire (voir également point 2.4, second paragraphe). Si cet essai s'avère nécessaire, il peut être utile de faire varier les conditions expérimentales, et notamment la plage ou l'espacement des concentrations, le temps de préincubation, et le temps d'exposition à l'irradiation. Une réduction de cette durée d'exposition peut présenter un intérêt pour les produits chimiques instables dans l'eau.
2.2. ÉVALUATION DES RÉSULTATS
Pour procéder à l'évaluation des données, il peut être nécessaire de calculer un facteur de photo-irritation (PIF) ou un photo-effet moyen (MPE).
Pour le calcul des mesures de photocytotoxicité (voir ci-après), l'ensemble des valeurs discrètes de concentration-effet doit être déterminé par une courbe concentration-effet continue appropriée (modèle). On fait généralement concorder la courbe aux données en appliquant une méthode de régression non linéaire (18). Pour évaluer l'influence de la variabilité des données sur la courbe ajustée, il est recommandé d'employer une procédure de type «bootstrap».
On calcule un facteur de photo-irritation (PIF) à l'aide de la formule suivante:

S'il n'est pas possible de calculer une CE50 en présence ou en l'absence de lumière, il ne sera pas possible de déterminer un PIF pour la substance d'essai. Le photo-effet moyen (MPE) est une mesure basée sur une comparaison des courbes concentration-effet complètes (19). Il correspond à la moyenne pondérée d'un ensemble représentatif de valeurs du photo-effet:

Le photo-effet PEc à une concentration C est défini comme le produit de la réponse-effet REC et de la dose-effet DEC, soit PEC = REC × DEC. La réponse-effet REC correspond à la différence entre les réponses observées en absence et en présence de lumière, soit REC = RC (-Irr) — RC (+Irr). La dose-effet est donnée par la formule suivante

où C* représente la concentration équivalence, c'est-à-dire la concentration à laquelle la réponse +Irr est équivalente à la réponse -Irr à la concentration C. S'il n'est pas possible de déterminer C* parce que les valeurs de la courbe +Irr sont systématiquement supérieures ou inférieures à RC (-Irr), la dose-effet est fixée à 1. Les facteurs de pondération w1 sont donnés par la valeur la plus élevée, soit wi = MAX {Ri (+Irr), Ri (-Irr)}. La grille de concentration Ci est choisie de façon à ce que le même nombre de points figure dans chaque intervalle de concentration défini par les valeurs de concentration utilisées dans l'expérience. Le calcul du MPE est limité par la valeur de concentration maximale à laquelle au moins une des deux courbes montre une valeur de réponse d'au moins 10 pour cent. Si cette concentration maximale est supérieure à la concentration la plus élevée utilisée dans l'expérience +Irr, la partie résiduelle de la courbe +Irr est ajustée à la valeur de réponse «0». La substance chimique est ensuite classée ou non comme étant phototoxique, selon que la valeur MPE est supérieure ou non à une valeur de seuil correctement choisie (MPEC = 0,15).
Un logiciel de calcul du PIF et du MPE est disponible (20).
2.3. INTERPRÉTATION DES RÉSULTATS
Sur la base de l'étude de validation (8), une substance d'essai dont le PIF est < 2 ou le MPE < 0,1 ne présente «aucune phototoxicité». Un PIF > 2 et < 5 ou un MPE > 0,1 et < 0,15 indique une «phototoxicité probable» et un PIF > 5 ou un MPE > 0,15 indique une «phototoxicité».
Pour les laboratoires qui entreprennent cet essai pour la première fois, il est préconisé de procéder à un essai sur les substances de référence données dans le Tableau 1, avant d'entreprendre l'évaluation phototoxique de substances d'essai. Les valeurs PIF ou MPE doivent être proches des valeurs données dans le tableau 1.
Tableau 1
|
Nom de la substance chimique
|
No EINECS
|
No CAS
|
PIF
|
MPE
|
Pic d'absorption
|
Solvant (9)
|
|
Amiodarone chlorhydrate
|
243-293-2
|
[19774-82-4]
|
>3,25
|
0,27-0,54
|
242 nm
300 nm
(épaulement)
|
éthanol
|
|
Chlorpromazine chlorhydrate
|
200-701-3
|
[69-09-0]
|
>14,4
|
0,33-0,63
|
309 nm
|
éthanol
|
|
Norfloxacine
|
274-614-4
|
[70458-96-7]
|
>71,6
|
0,34-0,90
|
316 nm
|
acétonitrile
|
|
Anthracène
|
204-371-1
|
[120-12-7]
|
>18,5
|
0,19-0,81
|
356 nm
|
acétonitrile
|
|
Protoporphyrine IX, disodium
|
256-815-9
|
[50865-01-5]
|
>45,3
|
0,54-0,74
|
402 nm
|
éthanol
|
|
L — Histidine
|
|
[7006-35-1]
|
pas de PIF
|
0,05-0,10
|
211 nm
|
eau
|
|
Hexachlorophene
|
200-733-8
|
[70-30-4]
|
1,1-1,7
|
0,00-0,05
|
299 nm
317nm
(épaulement)
|
éthanol
|
|
Sulfate de sodium et de dodécyle
|
205-788-1
|
[151-21-3]
|
1,0-1,9
|
0,00-0,05
|
pas d'absorption
|
eau
|
2.4. INTERPRÉTATION DES RÉSULTATS
Si des effets phototoxiques ne sont observés qu'à la concentration d'essai maximale (en particulier pour les substances d'essai solubles dans l'eau), d'autres investigations peuvent s'avérer nécessaires pour évaluer les risques. Il peut s'agir notamment d'étudier l'absorption cutanées et l'accumulation du produit chimique dans la peau, et/ou de soumettre le produit à un autre type d'essai, en recourant par exemple à des essais in vitro sur peau humaine ou animale, ou sur modèle de peau.
En revanche, si aucune toxicité n'a été mise en évidence (+Irr et -Irr) et si la solubilité faible du produit dans l'eau a limité les concentrations d'essai, il faut peut-être s'interroger sur l'adéquation du système d'essai pour la substance à tester, et envisager un essai de confirmation avec un autre modèle.
3. RAPPORT
RAPPORT D'ESSAI
Le rapport d'essai doit contenir au minimum les informations suivantes :
|
|
Substance d'essai:
|
—
|
données d'identification, nom générique commun et numéros IUPAC et CAS si connus
|
|
—
|
nature physique et pureté
|
|
—
|
propriétés physico-chimiques importantes pour la réalisation de l'étude
|
|
—
|
spectre d'absorption UV/lumière visible
|
|
—
|
stabilité et photostabilité si connues
|
|
|
|
Solvant:
|
—
|
justification du choix du solvant
|
|
—
|
solubilité de la substance d'essai dans le solvant
|
|
—
|
pourcentage de solvant présent dans le milieu de traitement
|
|
|
|
Cellules:
|
—
|
type et provenance des cellules
|
|
—
|
nombre de passages cellulaires, si connu
|
|
—
|
sensibilité des cellules aux radiations, déterminée avec l'appareil d'irradiation utilisé dans l'essai de phototoxicité in vitro 3T3 NRU.
|
|
|
|
Conditions expérimentales (1) incubation avant et après traitement:
|
—
|
type et composition du milieu de culture
|
|
—
|
conditions d'incubation (concentration de CO2, température, humidité)
|
|
—
|
durée de l'incubation (avant traitement et après traitement)
|
|
|
|
Conditions expérimentales (2) traitement par la substance chimique:
|
—
|
justification du choix des concentrations de substance d'essai utilisées, en présence et en absence de rayonnement
|
|
—
|
en cas de solubilité limitée du produit chimique et d'absence de cytotoxicité, justification de la concentration maximale utilisée
|
|
—
|
type et composition du milieu de traitement (solution saline tamponnée)
|
|
—
|
durée du traitement chimique.
|
|
|
|
Conditions expérimentales (3) Irradiation:
|
—
|
justification de la source de lumière utilisée
|
|
—
|
fabricant et type de source de lumière et de radiomètre
|
|
—
|
caractéristiques d'irradiance spectrale de la source de lumière
|
|
—
|
caractéristiques de transmission/absorption du (des) filtre(s) utilisé(s)
|
|
—
|
caractéristiques du radiomètre et modalités d'étalonnage
|
|
—
|
distance entre la source de lumière et le système d'essai
|
|
—
|
irradiance UVA à cette distance, exprimée en mW/cm2
|
|
—
|
durée de l'exposition UV/lumière visible
|
|
—
|
dose d'UVA (irradiance × temps), exprimée en J/cm2
|
|
—
|
température appliquée aux cultures cellulaires durant l'irradiation et aux cultures cellulaires maintenues concurremment dans l'obscurité.
|
|
|
|
Conditions expérimentales (4) test NRU:
|
—
|
composition du milieu de traitement NR
|
|
—
|
durée de l'incubation dans NR
|
|
—
|
conditions d'incubation (concentration de CO2, température, humidité)
|
—
|
d'extraction du NR (agent d'extraction, durée)
|
|
—
|
longueur d'ondes utilisée pour la lecture spectrophotométrique de la densité optique du NR
|
|
—
|
seconde longueur d'ondes (référence), le cas échéant
|
|
—
|
contenu de l'échantillon destiné à l'essai à blanc du spectrophotomètre, le cas échéant.
|
|
|
|
|
Résultats:
|
—
|
viabilité cellulaire obtenue pour chaque concentration de la substance d'essai, exprimée en pour cent de la viabilité moyenne des témoins de solvant
|
|
—
|
courbes de concentration-effet (concentration de la substance chimique — viabilité cellulaire relative) obtenues dans les expériences +Irr et -Irr parallèles
|
|
—
|
analyse des courbes concentration-effet: si possible, calcul/détermination des CE50 (+Irr) et CE50 (-Irr)
|
|
—
|
comparaison des deux courbes concentration-effet obtenues en présence et en l'absence de rayonnement, soit par le calcul du facteur de photo-irritation (PIF), soit par le calcul du photo-effet moyen (MPE)
|
|
—
|
critères d'acceptation de l'essai, témoin négatif (solvant) simultané:
|
|
—
|
viabilité absolue (densité optique de l'extrait de NR) des cellules irradiées et des cellules non irradiées
|
|
—
|
données historiques sur les témoins négatif et de solvant, moyennes et écarts types
|
|
—
|
critères d'acceptation de l'essai, témoin positif simultané:
|
|
—
|
CE50 (+Irr) et CE50 (-Irr) et PIF/MPE du produit chimique témoin positif
|
|
—
|
données historiques sur le produit chimique témoin positif: CE50 (+Irr) et CE50 (-Irr) et PIF/MPE moyennes et écarts types.
|
|
4. BIBLIOGRAPHIE
|
(1)
|
Lovell W.W. (1993). A scheme for in vitro screening of substances for photoallergenic potential. Toxic. In Vitro 7, p. 95-102.
|
|
(2)
|
Santamaria, L. and Prino, G. (1972). List of the photodynamic substances. In «Research Progress in Organic, Biological and Medicinal Chemistry» Vol. 3 part 1. North Holland Publishing Co. Amsterdam, p. XI-XXXV.
|
|
(3)
|
Spielmann, H., Lovell, W.W., Hölzle, E., Johnson, B.E., Maurer, T., Miranda, M.A., Pape, W.J.W., Sapora, O., and Sladowski, D. (1994). In vitro phototoxicity testing: The report and recommendations of ECVAM Workshop 2. ATLA, 22, p. 314-348.
|
|
(4)
|
Spikes, J.D. (1989). Photosensitization. In «The science of Photobiology» Edited by K.C. Smith. Plenum Press, New York. 2nd edition, p. 79-110.
|
|
(5)
|
OECD (1997). Environmental Health and Safety Publications, Series on Testing and Assessment No7 «Guidance Document On Direct Phototransformation Of Chemicals In Water» Environment Directorate, OECD, Paris.
|
|
(6)
|
Spielmann, H., Balls, M., Döring, B., Holzhütter, H.G., Kalweit, S., Klecak, G., L'Eplattenier, H., Liebsch, M., Lovell, W.W., Maurer, T., Moldenhauer. F. Moore. L., Pape, W., Pfannbecker, U., Potthast, J., De Silva, O., Steiling, W., and Willshaw, A. (1994). EEC/COLIPA project on in vitro phototoxicity testing: First results obtained with a Balb/c 3T3 cell phototoxicity assay. Toxic. In Vitro 8, p. 793-796.
|
|
(7)
|
Anon (1998). Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for phototoxicity), European Commission, Joint Research Centre: ECVAM and DGXI/E/2, 3 November 1997, ATLA, 26, p. 7-8.
|
|
(8)
|
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., Pechovitch, G. De Silva, O., Holzhütter, H.G., Clothier, R., Desolle, P., Gerberick, F., Liebsch, M., Lovell, W.W., Maurer, T., Pfannenbecker, U., Potthast, J. M., Csato, M., Sladowski, D., Steiling, W., and Brantom, P. (1998). The international EU/COLIPA In vitro phototoxicity validation study: results of phase II (blind trial), part 1: the 3T3 NRU phototoxicity test. Toxic. In Vitro 12, p. 305-327.
|
|
(9)
|
OECD (2002) Extended Expert Consultation Meeting on The In Vitro 3T3 NRU Phototoxicity Test Guideline Proposal, Berlin, 30th-31th October 2001, Secretariat's Final Summary Report, 15th March 2002, OECD ENV/EHS, available upon request from the Secretariat.
|
|
(10)
|
Borenfreund, E., and Puerner, J.A. (1985). Toxicity determination in vitro by morphological alterations and neutral red absorption. Toxicology Lett., 24 p. 119-124.
|
|
(11)
|
Hay, R.J. (1988). The seed stock concept and quality control for cell lines. Analytical Biochemistry 171, p. 225-237.
|
|
(12)
|
Lambert L.A, Warner W.G., and Kornhauser A. (1996) Animal models for phototoxicity testing. In «Dermatotoxicology», edited by F.N. Marzulli and H.I. Maibach. Taylor & Francis, Washington DC. 5th Edition, p. 515-530.
|
|
(13)
|
Tyrrell, R.M., Pidoux, M. (1987). Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet and violet regions of sunlight on epidermal keratinocytes. Cancer Res., 47, p. 1825-1829.
|
|
(14)
|
ISO 10977. (1993). Photography — Processed photographic colour films and paper prints — Methods for measuring image stability.
|
|
(15)
|
Sunscreen Testing (UV.B) TECHNICALREPORT, CIE, International Commission on Illumnation, Publication No 90, Vienna, 1993, ISBN 3900734275
|
|
(16)
|
ZEBET/ECVAM/COLIPA — Standard Operating Procedure: In Vitro 3T3 NRU Phototoxicity Test. Final Version, 7 September, 1998, p. 18.
|
|
(17)
|
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., De Silva, O., Holzhütter, H.G., Gerberick, F., Liebsch, M., Lovell, W.W., and Pfannenbecker, U. (1998) A study on UV filter chemicals from Annex VII of the European Union Directive 76/768/EEC, in the in vitro 3T3 NRU phototoxicity test. ATLA 26, p. 679-708.
|
|
(18)
|
Holzhütter, H.G., and Quedenau, J. (1995) Mathematical modeling of cellular responses to external signals. J. Biol. Systems 3, p. 127-138.
|
|
(19)
|
Holzhütter, H.G. (1997). A general measure of in vitro phototoxicity derived from pairs of dose-response curves and its use for predicting the in vivo phototoxicity of chemicals. ATLA, 25, p. 445-462.
|
|
(20)
|
http://www.oecd.org/document/55/0,2340,en_2649_34377_2349687_l_l_l_l,00.html
|
ANNEXE 1
Rôle de l'essai de phototoxicité 3T3 NRU dans une approche séquentielle des essais de phototoxicité des substances chimiques

ANNEXE 2
Figure 1
Distribution spectrale énergétique d'un simulateur solaire équipé de filtres

(voir point 1.4.1.5, second paragraphe)
La figure 1 donne un exemple de distribution acceptable de l'énergie spectrale d'un simulateur solaire équipé de filtres. Elle correspond à la source aux halogénures de métaux dopés utilisée dans l'essai de validation du test 3T3 NRU (6) (8) (17). Cette figure fait apparaître l'effet des deux filtres distincts, ainsi que l'effet de filtration de la plaque à 96 puits. Le filtre H2 est utilisé uniquement avec les systèmes d'essai qui peuvent supporter une quantité plus importante d'UVB (essai sur modèle de peau et essai de photo-hémolyse des globules rouges). Dans l'essai 3T3 NRU, le filtre H1 a été utilisé. La figure montre que l'effet de filtration supplémentaire de la plaque est principalement observé dans la plage des UVB, laissant néanmoins suffisamment d'UVB dans le spectre d'irradiation pour exciter les substances chimiques qui absorbent généralement la lumière dans la plage des UVB, telles que l'amiodarone (voir le tableau 1).
Figure 2
Sensibilité des cellules Balb/c 3T3 à l'irradiation (mesurée dans la plage des UVA)
Viabilité cellulaire ( % de fixation du rouge neutre des témoins dans l'obscurité)

Temps d'irradiation (minutes) (10 min = 1 joule UVA/cm2)
(voir point 1.4.1.5.2, second paragraphe, et points 1.4.2.2.1, 1.4.2.2.2)
Sensibilité des cellules Balb/c 3T3 à l'irradiation avec le simulateur solaire utilisé dans l'essai de validation du test de phototoxicité 3T3 NRU, mesurée dans la plage des UVA. La figure montre les résultats obtenus dans sept laboratoires différents au cours de l'étude de prévalidation (1). Les deux courbes avec des symboles transparents ont été obtenues avec des cellules âgées (nombre de passages élevés) qu'il a fallu remplacer par de nouvelles cellules, tandis que les courbes aux symboles pleins sont associées à des cellules montrant une tolérance acceptable à l'irradiation.
C'est à partir de ces données qu'on a dérivé la dose maximale d'irradiation non cytotoxique de 5 J/cm2 (ligne discontinue verticale). La ligne de pointillés horizontale montre en outre l'effet maximal d'irradiation acceptable (point 1.4.2.2).
B.42. SENSIBILISATION CUTANÉE: ESSAI DE STIMULATION LOCALE DES GANGLIONS LYMPHATIQUES
1. MÉTHODE
Cette méthode est équivalente à la ligne directrice TG 429 (2002) de l'OCDE.
1.1. INTRODUCTION
L'essai de stimulation locale des ganglions lymphatiques (ELGL) a été suffisamment validé et accepté pour pouvoir être adopté en tant que nouvelle méthode (1) (2) (3). Il s'agit, en l'occurrence, de la deuxième méthode permettant d'évaluer le pouvoir de sensibilisation cutanée des substances chimiques chez les animaux. L'autre méthode (B.6) fait appel à des essais sur cobayes, notamment l'essai de maximisation sur le cobaye et l'essai de Buehler (4).
L'ELGL offre un autre moyen d'identifier les produits chimiques qui exercent une action sensibilisante sur la peau et de confirmer que certaines substances sont dépourvues de pouvoir sensibilisant significatif. Cela ne signifie pas pour autant que l'ELGL doive systématiquement remplacer l'essai sur le cobaye, mais plutôt qu'il s'agit d'une méthode tout aussi valable, pouvant être utilisée à la place de ce dernier, et dont les résultats, qu'ils soient positifs ou négatifs, n'ont généralement pas besoin d'être reconfirmés.
L'ELGL présente certains avantages en ce qui concerne le progrès scientifique et la protection des animaux. Il étudie la phase d'induction de la sensibilisation cutanée et fournit des données quantitatives permettant d'évaluer l'effet en fonction de la dose. Les détails de la validation de l'ELGL et une synthèse des travaux qui y sont associés ont été publiés (5) (6) (7) (8). Il est également utile de noter que les sensibilisants légers à modérés recommandés comme témoins positifs dans les essais sur les cobayes conviennent aussi à l'ELGL (6) (8) (9).
L'ELGL, qui est une méthode in vivo, ne met pas un terme à l'utilisation des animaux dans l'évaluation de la sensibilisation par contact. Il peut, en revanche, diminuer le nombre d'animaux mobilisés à cette fin. De plus, l'ELGL améliore nettement la façon dont les animaux sont utilisés pour éprouver la sensibilisation par contact. L'ELGL se fonde sur l'observation de phénomènes immunologiques stimulés par des produits chimiques durant la phase d'induction de la sensibilisation. Avec l'ELGL, contrairement aux essais sur les cobayes, il n'est pas nécessaire de déclencher des réactions d'hypersensibilité cutanée par une exposition provocatrice. En outre, l'ELGL ne requiert pas d'adjuvant, à la différence de l'essai de maximisation sur les cobayes. Ainsi, l'ELGL diminue la souffrance infligée aux animaux. Malgré les avantages de l'ELGL par rapport aux essais classiques sur les cobayes, il faut reconnaître qu'il présente certaines limites imposant parfois le recours aux essais classiques sur cobayes (par exemple, des résultats faussement négatifs avec certains métaux, des résultats faussement positifs avec certains produits irritants pour la peau) (10).
Voir également Introduction, partie B.
1.2. PRINCIPE DE LA MÉTHODE D'ESSAI
L'ELGL repose sur le principe que les sensibilisants induisent une prolifération primaire de lymphocytes dans le ganglion lymphatique qui draine le site de l'application de la substance chimique. Cette prolifération est proportionnelle à la dose appliquée (et à la puissance de l'allergène) et permet d'obtenir facilement une mesure objective et quantitative de la sensibilisation. L'ELGL évalue cette prolifération d'après l'effet produit en fonction de la dose, en comparant les groupes d'essai aux groupes témoins traités par le véhicule. On détermine le rapport de la prolifération dans les groupes d'essai et de la prolifération observée dans les groupes témoins traités par le véhicule, dit indice de stimulation, lequel doit être au moins égal à trois pour que la substance d'essai continue d'être évaluée en tant que sensibilisant cutané potentiel. Les méthodes décrites ici mesurent la prolifération cellulaire à l'aide d'un marquage radioactif. Cependant, il est possible d'employer d'autres critères pour évaluer la prolifération, à condition que ce choix soit étayé par des données scientifiques pertinentes, notamment la citation de passages complets et une description de la méthode.
1.3. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.3.1. Préparations
1.3.1.1. Conditions d'hébergement et d'alimentation
Les animaux sont placés dans des cages individuelles. La température du local expérimental doit être de 22 oC (± 3 oC). L'humidité relative doit atteindre au moins 30 % et rester de préférence inférieure à 70 %, mais en dehors des heures de nettoyage du local, on s'efforcera de maintenir le taux d'humidité autour de 50 à 60 %. On appliquera un éclairage artificiel, alternant 12 heures de lumière et 12 heures d'obscurité. Les animaux seront nourris avec un mélange classique pour animaux de laboratoire et boiront de l'eau potable à volonté.
1.3.1.2. Préparation des animaux
Les animaux sont sélectionnés au hasard, marqués pour permettre leur identification individuelle (mais jamais sur l'oreille) et gardés dans leurs cages pendant au moins cinq jours avant le début du traitement, afin qu'ils s'acclimatent aux conditions du laboratoire. Avant le traitement, on examine tous les animaux pour vérifier qu'ils ne présentent pas de lésions cutanées observables.
1.3.2. Conditions expérimentales
1.3.2.1. Animaux d'expérience
L'espèce retenue pour cet essai est la souris. On utilise de jeunes femelles adultes de la souche CBA/Ca ou CBA/J, nullipares et non gravides. Au début de l'étude, les animaux doivent être âgés de 8 à 12 semaines; l'écart de poids entre les animaux doit être minime et ne doit pas dépasser 20 % du poids moyen. D'autres souches ainsi que des mâles pourront être utilisés s'il existe suffisamment d'informations démontrant l'absence de différences significatives entre les souches et/ou les sexes en ce qui concerne la réaction à l'ELGL.
1.3.2.2. Test de fiabilité
Les témoins positifs servent à démontrer le bon fonctionnement de l'essai et la capacité du laboratoire à réaliser correctement cet essai. Le témoin positif devrait réagir positivement à l'ELGL à un niveau d'exposition supposé accroître l'indice de stimulation (IS) > 3 par rapport au groupe de témoins négatifs. La dose administrée aux témoins positifs devrait être choisie de telle sorte que l'induction soit claire mais pas excessive. Les substances préférées sont l'aldéhyde hexylcinnamique (No CAS: 101-86-0, No E1NECS: 202-983-3) et le mercaptobenzothiazole (No CAS: 149-30-4, No EINECS: 205-736-8). Dans certains cas, d'autres substances témoins répondant aux critères susmentionnés pourront être employées, à condition que ce choix soit correctement justifié. Bien qu'il faille, en principe, inclure un groupe de témoins positifs dans chaque essai, certains laboratoires d'essai disposent de données antérieures sur des témoins positifs, qui permettent de montrer la constance d'une réaction satisfaisante sur une période de six mois ou plus. Dans ce cas, l'expérimentateur pourra espacer l'incorporation des témoins positifs, en respectant un intervalle maximal de six mois. Quoique la substance utilisée comme témoin positif doive être testée dans un véhicule déclenchant une réaction constante (par exemple acétone, huile d'olive), certaines situations réglementaires exigeront aussi l'essai d'un véhicule moins courant (mélange cliniquement ou chimiquement pertinent). Dans ces circonstances, il y a lieu de tester également l'interaction éventuelle entre une substance servant de témoin positif et ce véhicule inhabituel.
1.3.2.3. Nombre d'animaux, choix des doses et du véhicule
Chaque groupe d'essai comprend au moins quatre animaux sur lesquels on teste au moins trois concentrations de la substance d'essai, ainsi qu'un groupe de témoins négatifs traités seulement avec le véhicule de la substance d'essai et, au besoin, un témoin positif. S'il faut recueillir des données individuelles sur les animaux, les groupes d'essai compteront au moins cinq animaux. Mis à part l'absence de traitement par la substance d'essai, les animaux des groupes témoins doivent être manipulés et traités de la même manière que les animaux des groupes d'essai.
La sélection des doses et du véhicule s'effectue conformément aux recommandations données en référence (1). Les doses sont choisies parmi la série de concentrations suivante: 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 %, etc. Le cas échéant, on tiendra compte des données existantes concernant la toxicité aiguë et l'irritation cutanée en sélectionnant les trois concentrations consécutives, de telle façon que la concentration la plus élevée maximise l'exposition tout en évitant la toxicité systémique et une irritation cutanée locale excessive (2) (11).
On choisira le véhicule en fonction de sa capacité à maximiser les concentrations d'essai et la solubilité ainsi qu'à produire une solution ou une suspension se prêtant à l'application de la substance d'essai. Par ordre de préférence, les véhicules recommandés sont le mélange acétone/huile d'olive (4:1 v/v), le diméthyl-formamide, la méthyléthylcétone, le propylèneglycol et le diméthylsulfoxyde (2) (10), mais d'autres véhicules pourront également être utilisés à condition que ce choix soit suffisamment étayé sur le plan scientifique. Certaines situations réclameront un témoin supplémentaire, à savoir un solvant qui se justifie sur le plan clinique ou le mélange dans lequel la substance d'essai est commercialisée. L'expérimentateur veillera tout particulièrement à ce que les matières hydrophiles soient incorporées à un véhicule qui humidifie la peau et ne ruisselle pas immédiatement. Il conviendra donc d'éviter les véhicules totalement aqueux.
1.3.3. Mode opératoire
1.3.3.1. Programme expérimental
Le programme expérimental se déroule comme suit:
|
|
Premier jour:
Mesurer et consigner le poids de chaque animal. Application de 25 μL de la dilution appropriée de la substance d'essai, du véhicule seul, ou du témoin positif (le cas échéant), sur le dos de chaque oreille.
|
|
|
Deuxième et troisième jours:
Répéter la procédure d'application pratiquée le premier jour.
|
|
|
Quatrième et cinquième jours:
Pas de traitement.
|
|
|
Sixième jour:
Noter le poids de chaque animal. Injecter 250 μL de tampon phosphate contenant 20 µCi (7,4e + 8Bq) de 3H-méthylthymidine dans la veine caudale de toutes les souris traitées et témoins. Il est également possible d'injecter 250 μl de solution tampon phosphate contenant 2 μCi (7,4e + 7Bq) de 125I-iodo-déoxyuridine et de la fluorodéoxyuridine 10-5M dans la veine caudale de toutes les souris.
Cinq heures plus tard, les animaux sont sacrifiés. Après avoir excisé les ganglions rétro-auriculaires de chaque oreille de tous les animaux d'un groupe expérimental, on réunit ces ganglions dans un tampon phosphate (approche collective au niveau d'un groupe d'essai); on peut également exciser les paires de ganglions rétro-auriculaires de chaque animal et les mettre en suspension individuellement dans un tampon phosphate (méthode individuelle au niveau d'un animal). Les détails et les diagrammes relatifs à l'identification et à la dissection des ganglions sont repris à l'annexe I de la référence 10.
|
1.3.3.2. Préparation des suspensions cellulaires
L'expérimentateur préparera une suspension contenant les cellules des ganglions lymphatiques de tout un groupe ou d'un seul animal en pratiquant une séparation mécanique douce à travers une toile en acier inoxydable dont les mailles mesurent 200 μm. Les cellules de ganglions lymphatiques sont lavées deux fois avec un excès de tampon phosphate et précipitées avec de l'acide trichloracétique à 5 % à 4 oC pendant 18 heures (1). Les culots sont soit remis en suspension dans 1 mL d'acide trichloracétique puis transférés dans des flacons à scintillation contenant 10 mL de scintillateur liquide pour le comptage du tritium 3H, soit directement transférés dans des tubes compteurs de rayons gamma pour le comptage de l'iode 125I.
1.3.3.3. Mesure de la prolifération cellulaire (radioactivité incorporée)
L'incorporation de 3H-méthylthymidine se mesure par comptage de scintillations ß, en désintégrations par minute (DPM). L'incorporation de 125I-iododéoxyuridine se mesure par comptage de l'Iode 125I et s'exprime également en DPM. Suivant la méthode choisie, l'incorporation sera exprimée en DPM/groupe d'essai (méthode collective au niveau d'un groupe) ou en DPM/animal traité (méthode individuelle au niveau d'un animal).
1.3.3.4. Observations
1.3.3.4.1. Observations cliniques
Une fois par jour, l'expérimentateur examinera attentivement les animaux afin de déceler d'éventuels signes cliniques, se traduisant par une irritation locale sur le site d'application ou par une toxicité systémique. Toutes les observations sont systématiquement consignées, et ce pour chaque animal séparément.
1.3.3.4.2. Poids corporel
Comme indiqué au paragraphe 1.3.3.1, le poids corporel de chaque animal est relevé au début de l'essai et au moment prévu du sacrifice.
1.3.4. Calcul des résultats
Les résultats sont exprimés par l'indice de stimulation (IS). Si l'on utilise la méthode collective, l'indice de stimulation s'obtient en divisant la radioactivité incorporée globalement pour chaque groupe traité par la substance d'essai par la radioactivité incorporée globalement pour le groupe témoin traité par le véhicule; ce rapport livre un indice de stimulation moyen. Dans le cas de la méthode individuelle, l'IS est obtenu en divisant la moyenne DPM/animal de chaque groupe traité par la substance d'essai et celle du groupe témoin positif par la moyenne DPM/animal du groupe témoin traité par le véhicule. L'indice de stimulation moyen pour les témoins traités par le véhicule est alors égal à 1.
L'utilisation de la méthode individuelle pour calculer l'IS permet d'effectuer une analyse statistique des données. Pour choisir une méthode d'analyse statistique appropriée, l'expérimentateur doit être conscient du risque d'inégalité des variances et des autres problèmes connexes qui pourraient nécessiter une transformation des données ou une analyse statistique non paramétrique. Une bonne façon d'interpréter les données consiste à évaluer toutes les données individuelles des groupes d'essai et des groupes traités par le véhicule, afin d'en tirer la meilleure courbe d'ajustement de la relation dose-effet, compte tenu des intervalles de confiance (8) (12) (13). L'expérimentateur doit toutefois être attentif aux réactions «atypiques» possibles de certains animaux au sein d'un groupe, qui pourraient nécessiter le recours à une autre mesure de la réaction (par exemple la médiane au lieu de la moyenne) ou l'élimination de la réaction atypique.
Pour qu'une réaction puisse être considérée comme positive, il faut que l'indice de stimulation soit ≥ 3, tenir compte de la relation dose-effet et, s'il y a lieu, de la signification statistique (3) (6) (8) (12) (14).
Si les résultats obtenus ne sont pas assez concluants, on examinera les diverses propriétés de la substance d'essai, notamment afin de savoir si elle présente une similitude structurelle avec des sensibilisants cutanés connus, si elle déclenche une irritation cutanée excessive, ainsi que la nature de la relation dose-effet observée. Ces considérations, ainsi que d'autres, sont débattues en détail dans un autre document (7).
2. RÉSULTATS
Les données doivent être récapitulées sous forme de tableaux montrant les valeurs de DPM moyennes et individuelles et les indices de stimulation pour chaque groupe d'essai (y compris le groupe de témoins traités par le véhicule).
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit fournir les informations suivantes:
|
|
Substance d'essai:
|
—
|
données d'identification (par exemple numéro CAS, le cas échéant; source; pureté; impuretés connues; numéro de lot),
|
|
—
|
état physique et propriétés physico-chimiques (par exemple volatilité, stabilité, solubilité),
|
|
—
|
s'il s'agit d'un mélange: composition et pourcentages relatifs des constituants.
|
|
|
|
Véhicule:
|
—
|
données d'identification (pureté; concentration, s'il y a lieu; volume utilisé),
|
|
—
|
justification du choix du véhicule.
|
|
|
|
Animaux d'expérience:
|
—
|
souche de souris utilisée,
|
|
—
|
état microbiologique des animaux, s'il est connu,
|
|
—
|
source des animaux, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
nombre, âge et sexe des animaux;
|
|
|
|
Conditions expérimentales:
|
—
|
détails concernant la préparation et l'application de la substance d'essai,
|
|
—
|
justification du choix de la dose, y compris résultats d'une étude d'établissement de la gamme de doses, le cas échéant; concentrations du véhicule et de la substance d'essai et quantité totale de substance appliquée,
|
|
—
|
détails concernant la qualité de la nourriture et de l'eau de boisson (notamment le type de régime alimentaire, sa source et la source d'eau de boisson).
|
|
|
|
Test de fiabilité:
|
—
|
résumé des résultats du dernier test de fiabilité, notamment les informations sur la substance, sa concentration et le véhicule utilisé,
|
|
—
|
données antérieures ou concomitantes relatives aux témoins positifs et négatifs utilisés dans les laboratoires d'essai.
|
|
|
|
Résultats:
|
—
|
poids individuel des animaux au début de l'essai et au moment programmé du sacrifice,
|
|
—
|
tableau des valeurs de DPM moyennes (méthode collective) ou individuelles (méthode individuelle) ainsi que les échelles des valeurs pour les deux méthodes et indices de stimulation pour chaque groupe d'essai (y compris le groupe de témoins traités par le véhicule),
|
|
—
|
analyse statistique, si nécessaire,
|
|
—
|
moment du déclenchement des effets et signes de toxicité, y compris irritation cutanée au niveau du site d'application, pour chaque animal.
|
|
|
|
Discussion des résultats:
|
—
|
bref commentaire sur les résultats, analyse de la relation dose-effet et analyses statistiques, s'il y a lieu, et conclusion quant au fait de savoir si la substance doit être considérée comme un sensibilisant cutané.
|
|
4. BIBLIOGRAPHIE
|
(1)
|
Kimber, I. and Basketter, D.A. (1992). The murine local lymph node assay; collaborative studies and new directions: A commentary. Food and Chemical Toxicology 30, p. 165-169.
|
|
(2)
|
Kimber, I, Derman, R.J., Scholes E.W, and Basketter, D.A. (1994). The local lymph node assay: developments and applications. Toxicology, 93, p. 13-31.
|
|
(3)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E., Hastings, K.L. (1998). Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. Journal of Toxicology and Environmental Health, 53, p. 563-79.
|
|
(5)
|
Chamberlain, M. and Basketter, D.A. (1996). The local lymph node assay: status of validation. Food and Chemical Toxicology 34, 999-1002.
|
|
(6)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E (1996). The local lymph node assay. A viable alternative to currently accepted skin sensitisation tests. Food and Chemical Toxicology, 34, p. 985-997.
|
|
(7)
|
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998). Strategies for identifying false positive responses in predictive sensitisation tests. Food and Chemical Toxicology. 36, p. 327-33.
|
|
(8)
|
Van Och, F.M.M, Slob, W., De Jong, W.H., Vandebriel, R.J., Van Loveren, H. (2000). A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employement of a regression method that includes determination of uncertainty margins. Toxicology, 146, 49-59.
|
|
(9)
|
Dearman, R.J., Hilton,}., Evans, P., Harvey, P., Basketter, D.A. and Kimber, I. (1998). Temporal stability of local lymph node assay responses to hexyl cinnamic aldehyde. Food and Chemical Toxicology, 18, p. 281-4.
|
|
(10)
|
National Institute of Environmental Health Sciences (1999). The Murine Local Lymph Node Assay: A Test Method for Assessing the Allergic Contact Dermatitis Potential of Chemicals/Compounds: The Results of an Independent Peer Review Evaluation Coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99- 4494, Research Triangle Park, N.C. (http://iccvam.niehs.nih.gov).
|
|
(11)
|
Méthode d'essai B.4.
|
|
(12)
|
Basketter, D.A., Selbie, E., Scholes, E.W. Lees, D. Kimber, I. and Botham, P.A. (1993) Results with OECD recommended positive control sensitisers in the maximisation, Buehler and local lymph node assays. Food and Chemical Toxicology, 31, p. 63-67.
|
|
(13)
|
Basketter D.A., Lea L.J., Dickens A., Briggs D., Pate I., Dearman R.J., Kimber I. (1999). A comparison of statistical approaches to the derivation of EC3 values from local lymph node assay dose responses J. Appl. Toxicology, 19, p. 261-266.
|
|
(14)
|
Basketter DA, Blaikie L, Derman RJ, Kimber I, Ryan CA, Gerberick GF, Harvey P, Evans P, White IR and Rycroft RTG (2000). Use of local lymph node assay for the estimation of relative contact allergenic potency. Contact Dermatitis 42, p. 344-48.
|
B.43. ÉTUDE DE NEUROTOXICITÉ CHEZ LES RONGEURS
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 424 (1997) de l'OCDE.
Cette méthode d'essai a été conçue dans le but de recueillir des données permettant de confirmer ou de mieux caractériser la neurotoxicité potentielle d'une substance pour des animaux adultes. Elle peut être utilisée en association avec d'autres méthodes d'essai dans le cadre d'études de toxicité à dose répétée ou seule, en tant qu'étude indépendante. Il est recommandé de consulter le document d'orientation de l'OCDE sur les stratégies d'essais en matière de neurotoxicité (1). Cette recommandation est particulièrement importante lorsqu'on envisage de s'écarter des observations et des protocoles d'essai préconisés dans cette méthode. Le document d'orientation est également utile pour choisir un protocole d'essai adapté à un cas spécifique.
L'évaluation de la neurotoxicité développementale n'est pas l'objet de la présente méthode.
1.1. INTRODUCTION
Dans l'évaluation des propriétés toxiques d'un produit chimique, il est important de prendre en considération la possibilité d'effets neurotoxiques. La méthode d'essai concernant la toxicité systémique à dose répétée permet déjà un premier tri des substances potentiellement neurotoxiques. La présente méthode peut être utilisée pour obtenir des informations complémentaires sur les effets neurotoxiques observés dans les études de toxicité systémique à dose répétée, et éventuellement pour confirmer ces effets. Cependant, la neurotoxicité potentielle de substances appartenant à certaines catégories pourra être évaluée de façon plus appropriée en appliquant directement la présente méthode, sans recueillir au préalable les indications qui peuvent être fournies par les études de toxicité systémique à dose répétée. Ce sera notamment le cas lorsque:
|
—
|
des signes de neurotoxicité ou des lésions neuropathologiques sont observés dans des études de toxicité autres que des études de toxicité systémique à dose répétée, ou
|
|
—
|
lorsque les substances présentent des similitudes de structure ou ont d'autres caractéristiques en commun avec des substances neurotoxiques connues.
|
Cette méthode peut aussi être utile dans d'autres cas; voir référence (1) pour plus de détails.
Cette méthode a été conçue de manière à pouvoir être adaptée, en fonction des besoins spécifiques, aussi bien pour confirmer la neurotoxicité histopathologique d'une substance chimique ou sa neurotoxicité sur le plan du comportement que pour caractériser et quantifier les neurotoxiques.
Autrefois, la neurotoxicité était assimilée à une forme de neuropathie englobant des lésions neuropathologiques ou des dysfonctionnements neurologiques tels que apoplexie, paralysie ou tremblements. Bien que la neuropathie soit une manifestation importante de la neurotoxicité, il apparaît aujourd'hui clairement qu'il existe de nombreux autres signes de toxicité pour le système nerveux (par exemple, perte de la coordination motrice, déficits sensoriels, diminution de la faculté d'apprentissage et de la mémoire) qui ne sont pas toujours mis en évidence par les études neuropathologiques ou autres.
La présente méthode d'essai de neurotoxicité vise à détecter, chez les rongeurs adultes, les effets importants sur le comportement et les effets neuropathologiques. Bien que des effets sur le comportement, même s'ils ne sont pas accompagnés de changements morphologiques, puissent révéler un impact néfaste sur l'organisme, les changements de comportement ne sont pas tous spécifiques du système nerveux. Par conséquent, tout changement observé doit être évalué par rapport aux données histopathologiques, hématologiques ou biochimiques correspondantes et aux résultats d'autres études de toxicité systémique. Les essais préconisés par la présente méthode pour caractériser et quantifier les réponses neurotoxiques comprennent des procédures histopathologiques et des procédures spécifiquement axées sur le comportement, qui pourront être étayées par des études électrophysiologiques et/ou biochimiques (1) (2) (3) (4).
Les agents neurotoxiques peuvent agir sur différentes cibles dans le système nerveux, et cela par différents mécanismes. Comme il est impossible de concevoir une seule série d'essais permettant d'évaluer de manière approfondie le potentiel neurotoxique de toutes les substances, il peut s'avérer nécessaire de mettre en œuvre d'autres essais in vivo ou in vitro spécifiques du type de neurotoxicité observé ou escompté.
La présente méthode d'essai peut également servir, en association avec le document d'orientation de l'OCDE sur les stratégies et les méthodes d'essais en matière de neurotoxicité, à concevoir des études destinées à mieux caractériser la relation dose-effet ou à en améliorer la sensibilité, en vue d'obtenir une meilleure estimation de la concentration sans effet nocif observé ou de mettre en évidence les dangers connus ou suspectés de la substance. On peut ainsi concevoir des études pour identifier et évaluer le ou les mécanisme(s) neurotoxiques ou pour compléter des données déjà fournies par l'application de protocoles de base d'observations du comportement et d'observations neuropathologiques. Lors de ces études, il est inutile de chercher à obtenir en double des données qui seraient de toute façon obtenues en suivant les protocoles préconisés par cette méthode, si ces données sont déjà disponibles et ne sont pas nécessaires pour l'interprétation des résultats de l'étude.
Les informations recueillies dans cette étude de neurotoxicité, qu'elle soit réalisée de façon indépendante ou couplée à d'autres études, permettent de:
|
—
|
déterminer si les effets de la substance chimique sur le système nerveux sont permanents ou réversibles,
|
|
—
|
mieux caractériser les altérations du système nerveux qui sont liées à l'exposition à la substance et faciliter la compréhension du mécanisme sous-jacent,
|
|
—
|
déterminer les relations entre dose et effet et entre temps et effet afin de pouvoir estimer la concentration sans effet nocif observé (laquelle pourra servir à établir les critères de sécurité de la substance).
|
Dans cette méthode, la substance d'essai est administrée par voie orale. Si d'autres voies, comme la voie dermique ou l'inhalation, paraissent plus appropriées, des modifications des procédures recommandées s'imposent. Le choix de la voie d'administration est dicté par les circonstances de l'exposition humaine et par les informations toxicologiques ou cinétiques disponibles.
1.2. DÉFINITIONS
Effet nocif: toute altération par rapport à une situation de référence, qui est due au traitement et qui diminue l'aptitude d'un organisme à survivre, à se reproduire ou à s'adapter à l'environnement.
Dose: la quantité de substance d'essai administrée. La dose s'exprime en poids (g, mg) de substance d'essai ou en poids de substance d'essai par unité de poids de l'animal d'expérience (par exemple mg/kg) ou en concentration dans le régime (ppm).
Dosage: un terme général qui comprend la dose, la fréquence et la durée de l'administration.
Neurotoxicité: une altération de la structure ou de la fonction du système nerveux qui est la conséquence d'une exposition à un agent chimique, biologique ou physique.
Agent neurotoxique: tout agent chimique, biologique ou physique capable d'induire une neurotoxicité.
CSENO: concentration maximale dans effet nocif observé, c'est-à-dire la dose la plus élevée à laquelle aucun effet nocif imputable au traitement n'est observé.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance à tester est administrée par voie orale à différents niveaux de dose à plusieurs groupes de rongeurs. L'administration se fait généralement à doses répétées sur une période qui peut être de 28 jours, de 90 jours (étude subchronique) ou d'une année ou plus (étude chronique). Les procédures décrites dans cette méthode peuvent également être appliquées dans le cas d'une étude de neurotoxicité aiguë. Les animaux sont soumis à l'essai afin de détecter ou de caractériser des anomalies de comportement ou d'ordre neurologique. Au cours de chaque période d'observation, différents aspects du comportement qui pourraient être indicatifs d'une atteinte neurotoxique sont évalués. À la fin de l'essai, une partie des animaux de chaque groupe et de chaque sexe sont perfusés in situ et des coupes du cerveau, de la moelle épinière et des nerfs périphériques sont préparées et examinées.
Lorsque l'étude est conduite de façon indépendante pour dépister une neurotoxicité ou caractériser des effets neurotoxiques, les animaux de chaque groupe qui ne sont pas utilisés pour la perfusion et l'examen histopathologique (voir tableau 1) peuvent servir pour des examens du comportement et des examens neuropathologiques, neurochimiques ou électrophysiologiques qui permettent de compléter les données recueillies dans les observations de base préconisées par la présente méthode (1). Ces examens complémentaires peuvent être particulièrement utiles lorsque des observations empiriques ou des effets escomptés laissent prévoir un type spécifique de neurotoxicité ou une cible spécifique pour cette neurotoxicité. Une autre possibilité est d'utiliser les animaux restants dans des évaluations telles que celles qui sont préconisées par les méthodes d'essai de toxicité à doses répétées chez les rongeurs.
Lorsque les procédures de la présente méthode d'essai sont combinées avec celles d'autres méthodes, il faut prévoir un nombre suffisant d'animaux pour pouvoir effectuer les observations requises par les deux études.
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Choix de l'espèce animale
Le rat est l'espèce préférée, mais d'autres espèces de rongeurs peuvent être utilisées moyennant justification du choix. Il convient de recourir aux souches couramment utilisées en laboratoire. Les animaux doivent être adultes, jeunes et sains. Les femelles doivent être nullipares et non gravides. L'administration des doses doit débuter aussitôt après le sevrage, de préférence avant que les animaux n'atteignent l'âge de six semaines et en tout état de cause avant neuf semaines. Cependant, lorsque la présente étude est couplée à d'autres études, cette limite d'âge peut faire l'objet d'un ajustement. Au début de l'étude, les variations de poids entre animaux ne doivent pas dépasser plus ou moins 20 % du poids moyen des animaux du même sexe. Si une étude de toxicité orale à dose répétée de faible durée est réalisée en tant qu'essai préliminaire avant une étude à long terme, il faut utiliser des animaux de même souche et de même provenance dans les deux études.
1.4.2. Conditions d'hébergement et d'alimentation
La température du local des animaux d'expérience doit être de 22 oC (± 3 oC). L'humidité relative doit atteindre au moins 30 % et rester de préférence inférieure à 70 %, mais en dehors des heures de nettoyage du local, on s'efforcera de maintenir le taux d'humidité autour de 50 à 60 %. On appliquera un éclairage artificiel alternant 12 heures de lumière et 12 heures d'obscurité. Les bruits forts intermittents doivent être réduits au minimum. Pour l'alimentation des animaux, on pourra utiliser la nourriture classique de laboratoire avec de l'eau potable à satiété. Le choix du régime peut être influencé par la nécessité d'assurer un mélange convenable de la substance d'essai lorsqu'elle est administrée dans la nourriture. Les animaux peuvent être placés dans des cages soit individuellement, soit par petits groupes du même sexe.
1.4.3. Préparation des animaux
Des animaux jeunes et sains sont choisis au hasard pour être répartis entre les groupes de contrôle et de traitement. Les cages sont disposées de manière à minimiser les effets possibles dus à l'agencement des cages. Les animaux sont marqués individuellement afin de permettre leur identification. Ils sont maintenus dans les cages pendant cinq jours au moins avant le début de l'étude pour leur permettre de s'acclimater aux conditions du laboratoire.
1.4.4. Voies d'administration et préparation des doses
Dans cette méthode, la substance d'essai est administrée par voie orale. La substance à tester peut être administrée par gavage ou en capsules ou incorporée dans la nourriture ou l'eau de boisson. D'autres voies d'administration (par exemple voie dermique ou inhalation) peuvent être utilisées, mais nécessiteront le cas échéant des modifications du mode opératoire. Le choix de la voie d'administration est dicté par les circonstances de l'exposition humaine et par les informations toxicologiques ou cinétiques disponibles. Le choix de la voie d'administration ainsi que les éventuelles modifications du mode opératoire doivent être justifiés.
Si nécessaire, la substance d'essai peut être dissoute ou mise en suspension dans un véhicule approprié. Il est recommandé de privilégier les solutions ou suspensions aqueuses. À défaut, on peut utiliser une solution dans l'huile (huile de maïs, par exemple) et, en dernier recours, une solution dans d'autres véhicules. Lorsque le véhicule n'est pas aqueux, il faut en connaître les propriétés toxiques. D'autres caractéristiques du véhicule peuvent avoir de l'importance, notamment des éventuels effets sur l'absorption, la distribution, le métabolisme ou la rétention de la substance d'essai, pouvant agir sur les propriétés toxiques de celle-ci, et des effets sur la consommation de nourriture ou d'eau ou sur l'état nutritionnel des animaux.
1.5. MODE OPÉRATOIRE
1.5.1. Nombre et sexe des animaux
Lorsqu'il s'agit d'une étude indépendante, il faut utiliser au moins vingt animaux (dix femelles et dix mâles) dans chaque groupe de traitement et dans chaque groupe témoin pour les observations cliniques et fonctionnelles. À la fin de l'étude, au moins cinq mâles et cinq femelles sont prélevés parmi ces dix mâles et ces dix femelles pour être perfusés in situ et soumis à un examen neurohistopathologique. Lorsque dans un groupe de dosage, les observations visant à détecter les effets neurotoxiques ne sont réalisées que sur un nombre limité d'animaux, il convient que ces animaux fassent partie de ceux qui seront choisis pour être perfusés. Si l'étude est combinée avec une étude de toxicité à doses répétées, le nombre des animaux doit être suffisant pour que les objectifs des deux études puissent être atteints. Le tableau 1 donne les nombres minimaux d'animaux par groupe pour différentes combinaisons d'études. S'il est prévu de sacrifier des animaux avant le terme de l'étude ou de constituer des groupes pour observer la réversibilité des effets, leur persistance ou l'apparition différée d'effets toxiques après traitement, ou si des observations complémentaires sont envisagées, il faut augmenter le nombre d'animaux pour s'assurer que le nombre requis pour les observations et les examens histopathologiques sera disponible.
1.5.2. Groupes de traitement et groupes témoins
En règle générale, on doit disposer d'au moins trois groupes d'animaux traités et d'un groupe témoin. Cependant, s'il ressort de l'évaluation d'autres données qu'aucun effet n'est à attendre d'une administration répétée à la dose de 1 000 mg/kg de poids corporel/jour, on peut procéder à un essai limite. Si l'on ne dispose pas de données adéquates, on peut mener une étude préliminaire visant à délimiter la gamme des niveaux de doses à utiliser. À l'exception de l'exposition à la substance à tester, les animaux du groupe témoin doivent être traités exactement de la même manière que les animaux des groupes de traitement. Si la substance à tester est incorporée dans un véhicule, le groupe témoin doit en recevoir un volume égal au plus grand volume utilisé dans les groupes de traitement.
1.5.3. Contrôle de la fiabilité
Le laboratoire chargé de l'étude doit présenter des données attestant de sa capacité à réaliser l'étude et prouvant la sensibilité des méthodes employées. Ces données doivent constituer une preuve évidente de la faculté du laboratoire à détecter et, le cas échéant, à quantifier les changements observés pour les différents critères d'évaluation, notamment réactions neurovégétatives, réactivité sensorielle, force de préhension et activité motrice. Les références 2 à 9 contiennent des informations sur les substances qui provoquent différents types de réponses neurotoxiques et qui peuvent servir de témoins positifs. Des données antérieures peuvent également être utilisées à condition que les principaux éléments des procédures expérimentales soient restés les mêmes. Des mises à jour périodiques de ce type de données sont recommandées. Chaque fois que le laboratoire modifie un élément essentiel du mode opératoire ou des méthodes employées, de nouvelles données doivent être réunies pour démontrer que la sensibilité des procédures est maintenue.
1.5.4. Choix des doses
Les niveaux de dose doivent être choisis en fonction des données disponibles concernant la toxicité et la toxico-cinétique de la substance d'essai ou de substances apparentées. La dose la plus élevée est choisie dans le but de provoquer des effets neurotoxiques ou des effets de toxicité systémique manifeste. Il faut ensuite choisir une série de doses décroissantes de façon à mettre en évidence une éventuelle relation dose-effet et l'absence d'effets nocifs observables au niveau de la dose la plus faible (CSENO). En principe, les niveaux de doses doivent être choisis de telle manière que l'on puisse distinguer les effets neurotoxiques primaires des effets liés à la toxicité systémique. Deux à trois intervalles entre niveaux successifs représentent fréquemment la meilleure solution et il est souvent préférable d'ajouter un quatrième groupe d'essai plutôt que de fixer des intervalles trop espacés (par exemple dépassant un facteur dix) entre les niveaux de dose. Lorsque des estimations réalistes de l'exposition humaine existent, il faut en tenir compte.
1.5.5. Essai limite
Si une étude réalisée à une dose d'au moins 1 000 mg/kg de poids corporel/jour selon la méthode décrite ci-dessus ne provoque aucun effet toxique observable, et que d'après les données obtenues pour des composés de structure analogue il est peu probable que la substance testée soit toxique, on peut considérer qu'il est inutile d'effectuer une étude complète sur trois niveaux de dose. En fonction de l'exposition humaine escomptée, il pourra s'avérer nécessaire d'administrer une dose plus forte par voie orale pour l'essai limite. Pour les autres voies d'administration, comme l'inhalation ou l'application cutanée, ce sont souvent les propriétés physiques et chimiques de la substance qui déterminent le niveau d'exposition maximal. Dans le cas d'une étude orale aiguë, la dose de l'essai limite doit être au moins 2 000 mg/kg.
1.5.6. Administration des doses
La substance à tester est administrée aux animaux quotidiennement, sept jours par semaine, sur une période d'au moins 28 jours. L'administration à raison de cinq jours par semaine ou l'adoption d'une période d'exposition plus courte demandent à être justifiées. Lorsque la substance est administrée par gavage, les animaux doivent recevoir une dose unique introduite au moyen d'une sonde gastrique ou d'une canule d'intubation appropriée. Le volume maximal de liquide pouvant être administré en une fois dépend de la taille de l'animal. Ce volume ne doit pas dépasser 1 ml/100 g de poids corporel. Toutefois, dans le cas d'une solution aqueuse, il est possible d'utiliser jusqu'à 2 ml/100 g de poids corporel. Exception faite pour les substances irritantes ou corrosives, qui donneraient normalement lieu à des effets fortement amplifiés à des concentrations plus élevées, il convient de minimiser les variations du volume administré en ajustant la concentration de façon à maintenir un volume constant à tous les niveaux de dose.
Lorsque la substance est administrée dans la nourriture ou l'eau de boisson, il importe que les quantités de substance utilisées ne modifient pas les bilans nutritionnels ou hydriques normaux. Lorsque la substance est administrée dans la nourriture, deux possibilités sont offertes: soit le maintien d'une concentration constante (exprimée en ppm), soit le maintien d'un niveau de dose constant par rapport au poids corporel de l'animal. L'option choisie doit être précisée. Lorsque la substance est administrée par gavage, la dose doit être administrée chaque jour à la même heure et la quantité doit être ajustée de manière à maintenir un niveau de dose constant par rapport au poids corporel de l'animal. Lorsqu'un essai à dose répétée sert d'étude préliminaire à une étude à long terme, il faut appliquer le même régime alimentaire dans les deux études. Pour les études de toxicité aiguë, si la dose ne peut être administrée en une seule fois, il est possible de la fractionner sur période n'excédant pas 24 heures.
1.6. OBSERVATIONS
1.6.1. Fréquence des observations et essais
Dans les études à doses répétées la période d'observation doit s'étendre sur toute la période de traitement. Dans les études de toxicité aiguë, les observations sont poursuivies pendant 14 jours après le traitement. Les animaux de groupes satellites (qui ne sont pas exposés pendant une certaine période après le traitement) sont observés également pendant 14 jours.
Les observations doivent être suffisamment fréquentes pour favoriser la détection de toute anomalie de comportement ou d'ordre neurologique. Les observations se font de préférence aux mêmes moments chaque jour, en tenant compte de la période au cours de laquelle les effets escomptés du traitement seront les plus marqués. Le tableau 2 récapitule la fréquence des observations cliniques et des tests fonctionnels. Si des données de cinétique et autres, obtenues dans des études antérieures, indiquent que d'autres moments de la journée sont plus propices aux observations, essais ou observations après traitement, il y a lieu d'adopter un échéancier différent afin de recueillir le plus d'informations possible. Les modifications de l'échéancier doivent être justifiées.
1.6.1.1. Surveillance de l'état de santé général et relevés de mortalité/morbidité
Tous les animaux font l'objet d'un examen minutieux au moins une fois par jour pour vérifier leur état de santé, et des relevés de la morbidité et de la mortalité sont effectués au moins deux fois par jour.
1.6.1.2. Observations cliniques détaillées
Tous les animaux choisis pour être soumis à un examen clinique approfondi (voir tableau 1) subissent cet examen une fois avant la première exposition (pour permettre des comparaisons sur un même individu) et à divers intervalles par la suite, en fonction de la durée de l'étude (voir tableau 2). Les groupes satellites destinés à l'observation de la réversibilité des effets sont examinés à la fin de la période de récupération. Ces examens doivent être effectués hors de la cage habituelle, sur une aire standard. Les résultats doivent être soigneusement consignés, de préférence à l'aide de systèmes de cotation utilisant des critères ou d'échelles de cotation, pour chaque mesure effectuée. Les critères ou échelles employés doivent être explicitement définis par le laboratoire. Il faut s'efforcer de minimiser les variations des conditions d'essai (mis à part celles qui sont liées au traitement) et prendre les dispositions nécessaires pour que les examens soient effectués par des observateurs expérimentés n'ayant pas connaissance du traitement administré.
Il est recommandé de procéder de manière structurée en appliquant systématiquement, pour chaque animal et à chaque moment d'observation, des critères bien définis. Les données utilisées pour définir le niveau normal doivent être présentées. Tous les signes observés doivent être consignés. L'ampleur des signes observés est également consignée chaque fois que cela est possible. Les observations cliniques devraient notamment porter sur les modifications de l'état de la peau, de la fourrure, des yeux, des membranes muqueuses, l'apparition de sécrétions et d'excrétions et de réactions neurovégétatives (sécrétion de larmes, horripilation, variation du diamètre pupillaire, rythme respiratoire inhabituel, respiration par la bouche, tous signes inhabituels de miction ou défécation, urine décolorée, par exemple).
Il convient également de consigner toute réaction inhabituelle en ce qui concerne la position du corps, le niveau d'activité (par exemple exploration accrue ou diminuée de l'aire standard) et la coordination des mouvements. Il convient également de consigner les modifications dans la démarche (dandinement, ataxie, par exemple), dans la posture (dos arrondi, par exemple) et la réaction à la manipulation, au placement et autres stimuli, ainsi que la présence de mouvements cloniques ou toniques, les convulsions et tremblements, les comportements stéréotypés (par exemple, toilettage excessif, parcours circulaires répétitifs) ou les comportements bizarres (par exemple, tendance à mordre, léchage excessif, automutilation, marche à reculons, vocalisation) ou l'agressivité.
1.6.1.3. Tests fonctionnels
Comme dans le cas des observations cliniques, les tests fonctionnels sont réalisés sur tous les animaux sélectionnés à cet effet (voir tableau 1) une fois avant l'exposition et fréquemment ensuite. La fréquence des tests fonctionnels dépend également de la durée de l'étude (voir tableau 2). En plus des périodes d'observation stipulées dans le tableau 2, des observations fonctionnelles sont réalisées sur des groupes satellites aussi près que possible du sacrifice final. Les tests fonctionnels explorent notamment la réactivité sensorielle à divers stimuli [par exemple, stimuli auditifs, visuels et proprioceptifs (5) (6) (7)], la force de préhension (8) et l'activité motrice (9). Cette dernière doit être mesurée à l'aide d'un dispositif automatique pouvant détecter aussi bien les hausses que les baisses d'activité. Si un autre système défini est utilisé, il doit être quantitatif et de sensibilité et de fiabilité démontrées. Chaque dispositif doit être testé afin de garantir sa fiabilité au cours du temps et la cohérence des résultats d'un dispositif à l'autre. On trouvera dans les références bibliographiques susmentionnées une description plus détaillée des modes opératoires. En l'absence d'informations sur le potentiel neurotoxique (par exemple, informations sur la relation structure-activité, données épidémiologiques, autres études toxicologiques), il convient d'envisager des tests plus spécialisés d'exploration des fonctions sensorielles et motrices ou de la mémoire et de la faculté d'apprentissage. La référence (1) fournit de plus amples informations sur les tests plus spécialisés et leur utilisation.
Exceptionnellement, les animaux qui montrent des signes de toxicité marqués qui risqueraient de fausser considérablement les résultats du test fonctionnel peuvent ne pas être soumis à ce test. La décision d'écarter des animaux d'un test fonctionnel doit être justifiée.
1.6.2. Poids corporel et consommation de nourriture et d'eau
Dans les études allant jusqu'à 90 jours, tous les animaux doivent être pesés au moins une fois par semaine. Il faut également mesurer la quantité de nourriture consommée (ou d'eau consommée, lorsque la substance est administrée par cette voie), au moins une fois par semaine. Dans les études à long terme, tous les animaux sont pesés au moins une fois par semaine pendant les treize premières semaines et au moins une fois toutes les quatre semaines par la suite. La quantité de nourriture consommée (ou d'eau, si la substance est administrée par cette voie) doit être mesurée au moins une fois par semaine pendant les treize premières semaines et à peu près tous les trois mois par la suite, à moins qu'une détérioration de l'état général ou un amaigrissement n'imposent d'autres conditions.
1.6.3. Ophtalmologie
Dans les études de plus de 28 jours, un examen ophtalmologique à l'aide d'un ophtalmoscope ou d'un autre appareil approprié doit être effectué avant l'administration de la substance et au terme de l'étude. Cet examen est effectué de préférence sur tous les animaux et en tout état de cause au moins sur ceux du groupe ayant reçu la dose la plus forte ainsi que sur les animaux des groupes témoins. Si des altérations sont détectées dans les yeux, ou si des signes cliniques y incitent, l'examen est étendu à tous les animaux. Dans les études à long terme, un examen ophtalmologique est prévu après treize semaines. Des examens ophtalmologiques ne sont pas nécessaires si les informations pertinentes ont déjà été fournies par d'autres études de durée similaire réalisées à des doses comparables.
1.6.4. Hématologie et biochimie clinique
Lorsque l'étude de neurotoxicité est conduite en combinaison avec une étude de toxicité systémique à doses répétées, les examens hématologiques et les déterminations de biochimie clinique doivent être effectués comme prescrit par la méthode correspondante de l'étude de toxicité systémique. Les échantillons sont à prélever de telle façon que les effets possibles sur le comportement neurologique soient réduits au minimum.
1.6.5. Histopathologie
L'examen neuropathologique doit être conçu dans le but de compléter et d'approfondir les observations faites pendant la phase in vivo de l'étude. Les tissus d'au moins cinq animaux de chaque sexe et de chaque groupe (voir le tableau 1 et le paragraphe suivant) doivent être fixés in situ à l'aide des techniques de perfusion et de fixation usuelles (voir le chapitre 5 de la référence 3 et le chapitre 50 de la référence 4). Tout changement important observable doit être consigné. S'il s'agit d'une étude indépendante, réalisée pour dépister une neurotoxicité ou pour caractériser des effets neurotoxiques, les animaux restants peuvent être utilisés pour des examens spécifiques du comportement neurologique (10) (11), des examens neuropathologiques (10) (11) (12) (13), neurochimiques (10) (11) (14) (15) ou électrophysiologiques (10) (11) (16) (17) pouvant éventuellement compléter les procédures et examens décrits ici, ou bien venir s'ajouter aux animaux soumis à l'examen histopathologique. Ces procédures complémentaires sont particulièrement utiles lorsque, à la suite d'observations empiriques ou sur la base des effets escomptés, on peut s'attendre à un type spécifique de neurotoxicité ou à une cible spécifique de celle-ci (2) (3). Ces animaux restants peuvent également servir dans les évaluations pathologiques de routine qui sont décrites dans la méthode à doses répétées.
Les échantillons de tissus sont colorés par une méthode usuelle, par exemple par de l'hématoxyline et de l'éosine, inclus dans de la paraffine et examinés sous le microscope. Si des signes de neuropathie périphérique sont observés ou en cas de suspicion de tels effets, il conviendra d'examiner des échantillons de tissu nerveux périphérique inclus dans du plastique. Certains signes cliniques peuvent également inciter à l'examen d'autres sites ou à l'utilisation de méthodes de coloration spéciales. Les références (3) et (4) fournissent des indications sur les sites supplémentaires à examiner. Des colorants spéciaux peuvent aussi être utiles pour mettre en évidence des types spécifiques d'altérations pathologiques (18).
Un examen histologique doit être réalisé sur des coupes représentatives du système nerveux central et périphérique (voir le chapitre 5 de la référence 3 et le chapitre 50 de la référence 4). Les sites examinés doivent normalement comprendre le cerveau antérieur, le centre des hémisphères cérébraux, comprenant une coupe de l'hippocampe, le cerveau central, le cervelet, la protubérance annulaire, le bulbe rachidien, l'œil avec le nerf optique et la rétine, la moelle épinière et les renflements cervicaux et lombaires, les ganglions de la chaîne dorsale, les fibres de la chaîne dorsale et ventrale, le nerf sciatique proximal, le nerf tibial proximal (au genou) et les ramifications du nerf tibial aux muscles du mollet. Les sections de la moelle épinière et des nerfs périphériques doivent comprendre des coupes transversales et longitudinales. Il faut accorder une attention particulière à la vascularisation du système nerveux. Un échantillon de muscles squelettiques, en particulier du mollet, devrait également être examiné. Une attention particulière devrait être accordée aux régions à structure cellulaire et fibreuse du système nerveux central et périphérique, qui sont connues pour être particulièrement sujettes aux attaques neurotoxiques.
Les références (3) et (4) fournissent des informations sur les altérations neuropathologiques typiquement liées aux expositions à une substance neurotoxique. Il est recommandé de procéder par étapes pour l'examen des échantillons de tissus, en comparant d'abord des coupes du groupe ayant reçu la forte dose avec des coupes du groupe témoin. Si aucune altération neuropathologique n'est constatée dans les échantillons de ces groupes, il n'est pas nécessaire de poursuivre l'analyse. Si des altérations neuropathologiques apparaissent dans le groupe traité à la forte dose, il faut alors examiner successivement des échantillons de tous les tissus potentiellement affectés des groupes traités à la dose intermédiaire et à la dose faible.
Si l'examen qualitatif met en évidence des signes d'altérations neuropathologiques, il convient de procéder à un second examen de toutes les régions du système nerveux qui présentent ces altérations. Dans tous les groupes de dosage, il convient de réaliser des coupes de toutes les régions potentiellement affectées, qui seront ensuite codées, puis examinées à l'aveugle dans un ordre aléatoire. La fréquence et la gravité de chaque lésion sont consignées. Quand toutes les régions de tous les groupes ont été cotées, le code est cassé et une analyse statistique est faite afin de déterminer la relation dose-réponse. Il faut donner la description des différents degrés de gravité de chaque lésion.
Les résultats de l'examen neuropathologique doivent être évalués à la lumière des observations et déterminations fonctionnelles, ainsi que des résultats des études de toxicité systémique réalisées sur la substance.
2. RÉSULTATS
2.1. TRAITEMENT DES RÉSULTATS
Il faut présenter les résultats relatifs à chaque individu. En outre, toutes les données doivent être résumées sous forme de tableaux montrant pour chaque groupe d'essai ou groupe témoin le nombre d'animaux au début de l'essai, le nombre d'animaux trouvés morts ou euthanasiés au cours de l'essai, le moment de chaque décès ou sacrifice, le nombre d'animaux manifestant des signes de toxicité, une description de ces signes, y compris le moment de leur apparition, leur durée, leur type et leur gravité, le nombre d'animaux présentant des lésions, le type de ces dernières et leur gravité.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Les résultats de l'étude doivent être évalués sur les plans de la fréquence, de la gravité et de la corrélation des effets neuropathologiques et de comportement (le cas échéant, neurochimiques et électrophysiologiques si des examens supplémentaires ont été pratiqués) avec d'autres effets nocifs observés. Dans la mesure du possible, les résultats numériques devraient être évalués au moyen d'une méthode statistique adéquate et communément acceptée. Les méthodes statistiques doivent être choisies au moment de la conception de l'étude.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit comporter les informations ci-après:
|
|
Substance d'essai:
|
—
|
nature physique (y compris isomérisation, pureté et propriétés physico-chimiques),
|
|
—
|
données permettant l'identification.
|
|
|
|
Véhicule (le cas échéant):
|
—
|
justification du choix du véhicule.
|
|
|
|
Animaux d'expérience:
|
—
|
espèce/souche utilisée,
|
|
—
|
nombre, âge et sexe des animaux,
|
|
—
|
source, conditions d'encagement, acclimatation, alimentation, etc.,
|
|
—
|
source, conditions d'encagement, acclimatation, alimentation, etc.
|
|
|
|
Conditions de l'essai:
|
—
|
description détaillée de la formulation de la substance à tester et/ou de la préparation alimentaire, concentration atteinte, stabilité et homogénéité de la préparation,
|
|
—
|
précisions sur les doses administrées, le véhicule, le volume et la forme physique du mélange administré,
|
|
—
|
précisions sur le mode d'administration de la substance à tester,
|
|
—
|
justification du choix des niveaux de dose,
|
|
—
|
justification du choix de la voie et de! a durée d'exposition,
|
|
—
|
conversion de la concentration (en ppm) de la substance à tester dans la nourriture ou dans l'eau de boisson en dose (en mg/kg de poids corporel/jour), le cas échéant,
|
|
—
|
précisions sur la qualité des aliments et de l'eau.
|
|
|
|
Procédures d'observation et d'essais:
|
—
|
précisions sur l'affectation des animaux de chaque groupe aux sous-groupes de perfusion,
|
|
—
|
précisions sur les systèmes de cotation, les critères et les échelles utilisés pour chaque détermination lors des observations cliniques détaillées,
|
|
—
|
précisions sur les tests fonctionnels d'exploration de la réactivité sensorielle à divers stimuli (par exemple stimuli auditifs, visuels et proprioceptifs); précisions sur l'évaluation de la force de préhension; précisions sur l'évaluation de l'activité motrice (y compris détails sur les dispositifs automatiques de détection de l'activité) et sur les autres procédures,
|
|
—
|
précisions sur les examens ophtalmologiques et, le cas échéant, hématologiques et sur les tests de biochimie clinique, avec indication des valeurs de référence,
|
|
—
|
précisions sur les procédures spécifiques de recherche des modifications du comportement et des altérations neuropathologiques, neurochimiques et électrophysiologiques.
|
|
|
|
Résultats:
|
—
|
poids corporel/variations du poids corporel et poids au moment du sacrifice,
|
|
—
|
consommation d'aliments et d'eau, le cas échéant,
|
|
—
|
informations sur les réactions de toxicité, par sexe et par niveau de dose, y compris les symptômes de toxicité ou mortalité,
|
|
—
|
nature, gravité et durée (moment du début et évolution observée) des observations cliniques détaillées (préciser si les effets sont réversibles ou non),
|
|
—
|
description détaillée des résultats de tous les tests fonctionnels,
|
|
—
|
résultats de l'autopsie,
|
|
—
|
description détaillée de toutes les observations concernant le comportement, les modifications neuropathologiques, neurochimiques et électrophysiologiques, le cas échéant,
|
|
—
|
données concernant l'absorption et le métabolisme, si disponibles,
|
|
—
|
traitement statistique des résultats, le cas échéant.
|
|
|
|
Discussion des résultats:
|
—
|
informations concernant la relation dose-réponse,
|
|
—
|
rapport entre tout autre effet toxique et une conclusion quant au potentiel neurotoxique de la substance,
|
|
—
|
concentration sans effet nocif observé.
|
|
|
|
Conclusions:
|
—
|
une appréciation sur la neurotoxicité globale de la substance est demandée.
|
|
4. BIBLIOGRAPHIE
|
(1)
|
OECD Guidance Document on Neurotoxicity Testing Strategies and Test Methods. OECD, Paris In Preparation.
|
|
(2)
|
Test Guidline for a Developmental Neurotoxicitu Study, OECD Guidelines for the Testing of Chemicals. In preparation.
|
|
(3)
|
World Health Organization (WHO) (1986). Environmental Health Criteria Document 60: Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals.
|
|
(4)
|
Spencer, P.S. and Schaumburg, H.H. (1980). Spencer, P.S. and Schaumburg, H.H. eds., Williams and Wilkins Co., Baltimore/London, p. 726-742.
|
|
(5)
|
Tupper D.E. and Wallace, R.B. (1980). Utility of the Neurologic Examination in Rats. Acta Neurobiol. Exp., 40, p. 999-1003.
|
|
(6)
|
Gad, S.C. (1982). A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol. Environ. Health, 9, p. 691-704.
|
|
(7)
|
Moser, V.C., McDaniel, K.M. and Phillips, P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of amitraz. Toxic. Appl. Pharmacol., 108, p. 267-283.
|
|
(8)
|
Meyer, O.A., Tilson, H.A., Byrd, W.C and Riley, M.T. (1979). A Method for the Routine Assessment of Fore- and Hind- limb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, p. 233-236.
|
|
(9)
|
Crofton, K.M., Haward, J.L, Moser, V.C., Gill, M.W., Reirer, L.W., Tilson, H.A. and MacPhail, R.C. (1991) Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, p. 599-609.
|
|
(10)
|
Tilson, H.A., and Mitchell, C.L. eds. (1992). Neurotoxicology Target Organ Toxicology Series. Raven Press, New York.
|
|
(11)
|
Chang, L.W., ed. (1995). Principles of Neurotoxicology. Marcel Dekker, New York.
|
|
(12)
|
Broxup, B. (1991). Neuopathology as a screen for Neurotoxicity Assessment. J. Amer. Coll. Toxicol., 10, p. 689-695.
|
|
(13)
|
Moser, V.C., Anthony, D.C., Sette, W.F. and MacPhail, R.C. (1992). Comparison of Subchronic Neurotoxicity of 2-Hydroxyethyl Acrylate and Acrylamide in Rats. Fund. Appl.Toxicol., 18, p. 343-352.
|
|
(14)
|
O'Callaghan, J.P. (1988). Neurotypic and Gliotypic Proteins as Biochemical Markers of Neurotoxicity. Eurotoxicol. Teratol., 10, p. 445-452.
|
|
(15)
|
O'Callaghan, J.P. and Miller, D.B. (1988). Acute Exposure of the Neonatal Rat to Triethyltin Results in Persistent Changes in Neurotypic and Gliotypic Proteins. J. Pharmacol. Exp. Ther., 244, p. 368-378.
|
|
(16)
|
Fox, D.A., Lowndes, H.E. and Birkamper, G.G. (1982). Electrophysiological Techniques in Neurotoxicology. In: Nervous System Toxicology. Mitchell, C.L. ed. Raven Press, New York, p. 299-335.
|
|
(17)
|
Johnson, B.L. (1980). Electrophysiological Methods in Neurotoxicity Testing. In: Experimental and Clinical Neurotoxicology. Spencer, P.S. and Schaumburg, H.H. eds, Williams and Wilkins Co., Baltimore/London, p. 726-742.
|
|
(18)
|
Bancroft, J.D. and Steven A. (1990). Theory and Pratice of Histological Techniques. Chapter 17, Neuropathological Techniques. Lowe, James and Cox, Gordon eds. Churchill Livingstone.
|
Tableau 1
Nombres minimaux d'animaux requis par groupe selon que l'étude de neurotoxicité est conduite séparément ou en combinaison avec d'autres études
|
|
ÉTUDE DE NEUROTOXICITÉ CONDUITE COMME:
|
|
Étude séparée
|
Étude combinée avec une étude sur 28 jours
|
Étude combinée avec une étude sur 90 jours
|
Étude combinée avec une étude de toxicité chronique
|
|
Nombre total d'animaux par groupe
|
10 mâles et 10 femelles
|
10 mâles et 10 femelles
|
15 mâles et 15 femelles
|
25 mâles et 25 femelles
|
|
Nombre d'animaux sélectionnés pour les tests fonctionnels, y compris les observations cliniques détaillées
|
10 mâles et 10 femelles
|
10 mâles et 10 femelles
|
10 mâles et 10 femelles
|
10 mâles et 10 femelles
|
|
Nombre d'animaux sélectionnés pour la perfusion in situ et l'examen neurohistopathologique
|
5 mâles et 5 femelles
|
5 mâles et 5 femelles
|
5 mâles et 5 femelles
|
5 mâles et 5 femelles
|
|
Nombre d'animaux sélectionnés pour les essais de toxicité à dose répétée, de toxicité subchronique et chronique, les études hématologiques, de biochimie clinique, histopathologiques, etc., comme indiqué dans les lignes directrices correspondantes
|
|
5 mâles et 5 femelles
|
10 mâles (10) et 10 femelles (10)
|
20 mâles (10) et 20 femelles (10)
|
|
Observations complémentaires, le cas échéant
|
5 mâles et 5 femelles
|
|
|
|
Tableau 2
Fréquence des observations cliniques et périodicité des tests fonctionnels
|
Type d'observation
|
Durée de l'étude
|
|
Étude de toxicité aiguë
|
28 jours
|
90 jours
|
Étude de toxicité chronique
|
|
Sur tous les animaux
|
État de santé général
|
quotidiennement
|
quotidiennement
|
quotidiennement
|
quotidiennement
|
|
Mortalité/morbidité
|
deux fois par jour
|
deux fois par jour
|
deux fois par jour
|
deux fois par jour
|
|
Sur les animaux sélectionnés pour les observations fonctionnelles
|
Observations cliniques détaillées
|
|
—
|
avant la première exposition
|
|
—
|
dans les huit heures suivant l'administration, au moment où l'effet est censé être le plus prononcé
|
|
—
|
aux jours 7 et 14 après administration
|
|
|
—
|
avant la première exposition
|
|
—
|
une fois par semaine ensuite
|
|
|
—
|
avant la première exposition
|
|
—
|
une fois pendant la première ou la deuxième semaine d'exposition
|
|
—
|
une fois par mois ensuite
|
|
|
—
|
avant la première exposition
|
|
—
|
une fois à la fin du premier mois d'exposition
|
|
—
|
tous les trois mois ensuite
|
|
|
Tests fonctionnels
|
|
—
|
avant la première exposition
|
|
—
|
dans les huit heures suivant l'administration, au moment où l'effet est censé être le plus prononcé
|
|
—
|
aux jours 7 et 14 après administration
|
|
|
—
|
avant la première exposition
|
|
—
|
pendant la quatrième semaine du traitement le plus près possible de la fin de la période d'exposition
|
|
|
—
|
avant la première exposition
|
|
—
|
une fois pendant la première ou la deuxième semaine d'exposition
|
|
—
|
une fois par mois ensuite
|
|
|
—
|
avant la première exposition
|
|
—
|
une fois à la fin du premier mois d'exposition
|
|
—
|
tous les trois mois ensuite
|
|
B.44. ABSORPTION CUTANÉE: MÉTHODE IN VIVO
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 427 (2004) de l'OCDE.
1.1. INTRODUCTION
La voie d'exposition à de nombreux produits chimiques est essentiellement transcutanée, c'est-à-dire via la peau. Or, la majorité des études toxicologiques réalisées sur des animaux de laboratoire s'appuient sur une administration des produits par voie orale. L'étude d'absorption cutanée in vivo décrite dans la présente méthode offre donc les éléments nécessaires pour extrapoler les résultats des études par administration orale, lors des évaluations en matière de sécurité réalisées après exposition cutanée.
Avant de passer dans la circulation sanguine, une substance doit d'abord traverser un grand nombre de couches cellulaires de la peau. Pour la plupart des substances, c'est le stratum corneum, composé de cellules mortes, qui est la couche déterminante sur le plan cinétique. La perméabilité de la peau dépend à la fois de la lipophilicité de la substance et de l'épaisseur de la couche externe de l'épiderme, ainsi que d'autres facteurs tels que le poids moléculaire et la concentration de la substance. De manière générale, la peau des rats et des lapins est plus perméable que celle de l'homme, tandis que celle des cobayes, des porcs et des singes se rapproche de la peau humaine.
Les méthodes de mesure de l'absorption percutanée sont de deux types: in vivo et in vitro. La méthode in vivo fournit de bonnes informations sur l'absorption cutanée chez différentes espèces de laboratoire. Mises au point plus récemment, les méthodes in vitro se fondent sur le transport d'une substance à travers tout ou partie de l'épaisseur de la peau humaine ou animale jusqu'à un réservoir de fluide. La méthode in vitro est décrite dans une autre méthode d'essai (1). Pour choisir la méthode la mieux appropriée en fonction des situations données, il est recommandé de consulter le document d'orientation de l'OCDE pour la conduite des études d'absorption cutanée (2) qui examine en détails la pertinence respective des méthodes in vivo et in vitro.
La méthode in vivo décrite dans la présente méthode permet de déterminer que la substance d'essai a traversé la peau pour pénétrer dans le compartiment systémique. La technique est largement utilisée depuis de nombreuses années (3) (4) (5) (6) (7). Alors que les études in vitro d'absorption percutanée conviennent dans de nombreux cas, il existe certaines situations dans lesquelles seule une étude in vivo permet d'obtenir les données voulues.
Les avantages de la méthode in vivo sont les suivants: utilisation d'un système intact sur les plans physiologique et métabolique, utilisation d'une espèce commune à bon nombre d'études de toxicité, et possibilité de modification pour une utilisation sur d'autres espèces. En revanche, elle présente certains inconvénients: utilisation d'animaux vivants, utilisation de matière radiomarquée pour garantir la fiabilité des résultats, difficultés à déterminer la phase d'absorption précoce, et différences de perméabilité entre la peau humaine et celle des espèces les plus communément utilisées (rat). En effet, la peau animale est généralement plus perméable, ce qui peut conduire à une surestimation de l'absorption percutanée chez l'homme (6) (8) (9). Les substances caustiques et corrosives ne doivent pas être testées sur des animaux vivants.
1.2. DÉFINITIONS
Dose non absorbée: désigne la quantité de substance éliminée par le nettoyage de la surface de la peau après exposition, et toute quantité présente sur le pansement non occlusif, y compris toute quantité volatilisée au cours de l'exposition.
Dose absorbée (in vivo): désigne les quantités de substance présentes dans l'urine, les résidus ramassés dans la cage après nettoyage, les fèces, l'air expiré (s'il est mesuré), le sang, les tissus (s'ils sont collectés) et la carcasse, après récupération de la peau du site d'application.
Dose absorbable: désigne la quantité de substance présente sur ou dans la peau après nettoyage.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance d'essai, de préférence radiomarquée, est appliquée sur la peau rasée des animaux, à la dose ou aux différentes doses appropriée(s), sous la forme d'une préparation d'usage représentative. La préparation d'essai est maintenue au contact de la peau pendant une période donnée, sous un pansement adapté (non occlusif, semi-occlusif ou occlusif) de façon à prévenir l'ingestion de la préparation. Au terme de la période d'exposition, le pansement est retiré et la peau est nettoyée à l'aide d'un agent lavant approprié. Le pansement et le matériel utilisé pour appliquer l'agent lavant sont conservés pour analyse, et un nouveau pansement est appliqué. Avant, pendant et après la période d'exposition, les animaux sont hébergés dans des cages métaboliques individuelles, et les excrétions et l'air expiré au cours de ces périodes sont collectés pour analyse. Le cas échéant, si l'on dispose d'informations suffisantes confirmant l'absence ou la formation limitée de métabolites radioactifs volatils, la collecte de l'air expiré peut être omise. Normalement, chaque étude porte sur plusieurs groupes d'animaux exposés à la préparation d'essai. Un groupe est euthanasié à la fin de la période d'exposition, et les autres groupes sont euthanasiés par la suite à des intervalles déterminés (2). À la fin de la période d'échantillonnage, les animaux restants sont euthanasiés, le sang est collecté pour analyse, la zone d'application est prélevée pour analyse, et la carcasse est analysée pour rechercher toute matière non excrétée. Les échantillons sont analysés selon les méthodes appropriées et on estime le degré d'absorption percutanée (6) (8) (9).
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Sélection de l'espèce animale
Le rat est l'espèce la plus communément utilisée, mais des souches et espèces sans poils présentant un taux d'absorption cutanée plus proche de celui de l'homme peuvent aussi être utilisées (3) (6) (7) (8) (9). Des jeunes adultes sains du même sexe (mâles par défaut) d'une souche communément utilisée en laboratoire sont utilisés. Au début de l'étude, les animaux retenus ne doivent pas présenter un écart de poids de ± 20 % par rapport au poids moyen. Par exemple, des rats mâles de 200 à 250 grammes conviennent tout à fait, en particulier ceux présentant un poids dans la moitié supérieure de cette plage.
1.4.2. Nombre et sexe des animaux
Un groupe d'au moins quatre animaux du même sexe doit être utilisé pour chaque préparation d'essai et chaque durée programmée. Chacun des groupes est euthanasié à un intervalle de temps différent, par exemple à la fin de la période d'exposition (généralement de 6 à 24 heures) et à différentes échéances suivantes (48 et 72 heures par exemple). Dans le cas où des données disponibles démontrent une différence importante de sensibilité toxicologique dermique entre les mâles et les femelles, c'est le sexe le plus sensible qu'il convient d'utiliser. En l'absence de telles données, l'un ou l'autre des sexes peut être utilisé.
1.4.3. Conditions d'hébergement et d'alimentation
La température du local expérimental doit être de 22 oC (± 3 oC). Si l'humidité relative doit atteindre au moins 30 % sans excéder de préférence 70 %, en dehors des heures de nettoyage du local, on s'efforce de maintenir le taux d'humidité autour de 50 à 60 %. On applique un éclairage artificiel, alternant 12 heures de lumière et 12 heures d'obscurité. Les animaux sont nourris avec un mélange classique pour animaux de laboratoire disponible à volonté et boivent de l'eau potable à volonté. Pendant l'étude, et de préférence également pendant la phase d'acclimatation, les animaux sont placés dans des cages métaboliques individuelles. Les déversements d'eau ou de nourriture étant susceptibles de compromettre les résultats de l'étude, les risques de tels incidents doivent être minimisés.
1.4.4. Préparation des animaux
Les animaux sont marqués pour permettre une identification individuelle, et placés dans leurs cages cinq jours au moins avant le début de l'essai, pour qu'ils s'acclimatent aux conditions du laboratoire.
À l'issue de la période d'acclimatation et 24 heures environ avant l'application de la substance d'essai, la région dorsale des épaules de chaque animal est tondue à ras. La perméabilité d'une peau endommagée étant différente de celle d'une peau saine, un soin tout particulier doit être apporté pour ne pas «égratigner» la peau. Après la tonte et 24 heures environ avant l'application de la substance d'essai sur la peau (voir point 1.4.7), la surface de la peau est délicatement nettoyée à l'acétone pour retirer le sébum. Il n'est pas recommandé de procéder à un nettoyage supplémentaire à l'eau et au savon dans la mesure où les résidus de savon sont susceptibles de favoriser l'absorption de la substance d'essai. La surface tondue doit être suffisamment grande pour permettre un calcul fiable de la quantité de substance d'essai absorbée par centimètre carré de peau, soit de préférence au moins 10 centimètres carrés. Des rats de 200 à 250 grammes sont compatibles avec une telle surface. Après préparation, les animaux sont replacés dans leurs cages métaboliques.
1.4.5. Substance d'essai
La substance d'essai est le produit dont on étudie les caractéristiques de pénétration. Dans l'idéal, cette substance d'essai doit être radiomarquée.
1.4.6. Préparation de l'essai
La préparation de la substance d'essai (c'est-à-dire la préparation pure, diluée ou le mélange contenant la substance d'essai à appliquer sur la peau) doit être identique (ou représenter un substitut réaliste) aux produits auxquels peuvent être exposés l'homme ou les autres espèces cibles. Toute modification par rapport à la préparation «d'usage» doit être justifiée. Le cas échéant, la substance d'essai est dissoute ou mise en suspension dans un véhicule adapté. Lorsqu'un véhicule autre que l'eau est utilisé, ses caractéristiques d'absorption et son interaction potentielle avec la substance d'essai doivent être connues.
1.4.7. Application sur la peau
Une zone (site) d'application d'une superficie spécifique est définie sur la surface de la peau. Une quantité connue de la préparation d'essai est ensuite uniformément appliquée sur ce site. Normalement, cette quantité doit correspondre à une exposition humaine potentielle, soit généralement 1 à 5 mg/cm2 pour une substance solide ou jusqu'à 10 ml/cm2 pour les liquides. L'application de toute autre quantité doit être justifiée par les conditions d'utilisation attendues, les objectifs fixés pour l'étude, ou les caractéristiques physiques de la préparation d'essai. Après l'application, le site doit être protégé contre les frottements. La figure 1 présente un exemple de dispositif classiquement employé dans ce contexte. Généralement, on protège le site d'application par un pansement non occlusif (par exemple une compresse de gaze nylon perméable). Cela étant, pour des applications infinies, le site d'application doit être recouvert d'un pansement occlusif. Dans les cas où l'évaporation d'une substance d'essai semi-volatile réduit dans une trop grande mesure le taux de récupération de la substance d'essai (voir également le point 1.4.10, premier paragraphe), il convient de piéger la substance évaporée dans un filtre à charbon monté sur le dispositif d'application (voir la figure 1). Il est essentiel que les dispositifs d'application utilisés n'endommagent pas la peau, et qu'ils n'absorbent pas non plus la préparation d'essai ni n'entrent en réaction avec elle. Les animaux sont ensuite replacés dans leurs cages métaboliques individuelles pour la collecte de leurs excrétions.
1.4.8. Durée de l'exposition et échantillonnage
La durée de l'exposition correspond à l'intervalle de temps entre l'application de la préparation d'essai et son retrait par nettoyage de la peau. Une période d'exposition pertinente (généralement de 6 à 24 heures) doit être retenue, en fonction de la durée éventuelle de l'exposition humaine. À l'issue de la période d'exposition, les animaux sont maintenus dans leurs cages métaboliques jusqu'au terme prévu. Pendant toute la durée de l'étude, il convient d'observer les animaux à intervalles réguliers, pour détecter tout signe de toxicité ou toute réaction anormale. Après la période d'exposition, les éventuels signes visibles d'irritation doivent être observés sur la peau traitée.
Les cages métaboliques doivent permettre la collecte séparée des fèces et de l'urine pendant toute la durée de l'étude, ainsi que la collecte du dioxyde de carbone marqué au C14 et des composés volatils C14 qu'il convient d'analyser lorsqu'ils sont produits en quantité (> 5 %). L'urine, les fèces et autres fluides (par exemple, le dioxyde de carbone C14 et les composés volatils Cl4) doivent être collectés individuellement pour chaque groupe et à chaque étape d'échantillonnage. Sur la base d'informations suffisantes confirmant l'absence ou la formation limitée de métabolites radioactifs volatils, on peut le cas échéant utiliser des cages ouvertes.
Les excrétions sont collectées pendant la période d'exposition, soit 24 heures maximum après la première application sur la peau, puis ensuite chaque jour jusqu'à la fin de l'expérience. Si trois intervalles de collecte des excrétions suffisent normalement, des points de collecte supplémentaires ou plus appropriés peuvent être définis pour une étude, selon l'objectif envisagé pour la préparation d'essai ou en fonction de données cinétiques existantes.
À la fin de la période d'exposition, le dispositif de protection est retiré de chaque animal et conservé individuellement aux fins d'analyse. La peau traitée de tous les animaux doit être nettoyée au moins trois fois à l'aide d'un agent et de tampons nettoyants appropriés. Un soin particulier doit être apporté de façon à ne pas contaminer d'autres zones du corps des animaux. L'agent lavant doit être représentatif des pratiques d'hygiène normales (par exemple une solution d'eau savonneuse). Enfin, la peau doit être séchée. Tous les tampons et matériel de nettoyage utilisés doivent être conservés pour analyse. Un nouveau pansement doit être appliqué sur le site traité des animaux formant les derniers groupes, avant leur retour dans les cages métaboliques individuelles.
1.4.9. Procédures terminales
Pour chaque groupe, les animaux doivent être euthanasiés à l'échéance préétablie et leur sang collecté pour analyse. Le pansement ou dispositif de protection doit être retiré pour analyse. La peau du site d'application et une surface de peau équivalente mais provenant d'une zone rasée non traitée doivent être prélevées sur chaque animal, et des analyses distinctes doivent être conduites. Le cas échéant, le site d'application peut être fractionné pour séparer le stratum corneum de l'épiderme sous-jacent, de façon à fournir des informations supplémentaires sur l'évacuation et le comportement de la substance chimique d'essai. En effet, déterminer le comportement/élimination du produit au cours d'un laps de temps donné après la période d'exposition donne une indication sur le devenir de n'importe quelle substance chimique dans le stratum corneum. Pour faciliter le fractionnement de la peau (après le dernier nettoyage et l'euthanasie des animaux), il faut d'abord retirer tous les dispositifs de protection. Ensuite, on prélève la peau du site d'application sur le rat, plus une bande circulaire autour du site, puis on épingle le tout sur une planche. On applique alors, par quelques pressions légères, une bande de ruban adhésif à la surface de la peau. Cette bande est ensuite retirée, avec une partie du stratum corneum. On renouvelle l'opération jusqu'à ce que la bande n'adhère plus à la surface de la peau, lorsque tout le stratum corneum a été retiré. Pour chaque animal, on peut combiner toutes les bandes d'adhésif dans un seul récipient, dans lequel on ajoute un digestif pour solubiliser le stratum corneum. Le cas échéant, on récupère tout éventuel tissu cible pour procéder à des mesures séparées, avant analyse de la carcasse résiduelle et mesure de la dose absorbée par celle-ci. Les carcasses des animaux doivent être conservées pour analyse. En règle générale, l'analyse du contenu total de la carcasse est suffisante, mais certains organes peuvent être retirés pour des analyses séparées (par exemple si cela est indiqué par d'autres études). L'urine présente dans la vessie au moment de l'euthanasie doit être ajoutée à celle collectée auparavant. Après collecte des excrétions dans les cages métaboliques au moment de l'euthanasie, les cages et leurs trappes doivent être nettoyées avec un solvant approprié. Les autres équipements potentiellement contaminés doivent également être analysés.
1.4.10. Analyse
Dans toutes les études, un niveau de récupération approprié doit être atteint (à savoir une moyenne de 100 ± 10 % de la radioactivité). Les récupérations en dehors de cette plage doivent faire l'objet de justification. Le volume de la dose administrée dans chaque échantillon doit être analysé selon des procédures suffisamment validées.
L'analyse statistique doit inclure une mesure de la variance des essais répétés de chaque application.
2. RÉSULTATS
Les mesures suivantes de la substance d'essai et/ou des métabolites doivent être effectuées sur chaque animal et à chaque point d'échantillonnage. Tous les résultats doivent être présentés individuellement, mais aussi groupés par point d'échantillonnage sous forme de moyenne:
|
—
|
quantité associée aux dispositifs de protection,
|
|
—
|
quantité pouvant être récupérée de la peau,
|
|
—
|
quantité présente sur ou dans la peau et impossible à retirer par nettoyage,
|
|
—
|
quantité dans les échantillons de sang,
|
|
—
|
quantité dans les excrétions et l'air expiré (le cas échéant),
|
|
—
|
quantité restant dans la carcasse et tout organe éventuellement retiré pour analyse.
|
Les quantités de substance d'essai et/ou de métabolites trouvées dans les excrétions, l'air expiré, le sang et la carcasse permettent de déterminer les quantités totales absorbées à chaque point. À partir de ces données, on peut calculer la quantité de substance d'essai absorbée par centimètre carré de peau exposée à la substance au cours de la période d'exposition.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit préciser les conditions établies dans le protocole, justifier le choix du système d'essai utilisé, et également contenir les informations suivantes:
|
|
Substance d'essai:
|
—
|
données d'identification [par exemple, numéro CAS si disponible; source; pureté (pureté radiochimique); impuretés connues; numéro du lot],
|
|
—
|
état physique; propriétés physico-chimiques (par exemple, pH, volatilité, solubilité, stabilité, poids moléculaire et log Poe).
|
|
|
|
Préparation de l'essai:
|
—
|
formulation et justification de l'utilisation,
|
|
—
|
détails de la préparation d'essai, quantité appliquée, concentration atteinte, véhicule, stabilité et homogénéité.
|
|
|
|
Animaux d'essai:
|
—
|
espèce/souche utilisée,
|
|
—
|
nombre, âge et sexe des animaux utilisés,
|
|
—
|
source des animaux, conditions d'encagement, régime alimentaire, etc.,
|
|
—
|
poids de chaque animal au début de l'essai.
|
|
|
|
Conditions d'essai:
|
—
|
détails concernant l'administration de la préparation d'essai (site d'application, méthodes d'essai, occlusion/non-occlusion, volume, extraction, détection),
|
|
—
|
détails concernant la qualité de l'eau et la nourriture.
|
|
|
|
Résultats:
|
—
|
tout signe de toxicité,
|
|
—
|
tableau indiquant les données d'absorption (sous forme de taux, de quantités ou de pourcentage),
|
|
—
|
récupérations totales de l'expérience,
|
|
—
|
interprétation des résultats, avec comparaison avec toutes les autres données éventuellement disponibles concernant l'absorption de la substance d'essai.
|
|
4. BIBLIOGRAPHIE
|
1.
|
Méthode d'essai B.45. Absorption cutanée: méthode in vitro.
|
|
2.
|
OCDE (2002). Guidance Document for the Conduct of Skin Absorption Studies. OCDE, Paris.
|
|
3.
|
ECETOC (1993). Percutaneous Absorption. Centre européen d'écotoxicologie et de toxicologie des produits chimiques, Monographie No 20.
|
|
4.
|
Zendzian, RP (1989). Skin Penetration Method suggested for Environmental Protection Agency Requirements. J. Am. Coll Toxicol. 8(5), p. 829-835.
|
|
5.
|
Kemppainen, BW, Reifenrath, WG (1990). Methods for skin absorption. CRC Press Boca Raton, FL, USA.
|
|
6.
|
EPA (1992). Dermal Exposure Assessment: Principles and Applications. Exposure Assessment Group, Office of Health and Environmental Assessment.
|
|
7.
|
EPA (1998). Health Effects Test Guidelines, OPPTS 870-7600, Dermal Penetration. Office of Prevention, Pedsticides and Toxic Substances.
|
|
8.
|
Bronaugh, RL, Wester, RC, Bucks, D, Maibach, HI and Sarason R (1990) In vivo percutaneous absorption of fragrance ingredients in reshus monkeys and humans. Fd. Chem. Toxic. 28, p. 369-373.
|
|
9.
|
Feldman, RJ and Maibach HI (1970) Absorption of some organic compounds through the skin in man. J. Invest Dermatol. 54. p. 399-404.
|
Figure 1
Exemple de dispositif classiquement utilisé pour délimiter et protéger un site d'application cutanée au cours d'études d'absorption percutanée in vivo

B.45. ABSORPTION CUTANÉE: MÉTHODE IN VITRO
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 428 (2004) de l'OCDE.
1.1. INTRODUCTION
Cette méthode a été élaborée pour fournir des informations sur l'absorption d'une substance d'essai appliquée sur un échantillon de peau excisée. Elle peut être combinée à la méthode d'absorption cutanée: méthode in vivo (1), ou être menée séparément. Il est recommandé de consulter le document d'orientation de l'OCDE pour la conduite des études d'absorption cutanée (2) pour élaborer des études reposant sur cette méthode. Ce document d'orientation a été préparé pour faciliter la sélection des procédures in vitro appropriées, utilisables selon les circonstances spécifiques, et dans le but de garantir la fiabilité des résultats obtenus avec cette méthode.
Les méthodes de mesure de l'absorption cutanée se divisent en deux grandes catégories: in vivo et in vitro. Les méthodes in vivo d'étude de l'absorption cutanée sont bien connues et fournissent des informations pharmacocinétiques sur un large éventail d'espèces animales. Une méthode in vivo est décrite dans une autre méthode d'essai (1). Les méthodes in vitro sont utilisées depuis de nombreuses années pour mesurer l'absorption cutanée. Même s'il n'y a pas eu d'étude de validation formelle des méthodes in vitro présentées dans cette méthode d'essai, les experts de l'OCDE ont estimé en 1999 que le volume des données évaluées suffisait pour étayer la méthode in vitro (3). Le document d'orientation de l'OCDE (2) présente des détails supplémentaires appuyant cette décision, dont un certain nombre de comparaisons directes des méthodes in vitro et in vivo. De même, de nombreuses monographies examinent cette question et offrent un contexte de référence détaillé sur l'utilisation de la méthode in vitro (4) (5) (6) (7) (8) (9) (10) (11) (12). Les méthodes in vitro mesurent la diffusion d'une substance dans et à travers la peau jusqu'à un réservoir de fluide; elles utilisent des échantillons de peau non viable pour mesurer la diffusion seule, ou des échantillons de peau fraîche et métaboliquement active pour mesurer simultanément la diffusion et le métabolisme cutané. Ces méthodes conviennent particulièrement pour comparer l'absorption cutanée et percutanée de différentes formulations d'une substance chimique, mais elles proposent aussi des modèles utiles pour l'évaluation de l'absorption percutanée chez l'homme.
La méthode in vitro ne convient pas nécessairement pour toutes les situations et classes de substances, mais on peut l'utiliser pour une première évaluation qualitative de la pénétration cutanée. Dans certains cas, il peut s'avérer nécessaire de compléter l'étude par des données in vivo. Pour des informations complémentaires sur les situations où la méthode in vitro conviendrait, il est recommandé de consulter le document d'orientation de l'OCDE (2). La référence (3) fournit d'autres précisions qui peuvent faciliter la prise de décision.
La présente méthode présente des principes généraux pour mesurer l'absorption cutanée et la diffusion d'une substance d'essai à partir d'un échantillon de peau excisée. Dans ce contexte, la peau de nombreuses espèces de mammifères, y compris l'homme, peut être utilisée. Les propriétés de perméabilité de la peau sont conservées après le prélèvement de l'échantillon de peau, dans la mesure où c'est le stratum corneum non viable qui constitue le principal obstacle à la diffusion; aucune forme de transport actif des substances chimiques à travers la peau n'a jamais été identifiée. En revanche, il a été établi que la peau pouvait métaboliser certaines substances au cours de l'absorption percutanée (6), mais ce processus ne limite pas la dose réellement absorbée, même si elle est susceptible d'affecter la nature du produit qui passe ensuite dans le sang.
1.2. DÉFINITIONS
Dose non absorbée: désigne la quantité retirée par nettoyage de la surface de la peau après exposition et toute quantité présente sur le timbre non occlusif, y compris toute quantité volatilisée au cours de l'exposition.
Dose absorbée (in vitro): masse de substance d'essai atteignant le fluide receveur (ou la circulation systémique) dans une période de temps donnée.
Dose absorbable (in vitro): désigne la quantité présente sur ou dans la peau après nettoyage.
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
La substance d'essai, le cas échéant radiomarquée, est appliquée à la surface d'un échantillon de peau séparant les deux chambres d'une cellule à diffusion. La substance chimique reste sur la peau pendant un temps donné et dans des conditions spécifiques, avant d'être retirée selon une procédure de nettoyage appropriée. Au cours de l'étude, on échantillonne le fluide receveur à des moments donnés, puis on l'analyse pour y rechercher la substance d'essai et/ou des métabolites.
Lorsque des systèmes métaboliquement actifs sont utilisés, les éventuels métabolites de la substance d'essai peuvent être analysés selon les méthodes appropriées. À la fin de l'expérience, la distribution de la substance d'essai est déterminée et ses métabolites sont quantifiés, le cas échéant.
Dans les conditions appropriées, décrites dans cette méthode et dans le document d'orientation (2), on mesure l'absorption d'une substance d'essai au cours d'une période de temps donnée en analysant le fluide receveur et la peau traitée. La substance d'essai restant sur la peau doit être considérée comme étant absorbée, à moins qu'il soit démontré que l'on puisse déterminer l'absorption sur la seule base des valeurs relatives au fluide receveur. L'analyse des autres éléments (matières récupérées par nettoyage de la peau et matières restant à l'intérieur des couches dermiques) permet d'enrichir l'évaluation des données, en précisant notamment l'évacuation totale de la substance d'essai et le pourcentage de récupération.
Pour démontrer les performances et la fiabilité du système d'essai dans le laboratoire chargé de l'étude, les résultats relatifs aux substances chimiques de référence pertinentes doivent être tenus disponibles et être conformes à la littérature publiée pour la méthode utilisée. Pour répondre à cette exigence, on peut procéder à un essai simultané de la substance d'essai et d'une substance de référence appropriée (présentant de préférence une lipophilicité proche de celle de la substance d'essai), ou proposer un historique approprié relatif à un certain nombre de substances de référence présentant une lipophilicité différente (par exemple, la caféine, l'acide benzoïque et la testostérone).
1.4. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.4.1. Cellule à diffusion
Une cellule à diffusion se compose d'un compartiment donneur et d'un compartiment receveur entre lesquels on place la peau (la figure 1 présente une cellule à diffusion de conception classique). La cellule doit répondre à plusieurs objectifs: assurer une bonne étanchéité sur le pourtour de la peau, permettre un échantillonnage aisé et un bon mélange de la solution dans le compartiment receveur en contact avec le dessous de la peau, et garantir le maintien de la température voulue au niveau de la cellule et de son contenu. Des cellules à diffusion statique ou dynamique peuvent indifféremment être utilisées. Normalement, les compartiments donneurs sont laissés sans occlusion pendant l'exposition à une dose finie d'une préparation d'essai. Toutefois, pour les applications infinies et pour certains scénarios avec des doses finies, l'occlusion du compartiment donneur peut être envisagée.
1.4.2. Fluide receveur
L'emploi d'un fluide receveur compatible avec des éléments physiologiques doit être privilégié, mais d'autres fluides peuvent être utilisés à condition que ce choix soit justifié. La composition précise du fluide receveur doit être fournie. La bonne solubilité de la substance d'essai dans le fluide receveur doit être démontrée, de façon à garantir qu'il ne fait pas obstacle à l'absorption. En outre, le fluide receveur ne doit pas affecter l'intégrité de la préparation cutanée. Dans un système dynamique, la vitesse de l'écoulement ne doit pas gêner la diffusion de la substance d'essai dans le fluide receveur. Dans une cellule statique, le fluide doit être remué en permanence et échantillonné régulièrement. Si le métabolisme entre également dans le cadre de l'étude, le fluide receveur doit garantir la viabilité de la peau pendant toute la durée de l'expérience.
1.4.3. Préparations de peau
On peut utiliser indifféremment de la peau d'origine humaine ou animale. Il est entendu que l'utilisation de la peau humaine est soumise à des conditions et considérations d'éthique admises d'ordre national et international. L'utilisation d'une peau viable est préférable, mais une peau non viable peut aussi être utilisée dès lors que son intégrité est démontrée. Des membranes d'épiderme (séparées par procédé enzymatique, chimique ou à la chaleur) ou certaines strates de peau (d'une épaisseur généralement comprise entre 200 et 400 μm) préparées avec un dermatome peuvent être utilisées. Des épaisseurs de peau complètes peuvent être utilisées, mais il convient d'éviter les épaisseurs excessives (environ > 1 mm), sauf si elles sont spécifiquement requises pour examiner la substance d'essai dans les couches de la peau. Les différents choix effectués (espèce retenue, site anatomique et technique de préparation) doivent être justifiés. Des données acceptables pour un minimum de quatre essais répétés par préparation d'essai doivent être fournies.
1.4.4. Intégrité de la préparation cutanée
Il est essentiel que la peau soit correctement préparée. Toute manipulation inappropriée peut endommager le stratum corneum; il convient donc de contrôler l'intégrité de la peau préparée. Si le champ de l'étude englobe le métabolisme de la peau, une peau fraîchement excisée doit être utilisée dans les plus courts délais, et dans des conditions propres à soutenir l'activité métabolique. De manière générale, la peau fraîchement excisée doit être utilisée dans les 24 heures, mais la période de stockage acceptable peut varier selon le système enzymatique impliqué dans la métabolisation et les températures de stockage (13). Lorsque les préparations de peau ont été stockées avant utilisation, des éléments établissant qu'elles ont conservé toutes leurs fonctions doivent être présentés.
1.4.5. Substance d'essai
La substance d'essai est le produit dont on étudie les caractéristiques de pénétration. Dans l'idéal, cette substance d'essai doit être radiomarquée.
1.4.6. Préparation d'essai
La préparation de la substance d'essai (c'est-à-dire la préparation pure, diluée ou le mélange contenant la substance d'essai à appliquer sur la peau) doit être identique (ou représenter un substitut réaliste) aux produits auxquels peuvent être exposés l'homme ou les autres espèces cibles. Toute modification par rapport à la préparation «d'usage» doit être justifiée.
1.4.7. Concentrations et formulations des substances d'essai
Normalement, plusieurs concentrations de la substance d'essai sont utilisées, de façon à couvrir le haut de la gamme des expositions humaines potentielles. De la même manière, on peut envisager d'étendre l'étude à un éventail de formulations classiques.
1.4.8. Application sur la peau
Dans les conditions normales de l'exposition humaine aux produits chimiques, des doses limites sont généralement rencontrées. Par conséquent, il convient d'utiliser des doses correspondant à l'exposition humaine potentielle, soit généralement 1 à 5 mg/cm2 pour une substance solide et jusqu'à 10 μl/cm2 pour les liquides. La quantité retenue doit être justifiée par les conditions d'utilisation attendues, les objectifs fixés pour l'étude, ou les caractéristiques physiques de la préparation d'essai. Par exemple, des applications infinies sur la surface de la peau peuvent être envisagées s'il s'agit d'étudier une exposition à des volumes importants par unité de surface.
1.4.9. Température
La température a une incidence sur la diffusion passive des produits chimiques (et ainsi sur leur absorption cutanée). Par conséquent, la cellule à diffusion et la peau doivent être maintenues à une température constante proche de la température normale de la peau, soit 32 +/- 1 oC. Selon la conception de la cellule, la température du bain ou du bloc de chauffage pourra varier, l'objectif étant de maintenir la peau et le compartiment receveur à leur norme physiologique. De préférence, l'humidité doit être maintenue entre 30 et 70 %.
1.4.10. Durée de l'exposition et échantillonnage
L'exposition de la peau à la préparation d'essai peut durer tout le temps de l'expérience, ou moins longtemps (par exemple pour imiter un type spécifique d'exposition humaine). La peau doit être lavée, à l'aide d'un agent lavant approprié, pour éliminer les excédents de la préparation d'essai, et les produits de rinçage utilisés doivent être collectés pour analyse. La procédure de retrait de la préparation, qui dépend des conditions d'utilisation envisagées, doit être justifiée. Normalement, une période d'échantillonnage de 24 heures est nécessaire pour permettre une caractérisation adéquate du profil d'absorption. Sachant que l'intégrité de la peau peut commencer à se détériorer au-delà de 24 heures, la durée d'échantillonnage ne doit normalement pas excéder 24 heures. Cette durée n'est pas nécessaire pour les substances d'essai qui pénètrent rapidement la peau, mais pour celles qui pénètrent lentement, des temps plus importants peuvent se révéler nécessaires. La fréquence d'échantillonnage du fluide receveur doit permettre d'établir une représentation graphique du profil d'absorption de la substance d'essai.
1.4.11. Procédures terminales
Tous les éléments du système d'essai (compartiment donneur, produits de rinçage de la surface de la peau, préparation cutanée et compartiment/fluide receveur) doivent être analysés et le niveau de récupération déterminé. Dans certains cas, il peut être nécessaire de fractionner la peau en zone de peau exposée et zone de peau sous le rebord de la cellule, et en couches correspondant au stratum corneum, à l'épiderme et aux différentes strates du derme, pour procéder à des analyses séparées.
1.4.12. Analyse
Dans toutes les études, un niveau de récupération approprié doit être atteint (à savoir une moyenne de 100 +/- 10 % de la radioactivité, tous les écarts devant être justifiés). La quantité de substance d'essai dans le fluide receveur, la préparation cutanée, les produits de rinçage de la surface de la peau et des appareils doivent être analysés selon des procédures appropriées.
2. RÉSULTATS
L'analyse du fluide receveur, la distribution de la substance d'essai dans le système d'essai et le profil d'absorption dans le temps doivent être présentés. Dans des conditions d'exposition à une dose finie, la quantité de substance éliminée lors du nettoyage de la peau, la quantité présente dans la peau (et dans les différentes couches dermiques si celles-ci ont fait l'objet d'une analyse) et la quantité présente dans le fluide receveur (taux et quantité ou pourcentage de la dose appliquée) doivent être calculées. Parfois, l'absorption cutanée peut être exprimée à partir des seules données relatives au fluide receveur. Toutefois, lorsque de la substance d'essai reste dans la peau à la fin de l'étude, il peut être nécessaire de l'inclure dans la quantité totale absorbée [voir paragraphe 66 de la référence (3)]. Dans des conditions d'exposition à une dose infinie, les données peuvent permettre de calculer une constante de perméabilité (Kp). Dans ce dernier cas, le pourcentage absorbé n'est pas pertinent.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit préciser les conditions établies dans le protocole, justifier le choix du système d'essai utilisé, et également contenir les informations suivantes:
|
|
Substance d'essai:
|
—
|
état physique; propriétés physico-chimiques (au minimum, poids moléculaire et log Poe), pureté (pureté radiochimique),
|
|
—
|
informations d'identification (par exemple numéro de lot),
|
|
—
|
solubilité dans le fluide receveur.
|
|
|
|
Préparation de la substance d'essai:
|
—
|
formulation et justification de l'utilisation,
|
|
|
|
Conditions d'essai:
|
—
|
sources et site de la peau, méthode de préparation, conditions de stockage avant utilisation, prétraitement éventuel (nettoyage, traitement antibiotique, etc.), mesures de l'intégrité de la peau, état métabolique, justification de l'utilisation,
|
|
—
|
conception de la cellule à diffusion, composition du fluide receveur, vitesse d'écoulement du fluide receveur ou temps et procédures d'échantillonnage,
|
|
—
|
détails de l'application de la préparation d'essai et quantification de la dose appliquée,
|
|
—
|
détails sur l'élimination de la préparation d'essai présente sur la peau (par exemple rinçage de la peau),
|
|
—
|
détails sur l'analyse de la peau et les techniques de fractionnement employées pour mettre en évidence la distribution dans la peau,
|
|
—
|
procédures de nettoyage de la cellule et du matériel,
|
|
—
|
méthodes d'essai, techniques d'extraction, seuils de détection et validation de la méthode analytique.
|
|
|
|
Résultats:
|
—
|
récupération totale de l'expérience (dose appliquée = produits de rinçage de la peau + peau + fluide receveur + produits de rinçage de la cellule),
|
|
—
|
tableau indiquant la récupération de chaque cellule dans chaque compartiment,
|
|
—
|
tableau indiquant les données relatives à l'absorption (sous forme de taux, de quantités ou de pourcentages).
|
|
4. BIBLIOGRAPHIE
|
1.
|
Méthode d'essai B.44. Absorption cutanée: méthode in vivo.
|
|
2.
|
OCDE (2002). Document d'orientation de l'OCDE pour la conduite des études d'absorption cutanée, OCDE, Paris.
|
|
3.
|
OCDE (2000). Report of the Meeting of the OECD Extended Steering Committee for Percutaneous Absorption Testing, Annex 1 to ENV/JM/TG(2000)5. OCDE, Paris.
|
|
4.
|
Kemppainen, BW and Reifenrath, WG. (1990). Methods for skin absorption. CRC Press, Boca Raton.
|
|
5.
|
Bronaugh, RL and Collier, SW. (1991). Protocol for In vitro Percutaneous Absorption Studies, in In vitro Percutaneous Absorption: Principles, Fundamentals and Applications, RL Bronaugh and HI Maibach, Eds., CRC Press, Boca Raton, p. 237-241.
|
|
6.
|
Bronaugh, RL and Maibach HI. (1991). In vitro Percutaneous Absorption: Principles, Fundamentals and Applications. CRC Press, Boca Raton.
|
|
7.
|
Centre européen d'écotoxicologie et de toxicologie des produits chimiques (1993). Monograph No 20, Percutaneous Absorption, ECETOC, Brussels.
|
|
8.
|
Diembeck, W, Beck, H, Benech-Kieffer, F, Courtellemont, P, Dupuis, J, Lovell, W, Paye, M, Spengler, J, Steiling, W (1999). Test Guidelines for In Vitro Assessment of Dermal Absorption and Percutaneous Penetration of Cosmetic Ingredients, Fd Chem Tox, 37, p. 191-205.
|
|
9.
|
Recommended Protocol for In vitro Percutaneous Absorption Rate Studies (1996). US Federal Register, Vol. 61, No 65.
|
|
10.
|
Howes, D, Guy, R, Hadgraft, J, Heylings, JR et al. (1996). Methods for assessing Percutaneous absorption. ECVAM Workshop Report ATLA 24, 81 R10.
|
|
11.
|
Schaefer, H and Redelmeier, TE. (1996). Skin barrier: principles of percutaneous absorption. Karger, Basel.
|
|
12.
|
Roberts, MS and Walters, KA. (1998). Dermal absorption and toxicity assessment. Marcel Dekker, New York.
|
|
13.
|
Jewell, C, Heylings, JR., Clowes, HM. and Williams, FM. (2000). Percutaneous absorption and metabolism of dinitrochlorobenzene in vitro. Arch Toxicol 74, p. 356-365.
|
Figure 1
Exemple de cellule à diffusion statique classiquement utilisée pour la conduite d'études d'absorption percutanée in vitro
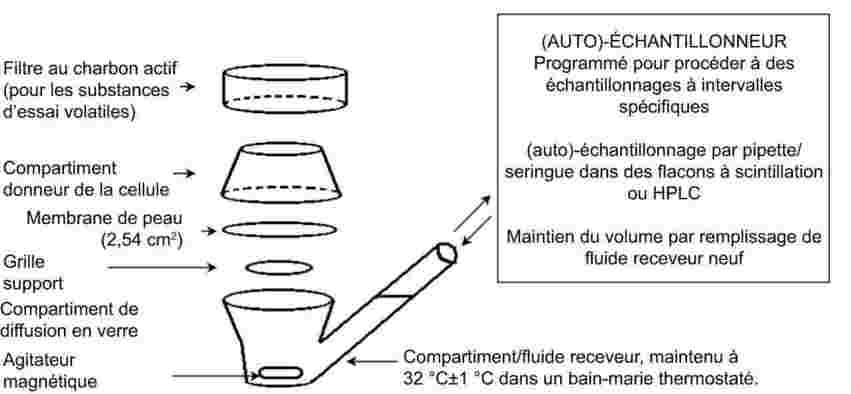
(1) L'étendue de l'opacité cornéenne doit être précisée.
(2) Pour un certain nombre de déterminations dans le sérum et le plasma, et principalement pour celle du glucose, il est préférable que les animaux soient à jeun depuis la veille. En l'absence de jeûne, la variabilité des résultats est en effet plus grande, et risque de masquer les effets les plus subtils et de rendre l'interprétation plus difficile. En revanche, le jeûne peut modifier le métabolisme général des animaux et, en particulier dans les études d'alimentation, perturber l'exposition quotidienne à la substance à tester. Si les prélèvements sont réalisés à jeun, les déterminations doivent être effectuées après les observations fonctionnelles de la quatrième semaine.
(3) Pour un certain nombre de paramètres mesurés dans le sérum ou le plasma, et particulièrement pour le glucose, il est préférable de faire jeûner les animaux durant la nuit qui précède la prise de sang, et ce essentiellement afin d'éviter l'accroissement de variabilité qui découlerait inévitablement de la prise de nourriture et qui aurait tendance à masquer des effets plus subtils, rendant de ce fait l'interprétation difficile. Cependant, le jeûne des animaux durant une nuit entière peut influer sur leur métabolisme général et risque, notamment dans les études où la substance d'essai est administrée dans la nourriture, de perturber l'exposition quotidienne à la substance d'essai. Si on a choisi de faire jeûner les animaux toute la nuit, les analyses de biochimie clinique doivent être réalisées après les observations fonctionnelles de l'étude.
(4) Connue actuellement sous le nom d'alanine transaminase serique.
(5) Connue actuellement sous le nom d'aspartate transaminase sérique.
(6) Ces organes prélevés sur 10 animaux par sexe et par groupe dans le cas des rongeurs et sur tous les non-rongeurs seront pesés de même que la thyroïde (et la parathyroïde) chez tous les non-rongeurs.
(7) Ces organes, prélevés sur 10 animaux par sexe et par lot dans le cas des rongeurs, seront pesés.
(8) Dans cette méthode, on entend par groupe de référence un groupe auquel la substance à tester est administrée par une autre voie garantissant une disponibilité complète de la dose.
(9) Solvant utilisé pour mesurer l'absorption.
(10)
|
†
|
Ce nombre comprend cinq animaux sélectionnés pour les tests fonctionnels et les observations cliniques détaillées faisant partie de l'étude de neurotoxicité.
|
PARTIE C: MÉTHODES DE DÉTERMINATION DE L'ÉCOTOXICITÉ
TABLE DES MATIÈRES
|
C.1.
|
TOXICITÉ AIGUË VIS-À-VIS DES POISSONS |
|
C.2.
|
DAPHNIA SP., ESSAI D'IMMOBILISATION IMMÉDIATE |
|
C.3.
|
ESSAI D'INHIBITION DES ALGUES |
|
C.4.
|
DÉTERMINATION DE LA BIODÉGRADABILITÉ «FACILE» |
|
PARTIE II.
|
ESSAI DE DISPARITION DU COD (Méthode C.4-A) |
|
PARTIE III.
|
ESSAI DE SCREENING MODIFIÉ DE L'OCDE (Méthode C.4-B) |
|
PARTIE IV.
|
ESSAI DE DÉGAGEMENT DE CO2 (Méthode C.4-C) |
|
PARTIE V.
|
ESSAI DE RESPIROMÉTRIE MANOMÉTRIQUE (Méthode C.4-D) |
|
PARTIE VI.
|
ESSAI EN FIOLES FERMÉES (Méthode C.4-E) |
|
PARTIE VII.
|
ESSAI MITI (Méthode C.4-F) |
|
C.5
|
DÉGRADATION — DEMANDE BIOCHIMIQUE EN OXYGÈNE |
|
C.6.
|
DÉGRADATION — DEMANDE CHIMIQUE EN OXYGÈNE |
|
C.7.
|
DÉGRADATION — DÉGRADATION ABIOTIQUE: HYDROLYSE EN FONCTION DU PH |
|
C. 8.
|
TOXICITÉ POUR LES VERS DE TERRE |
|
C. 9.
|
BIODÉGRADATION — ESSAI DE ZAHN ET WELLENS |
|
C. 10.
|
BIODÉGRADATION — ESSAIS DE SIMULATION DE BOUES ACTIVÉES |
|
C.11.
|
BIODÉGRADATION — BOUES ACTIVÉES: ESSAI D'INHIBITION DE LA RESPIRATION |
|
C. 12.
|
BIODÉGRADATION — TEST S.C.A.S. MODIFIÉ |
|
C.13
|
BIOCONCENTRATION: ESSAI AVEC RENOUVELLEMENT CONTINU SUR LES POISSONS |
|
C. 14.
|
POISSON, ESSAI SUR LA CROISSANCE DES JUVÉNILES |
|
C. 15.
|
POISSON, ESSAI DE TOXICITÉ À COURT TERME AUX STADES DE L'EMBRYON ET DE L'ALEVIN |
|
C.16.
|
ABEILLE DOMESTIQUE — ESSAI DE TOXICITÉ AIGUË PAR VOIE ORALE |
|
C.17.
|
ABEILLE DOMESTIQUE — ESSAI DE TOXICITÉ AIGUË PAR CONTACT |
|
C. 18.
|
DÉTERMINATION DE L'ADSORPTION/DÉSORPTION AU MOYEN DE LA MÉTHODE PAR AGITATION |
|
C. 19.
|
ESTIMATION DU COEFFICIENT D'ADSORPTION (KOC) SUR LE SOL ET LES BOUES D'ÉPURATION PAR CHROMATOGRAPHIE LIQUIDE HAUTE PERFORMANCE (HPLC) |
|
C.20
|
DAPHNIA MAGNA, ESSAI DE REPRODUCTION |
|
C.21.
|
MICRO-ORGANISMES DU SOL: ESSAI DE TRANSFORMATION DE L'AZOTE |
|
C.22.
|
MICRO-ORGANISMES DU SOL: ESSAI DE TRANSFORMATION DU CARBONE |
|
C.23.
|
TRANSFORMATION AÉROBIE ET ANAÉROBIE DANS LE SOL |
|
C.24.
|
TRANSFORMATION AÉROBIE ET ANAÉROBIE DANS LES SYSTÈMES SÉDIMENTAIRES AQUATIQUES |
C.1. TOXICITÉ AIGUË VIS-À-VIS DES POISSONS
1. MÉTHODE
1.1. INTRODUCTION
Cet essai a pour objet de déterminer la toxicité aiguë létale d'une substance vis-à-vis des poissons en eau douce. Dans la mesure du possible, il est souhaitable de disposer d'informations sur l'hydrosolubilité, la pression de vapeur, la stabilité chimique, les constantes de dissociation et la biodégradabilité de la substance d'essai en vue du choix de la méthode d'essai la plus appropriée (statique, semi-statique ou dynamique), permettant d'assurer des concentrations constantes satisfaisantes de la substance d'essai pendant la période expérimentale.
Des informations supplémentaires (par exemple, la formule développée, le degré de pureté, la nature et le pourcentage des impuretés significatives, la présence et la quantité d'additifs, le coefficient de partage n-octanol/eau) doivent être prises en considération lors de la conception de l'essai et de l'interprétation des résultats.
1.2. DÉFINITIONS ET UNITÉS
La toxicité aiguë est l'effet préjudiciable et observable provoqué dans un organisme pendant une courte durée (jours) d'exposition à une substance donnée. Dans le présent essai, la toxicité aiguë est exprimée comme la concentration létale médiane (CL50), c'est-à-dire la concentration qui, dans l'eau, est responsable de la mort de 50 % des poissons d'un lot soumis aux essais pendant une période d'exposition continue qui est à indiquer.
Toutes les concentrations de la substance d'essai sont indiquées en poids par volume (mg/l). Elles peuvent également être exprimées en poids par poids (mg/kg-1).
1.3. SUBSTANCES DE RÉFÉRENCE
On pourra soumettre à essai une substance de référence afin de démontrer que, dans les conditions d'essai en laboratoire, la réponse de l'espèce utilisée pour l'essai n'a pas varié de façon significative.
Aucune substance de référence n'est indiquée pour cet essai.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Un essai limite peut être effectué à 100 mg/l afin de démontrer que la CL50 est supérieure à cette concentration.
Les poissons sont exposés à la substance d'essai ajoutée à l'eau, dans une série de concentrations pendant une période de 96 heures. Les mortalités sont notées au moins toutes les 24 heures et les concentrations responsables de la mort de 50 % des poissons (CL50) sont calculées si possible à chaque moment d'observation.
1.5. CRITÈRES DE QUALITÉ
Les critères de qualité doivent s'appliquer à l'essai limite aussi bien qu'à l'essai complet.
La mortalité des témoins ne doit pas dépasser 10 % (ou un poisson, si le nombre d'animaux utilisés pour l'essai est inférieur à 10) à la fin de l'essai.
La concentration en oxygène doit être supérieure à 60 % de la concentration saturante pendant toute la durée de l'essai.
La concentration de la substance d'essai sera maintenue au moins à 80 % de la concentration initiale pendant toute la durée de l'essai.
Pour les substances qui se dissolvent facilement dans le milieu d'essai et qui donnent des solutions stables, c'est-à-dire les substances qui ne se volatilisent pas, ne se dégradent pas, ne s'hydrolysent pas ou ne s'adsorbent pas en quantité significative, on peut considérer que la concentration initiale est la même que la concentration nominale. Il faudra prouver que la concentration a été maintenue constante pendant la durée de l'essai et que les critères de qualité ont été respectés.
Pour les substances qui sont:
|
i)
|
peu solubles dans le milieu d'essai, ou
|
|
ii)
|
susceptibles de former des émulsions ou des dispersions stables, ou
|
|
iii)
|
instables en solution aqueuse,
|
la concentration initiale retenue sera la concentration mesurée dans la solution (ou si cela s'avère techniquement impossible, mesurée dans la colonne d'eau) au début de l'essai. La concentration doit être mesurée après une période d'équilibrage mais avant l'introduction des organismes d'essai.
Dans tous les cas cités ci-dessus, il y a lieu d'effectuer des mesures supplémentaires au cours de l'essai pour confirmer les concentrations réelles d'exposition et que les critères de qualité ont été respectés.
Le pH ne doit pas varier de plus d'une unité.
1.6. DESCRIPTION DE LA MÉTHODE
Trois types de systèmes peuvent être utilisés:
Essai en statique:
Essai de toxicité au cours duquel n'intervient pas de renouvellement de la solution à étudier (les solutions restent inchangées pendant toute la durée de l'essai.)
Essai en semi-statique:
Essai sans renouvellement continu de la solution, mais avec un renouvellement régulier de la solution d'essai après des périodes prolongées (par exemple toutes les 24 heures).
Essai en dynamique:
Essai de toxicité au cours duquel l'eau est constamment renouvelée dans les récipients d'essai, le produit chimique soumis à essai étant transporté par l'eau utilisée pour renouveler le milieu d'essai.
1.6.1. Réactifs
1.6.1.1. Solutions de substance à tester
Les solutions mères de concentration requise sont préparées par dissolution de la substance dans de l'eau désionisée, ou dans de l'eau répondant aux conditions fixées au point 1.6.1.2.
Les concentrations choisies pour l'essai sont préparées par dilution de la solution mère. Si l'on procède à des essais à concentrations élevées, la substance peut être dissoute directement dans l'eau de dilution.
Les substances ne doivent normalement être testées que jusqu'à la limite de solubilité. Pour certaines substances (par exemple, les substances qui sont peu hydrosolubles, ou qui ont un Pow élevé, ou qui forment dans l'eau une dispersion stable plutôt qu'une véritable solution), il est possible d'utiliser une concentration supérieure à la limite de solubilité de la substance pour être sûr que la concentration soluble/stable maximale a bien été atteinte. Il est toutefois important que cette concentration ne perturbe pas, par ailleurs, les conditions de l'essai (par exemple, formation d'un film de substance à la surface de l'eau empêchant l'oxygénation de l'eau, etc.).
Dans le cas de substances à faible hydrosolubilité, on peut avoir recours à la dispersion ultrasonique, à des solvants organiques, des émulsifiants ou des dispersants, pour préparer les solutions mères ou également pour faciliter la dispersion de ces substances dans le milieu d'essai. Quand des substances auxiliaires de ce type sont utilisées, toutes les concentrations d'essai doivent contenir la même quantité de substance auxiliaire, et un lot témoin supplémentaire de poissons doit être exposé à la même concentration de substance auxiliaire que celle qui est utilisée dans les séries d'essais. La concentration de tels auxiliaires doit être limitée et ne doit en aucun cas excéder 100 mg/l dans le milieu d'essai.
L'essai doit être effectué sans ajustement du pH. S'il apparaît un changement significatif du pH, il est souhaitable de répéter l'essai avec ajustement du pH et d'en consigner les résultats. Dans ce cas, la valeur du pH de la solution mère doit être ajustée à la valeur du pH de l'eau de dilution à moins que des raisons particulières ne s'y opposent. Pour ce faire, on utilisera de préférence HCl et NaOH. Cet ajustement doit être effectué de manière que la concentration de la substance d'essai dans la solution mère ne soit pas sensiblement modifiée. Si l'ajustement entraîne une réaction chimique ou une précipitation de la substance d'essai, il faut consigner ces observations dans le procès-verbal.
1.6.1.2. Eau d'élevage et de dilution
On peut utiliser de l'eau potable (non contaminée par des concentrations potentiellement nocives de chlore, de métaux lourds ou d'autres substances), de l'eau naturelle de bonne qualité ou de l'eau reconstituée (voir annexe 1). On utilisera de préférence des eaux dont la dureté totale est comprise entre 10 et 250 milligrammes par litre (en CaCO3), et dont le pH est compris entre 6,0 et 8,5.
1.6.2. Appareillage
Tout l'appareillage doit être en matériau chimiquement inerte:
|
—
|
système de dilution automatique (pour essai en dynamique),
|
|
—
|
dispositif de mesure de l'oxygène,
|
|
—
|
équipement pour la détermination de la dureté de l'eau,
|
|
—
|
appareillage approprié pour le contrôle de la température,
|
1.6.3. Poissons soumis à l'essai
Les poissons doivent être en bonne santé et ne présenter aucune malformation apparente.
Les espèces utilisées doivent être choisies sur la base de critères pratiques, tels que leur disponibilité toute l'année, leur facilité d'entretien, leur commodité pour l'essai, leur sensibilité relative aux produits chimiques et tous facteurs significatifs sur le plan économique, biologique ou écologique. Dans le choix des espèces de poissons, il faut également prendre en considération la nécessité de pouvoir comparer les résultats et l'harmonisation internationale existante (référence 1).
Une liste des espèces de poissons recommandées pour la réalisation de cet essai figure à l'annexe 2; le poisson zèbre et la truite arc-en-ciel sont les espèces à utiliser de préférence.
1.6.3.1. Élevage
Les poissons soumis à l'essai doivent provenir de préférence d'un seul et même lot, dont les individus ont la même longueur et le même âge. Ils doivent être conservés pendant au moins 12 jours dans les conditions suivantes:
charge biologique:
appropriée au système utilisé (avec recirculation ou en dynamique) et aux espèces de poissons,
eau:
voir point 1.6.1.2,
lumière:
photopériode de 12 à 16 heures par jour,
concentration en oxygène dissous:
au moins 80 % de la saturation en air,
alimentation:
quotidienne ou trois fois par semaine, avec arrêt de 24 heures avant le début de l'essai.
1.6.3.2. Mortalité
Après une période d'adaptation de 48 heures, enregistrer les morts et appliquer les critères suivants:
|
—
|
mortalité en 7 jours supérieure à 10 % de la population:
rejet de l'ensemble du lot,
|
|
—
|
mortalité en 7 jours comprise entre 5 et 10 % de la population:
prolonger la période d'observation pendant 7 jours.
Si l'on ne constate aucun autre cas de mortalité, le lot est acceptable, sinon, il doit être rejeté,
|
|
—
|
mortalité en 7 jours inférieure à 5 % de la population:
acceptation du lot.
|
1.6.4. Adaptation
Les poissons doivent être maintenus pendant au moins 7 jours avant leur utilisation dans les mêmes conditions que celles de l'essai (eau et température).
1.6.5. Mode opératoire
Avant l'essai définitif, on pourra procéder à un essai visant à déterminer l'intervalle de concentrations à utiliser lors de l'essai définitif.
Procéder, en plus de la série d'essais, à un essai témoin sans substance à étudier et, le cas échéant, à un essai témoin contenant le produit auxiliaire.
En fonction des propriétés physiques et chimiques de la substance à étudier, on choisira une méthode appropriée statique, semi-statique ou dynamique, qui réponde aux critères de qualité.
Les poissons sont exposés à la substance d'essai dans les conditions suivantes:
|
—
|
nombre d'animaux: au moins 7 par concentration,
|
|
—
|
récipients: d'une capacité appropriée, en fonction de la charge biologique recommandée,
|
|
—
|
charge biologique: charge maximale recommandée pour l'essai statique ou semi-statique: 1,0 gramme par litre; pour les essais dynamiques, une charge plus élevée peut être acceptable,
|
|
—
|
concentrations d'essai: au moins cinq concentrations qui diffèrent d'un facteur constant n'excédant pas 2,2 et couvrent, dans la mesure du possible, l'intervalle de mortalité de 0 à 100 %,
|
|
—
|
eau: voir point 1.6.1.2,
|
|
—
|
lumière: photopériode de 12 à 16 heures par jour,
|
|
—
|
température: convenant à l'espèce choisie (annexe 2), mais constante à ± 1 oC près, quel que soit le système d'essai,
|
|
—
|
concentration en oxygène dissous: au moins 60 % de la concentration saturante en air à la température choisie,
|
Les poissons sont examinés après les 2 à 4 premières heures et au moins à intervalles de 24 heures. Ils sont considérés comme morts si le fait de toucher le pédoncule caudal ne produit aucune réaction et si aucun mouvement respiratoire n'est visible. Les poissons morts sont éliminés à chaque observation et les mortalités enregistrées. Les anomalies visibles (par exemple, perte d'équilibre, perturbations au niveau de la nage, des fonctions respiratoires, de la pigmentation, etc.) seront consignées.
Les mesures du pH, de l'oxygène dissous et de la température doivent être effectuées quotidiennement.
Essai limite
Un essai limite peut être effectué à une concentration de 100 mg/l, en suivant les procédures décrites dans cette méthode d'essai, afin de montrer que la CL50 est supérieure à cette concentration.
Si la nature de la substance est telle qu'il est impossible d'obtenir une concentration de 100 mg/l dans l'eau d'essai, l'essai limite doit être effectué à une concentration égale à la solubilité de la substance (ou à la concentration maximale formant une dispersion stable) dans le milieu utilisé (voir également le point 1.6.1.1).
L'essai limite doit être effectué avec 7 à 10 poissons, et avec un nombre identique dans le(s) lot(s) témoin(s). (Selon la théorie binomiale, lorsque l'on utilise 10 poissons avec une mortalité nulle, il y a 99,9 % de chance que la CL50 soit supérieure à la concentration utilisée dans l'essai limite. Avec 7, 8 ou 9 poissons et une mortalité nulle, il y a 99 % de chance que la CL50 soit supérieure à la concentration utilisée.)
En cas de mortalité, il convient d'effectuer un essai complet. Si des effets sublétaux sont observés, ceux-ci doivent être consignés.
2. ÉVALUATION DES DONNÉES
Reporter, sur un papier log-probit, le pourcentage de mortalité pour chaque période d'exposition recommandée (24, 48, 72 et 96 heures) en fonction de la concentration.
Pour chaque temps d'observation, estimer, lorsque cela est possible, la CL50 et l'intervalle de confiance (p = 0,05) par les méthodes classiques; les valeurs obtenues doivent être arrondies à un ou deux (maximum) chiffres significatifs (exemples de nombres arrondis à deux chiffres: 170 pour 173,5; 0,13 pour 0,127; 1,2 pour 1,21).
Dans le cas où la pente de la courbe de pourcentage en fonction de la concentration est trop forte pour permettre de calculer la CL50, il suffit de procéder à une estimation graphique de cette valeur.
Lorsque deux concentrations consécutives dans un rapport de 2,2 donnent 0 et 100 % de mortalité, ces deux valeurs suffisent à indiquer l'intervalle dans lequel se situe la CL50.
Si l'on constate que la stabilité ou l'homogénéité de la substance d'essai ne peut pas être maintenue, cette observation doit être mentionnée et les résultats seront interprétés avec prudence.
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
des informations sur les poissons soumis à essai (nom scientifique, souche, fournisseur, tout prétraitement, taille et nombre utilisé pour chaque concentration d'essai),
|
|
—
|
origine et principales caractéristiques de l'eau de dilution (pH, dureté, température),
|
|
—
|
dans le cas de substances faiblement hydrosolubles, méthode de préparation de la solution mère et de la solution d'essai,
|
|
—
|
concentration de tout produit auxiliaire,
|
|
—
|
liste des concentrations utilisées et toute information disponible sur la stabilité de la substance d'essai dans la solution d'essai à ces concentrations,
|
|
—
|
si on procède à des analyses chimiques, méthodes utilisées et résultats,
|
|
—
|
éventuellement, résultats de l'essai limite,
|
|
—
|
motifs du choix et description détaillée de la méthode utilisée (par exemple système statique, semi-statique, taux d'administration, taux de renouvellement, aération ou non, charge en poisson, etc.),
|
|
—
|
description du matériel d'essai,
|
|
—
|
concentration en oxygène dissous, pH et température des solutions d'essai toutes les 24 heures,
|
|
—
|
preuves que les critères de qualité ont été respectés,
|
|
—
|
tableau contenant la mortalité cumulée pour chaque concentration et pour le témoin (et, au besoin, le témoin contenant la substance auxiliaire) à chaque temps d'observation recommandé,
|
|
—
|
représentation graphique du pourcentage de mortalité en fonction de la concentration à la fin de l'essai,
|
|
—
|
si possible, valeurs de la CL50 pour chacun des temps d'observation recommandés (avec l'intervalle de confiance à 95 %),
|
|
—
|
méthodes statistiques employées pour déterminer les valeurs de la CL50,
|
|
—
|
si une substance de référence est utilisée, les résultats obtenus,
|
|
—
|
la concentration d'essai la plus élevée qui ne cause pas de mortalité pendant la durée de l'essai,
|
|
—
|
la concentration d'essai la plus basse qui cause une mortalité de 100 % des poissons pendant la durée de l'essai.
|
4. RÉFÉRENCES
|
(1)
|
OCDE, Paris, 1981, ligne directrice no 203, décision du Conseil C(81) 30 final et mises à jours.
|
|
(2)
|
AFNOR — NFT 90-303, juin 1985 — Détermination de la toxicité aiguë d'une substance vis-à-vis du Brachydanio rerio — Méthode statique et dynamique.
|
|
(3)
|
AFNOR — NFT 90-305, juin 1985 — Détermination de la toxicité aiguë d'une substance vis-à-vis du Salmo gairdneri — Méthode statique et dynamique.
|
|
(4)
|
ISO 7346/1/2 and/3 — Water Quality — Determination of the acute lethal toxicity of substances to a fresh water fish (Brachydanio rerio Hamilton-Buchanan — Teleostei, Cyprinidae). Part 1: Static method. Part 2: Semi-static method. Part 3: Flow-through method.
|
|
(5)
|
Eidgenôssisches Department des Innern, Schweiz: Richtlinien fur Probenahme und Normung von Wasseruntersuchungsmethoden — Part II 1974.
|
|
(6)
|
DIN Testverfahren mit Wasserorganismen, 38 412 (L1) und L(15).
|
|
(7)
|
JIS K 0102, Acute toxicity test for fish.
|
|
(8)
|
NEN 6506 — Water — Bepaling van de akute toxiciteit met behulp van Poecilia reticulata, 1980.
|
|
(9)
|
Environmental Protection Agency, Methods for the acute toxicity tests with fish, macroinvertebrates and amphibians. The Committee on Methods for Toxicity Tests with Aquatic Organisms, Ecological Research Series EPA-660-75-009, 1975.
|
|
(10)
|
Environmental Protection Agency, Environmental monitoring and support laboratory, Office of Research and Development, EPA-600/4-78-012, January 1978.
|
|
(11)
|
Environmental Protection Agency, Toxic Substance Control, Part IV, 16 March 1979.
|
|
(12)
|
Standard methods for the examination of water and wastewater, 14th edition, APHA-AWWA-WPCF, 1975.
|
|
(13)
|
Commission of the European Communities, Inter-Laboratory test programme concerning the study of the ecotoxiciry of a chemical substance with respect to the fish. EEC Study D.8368, 22 March 1979.
|
|
(14)
|
Verfahrensvorschlag des Umweltbundesamtes zum akuten Fisch-Test. Rudolph, P. und Boje, R. Ökotoxikologie, Grundlagen für die Ökotoxikologische Bewertung von Umweltchemikalien nach dem Chemikaliengesetz, ecomed 1986.
|
|
(15)
|
Litchfield, J.T. and Wilcoxon, F., A simplified method for evaluating dose effects experiments, J. Pharm. and Exp. Therap., 1949, vol 96, p. 99.
|
|
(16)
|
Finney, D.J. Statistical Methods in Biological Assay. Griffin, Weycombe, U.K, 1978.
|
|
(17)
|
Sprague, J.B. Measurement of pollutant toxicity to fish. I Bioassay methods for acute toxicity. Water Res., 1969, vol. 3, p. 793-821.
|
|
(18)
|
Sprague, J.B. Measurement of pollutant toxicity to fish. II Utilising and applying bioassay results. Water Res., 1970, vol. 4, p. 3-32.
|
|
(19)
|
Stephan, C.E. Methods for calculating an LC50. In Aquatic Toxicology and Hasard Evaluation (edited by F.I. Mayer and J.L. Hamelink). American Society for Testing and Materials. ASTM STP 634, 1977, p. 65-84.
|
|
(20)
|
Stephan, C.E., Busch, K.A., Smith, R., Burke, J. and Andrews, R.W.A computer program for calculating an LC50. US EPA.
|
Annexe 1
Eau reconstituée
Exemple d'eau de dilution appropriée
Tous les produits chimiques doivent être de qualité analytique.
L'eau doit être une eau distillée de bonne qualité, ou de l'eau désionisée d'une conductivité inférieure à 5 μScm-1.
L'appareillage pour la distillation de l'eau ne doit contenir aucun élément en cuivre.
Solutions mères
|
CaCl2. 2 H2O (chlorure de calcium dihydraté):
dissoudre dans de l'eau, compléter à un litre
|
11,76 g
|
|
MgSO4. 7 H2O (sulfate de magnésium heptahydraté):
dissoudre dans de l'eau, compléter à un litre
|
4,93 g
|
|
NaHCO3 (hydrogénocarbonate de sodium):
dissoudre dans de l'eau, compléter à un litre
|
2,59 g
|
|
KCI (chlorure de potassium):
dissoudre dans de l'eau, compléter à un litre
|
0,23 g
|
Eau de dilution reconstituée
Mélanger 25 ml de chacune des quatre solutions mères et compléter à 1 litre avec de l'eau.
Aérer jusqu'à saturation en oxygène dissous 1qy.
Le pH doit être de 7,8±0,2.
Si nécessaire, ajuster le pH avec NaOH (hydroxyde de sodium) ou HC1 (acide chlorhydrique).
L'eau de dilution ainsi préparée est laissée au repos pendant environ 12 heures et ne doit pas être aérée ultérieurement.
La somme des ions Ca et Mg dans cette solution est égale à 2,5 mmol/l. Le rapport des ions Ca:Mg est de 4:1, celui des ions Na:K de 10:1. L'alcalinité totale de cette solution est égale à 0,8 mmol/l.
Les déviations éventuelles dans la préparation de l'eau de dilution ne doivent pas modifier la composition ou les propriétés de cette eau.
Annexe 2
Espèces de poissons recommandées pour l'essai
|
Espèces recommandées
|
Intervalles des températures d'essai recommandé (oC)
|
Longueur totale recommandée de l'animal soumis à l'essai (cm)
|
|
Brachydanio rerio (Teleostei, Cyprinidae) (Hamilton-Buchanan) Danio zébré
|
20 à 24
|
3,0±0,5
|
|
Pimephales promelas (Teleostei, Cyprinidae) (Rafinesque) Tête de boule
|
20 à 24
|
5,0±2,0
|
|
Cyprinus carpio (Teleostei, Cyprinidae} (Linnaeus 1758) Carpe commune
|
20 à 24
|
6,0±2,0
|
|
Oryzias latipes (Teleostei, Poeciliiae) Cyprinodonti-dae (Tomminck et Schlegel 1850) Nedaka
|
20 à 24
|
3,0±1,0
|
|
Poecilia reticulata (Teleostei, Poeciliidae) (Peters 1859) Guppy
|
20 à 24
|
3,0±1,0
|
|
Lepomis macrochirus (Teleostei, Centrarchidae) Rafinesque Linnaeus 1758) Crapet arlequin
|
20 à 24
|
5,0±2,0
|
|
Onchorhynchus mykiss (Teleostei, Salmonidae) (Walbaum 1988) Truite arc-en-ciel
|
12 à 17
|
6,0±2,0
|
|
Leuciscus idus (Teleostei, Cyprinidae) (Linnaeus 1758) Ide mélanote
|
20 à 24
|
6,0±2,0
|
Approvisionnement
Les poissons mentionnés ci-dessus sont faciles à élever et/ou largement disponibles pendant toute l'année. Ils peuvent se reproduire et se développer soit dans des exploitations piscicoles, soit en laboratoire, dans des conditions sanitaires contrôlées. L'animal soumis à essai doit être sain et d'origine connue. Ces poissons sont disponibles partout dans le monde.
Annexe 3
Exemple de courbe de pourcentage de mortalité en fonction de la concentration
Exemple de détermination de la CL50 sur papier log-probit

C.2. DAPHNIA SP., ESSAI D'IMMOBILISATION IMMÉDIATE
1. MÉTHODE
Cette méthode d'essai est équivalente à la ligne directrice 202 (2004) de l'OCDE.
1.1. INTRODUCTION
Cette méthode décrit un essai de toxicité aiguë visant à évaluer les effets des substances chimiques sur des daphnies. Les méthodes d'essai existantes ont été utilisées dans toute la mesure du possible (1) (2) (3).
1.2. DÉFINITIONS
Les définitions suivantes sont utilisées aux fins de la présente méthode:
|
|
CE50: estimation de la concentration capable d'immobiliser 50 % des daphnies après une période d'exposition définie. Si une autre définition est utilisée, elle doit être indiquée avec sa référence.
|
|
|
Immobilisation: les animaux incapables de nager au bout de 15 secondes après agitation douce du récipient utilisé pour l'essai sont considérés comme immobiles (même s'ils sont toujours capables de bouger leurs antennes).
|
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
De jeunes daphnies, âgées de moins de 24 heures au début de l'essai, sont exposées à la substance d'essai à différentes concentrations pendant une période de 48 heures. L'immobilisation est enregistrée à 24 et à 48 heures, puis comparée à des valeurs de contrôle. Les résultats sont analysés pour calculer la CE50 à 48 heures (voir point 1.2 pour les définitions). La détermination de la CE50 à 24 heures est facultative.
1.4. INFORMATION CONCERNANT LA SUBSTANCE D'ESSAI
La solubilité dans l'eau et la pression de vapeur de la substance d'essai doivent être connues. De même, il faut disposer d'une méthode d'analyse fiable pour doser la substance dans les solutions d'essai dont le rendement de récupération et la limite de détermination sont connus. Les informations utiles comprennent la formule structurale, la pureté de la substance, la stabilité dans l'eau ou à la lumière, le Poe et les résultats d'un essai de biodégradabilité immédiate (voir méthode C.4).
Remarque: Un document d'orientation (4) fournit des indications pour les substances qu'il est difficile de tester à cause de leurs propriétés physico-chimiques.
1.5. SUBSTANCES DE RÉFÉRENCE
Il est possible de déterminer la CE50 d'une substance de référence, de manière à s'assurer que les conditions d'essai sont fiables. Les toxiques utilisés dans les essais tournants internationaux (1) (5) sont recommandés à cet effet (1). Les essais avec une substance de référence doivent être effectués de préférence tous les mois et au moins deux fois par an.
1.6. CRITÈRES DE QUALITÉ
Pour qu'un essai soit valide, les critères de performance suivants s'appliquent:
|
—
|
pas plus de 10 % des daphnies ne doivent être immobilisées dans le groupe témoin, y compris le témoin contenant l'agent solubilisant,
|
|
—
|
la concentration d'oxygène dissous à la fin de l'essai doit être ≥ 3 mg/l dans les récipients d'essai et les récipients témoins.
|
Remarque: Pour le premier critère, pas plus de 10 % des daphnies du témoin ne doivent être immobilisées ou montrer d'autres signes de défaillance ou de stress, tels qu'une décoloration ou un comportement inhabituel (piégées à la surface de l'eau, par exemple).
1.7. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.7.1. Appareillage
Les récipients et autres équipements qui sont amenés à entrer en contact avec les solutions d'essai doivent être intégralement en verre, ou en un autre matériau chimiquement inerte. Les récipients d'essai sont le plus souvent des éprouvettes ou des béchers en verre. Avant chaque usage, ils doivent être nettoyés selon les procédures de laboratoire standard. Afin de réduire la déperdition d'eau par évaporation et de prévenir l'entrée de poussière dans les solutions, les récipients d'essai doivent être couverts. Les essais sur les substances volatiles doivent être réalisés dans des récipients complètement remplis et fermés, suffisamment grands pour éviter que la concentration d'oxygène ne soit trop faible ou ne devienne un facteur limitant (voir point 1.6 et premier paragraphe du point 1.8.3).
Par ailleurs, tout ou partie des équipements suivants est nécessaire: un appareil pour mesurer la concentration de l'oxygène (à l'aide d'une microélectrode ou d'un autre dispositif destiné à mesurer l'oxygène dissous dans des échantillons de faible volume); un pH-mètre; un thermostat adéquat; un appareil pour déterminer la concentration de carbone organique totale (COT); un appareil pour déterminer la demande chimique en oxygène (DCO); un appareil pour déterminer la dureté de l'eau, etc.
1.7.2. Organisme d'essai
L'espèce à utiliser de préférence dans cet essai est Daphnia magna Straus, mais d'autres espèces de Daphnia peuvent aussi être utilisées (Daphnia pulex, par exemple). Au début de l'essai, les animaux doivent être âgés de 24 heures au maximum, et, pour réduire la variabilité, il est fortement recommandé qu'ils ne proviennent pas d'une première génération de descendants. Ils doivent être issus d'un lot sain (c'est-à-dire qui ne présente pas de signes de stress, tels qu'une mortalité élevée, la présence de mâles et d'éphippies, un retard dans la production des premiers descendants, des animaux décolorés, etc.). Tous les organismes utilisés dans un essai donné doivent provenir de cultures établies à partir d'un même lot de daphnies. Le lot d'animaux doit être maintenu dans des conditions de culture (lumière, température, milieu) semblables à celles qui seront appliquées au cours de l'essai. Si le milieu utilisé dans l'essai est différent de celui où sont normalement élevées les daphnies, il convient de leur laisser une période d'acclimatation avant l'essai. En l'occurrence, il convient de les maintenir dans de l'eau de dilution à la température de l'essai pendant au moins 48 heures avant le début de l'essai.
1.7.3. Eau d'acclimatation et de dilution
On peut utiliser, comme eau d'acclimatation et de dilution, de l'eau naturelle (eau de surface ou eau souterraine), de l'eau reconstituée ou de l'eau du robinet déchlorée, du moment que les daphnies y survivent sans montrer de signes de stress pendant toute la durée de l'élevage, de l'acclimatation et de l'essai. Toute eau conforme aux caractéristiques chimiques d'une eau de dilution acceptable données dans la liste de l'annexe 1 convient pour cet essai. Sa qualité doit rester constante tout au long de l'essai. Les eaux reconstituées peuvent être composées d'eau distillée ou désionisée auxquelles on ajoute certaines quantités spécifiques de réactifs de qualité pour analyse. Des exemples d'eau reconstituée sont donnés dans les documents (1) (6) et dans l'annexe 2. Il convient de noter que les milieux contenant des agents chélatants connus, tels que les milieux M4 et M7 présentés dans l'annexe 2, doivent être évités pour les essais de substances contenant des métaux. Le pH doit être compris entre 6 et 9. Une dureté comprise entre 140 et 250 mg/l (en CaCO3) est recommandée pour les Daphnia magna, mais des duretés inférieures conviennent également pour d'autres espèces de Daphnia. L'eau de dilution peut être aérée avant utilisation dans l'essai, de façon que la concentration de l'oxygène dissous atteigne la saturation.
Lorsque de l'eau naturelle est utilisée, les paramètres de sa qualité doivent être mesurés au moins deux fois par an, ou chaque fois que ses caractéristiques sont susceptibles d'avoir été significativement modifiées (voir point précédent et annexe 1). Les métaux lourds (Cu, Pb, Zn, Hg, Cd, Ni, par exemple) doivent être mesurés. Si de l'eau du robinet déchlorée est utilisée, il est souhaitable de procéder chaque jour à une analyse de sa teneur en chlore. Si l'eau de dilution provient d'une eau souterraine ou d'une eau de surface, sa conductivité et son carbone organique total (COT) ou sa demande chimique en oxygène (DCO) doivent être mesurés.
1.7.4. Solutions d'essai
Les solutions d'essai sont généralement amenées à la concentration voulue par dilution d'une solution mère. De préférence, les solutions mères doivent être préparées par dissolution de la substance d'essai dans l'eau de dilution. Le recours à des solvants, émulsifiants ou dispersants doit être évité dans toute la mesure du possible. Néanmoins, ces additifs sont parfois nécessaires pour obtenir une solution mère à la concentration voulue. Le document guide (4) donne des informations sur le choix des solvants, émulsifiants et dispersants appropriés. En tout cas, la substance d'essai dans la solution d'essai ne doit pas excéder sa limite de solubilité dans l'eau de dilution.
L'essai doit être mené sans ajustement du pH. Si le pH ne reste pas dans la plage comprise entre 6 et 9, alors il convient de procéder à un deuxième essai, en ajustant le pH de la solution mère à celui de l'eau de dilution avant d'ajouter la substance d'essai. Lors de l'ajustement du pH, il faut que la concentration de la solution mère ne soit quasiment pas modifiée, ni qu'aucune réaction chimique, ni précipitation de la substance d'essai ne soit provoquée. De préférence, il convient d'employer du HCl et du NaOH.
1.8. PROCÉDURE
1.8.1. Conditions d'exposition
1.8.1.1. Témoins et groupes d'essai
On remplit les récipients d'essai avec les volumes voulus d'eau de dilution et de solutions de la substance d'essai. Le rapport air/volume d'eau à l'intérieur des récipients doit être identique pour les groupes témoins et les groupes d'essai. On place ensuite les daphnies dans les récipients. Il faut utiliser, pour chaque concentration d'essai et pour les témoins, au moins 20 animaux, répartis de préférence en quatre groupes de cinq animaux. Un volume d'au moins 2 ml de solution d'essai doit être prévu pour chaque animal (soit un volume de 10 ml pour cinq daphnies par récipient). Si la concentration de la substance d'essai n'est pas stable, un système de renouvellement semi-statique ou continu peut être utilisé pour l'essai.
Parallèlement aux séries traitées, il faut tester une série de témoins de l'eau de dilution et, le cas échéant, une série de témoins contenant l'agent solubilisant (solvant) en quantité utilisée dans les séries traitées.
1.8.1.2. Concentrations d'essai
À moins qu'une information ne soit déjà disponible sur la toxicité de la substance d'essai, un essai peut le cas échéant être mené pour déterminer la plage des concentrations de l'essai définitif. À cette fin, les daphnies sont exposées à une série de concentrations de la substance d'essai largement espacées. Cinq daphnies sont exposées à chaque concentration pendant 48 heures ou moins. Aucun essai identique n'est nécessaire. La période d'exposition peut être raccourcie (24 heures ou moins, par exemple) s'il est possible d'obtenir plus rapidement les données voulues.
Il faut utiliser au moins cinq concentrations formant une série géométrique de concentrations successives séparées, de préférence, par un facteur inférieur ou égal à 2,2. L'utilisation de moins de cinq concentrations doit être justifiée. De préférence, la concentration la plus élevée doit provoquer 100 % d'immobilisation, et la moins élevée ne doit donner lieu à aucun effet observable.
1.8.1.3. Conditions d'incubation
La température doit être comprise entre 18 et 22 oC, et, pour chaque essai, elle doit être constante à ± 1 oC. Une alternance de 16 heures de lumière et 8 heures d'obscurité est recommandée. Une obscurité complète est également acceptable, en particulier pour les substances d'essai instables à la lumière.
Les récipients d'essai ne doivent pas être aérés durant l'essai. L'essai est mené sans ajustement du pH. Les daphnies ne doivent pas être nourries pendant l'essai.
1.8.1.4. Durée
La durée de l'essai est de 48 heures.
1.8.2. Observations
À échéances de 24 et 48 heures après le début de l'essai, la présence de daphnies immobiles doit être contrôlée dans chaque récipient d'essai (voir définitions au point 1.2). Outre l'immobilité, tout comportement ou signe anormal doit être signalé.
1.8.3. Mesures analytiques
L'oxygène dissous et le pH sont mesurés au début et à la fin de l'essai dans les récipients témoins et dans ceux contenant la concentration de la substance d'essai la plus élevée. La concentration de l'oxygène dissous dans les témoins doit être conforme au critère de validité (voir le point 1.6). Normalement, le pH ne doit pas varier de plus de 1,5 unité au cours d'un essai. En règle générale, on mesure la température dans les récipients témoins ou la température ambiante, et de préférence il y a lieu de l'enregistrer en continu au cours de l'essai ou, au minimum, au début et à la fin de l'essai.
La concentration de la substance d'essai doit être mesurée, au minimum, dans les récipients contenant la concentration la plus élevée et la concentration la plus basse, au début et à la fin de l'essai (4). Il est recommandé de fonder les résultats sur les concentrations mesurées. Toutefois, si l'on peut démontrer de façon suffisamment convaincante que la concentration de la substance d'essai a été maintenue tout au long de l'essai de manière satisfaisante dans un intervalle de ± 20 % de la concentration nominale ou de la concentration initiale mesurée, alors les résultats peuvent être fondés sur les valeurs nominales ou les valeurs initiales mesurées.
1.9. ESSAI LIMITE
Sur la base du mode opératoire décrit dans la présente méthode, un essai limite peut être conduit à 100 mg/l de substance d'essai ou jusqu'à sa limite de solubilité dans le milieu d'essai (selon la valeur la plus basse), de façon à démonter que la CE50 est supérieure à cette concentration. L'essai limite doit être mené sur 20 daphnies (de préférence réparties en quatre groupes de cinq), avec un nombre équivalent d'animaux dans les récipients témoins. Si des daphnies sont immobilisées, il y a lieu de mener une étude complète. Tout comportement anormal observé doit être enregistré.
2. RÉSULTATS
Les résultats doivent être synthétisés dans des tableaux mettant en évidence, pour chaque groupe d'essai et chaque témoin, le nombre de daphnies utilisées et l'immobilisation à chaque observation. En outre, une représentation graphique doit montrer les pourcentages immobilisés à 24 et 48 heures par concentration d'essai. Les résultats sont analysés au moyen des méthodes statistiques appropriées (analyse probit, par exemple) permettant de calculer les pentes des courbes et la CE50 avec des limites de confiance de 95 % (p = 0,05) (7) (8).
Lorsque les données obtenues ne se prêtent pas au calcul de la CE50 par les méthodes standard, on doit utiliser la concentration la plus élevée qui ne donne aucune immobilisation et la concentration la plus faible qui donne 100 % d'immobilisation pour en déduire une valeur approximative de la CE50 (en prenant la moyenne géométrique de ces deux concentrations).
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit mentionner les informations suivantes:
Substance d'essai:
|
—
|
état physique et propriétés physico-chimiques pertinentes,
|
|
—
|
données permettant l'identification chimique, y compris la pureté.
|
Espèce d'essai:
|
—
|
source et espèce de Daphnia, fournisseur (s'il est connu) et conditions de culture appliquées (notamment source, type et quantités de nourriture, fréquence de l'alimentation).
|
Conditions d'essai:
|
—
|
description des récipients d'essai: type et volume des récipients, volume de la solution, nombre de daphnies par récipient, nombre de récipients d'essai (essais identiques) par concentration,
|
|
—
|
méthodes de préparation des solutions mères et d'essai, et notamment utilisation éventuelle de tout solvant ou agent dispersant, et concentrations utilisées,
|
|
—
|
détails concernant l'eau de dilution: source et caractéristiques (pH, dureté, rapport Ca/Mg, rapport Na/K, alcalinité, conductivité, etc.); le cas échéant, composition de l'eau reconstituée,
|
|
—
|
conditions d'incubation: température, intensité et périodicité de la lumière, oxygène dissous, pH, etc.
|
Résultats:
|
—
|
nombre et pourcentage de daphnies immobilisées ou ayant manifesté un effet adverse quelconque (y compris un comportement anormal) parmi les témoins et les groupes traités à chaque période d'observation, et une description de la nature des effets constatés,
|
|
—
|
résultats et date de l'essai effectué avec une substance de référence, le cas échéant,
|
|
—
|
concentrations d'essai nominales et résultat de toutes les analyses visant à déterminer la concentration de la substance d'essai dans les récipients d'essai; l'efficacité de la méthode de récupération et la limite de détermination doivent également être précisées,
|
|
—
|
toutes les mesures physico-chimiques (température, pH et oxygène dissous) effectuées au cours de l'essai,
|
|
—
|
CE50 à 48 heures (immobilisation) avec les intervalles de confiance et les graphiques du modèle utilisé pour le calcul, les pentes des courbes dose-effet et leur erreur standard; procédures statistiques utilisées pour déterminer la CE50 (ces informations doivent aussi être précisées pour l'immobilisation à 24 heures lorsque les mesures ont été effectuées),
|
|
—
|
explication de tout écart par rapport à la méthode d'essai, et conséquences éventuelles sur les résultats de l'essai.
|
4. BIBLIOGRAPHIE
|
(21)
|
ISO 6341 (1996). Qualité de l'eau — Détermination de l'inhibition de la mobilité de Daphnia magna Straus (Cladocera, Crustacea) — Essai de toxicité aiguë. Troisième édition, 1996.
|
|
(22)
|
EPA OPPTS 850.1010 (1996). Ecological Effects Test Guidelines — Aquatic Invertebrate Acute Toxicity Test, Freshwater Daphnids.
|
|
(23)
|
Environnement Canada (1996). Méthode d'essai biologique. Essai de létalité aiguë sur Daphnia spp. EPS 1/RM/1 1. Environment Canada, Ottawa, Ontario, Canada.
|
|
(24)
|
Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publication. Series on Testing and Assessment. No 23. Paris 2000.
|
|
(25)
|
Commission des Communautés européennes. Étude D8369 (1979). Programme d'essais interlaboratoires concernant l'étude de l'écotoxicité d'une substance chimique vis-à-vis de la Daphnia.
|
|
(26)
|
OCDE, ligne directrice pour les essais de produits chimiques. Ligne directrice 211: Daphnia magna, essai de reproduction, adoptée en septembre 1998.
|
|
(27)
|
Stephan, C.E. (1977). Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F.I. Mayer and J.L. Hamelink). ASTM STP 634 — American Society for Testing and Materials, p. 65-84.
|
|
(28)
|
Finney, D.J. (1978). Statistical Methods in Biological Assay. 3rd ed. London. Griffin, Weycombe, UK.
|
Annexe 1
QUELQUES CARACTÉRISTIQUES CHIMIQUES D'UNE EAU DE DILUTION ACCEPTABLE
|
Substance
|
Concentration
|
|
Particules
|
<20 mg/l
|
|
Carbone organique total
|
< 2 mg/l
|
|
Ammoniac non ionisé
|
< 1 μg/l
|
|
Chlore résiduel
|
<10 μg/l
|
|
Total des pesticides organophosphorés
|
<50 ng/l
|
|
Total des pesticides organochlorés plus biphényles polychlorés
|
<50 ng/l
|
|
Chlore organique total
|
<25 ng/l
|
Annexe 2
EXEMPLES D'EAU D'ESSAI RECONSTITUÉE ACCEPTABLE
Eau d'essai ISO (1)
|
Solutions mères (substance unique)
|
Pour préparer l'eau reconstituée, ajouter les volumes suivants de solutions mères à 1 litre d'eau (2)
|
|
Substance
|
Quantité ajoutée à 1 litre d'eau (2)
|
|
Chlorure de calcium
CaCl2, 2H2O
|
11,76 g
|
25 ml
|
|
Sulfate de magnésium
MgSO4, 7H2O
|
4,93 g
|
25 ml
|
|
Bicarbonate de sodium
NaHCO3
|
2,59 g
|
25 ml
|
|
Chlorure de potassium
KCl
|
0,23 g
|
25 ml
|
Milieux Elendt M7 et M4
Acclimatation aux milieux Elendt M4 et M7
Certains laboratoires ont rencontré des difficultés pour transférer directement les Daphnia dans les milieux M4 et M7. Ils sont toutefois parvenus à un certain résultat en les acclimatant progressivement, c'est-à-dire en les transférant de leur milieu vers un milieu à 30 % d'Elendt, puis à 60 % d'Elendt et enfin à 100 % d'Elendt. Les périodes d'acclimatation peuvent prendre jusqu'à un mois.
Préparation
Éléments en traces
Différentes solutions mères (I), contenant chacune une seule substance en traces, sont tout d'abord préparées avec une eau d'une pureté adéquate, par exemple de l'eau désionisée, distillée ou obtenue par osmose inverse. À partir de ces différentes solutions mères (I), on prépare une deuxième solution mère unique (II) renfermant toutes les substances en traces (solution combinée), à savoir:
|
Solution(s) mère(s) I (substance unique)
|
Quantité ajoutée à l'eau (mg/l)
|
Concentration (par rapport au milieu M4)
|
Pour préparer la solution mère combinée II, ajouter la quantité suivante de solution mère I à l'eau (ml/l)
|
|
M4
|
M7
|
|
H3BO3
|
57 190
|
20 000 fois
|
1,0
|
0,25
|
|
MnCl2.4H2O
|
7 210
|
20 000 fois
|
1,0
|
0,25
|
|
LiC1
|
6 120
|
20 000 fois
|
1,0
|
0,25
|
|
RbC1
|
1 420
|
20 000 fois
|
1,0
|
0,25
|
|
SrCl2.6H2O
|
3 040
|
20 000 fois
|
1,0
|
0,25
|
|
NaBr
|
320
|
20 000 fois
|
1,0
|
0,25
|
|
Na2 MoO4.2H2O
|
1 230
|
20 000 fois
|
1,0
|
0,25
|
|
CuCl2.2H2O
|
335
|
20 000 fois
|
1,0
|
0,25
|
|
ZnCl2
|
260
|
20 000 fois
|
1,0
|
1,0
|
|
CoCl2.6H2O
|
200
|
20 000 fois
|
1,0
|
1,0
|
|
KI
|
65
|
20 000 fois
|
1,0
|
1,0
|
|
Na2 SeO3
|
43,8
|
20 000 fois
|
1,0
|
1,0
|
|
NH4VO3
|
11,5
|
20 000 fois
|
1,0
|
1,0
|
|
Na2 EDTA.2H2O
|
5 000
|
2 000 fois
|
_
|
_
|
|
FeSO4.7H2O
|
1 991
|
2 000 fois
|
_
|
_
|
|
Les solutions de Na2EDTA et de FeSO4 sont préparées séparément, puis mélangées et immédiatement autoclavées.
|
|
Cela donne:
|
|
2 1 solution Fe-EDT
|
|
1 000 fois
|
20,0
|
5,0
|
Milieux M4 et M7
Les milieux M4 et M7 sont préparés à partir de la solution mère II, de macronutriments et de vitamines, de la façon suivante:
|
|
Quantité ajoutée à l'eau (mg/l)
|
Concentration (par rapport au milieu M4)
|
Quantité de solution mère II ajoutée pour préparer le milieu (ml/l)
|
|
M4
|
M7
|
|
Solution mère II (combinaison de substances en traces)
|
|
20 fois
|
50
|
50
|
|
Solutions mères contenant les macronutriments (une substance par solution)
|
|
|
|
|
|
CaCl2.2H20
|
293 800
|
1 000 fois
|
1,0
|
1,0
|
|
MgSO4.7H2O
|
246 600
|
2 000 fois
|
0,5
|
0,5
|
|
KCl
|
58 000
|
10 000 fois
|
0,1
|
0,1
|
|
NaHCO3
|
64 800
|
1 000 fois
|
1,0
|
1,0
|
|
Na2 SiO3.9H2O
|
50 000
|
5 000 fois
|
0,2
|
0,2
|
|
NaNO3
|
2 740
|
10 000 fois
|
0,1
|
0,1
|
|
KH2PO4
|
1 430
|
10 000 fois
|
0,1
|
0,1
|
|
K2HPO4
|
1 840
|
10 000 fois
|
0,1
|
0,1
|
|
Solution mère de vitamines combinées
|
_
|
10 000 fois
|
0,1
|
0,1
|
|
La solution mère de vitamines combinées est préparée en additionnant 3 vitamines à 1 litre d'eau, comme indiqué ci-dessous:
|
|
Chlorhydrate de thiamine
|
750
|
10 000 fois
|
|
|
|
Cyanocobalamine (B12)
|
10
|
10 000 fois
|
|
|
|
Biotine
|
7,5
|
10 000 fois
|
|
|
La solution mère de vitamines combinées est congelée par petites parties aliquotes. Les vitamines sont ajoutées au milieu peu avant son utilisation.
|
NB:
|
:
|
Afin d'éviter que les sels ne se précipitent lorsqu'on prépare le milieu complet, il faut ajouter les parties aliquotes de solution mère à quelque 500 à 800 ml d'eau désionisée et amener le volume à 1 litre.
|
|
NB:
|
:
|
La première publication relative au milieu M4 se trouve dans Elendt, B. P. (1990). Selenium deficiency in crustacea; an ultrastructual approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, p. 25-33.
|
C.3. ESSAI D'INHIBITION DES ALGUES
1. MÉTHODE
1.1. INTRODUCTION
Cet essai a pour objectif de déterminer les effets d'une substance sur la croissance d'algues vertes unicellulaires. Des essais de relativement courte durée (72 heures) peuvent permettre de mesurer les effets sur plusieurs générations. Cette méthode peut être adaptée de manière à être applicable à plusieurs espèces d'algues unicellulaires; dans ce cas, une description de la méthode utilisée doit figurer dans le procès-verbal d'essai.
Cette méthode est extrêmement facile à appliquer aux substances hydrosolubles, qui, dans les conditions de l'essai, sont susceptibles de rester dans l'eau.
Elle peut être utilisée pour des substances n'interférant pas directement avec la mesure de la croissance des algues.
Il est souhaitable de disposer, si possible, d'informations sur l'hydrosolubilité, la pression de vapeur, la stabilité chimique, les constantes de dissociation et la biodégradabilité de la substance à étudier avant de procéder à l'essai.
D'autres informations (par exemple, la formule développée, le degré de pureté, la nature et le pourcentage d'impuretés significatives, la présence et la quantité d'additifs, le coefficient de partage n-octanol/eau) doivent être prises en considération lors de la conception de l'essai et de l'interprétation des résultats.
1.2. DÉFINITIONS ET UNITÉS
Densité cellulaire: nombre de cellules par millilitre.
Croissance: augmentation de la densité cellulaire pendant la durée de l'essai.
Taux de croissance: augmentation de la densité cellulaire par unité de temps.
CE50: dans cette méthode, la concentration de la substance à étudier qui provoque une réduction de 50 %, soit de la croissance (CE50b), soit du taux de croissance (CE50r) par rapport au témoin.
CSEO (concentration sans effet observé): dans cette méthode, la concentration d'essai la plus élevée ne provoquant pas d'inhibition de croissance significative par rapport au témoin.
Toutes les concentrations de la substance à étudier sont données en poids par volume (mg/l). Elles peuvent aussi être exprimées en poids par poids (mg/kg-1).
1.3. SUBSTANCES DE RÉFÉRENCE
On peut soumettre à essai une substance de référence afin de démontrer que, dans les conditions d'essai en laboratoire, la sensibilité de l'espèce utilisée pour l'essai n'est pas sensiblement modifiée.
Si une substance de référence est utilisée, les résultats doivent être consignés dans le procès-verbal. Le bichromate de potassium peut être utilisé comme substance de référence, mais sa coloration peut affecter la qualité et l'intensité de la lumière parvenant aux cellules ainsi que les éventuelles mesures spectrophotomètriques. Le bichromate de potassium a été utilisé pour un essai interlaboratoires effectué à l'échelle internationale [voir référence (3) et annexe 2].
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Un essai limite peut être effectué à 100 mg/l afin de démontrer que la CE50 est supérieure à cette concentration.
Des cultures d'une espèce choisie d'algues vertes en phase de croissance exponentielle sont exposées à différentes concentrations de la substance à étudier pendant plusieurs générations dans des conditions définies.
Les solutions d'essai sont incubées pendant une période de 72 heures, au cours de laquelle on mesure la densité cellulaire dans chaque solution au moins toutes les 24 heures. L'inhibition de la croissance est déterminée par rapport à une culture témoin.
1.5. CRITÈRES DE QUALITÉ
Les critères de qualité doivent s'appliquer à l'essai limite aussi bien qu'à l'essai complet.
La densité cellulaire doit avoir augmenté d'au moins un facteur 16 en trois jours dans les cultures témoins.
La concentration de la substance à étudier doit être maintenue à 80 % au moins de la concentration initiale pendant la période correspondant à la durée de l'essai.
Pour les substances qui se dissolvent facilement dans le milieu d'essai et qui produisent des solutions stables, c'est-à-dire qui ne se volatilisent pas, ne se dégradent pas, ne s'hydrolysent pas ou ne s'adsorbent pas d'une manière significative, on peut considérer que la concentration initiale est équivalente à la concentration nominale. Il faudra montrer que la concentration a été maintenue pendant la durée de l'essai et que les critères de qualité ont été respectés.
Pour les substances qui sont:
|
i)
|
peu solubles dans le milieu d'essai, ou
|
|
ii)
|
susceptibles de former des émulsions ou des dispersions stables, ou
|
|
iii)
|
instables en solution aqueuse,
|
la concentration initiale sera la concentration mesurée dans la solution au début de l'essai. Elle devra être déterminée après une période d'équilibrage.
Dans tous les cas cités ci-dessus, il y a lieu d'effectuer des mesures supplémentaires au cours de l'essai pour confirmer les concentrations réelles d'exposition et que les critères de qualité ont été respectés.
Il est admis que des quantités significatives de la substance à étudier peuvent être incorporées dans la biomasse d'algues pendant la durée de l'essai. C'est pourquoi il convient, pour montrer que les critères de qualité cités ci-dessus sont respectés, de prendre en compte la substance incorporée dans la biomasse d'algues ainsi que la substance en solution (ou, en cas d'impossibilité technique, mesurée dans la colonne d'eau). La mesure de la concentration de la substance dans la biomasse d'algues pouvant, cependant, poser des problèmes techniques importants, on peut montrer que les critères de qualité sont respectés en effectuant un essai avec la concentration de la substance à étudier la plus élevée, mais avec l'absence d'algues, et en mesurant les concentrations dans la solution (ou, en cas d'impossibilité technique, mesurée dans la colonne d'eau) au début et à la fin de la période de l'essai.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Réactifs
1.6.1.1. Solutions de substance d'essai
Des solutions mères de concentration requise sont préparées en dissolvant la substance dans de l'eau désionisée ou dans de l'eau répondant aux conditions fixées au point 1.6.1.2.
Les concentrations d'essai choisies sont préparées en ajoutant la quantité appropriée de solution mère à des pré-cultures d'algues (voir annexe 1).
Les substances ne doivent normalement pas être testées au-delà de leur limite de solubilité. Pour certaines substances (par exemple les substances faiblement hydrosolubles, ou dont le pow est élevé, ou encore pour celles qui forment une dispersion stable plutôt qu'une vraie solution dans l'eau), on peut effectuer un essai à une concentration supérieure à la limite de solubilité de la substance afin de s'assurer que la concentration soluble/stable maximale a été atteinte. Il est toutefois important que cette concentration ne modifie pas, par ailleurs, les conditions de l'essai (par exemple par formation d'un film de substance à la surface de l'eau empêchant l'oxygénation de l'eau, etc.).
Dans le cas de substances à faible hydrosolubilité, on peut avoir recours à la dispersion ultrasonique, à des solvants organiques, des émulsifiants ou des dispersants, pour préparer les solutions mères ou également pour faciliter la dispersion de ces substances dans le milieu d'essai. Si on fait appel à ces substances auxiliaires, toutes les concentrations d'essai doivent contenir la même quantité de substances auxiliaires et des témoins supplémentaires doivent être exposés à une concentration de produit auxiliaire identique à celle utilisée pour les séries d'essais. La concentration de ces auxiliaires doit être limitée et ne doit en aucun cas dépasser 100 mg/l de milieu d'essai.
L'essai doit être effectué sans ajustement de pH. En cas de modification significative du pH, il est souhaitable de répéter l'essai en ajustant le pH et d'enregistrer les résultats. Dans ce cas, la valeur du pH de la solution mère doit être ajustée à la valeur du pH de l'eau de dilution à moins que des raisons particulières ne s'y opposent. Pour ce faire, on utilisera de préférence HCl ou NaOH. Cet ajustement du pH doit être effectué de manière à ne pas modifier sensiblement la concentration en substance d'essai de la solution mère. Si l'ajustement provoquait une réaction chimique ou une précipitation de la substance d'essai, il conviendrait de consigner ces observations au procès-verbal.
1.6.1.2. Milieu d'essai
L'eau doit être de l'eau distillée de bonne qualité, ou de l'eau désionisée d'une conductivité inférieure à 5 μS cm-1. L'appareil servant à la distillation de l'eau ne doit contenir aucun élément de cuivre.
Le milieu suivant est recommandé:
On prépare quatre solutions mères, conformément au tableau ci-après. Les solutions mères sont stérilisées par filtration sur membrane ou par autoclavage et stockées dans l'obscurité à 4 oC. La solution mère no 4 doit être stérilisée uniquement par filtration sur membrane. Ces solutions mères sont diluées pour atteindre les concentrations finales en substances nutritives dans les solutions d'essai.
|
Substance nutritive
|
Concentration dans la solution mère
|
Concentration finale dans la solution d'essai
|
|
Solution mère 1: macronutriments
|
|
NH4C1
|
1,5 g/1
|
15 mg/l
|
|
MgCl2.6H2O
|
1,2 g/1
|
12 mg/l
|
|
CaCl2.2H2O
|
1,8 g/1
|
18 mg/l
|
|
MgSO4.7H2O
|
1,5 g/1
|
15 mg/l
|
|
KH2 PO4
|
0,16 g/1
|
1,6 mg/l
|
|
Solution mère 2: Fe-EDTA
|
|
FeCl3.6H2O
|
80 mg/l
|
0,08 mg/l
|
|
Na2EDTA.2H2O
|
100 mg/l
|
0,1 mg/l
|
|
Solution mère 3: oligoéléments
|
|
H3BO3
|
185 mg/l
|
0,185 mg/l
|
|
MnCl2.4H2O
|
415 mg/l
|
0,415 mg/l
|
|
ZnClj
|
3 mg/l
|
3 × 10-3 mg/l
|
|
CoCl2.6H2O
|
1,5 mg/l
|
1,5 × 10-3 mg/l
|
|
CuCl2.2H2O
|
0,01 mg/l
|
10-5 mg/l
|
|
Na2MoO4.2H2O
|
7 mg/l
|
7 × 10-3 mg/l
|
|
Solution mère 4: NaHCO3
|
|
NaHCO3
|
50 g/1
|
50 mg/l
|
Après équilibrage avec l'air, le pH du milieu est environ de 8.
1.6.2. Appareillage
|
—
|
Matériel courant de laboratoire.
|
|
—
|
Flacons d'essai d'un volume approprié (on peut, par exemple, utiliser des flacons coniques de 250 ml pour un volume de solution d'essai de 100 ml). Tous les flacons d'essai doivent être identiques en ce qui concerne la matière et les dimensions.
|
|
—
|
Matériel de culture: une enceinte ou une pièce dans laquelle la température peut être maintenue dans un intervalle compris entre 21 et 25 oC avec une précision de ± 2 oC et munie d'un dispositif d'éclairage continu et uniforme dont le spectre d'émission est compris entre 400 et 700 nm. Si les algues des cultures témoins atteignent le taux de croissance recommandé, on peut estimer que les conditions de croissance, y compris l'intensité lumineuse, sont appropriées.
Il est recommandé d'utiliser, au niveau moyen des solutions d'essai, une intensité lumineuse comprise entre 60 et 120 μE m-2 s-1 (de 35 à 70 × 1018 photons m-2 s-1) mesurée dans la gamme de 400 à 700 nm à l'aide d'un récepteur approprié. Pour les appareils de mesure de la lumière étalonnés en lux, un intervalle équivalent, de 6 000 à 10 000 lux, est acceptable.
Cette intensité lumineuse peut être fournie par quatre à sept ampoules fluorescentes de 30 W, du type blanc classique (température de couleur d'environ 4 300 K), placées à une distance de 0,35 m de la culture d'algues.
|
|
—
|
Les mesures de la densité cellulaire doivent être effectuées par comptage direct des cellules vivantes, par exemple à l'aide d'un microscope et de cellules de comptage. Toutefois, on peut utiliser d'autres méthodes (photométrie, turbidimétrie...) à condition qu'elles soient suffisamment sensibles et s'il est montré qu'elles donnent une bonne corrélation avec la densité cellulaire.
|
1.6.3. Organismes soumis à l'essai
Il est conseillé d'utiliser des espèces d'algues vertes à croissance rapide qui soient commodes, tant pour la culture que pour l'essai. On utilise de préférence les espèces suivantes:
|
—
|
Selenastrum capricomutum, par exemple ATCC 22662 ou CCAP 278/4,
|
|
—
|
Scenedesmus subspicatus, par exemple 86.81 SAG.
|
Note:
|
ATCC
|
=
|
American Type Culture Collection (États-Unis)
|
|
CCAP
|
=
|
Culture Centre of Algae and Protozoa (Royaume-Uni)
|
|
SAC
|
=
|
Collection de cultures d'algues (Göttingen, RFA).
|
Si d'autres espèces sont utilisées, la souche doit être mentionnée.
1.6.4. Mode opératoire
Des essais sont effectués afin de déterminer la gamme de concentrations dans laquelle des effets sont susceptibles de se produire.
Les deux mesures de la croissance (biomasse et taux de croissance) peuvent fournir des valeurs très différentes de l'inhibition de la croissance; ces deux types de mesure devraient être utilisés dans les essais de recherche de l'intervalle de concentrations afin de s'assurer que la progression géométrique des concentrations permettra d'estimer à la fois la CE50b et la CE50r.
Densité cellulaire initiale
La densité cellulaire initiale recommandée dans les cultures d'essai est de 104 cellules/ml environ pour Selenastrum capricomutum et Scenedesmus subspicatus. Si l'on utilise d'autres espèces, la biomasse doit être comparable.
Concentrations de la substance à tester
Pour l'essai, on choisit au moins cinq concentrations en série géométrique avec un facteur n'excédant pas 2,2. La concentration la plus faible testée ne doit avoir aucun effet visible sur la croissance des algues. La concentration la plus élevée doit inhiber la croissance d'au moins 50 % par rapport au témoin et de préférence l'arrêter complètement.
Répétitions et témoins
Le protocole d'essai doit comporter trois répétitions de chaque concentration d'essai. Trois témoins sans substance à étudier doivent être soumis à l'essai ainsi que, s'il y a lieu, trois témoins contenant la substance auxiliaire. Le protocole d'essai peut, si cela se justifie, être modifié afin d'augmenter le nombre de concentrations et de réduire le nombre de répétitions par concentration.
Mode opératoire
Des cultures d'essai contenant les concentrations choisies de substance à étudier et la quantité désirée d'inoculum d'algues sont préparées par addition de quantités déterminées de solutions mères de substance d'essai à des quantités appropriées de précultures d'algues (voir annexe 1).
Les flacons de culture sont agités et placés dans l'appareil de culture. Les algues sont maintenues en suspension par agitation, en les remuant ou par barbotage d'air, afin d'augmenter les échanges gazeux et de réduire les variations de pH dans les solutions d'essai. Les cultures doivent être maintenues à une température se situant entre 21 et 25 oC, avec une précision de ± 2 oC.
La densité cellulaire est mesurée dans chaque flacon au moins 24, 48 et 72 heures après le début de l'essai. Lorsque la densité cellulaire est estimée à l'aide d'une méthode autre que le comptage direct, on mesure le bruit de fond à l'aide d'un milieu de culture d'algues filtré contenant la concentration appropriée du produit chimique à étudier.
Le pH est mesuré au début de l'essai et 72 heures plus tard.
Le pH des témoins ne devrait normalement pas varier de plus de 1,5 unité au cours de l'essai.
Essais concernant des substances volatiles
À l'heure actuelle, il n'existe aucune méthode généralement reconnue pour étudier les substances volatiles. Lorsque l'on sait qu'une substance a tendance à s'évaporer, on peut utiliser des flacons d'essai fermés avec un volume mort plus important. La possibilité d'un manque de CO2 doit être prise en considération lorsqu'on calcule le volume mort des flacons fermés. Des variantes de cette méthode ont été proposées [voir référence (4)].
Il convient d'essayer de mesurer la quantité de substance qui reste en solution et d'interpréter les résultats des essais réalisés avec les substances volatiles à l'aide de systèmes clos avec la plus grande prudence.
Essai limite
Un essai limite peut être effectué avec 100 mg/l, en suivant les procédures décrites dans cette méthode d'essai, afin de montrer que la CE50 est supérieure à cette concentration.
Si la nature de la substance est telle qu'il est impossible d'obtenir une concentration de 100 mg/l dans l'eau d'essai, l'essai limite doit être effectué à une concentration égale à la solubilité de la substance (ou à la concentration maximale formant une dispersion stable) dans le milieu utilisé (voir également le point 1.6.1.1).
L'essai limite doit être effectué au moins avec trois répétitions, avec le même nombre de témoins. Les deux mesures de la croissance (biomasse et taux de croissance) doivent être utilisées pour l'essai limite.
Si, dans un essai limite, on trouve une diminution moyenne de 25 % ou plus de la biomasse ou du taux de croissance par rapport au témoin, il convient d'effectuer un essai complet.
2. ÉVALUATION DES DONNÉES
Les densités cellulaires mesurées dans les cultures d'essai et les cultures témoins sont regroupées dans un tableau en même temps que les concentrations de la substance à étudier et les temps de mesures. La valeur moyenne de la densité cellulaire pour chaque concentration de substance à étudier et pour les témoins est reportée sur un graphique en fonction du temps (0-72 h) afin d'obtenir des courbes de croissance.
Pour déterminer la relation concentration/effet, on peut utiliser les deux méthodes suivantes. Certaines substances peuvent stimuler la croissance à de faibles concentrations. Seuls les points expérimentaux indiquant une inhibition comprise entre 0 et 100 % doivent être pris en considération.
2.1. COMPARAISON DES SURFACES DÉLIMITÉES PAR LES COURBES DE CROISSANCE
La surface qui est comprise entre la courbe de croissance et la ligne horizontale N = No peut être calculée à l'aide de la formule suivante:

où:
|
A
|
=
|
surface,
|
|
N0
|
=
|
nombre de cellules/ml mesuré au temps t0 (début de l'essai),
|
|
N1
|
=
|
nombre de cellules/ml mesuré au temps tt,
|
|
Nn
|
=
|
nombre de cellules/ml mesuré au temps tn,
|
|
tl
|
=
|
temps de la première mesure après le début de l'essai,
|
|
tn
|
=
|
temps de la ne mesure depuis le début de l'essai,
|
|
n
|
=
|
nombre de mesures effectuées depuis le début de l'essai.
|
Le pourcentage d'inhibition de la croissance cellulaire est calculé, pour chaque concentration de substance à étudier (IA), à l'aide de la formule suivante:

où:
|
Ac
|
= surface comprise entre la courbe de croissance témoin et la ligne horizontale N = N0,
|
|
At
|
= surface comprise entre la courbe de croissance à une concentration t et la ligne horizontale N = N0.
|
Les valeurs de IA sont portées sur du papier semi-logarithmique ou sur du papier semi-logarithmique-probit en fonction des concentrations correspondantes. Si l'on utilise du papier probit, on relie les points entre eux par une ligne droite, soit à l'œil soit par une régression calculée.
La CE50 est estimée à partir de la droite de régression par la concentration qui correspond à une inhibition de 50 % (IA = 50 %). Afin d'éviter toute ambiguïté, il est proposé de désigner la valeur de la CE50 obtenue avec cette méthode de calcul par le symbole CE50b. Il est important que la durée de l'exposition soit précisée avec la CE50b: CE50b (0-72 h).
2.2. COMPARAISON DES TAUX DE CROISSANCE
Le taux de croissance spécifique moyen (μ) pour des cultures en croissance exponentielle peut être calculé comme suit:
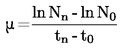
où t0 correspond au début de l'essai.
Le taux de croissance spécifique moyen peut également être déduit de la pente de la droite de régression dans un graphe de lnN en fonction de temps.
Le pourcentage d'inhibition du taux de croissance spécifique pour chaque concentration de substance d'essai (Iμt) est calculé à l'aide de la formule suivante:

où:
|
μc
|
=
|
taux de croissance spécifique moyen des témoins,
|
|
μt
|
=
|
taux de croissance spécifique moyen à la concentration d'essai t.
|
Le pourcentage de réduction du taux de croissance spécifique moyen pour chaque concentration de substance d'essai comparé à la valeur du témoin est porté sur un graphique en fonction du logarithme de la concentration. La CE50 peut être lue sur le graphique ainsi établi. Afin d'éviter toute ambiguïté, il est proposé de désigner la valeur de la CE50 obtenue avec cette méthode par le symbole CEr50. Les temps où sont effectuées les mesures doivent être précisés; par exemple, si la valeur se rapporte aux temps 0 et 72 heures, le symbole s'écrira CEr50 (0-72 h).
Note: Le taux de croissance spécifique est un terme logarithmique et de faibles variations du taux de croissance peuvent donner lieu à des changements importants dans la biomasse. Les valeurs de CEb et de CEr ne sont donc pas comparables numériquement.
2.3. CALCUL DE LA CSEO
La concentration sans effet observé est déterminée à l'aide d'une méthode statistique appropriée à la comparaison d'échantillons multiples (par exemple l'analyse de la variance et le test de Dunnett) en utilisant les valeurs individuelles des surfaces délimitées par les courbes de croissance A (voir point 2.1) ou par les taux de croissance spécifiques μ (voir point 2.2).
3. RÉSULTATS
Le procès-verbal d'essai contiendra, si possible, les renseignements suivants:
|
—
|
substance à étudier: données concernant son identification chimique,
|
|
—
|
organismes utilisés pour l'essai: origine, culture en laboratoire, numéro de la souche, méthode de culture,
|
|
—
|
conditions d'essai:
|
—
|
date du début et de la fin de l'essai, et sa durée,
|
|
—
|
pH des solutions au début et à la fin de l'essai (des variations du pH supérieures à 1,5 unité doivent faire l'objet d'une explication),
|
|
—
|
adjuvant et méthode utilisée pour solubiliser la substance à étudier, et concentrations de l'adjuvant dans les solutions d'essai,
|
|
—
|
intensité et qualité de la lumière,
|
|
—
|
concentrations étudiées (mesurées ou nominales),
|
|
|
—
|
résultats:
|
—
|
densité cellulaire pour chaque flacon à chaque point de mesure et méthode de mesure de la densité cellulaire,
|
|
—
|
valeurs moyennes de la densité cellulaire,
|
|
—
|
représentation graphique de la relation concentration/effet,
|
|
—
|
valeurs de la CE et méthode de calcul,
|
|
—
|
autres effets observés.
|
|
4. RÉFÉRENCES
|
(1)
|
OCDE, Paris, 1981, ligne directrice no 201, décision du Conseil C(81) 30 final.
|
|
(2)
|
Umweltbundesamt, Berlin, 1984, Verfahrensvorschlag «Hemmung der Zellvermehrung bei der Grùnalge Scenedesmus subpicatus», in: Rodolph/Boje: Okotoxikologie, ecomed, Landsberg, 1986.
|
|
(3)
|
ISO 8692 — Water quality — Fresh water algal growth inhibition test with Scenedesmus subspicatus and Selenastrum capricornutum.
|
|
(4)
|
S. Galassi and M. Vighi — Chemosphere, 1981, vol. 10, p. 1123-1126.
|
Annexe 1
Exemple de méthode de culture des algues
Observations générales
La présente méthode de culture a pour objectif de fournir des cultures d'algues permettant d'effectuer des essais de toxicité.
Il faut s'assurer, à l'aide de méthodes appropriées, que les cultures d'algues ne sont pas infectées par des bactéries (ISO 4833). Il peut être souhaitable d'utiliser des cultures axéniques mais il est essentiel de disposer de cultures unialguales.
Toutes les opérations doivent être effectuées dans des conditions stériles, afin d'éviter toute contamination aussi bien par des bactéries que par d'autres algues. Les cultures contaminées doivent être écartées.
Mode opératoire pour l'obtention de cultures d'algues
Préparation de solutions nutritives (milieux):
Le milieu peut être préparé par dilution de solutions mères concentrées de substances nutritives. Pour les milieux solides, ajouter 0,8 % de gélose. Le milieu utilisé doit être stérile. La stérilisation par autoclavage peut provoquer une perte de NH3.
Cultures mères:
Les cultures mères sont de petites cultures d'algues régulièrement transférées dans du milieu frais destinées à servir de matériel d'essai initial. Si les cultures ne sont pas utilisées régulièrement, elles sont étalées sur des tubes de gélose inclinés. Ces dernières sont transférées dans un milieu frais au moins tous les deux mois.
Les cultures mères sont cultivées dans des fioles coniques contenant le milieu approprié (volume de 100 ml environ). Lorsque les algues sont incubées à 20 oC et éclairées en permanence, il est nécessaire de les repiquer toutes les semaines.
Le transfert d'un prélèvement d'une «vieille» culture dans une fiole de milieu frais est effectué à l'aide de pipettes stériles, et de telle manière que, avec les espèces à croissance rapide, la concentration initiale soit environ 100 fois plus faible que celle de la «vieille» culture.
Le taux de croissance d'une espèce, qui peut être déterminé à partir de la courbe de croissance, permet d'estimer la densité à laquelle il convient de transférer la culture dans un nouveau milieu. Cela doit être fait avant que la culture n'atteigne la phase létale.
Préculture:
La préculture sert à fournir une quantité d'algues appropriée pour l'inoculation des cultures d'essai. La préculture est incubée dans les conditions de l'essai et elle est utilisée alors qu'elle est encore en croissance exponentielle, généralement après une période d'incubation de trois jours environ. Il faut écarter les cultures d'algues qui contiennent des cellules déformées ou anormales.
Annexe 2
La norme ISO 8692 — Qualité de l'eau — essai d'inhibition de la croissance des algues en eau douce effectué avec Scenedesmus subspicatus et Selenastrum capricornutum a permis d'obtenir les résultats suivants lors d'un essai interlaboratoires portant sur le bichromate de potassium auquel ont participé 16 laboratoires:
|
|
Moyenne (mg/I)
|
Intervalle (mg/1)
|
|
ErC50 (0 — 72 h)
|
0,84
|
de 0,60 à 1,03
|
|
EbC50 (0 — 72 h)
|
0,53
|
de 0,20 à 0,75
|
C.4. DÉTERMINATION DE LA BIODÉGRADABILITÉ «FACILE»
PARTIE I. GÉNÉRALITÉS
I.1. INTRODUCTION
Six méthodes d'essai permettant d'évaluer la biodégradabilité facile de produits chimiques en milieu aqueux en aérobiose sont décrites:
|
a)
|
Disparition du carbone organique dissous (COD) (Méthode C.4-A)
|
|
b)
|
Essai de screening modifié de l'OCDE — Disparition du COD — (Méthode C.4-B)
|
|
c)
|
Dégagement de dioxyde de carbone (CO2) (Essai Sturm modifié) (Méthode C.4-C)
|
|
d)
|
Respirométrie manométrique (Méthode C.4-D)
|
|
e)
|
Fiole fermée (Méthode C.4-E)
|
|
f)
|
MITI (ministère de l'industrie et du commerce international — Japon) (Méthode C.4-F)
|
La partie I contient des considérations d'ordre général ainsi que des considérations communes aux six essais. Les particularités propres à chaque méthode sont décrites dans les parties II à VII. Les annexes contiennent définitions, formules et données de référence.
Un exercice comparatif interlaboratoires de l'OCDE, réalisé en 1988, a montré que ces méthodes donnent des résultats comparables. Toutefois, l'une ou l'autre de ces méthodes peut être préférable en fonction des caractéristiques physiques de la substance.
I.2. CHOIX DE LA MÉTHODE APPROPRIÉE
Il est essentiel, pour choisir la méthode la mieux appropriée, de disposer d'informations concernant la solubilité, la pression de vapeur et les caractéristiques d'adsorption de la substance à étudier. Pour calculer la valeur théorique et/ou vérifier les valeurs mesurées d'un paramètre tel que la DThO, le CO2Th, le COD, le COT et la DCO (voir annexes I et II), il est nécessaire de connaître la structure chimique ou la formule brute du produit.
Toutes ces méthodes sont applicables aux substances d'essai qui peuvent être dissoutes dans l'eau à des concentrations de 100 mg/l au moins, pourvu qu'elles ne soient ni volatiles ni adsorbables. Dans le cas de produits peu hydrosolubles, volatiles ou adsorbables, les méthodes appropriées sont indiquées dans le tableau 1. La manière de procéder avec les produits faiblement hydrosolubles et les substances volatiles est décrite à l'annexe III. Les substances modérément volatiles peuvent être étudiées à l'aide de la méthode de disparition du COD pourvu que les récipients d'essai contiennent un volume gazeux suffisant et qu'ils soient fermés de manière appropriée. Dans ce cas, un contrôle abiotique doit toutefois être prévu afin de tenir compte de toute perte physique.
Tableau 1
Limites d'application des méthodes d'essai
|
Essai
|
Méthode analytique
|
Approprié pour les substances qui sont:
|
|
faiblement solubles
|
volatiles
|
adsorbables
|
|
Disparition du COD
|
Carbone organique dissous
|
—
|
—
|
+/-
|
|
Screening modifié de l'OCDE
|
Carbone organique dissous
|
—
|
—
|
+/-
|
|
Dégagement de CO2
|
Respirométrie: dégagement de CO2
|
+
|
—
|
+
|
|
Respirométrie manométrique
|
Respirométrie manométrique: consommation d'oxygène
|
+
|
+/-
|
+
|
|
Fiole fermée
|
Respirométrie: oxygène dissous
|
+/-
|
+
|
+
|
|
MITI
|
Respirométrie: consommation d'oxygène
|
+
|
+/-
|
+
|
L'interprétation des résultats obtenus nécessite des informations sur la pureté de la substance à étudier ou sur les proportions relatives de ses principaux composants, en particulier lorsque ces résultats sont faibles ou marginaux.
Il peut être extrêmement utile de disposer d'informations sur la toxicité du produit à étudier vis-à-vis des bactéries (annexe IV) pour choisir les concentrations d'essai appropriées et interpréter les résultats de biodégradation, notamment lorsque les valeurs sont peu élevées.
I.3. SUBSTANCES DE RÉFÉRENCE
La procédure est contrôlée à l'aide de substances de référence répondant aux critères de biodégradabilité facile. Ces substances sont étudiées parallèlement aux substances d'essai en ajoutant une fiole appropriée à la série normale d'essais.
Les produits chimiques adéquats sont l'aniline (fraîchement distillée), l'acétate de sodium et le benzoate de sodium. Ces produits de référence sont tous dégradés dans les conditions de ces méthodes, même si aucun inoculum n'est délibérément ajouté.
Il avait été suggéré de retenir un produit de référence qui soit facilement biodégradable, mais qui nécessite l'addition d'un inoculum. L'hydrogéno-phtalate de potassium a été proposé mais le bien-fondé de son utilisation reste à démontrer, avant de l'accepter comme substance de référence.
Dans le cas des essais respirométriques, les composés azotés peuvent influencer la consommation d'oxygène du fait de la nitrification (voir les annexes II et V).
I.4. PRINCIPE DES MÉTHODES D'ESSAI
Une solution ou une suspension de la substance à étudier dans un milieu minéral est ensemencée et incubée en aérobiose, dans l'obscurité ou sous lumière diffuse. La quantité de COD due à l'inoculum présente dans la solution d'essai doit être aussi faible que possible par rapport à la quantité de COD provenant de la substance à étudier. L'activité endogène de l'inoculum est estimée en effectuant parallèlement des essais à blanc avec l'inoculum mais sans addition de substance à étudier, quoique l'activité endogène des cellules en présence de la substance ne correspond pas exactement à celle de ce contrôle endogène. Un essai avec une substance de référence est effectué en parallèle pour vérifier le fonctionnement de ces procédures.
La dégradation est en général suivie par la détermination de paramètres comme le COD, la production de CO2 ou la consommation d'oxygène et les mesures sont effectuées avec une fréquence suffisante pour permettre l'identification du commencement et de la fin de la biodégradation. Les respiromètres automatiques permettent d'effectuer des mesures en continu. Le COD est parfois mesuré en plus d'un autre paramètre, mais cela n'est généralement le cas qu'au début et à la fin de l'essai. Une analyse chimique spécifique peut également servir à évaluer la dégradation primaire de la substance à étudier et à déterminer la concentration de toutes les substances intermédiaires formées (obligatoire dans l'essai MITI).
L'essai dure normalement 28 jours. Un essai peut cependant être interrompu avant, dès lors que la courbe de biodégradation atteint un plateau qui se prolonge au moins sur trois mesures. Un essai peut également être prolongé au-delà de 28 jours si la courbe montre que la biodégradation a manifestement commencé sans toutefois avoir atteint un palier le 28e jour.
I.5. CRITÈRES DE QUALITÉ
I.5.1. Reproductibilité
En raison de la nature de la biodégradation et de l'hétérogénéité des populations bactériennes utilisées comme inoculum, les mesures doivent être effectuées au moins en double.
On observe souvent que plus la concentration initiale des micro-organismes ajoutés au milieu d'essai est élevée, plus la variation entre les répétitions est faible. Des essais d'intercomparaison ont également montré que des variations importantes peuvent apparaître entre les résultats obtenus dans différents laboratoires, mais les résultats relatifs aux produits de référence facilement biodégradables sont généralement en bonne harmonie.
I.5.2. Validité de l'essai
Un essai est considéré comme valable si la différence entre les valeurs extrêmes en double de la mesure de la disparition du produit à étudier, au niveau du plateau, à la fin de l'essai ou à la fin de la fenêtre de 10 jours, est inférieure à 20 % et si le pourcentage de dégradation du produit de référence a atteint le niveau correspondant à une biodégradabilité facile en 14 jours. Si l'une de ces conditions n'est pas remplie, l'essai devra être recommencé. En raison du caractère rigoureux de ces méthodes, un résultat peu élevé ne signifie pas nécessairement que la substance soumise à l'essai n'est pas biodégradable dans les conditions de l'environnement; il indique seulement que d'autres études seront nécessaires pour en faire la preuve.
Si, lors d'un essai de toxicité effectué avec une solution contenant à la fois la substance à étudier et un produit de référence, la dégradation est inférieure à 35 % (établie d'après le COD) ou à 25 % (établie d'après la DThO ou le CO2Th) au cours des 14 premiers jours, on peut estimer que le produit a un effet inhibiteur (voir également l'annexe IV). La série d'essais doit être recommencée, si possible en utilisant une concentration de substance à étudier plus faible et/ou une concentration d'inoculum plus élevée, sans toutefois dépasser 30 mg/l de matière en suspension.
I.6. PROCÉDURES ET PRÉPARATIONS GÉNÉRALES
Les conditions générales qui s'appliquent aux essais sont résumées dans le tableau 2. Les appareils et les conditions expérimentales spécifiques d'un essai particulier sont décrits ci-après dans les chapitres consacrés à chaque essai.
Tableau 2
conditions d'essai
|
Essai
|
Disparition du COD
|
Dégagement de CO2
|
Respirométrie manométrique
|
Screening modifié de l'OECD
|
Fiolé fermée
|
MITI (1)
|
|
Concentration de la substance à étudier
|
|
|
|
|
|
|
|
en: mg/l C
|
|
|
100
|
|
2-10
|
100
|
|
mg/l COD
|
10-40
|
10-20
|
|
10-40
|
|
|
|
mg/l DThOD
|
|
|
50-100
|
|
5-10
|
|
|
Concentration de l'inoculum (approximative, en cellules/l)
|
≤ le; 30 mg/l MES
ou ≤ le; 100 ml/l d'effluent
(107 — 108)
|
0,5 ml/l d'effluent secondaire
(105)
|
jusqu'a 5 ml/l d'effluent
(104 — 106)
|
30 mg/l MES
(107 — 108)
|
|
Concentration d'éléments dans le milieu minéral (en mg/l):
|
|
|
|
|
|
|
|
P
|
116
|
11,6
|
29
|
|
N
|
1,3
|
0,13
|
1,3
|
|
Na
|
86
|
8,6
|
17,2
|
|
K
|
112
|
12,2
|
36,5
|
|
Mg
|
2,2
|
2,2
|
6,6
|
|
Ca
|
9,9
|
9,9
|
29,7
|
|
Fe
|
0,05-0,1
|
0,05-0,1
|
0,15
|
|
pH
|
7,4±0,2
|
de préférence 7,0
|
|
Température
|
22 ± oC
|
25 ± 1 oC
|
|
COD
|
=
|
Carbone Organique Dissout
|
|
|
DThOD
|
=
|
Demande Théorique en Oxygène
|
|
|
MES
|
=
|
Matières en Suspension
|
|
I.6.1. Eau de dilution
De l'eau désionisée ou distillée, exempte de substances toxiques à des concentrations inhibitrices (tels que des ions Cu + +), est utilisée. Elle ne contient pas plus de 10 % de la quantité de carbone organique introduite par la substance à étudier. L'eau d'essai doit être d'un haut degré de pureté afin d'éviter des valeurs élevées dans les essais à blanc. Des contaminations peuvent provenir non seulement des impuretés inhérentes, mais également des résines échangeuses d'ions et de matières provenant de la lyse des bactéries et des algues. Pour chaque série d'essais, n'utiliser qu'un même lot d'eau dont la teneur en COD doit être analysée au préalable. Une telle analyse n'est pas nécessaire pour l'essai en fiole fermée, mais la consommation en oxygène à partir de l'eau doit être faible.
I.6.2. Solutions mères de sels minéraux
Les solutions de sels minéraux utilisées pour l'essai sont préparées à partir de solutions mères de concentration appropriée. Les solutions mères décrites ci-après peuvent être utilisées (avec différents facteurs de dilution) dans les méthodes de disparition du COD, de screening modifié de l'OCDE, de dégagement de CO2, de respirométrie manométrique et dans l'essai en fiole fermée.
Les facteurs de dilution et, pour l'essai MITI, la préparation spécifique du milieu minéral sont mentionnés dans les chapitres consacrés à chaque essai.
Solutions mères:
Les solutions mères suivantes sont préparées en utilisant des réactifs de qualité analytique:
|
a)
|
Dihydrogénophosphate de potassium KH2PO4
|
8,50 g
|
|
Monohydrogénophosphate de potassium K2HPO4
|
21,75 g
|
|
Monohydrogénophosphate de sodium dihydraté Na2HPO4 2H20
|
33,40 g
|
|
Chlorure d'ammonium NH4Cl
|
0,50 g
|
|
Dissoudre dans de l'eau et compléter le volume à 1 litre. Le pH de la solution doit être égal à 7,4.
|
|
|
b)
|
Chlorure de calcium, anhydre CaCl2
|
27,50 g
|
|
ou chlorure de calcium dihydraté CaCl2, 2 H2O
|
36,40 g
|
|
Dissoudre dans de l'eau et compléter le volume à 1 litre
|
|
|
c)
|
Sulfate de magnésium heptahydraté, MgSO4,7 H2O
|
22,50 g
|
|
Dissoudre dans de l'eau et compléter le volume à 1 litre
|
|
|
d)
|
Chlorure de fer (III) hexahydraté, FeCl3, 6 H2O
|
0,25 g
|
|
Dissoudre dans de l'eau et compléter le volume à 1 litre
|
|
Note: Afin d'éviter d'avoir à préparer cette solution immédiatement avant usage, ajouter une goutte d'HCl concentré ou 0,4 g/1 de sel disodique d'acide éthylènediaminetétra-acétique (EDTA).
I.6.3. Solutions mères de produits chimiques
Dissoudre, si la solubilité excède 1 g/1, par exemple, entre 1 et 10 g, selon les cas, de substance à étudier ou de substance de référence dans de l'eau désionisée et compléter le volume à 1 litre. Dans le cas contraire, préparer des solutions mères dans du milieu minéral ou ajouter directement le produit au milieu minéral. Dans le cas de substances de plus faible solubilité, se référer à l'annexe III, mais noter que, dans l'essai MITI (Méthode C.4-F), aucun solvant ni émulsifiant ne doit être utilisé.
I.6.4. Inoculums
L'inoculum peut provenir de plusieurs sources: boues activées, effluents non chlorés, eaux de surface, sols, ou encore un mélange de ceux-ci. Pour les essais de disparition du COD, de dégagement de CO2 et de respirométrie manométrique, si des boues activées sont utilisées, elles doivent provenir d'une station d'épuration ou d'une unité pilote traitant principalement des eaux ménagères. Il a été montré que les inoculums provenant d'autres sources donnent des résultats plus dispersés. Pour l'essai de screening modifié de l'OCDE et l'essai en fiole fermée, il convient d'utiliser un inoculum plus dilué ne contenant pas de flocs de boue et la source la plus appropriée consiste en un effluent secondaire d'une station d'épuration des eaux résiduaires urbaines ou d'une installation à l'échelle du laboratoire. Pour l'essai MITI, l'inoculum, qui provient d'un mélange de plusieurs origines, est décrit dans le chapitre consacré à cet essai.
I.6.4.1. lnoculum provenant de boues activées
Recueillir un échantillon de boue activée dans le compartiment d'aération d'une station d'épuration traitant des eaux résiduaires ou d'une installation à l'échelle du laboratoire traitant principalement les eaux ménagères. Éliminer, si nécessaire, les grosses particules par filtration à travers un tamis fin et conserver ensuite la boue en aérobiose.
Une autre possibilité consiste à laisser la boue sédimenter ou à la centrifuger (par exemple à 1 100 g pendant 10 mn) après avoir éliminé toutes les grosses particules. Écarter le surnageant. La boue peut être lavée dans le milieu minéral. Mettre la boue concentrée en suspension dans un milieu minéral de telle sorte que la concentration soit comprise entre 3 et 5 g/1 de matière en suspension. Aérer la suspension jusqu'à son utilisation.
Les boues doivent provenir d'une station conventionnelle fonctionnant bien. Si elles proviennent d'une station d'épuration dont le débit est important ou si elles sont susceptibles de contenir des inhibiteurs, elles doivent être lavées. Laisser sédimenter ou centrifuger la boue remise en suspension par une agitation vigoureuse, écarter le surnageant et remettre la boue lavée en suspension dans un milieu minéral frais. Répéter cette opération jusqu'à ce que l'on puisse considérer que la boue est exempte d'inhibiteur et de substrat en excès.
Prélever un échantillon de boue remise en suspension ou de boue non traitée juste avant son utilisation afin de déterminer le poids sec des matières solides en suspension.
Une autre possibilité consiste à homogénéiser la boue activée (entre 3 et 5 g/l de matière solide en suspension) à l'aide d'un agitateur mécanique pendant 2 minutes à vitesse moyenne. Laisser décanter la boue homogénéisée pendant 30 minutes ou plus si besoin est, et prélever la phase liquide, qui sera utilisée comme inoculum à raison de 10 ml/1 de milieu minéral.
I.6.4.2. Autres sources d'inoculum
L'inoculum peut provenir de l'effluent secondaire d'une station d'épuration ou d'une installation à l'échelle du laboratoire qui reçoit principalement des eaux ménagères. Recueillir un échantillon frais qui doit être maintenu en aérobiose pendant le transport. Laisser reposer pendant une heure ou filtrer à l'aide d'un papier filtre grossier et conserver le surnageant transvasé ou le filtrat en aérobiose jusqu'au moment de son utilisation. On peut utiliser jusqu'à 100 ml de ce type d'inoculum par litre de milieu.
L'eau de surface peut constituer une autre source d'inoculum. Dans ce cas, recueillir un échantillon d'une eau de surface appropriée, par exemple de rivière ou de lac, et le maintenir en aérobiose jusqu'au moment de son utilisation. Si nécessaire, concentrer l'inoculum par filtration ou par centrifugation.
I.6.5. Préconditionnement des inoculums
Les inoculums peuvent être préconditionnés aux conditions expérimentales, mais non préadaptés au produit à étudier. Le préconditionnement consiste à aérer la boue activée dans un milieu minéral, ou l'effluent secondaire pendant une durée de 5 à 7 jours à la température de l'essai. Le préconditionnement permet parfois d'améliorer la précision des méthodes d'essai en réduisant les valeurs dans les essais à blanc. Il est inutile de préconditionner les inoculums destinés à l'essai MITI.
I.6.6. Contrôles abiotiques
Rechercher, si nécessaire, une dégradation abiotique éventuelle de la substance à étudier en déterminant la disparition du COD, la consommation d'oxygène ou le dégagement de dioxyde de carbone dans des témoins stériles ne contenant pas d'inoculum. La stérilisation est effectuée par filtration à travers une membrane (0,2 à 0,45 μm) ou par addition d'une substance toxique adéquate à une concentration appropriée. Si la filtration sur membrane est utilisée, prélever les échantillons de façon aseptique pour maintenir la stérilité. À moins que l'adsorption de la substance à étudier ait pu être exclue a priori, des essais où la biodégradation est mesurée par l'élimination de COD doivent comporter, particulièrement lorsqu'il s'agit d'inoculums provenant de boues activées, un témoin abiotique qui sera inoculé et additionné de l'agent stérilisant.
I.6.7. Nombre de fioles
Le nombre de fioles d'une série type est précisé dans les chapitres consacrés à ces essais.
Des fioles de type suivant peuvent être utilisées:
|
—
|
pour la suspension d'essai: contenant la substance à étudier et l'inoculum,
|
|
—
|
pour le blanc inoculum: ne contenant que l'inoculum,
|
|
—
|
pour le contrôle de la procédure: contenant la substance de référence et l'inoculum,
|
|
—
|
pour le témoin abiotique stérile: stérile et ne contenant que le produit à étudier (voir point I.6.6),
|
|
—
|
pour le contrôle de l'adsorption: contenant la substance à étudier, l'inoculum et l'agent stérilisant,
|
|
—
|
pour le contrôle de toxicité: contenant le produit à étudier, le produit de référence et l'inoculum.
|
Il est obligatoire de suivre le COD et/ou les autres paramètres parallèlement dans la suspension d'essai et dans l'essai à blanc contenant l'inoculum. Il est souhaitable de suivre également le COD en parallèle dans les autres fioles.
Toutefois, il se peut que cela ne soit pas toujours possible. S'assurer que le nombre d'échantillons ou de lectures est suffisant pour permettre de déterminer le pourcentage de disparition au cours de l'intervalle de temps de 10 jours.
I.7. ÉVALUATION DES DONNÉES
Le pourcentage de dégradation, Dt, est calculé à l'aide des valeurs moyennes des doubles déterminations du paramètre dans le récipient d'essai et dans le blanc contenant l'inoculum. Les formules sont décrites dans les chapitres consacrés à chaque essai. L'évolution de la dégradation est représentée graphiquement sur un diagramme indiquant l'intervalle de temps de 10 jours. Calculer et mentionner dans le rapport le pourcentage de disparition au terme de l'intervalle de temps de 10 jours et la valeur obtenue au plateau ou à la fin de l'essai, selon les cas.
Dans les essais de respirométrie, des composés contenant de l'azote peuvent affecter la consommation d'oxygène en raison de la nitrification (voir annexes II et V).
I.7.1. Estimation de la dégradation à l'aide de la détermination du COD
Pour pouvoir vérifier la validité de l'essai (voir I.5.2), le pourcentage de dégradation (Dt) à chaque instant t de prise d'un échantillon doit être calculé séparément pour les fioles contenant la substance à étudier en utilisant les valeurs moyennes de mesures en double du COD. Il se calcule à l'aide de la formule suivante:

où:
|
Dt
|
=
|
% de dégradation au temps t,
|
|
Co
|
=
|
concentration initiale moyenne de COD dans le milieu de culture ensemencé contenant la substance à étudier (mg COD/1),
|
|
Ct
|
=
|
concentration moyenne de COD dans le milieu de culture ensemencé contenant la substance à étudier, au temps t (mg COD/I),
|
|
Cbo
|
=
|
concentration initiale moyenne de COD dans l'essai à blanc (milieu minéral ensemencé) (mg COD/1),
|
|
Cbt
|
=
|
concentration moyenne de COD dans l'essai à blanc (milieu minéral ensemencé) au temps t (mg COD/1).
|
Toutes les concentrations sont mesurées expérimentalement.
I.7.2. Estimation de la dégradation à l'aide d'analyses spécifiques
Lorsque l'on dispose de données analytiques spécifiques, la biodégradation primaire est calculée grâce à la formule suivante:
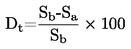
où:
|
Dt
|
=
|
% de dégradation au temps t, soit normalement le 28e jour,
|
|
Sa
|
=
|
quantité résiduelle de produit soumis à l'essai dans le milieu ensemencé à l'issue de l'essai (mg),
|
|
Sb
|
=
|
quantité résiduelle de produit à étudier dans l'essai à blanc constitué d'eau/milieu auquel on aura uniquement ajouté le produit à étudier (mg).
|
I.7.3. Dégradation abiotique
Lorsqu'on utilise un témoin abiotique stérile, le pourcentage de dégradation abiotique se calcule par la formule:

où:
|
Cs(o)
|
=
|
concentration en COD dans le témoin stérile au temps 0,
|
|
Cs(t)
|
=
|
concentration en COD dans le témoin stérile au temps t.
|
I.8. RAPPORT
Le procès-verbal d'essai devra, si possible, comporter les renseignements suivants:
|
—
|
produits à étudier et produits de référence ainsi que leur degré de pureté,
|
|
—
|
inoculum: nature et lieu(x) de prélèvement, concentration ainsi que tout traitement de préconditionnement,
|
|
—
|
proportion et nature des déchets industriels présents dans les eaux d'égout, si elles sont connues,
|
|
—
|
durée et température de l'essai,
|
|
—
|
dans le cas où les produits à étudier sont faiblement solubles, le traitement appliqué,
|
|
—
|
méthode d'essai appliquée; il convient de fournir les raisons scientifiques et l'explication de toute modification du mode opératoire,
|
|
—
|
tout phénomène d'inhibition observé,
|
|
—
|
toute dégradation abiotique observée,
|
|
—
|
les données concernant les analyses chimiques spécifiques, si elles sont disponibles,
|
|
—
|
les données concernant les analyses de substances intermédiaires, si elles sont disponibles,
|
|
—
|
la courbe représentant le pourcentage de dégradation en fonction du temps pour les substances soumises à l'essai et les substances de référence; la phase de latence, la phase de dégradation, l'intervalle de temps de 10 jours et la pente doivent être clairement indiqués (annexe I). Si l'essai a satisfait les critères de validité, on peut utiliser la moyenne des pourcentages de dégradation des fioles contenant la substance à étudier pour la courbe,
|
|
—
|
le pourcentage de disparition à l'issue de l'intervalle de temps de 10 jours et au plateau ou à la fin de l'essai.
|
PARTIE II. ESSAI DE DISPARITION DU COD (Méthode C.4.-A)
II.1. PRINCIPE DE LA MÉTHODE
Un volume mesuré de milieu minéral ensemencé, contenant une concentration connue de substance à étudier (10 à 40 mg/l de COD) comme seule source de carbone organique, est aéré dans l'obscurité ou sous une lumière diffuse, à 22 ± 2 oC.
La dégradation est suivie en analysant le COD à des intervalles fréquents pendant une durée de 28 jours. Le degré de biodégradation est calculé en exprimant la concentration de COD disparu (à laquelle on apportera une correction correspondant à la concentration de COD disparu dans l'essai à blanc contenant l'inoculum) en pourcentage de la concentration initiale. Le taux de biodégradation primaire peut également être calculé à partir d'analyses chimiques supplémentaires effectuées au début et à la fin de l'incubation.
II.2. DESCRIPTION DE LA MÉTHODE
II.2.1 Appareillage
|
a)
|
des fioles coniques, d'une contenance comprise, par exemple, entre 250 ml et 2 1, selon le volume requis pour l'analyse du COD;
|
|
b)
|
un dispositif d'agitation adapté aux fioles coniques, soit muni d'un système automatique de contrôle de température, soit placé dans une pièce thermostatée, et permettant de maintenir des conditions d'aérobiose dans toutes les fioles;
|
|
c)
|
un dispositif de filtration équipé des membranes adéquates;
|
|
d)
|
un analyseur du carbone organique dissous;
|
|
e)
|
un appareil de mesure de l'oxygène dissous;
|
II.2.2. Préparation du milieu minéral
Pour la préparation de la solution mère, voir I.6.2.
Mélanger 10 ml de solution a) avec 800 ml d'eau de dilution, ajouter 1 ml des solutions b) à d) et compléter le volume à 1 l avec l'eau de dilution.
II.2.3. Préparation et préconditionnement de l'inoculum
L'inoculum peut provenir de plusieurs sources: boues activées, effluents, eaux de surface, sols ou d'un mélange de celles-ci.
Voir I.6.4, I.6.4.1, I.6.4.2 et I.6.5.
II.2.4. Préparation des fioles
Exemple: verser un volume de 800 ml de milieu minéral par fiole conique de 2 1 et ajouter dans des fioles distinctes un volume suffisant de solution mère de substance d'essai ou de substance de référence pour que la concentration de la substance corresponde à une concentration en COD comprise entre 10 et 40 mg/l. Vérifier le pH et l'ajuster, si nécessaire, à une valeur de 7,4. Ensemencer les flacons avec de la boue activée ou avec un inoculum provenant d'une autre source (voir I.6.4.), de telle sorte que la concentration finale n'excède pas 30 mg/l de matière solide en suspension. Préparer également des témoins constitués de milieu minéral contenant l'inoculum mais sans substance d'essai ni substance de référence.
Ajouter, si nécessaire, une fiole pour la détection d'un éventuel effet inhibiteur de la substance d'essai. Pour cela, ensemencer une solution contenant des concentrations, à la fois de produit à étudier et de produit de référence, identiques à celles des autres fioles.
Ajouter, si besoin est, une fiole supplémentaire stérile, contenant une solution de produit non ensemencée afin de vérifier si la substance soumise à l'essai subit une dégradation abiatique (voir I.6.6).
De surcroît, si le produit à étudier est suspecté de s'adsorber de manière importante sur le verre, la boue, etc., il convient d'effectuer des mesures préliminaires permettant d'évaluer l'importance probable de cette adsorption et par conséquent de déterminer si l'essai convient pour la substance concernée (voir tableau 1). Préparer une fiole contenant la substance à étudier, l'inoculum et l'agent stérilisant.
Compléter le volume à 1 l dans toutes les fioles, avec le milieu minéral, mélanger, puis prélever un échantillon dans chaque fiole afin de déterminer la concentration initiale de COD (voir annexe II.4). Couvrir l'ouverture des fioles, par exemple avec du papier d'aluminium, de telle sorte que les échanges gazeux puissent s'effectuer librement entre la fiole et l'atmosphère environnante. Commencer ensuite l'essai en plaçant les fioles dans le dispositif d'agitation.
II.2.5. Nombre de fioles d'une série type
Fioles 1 et 2: suspension, d'essai
Fioles 3 et 4: blanc inoculum
Fiole 5: contrôle de la procédure
et de préférence et si nécessaire:
Fiole 6: témoin abiotique stérile
Fiole 7: contrôle de l'adsorption
Fiole 8: témoin de toxicité
Voir également I.6.7.
II.2.6. Déroulement de l'essai
Pendant l'essai, les concentrations de COD font l'objet de doubles déterminations à des intervalles de temps définis, suffisamment fréquents pour permettre de déterminer le début de l'intervalle de temps de 10 jours et le pourcentage de disparition au terme de cette période. Pour chaque détermination, il ne faut prélever que le volume de suspension d'essai strictement nécessaire.
Avant chaque prélèvement, le cas échéant, compenser les pertes dues à l'évaporation dans les fioles par ajout de la quantité nécessaire d'eau de dilution (I.6.1). Avant de prélever un échantillon, le milieu de culture est soigneusement mélangé et les substances adhérant aux parois du récipient doivent être dissoutes ou mises en suspension. Les échantillons sont immédiatement filtrés à travers une membrane ou centrifugés (voir annexe II.4); ils sont analysés le jour même; si tel n'est pas le cas, il faut les conserver entre 2 et 4 oC pendant un maximum de 48 heures, ou au-dessous de — 18 oC au-delà de 48 heures.
II.3. RÉSULTATS ET RAPPORT
II.3.1. Traitement des résultats
Calculer le pourcentage de dégradation au temps t, selon les indications figurant au point I.7.1 (détermination du COD) et, éventuellement, au point I.7.2 (analyse spécifique facultative).
Reporter tous les résultats sur les fiches de données fournies.
II.3.2. Validité des résultats
Voir I.5.2.
II. 3.3. Rapport
Voir I.8.
II.4. FICHE DE DONNÉES
Un exemple de fiche de données est fourni ci-dessous.
ESSAI DE DISPARITION DU COD
|
2.
|
DATE DU DÉBUT DE L'ESSAI
|
3. SUBSTANCE À ÉTUDIER
Nom:
Concentration de la solution mère: …mg/l produit
Concentration initiale dans le milieu, to: …mg/l produit
4. INOCULUM
Origine:
Traitement:
Préconditionnement éventuel:
Concentration des matières solides en suspension dans le mélange réactionnel: mg/l
5. DÉTERMINATION DU CARBONE
Analyseur de carbone:
|
|
Fiole no
|
|
COD après n jours (mg/l)
|
|
0
|
n1
|
n2
|
n3
|
nx
|
|
Substance à étudier plus inoculum
|
1
|
a1
|
|
|
|
|
|
|
a2
|
|
|
|
|
|
|
a, moyenne
Ca(t)
|
|
|
|
|
|
|
2
|
b1
|
|
|
|
|
|
|
b2
|
|
|
|
|
|
|
b, moyenne
Cb(t)
|
|
|
|
|
|
|
Essai à blanc avec l'inoculum et sans substance à étudier
|
3
|
C1
|
|
|
|
|
|
|
C2
|
|
|
|
|
|
|
c, moyenne Cc(t)
|
|
|
|
|
|
|
4
|
d1
|
|
|
|
|
|
|
d2
|
|
|
|
|
|
|
d, moyenne
Cd(t)
|
|
|
|
|
|
|

|
|
|
|
|
|
6. ÉVALUATION DES DONNÉES BRUTES
|
Fiole no
|
|
% de dégradat. après n jours
|
|
0
|
n1
|
n2
|
n3
|
nx
|
|
1
|

|
0
|
|
|
|
|
|
2
|
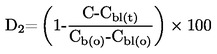
|
0
|
|
|
|
|
|
Moyenne (3)
|
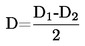
|
0
|
|
|
|
|
Note: Des tableaux similaires peuvent être utilisés pour les produits de référence et les témoins de toxicité.
7. CONTRÔLE ABIOTIQUE (facultatif)
|
|
Durée (en jours)
|
|
0
|
t
|
|
Concentration de COD (mg/l) dans le témoin stérile
|
Cs(o)
|
Cs(t)
|
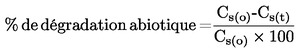
8. ANALYSE SPÉCIFIQUE (facultative)
|
|
quantité résiduelle de substance à étudier
|
% de dégradation primaire
|
|
Témoin stérile
|
Sb
|
|
|
Milieu d'essai contaminé
|
Sa
|

|
PARTIE III. ESSAI DE SCREENING MODIFIÉ DE L'OCDE (Méthode C.4-B)
III.1. PRINCIPE DE LA MÉTHODE
Un volume mesuré de milieu minéral contenant une concentration connue de substance à étudier (de 10 à 40 mg/l COD) comme unique source de carbone organique est ensemencé avec un volume de 0,5 ml d'effluent par litre de milieu. Le mélange est aéré à 22 ± 2 oC dans l'obscurité ou sous une lumière diffuse.
La dégradation est suivie grâce à l'analyse du COD à des intervalles rapprochés pendant une période de 28 jours. Le taux de biodégradation est calculé en exprimant la concentration en COD disparu (corrigé en fonction de la quantité de COD disparu dans l'essai à blanc avec l'inoculum) en pourcentage de la concentration initiale. Le taux de dégradation primaire peut également être calculé à partir d'analyses chimiques supplémentaires effectuées au début et à la fin de l'incubation.
III.2. DESCRIPTION DE LA MÉTHODE
III.2.1. Appareillage
|
a)
|
des fioles coniques, d'une contenance comprise, par exemple, entre 250 ml et 2 1, selon le volume requis pour l'analyse du COD;
|
|
b)
|
un dispositif d'agitation adapté aux fioles coniques, soit muni d'un système automatique de contrôle de température, soit placé dans une pièce thermostatée, et permettant de maintenir des conditions d'aérobiose dans toutes les fioles;
|
|
c)
|
un appareillage de filtration, accompagné des membranes adéquates;
|
|
d)
|
un analyseur du carbone organique dissous;
|
|
e)
|
un appareil de mesure de l'oxygène dissous;
|
III.2.2. Préparation du milieu minéral
Pour la préparation de la solution mère, voir I.6.2.
Mélanger 10 ml de solution a) avec 800 ml d'eau de dilution, ajouter 1 ml des solutions b) à d) et compléter le volume à 1 1 avec de l'eau de dilution.
Cette méthode ne nécessitant que 0,5 ml/1 d'effluent comme inoculum, le milieu doit par conséquent être enrichi en oligoéléments et facteurs de croissance. On ajoute pour cela 1 ml de chacune des solutions ci-après par litre de milieu final.
Solution d'oligoéléments:
|
Sulfate de manganèse tétrahydraté, MnSO4. 4H2O
|
39,9 mg
|
|
Acide borique, H3BO3
|
57,2 mg
|
|
Sulfate de zinc heptahydraté, ZnSO4. 7H2O
|
42,8 mg
|
|
Heptamolybdate d'ammonium (NH4)6 MO7O24
|
34,7 mg
|
|
Fer-chélaté (FeCl3, EDTA)
|
100,0 mg
|
|
Dissoudre et compléter le volume à 1 l avec l'eau de dilution.
|
|
|
Solution de vitamines:
|
|
|
Extrait de levure
|
15,0 mg
|
Dissoudre l'extrait de levure dans 100 ml d'eau. Stériliser par passage à travers une membrane de porosité de 0,2 nm ou préparer une solution fraîche.
III.2.3. Préparation et préconditionnement de l'inoculum
L'inoculum provient de l'effluent secondaire d'une station d'épuration ou d'une unité pilote qui reçoit essentiellement des eaux ménagères. Voir I.6.4.2 et I.6.5.
On en utilise 0,5 ml par litre de milieu minéral.
III.2.4. Préparation des fioles
Exemple: verser un volume de 800 ml de milieu minéral par fiole conique de 2 1 et ajouter dans des fioles distinctes des volumes de solution mère de substance à étudier et de substance de référence tels que la concentration du produit soit comprise entre 10 et 40 mg/l de COD. Vérifier le pH et l'ajuster, si nécessaire, à une valeur de 7,4. Ensemencer les fioles avec un volume de 0,5 ml/1 d'effluent d'eau d'égout (voir I.6.4.2). Préparer également des témoins en ajoutant l'inoculum au milieu minéral, mais sans addition de substance d'essai ou de substance de référence.
Ajouter, si nécessaire, une fiole pour mesurer un éventuel effet inhibiteur de la substance à étudier. Pour cela, ensemencer une solution contenant des concentrations de produit à étudier et de produit de référence identiques à celles des autres fioles.
Ajouter également, si besoin est, une fiole supplémentaire, stérile, contenant une solution de substance non ensemencée afin de vérifier si la substance à étudier subit une dégradation abiotique (voir I.6.6).
De surcroît, si le produit à étudier est suspecté de s'adsorber de manière importante sur le verre, la boue, etc., il convient d'effectuer des mesures préliminaires permettant de déterminer l'importance vraisemblable de l'adsorption et d'évaluer par conséquent si l'essai convient pour la substance concernée (voir tableau 1). Préparer une fiole contenant la substance à étudier, l'inoculum et l'agent stérilisant.
Compléter le volume à 1 l dans toutes les fioles, avec le milieu minéral, mélanger, puis prélever un échantillon dans chaque flacon afin de déterminer la concentration initiale de COD (voir annexe II.4). Recouvrir les fioles avec une feuille d'aluminium, par exemple, de telle sorte que les échanges gazeux puissent s'effectuer librement entre la fiole et l'atmosphère environnante. Commencer ensuite l'essai en plaçant les récipients dans le dispositif d'agitation.
III.2.5. Nombre de fioles d'une série type
Fioles 1 et 2: suspension d'essai
Fioles 3 et 4: blanc inoculum
Fiole 5: contrôle de la procédure
et de préférence et si besoin est:
Fiole 6: témoin abiotique stérile
Fiole 7: contrôle de l'adsorption
Fiole 8: témoin de toxicité
Voir également I.6.7.
III.2.6. Déroulement de l'essai
Au cours de l'essai, les concentrations de COD dans chaque fiole font l'objet d'une double détermination à des intervalles de temps définis, suffisamment fréquents pour permettre de déterminer le début de l'intervalle de temps de 10 jours et le pourcentage de disparition au terme de cette période. Pour chaque détermination, il ne faut prélever que le volume strictement nécessaire.
Avant chaque prélèvement, le cas échéant, compenser les pertes dues à l'évaporation dans les fioles par ajout de la quantité nécessaire d'eau de dilution (I.6.1). Avant de prélever un échantillon, le milieu de culture est soigneusement mélangé et les substances adhérant aux parois des récipients sont dissoutes ou mises en suspension. Les échantillons sont immédiatement filtrés à travers une membrane ou centrifugés (voir annexe II.4). Ils sont analysés le jour même, si tel n'est pas le cas, il faut les conserver entre 2 et 4 oC pendant un maximum de 48 heures, ou au-dessous de — 18 oC au-delà de 48 heures.
III.3. RÉSULTATS ET RAPPORT
III.3.1. Traitement des résultats
Calculer le pourcentage de dégradation au temps t, selon les indications figurant au point I.7.1 (détermination du COD) et, éventuellement, au point I.7.2 (analyse spécifique facultative).
Reporter tous les résultats sur les fiches de données fournies.
III.3.2. Validité des résultats
Voir I.5.2.
III. 3.3. Rapport
Voir I.8.
III.4. FICHE DE DONNÉES
Un exemple de fiche de données est présenté ci-après.
ESSAI DE TRIAGE DE L'OCDE MODIFIÉ
|
2.
|
DATE DU DÉBUT DE L'ESSAI
|
3. SUBSTANCE À ÉTUDIER
Nom:
Concentration de la solution mère: mg/l produit
Concentration initiale dans le milieu, to: mg/l produit
4. INOCULUM
Origine:
Traitement:
Préconditionnement éventuel:
Concentration des matières solides en suspension dans le mélange réactionnel: mg/l
5. DÉTERMINATION DU CARBONE
Analyseur de carbone:
|
|
Fiole no
|
|
COD après n jours (mg/l)
|
|
0
|
n1
|
n2
|
n3
|
nx
|
|
Substance à étudier plus inoculum
|
1
|
a1
|
|
|
|
|
|
|
a2
|
|
|
|
|
|
|
a, moyenne
Ca(t)
|
|
|
|
|
|
|
2
|
b1
|
|
|
|
|
|
|
b2
|
|
|
|
|
|
|
b, moyenne
Cb(t)
|
|
|
|
|
|
|
Essai à blanc avec l'inoculum et sans substance à étudier
|
3
|
C1
|
|
|
|
|
|
|
C2
|
|
|
|
|
|
|
c, moyenne Cc(t)
|
|
|
|
|
|
|
4
|
d1
|
|
|
|
|
|
|
d2
|
|
|
|
|
|
|
d, moyenne
Cd(t)
|
|
|
|
|
|
|

|
|
|
|
|
|
6. ÉVALUATION DES DONNÉES BRUTES
|
Fiole no
|
|
% de dégradat. après n jours
|
|
0
|
n1
|
n2
|
n3
|
nx
|
|
1
|

|
0
|
|
|
|
|
|
2
|
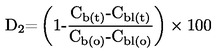
|
0
|
|
|
|
|
|
Moyenne (4)
|
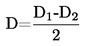
|
0
|
|
|
|
|
Note: Des tableaux similaires peuvent être utilisés pour les produits de référence et les témoins de toxicité.
7. CONTRÔLE ABIOTIQUE (facultatif)
|
|
Durée (en jours)
|
|
0
|
t
|
|
Concentration de COD (mg/l) dans le témoin stérile
|
Cs(o)
|
Cs(t)
|

8. ANALYSE SPÉCIFIQUE (facultative)
|
|
quantité résiduelle de substance à étudier
|
% de dégradation primaire
|
|
Témoin stérile
|
Sb
|
|
|
Milieu d'essai contaminé
|
Sa
|

|
PARTIE IV. ESSAI DE DÉGAGEMENT DE CO2 (Méthode C.4-C)
IV.1. PRINCIPE DE LA MÉTHODE
Un volume mesuré de milieu minéral ensemencé, contenant une concentration connue de la substance à étudier (10 à 20 mg/l de COD ou de COT) comme seule source de carbone organique, est aéré par le passage d'air exempt de dioxyde de carbone, à un débit contrôlé et dans l'obscurité ou en lumière diffuse. La dégradation est suivie pendant une durée de 28 jours en déterminant le dioxyde de carbone produit, qui est piégé par de l'hydroxyde de sodium ou de baryum et mesuré par titration de l'hydroxyde résiduel ou en tant que carbone inorganique. La quantité de dioxyde de carbone produite à partir de la substance à étudier (corrigée en fonction de la quantité produite dans l'essai à blanc avec l'inoculum) est exprimée en pourcentage du CO2Th. Le taux de biodégradation peut également être calculé à partir d'analyses supplémentaires du COD effectuées au début et à la fin de l'incubation.
IV.2. DESCRIPTION DE LA MÉTHODE
IV. 2.1. Appareillage
|
a)
|
des fioles d'une contenance de 2 à 5 litres, munies d'un tube d'aération atteignant presque le fond du récipient et d'un orifice de sortie;
|
|
b)
|
des agitateurs magnétiques pour les essais avec des substances peu solubles;
|
|
c)
|
des flacons absorbeurs de gaz;
|
|
d)
|
un dispositif de contrôle et de mesure du débit d'air;
|
|
e)
|
un appareil de lavage du dioxyde de carbone, permettant d'obtenir de l'air exempt de dioxyde de carbone; une autre possibilité consiste à utiliser un mélange d'oxygène et d'azote exempts de CO2, provenant de bouteilles de gaz, dans des proportions adéquates (20 % O2, 80 % N2);
|
|
f)
|
un dispositif de mesure du dioxyde de carbone, soit par titrimétrie, soit à l'aide d'un analyseur de carbone inorganique quelconque;
|
|
g)
|
un dispositif de filtration sur membrane (facultatif);
|
|
h)
|
un analyseur de COD (facultatif).
|
IV.2.2. Préparation du milieu minéral
Pour la préparation des solutions mères, voir I.6.2.
Mélanger 10 ml de solution a) et 800 ml d'eau de dilution, ajouter 1 ml des solutions b) à d) et compléter le volume à 1 l avec l'eau de dilution.
IV.2.3. Préparation et préconditionnement de l'inoculum
L'inoculum peut provenir de plusieurs sources: boues activées — effluents — eaux de surface — sols — ou d'un mélange de celles-ci.
Voir I.6.4, I.6.4.1, I.6.4.2 et I.6.5.
IV.2.4. Préparation des fioles
Les volumes et poids cités ci-après à titre d'exemple indiquent des valeurs correspondant à des fioles de 5 l contenant 3 1 de suspension. Si de plus petits volumes sont utilisés, il convient de modifier les valeurs en conséquence, mais en s'assurant que le dioxyde de carbone formé peut être mesuré avec précision.
Verser 2 400 ml de milieu minéral dans chaque fiole de 5 1. Ajouter un volume approprié de boue activée préparée au préalable (voir I.6.4.1 et I.6.5) tel que la concentration des matières en suspension n'excède pas 30 mg/l dans le volume final de 3 1 de mélange ensemencé. Une autre possibilité consiste à diluer d'abord la boue préparée jusqu'à obtention d'une suspension de 500 à 1 000 mg/l dans le milieu minéral, puis à ajouter une partie aliquote de cette suspension dans la fiole de 5 1, de manière à atteindre une concentration de 30 mg/l; cette méthode permet une plus grande précision. On peut utiliser des inoculums d'origines différentes (voir I.6.4.2).
Éliminer le dioxyde de carbone du mélange ensemencé par barbotage d'air exempt de CO2, pendant une nuit.
Ajouter séparément des volumes connus de solutions mères de la substance d'essai et de la substance de référence dans des fioles préparées en double, de telle sorte que la concentration de COD ou de COT, provenant des substances ajoutées, soit comprise entre 10 et 20 mg/l; conserver quelques fioles sans addition de substance comme témoins d'ensemencement. Si la substance à étudier est peu soluble, il convient d'ajouter directement un poids ou un volume défini, ou de procéder selon les indications données à l'annexe III.
Ajouter, si nécessaire, une fiole pour mesurer un éventuel effet inhibiteur de la substance à étudier. Pour cela, ensemencer une solution contenant des concentrations du produit à étudier et du produit de référence identiques à celles des autres fioles.
Ajouter également, si besoin est, une fiole supplémentaire stérile, contenant une solution de produit non ensemencée afin de vérifier si la substance d'essai subit une dégradation abiotique (voir I.6.6). Stériliser par addition d'une substance toxique à une concentration appropriée.
Compléter le volume des suspensions à 3 litres, dans toutes les fioles, avec le milieu minéral aéré au préalable au moyen d'air exempt de CO2. Des échantillons peuvent (facultatif) être prélevés afin d'analyser le COD (voir annexe II.4) et/ou d'effectuer une analyse spécifique. Relier les flacons absorbeurs à l'orifice de sortie d'air des fioles.
Si l'on utilise de l'hydroxyde de baryum, à chacune des fioles de 5 litres, relier en série trois flacons absorbeurs, contenant chacun 100 ml d'une solution 0,0125 M d'hydroxyde de baryum. La solution ne doit contenir ni sulfate ni carbonate précipité et il convient d'en déterminer la teneur juste avant son utilisation. Si l'on utilise de l'hydroxyde de sodium, relier deux pièges, le second servant de contrôle pour montrer que tout le dioxyde de carbone a été absorbé dans le premier. On pourra utiliser des flacons absorbeurs munis de systèmes de fermeture pour flacons de sérum. Ajouter dans chaque flacon 200 ml d'hydroxyde de sodium 0,05 M, ce qui suffit à absorber la totalité du dioxyde de carbone dégagé lors de la dégradation complète de la substance à étudier. La solution d'hydroxyde de sodium, même fraîchement préparée, contiendra des traces de carbonate; il convient donc d'effectuer une correction en déduisant la quantité de carbonate présente dans le témoin.
IV.2.5. Nombre de fioles d'une série type
Fioles 1 et 2: suspension d'essai
Fioles 3 et 4: blanc inoculum
Fiole 5: contrôle de la procédure
et de préférence et si nécessaire:
Fiole 6: témoin abiotique stérile
Fiole 7: témoin de toxicité
Voir également I.6.7.
IV.2.6. Déroulement de l'essai
Commencer l'essai en faisant barboter de l'air exempt de CO2 dans les suspensions à un débit de 30-100 ml/min. Prélever régulièrement des échantillons de l'absorbant de dioxyde de carbone pour en analyser la teneur en CO2. Il est conseillé d'effectuer des analyses tous les deux ou trois jours pendant les dix premiers jours, puis ensuite tous les cinq jours jusqu'au 28e jour de telle sorte que l'intervalle de temps de 10 jours puisse être identifié.
Le 28e jour, prélever des échantillons (facultatif) pour analyser le COD et/ou effectuer une analyse spécifique, mesurer le pH des suspensions et ajouter 1 ml d'acide chlorhydrique concentré à chaque fiole; aérer la fiole pendant une nuit pour éliminer le dioxyde de carbone présent dans la suspension d'essai. Le 29e jour, analyser pour la dernière fois le dégagement de dioxyde de carbone.
Les jours où l'on effectue les mesures de CO2, détacher le flacon absorbeur contenant de l'hydroxyde de baryum le plus proche de la fiole d'essai et titrer la solution d'hydroxyde de baryum à l'aide d'HCl 0,05 M en utilisant la phénolphtaléine comme indicateur. Rapprocher les autres flacons absorbeurs en les décalant d'une place et introduire un nouveau flacon absorbeur contenant 100 ml d'hydroxyde de baryum 0,0125 M frais à l'extrémité de la série. Effectuer les titrations nécessaires, par exemple lorsqu'une précipitation importante se produit dans le premier piège et avant qu'elle n'apparaisse dans le second, ou au moins une fois par semaine. Une autre possibilité consiste à utiliser le NaOH comme absorbant et à prélever à l'aide d'une seringue un échantillon de faible volume (en fonction du type d'analyseur de carbone utilisé) de la solution d'hydroxyde de sodium qui se trouve dans le flacon absorbeur le plus proche de la fiole. Injecter l'échantillon dans la partie CI de l'analyseur de carbone pour analyser directement le dioxyde de carbone dégagé.
N'analyser le contenu du second piège qu'à la fin de l'essai afin d'apporter une correction correspondant à tout passage de dioxyde de carbone dans ce piège.
IV.3. RÉSULTATS ET RAPPORT
IV.3.1. Traitement des résultats
Lorsqu'une titration est effectuée, la quantité de CO2 piégée dans un flacon absorbeur est calculée à l'aide de la formule suivante:
mg CO2 = (100 × CB-0,5 × V × CA) × 44
où:
|
V
|
=
|
volume de HC1 utilisé pour la titration des 100 ml contenus dans le flacon absorbeur (ml),
|
|
CB
|
=
|
concentration de la solution d'hydroxyde de baryum (M),
|
|
CA
|
=
|
concentration de la solution d'acide chlorhydrique (M),
|
si CB est 0,0125 M et CA est 0,05 M, la titration d'un volume de 100 ml d'hydroxyde de baryum est de 50 ml et le poids de CO2 est donné par la formule suivante:

Ainsi, dans ce cas, le facteur permettant de convertir le volume d'HCl titré en mg de CO2 est égal à 1,1.
Calculer le poids de CO2 produit par l'inoculum seul et par l'inoculum plus la substance à étudier à l'aide des valeurs de titration obtenues dans les deux cas; la différence correspond au poids de CO2 produit à partir de la substance à étudier seule.
Exemple: si la titration du flacon absorbeur correspondant à l'inoculum seul nécessite un volume de 48 ml et celui du flacon correspondant à l'inoculum additionné à la substance, un volume de 45 ml:
CO2 de l'inoculum = 1,1 × (50-48) = 2,2 mg
CO2 de l'inoculum plus substance à étudier = 1,1 × (50-45) = 5,5 mg
et par conséquent le poids de CO2 produit à partir de la substance à étudier est égal à 3,3 mg.
Le pourcentage de dégradation est calculé à l'aide de la formule suivante:
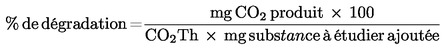
ou
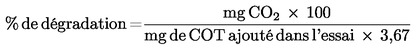
où 3,67 est le facteur de conversion (44/12) du carbone en dioxyde de carbone.
Calculer le pourcentage de dégradation après un temps déterminé en effectuant la somme des pourcentages de CO2Th calculés pour chaque jour où des mesures ont été effectuées pendant la période considérée.
Lorsque l'absorbant utilisé est de l'hydroxyde de sodium, calculer la quantité de dioxyde de carbone produite, exprimée en CI (mg), en multipliant la concentration en CI dans l'absorbant par le volume de ce dernier.
Calculer le pourcentage de dégradation à l'aide de la formule suivante:

Calculer la disparition du COD (facultatif) comme décrit au point I.7. Consigner ces résultats, ainsi que tout autre résultat, sur les fiches de données fournies.
IV.3.2 Validité des résultats
La teneur en CI de la suspension de substance à étudier en milieu minéral au début de l'essai doit être inférieure à 5 % du CT, et le dégagement total de CO2 dans l'essai à blanc contenant l'inoculum ne doit normalement pas excéder 40 mg/l de milieu à la fin de l'essai. Si les valeurs obtenues sont supérieures à 70 mg/l de CO2, les données et le protocole expérimental doivent être soumis à un examen critique.
Voir également I.5.2.
IV. 3.3 Rapport
Voir I.8.
IV.4. FICHE DE DONNÉES
Un exemple de fiche de données est présenté ci-dessous.
ESSAI DE DÉGAGEMENT DE DIOXYDE DE CARBONE
|
2.
|
DATE DU DÉBUT DE L'ESSAI
|
3. SUBSTANCE À ÉTUDIER
Nom:
Concentration de la solution mère: mg/l produit
Concentration initiale dans le milieu: mg/l produit
Carbone total ajouté dans la fiole: mg/l
CO2Th: mg CO2
4. INOCULUM
Origine:
Traitement:
Préconditionnement éventuel:
Concentration des matières solides en suspension dans le mélange réactionnel: mg/l
|
5.
|
PRODUCTION DE DIOXYDE DE CARBONE ET DÉGRADABILITÉ
Méthode: Ba(OH)2NaOH/autre
|
|
Temps
(jours)
|
CO2 formé
Essai (mg)
|
CO2 formé
blanc (mg)
|
CO2 formé cumulé (mg)
(essai moins blanc)
|
% CO2Th
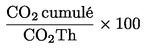
|
|
1
2
|
moy.
|
3
4
|
moy.
|
1
|
2
|
1
|
2
|
moy.
|
|
0
|
|
|
|
|
|
|
|
|
|
|
n1
|
|
|
|
|
|
|
|
|
|
|
n2
|
|
|
|
|
|
|
|
|
|
|
n3
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
28
|
|
|
|
|
|
|
|
|
|
Note: Des tableaux de ce type peuvent être utilisés pour le produit de référence et les témoins de toxicité.
6. DÉTERMINATION DU CARBONE (facultative)
Analyseur de carbone: ...
|
Temps (jours)
|
Blanc mg/l
|
Produit à étudier mg/l
|
|
0
|
Cb(o)
|
co
|
|
28 (5)
|
Cb(t)
|
ct
|

7. DÉGRADATION ABIOTIQUE (facultative)
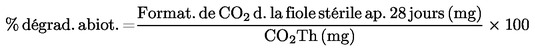
PARTIE V. ESSAI DE RESPIROMÉTRIE MANOMÉTRIQUE (Méthode C.4-D)
V.l. PRINCIPE DE LA MÉTHODE
Un volume mesuré de milieu minéral ensemencé, contenant une concentration connue de la substance d'essai (100 mg/l de substance d'essai, de telle sorte que la DThO soit au moins comprise entre 50 et 100 mg/l au moins) comme seule source de carbone organique, est agité en fiole fermée, à température constante (± 1 oC au maximum) pendant une durée maximale de 28 jours. La consommation d'oxygène est déterminée soit en mesurant la quantité d'oxygène (produit par électrolyse) nécessaire pour maintenir un volume gazeux constant dans la fiole respirométrique, soit en se basant sur les modifications de volume ou de pression (ou sur une combinaison des deux) dans l'appareil. Le dioxyde de carbone produit est absorbé par une solution d'hydroxyde de potassium ou par un autre absorbant approprié. La quantité d'oxygène consommée par la substance à étudier (après une correction correspondant à la quantité d'oxygène consommée par le témoin inoculum, analysée parallèlement) est exprimée en pourcentage de DThO ou de DCO. Facultativement, la biodégradation primaire peut également être calculée à partir d'analyses supplémentaires du COD effectuées au début et à la fin de l'incubation, et la biodégradation ultime par la mesure du COD.
V.2. DESCRIPTION DE LA MÉTHODE
V.2.1. Appareillage
|
a)
|
un respiromètre approprié,
|
|
b)
|
un dispositif de régulation de température d'une précision de ± 1 oC au moins,
|
|
c)
|
un dispositif de filtration sur membrane (facultatif),
|
|
d)
|
un analyseur de carbone (facultatif).
|
V.2.2. Préparation du milieu minéral
Pour la préparation des solutions mères, voir I.6.2.
Mélanger 10 ml de solution (a) et 800 ml d'eau de dilution, ajouter 1 ml des solutions (b) à (d) et compléter le volume à un litre avec l'eau de dilution.
V.2.3. Préparation et préconditionnement de l'inoculum
L'inoculum peut provenir de plusieurs sources: boues activées — effluents — eaux de surface — sols — ou d'un mélange de celles-ci.
Voir I.6.4., I.6.4.1., I.6.4.2. et I.6.5.
V.2.4. Préparation des fioles
À partir des solutions mères, préparer différents lots de solutions de substance d'essai et de substance de référence dans du milieu minéral, normalement à une concentration de 100 mg/l de substance (ce qui donne une DThO comprise, au moins, entre 50 et 100 mg/l).
Calculer la DThO à partir de la formation de sels d'ammonium, à moins qu'un processus de nitrification ne soit prévisible, auquel cas le calcul reposera sur la formation de nitrate (voir annexe II.2.)
Mesurer le pH et, le cas échéant, l'ajuster à 7,4±0,2.
Les substances faiblement solubles doivent être ajoutées lors d'une étape ultérieure (voir ci-après).
S'il convient de déterminer la toxicité de la substance à étudier, préparer une solution de milieu minéral supplémentaire contenant à la fois la substance de référence et le produit à étudier aux mêmes concentrations que dans les solutions ne contenant que l'un des produits.
Dans le cas où il est nécessaire de mesurer la consommation physico-chimique d'oxygène, préparer une solution de produit à étudier, dont la concentration corresponde normalement à une DThO de 100 mg/l, stérilisée par l'addition d'une substance toxique appropriée (voir I.6.6).
Ajouter dans deux fioles, au moins, le volume requis de solution d'essai et dans deux autres, au moins, le volume adéquat de solution de référence. Préparer d'autres fioles ne contenant que du milieu minéral (pour les témoins inoculum) et, le cas échéant, la solution de mélange produit à étudier/substance de référence et la solution stérile.
Si la substance à étudier est peu soluble, en ajouter un poids ou un volume déterminé, directement à ce stade, ou suivre le mode opératoire décrit à l'annexe III. Ajouter de l'hydroxyde de potassium, de la chaux sodée ou un autre absorbant dans les compartiments d'absorption du CO2.
V.2.5. Nombre de fioles d'une série type
Fioles 1 et 2: suspension d'essai
Fioles 3 et 4: blanc inoculum
Fiole 5: contrôle de la procédure de préférence,
et si nécessaire:
Fiole 6: témoin stérile
Fiole 7: témoin de toxicité
Voir également I.6.7.
V.2.6. Déroulement de l'essai
Laisser les récipients atteindre la température souhaitée. Ensemencer les récipients appropriés avec les boues activées préparées au préalable ou avec un inoculum provenant d'une autre source, de telle sorte que la concentration des matières solides en suspension ne dépasse pas 30 mg/l. Assembler le matériel, mettre l'agitateur en marche, vérifier l'étanchéité à l'air et commencer à mesurer la consommation d'oxygène. Le déroulement de l'essai ne nécessite normalement aucune attention particulière en dehors des lectures et des vérifications quotidiennes permettant de s'assurer que la température et l'agitation appropriées sont maintenues.
Calculer la consommation d'oxygène à partir des lectures effectuées à intervalles réguliers et rapprochés en suivant la méthode fournie par le fabriquant du matériel utilisé. A la fin de l'incubation, qui est normalement de 28 jours, mesurer le pH dans les fioles, en particulier si la consommation d'oxygène est faible ou supérieure à la DThONH4 (pour les composés azotés).
Prélever, si nécessaire, des échantillons dans les fioles du respiromètre, au début et à la fin de l'essai, pour analyser le COD ou effectuer un dosage spécifique (voir annexe II.4.). Lors du premier prélèvement, noter le volume de suspension d'essai restant dans la fiole. Si la substance à étudier contient de l'azote, déterminer l'augmentation de la concentration de nitrite et de nitrate au cours de ces 28 jours et calculer la correction relative à la consommation d'oxygène due à la nitrification (annexe V).
V.3. RÉSULTATS ET RAPPORT
V.3.1. Traitement des résultats
Diviser la consommation d'oxygène (mg) de la substance à étudier après une période donnée (après correction due à l'oxygène consommé dans l'essai à blanc contenant l'inoculum pendant une période identique) par le poids de substance utilisé. Le résultat, exprimé en mg d'oxygène par mg de substance, correspond à la DBO, à savoir:

= mg d'O2 par mg de produit à étudier
Calculer le pourcentage de biodégradation, soit à partir de la formule suivante:

soit à l'aide de celle-ci:
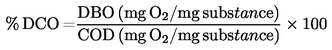
Il faut noter que ces deux méthodes n'aboutissant pas nécessairement au même résultat, il est préférable d'utiliser la première.
Pour les substances à étudier azotées, choisir la DThO appropriée (NH4 ou NO3) en fonction de ce que l'on sait ou suppose quant à la nitrification (annexe II.2.). Si la nitrification a lieu mais de manière incomplète, calculer la correction pour l'oxygène consommé par la nitrification à partir des modifications des concentrations de nitrite et de nitrate (annexe V).
Lorsque les mesures facultatives du carbone organique et/ou d'un produit spécifique sont effectuées, calculer le pourcentage de dégradation, comme décrit au paragraphe 1.7.
Consigner tous les résultats sur les fiches de données fournies.
V.3.2. Validité des résultats
La consommation d'oxygène dans l'essai à blanc contenant l'inoculum est normalement comprise entre 20 et 30 mg/l d'O2 et ne doit pas dépasser 60 mg/l en 28 jours. Des valeurs supérieures à 60 mg/l nécessitent un examen critique des données et des méthodes expérimentales. Si la valeur du pH se situe en dehors de l'intervalle compris entre 6 et 8,5 et si la consommation d'oxygène par le produit à étudier est inférieure à 60 %, l'essai doit être recommencé avec une concentration en produit à étudier plus faible.
Voir également 1.5.2.
V.3.3. Rapport
Voir 1.8.
V.4. FICHE DE DONNÉES
Un exemple de fiche de données est présenté ci-après.
ESSAI DE RESPIROMÉTRIE MANOMÉTRIQUE
|
2.
|
DATE DU DÉBUT DE L'ESSAI
|
3. SUBSTANCE À ÉTUDIER
Nom:
Concentration de la solution mère: … mg/l
Concentration initiale dans le milieu, Co: … mg/l
Volume dans la fiole d'essai (V): … ml
DThO/DCO: … mg O2/mg de produit à étudier (NH4, NO3)
4. INOCULUM
Origine: …
Traitement: …
Préconditionnement éventuel: …
Concentration des matières solides en suspension dans le mélange réactionnel: … mg/l
5. CONSOMMATION D'OXYGÈNE: BIODÉGRADABILITÉ
|
|
Temps (jours)
|
|
0
|
|
7
|
|
14
|
|
|
21
|
|
|
28
|
|
|
Cons. O2 (mg) du produit à étudier
|
1
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a, moy.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cons. O2 (mg) du blanc
|
3
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b, moy.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DBO corr. (mg)
|
(a1 — bm)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(a2 — bm)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DBO par mg de substance d'essai
|

|
|
|
|
|
|
|
|
|
|
|
|
|
|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
% de dégrad.

|
D1 (a1)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D2 (a2)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Moyenne (6)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V = volume de milieu dans la fiole d'essai
|
Note: Des tableaux de ce type peuvent être utilisés pour le produit de référence et les témoins de toxicité.
6. CORRECTION TENANT COMPTE DE LA NITRIFICATION (voir annexe V)
|
Jour
|
0
|
28
|
Différence
|
|
i)
|
Concentration de nitrate (mg N/l)
|
|
|
|
(N)
|
|
ii)
|
Équivalent oxygène (4,57 × N × V) (mg)
|
|
—
|
—
|
|
|
iii)
|
Concentration de nitrite (mg N/l)
|
|
|
|
(N)
|
|
(iv)
|
Équivalent oxygène (3,43 × N × V) (mg)
|
|
—
|
—
|
|
|
(ii + iv)
|
Total équivalent oxygène
|
|
—
|
—
|
|
7. ANALYSE DU CARBONE (facultative)
Analyseur de carbone: ...
|
Temps (jour)
|
Blanc mg/l
|
Produit à étudier mg/l
|
|
0
|
(Cblo)
|
(Co)
|
|
28 (7)
|
(Cblt)
|
(Ct)
|

8. ANALYSE SPÉCIFIQUE (facultative)
|
Sb
|
=
|
concentration dans le témoin physico-chimique (stérile), le 28e jour.
|
|
Sa
|
=
|
concentration dans la fiole ensemencée, le 28e jour.
|

9. DÉGRADATION ABIOTIQUE (facultative)
|
a
|
=
|
consommation d'oxygène dans les fioles stériles au bout de 28 jours (mg).
|

(Voir paragraphe 1 et 3)
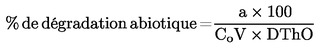
PARTIE VI. ESSAI EN FIOLES FERMÉES (Méthode C.4-E)
VI.1. PRINCIPE DE LA MÉTHODE
La solution de substance à étudier en milieu minéral, généralement d'une concentration de 2 à 5 mg/l, est ensemencée avec un nombre relativement faible de micro-organismes provenant d'une population hétérogène et conservée dans des fioles fermées et remplies complètement dans l'obscurité et à température constante. La dégradation est suivie par l'analyse de l'oxygène dissous pendant une période de 28 jours. La quantité d'oxygène consommé par la substance à étudier, corrigée en fonction de la consommation dans l'essai à blanc contenant l'inoculum réalisé dans la même série, est exprimée en pourcentage de DThO ou de DCO.
VI.2. DESCRIPTION DE LA MÉTHODE
VI.2.1. Appareillage
|
a)
|
des flacons pour analyse de la DBO, munis de bouchons de verre, d'un volume de 250 à 300 ml,
|
|
b)
|
un bain-marie ou un incubateur permettant de maintenir les fioles à température constante (± 1 oC au maximum) à l'abri de la lumière,
|
|
c)
|
de grands flacons de verre (2 à 5 l) pour la préparation des milieux et le remplissage des flacons à DBO,
|
|
d)
|
une électrode à oxygène couplée à un appareil de mesure, ou l'équipement et les réactifs nécessaires à la méthode de titration de Winkler.
|
VI.2.2. Préparation du milieu minéral
Pour la préparation des solutions mères, voir I.6.2.
Mélanger 1 ml des solutions a) à d) et compléter le volume à un litre avec l'eau de dilution.
VI.2.3. Préparation de l'inoculum
Normalement, l'inoculum provient de l'effluent secondaire d'une station d'épuration ou d'une unité pilote qui reçoit principalement des eaux ménagères. L'eau de surface peut constituer une autre source d'inoculum. Utiliser normalement une goutte (0,05 ml) à 5 ml de filtrat par litre de milieu; il peut être nécessaire de procéder à des essais afin de déterminer le volume optimal pour un effluent donné (voir I.6.4.2. et I.6.5.)
VI.2.4. Préparation des fioles
Aérer fortement le milieu minéral pendant au moins 20 mn. Chaque série d'essais doit être effectuée avec du milieu minéral provenant d'un lot unique. Le milieu est généralement prêt à l'emploi après être resté pendant 20 heures à la température de l'essai. Déterminer la concentration d'oxygène dissous à des fins de contrôle; la valeur obtenue doit être proche de 9 mg/l à 20 oC. Effectuer toutes les opérations de transfert et de remplissage avec le milieu saturé d'air en évitant la formation de bulles, par exemple à l'aide de siphons.
Préparer des lots parallèles de flacons à DBO pour les déterminations avec la substance d'essai et la substance de référence dans des séries expérimentales simultanées. Réunir un nombre suffisant de flacons à DBO, comprenant des essais à blanc avec l'inoculum, pour pouvoir effectuer, au moins en double, les mesures de la consommation d'oxygène aux intervalles de temps souhaités, par exemple après 0, 7, 14, 21 et 28 jours. Un plus grand nombre de flacons peut être nécessaire pour être sûr de pouvoir identifier l'intervalle de temps de 10 jours.
Transférer le milieu minéral parfaitement aéré dans les grands flacons de manière à remplir à peu près le tiers de leur volume. Ajouter ensuite dans de grands flacons distincts une quantité de solution mère du produit à étudier ou du produit de référence telle que la concentration finale ne dépasse pas, normalement, 10 mg/l. N'ajouter aucune substance dans le grand flacon destiné à l'essai à blanc.
Pour s'assurer que l'activité de l'inoculum n'est pas limitée, la concentration d'oxygène dissous ne doit pas descendre en-dessous de 0,5 mg/l dans les flacons à DBO. La concentration de produit à étudier se trouve ainsi limitée à environ 2 mg/l. Toutefois, on peut utiliser une concentration comprise entre 5 et 10 mg/l pour les composés peu dégradables et les composés dont la DThO est faible. Dans certains cas, il est préférable d'effectuer des séries d'essais en parallèle avec deux concentrations différentes, par exemple 2 et 5 mg/l. La DThO est normalement calculée à partir de la formation de sels d'ammonium, mais si l'on suppose ou si l'on est sûr qu'une nitrification doit avoir lieu, il convient alors de calculer la DThONO3 en se basant sur la formation de nitrate (voir annexe II.2). Toutefois, si une nitrification se produit mais sans être complète, il faut effectuer une correction tenant compte des modifications de la concentration de nitrite et de nitrate déterminées par l'analyse (voir annexe V).
Si la toxicité du produit à étudier doit être recherchée (dans le cas, par exemple, où une faible biodégradabilité aurait été observée au préalable), une autre série de flacons est nécessaire.
Préparer un autre grand flacon contenant du milieu minéral aéré (jusqu'au tiers de son volume environ) auquel on ajoute la substance à étudier et le produit de référence à des concentrations finales normalement égales à celles des autres grands flacons.
Ensemencer les solutions contenues dans les grands flacons avec un effluent secondaire (dont le volume est compris entre une goutte, soit environ 0,05 ml, et 5 ml) ou avec un inoculum d'une autre origine, comme de l'eau de rivière (voir I.6.4.2.). Enfin, compléter le volume des solutions avec du milieu minéral aéré à l'aide d'un tuyau touchant le fond du flacon pour permettre un mélange adéquat.
VI.2.5. Nombre de fioles d'une série type
Une série type comporte les flacons suivants:
|
—
|
au moins 10 flacons contenant la substance à étudier et l'inoculum (suspension d'essai),
|
|
—
|
au moins 10 flacons ne contenant que l'inoculum (essai à blanc avec l'inoculum),
|
|
—
|
au moins 10 flacons contenant le produit de référence et l'inoculum (contrôle de la procédure),
|
|
—
|
et, si nécessaire, 6 flacons contenant la substance à étudier, le produit de référence et l'inoculum (contrôle de toxicité). Néanmoins, pour être certain de pouvoir identifier l'intervalle de temps de 10 jours, il faudrait environ deux fois plus de flacons.
|
VI.2.6. Déroulement de l'essai
Toutes les solutions préparées sont immédiatement transférées dans le lot correspondant de flacons à DBO à l'aide d'un tuyau plongeant aux trois quarts (pas jusqu'au fond) du grand flacon approprié de telle sorte que tous les flacons à DBO soient complètement remplis. Tapoter doucement pour éliminer toutes les bulles d'air. L'oxygène dissous est analysé immédiatement dans les flacons destinés à la mesure au temps 0, par la méthode de Winkler ou avec une électrode. Le contenu des flacons peut être conservé en vue d'une analyse ultérieure par la méthode de Winkler si on lui ajoute du sulfate de manganèse (II) et de l'hydroxyde de sodium (le premier réactif de la méthode de Winkler). Les flacons dont le contenu en oxygène est fixé sous forme d'hydroxyde de manganèse (III) brun, sont conservés soigneusement fermés à l'obscurité, et à une température comprise entre 10 et 20 oC. Ne pas attendre plus de 24 heures avant de procéder aux étapes suivantes de la méthode de Winkler. Fermer les autres flacons préparés parallèlement en s'assurant qu'ils ne contiennent pas de bulles d'air et les incuber à 20 oC a l'obscurité. Chaque série doit être accompagnée d'une série complète d'essais à blanc ensemencés pour la détermination du témoin inoculum. Prélever régulièrement (au moins chaque semaine) deux flacons au moins dans chaque série, afin d'analyser l'oxygène dissous pendant la durée de l'incubation qui est de 28 jours.
Les prélèvements hebdomadaires d'échantillons permettent de déterminer le pourcentage de disparition dans un intervalle de 14 jours, alors que les prélèvements effectués tous les 3 ou 4 jours, qui nécessitent environ deux fois plus de flacons, permettent d'identifier l'intervalle de temps de 10 jours.
Si la substance à étudier contient de l'azote, il convient d'effectuer une correction pour tenir compte de la consommation d'oxygène due à la nitrification. Pour cela, déterminer la concentration d'oxygène dissous par la méthode de l'électrode à O2 et, ensuite, analyser la teneur en nitrite et en nitrate sur un échantillon prélevé dans le flacon DBO. Calculer la quantité d'oxygène utilisée sur la base de l'augmentation de la concentration de nitrite et de nitrate (voir annexe V).
VI.3. RÉSULTATS ET RAPPORT
VI.3.1. Traitement des résultats
En premier lieu, calculer la DBO après chaque période de temps en soustrayant la perte en oxygène (mg O2/l) dans l'essai à blanc contenant l'inoculum de celle que l'on observe avec la substance à étudier. Diviser cette perte en oxygène ainsi corrigée par la concentration (mg/l) de la substance à étudier. Le résultat obtenu correspond à la DBO spécifique exprimée en mg d'oxygène par mg de substance d'essai. Calculer le pourcentage de biodégradabilité en divisant la DBO spécifique par la DThO spécifique (calculée selon les indications données à l'annexe II.2) ou par la DCO (déterminée par analyse, voir annexe II.3).
Ainsi:
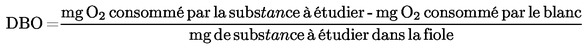
= mg d'O2 par mg de produit à étudier


Il faut noter que ces deux méthodes ne conduisant pas nécessairement au même résultat, il est préférable d'utiliser la première.
Dans le cas où la substance d'essai contient de l'azote, il convient d'utiliser la DThO appropriée (NH4 ou NO3) en fonction des connaissances ou des prévisions concernant l'éventualité de la nitrification (annexe II.2.). Si une nitrification a lieu sans toutefois être complète, corriger pour tenir compte de la consommation d'oxygène due à la nitrification à partir des modifications de la concentration de nitrite et de nitrate (annexe V).
VI.3.2. Validité des résultats
La perte d'oxygène dans l'essai à blanc contenant l'inoculum ne doit pas dépasser 1,5 mg/l d'oxygène dissous après 28 jours. Des valeurs supérieures nécessitent une vérification des méthodes expérimentales. La concentration résiduelle d'oxygène dans les flacons d'essai ne doit à aucun moment descendre au-dessous de 0,5 mg/l. Une teneur en oxygène aussi basse n'est acceptable que si la méthode utilisée pour mesurer l'oxygène dissous est capable de mesurer de telles concentrations avec précision.
Voir également I.5.2.
VI.3.3. Rapport
Voir 1.8.
VI.4. FICHE DE DONNÉES
Un exemple de fiche de données est présenté ci-dessous.
ESSAI EN FIOLES FERMÉES
|
2.
|
DATE DU DÉBUT DE L'ESSAI
|
3. SUBSTANCE À ÉTUDIER
Nom:
Concentration de la solution mère: ... mg/l
Concentration initiale dans la fiole: ... mg/l
DThO/DCO: ... mg O2/mg de produit à étudier
4. INOCULUM
Origine: ...
Traitement: ...
Préconditionnement éventuel: ...
Concentration dans le mélange réactionnel: ... mg/l
5. DÉTERMINATION DE L'OD
Méthode de Winkler ou de l'électrode.
Analyses des fioles
|
Temps d'incubation (d)
|
OD (mg/l)
|
|
0
|
n1
|
n2
|
|
|
Blanc (sans produit)
|
1
|
C1
|
|
|
|
|
|
2
|
C2
|
|
|
|
|
|
Moyenne
|

|
|
|
|
|
|
Produit à étudier
|
1
|
a1
|
|
|
|
|
|
|
2
|
a2
|
|
|
|
|
|
Moyenne
|

|
|
|
|
|
Note: Des tableaux de ce type peuvent être utilisés pour le produit de référence et le témoin de toxicité.
6. CORRECTION TENANT COMPTE DE LA NITRIFICATION (annexe V)
|
Temps d'incubation (d)
|
0
|
n1
|
n2
|
n3
|
|
(i)
|
Concentration de nitrate (mg N/l)
|
|
|
|
|
|
|
(ii)
|
Modif. de la conc. de nitrate (mg N/l)
|
|
—
|
|
|
|
|
(iii)
|
Équivalent oxygène (mg/l)
|
|
—
|
|
|
|
|
(iv)
|
Concentration de nitrite (mg N/l)
|
|
|
|
|
|
|
(v)
|
Modif. de la conc. de nitrite (mg N/l)
|
|
—
|
|
|
|
|
(vi)
|
Équivalent oxygène (mg/l)
|
|
—
|
|
|
|
|
(iii + vi)
|
Total équivalent oxygène (mg/l)
|
|
—
|
|
|
|
7. PERTE D'OD: % DE DÉGRADATION
|
|
Perte après n jours (mg/l)
|
|
n1
|
n2
|
n3
|
|
|
FIOLE 1: (mto — mtx) — (mbo — mbx)
|
|
|
|
|
|
FIOLE 2: (mto — mtx) — (mbo — mbx)
|
|
|
|
|
|
FIOLE 1:

|
|
|
|
|
|
FIOLE 2:

|
|
|
|
|
| % D moyen (8) =

|
|
|
|
|
|
mto
|
=
|
valeur dans la fiole d'essai au temps 0
|
|
mtx
|
=
|
valeur dans la fiole d'essai au temps x
|
|
mbo
|
=
|
valeur moyenne du blanc au temps 0
|
|
mbx
|
=
|
valeur moyenne du blanc au temps x
|
Appliquer également la correction tenant compte de la nitrification mentionnée aux points (iii) et (vi) du paragraphe 6.
8. PERTE D'OD DANS L'ESSAI À BLANC
Consommation d'oxygène dans l'essai à blanc: (mbo — mb28) mg/l. Cette consommation est importante pour la validité de l'essai. Elle doit être inférieure à 1,5 mg/l.
PARTIE VII. ESSAI M.I.T.I. (Méthode C.4-F)
VII.1. PRINCIPE DE LA MÉTHODE
La consommation d'oxygène d'une solution ou d'une suspension agitée de substance à étudier dans un milieu minéral, ensemencée avec des micro-organismes cultivés à cet effet sans adaptation préalable, est établie à l'aide d'un respiromètre automatique en circuit fermé, à l'obscurité, pendant une période de 28 jours à 25 ± 1 oC. Le dioxyde de carbone dégagé est absorbé par de la chaux sodée. La biodégradabilité est exprimée par le pourcentage de la consommation d'oxygène (corrigé en fonction de la consommation dans l'essai à blanc) par rapport à la consommation théorique (DThO). Le pourcentage de biodégradabilité primaire est également calculé d'après les analyses chimiques spécifiques supplémentaires effectuées au début et à la fin de l'incubation et, éventuellement, à l'aide de l'analyse du COD.
VII.2. DESCRIPTION DE LA MÉTHODE
VII.2.1. Appareillage
|
a)
|
appareil de mesure automatique de la DBO par électrolyse ou respiromètre normalement équipé de 6 fioles d'une contenance de 300 ml chacune et de récipients contenant l'absorbant de CO2;
|
|
b)
|
pièce thermostatée et/ou bain-marie à 25 oC (± 1 oC au maximum);
|
|
c)
|
dispositif de filtration sur membrane (facultatif);
|
|
d)
|
analyseur de carbone (facultatif).
|
VII.2.2. Préparation du milieu minéral
Préparer les solutions mères suivantes, à l'aide de réactifs de qualité analytique et d'eau (1.6.1.):
|
a)
|
Dihydrogénophosphate de potassium, KH2PO4
|
8,50 g
|
|
Monohydrogénophosphate de potassium, K2HPO4
|
21,75 g
|
|
Monohydrogénophosphate de sodium dodécahydraté, Na2HPO4.12 H2O
|
44,60 g
|
|
Chlorure d'ammonium, NH4Cl
|
1,70 g
|
|
Dissoudre dans de l'eau et compléter le volume à 1 litre
|
|
|
Le pH de la solution doit être égal à 7,2
|
|
|
b)
|
Sulfate de magnésium heptahydraté, MgSO4 7 H2O
|
22,50 g
|
|
Dissoudre dans de l'eau et compléter le volume à 1 litre
|
|
|
c)
|
Chlorure de calcium, anhydre CaCl2
|
27,50 g
|
|
Dissoudre dans de l'eau et compléter le volume à 1 litre
|
|
|
d)
|
Chlorure de fer (III) hexahydraté, FeCl3.6 H2O
|
0,25 g
|
|
Dissoudre dans de l'eau et compléter le volume à 1 litre
|
|
Prélever 3 ml de chacune des solutions a), b) c) et d) et compléter le volume à 1 litre.
VII.2.3. Préparation de l'inoculum
Recueillir des échantillons frais dans 10 emplacements au moins, principalement dans des zones où divers produits chimiques sont utilisés et rejetés dans l'environnement. Recueillir des échantillons d'un litre de boue, de sols superficiels, d'eau, etc., dans des sites tels que les stations d'épuration des eaux d'égout, les stations d'épuration industrielle, les rivières, les lacs et les mers et bien les mélanger. Après avoir enlevé les matières flottantes, laisser reposer le mélange, puis ajuster le pH du surnageant à 7 ± 1 au moyen d'hydroxyde de sodium ou d'acide phosphorique.
Utiliser un volume de surnageant filtré suffisant pour remplir un récipient de traitement des boues activées et aérer le liquide pendant environ 23 heures 30. Trente minutes après avoir interrompu l'aération, retirer environ un tiers du volume total du liquide surnageant et ajouter à la partie décantée un volume égal d'une solution (pH 7) contenant 0,1 % de chacun des produits suivants: glucose, peptone, monophosphate de potassium, puis aérer à nouveau le mélange. Répéter cette opération chaque jour. La réserve de boue doit être préparée selon les bonnes pratiques: l'effluent doit être clair, la température doit être maintenue à 25 ± 2 oC, son pH égal à 7 ± 1, la boue doit bien décanter, l'aération doit être suffisante pour que le mélange soit en permanence en aérobiose, des protozoaires doivent être présents et l'activité de la boue doit être testée à l'aide d'une substance de référence au moins tous les trois mois. Ne pas utiliser la boue comme inoculum avant au moins un mois après sa préparation ni après plus de quatre mois. Il convient donc d'effectuer périodiquement des prélèvements d'échantillons, tous les trois mois, en 10 emplacements différents au moins.
Afin de conserver la même activité aux boues fraîches et anciennes, le surnageant filtré d'une boue activée utilisée pour l'essai doit être mélangé en volume égal au surnageant filtré d'une boue fraîchement recueillie provenant de dix sources différentes. Le mélange ainsi obtenu est mis en culture comme décrit ci-avant. Prélever la boue pour l'utiliser comme inoculum 18 à 24 heures après cette opération.
VII.2.4. Préparation des fioles
Préparer les six fioles suivantes:
No 1: eau de dilution contenant 100 mg/l de substance à étudier,
Nos 2, 3 et 4: milieu minéral contenant 100 mg/l de substance à étudier,
No 5: milieu minéral contenant 100 mg/l de produit de référence (par exemple de l'aniline),
No 6: milieu minéral seul.
Si la substance à étudier est peu soluble, il convient d'en ajouter directement un poids ou un volume défini, ou de procéder selon les instructions figurant à l'annexe III, en n'utilisant toutefois ni solvant ni émulsifiant. Placer l'absorbant de CO2 pour chaque fiole, dans la coupelle spécialement prévue à cet effet. Ajuster à 7,0 le pH de la solution contenue dans les fioles nos 2, 3 et 4.
VII.2.5. Déroulement de l'essai
Ensemencer les fioles nos 2, 3 et 4 (suspensions d'essai), no 5 (témoin d'activité) et no 6 (blanc contenant l'inoculum) avec un petit volume d'inoculum de manière à ce que la concentration des matières solides en suspension soit égale à 30 mg/l. Ne pas ajouter d'inoculum dans la fiole no 1 qui sert de témoin abiotique. Assembler le matériel, vérifier l'étanchéité à l'air, brancher les agitateurs et commencer à mesurer la consommation d'oxygène dans l'obscurité. Vérifier chaque jour la température et le fonctionnement des agitateurs et de l'enregistreur de l'appareil de mesure coulométrique de la consommation d'oxygène et noter toute modification de couleur du milieu contenu dans les fioles. Lire la consommation d'oxygène dans les six fioles directement avec une méthode appropriée, par exemple sur l'enregistreur à six points, qui fournit une courbe de la DBO. À la fin de l'incubation, dont la durée normale est de 28 jours, mesurer le pH du contenu des fioles et déterminer la concentration résiduelle de la substance à étudier et de toute substance intermédiaire et dans le cas d'une substance hydrosoluble, la concentration de COD (annexe II.4). Des précautions particulières doivent être prises pour étudier les produits volatiles. Si un processus de nitrification est prévisible, déterminer, si possible, la concentration en nitrates et nitrites.
VII.3. PRÉSENTATION DES DONNÉES
VII.3.1. Traitement des résultats
La quantité d'oxygène consommée (mg) par la substance d'essai après un temps donné, corrigée de façon à tenir compte de la quantité d'O2 consommée dans l'essai à blanc contenant l'inoculum après la même période de temps, est divisée par le poids de substance à étudier utilisé pour l'essai. Le résultat ainsi obtenu correspond à la DBO exprimée en mg d'oxygène/mg de substance d'essai, c'est-à-dire:

= mg d'O2/mg de substance d'essai
Le pourcentage de biodégradation est alors obtenu à l'aide de la formule suivante:
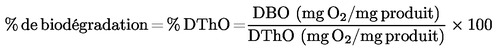
Pour les mélanges, calculer la DThO à partir de l'analyse élémentaire, comme pour un composé simple. Choisir la DThO appropriée (DThONH4 ou DThONO3) selon que la nitrification est absente ou complète (annexe II.2). Cependant, si la nitrification se produit mais sans toutefois être complète, il convient d'effectuer une correction correspondant à la quantité d'oxygène consommée par la nitrification à partir des modifications de la concentration en nitrites et en nitrates (annexe V).
Calculer le pourcentage de biodégradation primaire à partir de la perte de produit chimique spécifique (d'origine) (voir 1.7.2).
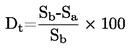
Si la mesure de la disparition physico-chimique révèle une perte de produit d'essai dans la fiole no 1, il convient de le consigner et de calculer le pourcentage de biodégradation en utilisant la concentration de substance d'essai (Sb) dans la fiole au bout de 28 jours.
Si la concentration de COD est déterminée (facultatif), calculer le pourcentage de biodégradation ultime à l'aide de la formule suivante:

décrite au paragraphe I.7.1. Si la mesure de la disparition physico-chimique révèle une perte de COD dans la fiole no 1, il convient de calculer le pourcentage de dégradation en utilisant la concentration de COD dans cette fiole.
Consigner tous les résultats sur les fiches de données fournies.
VII.3.2. Validité des résultats
La consommation d'oxygène dans l'essai à blanc contenant l'inoculum est normalement comprise entre 20 et 30 mg O2/l et ne doit pas excéder 60 mg/l en 28 jours. Des valeurs supérieures à 60 mg/l nécessitent un examen critique des données et des méthodes expérimentales. Si la valeur du pH se trouve en dehors de l'intervalle compris entre 6 et 8,5 et si la consommation d'oxygène par la substance d'essai est inférieure à 60 %, l'essai doit être répété avec une concentration plus faible de substance à étudier.
Voir également I.5.2.
Si le pourcentage de dégradation de l'aniline calculé à partir de la consommation d'oxygène ne dépasse pas 40 % après 7 jours et 65 % après 14 jours, l'essai est considéré comme non valable.
VII. 3.3. Rapport
Voir I.8.
VII.4. FICHE DE DONNÉES
Un exemple de fiche de données est présenté ci-dessous.
ESSAI MITI (I)
|
2.
|
DATE DU DÉBUT DE L'ESSAI
|
3. SUBSTANCE À ÉTUDIER
Nom:
Concentration de la solution mère: ... mg/l de produit
Conc. initiale dans le milieu, Co: ... mg/l de produit
Volume du mélange réactionnel, V: ... ml
DThO: ... mg O2/l
4. INOCULUM
Emplacements d'échantillonnage des boues:
Concentration des matières en suspension dans la boue activée après acclimatation à l'aide d'une eau d'égout synthétique = mg/l
Volume de boue activée par litre de milieu final = ml
Concentration de boue dans le milieu final = mg/l
5. CONSOMMATION D'OXYGÈNE: BIODÉGRADABILITÉ
Type de respiromètre utilisé:
|
|
Temps (jours)
|
|
0
|
7
|
14
|
21
|
28
|
|
Cons. d'O2 (mg) du produit
|
a1
|
|
|
|
|
|
|
a2
|
|
|
|
|
|
|
a3
|
|
|
|
|
|
|
Cons. d'O2 (mg) du blanc
|
b
|
|
|
|
|
|
|
Cons. d'O2 corrigée (mg)
|
(a1 - b)
(a2 - b)
(a3 - b)
|
|
|
|
|
|
|
DBO par mg de produit
|

|
Fiole 1
|
|
|
|
|
|
|
Fiole 2
|
|
|
|
|
|
|
Fiole 3
|
|
|
|
|
|
|
% dégradation

|
|
1
|
|
|
|
|
|
|
2
|
|
|
|
|
|
|
3
|
|
|
|
|
|
|
moy (9)
|
|
|
|
|
|
Note: Des tableaux de ce type peuvent être utilisés pour le produit de référence et les témoins de toxicité.
6. ANALYSE DE CARBONE (facultative)
Analyseur de carbone: ...
|
Fiole
|
COD
|
% COD éliminé
|
Moy.
|
|
Mesuré
|
Corrigé
|
|
Eau + subst. d'essai
|
a
|
|
|
|
—
|
—
|
|
Boue + subst. d'essai
|
b1
|
|
b1 — c
|
|
|
|
|
Boue + subst. d'essai
|
b2
|
|
b2 — c
|
|
|
|
|
Boue + subst. d'essai
|
b3
|
|
b3 — c
|
|
|
|
|
Essai à blanc
|
c
|
|
—
|
|
—
|
—
|

7. DONNÉES DE L'ANALYSE SPÉCIFIQUE CHIMIQUE
|
|
Quantité résiduelle de produit à la fin de l'essai
|
% de dégradation
|
|
essai à blanc avec de l'eau
|
Sb
|
|
|
milieu inoculé
|
Sal
|
|
|
Sa2
|
|
|
Sa3
|
|
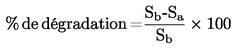
Calculer le % de dégradation pour les fioles a1 a2 et a3 respectivement.
8. OBSERVATIONS
La courbe de la DBO en fonction du temps doit, le cas échéant, être jointe.
Annexe 1
ABRÉVIATIONS ET DÉFINITIONS
|
OD
|
:
|
oxygène dissous (mg/l), c'est-à-dire la concentration d'oxygène dissous dans un échantillon aqueux.
|
|
DBO
|
:
|
demande biochimique en oxygène (g), soit la quantité d'oxygène consommée par des micro-organismes au moment où ils métabolisent un produit soumis à l'essai; la DBO est également exprimée en g d'oxygène consommé par g de produit à étudier (voir la méthode C.5).
|
|
DCO
|
:
|
demande chimique d'oxygène (g), c'est-à-dire la quantité d'oxygène consommée au cours de l'oxydation d'un produit à étudier en présence de bichromate acide chaud; la DCO fournit une mesure de la quantité de matière oxydable présente; la DCO est également exprimée en g d'oxygène consommé par g de substance à étudier (voir méthode C.6).
|
|
COD
|
:
|
carbone organique dissous. Il s'agit du carbone organique présent dans une solution ou qui traverse un filtre d'une porosité de 0,45 micromètre ou encore qui reste dans le surnageant après une centrifugation de 15 min à 40 000 m.s-2 (±4 000 g).
|
|
DThO
|
:
|
demande théorique en oxygène (mg), c'est-à-dire la quantité totale d'oxygène nécessaire pour parvenir à l'oxydation complète d'un produit chimique. Elle est calculée à partir de la formule moléculaire (voir annexe II.2). La DThO est également exprimée en mg d'oxygène nécessaire par mg de produit à étudier.
|
|
CO2Th
|
:
|
dioxyde de carbone théorique (mg). Il s'agit de la quantité calculée de dioxyde de carbone pouvant être produite à partir du contenu en carbone mesuré ou connu dans le produit à étudier, lors de sa minéralisation totale. Le CO2Th est également exprimé en mg de dioxyde de carbone qui se dégage par mg de produit à étudier.
|
|
COT
|
:
|
carbone organique total. Le COT d'un échantillon est égal à la somme du carbone organique en solution et en suspension.
|
|
CI
|
:
|
carbone inorganique.
|
|
CT
|
:
|
carbone total. Le CT est égal à la somme du carbone organique et du carbone inorganique présents dans l'échantillon.
|
Biodégradation primaire:
Altération de la structure chimique d'une substance obtenue par une action biologique et entraînant la perte de propriétés spécifiques de cette substance.
Biodégradation ultime (en aérobiose):
Taux de dégradation atteint lorsque la totalité de la substance à étudier a été utilisée par des micro-organismes pour produire du dioxyde de carbone, de l'eau, des sels minéraux et de nouveaux constituants cellulaires microbiens (biomasse).
Facilement biodégradable:
Il s'agit d'un classement arbitraire des produits chimiques qui ont répondu positivement à des essais portant sur la biodégradabilité ultime; du fait de la rigueur de ces essais, on suppose que ces composés se dégradent rapidement et complètement en milieu aquatique dans des conditions d'aérobiose.
Intrinsèquement biodégradable:
Il s'agit d'une classification des produits chimiques donc la biodégradation (primaire ou ultime) a été prouvée sans ambiguïté au cours d'un essai de biodégradabilité reconnu internationalement.
Traitabilité:
Aptitude des composés à disparaître au cours du traitement biologique des eaux résiduaires sans effet indésirable sur le déroulement normal du traitement. D'une manière générale, les composés facilement biodégradables sont traitables, ce qui n'est pas le cas de tous les composés intrinsèquement biodégradables. Des processus abiotiques peuvent également se produire.
Phase de latence
correspond à la période qui, au cours d'un essai de disparition, sépare le moment de l'ensemencement de celui où le pourcentage de dégradation a augmenté d'au moins 10 %. La durée de la phase de latence est souvent extrêmement variable et peu reproductible.
Phase de dégradation
correspond à la période qui commence à la fin de la phase de latence et se termine au moment où 90 % du taux maximal de dégradation a été atteint.
Intervalle de 10 jours
Il s'agit des 10 jours immédiatement consécutifs au moment où le taux de dégradation atteint 10 %.
Annexe II
CALCUL ET DÉTERMINATION DE PARAMÈTRES APPROPRIÉS
Certains paramètres seront requis en fonction de la méthode choisie. Le chapitre suivant décrit le calcul de ces valeurs. L'utilisation de ces paramètres est décrite dans les chapitres consacrés à chaque méthode.
1. Teneur en carbone
La teneur en carbone est calculée à partir de la composition élémentaire connue de la substance à étudier, ou déterminée par l'analyse élémentaire de cette substance.
2. Demande théorique en oxygène (DThO)
La demande théorique en oxygène (DThO) peut être calculée si la composition élémentaire est connue ou déterminée par analyse élémentaire. Par exemple la DThO du composé suivant:
CcHhClclNnNanaOoPpSs
sera, en l'absence de nitrification, égale à:
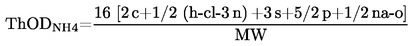 mg/mg
mg/mg
et si une nitrification a lieu, elle sera égale à:
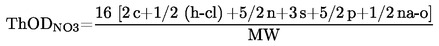 mg/mg
mg/mg
3. Demande chimique en oxygène (DCO)
La demande chimique en oxygène (DCO) est déterminée à l'aide de la méthode C.6.
4. Carbone organique dissous (COD)
Le carbone organique dissous (COD) est, par définition, le carbone organique présent dans la solution aqueuse d'un produit chimique ou d'un mélange, qui traverse un filtre d'une porosité de 0,45 micromètre.
Prélever des échantillons dans les récipients d'essai et les filtrer immédiatement à l'aide d'un dispositif de filtration en utilisant une membrane filtrante appropriée. Les 20 premiers ml de filtrat (la quantité peut être réduite si on utilise des filtres de petite taille) sont éliminés. Un volume de filtrat compris entre 10 et 20 ml, ou moins s'il doit être injecté (le volume dépend de la quantité requise pour l'analyseur de carbone), est retenu afin d'en analyser la teneur en carbone. La concentration en COD est déterminée à l'aide d'un analyseur de carbone organique capable de mesurer avec précision une concentration de carbone égale ou inférieure à 10 % de la concentration initiale de COD utilisée pour l'essai.
Les échantillons filtrés qui ne sont pas analysés le jour même peuvent être conservés pendant 48 heures dans un réfrigérateur entre 2 et 4 oC ou en-dessous de — 18 oC pour des périodes plus longues.
Remarque:
Les membranes filtrantes étant souvent imprégnées d'agents tensio-actifs pour l'hydrophilisation, sont susceptibles de contenir plusieurs milligrammes de carbone organique soluble qui interfèrent avec les déterminations de la biodégradabilité. Pour débarrasser les filtres des agents tensio-actifs ainsi que d'autres composés organiques solubles, il convient de les faire bouillir trois fois pendant une heure dans de l'eau déionisée. Ils peuvent ensuite être conservés dans de l'eau pendant une semaine. Si l'on utilise des cartouches filtrantes à usage unique, chaque lot doit être vérifié afin de s'assurer qu'il ne libère pas de carbone organique soluble.
La substance à étudier peut être retenue par adsorption sur certains types de membranes filtrantes. C'est pourquoi il est conseillé de s'assurer que le produit à étudier n'est pas retenu par le filtre.
Une centrifugation à 40 000 m.sec-2 (4 000 g) pendant 15 minutes peut remplacer la filtration pour distinguer le COT du COD. Cette méthode n'est pas fiable si la concentration initiale en COD est inférieure à 10 mg/l, soit parce que toutes les bactéries ne sont pas éliminées, soit parce que du carbone faisant partie intégrante du plasma bactérien est redissous.
BIBLIOGRAPHIE
|
(1)
|
Standard Methods for the Examination of Water and Wastewater, 12th ed, Am. Publ. Hlth. Ass., Am. Wat. Poll. Control Fed., Oxygen Demand, 1965, p 65.
|
|
(2)
|
Wagner, R. Von Wasser, 1976, Vol. 46, 139.
|
|
(3)
|
DIN-Entwurf 38 409 Teil 41 — Deutsches Einheitsverfahren zur Wasser-, Abwasser- und Schlammuntersuchung, Summarische Wirkungs- und Stoffkenngrößen (Gruppe H). Bestimmung des chemischen Sauerstoffbedarfs (CSB) (H 41), Normenausschuß Wasserwesen (NAW) in DIN Deutsches Institut für Normung e.V.
|
|
(4)
|
Gerike, P., The biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, Vol. 13 (1), 169.
|
Annexe III
ÉVALUATION DE LA BIODÉGRADABILITÉ DES SUBSTANCES FAIBLEMENT SOLUBLES
Lors des essais de biodégradabilité concernant des substances faiblement solubles, il faut prêter une attention particulière aux éléments énoncés ci-après.
Alors que les liquides homogènes posent rarement des problèmes d'échantillonnage, il est recommandé d'homogénéiser les matières solides par des moyens appropriés afin d'éviter les erreurs dues au manque d'homogénéité. Il convient de prendre des précautions particulières pour prélever, dans des mélanges de produits chimiques ou dans des substances riches en impuretés, des échantillons de quelques milligrammes qui soient représentatifs.
Différents types d'agitation peuvent être utilisés au cours de l'essai, mais il faut veiller à ce que l'agitation soit juste suffisante pour maintenir la dispersion du produit tout en évitant une surchauffe, la formation excessive de mousse ou des forces de cisaillement excessives.
Il est possible d'utiliser un émulsifiant qui produise une dispersion stable du produit. Il ne devra ni être toxique pour les bactéries, ni être biodégradé, ni former de la mousse dans les conditions d'essai.
Les mêmes critères s'appliquent aux solvants et aux émulsifiants.
Il n'est pas conseillé d'utiliser des supports solides pour les produits solides; leur utilisation peut, par contre, être appropriée dans le cas des substances huileuses.
Lorsqu'une substance auxiliaire, telle qu'un émulsifiant, un solvant ou un support, est utilisée, il convient d'effectuer un essai à blanc avec cette substance.
Les trois essais de respirométrie, CO2, DBO ou MITI, conviennent pour étudier la biodégradabilité des composés faiblement solubles.
BIBLIOGRAPHIE
|
—
|
de Morsier, A. et al., Biodegradation tests for poorly soluble compounds. Chemosphere, 1987, Vol. 16, 833.
|
|
—
|
Gerike, P. The Biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, Vol. 13, 169.
|
Annexe IV
ÉVALUATION DE LA BIODÉGRADABILITÉ DES PRODUITS CHIMIQUES SUSPECTÉS DE TOXICITÉ VIS-À-VIS DE L'INOCULUM
Lorsqu'un produit est soumis à des essais de biodégradabilité facile et semble ne pas être biodégradable, il est recommandé de suivre la procédure suivante si l'on souhaite effectuer une distinction entre inhibition et inertie (Reynolds et al., 1987).
Il convient de procéder aux essais de toxicité et de biodégradation avec des inoculums semblables ou identiques.
Pour évaluer la toxicité des produits soumis aux essais de biodégradabilité facile, il semble approprié d'appliquer soit l'une des méthodes suivantes soit une combinaison de ces méthodes: inhibition du taux de respiration de la boue (essai d'inhibition de la respiration des boues activées — directive 88/302/CEE), DBO, et/ou inhibition de la croissance.
S'il convient d'éviter l'inhibition due à la toxicité, il est suggéré d'utiliser pour les essais de biodégradabilité facile des concentrations de la substance d'essai inférieures au 1/10 de la valeur de la CE50 obtenue lors des essais de toxicité (ou inférieures à la valeur de la CE20). Il est peu vraisemblable que les composés dont la CE50 a une valeur supérieure à 300 mg/l produisent un effet toxique lors des tests de biodégradabilité facile. Il est vraisemblable que des valeurs de CE50 inférieures à 20 mg/l poseront de sérieux problèmes lors des essais ultérieurs. Il convient dans ce cas d'employer de faibles concentrations d'essai nécessitant l'utilisation de l'essai en fioles fermées, qui est rigoureux et sensible, ou de produit marqué au 14C. Une autre manière de procéder consiste à utiliser un inoculum acclimaté permettant d'utiliser des concentrations plus élevées de substance d'essai. Toutefois, dans ce cas, on perd le critère spécifique de l'essai de biodégradation facile.
BIBLIOGRAPHIE
Reynolds, L. et al. Evaluation of the toxicity of substances to be assessed for biodegradability. Chemosphere, 1987, Vol. 16,2259.
Annexe V
CORRECTION TENANT COMPTE DE LA CONSOMMATION D'OXYGÈNE DUE À L'INTERFÉRENCE DE LA NITRIFICATION
Les erreurs commises en ne tenant pas compte de la nitrification lors de la détermination de la biodégradabilité par mesure de la consommation d'oxygène sont mineures (inférieures a 5 %) lorsque les substances à étudier ne contiennent pas d'azote, même si l'oxydation de l'azote ammoniacal dans le milieu se produit occasionnellement dans les récipients d'essai ainsi que dans les essais à blanc. Par contre, des erreurs importantes peuvent se produire avec les substances qui contiennent de l'azote.
Si la nitrification se produit, mais n'est pas complète, la consommation d'oxygène observée dans le mélange réactionnel peut être corrigée d'une valeur correspondant à la quantité d'oxygène utilisée pour oxyder l'ammonium en nitrite et en nitrate, si les modifications de la concentration de nitrite et de nitrate au cours de l'incubation sont déterminées par les équations suivantes:
|
2 NH4C1 + 3 O2 = 2 HNO2 + 2 HCl + 2 H2O
|
(1)
|
|
2 HNO2 + O2 = 2 HNO3
|
(2)
|
|
Au total:
|
|
|
2 NH4Cl + 4 O2 = 2 HNO3 + 2 HCl + 2 H2O
|
(3)
|
D'après l'équation (1), la quantité d'oxygène consommée par l'oxydation de 28 g d'azote contenus dans du chlorure d'ammonium (NH4Cl) pour former des nitrites est égale à 96 g, ce qui correspond à un facteur 3,43 (96/28). De même, d'après l'équation (3), la quantité d'oxygène consommée par l'oxydation de 28 g d'azote pour former des nitrates est de 128 g, ce qui correspond à un facteur 4,57 (128/28).
Les réactions étant séquentielles et effectuées par différentes espèces bactériennes distinctes, la concentration en nitrite peut augmenter ou diminuer; dans ce dernier cas, la concentration de nitrate est équivalente. La quantité d'oxygène consommée par la formation de nitrate est donc égale a 4,57 multiplié par l'augmentation de la concentration de nitrate, alors que la quantité d'oxygène associée à la formation de nitrite est égale à 3,43 multiplié par l'augmentation de la concentration en nitrite ou dans le cas d'une réduction de sa teneur, la perte en oxygène est égale à -3,43 multiplié par la diminution de concentration.
Ainsi:
|
O2 consommé par la formation de nitrate = 4,57 × augmentation de la concentration de N-nitrate
|
(4),
|
|
.
|
|
|
O2 consommé par la formation de nitrite = 3,43 × augmentation de la concentration de N-nitrite
|
(5)
|
|
et donc
|
|
|
O2 perdu lors de la disparition de nitrite = -3,43 × réduction de la concentration de N-nitrite
|
(6)
|
|
De telle sorte que:
|
|
|
la consommation d'O2 due à la nitrification = ±3,43 × modif. de la conc. de N-nitrite + 4,57 × augm. de la conc. de N-nitrate
|
(7)
|
|
et donc:
|
|
|
la consommation d'O2 due à l'oxydation du C = consommation totale observée — consommation due à la nitrification
|
(8).
|
Une autre possibilité consiste à estimer que, si seul l'azote oxydé total est déterminé, la consommation d'oxygène due à la nitrification sera, en première approximation, égale à 4,57 × l'augmentation de la teneur en azote oxydé.
La valeur corrigée de la consommation d'oxygène due à l'oxydation du C est ensuite comparée à la DThONH3 calculée selon les instructions figurant à l'annexe II.
C.5. DÉGRADATION — DEMANDE BIOCHIMIQUE EN OXYGÈNE
1. MÉTHODE
1.1. INTRODUCTION
La méthode a pour objet la mesure de la demande biochimique en oxygène (DBO) de substances organiques, liquides ou solides.
Les données fournies par cet essai concernent les composés hydrosolubles; toutefois, les composes volatils et les composés à faible hydrosolubilité peuvent également être étudiés, du moins en principe.
La méthode est applicable uniquement aux substances organiques, non inhibitrices vis-à-vis des bactéries à la concentration utilisée au cours de l'essai. Si la substance d'essai n'est pas soluble à la concentration d'essai, des procédures particulières telles que le recours à la dispersion ultrasonique pourraient être utilisées, afin d'obtenir une bonne dispersion de la substance.
Des données sur la toxicité de la substance chimique peuvent être utiles pour l'interprétation de résultats faibles et pour le choix de concentrations d'essai appropriées.
1.2. DÉFINITION ET UNITÉS
La DBO est définie comme la masse d'oxygène dissoute nécessaire pour assurer, dans des conditions définies, l'oxydation biochimique d'un volume défini d'une solution de la substance soumise aux essais.
Les résultats sont exprimés en grammes de DBO par gramme de substance soumise à l'essai.
1.3. SUBSTANCE DE RÉFÉRENCE
Il est souhaitable de vérifier l'activité de l'inoculum à l'aide d'une substance de référence appropriée.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Ensemencer avec des micro-organismes et incuber à une température ambiante constante, définie, dans l'obscurité, une quantité prédéterminée de substance, dissoute ou dispersée en milieu aéré approprié.
La DBO est déterminée par la différence entre la teneur en oxygène dissous au début et à la fin de l'essai. La durée de l'essai est d'au moins cinq jours et ne dépassera pas 28 jours.
Un blanc doit être déterminé dans un essai parallèle ne contenant pas de substance d'essai.
1.5. CRITÈRES DE QUALITÉ
La détermination de la DBO ne peut être considérée comme représentant valablement la biodégradabilité d'une substance et ne constitue qu'un essai de sélection.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparer une solution ou une dispersion préliminaire de la substance, afin d'obtenir une concentration de DBO compatible avec la méthode utilisée. La DBO est alors déterminée par application de toute méthode appropriée, normalisée, nationale ou internationale.
2. ÉVALUATION DES DONNÉES
La DBO contenue dans la solution préliminaire est calculée suivant la méthode normalisée choisie et convertie en grammes de DBO par gramme de substance d'essai.
3. RÉSULTATS
La méthode utilisée doit être indiquée.
La demande biochimique en oxygène doit être la moyenne d'au moins trois mesures valables.
Toutes les données et observations pertinentes aidant à l'interprétation des résultats doivent être consignées, notamment en ce qui concerne les impuretés, l'état physique, les effets toxiques et la composition intrinsèque de la substance susceptibles d'affecter les résultats.
L'utilisation de tout additif inhibiteur de la nitrification biologique doit être consignée.
4. RÉFÉRENCES
Exemples de méthodes normalisées:
|
|
NF T 90-103: Détermination de la demande biochimique en oxygène.
|
|
|
NBN 407: Biochemical oxygen demand.
|
|
|
NEN 3235 5.4: Bepaling van net biochemish zuurstofverbruik (BZV).
|
|
|
The determination of biochemical oxygen demand, Methods for the examination of water and associated materials, HMSO, London.
|
|
|
ISO 5815: Determination of biochemical oxygen demand after n days.
|
C.6. DÉGRADATION — DEMANDE CHIMIQUE EN OXYGÈNE
1. MÉTHODE
1.1. INTRODUCTION
La méthode a pour objet la mesure de la demande chimique en oxygène (DCO) de substances organiques, liquides ou solides, par application de tout mode opératoire normalisé, dans des conditions de laboratoire déterminées.
Le fait de disposer de données sur la formule de la substance facilitera la réalisation de cet essai ainsi que l'interprétation des résultats obtenus (par exemple, sels halogénés, sels ferreux de composés organiques, composés organochlorés).
1.2. DÉFINITIONS ET UNITÉS
La demande chimique en oxygène est la mesure de l'oxydabilité d'une substance, définie comme la quantité d'oxygène d'un réactif oxydant, consommée par la substance dans des conditions de laboratoire déterminées.
Le résultat est exprimé en grammes de DCO par gramme de substance d'essai.
1.3. SUBSTANCES DE RÉFÉRENCE
L'emploi de substances de référence n'est pas requis dans tous les cas où l'on analyse une nouvelle substance. Ces substances de référence doivent permettre d'étalonner périodiquement la méthode et de permettre la comparaison des résultats en cas d'application de méthodes différentes.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Une quantité déterminée de substance dissoute ou dispersée dans de l'eau est oxydée à l'aide de bichromate de potassium en présence d'acide sulfurique concentré (le catalyseur étant du sulfate d'argent) à reflux pendant deux heures. Le bichromate résiduel est dosé par titration à l'aide de sulfate ferreux ammoniacal titré.
Au cas où les substances contiennent du chlore, ajouter du sulfate mercurique (10) pour réduire l'interférence des chlorures.
1.5. CRITÈRE DE QUALITÉ
En raison des conditions arbitraires de détermination de la DCO, cette dernière est un «indicateur d'oxydabilité» à utiliser en tant que tel comme méthode pratique de mesure de matière organique.
Les chlorures peuvent interférer au cours de cet essai; des substances inorganiques réductrices ou oxydantes peuvent également interférer avec la détermination de la DCO.
Certains composés cycliques et un grand nombre de produits volatils (par exemple les acides gras inférieurs) ne sont pas totalement oxydés lors de l'essai.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
Préparer une solution ou une dispersion préliminaire de la substance pour obtenir une DCO de 250 à 600 milligrammes par litre.
Remarque:
Dans le cas des substances peu solubles et non dispersables, peser une quantité de substance finement pulvérisée ou à l'état liquide, correspondant à environ 5 milligrammes de DCO, et l'introduire dans l'appareil d'expérimentation avec addition d'eau.
Il est souvent avantageux, en particulier lorsqu'on a affaire à des substances peu solubles, de déterminer la demande chimique en oxygène (DCO) à l'aide d'une variante de la méthode, c'est-à-dire dans un système clos muni d'un régulateur de pression (H. Kelkenberg, 1975). Cette modification permet souvent de parvenir à quantifier des substances qui le sont difficilement en utilisant la méthode classique — par exemple l'acide acétique. Toutefois cette méthode échoue elle aussi dans le cas de la pyridine. Le fait d'augmenter jusqu'à une valeur 0,25 N (0,0416 M) la concentration de bichromate de potassium prescrite dans la référence (1) facilite la pesée directe d'une quantité de substance comprise entre 5 et 10 mg, ce qui est essentiel pour déterminer la DCO de substances faiblement hydrosolubles [référence (2)].
Dans les autres cas, déterminer la DCO en suivant toute méthode normalisée nationale ou internationale.
2. ÉVALUATION DES DONNÉES
Calculer la DCO contenue dans le flacon expérimental, suivant la méthode normalisée choisie, et convertir en grammes de DCO par gramme de substance d'essai.
3. RÉSULTATS
La méthode de référence utilisée doit être indiquée.
La demande chimique en oxygène sera la moyenne d'au moins trois mesures. Il sera fait état de toutes données et observations présentant de l'intérêt pour l'interprétation des résultats, notamment en ce qui concerne les impuretés, l'état physique et les propriétés spécifiques de la substance (si elles sont connues) susceptibles d'affecter les résultats.
Mentionner si l'on a fait usage de sulfate mercurique pour réduire l'interférence des chlorures.
4. RÉFÉRENCES
|
(1)
|
Kelkenberg, H.,Z. von Wasser und Abwasserforschung, 1975, vol. 8, 146.
|
|
(2)
|
Gerike, P., The biodegradability testing of poorly water soluble compounds, Chemosphere, 1984, vol. 13,169.
Exemples de méthodes normalisées:
|
|
NBN T 91-201 Determination of the chemical oxygen demand.
|
|
|
ISBN O11 7512494 Chemical oxygen demand (dichromate value) of polluted and waste waters.
|
|
|
NF T 90-101 Determination of the chemical oxygen demand.
|
|
|
DS 217 = water analysis Determination of the chemical oxygen demand.
|
|
|
DIN 38409-H-41 Determination of the chemical oxygen demand (COD) within the range above 15 mg per litre.
|
|
|
NEN 3235 5.3 Bepaling van het chemisch zuurstofverbruik.
|
|
|
ISO 6060 Water quality: chemical oxygen demand dichromate methods.
|
|
C.7. DÉGRADATION — DÉGRADATION ABIOTIQUE: HYDROLYSE EN FONCTION DU PH
1. MÉTHODE
Cette méthode est équivalente à la ligne directrice 111 (2004) de l'OCDE.
1.1. INTRODUCTION
Les substances chimiques qui pénètrent dans les eaux de surface par différentes voies (applications directes, déperditions lors de l'épandage, drainages, élimination de déchets, effluents industriels, domestiques ou agricoles et dépôts atmosphériques) peuvent y être transformées sous l'action de processus chimiques (hydrolyse ou oxydation, par exemple), photochimiques et/ou microbiens. La ligne directrice 111 de l'OCDE décrit une méthode d'essai en laboratoire permettant d'évaluer les transformations hydrolytiques abiotiques des substances chimiques en milieux aquatiques à des valeurs de pH normalement rencontrées dans l'environnement (pH de 4 à 9), et s'appuie sur des lignes directrices existantes (1) (2) (3) (4) (5) (6) (7).
Des expériences sont menées pour déterminer: (i) le taux d'hydrolyse de la substance d'essai en fonction du pH; et (ii) l'identité ou la nature et les taux de formation et de disparition des produits d'hydrolyse auxquels les organismes peuvent être exposés. De telles études peuvent être requises pour les substances chimiques qui sont appliquées directement dans l'eau ou qui sont susceptibles d'atteindre l'environnement par les différentes voies décrites ci-dessus.
1.2. DÉFINITION ET UNITÉS
Voir annexe 2.
1.3. APPLICABILITÉ DE LA MÉTHODE
Cette méthode est globalement applicable aux substances chimiques (marquées ou non) pour lesquelles il existe une méthode analytique suffisamment sensible et précise. Elle convient également pour les composés légèrement volatils et non volatils suffisamment solubles dans l'eau. En revanche, cet essai ne peut pas être appliqué aux produits chimiques hautement volatils dans l'eau (fumigants et solvants organiques, par exemple), impossibles à maintenir en solution dans les conditions expérimentales de l'essai. De même, avec les substances trop peu solubles dans l'eau, la réalisation de cet essai peut s'avérer difficile (8).
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Des solutions aqueuses tampon stériles de différents pH (pH 4, 7 et 9) sont traitées avec la substance d'essai et incubées dans l'obscurité dans des conditions expérimentales contrôlées (à des températures constantes). Après des intervalles de temps appropriés, on analyse les solutions tampon pour déterminer la présence de la substance d'essai et des produits d'hydrolyse. Avec une substance d'essai marquée (par exemple, au 14C), il est plus facile d'établir un bilan massique.
La présente méthode décrit une approche à plusieurs niveaux, dont le détail est donné dans l'annexe 1. Chaque niveau est déclenché par les résultats du niveau précédent.
1.5. INFORMATIONS SUR LA SUBSTANCE D'ESSAI
Des substances d'essai marquées ou non peuvent être utilisées pour mesurer le taux d'hydrolyse. Les matières marquées conviennent généralement mieux pour étudier le chemin réactionnel de l'hydrolyse et établir un bilan massique. Toutefois, dans certains cas particuliers, le marquage n'est pas impératif. Le marquage au 14C est recommandé, mais l'utilisation d'autres isotopes (13C, 15N, 3H, par exemple) peut aussi convenir. Autant que possible, le marquage doit être placé dans la partie la plus stable de la molécule. Par exemple, si la substance d'essai comporte un cycle, le marquage de celui-ci est requis. En revanche, pour les substances comportant deux cycles ou plus, il peut être nécessaire de procéder à des études distinctes pour évaluer le sort de chaque cycle marqué et obtenir les informations requises sur la formation des produits d'hydrolyse. La pureté de la substance d'essai doit être au moins de 95 %.
Avant de procéder à un essai d'hydrolyse, il convient de rassembler les informations suivantes sur la substance d'essai:
|
a)
|
solubilité dans l'eau [méthode d'essai A.6];
|
|
b)
|
solubilité dans les solvants organiques;
|
|
c)
|
pression de vapeur [Méthode d'essai A.4] et/ou constante de la loi de Henry;
|
|
d)
|
coefficient de partage n-octanol/eau [méthode d'essai A.8];
|
|
e)
|
constante de dissociation (pKa) [ligne directrice 112 de l'OCDE] (9);
|
|
f)
|
vitesse de phototransformation directe et indirecte dans l'eau, le cas échéant.
|
Des méthodes d'analyse doivent être disponibles pour quantifier la substance d'essai et, le cas échéant, identifier et quantifier les produits d'hydrolyse dans les solutions aqueuses (voir également le point 1.7.2).
1.6. SUBSTANCE DE RÉFÉRENCE
Lorsque cela est possible, il convient d'utiliser des substances de référence pour identifier et quantifier les produits d'hydrolyse par analyse spectroscopique et chromatographique, ou autres méthodes suffisamment fines.
1.7. CRITÈRES DE QUALITÉ
1.7.1. Récupération
L'analyse d'au moins deux solutions tampon ou de leurs extraits, immédiatement après l'ajout de la substance d'essai donne une première indication de la répétabilité de la méthode analytique et de l'uniformité de la procédure d'application de la substance d'essai. Ce sont les bilans massiques (dans le cas d'une matière marquée) qui permettent ensuite d'établir aux étapes suivantes la récupération des substances testées. La récupération des substances chimiques, marquées ou non, doit être comprise entre 90 et 110 % (7). S'il est techniquement difficile d'atteindre cette plage de valeurs, une récupération de 70 % des substances chimiques non marquées est acceptable, mais celle-ci doit faire l'objet d'une justification.
1.7.2. Répétabilité et sensibilité de la méthode analytique
Pour contrôler la répétabilité de la (ou des) méthode(s) analytique(s) utilisée(s) pour quantifier la substance d'essai et les produits d'hydrolyse, on peut procéder à deux analyses des mêmes solutions tampon (ou de leurs extraits) après que des produits d'hydrolyse ont été formés en quantités suffisantes pour permettre une quantification.
La méthode analytique doit être suffisamment sensible pour permettre de quantifier des concentrations de la substance d'essai jusqu'à 10 %, ou moins, de la concentration initiale. Le cas échéant, les méthodes analytiques doivent également permettre de quantifier tout produit d'hydrolyse représentant 10 % ou plus de la substance appliquée (à n'importe quel moment au cours de l'étude), et jusqu'à 25 % ou moins de sa concentration maximale.
1.7.3. Intervalles de confiance pour les données cinétiques de l'hydrolyse
Des intervalles de confiance doivent être calculés et présentés pour tous les coefficients de régression, constantes de vitesse, demi-vies et autres paramètres cinétiques (DT50, par exemple).
1.8. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.8.1. Matériel et appareillage
L'étude doit être conduite dans des récipients en verre (tubes à essai, petits flacons, par exemple), et le cas échéant dans l'obscurité et dans des conditions de stérilité, sauf s'il ressort d'une information préliminaire (telle que le coefficient de partage n-octanol/eau) que la substance d'essai est susceptible d'adhérer au verre. Dans ce cas, une autre matière (le Téflon, par exemple) doit être envisagée. Les méthodes suivantes permettent également de contourner le problème de l'adhérence au verre:
|
—
|
détermination de la masse de la substance d'essai et des produits d'hydrolyse fixés par adsorption au récipient d'essai,
|
|
—
|
utilisation d'un bain à ultrasons,
|
|
—
|
nettoyage de toute la verrerie à l'aide d'un solvant à chaque intervalle d'échantillonnage,
|
|
—
|
utilisation de produits formulés,
|
|
—
|
utilisation d'une quantité plus importante de cosolvant à ajouter à la substance d'essai. Dans ce dernier cas, il convient d'employer un cosolvant qui n'entraîne pas de réaction d'hydrolyse de la substance d'essai.
|
L'incubation des différentes solutions d'essai doit être effectuée avec des agitateurs mécaniques à bain thermorégulé ou des incubateurs à commande thermostatique.
Cette expérience nécessite du matériel courant de laboratoire, et en particulier:
|
—
|
des instruments d'analyse [équipements de chromatographie en phase gazeuse, chromatographie en phase liquide à haute performance (HPLC), chromatographie en couche mince, par exemple], et notamment des systèmes de détection appropriés pour l'analyse des substances radiomarquées ou non, l'application de la méthode de dilution isotopique inverse,
|
|
—
|
des instruments d'identification (spectrométrie de masse, spectrométrie de masse couplée à la chromatographie en phase gazeuse, spectrométrie de masse couplée à la chromatographie en phase liquide à haute performance, résonance magnétique nucléaire, etc.),
|
|
—
|
un compteur à scintillation liquide;
|
|
—
|
des entonnoirs d'extraction liquide-liquide,
|
|
—
|
des instruments pour la concentration de solutions et d'extraits (évaporateur rotatif, par exemple),
|
|
—
|
des dispositifs de contrôle de la température (bain d'eau, par exemple).
|
Les réactifs chimiques sont notamment:
|
—
|
des solvants organiques, de pureté analytique, tels que l'hexane, le dichlorométhane, etc.,
|
|
—
|
du liquide de scintillation,
|
|
—
|
des solutions tampon (voir détails point 1.8.3).
|
Toute la verrerie, l'eau de qualité «réactif» et les solutions tampon utilisées dans les essais d'hydrolyse doivent être stérilisées.
1.8.2. Application de la substance d'essai
La substance d'essai doit être appliquée sous forme de solution aqueuse dans les différentes solutions tampon (voir annexe 3). Le cas échéant, pour obtenir une dissolution adéquate, l'utilisation de petites quantités de solvants miscibles dans l'eau (acétonitrile, acétone, éthanol, par exemple) est autorisée pour assurer une application et une distribution correctes de la substance d'essai, mais la proportion ne doit normalement pas dépasser 1 % v/v. Si une concentration supérieure en solvants est envisagée (par exemple dans le cas de substances d'essai faiblement solubles), cela ne peut être autorisé que s'il est démontré que le solvant n'a aucune incidence sur l'hydrolyse de la substance d'essai.
Le recours à un produit formulé n'est pas recommandé, dans la mesure où on ne peut exclure que les ingrédients de la formulation interfèrent avec le processus d'hydrolyse. Toutefois, pour les substances d'essai faiblement solubles dans l'eau, ou celles qui adhèrent au verre (voir point 1.8.1), le recours à un produit formulé peut constituer une solution acceptable.
Une seule concentration de la substance d'essai doit être utilisée; elle ne doit pas dépasser 0,01 M ou la moitié de la concentration de saturation (voir annexe 1).
1.8.3. Solutions tampon
L'essai d'hydrolyse doit être réalisé aux valeurs de pH suivantes: 4, 7 et 9. À cet effet, il convient de préparer des solutions tampon avec des produits chimiques de pureté analytique et de l'eau. L'annexe 3 présente quelques systèmes tampon utiles. Il faut noter que le système tampon utilisé peut influencer la vitesse d'hydrolyse. Lorsque c'est le cas, il faut utiliser un autre système tampon (11).
Le pH de chaque solution tampon doit être vérifié avec un pH-mètre étalonné à la température désirée avec une précision d'au moins 0,1.
1.8.4. Conditions d'essai
1.8.4.1. Température d'essai
Les expériences d'hydrolyse doivent être menées à des températures constantes. Pour permettre l'extrapolation, il est important de maintenir la température constante à ±0,5 oC.
Si on ne connaît pas le comportement de la substance d'essai à l'hydrolyse, il est nécessaire d'effectuer un essai préliminaire (niveau 1) à une température de 50 oC. Les essais cinétiques suivants doivent être menés à un minimum de trois températures différentes (y compris l'essai à 50 oC), à moins que l'essai préliminaire de niveau 1 n'ait mis en évidence la stabilité de la substance à l'hydrolyse. Une plage de températures allant de 10 à 70 oC est préconisée (avec de préférence au moins une température inférieure à 25 oC). Cette plage englobe la température utilisée dans le rapport d'essai (25 oC) et la plupart des températures rencontrées sur le terrain.
1.8.4.2. Lumière et oxygène
Tous les essais d'hydrolyse doivent être effectués à l'aide d'une méthode adéquate pour éviter les effets de photolyse. Toutes les mesures appropriées doivent être prises afin d'éviter l'oxygène (par exemple, par barbotage d'hélium, d'azote ou d'argon pendant 5 minutes avant la préparation de la solution).
1.8.4.3. Durée de l'essai
L'essai préliminaire doit être mené pendant 5 jours, tandis que les essais des niveaux supérieurs doivent être conduits jusqu'à 90 % de la substance d'essai, ou pendant 30 jours, au premier des deux termes échus.
1.8.5. Exécution de l'essai
1.8.5.1. Essai préliminaire (niveau 1)
L'essai préliminaire doit être réalisé à 50 ±0,5 oC et aux pH suivants: 4, 7 et 9. Si au bout de 5 jours, on observe un taux d'hydrolyse inférieur à 10 % (t0,5 25 oC > 1 an), la substance est considérée comme stable dans l'eau et il n'est normalement pas nécessaire d'effectuer d'autres essais. Si on sait que la substance est instable aux températures observées dans l'environnement (12), il n'est pas obligatoire de réaliser un essai préliminaire. La méthode analytique employée doit être suffisamment précise et sensible pour détecter une diminution de 10 % de la concentration initiale.
1.8.5.2. Hydrolyse des substances instables (niveau 2)
L'essai de niveau supérieur (avancé) doit être exécuté aux valeurs de pH auxquelles la substance d'essai est apparue instable selon la procédure définie pour l'essai préliminaire ci-avant. Les solutions tamponnées de la substance d'essai doivent alors être thermostatées aux températures choisies. Pour savoir si la réaction est du premier ordre, chaque solution doit être analysée à des intervalles de temps qui fournissent un minimum de six points espacés, se situant normalement entre 10 et 90 % d'hydrolyse de la substance étudiée. Des échantillons répétés (un minimum de deux échantillons identiques contenus dans des cuves de réaction distinctes) doivent être prélevés et leur contenu analysé à chacune des six échéances d'échantillonnage (pour un minimum de 12 points identiques). L'option consistant à utiliser un seul échantillon global d'où serait tirée chacune des aliquotes de la solution d'essai à chaque intervalle d'échantillonnage n'est pas considérée comme appropriée du fait qu'elle ne permet pas d'analyser la variabilité des données, et qu'elle peut poser des problèmes de contamination de la solution d'essai. Des essais de confirmation de la stérilité doivent être effectués à la fin de l'essai de niveau supérieur (c'est-à-dire à 90 % d'hydrolyse ou à 30 jours). Toutefois, si aucune dégradation (c'est-à-dire aucune transformation) n'est observée, les essais de stérilité ne sont pas nécessaires.
1.8.5.3. Identification des produits d'hydrolyse (niveau 3)
Tout produit d'hydrolyse majeur, et au minimum ceux représentant > 10 % de la dose appliquée, doit être identifié selon les méthodes analytiques appropriées.
1.8.5.4. Essais facultatifs
Des essais supplémentaires à des pH autres que 4, 7 et 9 peuvent être nécessaires pour les substances d'essai instables sur le plan hydrolytique. Par exemple, à des fins physiologiques, il peut s'avérer nécessaire d'effectuer un essai dans des conditions plus acides (par exemple à un pH de 1,2) à une seule température pertinente du point de vue physiologique (37 oC).
2. RÉSULTATS
Les quantités de la substance d'essai et des produits d'hydrolyse, le cas échéant, doivent être exprimées en pourcentage de la concentration initiale appliquée et, lorsque cela est possible, en mg/l pour chaque intervalle d'échantillonnage, ainsi que pour chaque pH et température d'essai. En outre, lorsqu'une substance d'essai marquée a été utilisée, un bilan massique doit être fourni en pourcentage de la concentration initiale appliquée.
Une présentation graphique des logarithmes des concentrations de la substance d'essai en fonction du temps doit être présentée. Tous les produits d'hydrolyse majeurs, et au minimum ceux représentant > 10 % de la dose appliquée, doivent être identifiés, et les logarithmes de leurs concentrations doivent être présentés de la même manière que pour la substance mère, de façon à mettre en évidence leurs vitesses de formation et de disparition.
2.1. TRAITEMENT DES RÉSULTATS
Des déterminations plus précises des demi-vies ou des valeurs de DT50 doivent être obtenues par application des calculs de modèles cinétiques appropriés. Les valeurs de demi-vie et/ou de DT50 (y compris les limites de confiance) doivent être précisées pour chaque pH et chaque température, avec une description du modèle utilisé, l'ordre de réaction cinétique et le coefficient de détermination (r2). Le cas échéant, ces calculs doivent être appliqués aux produits d'hydrolyse.
Pour les études menées à différentes températures, les constantes de la vitesse d'hydrolyse de pseudo-premier ordre (kobs) doivent être décrites comme une fonction de température. Le calcul doit être basé sur une séparation des kobs en constantes de vitesse pour les catalyses acides, neutres et basiques (respectivement kH, kneutre, et kOH) et sur l'équation d'Arrhenius:
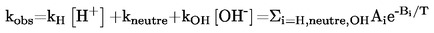
où Ai et Bi sont des constantes de régression et d'interception, respectivement, des lignes de meilleur ajustement générées à partir de la régression linéaire ln ki sur la réciproque de la température absolue en Kelvin (T). En utilisant la relation d'Arrhenius pour les catalyses acides, neutres et basiques, il est possible de calculer les constantes de pseudo-premier ordre, et ainsi les demi-vies, pour d'autres températures pour lesquelles la détermination expérimentale directe d'une constante de vitesse ne peut être pratiquée (10).
2.2. ÉVALUATION ET INTERPRÉTATIONS DES RÉSULTATS
La plupart des réactions d'hydrolyse affichent une vitesse de réaction apparente de premier ordre, et par conséquent les demi-vies sont indépendantes de la concentration (voir l'équation 4 dans l'annexe 2). Cette caractéristique permet généralement d'appliquer les résultats obtenus en laboratoire, déterminés entre 10-2 et 10-3 M, aux conditions dans l'environnement (≤ 10-6 M) (10). Mabey et Mill (11) font état de plusieurs exemples de bonne concordance entre les taux d'hydrolyse mesurés dans l'eau pure et dans les eaux naturelles, pour un ensemble de substances chimiques, à condition que le pH et la température aient bien été mesurés.
3. RAPPORTS
3.1. RAPPORT D'ESSAI
Le rapport d'essai doit contenir au moins les informations suivantes:
Substance d'essai:
|
—
|
nom commun, nom chimique, numéro CAS, formule structurale (montrant la position du radiomarqueur, le cas échéant) et propriétés physico-chimiques pertinentes (voir point 1.5),
|
|
—
|
pureté (impuretés) de la substance d'essai,
|
|
—
|
pureté radiochimique des substances marquées et activité molaire (s'il y a lieu).
|
|
—
|
dates et modalités de préparation,
|
|
—
|
tampons et eaux utilisés,
|
|
—
|
molarité et pH des solutions tampon.
|
Conditions d'essai:
|
—
|
dates de réalisation des études,
|
|
—
|
quantité de substance d'essai appliquée,
|
|
—
|
méthodes et solvants utilisés (types et quantité) pour l'application de la substance d'essai,
|
|
—
|
volume des solutions tamponnées de substances d'essai incubées,
|
|
—
|
description du système d'incubation utilisé,
|
|
—
|
pH et température au cours de l'étude,
|
|
—
|
méthode(s) d'extraction,
|
|
—
|
méthodes de quantification et d'identification de la substance d'essai et de ses produits d'hydrolyse dans les solutions tampon,
|
|
—
|
nombre d'essais identiques.
|
Résultats:
|
—
|
répétabilité et sensibilité des méthodes analytiques utilisées,
|
|
—
|
récupérations (les pourcentages pour une étude correcte sont données au point 1.7.1),
|
|
—
|
résultats et moyennes des essais identiques, présentés sous forme de tableaux,
|
|
—
|
bilan massique au cours et à la fin des études (en cas d'utilisation d'une substance marquée),
|
|
—
|
résultats de l'essai préliminaire,
|
|
—
|
discussion et interprétation des résultats,
|
|
—
|
intégralité des données et figures originales.
|
Les informations suivantes sont obligatoires uniquement si une vitesse d'hydrolyse est déterminée:
|
—
|
tracés des concentrations en fonction du temps pour les substances d'essai et, le cas échéant, pour les produits d'hydrolyse à chaque valeur de pH et température,
|
|
—
|
tableaux des résultats de l'équation d'Arrhenius pour la température 20 oC/25 oC, avec pH, constante [h-1 ou jour-1], demi-vie ou DT50, températures [en oC] y compris les limites de confiance et les coefficients de corrélation (r2) ou toute information comparable,
|
|
—
|
chemin réactionnel d'hydrolyse proposé.
|
4. BIBLIOGRAPHIE
|
(1)
|
OCDE (1981). Hydrolyse en fonction du pH. Ligne directrice de l'OCDE pour les essais de produits chimiques, No 111, adoptée le 12 mai 1981.
|
|
(2)
|
US-Environmental Protection Agency (1982). 40 CFR 796.3500, Hydrolysis as a Function of pH at 25 oC. Pesticide Assessment Guidelines, Subdivision N. Chemistry: Environmental Fate.
|
|
(3)
|
Agriculture Canada (1987). Guide d'homologation des pesticides au Canada: chimie et devenir dans l'environnement.
|
|
(4)
|
Union européenne (UE) (1995), directive 2001/36/CE de la Commission du 16 mai 2001 portant modification de la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques. Annexe V: Devenir et comportement dans l'environnement
|
|
(5)
|
Commission néerlandaise pour l'enregistrement des pesticides (1991). Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air.
|
|
(6)
|
BBA (1980). Merkblatt Nr. 55, Teil I und II: Prüfung des Verhaltens von Pflanzenbehandlungsmitteln im Wasser (October 1980).
|
|
(7)
|
SETAC (1995). Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides. Mark R. Lynch, Ed.
|
|
(8)
|
OECD (2000). Guidance document on aquatic toxicity testing of difficult substances and mixtures, OECD Environmental Health and Safety Publications Series on Testing and Assessment Nr.23.
|
|
(9)
|
OCDE (1993). Lignes directrices de l'OCDE pour les essais de produits chimiques. Paris. OCDE (1994-2000) : 6e au 1 1e addenda aux Lignes directrices pour les essais de produits chimiques.
|
|
(10)
|
Nelson, H, Laskowski D, Thermes S, and Hendley P. (1997) Recommended changes in pesticide fate study guidelines for improving input to computer models. (Text version of oral presentation at the 14th Annual Meeting of the Society of Environmental Toxicology and Chemistry, Dallas TX, November 1993).
|
|
(11)
|
Mabey, W. and Mill, T. (1978). Critical review of hydrolysis of organic compounds in water under environmental conditions. J. Phys. Chem. Ref. Data 7, 383-415.
|
Annexe 1
Plan de l'essai d'hydrolyse par niveaux

Annexe 2
Définitions et unités
Les unités du système international d'unités (SI) doivent être utilisées.
Substance d'essai: toute substance, qu'il s'agisse du composé parent ou des produits de transformation.
Produits de transformation: toutes les substances résultant des réactions de transformation biotique ou abiotique de la substance d'essai.
Produits d'hydrolyse: toutes les substances résultant des réactions de transformation hydrolytique de la substance d'essai.
L'hydrolyse consiste en la réaction d'une substance d'essai RX avec l'eau, avec échange net du groupe X contre le groupe OH selon la réaction:
Dans ce processus simplifié, la vitesse à laquelle la concentration de RX décroît est donnée par:
|
vitesse = k [H2O] [RX]
|
réaction du second ordre
|
|
ou
|
|
vitesse = k [RX]
|
réaction du premier ordre
|
selon la phase qui détermine la vitesse. Du fait de la présence d'eau en large excès par rapport à la substance d'essai, ce type de réaction est habituellement décrit comme une réaction de premier ordre, dans laquelle la constante de vitesse observée est donnée par la relation:
et peut être déterminée à partir de l'expression (13)
|
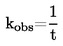
ln

|
[3]
|
où
t= temps
et Co, Ct= concentrations de RX au temps 0 et au temps t.
Les unités de cette constante ont les dimensions de (temps)-1 et la demi-vie de la réaction (temps au bout duquel 50 % de RX ont réagi) est donnée par:
|
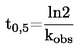
|
[4]
|
La demi-vie (t0,5) est le temps requis pour obtenir 50 % d'hydrolyse d'une substance d'essai lorsque la réaction peut être décrite comme une cinétique du premier ordre. La demi-vie est indépendante de la concentration.
La DT50 (Disappearance time 50) correspond au laps de temps au bout duquel la concentration de la substance d'essai est réduite de 50 pour cent. Cette valeur est différente de la demi-vie (t0,5) lorsque la réaction ne suit pas une cinétique du premier ordre.
Estimation de k à différentes températures
Lorsque les constantes de vitesse sont connues pour deux températures, les constantes de vitesse à d'autres températures peuvent être calculées en utilisant l'équation d'Arrhenius:
 or
or 
Un tracé de ln k en fonction de 1/T donne une ligne droite de pente -E/R
où:
|
k
|
=
|
constante de vitesse, mesurée à différentes températures
|
|
E
|
=
|
énergie d'activation [kJ/mol]
|
|
T
|
=
|
température absolue [K]
|
|
R
|
=
|
constante des gaz [8,314 J/mol*K]
|
L'énergie d'activation est calculée par analyse de régression ou par l'équation suivante:
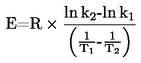
où: T2>T1.
Annexe 3
Systèmes tampon
A. CLARK ET LUBS:
Mélanges tampons de CLARK et LUBS
(14)
|
Composition
|
pH
|
|
HCl 0,2 N et KCl 0,2 N À 20 oC
|
|
47,5 ml HCl + 25 ml KCl dil. à 100 ml
|
1,0
|
|
32,25 ml HCl + 25 ml KCl dil. à 100 ml
|
1,2
|
|
20,75 ml HCl + 25 ml KCl dil. to 100 ml
|
1,4
|
|
13,15 ml HCl + 25 ml KCl dil. à 100 ml
|
1,6
|
|
8,3 ml HCl + 25 ml KCl dil. à 100 ml
|
1,8
|
|
5,3 ml HCl + 25 ml KCl dil. à 100 ml
|
2,0
|
|
3,35 ml HCl + 25 ml KCl dil. à 100 ml
|
2,2
|
|
Biphtalate de potassium 0,1 M + HCl 0,1 N à 20 oC
|
|
46,70 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
2,2
|
|
39,60 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
2,4
|
|
32,95 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
2,6
|
|
26,42 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
2,8
|
|
20,32 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
3,0
|
|
14,70 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
3,2
|
|
9,90 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
3,4
|
|
5,97 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
3,6
|
|
2,63 ml HCl 0,1 N + 50 ml biphtalate à 100 ml
|
3,8
|
|
Biphtalate de potassium 0,1 M + NaOH 0,1 N à 20 oC
|
|
0,40 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
4,0
|
|
3,70 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
4,2
|
|
7,50 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
4,4
|
|
12,15 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
4,6
|
|
17,70 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
4,8
|
|
23,85 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
5,0
|
|
29,95 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
5,2
|
|
35,45 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
5,4
|
|
39,85 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
5,6
|
|
43,00 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
5,8
|
|
45,45 ml NaOH 0,1 N + 50 ml biphtalate à 100 ml
|
6,0
|
|
Phosphate monopotassique 0,1 M + NaOH 0,1 N à 20 oC
|
|
5,70 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
6,0
|
|
8,60 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
6,2
|
|
12,60 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
6,4
|
|
17,80 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
6,6
|
|
23,45 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
6,8
|
|
29,63 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
7,0
|
|
35,00 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
7,2
|
|
39,50 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
7,4
|
|
42,80 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
7,6
|
|
45,20 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
7,8
|
|
46,80 ml NaOH 0,1 N + 50 ml phosphate à 100 ml
|
8,0
|
|
H3B03 0,1 M dans KCl 0,1 M + NaOH 0,1 N à 20 oC
|
|
2,61 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
7,8
|
|
3,97 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
8,0
|
|
5,90 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
8,2
|
|
8,50 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
8,4
|
|
12,00 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
8,6
|
|
16,30 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
8,8
|
|
21,30 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
9,0
|
|
26,70 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
9,2
|
|
32,00 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
9,4
|
|
36,85 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
9,6
|
|
40,80 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
9,8
|
|
43,90 ml NaOH 0,1 N + 50 ml acide borique à 100 ml
|
10,0
|
B. KOLTHOFF ET VLEESCHHOUWER:
Tampons citrate de KOLTHOFF et VLEESCHHOUWER
|
Composition
|
pH
|
|
Citrate monopotassique 0,1 M et HCl 0,1 N à 18 oC (15)
|
|
49,7 ml HCl 0,1 N + 50 ml citrate à 100 ml
|
2,2
|
|
43,4 ml HCl 0,1 N + 50 ml citrate à 100 ml
|
2,4
|
|
36,8 ml HCl 0,1 N + 50 ml citrate à 100 ml
|
2,6
|
|
30,2 ml HCl 0,1 N + 50 ml citrate à 100 ml
|
2,8
|
|
23,6 ml HCl 0,1 N + 50 ml citrate à 100 ml
|
3,0
|
|
17,2 ml HCl 0,1 N + 50 ml citrate à 100 ml
|
3,2
|
|
10,7 ml HCl 0,1 N + 50 ml citrate à 100 ml
|
3,4
|
|
4,2 ml HCl 0,1 N + 50 ml citrate à 100 ml
|
3,6
|
|
Citrate monopotassique 0,1 M et NaOH 0,1 N à 18 oC (15)
|
|
2,0 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
3,8
|
|
9,0 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
4,0
|
|
16,3 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
4,2
|
|
23,7 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
4,4
|
|
31,5 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
4,6
|
|
39,2 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
4,8
|
|
46,7 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
5,0
|
|
54,2 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
5,2
|
|
61,0 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
5,4
|
|
68,0 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
5,6
|
|
74,4 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
5,8
|
|
81,2 ml NaOH 0,1 N + 50 ml citrate à 100 ml
|
6,0
|
C. SÖRENSEN:
Mélanges de borate de SÖRENSEN
|
Composition
|
Sörensen
18 oC
|
Walbum, pH à
|
|
ml Borax
|
ml HCl/NaOH
|
10 oC
|
40 oC
|
70 oC
|
|
Borax 0,05 M + HCl 0,1 N
|
|
|
4,75
|
7,62
|
7,64
|
7,55
|
7,47
|
|
|
4,50
|
7,94
|
7,98
|
7,86
|
7,76
|
|
|
4,25
|
8,14
|
8,17
|
8,06
|
7,95
|
|
|
4,00
|
8,29
|
8,32
|
8,19
|
8,08
|
|
|
3,50
|
8,51
|
8,54
|
8,40
|
8,28
|
|
|
3,00
|
8,08
|
8,72
|
8,56
|
8,40
|
|
|
2,50
|
8,80
|
8,84
|
8,67
|
8,50
|
|
|
2,00
|
8,91
|
8,96
|
8,77
|
8,59
|
|
|
1,50
|
9,01
|
9,06
|
8,86
|
8,67
|
|
|
1,00
|
9,09
|
9,14
|
8,94
|
8,74
|
|
|
0,50
|
9,17
|
9,22
|
9,01
|
8,80
|
|
|
0,00
|
9,24
|
9,30
|
9,08
|
8,86
|
|
Borax 0,05 M + NaOH 0,1 N
|
|
10,0
|
0,0
|
9,24
|
9,30
|
9,08
|
8,86
|
|
|
1,0
|
9,36
|
9,42
|
9,18
|
8,94
|
|
|
2,0
|
9,50
|
9,57
|
9,30
|
9,02
|
|
|
3,0
|
9,68
|
9,76
|
9,44
|
9,12
|
|
|
4,0
|
9,97
|
10,06
|
9,67
|
9,28
|
Mélanges de phosphates de SÖRENSEN
|
Composition
|
pH
|
|
Phosphate monopotassique 0,0667 M + phosphate disodique 0,0667 M à 20 oC
|
|
99,2 ml KH2PO4+ 0,8 ml Na2HPO4
|
5,0
|
|
98,4 ml KH2PO4+ 1,6 ml Na2HPO4
|
5,2
|
|
97,3 ml KH2PO4+ 2,7 ml Na2HPO4
|
5,4
|
|
95,5 ml KH2PO4+ 4,5 ml Na2HPO4
|
5,6
|
|
92,8 ml KH2PO4+ 7,2 ml Na2HPO4
|
5,8
|
|
88,9 ml KH2PO4+ 11,1 ml Na2HPO4
|
6,0
|
|
83,0 ml KH2PO4+ 17,0 ml Na2HPO4
|
6,2
|
|
75,4 ml KH2PO4+ 24,6 ml Na2HPO4
|
6,4
|
|
65,3 ml KH2PO4+ 34,7 ml Na2HPO4
|
6,6
|
|
53,4 ml KH2PO4+ 46,6 ml Na2HPO4
|
6,8
|
|
41,3 ml KH2PO4+ 58,7 ml Na2HPO4
|
7,0
|
|
29,6 ml KH2PO4+ 70,4 ml Na2HPO4
|
7,2
|
|
19,7 ml KH2PO4+ 80,3 ml Na2HPO4
|
7,4
|
|
12,8 ml KH2PO4+ 87,2 ml Na2HPO4
|
7,6
|
|
7,4 ml KH2PO4+ 92,6 ml Na2HPO4
|
7,8
|
|
3,7 ml KH2PO4+ 96,3 ml Na2HPO4
|
8,0
|
C.8 TOXICITÉ POUR LES VERS DE TERRE
ESSAI SUR SOL ARTIFICIEL
1. MÉTHODE
1.1. INTRODUCTION
Dans cet essai de laboratoire, la substance d'essai est ajoutée à un sol artificiel dans lequel les vers sont placés pendant 14 jours. Au bout de cette période (et facultativement au bout de 7 jours), l'effet létal de la substance sur les vers de terre est examiné. La méthode permet de déterminer assez rapidement l'effet de l'absorption dermique et de l'ingestion des produits chimiques sur les vers de terre.
1.2. DÉFINITION ET UNITÉ
CL50: concentration d'une substance considérée comme responsable de la mort de 50 % des animaux d'expérience pendant la durée de l'essai.
1.3. SUBSTANCE DE RÉFÉRENCE
Une substance de référence est utilisée périodiquement pour démontrer que la sensibilité du système n'a pas changé de façon significative.
La substance de référence recommandée est le chloracétamide de qualité analytique.
1.4. PRINCIPE DE L'ESSAI
Le sol étant un milieu variable, on utilise pour cet essai un sol limoneux artificiel soigneusement défini. Des vers de terre adultes de l'espèce Eisenia foetida (voir note en appendice) sont placés dans un sol artificiel défini, traité par différentes concentrations de la substance d'essai. Le contenu des récipients est étalé sur un plateau 14 jours (et facultativement 7 jours) après le début de l'essai et, pour chaque concentration, les vers de terre survivants sont comptés.
1.5. CRITÈRES DE QUALITÉ
L'essai est conçu pour être aussi reproductible que possible au point de vue du substrat et de l'organisme. Si la mortalité des témoins dépasse 10 % à la fin de l'essai, celui-ci n'est pas valide.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Matériel
1.6.1.1. Substrat
On utilise comme substrat de base un sol artificiel bien défini.
|
a)
|
Substrat de base (les pourcentages sont exprimés en poids sec):
|
—
|
10 % de tourbe à sphaignes (de pH aussi proche que possible de 5,5 à 6,0, ne contenant pas de restes de plante visibles et finement broyée),
|
|
—
|
20 % d'argile kaolinique contenant si possible plus de 50 % de kaolinite,
|
|
—
|
environ 69 % de sable quartzique industriel (essentiellement du sable fin contenant plus de 50 % de particules de granulométrie comprise entre 0,05 et 0,2 mm). Si la substance n'est pas suffisamment dispersible dans l'eau, 10 g par récipient doivent être mis de côté pour être mélangés ultérieurement à la substance à tester,
|
|
—
|
environ 1 % de carbonate de calcium (CaCO3) pulvérisé, chimiquement pur, pour ajuster le pH à 6,0±0,5.
|
|
|
b)
|
Substrat d'essai:
Le substrat pour les essais contient le substrat de base, la substance d'essai et de l'eau désionisée.
Le teneur en eau est d'environ 25 à 42 % du poids sec du substrat de base. La teneur en eau du substrat est déterminée par séchage d'un échantillon à 105 oC, jusqu'à ce que le poids reste constant. Le critère clé est que le sol artificiel doit être humidifié jusqu'à saturation. Le mélange doit être effectué soigneusement, de manière à obtenir une répartition uniforme de la substance d'essai et du substrat. La façon dont la substance d'essai est introduite dans le substrat doit être indiquée.
|
|
c)
|
Substrat témoin:
Le substrat témoin contient le substrat de base et de l'eau. Si l'on utilise un additif, un substrat témoin supplémentaire doit contenir la même quantité d'additif.
|
1.6.1.2. Récipients
Récipients en verre d'une capacité d'environ 1 litre (convenablement recouverts de couvercles en plastique, de coupelles ou d'une feuille de plastique avec trous d'aération), remplis d'une quantité de substrat d'essai ou de substrat témoin humide équivalant à 500 g de substrat sec.
1.6.2. Conditions expérimentales
Les récipients doivent être déposés dans des locaux climatisés dont la température est maintenue à 20 oC ± 2 oC, sous éclairement continu compris entre 400 et 800 lux.
La durée de l'essai est de 14 jours, mais la mortalité peut être déterminée 7 jours après le début de l'essai.
1.6.3. Méthode
Concentrations
Les concentrations de la substance d'essai sont exprimées en poids de substance par unité de substrat de base sec (mg/kg).
Essai de sélection des concentrations à utiliser
La gamme des concentrations correspondant à des taux de mortalité allant de 0 à 100 % peut être déterminée par un essai renseignant sur l'intervalle de concentrations à utiliser dans l'essai définitif.
La substance doit être testée aux concentrations suivantes: 1 000, 100, 10, 1, 0,1 mg de substance/kg de substrat (poids sec).
Si un essai définitif complet est effectué, un lot par concentration et un lot témoin contenant chacun dix vers devraient être suffisants pour l'essai de sélection des concentrations à utiliser.
Essai définitif
Les résultats de l'essai de sélection des concentrations à employer sont utilisés pour choisir au moins 5 concentrations dans une série géométrique couvrant exactement l'intervalle de 0 à 100 % de mortalité et dans un rapport constant inférieur ou égal à 1,8.
Un essai utilisant cette série de concentrations devrait permettre d'évaluer avec la plus grande précision possible la valeur de la CL50 et de ses limites de confiance.
Pour l'essai définitif, on utilise au moins 4 lots par concentration et 4 lots témoins contenant chacun 10 vers. Les résultats de ces lots en parallèle sont exprimés sous la forme de moyenne et d'écart type.
Lorsque deux concentrations consécutives dans un rapport de 1,8 ne donnent que 0 % et 100 % de mortalité, ces deux valeurs suffisent à indiquer l'intervalle dans lequel se situe la CL50.
Mélange du substrat de base et de la substance d'essai
Le substrat ne devrait pas, si possible, contenir d'autres additifs que de l'eau. Immédiatement avant le début de l'essai, une émulsion ou une dispersion de la substance d'essai dans de l'eau désionisée ou dans un autre solvant est mélangée au substrat de base ou uniformément projetée à sa surface à l'aide d'un pulvérisateur de chromatographie ou du même type.
Si elle est insoluble dans l'eau, la substance d'essai peut être dissoute dans le plus petit volume possible d'un solvant organique approprié (par exemple hexane, acétone ou chloroforme).
Seuls des agents très volatils peuvent être utilisés pour solubiliser, disperser ou émulsifier la substance d'essai. Avant emploi, le substrat doit être aéré. La quantité d'eau évaporée doit être remplacée. Le substrat témoin doit contenir la même quantité de tout additif.
Si la substance d'essai n'est pas soluble, dispersible ou émulsifiable dans des solvants organiques, 10 g d'un mélange de sable quartzique finement broyé et d'une quantité de substance d'essai correspondant à la dose nécessaire pour traiter 500 g de sol artificiel sec sont mélangés à 490 g de substance d'essai sèche.
Pour chaque lot, une quantité de substrat humide équivalant à 500 g de poids sec est placée dans chaque récipient en verre et 10 vers de terre ayant été conditionnés pendant 24 heures dans un substrat de base humide analogue, puis rincés rapidement, l'eau excédentaire étant absorbée sur un papier-filtre sont placés à la surface du substrat.
Les récipients sont recouverts de couvercles, de coupelles ou d'une feuille en plastique perforé afin d'éviter le dessèchement du substrat et ils sont maintenus dans les conditions d'essai pendant 14 jours.
Les évaluations doivent être faites au bout de 14 jours (et facultativement de 7 jours). Le substrat est étalé sur une plaque de verre ou d'acier inoxydable. Les vers de terre sont examinés et le nombre de vers de terre survivants est déterminé. Les vers de terre sont considérés comme morts s'ils ne réagissent pas à un léger stimulus mécanique appliqué à leur extrémité antérieure.
Lorsque l'examen est fait au bout de 7 jours, le récipient est à nouveau rempli de substrat et les vers de terre survivants sont replacés sur le même substrat.
1.6.4. Organismes d'expérience
Les organismes d'expérience doivent être des Eisenia foetida adultes (voir note en appendice) (âgés d'au moins 2 mois, avec clitellum) — poids humide: 300 à 600 mg (pour la méthode d'élevage, voir appendice).
2. RÉSULTATS
2.1. TRAITEMENT ET ÉVALUATION DES RÉSULTATS
Les concentrations de la substance d'essai sont consignées avec les pourcentages correspondants de vers de terre morts.
Si les données sont adéquates, la valeur de la CL50 et les intervalles de confiance (p = 0,05) sont déterminées à l'aide de méthodes standards (Litchfield et Wilcoxon, 1949 ou méthode équivalente). La CL50 doit être exprimée en milligrammes de la substance d'essai par kilogramme de substrat (poids sec).
Dans le cas où la pente de la courbe est trop accentuée pour permettre le calcul de la CL50, il suffira de procéder à une estimation graphique de cette valeur.
Lorsque deux concentrations consécutives dans un rapport de 1,8 ne donnent que 0 ou 100 % de mortalité, ces deux valeurs suffisent à indiquer l'intervalle dans lequel se situe la CL50.
3. PRÉSENTATION DES RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
attestation de conformité de l'essai aux critères de qualité énoncés ci-dessus,
|
|
—
|
essai effectué (essai de détermination des ordres de grandeur et/ou essai définitif),
|
|
—
|
description précise des conditions d'essai ou attestation de conformité de l'essai, tout écart par rapport à la méthode étant indiqué,
|
|
—
|
description précise de la manière dont la substance d'essai a été mélangée au substrat de base,
|
|
—
|
information sur les organismes soumis à l'essai (espèce, âge, poids moyen et gamme de poids, conditions d'élevage, fournisseur),
|
|
—
|
méthode appliquée pour déterminer la CL50,
|
|
—
|
résultat de l'essai, avec toutes les données utilisées,
|
|
—
|
description des symptômes observés ou des modifications du comportement des organismes soumis à l'essai,
|
|
—
|
mortalité chez les témoins,
|
|
—
|
CL50 ou concentration maximale testée n'ayant pas causé de mortalité et concentration minimale testée ayant causé 100 % de mortalité au bout de 14 jours (et facultativement de 7 jours),
|
|
—
|
tracé de la courbe concentrations/réponses,
|
|
—
|
résultats obtenus avec la substance de référence, au cours du présent essai ou de contrôles antérieurs de la qualité.
|
4. BIBLIOGRAPHIE
|
(1)
|
OCDE, Paris, 1981, Ligne directrice 207, décision du Conseil C(81) 30 final.
|
|
(2)
|
Edwards, C, A. and Lofty, 1977, Biology of Earthworms, Chapman and Hall, London, 331 pages.
|
|
(3)
|
Bouche, M- B., 1972, Lombriciens de France, Écologie et Systématique, Institut national de la recherche agronomique, 671 pages.
|
|
(4)
|
Litchfield, J. T. and Wilcoxon, F., A simplified method of evaluating dose-effect experiments, J. Pharm. Exp. Therap., vol. 96, 1949, 99 pages.
|
|
(5)
|
Commission des Communautés européennes, Development of a standardized laboratory method for assessing the toxicity of chemical substances to earthworms, Report EUR 8714 EN, 1983.
|
|
(6)
|
Umweltbundesamt/Biologische Bundesanstalt fur Land- und Forsrwirtschaft, Berlin, 1984, Verfahrensvorschlag «Toxizitätstest am Regenwurm Eisenia foetida in künstlichem Boden», in: Rudolph/Boje: Ökotoxikologie, ecomed, Landsberg, 1986.
|
Appendice
Élevage des vers avant les essais
Trente à cinquante vers adultes sont introduits dans un récipient d'élevage contenant du substrat frais et en sont retirés au bout de 14 jours. Ces animaux peuvent être utilisés pour de nouveaux lots d'élevage. Les vers de terre sortis des cocons sont utilisés pour les essais lorsqu'ils sont arrivés à maturité (soit au bout de deux ou trois mois dans les conditions prescrites).
Conditions d'élevage
|
Local climatisé
|
:
|
température: 20 oC ± 2 oC, de préférence sous éclairement continu (intensité 400 à 800 lux).
|
|
Récipient d'élevage
|
:
|
récipients creux d'une capacité de 10 à 20 litres.
|
|
Substrat
|
:
|
l'Eisenia foetida peut être élevé avec différents excréments animaux. Il est recommandé d'utiliser comme milieu d'élevage un mélange en volume de 50 % de fumier de vache ou de cheval et de 50 % de tourbe. Le milieu doit avoir un pH d'environ 6 ou 7 (ajusté avec du carbonate de calcium) et une faible conductivité ionique (moins de 6 mmhos ou une concentration de sel de 0,5 %). Le substrat doit être humide, mais ne doit pas être trop mouillé.
Outre la méthode décrite ci-dessus, d'autres méthodes peuvent être utilisées.
|
Note: Il existe deux races d'Eisenia foetida que certains taxonomistes ont séparées en espèces (Bouche, 1972). Celles-ci sont morphologiquement semblables, mais l'une, Etsenia foetida foetida, se distingue par des rayures ou des bandes sur les segments, tandis que l'autre, Eisenia foetida andrei, n'en a pas et a une couleur rougeâtre aux nuances variées. Il convient d'utiliser si possible Eisenia foetida andrei. D'autres espèces peuvent être utilisées si l'on dispose de la méthodologie nécessaire.
C.9 BIODÉGRADATION
ESSAI DE ZAHN ET WELLENS
1. MÉTHODE
1.1. INTRODUCTION
La présente méthode a pour but d'évaluer la biodégradabilité totale potentielle de substances organiques non volatiles solubles dans l'eau lorsqu'elles sont exposées à des concentrations relativement élevées de micro-organismes lors d'un essai statique.
Il peut y avoir adsorption physico-chimique sur les solides en suspension, et il convient d'en tenir compte lors de l'interprétation des résultats (voir 3.2).
Les substances à étudier sont utilisées dans des concentrations correspondant à des valeurs de COD allant de 50 à 400 mg/litre ou à des valeurs de DCO situées entre 100 et 1 000 mg/litre (COD = carbone organique dissous; DCO = demande chimique en oxygène). Ces concentrations relativement élevées assurent une bonne fiabilité analytique. Les composés possédant des propriétés toxiques peuvent ralentir ou empêcher le processus de dégradation.
Dans cette méthode, on emploie la mesure de la concentration du carbone organique dissous ou la demande chimique en oxygène pour évaluer la biodégradation finale de la substance d'essai.
L'emploi simultané d'une méthode analytique spécifique peut permettre d'évaluer la biodégradation primaire de la substance (disparition de la structure chimique initiale).
La méthode n'est applicable qu'aux substances organiques qui, à la concentration utilisée dans l'essai:
|
—
|
sont solubles dans l'eau dans les conditions de l'essai,
|
|
—
|
ont une pression de vapeur négligeable dans les conditions de l'essai,
|
|
—
|
n'ont pas d'effets inhibiteurs vis-à-vis des bactéries,
|
|
—
|
ne font pas l'objet d'une adsorption importante sur l'appareillage,
|
|
—
|
ne disparaissent pas de la solution par formation de mousse.
|
Il est utile de connaître les proportions relatives des principaux éléments constitutifs de la substance d'essai pour interpréter les résultats obtenus, notamment lorsque ceux-ci sont peu élevés ou marginaux.
La connaissance de la toxicité de la substance vis-à-vis des micro-organismes peut être intéressante pour interpréter les résultats peu élevés et pour choisir les concentrations d'essai appropriées.
1.2. DÉFINITIONS ET UNITÉS
Le taux de dégradation obtenu à la fin de l'essai constitue la «biodégradabilité en essai de Zahn et Wellens»:

dans laquelle:
|
DT
|
=
|
biodégradation (%) au temps T,
|
|
CA
|
=
|
valeurs du COD (ou de la DCO) du mélange d'essai mesurées 3 heures après le début de l'essai (mg/l) (COD = carbone organique dissous; DCO = demande chimique en oxygène),
|
|
CT
|
=
|
valeurs du COD ou de la DCO du mélange d'essai au moment du prélèvement de l'échantillon (mg/l),
|
|
CB
|
=
|
valeur du COD ou de la DCO du témoin au moment du prélèvement (mg/l),
|
|
CBA
|
=
|
valeur du COD ou de la DCO du témoin mesurée 3 heures après le début de l'essai (mg/l).
|
Le taux de dégradation obtenu est arrondi au pourcentage entier le plus proche.
Le pourcentage de dégradation est défini comme étant le pourcentage de disparition du COD (ou de la DCO) de la substance d'essai.
La différence entre la valeur mesurée après 3 heures et la valeur initiale calculée ou, mieux, mesurée, peut donner des renseignements intéressants sur l'élimination de la substance (voir 3.2: Interprétation des résultats).
1.3. SUBSTANCES DE RÉFÉRENCE
Des substances de référence peuvent parfois être utiles lors de l'étude de substances nouvelles; toutefois, des substances de référence spécifiques ne peuvent pas encore être recommandées.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
On introduit la boue activée, les substances nutritives minérales et une solution aqueuse de la substance à examiner constituant la seule source de carbone dans un récipient en verre de 1 à 4 litres muni d'un agitateur et d'un aérateur. Le mélange est agité et aéré à une température de 20 à 25 oC, en lumière diffuse ou en chambre noire pendant une période pouvant aller jusqu'à 28 jours. Le processus de dégradation est suivi en déterminant la valeur du COD (ou de la DCO) dans la solution filtrée, quotidiennement ou selon une autre périodicité appropriée. Après chaque période, le COD (ou la DCO) éliminé est rapporté à la valeur constatée 3 heures après le début de l'essai et exprimé en pourcentage de biodégradation; cela constitue la mesure du taux de dégradation à ce moment. Le résultat rapporté graphiquement en fonction du temps donne la courbe de biodégradation.
Si on utilise une méthode analytique spécifique, on peut mesurer les variations de la concentration de la molécule originale dues à la biodégradation (biodégradabilité primaire).
1.5. CRITÈRES DE QUALITÉ
La reproductibilité de la méthode a été reconnue satisfaisante lors d'un essai d'intercomparaison
La sensibilité de la méthode est largement déterminée par la variabilité du témoin et, dans une moindre mesure, par la précision de la détermination du carbone organique dissous et de la concentration de la substance dans le milieu d'essai.
1.6. DESCRIPTION
1.6.1. Préparations
1.6.1.1. Réactifs
Eau: eau potable contenant moins de 5 mg/l de carbone organique. La concentration totale d'ions calcium et magnésium ne doit pas dépasser 2,7 mmoles/l; si ce n'est pas le cas, corriger la dilution par addition d'eau déionisée ou distillée.
|
Acide sulfurique, pureté analytique (P.A.):
|
50 g/l
|
|
Solution d'hydroxyde de soude P.A.:
|
40 g/l
|
|
Solution nutritive minérale: dissoudre dans un litre d'eau déionisée:
|
|
|
Chlorure d'ammonium, NH4C1, P.A.:
|
38,40 g
|
|
Dihydrogénophosphate de sodium, NaH2PO4.2H2O, P.A.:
|
33,40 g
|
|
Dihydrogénophosphate de potassium, KH2PO4, P.A.:
|
8,50 g
|
|
Monohydrogénophosphate de potassium, K2HPO4, P.A.:
|
21,75 g
|
Le mélange sert à la fois de milieu nutritif et de solution tampon.
1.6.1.2. Appareillage
Récipients de verre contenant entre 1 et 4 litres (par exemple récipients cylindriques).
Dispositif d'agitation comprenant un agitateur en verre ou en métal fixé à une tige appropriées (l'agitateur doit décrire un mouvement rotatif à environ 5 à 10 cm au-dessus du fond du récipient). On peut utiliser également un agitateur magnétique muni d'un barreau de 7 à 10 cm de long.
Tube d'aération en verre de 2 à 4 mm de diamètre intérieur. L'ouverture du tube doit se trouver à environ 1 cm au-dessus du fond du récipient.
Centrifugeuse (environ 3 550 g).
pH-mètre.
Appareil pour la mesure de l'oxygène dissous.
Filtres papier.
Appareil de filtration sur membranes.
Membranes filtrantes, porosité: 0,45 μm. Les membranes filtrantes conviennent si elles ne libèrent pas de carbone et n'absorbent pas la substance au stade de la filtration.
Matériel d'analyse pour le dosage du carbone organique et la détermination de la demande chimique en oxygène.
1.6.1.3. Préparation de l'inoculum
Laver la boue activée provenant d'une station d'épuration biologique par centrifugations ou décantations successives dans l'eau utilisée pour l'essai (voir supra).
La boue activée doit avoir les caractéristiques requises. Cette boue peut être obtenue dans une station d'épuration en bon état de fonctionnement. Pour disposer d'autant d'espèces et de souches différentes de bactéries que possible, il peut être préférable de mélanger des inoculums provenant de sources différentes (par exemple de différentes stations d'épuration, d'extraits de sol, des eaux de rivière, etc.). Le mélange doit être traité comme indiqué plus haut.
Pour le contrôle d'activité de la boue activée, voir infra «Contrôle fonctionnel».
1.6.1.4. Préparation des solutions
Dans le récipient d'essai, ajouter 500 ml d'eau (voir supra), 2,5 ml/litre de solution minérale nutritive et une quantité de boues activées correspondant à 0,2-1,0 g/l de matière sèche dans le mélange final. Ajouter suffisamment de solution stock de la substance à tester de façon à obtenir dans le mélange final une concentration de COD de 50 à 400 mg/l. Les valeurs correspondantes de DCO sont 100-1 000 mg/l. Ajouter de l'eau jusqu'à un volume total de 1 à 4 litres. Le volume total choisi dépend du nombre d'échantillons à prélever pour la détermination du COD ou de la DCO et du volume nécessaire pour l'analyse.
Normalement, on considère 2 litres comme un volume satisfaisant. Pour chaque série d'essais, préparer au moins un récipient témoin (blanc) contenant uniquement de la boue activée et la solution minérale nutritive, diluées avec de l'eau jusqu'au même volume total que dans les récipients d'essai.
1.6.2. Mode opératoire
Les récipients d'essai sont agités au moyen d'agitateurs magnétiques ou à hélice, en lumière diffuse ou en chambre noire, a une température de 20 à 25 oC. L'aération est assurée par injection d'air comprimé purifié par un filtre d'ouate et si nécessaire par un flacon laveur. Veiller à ce que la boue ne décante pas et que la concentration d'oxygène ne tombe pas au-dessous de 2 mg/l.
Vérifier le pH à intervalles réguliers (par exemple quotidiennement) et l'amener le cas échéant à pH 7-8.
Les pertes dues à l'évaporation sont compensées juste avant chaque prélèvement d'échantillon au moyen d'eau déionisée ou distillée. Une bonne méthode consiste à marquer le niveau du liquide sur le récipient avant de commencer l'essai. De nouvelles marques sont faites après chaque prélèvement (sans aération ni agitation). Les premiers échantillons sont toujours prélevés 3 heures après le début de l'essai de façon à permettre de détecter une adsorption de la substance d'essai sur la boue activée.
Pour suivre la dégradation de la substance d'essai, effectuer un dosage de COD ou de DCO quotidiennement ou à tout autre intervalle régulier. Filtrer les échantillons provenant du récipient d'essai et du récipient témoin sur un papier filtre soigneusement lavé. Éliminer les 5 premiers millilitres du filtrat de la solution d'essai. Les boues difficilement filtrables peuvent être éliminées au préalable par une centrifugation de 10 minutes. Les déterminations de COD et de DCO sont faites au moins en double. La durée des essais va jusqu'à 28 jours.
Note: Les échantillons restant troubles sont filtrés sur membranes. Celles-ci ne doivent ni libérer ni adsorber de matières organiques.
Contrôle fonctionnel des boues activées
Pour chaque série d'essais, on peut prévoir un récipient contenant une substance connue, de façon à pouvoir vérifier la capacité fonctionnelle de la boue activée. Le diéthylèneglycol s'est révélé utile à cette fin.
Adaptation
Si des analyses sont effectuées à des intervalles relativement courts (par exemple quotidiennement), la courbe de dégradation peut faire clairement apparaître un phénomène d'adaptation (voir figure 2). L'essai ne devrait donc pas être commencé immédiatement avant un week-end.
Si l'adaptation se produit dans les derniers jours de l'essai, celui-ci peut être prolongé jusqu'à dégradation complète.
Note: Si une meilleure connaissance du comportement de la boue adaptée est nécessaire, on exposera à nouveau la même boue activée à la même substance d'essai suivant le mode opératoire ci-après:
Arrêter l'agitateur et l'aérateur et laisser décanter la boue activée. Éliminer le surnageant, remplir d'eau jusqu'à atteindre 2 litres, agiter pendant 15 minutes et laisser à nouveau décanter. Éliminer à nouveau le surnageant, et utiliser la boue restante pour répéter l'essai avec la même substance conformément aux paragraphes 1.6.1.4 et 1.6.2 ci-dessus. La boue activée peut également être séparée par centrifugation plutôt que par décantation.
La boue adaptée peut être mélangée avec de la boue fraîche pour atteindre 0,2 à 1 g de matière sèche par litre.
Analyses
En général, on filtre les échantillons sur papier-filtre soigneusement lavé à l'eau déionisée.
Les échantillons qui restent troubles sont filtrés sur membranes (0,45 μm).
Déterminer la concentration de COD en double dans les filtrats des échantillons (éliminer les cinq premiers millilitres), à l'aide d'un appareil de mesure du COT. Si l'analyse du filtrat ne peut être faite le jour même, le conserver au réfrigérateur jusqu'au lendemain. Il est déconseillé de la conserver plus longtemps.
Déterminer la concentration de DCO dans les filtrats d'échantillon à l'aide du dispositif d'analyse de la DCO conformément au mode opératoire décrit dans la référence (2) ci-après.
2. ÉVALUATION DES DONNÉES
Procéder au moins à deux déterminations de la concentration de COD et de DCO dans les échantillons, conformément aux indications fournies ci-dessus au paragraphe 1.6.2. Calculer le pourcentage de dégradation au temps T suivant la formule (avec ses définitions) donnée au paragraphe 1.2 ci-dessus.
Arrondir le taux de dégradation à l'unité de pourcentage la plus proche. Le taux de dégradation atteint à la fin de l'essai constitue la «biodégradabilité dans l'essai de Zahn-Wellens».
Note: Si la dégradation complète est réalisée avant la fin de la durée de l'essai et si ce résultat est confirmé par une seconde analyse effectuée le lendemain, il peut être mis fin à l'essai.
3. PRÉSENTATION DES RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal de l'essai contiendra, si possible:
|
—
|
la concentration initiale de la substance,
|
|
—
|
toutes autres indications et les résultats expérimentaux relatifs à la substance testée, à la substance de référence éventuelle, et au témoin,
|
|
—
|
la concentration après trois heures,
|
|
—
|
la courbe de biodégradation avec description,
|
|
—
|
la date et l'endroit du prélèvement de l'inoculum, stade d'adaptation, concentration utilisée, etc.,
|
|
—
|
les raisons scientifiques d'éventuelles modifications de la procédure d'essai.
|
3.2. INTERPRÉTATION DES RÉSULTATS
L'élimination du COD (ou de la DCO) qui se produit graduellement pendant un certain nombre de jours ou de semaines indique que la substance testée se dégrade biologiquement.
Cependant, une adsorption physico-chimique peut jouer un rôle dans certains cas: ceci est montré lorsqu'il y a élimination totale ou partielle de la substance dès le début, lors des 3 premières heures, et que la différence entre le surnageant des milieux témoins et d'essai reste faible.
Des essais supplémentaires sont nécessaires si l'on doit faire la distinction entre biodégradation (ou biodégradabilité partielle) et adsorption.
Il y a pour cela plusieurs méthodes, la meilleure étant d'employer le surnageant comme inoculum dans un essai du niveau de base (de préférence un essai respirométrique).
Les substances donnant dans cet essai une élimination élevée, non liée a l'adsorption du COD (ou de la DCO) devraient être considérées comme potentiellement biodégradables. Une élimination partielle, non liée à l'adsorption, indique que le produit chimique est au moins biodégradable dans une certaine mesure. Une élimination faible ou nulle de COD (DCO) peut être due à l'inhibition des micro-organismes par la substance testée; ceci peut être révélé par lyse et perte de boue donnant des surnageants troubles. L'essai devrait être répété avec une concentration plus faible de substance.
L'emploi d'une méthode analytique spécifique ou d'une substance d'essai marquée au 14C peut permettre une plus grande sensibilité. Dans le cas de composés marqués au 14C, la récupération de 14CO2 confirmera qu'il y a eu biodégradation.
Quand les résultats sont exprimés en termes de biodégradation primaire, il conviendrait de fournir dans la mesure du possible une explication concernant le changement de structure chimique qui a conduit à une diminution de réponse de la substance d'origine.
La validation de la méthode analytique doit être accompagnée de l'indication de la réponse trouvée avec le témoin.
4. RÉFÉRENCES
|
(1)
|
OCDE, Paris, 1981, Ligne directrice 302 B, décision du Conseil C(81) 30 final.
|
Appendice
EXEMPLE D'ÉVALUATION
|
Composé organique:
|
acide 4-éthoxybenzoïque
|
|
Concentration théorique:
|
600 mg/l
|
|
COD théorique:
|
390 mg/l
|
|
Inoculum:
|
Station d'épuration des eaux d'égout de ...
|
|
Concentration;
|
1 gramme de matière sèche par litre
|
|
État d'adaptation:
|
non adapté
|
|
Analyse:
|
détermination du COD
|
|
Volume de l'échantillon:
|
3 ml
|
|
Substance de contrôle:
|
diethylèneglycol
|
|
Toxicité du composé:
|
sans effet toxique au-dessous de 1 000 mg/l
Essai pratique: essai des tubes de fermentation.
|
|
Temps
|
Substance de contrôle
|
Substance à étudier
|
|
Blanc
COD (16)
mg/l
|
COD (16)
mg/l
|
COD net
mg/l
|
Dégradation
%
|
COD (16)
mg/l
|
COD net
mg/l
|
Dégradation
%
|
|
0
|
—
|
—
|
300,0
|
—
|
—
|
390,0
|
—
|
|
3 heures
|
4,0
|
298,0
|
294,0
|
2
|
371,6
|
367,6
|
6
|
|
1 jour
|
6,1
|
288,3
|
282,2
|
6
|
373,3
|
367,2
|
6
|
|
2 jours
|
5,0
|
281,2
|
276,2
|
8
|
360,0
|
355,0
|
9
|
|
5 jours
|
6,3
|
270,5
|
264,2
|
12
|
193,8
|
187,5
|
52
|
|
6 jours
|
7,4
|
253,3
|
245,9
|
18
|
143,9
|
136,5
|
65
|
|
7 jours
|
11,3
|
212,5
|
201,2
|
33
|
104,5
|
93,2
|
76
|
|
8 jours
|
7,8
|
142,5
|
134,7
|
55
|
58,9
|
51,1
|
87
|
|
9 jours
|
7,0
|
35,0
|
28,0
|
91
|
18,1
|
11,1
|
97
|
|
10 jours
|
18,0
|
37,0
|
19,0
|
94
|
20,0
|
2,0
|
99
|
Figure 1
Exemples de courbes de biodégradation

Figure 2
Exemples d'adaptation d'une boue
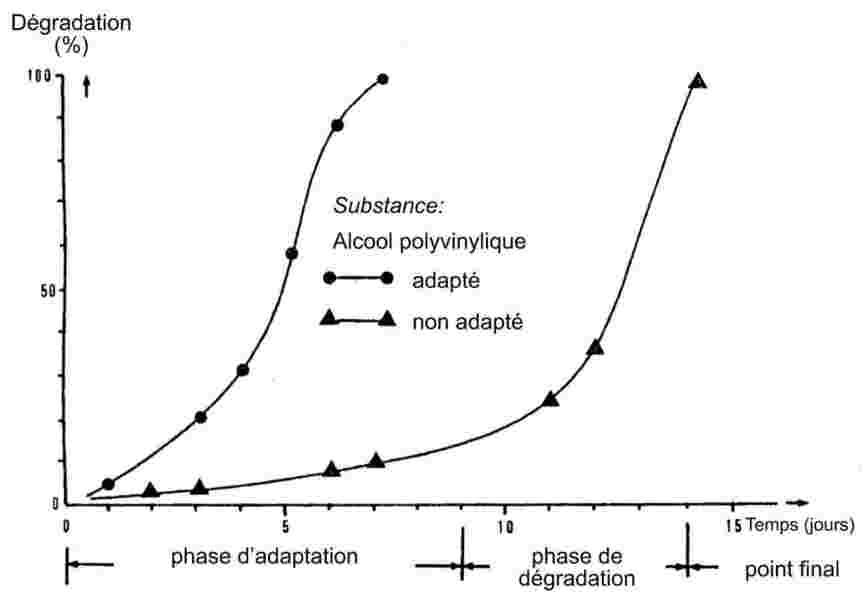
C. 10 BIODÉGRADATION
ESSAIS DE SIMULATION DE BOUES ACTIVÉES
1. MÉTHODE
1.1. INTRODUCTION
1.1.1. Remarques générales
Cette méthode n'est applicable qu'aux substances organiques qui, à la concentration d'essai:
|
—
|
sont suffisamment solubles dans l'eau pour permettre la préparation des solutions d'essai,
|
|
—
|
ont une pression de vapeur négligeable dans les conditions de l'essai,
|
|
—
|
n'ont pas d'effet inhibiteur vis-à-vis des bactéries.
|
Il est utile de connaître les proportions relatives des principaux éléments constitutifs de la substance d'essai pour interpréter les résultats obtenus, notamment lorsque ceux-ci sont peu élevés ou marginaux.
Il est souhaitable de connaître le seuil de toxicité de la substance vis-à-vis des micro-organismes pour interpréter les résultats peu élevés et pour choisir les concentrations d'essai appropriées.
1.1.2. Détermination de la biodégradabilité totale (analyse COD/DCO)
Le but de la méthode est de déterminer la biodégradabilité totale en mesurant la disparition de la substance et de tout métabolite de la substance dans une maquette d'installation de traitement de boues activées à une concentration correspondant à > 12 mg de COD/L (ou approximativement 40 mg de DCO/l). 20 mg de COD/l semble une chiffre optimal (COD = carbone organique dissout, DCO = demande chimique en oxygène).
La teneur en carbone organique (ou la demande chimique en oxygène) de la substance d'essai doit être établie.
1.1.3. Détermination de la biodégradabilité primaire (analyse spécifique)
Le but de la méthode est de déterminer la biodégradabilité primaire d'une substance dans une maquette d'installation de traitement de boues activées à une concentration d'environ 20 mg/l, à l'aide d'une méthode d'analyse spécifique (une concentration plus faible ou plus élevée peut être utilisée si la méthode d'analyse et les limites de toxicité le permettent). Cela permet d'évaluer la biodégradabilité primaire de la substance (disparition de la structure chimique initiale).
Le but de cette méthode n'est pas de déterminer le degré de minéralisation de la substance d'essai.
Une méthode adéquate d'analyse doit être disponible pour la détermination de la substance d'essai.
1.2. DÉFINITIONS ET UNITÉS
1.2.1. Analyse COD/DCO
Le taux de dégradation de la substance est donné par la formule:
|

|
[1 (a)]
|
où:
|
TD
|
=
|
taux de dégradation en pourcentage de COD (ou de DCO) pendant le temps de rétention moyen donné de la substance d'essai,
|
|
T
|
=
|
concentration de la substance d'essai dans le milieu entrant dans l'unité d'essai en mg de COD/l (ou mg de DCO/l),
|
|
E
|
=
|
concentration de COD (ou DCO) dans le milieu sortant de l'unité d'essai en mg de COD/l (ou de DCO/l),
|
|
Eo
|
=
|
concentration de COD (ou DCO) dans le milieu sortant de l'unité témoin en mg de COD/l (ou DCO/l).
|
La dégradation est exprimée en pourcentage de disparition du COD (ou de la DCO) pendant le temps de rétention donné de la substance d'essai.
1.2.2. Analyse spécifique
Le pourcentage d'élimination de la substance d'essai de la phase aqueuse (Rw) pendant le temps de rétention moyen donné s'obtient à l'aide de la formule
|

|
[1 (b)]
|
où:
|
C1
|
=
|
concentration de la substance dans le milieu entrant dans l'unité d'essai (mg de substance/l, déterminée par l'analyse spécifique),
|
|
C0
|
=
|
concentration de la substance dans le milieu sortant de l'unité d'essai (mg de substance/1, déterminée par l'analyse spécifique).
|
1.3. SUBSTANCES DE RÉFÉRENCE
Quand on étudie une nouvelle substance, des produits de référence peuvent parfois se révéler utiles; on ne peut cependant pas encore recommander de produits de référence particuliers.
1.4. PRINCIPE DES MÉTHODES D'ESSAI
Pour la détermination de la biodégradabilité totale, deux unités pilotes de traitement de boues activées sont utilisées en parallèle [unités d'essai de confirmation de l'Organisation de coopération et de développement économiques (OCDE) ou vases poreux]. Dans l'une des unités, la substance d'essai est ajoutée au milieu entrant (eaux résiduaires synthétiques ou domestiques), tandis que l'autre est uniquement alimenté avec le milieu. Pour la détermination de la biodégradabilité primaire avec analyse spécifique dans les milieux d'entrées et de sortie, seule une unité est utilisée.
On mesure les concentrations de COD (ou la DCO) dans les effluents ou bien on détermine les concentrations de la substance par analyse spécifique.
Le COD dû à la substance d'essai n'est pas mesuré, mais simplement mentionné.
Lorsque l'on mesure le COD (ou la DCO), la différence de concentration moyenne entre l'effluent d'essai et l'effluent témoin est censée être due à la substance d'essai non dégradée.
Lorsque l'on effectue des analyses spécifiques, la modification de la concentration de la molécule mère peut être mesurée (biodégradation primaire).
Les unités peuvent fonctionner en «couplage», selon un procédé de transinoculation.
1.5. CRITÈRES DE QUALITÉ
La concentration initiale de la substance dépend du type d'analyse effectué et de ses limites.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Préparation
1.6.1.1. Appareillage
Deux unités de même type sont nécessaires, sauf pour les analyses spécifiques. Deux types de dispositif peuvent être utilisés:
Essai de confirmation de l'OCDE:
L'équipement (appendice 1) se compose d'un récipient (A) pour stocker les eaux résiduaires synthétiques, d'une pompe doseuse(B), d'une cuve d'aération (C), d'un décanteur (D), d'une pompe à air comprimé (E) pour recycler la boue activée et d'un récipient (F) pour recueillir l'effluent traité.
Les récipients (A) et (F) doivent être en verre ou en matière plastique appropriée et contenir au moins 24 litres. La pompe (B) doit assurer une alimentation régulière de la cuve d'aération en effluent synthétique; tout système approprié peut être utilisé, à condition que le débit d'entrée et la concentration soient assurés.
En fonctionnement normal, le décanteur (D) est fixé à une hauteur telle que la cuve d'aération contienne 3 litres de liqueur mixte. Un aérateur fritte (G) est suspendu dans la cuve (C) au-dessus du cône. La quantité d'air insufflée par le dispositif d'aération doit être contrôlée par un débitmètre. La pompe à air comprimé (E) est réglée de façon que la boue activée provenant du décanteur soit continuellement et régulièrement recyclée dans la cuve d'aération (C).
«Vase poreux»:
Le vase poreux est réalisé à partir de feuilles de polyéthylène poreux (2 mm d'épaisseur, taille maximale des pores: 95 μm) formant des cylindres de 14 cm de diamètre à base conique de 45o (figures 1 et 2 de l'appendice 27). Le vase poreux se trouve dans une cuve étanche en matière plastique appropriée de 15 cm de diamètre, pourvue dans la partie cylindrique, à la hauteur de 17,2 cm, d'un orifice qui détermine la capacité du vase (3 1), Un anneau de support rigide réalisé en matière plastique appropriée entoure le sommet du vase, de sorte qu'il y a un espace de 0,5 cm entre le vase et la cuve.
Les vases poreux peuvent être montés à la base d'un bain-marie thermostatisé. À la base du vase est prévue une alimentation en air sur laquelle sont placés des diffuseurs.
Les récipients (A) et (E) doivent être en verre ou en matière plastique appropriée et avoir une capacité d'au moins 24 litres. La pompe (B) doit assurer une alimentation régulière de la cuve d'aération en effluent synthétique; tout système approprié peut être utilisé, à condition que le débit d'entrée et la concentration soient assurés.
Des vases poreux de rechange doivent être disponibles pour remplacer ceux qui pourraient se boucher à l'usage; les vases bouchés sont nettoyés par immersion pendant 24 heures dans une solution d'hypochlorite suivie d'un rinçage à l'eau du robinet.
1.6.1.2. Filtration
Appareil de filtration sur membranes filtrantes avec des pores de 0,45 μm. Les membranes filtrantes conviennent s'il a été prouvé qu'elles ne libèrent pas de carbone et qu'elles n'absorbent pas la substance d'essai au cours de la filtration.
1.6.1.3. Eaux résiduaires
On peut utiliser soit des effluents synthétiques appropriés, soit des eaux résiduaires domestiques.
Exemple d'effluent synthétique:
Dissoudre par litre d'eau du robinet:
|
Peptone:
|
160 mg,
|
|
Extrait de viande:
|
110 mg,
|
|
Urée:
|
30 mg,
|
|
NaCL:
|
7 mg,
|
|
CaCl2.2H2O:
|
4 mg,
|
|
MgS02.7H2O:
|
2 mg,
|
|
K2HPO4:
|
28 mg.
|
Eaux résiduaires domestiques:
Les eaux résiduaires domestiques doivent être recueillies chaque jour au niveau du trop-plein de la cuve de décantation primaire d'une installation de traitement d'eaux résiduaires essentiellement domestiques.
1.6.1.4. Solution de réserve de la substance d'essai
Une solution de la substance d'essai, par exemple à 1 %, doit être préparée pour être introduite dans l'unité d'essai. La concentration de la substance doit être déterminée de façon que soit connu le volume approprié à ajouter aux eaux résiduaires ou directement à l'unité par le moyen d'une pompe secondaire afin d'obtenir la concentration d'essai requise.
1.6.1.5. Inoculum
Remarque: Avec des eaux résiduaires domestiques, il serait superflu d'utiliser un inoculum à faible concentration bactérienne, mais on peut utiliser des boues activées.
On peut utiliser plusieurs types d'inoculum.
Nous donnons ici trois exemples d'inoculum appropriés:
|
a)
|
Inoculum à base d'effluent secondaire:
L'inoculum doit être obtenu à partir d'un effluent secondaire de bonne qualité, provenant d'une installation de traitement d'eaux résiduaires essentiellement domestiques. Cet effluent doit être aéré depuis le prélèvement jusqu'à l'utilisation. Il est filtré sur un filtre grossier, les 200 premiers millilitres étant éliminés. Le filtrat, convenablement aéré, doit être utilisé le jour même. Il faut utiliser au moins 3 ml pour l'inoculation.
|
|
b)
|
Inoculum composite:
Inoculum à base d'effluent secondaire:
Voir description ci-dessus.
Inoculum à base de terre:
100 g de terre de jardin (fertile, non stérile) sont mis en suspension dans 1 000 ml d'eau potable exempte de chlore (les terres à très forte teneur en argile, sable ou humus ne peuvent être utilisées). Après agitation, il y a lieu de laisser reposer la suspension durant 30 minutes. Le surnageant est alors filtré sur un papier filtre grossier, les 200 premiers millilitres étant éliminés. Le filtrat est aéré immédiatement, et cela jusqu'à son utilisation. 11 doit être utilisé le jour même.
Inoculum à base d'eau de surface:
Un échantillon d'eau de surface mésosaprobe est filtré sur un papier filtre grossier, les 200 premiers millilitres étant éliminés. Le filtrat, convenablement aéré jusqu'à son utilisation, doit être utilisé le jour même.
Pour obtenir l'inoculum final, on mélange avec soin des volumes égaux de ces trois types d'échantillons d'inoculum. Il faut utiliser au moins 3 ml pour l'inoculation.
|
|
c)
|
Inoculum à base de boue activée:
On peut utiliser comme inoculum un volume (ne dépassant pas 3 l) de boue activée (dont la teneur en solides en suspension peut atteindre jusqu'à 2,5 g/l), prélevé dans le bassin d'aération d'une installation de traitement d'eaux résiduaires essentiellement domestiques.
|
1.6.2. Mode opératoire
L'essai est effectué à la température ambiante; celle-ci doit être maintenue entre 18 oC et 25 oC.
L'essai peut être effectué s'il y a lieu à une température inférieure (qui peut descendre jusqu'à 10 oC): si la substance est dégradée, on peut s'en tenir là. Si toutefois la substance n'est pas dégradée, l'essai doit être effectué à une température constante située entre 18 oC et 25 oC.
1.6.2.1. Période initiale: formation des boues/stabilisation des unités
La période de formation des boues/stabilisation est la période pendant laquelle la concentration des solides en suspension dans les boues activées et les performances des unités augmentent jusqu'à atteindre un état stationnaire dans les conditions de fonctionnement utilisées.
La période initiale est la période qui s'écoule entre la première addition de substance d'essai et le moment où la dégradation atteint un plateau (valeur relativement constante). Cette période ne doit pas dépasser 6 semaines.
La période d'évaluation est une période de 3 semaines à compter du moment où la dégradation de la substance d'essai atteint une valeur relativement constante, habituellement élevée. Pour les substances qui ne se dégradent pas ou se dégradent peu au cours des 6 premières semaines, on prend comme période d'évaluation les 3 semaines suivantes.
Tout d'abord, remplir les unités nécessaires pour un essai avec l'inoculum mélangé au liquide entrant.
Mettre ensuite en marche le dispositif d'admission d'air [et la pompe à air comprimé (E) dans le cas des unités d'essai de confirmation de l'OCDE], ainsi que le doseur (B).
Le liquide entrant ne contenant pas la substance d'essai doit traverser la cuve d'aération à un débit horaire de 1 l ou de ½ l, ce qui donne un temps moyen de rétention de 3 ou 6 heures.
11 faut régler le débit d'air de façon que le contenu de la cuve (C) reste constamment en suspension et que le taux d'oxygène dissout soit au minimum de 2 mg/l.
La formation de mousse doit être empêchée par des moyens appropriés. On n'utilisera cependant pas d'agents antimousse ayant une action inhibitrice sur la boue activée.
La boue qui s'est accumulée au sommet de la cuve d'aération (C) [et, dans le cas des unités d'essai de confirmation de l'OCDE, au fond du décanteur (D) et dans le circuit] doit être renvoyée dans la liqueur mixte au moins une fois par jour par brossage ou par tout autre moyen approprié.
Quand la boue ne décante pas, on peut en augmenter la densité par addition, répétée si nécessaire, de fractions de 2 ml d'une solution à 5 % de chlorure ferrique.
L'effluent est recueilli dans le récipient (E ou F) pendant 20 à 24 heures et on prélève un échantillon après avoir procédé à l'homogénéisation du mélange. Le récipient (E ou F) doit être nettoyé soigneusement.
Pour vérifier l'efficacité du procédé, on mesure au moins deux fois par semaine la demande chimique en oxygène (DCO) ou le carbone organique dissout (COD) du filtrat de l'effluent collecté, ainsi que du liquide entrant filtré [à l'aide d'une membrane dont les pores ont 0,45 μm de diamètre, les 20 premiers millilitres (environ) du filtrat étant éliminés].
La diminution de la DCO ou du COD doit se stabiliser lorsque la dégradation journalière est à peu près régulière.
La teneur en matières sèches de la boue activée contenue dans la cuve d'aération doit être déterminée deux fois par semaine (en g/l). On peut faire fonctionner les installations de deux manières: ou bien la teneur en matières sèches de la boue activée est déterminée deux fois par semaine et si elle est supérieure à 2,5 g/l, il faut éliminer l'excès de boue activée; ou bien on enlève 500 ml de liqueur mixte de chaque vase tous les jours pour obtenir un temps moyen de rétention de la boue de six jours.
Quand les paramètres mesurés et estimés [efficacité du procédé (disparition dé la DCO ou du COD), concentration de la boue, sédimentabilité de la boue, turbidité des effluents, etc.) des deux unités sont suffisamment stables, la substance d'essai peut être introduite dans le liquide entrant dans l'une des unités suivant 1.6.2.2.
Une autre solution consiste à ajouter la substance d'essai au début de la période de formation de la boue (1.6.2.1.), notamment lorsque la boue est ajoutée comme inoculum.
1.6.2.2. Essai
Les conditions de fonctionnement de la période initiale sont maintenues et une quantité suffisante de solution de réserve de la substance d'essai (à environ 1 %) est ajoutée au liquide entrant dans l'unité d'essai, de façon à obtenir la concentration désirée de la substance d'essai (approximativement 10 ou 20 mg de COD/l ou 40 mg de DOC/l) dans les eaux résiduaires. Pour ce faire, on peut soit mélanger quotidiennement la solution de réserve aux eaux résiduaires, soit utiliser un système de pompage séparé. Cette concentration peut être atteinte progressivement. Si la substance d'essai n'a pas d'effets toxiques sur la boue activée, des concentrations plus fortes peuvent être essayées.
Dans l'unité témoin ne sont injectées que les eaux résiduaires. Des volumes adéquats des effluents sont prélevés pour analyse et filtrés sur membrane (0,45 μm), les 20 premiers millilitres (environ) du filtrat étant éliminés.
Les échantillons filtrés doivent être analysés le jour même; sinon, ils doivent être conservés d'une manière adéquate, par exemple à l'aide de 0,05 ml d'une solution de chlorure mercurique à 1 % (HgCl2) pour 10 ml de filtrat ou par stockage à 2 à 4 oC pendant 24 heures ou à moins de — 18 oC pendant plus longtemps,
La période initiale, qui comprend l'addition de la substance d'essai, ne doit pas dépasser 6 semaines et la période d'évaluation ne doit pas être inférieure à 3 semaines, c'est-à-dire qu'environ 14 à 20 déterminations doivent être pratiquées pour le calcul du résultat final.
Couplage des unités:
On réalise le couplage des unités en interchangeant une fois par jour 1,5 l de liqueur mixte (y compris la boue) provenant des cuves d'aération des boues activées entre les deux unités. Dans le cas de substances d'essai très absorbantes, on prélève seulement un volume de 1,5 1 de surnageant dans les cuves de décantation pour le verser dans le récipient de boue activée de l'autre unité.
1.6.2.3. Analyse
Deux types d'analyses peuvent être effectuées en vue de suivre le comportement de la substance:
COD et DCO:
Les concentrations de COD sont déterminées en double avec l'analyseur de carbone et/ou les valeurs de la DCO sont déterminées conformément à la référence (2).
Analyse spécifique:
Les concentrations de la substance d'essai sont déterminées par une méthode d'analyse appropriée. On doit si possible procéder à une détermination spécifique de la substance absorbée sur la boue.
2. DONNÉES ET ÉVALUATION
2.1. COUPLAGE D'UNITÉS
Dans le cas du «couplage des unités», les pourcentages quotidiens de dégradation TD sont calculés conformément à 1.2.1.
Ces taux journaliers de dégradation TD sont, en raison du transfert de matière dû au procédé de transinoculation, corrigés en TDc à l'aide de l'équation (2) pour un temps moyen de rétention de 3 heures ou à l'aide de l'équation (3) pour un temps moyen de rétention de 6 heures.
|

|
(2)
|
|

|
(3)
|
On calcule la moyenne de la série des valeurs de TDc et, en plus, l'écart type selon d'équation (4).
|

|
(4)
|
où:
|
STDc
|
=
|
écart type de la série des valeurs de TDc,
|
|

|
=
|
moyenne des valeurs de TDc,
|
|
n
|
=
|
nombre de déterminations,
|
On élimine les valeurs extrêmes de la série des TDc conformément à une méthode de statistique appropriée, par exemple celle de Nalimov (6), au niveau de probabilité de 95 % et on recalcule la moyenne et l'écart type de la série des données TDc moins ces valeurs extrêmes.
Le résultat final est donné par l'équation (5):
|

|
(5)
|
où:
|
tn –1;α
|
=
|
valeur du tableau de t pour n paires de valeurs de E et E0 et le niveau de confiance statistique P (P = 1-α), où P est fixé à 95 % (1).
|
Le résultat est exprimé par la moyenne avec les limites de tolérance au niveau de probabilité de 95 %, l'écart type et le nombre de données de la série des TDc, moins les valeurs extrêmes, par exemple:
|
TDc
|
=
|
98,6±2,3 % de disparition de COD,
|
|
s
|
=
|
4,65 % de disparition de COD,
|
|
n
|
=
|
18,
|
|
x
|
=
|
nombre de valeurs extrêmes.
|
2.2. UNITÉS NON COUPLÉES
La performance des unités peut être vérifiée comme suit:
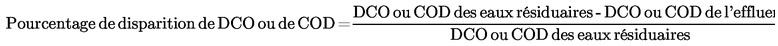
Cette dégradation quotidienne peut être portée sur un graphique montrant par exemple les tendances à l'acclimatation.
2.2.1. Détermination de la DCO/du COD
On calcule le taux quotidien de dégradation TD conformément à 1.2.1.
On calcule la moyenne de la série des valeurs TD et, en plus, son écart type conformément à la formule suivante:
|

|
(6)
|
où
|
STD
|
=
|
écart type de la série des valeurs TDi,
|
|

|
=
|
moyenne des valeurs TDi,
|
|
n
|
=
|
nombre de déterminations.
|
On élimine les valeurs extrêmes de la série des TD conformément à une méthode de statistique appropriée, par exemple celle de Nalimov (6), au niveau de probabilité de 95 % et on recalcule la moyenne et l'écart type de la série des données TD, moins les valeurs extrêmes.
Le résultat final est donné par l'équation (7):
|

|
(7)
|
où:
|
tn - 1;α
|
=
|
valeur du tableau de t pour n paires de valeurs de E et de E0 et le niveau de confiance statistique P (P = 1 — α), où est fixé à 95 % (1).
|
Le résultat est exprimé par la moyenne avec les limites de tolérance au niveau de probabilité de 95 %, l'écart type et le nombre de données de la série des TD moins les valeurs extrêmes, par exemple:
|
TD
|
=
|
(98,6±2,3 %) de disparition de COD,
|
|
s
|
=
|
4,65 % de disparition de COD,
|
|
n
|
=
|
18,
|
|
x
|
=
|
nombre de valeurs extrêmes.
|
2.2.2. Analyse spécifique
On calcule le pourcentage d'élimination de la substance d'essai et de la phase aqueuse (Rw) conformément à 1.2.2.
3. PRÉSENTATION DES RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible:
|
—
|
le formulaire donné à l'annexe III, indiquant les conditions de fonctionnement de l'essai,
|
|
—
|
l'appareillage choisi (essai de confirmation de l'OCDE ou vase poreux),
|
|
—
|
le mode de fonctionnement choisi: unités couplées ou non,
|
|
—
|
les eaux résiduaires: synthétiques ou domestiques. Dans le cas d'eaux résiduaires domestiques: date et lieu de prélèvement de l'échantillon,
|
|
—
|
l'inoculum, avec date et lieu de prélèvement de l'échantillon,
|
|
—
|
une description de la méthode d'analyse si des analyses spécifiques ont été effectuées,
|
|
—
|
le diagramme de la disparition de la DCO ou du COD en fonction du temps pendant la période initiale et la période d'évaluation,
|
|
—
|
une mesure analytique de la substance d'essai sous la forme de DCO ou de COD dans la solution de réserve,
|
|
—
|
si des analyses spécifiques ont été effectuées, le diagramme du pourcentage d'élimination de la substance d'essai de la phase aqueuse en fonction du temps (période initiale et période d'évaluation),
|
|
—
|
la disparition moyenne du COD ou de la DCO de la substance d'essai et l'écart type sont calculés à partir des résultats de la période d'évaluation, c'est-à-dire lorsqu'il y a disparition régulière de la substance d'essai ou qu'on se trouve en période de fonctionnement stationnaire,
|
|
—
|
un diagramme de la concentration des boues activées en fonction du temps,
|
|
—
|
toute remarque concernant les boues activées (rejet d'un excès de boues, présence d'un agglomérat, FeCl3, etc.),
|
|
—
|
la concentration de la substance utilisée pour l'essai,
|
|
—
|
tous les résultats concernant l'analyse faite sur la boue,
|
|
—
|
toutes informations et tous résultats expérimentaux concernant la substance d'essai et, les cas échéant, la substance de référence,
|
|
—
|
les raisons scientifiques de toutes modifications de mode opératoire.
|
3.2. INTERPRÉTATION DES RÉSULTATS
Une faible élimination de la substance d'essai de la phase aqueuse peut être due à une inhibition des micro-organismes par la substance d'essai. Celle-ci peut également être révélée par une lyse et une perte de boue, donnant un surnageant turbide, et par une diminution de l'efficacité de l'installation pilote au point de vue de l'élimination de la DCO (ou du COD).
L'adsorption physico-chimique peut parfois jouer un rôle. Des différences entre l'action biologique sur la molécule et l'adsorption physico-chimique peuvent être révélées par une analyse effectuée sur la boue après une désorption adéquate.
Des essais complémentaires sont nécessaires si l'on veut faire une distinction entre biodégradation (ou biodégradation partielle) et adsorption.
On peut y procéder de différentes façons, mais la plus convaincante consiste à utiliser le surnageant comme inoculum dans un essai du niveau de base de préférence (essai respirométrique).
Les fortes pertes de COD et de DCO éventuellement observées sont dues à la biodégradation, tandis que pour de faibles pertes, on ne peut distinguer la biodégradation de l'élimination. Par exemple, si un composé soluble présente une constante d'adsorption de 98 % et si le taux d'enlèvement de boues excédentaires est de 10 % par jour, une élimination atteignant jusqu'à 40 % est possible; pour un taux d'enlèvement de boues excédentaires de 30 %, l'élimination due à l'adsorption et à l'enlèvement e boues excédentaires peut atteindre jusqu'à 65 % (4).
Dans le cas d'une analyse spécifique, il convient d'accorder une grande attention à la relation entre la structure de la substance et l'analyse spécifique effectuée. Dans ce cas, le phénomène observé ne peut être interprété comme une minéralisation de la substance.
4. BIBLIOGRAPHIE
|
(1)
|
OCDE, Paris, 1981, Ligne directrice 303 A, décision du Conseil C(81) 30 final.
|
|
(3)
|
Painter, H. A., King, E. F., WRC Porous-Pot method for assessmg biodegradability, Technical Report TR70, June 1978, Water Research Center, United Kingdom.
|
|
(4)
|
Wierich, P.,Gerike, P.,Thefateofsoiuble, récalcitrant, and adsorbingcompounds in activated sludge plants, Ecotoxiology and Environmental Safety, vol. 5, No 2, June 1981, p. 161 à 171.
|
|
(6)
|
Strculi, H., Fehlerhafte Interprétation und Anwendung von Ausreifiertests, insbesondere bei Ringversuchen zur Ûberprùfung analytisch-chemischer Untersuchungsmethoden, Fresenius-Zeitschrift fur Analytische Chemie, 303 (1980), p. 406 à 409.
|
Appendice 1
Figure 1

Figure 2
Appendice 2
Figure 1
Équipement utilise pour évaluer la biodégradabilité

Figure 2
Détail du vase d'aération poreux de 3 litres

Appendice 3
Conditions de l'essai de simulation des boues activées
Vérifier dans chaque groupe
Appareillage
|
Confirmation OCDE
|
|
|
Vase poreux
|
|
Mode de fonctionnement
|
Unité simple
|
|
|
Unités couplées
|
|
|
Unités non couplées
|
|
Transinoculation
|
Néant
|
|
|
Boues activées
|
|
|
Surnageant
|
|
Temps moyen de rétention
Élément nutritif de base
|
Eaux résiduaires domestiques
|
|
|
Eaux résiduaires synthétiques
|
|
Inoculum
|
Effluent secondaire
|
|
|
Composite
|
|
|
Boue activée
|
|
Addition de la substance d'essai
|
Dès le début
|
|
|
Augmentation progressive
|
|
|
Après formation de la boue
|
|
Analyse
C. 11 BIODÉGRADATION
BOUES ACTIVÉES; ESSAI D'INHIBITION DE LA RESPIRATION
1. MÉTHODE
1.1. INTRODUCTION
La méthode décrite évalue l'effet d'une substance d'essai sur les micro-organismes en mesurant le rythme de respiration dans des conditions déterminées, en présence de différentes concentrations de la substance.
L'objectif de cette méthode est de fournir un procédé de sélection rapide permettant d'identifier les substances d'essai qui peuvent nuire au fonctionnement des installations de traitement biologique aérobie et de faire une estimation des concentrations adéquates non inhibitrices des substances d'essai à utiliser dans les expériences de biodégradabilité.
Un essai de détermination de l'ordre de grandeur peut précéder l'essai plus complet. Il fournit des informations sur la gamme de concentrations à utiliser dans l'essai principal.
En plus des essais avec la substance étudiée, on effectue deux essais témoins, l'un au début et l'autre à la fin de série expérimentale. Chaque lot de boue activée doit également être vérifié à l'aide d'une substance de référence.
Cette méthode s'applique surtout aux substances qui, en raison de leur solubilité dans l'eau et de leur faible volatilité, sont susceptibles de demeurer dans l'eau.
Pour les substances dont la solubilité dans le milieu d'essai est limitée, il peut ne pas être possible de déterminer la CE50.
Les résultats basés sur la consommation d'oxygène peuvent mener à des conclusions erronées lorsqu'une substance d'essai a tendance à interférer sur la phosphorylation oxydative.
Il est utile de disposer des informations suivantes pour faire l'essai:
|
—
|
pureté de la substance d'essai.
|
Recommandation:
Les boues activées peuvent contenir des organismes pathogènes et doivent être manipulées avec prudence.
1.2. DÉFINITIONS ET UNITÉS
Le taux de respiration est la consommation d'oxygène des micro-organismes aérobies contenus dans les boues ou dans les eaux usées, exprimée généralement en mg 02 par milligramme de boue, par heure.
Pour calculer l'effet inhibiteur d'une substance d'essai, à une concentration donnée, le taux de respiration est exprimé en pourcentage de la moyenne des taux de respiration des deux témoins:

ou:
|
Rs
|
=
|
taux de consommation de l'oxygène à une concentration donnée en substance étudiée,
|
|
Rc1
|
=
|
taux de consommation d'oxygène du Témoin C1,
|
|
Rc2
|
=
|
taux de consommation d'oxygène du témoin C2.
|
Dans cette méthode, la CE50 est la concentration de la substance d'essai pour laquelle le taux de respiration est égal à 50 % de celui des témoins dans les conditions décrites.
1.3. SUBSTANCE DE RÉFÉRENCE
Il est recommandé d'utiliser comme substance de référence le 3,5-dichlorophénol, qui est un inhibiteur connu de la respiration. Pour chaque lot de boue activée, on mesure la CE50 de cette substance, afin de déterminer si la sensibilité de la boue n'est pas anormale.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
On mesure le taux de respiration d'une boue activée alimentée en une quantité donnée d'effluent synthétique après un temps de contact de 30 minutes ou de 3 heures, ou à ces deux moments. On mesure également dans des conditions identiques le taux de respiration de la même boue activée en présence de la substance d'essai à différentes concentrations. L'effet inhibiteur de la substance d'essai à une concentration donnée s'exprime en pourcentage de la valeur moyenne des taux de respiration des deux témoins. À partir des déterminations faites à différentes concentrations, on calcule une valeur de la CE50.
1.5. CRITÈRES DE QUALITÉ
Les résultats sont valables si:
|
—
|
les taux de respiration des deux témoins ne différent pas l'un de l'autre de plus de 15 %,
|
|
—
|
la CE50 (30 minutes et/ou 3 heures) du 3,5-dichlorophénol se situe dans l'intervalle 5 à 30 mg/l.
|
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Réactifs
1.6.1.1. Solutions de la substance d'essai
Des solutions de la substance d'essai sont préparées au début de l'essai à partir d'une solution de réserve. Une solution de réserve à 0,5 g/l convient quand le mode opératoire indiqué ci-dessous est suivi.
1.6.1.2. Solution de la substance de référence
On peut préparer par exemple une solution de 3,5-dichlorophénol en dissolvant 0,5 g de 3,5-dichlorophénol dans 10 ml de 1M NaOH, en diluant à 30 ml à l'eau distillée, en ajoutant, tout en agitant, du 0,5M H2SO4 jusqu'au point où la précipitation commence — environ 8 ml sont nécessaires — et, enfin, en diluant à 1 litre avec de l'eau distillée. Le pH devrait alors être compris dans l'intervalle 7 à 8.
1.6.1.3. Effluent synthétique
On prépare un effluent synthétique en dissolvant les quantités suivantes de substance dans 1 litre d'eau:
|
—
|
11g d'extrait de viande,
|
Note 1: Cet effluent synthétique est 100 fois plus concentré que celui décrit dans le rapport technique de l'OCDE «Méthode proposée pour la détermination de la biodégradabilité des agents tensio-actifs utilisés dans les détergents synthétiques» du 11 juin 1976, et contient en outre de l'hydrogénophosphate de potassium.
Note 2: Si le milieu ainsi préparé n'est pas immédiatement utilisé, il sera stocké à l'obscurité, entre 0 et 4 oC pendant une durée maximale d'une semaine, dans des conditions qui ne conduiront à aucun changement en ce qui concerne sa composition. Le milieu peut aussi être stérilisé avant son stockage, ou bien la peptone et l'extrait de viande peuvent être ajoutés immédiatement avant le début de l'essai. Avant son emploi, le milieu sera agité avec soin et son pH sera ajusté.
1.6.2. Équipement
Appareil de mesure: aucune prescription n'est fournie à ce sujet. Toutefois, il faut veiller à ce qu'il n'y ait pas de volume libre au-dessus du liquide et à ce que l'électrode s'adapte parfaitement au récipient.
Un équipement normal de laboratoire et notamment les articles suivants sont nécessaires:
|
—
|
électrode et appareil de mesure pour le pH,
|
1.6.3. Préparation de l'inoculum
Comme inoculum pour l'essai, on utilise de la boue activée provenant d'une installation de traitement des eaux usées traitant un effluent à dominance domestique.
En cas de nécessité, à l'arrivée au laboratoire, les grosses particules pourront être éliminées par sédimentation pendant une courte période, par exemple pendant 15 minutes, puis par décantation des particules fines de la couche supérieure. Comme solution alternative, la boue peut être agitée quelques secondes au moyen d'un mixeur.
Si des produits inhibiteurs sont présents la boue peut être lavée avec de l'eau du robinet ou avec la solution isotonique. Après centrifugation, le surnageant est décanté (cette manipulation est répétée 3 fois).
Une petite quantité de boue est pesée et séchée. À partir de ce résultat, il est possible de déterminer la quantité de boue humide qui doit être mise en suspension dans l'eau dans le but d'obtenir une boue activée renfermant 2 à 4 g/l de matières en suspension. Cette concentration permet d'obtenir une teneur en matières en suspension comprise entre 0,8 et 1,6 g/l dans le milieu d'essai, si le protocole expérimental recommandé est appliqué.
Si la boue ne peut être utilisée le jour même de sa collecte, on ajoute 50 ml d'effluent synthétique à chaque litre de boue activée préparée de la façon décrite ci-dessus; elle est ensuite aérée toute la nuit à 20 oC ± 2 oC. Elle est alors conservée sous aération en vue de son emploi dans la journée. Avant de l'utiliser, on vérifie le pH et, si nécessaire, on l'ajuste de façon à atteindre un pH se situant entre 6,0 et 8,0. La teneur en matière en suspension du mélange peut être déterminée selon la méthode décrite dans le précédent paragraphe.
Si l'on doit utiliser le même lot de boue pendant plusieurs jours consécutifs (4 au maximum), on ajoute un autre volume de S0 ml d'effluent synthétique par litre de boue à la fin de chaque jour de travail.
1.6.4. Exécution de l'essai
|
Durée/temps de contact:
|
30 minutes et/ou 3 heures, pendant lesquelles le milieu d'essai est aéré
|
|
Eau:
|
Eau potable (déchlorurée, si nécessaire)
|
|
Apport d'air:
|
Air propre, exempt d'huile. Débit d'air de 0,5 à 1 litre par minute
|
|
Appareil de mesure:
|
Fiole a fond plat du type DBO
|
|
Mesure de l'oxygène:
|
Électrode à oxygène adéquate, avec enregistrement
|
|
Solution nutritive:
|
Effluent synthétique (voir ci-dessus)
|
|
Substance d'essai:
|
Préparée au début de l'essai
|
|
Substance de référence:
|
Par exemple 3,5-dichIorophénol (au moins 3 concentrations)
|
|
Témoins:
|
Échantillon inoculé, sans substance d'essai
|
|
Température:
|
20 oC ± 2 oC
|
On trouvera ci-après une méthode expérimentale pouvant être appliquée à la fois pour la substance d'essai et pour la substance de référence pendant une période de 3 heures:
On utilise plusieurs récipients (par exemple béchers d'un litre). On peut utiliser au moins 5 concentrations croissantes, selon une progression de raison n'excédant pas 3,2.
Au temps «O», on verse 16 ml d'effluent synthétique dans un verre cylindrique graduée de 500 ml et l'on complète à 300 ml avec de l'eau. On ajoute 200 ml d'inoculum bactérien et l'on verse l'ensemble du mélange dans le premier récipient (premier témoin C1).
Les récipients d'essai doivent être aérés en continu de façon à être certain que les concentrations en oxygène dissout ne seront pas inférieures à 2,5 mg/l et qu'immédiatement avant détermination du taux de respiration, la concentration en oxygène dissout sera au moins égale à 6,5 mg/l.
Au temps «15 minutes» (15 minutes représentant un intervalle de temps arbitraire, mais commode), on répète l'opération précédente, sauf que l'on ajoute 100 ml de la solution de réserve de la substance d'essai aux 16 ml d'effluent synthétique avant de procéder à l'addition d'eau jusqu'à 300 ml et d'inoculum jusqu'à 500 ml. Ce mélange est ensuite versé dans le deuxième récipient, puis aéré comme ci-dessus. On recommence cette opération toutes les 15 minutes avec différents volumes de la solution de réserve de la substance d'essai, afin d'obtenir une série de récipients avec des concentrations différentes en substance d'essai. Enfin, on prépare un deuxième témoin (C2).
Au bout de trois heures, on mesure et note le pH; un échantillon homogène du contenu du premier récipient est versé dans l'appareil de mesure et le taux de respiration est mesuré pendant une période allant jusqu'à 10 minutes.
Cette détermination est répétée sur le contenu de chaque récipient à des intervalles de 15 minutes, de manière à obtenir un temps de contact de 3 heures pour chaque récipient.
La substance de référence est testée de la même façon sur chaque lot d'inoculum bactérien.
Une organisation différente (par exemple plus d'un appareil de mesure d'oxygène) est nécessaire, si les mesures doivent être faites après un contact de 30 minutes.
S'il est nécessaire de mesurer la consommation chimique en oxygène, on ajoute des récipients supplémentaires contenant la substance d'essai, l'effluent synthétique, de l'eau, mais pas de boue activée.
La consommation d'oxygène est mesurée et enregistrée après un temps d'aération de 30 minutes et/ou de 3 heures (temps de contact).
2. TRAITEMENT ET ÉVALUATION DES DONNÉES
Le taux de respiration est calculé à partir du tracé de l'enregistreur en mg 02/l.h pour les concentrations comprises approximativement entre 6,5 mg de 02/l et 2,5 mg de 02/l, ou pendant une période de dix minutes quand le taux de respiration est faible. La partie de la courbe à partir de laquelle on mesure le taux de respiration doit être linéaire.
Si les taux de respiration des deux témoins diffèrent l'un de l'autre de plus de 15 % ou si la CE50 (30 minutes et/ou 3 heures) de la substance de référence n'est pas dans l'intervalle requis (5 à 30 mg/l pour le 3,5-dichlorophénol), l'essai n'est pas valable et doit être recommencé.
Le pourcentage d'inhibition est calculé pour chaque concentration d'essai (voir 1.2). On porte le pourcentage d'inhibition en fonction de la concentration sur du papier log-normal (ou log-probabilité) et on en déduit une valeur de la CE50.
Des limites de confiance à 95 % pour les valeurs de la CE50 peuvent être déterminées par application de méthodes normalement utilisées.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le procès-verbal d'essai contiendra si possible les renseignements suivants:
|
—
|
substance d'essai: données permettant son identification,
|
|
—
|
système d'essai: origine, concentration et traitement préalable éventuel de la boue activée,
|
|
—
|
conditions d'essai:
|
—
|
pH du mélange avant mesure de la consommation d'oxygène,
|
|
—
|
substance de référence et CE50 mesurée,
|
|
—
|
consommation chimique d'oxygène (s'il y a lieu),
|
|
|
—
|
résultats:
|
—
|
toutes les données obtenues,
|
|
—
|
courbe d'inhibition et méthode de calcul de la CE50,
|
|
—
|
CE50 et, si possible, limites de confiance à 95 %, CE20 et CE50,,
|
|
—
|
toutes les observations et tous les écarts par rapport à la méthode d'essai qui auraient pu influencer les résultats.
|
|
3.2. INTERPRÉTATION DES DONNÉES
La valeur de la CE50 doit simplement être considérée comme une indication de la toxicité probable de la substance d'essai pour le traitement des eaux usées par des boues activées ou pour les micro-organismes des eaux usées. En effet, les interactions complexes qui se produisent dans l'environnement ne peuvent pas être simulées avec précision dans un essai de laboratoire. En outre, les substances testées qui pourraient avoir des effets inhibiteurs sur l'oxydation de l'ammoniaque peuvent fournir des courbes d'inhibition atypiques. En conséquence, de telles courbes doivent être interprétées avec précaution.
4. BIBLIOGRAPHIE
|
(1)
|
International Standard ISO 8192-1986.
|
|
(2)
|
Broecker, B., Zahn, R., Water Research 11,1977, p. 165.
|
|
(3)
|
Brown, D., Hitz, H. R., Schaefer, L., Chemosphere 10, 1981, p. 245.
|
|
(4)
|
ETAD (Ecological and Tox.icological Association of Dyestuffs Manufacturing Industries), Recommended Method no 103, also described by:
|
|
(5)
|
Robra, B., Wasserl Abwasser 117, 1976, p. 80.
|
|
(6)
|
Schefer, W., Textilveredlung 6,1977, p. 247.
|
|
(7)
|
OECD, Paris, 1981, Ligne directrice 209, décision du Conseil C(84) 30 final.
|
C. 12. BIODÉGRADATION
TEST S.C.A.S. MODIFIÉ
1. MÉTHODE
1.1. INTRODUCTION
Le but de la méthode est de mesurer la biodégradabilité totale potentielle de composés organiques non volatils et solubles dans l'eau, lorsqu'ils sont exposés à des concentrations relativement élevées de micro-organismes pendant une longue période. La viabilité des micro-organismes est maintenue tout au long de cette période par un apport journalier d'eaux résiduaires décantées. (Pendant le week-end, les eaux résiduaires peuvent être stockées à 4 oC. Les eaux résiduaires synthétiques de l'essai de confirmation de l'OCDE peuvent également être utilisées.)
Une adsorption physico-chimique sur les particules solides en suspension peut se produire et doit être prise en considération lors de l'interprétation des résultats (voir 3.2).
Étant donné la longue période de rétention de la phase aqueuse (36 heures) et l'ajout régulier de substances nutritives, l'essai n'est pas une simulation des conditions rencontrées dans une station d'épuration des eaux usées. Les résultats obtenus avec diverses substances d'essai indiquent que cet essai a un potentiel élevé de biodégradation.
Les conditions dans lesquelles se déroule l'essai facilitent la sélection et/ou l'acclimatation de micro-organismes susceptibles de dégrader la solution d'essai (la procédure peut également être utilisée en vue de préparer un inoculum acclimaté pour d'autres essais).
Dans cette méthode, on mesure la concentration de carbone organique dissous (COD) pour évaluer la biodégradabilité finale des substances d'essai. Il est préférable de déterminer le COD après acidification et purge plutôt qu'en établissant la différence Ctotal—Cminéral.
Le recours simultané à une analyse spécifique permet de déterminer la biodégradabilité primaire de la substance (disparition de la structure chimique initiale).
La méthode n'est applicable qu'aux substances organiques qui, à la concentration d'essai:
|
—
|
sont solubles dans l'eau (au moins 20 mg de COD par litre),
|
|
—
|
ont une pression de vapeur négligeable,
|
|
—
|
n'ont pas d'effet inhibiteur vis-à-vis des bactéries,
|
|
—
|
ne font pas l'objet d'une adsorption importante au cours de l'essai,
|
|
—
|
ne sont pas éliminées par moussage de la solution d'essai.
|
La teneur en carbone organique de la substance d'essai doit être déterminée.
Il est utile de connaître les proportions relatives des principaux éléments constitutifs de la substance d'essai pour interpréter les résultats obtenus, notamment lorsque ceux-ci sont peu élevés ou marginaux.
Il est souhaitable de connaître le seuil de toxicité de la substance vis-à-vis des micro-organismes pour interpréter les résultats peu élevés et pour choisir les concentrations d'essai appropriées.
1.2. DÉFINITIONS ET UNITÉS
|
CT
|
=
|
concentration de la substance d'essai en carbone organique contenu dans ou ajouté à l'effluent décanté au début de la période d'aération (mg par litre)
|
|
Ct
|
=
|
concentration en COD du liquide surnageant de la substance d'essai à la fin de la période d'aération (mg par litre)
|
|
Cc
|
=
|
concentration en COD du liquide surnageant du témoin à la fin de la période d'aération (mg par litre)
|
Dans cette méthode, la biodégradation est déterminée par la disparition du carbone organique. La dégradation biotique peut être exprimée en:
|
1)
|
pourcentage d'élimination de Dda de l'apport journalier de la substance:
|

|
(1)
|
où:
|
Dda
|
=
|
dégradation/apport journalier;
|
|
|
2)
|
pourcentage d'élimination des substances présentes en début de journée:
|
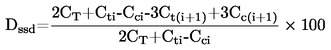
|
[2(a)]
|
|
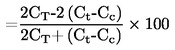
|
[2(b)]
|
où:
|
Dssd
|
=
|
dégradation/substance en début de journée.
|
Les facteurs i et (i + 1) se réfèrent au jour où la mesure a été effectuée.
Il est recommandé d'utiliser l'équation 2(a) si l'effluent COD varie d'un jour à l'autre et l'équation 2(b) lorsque l'effluent COD reste relativement constant d'un jour à l'autre.
|
1.3. SUBSTANCES DE RÉFÉRENCE
Quand on étudie une nouvelle substance, les produits de référence peuvent parfois se révéler utiles; on ne peut cependant pas encore recommander de produits de référence particuliers.
Il est fait mention de données relatives à plusieurs composés qui ont fait l'objet d'essais d'intercomparaison (voir appendice 1), notamment pour pouvoir procéder à l'étalonnage périodique de la méthode et pour permettre de comparer des résultats obtenus par une autre méthode.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
Placer des boues activées provenant d'une station d'épuration des eaux usées dans une unité de traitement de boue activée à alimentation semi-continue (SCAS). Ajouter la substance d'essai et des eaux domestiques décantées et aérer le mélange pendant 23 heures. Arrêter ensuite l'aération, mettre la boue à décanter et retirer le liquide surnageant.
Puis, mélanger les boues restées dans l'aérateur à une autre partie aliquote de substance d'essai et d'eaux résiduaires et répéter le cycle.
La biodégradabilité est mesurée en déterminant la teneur en carbone organique dissous du liquide surnageant. Cette valeur est comparée à celle du liquide provenant d'un témoin alimenté uniquement avec des eaux domestiques décantées.
Lorsqu'on effectue une analyse spécifique, la modification de la concentration de la molécule mère en fonction de la biodégradabilité peut être mesurée (biodégradation primaire).
1.5. CRITÈRES DE QUALITÉ
La reproductibilité de cette méthode basée sur l'élimination de COD n'a pas encore été établie (en ce qui concerne la biodégradation primaire, on obtient des données très précises pour les substances qui sont fortement dégradées).
La sensibilité de la méthode est en grande partie conditionnée par la variabilité du témoin et, dans une moindre mesure, par la précision de la détermination de COD et par la concentration de la substance d'essai dans le liquide au début de chaque cycle.
1.6. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.6.1. Préparations
Réunir un nombre suffisant d'unités d'aération (l'unité d'essai SCAS de 1,5 litre peut également être utilisée) et de tuyaux d'aération (figure 1) pour chaque substance d'essai et de référence. L'air comprimé fourni aux unités d'essai, nettoyées au moyen d'un filtre de coton hydrophile, ne doit pas contenir de carbone organique et doit être saturé préalablement à l'eau pour réduire les pertes dues à l'évaporation.
Prélever un échantillon de liqueur mixte, contenant de 1 à 4 grammes de solides en suspension par litre dans une station d'épuration de boues activées traitant plus particulièrement les eaux ménagères. Prévoir environ 150 ml de liqueur mixte par aérateur.
Préparer la solution mère de la substance d'essai dans de l'eau distillée; la concentration normalement requise est de 400 mg/l de carbone organique, ce qui donne une concentration en carbone de la substance d'essai de 20 mg/l au début de chaque cycle d'aération s'il n'y a pas de biodégradation.
Des concentrations plus élevées peuvent être utilisées si la toxicité vis-à-vis des micro-organismes le permet.
On mesure la teneur en carbone organique de la solution mère.
1.6.2. Mode opératoire
L'essai doit être effectué à une température comprise entre 20 et 25 oC.
Utiliser une concentration élevée de micro-organismes aérobies (de 1 à 4 g/l de matières en suspension) et respecter une période de rétention effective de 36 heures. Largement oxyder les substances carbonées dans l'effluent d'alimentation, normalement pendant une période de 8 heures suivant le début de chaque cycle d'aération. Laisser ensuite respirer la boue en façon endogène pendant le reste de la période d'aération. Le seul substrat disponible est la substance d'essai, à moins qu'elle n'ait été également métabolisée. Ces conditions, combinées à une réinoculation journalière de l'essai lorsque des eaux domestiques sont utilisées comme milieu, sont extrêmement favorables à l'acclimatation et à une biodégradation rapide.
1.6.3. Réalisation de l'essai
Prélever un échantillon de liqueur mixte dans une station d'épuration de boues activées traitant plus particulièrement les eaux domestiques ou dans une unité de laboratoire et le maintenir en aérobiose jusqu'à son utilisation en laboratoire. Chaque aérateur ainsi que l'unité de référence sont remplis avec 150 ml (en cas d'utilisation de l'unité d'essai SCAS originelle, il faut multiplier les volumes donnés par 10) de liqueur mixte et l'aération commence. Après 23 heures, l'aération est arrêtée et on laisse reposer les boues pendant 45 minutes. Ouvrir un à un les robinets de chaque récipient et retirer 100 ml du liquide surnageant. Prélever un échantillon des eaux domestiques décantées immédiatement avant l'utilisation et ajouter 100 ml aux boues demeurant dans chaque aérateur. L'aération est répétée. À ce stade, ne plus ajouter aucune substance d'essai et alimenter journellement les unités avec des eaux ménagères jusqu'à l'obtention d'un liquide surnageant limpide lors de la précipitation. Cette opération prend normalement deux semaines. À ce moment, le COD dans le liquide surnageant à la fin de chaque cycle d'aération tend vers une valeur constante.
À la fin de cette période, mélanger les boues décantées individuellement et ajouter 50 ml de boues composites à chaque unité.
Ajouter 95 ml d'eaux résiduaires décantées et 5 ml d'eau aux unités de référence et 95 m) d'eaux résiduaires décantées et 5 ml de solution mère de la substance d'essai appropriée (400 mg par litre) aux unités d'essai. Procéder à une nouvelle aération pendant 23 heures. Laisser les boues décanter pendant 45 minutes, retirer le liquide surnageant et l'analyser pour déterminer sa teneur en COD.
Cette procédure de prélèvement et de remplissage décrite ci dessus est répétée tous les jours pendant la période de l'essai.
Avant la décantation, il peut s'avérer nécessaire de nettoyer les parois des unités afin d'empêcher l'accumulation de solides au-dessus du niveau du liquide. Pour nettoyer chaque unité, un hérisson ou une brosse séparée sont utilisés afin d'empêcher la contamination croisée.
Théoriquement, il faudrait déterminer journellement le COD dans les liquides surnageants, mais des analyses moins fréquentes sont autorisées. Avant d'être analysées, les liqueurs doivent être filtrées sur une membrane de 0,45 μm ou centrifugées. Les membranes filtrantes conviennent s'il a été prouvé qu'elles ne libèrent pas de carbone et qu'elles n'absorbent pas la substance d'essai au cours de la filtration. La température de l'échantillon ne doit pas dépasser 40 oC lorsqu'il se trouve dans la centrifugeuse.
La durée de l'essai pour les composés peu ou non biodégradables est indéterminée, mais l'expérience démontre qu'il faut compter au moins 12 semaines et qu'il ne faut pas dépasser 26 semaines.
2. DONNÉES ET ÉVALUATION
La courbe de la teneur en COD des liquides surnageants des unités d'essai et des unités de référence est établie en fonction du temps.
Lorsque la biodégradation est terminée, le niveau constaté dans la substance d'essai est voisin de celui du témoin. Lorsque la différence entre ces deux niveaux ne varie plus sur trois mesures consécutives, il suffit alors d'obtenir un nombre de mesures complémentaires suffisantes pour permettre l'analyse statistique des données et de calculer le pourcentage de biodégradation de la substance d'essai (Dda ou Dssd: voir 1.2).
3. PRÉSENTATION DES RÉSULTATS
3.1. PROCÈS-VERBAL D'ESSAI
Le procès-verbal d'essai contiendra si possible:
|
—
|
toutes les informations relatives à la nature des eaux résiduaires, au type d'unité utilisé et aux résultats expérimentaux concernant la substance d'essai, a la substance de référence s'il y a lieu et au témoin,
|
|
—
|
la courbe de disparition avec description, mode de calcul (voir 1.2),
|
|
—
|
la date et l'endroit du prélèvement des échantillons de boues activées et d'eaux résiduaires, le degré d'acclimatation, de concentration etc.,
|
|
—
|
les raisons scientifiques de toute modification du mode opératoire,
|
|
—
|
la signature et la date.
|
3.2. INTERPRETATION DES RESULTATS
Étant donné que, en l'occurrence, la substance d'essai n'est pas facilement biodégradable, toute disparition de COD due uniquement à la biodégradation devrait se faire graduellement sur une période de quelques jours ou de quelques semaines, sauf dans les cas où l'acclimatation est soudaine, ce qui se traduit par une brusque disparition après quelques semaines.
Toutefois, l'adsorption physico-chimique peut parfois jouer un rôle important; cela se manifeste par la disparition partielle ou complète du COD ajouté au départ. Ce qui se passe ultérieurement dépend de facteurs tels que les degrés d'adsorption et de concentration des solides en suspension dans l'effluent écarté. Généralement, la différence de concentration en COD dans les liquides surnageants d'essai et de référence augmente graduellement à partir d'une valeur initiale peu élevée et se maintient au niveau de la nouvelle valeur pour la durée restante de l'expérience, à moins qu'une acclimatation n'intervienne.
Pour distinguer la biodégradation (ou la biodégradation partielle) de l'adsorption, il faut procéder à d'autres essais. Il existe plusieurs méthodes mais la plus probante consiste à utiliser les liquides surnageants ou les boues comme inoculum dans un test du dossier de base (de préférence un test respirométrique).
Les substances d'essai faisant disparaître de grandes quantités de COD sans adsorption doivent être considérées comme potentiellement biodégradables. Une disparition partielle, qui n'est pas due à l'adsorption, indique que le produit est susceptible de se dégrader partiellement.
Des disparitions de COD peu importantes ou nulles peuvent être dues à l'inhibition de micro-organismes par des substances d'essai, ce qui peut également se traduire par lyses et perte de boues; dans ce cas, les liquides surnageants sont troubles. Le test doit alors être répété en utilisant une concentration moins élevée de substances d'essai.
L'utilisation d'une analyse spécifique ou d'une substance d'essai marquée au 14C peut permettre une plus grande sensibilité. Dans le cas d'un composé d'essai marqué au 14C, la récupération de 14C02 confirmera la biodégradation.
Lorsque les résultats sont également exprimés en termes de biodégradation primaire, il convient de donner, dans la mesure du possible, des explications sur les modifications intervenues dans les structures chimiques aboutissant au défaut de réponse de la substance d'essai mère.
La validation de l'analyse doit être indiquée avec! e résultat obtenu dans le milieu de l'essai témoin.
4. BIBLIOGRAPHIE
|
(1)
|
OCDE, Paris, Lignes directrices 302 A, décision du Conseil C(81) 30 final.
|
Appendice 1
Test S.C.A.S.: exemple de résultats
|
Substance
|
CT
(mg/l)
|
Ct — Cc
(mg/l)
|
Pourcentage biodégradation
Dda
|
Durée de l'essai
(jours)
|
|
4-acétyl aminobenzène sulphonate
|
17,2
|
2,0
|
85
|
40
|
|
Tetra propylène benzène sulphonate
|
17,3
|
8,4
|
51,4
|
40
|
|
4-nitrophénol
|
16,9
|
0,8
|
95,3
|
40
|
|
Diéthylène glycol
|
16,5
|
0,2
|
98,8
|
40
|
|
Aniline
|
16,9
|
1,7
|
95,9
|
40
|
|
Cyclopentane tétra carboxylate
|
17,9
|
3,2
|
81,1
|
120
|
Appendice 2
Exemple d'appareil d'essai

C.13 BIOCONCENTRATION: ESSAI AVEC RENOUVELLEMENT CONTINU SUR LES POISSONS
1. MÉTHODE
La présente méthode de bioconcentration reproduit les directives OECD TG 305 (1996).
1.1. INTRODUCTION
La présente méthode décrit une procédure de caractérisation du potentiel de bioconcentration dans le cas de poissons soumis à un renouvellement continu de certaines substances. Les régimes d'essais avec renouvellement continu seront très largement préférés, mais des régimes semi-statiques sont également acceptables dans la mesure où les critères de validité sont satisfaits.
La méthode donne toutes informations nécessaires à l'exécution de fessai mais laisse les libertés indispensables à l'adaptation du concept expérimental aux conditions spécifiques de chaque laboratoire et n'impose pas strictement les caractéristiques des substances à tester. Elle convient particulièrement bien aux produits organiques stables dont les valeurs log Pow sont comprises entre 1,5 et 6,0 (1) mais reste applicable aux substances superlipophiles (log Pow> 6,0). Pour ces dernières, l'estimation préalable du facteur de bioconcentration (BCF), parfois dénommé KB, sera vraisemblablement supérieure à la valeur du facteur de bioconcentration à l'état stable (BCFSS) auquel on peut s'attendre dans une expérience de laboratoire. Les évaluations préliminaires du facteur de bioconcentration des produits organiques dont les valeurs log Pow vont jusqu'à 9,0 environ sont calculables à l'aide de l'équation de Bintein et al (2). Le potentiel de bioconcentration se caractérise par certains paramètres, dont la constante de vitesse d'absorption (k1), la constante de vitesse d'élimination (k2) et le BCFSS.
Les substances d'essai radiomarquées peuvent faciliter l'analyse des échantillons d'eau et de poissons et peuvent aussi servir à définir s'il est souhaitable d'identifier et quantifier la dégradation. Si les résidus radioactifs totaux sont mesurés (par exemple par combustion ou solubilisation des tissus), le BCF est basé sur le composé d'origine, tous les métabolites retenus ainsi que le carbone assimilé. Les BCF basés sur les résidus radioactifs totaux ne peuvent donc pas être directement comparables à un BCF dérivé d'une analyse chimique particulière du composé d'origine seul.
On pourra mettre en œuvre des procédures d'assainissement dans les études radiomarquées pour déterminer le BCF sur ta base du composé d'origine et les principaux métabolites seront déterminés si on le juge nécessaire. Il est possible aussi de combiner une étude du métabolisme du poisson avec une étude de bioconcentration par analyse et identification des résidus tissulaires.
1.2. DÉFINITIONS ET UNITÉS
Bioconcentration/Bioaccumulation: augmentation de la concentration de la substance à tester dans ou sur un organisme (des tissus spécifiques de celui-ci) par rapport à la concentration de cette substance dans le milieu ambiant.
Facteur de bioconcentration (BCF ou Kb): à tout moment pendant la phase d'absorption de l'essai d'accumulation, concentration de la substance à tester dans/sur le poisson, ou des tissus déterminés de celui-ci, [Ct en μg/g (ppm)] divisée par la concentration de la substance chimique dans le milieu ambiant [Cw en μg/ml (ppm)].
Facteur de bioconcentration à l'état stable (BCFSS ou Kb): il ne change pas notablement pendant un laps de temps prolongé, la concentration de la substance d'essai dans le milieu ambiant étant constante pendant cette même période.
Plateau ou état stable; sur la représentation graphique de la substance d'essai dans le poisson (Ct) en fonction du temps, il est atteint lorsque la courbe devient parallèle à l'axe du temps et que trois analyses successives du Cf réalisées sur des échantillons prélevés à des intervalles de deux jours au moins restent dans une plage de 20 % les unes des autres, et qu'il n'y a pas de différences significatives entre les trois périodes d'échantillonnage. Lorsqu'on analyse des échantillons mis en commun, il faut procéder à quatre analyses successives au moins. Si les substances d'essais ont des caractéristiques d'absorption lentes, on optera de préférence pour des intervalles hebdomadaires.
Facteurs de bioconcentration: calculés directement à partir des constantes cinétiques (kl/k2) on les appelle facteurs de concentration cinétique, BCFk.
Coefficient de partage octanol-eau (Pow): rapport de la solubilité d'un produit chimique dans le n-octanol et dans l'eau, à l'équilibre (Méthode A.8), également désigné par Kow. Le logarithme de Pow indique le potentiel de bioconcentration d'un produit chimique par les organismes aquatiques.
Phase d'exposition ou d'absorption: temps pendant lequel les poissons sont exposés au produit chimique teste.
Constante de vitesse d'absorption (k1): valeur numérique définissant la vitesse d'accroissement de la concentration dans la substance à tester dans/sur le poisson (ou des tissus spécifiques de celui-ci) lorsqu'il est exposé à ce produit chimique (k1 est exprimé en jours – 1).
Phase de post-exposition ou d'élimination (perte), à la suite du transfert des poissons d'un milieu contenant la substance à tester à un milieu exempt de celle-ci, temps pendant lequel l'élimination (ou perte nette) de la substance par les poissons de l'essai (ou les tissus spécifiques) est étudiée.
Constante de vitesse d'élimination (perte) (k2: valeur numérique définissant la vitesse de diminution de la concentration de la substance à tester dans les poissons de l'essai (ou les tissus spécifiques) à la suite de leur transfert d'un milieu contenant la substance à tester à un milieu qui en est exempt (k2 exprimé en jours – 1).
1.3. PRINCIPE DE LA MÉTHODE D'ESSAI
L'essai se décompose en deux phases: la phase d'exposition (absorption) et de post-exposition (élimination). Pendant la phase d'absorption, des groupes distincts de poissons d'une espèce sont exposés à au moins deux concentrations de la substance d'essai. Puis ils sont transférés dans un milieu exempt de cette substance, pour la phase d'élimination. Cette dernière est toujours nécessaire sauf lorsque l'absorption de la substance pendant la phase d'absorption a été insignifiante (par exemple si le BCF est inférieur à 10). La concentration de la substance à tester dans/sur le poisson (ou des tissus spécifiques) est suivie pendant les deux phases de l'essai. Outre les deux concentrations de l'essai, un groupe témoin de poissons est maintenu dans des conditions identiques hormis la substance à tester, qui est absente. Ceci pour établir les éventuelles relations entre les effets néfastes observés dans le test de bioconcentration et ce groupe témoin assorti, et déduire les concentrations de fond de la substance à tester.
La phase d'absorption dure 28 jours sauf s'il est démontré que l'équilibre a été atteint plus tôt. On peut prévoir la durée de la phase d'absorption et le temps nécessaire à l'obtention de l'état stable grâce à l'équation donnée à l'annexe 3- La période de dépuration commence alors par le transfert des poissons dans un autre récipient propre contenant le même milieu, exempt cette fois de la substance à tester. Lorsque cela est possible, on calcule le facteur de bioconcentration de préférence à la fois en tant que rapport (BCFss) de concentration des poissons (Cf) et dans l'eau (C .) à l'état d'équilibre apparent et en tant que facteur de bioconcentration cinétique; et BCFk en tant que rapport des constantes de vitesse d'absorption (k1) et de dépuration (k2), en supposant une cinétique de premier ordre. Si, à l'évidence, on n'entre pas dans une cinétique de premier ordre, des modèles plus complexes devront être mis en œuvre (annexe 5).
Si l'on ne parvient pas à un état stable dans les 28 jours, la phase d'absorption sera prolongée jusqu'à cet état stable, ou bien pendant 60 jours, l'alternative la plus courte prévalant; puis commence la phase d'élimination.
La constante de vitesse d'absorption, la constante de vitesse de dépuration (perte) (ou les constantes, lorsque des modèles plus complexes entrent en jeu), le facteur de bioconcentration et, si possible, les limites de confiance de chacun de ces paramètres, sont calculés à partir du modèle décrivant le plus fidèlement les mesures de concentrations de la substance à tester dans les poissons et l'eau.
Le BCF est exprimé en fonction du poids frais total du poisson. Cependant, pour certaines études, des tissus ou organes spécifiques (par exemple muscles, foie) peuvent être utilisés si les poissons sont suffisamment gros ou s'il est possible d'en séparer les parties comestibles (filets) et non comestibles (viscères). La relation claire liant le potentiel de bioconcentration et la lipophilie pour de nombreuses substances organiques, entraîne une relation correspondante entre la teneur en lipides des poissons de l'essai et des bioconcentrations observées de ces substances. Donc, pour réduire cette source de variabilité dans les résultats des essais concernant les substances à forte lipophilie (c'est-à-dire ayant un log Pow > 3), la bioconcentration devrait être exprimée en fonction de la masse corporelle totale, mais aussi de la teneur lipidique.
La teneur lipidique sera établie si possible sur les mêmes matériels biologiques qui auront servi à déterminer la concentration de la substance à tester.
1.4. INFORMATIONS SUR LA SUBSTANCE D'ESSAI
Avant d'entreprendre l'essai de bioconcentration, les informations suivantes devront être réunies au sujet de la substance à tester:
|
a)
|
solubilité dans l'eau;
|
|
b)
|
coefficient de partage octanol-eau Pow, (noté également Kow, déterminé par une méthode HPLC en A.8);
|
|
d)
|
caractéristiques de la phototransformation dans l'eau sous rayonnement solaire ou solaire artificiel et dans les conditions de rayonnement de l'essai de bioconcentration (3),
|
|
e)
|
tension superficielle (c'est-à-dire pour les substances dont le log Pow ne peut être établi;
|
|
g)
|
biodégradabilité dite «facile» (le cas échéant).
|
Il faudra aussi connaître la toxicité relative à l'espèce de poissons utilisée dans l'essai, de préférence la LC50 à l'asymptote (c'est-à-dire indépendante du temps). Il est indispensable de disposer d'une méthode analytique correcte, d'une exactitude, d'une précision et d'une sensibilité connues pour la quantification de la substance à tester dans les solutions d'essai et dans le matériel biologique, ainsi que d'informations précises sur la préparation et le stockage des échantillons. Il faut aussi connaître la limite de détection analytique de la substance à tester tant dans l'eau que dans les tissus des poissons. Si une substance à tester marquée au 14C est utilisée, le pourcentage de radioactivité associée aux impuretés devra être connu.
1.5. CONDITIONS DE VALIDITÉ DE L'ESSAI
Pour qu'un essai soit valide, les conditions suivantes devront être vérifiées:
|
—
|
la variation de température est inférieure à ± 2 oC,
|
|
—
|
la concentration d'oxygène dissous ne tombe pas au-dessous de 60 o/o du niveau de saturation
|
|
—
|
la concentration de la substance à tester dans les enceintes est maintenue à ± 20 % de la moyenne des valeurs mesurées pendant la phase d'absorption,
|
|
—
|
la mortalité et autres effets/maladies indésirables tant parmi les poissons témoins que chez ceux traités sont inférieures à 10 % à la fin de l'essai; lorsque l'essai est prolongé sur plusieurs semaines ou mois, la mortalité ou les autres effets indésirables dans les deux ensembles de poissons devront être inférieurs à 5 % par mois et ne pas excéder 30 % au total.
|
1.6. COMPOSÉS DE RÉFÉRENCE
L'utilisation de composés de référence ayant un potentiel de bioconcentration connu pourra aider au contrôle de la procédure expérimentale, si nécessaire. On ne peut cependant encore recommander de substances spécifiques.
1.7. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.7.1. Appareillage
On prendra soin d'éviter les matériaux susceptibles de présenter un effet indésirable d'adsorption, de dissolution ou de lessivage sur les poissons, et ce pour toutes les pièces de l'équipement. Des réservoirs standards rectangulaires ou cylindriques, en matériaux chimiquement inertes et d'une capacité adaptée au régime de remplissage seront utilisés. On réduira au minimum l'usage de tuyaux en plastique souple. Les tubages en Téflon ®, acier inoxydable et/ou verre auront la préférence. L'expérience a montré que pour les substances présentant de forts coefficients d'adsorption, tels que les pyréthrines de synthèse, le verre silanisé peut être nécessaire. Il faudra, dans ces cas-là, se débarrasser des équipements après usage.
1.7.2. Eau
On utilise généralement pour les essais une eau naturelle provenant d'une source non polluée et de qualité uniforme. L'eau de dilution doit être d'une qualité permettant la survie de l'espèce de poissons choisie, pour la durée des périodes d'acclimatation et d'essai, sans qu'apparaissent aucun comportement ni aspect anormaux. Dans l'idéal, il faudrait démontrer que les espèces soumises à l'essai peuvent survivre, grandir et se reproduire dans l'eau de dilution (par exemple en élevage de laboratoire ou par une étude de toxicité sur un cycle biologique). L'eau sera caractérisée au minimum selon son pH, sa dureté, ses matières solides totales, son carbone organique total et, de préférence aussi, ses teneurs ammoniacales, en nitrates et son alcalinité et, pour les espèces marines, sa salinité. On connaît parfaitement les paramètres importants du bien-être optimal chez les poissons, mais l'annexe 1 indique les concentrations maximales recommandées d'un certain nombre de paramètres concernant les eaux d'essais, douces et marines.
Pendant toute la durée d'un essai, l'eau sera d'une qualité constante. Le pH se tiendra entre 6,0 et 8,5, mais pour un essai donné il restera à l'intérieur d'une plage de ±0,5 unité de pH. Pour s'assurer que l'eau de dilution n'influencera pas indûment les résultats de l'étude (par exemple par complexation de la substance à tester) ou n'affectera pas négativement les performances du stock de poissons, on prélèvera régulièrement des échantillons pour analyses. Il conviendra par exemple de procéder tous les trois mois, lorsqu'une eau de dilution est connue pour être relativement constante en qualité, à la détermination des métaux lourds (par exemple Cu, Pb, Zn, Hg, Cd, Ni), des anions et cations principaux (par exemple Ca, Mg, Na, K, CI, S04), des pesticides (par exemple organophosphorés totaux et organochlorés totaux), du carbone organique total et des solides en suspension. S'il est démontré que la qualité de l'eau est constante pendant une année au moins, ces analyses pourront être moins fréquentes et leurs intervalles espacés (par exemple tous les six mois).
La teneur en particules naturelles ainsi que le carbone organique total (TOC) de l'eau de dilution seront aussi faibles que possible pour éviter l'adsorption de la substance à tester sur les matières organiques, qui pourrait réduire sa biodisponibilité (4). La valeur maximale admissible est de 5 mg/l pour les particules de matière (matière sèche ne traversant pas un filtre de 0,45 μm) et 2 mg/l pour le carbone organique total (voir annexe 1). Si nécessaire, l'eau sera filtrée avant usage. La contribution des poissons de l'expérience (excréta) et des résidus alimentaires, et la teneur de l'eau en carbone organique total devra être aussi faible que possible. Tout au long de l'essai, la concentration en carbone organique dans le récipient concerné ne devra pas dépasser de plus de 10 mg/l (± 20 %) celle du carbone organique provenant de la substance à tester et, le cas échéant, de l'agent de dissolution.
1.7.3. Solutions d'essai
On prépare une solution stock de la substance à tester, à la concentration voulue. La solution stock sera de préférence préparée par simple mélange ou agitation de la substance à tester dans l'eau de dilution. L'utilisation de solvants ou de dispersants (agents de dissolution) n'est pas recommandée; on peut y recourir cependant dans certains cas pour produire une solution stock à la concentration nécessaire. Les solvants susceptibles d'être utilisés sont l'éthanol, le méthanol, l'éther monométhylique d'éthylène glycol, l'éther diméthylique d'éthylène glycol, le diméthylformamide et le triéthylène glycol. Les dispersants utilisables sont le Cremophor RH40, le Tween 80, la méthylcellulose à 0,01 % et le HCO-40. Des précautions seront prises en cas d'utilisation d'agents facilement biodégradables, ces derniers pouvant provoquer des problèmes de croissance bactérienne dans les tests avec renouvellement continu. La substance d'essai peut être radiomarquée et devra être du plus haut niveau de pureté (par ex. de préférence > 98 %).
Pour les essais avec renouvellement continu, un appareillage apportera et diluera en permanence la solution stock de la substance à tester (par exemple pompe doseuse, dilueur proportionnel, système saturateur) pour amener les concentrations d'essai jusqu'aux enceintes. Il sera préférable de prévoir au moins cinq volumes de remplacement par jour pour chaque enceinte d'essai. Le mode avec renouvellement continu sera préféré, mais en cas d'impossibilité (par exemple les organismes de l'essai subissent des effets néfastes) une technique semistatique pourra être mise en œuvre si les critères de validité restent satisfaits. Les régimes d'écoulement des solutions stocks et de l'eau de dilution seront contrôlés 48 heures avant l'essai puis au moins quotidiennement pendant celui-ci. Ce contrôle comportera la détermination du débit dans chaque enceinte d'essai et l'on s'assurera qu'il ne s'écarte pas de plus de 20 % pour chacune d'elles ou bien entre elles et les autres.
1.7.4. Sélection des espèces
Parmi les critères importants de sélection des espèces figurent la facilité de se les procurer, leur taille, et la possibilité d'un entretien aisé en laboratoire. On sélectionnera aussi les espèces de poissons en fonction de leur importance en matière de loisirs, commerce ou écologie; ils doivent également être de sensibilités comparables, avoir produit de bons résultats dans le passé, etc.
On trouvera à l'annexe 2 une liste d'espèces recommandées pour les essais. D'autres espèces peuvent être utilisées mais la procédure d'essai peut devoir être adaptée pour établir des conditions expérimentales convenables. Dans un tel cas, on exposera les motivations du choix des espèces et la méthode expérimentale retenue.
1.7.5. Conservation des poissons
Il faut acclimater la population de poissons pendant deux semaines au moins dans une eau à la température de l'essai et apporter une quantité de nourriture suffisante et du même type que celle qui sera utilisée pendant l'essai.
Après une période d'installation de 48 heures, on enregistre les taux de mortalité et les critères suivants sont appliques:
|
—
|
mortalité supérieure à 10 % de la population en sept jours: rejeter le lot entier,
|
|
—
|
mortalité entre 5 et 10 % de la population en sept jours: prolonger l'acclimatation pendant sept jours de plus,
|
|
—
|
mortalité inférieure à 5 % de la population en sept jours: accepter le lot — si la mortalité dépasse 5 % pendant les sept jours suivants, rejeter le lot entier.
|
S'assurer que les poissons utilisés pour les essais ne présentent pas de maladies ni d'anomalies observables. Écarter tout poisson malade. Les poissons ne sont pas censés être traités contre une quelconque maladie pendant les deux semaines précédant l'essai, ni pendant celui-ci.
1.8. EXÉCUTION DE L'ESSAI
1.8.1. Essai préliminaire
Il peut être utile de procéder à une expérimentation préliminaire pour optimiser les conditions d'exécution de l'essai réel, en ce qui concerne par ex. la sélection de la/des concentration(s) de la substance à tester, la durée des phases d'absorption et d'élimination.
1.8.2. Conditions d'exposition
1.8.2.1. Durée de la phase d'absorption
On peut prévoir la durée de la phase d'absorption en s'appuyant sur une expérience concrète (par ex. à partir d'une étude antérieure ou d'un produit chimique ayant des propriétés d'accumulation) ou à partir de certaines relations empiriques fondées sur ce que l'on sait de la solubilité dans l'eau ou bien du coefficient de partage octanol/eau de la substance à tester (voir annexe 3).
La phase d'absorption durera 28 jours sauf s'il peut être démontré qu'un équilibre a été atteint plus tôt. Si l'on ne parvient pas à un état stable dans les 28 jours, la phase d'équilibre sera prolongée et l'on procédera à d'autres mesures, jusqu'à état stable ou pendant 60 jours, l'alternative la plus courte prévalant.
1.8.2.2. Durée de la phase d'élimination
La moitié de la durée de la phase d'absorption convient généralement pour que se produise une réduction convenable (par exemple 95 %) de la charge corporelle en substance d'essai (voir les explications de l'estimation à l'annexe 3). Si le temps nécessaire pour parvenir à une perte de 95 % est démesurément long, dépassant par exemple de deux fois la durée normale de la phase d'absorption (à savoir plus de 56 jours), on pourra en rester à une période plus courte (c'est-à-dire jusqu'à ce que la concentration de la substance à tester soit inférieure à 10 % de la concentration d'état stable). Cependant, pour les substances ayant des schémas d'absorption et d'élimination plus complexes que le modèle de poissons à compartiment unique, donnant une cinétique de premier ordre, on autorisera des phases de dépuration plus longues pour déterminer les constantes de vitesse d'élimination. Ce laps de temps peut néanmoins être régi par la période durant laquelle la concentration dans les poissons de la substance à tester demeure au-dessus de la limite de détection analytique.
1.8.2.3. Nombre de poissons de l'essai
Choisir les nombres de poissons par concentrations d'essai de telle sorte qu'au minimum quatre poissons par échantillon seront disponibles à chaque échantillonnage. Si l'on souhaite une statistique plus fine, le nombre de poissons par échantillon devra augmenter.
Si on utilise des poissons adultes, indiquer s'ils sont mâles ou femelles ou si les deux sexes servent à l'expérience. Dans ce dernier cas, avant de commencer l'exposition, il faudra vérifier que les différences de teneurs lipidiques ne sont pas significatives; il pourra être nécessaire de faire des lots distincts de mâles et de femelles.
Tous les essais se feront avec un choix de poissons de poids similaires, de sorte que les plus petits ne soient pas inférieurs aux deux tiers du poids des plus gros. Ils appartiendront tous à la même classe d'âge et proviendront de la même source. Le poids et l'âge d'un poisson ayant apparemment parfois des effets notables sur les valeurs BCF (1), ces informations seront enregistrées avec précision. Il est souhaitable qu'un sous-échantillon de la population de poissons soit pesé avant l'essai, pour en estimer le poids moyen.
1.8.2.4. Chargement
Les rapports eau/poissons seront élevés afin de minimiser la réduction du Cw due à l'ajout des poissons au début de l'essai, mais aussi pour éviter les diminutions de concentration en oxygène dissous. Il importe que le régime de chargement soit adapté à l'espèce utilisée pour l'essai. Un régime de chargement de 0,1-1,0 g de poisson (poids humide) par litre d'eau et par jour est recommandé en tout état de cause. On peut réaliser des chargements à hauts régimes s'il est démontré que la concentration voulue en substance à tester est maîtrisable dans des limites de ± 20 % et que la concentration d'oxygène dissous ne tombe pas au-dessous de 60 % du niveau de saturation.
On tiendra compte de l'habitat normal de l'espèce dans le choix du régime de chargement. Les poissons benthiques par exemple peuvent demander un aquarium ayant davantage de surface de fond que les espèces pélagiques, pour un même volume d'eau.
1.8.2.5. Alimentation
Pendant les périodes d'alimentation et d'essai, les poissons sont nourris selon un régime approprié ayant des teneurs en lipides et protéines totales déterminées, en quantités suffisantes pour les maintenir en bonne santé et conserver leur poids corporel. Les poissons sont nourris quotidiennement pendant les périodes d'acclimatation et d'essai à raison d'environ 1 à 2 % de leur poids corporel chaque jour; on maintient ainsi pour la plupart des espèces une concentration lipidique d'un niveau relativement constant pendant l'essai. La quantité de nourriture devra être recalculée une fois par semaine par exemple, afin de maintenir un poids corporel et des teneurs lipidiques cohérents. Pour ce calcul, le poids des poissons de chaque enceinte d'essai sera estimé à partir du poids des poissons échantillonnés le plus récemment dans cette enceinte. Ne pas peser les poissons restant dans l'enceinte.
La nourriture non consommée et les excréments sont évacués par siphonage quotidien des enceintes d'essai peu après le nourrissage (30 minutes à une heure). Les enceintes seront tenues aussi propres que possible tout au long de l'essai de sorte que la concentration de matière organique reste au plus bas niveau envisageable, car la présence de carbone organique peut limiter la biodisponibilité de la substance à tester. (1)
Nombre d'aliments étant des dérivés de farines de poissons, ils seront analysés pour déterminer leur teneur en substance d'essai. Il est préférable aussi d'analyser les teneurs de ces nourritures en pesticides et métaux lourds.
1.8.2.6. Éclairage et température
La photopériode est généralement de 12 à 16 heures et la température (+ 2 oC) correspondra à l'espèce utilisée (voir annexe 2). Le type et les caractéristiques de l'éclairage seront nettement définis. Il faudra prendre garde aux phototransformations éventuelles de la substance à tester dans les conditions de rayonnement de l'étude. L'éclairage ne devra pas exposer les poissons à des photoproduits non naturels. Dans certains cas, il peut être essentiel d'utiliser un filtre pour bloquer les rayons UV inférieurs à 290 nm.
1.8.2.7. Concentrations de l'essai
Les poissons sont exposés a un renouvellement continu de deux concentrations aqueuses au moins de la substance à tester. Normalement, la concentration haute (ou la plus haute) de la substance à tester sera d'environ 1 % de sa LC50 aiguë à l'asymptote, et au moins 10 fois supérieure à sa limite de détection dans l'eau par la méthode d'analyse retenue.
La plus forte concentration de l'essai peut aussi être établie en divisant la LC50 à 96 heures par un ratio approprié aigu/chronique (les ratios adéquats de certains produits chimiques peuvent aller de 3 à 100). Si possible, choisir l'autre (les autres) concentration(s) de sorte que la différence avec celle qui lui est supérieure atteigne un facteur dix. En cas d'impossibilité due au critère de 1 % de la LC.50 et de la limite analytique, un facteur inférieur à dix pourra être utilisé ou bien on songera à l'intervention d'une substance radiomarquée du 14C. Aucune concentration mise en œuvre ne devrait dépasser la solubilité de la substance d'essai.
Lorsqu'un agent de dissolution est utilisé sa concentration ne doit pas dépasser 0,1 ml/1 et doit être identique dans tous les récipients de l'essai. Sa contribution, ajoutée à celle de la substance à tester, à la teneur globale en carbone organique dans l'eau de l'expérience doivent être connues. Tout sera néanmoins fait pour éviter le recours à ce type de matériels.
1.8.2.8. Témoins
Une eau de dilution témoin ou, si nécessaire, un témoin contenant l'agent de dissolution sera disponible en marge de la série des tests, dans la mesure où il a été établi que l'agent n'a aucun effet sur les poissons. Dans le cas contraire, on mettra en place les deux témoins.
1.8.3. Fréquence des mesures de la qualité de l'eau
Pendant l'essai, l'oxygène dissous, le TOC, le pH et la température seront mesurés dans tous les récipients. La dureté totale, et la salinité éventuellement, seront mesurées dans les témoins et un récipient contenant la concentration haute (la plus haute). Au minimum, l'oxygène dissous et éventuellement la salinité seront mesurés trois fois — au début, vers le milieu et à la fin de la période d'absorption — et une fois par semaine pendant la période d'élimination. Le TOC sera mesuré au début de l'essai (24 et 48 heures avant le démarrage de la phase d'absorption), avant l'ajout des poissons et au moins une fois par semaine pendant les périodes d'absorption et d'élimination. La température sera mesurée quotidiennement, le pH au début et à la fin de chaque période et la dureté une fois pour chaque essai. La température sera de préférence surveillée en continu dans un récipient au moins.
1.8.4. Échantillonnage et analyse des poissons et de l'eau
1.8.4.1. Calendrier d'échantillonnage des poissons et de l'eau
De l'eau sera prélevée dans les enceintes d'essai pour établir la concentration de la substance a tester avant l'ajout des poissons et pendant les phases d'absorption et de dépuration. Au minimum, l'eau est prélevée en même temps que les poissons et avant leur alimentation. Pendant la phase d'absorption, les concentrations de substance à tester sont déterminées pour vérifier qu'elles satisfont aux critères de validité.
Les poissons sont échantillonnés en cinq occasions au moins pendant la phase d'absorption et en quatre occasions au moins pendant la phase de dépuration. Il est parfois difficile de calculer une valeur estimative raisonnablement précise du BCF sur la base de ce nombre d'échantillons, en particulier lorsque l'on sort du cadre d'une simple cinétique de dépuration de premier ordre. Dans ce cas, on pourra alors échantillonner à des rythmes plus rapides dans les deux périodes (voir annexe 4). Les échantillons supplémentaires sont stockés et analysés uniquement si les résultats de la première série d'analyses se révèlent insuffisants au calcul du BCF avec la précision voulue.
On trouvera à l'annexe 4 un exemple de calendrier d'échantillonnage acceptable. D'autres peuvent être facilement établis sur la base d'autres valeurs supposées du POW pour calculer le temps d'exposition correspondant à une absorption de 95 %.
On poursuit l'échantillonnage pendant la phase d'absorption jusqu'à établissement d'un état stable ou pendant 28 jours, l'alternative la plus courte prévalant. Si l'on ne parvient pas a un état stable en 28 jours, l'échantillonnage continue jusqu'à obtention d'un état stable ou pendant 60 jours, l'alternative la plus courte prévalant. Avant de commencer la phase d'élimination, les poissons sont transférés dans des réservoirs propres.
1.8.4.2. Échantillonnage et préparation des échantillons
On prélève les échantillons d'eau destinés aux analyses, par exemple en siphonnant à l'aide d'un tube inerte un point central de l'enceinte d'essai. Puisqu'il semble que l'on ne peut séparer ni par centrifugation ni par filtration la fraction non biodisponible de la substance à tester de celle qui est biodisponible (en particulier dans le cas des produits chimiques super-lipophiles, c'est-à-dire ceux ayant un log POw > 5) (1) (5), on peut s'abstenir de soumettre les échantillons à ces traitements.
Par contre, il faudra veiller à ce que les réservoirs restent aussi propres que possible et à ce que la teneur en carbone organique total soit surveillée tout au long des phases d'absorption et d'élimination.
Un nombre convenable de poissons (normalement quatre au minimum) est prélevé dans chaque enceinte d'essai pour chaque échantillonnage. Les poissons prélevés sont rapidement rincés à l'eau, séchés avec un papier absorbant, et sacrifiés instantanément de la manière la plus humaine possible, puis pesés.
Il est préférable d'analyser les poissons et l'eau immédiatement après l'échantillonnage pour éviter toute dégradation ou autres pertes et pour calculer approximativement les régimes d'absorption et de dépuration pendant que l'essai se poursuit. L'analyse immédiate évite aussi de ne pas retarder le constat qu'un plateau a été atteint.
Faute d'une analyse immédiate, les échantillons sont conservés selon une méthode convenable. Avant le début de l'étude, on réunira toutes informations sur la méthode de stockage adéquate de cette substance à tester, par exemple congélation, maintien à 4 oC, durée du stockage, extraction, etc.
1.8.4.3. Qualité de la méthode analytique
L'intégralité de la procédure étant régie principalement par l'exactitude, la précision et la sensibilité de la méthode analytique mise en œuvre pour la substance à tester, il faut contrôler expérimentalement que la précision et la reproductibilité de l'analyse chimique, ainsi que la récupération de la substance à tester tant dans l'eau que dans les poissons donnent toute satisfaction dans le cadre particulier de cette méthode. Vérifier aussi que la substance à tester n'est pas détectable dans l'eau de dilution utilisée.
Si nécessaire, les valeurs de Cw et Cf dérivées des essais seront corrigées au vu des valeurs fournies par les témoins et la concentration naturelle de la substance d'essai. Les échantillons de poissons et d'eau sont manipulés en permanence de manière à minimiser les pollutions et les pertes (résultant par exemple de l'absorption par le matériel d'échantillonnage).
1.8.4.4. Analyse des poissons échantillonnés
Si l'on utilise pour l'essai des matériels radiomarqués, on peut analyser le marquage radioactif total (c.à.d. composés d'origine et métabolites) ou purifier les échantillons de sorte que le composé d'origine puisse être analysé séparément. En outre, les principaux métabolites peuvent être caractérisés soit à l'état stable soit à la fin de la phase d'absorption, l'alternative la plus courte prévalant. Si le BCF en termes de résidus radiomarqués totaux est > 1 000 %, il peut être conseillé (et, pour certaines catégories de produits chimiques tels que les pesticides, fortement recommandé), d'identifier et quantifier les produits de dégradation représentant ≥10 % des résidus totaux dans les tissus des poissons à l'état stable. Si ces produits représentant ≥ 10 % des résidus totaux radiomarqués dans les tissus de poissons sont identifies et quantifiés, il est alors recommandé de les identifier et les quantifier aussi dans l'eau de l'essai.
La concentration de la substance à tester devrait généralement être déterminée pour chaque poisson pesé. Si cela n'est pas possible, on pourra mettre en commun les échantillons à chaque échantillonnage, mais cette mise en commun restreint véritablement les procédures statistiques dont les données pourraient bénéficier. Si une procédure et une finesse statistiques spécifiques sont souhaitées, il conviendra alors de faire participer à l'essai un nombre adéquat de poissons pour satisfaire à la procédure de regroupement et à la précision statistique désirées (6) (7).
Le BCF sera exprimé à la fois en fonction du poids frais total et, pour les substances fortement lipophiles, en fonction de la teneur lipidique. La teneur lipidique des poissons est établie si possible lors de chaque échantillonnage. Des méthodes bien adaptées seront utilisées pour déterminer la teneur en lipides (réf. 8 et 2 de l'annexe 3). On recommande les techniques d'extraction par chloroforme/méthanol comme méthode standard (9). Toutes les méthodes ne donnant pas des valeurs identiques (10), il importe de bien préciser celle utilisée. L'analyse des lipides sera si possible faite sur les mêmes extraits que ceux consacrés à l'analyse de la substance à tester, puisque les lipides doivent souvent être enlevés de l'extrait avant qu'on puisse l'analyser par chromatographie. La teneur lipidique des poissons (en mg/kg de poids frais) à la fin de l'expérience ne devrait pas s'écarter de celle du début de ± 25 %. Le pourcentage de solides dans les tissus sera lui aussi indique pour permettre la conversion de la concentration lipidique de la base humide à la base sèche.
2. DONNÉES
2.1. TRAITEMENT DES RÉSULTATS
La courbe d'absorption de la substance à tester résultera du tracé de sa concentration, en fonction du temps, dans/sur les poissons (ou des tissus spécifiques) pendant la phase d'absorption, sur une échelle arithmétique. Si la courbe a atteint un plateau, c'est-à-dire adopte approximativement un tracé asymptotique à Taxe du temps, l'état stable BCFss sera calculé comme suit:

Lorsqu'on ne parvient à aucun état stable, il reste possible de calculer un BCFss d'une précision suffisante pour l'évaluation des risques à partir d'un «état stable» à 80 % (1,6/k2) ou 9,5 % (3,0/k2) de l'équilibre.
En outre, le facteur de concentration (BCFk) sera défini comme le rapport k1/k2, des deux constantes cinétiques de premier ordre. La constante de vitesse d'élimination (k2) est généralement déterminée à partir de la courbe d'élimination (c.a.d. un tracé de la décroissance en fonction du temps de la concentration de la substance à tester dans le poisson). La constante de vitesse d'absorption (kt) est alors calculée en fonction de k2 et d'une valeur de C1 dérivée de la courbe d'absorption (voir aussi annexe 5). La méthode de choix pour obtenir BCFk et les constantes de vitesse k1 et k2, est de recourir à des méthodes informatiques non linéaires d'estimation des paramètres (11). On peut alternativement utiliser des méthodes graphiques pour calculer kt et k2. Si la courbe de dépuration est à l'évidence autre que de premier ordre, alors il conviendra d'utiliser des modèles plus complexes (voir références à l'annexe 3) et de demander conseil à un biostatisticien.
2.2. INTERPRÉTATION DES RÉSULTATS
Les résultats seront interprétés avec précaution lorsque les concentrations de solution d'essai mesurées se trouvent à des niveaux proches de la limite de détection de la méthode d'analyse.
La clarté de définition des courbes d'absorption et d'élimination témoigne de la bonne qualité des données de bioconcentration. La variation des constantes absorption/élimination entre les deux concentrations d'essai doit être inférieure à 20 %. Les différences notables observées dans les taux d'absorption/élimination entre les deux essais de concentration seront enregistrées et expliquées si possible. En général, la limite de confiance des BCF approche ± 20 % dans les études bien connues.
3. RAPPORT
Le procès-verbal d'essai doit comporter les informations suivantes:
3.1. SUBSTANCE D'ESSAI
|
—
|
nature physique et, le cas échéant, propriétés physico-chimiques,
|
|
—
|
données d'identification chimique (y compris la teneur en carbone organique, si besoin),
|
|
—
|
en cas de marquage radioactif, position précise du/des atome(s) marqué(s) et pourcentage de radioactivité associée aux impuretés.
|
3.2. ESPÈCES UTILISÉES
|
—
|
Dénomination scientifique, souche, source, tout traitement préalable éventuel, acclimatation, âge, gamme des tailles, etc.
|
3.3. CONDITIONS DE L'ESSAI
|
—
|
procédure mise en œuvre (par exemple renouvellement continu ou semi-statique),
|
|
—
|
type et caractéristiques de l'éclairage utilisé et photopériode(s),
|
|
—
|
concept de l'essai (par exemple nombre et taille des enceintes d'essai, régime de remplacement des volumes d'eau, nombre de sous-échantillons et de poissons par sous-échantillon, nombre de concentrations testées, durée des phases d'absorption et de dépuration, fréquence des échantillonnages pour les poissons et l'eau),
|
|
—
|
méthode de préparation des solutions mères et fréquence de renouvellement (l'agent de dissolution, sa concentration et sa contribution à la teneur en carbone organique de l'eau de l'essai seront indiqués, le cas échéant),
|
|
—
|
les concentrations d'essai nominales, les moyennes des valeurs mesurées et leurs écarts-types dans les récipients d'essai et leur méthode d'obtention,
|
|
—
|
source de l'eau de dilution, description de tout traitement préalable, résultats de toute démonstration de l'aptitude des poissons utilisés à vivre dans cette eau et caractéristiques de celle-ci: pH, dureté, température, concentration en oxygène dissous, niveaux de chlore résiduel (si mesuré) carbone organique total, solides en suspension, salinité du milieu d'essai (le cas échéant) et toutes autres mesures réalisées,
|
|
—
|
qualité de l'eau dans les récipients d'essai, pH, dureté, TOC, température et concentration en oxygène dissous,
|
|
—
|
renseignements précis sur l'alimentation (par exemple type de nourriture, source, composition — au moins teneurs en lipides et protéines si possible, quantité donnée et fréquence),
|
|
—
|
renseignements sur le traitement des poissons et de l'eau échantillonnés, y compris détails de préparation, stockage, extraction, procédures (et précision) de l'analyse de la substance à tester et teneur en lipides (si mesurée).
|
3.4. RÉSULTATS
|
—
|
résultats de toute étude préliminaire effectuée,
|
|
—
|
mortalité des poissons témoins et des poissons dans chaque enceinte d'essai et tous comportements anormaux observés,
|
|
—
|
teneur lipidique des poissons (si détermination à l'occasion de l'essai),
|
|
—
|
courbes (dont toutes données mesurées) montrant l'absorption et l'élimination des produits chimiques de l'essai dans les poissons, le temps d'accès à l'état stable,
|
|
—
|
Cf et Cw (avec écart-type et plage, le cas échéant) au moment de chaque échantillonnage (Cf exprimé en μg/g de poids frais (ppm) de tout l'animal ou de certains de ses tissus, par exemple lipides et Cw en μg/ml (ppm). Valeurs Cw de la série témoin (indiquer aussi la concentration de fond),
|
|
—
|
facteur de bioconcentration à l'état stable (BCFss) et/ou facteur de concentration cinétique (BCFk) et, le cas échéant, limites de confiance à 95 % des constantes de vitesse d'absorption et d'élimination (perte) toutes exprimées par rapport au corps entier et de la teneur totale en lipide, si elle est mesurée, de l'animal (ou certains de ses tissus), limites de confiance et écart-type (si disponible), méthodes de calcul/analyse des données pour chaque concentration de substance d'essai utilisée,
|
|
—
|
lorsqu'on utilise des substances radiomarquées, l'accumulation de tous les métabolites détectés pourra être signalée si cela est nécessaire,
|
|
—
|
toute situation inhabituelle concernant l'essai, tout écart de ces procédures et toutes autres informations pertinentes.
|
Minimiser les résultats du type «non détecté à la limite de détection» par la mise en place d'une méthode d'essai préliminaire et l'élaboration d'un concept expérimental, car ces résultats sont inutilisables pour les calculs des constantes de vitesse.
4 BIBLIOGRAPHIE
|
(1)
|
Connell D.W. (1988). Bioaccumulation behaviour of persistent chemicals with aquatic organisms. Rev. Environ. Contam. Toxicol. 102, pp 117-156.
|
|
(2)
|
Bintein S., Devillers, J. and Karcher W. (1993). Nonlinear dependence of fish bioconcentration on n-octanol/water partition coefficient. SAR and QSAR in Environmental Research, 1, 29-390.
|
|
(3)
|
OECD, Paris (1996). Direct Phototransformation of chemicals in water. Environmental Health and Safety Guidance Document Séries on Testing and Assessment of Chemicals No 3.
|
|
(4)
|
Kristensen P. (1991). Bioconcentration in fish: Comparison of bioconcentration factors derived from OECD and ASlTM testing methods; influence of particulate organic matter to the bioavaila-bility of chemicals. Water Quality Institute, Denmark.
|
|
(5)
|
US EPA 822-R-94-002 (1994) Great Lake Water Quality Initiative Technical Support Doc. for the Procédure to Détermine Bioaccumulation Factors. July 1994.
|
|
(6)
|
US FDA (Food and Drug Administration) Revision. Pesticide analytical manual, 1, 5600 Fisher's Lane, Rockville, MD 20852, July 1975.
|
|
(7)
|
US EPA (1974). Section 5, A(l) Analysis of Human or Animal Adipose Tissue, in Analysis of Pesticide Residues in Human and Environmental Samples, Thompson J.F. (ed.) Research Triangle Park, N.C. 27711.
|
|
(8)
|
Compaan H. (1980) in The détermination of the possible effects of chemicals and wastes on the aquatic environ ment: dégradation, toxicity, bioaccumulation' Ch. 2.3, Part II. Government Publishing Office, The Hague, The Netherlands.
|
|
(9)
|
Gardner et al, (1995) Limn. & Oceanogr. 30, 1099-1105.
|
|
(10)
|
Randall R.C., Lee H., Ozretich R.J., Lake J.L. and Pruell R.J. (1991). Evaluation of selected lipid methods for normalising pollutant bioaccumulation. Envir. Toxicol. Chem. 10, pp 1431-1436.
|
|
(11)
|
CEC, Bioconcentration of chemical substances in fish: the flow-through method-Ring Test Programme, 1984-1985. Final report March 1987. Authors: P. Kristensen and N. Nyholm.
|
|
(12)
|
ASTM E-1022-84 (Reapproved 1988) Standard Practice for conducting Bioconcentration Tests with Fishes and Saltwater Bivalve Molluscs.
|
Annexe 1
Caractéristiques chimiques acceptables pour l'eau de dilution
|
|
Substance
|
Limite de concentration
|
|
1
|
Particules de matière
|
5 mg/l
|
|
2
|
Carbone organique total
|
2 mg/l
|
|
3
|
Ammoniaque non ionisé
|
1 μg/l
|
|
4
|
Chlore résiduel
|
10 μg/l
|
|
5
|
Pesticides organophosphorés totaux
|
50 μg/l
|
|
6
|
Pesticides organochlorés totaux plus polychlorobiphényles
|
50 ng/l
|
|
7
|
Chlore organique total
|
25 ng/l
|
|
8
|
Aluminium
|
1 μg/l
|
|
9
|
Arsenic
|
1 μg/l
|
|
10
|
Chrome
|
1 μg/l
|
|
11
|
Cobalt
|
1 μg/l
|
|
12
|
Cuivre
|
1 μg/l
|
|
13
|
Fer
|
1 μg/l
|
|
14
|
Plomb
|
1 μg/l
|
|
15
|
Nickel
|
1 μg/l
|
|
16
|
Zinc.
|
1 μg/l
|
|
17
|
Cadmium
|
100 ng/l
|
|
18
|
Mercure
|
100 ng/l
|
|
19
|
Argent
|
100 ng/l
|
Annexe 2
Espèces de poissons recommandées pour les essais
|
|
Espèces recommandées
|
Plages de températures recommandées pour les essais (oC)
|
Longueur totale recommandée des animaux utilisés (cm)
|
|
1
|
Danio rerio (17) (Teleostei, Cyprinidae) (Hamilton-Buchanan) Petit Danio
|
20-25
|
3,0±0,5
|
|
2
|
Pimephales promelas (Teleostei, Cyprinidae) (Rafinesque) Tète de boule
|
20-25
|
5,0±2,0
|
|
3
|
Cyprinus carpio (Teleostei, Cyprinidae) (Linnaeus) Carpe
|
20-25
|
5,0±3,0
|
|
4
|
Oryzias latipes (Teleostei, Poeciliidae) (Temminck and Schlegel)
|
20-25
|
4,0±1,0
|
|
5
|
Poecilia reticulata (Teleostei, Poeciliidae) (Peters) Guppy
|
20-25
|
3,0±1,0
|
|
6
|
Lepomis macrochirus (Teleostei, Centrarchidae) (Rafinesque) Crapet arlequin
|
20-25
|
5,0±2,0
|
|
7
|
Oncorhynchus mykiss (Teleostei, Salmonidae) (Walbaum) Truite de Kamloops
|
13-17
|
8,0±4,0
|
|
8
|
Gasterosteus aculeatus (Teleostei, Gasterosteidae) (Linnaeus) Epinoche épineuse
|
18-20
|
3,0±1,0
|
Diverses espèces d'estuaires et marines ont été utilisées dans certains pays par exemple:
|
Tambour croca
|
Leiostomus xanthurus
|
|
Pétote
|
Cyprinodon variegatus
|
|
Siouclet
|
Menidia beryllina
|
|
Perche-méné
|
Cymatogaster aggregata
|
|
Carlottin anglais
|
Parophrys vetulus
|
|
Chabot
|
Leptocottus armatus
|
|
Epinoche épineuse
|
Gasterosteus aculeatus
|
|
Bar
|
Dicentracus labrax
|
|
Ablette
|
Alburnus alburnus
|
Récolte
Les poissons d'eau douce énumérés dans le tableau ci-dessus sont faciles à élever et/ou largement disponibles tout au long de l'année, alors que la disponibilité des espèces marines ou d'estuaires est en partie limitée à certains pays. Ils peuvent se reproduire et vivre soit dans des fermes piscicoles soit en laboratoire, dans des conditions où les maladies et parasites sont sous contrôle; les animaux utilisés seront donc sains et de lignées connues. On les trouve à peu près partout dans le monde.
Annexe 3
Prévision de la durée des phases d'absorption et d'élimination
1. Prévision de la durée de la phase d'absorption
Avant d'exécuter l'essai, on pourra estimer k2 et donc un pourcentage du temps nécessaire pour parvenir à l'état stable, à partir de relations empiriques entre k2 et le coefficient de partage n-octanol/eau (Pow) ou k2 et la solubilité dans l'eau (s).
On estimera k2 (jour-1) à partir, par exemple, de la relation empirique suivante (1):
|
log10 k2 = -0,414 log10(Pow) + 1,47 (r2 = 0,95)
|
(equation 1)
|
Pour les autres relations, voir réf. (2).
Si le coefficient de partage (Pow) est inconnu, on procédera à une estimation (3) à partir de la solubilité dans l'eau (s) de la substance d'après:
|
log10 (Pow) = 0,862 log10(s) + 0,710 (r2 = 0,994)
|
(equation 2)
|
où
s= solubilité (moles/l): (n = 36).
Ces relations ne s'appliquent qu'aux produits chimiques dont les valeurs log Pow se situent entre 2 et 6,5 (4).
Le temps nécessaire pour atteindre un certain pourcentage de l'état stable résultera de l'équation cinétique générale illustrant l'absorption et l'élimination (cinétique de premier ordre), par application du k2 estimé:

où, si Cw est constant:
|

|
(equation 3)
|
Lorsque l'on se rapproche de l'état stable (t → ∞), l'équation 3 se réduit (5) (6) à:
 or Cf/Cw = k1/k2
= BCF.
or Cf/Cw = k1/k2
= BCF.
Le rapport k1/k2 . Cw approche alors la concentration dans le poisson à «l'état stable» (Cfs).
L'équation 3 peut être ainsi transcrite:
|

ou

|
(equation 4)
|
Le temps nécessaire pour atteindre un certain pourcentage de l'état stable peut être prévu grâce à l'équation 4 lorsque k2, est estimé au préalable à l'aide des équations 1 ou 2.
À titre indicatif, la durée optimale statistique de la phase d'absorption permettant d'obtenir des données statistiquement acceptables (BCFk) est la période nécessaire pour que la courbe du logarithme de la concentration de la substance à tester dans le poisson, portée en temps linéaire, parvienne à son point médian, soit à 1,6/k2, ou à 80 % de l'état stable, mais ne dépasse pas 3,0/k, ou 95 % de l'état stable (7).
Le temps d'accession à 80 % de l'état stable est (équation 4):
|

ou

|
(equation 5)
|
De même, pour 95 % de l'état stable:
|

|
(equation 6)
|
Par exemple, la durée de la phase d'absorption (abs.) d'une substance à tester ayant un log Pow = 4 serait (avec les équations 1, 5 et 6):
|
log10k2 = -0,414.(4) + 1,47
|
k2 = 0,652 jours-1
|
abc. (80 %) = 1,6/0,652, c'est à dire. 2,45 jours (59 heures)
ou abc. (95 %) = 3,0/0,652, c'est à dire. 4,60 jours (110 heures)
De même, pour une substance à tester avec s = 10 - 5 mol/l (log(s) = -5,0), la durée de l'absorption serait (avec les équations 1, 2, 5 et 6):
log10 (Pow) = 0,862 (-5,0) + 0,710 = 5,02
log10 K2 = 0,414 (5,02) + 1,47
k2 = 0,246 jours -1
abc. (80 %) = 1,6/0,246, c'est à dire 6,5 jours (156 heures)
ou abc. (95 %) = 3,0/0,246, c'est à dire 12,2 jours (293 heures)
On utilisera aussi, à titre d'alternative, l'expression:
teq = 6,54 × 10-3 Pow + 55,31 (heures)
pour calculer le temps d'accession effective à l'état stable (4).
2. Prévision de la durée de la phase d'élimination
On pourra également prévoir le temps nécessaire pour que la charge corporelle se réduise à un certain pourcentage de la concentration initiale grâce à l'équation générale illustrant l'absorption et l'élimination (cinétique de premier ordre) (1) (8).
Pour la phase d'élimination, Cw est supposé nul. L'équation peut être réduite à:
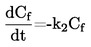 ou
ou 
où Cfo est la concentration au début de la période d'élimination. On parviendra ensuite à une élimination de 50 % à l'instant (t50):
 ou
ou

De même, 95 % d'élimination seront atteints à:

Si l'on se place à 80 % de l'absorption pour la première période (1,6/k2) et 95 % de perte dans la phase d'élimination (3,0/k2), alors la phase d'élimination est approximativement le double de la durée de la phase d'absorption.
Il importe cependant de noter que ces estimations sont fondées sur l'hypothèse que les schémas d'absorption et d'élimination suivent une cinétique de premier ordre. Si l'on n'est pas, à l'évidence, dans ce cadre, des modèles plus complexes devront être employés [par exemple réf (1)].
Bibliographie (de l'annexe 3)
|
(1)
|
Spacie A. and Hamelink J.L. (1982) Alternative models for describing the bioconcentration of organics in fish. Environ. Toxicol. and Chem. 1, pp 309-320.
|
|
(2)
|
Kristensen P. (1991) Bioconcentration in fish: comparison of BCF's derived from OECD and ASTM testing methods; influence of particulate matter to the bioavailability of chemicals. Danish Water Quality Institute.
|
|
(3)
|
Chiou C.T. and Schmedding D.W. (1982) Partitioning of organic compounds in octanol-water systems. Environ. Sci. Technol. 16 (1), pp 4-10.
|
|
(4)
|
Hawker D.W. and Connell D.W. (1988) Influence of partition coefficient of lipophilic compounds on bioconcentration kinetics with fish. Wat. Res. 22 (é), pp 701-707.
|
|
(5)
|
Branson D.R., Blau G.E., Alexander H.C. and Neely W.B. (1975) Transactions of the American Fisheries Society, 104 (4), pp 785-792.
|
|
(6)
|
Ernst W. (1985) Accumulation in Aquatic organisms. In: Appraisal of tests to predict the environmental behaviour of chemicals. Ed. by Sheehman P., Korte F., Klein W. and Bourdeau P.H. Part 4.4 pp 243-255. SCOPE, 1985, John Wiley & Sons Ltd. N.Y.
|
|
(7)
|
Reilly P.M., Bajramovic R., Blau G.E., Branson D.R. and Sauerhoff M.W. (1977) Guidelines for the optimal design of experiments to estimate parameters in first order kenetic models, Can. J. Chem. Eng. 55, pp 614-622.
|
|
(8)
|
Könemann H. and Van Leeuwen K. (1980) Toxicokinetics in fish: Accumulation and Elimination of six Chlorobenzenes by Guppies. Chemosphere, 9, pp 3-19.
|
Annexe 4
Exemples théoriques de calendriers d'échantillonnage pour les tests de bioconcentration des substances ayant un log Pow = 4
|
Prélèvements de poissons
|
Calendrier d'échantillonnage
|
Nombre d'échantillons d eau
|
Nombre de poissons par échantillon
|
|
Fréquence minimale requise (jours)
|
Échantillonnage supplémentaire
|
|
Phase d'absorption
|
– 1
0
|
|
2 (18)
2
|
ajouter 45-80 poissons
|
|
1er
|
0,3
|
0,4
|
2
(2)
|
4
(4)
|
|
2e
|
0,6
|
0,9
|
2
(2)
|
4
(4)
|
|
3e
|
1,2
|
1,7
|
2
(2)
|
4
(4)
|
|
4e
|
2,4
|
3,3
|
2
(2)
|
4
(4)
|
|
5e
|
4,7
|
|
2
|
6
|
|
Phase de dépuration
|
|
|
|
Transférer les poissons dans une eau exempte du produit à tester
|
|
6e
|
5,0
|
5,3
|
|
4
(4)
|
|
7e
|
5,9
|
7,0
|
|
4
(4)
|
|
8e
|
9,3
|
11,2
|
|
4
(4)
|
|
9e
|
14,0
|
17,5
|
|
6
(4)
|
|
Les valeurs entre parenthèses sont les nombres d'échantillons (eau, poissons) à prélever si l'on procède a un échantillonnage supplémentaire.
|
Note
|
:
|
L'estimation avant essai de k2, pour un log Pow de 4,0 est de 0,652 jour –1. La durée totale de l'expérience est fixée à: 3 × abs. = 3 × 4,6 jours, soit 14 jours. Se reporter a l'annexe 3 pour l'estimation de l'«absorption».
|
|
Annexe 5
Discrimination des modèles
On suppose que la plupart des données de bioconcentration sont «raisonnablement» bien décrites par un modèle simple à deux compartiments/deux paramètres, selon la courbe rectiligne qui reprend approximativement les points de mesure des concentrations dans les poissons pendant la phase d'élimination, sur papier semilogarithmique. (Lorsque ces points ne peuvent se ramener à une droite, il convient de recourir à des modèles plus complexes, voir par exemple Spacie and Hamelink, réf. 1 à l'annexe 3.)
Méthode graphique de détermination de la constante de vitesse d'élimination (perte) k2
Porter sur du papier semilogarithmique la concentration de la substance à tester trouvée dans chaque échantillon de poisson, selon le calendrier d'échantillonnage. La pente de cette droite est k2


Attention: les écarts par rapport à cette ligne droite peuvent indiquer un schéma d'élimination plus complexe qu'une cinétique de premier ordre. Une méthode graphique peut résoudre des types d'élimination s'écartant de la cinétique de premier ordre.
Méthode graphique de détermination de la constante de vitesse d'absorption kt
K2 étant établi, on calcule k1 comme suit:
|
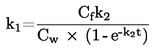
|
[equation 1]
|
On lit la valeur de Cf au point médian de la courbe d'absorption lissée produite par les données lorsque la concentration logarithmique est portée en fonction du temps (sur une échelle arithmétique).
Méthode de calcul informatique des constantes de vitesse d'absorption et d'élimination (perte)
Pour obtenir le facteur de bioconcentration et les constantes de vitesse k1 et k2 on utilisera de préférence des méthodes informatiques non linéaires d'estimation paramétriques. Ces programmes établissent les valeurs de k1 et k2 en fonction d'un ensemble de données séquentielles de concentration dans le temps et du modèle:
|

0 < t < tc
|
[equation 2]
|
|

t < tc
|
[equation 3]
|
où te = temps à la fin de la phase d'absorption.
Cette démarque débouche sur des estimations de l'écart-type pour k1 et k2
Puisqu'on peut dans la plupart des cas estimer k2, à partir de la courbe d'élimination et ce avec une précision relativement grande, et puisqu'il existe une forte corrélation entre les deux paramètres k1 et k2, il peut être souhaitable de calculer d'abord k2, à partir des données d'élimination uniquement, puis de calculer k1, à partir des données d'absorption à l'aide d'une régression non linéaire.
C.14. POISSON, ESSAI SUR LA CROISSANCE DES JUVÉNILES
1. MÉTHODE
La méthode décrite pour cet essai de toxicité sur la croissance des juvéniles reprend la ligne directrice no 215 de l'OCDE (2000).
1.1. INTRODUCTION
Cet essai vise à évaluer les effets d'une exposition prolongée à des produits chimiques sur la croissance des poissons au stade juvénile. Il s'appuie sur une méthode mise au point et soumise à des essais tournants (1) (3) dans l'Union européenne, qui a pour objet d'évaluer les effets de substances chimiques sur la croissance de la truite arc-en-ciel (Oncorynchus mvkiss) au stade juvénile dans des conditions dynamiques. D'autres espèces de poissons bien étudiées peuvent être utilisées. Par exemple, on possède une certaine expérience des essais de croissance sur le danio (Danio rerio) (19) (2) (4) (5) et sur le medaka (Oryzias latipes) (6) (7) (8).
Voir aussi introduction générale, partie C.
1.2. DÉFINITIONS
Concentration minimale avec effet observé (CMEO): la plus faible concentration de la substance d'essai à laquelle on observe un effet significatif (à p < 0,05) par rapport au témoin. Cependant, toutes les concentrations d'essai supérieures à la CMEO doivent exercer un effet nocif égal ou supérieur à celui observé à la CMEO.
Concentration sans effet observé (CSEO): la concentration d'essai immédiatement inférieure à la CMEO.
CEX
: dans la méthode décrite pour cet essai, la concentration de la substance d'essai qui provoque une variation de x % du taux de croissance des poissons par rapport aux témoins.
Taux de charge: le poids frais des poissons par unité de volume d'eau.
Densité de peuplement: le nombre de poissons par unité de volume d'eau.
Taux de croissance spécifique de chaque poisson: le taux de croissance d'un individu par rapport à son poids initial.
Taux de croissance spécifique moyen par récipient: le taux de croissance moyen de la population d'un récipient (cuve) à une concentration donnée.
Taux de croissance pseudo-spécifique: le taux de croissance individuel comparé au poids initial moyen de la population du récipient.
1.3. PRINCIPE DE L'ESSAI
Des juvéniles en phase de croissance exponentielle sont pesés, puis placés dans des enceintes expérimentales où ils sont exposés à une gamme de concentrations sublétales de la substance d'essai dissoute dans l'eau, de préférence dans des conditions dynamiques ou, à défaut, dans des conditions semi-statiques (renouvellement discontinu) adéquates. L'essai dure 28 jours. Les poissons sont nourris quotidiennement. La ration alimentant est déterminée en fonction du poids initial des poissons et peut être recalculée après 14 jours. On pèse à nouveau les poissons à la fin de l'essai. Les effets sur le taux de croissance sont analysés à l'aide d'un modèle de-régression afin d'estimer la concentration qui provoquerait une variation de x % du taux de croissance, soit CEX (CE10, CE20 ou CE30 par exemple). Les données peuvent aussi être comparées aux valeurs des témoins pour déterminer la concentration minimale avec effet observé (CMEO) et, de là, la concentration (maximale) sans effet observé (CSEO).
1.4. INFORMATIONS SUR LA SUBSTANCE D'ESSAI
Il faudrait disposer des résultats d'un essai de toxicité aigué (voir méthode d'essai C.1) réalisé de préférence sur l'espèce choisie pour le présent essai. Cela implique que la solubilité dans l'eau et la pression de vapeur de la substance d'essai soient connues et que l'on dispose d'une méthode d'analyse fiable pour déterminer la quantité de substance dans les solutions d'essai avec une précision et une limite de détection connues et mentionnées dans le rapport.
Il est utile de connaître la formule structurale, la pureté de la substance, la stabilité dans l'eau et à la lumière, le pKa, le Poe et les résultats d'un essai de biodégradabilité immédiate (voir méthode d'essai C.4).
1.5. VALIDITÉ DE L'ESSAI
Pour qu'un essai soit valide, les conditions suivantes doivent être remplies:
|
—
|
la mortalité dans le(s) groupe(s) témoin(s) ne doit pas dépasser 10 % a la fin de l'essai,
|
|
—
|
le poids moyen des poissons du (des) groupe(s) témoin(s) doit avoir augmenté suffisamment pour que la variation minimale du taux de croissance qui est jugée significative soit détectable. Un essai tournant (2) a montré que, dans le cas de la truite arc-en-ciel, le poids moyen des poissons dans les groupes témoins doit avoir au moins doublé (soit 50 %) par rapport à leur poids moyen initial sur 28 jours; par exemple, poids initial: 1 g/poisson (= 100 %), poids final après 28 jours: > 1,5 g/poisson (> 150 %),
|
|
—
|
la concentration de l'oxygène dissous doit se situer à au moins 60 % de la valeur de saturation en air tout au long de l'essai,
|
|
—
|
à aucun moment, durant l'essai, la température de l'eau ne doit varier de plus de ± 1 oC entre les enceintes expérimentales et devrait être maintenue dans un intervalle de 2 oC compris dans la plage spécifiée pour l'espèce testée (voir appendice 1).
|
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Appareillage
On utilise du matériel courant de laboratoire et notamment:
|
a)
|
un pH-mètre et un appareil à mesurer l'oxygène;
|
|
b)
|
un instrument pour mesurer la dureté et l'alcalinité de l'eau;
|
|
c)
|
un dispositif adéquat de régulation de la température avec, de préférence, une surveillance en continu;
|
|
d)
|
des récipients (cuves) fabriqués en un matériau chimiquement inerte et d'une capacité adaptée à la charge et à la densité de peuplement recommandées (voir point 1.8.5 et appendice 1);
|
|
e)
|
une balance suffisamment précise (précision de ±0,5 %).
|
1.6.2. Eau
Toute eau dans laquelle l'espèce testée présente des taux de survie et de croissance à long terme adéquates peut être utilisée pour l'essai. Sa qualité doit demeurer constante pendant toute la durée de l'essai. Le pH de l'eau sera compris dans un intervalle de 6,5 à 8,5 unités, mais sans varier de plus de ±0,5 unité (de pH) au cours d'un même essai. Une dureté supérieure à 140 mg/l (CaC03) est recommandée, Pour s'assurer que l'eau de dilution n'influencera pas indûment le résultat de l'essai (par exemple en complexant la substance d'essai), on prélèvera des échantillons à différents intervalles pour analyse. Il y a lieu de mesurer la teneur en métaux lourds (Cu, Pb, Zn, Hg, Cd, Ni, par exemple), en principaux anions et cations (Ca, Mg, Na, K, Cl, S04, par exemple), en pesticides (total des pesticides organophosphorés et organochlorés, par exemple), en carbone organique total et en solides en suspension. Ces mesures devraient être effectuées tous les trois mois, par exemple, pour une eau de dilution dont on sait que la qualité est relativement constante. Si la qualité de l'eau s'est avérée constante durant au moins un an, on pourra espacer les déterminations (par exemple tous les 6 mois). Certaines caractéristiques chimiques requises pour une eau de dilution acceptable sont énumérées a l'appendice 2.
1.6.3. Solutions d'essai
Par dilution d'une solution mère, on prépare des solutions d'essai aux concentrations choisies.
La solution mère devrait, de préférence, être préparée par simple mélange ou agitation de la substance d'essai dans l'eau de dilution par des moyens mécaniques (agitateur ou ultrasons, par exemple). Des colonnes de saturation (colonnes de solubilité) peuvent être utilisées pour amener la solution mère à la concentration souhaitée.
L'emploi de solvants ou de dispersants (agents solubilisants) peut être indiqué dans certains cas pour obtenir une solution mère à la concentration voulue. Les solvants qui conviennent sont, par exemple, l'acétone, l'éthanol, le méthanol, le diméthylsulfoxyde, le dimethylformamide et le triethylèneglycol. Parmi les dispersants adéquats. citons le Cremophor RH40. le Tween 80, la méthylcellulose à 0,01 % et le HCO-40. Il convient d'être vigilant lorsqu'on utilise des agents facilement biodégradables (comme l'acétone) et/ou des composés très volatils car ils peuvent entraîner une prolifération bactérienne dans les essais dynamiques. Lorsqu'on utilise un agent solubilisant, il ne doit pas avoir d'effet significatif sur la croissance des poissons ni d'effets nocifs visibles sur les juvéniles, ce que révélera l'observation d'un témoin ne comprenant que le solvant.
Les essais dynamiques nécessitent un système qui délivre et dilue en continu une solution mère de la substance d'essai (par exemple, une pompe doseuse, un dilueur proportionnel, un système de saturation), pour distribuer une série de concentrations dans les enceintes d'essai. Les débits des solutions mères et de l'eau de dilution doivent être contrôlés à intervalles quotidiens de préférence, au cours de l'essai, et ne devraient pas varier de plus de 10 % tout au long de celui-ci. Un essai tournant (3) a montré que, pour la truite arc-en-ciel, une fréquence de renouvellement de l'eau au cours de l'essai de 6 litres/g de poisson/jour était acceptable (voir point 1.8.2.2).
S'agissant des essais semi-statiques (essais avec renouvellement), la fréquence de renouvellement du milieu dépendra de la stabilité de la substance d'essai, mais un renouvellement quotidien de l'eau est recommandé. Si des essais de stabilité préliminaires (voir point 1.4) révèlent que la concentration de la substance n'est pas stable (c'est-à-dire qu'elle sort d'un intervalle de 80 à 120 % de la concentration nominale ou qu'elle tombe en dessous de 80 % de la concentration mesurée initialement) au cours de la période de renouvellement, il faudra envisager de pratiquer un essai dynamique.
1.6.4. Sélection de l'espèce
La truite arc-en-ciel (Oncorhynchas mykiss) est recommandée dans cet essai car c'est à propos de cette espèce que l'on a acquis la plus grande expérience au cours d'essais tournants (1) (3). D'autres espèces bien étudiées peuvent cependant être utilisées, mais le mode opératoire devra éventuellement être adapté afin d'offrir les conditions adéquates. Par exemple, on dispose également d'une certaine expérience au sujet du danio (Danio rerio) (4) (5) et du medaka (Oryzias latipes) (6) (7) (8). Dans ce cas, le choix de l'espèce et de la méthode expérimentale doit être justifié.
1.6.5. Soins des poissons
Les poissons d'essai seront sélectionnes au sein d'une même population, issue de préférence du même frai, qui aura été gardée pendant au moins deux semaines avant l'essai dans des conditions de qualité de l'eau et d'illumination similaires à celles de l'essai. Ils devraient recevoir une ration alimentaire quotidienne atteignant au moins 2 % de leur poids corporel et de préférence 4 % pendant toute la durée des soins et de l'essai.
Après une période de mise en condition de 48 heures, on mesure la mortalité et on applique les critères suivants:
|
—
|
mortalité supérieure à 10 % de la population en sept jours: le lot entier est rejeté,
|
|
—
|
mortalité comprise entre 5 et 10 % de la population: période d'acclimatation prolongée de sept jours, si la mortalité dépasse 5 % pendant la deuxième période de sept jours, le lot entier est rejeté,
|
|
—
|
mortalité inférieure à 5 % de la population en sept jours: le lot est accepté.
|
Les poissons ne devraient pas recevoir un traitement pour une maladie durant les deux semaines précédant l'essai ou pendant l'essai.
1.7. CONCEPTION DE L'ESSAI
Par «conception de l'essai», on entend le choix du nombre et de l'espacement des concentrations d'essai, le nombre de récipients par concentration et le nombre de poissons par récipient. Idéalement, l'essai devrait être conçu en fonction de:
|
a)
|
l'objectif de l'étude:
|
|
b)
|
la méthode d'analyse statistique qui sera utilisée;
|
|
c)
|
la disponibilité et le coût des ressources expérimentales.
|
L'énoncé de l'objectif devrait, si possible, spécifier la puissance statistique que requiert une différence donnée (par exemple, dans le taux de croissance) pour être détectée, ou encore la précision avec laquelle la CE, doit être fournie (avec x = 10, 20 ou 30, par exemple et de préférence pas au-dessous de 10) pour être estimée. Faute de quoi il est impossible de donner une indication précise de l'échelle de l'étude.
Il importe de reconnaître qu'une conception qui est optimale (qui tire le meilleur parti des ressources) lorsqu'on utilise une certaine méthode d'analyse statistique ne l'est pas nécessairement avec une autre méthode. La conception recommandée pour l'estimation d'une CMEO/CSEO ne sera donc pas identique à celle recommandée pour une analyse par régression.
Dans la plupart des cas, l'analyse par régression est préférable à l'analyse de la variance, pour des raisons exposées par Stephan et Rogers (9) Cependant, si on ne trouve aucun modèle de régression adéquat (r2 < 0,9), il faut recourir à la CSEO/CMEO.
1.7.1. Conception pour l'analyse par régression
Les considérations importantes pour la conception d'un essai à analyser par régression sont les suivantes:
|
a)
|
La concentration avec effet (par exemple, CE10, 20, 30) et la gamme de concentrations dans laquelle la substance d'essai produit un effet intéressant doivent nécessairement être couvertes par les concentrations incluses dans l'essai. La précision avec laquelle les concentrations produisant un effet peuvent être estimées sera la meilleure lorsque la concentration avec effet se trouve au milieu de la plage des concentrations testées. Un essai préliminaire de détermination de l'ordre de grandeur peut s'avérer utile pour sélectionner les concentrations d'essai appropriées.
|
|
b)
|
Pour satisfaire les conditions de la modélisation statistique, l'essai doit comporter au moins un récipient témoin et cinq autres à différentes concentrations. Le cas échéant, lorsqu'on utilise un agent solubilisant, il faudrait étudier un groupe témoin contenant l'agent solubilisant à la concentration d'essai la plus élevée en plus des groupes d'essai (voir paragraphes 1.8.3 et 1.8.4).
|
|
c)
|
Une série géométrique ou logarithmique appropriée (10) (voir appendice 3) peut être utilisée. Un espacement logarithmique entre les concentrations d'essai est préférable.
|
|
d)
|
Si on dispose de plus de six récipients, les récipients supplémentaires devraient servir de répliques ou être répartis dans la gamme des concentrations de manière à diminuer l'espacement entre les concentrations. Ces deux mesures sont aussi valables l'une que l'autre.
|
1.7.2. Conception pour l'estimation de la CSEO/CMEO à l'aide de l'analyse de la variance
Il serait préférable d'avoir plusieurs récipients répliques pour chaque concentration, et d'effectuer l'analyse statistique au niveau du récipient (11). Sans récipients répliques, il est impossible de prendre en compte la variabilité entre les récipients en plus de celle qui existe d'un poisson à l'autre. L'expérience a cependant montré (12) que la variabilité entre les récipients était très inférieure à celle qui existe au sein de chaque récipient (autrement dit, entre les poissons) dans le cas examiné. C'est pourquoi une alternative relativement acceptable consiste à effectuer l'analyse statistique au niveau de chaque poisson.
On doit normalement utiliser au moins cinq concentrations d'essai selon une série géométrique obéissant à un facteur qui ne dépasse pas de préférence 3.2.
Généralement, lorsque les essais sont conduits dans des récipients répliques, le nombre de récipients témoins répliques, et par conséquent le nombre de poissons, devrait être le double du nombre choisi à chaque concentration d'essai, qui devrait être toujours le même (13) (14) (15). A contrario, sans récipients répliques, le nombre de poissons dans le groupe témoin devrait être le même que dans chaque concentration d'essai.
Si l'analyse de la variance est conduite au niveau des récipients plutôt qu'à celui des poissons [ce qui exigerait un marquage individuel des poissons ou l'utilisation de taux de croissance «pseudo-spécifiques» (voir paragraphe 2.1.2)], il faut un nombre suffisant de récipients répliques pour pouvoir déterminer l'écart-type entre les «récipients de même concentration». Ce qui signifie que l'erreur dans l'analyse de la variance ait au moins 5 degrés de liberté (1l). Si seuls les témoins ont des répliques, la variabilité de l'erreur risque d'être faussée puisqu'elle pourrait s'accroître avec la valeur moyenne du taux de croissance en question. Comme le taux de croissance a des chances de diminuer lorsque la concentration augmente, on aura tendance à surestimer la variabilité.
1.8. MODE OPÉRATOIRE
1.8.1. Sélection et pesée des poissons d'essai
Il importe que le poids des poissons varie le moins possible au début de l'essai. On trouvera à l'appendice 1 les gammes de poids qui conviennent aux différentes espèces recommandées pour cet essai. Idéalement, au début de l'essai, la gamme des poids de l'ensemble du lot de poissons utilisés dans l'essai devrait rester comprise dans un intervalle de ± 10 % de la moyenne arithmétique et en aucun cas excéder 25 %. Il est recommandé de peser un sous-échantillon de poissons avant l'essai afin d'estimer le poids moyen.
Les poissons doivent être privés de nourriture pendant les 24 heures qui précèdent le début de l'essai. Ils sont ensuite choisis au hasard. À l'aide d'un anesthésique général [par exemple, une solution aqueuse de 100 mg/litre de méthanesulphonate de tricaïne (MS 222) neutralisée par l'adjonction de deux parts de bicarbonate de sodium par part de MS 222], les poissons doivent être pesés individuellement en poids frais (blolted dry, poissons essuyés) avec la précision indiquée à l'appendice 1. Les poissons dont le poids se situe à l'intérieur de l'intervalle voulu seront retenus puis répartis au hasard entre les récipients d'essai. On notera le poids frais (wet weighl) total des poissons dans chaque récipient, l'utilisation d'anesthésique de même que la manipulation des poissons (y compris le séchage et la pesée) peuvent stresser et blesser les jeunes poissons, en particulier les espèces de petite taille. Les juvéniles doivent donc être manipulés avec la plus grande précaution afin d'éviter de stresser et de blesser les animaux testés.
Les poissons sont pesés à nouveau le 28e jour de l'essai (voir point 1.8.6). Si toutefois on juge nécessaire de recalculer la ration alimentaire, on pèsera à nouveau les poissons au 14e jour de l'essai (voir point 1.8.2.3). On pourra recourir à une autre méthode telle que la technique photographique par exemple pour évaluer les variations de taille des poissons à partir de quoi la ration alimentaire pourra être ajustée.
1.8.2. Conditions d'exposition
1.8.2.1. Durée
La durée de l'essai est de ≥ 28 jours.
1.8.2.2. Taux de charge et densité de peuplement
Il importe que le taux de charge et la densité soient adaptés à l'espèce testée (voir appendice 1). Si la densité de peuplement est trop élevée, le stress engendré par la surpopulation entraînera une diminution des taux de croissance, et probablement la maladie. Si elle est trop faible, elle peut induire un comportement territorial susceptible d'affecter également la croissance. En tout état de cause, le taux de charge doit être suffisamment bas pour que la concentration de l'oxygène dissous puisse être maintenue à au moins 60 % de la valeur de saturation en air, sans aération. Un essai tournant (3) a montré que, dans le cas de la truite arc-en-ciel, un taux de charge de 16 truites de 3 à 5 grammes dans un volume de 40 litres était acceptable. La fréquence de renouvellement de l'eau recommandée durant l'essai est de 6 litres/g de poissons par jour.
1.8.2.3. Alimentation
Les poissons doivent recevoir une alimentation adéquate (voir appendice 1) en quantité suffisante pour permettre un taux de croissance acceptable. Il faut veiller à éviter la prolifération microbienne et la turbidité de l'eau. Dans le cas de la truite arc-en-ciel, une ration quotidienne de 4 % de son poids corporel par jour devrait remplir ces conditions (3) (16) (17) (18). La ration quotidienne peut être divisée en deux parts égales et administrée aux poissons en deux fois, à au moins 5 heures d'intervalle. La ration est fonction du poids total initial des poissons dans chaque récipient d'essai. Si les poissons sont pesés à nouveau le 14e jour, on recalcule la ration. Les poissons doivent être privés de nourriture pendant les 24 heures qui précèdent leur pesée.
Les aliments non consommés et les matières fécales doivent être enlevés quotidiennement des récipients d'essai par un nettoyage soigneux du fond de chaque récipient à l'aide d'un suceur.
1.8.2.4. Lumière et température
La photopériode et la température de l'eau d'essai doivent être adaptées à l'espèce testée (voir appendice 1).
1.8.3. Concentrations d'essai
On doit normalement utiliser cinq concentrations de la substance d'essai, quelle que soit la conception de l'essai (voir point 1.7.2). Une connaissance préalable de la toxicité de la substance d'essai (tirée par exemple d'un essai de toxicité aiguë et/ou d'études de détermination de l'ordre de grandeur) devrait faciliter le choix des concentrations d'essai adéquates. L'utilisation de moins de cinq concentrations doit être justifiée. La concentration d'essai la plus élevée ne doit pas dépasser la limite de solubilité de la substance dans l'eau.
Si un agent solubilisant est utilisé pour faciliter la préparation de la solution mère, sa concentration finale ne doit pas dépasser 0,1 ml/1 et doit, de préférence, être identique dans tous les récipients d'essai (voir point 1.6.3). Le recours à ce genre de produit devrait cependant être évité dans toute la mesure du possible.
1.8.4. Témoins
Le nombre de récipients témoins contenant l'eau de dilution dépend de la conception de l'essai (voir points 1.7-1.7.2). Si l'on utilise un agent solubilisant, on inclura le même nombre de témoins pour l'eau de dilution que pour l'agent solubilisant.
1.8.5. Fréquence des analyses quantitatives et des mesures
Au cours de l'essai, les concentrations de la substance à tester sont déterminées à intervalles réguliers (voir ci-après).
Dans les essais dynamiques, les débits du diluant et de la solution mère de substance toxique seront vérifiés périodiquement, de préférence chaque jour. Ces débits ne devraient pas varier de plus de 10 % tout au long de l'essai. Lorsque les concentrations de la substance d'essai sont censées ne pas s'écarter de ± 20 % des valeurs nominales (autrement dit rester comprises dans une plage de 80 à 120 %; voir points 1.6.2 et 1.6.3), on recommande d'analyser au moins la concentration la plus basse et la plus élevée au début de l'essai et périodiquement toutes les semaines par la suite. Pour les essais dans lesquels la concentration de la substance d'essai n'est pas censée rester comprise dans un intervalle de ± 20 % des valeurs nominales (d'après les données sur la stabilité de la substance d'essai), il est nécessaire d'analyser toutes les concentrations d'essai, mais selon le même régime.
Dans les essais semi-statiques (avec renouvellement) où la concentration de la substance d'essai est censée rester comprise dans un intervalle de ± 20 % des valeurs nominales, il est recommandé, au minimum, d'analyser la concentration la plus basse et la plus élevée juste après leur préparation et juste avant le renouvellement au début de l'essai et toutes les semaines par la suite. Pour les essais dans lesquels la concentration de la substance d'essai n'est pas censée rester comprise dans un intervalle de ± 20 % des valeurs nominales, il est nécessaire d'analyser toutes les concentrations selon le même régime que pour les substances plus stables.
On préconise de fonder les résultats sur les concentrations mesurées. Toutefois, si les données disponibles démontrent que la concentration de la substance d'essai en solution a été correctement maintenue, tout au long de l'essai, dans un intervalle de ± 20 % autour de la concentration nominale ou de la concentration mesurée au départ, les résultats peuvent s'appuyer sur les valeurs nominales ou mesurées.
Il peut s'avérer nécessaire de filtrer (par exemple, à l'aide d'un filtre à porcs de 0,45 μm) ou de centrifuger les échantillons. La centrifugation est la procédure recommandée. Toutefois, si le milieu d'essai ne s'absorbe pas sur les filtres, la filtration est également acceptable.
Pendant l'essai, l'oxygène dissous, le pH et la température doivent être mesurés dans tous les récipients d'essai. La dureté totale, l'alcalinité et la salinité, s'il y a lieu, doivent être mesurées dans les récipients témoins et dans celui qui contient la concentration la plus forte. L'oxygène dissous et la salinité, s'il y a lieu, doivent être mesurés au moins trois fois: au début, au milieu et à la fin de l'essai. Dans les essais semi-statiques, on recommande de mesurer l'oxygène dissous plus souvent, de préférence avant et après chaque renouvellement de l'eau ou au moins une fois par semaine. Il faut mesurer le pH au début et à la fin de chaque période de renouvellement de l'eau dans les essais à renouvellement statique et au moins une fois par semaine dans les essais dynamiques. La dureté et l'alcalinité devraient être mesurées une fois au cours de chaque essai. 11 est préférable de surveiller la température en continu dans au moins un récipient d'essai.
1.8.6. Observations
Poids: à la fin de l'essai tous les poissons survivants seront pesés en poids frais (poissons essuyés, blotted dry) soit en groupes par récipient d'essai soit individuellement. Il est préférable de peser les animaux par récipient d'essai plutôt qu'individuellement, cette dernière méthode obligeant à marquer chaque poisson. Si l'on mesure le taux de croissance spécifique de chaque poisson (pesée individuelle), la technique de marquage devra être choisie de façon à perturber le moins possible les animaux [on peut utiliser une technique différente du cryo-marquage (par congélation), par exemple un mince fil de pêche coloré].
Il faut examiner quotidiennement les poissons durant l'essai et relever toutes les anomalies externes éventuelles (hémorragie, décoloration, par exemple) et les comportement anormaux. Les décès doivent être comptés et les poissons morts retirés du récipient dès que possible. Les poissons morts ne seront pas remplacés, le taux de charge et la densité de peuplement étant suffisamment élevés pour éviter que la modification du nombre de poissons ait des effets sur la croissance. Cependant, la ration alimentaire devra être ajustée.
2. RÉSULTATS ET RAPPORT
2.1. TRAITEMENT DES RÉSULTATS
Il est recommandé qu'un statisticien participe à la fois à la conception et à l'analyse de l'essai, car cette méthode autorise des variations considérables dans la procédure expérimentale, par exemple en ce qui concerne le nombre d'enceintes expérimentales, de concentrations d'essai, de poissons etc. Compte tenu des choix possibles dans la conception de l'essai, aucune orientation ou méthode statistique précise n'est proposée ici.
Il n'y a pas lieu de calculer les taux de croissance dans les récipients d'essai où la mortalité dépasse 10 %. Le taux de mortalité doit cependant être spécifié pour toutes les concentrations d'essai.
Quelle que soit la méthode utilisée pour analyser les données, le concept central est le taux de croissance spécifique r entre l'instant t1 et l'instant t2. Il peut être défini de diverses manières selon que les poissons ont été ou non marqués individuellement ou selon que l'on recherche une moyenne par récipient.


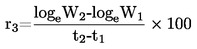
où:
|
r1
|
=
|
taux de croissance spécifique de chaque poisson
|
|
r2
|
=
|
taux de croissance spécifique moyen par récipient
|
|
r3
|
=
|
taux de croissance «pseudo»-spécifique
|
|
w1, w2
|
=
|
poids d'un poisson donné aux instants t1 et t2, respectivement
|
|
loge w1
|
=
|
logarithme du poids d'un poisson donné au début de la période d'étude
|
|
loge w2
|
=
|
logarithme du poids d'un poisson donné à la fin de la période d'étude
|
|
loge W1
|
=
|
moyenne des logarithmes des valeurs w1 des poissons du récipient à la fin de la période d'étude
|
|
loge W2
|
=
|
moyenne des logarithmes des valeurs w2 des poissons du récipient à la fin de la période d'étude
|
|
t1, t2
|
=
|
moments (jours) du début et de la fin de la période d'étude
|
r1, r2, r3 peuvent être calculés pour la période comprise entre le jour 0 et le jour 28 et, s'il y a lieu (c'est-à-dire lorsqu'une mesure a été effectuée au 14e jour), pour les périodes entre 0 et 14 jours et 14 et 28 jours.
2.1.1. Analyse des résultats par régression (modélisation concentration-effet)
Cette méthode d'analyse établit une relation mathématique adéquate entre le taux de croissance spécifique et la concentration, ce qui permet d'estimer la «CEX» c'est-à-dire toute valeur de CE requise. Si l'on utilise cette méthode, il est inutile de calculer r pour chaque poisson (r1) et l'analyse peut alors s'appuyer sur la valeur moyenne de r pour le récipient (r2). Cette dernière méthode est préférable. En outre, elle se prête mieux à l'utilisation d'espèces plus petites.
Pour étudier la relation concentration-effet, on porte sur un graphique les taux de croissance spécifiques moyens par récipient (r2) en fonction de la concentration.
Pour exprimer la relation entre r2 et la concentration, il convient de choisir un modèle adéquat et d'étayer ce choix par un raisonnement pertinent.
Si le nombre de poissons survivants varie suivant le récipient, le processus d'ajustement du modèle, qu'il soit simple ou non linéaire, doit être pondéré pour tenir compte de la taille inégale des groupes.
La méthode d'ajustement du modèle doit permettre d'estimer, par exemple, la CE20 et de déduire sa dispersion (écart-type ou intervalle de confiance). Le graphique du modèle ajusté devrait être montré en regard des données pour permettre d'apprécier l'adéquation de l'ajustement du modèle (9) (19) (20) (21).
2.1.2. Analyse des résultats pour l'estimation de la CMEO
Si le test porte sur plusieurs récipients d'essai répliques (identiques) pour toutes les concentrations, l'estimation de la CMEO pourrait s'appuyer sur une analyse de la variance du taux de croissance spécifique moyen de chaque récipient (voir point 2.1), suivie de l'utilisation d'une méthode pertinente [par exemple, un test de Dunnett ou de Williams (13) (14) (15) (22)] consistant à comparer la moyenne r à chaque concentration avec la moyenne r des témoins afin d'identifier la concentration minimale à laquelle cette différence est significative avec un seuil de probabilité de 0,05. Si les hypothèses requises concernant les méthodes paramétriques ne sont pas remplies — distribution non normale (par exemple, test de Shapiro-Wilk) ou variance hétérogène (test de Bartlett) — il faudra envisager de transformer les données pour homogénéiser les variances avant de mener l'analyse de la variance ou de réaliser une analyse pondérée de la variance.
Si l'essai ne porte pas sur plusieurs récipients par concentration, l'analyse de la variance fondée sur les récipients sera insensible ou impossible. Dans ces circonstances, on peut parvenir à un compromis acceptable en fondant l'analyse de la variance sur le taux de croissance «pseudo» spécifique r3 de chaque poisson.
La moyenne r3 pour chaque concentration d'essai peut ensuite être comparée à la moyenne r3 des témoins. Et la CMEO peut alors être déterminée comme précédemment. Il faut admettre que cette méthode ne tient absolument pas compte de la variabilité entre les récipients, en dehors de celle imputable à la variabilité entre les poissons, et n'offre aucune protection à cet égard. L'expérience a cependant montre (9) que la variabilité entre les récipients était très faible par rapport à celle qui existe dans les récipients (c'est-à-dire entre les poissons). Si l'analyse n'englobe pas les données concernant chaque poisson, il convient d'indiquer la méthode d'identification des valeurs aberrantes (erronées) et de justifier son emploi.
2.2. INTERPRÉTATION DES RÉSULTATS
Les résultats doivent être interprétés avec prudence lorsque les concentrations mesurées des produits toxiques dans les solutions d'essai sont proches des limites de détection de la méthode d'analyse ou, dans les essais semi-statiques, lorsque la concentration de la substance d'essai diminue entre le moment où la solution vient d'être préparée et le moment qui précède le renouvellement.
2.3. RAPPORT D'ESSAI
Le rapport d'essai doit fournir les informations suivantes:
2.3.1. Substance d'essai
|
—
|
État physique et propriétés physico-chimiques pertinentes,
|
|
—
|
données relatives à l'identification chimique, notamment la pureté et la méthode d'analyse quantitative de la substance d'essai, s'il y a lieu.
|
2.3.2. Espèce testée
|
—
|
souche éventuelle, taille, fournisseur, traitement préalable éventuel, etc.
|
2.3.3. Conditions d'essai
|
—
|
Méthode utilisée (par exemple, semi-statique/avec renouvellement, dynamique, charge, densité de peuplement, etc.),
|
|
—
|
conception de l'essai (par exemple, nombre d'enceintes expérimentales, de concentrations d'essai et de répliques, nombre de poissons par récipient),
|
|
—
|
méthode de préparation des solutions mères et fréquence de renouvellement (le solubilisant et sa concentration doivent être indiqués, le cas échéant),
|
|
—
|
concentrations d'essai nominales, moyennes des valeurs mesurées avec leurs écarts-types dans les récipients d'essai, méthode de détermination et données montrant que les mesures se réfèrent aux concentrations de la substance d'essai en solution vraie,
|
|
—
|
caractéristiques de l'eau de dilution: pH. dureté, alcalinité, température, concentration de l'oxygène dissous, teneurs résiduelles en chlore (si mesurées), carbone organique total, solides en suspension, salinité du milieu d'essai (si mesurée) et toute autre mesure effectuée,
|
|
—
|
qualité de l'eau dans les récipients d'essai: pH, dureté, température et concentration de l'oxygène dissous,
|
|
—
|
informations détaillées sur l'alimentation, [par exemple, le type d'aliment{s), la source, la quantité donnée et la fréquence].
|
2.3.4. Résultats
|
—
|
Données montrant que les témoins remplissent les critères de validité relatifs à la survie, et données sur la mortalité à toutes les concentrations d'essai,
|
|
—
|
techniques d'analyse statistique appliquées, statistiques fondées sur les répliques ou les poissons, traitement des données et justification des méthodes utilisées,
|
|
—
|
tableaux donnant les poids individuels et moyens des poissons aux jours 0, 14 (si mesurés) et 28, valeurs des moyennes par récipient ou taux de croissance pseudo-spécifiques (s'il y a lieu) pour les périodes de 0 à 28 jours, ou éventuellement de 0 à 14 et de 14 à 28 jours,
|
|
—
|
résultats de l'analyse statistique (analyse par régression ou analyse de la variance) fournis de préférence sous forme de tableau ou de graphique, CMEO (p = 0,05) et CSEO ou CEX avec leurs écarts-types si possible,
|
|
—
|
incidence de toute réaction inhabituelle des poissons et de tout effet visible produit par la substance d'essai.
|
3. BIBLIOGRAPHIE
|
(1)
|
Solbe J.F. de LG (1987). Environmental Effects of Chemicals (CFM 9350 SLD). Report on a UK Ring Test of a Method for Studying the Effects of Chemicals on the Growth rate of Fish. WRc Report No. PRD 1388-M/2.
|
|
(2)
|
Ashley S., Mallett M.J. and Grandy N.J. (1990). EEC Ring Test of a Method for Determining the Effects of Chemicals on the Growth Rate of Fish. Final Report to the Commission of the European Communities. WRc Report No EEC 2600-M.
|
|
(3)
|
Crossland N.O. (1985). A method to evaluate effects of toxic chemicals on fish growth. Chemosphere, 14, pp 1855-1870.
|
|
(4)
|
Nagel R., Bresh H., Caspers N., Hansen P.D., Market M., Munk R., Scholz N. and Höfte B.B. (1991). Effect of 3,4-dichloroaniline on the early life stages of the Zebrafish (Brachydanio rerio): results of a comparative laboratory study. Ecotox. Environ. Safety, 21, pp 157-164.
|
|
(5)
|
Yamamoto, Tokio. (1975). Series of stock cultures in biological field. Medaka (killifish) biology and strains. Keigaku Publish. Tokio, Japan.
|
|
(6)
|
Holcombe, G.W., Benoit D.A., Hammermeister, D.E., Leonard, E.N. and Johnson, R.D. (1995). Acute and long-term effects of nine chemicals on the Japanese medaka (Oryzias latipes).Arch. Environ. Conta. Toxicol. 28, pp 287-297.
|
|
(7)
|
Benoit, D.A., Holcombe, G.W. and Spehar, R.L. (1991). Guidelines for conducting early life toxicity tests with Japanese medaka (Oryzias latipes). Ecological Research Series EPA-600/3-91-063. U. S. Environmental Protection Agency, Duluth, Minesota.
|
|
(8)
|
Stephan C.E. and Rogers J.W. (1985). Advantages of using regression analysis to calculate results of chronic toxicity tests. Aquatic Toxicology and Hazard Assessment: Eighth Symposium, ASTM STP 891, R C Bahner and D J Hansen, Eds., American Society for Testing and Materials, Philadelphia, pp 328-338.
|
|
(9)
|
Environment Canada (1992). Biological test method: toxicity tests using early life stages of salmonid fish (rainbow trout, coho salmon, or atlantic salmon). Conservation and Protection, Ontario, Report EPS 1/RM/28, 81 p.
|
|
(10)
|
Cox D.R. (1958). Planning of experiments. Wiley Edt.
|
|
(11)
|
Pack S. (1991). Statistical issues concerning the design of tests for determining the effects of chemicals on the growth rate of fish. Room Document 4, OECD Ad Hoc Meeting of Experts on Aquatic Toxicology, WRc Medmenham, UK, 10-12 December 1991.
|
|
(12)
|
Dunnett C.W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with a Control, J. Amer. Statist. Assoc., 50, pp 1096-1121.
|
|
(13)
|
Dunnett C.W. (1964). New tables for multiple comparisons with a control. Biometrics, 20, pp 482-491.
|
|
(14)
|
Williams D.A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, pp 103-117.
|
|
(15)
|
Johnston, W.L., Atkinson, J.L., Glanville N.T. (1994). A technique using sequential feedings of different coloured food t o determine food intake by individual rainbow trout, Oncorhynchus mykiss: effect of feeding level. Aquaculture 120, 123-133.
|
|
(16)
|
Quinton, J. C. and Blake, R.W. (1990). The effect of feed cycling and ration level on the compensatory growth response in rainbow trout, Oncorhynchus mykiss. Journal of Fish Biology, 37, 33-41.
|
|
(17)
|
Post, G. (1987). Nutrition and Nutritional Diseases of Fish. Chapter IX in Testbook of Fish Health. T.F.H. Publications, Inc. Neptune City, New Jersey, USA. 288 P.
|
|
(18)
|
Bruce, R.D. and Versteeg D.J. (1992). A statistical procedure for modelling continuous toxicity data . Environ. Toxicol. Chem. 11, 1485-1494.
|
|
(19)
|
DeGraeve, G.M., Cooney, J.M., Pollock, T.L., Reichenbach, J.H., Dean, Marcus, M.D. and McIntyre, D.O. (1989). Precision of EPA seven-day fathead minnow larval survival and growth test; intra and interlaboratory study. report EA-6189 (American Petroleum Institute Publication, n. 4468). Electric Power Research Institute, Palo alto, CA.
|
|
(20)
|
Norbert-King T.J. (1988). An interpolation estimate for chronic toxicity: the ICp approach. US Environmental Protection Agency. Environmental Research Lab., Duluth, Minesota. Tech. Rep. No 05-88 of National Effluent Toxicity Assesment Center. Sept. 1988. 12 pp.
|
|
(21)
|
Williams D.A. (1972). The comparison of several dose levels with a zero dose control. Biometrics 28, pp 510-531.
|
Appendice 1
ESPÈCES DE POISSONS RECOMMANDÉES POUR L'ESSAI ET CONDITIONS D'ESSAI APPROPRIÉES
|
Espèce
|
Gamme de températures recommandée
( oC)
|
Photopériode
(heures)
|
Gamme recommandée pour le poids initial des poissons
(g)
|
Précision requise de la mesure
|
Taux de charge
(g/l)
|
Densité de peuplement
(par litre)
|
Alimentation
|
Durée de l'essai
(jours)
|
|
Espèce recommandée:
|
|
|
|
|
|
|
|
|
|
Oncorhynchus mykiss
Truite arc-en-ciel
|
12,5-16,0
|
12-16
|
1-5
|
à 100 mg près
|
1,2-2,0
|
4
|
Spécialité alimentaire sèche pour frai de salmonidé
|
≥ 28
|
|
Autres espèces bien étudiées:
|
|
|
|
|
|
|
|
|
|
Danio rerio
Danio
|
21-25
|
12-16
|
0,050-0,100
|
à 1 mg près
|
0,2-1,0
|
5-10
|
nourriture vivante (Brachionus Artemia)
|
≥ 28
|
|
Oryzias latipes
Medaka
|
21-25
|
12-16
|
0,050-0,100
|
à 1 mg près
|
0,2-1,0
|
5-20
|
nourriture vivante (Brachionus Artemia)
|
≥ 28
|
Appendice 2
QUELQUES CARACTÉRISTIQUES CHIMIQUES D'UNE EAU DE DILUTION ACCEPTABLE
|
Substance
|
Concentrations
|
|
Matières particulaires (particules)
|
< 20 mg/l
|
|
Carbone organique total
|
< 2 mg/l
|
|
Ammoniac non ionisé
|
< 1 μg/l
|
|
Chlore résiduel
|
< 10 μg/l
|
|
Pesticides organophosphorés totaux
|
< 50 ng/l
|
|
Pesticides organochlorés totaux et biphényles polychlorés
|
< 50 ng/l
|
|
Chlore organique total
|
< 25 ng/l
|
Appendice 3
Série logarithmique de concentrations convenant à un essai de toxicité (9)
|
Colonne (nombre de concentrations entre 100 et 10, ou entre 10 et 1) (20)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
|
100
|
100
|
100
|
100
|
100
|
100
|
100
|
|
32
|
46
|
56
|
63
|
68
|
72
|
75
|
|
10
|
22
|
32
|
40
|
46
|
52
|
56
|
|
3,2
|
10
|
18
|
25
|
32
|
37
|
42
|
|
1,0
|
4,6
|
10
|
16
|
22
|
27
|
32
|
|
|
2,2
|
5,6
|
10
|
15
|
19
|
24
|
|
|
1,0
|
3,2
|
6,3
|
10
|
14
|
18
|
|
|
|
1,8
|
4,0
|
6,8
|
10
|
13
|
|
|
|
1,0
|
2,5
|
4,6
|
7,2
|
10
|
|
|
|
|
1,6
|
3,2
|
5,2
|
7,5
|
|
|
|
|
1,0
|
2,2
|
3,7
|
5,6
|
|
|
|
|
|
1,5
|
2,7
|
4,2
|
|
|
|
|
|
1,0
|
1,9
|
3,2
|
|
|
|
|
|
|
1,4
|
2,4
|
|
|
|
|
|
|
1,0
|
1,8
|
|
|
|
|
|
|
|
1,3
|
|
|
|
|
|
|
|
1,0
|
C.15. POISSON, ESSAI DE TOXICITÉ À COURT TERME AUX STADES DE L'EMBRYON ET DE L'ALEVIN
1. MÉTHODE
La méthode décrite pour cet essai de toxicité à court terme reprend la ligne directrice no 212 de l'OCDE (1998).
1.1. INTRODUCTION
Cet essai de toxicité à court terme chez le poisson aux stades de l'embryon et de l'alevin consiste à exposer l'animal depuis le stade de l'œuf qui vient d'être fécondé jusqu'à la fin du stade de l'alevin. Aucune alimentation n'est fournie durant l'essai sur les embryons et les alevins, et l'essai devrait donc prendre fin alors que les alevins se nourrissent encore grâce à leur sac vitellin.
Cet essai vise à déterminer les effets létaux et, dans une moindre mesure, sublétaux de produits chimiques à des stades définis de la vie des espèces testées. Il devrait fournir des informations utiles en ce sens qu'il pourrait a) permettre de faire le lien entre les essais létaux et sublétaux, b) servir d'essai de sélection en vue soit d'un essai (complet) de toxicité aux premiers stades de la vie, soit d'un essai de toxicité chronique, et c) être utilisé pour tester des espèces pour lesquelles les techniques d'élevage ne sont pas suffisamment au point pour couvrir la période de transition entre alimentation endogène et alimentation exogène. Il faut savoir que seuls les essais couvrant tous les stades de la vie du poisson sont généralement susceptibles de donner une estimation précise de la toxicité chronique de produits chimiques pour les poissons et que toute limitation de l'exposition écartant tel ou tel stade de la vie risque de diminuer la sensibilité et donc de donner lieu à une sous-estimation de la toxicité chronique. L'essai aux stades de l'embryon et de l'alevin devrait donc être moins sensible que l'essai (complet) de toxicité aux premiers stades de la vie, notamment en ce qui concerne les produits chimiques fortement lipophiles (log Poe > 4) et les substances possédant un mode d'action toxique particulier. On s'attend toutefois à ce que la différence de sensibilité entre les deux essais soit moindre pour les produits chimiques à mode d'action narcotique non spécifique (1).
Avant la publication du présent essai, l'essai aux stades de l'embryon et de l'alevin a surtout été réalisé sur le poisson d'eau douce Danio rerio Hamilton-Buchanan (téléostéens, cyprinidés — nom commun: danio). C'est la raison pour laquelle l'appendice 1 donne des indications détaillées pour la réalisation de l'essai sur cette espèce. Cela n'exclut pas pour autant l'utilisation d'autres espèces de poissons pour lesquelles on dispose déjà d'une certaine expérience (tableaux A et B).
1.2. DÉFINITIONS
Concentration minimale avec effet observé (CMEO): concentration la plus basse d'une substance d'essai à laquelle on observe un effet significatif (à p ≤ 0,05) par rapport au témoin. Cependant, toutes les concentrations d'essai supérieures à la CMEO doivent exercer un effet nocif supérieur ou égal à celui observé à la CMEO.
Concentration (maximale) sans effet observé (CSEO): concentration d'essai immédiatement inférieure à la CMEO.
1.3. PRINCIPE DE L'ESSAI
Les poissons aux stades de l'embryon et de l'alevin sont exposés à une gamme de concentrations de la substance d'essai dissoute dans l'eau. Le protocole permet de choisir entre un essai semi-statique ou dynamique. Le choix dépend de la nature de la substance d'essai. L'essai débute au moment où l'on place les œufs fécondés dans les enceintes expérimentales et prend fin juste avant que le sac vitellin d'une quelconque larve d'une quelconque enceinte expérimentale ait été complètement absorbé ou avant que des poissons témoins ne meurent d'inanition. Les effets létaux et sublétaux sont évalués et comparés aux valeurs des témoins, en vue de déterminer la concentration minimale avec effet observé (CMEO) et, de là. la concentration (maximale) sans effet observé (CSEO). Une autre possibilité consiste à analyser ces effets à l'aide d'un modèle de régression permettant d'estimer la concentration qui provoquerait un certain pourcentage d'effet (CL/CEx) où x représente un pourcentage d'effet défini).
1.4. INFORMATIONS CONCERNANT LA SUBSTANCE D'ESSAI
Il faudrait disposer des résultats d'un essai de toxicité aiguë réalisé de préférence sur l'espèce choisie pour le présent essai. Ces résultats pourront s'avérer utiles pour sélectionner une plage de concentrations appropriée pour l'essai aux premiers stades de la vie. La solubilité dans l'eau (y compris la solubilité dans l'eau de l'essai) et la pression de vapeur de la substance d'essai doivent être connues. Pour déterminer la concentration de la substance dans les solutions d'essai, il convient d'appliquer une méthode d'analyse fiable, dont la précision et la limite de détection sont connues et mentionnées dans le rapport.
Les informations sur la substance d'essai qui peuvent être utiles pour établir les conditions de l'essai comprennent la formule structurale, la pureté de la substance, la stabilité à la lumière, la stabilité dans les conditions de l'essai, le pKa, le coefficient de partage n-octanol/eau (Poe) et les résultats d'un essai de biodégradabilité immédiate (voir méthode C.4).
1.5. VALIDITÉ DE L'ESSAI
La validité de l'essai repose sur les conditions suivantes:
|
—
|
le taux global de survie des œufs fécondés dans les groupes témoins et, s'il y a lieu, dans les récipients ne contenant que le solvant, doit être supérieur ou égal aux limites définies dans les appendices 2 et 3,
|
|
—
|
la concentration de l'oxygène dissous doit se situer entre 60 et 100 % de la valeur de saturation en air tout au long de l'essai,
|
|
—
|
à aucun moment au cours de l'essai, la température de l'eau ne doit varier de plus de ±1,5 oC entre les différentes enceintes expérimentales ou entre deux jours consécutifs, et devrait être maintenue dans la plage spécifiée pour l'espèce testée (appendices 2 et 3).
|
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Enceintes d'essai
N'importe quel récipient en verre ou fabriqué dans un matériau chimiquement inerte peut être utilisé. Les récipients doivent être suffisamment grands pour répondre aux critères de charge (voir point 1.7.1.2). Il est recommandé de placer les enceintes expérimentales au hasard dans la partie du laboratoire où se déroule l'essai. Il est souhaitable de disposer les enceintes expérimentales selon un schéma randomisé par blocs, chaque traitement étant représenté dans chaque bloc, plutôt que de les placer de façon complètement aléatoire, lorsqu'il existe des effets systématiques dans le laboratoire qui peuvent être contrecarrés par la randomisation par blocs. Si celle-ci est utilisée, elle doit être prise en compte dans l'analyse ultérieure des résultats. Les enceintes expérimentales doivent être protégées de toute perturbation indésirable.
1.6.2. Sélection des espèces de poissons
Les espèces de poissons recommandées sont énumérées dans le tableau 1A. Cela n'exclut pas l'utilisation d'autres espèces (des exemples figurent dans le tableau 1B), mais le mode opératoire devra éventuellement être adapté afin d'offrir les conditions adéquates. Dans ce cas, les choix de l'espèce et de la méthode expérimentale doivent être justifiés.
1.6.3. Soin des poissons géniteurs
La ligne directrice 210 de l'OCDE (21) et les références (2) (3) (4) (5) (6) donnent des indications détaillées sur la façon de maintenir les poissons géniteurs dans des conditions satisfaisantes.
1.6.4. Manipulation des embryons et des larves
Les embryons et les larves peuvent être exposés, au sein de la cuve principale, dans des récipients plus petits pourvus de côtés ou d'extrémités en filet, permettant le passage de la solution d'essai. Il est possible d'induire un écoulement non turbulent dans ces petits récipients en les suspendant à un bras qui déplace le récipient verticalement, en gardant toujours les organismes submergés; un système de siphon peut aussi être employé. Les œufs fécondés des salmonidés peuvent être posés sur des supports ou des grilles ayant des ouvertures suffisantes pour permettre aux larves de passer au travers après l'éclosion. Les pipettes Pasteur conviennent au prélèvement des embryons et des larves dans les essais semi-statiques où le milieu est entièrement renouvelé chaque jour (voir point 1.6.6).
Quand on utilise des récipients, des grilles ou des filets pour maintenir les œufs à l'intérieur de la cuve d'essai principale, ces dispositifs doivent être enlevés après l'éclosion des larves (21), mais il faut conserver des filets pour empêcher les poissons de s'échapper. Si les larves doivent être transférées, il ne faut pas les exposer à l'air ni utiliser de filets pour enlever les poissons des récipients contenant les œufs (ces précautions sont superflues pour des espèces moins fragiles, comme la carpe). Le moment de ce transfert varie avec l'espèce et il n'est pas toujours nécessaire. Dans les essais semi-statiques, des béchers ou des récipients peu profonds peuvent être utilisés, munis, si nécessaire, d'une grille placée un peu au-dessus du fond du récipient. Si le volume de ces récipients est suffisant pour satisfaire aux critères de charge (voir point 1.7.1.2), il n'est pas nécessaire de transférer les embryons ou les larves.
1.6.5. Eau
Toute eau répondant aux caractéristiques chimiques d'une eau de dilution acceptable (énumérées à l'appendice 4) et dans laquelle les témoins de l'espèce testée ont un taux de survie au moins aussi satisfaisant que celui décrit dans les appendices 2 et 3 peut être utilisée pour l'essai. La qualité de cette eau devrait rester constante pendant toute la durée de l'essai. La variation du pH devrait rester comprise dans un intervalle de ±0,5 unités. Pour s'assurer que l'eau de dilution n'influencera pas indûment les résultats de l'essai (par exemple en complexant la substance d'essai) ou ne nuira pas au comportement des poissons géniteurs, des échantillons doivent être prélevés périodiquement en vue d'être analysés. Il y a lieu de mesurer la teneur en métaux lourds (par exemple, Cu, Pb, Zn, Hg, Cd et Ni), en principaux anions et cations (par exemple, Ca, Mg, Na, K, Cl et SO4), en pesticides (par exemple, la totalité des organophosphorés et la totalité des organochlorés), en carbone organique total et en solides en suspension. Ces mesures devraient être effectuées tous les trois mois, par exemple, lorsqu'on sait que l'eau de dilution présente une qualité relativement constante. Si la qualité de l'eau s'est avérée constante sur une période d'au moins un an, les déterminations pourront être plus espacées (par exemple, tous les six mois).
1.6.6. Solutions d'essai
Par dilution d'une solution mère, on prépare des solutions d'essai aux concentrations choisies.
La solution mère devrait, de préférence, être préparée par simple mélange ou agitation mécanique de la substance d'essai dans l'eau de dilution (par exemple, au moyen d'un agitateur ou par ultrasons). Des colonnes de saturation (colonnes de solubilité) peuvent être utilisées pour amener la solution mère à la concentration souhaitée. Le recours à des solvants ou dispersants (agents solubilisants) doit être évité dans toute la mesure du possible; il peut toutefois s'avérer nécessaire d'utiliser ces produits pour obtenir une solution mère à la concentration voulue. Les solvants qui conviennent sont, par exemple, l'acétone, l'éthanol, le méthanol, le diméthyl-formamide et le triéthylèneglycol. Parmi les dispersants adéquats, citons le Cremophor RH40, le Tween 80, la méthylcellulose à 0,01 % et le HCO-40. Il convient d'être vigilant lorsque l'on utilise des agents facilement biodégradables (par exemple, l'acétone) et/ou volatils, car ils peuvent entraîner une prolifération bactérienne dans les essais dynamiques. L'agent solubilisant, le cas échéant, ne doit pas exercer d'effet significatif sur la survie ni nuire de façon visible aux premiers stades de la vie. d'après l'observation d'un témoin ne comportant que le solvant. Cependant, l'usage de ces substances doit être évité dans toute la mesure du possible.
S'agissant des essais semi-statiques, deux méthodes de renouvellement peuvent être adoptées: soit i) les nouvelles solutions d'essai sont préparées dans des récipients propres et l'on transvase avec précaution les œufs et les larves survivants dans les nouveaux récipients avec un petit volume de solution ancienne, en évitant de les exposer à l'air, soit ii) les organismes testés sont laissés dans leur récipient d'essai pendant que l'on renouvelle au moins les trois-quarts de leur eau. La fréquence de renouvellement du milieu dépend de la stabilité de la substance d'essai, mais un renouvellement quotidien de l'eau est recommandé. Si les tests préliminaires de stabilité (voir point 1.4) révèlent que la concentration de la substance n'est pas stable (sort de l'intervalle de 80 à 120 % de la concentration nominale ou tombe en dessous de 80 % de la concentration mesurée initialement) au cours de la période de renouvellement, il faudrait envisager de pratiquer un essai dynamique. Quoi qu'il en soit, il convient d'éviter de stresser les larves durant le renouvellement de l'eau.
Les essais dynamiques nécessitent un système qui délivre et dilue en continu la solution mère de la substance d'essai (par exemple, une pompe doseuse, un dilueur proportionnel, un système de saturation), pour distribuer une série de concentrations vers les enceintes d'essai. Les débits des solutions mères et de l'eau de dilution doivent être contrôlés à intervalles quotidiens de préférence, et ne devraient pas varier de plus de 10 % tout au long de l'essai. Un débit équivalent à au moins cinq volumes d'enceintes expérimentales par 24 heures s'est avéré adéquat (2).
1.7. MODE OPÉRATOIRE
Des informations utiles concernant la marche à suivre pour les essais de toxicité sur les embryons de poisson et les alevins se trouvent dans les publications; la bibliographie de ce texte en donne quelques références (7) (8) (9).
1.7.1. Conditions d'exposition
1.7.1.1. Durée
L'essai devrait débuter de préférence dans les 30 minutes qui suivent la fécondation des œufs. Les embryons sont immergés dans la solution d'essai avant, ou aussitôt que possible après, le commencement de la phase de segmentation du blastodisque et, dans tous les cas. avant le début du stade de la gastrula. Quand les œufs proviennent d'un fournisseur commercial, il n'est pas toujours possible de débuter l'essai juste après la fécondation. Comme la sensibilité de l'essai peut être fortement influencée par un retard dans son lancement, l'essai devrait débuter dans les 8 heures qui suivent la fécondation. Les larves n'étant pas nourries durant la période d'exposition, l'essai doit s'achever juste avant que le sac vitellin d'une quelconque larve d'une quelconque enceinte expérimentale soit complètement absorbé ou avant que les témoins ne meurent d'inanition. La durée de l'essai dépend de l'espèce utilisée. Certaines durées sont recommandées aux appendices 2 et 3.
1.7.1.2. Charge
Le nombre d'œufs fécondés au début de l'essai doit être suffisant pour répondre aux critères statistiques. Il y a lieu de les répartir au hasard entre les différents traitements et d'utiliser au moins 30 œufs fécondés répartis en nombre égal (ou aussi proche de l'égalité que possible, car il peut être difficile d'obtenir des lots identiques avec certaines espèces) entre au moins trois enceintes d'essai identiques pour chaque concentration. Le taux de charge (biomasse par volume de solution d'essai) doit être suffisamment bas pour qu'une concentration d'oxygène dissous d'au moins 60 % de la valeur de saturation en air puisse être maintenue sans aération. Dans le cas des essais dynamiques, un taux de charge n'excédant pas 0,5 g/l par 24 heures et ne dépassant pas 5 g/l de solution, à tout moment, a été recommandé (2).
1.7.1.3. Lumière et température
La photopériode et la température de l'eau d'essai doivent être adaptées à l'espèce testée (voir appendices 2 et 3). En vue de surveiller la température, il peut être approprié d'utiliser un récipient d'essai supplémentaire.
1.7.2. Concentrations d'essai
On doit normalement utiliser cinq concentrations de la substance d'essai espacées par un facteur constant ne dépassant pas 3,2. La courbe de la CL50 en fonction de la période d'exposition, obtenue au cours de l'étude de toxicité aiguë, doit être prise en considération lors de la sélection de la plage des concentrations d'essai. Il peut être judicieux, dans certaines circonstances, d'utiliser moins de cinq concentrations, par exemple, dans les essais limites, et un intervalle plus faible entre les concentrations. L'utilisation de moins de cinq concentrations doit être justifiée. Il n'est pas nécessaire de tester les concentrations supérieures à la CL50 après 96 heures ou supérieures à 100 mg/l lorsque la CL50 est supérieure à cette concentration. Les substances ne devraient pas être testées au-delà de leur limite de solubilité dans l'eau d'essai.
Lorsqu'un agent solubilisant est utilisé pour faciliter la préparation des solutions d'essai (voir point 1.6.6), sa concentration finale dans les récipients d'essai ne doit pas dépasser 0,1 ml/1 et doit être identique dans tous les récipients d'essai.
1.7.3. Témoins
Un témoin contenant l'eau de dilution (en autant d'exemplaires que de besoin) et, le cas échéant, un témoin contenant l'agent solubilisant (en autant d'exemplaires que de besoin) doivent être étudiés parallèlement aux séries traitées avec la substance.
1.7.4. Fréquence des analyses quantitatives et des mesures
Au cours de l'essai, les concentrations de la substance à tester sont déterminées à intervalles réguliers.
Dans les essais semi-statiques où la concentration de la substance d'essai est censée rester comprise dans un intervalle de ± 20 % autour de la concentration nominale (dans une plage de 80 à 120 %, voir points 1.4 et 1.6.6), il est recommandé, au minimum, d'analyser la concentration la plus basse et la plus élevée, dès leur préparation et juste avant le renouvellement du milieu, à au moins trois occasions régulièrement espacées au cours de l'essai (les analyses devraient être pratiquées sur des échantillons prélevés dans la même solution, dès leur préparation et au moment du renouvellement).
Pour les essais dans lesquels la concentration de la substance d'essai n'est pas censée rester comprise dans un intervalle de ± 20 % autour de la concentration nominale (d'après les résultats concernant la stabilité de la substance), ii est nécessaire d'analyser toutes les concentrations, dès leur préparation et au moment du renouvellement, mais selon le même régime, c'est-à-dire à au moins trois occasions, régulièrement espacées au cours de l'essai. La détermination des concentrations de la substance d'essai avant le renouvellement ne doit être réalisée que sur un seul récipient par concentration. Les déterminations ne devraient pas être espacées de plus de sept jours. On préconise de baser les résultats sur les concentrations mesurées. Toutefois, si les données disponibles montrent que la concentration de la substance d'essai dans la solution s'est maintenue correctement dans un intervalle de ± 20 % autour de la concentration nominale ou mesurée au départ, tout au long de l'essai, les résultats peuvent s'appuyer sur la concentration nominale ou les valeurs mesurées au départ.
Dans le cas des essais dynamiques, il convient d'appliquer un régime de prélèvement des échantillons similaire à celui décrit pour les essais semi-statiques (mais le dosage des solutions juste avant leur renouvellement ne s'applique pas ici). Néanmoins, si l'essai dure plus de sept jours, il peut être souhaitable d'augmenter le nombre de prélèvements au cours de la première semaine (par exemple, trois séries de mesures), afin de s'assurer que les concentrations d'essai restent stables.
Il peut, dans certains cas, s'avérer nécessaire de filtrer (par exemple, à l'aide d'un filtre à pores de 0,45 μm de diamètre) ou de centrifuger les échantillons. Toutefois, comme la centrifugation ou la filtration ne permettent pas toujours de séparer la fraction non biodisponible de la substance d'essai de sa fraction biodisponible, il peut être inutile de soumettre les échantillons à ces traitements.
Pendant l'essai, l'oxygène dissous, le pH et la température doivent être mesurés dans tous les récipients d'essai. La dureté totale et la salinité, le cas échéant, doivent être déterminées dans les témoins et dans un des récipients contenant la concentration la plus élevée. L'oxygène dissous et la salinité, le cas échéant, doivent être mesurés au moins trois fois: au début, au milieu et à la fin de l'essai. Dans les essais semi-statiques, il est recommandé de mesurer l'oxygène dissous plus fréquemment, de préférence avant et après chaque renouvellement de l'eau ou au moins une fois par semaine. Le pH devrait être mesuré au début et à la fin de chaque période de renouvellement de l'eau dans les essais semi-statiques et au moins une fois par semaine dans les essais dynamiques. Il faudrait déterminer la dureté une fois dans chaque essai. La température devrait être mesurée quotidiennement et, de préférence, surveillée en continu dans au moins un récipient d'essai.
1.7.5. Observations
1.7.5.1. Stade de développement embryonnaire
Le stade embryonnaire (stade de la gastrula) au début de l'exposition à la substance d'essai doit être vérifié aussi précisément que possible. Cela peut se faire sur un échantillon représentatif d'œufs bien conservés et rendus translucides. Des descriptions et des illustrations des stades embryonnaires peuvent être consultées dans les publications (2) (5) (10) (11).
1.7.5.2. Éclosion et survie
Il faut observer et dénombrer les éclosions et les survivants au moins une fois par jour. Il peut être souhaitable d'effectuer des observations plus fréquentes au début de l'essai (par exemple, toutes les 30 minutes au cours des trois premières heures), puisque dans certains cas, les temps de survie peuvent être plus significatifs que le seul nombre de morts (notamment en cas d'effets toxiques aigus). Les larves et les embryons morts doivent être retirés dès qu'ils sont repérés, car ils peuvent se décomposer rapidement. Il convient d'être extrêmement attentif, lorsque l'on retire les individus morts, à ne pas heurter ou léser physiquement les œufs et les larves adjacents, qui sont très délicats et sensibles. Les critères indiquant la mort varient en fonction du stade de développement:
|
—
|
pour les œufs: en particulier au début de leur cycle, une diminution marquée de la transparence et un changement de coloration, dus à la coagulation et/ou à la précipitation de protéines et conduisant à un aspect blanc opaque,
|
|
—
|
pour les embryons: absence de mouvement corporel et/ou absence de battement du cœur et/ou décoloration opaque chez les espèces dont les embryons sont normalement transparents,
|
|
—
|
pour les larves: immobilité et/ou absence de mouvement respiratoire et/ou absence de battement du cœur et/ou coloration blanche opaque du système nerveux central et/ou absence de réaction aux stimuli mécaniques.
|
1.7.5.3. Aspect anormal
On doit noter le nombre de larves présentant une anomalie corporelle et/ou de la pigmentation, ainsi que le stade de résorption du sac vitellin, à des intervalles de temps adéquats, en fonction de la durée de l'essai et de la nature de l'anomalie décrite. Il faut savoir que des larves et des embryons anormaux surviennent de façon naturelle et que leur proportion peut atteindre quelques pour cent chez le ou les témoins de certaines espèces. Les animaux anormaux ne doivent être retirés des récipients d'essai qu'à leur mort.
1.7.5.4. Comportement anormal
Des anomalies telles qu'une hyperventilation, une nage mal coordonnée et une immobilité atypique doivent être notées à des intervalles de temps qui dépendent de la durée de l'essai. Ces effets, bien que difficiles à quantifier, peuvent le cas échéant contribuer à l'interprétation des données de mortalité, en fournissant des informations sur le mode d'action toxique de la substance.
1.7.5.5. Longueur
À la fin de l'essai, il est recommandé de mesurer la longueur de chaque individu en utilisant la longueur standard, la longueur à la fourche ou la longueur totale. Cependant, si l'on constate une putréfaction de la nageoire caudale ou une usure des nageoires, on doit employer la longueur standard. Habituellement, dans un essai correctement exécuté, le coefficient de variation de la longueur entre les différents récipients des témoins doit être ≤ 20 %.
1.7.5.6. Poids
À la fin de l'essai, chaque individu peut être pesé; le poids sec (24 heures à 60o C) est préférable au poids frais (poissons essuyés). Généralement, dans un essai correctement exécuté, le coefficient de variation du poids entre les différents récipients des témoins doit être ≤ 20 %.
Ces observations permettront de livrer une partie ou la totalité des données suivantes à l'analyse statistique:
|
—
|
nombre de larves saines à la fin de l'essai,
|
|
—
|
moment du commencement de l'éclosion et de la fin de celle-ci (90 % d'œufs éclos dans chaque enceinte de concentration identique),
|
|
—
|
nombre de larves écloses chaque jour,
|
|
—
|
longueur (et poids) des animaux survivants à la fin de l'essai,
|
|
—
|
nombre de larves déformées ou d'aspect anormal,
|
|
—
|
nombre de larves présentant un comportement anormal.
|
2. RÉSULTATS ET RAPPORT
2.1. TRAITEMENT DES RÉSULTATS
Il est recommandé qu'un statisticien participe à la fois à la conception et à l'analyse de l'essai, puisque cette méthode autorise des variations considérables dans la procédure expérimentale, par exemple en ce qui concerne le nombre d'enceintes expérimentales, le nombre de concentrations d'essai, le nombre initial d'œufs fécondés et les paramètres mesurés. Compte tenu des choix possibles dans la conception de l'essai, on ne donnera pas ici d'orientations précises sur les méthodes statistiques.
Si la CMEO et la CSEO doivent être estimées, il sera nécessaire d'analyser les variations au sein de chaque série d'enceintes de concentration identique par l'analyse de la variance ou les méthodes avec tableau de contingence. La méthode de Dunnett peut être utile pour procéder à des comparaisons multiples entre les résultats obtenus à chaque concentration et ceux des témoins (12) (13). Il existe d'autres exemples pertinents (14) (15). La grandeur de l'effet détectable par l'analyse de la variance ou par d'autres méthodes (c'est-à-dire la puissance de l'essai) devrait être calculée et mentionnée dans le rapport. Il faut savoir que les observations énumérées au point 1.7.5.6 ne se prêtent pas toutes au traitement statistique par l'analyse de la variance. À titre d'exemple, la mortalité cumulée et le nombre de larves saines à la fin de l'essai pourraient être analysés à l'aide de méthodes des probits.
S'il y a lieu d'estimer la CL et la CEX, une courbe adéquate, telle que la courbe logistique, doit être ajustée aux résultats étudiés par une méthode statistique telle que la méthode des moindres carrés ou des moindres carrés non linéaires. La ou les courbes doivent être paramétrées de façon que la CL et la CE, recherchées et leur écart-type puissent être estimés directement. Cela facilitera beaucoup le calcul des limites de confiance autour de la CL et de la CEX. À moins d'avoir de bonnes raisons de préférer d'autres niveaux de confiance, des limites de confiance de 95 % de part et d'autre devraient être choisies. La procédure d'ajustement devrait, de préférence, permettre d'évaluer la signification du manque d'ajustement. Des méthodes graphiques d'ajustement des courbes peuvent être utilisées. Toutes les observations énumérées au point 1.7.5.6 se prêtent à l'analyse de régression.
2.2. INTERPRÉTATION DES RÉSULTATS
Les résultats doivent être interprétés avec prudence, lorsque les concentrations mesurées des produits toxiques dans les solutions d'essai sont proches des limites de détection de la méthode d'analyse. L'interprétation des résultats concernant les concentrations supérieures à la solubilité de la substance dans l'eau doit aussi se faire avec prudence.
2.3. RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
2.3.1. Substance d'essai
|
—
|
État physique et propriétés physico-chimiques pertinentes,
|
|
—
|
données relatives à l'identification chimique, notamment la pureté et la méthode d'analyse quantitative de la substance d'essai, s'il y a lieu.
|
2.3.2. Espèce d'expérience
|
—
|
Nom scientifique, variété, nombre de poissons parents (c'est-à-dire nombre de femelles utilisées pour fournir le nombre d'œufs requis par l'essai), source et méthode de collecte des œufs fécondés et manipulations ultérieures.
|
2.3.3. Conditions d'essai
|
—
|
Méthode utilisée (par exemple, semi-statique ou dynamique, temps écoulé entre la fécondation et le début de l'essai, charge, etc.),
|
|
—
|
conception de l'essai (par exemple, nombre d'enceintes expérimentales et d'exemplaires de même concentration, nombre d'embryons par exemplaire),
|
|
—
|
méthode de préparation des solutions mères et fréquence de renouvellement (l'agent solubilisant et sa concentration doivent être spécifiés, le cas échéant),
|
|
—
|
concentrations nominales de l'essai, valeurs mesurées dans les récipients d'essai, moyennes et écarts-types de celles-ci et méthode de détermination; si la substance d'essai est soluble dans l'eau à des concentrations inférieures à celles qui ont été testées, il faut démontrer que les mesures se réfèrent aux concentrations de la substance d'essai dans la phase dissoute,
|
|
—
|
caractéristiques de l'eau de dilution: pH, dureté, température, concentration d'oxygène dissous, teneur en chlore résiduel (si elle a été mesurée), carbone organique total, solides en suspension, salinité du milieu d'essai (si elle a été mesurée) et toute autre mesure effectuée,
|
|
—
|
qualité de l'eau dans les récipients d'essai, pH, dureté, température et concentration d'oxygène dissous.
|
2.3.4. Résultats
|
—
|
Résultats des éventuelles études préliminaires sur la stabilité de la substance d'essai,
|
|
—
|
données montrant que les témoins répondent à la norme générale d'acceptabilité relative à la survie de l'espèce (appendices 2 et 3),
|
|
—
|
résultats concernant la mortalité et la survie aux stades de l'embryon et de la larve et taux de mortalité et de survie globaux,
|
|
—
|
délais d'éclosion et nombre d'œufs éclos,
|
|
—
|
description et fréquence des anomalies morphologiques, le cas échéant,
|
|
—
|
description et fréquence des effets sur le comportement, le cas échéant,
|
|
—
|
analyse statistique et traitement des données,
|
|
—
|
pour les essais soumis à l'analyse de la variance, la concentration minimale avec effet observé (CMEO) à p = 0,05 et la concentration sans effet observé (CSEO) pour chaque réponse évaluée, ainsi qu'une description des méthodes statistiques utilisées et une indication de la grandeur de l'effet qui peut être détecté.
|
|
—
|
pour les essais analysés à l'aide de techniques de régression, la CL et la CEx et leurs intervalles de confiance et un graphique du modèle ajusté utilisé pour les calculer,
|
|
—
|
justification de tout écart par rapport à la présente méthode d'essai.
|
3. BIBLIOGRAPHIE
|
(1)
|
Kristensen P. (1990). Evaluation of the Sensitivity of Short Term Fish Early Life Stage Tests in Relation to other FEES Test Methods. Final report to the Commission of the European Communities, 60 pp. June 1990.
|
|
(2)
|
ASTM (1988). Standard Guide for Conducting Early Life-Stage Toxicity Tests with Fishes. American Society for Testing and Materials. E 1241-88. 26 pp.
|
|
(3)
|
Brauhn J. L. and Schoettger R. A. (1975). Acquisition and Culture of Research Fish: Rainbow trout, Fathead minnows, Channel Catfish and Bluegills. p. 54, Ecological Research Series, EPA-660/3-75-011, Duluth, Minnesota.
|
|
(4)
|
Brungs W. A. and Jones B. R. (1977). Temperature Criteria for Freshwater Fish: Protocol and Procedures, p. 128, Ecological Research Scries EPA-600/3-77-061, Duluth, Minnesota.
|
|
(5)
|
Laale H. W. (1977). The Biology and Use of the Zebrafish (Brachydanio rerio) in Fisheries Research. A Literature Review. J. Biol. 10, pp. 121-173.
|
|
(6)
|
Legault R. (1958). A Technique for Controlling the Time of Daily Spawning and Collecting Eggs of the Zebrafish, Brachydanio rerio (Hamilton-Buchanan) Copeia, 4, pp. 328-330.
|
|
(7)
|
Dave G., Damgaard B., Grande M., Martelin J. E., Rosander B. and Viktor T. (1987). Ring Test of an Embryo-larval Toxicity Test with Zebrafish (Brachydanio rerio) Using Chromium and Zinc as Toxicants. Environmental Toxicology and Chemistry. 6, pp. 61-71.
|
|
(8)
|
Birge J. W., Black J. A. and Westerman A. G. (1985). Short-term Fish and Amphibian Embryo-larval Tests for Determining the Effects of Toxicant Stress on Early Life Stages and Estimating Chronic Values for Single Compounds and Complex Effluents. Environmental Toxicology and Chemistry 4, pp. 807-821.
|
|
(9)
|
Van Leeuwen C. J., Espeldoorn A. and Mol F. (1986). Aquatic Toxicological Aspects of Dithiocarbamates and Related Compounds. III. Embryolarval Studies with Rainbow Trout (Salmo gairdneri). J. Aquatic Toxicology, 9, pp. 129-145.
|
|
(10)
|
Kirchen R. V. and W. R. West (1969). Teleostean Development. Carolina Tips 32(4): 1-4. Carolina Biological Supply Company.
|
|
(11)
|
Kirchen R. V. and W. R. West (1976). The Japanese Medaka. Its care and Development. Carolina Biological Supply Company, North Carolina. 36 pp.
|
|
(12)
|
Dunnett C. W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with Control. J. Amer. Statist. Assoc, 50, pp. 1096-1121.
|
|
(13)
|
Dunnett C. W. (1964). New Tables for Multiple Comparisons with a Control. Biometrics, 20, pp. 482-491.
|
|
(14)
|
Mc Clave J. T., Sullivan J. H. and Pearson J.G. (1980). Statistical Analysis of Fish Chronic Toxicity Test Data. Proceedings of 4th Aquatic Toxicology Symposium, ASTM, Philadelphia.
|
|
(15)
|
Van Leeuwen C. J., Adema D. M. M. and Hermes J. (1990). Quantitative Structure-Activity Relationships for Fish Early Life Stage Toxicity. Aquatic Toxicology, 16, pp. 321-334.
|
|
(16)
|
Environment Canada. (1992). Toxicity Tests Using Early Life Stages of Salmonid Fish (Rainbow Trout, Coho Salmon or Atlantic Salmon). Biological Test Method Series. Report EPS 1/RM/28, December 1992, 81 pp.
|
|
(17)
|
Dave G. and Xiu R. (1991). Toxicity of Mercury. Nickel. Lead and Cobalt to Embryos and Larvae of Zebrafish, Brachydanio rerio. Arch, of Environmental Contamination and Toxicology, 21, pp. 126-134.
|
|
(18)
|
Meyer A., Bierman C. H. and Orti G. (1993). The phylogenetic position of the Zebrafish (Danio rerio), a model system in developmental biology — an invitation to the comperative methods. Proc. Royal Society of London, Series B, 252: pp. 231-236.
|
|
(19)
|
Ghillebaert F., Chaillou C, Deschamps F. and Roubaud P. (1995). Toxic Effects, at Three pH Levels, of Two Reference Molecules on Common Carp Embryo. Ecotoxicology and Environmental Safety 32, pp. 19-28.
|
|
(20)
|
US EPA, (1991). Guidelines for Culturing the Japanese Medaka, Oryzias latipes. EPA report EPA/600/3-91/064, Dec. 1991, EPA, Duluth.
|
|
(21)
|
US EPA, (1991). Guidelines for Conducting Early Life Stage Toxicity Tests with Japanese Medaka, (Oryzias latipes). EPA report EPA/600/3-91/063, Dec. 1991, EPA, Duluth.
|
|
(22)
|
De Graeve G. M., Cooney J. D., McIntyre D. O., Poccocic T. L., Reichenbach N. G., Dean J. H. and Marcus M. D. (1991). Validity in the performance of the seven-day Fathead minnow {Pimephales pwmelas) larval survival and growth test: an intra- and interlaboratory study. Environ. Tox. Chem. 10, pp. 1189-1203.
|
|
(23)
|
Calow P. (1993). Handbook of Ecotoxicology, Blackwells, Oxford. Vol. 1, Chapter 10: Methods for spawning, culturing and conducting toxicity tests with Early Life stages of Estuarine and Marine fish.
|
|
(24)
|
Balon E. K. (1985). Early life history of fishes: New developmental, ecological and evolutionary perspectives, Junk Publ., Dordrecht, 280 pp.
|
|
(25)
|
Blaxter J. H. S. (1988). Pattern and variety in development, in: W. S. Hoar and D. J. Randall eds., Fish Physiology, Vol. XIA, Academic press, pp. 1-58.
|
Tableau 1A
Espèces de poissons recommandées pour l'essai
|
Eau douce
|
|
Oncorhynchus mykiss
Truite arc-en-ciel (9) (16)
|
|
Danio rerio
Danio (7) (17) (18)
|
|
Cyprinus caprio
Carpe commune (8) (19)
|
|
Oryzias latipes
Medaka (20) (21)
|
|
Pimephales promelas
Tête-de-boule (8) (22)
|
Tableau 1B
Exemples d'autres espèces également utilisées et sur lesquelles on possède une bonne documentation
|
Eau douce
|
Eau salée
|
|
Carassius auratus
Cyprin doré (8)
|
Menidia peninsulae
Capucette nord-américaine (23) (24) (25)
|
|
Lepomis macrochirus
Crapet arlequin (8)
|
Clupea harengus
Hareng (24) (25)
|
|
Gadus morhua
Morue (24) (25)
|
|
Cyprinodon variegalus
Fondule tête de mouton (23) (24) (25)
|
Annexe 1
ORIENTATIONS POUR LA RÉALISATION D'UN ESSAI DE TOXICITÉ SUR LES EMBRYONS ET LES ALEVINS DU DANIO (BRACHYDANIO RERIO)
INTRODUCTION
Le danio est originaire de la côte de Coromandel. en Inde, où il peuple les rivières à courant rapide. C'est une espèce courante en aquarium, appartenant à la famille des carpes. Des informations sur les méthodes d'élevage et les soins à lui apporter se trouvent dans des manuels de référence sur les poissons tropicaux. Sa biologie et son utilisation dans la recherche liée à la pêche ont été passées en revue par Laale (1).
Ce poisson dépasse rarement 45 mm de longueur. Son corps cylindrique arbore 7 à 9 rayures horizontales bleu foncé et argentées. Ces rayures se prolongent dans les nageoires caudales et anales. Son dos est vert olive. Les mâles sont plus minces que les femelles. Les femelles sont plus argentées et leur abdomen est distendu, en particulier avant le frai.
Les poissons adultes sont capables de supporter de grandes fluctuations de température, de pH et de dureté. Toutefois, afin de disposer de poissons sains qui produisent des œufs de bonne qualité, il faut leur fournir des conditions optimales.
Pendant la parade nuptiale, le mâle poursuit la femelle et lui donne des coups de tête; les œufs sont fécondés dès qu'ils sont expulsés. Les œufs, qui sont transparents et non adhérents, tombent au fond où ils peuvent être mangés par les parents. Le frai est influencé par la lumière. Si la lumière du matin est suffisante, le poisson fraie habituellement au cours des premières heures qui suivent le lever du soleil.
Une femelle peut produire des pontes de plusieurs centaines d'œufs à des intervalles d'une semaine.
ÉTAT DES POISSONS PARENTS, REPRODUCTION ET PREMIERS STADES DE LA VIE
Un nombre adéquat de poissons sains est sélectionné et gardé dans une eau appropriée (voir à l'appendice 4 par exemple) pendant au moins deux semaines avant le frai prévu. Le groupe de poissons devrait s'être reproduit au moins une fois avant d'engendrer la série d'œufs utilisés dans l'essai. La densité des poissons durant cette période ne devrait pas excéder 1 gramme de poissons par litre. La densité pourra être plus élevée si l'eau est renouvelée régulièrement ou si on a recours à des systèmes de purification. La température des aquariums devrait être maintenue à 25 ± 2 oC. Les poissons devraient recevoir un régime alimentaire varié, qui pourrait se composer, par exemple, d'aliments déshydratés appropriés trouvés dans le commerce, d'Artemia vivants récemment éclos, de chironomidés, de Daphnia et de vers blancs de la famille des enchytréidés.
Deux méthodes ayant permis d'obtenir un lot suffisant d'œufs fécondés sains, en vue de l'essai, sont résumées ci-dessous:
|
i)
|
Huit femelles et seize mâles sont placés dans un aquarium contenant 50 litres d'eau de dilution, protégé de la lumière directe. On évitera le plus possible de les perturber pendant au moins 48 heures. Un support de frai est disposé au fond de l'aquarium, pendant l'après-midi du jour qui précède le début de l'essai. Le support de frai se compose d'un cadre (en plexiglas ou dans un autre matériau adapté) de 5 à 7 cm de haut, muni d'un filet à grosse; mailles (2-5 mm) à son sommet et d'un filet à mailles fines (10-30 um) à sa base. Plusieurs «arbres à frai», consistait en une corde de nylon non torsadée, sont attachés au filet à grosses mailles du support. Après que les poissons on été laissés dans l'obscurité pendant 1 2 heures, on allume une faible lumière qui va déclencher le frai. 2 à 4 heure après le frai, le support de frai est retiré et les œufs sont récupérés. Le support de frai empêchera les poissons d manger les œufs et facilitera en même temps la collecte des œufs. Le groupe de poissons devra avoir frayé au moin une fois avant le frai qui donnera les oeufs destinés à l'essai.
|
|
ii)
|
Cinq à dix poissons mâles et femelles sont gardés dans des aquariums individuels pendant au moins deux semaint avant le frai prévu. Après 5 à 10 jours, les abdomens des femelles seront distendus et leurs papilles génitales app, raîtront visiblement. Les poissons mâles n'ont pas de papilles. Le frai se déroule dans des cuves équipées d'un fat fond en filet (voir ci-dessus}. La cuve est remplie d'eau de dilution jusqu'à une hauteur de 5 à 10 cm au-dessus c filet. Une femelle et deux mâles sont placés dans la cuve un jour avant le frai prévu. La température de l'eau (portée progressivement à un degré au-dessus de la température d'acclimatation. On éteint la lumière et on évi autant que possible toute perturbation aux poissons. Au matin, une faible lumière est allumée, qui déclenchera frai. Après 2 à 4 heures, les poissons sont enlevés et les œufs récoltés. Si le nombre d'œufs nécessaire dépasse capacité de production d'une femelle, un nombre suffisant de cuves de frai peut être disposé en parallèle. En nota la capacité de reproduction de chaque femelle avant l'essai (taille de la ponte et qualité des œufs), on peut sélectio ner les femelles qui possèdent la capacité de reproduction la plus avantageuse pour l'essai.
|
Les œufs doivent être transférés vers les récipients d'essai dans des tubes en verre (diamètre intérieur supérieur ou égal à 4 mm) dotés d'une poire aspirante souple. La quantité d'eau accompagnant les œufs lors de leur transfert devrait être aussi faible que possible. Les œufs sont plus denses que l'eau et tombent hors du tube. Il faut veiller à ce que les œufs (et les larves) n'entrent pas en contact avec l'air. On procédera à un examen microscopique d'un ou de plusieurs échantillons de pontes, pour vérifier s'il n'y a pas d'anomalies aux premiers stades de développement. La désinfection des œufs n'est pas autorisée.
Le taux de mortalité des œufs est le plus élevé dans les 24 heures qui suivent la fécondation. On observe souvent une mortalité de 5 à 40 % durant cette période. Les œufs dégénèrent parce que la fécondation a échoué ou le développement ne se déroule pas normalement. La qualité des œufs d'une ponte semble dépendre de la femelle, certaines femelles produisant systématiquement des œufs de bonne qualité, tandis que d'autres n'y arrivent jamais. La vitesse de développement et d'éclosion varie aussi d'une ponte à l'autre. Les œufs dont la fécondation s'est bien déroulée et les alevins présentent un taux de survie élevé, normalement supérieur à 90 % pour cent. À 25 oC, les œufs éclosent 3 à 5 jours après la fécondation et le sac vitellin est absorbé environ 13 jours après la fécondation.
Le développement embryonnaire a été bien caractérisé par Hisaoka et Battle (2). Grâce à la transparence des œufs et des larves écloses, il est possible de suivre le développement des poissons et d'observer la présence de malformations. Environ 4 heures après le frai, les œufs fécondés peuvent être distingués des œufs non fécondés (3). Les œufs et les larves sont placés dans des récipients d'essai de petit volume puis examinés au microscope.
Les conditions d'essai qui s'appliquent aux premiers stades de la vie sont énumérées à l'appendice 2. Les valeurs optimales pour le pH et la dureté de l'eau de dilution sont respectivement de 7,8 et de 250 mg de CaCO3/l.
CALCULS ET STATISTIQUES
Une démarche en deux étapes est proposée. Tout d'abord, on procède à l'analyse statistique des données concernant la mortalité, les anomalies du développement et le moment de l'éclosion. Ensuite, on évalue statistiquement la longueur corporelle des poissons pour les concentrations auxquelles aucun effet nuisible n'a été détecté sur l'ensemble de ces trois premiers paramètres. Cette démarche est souhaitable étant donné que le produit toxique peut sélectivement tuer les plus petits poissons, retarder l'éclosion et induire des malformations évidentes, et par conséquent fausser les mesures de la longueur. En outre, il y aura à peu près le même nombre de poissons à mesurer par traitement, ce qui garantira la validité de l'analyse statistique de l'essai.
DÉTERMINATION DE LA CL50 ET DE LA CE50
Le pourcentage d'œufs et de larves survivants est calculé et corrigé en fonction de la mortalité chez les témoins à l'aide de la formule d'Abbott (4):

où:
|
P
|
=
|
pourcentage de survivants corrigé
|
|
P'
|
=
|
pourcentage de survivants observé dans la concentration d'essai
|
|
C
|
=
|
pourcentage de survivants chez les témoins
|
Si possible, la CL50 devra être déterminée par une méthode appropriée à la fin de l'essai.
Si l'on souhaite intégrer les données relatives aux malformations morphologiques dans le traitement statistique de In CE50 on trouvera des indications à ce sujet dans l'article de Stephan (5).
ESTIMATION DE LA CMEO ET DE LA CSEO
L'essai de toxicité aux stades de l'embryon et de l'alevin vise notamment à comparer les expériences menées à des concentrations non nulles avec les témoins afin de déterminer la CMEO. Il y a donc lieu d'appliquer les méthodes de comparaisons multiples (6) (7) (8) (9) (10).
BIBLIOGRAPHIE
|
(1)
|
Laale H. W. (1977) The Biology and Use of the Zebrafish (Brachydanïo rerio) in Fisheries Research. A Literature Review. J. Fish Biol. 10. pp. 121-173.
|
|
(2)
|
Hisaoka K. K. and Battle H. I. (1958). The Normal Development Stages of the Zebrafish Brachydanio reno (Hamilton-Buchanan) J. Morph., 102. 311 pp.
|
|
(3)
|
Nagel R. (1986). Untersuchungen zur Eiproduktion beim Zebrabärbling (Brachydanio rerio Hamilton-Buchanan). Journal of Applied Ichthyology, 2, pp. 173-181.
|
|
(4)
|
Finney D. J. (1971). Probit Analysis, 3rd ed., Cambridge University Press, Great Britain, pp. 1-333.
|
|
(5)
|
Stephan C. E. (1982). Increasing the Usefulness of Acute Toxicity Tests. Aquatic Toxicology and Hazard Assessment: Fifth Conference, ASTM STP 766, J. G. Pearson, R. B. Foster and W. E. Bishop, Eds., American Society for Testing and Materials, pp. 69-81.
|
|
(6)
|
Dunnett C. W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with a Control.). Amer. Statist. Assoc, 50, pp. 1096-1121.
|
|
(7)
|
Dunnett C. W. (1964) New Tables for Multiple Comparisons with a Control. Biometrics, 20, pp. 482-491.
|
|
(8)
|
Williams D. A. (1971). A Test for Differences Between Treatment Means when Several Dose Levels are Compared with a Zero Dose Control. Biometrics, 27, pp. 103-117.
|
|
(9)
|
Williams D. A. (1972). The Comparison of Several Dose Levels with a Zero Dose Control. Biometrics 28, pp. 519-531.
|
|
(10)
|
Sokal R. R. and Rohlf F. J. (1981). Biometry, the Principles and Practice of Statistics in Biological Research, W. H. Freeman and Co., San Francisco.
|
Appendice 2
CONDITIONS ET DURÉE DE L'ESSAI ET CRITÈRES DE SURVIE POUR LES ESPÈCES RECOMMANDÉES
|
Espèce
|
Température
(degrés C)
|
Salinité
( %o)
|
Photopériode
(heures)
|
Durée des stades
(jours)
|
Durée habituelle de l'essai
|
Survie des témoins
( % minimum)
|
|
Embryon
|
Alevin
|
Taux de réussite de l'éclosion
|
Après éclosion
|
|
EAU DOUCE
|
|
Brachydanio rerio
Danio
|
25 ± 1
|
—
|
12-16
|
3-5
|
8-10
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 5 jours après l'éclosion (8-10 jours)
|
80
|
90
|
|
Oncorhynchus mykiss
Truite arc-en-ciel
|
10 ± 1 (22)
12 ± 1 (23)
|
—
|
0 (24)
|
30-35
|
25-30
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 20 jours après l'éclosion (50-55 jours)
|
66
|
70
|
|
Cyprinus carpio
Carpe commune
|
21-25
|
—
|
12-16
|
5
|
> 4
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 4 jours après l'éclosion (8-9 jours)
|
80
|
75
|
|
Oryzias latipes
Medaka
|
24 ± 1 (22)
23 ± 1 (23)
|
—
|
12-16
|
8-11
|
4-8
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 5 jours après l'éclosion (13-16 jours)
|
80
|
80
|
|
Pimephales promelas
Tête-de-boule
|
25 ± 2
|
—
|
16
|
4-5
|
5
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 4 jours après l'éclosion (8-9 jours)
|
60
|
70
|
Appendice 3
Conditions de l'essai, durée et critères de survie pour d'autres espèces sur lesquelles on possède une bonne documentation
|
Espèce
|
Température (degrés C)
|
Salinité (% 0)
|
Photopériode (heures)
|
Durée des stades (jours)
|
Durée habituelle de l'essai sur l'embryon et l'alevin
|
Survie des témoins ( % minimum)
|
|
Embryon
|
Alevin
|
Taux de réussite de l'éclosion
|
Après éclosion
|
|
EAU DOUCE
|
|
Carassius auratus
Cyprin doré
|
24 ± 1
|
—
|
—
|
3-4
|
> 4
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 4 jours après l'éclosion (7 jours)
|
_
|
80
|
|
Léopomis macrochirus
Crapet arlequin
|
21 ± 1
|
—
|
16
|
3
|
> 4
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 4 jours après l'éclosion (7 jours)
|
—
|
75
|
|
EAU SALÉE
|
|
Menidia peninsulae
Capucette nord-américaine
|
22-25
|
15-22
|
12
|
1,5
|
10
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 5 jours après l'éclosion (6-7 jours)
|
80
|
60
|
|
Clupea harengus
Hareng
|
10 ± 1
|
8-15
|
12
|
20-25
|
3-5
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 3 jours après l'éclosion (23-27 jours)
|
60
|
80
|
|
Gadus morhua
Morue
|
5 ± 1
|
5-30
|
12
|
14-16
|
3-5
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 3 jours après l'éclosion (18 jours)
|
60
|
80
|
|
Cyprinodon variegatus
Fondule tête de mouton
|
25 ± 1
|
15-30
|
12
|
—
|
—
|
Dès que possible après la fécondation (début du stade gastrula) jusqu'à 4/7 jours après l'éclosion (28 jours)
|
> 75
|
80
|
APPENDICE 4
QUELQUES CARACTÉRISTIQUES CHIMIQUES D'UNE EAU DE DILUTION ACCEPTABLE
|
Substance
|
Concentrations
|
|
Matières particulaires
|
< 20 mg/l
|
|
Carbone organique total
|
< 2 mg/l
|
|
Ammoniac non ionisé
|
< 1 μg/l
|
|
Chlore résiduel
|
< 10 μg/l
|
|
Pesticides organophosphorés totaux
|
< 50 ng/l
|
|
Pesticides organochlorés totaux et biphényles polychlorés
|
< 50 ng/l
|
|
Chlore organique total
|
< 25 ng/l
|
C.16. ABEILLE DOMESTIQUE — ESSAI DE TOXICITÉ AIGUË PAR VOIE ORALE
1. MÉTHODE
La méthode décrite pour cet essai de toxicité aiguë reprend la ligne directrice no 213 de l'OCDE (1998).
1.1. INTRODUCTION
Le présent essai de toxicité est une méthode d'essai en laboratoire destinée à évaluer la toxicité aiguë par voie orale des pesticides et d'autres produits chimiques pour des abeilles domestiques ouvrières adultes.
Lorsqu'on apprécie et évalue les caractéristiques toxiques d'une substance, il peut être nécessaire de déterminer sa toxicité aiguë par voie orale pour les abeilles domestiques, par exemple, lorsque des abeilles risquent d'être exposées à cette substance. L'essai de toxicité aiguë par voie orale vise à déterminer la toxicité intrinsèque des pesticides et d'autres produits chimiques pour les abeilles. Les résultats de cet essai devraient être mis à profit pour établir la nécessité d'une évaluation plus poussée. Cette méthode peut être utilisée, en particulier, dans des programmes séquentiels destinés à évaluer les risques des pesticides pour les abeilles, selon la progression suivante: essais de toxicité en laboratoire, essais en conditions semi-naturelles et essais sur le terrain (1). Les pesticides peuvent être testés en tant que substances actives ou en tant que préparations chimiques.
Un étalon de toxicité doit être utilisé pour vérifier la sensibilité des abeilles et la précision du protocole d'essai.
1.2. DÉFINITIONS
Toxicité aiguë par voie orale: effets nocifs se produisant dans un délai maximal de 96 heures après l'administration d'une dose unique de la substance d'essai par voie orale.
Dose: de substance d'essai consommée. La dose est exprimée en masse (μg) de substance d'essai par animal testé (μg/abeille). La dose réellement consommée par chaque abeille ne peut être calculée, étant donné que les abeilles sont nourries collectivement, mais il est possible d'estimer une dose moyenne (quantité totale de substance d'essai consommée/nombre d'abeilles testées par cage).
DL50 (dose létale 50 %) orale: dose unique d'une substance, obtenue par calcul statistique, susceptible d'entraîner la mort de 50 % des animaux lorsqu'elle est administrée par voie orale. La DL50 s'exprime en μg de substance d'essai par abeille. Pour les pesticides, la substance d'essai peut être une substance active ou une préparation chimique contenant une ou plusieurs substances actives.
Mortalité: un animal est noté comme mort lorsqu'il est complètement immobile.
1.3. PRINCIPE DE LA MÉTHODE
Des abeilles domestiques ouvrières adultes (Apis mellifera) sont exposées à une gamme de doses de la substance d'essai dispersée dans une solution de saccharose. Elles reçoivent ensuite la même alimentation, mais sans la substance d'essai. La mortalité est notée quotidiennement durant au moins 48 heures et comparée aux valeurs des témoins. Si le taux de mortalité augmente entre 24 heures et 48 heures, tandis que la mortalité des témoins demeure à un niveau acceptable, c'est-à-dire ≤ 10 %, il convient d'allonger la durée de l'essai jusqu'à 96 heures au maximum. On analyse les résultats afin de calculer la DL50 à 24 heures et à 48 heures et, si l'étude est prolongée, à 72 heures et à 96 heures.
1.4. VALIDITÉ DE L'ESSAI
La validité de l'essai repose sur les conditions suivantes:
|
—
|
le taux moyen de mortalité de l'ensemble des témoins ne doit pas dépasser 10 % à la fin de l'essai,
|
|
—
|
la DL50 de l'étalon de toxicité se trouve dans la gamme spécifiée.
|
1.5. DESCRIPTION DE LA MÉTHODE
1.5.1. Collecte des abeilles
Il convient d'utiliser de jeunes abeilles ouvrières adultes de la même race, autrement dit des abeilles du même âge, nourries de la même façon, etc. Elles doivent provenir de colonies saines, correctement nourries, dépourvues autant que possible d'individus malades, possédant une reine dans la ruche, et dont on connaît l'histoire et l'état physiologique. Elles peuvent être collectées le matin du jour où elles seront utilisées ou la veille au soir et gardées dans les conditions de l'essai jusqu'au jour suivant. Les abeilles récoltées sur des cadres dépourvus de couvain conviennent. Il faut éviter de collecter les abeilles au début du printemps ou à la fin de l'automne parce qu'elles subissent un changement physiologique à ces époques. Si les essais doivent être effectués au début du printemps ou à la fin de l'automne, on peut faire éclore les abeilles dans une étuve et elles seront ensuite élevées pendant une semaine avec du pain d'abeille (pollen récolté sur les rayons) et une solution de saccharose. Les abeilles traitées avec des substances chimiques, telles que des antibiotiques, des antivarroa, etc., ne doivent pas être soumises à des essais de toxicité durant 4 semaines à partir de la fin du dernier traitement.
1.5.2. Conditions d'encagement et d'alimentation
On utilise des cages faciles à nettoyer et bien ventilées. Tout matériau approprié peut être employé, par exemple des cages en acier inoxydable, en toile métallique, en plastique, des cages jetables en bois, etc. Il est préférable de répartir les abeilles en groupes de dix par cage. La dimension des cages d'essai doit être adaptée au nombre d'abeilles, c'est-à-dire leur fournir un espace suffisant.
Les abeilles doivent être gardées à l'obscurité dans une salle d'expérience chauffée à une température de 25 ± 2 oC. L'humidité relative, comprise normalement entre 50 et 70 %, doit être enregistrée tout au long de l'essai. Les manipulations, et notamment le traitement et les observations, peuvent s'effectuer à la lumière (du jour). Les abeilles sont nourries avec une solution de saccharose dans l'eau dont la concentration finale s'élève à 500 g/1 (50 % en poids par unité de volume). Après que les doses d'essai ont été administrées, l'alimentation doit être fournie ad libitum. Le système d'alimentation doit permettre l'enregistrement de la prise de nourriture dans chaque cage (voir point 1.6.3.1). Un tube de verre (de quelque 50 mm de long et 10 mm de large, dont le diamètre de l'extrémité ouverte est rétréci à environ 2 mm) peut être utilisé.
1.5.3. Préparation des abeilles
Les abeilles récoltées sont réparties au hasard dans les cages d'essai qui, elles-mêmes, sont disposées au hasard dans la salle d'expérience.
Les abeilles peuvent être privées de nourriture pendant une durée maximale de 2 heures avant le début de l'essai. On recommande de faire jeûner les abeilles avant le traitement pour que le niveau de remplissage de leur tube digestif soit identique au début de l'essai. Les abeilles moribondes doivent être enlevées et remplacées par des abeilles saines avant le début de l'essai.
1.5.4. Préparation des doses
Si la substance d'essai est miscible dans l'eau, elle peut être dispersée directement dans une solution de saccharose à 50 %. Pour les produits de qualité technique et les substances peu solubles dans l'eau, des véhicules tels que des solvants organiques, des émulsifiants ou des dispersants peu toxiques pour les abeilles peuvent être utilisés (par exemple, de l'acétone, du diméthylformamide, du diméthylsulfoxyde). La concentration du véhicule dépend de la solubilité de la substance d'essai et doit être identique pour toutes les concentrations testées. Cependant, il convient généralement d'appliquer une concentration de 1 % pour le véhicule et de ne pas la dépasser.
Des solutions témoins appropriées doivent être préparées; autrement dit, si un solvant ou un dispersant ont été employés pour solubiliser la substance d'essai, deux groupes de témoins doivent être utilisés: une solution aqueuse et une solution de saccharose renfermant le solvant ou le véhicule à la même concentration que dans les solutions d'essai.
1.6. MODE OPÉRATOIRE
1.6.1. Groupes testés et groupes témoins
Le nombre de doses et de groupes d'essai testés par dose doit se prêter aux exigences statistiques de la détermination de la DL50, avec des limites de confiance de 95 %. L'essai demande normalement 5 doses appartenant à une série géométrique dont le facteur n'excède pas 2,2 et qui englobe la DL50. Toutefois, le facteur de dilution et le nombre de concentrations du traitement doivent être déterminés en fonction de la pente de la courbe de toxicité (dose en fonction de la mortalité) et en tenant compte de la méthode statistique choisie pour analyser les résultats. Un test de détermination de l'ordre de grandeur permet de choisir les concentrations appropriées pour le dosage.
Il faut tester au moins 3 groupes d'essai identiques, comportant chacun 10 abeilles, par concentration d'essai. Pas moins de 3 groupes témoins, comportant chacun 10 abeilles, devraient être testés parallèlement aux groupes traités. Des séries de témoins devraient également être incluses pour les solvants ou les véhicules utilisés (voir point 1.5.4).
1.6.2. Étalon de toxicité
Un étalon de toxicité doit être testé parallèlement aux groupes traités. Il faut sélectionner au moins 3 doses afin d'englober la valeur supposée de la DL50. Chaque dose doit être testée dans au moins 3 cages contenant chacune 10 abeilles. L'étalon de toxicité préféré est le diméthoate, dont la DL50 par voie orale après 24 heures se situe entre 0,10 et 0,35 μg de substance active/abeille (2). D'autres étalons de toxicité pourraient toutefois être acceptables, s'il existe suffisamment de données qui permettent de vérifier la relation dose/effet escomptée (par exemple le parathion).
1.6.3. Exposition
1.6.3.1. Administration des doses
Il faut fournir à chaque groupe d'abeilles testé 100 à 200 μl d'une solution aqueuse de saccharose à 50 % contenant la substance d'essai à la concentration appropriée. Il est nécessaire d'administrer un volume plus grand dans le cas des produits peu solubles, peu toxiques ou peu concentrés dans la préparation, étant donné qu'il faut utiliser des proportions plus élevées dans la solution de saccharose. La quantité de nourriture traitée consommée par groupe doit être surveillée. Une fois vidé (généralement en 3 à 4 heures), le tube contenant la solution alimentaire traitée doit être retiré de la cage et remplacé par un tube contenant une solution de saccharose pure. Les solutions de saccharose sont ensuite fournies ad libitum. Pour certains composés, la nourriture traitée avec des concentrations élevées peut être rejetée par les abeilles, si bien que la quantité de nourriture consommée risque d'être faible ou nulle. Après 6 heures au maximum, la nourriture traitée non consommée doit être remplacée par une solution de saccharose pure. La quantité de nourriture traitée consommée doit être évaluée {par exemple, en mesurant le volume ou le poids de la nourriture traitée restante).
1.6.3.2. Durée
L'essai dure 48 heures après que la solution d'essai a été remplacée par une solution de saccharose pure. Si! a mortalité continue de s'accroître de plus de 10 % après les premières 24 heures, la durée de l'essai doit être prolongée jusqu'à 96 heures au maximum, à condition que la mortalité des témoins n'excède pas 10 %.
1.6.4. Observations
La mortalité est relevée 4 heures après le début de l'essai et ensuite 24 heures et 48 heures après que la dose a été administrée. Si une période d'observation prolongée est requise, d'autres évaluations devront être pratiquées à des intervalles de 24 heures jusqu'à 96 heures au maximum, à condition que la mortalité des témoins ne dépasse pas 10 %.
La quantité de solution alimentaire consommée par groupe doit être estimée. La comparaison entre les taux de consommation des solutions traitées et non traitées en l'espace de 6 heures peut donner une idée des qualités organoleptiques de la nourriture traitée.
Tous les effets se traduisant par des anomalies du comportement pendant la période d'essai doivent être notés.
1.6.5. Essai limite
Dans certains cas (par exemple, lorsqu'on s'attend à ce qu'une substance d'essai soit peu toxique), un essai limite peut être exécuté avec 100 μg de substance active/abeille afin de démontrer que la DL50 est supérieure à cette valeur. Le même protocole doit être appliqué, et notamment les 3 groupes d'essai identiques pour la dose testée, les témoins nécessaires, l'évaluation de la quantité de nourriture traitée consommée et l'utilisation d'un étalon de toxicité. Si on constate une mortalité, il convient de mener une étude complète. Si on observe des effets sublétaux (voir point 1.6.4), il faut les noter.
2. RÉSULTATS ET RAPPORT
2.1. RÉSULTATS
Les résultats doivent être récapitulés sous la forme de tableaux, montrant pour chaque groupe traité avec la substance d'essai ainsi que pour les témoins et les groupes traités avec l'étalon de toxicité, le nombre d'abeilles employé, la mortalité à chaque heure d'observation et le nombre d'abeilles présentant un comportement anormal. Les données de mortalité seront analysées par les méthodes statistiques appropriées (par exemple, la méthode des probits, la moyenne mobile, la probabilité binomiale) (3) (4). On trace les courbes dose/effet pour chaque moment d'observation recommandé (24 heures, 48 heures, et, si nécessaire, 72 heures, 96 heures) et on calcule les pentes des courbes et les doses létales 50 % (DL50) avec des limites de confiance de 95 %. Les corrections relatives à la mortalité des témoins doivent s'effectuer selon la méthode d'Abbott (4) (5). Lorsque la nourriture traitée n'est pas complètement consommée, la dose de la substance d'essai consommée par groupe doit être déterminée. La DL50 doit être exprimée en μg de substance d'essai par abeille.
2.2. RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
2.2.1. Substance d'essai
|
—
|
État physique et propriétés physico-chimiques pertinentes (par exemple, stabilité dans l'eau, pression de vapeur),
|
|
—
|
données relatives à l'identification chimique, notamment la formule structurale, la pureté (dans le cas des pesticides, l'identité et la concentration de la ou des substances actives).
|
2.2.2. Espèce d'expérience
|
—
|
Nom scientifique, race, âge approximatif (en semaines), méthode et date de collecte,
|
|
—
|
informations sur les colonies dans lesquelles les abeilles ont été récoltées, en particulier l'état de santé, toute maladie des adultes, les prétraitements éventuels, etc.
|
2.2.3. Conditions d'essai
|
—
|
Température et humidité relative de la salle d'expérience,
|
|
—
|
conditions d'encagement, en particulier le type, la dimension et le matériau des cages,
|
|
—
|
méthodes de préparation des solutions mères et des solutions d'essai (le solvant et sa concentration doivent être indiqués, le cas échéant),
|
|
—
|
conception de l'essai, par exemple, nombre et niveau des concentrations d'essai appliquées, nombre de témoins; pour chaque concentration d'essai et témoin: nombre de cages et nombre d'abeilles par cage,
|
2.2.4. Résultats
|
—
|
Résultats d'une étude préliminaire de détermination de l'ordre de grandeur, le cas échéant,
|
|
—
|
données brutes: mortalité à chaque dose testée et à chaque temps d'observation.
|
|
—
|
courbes dose/effet à la fin de l'essai,
|
|
—
|
valeurs de la DL50 avec des limites de confiance de 95 %, à chaque temps d'observation recommandé, pour la substance d'essai et l'étalon de toxicité,
|
|
—
|
méthodes statistiques utilisées pour calculer la DL50,
|
|
—
|
mortalité chez les témoins,
|
|
—
|
autres effets biologiques observes ou mesurés, par exemple, comportement anormal des abeilles (y compris rejet de la dose d'essai), vitesse de consommation de la nourriture dans les groupes traités et non traités,
|
|
—
|
tout écart par rapport au protocole décrit ici et toute autre information utile.
|
3. BIBLIOGRAPHIE
|
(1)
|
EPPO/Council of Europe (1993). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products — Honeybees. EPPO Bulletin, Vol. 23, N.1, pp. 151-165. March 1993.
|
|
(2)
|
Gough, H. J., McIndoe, E.C., Lewis, G.B. (1994). The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L.) 1981-1992. Journal of Apicultural Research, 22, pp. 119-125.
|
|
(3)
|
Litchfield, J. T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, pp. 99-113.
|
|
(4)
|
Finney, D. J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New York.
|
|
(5)
|
Abbott, W. S. (1925). A method for computing the effectiveness of an insecticide. Jour. Econ. Entomol., 18, pp. 265-267.
|
C.17. ABEILLE DOMESTIQUE — ESSAI DE TOXICITÉ AIGUË PAR CONTACT
1. MÉTHODE
La méthode décrite pour cet essai de toxicité aiguë reprend la ligne directrice no 214 de l'OCDE (1998).
1.1. INTRODUCTION
Le présent essai de toxicité est une méthode d'essai en laboratoire destinée à évaluer la toxicité aiguë par contact des pesticides et d'autres produits chimiques pour des abeilles domestiques ouvrières adultes.
Lorsqu'on apprécie et évalue les caractéristiques toxiques d'une substance, il peut être nécessaire de déterminer sa toxicité aiguë par contact pour les abeilles domestiques, par exemple lorsque des abeilles risquent d'être exposées à cette substance. L'essai de toxicité aiguë par contact vise à déterminer la toxicité intrinsèque des pesticides et d'autres produits chimiques pour les abeilles. Les résultats de cet essai devraient être mis à profit pour établir la nécessité d'une évaluation plus poussée. Cette méthode peut être utilisée, en particulier, dans des programmes séquentiels destinés à évaluer les risques des pesticides pour les abeilles, selon la progression suivante: essais de toxicité en laboratoire, essais en conditions semi-naturelles et essais sur le terrain (1). Les pesticides peuvent être testés en tant que substances actives ou en tant que préparations chimiques.
Un étalon de toxicité doit être utilisé pour vérifier la sensibilité des abeilles et la précision du protocole d'essai.
1.2. DÉFINITIONS
Toxicité aiguë par contact: effets nocifs se produisant dans un délai maximal de 96 heures après l'application locale d'une dose unique de la substance d'essai.
Dose: quantité de substance d'essai appliquée. La dose est exprimée en masse (μg) de substance d'essai par animal testé (μg/abeille).
DL50 (dose létale 50 %) par contact: dose unique d'une substance, obtenue par calcul statistique, susceptible d'entraîner la mort de 50 % des animaux lorsqu'elle est administrée par contact. La DL50 est exprimée en μg de substance d'essai par abeille. Pour les pesticides, la substance d'essai peut être une substance active ou une préparation chimique contenant une ou plusieurs substances actives.
Mortalité: un animal est noté comme mort lorsqu'il est complètement immobile.
1.3. PRINCIPE DE LA MÉTHODE
Des abeilles domestiques ouvrières adultes sont exposées à une gamme de doses de la substance d'essai dissoute dans un véhicule approprié, par application directe sur le thorax (aérosol). L'essai dure 48 heures. Si le taux de mortalité augmente entre 24 heures et 48 heures, alors que la mortalité des témoins reste à un niveau acceptable, c'est-à-dire ≤ 10 %, il convient de prolonger l'essai jusqu'à 96 heures au maximum. La mortalité est notée tous les jours et comparée avec les valeurs des témoins. On analyse les résultats afin de calculer la DL50 à 24 heures et à 48 heures et, au cas où l'étude est prolongée, à 72 heures et à 96 heures.
1.4. VALIDITÉ DE L'ESSAI
La validité de l'essai repose sur les conditions suivantes:
|
—
|
le taux moyen de mortalité de l'ensemble des témoins ne doit pas dépasser 10 % à la fin de l'essai,
|
|
—
|
la DL50 de l'étalon de toxicité se trouve dans la gamme spécifiée.
|
1.5. DESCRIPTION DE LA MÉTHODE
1.5.1. Collecte des abeilles
Il convient d'utiliser de jeunes abeilles ouvrières adultes de la même race, autrement dit des abeilles du même âge, nourries de la même façon, etc. Elles doivent provenir de colonies saines, correctement nourries, dépourvues autant que possible d'individus malades, possédant une reine dans la ruche, et dont on connaît l'histoire et l'état physiologique. Elles peuvent être collectées le matin du jour où elles seront utilisées ou la veille au soir et gardées dans les conditions de l'essai jusqu'au jour suivant. Les abeilles récoltées sur des cadres dépourvus de couvain conviennent. Il faut éviter de collecter les abeilles au début du printemps ou à la fin de l'automne parce qu'elles subissent un changement physiologique à ces époques. Si les essais doivent être effectués au début du printemps ou à la fin de l'automne, on peut faire éclore les abeilles dans un incubateur et elles seront ensuite élevées pendant une semaine avec du pain d'abeille (pollen récolté sur les rayons) et une solution de saccharose. Les abeilles traitées avec des substances chimiques, telles que des antibiotiques, des antivarroa, etc., ne doivent pas être soumises à des essais de toxicité durant 4 semaines à partir de la fin du dernier traitement.
1.5.2. Conditions d'encagement et d'alimentation
On utilise des cages faciles à nettoyer et bien ventilées. Tout matériau approprié peut être employé, par exemple des cages en acier inoxydable, en toile métallique, en plastique, des cages jetables en bois, etc. La dimension des cages d'essai doit être adaptée au nombre d'abeilles, c'est-à-dire leur fournir un espace suffisant. Il est préférable de répartir les abeilles en groupes de dix par cage.
Les abeilles doivent être gardées à l'obscurité dans une salle d'expérience chauffée à 25 ± 2 oC. L'humidité relative, généralement comprise entre 50 et 70 %, doit être enregistrée tout au long de l'essai. Les manipulations, et notamment le traitement et les observations, peuvent s'effectuer à la lumière (du jour). La nourriture se compose d'une solution de saccharose dans l'eau dont la concentration finale s'élève à 500 g/l (50 % en poids par unité de volume), fournie ad libitum durant toute la durée de l'essai à l'aide d'une mangeoire pour abeilles. Celle-ci peut consister en un tube de verre (d'environ 50 mm de long et 10 mm de large, dont le diamètre de l'extrémité ouverte est rétréci à environ 2 mm).
1.5.3. Préparation des abeilles
Les abeilles récoltées peuvent être anesthésiées avec du dioxyde de carbone ou de l'azote en vue de l'application de la substance d'essai. La quantité d'anesthésiant utilisée et la durée de l'exposition doivent être réduites au minimum. Les abeilles moribondes doivent être enlevées et remplacées par des abeilles saines avant le début de l'essai.
1.5.4. Préparation des doses
La substance d'essai doit être appliquée sous la forme de solution dans un véhicule, à savoir un solvant organique ou de l'eau avec un agent mouillant. Parmi les solvants organiques, on préfère l'acétone, mais d'autres solvants organiques peu toxiques pour les abeilles peuvent être utilisés (par exemple, le diméthylformamide, le diméthylsulfoxyde). Dans le cas des préparations chimiques dispersées dans l'eau et des substances organiques fortement polaires non solubles dans les solvants organiques, les solutions peuvent être plus faciles à appliquer si elles sont préparées dans une solution faible d'un agent mouillant commercial (par exemple, Agral, Citowett, Lubrol, Triton, Tween).
Des solutions témoins appropriées doivent être préparées, autrement dit, si un solvant ou un dispersant ont été employés pour solubiliser la substance d'essai, deux groupes de témoins doivent être utilisés, l'un étant traité avec de l'eau et l'autre avec le solvant ou le dispersant.
1.6. MODE OPÉRATOIRE
1.6.1. Groupes testés et groupes témoins
Le nombre de doses et de groupes d'essai testes par dose doit se prêter aux exigences statistiques de la détermination de la DL50, avec des limites de confiance de 95 %. L'essai demande normalement cinq doses appartenant à une série géométrique dont le facteur n'excède pas 2,2 et qui englobe la DL50. Toutefois, le nombre de doses doit être déterminé en fonction de la pente de la courbe de toxicité (dose en fonction de la mortalité) et en tenant rompre de la méthode statistique choisie pour analyser les résultats. Un test de détermination de l'ordre de grandeur permet de choisir les doses appropriées.
Il faut tester au moins 3 groupes d'essai identiques, comportant chacun 10 abeilles, par concentration d'essai.
Pas moins de 3 groupes témoins, comportant chacun 10 abeilles, devraient être testes parallèlement aux groupes traités. Si on utilise un solvant organique ou un agent mouillant, 3 groupes supplémentaires de 10 abeilles chacun doivent être inclus pour le solvant ou l'agent mouillant.
1.6.2. Étalon de toxicité
Un étalon de toxicité doit être testé parallèlement aux groupes traités. Il faut sélectionner au moins 3 doses afin d'englober la valeur supposée de la DL50. Chaque dose doit être testée dans au moins 3 cages contenant chacune 10 abeilles. L'étalon de toxicité préféré est le diméthoate, dont la DL50 par contact après 24 heures se situe entre 0,10 et 0,30 μg de substance active/abeille (2). D'autres étalons de toxicité pourraient toutefois être acceptables, s'il existe suffisamment de données qui permettent de vérifier la relation dose/effet escomptée (par exemple, le parathion).
1.6.3. Exposition
1.6.3.1. Administration des doses
Les abeilles anesthésiées sont traitées individuellement par application locale. Les abeilles sont réparties au hasard entre les groupes traites aux différentes doses et les groupes témoins. Un volume de 1 μl de solution contenant la substance d'essai à la concentration appropriée doit être appliqué à l'aide d'un micro-applicateur sur la face dorsale du thorax de chaque abeille. D'autres volumes peuvent être utilisés si cela se justifie. Après l'application, les abeilles sont réparties entre les cages d'essai dans lesquelles on distribue une solution de saccharose.
1.6.3.2. Durée
L'essai dure 48 heures. Si la mortalité augmente de plus de 10 % entre 24 heures et 48 heures, la durée de l'essai doit être prolongée jusqu'à 96 heures, à condition que la mortalité des témoins ne dépasse pas 10 %.
1.6.4. Observations
La mortalité est relevée 4 heures après l'application de la dose, puis 24 heures et 48 heures après celle-ci. Si une période d'observation prolongée est requise, d'autres évaluations devront être pratiquées à des intervalles de 24 heures pendant 96 heures au maximum, à condition que la mortalité des témoins ne dépasse pas 10 %.
Tous les effets se traduisant par des anomalies du comportement pendant la période d'essai doivent être notés.
1.6.5. Essai limite
Dans certains cas (par exemple lorsqu'on s'attend à ce qu'une substance d'essai soit peu toxique), un essai limite peut être exécuté avec 100 μg de substance active/abeille afin de démontrer que la DL50 est supérieure à cette valeur. Le même protocole doit être appliqué, et notamment les 3 groupes d'essai identiques pour la dose testée, les témoins nécessaires et l'utilisation d'un étalon de toxicité. Si on constate une mortalité, il convient de mener une étude complète. Si on observe des effets sublétaux (voir point 1.6.4), il faut les noter.
2. RÉSULTATS ET RAPPORT
2.1. RÉSULTATS
Les résultats doivent être récapitulés sous la forme de tableaux, montrant pour chaque groupe traité avec la substance d'essai, ainsi que pour les témoins et les groupes traites avec l'étalon de toxicité, le nombre d'abeilles employé, la mortalité à chaque heure d'observation et le nombre d'abeilles présentant un comportement anormal. Les données de mortalité seront analysées par les méthodes statistiques appropriées (par exemple, la méthode des probits, la moyenne mobile, la probabilité binomiale) (3) (4). On trace les courbes dose-effet pour chaque moment d'observation recommandé (24 heures, 48 heures, et, si nécessaire, 72 heures, 96 heures) et on calcule les pentes des courbes et les doses létales 50 % DL50) avec des limites de confiance de 95 %. Les corrections relatives à la mortalité des témoins doivent s'effectuer selon la méthode d'Abbott (4) (5). La DL50 doit être exprimée en μg de substance d'essai par abeille.
2.2. RAPPORT D'ESSAI
Le rapport d'essai doit contenir les informations suivantes:
2.2.1. Substance d'essai
|
—
|
État physique et propriétés physico-chimiques pertinentes (par exemple, stabilité dans l'eau, pression de- vapeur),
|
|
—
|
données relatives à l'identification chimique, notamment la formule structurale, la pureté (dans le cas des pesticides, l'identité et la concentration de la ou des substances actives).
|
2.2.2. Espèce d'expérience
|
—
|
Nom scientifique, race, âge approximatif (en semaines), méthode et date de collecte,
|
|
—
|
informations sur les colonies dans lesquelles les abeilles ont été récoltées, en particulier l'état de santé, toute maladie des adultes, les prétraitements éventuels, etc.
|
2.2.3. Conditions d'essai
|
—
|
Température et humidité relative de la salle d'expérience,
|
|
—
|
conditions d'encagement, en particulier le type, la dimension et le matériau des cages,
|
|
—
|
modalités d'administration de la substance d'essai, par exemple, solvant utilisé, volume de solution d'essai appliqué, anesthésiants utilisés,
|
|
—
|
conception de l'essai, par exemple, nombre et niveau des concentrations d'essai appliquées, nombre de témoins; pour chaque concentration d'essai et témoin: nombre de cages et nombre d'abeilles par cage,
|
2.2.4. Résultats
|
—
|
Résultats d'une étude préliminaire de détermination de l'ordre de grandeur, le cas échéant,
|
|
—
|
données brutes: mortalité à chaque concentration testée et à chaque temps d'observation,
|
|
—
|
courbes dose/effet à la fin de l'essai,
|
|
—
|
valeurs de la DL50 avec des limites de confiance de 95 %, à chaque temps d'observation recommandé, pour la substance d'essai et l'étalon de toxicité,
|
|
—
|
méthodes statistiques utilisées pour calculer la DL50,
|
|
—
|
mortalité chez les témoins,
|
|
—
|
autres effets biologiques observés et toute réaction anormale des abeilles,
|
|
—
|
tout écart par rapport au protocole décrit ici et toute autre information utile.
|
3. BIBLIOGRAPHIE
|
(1)
|
EPPO/Council of Europe (1993). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products — Honeybees. EPPO bulletin, Vol. 23, N.1, pp. 151-165. March 1993.
|
|
(2)
|
Cough, H. J., McIndoe, E. C, Lewis, G. B. (1994). The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L.), 1981-1992. Journal of Apicultural Research 22, pp. 119-125.
|
|
(3)
|
Litchfield, J. T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, pp. 99-113.
|
|
(4)
|
Finney, D. J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New York.
|
|
(5)
|
Abbott, W. S. (1925). A method for computing the effectiveness of an insecticide. Jour. Econ. Entomol. 18, pp. 265-267.
|
C.18. DÉTERMINATION DE L'ADSORPTION/DÉSORPTION AU MOYEN DE LA MÉTHODE PAR AGITATION
1. MÉTHODE
La méthode décrite reprend la ligne directrice no 106 de l'OCDE sur l'adsorption/désorption dans les sols fondée sur la méthode par agitation (2000).
1.1. INTRODUCTION
La méthode s'inspire d'un test circulaire, d'un séminaire sur la sélection des sols pour le développement d'un essai d'adsorption (1) (2) (3) (4) et de certaines lignes directrices nationales (5) (6) (7) (8) (9) (10) (11).
Les études d'adsorption/désorption donnent des informations précieuses sur la mobilité des substances chimiques et leur répartition dans les trois compartiments de la biosphère (lithosphère, hydrosphère, atmosphère) (12) (13) (14) (15) (16) (17) (18) (19) (20) (21). Ces informations servent à prédire et à évaluer certaines caractéristiques d'une substance: la capacité de dégradation (22) (23), de transformation et d'absorption par les organismes (24), la lixiviation à travers le sol (16) (18) (19) (21) (25) (26) (27) (28), la volatilité à partir du sol (21) (29) (30), le ruissellement à la surface du sol vers les eaux naturelles (18) (31) (32). Les données sur l'adsorption peuvent être utilisées à des fins de comparaison ou de modélisation (19) (33) (34) (35).
La distribution d'une substance chimique entre les phases solides et aqueuses est un processus complexe qui dépend d'un grand nombre de facteurs: nature chimique de la substance (12) (36) (37) (38) (39) (40), caractéristiques du sol (4) (12) (13) (14) (41) (42) (43) (44) (45) (46) (47) (48) (49), mais aussi des conditions climatiques, telles que précipitations, température, ensoleillement, vent. La méthode présentée ici, qui est un modèle de laboratoire simplifié, ne permet pas d'expliquer entièrement les phénomènes et les mécanismes impliqués dans l'adsorption d'une substance chimique par le sol. Mais si elle n'est pas exhaustive, elle donne tout de même d'utiles informations sur les effets environnementaux de l'adsorption d'un produit chimique.
Voir également l'introduction générale.
1.2. OBJET DE LA MÉTHODE
La méthode vise à évaluer le comportement d'adsorption/désorption d'une substance dans les sols. Il s'agit d'obtenir une valeur de sorption permettant de prédire le partage d'une substance sous diverses conditions environnementales. Les coefficients d'adsorption à l'équilibre d'une substance chimique sur différents sols sont déterminés en fonction des caractéristiques du sol (par exemple, teneur en carbone organique, teneur en argile, texture, pH). Il faut utiliser plusieurs types de sols pour couvrir le plus grand nombre possible d'interactions d'une substance donnée avec des sols naturels.
La présente méthode considère l'adsorption comme la liaison d'une substance chimique à la surface des sols. Elle ne fait pas de distinction entre les différents processus d'adsorption (adsorption physique et chimique) et d'autres processus tels que la dégradation catalysée par la surface, l'adsorption de masse ou la réaction chimique. Elle ne tient pas compte de l'adsorption sur les particules colloïdales (diamètre < 0,2 μm) générées par les sols.
Les paramètres considérés comme les plus importants pour l'adsorption sont: la teneur en carbone organique (3) (4) (12) (13) (14) (41) (43) (44) (45) (46) (47) (48), la teneur en argile, la texture (3) (4) (41) (42) (43) (44) (45) (46) (47) (48), le pH (pour les composés ionisables) (3) (4) (42). D'autres sont susceptibles d'avoir une incidence sur l'adsorption: la capacité d'échange cationique réelle (CECR), la teneur en fer amorphe et en oxydes d'aluminium, notamment dans le cas des sols volcaniques et tropicaux (4), ainsi que la surface spécifique (49).
L'essai permet d'évaluer l'adsorption d'une substance chimique sur des sols de texture et de pH différents et n'ayant pas la même teneur en carbone organique et en argile. Il comprend trois phases:
|
Phase 1:
|
Étude préliminaire servant à déterminer:
|
—
|
le rapport sol/solution,
|
|
—
|
le temps d'équilibre de l'adsorption et la quantité de substance adsorbée à l'équilibre,
|
|
—
|
l'adsorption de la substance sur la surface des récipients d'essai et la stabilité de la substance durant l'essai.
|
|
|
Phase 2:
|
Essai de sélection: l'adsorption est étudiée sur cinq sols différents. On détermine la cinétique d'adsorption avec une concentration unique ainsi que le coefficient de distribution Kd et Koc.
|
|
Phase 3:
|
Détermination des isothermes d'adsorption de Freundlich: cet essai sert à évaluer l'effet de la concentration sur le degré d'adsorption sur les sols.
Étude de la désorption par l'évaluation de la cinétique et des isothermes de désorption de Freundlich (appendice 1).
|
1.3. DÉFINITIONS ET UNITÉS
|
Symbole
|
Définition
|
Unités
|
|

|
pourcentage d'adsorption au temps ti
|
%
|
|
Aeq
|
pourcentage d'adsorption à l'équilibre
|
%
|
|

|
masse de substance adsorbée sur le sol à l'instant ti
|
μg
|
|
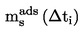
|
masse de substance adsorbée sur le sol durant l'intervalle de temps Δti
|
μg
|
|

|
masse de substance adsorbée sur le sol à l'équilibre d'adsorption
|
μg
|
|
m0
|
masse de substance dans le tube au début de l'essai
|
μg
|
|

|
masse de substance mesurée dans une aliquote (

) au temps ti
|
μg
|
|

|
masse de substance dans la solution à l'équilibre d'adsorption
|
μg
|
|
msoil
|
quantité de phase de sol exprimée en masse sèche du sol
|
g
|
|
Cst
|
concentration massique de la solution de réserve
|
μg cm-3
|
|
C0
|
concentration massique initiale de la solution en contact avec le sol
|
μg cm-3
|
|
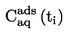
|
concentration massique de la substance dans la phase aqueuse au temps ti, lorsque l'analyse est effectuée
|
μg cm-3
|
|

|
concentration de la substance adsorbée sur le sol à l'équilibre d'adsorption
|
μg g-1
|
|
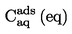
|
concentration massique de la substance dans la phase aqueuse à l'équilibre d'adsorption
|
μg cm-3
|
|
V0
|
volume initial de la phase aqueuse en contact avec le sol durant l'essai d'adsorption
|
cm3
|
|

|
volume de l'aliquote dans laquelle la substance est mesurée
|
cm3
|
|
Kd
|
coefficient de répartition de l'adsorption
|
cm-3 g-1
|
|
Koc
|
coefficient d'adsorption normalisé basé sur la teneur en carbone organique
|
cm3 g-1
|
|
Kom
|
coefficient de la répartition normalisé basé sur la teneur en matière organique
|
cm3 g-1
|
|

|
coefficient d'adsorption de Freundlich
|
μg1-1
/n (cm3)1
/ng-1
|
|
1/n
|
exposant de Freundlich
|
|
|
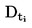
|
pourcentage de désorption à l'instant ti
|
%
|
|

|
pourcentage de désorption durant l'intervalle de temps Δti
|
%
|
|
Kdes
|
coefficient de désorption apparente
|
cm3 g-1
|
|
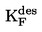
|
coefficient de désorption de Freundlich
|
μg-1
/n (cm3)1
/n g1
|
|
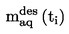
|
masse de substance désorbée à partir du sol à l'instant tj
|
μg
|
|
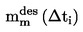
|
masse de substance désorbée à partir du sol durant l'intervalle de temps Δti
|
μg
|
|

|
masse de substance déterminée par analyse dans la phase aqueuse à l'équilibre de désorption
|
μg
|
|

|
masse totale de substance désorbée à l'équilibre de désorption
|
μg
|
|

|
masse de substance restant adsorbée sur le sol après l'intervalle de temps Δti
|
μg
|
|

|
masse de substance résiduelle après atteinte de l'équilibre d'adsorption due à un volume de remplacement insuffisant
|
μg
|
|

|
concentration de la substance restant adsorbée dans le sol à l'équilibre de désorption
|
μg g-1
|
|

|
concentration massique de la substance dans la phase aqueuse à l'équilibre de désorption
|
μg cm-3
|
|
VT
|
volume total de la phase aqueuse en contact avec le sol durant l'essai de cinétique de désorption réalisé avec la méthode séquentielle
|
cm3
|
|
VR
|
volume de surnageant extrait du tube après obtention de l'équilibre d'adsorption et remplacé par un volume identique d'une solution de 0,01 M de CaCl2
|
cm3
|
|
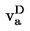
|
volume de l'aliquote extrait du tube (i) pour analyse lors de l'essai de cinétique de désorption réalisé avec la méthode séquentielle
|
cm3
|
|

|
volume de solution extrait du tube (i) afin de mesurer la substance lors de l'essai de cinétique de désorption (méthode parallèle)
|
cm3
|
|

|
volume de solution extrait du tube afin de mesurer la substance à l'équilibre de désorption
|
cm3
|
|
MB
|
bilan matière
|
%
|
|
mt
|
masse totale de substance extraite en deux étapes du sol et des parois du tube d'essai
|
μg
|
|
Vrec
|
volume de surnageant récupéré après l'équilibre d'adsorption
|
cm3
|
|
Pow
|
coefficient de partage octanol/eau
|
|
|
pKa
|
constante de dissociation
|
|
|
Sw
|
solubilité dans l'eau
|
g l-1
|
1.4. PRINCIPE DE LA MÉTHODE
Des volumes connus de la substance, non marquée ou radiologiquement marquée, mélangée à des concentrations connues de CaCl20,01 M, sont ajoutés à des échantillons de sol d'un poids sec connu, préalablement équilibrés dans du CaCl20,01 M. Le mélange est agité pendant le temps requis. Les particules du sol en suspension sont séparées par centrifugation et, le cas échéant, par filtration, puis la phase aqueuse est analysée. La quantité de substance adsorbée sur le sol correspond à la différence entre la quantité de substance initialement présente dans la solution et la quantité restant à la fin de l'expérience (méthode indirecte).
La quantité de substance adsorbée peut également être directement calculée par l'analyse du sol (méthode directe). Cette méthode, qui consiste à extraire progressivement le sol au moyen d'un solvant approprié, est recommandée lorsque l'on ne peut pas déterminer avec précision les écarts de concentration de la substance dans la solution, par exemple dans les cas suivants: adsorption de la substance sur la surface des tubes d'essai, instabilité de la substance au cours de l'expérience, faible adsorption modifiant peu la concentration de la substance dans la solution, forte adsorption entraînant une faible concentration ne pouvant être mesurée avec précision. L'utilisation d'une substance radioactivement marquée permet d'éviter d'extraire le sol. On analyse dans ce cas la phase du sol par combustion et comptage à scintillation liquide. Cette dernière méthode, non spécifique, ne permet cependant pas de distinguer les produits primaires des produits de transformation. Elle ne devrait donc être utilisée que si la substance à tester reste stable pendant toute la durée de l'étude.
1.5. INFORMATION SUR LA SUBSTANCE
Les réactifs chimiques doivent être purs. On recommande l'emploi de substances non marquées de composition connue et présentant de préférence un degré de pureté d'au moins 95 % ou de substances radioactivement marquées de composition connue et radioactivement pures. Il faut appliquer des corrections pour tenir compte de la désintégration lorsque l'on emploie des traceurs à demi-vie courte.
Les paramètres suivants doivent être connus avant de réaliser un essai d'adsorption/désorption:
|
a)
|
solubilité dans l'eau (A.6);
|
|
b)
|
pression de vapeur (A.4) et/ou constante de la loi de Henry;
|
|
c)
|
dégradation non biologique: hydrolyse en fonction du pH (C.7);
|
|
d)
|
coefficient de partage (A.8);
|
|
e)
|
biodégradabilité facile (C.4) ou transformation aérobie et anaérobie dans le sol;
|
|
f)
|
pKa des substances ionisables;
|
|
g)
|
photolyse directe dans l'eau (c'est-à-dire spectre d'absorption UV-Vis dans l'eau, rendement quantique, par exemple) et photodégradation sur le sol.
|
1.6. APPLICABILITÉ DE L'ESSAI
L'essai est applicable aux substances chimiques pour lesquelles on dispose d'une méthode analytique suffisamment précise. La stabilité de la substance durant l'essai est un paramètre important qui peut influencer la fiabilité des résultats, notamment avec la méthode indirecte. Il faut donc vérifier la stabilité par une étude préliminaire. Si l'on observe une transformation durant l'essai, il est recommandé d'analyser le sol et les phases aqueuses lors de l'étude principale.
Des difficultés peuvent survenir avec des substances faiblement solubles dans l'eau (Sw < 10-4 g l-1) ou avec des substances fortement chargées du fait que la concentration de la phase aqueuse ne peut pas être mesurée avec suffisamment de précision avec la méthode analytique. Il faut prendre d'autres mesures dans ces cas. Les moyens d'aborder ces problèmes sont traités dans les points correspondants.
Il faut veiller à éviter les pertes si l'on analyse des substances volatiles.
1.7. MODE OPÉRATOIRE
1.7.1. Appareillage, réactifs chimiques
Le laboratoire doit être équipé de l'équipement standard suivant:
|
a)
|
tubes ou récipients d'essai. Il est important qu'ils soient:
|
—
|
adaptés à la centrifugeuse afin de diminuer les erreurs de manipulation et de transfert,
|
|
—
|
constitués d'un matériau inerte qui diminue l'adsorption de la substance sur les parois:
|
|
|
b)
|
agitateur: agitateur vertical ou équipement équivalent: le sol doit être maintenu en suspension pendant l'agitation;
|
|
c)
|
centrifugeuse: elle doit être de préférence à vitesse de rotation élevée (forces de centrifugation > 3 000 g, par exemple), à température contrôlée et capable d'éliminer de la solution aqueuse les particules d'un diamètre supérieur à 0,2 μm. Les récipients doivent être fermés durant l'agitation et la centrifugation afin d'éviter les pertes par volatilité et les pertes en eau; il est recommandé d'utiliser des couvercles désactivés (couvercles filetés recouverts de téflon, par exemple) afin de diminuer l'adsorption sur leur surface;
|
|
d)
|
équipement facultatif; filtres stériles jetables d'une porosité de 0,2 μm. Le matériau filtrant doit être choisi avec soin afin d'éviter l'adsorption de la substance à sa surface; les filtres organiques sont déconseillés pour les substances peu solubles;
|
|
e)
|
instrumentation analytique permettant de mesurer la concentration de la substance;
|
|
f)
|
four de laboratoire permettant de maintenir une température de 103 à 110 oC.
|
1.7.2. Caractérisation et sélection des sols
Les sols doivent être caractérisés par les trois paramètres principalement responsables de la capacité d'adsorption: le carbone organique, la teneur en argile et la texture, le pH. Comme nous l'avons dit plus haut (voir le point «Objet de la méthode»), d'autres caractéristiques physico-chimiques peuvent influer sur l'adsorption/désorption d'une substance et elles doivent donc être prises en compte.
Les méthodes de caractérisation des sols revêtent une extrême importance car elles peuvent influencer les résultats de manière significative. Il est donc recommandé de mesurer le pH du sol dans une solution de CaCl2, 0,01 M (la solution utilisée lors de l'essai d'adsorption/désorption) selon la méthode ISO correspondante (ISO 10390-1). Il est également conseillé de déterminer certaines autres propriétés du sol selon les méthodes standard (par exemple, manuel ISO de l'analyse des sols, «Handbook of Soil Analysis»). On peut ainsi faire reposer l'analyse des données relatives à la sorption sur des paramètres de sol harmonisés. La bibliographie donne quelques références de méthodes harmonisées d'analyse et de caractérisation des sols (50-52). L'utilisation de sols de référence est recommandée pour calibrer les méthodes d'essai des sols.
Le tableau 1 indique comment choisir les sols pour les essais d'adsorption/désorption. Les sept types de sols choisis se rencontrent dans des zones géographiques tempérées. Pour les substances ionisables, les sols sélectionnés doivent couvrir un large spectre de pH pour permettre d'évaluer l'adsorption de la substance sous sa forme ionisée et non ionisée. Le point 1.9 («Réalisation de l'essai») indique combien de sols différents doivent être utilisés durant les diverses phases de l'essai.
Si d'autres types de sols sont choisis, il faut les caractériser au moyen des mêmes paramètres et ils doivent présenter des propriétés aussi diverses que celles décrites dans le tableau 1, même s'ils ne remplissent pas tout à fait les critères.
Tableau 1
Aide à la sélection des échantillons de sol pour les essais d'adsorption/désorption
|
Type de sol
|
pH (dans le CaCl20,01 M)
|
Teneur en carbone organique (%)
|
Teneur en argile (%)
|
Texture du sol (25)
|
|
1
|
4,5-5,5
|
1,0-2,0
|
65-80
|
argile
|
|
2
|
> 7,5
|
3,5-5,0
|
20-40
|
limon argileux
|
|
3
|
5,5-7,0
|
1,5-3,0
|
15-25
|
limons fins
|
|
4
|
4,0-5,5
|
3,0-4,0
|
15-30
|
limon
|
|
5
|
< 4,0-6,0 (26)
|
< 0,5-1,5 (26)
(27)
|
< 10-15 (26)
|
sable limoneux
|
|
6
|
> 7,0
|
< 0,5-1,0 (26)
(27)
|
40-65
|
limon argileux/argile
|
|
7
|
< 4,5
|
> 10
|
< 10
|
sable/sable glaiseux
|
1.7.3. Collecte et stockage des échantillons de sols
1.7.3.1. Collecte
Aucune technique ou instrument d'échantillonnage particulier n'est recommandée; la technique d'échantillonnage dépend des objectifs de l'étude (53) (54) (55) (56) (57) (58).
Il convient de considérer les aspects suivants:
|
a)
|
il faut disposer d'informations détaillées sur le site d'essai: localisation, couverture végétale, traitement aux pesticides et/ou aux engrais, adjuvants biologiques ou contamination accidentelle. Le site doit être décrit selon les recommandations de la norme ISO sur l'échantillonnage des sols (ISO 10381-6);
|
|
b)
|
le site d'échantillonnage doit être défini par l'UTM (Universal Transversal Mercator-Projection/European Horizontal Datum) ou par des coordonnées géographiques, de manière à pouvoir reconnaître ultérieure ment un sol particulier ou à définir un sol en fonction des divers systèmes de classification utilisés dans les différents pays. Il est également recommandé de prélever dans l'horizon A jusqu'à une profondeur maximale de 20 cm. Si un horizon Oh est présent dans le sol no 7 en particulier, il doit être inclus dans l'échantillon.
|
Les échantillons sont transportés dans des conteneurs et dans des conditions thermiques propres à préserver au mieux les propriétés initiales du sol.
1.7.3.2. Stockage
Il est préférable d'utiliser des sols fraîchement prélevés. Si cela n'est pas possible, le sol peut être stocke à la température ambiante et maintenu au sec. Aucun temps de stockage maximal n'est recommandé, mais au-delà de trois ans, les sols doivent être à nouveau analyses avant l'emploi afin de vérifier leur teneur en carbone-organique, leur pH et leur CEC.
1.7.3.3. Manipulation et préparation des échantillons d'essai
Les sols sont séchés à l'air à la température ambiante (de préférence entre 20 et 25 oC). Ils sont désagrégés en appliquant des forces minimes de manière à modifier aussi peu que possible la texture originale, puis tamises pour ne conserver que les particules ≤ 2 mm. Le tamisage doit respecter les recommandations de la norme ISO sur l'échantillonnage des sols (ISO 10381-6). Une bonne homogénéisation est recommandée car elle améliore la reproductibilité des résultats. La teneur en eau de chaque sol est déterminée à partir de trois aliquotes par un réchauffement à 105 oC jusqu'à stabilisation du poids (environ 12 heures). Pour tous les calculs, la masse du sol se réfère à la masse sèche à l'étuve, c'est-à-dire au poids de sol diminué de la teneur en eau.
1.7.4. Préparation de la substance d'essai devant être appliquée sur le sol
La substance d'essai est dissoute dans une solution de CaCl20,01 M dans de l'eau distillée ou désionisée; la solution de CaCl2 est utilisée comme solvant aqueux pour améliorer la centrifugation et diminuer les échanges de cations. La concentration de la solution mère doit être supérieure de 3 ordres de grandeur à la limite de détection de la méthode d'analyse utilisée. Ce seuil permet de faire des mesures précises avec la méthodologie employée lors de l'essai. Enfin, la concentration de la solution de réserve doit être inférieure à la solubilité dans l'eau de la substance.
Il est conseillé de préparer la solution de réserve immédiatement avant de l'appliquer sur le sol et de la conserver dans un récipient hermétique à l'abri de la lumière à une température de 4 oC. La durée de stockage dépend de la stabilité de la substance et de sa concentration dans la solution.
Un agent solubilisant peut être utilisé pour les substances peu solubles (Sw < 10 4 g l-1) qui se dissolvent difficilement. Il doit être miscible avec l'eau, comme le méthanol ou l'acétonitrile: sa concentration ne doit pas dépasser 1 % du volume total de la solution de réserve et elle doit être inférieure à celle de la substance dans la solution qui entrera en contact avec le sol (de préférence moins de 0,1 %); il ne doit pas être un surfactant ou subir des réactions solvolytiques avec la substance d'essai. Le procès-verbal d'essai doit indiquer qu'un tel agent a été employé et en donner les raisons.
Un autre moyen de traiter les substances peu solubles consiste à introduire la substance d'essai dans le système d'essai à l'aide d'un solvant auxiliaire: la substance est dissoute dans un solvant organique, dont une fraction est ajoutée au système constitué par le sol et par une solution 0,01 M de CaCl2 dans de l'eau distillée ou désionisée. La concentration du solvant organique dans la phase aqueuse devrait être aussi faible que possible et ne pas excéder en principe 0,1 %. L'expérimentateur devra toutefois tenir compte du fait que l'introduction de la substance à l'aide d'une solution organique est susceptible de nuire à la reproductibilité des volumes. La concentration de la substance d'essai et du solvant auxiliaire risquent donc de varier légèrement entre les essais et d'introduire une erreur supplémentaire.
1.8. CONDITIONS PRÉALABLES À LA RÉALISATION DE L'ESSAI D'ADSORPTION/DÉSORPTION
1.8.1. Méthode analytique
Un certain nombre de paramètres peuvent influencer la précision des mesures: la précision de la méthode d'analyse de la solution et des phases adsorbées, la stabilité et la pureté de la substance, le temps pour atteindre l'équilibre de sorption, l'étendue des variations de concentration de la solution, le rapport sol/solution et les modifications de la structure du sol au cours du processus d'équilibrage (35) (59-62). L'appendice 2 donne quelques exemples à ce propos.
La fiabilité de la méthode d'analyse doit être vérifiée selon la gamme de concentration susceptible d'être celle rencontrée durant l'essai. L'expérimentateur est libre d'élaborer une méthode appropriée présentant toutes les qualités requises en matière de précision, d'exactitude, de reproductibilité, de limites de détection et de récupération. L'expérience ci-dessous montre comment effectuer un tel essai.
Un volume approprié de CaCl2, 0,01 M (100 cm3, par exemple), est agité pendant 4 heures avec un certain poids de sol (20 g, par exemple) hautement adsorbant, c'est-à-dire riche en carbone organique et en argile. Le poids et le volume varient selon les besoins de l'analyse mais un rapport sol/solution de 1:5 constitue un bon point de départ. Le mélange est centrifugé et la phase aqueuse filtrée. Une partie de la solution de réserve de la substance est ajoutée à cette dernière afin d'obtenir une concentration nominale comprise dans la gamme de concentration correspondant à celle de l'essai. Ce volume ne doit pas excéder 10 % du volume final de la phase aqueuse afin de modifier aussi peu que possible la nature de la solution de prééquilibrage. La solution est ensuite analysée.
Un témoin constitué du système du sol et de la solution de CaCl2 (sans substance d'essai) doit être ajouté afin de vérifier les artéfacts de la méthode d'analyse et les effets de matrice induits par le sol.
La chromatographie gaz-liquide (CGL), la chromatographie liquide à haute performance (HPLC), la spectrométrie (CG/spectrométrie de masse, HPLC/spectrométrie de masse) et le comptage à scintillation liquide (pour les substances radiologiquement marquées) figurent parmi les méthodes de mesure de la sorption. Un taux de récupération de 90 à 110 % de la valeur nominale est jugé satisfaisant quelle que soit la méthode d'analyse utilisée. La limite de détection de la méthode d'analyse doit être d'au moins de deux ordres de grandeur inférieure à la concentration nominale afin de permettre la détection et l'évaluation une fois que le partage est effectué.
Les caractéristiques et la limite de détection de la méthode utilisée pour effectuer les études d'adsorption déterminent les conditions d'essai et les résultats expérimentaux. La méthode présentée ici suit un protocole expérimental général et propose des solutions de rechange lorsque la méthode analytique ou les équipements de laboratoire imposent des limites.
1.8.2. Choix de rapports sol/solution optimaux
Les rapports sol/solution appropriés à l'étude de la sorption sont choisis en fonction du coefficient de répartition Kd et du degré relatif d'adsorption souhaité. Le changement de la concentration de la substance dans la solution détermine la précision statistique de la mesure, fondée sur la forme de l'équation d'adsorption et sur la limite de la méthode d'analyse, ainsi que l'exactitude avec laquelle elle permettra de détecter la substance contenue dans la solution. Il est donc utile de déterminer plusieurs rapports fixes présentant un pourcentage adsorbé supérieur à 20 % ou, de préférence, à 50 % (62), tout en veillant a ce que la concentration de la substance dans la phase aqueuse reste suffisamment élevée pour pouvoir être mesurée avec précision, notamment lorsque les pourcentages d'adsorption sont importants.
Une solution pratique consiste à choisir les rapports sol/solution en estimant la valeur du coefficient de répartition Kd lors d'études préliminaires ou par des techniques d'estimation bien établies (voir l'appendice 3). On peut ensuite porter sur un graphique le rapport sol/solution en fonction du coefficient de distribution pour différents pourcentages fixes d'adsorption (figure 1). Dans le graphique ci-dessous, on estime que l'équation d'adsorption est linéaire (28). La relation est obtenue en réécrivant l'équation 4 du Kd sous la forme suivante 1:
|
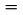
|
(1)
|
ou sous sa forme logarithmique, en supposant que R = msoil/V0 et Aeq %/100 =  :
:
|

|
(2)
|

La figure 1 montre le rapport sol/solution requis en fonction de Kd pour divers niveaux d'adsorption. Par exemple, avec un rapport sol/solution de 1:5 et un Kd de 20, l'adsorption serait de 80 % environ. Pour obtenir un pourcentage d'adsorption de 50 % avec le même Kd, il faudrait un rapport de 1:25. Cette méthode souple permet de choisir les rapports sol/solution en fonction des besoins de l'expérience.
Les zones où la substance est très fortement ou très légèrement adsorbée sont plus difficiles à maîtriser. En cas de faible absorption, un rapport sol/solution de 1:1 est recommandé, mais des ratios plus faibles peuvent être nécessaires avec certains types de sols très organiques afin d'obtenir une boue. Avec la méthode analytique, il faut veiller à mesurer les légères variations de concentration de la substance, faute de quoi la mesure d'adsorption sera inexacte. Avec un coefficient de distribution Kd très élevé, on peut aller jusqu'à un rapport de 1:100 de manière à conserver une quantité importante de substance en solution. Il faut cependant bien mélanger et donner au système le temps de s'équilibrer. On peut aussi prédire la valeur Kj au moyen de techniques d'estimation fondées par exemple sur la valeur Pow (voir l'appendice 3). Cette méthode peut être utile pour les substances chimiques polaires ou faiblement adsorbées ayant un Pow < 20 et pour les substances lipophiles ou fortement sorbantes dotées d'un Pow > 104.
1.9. RÉALISATION DE L'ESSAI
1.9.1. Conditions de l'essai
Les essais sont réalisés à température ambiante et, si possible, à une température constante comprise entre 20 et 25 oC.
La centrifugation doit permettre d'éliminer de la solution les particules supérieures à 0,2 μm. Cette valeur correspond à la plus petite particule considérée comme une particule solide et constitue la limite entre les particules solides et les colloïdes. L'appendice 4 explique comment déterminer les paramètres de la centrifugation.
Si l'équipement de centrifugation ne permet pas d'éliminer à coup sûr les particules supérieures à 0,2 μm, on peut combiner la centrifugation et la filtration avec des filtres de 0,2 μm. Ces filtres doivent être fabriqués en matériau inerte afin d'éviter les pertes de substance. Il faut dans tous les cas apporter la preuve qu'il n'y a aucune perte de substance au cours de la filtration.
1.9.2. Phase 1 — Étude préliminaire
Le but de l'étude préliminaire a déjà été expliqué dans le point «Objet de la méthode». La réalisation de l'essai est exposée ci-après.
1.9.2.1. Sélection de rapports sol/solution optimaux
On utilise deux types de sols et trois rapports sol/solution (six essais). Un sol a une teneur en carbone organique élevée et une faible teneur en argile, l'autre une faible teneur en carbone organique et une teneur élevée en argile. Les rapports suivants sont proposés:
|
—
|
50 g de sol et 50 cm3 de solution aqueuse de la substance d'essai (rapport 1:1),
|
|
—
|
10 g de sol et 50 cm3 de solution aqueuse de la substance d'essai (rapport 1:5),
|
|
—
|
2 g de sol et 50 cm3 de solution aqueuse de la substance d'essai (rapport 1:25).
|
La quantité minimale de sol qui va servir à l'expérience dépend de l'équipement de laboratoire dont on dispose et des performances des méthodes analytiques utilisées. Il est cependant recommandé d'utiliser au moins 1 g, de préférence 2 g, afin d'obtenir des résultats d'essai fiables.
Un échantillon de contrôle contenant uniquement la substance d'essai dans une solution de CaCl20,01 M (sans sol) est soumise exactement aux mêmes opérations que le système d'essai afin de vérifier la stabilité de la substance dans une solution de CaCl2 et son adsorption éventuelle à la surface des récipients d'essai.
Un témoin par sol contenant la même quantité de sol dans un volume total de 50 cm3 de solution de CaCl20,01 M (sans substance) est soumis à la même procédure d'essai. Il permet de détecter les substances étrangères ou les sols contaminés.
Tous les essais, contrôles et témoins sont réalisés au moins en double. Le nombre total d'échantillons devant être préparés pour l'étude est calculé selon la méthode suivie.
L'étude préliminaire et l'étude principale reposent sur les mêmes méthodes, sauf indication contraire.
Les échantillons de sols séchés à l'air sont mélangés à un volume minimal de 45 cm3 de CaCl20,01 M et agités pendant 12 heures la nuit précédant l'expérience. On y ajoute par la suite de la solution de réserve de la substance d'essai afin d'obtenir un volume final de 50 cm3. Ce volume ajouté ne doit pas excéder 10 % du volume final de la phase aqueuse (50 cm3) afin de modifier aussi peu que possible la nature de la solution de prééquilibrage. Par ailleurs, la concentration initiale de la substance d'essai en contact avec le sol (C0) doit être supérieure d'au moins deux ordres de grandeur à la limite de détection de la méthode analytique. Ce seuil permet d'obtenir des mesures fiables, même en cas d'adsorption élevée (> 90 %) et de déterminer ultérieurement les isothermes d'adsorption. La concentration (C0) de la substance initiale ne devrait pas non plus excéder la moitié de sa limite de solubilité.
Voici comment calculer la concentration de la solution de réserve (CM). Si la limite de détection est de 0,01 μg cm-3 et l'adsorption de 90 %, la concentration initiale de la substance en contact avec le sol sera de préférence de 1 μg cm-3 (soit de deux ordres de grandeur plus élevée que celle de la limite de détection). En supposant que l'on ajoute le volume maximal recommandé de la solution de réserve, soit 5 cm3, aux 45 cm3 de la solution d'équilibrage de CaCl20,01 M (c'est-à-dire 10 % de la solution de réserve au volume total de la phase aqueuse), la concentration de la solution de réserve sera de 10 μg cm-3, ce qui est de trois ordres de grandeur supérieur à la limite de détection de la méthode analytique.
Il faut mesurer le pH de la phase aqueuse avant et après le contact avec le sol, compte tenu de son rôle dans le processus d'adsorption, notamment dans le cas des substances ionisables.
Le mélange est agité jusqu'à ce que le point d'équilibre soit atteint. Le temps d'équilibre dans les sols est très variable. Il dépend du produit chimique et du sol. Une période de 24 heures est généralement suffisante (77). Lors de l'étude préliminaire, des échantillons peuvent être prélevés de manière séquentielle pendant une période de mélange de 48 heures (à la 4e, 8e, 24e et 48e heure, par exemple). Les temps d'analyse ne sont pas rigides et doivent être considérés en fonction du programme de travail du laboratoire.
La substance dans la solution aqueuse peut être mesurée par la méthode parallèle ou par la méthode séquentielle. La méthode parallèle est plus difficile à réaliser sur le plan expérimental mais le traitement mathématique des résultats est plus simple (voir l'appendice 5). Le choix de la méthode est laissée à l'appréciation de l'expérimentateur, qui décide en fonction des équipements de laboratoire et des ressources disponibles.
|
a)
|
Méthode parallèle: on prépare des échantillons avec un rapport sol/solution identique, leur nombre étant égal aux intervalles de temps auxquels on souhaite étudier la cinétique d'adsorption. Après centrifugation et, le cas échéant, filtration, la phase aqueuse du premier tube est récupérée aussi complètement que possible et mesurée au bout de 4 heures, par exemple, celle du deuxième tube au bout de 8 heures, celle du troisième au bout de 24 heures, etc.
|
|
b)
|
Méthode séquentielle: on prépare uniquement un double échantillon pour chaque rapport sol/solution. Le mélange est centrifugé à des intervalles de temps définis afin de séparer les phases. Une petite aliquote de la phase aqueuse est immédiatement analysée afin de rechercher la substance, puis l'expérience se poursuit avec le mélange original. S'il y a filtration après centrifugation, le laboratoire doit disposer d'équipements permettant de filtrer de petites aliquotes en phase aqueuse. Le volume total des aliquotes ne devrait pas dépasser 1 % du volume total de la solution afin de ne pas modifier de manière significative le rapport sol/ solution et de diminuer la masse de soluté susceptible d'être adsorbée durant l'essai.
|
Le pourcentage d'adsorption  est calculé à chaque instant (tj) sur la base de la concentration initiale nominale et de la concentration mesurée au temps d'échantillonnage (ti), corrigé de la valeur du témoin. Des graphes
est calculé à chaque instant (tj) sur la base de la concentration initiale nominale et de la concentration mesurée au temps d'échantillonnage (ti), corrigé de la valeur du témoin. Des graphes 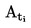 représentant en fonction du temps (figure 1, appendice 5) sont tracés afin de savoir à quel moment le plateau d'équilibre est atteint (29). On calcule également la valeur Kd à l'équilibre. On choisit à partir de cette valeur le rapport sol/solution approprié de la figure 1, de manière que le pourcentage d'adsorption dépasse 20 % et, de préférence, 50 % (61). Les équations et les principes relatifs au tracé sont exposés au point «Présentation des données» ainsi qu'à l'appendice 5.
représentant en fonction du temps (figure 1, appendice 5) sont tracés afin de savoir à quel moment le plateau d'équilibre est atteint (29). On calcule également la valeur Kd à l'équilibre. On choisit à partir de cette valeur le rapport sol/solution approprié de la figure 1, de manière que le pourcentage d'adsorption dépasse 20 % et, de préférence, 50 % (61). Les équations et les principes relatifs au tracé sont exposés au point «Présentation des données» ainsi qu'à l'appendice 5.
1.9.2.2. Détermination du temps d'adsorption et de la quantité de substance adsorbée à l'équilibre
Comme nous l'avons déjà dit, le graphe représentant  ou
ou  en fonction du temps permet d'estimer l'adsorption et la quantité de substance adsorbée à l'équilibre (voir les figures 1 et 2 de l'appendice 5). Le temps d'équilibre correspond au temps que le système met pour atteindre un plateau.
en fonction du temps permet d'estimer l'adsorption et la quantité de substance adsorbée à l'équilibre (voir les figures 1 et 2 de l'appendice 5). Le temps d'équilibre correspond au temps que le système met pour atteindre un plateau.
L'absence de plateau ou l'accroissement progressif de la courbe peut être dû à des facteurs complexes tels que b. biodégradation ou une diffusion lente. La biodégradation peut être mise en évidence en répétant l'expérience avec un échantillon de sol stérile. Si aucun plateau n'est atteint même dans ce cas, l'expérimentateur doit chercher d'autres causes inhérentes à l'étude. Il peut par exemple modifier les conditions de l'essai (température, temps d'agitation, rapport sol/solution). Lui seul décide de poursuivre la procédure d'essai, même s'il court le risque de ne pas atteindre un équilibre.
1.9.2.3. Adsorption sur les parois du récipient et stabilité de la substance
L'analyse des échantillons de contrôle peut fournir quelques informations sur l'adsorption de la substance sur les parois des récipients d'essai ainsi que sur sa stabilité. Une déplétion supérieure à l'écart type de la méthode analytique peut être due à une dégradation abiotique et/ou à l'adsorption sur les parois du récipient. On peut individualiser l'un ou l'autre de ces phénomènes en lavant soigneusement les parois du récipient avec un volume connu de solvant, puis en recherchant la substance dans la solution de lavage. L'absence d'adsorption sur les parois du récipient prouve l'instabilité abiotique de la substance. En cas d'adsorption, en revanche, il convient de modifier le matériau du récipient d'essai. Ces données sur l'adsorption sur les parois des récipients d'essai ne peuvent cependant pas être directement extrapolées à l'essai sol/solution, dans la mesure où la présence de sol modifie l'adsorption.
On peut obtenir des informations supplémentaires sur la stabilité de la substance d'essai en déterminant le bilan matière dans le temps. La substance est recherchée dans la phase aqueuse, dans les extraits de sol et sur les parois des récipients d'essai. La différence entre la masse de la substance ajoutée et la somme des masses de substance présentes dans la phase aqueuse, les extraits de sol et sur les parois des récipients correspond à la masse dégradée, volatilisée et/ou non extraite. Pour effectuer un bilan matière, l'équilibre d'adsorption doit être atteint durant l'essai.
Le bilan matière est effectué sur les deux sols et avec un rapport sol/solution par sol générant une déplétion à l'équilibre supérieure à 20 % et, de préférence, à 50 %. Une fois que l'essai consistant à trouver le rapport sol/solution est terminé (analyse du dernier échantillon de phase aqueuse au bout de 48 heures), les phases sont séparées par centrifugation et, le cas échéant, par filtration. Le maximum de phase aqueuse est récupérée puis un solvant d'extraction approprié (c'est-à-dire doté d'un coefficient d'extraction d'au moins 95 %) est ajouté au sol afin d'extraire la substance. Deux extractions successives au moins sont recommandées. On détermine ensuite la quantité de substance présente dans les extraits de sol et dans le récipient afin de calculer le bilan matière (équation 10 du chapitre «Présentation des données»). S'il est inférieur à 90 %, la substance est jugée instable durant la durée de l'essai. Les études peuvent être cependant poursuivies en tenant compte de cette instabilité. Il est conseillé dans ce cas d'analyser les deux phases lors de l'étude principale.
1.9.2.4 Phase 2 — Cinétique d'adsorption avec une concentration unique de substance
Cinq sols choisis dans le tableau 1 sont utilisés à cet effet. On a intérêt à y inclure tout ou partie des sols utilisés dans l'étude préliminaire. Dans ce cas, ils ne sont pas soumis aux essais de la phase 2.
Le temps d'équilibrage, le rapport sol/solution, le poids de l'échantillon de sol, le volume de la phase aqueuse en contact avec le sol et la concentration de la substance d'essai dans la solution sont choisis en fonction des résultats de l'étude préliminaire. L'analyse doit être effectuée de préférence après un temps de contact de 2, 4, 6, 8 (éventuellement 10) et 24 heures. Le temps d'agitation peut être étendu à 48 heures maximum lorsque les résultats sur le rapport sol/solution font ressortir un temps d'équilibrage plus long. Les temps d'analyse doivent être considérés avec une certaine souplesse.
Chaque expérience (un sol et une solution) est effectuée au moins en double afin d'évaluer la variance des résultats. Un témoin est préparé pour chacune d'elle avec du sol et de la solution de CaCl20,01 M, sans substance d'essai, d'un poids et d'un volume identiques aux échantillons d'essai. Un échantillon de contrôle préparé uniquement à partir de la substance d'essai contenue dans une solution de CaCl20,01 M (sans sol) est soumis à la même procédure d'essai afin de se prémunir contre l'imprévu.
Le pourcentage d'adsorption est calculé à chaque instant 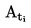 et/ou intervalle de temps
et/ou intervalle de temps 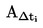 (selon les besoins). Il est porté sur un graphe en fonction du temps. Le coefficient de distribution Kd à l'équilibre ainsi que le coefficient d'adsorption normalisé basé sur la teneur en carbone organique Koc (pour les substances chimiques non polaires) sont également calculés.
(selon les besoins). Il est porté sur un graphe en fonction du temps. Le coefficient de distribution Kd à l'équilibre ainsi que le coefficient d'adsorption normalisé basé sur la teneur en carbone organique Koc (pour les substances chimiques non polaires) sont également calculés.
Résultats de l'essai de cinétique d'adsorption
La valeur linéaire Kd, qui exprime la mobilité inhérente des substances chimiques dans le sol, est généralement suffisamment précise pour décrire le comportement de sorption du sol (35)(78). Les substances chimiques dont Kd est ≤ 1 cm3 g-1 sont en général considérées comme étant mobiles. Mac Gall et coll. ont élaboré un système de classification de la mobilité fondé sur les valeurs Koc (16). Il existe également des systèmes de classification de la lixiviation fondés sur la relation entre Koc et DT-50 (30) (32) (79).
Selon les études d'analyse d'erreurs (61), les valeurs Kd inférieures à 0,3 cm3 g-1 ne peuvent pas être estimées avec précision à partir d'une diminution de la concentration dans la phase aqueuse, même si l'on applique le rapport sol/solution le plus favorable (sur le plan de la précision), c'est-à-dire 1:1. Dans ce cas, l'analyse des deux phases (sol et solution) est recommandée.
En ce qui concerne les remarques précédentes, il est conseillé de poursuivre l'étude du comportement de sorption d'un produit chimique dans le sol et de sa mobilité potentielle en déterminant les isothermes d'adsorption de Freundlich des systèmes dont on peut déterminer précisément Kd grâce au protocole expérimental suivi dans la présente méthode. Il suffit pour cela que la valeur obtenue en multipliant Kd avec le rapport sol/solution soit > 0,3 — lorsque les mesures reposent sur la baisse de concentration de la phase aqueuse (méthode indirecte) — ou > 0,1 lorsque les deux phases sont analysées (méthode directe) (61).
1.9.2.5 Phase 3 — Isothermes d'adsorption, cinétique de désorption et isothermes de désorption
1.9.2.5.1 Isothermes d'adsorption
On utilise cinq concentrations de substances d'essai couvrant de préférence deux ordres de grandeur. La solubilité dans l'eau et les concentrations à l'équilibre de la phase aqueuse qui en résultent sont prises en compte lors du choix de ces concentrations. Il convient de garder le même rapport sol/solution par sol tout au long de l'étude. L'essai d'adsorption est effectué selon la description ci-dessus, à la différence près que la phase aqueuse est analysée une seule fois, au moment où le point d'équilibre déterminé au cours de la phase 2 est atteint. Les concentrations à l'équilibre dans la solution sont déterminées et la quantité adsorbée est calculée à partir de la déplétion de la substance dans la solution ou avec la méthode directe. La masse adsorbée par unité de masse de sol est portée sur un graphe en fonction de la concentration à l'équilibre de la substance (voir le chapitre «Présentation des données»).
Résultats de l'essai relatif aux isothermes d'adsorption
De tous les modèles mathématiques d'adsorption proposés jusqu'à présent, l'isotherme de Freundlich est le plus fréquemment utilisé pour décrire les processus d'adsorption. On peut trouver de plus amples informations sur l'interprétation et l'importance des modèles d'adsorption dans la bibliographie (41) (45) (80) (81) (82).
Remarque: il est bon de mentionner qu'une comparaison des valeurs KF (coefficient d'adsorption de Freundlich) de différentes substances n'est possible que si ces valeurs sont exprimées en unités identiques (83).
1.9.2.5.2 Cinétique de désorption
Cet essai vise à évaluer le caractère réversible ou irréversible de l'adsorption d'une substance sur un sol. C'est une information importante dans la mesure où le processus de désorption joue un rôle non négligeable dans le comportement d'une substance chimique dans un sol. Ces données sur la désorption peuvent également servir à élaborer des modèles informatisés de simulation du lessivage et de l'écoulement des substances dissoutes. Si l'on souhaite effectuer une étude de désorption, il est conseillé d'effectuer l'étude ci-après pour tous les systèmes dont on aura pu déterminer Kd au cours de l'essai sur la cinétique d'adsorption précédent.
Comme pour l'étude sur la cinétique d'adsorption, l'essai sur la cinétique de désorption peut se faire selon la méthode parallèle ou la méthode séquentielle. Le choix de la méthode est laissé à l'appréciation de l'expérimentateur, qui devra considérer les disponibilités en équipements de laboratoire et en ressources.
|
a)
|
méthode parallèle: on prépare, pour chaque sol inclus dans l'étude de désorption, des échantillons ayant un rapport sol/solution identique, leur nombre étant égal aux intervalles de temps auxquels on souhaite étudier la cinétique de désorption. Il est préférable d'utiliser les mêmes intervalles de temps que pour l'étude sur la cinétique d'adsorption. Le temps total peut cependant être étendu de manière que le système parvienne à l'équilibre de désorption. On prépare un témoin pour chaque expérience (un sol, une solution) à partir de sol et d'une solution de CaCl20,01 M (sans substance d'essai), d'un poids et d'un volume identiques à ceux de l'expérience. La substance d'essai dans une solution de CaCl20,01 M (sans sol) est soumise à la même procédure d'essai en tant qu'échantillon de contrôle. Tous les mélanges sol/solution sont agités jusqu'à l'équilibre d'adsorption (tel qu'il a été déterminé dans la phase 2). Les phases sont ensuite séparées par centrifugation et les phases aqueuses sont extraites le plus complètement possible. Le volume de solution extrait est remplacé par un volume égal de CaCl20,01 M ne contenant pas de substance d'essai et ces mélanges sont à nouveau agités. La phase aqueuse du premier tube est récupérée le plus complètement possible puis mesurée après 2 h, par exemple, celle du deuxième tube après 4 h, celle du troisième après 6 h, etc., jusqu'à ce que l'équilibre de désorption soit atteint.
|
|
b)
|
méthode séquentielle: après l'essai de cinétique d'adsorption, le mélange est centrifugé et la phase aqueuse est extraite le plus complètement possible. Le volume de solution extrait est remplacé par un volume égal de CaCl20,01 M sans substance d'essai. Ce mélange est agité jusqu'à ce que l'équilibre de désorption soit atteint. Pendant ce temps, le mélange est centrifugé à des intervalles de temps définis afin de séparer les phases. Une petite aliquote de la phase aqueuse est immédiatement analysée afin de rechercher la substance d'essai, puis on poursuit l'expérience avec le mélange original. Le volume de chaque aliquote ne devrait pas dépasser 1 % du volume total. La même quantité de solution fraîche de CaCl20,01 M est ajoutée au mélange afin de maintenir le rapport sol/solution, puis celui-ci est agité jusqu'au prochain intervalle de temps.
|
Le pourcentage de désorption est calculé à chaque temps ( ) et/ou intervalle de temps (
) et/ou intervalle de temps (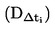 ) (selon les besoins de l'étude), puis il est porté sur un graphique en fonction du temps. Le coefficient de désorption Kdes à l'équilibre est également calculé. Toutes les équations applicables sont données dans le chapitre «Présentation des données» de l'annexe 5.
) (selon les besoins de l'étude), puis il est porté sur un graphique en fonction du temps. Le coefficient de désorption Kdes à l'équilibre est également calculé. Toutes les équations applicables sont données dans le chapitre «Présentation des données» de l'annexe 5.
Résultats de l'essai de cinétique de désorption
L'inscription sur un même graphe du pourcentage de désorption  et d'adsorption
et d'adsorption  en fonction du temps permet d'estimer la réversibilité du processus d'adsorption. L'adsorption est jugée réversible si l'équilibre de désorption est atteint avant le double de temps nécessaire à l'équilibre d'adsorption et si la désorption totale est supérieure à 75 % de la quantité adsorbée.
en fonction du temps permet d'estimer la réversibilité du processus d'adsorption. L'adsorption est jugée réversible si l'équilibre de désorption est atteint avant le double de temps nécessaire à l'équilibre d'adsorption et si la désorption totale est supérieure à 75 % de la quantité adsorbée.
1.9.2.5.3 Isothermes de désorption
Les isothermes de désorption de Freundlich sont déterminés sur les sols utilisés lors de l'expérience relative aux isothermes d'adsorption. L'essai de désorption est réalisé selon les modalités décrites dans le chapitre «Cinétique de désorption», à la différence près que la phase aqueuse n'est analysée qu'une fois, à l'équilibre de désorption. On calcule ensuite la quantité de substance désorbée. La quantité de substance restant adsorbée sur le sol à l'équilibre de désorption est portée sur un graphe en fonction de la concentration d'équilibre de la substance d'essai en solution (voir le chapitre «Présentation des données» et l'annexe 5).
2. PRÉSENTATION DES DONNÉES
Les données analytiques sont présentées sous forme de tableaux (voir l'annexe 6) dans lesquels figurent les mesures et les moyennes calculées. Les isothermes d'adsorption sont représentés sous forme de graphiques. Les calculs sont effectués selon les modalités présentées ci-après.
Pour les besoins de l'essai, on estime que le poids de 1 cm3 de solution aqueuse est de 1 g. Le rapport sol/solution peut être exprimé en M/M ou M/vol.
2.1 ADSORPTION
L'adsorption ( ) est définie comme le pourcentage de substance adsorbée sur le sol en fonction de la quantité présente au début de l'essai, dans les conditions de l'essai. Si la substance est stable et qu'elle n'adsorbe pas de manière significative sur la paroi du récipient,
) est définie comme le pourcentage de substance adsorbée sur le sol en fonction de la quantité présente au début de l'essai, dans les conditions de l'essai. Si la substance est stable et qu'elle n'adsorbe pas de manière significative sur la paroi du récipient,  est calculé à chaque instant ti selon l'équation suivante:
est calculé à chaque instant ti selon l'équation suivante:
|
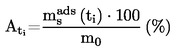
|
(3)
|
où:
|

|
=
|
est le pourcentage d'adsorption à l'instant ti (%);
|
|
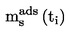
|
=
|
est la masse de substance d'essai adsorbée sur le sol durant le temps ti (μg);
|
|
m0
|
=
|
est la masse de substance dans le tube au début de l'essai (μg).
|
L'annexe 5 donne des informations détaillées sur le mode de calcul du pourcentage d'adsorption  avec les méthodes parallèles et séquentielles.
avec les méthodes parallèles et séquentielles.
Le coefficient de répartition Kd est le rapport entre le contenu de la substance dans le sol et sa concentration massique dans la solution aqueuse, dans les conditions d'essai, lorsque l'équilibre d'adsorption est atteint.
Soit:
|

(cm3 g-1) |
(4)
|
où:
|

|
=
|
est la concentration de la substance adsorbée sur le sol à l'équilibre d'adsorption (μg g-1);
|
|
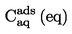
|
=
|
est la concentration massique de la substance dans la phase aqueuse à l'équilibre d'adsorption (μg cm-3). Elle est déterminée par analyse en tenant compte des valeurs données par les témoins.
|
|

|
=
|
est la masse de substance adsorbée sur le sol à l'équilibre d'adsorption (μg);
|
|

|
=
|
est la masse de substance dans la solution à l'équilibre d'adsorption (μg);
|
|
msoil
|
=
|
est la quantité de phase de sol exprimée en masse sèche de sol (g);
|
|
V0
|
=
|
est le volume initial de la phase aqueuse en contact avec le sol (cm3).
|
La relation entre Aeq et Kd est donnée par l'équation suivante:
|
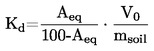
(cm3 g-1) |
(5)
|
où:
|
Aeq
|
=
|
est le pourcentage d'adsorption à l'équilibre d'adsorption (%).
|
Le coefficient normalisé basé sur la teneur en carbone organique Koc lie le coefficient de répartition Kd à la teneur en carbone organique de l'échantillon de sol:
soit:
|

(cm3 g-1) |
(6)
|
où:
|
%OC
|
=
|
est le pourcentage de carbone organique dans l'échantillon de sol (g g-1).
|
Le coefficient Koc représente une valeur unique qui caractérise principalement le partage de substances chimiques non polaires entre le carbone organique dans le sol ou sédiment et eau. L'adsorption de ces substances est liée à la teneur organique du solide adsorbant (7); les valeurs Koc dépendent donc des caractéristiques spécifiques des fractions humiques dont la capacité de sorption varie considérablement selon l'origine, la genèse, etc.
2.1.1 Isothermes d'adsorption
L'équation des isothermes d'adsorption de Freundlich lie la quantité de substance adsorbée à la concentration de substance en solution à l'équilibre (équation 8).
Les données sont traitées comme dans le chapitre «Adsorption». On calcule pour chaque tube d'essai la quantité de la substance adsorbée sur le sol après l'essai d'adsorption (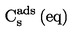 , noté ailleurs x/m). On suppose que l'équilibre a été atteint et que
, noté ailleurs x/m). On suppose que l'équilibre a été atteint et que  représente la valeur d'équilibre:
représente la valeur d'équilibre:
|

(μg g-1) |
(7)
|
L'adsorption de Freundlich est donnée par l'équation suivante (8):
|
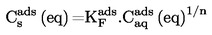
(μg g-1) |
(8)
|
ou, sous sa forme linéaire, par:
|

|
(9)
|
où:
|
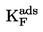
|
=
|
est le coefficient d'adsorption de Freundlich; il se mesure en cm3 g-1 uniquement si 1/n = 1; dans tous les autres cas, la pente l/n s'écrit 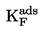 (μg1-1
/n(cm3)1/ng-1); (μg1-1
/n(cm3)1/ng-1);
|
|
n
|
=
|
est la constante de régression; l/n est généralement compris entre 0,7 et 1,0, ce qui indique que la donnée de sorption est fréquemment légèrement non linéaire.
|
Les équations (8) et (9) sont portées sur un graphique et les valeurs 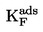 et l/n sont calculées au moyen d'une analyse de régression utilisant l'équation 9. Le coefficient de corrélation r2 de l'équation logarithmique est également calculé. La figure 2 donne des exemples de graphes.
et l/n sont calculées au moyen d'une analyse de régression utilisant l'équation 9. Le coefficient de corrélation r2 de l'équation logarithmique est également calculé. La figure 2 donne des exemples de graphes.

2.1.2 Bilan matière
Le bilan matière (MB) correspond au pourcentage de substance récupéré par analyse après un essai d'adsorption par rapport à la quantité nominale de substance présente au début de l'essai.
Le traitement des données est différent si le solvant est complètement miscible avec l'eau. On peut dans ce cas appliquer le traitement des données visé au chapitre «Désorption» pour déterminer la quantité de substance extraite par solvant. Si celui-ci est moins miscible avec l'eau, il convient de déterminer la quantité récupérée.
Le bilan matière de l'adsorption est calculé de la manière suivante: on suppose que le terme (mE) correspond à la somme des masses de substance extraites du sol et de la surface du récipient d'essai au moyen d'un solvant organique.
soit:
|

|
(10)
|
où:
|
MB
|
=
|
est le bilan matière (%);
|
|
mE
|
=
|
est la masse totale de substance extraite en deux étapes du sol et des parois du récipient (μg);
|
|
Co
|
=
|
est la concentration massique initiale de la solution en contact avec le sol (μg cm-3);
|
|
Vrec
|
=
|
est le volume de surnageant récupéré à l'équilibre d'adsorption (cm-3).
|
2.2 DÉSORPTION
La désorption (D) est définie comme le pourcentage de substance désorbée, rapporté à la quantité de substance préalablement adsorbée, dans les conditions d'essai:
soit:
|
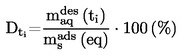
|
(11)
|
où:
|

|
=
|
est le pourcentage de désorption à l'instant ti (%);
|
|
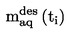
|
=
|
est la masse de substance désorbée à partir du sol à l'instant ti, (μg);
|
|
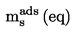
|
=
|
est la masse de substance adsorbée sur le sol à l'équilibre d'adsorption, (μg).
|
L'annexe 5 donne des informations détaillées sur la manière de calculer le pourcentage de désorption  par la méthode parallèle et séquentielle.
par la méthode parallèle et séquentielle.
Le coefficient de désorption apparente (Kdes) correspond, dans les conditions d'essai, au rapport entre le contenu de la substance restant dans la phase du sol et la concentration massique de la substance désorbée dans la solution aqueuse, lorsque l'équilibre de désorption est atteint:
soit:
|
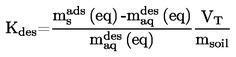
(cm3 g-1) |
(12)
|
où:
|
Kdes
|
=
|
est le coefficient de désorption (cm3 g-1);
|
|

|
=
|
est la masse totale de substance désorbée à partir du sol à l'équilibre de désorption (μg);
|
|
VT
|
=
|
est le volume total de la phase aqueuse en contact avec le sol durant l'essai de cinétique de désorption (cm3).
|
Le chapitre «Désorption» de l'annexe 5 explique comment calculer 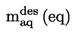 .
.
Remarque
Si l'essai d'adsorption a été réalisé avec la méthode parallèle, le volume VT de l'équation (12) est estimé égal à V0.
2.2.1 Isothermes de désorption
L'équation des isothermes de désorption de Freundlich relie la quantité de la substance restant adsorbée sur le sol à la concentration de substance en solution à l'équilibre de désorption (équation 16).
La quantité de substance restant adsorbée sur le sol à l'équilibre de désorption est calculé pour chaque tube de la manière suivante:
|

(µg g-1) |
(13)
|
 est défini comme:
est défini comme:
|

(μg) |
(14)
|
|

|
=
|
est la quantité de la substance restant adsorbée sur le sol à l'équilibre de désorption (μg g-1);
|
|

|
=
|
est la masse de substance déterminée par analyse dans la phase aqueuse à l'équilibre de désorption (μg);
|
|

|
=
|
est la masse de substance restant après l'équilibre d'adsorption en raison d'un remplacement volumique incomplet μg);
|
|

|
=
|
est la masse de substance en solution à l'équilibre d'adsorption (μg);
|
|

|
(15)
|
|
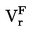
|
=
|
est le volume de solution prélevé du tube afin de mesurer la substance, à l'équilibre de désorption (cm3)
|
|
VR
|
=
|
est le volume de surnageant extrait du tube après obtention de l'équilibre d'adsorption et remplacé par le même volume de CaCl20,01 M (cm3);
|
L'équation de la désorption de Freundlich s'écrit (16):
|
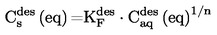
(μg g-1) |
(16)
|
ou, sous une forme linéaire:
|

|
(17)
|
où:
|
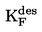
|
=
|
est le coefficient de désorption de Freundlich;
|
|
n
|
=
|
est la constante de régression;
|
|
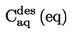
|
=
|
est la concentration massique de substance dans la phase aqueuse à l'équilibre de désorption (μg cm-3).
|
Les équations (16) et (17) peuvent être portées sur un graphique et les valeurs  et 1/n sont calculées au moyen d'une analyse de régression en utilisant l'équation 17.
et 1/n sont calculées au moyen d'une analyse de régression en utilisant l'équation 17.
Remarque:
Si l'exposant d'adsorption ou de désorption de Freundlich 1/n est égal à 1, les constantes d'adsorption ou de désorption de Freundlich (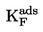 et
et  ) sont identiques aux constantes d'adsorption ou de désorption à l'équilibre (Kd et Kdes) et les graphiques de Cs en fonction de Caq seront linéaires. Si les exposants ne sont pas égaux à 1, les graphiques de Cs en fonction de Caq seront non linéaires et les constantes d'adsorption et de désorption varieront selon les isothermes.
) sont identiques aux constantes d'adsorption ou de désorption à l'équilibre (Kd et Kdes) et les graphiques de Cs en fonction de Caq seront linéaires. Si les exposants ne sont pas égaux à 1, les graphiques de Cs en fonction de Caq seront non linéaires et les constantes d'adsorption et de désorption varieront selon les isothermes.
2.2.2 Procès-verbal d'essai
Le procès-verbal doit comprendre les informations suivantes:
|
—
|
Identification complète des échantillons de sol utilisés, à savoir:
|
|
—
|
références géographiques du site (latitude, longitude);
|
|
—
|
date de l'échantillonnage;
|
|
—
|
origine (sol agricole, forêt, etc.);
|
|
—
|
profondeur de l'échantillonnage;
|
|
—
|
contenu en sable/limon/argile;
|
|
—
|
valeurs du pH (dans CaCl20,01M);
|
|
—
|
teneur en carbone organique;
|
|
—
|
teneur en matière organique;
|
|
—
|
capacité d'échange cationique (mmol/kg);
|
|
—
|
toutes les informations sur la collecte et le stockage des échantillons de sol;
|
|
—
|
le cas échéant, toutes les informations utiles à l'interprétation de l'adsorption et de la désorption de la substance testée;
|
|
—
|
la référence aux méthodes utilisées pour déterminer chaque paramètre;
|
|
—
|
le cas échéant, des informations sur la substance à tester;
|
|
—
|
la température des essais;
|
|
—
|
les conditions de centrifugation;
|
|
—
|
le procédé utilisé pour analyser la substance;
|
|
—
|
les raisons motivant l'emploi d'un agent solubilisant pour préparer la solution de substance de réserve;
|
|
—
|
les raisons expliquant les corrections de calcul, le cas échéant;
|
|
—
|
les données relatives au formulaire (annexe 6) et à la présentation graphique;
|
|
—
|
toutes les informations et observations utiles pour interpréter les résultats des essais.
|
3. BIBLIOGRAPHIE
|
1.
|
Kukowski H. and Brümmer G., (1987). Investigations on the Adsorption and Desorption of Selected Chemicals in Soils. UBA Report 106 02 045, Part II.
|
|
2.
|
Fränzle O., Kuhnt G. and Vetter L., (1987). Selection of Representative Soils in the EC-Territory. UBA Report 106 02 045, Part I.
|
|
3.
|
Kuhnt G. and Muntau H. (Eds.) EURO-Soils: Identification, Collection, Treatment, Characterisation. Special Publication no. 1.94.60, Joint Research Centre. European Commission, ISPRA, December 1994.
|
|
4.
|
OECD Test Guidelines Programme, Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18-20 January 1995 (June 1995).
|
|
5.
|
US-Environment Protection Agency: Pesticide Assessment Guidelines, Subdivision N, Chemistry: Environmental Fate, Series 163-1, Leaching and Adsorption/Desorption Studies, Addendum 6 on Data Reporting, 540/09-88-096, Date: 1/1988.
|
|
6.
|
US-Environment Protection Agency: Prevention, Pesticides and Toxic Substances, OPPTS Harmonized Test Guidelines, Series 835-Fate, Transport and Transformation Test Guidelines, 0PPTS No: 835.1220 Sediment and Soil Adsorption/Desorption Isotherm. EPA No: 712-C-96-048, April 1996.
|
|
7.
|
ASTM Standards, E 1195-85, Standard Test Method for Determining a Sorption Constant (Koc) for an Organic Chemical in Soil and Sediments.
|
|
8.
|
Agriculture Canada: Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada, 15 July 1987.
|
|
9.
|
Netherlands Commission Registration Pesticides (1995): Application for registration of a pesticide. Section G. Behaviour of the product and its metabolites in soil, water and air.
|
|
10.
|
Danish National Agency of Environmental Protection (October 1988): Criteria for registration of pesticides as especially dangerous to health or especially harmful to the environment.
|
|
11.
|
BBA (1990), Guidelines for the Official Testing of Plant Protection Products, Biological Research Centre for Agriculture and Forestry, Braunschweig, Germany.
|
|
12.
|
Calvet R., (1989), «Evaluation of adsorption coefficients and the prediction of the mobilities of pesticides in soils», in Methodological Aspects of the Study of Pesticide Behaviour in Soil (ed. P. Jamet), INRA, Paris, (Review).
|
|
13.
|
Calvet R., (1980) «Adsorption-Desorption Phenomena» in Interactions between herbicides and the soil. (R.J. Hance ed.), Academic Press, London, pp. 83-122.
|
|
14.
|
Hasset J.J., and Banwart W.L., (1989), «The sorption of nonpolar organics by soils and sediments» in Reactions and Movement of Organic Chemicals in Soils. Soil Science Society of America (S.S.S.A), Special Publication no. 22, pp 31-44.
|
|
15.
|
van Genuchten M. Th., Davidson J.M., and Wierenga P.J., (1974), «An evaluation of kinetic and equilibrium equations for the prediction of pesticide movement through porous media». Soil Sci. Soc. Am. Proc, Vol. 38(1), 29-35.
|
|
16.
|
McCall P.J., Laskowski D.A., Swann R.L., and Dishburger H.J., (1981), «Measurement of sorption coefficients of organic chemicals and their use, in environmental fate analysis», in Test Protocols for Environmental Fate and Movement of Toxicants. Proceedings of AOAC Symposium, AOAC, Washington DC.
|
|
17.
|
Lambert S.M., Porter P.E., and Schieferrstein R.H., (1965), «Movement and sorption of chemicals applied to the soil». Weeds, 13, 185-190.
|
|
18.
|
Rhodes R.C., Belasco I.J., and Pease H.L., (1970) «Determination of mobility and adsorption of agrochemicals in soils». J.Agric.Food Chem., 18, 524-528.
|
|
19.
|
Russell M.H., (1995), «Recommended approaches to assess pesticide mobility in soil», in Environmental Behavior of Agrochemicals (ed. T.R. Roberts and P.C. Kearney). John Wiley & Sons Ltd.
|
|
20.
|
Esser H.O., Hemingway R.J., Klein W., Sharp D.B., Vonk J.W. and Holland P.T., (1988), «Recommended approach to the evaluation of the environmental behavior of pesticides», IUPAC Reports on Pesticides (24). Pure Appl. Chem., 60, 901-932.
|
|
21.
|
Guth J.A., Burkhard N., and D.O. Eberle, (1976), «Experimental models for studying the persistence of pesticides in soils». Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, pp 137-157, BCPC, Surrey, UK.
|
|
22.
|
Furminge C.G.L., and Osgerby J.M., (1967), «Persistence of herbicides in soil». J. Sci. Fd Agric, 18, 269-273.
|
|
23.
|
Burkhard N., and Guth J.A., (1981), «Chemical hydrolysis of 2-Chloro-4,6-bis(alkylamino)-1,3,5-triazine herbicides and their breakdown in soil under the influence of adsorption». Pestic. Sci. 12, 45-52.
|
|
24.
|
Guth J.A., Gerber H.R., and Schlaepfer T., (1977). «Effect of adsorption, movement and persistence on the biological availability of soil-applied pesticides». Proc. Br. Crop Prot. Conf., 3, 961-971.
|
|
25.
|
Osgerby J.M., (1973), «Process affecting herbicide action in soil». Pestic. Sci., 4, 247-258.
|
|
26.
|
Guth J.A., (1972), «Adsorptions- und Einwascheverhalten von Pflanzenschutzmitteln in Böden». Schr. Reihe Ver. Wass. -Boden-Lufthyg. Berlin-Dahlem, Heft 37, 143-154.
|
|
27.
|
Hamaker J.W., (1975), «The interpretation of soil leaching experiments», in Environmental Dynamics of Pesticides (eds R. Haque and V.H. freed), pp. 135-172, Plenum Press, NY.
|
|
28.
|
Helling C.S., (1971), «Pesticide mobility in soils». Soil Sci. Soc. Amer. Proc, 35, 732-210.
|
|
29.
|
Hamaker J.W., (1972), «Diffusion and volatilization» in Organic chemicals in the soil environment (C.A.I. Goring and J.W. Hamaker eds), Vol. I, 49-143.
|
|
30.
|
Burkhard N. and Guth J.A., (1981), «Rate of volatilisation of pesticides from soil surfaces; Comparison of calculated results with those determined in a laboratory model system». Pestic. Sci. 12, 37-44.
|
|
31.
|
Cohen S.Z., Creeger S.M., Carsel R.F., and Enfield C.G., (1984), «Potential pesticide contamination of groundwater from agricultural uses», in Treatment and Disposal of Pesticide Wastes, pp. 297-325, Acs Symp. Ser. 259, American Chemical Society, Washington, DC.
|
|
32.
|
Gustafson D.I., (1989), «Groundwater ubiquity score: a simple method for assessing pesticide teachability». J. Environ. Toxic. Chem., 8(4), 339-357.
|
|
33.
|
Leistra M., and Dekkers W.A., (1976). «Computed effects of adsorption kinetics on pesticide movement in soils». J. of Soil Sci., 28, 340-350.
|
|
34.
|
Bromilov R.H., and Leistra M., (1980), «Measured and simulated behavior of aldicarb and its oxydation products in fallow soils». Pest. Sci., 11, 389-395.
|
|
35.
|
Green R.E., and Karickoff S.W., (1990), «Sorption estimates for modeling», in Pesticides in the Soil Environment: Process, Impacts and Modeling (ed. H.H. Cheng). Soil Sci. Soc. Am., Book Series no. 2, pp. 80- 101,
|
|
36.
|
Lambert S.M., (1967), «Functional relationship between sorption in soil and chemical structure». J. Agri. Food Chem., 15, 572-576.
|
|
37.
|
Hance R.J., (1969), «An empirical relationship between chemical structure and the sorption of some herbicides by soils». J. Agri. Food Chem., 17, 667-668.
|
|
38.
|
Briggs G.G. (1969), «Molecular structure of herbicides and their sorption by soils». Nature, 223, 1288.
|
|
39.
|
Briggs G.G. (1981). «Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor». J. Agric. Food Chem., 29, 1050-1059.
|
|
40.
|
Sabljic A., (1984), «Predictions of the nature and strength of soil sorption of organic polutance by molecular topology». J. Agric. Food Chem., 32, 243-246.
|
|
41.
|
Bailey G.W., and White J.L., (1970), «Factors influencing the adsorption, desorption, and movement of pesticides in soil». Residue Rev., 32, 29-92.
|
|
42.
|
Bailey G.W., J.L. White and Y. Rothberg., (1968), «Adsorption of organic herbicides by montomorillonite: Role of pH and chemical character of adsorbate». Soil Sci. Soc. Amer. Proc. 32:222-234.
|
|
43.
|
Karickhoff S.W., (1981) «Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils». Chemosphere 10, 833-846.
|
|
44.
|
Paya-Perez A., Riaz M. and Larsen B., (1989), «Soil Sorption of 6 Chlorobenzenes and 20 PCB Congeners». Environ. Toxicol. Safety 21, 1-17.
|
|
45.
|
Hamaker J.W., and Thompson J.M., (1972). «Adsorption in organic chemicals» in Organic Chemicals in the Soil Environment (Goring C.A.I, and Hamaker J.W., eds), Vol I and II, Marcel Dekker, Inc., New York, NY, 1972, pp. 49-143.
|
|
46.
|
Deli J., and Warren G.F., 1971, «Adsorption, desorption and leaching of diphenamid in soils». Weed Sci. 19:67-69.
|
|
47.
|
Chu-Huang Wu, Buehring N., Davinson J.M. and Santelmann, (1975), «Napropamide Adsorption, desorption and Movement in soils». Weed Science, Vol. 23, 454-457.
|
|
48.
|
Haues M.H.B., Stacey M., and Thompson J.M., (1968) «Adsorption of s-triazine herbicides by soil organic preparations» in Isotopes and Radiation in Soil Organic Studies, p. 75, International. Atomic Energy Agency, Vienna.
|
|
49.
|
Pionke H.B., and Deangelis R.J., (1980), «Methods for distributing pesticide loss in field run-off between the solution and adsorbed phase», CREAMS, in A Field Scale Model for Chemicals, Run-off and Erosion from Agricultural Management Systems, Chapter 19, Vol. III: Supporting Documentation, USDA Conservation Research report.
|
|
50.
|
ISO Standard Compendium Environment: Soil Quality — General aspects; chemical and physical methods of analysis; biological methods of analysis. First Edition (1994).
|
|
51.
|
Scheffer F., and Schachtschabel, Lehrbuch der Bodenkunde, F. Enke Verlag, Stuttgart (1982), 1 1th edition.
|
|
52.
|
Black, Evans D.D., White J.L., Ensminger L.E., and Clark F.E., eds. «Methods of Soil Analysis», Vol 1 and 2, American Society of Agronomy, Madison, WI, 1982.
|
|
53.
|
ISO/DIS 10381-1 Soil Quality — Sampling — Part 1: Guidance on the design of sampling programmes.
|
|
54.
|
ISO/DIS 10381-2 Soil Quality — Sampling — Part 2: Guidance on sampling techniques.
|
|
55.
|
ISO/DIS 10381-3 Soil Quality — Sampling — Part 3: Guidance on safety of sampling.
|
|
56.
|
ISO/DIS 10381-4 Soil Quality — Sampling — Part 4: Guidance on the investigation of natural and cultivated soils.
|
|
57.
|
ISO/DIS 10381-5 Soil Quality — Sampling — Part 5: Guidance on the investigation of soil contamination of urban and industrial sites.
|
|
58.
|
ISO 10381-6, 1993: Soil Quality — Sampling — Part 6: Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory.
|
|
59.
|
Green R.E., and Yamane V.K., (1970) «Precision in pesticide adsorption measurements». Soil Sci. Am. Proa, 34, 353-354.
|
|
60.
|
Grover R., and Hance RJ. (1970), «Effect of ratio of soil to water on adsorption of linuron and atrazine». Soil Sci., 109-138.
|
|
61.
|
Boesten, J J.T.I, «Influence of soil/liquid ratio on the experimental error of sorption coefficients in pesticide/soil system». Pest. Sci. 1990, 30, 31-41.
|
|
62.
|
Boesten, J J.T.I. «Influence of soil/liquid ratio on the experimental error of sorption coefficients in relation to OECD guideline 106» Proceedings of 5th international workshop on environmental behaviour of pesticides and regulatory aspects, Brussels, 26-29 April 1994.
|
|
63.
|
Bastide J., Cantier J.M., et Coste C, (1980), «Comportement de substances herbicides dans le sol en fonction de leur structure chimique». Weed Res. 21, 227-231.
|
|
64.
|
Brown D.S., and Flagg E.W., (1981), «Empirical prediction of organic pollutants sorption in natural sediments». J. Environ.QuaL, 10(3), 382-386.
|
|
65.
|
Chiou C.T., Porter P.E., and Schmedding D.W., (1983), «Partition equilibria of non-ionic organic compounds between soil organic matter and water». Environ. Sci. Technol., 17(4), 227-231.
|
|
66.
|
Gerstl Z., and Mingelgrin U., (1984), «Sorption of organic substances by soils and sediments». J. Environm. Sci. Health, B19 (3), 297-312.
|
|
67.
|
Vowles P.D., and Mantoura R.F.C., (1987), «Sediment-water partition coefficient and HPLC retention factors of aromatic hydrocarbons». Chemosphere, 16(1), 109-116.
|
|
68.
|
Lyman WJ., Reehl W.F.and Rosenblatt D.H. (1990). Handbook of Chemical Property Estimation Methods. Environmental Behaviour of Organic Compounds. American Chemical Society, Washington DC.
|
|
69.
|
Keniga E.E., and Goring, C.A.I. (1980). «Relationship between water solubility, soil sorption, octanol-water partitioning and concentration of chemicals in the biota» in Aquatic Toxicology (eds J.G. Eaton, et al.), pp. 78- 115, ASTM STP 707, Philadelphia.
|
|
70.
|
Chiou C.T., Peters L.J., and Freed V.H., (1979), «A physical concept of soil-water equilibria for non-ionic organic compounds». Science, Vol. 206, 831-832.
|
|
71.
|
Hassett J.J., Banwart W.I., Wood S.G., and Means J.C., (1981), «Sorption of/-Naphtol: implications concerning the limits of hydrophobic sorption». Soil Sci. Soc. Am. J. 45, 38-42.
|
|
72.
|
Karickhoff S.W., (1981), «Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils». Chemosphere, Vol. 10(8), 833-846.
|
|
73.
|
Moreale A., van Bladel R., (1981), "Adsorption de 13 herbicides et insecticides par le sol. Relation solubilité - reactivité. Revue de l'Agric, 34 (4), 319-322.
|
|
74.
|
Millier M., Kôrdel W. (1996), «Comparison of screening methods for the determination/estimation of adsorption coefficients on soil». Chemosphere, 32(12), 2493-2504.
|
|
75.
|
Kôrdel W., Kotthoff G., Millier M. (1995), «HPLC — screening method for the determination of the adsorption coefficient on soil — results of a ring test». Chemosphere 30 (7), 1373-1384.
|
|
76.
|
Kôrdel W., Stutte J., Kotthoff G. (1993), "HPLC — screening method for the determination of the adsorption coefficient on soil — comparison of different stationary phases. Chemosphere 27 (12), 2341-2352.
|
|
77.
|
Hance, R.J., (1967), «The speed of Attainment of Sorption Equilibria in Some Systems Involving Herbicides». Weed Research, Vol. 7, pp. 29-36.
|
|
78.
|
Koskinen W.C., and Harper S.S., (1990), «The retention processes: mechanisms» in Pesticides in the Soil Environment: Processes, Impacts and Modelling (ed. H.H. Cheng). Soil Sci. Soc. Am. Book Series, No. 2, Madison, Wisconsin.
|
|
79.
|
Cohen S.Z., Creeger S.M., Carsel R.F., and Enfield C.G. (1984), «Potential pesticide contamination of groundwater from agricultural uses», in Treatment and Disposal of Pesticide Wastes, pp. 297-325, ACS Symp. Ser. 259, American Chemical Society, Washington, DC.
|
|
80.
|
Giles C.H., (1970), «Interpretation and use of sorption isotherms» in Sorption and Transport Processes in Soils. S.C.I. Monograph No. 37, 14-32.
|
|
81.
|
Giles, C.H.; McEwan J.H.; Nakhwa, S.N. and Smith, D, (1960), «Studies in adsorption: XI. A system of classification of solution adsorption isotherms and its use in the diagnosis of adsorption mechanisms and in measurements of pesticides surface areas of soils». J. Chem. Soc, 3973-93.
|
|
82.
|
Calvet R., Tercé M., and Arvien J.C., (1980), «Adsorption des pesticides par les sols et leurs constituants: 3. Caractéristiques générales de l'adsorption». Ann. Agron. 31: 239-251.
|
|
83.
|
Bedbur E., (1996), «Anomalies in the Freundlich equation», Proc. COST 66 Workshop, Pesticides in soil and the environment, 13-15 May 1996, Stratford-upon-Avon, U.K.
|
|
84.
|
Guth, J.A., (1985), «Adsorption/desorption», in Joint International Symposium, Physicochemical Properties and their Role in Environmental Hazard Assessment, July 1-3, Canterbury, UK.
|
|
85.
|
Soil Texture Classification (US and FAO systems): Weed Science, 33, Suppl. 1 (1985) and Soil Sci. Soc. Amer. 26:305 (1962).
|
Annexe 1
Organigramme d'essai

Appendice 2
LES EFFETS DE LA PRÉCISION DE LA MÉTHODE ANALYTIQUE ET DES VARIATIONS DE CONCENTRATION SUR LA PRÉCISION DES RÉSULTATS DE L'ADSORPTION
II ressort clairement du tableau suivant (84) que si la différence entre la masse initiale (m0 = 110 μg) et la masse ( = 100 μg) de la substance dans la solution à l'équilibre est très faible, une erreur de 5 % dans la mesure de la concentration à l'équilibre fausse de 50 % le calcul de la quantité de la substance adsorbée sur le sol (
= 100 μg) de la substance dans la solution à l'équilibre est très faible, une erreur de 5 % dans la mesure de la concentration à l'équilibre fausse de 50 % le calcul de la quantité de la substance adsorbée sur le sol ( ) et de 52,4 % le calcul de Kd.
) et de 52,4 % le calcul de Kd.
|
Quantité de sol
|
msol
|
= 10g
|
|
Volume de solution
|
V0
|
= 100 cm3
|
|
|
FOR-L_2008142FR.01044401.notes.0187.xml.jpg (μg)
|
FOR-L_2008142FR.01044401.notes.0188.xml.jpg (μg cm-3)
|
R
|
FOR-L_2008142FR.01044401.notes.0189.xml.jpg (μg)
|
FOR-L_2008142FR.01044401.notes.0190.xml.jpg (μg g-1)
|
R‡
|
Kd
(31)
|
R‡
|
|
|
POUR A = 9 %
|
|
m0 = 110 μg ou C0 = 1,100 μg/cm3
|
100
|
1,000
|
valeur réelle
|
10
|
1,00
|
valeur réelle
|
1
|
|
|
101
|
1,010
|
1 %
|
9
|
0,90
|
10 %
|
0,891
|
10,9 %
|
|
105
|
1,050
|
5 %
|
5
|
0,50
|
50 %
|
0,476
|
52,4 %
|
|
109
|
1,090
|
9 %
|
1
|
0,10
|
90 %
|
0,092
|
90,8 %
|
|
|
POUR A = 55 %
|
|
m0 = 110 μg ou C0 = 1,100 μg/cm3
|
50,0
|
0,500
|
valeur réelle
|
60,0
|
6,00
|
valeur réelle
|
12,00
|
|
|
50,5
|
0,505
|
1 %
|
59,5
|
5,95
|
0,8 %
|
11,78
|
1,8 %
|
|
52,5
|
0,525
|
5 %
|
57,5
|
5,75
|
4,0 %
|
10,95
|
8,8 %
|
|
55,0
|
0,550
|
10 %
|
55,0
|
5,50
|
8,3 %
|
10,00
|
16,7 %
|
|
|
POUR A = 99 %
|
|
m0 = 110 μg ou C0 = 1,100 μg/cm3
|
1,100
|
0,011
|
valeur réelle
|
108,9
|
10,89
|
valeur réelle
|
990
|
|
|
1,111
|
0,01111
|
1 %
|
108,889
|
10,8889
|
0,01 %
|
980
|
1,0 %
|
|
1,155
|
0,01155
|
5 %
|
108,845
|
10,8845
|
0,05 %
|
942
|
4,8 %
|
|
1,21
|
0,0121
|
10 %
|
108,790
|
10,8790
|
0,10 %
|
899
|
9,2 %
|
où:
|
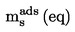
|
=
|
est la masse de substance dans la phase de sol a l'équilibre (μg);
|
|

|
=
|
est la masse de substance dans la phase aqueuse à l'équilibre (μg);
|
|

|
=
|
est la concentration de substance dans la phase de sol à l'équilibre (μg g-1);
|
|
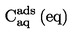
|
=
|
est la concentration massique de la substance dans la phase aqueuse à l'équilibre (μg cm-3);
|
|
R
|
=
|
est l'erreur analytique lors du calcul de  ; ;
|
|
R‡
|
=
|
est l'erreur calculée due à l'erreur analytique R.
|
Appendice 3
TECHNIQUES D'ESTIMATION DE Kd
|
1.
|
Les techniques d'estimation permettent de prédire Kd en se fondant sur des corrélations avec des valeurs Pow (12) (39) (63-68), avec des données sur l'hydrosolubilité (12) (19) (21) (39) (68-73) ou avec des données sur la polarité obtenues par HPLC en phase inversée (74-76). Comme le montrent les tableaux 1 et 2, on calcule Koc ou Kom à partir de ces équations puis on obtient, indirectement, Kd à partir des équations suivantes:
|
|
2.
|
Ces corrélations se fondent sur deux hypothèses: 1) c'est la matière organique du sol qui influence principalement l'adsorption d'une substance: 2) les interactions en jeu sont essentiellement non polaires. Il en résulte que ces corrélations: 1) ne sont pas applicables aux substances polaires, ou uniquement de manière limitée et: 2) qu'elles ne s'appliquent pas lorsque la teneur en matière organique du sol est très faible (12). Par ailleurs, si l'on a trouvé des corrélations satisfaisantes entre Pow et l'adsorption (19), on ne peut pas en dire autant des relations entre l'hydrosolubilité et l'étendue de l'adsorption (19) (21); les études sont par conséquent très contradictoires.
|
|
3.
|
Les tableaux 1 et 2 donnent quelques exemples de corrélations entre le coefficient d'adsorption et, respectivement, le coefficient de partage octanol-eau et l'hydrosolubilité.
Tableau 1
Exemples de corrélations entre le coefficient de distribution de l'adsorption et le coefficient de partage octanol-eau: pour d'autres exemples voir (12) (68)
|
Substances
|
Corrélations
|
Auteurs
|
|
Urée substituée
|
log Kom = 0,69 + 0,52 log Pow
|
Briggs (1981) (39)
|
|
Substances aromatiques chlorées
|
log Koc = -0,779 + 0,904 log Pow
|
Chiou et al. (1983) (65)
|
|
Divers pesticides
|
log Kom = 4,4 + 0,72 log Pow
|
Gerstl et Mingelgrin (1984) (66)
|
|
Hydrocarbures aromatiques
|
log Koc = -2,53 + 1,15 log Pow
|
Vowles et Mantoura (1987) (67)
|
Tableau 2
Exemples de corrélations entre le coefficient de distribution de l'adsorption et l'hydrosolubilité; pour d'autres exemples voir (68) (69)
|
Composés
|
Corrélations
|
Auteurs
|
|
Divers pesticides
|
log Kom = 3,8 - 0,561 log Sw
|
Gerstl et Mingelgrin (1984) (66)
|
|
Substances aliphatiques aromatiques chlorées
|
log Kom = (4,040 +/– 0,038) – (0,557 +/– 0,012) log Sw
|
Chiou et al. (1979) (70)
|
|
α-naphtol
|
log Koc = 4,273 - 0,686 log Sw
|
Hasset et al. (1981) (71)
|
|
Substances cycliques aliphatiques chlorées
|
log Koc = -1,405 - 0,921 log Sw-0,00953 (mp-25)
|
Karickhoff (1981) (72)
|
|
Divers composés
|
log Kom = 2,75 - 0,45 log Sw
|
Moreale van Blade (1982) (73)
|
|
Appendice 4
CALCUL DES CONDITIONS DE CENTRIFUGATION
|
1.
|
Le temps de centrifugation est donné par la formule suivante (on suppose que les particules sont sphériques):
|

|
(1)
|
Par commodité, tous les paramètres sont indiqués en unités n'appartenant pas au SI (g, cm)
où:
|
ω
|
=
|
vitesse de rotation (= 2 π rpm/60), rad s-1
|
|
rpm
|
=
|
tours par minute;
|
|
η
|
=
|
viscosité de la solution, g s-1 cm-1
|
|
rp
|
=
|
rayon des particules, cm
|
|
ps
|
=
|
densité du sol, g cm-3
|
|
paq
|
=
|
densité de la solution, g cm-3
|
|
Rt
|
=
|
distance entre le centre du rotor de la centrifugeuse et le haut de la solution dans le tube de la centrifugeuse, cm
|
|
Rb
|
=
|
distance entre le centre du rotor de la centrifugeuse et la base du tube de la centrifugeuse, cm
|
|
Rb-Rt
|
=
|
longueur du mélange sol/solution dans le tube de la centrifugeuse, cm
|
Dans la pratique, le double du temps calculé est nécessaire pour obtenir une séparation complète.
|
|
2.
|
On peut simplifier l'équation (1) si l'on considère que la viscosité (η) et la densité (paq) de la solution sont égales à la viscosité et à la densité de l'eau à 25 oC; dans ce cas, η = 8,95 x 10-3 g s-1 cm-1 et paq = 1,0 g cm3.
Le temps de centrifugation est donné par l'équation 2:
|
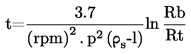
|
(2)
|
|
|
3.
|
L'équation 2 montre l'importance des paramètres du temps (t) et de la vitesse (rpm} pour définir les conditions de centrifugation permettant de séparer des particules d'une taille définie (c'est-à-dire d'un rayon de 0,1 μm dans notre cas): 1) la densité du sol et 2) la longueur du mélange dans le tube de la centrifugeuse (Rb-Rt), déterminent la distance qu'une particule de sol doit parcourir entre le haut de la solution et la base du tube; il apparaît que, pour un volume fixe, la longueur du mélange dans le tube dépend du carré du rayon du tube.
|
|
4.
|
La figure 1 présente les différents temps de centrifugation (t) en fonction de la vitesse de centrifugation (rpm) pour différentes densités du sol (ps) (figure la) et pour différentes longueurs du mélange dans les tubes de la centrifugeuse (figure 2a). La figure la montre nettement l'influence de la densité du sol. Pour une centrifugation classique de 3 000 rpm, le temps de centrifugation est d'environ 240 min pour une densité du sol de 1,2 g cm1, alors qu'il n'est que de 50 min pour 2,0 g cm3. La figure lb montre d'une manière similaire que, pour une centrifugation classique de 3 000 rpm, le temps de centrifugation est d'environ 50 min pour un mélange de 10 cm de long et de seulement 7 min pour une longueur de 1 cm. Il convient cependant de trouver un compromis optimal entre un mode de centrifugation exigeant le moins de longueur possible et la séparation commode des phases après la centrifugation.
|
|
5.
|
De plus, lorsque l'on définit les conditions expérimentales de la séparation sol/solution, il importe de considérer l'existence d'une troisième «pseudo-phase», les colloïdes. Ces particules, d'une taille inférieure à 0,2 μm, peuvent influencer de manière significative le mécanisme d'adsorption d'une substance dans une suspension de sol. Après la centrifugation, réalisée comme cela est décrit ci-dessus, les colloïdes restent dans la phase aqueuse et sont analysés en même temps que celle-ci. On perd donc les informations sur leur impact.
Si le laboratoire dispose d'équipements d'ultracentrifugation ou d'ultrafiltration, il peut étudier plus en détail l'adsorption/désorption d'une substance dans le sol et obtenir des informations sur l'adsorption de la substance sur les colloïdes. Il faut effectuer dans ce cas une ultracentrifugation à 60 000 rpm/min ou une ultrafiltration avec des filtres d'une porosité de 100 000 dalton pour séparer les trois phases, sol, colloïdes, solution. Le protocole d'essai doit être modifié en conséquence afin que les trois phases soient analysées.
|
Fig. la.

Fig. lb.

Appendice 5
CALCUL DE L'ADSORPTION A (%) ET DE LA DÉSORPTION D (%)
Organisation de l'essai dans le temps:

On suppose pour tous les calculs que la substance d'essai est stable et qu'elle n'adsorbe pas de manière significative sur les parois du récipient.
ADSORPTION A (A %)
a) Méthode parallèle
Le pourcentage d'adsorption est calculé pour chaque tube d'essai (i) au temps (ti) selon l'équation suivante:
On peut calculer ainsi les termes de l'équation:
|
m0 = C0 · V0 (μg)
|
(2)
|
|

(μg) |
(3)
|
où
|

|
=
|
pourcentage d'adsorption ( %) au temps ti
|
|
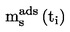
|
=
|
masse de substance d'essai sur le sol au moment ti où l'analyse est effectuée (μg)
|
|
m0
|
=
|
masse de la substance d'essai dans le tube au début de l'essai (μg)
|
|
Co
|
=
|
concentration massique initiale de la solution en contact avec le sol (μg cm-3)
|
|

|
=
|
concentration massique de la substance dans la phase aqueuse au temps ti où l'analyse est effectuée (μg cm -3); cette concentration est déterminée par analyse en tenant compte des valeurs données par les témoins
|
|
VO
|
=
|
volume initial de la solution en contact avec le sol (cm3).
|
Les valeurs du pourcentage d'adsorption 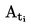 ou
ou  sont portées sur un graphe en fonction du temps, puis on détermine le temps mis pour atteindre l'équilibre de sorption (voir les figures 1 et 2).
sont portées sur un graphe en fonction du temps, puis on détermine le temps mis pour atteindre l'équilibre de sorption (voir les figures 1 et 2).


b) Méthode séquentielle
Les équations suivantes tiennent compte du fait que le procédé d'adsorption s'opère en mesurant la substance d'essai dans de petites aliquotes de la phase aqueuse à des intervalles de temps précis.
|
—
|
Durant chaque intervalle de temps, la quantité de substance adsorbée sur le sol est calculée comme suit:
|
—
|
pour le premier intervalle de temps Δti = ti — t0
|

|
(4)
|
|
|
—
|
pour le deuxième intervalle de temps Δt2 = t2 — t1
|

|
(5)
|
|
|
—
|
pour le troisième intervalle de temps Δt3 = t3 — t2
|

|
(6)
|
|
|
—
|
pour le nième intervalle de temps Δtn = tn- tn-1
|

|
(7)
|
|
|
|
—
|
le pourcentage d'adsorption à chaque intervalle de temps  est calculée selon l'équation suivante: est calculée selon l'équation suivante:
tandis que le pourcentage d'adsorption (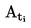 ) au temps ti est donné par l'équation: ) au temps ti est donné par l'équation:
Les valeurs de l'adsorption  or or  (selon les besoins de l'étude) sont portées sur un graphe en fonction du temps, puis on détermine le temps mis pour atteindre l'équilibre de sorption. (selon les besoins de l'étude) sont portées sur un graphe en fonction du temps, puis on détermine le temps mis pour atteindre l'équilibre de sorption.
|
|
—
|
Au temps d'équilibre teq:
|
—
|
la masse de substance adsorbée sur le sol est égale à:
|
|
—
|
la masse de substance dans la soulution est égale à:
|
|
—
|
et le pourcentage d'adsorption à l'équilibre est égal à:
|
|
Les paramètres ci-dessus sont définis de la manière suivante:
|
|
=
|
masse de substance adsorbée sur le sol durant les intervalles de temps Δt1. Δt2,..., Δtn (μg)
|
|

|
=
|
masse de substance mesurée dans une aliquote 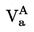 au temps t1, t2, ..., tn (μg) au temps t1, t2, ..., tn (μg)
|
|

|
=
|
masse de substance adsorbée sur le sol à l'équilibre d'adsorption (μg)
|
|
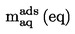
|
=
|
masse de substance dans la solution à l'équilibre d'adsorption (μg)
|
|

|
=
|
volume de l'aliquote dans laquelle la substance est mesurée (cm3)
|
|

|
=
|
pourcentage d'adsorption correspondant à l'intervalle de temps Δti ( %)
|
|

|
=
|
pourcentage d'adsorption à l'équilibre d'adsorption ( %).
|
DÉSORPTION D (%)
Le temps t0 qui marque le début de l'essai de cinétique de désorption correspond au moment où le volume maximal de substance récupérée (une fois que l'équilibre d'adsorption a été atteint) est remplacé par un volume égal de solution de CaCl20,01 M.
a) Méthode parallèle
Au temps ti, on mesure la concentration massique de la substance dans la phase aqueuse prélevée dans le tube i ( ) et la masse désorbée est calculée selon l'équation suivante:
) et la masse désorbée est calculée selon l'équation suivante:
|

|
(13)
|
À l'équilibre de désorption ti — teq de sorte que  =
= 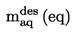
La masse de substance désorbée durant l'intervalle de temps (Δti) est donnée par l'équation suivante:
|

|
(14)
|
Le pourcentage de désorption est calculé comme suit:
au temps ti avec l'équation:
|

|
(15)
|
durant l'intervalle de temps (Δti) avec l'équation:
|

|
(16)
|
où:
|
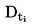
|
=
|
pourcentage de désorption au temps ti ( %)
|
|

|
=
|
pourcentage de désorption à l'intervalle de temps Δti ( %)
|
|

|
=
|
masse de substance désorbée au temps ti, (μg)
|
|

|
=
|
masse de substance désorbée durant l'intervalle de temps Δti (μg)
|
|

|
=
|
masse de substance mesurée par analyse au temps ti dans un volume de solution 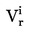 prélevé pour l'analyse (μg); prélevé pour l'analyse (μg);
|
|

|
=
|
masse de substance subsistant après l'équilibre d'adsorption en raison d'un volume de remplacement insuffisant (μg);
|
|
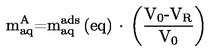
|
(17)
|
|

|
=
|
masse de substance dans la solution à l'équilibre d'adsorption (μg)
|
|
VR
|
=
|
volume de surnageant prélevé du tube après obtention de l'équilibre d'adsorption et remplacé par un volume égal de solution de CaCl20,01 M (cm3)
|
|
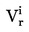
|
=
|
volume de solution prélevé du tube (i) afin de mesurer la substance lors de l'essai de cinétique de désorption (cm3).
|
Les valeurs de désorption  ou
ou  (selon les besoins de l'étude) sont portées sur un graphe en fonction du temps, puis on calcule le temps mis pour atteindre l'équilibre de désorption.
(selon les besoins de l'étude) sont portées sur un graphe en fonction du temps, puis on calcule le temps mis pour atteindre l'équilibre de désorption.
b) Méthode séquentielle
Les équations ci-après tiennent compte du fait que le procédé d'adsorption a été réalisé en mesurant la substance d'assai dans de petites aliquotes (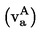 ) de la phase aqueuse (voir le chapitre sur la méthode séquentielle au point 1.9 «Réalisation de l'essai»). On suppose a) que le volume de surnageant prélevé du tube après l'essai de cinétique d'adsorption a été remplacé par un volume égal de solution de CaCl20,01 M (VR) et b) que le volume total de la phase aqueuse en contact avec le sol (VT) durant l'essai de cinétique de désorption reste constant et égal à l'équation suivante:
) de la phase aqueuse (voir le chapitre sur la méthode séquentielle au point 1.9 «Réalisation de l'essai»). On suppose a) que le volume de surnageant prélevé du tube après l'essai de cinétique d'adsorption a été remplacé par un volume égal de solution de CaCl20,01 M (VR) et b) que le volume total de la phase aqueuse en contact avec le sol (VT) durant l'essai de cinétique de désorption reste constant et égal à l'équation suivante:
|

|
(18)
|
Au temps ti:
|
—
|
La masse de substance est mesurée dans une petite aliquote  et la masse désorbée est calculée selon l'équation suivante: et la masse désorbée est calculée selon l'équation suivante:
|

|
(19)
|
|
|
—
|
À l'équilibre de désorption ti = teq de sorte que  = =  . .
|
|
—
|
Le pourcentage de désorption 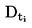 est calculé selon l'équation suivante: est calculé selon l'équation suivante:
|
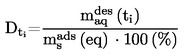
|
(20)
|
|
À l'intervalle de temps (Δti):
La quantité de substance désorbée durant chaque intervalle de temps est calculée de la manière suivante:
|
—
|
pour le premier intervalle de temps Δt1 = t1 — t0
|

and
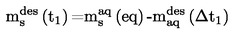
|
(21)
|
|
|
—
|
pour le deuxième intervalle de temps Δt2 = t2 — t1
|
 et et

|
(22)
|
|
|
—
|
pour le nième intervalle de temps Δtn = tn — tn-1
|
 et et

|
(23)
|
|
Enfin, le pourcentage de désorption à chaque intervalle de temps  est calculé au moyen de l'équation suivante:
est calculé au moyen de l'équation suivante:
|
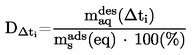
|
(24)
|
tandis que le pourcentage de désorption 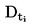 à un moment ti est donné par l'équation ci-dessous:
à un moment ti est donné par l'équation ci-dessous:
|

|
(25)
|
Les paramètres ci-dessus sont définis de la manière suivante:
|

,
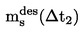
,... ,

|
=
|
masse de substance restant adsorbée sur le sol après les intervalles de temps Δt1, Δt2, ..., Δtn (μg)
|
|

,
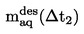
,... ,

|
=
|
masse de substance désorbée durant les intervalles de temps Δt1, Δ t2, …, Δtn (μg)
|
|

,

,... ,

|
=
|
masse de substance mesurée dans une aliquote  au temps t1, t2, …, tn, (μg); au temps t1, t2, …, tn, (μg);
|
|
VT
|
=
|
volume total de la phase aqueuse en contact avec le sol durant l'essai de cinétique de désorption réalisé avec la méthode séquentielle (cm3)
|
|
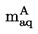
|
=
|
masse de substance subsistant après l'équilibre d'adsorption en raison d'un volume de remplacement insuffisant (μg);
|

|
(26)
|
|
où:
|
VR
|
=
|
volume de surnageant extrait du tube après l'obtention de l'équilibre d'adsorption et remplacé par un volume égale de solution de CaCl20,01 M (cm3)
|
|
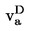
|
=
|
volume d'aliquote extrait du tube (i) pour analyse durant l'essai de cinétique de désorption réalisé avec la méthode séquentielle (cm3)
|
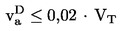
|
(27)
|
|
Appendice 6
ADSORPTION-DÉSORPTION DANS LES SOLS: PRÉSENTATION DES DONNÉES (FORMULAIRES)
Substance testée:
Sol testé:
Teneur du sol en matière sèche (105 oC, 12 h): … %
Température: … oC
Adéquation de la méthode analytique
|
Poids du sol
|
g
|
|
|
Sol: matière sèche
|
g
|
|
|
Volume de la solution de CaCl2
|
cm3
|
|
|
Concentration nominale de la solution finale
|
μg cm-3
|
|
|
Concentration d'analyse de la solution finale
|
μg cm-3
|
|
Méthode analytique utilisée:
Calibration de la méthode analytique:
Substance testée:
Sol testé:
Teneur du sol en matière sèche (105 oC, 12 h): … %
Température: … oC
|
Méthode analytique utilisée:
|
Indirecte
|
|
Parallèle
|
|
Séquentielle
|
|
|
|
Directe
|
|
|
|
|
|
Essai d'adsorption: échantillons d'essai
|
|
Symbole
|
Unités
|
Temps d'équilibrage
|
Temps d'équilibrage
|
Temps d'équilibrage
|
Temps d'équilibrage
|
|
Tube no
|
|
|
|
|
|
|
|
|
|
|
|
Poids du sol
|
—
|
g
|
|
|
|
|
|
|
|
|
|
Sol: matière sèche
|
msol
|
g
|
|
|
|
|
|
|
|
|
|
Volume d'eau dans le sol pesé (calculé)
|
Vws
|
cm3
|
|
|
|
|
|
|
|
|
|
Volume de CaCl20,01 M pour équilibrer le sol
|
|
cm3
|
|
|
|
|
|
|
|
|
|
Volume de solution de réserve
|
|
cm3
|
|
|
|
|
|
|
|
|
|
Volume total de phase aqueuse en contact avec le sol
|
V0
|
cm3
|
|
|
|
|
|
|
|
|
|
Concentration initiale de la solution à tester
|
C0
|
μg cm-3
|
|
|
|
|
|
|
|
|
|
Masse de substance au début de l'essai
|
m0
|
μg
|
|
|
|
|
|
|
|
|
|
Après agitation et centrifugation
|
|
MÉTHODE INDIRECTE
|
|
Méthode parallèle
|
|
Concentration de la substance dans la phase aqueuse, correction témoins incluse
|
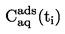
|
μg cm-3
|
|
|
|
|
|
|
|
|
|
Méthode séquentielle
|
|
Masse de substance mesurée dans l'aliquote Va
A
|

|
μg
|
|
|
|
|
|
|
|
|
|
MÉTHODE DIRECTE
|
|
Masse de substance adsorbée sur le sol
|

|
μg
|
|
|
|
|
|
|
|
|
|
Calcul de l'adsorption
|
|
Adsorption
|
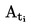
|
%
|
|
|
|
|
|
|
|
|
|

|
%
|
|
|
|
|
|
|
|
|
|
Moyennes
|
|
|
|
|
|
|
|
Coefficient d'adsorption
|
Kd
|
cm3 g-1
|
|
|
|
|
|
|
|
|
|
Moyennes
|
|
|
|
|
|
|
|
Coefficient d'adsorption
|
Koc
|
cm3 g-1
|
|
|
|
|
|
|
|
|
|
Moyennes
|
|
|
|
|
|
|
Substance testée:
Sol testé:
Teneur du sol en matière sèche (105 oC, 12 h): … %
Température: … oC
Essai d'adsorption: témoins et contrôles
|
|
Symbole
|
Unités
|
Témoin
|
Témoin
|
Contrôle
|
|
Tube no
|
|
|
|
|
|
|
|
|
|
Poids du sol
|
|
g
|
|
|
|
|
0
|
0
|
|
Volume d'eau dans le sol pesé (calculé)
|
|
cm3
|
|
|
|
|
—
|
—
|
|
Volume de solution de CaCl20,01 M ajouté
|
|
cm3
|
|
|
|
|
|
|
|
Volume ajouté de solution de réserve de la substance
|
|
cm3
|
0
|
0
|
|
|
|
|
|
Volume total de la phase aqueuse (calculé)
|
|
cm3
|
|
|
|
|
—
|
—
|
|
Concentration initiale de la substance dans la phase aqueuse
|
|
μg cm-3
|
|
|
|
|
|
|
|
Après agitation et centrifugation
|
|
Concentration dans la phase aqueuse
|
|
μg cm-3
|
|
|
|
|
|
|
Remarque: ajouter des colonnes si nécessaire.
Substance testée:
Sol testé:
Teneur du sol en matière sèche (105 oC, 12 h): … %
Température: … oC
Bilan matière
|
|
Symbole
|
Unités
|
|
|
|
|
|
Tube no
|
|
|
|
|
|
|
|
Poids du sol
|
—
|
g
|
|
|
|
|
|
Sol: matière sèche
|
msol
|
g
|
|
|
|
|
|
Volume d'eau dans le sol pesé (calculé)
|
Vws
|
ml
|
|
|
|
|
|
Volume de CaCl20,01 M pour équilibrer le sol
|
|
ml
|
|
|
|
|
|
Volume de solution de réserve
|
|
cm3
|
|
|
|
|
|
Volume total de phase aqueuse en contact avec le sol
|
V0
|
cm3
|
|
|
|
|
|
Concentration initiale de la solution à tester
|
C0
|
μg cm-3
|
|
|
|
|
|
Temps d'équilibrage
|
—
|
h
|
|
|
|
|
|
Après agitation et centrifugation
|
|
Concentration de la substance dans la phase aqueuse à l'équilibre d'adsorption, correction témoin incluse
|

|
μg cm-3
|
|
|
|
|
|
Temps d'équilibrage
|
teq
|
h
|
|
|
|
|
|
1re dilution au solvant
|
|
Volume extrait de phase aqueuse
|
Vrec
|
cm3
|
|
|
|
|
|
Volume ajouté de solvant
|
ΔV
|
cm3
|
|
|
|
|
|
1re extraction au solvant
|
|
Signal analysé dans le solvant
|
SE1
|
var.
|
|
|
|
|
|
Concentration de substance dans le solvant
|
CE1
|
μg cm-3
|
|
|
|
|
|
Masse de substance extraite du sol et des parois du récipient
|
m E1
|
μg
|
|
|
|
|
|
2e dilution au solvant
|
|
Volume de solvant extrait
|
ΔVS
|
cm3
|
|
|
|
|
|
Volume de solvant ajouté
|
ΔV'
|
cm3
|
|
|
|
|
|
2e extraction au solvant
|
|
Signal analysé dans le solvant
|
SE2
|
var.
|
|
|
|
|
|
Concentration de substance dans le solvant
|
CE2
|
μg cm-3
|
|
|
|
|
|
Masse de substance extraite du sol et des parois lu récipient
|
mE2
|
μg
|
|
|
|
|
|
Masse totale de substance: extraction en deux tapes
|
mE
|
μg
|
|
|
|
|
|
Bilan matière
|
MB
|
%
|
|
|
|
|
Substance testée:
Sol testé:
Teneur du sol en matière sèche (105 oC, 12 h): … %
Température: … oC
Isothermes d'adsorption
|
|
Symbole
|
Unités
|
|
|
|
|
|
|
|
|
|
Tube no
|
|
|
|
|
|
|
|
|
|
|
|
Poids du sol
|
—
|
g
|
|
|
|
|
|
|
|
|
|
Sol: matière sèche
|
E
|
g
|
|
|
|
|
|
|
|
|
|
Volume d'eau dans le sol pesé (calculé)
|
VWS
|
cm3
|
|
|
|
|
|
|
|
|
|
Volume de CaCl20,01 M pour équilibrer le sol
|
|
cm3
|
|
|
|
|
|
|
|
|
|
Volume de solution de réserve ajouté
|
|
cm3
|
|
|
|
|
|
|
|
|
|
Volume total de phase aqueuse en contact avec le sol (calculé)
|
V0
|
cm3
|
|
|
|
|
|
|
|
|
|
Concentration de la solution
|
C0
|
μg cm-3
|
|
|
|
|
|
|
|
|
|
Temps d'équilibrage
|
—
|
h
|
|
|
|
|
|
|
|
|
|
Après agitation et centrifugation
|
|
Concentration de la substance dans la phase aqueuse, correction témoin incluse
|

|
μg cm-3
|
|
|
|
|
|
|
|
|
|
Température
|
|
oC
|
|
|
|
|
|
|
|
|
|
Masse adsorbée par unité de sol
|

|
μg g-1
|
|
|
|
|
|
|
|
|
Analyse de régression:
valeur de KF
ads:
valeur de l/n:
coefficient de régression r2:
Substance testée:
Sol testé:
Teneur du sol en matière sèche (105 oC 12 h): … %
Température: … oC
|
Méthode analytique utilisée:
|
Indirecte
|
|
Parallèle
|
|
Séquentielle
|
|
Essai de désorption
|
|
Symbole
|
Unités
|
Intervalle de temps
|
Intervalle de temps
|
Intervalle de temps
|
Intervalle de temps
|
|
Tube no provenant de l'essai d'adsorption
|
|
|
|
|
|
|
|
Masse de substance adsorbée sur le sol à l'équilibre d'adsorption
|

|
μg
|
|
|
|
|
|
Volume extrait de phase aqueuse remplacé par CaCl20,01 M
|
VR
|
cm3
|
|
|
|
|
|
Volume total de phase aqueuse en contact avec le sol
|
PM
|
V0
|
cm3
|
|
|
|
|
|
SM
|
VT
|
cm3
|
|
|
|
|
|
Masse de substance restant après l'équilibre d'adsorption en raison d'un volume de remplacement insuffisant
|

|
μg
|
|
|
|
|
|
Cinétique de désorption
|
|
Masse mesurée de substance désorbée du sol au temps ti
|
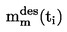
|
μg
|
|
|
|
|
|
Volume de solution extrait du tube (i) afin
|
PM
|
Vf
i
|
cm3
|
|
|
|
|
|
SM
|
Va
D
|
cm3
|
|
|
|
|
|
Masse de substance désorbée du sol du sol au temps ti (calculé)
|

|
μg
|
|
|
|
|
|
Masse de substance désorbée du sol durant l'intervalle de temps Δti (calculé)
|
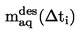
|
μg
|
|
|
|
|
|
Pourcentage de désorption
|
|
Désorption au temps ti
|
Dti
|
%
|
|
|
|
|
|
Désorption à l'intervalle de temps Δti
|

|
%
|
|
|
|
|
|
Coefficient de désorption apparente
|
Kdes
|
|
|
|
|
|
PM: Méthode parallèle.
SM: Méthode séquentielle.
C.19. ESTIMATION DU COEFFICIENT D'ADSORPTION (KOC) SUR LE SOL ET LES BOUES D'ÉPURATION PAR CHROMATOGRAPHIE LIQUIDE HAUTE PERFORMANCE (HPLC)
1. MÉTHODE
La méthode décrite reprend la ligne directrice no 121 de l'OCDE (2001).
1.1. INTRODUCTION
On peut décrire le comportement de sorption des substances dans les sols ou les boues d'épuration à l'aide de paramètres déterminés expérimentalement selon la méthode d'essai C18. Un des paramètres essentiels est le coefficient d'adsorption, défini comme le rapport entre la concentration d'une substance dans le sol ou les boues et sa concentration dans la phase aqueuse à l'équilibre d'adsorption. Le coefficient d'adsorption normalisé basé sur la teneur en carbone organique du sol (Koc) est un bon indicateur de la capacité de liaison d'une substance chimique à la matière organique du sol ou des boues d'épuration, et permet de comparer les substances chimiques entre elles. On peut estimer ce paramètre à partir de corrélations entre la solubilité dans l'eau et le coefficient de partage n-octanol/eau (1) (2) (3) (4) (5) (6) (7).
La méthode expérimentale décrite dans cet essai utilise la HPLC pour estimer le coefficient d'adsorption Koc dans le sol ou les boues d'épuration (8). Elle donne des résultats beaucoup plus fiables que ceux obtenus par les calculs de la méthode QSAR (9). En tant que méthode d'estimation, elle ne peut pas être totalement substituée aux essais par agitation conduits dans le cadre de la méthode d'essai C.18. Toutefois, le Koc estimé peut servir à sélectionner des paramètres d'essai pertinents pour l'étude des processus d'adsorption/désorption suivant la méthode précitée, grâce au calcul du coefficient de distribution (Kd) ou du coefficient d'adsorption de Freundlich (Kf) selon l'équation 3 (voir paragraphe 1.2).
1.2. DÉFINITIONS
Kd
est le coefficient de distribution défini comme le rapport des concentrations à l'équilibre (C) d'une substance d'essai dissoute dans un système à deux phases dont une sorbante (sol ou boues d'épuration) et l'autre aqueuse. Il est sans unité lorsque les concentrations dans les deux phases sont exprimées en termes de poids par poids. Si la concentration dans la phase aqueuse est exprimée en poids par volume, les unités sont alors en ml· g-1. Kd est susceptible de varier en fonction des propriétés du sorbant et peut dépendre de la concentration.
|

|
(1)
|
où:
|
Csoil
|
=
|
est la concentration de la substance d'essai dans le sol à l'équilibre d'adsorption (μg· g-1);
|
|
Cslud
|
=
|
est la concentration de la substance d'essai dans les boues à l'équilibre d'adsorption (μg· g-1);
|
|
Caq
|
=
|
est la concentration de la substance d'essai dans la phase aqueuse à l'équilibre d'adsorption (μg· g-1, μg· ml-1).
|
Kf
est le coefficient d'adsorption de Freundlich, défini comme la concentration de la substance d'essai dans le sol ou les boues d'épuration (x/m) lorsque la concentration à l'équilibre (Caq) dans la phase aqueuse est égale à un. Il est exprimé en μg.g-1 de sorbant. Il se calcule selon l'équation suivante.
|

|
(2)
|
où:
|
x/m
|
=
|
est la quantité de sorbant;
|
|
1/n
|
=
|
est la pente de l'isotherme d'adsorption de Freundlich;
|
|
Caq
|
=
|
est la concentration de la substance d'essai dans la phase aqueuse à l'équilibre d'adsorption (μg . ml- 1).
|
Lorsque 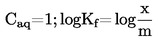
Koc
est le coefficient de distribution (Kd) ou le coefficient d'adsorption de Freundlich (Kf) normalisé basé sur la teneur en carbone organique (foc) du sorbant. Plus particulièrement dans le cas des substances non ionisées, il donne une approximation du degré d'adsorption d'une substance sur le sorbant et permet de faire des comparaisons entre différents produits chimiques. Dépendant de Kd et de Kf, il peut être sans unité ou s'exprimer en ml . g-1 ou en μg . g-1 de matière organique.
|

|
(3)
|
Comme la relation entre Koc et Kd n'est pas toujours linéaire, les valeurs de Koc peuvent donc varier d'un sol à l'autre, mais leur variabilité est très réduite par rapport aux valeurs de Kd ou de Kf.
Le coefficient d'adsorption (Koc) se déduit du facteur de capacité (k') à partir de la courbe d'étalonnage de log k' en fonction de log Koc, tracée pour les composés de référence sélectionnés. L'équation est la suivante:
|

|
(4)
|
où:
|
tR
|
=
|
est le temps de rétention de la substance d'essai et de la substance de référence dans la colonne de HPLC (en minutes)
|
|
t0
|
=
|
est le temps mort dans la colonne de HPLC (en minutes) (voir point 1.8.2).
|
Pow
est le coefficient de partage octanol/eau. défini comme le rapport des concentrations d'une substance dissoute dans l'eau et le n-octanol. Il est sans dimension.
|

|
(5)
|
1.3. SUBSTANCES DE RÉFÉRENCE
Avant d'utiliser cette méthode, il convient de connaître la formule développée, la pureté et, le cas échéant, la constante de dissociation des substances de référence. Il est également utile d'être renseigné sur leur solubilité dans l'eau et les solvants organiques, leur coefficient de partage octanol/eau ainsi que sur leurs caractéristiques d'hydrolyse.
Pour établir la corrélation entre les résultats expérimentaux de rétention en HPLC d'une substance d'essai et son coefficient d'adsorption Koc, on doit tracer la courbe d'étalonnage de log Koc en fonction de log k'. Il faudrait utiliser au minimum six points dont au moins un supérieur et un inférieur à la valeur supposée de la substance d'essai. La précision de la méthode sera d'autant plus grande que les substances de référence employées présenteront une structure chimique analogue à celle de la substance d'essai. S'il n'est pas possible d'avoir ces données, l'utilisateur est libre de sélectionner les substances d'étalonnage les mieux adaptées. Il devrait alors opter pour une série plus générale de substances présentant une hétérogénéité de structure. Les substances et les valeurs de Koc recommandées à l'usage sont indiquées en annexe, au tableau 1 pour les boues d'épuration et au tableau 3 pour les sols. Il y aura donc lieu de justifier le choix de toute autre substance d'étalonnage.
1.4. PRINCIPE DE LA MÉTHODE D'ESSAI
La HPLC est réalisée sur des colonnes d'analyse dont la phase solide est composée de résines commerciales cyanopropyliques contenant des radicaux lipophiles et polaires. On utilise une phase stationnaire modérément polaire basée sur une matrice de silice:
|
– O – Si
|
– CH2 — CH2 — CH2
|
– CN
|
|
silice
|
espaceur apolaire
|
radical polaire
|
Le principe de cette méthode d'essai est analogue à celui de la méthode d'essai A.8 (coefficient de partage, méthode par HPLC). La substance d'essai, en migrant dans la colonne contenant la phase mobile, interagit avec la phase stationnaire. La répartition de la substance entre la phase mobile et la phase stationnaire contribue à en ralentir la progression. La composition de la phase stationnaire qui comporte à la fois des sites polaires et apolaires fait qu'une interaction est possible entre groupements polaires et radicaux apolaires d'une molécule — à l'instar de la matière organique dans des matrices de sol ou de boues d'épuration. On peut ainsi établir une relation entre le temps de rétention sur la colonne et le coefficient d'adsorption sur la matière organique.
Le pH exerce une influence significative, en particulier, sur le comportement de sorption des substances polaires. En règle générale, le pH varie entre 5,5 et 7,5 dans les sols agricoles et les bassins des stations de traitement des eaux usées. Les substances ionisables devraient être testées deux fois dans des solutions tampons appropriées, à savoir sous leur forme ionisée et non ionisée, mais seulement si la dissociation du composé chimique atteint au moins 10 %, à des valeurs de pH comprises entre 5,5 et 7,5.
Comme l'évaluation se fonde exclusivement sur la relation entre la rétention sur la colonne de HPLC et le coefficient d'adsorption, il est inutile de faire appel à une méthode d'analyse quantitative, car seule la détermination du temps de rétention est nécessaire. À condition de disposer d'une série de substances de référence appropriées et d'appliquer des conditions expérimentales normalisées, cette méthode constitue un moyen rapide et efficace d'estimer le coefficient d'adsorption Koc.
1.5. APPLICABILITÉ DE L'ESSAI
La méthode par HPLC convient aux substances chimiques (marquées ou non) pour lesquelles il existe un système de détection approprié (spectrophotomètre ou détecteur de radioactivité, par exemple) et qui restent suffisamment stables tout au long de l'essai. Elle peut s'avérer particulièrement utile pour les substances difficiles à étudier dans d'autres systèmes expérimentaux (à savoir, les substances volatiles, les substances insolubles dans l'eau à une concentration mesurable par analyse et les substances présentant une très grande affinité avec la surface des récipients d'incubation). On peut également l'appliquer à des mélanges qui donnent des bandes d'élution non résolues. Dans ce cas, il faut déterminer les valeurs limites supérieures et inférieures du log Koc des composants du mélange d'essai.
Les impuretés risquent parfois de compliquer l'interprétation des résultats de la HPLC, mais leur importance restera négligeable si la substance d'essai peut être clairement identifiée et séparée des impuretés par une méthode analytique.
Après avoir été validée pour les substances énumérées au tableau 1 de l'appendice, cette méthode a été appliquée à toute une série d'autres composés chimiques répertoriés dans les classes chimiques suivantes:
|
—
|
amines aromatiques (exemples: trifluraline, 4-chloroaniline, 3,5-dinitro-aniline, 4-méthylaniline, N-méthvl- aniline, 1-naphthylamine),
|
|
—
|
esters d'acides carboxyliques aromatiques (exemples: ester méthylique de l'acide benzoïque, 3,5-dinitroben- zoate d'éthyle),
|
|
—
|
hydrocarbures aromatiques (exemples: toluène, xylène, éthylbenzène, nitrobenzène),
|
|
—
|
esters de l'acide aryloxyphénoxypropionique (exemples: didofop-méthyle, fénoxaprop-éthyle, fénoxaprop- P-éthyle).
|
|
—
|
fongicides à base de benzimidazole ou d'imidazole (exemples: carbendazime, fubéridazole, triazoxyde),
|
|
—
|
amides de l'acide carboxilique (exemples: 2-chlorobenzamide, N,N-diméthylbenzamide, 3,5-dinitrobenzamide, N-méthylbenzamide, 2-nitrobenzamide, 3-nitrobenzamide),
|
|
—
|
hydrocarbures chlorés (exemples: endosulfan, DDT, hexachlorobenzène, quintozène, 1,2,3-trichlorobenzène),
|
|
—
|
insecticides organo-phosphorés (exemples: azinphos-méthyle, disulfoton, phénamiphos, isophenphos, pyrazophos, sulprophos, triazophos),
|
|
—
|
phénols (exemples: phénol, 2-nitrophénol, 4-nitrophénol, pentachlorophénol, trichloro-2,4,6-phénol, 1-naphtol),
|
|
—
|
dérivés de la phénylurée (exemples: isoproturon, monolinuron, pencycuron),
|
|
—
|
colorants pigmentaires (exemples: Acid yellow 219, Basic Blue 41, Direct Red 81),
|
|
—
|
hydrocarbures aromatiques polycycliques (exemples: acénaphthène, naphthalène),
|
|
—
|
herbicides à base de triazine-1,3,5 (exemples: prométryne, propazine, simazine, terbutryne),
|
|
—
|
dérivés de triazole (exemples: tébuconazole, triadiméfon, tradiménol, triapenthénol).
|
Cette méthode ne convient pas aux substances réagissant avec l'éluant ou la phase stationnaire. Par ailleurs, elle n'est pas applicable aux substances qui interagissent de manière spécifique avec des constituants inorganiques (formation de complexes en grappe avec les minéraux argileux, par exemple). Elle risque d'être inopérante pour les produits tensio-actifs, les substances inorganiques ainsi que les acides et bases modérés à forts. Elle permet de déterminer des valeurs de log Koc comprises entre 1,5 et 5,0. Les substances ionisantes doivent être mesurées dans une phase mobile tamponnée, en prenant toutes les précautions nécessaires pour éviter la précipitation des composants du tampon ou de la substance d'essai.
1.6. CRITÈRES DE QUALITÉ
1.6.1. Précision
En règle générale, le coefficient d'adsorption d'une substance d'essai peut être estimé à ±0,5 unité logarithmiques de la valeur déterminée d'après la méthode par agitation (voir au tableau 1 de l'appendice). Il est possible d'atteindre une plus grande précision si les substances de référence utilisées présentent une structure chimique analogue à celle de la substance d'essai.
1.6.2. Répétabilité
Toutes les analyses doivent être répétées au moins deux fois. Les valeurs du log Koc calculées à partir de résultats expérimentaux doivent être comprises dans un intervalle de 0,25 unité log.
1.6.3. Reproductibilité
L'expérience acquise jusqu'à présent en appliquant la méthode d'essai par HPLC vient en corroborer la validité. À l'issue d'une étude expérimentant 48 substances (en majorité des pesticides) pour lesquelles existaient des données fiables concernant le Koc sur des sols, on a obtenu un coefficient de corrélation: R = 0,95 (10) (11).
Un essai comparatif interlaboratoires conduit par 11 laboratoires a permis d'améliorer et de valider la méthode (12). Tous les résultats sont reproduits au tableau 2 de l'appendice.
1.7. DESCRIPTION DE LA MÉTHODE D'ESSAI
1.7.1. Estimation préliminaire du coefficient d'adsorption
Le coefficient de partage octanol/eau Pow (= Kow) et, dans une certaine mesure, l'hydrosolubilité peuvent servir d'indicateurs du degré d'adsorption, en particulier, pour les substances non ionisées. Ils permettent donc une première approximation des ordres de grandeur. Une série de corrélations utiles ont été publiées concernant plusieurs classes de produits chimiques (1) (2) (3) (4) (5) (6) (7).
1.7.2. Appareillage
L'appareillage doit nécessairement comprendre un chromatographe en phase liquide équipé d'une pompe sans pulsations et d'un système de détection adapté. Il est recommandé d'utiliser une boucle d'injection munie d'une valve d'injection. Il faut employer des résines commerciales cyanopropyliques liées chimiquement sur une base de silice (Hypersil ou Zorbax CN, par exemple). Une colonne de garde du même matériau peut être insérée entre le système d'injection et la colonne d'analyse. La puissance de séparation des colonnes d'analyse est susceptible de varier énormément d'un fournisseur à l'autre. À titre indicatif, il faudrait atteindre les facteurs de capacité (k') suivants: log k' > 0,0 pour log Koc = 3,0 et log k' > — 0,4 pour log Koc = 2,0, en utilisant une phase mobile méthanol/eau 55/45 %.
1.7.3. Phases mobiles
Après avoir testé différentes phases mobiles, on recommande les deux suivantes:
|
—
|
méthanol/eau (55/45 % v/v),
|
|
—
|
méthanol/solution tampon de citrate 0,01 M à pH 6,0 (55/45 % v/v).
|
Le solvant d'élution est préparé à partir de méthanol de qualité HPLC et d'eau distillée ou d'un tampon citrate. Le mélange est dégazé avant emploi. Il est préférable d'effectuer une élution isocratique. Si les mélanges méthanol/eau sont contre-indiqués, on peut essayer d'autres mélanges de solvant organique/eau, à savoir éthanol/eau ou acétonitrile/eau. Dans le cas de composés ionisants, il est recommandé d'utiliser une solution tampon afin de stabiliser le pH. Il faut veiller à éviter que les sels ne précipitent ou que la colonne ne se détériore, ce qui risque de se produire avec certains mélanges de phase organique/tampon.
On s'abstiendra d'utiliser des additifs tels que des réactifs à paires d'ions, car ils risquent d'affecter les propriétés sorbantes de la phase stationnaire. Vu que des modifications de cette nature dans la phase stationnaire risquent d'être irréversibles, il est impératif d'effectuer les analyses comportant ce type d'additifs sur des colonnes séparées.
1.7.4. Solutés
Les substances de référence et d'essai doivent être dissoutes dans la phase mobile.
1.8. RÉALISATION DE L'ESSAI
1.8.1. Conditions expérimentales
On doit enregistrer la température pendant les mesures. Il est fortement recommandé d'utiliser une colonne placée dans une enceinte thermorégulée afin de garantir des conditions de température constantes pendant les différents cycles d'étalonnage et d'évaluation et la mesure de la substance d'essai.
1.8.2. Détermination du temps mort to
On peut utiliser deux méthodes pour déterminer le temps mort to (voir également point 1.2).
1.8.2.1. Détermination du temps mon to au moyen d'une série homologue
Cette procédure a montré qu'il est possible d'obtenir des valeurs de to harmonisées et fiables. Pour sa description, il faut se reporter à la méthode d'essai A.8 «Coefficient de partage n-octanol/eau et méthode d'analyse par HPLC».
1.8.2.2. Détermination du temps mort to par des substances inertes non retenues sur la colonne
Cette technique met en jeu l'injection de solutions de formamide, d'urée ou de nitrate de sodium. Il convient de répéter les mesures au moins deux fois.
1.8.3. Détermination des temps de rétention tR
Les substances de référence doivent être sélectionnées selon les indications du point 1.3. Pour déterminer leur temps de rétention, on peut les injecter sous la forme d'un étalon composite, à condition d'avoir vérifié au préalable que le temps de rétention de chaque étalon n'est pas influencé par la présence des autres étalons de référence. Il faudrait procéder à un étalonnage à intervalles réguliers, au moins deux fois par jour, pour tenir compte de toute variation imprévue dans le fonctionnement de la colonne. Pour la bonne règle, il est préférable d'injecter les étalons avant et après la substance d'essai pour s'assurer que les temps de rétention n'ont pas évolué. On injecte chaque substance d'essai séparément dans des proportions aussi faibles que possible (ce qui évite de surcharger la colonne}, puis on détermine son temps de rétention.
Afin d'accroître la fiabilité des mesures, toutes les déterminations doivent être effectuées au moins en double. Les valeurs de log Koc déduites des résultats expérimentaux doivent être comprises dans un intervalle de 0,25 unité log.
1.8.4. Évaluation
Les facteurs de capacité k' sont calculés à partir du temps mort to et des temps de rétention tR des substances de référence selon l'équation 4 (voir point 1.2). Les valeurs de log k' des substances de référence sont ensuite portées sur une courbe graphique en fonction des valeurs de leur log Koc extraites des essais par agitation dont les résultats figurent aux tableaux 1 et 3 de l'appendice. À l'aide de cette courbe, la valeur du log Koc d'une substance d'essai est calculée à partir de son log k'. Si les résultats expérimentaux montrent que le log Koc de la substance sort de l'intervalle des valeurs d'étalonnage, il convient de recommencer l'essai en employant d'autres substances de référence plus appropriées.
2. PRÉSENTATION DES DONNÉES
Le rapport d'essai doit comporter les informations suivantes:
|
—
|
identité, degré de pureté et. le cas échéant, valeurs de pKd des substances d'essai et de référence.
|
|
—
|
description des matériels utilisés et des conditions expérimentales, en indiquant le type et les dimensions de la colonne d'analyse (et de la colonne de garde), du dispositif de détection, de la phase mobile (rapport des composants et pH), de la plage des températures pendant les mesures,
|
|
—
|
temps mort et méthode appliquée pour l'évaluer,
|
|
—
|
quantités de substances de référence et d'essai injectées dans la colonne,
|
|
—
|
temps de rétention des composés de référence employés pour l'étalonnage,
|
|
—
|
détails de la courbe de régression ajustée (log k' en fonction de log Koc) et représentation graphique de cette courbe,
|
|
—
|
valeurs moyennes de rétention et valeur estimée du log Koc de la substance d'essai,
|
3. BIBLIOGRAPHIE
|
(1)
|
W. J. Lyman, W. F. Reehl, D. H. Rosenblatt (ed). (1990). Handbook of chemical property estimation methods, Chap. 4, McGraw-Hill, New York.
|
|
(2)
|
J. Hodson, N. A. Williams (1988). The estimation of the adsorption coefficient (Koc) for soils by HPLC. Chemosphere, 17, 1 67.
|
|
(3)
|
G. G. Briggs (1981). Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor. J. Agric. Food Chem., 29, pp. 1050-1059.
|
|
(4)
|
C. T. Chiou, P. E. Porter, D.W. Schmedding (1983). Partition equilibria of nonionic organic compounds between soil organic matter and water. Environ. Sci. Technol., 17, pp. 227-231.
|
|
(5)
|
Z. Gerstl, U. Mingelgrin (1984). Sorption of organic substances by soils and sediment. J. Environm. Sci. Health, B19, pp. 297-312.
|
|
(6)
|
C. T. Chiou, L. J. Peters, V. H. Freed (1979). A physical concept of soil water equilibria for nonionic organic compounds, Science, 106, pp. 831-832.
|
|
(7)
|
S. W. Karickhoff (1981). Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils. Chemosphere, 10, pp. 833-846.
|
|
(8)
|
W. Kördel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), pp. 121-128.
|
|
(9)
|
M. Mueller, W. Kördel (1996). Comparison of screening methods for the estimation of adsorption coefficients on soil. Chemosphere, 32(12), pp. 2493-2504.
|
|
(10)
|
W. Kördel, J. Stutte, G. Kotthoff (1993). HPLC-screening method for the determination of the adsorption coefficient in soil-comparison of different stationary phases, Chemosphere, 27(12), pp. 2341-2352.
|
|
(11)
|
B. von Oepen, W. Kördel, W. Klein (1991). Sorption of nonpolar and polar compounds to soils: Processes, measurements and experience with the applicability of the modified OECD Guideline 106, Chemosphere, 22, pp. 28 5-304.
|
|
(12)
|
W. Kördel, G. Kotthoff, J. Müller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere, 30(7), pp. 1373-1384.
|
Appendice
Tableau 1
Comparaison entre les valeurs de Koc sur les sols et les boues d'épuration et les valeurs calculées suivant la méthode de sélection par HPLC (34)
(35)
|
Type de substance
|
No CAS
|
Log Koc pour boues d'épuration
|
Lo Log Koc par HPLC
|
Δ
|
Log Log Koc pour sols
|
Lo Log Koc par HPLC
|
Δ
|
|
Atrazine
|
1912-24-9
|
1,66
|
2,14
|
0,48
|
1,81
|
2,20
|
0,39
|
|
Linuron
|
330-55-2
|
2,43
|
2,96
|
0,53
|
2,59
|
2,89
|
0,30
|
|
Fenthion
|
55-38-9
|
3,75
|
3,58
|
0,17
|
3,31
|
3,40
|
0,09
|
|
Monuron
|
150-68-5
|
1,46
|
2,21
|
0,75
|
1,99
|
2,26
|
0,27
|
|
Phénanthrène
|
85-01-8
|
4,35
|
3,72
|
0,63
|
4,09
|
3,52
|
0,57
|
|
Benzoate de phényle
|
93-99-2
|
3,26
|
3,03
|
0,23
|
2,87
|
2,94
|
0,07
|
|
Benzamide
|
55-21-0
|
1,60
|
1,00
|
0,60
|
1,26
|
1,25
|
0,01
|
|
4-Nitrobenzamide
|
619-80-7
|
1,52
|
1,49
|
0,03
|
1,93
|
1,66
|
0,27
|
|
Acétanilide
|
103-84-4
|
1,52
|
1,53
|
0,01
|
1,26
|
1,69
|
0,08
|
|
Aniline
|
62-53-3
|
1,74
|
1,47
|
0,27
|
2,07
|
1,64
|
0,43
|
|
2,5-Dichloroaniline
|
95-82-9
|
2,45
|
2,59
|
0,14
|
2,55
|
2,58
|
0,03
|
Tableau 2
Résultats des essais comparatifs inter-laboratoires (11 laboratoires participants) en vue d'améliorer et de valider la méthode par HPLC (36)
|
Type de substance
|
No CAS
|
Log Koc
|
|
Log Koc
|
|
(OCDE 106)
|
[Méthode par HPLC|
|
|
Atrazine
|
1912-24-9
|
1,81
|
78 ± 16
|
1,89
|
|
Monuron
|
150-68-5
|
1,99
|
100 ± 8
|
2,00
|
|
Triapenthénol
|
77608-88-3
|
2,37
|
292 ± 58
|
2,47
|
|
Linuron
|
330-55-2
|
2,59
|
465 ± 62
|
2,67
|
|
Fenthion
|
55-38-9
|
3,31
|
2062 ± 648
|
3,31
|
Tableau 3
Substances de référence recommandées pour des essais de sélection par HPLC d'après des données relatives à l'adsorption sur les sols
|
Substance de référence
|
No CAS
|
Valeurs moyennes de log Koc d'après la méthode par agitation
|
Nombre de résultats pour le Koc
|
Écart type du log
|
Source
|
|
Acétanilide
|
103-84-4
|
1,25
|
4
|
0,48
|
(37)
|
|
Phénol
|
108-95-2
|
1,32
|
4
|
0,70
|
(37)
|
|
2-Nitrobenzamide
|
610-15-1
|
1,45
|
3
|
0,90
|
(38)
|
|
N.N-diméthylbenzamide
|
611-74-5
|
1,52
|
2
|
0,45
|
(37)
|
|
4-Méthylbenzamide
|
619-55-6
|
1,78
|
3
|
1,76
|
(37)
|
|
Benzoate de méthyle
|
93-58-3
|
1,80
|
4
|
1,08
|
(37)
|
|
Atrazine
|
1912-24-9
|
1,81
|
3
|
1,08
|
(39)
|
|
Isoproturon
|
34123-59-6
|
1,86
|
5
|
1,53
|
(39)
|
|
3-Nitrobenzamidc
|
645-09-0
|
1,95
|
3
|
1,31
|
(38)
|
|
Aniline
|
62-53-3
|
2,07
|
4
|
1,73
|
(37)
|
|
3,5-Dinitrobenzamide
|
121-81-3
|
2,31
|
3
|
1,27
|
(38)
|
|
Carbendazime
|
10605-21-7
|
2,35
|
3
|
1,37
|
(39)
|
|
Triadiménol
|
55219-65-3
|
2,40
|
3
|
1,85
|
(39)
|
|
Triazoxyde
|
72459-58-6
|
2,44
|
3
|
1,66
|
(39)
|
|
Triazophos
|
24017-47-8
|
2,55
|
3
|
1,78
|
(39)
|
|
Linuron
|
330-55-2
|
2,59
|
3
|
1,97
|
(39)
|
|
Naphtalène
|
91-20-3
|
2,75
|
4
|
2,20
|
(37)
|
|
Endosulfane-diol
|
2157-19-9
|
3,02
|
5
|
2,29
|
(39)
|
|
Méthiocarbe
|
2032-65-7
|
3,10
|
4
|
2,39
|
(39)
|
|
Acid Yellow 219
|
63405-85-6
|
3,16
|
4
|
2,83
|
(37)
|
|
1,2,3-Trichlorobenzène
|
87-61-6
|
3,16
|
4
|
1,40
|
(37)
|
|
γ-HCH
|
58-89-9
|
3,23
|
5
|
2,94
|
(37)
|
|
Fenchion
|
55-38-9
|
3,31
|
3
|
2,49
|
(39)
|
|
Direct Red 8 1
|
2610-11-9
|
3,43
|
4
|
2,68
|
(37)
|
|
Pyrazophos
|
13457-18-6
|
3,65
|
3
|
2,70
|
(39)
|
|
α-Endosulfane
|
959-98-8
|
4,09
|
5
|
3,74
|
(39)
|
|
Diclofop-méthyle
|
i 51338-27-3
|
4,20
|
3
|
3,77
|
(39)
|
|
Phénanthrène
|
85-01-8
|
4,09
|
4
|
3,83
|
(37)
|
|
Basic Blue 41 (mix)
|
26850-47-5
12270-13-2
|
4,89
|
4
|
4,46
|
(37)
|
|
DDT
|
50-29-3
|
5,63
|
1
|
|
(38)
|
C.20. DAPHNIA MAGNA, ESSAI DE REPRODUCTION
1. MÉTHODE
La méthode décrite pour cet essai de reproduction reprend la ligne directrice no 211 de l'OCDE (1998).
1.1. INTRODUCTION
Le principal objectif de l'essai consiste à évaluer l'effet de produits chimiques sur la capacité reproductrice de Daphnia magna.
1.2. DÉFINITIONS ET UNITÉS
Animaux parents: Daphnia femelles présentes au début de l'essai et dont la capacité reproductrice représente l'objet de cette étude.
Descendants: jeunes Daphnia engendrées par les animaux parents au cours de l'essai.
Concentration minimale avec effet observé (CMEO): concentration d'essai la plus basse à laquelle on a observé un effet statistiquement significatif de la substance sur la reproduction et la mortalité des animaux parents (à p < 0,05) par rapport au témoin, durant une période d'exposition définie. Cependant, toutes les concentrations d'essai supérieures à ta CMEO doivent exercer un effet nocif supérieur ou égal à celui observé à la CMEO. Si ces deux conditions ne peuvent être remplies, il convient de justifier de façon détaillée le choix de la CMEO (et donc de la CSEO).
Concentration (maximale) sans effet observé (CSEO): concentration d'essai immédiatement inférieure à la CMEO qui, comparée au témoin, n'a pas d'effet statistiquement significatif (à p < 0,05), durant une période d'exposition définie.
CEx: concentration de la substance d'essai dissoute dans l'eau qui entraîne une diminution de x % de la reproduction chez Daphnia magna, durant une période d'exposition définie.
Taux intrinsèque d'accroissement: mesure de l'accroissement de la population qui intègre la capacité reproductrice et la mortalité par tranche d'âge (20) (21) (22). Cette valeur est nulle dans les populations à l'état sta-tionnaire, positive dans les populations en croissance et négative dans les populations qui régressent. Cette dernière catégorie de population n'est évidemment pas durable et est vouée en fin de compte à l'extinction.
Limite de détection: la plus basse concentration susceptible d'être détectée, mais non chiffrée.
Limite de détermination: la plus basse concentration susceptible d'être mesurée quantitativement.
Mortalité: un animal est noté comme mort lorsqu'il est immobile, autrement dit lorsqu'il n'est pas capable de nager ou si aucun mouvement des appendices ou du postabdomen n'est observé dans les 15 secondes qui suivent l'agitation douce du récipient d'essai. (Si on utilise une autre définition, celle-ci doit être stipulée avec sa référence.)
1.3. PRINCIPE DE LA MÉTHODE
De jeunes femelles Daphnia (les animaux parents), âgées de moins de 24 heures au début de l'essai, sont expo-secs à la substance d'essai ajoutée à l'eau à différentes concentrations. L'essai dure 21 jours. À la fin de l'essai, le nombre total de descendants vivants produits par animal parent survivant à la fin de l'essai est évalué. La capacité reproductrice des animaux parents peut s'exprimer autrement (par exemple, par le nombre de descendants vivants produits par animal et par jour, à partir du premier jour où des descendants ont été observés), mais ces résultats doivent être fournis en plus du nombre total de juvéniles engendrés par parent survivant à la fin de l'essai. La capacité reproductrice des animaux exposés à la substance d'essai est comparée à celle des témoins afin de déterminer la concentration minimale avec effet observé (CMEO) et, de là, la concentration (maximale) sans effet observé (CSEO). De plus, dans toute la mesure du possible, les résultats sont analysés à l'aide d'un modèle de régression, en vue d'estimer la concentration qui entraînerait une réduction de x % de la capacité reproductrice, c'est-à-dire la CEx (par exemple, CE50, CE20 ou CE10).
Il faut aussi indiquer le taux de survie des animaux parents et le moment de la première portée. D'autres effets liés à la substance sur des paramètres tels que la croissance (par exemple, la longueur) et éventuellement le taux intrinsèque d'accroissement de la population peuvent aussi être examinés.
1.4. INFORMATIONS CONCERNANT LA SUBSTANCE D'ESSAI
Les résultats d'un essai de toxicité aiguë (voir méthode C.2, partie I) réalisé sur Daphnia magna devraient être disponibles. Ces résultats peuvent être utiles pour sélectionner une plage de concentrations d'essai adaptée aux essais de reproduction. La solubilité dans l'eau et la pression de vapeur de la substance d'essai doivent être connues. Il faudrait pouvoir disposer d'une méthode d'analyse fiable, dont le rendement et la limite de détermination sont connus, pour doser la substance dans les solutions d'essai.
Des informations sur la substance d'essai qui peuvent être utiles pour établir les conditions de l'essai comprennent la formule structurale, la pureté de la substance, la stabilité à la lumière, la stabilité dans les conditions de l'essai, le pKa, le coefficient de partage n-octanol/eau (Poe) et les résultats d'un essai de biodégradabilité immédiate (voir méthode C.4).
1.5. VALIDITÉ DE L'ESSAI
Pour que l'essai soit valable, les témoins doivent remplir les critères de performance suivants:
|
—
|
la mortalité des animaux parents (Daphnia femelles) ne dépasse pas 20 % à la fin de l'essai,
|
|
—
|
le nombre moyen de descendants vivants produits par animal parent survivant à la fin de l'essai est ≥ 60.
|
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Appareillage
Les récipients d'essai et les autres dispositifs qui entreront en contact avec les solutions d'essai doivent être entièrement en verre ou constitués d'une autre matière chimiquement inerte. On utilisera en principe des béchers en verre.
En outre, il sera nécessaire d'employer une partie ou la totalité du matériel suivant:
|
—
|
un appareil pour mesurer la concentration de l'oxygène (à l'aide d'une microélectrode ou d'un autre dispositif destiné à mesurer l'oxygène dissous dans des échantillons de faible volume),
|
|
—
|
un appareil pour mesurer la dureté de l'eau,
|
|
—
|
un appareil pour déterminer la concentration de carbone organique total (COT) dans l'eau ou la demande chimique en oxygène (DCO),
|
|
—
|
un dispositif approprié pour régler le régime d'éclairage et mesurer l'intensité lumineuse.
|
1.6.2. Organisme d'essai
L'espèce à utiliser dans cet essai est Daphnia magna Straus. D'autres espèces de Daphnia peuvent être utilisées à condition qu'elles remplissent les critères de validité pertinents (ceux qui concernent la capacité reproductrice des témoins doivent s'appliquer aux espèces de Daphnia). Si d'autres espèces de Daphnia sont utilisées, il y a lieu de les identifier clairement et de justifier leur utilisation.
Le clone devrait de préférence avoir été identifié d'après son génotype. La recherche (1) a montré que la capacité reproductrice du clone A (originaire de l'IRCHA, en France) (3) répond de façon stable au critère de validité qui stipule une moyenne de ≥ 60 descendants par animal parent survivant, lorsqu'il est élevé dans les conditions décrites dans la présente méthode. D'autres clones sont toutefois acceptables à condition de prouver que la culture de Daphnia remplit les critères de validité pour l'essai.
Au début de l'essai, les animaux doivent être âgés de moins de 24 heures et ne peuvent pas provenir d'une première génération de descendants. Ils doivent être issus d'un lot sain (c'est-à-dire qui ne présente pas de signes de stress, tels qu'une mortalité élevée, la présence de mâles et d'éphippies, un retard dans la production des premiers descendants, des animaux décolorés, etc.). Le lot d'animaux doit être maintenu dans des conditions de culture (lumière, température, milieu, alimentation et nombre d'animaux par unité de volume) semblables à celles qui seront appliquées au cours de l'essai. Si le milieu utilisé dans l'essai est différent de celui où sont normalement élevées les Daphnia, il convient de laisser aux Daphnia une période d'acclimatation, avant l'essai, qui dure habituellement quelque 3 semaines (c'est-à-dire une génération), afin d'éviter de stresser les animaux parents.
1.6.3. Milieu d'essai
Il est recommandé d'utiliser un milieu entièrement défini dans cet essai. Cela permet d'éviter le recours aux additifs (par exemple, algues, extraits de sol, etc.), qui sont difficiles à caractériser, et d'améliorer ainsi les possibilités de normalisation entre les laboratoires. Les milieux Elendt M4 (4) et M7 (voir appendice 1) se sont avérés pertinents à cette fin. D'autres milieux sont cependant acceptables [par exemple (5) et (6)], à condition que les Daphnia élevées dans ces milieux satisfassent aux critères de validité établis pour l'essai.
Si le milieu utilisé contient des additifs non définis, ceux-ci devraient être spécifiés clairement et le rapport d'essai devrait comporter des informations sur la composition, notamment la teneur en carbone, étant donné qu'elle peut contribuer au régime alimentaire fourni. On préconise de déterminer le carbone organique total (COT) et/ou la demande chimique en oxygène (DCO) de la solution mère de l'additif organique et d'estimer leur incidence sur le COT et la DCO du milieu d'essai. En outre, il est souhaitable que les concentrations de COT du milieu (c'est-à-dire avant l'ajout des algues) soient inférieures à 2 mg/l (7).
Lorsque l'on teste des substances contenant des métaux, il est important de savoir que les propriétés du milieu d'essai (par exemple, la dureté, le pouvoir de chélation) peuvent influer sur leur toxicité. C'est pourquoi il est souhaitable d'opérer dans un milieu entièrement défini. Néanmoins, dans l'état actuel des connaissances, les seuls milieux entièrement définis qui conviennent aux cultures à long terme de Daphnia magna sont Elendt M4 et M7. Ces deux milieux contiennent l'agent chélateur EDTA. Des travaux ont montré (2) que la «toxicité apparente» du cadmium est généralement inférieure lorsque l'essai de reproduction est effectué dans les milieux M4 et M7, que lorsqu'il est effectué dans des milieux ne contenant pas d'EDTA. M4 et M7 ne sont donc pas recommandés pour tester des substances contenant des métaux, de même que d'autres milieux contenant des agents chélateurs connus. Il est souhaitable d'utiliser un autre milieu pour les substances renfermant des métaux, par exemple l'eau douce calcaire reconstituée d'ASTM (7), qui ne contient pas d'EDTA, additionnée d'un extrait d'algues (8). La combinaison de l'eau douce calcaire reconstituée d'ASTM et de l'extrait d'algues convient également aux essais et aux cultures à long terme de Daphnia magna (2), bien qu'elle exerce encore une légère action chélatante à cause des matières organiques contenues dans l'extrait d'algues.
Au début de l'essai et durant celui-ci, la concentration d'oxygène dissous devrait être supérieure à 3 mg/l. Le pH devrait être compris entre 6 et 9, et ne devrait normalement pas varier de plus de 1,5 unité au cours d'un même essai. Une dureté supérieure à 140 mg/l (en CaCO3) est recommandée. La capacité reproductrice des animaux dans les essais pratiqués à un niveau au moins égal à ce seuil s'est avérée conforme aux critères de validité (9) (10).
1.6.4. Solutions d'essai
Les solutions d'essai sont généralement amenées à la concentration voulue par dilution d'une solution mère. Les solutions mères devraient, de préférence, être préparées par dissolution de la substance dans le milieu d'essai.
Le recours à des dispersants ou solvants organiques peut quelquefois s'avérer nécessaire pour obtenir une solution mère à la concentration voulue, mais ces additifs doivent être évités dans toute la mesure du possible. Voici quelques exemples de solvants adéquats: acétone, éthanol, méthanol, diméthylformamide et triéthylè-neglycol. Parmi les dispersants recommandables, citons le Cremophor RH40, la méthylcellulose à 0,01 % et le HCO-40. En tout état de cause, la substance d'essai ne doit pas être testée au-delà de sa limite de solubilité dans le milieu d'essai.
Les solvants sont employés pour confectionner une solution mère qui peut être dosée avec précision dans l'eau. Les solvants énumérés ci-dessus, utilisés à la concentration recommandée dans le milieu d'essai final (≤ 0,1 ml/1), ne seront pas toxiques et n'augmenteront pas la solubilité de la substance dans l'eau.
Les dispersants peuvent faciliter le dosage précis et la dispersion. À la concentration recommandée dans le milieu d'essai final (≤ 0,1 ml/1), les dispersants mentionnés ci-dessus ne seront pas toxiques et n'augmenteront pas la solubilité de la substance dans l'eau.
1.7. CONCEPTION DE L'ESSAI
Les traitements sont répartis dans les récipients d'essai et toutes les manipulations ultérieures de ces récipients sont réalisées dans un ordre aléatoire, faute de quoi, les résultats pourraient présenter un biais qui risquerait d'être interprété comme un effet de la concentration. Notamment, si les unités expérimentales sont manipulées par ordre de traitement ou de concentration, certains effets liés au temps, comme la fatigue du manipulateur ou d'autres erreurs, pourraient avoir un impact plus prononcé aux concentrations supérieures. En outre, si les résultats de l'essai sont susceptibles d'être affectés par une condition initiale ou liée à l'environnement, comme la situation dans le laboratoire, il conviendra d'envisager un plan en blocs.
1.8. MODE OPÉRATOIRE
1.8.1. Conditions d'exposition
1.8.1.1. Durée
L'essai dure 21 jours.
1.8.1.2. Charge
Les animaux parents sont répartis individuellement dans les récipients d'essai contenant chacun 50-100 ml de milieu.
Il est parfois nécessaire d'avoir recours à des volumes plus importants afin de pouvoir appliquer la méthode d'analyse utilisée pour déterminer la concentration de la substance d'essai, bien qu'il soit aussi possible de mélanger les récipients de même concentration aux fins de l'analyse chimique. Si des volumes supérieurs à 100 ml sont employés, il faudra peut-être augmenter la ration distribuée aux Daphnia, afin que la nourriture disponible soit suffisante et que les critères de validité soient satisfaits. S'agissant des essais dynamiques, d'autres protocoles peuvent être envisagés pour des raisons techniques (par exemple, 4 groupes de 10 animaux dans des volumes expérimentaux plus élevés), mais toute modification du mode opératoire doit être signalée.
1.8.1.3. Nombre d'animaux
Pour les essais semi-statiques, on utilisera au moins 10 animaux répartis individuellement à chaque concentration d'essai et au moins 10 animaux répartis individuellement dans la série de témoins.
Pour les essais dynamiques, il s'avère approprié d'utiliser 40 animaux répartis en 4 groupes de 10 à chaque concentration d'essai (1). Un plus petit nombre d'animaux d'expérience peut être utilisé, mais on recommande d'employer au moins 20 animaux par concentration, répartis dans au moins 2 récipients contenant le même nombre d'animaux (par exemple, 4 récipients contenant 5 daphnies chacun). Notons que pour les essais où les animaux sont maintenus en groupe, il ne sera pas possible d'exprimer la capacité reproductrice en fonction du nombre total de descendants vivants produits par animal parent survivant à la fin de l'essai, si des animaux parents meurent. Auquel cas, il faudra exprimer la capacité reproductrice par «le nombre total de descendants vivants produits par parent présent au début de l'essai».
1.8.1.4. Alimentation
Dans les essais semi-statiques, on préconise d'administrer une ration quotidienne, ou au moins tri-hebdomadaire (ce qui correspond au renouvellement du milieu). Les écarts par rapport à cette fréquence (par exemple, dans les essais dynamiques) doivent être signalés.
Au cours de l'essai, le régime alimentaire des animaux parents devrait, de préférence, se composer d'algues unicellulaires vivantes appartenant à une ou plusieurs des espèces suivantes: Chlorella sp., Selenastrum capricor-nutum [renommée Pseudokirchneriella subcapitata (11)] et Scenedesmus subspicatus. La nourriture devrait être dispensée en fonction de la teneur en carbone organique (C) fournie à chaque animal parent. La recherche (12) a montré que pour Daphnia magna, des teneurs comprises entre 0,1 et 0,2 mg de C/Daphnia/jour dans la ration alimentaire suffisaient pour produire le nombre de descendants requis selon les critères de validité. On peut fournir des rations à teneur constante tout au long de l'essai ou utiliser des teneurs plus faibles au début, que l'on augmentera afin de tenir compte de la croissance des animaux parents. Dans ce cas, la ration devrait toujours contenir entre 0,1 et 0,2 mg de C/Daphnia/jour, qui est la teneur recommandée.
Si des paramètres de remplacement, comme le nombre de cellules d'algues ou l'absorbance de la lumière, sont utilisés pour doser la teneur en carbone requise dans la ration alimentaire (pour des raisons pratiques, car la mesure de la teneur en carbone prend beaucoup de temps), chaque laboratoire est tenu de produire son propre nomogramme mettant en relation le paramètre de remplacement et la teneur en carbone de la culture d'algues (des conseils concernant l'établissement d'un nomogramme sont donnés à l'appendice 2). Les nomogrammes devraient être vérifiés au moins une fois par an, et plus souvent si les conditions de culture des algues ont changé. Il a été démontré que l'absorbance de la lumière donne une meilleure indication de la teneur en carbone que le nombre de cellules (13).
Il convient d'administrer une suspension d'algues concentrée aux Daphnia, afin de réduire au minimum le volume du milieu de culture des algues transféré dans les récipients d'essai. On peut concentrer les algues par centrifugation, puis remise en suspension dans de l'eau distillée, de l'eau désionisée ou le milieu de culture des Daphnia.
1.8.1.5. Lumière
16 heures de lumière à une intensité ne dépassant pas 15-20 μE· m-2 · s-1.
1.8.1.6. Température
La température des milieux d'essai devrait être comprise entre 18 et 22 oC. Néanmoins, dans un même essai, la température ne devrait pas, si possible, varier de plus de 2 oC au sein de cet intervalle (par exemple, 18-20, 19-21 ou 20-22 oC). Il peut être utile d'utiliser un récipient d'essai supplémentaire pour surveiller la température.
1.8.1.7. Aération
Les récipients d'essai ne doivent pas être aérés durant l'essai.
1.8.2. Concentrations d'essai
Il faudrait normalement utiliser au moins 5 concentrations d'essai formant une série géométrique, de préférence espacées d'un facteur n'excédant pas 3,2. Il convient d'utiliser un nombre approprié de répliques pour chaque concentration d'essai (voir point 1.8.1.3). L'utilisation de moins de 5 concentrations doit être justifiée. Les substances ne devraient pas être testées au-delà de leur limite de solubilité dans le milieu d'essai.
Lors de la détermination de la plage de concentrations, il convient de tenir compte des éléments suivants:
|
i)
|
si l'on cherche à déterminer la CMEO et la CSEO, la plus faible concentration testée doit être suffisamment basse pour que le taux de fécondité à cette concentration ne soit pas significativement plus bas que celui du groupe témoin. Dans le cas contraire, il faudra recommencer l'essai en abaissant la concentration la plus basse;
|
|
ii)
|
si l'on cherche à déterminer la CMEO et la CSEO, la plus forte concentration testée devra être suffisamment élevée pour que le taux de fécondité à cette concentration soit significativement inférieur à celui du groupe témoin. Dans le cas contraire, il faudra recommencer l'essai en augmentant la concentration la plus forte;
|
|
iii)
|
si l'on veut estimer la CEx relative aux effets sur la reproduction, il est conseillé d'utiliser des concentrations suffisantes pour définir cette CEx avec un niveau de confiance satisfaisant. Si l'on évalue la CE50 relative aux effets sur la reproduction, il est souhaitable que la concentration d'essai la plus forte soit supérieure à cette CE50. Si tel n'est pas le cas, on pourra toujours estimer la CE50, mais l'intervalle de confiance sera très large et on risque de ne pas être en mesure d'évaluer la pertinence du modèle;
|
|
iv)
|
il vaut mieux éviter d'inclure dans la plage des concentrations d'essai celles qui ont un effet statistiquement significatif sur la survie des adultes parce que, ce faisant, on modifierait la nature de l'essai qui passerait d'un simple essai de reproduction à un essai combiné de reproduction et de mortalité, ce qui exige une analyse statistique bien plus complexe.
|
Une connaissance préalable de la toxicité de la substance d'essai (résultant d'une étude de toxicité aiguë et/ou d'études de détermination de l'ordre de grandeur) devrait faciliter la sélection de concentrations d'essai pertinentes.
Lorsqu'un solvant ou un dispersant est utilisé pour faciliter la préparation des solutions d'essai (voir point 1.6.4), sa concentration finale dans les récipients d'essai ne doit pas dépasser 0,1 ml/l et doit être identique dans tous les récipients d'essai.
1.8.3. Témoins
Il faudrait tester une série de témoins du milieu d'essai et, le cas échéant, une série de témoins contenant le solvant ou le dispersant, parallèlement aux séries traitées avec la substance d'essai. Le cas échéant, la concentration du solvant ou du dispersant doit être identique à celle utilisée dans les récipients contenant la substance d'essai. Il convient d'utiliser un nombre approprié de répliques (voir point 1.8.1.3).
Généralement, dans un essai correctement mené, le coefficient de variation autour du nombre moyen de descendants vivants produits par animal parent dans le ou les groupes témoins devrait être ≤ 25 %, et cette information doit être communiquée pour les essais où les animaux sont maintenus dans des récipients individuels.
1.8.4. Renouvellement du milieu d'essai
La fréquence de renouvellement du milieu dépend de la stabilité de la substance d'essai, mais devrait être au moins trihebdomadaire. Si les essais préliminaires de stabilité (voir point 1.4) montrent que la concentration de la substance d'essai n'est pas stable (c'est-à-dire en dehors de la fourchette de 80-120 % de la concentration nominale ou inférieure à 80 % de la concentration initiale mesurée) durant la période de renouvellement la plus longue (3 jours), il faut envisager de renouveler le milieu plus fréquemment ou de pratiquer un essai dynamique.
Lors du renouvellement du milieu dans les essais semi-statiques, on prépare une deuxième série de récipients d'essai en vue d'y transférer les animaux parents avec, par exemple, une pipette en verre d'un diamètre approprié. Le volume de milieu transféré avec les Daphnia doit être le plus petit possible.
1.8.5. Observations
Les résultats des observations effectuées durant l'essai doivent être consignés dans des fiches de données (voir exemples aux appendices 3 et 4). Si d'autres mesures s'imposent (voir points 1.3 et 1.8.8), des observations supplémentaires pourront être requises.
1.8.6. Descendants
Les descendants engendrés par chaque animal parent devraient de préférence être retirés et comptés quotidiennement, dès la première portée, pour ne pas qu'ils consomment la nourriture destinée aux adultes. Aux fins de la présente méthode, il ne faut compter que les descendants vivants, mais la présence d'œufs avortés ou de descendants morts doit être signalée.
1.8.7. Mortalité
La mortalité des animaux parents doit être notée, de préférence quotidiennement, et au moins à chaque comptage des descendants.
1.8.8. Autres paramètres
Bien que la présente méthode vise avant tout à évaluer les effets sur la reproduction, il se peut que d'autres effets soient suffisamment quantifiés pour se prêter à l'analyse statistique. La mesure de la croissance est très intéressante, puisqu'elle fournit des indications sur les éventuels effets sublétaux, ce qui peut se révéler plus utile que les seules données de reproduction. On préconise de mesurer la longueur des animaux parents (la longueur du corps sans l'épine anale) à la fin de l'essai. D'autres paramètres peuvent être mesurés ou calculés: le moment de la première portée (et des portées suivantes), le nombre et la taille des portées par animal, le nombre de portées avortées, la présence de mâles ou d'éphippies et, éventuellement, le taux intrinsèque d'accroissement de la population.
1.8.9. Fréquence des analyses quantitatives et des mesures
La concentration d'oxygène, la température, la dureté et le pH doivent être mesurés au moins une fois par semaine, avant et après le renouvellement des milieux, chez les témoins et dans les récipients qui renferment la concentration la plus élevée de la substance d'essai.
Au cours de l'essai, les concentrations de la substance à tester sont déterminées à intervalles réguliers.
Dans les essais semi-statiques, où l'on suppose que la concentration de la substance d'essai ne s'écartera pas de plus de 20 % de la concentration nominale (c'est-à-dire qu'elle restera comprise dans un intervalle de 80 à 120 % — voir points 1.4 et 1.8.4), on recommande, au minimum, d'analyser les concentrations d'essai la plus élevée et la plus basse juste après leur préparation et au moment de leur renouvellement au cours de la première semaine de l'essai (les analyses devraient être pratiquées sur des échantillons prélevés dans la même solution, juste après la préparation de la solution et au moment du renouvellement). Ces déterminations devraient ensuite être répétées selon une fréquence au moins hebdomadaire.
S'agissant des essais où la concentration n'est pas supposée demeurer dans un intervalle de ± 20 % de la concentration nominale, il est nécessaire d'analyser toutes les concentrations d'essai, juste après leur préparation et au moment du renouvellement. Cependant, pour les essais où la concentration initiale mesurée de la substance d'essai sort de l'intervalle de ± 20 % de la concentration nominale, mais pour lesquels on peut montrer de façon suffisamment convaincante que les concentrations initiales sont répétables et stables (c'est-à-dire comprises dans un intervalle de 80 à 120 % des concentrations initiales), les déterminations chimiques pourraient se limiter durant les deuxième et troisième semaines aux concentrations d'essai la plus élevée et la plus basse. En tout état de cause, la détermination de la concentration de la substance d'essai avant le renouvellement ne doit être effectuée que sur une seule réplique de chaque concentration d'essai.
Si on pratique un essai dynamique, il convient de recourir à un régime de prélèvements identique à celui décrit pour les essais semi-statiques (mais l'analyse des solutions «anciennes» ne s'applique pas ici). Toutefois, il peut être souhaitable d'augmenter le nombre de prélèvements durant la première semaine (par exemple, trois séries de mesures) afin de vérifier la stabilité des concentrations d'essai. Dans ces types d'essai, le débit du diluant et de la substance d'essai doivent être contrôlés chaque jour.
S'il s'avère que la concentration de la substance d'essai a pu être correctement maintenue tout au long de l'essai dans un intervalle de ± 20 % de la concentration initiale mesurée ou nominale, les résultats peuvent être déduits des valeurs initiales mesurées ou nominales. Si l'écart par rapport à la concentration initiale mesurée ou nominale est supérieur à ± 20 %, les résultats devraient être exprimés par rapport à la moyenne pondérée en fonction du temps (voir appendice 5).
2. RÉSULTATS ET RAPPORT
2.1. TRAITEMENT DES RÉSULTATS
L'objectif de cet essai est de déterminer l'effet de la substance à tester sur le nombre total de descendants vivants produits par animal parent survivant à la fin de l'essai. Le nombre total de descendants par animal parent doit être calculé pour chaque récipient d'essai (autrement dit, pour chaque récipient de même concentration). Si, dans un récipient, un animal parent meurt durant l'essai ou se révèle être un mâle, ce récipient est exclu de l'analyse. L'analyse reposera alors sur un nombre réduit de récipients de même concentration.
Pour l'estimation de la CMEO et, par conséquent, de la CSEO relatives aux effets du produit chimique sur la capacité reproductrice, il est nécessaire de calculer la moyenne de la capacité reproductrice à chaque concentration pour tous les récipients, ainsi que l'écart type résiduel groupé, et ce calcul peut être effectué à l'aide de l'analyse de la variance. La moyenne pour chaque concentration doit ensuite être comparée à la moyenne pour les témoins, par une méthode appropriée de comparaisons multiples. Les tests de Dunnett ou de Williams peuvent convenir (14) (15) (16) (17). 11 est nécessaire de vérifier l'hypothèse (de l'analyse de la variance) concernant l'homogénéité de la variance. On recommande d'effectuer cette vérification sur un graphique plutôt que par un test formel de signification (18); le test de Barlett convient également. Si cette hypothèse n'est pas vérifiée, il faut envisager de transformer les résultats afin d'homogénéiser les variances avant d'effectuer l'analyse de la variance ou de réaliser une analyse de la variance pondérée. La grandeur de l'effet détectable par l'analyse de la variance (c'est-à-dire la plus petite différence significative) devrait être calculée et figurer dans le rapport.
Four estimer la concentration qui provoquerait une réduction de 50 % de la capacité reproductrice (CE50), une courbe adéquate, comme la courbe logistique, devrait être ajustée aux résultats au moyen d'une méthode statistique telle que les moindres carrés. La courbe pourrait être paramétrée de manière que la CE50 et son écart type puissent être estimés directement. Cela faciliterait considérablement le calcul des limites de confiance de la CE50. À moins d'avoir de bonnes raisons de préférer d'autres niveaux de confiance, il convient d'opter pour des limites de confiance de 95 % de part et d'autre. La procédure d'ajustement devrait, de préférence, permettre d'évaluer la signification du manque d'ajustement. Cela peut se faire graphiquement ou en recherchant la part du manque d'ajustement et la part des erreurs proprement dites dans la somme des carrés des résidus et en pratiquant un test de signification pour le manque d'ajustement. Comme les traitements qui entraînent un taux de fécondité élevé sont susceptibles de donner lieu à une plus grande variance dans le nombre de juvéniles produits que les traitements diminuant le taux de fécondité, il faudrait envisager de pondérer les valeurs observées afin de refléter les diverses variances dans les différents groupes traités (voir référence 18).
Dans l'analyse des données obtenues à partir de l'essai tournant final (2), une courbe logistique a été ajustée à l'aide du modèle suivant, mais d'autres modèles conviennent également:
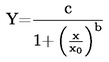
où:
|
Y
|
=
|
est le nombre total de juvéniles par anima! paient survivant à la fin de l'essai (calculé pour chaque récipient)
|
|
x
|
=
|
est la concentration de la substance
|
|
c
|
=
|
est le nombre escompté de juvéniles lorsque x = 0
|
|
x0
|
=
|
est la CE50 dans la population
|
|
b
|
=
|
est le paramètre caractérisant la pente
|
Ce modèle devrait convenir dans un grand nombre de situations, mais pas dans toutes. Il faudrait vérifier la validité du modèle comme indiqué ci-dessus. Dans certains cas. il pourrait être approprié d'utiliser un modèle d'hormèse dans lequel les basses concentrations donnent des effets plus marqués (19).
D'autres effets liés à la concentration, comme la CE10 ou la CE20, peuvent également être estimés, mais il faudra probablement paramétrer le modèle autrement que dans le cas de la CE50.
2.2. RAPPORT D'ESSAI
Le rapport d'essai doit mentionner les informations suivantes:
2.2.1. Substance d'essai
|
—
|
État physique et propriétés physico-chimiques pertinentes,
|
|
—
|
données permettant l'identification chimique, y compris la pureté.
|
2.2.2. Espèce d'expérience
|
—
|
Clone (préciser si son génotype a été déterminé), fournisseur ou source (quand elle est connue) et conditions de culture appliquées. Si l'on utilise une espèce autre que Daphnia magna, il faut le signaler et le justifier.
|
2.2.3. Conditions d'essai
|
—
|
Mode opératoire utilisé (par exemple, semi-statique ou dynamique, volume, charge en nombre de Daphnia par litre),
|
|
—
|
photopériode et intensité lumineuse,
|
|
—
|
plan de l'essai (par exemple, nombre de récipients de même concentration, nombre de parents par récipient),
|
|
—
|
détails du milieu de culture employé,
|
|
—
|
le cas échéant, ajout de matière organique (préciser la composition, la source, la méthode de préparation, le COT et la DCO des solutions mères, une estimation du COT et de la DCO résultants dans le milieu d'essai,
|
|
—
|
informations détaillées concernant l'alimentation, notamment quantité (en mg de C/Daphnia/jour) et programme (par exemple, type d'aliment(s), y compris, pour les algues, le nom de l'espèce et, si elles sont connues, la variété et les conditions de culture),
|
|
—
|
méthode de préparation des solutions mères et fréquence de renouvellement (la nature et la concentration du solvant ou du dispersant doivent être indiquées, le cas échéant).
|
2.2.4. Résultats
|
—
|
Résultats des éventuelles études préliminaires sur la stabilité de la substance d'essai,
|
|
—
|
concentrations d'essai nominales et résultats de toutes les analyses permettant de déterminer la concentration de la substance d'essai dans les récipients d'essai (l'appendice 4 fournit des exemples de fiches de données); le rendement de la méthode et sa limite de détermination devraient aussi être mentionnés,
|
|
—
|
qualité de l'eau dans les récipients d'essai (pH, température, concentration d'oxygène dissous, COT et/ou DCO et dureté, le cas échéant) (l'appendice 3 fournit un exemple de fiche de données),
|
|
—
|
dénombrement complet des descendants vivants de chaque animal parent (voir exemple de fiche de données à l'appendice 3),
|
|
—
|
nombre et dates des décès chez les animaux parents (voir exemple de fiche de données à l'appendice 3),
|
|
—
|
coefficient de variation de la fécondité chez les témoins (en fonction du nombre total de descendants vivants par animal parent survivant à la fin de l'essai),
|
|
—
|
graphique représentant le nombre total de descendants vivants par animal parent survivant (pour chaque récipient de même concentration) à la fin de l'essai, en fonction de la concentration de la substance d'essai,
|
|
—
|
concentration minimale avec effet observé (CMEO) sur la reproduction, y compris une description des méthodes statistiques utilisées et une indication de la grandeur de l'effet qui a pu être détecté, et concentration (maximale) sans effet observé (CSEO) sur la reproduction; s'il y a lieu, mentionner également la CMEO et la CSEO sur la mortalité des animaux parents,
|
|
—
|
s'il y a lieu, CEx pour la reproduction et intervalles de confiance, ainsi qu'un graphique du modèle ajusté employé pour ces calculs, la pente de la courbe dose-effet et son écart type,
|
|
—
|
autres effets biologiques observés ou mesurés: indiquer tout autre effet biologique observé ou mesuré (par exemple, croissance des animaux parents) avec justification appropriée, le cas échéant,
|
|
—
|
justification de tout écart par rapport à la présente méthode d'essai.
|
3. BIBLIOGRAPHIE
|
(1)
|
OECD Test Guideline Programme. Report of the Workshop on the Daphnia magna Pilot Ring Test, Sheffield University, UK, 20-21 March 1993.
|
|
(2)
|
OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 6. Report of the Final Ring Test of the Daphnia magna Reproduction Test Paris. 1997.
|
|
(3)
|
Baird D. J., Barber J., Bradley M. C., Soares A. M. V. M. and Calow P. (1991). A comparative study of genotype sensitivity to acute toxic stress using clones of Daphnia magna Strauss. Ecotoxicology and Environmental Safety, 21, pp. 257-265.
|
|
(4)
|
Elendt B. P., (1990). Selenium deficiency in Crustacea; An ultrastructural approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, pp. 25-33.
|
|
(5)
|
EPA (1993). Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms. (Fourth éd.). EPA/600/4-90/027F. C. I. Weber (ed), USEPA, Cincinnati, Ohio.
|
|
(6)
|
Vigano L., (1991) Suitability of commercially available spring waters as standard medium for culturing Daphnia magna. Bull. Environ. Contam. Toxicol, 47, pp. 775-782.
|
|
(7)
|
ASTM (1988). Standard Guide for Conducting Acute Toxicity Tests with Fishes, Macroinvertebrates and Amphibians. E729-88a. American Society for Testing and Materials, Philadelphia P.A. 20 pp.
|
|
(8)
|
Baird D. J., Soares A. M. V. M., Girling A., Barber J., Bradley M. C. and Calow P. (1989). The long term maintenance of Daphnia magna Straus for use in ecotoxicological tests; problems and prospects. In: Proceedings of the lst European Conference on Ecotoxicology. Copenhagen 1988 (H. Løkke, H. Tyle & F. Bro-Rasmussen. Eds.), pp. 144-148.
|
|
(9)
|
Parkhurst B. R., Forte J. L. and Wright G. P. (1981). Reproducibility of a life-cycle toxicity test with Daphnia magna. Bull. Environ. Contam. and Toxicol., 26, pp. 1-8.
|
|
(10)
|
Cowgill U. M. and Milazzo D. P. (1990) The sensitivity of two cladocerans to water quality variables: salinity and hardness. Arch. Hydrobiol., 120(2), pp. 185-196.
|
|
(11)
|
Korshikov (1990). Pseudokirchneriella subcapitata Hindak, F-1990. Biologice Prace, 36, 209.
|
|
(12)
|
Sims I. R., Watson S. and Holmes D. (1993). Toward a standard Daphnia juvenile production test. Environmental Toxicology and Chemistry, 12, pp. 2053-2058.
|
|
(13)
|
Sims I. (1993). Measuring the growth of phytoplankton: the relationship between total organic carbon with three commonly used parameters of algal growth. Arch. Hydrobiol., 128, pp. 459-466.
|
|
(14)
|
Dunnett C. W., (1955). A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc., 50, pp. 1096-1121.
|
|
(15)
|
Dunnett C. W., (1964). New tables for multiple comparisons with a control. Biometrics, 20, pp. 482-491.
|
|
(16)
|
Williams D. A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, pp. 103-117.
|
|
(17)
|
Williams D. A. (1972). The comparison of several dose levels with a zero dose control. Biometrics, 28, pp. 510-531.
|
|
(18)
|
Draper N. R. and Smith H. (1981). Applied Regression Analysis, second edition, Wiley, N.Y.
|
|
(19)
|
Brain P. and Cousens R. (1989). An equation to describe dose responses where there is stimulation of growth at low doses. Weed Research, 29, pp. 93-96.
|
|
(20)
|
Wilson E. O. and Bossert, W. H. (1971). A Primer of Population Biology. Sinauer Associates Inc. Publishers.
|
|
(21)
|
Poole R. W. (1974). An Introduction to quantitative Ecology. McGraw-Hill Series in Population Biology, New York, pp. 5 32.
|
|
(22)
|
Meyer J. S., Ingersoll C. G., McDonald L. L. and Boyce M. S. (1986). Estimating uncertainty in population growth rates: Jackknife vs bootstrap techniques. Ecology, 67, pp. 1156-1166.
|
Appendice 1
PRÉPARATION DE MILIEUX ELENDT M7 ET M4 ENTIÈREMENT DÉFINIS
Acclimatation aux milieux Elendt M7 et M4
Certains laboratoires ont éprouvé des difficultés à transférer directement les Daphnia dans les milieux M4 (1) et M7. Ils ont toutefois obtenu certains résultats en procédant à une acclimatation progressive, c'est-à-dire en transférant les animaux de leur milieu vers un milieu à 30 % d'Elendt, puis à 60 % d'Elendt et enfin à 100 % d'Elendt. La période d'acclimatation peut prendre jusqu'à un mois.
PRÉPARATION
Éléments en traces
Différentes solutions mères (1), contenant chacune une seule substance en traces, sont tout d'abord préparées avec une eau d'une pureté adéquate, par exemple de l'eau désionisée, distillée ou obtenue par osmose inverse. À partir de ces différentes solutions mères (1), on prépare une deuxième solution mère unique (II) renfermant toutes les substances en traces (solution combinée), à savoir:
|
Solutions mères I
(substance unique)
|
Quantité ajoutée à l'eau
(mg/l)
|
Concentration (par rapport au milieu M4)
(fois)
|
Pour préparer la solution mère combinée II, ajouter la quantité suivante de solution mère I à l'eau
(ml/l)
|
|
M 4
|
M 7
|
|
H3BO3
|
57 190
|
20 000
|
1,0
|
0,25
|
|
MnCl2 * 4 H2O
|
7 210
|
20 000
|
1,0
|
0,25
|
|
LiCl
|
6 120
|
20 000
|
1,0
|
0,25
|
|
RbCl
|
1 420
|
20 000
|
1,0
|
0,25
|
|
SrCl2 * 6 H2O
|
3 040
|
20 000
|
1,0
|
0,25
|
|
NaBr
|
320
|
20 000
|
1,0
|
0,25
|
|
Na2MoO4 * 2 H2O
|
1 260
|
20 000
|
1,0
|
0,25
|
|
CuCl2 * 2 H2O
|
335
|
20 000
|
1,0
|
0,25
|
|
ZnCl2
|
260
|
20 000
|
1,0
|
1,0
|
|
CoCl2 * 6 H2O
|
200
|
20 000
|
1,0
|
1,0
|
|
Kl
|
65
|
20 000
|
1,0
|
1,0
|
|
Na2SeO3
|
43,8
|
20 000
|
1,0
|
1,0
|
|
NH4VO3
|
11,5
|
20 000
|
1,0
|
1,0
|
|
Na2EDTA * 2 H2O
|
5 000
|
2 000
|
—
|
—
|
|
FeSO4 * 7 H2O
|
1 991
|
2 000
|
—
|
—
|
|
Les solutions de Na2EDTA et de FeSO4 sont préparées séparément, puis mélangées et immédiatement autoclavées. Cela donne:
|
|
Solution 21 Fe-EDTA
|
|
1 000
|
20,0
|
5,0
|
Milieux M4 et M7
Les milieux M4 et M7 sont préparés à partir de la solution mère II, des macronutriments et des vitamines suivants:
|
|
Quantité ajoutée à l'eau
(mg/l)
|
Concentration (par rapport au milieu M4)
(fois)
|
Quantité de solution mère ajoutée pour préparée le milieu
(ml/l)
|
|
M 4
|
M 7
|
|
Solution mère II (combinaison de substances en traces)
|
|
20
|
50
|
50
|
|
Solutions mères contenant les macronutriments (une substance par solution)
|
|
CaCl2 * 2 H2O
|
293 800
|
1 000
|
1,0
|
1,0
|
|
MgSO4 * 7 H2O
|
246 600
|
2 000
|
0,5
|
0,5
|
|
KC1
|
58 000
|
10 000
|
0,1
|
0,1
|
|
NaHCO3
|
64 800
|
1 000
|
1,0
|
1,0
|
|
Na2SiO3 * 9 H2O
|
50 000
|
5 000
|
0,2
|
0,2
|
|
NaNO3
|
2 740
|
10 000
|
0,1
|
0,1
|
|
KH2PO4
|
1 430
|
10 000
|
0,1
|
0,1
|
|
K2HPO4
|
1 840
|
10 000
|
0,1
|
0,1
|
|
Solution mère de vitamines combinées
|
—
|
10 000
|
0,1
|
0,1
|
|
La solution mère de vitamines combinées est préparée en additionnant 3 vitamines à 1 litre d'eau, comme indiqué ci-dessous:
|
|
Chlorhydrate de thyamine
|
750
|
10 000
|
—
|
—
|
|
Cyanocobalamine (B12)
|
10
|
10 000
|
—
|
—
|
|
Biotine
|
7,5
|
10 000
|
—
|
—
|
|
La solution mère de vitamines combinées est congelée par petites parties aliquotes. Les vitamines sont ajoutées au milieu peu de temps avant son utilisation.
Notes: Afin d'éviter la précipitation des sels lors de la préparation du milieu complet, il faut ajouter les parties aliquotes de solution mère à quelque 500-800 ml d'eau désionisée et amener le volume à 1 litre.
La première publication relative au milieu M4 se trouve dans Elendt, B. P. (1990). Selenium deficiency in crustacea; an ultra-structural approach to antennal damage in Daphnia magna Straus. Protoplasma. 154, pp. 25-33.
|
Appendice 2
ANALYSE DE LA TENEUR EN CARBONE ORGANIQUE TOTAL (COT) ET ÉTABLISSEMENT D'UN NOMOGRAMME POUR LA TENEUR EN COT DES ALIMENTS À BASE D'ALGUES
Il est admis que la teneur en carbone des algues alimentaires n'est généralement pas mesurée directement, mais déduite de corrélations (par nomogramme) avec des paramètres de remplacement comme le nombre de cellules d'algues ou l'ab-sorbanec de la lumière.
Il faudrait mesurer le COT par oxydation à haute température plutôt que par les méthodes aux UV ou au persulfate (voir: The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, HMSO 1980; 49 High Holborn, London WCIV 6HB).
Pour établir le nomogramme, il y a lieu d'isoler les algues du milieu de croissance par une centrifugation suivie d'une remise en suspension dans de l'eau distillée. On mesure le paramètre de remplacement et la teneur en COT dans chaque échantillon, produit en triple exemplaire. Les témoins contenant uniquement de l'eau distillée devraient être analysés et la teneur en COT déduite de celle de l'échantillon contenant les algues.
Les nomogrammes devraient être linéaires sur la plage prescrite des concentrations de carbone. Des exemples sont présentés ci-dessous.
NB: Ne pas utiliser pour les conversions; il est essentiel que les laboratoires établissent leurs propres nomogrammes.



Appendice 3
EXEMPLE DE FICHE POUR CONSIGNER LE RENOUVELLEMENT DU MILIEU, LES DONNÉES DE SURVEILLANCE PHYSICO-CHIMIQUES, L'ALIMENTATION, LA REPRODUCTION DES DAPHNIA ET LA MORTALITÉ DES ADULTES
|
Expérience n0:
|
Date de départ:
|
Clone:
|
Milieu:
|
Type d'alimentation:
|
Substance d'essai:
|
Concentration nominale
|
|
Jour
|
0
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
13
|
14
|
15
|
16
|
17
|
18
|
19
|
20
|
21
|
|
|
|
Renouvellement du milieu (cocher)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PH (40)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nouveau
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ancien
|
|
|
O2 mg/l (40)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nouveau
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ancien
|
|
|
Température ( oC) (40)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nouveau
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ancien
|
|
|
Alimentation fournie (cocher)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nombre de descendants vivants (41)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
Récipient 1
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
9
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
Mortalité cumulée des adultes (42)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Appendice 4
EXEMPLE DE FICHE DE DONNÉES POUR CONSIGNER LES RÉSULTATS DES ANALYSES CHIMIQUES
a) Concentrations mesurées
|
Concentration nominale
|
Échantillon prélevé durant la première semaine
|
Échantillon prélevé durant la deuxième semaine
|
Échantillon prélevé durant la troisième semaine
|
|
Après renouvellement
|
Avant renouvellement
|
Après renouvellement
|
Avant renouvellement
|
Après renouvellement
|
Avant renouvellement
|
|
|
|
|
|
|
|
|
b) Concentrations mesurées en pourcentage de la concentration nominale
|
Concentration nominale
|
Échantillon prélevé durant la première semaine
|
Échantillon prélevé durant la deuxième semaine
|
Échantillon prélevé durant la troisième semaine
|
|
Après renouvellement
|
Avant renouvellement
|
Après renouvellement
|
Avant renouvellement
|
Après renouvellement
|
Avant renouvellement
|
|
|
|
|
|
|
|
|
Appendice 5
CALCUL D'UNE MOYENNE PONDÉRÉE DANS LE TEMPS
Moyenne pondérée dans le temps
Étant donné que la concentration de la substance d'essai peut diminuer au cours de la période comprise entre deux renouvellements du milieu, il est nécessaire de rechercher la concentration qui devrait être considérée comme représentative de la plage des concentrations appliquées aux Daphnia parents. Le choix devrait être guidé par des considérations tant biologiques que statistiques. Si on estime, par exemple, que les effets sur la reproduction sont surtout sensibles à la concentration maximale, il faut utiliser la concentration maximale. Si, par contre, on juge que c'est l'effet cumulé ou à long terme de la substance toxique qui est prépondérant, il est plus pertinent d'utiliser une concentration moyenne. Dans ce cas, la concentration moyenne pondérée dans le temps convient, puisqu'elle tient compte de la variation de la concentration instantanée en fonction du temps.
Figure 1
Exemple de moyenne pondérée dans le temps

La figure 1 représente un essai (simplifié), d'une durée de 7 jours, avec renouvellement du milieu aux jours 0, 2 et 4.
|
—
|
La ligne fine en zigzag représente la concentration en fonction du temps. La concentration est censée diminuer sui vant un schéma de décroissance exponentielle.
|
|
—
|
Les six points portés sur le graphique représentent les concentrations mesurées au début et à la fin de chaque période de renouvellement.
|
|
—
|
La ligne épaisse représente la moyenne pondérée dans le temps.
|
La moyenne pondérée dans le temps est calculée de façon que l'aire sous la moyenne pondérée dans le temps soit égale à l'aire sous la courbe de concentration. Le calcul correspondant a l'exemple Ci-dessus est illustré dans le tableau 1.
Tableau 1
Calcul de la moyenne pondérée dans le temps
|
Renouvellement no
|
Jours
|
Conc. 0
|
Conc. 1
|
Ln (Conc. 0)
|
Ln (Conc. 1)
|
Aire
|
|
1
|
2
|
10,000
|
4,493
|
2,303
|
1,503
|
13,767
|
|
2
|
2
|
11,000
|
6,037
|
2,398
|
1,798
|
16,544
|
|
3
|
3
|
10,000
|
4,066
|
2,303
|
1,403
|
19,781
|
|
Nombre total de jours: 7
|
Aire totale
|
50,091
|
|
Moyenne PT
|
7,156
|
|
«Jours» correspond au nombre de jours de la période de renouvellement.
«Conc. 0» est la concentration mesurée au début de chaque période de renouvellement.
«Conc. 1» est la concentration mesurée à la fin de chaque période de renouvellement,
«Ln (Conc. 0)» est le logarithme népérien de Conc. 0.
«Ln (Conc. 1)» est le logarithme népérien de Conc. 1.
«Aire» est l'aire sous la courbe exponentielle correspondant à chaque période de renouvellement. Elle se calcule par la formule suivante:

|
La moyenne pondérée dans «moyenne PT» est l'«aire totale» divisée par le «nombre total de jours».
Bien entendu, dans le cas de l'essai de reproduction chez Daphnia, le tableau devrait être complété jusqu'à 21 jours.
Il est clair que lorsque les observations ne sont faites qu'au début et à la fin de chaque période de renouvellement, il n'est pas possible d'affirmer avec certitude que la baisse de concentration est effectivement exponentielle. Une courbe différente donnerait lieu à un calcul différent de l'aire. Cependant, une baisse de concentration exponentielle est plausible et constitue probablement la meilleure courbe à utiliser en l'absence d'autres informations.
Il convient toutefois d'être prudent si l'analyse chimique ne détecte aucune substance à la fin de la période de renouvellement. À moins de pouvoir estimer la vitesse à laquelle la substance a disparu de la solution, il est impossible d'obtenir une aire sous la courbe réaliste et, partant, une moyenne pondérée dans le temps qui soit raisonnable.
C.21. MICRO-ORGANISMES DU SOL: ESSAI DE TRANSFORMATION DE L'AZOTE
1. MÉTHODE
La présente méthode d'essai reprend l'essai TG 216 (2000) de l'OCDE.
1.1. INTRODUCTION
La présente méthode d'essai décrit une méthode de laboratoire conçue pour étudier les effets à long terme des substances chimiques, après une exposition unique, sur l'activité de transformation de l'azote par les micro-organismes du sol. L'essai s'appuie principalement sur les recommandations de l'Organisation européenne et méditerranéenne pour la protection des plantes (1). Toutefois, d'autres lignes directrices, notamment celles du Biologische Bundesanstalt allemand (2), de l'agence de protection de l'environnement des États-Unis (3) de la SETAC (4) et de l'Organisation internationale de normalisation (ISO) (5), ont également été prises en considération. Lors d'un atelier de l'OCDE sur la sélection des sols et des sédiments, qui s'est tenu à Belgirate, Italie, en 1995 (6), le nombre et le type de sols à utiliser dans le cadre de cet essai ont été définis. Les recommandations relatives au prélèvement, à la manipulation et au stockage des échantillons de sol s'appuient sur des lignes directrices de l'ISO (7) et sur les recommandations de l'atelier de Belgirate. Pour déterminer et évaluer les caractéristiques toxiques des substances d'essai, il peut être nécessaire de déterminer leurs effets sur l'activité microbienne du sol, par exemple lorsqu'il est nécessaire de disposer de données sur les effets secondaires potentiels des produits phytosanitaires sur la microflore du sol ou bien en cas de risque d'exposition des micro-organismes du sol à des substances chimiques autres que les produits phytosanitaires. L'essai de transformation de l'azote est effectué pour déterminer les effets de ces substances chimiques sur la microflore du sol. Si l'essai porte sur des produits agrochimiques (produits phytosanitaires, engrais, produits chimiques sylvicoles), deux essais sont effectués: l'essai de transformation de l'azote et l'essai de transformation du carbone. S'il s'agit d'autres substances que les produits agrochimiques, l'essai de transformation de l'azote suffit. Toutefois, si les valeurs CE50 de l'essai de transformation de l'azote relatives à ces substances chimiques correspondent à celles des inhibiteurs de la nitrification commerciaux (nitrapyrine), un essai de transformation du carbone peut être effectué afin d'obtenir des informations complémentaires.
Les sols sont formés de composés vivants et non vivants qui existent sous forme de mélanges complexes et hétérogènes. Les micro-organismes jouent un rôle important dans la décomposition et la transformation de la matière organique en sol fertile et un grand nombre d'espèces contribuent aux différents aspects de la fertilisation des sols. Toute interférence à long terme avec ces processus biochimiques risque de perturber le cycle des éléments nutritifs et par conséquent d'altérer la fertilité du sol. La transformation du carbone et de l'azote se produit dans tous les sols fertiles. Bien que les colonies microbiennes responsables de ces processus diffèrent d'un sol à un autre, les voies de transformation sont identiques pour l'essentiel.
La méthode d'essai décrite ici est conçue pour détecter les effets nocifs à long terme d'une substance sur le processus de transformation de l'azote à la surface aérobie des sols. La méthode d'essai permet également d'estimer les effets des substances sur la transformation du carbone par la microflore du sol. La formation de nitrate se produit par suite de la dégradation des liaisons carbone-azote. Par conséquent, si l'on trouve des taux identiques de production de nitrate dans des sols traités et dans les sols témoins, il est très probable que les voies de dégradation principales du carbone soient intactes et fonctionnelles. Le substrat choisi pour l'essai (farine de luzerne en poudre) présente un bon rapport carbone-azote (généralement entre 12/1 et 16/1). De ce fait, la carence en carbone est réduite pendant l'essai et si la population microbienne est endommagée par une substance chimique, elle peut se reconstituer dans un délai de 100 jours.
Les essais à partir desquels la présente méthode d'essai a été mise au point étaient conçus avant tout pour des substances dont il est possible d'estimer la quantité qui pénètre dans le sol. C'est le cas par exemple, des produits phytosanitaires dont la dose d'application dans le champ est connue. Pour les produits agrochimiques, il suffit de pratiquer un essai sur deux doses correspondant au taux d'application prévu ou estimé. Les produits agrochimiques peuvent être testés sous forme de principes actifs (p.a.) ou de produits formulés. Toutefois, l'essai n'est pas limité aux produits agrochimiques. En modifiant la quantité de substance-d'essai appliquée au sol, et le mode d'évaluation des données, on détermine les effets d'une série de concentrations sur la transformation de l'azote pour les substances chimiques autres que les produits agrochimiques L'essai peut également être utilisé pour les substances chimiques dont on ne connaît pas la quantité qui pénètre dans le sol.. Les résultats de ces essais sont utilisés pour préparer une courbe dose-réponse et calculer les valeurs CEx, où x est défini comme le pourcentage d'effet.
1.2. DÉFINITIONS
Transformation de l'azote: dégradation par des micro-organismes de la matière organique contenant de l'azote, par le processus d'ammonification et de nitrification, en son produit final, le nitrate inorganique.
CE
x
(concentration produisant x % d'effet): concentration de la substance d'essai dans le sol qui produit x pour cent d'inhibition de la transformation de l'azote en nitrate.
CE
50
(concentration produisant 50 % d'effet): concentration de la substance d'essai dans le sol qui produit 50 pour cent (50 %) d'inhibition de la transformation de l'azote en nitrate.
1.3. SUBSTANCES DE RÉFÉRENCE
Aucune.
1.4. PRINCIPE DE LA MÉTHODE
Le sol tamisé est amendé avec de la farine végétale en poudre puis traité avec la substance d'essai ou laissé non traité (témoin). Si l'essai porte sur des produits agrochimiques, il est recommandé d'utiliser au moins deux concentrations d'essai et celles-ci doivent être choisies en fonction de la concentration maximale prévisible dans le champ. Au bout de 0, 7, 14 et 28 jours d'incubation, des échantillons de sol traité et des échantillons témoins sont extraits avec un solvant approprié, et les quantités de nitrate présentes dans les extraits sont déterminées. Le taux de formation de nitrate dans les échantillons traités est comparé au taux des échantillons témoins, et l'écart en pourcentage entre l'échantillon traité et l'échantillon témoin est calculé. Tous les essais durent au moins 28 jours. Si, au 28e jour, les différences entre les sols traités et les sols non traités sont égales ou supérieures à 25 %, les mesures sont poursuivies pendant une durée maximale de 100 jours. Lorsque l'essai porte sur des produits non agrochimiques, une série de concentrations de substance d'essai sont ajoutées aux échantillons de sol, et les quantités de nitrate formées dans les échantillons traités et les échantillons témoins sont mesurées au bout de 28 jours d'incubation. Les résultats des essais avec une série de concentrations sont analysés à l'aide d'un modèle de régression, et les valeurs CEx sont calculées (CE50, CE25 et/ou CE10). Voir définitions.
1.5. VALIDITÉ DE L'ESSAI
L'évaluation des résultats des essais effectués avec des produits agrochimiques repose sur des différences relativement faibles (valeur moyenne ± 25 %) entre les concentrations en nitrate dans les échantillons témoins et les échantillons traités, de sorte qu'une variabilité importante des témoins peut entraîner des résultats erronés. C'est pourquoi la variation entre les témoins doit être inférieure à ± 15 %.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Appareillage
Des récipients d'essai en matériau chimiquement inerte sont utilisés. Ces récipients doivent avoir une contenance adaptée à la méthode d'incubation utilisée, c'est-à-dire: incubation en vrac ou avec une série d'échantillons individuels (voir le paragraphe 1.7.1.2). Il convient de veiller à la fois à minimiser l'évaporation et à permettre l'échangé gazeux pendant l'essai (les récipients d'essai peuvent par exemple être couverts d'une feuille de polyéthylène perforée). Lorsque l'essai porte sur des substances volatiles, il convient d'utiliser des récipients hermétiques et étanches au gaz. La taille de ces récipients doit être telle qu'ils soient remplis environ au quart de leur contenance avec l'échantillon de sol.
Le matériel courant de laboratoire suivant est utilisé:
|
—
|
agitateur: agitateur mécanique ou équivalent;
|
|
—
|
centrifugeuse (3 000 g) ou appareil de filtration (avec du papier filtre sans nitrate);
|
|
—
|
instrument de sensibilité et de reproductibilité adapté à l'analyse des nitrates.
|
1.6.2. Sélection et nombre de sols
Un seul sol est utilisé. Les caractéristiques recommandées sont les suivantes:
|
—
|
teneur en sable: supérieure à 50 % et inférieure à 75 %,
|
|
—
|
teneur en carbone organique: 0,5 — 1,5 %,
|
|
—
|
la biomasse microbienne doit être mesurée (8) (9) et sa teneur en carbone doit être égale à 1 % au moins du carbone organique total du sol.
|
Dans la plupart des cas, un sol possédant ces caractéristiques représente les conditions les plus défavorables, puisque l'adsorption de la substance chimique de l'essai est minimale et que l'exposition de la microflore est maximale. Il est donc généralement inutile d'effectuer des essais avec d'autres sols. Toutefois, dans certains cas, par exemple lorsque l'utilisation principale prévue de la substance d'essai concerne des sols particuliers comme les sols forestiers acides, ou dans le cas de substances chimiques chargées électrostatiquement, il peut être nécessaire d'utiliser un sol différent.
1.6.3. Prélèvement et stockage des échantillons de sol
1.6.3.1. Prélèvement
Il est nécessaire de fournir des informations détaillées sur l'historique du site de prélèvement. Ces détails doivent comprendre la localisation exacte, le couvert végétal, les dates de traitement aux produits phytosanitaires, les traitements aux engrais organiques et inorganiques, les apports biologiques ou les contaminations accidentelles. Le site choisi pour le prélèvement de sol doit être de nature à permettre une utilisation de longue durée. Les pâturages permanents, les champs plantés de céréales annuelles (à l'exception du maïs) ou à ensemencement dense en engrais verts conviennent. Le site de prélèvement retenu ne doit pas avoir été traité avec des produits phytosanitaires pendant un an au moins avant le prélèvement des échantillons. De même, aucun engrais organique ne doit avoir été appliqué pendant au moins six mois. L'utilisation d'engrais minéraux est acceptable uniquement si elle satisfait aux exigences de la culture et aucun échantillon de sol ne doit être prélevé que trois mois au moins après l'application d'engrais. L'utilisation de sol traité avec des engrais ayant des effets biocides connus (comme le cyanamide calcique) doit être évitée.
Il convient d'éviter de prélever des échantillons pendant ou immédiatement après de longues périodes (plus de 30 jours) de sécheresse ou de saturation en eau. En ce qui concerne les sols labourés, les échantillons doivent être prélevés à une profondeur de 0 à 20 cm. En ce qui concerne les herbages (pâture) ou les sols qui ne sont pas labourés pendant de longues périodes (au moins une période végétative), la profondeur maximale du prélèvement peut être légèrement supérieure à 20 cm (25 cm par exemple).
Les échantillons de sol doivent être transportés dans des récipients et dans des conditions de température de nature à garantir que les propriétés initiales du sol ne soient pas altérées de manière significative.
1.6.3.2. Stockage
Il est préférable d'utiliser des sols qui viennent d'être extraits du site. S'il n'est pas possible d'éviter le stockage dans le laboratoire, les sols peuvent être entreposés dans le noir à 4 ± 2 oC pendant une durée maximale de trois mois. Pendant la durée de stockage des sols, des conditions aérobies doivent être assurées. Si les sols sont prélevés dans une zone où il gèle au moins trois mois par an, il peut être envisagé de stocker le sol pendant six mois entre moins 18 oC et moins 22 oC. La biomasse microbienne des sols stockés est mesurée avant chaque expérience et le carbone présent dans la biomasse doit être égal à 1 % au moins de la teneur totale en carbone organique du sol (voir le paragraphe 1.6.2).
1.6.4 Manipulation et préparation du sol pour l'essai
1.6.4.1 Pré-incubation
Si le sol a été stocké (voir le paragraphe 1.6.3.2), il est recommandé de procéder à une pré-incubation pendant 2 à 28 jours. La température et la teneur en eau du sol pendant la pré-incubation doivent être les mêmes que celles utilisées pendant l'essai (voir les paragraphes 1.6.4.2 et 1.7.1.3).
1.6.4.2 Propriétés physico-chimiques
Le sol est débarrassé manuellement des gros objets (pierres, débris végétaux, etc.) puis tamisé humide sans séchage excessif jusqu'à obtention d'une granulométrie inférieure ou égale à 2 mm. La teneur en eau de l'échantillon de sol doit être ajustée avec de l'eau distillée ou désionisée à une valeur située entre 40 % et 60 % de la capacité maximale de rétention d'eau.
1.6.4.3 Amendement à l'aide d'un substrat organique
Le sol doit être amendé à l'aide d'un substrat organique approprié, comme de la farine de luzerne verte en poudre (composant principal: Medicago sativa) avec un rapport C/N situé entre 12/1 et 16/1. Le rapport luzerne-sol recommandé est de 5 g de luzerne par kilogramme de sol (poids sec).
1.6.5 Préparation de la substance d'essai à appliquer au sol
La substance d'essai est généralement appliquée à l'aide d'un excipient. L'excipient peut être de l'eau (pour les substances solubles dans l'eau) ou un solide inerte comme du sable de quartz fin (granulométrie: 0,1-0,5mm). Il convient d'éviter les excipients liquides autres que l'eau (solvants organiques tels que l'acétone ou le chloroforme) car ils peuvent endommager la microflore. Si l'on utilise du sable comme excipient, celui-ci peut être traité par la substance d'essai dissoute ou en suspension dans un solvant approprié. Dans ce cas, le solvant doit être éliminé par évaporation avant de mélanger l'excipient avec le sol. Pour obtenir une répartition optimale de la substance d'essai dans le sol, un rapport de 10 g de sable par kilogramme de sol (poids sec) est recommandé. Les échantillons témoins sont traités avec une quantité équivalente d'eau et/ou de sable de quartz uniquement.
Lorsque l'on teste des substances chimiques volatiles, il convient d'éviter dans la mesure du possible les pertes pendant le traitement et il convient d'essayer de veiller à la répartition homogène dans l'échantillon de sol (la substance d'essai doit être injectée dans le sol en plusieurs endroits).
1.6.6 Concentrations d'essai
Si l'essai porte sur des produits agrochimiques, il convient d'utiliser au moins deux concentrations. La concentration la plus faible doit correspondre au moins à la quantité maximale susceptible de pénétrer dans le sol dans des conditions réelles alors que la concentration la plus élevée doit être un multiple de la concentration la plus faible. Les concentrations de substance d'essai ajoutées au sol sont calculées en supposant une incorporation uniforme à une profondeur de 5 cm et une densité apparente du sol de 1,5. Dans le cas des produits agrochimiques appliqués directement sur le sol, ou des substances chimiques dont on peut prévoir la quantité qui pénètre dans le sol, les concentrations d'essai recommandées sont égales à la concentration maximale prévisible dans l'environnement (CPE) et cinq fois cette concentration. Les substances qui doivent être appliquées sur le sol plusieurs fois au cours d'une même saison doivent être testées à des concentrations calculées en multipliant la CPE par le nombre maximal d'applications prévu. La concentration la plus élevée testée ne doit toutefois pas dépasser dix fois la dose maximale d'une application. Si l'essai porte sur des produits non agrochimiques, une série géométrique de cinq concentrations au moins est utilisée. Les concentrations testées doivent couvrir la gamme nécessaire pour déterminer les valeurs CEx.
1.7. EXÉCUTION DE L'ESSAI
1.7.1. Conditions expérimentales
1.7.1.1. Traitement et contrôle
Si l'essai porte sur des produits agrochimiques, le sol est divisé en trois fractions de poids égal. Deux fractions sont mélangées à l'excipient contenant le produit, et la troisième avec l'excipient sans produit (témoin). Il est recommandé de préparer un minimum de trois réplicats de sol traité et non traité. Si l'essai porte sur des produits non agrochimiques, le sol est divisé en six fractions de poids égal. Cinq échantillons sont mélangés avec l'excipient contenant la substance d'essai, et le sixième est mélangé avec l'excipient sans la substance chimique. Il est recommandé de préparer trois réplicats de sol traité et de témoins. Il convient de veiller à la répartition homogène de la substance d'essai dans les échantillons de sol traité. Pendant le mélange, il convient d'éviter la compression ou le compactage de l'échantillon de sol.
1.7.1.2. Incubation des échantillons de sol
L'incubation des échantillons de sol peut être effectuée de deux façons: sous forme d'échantillons en vrac de sol traité et de sol non traité ou sous forme d'une série de sous-échantillons individuels et de taille identique de chaque sol traité et non traité. Toutefois, lorsque l'essai porte sur des substances volatiles, l'essai doit être effectué uniquement avec une série de sous-échantillons individuels. Lorsque les sols sont incubés en vrac, de grandes quantités de chaque sol traité et non traité sont préparées et les sous-échantillons à analyser sont prélevés en fonction des besoins pendant l'essai. La quantité préparée initialement pour chaque traitement et pour le contrôle dépend de la taille des sous-échantillons, du nombre de réplicats utilisés pour l'analyse et du nombre maximal de temps de prélèvement. Les sols incubés en vrac doivent être soigneusement mélangés avant de procéder au sous-échantillonnage. Lorsque les sols sont incubés sous forme d'échantillons individuels, on divise chaque sol en vrac traité et non traité dans le nombre requis de sous-échantillons, et ceux-ci sont utilisés selon les besoins. Dans les expériences où l'on peut prévoir plus de deux temps de prélèvement, une quantité suffisante de sous-échantillons doit être préparée pour tenir compte de tous les réplicats et de tous les temps de prélèvement. Au moins trois exemplaires d'échantillons du sol d'essai doivent être incubés en milieu aérobie (voir le paragraphe 1.7.1.1). Pendant tous les essais, il convient d'utiliser des récipients appropriés comportant-un espace libre suffisant afin d'éviter le développement de conditions anaérobies. Lorsque l'essai porte sur des substances volatiles, il doit être effectué uniquement avec une série de sous-échantillons individuels.
1.7.1.3. Conditions et durée de l'essai
L'essai est effectué dans le noir à température ambiante de 20 ± 2 oC. Pendant l'essai, la teneur en eau des échantillons de sol doit être maintenue entre 40 % et 60 % de la capacité maximale de rétention d'eau du sol (voir le paragraphe 1.6.4.2) avec une marge de ± 5 %. De l'eau distillée, désionisée peut être ajoutée si nécessaire.
La durée minimale de l'essai est de 28 jours. Si l'essai porte sur des produits agrochimiques, on compare les taux de formation de nitrate dans les échantillons traités et dans les échantillons témoins. Si ceux-ci diffèrent de plus de 25 % au jour 28, on poursuit l'essai jusqu'à obtenir une différence égale ou inférieure à 25 %, ou pendant une durée maximale de 100 jours. En ce qui concerne les produits non agrochimiques, l'essai est arrêté au bout de 28 jours. Au jour 28, les quantités de nitrate dans les échantillons traités et les échantillons témoins sont déterminées et les valeurs CEx sont calculées.
1.7.2. Prélèvement et analyse des sols
1.7.2.1. Échelonnement des prélèvements
Si l'essai porte sur des produits agrochimiques, les échantillons de sol font l'objet d'une analyse de mesure du nitrate aux jours 0, 7, 14 et 28. S'il est nécessaire de prolonger l'essai, de nouvelles mesures doivent être effectuées tous les 14 jours après le jour 28.
Lorsque l'essai porte sur des produits non agrochimiques, au moins cinq concentrations d'essai sont utilisées et les échantillons de sol sont analysés pour mesurer le nitrate au début (jour 0) et à la fin de la période d'exposition (28 jours). Une mesure intermédiaire, par exemple au jour 7, peut être ajoutée si nécessaire. Les données obtenues au jour 28 sont utilisées pour déterminer la valeur CEx de la substance chimique. Si on le souhaite, on peut utiliser les données des témoins du jour 0 afin de mesurer la quantité initiale de nitrate dans le sol.
1.7.2.2. Analyse des échantillons de sol
La quantité de nitrate formé dans chaque échantillon traité et dans chaque témoin est déterminée à chaque temps de prélèvement. Le nitrate est extrait du sol en agitant les échantillons en présence d'un solvant d'extraction, par exemple, une solution de 0,1 M de chlorure de potassium. Un rapport de 5 ml de solution de KCl par gramme d'équivalent poids sec de sol est recommandé. Afin d'optimiser l'extraction, les récipients contenant le sol et la solution d'extraction ne doivent pas être remplis à plus de la moitié de leur contenance. Les mélanges sont agités à 150 tpm pendant 60 minutes. Les mélanges sont centrifugés ou filtrés et les phases liquides sont analysées pour mesurer le nitrate. Les extraits liquides débarrassés de particules peuvent être stockés à moins 20 ± 5 oC pendant une durée pouvant aller jusqu'à six mois avant d'être analysés.
2. DONNÉES
2.1. TRAITEMENT DES RÉSULTATS
Si l'essai porte sur des produits agrochimiques, la quantité de nitrate formé dans chaque échantillon de sol doit être enregistrée, et les valeurs moyennes de tous les réplicats doivent être présentées sous forme de tableau. Les taux de transformation de l'azote doivent être évalués à l'aide de méthodes statistiques appropriées et généralement reconnues (F-test, seuil de signification de 5 %). Les quantités de nitrate formé sont exprimées en mg nitrate/kg poids sec/jour. On compare le taux de formation de nitrate dans chaque échantillon traité avec le taux dans le témoin, et on calcule l'écart en pourcentage par rapport au témoin.
Si l'essai porte sur des produits non agrochimiques, la quantité de nitrate formé dans chaque réplicat est déterminée, et une courbe dose-réponse est établie pour déterminer les valeurs CEx. Les quantités de nitrate (mg nitrate/kg poids sec de sol) trouvées dans les échantillons traités au bout de 28 jours sont comparées aux quantités observées dans les échantillons témoins. À partir de ces résultats, on calcule le pourcentage des valeurs d'inhibition pour chaque concentration d'essai. Une courbe de pourcentages en fonction de la concentration est établie, et on utilise ensuite une méthode statistique pour calculer les valeurs CEx. Les seuils de confiance (p = 0,95) des CEx calculées sont également déterminés à l'aide des méthodes standards (10) (11) (12).
Les substances d'essai qui contiennent d'importantes quantités d'azote peuvent être à l'origine des quantités de nitrate qui se forment pendant l'essai. Si ces substances sont testées à une concentration élevée (par exemple, dans le cas des substances chimiques destinées à être utilisées en applications répétées), des contrôles appropriés doivent être prévus dans l'essai (sol plus substance d'essai mais sans farine végétale). Les résultats de ces contrôles doivent être pris en compte dans le calcul des valeurs CEx.
2.2. INTERPRÉTATION DES RÉSULTATS
Lorsqu'on évalue les résultats d'essais avec des produits agrochimiques, et que la différence de taux de formation de nitrate entre l'échantillon le moins traité (concentration maximale prévisible) et le témoin est égale ou inférieure à 25 % quel que soit le temps de prélèvement après le jour 28, le produit peut être considéré comme n'ayant aucune influence à long terme sur la transformation de l'azote dans le sol. Lorsqu'on évalue les résultats d'essais avec des substances chimiques autres que les produits agrochimiques, on utilise les valeurs CE50, CE25 et/ou CE10.
3. RAPPORT D'ESSAI
Le rapport d'essai doit comporter les informations suivantes:
|
|
Identification complète du sol utilisé à savoir:
|
—
|
coordonnées géographiques du site (latitude, longitude),
|
|
—
|
information sur l'historique du site (couvert végétal, traitement aux produits phytosanitaires, traitement aux engrais, contamination accidentelle, etc.),
|
|
—
|
type d'utilisation (sol agricole, forestier, etc.),
|
|
—
|
profondeur du prélèvement (cm),
|
|
—
|
teneur en sable/limon/argile (% poids sec),
|
|
—
|
valeurs du pH (dans l'eau),
|
|
—
|
teneur en carbone organique (% poids sec),
|
|
—
|
teneur en azote (% poids sec),
|
|
—
|
concentration initiale en nitrate (mg nitrate/kg poids sec),
|
|
—
|
capacité d'échange cationique (mmol/kg),
|
|
—
|
biomasse microbienne en pourcentage du carbone organique total,
|
|
—
|
référence des méthodes utilisées pour la détermination de chaque paramètre,
|
|
—
|
toutes les informations relatives au prélèvement et au stockage des échantillons de sol,
|
|
—
|
détails de la pré-incubation du sol s'il y a lieu.
|
|
|
|
Substance d'essai:
|
—
|
nature physique et, s'il y a lieu, propriétés physico-chimiques,
|
|
—
|
données d'identification des substances chimiques, s'il y a lieu, comprenant la formule structurale, la pureté (pour les produits phytosanitaires, le pourcentage de produit actif), la teneur en azote.
|
|
|
|
Substrat:
|
—
|
composition (farine de luzerne, farine de luzerne verte),
|
|
—
|
teneur en carbone, azote (% poids sec),
|
|
|
|
Conditions de l'essai:
|
—
|
détails de la modification du sol avec un substrat organique,
|
|
—
|
nombre de concentrations de la substance d'essai utilisée et, le cas échéant, justification des concentrations sélectionnées,
|
|
—
|
procédure d'application de la substance d'essai au sol,
|
|
—
|
température d'incubation,
|
|
—
|
teneur en eau du sol au début et pendant le déroulement de l'essai,
|
|
—
|
méthode d'incubation du sol utilisée (en vrac ou en série de sous-échantillons individuels),
|
|
—
|
méthode utilisée pour l'extraction du nitrate du sol.
|
|
|
|
Résultats:
|
—
|
procédure analytique et matériel utilisé pour analyser le nitrate,
|
|
—
|
tableaux indiquant les valeurs individuelles et moyennes des mesures du nitrate,
|
|
—
|
variation entre réplicats dans les échantillons traités et dans les échantillons témoins,
|
|
—
|
explications des corrections apportées aux calculs, le cas échéant,
|
|
—
|
la variation en pourcentage des taux de formation de nitrate à chaque temps de prélèvement ou, le cas échéant, la valeur CE50 avec une limite de confiance de 95 pour cent, les autres valeurs CEx (CE25 ou CE10) avec des intervalles de confiance, et un graphique de la courbe dose-réponse,
|
|
—
|
traitement statistique des résultats,
|
|
—
|
toutes informations et observations utiles pour l'interprétation des résultats.
|
|
4 RÉFÉRENCES
|
(1)
|
EPPO (1994). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora. EPPO Bulletin 24, p. 1-16, 1994.
|
|
(2)
|
BBA (1990). Effects on the Activity of the Soil Microflora. BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1-1 (2nd eds., 1990).
|
|
(3)
|
EPA (1987). Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule. September 28, 1987.
|
|
(4)
|
SET AC-Europe (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides, Ed. M.R. Lynch, Pub. SETAC-Europe, Bruxelles.
|
|
(5)
|
ISO/DIS 14238 (1995). Soil Quality — Determination of Nitrogen Mineralisation and Nitrification in Soils and the Influence of Chemicals on these Processes. Technical Committee ISO/TC 190/SC 4: Soil Quality - Biological Methods.
|
|
(6)
|
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italie, p. 18-20, January 1995.
|
|
(7)
|
ISO 10381-6 (1993). Soil quality — Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory.
|
|
(8)
|
ISO 14240-1 (1997). Soil quality — Determination of soil microbial biomass — Part 1: Substrate-induced respiration method.
|
|
(9)
|
ISO 14240-2 (1997). Soil quality — Determination of soil microbial biomass — Part 2: Fumigation-extraction method.
|
|
(10)
|
Litchfield, J.T. and WUcoxon F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol, and Exper. Ther., 96, p. 99-113.
|
|
(11)
|
Finney, D.J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New-York.
|
|
(12)
|
Finney, D.J. (1978). Statistical Methods in biological Assay. Griffin, Weycombe, UK.
|
C.22. MICRO-ORGANISMES DU SOL: ESSAI DE TRANSFORMATION DU CARBONE
1. MÉTHODE
La présente méthode d'essai reprend l'essai TG 217 (2000) de l'OCDE.
1.1. INTRODUCTION
La présente méthode d'essai décrit une méthode de laboratoire conçue pour étudier les effets à long terme des substances chimiques, après une exposition unique, sur l'activité de transformation du carbone par les micro-organismes du sol. L'essai s'appuie principalement sur les recommandations de l'Organisation européenne et méditerranéenne pour la protection des plantes (1). Toutefois, d'autres lignes directrices, notamment celles du Biologische Bundesaristalt allemand (2), de l'agence de protection de l'environnement des États-Unis (3) et de la SETAC (4), ont également été prises en considération. Lors d'un atelier de l'OCDE sur la sélection des sols et des sédiments, qui s'est tenu à Belgirate, Italie, en 1995 (5), le nombre et le type de sols à utiliser dans le cadre de cet essai ont été définis. Les recommandations relatives au prélèvement, à la manipulation et au stockage des échantillons de sol s'appuient sur des lignes directrices de l'ISO (6) et sur les recommandations de l'atelier de Belgirate.
Pour déterminer et évaluer les caractéristiques toxiques des substances d'essai, il peut être nécessaire de déterminer leurs effets sur l'activité microbienne du sol, par exemple lorsqu'il est nécessaire de disposer de données sur les effets secondaires potentiels des produits phytosanitaires sur la microflore du sol ou bien en cas de risque d'exposition des micro-organismes du sol à des substances chimiques autres que les produits phytosanitaires. L'essai de transformation du carbone est effectué pour déterminer les effets de ces substances chimiques sur la microflore du sol. Si l'essai porte sur des produits agrochimiques (produits phytosanitaires, engrais, produits chimiques sylvicoles), deux essais sont effectués: l'essai de transformation du carbone et l'essai de transformation de l'azote. S'il s'agit d'autres substances que les produits agrochimiques, l'essai de transformation de l'azote suffit. Toutefois, si les valeurs CE50 de l'essai de transformation de l'azote relatives à ces substances chimiques correspondent à celles des inhibiteurs de la nitrification (nitrapyrine) commerciaux, un essai de transformation du carbone peut être effectué afin d'obtenir des informations complémentaires.
Les sols sont formés de composés vivants et non vivants qui existent sous forme de mélanges complexes et hétérogènes. Les micro-organismes jouent un rôle important dans la décomposition et la transformation de la matière organique en sol fertile et un grand nombre d'espèces contribuent aux différents aspects de la fertilisation des sols. Toute interférence à long terme avec ces processus biochimiques risque de perturber le cycle des éléments nutritifs et par conséquent d'altérer la fertilité du sol. La transformation du carbone et de l'azote se produit dans tous les sols fertiles. Bien que les colonies microbiennes responsables de ces processus diffèrent d'un sol à un autre, les voies de transformation sont identiques pour l'essentiel.
La méthode d'essai décrite ici est conçue pour détecter les effets nocifs à long terme d'une substance sur le processus de transformation du carbone à la surface aérobie des sols. L'essai est sensible aux changements de taille et d'activité des colonies microbiennes responsables de la transformation du carbone puisqu'il soumet ces colonies à la fois au stress chimique et à la carence en carbone. Un sol sableux pauvre en matière organique est utilisé. Ce sol est traité avec la substance d'essai et il est incubé dans des conditions permettant un métabolisme microbien rapide. Dans ces conditions, les sources de carbone présentes dans le sol sont rapidement épuisées. Cela provoque la carence en carbone qui tue à la fois les cellules microbiennes et qui induit la dormance et/ou la sporulation. Si la durée de l'essai est supérieure à 28 jours, on peut mesurer la somme de ces réactions dans les témoins (sol non traité) en mesurant la perte progressive de biomasse microbienne active métaboliquement (7). Si la biomasse d'un sol carence en carbone, dans les conditions de l'essai, est affectée par la présence d'une substance chimique, celle-ci peut ne pas retrouver le même niveau que l'échantillon témoin. C'est pourquoi la perturbation provoquée par la substance d'essai à n'importe quel moment de l'essai perdurera souvent jusqu'à la fin de l'essai.
Les essais à partir desquels la présente méthode d'essai a été mise au point étaient conçus principalement pour des substances dont il est possible d'estimer la quantité qui pénètre dans le sol. C'est le cas par exemple des produits phytosanitaires dont la dose d'application dans le champ est connue. Pour les produits agrochimiques, il suffit de pratiquer un essai sur deux doses correspondant au taux d'application prévu ou estimé. Les produits agrochimiques peuvent être testés sous forme de principes actifs (p.a.) ou de produits formulés. Toutefois, l'essai n'est pas limité aux substances chimiques dont les concentrations dans l'environnement peuvent être estimées. En modifiant à la fois la quantité de substance d'essai appliquée au sol, et le mode d'évaluation des données, on peut également utiliser l'essai pour les substances chimiques dont on ne connaît pas la quantité susceptible d'atteindre le sol. C'est pourquoi, dans le cas des produits non agrochimiques, les effets d'une série de concentrations sur la transformation du carbone sont déterminés. Les résultats de ces essais sont utilisés pour préparer une courbe dose-réponse et calculer les valeurs CEX, où x est défini comme le pourcentage d'effet.
1.2. DÉFINITIONS
Transformation du carbone: dégradation par des micro-organismes de la matière organique en son produit final, le dioxyde de carbone inorganique.
CE
X
(concentration produisant x % d'effet): concentration de la substance d'essai dans le sol qui produit x % d'inhibition de la transformation du carbone en dioxyde de carbone.
CE
50
(concentration produisant 50 % d'effet): concentration de la substance d'essai dans le sol qui produit 50 % d'inhibition de la transformation du carbone en dioxyde de carbone.
1.3. SUBSTANCES DE RÉFÉRENCE
Aucune.
1.4. PRINCIPE DE LA MÉTHODE
Le sol tamisé est traité avec la substance d'essai ou laissé non traité (témoin). Si l'essai porte sur des produits agrochimiques, il est recommandé d'utiliser au moins deux concentrations d'essai et celles-ci doivent être choisies en fonction de la concentration maximale prévisible dans le champ. Au bout de 0, 7, 14 et 28 jours d'incubation, les échantillons de sol traité et les échantillons témoins sont mélangés avec du glucose, et les taux de respiration induits par le glucose sont mesurés pendant 12 heures consécutives. Les taux de respiration sont exprimés en dioxyde de carbone dégagé (mg dioxyde de carbone/kg sol sec/h) ou oxygène consommé (mg oxygène/kg sol/h). On compare le taux moyen de respiration dans les échantillons de sol traité avec celui des échantillons témoins et on calcule l'écart en pourcentage entre l'échantillon traité et l'échantillon témoin. Tous les essais ont une durée de 28 jours au moins. Si, au 28e jour, les différences entre les sols traités et les sols non traités sont égales ou supérieures à 25 %, les mesures se poursuivent à intervalles de 14 jours pendant une durée maximale de 100 jours. Si l'essai porte sur des substances chimiques autres que les produits agrochimiques, une série de concentrations de la substance d'essai est ajoutée aux échantillons de sol, et les taux de respiration induits par le glucose (moyenne des quantités de dioxyde de carbone formé ou l'oxygène consommé) sont mesurés au bout de 28 jours. Les résultats des essais effectués avec une série de concentrations sont analysés à l'aide d'un modèle de régression, et les valeurs CEx sont calculées (CE50, CE25 et/ou CE10). Voir définitions.
1.5. VALIDITÉ DE L'ESSAI
L'évaluation des résultats des essais effectués avec des produits agrochimiques repose sur des différences relativement faibles (valeur moyenne ± 25 %) entre le dioxyde de carbone dégagé ou l'oxygène consommé dans (ou par) les échantillons témoins et les échantillons de sol traité, de sorte qu'une variabilité importante des témoins peut entraîner des résultats erronés. C'est pourquoi la variation entre les témoins doit être inférieure à ± 15 %.
1.6. DESCRIPTION DE LA MÉTHODE
1.6.1. Appareillage
Des récipients d'essai en matériau chimiquement inerte sont utilisés. Ces récipients doivent avoir une contenance adaptée à la méthode d'incubation utilisée, c'est-à-dire incubation en vrac ou avec une série d'échantillons individuels (voir le point 1.7.1.2). Il convient de veiller à la fois à minimiser l'évaporation et à permettre l'échange gazeux pendant l'essai (les récipients d'essai peuvent par exemple être couverts d'une feuille de polyéthylène perforée). Lorsque l'essai porte sur des substances volatiles, il convient d'utiliser des récipients hermétiques et étanches. La taille de ces récipients doit être telle qu'ils soient remplis environ au quart de leur contenance avec l'échantillon de sol.
Pour déterminer la respiration induite par le glucose, des systèmes d'incubation et des instruments de mesure de la production de dioxyde de carbone ou de la consommation d'oxygène sont nécessaires. On trouve des exemples de ces systèmes et de ces instruments dans la littérature (8) (9) (10) (11).
1.6.2. Sélection et nombre de sols
Un seul sol est utilisé. Les caractéristiques recommandées sont les suivantes:
|
—
|
teneur en sable: supérieure à 50 % et inférieure à 75 %,
|
|
—
|
teneur en carbone organique: 0,5-1,5 %,
|
|
—
|
la biomasse microbienne doit être mesurée (12) (13) et sa teneur en carbone doit être égale à 1 % au moins du carbone organique total du sol.
|
Dans la plupart des cas, un sol possédant ces caractéristiques correspond aux conditions les plus défavorables, puisque l'adsorption de la substance chimique de l'essai est minimale et que l'exposition de la microflore est maximale. Il est donc généralement inutile d'effectuer des essais avec d'autres sols. Toutefois, dans certains cas, par exemple lorsque l'utilisation principale prévue de la substance d'essai concerne des sols particuliers comme les sols forestiers acides, ou dans le cas de substances chimiques chargées électrostatiquement, il peut être nécessaire d'utiliser un sol différent.
1.6.3. Prélèvement et stockage des échantillons de sol
1.6.3.1. Prélèvement
Il est nécessaire de fournir des informations détaillées sur l'historique du site de prélèvement. Ces détails doivent comprendre la localisation exacte, le couvert végétal, les dates de traitement avec des produits phytosanitaires, les traitements aux engrais organiques et inorganiques, les apports biologiques ou les contaminations accidentelles. Le site choisi pour le prélèvement de sol doit être de nature à permettre une utilisation de longue durée. Les pâturages permanents, les champs plantés de céréales annuelles (à l'exception du maïs) ou à ensemencement dense en engrais verts conviennent. Le site de prélèvement retenu ne doit pas avoir été traité avec des produits phytosanitaires pendant un an au moins avant les prélèvements. De même, aucun engrais organique ne doit avoir été appliqué pendant au moins six mois. L'utilisation d'engrais minéraux est acceptable uniquement si elle satisfait aux exigences de la culture, et les échantillons de sol ne doivent être prélevés qu'au moins trois mois après l'application d'engrais. L'utilisation de sol traité avec des engrais aux effets biologiques connus (cyanamide calcique) doit être évitée.
Il convient d'éviter de prélever des échantillons pendant ou immédiatement après de longues périodes (plus de 30 jours) de sécheresse ou de saturation en eau. En ce qui concerne les sols labourés, les échantillons doivent être prélevés à une profondeur de 0 à 20 cm. En ce qui concerne les herbages (pâture) ou les sols qui ne sont pas labourés pendant de longues périodes (au moins une période végétative), la profondeur maximale du prélèvement peut être légèrement supérieure à 20 cm (25 cm par exemple). Les échantillons de sol doivent être transportés dans des récipients et dans des conditions de température de nature à garantir que les propriétés initiales du sol ne soient pas altérées de manière significative.
1.6.3.2. Stockage
Il est préférable d'utiliser des sols qui viennent d'être extraits du site. S'il n'est pas possible d'éviter le stockage dans le laboratoire, les sols peuvent être entreposés dans le noir à 4 ± 2 oC pendant une durée maximale de trois mois. Pendant la durée de stockage des sols, des conditions aérobies doivent être assurées. Si les sols sont prélevés dans une zone où il gèle au moins trois mois par an, il peut être envisagé de stocker le sol pendant six mois à — 18 oC. La biomasse microbienne des sols stockés est mesurée avant chaque expérience et le carbone présent dans la biomasse doit être égal à 1 % au moins de la teneur totale en carbone organique du sol (voir le point 1.6.2).
1.6.4. Manipulation et traitement du sol pour l'essai
1.6.4.1. Préincubation
Si le sol a été stocké (voir les points 1.6.4.2 et 1.7.1.3), il est recommandé de procéder à une préincubation pendant 2 à 28 jours. La température et la teneur en eau du sol pendant la préincubation doivent être les mêmes que celles utilisées pendant l'essai (voir les points 1.6.4.2 et 1.7.1.3).
1.6.4.2. Propriétés physico-chimiques
Le sol est débarrassé manuellement des gros objets (pierres, débris végétaux, etc.) puis tamisé humide sans séchage excessif jusqu'à obtention d'une granulométrie inférieure ou égale à 2 mm. La teneur en eau de l'échantillon doit être ajustée avec de l'eau distillée ou désionisée à une valeur située entre 40 % et 60 % de la capacité maximale de rétention d'eau.
1.6.5. Préparation de la substance d'essai à appliquer au sol
La substance d'essai est généralement appliquée à l'aide d'un excipient. L'excipient peut être de l'eau (pour les substances solubles dans l'eau) ou un solide inerte comme du sable de quartz fin (granulométrie: 0,1-0,5 mm). Il convient d'éviter les excipients liquides autres que l'eau (solvants organiques tels que l'acétone ou le chloroforme), car ils peuvent endommager la microflore. Si l'on utilise du sable comme excipient, celui-ci peut être traité par la substance d'essai dissoute ou en suspension dans un solvant approprié. Dans ce cas, le solvant doit être éliminé par évaporation avant de mélanger l'excipient avec le sol. Pour obtenir une répartition optimale de la substance d'essai dans le sol, un rapport de 10 g de sable par kilogramme de sol (poids sec) est recommandé. Les échantillons témoins sont traités avec une quantité équivalente d'eau et/ou de sable de quartz uniquement.
Lorsque l'on teste des substances chimiques volatiles, il convient d'éviter dans la mesure du possible les pertes pendant le traitement et il convient d'essayer de veiller à la répartition homogène dans l'échantillon de sol (la substance d'essai doit être injectée dans le sol en plusieurs endroits).
1.6.6. Concentrations d'essai
Si l'essai porte sur des produits phytosanitaires ou sur d'autres substances chimiques dont les concentrations dans l'environnement peuvent être estimées, il convient d'utiliser au moins deux concentrations. La concentration la plus faible doit correspondre au moins à la quantité maximale susceptible de pénétrer dans le sol dans des conditions réelles alors que la concentration la plus élevée doit être un multiple de la concentration la plus faible. Les concentrations de substance d'essai ajoutées au sol sont calculées en supposant une incorporation uniforme à une profondeur de 5 cm et une densité apparente du sol de 1,5. Dans le cas des produits agrochimiques appliqués directement sur le sol, ou des substances chimiques dont on peut prévoir la quantité qui pénètre dans le sol, les concentrations d'essai recommandées sont égales à la concentration maximale prévisible dans l'environnement (CPE) et cinq fois cette concentration. Les substances qui doivent être appliquées sur le sol plusieurs fois au cours d'une même saison doivent être testées à des concentrations calculées en multipliant la CPE par le nombre maximal d'applications prévues. La concentration la plus élevée testée ne doit toutefois pas dépasser dix fois la dose maximale d'application unique.
Si l'essai porte sur des produits non agrochimiques, une série géométrique de cinq concentrations au moins est utilisée. Les concentrations testées doivent couvrir la gamme nécessaire pour déterminer les valeurs CEx.
1.7. EXÉCUTION DE L'ESSAI
1.7.1. Conditions expérimentales
1.7.1.1. Traitement et contrôle
Si l'essai porte sur des produits agrochimiques, le sol est divisé en trois fractions de poids égal. Deux fractions sont mélangées avec l'excipient contenant le produit, et la troisième avec l'excipient sans produit (témoin). Il est recommandé de préparer un minimum de trois réplicats de sol traité et non traité. Si l'essai porte sur des produits non agrochimiques, le sol est divisé en six fractions de poids égal. Cinq échantillons sont mélangés avec l'excipient contenant la substance d'essai, et le sixième est mélangé avec l'excipient sans la substance chimique. Il est recommandé de préparer trois réplicats de sol traité et de témoins. Il convient de veiller à la répartition homogène de la substance d'essai dans les échantillons de sol traité. Pendant le mélange, il convient d'éviter la compression ou le compactage du sol.
1.7.1.2 Incubation des échantillons de sol
L'incubation des échantillons de sol peut être effectuée de deux façons: sous forme d'échantillons en vrac de sol traité et de sol non traité ou sous forme d'une série de sous-échantillons individuels et de taille identique de chaque sol traité et non traité. Toutefois, lorsque l'essai porte sur des substances volatiles, l'essai doit être effectué uniquement avec une série de sous-échantillons individuels. Lorsque les sols sont incubés en vrac, de grandes quantités de chaque sol traité et non traité sont préparées et les sous-échantillons à analyser sont prélevés en fonction des besoins pendant l'essai. La quantité préparée initialement pour chaque traitement et pour le contrôle dépend de la taille des sous-échantillons, du nombre de réplicats utilisés pour l'analyse et du nombre maximal de temps de prélèvement. Les sols incubés en vrac doivent être soigneusement mélangés avant de procéder au sous-échantillonnage. Lorsque les sols sont incubés sous forme d'échantillons individuels, on divise chaque sol en vrac traité et non traité dans le nombre requis de sous-échantillons, et ceux-ci sont utilisés selon les besoins. Dans les expériences où l'on peut prévoir plus de deux temps de prélèvement, une quantité suffisante de sous-échantillons doit être préparée pour tenir compte de tous les réplicats et de tous les temps de prélèvement. Au moins trois exemplaires de sol d'essai doivent être incubés en milieu aérobie (voir le point 1.7.1.1). Pendant tous les essais, il convient d'utiliser des récipients appropriés comportant un espace libre suffisant afin d'éviter le développement de conditions anaérobies. Lorsque l'essai porte sur des substances volatiles, il doit être effectué uniquement avec une série de sous-échantillons individuels.
1.7.1.3. Conditions et durée de l'essai
L'essai est effectué dans le noir à température ambiante de 20 ± 2 oC. Pendant l'essai, la teneur en eau des échantillons de sol doit être maintenue entre 40 % et 60 % de la capacité maximale de rétention d'eau du sol (voir le point 1.6.4.2) avec une marge de ± 5 %. De l'eau distillée, désionisée peut être ajoutée si nécessaire.
La durée minimale de l'essai est de 28 jours. Si l'essai porte sur des produits agrochimiques, on compare les quantités de dioxyde de carbone dégagé ou d'oxygène consommé dans les échantillons traités et dans les échantillons témoins. Si celles-ci diffèrent de plus de 25 % au jour 28, on poursuit l'essai jusqu'à obtenir une différence égale ou inférieure à 25 %, ou pendant une durée maximale de 100 jours, si celle-ci est la plus courte. Si l'essai porte sur des produits non agrochimiques, l'essai est arrêté au bout de 28 jours. Au jour 28, les quantités de dioxyde de carbone dégagé ou d'oxygène consommé dans les échantillons traités et les échantillons témoins sont déterminées et les valeurs CEx sont calculées.
1.7.2. Prélèvement et analyse des sols
1.7.2.1. Échelonnement des prélèvements
Si l'essai porte sur des produits agrochimiques, les échantillons de sol font l'objet d'une analyse de mesure des taux de respiration induits par le glucose aux jours 0, 7, 14 et 28. S'il est nécessaire de prolonger l'essai, de nouvelles mesures doivent être effectuées tous les 14 jours après le jour 28.
Lorsque l'essai porte sur des produits non agrochimiques, au moins cinq concentrations d'essai sont utilisées et les échantillons de sol sont analysés pour mesurer la respiration induite par le glucose au début (jour 0) et à la fin de la période d'exposition (28 jours). Une mesure intermédiaire, par exemple au jour 7, peut être ajoutée si nécessaire. Les données obtenues au jour 28 sont utilisées pour déterminer la valeur CEx de la substance chimique. Si on le souhaite, on peut utiliser les données des témoins du jour 0 afin d'estimer les quantités initiales de biomasse microbienne métaboliquement active dans le sol (12).
1.7.2.2. Mesure des taux de respiration induits par le glucose
Le taux de respiration induit par le glucose est déterminé dans chaque échantillon de sol traité et dans chaque témoin à chaque temps de prélèvement. Les échantillons de sol sont mélangés avec une quantité de glucose suffisante pour entraîner une réaction respiratoire maximale immédiate. La quantité de glucose nécessaire pour provoquer une réaction respiratoire maximale dans un sol donné peut être déterminée dans un essai préliminaire à l'aide d'une série de concentrations de glucose (14). Toutefois, dans le cas des sols sableux contenant 0,5-1,5 % de carbone organique, 2 000 mg à 4 000 mg de glucose par kg de poids sec sont généralement suffisants. Le glucose peut être réduit en poudre avec du sable de quartz propre (10 g sable/kg poids sec) et mélangé de façon homogène avec le sol.
Les échantillons de sol amendés avec le glucose sont incubés dans un appareil adapté pour mesurer les taux de respiration en continu, toutes les heures, ou toutes les deux heures (voir point 1.6.1) à 20 ± 2 oC. Le dioxyde de carbone dégagé ou l'oxygène consommé est mesuré pendant 12 heures consécutives et les mesures doivent commencer le plus rapidement possible, c'est-à-dire 1 à 2 heures après l'adjonction de glucose. On mesure la quantité totale de dioxyde de carbone dégagé ou d'oxygène consommé pendant les 12 heures et on détermine le taux moyen de respiration.
2 DONNÉES
2.1. TRAITEMENT DES RÉSULTATS
Si l'essai porte sur des produits agrochimiques, le dioxyde de carbone dégagé ou l'oxygène consommé par chaque réplicat doit être enregistré, et les valeurs moyennes de tous les réplicats doivent être présentées sous forme de tableau. Les résultats doivent être évalués à l'aide de méthodes statistiques appropriées et généralement reconnues (F-test, seuil de signification de 5 %). Les taux de respiration induits par le glucose sont exprimés en mg de dioxyde de carbone/kg poids sec/h ou mg oxygène/poids sec/h. Le taux moyen de formation de dioxyde de carbone ou le taux moyen de consommation d'oxygène de chaque échantillon traité est comparé à celui du témoin, et l'écart en pourcentage par rapport au témoin est calculé.
Si l'essai porte sur des produits non agrochimiques, les quantités de dioxyde de carbone dégagé ou d'oxygène consommé dans chaque réplicat sont déterminées, et une courbe dose-réponse est établie pour déterminer les valeurs CEx. Les taux de respiration induits par le glucose (mg de dioxyde de carbone/kg poids sec/h ou mg oxygène/poids sec/h) observés dans les échantillons traités au bout de 28 jours sont comparés à ceux des échantillons témoins. À partir de ces résultats, on calcule le pourcentage des valeurs d'inhibition pour chaque concentration d'essai. Une courbe de pourcentages en fonction de la concentration est établie, et on utilise ensuite une méthode statistique pour calculer les valeurs CEx. Les seuils de confiance (p = 0,95) des CEx calculées sont également déterminés à l'aide des méthodes standard (15) (16) (17).
2.2. INTERPRÉTATION DES RÉSULTATS
Lorsqu'on évalue les résultats d'essais avec des produits agrochimiques, et que la différence de taux de respiration entre l'échantillon le moins traité (concentration maximale prévisible) et l'échantillon témoin est égale ou inférieure à 25 % quel que soit le temps de prélèvement après le jour 28, le produit peut être considéré comme n'ayant aucune influence à long terme sur la transformation du carbone dans le sol. Lorsqu'on évalue les résultats d'essais avec des substances chimiques autres que les produits agrochimiques, on utilise les valeurs CE50, CE25 et/ou CE10.
3. RAPPORT D'ESSAI
RAPPORT DE L'ESSAI
Le rapport de l'essai doit comporter les informations suivantes:
|
|
Identification complète du sol utilisé, à savoir:
|
—
|
coordonnées géographiques du site (latitude, longitude),
|
|
—
|
information sur l'historique du site (couvert végétal, traitement aux produits phytosanitaires, traitement aux engrais, contamination accidentelle, etc.),
|
|
—
|
type d'utilisation (sol agricole, forestier, etc.),
|
|
—
|
profondeur du prélèvement (cm),
|
|
—
|
teneur en sable/limon/argile (% poids sec),
|
|
—
|
teneur en carbone organique (% poids sec),
|
|
—
|
teneur en azote (% poids sec),
|
|
—
|
capacité d'échange cationique (mmol/kg),
|
|
—
|
biomasse microbienne initiale en pourcentage du carbone organique total,
|
|
—
|
référence des méthodes utilisées pour la détermination de chaque paramètre,
|
|
—
|
toutes les informations relatives au prélèvement et au stockage des échantillons de sol,
|
|
—
|
détails de la préincubation du sol s'il y a lieu.
|
|
|
|
Substance d'essai:
|
—
|
nature physique et, s'il y a lieu, propriétés physico-chimiques,
|
|
—
|
données d'identification des substances chimiques, s'il y a lieu, notamment la formule structurale, la pureté (pour les produits phytosanitaires, le pourcentage de produit actif), la teneur en azote.
|
|
|
|
Conditions de l'essai:
|
—
|
détails de la modification du sol avec un substrat organique,
|
|
—
|
nombre de concentrations de la substance d'essai utilisée et, le cas échéant, justification des concentrations sélectionnées,
|
|
—
|
procédure d'application de la substance d'essai au sol,
|
|
—
|
température d'incubation,
|
|
—
|
teneur en eau du sol au début et pendant le déroulement de l'essai,
|
|
—
|
méthode d'incubation du sol utilisée (en vrac ou en série de sous-échantillons individuels),
|
|
|
|
Résultats:
|
—
|
méthode et matériel utilisés pour mesurer les taux de respiration,
|
|
—
|
tableaux indiquant les valeurs individuelles et les valeurs moyennes des quantités de dioxyde de carbone ou d'oxygène,
|
|
—
|
variation entre réplicats dans les échantillons traités et dans les échantillons témoins,
|
|
—
|
explications des corrections apportées aux calculs, s'il y a lieu,
|
|
—
|
la variation en pourcentage des taux de respiration induits par le glucose à chaque temps de prélèvement ou, le cas échéant, la valeur CE50 avec une limite de confiance de 95 %, les autres valeurs CEX (CE25 ou CE10) avec des intervalles de confiance, et un graphique de la courbe dose-réponse,
|
|
—
|
traitement statistique des résultats, s'il y a lieu,
|
|
—
|
toutes informations et observations utiles pour l'interprétation des résultats.
|
|
4. RÉFÉRENCES
|
(1)
|
EPPO (1994). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora. EPPO Bulletin 24, p. 1-16, 1994.
|
|
(2)
|
BBA (1990). Effects on the Activity of the Soil Microflora. BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1-1 (2nd eds., 1990).
|
|
(3)
|
EPA (1987). Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule. September 28, 1987.
|
|
(4)
|
SETAC-Europe (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides, Ed. M.R. Lynch, Pub. SETAC-Europe, Brussels.
|
|
(5)
|
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18-20 January 1995.
|
|
(6)
|
ISO 10381-6 (1993). Soil quality — Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory.
|
|
(7)
|
Anderson, J.P.E. (1987). Handling and Storage of Soils for Pesticide Experiments, in «Pesticide Effects on Soil Microflora». Eds. L. Somerville and M.P. Greaves, Chap. 3, p. 45-60.
|
|
(8)
|
Anderson, J.P.E. (1982). Soil Respiration, in «Methods of Soil Analysis — Part 2: Chemical and Microbiological Properties». Agronomy Monograph No 9. Eds. A.L. Page, R.H. Miller and D.R. Keeney. 41, p. 831-871.
|
|
(9)
|
ISO 11266-1. (1993). Soil Quality — Guidance on Laboratory Tests, for Biodegradation in Soil: Part 1. Aerobic Conditions.
|
|
(10)
|
ISO 14239 (1997E). Soil Quality — Laboratory incubation systems for measuring the mineralization of organic chemicals in soil under aerobic conditions.
|
|
(11)
|
Heinemeyej, O., Insam, H., Kaiser, E.A, and Walenzik, G. (1989). Soil microbial biomass and respiration measurements; an automated technique based on infrared gas analyses. Plant and Soil, 116, p. 77-81.
|
|
(12)
|
ISO 14240-1 (1997). Soil quality — Determination of soil microbial biomass — Part 1: Substrate-induced respiration method.
|
|
(13)
|
ISO 14240-2 (1997). Soil quality — Determination of soil microbial biomass — Part 2: Fumigation-extraction method.
|
|
(14)
|
Malkomes, H.-P. (1986). EinfluB von Glukosemenge auf die Reaktion der Kurzzeit-Atmung im Boden Gegenuber Pflanzenschutzmitteln, Dargestellt am Beispiel eines Herbizide. (Influence of the Amount of Glucose Added to the Soil on the Effect of Pesticides in Short-Term Respiration, using a Herbicide as an Example). Nachrichtenbl. Deut. Pflanzenschutzd., Braunschweig, 38, p. 113-120.
|
|
(15)
|
Litchfield, J.T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol, and Exper. Ther., 96, p. 99-113.
|
|
(16)
|
Finney, D.J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New-York.
|
|
(17)
|
Finney D.J. (1978). Statistical Methods in biological Assay. Griffin, Weycombe, UK.
|
C.23. TRANSFORMATION AÉROBIE ET ANAÉROBIE DANS LE SOL
1. MÉTHODE
La présente méthode d'essai reprend la méthode TG 307 (2002) de l'OCDE.
1.1. INTRODUCTION
La présente méthode d'essai s'appuie sur les lignes directrices existantes (1) (2) (3) (4) (5) (6) (7) (8) (9). La méthode décrite ici est conçue pour mesurer la transformation aérobie et anaérobie des substances chimiques dans le sol. Les expériences ont pour but de déterminer i) le taux de transformation de la substance d'essai, et ii) la nature des produits de transformation auxquels les végétaux et les organismes du sol peuvent être exposés, ainsi que les taux de formation et de déplétion de ces produits. Ces études sont nécessaires pour les substances chimiques qui sont appliquées directement sur le sol ou qui sont susceptibles d'atteindre l'environnement du sol. Les résultats de ces études de laboratoire peuvent également être utilisés pour mettre au point des protocoles d'échantillonnage et d'analyse destinés à des études dans des domaines voisins.
Il est généralement suffisant d'effectuer des études aérobies et anaérobies avec un seul type de sol pour déterminer les voies de transformation (8) (10) (11). Les taux de transformation doivent être déterminés dans trois autres sols au moins (8) (10).
Un atelier de l'OCDE sur la sélection des sols et des sédiments, organisé à Belgirate, en Italie, en 1995 (10), a défini notamment le nombre et le type de sols à utiliser dans le cadre de cet essai. Les types de sol testés doivent être représentatifs des conditions environnementales d'utilisation ou de rejet prévus. Par exemple, les substances chimiques devant être appliquées sous des climats subtropicaux ou tropicaux doivent être testées avec des Ferrasols ou des Nitosols (système FAO). Cet atelier a également formulé des recommandations relatives au prélèvement, à la manipulation et au stockage des échantillons de sol, sur la base des lignes directrices de l'ISO (15). L'utilisation de sols de rizières est également étudiée dans le cadre de cette méthode.
1.2. DÉFINITIONS
Substance d'essai: toute substance, qu'il s'agisse de la substance mère ou des produits de transformation correspondants.
Produits de transformation: toute substance résultant de réactions de transformations biotiques ou abiotiques de la substance d'essai et en particulier le CO2 et les produits qui se trouvent dans les résidus liés.
Résidus liés: les «résidus liés» désignent des composés du sol, végétaux ou animaux, qui subsistent dans la matrice après extraction, sous forme de la substance mère ou de ses métabolite(s)/produits de transformation. La méthode d'extraction ne doit pas modifier de manière substantielle les composés eux-mêmes ni la structure de la matrice. La nature de la liaison peut être clarifiée en partie par des méthodes d'extraction qui modifient la matrice et par des techniques analytiques complexes. C'est de cette façon que l'on a déterminé jusqu'ici la nature de la liaison, covalente, ionique et de la liaison par sorption ou piégeage. De manière générale, la formation de résidus liés abaisse sensiblement la bioaccessibilité et la biodisponibilité (12) [modifié d'après l'UICPA 1984 (13)].
Transformation aérobie: réaction se produisant en présence d'oxygène moléculaire (14).
Transformation anaérobie: réaction se produisant en l'absence d'oxygène moléculaire (14).
Sol: mélange de constituants chimiques minéraux et organiques, ces derniers contenant des composés à haute teneur en carbone et en azote, et à poids moléculaire élevé, contenant de petits (principalement micro) organismes. Le sol peut être manipulé sous deux formes:
|
a)
|
non brassé, tel qu'il s'est formé au cours du temps, en couches caractéristiques d'un grand nombre de types de sol;
|
|
b)
|
brassé, tel qu'on le trouve habituellement dans les terres arables ou qu'il est recueilli en creusant et tel qu'il est utilisé dans la présente méthode d'essai (14).
|
Minéralisation: dégradation complète d'un composé organique en CO2 et H2O dans des conditions aérobies, et en CH4, CO2 et H2O dans des conditions anaérobies. Dans le cadre de la présente méthode d'essai, lorsqu'on utilise un composé marqué au 14C, la minéralisation désigne la dégradation importante au cours de laquelle un atome de carbone marqué est oxydé quantitativement avec dégagement de la quantité correspondante de 14CO2 (14).
Demi-vie: t0,5, temps nécessaire à la transformation de 50 % d'une substance d'essai lorsque la transformation peut être décrite par une cinétique du premier ordre; la demi-vie est indépendante de la concentration.
DT
50
(temps de dégradation 50): période au cours de laquelle la concentration de la substance d'essai diminue de 50 %; elle est différente de la demi-vie t0,5 lorsque la transformation ne suit pas une cinétique du premier ordre.
DT
75
(temps de dégradation 75): période au cours de laquelle la concentration de la substance d'essai diminue de 75 %.
DT
90
(temps de dégradation 90): période au cours de laquelle la concentration de la substance d'essai diminue de 90 %.
1.3. SUBSTANCES DE RÉFÉRENCE
Des substances de référence doivent être utilisées pour caractériser et/ou identifier les produits de transformation par des méthodes spectroscopiques et chromatographiques.
1.4. APPLICABILITÉ DE L'ESSAI
La méthode s'applique à toutes les substances chimiques (non marquées ou radiomarquées) pour lesquelles il existe une méthode analytique présentant une précision et une sensibilité suffisantes. Elle peut être appliquée aux composés légèrement volatils, non volatils, solubles ou insolubles dans l'eau. L'essai ne doit pas être appliqué aux substances chimiques fortement volatiles à partir du sol (fumigants, solvants organiques) et qui ne peuvent donc pas être maintenues dans le sol dans les conditions expérimentales de cet essai.
1.5. INFORMATIONS RELATIVES À LA SUBSTANCE D'ESSAI
Une substance d'essai marquée ou non marquée peut être utilisée pour mesurer le taux de transformation. Il est nécessaire d'utiliser du matériel marqué pour étudier les voies de transformation et établir un bilan massique. Le marquage au 14C est recommandé mais l'utilisation d'autres isotopes, comme 13C, 15N, 3H, 32P, peut aussi être utile. Dans la mesure du possible, le marquage doit être situé dans la(les) partie(s) la(les) plus stable(s) de la molécule (43). La pureté de la substance d'essai doit être d'au moins 95 %.
Avant de procéder à un essai sur la transformation aérobie et anaérobie dans le sol, il convient de disposer des informations suivantes sur la substance d'essai:
|
a)
|
solubilité dans l'eau (Méthode A.6)
|
|
b)
|
solubilité dans les solvants organiques;
|
|
c)
|
pression de vapeur (Méthode A.4) et constante de la loi de Henry;
|
|
d)
|
coefficient de partage n-octanol/eau (Méthode A.8);
|
|
e)
|
stabilité chimique dans le noir (hydrolyse) (Méthode C.7);
|
|
f)
|
coefficient pKa si une molécule est susceptible de subir une protonation ou une déprotonation (ligne directrice 112 de l'OCDE) (16).
|
Il peut également être utile de disposer d'informations relatives à la toxicité de la substance d'essai sur les micro-organismes du sol (Méthodes d'essai C.21 et C.22) (16).
Il faut disposer des méthodes analytiques (dont des méthodes d'extraction et de purification) nécessaires à la quantification et à l'identification de la substance d'essai et de ses produits de transformation.
1.6. PRINCIPE DE LA MÉTHODE
Les échantillons de sol sont traités avec la substance d'essai et incubés dans le noir dans des flacons de type biomètre ou dans des systèmes à circulation continue dans des conditions de laboratoire contrôlées (à température et humidité constantes). Après un intervalle de temps approprié, les échantillons de sol sont extraits et analysés pour mesurer la substance mère et les produits de transformation. Les produits volatils sont aussi collectés pour analyse au moyen de dispositifs d'absorption appropriés. À l'aide de produit marqué au 14C, il est possible de mesurer les différents taux de minéralisation de la substance d'essai en piégeant le 4CO2 dégagé, et un bilan massique, notamment la formation de résidus liés au sol, peut être établi.
1.7. CRITÈRES DE QUALITÉ
1.7.1. Récupération
L'extraction et l'analyse, en double exemplaire au moins, des échantillons de sol immédiatement après l'addition de la substance d'essai donnent une première indication de la reproductibilité de la méthode analytique et de l'uniformité de la procédure d'application de la substance d'essai. Les taux de récupération concernant les étapes ultérieures des expériences sont fournis par les bilans massiques respectifs. Ces taux de récupération doivent osciller entre 90 % et 110 % pour les substances chimiques marquées (8) et entre 70 % et 110 % pour les substances chimiques non marquées (3).
1.7.2. Reproductibilité et sensibilité de la méthode analytique
La reproductibilité de la méthode analytique (à l'exception du rendement d'extraction initial) servant à quantifier la substance d'essai et les produits de transformation peut être contrôlée en effectuant en double les analyses du même extrait de sol, incubé suffisamment longtemps pour permettre la formation de produits de transformation.
Le seuil de détection de la méthode d'analyse de la substance d'essai et des produits de transformation doit être au moins égal à 0,01 mg.kg-1 de sol (substance d'essai) ou 1 % de la dose appliquée si celle-ci est inférieure. Le seuil de quantification doit également être spécifié.
1.7.3. Précision des données de transformation
L'analyse de régression des concentrations de la substance d'essai en fonction du temps permet d'obtenir les informations sur la précision de la courbe de transformation et de calculer les limites de confiance des demi-vies (en cas de cinétique du pseudo premier ordre) ou des valeurs DT50 et, le cas échéant, des valeurs DT75 et DT90.
1.8. DESCRIPTION DE LA MÉTHODE
1.8.1. Appareils et réactifs chimiques
Les incubateurs sont composés de circuits fermés statiques ou de systèmes à circulation continue adaptés (7) (17). Les figures 1 et 2, respectivement, présentent des exemples d'incubateur à circulation continue et de flacons de type biomètre. Les deux types d'incubateur présentent des avantages et des inconvénients (7) (17).
Matériel courant de laboratoire, notamment:
|
—
|
instruments d'analyse: chromatographie gaz-liquide, chromatographie en phase liquide à haute performance (HPLC), chromatographie en couche mince, y compris les systèmes de détection appropriés pour analyser les substances radiomarquées ou non marquées ou la méthode de dilution isotopique inverse,
|
|
—
|
instruments destinés à l'identification (spectromètre de masse, chromatographie en phase gazeuse couplée à la spectrométrie de masse (GC-MS, HPLC-MS, RMN, etc.),
|
|
—
|
compteur à scintillation liquide,
|
|
—
|
appareillage d'oxydation pour la combustion des produits radioactifs,
|
|
—
|
appareil d'extraction (par exemple, tubes de centrifugation pour extraction à froid et appareil pour extraction en continu sous reflux de type soxhlet),
|
|
—
|
instrumentation pour concentrer les solutions et les extraits (évaporateur rotatif),
|
|
—
|
mélangeurs mécaniques (pétrin, mélangeur rotatif).
|
Les réactifs chimiques utilisés sont, par exemple:
|
—
|
NaOH, de pureté analytique, 2 mol . dm-3, ou une autre base appropriée (par exemple, KOH, éthanolamine),
|
|
—
|
H2SO4, de pureté analytique, 0,05 mol . dm-3,
|
|
—
|
Éthylène glycol, de pureté analytique,
|
|
—
|
matériaux d'absorption solide tels que chaux sodée et tampons de polyuréthane,
|
|
—
|
solvants organiques, de pureté analytique, tels qu'acétone, méthanol, etc.,
|
|
—
|
liquide de scintillation.
|
1.8.2. Application de la substance d'essai
Pour l'incorporer et la répartir dans le sol, on peut dissoudre la substance d'essai dans l'eau (désionisée ou distillée) ou, si nécessaire, dans une quantité minimale d'acétone ou d'autres solvants organiques (6) dans lesquels la substance d'essai est suffisamment soluble et stable. Toutefois, la quantité de solvant sélectionnée ne doit pas avoir une influence significative sur l'activité microbienne du sol (voir les points 1.5 et 1.9.2-1.9.3). Il convient d'éviter d'utiliser des solvants qui inhibent l'activité microbienne, comme le chloroforme, le dichlorométhane et autres solvants halogénés.
La substance d'essai peut également être ajoutée sous forme solide, mélangée avec du sable de quartz (6) ou dans un petit sous-échantillon de sol séché à l'air et stérilisé. Si la substance d'essai est ajoutée à l'aide d'un solvant, le solvant doit pouvoir s'évaporer avant que le sous-échantillon chargé soit ajouté à l'échantillon original de sol non stérile.
Pour les substances chimiques courantes, dont la principale voie de pénétration dans le sol passe par les boues d'épuration ou le traitement agricole, il convient de commencer par ajouter la substance d'essai dans la boue avant de l'introduire dans l'échantillon de sol (voir les points 1.9.2 et 1.9.3).
Il n'est pas recommandé d'utiliser systématiquement des produits formulés. Toutefois, pour les substances peu solubles, l'utilisation de produit formulé peut être une solution appropriée.
1.8.3. Sols
1.8.3.1. Sélection du sol
Pour déterminer la voie de transformation, on peut utiliser un sol représentatif; un limon sableux, un limon fin, un limon ou un sable limoneux [selon la classification de la FAO et de l'USDA (18)] avec un pH de 5,5-8,0, une teneur en carbone organique de 0,5-2,5 % et une biomasse microbienne d'au moins 1 % de carbone organique total est recommandée (10).
Pour les études des taux de transformation, il convient d'utiliser au moins trois sols supplémentaires représentatifs de la gamme de sols concernés. La teneur en carbone organique, le pH, la teneur en argile et la biomasse microbienne des sols doivent varier (10).
Pour tous les sols, il convient d'établir au moins les caractéristiques suivantes: texture (% sable, % limon, % argile) [selon la classification de la FAO et de l'USDA (18)], pH, capacité d'échange cationique, carbone organique, densité apparente, caractéristiques de rétention d'eau (44) et biomasse microbienne (pour les études aérobies uniquement). Des informations complémentaires sur les propriétés du sol peuvent être utiles pour interpréter les résultats. Les méthodes recommandées dans les références (19) (20) (21) (22) (23) peuvent être utilisées pour déterminer les caractéristiques du sol. La biomasse microbienne doit être déterminée à l'aide de la méthode de respiration induite par le substrat (SIR) (25) (26) ou d'autres méthodes (20).
1.8.3.2. Prélèvement, manipulation et stockage des sols
Il est nécessaire de fournir des informations détaillées sur l'historique du site de prélèvement, qui englobent la localisation, le couvert végétal, les traitements aux substances chimiques, les traitements aux engrais organiques et inorganiques, les apports biologiques ou d'autres contaminations. Si les sols ont été traités avec la substance d'essai ou ses analogues structuraux au cours des quatre années précédentes, ils ne doivent pas être utilisés pour les études de transformation (10) (15).
Le sol doit être fraîchement extrait du site (de l'horizon A ou de la couche supérieure de 20 cm) avec une teneur en eau facilitant le tamisage. Pour les sols autres que les sols de rizières, il faut éviter de prélever les échantillons pendant ou immédiatement après de longues périodes (> 30 jours) de sécheresse, de gel ou d'inondations (14). Les échantillons doivent être transportés de façon à minimiser les modifications de la teneur en eau du sol et conservés dans le noir avec libre circulation d'air, dans la mesure du possible. Un sac en polyéthylène à fermeture non étanche est généralement indiqué à cet effet.
Le sol doit être traité le plus rapidement possible après le prélèvement. Il convient de retirer les gros débris végétaux, animaux et les pierres avant de passer le sol à travers un tamis de 2 mm pour retirer les petits débris de pierres, et les débris animaux et végétaux. Il convient d'éviter de sécher et de broyer le sol de manière importante avant de le tamiser (15).
Lorsqu'il est difficile de prélever des échantillons en hiver (sol gelé ou recouvert d'une couche de neige), ceux-ci peuvent être prélevés sur un lot de sol stocké dans une serre sous couvert végétal (herbe ou mélange d'herbe et de trèfle). Il est nettement préférable d'effectuer des études avec des sols qui viennent d'être extraits du site, mais si le sol prélevé et traité doit être stocké avant le début de l'étude, les conditions de stockage doivent être appropriées et leur durée doit être limitée (4 + 2 oC pendant une durée maximale de trois mois) afin de préserver l'activité microbienne (45). On trouvera des instructions détaillées sur le prélèvement, la manipulation et le stockage des sols à utiliser pour les expériences de biotransformation sous les références (8) (10) (15) (26) (27).
Avant son utilisation dans le cadre du présent essai, le sol traité doit être préincubé afin de permettre la germination et l'élimination des semences, et de rétablir l'équilibre du métabolisme microbien après le passage des conditions de prélèvement ou de stockage aux conditions d'incubation. Une période de préincubation de 2 à 28 jours approchant les conditions de température et d'humidité de l'essai réel est généralement indiquée (15). La durée du stockage et de la préincubation ne doit pas dépasser trois mois au total.
1.9. EXÉCUTION DE L'ESSAI
1.9.1. Conditions de l'essai
1.9.1.1. Température de l'essai
Au cours de toute la période d'essai, les sols doivent être incubés dans le noir, à température constante, respectant les conditions climatiques dans lesquelles l'utilisation ou le rejet interviendra. Une température de 20 ± 2 oC est recommandée pour toutes les substances d'essai susceptibles d'entrer en contact avec le sol dans les climats tempérés. La température doit être enregistrée.
Pour les substances chimiques appliquées ou libérées dans les climats froids (par exemple dans les pays nordiques, pendant l'automne/hiver), des échantillons de sol supplémentaires doivent également être incubés mais à une température plus basse (par exemple à 10 ± 2 oC).
1.9.1.2. Teneur en humidité
Pour les essais de transformation en milieu aérobie, la teneur en humidité du sol (46) doit être ajustée et maintenue à une pF située entre 2,0 et 2,5 (3). La teneur en humidité du sol est exprimée en masse d'eau par masse de sol sec et elle doit être contrôlée régulièrement (toutes les 2 semaines par exemple) en pesant les flacons d'incubation et les pertes en eau doivent être compensées par l'adjonction d'eau (de préférence de l'eau du robinet stérilisée par filtration). Il convient de veiller à éviter ou à réduire les pertes de substance d'essai et/ou des produits de transformation par évaporation et/ou photodégradation (le cas échéant) pendant l'ajout d'eau.
Pour les essais de transformation dans des conditions anaérobies et de rizière, le sol est saturé en eau par submersion.
1.9.1.3. Conditions d'incubation aérobies
Dans les systèmes à circulation continue, les conditions aérobies sont maintenues par lessivage périodique ou par aération continue avec de l'air humidifié. Dans les flacons de type biomètre, l'échange d'air est maintenu par diffusion.
1.9.1.4. Conditions aérobies stériles
Pour obtenir des informations sur l'importance de la transformation abiotique d'une substance d'essai, les échantillons de sol peuvent être stérilisés (pour les méthodes de stérilisation, voir les références 16 et 29), traités avec une substance d'essai stérile (par exemple l'addition de solution à travers un filtre stérile) et aérés avec de l'air stérile humidifié comme décrit au point 1.9.1.3. Pour les sols de rizière, le sol et l'eau doivent être stérilisés et l'incubation doit être effectuée comme décrit au point 1.9.1.6.
1.9.1.5. Conditions d'incubation anaérobies
Afin de créer et de maintenir des conditions anaérobies, le sol traité avec la substance d'essai et incubé dans des conditions aérobies pendant 30 jours ou une demi-vie ou DT50 (si celle-ci est plus courte) est ensuite recouvert d'eau (couche de 1-3 cm d'eau) et le système d'incubation est balayé avec un gaz inerte (azote ou argon) (47). Le système d'essai doit permettre de mesurer le pH, la concentration en oxygène et le potentiel redox et comprendre des dispositifs de piégeage des produits volatiles. Le système de type biomètre doit être fermé pour éviter toute entrée d'air par diffusion.
1.9.1.6. Conditions d'incubation dans les sols de rizière
Pour étudier la transformation dans les sols de rizière, le sol est recouvert d'une couche d'eau de 1-5 cm environ et la substance d'essai appliquée à la phase aqueuse (9). Une profondeur de couche de sol de 5 cm au moins est recommandée. Le système est ventilé avec de l'air comme en situation aérobie. Le pH, la concentration en oxygène et le potentiel redox de la couche aqueuse doivent être surveillés et enregistrés. Une période de préincubation de deux semaines au moins est nécessaire avant de commencer les études de transformation (voir le point 1.8.3.2).
1.9.1.7. Durée de l' essai
En règle générale, les études des taux et des voies de transformation ne doivent pas dépasser 120 jours (48) (3) (6) (8), parce qu'au-delà, dans un système artificiel en laboratoire qui ne peut se reconstituer naturellement, on peut attendre une diminution de l'activité microbienne du sol avec le temps. S'il est nécessaire de caractériser la déplétion de la substance d'essai et la formation et la déplétion des principaux produits de transformation, les études peuvent être poursuivies pendant une période plus longue (de 6 à 12 mois) (8). Des périodes d'incubation plus longues doivent être justifiées dans le rapport d'essai et accompagnées de mesures de la biomasse pendant et à la fin de ces périodes.
1.9.2. Exécution de l'essai
On place 50 à 200 g de sol environ (sur la base du poids sec) dans chaque flacon d'incubation (voir les figures 1 et 2 à l'annexe 3) et on traite le sol avec la substance d'essai en utilisant l'une des méthodes décrites au point 1.8.2. Si l'on utilise des solvants organiques pour appliquer la substance d'essai, il convient de les éliminer du sol par évaporation. Puis, le sol est soigneusement mélangé à l'aide d'une spatule et/ou en agitant le flacon. Si l'étude est menée dans des conditions de sol de rizière, le sol et l'eau doivent être soigneusement mélangés après l'application de la substance d'essai. De petits aliquots (1 g par exemple) des sols traités doivent être analysés pour mesurer la substance d'essai afin de vérifier l'uniformité de la répartition. D'autres méthodes sont décrites ci-dessous.
Le taux de traitement doit correspondre à la dose d'application la plus élevée d'un produit phytosanitaire recommandée dans les instructions d'utilisation et à l'incorporation uniforme à une profondeur appropriée dans le champ [couche supérieure du sol sur 10 cm (49) ]. Par exemple, pour les substances chimiques appliquées sur le feuillage ou sans enfouissement dans le sol, la profondeur appropriée pour calculer la quantité de substance chimique à ajouter à chaque flacon est de 2,5 cm. Pour les substances chimiques enfouies dans le sol, la profondeur appropriée est la profondeur d'enfouissement spécifiée dans les instructions d'utilisation. Pour les substances chimiques courantes, la dose d'application doit être estimée sur la base de la voie de pénétration la plus pertinente; par exemple, lorsque les boues d'épuration sont la principale voie de pénétration dans le sol, la substance chimique doit être dosée dans la boue à une concentration correspondant à la concentration prévue et la quantité de boue ajoutée au sol doit correspondre à la charge normale en boue des sols agricoles. Si cette concentration n'est pas suffisamment élevée pour identifier les principaux produits de transformation, l'incubation d'échantillons de sol contenant des taux plus élevés peut être utile, mais il convient d'éviter des taux excessifs qui influencent les fonctions microbiennes du sol (voir les points 1.5 et 1.8.2).
Un autre moyen consiste à traiter un lot plus important (de 1 à 2 kg) de sol avec la substance d'essai, en le mélangeant soigneusement dans un mélangeur adapté puis en le transférant en petites fractions de 50 à 200 g dans les flacons d'incubation (par exemple à l'aide de répartiteurs). De petits aliquots (1 g par exemple) du lot de sol traité doivent être analysés afin de vérifier l'uniformité de la répartition de la substance d'essai. Cette procédure est préférable car elle permet une répartition plus uniforme de la substance d'essai dans le sol.
De même, les échantillons de sol non traités sont incubés dans les mêmes conditions (aérobies) que les échantillons traités avec la substance d'essai. Ces échantillons sont utilisés pour mesurer la biomasse pendant le déroulement et à la fin des essais.
Lorsque la substance d'essai est appliquée dans le sol dissous dans un (ou plusieurs) solvant(s) organique(s), des échantillons de sol traité avec la même quantité de solvant(s) sont incubés dans les mêmes conditions (aérobies) que les échantillons traités avec la substance d'essai. Ces échantillons sont utilisés pour mesurer la biomasse au début, pendant le déroulement et à la fin des essais afin de contrôler les effets du(des) solvant(s) sur la biomasse microbienne.
Les flacons contenant le sol traité sont attachés au système à circulation continue décrit à la figure 1 ou fermés par la colonne d'absorption présentée dans la figure 2 (voir l'annexe 3).
1.9.3. Échantillonnage et mesures
Les flacons d'incubation en deux exemplaires sont retirés à des intervalles appropriés et les échantillons de sol extraits à l'aide de solvants appropriés de polarité différente et analysés pour mesurer la substance d'essai et/ou les produits de transformation. Une étude bien conçue comprend un nombre suffisant de flacons de façon à pouvoir sacrifier deux flacons à chaque prélèvement. De plus, des solutions d'absorption ou des matériaux d'absorption solides sont retirés à différents intervalles de temps (tous les 7 jours au cours du premier mois et, au bout d'un mois, tous les 17 jours) pendant et à la fin de l'incubation de chaque échantillon de sol et analysés pour mesurer les produits volatils. Par ailleurs, au moins 5 points supplémentaires de prélèvement devraient être prévus en plus de l'échantillon de sol prélevé directement après application (échantillon de 0 jour). Les intervalles de temps doivent être choisis de façon à pouvoir établir la courbe de déplétion de la substance d'essai et les courbes de formation et de déplétion des produits de transformation (par exemple 0, 1, 3, 7 jours; 2, 3 semaines; 1, 2, 3 mois, etc.).
Lorsqu'on utilise une substance d'essai marquée au 14C, la radioactivité non extractible est quantifiée par combustion et un bilan massique est calculé pour chaque intervalle de prélèvement.
Dans le cas de l'incubation anaérobie et des sols de rizière, la phase sol et la phase aqueuse peuvent être analysées ensemble pour mesurer la substance d'essai et les produits de transformation ou séparées par filtration ou centrifugation avant extraction et analyse.
1.9.4. Essais facultatifs
Il peut être utile d'effectuer des études aérobies non stériles à d'autres températures et humidités de sol pour estimer l'influence de la température et de l'humidité du sol sur le taux de transformation d'une substance d'essai et/ou de ses produits de transformation dans le sol.
On peut essayer d'effectuer une caractérisation plus poussée de la radioactivité non extractible en utilisant, par exemple, l'extraction par un fluide supercritique.
2. DONNÉES
2.1. TRAITEMENT DES RÉSULTATS
Les quantités de substance d'essai, de produits de transformation, de substances volatiles (en % uniquement) et non extractibles doivent être données en pourcentage de la concentration initiale appliquée et, s'il y a lieu, en mg.kg-1 de sol (sur la base du poids sec de sol) à chaque intervalle de prélèvement. Pour chaque intervalle de prélèvement, un bilan massique doit être donné en pourcentage de la concentration initiale appliquée. La représentation graphique des concentrations de la substance d'essai en fonction du temps permet d'estimer sa durée de demi-vie de la transformation ou sa DT50. Les produits de transformation principaux doivent être identifiés et leurs concentrations doivent également être présentées graphiquement en fonction du temps afin de montrer leurs taux de formation et de déplétion. On entend par produit de transformation principal tout produit représentant > 10 % de la dose appliquée à n'importe quel moment de l'étude.
Les produits volatils piégés donnent une indication du potentiel de volatilité d'une substance d'essai et de ses produits de transformation à partir du sol.
Il est possible de déterminer avec plus de précision les demi-vies ou les valeurs DT50 et, s'il y a lieu, les valeurs DT75 et DT90 en utilisant des méthodes de calcul fondées sur des modèles cinétiques appropriés. La durée de demi-vie et les valeurs DT50 doivent être notées dans le rapport avec la description du modèle cinétique utilisé, l'ordre de la cinétique et le coefficient de détermination (r2). La cinétique du premier ordre est préférable sauf si r2 < 0,7. Si nécessaire, les calculs doivent aussi être appliqués aux principaux produits de transformation. Des exemples des modèles appropriés sont décrits sous les références 31 à 35.
Dans le cas des études de taux effectuées à des températures différentes, les taux de transformation doivent être décrits comme fonction de la température à l'intérieur de l'intervalle de température de l'expérience en appliquant la formule de la relation d'Arrhenius:
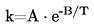 or
or 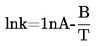
où In A et B sont des constantes de régression, respectivement, l'intersection et la pente de la droite d'interpolation produite par régression linéaire de In k par rapport à 1/T, où k est la constante de vitesse à la température T et T la température en degrés Kelvin. Il convient de prendre soin à l'intervalle limité de température à l'intérieur de laquelle la relation d'Arrhenius est valable dans le cas où la transformation dépend de l'activité microbienne.
2.2. ÉVALUATION ET INTERPRÉTATION DES RÉSULTATS
Bien que les études soient effectuées dans un système artificiel en laboratoire, les résultats permettent d'estimer le taux de transformation de la substance d'essai ainsi que les taux de formation et de déplétion des produits de transformation dans des conditions de terrain (36) (37).
L'étude des voies de transformation d'une substance d'essai donne des informations sur la façon dont la structure de la substance appliquée est modifiée dans le sol par des réactions chimiques et microbiennes.
3. RAPPORT
RAPPORT D'ESSAI
Le rapport d'essai doit comporter les informations suivantes:
|
|
Substance d'essai:
|
—
|
nom courant, nom chimique, numéro de fichier CAS, formule structurale [indiquant la(les) position (s) du(des) marquage(s) lorsqu'on utilise un produit radiomarqué] et propriétés physico-chimiques pertinentes (voir le point 1.5),
|
|
—
|
pureté (impuretés) de la substance d'essai,
|
|
—
|
pureté radiochimique de la substance marquée et activité spécifique (s'il y a lieu).
|
|
|
|
Substances de référence:
|
—
|
nom et structure chimique des substances de référence utilisées pour caractériser et/ou identifier le produit de transformation.
|
|
|
|
Sol d'essai:
|
—
|
détails du site de prélèvement,
|
|
—
|
date et procédure d'échantillonnage du sol,
|
|
—
|
propriétés des sols: pH, teneur en carbone organique, texture (% sable, % limon, % argile), capacité d'échange cationique, densité apparente, caractéristique de rétention d'eau, et biomasse microbienne,
|
|
—
|
durée et conditions de stockage du sol (s'il y a lieu).
|
|
|
|
Conditions d'essai:
|
—
|
dates de réalisation des études,
|
|
—
|
quantité de substance d'essai appliquée,
|
|
—
|
solvants utilisés et méthode d'application de la substance d'essai,
|
|
—
|
poids de sol traité initialement et prélevé à chaque intervalle pour analyse,
|
|
—
|
description du système d'incubation utilisé,
|
|
—
|
débit d'écoulement de l'air (pour les systèmes à circulation continue uniquement),
|
|
—
|
température au début de l'expérience,
|
|
—
|
teneur en humidité du sol pendant l'incubation,
|
|
—
|
biomasse microbienne au début, pendant le déroulement et à la fin des études aérobies,
|
|
—
|
pH, concentration en oxygène et potentiel redox au début, pendant le déroulement et à la fin des études anaérobies et des sols de rizière,
|
|
—
|
méthode(s) d'extraction,
|
|
—
|
méthodes de quantification et d'identification de la substance d'essai et des principaux produits de transformation dans le sol et matériau d'absorption,
|
|
—
|
nombre de réplicats et nombre de témoins.
|
|
|
|
Résultats:
|
—
|
résultat de la détermination de l'activité microbienne,
|
|
—
|
reproductibilité et sensibilité des méthodes analytiques utilisées,
|
|
—
|
taux de récupération (les pourcentages acceptables pour que l'étude soit valable sont présentés au point 1.7.1),
|
|
—
|
tableaux de résultats exprimés en % de la dose initiale appliquée et, s'il y a lieu, en mg.kg-1 de sol (sur la base de poids sec),
|
|
—
|
bilan massique pendant et à la fin des études,
|
|
—
|
caractérisation de la radioactivité non extractible (liée) ou des résidus dans le sol,
|
|
—
|
quantification du CO2 dégagé et des autres composés volatiles,
|
|
—
|
graphes représentant la concentration de la substance d'essai dans le sol en fonction du temps et, s'il y a lieu, des principaux produits de transformation,
|
|
—
|
demi-vie ou DT50, DT75 et DT90 pour la substance d'essai et, s'il y a lieu, des principaux produits de transformation, ainsi que les limites de confiance,
|
|
—
|
estimation de la vitesse de dégradation abiotique dans des conditions stériles,
|
|
—
|
évaluation de la cinétique de transformation de la substance d'essai et, s'il y a lieu, des principaux produits de transformation,
|
|
—
|
voies de transformation proposées, s'il y a lieu,
|
|
—
|
discussion et interprétation des résultats,
|
|
—
|
données brutes (chromatogramme des échantillons, calcul des vitesses de transformation des échantillons et moyens utilisés pour identifier les produits de transformation).
|
|
4. RÉFÉRENCES
|
(1)
|
US- Environmental Protection Agency (1982). Pesticide Assessment Guidelines, Subdivision N. Chemistry: Environmental Fate.
|
|
(2)
|
Agriculture Canada (1987). Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada.
|
|
(3)
|
Union européenne (UE) (1995). Directive 95/36/CE de la Commission du 14 juillet 1995 modifiant la directive 91/414/CEE du Conseil concernant la mise sur le marché des produits phytopharmaceutiques. Annexe II, partie A, et annexe III, partie A: Devenir et comportement dans l'environnement.
|
|
(4)
|
Dutch Commission for Registration of Pesticides (1995). Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air.
|
|
(5)
|
BBA (1986). Richtlinie fur die amtliche Prüfung von Pflanzenschutzmitteln, Teil IV, 4-1. Verbleib von Pflanzenschutzmitteln im Boden — Abbau, Umwandlung und Metabolismus.
|
|
(6)
|
ISO/DIS 11266-1 (1994). Soil Quality -Guidance on laboratory tests for biodegradation of organic chemicals in soil — Part 1: Aerobic conditions.
|
|
(7)
|
ISO 14239 (1997). Soil Quality — Laboratory incubation Systems for measuring the mineralization of organic chemicals in soil under aerobic conditions.
|
|
(8)
|
SETAC (1995). Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides. Mark R. Lynch, Ed.
|
|
(9)
|
MAFF — Japon 2000 — Draft Guidelines for transformation studies of pesticides in soil — Aerobic metabolism study in soil under paddy field conditions (flooded).
|
|
(10)
|
OCDE (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments. Belgirate, Italie, 18-20 January 1995.
|
|
(11)
|
Guth, J.A. (1980). The study of transformations. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academie Press, p. 123-157.
|
|
(12)
|
DFG: Pesticide Bound Residues in Soil. Wiley — VCH (1998).
|
|
(13)
|
T.R. Roberts: Non extractable pesticide residue in soils and plants. Pure Appl. Chem. 56, 945-956 (IUPAC 1984).
|
|
(14)
|
Ligne directrice de l'OCDE 304 A: Biodégradabilité intrinsèque dans le sol (adoptée le 12 mai 1981).
|
|
(15)
|
ISO 10381-6 (1993). Qualité des sols — Échantillonnage — Partie 6: Guide du prélèvement, de la manipulation et du stockage des sols pour l'évaluation des processus microbiens en laboratoire.
|
|
(16)
|
Annexe V de la directive 67/548/CEE.
|
|
(17)
|
Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry. D.H. Hutson, T.R. Roberts, Eds. J. Wiley & Sons. Vol 1, p. 85-114.
|
|
(18)
|
Soil Texture Classification (US and FAO Systems): Weed Science, 33, Suppl. 1 (1985) and Soil Sci. Soc. Amer. Proc. 26:305 (1962).
|
|
(19)
|
Methods of Soil Analysis (1986). Part 1, Physical and Mineralogical Methods. A. Kliite, Ed.) Agronomy Series No 9, 2nd Edition.
|
|
(20)
|
Methods of Soil Analysis (1982). Part 2, Chemical and Microbiological Properties. A.L. Page, R.H. Miller and D.R. Kelney, Eds. Agronomy Series No 9, 2nd Edition.
|
|
(21)
|
ISO Standard Compendium Environment (1994). Soil Quality — General aspects; chemicai and physical méthodes of analysis; biological methods of analysis. First Edition.
|
|
(22)
|
Mückenhausen, E. (1975). Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen. DLG-Verlag, Frankfurt, Main.
|
|
(23)
|
Schefïer, F., Schachtschabel, P. (1975). Lehrbuch der Bodenkunde. F. Enke Verlag, Stuttgart.
|
|
(24)
|
Anderson, J.P.E., Domsch, K.H. (1978) A physiological method for the quantitative measurement of microbial biomass in soils. Soil Biol. Biochem. 10, p. 215-221.
|
|
(25)
|
ISO 14240-1 and 2 (1997). Soil Quality — Determination of soil microbial biomass — Part 1: Substrate-induced respiration method. Part 2: fumigation-extraction method.
|
|
(26)
|
Anderson, J.P.E. (1987). Handling and storage of soils for pesticide experiments. In Pesticide Effects on Soil Microflora. L. Somerville, M.P. Greaves, Eds. Taylor & Francis, p. 45-60.
|
|
(27)
|
Kato, Yasuhiro. (1998). Mechanism of pesticide transformation in the environment: Aerobic and bio- transformation of pesticides in aqueous environment. Proceedings of the 16th Symposium on Environmental Science of Pesticide, p. 105-120.
|
|
(28)
|
Keuken O., Anderson J.P.E. (1996). Influence of storage on biochemical processes in soil. In Pesticides, Soil Microbiology and Soil Quality, 59-63 (SETAC-Europe).
|
|
(29)
|
Stenberg B., Johansson M., Pell M., Sjödahl-Svensson K., Stenström J., Torstensson L. (1996). Effect of freeze and cold storage of soil on microbial activities and biomass. In Pesticides, Soil Microbiology and Soil Quality, 68-69 (SETAC-Europe).
|
|
(30)
|
Gennari, M., Negre, M., Ambrosoli, R. (1987). Effects of ethylene oxide on soil microbial content and some chemical characteristics. Plant and Soil 102, p. 197-200.
|
|
(31)
|
Anderson, J.P.E. (1975). Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden. Z. PflKrankh Pflschutz, Sonderheft VII, p. 141-146.
|
|
(32)
|
Hamaker, J.W. (1976). The application of mathematical modelling to the soil persistence and accumulation of pesticides. Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, p. 181-199.
|
|
(33)
|
Goring, C.A.I., Laskowski, D.A., Hamaker, J.W., Meikle, R.W. (1975). Principles of pesticide degradation in soil. In «Environmental Dynamics of Pesticides». R. Haque and V.H. Freed, Eds., p. 135-172.
|
|
(34)
|
Timme, G., Frehse, H., Laska, V. (1986). Statistical interpretation and graphic representation of the degradational behaviour of pesticide résidus. II. Pflanzenschutz — Nachrichten Bayer 39, p. 188-204.
|
|
(35)
|
Timme, G., Frehse, H. (1980). Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues. I. Pflanzenschutz — Nachrichten Bayer 33, p. 47-60.
|
|
(36)
|
Gustafson D.I., Holden L.R. (1990). Non-linear pesticide dissipation in soil; a new model based on spatial variability. Environm. Sci. Technol. 24, p. 1032-1041.
|
|
(37)
|
Hurle K., Walker A. (1980). Persistence and its prediction. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academic Press, p. 83-122.
|
Annexe 1
SUCCION, CAPACITÉ AU CHAMP (FC) ET CAPACITÉ DE RÉTENTION D'EAU (WHC) (50)
|
Hauteur de la colonne d'eau
[cm]
|
pF (51)
|
bar (52)
|
Remarques
|
|
107
|
7
|
104
|
Sol sec
|
|
1,6 · 104
|
4,2
|
16
|
Point de flétrissement
|
|
104
|
4
|
10
|
|
|
103
|
3
|
1
|
|
|
6· 102
|
2,8
|
0,6
|
|
|
3,3 · 102
|
2,5
|
0,33 (53)
|
Échelle de la
capacité au champ (54)
|
|
102
|
2
|
0,1
|
|
60
|
1,8
|
0,06
|
|
33
|
1,5
|
0,033
|
|
10
|
1
|
0,01
|
Capacité de rétention d'eau (approximation)
|
|
1
|
0
|
0,001
|
Sol saturé en eau
|
La succion est mesurée en cm de colonne d'eau ou en bar. Comme l'échelle de valeur de la succion est très large, celle-ci est exprimée simplement en pF, qui est égal au logarithme de la colonne d'eau en cm.
La capacité au champ (FC) est définie comme la quantité d'eau qui peut être stockée contre la gravité par un sol naturel 2 jours après une longue période de pluie ou après une irrigation suffisante. Elle est déterminée dans un sol non brassé in situ dans le champ. Cette mesure ne peut donc pas s'appliquer aux échantillons de sol brassé. Les valeurs de capacité au champ déterminées dans les sols brassés peuvent présenter une variabilité systématique importante.
La capacité de rétention d'eau (WHC) est déterminée en laboratoire avec un sol non brassé et un sol brassé en saturant une colonne de sol avec de l'eau par capillarité. Elle est particulièrement utile pour les sols brassés et peut être jusqu'à 30 % supérieure à la capacité au champ (1). Elle est également plus facile à déterminer expérimentalement que des valeurs de capacité au champ fiables.
Notes
Annexe 2
TENEUR EN HUMIDITÉ (g d'eau pour 100 g de sol sec) DE DIFFÉRENTS TYPES DE SOL PROVENANT DE DIFFÉRENTS PAYS
|
|
|
Teneur en humidité à
|
|
Type de sol
|
Pays
|
|
|
|
WHC (55)
|
pF = 1,8
|
pF = 2,5
|
|
Sable
|
Allemagne
|
28,7
|
8,8
|
3,9
|
|
Sable limoneux
|
Allemagne
|
50,4
|
17,9
|
12,1
|
|
Sable limoneux
|
Suisse
|
44,0
|
35,3
|
9,2
|
|
Limon fin
|
Suisse
|
72,8
|
56,6
|
28,4
|
|
Limon argileux
|
Brésil
|
69,7
|
38,4
|
27,3
|
|
Limon argileux
|
Japon
|
74,4
|
57,8
|
31,4
|
|
Limon sableux
|
Japon
|
82,4
|
59,2
|
36,0
|
|
Limon fin
|
États-Unis
|
47,2
|
33,2
|
18,8
|
|
Limon sableux
|
États-Unis
|
40,4
|
25,2
|
13,3
|
Annexe 3
Figure 1
Exemple d'appareil à circulation continue pour étudier la transformation des substances chimiques dans le sol
(56)
(57)

Figure 2
Exemple de flacon de type biomètre pour l'étude de la transformation des substances chimiques dans le sol
(58)

C.24. TRANSFORMATION AÉROBIE ET ANAÉROBIE DANS LES SYSTÈMES SÉDIMENTAIRES AQUATIQUES
1. MÉTHODE
La présente méthode d'essai reprend l'essai TG 308 (2002) de l'OCDE.
1.1. INTRODUCTION
Les substances chimiques peuvent pénétrer dans les eaux de surface peu ou très profondes par des voies telles que l'application directe, la déperdition lors de l'épandage, le ruissellement, le drainage, l'élimination des déchets, les effluents industriels, domestiques ou agricoles et le dépôt atmosphérique. La présente méthode d'essai décrit une méthode de laboratoire conçue pour évaluer la transformation aérobie et anaérobie des substances chimiques organiques dans les systèmes sédimentaires aquatiques. Elle s'appuie sur les lignes directrices existantes (1) (2) (3) (4) (5) (6). Un atelier de l'OCDE sur la sélection des sols et des sédiments, qui s'est tenu à Belgirate, en Italie, en 1995 (7), a défini notamment le nombre et le type de sédiments à utiliser dans le cadre de cet essai. Il a également formulé des recommandations relatives au prélèvement, à la manipulation et au stockage des échantillons de sédiments, sur la base des lignes directrices de l'ISO (8). Ces études sont nécessaires pour les substances chimiques qui sont introduites directement dans l'eau ou qui sont susceptibles de pénétrer dans le milieu aquatique par les voies décrites ci-dessus.
La phase aqueuse supérieure des systèmes sédimentaires aquatiques présente souvent des conditions aérobies. La couche superficielle du sédiment peut être aérobie ou anaérobie, alors que, en profondeur, le sédiment est généralement anaérobie. Afin de tenir compte de toutes ces possibilités, le présent document décrit des essais aérobies et anaérobies. L'essai aérobie simule une colonne d'eau aérobie sur une couche de sédiment aérobie et une sous-couche avec un gradient anaérobie. L'essai anaérobie simule un système eau-sédiment complètement anaérobie. Si, selon les circonstances, il est nécessaire de s'écarter sensiblement de ces recommandations, par exemple en utilisant des carottes de sédiment intact ou des sédiments qui ont pu être exposés à la substance d'essai, il existe d'autres méthodes à cette fin (9).
1.2. DÉFINITIONS
Il convient d'utiliser dans tous les cas les unités standard internationales (SI).
Substance d'essai: toute substance, qu'il s'agisse de la substance mère ou des produits de transformation correspondants.
Produits de transformation: toutes les substances résultant des réactions de transformation biotiques et abiotiques de la substance d'essai, et notamment le CO2 et les résidus liés.
Résidus liés: les «résidus liés» désignent des composés du sol, végétaux ou animaux, qui subsistent dans la matrice après extraction, sous la forme de la substance mère ou de ses métabolite(s). La méthode d'extraction ne doit pas modifier de manière substantielle les composés eux-mêmes ni la structure de la matrice. La nature de la liaison peut être clarifiée en partie par les méthodes d'extraction qui modifient la matrice et des techniques analytiques sophistiquées. C'est de cette façon que l'on a déterminé jusqu'ici la nature de la liaison, covalente ionique, par sorption ou piégeage. De manière générale, la formation de résidus liés abaisse sensiblement la bioaccessibilité et la biodisponibilité (10) [modifié d'après l'UICPA 1984 (11)].
Transformation aérobie: (oxydation): réaction se produisant en présence d'oxygène moléculaire (12).
Transformation anaérobie: (réduction): réaction se produisant en l'absence d'oxygène moléculaire (12).
Eaux naturelles: eaux superficielles provenant de mares, rivières, fleuves, etc.
Sédiment: mélange de constituants chimiques minéraux et organiques, ces derniers contenant des composés à haute teneur en carbone et en azote et à masse moléculaire élevée. Il est déposé par les eaux naturelles avec lesquelles il forme une interface.
Minéralisation: dégradation complète d'un composé organique en CO2 et H2O dans des conditions aérobies, et en CH4, CO2 et H2O dans des conditions anaérobies. Dans le cadre de la présente méthode d'essai, lorsqu'on utilise un composé radiomarqué, la minéralisation désigne la dégradation importante au cours de laquelle un atome de carbone marqué est oxydé ou réduit quantitativement avec dégagement de la quantité correspondante de 14CO2 ou de 14CH4, respectivement.
Demi-vie, t0,5, temps nécessaire à la transformation de 50 % d'une substance d'essai lorsque la transformation peut être décrite par une cinétique du premier ordre; elle est indépendante de la concentration initiale.
DT
50
(temps de dégradation 50): période au cours de laquelle la concentration initiale de la substance d'essai diminue de 50 %.
DT
75
(temps de dégradation 75): période au cours de laquelle la concentration initiale de la substance d'essai diminue de 75 %.
DT
90
(temps de dégradation 90): période au cours de laquelle la concentration initiale de la substance d'essai diminue de 90 %.
1.3. SUBSTANCES DE RÉFÉRENCE
Des substances de référence doivent être utilisées pour caractériser et/ou identifier les produits de transformation par des méthodes spectroscopiques et chromatographiques.
1.4. INFORMATIONS RELATIVES À LA SUBSTANCE D'ESSAI
Une substance d'essai marquée ou non marquée peut être utilisée pour mesurer le taux de transformation, bien qu'il soit préférable d'utiliser du matériel marqué. Il est nécessaire d'utiliser du matériel marqué pour étudier les voies de transformation et établir un bilan massique, le marquage au 14C est recommandé mais l'utilisation d'autres isotopes, comme 13C, 15N, 3H, 32P, peut aussi être utile. Dans la mesure du possible, le-marquage doit être situé dans la partie(s) la plus stable de la molécule (59). La substance d'essai doit avoir une pureté chimique et/ou radiochimique d'au moins 95 %.
Avant de procéder à un essai, il convient de disposer des informations suivantes sur la substance d'essai:
|
a)
|
solubilité dans l'eau (Méthode A.6);
|
|
b)
|
solubilité dans les solvants organiques;
|
|
c)
|
pression de vapeur (Méthode A.4) et constante de la loi de Henry;
|
|
d)
|
coefficient de partage n-octanol/eau (Méthode A.8);
|
|
e)
|
coefficient d'adsorption (Kd, Kr ou Koc, s'il y a lieu) (Méthode C.18);
|
|
f)
|
hydrolyse (Méthode C.7);
|
|
g)
|
constante de dissociation (pKa) [ligne directrice 112 de l'OCDE] (13);
|
|
h)
|
structure chimique de la substance d'essai et position des marqueurs isotopiques, s'il y a lieu.
|
Note: Il convient d'indiquer la température à laquelle ces mesures ont été effectuées.
Il peut également être utile de disposer d'informations sur la toxicité de la substance d'essai sur les micro-organismes, sur la biodégradabilité immédiate et/ou intrinsèque, ainsi que sur la transformation aérobie et anaérobie dans le sol.
Il faut disposer des méthodes analytiques (dont des méthodes d'extraction et de purification) nécessaires à la quantification et à l'identification de la substance d'essai et de ses produits de transformation dans l'eau et dans les sédiments (voir le point 1.7.2).
1.5. PRINCIPE DE LA MÉTHODE
La méthode décrite ici emploie un système sédimentaire aquatique aérobie et anaérobie (voir l'annexe 1) qui permet:
|
i)
|
de mesurer le taux de transformation de la substance d'essai dans le sédiment;
|
|
ii)
|
de mesurer le taux de transformation de la substance d'essai dans le sédiment;
|
|
iii)
|
de mesurer le taux de minéralisation de la substance d'essai et/ou de ses produits de transformation (lorsqu'on utilise une substance d'essai marquée au 14C);
|
|
iv)
|
d'identifier et de quantifier les produits de transformation dans les phases aqueuse et sédimentaire et notamment le bilan massique (lorsqu'on utilise une substance d'essai marquée);
|
|
v)
|
de mesurer la répartition de la substance d'essai et de ses produits de transformation entre les deux phases pendant une période d'incubation dans le noir (pour éviter, par exemple, la prolifération d'algues) à température constante. Les durées de demi-vie, les valeurs DT50, DT75 et DT90 sont déterminées lorsque les données le permettent, mais elles ne doivent pas être extrapolées bien au-delà de la période expérimentale (voir le point 1.2).
|
Au moins deux sédiments et les phases aqueuses associées sont nécessaires pour les études aérobies et anaérobies respectivement (7). Il peut toutefois être nécessaire dans certains cas d'utiliser plus de deux sédiments aquatiques, par exemple, pour une substance chimique pouvant être présente dans l'eau douce et/ou le milieu marin.
1.6. APPLICABILITÉ DE L'ESSAI
La méthode peut être appliquée aux substances chimiques (marquées ou non marquées) pour lesquelles il existe une méthode analytique de précision et de sensibilité suffisantes. Elle s'applique à des composés faiblement volatils, non volatils, hydrosolubles ou peu hydrosolubles. L'essai ne doit pas être appliqué aux substances chimiques fortement volatiles dans l'eau (fumigants, solvants organiques) et qui ne peuvent donc pas être gardées dans l'eau et/ou le sédiment dans les conditions expérimentales de cet essai.
La méthode a été appliquée jusqu'ici pour étudier la transformation des substances chimiques dans les eaux douces et dans les sédiments, mais, en principe, elle peut également être appliquée aux systèmes estuariens/marins. Elle n'est pas adaptée à la simulation des conditions de l'eau courante (rivières) ou de pleine mer.
1.7. CRITÈRES DE QUALITÉ
1.7.1. Récupération
L'extraction et l'analyse, au moins en double exemplaire, des échantillons d'eau et de sédiment immédiatement après l'addition de la substance d'essai donnent une première indication de la reproductibilité de la méthode analytique et de l'uniformité de la procédure d'application de la substance d'essai. Les taux de récupération des stades ultérieurs des expériences sont fournis par les bilans massiques respectifs (lorsqu'on utilise une substance marquée). Les taux de récupération doivent osciller entre 90 % et 110 % pour les substances chimiques marquées (6) et entre 70 % et 110 % pour les substances chimiques non marquées.
1.7.2. Reproductibilité et sensibilité de la méthode analytique
La reproductibilité de la méthode analytique (à l'exception du rendement d'extraction initial) servant à quantifier la substance d'essai et les produits de transformation peut être contrôlée en effectuant en double les analyses du même extrait d'échantillons d'eau ou de sédiments, incubés suffisamment longtemps pour permettre la formation de produits de transformation.
La limite de détection de la méthode d'analyse de la substance d'essai et ses produits de transformation doit être au moins égale à 0,01 mg.kg-1 dans l'eau ou le sédiment (substance d'essai) ou à 1 % de la quantité initiale appliquée à un système d'essai si cette quantité est inférieure. La limite de quantification doit également être spécifiée.
1.7.3. Précision des données de transformation
L'analyse de régression des concentrations de la substance d'essai en fonction du temps permet d'obtenir les informations appropriées sur la précision de la courbe de transformation et de calculer les limites de confiance des demi-vies (en cas de cinétique du pseudo premier ordre) ou des valeurs DT50 et, le cas échéant, des valeurs DT75 et DT90.
1.8. DESCRIPTION DE LA MÉTHODE
1.8.1. Système et appareillage d'essai
L'étude doit être effectuée dans des récipients en verre (bouteilles, tubes de centrifugation), à moins que les informations préliminaires (coefficient de partage n-octanol/eau, données de sorption, etc.) indiquent que la substance d'essai risque d'adhérer au verre, auquel cas on peut envisager l'utilisation d'un autre matériau (Téflon). Lorsque l'on sait que la substance d'essai adhère au verre, il est possible de contourner le problème en utilisant l'une des méthodes suivantes:
|
—
|
déterminer la masse de la substance d'essai et de ses produits de transformation sorbés sur le verre,
|
|
—
|
veiller à nettoyer toute la verrerie à l'aide d'un solvant à la fin de l'essai,
|
|
—
|
utilisation de produits formulés (voir également le point 1.9.2),
|
|
—
|
utilisation d'une quantité croissante de cosolvant pour ajouter la substance d'essai au système; si l'on utilise un cosolvant, celui-ci ne doit pas entraîner de réaction de solvolyse avec la substance d'essai.
|
Des exemples d'appareillage d'essai courant (systèmes à flux continu et biomètres) sont présentés dans les annexes 2 et 3, respectivement (14). On trouvera d'autres systèmes d'incubation sous la référence 15. L'appareil utilisé pour l'expérience doit permettre l'échange d'air ou d'azote et le piégeage des produits volatils. Les dimensions de l'appareillage doivent permettre de satisfaire aux exigences de l'essai (voir le point 1.9.1). La ventilation peut être effectuée soit par léger barbotage soit en faisant circuler de l'air ou de l'azote à la surface de l'eau. Dans ce cas, il est conseillé de remuer doucement la surface de l'eau pour obtenir une meilleure répartition de l'oxygène ou de l'azote dans l'eau. Il ne faut pas utiliser d'air sans CO2 car cela pourrait entraîner une augmentation du pH de l'eau. Dans les deux cas, il n'est pas souhaitable de toucher au sédiment et il convient de l'éviter dans toute la mesure du possible. Les substances chimiques faiblement volatiles doivent être testées dans un système de type biomètre en remuant légèrement la surface de l'eau. Des récipients fermés avec un espace libre d'air atmosphérique ou d'azote et des fioles à l'intérieur pour le piégeage des produits volatils peuvent également être utilisés (16). Un bon échange du gaz de surface est nécessaire dans l'essai aérobie afin de compenser la consommation d'oxygène par la biomasse.
La liste non restrictive des pièges adaptés à la collecte des produits de transformation volatils est la suivante: solutions à 1 mol.dm-3 d'hydroxyde de potassium ou d'hydroxyde de sodium pour le dioxyde de carbone (60) et éthylène glycol, éthanolamine ou 2 % paraffine dans le xylène pour les composés organiques. Les produits volatils formés en anaérobie, comme le méthane, peuvent être récupérés, par exemple, à l'aide de tamis moléculaires. Ces produits volatils peuvent être brûlés, par exemple, en CO2 en faisant passer le gaz à travers un tube de quartz rempli de CuO à la température de 900 oC et en piégeant le CO2 formé dans une colonne d'absorption contenant un produit alcalin (17).
Des instruments de laboratoire pour l'analyse chimique de la substance d'essai et des produits de transformation sont nécessaires [chromatographie gaz-liquide (GLQ), chromatographie en phase liquide à haute performance (HPLC), chromatographie en couche mince (TLC), spectroscopie de masse (MS), chromatographie en phase gazeuse couplée à la spectrométrie de masse (GC-MS), chromatographie en phase liquide couplée à la spectrométrie de masse (LC-MS), résonance magnétique nucléaire (RMN), etc.], ainsi que, le cas échéant, des dispositifs de détection des substances chimiques radiomarquées ou non marquées. Lorsqu'on utilise des substances radiomarquées, un compteur à scintillation liquide et un appareil d'oxydation pour la combustion (pour la combustion des échantillons de sédiment avant l'analyse de radioactivité) sont également nécessaires.
D'autres équipements courants de laboratoire peuvent s'avérer nécessaires selon le cas pour effectuer des analyses physico-chimiques et biologiques (voir le tableau 1 du point 1.8.2.2), ainsi que de la verrerie, des produits chimiques et des réactifs.
1.8.2. Sélection et nombre de sédiments aquatiques
Les sites de prélèvement doivent être choisis en fonction de la finalité de l'essai dans une situation donnée. Pour choisir les sites de prélèvement, il convient de prendre en compte l'historique des éventuels apports agricoles, industriels ou domestiques dans le bassin versant et les eaux en amont. Les sédiments ne doivent pas être utilisés s'ils ont été contaminés par la substance d'essai ou ses analogues structuraux au cours des quatre années précédentes.
1.8.2.1. Sélection des sédiments
On utilise généralement deux sédiments pour les études aérobies (7). Les deux sédiments sélectionnés doivent différer en texture et en teneur en carbone organique. Un sédiment doit avoir une teneur élevée en carbone organique (2,5-7,5 %) et une texture fine, et l'autre doit avoir une teneur en carbone organique peu élevée (0,5-2,5 %) et une texture grossière. La différence de teneur en carbone organique doit être normalement égale ou supérieure à 2 %. On entend par «texture fine» une teneur en [argile + limon] (61) > 50 % et par «texture grossière» une teneur en [argile + limon] < 50 %. La différence de teneur [argile + limon] entre les deux sédiments doit être normalement égale ou supérieure à 20 %. Dans le cas où des substances chimiques risquent d'entrer également en contact avec les eaux de mer, au moins un des deux systèmes eau-sédiment doit être d'origine marine.
Pour l'étude strictement anaérobie, deux échantillons de sédiments (ainsi que la phase aqueuse associée) doivent être prélevés dans les zones anaérobies des systèmes à eaux de surface (7). Les sédiments comme les phases aqueuses doivent être manipulés et transportés avec précaution en évitant tout contact avec l'oxygène.
D'autres paramètres peuvent présenter de l'importance dans la sélection des sédiments et ils doivent être envisagés cas par cas. Par exemple, le pH des sédiments est important pour tester les substances chimiques dont la transformation et/ou la sorption peuvent être dépendantes du pH. La dépendance au pH de la sorption peut être une conséquence du pKa de la substance d'essai.
1.8.2.2. Caractérisation des échantillons eau-sédiment
Les paramètres clés qui doivent être mesurés et notés dans le rapport (en indiquant la méthode utilisée) pour l'eau et pour le sédiment, ainsi que l'étape de l'essai à laquelle ces paramètres doivent être déterminés, sont résumés dans le tableau ci-dessous. Pour information, les méthodes de détermination de ces paramètres sont fournies sous les références (18) (19) (20) (21).
Par ailleurs, il peut être nécessaire de mesurer et d'enregistrer d'autres paramètres selon le cas [eau douce: particules, alcalinité, dureté, conductivité, NO3/PO4 (rapport et valeurs individuelles); pour les sédiments: capacité d'échange cationique, capacité de rétention d'eau, carbonate, azote total et phosphore; pour les systèmes marins: salinité]. Il peut également être utile d'analyser les sédiments et l'eau pour mesurer le nitrate, le sulfate, le fer biodisponible, et d'autres accepteurs d'électrons peuvent aussi être utiles pour évaluer les conditions redox, notamment en ce qui concerne la transformation anaérobie.
Paramètres mesurés pour la caractérisation des échantillons d'eau et de sédiment (7) (22) (23)
|
Paramètre
|
Étape de la procédure d'essai
|
|
prélèvement sur site
|
post-manipulation
|
début de l'acclimatation
|
début de l'essai
|
pendant l'essai
|
à la fin de l'essai
|
|
Eau
|
|
Origine/source
|
x
|
|
|
|
|
|
|
Température
|
x
|
|
|
|
|
|
|
pH
|
x
|
|
x
|
x
|
x
|
x
|
|
Carbone organique total
|
|
|
x
|
x
|
|
x
|
|
Concentration O2*
|
x
|
|
x
|
x
|
x
|
x
|
|
Potentiel redox*
|
|
|
x
|
x
|
x
|
x
|
|
Sédiment
|
|
Origine/source
|
x
|
|
|
|
|
|
|
Profondeur de la couche
|
x
|
|
|
|
|
|
|
pH
|
|
x
|
x
|
x
|
x
|
x
|
|
Répartition de la taille des particules
|
|
x
|
|
|
|
|
|
Carbone organique total
|
|
x
|
x
|
x
|
|
x
|
|
Biomasse microbienne (63)
|
|
x
|
|
x
|
|
x
|
|
Potentiel redox (62)
|
Observation (couleur/ odeur)
|
|
x
|
x
|
x
|
x
|
1.8.3. Prélèvement, manipulation et stockage
1.8.3.1. Prélèvement
Il convient de se référer au projet de ligne directrice ISO sur l'échantillonnage des fonds sédimentaires (8) pour prélever les échantillons de sédiment. Les échantillons de sédiment doivent être prélevés sur la totalité de la couche supérieure de 5 à 10 cm du sédiment. L'eau correspondante doit être collectée sur le même site ou dans le même lieu et en même temps que le sédiment. Pour l'étude anaérobie, le sédiment et l'eau correspondante doivent être prélevés et transportés en évitant tout contact avec l'oxygène (28) (voir le point 1.8.2.1). Quelques dispositifs d'échantillonnage sont décrits dans la littérature (8) (23).
1.8.3.2. Manipulation
Le sédiment est séparé de l'eau par filtration et passé au travers d'un tamis de 2 mm en utilisant l'eau excédentaire prélevée sur le même site qui est ensuite éliminée. Des quantités connues de sédiments et d'eau sont ensuite mélangées dans le rapport désiré (voir le point 1.9.1) dans des flacons d'incubation et préparées pour la période d'acclimatation (voir le point 1.8.4). Pour l'étude anaérobie, toutes les étapes de la manipulation doivent être effectuées en l'absence d'oxygène (29) (30) (31) (32) (33).
1.8.3.3. Stockage
Il est vivement recommandé d'utiliser des sédiments et de l'eau fraîchement prélevés, mais si le stockage est nécessaire, le sédiment et l'eau doivent être tamisés comme décrit ci-dessus et stockés ensemble, recouverts d'eau (couche d'eau de 6-10 cm), dans l'obscurité, à 4 ± 2 oC4 pendant une durée maximale de quatre semaines (7) (8) (23). Les échantillons destinés aux études aérobies doivent être stockés en laissant un accès libre à l'air (dans des récipients ouverts), alors que les échantillons destinés aux études anaérobies doivent être conservés en l'absence d'oxygène. Il ne faut pas que le sédiment et l'eau soient congelés ni que le sédiment sèche pendant le transport et le stockage.
1.8.4. Préparation des échantillons de sédiment/eau pour l'essai
Une période d'acclimatation doit être respectée avant d'ajouter la substance d'essai, au cours de laquelle chaque échantillon de sédiment/eau est placé dans le récipient d'incubation à utiliser dans l'essai principal, et l'acclimatation est réalisée exactement dans les mêmes conditions que l'incubation de l'essai (voir le point 1.9.1). La période d'acclimatation est le temps nécessaire pour atteindre une stabilité suffisante du système, en termes de pH, de concentration de l'oxygène dans l'eau, de potentiel redox du sédiment et de l'eau, et de séparation macroscopique des phases. La période d'acclimatation doit durer normalement de une à deux semaines et elle ne doit pas excéder quatre semaines. Le résultat des déterminations effectuées pendant cette période doit être enregistré.
1.9. RÉALISATION DE L'ESSAI
1.9.1. Conditions de l'essai
L'essai doit être effectué dans le dispositif d'incubation (voir le point 1.8.1) avec un rapport de volume eau/sédiment situé entre 3:1 et 4:1, et une couche sédimentaire de 2,5 cm (±0,5 cm). (64) On recommande une quantité minimale de 50 g de sédiment (sur une base de poids sec) par récipient d'incubation.
L'essai doit être effectué dans le noir à température constante dans la fourchette de 10 à 30 oC. La température indiquée est de 20 ± 2 oC. Le cas échéant, une température supplémentaire plus basse (10 oC par exemple) peut être envisagée selon le cas, en fonction de l'information que l'on souhaite retirer de l'essai. La température d'incubation doit être surveillée et enregistrée.
1.9.2. Traitement et application de la substance d'essai
Une seule concentration d'essai de la substance chimique est utilisée (65) Pour les produits phytosanitaires appliqués directement dans le milieu aquatique, le dosage maximal indiqué sur le mode d'emploi doit être pris comme taux maximal d'application calculé sur la base de la surface de l'eau dans le récipient d'essai. Dans tous les autres cas, la concentration à utiliser doit s'appuyer sur les estimations des émissions dans l'environnement. Il convient de veiller à appliquer une concentration de substance d'essai adéquate afin de caractériser la voie de transformation et la formation et le déclin des produits de transformation. Il peut être nécessaire d'appliquer des doses plus élevées (10 fois par exemple) dans le cas où les concentrations de la substance d'essai sont proches de la limite de détection au début de l'étude et/ou les principaux produits de transformation n'ont pas pu être facilement détectés à un taux égal à 10 % du taux d'application de la substance d'essai. Toutefois, si des concentrations d'essai plus élevées sont utilisées, celles-ci ne doivent pas avoir un effet négatif important sur l'activité microbienne du système eau-sédiment. Afin d'obtenir une concentration constante de la substance d'essai dans des récipients de dimensions différentes, il peut être indiqué d'ajuster la quantité de produit appliqué, en fonction de la profondeur de la colonne d'eau dans le récipient par rapport à la profondeur de l'eau dans le champ (que l'on suppose égale à 100 cm, mais on peut prendre une autre profondeur comme base). Voir l'annexe 4 pour un exemple de calcul.
Théoriquement, la substance d'essai doit être appliquée sous forme de solution aqueuse dans la phase aqueuse du système d'essai. S'il n'est pas possible de procéder autrement, on peut utiliser de faibles quantités de solvants miscibles avec l'eau (comme l'acétone ou l'éthanol) pour appliquer et répartir la substance d'essai, mais ce solubilisant ne doit pas dépasser 1 % v/v ni avoir d'effets négatifs sur l'activité microbienne du système d'essai. La solution aqueuse de la substance d'essai doit être préparée avec soin — afin d'assurer une homogénéité totale, on peut effectuer un prémélange et utiliser des colonnes de générateur. Après l'addition de la solution aqueuse au système d'essai, il est recommandé de remuer doucement la phase aqueuse en brassant le moins possible le sédiment.
Il n'est pas recommandé d'utiliser de manière routinière des produits formulés car les ingrédients de formulation risquent d'affecter la répartition de la substance d'essai et/ou des produits de transformation entre la phase aqueuse et la phase sédimentaire. Toutefois, dans le cas de substances peu solubles dans l'eau, l'utilisation de matériau formulé peut être une solution de remplacement appropriée.
Le nombre de récipients d'incubation dépend du nombre de temps de prélèvement (voir le point 1.9.3). Un nombre suffisant de systèmes d'essai doit être prévu de façon à pouvoir sacrifier deux systèmes à chaque temps de prélèvement. Lorsqu'on utilise des unités témoins de chaque système sédimentaire aquatique, ces unités ne doivent pas être traitées avec la substance d'essai. Les unités témoins peuvent être utilisées pour déterminer la biomasse microbienne du sédiment et le carbone organique total de l'eau et du sédiment à la fin de l'étude. Deux unités témoins (une unité de chaque sédiment aquatique) peuvent être utilisées pour surveiller les paramètres requis dans le sédiment et dans l'eau pendant la période d'acclimatation (voir le tableau du point 1.8.2.2). Deux témoins supplémentaires doivent être ajoutés lorsque la substance d'essai est appliquée au moyen d'un solvant afin de mesurer les effets négatifs sur l'activité microbienne du système d'essai.
1.9.3. Durée de l'essai et échantillonnage
L'expérience ne doit pas durer plus de 100 jours (6), et elle doit se poursuivre jusqu'à ce que les voies de dégradation et le profil de répartition eau/sédiment se soient établis ou lorsque 90 % de la substance d'essai s'est dissipée par transformation et/ou volatilisation. Le nombre de temps de prélèvement doit être au moins égal à six (y compris le temps zéro), et une étude préliminaire facultative (voir le point 1.9.4) est effectuée pour déterminer le régime de prélèvement et la durée de l'essai, à moins que l'on ne dispose de données suffisantes sur la substance d'essai d'après les études antérieures. Pour les substances hydrophobes, il peut être nécessaire d'effectuer des points de prélèvement complémentaires pendant la période initiale afin de déterminer le taux de répartition entre la phase aqueuse et la phase sédimentaire.
À chaque temps de prélèvement, les récipients d'incubation (réplicats) sont retirés pour analyse. Le sédiment et l'eau qui le recouvre sont analysés séparément (66). L'eau de surface doit être retirée avec précaution en évitant autant que possible de toucher au sédiment. L'extraction et la caractérisation de la substance d'essai et des produits de transformation doivent suivre les procédures analytiques appropriées. Il faut prendre soin d'éliminer les matières adsorbées sur les parois du récipient d'incubation et dans les tuyaux utilisés pour piéger les produits volatils.
1.9.4. Essai préliminaire facultatif
S'il n'est pas possible de prévoir la durée et le régime d'échantillonnage à partir d'autres études analogues sur la substance d'essai, il peut être indiqué d'effectuer un essai préliminaire, dans les mêmes conditions d'essai que les conditions proposées pour l'étude définitive. Si cet essai préliminaire est effectué, les conditions expérimentales et les résultats de l'essai doivent être décrits brièvement.
1.9.5. Mesures et analyse
La concentration de la substance d'essai et des produits de transformation à chaque temps de prélèvement dans l'eau et le sédiment doit être mesurée et enregistrée (en concentration et en pourcentage de la substance appliquée). En règle générale, tout produit de transformation détecté à > 10 % de la radioactivité totale appliquée au système eau/sédiment, quel que soit le temps de prélèvement, doit être identifié, à moins d'une justification suffisante. Les produits de transformation dont les concentrations sont en augmentation constante pendant la durée de l'étude doivent également être identifiés, même si leurs concentrations ne dépassent pas les limites indiquées ci-dessus, car cela peut indiquer une persistance. Des justifications doivent être fournies dans le rapport.
Les résultats des systèmes de piégeage des gaz/volatils (CO2 et autres, à savoir les composés organiques volatils) doivent être enregistrés à chaque temps de prélèvement. Les taux de minéralisation doivent être enregistrés. Les résidus (liés) non extractibles dans le sédiment doivent être notés à chaque temps de prélèvement.
2. DONNÉES
2.1. TRAITEMENT DES RÉSULTATS
Le bilan massique total ou la récupération (voir le point 1.7.1) de la radioactivité ajoutée doit être calculé à chaque temps de prélèvement. Les résultats doivent être transcrits en pourcentage de radioactivité ajoutée. La répartition de la radioactivité entre l'eau et le sédiment doit être transcrite sous la forme de concentration et de pourcentage, à chaque temps de prélèvement.
La demi-vie, les valeurs DT50 et, le cas échéant, DT75 et DT90 de la substance d'essai doivent être calculées avec leurs limites de confiance (voir le point 1.7.3). Différents outils d'évaluation permettent d'obtenir des informations sur le taux de dégradation de la substance d'essai dans l'eau et le sédiment. Il est possible d'appliquer des cinétiques du pseudo premier ordre, des techniques d'interpolation empiriques utilisant des solutions graphiques ou numériques ou d'autres méthodes d'évaluation utilisant par exemple des modèles à un ou plusieurs compartiments. On trouvera plus de détails dans la littérature spécialisée (35) (36) (37).
Toutes les méthodes ont leurs avantages et leurs inconvénients et leur complexité varie considérablement. L'hypothèse d'une cinétique du premier ordre peut être une simplification trop poussée des processus de dégradation et de répartition, mais cette hypothèse, lorsque c'est possible, donne un terme (la constante de vitesse ou la demi-vie) facilement compréhensible et très utile en modélisation par simulation et pour calculer les concentrations prévisibles dans l'environnement. Les méthodes empiriques ou les transformations linéaires peuvent produire une meilleure interpolation entre les courbes et les données et permettre ainsi une meilleure estimation des demi-vies, des valeurs DT50 et, le cas échéant, DT75 et DT90. L'utilisation des constantes dérivées est toutefois limitée. Les modèles à compartiments servent à établir des constantes utiles pour l'évaluation des risques qui décrivent la vitesse de dégradation dans les différents compartiments et la répartition de la substance chimique. Ils doivent également être utilisés pour estimer les constantes de vitesse de formation et de dégradation des principaux produits de transformation. Dans tous les cas, la méthode choisie doit être justifiée et l'expérimentateur doit démontrer graphiquement et/ou statistiquement la qualité de l'ajustement.
3. RAPPORT
3.1. RAPPORT D'ESSAI
Le rapport doit comprendre les informations suivantes:
|
|
Substance d'essai:
|
—
|
nom courant, nom chimique, numéro de fichier CAS, formule structurale (indiquant la position du marquage lorsqu'on utilise un produit radiomarqué) et propriétés physico-chimiques pertinentes,
|
|
—
|
pureté (impuretés) de la substance d'essai,
|
|
—
|
pureté radiochimique de la substance marquée et activité molaire (s'il y a lieu).
|
|
|
|
Substances de référence:
|
—
|
nom et structure chimique des substances de référence utilisées pour caractériser et/ou identifier les produits de transformation.
|
|
|
|
Sédiments et eaux d'essai:
|
—
|
localisation et description du (ou des) site(s) de prélèvement des sédiments aquatiques avec, si possible, l'historique de la contamination,
|
|
—
|
toute information relative au prélèvement, au stockage (s'il y a lieu) et à l'acclimatation des systèmes eau-sédiment,
|
|
—
|
caractéristiques des échantillons eau-sédiment telles qu'indiquées dans le tableau du point 1.8.2.2.
|
|
|
|
Conditions de l'essai:
|
—
|
système d'essai utilisé (circulation continue, biomètre, mode de ventilation, méthode de remuage, volume d'eau, masse sédimentaire, épaisseur de la couche d'eau et de la couche de sédiment, dimension des récipients d'essai, etc.),
|
|
—
|
application de la substance d'essai au système: concentration utilisée, nombre de réplicats et de témoins, mode d'application de la substance d'essai (usage éventuel de solvant), etc.,
|
|
—
|
température d'incubation,
|
|
—
|
méthodes d'extraction, rendements et limites de détection des méthodes analytiques,
|
|
—
|
méthodes de caractérisation/identification des produits de transformation,
|
|
—
|
modification du protocole d'essai ou des conditions de l'essai pendant l'étude.
|
|
|
|
Résultats:
|
—
|
données brutes des analyses représentatives (toutes les données brutes doivent être conservées dans les archives BPL),
|
|
—
|
reproductibilité et sensibilité des méthodes analytiques utilisées,
|
|
—
|
taux de récupération (les % acceptables pour valider une étude sont présentés au point 1.7.1),
|
|
—
|
tableaux des résultats exprimés en % de la dose appliquée et en mg.kg-1 dans l'eau, le sédiment et le système total (% uniquement) de la substance d'essai et, s'il y a lieu, des produits de transformation et de la radioactivité non extractible,
|
|
—
|
bilan massique pendant la durée et à la fin des expériences,
|
|
—
|
représentation graphique de la transformation dans les fractions eau/sédiment et dans le système total (y compris la minéralisation),
|
|
—
|
taux de minéralisation,
|
|
—
|
durée de demi-vie, DT50 et, le cas échéant, valeurs DT75 et DT90 de la substance d'essai et, s'il y a lieu, des principaux produits de transformation comprenant les limites de confiance dans l'eau, le sédiment et dans le système total,
|
|
—
|
une évaluation de la cinétique de transformation de la substance d'essai et, le cas échéant, des principaux produits de transformation,
|
|
—
|
voie de transformation proposée, le cas échéant,
|
|
—
|
discussion des résultats.
|
|
4. RÉFÉRENCES
|
(1)
|
BBA-Guidelines for the examination of plant protectors in the registration process. (1990). Part IV, Section 5-1: Degradability and fate of plant protectors in the water/sediment System. Germany.
|
|
(2)
|
Commission for registration of pesticides: Application for registration of a pesticide. (1991). Part G. Behaviour of the product and its metabolites in soil, water and air, Section G.2.1 (a). The Netherlands.
|
|
(3)
|
MAFF Pesticides Safety Directorate. (1992). Preliminary guideline for the conduct of biodegradability tests on pesticides in natural sediment/water Systems. Ref No SC 9046. United Kingdom.
|
|
(4)
|
Agriculture Canada: Environmental chemistry and fate. (1987). Guidelines for registration of pesticides in Canada. Aquatic (Laboratory) — Anaerobic and aerobic. Canada. p. 35-37.
|
|
(5)
|
US-EPA: Pesticide assessment guidelines, Subdivision N. Chemistry: Environmental fate (1982). Section 162-3, Anaerobic aquatic metabolism.
|
|
(6)
|
SETAC-Europe publication. (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides. Ed. Dr Mark R. Lynch. SETAC-Europe, Brussels.
|
|
(7)
|
OECD Test Guidelines Programme. (1995). Final Report of the OECD Workshop on Selection of Soils/sediments, Belgirate, Italy, 18-20 January 1995.
|
|
(8)
|
ISO/DIS 5667-12. (1994). Water quality — Sampling — Part 12: Guidance on sampling of bottom sediments.
|
|
(9)
|
US-EPA (1998a). Sediment/water microcosm biodegradation test. Harmonised Test Guidelines (OPPTS 835.3180). EPA 712-C-98-080.
|
|
(10)
|
DFG: Pesticide Bound Residues in Soil. Wiley-VCH (1998).
|
|
(11)
|
T.R. Roberts: Non-extractable pesticide residues in soils and plants. Pure Appl. Chem. 56, p. 945-956 (IUPAC 1984).
|
|
(12)
|
OECD Test Guideline 304A: Inherent Biodegradability in Soil (adopted 12 May 1981).
|
|
(13)
|
OECD (1993): Guidelines for Testing of Chemicals. Paris. OECD (1994-2000): Addenda 6-11 to Guidelines for the Testing of Chemicals.
|
|
(14)
|
Scholz, K., Fritz R., Anderson C. and Spiteller M. (1988). Degradation of pesticides in an aquatic model ecosystem.BCPC — Pests and Diseases, 3B-4, p. 149-158.
|
|
(15)
|
Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry (D.H. Hutson, T.R. Roberts, Eds.), Vol. 1, 85-114. J. Wiley & Sons.
|
|
(16)
|
Madsen, T., Kristensen, P. (1997). Effects of bacterial inoculation and non-ionic surfactants on degradation of polycyclic aromatic hydrocarbons in soil. Environ. Toxicol. Chem. 16, p. 631-637.
|
|
(17)
|
Steber, J., Wierich, P. (1987). The anaerobic degradation of detergent range fatty alcohol ethoxylates. Studies with 14C-labelled model surfactants. Water Research 21, p. 661-667.
|
|
(18)
|
Black, C.A. (1965). Methods of Soil Analysis. Agronomy Monograph No. 9. American Society of Agronomy, Madison.
|
|
(19)
|
APHA (1989). Standard Methods for Examination of Water and Wastewater (17th edition). American Public Health Association, American Water Works Association and Water Pollution Control Federation, Washington D.C.
|
|
(20)
|
Rowell, D.L. (1994). Soil Science Methods and Applications. Longman.
|
|
(21)
|
Light, T.S. (1972). Standard solution for redox potential measurements. Anal. Chemistry 44, p. 1038- 1039.
|
|
(22)
|
SETAC-Europe publication (1991). Guidance document on testing procedures for pesticides in fresh water mesocosms. From the Workshop «A Meeting of Experts on Guidelines for Static Field Mesocosms Tests», 3-4 July 1991.
|
|
(23)
|
SETAC-Europe publication. (1993). Guidance document on sediment toxicity tests and bioassays for freshwater and marine environments. From the Workshop On Sediment Toxicity Assessment (WOSTA), 8-10 November 1993. Eds.: I.R. Hill, P. Matthiessen and F. Heimbach.
|
|
(24)
|
Vink, J.P.M., van der Zee, S.E.A.T.M. (1997). Pesticide biotransformation in surface waters: multivariate analyses of environmental factors at field sites. Water Research 31, p. 2858-2868.
|
|
(25)
|
Vink, J.P.M., Schraa, G., van der Zee, S.E.A.T.M. (1999). Nutrient effects on microbial transformation of pesticides in nitrifying waters. Environ. Toxicol, p. 329-338.
|
|
(26)
|
Anderson, T.H., Domsch, K.H. (1985). Maintenance carbon requirements of actively-metabolising microbial populations under in-situ conditions. Soil Biol. Biochem. 17, p. 197-203.
|
|
(27)
|
ISO-14240-2. (1997). Soil quality — Determination of soil microbial biomass — Part 2: Fumigation-extraction method.
|
|
(28)
|
Beelen, P. Van and F. Van Keulen, (1990), The Kinetics of the Degradation of Chloroform and Benzene in Anaerobic Sediment from the River Rhine. Hydrobiol. Bull. 24 (1), p. 13-21.
|
|
(29)
|
Shelton, D.R. and Tiedje, J.M. (1984). General method for determining anaerobic biodegradation potential. App. Environ. Microbiol. 47, p. 850-857.
|
|
(30)
|
Birch, R.R., Biver, C, Campagna, R., Gledhill, W.E., Pagga, U., Steber, J., Reust, H. and Bontinck, W.J. (1989). Screening of chemicals for anaerobic biodegradation. Chemosphere 19, 1527-1550.
|
|
(31)
|
Pagga, U. and Beimborn, D.B. (1993). Anaerobic biodegradation tests for organic compounds. Chemoshpere 27, p. 1499-1509.
|
|
(32)
|
Nuck, B.A. and Federle, T.W. (1986). A batch test for assessing the mineralisation of 14C-radiolabelled compounds under realistic anaerobic conditions. Environ. Sci. Technol. 30, p. 3597-3603.
|
|
(33)
|
US-EPA (1998b). Anaerobic biodegradability of organic chemicals. Harmonised Test Guidelines (OPPTS 835.3400). EPA 712-C-98-090.
|
|
(34)
|
Sijm, Haller and Schrap (1997). Influence of storage on sediment characteristics and drying sediment on sorption coefficients of organic contaminants. Bulletin Environ. Contain. Toxicol. 58, p. 961-968.
|
|
(35)
|
Timme, G., Frehse H. and Laska V. (1986) Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues II. Pflanzenschutz — Nachrichten Bayer, 39, p. 187-203.
|
|
(36)
|
Timme, G., Frehse, H. (1980) Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues I. Pflanzenschutz — Nachrichten Bayer, 33, p. 47-60.
|
|
(37)
|
Carlton, R.R. and Allen, R. (1994). The use of a compartment model for evaluating the fate of pestici- des in sediment/water systems. Brighton Crop Protection Conference — Pest and Diseases, p. 1349- 1354.
|
Annexe 1
INFORMATIONS SUR LES SYSTÈMES D'ESSAI AÉROBIES ET ANAÉROBIES
Système d'essai aérobie
Le système d'essai aérobie décrit dans la présente méthode d'essai se compose d'une couche d'eau aérobie (généralement la concentration d'oxygène varie entre 7 et 10 mg.1–1) et une couche de sédiment, aérobie à la surface et anaérobie sous la surface [généralement, le potentiel redox moyen (Eh) dans la zone anaérobie du sédiment est situé entre –80 et –190 mV]. On fait passer de l'air humide à la surface de l'eau dans chaque unité d'incubation afin de maintenir suffisamment d'oxygène dans l'espace libre.
Système d'essai anaérobie
Dans le système d'essai anaérobie, la méthode d'essai est pratiquement la même que celle du système aérobie, à l'exception que l'on fait passer de l'azote humide à la surface de l'eau dans chaque unité d'incubation afin de maintenir un espace libre d'azote. Le sédiment et l'eau sont considérés comme anaérobies lorsque le potentiel redox (Eh) est inférieur à –100 mV.
Dans l'essai anaérobie, l'évaluation de la minéralisation comprend la mesure du dioxyde de carbone et du méthane dégagé.
Annexe 2
EXEMPLE D'APPAREIL À CIRCULATION CONTINUE

Annexe 3
EXEMPLE DE BIOMÈTRE

Annexe 4
EXEMPLE DE CALCUL DE LA DOSE À APPLIQUER AUX RÉCIPIENTS D'ESSAI
|
Diamètre interne du cylindre:
|
= 8 cm
|
|
Profondeur de la colonne d'eau excluant le sédiment:
|
= 12 cm
|
|
Surface: 3,142 × 42
|
= 50,3 cm2
|
|
Taux d'application: 500 g de substance d'essai/ha correspondant à 5 μg/cm2
|
|
|
Total en μg: 5 × 50,3
|
= 251,5 μg
|
|
Ajuster la quantité par rapport à la quantité correspondant à une profondeur de 100 cm: 12 × 251,5 ÷100
|
= 30,18 μg
|
|
Volume de la colonne d'eau: 50,3 × 12
|
= 603 ml
|
|
Concentration dans l'eau: 30,18 ÷ 603
|
= 0,050 μg/ml ou 50 μg/l
|
(1) Les résultats de ces essais interlaboratoires, ainsi qu'un corrigendum technique à l'ISO 6341, donnent une CE50 24 h du dichromate de potassium (K2Cr2O7) comprise entre 0,6 mg/l et 1,7 mg/l.
(2) Eau de pureté adéquate, par exemple, eau désionisée, distillée ou obtenue par osmose inverse, de conductivité de préférence inférieure à 10 μS.cm-1.
(3) Lorsque la différence entre les valeurs de D1 et D2 est trop grande, il ne faut pas calculer leur moyenne.
(4) Il ne faut pas calculer la moyenne de D1 et D2 lorsque la différence entre leurs valeurs est trop grande.
(5) ou à la fin de l'incubation
(6) Il ne faut pas calculer la moyenne de D1 et D2 lorsque la différence entre leurs valeurs est trop grande.
(7) ou à la fin de l'incubation
(8) Ne pas utiliser la moyenne si tes résultats des essais en double varient considérablement.
(9) Ne pas calculer la moyenne lorsque la différence entre les réplicats est trop grande.
(10) Afin d'éviter la dissémination de mercure dans l'environnement, les solutions contenant des sels de mercure doivent être traitées après utilisation.
(11) Mabey et Mill recommandent l'utilisation de tampons borate ou acétate de préférence à des tampons phosphate (11).
(12) Cette information peut provenir d'autres sources, notamment des résultats d'hydrolyse de composés de structure comparable fournis par la littérature ou par d'autres essais d'hydrolyse préliminaires ou semi-quantitatifs effectués sur la substance d'essai à une étape plus précoce de son développement.
(13) Si le tracé des transformations logarithmiques en fonction du temps n'indique pas une fonction linéaire (ce qui équivaut à une vitesse de réaction du premier ordre), alors l'utilisation de l'équation [3] ne convient pas pour déterminer la constante de vitesse d'hydrolyse de la substance d'essai.
(14) Ajouter de petits cristaux de thymol ou d’une substance comparable pour éviter la croissance de moisissures.
(15) Ajouter de petits cristaux de thymol ou d’une substance comparable pour éviter la croissance de moisissures.
(16) Valeurs moyennes de trois déterminations.
(17) Meyer A., Orti G. (1993) Proc. Royal Society of London, Séries B., Vol. 252, p. 231.
(18) Prélever l'eau après apport de 3 «volumes d'enceintes» au moins.
Les valeurs entre parenthèses sont les nombres d'échantillons (eau, poissons) à prélever si l'on procède a un échantillonnage supplémentaire.
|
Note
|
:
|
L'estimation avant essai de k2, pour un log Pow de 4,0 est de 0,652 jour –1. La durée totale de l'expérience est fixée à: 3 × abs. = 3 × 4,6 jours, soit 14 jours. Se reporter a l'annexe 3 pour l'estimation de l'«absorption».
|
(19) Meyer, A., Bierman, C.H. and Orti, G. (1993). The phylogenetic position of the zebrafish (Danio rerio), a model system in developmental biology: an invitation to the comparative method. Proc. R. Soc. Lond. B. 252, p. 231-236.
(20) Une série de cinq concentrations successives (ou plus) peut être choisie dans une colonne. Les points intermédiaires entre les concentrations d'une colonne (x) se trouvent dans la colonne (2x + 1). Les valeurs énumérées peuvent représenter des concentrations exprimées sous forme de pourcentage en volume ou en poids (mg/l ou μg/l). Les valeurs peuvent être multipliées ou divisées par la puissance de 10, adéquate. On pourra utiliser la première colonne si le degré de toxicité est très incertain.
(21) OCDE. Paris, 1992, ligne directrice 210, «Poisson, essai de toxicité aux premiers stades de la vie».
(22) Pour les embryons.
(23) Pour les larves.
(24) Obscurité pour les embryons et les larves jusqu'à une semaine après l'éclosion, sauf pendant qu'on les examine. Une lumière tamisée est ensuite appliquée jusqu'à la fin de l'essai.
(25) Selon la FAO et le système américain (85).
(26) Les valeurs des variables doivent être comprises de préférence dans les limites du spectre indiqué. En cas de difficulté à trouver un sol approprié, des valeurs inférieures au minimum indiqué sont acceptées.
(27) Les sols contenant moins de 0,3 % de carbone organique risquent de perturber la corrélation entre la teneur en carbone organique et l'adsorption. Il est donc recommandé d'utiliser des sols présentant une teneur supérieure à 0,3 %.
(28) 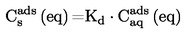
(29) On peut également utiliser des graphiques de concentration de la substance en phase aqueuse ( ) en fonction du temps pour estimer l'obtention du plateau d'équilibre (voir la figure 2 de l'appendice 5).
) en fonction du temps pour estimer l'obtention du plateau d'équilibre (voir la figure 2 de l'appendice 5).
(30) DT-50: temps de dégradation de 50 % de la substance d'essai.
(31)

(32) Équation applicable à la méthode directe et indirecte. Toutes les autres équations s'appliquent uniquement à la méthode indirecte.
(33) Équations applicables à la méthode directe et indirecte. Toutes les autres équations s'appliquent uniquement à la méthode indirecte.
(34) W. Kördel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35 (1/2), pp. 121-128.
(35) W. Kördel, D. Hennecke, C. Franke (1997). Determination of the adsorption coefficients of organic substances on sewage sludges. Chemosphere, 35 (1/2), pp. 107-119.
(36) W. Kördel, G. Kotthoff, J. Müller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere, 30 (7), pp. 1373-1384.
(37) W. Kördel, J. Müller (1994). Bestimmug; des orptionskoeffizienten organischer Chemikalien mit der HPLC. UBA R & D Report No 106 01044 (1994).
(38) B.V. Oepen, W. Kördel, W. Klein (1991). Chemosphere, 22, pp. 285-304.
(39) Données communiquées par les industriels.
(40) Indiquer quel récipient a été utilisé pour l'expérience.
(41) Indiquer la mortalité de tout animal adulte par la lettre «M» dans la case appropriée.
(42) Indiquer les portées avortées par les lettres «AB» dans la case appropriée.
(43) Par exemple, si la substance d'essai contient un cycle, il est nécessaire de marquer ce cycle; si la substance d'essai contient deux cycles ou plus, il peut être nécessaire d'effectuer des études distinctes pour évaluer le devenir de chaque cycle marqué et obtenir des informations fiables sur la formation de produits de transformation.
(44) La caractéristique de rétention d'eau d'un sol peut être mesurée comme capacité au champ, capacité de rétention d'eau ou comme potentiel de succion (pF). Pour les explications, voir l'annexe 1. Il convient d'indiquer dans le rapport d'essai si les caractéristiques de rétention d'eau et la densité apparente des sols ont été déterminées sur des échantillons de sol non brassé ou avec des échantillons brassés (transformés).
(45) Des résultats récents indiquent que les sols des zones tempérées peuvent également être stockés à — 20 oC pendant plus de trois mois (28) (29) sans perte significative de l'activité microbienne.
(46) Le sol ne doit jamais être trop humide ni trop sec afin de maintenir des conditions adéquates d'aération et de nutrition de la microflore du sol. Les teneurs en eau recommandées pour une croissance microbienne optimale vont de 40 à 60 % de capacité de rétention d'eau (WHC) et de 0,1 à 0,33 bar (6). Cette fourchette est équivalente à un intervalle de pF de 2,0-2,5. L'annexe 2 présente la teneur habituelle en eau de plusieurs types de sol.
(47) Les conditions aérobies sont prédominantes dans les sols de surface et même dans les sols subsurfaciques comme le montre un projet de recherche financé par PUE [K. Takagi et al. (1992). Microbial diversity and activity in subsoils: Methods, field site, seasonal variation in subsoil températures and oxygen contents. Proc. Internat. Symp. Environm. Aspects Pesticides Microbiol., p. 270-277, 17-21 août 1992, Sigtuna, Suède]. Les conditions anaérobies ne peuvent se produire qu'occasionnellement sur des sols inondés après d'importantes chutes de pluie ou sur des sols de rizières submergées.
(48) Les études aérobies doivent être terminées en moins de 120 jours sous réserve que la voie de transformation et la minéralisation se soient effectivement produites à cette date. Il est possible de mettre fin à l'essai au bout de 120 jours, ou lorsque 90 % au moins de la substance d'essai sont transformés, mais seulement s'il y a formation de 5 % de CO2 au moins.
(49) Calcul de la concentration initiale en fonction de la superficie à l'aide de l'équation suivante:

|
Csol
|
=
|
concentration initiale dans le sol [mg.kg-1]
|
|
A
|
=
|
Taux d'application [kg.ha-1]; 1 = épaisseur de la couche de sol du champ [m]; d = densité apparente du sol sec [kg-m-3].
|
En règle générale, si on applique un taux de 1 kg.ha-1, on obtient une concentration de 1 mg.kg-1 environ dans une couche de 10 cm (en supposant une densité apparente de 1 g.cm-3).
(50) Mückenhausen, E. (1975). Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen. DLG-Verlag, Frankfurt, Main.
(51) pF = log de la colonne d'eau en cm.
(52) 1 bar = 105 Pa.
(53) Correspond à une teneur en eau approximative de 10 % dans le sable, de 35 % dans le limon et de 45 % dans l'argile.
(54) La capacité au champ n'est pas constante mais varie selon le type de sol de pF 1,5 à 2,5.
(55) Capacité de rétention d'eau
(56) Guth, J.A. (1980). The study of transformations. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academie Press, p. 123-157.
(57) Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry. D.H. Hutson, T.R. Roberts, Eds. J. Wiley & Sons. Vol 1, p. 85-114.
(58) Anderson, J.P.E. (1975). Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden. Z. PflKrankh Pflschutz, Sonderheft VII, p. 141-146.
(59) Par exemple, si la substance d'essai contient un cycle, il est nécessaire de marquer ce cycle; si la substance d'essai contient deux cycles ou plus, il peut être nécessaire d'effectuer des études distinctes pour évaluer le devenir de chaque cycle marqué et obtenir des informations fiables sur la formation de produits de transformation.
(60) Comme ces solutions d'absorption alcalines absorbent également le dioxyde de carbone de l'air de ventilation et de l'air formé par la respiration dans les expériences aérobies, il convient de les changer à intervalles réguliers afin d'éviter leur saturation et par conséquent la perte de leur capacité d'absorption.
(61) [Argile + limon] est la fraction minérale du sédiment de granulométrie < 50 μm.
(62) Selon des résultats récents, les mesures des concentrations d'oxygène dans l'eau et des potentiels redox n'ont de valeur ni mécanique ni prédictive en ce qui concerne la croissance et le développement de colonies microbiennes dans les eaux superficielles (24) (25). La détermination de la demande biochimique en oxygène (lors du prélèvement, au début et à la fin de l'essai) et des concentrations de micro/macroéléments nutritifs Ca, Mg et Mn (au début et à la fin de l'essai) dans l'eau et la mesure du N total et du P total dans les sédiments (lors du prélèvement et à la fin de l'essai) peuvent être de meilleurs outils pour interpréter et évaluer les taux et les voies de biotransformation aérobie.
(63) Méthode du taux de respiration microbienne (26), méthode de fumigation (27) ou mesures de numération (bactéries, actinomycètes, champignons et colonies totales) pour les études aérobies; taux de méthanogenèse pour les études anaérobies.
(64) Selon des études récentes, un stockage à 4 oC peut entraîner une diminution de la teneur en carbone organique du sédiment, ce qui risque d'entraîner une diminution de l'activité microbienne (34).
(65) Il peut être utile d'effectuer un essai avec une deuxième concentration pour les substances chimiques qui atteignent les eaux superficielles par différentes voies d'entrée, ce qui entraîne des concentrations très différentes, dans la mesure où la concentration la plus faible peut être analysée avec une précision suffisante.
(66) Dans le cas où les produits de transformation anaérobie peuvent se réoxyder très rapidement, les conditions anaérobies doivent être maintenues pendant l'échantillonnage et l'analyse.