

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 31985L0490
Fourth Commission Directive 85/490/EEC of 11 October 1985 on the approximation of the laws of the Member States relating to methods of analysis necessary for checking the composition of cosmetic products
Quatrième directive 85/490/CEE de la Commission du 11 octobre 1985 concernant le rapprochement des législations des États membres relatives aux méthodes d'analyse nécessaires au contrôle de la composition des produits cosmétiques
Quatrième directive 85/490/CEE de la Commission du 11 octobre 1985 concernant le rapprochement des législations des États membres relatives aux méthodes d'analyse nécessaires au contrôle de la composition des produits cosmétiques
OJ L 295, 7.11.1985, p. 30–45
(DA, DE, EL, EN, FR, IT, NL)
Spanish special edition: Chapter 15 Volume 006 P. 115 - 130
Portuguese special edition: Chapter 15 Volume 006 P. 115 - 130
Special edition in Finnish: Chapter 13 Volume 014 P. 192 - 207
Special edition in Swedish: Chapter 13 Volume 014 P. 192 - 207
Special edition in Czech: Chapter 13 Volume 008 P. 46 - 61
Special edition in Estonian: Chapter 13 Volume 008 P. 46 - 61
Special edition in Latvian: Chapter 13 Volume 008 P. 46 - 61
Special edition in Lithuanian: Chapter 13 Volume 008 P. 46 - 61
Special edition in Hungarian Chapter 13 Volume 008 P. 46 - 61
Special edition in Maltese: Chapter 13 Volume 008 P. 46 - 61
Special edition in Polish: Chapter 13 Volume 008 P. 46 - 61
Special edition in Slovak: Chapter 13 Volume 008 P. 46 - 61
Special edition in Slovene: Chapter 13 Volume 008 P. 46 - 61
Special edition in Bulgarian: Chapter 13 Volume 007 P. 43 - 58
Special edition in Romanian: Chapter 13 Volume 007 P. 43 - 58
Special edition in Croatian: Chapter 13 Volume 009 P. 113 - 128
 In force
In force
|
7.11.1985 |
FR |
Journal officiel de l'Union européenne |
L 295/30 |
QUATRIÈME DIRECTIVE DE LA COMMISSION
du 11 octobre 1985
concernant le rapprochement des législations des États membres relatives aux méthodes d'analyse nécessaires au contrôle de la composition des produits cosmétiques
(85/490/CEE)
LA COMMISSION DES COMMUNAUTÉS EUROPÉENNES,
vu le traité instituant la Communauté économique européenne,
vu la directive 76/768/CEE du Conseil, du 27 juillet 1976, concernant le rapprochement des législations des États membres relatives aux produits cosmétiques (1), modifiée en dernier lieu par la directive 85/391/CEE (2), et notamment son article 8 paragraphe 1,
considérant que la directive 76/768/CEE prévoit des contrôles officiels des produits cosmétiques visant à constater que les conditions prévues par les dispositions communautaires concernant la composition des produits cosmétiques sont respectées;
considérant qu'il convient d'établir le plus rapidement possible toutes les méthodes d'analyse nécessaires et que, trois étapes pour atteindre ce but ayant été réalisées par la fixation de certaines méthodes dans les directives 80/1335/CEE (3), 82/434/CEE (4) et 83/514/CEE (5) de la Commission, la fixation des méthodes d'identification et de dosage du 1-(4-aminobenzoate) de glycérol, de dosage du chlorobutanol, d'identification et de dosage de la quinine, d'identification et de dosage des sulfites inorganiques et bisulfites inorganiques, d'identification et de dosage des chlorates de métaux alcalins, d'identification et de dosage de l'iodate sodique constituent une quatrième étape;
considérant que les mesures prévues à la présente directive sont conformes à l'avis du comité pour l'adaptation au progrès technique de la directive 76/768/CEE,
A ARRÊTÉ LA PRÉSENTE DIRECTIVE:
Article premier
Les États membres prennent toutes les mesures utiles pour que, lors des contrôles officiels des produits cosmétiques:
|
— |
l'identification et le dosage du 1-(4-aminobenzoate) de glycérol, |
|
— |
le dosage du chlorobutanol, |
|
— |
l'identification et le dosage de la quinine, |
|
— |
l'identification et le dosage des sulfites inorganiques et bisulfites inorganiques, |
|
— |
l'identification et le dosage des chlorates de métaux alcalins, |
|
— |
l'identification et le dosage de l'iodate sodique soient effectués selon les méthodes décrites à l'annexe. |
Article 2
Les États membres mettent en vigueur les dispositions législatives, réglementaires ou administratives nécessaires pour se conformer aux dispositions de la présente directive, au plus tard le 31 décembre 1986.
Ils en informent immédiatement la Commission.
Article 3
Les États membres sont destinataires de la présente directive.
Fait à Bruxelles, le 11 octobre 1985.
Par la Commission
Stanley CLINTON DAVIS
Membre de la Commission
(1) JO no L 262 du 27. 9. 1976, p. 169.
(2) JO no L 224 du 22. 8. 1985, p. 40.
(3) JO no L 383 du 31. 12. 1980, p. 27.
(4) JO no L 185 du 30. 6. 1982, p. 1.
(5) JO no L 291 du 24. 10. 1983, p. 9.
ANNEXE
IDENTIFICATION ET DOSAGE DU 1-(4-AMINOBENZOATE) DE GLYCÉROL
A. IDENTIFICATION
1. OBJET ET CHAMP D'APPLICATION
Cette méthode est destinée à mettre en évidence le l-(4-aminobenzoate) de glycérol ou 4-aminobenzoate d'α-monoglycéryle. Elle permet également d'identifier le 4-aminobenzoate d'éthyle (benzocaïne DCI) éventuellement présent comme impureté.
2. PRINCIPE
L'identification se fait par chromatographie sur couche mince de silicagel avec indicateur fluorescent et mise en évidence de la fonction amine primaire libre par formation sur la plaque d'un colorant diazoïque.
3. RÉACTIFS
Tous les réactifs doivent être de qualité analytique.
|
3.1. |
Mélange solvant: cyclohexane/isopropanol/dichlorométhane stabilisé: 48/64/9 (v/v/v). |
|
3.2. |
Solvant de développement: éther de pétrole/benzène/acétone ammoniaque (minimum 25 % NH3): 35/35/35/1 (v/v/v/v). |
|
3.3. |
|
|
3.4. |
Solutions étalons:
|
|
3.5. |
Plaque de silicagel 60 F254 d'épaisseur 0,25 mm et du format 20 x 20 cm. |
4. APPAREILLAGE
|
4.1. |
Équipement courant de chromatographie sur couche mince. |
|
4.2. |
Appareil vibreur à ultra-sons. |
|
4.3. |
Filtre Millipore FH 0,5 μm ou équivalent. |
5. MODE OPÉRATOIRE>
5.1. Préparation de l'échantillon
Peser 1,5 g d'échantillon dans une fiole jaugée bouchée à l'émeri de 10 ml. Porter à 10 ml avec le mélange solvant (3.1). Boucher et laisser pendant 1 heure à température ambiante dans un appareil vibreur à ultra-sons (4.2). Filtrer sur filtre millipore (4.3). Utiliser le filtrat pour la chromatographie.
5.2. Chromatographie sur couche mince
Déposer sur la plaque (3.5) 10 μl du filtrat 5.1 et 10 ul de chaque solution étalon (3.4). Développer le chromatogramme sur 15 cm dans une cuve préalablement saturée avec le solvant (3.2). Laisser sécher à la température ambiante.
5.3. Révélation
|
5.3.1. |
Observer la plaque sous ultraviolets à 254 nm. |
|
5.3.2. |
Sur la plaque parfaitement sèche, vaporiser la solution (3.3 a). Laisser sécher à la température ambiante pendant 1 mn et vaporiser immédiatement la solution (3.3 b). Sécher la plaque à l'étuve à 60 oC. Les tâches apparaissent colorées en orange avec les Rf suivants: 4-aminobenzoate d'α-monoglycéryle: 0,07; 4-aminobenzoate d'éthyle: 0,55. |
B. DOSAGE
1. OBJET ET CHAMP D'APPLICATION
Cette méthode décrit le dosage du 1-(4-aminobenzoate) de glycérol (4-aminobenzoate d'α-monoglycéryle). Elle permet également le dosage du 4-aminobenzoate d'éthyle. Elle convient pour doser au maximum 5 % (m/m) de 4-aminobenzoate d'α-monoglycéryle et 1 % (m/m) de 4-aminobenzoate d'éthyle.
2. DÉFINITION
La teneur en 4-aminobenzoate d'α-monoglycéryle et en 4-aminobenzoate d'éthyle mesurée par cette méthode est exprimée en pourcentage de masse (% m/m) du produit.
3. PRINCIPE
Le produit à analyser est mis en suspension dans du méthanol et, après traitement approprié, le dosage est effectué par chromatographie liquide à haute pression (CLHP).
4. RÉACTIFS
Tous les réactifs doivent être de qualité analytique et, notamment, convenir pour la chromatographie liquide à haute pression.
|
4.1. |
Méthanol. |
|
4.2. |
Dihydrogénoorthophosphate de potassium KH2PO4 |
|
4.3. |
Di(acétate) de zinc Zn(CH3C00)2·2H2O |
|
4.4. |
Acide acétique:
|

|
4.5. |
Hexacyanoferrate de tétrapotassium: K4(Fe(CN)6)·3H2O |
|
4.6. |
4-hydroxybenzoate d'éthyle. |
|
4.7. |
4-aminobenzoate d'α-monoglycéryle. |
|
4.8. |
4-aminobenzoate d'éthyle (benzocaïne). |
|
4.9. |
Solution tampon (0,02 M): dissoudre 2,72 g de dihydrogénoorthophosphate de potassium (4.2) dans 1 l d'eau. |
|
4.10. |
Éluant: solution tampon (4.9) méthanol (4.1): 61/39 (v/v). La composition de cette phase mobile peut être modifiée de telle manière que le facteur de résolution R soit égal ou supérieur à 1,5:
où:
|

|
4.11. |
Solution-mère de 4-aminobenzoate d'α-monoglycéryle: peser exactement environ 40 mg de 4-aminobenzoate d'α-monoglycéryle. Les introduire dans une fiole jaugée de 100 ml. Dissoudre dans 40 ml de méthanol (4.1). Compléter jusqu'au trait avec la solution tampon (4.9) et mélanger. |
|
4.12. |
Solution-mère de 4-aminobenzoate d'éthyle: peser exactement environ 40 mg de 4-aminobenzoate d'éthyle. Les introduire dans une fiole jaugée de 100 ml. Dissoudre dans 40 ml de méthanol (4.1). Compléter jusqu'au trait avec la solution tampon (4.9) et mélanger. |
|
4.13. |
Solution étalon interne: peser exactement environ 50 mg de 4-hydroxybenzoate d'éthyle (4.6), les dissoudre par 40 ml de méthanol (4.1). Introduire la solution dans une fiole jaugée de 100 ml, compléter jusqu'au trait avec la solution tampon (4.9) et mélanger. |
|
4.14. |
Solutions étalons: par dissolution dans 100 ml d'éluant (4.10), préparer quatre solutions étalons selon le tableau suivant:
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
4.15 |
Solution de Carrez I: dissoudre 26,5 g de hexacyanoferrate de tétrapotassium (4.5) dans l'eau et compléter à 250 ml. |
|
4.16 |
Solution de Carrez II: dissoudre 54,9 g de di(acétate) de zinc (4.3) et 7,5 ml d'acide acétique (4.4) dans l'eau et compléter à 250 ml. |
|
4.17 |
Lichrosorb RP-18, ou équivalent, de 5 μm. |
5. APPAREILLAGE
|
5.1. |
Matériel courant de laboratoire. |
|
5.2. |
Chromatographe en phase liquide, à haute pression avec détecteur ultraviolets, à longueur d'onde variable et chambre thermostatique à 45 oC. |
|
5.3. |
Colonne en acier inoxydable: longueur 250 mm, diamètre intérieur 4,6 mm. La colonne est remplie de Lichrosorb RP-18 (4.17). |
|
5.4. |
Bain à ultra-sons. |
6. MODE OPÉRATOIRE
6.1. Préparation de l'échantillon
|
6.1.1. |
Peser exactement environ 1 g d'échantillon dans un bécher de 100 ml et ajouter 10 ml de méthanol (4.1). |
|
6.1.2. |
Placer le bécher pendant 20 minutes dans un bain à ultra-sons (5.4). Transvaser quantitativement la suspension ainsi obtenue dans un ballon jaugé de 100 ml avec 75 ml d'éluant (4.10) au maximum. Ajouter successivement 1 ml de solution de Carrez I (4.15) et 1 ml de solution de Carrez II (4.16) en mélangeant après chaque opération. Compléter jusqu'au trait avec l'éluant (4.10), mélanger de nouveau et filtrer sur un filtre plissé. |
|
6.1.3. |
Au moyen d'une pipette, introduire, dans une fiole jaugée de 50 ml, 3,0 ml du filtrat obtenu en 6.1.2 et 5,0 ml de la solution étalon interne (4.13). Compléter au trait avec l'éluant (4.10) et mélanger. Utiliser la solution ainsi obtenue pour procéder à l'analyse chromatographique décrite au point 6.2. |
6.2. Chromatographie
|
6.2.1. |
Régler le débit de la phase mobile (4.10) à 1,2 ml/mn et fixer la température de la colonne à 45 oC. |
|
6.2.2. |
Régler le détecteur (5.2) sur 274 nm. |
|
6.2.3. |
Au moyen d'une microseringue, faire au moins deux injections de 20 μl de la solution (6.1.3) et mesurer la surface des pics. |
6.3. Courbes d'étalonnage
|
6.3.1. |
Injecter 20 μl de chacune des solutions étalons (4.14) et mesurer la surface des pics. |
|
6.3.2. |
Pour chaque concentration, calculer le rapport entre la surface du pic de 4-aminobenzoate d'α-monoglycéryle et la surface du pic de l'étalon interne. Tracer la courbe d'étalonnage en plaçant ce rapport en ordonnée et le rapport des masses correspondantes en abscisse. |
|
6.3.3. |
Procéder de la même manière pour le 4-aminobenzoate d'éthyle. |
7. CALCUL
|
7.1. |
Sur les courbes d'étalonnage obtenues au point 6.3, lire les rapports de masse RP1 et RP2 correspondant aux rapports entre les surfaces des pics calculés au point 6.2.3 où:
|
|
7.2. |
À partir des rapports de masse ainsi obtenus, calculer la teneur en 4-aminobenzoate d'α-monoglycéryle et en 4-aminobenzoate d'éthyle, en pourcentage de masse (% m/m), au moyen des formules:
où:
|
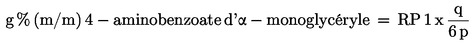

8. RÉPÉTABILITÉ (2)
|
8.1. |
Pour une teneur de 5 % (m/m) en 4-aminobenzoate d'α-monoglycéryle, la différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne doit pas dépasser 0,25 %. |
|
8.2. |
Pour une teneur de 1 % (m/m) de 4-aminobenzoate d'éthyle, la différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne doit pas dépasser 0,10 %. |
9. REMARQUES
|
9.1. |
Avant d'entreprendre l'analyse proprement dite, il convient de déterminer si l'échantillon ne contient pas de composé susceptible de coïncider avec le pic de l'étalon interne (4-aminobenzoate d'éthyle) sur le chromatogramme. |
|
9.2. |
Pour vérifier l'absence d'interférences éventuelles, répéter le dosage en modifiant de plus ou moins 10 % la proportion de méthanol dans la phase mobile. |
DOSAGE DU CHLOROBUTANOL
1. OBJET ET CHAMP D'APPLICATION
La présente méthode convient pour le dosage du chlorobutanol à la concentration maximale de 0,5 % (m/m) pour tous les produits cosmétiques, à l'exception des aérosols.
2. DÉFINITION
La teneur en chlorobutanol mesurée par cette méthode est exprimée en pourcentage de masse (% m/m) du produit.
3. PRINCIPE
Après traitement approprié du produit à analyser, le dosage est effectué par chromatographie en phase gazeuse en utilisant le 2,2,2-trichloroéthanol comme étalon interne.
4. RÉACTIFS
Tous les réactifs doivent être de qualité analytique.
|
4.1. |
Chlorobutanol l,l,l-trichloro-2-méthyl propanol-2. |
|
4.2. |
2,2,2-trichloroéthanol. |
|
4.3. |
Éthanol absolu. |
|
4.4. |
Solution étalon de chlorobutanol: 0,025 g dans 100 ml d'éthanol (4.3) (m/v). |
|
4.5. |
Solution étalon de 2,2,2-trichloroéthanol: 0,004 g dans 100 ml d'éthanol (4.3) (m/v). |
5. APPAREILLAGE
|
5.1. |
Matériel courant de laboratoire. |
|
5.2. |
Chromatographe en phase gazeuse avec détecteur à capture d'électrons 63Ni. |
6. MODE OPÉRATOIRE
6.1. Préparation de l'échantillon
Peser exactement de 0,1 g à 0,3 g d'échantillon (p g). L'introduire dans une fiole jaugée de 100 ml, le dissoudre dans l'éthanol (4.3), ajouter 1 ml de la solution étalon interne (4.5) et compléter au trait avec de l'éthanol (4.3).
6.2. Conditions de la chromatographie en phase gazeuse
|
6.2.1. |
Les conditions opératoires doivent être telles que le facteur de résolution R de la colonne soit égal ou supérieur à 1,5:
où:
|

|
6.2.2. |
À titre d'exemple, les conditions opératoires suivantes fournissent le résultat recherché:
|
6.3. Courbe étalon
Dans 5 fioles jaugées de 100 ml, ajouter à 1 ml de la solution étalon interne (4.5) respectivement 0,20, 0,30, 0,40, 0,50 et 0,60 ml de la solution (4.4) et compléter à 100 ml avec de l'éthanol (4.3). Injecter 1 μl de chacune de ces solutions dans le chromatographe selon les conditions opératoires décrites au point 6.2.2 et tracer la courbe étalon en portant en abscisse le rapport des masses du chlorobutanol au 2,2,2-tri-chloroéthanol et en ordonnée le rapport des surfaces correspondantes.
|
6.4. |
Injecter 1 μl de la solution obtenue en 6.1 et opérer selon les conditions décrites au point 6.2.2. |
7. CALCUL
|
7.1. |
Calculer à partir de la courbe étalon (6.3) la quantité « a » exprimée en μg de chlorobutanol de la solution (6.1). |
|
7.2. |
La teneur en chlorobutanol de l'échantillon % m/m est calculée selon la formule:
|

8. RÉPÉTABILITÉ (3)
Pour une teneur en chlorobutanol de 0,5 % (m/m), la différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne doit pas dépasser 0,01 %.
Note
Si le résultat est égal ou supérieur à la concentration maximale autorisée, il convient de vérifier l'absence d'interférences.
IDENTIFICATION ET DOSAGE DE LA QUININE
A. IDENTIFICATION
1. OBJET ET CHAMP D'APPLICATION
Cette méthode est destinée à mettre en évidence la présence de quinine dans les shampooings et lotions capillaires.
2. PRINCIPE
L'identification s'effectue par chromatographie sur couche mince de silicagel et mise en évidence de la fluorescence bleue de la quinine en milieu acide à 360 nm.
Pour confirmation ultérieure, on peut supprimer cette fluorescence au moyen de vapeurs de brome et faire apparaître une fluorescence jaunâtre par les vapeurs d'ammoniac.
3. RÉACTIFS
Tous les réactifs doivent être de qualité analytique.
|
3.1. |
Plaques de silicagel d'une épaisseur de 0,25 mm sans indicateur de fluorescence, format 20 x 20 cm. |
|
3.2. |
Solvant de développement: toluène/éther diéthylique/dichlorométhane/diéthylamine: 20/20/20/8 (v/v/v/v). |
|
3.3. |
Méthanol. |
|
3.4. |
Acide sulfurique 96 % (
|

|
3.5. |
Éther diéthylique. |
|
3.6. |
Réactif révélateur: ajouter avec précaution 5 ml d'acide sulfurique (3.4) à 95 ml d'éther diéthylique (3.5) dans un récipient réfrigéré. |
|
3.7. |
Brome. |
|
3.8. |
Ammoniaque à 28 % (
|

|
3.9. |
Quinine anhydre. |
|
3.10. |
Solution étalon: peser exactement environ 100 mg de quinine anhydre (3.9) et les dissoudre avec du méthanol (3.3) dans une fiole jaugée de 100 ml. |
4. APPAREILLAGE
|
4.1. |
Équipement courant de chromatographie sur couche mince. |
|
4.2. |
Bain à ultra-sons. |
|
4.3. |
Filtres Millipore FH 0,5 μm ou équivalent avec appareillage de filtration adéquat. |
5. MODE OPÉRATOIRE
5.1. Préparation de l'échantillon
Peser avec précision une quantité de produit cosmétique susceptible de contenir environ 100 mg de quinine. L'introduire dans une fiole jaugée de 100 ml, dissoudre et porter au trait de jauge avec du méthanol (3.3). Boucher et laisser pendant 1 heure à température ambiante dans le bain à ultra-sons (4.2). Filtrer sur filtre (4.3) et utiliser ce filtrat pour la chromatographie.
5.2. Chromatographie sur couche mince
Déposer sur la plaque de silicagel (3.1) 1,0 μl de solution étalon (3.10) et 1,0 μl de solution échantillon (5.1). Développer le chromatogramme sur 15 cm dans une cuve préalablement saturée aux vapeurs du solvant (3.2).
5.3. Révélation
|
5.3.1. |
Sécher la plaque à température ambiante. |
|
5.3.2. |
Pulvériser le réactif (3.6). |
|
5.3.3. |
Laisser sécher la plaque pendant 1 heure à température ambiante. |
|
5.3.4. |
Observer la plaque sous rayonnement ultraviolet à 360 nm. La quinine apparaît sous forme d'une tache fluorescente bleu intense. À titre d'exemple, le tableau ci-après reproduit les Rf des principaux alcaloïdes du quinquina, développés au moyen du solvant (3.2).
|
|
5.3.5. |
Pour confirmation ultérieure de l'identification de la quinine, on expose pendant environ 1 heure la plaque aux vapeurs de brome (3.7); la fluorescence disparaît. En exposant ensuite la même plaque aux vapeurs d'ammoniaque (3.8), les taches réapparaissent avec une coloration brune et, si l'on réexamine la plaque sous ultraviolets à 360 nm, on constate une fluorescence jaunâtre. Limite d'identification: 0,1 μg de quinine. |
B. DOSAGE
1. OBJET ET CHAMP D'APPLICATION
Cette méthode décrit le dosage de la quinine dans les shampooings et les lotions capillaires. Elle convient pour doser au maximum 0,5 % m/m dans les shampooings et 0,2 % m/m dans les lotions.
2. DÉFINITION
La teneur en quinine mesurée par cette méthode est exprimée en pourcentage de masse (% m/m) du produit.
3. PRINCIPE
Après traitement approprié du produit à analyser, le dosage est effectué par chromatographie liquide à haute pression (CLHP).
4. RÉACTIFS
Tous les réactifs doivent être de qualité analytique. Ils doivent notamment convenir pour la chromatographie liquide à haute pression.
|
4.1. |
Acétonitrile. |
|
4.2. |
Dihydrogénoorthophosphate de potassium KH2PO4 |
|
4.3. |
Acide orthophosphorique à 85 % (
|

|
4.4. |
Bromure de tétraméthylammonium. |
|
4.5. |
Quinine anhydre. |
|
4.6. |
Méthanol. |
|
4.7. |
Solution d'acide orthophosphorique 0,1 M: dissoudre 11,53 g d'acide orthophosphorique (4.3) dans 1 000 ml d'eau distillée dans une fiole jaugée. |
|
4.8. |
Solution de dihydrogénoorthophosphate de potassium 0,1 M: dissoudre 13,6 g de dihydrogénoorthophosphate de potassium (4.2) dans 1 000 ml d'eau distillée dans une fiole jaugée. |
|
4.9. |
Solution de bromure de tétraméthylammonium 0,1 M: dissoudre 15,40 g de bromure de tétraméthylammonium (4.4) dans 1 000 ml d'eau distillée dans une fiole jaugée. |
|
4.10. |
Éluant: acide orthophosphorique 0,1 M (4.7) /dihydrogénoorthophosphate de potassium 0,1 M (4.8) /bromure de tétraméthylammonium 0,1 M (4.9) /eau pour CLHP /acétonitrile (4.1): 10/50/100/340/90 (v/v/v/v/v). La composition de la phase mobile peut être modifiée de telle manière que le facteur de résolution R soit égal ou supérieur à 1,5:
où:
|
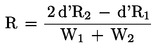
|
4.11. |
Silice octadécylsilanisée de granulométrie 10 μm. |
|
4.12. |
Solution étalon: dans une série de fioles jaugées de 100 ml, peser successivement avec précision 5,0, 10,0 15,0 et 20,0 mg de quinine anhydre (4.5). Ajuster avec du méthanol (4.6) et agiter jusqu'à dissolution. Filtrer chaque solution sur filtre (5.5) de 0,5 μm. |
5. APPAREILLAGE
|
5.1. |
Matériel courant de laboratoire. |
|
5.2. |
Bain à ultra-sons. |
|
5.3. |
Chromatographe en phase liquide à haute pression avec détecteur ultraviolets à longueur d'onde variable. |
|
5.4. |
Colonne en acier inoxydable: longueur 25 cm, diamètre intérieur: 4,6 mm, remplissage: silice octadécylsilanisée (4.11). |
|
5.5. |
Filtre Millipore FH 0,5 μm ou équivalent avec appareil de filtration adéquat. |
6. MODE OPÉRATOIRE
6.1. Préparation de l'échantillon
Peser avec précision dans une fiole jaugée de 100 ml une quantité d'échantillon correspondant à environ 10 mg de quinine anhydre. Ajouter 20 ml de méthanol (4.6) et placer la fiole pendant 20 minutes dans un bain à ultra-sons (5.2). Compléter au trait au moyen de méthanol (4.6). Homogénéiser la solution et filtrer une partie aliquote sur le filtre (5.5).
6.2. Conditions de la chromatographie
|
— |
Débit de la phase mobile (4.10): 1,0 ml/mn. |
|
— |
Détection: 332 nm. |
|
— |
Quantité à injecter: 10,0 μl de la solution filtrée (6.1). |
|
— |
Mesure de la surface du pic. |
6.3. Courbe d'étalonnage
Introduire au moins trois fois 10,0 μl de chacune des solutions étalons (4.12). Mesurer la surface du pic et en calculer la valeur moyenne pour chaque concentration.
Tracer la droite d'étalonnage.
7. CALCUL
|
7.1. |
À partir de la courbe d'étalonnage (6.3), calculer la quantité de quinine anhydre, exprimée en μg, contenue dans le volume injecté. |
|
7.2. |
La concentration en quinine anhydre dans l'échantillon, en pourcentage de masse, est obtenue par la formule suivante:
où:
|

8. RÉPÉTABILITÉ (4)
Pour une teneur en quinine anhydre de l'ordre de 0,5 % (m/m), la différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne doit pas être supérieure à 0,02 %.
Pour une teneur en quinine anhydre de l'ordre de 0,2 % (m/m), la différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne doit pas être supérieure à 0,01 %.
IDENTIFICATION ET DOSAGE DES SULFITES ET BISULFITES INORGANIQUES
OBJET ET CHAMP D'APPLICATION
La méthode décrit l'identification et le dosage des sulfites et des hydrogénosulfites inorganiques dans les produits cosmétiques. Elle est applicable seulement aux produits contenant une phase aqueuse ou alcoolique et pour des teneurs jusqu'à 0,2 % exprimé en SO2.
A. IDENTIFICATION
1. PRINCIPE
L'échantillon est chauffé dans de l'acide chlorhydrique et l'anhydride sulfureux libéré est identifié par son odeur ou à l'aide d'un papier indicateur.
2. RÉACTIFS
Tous les réactifs doivent être de qualité analytique.
|
2.1. |
Acide hydrochlorique (4 M). |
|
2.2. |
Papier indicateur à l'iodate de potassium-amidon, ou autre papier indicateur. |
3. APPAREILLAGE
|
3.1. |
Matériel courant de laboratoire. |
|
3.2. |
Ballon de 25 ml pourvu d'un réfrigérant à reflux court. |
4. MODE OPÉRATOIRE
|
4.1. |
Introduire dans le flacon (3.2) 2,5 g d'échantillon et 10 ml d'acide hydrochlorique (2.1). |
|
4.2. |
Mélanger et porter à ébullition. |
|
4.3. |
Détecter l'anhydride sulfureux à l'odeur ou au moyen du papier indicateur (2.2). |
B. DOSAGE
1. DÉFINITION
La teneur de l'échantillon en sulfite ou en hydrogénosulfite déterminée selon cette méthode est exprimée en pourcentage de la masse de dioxyde de soufre.
2. PRINCIPE
Après acidification de l'échantillon, le dioxyde de soufre libéré est distillé et recueilli dans une solution de peroxyde d'hydrogène. L'acide sulfurique formé est titré à l'aide d'une solution titrée d'hydroxyde de sodium.
3. RÉACTIFS
Tous les réactifs doivent être de qualité analytique.
|
3.1. |
Peroxyde d'hydrogène 0,2 % (m/v). Ce réactif doit être préparé quotidiennement. |
|
3.2. |
Acide orthophosphorique (
|

|
3.3. |
Méthanol. |
|
3.4. |
Solution titrée d'hydroxyde de sodium: 0,01 M. |
|
3.5. |
Azote. |
|
3.6. |
Indicateur: mélange 1: 1 (v/v) de rouge de méthyle (0,03 % m/v dans de l'éthanol) et de bleu de méthylène (0,05 % m/v dans de l'éthanol). Filtrer la solution. |
4. APPAREILLAGE
|
4.1. |
Matériel courant de laboratoire. |
|
4.2. |
Appareil à distiller (voir schéma). |
5. MODE OPÉRATOIRE
|
5.1. |
Peser avec précision environ 2,5 g d'échantillon et les introduire dans le ballon de distillation A (voir schéma). |
|
5.2. |
Ajouter 60 ml d'eau et 50 ml de méthanol (3.3), et mélanger. |
|
5.3. |
Introduire 10 ml de peroxyde d'hydrogène (3.1), 60 ml d'eau et quelques gouttes d'indicateur (3.6) dans le récipient D destiné à recevoir le distillat (voir schéma). Ajouter quelques gouttes d'hydroxyde de sodium (3.4) jusqu'à ce que l'indicateur vire au vert. |
|
5.4. |
Procéder de même dans le flacon laveur E (voir schéma). |
|
5.5. |
Assembler l'appareil et régler le débit d'azote (3.5) à environ 60 bulles par minute. |
|
5.6. |
Introduire par l'entonnoir 15 ml d'acide orthophosphorique (3.2) dans le flacon de distillation A. |
|
5.7. |
Porter rapidement à ébullition et laisser bouillir régulièrement pendant 30 minutes. |
|
5.8. |
Déconnecter le récipient D contenant le distillat. Rincer le tube et, ensuite, titrer au moyen d'une solution d'hydroxyde de sodium (3.4) jusqu'à ce que l'indicateur (3.6) vire au vert. |
6. CALCUL
Calculer la teneur en sulfite ou en hydrogénosulfite de l'échantillon en pourcentage de masse à l'aide de la formule suivante:

où:
|
M |
= |
concentration molaire de la solution d'hydroxyde de sodium (3.4), |
|
V |
= |
volume (en ml) d'hydroxyde de sodium (3.4) nécessaire pour le titrage (5.8), |
|
m |
= |
masse (en g) de l'échantillon (5.1). |
7. RÉPÉTABILITÉ (5)
Pour une teneur en dioxyde de soufre de 0,2 % m/m, la différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne doit pas dépasser 0,006 %.
Appareil à distillation de l'anhydride sulfureux selon Tanner
Toutes les dimensions sont données en millimètres

IDENTIFICATION ET DOSAGE DES CHLORATES DE MÉTAUX ALCALINS
OBJET ET CHAMP D'APPLICATION
La méthode décrit l'identification et le dosage des chlorates dans les dentifrices et autres produits cosmétiques.
A. IDENTIFICATION
1. PRINCIPE
Les chlorates sont séparés des autres halogénates par chromatographie sur couche mince et détectés par formation de l'iode obtenue par oxydation de l'iodure de potassium.
2. RÉACTIFS
Tous les réactifs doivent être de qualité analytique.
|
2.1. |
Solution de référence: solutions aqueuses de chlorate, bromate et iodate de potassium (0,2 % m/v) fraîchement préparées. |
|
2.2. |
Solvant de développement: solution d'ammoniaque (28 % m/v)/acétone/butanol (60/130/30 v/v/v). |
|
2.3. |
Solution aqueuse d'iodure de potassium (5 % m/v). |
|
2.4. |
Solution d'amidon (1 à 5 % m/v). |
|
2.5. |
Acide chlorhydrique M. |
|
2.6. |
Plaques de chromatographie sur couche mince prêtes à l'emploi, revêtues de cellulose (épaisseur: 0,25 mm). |
3. APPAREILLAGE
Matériel courant de laboratoire pour chromatographie sur couche mince.
4. MODE OPÉRATOIRE
|
4.1. |
Extraire environ 1 g de l'échantillon avec de l'eau, filtrer et diluer jusqu'à 25 ml environ. |
|
4.2. |
Déposer sur la plaque (2.6) 2 μl de la solution (4.1) et 2 μl de chacune des trois solutions de référence (2.1). |
|
4.3. |
Introduire la plaque dans une cuve et développer par chromatographie ascendante sur trois quarts environ de la longueur de la plaque à l'aide du solvant (2.2). |
|
4.4. |
Retirer la plaque de la cuve et laisser évaporer le solvant (2 heures environ). |
|
4.5. |
Pulvériser sur la plaque la solution d'iodure de potassium (2.3) et laisser sécher pendant environ 5 minutes. |
|
4.6. |
Pulvériser sur la plaque la solution d'amidon (2.4) et laisser sécher pendant environ 5 minutes. |
|
4.7. |
Pulvériser sur la plaque de l'acide chlorhydrique (2.5). |
5. LECTURE
En présence de chlorate, il apparaît une tache bleue (éventuellement brune) après une demi-heure.
Les valeurs de Rf sont les suivantes:
|
|
Rf |
|
Iodate |
0 — 0,2 |
|
Bromate |
0,5 — 0,6 |
|
Chlorate |
0,7 — 0,8 |
On notera que les bromates et les iodates produisent des réactions immédiates et on veillera à ne pas confondre les taches de bromate et de chlorate.
B. DOSAGE
1. DÉFINITION
La teneur de l'échantillon en chlorate déterminée selon cette méthode est exprimée en pourcentage de masse de chlorate.
2. PRINCIPE
Le chlorate est réduit par du zinc en poudre en milieu acide. Le chlorure formé est titré potentiométriquement par le nitrate d'argent. Un dosage similaire avant la réduction permet de déceler la présence éventuelle d'halogénures.
3. RÉACTIFS
Tous les réactifs doivent être de qualité analytique.
|
3.1. |
Acide acétique 80 % m/m. |
|
3.2. |
Zinc en poudre. |
|
3.3. |
Solution titrée de nitrate d'argent 0,1 M. |
4. APPAREILLAGE
|
4.1. |
Matériel courant de laboratoire. |
|
4.2. |
Potentiographe équipé d'une électrode indicatrice d'argent. |
5. MODE OPÉRATOIRE
5.1. Préparation de l'échantillon
Peser avec précision une quantité (m) de 2 g environ dans un tube à centrifuger. Ajouter environ 15 ml d'acide acétique (3.1) et mélanger soigneusement. Attendre 30 minutes et centrifuger pendant 15 minutes à 2 000 tours/mn. Transvaser la solution surnageante dans une fiole jaugée de 50 ml. Répéter deux fois la centrifugation en ajoutant 15 ml d'acide acétique (3.1) au résidu. Recueillir les solutions contenant le chlorate dans la même fiole jaugée. Compléter au trait de jauge avec de l'acide acétique (3.1).
5.2. Réduction du chlorate
Prélever 20 ml de la solution (5.1) et ajouter 0,6 g de zinc en poudre (3.2). Porter à ébullition dans un ballon surmonté d'un réfrigérant ascendant. Après 30 minutes d'ébullition, laisser refroidir et filtrer. Rincer le ballon avec de l'eau, filtrer et combiner le filtrat et les eaux de rinçage.
5.3. Dosage du chlorure
Titrer la solution (5.2) avec du nitrate d'argent (3.3) au moyen du potentiographe (4.2). Titrer de la même manière 20 ml de solution (5.1) avec du nitrate d'argent (3.3).
Si le produit contient des dérivés de brome ou d'iode susceptibles de libérer des bromures ou des iodures après la réduction, la courbe de titrage présente plusieurs points d'inflexion. Dans ce cas, le volume de la solution titrée (3.3) correspondant au chlorure est représenté par la différence entre les volumes correspondant aux dernier et avant-dernier points d'inflexion.
6. CALCUL
La teneur en chlorate de l'échantillon (% m/m) est calculée selon la formule:

où:
|
V |
= |
volume en ml de la solution de nitrate d'argent (3.3) utilisée pour le titrage de la solution (5.2), |
|
V' |
= |
volume en ml de la solution de nitrate d'argent (3.3) utilisée pour le titrage de la solution (5.1), |
|
M |
= |
molarité de la solution de nitrate d'argent (3.3), |
|
m |
= |
masse en g de l'échantillon (5.1). |
7. RÉPÉTABILITÉ (6)
Pour une teneur en chlorate de 3 à 5 % m/m, la différence entre les résultats des deux dosages parallèles effectués sur le même échantillon ne doit pas dépasser 0,07 % m/m.
IDENTIFICATION ET DOSAGE DE L'IODATE SODIQUE
OBJET ET CHAMP D'APPLICATION
La méthode convient à l'identification et au dosage de l'iodate sodique dans les produits cosmétiques rincés après usage.
A. IDENTIFICATION
1. PRINCIPE
L'iodate sodique est séparé des autres halogénates par chromatographie sur couche mince et identifié par l'oxydation de l'iodure en iode.
2. RÉACTIFS
Tous les réactifs doivent être de qualité analytique.
|
2.1. |
Solutions de référence: solutions aqueuses de chlorate, de bromate et d'iodate de potassium (0,01 % m/v) fraîchement préparées. |
|
2.2. |
Solvant de développement: solution d'ammoniaque (28% m/v)/acétone/butanol (60/130/30 v/v/v). |
|
2.3. |
Solution aqueuse d'iodure de potassium (5 % m/v). |
|
2.4. |
Solution d'amidon (1 à 5 % m/v). |
|
2.5. |
Acide chlorhydrique M. |
3. APPAREILLAGE
|
3.1. |
Plaques de chromatographie sur couche mince prêtes à l'emploi revêtues de cellulose (0,25 mm). |
|
3.2. |
Matériel courant de chromatographie sur couche mince. |
4. MODE OPÉRATOIRE
|
4.1. |
Extraire environ 1 g de l'échantillon avec de l'eau, filtrer et diluer à environ 10 ml. |
|
4.2. |
Déposer 2 μl de cette solution sur la ligne de base de la plaque (3.1) ainsi que 2 μl de chacune des trois solutions de référence (2.1). |
|
4.3. |
Placer la plaque dans une cuve de développement et développer par chromatographie ascendante sur les trois quarts environ de la longueur de la plaque à l'aide du solvant (2.2). |
|
4.4. |
Retirer la plaque de la cuve et laisser s'évaporer le solvant à la température ambiante. (Note: L'opération peut demander 2 heures.) |
|
4.5. |
Pulvériser la plaque avec de l'iodure de potassium (2.3) et laisser sécher pendant environ 5 minutes. |
|
4.6. |
Pulvériser ensuite avec la solution d'amidon (2.4) et laisser sécher pendant environ 5 minutes. |
|
4.7. |
Pulvériser finalement avec de l'acide chlorhydrique (2.5). |
5. ÉVALUATION
En présence d'iodate, une tache bleue (éventuellement brune ou le devenant) apparaît immédiatement, la valeur Rf étant approximativement de 0 à 0,2.
On notera que les bromates réagissent immédiatement à des valeurs Rf de 0,5 à 0,6 et que les chlorates réagissent, après 30 minutes environ, à des valeurs de Rf correspondant à 0,7 à 0,8.
B. DOSAGE
1. DÉFINITION
La teneur de l'échantillon en iodate sodique déterminée par cette méthode est exprimée en pourcentage de la masse d'iodate sodique.
2. PRINCIPE
L'iodate sodique est dissous dans l'eau et dosé par chromatographie liquide à haute pression, en utilisant (en série) une colonne phase inverse C 18 et une colonne échangeuse d'anions.
3. RÉACTIFS
Tous les réactifs doivent être de qualité analytique. Ils doivent notamment convenir pour la chromatographie liquide à haute pression.
|
3.1. |
Acide chlorhydrique, 4 M. |
|
3.2. |
Solution aqueuse de sulfite sodique, 5 % m/v. |
|
3.3. |
Solution-mère d'iodate sodique: préparer une solution contenant 50 mg d'iodate de sodium par 100 ml d'eau. |
|
3.4. |
Dihydrogénoorthophosphate de potassium. |
|
3.5. |
Hydrogénoorthophosphate disodique 2H2O. |
|
3.6. |
Phase mobile de chromatographie en phase liquide à haute pression. Dissoudre 3,88 g de dihydrogénoorthophosphate de potassium (3.4) et 1,19 g d'hydrogénoorthophosphate disodique 2H2O (3.5) dans 1 l d'eau. Le pH de la solution obtenue est 6,2. |
|
3.7. |
Papier indicateur universel, pH 1-11. |
4. APPAREILLAGE
Matériel courant de laboratoire.
|
4.1. |
Papier filtre de 110 mm de diamètre, Schleicher et Schull no 575 ou équivalent. |
|
4.2. |
Chromatographe liquide à haute pression muni d'un détecteur à longueur d'onde variable. |
|
4.3. |
Colonnes: d'une longueur de 120 mm et d'un diamètre interne de 4,6 mm, au nombre de 2, en série, première colonne conditionnée en nucléosil (R) 5 C18 ou équivalent, deuxième colonne conditionnée en Vydac, TM — 301-SB ou équivalent. |
5. MODE OPÉRATOIRE
5.1. Préparation des échantillons
5.1.1. Échantillons fluides (shampooings)
Peser avec précision une prise d'essai d'environ 1,0 g dans un tube de verre gradué et bouché à l'émeri ou dans une fiole jaugée de 10 ml. Compléter au trait avec de l'eau et mélanger. Si besoin, filtrer la solution. Déterminer la quantité d'iodate dans la solution au moyen de la chromatographie liquide à haute pression comme décrit au point 5.2.
5.1.2. Échantillons solides (savon)
Diviser finement une partie de l'échantillon et peser avec précision une prise d'essai d'environ 1,0 g, et l'introduire dans un tube en verre gradué et bouché à l'émeri de 100 ml.
Remplir d'eau jusqu'à 50 ml et agiter énergiquement pendant 1 minute; centrifuger ou filtrer à travers un filtre en papier ou laisser reposer le mélange pendant une nuit au moins, agiter énergiquement la solution gélatineuse et filtrer celle-ci à travers un filtre en papier (4.1).
Doser l'iodate dans le filtrat par chromatographie liquide à haute pression comme décrit au point 5.2.
5.2. Chromatographie
Débit: 1 ml/mn.
Longueur d'onde de détecteur: 210 nm.
Volume injecté: 10 μl
Mesure: surface du pic.
5.3. Étalonnage
Pipeter respectivement 1,0, 2,0, 5,0, 10,0 et 20,0 ml de la solution-mère d'iodate sodique (3.3) dans des fioles jaugées de 50 ml. Porter au trait et agiter. Les solutions ainsi obtenues contiennent respectivement 0,01, 0,02, 0,05, 0,10 et 0,20 mg d'iodate sodique par ml. Injecter 10 μl de chaque solution de référence dans le chromatographe liquide (4.3). Déterminer la surface du pic pour l'iodate et établir une courbe d'étalonnage: surface de pic — concentration d'iodate.
6. CALCUL
Calculer en iodate sodique, en pourcentage de la masse (% m/m) en utilisant la formule:

ou:
|
m |
= |
la masse, exprimée en g, de la prise d'essai (5.1), |
|
V |
= |
le volume total de la solution d'échantillon, exprimé en ml, obtenu comme décrit au point 5.1, |
|
c |
= |
la concentration, exprimée en mg/ml, d'iodate sodique obtenue à partir de la courbe d'étalonnage. |
7. RÉPÉTABILITÉ (7)
Pour une teneur en iodate sodique de 0,1 % (m/m), la différence entre les résultats de deux dosages effectués en parallèle sur le même échantillon ne doit pas dépasser 0,002 %.
8. CONFIRMATION
8.1. Principe
En solution acide, l'iodate (IO3—) est réduit en iodure (I —) par le sulfite et la solution obtenue est examinée par chromographie en phase liquide à haute pression. Si un pic ayant un temps de rétention correspondant au temps de rétention de l'iodate disparaît après traitement au sulfite, le pic original peut être considéré comme de l'iodate.
8.2. Mode opératoire
Pipeter dans une fiole conique une prise d'essai de 5 ml de la solution d'échantillon obtenue au point 5.1. Ajuster le pH de la solution à une valeur inférieure ou égale à 3 à l'aide d'acide chlorhydrique (3.1) et du papier universel indicateur de pH (3.7) Ajouter trois gouttes de la solution de sulfite sodique (3.2) et agiter. Injecter 10 μl de la solution dans le chromatographe liquide (4.2). Comparer le chromatogramme avec celui obtenu de la manière décrite au point 5 pour le même échantillon.
(1) Ces valeurs sont données à titre indicatif et correspondent à une pesée exacte des solutions 4.11, 4.12 et 4.13.
Note: Ces solutions peuvent être obtenues de façon différente.
(2) Selon la norme ISO 5725.
(3) Selon la norme ISO 5725.
(4) Selon la norme ISO 5725.
(5) Selon la norme ISO 5725.
(6) Selon la norme ISO 5725.
(7) Selon la norme ISO 5725.