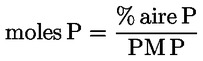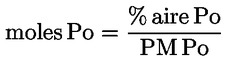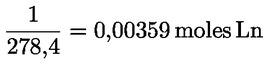01991R2568 — FR — 04.12.2016 — 031.005
Ce texte constitue seulement un outil de documentation et n’a aucun effet juridique. Les institutions de l'Union déclinent toute responsabilité quant à son contenu. Les versions faisant foi des actes concernés, y compris leurs préambules, sont celles qui ont été publiées au Journal officiel de l’Union européenne et sont disponibles sur EUR-Lex. Ces textes officiels peuvent être consultés directement en cliquant sur les liens qui figurent dans ce document
|
RÈGLEMENT (CEE) No 2568/91 DE LA COMMISSION du 11 juillet 1991 ►C1 relatif aux caractéristiques des huiles d'olive et des huiles de grignons d'olive ainsi qu'aux méthodes d'analyse y afférentes ◄ (JO L 248 du 5.9.1991, p. 1) |
Modifié par:
Rectifié par:
RÈGLEMENT (CEE) No 2568/91 DE LA COMMISSION
du 11 juillet 1991
►C1 relatif aux caractéristiques des huiles d'olive et des huiles de grignons d'olive ainsi qu'aux méthodes d'analyse y afférentes ◄
Article premier
1. Sont considérées comme huiles d'olive vierges, au sens du point 1 a), et b) de l'annexe du règlement no 136/66/CEE, les huiles dont les caractéristiques respectives sont conformes à celles indiquées à l'annexe I, points 1 et 2, du présent règlement.
2. Est considérée comme huile d'olive lampante, au sens du point 1 c) de l'annexe du règlement no 136/66/CEE, l'huile dont les caractéristiques sont conformes à celles indiquées à l'annexe I, point 3, du présent règlement.
3. Est considérée comme huile d'olive raffinée, au sens du point 2 de l'annexe du règlement no 136/66/CEE, l'huile dont les caractéristiques sont conformes à celles indiquées à l'annexe I, point 4, du présent règlement.
4. Est considérée comme huile d'olive composée d'huile d'olive raffinée et d'huiles d'olive vierges, au sens du point 3 de l'annexe du règlement no 136/66/CEE, l'huile dont les caractéristiques sont conformes à celles indiquées à l'annexe I, point 5, du présent règlement.
5. Est considérée comme huile de grignons d'olive brute, au sens du point 4 de l'annexe du règlement no 136/66/CEE, l'huile dont les caractéristiques sont conformes à celles indiquées à l'annexe I, point 6, du présent règlement.
6. Est considérée comme huile de grignons d'olive raffinée, au sens du point 5 de l'annexe du règlement no 136/66/CEE, l'huile dont les caractéristiques sont conformes à celles indiquées à l'annexe I, point 7, du présent règlement.
7. Est considérée comme huile de grignons d'olive, au sens du point 6 de l'annexe du règlement no 136/66/CEE, l'huile dont les caractéristiques sont conformes à celles indiquées à l'annexe I, point 8, du présent règlement.
Article 2
1. Les caractéristiques des huiles définies à l'annexe I sont déterminées selon les méthodes d'analyse suivantes:
a) pour la détermination des acides gras libres, exprimés en pourcentage d'acide oléique, la méthode figurant à l'annexe II;
b) pour la détermination de l'indice de peroxyde, la méthode figurant à l'annexe III;
c) pour la détermination de la teneur en cires, la méthode figurant à l'annexe IV;
d) pour la détermination de la composition et de la teneur en stérols et en diols triterpéniques par chromatographie en phase gazeuse sur colonne capillaire, la méthode figurant à l’annexe V;
e) pour la détermination du pourcentage de 2-glycéril monopalmitate, la méthode figurant à l’annexe VII;
f) pour l'analyse spectrophotométrique, la méthode figurant à l'annexe IX;
g) pour la détermination de la composition en acides gras, la méthode figurant à l'annexe X;
h) pour la détermination des solvants halogénés volatils, la méthode figurant à l'annexe XI;
i) pour l'évaluation des caractéristiques organoleptiques des huiles d'olive vierges, la méthode figurant à l'annexe XII;
j) pour la détermination des stigmastadiènes, la méthode figurant à l'annexe XVII;
k) pour la détermination de la composition des triglycérides à ECN 42, la méthode figurant à l'annexe XVIII;
l) pour la détermination du contenu en alcools aliphatiques et triterpéniques, la méthode figurant à l'annexe XIX;
m) pour la détermination de la teneur en cires et en esters méthyliques ou éthyliques d'acides gras, la méthode figurant à l'annexe XX.
▼M28 —————
2. La vérification par les autorités nationales ou leurs représentants des caractéristiques organoleptiques des huiles d'olive vierges est réalisée par des jurys de dégustateurs agréés par les États membres.
Les caractéristiques organoleptiques d'une huile d'olive visée au premier alinéa sont considérées comme conformes à la catégorie d'huile d'olive déclarée, si un jury agréé par l'État membre concerné confirme le classement à cet égard.
Dans le cas où le jury agréé ne confirme pas la catégorie déclarée en ce qui concerne les caractéristiques organoleptiques, les autorités nationales ou leurs représentants font procéder sans tarder, à la demande de l'intéressé, à deux contre-analyses par d'autres jurys agréés, dont au moins une est effectuée par un jury agréé par l'État membre producteur concerné. Les caractéristiques en question sont considérées comme conformes à celles qui sont déclarées si les deux contre-analyses confirment le classement déclaré. Dans le cas contraire, les frais des contre-analyses sont à la charge de l'intéressé.
3. Pour la vérification des caractéristiques des huiles par les autorités nationales ou leurs représentants suivant les dispositions du paragraphe 1, le prélèvement des échantillons s'effectue conformément aux normes internationales EN ISO 661 et EN ISO 5555 concernant respectivement la préparation des échantillons pour essai et l'échantillonnage. Toutefois, par dérogation au point 6.8 de la norme EN ISO 5555, dans le cas de lots desdites huiles, en conditionnement primaire, le prélèvement des échantillons s'effectue conformément à l'annexe I bis du présent règlement. Dans le cas des huiles en vrac où l’échantillonnage ne peut être réalisé conformément à la norme EN ISO 5555, les échantillons sont prélevés conformément aux instructions fournies par l’autorité compétente de l’État membre.
Sans préjudice des dispositions de la norme EN ISO 5555 et du chapitre 6 de la norme EN ISO 661, les échantillons sont mis à l’abri de la lumière et des fortes chaleurs dans les plus brefs délais et sont envoyés au laboratoire pour les analyses au plus tard le cinquième jour ouvrable suivant celui de leur prélèvement, ou bien sont conservés de manière à éviter leur dégradation ou leur endommagement lors du transport ou du stockage avant d'être envoyés au laboratoire.
4. Aux fins de la vérification prévue au paragraphe 3, les analyses visées aux annexes II, III, IX, XII et XX, ainsi que, le cas échéant, les contre-analyses requises en vertu de la législation nationale, sont effectuées, dans le cas des produits conditionnés, avant la date de durabilité minimale. En cas d'échantillonnage d'huiles en vrac, ces analyses sont effectuées au plus tard le sixième mois suivant celui du prélèvement.
Aucun délai ne s'applique pour les autres analyses prévues par ledit règlement.
Sauf si l'échantillon a été prélevé moins de deux mois avant la date de durabilité minimale, lorsque les résultats des analyses ne correspondent pas aux caractéristiques de la catégorie d'huile d'olive ou de grignons d'olive déclarée, l'intéressé en est informé au plus tard un mois avant la fin du délai visé au premier alinéa.
5. Pour la détermination des caractéristiques des huiles d'olive par les méthodes prévues au paragraphe 1, premier alinéa, les résultats des analyses sont directement comparés avec les limites fixées par le présent règlement.
Article 2 bis
1. Aux fins du présent article, on entend par «huile d’olive commercialisée», la quantité totale d’huile d’olive et d’huile de grignons d’olive d’un État membre qui est consommée dans cet État membre ou exportée à partir de cet État membre.
2. Les États membres veillent à ce que des contrôles de conformité soient réalisés de manière sélective, sur la base d’une analyse de risques et à une fréquence appropriée, afin de garantir que l’huile d’olive commercialisée correspond à la catégorie déclarée.
3. Les critères d’évaluation du risque peuvent porter sur:
a) la catégorie de l’huile, la période de production, le prix des huiles par rapport à celui des autres huiles végétales, les opérations de mélange et de conditionnement, les installations et conditions de stockage, le pays d’origine, le pays de destination, le moyen de transport ou le volume du lot;
b) la position des opérateurs dans la chaîne de commercialisation, le volume et/ou la valeur, ainsi que la gamme de catégories d’huiles qu’ils commercialisent, le type d’activité économique menée telle que le pressage, le stockage, le raffinage, le mélange, le conditionnement ou la vente au détail;
c) les constatations faites lors des contrôles précédents, notamment en ce qui concerne le nombre et le type d’irrégularités constatés, la qualité habituelle des huiles commercialisées et le niveau de performance de l’équipement technique utilisé;
d) la fiabilité des systèmes d’assurance qualité des opérateurs ou de leurs systèmes d’autocontrôle, au regard de la conformité aux normes de commercialisation;
e) le lieu où le contrôle est effectué, en particulier s’il s’agit du premier point d’entrée dans l’Union, du dernier point de sortie de l’Union ou du lieu où les huiles sont produites, conditionnées, chargées ou vendues au consommateur final;
f) toute autre information susceptible d’indiquer un risque de non-conformité.
4. Les États membres arrêtent à l’avance:
a) les critères d’évaluation du risque de non-conformité des lots;
b) sur la base d’une analyse des risques portant sur chaque catégorie de risques, le nombre minimal d’opérateurs ou de lots et/ou les quantités minimales qu’il y a lieu de soumettre à un contrôle de conformité.
Au moins un contrôle annuel de conformité est effectué pour mille tonnes d’huile d’olive commercialisée dans l’État membre.
5. Les États membres vérifient la conformité:
a) en procédant, dans n’importe quel ordre, aux analyses prévues à l’annexe I, ou
b) dans l’ordre prévu par l’arbre décisionnel figurant à l’annexe I ter, jusqu’à aboutir à l’une des décisions prévues par ledit arbre.
▼M19 —————
Article 3
Lorsqu’il est constaté qu’une huile ne correspond pas à la description de sa catégorie, les États membres concernés appliquent, sans préjudice d’autres sanctions éventuelles, des sanctions effectives, proportionnées et dissuasives arrêtées en fonction de la gravité de l’irrégularité constatée.
Si les contrôles font apparaître des irrégularités importantes, les États membres renforcent la fréquence des contrôles portant sur le stade de commercialisation, la catégorie de l’huile, son origine ou d’autres critères.
Article 4
1. Aux fins de l'appréciation et du contrôle par les autorités nationales ou leur représentant des caractéristiques organoleptiques, les États membres peuvent agréer des jurys de dégustateurs.
Les conditions d'agrément sont établies par les États membres, notamment de façon à:
— répondre aux conditions de l'annexe XII, point 4.
— assurer que la formation du chef du jury soit effectuée par un établissement et dans des conditions, reconnus à cet effet par l'État membre.
— soumettre la validité de l'agrément aux résultats des performances obtenues dans le cadre d'un système de contrôle annuel institué par l'État membre.
Chaque État membre communique à la Commission la liste des jurys agréés ainsi que les mesures prises en conformité avec le présent paragraphe.
2. Au cas où un État membre rencontre des difficultés pour instituer un jury de dégustateurs sur son territoire, il peut recourir à un jury de dégustateurs agréé dans un autre État membre.
3. Chaque État membre établit la liste des jurys de dégustateurs institués par des organisations professionnelles ou interprofessionnelles conformément aux conditions décrites au paragraphe 1 et veille au respect de ces conditions.
▼M19 —————
Article 6
1. La teneur en huile des grignons et des autres résidus de l'extraction de l'huile relevant des codes NC 2306 90 11 et 2306 90 19 est déterminée conformément à la méthode figurant à l'annexe XV.
2. la teneur en huile visée au paragraphe 1 est exprimée en pourcentage de son poids rapporté à celui de la matière sèche.
Article 7
Les dispositions communautaires concernant la présence de contaminants sont applicables.
En ce qui concerne la teneur en solvants halogénés les limites pour toutes les catégories d'huiles d'olive sont les suivantes:
— teneur maximale de chaque solvant halogéné détecté: 0,1 mg/kg,
— teneur maximale de la somme des solvants halogénés détectés: 0,2 mg/kg
Article 7 bis
Les personnes et groupes de personnes physiques ou morales qui détiennent, à quelque titre que ce soit, pour l’exercice de leur profession ou à des fins commerciales, de l’huile d’olive et de l’huile de grignons d’olive, depuis le stade de l’extraction au pressoir jusqu’à l’embouteillage inclus, sont tenus de tenir des registres d’entrée et de sortie pour chaque catégorie de ces huiles.
Les États membres veillent à ce que l’obligation prévue au premier paragraphe soit respectée.
Article 8
1. Les États membres communiquent à la Commission les mesures qui mettent en œuvre le présent règlement. Ils informent la Commission de toute modification ultérieure de ces mesures.
2. Le 31 mai de chaque année au plus tard, les États membres transmettent à la Commission un rapport sur l’application du présent règlement au cours de l’année civile écoulée. Ce rapport contient au moins les résultats des contrôles de conformité réalisés sur les huiles d’olive, présentés suivant les tableaux figurant à l’annexe XXI.
3. Les notifications visées dans le présent règlement sont effectuées conformément au règlement (CE) no 792/2009 de la Commission ( 1 ).
Article 9
Le règlement (CEE) no 1058/77 est abrogé.
Article 10
1. Le présent règlement entre en vigueur le jour suivant celui de sa publication au Journal officiel des Communautés européennes. Toutefois, la méthode reprise à l'annexe XII est mise en application à partir du ►M1 1er novembre 1992 ◄ , sauf en ce qui concerne les opérations liées à l'intervention.
Cette méthode ne s'applique pas aux huiles d'olive vierges conditionnées avant le 1er novembre 1992.
2. Le présent règlement ne s'applique pas aux huiles d'olive et de grignons d'olive conditionnées avant la date d'entrée en vigueur du présent règlement et commercialisées jusqu'au 31 octobre 1992.
Le présent règlement est obligatoire dans tous ses éléments et directement applicable dans tout État membre.
ANNEXES
Sommaire
|
Annexe I: |
Caractéristiques des huiles d'olive |
|
Annexe I bis: |
Échantillonnage des huiles d'olive ou des huiles de grignons d'olive livrées en conditionnement primaire |
|
Annexe I ter: |
Schéma décisionnel pour la vérification de la conformité d'un échantillon d'huile d'olive à la catégorie déclarée |
|
Annexe II: |
Détermination des acides gras libres, méthode à froid |
|
Annexe III: |
Détermination de l'indice de peroxyde |
|
Annexe IV: |
Détermination de la teneur en cires au moyen de la chromatographie en phase gazeuse avec colonne capillaire |
|
Annexe V: |
Détermination de la composition et de la teneur en stérols et en diols triterpéniques par chromatographie en phase gazeuse sur colonne capillaire |
|
Annexe VII: |
►M21 Détermination du pourcentage du 2-glycéril monopalmitate ◄ |
|
▼M20 ————— |
|
|
Annexe IX: |
Analyse spectrophotométrique dans l’ultraviolet |
|
Annexe X: |
Détermination des esters méthyliques d'acides gras par chromatographie en phase gazeuse |
|
Annexe XI: |
Détermination de la teneur en solvants halogénés volatils dans l'huile d'olive |
|
Annexe XII: |
Méthode du conseil oléicole international pour l'évaluation organoleptique des huiles d'olive vierges |
|
▼M20 ————— |
|
|
▼M19 ————— |
|
|
Annexe XV: |
Teneur en huile des grignons d'olive |
|
Annexe XVI: |
Détermination de l'indice d'iode |
|
Annexe XVII: |
Méthode de détermination des stigmastadiènes dans les huiles végétales |
|
Annexe XVIII: |
Détermination de la différence entre la composition réelle et la composition théorique des triglycérides à ECN 42 |
|
Annexe XIX: |
►M28 Détermination du contenu en alcools aliphatiques et triterpéniques par chromatographie en phase gazeuse avec colonne capillaire ◄ |
|
Annexe XX: |
méthode de détermination de la teneur en cires et en esters méthyliques et éthyliques d'acides gras par chromatographie en phase gazeuse sur colonne capillaire |
|
▼M28 ————— |
|
|
Annexe XXI: |
Résultats des contrôles de conformité réalisés sur les huiles d’olive visés à l’article 8, paragraphe 2 |
ANNEXE I
CARACTÉRISTIQUES DES HUILES D'OLIVE
Caractéristiques de qualité
|
Catégorie |
Acidité (%) (*) |
Indice de peroxyde mEq O2/kg (*) |
K232 (*) |
K268 ou K270 (*) |
Delta-K (*) |
Évaluation organoleptique |
Esters éthyliques d'acides gras mg/kg (*) |
|
|
Médiane du défaut (Md) (*) |
Médiane du fruité (Mf) (*) |
|||||||
|
1. Huile d'olive vierge extra |
≤ 0,8 |
≤ 20 |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0 |
Mf > 0 |
≤ 35 |
|
2. Huile d'olive vierge |
≤ 2,0 |
≤ 20 |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0 |
— |
|
3. Huile d'olive lampante |
> 2,0 |
— |
— |
— |
— |
Md > 3,5 (1) |
— |
— |
|
4. Huile d'olive raffinée |
≤ 0,3 |
≤ 5 |
— |
≤ 1,25 |
≤ 0,16 |
— |
— |
— |
|
5. Huile d'olive composée d'huile d'olive raffinée et d'huile d'olive vierge |
≤ 1,0 |
≤ 15 |
— |
≤ 1,15 |
≤ 0,15 |
— |
— |
— |
|
6. Huile de grignons d'olive brute |
— |
— |
— |
— |
— |
— |
— |
— |
|
7. Huile de grignons d'olive raffinée |
≤ 0,3 |
≤ 5 |
— |
≤ 2,00 |
≤ 0,20 |
— |
— |
— |
|
8. Huile de grignons d'olive |
≤ 1,0 |
≤ 15 |
— |
≤ 1,70 |
≤ 0,18 |
— |
— |
— |
|
(1) La médiane du défaut peut être inférieure ou égale à 3,5 lorsque la médiane du fruité est égale à 0. |
||||||||
Caractéristiques de pureté
|
Catégorie |
Teneur en acides gras (1) |
Sommes des isomères transoléiques (%) |
Sommes des isomères translinoléiques + translinoléniques (%) |
Stigmastadiènes mg/kg (2) |
►C6 Différence: ECN42 (HPLC) et ECN42 (calcul théorique) ◄ |
2-glycéril monopalmitate (%) |
|||||
|
Myristique (%) |
Linolénique (%) |
Arachidique (%) |
Eicosénoïque (%) |
Béhénique (%) |
Lignocérique (%) |
||||||
|
1. Huile d'olive vierge extra |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,05 |
►C6 ≤ |0,2| ◄ |
≤ 0,9 si acide palmitique total en % ≤ 14 % |
|
≤ 1,0 si acide palmitique total en % > 14 % |
|||||||||||
|
2. Huile d'olive vierge |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,05 |
►C6 ≤ |0,2| ◄ |
≤ 0,9 si acide palmitique total en % ≤ 14 % |
|
≤ 1,0 si acide palmitique total en % > 14 % |
|||||||||||
|
3. Huile d'olive lampante |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,10 |
≤ 0,50 |
►C6 ≤ |0,3| ◄ |
≤ 0,9 si acide palmitique total en % ≤ 14 % |
|
≤ 1,1 si acide palmitique total en % > 14 % |
|||||||||||
|
4. Huile d'olive raffinée |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
— |
►C6 ≤ |0,3| ◄ |
≤ 0,9 si acide palmitique total en % ≤ 14 % |
|
≤ 1,1 si acide palmitique total en % > 14 % |
|||||||||||
|
5. Huile d'olive composée d'huile d'olive raffinée et d'huile d'olive vierge |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
— |
►C6 ≤ |0,3| ◄ |
≤ 0,9 si acide palmitique total en % ≤ 14 % |
|
≤ 1,0 si acide palmitique total en % > 14 % |
|||||||||||
|
6. Huile de grignons d'olive brute |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
— |
►C6 ≤ |0,6| ◄ |
≤ 1,4 |
|
7. Huile de grignons d'olive raffinée |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
— |
►C6 ≤ |0,5| ◄ |
≤ 1,4 |
|
8. Huile de grignons d'olive |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
— |
►C6 ≤ |0,5| ◄ |
≤ 1,2 |
|
Catégorie |
Composition en stérols |
Stérols totaux (mg/kg) |
Érythrodiol et uvaol (%) (**) |
Cires mg/kg (**) |
|||||
|
Cholestérol (%) |
Brassicastérol (%) |
Campestérol (3) (%) |
Stigmastérol (%) |
Sitostérol αππ. (%) (4) |
Delta-7-stigmasténol (3) (%) |
||||
|
1. Huile d'olive vierge extra |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C42 + C44 + C46 ≤ 150 |
|
2. Huile d'olive vierge |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C42 + C44 + C46 ≤ 150 |
|
3. Huile d'olive lampante |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (5) |
C40 + C42 + C44 + C46 ≤ 300 (5) |
|
4. Huile d'olive raffinée |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C40 + C42 + C44 + C46 ≤ 350 |
|
5. Huile d'olive composée d'huile d'olive raffinée et d'huile d'olive vierge |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C40 + C42 + C44 + C46 ≤ 350 |
|
6. Huile de grignons d'olive brute |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (6) |
C40 + C42 + C44 + C46 > 350 (6) |
|
7. Huile de grignons d'olive raffinée |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
C40 + C42 + C44 + C46 > 350 |
|
8. Huile de grignons d'olive |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
C40 + C42 + C44 + C46 > 350 |
(1) Teneur en autres acides gras (%): palmitique: 7,50-20,00; palmitoléique: 0,30-3,50; heptadécanoïque: ≤ 0,40; heptadécénoïque: ≤ 0,60; stéarique: 0,50-5,00; oléique: 55,00-83,00; linoléique: 2,50-21,00.
(2) Sommes des isomères qui pourraient (ou pas) être séparés par colonne capillaire.
(3) Voir l'appendice de la présente annexe.
(4) Sitostérol β-app.: delta-5,23-stigmastadiénol+clérostérol+bêta-sitostérol+sitostanol+delta-5-avenastérol+delta-5,24-stigmastadiénol.
(5) Les huiles dont la teneur en cires est comprise entre 300 et 350 mg/kg sont considérées comme des huiles d'olive lampantes si leur teneur totale en alcools aliphatiques est inférieure ou égale à 350 mg/kg ou si la proportion d'érythrodiol et d'uvaol est inférieure ou égale à 3,5 %.
(6) Les huiles ayant une teneur en cires comprise entre 300 et 350 mg/kg sont considérées comme des huiles de grignons d'olive brutes si leur teneur totale en alcools aliphatiques est supérieure à 350 mg/kg et si la proportion d'érythrodiol et d'uvaol est supérieure à 3,5 %.
Remarques:
a) Les résultats des analyses doivent être exprimés en indiquant le même nombre de décimales que pour les valeurs de chaque caractéristique. Le dernier chiffre doit être augmenté d'une unité si le chiffre suivant dépasse 4.
b) Il suffit qu'une seule caractéristique ne corresponde pas aux valeurs indiquées pour que l'huile change de catégorie ou soit déclarée non conforme quant à sa pureté aux fins du présent règlement.
c) Les caractéristiques marquées d'un astérisque (*), se référant à la qualité de l'huile, indiquent: dans le cas de l'huile d'olive lampante, qu'il est possible que les deux limites pertinentes diffèrent simultanément des valeurs indiquées; dans le cas des huiles d'olive vierges, que si au moins une de ces limites diffère des valeurs indiquées, l'huile change de catégorie tout en restant classée dans une des catégories d'huiles d'olive vierges.
d) Les caractéristiques marquées de deux astérisques (**) indiquent, pour tous les types d'huiles de grignons d'olive, qu'il est possible que les deux limites pertinentes diffèrent simultanément des valeurs indiquées.
Appendice
SCHÉMA DÉCISIONNEL
Schéma décisionnel du campestérol pour l'huile d'olive vierge et l'huile d'olive vierge extra:
Les autres paramètres sont conformes aux limites fixées dans le présent règlement.
Schéma décisionnel du delta-7-stigmasténol pour:
— les huiles d'olive vierges et vierges extra
—

Les autres paramètres sont conformes aux limites fixées dans le présent règlement.
— les huiles de grignons d'olive (brutes et raffinées)
—

ANNEXE I bis
ÉCHANTILLONNAGE DES HUILES D'OLIVE OU DES HUILES DE GRIGNONS D'OLIVE LIVRÉES EN CONDITIONNEMENT PRIMAIRE
La présente méthode d’échantillonnage s’applique aux lots d’huile d’olive ou de grignons d’olive en conditionnement primaire. Des méthodes d’échantillonnage différentes s’appliquent selon que le conditionnement primaire dépasse ou non 5 litres.
On entend par «lot», un ensemble d'unités de vente qui sont produites, fabriquées et conditionnées dans des circonstances telles que l'huile contenue dans chacune de ces unités de vente est considérée comme homogène pour toutes les caractéristiques analytiques. L'individualisation d’un lot doit être réalisée conformément à la directive 2011/91/UE du Parlement européen et du Conseil ( 8 ).
On entend par «prélèvement élémentaire», la quantité d’huile contenue dans un conditionnement primaire et prélevée en un point aléatoire du lot.
1. CONTENU D'UN ÉCHANTILLON ÉLÉMENTAIRE
1.1. Conditionnement primaire ne dépassant pas 5 litres
on entend par «échantillon élémentaire», dans le cas d'un conditionnement primaire ne dépassant pas 5 litres, le nombre de prélèvements élémentaires effectués sur un lot et conformes au tableau 1.
Tableau 1
Taille minimale des prélèvements élémentaires
|
Si le conditionnement primaire a une capacité |
L'échantillon primaire doit être constitué de l'huile provenant de |
|
a) égale ou supérieure à 1 litre |
a) 1 conditionnement primaire; |
|
b) inférieure à 1 litre |
b) le nombre minimal de conditionnements donnant une capacité totale d’au moins 1,0 litre |
Le nombre de conditionnements visé au tableau 1, constituant un échantillon primaire, peut être augmenté par chaque État membre, en fonction de ses propres besoins (par exemple, évaluation organoleptique réalisée par un laboratoire différent de celui qui a effectué les analyses chimiques, les contre-analyses, etc.).
1.2. Conditionnement primaire de plus de 5 litres
On entend par «échantillon élémentaire», dans le cas d'un conditionnement primaire de plus de 5 litres, une partie représentative de l'ensemble des prélèvements élémentaires, obtenue par un processus de réduction et conforme au tableau 2. L’échantillon élémentaire doit être composé de plusieurs exemplaires.
On entend par «exemplaire» d’un échantillon élémentaire, chacun des conditionnements composant l’échantillon élémentaire.
Tableau 2
Nombre minimal de prélèvements élémentaires à sélectionner
|
Nombre de conditionnements compris dans le lot |
Nombre minimal de prélèvements élémentaires à sélectionner |
|
Jusqu’à 10 |
1 |
|
Entre … 11 et 150 |
2 |
|
Entre … 151 et 500 |
3 |
|
Entre … 501 et 1 500 |
4 |
|
Entre … 1 501 et 2 500 |
5 |
|
> 2 500 pour 1 000 conditionnements |
1 prélèvement élémentaire supplémentaire |
Afin de réduire le volume d'échantillonnage des conditionnements primaires, le contenu des prélèvements élémentaires est homogénéisé pour préparer l’échantillon élémentaire. Les fractions des différents prélèvements élémentaires sont versées dans un même conteneur en vue d'une homogénéisation par agitation, de façon à garantir une protection optimale contre l'air.
Le contenu de l'échantillon élémentaire doit être versé dans une série de conditionnements d'une capacité minimale de 1,0 litre, constituant chacun un exemplaire de l’échantillon élémentaire.
Le nombre d'échantillons élémentaires peut être augmenté par chaque État membre, en fonction de ses propres besoins (par exemple, évaluation organoleptique réalisée par un laboratoire différent de celui qui a effectué les analyses chimiques, les contre-analyses, etc.).
Chaque conditionnement doit être rempli de manière à réduire au minimum la couche d’air superficielle, puis convenablement fermé et scellé afin de protéger le produit contre toute falsification.
Ces exemplaires doivent être étiquetés afin de permettre leur identification.
2. ANALYSES ET RÉSULTATS
2.1. Chaque échantillon élémentaire doit être subdivisé en échantillons de laboratoire, conformément au point 2.5 de la norme EN ISO 5555, puis soumis à des analyses dans l’ordre indiqué par le schéma décisionnel figurant à l’annexe IB ou dans un ordre aléatoire.
2.2. Lorsque tous les résultats des analyses concordent avec les caractéristiques de la catégorie d'huile déclarée, l'ensemble du lot est déclaré conforme.
Il suffit qu'un seul résultat d'analyse ne concorde pas avec les caractéristiques de la catégorie d'huile déclarée pour que l'ensemble du lot soit déclaré non conforme.
3. VÉRIFICATION DE LA CATÉGORIE DU LOT
3.1. Afin de vérifier la catégorie du lot, l’autorité compétente peut augmenter le nombre d’échantillons élémentaires prélevés en différents points du lot conformément au tableau suivant:
Tableau 3
Nombre d'échantillons élémentaires, déterminé par la taille du lot
|
Taille du lot (en litres) |
Nombre d'échantillons élémentaires |
|
moins de 7 500 |
2 |
|
entre 7 500 et moins de 25 000 |
3 |
|
entre 25 000 et moins de 75 000 |
4 |
|
entre 75 000 et moins de 125 000 |
5 |
|
125 000 et plus |
6 + 1 tous les 50 000 litres supplémentaires |
Chaque prélèvement élémentaire constituant un échantillon élémentaire doit être prélevé au même endroit dans le lot; il est nécessaire de relever l'emplacement de chaque échantillon élémentaire et de repérer celui-ci sans ambiguïté.
Chaque échantillon élémentaire doit être constitué conformément aux procédures visées aux points 1.1 et 1.2.
Chaque échantillon élémentaire est ensuite soumis aux analyses visées à l’article 2, paragraphe 1.
3.2. Lorsqu’un des résultats des analyses visées à l’article 2, paragraphe 1, d’au moins un échantillon élémentaire ne correspond pas aux caractéristiques de la catégorie d’huile déclarée, l’ensemble du lot est déclaré non conforme.
ANNEXE I ter
SCHÉMA DÉCISIONNEL POUR LA VÉRIFICATION DE LA CONFORMITÉ D'UN ÉCHANTILLON D'HUILE D'OLIVE À LA CATÉGORIE DÉCLARÉE
Tableau 1

Tableau 2

Tableau 3

Appendice 1
Correspondance entre les annexes du présent règlement et les analyses indiquées dans le schéma décisionnel
|
— Acidité |
Annexe II |
Détermination des acides gras libres, méthode à froid |
|
— Indice de peroxyde |
Annexe III |
Détermination de l'indice de peroxyde |
|
— Spectrométrie UV |
Annexe IX |
Analyse spectrophotométrique |
|
— Évaluation organoleptique |
Annexe XII |
Évaluation organoleptique de l'huile d'olive vierge |
|
— Esters éthyliques |
Annexe XX |
Méthode de détermination de la teneur en cires et en esters méthyliques et éthyliques d'acides gras par chromatographie gazeuse sur colonne capillaire |
|
— 3,5-stigmastadiènes |
Annexe XVII |
Méthode de détermination des stigamastadiènes dans les huiles végétales |
|
— Isomères trans des acides gras |
Annexe X |
Détermination des esters méthyliques d'acides gras par chromatographie en phase gazeuse |
|
— Teneur en acides gras |
Annexe X |
Détermination des esters méthyliques d'acides gras par chromatographie en phase gazeuse |
|
— ΔECN42 |
Annexe XVIII |
Détermination de la composition des triglycérides à ECN42 (différence entre les résultats en CLHP et composition théorique) |
|
— Composition en stérols et stérols totaux — Érythrodiol et uvaol |
Annexe V |
Détermination de la composition et de la teneur en stérols et en diols triterpéniques par chromatographie en phase gazeuse sur colonne capillaire |
|
— Cires |
Annexe IV |
Détermination de la teneur en cires au moyen de la chromatographie en phase gazeuse avec colonne capillaire |
|
— Alcools aliphatiques et triterpéniques |
Annexe XIX |
Détermination du contenu en alcools aliphatiques et triterpéniques par chromatographie en phase gazeuse avec colonne capillaire |
|
— Acides gras saturés en position 2 |
Annexe VII |
Détermination du pourcentage de 2-glycéryl monopalmitate |
ANNEXE II
DÉTERMINATION DES ACIDES GRAS LIBRES, MÉTHODE À FROID
1. OBJET ET DOMAINE D'APPLICATION
Cette méthode décrit la détermination des acides gras libres dans les huiles d'olive et les huiles de grignons d'olive. La teneur en acides gras libres est exprimée par l'acidité calculée en pourcentage de l'acide oléique.
2. PRINCIPE
Mise en solution d'une prise d'essai dans un mélange de solvants, puis titrage des acides gras libres présents à l'aide d'une solution d'hydroxyde de potassium ou d'hydroxyde de sodium.
3. RÉACTIFS
Tous les réactifs doivent être de qualité analytique reconnue et l'eau utilisée doit être de l'eau distillée ou de pureté équivalente.
|
3.1. |
Éther diéthylique; oxyde diéthylique éthanol à 95 % (V/V), mélange 1-1 en volume. Neutraliser exactement au moment de l'emploi avec la solution d'hydroxyde de potassium (3.2) en présence de 0,3 millilitre de la solution de phénolphtaléine (3.3) pour 100 millilitres de mélange. Note 1: l'oxyde diéthylique est très inflammable et peut former des peroxydes explosifs. Il doit être utilisé en prenant des précautions particulières. Note 2: s'il n'est pas possible d'utiliser l'oxyde diéthylique, un mélange de solvants formé d'éthanol et de toluène peut être utilisé. Si nécessaire, l'éthanol peut être remplacé par le propanol 2. |
|
3.2. |
Hydroxyde de potassium ou hydroxyde de sodium, solution éthanolique titrée ou solution aqueuse, c(KOH) [ou c(NaOH)] – 0,1 mole par litre environ ou, si nécessaire, c(KOH) [ou c(NaOH)] – 0,5 mole par litre environ. Il existe dans le commerce des solutions prêtes à l'emploi. La concentration exacte de la solution d'hydroxyde de potassium (ou de solution d'hydroxyde de sodium) doit être connue et vérifiée avant l'emploi. Utiliser une solution préparée au moins cinq jours avant emploi et décantée dans un flacon en verre brun fermé avec un bouchon de caoutchouc. La solution doit être incolore ou jaune paille. Si une séparation de phases est observée en utilisant une solution aqueuse d'hydroxyde de potassium (ou d'hydroxyde de sodium), remplacer la solution aqueuse par une solution éthanolique. Note 3: une solution incolore stable d'hydroxyde de potassium (ou d'hydroxyde de sodium) peut être préparée de la façon suivante. Porter et maintenir durant une heure à ébullition à reflux 1 000 millilitres d'éthanol ou d'eau avec 8 grammes d'hydroxyde de potassium (ou d'hydroxyde de sodium) et 0,5 gramme de rognures d'aluminium. Distiller immédiatement. Dissoudre dans le distillat la quantité requise d'hydroxyde de potassium (ou d'hydroxyde de sodium). Laisser reposer durant plusieurs jours et décanter le liquide clair surnageant du précipité de carbonate de potassium (ou du carbonate de sodium). La solution peut aussi être préparée sans distillation de la façon suivante: à 1 000 millilitres d'éthanol (ou d'eau), ajouter 4 millilitres de butylite d'aluminium et laisser reposer le mélange durant quelques jours. Décanter le liquide surnageant et y dissoudre la quantité requise d'hydroxyde de potassium (ou d'hydroxyde de sodium). Cette solution est prête pour l'emploi. |
|
3.3. |
Phénolphtaléine, solution à 10 grammes par litre dans l'éthanol à 95-96 % (V/V) ou bleu alcalin 6B ou thymolphtaléine, solution à 20 grammes par litre dans l'éthanol à 95-96 % (V/V). Dans le cas d'huiles fortement colorées, du bleu alcalin ou de la thymolphthaléine doit être utilisé. |
4. APPAREILLAGE
Matériel courant de laboratoire, et notamment:
4.1. Balance analytique
4.2. Fiole conique de 250 millilitres de capacité
4.3. Burette de 10 millilitres de capacité classe A, graduée en 0,05 millilitre, ou burette automatique équivalente.
5. MODE OPÉRATOIRE
5.1. Préparation de l'échantillon pour essai
Lorsque l'échantillon est trouble, il doit être filtré.
5.2. Prise d'essai
Prélever une prise d'essai, selon l'acidité présumée, d'après les indications du tableau suivant.
|
Acidité attendue (acidité oléique en g/100 g) |
Masse de la prise d'essai (en g) |
Précision de la pesée de la prise d'essai (en g) |
|
0 à 2 |
10 |
0,02 |
|
> 2 à 7,5 |
2,5 |
0,01 |
|
> 7,5 |
0,5 |
0,001 |
Peser la prise d'essai dans la fiole conique (4.2).
5.3. Détermination
Dissoudre la prise d'essai (5.2) dans 50 à 100 millilitres du mélange oxyde diéthylique/éthanol (3.1), préalablement neutralisé.
Titrer, en agitant, avec la solution d'hydroxyde de potassium (ou d'hydroxyde de sodium) à 0,1 mole par litre (3.2) (voir note 4) jusqu'à virage de l'indicateur (la couleur de l'indicateur persistant durant au moins 10 secondes).
Note 4: si la quantité nécessaire de solution d'hydroxyde de potassium (ou d'hydroxyde de sodium) à 0,1 mole par litre dépasse 10 millilitres, utiliser une solution à 0,5 mole par litre ou modifier la masse de l'échantillon en fonction de l'acidité libre escomptée et du tableau proposé.
Note 5: si la solution devient trouble pendant le titrage, ajouter une quantité suffisante du mélange de solvants (3.1) pour donner une solution claire.
Effectuer une deuxième détermination uniquement si le premier résultat est supérieur à la valeur limite spécifiée pour la catégorie de l'huile.
6. EXPRESSION DES RÉSULTATS
L'acidité, exprimée en pourcentage d'acide oléique, en poids, est égale à:
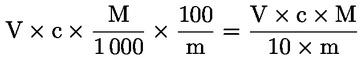
dans laquelle:
|
V |
= |
volume, en millilitres, de la solution titrée d'hydroxyde de potassium (ou d'hydroxyde de sodium) utilisée; |
|
c |
= |
concentration exacte, en moles par litre, de la solution titrée d'hydroxyde de potassium (ou d'hydroxyde de sodium) utilisée; |
|
M |
= |
282 g/mol, masse molaire, en grammes par mole, de l'acide oléique; |
|
m |
= |
masse de la prise d'essai, en grammes. |
L'acidité oléique est déclarée comme suit:
a) à la deuxième décimale pour les valeurs allant de 0 à 1 compris;
b) à la première décimale pour les valeurs allant de 1 à 100 compris.
ANNEXE III
DÉTERMINATION DE L'INDICE DE PEROXYDE
1. Champ d'application
La présente annexe décrit une méthode de détermination de l'indice de peroxyde dans les huiles et matières grasses animales et végétales.
2. Définition
L'indice de peroxyde représente la quantité des substances de l'échantillon (exprimée en milliéquivalents d'oxygène actif par kilogramme) qui oxydent l'iodure de potassium dans les conditions de travail décrites.
3. Principe
La prise d'essai en solution dans un mélange acide acétique et chloroforme est traitée par une solution d'iodure de potassium. L'iode libéré est titré avec une solution de thiosulfate de sodium.
4. Appareillage
Les équipements utilisés doivent être exempts de toute trace de substances oxydantes ou réductrices.
Nota bene: ne pas graisser les surfaces rodées.
4.1. Cuiller en verre de 3 millilitres.
4.2. Fioles d'environ 250 millilitres, avec col et bouchons rodés, séchées au préalable et remplies d'un gaz inerte, sec et pur (azote ou, de préférence, dioxyde de carbone).
4.3. Burette d'une capacité de 5, 10 ou 25 millilitres, avec graduations de 0,05 millilitre au minimum, de préférence avec mise à zéro automatique, ou burette automatique équivalente.
4.4. Balance analytique.
5. Réactifs
5.1. Chloroforme de qualité analytique, exempt d'oxygène (ce dernier ayant été éliminé par barbotage d'un courant de gaz inerte, sec et pur).
5.2. Acide acétique glacial de qualité analytique, exempt d'oxygène (ce dernier ayant été éliminé par barbotage d'un courant de gaz inerte, sec et pur).
5.3. Iodure de potassium en solution aqueuse saturée de préparation récente, exempte d'iode et d'iodates. Dissoudre environ 14 g d'iodure de potassium dans environ 10 ml d'eau à température ambiante.
5.4. Solution aqueuse de thiosulfate de sodium à 0,01 mole par litre (équivalant à 0,01 N), soigneusement normalisée juste avant l'emploi.
Préparer, chaque jour et avant emploi, la nouvelle solution de thiosulfate de sodium à 0,01 mole par litre à partir d'une solution étalon de thiosulfate de sodium à 0,1 mole par litre, ou déterminer la molarité exacte. Comme l'expérience le montre, la stabilité est limitée et dépend de la valeur du pH et de la teneur en dioxyde de carbone libre. Pour la dilution, utiliser exclusivement de l'eau fraîchement portée à ébullition, éventuellement purgée à l'azote.
La procédure suivante est recommandée pour déterminer la molarité exacte de la solution de thiosulfate de sodium:
peser, à 0,001 g près, 0,27 g à 0,33 g d'iodate de potassium (mKIO3) dans une fiole jaugée (de 250 ml ou 500 ml) et compléter au volume avec de l'eau fraîchement portée à ébullition (V2), refroidie à température ambiante. Au moyen d'une pipette, transvaser 5 ml ou 10 ml de cette solution d'iodate de potassium (V1) dans un Erlenmeyer de 250 ml. Ajouter 60 ml d'eau fraîchement portée à ébullition, 5 ml d'acide chlorhydrique à 4 moles par litre, et 25 à 50 mg d'iodure de potassium ou 0,5 ml de la solution aqueuse saturée d'iodure de potassium. Titrer ensuite cette solution avec la solution de thiosulfate de sodium (V3) pour déterminer la molarité exacte de celle-ci.

où:
mKIO3 est la masse d'iodate de potassium, en grammes
V1 est le volume de la solution d'iodate de potassium, en millilitres (5 ml ou 10 ml)
V2 est le volume total de la solution d'iodate de potassium, en millilitres (250 ml ou 500 ml)
V3 est le volume de la solution de thiosulfate de sodium, en millilitres
wKIO3 est la pureté de l'iodate de potassium, en grammes/100 g
MKIO3 est la masse moléculaire de l'iodate de potassium (214 g/mol)
T désigne la molarité exacte de la solution de thiosulfate de sodium (mol/l).
5.5. Solution d'amidon (dispersion aqueuse de 10 grammes par litre) récemment préparée à partir d'amidon naturel soluble. Des réactifs équivalents peuvent également être utilisés.
6. Échantillon
Veiller à ce que l'échantillon soit prélevé et stocké hors de la lumière, conservé au frais et enfermé dans des conteneurs de verre remplis entièrement et fermés hermétiquement à l'aide de bouchons de liège ou de verre rodé.
7. Mode opératoire
L'essai doit être réalisé sous une lumière diffuse (lumière du jour) ou artificielle. Dans une cuiller en verre (4.1) ou, à défaut, dans une fiole (4.2), peser, à 0,001 gramme près, une des masses de l'échantillon visées dans le tableau ci-après,en fonction de l'indice de peroxyde prévu.
|
Indice de peroxyde prévu (en meq) |
Poids de la prise d'essai (en g) |
|
de 0 à 12 |
de 5,0 à 2,0 |
|
de 12 à 20 |
de 2,0 à 1,2 |
|
de 20 à 30 |
de 1,2 à 0,8 |
|
de 30 à 50 |
de 0,8 à 0,5 |
|
de 50 à 90 |
de 0,5 à 0,3 |
Déboucher une fiole (4.2) et introduire la cuiller en verre contenant la prise d'essai. Ajouter 10 millilitres de chloroforme (5.1). Dissoudre rapidement la prise d'essai en agitant. Ajouter 15 millilitres d'acide acétique (5.2) puis 1 millilitre de solution d'iodure de potassium (5.3). Remettre le bouchon rapidement, agiter pendant une minute et laisser reposer pendant exactement 5 minutes à l'abri de la lumière et à une température de 15 à 25 °C.
Ajouter environ 75 millilitres d'eau distillée. Titrer l'iode libéré avec la solution de thiosulfate de sodium (5.4) en agitant vigoureusement et en employant la solution d'amidon (5.5) comme indicateur.
Effectuer deux déterminations sur le même échantillon.
Effectuer simultanément un essai à blanc. Si le résultat de ce dernier excède 0,05 millilitre de solution de thiosulfate de sodium 0,01 N (5.4), remplacer les réactifs impurs.
8. Expression des résultats
L'indice de peroxyde (IP), exprimé en milliéquivalents d'oxygène actif par kilogramme, est fourni par la formule:

où:
|
V |
= |
nombre de millilitres de solution de thiosulfate de sodium normalisée (5.4) utilisé pour l'essai, corrigé en fonction des résultats de l'essai à blanc; |
|
T |
= |
molarité exacte de la solution de thiosulfate de sodium utilisée (5.4), en mol/l. |
|
m |
= |
poids (en grammes) de la prise d'essai. |
Prendre comme résultat la moyenne arithmétique des deux déterminations.
Indiquer le résultat de la détermination à la première décimale.
ANNEXE IV
DÉTERMINATION DE LA TENEUR EN CIRES AU MOYEN DE LA CHROMATOGRAPHIE EN PHASE GAZEUSE AVEC COLONNE CAPILLAIRE
1. OBJET
Cette méthode décrit un procédé pour la détermination de la teneur en cires des huiles d’olive. Les cires sont séparées en fonction du nombre d’atomes de carbone. La méthode peut être employée notamment pour différencier l’huile d’olive de pression de celle d’extraction (huile de grignons).
2. PRINCIPE
La matière grasse, additionnée d’un étalon interne approprié, est fractionnée par chromatographie sur colonne de gel de silice hydraté; la fraction éluée en premier dans les conditions de l’essai (à polarité inférieure à celle des triglycérides) est récupérée puis analysée directement par chromatographie en phase gazeuse sur colonne capillaire.
3. MATÉRIEL
|
3.1. |
Erlenmeyer de 25 ml. |
|
3.2. |
Colonne en verre pour chromatographie en phase gazeuse, diamètre intérieur 15,0 mm, hauteur 30 à 40 cm, équipée d’un robinet. |
|
3.3. |
Appareil de chromatographie en phase gazeuse approprié pour le fonctionnement avec colonne capillaire, équipé d’un système d’introduction directe dans la colonne, constitué par:
|
|
3.4. |
Microseringue pour introduction directe dans la colonne de 10 μl, équipée d’une aiguille cémentée. |
|
3.5. |
Vibrateur électrique. |
|
3.6. |
Évaporateur rotatif. |
|
3.7. |
Four à moufle. |
|
3.8. |
Balance analytique garantissant une précision de la mesure de ± 0,1 mg. |
|
3.9. |
Verrerie normale de laboratoire. |
4. RÉACTIFS
|
4.1. |
Gel de silice d’une granulométrie comprise entre 60 et 200 μm. Le gel de silice doit être placé dans le four à 500 °C pendant au moins 4 heures. Après refroidissement, y ajouter 2 % d’eau par rapport à la quantité de gel de silice prélevée. Agiter convenablement afin d’homogénéiser la masse. Conserver à l’obscurité pendant au moins 12 heures avant emploi. |
|
4.2. |
n-hexane, pour chromatographie. |
|
4.3. |
Éther éthylique, pour chromatographie. |
|
4.4. |
n-Heptane, pour chromatographie. |
|
4.5. |
Solution étalon d’arachidate laurique, solution à 0,1 % (m/V) dans l’hexane (étalon interne). (Il est également possible d’utiliser du palmitate de palmityle ou du stéarate de myristyle)
|
|
4.6. |
Gaz vecteur: hydrogène ou hélium pur, pour chromatographie en phase gazeuse. |
|
4.7. |
Gaz auxiliaires: — hydrogène pur, pour chromatographie en phase gazeuse, — air pur, pour chromatographie en phase gazeuse. |
5. MODE OPÉRATOIRE
5.1. Préparation de la colonne chromatographique
Suspendre 15 g de gel de silice (4.1) dans le n-hexane (4.2) et l’introduire dans la colonne (3.2). Après tassement spontané, le compléter à l’aide d’un agitateur électrique (3.5) pour rendre la couche chromatographique plus homogène. Percoler 30 ml de n-hexane afin d’éliminer les impuretés éventuelles. Peser exactement à l’aide de la balance (3.8) 500 mg de l’échantillon dans l’Erlenmeyer de 25 ml (3.1), ajouter la quantité appropriée d’étalon interne (4.5), en fonction du contenu présumé de cires. Par exemple, ajouter 0,1 mg d’arachidate laurique dans le cas de l’huile d’olive et 0,25 à 0,5 mg dans le cas de l’huile de grignons. Transférer l’échantillon ainsi préparé dans la colonne chromatographique à l’aide de deux portions de 2 ml chacune de n-hexane (4.2).
Laisser s’écouler le solvant jusqu’à 1 mm au-dessus du niveau supérieur de l’absorbant puis percoler 70 ml de n-hexane supplémentaires afin d’éliminer les n-alcanes naturellement présents. Commencer alors l’élution chromatographique en recueillant 180 ml du mélange n-hexane/éther éthylique, rapport 99:1, tout en respectant un débit d’environ 15 gouttes toutes les 10 secondes. L’élution de l’échantillon doit être effectuée à une température ambiante de 22 °C ± 4.
Notes:
— Le mélange n-hexane/éther éthylique (99:1) doit être préparé chaque jour.
— Pour contrôler visuellement l’élution correcte des cires, il est possible d’ajouter à l’échantillon en solution 100 μl de soudan à 1 % dans le mélange d’élution. Le colorant ayant une rétention intermédiaire entre les cires et les triglycérides, lorsque la coloration atteint le fonds de la colonne chromatographique, il convient de suspendre l’élution car toutes les cires ont été éluées.
La fraction ainsi obtenue est séchée dans l’évaporateur rotatif (3.6) jusqu’à élimination pratiquement totale du solvant. Les deux derniers ml du solvant sont éliminés à l’aide d’un faible courant d’azote; ajouter ensuite 2-4 ml de n-heptane
5.2. Analyse par chromatographie en phase gazeuse
5.2.1. Opérations préliminaires
Installer la colonne dans le chromatographe en phase gazeuse (3.3), en branchant le terminal d’entrée au système «on-column» et le terminal de sortie au révélateur. Effectuer les contrôles généraux de l’appareillage de chromatographie en phase gazeuse (tenue des circuits des gaz, efficience du révélateur et du système d’enregistrement, etc.).
Si la colonne est utilisée pour la première fois, il est recommandé de procéder à son conditionnement. Laisser s’écouler un léger débit de gaz à travers la colonne, puis allumer l’appareillage de chromatographie en phase gazeuse. Chauffer graduellement jusqu’à atteindre, au bout d’environ 4 heures, une température de 350 °C. Maintenir cette température pendant au moins 2 heures, puis procéder au réglage de l’appareillage aux conditions de fonctionnement [réglage du débit des gaz, allumage de la flamme, branchement à l’enregistreur électronique (3.3.4), réglage de la température de la chambre pour la colonne, du révélateur, etc.] et enregistrer le signal à une sensibilité au moins 2 fois supérieure à celle prévue pour l’exécution de l’analyse. Le tracé de la ligne de base doit être linéaire, exempt de pics de toute nature, et ne doit pas présenter de déviation.
Une déviation rectiligne négative indique une tenue imparfaite des connexions de la colonne; une déviation positive indique un conditionnement insuffisant de la colonne.
5.2.2. Choix des conditions opératoires
D’une manière générale, les conditions opératoires à observer sont les suivantes:
— température de la colonne:
—
|
|
20 °C/minute |
|
5 °C/minute |
|
20 °C/minute |
|
|
au départ 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— température du révélateur: 350 °C,
— quantité de matière injectée: 1 μl de la solution (2-4 ml) de n-heptane,
— gaz vecteur: hélium ou hydrogène à la vitesse linéaire optimale pour le gaz sélectionné (voir appendice),
— sensibilité instrumentale: en mesure de répondre aux conditions ci-dessous:
Ces conditions peuvent être modifiées en fonction des caractéristiques de la colonne et de l’appareil de chromatographie en phase gazeuse, de manière à obtenir une séparation de toutes les cires, une résolution satisfaisante des pics (voir figure) et un temps de rétention de l’étalon interne C32 qui doit être de 18 ± 3 minutes. Le pic des cires le plus représentatif doit avoir mesuré au moins 60 % du fond de l’échelle.
Les paramètres d’intégration des pics doivent être déterminés de façon à obtenir une évaluation correcte des aires des pics pris en considération.
Note:Vu la température finale élevée, on admet une dérive positive qui de doit pas être supérieure à 10 % du fonds de l’échelle.
5.3. Exécution de l’analyse
Prélever 1 μl de la solution à l’aide de la microseringue de 10 μl; retirer le piston de la seringue de manière à ce que l’aiguille soit vide. Introduire l’aiguille dans le dispositif d’injection et, après 1-2 secondes, injecter rapidement; au bout d’environ 5 secondes, extraire lentement l’aiguille.
Effectuer l’enregistrement jusqu’à élution complète des cires.
La ligne de base doit toujours répondre aux conditions requises.
5.4. Identification des pics
L’identification des différents pics doit être effectuée à partir des temps de rétention et par comparaison avec des mélanges de cires aux temps de rétention connus, analysés dans les mêmes conditions.
La figure ci-après représente un chromatogramme des cires d’une huile d’olive vierge.
5.5. Évaluation quantitative
Procéder au calcul des aires des pics de l’étalon interne et des esters aliphatiques de C40 à C46 à l’aide de l’intégrateur.
Calculer la teneur en cires de chacun des esters, en mg/kg de matière grasse, par la formule:
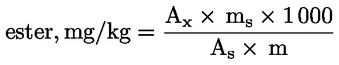
Où:
|
Ax |
= |
aire du pic de chaque ester, en millimètres carrés; |
|
As |
= |
aire du pic de l’étalon interne, en millimètres carrés; |
|
ms |
= |
masse d’étalon interne ajoutée, en milligrammes; |
|
m |
= |
masse de l’échantillon prélevé pour la détermination, en grammes. |
6. EXPRESSION DES RÉSULTATS
Indiquer la somme des teneurs des différentes cires de C40 à C46, en mg/kg de matière grasse (ppm).
Note:Les composants à quantifier font référence aux pics à nombre de carbone paires compris entre les esters C40 et C46, selon l’exemple de chromatogramme des cires de l’huile d’olive reporté dans la figure ci-après. Si l’ester C46 apparaît en double, il est conseillé, pour l’identifier, d’analyser la fraction des cires d’une huile de grignons d’olive où le pic C46 est facilement identifiable car nettement majoritaire.
Les résultats sont exprimés avec une décimale.
Figure
Chromatogramme des cires d’une huile d’olive ( 9 )

Légende:
|
I.S. |
= |
Arachidate laurique |
|
1. |
= |
Esters diterpéniques |
|
2 + 2′ |
= |
Esters C40 |
|
3 + 3′ |
= |
Esters C42 |
|
4 + 4′ |
= |
Esters C44 |
|
5. |
= |
Esters C46 |
|
6. |
= |
Esters stérols et alcool triterpéniques |
APPENDICE
Détermination de la vitesse linéraire du gaz
Injecter de 1 à 3 μl de méthane (ou propane) dans l'appareil de chromatographie en phase gazeuse, après son réglage aux conditions opératoires normales. Chronométrer le temps employé par le gaz pour parcourir la colonne, à partir du moment de son injection jusqu'au moment de la sortie du pic (tM).
La vitesse linéaire, en cm/s, est donnée par la formule L/tM, où L est la longueur de la colonne en centimètres et tM le temps chronométré en secondes.
ANNEXE V
DÉTERMINATION DE LA COMPOSITION ET DE LA TENEUR EN STÉROLS ET EN DIOLS TRITERPÉNIQUES PAR CHROMATOGRAPHIE EN PHASE GAZEUSE SUR COLONNE CAPILLAIRE
1. CHAMP D’APPLICATION
La méthode décrit le procédé de détermination de la teneur en chaque stérol et en stérols totaux ainsi qu'en diols triterpéniques des huiles d'olive et de grignons d'olive.
2. PRINCIPE
L'huile, additionnée d'α-cholestanol en tant qu'étalon interne, est saponifiée avec de l'hydroxyde de potassium en solution dans de l'éthanol, puis l'insaponifiable est extrait avec de l'éther éthylique.
La fraction des stérols et des diols triterpéniques est séparée de l'insaponifiable par chromatographie en couche mince sur une plaque de gel de silice basique; les fractions récupérées dans le gel de silice sont transformées en triméthylsilyléthers qui sont ensuite analysés par chromatographie en phase gazeuse sur colonne capillaire.
3. APPAREILLAGE
Matériel courant de laboratoire, et notamment les éléments suivants:
3.1. Ballon de 250 millilitres muni d'un réfrigérant à reflux avec embouts rodés.
3.2. Ampoule à décanter de 500 ml
3.3. Ballons de 250 ml.
3.4. Équipement complet pour chromatographie en couche mince, avec plaques de verre de 20 × 20 cm.
3.5. Lampe à lumière ultraviolette d'une longueur d'onde de 254 ou 366 nm.
3.6. Microseringues de 100 et 500 μl.
3.7. Ampoule cylindrique filtrante à filtre poreux G 3 (porosité 15-40 μm) d'environ 2 cm de diamètre et 5 cm de hauteur, appropriée pour la filtration sous vide, avec embout rodé mâle.
3.8. Fiole à vide conique de 50 ml avec embout rodé femelle, pouvant s'adapter sur l'ampoule filtrante (point 3.7).
3.9. Tube à fond conique de 10 millilitres, avec bouchon hermétique en verre.
3.10. Appareil de chromatographie en phase gazeuse pouvant être utilisé sur une colonne capillaire avec dispositif d'injection à débit divisé, composé des éléments suivants:
3.10.1. un four thermostaté pour les colonnes, pouvant maintenir la température souhaitée avec une précision de ± 1 °C;
3.10.2. un ensemble d'injection thermoréglable avec élément vaporisateur en verre persilylaté et système à débit divisé;
3.10.3. Détecteur à ionisation de flamme;
3.10.4. Système d’acquisition des données pouvant être utilisé avec l’analyseur FID (point 3.10.3.), avec possibilité d'intégration manuelle.
3.11. Colonne capillaire en silice fondue d'une longueur de 20 à 30 m, d'un diamètre interne compris entre 0,25 et 0,32 mm, recouverte de 5 % diphényle - 95 % diméthylpolysiloxane (phase stationnaire SE-52 ou SE-54 ou équivalent) jusqu'à obtention d'une épaisseur uniforme comprise entre 0,10 et 0,30 μm.
3.12. Microseringue d'une capacité de 10 μl, pour chromatographie en phase gazeuse, avec aiguille soudée, convenant pour l'injection à débit divisé.
3.13. Dessiccateur au dichlorure de calcium
4. RÉACTIFS
4.1. Hydroxyde de potassium (titre minimum 85 %).
4.2. Hydroxyde de potassium en solution éthanolique à environ 2 N.
Dissoudre, tout en refroidissant, 130 g d'hydroxyde de potassium (point 4.1) dans 200 ml d'eau distillée, puis compléter à un litre avec de l'éthanol (point 4.10). Conserver cette solution dans des bouteilles en verre sombre bien fermées pendant deux jours au maximum.
4.3. Éther éthylique, de qualité analytique.
4.4. Hydroxyde de potassium en solution éthanolique à environ 0,2 N.
Dissoudre 13 g d'hydroxyde de potassium (point 4.1) dans 20 ml d'eau distillée, puis compléter à un litre avec de l'éthanol (point 4.10).
4.5. Sulfate de sodium anhydre, de qualité analytique.
4.6. Plaques de verre (20 x 20 cm) recouvertes d'une couche de gel de silice, sans indicateur de fluorescence, d'une épaisseur de 0,25 millimètre d'épaisseur (disponibles dans le commerce déjà prêtes à l'emploi).
4.7. Toluène, de qualité chromatographique.
4.8. Acétone, de qualité chromatographique.
4.9. n-Hexane, de qualité chromatographique.
4.10. Éther éthylique, de qualité chromatographique.
4.11. Éthanol, de qualité analytique.
4.12. Acétate d’éthyle, de qualité analytique.
4.13. Solution de référence pour la chromatographie en couche mince: cholestérol ou phytostérols, et solution d'érythrodiol à 5% dans de l'acétate d'éthyle (point 4.11).
4.14. Dichloro-2′ -7′ fluorescéine, solution éthanolique à 0,2 %. Elle est rendue légèrement basique par addition de quelques gouttes d'une solution alcoolique 2N d'hydroxyde de potassium (point 4.2).
4.15. Pyridine anhydre, de qualité chromatographique (voir note 5).
4.16. Disilazane d'hexaméthyle, de qualité analytique.
4.17. Triméthylchlorosilane, de qualité analytique.
4.18. Solutions échantillons des triméthylsilyléthers des stérols.
À préparer au moment de l'emploi à partir des stérols et de l'érythrodiol tirés des huiles qui les contenaient.
4.19. α-cholestanol, de pureté supérieure à 99% (la pureté doit être vérifiée par analyse chromatographique en phase gazeuse).
4.20. Solution étalon interne d'α-cholestanol à 0,2 % (m/V) dans de l'acétate d'éthyle (point 4.11).
4.21. Solution de phénolphtaléine, 10 g/l dans l’éthanol (point 4.10).
4.22. Gaz vecteur: hydrogène ou hélium pur, pour chromatographie en phase gazeuse.
4.23. Gaz auxiliaires: hydrogène, hélium, azote et air, pour chromatographie en phase gazeuse.
4.24. Mélange 65:35 (V/V) n-Hexane (point 4.9)/éther éthylique (point 4.10).
4.25. Réactif de silylation, constitué d’un mélange 9: 3: 1 (V/V/V) de pyridine/hexaméthyldisilazane/triméthylchlorosilane.
5. MODE OPÉRATOIRE
|
5.1. |
Préparation de l'insaponifiable:
|
|
5.2. |
Séparation de la fraction des stérols et diols triterpéniques (érytrodiol + uvaol)
|
|
5.3. |
Préparation des triméthylsilyléthers (TMSE)
|
|
5.4. |
Analyse par chromatographie en phase gazeuse
|
6. EXPRESSION DES RÉSULTATS
6.1. La concentration de chaque stérol est exprimée en mg/kg de matière grasse, et leur somme correspond aux «stérols totaux».
La composition de chacun des stérols ainsi que la teneur en érytrodiol et en uvaol sont exprimées par des nombres à une décimale.
La teneur en stérols totaux doit être exprimée par un nombre entier.
6.2. Calculer le pourcentage de chaque stérol à partir du rapport entre l'aire du pic correspondant et la somme des aires des pics des stérols:

où:
|
Ax |
= |
aire du pic de x; |
|
ΣA |
= |
somme des aires des pics des stérols. |
6.3. β-sitostérol apparent: Δ5-23-stigmastadiénol + clérostérol + β-sitostérol + sitostanol + Δ5-avenastérol + Δ5-24-stigmastadiénol.
6.4. Calculer le pourcentage d'érytrodiol et d'uvaol:
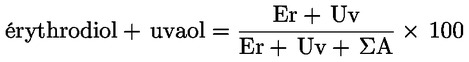
où:
|
ΣA |
= |
somme des aires des pics des stérols, en unités d'intégration; |
|
Er |
= |
aire du pic d'érythrodiol, en unités d'intégration; |
|
Uv |
= |
aire du pic d'uvaol, en unités d'intégration; |
Appendice
Détermination de la vitesse linéaire du gaz
Dans l'appareil de chromatographie en phase gazeuse, réglé aux conditions opératoires normales, injecter 1 à 3 μl de méthane (ou de propane) et chronométrer le temps que met le gaz pour parcourir la colonne, entre le moment de l'injection et celui de la sortie du pic (tM).
La vitesse linéaire, exprimée en cm/s, est donnée par L/tM, où L est la longueur de la colonne, en centimètres, et tM le temps chronométré, en secondes.
Tableau 1
Temps de rétention relatifs des stérols
|
Pic |
Identification |
Temps de rétention relatif |
||
|
SE 54 colonne |
SE 52 colonne |
|||
|
1 |
Cholestérol |
Δ-5-cholestén-3ß-ol |
0,67 |
0,63 |
|
2 |
Cholestanol |
5α-cholestan-3ß-ol |
0,68 |
0,64 |
|
3 |
Brassicastérol |
[24S]-24-méthyl-Δ-5,22-cholestadién-3ß-ol |
0,73 |
0,71 |
|
* |
Ergostérol |
[24S] 24 méthyl Δ5-7-22 cholestatrién 3ß-ol |
0,78 |
0,76 |
|
4 |
24-méthylène-cholestérol |
24-méthylène-Δ-5,24-cholestadién-3ß-o1 |
0,82 |
0,80 |
|
5 |
Campestérol |
(24R)-24-méthyl-Δ-5-cholestén-3ß-ol |
0,83 |
0,81 |
|
6 |
Campestanol |
(24R)-24-méthyl-cholestan-3ß-ol |
0,85 |
0,82 |
|
7 |
Stigmastérol |
(24S)-24-éthyl-Δ-5,22-cholestadién-3ß-ol |
0,88 |
0,87 |
|
8 |
Δ-7-campestérol |
(24R)-24-méthyl-Δ-7-cholestén-3ß-ol |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadiénol |
(24R,S)-24-éthyl-Δ-5,23-choIestadién-3ß-ol |
0,95 |
0,95 |
|
10 |
Clérostérol |
(24S)-24-éthyl-Δ-5,22-cholestadién-3ß-ol |
0,96 |
0,96 |
|
11 |
ß-sitostérol |
(24R)-24-éthyl-Δ-5-cholestén-3ß-ol |
1,00 |
1,00 |
|
12 |
Sitostanol |
24-éthyl-cholestan-3ß-ol |
1,02 |
1,02 |
|
13 |
Δ-5-avénastérol |
(24Z)-24-éthylidène-Δ-cholestén-3ß-ol |
1,03 |
1,03 |
|
14 |
Δ-5-24-stigmastadiénol |
(24R,S)-24-éthyl-Δ-5,24-cholestadién-3ß-ol |
1,08 |
1,08 |
|
15 |
Δ-7-stigmasténol |
(24R,S)-24-éthyl-Δ-7-cholestén-3ß-ol |
1,12 |
1,12 |
|
16 |
Δ-7-avénastérol |
(24Z)-24-éthylidène-Δ-7-cholestén-3ß-ol |
1,16 |
1,16 |
|
17 |
Érythrodiol |
5α oléan-12en-3bß8 diol |
1,41 |
1,41 |
|
18 |
Uvaol |
Δ12-ursen-3ß28 diol |
1,52 |
1,52 |
Figure 1
Chromatogramme (en phase gazeuse) de la fraction des stérols et des diols triterpéniques d'une huile d’olive lampante (avec étalon interne)

Figure 2
Chromatogramme (en phase gazeuse) de la fraction des stérols et des diols triterpéniques d'une huile d’olive raffinée (avec étalon interne)
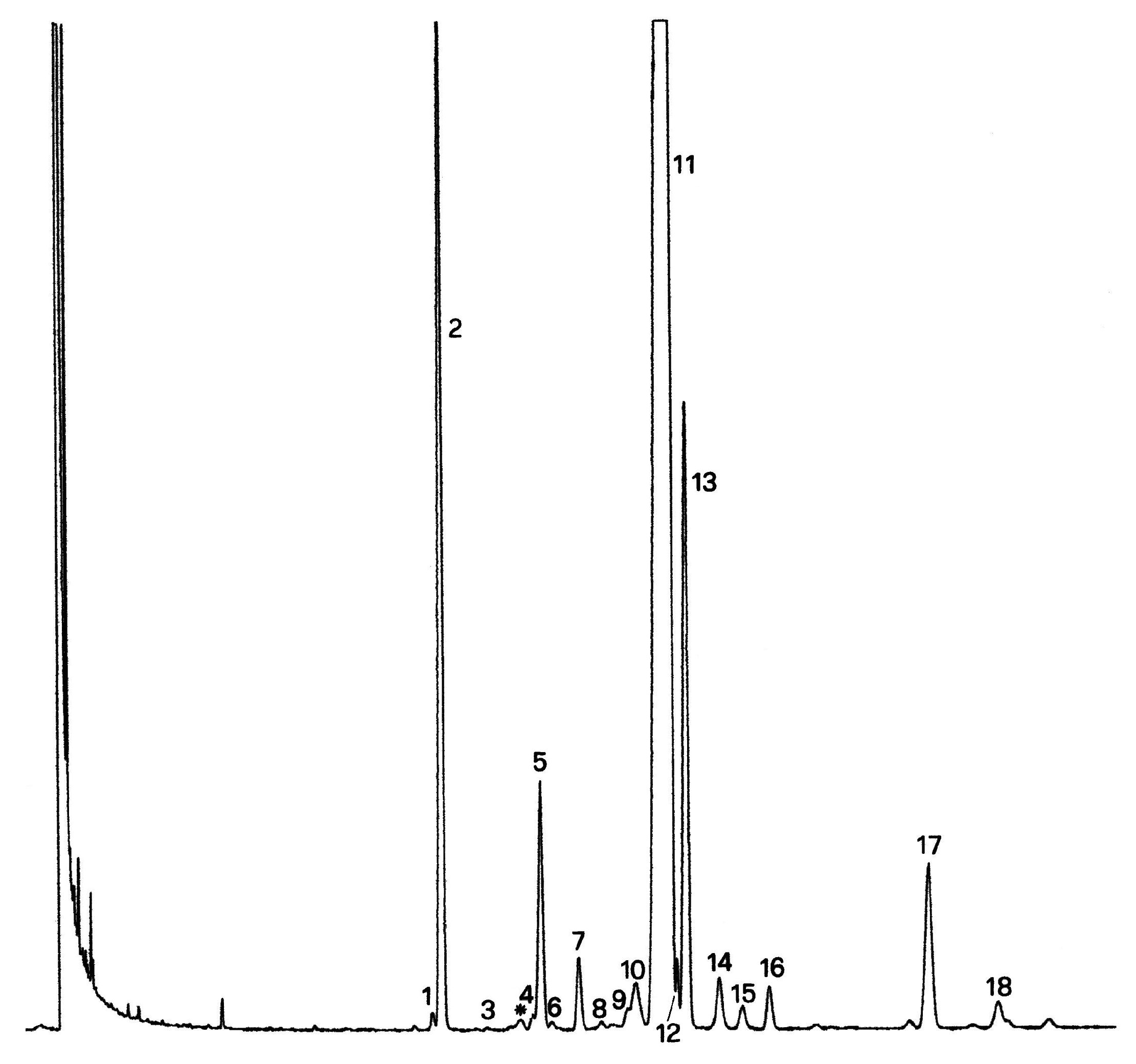
Figure 3
Plaque de chromatographie en couche mince d'une huile de grignons d’olive, montrant la zone à racler pour la détermination des stérols et des diols triterpéniques

1 — Squalène
2 — Alcools triterpéniques et aliphatiques
3 — Stérols et diols triterpéniques
4 — Acides gras initiaux et acides gras libres
▼M26 —————
ANNEXE VII
DÉTERMINATION DU POURCENTAGE DU 2-GLYCÉRIL MONOPALMITATE
1. OBJET ET DOMAINE D’APPLICATION
Cette méthode décrit la procédure analytique pour la détermination du pourcentage d’acide palmitique en position 2 des triglycérides au moyen de l’évaluation du 2-glycéril monopalmitate.
Cette méthode est applicable aux huiles végétales liquides à température ambiante (20 °C).
2. PRINCIPE
Après préparation, l’échantillon d’huile est soumis à l’action de la lipase pancréatique: une hydrolyse partielle et spécifique dans les positions 1 et 3 de la molécule de triglycéride entraîne l’apparition des monoglycérides en position 2. Le pourcentage de 2-glycéril monopalmitate dans la fraction monoglycéridique est déterminé, après silylation, par chromatographie en phase gazeuse sur colonne capillaire.
3. APPAREILLAGE ET MATÉRIEL COURANT
|
3.1. |
Erlenmeyer, 25 ml |
|
3.2. |
Béchers 100, 250 et 300 ml |
|
3.3. |
Colonne en verre pour chromatographie, diamètre intérieur 21-23 mm, longueur 400 mm, équipée d’une plaque en verre fritté et d’un robinet |
|
3.4. |
Éprouvettes graduées de 10, 50, 100 et 200 ml |
|
3.5. |
Ballons de 100 et 250 ml |
|
3.6. |
Évaporateur rotatif |
|
3.7. |
Tubes de centrifugeuse à fond conique de 10 ml, avec bouchon rodé |
|
3.8. |
Centrifugeuse pour des tubes de 10 et 100 ml |
|
3.9. |
Thermostat permettant de maintenir la température à 40 ± 0,5 °C |
|
3.10. |
Pipettes graduées de 1 et 2 ml |
|
3.11. |
Seringue hypodermique de 1 ml |
|
3.12. |
Microseringue de 100 μl |
|
3.13. |
Entonnoir, 1 000 ml |
|
3.14. |
Chromatographe en phase gazeuse pour colonnes capillaires, équipé d’un dispositif d’injection «on-column» à froid pour l’introduction directe de l’échantillon dans la colonne et d’un four susceptible de maintenir la température choisie à 1 °C près |
|
3.15. |
Injecteur «on-column» à froid pour l’introduction directe de l’échantillon dans la colonne |
|
3.16. |
Détecteur à ionisation de flamme et électromètre |
|
3.17. |
Enregistreur-intégrateur adapté à l’électromètre avec une vitesse de réponse non supérieure à 1 seconde et une vitesse variable de déroulement du papier |
|
3.18. |
Colonne capillaire en verre ou en silice fondue de 8 à 12 mètres, d’un diamètre intérieur de 0,25 à 0,32 mm, recouverte de methylpolysiloxane ou de phényl méthylpolysiloxane 5 %, d’une épaisseur de 0,10-0,30 μm, pouvant être utilisée à 370 °C |
|
3.19. |
Microseringue de 10 μl munie d’une aiguille cémentée, d’au moins 7,5 cm de long, pour injection directe sur colonne |
4. RÉACTIFS
|
4.1. |
Gel de silice ayant une granulométrie comprise entre 0,063 et 0,200 mm (70/280 mesh), préparé comme suit: mettre le gel de silice dans une capsule de porcelaine, sécher à l’étuve à 160 °C pendant 4 heures, puis laisser refroidir à température ambiante dans un dessiccateur. Ajouter un volume d’eau équivalent à 5 % du poids du gel de silice, comme suit: dans un Erlenmeyer de 500 ml, peser 152 g de gel de silice et ajouter 8 g d’eau distillée, boucher et agiter délicatement pour obtenir une répartition uniforme de l’eau. Laisser reposer au moins 12 heures avant l’emploi. |
|
4.2. |
n-hexane (pour chromatographie) |
|
4.3. |
Isopropanol |
|
4.4. |
Isopropanol, solution aqueuse 1/1 (V/V) |
|
4.5. |
Lipase pancréatique. La lipase utilisée doit avoir une activité comprise entre 2,0 et 10 unités de lipase par mg. (Il existe dans le commerce des lipases pancréatiques ayant une activité comprise entre 2 et 10 unités par mg d’enzyme.) |
|
4.6. |
Solution tampon de tris-hydroxy-méthylaminométhane: solution aqueuse 1 M amenée jusqu’à pH 8 (contrôle potentiométrique) par du HCI concentré (1/1 V/V) |
|
4.7. |
Cholate de sodium, qualité enzymatique, solution aqueuse à 0,1 % (cette solution doit être utilisée dans les 15 jours suivant sa préparation) |
|
4.8. |
Chlorure de calcium, solution aqueuse à 22 % |
|
4.9. |
Éther diéthylique pour chromatographie |
|
4.10. |
Solvant de développement: mélange n-hexane/éther diéthylique (87/13) (V/V) |
|
4.11. |
Hydroxyde de sodium, solution à 12 % en poids |
|
4.12. |
Phénolphtaléine, solution à 1 % dans l’éthanol |
|
4.13. |
Gaz vecteur: hydrogène ou hélium, pour chromatographie en phase gazeuse |
|
4.14. |
Gaz auxiliaires: hydrogène, à 99 % minimum, exempt d’humidité et de substances organiques; et air, pour chromatographie en phase gazeuse de la même pureté |
|
4.15. |
Réactif de silanisation: mélange pyridine/hexamethyldisilazane, trimethylchlorosilane 9/3/1 (V/V/V). (Des solutions prêtes à l'emploi existent dans le commerce. D’autres réactifs de silylation peuvent être employés, notamment bis-trimethylsilyl trifluoracetamide + 1 % trimethylchlorosilane, dilués avec un volume identique de pyridine anhydre.) |
|
4.16. |
Échantillons de référence: monoglycérides purs ou mélanges de monoglycérides ayant une composition en pourcentage connue similaire à celle de l’échantillon. |
5. PROCÉDURE
5.1. Préparation de l’échantillon
|
5.1.1. |
Les huiles ayant une acidité libre inférieure à 3 % n’ont pas besoin d’être neutralisées avant la chromatographie sur colonne de gel de silice. Les huiles ayant une acidité libre supérieure à 3 % devront être soumises à la neutralisation conformément au point 5.1.1.1.
|
|
5.1.2. |
Introduire 1,0 g d’huile préparée comme indiqué ci-dessus dans un Erlenmeyer de 25 ml (3.1) et dissoudre dans 10 ml de mélange de développement (4.10). Laisser reposer la solution pendant au moins 15 minutes avant la chromatographie sur colonne de gel de silice. Si la solution est trouble, la centrifuger pour garantir des conditions optimales pour la chromatographie. (Des cartouches de gel de silice SPE de 500 mg prêtes à l’emploi peuvent être utilisées.) |
|
5.1.3. |
Préparation de la colonne chromatographique Verser dans la colonne (3.3) environ 30 ml du solvant de développement (4.10), introduire un morceau de coton dans la partie inférieure de la colonne à l’aide d’une baguette de verre; presser pour éliminer l’air. Dans un bécher, préparer une suspension de 25 g de gel de silice (4.1) dans environ 80 ml de solvant de développement et le verser dans la colonne à l’aide d’un entonnoir. Vérifier que tout le gel de silice a été introduit dans la colonne; laver avec le solvant de développement (4.10), ouvrir le robinet et laisser le niveau du liquide atteindre environ 2 mm au-dessus du niveau supérieur du gel de silice. |
|
5.1.4. |
Chromatographie sur colonne Dans un Erlenmeyer de 25 ml (3.1), peser exactement 1,0 g d’échantillon préparé conformément au point 5.1. Dissoudre l’échantillon dans 10 ml de solvant de développement (4.10). Verser la solution dans la colonne chromatographique préparée conformément au point 5.1.3. Éviter de remuer la surface de la colonne. Ouvrir le robinet et laisser s’écouler la solution de l’échantillon jusqu’à ce qu’elle atteigne le niveau du gel de silice. Développer avec 150 ml de solvant de développement. Ajuster le débit à 2 ml/min (de façon à ce que 150 ml s’écoulent dans la colonne en 60-70 minutes environ). Récupérer l’éluat dans un ballon de 250 ml préalablement taré. Évaporer le solvant sous vide et enlever les dernières traces de celui-ci sous un courant d’azote. Peser le ballon et calculer l’extrait récupéré [En cas d’utilisation de cartouches SPE de silice prêtes à l’emploi, procéder comme suit: introduire 1 ml de solution (5.1.2) dans les cartouches préalablement préparées avec 3 ml de n-hexane. Après percolation de la solution, développer avec 4 ml de n-hexane/éther diéthylique 9/1 (V/V). Récupérer l’éluat dans un tube de 10 ml et le soumettre à évaporation sous un courant d’azote jusqu’à siccité. Soumettre le résidu sec à l’action de la lipase pancréatique (5.2). Il est fondamental de vérifier la composition en acide gras avant et après passage sur cartouche SPE]. |
5.2. Hydrolyse par la lipase pancréatique
|
5.2.1. |
Dans le tube de la centrifugeuse, peser 0,1 g de l’huile préparée conformément au point 5.1. Ajouter 2 ml de solution tampon (4.6), 0,5 ml de la solution de cholate de sodium (4.7) et 0,2 ml de la solution de chlorure de calcium, en agitant bien après chaque addition. Fermer le tube par le bouchon rodé et le placer dans le thermostat à 40 ± 0,5 °C. |
|
5.2.2. |
Ajouter 20 mg de lipase, agiter soigneusement (en évitant de mouiller le bouchon) et mettre le tube dans le thermostat pendant exactement 2 minutes, puis le retirer, agiter énergiquement pendant 1 minute exactement et laisser refroidir. |
|
5.2.3. |
Ajouter 1 ml d’éther diéthylique, boucher et agiter énergiquement, puis centrifuger et transférer la solution d’éther dans un tube propre et sec, à l’aide d’une microseringue. |
5.3. Préparation des dérivés silanisés et de la chromatographie en phase gazeuse
|
5.3.1. |
À l’aide d’une microseringue, introduire 100 μl de solution (5.2.3) dans un tube à fond conique de 10 ml. |
|
5.3.2. |
Éliminer le solvant sous un léger courant d’azote, ajouter 200 μl de réactif de silanisation (4.15), boucher le tube et laisser reposer pendant 20 minutes. |
|
5.3.3. |
Après 20 minutes, ajouter 1 à 5 ml de n-hexane (en fonction des conditions chromatographiques): la solution résultant est prête pour la chromatographie en phase gazeuse. |
5.4. Chromatographie en phase gazeuse
Les conditions d’opération sont les suivantes:
— température de l’injecteur (injecteur «on-column») inférieure à la température d’ébullition du solvant (68 °C),
— température du détecteur: 350 °C,
— température de la colonne: programmation de la température du four: 60 °C pendant 1 minute, en augmentant de 15 °C par minute jusqu’à 180 °C, puis de 5 °C par minute jusqu’à 340 °C, puis 340 °C pendant 13 minutes,
— gaz vecteur: hydrogène ou hélium, réglé à la vitesse linéaire adéquate en vue d’obtenir la résolution reflétée dans la figure 1. Le temps de rétention du triglycéride C54 doit être de 40 ± 5 minutes (voir figure 2). (Les conditions d’opérations indiquées ci-dessus sont proposées à titre indicatif. Chaque opérateur devra les optimiser pour atteindre la résolution désirée. La hauteur du pic correspondant au 2-glycéryl monopalmitate doit avoir une hauteur minimale égale à 10 % de l’échelle de l’enregistreur),
— quantité de substance injectée: 0,5-1 μl de la solution (5 ml) de n-hexane (5.3.3).
5.4.1. Identification des pics
Les monoglycérides individuels sont identifiés en fonction des temps de rétention obtenus et par rapport à ceux obtenus avec les mélanges standards de monoglycérides analysés dans les mêmes conditions.
5.4.2. Évaluation quantitative
L’aire de chaque pic est calculée au moyen d’un intégrateur électronique.
6. EXPRESSION DES RÉSULTATS
Le pourcentage de glycéril monopalmitate est calculé à partir du rapport entre l’aire du pic correspondant et la somme des aires des pics de tous les monoglycérides (voir figure 2), selon la formule:
Glycéryl monopalmitate (%):

Où:
|
Ax |
= |
aire du pic correspondant au glycéryl monopalmitate; |
|
ΣA |
= |
somme des aires de la totalité des pics des monoglycérides. |
Le résultat doit être donné avec une décimale.
7. COMPTE RENDU DE L’ANALYSE
Le compte rendu de l’analyse devra spécifier:
— la référence à cette méthode,
— toute information nécessaire à l’identification complète de l’échantillon,
— le résultat de l’analyse,
— tout écart de cette méthode, qu’il s’agisse d’une décision des parties concernées ou pour une autre raison,
— les détails d’identification du laboratoire, la date de l’analyse et la signature des responsables de celle-ci.
Figure 1
Chromatogramme des produits de la réaction de silanisation obtenus par l’action de la lipase sur une huile d’olive raffinée additionnée de 20 % d’huile estérifiée (100 %)
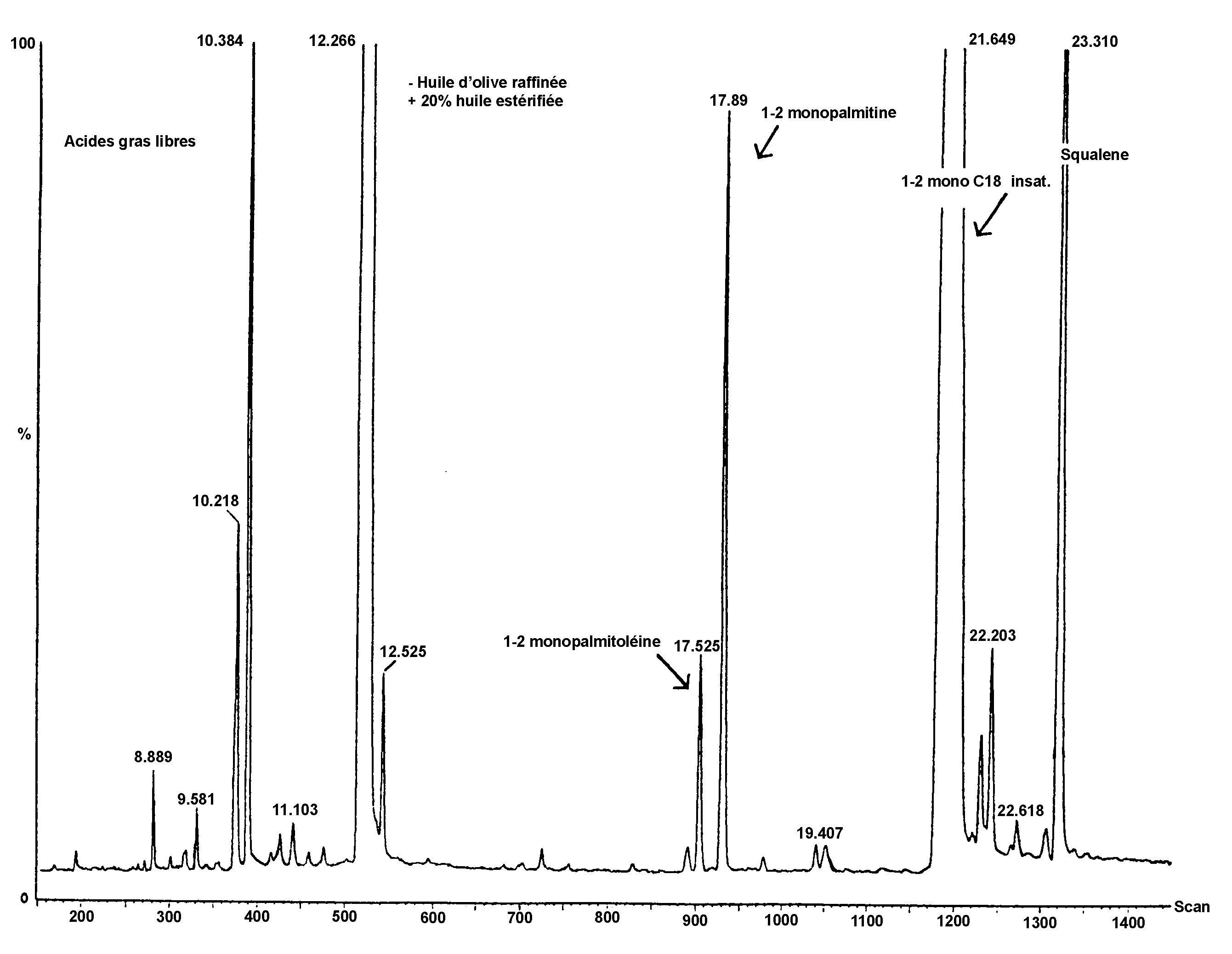
Figure 2
Chromatogramme de:
|
A) |
huile d’olive non estérifiée, après lipase; après silanisation; dans ces conditions (colonne capillaire 8-12 m), la fraction de cire est éluée en même temps que la fraction de diglycéride ou peu de temps après. Après lipase, la teneur en triglycérides ne devrait pas excéder 15 %. 
Légende:
|
Chromatogramme de:
|
B) |
huile estérifiée après lipase; après silanisation; dans ces conditions (colonne capillaire 8-12 m), la fraction de cire est éluée en même temps que la fraction de diglycéride ou peu de temps après. Après lipase, la teneur en triglycérides ne devrait pas excéder 15 %. 
Légende:
|
8. NOTES
PRÉPARATION DE LA LIPASE
Il existe dans le commerce des lipases ayant une activité satisfaisante. Il est également possible de les préparer au laboratoire de la façon suivante:
Refroidir à 0 °C 5 kg de pancréas frais de porc. Débarrasser de la graisse solide et du tissu conjonctif qui les entourent et les triturer dans un moulin à lames jusqu’à obtention d’une pâte fluide. Agiter cette pâte pendant 4 à 6 heures avec 2,5 litres d’acétone anhydre puis centrifuger. Extraire le résidu trois autres fois avec le même volume d’acétone anhydre, puis deux fois avec un mélange acétone/éther diéthylique (1/1) (V/V) et deux fois avec de l’éther diéthylique.
Sécher le résidu pendant 48 heures sous vide pour obtenir une poudre stable qui se conservera longtemps au réfrigérateur et à l’abri de l’humidité.
CONTRÔLE DE L’ACTIVITÉ LIPASIQUE
Préparer une émulsion d’huile d’olive comme suit:
Agiter pendant 10 minutes dans un mélangeur un mélange constitué de 165 ml d’une solution de gomme arabique à 100 g/l, 15 g de glace pilée et 20 ml d’une huile d’olive préalablement neutralisée.
Introduire successivement 10 ml de cette émulsion dans un bécher de 50 ml puis 0,3 ml d’une solution de cholate de sodium à 0,2 g/ml et 20 ml d’eau distillée.
Placer le bécher dans un thermostat régulé à 37 °C; introduire les électrodes du pH-mètre et l’agitateur à hélice.
Ajouter goutte à goutte, à l’aide d’une burette, une solution d’hydroxyde de sodium 0,1 N jusqu’à obtention d’un pH de 8,3.
Ajouter un volume de suspension de poudre de lipase dans l’eau (0,1 g/ml de lipase). Dès que le pH-mètre indique un pH de 8,3, mettre en marche le chronomètre et ajouter la solution d’hydroxyde de sodium, goutte à goutte, au rythme nécessaire pour maintenir le pH à la valeur de 8,3. Noter chaque minute le volume de solution consommé.
Reporter les données dans un système d’axes de coordonnées en portant en abscisses les temps et en ordonnées les millilitres de solution alcaline 0,1 N consommés pour maintenir le pH constant. Un graphique linéaire doit être obtenu.
L’activité de la lipase, mesurée en unités lipase par mg, est donnée par la formule suivante:
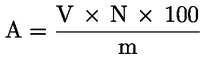
où:
|
A |
est l’activité en unités lipase/mg; |
|
V |
est le nombre de millilitres de solution d’hydroxyde de sodium 0,1 N par minute (calculé à partir du graphique); |
|
N |
est la normalité de la solution d’hydroxyde de sodium; |
|
m |
est la masse en mg de la lipase d’essai. |
L’unité lipase est définie comme la quantité d’enzyme qui libère 10 micro-équivalents d’acide par minute.
▼M20 —————
ANNEXE IX
ANALYSE SPECTROPHOTOMÉTRIQUE DANS L'ULTRAVIOLET
AVANT-PROPOS
L'analyse spectrophotométrique dans l'ultraviolet peust fournir des indications sur la qualité d'une matière grasse, sur son état de conservation et sur les modifications subies du fait de processus technologiques. L'absorption aux longueurs d'onde spécifiées dans la méthode est due à la présence de systèmes de diènes et triènes conjugués résultant de processus d'oxydation et/ou de pratiques de raffinage. Les valeurs de cette absorption sont exprimées comme l'extinction spécifique
![]()
(l'extinction d'une solution de matière grasse à 1 % m/v dans le solvant spécifié, dans une cuve de 10 mm), notée de façon conventionnelle K (également dénommé «coefficient d'extinction»).
1. CHAMP D'APPLICATION
La présente annexe décrit la procédure à suivre pour réaliser une analyse spectrophotométrique dans le domaine de l'ultraviolet de l'huile d'olive.
2. PRINCIPE DE LA MÉTHODE
Un échantillon est dissous dans le solvant requis, puis l'absorbance de la solution est mesurée aux longueurs d'onde spécifiées, par rapport au solvant pur.
Les valeurs de l'extinction spécifique à 232 nm et à 268 nm dans de l'iso-octane ou à 232 nm et 270 nm dans du cyclohexane sont calculées pour une concentration de 1 % m/v dans une cuve de 10 mm.
3. APPAREILLAGE
|
3.1. |
Spectrophotomètre permettant d'effectuer des mesures à des longueurs d'onde UV (220 nm à 360 nm), avec capacité de lecture nanométrique. Un contrôle régulier est recommandé pour la précision et la reproductibilité des échelles des longueurs d'onde et d'absorbance ainsi que pour la lumière parasite.
|
|
3.2. |
Cuves en quartz rectangulaires, avec couvercle, pour les mesures aux longueurs d'onde UV (220 à 360 nm) et d'une longueur optique de 10 mm. Lorsqu'elles sont remplies d'eau ou d'un autre solvant approprié, les cuves ne doivent pas présenter entre elles de différence supérieure à 0,01 unité d'extinction. |
|
3.3. |
Fioles jaugées à un trait, d'une capacité de 25 ml, classe A. |
|
3.4. |
Balance analytique, permettant une lecture au 0,0001 g près. |
4. RÉACTIFS
Au cours de l'analyse, sauf indication contraire, n'utiliser que des réactifs de qualité analytique reconnue et de l'eau distillée ou déminéralisée ou de l'eau de pureté équivalente.
Solvant: iso-octane (2,2,4-triméthylpentane) pour les mesures à 232 nm et à 268 nm et cyclohexane pour les mesures à 232 nm et à 270 nm, présentant une absorbance inférieure à 0,12 à 232 nm et inférieure à 0,05 à 270 nm par rapport à l'eau distillée, mesurée dans une cuve de 10 mm.
5. PROCÉDURE
|
5.1. |
L'échantillon doit être parfaitement homogène et exempt d'impuretés en suspension. Dans le cas contraire, il doit être filtré sur papier à une température d'environ 30 °C. |
|
5.2. |
Introduire environ 0,25 g (au mg près) de l'échantillon ainsi préparé dans une fiole jaugée de 25 ml, compléter avec le solvant spécifié et homogénéiser. La solution obtenue doit être parfaitement limpide. Au cas où la solution présenterait une opalescence ou un trouble, filtrer rapidement sur papier. REMARQUE: en général, une masse de 0,25 à 0,30 g est suffisante pour des mesures d'absorbance d'huiles d'olive vierge et vierge extra à 268 nm et 270 nm. Pour des mesures à 232 nm, 0,05 g d'échantillon est habituellement nécessaire, de sorte que deux solutions distinctes sont généralement préparées. Pour les mesures d'absorbance d'huiles de grignons d'olive, d'huiles d'olive raffinées et d'huiles d'olive frelatées, une plus petite partie de l'échantillon (0,1 g) est généralement nécessaire du fait de leur plus forte absorbance. |
|
5.3. |
Le cas échéant, corriger les valeurs de base (220-290 nm) avec un solvant dans les deux cuves de quartz (échantillon et référence), puis remplir la cuve en quartz de l'échantillon avec la solution d'essai et mesurer les valeurs d'extinction à 232, 268 ou 270 nm par comparaison avec le solvant servant de référence. Les valeurs d'extinction enregistrées doivent être comprises dans l'intervalle 0,1 à 0,8 ou dans la plage de linéarité du spectrophotomètre qui doit être vérifiée. Dans le cas contraire, il faut répéter les mesures en utilisant des solutions plus concentrées ou plus diluées, selon le cas. |
|
5.4. |
Après la mesure de l'absorbance à 268 nm ou à 270 nm, mesurer l'absorbance à λmax, λmax + 4 et λmax – 4. Ces valeurs d'absorbance sont utilisées pour déterminer la variation de l'extinction spécifique (ΔΚ). REMARQUE: λmax est fixé à 268 nm pour l'iso-octane utilisé comme solvant et à 270 nm pour le cyclohexane. |
6. EXPRESSION DES RÉSULTATS
|
6.1. |
Relever les extinctions spécifiques (coefficients d'extinction) aux diverses longueurs d'onde, calculées comme suit:
où:
Les résultats sont exprimés avec deux décimales. |
|
6.2. |
Variation de l'extinction spécifique (ΔΚ) La variation de la valeur absolue de l'extinction (ΔΚ) est donnée par la formule:
où Km est l'extinction spécifique à la longueur d'onde correspondant à l'absorption maximale à 270 nm et 268 nm en fonction du solvant utilisé. Les résultats sont exprimés avec deux décimales. |
ANNEXE X
DÉTERMINATION DES ESTERS MÉTHYLIQUES D'ACIDES GRAS PAR CHROMATOGRAPHIE EN PHASE GAZEUSE
1. CHAMP D'APPLICATION
La présente annexe fournit des directives pour la détermination par chromatographie en phase gazeuse des acides gras libres et liés dans les matières grasses et les huiles végétales après leur conversion en esters méthyliques d'acides gras (EMAG).
Les acides gras liés des triglycérides (TG) et, en fonction de la méthode d'estérification, les acides gras libres (AGL), sont convertis en esters méthyliques d'acides gras (EMAG), qui sont déterminés par chromatographie en phase gazeuse sur colonne capillaire.
La méthode décrite dans la présente annexe permet la détermination des EMAG de C12 à C24, y compris les esters méthyliques d'acides gras saturés, mono-insaturés cis et trans et poly-insaturés cis et trans.
2. PRINCIPE
La chromatographie en phase gazeuse (CG) est utilisée pour l'analyse quantitative des EMAG. Les EMAG sont préparés conformément à la partie A. Ils sont ensuite injectés et vaporisés dans l'injecteur. La séparation des EMAG est effectuée sur des colonnes analytiques de polarité et de longueur particulières. Un détecteur à ionisation de flamme (FID) est utilisé pour la détection des EMAG. Les conditions d'analyse sont décrites dans la partie B.
L'hydrogène ou l'hélium peuvent être utilisés comme gaz vecteur (phase mobile) dans la chromatographie en phase gazeuse des EMAG avec FID. L'hydrogène accélère la séparation et fournit des pics plus marqués. La phase stationnaire est une couche microscopique d'un mince film liquide sur une surface solide inerte de silice fondue.
Lors de leur passage le long de la colonne capillaire, les composés volatiles en cours d'analyse interagissent avec la phase stationnaire tapissant la surface interne de la colonne. En raison de cette interaction différente de divers composés, l'élution de ces derniers s'effectue à des moments différents, qui représentent chacun le temps de rétention du composé pour un ensemble de paramètres d'analyse donné. La comparaison des temps de rétention sert à l'identification des différents composés.
PARTIE A
PRÉPARATION DES ESTERS MÉTHYLIQUES D'ACIDES GRAS DE L'HUILE D'OLIVE ET DE L'HUILE DE GRIGNONS D'OLIVE
1. CHAMP D'APPLICATION
La présente partie définit la préparation des esters méthyliques d'acides gras. Elle présente des méthodes de préparation des esters méthyliques d'acides gras des huiles d'olive et des huiles de grignons d'olive.
2. DOMAINE D'APPLICATION
La préparation des esters méthyliques d'acides gras des huiles d'olive et des huiles de grignons d'olive est effectuée par transestérification avec une solution méthanolique d'hydroxyde de potassium à température ambiante. La nécessité de purification de l'échantillon avant la transestérification dépend de la teneur de l'échantillon en acides gras libres et du paramètre analytique à déterminer; la méthode peut être choisie conformément au tableau ci-après:
|
Catégorie d'huile |
Méthode |
|
Huile d'olive vierge avec une acidité inférieure ou égale à 2,0 %. |
1. Acides gras 2. Acides gras trans 3. ΔECN42 (après purification par SPE avec gel de silice) |
|
Huile d'olive raffinée |
|
|
Huile d'olive composée d'huile d'olive raffinée et d'huiles d'olive vierges |
|
|
Huile de grignons d'olive raffinée |
|
|
Huile de grignons d'olive |
|
|
Huile d'olive vierge avec une acidité supérieure à 2,0 %. Huile de grignons d'olive brute |
1. Acides gras (après purification par SPE avec gel de silice) 2. Acides gras trans (après purification par SPE avec gel de silice) 3. ΔECN42 (après purification par SPE avec gel de silice) |
3. MÉTHODOLOGIE
3.1. Transestérification dans une solution méthanolique d'hydroxyde de potassium à température ambiante
3.1.1. Principe
Les esters méthyliques se forment par transestérification dans une solution méthanolique d'hydroxyde de potassium comme phase intermédiaire avant la saponification.
3.1.2. Réactifs
|
3.1.2.1. |
Méthanol ne contenant pas plus de 0,5 % (m/m) d'eau. |
|
3.1.2.2. |
Hexane pour chromatographie. |
|
3.1.2.3. |
Heptane pour chromatographie. |
|
3.1.2.4. |
Éther diéthylique, stabilisé pour analyses. |
|
3.1.2.5. |
Acétone pour chromatographie. |
|
3.1.2.6. |
Solvant d'élution pour la purification de l'huile par chromatographie sur colonne ou par SPE, mélange hexane/éther diéthylique dans les proportions 87/13 (v/v). |
|
3.1.2.7. |
Hydroxyde de potassium, solution méthanolique d'environ 2 M: dissoudre 11,2 g d'hydroxyde de potassium dans 100 ml de méthanol. |
|
3.1.2.8. |
Cartouches de gel de silice, 1 g (6 ml), pour l'extraction en phase solide. |
3.1.3. Appareillage
|
3.1.3.1. |
Éprouvettes à bouchon vissant (de 5 ml de capacité) avec un bouchon muni d'un joint de PTFE. |
|
3.1.3.2. |
Pipettes graduées ou automatiques de 2 ml et 0,2 ml. |
3.1.4. Purification des échantillons d'huile
Si nécessaire, les échantillons seront purifiés par passage de l'huile dans une cartouche de gel de silice pour l'extraction en phase solide. Placer une cartouche de gel de silice (3.1.2.8) dans un appareil d'élution à vide et laver avec 6 ml d'hexane (3.1.2.2); le lavage est effectué sans vide. Charger ensuite dans la colonne une solution d'huile (environ 0,12 g) dans 0,5 ml d'hexane (3.1.2.2). Faire descendre la solution pour élution avec 10 ml d'hexane/d'éther diéthylique (87:13 v/v) (3.1.2.6). Homogénéiser la totalité des éluats et diviser en deux volumes similaires. Faire évaporer un des volumes jusqu'à dessiccation dans un évaporateur rotatif sous pression réduite et à température ambiante. Dissoudre le résidu dans 1 ml d'heptane. La solution obtenue est prête pour l'analyse des acides gras par CG. Faire évaporer le second volume et dissoudre le résidu dans 1 ml d'acétone pour l'analyse des triglycérides par CLHP si nécessaire.
3.1.5. Procédure
Dans une éprouvette à bouchon vissant de 5 ml (3.1.3.1), peser environ 0,1 g de l'échantillon d'huile. Ajouter 2 ml d'heptane (3.1.2.2) et agiter. Ajouter 0,2 ml de la solution méthanolique d'hydroxyde de potassium (3.1.2.7), boucher à l'aide du bouchon muni d'un joint en PTFE, bien fermer et agiter énergiquement pendant 30 secondes. Laisser reposer jusqu'à ce que la partie supérieure de la solution devienne claire. Décanter la couche supérieure, qui est celle qui contient les esters méthyliques. La solution d'heptane est prête pour injection dans le chromatographe. Il est conseillé de maintenir la solution au réfrigérateur jusqu'au moment de l'analyse chromatographique. Il n'est pas recommandé de stocker la solution pendant plus de 12 heures.
PARTIE B
ANALYSE DES ESTERS MÉTHYLIQUES D'ACIDES GRAS PAR CHROMATOGRAPHIE EN PHASE GAZEUSE
1. CHAMP D'APPLICATION
La présente partie donne des directives générales pour la détermination par chromatographie en phase gazeuse par colonne capillaire de la composition qualitative et quantitative d'un mélange d'esters méthyliques d'acides gras obtenu selon la méthode spécifiée dans la partie A.
La partie ne s'applique pas aux acides gras polymérisés.
2. RÉACTIFS
2.1. Gaz vecteur
Gaz inerte (hélium ou hydrogène) soigneusement desséché et contenant moins de 10 mg/kg d'oxygène.
Note 1: l'hydrogène peut doubler la vitesse d'analyse mais présente des dangers. Il existe cependant des dispositifs de sécurité.
2.2. Gaz auxiliaires
|
2.2.1. |
Hydrogène (pureté ≥ 99,9 %), ne contenant pas d'impuretés organiques. |
|
2.2.2. |
Air ou oxygène ne contenant pas d'impuretés organiques. |
|
2.2.3. |
Azote (pureté > 99 %). |
2.3. Produits d'étalonnage
Mélange d'esters méthyliques d'acides gras purs, ou esters méthyliques d'un corps gras, de composition connue, si possible voisine de celle du corps gras à analyser. Les isomères cis et trans d'esters méthyliques octadécénoïques, octadécadiénoïques et octadécatriénoïques sont utiles pour l'identification des isomères trans des acides insaturés.
Prendre toutes précautions afin d'éviter l'oxydation des acides gras polyinsaturés.
3. APPAREILLAGE
Les prescriptions fournies concernent les appareils usuels de chromatographie en phase gazeuse utilisant des colonnes capillaires et un détecteur à ionisation de flamme.
3.1. Chromatographie en phase gazeuse
L'appareil de chromatographie en phase gazeuse comprend les éléments suivants.
3.1.1. Système d'injection
Utiliser un système d'injection avec des colonnes capillaires, auquel cas il importe que le dispositif soit spécialement conçu pour l'utilisation de telles colonnes. Il peut s'agir d'un système d'injection à division de flux ou d'un système d'injection sans division dans la colonne (injecteur on-column).
3.1.2. Four
Le four doit être en mesure de porter la colonne capillaire à une température d'au moins 260 °C et de maintenir la température choisie à 0,1 °C près. Cette dernière exigence est particulièrement importante lorsqu'un tube en silice fondue est utilisé.
L'utilisation d'un appareil équipé d'un programmateur de température est recommandée dans tous les cas et, en particulier, en présence d'acides gras à moins de 16 atomes de carbone.
3.1.3. Colonne capillaire
|
3.1.3.1. |
Tube en matériau inerte vis-à-vis des corps à analyser, généralement en verre ou en silice fondue. Le diamètre interne doit être compris entre 0,20 et 0,32 mm. Intérieurement, il devra subir des traitements appropriés (préparation de l'état de surface, inactivation) avant de recevoir le film de phase stationnaire. Une longueur de 60 m est suffisante pour les acides gras et les isomères cis et trans des acides gras. |
|
3.1.3.2. |
Phase stationnaire, polysiloxanes polaires (cyanopropylsilicone). Les colonnes greffées ou réticulées conviennent. Note 2: toutefois les polysiloxanes polaires peuvent compliquer l'identification et la séparation de l'acide linolénique et des acides en C20. Les épaisseurs de film doivent être faibles: 0,1 à 0,2 μm. |
|
3.1.3.3. |
Montage et conditionnement de la colonne Respecter les précautions habituelles de montage des colonnes capillaires, c'est-à-dire la disposition de la colonne dans le four (support), le choix et le montage des joints (étanchéité), le positionnement des extrémités de la colonne dans l'injecteur et le détecteur (réduction des volumes morts). Mettre la colonne sous gaz vecteur [par exemple, 0,3 bar (30 kPa) pour une colonne de 25 mètres de longueur et d'un diamètre intérieur de 0,3 millimètre]. Conditionner la colonne par programmation de la température du four à 3 °C par minute depuis la température ambiante jusqu'à une température inférieure de 10 °C à la limite de décomposition de la phase stationnaire. Maintenir à cette température 1 heure, jusqu'à stabilisation de la ligne de base. Revenir à 180 °C pour travailler dans des conditions isothermes. Note 3: des colonnes préconditionnées adéquates sont disponibles dans le commerce. |
3.1.4. Détecteur à ionisation de flamme et convertisseur-amplificateur
3.2. Seringue
La seringue doit avoir une capacité de 10 microlitres au maximum et être graduée en 0,1 μl.
3.3. Système d'acquisition des données
Système d'acquisition des données connecté en ligne avec les détecteurs et utilisé avec un logiciel adapté pour l'intégration et la normalisation des pics.
4. PROCÉDURE
Les détails opératoires présentés aux points 4.1 à 4.3 concernent l'utilisation d'un détecteur à ionisation de flamme.
4.1. Conditions d'essai
4.1.1. Choix des conditions opératoires optimales pour colonnes capillaires
Compte tenu des caractéristiques d'efficacité et de perméabilité des colonnes capillaires, la séparation entre les constituants et la durée de l'analyse dépendent en grande partie du débit du gaz vecteur dans la colonne. Il sera donc nécessaire d'optimiser les conditions opératoires en ajustant ce paramètre (ou plus simplement sur la perte de charge en tête de colonne) selon que l'on cherche à améliorer la séparation ou à accélérer l'analyse.
Les conditions suivantes se sont révélées utilisables pour la séparation des EMAG (C4 à C26). Des exemples de chromatogrammes figurent à l'appendice B:
|
Température de l'injecteur: |
250 °C |
|
Température du détecteur: |
250 °C |
|
Température du four: |
de 165 °C (8 min) à 210 °C à 2 °C/min |
|
Gaz vecteur hydrogène: |
pression de la tête de colonne: 179 kPa |
|
Débit total: |
154,0 ml/min |
|
Ratio de la division: |
1:100 |
|
Volume d'injection: |
1 μl |
4.1.2. Détermination de la résolution (voir appendice A)
Calculer la résolution R, de deux pics voisins I et II, au moyen de la formule suivante:
R = 2 × ((dr(II) – dr(I) )/(ω(I) + ω(II))) ou R = 2 × ((tr(II) – t r(I))/(ω(I) + ω(II))) (USP) (United States Pharmacopeia),
ou
R = 1,18 × ((tr(II) – tr(I))/(ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB), (JP (Japanese Pharmacopeia), EP (Pharmacopée européenne), BP (British Pharmacopeia))
où:
d r(I) est la distance de rétention du pic I;
d r(II) est la distance de rétention du pic II;
t r(I) est le temps de rétention du pic I;
t r(II) est le temps de rétention du pic II;
ω(I) est la largeur de la base du pic I;
ω(II) est la largeur de la base du pic II;
ω0,5 est la largeur du pic du composé donné, à mi-hauteur du pic;
Si ω(I) ≈ ω(II), calculer R en utilisant les formules suivantes:
R = (dr(II) – d r(I))/ω = (dr(II) – d r(I))/4σ
où:
σ est l'écart type (voir appendice A, figure 1).
Si la distance dr entre les deux pics d r(II) – d r(I) est égale à 4σ, le facteur de résolution R = 1.
Si deux pics ne sont pas complètement séparés, les tangentes aux points d'inflexion des deux pics se coupent au point C. Afin de séparer complètement les deux pics, la distance entre eux doit être égale à:
d r(II) – d r(I) = 6 σ d'où R = 1,5 (voir appendice A, figure 3).
5. EXPRESSION DES RÉSULTATS
5.1. Analyse qualitative
Identifier les pics d'ester méthylique de l'échantillon à partir du chromatogramme de l'appendice B, figure 1, si nécessaire par interpolation ou par comparaison avec ceux des mélanges d'esters méthyliques de référence (comme indiqué au point 2.3).
5.2. Évaluation quantitative
5.2.1. Détermination de la composition
Calculer la fraction massique wi des esters méthyliques d'acides gras, exprimée en pourcentage en masse des esters méthyliques, comme suit:
5.2.2. Méthode de calcul
5.2.2.1. Cas général
Calculer la teneur en un constituant donné i, exprimée en pourcentage en masse, des esters méthyliques, en déterminant le pourcentage représenté par le rapport de l'aire du pic correspondant à la somme des aires de la totalité des pics, à l'aide de la formule:
wi = (Ai/ΣA) × 100
où:
Ai est l'aire du pic de chaque ester méthylique d'acide gras i;
ΣA est la somme des aires de la totalité des pics de tous les esters méthyliques d'acides gras.
Les résultats sont exprimés avec deux décimales.
Note 4: pour les matières grasses et les huiles, la fraction massique des esters méthyliques d'acides gras est égale à la fraction massique des triglycérides en grammes par 100 g. Pour le cas où cette hypothèse n'est pas permise, voir le point 5.2.2.2.
5.2.2.2. Cas de l'emploi des facteurs de correction
Dans certains cas, par exemple en présence d'acides gras à moins de huit atomes de carbone ou d'acides avec des groupes secondaires, les aires doivent être corrigées à l'aide de facteurs de correction spécifiques (Fci). Ces facteurs sont déterminés pour chaque instrument. À cet effet, des matériaux de référence appropriés, avec une composition en acides gras certifiée dans la fourchette correspondante, doivent être utilisés.
Note 5: ces facteurs de correction ne sont pas identiques aux facteurs de correction FID théoriques, qui sont indiqués dans l'appendice A, étant donné qu'ils comprennent également les performances du système d'injection, etc. Toutefois, dans le cas de plus grandes différences, le système dans son ensemble devrait faire l'objet d'un contrôle de performance.
Pour ce mélange témoin, le pourcentage en masse de l'EMAG i est donné par la formule:
wi = (mi /Σm) × 100
où
mi est la masse de l'EMAG i dans le mélange témoin;
Σm est la somme des masses des divers constituants comme les EMAG du mélange témoin.
À partir du chromatogramme du mélange témoin, calculer le pourcentage par aire de l'EMAG i comme suit:
wi = (Ai/ΣA) × 100
où:
Ai est l'aire de l'EMAG i dans le mélange témoin;
ΣA est la somme de toutes les aires de tous les EMAG du mélange témoin.
Le facteur de correction Fc est alors
Fc = (mi × ΣA)/(Ai/Σm)
Pour l'échantillon, le pourcentage en masse de chaque EMAG i est:
wi = (Fi × Ai)/Σ (Fi × Ai)
Les résultats sont exprimés avec deux décimales.
Note 6: la valeur calculée correspond au pourcentage en masse de chaque acide gras calculé en triglycérides par 100 g de matières grasses.
5.2.2.3. Cas de l'emploi d'un étalon interne
Dans certaines analyses (par exemple, lorsque tous les acides gras ne sont pas quantifiés, et que des acides en C4 et en C6 sont présents à côté d'acides en C16 et en C18, ou bien lorsqu'il est nécessaire de déterminer la quantité absolue d'acides gras dans un échantillon), il est nécessaire d'utiliser un étalon interne. Des acides gras en C5, C15 ou C17 sont utilisés fréquemment. Le facteur de correction de l'étalon interne doit être déterminé (s'il y a lieu).
Le pourcentage en masse du composé i, exprimé en esters méthyliques, est par suite donné par la formule:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
où:
Ai est l'aire de l'EMAG i;
A IS est l'aire de l'étalon interne;
F i est le facteur de correction de l'acide gras i, exprimé en EMAG;
F IS est le facteur de correction de l'étalon interne;
m est la masse, en milligrammes, de la prise d'essai;
m IS est la masse, en milligrammes, de l'étalon interne;
Les résultats sont exprimés avec deux décimales.
6. RAPPORT D'ANALYSE
Le rapport d'essai doit indiquer les méthodes utilisées pour la préparation des esters méthyliques et pour l'analyse par chromatographie en phase gazeuse. Il doit, en outre, mentionner tous les détails opératoires non prévus dans la présente méthode standard, ou facultatifs, ainsi que les incidents éventuels susceptibles d'avoir influé sur les résultats.
Le rapport d'analyse doit comporter toutes les informations permettant une identification complète de l'échantillon.
7. PRÉCISION
7.1. Résultats de l'essai interlaboratoire
Les détails d'un essai interlaboratoire sur la précision de la méthode sont présentés à l'annexe C de la norme COI/T.20/Doc. no 33. Les valeurs dérivées de l'essai interlaboratoire peuvent ne pas être applicables aux gammes de concentration et aux matrices autres que celles données.
7.2. Répétabilité
La différence absolue entre les résultats de deux tests distincts, obtenus selon la même méthode, sur du matériel d'essai identique, dans le même laboratoire, par un seul opérateur utilisant le même équipement et effectués dans un bref intervalle de temps sera supérieure, dans 5 % des cas au maximum, à la valeur R présentée dans les tableaux 1 à 14 de l'annexe C de la norme COI/T.20/Doc. no 33.
7.3. Reproductibilité
La différence absolue entre les résultats de deux tests individuels, obtenus selon la même méthode, sur du matériel d'essai identique, dans des laboratoires différents, par des opérateurs différents utilisant un équipement différent sera supérieure, dans 5 % des cas au maximum, à la valeur R présentée dans les tableaux 1 à 14 de l'annexe C de la norme COI/T.20/Doc. no 33.
Appendice A
Figure 1
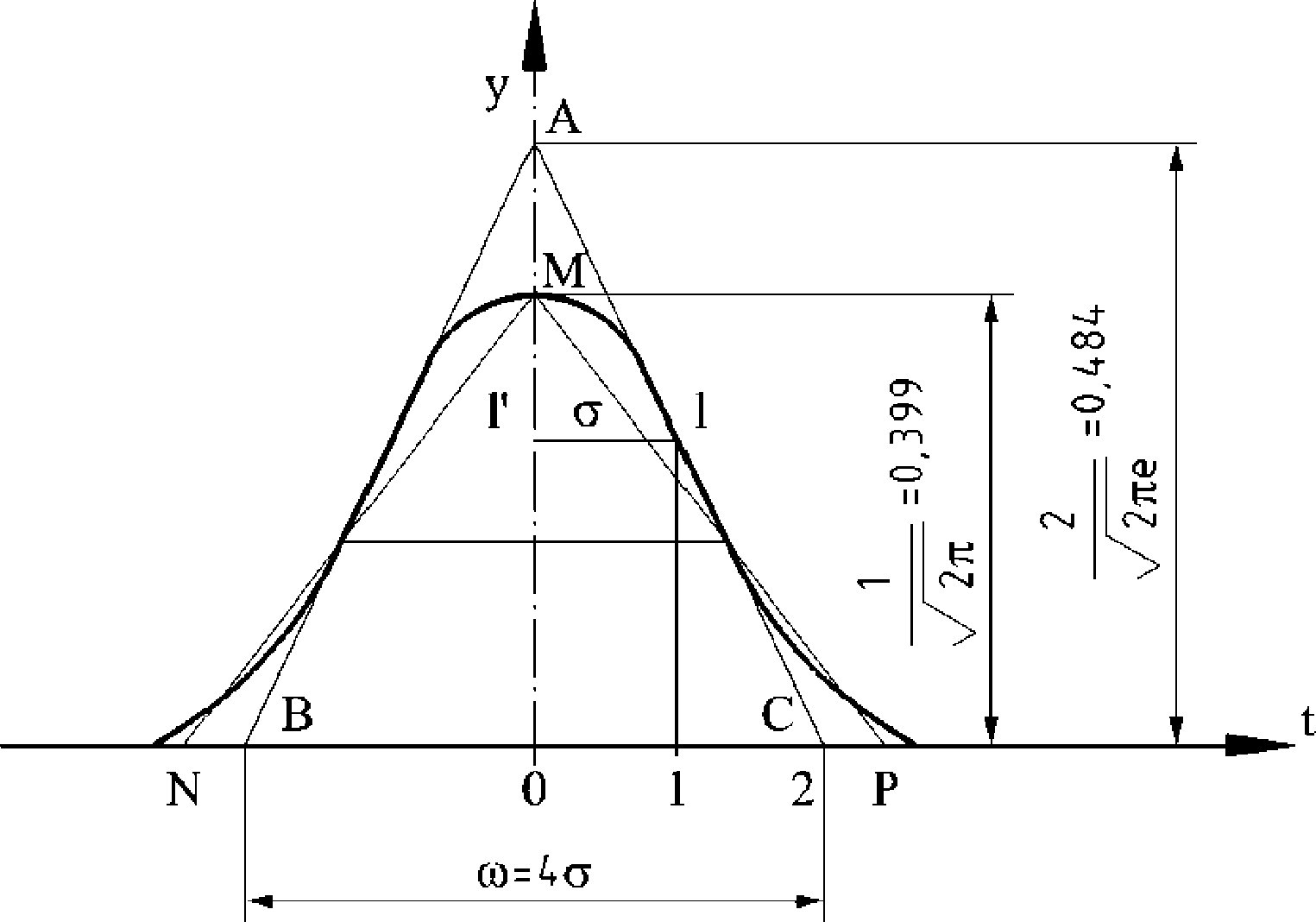
Avec ω0,5 pour la largeur à mi-hauteur du triangle (ABC) et b pour la largeur à mi-hauteur du triangle (NPM).
|
Figure 2 |
Figure 3 |
|
|
|
Appendice B
Figure 1
Profil chromatographique en phase gazeuse d'une huile de grignons d'olive, obtenu par la méthode de méthylation à froid.
Les pics chromatographiques correspondent aux esters méthyliques et éthyliques, sauf indication contraire.
ANNEXE XI
DÉTERMINATION DE LA TENEUR EN SOLVANTS HALOGÉNÉS VOLATILS DANS L'HUILE D'OLIVE
1. PRINCIPE
Analyse par chromatographie en phase gazeuse selon la technique de l'espace de tête (head space).
2. APPAREILLAGE
|
2.1. |
Appareil de chromatographie en phase gazeuse, équipé d'un détecteur à capture d'électrons (ECD). |
|
2.2. |
Appareillage pour espace de tête (head space). |
|
2.3. |
Colonne de chromatographie en phase gazeuse en verre de 2 mètres de long et de 2 millimètres de diamètre, phase stationnaire. OV101 à 10 % ou équivalent, imprégnant une terre de diatomée calcinée, lavée aux acides et silanisée, de granulométrie de 80 à 100 Mesh. |
|
2.4. |
Gaz vecteur et gaz auxiliaire: azote pour chromatographie en phase gazeuse, adaptée à la détection par capture d'électrons. |
|
2.5. |
Flacons en verre de 10 à 15 millimètres munis d'une garniture en téflon et d'un bouchon en aluminium muni d'un office pour prélèvement par seringue. |
|
2.6. |
Pinces à fermeture hermétique. |
|
2.7. |
Seringue pour gaz de 0,5 à 2 millilitres. |
3. RÉACTIFS
Standard: solvants halogénés volatils à un degré de pureté approprié à un usage de chromatographie en phase gazeuse.
4. PROCÉDURE D'ANALYSE
|
4.1. |
Peser exactement environ 3 grammes d'huile dans un flacon en verre (à ne pas réutiliser), boucher le flacon jusqu'à fermeture hermétique. Introduire le flacon dans un thermostat à 70 °C pendant 1 heure. Prélever avec précision au moyen de la seringue un volume de 0,2 à 0,5 millilitre de l'espace de tête. L'injecter dans la colonne de l'appareil de chromatographie en phase gazeuse réglé comme suit: — température injecteur: 150 °C, — température colonne: 70 à 80 °C, — température détecteur: 200 à 250 °C. |
|
4.2. |
Solutions de référence. Préparer des solutions standard, en utilisant de l'huile d'olive raffinée sans trace de solvants, à des concentrations variables entre 0,05 et 1 milligramme par kilogramme et en rapport à la teneur présumée de l'échantillon. La dilution éventuelle doit être effectuée avec du pentane. |
|
4.3. |
Évaluation quantitative. Faire le rapport entre les surfaces ou les hauteurs des pics de l'échantillon et de la solution standard ayant la concentration présumée le plus proche. Si l'écart relatif est supérieur à 10 %, il est nécessaire de refaire l'analyse par comparaison avec une nouvelle solution standard jusqu'à ce que sa concentration respecte l'écart relatif susmentionné. La teneur est établie sur la base d'une moyenne d'injections élémentaires. |
|
4.4. |
Expression des résultats. Les résultats sont exprimés en milligrammes par kilogramme (ppm). La limite de détection de la méthode est de 0,01 milligramme par kilogramme. |
ANNEXE XII
MÉTHODE DU CONSEIL OLÉICOLE INTERNATIONAL POUR L'ÉVALUATION ORGANOLEPTIQUE DES HUILES D'OLIVE VIERGES
1. OBJET ET CHAMP D'APPLICATION
L'objet de la méthode internationale décrite dans la présente annexe est de déterminer la procédure d'évaluation des caractéristiques organoleptiques des huiles d'olive vierges au sens de l'annexe VII, partie VIII, point 1, du règlement (UE) no 1308/2013 du Parlement européen et du Conseil ( 10 ) et d'établir la méthode à utiliser pour le classement de ces huiles sur la base de ces caractéristiques. Elle fournit également des indications concernant l'étiquetage facultatif.
La méthode décrite ne s'applique qu'aux huiles d'olive vierges et à leur classement ou à leur étiquetage en fonction de l'intensité des défauts perçus et du fruité, déterminés par un groupe de dégustateurs sélectionnés, entraînés et testés, constitués en jury.
Les normes COI mentionnées dans la présente annexe sont utilisées dans leur dernière version disponible.
2. VOCABULAIRE GÉNÉRAL DE BASE POUR L’ANALYSE SENSORIELLE
Voir la norme COI/T.20/Doc. no 4 «Analyse sensorielle: vocabulaire général de base»
3. VOCABULAIRE SPÉCIFIQUE
3.1. Attributs négatifs
Chômé/lies Flaveur caractéristique de l’huile tirée d’olives entassées ou stockées dans des conditions telles qu'elles se trouvent dans un état avancé de fermentation anaérobie, ou de l'huile restée en contact avec les boues de décantation, ayant elles aussi subi un processus de fermentation anaérobie, dans les piles et les cuves.
Moisi – humidité-terre Flaveur caractéristique des huiles obtenues à partir de fruits attaquées par des moisissures et des levures par suite d'un stockage des fruits pendant plusieurs jours dans l’humidité, ou des huiles obtenues à partir d’olives ayant été ramassées avec de la terre, ou boueuses et non lavées.
Vineux-vinaigré-acide-aigre Flaveur caractéristique de certaines huiles, rappelant le vin ou le vinaigre. Cette flaveur est due fondamentalement à un processus de fermentation aérobie des olives ou des restes de pâte d'olive dans des scourtins qui n'auraient pas été correctement lavés, qui donne lieu à la formation d'acide acétique, d'acétate d'éthyle et d'éthanol.
Rance Flaveur des huiles ayant subi un processus d'oxydation intense.
Olives gelées (bois humide) Flaveur caractéristique des huiles extraites d’olives endommagées par le gel, sur l’arbre.
3.1.1 Autres attributs négatifs
|
Cuit ou brûlé |
Flaveur caractéristique des huiles qui tire son origine d'un réchauffement excessif et/ou prolongé au cours de la transformation et tout particulièrement pendant le thermo-malaxage de la pâte, si celui-ci est réalisé dans des conditions thermiques inappropriées. |
|
Foin-bois |
Flaveur caractéristique de certaines huiles provenant d'olives sèches. |
|
Grossier |
Sensation bucco-tactile dense et pâteuse produite par certaines huiles vieilles. |
|
Lubrifiants |
Flaveur de l'huile qui rappelle celle du gazole, de la graisse ou de l'huile minérale. |
|
Margines |
Flaveur acquise par l'huile à la suite d'un contact prolongé avec les eaux de végétation qui ont subi des processus de fermentation. |
|
Saumure |
Flaveur de l'huile obtenue à partir d'olives conservées en saumure. |
|
Métallique |
Flaveur qui rappelle les métaux. Elle est caractéristique de l'huile qui est demeurée longtemps en contact avec des surfaces métalliques, au cours des processus de broyage, de malaxage, de pression ou de stockage. |
|
Sparte |
Flaveur caractéristique de l'huile obtenue à partir d'olives pressées dans des scourtins en sparte neufs. La flaveur peut être différente selon qu'il s'agit de scourtins fabriqués à partir de sparte vert ou de sparte sec. |
|
Ver |
Flaveur de l'huile issue d'olives ayant subi une forte attaque de larves de la mouche de l'olive (Bactrocera Oleae). |
|
Concombre |
Flaveur acquise par l'huile à la suite d'un conditionnement hermétique excessivement prolongé, notamment dans des récipients en fer-blanc, et qui est attribuée à la formation de 2-6 nonadiénal. |
3.2. Attributs positifs
|
Fruité |
Ensemble des sensations olfactives caractéristiques de l'huile, dépendant de la variété des olives et provenant de fruits sains et frais, verts ou mûrs, perçues par voie directe et/ou rétronasale. |
|
Amer |
Goût élémentaire caractéristique de l'huile obtenue à partir d'olives vertes ou au stade de la véraison. Il est perçu par les papilles caliciformes formant le V lingual. |
|
Piquant |
Sensation tactile de picotement, caractéristique des huiles produites au début de la campagne, principalement à partir d'olives encore vertes. Il peut être perçu dans toute la cavité buccale, en particulier dans la gorge. |
3.3. Terminologie facultative aux fins de l'étiquetage
Sur demande, le chef de jury peut certifier que les huiles qui ont été évaluées répondent aux définitions et aux palettes de sensation correspondant uniquement aux adjectifs ci-après, en fonction de l'intensité et de la perception des attributs:
Attributs positifs (fruité, amer et piquant): en fonction de l'intensité de la perception:
— intense, lorsque la médiane de l'attribut est supérieure à 6,
— moyen, lorsque la médiane de l'attribut est comprise entre 3 et 6,
— léger, lorsque la médiane de l'attribut est inférieure à 3.
|
Fruité |
Ensemble des sensations olfactives caractéristiques de l'huile, dépendant de la variété des olives, provenant de fruits sains et frais, au sein desquelles ne prédominent ni le fruité vert ni le fruité mûr, et perçues par voie directe et/ou rétronasale. |
|
Fruité vert |
Ensemble des sensations olfactives caractéristiques de l'huile rappelant les fruits verts, dépendant de la variété des olives, provenant de fruits verts, sains et frais, et perçues par voie directe et/ou rétronasale. |
|
Fruité mûr |
Ensemble des sensations olfactives caractéristiques de l'huile rappelant les fruits mûrs, dépendant de la variété des olives, provenant de fruits sains et frais, et perçues par voie directe et/ou rétronasale. |
|
Équilibré |
Huile qui n'est pas déséquilibrée. On entend par «déséquilibre» la sensation olfactogustative et tactile de l'huile dans laquelle la médiane de l'attribut amer ou la médiane de l'attribut piquant est supérieure de deux points à la médiane du fruité. |
|
Huile douce |
Huile dans laquelle la médiane des attributs amer et piquant est inférieure ou égale à 2. |
Liste de termes en fonction de l'intensité de la perception:
|
Termes subordonnés à la présentation d'un certificat d'essai organoleptique |
Médiane de l'attribut |
|
Fruité |
— |
|
Fruité mûr |
— |
|
Fruité vert |
— |
|
Fruité léger |
Inférieure à 3 |
|
Fruité moyen |
Entre 3 et 6 |
|
Fruité intense |
Supérieure à 6 |
|
Fruité mûr léger |
Inférieure à 3 |
|
Fruité mûr moyen |
Entre 3 et 6 |
|
Fruité mûr intense |
Supérieure à 6 |
|
Fruité vert léger |
Inférieure à 3 |
|
Fruité vert moyen |
Entre 3 et 6 |
|
Fruité vert intense |
Supérieure à 6 |
|
Amer léger |
Inférieure à 3 |
|
Amer moyen |
Entre 3 et 6 |
|
Amer intense |
Supérieure à 6 |
|
Piquant léger |
Inférieure à 3 |
|
Piquant moyen |
Entre 3 et 6 |
|
Piquant intense |
Supérieure à 6 |
|
Huile équilibrée |
La médiane de l'attribut amer et la médiane de l'attribut piquant ne dépassent pas de plus de 2 points la médiane du fruité |
|
Huile douce |
La médiane de l'attribut amer et la médiane de l'attribut piquant n'excèdent pas 2 |
4. VERRE DE DÉGUSTATION DE L'HUILE
Voir la norme COI/T.20/Doc. No5, «Verre pour la dégustation des huiles»
5. SALLE DE DÉGUSTATION
Voir la norme COI/T.20/Doc. No6 «Guide pour l'installation d'une salle de dégustation»
6. ACCESSOIRES
Les accessoires suivants sont mis à la disposition des dégustateurs, dans chaque cabine, afin de leur permettre de remplir convenablement leur tâche. Ces accessoires sont à portée de main des dégustateurs:
— verres (normalisés) contenant les échantillons, identifiés par un numéro de code, recouverts d’un verre de montre et maintenus à 28 °C ± 2 °C;
— feuille de profil (voir figure 1) sur support papier ou informatique, pour autant que les conditions soient respectées, assortie d'instructions d'utilisation, si nécessaire;
— stylo ou encre indélébile;
— plateaux avec tranches de pomme et/ou eau, eau gazeuse et/ou biscottes;
— verre d’eau à la température ambiante;
— document rappelant les règles générales énoncées aux points 8.4 et 9.1.1;
— crachoirs.
7. CHEF DE JURY ET DÉGUSTATEURS
7.1. Chef de jury
Le chef de jury doit jouir d’une formation solide, tout en étant un connaisseur et un expert averti de tous les types d’huile auxquels il aura affaire au cours de son travail. Il est le personnage clé du jury et est responsable de l’organisation et de la gestion de celui-ci.
Le travail du chef de jury requiert une formation de base aux outils de l'analyse sensorielle, des compétences dans le domaine de l'analyse sensorielle, de la méticulosité dans la préparation, l’organisation et la réalisation des essais, ainsi que de l’habileté et de la patience pour planifier et exécuter les essais de manière scientifique.
Il est également du ressort exclusif du chef de jury de veiller à la sélection, à la formation et à la supervision des dégustateurs, afin de s’assurer de leur niveau d’aptitude. Le chef de jury est donc responsable de l'évaluation des dégustateurs, qui doit toujours être objective. À cet effet, il doit élaborer des procédures spécifiques basées sur des essais et des critères dûment fondés d'acceptation et de rejet. Voir la norme COI/T.20/Doc. No 14 «Guide pour la sélection, l'entraînement et le contrôle des dégustateurs qualifiés d'huile d'olive vierge».
Le chef de jury est responsable de la performance du jury et partant, des évaluations qu'il produit, et il doit en attester de façon fiable et objective. En tout état de cause, il doit pouvoir démontrer à tout moment que la méthode et les dégustateurs sont placés sous son contrôle. Un étalonnage périodique du jury est recommandé (COI/T.20/Doc. No 14, point 5).
Le chef de jury est responsable, en dernier ressort, de la tenue des registres du jury. Ces registres doivent toujours être traçables. Ils doivent être conformes aux exigences d'assurance de qualité énoncées dans les normes internationales d'analyse sensorielle et garantir l’anonymat des échantillons à tout moment.
Le chef de jury est responsable de l'inventaire des ustensiles et du matériel nécessaires au respect des spécifications de la présente méthode et doit veiller à ce qu'ils soient correctement nettoyés et entretenus; il fera un compte-rendu écrit de tout ce qui précède, spécifiant que les conditions de l’essai ont été respectées.
Le chef de jury est responsable de la réception et du stockage des échantillons à leur arrivée au laboratoire ainsi que de leur conservation après analyse. Dans ce contexte, il veille à garantir à tout moment l'anonymat des échantillons et leur conservation adéquate. À cet effet, il doit élaborer des procédures écrites en vue de garantir la traçabilité de l'ensemble du processus.
Le chef de jury est en outre responsable de la préparation, de la codification et la présentation des échantillons aux dégustateurs selon le schéma expérimental adéquat conformément au protocole préalablement établi, ainsi que de la collecte des résultats auprès des dégustateurs et de leur traitement statistique.
Il est responsable de la mise au point et de la rédaction de toutes les autres procédures qui pourraient être nécessaires pour compléter cette norme et qui seraient nécessaires pour le fonctionnement adéquat du jury.
Il doit trouver des moyens pour comparer les résultats du jury avec ceux d’autres jurys d’huile d’olive vierge afin de s’assurer que le fonctionnement de son jury est adéquat.
Le chef de jury a en outre pour mission de motiver les membres du groupe en stimulant leur intérêt, leur curiosité et leur esprit de compétition. C’est la raison pour laquelle il est fortement recommandé d’assurer un échange fluide d’informations avec les membres du groupe en les impliquant dans tout le travail qu’ils réalisent ainsi que dans les résultats obtenus. Le chef de jury doit éviter d'exprimer son point de vue et doit veiller à ce que certains membres du jury n'imposent pas leurs critères aux autres dégustateurs.
Il doit convoquer les dégustateurs suffisamment à l’avance et répondre à toutes les questions concernant la réalisation des essais, tout en s’abstenant d'émettre un avis, quel qu’il soit, sur l’échantillon.
7.1.1. Chef de jury adjoint
Pour des raisons dûment justifiées, le chef de jury peut être remplacé dans ses fonctions relatives à la réalisation des essais par un chef de jury adjoint. Le substitut doit posséder l'expérience requise pour la figure du chef de jury.
7.2. Dégustateurs
Les personnes intervenant en qualité de dégustateurs dans les essais organoleptiques d'huiles d'olive doivent le faire de manière volontaire. Il est donc recommandé d'exiger une demande écrite des candidats. Ceux-ci devront être sélectionnés, entraînés et testés par le chef de jury en en relation avec leur habileté à faire la distinction entre échantillons proches. Il y a lieu de ne pas perdre de vue le fait que la précision du dégustateur s'améliore avec l'entraînement.
Le dégustateur doit se comporter comme un véritable observateur sensoriel, en laissant de côté ses goûts personnels et en ne rendant compte que des sensations qu'il perçoit. À cet effet, il doit toujours réaliser son travail en silence, être détendu et ne pas être pressé. Il doit prêter toute l'attention sensorielle possible à l'échantillon qu'il déguste.
Pour chaque essai, il faut disposer de 8 à 12 dégustateurs. Toutefois, il convient de prévoir quelques dégustateurs supplémentaires auxquels on peut faire appel en cas d'absences éventuelles.
8. CONDITIONS DE L’ESSAI
8.1. Présentation de l'échantillon
L’échantillon d’huile à analyser sera présenté dans les verres de dégustation normalisés conformément à la Norme COI/T.20/Doc. No 5 «Verre pour la dégustation des huiles».
Le verre doit contenir entre 14 et 16 ml d’huile, ou entre 12,8 et 14,6 g si les échantillons sont pesés, et être recouvert d’un verre de montre.
Chaque verre doit être marqué, au moyen d’un système inodore, d’un code composé de chiffres ou de chiffres et de lettres pris au hasard.
8.2. Température de l'essai et de l’échantillon
Les échantillons d’huile à déguster doivent être maintenus dans les verres à une température de 28 °C ± 2 °C durant tout l’essai. Cette température a été retenue car elle permet de relever des différences organoleptiques plus aisément qu’à température ambiante, et parce que des températures plus basses produisent une faible volatilisation des composés aromatiques propres à ces huiles, tandis que des températures plus élevées amènent la formation de composés volatils propres aux huiles chauffées. Voir la norme COI/T.20/doc. No 5 «Verre pour la dégustation des huiles» en ce qui concerne le système de réchauffement des échantillons qui doit être utilisé une fois l’échantillon introduit dans le verre.
La température de la salle de dégustation doit être comprise entre 20 et 25 °C (voir COI/T. 20/Doc. no6).
8.3. Horaire des essais
La matinée est le moment le plus opportun pour la dégustation des huiles. Il est prouvé qu’il existe, pendant la journée, des périodes de perception optimale pour ce qui est du goût et de l'odeur. Une période d’acuité olfactogustative accrue précède les repas, qui sont suivis par une diminution de cette acuité.
Toutefois, ce critère ne doit pas être poussé à l’extrême, au point que la faim puisse constituer un facteur de distraction chez les dégustateurs et réduire leur capacité de discrimination. Par conséquent, il est recommandé d’organiser les séances de dégustations entre dix heures du matin et midi.
8.4. Dégustateurs: règles générales de conduite
Les recommandations suivantes visent le comportement devant être observé par les dégustateurs au cours de leur travail.
Dès réception de la convocation du responsable du jury l’invitant à participer à un essai organoleptique, le dégustateur doit être en mesure d'intervenir à l'heure préalablement fixée et est tenu au respect des règles ci-après:
— s’abstenir de fumer et de boire du café pendant au moins 30 minutes avant l’heure fixée pour l’essai;
— ne pas avoir utilisé un parfum, un cosmétique ou un savon dont l’odeur pourrait persister au moment de l’essai. Les mains doivent être lavées avec un savon non parfumé, puis rincées et séchées autant de fois que nécessaire pour éliminer toute trace d’odeur;
— ne rien manger pendant au moins une heure avant la dégustation;
— dans l’hypothèse où ses fonctions physiologiques seraient affectées, notamment l’odorat ou le goût, ou s’il était confronté à un problème psychologique quelconque qui l’empêcherait de se concentrer, le dégustateur devrait s’abstenir de déguster et prévenir le chef de jury;
— après s'être conformé aux règles précitées, le dégustateur doit s’installer dans la cabine qui lui a été assignée, de façon disciplinée et silencieuse;
— il doit lire attentivement les instructions figurant sur la feuille de profil et ne commencer l’examen de l’échantillon que lorsqu’il est tout à fait prêt pour la tâche dont il doit s’acquitter (calme et détendu). En cas de doute, il doit s’adresser au responsable du jury pour discuter en privé avec lui des difficultés rencontrées;
— il doit rester silencieux pendant qu'il accomplit sa tâche;
— le cas échéant, son téléphone portable devra toujours être coupé afin de ne pas gêner la concentration et le travail de ses collègues.
9. PROCÉDURE D'ÉVALUATION ORGANOLEPTIQUE ET DE CLASSEMENT DE L'HUILE D’OLIVE VIERGE
9.1. Dégustation technique
9.1.1. Le dégustateur doit prendre le verre, en le maintenant couvert avec le verre de montre, puis l'incliner légèrement et, dans cette position, le faire tourner sur lui-même afin d'en mouiller le plus possible la surface intérieure. Après cette opération, le dégustateur doit retirer le verre de montre et flairer l'échantillon par des inspirations lentes et profondes, pour évaluer l'huile. La durée du test olfactif ne devrait pas dépasser 30 secondes. Si le dégustateur n'est parvenu à aucune conclusion au terme de ce délai, il doit faire une pause avant de procéder à une nouvelle tentative.
Une fois l'essai olfactif terminé, le dégustateur doit procéder à l'évaluation des sensations buccales (ensemble des sensations olfactogustatives par voie rétronasale et tactile). Pour ce faire, prendre une petite gorgée d'huile, de 3 ml environ. Il est très important de distribuer l'huile sur toute la cavité buccale, depuis la partie antérieure de la bouche et la langue, en passant par les parties latérales et la partie postérieure jusqu'au voile du palais et la gorge; comme chacun sait, les saveurs et les sensations tactiles sont en effet perçues avec une intensité variable selon les différentes zones de la langue, du palais et de la gorge.
Il y a lieu d'insister sur la nécessité de répandre l'huile en quantité suffisante et très lentement sur la partie postérieure de la langue jusqu'au voile du palais et la gorge, en concentrant l'attention sur l'ordre d'apparition des stimuli amer et piquant; si on ne procède pas de cette façon, pour certaines huiles, ces deux stimuli peuvent passer inaperçus, ou encore le stimulus amer peut être masqué par le stimulus piquant.
Des inspirations brèves et successives, en faisant pénétrer de l'air par la bouche, permettent non seulement de répandre l'échantillon sur toute la cavité buccale, mais également de percevoir les composés aromatiques volatils par voie rétronasale puisque l'usage de cette voie est forcé.
NB: Lorsque le dégustateur ne perçoit pas le fruité dans un échantillon et que l'intensité de l'attribut négatif de classification est égale ou inférieure à 3,5, le chef du jury peut décider de faire procéder à l'analyse des échantillons par les dégustateurs à température ambiante (COI/T.20/Doc. No 6/Rév. 1er septembre 2007, section 3 — Guide pour l'installation d'une salle de dégustation) en précisant le contexte et le concept de la température ambiante. Lorsque l'échantillon atteint la température ambiante, le dégustateur doit réévaluer celui-ci afin de vérifier uniquement si le fruité est perçu. Si c'est le cas, il doit noter l'intensité sur l'échelle.
La sensation tactile de piquant doit être prise en considération. À cette fin, il convient d'avaler l'huile.
9.1.2. Il est recommandé que l’évaluation organoleptique d’une huile d’olive vierge porte au maximum sur QUATRE ÉCHANTILLONS par séance avec un maximum de 3 séances par jour, dans le souci d’éviter l’effet de contraste que pourrait provoquer la dégustation immédiate d’autres échantillons.
Étant donné que des dégustations successives entraînent de la fatigue ou une perte d'acuité due aux échantillons précédents, il est nécessaire d’utiliser un produit capable d'éliminer de la bouche les restes d'huile de la précédente dégustation.
Il est recommandé d'utiliser une petite tranche de pomme, qui peut être rejetée dans le crachoir après mastication. Il convient ensuite de se rincer la bouche avec un peu d’eau à température ambiante. Au minimum 15 minutes doivent s'être écoulées entre la fin d’une séance et le début de la suivante.
9.2. Utilisation de la feuille de profil par le dégustateur
La feuille de profil destinée aux dégustateurs est reproduite sur la figure 1 de la présente annexe.
Chaque dégustateur faisant partie du jury doit flairer, puis déguster ( 11 ) l'huile soumise à examen. Il doit ensuite indiquer, sur les échelles de 10 cm figurant sur la feuille de profil à sa disposition, l’intensité avec laquelle il perçoit chacun des attributs négatifs et positifs.
Si le dégustateur perçoit des attributs négatifs non énumérés au point 4, il doit le consigner dans la rubrique «autres», en employant le ou les termes qui décrivent avec le plus de précision ces attributs.
9.3. Utilisation des données par le chef de jury
Le chef du jury doit recueillir les feuilles de profil remplies par chacun des dégustateurs; il doit contrôler les intensités assignées aux différents attributs; s'il constate une anomalie, il doit inviter le dégustateur à réviser sa feuille de profil et, si nécessaire, à répéter l'essai.
Le chef de jury entre les données d'évaluation de chaque membre du jury dans un programme informatique tel que celui préconisé par la norme COI/T.20/Doc. no 15, en vue du calcul statistique des résultats de l'analyse, sur la base du calcul de la médiane. Voir point 9.4 et l'appendice de la présente annexe. Les données correspondant à un échantillon donné sont saisies à l'aide d'une matrice à 9 colonnes, représentant les 9 attributs sensoriels, et à n lignes, représentant les n membres du jury concerné.
Lorsqu'un défaut est perçu et consigné dans la rubrique «Autres» par au moins 50 % des membres du jury, le chef de jury calcule la médiane de ce défaut et en déduit le classement correspondant.
La valeur du coefficient de variation robuste qui définit le classement (défaut perçu avec la plus forte intensité et attribut fruité) ne doit pas dépasser 20 %.
Dans le cas contraire, le chef de jury devra répéter l'évaluation de l'échantillon en question lors d'une autre séance de dégustation.
Si cette situation se reproduit fréquemment, il est recommandé au chef de jury de prévoir une formation additionnelle spécifique des dégustateurs du jury (COI/T.20/Doc. no 14, point 5) et d'utiliser l'indice de répétabilité et l'indice d'écart pour vérifier la performance du dégustateur (COI/T.20/Doc. no14, point 6).
9.4. Classement de l'huile
L'huile est classée dans les catégories ci-après, en fonction de la médiane des défauts et de la médiane de l'attribut fruité. La médiane des défauts est définie comme la médiane du défaut perçu avec la plus grande intensité. La médiane des défauts et la médiane de l'attribut fruité sont exprimées avec une seule décimale.
Le classement de l'huile est effectué par comparaison de la valeur de la médiane des défauts et de la médiane du fruité avec les plages de référence indiquées ci-après. Les limites de ces plages ayant été établies en tenant compte de l'erreur de la méthode, elles sont considérées comme absolues. Les logiciels informatiques permettent de visualiser le classement sous la forme d'un tableau de données statistiques ou d'un graphique.
a) Huile d'olive vierge extra: la médiane des défauts est égale à 0 et la médiane du fruité est supérieure à 0;
b) Huile d'olive vierge: la médiane des défauts est supérieure à 0 mais ne dépasse pas 3,5, et la médiane du fruité est supérieure à 0;
c) Huile d'olive vierge lampante: la médiane des défauts est supérieure à 3,5, ou la médiane des défauts est inférieure ou égale à 3,5 et la médiane du fruité est égale à 0.
Note 1: lorsque la médiane de l'attribut amer et/ou piquant est supérieure à 5,0, le chef de jury le précise sur le certificat d'analyse.
Dans le cas des analyses effectuées dans le cadre de contrôles de conformité, un essai est réalisé. Dans le cas d'analyses contradictoires, l'analyse doit être effectuée en double au cours de séances distinctes. Les résultats de la contre-analyse doivent être statistiquement homogènes. (Voir point 9.5.) Si tel n'est pas le cas, l'échantillon doit à nouveau être analysé deux fois. La valeur finale de la médiane des attributs de classement sera calculée à l'aide de la moyenne des deux médianes.
9.5. Critères d'acceptation et de rejet des doublons
L'erreur standard, définie ci-dessous, doit être utilisée pour déterminer si les deux résultats d'une double analyse sont homogènes ou statistiquement acceptables:

Lorsque Me1 et Me2 sont les médianes des deux doubles (respectivement à la première et à la deuxième analyse) et que U1 et U2 sont les incertitudes élargies obtenues pour les deux valeurs, calculées comme suit et spécifié à l'appendice:
U
1 = c × s* and

Pour l'incertitude élargie, c = 1,96; soit
U1 = 0,0196 × CVr × Me1
si CVr est le coefficient de variation robuste.
Qu'il soit précisé que les deux valeurs obtenues ne sont pas statistiquement différentes, En doit être inférieure ou égale à 1,0.
Appendice
Méthode de calcul de la médiane et des intervalles de confiance
Médiane
![]()
La médiane est définie comme le nombre réel Xm caractérisé par le fait que la probabilité (p) que les valeurs de la distribution (X) soient inférieures à ce nombre (Xm) est inférieure ou égale à 0,5 et que, simultanément, la probabilité (p) que les valeurs de la distribution (X) soient inférieures ou égales à Xm est supérieure ou égale à 0,5. Il est plus pratique de définir la médiane comme le 50e percentile d’une distribution de nombres classés par ordre croissant. En termes plus simples, il s'agit de la valeur centrale d’un ensemble ordonné de nombres impairs, ou bien de la moyenne de deux valeurs centrales d’un ensemble ordonné de nombres pairs.
Écart type robuste
Pour obtenir une estimation fiable de la variabilité autour de la moyenne, il convient de se référer à l’estimation de l’écart type robuste selon Stuart et Kendall (4). La formule donne l'écart-type asymptotique, c'est-à-dire l'estimation robuste de la variabilité des données considérées, où N est le nombre d'observations et IQR l'intervalle interquartile, qui couvre exactement 50% des cas d'une distribution de probabilité quelconque.
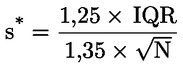
Le calcul de l'intervalle interquartile s'effectue en calculant l'amplitude de l'écart entre le soixante-quinzième et le vingt-cinquième percentiles.
![]()
où le percentile est la valeur Xpc caractérisée par le fait que la probabilité (p) que les valeurs de la distribution soient inférieures à Xpc est inférieure ou égale à un centième déterminé et que, simultanément, la probabilité (p) que les valeurs de la distribution soient inférieures ou égales à Xpc est supérieure ou égale audit centième. Le centième indique la fraction de distribution retenue. Dans le cas de la médiane, celle-ci est égale à 50/100.

Dans la pratique, le percentile est la valeur de distribution correspondant à une aire déterminée tracée à partir de la courbe de distribution ou de densité. A titre d'exemple, le 25ème percentile représente la valeur de distribution correspondant à une aire égale à 0,25 ou 25/100.
Dans cette méthode, les percentiles sont calculés à partir des valeurs réelles figurant dans la matrice des données (procédure de calcul des percentiles).
Coefficient de variation robuste (%)
Le CVr% représente un nombre adimensionnel qui indique le pourcentage de variabilité de la série de nombres analysée. Ce coefficient est donc très utile pour vérifier la fiabilité des membres du jury.

Intervalles de confiance à 95 % sur la médiane
Les intervalles de confiance à 95 % (valeur de l'erreur de première espèce égale à 0,05 ou à 5 %) représentent l'intervalle au sein duquel la valeur de la médiane pourrait varier dans l'hypothèse où il serait possible de répéter une expérience un nombre infini de fois. Dans la pratique, il s'agit de l'intervalle de variabilité de l'essai dans les conditions opératoires retenues, si l'on part de l'hypothèse que l'essai pourrait être répété plusieurs fois. À l'instar du CVr%, cet intervalle aide à évaluer la fiabilité de l'essai.


Où C = 1,96 pour l’intervalle de confiance à 95 %.
Un exemple de la feuille de calcul est présenté à l’annexe I de la norme COI/T 20/Doc. No 15.
Bibliographie
(1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.
(2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
(3) Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.
(4) Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
(5) McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
(6) COI/T.28/Doc. No 1, septembre 2007, Lignes directrices pour l’accréditation des laboratoires d’analyse sensorielle de l’huile d’olive vierge en particulier, selon la norme ISO/IEC 17025:2005.
(7) COI/T.20/Doc. No 14
(8) COI/T.20/Doc. No 15
(9) ISO/IEC 17025: 05.
▼M20 —————
▼M19 —————
ANNEXE XV
1. TENEUR EN HUILE DES GRIGNONS D'OLIVE
1.1. Matériel
— appareil d'extraction approprié muni d'un ballon de 200 à 250 millilitres,
— bain à chauffage électrique (bain de sable, bain d'eau, etc.) ou plaque chauffante,
— balance analytique,
— étuve réglée à 80 °C au maximum,
— étuve à chauffage électrique muni d'un dispositif de thermorégulation réglé à 103 °C ± 2 °C et permettant de réaliser une insufflation d'air ou une pression réduite,
— broyeur mécanique facile à nettoyer et permettant le broyage sans échauffement et sans diminution sensible de leur teneur en eau et en huile,
— cartouche d'extraction et coton hydrophile ou papier filtre, exempts de produits extractibles à l'hexane,
— dessiccateur,
— tamis à trous de 1 millimètre de diamètre,
— pierre ponce en petits grains, préalablement séchée.
1.2. Réactif
n-hexane technique dont le résidu à l'évaporation complète doit être inférieur à 0,002 gramme pour 100 millilitres.
2. MODE OPÉRATOIRE
2.1. Préparation de l'échantillon pour essai
Broyer l'échantillon pour laboratoire, si nécessaire, dans le broyeur mécanique préalablement bien nettoyé afin de le réduire en particules pouvant traverser complètement le tamis.
Utiliser un vingtième environ de l'échantillon pour parfaire le nettoyage du broyeur, rejeter cette mouture, broyer le reste, le recueillir, le mélanger avec soin et l'analyser sans délai.
2.2. Prise d'essai
Peser, à 0,01 gramme près, dès la fin du broyage, environ 10 grammes de l'échantillon pour essai.
2.3. Préparation de la cartouche d'extraction
Placer la prise d'essai dans la cartouche et boucher celle-ci avec le tampon de coton hydrophile. Dans le cas où on a utilisé un papier filtre, emballer la mouture dans ce papier.
2.4. Préséchage
Si le grignon est très humide (teneur en eau et en matières volatiles supérieure à 10 %), effectuer un préséchage en plaçant pendant un temps convenable la cartouche remplie (ou le papier filtre) dans l'étuve chauffée à 80 °C au maximum, pour ramener la teneur en eau et en matières volatiles au-dessous de 10 %.
2.5. Préparation du ballon
Peser, à 1 milligramme près, le ballon contenant 1 à 2 grains de pierre ponce, préalablement séché à l'étuve à 103 °C ± 2 °C puis refroidi pendant au moins une heure dans un dessiccateur.
2.6. Première extraction
Placer dans l'appareil d'extraction la cartouche (ou le papier filtre) contenant la prise d'essai. Verser dans le ballon la quantité nécessaire d'hexane. Adapter le ballon à l'appareil d'extraction et placer le tout sur le bain à chauffage électrique. Conduire le chauffage dans des conditions telles que le débit du reflux soit au moins de trois gouttes à la seconde (ébullition modérée, non tumultueuse).
Après quatre heures d'extraction, laisser refroidir. Enlever la cartouche de l'appareil d'extraction et la placer dans un courrant d'air afin d'éliminer la majeure partie du solvant qui l'imprègne.
2.7. Deuxième extraction
Vider la cartouche dans le microbroyeur et broyer aussi finement que possible. Replacer quantitativement le mélange dans la cartouche et celle-ci dans l'appareil d'extraction.
Recommencer l'extraction pendant encore deux heures en utilisant le même ballon contenant la première extraction.
La solution obtenue dans le ballon d'extraction doit être limpide. À défaut, la filtrer sur un papier filtre en lavant plusieurs fois le premier ballon et le papier filtre avec de l'hexane. Recueillir le filtrat et le solvant de lavage dans un deuxième ballon préalablement séché et taré à 1 milligramme près.
2.8. Élimination du solvant et pesée de l'extrait
Chasser par distillation sur bain à chauffage électrique la majeure partie du solvant. Éliminer les dernières traces de solvant en chauffant le ballon à l'étuve à 103 °C ± 2 °C pendant 20 minutes. Faciliter cette élimination, soit en insufflant de l'air de temps à autre ou de préférence un gaz inerte, soit en opérant sous pression réduite.
Laisser refroidir le ballon dans un dessiccateur pendant au moins une heure, et le peser à 1 milligramme près.
Chauffer à nouveau 10 minutes dans les mêmes conditions, refroidir au dessiccateur et peser.
La différence entre les résultats de ces deux pesées doit être inférieure ou égale à 10 milligrammes. Sinon, chauffer à nouveau pendant des périodes de dix minutes suivies du refroidissement et de la pesée, jusqu'à ce que la différence de masse soit au plus égale à 10 milligrammes. Retenir la dernière pesée du ballon.
Effectuer deux déterminations sur le même échantillon pour essai.
3. EXPRESSION DES RÉSULTATS
3.1. Mode de calcul et formule
a) L'extrait exprimé en pourcentage en masse du produit tel quel, est égal à:
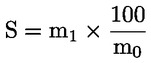
|
où: |
S est le pourcentage en masse d'extrait du produit tel quel, m0 est la masse, en grammes, de la prise d'essai, m1 est la masse, en grammes, de l'extrait après séchage. |
Prendre comme résultat la moyenne arithmétique des deux déterminations, si les conditions de répétabilité sont remplies.
Exprimer le résultat avec une seule décimale.
b) L'extrait est rapporté à la matière sèche en utilisant la formule suivante:

|
où: |
S est le pourcentage en masse d'extrait du produit tel quel [voir sous a)], U est sa teneur en eau et en matières volatiles. |
3.2. Répétabilité
La différence entre les résultats des deux déterminations, effectuées simultanément ou rapidement l'une après l'autre par le même analyste, ne doit pas être supérieure à 0,2 gramme d'extrait à l'hexane pour 100 grammes d'échantillon.
Dans le cas contraite, répéter l'analyse sur deux autres prises d'essai. Si cette fois encore la différence dépasse 0,2 gramme, prendre comme résultat la moyenne arithmétique des quatre déterminations effectuées.
ANNEXE XVI
DÉTERMINATION DE L'INDICE D'IODE
1. OBJET
La présente norme internationale décrit une méthode destinée à la détermination de l'indice d'iode dans les corps gras d'origine animale et végétale.
2. DÉFINITION
Aux fins de la présente norme internationale, les définitions suivantes sont applicables.
|
2.1. |
Indice d'iode: la masse d'iode absorbée par l'échantillon dans les conditions opératoires spécifiées par la présente norme internationale. L'indice d'iode est exprimé en nombre de grammes d'iode par 100 grammes d'échantillon. |
3. PRINCIPE
Mise en solution d'une prise d'essai dans un solvant et addition du réactif de Wijs. Au terme d'un laps de temps déterminé, addition d'une solution d'iodure de potassium et d'eau; titrage de l'iode libéré par une solution de thiosulfate de sodium.
4. RÉACTIFS
Tous les réactifs sont de qualité analytique reconnue.
|
4.1. |
Iodure de potassium, solution à 100 grammes par litre, exempte d'iode ou d'iodate. |
|
4.2. |
Solution d'amidon. Mélanger 5 grammes d'amidon soluble dans 30 millilitres d'eau, ajouter ce mélange à 1 000 millilitres d'eau bouillante, bouillir 3 minutes et laisser refroidir. |
|
4.3. |
Solution volumétrique standard de thiosulfate de sodium. c(Na2S303.5H20) = 0,1 mole par litre, normalisé pas plus de sept jours avant l'usage. |
|
4.4. |
Solvant préparé en mélangeant des volumes égaux de cyclohexane et d'acide acétique. |
|
4.5. |
Réactif de Wijs contenant du monochlorure d'iode dans de l'acide acétique. Utiliser le réactif de Wijs se trouvant dans le commerce. Note: Le réactif contient 9 grammes de ICI3 + 9 grammes de I dans l'acide acétique. |
5. APPAREILLAGE
Le matériel courant de laboratoire et notamment:
|
5.1. |
Nacelles de pesée en verre, appropriées à la prise d'essai et pouvant être introduites dans les fioles (5.2). |
|
5.2. |
Fioles coniques, d'une capacité de 500 millilitres, pourvues de bouchons en verre et complètement sèches. |
6. PRÉPARATION DE L'ÉCHANTILLON À ANALYSER. L'ÉCHANTILLON HOMOGÉNÉISÉ EST SÉCHÉ SUR SULFATE DE SODIUM ET FILTRÉ.
7. MODE OPÉRATOIRE
|
7.1. |
Prise d'essai La masse de la prise d'essai varie selon l'indice d'iode présumé, comme l'indique le tableau 1.
Tableau 1
Peser la prise d'essai à 0,1 milligramme près dans une nacelle de pesée en verre (5.1). |
|
7.2. |
Détermination Introduire la prise d'essai dans une fiole de 500 millilitres (5.2). Ajouter 20 millilitres de solvant (4.4) pour dissoudre les corps gras. Ajouter exactement 25 millilitres du réactif de Wijs (4.5), boucher, agiter le contenu et placer la fiole dans un endroit sombre. Ne pas utiliser de pipette pour le réactif de Wijs. Préparer un essai à blanc avec le solvant et le réactif mais sans la prise d'essai. Pour les échantillons ayant un indice d'iode inférieur à 150, laisser les fioles dans un endroit sombre pendant 1 heure; pour ceux ayant un indice d'iode supérieur à 150 et pour les produits polymérisés ou des produits oxydés dans une mesure considérable, les laisser pendant 2 heures. Une fois ce laps de temps écoulé, ajouter 20 millilitres de la solution d'iodure de potassium (4.1) et 150 millilitres d'eau dans chaque fiole. Titrer avec la solution de thiosulfate de sodium volumétrique standard (4.3) jusqu'à ce que la couleur jaune due à l'iode ait pratiquement disparu. Ajouter quelques gouttes de la solution d'amidon (4.2) et poursuivre la titration jusqu'au moment où la couleur bleue disparaît après avoir agité vigoureusement le contenu. Note: La détermination potentiométrique du point final est tolérée. |
|
7.3. |
Nombre de déterminations Effectuer deux déterminations sur le même échantillon. |
8. EXPRESSION DES RÉSULTATS
L'indice d'iode est égal à:

où:
c : valeur numérique de la concentration exacte, en moles par litre, de la solution de thiosulfate de sodium volumétrique standard (4.3) utilisée;
V1 : valeur numérique du volume, en millilitres, de la solution de thiosulfate de sodium volumétrique standard (4.3) utilisée pour l'essai à blanc;
V2 : valeur numérique du volume, en millilitres, de la solution de thiosulfate de sodium volumétrique standard (4.3) utilisée pour la détermination;
m : valeur numérique de la masse, en grammes, de la prise d'essai (7.1).
Prendre comme résultat la moyenne arithmétique des deux déterminations, à condition que le critère de répétabilité soit rempli.
ANNEXE XVII
MÉTHODE DE DÉTERMINATION DES STIGMASTADIÈNES DANS LES HUILES VÉGÉTALES
1. OBJET
Détermination des stigmastadiènes dans les huiles végétales contenant de faibles concentrations de ces hydrocarbures, en particulier dans les huiles d'olive vierge et les huiles de grignons d'olive.
2. CHAMP D'APPLICATION
La méthode est utilisable pour toutes les huiles végétales, mais les mesures ne sont fiables que lorsque la teneur en hydrocarbures est comprise entre 0,01 et 4 mg/kg. Cette méthode est particulièrement adaptée pour détecter la présence d'huiles végétales raffinées (olive, grignons d'olive, tournesol, palme, etc) dans l'huile d'olive vierge, étant donné que les huiles raffinées contiennent des stigmastadiènes, alors que les huiles vierges n'en contiennent pas.
3. PRINCIPE
Isolement de l'insaponifiable. Séparation de la fraction d'hydrocarbures stéroïdes par chromatographie sur colonne sur gel de silice et analyse par chromatographie capillaire en phase gazeuse.
4. APPAREILLAGE
|
4.1. |
Flacons appropriés de 250 millilitres avec condenseur à reflux. |
|
4.2. |
Ampoules à décanter de 500 millilitres. |
|
4.3. |
Flacons à fond rond de 100 millilitres. |
|
4.4. |
Évaporateur rotatif. |
|
4.5. |
Colonne de chromatographie en verre (de 1,5 à 2,0 centimètres de diamètre interne sur 50 centimètres de longueur) avec bouchon en Teflon et tampon de laine de verre ou disque de verre fritté dans le fond. Pour préparer la colonne de gel de silice, verser de l'hexane dans la colonne de chromatographie sur une hauteur d'environ 5 centimètres, puis compléter avec une suspension de gel de silice dans de l'hexane (15 grammes dans 40 millilitres) en utilisant des fractions d'hexane. Laisser reposer, puis soumettre à de légères vibrations. Ajouter du sulfate de sodium anhydre sur environ 0,5 centimètre de hauteur, puis éluer l'hexane en excès. |
|
4.6. |
Appareil de chromatographie en phase gazeuse avec détecteur d'ionisation à flamme, injecteur-diviseur ou sur colonne refroidie («on -column») et four programmable à ± 1 °C près. |
|
4.7. |
Colonne capillaire en silice fondue pour chromatographie en phase gazeuse (0,25 ou 0,5 millimètre de diamètre interne sur 25 mètres de longueur) recouvertes d'une phase de phénylméthylsilicone à 5 % formant un film de 0,25 micron d'épaisseur. Remarque 1. Il est possible d'utiliser d'autres colonnes de polarité similaire ou inférieure. |
|
4.8. |
Intégrateur-enregistreur permettant une intégration de vallée à vallée. |
|
4.9. |
Microseringue de 5 à 10 μl (microlitres) pour chromatographie en phase gazeuse avec aiguille cémentée. |
|
4.10. |
Chauffe-ballon électrique ou plaque chauffante. |
5. RÉACTIFS
Sauf indication contraire, tous les réactifs doivent être purs. Il convient d'utiliser de l'eau distillée ou de l'eau d'une pureté au moins équivalente.
|
5.1. |
Hexane ou mélange d'alcanes de point d'ébullition compris entre 65 et 70 °C, distillés à l'aide d'une colonne de fractionnement. Remarque 2. Le solvant doit être distillé pour éliminer les impuretés. |
|
5.2. |
Éthanol à 96 v/v. |
|
5.3. |
Sulfate de sodium anhydre. |
|
5.4. |
Solution d'hydroxyde de potassium alcoolique à 10 %. Ajouter 10 millilitres d'eau à 50 grammes d'hydroxyde de potassium, mélanger, puis dissoudre le mélange dans de l'éthanol jusqu'à obtention de 500 millilitres de solution. Remarque 3. La potasse alcoolique brunit au repos. Elle doit être préparée fraîchement chaque jour et conservée dans des flacons en verre brun bin fermés. |
|
5.5. |
Gel de silice 60 pour colonne de chromatographie, maille 70 à 230 (Merck réf. 7734 ou similaire). Remarque 4. En général, le gel de silice peut être utilisé directement tel qu'il se présente dans le conteneur, sans traitement préalable. Toutefois, certains lots de silice peuvent présenter une faible activité, ce qui se traduit par une mauvaise séparation chromatographique. Dans ce cas, il convient de traiter le gel de silice de la façon suivante: désactiver le gel de silice en le chauffant à 550 °C pendant 4 heures au minimum. Après chauffage, placer le gel de silice dans un déssicateur jusqu'à refroidissement, puis le transvaser dans un flacon fermé. Ajouter 2 % d'eau et secouer jusqu'à disparition des grumeaux et obtention d'une poudre flottant librement. Si certains lots de gel de silice donnent des chromatogrammes présentant des pics parasites, le gel de silice doit être traité comme indiqué ci-dessus. Une autre solution consiste à utiliser un autre gel de silice pur (Merck, réf. 7754). |
|
5.6. |
Solution mère (200 ppm) de cholesta-3,5-diène (Sigma, pureté de 99 %) dans de l'hexane. (10 milligrammes dans 50 millilitres). |
|
5.7. |
Solution standard de cholesta-3,5-diène dans de l'hexane à une concentration de 20 ppm, obtenue par dilution de la solution précédente. Remarque 5. Conservées à une température inférieure à 4 °C, les solutions désignées aux points 5.6 et 5.7 se conserveront pendant au moins 4 mois. |
|
5.8. |
Solution de n-nonacosane dans de l'hexane à une concentration d'environ 100 ppm. |
|
5.9. |
Gaz vecteur pour la chromatographie: hélium ou hydrogène d'une pureté de 99,9990 %. |
|
5.10. |
Gaz auxiliaires pour le détecteur d'ionisation à flamme: hydrogène d'une pureté de 99,9990 % et air purifié. |
6. MÉTHODE
6.1. Préparation de l'insaponifiable:
|
6.1.1. |
Peser 20 ± 0,1 grammes d'huile dans un flacon de 250 millilitres (point 4.1), ajouter 1 millilitre de la solution standard de cholesta-3,5-diène (20 microgrammes) et 75 millilitres de potasse alcoolique à 10 %, mettre en place le condenseur à reflux et chauffer en maintenant en légère ébullition pendant 30 minutes. Éloigner le flacon contenant l'échantillon de la source de chaleur et laisser refroidir légèrement (ne pas laisser refroidir complètement, sinon l'échantillon figerait). Ajouter 100 millilitres d'eau et transvaser la solution dans une ampoule à décanter (point 4.2) avec 100 millilitres d'hexane. Secouer le mélange énergiquement pendant 30 secondes et laisser les différentes couches se former. Remarque 6. S'il se forme une émulsion qui ne disparaît pas rapidement, ajouter de petites quantités d'éthanol. |
|
6.1.2. |
Transférer la phase aqueuse du dessous dans une seconde ampoule à décanter et extraire à nouveau avec 100 millilitres d'hexane. Récupérer à nouveau la phase inférieure et laver les extraits d'hexane (regroupés dans une autre ampoule à décanter) trois fois avec chaque fois 100 millilitres d'un mélange éthanol-eau (1: 1) jusqu'à obtention d'un pH neutre. |
|
6.1.3. |
Faire passer la solution d'hexane sur du sulfate de sodium anhydre (50 grammes), laver avec 20 millilitres d'hexane et faire sécher dans un évaporateur rotatif à 30 °C et à une faible pression. |
6.2. Séparation de la fraction d'hydrocarbures stéroïdes:
|
6.2.1. |
Placer le résidu dans la colonne de fractionnement avec deux fractions d'1 millilitre d'hexane, faire s'écouler l'échantillon le long de la colonne en amenant le niveau de la solution au-dessus du sulfate de sodium et commencer l'élution chromatographique avec l'hexane à un débit de 1 millilitre par minute environ. Éliminer le premier éluat de 25 à 30 millilitres, puis recueillir la fraction de 40 millilitres suivante. Ensuite, transférer cette fraction dans un flacon à fond rond de 100 millilitres (point 4.3). Remarque 7. La première fraction contient les hydrocarbures saturés (figure 1a) et la seconde, les hydrocarbures stéroïdes. En poursuivant l'élution, on obtient du squalène et des composés apparentés. Pour obtenir une bonne séparation entre les hydrocarbures saturés et les hydrocarbures stéroïdes, le volume des fractions doit être optimal. À cet effet, il convient d'ajuster le volume de la première fraction de manière que, lors de l'analyse de la seconde fraction, les pics représentant les hydrocarbures saturés soient faibles (figure 1c); si ces pics ne se forment pas, mais que l'intensité du pic standard est faible, il faut réduire le volume. En tout état de cause, il est inutile de séparer complètement les composants de la première et de la seconde fractions, étant donné qu'il n'y a pas de chevauchement des pics lors de l'analyse par chromatographie en phase gazeuse si les conditions de la CG sont ajustées conformément au point 6.3.1. En général, il est inutile d'optimaliser le volume de la seconde fraction car on obtient une bonne séparation avec les composants suivants. Quoi qu'il en soit. la formation d'un grand pic à un temps de rétention inférieur d'environ 1,5 minute à celui du pic standard est due au squalène et témoigne d'une mauvaise séparation. |
|
6.2.2. |
Faire évaporer la seconde fraction dans un évaporateur à 30 °C et à une faible pression jusqu'à séchage, puis dissoudre immédiatement le résidu dans 0,2 millilitre d'hexane. Conserver la solution au réfrigérateur jusqu'à analyse. Remarque 8. Les résidus désignés aux points 6.1.3 et 6.2.2 ne doivent pas être conservés secs et à température ambiante. Dès leur obtention, il convient d'ajouter le solvant et de conserver les solutions au réfrigérateur. |
6.3. Chromatographie en phase gazeuse
|
6.3.1. |
Conditions opératoires applicables à l'injection: — température de l'injecteur: 300 °C, — température du détecteur: 320 °C, — intégrateur-enregistreur: les paramètres d'intégration doivent être fixés de manière à permettre une évaluation correcte des aires. Le mode d'intégration de vallée à vallée est recommandé, — sensibilité: environ 16 fois l'atténuation minimale, — quantité de solution injectée: 1 microlitre, — températures de programmation du four: température initiale de 235 °C pendant 6 minutes, puis élévation de 2 °C par minute jusqu'à 285 °C, — injecteur avec diviseur de débit 1: 15, — vecteur: hélium ou hydrogène à une pression d'environ 120 kPa. Ces conditions peuvent être modifiées en fonction des caractéristiques du chromatographe et de la colonne, de manière que les chromatogrammes répondent aux exigences suivantes: formation du pic standard interne à plus ou moins 5 minutes du temps indiqué au point 6.3.2; le pic standard interne doit s'étirer sur au moins 80 % de l'échelle totale. Il y a lieu de vérifier le système de chromatographie en phase gazeuse en injectant un mélange de solution mère de cholestadiène (point 5.6) et de solution de n-nonacosane (point 5.8). Le pic du cholesta-3,5-diène doit se former avant celui du n-nonacosane (figure 1c); si cela ne se produit pas, deux mesures peuvent être prises: réduire la température du four et/ou utiliser une colonne moins polaire. |
|
6.3.2. |
Identification des pics Le pic standard interne se forme à environ 19 minutes et le stigmasta-3,5-diène à un temps de rétention relatif d'environ 1,29 (voir figure 1b). Le stigmastadiène s'accompagne de faibles quantités d'isomère et, généralement, ils produisent un pic chromatographique unique. Néanmoins, si la colonne est trop polaire ou si elle présente un grand pouvoir de résolution, l'isomère peut former un petit pic avant celui du stigmasta-3,5-diène et très près de lui (figure 2). Pour garantir que les stigmastadiènes produisent un pic unique, il est conseillé de remplacer la colonne par une autre moins polaire ou à diamètre interne plus large. Remarque 9. La méthode de détermination des hydrocarbures stéroïdes appliquée à l'analyse d'une huile végétale raffinée permet d'obtenir un pic témoin pour les stigmastadiènes. La substance fait apparaître un pic de hauteur appréciable, facilement identifiable. |
|
6.3.3. |
Analyse quantitative La teneur en stigmastadiène est déterminée par la formule suivante: mg/kg de stigmastadiènes =
Limite de détection: 0,01 mg/kg environ. |
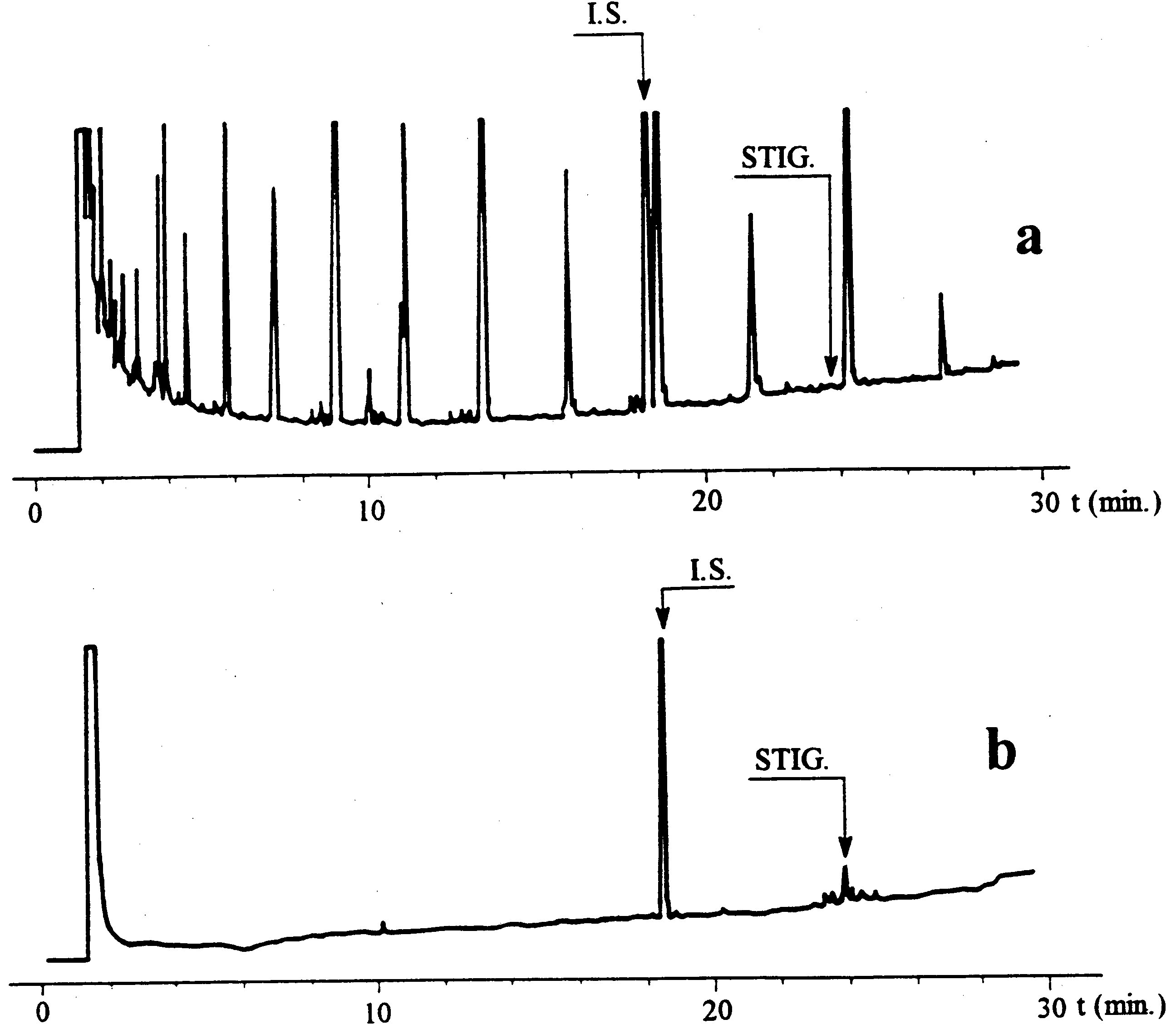

Figure 1
Chromatogrammes (chromatographie en phase gazeuse) obtenus par analyse d'échantillons d'huile d'olive sur une colonne capillaire en silice fondue (0,25 millimètre de diamètre interne sur 25 mètres de longueur) recouverte d'un film de 0,25 micron d'épaisseur de phénylméthylsilicone à 5 %.
a) Première fraction (30 millilitres) d'une huile vierge, éluée avec le standard.
b) Seconde fraction (40 millilitres) d'une huile d'olive contenant 0,10 mg/kg de stigmastadiènes.
c) Seconde fraction (40 millilitres) contenant une petite proportion de la première fraction.

Figure 2
Chromatogramme en phase gazeuse obtenu à partir d'un échantillon d'huile d'olive raffinée analysé sur une colonne DB-5 sur laquelle figure l'isomère de stigmasta-3,5-diène.
ANNEXE XVIII
DÉTERMINATION DE LA DIFFÉRENCE ENTRE LA COMPOSITION RÉELLE ET LA COMPOSITION THÉORIQUE DES TRIGLYCÉRIDES À ECN 42
1. CHAMP D’APPLICATION
Détermination de la différence absolue entre les valeurs expérimentales des triglycérides (TG) à nombre équivalent d’atomes de carbone égal à 42 (ECN 42CLHP) obtenues par détermination dans l’huile par chromatographie liquide haute performance (CLHP) et la valeur théorique des TG à nombre équivalent d’atomes de carbone égal à 42 (ECN 42théorique) calculée d’après la composition en acides gras.
2. DOMAINE D’APPLICATION
La méthode s’applique aux huiles d’olive. Elle vise à détecter la présence de faibles quantités d’huiles de graines (riches en acide linoléique) dans chaque catégorie d’huile d’olive.
3. PRINCIPE
Dans le cas des huiles d’olive pures, la composition des triglycérides à ECN 42 déterminée par CLHP est plus ou moins équivalente à la composition théorique des triglycérides à ECN 42 (calculée à partir de la composition en acides gras déterminée par chromatographie en phase gazeuse). Une différence supérieure aux valeurs adoptées pour chaque catégorie d’huile indique que l’huile contient des huiles de graines.
4. MÉTHODE
La méthode permettant de calculer la composition théorique des triglycérides à ECN 42 ainsi que la différence par rapport aux données CLHP consiste essentiellement en la coordination des résultats d’analyse obtenus par d’autres méthodes. On distingue trois phases: la détermination de la composition en acides gras par chromatographie en phase gazeuse (CPG) sur colonne capillaire, le calcul de la composition théorique des triglycérides à ECN 42 et la détermination des triglycérides à ECN 42 par CLHP.
4.1. Appareillage
|
4.1.1. |
Ballons à fond rond de 250 et 500 ml. |
|
4.1.2. |
Béchers de 100 ml. |
|
4.1.3. |
Colonne de chromatographie en verre (diamètre intérieur: 21 mm; longueur: 450 mm) avec robinet et cône normalisé (femelle) au sommet. |
|
4.1.4. |
Ampoules à décanter de 250 ml avec cône normalisé (mâle) à la base, pouvant s’adapter au sommet de la colonne. |
|
4.1.5. |
Baguette en verre de 600 mm de longueur. |
|
4.1.6. |
Entonnoir en verre de 80 mm de diamètre. |
|
4.1.7. |
Fioles jaugées de 50 ml |
|
4.1.8. |
Fioles jaugées de 20 ml |
|
4.1.9. |
Évaporateur rotatif. |
|
4.1.10. |
Chromatographe en phase liquide à haute performance, équipé d’un réglage thermostatique de la température de la colonne. |
|
4.1.11. |
Vannes d’injection pour 10 μl. |
|
4.1.12. |
Détecteur: réfractomètre différentiel. La sensibilité pleine échelle doit atteindre au moins 10–4 unités d’indice de réfraction. |
|
4.1.13. |
Colonne: tube en acier inoxydable de 250 mm de longueur et de 4,5 mm de diamètre intérieur, rempli de particules de silice de 5 μm de diamètre contenant 22 à 23 % de carbone sous forme d’octadécylsilane. |
|
4.1.14. |
Logiciel de traitement des données. |
|
4.1.15. |
Flacons d’environ 2 ml, avec bouchon à vis et septa en téflon. |
4.2. Réactifs
Les réactifs doivent être de pureté analytique. Les solvants d’élution doivent être dégazés et peuvent être recyclés plusieurs fois sans que cela ait une incidence sur les séparations.
|
4.2.1. |
éther de pétrole 40-60 °C pour chromatographie ou hexane. |
|
4.2.2. |
Éther éthylique, exempt de peroxydes, fraîchement distillé. |
|
4.2.3. |
Solvant d’élution pour la purification de l’huile par chromatographie sur colonne: mélange d’éther de pétrole/éther éthylique dans les proportions 87/13 (v/v). |
|
4.2.4. |
Gel de silice, granulométrie 70-230, type Merck 7734, à teneur en eau normalisée de 5 % (m/m). |
|
4.2.5. |
Laine de verre. |
|
4.2.6. |
Acétone pour CLHP. |
|
4.2.7. |
Acétonitrile ou propionitrile pour CLHP. |
|
4.2.8. |
Solvant d’élution pour CLHP: acétonitrile + acétone (proportions à ajuster pour obtenir la séparation souhaitée; commencer avec le mélange 50:50) ou propionitrile. |
|
4.2.9. |
Solvant de solubilisation: acétone. |
|
4.2.10. |
Triglycérides de référence: soit des triglycérides que l’on trouve dans le commerce (tripalmitine, trioléine, etc.); dans ce cas, les temps de rétention sont reportés sur un diagramme en fonction du nombre équivalent d’atomes de carbone; soit des chromatogrammes de référence obtenus pour de l’huile de soja, pour un mélange 30:70 d’huile de soja et d’huile d’olive, et pour de l’huile d’olive pure (voir notes 1 et 2 et figures 1 à 4). |
|
4.2.11. |
Colonne d’extraction en phase solide avec phase de silice 1 g, 6 ml. |
4.3. Préparation des échantillons
Étant donné qu’un certain nombre de substances peuvent interférer et donner lieu à des résultats faux positifs, l’échantillon doit toujours être purifié selon la méthode IUPAC 2.507 utilisée pour la détermination des composés polaires dans les graisses de friture.
4.3.1. Préparation de la colonne de chromatographie
Remplir la colonne (4.1.3) avec 30 ml environ de solvant d’élution (4.2.3); introduire ensuite un tampon de laine de verre (4.2.5) dans la colonne et l’enfoncer jusqu’au fond de la colonne au moyen de la baguette en verre (4.1.5).
Préparer dans un bécher de 100 ml une suspension avec 25 g de gel de silice (4.2.4) dans 80 ml de mélange d’élution (4.2.3); la transférer ensuite dans la colonne au moyen d’un entonnoir en verre (4.1.6).
Afin de s’assurer que la totalité du gel de silice a été transférée dans la colonne, laver le bécher avec le mélange d’élution et transférer également le liquide de lavage dans la colonne.
Ouvrir le robinet et laisser le solvant s’écouler jusqu’à ce que son niveau se situe à environ 1 cm au-dessus du gel de silice.
4.3.2. Chromatographie sur colonne
Peser, avec un degré de précision de 0,001 g, 2,5 ± 0,1 g d’huile, préalablement filtrée, homogénéisée et, si nécessaire, déshydratée dans une fiole jaugée de 50 ml (4.1.7).
Diluer dans environ 20 ml de solvant d’élution (4.2.3); si nécessaire, chauffer légèrement pour faciliter la dissolution. Refroidir à température ambiante et porter au volume avec du solvant d’élution.
À l’aide d’une pipette jaugée, introduire 20 ml de solution dans la colonne préparée conformément au point 4.3.1, ouvrir le robinet et laisser le solvant s’écouler jusqu’au niveau de la couche de gel de silice.
Éluer ensuite avec 150 ml de solvant d’élution (4.2.3), en réglant le débit du solvant à 2 ml par minute environ (de telle sorte que 150 ml s’écoulent dans la colonne en 60-70 minutes).
Recueillir l’éluat dans un ballon à fond rond de 250 ml (4.1.1) préalablement taré et placé dans un four et le peser avec précision. Éliminer le solvant sous pression réduite dans un évaporateur rotatif (4.1.9) et peser le résidu qui sera utilisé pour préparer la solution pour l’analyse CLHP et pour la préparation des esters méthyliques.
Après passage dans la colonne, l’échantillon doit être récupéré à 90 % au moins pour les catégories d’huile d’olive vierge extra, vierge et raffinée normalement, et à 80 % au moins pour les huiles lampantes et les huiles de grignons.
4.3.3. Purification par extraction en phase solide
Activer la colonne d’extraction en phase solide sur silice en passant 6 ml d’hexane (4.2.3) sous vide, en évitant la dessiccation.
Peser 0,12 g avec un degré de précision de 0,001 g dans un flacon de 2 ml (4.1.15) et dissoudre dans 0,5 ml d’hexane (4.2.3).
Charger la colonne d’extraction en phase solide avec la solution et éluer avec 10 ml de mélange hexane-éther diéthylique (87:13 v/v) (4.2.3) sous vide.
La fraction recueillie est évaporée à sec dans un évaporateur rotatif (4.1.9) à pression réduite et à température ambiante. Le résidu est dissous dans 2 ml d’acétone (4.2.6) en vue de l’analyse des triglycérides (TG).
4.4. Analyse CLHP
4.4.1. Préparation des échantillons pour l’analyse chromatographique
Préparer une solution à 5 % de l’échantillon à analyser en pesant 0,5 ± 0,001 g de l’échantillon dans une fiole jaugée de 10 ml et compléter à 10 ml avec le solvant de solubilisation (4.2.9).
4.4.2. Procédure
Régler le système chromatographique. Pomper du solvant d’élution (4.2.8) à un débit de 1,5 ml par minute de façon à purger l’ensemble du système. Attendre d’avoir une ligne de base stable.
Injecter 10 μl de l’échantillon préparé selon le point 4.3.
4.4.3. Calcul et expression des résultats
Utiliser la méthode de normalisation des aires des pics, qui consiste à admettre que la somme des aires des pics correspondant aux triglycérides (TG) de ECN 42 à ECN 52 est égale à 100 %.
Calculer le pourcentage relatif de chaque triglycéride selon la formule:
![]()
.
Les résultats doivent comporter au moins deux chiffres après la virgule.
Voir notes 1 et 4.
4.5. Calcul de la composition des triglycérides (% de moles) à partir de la composition en acides gras (% de l’aire)
4.5.1. Détermination de la composition en acides gras
La composition en acides gras est déterminée par la norme ISO 5508 au moyen d’une colonne capillaire. Les esters méthyliques sont préparés selon la méthode COI/T.20/doc. no 24.
4.5.2. Acides gras pris en considération dans le calcul
Les glycérides sont regroupés selon leur nombre équivalent d’atomes de carbone (ECN — Equivalent Carbon Number), compte tenu des équivalences suivantes entre ECN et acides gras. Seuls les acides gras à 16 ou 18 atomes de carbone ont été pris en considération, car ce sont les seuls qui sont importants pour l’huile d’olive. Les acides gras doivent être normalisés à 100 %.
|
Acide gras (AG) |
Abréviation |
Poids moléculaire (PM) |
ECN |
|
Acide palmitique |
P |
256,4 |
16 |
|
Acide palmitoléique |
Po |
254,4 |
14 |
|
Acide stéarique |
S |
284,5 |
18 |
|
Acide oléique |
O |
282,5 |
16 |
|
Acide linoléique |
L |
280,4 |
14 |
|
Acide linolénique |
Ln |
278,4 |
12 |
4.5.3. Conversion en moles du % de l’aire pour tous les acides gras (1)
|
|
|
|
|
|
|
|
4.5.4. Normalisation à 100 % des moles d’acides gras (2)




Le résultat indique le pourcentage de chaque acide gras, en moles, dans la position globale (1,2,3) des TG.
On calcule ensuite la somme des acides gras saturés P et S (AGS) et des acides gras insaturés Po, O, L et Ln (AGI):
![]()
![]()
4.5.5. Calcul de la composition des acides gras en position 2 et en positions 1, 3 des TG
Les acides gras sont répartis en trois groupes, comme suit: un groupe pour la position 2 et deux groupes identiques pour les positions 1 et 3, avec des coefficients différents pour les acides saturés (P et S) et les acides insaturés (Po, O, L et Ln).
4.5.5.1. Acides gras saturés en position 2 [P(2) et S(2)] (4):
![]()
![]()
4.5.5.2. Acides gras insaturés en position 2 [Po(2), O(2), L(2) et Ln(2)] (5):




4.5.5.3. Acides gras en positions 1 et 3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) et Ln(1,3)] (6):




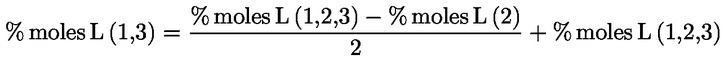

4.5.6. Calcul des triglycérides
4.5.6.1. TG à un acide gras (AAA, ici LLL, PoPoPo) (7)
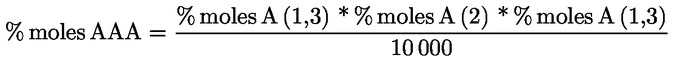
4.5.6.2. TG à deux acides gras (AAB, ici PoPoL, PoLL) (8)


4.5.6.3. TG à trois acides gras distincts (ABC, ici OLLn, PLLn, PoOLn, PPoLn) (9)



4.5.6.4. Triglycérides à ECN 42
Les triglycérides à ECN 42 sont calculés selon les équations 7, 8 et 9, et sont ensuite indiqués par ordre d’élution attendu en CLHP (en général, on observe seulement trois pics).
LLL
PoLL et l’isomère de position LPoL
OLLn et les isomères de position OLnL et LnOL
PoPoL et l’isomère de position PoLPo
PoOLn et les isomères de position OPoLn et OLnPo
PLLn et les isomères de position LLnP et LnPL
PoPoPo
SLnLn et l’isomère de position LnSLn
PPoLn et les isomères de position PLnPo et PoPLn
Les triglycérides à ECN 42 s’obtiennent en additionnant les neuf triglycérides, y compris leurs isomères de position. Les résultats doivent comporter au moins deux chiffres après la virgule.
5. ÉVALUATION DES RÉSULTATS
On compare la composition théorique calculée et celle déterminée par CLHP. Si, en valeur absolue, la différence «données CLHP moins données théoriques» est supérieure aux valeurs spécifiées pour la catégorie d’huile concernée dans la norme de commercialisation, l’échantillon contient de l’huile de graines.
Les résultats sont exprimés avec deux décimales.
6. EXEMPLE (LA NUMÉROTATION RENVOIE AUX SECTIONS DU TEXTE DE LA MÉTHODE)
— 4.5.1. Calcul du % de moles d’acides gras à partir des données de la CPG (% de l’aire normalisé)
La détermination de la composition en acides gras par CPG donne les valeurs suivantes:
|
AG |
P |
S |
Po |
O |
L |
Ln |
|
PM |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
% aire |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3 Conversion en moles du % de l’aire pour tous les acides gras [voir formule (1)]
|
moles P |
= |
|
|
moles S |
= |
|
|
moles Po |
= |
|
|
moles O |
= |
|
|
moles L |
= |
|
|
moles Ln |
= |
|
|
Total |
= |
0,35821 moles TG |
— 4.5.4 Normalisation à 100 % des moles d’acides gras [voir formule (2)]
|
% moles P(1,2,3) |
= |
|
|
% moles S(1,2,3) |
= |
|
|
% moles Po(1,2,3) |
= |
|
|
% moles O(1,2,3) |
= |
|
|
% moles L(1,2,3) |
= |
|
|
% moles Ln(1,2,3) |
= |
|
|
Total % moles |
= |
100 % |
Somme des acides gras saturés et insaturés en position 1, 2 et 3 des TG [voir formule (3)]:
![]()
![]()
— 4.5.5 Calcul de la composition en acides gras en position 2 et en position 1 et 3 des TG
— 4.5.5.1 Acides gras saturés en position 2 [P(2) et S(2)] [voir formule (4)]
![]()
![]()
— 4.5.5.2 Acides gras insaturés en position 2 [Po(1,3), O(1,3), L(1,3) et Ln(1,3)] [voir formule (5)]
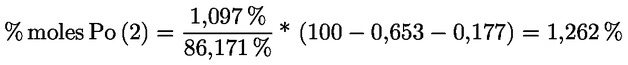


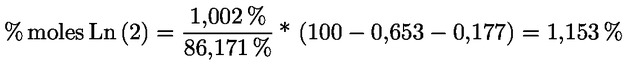
— 4.5.5.3 Acides gras en position 1 et 3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) et Ln(1,3)] [voir formule (6)]
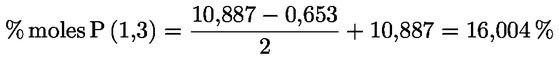



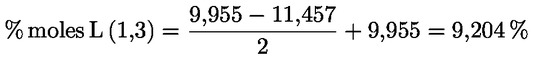

— 4.5.6. Calcul des triglycérides
À partir de la composition calculée en acides gras en position 2 et en position 1 et 3 (voir tableau suivant):
|
AG en |
positions 1 et 3 |
position 2 |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Total |
100,0 % |
100,0 % |
Les triglycérides suivants sont calculés:
LLL
PoPoPo
PoLL avec un isomère de position
SLnLn avec un isomère de position
PoPoL avec un isomère de position
PPoLn avec 2 isomères de position
OLLn avec 2 isomères de position
PLLn avec 2 isomères de position
PoOLn avec 2 isomères de position
— 4.5.6.1. TG à un acide gras (LLL, PoPoPo) [voir formule (7)]
 , = 0,09706 mol LLL
, = 0,09706 mol LLL

— 4.5.6.2 TG à deux acides gras (PoLL, SLnLn, PoPoL) (voir formule (8)]
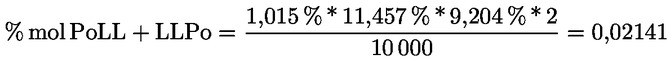
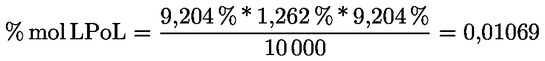
0,03210 mol PoLL

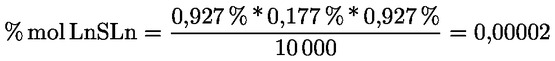
0,00094 mol SLnLn

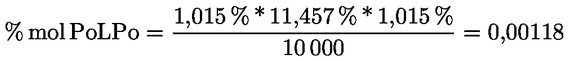
0,00354 mol PoPoL
— 4.5.6.3 TG à trois acides gras distincts (PoPLn, OLLn, PLLn, PoOLn) [voir formule (9)]
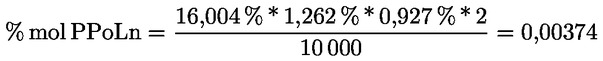


0,00761 mol PPoLn



0,43655 mol OLLn

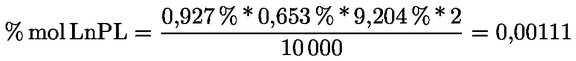

0,06907 mol PLLn


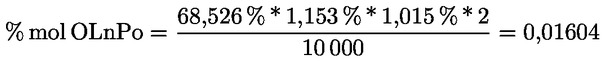
0,04812 mol PoOLn
ECN 42 = 0,69512 mol TG
Note 1: Il est possible de déterminer l’ordre d’élution en calculant le nombre équivalent d’atomes de carbone, souvent défini par la relation
![]() , dans laquelle CN est le nombre d’atomes de carbone et n le nombre de doubles liaisons; on peut affiner le calcul en tenant compte de l’origine de la double liaison. Si no, nl et nln sont les nombres de doubles liaisons attribués respectivement aux acides oléique, linoléique et linolénique, le nombre équivalent d’atomes de carbone peut être calculé selon la formule suivante:
, dans laquelle CN est le nombre d’atomes de carbone et n le nombre de doubles liaisons; on peut affiner le calcul en tenant compte de l’origine de la double liaison. Si no, nl et nln sont les nombres de doubles liaisons attribués respectivement aux acides oléique, linoléique et linolénique, le nombre équivalent d’atomes de carbone peut être calculé selon la formule suivante:
![]()
dans laquelle les coefficients do, dl et dln peuvent être calculés à l’aide des triglycérides de référence. Dans les conditions spécifiées dans la présente méthode, la relation obtenue sera voisine de:
![]()
Note 2: Avec plusieurs triglycérides de référence, il est également possible de calculer la résolution par rapport à la trioléine:
![]()
de la trioléineen utilisant le temps de rétention réduit
![]()
La représentation graphique de log α en fonction de f (nombre de doubles liaisons) permet de déterminer les valeurs de rétention pour tous les triglycérides à acides gras contenus dans les triglycérides de référence — voir figure 1.
Note 3: L’efficacité de la colonne doit permettre de séparer nettement le pic de la trilinoléine des pics des triglycérides dont le TR est proche. L’élution est effectuée jusqu’au pic ECN 52.
Note 4: Une mesure correcte des aires de tous les pics intéressants pour la présente détermination est garantie si le deuxième pic correspondant à ECN 50 est égal à 50 % du maximum de l’échelle.
Figure 1
Représentation graphique de log α en fonction de f (nombre de doubles liaisons)
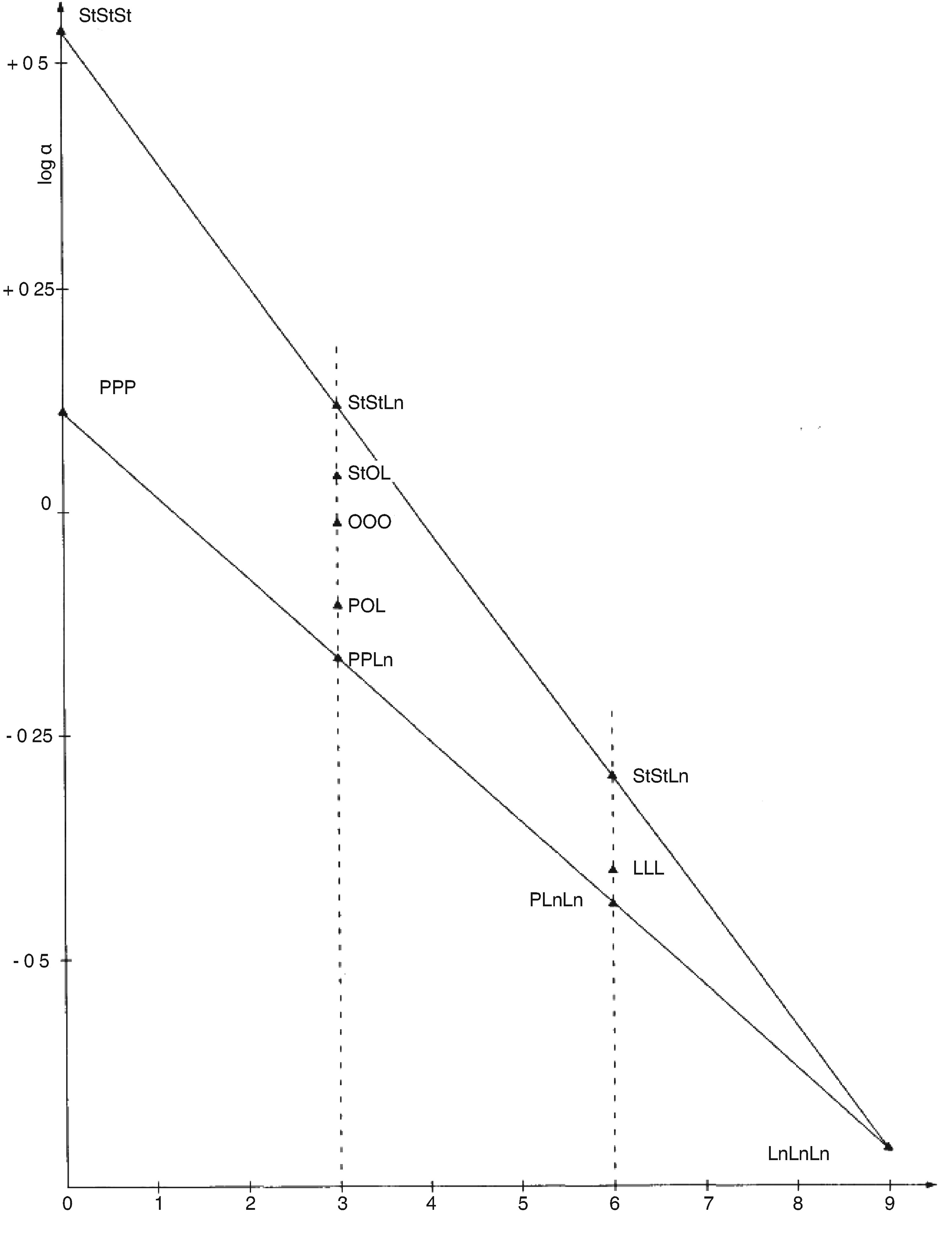
Nombre de doubles liaisons
La: acide laurique; My: acide myristique; P: acide palmitique; S: acide stéarique; O: acide oléique; L: acide linoléique; Ln: acide linolénique
Figure 2
Huile d’olive à faible teneur en acide linoléique
a)
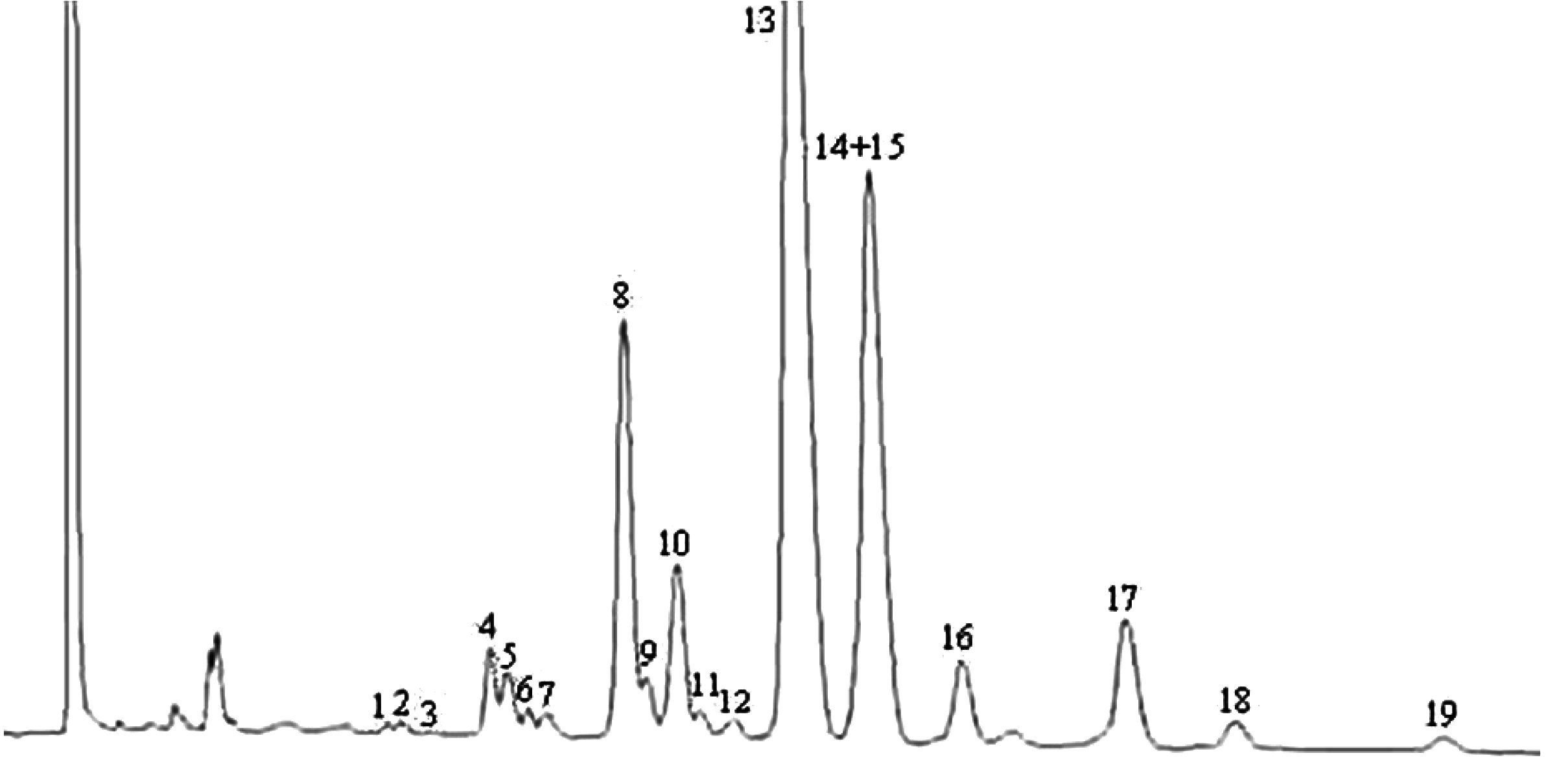
Solvant: acétone/acétonitrile.
Tracé a: Principaux composants des pics chromatographiques: ECN 42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN 44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN 46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN 48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN 50: (17) SOO; (18) POS + SLS.
b)
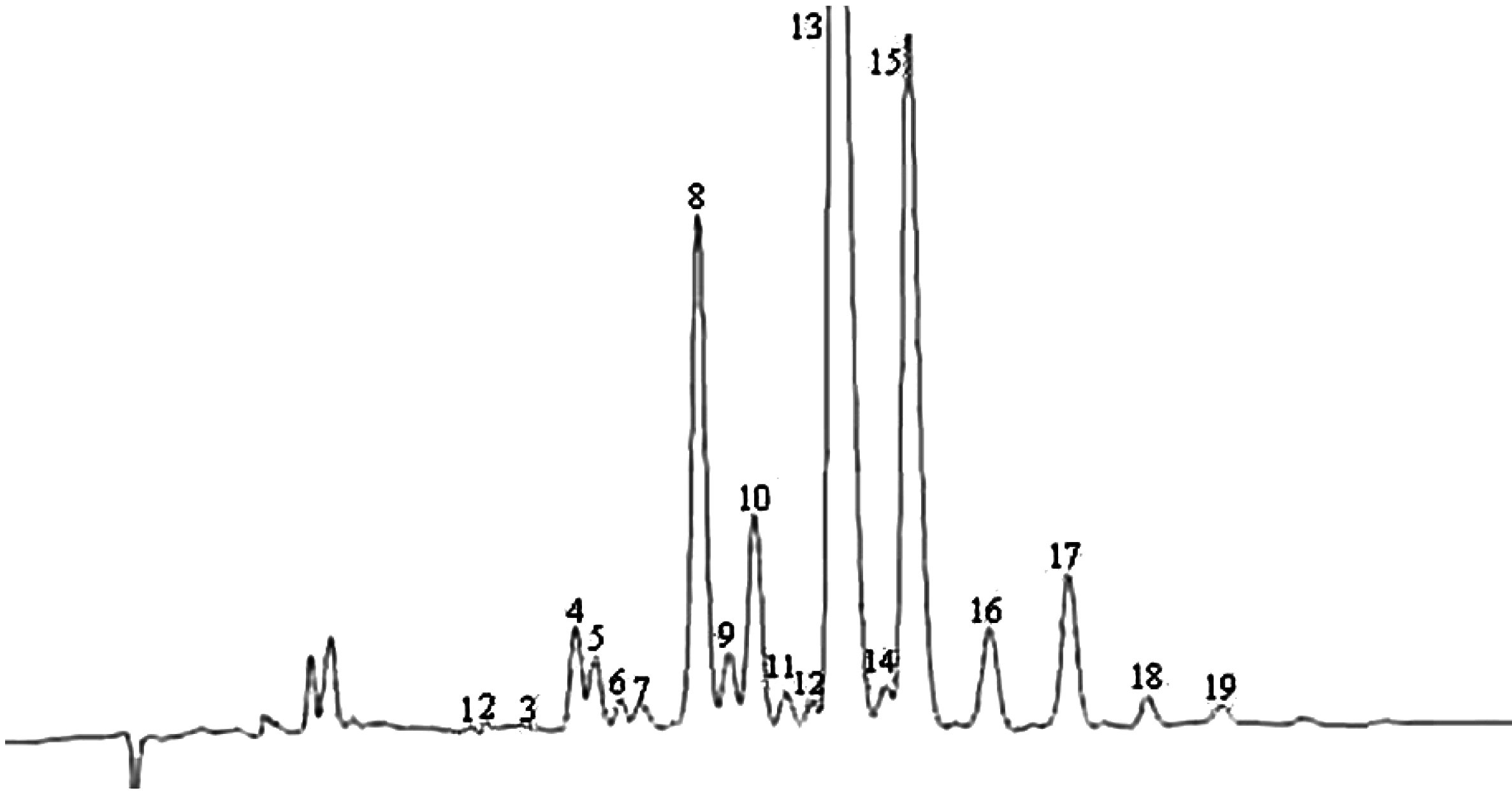
Solvant: propionitrile.
Tracé b: Principaux composants des pics chromatographiques: ECN 42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN 44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN 46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN 48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN 50: (17) SOO; (18) POS + SLS
Figure 3
Huile d’olive à teneur élevée en acide linoléique
a)
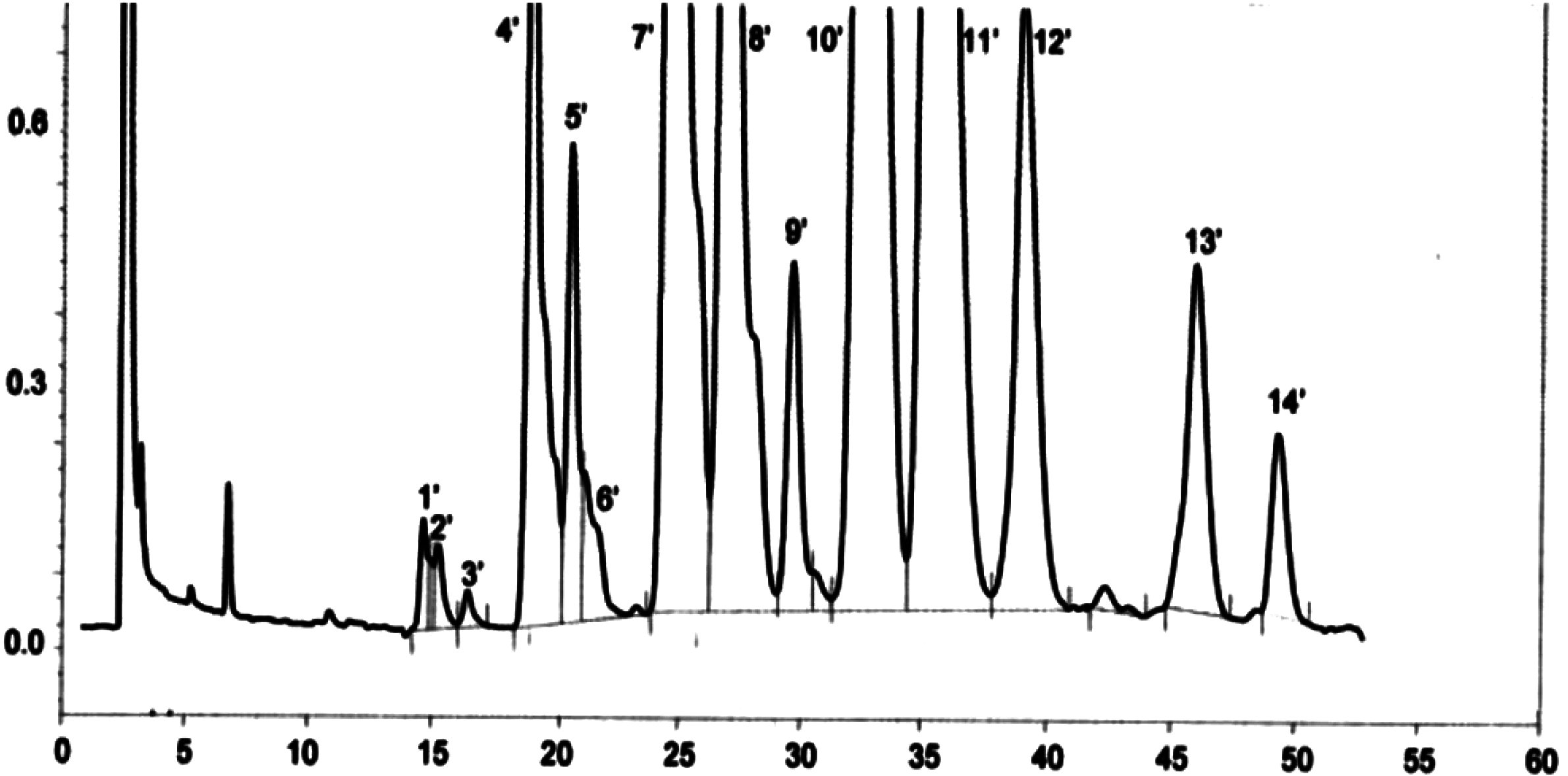
Solvant: acétone/acétonitrile (50:50).
Tracé a: Principaux composants des pics chromatographiques: ECN 42: (1’) LLL + PoLL; (2’) OLLn + PoOLn; (3’) PLLn; ECN 44: (4’) OLL + PoOL; (5’) OOLn + PLL; (6’) POLn + PPoPo; ECN 46: (7’) OOL + PoOO; (8’) PLO + SLL + PoOP; (9’) PLP + PoPP; ECN 48: (10’) OOO; (11’) POO + SLL + PPoO; (12’) POP + PLS; ECN 50: (13’) SOO; (14’) POS + SLS
b)

Solvant: propionitrile.
Tracé b: Principaux composants des pics chromatographiques: ECN 42: (1) LLL; (2 + 2’) OLLn + PoLL; (3) PLLn; ECN 44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN 46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN 48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN 50: (17) SOO; (18) POS + SLS; ECN 52: (19) AOO.
ANNEXE XIX
DÉTERMINATION DU CONTENU EN ALCOOLS ALIPHATIQUES ET TRITERPÉNIQUES PAR CHROMATOGRAPHIE EN PHASE GAZEUSE AVEC COLONNE CAPILLAIRE
1. OBJET
La présente annexe décrit un procédé de détermination du contenu en alcools aliphatiques et triterpéniques des huiles et des matières grasses.
2. PRINCIPE DE LA MÉTHODE
La matière grasse, additionnée de 1-eicosanol comme standard interne, est saponifiée avec de l'hydroxyde de potassium en solution dans l'éthanol, puis l'insaponifiable est extrait avec de l'éther éthylique. La fraction des alcools est séparée de l'extrait insaponifiable par chromatographie sur plaque de gel de silice basique; les alcools récupérés dans le gel de silice sont transformés en triméthylsilyléthers et analysés par chromatographie en phase gazeuse en colonne capillaire.
3. APPAREILLAGE
3.1. Ballon de 250 millilitres muni d'un réfrigérant à reflux avec embouts rodés.
3.2. Ampoule à décanter de 500 millilitres.
3.3. Ballons de 250 millilitres.
3.4. Équipement complet pour chromatographie en phase solide, avec plaques de verre de 20 × 20, centimètres.
3.5. Lampe à lumière ultraviolette, de longueur d'onde de 366 ou 254 nm.
3.6. Microseringues de 100 et 500 microlitres.
3.7. Ampoule cylindrique filtrante à filtre poreux G 3 (porosité 15 à 40 micromètres) de 2 centimètres de diamètre environ et de 5 centimètres de hauteur, avec embout approprié pour filtration sous vide et embout rodé mâle 12/21.
3.8. Fiole à vide de 50 millilitres avec embout rodé femelle 12/21 adaptable à l'ampoule filtrante (3.7).
3.9. Tube à fond conique, de 10 millilitres, avec bouchon hermétique.
3.10. Chromatographe en phase gazeuse approprié au fonctionnement avec colonne capillaire, équipé d'un système de fractionnement, constitué de:
3.10.1. Enceinte thermostatée pour la colonne, permettant de maintenir la température désirée avec une précision d'environ 1 °C.
3.10.2. Ensemble d'injection thermoréglable avec élément vaporisateur en verre persilanisé.
3.10.3. Détecteur à ionisation de flamme et convertisseur-amplificateur.
3.10.4. Enregistreur-intégrateur approprié au fonctionnement avec un convertisseur-amplificateur avec un temps de réponse non supérieur à 1 seconde et avec une vitesse de papier variable.
3.11. Colonne capillaire en verre ou en silice fondue, de 20 à 30 mètres de long, de 0,25 à 0,32 millimètre de diamètre intérieur, recouverte intérieurement de liquide SE-52 ou SE-54 ou équivalent, avec une épaisseur comprise entre 0,10 et 0,30 micromètre.
3.12. Microseringue pour chromatographie en phase gazeuse de 10 microlitres avec aiguille cémentée.
3.13. Balance de précision ayant une sensibilité de 1 mg (avec affichage 0.1 mg).
4. RÉACTIFS
4.1. Hydroxyde de potassium, solution éthanolique à environ 2N: dissoudre, tout en refroidissant, 130 grammes d'hydroxyde de potassium (titre minimum 85 %) dans 200 millilitres d'eau distillée, puis compléter à un litre avec de l'éthanol. La solution se conserve dans des bouteilles de verre opaque bien bouchées.
4.2. Éther éthylique, pour analyses.
4.3. Sulfate de sodium anhydre pur, pour analyses.
4.4. Plaques de verre recouvertes de gel de silice sans indicateur de fluorescence, de 0,25 millimètre d'épaisseur (elles sont disponibles dans le commerce déjà prêtes à l'emploi).
4.5. Hydroxyde de potassium, solution éthanolique à 0,2 N: dissoudre 13 grammes d'hydroxyde de potassium dans 20 millilitres d'eau distillée, puis compléter à un litre avec de l'éthanol.
4.6. Benzène, pour chromatographie (5.2.2).
4.7. Acétone, pour chromatographie (5.2.2).
4.8. Hexane pour chromatographie (5.2.2).
4.9. Éther éthylique, pour chromatographie (5.2.2).
4.10. Chloroforme pur, pour analyse.
4.11. Solution de référence pour la chromatographie en couche mince: alcools C20-C28 à 0,5 % dans le chloroforme, ou une fraction des alcools obtenue comme indiqué au point 5.2 de l'extrait insaponifiable d'une huile de grignons d'olive.
4.12. Dichloro-2'-7' fluorescéine, solution éthanolique à 0,2 %. Elle est rendue légèrement basique par addition de quelques gouttes d'une solution alcoolique 2N d'hydroxyde de potassium.
4.13. Pyridine anhydre, pour chromatographie.
4.14. Hexaméthyldisilazane.
4.15. Triméthylchlorosilane.
4.16. Solution étalon de triméthylsilyléthers des alcools aliphatiques de C20 à C28. À préparer au moment de l'emploi à partir de mélanges d'alcools purs.
4.17. 1-eicosanol, solution à 0,1 % (m/V) dans le chloroforme (standard interne).
4.18. Gaz vecteur: hydrogène ou hélium pur, pour chromatographie en phase gazeuse.
4.19. Gaz auxiliaire: nitrogène pur, pour chromatographie en phase gazeuse.
5. PROCÉDÉ
5.1. Préparation de l'insaponifiable
|
5.1.1. |
Introduire dans le ballon de 250 millilitres, au moyen de la microseringue de 500 microlitres, un volume de solution d'1-eicosanol à 0,1 % dans le chloroforme (4.17) qui contient une quantité d'1-eicosanol qui corresponde à environ 10 % du contenu en alcools aliphatiques dans l'aliquote de l'échantillon à prélever pour la détermination. Par exemple, pour 5 grammes d'échantillon, il faut ajouter 250 microlitres de la solution d'1-eicosanol à 0,1 % s'il s'agit d'un échantillon d'huile d'olive et 1 500 microlitres s'il s'agit d'huile de grignons d'olive. Evaporer dans un courant d'azote jusqu'à dessiccation, puis peser exactement 5 grammes d'échantillon sec et filtré dans le même ballon. |
|
5.1.2. |
Ajouter 50 millilitres de solution éthanolique d'hydroxyde de potassium à 2 N, mettre en marche le réfrigérant à reflux, chauffer au bain-marie jusqu'à légère ébullition tout en maintenant une agitation énergique jusqu'à ce que la saponification se soit produite (la solution devient limpide). Continuer à réchauffer pendant 20 minutes, puis ajouter 50 millilitres d'eau distillée que l'on fait descendre du haut du réfrigérant, débrancher le réfrigérant et refroidir le ballon à environ 30 °C. |
|
5.1.3. |
Transvaser le contenu du ballon, de façon quantitative, dans une ampoule à décanter de 500 millilitres, en s'aidant d'eau distillée à plusieurs reprises, en utilisant au total environ 50 millilitres. Ajouter environ 80 millilitres d'éther éthylique, agiter énergiquement durant environ 30 secondes et laisser la séparation se faire (note 1). Séparer la phase aqueuse inférieure en la recueillant dans une autre ampoule à décanter. Faire encore deux extractions sur la phase aqueuse, selon les mêmes modalités, en utilisant à chaque fois 60 à 70 millilitres d'éther éthylique. Note 1: Des éventuelles émulsions peuvent être éliminées en ajoutant, avec une pissette, une petite quantité d'alcool éthylique ou méthylique. |
|
5.1.4. |
Réunir les extraits éthérés dans une seule ampoule à décanter et laver à l'eau distillée (50 millilitres à chaque fois) jusqu'à réaction neutre de l'eau de lavage. Éliminer l'eau de lavage, dessécher au sulfate de sodium anhydre et filtrer, sur sulfate de sodium anhydre, dans un ballon de 250 millilitres pesé au préalable, en lavant ampoule et filtre avec de petites quantités d'éther éthylique. |
|
5.1.5. |
Distiller l'éther jusqu'à ce qu'il n'en reste qu'une petite quantité, puis porter à sec sous un léger vide ou dans un courant d'azote, parfaire le séchage à l'étuve à 100 °C durant un quart d'heure environ et peser après refroidissement dans un dessiccateur. |
5.2. Séparation de la fraction alcoolique
|
5.2.1. |
Préparation des plaques basiques: immerger les plaques au gel de silice (4.4), complètement, dans la solution éthanolique 0,2 N d'hydroxyde de potassium (4.5) durant 10 secondes, laisser agir ensuite; bien sécher sous hotte aspirante, pendant deux heures et mettre finalement à l'étuve à 100 °C pendant une heure. Retirer de l'étuve et conserver dans un dessiccateur à chlorure de calcium jusqu'au moment de l'emploi (les plaques ainsi traitées doivent être employées dans les quinze jours). Note 2: L'emploi des plaques de gel de silice basiques pour la séparation de la fraction alcoolique élimine le besoin du traitement de l'insaponifiable avec l'alumine. De cette manière, tous les composés de nature acide (acides gras et autres) sont retenus sur la ligne de dépôt. On obtient ainsi la bande des alcools aliphatiques et terpéniques nettement séparée de la bande des stérols. |
|
5.2.2. |
Introduire dans la cuve de développement un mélange hexane-éther éthylique 65/35 (V/V) jusqu'à une hauteur d'environ 1 centimètre ( 12 ). Fermer la cuve à l'aide d'un couvercle approprié et laisser ainsi pendant une demi-heure au moins, de façon à ce que l'équilibre liquide/vapeur s'établisse. Il est possible de fixer sur les surfaces intérieures de la cuve des bandes de papier filtre qui plongent dans l'éluant: cette précaution permet de réduire d'un tiers environ les temps de migration du front du liquide et d'obtenir une élution plus uniforme des composants. Note 3: Afin d'avoir des conditions d'élution parfaitement reproductibles, le mélange doit être changé à chaque essai. |
|
5.2.3. |
Préparer une solution à 5 % environ d'insaponifiable (5.1.5) dans le chloroforme et, avec la microseringue de 100 microlitres, déposer sur la plaque chromatographique (5.2.1) à 2 centimètres environ d'un bord, 0,3 millilitre de la solution susdite en une ligne continue, la plus fine et uniforme possible. Dans l'alignement de la ligne de dépôt, déposer, à une des extrémités de la plaque, 2 à 3 microlitres de la solution de référence des alcools aliphatiques (4.11), dans le but d'identifier la bande des alcools aliphatiques lors du dernier développement. |
|
5.2.4. |
Mettre la plaque dans la cuve de développement, préparée comme décrit au point 5.2.2. La température ambiante doit être maintenue entre 15 et 20 °C. Fermer aussitôt la cuve avec le couvercle et laisser éluer jusqu'à ce que le front de solvant arrive à environ 1 centimètre du bord supérieur de la plaque. Enlever ensuite la plaque de la cuve de développement et faire évaporer le solvant dans un courant d'air chaud ou bien en laissant la plaque sécher sous hotte aspirante pendant un petit moment. |
|
5.2.5. |
Vaporiser la plaque légèrement et uniformément avec la solution de dichloro-2′-7′ fluorescéine lorsque la plaque est observée à l'ultra-violet. Identifier la bande des alcools aliphatiques par alignement avec la tache obtenue avec la solution de référence; délimiter la bande avec un crayon noir, en indiquant l'ensemble de la bande des alcools aliphatiques et la bande immédiatement supérieure qui correspond aux alcools terpéniques (note 4). Note 4: les bandes des alcools aliphatiques et des alcools terpéniques doivent être regroupées du fait d'une migration éventuelle de certains alcools aliphatiques dans la bande des alcools triterpéniques. La figure 1 de l'appendice présente un exemple de la séparation par chromatographie en couche mince. |
|
5.2.6. |
Racler avec une spatule métallique le gel de silice compris dans la zone délimitée. Le matériau retiré, finement broyé, est introduit dans l'ampoule filtrante (3.7); ajouter 10 millilitres de chloroforme chaud, mélanger soigneusement avec la spatule métallique et filtrer à l'aide du vide, puis recueillir le filtrat dans la fiole conique (3.8), reliée à l'ampoule filtrante. Laver le gel de silice dans la fiole par trois fois à l'éther éthylique (environ 10 ml à chaque fois) et recueillir le filtrat dans la même fiole reliée à l'ampoule filtrante. Évaporer le filtrat jusqu'à l'amener à un volume d'environ 4 à 5 millilitres, transvaser la solution résiduelle dans le tube de 10 millilitres (3.9) pesé au préalable, porter à sec en chauffant légèrement dans un léger courant d'azote, reprendre avec quelques gouttes d'acétone, amener à nouveau à sec, mettre 10 minutes environ à l'étuve à 105 °C, puis laisser refroidir au dessiccateur et peser. Le résidu contenu dans le tube est constitué de la fraction alcoolique. |
5.3. Préparation des triméthylsilyléthers
|
5.3.1. |
Ajouter, dans le tube contenant la fraction alcoolique, le réactif de silylation, constitué d'un mélange de pyridine-hexaméthyldisilazane-triméthylchlorosilane 9/3/1 (V/V/V) (note 5) dans une proportion de 50 microlitres par milligramme d'alcools aliphatiques, en évitant toute absorption d'humidité (note 6). Note 5: Il existe dans le commerce des solutions prêtes à l'emploi; en outre, d'autres réactifs silanisants, tels que, par exemple, le bis-triméthylsilyltrifluoracétamide + 1 % de triméthylchlorosilane à diluer par un même volume de pyridine anhydre. Note 6: La formation éventuelle d'une légère opalescence est normale et n'est la cause d'aucun dérangement. La formation d'une floculation blanche ou l'apparition d'une coloration rose sont l'indice de la présence d'humidité ou d'altération du réactif. Dans ce cas, l'essai doit être répété. |
|
5.3.2. |
Boucher le tube, agiter soigneusement (sans retourner) jusqu'à complète solubilisation des alcools aliphatiques. Laisser reposer pendant au moins 15 minutes à température ambiante, puis centrifuger pendant quelques minutes: la solution limpide est prête pour l'analyse par chromatographie en phase gazeuse. |
5.4. Analyse par chromatographie en phase gazeuse
5.4.1. Opérations préliminaires, conditionnement de la colonne
|
5.4.1.1. |
Installer la colonne dans le chromatographe en phase gazeuse, en reliant l'extrémité d'entrée à l'injecteur connecté au système de fractionnement et l'extrémité de sortie au détecteur. Effectuer les contrôles généraux du complexe de chromatographie en phase gazeuse (étanchéité du circuit des gaz, efficacité du détecteur, efficacité du système de fractionnement et du système d'enregistrement, etc.). |
|
5.4.1.2. |
Si la colonne est utilisée pour la première fois, il est conseillé de procéder à son conditionnement. Faire passer un léger flux de gaz au travers de cette colonne, puis allumer le complexe de chromatographie en phase gazeuse et commencer un chauffage graduel jusqu'à atteindre une température d'au moins 20 °C supérieure à celle d'exercice (note 7). Maintenir cette température pendant au moins 2 heures, puis porter le complexe aux conditions de fonctionnement (régulation du flux gazeux et de la séparation, allumage de la flamme, jonction avec l'enregistreur électronique, régulation de la température de la chambre pour la colonne, du détecteur et de l'initiateur etc.) et enregistrer le signal à une sensibilité au moins deux fois supérieure à celle prévue pour l'exécution de l'analyse. Le tracé de la ligne de base obtenue doit être linéaire, exempt de pic de quelque nature que ce soit et ne doit pas présenter de dérive. Une dérive rectiligne négative indique une étanchéité imparfaite des connexions de la colonne, une dérive positive indique un conditionnement insuffisant de la colonne. Note 7: La température de conditionnement doit être toujours inférieure d'au moins 20 °C à la température maximale prévue pour le liquide de répartition employé. |
5.4.2. Choix des conditions opératoires
5.4.2.1. Les conditions opératoires indicatives sont les suivantes:
— température de la colonne: début isotherme 8 minutes à 180 °C, puis programme de 5 °C par minute jusqu'à 260 °C puis encore 15 minutes à 260 °C,
— température de l'évaporateur: 280 °C,
— température du détecteur: 290 °C,
— vitesse linéaire du gaz vecteur: hélium, 20 à 35 centimètres par seconde; hydrogène, 30 à 50 centimètres par seconde,
— rapport de division: de 1/50 à 1/100,
— sensibilité instrumentale: de 4 à 16 fois l'atténuation minimale,
— sensibilité d'enregistrement: 1 à 2 millivolts sur échelle de fond,
— vitesse du papier: 30 à 60 centimètres par heure,
— quantité de substance injectée: 0,5 à 1 microlitre de solution de TMSE.
Ces conditions peuvent être modifiées en fonction des caractéristiques de la colonne et du chromatographe en phase gazeuse de façon à obtenir des chromatogrammes qui satisfassent aux conditions suivantes:
— le temps de rétention de l'alcool en C26 doit être de 18 ± 5 minutes,
— le pic de l'alcool en C22 doit être 80 ± 20 % de l'échelle de fond pour l'huile d'olive et pour l'huile de semences 40 ± 20 % de l'échelle de fond.
5.4.2.2. Pour vérifier les conditions exigées ci-dessus, effectuer des injections répétées avec les échantillons de mélanges des TMSE des alcools et retoucher les conditions opératoires jusqu'à obtenir les meilleurs résultats.
5.4.2.3. Les paramètres d'intégration des pics doivent être imposés de façon à obtenir des valeurs correctes pour les pics qui sont pris en considération.
5.4.3. Exécution de l'analyse
5.4.3.1. Prélever, avec la microseringue de 10 microlitres, 1 microlitre d'hexane, aspirer 0,5 microlitre d'air et successivement 0,5 à 1 microlitre de la solution de l'échantillon; tirer encore le piston de la seringue de façon à ce que l'aiguille soit vide. Introduire l'aiguille au travers de la membrane du complexe d'injection et, après 1 à 2 secondes, injecter rapidement et extraire ensuite l'aiguille lentement, après 5 secondes environ.
5.4.3.2. Procéder à l'enregistrement jusqu'à élution complète des TMSE des alcools aliphatiques présents. La ligne de base doit toujours correspondre aux conditions requises (5.4.1.2):
5.4.4. Identification des pics
L'identification des pics individuels est effectuée sur la base des temps de rétention et par comparaison avec le mélange des TMSE des alcools aliphatiques, analysés dans les mêmes conditions.
Les figures 2 et 3 de l'appendice montrent un chromatogramme de la fraction alcoolique d'une huile d'olive raffinée.
5.4.5. Évaluation quantitative
5.4.5.1. Procéder, avec l'intégrateur, au calcul de l'aire des pics de l'1-eicosanol et des alcools aliphatiques C22, C24, C26 et C28.
5.4.5.2. Calculer le contenu en chaque alcool aliphatique individuel, en milligrammes pour
1 000
grammes de matière grasse, comme suit:
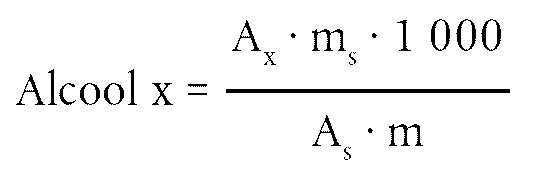
où:
Ax=aire du pic de l'alcools x
As=aire du pic d'1-eicosanol
ms=poids d'1-eicosanol ajouté, en milligrammes
m =poids de l'échantillon prélevé pour la détermination, en grammes.
6. EXPRESSION DES RÉSULTATS
Rapporter les contenus en alcools aliphatiques simples en milligrammes pour 1 000 grammes de matière grasse et leur somme comme «alcools aliphatiques totaux».
Appendice
Exemple de séparation par chromatographie en couche mince et exemples de chromatogramme
Figure 1
Chromatographie en couche mince de la fraction insaponifiable de l'huile d'olive éluée avec de l'hexane/éther éthylique (65/35)

|
1 |
Alcool C26 |
A |
Stérols |
|
2 |
Alcool C30 |
B |
Alcools aliphatiques |
|
3 |
Alcool C20 |
C |
Alcools triterpéniques |
|
4 |
Mélange alcools C20-22-26-30 |
D |
Squalène |
|
5 |
Vierge extra insaponifiable |
|
|
Figure 2
Chromatogramme de la fraction alcoolique d'une huile d'olive raffinée
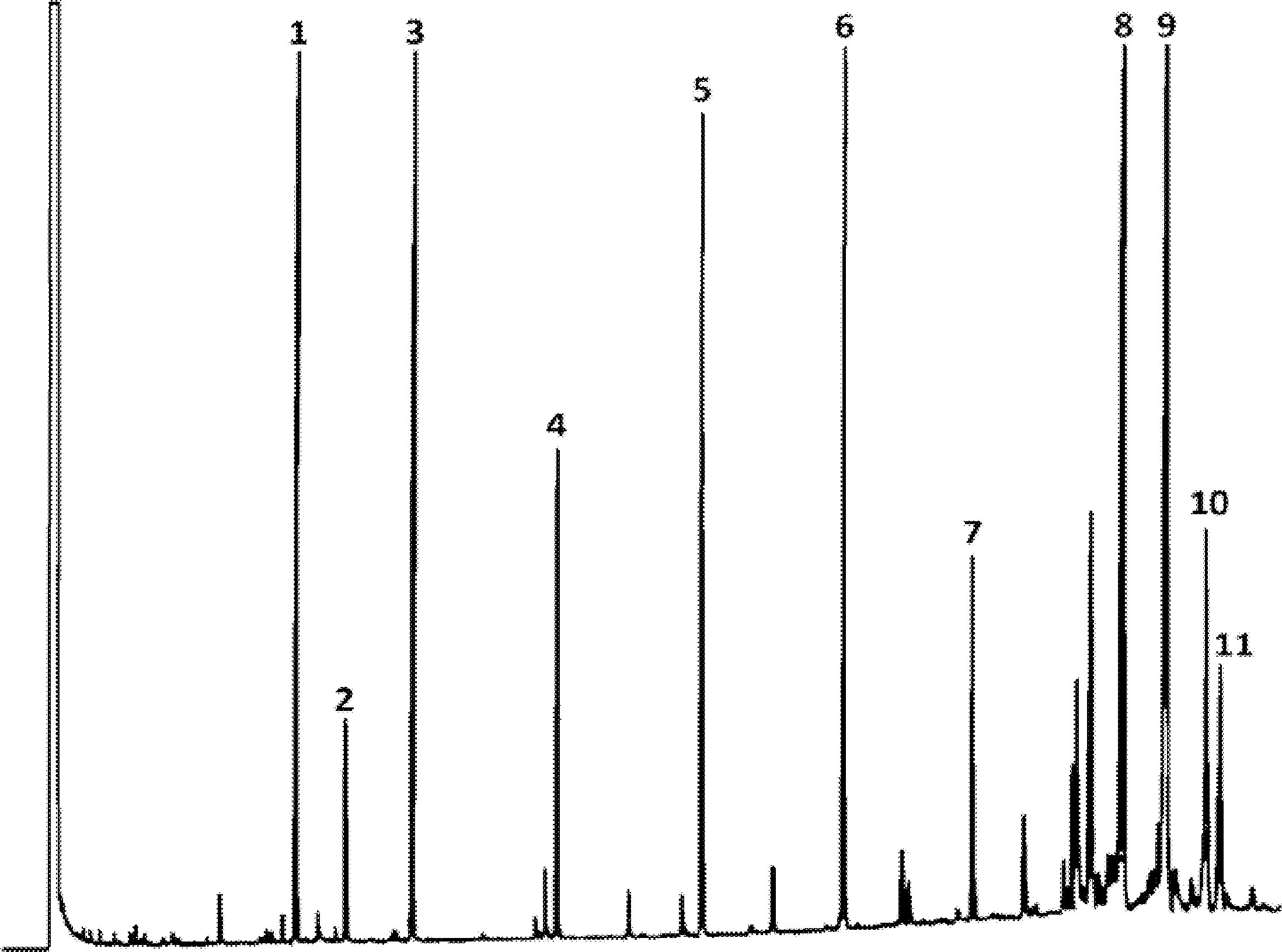
|
1 = Phytol |
5 = Alcool C24 |
9 = 24-Méthylène-cycloartenol |
|
2 = Géranyl géraniol |
6 = Alcool C26 |
10 = Citrostadiénol |
|
3 = Alcool C20 (IS) |
7 = Alcool C28 |
11 = Cyclobranol |
|
4 = Alcool C22 |
8 = Cycloarténol |
|
Figure 3
Alcools aliphatiques et triterpéniques d'une huile d'olive raffinée et d'une huile d'olive de deuxième centrifugation
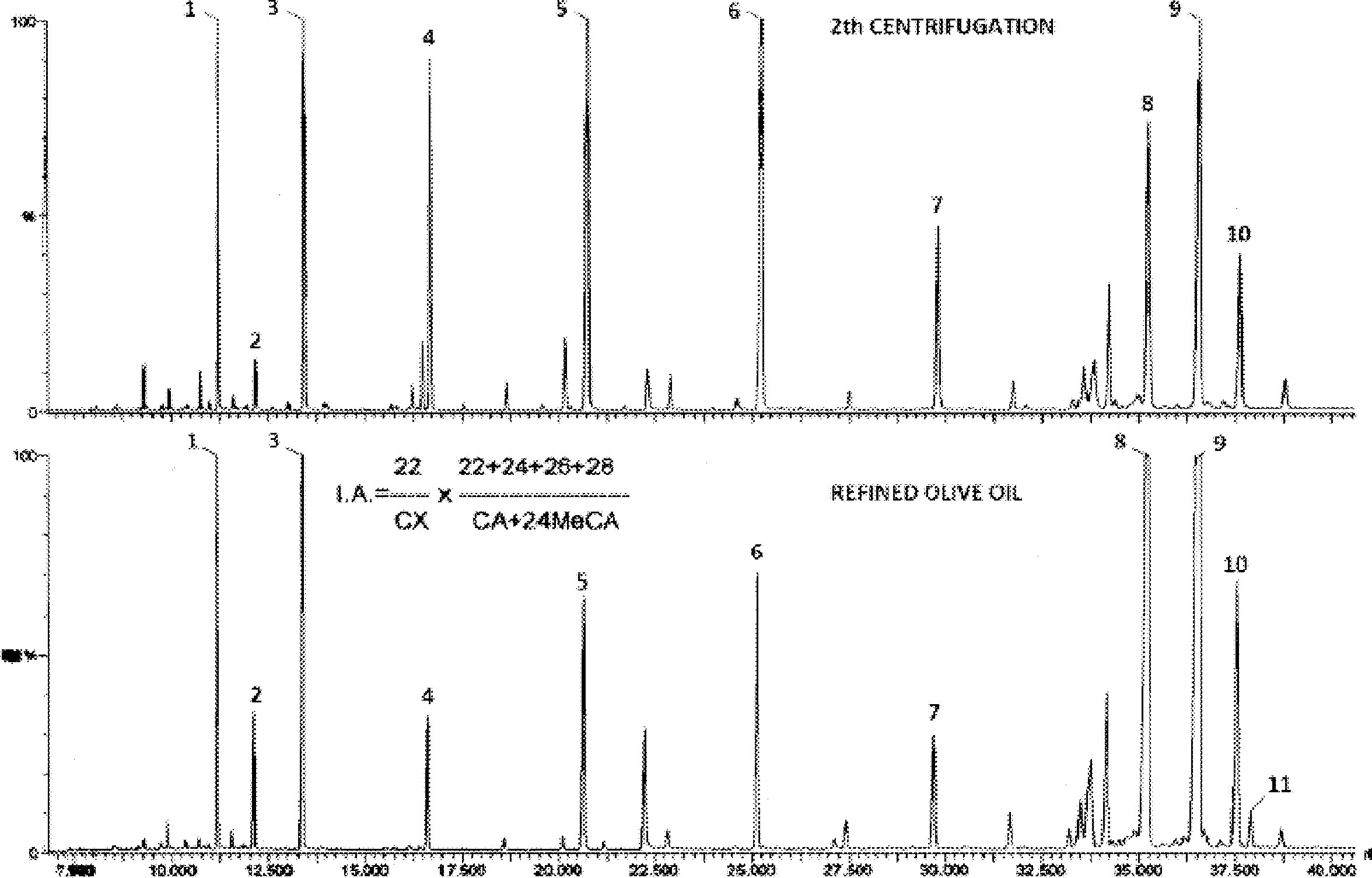
|
1 = Phytol |
5 = Alcool C24 |
9 = 24-Méthylène-cycloarténol (24MeCA) |
|
2 = Géranyl géraniol (CX) |
6 = Alcool C26 |
10 = Citrostadiénol |
|
3 = Alcool C20 |
7 = Alcool C28 |
11 = Cyclobranol |
|
4 = Alcool C22 |
8 = Cycloarténol (CA) |
|
ANNEXE XX
Méthode de détermination de la teneur en cires et en esters méthyliques et éthyliques d'acides gras par chromatographie gazeuse sur colonne capillaire
1. OBJET
Cette méthode décrit un procédé pour déterminer la teneur en cires et en esters méthyliques et éthyliques d'acides gras dans les huiles d’olive. Les cires et les alkyls esters sont séparés en fonction du nombre d’atomes de carbone. La méthode peut être employée notamment pour différencier l’huile d’olive de l’huile de grignons d’olive et comme paramètre de qualité pour les huiles d’olive vierges extra, dans la mesure où elle permet la détection des mélanges frauduleux d’huiles d’olive vierge extra avec des huiles de qualité inférieure, notamment les huiles d’olive vierges, lampantes ou désodorisées.
2. PRINCIPE
L'huile, additionnée d'étalons internes appropriés, est fractionnée par chromatographie sur colonne de gel de silice hydraté; la fraction éluée dans les conditions de l’essai (à polarité inférieure à celle des triglycérides) est recueillie puis analysée directement par chromatographie en phase gazeuse sur colonne capillaire.
3. APPAREILLAGE
|
3.1. |
Erlenmeyer de 25 ml. |
|
3.2. |
Colonne en verre pour chromatographie en phase liquide, diamètre intérieur 15,0 mm, hauteur 30 à 40 cm, équipée d’un robinet approprié. |
|
3.3. |
Appareil de chromatographie en phase gazeuse approprié pour le fonctionnement avec colonne capillaire, équipé d’un système d'injection directe sur colonne, constitué des éléments suivants:
|
|
3.4. |
Microseringue de 10 μl, équipée d’une aiguille en acier trempé, pour injection directe sur colonne. |
|
3.5. |
Agitateur électrique. |
|
3.6. |
Évaporateur rotatif. |
|
3.7. |
Four à moufle. |
|
3.8. |
Balance analytique garantissant une précision de la mesure de ± 0,1 mg. |
|
3.9. |
Verrerie normale de laboratoire. |
4. RÉACTIFS
|
4.1. |
Gel de silice, d’une granulométrie comprise entre 60 et 200 μm mesh [NdT: les deux unités de mesure coexistent dans l'original]. Le gel de silice doit être placé dans le four à moufle à 500 °C pendant au moins 4 h. Après refroidissement, y ajouter 2 % d’eau par rapport à la quantité de gel de silice utilisée. Agiter convenablement afin d’homogénéiser la suspension. Conserver dans le dessiccateur pendant au moins 12 heures avant emploi. |
|
4.2. |
n-hexane, pour chromatographie ou analyse de résidus (la pureté doit être vérifiée). AVERTISSEMENT: risques d’inflammation des vapeurs. Tenir à l’écart de sources de chaleur, étincelles ou flammes nues. Bien fermer les récipients et utiliser dans un local bien aéré. Éviter l’accumulation de vapeurs et éliminer toute cause possible d’incendie, telle que radiateurs et appareils électriques non antidéflagrants. Nocif par inhalation: peut nuire aux cellules du système nerveux. Éviter de respirer les vapeurs. Utiliser si nécessaire un appareil respiratoire adéquat. Éviter tout contact avec les yeux et la peau. |
|
4.3. |
Éther éthylique, pour chromatographie.
AVERTISSEMENT: extrêmement inflammable. Modérément toxique. Irritant pour la peau. Nocif par inhalation. Peut être nuisible pour les yeux. Les effets peuvent être différés. Risque de formation de peroxydes explosifs. Risques d’inflammation des vapeurs. Tenir à l’écart de sources de chaleur, étincelles ou flammes nues. Bien fermer les récipients et utiliser dans un local bien aéré. Éviter l’accumulation de vapeurs et éliminer toute cause possible d’incendie, telle que radiateurs et appareils électriques non antidéflagrants. Ne pas évaporer jusqu’à dessiccation totale ou quasi totale. L’adjonction d’eau ou d’un autre agent réducteur approprié peut réduire la formation des peroxydes. Ne pas ingérer. Éviter de respirer les vapeurs. Éviter le contact prolongé ou répété avec la peau. |
|
4.4. |
n-heptane, pour chromatographie, ou iso-octane. AVERTISSEMENT: inflammable. Nocif par inhalation. Tenir à l’écart de sources de chaleur, étincelles ou flammes nues. Bien fermer les récipients et utiliser dans un local bien aéré. Éviter de respirer les vapeurs. Éviter le contact prolongé ou répété avec la peau. |
|
4.5. |
Solution étalon d’arachidate laurique (Note 3), diluée à 0,05 % (m/V) dans de l’heptane (étalon interne pour cires). Note 3: Il est également possible d’utiliser du palmitate de palmityle, du stéarate de myristyle ou du lauréate d’arachidyle. |
|
4.6. |
Solution étalon d'heptadécanoate méthylique, diluée à 0,02 % (m/V) dans de l’heptane (étalon interne pour esters méthyliques et éthyliques). |
|
4.7. |
Soudan 1 (1-phényl-azo-2-naphthol) |
|
4.8. |
Gaz vecteur: hydrogène ou hélium, pur, pour chromatographie en phase gazeuse. AVERTISSEMENT: Hydrogène. extrêmement inflammable, sous pression. Tenir à l’écart de sources de chaleur, étincelles ou flammes nues ou d’appareils électriques non antidéflagrants. S’assurer que la soupape de la bouteille est bien fermée lorsqu’elle n’est pas utilisée. Utiliser exclusivement avec un réducteur de pression. Desserrer le ressort du réducteur avant l’ouverture de la soupape de la bouteille. S’éloigner du point de sortie du gaz de la bouteille au moment de l’ouverture de la soupape. Utiliser dans un local bien aéré. Ne pas transférer l’hydrogène d’une bouteille à une autre. Ne pas mélanger de gaz dans la bouteille. S’assurer que les bouteilles ne risquent pas de tomber. Éloigner les bouteilles des rayons du soleil ou de toute autre source de chaleur. Ne pas stocker dans des espaces corrosifs. Ne pas utiliser de bouteilles abîmées ou sans étiquette. Hélium. comprimé sous haute pression. Réduit l’oxygène disponible pour la respiration. Conserver le conteneur fermé. Utiliser dans un local bien aéré. Ne pas entrer dans les locaux de stockage s’ils ne sont pas bien ventilés. Utiliser exclusivement avec un réducteur de pression. Desserrer le ressort du réducteur avant l’ouverture de la soupape de la bouteille. Ne pas transférer le gaz d’une bouteille à une autre. S’assurer que les bouteilles ne risquent pas de tomber. S’éloigner du point de sortie du gaz de la bouteille au moment de l’ouverture de la soupape. Éloigner les bouteilles des rayons du soleil ou de toute autre source de chaleur. Ne pas stocker dans des espaces corrosifs. Ne pas utiliser de bouteilles abîmées ou sans étiquette. Ne pas inhaler. Utiliser exclusivement à des fins techniques. |
|
4.9. |
Gaz auxiliaires: — hydrogène pur, pour chromatographie en phase gazeuse. — air pur, pour chromatographie en phase gazeuse. AVERTISSEMENT: Air. Comprimé sous haute pression. Utiliser avec précaution en présence de substances combustibles car la température d’auto-allumage de la plupart des composés organiques dans l’air diminue fortement à haute pression. S’assurer que la soupape de la bouteille est bien fermée lorsqu’elle n’est pas utilisée. Utiliser exclusivement avec un réducteur de pression. Desserrer le ressort du réducteur avant l’ouverture de la soupape de la bouteille. S’éloigner du point de sortie du gaz de la bouteille au moment de l’ouverture de la soupape. Ne pas transférer le gaz d’une bouteille à une autre. Ne pas mélanger de gaz dans la bouteille. S’assurer que les bouteilles ne risquent pas de tomber. Éloigner les bouteilles des rayons du soleil ou de toute autre source de chaleur. Ne pas stocker dans des espaces corrosifs. Ne pas utiliser de bouteilles abîmées ou sans étiquette. Ne pas inhaler et ne pas utiliser pour des appareils respiratoires l’air destiné à des fins techniques. |
5. MODE OPÉRATOIRE
5.1. Préparation de la colonne chromatographique
Mettre 15 g de gel de silice (point 4.1) en suspension dans le n-hexane (point 4.2) et l’introduire dans la colonne (point 3.2). Laisser se déposer spontanément, puis utiliser un agitateur électrique pour rendre la couche chromatographique plus homogène. Filtrer 30 ml de n-hexane afin d’éliminer les impuretés éventuelles. Peser exactement à l’aide de la balance analytique (point 3.8) environ 500 mg de l’échantillon dans l’Erlenmeyer de 25 ml (point 3.1). Ajouter une quantité appropriée d’étalon interne (point 4.5), en fonction du contenu présumé de cires. Par exemple, ajouter 0,1 mg d’arachidate laurique dans le cas de l’huile d’olive et 0,25 à 0,5 mg dans le cas de l’huile de grignons d'olive et 0,05 mg de heptadécanoate méthylique dans le cas des huiles d’olive (point 4.6).
Introduire l’échantillon ainsi préparé dans la colonne chromatographique à l’aide de deux fractions de 2 ml de n-hexane (point 4.2.).
Laisser couler le solvant jusqu’à 1 mm au-dessus du niveau supérieur de l’absorbant. Filtrer [NdT: passage manquant dans l'original] supplémentaires de n-hexane/éther éthylique (99:1) et recueillir 220 ml à un débit d’environ 15 gouttes toutes les 10 secondes. (Cette fraction contient les esters méthyliques et éthyliques et les cires). (Note 4) (Note 5).
Note 4: Un nouveau mélange n-hexane/éther éthylique (99:1) doit être préparé chaque jour.
Note 5: Pour contrôler visuellement l’élution correcte des cires, il est possible d’ajouter à la solution échantillon 100μl de colorant soudan I dilué à 1 % dans le mélange d’élution.
Le colorant ayant un temps de rétention compris entre celui des cires et des triglycérides, il convient de suspendre l’élution lorsque le colorant atteint le fond de la colonne chromatographique, car toutes les cires ont alors été éluées.
Évaporer les fractions ainsi obtenues dans l’évaporateur rotatif jusqu’à l'évacuation pratiquement totale du solvant. Les 2 derniers ml sont évacués avec un faible flux d’azote. Recueillir la fraction contenant les esters méthyliques et éthyliques est diluée avec 2 à 4 ml de n-heptane ou d'iso-octane [NdT: cette phrase est entachée d'une erreur de syntaxe dans l'original].
5.2. Analyse par chromatographie en phase gazeuse
5.2.1. Opérations préliminaires
Connecter la colonne et le chromatographe en phase gazeuse (point 3.3), en raccordant, d'une part, le point d'admission au système sur colonne et, d'autre part, le point de sortie au détecteur. Vérifier le bon fonctionnement de l’appareillage de chromatographie en phase gazeuse (fonctionnement des circuits des gaz, efficacité du système de détection et d'enregistrement, etc.).
Si la colonne est utilisée pour la première fois, il est recommandé de procéder à son conditionnement. Laisser s’écouler un léger débit de gaz à travers la colonne, puis allumer l’appareil de chromatographie en phase gazeuse. Chauffer graduellement jusqu’à atteindre, au bout d’environ 4 heures, une température de 350°C.
Maintenir cette température pendant au moins 2 heures, puis régler l’appareillage sur les conditions opératoires (réglage du débit de gaz, allumage de la flamme, raccordement à l’enregistreur électronique (point 3.3.4), réglage de la température du four en fonction de la colonne, réglage du détecteur, etc.) et enregistrer le signal à une sensibilité au moins deux fois supérieure à celle requise pour l’analyse. Le tracé de la ligne de base doit être linéaire, exempt de tout pic, et ne doit présenter aucune déviation.
Une déviation rectiligne négative indique que les raccordements de la colonne sont mauvais; une déviation positive indique un conditionnement inadéquat de la colonne.
5.2.2. Choix des conditions opératoires pour les cires et les esters éthyliques et méthyliques (Note 6)
Les conditions opératoires sont en général les suivantes:
|
— |
température de la colonne : 20 °C/min 5 °C/min 80 °C au départ (1 minute) |
|
— |
température du détecteur : 350 °C. |
|
— |
quantité injectée : 1μl de solution de n-heptane (2-4 ml). |
|
— |
gaz vecteur : hélium ou hydrogène à la vitesse linéaire optimale pour le gaz choisi (voir Appendice A). |
|
— |
sensibilité instrumentale : telle qu'elle permet de satisfaire aux conditions ci-dessus. |
Note 6: En raison de la température finale élevée, on admet une déviation positive qui ne doit toutefois pas être supérieure à 10 % de la valeur réelle.
Ces conditions peuvent être modifiées de façon à les adapter aux caractéristiques de la colonne et de l’appareil de chromatographie en phase gazeuse, afin de séparer toutes les cires et tous les esters éthyliques et méthyliques d'acides gras et d'obtenir une séparation satisfaisante des pics (voir figures 2, 3 et 4) et un temps de rétention de l’étalon interne d'arachidate laurique de 18 ± 3 minutes. Le pic des cires le plus représentatif doit être supérieur à 60 % de l'amplitude réelle, tandis que l’étalon interne d'heptadécanoate méthylique pour les esters méthyliques et éthyliques doit atteindre l'amplitude réelle.
Les paramètres d’intégration des pics doivent être déterminés de façon à obtenir une évaluation correcte des aires des pics pris en considération.
5.3. Exécution de l’analyse
Prélever 10 μl de la solution à l’aide de la microseringue de 10 μl; tirer le piston de la seringue en arrière jusqu'à ce que l’aiguille soit vide. Introduire l’aiguille dans le dispositif d’injection et après 1-2 secondes, injecter rapidement. Au bout d’environ 5 secondes, extraire délicatement l’aiguille.
Effectuer l’enregistrement jusqu’à l'élution complète des cires ou des stigmastadiènes, selon la fraction analysée.
La ligne de base doit toujours satisfaire aux conditions requises.
5.4. Identification des pics
Les différents pics sont identifiés en comparant les temps de rétention et les mélanges de cires dont les temps de rétention sont connus du fait qu'ils ont été analysés dans les mêmes conditions. Les alkyl esters sont identifiés à partir de mélanges d’esters méthyliques et éthyliques des principaux acides gras contenus dans l’huile d’olive (palmitique et oléique).
La Figure 1 représente un chromatogramme des cires d’une huile d’olive vierge. Les figures 2 et 3 représentent les chromatogrammes de deux huiles d’olive vierges extra disponibles dans le commerce, l’une contenant des esters méthyliques et éthyliques et l’autre non. La figure 4 représente le chromatogramme d’une huile d’olive vierge extra de qualité optimale et le chromatogramme de la même huile, mais qui contient des pics correspondant à 20 % d’huile désodorisée.
5.5. Analyse quantitative des cires
Procéder au calcul de l'aire des pics correspondant à l’étalon interne d'arachidate laurique et aux esters aliphatiques de C40 à C46 à l’aide de l’intégrateur.
Calculer la teneur totale en cires en additionnant chaque cire, en mg/kg de matière grasse, par la formule:

où:
|
Ax |
= |
aire correspondant au pic de chaque ester, en données de comptage sous forme numérique |
|
As |
= |
aire correspondant au pic de l’étalon interne d'arachidate laurique, en données de comptage sous forme numérique |
|
ms |
= |
masse de l’étalon interne d'arachidate laurique ajouté, en milligrammes |
|
m |
= |
masse de l’échantillon prélevé pour la détermination, en grammes. |
5.5.1. Analyse quantitative des esters méthyliques et éthyliques
Procéder au calcul des aires des pics correspondant à l’étalon interne d'heptadécanoate méthylique, aux esters méthyliques des acides gras C16 et C18 et aux esters éthyliques des acides gras C16 et C18 à l’aide de l’intégrateur.
Calculer la teneur de chacun des alkyl esters, en mg/kg de matière grasse, par la formule:

où:
|
Ax |
= |
aire correspondant au pic de chaque ester C16 et C18, en données de comptage sous forme numérique |
|
As |
= |
aire correspondant au pic de l’étalon interne d'heptadécanoate méthylique, en données de comptage sous forme numérique |
|
ms |
= |
masse de l’étalon interne d'heptadécanoate méthylique ajouté, en milligrammes |
|
m |
= |
masse de l’échantillon prélevé pour la détermination, en grammes. |
6. EXPRESSION DES RÉSULTATS
Indiquer la somme des teneurs en cires de C40 à C46 (Note 7), en mg/kg de matière grasse.
Indiquer la somme des teneurs en esters méthyliques et éthyliques de C16 à C18, et le total des deux.
Il convient d'indiquer les résultats en arrondissant au mg/kg inférieur ou supérieur.
Note 7: Les constituants à quantifier correspondent aux pics des esters C40 à C46 ayant un nombre d'atomes de carbone pair, selon l’exemple de chromatogramme des cires contenues dans une huile d’olive reproduit dans la figure ci-après. À des fins d'identification, si l’ester C46 est scindé, il est conseillé d’analyser la fraction des cires d’une huile de grignons d’olive dont le pic C46 est clairement identifiable du fait qu'il prédomine.
Indiquer le ratio entre les esters éthyliques et les esters méthyliques.
Figure 1
Chromatogramme de la fraction de cires d’une huile d’olive, à titre d'exemple ( 13 )
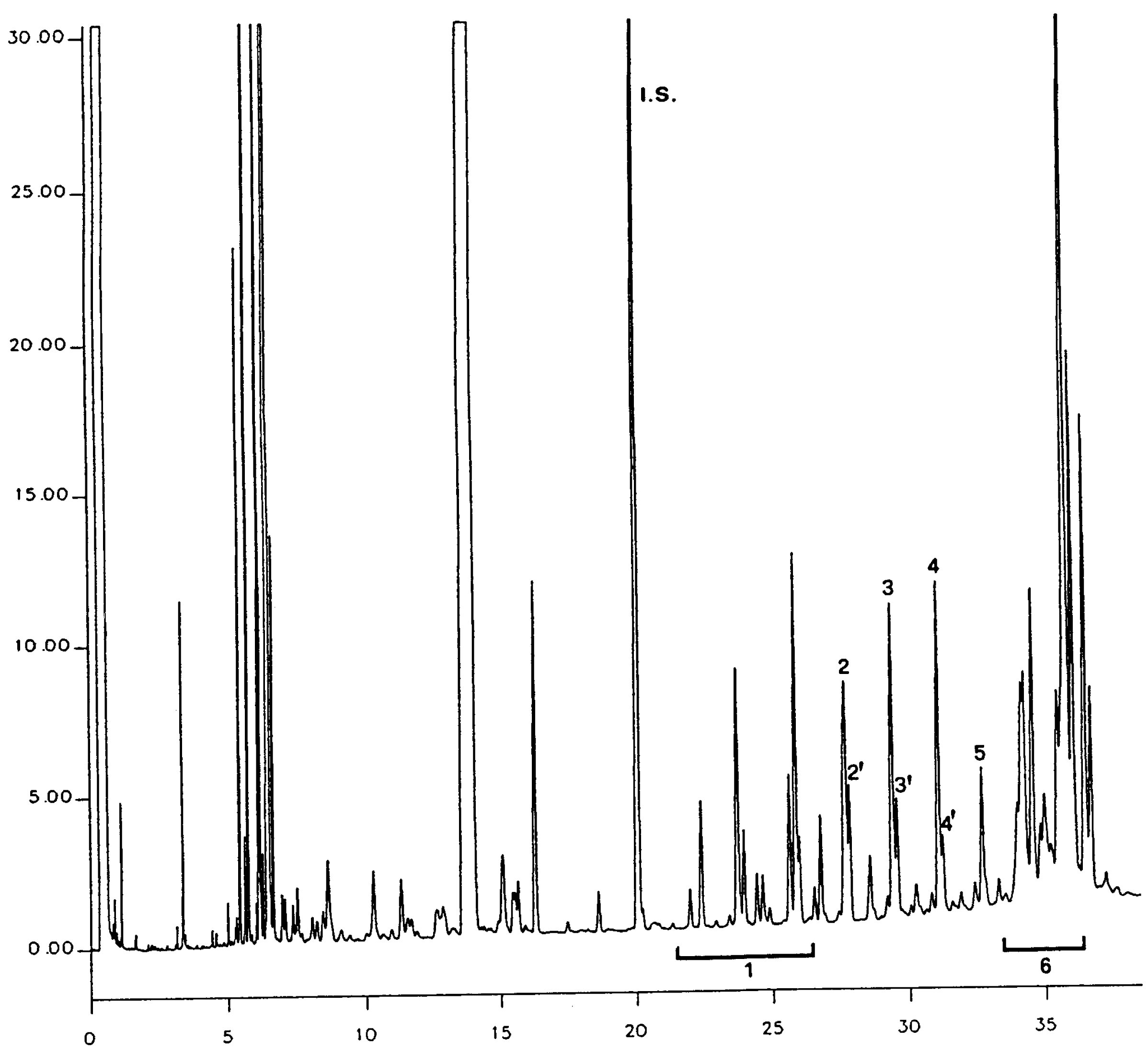
Pics avec un temps de rétention de 5 à 8 min des esters éthyliques et méthyliques d'acides gras
Légende:
|
I.S. |
= |
Arachidate laurique |
|
1 |
= |
Esters diterpéniques |
|
2+2′ |
= |
esters C40 |
|
3+3′ |
= |
esters C42 |
|
4+4′ |
= |
esters C44 |
|
5 |
= |
esters 46 |
|
6 |
= |
Esters stéroliques et alcools triterpéniques |
Figure 2
Esters méthyliques, esters éthyliques et cires dans une huile d’olive vierge

Légende:
|
1 |
– |
Méthyl C16 |
|
2 |
– |
Éthyl C16 |
|
3 |
– |
Heptadécanoate méthylique I.S. |
|
4 |
– |
Méthyl C18 |
|
5 |
– |
Éthyl C18 |
|
6 |
– |
Squalene |
|
7 |
– |
Arachidate laurique I.S. |
|
A |
– |
Esters diterpéniques |
|
B |
– |
Cires |
|
C |
– |
Esters stéroliques et esters triterpéniques |
Figure 3
Esters méthyliques, esters éthyliques et cires dans une huile d’olive vierge extra

Légende:
|
1 |
– |
Heptadécanoate méthylique I.S. |
|
2 |
– |
Méthyl C18 |
|
3 |
– |
Éthyl C18 |
|
4 |
– |
Squalène |
|
5 |
– |
Arachidate laurique I.S. |
|
A |
– |
Esters diterpéniques |
|
B |
– |
Cires |
|
C |
– |
Esters stéroliques et esters triterpéniques |
Figure 4
Partie d’un chromatogramme d’une huile d’olive vierge extra et de la même huile contenant des pics d’huile désodorisée

Légende:
|
1 |
– |
Myristate méthylique I.S. |
|
2 |
– |
Palmitate méthylique |
|
3 |
– |
Palmitate éthylique |
|
4 |
– |
Heptadécanoate méthylique I.S. |
|
5 |
– |
Linoléate méthylique |
|
6 |
– |
Oléate méthylique |
|
7 |
– |
Stéarate méthylique |
|
8 |
– |
Linoléate éthylique |
|
9 |
– |
Oléate éthylique |
|
10 |
– |
Stéarate éthylique |
Appendice A
Détermination de la vitesse linéaire du gaz
Injecter de 1 à 3 μl de méthane (ou propane) dans l’appareil de chromatographie en phase gazeuse, après l'avoir réglé sur les conditions opératoires normales. Chronométrer le temps mis par le gaz pour parcourir la colonne, depuis son injection jusqu’à l'apparition du pic (tM).
La vitesse linéaire, en cm/s, est donnée par la formule L/tM, où L est la longueur de la colonne, en centimètres, et tM le temps chronométré en secondes.
▼M28 —————
ANNEXE XXI
Résultats des contrôles de conformité réalisés sur les huiles d’olive visés à l’article 8, paragraphe 2
|
|
Étiquetage |
Paramètres chimiques |
Caractéristiques organoleptiques (4) |
Conclusion finale |
|||||||||||||
|
Échantillon |
Catégorie |
Pays d’origine |
Lieu du contrôle (1) |
Dénomination légale |
Appellation d’origine |
Conditions de stockage |
Informations erronées |
Lisibilité |
C/NC (3) |
Paramètres hors limites O/N |
Dans l’affirmative, veuillez indiquer le(s)quel(s) (2) |
C/NC (3) |
Médiane du défaut |
Médiane du fruité |
C/NC (3) |
Mesures requises |
Sanction |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Marché intérieur (pressoir, embouteillage, vente au détail), exportation, importation (2) Un code est attribué à chacune des caractéristiques de l’huile d’olive mentionnées à l’annexe I (3) Conforme/non conforme (4) Pas nécessaire pour l’huile d’olive et l’huile de grignons d’olive. |
|||||||||||||||||
( 1 ) JO L 228 du 1.9.2009, p. 3.
( 2 ) Teneur en autres acides gras (%): palmitique: 7,50-20,00; palmitoléique: 0,30-3,50; heptadécanoïque: ≤ 0,40; heptadécénoïque: ≤ 0,60; stéarique: 0,50-5,00; oléique: 55,00-83,00; linoléique: 2,50-21,00.
( 3 ) Sommes des isomères qui pourraient (ou pas) être séparés par colonne capillaire.
( 4 ) Voir l'appendice de la présente annexe.
( 5 ) Sitostérol β-app.: delta-5,23-stigmastadiénol+clérostérol+bêta-sitostérol+sitostanol+delta-5-avenastérol+delta-5,24-stigmastadiénol.
( 6 ) Les huiles dont la teneur en cires est comprise entre 300 et 350 mg/kg sont considérées comme des huiles d'olive lampantes si leur teneur totale en alcools aliphatiques est inférieure ou égale à 350 mg/kg ou si la proportion d'érythrodiol et d'uvaol est inférieure ou égale à 3,5 %.
( 7 ) Les huiles ayant une teneur en cires comprise entre 300 et 350 mg/kg sont considérées comme des huiles de grignons d'olive brutes si leur teneur totale en alcools aliphatiques est supérieure à 350 mg/kg et si la proportion d'érythrodiol et d'uvaol est supérieure à 3,5 %.
( 8 ) Directive 2011/91/UE du Parlement européen et du Conseil du 13 décembre 2011 relative aux mentions ou marques permettant d’identifier le lot auquel appartient une denrée alimentaire (JO L 334 du 16.12.2011, p. 1).
( 9 ) Après l’élution des esters des stérols, le tracé chromatographique ne doit pas présenter de pics significatifs (triglycérides).
( 10 ) Règlement (UE) no 1308/2013 du Parlement européen et du Conseil du 17 décembre 2013 portant organisation commune des marchés des produits agricoles et abrogeant les règlements (CEE) no 922/72, (CEE) no 234/79, (CE) no 1037/2001 et (CE) no 1234/2007 du Conseil (JO L 347 du 20.12.2013, p. 671).
( 11 ) Le dégustateur peut s'abstenir de déguster une huile s'il détecte par voie olfactive directe un quelconque attribut négatif extrêmement intense. En pareil cas, il consignera cette circonstance exceptionnelle sur la feuille de profil.
( 12 ) Dans ces cas en particuliers, il faut employer le mélange éluant benzène-acétona 95/5 (v/v) pour obtenir une bonne séparation de bandes.
( 13 ) Après l’élution des esters stéroliques, le chromatogramme ne devrait pas présenter de pics significatifs (triglycérides).