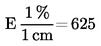ISSN 1725-261X
Euroopan unionin
virallinen lehti
L 54

Suomenkielinen laitos
Lainsäädäntö
52. vuosikerta
26. helmikuu 2009
|
ISSN 1725-261X |
||
|
Euroopan unionin virallinen lehti |
L 54 |
|
|
|
||
|
Suomenkielinen laitos |
Lainsäädäntö |
52. vuosikerta |
|
Sisältö |
|
I EY:n ja Euratomin perustamissopimuksia soveltamalla annetut säädökset, joiden julkaiseminen on pakollista |
Sivu |
|
|
|
ASETUKSET |
|
|
|
* |
Komission asetus (EY) N:o 152/2009, annettu 27 päivänä tammikuuta 2009, näytteenotto- ja määritysmenetelmistä rehujen virallista valvontaa varten ( 1 ) |
|
|
|
||
|
|
* |
|
|
|
|
|
(1) ETA:n kannalta merkityksellinen teksti |
|
FI |
Säädökset, joiden otsikot on painettu laihalla kirjasintyypillä, ovat maatalouspolitiikan alaan kuuluvia juoksevien asioiden hoitoon liityviä säädöksiä, joiden voimassaoloaika on yleensä rajoitettu. Kaikkien muiden säädösten otsikot on painettu lihavalla kirjasintyypillä ja merkitty tähdellä. |
I EY:n ja Euratomin perustamissopimuksia soveltamalla annetut säädökset, joiden julkaiseminen on pakollista
ASETUKSET
|
26.2.2009 |
FI |
Euroopan unionin virallinen lehti |
L 54/1 |
KOMISSION ASETUS (EY) N:o 152/2009,
annettu 27 päivänä tammikuuta 2009,
näytteenotto- ja määritysmenetelmistä rehujen virallista valvontaa varten
(ETA:n kannalta merkityksellinen teksti)
EUROOPAN YHTEISÖJEN KOMISSIO, joka
ottaa huomioon Euroopan yhteisön perustamissopimuksen,
ottaa huomioon rehu- ja elintarvikelainsäädännön sekä eläinten terveyttä ja hyvinvointia koskevien sääntöjen mukaisuuden varmistamiseksi suoritetusta virallisesta valvonnasta 29 päivänä huhtikuuta 2004 annetun Euroopan parlamentin ja neuvoston asetuksen (EY) N:o 882/2004 (1) ja erityisesti sen 11 artiklan 4 kohdan a, b ja c alakohdan,
sekä katsoo seuraavaa:
|
(1) |
Direktiivin 70/373/ETY täytäntöönpanemiseksi annettiin seuraavat säädökset, jotka ovat voimassa asetuksen (EY) N:o 882/2004 61 artiklan 2 kohdan mukaisesti:
|
|
(2) |
Koska direktiivi 70/373/ETY on korvattu asetuksella (EY) N:o 882/2004, on aiheellista korvata kyseisen direktiivin täytäntöönpanosäädökset yhdellä asetuksella. Samalla menetelmät olisi mukautettava tieteen ja tekniikan kehitykseen. Menetelmät, jotka eivät enää täytä alkuperäistä tarkoitustaan, on poistettava. Lainsäädännön mukaisesti näytteenottoa koskevat säännökset päivitetään sopivana ajankohtana, jotta otetaan huomioon rehujen valmistus-, varastointi-, kuljetus- ja markkinointitapojen viimeaikainen kehitys, mutta nykyiset näytteenottoa koskevat säännökset on aiheellista pitää toistaiseksi voimassa. |
|
(3) |
Sen vuoksi direktiivit 71/250/ETY, 71/393/ETY, 72/199/ETY, 73/46/ETY, 76/371/ETY, 76/372/ETY, 78/633/ETY, 81/715/ETY, 84/425/ETY, 86/174/ETY, 93/70/ETY, 93/117/EY, 98/64/EY, 1999/27/EY, 1999/76/EY, 2000/45/EY, 2002/70/EY ja 2003/126/EY olisi kumottava. |
|
(4) |
Tässä asetuksessa säädetyt toimenpiteet ovat elintarvikeketjua ja eläinten terveyttä käsittelevän pysyvän komitean lausunnon mukaiset, |
ON ANTANUT TÄMÄN ASETUKSEN:
1 artikla
Rehujen virallisessa tarkastuksessa suoritettava näytteenotto rehun sisältämien aineiden, lisäaineiden sekä haitallisten aineiden määrittämiseksi, lukuun ottamatta torjunta-ainejäämiä ja mikro-organismeja, on suoritettava tämän direktiivin liitteessä I esitettyjen menetelmien mukaisesti.
2 artikla
Näytteet on valmistettava määritystä varten ja tulokset on ilmoitettava liitteessä II esitettyjen menetelmien mukaisesti.
3 artikla
Rehujen virallista tarkastusta varten tehtävissä määrityksissä on noudatettava menetelmiä, jotka on vahvistettu liitteessä III (Rehuaineiden ja rehuseosten koostumuksen valvonnassa käytettävät määritysmenetelmät), liitteessä IV (Rehujen sisältämien sallittujen lisäaineiden valvonnassa käytettävät määritysmenetelmät), liitteessä V (Rehujen sisältämien haitallisten aineiden valvonnassa käytettävät määritysmenetelmät) ja liitteessä VI (Rehujen sisältämien eläinperäisten ainesosien määritysmenetelmät rehujen virallista valvontaa varten).
4 artikla
Siipikarjan rehuseosten energia-arvo on laskettava liitteen VII mukaisesti.
5 artikla
Liitteessä VIII vahvistettuja määritysmenetelmiä sellaisten laittomien lisäaineiden osoittamiseksi, jotka eivät enää ole sallittuja rehuissa, käytetään varmistustarkoituksiin.
6 artikla
Kumotaan direktiivit 71/250/ETY, 71/393/ETY, 72/199/ETY, 73/46/ETY, 76/371/ETY, 76/372/ETY, 78/633/ETY, 81/715/ETY, 84/425/ETY, 86/174/ETY, 93/70/ETY, 93/117/EY, 98/64/EY, 1999/27/EY, 1999/76/EY, 2000/45/EY, 2002/70/EY ja 2003/126/EY.
Viittauksia kumottuihin direktiiveihin pidetään viittauksina tähän asetukseen liitteessä IX olevien vastaavuustaulukoiden mukaisesti.
7 artikla
Tämä asetus tulee voimaan kahdentenakymmenentenä päivänä sen jälkeen, kun se on julkaistu Euroopan unionin virallisessa lehdessä.
Sitä sovelletaan 26 päivästä elokuuta 2009.
Tämä asetus on kaikilta osiltaan velvoittava, ja sitä sovelletaan sellaisenaan kaikissa jäsenvaltioissa.
Tehty Brysselissä 27 päivänä tammikuuta 2009.
Komission puolesta
Androulla VASSILIOU
Komission jäsen
(1) EUVL L 165, 30.4.2004, s. 1, oikaisu EUVL L 191, 28.5.2004, s. 1.
(2) EYVL L 155, 12.7.1971, s. 13.
(3) EYVL L 279, 20.12.1971, s. 7.
(4) EYVL L 123, 29.5.1972, s. 6.
(5) EYVL L 83, 30.3.1973, s. 21.
(6) EYVL L 102, 15.4.1976, s. 1.
(7) EYVL L 102, 15.4.1976, s. 8.
(8) EYVL L 206, 29.7.1978, s. 43.
(9) EYVL L 257, 10.9.1981, s. 38.
(10) EYVL L 238, 6.9.1984, s. 34.
(11) EYVL L 130, 16.5.1986, s. 53.
(12) EYVL L 234, 17.9.1993, s. 17.
(13) EYVL L 329, 30.12.1993, s. 54.
(14) EYVL L 257, 19.9.1998, s. 14.
(15) EYVL L 118, 6.5.1999, s. 36.
(16) EYVL L 207, 6.8.1999, s. 13.
(17) EYVL L 174, 13.7.2000, s. 32.
(18) EYVL L 209, 6.8.2002, s. 15.
(19) EUVL L 339, 24.12.2003, s. 78.
LIITE I
NÄYTTEENOTTOMENETELMÄT
1. TARKOITUS JA SOVELTAMISALA
Rehujen virallisessa tarkastuksessa käytettävät näytteet on otettava seuraavassa esitettyjen menetelmien mukaisesti. Näin saatujen näytteiden katsotaan edustavan koko tutkittavaa erää.
2. NÄYTTEENOTTAJAT
Näytteitä ottavat kyseisen jäsenvaltion tähän tehtävään valtuuttamat henkilöt.
3. MÄÄRITELMÄT
Tutkittava erä: tuoteyksikkömäärä, jonka katsotaan olevan ominaisuuksiltaan tasalaatuinen.
Osanäyte: tuotemäärä, joka on otettu tutkittavan erän yhdestä kohdasta.
Kokoomanäyte: tutkittavan erän eri kohdista otetuista osanäytteistä muodostettu yhteisnäyte.
Supistettu näyte: kokoomanäytteen edustava osa, joka on saatu kokoomanäytteestä jakomenettelyn avulla.
Lopullinen näyte: supistetun näytteen tai homogenoidun kokoomanäytteen osa.
4. VÄLINEISTÖ
|
4.1 |
Näytteenottovälineiden on oltava valmistettu materiaaleista, jotka eivät aiheuta kontaminaatiota tuotteista otettuihin näytteisiin. Nämä välineet voivat olla jäsenvaltioiden virallisesti hyväksymiä. |
4.2 Kiinteiden rehujen näytteenottoon suositellut välineet
4.2.1 Manuaalinen näytteenotto
|
4.2.1.1 |
Tasapohjainen kauha, jossa on pystysuorat sivuseinämät. |
|
4.2.1.2 |
Näytteenottokaira, jossa on pitkä ura tai osastoja. Kairan mittojen on oltava oikeassa suhteessa tutkittavaan erään (esim. astian syvyys, säkin mitat) sekä rehun raekokoon. |
4.2.2 Koneellinen näytteenotto
Hyväksyttyä koneellista laitetta voidaan käyttää otettaessa näytteitä liikkuvasta rehusta.
4.2.3 Näytteenjakolaite
Osanäytteiden ottamiseen sekä supistettujen ja lopullisten näytteiden valmistamiseen voidaan käyttää laitetta, joka on suunniteltu jakamaan näyte suunnilleen yhtä suuriin osiin.
5. MÄÄRÄÄ KOSKEVAT VAATIMUKSET
|
5.A |
Rehut, joissa aineet tai tuotteet ovat tasaisesti jakaantuneina |
|
|
5.A.1 |
Tutkittava erä Tutkittavan erän on oltava sen kokoinen, että näyte voidaan ottaa kaikista erän kohdista. |
|
|
5.A.2 |
Osanäytteet |
|
|
5.A.2.1 |
Irtorehu: |
Pienin vaadittu osanäytteiden lukumäärä: |
|
5.A.2.1.1 |
tutkittava erä enintään 2,5 tonnia: |
seitsemän |
|
5.A.2.1.2 |
tutkittava erä yli 2,5 tonnia |
√ 20 x tutkittavan erän tonnimäärä (1), kuitenkin enintään 40 osanäytettä |
|
5.A.2.2 |
Pakattu rehu: |
Pienin vaadittu määrä pakkauksia, joista otetaan näyte (2): |
|
5.A.2.2.1 |
Yli 1 kg:n pakkaukset: |
|
|
5.A.2.2.1.1 |
tutkittavassa erässä 1–4 pakkausta |
kaikki pakkaukset |
|
5.A.2.2.1.2 |
tutkittavassa erässä 5–16 pakkausta |
neljä |
|
5.A.2.2.1.3 |
tutkittavassa erässä yli 16 pakkausta |
√ tutkittavassa erässä (1) olevien pakkausten lukumäärä, kuitenkin enintään 20 pakkausta |
|
5.A.2.2.2 |
Pakkausten paino enintään 1 kg |
neljä |
|
5.A.2.3 |
Nestemäinen tai puolijuokseva rehu: |
Pienin vaadittu määrä tutkittavia säiliöitä, joista otetaan näyte (2): |
|
5.A.2.3.1 |
Säiliön tilavuus yli 1 litra: |
|
|
5.A.2.3.1.1 |
tutkittavassa erässä 1–4 säiliötä |
kaikki säiliöt |
|
5.A.2.3.1.2 |
tutkittavassa erässä 5–16 säiliötä |
neljä |
|
5.A.2.3.1.3 |
tutkittavassa erässä yli 16 säiliötä |
√ tutkittavassa erässä (1) olevien säiliöiden lukumäärä, kuitenkin enintään 20 säiliötä |
|
5.A.2.3.2 |
Säiliön tilavuus enintään yksi litra |
neljä |
|
5.A.2.4 |
Rehukakut ja nuolukivet |
Pienin vaadittu tutkittavien rehukakkujen tai nuolukivien lukumäärä, joista otetaan näyte (2): Yksi rehukakku tai yksi nuolukivi tutkittavan erän jokaista 25 yksikköä kohti, kuitenkin enintään 4 rehukakkua tai nuolukiveä |
|
5.A.3 |
Kokoomanäyte Tutkittavaa erää kohden riittää yksi kokoomanäyte. Yhden kokoomanäytteen muodostavien osanäytteiden kokonaismäärän on oltava vähintään seuraava: |
|
|
5.A.3.1 |
Irtorehu: |
4 kg |
|
5.A.3.2 |
Pakattu rehu: |
|
|
5.A.3.2.1 |
pakkauksen paino yli 1 kg |
4 kg |
|
5.A.3.2.2 |
pakkauksen paino enintään 1 kg |
neljän alkuperäisen pakkauksen sisällön paino |
|
5.A.3.3 |
Nestemäinen tai puolijuokseva rehu: |
|
|
5.A.3.3.1 |
säiliön tilavuus yli 1 litra: |
neljä litraa |
|
5.A.3.3.2 |
säiliön tilavuus enintään yksi litra |
neljän alkuperäisen säiliön sisällön paino |
|
5.A.3.4 |
Rehukakut ja nuolukivet |
|
|
5.A.3.4.1 |
yksikköpaino yli 1 kg |
4 kg |
|
5.A.3.4.2 |
yksikköpaino enintään 1 kg |
neljän alkuperäisen rehukakun tai nuolukiven paino |
|
5.A.4 |
Lopullinen näyte Lopullinen näyte on kokoomanäyte tarpeen mukaan supistettuna. Analyysi tehdään vähintään yhdestä lopullisesta näytteestä. Analyysia varten valmistetun lopullisen näytteen määrän on oltava vähintään: |
|
|
|
Kiinteä rehu |
500 g |
|
|
Nestemäinen tai puolijuokseva rehu |
500 ml |
|
5.B |
Rehut, jotka sisältävät sellaisia haitallisia aineita tai tuotteita, jotka todennäköisesti ovat epätasaisesti jakaantuneina rehuaineessa, kuten aflatoksiinit, torajyvä, risiinikasvi ja crotalaria (3). |
|
|
5.B.1 |
Tutkittava erä: katso 5.A.1 |
|
|
5.B.2 |
Osanäytteet |
|
|
5.B.2.1 |
Irtorehu: katso 5.A.2.1 |
|
|
5.B.2.2 |
Pakattu rehu: |
Pienin vaadittu lukumäärä tutkittavia pakkauksia, joista otetaan näyte: |
|
5.B.2.2.1 |
tutkittavassa erässä 1–4 pakkausta |
kaikki pakkaukset |
|
5.B.2.2.2 |
tutkittavassa erässä 5–16 pakkausta |
neljä |
|
5.B.2.2.3 |
tutkittavassa erässä yli 16 pakkausta |
√ tutkittavan erän (1) pakkausten lukumäärä, kuitenkin enintään 40 pakkausta |
|
5.B.3 |
Kokoomanäyte Kokoomanäytteiden lukumäärä vaihtelee tutkittavan erän koon mukaan. Seuraavassa annetaan pienin vaadittu kokoomanäytteiden lukumäärä tutkittavaa erää kohti. Yhden kokoomanäytteen muodostavien osanäytteiden yhteispainon on oltava vähintään seuraava: |
|
|
5.B.3.1 |
Irtorehu: |
|
|
|
Tutkittavan erän paino (tonnia): |
Pienin vaadittu kokoomanäytteiden lukumäärä tutkittavaa erää kohti: |
|
|
enintään 1 |
1 |
|
|
yli 1 ja enintään 10 |
2 |
|
|
yli 10 ja enintään 40 |
3 |
|
|
yli 40 |
4 |
|
5.B.3.2 |
Pakattu rehu |
|
|
|
Tutkittavan erän koko ilmaistuna pakkausten lukumääränä: |
Pienin vaadittu kokoomanäytteiden lukumäärä tutkittavaa erää kohti: |
|
|
1–16 |
1 |
|
|
17–200 |
2 |
|
|
201–800 |
3 |
|
|
yli 800 |
4 |
|
5.B.4 |
Lopullinen näyte Lopullinen näyte on kokoomanäyte supistettuna. Analyysi tehdään vähintään yhdestä lopullisesta näytteestä kutakin kokoomanäytettä kohti. Analyysia varten valmistetun lopullisen näytteen on oltava massaltaan vähintään 500 g. |
|
6. NÄYTTEIDEN OTTAMISTA, VALMISTAMISTA JA PAKKAAMISTA KOSKEVAT OHJEET
6.1 Yleistä
Näytteet on otettava ja valmistettava mahdollisimman nopeasti noudattaen varotoimenpiteitä, joilla varmistetaan, ettei tuote muutu tai saastu. Kaikkien näytteiden kanssa kosketuksiin joutuvien välineiden sekä pintojen ja säiliöiden on oltava puhtaita ja kuivia.
6.2 Osanäytteet
6.2.A Rehut, joissa aineet tai tuotteet ovat tasaisesti jakaantuneina
Osanäytteet on otettava satunnaisesti koko tutkittavasta erästä, ja niiden on oltava suunnilleen samansuuruisia.
6.2.A.1
Tutkittava erä kuvitellaan jaetuksi suunnilleen samankokoisiin osiin. Valitaan satunnaisesti sellainen määrä osia, joka vastaa 5.A.2 kohdan mukaista osanäytteiden määrää, ja jokaisesta osasta otetaan vähintään yksi näyte.
Näytteet voidaan tarvittaessa ottaa tutkittavaa erää siirrettäessä (lastauksen tai purkamisen yhteydessä).
6.2.A.2
Kun 5.A.2 kohdan mukainen lukumäärä pakkauksia on valittu näytteenottoa varten, kunkin pakkauksen sisällöstä otetaan osa kairaa tai kauhaa käyttäen. Tarvittaessa näytteet on otettava vasta sitten, kun pakkaukset on erikseen tyhjennetty. Kaikki kokkareet on hajotettava, jolloin ne erotetaan näytteestä ja pannaan hajotettuina takaisin näytteeseen.
6.2.A.3
Kun 5.A.2 kohdan mukainen lukumäärä säiliöitä on valittu näytteenottoa varten, sisältö on tarvittaessa homogenoitava ja vaadittava määrä näytettä otettava kustakin säiliöstä.
Osanäytteet voidaan ottaa säiliöitä tyhjennettäessä.
6.2.A.4
Kun 5.A.2 kohdan mukainen lukumäärä säiliöitä on valittu näytteenottoa varten, on otettava näytteet rehun eri kerroksista.
Näytteet voidaan ottaa myös säiliötä tyhjennettäessä, kuitenkin siten, että ensimmäisiä jakeita ei oteta talteen.
Kummassakin tapauksessa näytteen kokonaismäärän on oltava vähintään 10 litraa.
6.2.A.5
Kun 5.A.2 kohdan mukainen lukumäärä rehukakkuja tai nuolukiviä on valittu näytteenottoa varten, otetaan pala jokaisesta kakusta tai kivestä.
6.2.B Rehut, jotka sisältävät sellaisia haitallisia aineita tai tuotteita, jotka todennäköisesti ovat epätasaisesti jakaantuneina rehuaineessa, kuten aflatoksiinit, torajyvä, risiinikasvi ja crotalaria
Tutkittava erä kuvitellaan jaetuksi suunnilleen samankokoisiin osiin, joiden lukumäärä vastaa 5.B.3 kohdan mukaista kokoomanäytteiden lukumäärää. Jos kyseinen luku on suurempi kuin yksi, jaetaan tutkittava erä 5.B.2 kohdan mukaista osanäytteiden kokonaislukumäärää vastaavaan määrään suunnilleen samankokoisia osia. Jokaisesta osasta otetaan suunnilleen samankokoinen näyte (4) siten, että kustakin osasta otettujen näytteiden yhteispainon on oltava vähintään jokaiselle kokoomanäytteelle asetettu minimi 4 kg. Tutkittavan erän eri osista otettuja osanäytteitä ei saa yhdistää kokoomanäytteeksi.
6.3 Kokoomanäytteen valmistaminen
6.3.A Rehut, joissa aineet tai tuotteet ovat tasaisesti jakaantuneina
Kokoomanäyte muodostetaan sekoittamalla osanäytteet keskenään.
6.3.B Rehut, jotka sisältävät sellaisia haitallisia aineita tai tuotteita, jotka todennäköisesti ovat epätasaisesti jakaantuneina rehuaineessa, kuten aflatoksiinit, torajyvä, risiinikasvi ja crotalaria
Kustakin tutkittavan erän osasta otetut osanäytteet sekoitetaan ja niistä valmistetaan 5.B.3 kohdan mukainen määrä kokoomanäytteitä pitäen huolta siitä, että jokaisen kokoomanäytteen alkuperä on merkitty muistiin.
6.4 Lopullisen näytteen valmistaminen
Kunkin kokoomanäytteen sisältämä aines sekoitetaan huolellisesti, jotta näytteestä tulee homogeeninen. (5) Tarvittaessa kokoomanäyte on ensin pienennettävä vähintään 2 kg:n tai 2 litran suuruiseksi (supistettu näyte) joko käyttämällä koneellista tai automaattista näytteenjakolaitetta tai jakamalla näyte neljännesmenetelmällä.
Tämän jälkeen valmistetaan vähintään kolme lopullista näytettä, jotka ovat suunnilleen samankokoisia ja täyttävät 5.A.4 tai 5.B.4 kohdan määrälliset vaatimukset. Kukin näyte laitetaan asianmukaiseen astiaan. On toteutettava kaikki tarvittavat varotoimenpiteet, jotta vältetään näytteen koostumuksen muuttuminen, saastuminen tai turmeltuminen kuljetuksen tai varastoinnin aikana.
6.5 Lopullisen näytteen pakkaaminen
Näyteastiat tai -pakkaukset sinetöidään ja varustetaan etiketillä siten, ettei niitä voi avata sinettiä vahingoittamatta (koko etiketin on oltava kiinni sinetissä).
7. NÄYTTEENOTTOPÖYTÄKIRJA
Jokaisesta näytteenotosta on laadittava näytteenottopöytäkirja, josta jokainen tutkittava erä voidaan yksiselitteisesti tunnistaa.
8. NÄYTTEIDEN LÄHETTÄMINEN
Jokaisesta kokoomanäytteestä on lähetettävä vähintään yksi lopullinen näyte mahdollisimman nopeasti viralliseen laboratorioon yhdessä analyysin suorittajan tarvitsemien tietojen kanssa.
(1) Mikäli saatu luku ei ole kokonaisluku, se pyöristetään ylöspäin lähimpään kokonaislukuun.
(2) Jos pakkauksen tai säiliön sisältö ei ylitä yhtä kiloa tai yhtä litraa ja jos rehukakun tai nuolukiven paino ei ylitä yhtä kiloa, osanäyte on yhden alkuperäisen pakkauksen tai säiliön sisältö, yksi rehukakku tai yksi nuolukivi.
(3) 5.A kohdassa säädettyjä menetelmiä käytetään aflatoksiinien, torajyvän, risiinikasvin ja crotalarian valvontaan täysrehuissa ja täydennysrehuissa.
(4) Kun kyseessä on pakattu rehu, kunkin tutkittavan pakkauksen sisällöstä otetaan osa kairaa tai kauhaa käyttäen sen jälkeen, kun pakkaukset on tarvittaessa erikseen tyhjennetty.
(5) Kaikki kokkareet on hajotettava, jolloin ne erotetaan näytteestä ja pannaan hajotettuina takaisin näytteeseen.
LIITE II
REHUJEN MÄÄRITYSMENETELMIÄ KOSKEVAT YLEISET SÄÄNNÖKSET
A. NÄYTTEIDEN VALMISTAMINEN MÄÄRITYSTÄ VARTEN
1. Tarkoitus
Seuraavassa esitettävät menetelmät koskevat lopullisten näytteiden valmistusta määritystä varten; näytteet lähetetään tutkimuslaboratorioihin sen jälkeen, kun ne on otettu liitteessä I vahvistettujen säännösten mukaisesti.
Näytteet on valmistettava siten, että määritysmenetelmien mukaiset punnitut määrät ovat homogeenisia ja edustavia.
2. Varotoimenpiteet
Näytteiden valmistusmenettely riippuu käytettävistä määritysmenetelmistä. Tästä syystä on olennaisen tärkeää varmistaa, että näytteiden valmistusmenettely on käytettävien määritysmenetelmien kannalta asianmukainen.
Kaikki toimenpiteet on suoritettava siten, että vältetään mahdollisimman hyvin näytteen saastuminen ja sen koostumuksen muuttuminen.
Näyte on jauhettava, sekoitettava ja seulottava mahdollisimman nopeasti altistaen sitä ilmalle ja valolle mahdollisimman vähän. Sellaisia myllyjä ja jauhatuslaitteita, joissa näytteen huomattava lämpeneminen on todennäköistä, ei pidä käyttää.
On suositeltavaa jauhaa erityisen lämmönarat rehut käsin. Lisäksi on huolehdittava siitä, ettei itse välineistössä ole kontaminaatiota aiheuttavia hivenainejäämiä.
Jos näytettä ei voi valmistaa muuttamatta merkittävästi sen sisällön kosteuspitoisuutta, on kosteuspitoisuus määritettävä ennen valmistusta ja sen jälkeen liitteessä III olevassa A osassa säädetyn menetelmän mukaisesti.
3. Menettely
Näyte jaetaan sopiviksi eränäytteiksi määritystä ja vertailua varten käyttäen asianmukaisia jakotekniikoita, kuten vuorottelevaa kahtiointia taikka kiinteän tai pyörivän kourujakajan käyttöä. Kartiomurskaimen ja neljännesmenetelmän käyttöä ei suositella, koska ne aiheuttavat eränäytteisiin suuren jakovirheriskin. Vertailunäyte pannaan sopivaan puhtaaseen, kuivaan ja ilmatiiviisti suljettavaan näyteastiaan, ja määritykseen tarkoitetut, vähintään 100 gramman suuruiset eränäytteet valmistetaan jäljempänä esitetyllä tavalla.
3.1 Sellaisenaan jauhettavat rehut
Jollei määritysmenetelmissä toisin mainita, koko näyte seulotaan tarvittaessa jauhamisen jälkeen seulalla, jonka neliönmuotoisten reikien sivu on 1 mm (suosituksen ISO R565 mukaisesti). Liikajauhatusta on vältettävä.
Seulottu näyte sekoitetaan ja pannaan tarkoituksenmukaiseen puhtaaseen, kuivaan ja ilmatiiviisti suljettavaan näyteastiaan. Näyte sekoitetaan uudelleen välittömästi ennen punnitsemista määritystä varten.
3.2 Kuivauksen jälkeen jauhettavat rehut
Jollei määritysmenetelmissä toisin mainita, näyte kuivataan kosteuspitoisuuteen 8–12 % käyttäen esikuivatusmenetelmää, joka esitetään liitteessä III olevan A osan 4.3 kohdassa kuvatun kosteudenmääritysmenetelmän yhteydessä. Tämän jälkeen jatketaan edellä 3.1 kohdassa esitetyllä tavalla.
3.3 Nestemäiset tai puolijuoksevat rehut
Näyte kerätään sopivaan puhtaaseen, kuivaan ja ilmatiiviisti suljettavaan näyteastiaan. Näyte sekoitetaan perusteellisesti välittömästi ennen näytteen ottamista.
3.4 Muut rehut
Jos näytettä ei voi valmistaa jollain edellä esitetyllä tavalla, se on valmistettava millä tahansa muulla tavalla, jolla varmistetaan, että määritystä varten punnitut ainemäärät ovat homogeenisia ja edustavat lopullisia näytteitä.
4. Näytteiden säilytys
Näytteet on säilytettävä lämpötilassa, jossa niiden koostumus ei muutu. Vitamiinien tai valolle erityisen herkkien aineiden määritykseen tarkoitetut näytteet on säilytettävä ruskeasta lasista valmistetuissa näyteastioissa.
B. MÄÄRITYSMENETELMISSÄ KÄYTETTÄVIÄ REAGENSSEJA JA VÄLINEITÄ KOSKEVAT SÄÄNNÖKSET
|
1. |
Jollei määritysmenetelmissä toisin mainita, kaikkien reagenssien on oltava analyysilaatua (p.a.). Hivenainejäämiä määritettäessä reagenssien puhtaus on tarkistettava nollanäytteellä. Saatujen tuloksien mukaan voi reagenssien lisäpuhdistus olla tarpeen. |
|
2. |
Määritysmenetelmissä mainituissa toimenpiteissä, joihin liittyy liuosten valmistus, laimennus, huuhtelu tai pesu ja joissa käytettävän liuottimen tai laimentimen laatua ei mainita, on käytettävä vettä. Pääsääntöisesti on käytettävä demineralisoitua tai tislattua vettä. Määritysmenetelmissä mainituissa erityistapauksissa vesi on puhdistettava erityisillä puhdistusmenetelmillä. |
|
3. |
Tutkimuslaboratorioissa käytettävistä välineistä määritysmenetelmissä mainitaan ainoastaan erikoisinstrumentit ja -välineet tai erityiskäyttöön tarkoitetut instrumentit ja välineet. Niiden on oltava puhtaita etenkin määritettäessä hyvin pieniä ainemääriä. |
C. MÄÄRITYSMENETELMIEN KÄYTTÖ JA TULOSTEN ILMOITTAMINEN
1. Uuttomenettely
Useat menetelmät edellyttävät erityistä uuttomenetelmää. Pääsääntöisesti voidaan soveltaa myös muita uuttomenettelyjä kuin menetelmän kuvauksessa tarkoitettu menettely, jos voidaan osoittaa, että käytetyn menettelyn uuttoteho on analysoitavan materiaalin osalta samantasoinen kuin menetelmässä kuvatun menettelyn uuttoteho.
2. Puhdistusmenettely
Useat menetelmät edellyttävät erityistä puhdistusmenetelmää. Pääsääntöisesti voidaan soveltaa myös muita puhdistusmenettelyjä kuin menetelmän kuvauksessa tarkoitettu menettely, jos voidaan osoittaa, että käytetty puhdistusmenettely johtaa tutkittavaa materiaalia analysoitaessa analyysituloksiin, jotka vastaavat menetelmässä mainittua menettelyä käytettäessä saatuja tuloksia.
3. Käytetyn määritysmenetelmän ilmoittaminen
Yleensä kunkin rehussa olevan ainesosan määrittämiseksi vahvistetaan vain yksi määritysmenetelmä. Kun useita menetelmiä on mainittu, määritysselosteessa on mainittava tutkimuslaboratorion käyttämä menetelmä.
4. Määritysten lukumäärä
Määritysselosteessa annetun tuloksen on oltava keskiarvo, joka on saatu ainakin kahdesta määrityksestä, jotka on tehty erillisistä näytteistä ja joiden toistettavuus on tyydyttävä.
Jos kuitenkin haitallisten aineiden määrityksessä ensimmäisen määrityksen tulos on merkittävästi (> 50 %) valvottavaa arvoa alempi, lisämääritykset eivät ole tarpeen edellyttäen, että asianmukaisia laatumenettelyjä on sovellettu.
Kun kyse on aineen tai ainesosan ilmoitetun pitoisuuden valvonnasta ja jos ensimmäisen määrityksen tulos vahvistaa ilmoitetun pitoisuuden eli määrityksen tulos on ilmoitetun pitoisuuden hyväksyttävällä vaihteluvälillä, lisämääritykset eivät ole tarpeen edellyttäen, että asianmukaisia laatumenettelyjä on sovellettu.
Joissain tapauksissa hyväksyttävä vaihteluväli on määritelty lainsäädännössä, kuten neuvoston direktiivissä 79/373/ETY (1).
5. Määritystuloksen ilmoittaminen
Määritystulos on ilmaistava määritysmenetelmässä vahvistetulla tavalla asianmukaisella merkitsevien numeroiden tarkkuudella, ja tulos on tarvittaessa korjattava sen mukaan, mikä lopullisen näytteen kosteuspitoisuus oli ennen näytteen valmistamista.
6. Mittausepävarmuus ja saanto haitallisten aineiden määrityksessä
Direktiivissä 2002/32/EY tarkoitettujen haitallisten aineiden, kuten dioksiinien ja dioksiinin kaltaisten PCB-yhdisteiden, ollessa kyseessä eläinten rehuksi tarkoitettua tuotetta ei pidetä vahvistetun enimmäispitoisuuden mukaisena, jos määritystuloksen katsotaan ylittävän enimmäispitoisuuden, kun otetaan huomioon laajennettu mittausepävarmuus ja korjaus saannon suhteen. Vaatimustenmukaisuuden arvioinnissa käytetään määritettyä pitoisuutta, joka on korjattu saannon suhteen ja josta on vähennetty laajennettu mittausepävarmuus. Tätä menettelyä sovelletaan ainoastaan silloin, kun määritysmenetelmässä voidaan arvioida mittausepävarmuus ja korjata tulos saannon suhteen (esimerkiksi mikroskooppitutkimuksessa tämä ei ole mahdollista).
Määritystulos on esitettävä seuraavalla tavalla (sikäli kuin käytetty määritysmenetelmän mittausepävarmuus voidaan arvioida ja tulos korjata saannon suhteen):
|
a) |
saannon suhteen korjattuna, saantoaste ilmoitetaan. Korjaus saannon suhteen ei ole tarpeen, jos saantoaste on 90–110 %; |
|
b) |
muodossa x +/- U, jossa x on määritystulos ja U on mittaukseen liittyvä laajennettu epävarmuus, käyttäen kattavuuskerrointa 2, jolloin luotettavuustaso on noin 95 %. |
Kuitenkin jos määrityksen tulos on merkittävästi (> 50 %) valvottavaa arvoa alempi ja edellyttäen, että asianmukaisia laatumenettelyjä on sovellettu ja määrityksen ainoana tarkoituksena on todeta säännösten noudattaminen, määritystulos voidaan ilmoittaa ilman korjausta saannon suhteen, ja saantoaste ja mittausepävarmuus voidaan näissä tapauksissa jättää pois.
LIITE III
REHUAINEIDEN JA REHUSEOSTEN KOOSTUMUKSEN VALVONNASSA KÄYTETTÄVÄT MÄÄRITYSMENETELMÄT
A. KOSTEUDEN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehun kosteuspitoisuus. Jos rehu sisältää haihtuvia aineita, kuten orgaanisia happoja, on huolehdittava siitä, että kosteuspitoisuuden kanssa määritetään myös merkittäviä määriä haihtuvia aineita.
Menetelmä ei koske suoraan rehuina käytettäviä maitotuotteita, kivennäisaineita ja pääasiassa kivennäisaineista koostuvia seoksia, eläin- ja kasvirasvoja ja -öljyjä tai öljysiemeniä ja öljypitoisia hedelmiä.
2. Periaate
Näyte kuivataan määrätyissä olosuhteissa, jotka vaihtelevat rehun laadun mukaan. Painohäviö määritetään punnitsemalla. Hyvin kosteat, kiinteät rehunäytteet on syytä esikuivata ennen varsinaista kosteusmääritystä.
3. Välineistö
|
3.1 |
Kosteutta imemättömästä materiaalista valmistettu mylly, joka on helppo puhdistaa ja jolla saadaan aikaan nopea, tasainen jauhautuminen mutta jolla vältetään havaittavissa oleva kuumentuminen ja ulkopuolisen ilman vaikutus mahdollisimman hyvin ja joka on 4.1.1 ja 4.1.2 kohdassa säädettyjen vaatimusten mukainen (esimerkiksi vasara- tai vesijäähdytteiset pienoismyllyt, kartiomyllyt, hitaasti liikkuvat tai hammaspyörillä varustetut hienonnuslaitteet). |
|
3.2 |
Analyysivaaka, jonka tarkkuus on 1 mg. |
|
3.3 |
Lasisia tai korroosionkestävästä metallista valmistettuja kuivausastioita, jotka voidaan sulkea ilmatiiviisti kannella; astian tilavuuden on oltava sellainen, että näytettä voidaan levittää sille noin 0,3 g/cm2. |
|
3.4 |
Sähkölämmitteinen, tasalämpöinen ja nopeasti säädettävissä oleva lämpökaappi (± 2 oC), jossa on tuuletusjärjestelmä (1). |
|
3.5 |
Säädettävä sähkölämmitteinen vakuumilämpökaappi, jossa on öljypumppu ja johon voidaan johtaa kuumaa, kuivaa ilmaa tai jossa voidaan käyttää kuivausainetta (kuten kalsiumoksidia). |
|
3.6 |
Eksikkaattori, jossa on paksu rei’itetty metalli- tai posliinilevy ja joka sisältää tehokasta kuivausainetta. |
4. Menettely
|
Huomautus: |
Tässä jaksossa kuvatut toimenpiteet on suoritettava välittömästi näytelähetyksen avaamisen jälkeen. Määritykset on suoritettava ainakin kahtena rinnakkaismäärityksenä. |
4.1 Valmistaminen
4.1.1
Punnitaan vähintään 50 g näytettä. Tarvittaessa se jauhetaan tai jaetaan sopivalla tavalla kosteuspitoisuuden muuttumista välttäen (ks. 6 kohta).
4.1.2
Punnitaan vähintään 50 g näytettä. Se jauhetaan niin hienoksi, että vähintään 50 % näytteestä läpäisee seulan, jonka reikien koko 0,5 mm, eikä 1 mm:n pyöreäreikäistä seulaa jää läpäisemättä yli 10 % näytteestä.
4.1.3
Punnitaan noin 25 g näytettä 10 mg:n tarkkuudella, lisätään sopiva määrä 10 mg:n tarkkuudella punnittua vedetöntä hiekkaa ja sekoitetaan kunnes saadaan homogeeninen seos.
4.2 Kuivaus
4.2.1
Näyteastia (3.3) punnitaan kansineen 1 mg:n tarkkuudella. Siihen punnitaan 1 mg:n tarkkuudella noin 5 g hienonnettua näytettä, joka levitetään tasaisesti. Astia asetetaan ilman kantta 103 oC:ssa olevaan lämpökaappiin. Kaapin lämpötilan tarpeettoman laskun välttämiseksi astia viedään sen sisään mahdollisimman nopeasti. Annetaan kuivua neljän tunnin ajan laskettuna siitä hetkestä, kun lämpökaapin lämpötila on uudelleen saavuttanut 103 oC. Astia suljetaan kannella, otetaan pois lämpökaapista, annetaan jäähtyä 30–45 minuuttia eksikkaattorissa (3.6) ja punnitaan 1 mg:n tarkkuudella.
Pääasiassa öljyistä ja rasvoista koostuvia rehuja kuivataan lämpökaapissa 130 oC:ssa vielä 30 minuutin ajan. Kahden punnituksen välinen ero ei saa olla suurempi kuin 0,1 % kosteudesta.
4.2.2
Näyteastia (3.3) punnitaan kansineen 0,5 mg:n tarkkuudella. Siihen punnitaan 1 mg:n tarkkuudella noin 5 g hienonnettua näytettä, joka levitetään tasaisesti. Astia asetetaan ilman kantta 130 oC:ssa olevaan lämpökaappiin. Kaapin lämpötilan tarpeettoman laskun välttämiseksi astia viedään sen sisään mahdollisimman nopeasti. Annetaan kuivua kahden tunnin ajan laskettuna siitä hetkestä, kun lämpökaapin lämpötila on uudelleen saavuttanut 130 oC. Astia suljetaan kannella, otetaan pois lämpökaapista, annetaan jäähtyä 30–45 minuuttia eksikkaattorissa (3.6) ja punnitaan 1 mg:n tarkkuudella.
|
4.2.3 |
Rehuseokset, jotka sisältävät yli 4 % sakkaroosia tai laktoosia: rehuaineet, kuten johanneksenleipäpuun palot, hydrolysoidut viljatuotteet, maltaat, kuivatut juurikasleikkeet, kala ja liukoisia sokereita sisältävät rehut; rehuseokset, jotka sisältävät yli 25 % mineraalisuoloja kidevesi mukaan luettuna. Näyteastia (3.3) punnitaan kansineen 0,5 mg:n tarkkuudella. Siihen punnitaan 1 mg:n tarkkuudella noin 5 g hienonnettua näytettä, joka levitetään tasaisesti. Astia asetetaan ilman kantta vakuumilämpökaappiin (3.5), joka on kuumennettu 80–85 oC:seen. Kaapin lämpötilan tarpeettoman laskun välttämiseksi astia viedään sen sisään mahdollisimman nopeasti. Paine säädetään 100 torriksi ja annetaan kuivua vielä neljän tunnin ajan tässä paineessa joko kuumassa, kuivassa ilmavirrassa tai kuivausainetta käyttäen (noin 300 g 20:tä näytettä kohden). Jälkimmäisessä tapauksessa vakuumipumppu kytketään pois päältä, kun vaadittu paine on saavutettu. Kuivausaika lasketaan siitä hetkestä, kun lämpökaapin lämpötila on uudelleen saavuttanut 80–85 oC. Kuivausajan päätyttyä lämpökaapin paine palautetaan varovasti normaalipaineeseen. Lämpökaappi avataan, näyteastia suljetaan välittömästi kannella, otetaan pois lämpökaapista, annetaan jäähtyä 30–45 minuuttia eksikkaattorissa (3.6) ja punnitaan 1 mg:n tarkkuudella. Näytteen annetaan kuivua vielä 30 minuuttia vakuumilämpökaapissa 80–85 oC:ssa ja se punnitaan uudelleen. Kahden punnituksen välinen ero ei saa ylittää 0,1 %:a kosteudesta. |
4.3 Esikuivaus
4.3.1
Kiinteille, vaikeasti jauhettaville rehunäytteille, joiden kosteuspitoisuus on suuri, suoritetaan esikuivaus seuraavasti:
Punnitaan 50 g hienontamatonta näytettä (puristetut tai kokkareina olevat rehut voidaan tarvittaessa hienontaa karkeiksi) 10 mg:n tarkkuudella sopivaan astiaan (esim. 0,5 cm:n reunalla varustettu 20 x 12 cm:n alumiiniastia). Annetaan kuivua lämpökaapissa 60–70 oC:ssa, kunnes kosteuspitoisuus on laskenut 8–12 prosenttiin. Näyteastia otetaan pois lämpökaapista, annetaan jäähtyä avoimena laboratoriossa tunnin ajan ja punnitaan 10 mg:n tarkkuudella. Näyte jauhetaan välittömästi 4.1.1 kohdassa esitetyllä tavalla ja kuivataan rehutyypin mukaan 4.2.1 tai 4.2.3 kohdassa esitetyllä tavalla.
4.3.2
Jyvät, joiden kosteuspitoisuus on yli 17 %, on esikuivattava seuraavasti:
Punnitaan 50 g jauhamattomia jyviä 10 mg:n tarkkuudella sopivaan astiaan (esim. 0,5 cm:n reunalla varustettu 20 × 12 cm:n alumiiniastia). Annetaan kuivua lämpökaapissa 130 oC:ssa 5–7 minuuttia. Astia poistetaan lämpökaapista, annetaan jäähtyä avoimena laboratoriossa 2 tunnin ajan ja punnitaan 10 mg:n tarkkuudella. Jyvät jauhetaan välittömästi 4.1.2 kohdassa esitetyllä tavalla ja kuivataan 4.2.2 kohdan mukaisesti.
5. Tulosten laskeminen
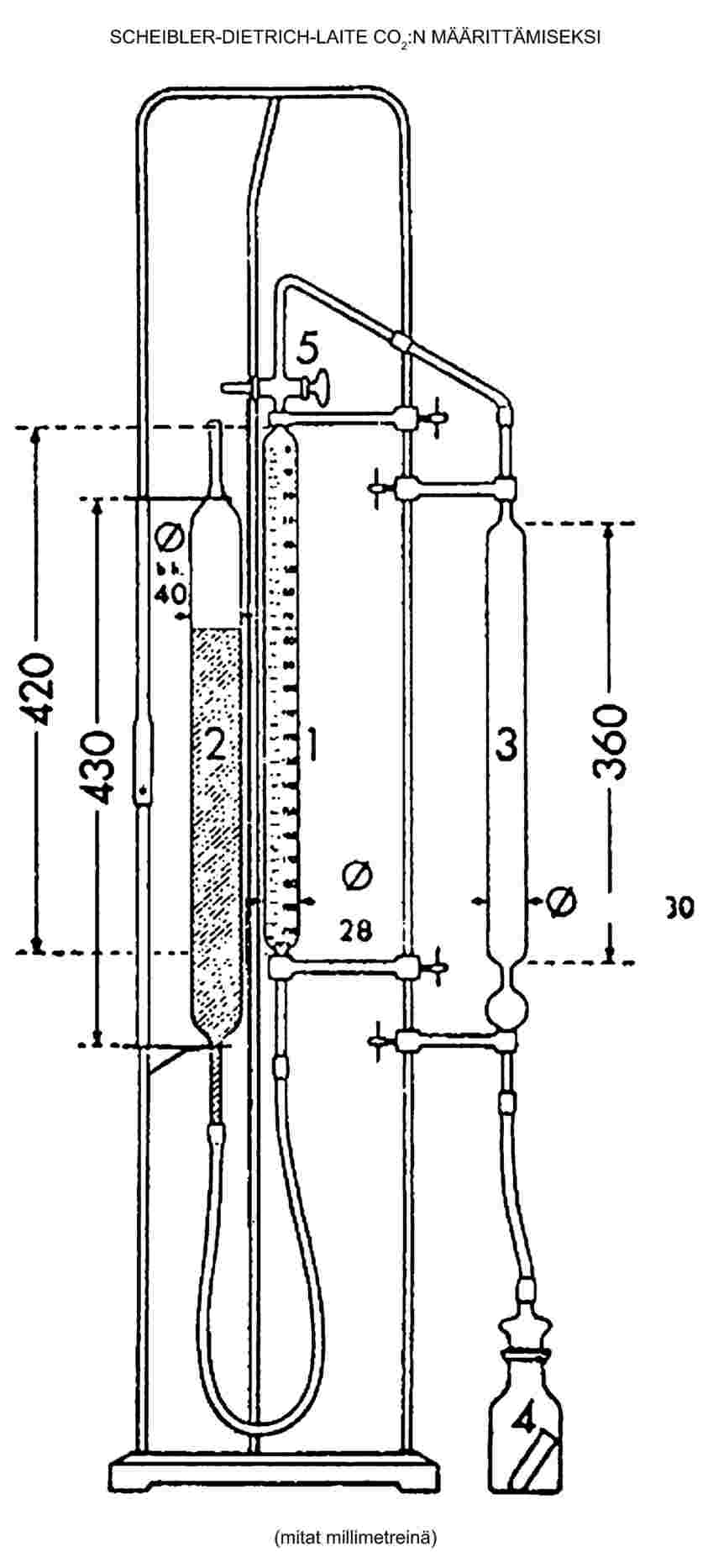
Kosteuspitoisuus (X), joka ilmoitetaan prosentteina näytteestä, lasketaan käyttäen seuraavia kaavoja:
5.1 Kosteuspitoisuus ilman esikuivausta

jossa:
|
m |
= |
näytteen alkuperäinen paino grammoina, |
|
m0 |
= |
näytteen paino grammoina kuivauksen jälkeen. |
5.2 Kosteuspitoisuus esikuivausta käyttäen
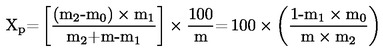
jossa:
|
m |
= |
näytteen alkuperäinen paino grammoina, |
|
m1 |
= |
näytteen paino grammoina esikuivauksen jälkeen, |
|
m2 |
= |
näytteen paino grammoina hienontamisen tai jauhamisen jälkeen, |
|
m0 |
= |
näytteen paino grammoina kuivauksen jälkeen. |
5.3 Toistettavuus
Samasta näytteestä tehdyn kahden rinnakkaismäärityksen tulosten välinen ero ei saa olla suurempi kuin 0,2 % kosteuden absoluuttisesta arvosta.
6. Huomautus
Jos jauhaminen on tarpeen ja sen havaitaan muuttavan tuotteen kosteuspitoisuutta, rehun komponenttien määritystulokset on korjattava alkuperäisen näytteen kosteuspitoisuuden perusteella.
B. ELÄIN- JA KASVIRASVOJEN JA ÖLJYJEN KOSTEUDEN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää eläin- ja kasvirasvojen ja öljyjen vesipitoisuus ja haihtuvien aineiden pitoisuus.
2. Periaate
Näyte kuivataan vakiopainoon 103 oC:ssa (kahden peräkkäisen punnituksen välisen painoeron on oltava alle 1 mg). Painohäviö määritetään punnitsemalla.
3. Välineistö
|
3.1 |
Korroosionkestävästä materiaalista valmistettu tasapohjainen astia, jonka läpimitta on 8–9 cm ja jonka korkeus on noin 3 cm. |
|
3.2 |
Lämpömittari, jossa on vahvistettu säiliö ja yläpäässä oleva paisuntaputki ja joka on varustettu asteikolla noin 80 oC:sta ainakin 110 oC:seen ja jonka pituus on noin 10 cm. |
|
3.3 |
Hiekkahaude tai sähkölevy. |
|
3.4 |
Eksikkaattori, jossa on tehokasta kuivausainetta. |
|
3.5 |
Analyysivaaka. |
4. Menettely
Punnitaan mg:n tarkkuudella noin 20 g homogenisoitua näytettä kuivaan punnittuun astiaan (3.1), jossa on lämpömittari (3.2). Näytettä kuumennetaan hiekkahauteella tai sähkölevyllä (3.3), sekoittaen jatkuvasti lämpömittarilla niin, että lämpötila saavuttaa 90 oC:n noin 7 minuutissa.
Lämpöä vähennetään pitäen silmällä lautasen pohjasta nousevien kuplien syntymisnopeutta. Lämpötila ei saa nousta yli 105 oC:n. Sekoitusta jatketaan astian pohjaa raaputtaen, kunnes kuplien muodostus lakkaa.
Kosteuden täydellisen poistumisen varmistamiseksi kuumennetaan useita kertoja 103 ± 2 oC:seen jäähdyttäen peräkkäisten kuumennusten välillä 93 oC:seen. Sen jälkeen astian sisältöineen annetaan jäähtyä huoneen lämpötilaan eksikkaattorissa (3.4) ja punnitaan. Tämä menettely toistetaan niin monta kertaa, että kahden peräkkäisen punnituksen välinen ero ei ole yli 2 mg:aa.
|
Huomautus |
: |
Näytteen painon kasvu toistettujen kuumennusten jälkeen viittaa rasvan hapettumiseen, ja tällaisessa tapauksessa tulos lasketaan siitä punnituksesta, joka on tehty välittömästi ennen kuin paino on alkanut kasvaa. |
5. Tulosten laskeminen
Kosteuspitoisuus (X), joka ilmoitetaan prosentteina näytteestä, saadaan seuraavasta kaavasta:
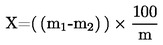
joissa:
|
m |
= |
näytteen paino grammoina, |
|
m1 |
= |
astian paino sisältöineen grammoina ennen kuumennusta, |
|
m2 |
= |
astian paino sisältöineen grammoina kuumennuksen jälkeen. |
Tulokset, jotka ovat pienempiä kuin 0,05 %, on varustettava merkinnällä ”alle 0,05 %”.
Toistettavuus
Samasta näytteestä suoritetun kahden samanaikaisen rinnakkaismäärityksen ero ei saa absoluuttisena arvona ilmoitettuna olla yli 0,05 %.
C. RAAKAVALKUAISPITOISUUDEN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan laskea Kjeldahlin menetelmän mukaan saadusta typpipitoisuudesta rehujen raakavalkuaispitoisuus.
2. Periaate
Näyte hajotetaan rikkihapolla katalysaattorin läsnä ollessa. Hapan liuos tehdään emäksiseksi natriumhydroksidiliuoksella. Ammoniakki tislataan ja kerätään mitattuun määrään rikkihappoa, ja ylimääräinen rikkihappo titrataan natriumhydroksidin standardiliuoksella.
Vaihtoehtoisesti vapautunut ammoniakki tislataan ylimäärään boorihappoliuosta, jonka jälkeen se titrataan kloorivetyhappo- tai rikkihappoliuoksella.
3. Reagenssit
|
3.1 |
Kaliumsulfaatti. |
|
3.2 |
Katalysaattori: kupari(II)oksidi CuO tai kupari(II)sulfaattipentahydraatti, CuSO45H2O. |
|
3.3 |
Sinkki, rakeinen. |
|
3.4 |
Rikkihappo, ρ20 = 1,84 g/ml. |
|
3.5 |
Rikkihappo, standardiliuos, c(H2SO4) = 0,25 mol/l. |
|
3.6 |
Rikkihappo, standardiliuos, c(H2SO4) = 0,10 mol/l. |
|
3.7 |
Rikkihappo, standardiliuos, c(H2SO4) = 0,05 mol/l. |
|
3.8 |
Metyylipunaindikaattori: 300 g metyylipunaista liuotetaan 100 ml:aan 95–96 % etanolia (v/v) |
|
3.9 |
Natriumhydroksidiliuos (teknisen laadun käyttö mahdollista) β = 40 g/100 ml (m/v: 40 %). |
|
3.10 |
Natriumhydroksidi, standardiliuos c(NaOH) = 0,25 mol/l. |
|
3.11 |
Natriumhydroksidi, standardiliuos c(NaOH) = 0,10 mol/l. |
|
3.12 |
Kiehumakiviä, suolahapossa pestyjä ja hehkutettuja. |
|
3.13 |
Asetanilidi (sulamispiste = 114 oC, typpipitoisuus = 10,36 %). |
|
3.14 |
Typetön sakkaroosi. |
|
3.15 |
Boorihappo (H3BO3). |
|
3.16 |
Metyylipunaindikaattoriliuos: liuotetaan 0,1 g metyylipunaa 100 ml:aan etanolia tai metanolia. |
|
3.17 |
Bromikresolivihreäliuos: liuotetaan 100 mg bromikresolivihreää 100 ml:aan etanolia tai metanolia. |
|
3.18 |
Boorihappoliuos (10–40 g/l, käytettävien laitteiden mukaan) Jos loppupisteen toteamiseksi käytetään kolorimetristä menetelmää, boorihappoliuoksiin on lisättävä metyylipuna- ja bromokresoli-indikaattoreita. Kun boorihappoliuosta valmistetaan 1 litra, siihen on ennen tilavuuden säätämistä lisättävä 7 ml metyylipunaindikaattoriliuosta (3.16) ja 10 ml bromikresolivihreäliuosta (3.17). Käytetystä vedestä johtuen boorihappoliuoksen pH saattaa vaihdella erien välillä. Usein on lisättävä vähän emästä, jotta nollanäyte saadaan positiiviseksi.
|
|
3.19 |
Kloorivetyhapon standardiliuos c(HCl) = 0,10 mol/l.
|
4. Välineistö
Välineet näytteen hajottamista, tislausta ja titrausta varten Kjeldahlin menetelmän mukaan.
5. Menettely
5.1 Hajotus
Punnitaan 1 g näytettä 1 mg:n tarkkuudella Kjeldahl-kolviin hajotuspolttoa varten. Lisätään 15 g kaliumsulfaattia (3.1), sopiva määrä katalysaattoria (3.2) (0,3–0,4 g kupari(II)oksidia tai 0,9–1,2 g kupari(II)sulfaattipentahydraattia, 25 ml rikkihappoa (3.4) ja tarvittaessa muutamia kiehumakiviä (3.12) ja sekoitetaan.
Kolvia kuumennetaan varovasti tarvittaessa ajoittain pyörittäen, kunnes massa on hiiltynyt ja kuohu hävinnyt. Tämän jälkeen kuumennetaan voimakkaammin, kunnes neste kiehuu tasaisesti. Kuumennus on sopiva, jos kiehuva happo tiivistyy kolvin seinämiin. Kolvin seinämien ylikuumennusta ja orgaanisen aineksen tarttumista kolvin seinämiin on vältettävä.
Kun liuos on muuttunut kirkkaaksi ja kirkkaanvihreäksi, keittämistä jatketaan vielä kahden tunnin ajan; tämän jälkeen kolvin annetaan jäähtyä.
5.2 Tislaus
Kolviin lisätään varovasti riittävä määrä vettä, jotta sulfaatit liukenisivat täysin. Annetaan jäähtyä ja lisätään tarvittaessa muutama sinkkirae (3.3). Jatketaan 5.2.1 tai 5.2.2 kohdan mukaisesti.
5.2.1
Tislauslaitteen keräilyastiaan lisätään oletetun typpipitoisuuden mukaan 25 ml rikkihappoa (3.5) tai (3.7) tarkasti mitattuna. Lisätään muutama pisara metyylipunaindikaattoria (3.8).
Kolvi yhdistetään tislauslaitteen jäähdyttimeen ja jäähdyttimen pää upotetaan keräilyastiassa olevaan liuokseen vähintään 1 cm:n syvyyteen (ks. 8.3 huomautus). Kolviin lasketaan hitaasti 100 ml natriumhydroksidiliuosta (3.9) siten, ettei ammoniakkia katoa (ks. 8.1 huomautus). Kolvia kuumennetaan siten, että ammoniakki tislautuu kokonaan.
5.2.2
Kun tisleestä titrataan ammoniakki manuaalisesti, noudatetaan alla olevaa menettelyä. Jos tislauslaite on täysin automaattinen ja suorittaa ammoniakin titraamisen tisleestä, noudatetaan valmistajan käyttöohjeita.
Keräilyastia, joka sisältää 25–30 ml boorihappoliuosta (3.18), asetetaan jäähdyttimen ulostulon alle siten, että poistoputki jää boorihappoliuoksen pinnan alapuolelle. Tislauslaite säädetään annostelemaan 50 ml natriumhydroksidiliuosta (3.9). Tislauslaitetta käytetään valmistajan ohjeiden mukaan ja tislataan pois natriumhydroksidiliuoksen lisäyksen tuloksena vapautunut ammoniakki. Tisle kerätään boorihappoa sisältävään vastaanottoliuokseen. Tisleen määrä (vesihöyrytislauksen aika) riippuu näytteen sisältämän typen määrästä. Valmistajan ohjeita noudatetaan.
|
Huomautus |
: |
Puoliautomaattisessa tislauslaitteessa natriumhydroksidiylimäärän lisäys ja vesihöyrytislaus tapahtuvat automaattisesti. |
5.3 Titraus
Jatketaan 5.3.1 tai 5.3.2 kohdan mukaisesti.
5.3.1
Keräilyastiassa oleva rikkihappoylimäärä titrataan käytetyn rikkihapon konsentraation mukaan natriumhydroksidiliuoksella (3.10 tai 3.11), kunnes loppupiste saavutetaan.
5.3.2
Keräilyastian sisältö titrataan kloorivetyhapon standardiliuoksella (3.19) tai rikkihapon standardiliuoksella (3.6) byrettiä käyttäen ja todetaan käytetyn titrausliuoksen määrä.
Jos loppupisteen toteamiseksi käytetään kolorimetristä menetelmää, loppupiste on saavutettu, kun sisältöön alkaa ilmestyä vaaleanpunaista väriä. Byretin lukema arvioidaan 0,05 ml:n tarkkuudella. Loppupisteen havaitseminen saattaa olla helpompaa valaistun magneettisekoittimen tai fotometrisen detektorin avulla.
Tämä voidaan tehdä automaattisesti vesihöyrytislaimella, jossa on automaattinen titraus.
Tislauslaitteen tai tislaus/titrauslaitteen käytössä noudatetaan valmistajan ohjeita.
|
Huomautus |
: |
Kun käytetään automaattista titrausjärjestelmää, titraus alkaa välittömästi tislauksen alettua ja kun 1-prosenttinen boorihappoliuos (3.18) on kulunut. Käytettäessä täysin automaattista tislausyksikköä ammoniakki voidaan titrata automaattisesti ja loppupiste detektoida käyttämällä potentiometristä pH-järjestelmää. Tässä tapauksessa käytetään automaattista titrauslaitetta, jossa on pH-mittari. pH-mittari on kalibroitava asianmukaisesti välille pH 4–7 noudattaen tavanomaisia laboratorioiden pH-kalibrointimenettelyjä. Titrauksen pH-loppupiste saavutetaan, kun pH on 4,6, mikä on titrauskäyrän jyrkin kohta (käännekohta). |
5.4 Nollakoe
Reagenssien typettömyyden varmistamiseksi suoritetaan nollakoe (hajotus, tislaus ja titraus) käyttämällä 1 g sakkaroosia (3.14) näytteen sijasta.
6. Tulosten laskeminen
Tulokset lasketaan 6.1 tai 6.2 kohdan mukaisesti.
6.1 Tuloksen laskeminen 5.3.1 kohdan mukaisessa titrauksessa
Raakavalkuaispitoisuus ilmaistuna painoprosentteina lasketaan seuraavaa kaavaa käyttäen:

jossa:
|
V0 |
= |
nollakokeessa kulunut NaOH:n (3.10 tai 3.11) määrä (ml), |
|
V1 |
= |
näytteen titrauksessa kulunut NaOH:n (3.10 tai 3.11) määrä (ml), |
|
c |
= |
natriumhydroksidin (3.10 tai 3.11) konsentraatio (mol/l), |
|
m |
= |
näytteen paino (g). |
6.2 Tuloksen laskeminen 5.3.2 kohdan mukaisessa titrauksessa
6.2.1
Raakavalkuaispitoisuus ilmaistuna painoprosentteina lasketaan seuraavalla kaavalla:
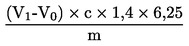
jossa:
|
m |
= |
näytteen paino (g), |
|
c |
= |
kloorivetyhapon standardiliuoksen (3.19) konsentraatio (mol/l), |
|
V0 |
= |
nollakokeessa kuluneen kloorivetyhapon määrä (ml), |
|
V1 |
= |
näytteen titrauksessa kuluneen kloorivetyhapon määrä (ml). |
6.2.2
Raakavalkuaispitoisuus ilmaistuna painoprosentteina lasketaan seuraavalla kaavalla:

jossa:
|
m |
= |
näytteen paino (g), |
|
c |
= |
rikkihapon standardiliuoksen (3.6) konsentraatio (mol/l), |
|
V0 |
= |
nollakokeessa kuluneen rikkihapon (3.6) määrä (ml), |
|
V1 |
= |
näytteen titrauksessa kuluneen rikkihapon (3.6) määrä (ml). |
7. Menetelmän varmistus
7.1 Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla suurempi kuin:
|
— |
0,2 % absoluuttisena arvona, kun raakavalkuaispitoisuus on alle 20 %, |
|
— |
1,0 %, suhteellisena arvona suuremmasta lukuarvosta, kun raakavalkuaispitoisuus on 20–40 %, |
|
— |
0,4 % absoluuttisena arvona, kun raakavalkuaispitoisuus on yli 40 %. |
7.2 Tarkkuus
Määritys (hajotus, tislaus ja titraus) tehdään 1,5–2,0 grammalla asetanilidia (3.13) siten, että läsnä on 1 grammaa sakkaroosia (3.14); 1 g asetanilidia kuluttaa 14,80 ml rikkihappoa (3.5). Saannon on oltava vähintään 99 %.
8. Huomautukset
|
8.1 |
Välineet voivat olla käsikäyttöisiä, puoliautomaattisia tai automaattisia. Jos liuos joudutaan siirtämään astiasta toiseen hajotuksen ja tislauksen välillä, siirto on suoritettava siten, ettei häviöitä tapahdu. Jos tislauslaitteen kolvissa ei ole tiputussuppiloa, lisätään natriumhydroksidi välittömästi ennen kolvin liittämistä jäähdyttimeen kaataen liuos hitaasti kolvin seinämää myöten. |
|
8.2 |
Jos hajotettu tuote kiinteytyy, määritys on aloitettava alusta käyttämällä suurempaa määrää rikkihappoa (3.4) kuin edellä on mainittu. |
|
8.3 |
Kun tuotteiden typpipitoisuus on alhainen, voidaan rikkihapon (3.7) määrä keräilyastiassa tarvittaessa vähentää 10–15 ml:aan ja täyttää 25 ml:ksi vettä lisäämällä. |
|
8.4 |
Raakavalkuaisen rutiinimäärityksissä voidaan käyttää vaihtoehtoisia menetelmiä, mutta vertailumenetelmänä on käsillä olevassa C osassa kuvailtu Kjeldahlin menetelmä. Vaihtoehtoisilla menetelmillä (kuten Dumas-menetelmällä) saatujen tulosten vastaavuus on osoitettava erikseen kunkin analysoitavan materiaalin osalta. Koska vaihtoehtoisilla menetelmillä saadut tulokset saattavat vastaavuuden varmistamisesta huolimatta hieman poiketa vertailumenetelmällä saaduista tuloksista, määritysselosteessa on mainittava, mitä menetelmää raakavalkuaisen määrityksessä on käytetty. |
D. UREAN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehujen ureapitoisuus.
2. Periaate
Näyte suspendoidaan veteen sellaisen aineen läsnä ollessa, joka saa suspension kirkastumaan. Suspensio suodatetaan. Suodoksen ureamäärä määritetään sen jälkeen kun siihen on lisätty 4-dimetyyliaminobentsaldehydiä (4-DMAB) mittaamalla liuoksen absorbanssi 420 nm:n aallonpituudella.
3. Reagenssit
|
3.1 |
4-dimetyyliaminobentsaldehydiliuos: 1,6 g 4-DMAB:ta liuotetaan 100 ml:aan 96-prosenttista etanolia ja siihen lisätään 10 ml kloorivetyhappoa (ρ201,19 g/ml). Tämä reagenssi säilyy korkeintaan kahden viikon ajan. |
|
3.2 |
Carrez-liuos I: veteen liuotetaan 21,9 g sinkkiasetaattia Zn(CH3COO)2 2H2O ja 3 g jääetikkaa. Tilavuus säädetään 100 ml:ksi vedellä. |
|
3.3 |
Carrez-liuos II: Liuotetaan veteen 10,6 g kaliumferrosyanidia K4Fe(CN)6 3H2O. Tilavuus säädetään 100 ml:ksi vedellä. |
|
3.4 |
Aktiivihiili, joka ei absorboi ureaa (tarkastettava). |
|
3.5 |
Urealiuos 0,1 % (w/v). |
4. Välineistö
|
4.1 |
Sekoitin: noin 35–40 kierr./min. |
|
4.2 |
Lasihiostulpallisia koeputkia 160 × 16 mm. |
|
4.3 |
Spektrofotometri. |
5. Menettely
5.1 Näytteen määritys
Punnitaan 1 mg:n tarkkuudella 2 g näytettä sekä 1 g aktiivihiiltä (3.4) 500 ml:n mittapulloon. Lisätään 400 ml vettä ja 5 ml Carrez I -liuosta (3.2), sekoitetaan noin 30 sekunnin ajan ja lisätään 5 ml Carrez II -liuosta (3.3). Pidetään sekoittimessa 30 minuutin ajan. Lisätään vettä merkkiin asti, sekoitetaan ja suodatetaan.
Läpinäkyvästä, värittömästä suodoksesta pipetoidaan 5 ml lasihiostulpallisiin koeputkiin, lisätään 5 ml 4-DMAB -liuosta (3.1) ja sekoitetaan. Putket asetetaan 20 C:ssa (+/– 4 C) olevaan vesihauteeseen. Viidentoista minuutin kuluttua mitataan näyteliuoksen absorbanssi spektrofotometrillä 420 nm:n aallonpituudella. Lukemaa verrataan reagensseista valmistettuun nollakoeliuokseen.
5.2 Kalibrointikäyrä
Pipetoidaan 100 ml:n mittapulloihin 1, 2, 4, 5 ja 10 ml urealiuosta (3.5) ja täytetään merkkiin asti vedellä. Kutakin liuosta pipetoidaan koeputkiin 5 ml, lisätään 5 ml 4-DMAB -liuosta (3.1), homogenoidaan ja niiden absorbanssi mitataan edellä kuvatulla tavalla verrattuna kontrolliliuokseen, joka sisältää 5 ml 4-DMAB:ta ja 5 ml vettä ja jossa ei ole ureaa. Piirretään kalibrointikäyrä.
6. Tulosten laskeminen
Urean määrä näytteessä määritetään kalibrointikäyrää käyttäen.
Tulokset ilmoitetaan prosentteina näytteestä.
7. Huomautukset
|
7.1 |
Kun urean määrä ylittää 3 %, näytteen määrä vähennetään 1 g:ksi tai alkuperäinen liuos laimennetaan siten, ettei siinä ole yli 50 mg ureaa 500 ml:ssa. |
|
7.2 |
Kun ureapitoisuus on alhainen, näytteen kokoa suurennetaan, kunhan suodos säilyy läpinäkyvänä ja värittömänä. |
|
7.3 |
Jos näyte sisältää yksinkertaisia typpiyhdisteitä, kuten aminohappoja, optinen tiheys on mitattava 435 nm:ssä. |
E. HAIHTUVIEN TYPPIPITOISTEN EMÄSTEN MÄÄRITYS
I MIKRODIFFUUSIO
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehuista haihtuvien typpipitoisten emästen pitoisuus ammoniakkina ilmoitettuna.
2. Periaate
Näyte uutetaan vedellä, liuos kirkastetaan ja suodatetaan. Haihtuvat typpipitoiset emäkset vapautetaan mikrodiffuusiolla käyttäen kaliumkarbonaattiliuosta, sidotaan boorihappoliuokseen ja titrataan rikkihapolla.
3. Reagenssit
|
3.1 |
Trikloorietikkahappoliuos, 20 % (w/v). |
|
3.2 |
Indikaattori: liuotetaan 33 mg bromikresolivihreää ja 65 mg metyylipunaista 100 ml:aan 95–96-prosenttista (v/v) etanolia. |
|
3.3 |
Boorihappoliuos: liuotetaan 10 g boorihappoa 1 litran mittapullossa 200 ml:aan 95–96-prosenttista (v/v) etanolia ja 700 ml:aan vettä. Lisätään 10 ml indikaattoria (3.2). Sekoitetaan ja liuoksen väri säädetään tarvittaessa vaaleanpunaiseksi lisäämällä natriumhydroksidiliuosta. 1 ml tätä liuosta sitoo enintään 300 μg NH3:a. |
|
3.4 |
Kyllästetty kaliumkarbonaattiliuos: liuotetaan 100 g kaliumkarbonaattia 100 ml:aan kiehuvaa vettä. Annetaan jäähtyä ja suodatetaan. |
|
3.5 |
Rikkihappo, 0,01 mol/l. |
4. Välineistö
|
4.1 |
Sekoitin: noin 35–40 kierr./min. |
|
4.2 |
Lasisia tai muovisia Conway-maljoja (ks. oheinen kaavio). |
|
4.3 |
1/100 ml:n asteikolla varustettuja mikrobyrettejä. |
5. Menettely
Punnitaan 10 g näytettä 1 mg:n tarkkuudella 200 ml:n mittapulloon, johon lisätään 100 ml vettä. Pidetään sekoittimessa 30 minuutin ajan. Lisätään 50 ml trikloorietikkahappoliuosta (3.1), täytetään merkkiin asti vedellä, ravistellaan voimakkaasti ja suodatetaan laskostetun suodatinpaperin läpi.
Pipetoidaan Conway-maljan keskiympyrään 1 ml boorihappoliuosta (3.3) ja reunaympyrään 1 ml näytemaljasuodosta. Peitetään osittain rasvatulla kannella. Reunaympyrään lisätään nopeasti 1 ml kyllästettyä kaliumkarbonaattiliuosta (3.4) ja kansi suljetaan ilmatiiviisti. Maljaa pyöritetään varovasti vaakasuorassa tasossa siten, että nämä kaksi reagenssia sekoittuvat. Inkuboidaan vähintään neljä tuntia huoneenlämmössä tai yksi tunti 40 oC:ssa.
Boorihappoliuoksessa olevat haihtuvat emäkset titrataan rikkihapolla (3.5) mikrobyrettiä (4.3) käyttäen.
Nollakoe suoritetaan samalla tavalla, mutta ilman määritettävää näytettä.
6. Tulosten laskeminen
1 ml H2SO4:ää, 0,01 mol/l, vastaa 0,34 mg:aa ammoniakkia.
Tulos ilmoitetaan prosentteina näytteestä.
Toistettavuus
Samasta näytteestä suoritetun kahden rinnakkaismäärityksen tulosten välinen ero ei saa olla yli:
|
— |
10 %:a, suhteellisena arvona ilmoitettuna, kun ammoniakkipitoisuus on alle 1,0 %, |
|
— |
0,1 %:a, absoluuttisena arvona ilmoitettuna, kun ammoniakkipitoisuus on vähintään 1,0 %. |
7. Huomautus
Jos näytteen ammoniakkipitoisuus ylittää 0,6 %, on alkuperäinen suodos laimennettava.
CONWAY CELL
Scale 1/1

II TISLAUSMENETELMÄ
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää haihtuvien typpipitoisten emästen pitoisuus ammoniakkina ilmoitettuna kalajauhosta, joka ei käytännöllisesti katsoen sisällä ureaa. Menetelmä soveltuu ainoastaan näytteisiin, joiden ammoniakkipitoisuus on alle 0,25 %.
2. Periaate
Näyte uutetaan vedellä, liuos kirkastetaan ja suodatetaan. Haihtuvat typpipitoiset emäkset vapautuvat keitettäessä näyteliuosta magnesiumoksidin kanssa ja ne sidotaan tunnettuun määrään rikkihappoa, jonka ylimäärä titrataan takaisin natriumhydroksidiliuoksella.
3. Reagenssit
|
3.1 |
Trikloorietikkahappoliuos, 20 % (w/v). |
|
3.2 |
Magnesiumoksidi. |
|
3.3 |
Kuohunestoaine (esim. silikoni). |
|
3.4 |
Rikkihappo, 0,05 mol/l. |
|
3.5 |
Natriumhydroksidiliuos, 0,1 mol/l. |
|
3.6 |
Metyylipunaliuos, 0,3 % (w/v), joka on valmistettu 95–96-prosenttiseen (v/v) etanoliin. |
4. Välineistö
|
4.1 |
Sekoitin: noin 35–40 kierr./min. |
|
4.2 |
Kjeldahl-tyyppinen tislauslaitteisto. |
5. Menettely
Punnitaan 10 g näytettä 1 mg:n tarkkuudella 200 ml:n mittapulloon, johon lisätään 100 ml vettä. Pidetään sekoittimessa 30 minuutin ajan. Lisätään 50 ml trikloorietikkahappoliuosta (3.1), täytetään merkkiin asti vedellä, ravistellaan voimakkaasti ja suodatetaan laskostetun suodatinpaperin läpi.
Otetaan kirkasta liuosta sopiva määrä haihtuvien typpipitoisten emästen oletetun pitoisuuden kannalta (100 ml on yleensä sopiva määrä). Laimennetaan 200 ml:ksi ja lisätään 2 g magnesiumoksidia (3.2) ja muutama pisara kuohunestoainetta (3.3). Liuoksen on oltava lakmuspaperilla määritettynä emäksinen; muussa tapauksessa siihen lisätään hieman magnesiumoksidia (3.2). Jatketaan raakavalkuaispitoisuuden määrityksessä käytettävän menetelmän 5.2 ja 5.3 kohdan mukaisesti (tämän liitteen C osa).
Nollakoe suoritetaan samalla tavalla, mutta ilman määritettävää näytettä.
6. Tulosten laskeminen
1 ml H2SO4:ää, 0,05 mol/l, vastaa 1,7 mg:aa ammoniakkia.
Tulos ilmoitetaan prosentteina näytteestä.
Toistettavuus
Kahden samasta näytteestä suoritetun rinnakkaismäärityksen tulosten välinen ero ei saa olla suhteellisena arvona ilmoitettuna suurempi kuin 10 % ammoniakkimäärästä.
F. AMINOHAPPOJEN MÄÄRITYS (TRYPTOFAANIA LUKUUN OTTAMATTA)
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehujen vapaat aminohapot (synteettiset ja luonnon aminohapot) ja kokonaisaminohapot (peptideihin kiinnittyneet ja vapaat) aminohappoanalysaattorilla. Se sopii seuraaviin aminohappoihin: kyst(e)iini, metioniini, lysiini, treoniini, alaniini, arginiini, asparagiinihappo, glutamiinihappo, glysiini, histidiini, isoleusiini, leusiini, fenyylialaniini, proliini, seriini, tyrosiini ja valiini.
Menetelmällä ei eroteta toisistaan aminohappojen suoloja eikä aminohappojen D- ja L-muotoja. Se ei sovellu tryptofaanin eikä aminohappojen hydroksianalogien määritykseen.
2. Periaate
2.1 Vapaat aminohapot
Vapaat aminohapot uutetaan laimealla kloorivetyhapolla. Mukana uuttuneet typpipitoiset makromolekyylit saostetaan sulfosalisyylihapolla ja poistetaan suodattamalla. Suodatetun liuoksen pH säädetään arvoon 2,20. Aminohapot erotetaan ioninvaihtokromatografialla ja määritetään fotometrillä ninhydriinijohdoksina aallonpituudella 570 nm.
2.2 Kokonaisaminohapot
Menetelmä valitaan tutkittavien aminohappojen mukaan. Kyst(e)iini ja metioniini on hapetettava kysteiinihapoksi ja metioniinisulfoniksi ennen hydrolyysiä. Tyrosiini on määritettävä näytteistä, joita ei hapeteta ennen hydrolyysiä. Kaikki muut 1 kohdassa mainitut aminohapot voidaan määrittää joko hapetetusta tai hapettamattomasta näytteestä.
Hapettaminen tapahtuu 0 oC:ssa permuurahaishapon ja fenolin seoksella. Hapetusreagenssin ylimäärä hajotetaan natriumdisulfiitin avulla. Hapetettu tai hapettamaton näyte hydrolysoidaan kloorivetyhapolla (3.20) avulla 23 tunnin ajan. Hydrolyysituotteen pH säädetään arvoon 2,20. Aminohapot erotetaan ioninvaihtokromatografialla ja määritetään fotometrillä ninhydriinijohdoksina aallonpituudella 570 nm (proliini 440 nm).
3. Reagenssit
Veden on oltava kaksoistislattua tai vastaavan laatuista (johtokyky < 10 μS).
|
3.1 |
Vetyperoksidi, w (w/w) = 30 %. |
|
3.2 |
Muurahaishappo, w (w/w) = 98–100 %. |
|
3.3 |
Fenoli. |
|
3.4 |
Natriumdisulfiitti. |
|
3.5 |
Natriumhydroksidi. |
|
3.6 |
5-Sulfosalisyylihappo-dihydraatti. |
|
3.7 |
Kloorivetyhappo, tiheys noin 1,18 g/ml. |
|
3.8 |
Trinatriumsitraatti-dihydraatti. |
|
3.9 |
2,2'-Tiodietanoli (tiodiglykoli). |
|
3.10 |
Natriumkloridi. |
|
3.11 |
Ninhydriini. |
|
3.12 |
Petrolieetteri, kiehumislämpötila 40–60 oC. |
|
3.13 |
Norleusiini tai muu yhdiste, jota voidaan käyttää sisäisenä standardina. |
|
3.14 |
Typpikaasu (<10 ppm happea). |
|
3.15 |
1-Oktanoli. |
|
3.16 |
Aminohapot. |
|
3.16.1 |
Edellä 1 kohdassa luetellut standardina käytettävät yhdisteet. Puhtaat yhdisteet, jotka eivät sisällä kidevettä. Kuivataan tyhjiössä P2O5:n tai H2SO4:n päällä viikon ajan ennen käyttöä. |
|
3.16.2 |
Kysteiinihappo. |
|
3.16.3 |
Metioniinisulfoni. |
|
3.17 |
Natriumhydroksidiliuos, c = 7,5 mol/l: liuotetaan 300 g NaOH:ta (3.5) veteen ja laimennetaan 1 litraksi. |
|
3.18 |
Natriumhydroksidiliuos, c = 1 mol/l: liuotetaan 40 g NaOH:ta (3.5) veteen ja laimennetaan 1 litraksi. |
|
3.19 |
Muurahaishappo-fenoliliuos: sekoitetaan 889 g muurahaishappoa (3.2) ja 111 g vettä ja lisätään 4,73 g fenolia (3.3). |
|
3.20 |
Hydrolyysiseos, c = 6 mol HCl/l ja 1 g fenolia litrassa: lisätään 1 g fenolia (3.3) 492 ml:aan HCl (3.7) ja laimennetaan vedellä 1 litraksi. |
|
3.21 |
Uuttoseos, c = 0,1 mol HCl /l ja 2 % tiodiglykolia litrassa: laimennetaan 8,2 ml HCl (3.7) noin 900 ml:aan vettä, lisätään 20 ml tiodiglykolia (3.9) ja täytetään vedellä yhdeksi litraksi (ei sekoiteta suoraan aineita 3.7 ja 3.9). |
|
3.22 |
5-Sulfosalisyylihappo, ß = 6 %: Liuotetaan 60 g 5-sulfosalisyylihappoa (3.6) veteen ja laimennetaan vedellä litraksi. |
|
3.23 |
Hapetusseos (permuurahaishappo-fenoli): sekoitetaan 0,5 ml vetyperoksidia (3.1) 4,5 ml:aan muurahaishappo-fenoliliuosta (3.19) pienessä dekantterilasissa. Inkuboidaan 20–30 oC:ssa tunnin ajan, jotta saadaan permuurahaishappoa, annetaan sitten jäähtyä jäävesihauteessa (15 min) ennen näytteeseen lisäämistä. Varoitus: vältettävä ihokosketusta ja käytettävä suojavaatteita. |
|
3.24 |
Sitraattipuskuriliuos, c = 0,2 mol Na+/l, pH 2,20: liuotetaan 19,61 g natriumsitraattia (3.8), 5 ml tiodiglykolia (3.9), 1 g fenolia (3.3) ja 16,50 ml HCl (3.7) noin 800 ml:aan vettä. Säädetään pH arvoon 2,20. Laimennetaan vedellä yhdeksi litraksi. |
|
3.25 |
Eluointipuskurit, valmistetaan analysaattorin vaatimusten mukaisesti (4.9). |
|
3.26 |
Ninhydriinireagenssi, valmistetaan analysaattorin vaatimusten mukaisesti (4.9). |
|
3.27 |
Aminohappojen standardiliuokset. Nämä liuokset säilytetään alle 5 oC:n lämpötilassa. |
|
3.27.1 |
Aminohappojen standardin kantaliuos (3.16.1). c = 2,5 μmol/ml kutakin aminohappoa kloorivetyhapossa. Saatavana kaupallisesti. |
|
3.27.2 |
Kysteiinihapon ja metioniinisulfonin standardin kantaliuos, c = 1,25 μmol/ml. Liuotetaan 0,2115 g kysteiinihappoa (3.16.2) ja 0,2265 g metioniinisulfonia (3.16.3) sitraattipuskuriin (3.24) litran mittapullossa ja täytetään merkkiin asti sitraattipuskurilla. Säilytetään alle 5 oC:n lämpötilassa enintään 12 kuukautta. Tätä liuosta ei tarvita, jos standardin kantaliuos (3.27.1) sisältää kysteiinihappoa ja metioniinisulfonia. |
|
3.27.3 |
Sisäisen standardin kantaliuos, esimerkiksi norleusiini, c = 20 μmol/ml. Liuotetaan 0,6560 g norleusiinia (3.13) sitraattipuskuriin (3.24) mittapullossa ja täytetään 250,0 ml:ksi sitraattipuskurilla (3.24). Säilytetään alle 5 oC:n lämpötilassa enintään 6 kuukautta. |
|
3.27.4 |
Aminohappojen kalibrointiliuos hydrolysaateille: kysteiinihappo ja metioniinisulfoni, c = 5 nmol/50 μl, muut aminohapot c = 10 nmol/50 μl. Liuotetaan 2,2 g natriumkloridia (3.10) 30 ml:aan sitraattipuskuria (3.24) 100 ml:n dekantterilasissa. Lisätään 4,00 ml aminohappojen standardin kantaliuosta (3.27.1), 4,00 ml kysteiinihapon ja metioniinisulfonin standardin kantaliuosta (3.27.2) ja tarvittaessa 0,50 ml sisäisen standardin kantaliuosta (3.27.3). Säädetään pH arvoon 2,20 natriumhydroksidilla (3.18). Siirretään kvantitatiivisesti 50 ml:n mittapulloon, täytetään merkkiin asti sitraattipuskurilla (3.24) ja sekoitetaan. Säilytetään alle 5 oC:n lämpötilassa enintään 3 kuukautta. Katso myös huomautukset 9.1 kohdassa. |
|
3.27.5 |
Aminohappojen kalibrointiliuos 5.3.3.1 kohdan mukaisesti valmistetuille hydrolysaateille ja uutteille (5.2). Kalibrointiliuos valmistetaan 3.27.4 kohdan mukaisesti kuitenkin jättäen natriumkloridi pois. Säilytetään alle 5 oC:n lämpötilassa enintään 3 kuukautta. |
4. Välineistö
|
4.1 |
100 ml:n tai 250 ml:n pyörökolvi, joka on varustettu palautusjäähdyttimellä. |
|
4.2 |
Borosilikaattilasinen 100 ml:n pullo, jossa on kumi/teflontiivisteinen kierrettävä uuninkestävä korkki (esimerkiksi Duran, Schott). |
|
4.3 |
Koneellisella ilmanvaihdolla varustettu ja lämpötilaltaan säädettävä kuivausuuni, jonka tarkkuus on parempi kuin ± 2 oC. |
|
4.4 |
pH-mittari (lukemat kolmen desimaalin tarkkuudella). |
|
4.5 |
Kalvosuodatin (0,2 μm). |
|
4.6 |
Sentrifugi. |
|
4.7 |
Vakuumipyöröhaihdutin. |
|
4.8 |
Mekaaninen ravistelija tai magneettisekoitin. |
|
4.9 |
Aminohappoanalysaattori tai HPLC-laitteisto, joissa on ioninvaihtokolonni, välineistö kolonnin jälkeistä ninhydriiniderivointia varten ja fotometri. Kolonni täytetään sellaisella sulfonoidulla polystyreenihartsilla, jolla voidaan erottaa eri aminohapot toisistaan ja muista ninhydriinin kanssa reagoivista yhdisteistä. Puskuri- ja ninhydriinipumpun virtausnopeuden stabiilisuuden tulee olla ±0,5 % ajanjaksona, jona analysoidaan sekä standardiliuos että näyte. Tietyissä aminohappoanalysaattoreissa voidaan käyttää hydrolyysimenetelmää, jossa hydrolysaatin natriumpitoisuus on c = 0,8 mol/l ja se sisältää näin kaiken hapetuksessa jäljelle jääneen muurahaishapon. Muut laitteet eivät erota tyydyttävästi tiettyjä aminohappoja, jos hydrolysaatti sisältää liikaa muurahaishappoa ja/tai natriumionipitoisuus on suuri. Tässä tapauksessa hapon määrää vähennetään haihduttamalla hydrolysaatti noin 5 ml:aan ennen pH:n säätämistä. Haihduttamisen on tapahduttava tyhjiössä enintään 40 oC:ssa. |
5. Menettely
5.1 Näytteen valmistaminen
Näyte jauhetaan siten, että se läpäisee 0,5 mm:n seulan. Näytteet, joiden kosteuspitoisuus on suuri, on ilmakuivattava korkeintaan 50 oC:n lämpötilassa tai kylmäkuivattava ennen jauhamista. Näytteet, joiden rasvapitoisuus on suuri, on uutettava petrolieetterillä (3.12) ennen jauhamista.
5.2 Vapaiden aminohappojen määrittäminen rehuista ja esiseoksista
Punnitaan 0,2 mg:n tarkkuudella sopiva määrä (1–5 g) valmistettua näytettä (5.1) erlenmeyerkolviin ja lisätään 100,0 ml uuttoseosta (3.21). Ravistellaan seosta 60 minuuttia mekaanisella ravistelijalla tai magneettisekoittimella (4.8). Annetaan sakan laskeutua ja pipetoidaan 10,0 ml supernatanttiliuosta dekantterilasiin.
Lisätään koko ajan sekoittaen 5,0 ml sulfosalisyylihappoliuosta (3.22) ja jatketaan sekoittamista 5 minuutin ajan magneettisekoittimella. Suodatetaan tai sentrifugoidaan liuos mahdollisen sakan erottamiseksi. Pipetoidaan 10,0 ml saatua liuosta 100 ml:n dekantterilasiin, säädetään pH arvoon 2,20 natriumhydroksidiliuoksella (3.18), huuhdotaan sopivan kokoiseen mittapulloon sitraattipuskurilla (3.24) ja täytetään merkkiin asti puskuriliuoksella (3.24).
Jos käytetään sisäistä standardia (3.27.3), sitä lisätään 1,00 ml lopullisen liuoksen kutakin 100 ml:aa kohden ja täytetään merkkiin asti purskuriliuoksella (3.24).
Siirrytään kromatografiavaiheeseen 5.4 kohdan mukaisesti.
Jos kromatografiaa ei tehdä samana päivänä, uutteet säilytetään alle 5 oC:n lämpötilassa.
5.3 Kokonaisaminohappojen määrittäminen
5.3.1
Punnitaan 0,2 mg:n tarkkuudella 0,1–1 g valmistettua näytettä (5.1):
|
— |
100 ml:n pyörökolviin (4.1) avointa hydrolyysiä varten (5.3.2.3), tai |
|
— |
250 ml:n pyörökolviin (4.1), jos tarvitaan alhaista natriumpitoisuutta (5.3.3.1), tai |
|
— |
100 ml:n kierrekorkilliseen pulloon (4.2) suljettua hydrolyysiä varten (5.3.2.4). |
Punnitun näytemäärän typpipitoisuuden on oltava noin 10 mg ja kosteuspitoisuuden enintään 100 mg.
Asetetaan pyörökolvi tai pullo jää-vesihauteeseen ja jäähdytetään 0 oC:seen, lisätään 5 ml hapetusseosta (3.23) ja sekoitetaan kaarevakärkisellä lasilastalla. Suljetaan kolvi/pullo lastoineen ilmatiiviin kalvon avulla, asetetaan jää-vesihaude näin suljettuine astioineen jääkaappiin, jonka lämpötila on 0 oC, ja jätetään sinne 16 tunniksi. Tämän jälkeen se otetaan jääkaapista ja hajotetaan hapetusreagenssin ylimäärä lisäämällä 0,84 g natriumdisulfiittia (3.4).
Siirrytään 5.3.2.1 kohtaan.
5.3.2
5.3.2.1
Edellä 5.3.1. kohdan mukaisesti valmistettuun hapetettuun näytteeseen lisätään 25 ml hydrolyysiseosta (3.20) huolehtien siitä, että astian seinämiin ja lastaan jäänyt näyte huuhdellaan pois.
Jatketaan valitun hydrolyysimenetelmän mukaan 5.3.2.3 tai 5.3.2.4 kohdasta.
5.3.2.2
Punnitaan 100 ml:n tai 200 ml:n pyörökolviin (4.1) tai 100 ml:n kierrekorkilliseen pulloon (4.2) 0,1–1,0 g valmistettua näytettä (5.1) 0,2 mg:n tarkkuudella. Punnitun näytemäärän typpipitoisuuden on oltava noin 10 mg. Lisätään varovasti 25 ml hydrolyysiseosta (3.20) ja sekoitetaan näytteeseen. Jatketaan joko 5.3.2.3 tai 5.3.2.4 kohdasta.
5.3.2.3
Lisätään kolme lasihelmeä kolvissa olevaan seokseen (valmistettu 5.3.2.1 tai 5.3.2.2 kohdan mukaisesti) ja keitetään tasaisesti kuplittaen palautusjäähdyttimen alla 23 tuntia. Hydrolyysin jälkeen huuhdellaan jäähdytin 5 ml:lla sitraattipuskuria (3.24). Kolvi irrotetaan ja jäähdytetään jäähauteessa.
Jatketaan 5.3.3 kohdan mukaisesti.
5.3.2.4
Asetetaan 5.3.2.1 tai 5.3.2.2 kohdan mukaisesti valmistettua näyteseosta sisältävä pullo uuniin (4.3) 110 oC:seen. Ensimmäisen tunnin aikana pidetään kierrekorkki sulkematta pullon päällä, jottei kaasun muodostuksen vuoksi syntyisi painetta ja jotta vältyttäisiin räjähdyksiltä. Pulloa ei saa sulkea korkilla. Tunnin kuluttua pullo suljetaan kierrekorkilla ja sen annetaan olla uunissa (4.3) 23 tunnin ajan. Hydrolyysin jälkeen pullo otetaan uunista, avataan korkki varovasti ja asetetaan pullo jää-vesihauteeseen. Annetaan jäähtyä.
Riippuen tavasta, jolla pH säädetään (5.3.3), siirretään pullon sisältö kvantitatiivisesti sitraattipuskurin avulla (3.24) 250 ml:n dekantterilasiin tai 250 ml:n pyörökolviin.
Jatketaan 5.3.3 kohdan mukaisesti.
5.3.3
Aminohappoanalysaattorin (4.9) natriuminsietokyvyn mukaan noudatetaan 5.3.3.1 tai 5.3.3.2 kohtaa pH:n säätämiseksi.
5.3.3.1
On suositeltavaa käyttää sisäisen standardin perusliuosta (3.27.3), jos käytetään aminohappoanalysaattoreita, jotka edellyttävät alhaista natriumpitoisuutta (kun hapon määrää on vähennettävä).
Tässä tapauksessa lisätään 2,00 ml sisäisen standardin perusliuosta (3.27.3) hydrolysaattiin ennen haihdutusta tai sen jälkeen.
Lisätään 2 tippaa 1-oktanolia (3.15) 5.3.2.3 tai 5.3.2.4 kohdan mukaisesti saatuun hydrolysaattiin.
Haihdutetaan näyte 5–10 ml:aan pyöröhaihduttimella (4.7), jonka hauteen lämpötila on 40 oC. Jos tilavuus pienenee vahingossa alle viiden millilitran, hydrolysaatti on hylättävä ja analyysi aloitettava uudelleen.
pH säädetään arvoon 2,20 natriumhydroksidiliuoksella (3.18) ja jatketaan kohdasta 5.3.4.
5.3.3.2
Kohdan 5.3.2.3 tai 5.3.2.4 mukaisesti saatu hydrolysaatti neutraloidaan osittain lisäämällä varovasti koko ajan sekoittaen 17 ml natriumhydroksidiliuosta (3.17) ja huolehditaan siitä, että lämpötila on alle 40 oC.
Lopullinen pH:n säätäminen 2,20:een tapahtuu huoneen lämpötilassa 3.17 kohdan mukaisen natriumhydroksidiliuoksen avulla ja lopuksi 3.18 kohdan mukaisella natriumhydroksidiliuoksella. Siirrytään 5.3.4 kohtaan.
5.3.4
Siirretään kvantitatiivisesti hydrolysaatti, jonka pH on säädetty (5.3.3.1 tai 5.3.3.2.), sitraattipuskurilla (3.24) 200 ml:n mittapulloon ja täytetään merkkiin asti puskurilla (3.24).
Jos sisäistä standardia ei ole vielä käytetty, lisätään sitä 2,00 ml (3.27.3) ja täytetään merkkiin asti sitraattipuskurilla (3.24). Sekoitetaan huolellisesti.
Siirrytään kromatografiavaiheeseen (5.4).
Jos kromatografiaa ei tehdä samana päivänä, näyteliuokset säilytetään alle 5 oC:n lämpötilassa.
5.4 Kromatografia
Annetaan uutteen (5.2) tai hydrolysaatin (5.3.4) tasoittua huoneenlämpöisiksi ennen kromatografiaa. Ravistellaan seosta ja suodatetaan sopiva määrä 0,2 μm:n kalvosuodattimen läpi (4.5). Saatu kirkas liuos analysoidaan aminohappoanalysaattorilla käyttäen ioninvaihtokromatografiaa (4.9).
Injektio voidaan suorittaa käsin tai automaattisesti. On tärkeää, että kolonniin lisätään aina sama määrä (±0,5 %) vertailu- tai näyteliuosta, paitsi silloin kun käytetään sisäistä standardia, ja että natriumin ja aminohappojen suhde standardi- ja näyteliuoksissa on mahdollisimman lähellä toisiaan.
Kalibrointiajojen tiheys riippuu ninhydriinireagenssin ja analysaattorin toiminnan stabiilisuudesta. Standardi- tai näyteliuosta laimennetaan sitraattipuskurilla (3.24), siten, että standardiliuoksen antama piikin pinta-ala on 30–200 % näytteen aminohapon antaman piikin pinta-alasta.
Aminohappojen kromatografia vaihtelee jonkin verran käytetyn analysaattorin tyypin ja käytetyn hartsin mukaan. Valitun menetelmän on mahdollistettava aminohappojen erottaminen toisistaan ja muista ninhydriinin kanssa reagoivista aineista. Pylvääseen ruiskutettujen yhdisteiden pitoisuuden muutosten on käyttöalueella annettava lineaarinen vaste.
Analysoitaessa aminohappojen ekvimolaarisia liuoksia sovelletaan seuraavassa kappaleessa määriteltyjä laakson ja piikin välisiä suhteita. Ekvimolaarisen liuoksen tulee sisältää vähintään 30 % kunkin aminohapon siitä enimmäismäärästä, joka voidaan määrittää tarkasti käytetyllä aminohappoanalysaattorilla (4.9).
Kromatogrammissa treoniinin ja seriinin osittain päällekkäisten piikkien välisen laakson suhde matalamman piikin korkeuteen ei saa olla suurempi kuin 2:10. (Jos määritetään ainoastaan kyst(e)iiniä, metioniiniä, treoniiniä ja lysiiniä, vierekkäisten piikkien riittämätön erottuminen toisistaan häiritsee määritystä.) Muiden aminohappojen osalta erottumisen on oltava parempi kuin 1:10.
Kromatografiajärjestelmän on pystyttävä erottamaan lysiini lysiiniartefaktoista ja ornitiinistä.
6. Tulosten laskeminen
Näytteen ja vertailuliuoksen piikkien pinta-ala mitataan kunkin yksittäisen aminohapon osalta ja määrä (X) lasketaan seuraavan kaavan mukaisesti; tulos ilmoitetaan grammoina aminohappoa näytekiloa kohden.

Jos sisäistä standardia käytetään, kerrotaan kaava vielä termillä 
|
A |
= |
analysoitavan piikin pinta-ala, hydrolysaatti tai uute |
|
B |
= |
analysoitavan piikin pinta-ala, standardiliuos |
|
C |
= |
sisäisen standardin piikin pinta-ala hydrolysaatissa tai uutteessa |
|
D |
= |
sisäisen standardin piikin pinta-ala kalibrointiliuoksessa |
|
M |
= |
analysoitavan aminohapon molekyylipaino |
|
c |
= |
standardin konsentraatio μmol/ml |
|
m |
= |
näytteen paino grammoina (korjataan alkuperäiseen painoon, jos kuivattu tai rasva poistettu) |
|
V |
= |
ml kokonaishydrolysaattia (5.3.4) tai ml uutteen laskettua kokonaislaimennosta (6.1). |
Sekä kystiini että kysteiini määritetään kysteiinihappona hapetetun näytteen hydrolysaateista, mutta ne lasketaan kystiiniksi (C6H12N2O4S2, M 240,30 g/mol) käyttäen molekyylipainoa 120,15 g/mol (= 0,5 × 240,30 g/mol).
Metioniini määritetään metioniinisulfonina hapetetun näytteen hydrolysaateista, mutta lasketaan metioniiniksi käyttäen metioniinin molekyylipainoa 149,21 g/mol.
Lisätty vapaa metioniini määritetään uuttamisen jälkeen metioniinina; laskennassa käytetään samaa molekyylipainoa.
|
6.1 |
Uutteiden kokonaislaimennoksen tilavuus (F) vapaiden aminohappojen määrittämistä varten (5.2) lasketaan seuraavasti: 
|
7. Menetelmän arviointi
Menetelmä on testattu vuonna 1990 tehdyssä kansainvälisessä laboratorioiden välisessä vertailussa käyttäen neljää eri rehua (sikojen täysrehu, broilereiden täysrehu, valkuaistiiviste ja esiseos). Tulosten keskiarvo ja standardipoikkeama poikkeavien arvojen poissulkemisen jälkeen on esitetty tämän kohdan taulukoissa.
Keskiarvot g/kg
|
Vertailuaine |
Aminohappo |
||||||
|
Treoniini |
Kyst(e)iini |
Metioniini |
Lysiini |
||||
|
Sikojen täysrehu |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|||
|
Broilereiden täysrehu |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|||
|
Valkuaistiiviste |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|||
|
Esiseos |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|||
|
|||||||
7.1 Toistettavuus
Toistettavuus edellä mainitussa laboratorioiden välisessä vertailussa on ilmaistu muodossa ”laboratorion sisäinen standardipoikkeama” alla olevassa taulukossa.
Laboratorion sisäinen standardipoikkeama (Sr) g/kg
|
Vertailuaine |
Aminohappo |
||||||
|
Treoniini |
Kyst(e)iini |
Metioniini |
Lysiini |
||||
|
Sikojen täysrehu |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|||
|
Broilereiden täysrehu |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|||
|
Valkuaistiiviste |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|||
|
Esiseos |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|||
|
|||||||
Variaatiokerroin (%) laskettuna laboratorion sisäisestä standardipoikkeamasta (Sr)
|
Vertailuaine |
Aminohappo |
||||||
|
Treoniini |
Kyst(e)iini |
Metioniini |
Lysiini |
||||
|
Sikojen täysrehu |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|||
|
Broilereiden täysrehu |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|||
|
Valkuaistiiviste |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|||
|
Esiseos |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|||
|
|||||||
7.2 Uusittavuus
Edellä mainitun vertailun laboratorioiden välisten standardipoikkeamien tulokset on esitetty seuraavassa taulukossa.
Standardointipoikkeama laboratorioiden välillä (SR) g/kg
|
Vertailuaine |
Aminohappo |
||||||
|
Treoniini |
Kyst(e)iini |
Metioniini |
Lysiini |
||||
|
Sikojen täysrehu |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|||
|
Broilereiden täysrehu |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|||
|
Valkuaistiiviste |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|||
|
Esiseos |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|||
|
|||||||
Laboratorioiden välisistä standardipoikkeamista (SR) laskettu variaatiokerroin (%)
|
Vertailuaine |
Aminohappo |
||||||
|
Treoniini |
Kyst(e)iini |
Metioniini |
Lysiini |
||||
|
Sikojen täysrehu |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|||
|
Broilereiden täysrehu |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|||
|
Valkuaistiiviste |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|||
|
Esiseos |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|||
|
|||||||
8. Vertailumateriaalin käyttö
Menetelmän oikea soveltaminen tarkistetaan sertifioidulla vertailumateriaalilla, jos sellainen on saatavilla, tehdyllä kaksoismäärityksellä. Kalibrointi sertifioidulla aminohappojen kalibrointiliuoksella on suotavaa.
9. Huomautukset
|
9.1 |
Aminohappoanalysaattoreiden välisten erojen vuoksi annettuja aminohappojen kalibrointiliuosten (3.27.4 ja 3.27.5) ja hydrolysaattien (5.3.4) lopullisia konsentraatioita on pidettävä suuntaa antavina. Laitteiston lineaarisen vasteen alue on tarkistettava kaikkien aminohappojen osalta. Standardiliuos laimennetaan sitraattipuskurilla siten, että piikit sijoittuvat alueen keskivaiheille. |
|
9.2 |
Kun hydrolysaattien analysointiin käytetään suuren erotuskyvyn nestekromatografialaitteistoa, koeolosuhteet on optimoitava valmistajan suositusten mukaisesti. |
|
9.3 |
Jos menetelmää sovelletaan rehuihin, jotka sisältävät enemmän kuin yhden prosentin kloridia (tiiviste, kivennäisaineita sisältävät ravinteet tai lisäravinteet), sillä saatetaan saada metioniinille liian pieniä arvoja, ja erityiskäsittely on tarpeen. |
G. TRYPTOFAANIN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmän avulla voidaan määrittää rehuissa olevan tryptofaanin kokonaismäärä ja vapaan tryptofaanin määrä. Menetelmällä ei voi erottaa D- ja L-muotoja toisistaan.
2. Periaate
Tryptofaanin kokonaismäärän määrittämiseksi näyte hydrolysoidaan emäksisissä olosuhteissa kyllästetyllä bariumhydroksidiliuoksella ja kuumennetaan 110 oC:ssa 20 tuntia. Hydrolyysin jälkeen lisätään sisäinen standardi.
Vapaan tryptofaanin määrittämiseksi näytettä uutetaan lievästi happamissa olosuhteissa yhdessä sisäisen standardin kanssa.
Tryptofaani ja sisäinen standardi hydrolysaatissa tai uutteessa määritetään HPLC:llä käyttäen fluoresenssidetektoria.
3. Reagenssit
|
3.1 |
Veden on oltava kaksoistislattua tai vastaavan laatuista (johtokyky < 10 μS/cm). |
|
3.2 |
Standardi: tryptofaani (puhtaus/pitoisuus ≥ 99 %), kuivattu tyhjiössä fosforipentoksidin päällä. |
|
3.3 |
Sisäinen standardi: α-metyylitryptofaani (puhtaus/pitoisuus ≥ 99 %), kuivattu tyhjiössä fosforipentoksidin päällä. |
|
3.4 |
Bariumhydroksidioktahydraatti (on varottava, ettei Ba(OH)2 8 H2O joudu liiaksi kosketuksiin ilman kanssa; tällöin muodostuisi BaCO3:a, joka voi häiritä määritystä) (ks. huomautus 9.3). |
|
3.5 |
Natriumhydroksidi. |
|
3.6 |
Ortofosforihappo, w (w/w) = 85 % |
|
3.7 |
Kloorivetyhappo, ρ201,19 g/ml. |
|
3.8 |
Metanoli, HPLC-laatua vastaava. |
|
3.9 |
Petrolieetteri, kiehumislämpötila 40–60 oC. |
|
3.10 |
Natriumhydroksidiliuos, c = 1 mol/l: Liuotetaan 40,0 g NaOH:ta (3.5) veteen ja täytetään 1 litraksi vedellä (3.1). |
|
3.11 |
Kloorivetyhappo, c = 6 mol/l: Mitataan 492 ml HCl:ää (3.7) ja täytetään 1 litraksi vedellä. |
|
3.12 |
Kloorivetyhappo, c = 1 mol/l: Mitataan 82 ml HCl:ää (3.7) ja täytetään 1 litraksi vedellä. |
|
3.13 |
Kloorivetyhappo, c = 0,1 mol/l: Mitataan 8,2 ml HCl:ää (3.7) ja täytetään 1 litraksi vedellä. |
|
3.14 |
Ortofosforihappo, c = 0,5 mol/l: Mitataan 34 ml ortofosforihappoa (3.6) ja täytetään 1 litraksi vedellä (3.1). |
|
3.15 |
Väkevä tryptofaaniliuos (3.2), c = 2,50 μmol/ml: Liuotetaan 500 ml:n mittapullossa 0,2553 g tryptofaania (3.2) kloorivetyhappoon (3.13) ja täytetään merkkiin asti kloorivetyhapolla (3.13). Säilyy –18 oC:ssa enintään 4 viikkoa. |
|
3.16 |
Konsentroitu sisäisen standardin liuos, c = 2,50 μmol/ml: Liuotetaan 500 ml:n mittapullossa 0,2728 g α-metyylitryptofaania (3.3) kloorivetyhappoon (3.13) ja täytetään merkkiin asti kloorivetyhapolla (3.13). Säilyy –18 oC:ssa enintään 4 viikkoa. |
|
3.17 |
Tryptofaanin ja sisäisen standardin kalibrointistandardiliuos: Mitataan 2,00 ml konsentroitua tryptofaaniliuosta (3.15) ja 2,00 ml konsentroitua sisäisen standardin (α-metyylitryptofaanin) liuosta (3.16). Laimennetaan vedellä (3.1) ja metanolilla (3.8) siten, että liuoksen tilavuus ja metanolipitoisuus on suunnilleen sama kuin valmiin hydrolysaatin (metanolia 10–30 %). Tämä liuos on valmistettava juuri ennen käyttöä. Liuos suojataan suoralta auringonvalolta valmistuksen aikana. |
|
3.18 |
Etikkahappo. |
|
3.19 |
1,1,1-Trikloori-2-metyyli-2-propanoli. |
|
3.20. |
Etanoliamiini w (w/w) > 98 %. |
|
3.21 |
Valmistetaan liuos, jossa on 1 g 1,1,1-trikloori-2-metyyli-2-propanolia (3.19) 100 ml:ssa metanolia (3.8). |
|
3.22 |
HPLC:ssä käytettävä liikkuva faasi: 3,00 g etikkahappoa (3.18) + 900 ml vettä (3.1) +50,0 ml 1,1,1-trikloori-2-metyyli-2-propanolia (3.19) metanolissa (3.21) (1g/100ml). pH säädetään arvoon 5,00 etanoliamiinilla (3.20). Täytetään 1 000 ml:ksi vedellä (3.1). |
4. Välineistö
|
4.1 |
HPLC-laitteisto, jossa on spektrofluorimetri-detektori. |
|
4.2 |
Nestekromatografiakolonni, 125 mm × 4 mm, C18, hartsin (pakkausaineen) hiukkaskoko 3 μm, tai vastaava. |
|
4.3 |
pH-mittari. |
|
4.4 |
Polypropyleenipullo, tilavuus 125 ml, jossa on leveä kaula ja kierretulppa. |
|
4.5 |
Kalvosuodatin 0,45 μm. |
|
4.6 |
Autoklaavi, 110 (± 2) oC, 1,4 (±0,1) bar. |
|
4.7 |
Mekaaninen ravistelija tai magneettisekoitin. |
|
4.8 |
Vortex-sekoitin. |
5. Menettely
5.1 Näytteiden valmistaminen
Näyte jauhetaan siten, että se läpäisee 0,5 mm:n seulan. Näytteet, joilla on suuri kosteuspitoisuus, on ilmakuivattava korkeintaan 50 oC:n lämpötilassa tai kylmäkuivattava ennen jauhamista. Näytteet, joilla on suuri rasvapitoisuus, on uutettava petrolieetterillä (3.9) ennen jauhamista.
5.2 Vapaan tryptofaanin määritys (uutteesta)
Punnitaan 1 mg:n tarkkuudella sopiva määrä (1–5 g) valmistettua näytettä (5.1) erlenmeyerkolviin. Lisätään 100,0 ml kloorivetyhappoa (3.13) ja 5,00 ml konsentroitua sisäisen standardin liuosta (3.16). Ravistellaan tai sekoitetaan 60 minuuttia mekaanisella ravistelijalla tai magneettisekoittimella (4.7). Annetaan sakan laskeutua ja pipetoidaan 10,0 ml supernatanttiliuosta dekantterilasiin. Lisätään 5 ml ortofosforihappoa (3.14). Säädetään pH arvoon 3 natriumhydroksidilla (3.10). Lisätään metanolia (3.8) niin että lopputilavuudessa on 10–30 % metanolia. Siirretään liuos sopivankokoiseen mittapulloon ja laimennetaan vedellä sopivaan tilavuuteen kromatografiaa varten (suunnilleen sama tilavuus kuin kalibrointistandardiliuoksella (3.17)).
Suodatetaan muutama ml liuosta 0,45 μm:n kalvosuodattimen (4.5) läpi ennen HPLC-kolonniin injektointia. Siirrytään kromatografiavaiheeseen 5.4 kohdan mukaisesti.
Standardiliuos ja uutteet suojataan suoralta auringonvalolta. Jos näyteliuoksia ei voida analysoida samana päivänä, uutteita voi säilyttää 5 oC:ssa enintään 3 päivää.
5.3 Tryptofaanin kokonaismäärän määritys (hydrolysaatista)
Punnitaan 0,2 mg:n tarkkuudella 0,1–1 g valmistettua näytettä (5.1) polypropyleenipulloon (4.4). Punnitun näytemäärän typpipitoisuuden on oltava noin 10 mg. Lisätään 8,4 g bariumhydroksidioktahydraattia (3.4) ja 10 ml vettä. Sekoitetaan vortex-sekoittimella (4.8) tai magneettisekoittimella (4.7). Teflon-päällystetty magneetti jätetään seokseen. Huuhdotaan pullon seinämät 4 ml:lla vettä. Pullo suljetaan löyhästi kierrekorkilla. Pullo pannaan autoklaaviin (4.6), jossa on kiehuvaa vettä, ja autoklavoidaan 30–60 minuutin ajan. Suljetaan autoklaavi ja autoklavoidaan 110 (± 2) oC:ssa 20 tuntia.
Lämpötila on laskettava juuri alle 100 oC:seen, ennen kuin autoklaavi avataan. Jotta Ba(OH)2 · 8 H2O ei kiteytyisi, lämpimään seokseen lisätään 30 ml huoneenlämpöistä vettä. Ravistellaan tai sekoitetaan varovasti. Lisätään 2,00 ml konsentroitua sisäisen standardin liuosta (α-metyylitryptofaania, 3.16). Jäähdytetään pulloja vesi/jäähauteessa 15 minuuttia.
Lisätään 5 ml ortofosforihappoa (3.14). Pullo pidetään jäähdytyshauteessa ja liuos neutraloidaan HCl:llä (3.11) sekoittaen samalla, ja pH säädetään arvoon 3,0 HCl:llä (3.12). Lisätään metanolia niin että lopputilavuudessa on 10–30 % metanolia. Siirretään liuos sopivankokoiseen mittapulloon ja laimennetaan vedellä tiettyyn sopivaan tilavuuteen kromatografiaa varten (esim. 100 ml). Metanolin lisääminen ei saa aiheuttaa saostumista.
Suodatetaan muutama ml liuosta 0,45 μm:n kalvosuodattimen (4.5) läpi ennen HPLC-kolonniin injektointia. Siirrytään kromatografiavaiheeseen 5.4 kohdan mukaisesti.
Standardiliuos ja hydrolysaatit suojataan suoralta auringonvalolta. Jos hydrolysaatteja ei voida analysoida samana päivänä, niitä voi säilyttää 5 oC:ssa enintään 3 päivää.
5.4 HPLC-määritys
Seuraavat isokraattisen erotuksen olosuhteet ovat ohjeellisia; ajo voidaan suorittaa muissa olosuhteissa, jos niillä saadaan samanlaiset tulokset (ks. myös huomautukset 9.1 ja 9.2):
|
Nestekromatografiakolonni (4.2): |
125 mm × 4 mm, C18, hiukkaskoko 3 μm, tai vastaava |
|
Kolonnin lämpötila: |
huoneenlämpötila |
|
Liikkuva faasi (3.22): |
3,00 g etikkahappoa (3.18) + 900 ml vettä (3.1) +50,0 ml liuosta (3.21), jossa on 1,1,1-trikloori-2-metyyli-2-propanolia (3.19) metanolissa (3.8) (1 g/100 ml). Säädetään pH arvoon 5,00 etanoliamiinilla (3.20). Täytetään 1 000 ml:ksi vedellä (3.1). |
|
Virtausnopeus: |
1 ml/min |
|
Kokonaisajoaika: |
noin 34 min |
|
Detektioaallonpituus: |
eksitaatio: 280 nm, emissio: 356 nm. |
|
Injektiotilavuus: |
20 μl. |
6. Tulosten laskeminen
Lasketaan tryptofaanin määrä (X) grammoina 100 grammassa näytettä.

|
A |
= |
sisäisen standardin piikin pinta-ala, kalibrointistandardiliuos (3.17) |
|
B |
= |
tryptofaanin piikin pinta-ala, uute (5.2) tai hydrolysaatti (5.3) |
|
V1 |
= |
kalibrointiliuokseen (3.17) lisätyn konsentroidun tryptofaaniliuoksen (3.15) tilavuus ml (2 ml) |
|
c |
= |
kalibrointiliuokseen (3.17) lisätyn konsentroidun tryptofaaniliuoksen (3.15) pitoisuus μmol/ml (= 2,50) |
|
V2 |
= |
uuttovaiheessa (5.2) lisätyn (= 5,00 ml) tai hydrolysaattiin (5.3) lisätyn (= 2,00 ml) konsentroidun sisäisen standardiliuoksen (3.16) tilavuus ml |
|
C |
= |
sisäisen standardin piikin pinta-ala, uute (5.2) tai hydrolysaatti (5.3) |
|
D |
= |
tryptofaanin piikin pinta-ala, kalibrointistandardiliuos (3.17) |
|
V3 |
= |
kalibrointiliuokseen (3.17) lisätyn konsentroidun sisäisen standardiliuoksen (3.16) tilavuus ml (= 2,00 ml) |
|
m |
= |
näytteen paino grammoina (korjataan alkuperäiseen painoon, jos kuivattu ja/tai rasva poistettu) |
|
M |
= |
tryptofaanin molekyylipaino (= 204,23 g/mol). |
7. Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla suurempi kuin 10 % suuremmasta tuloksesta.
8. Laboratorioiden välisen vertailun tulokset
Järjestetyssä EY-vertailussa (4. vertailututkimus) 12 laboratoriota analysoi kolme näytettä hydrolyysimenetelmän varmentamiseksi. Kustakin näytteestä tehtiin viisi rinnakkaismääritystä. Tulokset on esitetty seuraavassa taulukossa:
|
|
Näyte 1 Sian rehu |
Näyte 2 Sian rehu, johon on lisätty L-tryptofaania |
Näyte 3 Sian rehutiiviste |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Keskiarvo [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [%] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [%] |
6,3 |
6,0 |
2,2 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
keskiarvoon mukaan otettujen yksittäisten tulosten lukumäärä, kun on poistettu poikkeavat arvot (tunnistettu Cochranin ja Dixonin poikkeavien arvojen testillä) |
|
sr |
= |
toistettavuuden keskihajonta |
|
SR |
= |
uusittavuuden keskihajonta |
|
r |
= |
toistettavuus |
|
R |
= |
uusittavuus |
|
CVr |
= |
toistettavuuden variaatiokerroin, % |
|
CVR |
= |
uusittavuuden variaatiokerroin, %. |
Toisessa EY-vertailussa (3. vertailututkimus) 13 laboratoriota analysoi kaksi näytettä vapaan tryptofaanin uuttomenetelmän varmentamiseksi. Kustakin näytteestä tehtiin viisi rinnakkaismääritystä. Tulokset on esitetty seuraavassa taulukossa:
|
|
Näyte 4 Vehnä-soijaseos |
Näyte 5 Vehnä-soijaseos (= näyte 4), johon on lisätty tryptofaania (0,457 g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Keskiarvo [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [%] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [%] |
4,71 |
5,11 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
keskiarvoon mukaan otettujen yksittäisten tulosten lukumäärä, kun on poistettu suuresti poikkeavat arvot (tunnistettu Cochranin ja Dixonin poikkeavien arvojen testillä) |
|
sr |
= |
toistettavuuden keskihajonta |
|
SR |
= |
uusittavuuden keskihajonta |
|
r |
= |
toistettavuus |
|
R |
= |
uusittavuus |
|
CVr |
= |
toistettavuuden variaatiokerroin, % |
|
CVR |
= |
uusittavuuden variaatiokerroin, %. |
Vielä järjestettiin EY-vertailututkimus, jossa seitsemän laboratoriota analysoi neljä näytettä, tarkoituksena tryptofaanin varmentaminen hydrolyysissä. Tulokset on esitetty seuraavassa taulukossa. Kustakin näytteestä tehtiin viisi rinnakkaismääritystä.
|
|
Näyte 1 Sikarehuseos (CRM 117) |
Näyte 2 Vähärasvainen kalajauho (CRM 118) |
Näyte 3 Soijajauho (CRM 119) |
Näyte 4 Rasvaton maitojauhe (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Keskiarvo [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [%] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [%] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
keskiarvoon mukaan otettujen yksittäisten tulosten lukumäärä, kun on poistettu suuresti poikkeavat arvot (tunnistettu Cochranin ja Dixonin poikkeavien arvojen testillä) |
|
sr |
= |
toistettavuuden keskihajonta |
|
SR |
= |
uusittavuuden keskihajonta |
|
r |
= |
toistettavuus |
|
R |
= |
uusittavuus |
|
CVr |
= |
toistettavuuden variaatiokerroin, % |
|
CVR |
= |
uusittavuuden variaatiokerroin, %. |
9. Huomautukset
|
9.1 |
Seuraavilla erityisillä kromatografiaolosuhteilla voidaan erottaa paremmin tryptofaani ja α-metyylitryptofaani. Isokraattinen erotus, jonka jälkeen kolonni puhdistetaan gradienteilla:
|
||||||||||||||||||||||||||||||||||||||||||||
|
9.2 |
Kromatografia vaihtelee HPLC-laitetyypin ja käytetyn kolonnin täytemateriaalin mukaan. Valitun järjestelmän on pystyttävä erottamaan tryptofaanin piikki ja sisäisen standardin piikki pohjaviivalle asti. Lisäksi on tärkeää, että hajoamistuotteet eroavat hyvin tryptofaanista ja sisäisestä standardista. Hydrolysaatteja on ajettava myös ilman sisäistä standardia, jotta voidaan tarkistaa epäpuhtauksien esiintyminen sisäisen standardin kohdalla olevan pohjaviivan avulla. On tärkeää, että ajoaika on riittävän pitkä, jotta kaikki hajoamistuotteet eluoituvat; myöhään eluoituvat piikit saattavat muuten häiritä myöhempiä kromatografia-ajoja. Kromatografiajärjestelmän vasteen on oltava lineaarinen toiminta-alueella. Lineaarinen vaste on mitattava sisäisen standardin vakiopitoisuudella (normaalilla pitoisuudella) ja erilaisilla tryptofaanipitoisuuksilla. On tärkeää, että sekä tryptofaanin että sisäisen standardin piikkien koot ovat HPLC-fluoresenssijärjestelmän lineaarisella alueella. Jos joko tryptofaanin ja/tai sisäisen standardin piikki on liian pieni tai liian suuri (piikit ovat liian pienet tai suuret), analyysi on toistettava käyttäen erilaista näytemäärää ja/tai muuttaen lopputilavuutta. |
|
9.3 |
Bariumhydroksidi
Bariumhydroksidi tulee vanhetessaan vaikealiukoisemmaksi. HPLC-määritystä varten tehty liuos voi sen vuoksi olla samea, minkä vuoksi voidaan saada alhaisia tryptofaanituloksia. |
H. RAAKARASVAN JA RAAKAÖLJYN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä määritetään rehuissa olevan raakarasvan ja -öljyn määrä. Se ei koske öljysiementen ja öljypitoisten hedelmien määrityksiä.
Jäljempänä kuvattavan kahden menettelytavan käyttö riippuu rehun luonteesta ja koostumuksesta sekä määrityksen käyttötarkoituksesta.
1.1 Menettelytapa A — Suoraan uutettavissa oleva raakarasva ja -öljy
Tätä menetelmää voidaan käyttää kasviperäisiin rehuihin lukuun ottamatta niitä, jotka kuuluvat menettelytavan B soveltamisalaan.
1.2 Menettelytapa B — Raakarasvan ja -öljyn kokonaismäärä
Menetelmää voidaan soveltaa eläinperäisiin rehuihin sekä kaikkiin rehuseoksiin. Sitä käytetään kaikkiin sellaisiin rehuihin, joista rasvaa ja öljyä ei voida täysin uuttaa ilman sitä edeltävää hydrolysointia (esim. gluteenit, hiiva, perunaproteiinit ja tuotteet, joita on käsitelty muun muassa ekstrudoimalla, kuumentamalla tai hiutaleiksi tekemällä).
1.3 Tulosten tulkitseminen
Aina, kun menettelytapaa B käyttäen saadaan suurempi tulos kuin menettelytapaa A käyttäen, menettelytavalla B saatua tulosta pidetään todellisena arvona.
2. Periaate
2.1 Menettelytapa A
Näyte uutetaan petrolieetterillä. Liuotin tislataan pois ja jäännös kuivataan ja punnitaan.
2.2 Menettelytapa B
Näyte käsitellään kuumentamalla sitä kloorivetyhapon kanssa. Seos jäähdytetään ja suodatetaan. Jäännös pestään ja kuivataan ja määritystä jatketaan menettelytavan A mukaisesti.
3. Reagenssit
|
3.1 |
Petrolieetteri, kiehumislämpötila: 40–60 oC. Bromiluvun on oltava alle 1 ja haihdutusjäännöksen alle 2 mg/100 ml. |
|
3.2 |
Natriumsulfaatti, vedetön. |
|
3.3 |
Kloorivetyhappo, c = 3 mol /l. |
|
3.4 |
Suodatuksen apuaine, esim. Kieselgur, Hyflo-supercel. |
4. Välineistö
|
4.1 |
Uuttolaitteisto. Jos käytetään Soxhlet-laitteistoa (jossa on sifoni), refluksointinopeuden on oltava sellainen, että Soxhlet-väliosan täyttyminen liuottimella ja tyhjeneminen siitä tapahtuvat noin 10 kertaa tunnissa. Jos laitteistoon kuuluu suoramallinen väliosa, refluksointinopeuden on oltava noin 10 ml minuutissa. |
|
4.2 |
Uuttohylsyjä, joissa ei ole petrolieetteriin liukenevaa materiaalia ja joiden huokoisuus täyttää 4.1 kohdassa esitetyt vaatimukset. |
|
4.3 |
Lämpökaappi, joko vakuumikaappi, joka on säädetty 75 ± 3 oC:seen tai kiertoilmakaappi, joka on säädetty 100 ± 3 oC:seen. |
5. Menettely
5.1 Menettelytapa A (katso 8.1 kohta)
Näytettä punnitaan 5 g 1 mg:n tarkkuudella uuttohylsyyn (4.2) ja se peitetään rasvattomalla puuvillavanulla.
Hylsy asetetaan uuttolaitteeseen (4.1) ja sitä uutetaan kuusi tuntia petrolieetterillä (3.1). Petrolieetteriuute otetaan talteen kuivaan, punnittuun pulloon, joka sisältää kiehumakiviä (2).
Liuotin tislataan pois. Pullon ja sen sisältämän jäännöksen annetaan kuivua puolentoista tunnin ajan lämpökaapissa (4.3). Pullon jäännöksineen annetaan jäähtyä eksikkaattorissa ja se punnitaan. Kuivausta jatketaan edelleen 30 minuutin ajan sen varmistamiseksi, että rasvan ja öljyn paino pysyy vakiona (kahden peräkkäisen punnituksen välinen painoero saa olla enintään 1 mg).
5.2 Menettelytapa B
Näytettä punnitaan 2,5 g 1 mg:n tarkkuudella (ks. 8.2 kohta) 400 ml:n dekantterilasiin tai 300 ml:n erlenmeyerkolviin ja astiaan lisätään 100 ml kloorivetyhappoa (3.3) ja kiehumakiviä. Dekantterilasi peitetään kellonlasilla tai erlenmeyerkolvi yhdistetään palautusjäähdyttimeen. Seos saatetaan varovasti kiehuvaksi käyttäen pientä liekkiä tai sähkölevyä ja seoksen annetaan kiehua hiljalleen tunnin ajan. Näytteen tarttumista astian seinämiin tulee välttää.
Astian sisällön annetaan jäähtyä ja lisätään suodatuksen apuainetta (3.4) sen verran, ettei rasvaa ja öljyä häviä suodatuksessa. Suodatukseen käytetään kostutettua, rasvatonta, kaksinkertaista suodatinpaperia. Jäännös pestään kylmällä vedellä, kunnes saadaan neutraali suodos. Tarkastetaan, ettei suodos sisällä öljyä tai rasvaa. Jos sitä esiintyy, näyte on ennen hydrolysointia uutettava petrolieetterillä menettelytapaa A käyttäen.
Jäännöksen sisältävä kaksinkertainen suodatinpaperi asetetaan kellonlasille ja sen annetaan kuivua puolitoista tuntia 100 ± 3 oC:ssa lämpökaapissa (4.3).
Kuivan jäännöksen sisältävä kaksinkertainen suodatinpaperi pannaan uuttohylsyyn (4.2) ja peitetään rasvattomalla puuvillavanulla. Hylsy pannaan uuttolaitteeseen (4.1) ja määritystä jatketaan 5.1 kohdan toisessa ja kolmannessa kappaleessa esitetyllä tavalla.
6. Tuloksen ilmoittaminen
Jäännöksen paino ilmoitetaan prosentteina näytteen painosta.
7. Toistettavuus
Samasta näytteestä saman henkilön suorittaman kahden rinnakkaismäärityksen tulosten välinen ero ei saa olla yli:
|
— |
0,2 %:a absoluuttisena arvona, kun raakarasva- ja -öljypitoisuus on alle 5 %, |
|
— |
4,0 %:a suhteellisena arvona, kun raakarasva- ja -öljypitoisuus on 5–10 %, |
|
— |
0,4 %:a absoluuttisena arvona, kun raakarasva- ja -öljypitoisuus on yli 10 %. |
8. Huomautukset
|
8.1 |
Runsaasti rasvaa ja öljyä sisältäviä tuotteita, joita on hankala jauhaa tai joista on vaikea saada homogeenista näytettä, käsitellään seuraavasti: Näytettä punnitaan 20 g 1 mg:n tarkkuudella ja siihen sekoitetaan vähintään 10 g vedetöntä natriumsulfaattia (3.2). Sitä uutetaan petrolieetterillä (3.1) 5.1 kohdassa esitetyllä tavalla. Saadun uutteen tilavuus täydennetään 500 ml:ksi petrolieetterillä (3.1) ja se sekoitetaan. Liuosta otetaan 50 ml ja kaadetaan pieneen, kuivaan, punnittuun pulloon, joka sisältää kiehumakiviä. Liuotin tislataan pois, jäännös kuivataan ja määritystä jatketaan 5.1 kohdan viimeisessä kappaleessa kuvatulla tavalla. Liuottimen annetaan haihtua hylsyssä olevasta uuttojäännöksestä, jäännös jauhetaan 1 mm raekokoon ja siirretään takaisin hylsyyn (lisäämättä natriumsulfaattia) ja määritystä jatketaan 5.1 kohdan toisessa ja kolmannessa kappaleessa kuvatulla tavalla. Rasvan ja öljyn pitoisuus prosentteina näytteestä lasketaan seuraavalla kaavalla: (10m1 + m2) × 5 jossa:
|
|
8.2 |
Pieniä rasva- ja öljymääriä sisältävien näytteiden punnitusmäärä voidaan nostaa 5 grammaan. |
|
8.3 |
Lemmikkieläinten ruokiin, joiden vesipitoisuus on suuri, voi olla tarvetta sekoittaa vedetöntä natriumsulfaattia ennen hydrolysointia ja uuttoa, joka suoritetaan menettelytavassa B kuvatulla tavalla. |
|
8.4 |
Edellä 5.2 kohdassa voi olla tehokkaampaa käyttää kuumaa vettä kylmän veden sijaan jäännöksen pesemiseksi suodatuksen jälkeen. |
|
8.5 |
Mainittua 1,5 tunnin kuivatusaikaa saatetaan joutua pidentämään joidenkin rehujen osalta. Liiallista kuivaamista on kuitenkin vältettävä, sillä se voi johtaa alhaisempiin tuloksiin. Myös mikroaaltouunia voidaan käyttää. |
|
8.6 |
Ennen hydrolysointia tapahtuvaa menettelytavan A mukaista esiuuttoa ja menettelytavan B mukaista uudelleen uuttoa suositellaan, jos raakarasvan/öljyn pitoisuus on yli 15 %. Tämä riippuu jossakin määrin rehun tai rehuseoksen luonteesta ja siinä olevan rasvan/öljyn laadusta. |
I. RAAKAKUIDUN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehusta sen rasvaton orgaaninen aines, joka ei liukene happo- ja emäsliuoksen kanssa keitettäessä ja jota kutsutaan sovitusti raakakuiduksi.
2. Periaate
Näyte, josta tarvittaessa on poistettu rasva, käsitellään peräkkäin tietyn väkevyisillä kiehuvilla rikkihappo- ja kaliumhydroksidiliuoksilla. Jäännös erotetaan suodattamalla näyte lasisintterisuodattimella, se pestään, kuivataan, punnitaan ja poltetaan tuhkaksi 475–500 oC:ssa. Tuhkaamisesta aiheutuva painohäviö vastaa näytteen raakakuitupitoisuutta.
3. Reagenssit
|
3.1 |
Rikkihappo, c = 0,13 mol/l. |
|
3.2 |
Kuohunestoaine (esimerkiksi n-oktanoli). |
|
3.3 |
Suodatuksen apuaine (Celite 545 tai vastaava), jota on kuumennettu 500 oC:n lämpötilassa neljän tunnin ajan (8.6). |
|
3.4 |
Asetoni. |
|
3.5 |
Petrolieetteri, kiehumislämpötila: 40–60 oC. |
|
3.6 |
Kloorivetyhappo, c = 0,5 mol/1. |
|
3.7 |
Kalumhydroksidiliuos, c = 0,23 mol/l. |
4. Välineistö
|
4.1 |
Lämmityslaitteisto rikkihappoliuoksella tai kaliumhydroksidiliuoksella tehtävään kuumauuttoon; lämmityslaitteistoon kuuluu suodatinupokkaan (4.2) jalusta ja letku, jossa on ilmanpoisto- ja tyhjennysventtiili; tarvittaessa laitteistoon saadaan paineilmaa. Ennen kutakin päivittäistä käyttöönottoa laitteisto esilämmitetään keittämällä siinä vettä viiden minuutin ajan. |
|
4.2 |
Vetoisuudeltaan 50 ml:n suuruinen lasinen suodatinupokas, jossa on huokoisuudeltaan 40–90 μm:n lasisintterisuodatin. Ennen ensimmäistä käyttökertaa upokasta kuumennetaan 500 oC:ssa muutaman minuutin ajan ja jäähdytetään (8.6). |
|
4.3 |
Vetoisuudeltaan vähintään 270 ml oleva sylinteri, jossa on kiehumisen kestävä palautusjäähdytin. |
|
4.4 |
Termostaattisäätöinen lämpökaappi. |
|
4.5 |
Termostaattisäätöinen muhveliuuni. |
|
4.6 |
Uuttoyksikkö, johon kuuluu suodatinupokkaan jalusta (4.2) ja poistoletku, jossa on ilmanpoisto- ja tyhjennysventtiili. |
|
4.7 |
Osia lämmityslaitteiston (4.1), upokkaan (4.2) ja sylinterin (4.3) kokoamiseen sekä kylmäuuttoyksikön (4.6) ja upokkaan liittämiseen. |
5. Menettely
Punnitaan 1 mg:n tarkkuudella 1 g esikäsiteltyä näytettä upokkaaseen (4.2) (ks. huomautukset 8.1, 8.2 ja 8.3) ja lisätään 1 g suodatuksen apuainetta (3.3).
Liitetään suodatinupokas (4.2) lämmityslaitteistoon (4.1), ja tämän jälkeen liitetään sylinteri (4.3) upokkaaseen. Sylinteri-upokasyhdistelmä täytetään 150 ml:lla kiehuvaa rikkihappoa (3.1) ja tarvittaessa lisätään muutama pisara kuohunestoainetta (3.2).
Neste saatetaan kiehuvaksi 5 ± 2 minuutissa ja annetaan kiehua voimakkaasti tarkalleen 30 minuuttia.
Avataan poistoletkun venttiili (4.1), ja suodatetaan rikkihappo tyhjössä suodatinupokkaan läpi ja pestään jäännös kolme kertaa peräkkäin 30 ml:lla kiehuvaa vettä huolehtien siitä, että jäännös suodatetaan kuivaksi jokaisen pesukerran jälkeen.
Suljetaan tyhjennysventtiili, kaadetaan 150 ml kiehuvaa kaliumhydroksidiliuosta (3.7) sylinteriupokasyhdistelmään ja lisätään muutama pisara kuohunestoainetta (3.2). Neste saatetaan kiehuvaksi 5 ± 2 minuutissa ja annetaan kiehua voimakkaasti tarkalleen 30 minuuttia. Suodatetaan ja toistetaan pesumenettelyt samalla tavoin kuin rikkihapon kanssa.
Viimeisen pesun ja kuivauksen jälkeen upokas sisältöineen irrotetaan ja upokas yhdistetään uudelleen kylmäuuttoyksikköön (4.6). Poistetaan ilma ja pestään jäännös upokkaassa kolme kertaa peräkkäin 25 ml:lla asetonia (3.4) huolehtien siitä, että jäännös kuivuu jokaisen pesukerran jälkeen.
Annetaan upokkaan kuivua vakiopainoon lämpökaapissa, jonka lämpötila on 130 oC. Jokaisen kuivauksen jälkeen jäähdytetään eksikkaattorissa ja punnitaan nopeasti. Upokas asetetaan muhveliuuniin, jossa näytteen annetaan palaa tuhkaksi vakiopainoon (kahden peräkkäisen punnituksen välinen painoero saa olla enintään 2 mg) 475–500 oC:ssa vähintään 30 minuuttia.
Jokaisen kuumennuksen jälkeen upokkaan annetaan jäähtyä ensin uunissa ja tämän jälkeen eksikkaattorissa ennen punnitusta.
Nollakoe suoritetaan ilman näytettä. Tuhkaamisesta johtuva painohäviö ei saa olla yli 4 mg:aa.
6. Tulosten laskeminen
Raakakuitupitoisuus prosentteina näytteestä saadaan seuraavasti:

jossa:
|
m |
= |
näytteen paino grammoina, |
|
m0 |
= |
määrityksen aikana näytteen polttamisesta aiheutuva painohäviö grammoina, |
|
m1 |
= |
nollakokeessa polttamisesta aiheutuva painohäviö grammoina. |
7. Toistettavuus
Kahden samasta näytteestä suoritetun määrityksen tulosten välinen ero ei saa olla yli:
|
— |
0,6 %:a absoluuttisena arvona, kun raakakuitupitoisuus on alle 10 %, |
|
— |
6 %:a suhteellisena arvona suuremmasta tuloksesta, kun raakakuitupitoisuus on vähintään 10 %. |
8. Huomautukset
|
8.1 |
Rehuista, jotka sisältävät yli 10 % öljyjä ja rasvoja, on rasva ennen määritystä poistettava petrolieetterillä (3.5). Liitetään suodatinupokas (4.2) sisältöineen kylmäuuttoyksikköön (4.6), poistetaan ilma ja pestään jäännös kolme kertaa 30 ml:lla petrolieetteriä; varmistetaan, että jäännös on kuiva. Yhdistetään upokas sisältöineen lämmityslaitteistoon (4.1) ja jatketaan 5 kohdassa esitetyllä tavalla. |
|
8.2 |
Rehuista, jotka sisältävät öljyjä ja rasvoja, joita ei voida uuttaa suoraan petrolieetterillä (3.5), rasva on poistettava 8.1 kohdassa esitetyllä tavalla ja tämän jälkeen rasva on poistettava vielä uudelleen hapolla kiehuttamisen jälkeen. Hapolla kiehuttamisen ja sitä seuraavien pesujen jälkeen upokas sisältöineen yhdistetään kylmäuuttoyksikköön (4.6) ja jäännöstä pestään kolme kertaa 30 ml:lla asetonia ja tämän jälkeen kolme kertaa 30 ml:lla petrolieetteriä. Suodatetaan tyhjössä kunnes jäännös on kuiva ja jatketaan määritystä kaliumhydroksidikäsittelyllä 5 kohdassa esitetyllä tavalla. |
|
8.3 |
Jos näyte sisältää yli 5 % karbonaatteja kalsiumkarbonaattina ilmaistuna, upokas (4.2) yhdistetään punnittuine näytteineen lämmityslaitteistoon (4.1). Näyte pestään kolme kertaa 30 ml:lla kloorivetyhappoa (3.6). Jokaisen lisäämiskerran jälkeen odotetaan noin yksi minuutti ennen suodatuksen aloittamista. Näyte pestään kerran 30 ml:lla vettä ja tämän jälkeen jatketaan 5 kohdassa esitetyllä tavalla. |
|
8.4 |
Jos käytetään telineenmuotoista laitteistoa (useita upokkaita on liitetty samaan lämmityslaitteistoon), kahta koetta samasta näytteestä ei saa suorittaa samalla kertaa. |
|
8.5 |
Jos kiehuttamisen jälkeen happamien ja emäksisten liuosten suodatus osoittautuu vaikeaksi, käytetään lämmityslaitteiston tyhjennysletkun kautta annettavaa paineilmaa, minkä jälkeen jatketaan suodatusta. |
|
8.6 |
Tuhkauslämpötila ei saa olla yli 500:aa oC lasisten suodatinupokkaiden käyttöiän pidentämiseksi. Kuumennus- ja jäähdytysjaksojen aikana on vältettävä suuria lämpötilanvaihteluita. |
J. SOKERIN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää pelkistävien sokerien ja kokonaissokerien määrä invertoinnin jälkeen glukoosina, tai tarvittaessa sakkaroosina käyttäen muunnoskerrointa 0,95. Menetelmä soveltuu rehuseoksiin. Muita rehuja varten on säädetty erityismenetelmistä. Laktoosi on tarvittaessa mitattava erikseen ja otettava huomioon tuloksia laskettaessa.
2. Periaate
Sokerit uutetaan laimeaan etanoliin ja liuos kirkastetaan Carrez-liuoksilla I ja II. Etanolin poiston jälkeen määritykset suoritetaan ennen invertointia ja sen jälkeen saadut Luff-Schoorl-menetelmällä.
3. Reagenssit
|
3.1 |
Etanoli 40 % (v/v), tiheys: 0,948 g/ml 20 oC:ssa, neutraalia fenoliftaleiinilla määritettynä. |
|
3.2 |
Carrez-liuos I: liuotetaan veteen 21,9 g sinkkiasetaattia, Zn(CH3COO)2·2H2O, ja 3 g jääetikkaa. Täytetään 100 ml:ksi vedellä. |
|
3.3 |
Carrez-liuos II: liuotetaan veteen 10,6 g kaliumferrosyanidia K4Fe(CN)6·3H2O. Täytetään 100 ml:ksi vedellä. |
|
3.4 |
Metyylioranssiliuos 0,1 % (w/v). |
|
3.5 |
Kloorivetyhappo 4 mol/l. |
|
3.6 |
Kloorivetyhappo 0,1 mol/l. |
|
3.7 |
Natriumhydroksidiliuos 0,1 mol/l. |
|
3.8 |
Luff-Schoorl-reagenssi: Kaadetaan sitruunahappoliuos (3.8.2) natriumkarbonaattiliuokseen (3.8.3) varovasti sekoittaen. Tämän jälkeen lisätään kuparisulfaattiliuos (3.8.1) ja täytetään 1 litraksi vedellä. Annetaan seistä yön yli ja suodatetaan. Näin saadun reagenssin (Cu 0,05 mol/l; Na2 CO3 1 mol/l) konsentraatio tarkistetaan; ks. 5.4 kohdan viimeinen kappale. Liuoksen pH:n on oltava noin 9,4. |
|
3.8.1 |
Kuparisulfaattiliuos: liuotetaan 25 g raudatonta kuparisulfaattia, CuSO4·5H2O, 100 ml:aan vettä. |
|
3.8.2 |
Sitruunahappoliuos: liuotetaan 50 g sitruunahappoa, C6H8O7·H2O, 50 ml:aan vettä. |
|
3.8.3 |
Natriumkarbonaattiliuos: liuotetaan 143,8 g vedetöntä natriumkarbonaattia noin 300 ml:aan lämmintä vettä. Annetaan jäähtyä. |
|
3.9 |
Natriumtiosulfaattiliuos 0,1 mol/l. |
|
3.10 |
Tärkkelysliuos: 5 g liukoista tärkkelystä lisätään 30 ml:aan vettä, seos lisätään 1 litraan kiehuvaa vettä. Keitetään kolme minuuttia, annetaan jäähtyä ja tarvittaessa lisätään 10 mg elohopeajodidia säilyteaineeksi. |
|
3.11 |
Rikkihappo, 3 mol/l. |
|
3.12 |
Kaliumjodidiliuos, 30 % (w/v) |
|
3.13 |
Kiehumakiviä, jotka keitetään suolahapossa, pestään vedellä ja kuivataan. |
|
3.14 |
3-metyylibutan-1-oli. |
4. Välineistö
Sekoitin: noin 35–40 kierr./min.
5. Menettely
5.1 Näytteen uutto
Punnitaan 1 mg:n tarkkuudella 2,5 g näytettä 250 ml:n mittapulloon. Lisätään 200 ml etanolia (3.1) ja sekoitetaan sekoittajassa yhden tunnin ajan. Lisätään 5 ml Carrez-liuosta I (3.2) ja sekoitetaan noin 30 sekunnin ajan. Lisätään 5 ml Carrez-liuosta II (3.3) ja sekoitetaan uudelleen yhden minuutin ajan. Täytetään pullo etanolilla (3.1), homogenoidaan ja suodatetaan. Otetaan 200 ml suodosta ja haihdutetaan se noin puoleen tilavuuteen, jotta saadaan poistetuksi suurin osa etanolista. Haihdutusjäännös siirretään kvantitatiivisesti 200 ml:n mittapulloon käyttäen lämmintä vettä, jäähdytetään, täytetään vedellä merkkiin asti, homogenoidaan ja suodatetaan tarvittaessa. Tätä liuosta käytetään pelkistävien sokerien määrittämiseksi ja invertointireaktion jälkeen kokonaissokerien määrittämiseksi.
5.2 Pelkistävien sokerien määritys
Pipetoidaan enintään 25 ml liuosta, joka sisältää alle 60 mg glukoosina ilmoitettuja pelkistäviä sokereita. Tarvittaessa tilavuus säädetään 25 ml:ksi tislatulla vedellä ja pelkistävien sokerien pitoisuus määritetään Luff-Schoorl-menetelmällä. Tulos ilmoitetaan glukoosiprosenttina.
5.3 Kokonaissokerien määritys invertointireaktion jälkeen
Pipetoidaan 50 ml liuosta 100 ml:n mittapulloon. Lisätään muutama pisara metyylioranssiliuosta (3.4) ja tämän jälkeen lisätään varovasti ja jatkuvasti sekoittaen kloorivetyhappoa (3.5), kunnes neste muuttuu selvästi punaiseksi. Lisätään 15 ml kloorivetyhappoa (3.6), pullo upotetaan voimakkaasti kiehuvaan vesihauteeseen ja pidetään siinä 30 minuutin ajan. Jäähdytetään nopeasti noin 20 oC:seen ja lisätään 15 ml natriumhydroksidiliuosta (3.7). Tilavuus säädetään 100 ml:ksi vedellä ja homogenoidaan. Otetaan enintään 25 ml:n erä, joka sisältää alle 60 mg glukoosina ilmoitettuja pelkistäviä sokereita. Tarvittaessa tilavuus säädetään 25 ml:ksi tislatulla vedellä, ja pelkistävien sokerien pitoisuus määritetään Luff-Schoorl-menetelmällä. Tulos ilmoitetaan glukoosiprosenttina tai tarvittaessa sakkaroosina kertomalla kertoimella 0,95.
5.4 Titraus Luff-Schoorl-menetelmällä
Pipetoidaan 25 ml Luff-Schoorl-reagenssia (3.8) 300 ml:n erlenmeyerkolviin, lisätään tarkalleen 25 ml kirkastettua sokeriliuosta. Lisätään kaksi kiehumakiveä (3.13), lämmitetään keskikokoisen liekin yläpuolella käsin ravistaen ja kuumennetaan neste kiehumapisteeseen noin kahdessa minuutissa. Erlenmeyerkolvi asetetaan välittömästi asbestiverkolle, jossa on noin 6 cm:n läpimittainen reikä ja verkon alla on etukäteen sytytetty liekki. Säädetään liekki sellaiseksi, että erlenmeyerkolvi kuumenee ainoastaan pohjastaan. Tämän jälkeen erlenmeyerkolviin kiinnitetään palautusjäähdytin. Keitetään tarkalleen 10 minuutin ajan. Jäähdytetään välittömästi kylmässä vedessä ja noin viiden minuutin kuluttua titrataan seuraavasti:
Lisätään 10 ml kaliumjodidiliuosta (3.12) ja välittömästi tämän jälkeen (varoen, koska liuos voi vaahdota runsaasti) lisätään 25 ml rikkihappoa (3.11). Titrataan natriumtiosulfaattiliuoksella (3.9) kunnes väri muuttuu samean keltaiseksi, lisätään tärkkelysindikaattori (3.10) ja titraus suoritetaan loppuun.
Sama titraus suoritetaan liuoksesta, jossa on tarkasti mitattuna 25 ml Luff-Schoorl-reagenssia (3.8) ja 25 ml vettä, sen jälkeen kun 10 ml kaliumjodidiliuosta (3.12) ja 25 ml rikkihappoa (3.11) on lisätty kohottamatta lämpötilaa kiehumispisteeseen.
6. Tulosten laskeminen
Oheisen taulukon avulla määritetään milligrammoissa se glukoosimäärä, joka vastaa näiden kahden titrauksen erotusta ilmaistuna milligrammoina natriumtiosulfaattia, 0,1 mol/l. Tulos ilmoitetaan prosentteina näytteestä.
7. Menettely erityistapauksissa
|
7.1 |
Runsaasti melasseja sisältävistä rehuista ja rehuista, jotka eivät ole homogeenisia, punnitaan 20 g näyte 1 litran mittapulloon ja lisätään 500 ml vettä. Sekoitetaan 1 tunti sekoittajassa. Selkeytetään käyttäen Carrez I (3.2) ja II (3.3) -reagensseja 5.1 kohdassa kuvatulla tavalla kuitenkin tällä kertaa käyttäen nelinkertaiset määrät kutakin reagenssia. Täytetään merkkiin asti 80-prosenttisella (v/v) etanolilla. Homogenoidaan ja suodatetaan. Poistetaan etanoli 5.1 kohdan mukaisesti. Jos yhtään dekstrinoitua tärkkelystä ei esiinny, täytetään merkkiin asti tislatulla vedellä. |
|
7.2 |
Melassista ja runsaasti sokeria sisältävistä ja tärkkelysköyhistä rehuaineista (kuten johanneksenleipäpuun hedelmät, kuivatut sokerijuurikasleikkeet) punnitaan 5 gramman näyte 250 ml:n mittapulloon, lisätään 200 ml tislattua vettä ja sekoitetaan ravistelijassa yhden tunnin ajan tai tarvittaessa kauemmin. Kirkastetaan käyttäen Carrez-reagensseja I (3.2) ja II (3.3) 5.1 kohdassa kuvatulla tavalla. Mittapullo täytetään vedellä merkkiin, liuos homogenoidaan ja suodatetaan. Kokonaissokerien määrän toteamiseksi suoritusta jatketaan 5.3 kohdan mukaisesti. |
8. Huomautukset
|
8.1 |
Vaahdonmuodostuksen estämiseksi on suositeltavaa lisätä noin 1 ml 3-metyylibutan-1-olia (3.14) (riippumatta tilavuudesta) ennen Luff-Schoorl-reagenssin kanssa keittämistä. |
|
8.2 |
Invertoinnin jälkeen saadun glukoosina ilmoitetun kokonaissokeripitoisuuden ja glukoosina ilmoitetun pelkistävien sokerien pitoisuuden välisestä erosta 0,95:lla kerrottuna saadaan sakkaroosin prosentuaalinen pitoisuus. |
|
8.3 |
Pelkistävien sokerien, paitsi laktoosin, pitoisuuden määrittämiseksi voidaan käyttää kahta menetelmää: |
|
8.3.1 |
Likimääräisen arvion laskemiseksi laktoosipitoisuus, joka on saatu eri analyysimenetelmällä, kerrotaan 0,675:llä ja saatu tulos vähennetään pelkistävien sokerien pitoisuudesta. |
|
8.3.2 |
Pelkistävien sokerien, paitsi laktoosin, määrän tarkkaa laskemista varten on tehtävä kaksi lopullista määritystä samasta näytteestä. Toinen määrityksistä suoritetaan liuokselle, joka saadaan osasta 5.1 kohdan mukaisesti saatua liuosta, ja toinen määrityksistä suoritetaan osasta liuosta, joka saadaan laktoosimäärityksen yhteydessä tähän tarkoitukseen kehitetyn menetelmän mukaisesti (sen jälkeen kun muut sokerit on fermentoitu ja liuos kirkastettu). Molemmissa tapauksissa sokerin määrä määritetään Luff-Schoorl-menetelmällä ja lasketaan milligrammoina glukoosia. Arvot vähennetään toisistaan ja erotus ilmoitetaan prosentteina näytteestä. Esimerkki Kaksi tilavuutta vastaa 250 mg:n näytteestä tehtyjä määrityksiä. Ensimmäisessä tapauksessa on kulunut 17 ml natriumtiosulfaattiliuosta, 0,1 mol/l, joka vastaa 44,2 mg:aa glukoosia; toisessa tapauksessa 11 ml, joka vastaa 27,6 mg:aa glukoosia. Erotus on 16,6 mg glukoosia. Pelkistävien sokerien, laktoosia lukuun ottamatta, glukoosina laskettu määrä on:
|
Taulukko 25 ml:lle Luff-Schoorl-reagenssia
Na2S2O3, 0,1 mol/l, millilitroina, kahden minuutin kuumennus, 10 minuutin keitto
|
Na2S2O3 0,1 mol/l |
Glukoosi, fruktoosi, inverttisokerit C6H12O6 |
Laktoosi C12H22O11 |
Maltoosi C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
erotus |
mg |
erotus |
mg |
erotus |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. LAKTOOSIN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää laktoosin pitoisuus rehuissa, jotka sisältävät yli 0,5 % laktoosia.
2. Periaate
Sokerit liuotetaan veteen. Liuokselle suoritetaan fermentointi käyttäen Saccharomyces cerevisiae -hiivaa, joka ei kuluta laktoosia. Selkeyttämisen ja suodatuksen jälkeen määritetään suodoksen laktoosipitoisuus Luff-Schoorl-menetelmällä.
3. Reagenssit
|
3.1 |
Saccharomyces cerevisiae -suspensio: suspendoidaan 25 g tuoretta hiivaa 100 ml:aan vettä. Suspensio säilyy jääkaapissa enintään yhden viikon ajan. |
|
3.2 |
Carrez-liuos I: liuotetaan veteen 21,9 g sinkkiasetaattia Zn (CH3COO)2·2H2O ja 3 g jääetikkaa. Täytetään 100 ml:ksi vedellä. |
|
3.3 |
Carrez-liuos II: liuotetaan veteen 10,6 g kaliumferrosyanidia K4Fe(CN)6·3H2O. Täytetään 100 ml:ksi vedellä. |
|
3.4 |
Luff-Schoorl-reagenssi: Kaadetaan sitruunahappoliuos (3.4.2) natriumkarbonaattiliuokseen (3.4.3) sekoittaen varovasti lisäyksen aikana. Tämän jälkeen lisätään kuparisulfaattiliuos (3.4.1) ja täytetään 1 litraksi vedellä. Annetaan seistä yön yli ja suodatetaan. Näin saadun reagenssin (Cu 0,05 mol/l; Na2CO3 1 mol/l) konsentraatio tarkistetaan. Liuoksen pH:n on oltava noin 9,4. |
|
3.4.1 |
Kuparisulfaattiliuos: liuotetaan 25 g raudatonta kuparisulfaattia, CuSO4·5H2O, 100 ml:aan vettä. |
|
3.4.2 |
Sitruunahappoliuos: liuotetaan 50 g sitruunahappoa, C6H8O7·H2O, 50 ml:aan vettä. |
|
3.4.3 |
Natriumkarbonaattiliuos: liuotetaan 143,8 g vedetöntä natriumkarbonaattia noin 300 ml:aan lämmintä vettä. Annetaan jäähtyä. |
|
3.5 |
Kiehumakiviä, jotka keitetään suolahapossa, pestään vedellä ja kuivataan. |
|
3.6 |
Kaliumjodidiliuos, 30 % (w/v). |
|
3.7 |
Rikkihappo, 3 mol/l. |
|
3.8 |
Natriumtiosulfaattiliuos, 0,1 mol/l. |
|
3.9 |
Tärkkelysliuos: 5 g liukoista tärkkelystä lisätään 30 ml:aan vettä, seos lisätään 1 litraan kiehuvaa vettä. Keitetään kolme minuuttia, annetaan jäähtyä ja tarvittaessa lisätään 10 mg elohopeajodidia säilyteaineeksi. |
4. Välineistö
Vesihaude, jonka termostaatti on säädetty 38–40 oC:seen.
5. Menettely
Punnitaan 1 mg:n tarkkuudella 1 g näytettä 100 ml:n mittapulloon. Lisätään 25–30 ml vettä. Pullo asetetaan kiehuvaan vesihauteeseen 30 minuutin ajaksi ja tämän jälkeen se jäähdytetään noin 35 oC:seen. Lisätään 5 ml hiivasuspensiota (3.1) ja homogenoidaan. Annetaan pullon seistä kaksi tuntia vesihauteessa 38–40 oC:n lämpötilassa. Jäähdytetään noin 20 oC:seen.
Lisätään 2,5 ml Carrez-liuosta I (3.2) ja sekoitetaan 30 sekuntia, tämän jälkeen lisätään 2,5 ml Carrez-liuosta II (3.3) ja sekoitetaan uudelleen 30 sekuntia. Täytetään 100 ml:ksi vedellä, sekoitetaan ja suodatetaan. Suodosta pipetoidaan 300 ml:n erlenmeyerkolviin sellainen määrä, joka sisältää 40–80 mg laktoosia, korkeintaan 25 ml. Tarvittaessa täytetään vedellä 25 ml:ksi.
Nollakoe suoritetaan samalla tavoin käyttäen 5 ml hiivasuspensiota (3.1). Laktoosimäärä määritetään Luff-Schoorlin mukaan seuraavasti. Lisätään tarkalleen 25 ml Luff-Schoorl-reagenssia (3.4) ja kaksi kiehumakiveä (3.5). Lämmitetään keskikokoisen liekin yläpuolella käsin ravistellen ja kuumennetaan neste kiehumispisteeseen noin kahdessa minuutissa. Erlenmeyerkolvi asetetaan välittömästi asbestiverkolle, jossa on noin 6 cm:n läpimittainen reikä ja verkon alla on etukäteen sytytetty liekki. Säädetään liekki sellaiseksi, että erlenmeyerkolvi kuumenee ainoastaan pohjastaan. Tämän jälkeen erlenmeyerkolviin kiinnitetään pystyjäähdytin. Keitetään tarkalleen 10 minuutin ajan. Jäähdytetään välittömästi kylmässä vedessä ja noin viiden minuutin kuluttua titrataan seuraavasti:
Lisätään 10 ml kaliumjodidiliuosta (3.6) ja välittömästi tämän jälkeen (varovasti, koska liuos voi vaahdota voimakkaasti) lisätään 25 ml rikkihappoa (3.7). Titrataan natriumtiosulfaattiliuoksella (3.8) kunnes väri muuttuu samean keltaiseksi, lisätään tärkkelysindikaattori (3.9) ja titraus suoritetaan loppuun.
Sama titraus suoritetaan liuoksesta, johon pipetoidaan tarkalleen mitattuna 25 ml Luff-Schoorl-reagenssia (3.4) ja 25 ml vettä, sen jälkeen kun 10 ml kaliumjodidiliuosta (3.6) ja 25 ml rikkihappoa (3.7) on lisätty ilman keittämistä.
6. Tulosten laskeminen
Oheisen taulukon avulla määritetään milligrammoina se laktoosimäärä, joka vastaa näiden kahden titraustuloksen erotusta ilmaistuna natriumtiosulfaattia, 0,1 mol/l, millilitroissa.
Tulos ilmoitetaan vedettömän laktoosin prosentteina näytteestä.
7. Huomautus
Yli 40 % fermentoituvaa sokeria sisältäville tuotteille käytetään yli 5 ml hiivasuspensiota (3.1).
Taulukko 25 ml:lle Luff-Schoorl-reagenssia
ml Na2S2O3, 0,1 mol/l, kahden minuutin kuumennus, 10 minuutin keitto
|
Na2S2O3 0,1 mol/l |
Glukoosi, fruktoosi, inverttisokerit C6H12O6 |
Laktoosi C12H22O11 |
Maltoosi C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
erotus |
mg |
erotus |
mg |
erotus |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. TÄRKKELYKSEN MÄÄRITYS
POLARIMETRINEN MENETELMÄ
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää tärkkelyksen ja tärkkelyksen suurimolekyylisten pilkkoutumistuotteiden pitoisuus rehuissa tarkoituksena todeta, onko niiden ilmoitettu energiamäärä liitteen VII mukainen ja ovatko ne neuvoston direktiivin 96/25/EY (3) säännösten mukaisia.
2. Periaate
Menetelmä perustuu kahteen määritykseen. Ensimmäisessä määrityksessä näyte käsitellään laimealla kloorivetyhapolla. Kun liuos on saatu kirkkaaksi ja suodatettu, liuoksen optinen kiertokyky mitataan polarimetrisesti.
Toisessa määrityksessä näyte uutetaan 40-prosenttisella etanolilla. Kun suodos on tehty happamaksi kloorivetyhapolla ja se on saatu kirkkaaksi ja suodatettu, optinen kiertokyky mitataan kuten ensimmäisessä määrityksessä.
Kun näiden kahden mittauksen erotus kerrotaan tunnetulla kertoimella, saadaan näytteen tärkkelyspitoisuus.
3. Reagenssit
|
3.1 |
Kloorivetyhappoliuos 25 % (w/w) tiheys: 1,126 g/ml. |
|
3.2 |
Kloorivetyhappoliuos 1,13 % (w/v) Konsentraatio on tarkistettava titraamalla natriumhydroksidiliuoksella 0,1 mol/l, indikaattorina 0,1 % (w/v) metyylipuna 94 % (v/v) etanolissa. 10 ml:n neutralointiin tarvitaan 30,94 ml NaOH:ta, 0,1 mol/l. |
|
3.3 |
Carrez-liuos I: liuotetaan veteen 21,9 g sinkkiasetaattia, Zn(CH3COO)2·2H2O, ja 3 g jääetikkaa. Täytetään 100 ml:ksi vedellä. |
|
3.4 |
Carrez-liuos II: liuotetaan veteen 10,6 g kaliumferrosyanidia K4Fe(CN)6·3H2O. Täytetään 100 ml:ksi vedellä. |
|
3.5 |
Etanoli 40 % (v/v), tiheys: 0,948 g/ml 20 oC:ssa. |
4. Välineistö
|
4.1 |
250 ml:n vakiohioksellinen erlenmeyerkolvi, jossa on palautusjäähdytin. |
|
4.2 |
Polarimetri tai sakkarimetri. |
5. Menettely
5.1 Näytteen valmistaminen
Näyte jauhetaan niin hienoksi, että se läpäisee kokonaan 0,5 mm:n pyöreäreikäisen seulan.
5.2 Optisen kokonaiskiertokyvyn (P tai S) määritys (ks. huomautus 7.1)
Punnitaan 2,5 g hienoksi jauhettua näytettä 1 mg:n tarkkuudella 100 ml:n mittapulloon. Lisätään 25 ml kloorivetyhappoa (3.2), ravistellaan kunnes näyte on jakaantunut tasaisesti, sen jälkeen lisätään vielä 25 ml kloorivetyhappoa (3.2). Pullo pannaan kiehuvaan vesihauteeseen ja sitä ravistellaan voimakkaasti ja yhtäjaksoisesti ensimmäisten kolmen minuutin ajan kokkaroitumisen estämiseksi. Vesihauteen vesimäärän on oltava riittävän suuri, jotta haude pysyy kiehuvana kun pullo upotetaan siihen. Pulloa ravisteltaessa sitä ei saa ottaa pois hauteesta. Tarkalleen 15 minuutin kuluttua pullo otetaan hauteesta, lisätään 30 ml kylmää vettä ja jäähdytetään välittömästi 20o:seen.
Lisätään 5 ml Carrez-liuosta I (3.3) ja ravistellaan noin 30 sekunnin ajan. Lisätään 5 ml Carrez-liuosta II (3.4) ja ravistellaan uudestaan 30 sekunnin ajan. Täytetään merkkiin asti vedellä, sekoitetaan ja suodatetaan. Jos suodos ei ole täysin kirkas (mikä tapahtuu harvoin), määritys uusitaan käyttäen suurempaa määrää Carrez-liuoksia I ja II (esimerkiksi 10 ml).
Liuoksen optinen kiertokyky mitataan 200 mm:n putkessa polarimetrillä tai sakkarimetrillä.
5.3 40-prosenttiseen etanoliin liukoisten yhdisteiden optisen kiertokyvyn (P' tai S') määritys
Punnitaan 5 g näytettä 1 mg:n tarkkuudella 100 ml:n mittapulloon ja lisätään noin 80 ml etanolia (3.5) (ks. huomautus 7.2). Mittapullon annetaan seistä yhden tunnin ajan huoneenlämmössä; tänä aikana sitä ravistellaan voimakkaasti kuusi kertaa siten, että näyte sekoittuu perusteellisesti etanoliin. Täytetään merkkiin asti etanolilla (3.5), sekoitetaan ja suodatetaan.
Pipetoidaan 50 ml suodosta (vastaa 2,5:tä grammaa näytettä) 250 ml:n erlenmeyerkolviin, lisätään 2,1 ml kloorivetyhappoa (3.1) ja ravistellaan voimakkaasti. Erlenmeyerkolviin kiinnitetään pystyjäähdytin ja pullo pannaan kiehuvaan vesihauteeseen. Tarkalleen 15 minuutin kuluttua erlenmeyerkolvi otetaan hauteesta ja liuos siirretään 100 ml:n mittapulloon huuhtomalla pienellä määrällä kylmää vettä ja jäähdytetään 20 oC:seen.
Tämän jälkeen näyteliuos tehdään kirkkaaksi Carrez-liuoksilla I (3.3) ja II (3.4), täytetään merkkiin asti vedellä, sekoitetaan, suodatetaan ja mitataan optinen kiertokyky 5.2 kohdan toisessa ja kolmannessa alakohdassa esitetyllä tavalla.
6. Tulosten laskeminen
Tärkkelyspitoisuus (%) lasketaan seuraavalla tavalla:
6.1 Polarimetriset mittaukset
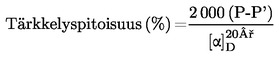
|
P |
= |
optinen kokonaiskiertokyky asteina |
||||||||||||||
|
P' |
= |
40-prosenttiseen etanoliin (V/V) liukoisten yhdisteiden optinen kiertokyky asteina |
||||||||||||||
|
|
= |
puhtaan tärkkelyksen ominaiskiertokyky. Tälle on sovittu seuraavat numeeriset arvot:
|
6.2 Sakkarimetriset mittaukset

|
S |
= |
optinen kokonaiskiertokyky sakkarimetrin asteina |
|
S' |
= |
40-prosenttiseen (v/v) etanoliin liukoisten yhdisteiden optinen kiertokyky asteina |
|
N |
= |
sellainen sakkaroosin määrä grammoina, jonka optinen kiertokyky on 100 sakkarimetrin astetta 100 ml:ssa vettä ja 200 mm:n putkessa mitattuna 16,29 g ranskalaisella sakkarimetrillä 26,00 g saksalaisella sakkarimetrillä 20,00 g muilla sakkarimetreillä.
|
6.3 Toistettavuus
Samasta näytteestä tehdyn kahden rinnakkaismäärityksen tulosten välinen ero ei saa ylittää 0,4:ää prosenttiyksikköä, jos tärkkelyspitoisuus on alle 40 %, eikä 1:tä prosenttia tärkkelyspitoisuudesta suhteellisena arvona, jos tärkkelyspitoisuus on 40 % tai suurempi.
7. Huomautukset
|
7.1 |
Jos näyte sisältää yli 6 % karbonaatteja, kalsiumkarbonaattina laskettuna, on ne ennen optisen kokonaiskiertokyvyn määritystä hajotettava juuri tarkalleen sopivalla määrällä laimeaa rikkihappoa. |
|
7.2 |
Paljon laktoosia sisältävien tuotteiden, kuten herajauheen tai rasvattoman maitojauheen, määritystä jatketaan 80 ml:n etanolilisäyksen (3.5) jälkeen seuraavasti. Palautusjäähdytin kiinnitetään pulloon ja pullo pannaan 50 oC:n vesihauteeseen 30 minuutin ajaksi. Annetaan jäähtyä ja jatketaan määritystä 5.3 kohdassa esitetyllä tavalla. |
|
7.3 |
Seuraavien rehuissa merkittävinä määrinä esiintyvien rehuaineiden tiedetään häiritsevän tärkkelyspitoisuuden määritystä polarimetrisellä menetelmällä ja näin ollen voidaan saada vääriä tuloksia:
|
M. RAAKATUHKAN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehujen raakatuhkapitoisuus.
2. Periaate
Näyte poltetaan tuhkaksi 550 oC:ssa, jäännös punnitaan.
3. Reagenssit
Ammoniumnitraattiliuos, 20 % (w/v).
4. Välineistö
|
4.1 |
Sähkölevy. |
|
4.2 |
Sähkökäyttöinen muhveliuuni, jossa on termostaatti. |
|
4.3 |
Hehkutusupokkaita, jotka on valmistettu kvartsista, posliinista tai platinasta, joko suorakulmaisia (noin 60 × 40 × 25 mm) tai pyöreitä (läpimitta 60–75 mm, korkeus 20–25 mm). |
5. Menettely
Noin 5 grammaa näytettä (2,5 g sellaisia tuotteita, joilla on pyrkimys turvota) punnitaan 1 mg:n tarkkuudella hehkutusupokkaaseen, joka on ensin hehkutettu 550 oC:seen, jäähdytetty ja taarattu. Upokas asetetaan sähkölevylle ja sitä kuumennetaan vähitellen kunnes aine hiiltyy. Tuhkataan 5.1 tai 5.2 kohdan mukaisesti.
|
5.1 |
Upokas asetetaan kalibroituun muhveliuuniin, joka on säädetty 550 oC:seen. Upokasta pidetään tässä lämpötilassa, kunnes saadaan valkea, vaaleanharmaa tai punertava tuhka, jossa ei näytä enää olevan orgaanista ainesta. Upokas asetetaan eksikkaattoriin, sen annetaan jäähtyä ja se punnitaan välittömästi. |
|
5.2 |
Upokas asetetaan kalibroituun muhveliuuniin, joka on säädetty 550 oC:seen. Poltetaan tuhkaksi 3 tunnin ajan. Upokas asetetaan eksikkaattoriin, sen annetaan jäähtyä ja se punnitaan välittömästi. Kuivausta jatketaan edelleen 30 minuutin ajan sen varmistamiseksi, että tuhkan paino pysyy vakiona (kahden peräkkäisen punnituksen välinen painoero saa olla enintään 1 mg). |
6. Tulosten laskeminen
Jäännöksen paino lasketaan vähentämällä taarapaino.
Tulos ilmoitetaan prosentteina näytteestä.
7. Huomautukset
|
7.1 |
Niille näytteille, joita on vaikea polttaa tuhkaksi, on alustava tuhkaksi polttaminen tehtävä ainakin kolmen tunnin ajan, jonka jälkeen näyte jäähdytetään ja siihen lisätään muutama pisara 20 % ammoniumnitraattiliuosta tai vettä (varovasti, välttäen tuhkan leviämistä tai paakkuuntumista). Hehkutusta jatketaan uunissa kuivaamisen jälkeen. Menettely toistetaan tarvittaessa kunnes näyte on tuhkaantunut täydellisesti. |
|
7.2 |
Ne aineet, joihin 7.1 kohdan käsittely ei vaikuta, käsitellään seuraavasti: Kolmen tunnin polttamisen jälkeen tuhkan joukkoon lisätään lämmintä vettä ja se suodatetaan pienen tuhkattoman suodattimen läpi. Suodatin ja sen sisältö poltetaan tuhkaksi alkuperäisessä upokkaassa. Suodos siirretään jäähtyneeseen upokkaaseen, haihdutetaan kuivaksi, poltetaan tuhkaksi ja punnitaan. |
|
7.3 |
Jos kyse on öljyistä ja rasvoista, punnitaan tarkasti 25 g näytettä sopivan kokoiseen upokkaaseen. Näytteen annetaan hiiltyä sytyttämällä se tuhkattomalla suodatinpaperiliuskalla. Palamisen jälkeen näytettä kostutetaan mahdollisimman pienellä määrällä vettä. Kuivataan ja poltetaan tuhkaksi 5 kohdan mukaisesti. |
N. KLOORIVETYHAPPOON LIUKENEMATTOMAN TUHKAN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää kloorivetyhappoon liukenemattomien kivennäisainesten pitoisuus rehuissa. Näytteen laadun mukaan voidaan käyttää kahta eri menetelmää.
|
1.1 |
Menetelmä A: soveltuu orgaanista ainetta oleviin rehuihin ja useimpiin rehuseoksiin; |
|
1.2 |
Menetelmä B: soveltuu kivennäisrehuihin ja kivennäisrehuseoksiin sekä rehuseoksiin, joiden kloorivetyhappoon liukenemattomien ainesten pitoisuus menetelmällä A määritettynä on suurempi kuin 1 %. |
2. Periaate
|
2.1 |
Menetelmä A: näyte poltetaan tuhkaksi, tuhka keitetään kloorivetyhapossa ja liukenematon jäännös suodatetaan ja punnitaan. |
|
2.2 |
Menetelmä B: näytettä käsitellään kloorivetyhapolla. Liuos suodatetaan, jäännös poltetaan tuhkaksi ja näin saatu tuhka käsitellään menetelmän A mukaan. |
3. Reagenssit
|
3.1 |
Kloorivetyhappo, 3 mol/l. |
|
3.2 |
Trikloorietikkahappoliuos, 20 % (w/v). |
|
3.3 |
Trikloorietikkahappoliuos, 1 % (w/v). |
4. Välineistö
|
4.1 |
Sähkölevy. |
|
4.2 |
Sähkökäyttöinen muhveliuuni, jossa on termostaatti. |
|
4.3 |
Hehkutusupokkaita, jotka on valmistettu kvartsista, posliinista tai platinasta, joko suorakulmaisia (noin 60 × 40 × 25 mm) tai pyöreitä (läpimitta 60–75 mm, korkeus 20–25 mm). |
5. Menettely
5.1 Menetelmä A:
Näyte poltetaan tuhkaksi raakatuhkan määritystä varten esitettyä menetelmää käyttäen. Myös tästä määrityksestä saatua tuhkaa voidaan käyttää.
Tuhka siirretään 250–400 ml:n dekantterilasiin käyttäen 75 ml kloorivetyhappoa (3.1). Liuos kuumennetaan hitaasti kiehumapisteeseen ja sitä keitetään varovasti 15 minuuttia. Lämmin liuos suodatetaan tuhkattoman suodatinpaperin läpi ja jäännös pestään lämpimällä vedellä kunnes pesuvesi ei enää ole hapan. Jäännöksen sisältävä suodatinpaperi kuivataan ja poltetaan tuhkaksi taaratussa upokkaassa 550–700 oC:n lämpötilassa. Sen annetaan jäähtyä eksikkaattorissa ja se punnitaan.
5.2 Menetelmä B:
Punnitaan 5 g näytettä 1 mg:n tarkkuudella 250–400 ml:n dekantterilasiin. Siihen lisätään ensin 25 ml vettä ja sen jälkeen 25 ml kloorivetyhappoa (3.1), sekoitetaan ja odotetaan kunnes kuohuminen lakkaa. Lisätään vielä 50 ml kloorivetyhappoa (3.1). Kun kaasuja ei enää muodostu, dekantterilasi asetetaan kiehuvaan vesihauteeseen ja pidetään siinä 30 minuuttia tai tarvittaessa pitempään, jotta mahdollinen jäljellä oleva tärkkelys tulisi kokonaan hydrolysoiduksi. Liuos suodatetaan lämpimänä käyttäen tuhkatonta suodatinpaperia, joka pestään 50 ml:lla lämmintä vettä (ks. 7 kohdan huomautus). Suodatinpaperi jäännöksineen siirretään hehkutusupokkaaseen, kuivataan ja poltetaan tuhkaksi 550–700 oC:n lämpötilassa. Tuhka siirretään 250–400 ml:n dekantterilasiin käyttäen 75 ml kloorivetyhappoa (3.1) ja jatketaan 5.1 kohdan toisen alakohdan mukaan.
6. Tulosten laskeminen
Jäännöksen paino lasketaan vähentämällä taarapaino. Tulos ilmoitetaan prosentteina näytteestä.
7. Huomautus
Jos suodatus osoittautuu vaikeaksi, suoritetaan määritys uudelleen korvaamalla 50 ml kloorivetyhappoa 50 ml:lla 20-prosenttista trikloorietikkahappoa (3.2) ja suodatinpaperin pesuun käytetään lämmintä 1-prosenttista trikloorietikkahappoliuosta (3.3).
O. KARBONAATTIEN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää useimpien rehujen karbonaatit, jotka tavanomaisesti ilmoitetaan kalsiumkarbonaattina.
Tietyissä tapauksissa (esim. rautakarbonaatin osalta) on kuitenkin käytettävä erityismenetelmää.
2. Periaate
Karbonaatit hajotetaan kloorivetyhapossa, vapautunut hiilidioksidi kerätään talteen asteikolla varustettuun putkeen ja sen tilavuutta verrataan tilavuuteen, jonka tunnettu määrä kalsiumkarbonaattia vapauttaa samoissa olosuhteissa.
3. Reagenssit
|
3.1 |
Kloorivetyhappo, tiheys 1,10 g/ml. |
|
3.2 |
Kalsiumkarbonaatti. |
|
3.3 |
Rikkihappo, noin 0,05 mol/l, värjätty metyylipunalla. |
4. Välineistö
Scheibler-Dietrich-laite (ks. kaavio) tai vastaava laite.
5. Menettely
Näytettä punnitaan karbonaattipitoisuuden mukaan seuraavasti:
|
— |
0,5 g tuotetta, joka sisältää 50–100 % karbonaattia, kalsiumkarbonaattina ilmoitettuna, |
|
— |
1 g tuotetta, joka sisältää 40–50 % karbonaattia, kalsiumkarbonaattina ilmoitettuna, |
|
— |
2–3 g muita tuotteita. |
Punnittu näyte pannaan laitteeseen kuuluvaan pulloon (4), jossa on pieni murtumattomasta materiaalista valmistettu koeputki, joka sisältää 10 ml kloorivetyhappoa (3.1), ja pullo liitetään laitteeseen. Käännetään kolmitiehanaa (5) siten, että putki (1) on auki laitteen ulkopuolelle. Käyttäen liikkuvaa putkea (2), joka on täytetty värjätyllä rikkihapolla (3.3) ja liitetty asteikolla varustettuun putkeen (1), säädetään nesteen taso nollamerkkiin. Käännetään hanaa (5) putkien (1) ja (3) yhdistämiseksi ja tarkastetaan, että taso säilyy nollassa.
Kaadetaan kloorivetyhappo (3.1) koeputkesta hitaasti näyte-erän päälle pulloa (4) kallistaen. Paine tasoitetaan laskemalla putki (2) alemmaksi. Pulloa (4) ravistellaan kunnes hiilidioksidin kehittyminen on kokonaan lakannut.
Paine palautetaan alkuperäiseksi tuomalla neste samalle tasolle putkissa (1) ja (2). Lukema otetaan muutaman minuutin kuluttua siitä, kun kaasutilavuus on saavuttanut vakiotason.
Kontrollikoe suoritetaan samoissa olosuhteissa käyttäen 0,5 g kalsiumkarbonaattia (3.2).
6. Tulosten laskeminen
Karbonaattipitoisuus kalsiumkarbonaattina ilmaistuna lasketaan kaavasta:

jossa:
|
X |
= |
karbonaatti prosentteina (w/w) näytteestä, ilmaistuna kalsiumkarbonaattina, |
|
V |
= |
ml CO2:ta, jonka punnittu näytemäärä on vapauttanut, |
|
V1 |
= |
ml CO2:ta, jonka 0,5 g CaCO3:a on vapauttanut, |
|
m |
= |
näytteen paino grammoina. |
7. Huomautukset
|
7.1 |
Jos näyte painaa yli 2 g, lisätään ensin 15 ml tislattua vettä pulloon (4) ja sekoitetaan ennen kokeen alkua. Kontrollikokeessa käytetään samaa vesitilavuutta. |
|
7.2 |
Jos käytetyssä laitteessa on eri tilavuus kuin Scheibler-Dietrich-laitteessa, on näytteestä ja kontrolliaineesta otettavia eriä ja tulosten laskua muutettava vastaavanlaisesti.
|
P. KOKONAISFOSFORIN MÄÄRITYS
FOTOMETRINEN MENETELMÄ
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehujen kokonaisfosforipitoisuus. Se soveltuu erityisesti alhaisten fosforipitoisuuksien analysoimiseen. Tietyissä tapauksissa (runsaasti fosforia sisältävät tuotteet) voidaan käyttää gravimetristä menetelmää.
2. Periaate
Näyte mineralisoidaan joko polttamalla kuivana ja liuottamalla happoon (orgaaniset rehut) tai happokeiton avulla (mineraaliyhdisteet ja nestemäiset rehut), ja se siirretään happamaan liuokseen. Liuos käsitellään molybdovanadaattireagenssilla. Muodostuneen keltaisen liuoksen optinen tiheys mitataan spektrofotometrissä 430 nm:ssä.
3. Reagenssit
|
3.1 |
Kalsiumkarbonaatti. |
|
3.2 |
Kloorivetyhappo, ρ20 = 1,10 g/ml (noin 6 mol/l). |
|
3.3 |
Typpihappo, ρ20 = 1,045 g/ml. |
|
3.4 |
Typpihappo, ρ201,38–1,42 g/ml. |
|
3.5 |
Rikkihappo, ρ201,84 g/ml. |
|
3.6 |
Molybdovanadaattireagenssi: sekoitetaan 200 ml ammoniumheptamolybdaattiliuosta (3.6.1), 200 ml ammoniummonovanadaattiliuosta (3.6.2) ja 134 ml typpihappoa (3.4) 1 litran mittapullossa. Täytetään merkkiin asti vedellä. |
|
3.6.1 |
Ammoniumheptamolybdaattiliuos: liuotetaan 100 g ammoniumheptamolybdaattia (NH4)6Mo7O24·4H2O kuumaan veteen. Lisätään 10 ml ammoniakkia (tiheys 0,91 g/ml) ja täytetään 1 litraksi vedellä. |
|
3.6.2 |
Ammoniumonovanadaattiliuos: liuotetaan 2,35 g ammoniummonovanadaattia NH4VO3 400 ml:aan kuumaa vettä. Lisätään hitaasti, samalla sekoittaen 20 ml laimeaa typpihappoa (7 ml HNO3 (3.4) + 13 ml H2O) ja täytetään 1 litraksi vedellä. |
|
3.7 |
Fosforistandardiliuos, 1 mg/ml: liuotetaan 4,387 g kaliumdivetyfosfaattia KH2PO4 veteen. Täytetään 1 litraksi vedellä. |
4. Välineistö
|
4.1 |
Kvartsi-, posliini- tai platinaupokkaita. |
|
4.2 |
Sähkölämmitteinen muhveliuuni, jossa on 550 oC:seen säädetty termostaatti. |
|
4.3 |
250 ml:n Kjeldahl-kolvi. |
|
4.4 |
Mittapulloja ja tarkkuuspipettejä. |
|
4.5 |
Spektrofotometri. |
|
4.6 |
Noin 16 mm:n läpimittaisia koeputkia, joissa on 14,5 mm:n läpimittaiset tulpat ja joiden vetoisuus on 25–30 ml. |
5. Menettely
5.1 Liuoksen valmistaminen
Näytteen pitoisuuden mukaan valmistetaan 5.1.1 tai 5.1.2 kohdassa esitetty liuos.
5.1.1
Punnitaan vähintään 1 g näytettä 1 mg:n tarkkuudella. Näyte pannaan Kjeldahl-kolviin, lisätään 20 ml rikkihappoa (3.5), ravistellaan, jotta happo imeytyisi aineeseen täydellisesti eikä aine tarttuisi pullon seinämiin, kuumennetaan ja pidetään kiehuvana 10 minuutin ajan. Annetaan jäähtyä hieman, lisätään 2 ml typpihappoa (3.4), kuumennetaan varovasti, annetaan jäähtyä hieman, lisätään uudelleen hieman typpihappoa (3.4) ja kohotetaan lämpötila jälleen kiehumispisteeseen. Tämä menettely toistetaan kunnes saadaan väritön liuos. Jäähdytetään, lisätään hieman vettä, neste dekantoidaan 500 ml:n mittapulloon huuhtomalla Kjeldahl-kolvi kuumalla vedellä. Annetaan jäähtyä, täytetään merkkiin asti vedellä, homogenoidaan ja suodatetaan.
5.1.2
Hehkutusupokkaaseen punnitaan noin 2,5 g näytettä 1 mg:n tarkkuudella. Näytteeseen lisätään 1 g kalsiumkarbonaattia (3.1) ja sekoitetaan kunnes se on täysin sekoittunut näytteeseen. Poltetaan tuhkaksi uunissa 550 oC:ssa kunnes saadaan valkoista tai harmaata tuhkaa (pieni määrä hiiltä ei häiritse). Tuhka siirretään 250 ml:n dekantterilasiin. Lisätään 20 ml vettä ja kloorivetyhappoa (3.2) kunnes kuohuminen lakkaa. Lisätään vielä 10 ml kloorivetyhappoa (3.2). Dekantterilasi asetetaan hiekkahauteelle ja haihdutetaan kuivaksi piin saostamiseksi. Jäännös liuotetaan uudelleen 10 ml:aan typpihappoa (3.3) ja keitetään hiekkahauteella tai sähkölevyllä 5 minuuttia haihduttamatta sitä kuivaksi. Neste dekantoidaan 500 ml:n mittapulloon huuhtomalla dekantterilasi useaan kertaan kuumalla vedellä. Annetaan jäähtyä, täytetään merkkiin asti vedellä, homogenoidaan ja suodatetaan.
5.2 Värin kehitys ja optisen tiheyden mittaus
Laimennetaan 5.1.1 tai 5.1.2 kohdassa saatu suodos siten, että fosforipitoisuus on enintään 40 μg/ml. Pipetoidaan 10 ml tätä liuosta koeputkeen (4.6) ja lisätään siihen 10 ml molybdovanadaattireagenssia (3.6). Homogenoidaan ja annetaan seistä ainakin 10 minuuttia 20 oC:ssa. Mitataan optinen tiheys spektrofotometrissä 430 nm:ssä liuosta vastaan, joka on saatu lisäämällä 10 ml molybdovanadaattireagenssia (3.6) 10 ml:aan vettä.
5.3 Kalibrointikäyrä
Standardiliuoksesta (3.7) valmistetaan liuokset, joissa on 5, 10, 20, 30 tai 40 μg fosforia/ml. Kaikista liuoksista otetaan 10 ml:n erä ja siihen lisätään 10 ml molybdovanadaattireagenssia (3.6). Homogenoidaan ja annetaan seistä ainakin 10 minuuttia 20 oC:ssa. Optinen tiheys mitataan 5.2 kohdan mukaisesti. Kalibrointikäyrä laaditaan piirtämällä optisia tiheyksiä ja näitä vastaavia fosforimääriä esittävä kuvaaja. Käyrä on lineaarinen välillä 0–40 μg/ml olevien konsentraatioiden osalta.
6. Tulosten laskeminen
Tutkittavassa näytteessä oleva fosforimäärä määritetään kalibrointikäyrää käyttäen.
Tulos ilmoitetaan prosentteina näytteestä.
Toistettavuus
Samasta näytteestä suoritetun kahden rinnakkaismäärityksen tulosten välinen ero ei saa olla yli:
|
— |
3 %:a, suhteessa korkeampaan tulokseen, alle 5 % olevien fosforipitoisuuksien osalta, |
|
— |
0,15 %:a, absoluuttisena arvona ilmoitettuna, vähintään 5 % olevien fosforipitoisuuksien osalta. |
Q. KLORIDIEN MÄÄRITYS REHUISTA
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää vesiliukoisten kloridien määrä; tulokset ilmoitetaan yleensä natriumkloridina. Menetelmä soveltuu käytettäväksi kaikkiin rehuihin.
2. Periaate
Kloridit liuotetaan veteen. Jos tuote sisältää orgaanista ainetta, se kirkastetaan. Liuos tehdään hieman happamaksi typpihapolla ja kloridit saostetaan hopeakloridin muodossa hopeanitraattiliuosta käyttäen. Hopeanitraatin ylimäärä titrataan ammoniumtiosyanaattiliuoksella Volhardin menetelmän mukaan.
3. Reagenssit
|
3.1 |
Ammoniumtiosyanaattiliuos 0,1 mol/l. |
|
3.2 |
Hopeanitraattiliuos 0,1 mol/l. |
|
3.3 |
Ammoniumferrisulfaattiliuos, kyllästetty liuos (NH4)Fe(SO4)2. |
|
3.4 |
Typpihappo, tiheys 1,38 g/ml. |
|
3.5 |
Dietyylieetteri. |
|
3.6 |
Asetoni. |
|
3.7 |
Carrez I -liuos: liuotetaan veteen 21,9 g sinkkiasetaattia, Zn(CH3COO)2·2H2O, ja 3 g jääetikkaa. Täytetään 100 ml:ksi vedellä. |
|
3.8 |
Carrez-liuos II: liuotetaan veteen 10,6 g kaliumferrosyanidia K4Fe(CN)6·3H2O. Täytetään 100 ml:ksi vedellä. |
|
3.9 |
Aktiivihiili, jossa ei ole klorideja ja joka ei absorboi niitä. |
4. Välineistö
Sekoittaja: noin 35–40 kierr./min.
5. Menettely
5.1 Liuoksen valmistaminen
Näytteen mukaan valmistetaan 5.1.1, 5.1.2 tai 5.1.3 kohdassa esitetty liuos.
Samanaikaisesti suoritetaan nollakoe, josta määritettävä näyte on jätetty pois.
5.1.1
Punnitaan milligramman tarkkuudella enintään 10 g näytettä, joka sisältää alle 3 g kloridien muodossa olevaa klooria. Lisätään 500 ml:n mittapulloon näyte ja 400 ml noin 20 oC:n lämpöistä vettä. Sekoitetaan 30 minuuttia sekoittajassa, täytetään merkkiin asti, homogenoidaan ja suodatetaan.
5.1.2
Punnitaan 1 mg:n tarkkuudella noin 5 g näytettä sekä 1 g aktiivihiiltä 500 ml:n mittapulloon. Lisätään 400 ml noin 20 oC:n lämpöistä vettä ja 5 ml Carrez I -liuosta (3.7), sekoitetaan 30 sekunnin ajan ja tämän jälkeen lisätään 5 ml Carrez II -liuosta (3.8). Sekoitetaan 30 minuuttia sekoittajassa, täytetään merkkiin asti, homogenoidaan ja suodatetaan.
5.1.3
Valmistetaan liuos 5.1.2 kohdassa esitetyllä tavalla, mutta sitä ei suodateta. Dekantoidaan (tarvittaessa sentrifugoidaan), siirretään 100 ml supernatanttia 200 ml:n mittapulloon. Sekoitetaan asetonin (3.6) kanssa ja täytetään merkkiin asti asetonilla, homogenoidaan ja suodatetaan.
5.2 Titraus
Pipetoidaan erlenmeyerkolviin 25–100 ml suodosta (oletetun klooripitoisuuden mukaan), joka on saatu 5.1.1, 5.1.2 tai 5.1.3 kohdassa esitetyllä tavalla. Pipetoitu erä saa sisältää korkeintaan 150 mg klooria (Cl). Laimennetaan tarvittaessa vähintään 50 ml:ksi vedellä, lisätään 5 ml typpihappoa (3.4), 20 ml kyllästettyä ammoniumferrisulfaattiliuosta (3.3) ja kaksi tippaa ammoniumtiosyanaattiliuosta (3.1), joka annostellaan nollamerkkiin täytetystä byretistä. Byrettiä käyttäen lisätään hopeanitraattiliuosta (3.2) siten, että saadaan 5 ml:n ylimäärä. Lisätään 5 ml dietyylieetteriä (3.5) ja ravistellaan voimakkaasti saostuman koaguloimiseksi. Ylimäärä hopeanitraattia titrataan ammoniumtiosyanaattiliuoksella (3.1) kunnes punaruskea väri säilyy noin yhden minuutin ajan.
6. Tulosten laskeminen
Kloorimäärä (X), joka ilmoitetaan natriumkloridiprosentteina, lasketaan seuraavaa kaavaa käyttäen:

jossa:
|
V1 |
= |
lisätyn hopeanitraattiliuoksen 0,1 mol/l määrä millilitroina |
|
V2 |
= |
itraukseen käytetyn ammoniumtiosyanaatin 0,1 mol/l määrä millilitroina. |
|
m |
= |
näytteen paino. |
Jos hopeanitraattiliuosta, 0,1 mol/l, on kulunut nollakoetta titrattaessa, kuluneen liuoksen määrä vähennetään tilavuudesta (V1 - V2).
7. Huomautukset
|
7.1 |
Titraus voidaan suorittaa myös potentiometrisesti. |
|
7.2 |
Jos määritettävät tuotteet sisältävät runsaasti öljyjä ja rasvoja, öljyt ja rasvat on ensin poistettava dietyylieetterillä tai petroolieetterillä. |
|
7.3 |
Kalajauhon osalta titraus voidaan suorittaa Mohrin menetelmällä. |
(1) Viljan, jauhojen, suurimoiden ja rouheiden kuivaamiseen lämpökaapin lämpökapasiteetin on oltava sellainen, että 13 oC:seen etukäteen säädettynä kaapin lämpötila palautuu ennalleen alle 45 minuutissa sen jälkeen, kun siellä on samanaikaisesti suurin mahdollinen määrä näytteitä. Kaapin tuuletuksen on oltava sellainen, että kun siinä kahden tunnin ajan kuivataan niin monta vehnänäytettä kuin sinne mahtuu, tulokset eroavat neljän tunnin ajan suoritetun kuivauksen tuloksista alle 0,15 %.
(2) Jos rasvalle tai öljylle tehdään vielä tämän jälkeen sen laatuun liittyviä määräyksiä, kiehumakivet korvataan lasihelmillä.
LIITE IV
REHUJEN SISÄLTÄMIEN SALLITTUJEN LISÄAINEIDEN VALVONNASSA KÄYTETTÄVÄT MÄÄRITYSMENTELMÄT
A. A-VITAMIININ MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää A-vitamiinin (retinolin) pitoisuus rehuissa ja esiseoksissa. A-vitamiini käsittää all-trans-retinyylialkoholin ja sen cis-isomeerit, jotka saadaan määritetyiksi tällä menetelmällä. A-vitamiinipitoisuus ilmoitetaan kansainvälisinä yksikköinä (ky) kilogrammaa kohti. Yksi ky vastaa aktiivisuutta, joka on 0,300 μg:lla all-trans-A-vitamiinialkoholia tai 0,344 μg:lla all-trans-A-vitamiiniasetaattia tai 0,550 μg:lla all-trans-A-vitamiinipalmitaattia.
Määritysraja on 2 000 ky A-vitamiinia/kg.
2. Periaate
Näyte hydrolysoidaan etanoli-kaliumhydroksidiliuoksella, ja A-vitamiini uutetaan petrolieetteriin. Liuotin haihdutetaan pois, haihdutusjäännös liuotetaan metanoliin ja laimennetaan tarvittaessa halutulle pitoisuusalueelle. A-vitamiinipitoisuus määritetään korkean erotuskyvyn käänteisfaasinestekromatografialla (RP-HPLC) UV- tai fluoresenssidetektoria käyttäen. Kromatografian ajo-olosuhteet valitaan siten, että all-trans-A-vitamiinialkoholi ja sen cis-isomeerit eivät erotu toisistaan.
3. Reagenssit
|
3.1 |
Etanoli, σ = 96 %. |
|
3.2 |
Petrolieetteri, kiehumislämpötila 40–60 oC. |
|
3.3 |
Metanoli. |
|
3.4 |
Kaliumhydroksidiliuos, c = 50 g/100 ml. |
|
3.5 |
Natriumaskorbaattiliuos, c = 10 g/100 ml (ks. 7.7 huomautukset). |
|
3.6 |
Natriumsulfidi, Na2S · × H2O (x = 7–9). |
|
3.6.1 |
Natriumsulfidiliuos, c = 0,5 mol/1 glyserolissa, ß = 120 g/l (kun x = 9) (ks. 7.8 huomautukset). |
|
3.7 |
Fenolftaleiiniliuos, c = 2 g/100 ml etanolissa (3.1). |
|
3.8 |
2-Propanoli. |
|
3.9 |
HPLC:ssä käytettävä liikkuva faasi: metanolin (3.3) ja veden seos, esim. 980 + 20 (v + v). Tarkka suhde määräytyy käytetyn kolonnin ominaisuuksien perusteella. |
|
3.10 |
Typpi, happivapaa. |
|
3.11 |
All-trans-A-vitamiiniasetaatti, erikoispuhdasta, jonka aktiivisuus on sertifioitu, esim. 2,80 × 106 ky/g. |
|
3.11.1 |
All-trans-A-vitamiiniasetaatin kantaliuos: Punnitaan 0,1 milligramman tarkkuudella 50 mg A-vitamiiniasetaattia (3.11) 100 ml:n mittapulloon. Liuotetaan 2-propanoliin (3.8) ja täytetään merkkiin asti samalla liuottimella. Tämän liuoksen nimellinen A-vitamiinipitoisuus on 1 400 ky/ml. Tarkka pitoisuus määritetään 5.6.3.1 kohdan mukaisesti. |
|
3.12 |
All-trans-A-vitamiinipalmitaatti, erikoispuhdasta, jonka aktiivisuus on sertifioitu, esim. 1,80 × 106 ky/g. |
|
3.12.1 |
All-trans-A-vitamiinipalmitaatin kantaliuos: Punnitaan 0,1 milligramman tarkkuudella 80 mg A-vitamiinipalmitaattia (3.12) 100 ml:n mittapulloon. Liuotetaan 2-propanoliin (3.8) ja täytetään merkkiin asti samalla liuottimella. Tämän liuoksen nimellinen A-vitamiinipitoisuus on 1 400 ky/ml. Tarkka pitoisuus määritetään 5.6.3.2 kohdan mukaisesti. |
|
3.13 |
2,6-Di-tert-butyyli-4-metyylifenoli (BHT) (ks. 7.5 huomautukset). |
4. Välineistö
|
4.1 |
Vakuumipyöröhaihdutin. |
|
4.2 |
Ruskeita lasiastioita. |
|
4.2.1 |
Tasapohjaisia tai kartionmuotoisia kolveja, 500 ml, joissa on lasihios. |
|
4.2.2 |
Mittapulloja, joissa on lasihiostulpat, kapeakaulaisia, 10, 25, 100 ja 500 ml. |
|
4.2.3 |
Erotussuppiloita, kartiomaisia, 1 000 ml, joissa on lasihiostulpat. |
|
4.2.4 |
Kartiopohjaisia kolveja, 250 ml, joissa on lasihios. |
|
4.3 |
Allihn-jäähdytin, vaipan pituus 300 mm, jossa on lasihios ja liitin kaasunsyöttöputkelle. |
|
4.4 |
Laskostettua suodatinpaperia faasien erotukseen, halkaisija 185 mm (esim. Schleicher & Schüll 597 HY 1/2). |
|
4.5 |
HPLC-laitteisto, jossa injektori. |
|
4.5.1 |
Nestekromatografiakolonni, 250 mm × 4 mm, C18, hiukkaskoko 5 tai 10 μm, tai vastaava (suorituskykyvaatimus: vain yksi piikki kaikille retinoli-isomeereille ko. HPLC-ajo-olosuhteilla). |
|
4.5.2 |
Vaihtuva-aallonpituuksinen UV- tai fluoresenssidetektori. |
|
4.6 |
Spektrofotometri ja 10 mm:n kvartsikyvettejä. |
|
4.7 |
Vesihaude, jossa on magneettisekoitin. |
|
4.8 |
Uuttolaitteisto, jossa on (ks. kuva 1): |
|
4.8.1 |
Lasisylinteri, tilavuus 1 l, jossa on lasihioskaula ja tulppa. |
|
4.8.2 |
Lasihiosliitin, jossa on sivuputki ja keskeltä läpi menevä liikuteltava putki. Liikuteltavassa putkessa on oltava U:n muotoinen alapää ja toisessa päässä nokka, niin että sylinterissä oleva ylempi nestekerros voidaan siirtää erotussuppiloon. |
5. Menettely
|
Huomautus: |
A-vitamiini on herkkä (UV-)valolle ja hapettumiselle. Kaikki toimenpiteet on suoritettava valolta suojattuna (käyttäen ruskeita tai alumiinifoliolla suojattuja lasitarvikkeita) ja hapelta suojattuna (typpivirtauksessa). Uuton aikana nestepinnan yläpuolella oleva ilma on korvattava typellä (ylipaineen muodostuminen estettävä raottamalla tulppaa silloin tällöin). |
5.1 Näytteen valmistaminen
Näyte jauhetaan niin että se läpäisee 1 mm:n seulan; näytteen lämpenemistä on vältettävä. Näytteen jauhaminen on tehtävä välittömästi ennen punnitusta ja saippuointia A-vitamiinin hajoamisen välttämiseksi.
5.2 Saippuointi
A-vitamiinipitoisuudesta riippuen punnitaan 1 mg:n tarkkuudella 2–25 g näytettä 500 ml:n tasapohjaiseen tai pyöreäpohjaiseen kolviin (4.2.1). Lisätään peräkkäin kolvia pyörittäen 130 ml etanolia (3.1), noin 100 mg BHT:tä (3.13), 2 ml natriumaskorbaattiliuosta (3.5) ja 2 ml natriumsulfidiliuosta (3.6). Kiinnitetään jäähdytin (4.3) kolviin, ja kolvi pannaan vesihauteeseen, jossa on magneettisekoitin (4.7). Kuumennetaan kiehumispisteeseen ja annetaan kiehua 5 minuuttia. Lisätään 25 ml kaliumhydroksidiliuosta (3.4) jäähdyttimen läpi (4.3) ja annetaan kiehua vielä 25 minuuttia sekoittaen hitaassa typpivirrassa. Jäähdytin huuhdellaan noin 20 ml:lla vettä ja kolvin sisältö jäähdytetään huoneenlämpötilaan.
5.3 Uutto
Saippuointiliuos siirretään dekantoimalla kvantitatiivisesti, huuhtomalla kaikkiaan 250 ml:lla vettä, 1 000 ml:n erotussuppiloon (4.2.3) tai uuttolaitteeseen (4.8). Saippuointikolvi huuhdotaan ensin 25 ml:lla etanolia (3.1) ja sitten 100 ml:lla petrolieetteriä (3.2) ja huuhteluliuos siirretään erotussuppiloon tai uuttolaitteeseen. Veden ja etanolin lopputilavuuksien suhteen on oltava noin 2:1. Ravistellaan voimakkaasti 2 minuuttia ja annetaan laskeutua 2 minuuttia.
5.3.1
Kun kerrokset ovat erottuneet (ks. huomautus 7.3), petrolieetterikerros siirretään toiseen erotussuppiloon (4.2.3). Uutto toistetaan kahdesti 100 ml:lla petrolieetteriä (3.2) ja kahdesti 50 ml:lla petrolieetteriä (3.2).
Yhdistetyt uutteet pestään erotussuppilossa, pyörittäen varovasti (emulsion muodostumisen välttämiseksi), kahdesti 100 ml:lla vettä ja sitten toistuvasti ravistelemalla edelleen 100 ml:lla vettä kerrallaan, kunnes pesuvesi on väritön lisättäessä fenolftaleiiniliuosta (3.7) (neljä pesukertaa riittää yleensä). Pesty uute suodatetaan suspendoitujen vesijäämien poistamiseksi kuivan laskostetun faasinerotussuodatinpaperin läpi (4.4) 500 ml:n mittapulloon (4.2.2). Erotussuppilo ja suodatinpaperi huuhdotaan 50 ml:lla petrolieetteriä (3.2), täytetään merkkiin asti petrolieetterillä (3.2) ja sekoitetaan hyvin.
5.3.2
Kun kerrokset ovat erottuneet (ks. huomautus 7.3), pannaan lasisylinterin (4.8.1) tulpan tilalle lasihiosliitin (4.8.2) ja asetetaan liikuteltavan putken U:n muotoinen alapää siten, että se on juuri faasien rajakohdan yläpuolella. Johtamalla paineistettua typpeä sivuputkeen siirretään ylempi petrolieetterikerros 1 000 ml:n erotussuppiloon (4.2.3). Lisätään 100 ml petrolieetteriä (3.2) lasisylinteriin, suljetaan tulpalla ja ravistellaan hyvin. Annetaan faasien erottua ja siirretään ylempi kerros erotussuppiloon kuten edellä. Uutto toistetaan vielä 100 ml:lla petrolieetteriä (3.2) ja kahdesti 50 ml:lla petrolieetteriä (3.2) ja petrolieetterikerrokset siirretään erotussuppiloon.
Yhdistetyt petrolieetteriuutteet pestään kuten 5.3.1 kohdassa ja jatketaan kuten kyseisessä kohdassa kerrottu.
5.4 Näyteliuoksen valmistaminen HPLC-ajoja varten
Pipetoidaan tietty määrä petrolieetteriliuosta (joka on saatu kohdassa 5.3.1 tai 5.3.2) 250 ml:n päärynänmuotoiseen kolviin (4.2.4). Liuotin haihdutetaan lähes kuiviin pyöröhaihduttimella (4.1) alennetussa paineessa; vesihauteen lämpötila saa olla korkeintaan 40 oC. Ilmanpaine palautetaan normaaliksi typellä (3.10) ja kolvi otetaan pois pyöröhaihduttimesta. Jäljellä oleva liuotin haihdutetaan typpivirrassa (3.10) ja jäännös liuotetaan välittömästi tunnettuun tilavuuteen (10–100 ml) metanolia (3.3) (A-vitamiinipitoisuuden on oltava välillä 5–30 ky/ml).
5.5 Määritys HPLC:llä
A-vitamiini erotetaan C18-käänteisfaasikolonnilla (4.5.1) ja sen pitoisuus mitataan UV-detektorilla (325 nm) tai fluoresenssidetektorilla (eksitaatio: 325 nm, emissio: 475 nm) (4.5.2).
Injektoidaan tietty määrä (esim. 20 μl) kohdassa 5.4 saatua metanoliliuosta ja eluoidaan liikkuvalla faasilla (3.9). Lasketaan useiden saman näyteliuoksen injektioiden sekä useiden kalibrointiliuosten (5.6.2) injektioiden piikkien korkeuksien (pinta-alojen) keskiarvo.
Seuraavat ajo-olosuhteet ovat ohjeellisia; muita ajo-olosuhteita voidaan käyttää, jos niillä saadaan vastaavat tulokset.
|
Nestekromatografiakolonni (4.5.1): |
250 mm × 4 mm, C18, hiukkaskoko 5 tai 10 μm, tai vastaava |
|
Liikkuva faasi (3.9): |
Metanolin (3.3) ja veden seos, esim. 980 + 20 (v + v). |
|
Virtausnopeus: |
1–2 ml/min |
|
Detektori (4.5.2): |
UV-detektori (325 nm) tai fluoresenssidetektori (eksitaatio: 325 nm / emissio: 475 nm) |
5.6 Kalibrointi
5.6.1
Pipetoidaan 20 ml A-vitamiiniasetaatin kantaliuosta (3.11.1) tai 20 ml A-vitamiinipalmitaatin kantaliuosta (3.12.1) 500 ml:n tasapohjaiseen tai pyöreäpohjaiseen kolviin 4.2.1) ja hydrolysoidaan kuten kohdassa 5.2, mutta ilman BHT-lisäystä. Tämän jälkeen uutetaan petrolieetterillä (3.2) kohdan 5.3 mukaisesti ja täytetään 500 ml:aan petrolieetterillä (3.2). Haihdutetaan 100 ml tätä uutetta pyöröhaihduttimella (ks. 5.4) lähes kuiviin, jäljellä oleva liuotin poistetaan typpivirralla (3.10) ja jäännös liuotetaan 10,0 ml:aan metanolia (3.3). Tämän liuoksen nimellinen A-vitamiinipitoisuus on 560 ky/ml. Tarkka pitoisuus on määritettävä kohdan 5.6.3.3 mukaisesti. Käyttöstandardiliuos valmistetaan juuri ennen käyttöä.
Pipetoidaan 2,0 ml tätä käyttöstandardiliuosta 20 ml:n mittapulloon, täytetään merkkiin asti metanolilla (3.3) ja sekoitetaan. Tämän laimennetun käyttöstandardiliuoksen nimellinen pitoisuus on 56 ky/ml A-vitamiinia.
5.6.2
Pipetoidaan 1,0, 2,0, 5,0 ja 10,0 ml laimennettua käyttöstandardiliuosta 20 ml:n mittapulloihin, täytetään merkkiin asti metanolilla (3.3) ja sekoitetaan. Näiden liuosten nimelliset pitoisuudet ovat 2,8, 5,6, 14,0 ja 28,0 ky/ml A-vitamiinia.
Injektoidaan 20 μl kutakin kalibrointiliuosta useita kertoja ja määritetään piikkien korkeuksien (pinta-alojen) keskiarvot. Piirretään kalibrointikäyrä käyttäen korkeuksien (pinta-alojen) keskiarvoja, ottaen huomioon UV-spektrin tarkistuksen tulokset (5.6.3.3).
5.6.3
5.6.3.1
Pipetoidaan 2,0 ml A-vitamiiniasetaattikantaliuosta (3.11.1) 50 ml:n mittapulloon (4.2.2) ja täytetään merkkiin asti 2-propanolilla (3.8). Tämän liuoksen nimellinen pitoisuus on 56 ky/ml A-vitamiinia. Pipetoidaan 3,0 ml tätä laimennettua A-vitamiiniasetaattiliuosta 25 ml:n mittapulloon ja täytetään merkkiin asti 2-propanolilla (3.8). Tämän liuoksen nimellinen pitoisuus on 6,72 ky/ml A-vitamiinia. Mitataan liuoksen UV-spektri 2-propanolia (3.8) vastaan spektrofotometrillä (4.6) välillä 300–400 nm. Ekstinktiomaksimin on oltava välillä 325–327 nm.
A-vitamiinipitoisuuden laskeminen:
ky/ml A-vitamiinia = E326 × 19,0
(A-vitamiiniasetaatin 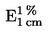 = 1 530 aallonpituudella 326 nm 2-propanolissa)
= 1 530 aallonpituudella 326 nm 2-propanolissa)
5.6.3.2
Pipetoidaan 2,0 ml A-vitamiinipalmitaattikantaliuosta (3.12.1) 50 ml:n mittapulloon (4.2.2) ja täytetään merkkiin asti 2-propanolilla (3.8). Tämän liuoksen nimellinen pitoisuus on 56 ky/ml A-vitamiinia. Pipetoidaan 3,0 ml tätä laimennettua A-vitamiinipalmitaattiliuosta 25 ml:n mittapulloon ja täytetään merkkiin asti 2-propanolilla (3.8). Tämän liuoksen nimellinen pitoisuus on 6,72 ky/ml A-vitamiinia. Mitataan liuoksen UV-spektri 2-propanolia (3.8) vastaan spektrofotometrillä (4.6) välillä 300–400 nm. Ekstinktiomaksimin on oltava välillä 325–327 nm.
A-vitamiinipitoisuuden laskeminen:
ky/ml A-vitamiinia = E326 × 19,0
(A-vitamiinipalmitaatin 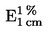 = 957 aallonpituudella 326 nm 2-propanolissa)
= 957 aallonpituudella 326 nm 2-propanolissa)
5.6.3.3
Pipetoidaan 3,0 ml laimentamatonta A-vitamiinin käyttöstandardiliuosta, joka on valmistettu kohdan 5.6.1 mukaisesti, 50 ml:n mittapulloon (4.2.2) ja täytetään merkkiin asti 2-propanolilla (3.8). Pipetoidaan 5,0 ml tätä liuosta 25 ml:n mittapulloon ja täytetään merkkiin asti 2-propanolilla (3.8). Tämän liuoksen nimellinen pitoisuus on 6,72 ky/ml A-vitamiinia. Mitataan liuoksen UV-spektri 2-propanolia (3.8) vastaan spektrofotometrillä (4.6) välillä 300–400 nm. Ekstinktiomaksimin on oltava välillä 325–327 nm.
A-vitamiinipitoisuuden laskeminen:
ky/ml A-vitamiinia = E325 × 18,3
(A-vitamiinialkoholin  = 1 821 aallonpituudella 325 nm 2-propanolissa)
= 1 821 aallonpituudella 325 nm 2-propanolissa)
6. Tulosten laskeminen
Näyteliuoksen A-vitamiinipitoisuus (ky/ml) lasketaan näyteliuoksen A-vitamiinipiikkien korkeuden (pinta-alan) keskiarvosta kalibrointikäyrän (5.6.2) avulla.
Näytteen A-vitamiinipitoisuus w (ky/kg) lasketaan seuraavan kaavan avulla:
 [IU/kg]
[IU/kg]
jossa:
|
c |
= |
näyteliuoksen (5.4) A-vitamiinipitoisuus (ky/ml), |
|
V1 |
= |
näyteliuoksen (5.4) tilavuus (ml), |
|
V2 |
= |
kohdassa 5.4 otetun näyteliuoksen tilavuus (ml), |
|
m |
= |
näyte-erän paino (g). |
7. Huomautukset
|
7.1 |
Jos näytteiden A-vitamiinipitoisuus on alhainen, voi olla hyödyllistä yhdistää kahden saippuointierän petrolieetteriuutteet (punnittu määrä: 25 g) yhdeksi näyteliuokseksi HPLC-määritystä varten. |
|
7.2 |
Analyysiä varten otetussa näytemäärässä ei saa olla yli 2 g:aa rasvaa. |
|
7.3 |
Jos faasit eivät erotu, lisätään noin 10 ml etanolia (3.1) emulsion hajottamiseksi. |
|
7.4 |
Kun on kyseessä kalanmaksaöljy ja muut puhtaat rasvat, saippuointiajan on oltava 45–60 minuuttia. |
|
7.5 |
Hydrokinonia voi käyttää BHT:n sijasta. |
|
7.6 |
Käytettäessä normaalifaasikolonnia retinoli-isomeerit voivat erottua toisistaan. Siinä tapauksessa kaikkien cis- ja trans-isomeeripiikkien korkeudet (pinta-alat) on laskettava yhteen. |
|
7.7 |
Natriumaskorbaattiliuoksen voi korvata noin 150 mg:lla askorbiinihappoa. |
|
7.8 |
Natriumsulfidiliuoksen voi korvata noin 50 mg:lla EDTA:ta. |
|
7.9 |
Maidonkorvikkeiden A-vitamiinin määrityksessä on kiinnitettävä erityistä huomiota
Ylimääräisellä näyte-erällä tehtävällä saantokokeella varmistetaan, että käytetty menetelmä tuottaa luotettavat tulokset tällä materiaalilla (maidonkorvikkeella). Jos saantoaste on alempi kuin 80 %, määrityksen tulosta on korjattava saannon suhteen. |
8. Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla suurempi kuin 15 % suuremmasta arvosta.
9. Laboratorioiden välisen vertailun tulokset (1)
|
|
Esiseos |
Esiseosvalmiste |
Kivennäisrehu |
Tiiviste |
Porsasrehu |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
keskiarvo [ky/kg] |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
sr [ky/kg] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [ky/kg] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
sR [ky/kg] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [ky/kg] |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
CVR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
yksittäisten arvojen lukumäärä |
|
sr |
= |
toistettavuuden keskihajonta |
|
sR |
= |
uusittavuuden keskihajonta |
|
r |
= |
toistettavuus |
|
R |
= |
uusittavuus |
|
CVr |
= |
toistettavuuden variaatiokerroin |
|
CVR |
= |
uusittavuuden variaatiokerroin |

B. E-VITAMIININ MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää E-vitamiinin pitoisuus rehuissa ja esiseoksissa. E-vitamiinipitoisuus ilmoitetaan mg/kg DL-α-tokoferoliasetaattia. 1 mg DL-α-tokoferoliasetaattia vastaa 0,91 mg:aa DL-α-tokoferolia (E-vitamiinia).
Määritysraja on 2 mg E-vitamiinia/kg. Tämä määritysraja voidaan saavuttaa ainoastaan fluoresenssi-detektorilla. UV-detektoria käytettäessä määritysraja on 10 mg/kg.
2. Periaate
Näyte hydrolysoidaan etanoli-kaliumhydroksidiliuoksella ja E-vitamiini uutetaan petrolieetteriin. Liuotin haihdutetaan pois, haihdutusjäännös liuotetaan metanoliin ja laimennetaan tarvittaessa halutulle pitoisuusalueelle. E-vitamiinipitoisuus määritetään korkean erotuskyvyn käänteisfaasinestekromatografialla (RP-HPLC) fluoresenssi- tai UV-detektoria käyttäen.
3. Reagenssit
|
3.1 |
Etanoli, σ = 96 %. |
|
3.2 |
Petrolieetteri, kiehumislämpötila 40–60 oC. |
|
3.3 |
Metanoli. |
|
3.4 |
Kaliumhydroksidiliuos, c = 50 g/100 ml. |
|
3.5 |
Natriumaskorbaattiliuos, c = 10 g/100 ml (ks. 7.7 huomautukset). |
|
3.6 |
Natriumsulfidi, Na2S · × H2O (x = 7–9). |
|
3.6.1 |
Natriumsulfidiliuos, c = 0,5 mol/1 glyserolissa, β = 120 g/l (kun x = 9) (ks. 7.8 huomautukset). |
|
3.7 |
Fenolftaleiiniliuos, c = 2 g/100 ml etanolissa (3.1). |
|
3.8 |
HPLC:ssä käytettävä liikkuva faasi: metanolin (3.3) ja veden seos, esim. 980 + 20 (v + v). Tarkka suhde määräytyy käytetyn kolonnin ominaisuuksien perusteella. |
|
3.9 |
Typpi, happivapaa. |
|
3.10 |
DL-α-tokoferoliasetaatti, erikoispuhdas, sertifioitu aktiivisuus. |
|
3.10.1 |
DL-α-tokoferoliasetaatin kantaliuos: Punnitaan 0,1 mg:n tarkkuudella 100 mg DL-α-tokoferoliasetaattia (3.10) 100 ml:n mittapulloon. Liuotetaan etanoliin (3.1) ja täytetään merkkiin asti samalla liuottimella. 1 ml tätä liuosta sisältää 1 mg DL-α-tokoferoliasetaattia. (UV-tarkistus, ks. 5.6.1.3; stabilointi, ks. 7.4 huomautukset). |
|
3.11 |
DL-α-tokoferoli, erikoispuhdas, sertifioitu aktiivisuus. |
|
3.11.1 |
DL-α-tokoferolin kantaliuos: Punnitaan 0,1 mg:n tarkkuudella 100 mg DL-α-tokoferolia (3.11) 100 ml:n mittapulloon. Liuotetaan etanoliin (3.1) ja täytetään merkkiin asti samalla liuottimella. 1 ml tätä liuosta sisältää 1 mg DL-α-tokoferolia. (UV-tarkistus, ks. 5.6.2.3; stabilointi, ks. 7.4 huomautukset). |
|
3.12 |
2,6-Di-tert-butyyli-4-metyylifenoli (BHT) (ks. 7.5 huomautukset). |
4. Välineistö
|
4.1 |
Pyörivä kalvohaihdutin. |
|
4.2 |
Ruskeita lasiastioita. |
|
4.2.1 |
Tasapohjaisia tai kartionmuotoisia keittokolveja, 500 ml, joissa on lasihios. |
|
4.2.2 |
Mittapulloja, joissa on lasihiostulpat, kapeakaulaisia, 10, 25, 100 ja 500 ml. |
|
4.2.3 |
Erotussuppiloita, kartiomaisia, 1 000 ml, joissa on lasihiostulpat. |
|
4.2.4 |
Päärynänmuotoisia kolveja, 250 ml, joissa on lasihios. |
|
4.3 |
Allihn-jäähdytin, vaipan pituus 300 mm, jossa on lasihios ja liitin kaasunsyöttöputkelle. |
|
4.4 |
Laskostettua suodatinpaperia faasien erotukseen, halkaisija 185 mm (esim. Schleicher & Schüll 597 HY 1/2). |
|
4.5 |
HPLC-laitteisto, jossa injektori. |
|
4.5.1 |
Nestekromatografiakolonni, 250 mm × 4 mm, C18, hiukkaskoko 5 tai 10 μm, tai vastaava. |
|
4.5.2 |
Vaihtuva-aallonpituuksinen fluoresenssi- tai UV- detektori. |
|
4.6 |
Spektrofotometri ja 10 mm:n kvartsikyvettejä. |
|
4.7 |
Vesihaude, jossa on magneettisekoitin. |
|
4.8 |
Uuttolaitteisto, jossa on (ks. kuva 1): |
|
4.8.1 |
Lasisylinteri, tilavuus 1 l, jossa on lasihioskaula ja tulppa. |
|
4.8.2 |
Lasihiosliitin, jossa on sivuputki ja keskeltä läpi menevä liikuteltava putki. Liikuteltavassa putkessa on oltava U:n muotoinen alapää ja toisessa päässä nokka, niin että sylinterissä oleva ylempi nestekerros voidaan siirtää erotussuppiloon. |
5. Menettely
|
Huomautus: |
E-vitamiini on herkkä (UV-)valolle ja hapettumiselle. Kaikki toimenpiteet on suoritettava valolta suojattuna (käyttäen ruskeita tai alumiinifoliolla suojattuja lasitarvikkeita) ja hapelta suojattuna (typpivirtauksessa). Uuton aikana nestepinnan yläpuolella oleva ilma on korvattava typellä (ylipaineen muodostuminen estettävä raottamalla tulppaa silloin tällöin). |
5.1 Näytteen valmistaminen
Näyte jauhetaan niin että se läpäisee 1 mm:n seulan; näytteen lämpenemistä on vältettävä. Näytteen jauhaminen on tehtävä välittömästi ennen punnitusta ja saippuointia E-vitamiinin hajoamisen välttämiseksi.
5.2 Saippuointi
E-vitamiinipitoisuudesta riippuen punnitaan 0,01 g:n tarkkuudella 2–25 g näytettä 500 ml:n tasapohjaiseen tai pyöreäpohjaiseen kolviin (4.2.1). Lisätään peräkkäin kolvia pyörittäen 130 ml etanolia (3.1), noin 100 mg BHT:tä (3.12), 2 ml natriumaskorbaattiliuosta (3.5) ja 2 ml natriumsulfidiliuosta (3.6). Kiinnitetään jäähdytin (4.3) kolviin ja kolvi pannaan vesihauteeseen, jossa on magneettisekoitin (4.7). Kuumennetaan kiehumispisteeseen ja annetaan kiehua 5 minuuttia. Lisätään 25 ml kaliumhydroksidiliuosta (3.4) jäähdyttimen läpi (4.3) ja annetaan kiehua vielä 25 minuuttia sekoittaen hitaassa typpivirrassa. Jäähdytin huuhdellaan noin 20 ml:lla vettä ja kolvin sisältö jäähdytetään huoneenlämpötilaan.
5.3 Uutto
Saippuointiliuos siirretään dekantoimalla kvantitatiivisesti, huuhtomalla kaikkiaan 250 ml:lla vettä, 1 000 ml:n erotussuppiloon (4.2.3) tai uuttolaitteeseen (4.8). Saippuointikolvi huuhdotaan ensin 25 ml:lla etanolia (3.1) ja sitten 100 ml:lla petrolieetteriä (3.2) ja huuhteluliuos siirretään erotussuppiloon tai uuttolaitteeseen. Veden ja etanolin lopputilavuuksien suhteen pitää olla noin 2:1. Ravistellaan voimakkaasti 2 minuuttia ja annetaan laskeutua 2 minuuttia.
5.3.1
Kun kerrokset ovat erottuneet (ks. huomautus 7.3), petrolieetterikerros siirretään toiseen erotussuppiloon (4.2.3). Uutto toistetaan kahdesti 100 ml:lla petrolieetteriä (3.2) ja kahdesti 50 ml:lla petrolieetteriä (3.2).
Yhdistetyt uutteet pestään erotussuppilossa, pyörittäen varovasti (emulsion muodostumisen välttämiseksi), kahdesti 100 ml:lla vettä ja sitten toistuvasti ravistelemalla edelleen 100 ml:lla vettä kerrallaan, kunnes pesuvesi on väritön lisättäessä fenolftaleiiniliuosta (3.7) (neljä pesukertaa riittää yleensä). Pesty uute suodatetaan suspendoitujen vesijäämien poistamiseksi kuivan laskostetun suodatinpaperin läpi (4.4) 500 ml:n mittapulloon (4.2.2). Erotussuppilo ja suodatinpaperi huuhdotaan 50 ml:lla petrolieetteriä (3.2), täytetään merkkiin asti petrolieetterillä (3.2) ja sekoitetaan hyvin.
5.3.2
Kun kerrokset ovat erottuneet (ks. huomautus 7.3), pannaan lasisylinterin (4.8.1) tulpan tilalle lasihiosliitin (4.8.2) ja asetetaan liikuteltavan putken U:n muotoinen alapää siten, että se on juuri faasien rajakohdan yläpuolella. Johtamalla paineistettua typpeä sivuputkeen siirretään ylempi petrolieetterikerros 1 000 ml:n erotussuppiloon (4.2.3). Lisätään 100 ml petrolieetteriä (3.2) lasisylinteriin, suljetaan tulpalla ja ravistellaan hyvin. Annetaan faasien erottua ja siirretään ylempi kerros erotussuppiloon kuten edellä. Uutto toistetaan vielä 100 ml:lla petrolieetteriä (3.2) ja kahdesti 50 ml:lla petrolieetteriä (3.2) ja petrolieetterikerrokset siirretään erotussuppiloon.
Yhdistetyt petrolieetteriuutteet pestään kuten 5.3.1 kohdassa ja jatketaan kuten kyseisessä kohdassa kerrottu.
5.4 Näyteliuoksen valmistaminen HPLC-ajoja varten
Pipetoidaan tietty määrä petrolieetteriliuosta (joka on saatu kohdassa 5.3.1 tai 5.3.2) 250 ml:n päärynänmuotoiseen kolviin (4.2.4). Liuotin haihdutetaan lähes kuiviin pyöröhaihduttimella (4.1) alennetussa paineessa; vesihauteen lämpötila saa olla korkeintaan 40 oC. Ilmanpaine palautetaan normaaliksi typellä (3.9) ja kolvi otetaan pois pyöröhaihduttimesta. Jäljellä oleva liuotin haihdutetaan typpivirrassa (3.9) ja jäännös liuotetaan välittömästi tunnettuun tilavuuteen (10–100 ml) metanolia (3.3) (DL-α-tokoferolipitoisuuden on oltava välillä 5–30 μg/ml).
5.5 Määritys HPLC:llä
E-vitamiini erotetaan C18-käänteisfaasikolonnilla (4.5.1) ja sen pitoisuus mitataan fluoresenssidetektorilla (eksitaatio: 295 nm, emissio: 330 nm) tai UV-detektorilla (292 nm) (4.5.2).
Injektoidaan tietty määrä (esim. 20 μl) kohdassa 5.4 saatua metanoliliuosta ja eluoidaan liikkuvalla faasilla (3.8). Lasketaan useiden saman näyteliuoksen injektioiden sekä useiden kalibrointiliuosten (5.6.2) injektioiden piikkien korkeuksien (pinta-alojen) keskiarvo.
Seuraavat ajo-olosuhteet ovat ohjeellisia; muita ajo-olosuhteita voidaan käyttää, jos niillä saadaan vastaavat tulokset.
|
Nestekromatografiakolonni (4.5.1): |
250 mm × 4 mm, C18, hiukkaskoko 5 tai 10 μm, tai vastaava |
|
Liikkuva faasi (3.8): |
Metanolin (3.3) ja veden seos, esim. 980 + 20 (v + v). |
|
Virtausnopeus: |
1–2 ml/min |
|
Detektori (4.5.2): |
Fluoresenssidetektori (eksitaatio: 295 nm / emissio: 330 nm) tai UV-detektori (292 nm) |
5.6 Kalibrointi (DL-α-tokoferoliasetaatti tai DL-α-tokoferoli)
5.6.1
5.6.1.1
Pipetoidaan 25 ml DL-α-tokoferoliasetaattikantaliuosta (3.10.1) 500 ml:n tasapohjaiseen tai pyöreäpohjaiseen kolviin (4.2.1) ja hydrolysoidaan kuten kohdassa 5.2. Tämän jälkeen uutetaan petrolieetterillä (3.2) kohdan 5.3 mukaisesti ja täytetään 500 ml:aan petrolieetterillä. Haihdutetaan 25 ml tätä uutetta pyöröhaihduttimella (ks. 5.4) lähes kuiviin, jäljellä oleva liuotin poistetaan typpivirralla (3.9) ja jäännös liuotetaan 25,0 ml:aan metanolia (3.3). Tämän liuoksen nimellinen pitoisuus on 45,5 μg/ml DL-α-tokoferolia, mikä vastaa 50 μg/ml:aa DL-α-tokoferoliasetaattia. Käyttöstandardiliuos valmistetaan juuri ennen käyttöä.
5.6.1.2
Pipetoidaan 1,0, 2,0, 4,0 ja 10,0 ml kantastandardiliuosta 20 ml:n mittapulloihin, täytetään merkkiin asti metanolilla (3.3) ja sekoitetaan. Näiden liuosten nimelliset pitoisuudet ovat 2,5, 5,0, 10,0 ja 25,0 μg/ml DL-α-tokoferoliasetaattia eli 2,28, 4,55, 9,10 μg/ml ja 22,8 μg/ml DL-α-tokoferolia.
Injektoidaan 20 μl kutakin kalibrointiliuosta useita kertoja ja määritetään piikkien korkeuksien (pinta-alojen) keskiarvot. Piirretään kalibrointikäyrä käyttäen piikkien korkeuksien (pinta-alojen) keskiarvoja.
5.6.1.3
Laimennetaan 5,0 ml DL-α-tokoferoliasetaattikantaliuosta (3.10.1) 25,0 ml:ksi etanolilla ja mitataan tämän liuoksen UV-spektri etanolia (3.1) vastaan spektrofotometrillä (4.6) välillä 250–320 nm.
Absorptiomaksimin pitää olla aallonpituudella 284 nm:
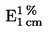 = 43,6 aallonpituudella 284 nm etanolissa
= 43,6 aallonpituudella 284 nm etanolissa
Tällä laimennuksella pitää saada ekstinktioarvo 0,84–0,88.
5.6.2
5.6.2.1
Pipetoidaan 2 ml DL-α-tokoferolikantaliuosta (3.11.1) 50 ml:n mittapulloon, liuotetaan metanoliin (3.3) ja täytetään merkkiin asti metanolilla. Tämän liuoksen nimellinen pitoisuus on 40 μg/ml DL-α-tokoferolia, mikä vastaa 44,0 μg/ml:aa DL-α-tokoferoliasetaattia. Käyttöstandardiliuos valmistetaan juuri ennen käyttöä.
5.6.2.2
Pipetoidaan 1,0, 2,0, 4,0 ja 10,0 ml kantastandardiliuosta 20 ml:n mittapulloihin, täytetään merkkiin asti metanolilla (3.3) ja sekoitetaan. Näiden liuosten nimelliset pitoisuudet ovat 2,0, 4,0, 8,0 ja 20,0 μg/ml DL-α-tokoferolia eli 2,20, 4,40, 8,79 μg/ml ja 22,0 μg/ml DL-α-tokoferoliasetaattia.
Injektoidaan 20 μl kutakin kalibrointiliuosta useita kertoja ja määritetään piikkien korkeuksien (pinta-alojen) keskiarvot. Piirretään kalibrointikäyrä käyttäen piikkien korkeuksien (pinta-alojen) keskiarvoja.
5.6.2.3
Laimennetaan 2,0 ml DL-α-tokoferolikantaliuosta (3.11.1) 25,0 ml:ksi etanolilla ja mitataan tämän liuoksen UV-spektri etanolia (3.1) vastaan spektrofotometrillä (4.6) välillä 250–320 nm. Absorptiomaksimin pitää olla aallonpituudella 292 nm:
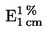 = 75,8 aallonpituudella 292 nm etanolissa
= 75,8 aallonpituudella 292 nm etanolissa
Tällä laimennuksella pitää saada ekstinktioarvo 0,6.
6. Tulosten laskeminen
Näyteliuoksen E-vitamiinipitoisuus μg/ml (laskettuna α-tokoferoliasetaattina) määritetään näyteliuoksen E-vitamiinipiikkien korkeuden (pinta-alan) keskiarvosta kalibrointikäyrän (5.6.1.2 tai 5.6.2.2) avulla.
Näytteen E-vitamiinipitoisuus w (mg/kg) lasketaan seuraavan kaavan avulla:
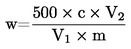 [mg/kg]
[mg/kg]
jossa:
|
c |
= |
näyteliuoksen (5.4) E-vitamiinipitoisuus (α-tokoferoliasetaattina) (μg/ml); |
|
V1 |
= |
näyteliuoksen (5.4) tilavuus (ml); |
|
V2 |
= |
näyteliuoksen (5.4) tilavuus (ml); |
|
m |
= |
näyte-erän paino (g). |
7. Huomautukset
|
7.1 |
Jos näytteiden E-vitamiinipitoisuus on alhainen, voi olla hyödyllistä yhdistää kahden saippuointierän petrolieetteriuutteet (punnittu määrä: 25 g) yhdeksi näyteliuokseksi HPLC-määritystä varten. |
|
7.2 |
Analyysiä varten otetussa näytemäärässä ei saa olla yli 2 g:aa rasvaa. |
|
7.3 |
Jos faasit eivät erotu, lisätään noin 10 ml etanolia (3.1) emulsion hajottamiseksi. |
|
7.4 |
Sen jälkeen, kun DL-α-tokoferoliasetaattiliuoksen pitoisuus on määritetty 5.6.1.3 kohdan mukaisesti tai DL-α-tokoferoliliuoksen pitoisuus 5.6.2.3 kohdan mukaisesti spektrofotometrisesti, lisätään noin 10 mg BHT:tä (3.12) liuokseen (3.10.1 tai 3.10.2); liuos säilytetään jääkaapissa (enintään 4 viikkoa). |
|
7.5 |
Hydrokinonia voi käyttää BHT:n sijasta. |
|
7.6 |
Käytettäessä normaalifaasikolonnia α-, β-, γ- ja δ-tokoferolit voivat erottua toisistaan. |
|
7.7 |
Natriumaskorbaattiliuoksen voi korvata noin 150 mg:lla askorbiinihappoa. |
|
7.8 |
Natriumsulfidiliuoksen voi korvata noin 50 mg:lla EDTA:ta. |
|
7.9 |
E-vitamiiniasetaatti hydrolysoituu hyvin nopeasti emäksisissä olosuhteissa, ja siksi se hapettuu herkästi eritoten hivenaineiden kuten raudan tai kuparin läsnä ollessa. Jos esiseoksessa on E-vitamiinia yli 5 000 mg/kg, E-vitamiini saattaa hajota. Tästä syystä varmistuksessa suositellaan käytettäväksi HPLC-menetelmää, johon kuuluu E-vitamiiniyhdisteen hajottaminen entsymaattisesti ilman emäksistä saippuointivaihetta. |
8. Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla suurempi kuin 15 % suuremmasta arvosta.
9. Laboratorioiden välisen vertailun tulokset (2)
|
|
Esiseos |
Esiseosvalmiste |
Kivennäisrehu |
Tiiviste |
Porsasrehu |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
keskiarvo [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [ %] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
sR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [ %] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
yksittäisten arvojen lukumäärä |
|
sr |
= |
toistettavuuden keskihajonta |
|
sR |
= |
uusittavuuden keskihajonta |
|
r |
= |
toistettavuus |
|
R |
= |
uusittavuus |
|
CVr |
= |
toistettavuuden variaatiokerroin |
|
CVR |
= |
uusittavuuden variaatiokerroin. |

C. RAUDAN, KUPARIN, MANGAANIN JA SINKIN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehuista hivenaineet rauta, kupari, mangaani ja sinkki. Määritysrajat ovat:
|
— |
rauta (Fe): 20 mg/kg |
|
— |
kupari (Cu): 10 mg/kg |
|
— |
mangaani (Mn): 20 mg/kg |
|
— |
sinkki (Zn): 20 mg/kg. |
2. Periaate
Näytteet liuotetaan kloorivetyhappoon sen jälkeen, kun siinä mahdollisesti esiintyvät orgaaniset aineet on tuhottu. Alkuaineet rauta, kupari, mangaani ja sinkki määritetään sopivan laimennuksen jälkeen atomiabsorbtiospektrometrillä.
3. Reagenssit
Alustavat huomautukset
Reagenssien ja analyysiliuosten valmistuksessa käytetään määritettävistä kationeista vapaata vettä, joka on saatu joko tislaamalla se kaksi kertaa borosilikaattilasista tai kvartsista valmistetussa tislauslaitteessa tai käsittelemällä se kaksi kertaa ioninvaihtohartsilla.
Reagenssien on oltava vähintään analyysilaatua. Nollakokeella tarkistetaan, ettei määritettävää alkuainetta esiinny. Tarvittaessa reagensseja on puhdistettava lisää.
Seuraavassa kuvattujen standardiliuosten tilalla voidaan käyttää kaupallisesti saatavilla olevia standardiliuoksia, jos ne ovat takuulaatua ja ne on tarkistettu ennen käyttöä.
|
3.1 |
Kloorivetyhappo, tiheys 1,19 g/ml. |
|
3.2 |
Kloorivetyhappo, 6 mol/l. |
|
3.3 |
Kloorivetyhappo, 0,5 mol/l. |
|
3.4 |
38–40 % (v/v) fluorivetyhappo, jonka rautapitoisuus on alle 1 mg Fe/l ja haihdutusjäännös alle 10 mg (sulfaattina)/litra. |
|
3.5 |
Rikkihappo, tiheys 1,84 g/ml. |
|
3.6 |
Vetyperoksidi, noin 100 tilavuusosaa happea (30 % (w/v)). |
|
3.7 |
Rautastandardiliuos (1 000 μg Fe/ml), joka on valmistettu seuraavasti, tai vastaava kaupallisesti saatavilla oleva liuos: liuotetaan 1 g rautalankaa 200 ml:aan kloorivetyhappoa, 6 mol/l, (3.2), lisätään 16 ml vetyperoksidia (3.6) ja täytetään 1 litraksi vedellä. |
|
3.7.1 |
Rautakäyttöstandardiliuos (100 μg Fe/ml), joka on valmistettu laimentamalla 1 osa standardiliuosta (3.7) 9 osalla vettä. |
|
3.8 |
Kuparistandardiliuos (1 000 μg Cu/ml), joka on valmistettu seuraavasti, tai vastaava kaupallisesti saatavilla oleva liuos:
|
|
3.8.1 |
Kuparikäyttöstandardiliuos (10 μg Cu/ml), joka on valmistettu laimentamalla 1 osa standardiliuosta (3.8) 9 osalla vettä ja sen jälkeen laimentamalla 1 osa saatua liuosta 9 osalla vettä. |
|
3.9 |
Mangaanistandardiliuos (1 000 μg Mn/ml), joka on valmistettu seuraavasti, tai vastaava kaupallisesti saatavilla oleva liuos:
|
|
3.9.1 |
Mangaanikäyttöstandardiliuos (10 μg Mn/ml), joka on valmistettu laimentamalla 1 osa standardiliuosta (3.9) 9 osalla vettä ja sen jälkeen laimentamalla 1 osa saatua liuosta 9 osalla vettä. |
|
3.10 |
Sinkkistandardiliuos (1 000 μg Zn/ml), joka on valmistettu seuraavasti, tai vastaava kaupallisesti saatavilla oleva liuos:
|
|
3.10.1 |
Sinkkikäyttöstandardiliuos (10 μg Zn/ml), joka on valmistettu laimentamalla 1 osa standardiliuosta (3.10) 9 osalla vettä ja sen jälkeen laimentamalla 1 osa saatua liuosta 9 osalla vettä. |
|
3.11 |
Lantaanikloridiliuos: liuotetaan 12 g lantaanioksidia 150 ml:aan vettä, lisätään 100 ml kloorivetyhappoa, 6 mol/l (3.2), ja täytetään 1 litraksi vedellä. |
4. Välineistö
|
4.1 |
Muhveliuuni, jossa on lämpötilan säätölaite ja mielellään myös piirturi. |
|
4.2 |
Lasitavaroiden on oltava kestävää borosilikaattityyppiä, ja on suositeltavaa käyttää välineitä, jotka on erityisesti tarkoitettu hivenaineiden määrityksiin. |
|
4.3 |
Atomiabsorbtiospektrofotometri, jonka herkkyys ja tarkkuus vaaditulla alueella täyttävät menetelmän vaatimukset. |
5. Menettely (3)
5.1 Orgaanista ainetta sisältävät näytteet
5.1.1 (4)
|
5.1.1.1 |
Punnitaan 5–10 g näytettä 0,2 mg:n tarkkuudella, pannaan kvartsi- tai platinaupokkaaseen (ks. huomautus b), kuivataan lämpökaapissa 105 oC:ssa ja pannaan upokas kylmään muhveliuuniin (4.1). Uuni suljetaan (ks. huomautus c) ja lämpötila nostetaan vähitellen 450–475 oC:seen noin 90 minuutin aikana. Lämpötila pidetään tässä 4–16 tuntia (esimerkiksi yön yli) hiilipitoisen materiaalin poistamiseksi ja uuni avataan ja sen annetaan jäähtyä (ks. huomautus d). Tuhka kostutetaan vedellä ja siirretään 250 ml:n dekantterilasiin. Upokas pestään yhteensä noin 5 ml:lla kloorivetyhappoa (3.1), happo lisätään hitaasti ja varovasti dekantterilasiin (tällöin voi esiintyä voimakas reaktio, joka johtuu CO2:n muodostumisesta). Lisätään kloorivetyhappoa (3.1) pisaroittain samalla sekoittaen kunnes kupliminen on kokonaan lakannut. Haihdutetaan kuivaksi ajoittain lasisauvalla sekoittaen. Jäännökseen lisätään 15 ml kloorivetyhappoa, 6 mol/l (3.2), ja noin 120 ml vettä. Sekoitetaan lasisauvalla, joka jätetään dekantterilasiin, ja peitetään astia kellonlasilla. Kuumennetaan varovasti kiehumispisteeseen ja pidetään kiehumispisteessä kunnes enempää tuhkaa ei havaita liukenevan. Suodatetaan tuhkattomalla suodatinpaperilla ja suodos kerätään talteen 250 ml:n mittapulloon. Dekantterilasi ja suodatin pestään 5 ml:lla kuumaa kloorivetyhappoa, 6 mol/l (3.2), sekä kaksi kertaa kiehuvalla vedellä. Mittapullo täytetään merkkiin asti vedellä (HCl-konsentraatio noin 0,5 mol/l). |
|
5.1.1.2 |
Jos suodatinpaperissa oleva jäännös on ulkonäöltään mustaa (hiiltä), se viedään takaisin uuniin ja poltetaan uudelleen tuhkaksi 450–475 oC:ssa. Tämä poltto kestää vain muutaman tunnin (noin 3–5 tuntia) ja on suoritettu loppuun silloin, kun tuhka on ulkonäöltään valkeaa tai lähes valkeaa. Jäännös liuotetaan noin 2 ml:aan kloorivetyhappoa (3.1), haihdutetaan kuivaksi ja lisätään 5 ml kloorivetyhappoa, 6 mol/l (3.2). Kuumennetaan, suodatetaan mittapulloon ja täytetään merkkiin asti vedellä (HCl-konsentraatio noin 0,5 mol/l). Huomautukset:
|
5.1.2
5.1.2.1
Kutakin määritettävää alkuainetta varten valmistetaan 3.7.1, 3.8.1, 3.9.1 ja 3.1.10 kohdassa mainituista käyttöstandardiliuoksista sarja standardiliuoksia siten, että kunkin standardiliuoksen HCl-konsentraatio on noin 0,5 mol/l ja lantaanikloridikonsentraatio sellainen, joka vastaa 0,1 %:a (w/v) lantaania.
Valittujen hivenainekonsentraatioiden on oltava käytetyn spektrofotometrin herkkyysalueella. Seuraavissa taulukoissa annetaan esimerkiksi erilaisia standardiliuoskoostumuksia; käytetyn spektrofotometrin tyypistä ja herkkyydestä riippuen voi kuitenkin olla tarpeen valita muita pitoisuuksia.
Rauta
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
Käyttöstandardiliuoksen (3.7.1) ml-määrä (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantaanikloridiliuosta (3.11) ja täytetään 100 ml:ksi vedellä |
|||||||
Kupari
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Käyttöstandardiliuoksen (3.8.1) ml-määrä (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
Mangaani
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Käyttöstandardiliuoksen (3.9.1) ml-määrä (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantaanikloridiliuosta (3.11) ja täytetään 100 ml:ksi vedellä |
|||||||
Sinkki
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
Käyttöstandardiliuoksen (3.10.1) ml-määrä (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantaanikloridiliuosta (3.11) ja täytetään 100 ml:ksi vedellä |
|||||||
5.1.2.2
Kuparin määritystä varten voidaan 5.1.1 kohdassa valmistettua liuosta käyttää yleensä suoraan. Tarvittaessa sen pitoisuus tuodaan kalibrointiliuosten alueelle, tästä otettu erä voidaan pipetoida 100 ml:n mittapulloon ja täyttää merkkiin asti kloorivetyhapolla, 0,5 mol/l (3.3).
Raudan, mangaanin ja sinkin määritystä varten pipetoidaan erä 5.1.1 kohdassa valmistettua liuosta 100 ml:n mittapulloon, lisätään 10 ml lantaanikloridiliuosta (3.11) ja täytetään merkkiin asti kloorivetyhapolla, 0,5 mol/l (3.3.) (ks. myös 8 kohdan huomautus).
5.1.2.3
Nollakoe suoritetaan samalla tavalla mutta ilman määritettävää näytettä. Standardiliuosta ’0’ ei saa käyttää nollanäytteenä.
5.1.2.4
Standardiliuosten ja määritettävän liuoksen atomiabsorptio mitataan hapettavalla ilma-asetyleeniliekillä seuraavilla aallonpituuksilla:
|
|
Fe: 248,3 nm |
|
|
Cu: 324,8 nm |
|
|
Mn: 279,5 nm |
|
|
Zn: 213,8 nm |
Kukin mittaus suoritetaan neljä kertaa.
5.2 Kivennäisrehut
Jos näyte ei sisällä orgaanista ainetta, sitä ei ole tarpeen polttaa tuhkaksi. Suoritusta jatketaan 5.1.1.1 kohdan toisesta kappaleesta. Haihdutus fluorivetyhapolla voidaan jättää pois.
6. Tulosten laskeminen
Määritettävässä liuoksessa oleva hivenainepitoisuus lasketaan kalibrointikäyrältä ja tulos ilmoitetaan milligrammoina hivenainetta kilogrammassa näytettä (ppm).
7. Toistettavuus
Samasta näytteestä saman henkilön tekemän kahden rinnakkaismäärityksen tulosten välinen ero ei saa olla yli:
|
— |
5:tä mg/kg, absoluuttisena arvona, kun hivenainepitoisuus on enintään 50 mg/kg, |
|
— |
10 %:a, suurempaan arvoon verrattuna, kun hivenainepitoisuus on 50–100 mg/kg, |
|
— |
10:tä mg/kg, absoluuttisena arvona, kun hivenainepitoisuus on 100–200 mg/kg, |
|
— |
5 %:a, suurempaan arvoon verrattuna, kun hivenainepitoisuus on yli 200 mg/kg. |
8. Huomautus
Suuret fosfaattimäärät voivat häiritä raudan, mangaanin ja sinkin määritystä. Tällainen häiriö on korjattava lantaanikloridiliuosta (3.11) lisäämällä. Jos näytteessä painosuhde Ca + Mg / P on > 2, voidaan lantaanikloridiliuoksen (3.11) lisäys määritettävään liuokseen ja standardiliuoksiin jättää pois.
D. HALOFUGINONIN MÄÄRITTÄMINEN
DL-trans-7-bromi-6-kloori-3-[3-(3-hydroksi-2-piperidyyli)asetonyyli]-kinatsoliini-4-(3H)-oni-hydrobromidi
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehujen halufuginonipitoisuus. Määritysraja on 1 mg/kg.
2. Periaate
Näytettä käsitellään kuumalla vedellä ja halofuginoni uutetaan tämän jälkeen vapaana emäksenä etyyliasetaattiin ja erotetaan hydrokloridina happamaan vesiliuokseen. Uute puhdistetaan ioninvaihtokromatografialla. Halofuginonipitoisuus määritetään suuren erotuskyvyn käänteisfaasinestekromatografialla (HPLC) UV-detektoria käyttäen.
3. Reagenssit
|
3.1 |
Asetonitriili, HPLC-laatua vastaava. |
|
3.2 |
Amberlite XAD-2 -hartsi. |
|
3.3 |
Ammoniumasetaatti. |
|
3.4 |
Etyyliasetaatti. |
|
3.5 |
Jääetikka. |
|
3.6 |
Halofuginonistandardi (DL-trans-7-bromi-6-kloori-3-[3-hydroksi-2-piperidyyli)asetonyyli]-kinatsoliini-4-(3H)-onihydrobromidi, E 764). |
|
3.6.1 |
Mitataan 500 ml:n mittapulloon 50 mg (0,1 mg:n tarkkuudella) halofuginonia (3.6), liuotetaan ammoniumasetaattipuskuriliuokseen (3.18), täytetään merkkiin asti puskuriliuoksella ja sekoitetaan. Tämä liuos säilyy 5 oC:ssa kolmen viikon ajan, jos sitä säilytetään valolta suojattuna. |
|
3.6.2 |
Mitataan sarjaan 100 ml:n mittapulloja 1,0, 2,0, 3,0, 4,0 ja 6,0 ml standardikantaliuosta (3.6.1). Täytetään merkkiin asti liikkuvalla faasilla (3.21) ja sekoitetaan. Näiden liuosten halofuginonipitoisuudet ovat 1,0, 2,0, 3,0, 4,0 ja 6,0 μg/ml. Liuokset on valmistettava juuri ennen käyttöä. |
|
3.7 |
Kloorivetyhappo (ρ20 noin 1,16 g/ml) |
|
3.8 |
Metanoli |
|
3.9 |
Hopeanitraatti. |
|
3.10 |
Natriumaskorbaatti. |
|
3.11 |
Natriumkarbonaatti. |
|
3.12 |
Natriumkloridi. |
|
3.13 |
EDTA (etyleenidiamiinitetraetikkahappo, dinatriumsuola). |
|
3.14 |
Vesi, HPLC-laatua vastaava. |
|
3.15 |
Natriumkarbonaattiliuos, c = 10 g/100 ml. |
|
3.16 |
Natriumkloridilla kyllästetty natriumkarbonaattiliuos, c = 5 g/100 ml. Liuotetaan 50 g natriumkarbonaattia (3.11) veteen, täytetään 1 litraksi ja lisätään natriumkloridia (3.12), kunnes liuos on kyllästetty. |
|
3.17 |
Kloorivetyhappo: noin 0,1 mol/l. Laimennetaan 10 ml kloorivetyhappoa (3.7) vedellä 1 litraksi. |
|
3.18 |
Ammoniumasetaattipuskuriliuos, noin 0,25 mol/l. Liuotetaan 19,3 g ammoniumasetaattia (3.3) ja 30 ml etikkahappoa (3.5) veteen (3.14) ja laimennetaan 1 litraksi. |
|
3.19 |
Amberlite XAD-2 -hartsin valmistaminen. Pestään sopivaa määrää Amberlitea (3.2) vedellä, kunnes pesuun käytetyssä vesifaasissa hopeanitraatilla (3.20) tehtävä koe osoittaa, että kloridi-ioneita ei enää ole. Tämän jälkeen hartsi pestään 50 ml:lla metanolia (3.8), tämä metanoli heitetään pois ja hartsia säilytetään puhtaassa metanolissa. |
|
3.20 |
Hopeanitraattiliuos, noin 0,1 mol/l. Liuotetaan 0,17 g hopeanitraattia (3.9) 10 ml:aan vettä. |
|
3.21 |
HPLC:n liikkuva faasi. Sekoitetaan 500 ml asetonitriiliä (3.1) 300 ml:aan ammoniumasetaattipuskuriliuosta (3.18) ja 1 200 ml:aan vettä (3.14). Säädetään pH arvoon 4,3 etikkahapolla (3.5). Suodatetaan 0,22 μm:n suodattimen (4.8) läpi ja poistetaan kaasut liuoksesta (esimerkiksi 10 minuutin ultraäänikäsittelyllä). Tämä liuos säilyy yhden kuukauden ajan, jos sitä säilytetään suljetussa astiassa valolta suojattuna. |
4. Välineistö
|
4.1 |
Ultraäänihaude. |
|
4.2 |
Pyörivä kalvohaihdutin. |
|
4.3 |
Sentrifugi. |
|
4.4 |
HPLC-laitteisto, jossa on vaihtuva-aallonpituuksinen UV-detektori tai diodirividetektori. |
|
4.4.1 |
Nestekromatografiakolonni, 300 mm × 4 mm, C18, hiukkaskoko 10 μm, tai vastaava kolonni. |
|
4.5 |
Lasikolonni (300 mm × 10 mm), joka on varustettu lasisintterisuodattimella ja hanalla. |
|
4.6 |
Lasikuitusuodattimia, halkaisija 150 mm. |
|
4.7 |
Kalvosuodattimia, 0,45 μm. |
|
4.8 |
Kalvosuodattimia, 0,22 μm. |
5. Menettely
|
Huomautus: |
Vapaana emäksenä halofuginoni on epästabiili alkalisissa liuoksissa ja etyyliasetaattiliuoksissa. Se ei saa olla etyyliasetaatissa yli 30 minuuttia. |
5.1 Yleistä
|
5.1.1 |
Nollanäyte on analysoitava, jotta varmistetaan, ettei se sisällä halofuginonia tai mittausta häiritseviä aineita. |
|
5.1.2 |
Saanto on määritettävä analysoimalla nollanäyte, johon on tehty näytteen halofuginonipitoisuutta vastaava halofuginonilisäys. Pitoisuuden 3 mg/kg aikaan saamiseksi lisätään 300 μl standardikantaliuosta (3.6.1) 10 grammaan nollarehua, sekoitetaan ja annetaan seistä 10 minuuttia ennen uuttamista (5.2).
|
5.2 Uutto
Punnitaan 0,1 gramman tarkkuudella 10 g valmistettua näytettä 200 ml:n sentrifugiputkeen, lisätään 0,5 g natriumaskorbaattia (3.10), 0,5 g EDTA:a (3.13) ja 20 ml vettä; sekoitetaan. Asetetaan putki vesihauteeseen (80 oC) 5 minuutiksi. Annetaan jäähtyä huoneen lämpötilaan, jonka jälkeen lisätään 20 ml natriumkarbonaattiliuosta (3.15) ja sekoitetaan. Lisätään viipymättä 100 ml etyyliasetaattia (3.4) ja ravistellaan käsin voimakkaasti 15 sekunnin ajan. Tämän jälkeen putki sijoitetaan kolmeksi minuutiksi ultraäänihauteeseen (4.1) ja korkkia höllennetään. Sentrifugoidaan 2 minuuttia ja dekantoidaan etyyliasetaattifaasi lasikuitusuodattimen (4.6) läpi 500 ml:n erotussuppiloon. Toistetaan näytteen uutto toisella 100 ml:n etyyliasetaattiannoksella. Pestään yhdistettyjä uutteita 1 minuutin ajan 50 ml:lla natriumkloridilla kyllästettyä natriumkarbonaattiliuosta (3.16) ja poistetaan vesifaasi.
Uutetaan orgaanista kerrosta 50 ml:lla kloorivetyhappoa (3.17) 1 minuutin ajan. Juoksutetaan alempi happokerros 250 ml:n erotussuppiloon. Uutetaan orgaanista kerrosta vielä 50 ml:lla kloorivetyhappoa 1,5 minuutin ajan ja yhdistetään uutteet. Pestään yhdistetyt happouutteet heiluttamalla niitä noin 10 sekunnin ajan 10 ml:n etyyliasetaattimäärän kanssa (3.4).
Siirretään vesikerros kvantitatiivisesti 250 ml:n pyörökolviin ja heitetään orgaaninen faasi pois. Haihdutetaan pyörivällä kalvohaihduttimella (4.2) kaikki etyyliasetaattijäämät happoliuoksesta. Vesihauteen lämpötila ei saa olla yli 40:tä oC. Noin 25 mbar:n tyhjössä etyyliasetaattijäämät poistuvat 5 minuutissa 38 oC:ssa.
5.3 Puhdistaminen
5.3.1
Kullekin näyteuutteelle valmistetaan XAD-2-kolonni. Siirretään metanolilla (3.8) 10 g esikäsiteltyä Amberlitea (3.19) lasikolonniin (4.5). Lisätään pieni lasivillatuppo hartsikerroksen päälle. Juoksutetaan metanoli kolonnista ja pestään hartsi 100 ml:lla vettä ja pysäytetään virtaus, kun neste yltää hartsikerroksen yläosaan. Annetaan kolonnin stabiloitua 10 minuuttia ennen sen käyttöä. Kolonnin ei saa koskaan antaa kuivua.
5.3.2
Siirretään uute (5.2) kvantitatiivisesti valmistetun Amberlite-kolonnin (5.3.1) yläosaan, eluoidaan ja heitetään eluaatti pois. Eluointinopeus ei saa olla yli 20 ml:aa minuutissa. Huuhdellaan pyörökolvi 20 ml:lla kloorivetyhappoa (3.17) ja käytetään tätä liuosta hartsikolonnin pesemiseen. Poistetaan happoliuosjäämä ilmavirralla. Heitetään pesuliuokset pois. Lisätään kolonniin 100 ml metanolia (3.8) ja annetaan eluoitua 5–10 ml ja kerätään eluaatti 250 ml:n pyörökolviin. Annetaan jäljelle jääneen metanolin stabiloitua hartsin kanssa 10 minuutin ajan, jatketaan eluointia alle 20 ml minuutissa olevalla nopeudella ja kerätään eluaatti samaan pyörökolviin. Haihdutetaan metanoli pyöröhaihduttimella (4.2) huolehtien siitä, että vesihauteen lämpötila ei ole yli 40 oC. Siirretään jäännös kvantitatiivisesti 10 ml:n mittapulloon käyttäen liikkuvaa faasia (3.21). Täytetään merkkiin asti liikkuvalla faasilla ja sekoitetaan. Suodatetaan näyte kalvosuodattimen (4.7) läpi. Säilytetään tämä liuos HPLC-määritystä (5.4) varten.
5.4 HPLC-määritys
5.4.1
Seuraavat edellytykset ovat ohjeellisia; muita määritysolosuhteita voidaan käyttää sillä edellytyksellä, että niillä saadaan vastaavat tulokset.
Nestekromatografiakolonni (4.4.1)
HPLC:n liikkuva faasi (3.21)
Virtausnopeus: 1,5–2 ml/min
Detektioaallonpituus: 243 nm
Injektiotilavuus: 40–100 μl.
Kromatografiajärjestelmän stabiilisuus on tarkistettava injektoimalla useita kertoja kalibrointiliuosta (3.6.2), jonka pitoisuus on 3,0 μg/ml, kunnes piikkien korkeudet (pinta-alat) ja retentioajat eivät muutu.
5.4.2
Injektoidaan jokaista kalibrointiliuosta (3.6.2) useita kertoja ja mitataan piikkien korkeudet (pinta-alat) jokaiselle pitoisuudelle. Kalibrointikäyrä piirretään siten, että y-akselilla ovat kalibrointiliuosten piikkien korkeuksien (pinta alojen) keskiarvot ja x-akselilla vastaavat konsentraatiot (μg/ml).
5.4.3
Injektoidaan useita kertoja näyteuutetta (5.3.2) käyttäen samaa tilavuutta kuin kalibrointiliuoksille ja määritetään halofuginonin piikkien korkeuden (pinta-alan) keskiarvo.
6. Tulosten laskeminen
Määritetään näyteliuoksen pitoisuus (μg/ml) näyteliuoksen halofuginonin piikkien korkeuden (pinta-alan) keskiarvosta kalibrointikäyrän (5.4.2) avulla.
Näytteessä olevan halofuginonin määrä w (mg/kg) lasketaan seuraavalla kaavalla:

jossa:
|
c |
= |
näyteliuoksen halofuginonipitoisuus, μg/ml, |
|
m |
= |
näyte-erän paino grammoina. |
7. Tulosten validointi
7.1 Tunnistaminen
Tutkittavan aineen tunnistaminen voidaan varmistaa yhteiskromatografialla tai käyttämällä diodirividetektoria, jolla voidaan verrata näyteuutteen ja halofuginonia 6,0 μg/ml sisältävän kalibrointiliuoksen (3.6.2) spektrejä.
7.1.1
Näyteuutteeseen lisätään sopiva määrä kalibrointiliuosta (3.6.2). Lisätyn halofuginonimäärän on vastattava näyteuutteesta todetun halofuginonin arvioitua määrää.
Ainoastaan halofuginonipiikin korkeus saa kasvaa, kun otetaan huomioon sekä lisätty määrä halofuginonia että näyteliuoksen laimeneminen. Piikin puolivälissä sen leveyden on oltava ± 10 % lähtöleveydestä.
7.1.2
Tulokset arvioidaan seuraavien arviointiperusteiden mukaisesti:
|
a) |
näytteen ja standardin spektrien maksimiabsorptioaallonpituuksien on kromatogrammin piikin huipun kohdalla mitattuna oltava samat ilmaisinjärjestelmän erotuskyvyn mukaan määräytyvissä rajoissa. Diodirivimäärityksessä se on yleensä ± 2 nm; |
|
b) |
välillä 225–300 nm kromatogrammin piikin huipun kohdalla ajetut näyte- ja standardispektrit eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat ja näiden kahden spektrin välillä havaittu poikkeama ei ole missään kohdassa yli 15 % standardina olevan tutkittavan aineen absorbanssista; |
|
c) |
välillä 225–300 nm näyteliuoksesta ajetut spektrit piikin nousun, huipun ja laskun kohdalla eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun samat maksimit esiintyvät ja kun kaikissa havaittavissa kohdissa spektrien välinen poikkeama ei ole suurempi kuin 15 % huipun spektrin absorbanssista. |
Jos jokin näistä edellytyksistä ei täyty, tutkittavan aineen esiintymistä ei ole vahvistettu.
7.2 Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla yli 0,5 mg/kg, kun halofuginonipitoisuudet ovat enintään 3 mg/kg.
7.3 Saanto
Sellaisen nollanäytteen saannon, johon on lisätty määritettävää ainetta, on oltava vähintään 80 %.
8. Laboratorioiden välisen vertailun tulokset
Laboratorioiden välisessä tutkimuksessa (5) analysoitiin kolme näytettä kahdeksassa laboratoriossa.
Tulokset
|
|
Näyte A (nollanäyte) vastaanotettaessa |
Näyte B (jauhona) |
Näyte C (rakeina) |
||
|
|
|
Vastaanotettaessa |
2 kuukauden kuluttua |
Vastaanotettaessa |
2 kuukauden kuluttua |
|
Keskiarvo [mg/kg] |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [ %] |
— |
16 |
18 |
14 |
17 |
|
Rec. [ %] |
|
86 |
74 |
88 |
75 |
|
ND |
= |
ei havaittu |
|
SR |
= |
uusittavuuden keskihajonta |
|
CVR |
= |
uusittavuuden variaatiokerroin, % |
|
Rec. |
= |
saanto ( %). |
E. ROBENIDIININ MÄÄRITTÄMINEN
1,3-bis[(4-kloori-bentsylideeni)amino]guanidiinihydrokloridi
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää robenidiinin pitoisuus rehuissa. Määritysraja on 5 mg/kg.
2. Periaate
Näyte uutetaan happamoidulla metanolilla. Uute kuivataan ja määräosa puhdistetaan alumiinioksidikolonnissa. Robenidiini eluoidaan kolonnista metanolilla, konsentroidaan ja saatetaan sopivaksi tilavuudeksi liikkuvalla faasilla. Robenidiinipitoisuus määritetään korkean erotuskyvyn käänteisfaasinestekromatografialla (HPLC) UV-detektoria käyttäen.
3. Reagenssit
|
3.1 |
Metanoli. |
|
3.2 |
Happamoitu metanoli. Siirretään 4,0 ml kloorivetyhappoa (ρ20 = 1,18 g/ml) 500 ml:n mittapulloon, täytetään merkkiin asti metanolilla (3.1) ja sekoitetaan. Tämä liuos on valmistettava juuri ennen käyttöä. |
|
3.3 |
Asetonitriili, HPLC-laatua vastaava. |
|
3.4 |
Molekyyliseula. Tyyppiä 3A, 8–12 meshin helmiä (1,6–2,5 mm:n helmiä, kiteistä alumiinisilikaattia, huokosten halkaisija 0,3 mm). |
|
3.5 |
Alumiinioksidi, hapan, aktiviteettiluokka I pylväskromatografiaa varten. Siirretään 100 grammaa alumiinioksidia sopivaan astiaan ja lisätään 2,0 ml vettä. Suljetaan ja ravistellaan noin 20 minuutin ajan. Säilytetään hyvin suljetussa astiassa. |
|
3.6 |
Kaliumdivetyfosfaattiliuos, c = 0,025 mol/l. Liuotetaan 3,40 g kaliumdivetyfosfaattia veteen (HPLC-laatua) 1 000 ml:n mittapullossa, täytetään merkkiin asti ja sekoitetaan. |
|
3.7 |
Dinatriumvetyfosfaattiliuos, c = 0,025 mol/l. Liuotetaan 3,55 g vedetöntä (tai 4,45 g dihydraattia tai 8,95 g dodekahydraattia) dinatriumvetyfosfaattia veteen (HPLC-laatua vastaava) litran mittapullossa, täytetään merkkiin asti ja sekoitetaan. |
|
3.8 |
HPLC:n liikkuva faasi. Sekoitetaan seuraavat reagenssit:
Suodatetaan 0,22 μm:n suodattimen (4.6) läpi ja poistetaan kaasut liuoksesta (esimerkiksi 10 minuutin ultraäänikäsittelyllä). |
|
3.9 |
Standardiyhdiste. Puhdas robenidiini: 1,3-bis[(4-klooribentsylideeni)amino]guanidiinihydrokloridi. |
|
3.9.1 |
Punnitaan 30 mg 0,1 mg:n tarkkuudella robenidiinin standardiyhdistettä (3.9). Liuotetaan happamoituun metanoliin (3.2) 100 ml:n mittapullossa, täytetään merkkiin asti samalla liuottimella ja sekoitetaan. Kääritään pullo alumiinifolioon ja säilytetään valolta suojattuna. |
|
3.9.2 |
Siirretään 10,0 ml standardikantaliuosta (3.9.1) 250 ml:n pulloon, täytetään merkkiin asti liikkuvalla faasilla (3.8) ja sekoitetaan. Kääritään pullo alumiinifolioon ja säilytetään valolta suojattuna. |
|
3.9.3 |
Siirretään sarjassa 50 ml:n mittapulloihin 5,0, 10,0, 15,0, 20,0 ja 25,0 ml standardin välimuotoliuosta (3.9.2). Täytetään merkkiin asti liikkuvalla faasilla (3.8) ja sekoitetaan. Näiden liuosten robenidiinipitoisuudet ovat vastaavasti 1,2, 2,4, 3,6, 4,8 ja 6,0 μg/ml. Liuokset on valmistettava juuri ennen käyttöä. |
|
3.10 |
Vesi, HPLC-laatua vastaava. |
4. Välineistö
|
4.1 |
Lasikolonni. Valmistettu ruskeasta lasista, varustettu hanalla ja säiliöllä, jonka tilavuus on noin 150 ml, sisähalkaisija 10–15 mm, pituus 250 mm. |
|
4.2 |
Mekaaninen ravistelija tai magneettisekoitin. |
|
4.3 |
Pyörivä kalvohaihdutin. |
|
4.4 |
Vaihtuva-aallonpituuksinen HPLC-laitteisto tai diodirividetektori, detektioaallonpituusalue 250–400 nm. |
|
4.4.1 |
Nestekromatografiakolonni: 300 mm × 4 mm, C18, hiukkaskoko 10 μm, tai vastaava |
|
4.5 |
Lasikuitusuodatinpaperia (Whatman GF/A tai vastaava). |
|
4.6 |
Kalvosuodattimia, 0,22 μm. |
|
4.7 |
Kalvosuodattimia, 0,45 μm. |
5. Menettely
|
Huomautus: |
Robenidiini on herkkä valolle. Kaikissa toiminnoissa on käytettävä ruskeaa lasia. |
5.1 Yleistä
|
5.1.1 |
Nollanäyte on analysoitava, jotta varmistetaan, ettei se sisällä robenidiinia tai mittausta häiritseviä aineita. |
|
5.1.2 |
Saanto on määritettävä analysoimalla nollanäyte (5.1.1), johon on lisätty näytteen robenidiinipitoisuutta vastaava määrä robenidiinia. Pitoisuuden 60 mg/kg aikaan saamiseksi siirretään 3,0 ml standardikantaliuosta (3.9.1) 250 ml:n erlenmeyerkolviin. liuos haihdutetaan noin 0,5 ml:ksi typpivirrassa. Lisätään 15 g nollarehua, sekoitetaan ja odotetaan 10 minuuttia ennen siirtymistä uuttovaiheeseen (5.2).
|
5.2 Uutto
Punnitaan 0,01 g:n tarkkuudella noin 15 g valmistettua näytettä. Siirretään 250 ml:n erlenmeyerkolviin ja lisätään 100,0 ml happamoitua metanolia (3.2), suljetaan astia ja ravistellaan tunnin ajan ravistelijalla (4.2). Suodatetaan liuos lasikuitusuodatinpaperin (4.5) läpi ja koko suodos kerätään talteen 150 ml:n erlenmeyerkolviin. Lisätään 7,5 g molekyyliseulaa (3.4), suljetaan astia ja ravistellaan viisi minuuttia. Suodatetaan välittömästi lasikuitusuodatinpaperin läpi. Tämä liuos säilytetään puhdistusta (5.3) varten.
5.3 Puhdistus
5.3.1
Lasikolonnin (4.1) alapäähän laitetaan pieni tuppo lasivillaa ja tiivistetään se lasisauvaa käyttäen. Punnitaan 11,0 g valmistettua alumiinioksidia (3.5) ja siirretään kolonniin. Tässä vaiheessa on pidettävä huolta siitä, että altistuminen ilmalle on mahdollisimman vähäistä. Taputellaan täytetyn kolonnin alapäätä kevyesti, jotta alumiinioksidi tiivistyy sopivasti.
5.3.2
Pipetoidaan kolonniin 5,0 ml valmistettua näyteuutetta (5.2). Annetaan pipetin kärjen levätä kolonnin seinää vasten ja liuoksen absorboitua alumiinioksidiin. Eluoidaan robenidiini kolonnista 100 ml:lla metanolia (3.1) virtausnopeudella 2–3 ml/min ja kerätään eluaatti 250 ml:n pyörökolviin. Haihdutetaan metanoliliuos kuiviin alipaineessa pyöröhaihduttimessa (4.3) lämpötilan ollessa 40 oC. Liuotetaan jäännös uudelleen 3–4 ml:aan liikkuvaa faasia (3.8) ja siirretään kvantitatiivisesti 10 ml:n mittapulloon. Huuhdellaan pullo usealla 1–2 ml:n annoksella liikkuvaa faasia ja siirretään nämä huuhteluliuokset mittapulloon. Täytetään merkkiin asti samalla liuottimella ja sekoitetaan. Suodatetaan näyte 0,45 μm:n kalvosuodattimen (4.7) läpi. Tämä liuos säilytetään HPLC-määritystä (5.4) varten.
5.4 HPLC-määritys
5.4.1
Seuraavat olosuhteet ovat ohjeellisia, ja myös muita olosuhteita voidaan käyttää, jos niillä saadaan vastaavat tulokset:
Nestekromatografiakolonni (4.4.1)
HPLC:n liikkuva faasi (3.8)
Virtausnopeus: 1,5–2 ml/min
Detektioaallonpituus: 317 nm
Injektiotilavuus: 20–50 μl.
Kromatografisen järjestelmän stabiilisuus tarkastetaan injektoimalla useita kertoja kalibrointiliuosta (3.9.3), jonka pitoisuus on 3,6 μg/ml, kunnes saadaan piikkejä, joiden korkeudet (pinta-alat) ja retentioajat pysyvät vakioina.
5.4.2
Injektoidaan useita kertoja kutakin kalibrointiliuosta (3.9.3) ja mitataan kunkin pitoisuuden piikkien korkeudet (pinta-alat). Kalibrointikäyrä piirretään siten, että y-akselilla ovat kalibrointiliuosten piikkien korkeuksien (pinta alojen) keskiarvot ja x-akselilla vastaavat konsentraatiot μg/ml.
5.4.3
Injektoidaan useita kertoja näyteuutetta (5.3.2) käyttäen samaa määrää kuin kalibrointiliuoksille, ja määritetään robenidiinipiikkien korkeuden (pinta-alan) keskiarvo.
6. Tulosten laskeminen
Näyteliuoksen robenidiinipitoisuus μg/ml lasketaan robenidiinipiikkien korkeuden (pinta-alan) keskiarvosta kalibrointikäyrän (5.4.2) avulla.
Näytteen robenidiinipitoisuus w (mg/kg) lasketaan seuraavalla kaavalla:

jossa:
|
c |
= |
näyteliuoksen robenidiinipitoisuus, μg/ml, |
|
m |
= |
näyte-erän paino grammoina. |
7. Tulosten validointi
7.1 Tunnistaminen
Määritettävän aineen tunnistaminen voidaan varmistaa yhteiskromatografialla tai diodirividetektorilla, joka mahdollistaa näyteuutteen ja 6 μg/ml robenidiinia sisältävän kalibrointiliuoksen (3.6.3) spektrien vertailun.
7.1.1
Näyteuutteeseen lisätään sopiva määrä kalibrointiliuosta (3.9.3). Lisätyn robenidiinin määrän on vastattava näyteuutteessa olevan robenidiinin arvioitua määrää.
Ainoastaan robenidiinipiikin korkeus saa kasvaa, kun otetaan huomioon sekä lisätty määrä että uutteen laimeneminen. Piikin leveyden, sen puolikorkeudella, on oltava noin 10 % sen alkuperäisestä leveydestä.
7.1.2
Tulokset arvioidaan seuraavien arviointiperusteiden mukaisesti:
|
a) |
kromatogrammin piikin huipun kohdalla näytteen ja standardin spektrien maksimiabsorptioaallonpituuksien on oltava samat ilmaisinjärjestelmän erotuskyvyn mukaan määräytyvissä rajoissa. Diodirividetektorimäärityksessä tämä on yleensä noin ± 2 nm; |
|
b) |
välillä 250–400 nm kromatogrammin piikin huipun kohdalla ajetut näyte- ja standardispektrit eivät saa erota niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat ja näiden kahden spektrin välillä havaittu poikkeama ei ole missään kohdassa yli 15 % standardina olevan tutkittavan aineen absorbanssista; |
|
c) |
välillä 250–400 nm näyteliuoksesta ajetut spektrit piikin nousun, huipun ja laskun kohdalla eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun samat maksimit esiintyvät ja kun kaikissa havaittavissa kohdissa spektrien välinen poikkeama ei ole suurempi kuin 15 % huipun spektrin absorbanssista. |
Jos jokin näistä edellytyksistä ei täyty, tutkittavan aineen esiintymistä ei ole vahvistettu.
7.2 Toistettavuus
Kahden rinnakkaisen, samasta näytteestä tehdyn määrityksen tulosten välinen ero ei saa olla suurempi kuin 10 % suuremmasta tuloksesta, kun robenidiinipitoisuus on yli 15 mg/kg.
7.3 Saanto
Sellaisen nollanäytteen saannon, johon on lisätty määritettävää ainetta, on oltava vähintään 85 %.
8. Laboratorioiden välisen vertailun tulokset
Euroopan yhteisön laboratorioiden välisessä tarkastelussa 12 laboratoriota määritti neljä jauhon tai rakeiden muodossa olevaa kani- ja siipikarjarehunäytettä. Kukin näyte määritettiin kaksi kertaa. Tulokset esitetään alla olevassa taulukossa:
|
|
Siipikarja |
Kanit |
||
|
|
Jauho |
Rakeet |
Jauho |
Rakeet |
|
Keskiarvo [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
Sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [ %] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [ %] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Saanto [ %] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
toistettavuuden keskihajonta |
|
CVr |
= |
toistettavuuden variaatiokerroin, % |
|
SR |
= |
uusittavuuden keskihajonta |
|
CVR |
= |
uusittavuuden variaatiokerroin, %. |
F. DIKLATSURIILIN MÄÄRITYS
(+)-4-kloorifenyyli-[2,6-dikloori-4-(2,3,4,5-tetrahydro-3,5-diokso-1,2,4-triatsin-2-yyli)fenyyli]asetonitriili
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää diklatsuriilin pitoisuus rehuissa ja esiseoksissa. Toteamisraja on 0,1 mg/kg ja määritysraja 0,5 mg/kg.
2. Periaate
Sisäisen standardin lisäyksen jälkeen näyte uutetaan happamoidulla metanolilla. Osa rehunäytteen uutteesta puhdistetaan C18-kiinteäfaasiuuttopylväällä. Diklatsuriili eluoidaan pylväästä happamoidun metanolin ja veden seoksella. Haihdutuksen jälkeen jäännös liuotetaan DMF-vesiseokseen. Esiseoksista saadut uutteet haihdutetaan ja jäännös liuotetaan DMF-vesiseokseen. Diklatsuriilipitoisuus määritetään korkean erotuskyvyn käänteisfaasinestekromatografialla (RP-HPLC) käyttäen ternaarigradienttia ja UV-detektoria.
3. Reagenssit
|
3.1 |
Vesi, HPLC-laatua vastaava. |
|
3.2 |
Ammoniumasetaatti. |
|
3.3 |
Tetrabutyyliammoniumvetysulfaatti (TBHS). |
|
3.4 |
Asetonitriili, HPLC-laatua vastaava. |
|
3.5 |
Metanoli, HPLC-laatua vastaava. |
|
3.6 |
N, N-dimetyyliformamidi (DMF). |
|
3.7 |
Kloorivetyhappo, ρ20 = 1,19 g/ml. |
|
3.8 |
Standardiyhdiste: diklatsuriili II-24: (+)-4-kloorifenyyli-[2,6-dikloori-4-(2,3,4,5-tetrahydro-3,5-diokso-1,2,4-triatsin-2-yyli)-fenyyli]asetonitriili, jonka puhtaus on taattu, E 771. |
|
3.8.1 |
Punnitaan 0,1 mg:n tarkkuudella noin 25 mg diklatsuriilistandardia (3.8) 50 ml:n mittapulloon. Liuotetaan DMF:ään (3.6), täytetään merkkiin asti DMF:llä (3.6) ja sekoitetaan. Pullo kääritään alumiinifolioon tai käytetään tummaa pulloa ja säilytetään jääkaapissa. Liuos säilyy ≤ 4 oC:n lämpötilassa yhden kuukauden. |
|
3.8.2 |
Pipetoidaan 5,00 ml standardin kantaliuosta (3.8.1) 50 ml:n mittapulloon, täytetään merkkiin asti DMF:llä (3.6) ja sekoitetaan. Pullo kääritään alumiinifolioon tai käytetään tummaa pulloa ja säilytetään jääkaapissa. Liuos säilyy ≤ 4 oC:n lämpötilassa yhden kuukauden. |
|
3.9 |
Sisäinen standardi: 2,6-dikloori-α-(4-kloorifenyyli)-4-(4,5-dihydro-3,5-diokso-1,2,4-triatsin-2-(3H)-yyli)-α-metyylibentseeniasetonitriili. |
|
3.9.1 |
Punnitaan 0,1 mg:n tarkkuudella noin 25 mg sisäistä standardia (3.9) 50 ml:n mittapulloon. Liuotetaan DMF:ään (3.6), täytetään merkkiin asti DMF:llä (3.6) ja sekoitetaan. Pullo kääritään alumiinifolioon tai käytetään tummaa pulloa ja säilytetään jääkaapissa. Liuos säilyy ≤ 4 oC:n lämpötilassa yhden kuukauden. |
|
3.9.2 |
Pipetoidaan 5,00 ml sisäisen standardin kantaliuosta (3.9.1) 50 ml:n mittapulloon, täytetään merkkiin asti DMF:llä (3.6) ja sekoitetaan. Pullo kääritään alumiinifolioon tai käytetään tummaa pulloa ja säilytetään jääkaapissa. Liuos säilyy ≤ 4 oC:n lämpötilassa yhden kuukauden. |
|
3.9.3 |
(p = diklatsuriilin nimellispitoisuus esiseoksessa, mg/kg) Punnitaan 0,1 mg:n tarkkuudella 10 mg sisäistä standardia 100 ml:n mittapulloon, liuotetaan DMF:ään (3.6) ultraäänihauteessa (4.6), täytetään merkkiin asti DMF:llä ja sekoitetaan. Pullo kääritään alumiinifolioon tai käytetään tummaa. pulloa ja säilytetään jääkaapissa. Liuos säilyy ≤ 4 oC:n lämpötilassa yhden kuukauden. |
|
3.10 |
Kalibrointiliuos, 2 μg/ml. Pipetoidaan 2,00 ml diklatsuriilin standardiliuosta (3.8.2) ja 2,00 ml sisäistä standardiliuosta (3.9.2) 50 ml:n mittapulloon. Lisätään 16 ml DMF:ää (3.6), täytetään merkkiin asti vedellä ja sekoitetaan. Tämä liuos on valmistettava juuri ennen käyttöä. |
|
3.11 |
C18-kiinteäfaasiuuttopylväs, esim. Bond Elut, 1 ml, sorbentin paino: 100 mg. |
|
3.12 |
Uuttoliuos: happamoitu metanoli. Pipetoidaan 5,0 ml suolahappoa (3.7) 1 000 ml:aan metanolia (3.5) ja sekoitetaan. |
|
3.13 |
HPLC:ssä käytettävä liikkuva faasi. |
|
3.13.1 |
Eluentti A: ammoniumasetaatti-tetrabutyyliammoniumvetysulfaattiliuos. Liuotetaan 5 g ammoniumasetaattia (3.2) ja 3,4 g TBHS:ää (3.3) 1 000 ml:aan vettä (3.1) ja sekoitetaan. |
|
3.13.2 |
Eluentti B: asetonitriili (3.4). |
|
3.13.3 |
Eluentti C: metanoli (3.5). |
4. Välineistö
|
4.1 |
Mekaaninen ravistelija. |
|
4.2 |
Ternaarigradientti HPLC -laitteisto. |
|
4.2.1 |
Nestekromatografiakolonni, 100 mm × 4,6 mm, Hypersil ODS, hiukkaskoko 3 μm, tai vastaava. |
|
4.2.2 |
Vaihtuva-aallonpituuksinen UV-detektori tai diodirividetektori. |
|
4.3 |
Pyörivä kalvohaihdutin. |
|
4.4 |
Kalvosuodatin, 0,45 μm. |
|
4.5 |
Tyhjiösuodatuslaite. |
|
4.6 |
Ultraäänihaude. |
5. Menettely
5.1 Yleistä
5.1.1
Nollanäyte on analysoitava, jotta varmistetaan, ettei se sisällä diklatsuriilia tai mittausta häiritseviä aineita. Nollanäytteen on oltava näytteen kaltainen, eikä siinä saa olla havaittavia määriä diklatsuriilia tai häiritseviä aineita.
5.1.2
Saanto on määritettävä analysoimalla nollanäyte, johon on lisätty näytteen diklatsuriilipitoisuutta vastaava määrä diklatsuriilia. Jotta saadaan konsentraatio, joka vastaa arvoa 1 mg/kg diklatsuriilia, siirretään 0,1 ml standardin kantaliuosta (3.8.1) 50 grammaan nollanäytettä, sekoitetaan perusteellisesti ja annetaan tasoittua 10 minuuttia sekoittaen useita kertoja ennen uuttovaiheeseen siirtymistä (5.2).
Jos saatavilla ei ole näytteen kaltaista nollanäytettä (katso 5.1.1), voidaan saantokoe suorittaa vaihtoehtoisesti standardinlisäysmenetelmällä. Tällöin analysoitavaan näytteeseen lisätään suunnilleen näytteiden jo sisältämä määrä diklatsuriilia. Tämä näyte analysoidaan yhdessä varsinaisen näytteen kanssa, ja saanto voidaan laskea vähennyslaskulla.
5.2 Uutto
5.2.1
Punnitaan noin 50 g näytettä 0,01 gramman tarkkuudella. Siirretään 500 ml:n erlenmeyerkolviin, lisätään 1,00 ml sisäistä standardiliuosta (3.9.2), 200 ml uuttoliuosta (3.12) ja suljetaan kolvi tulpalla. Seosta ravistellaan ravistelijalla (4.1) yli yön. Annetaan laskeutua 10 minuuttia. Siirretään 20 ml supernatantista sopivaan lasiastiaan ja laimennetaan 20 ml:lla vettä. Siirretään liuos uuttohylsyyn (3.11) ja imetään liuos sen läpi vakuumilla (4.5). Pestään hylsy 25 ml:lla uuttoliuoksen (3.12) ja veden seosta, 65 + 35 (V + V). Kerätyt fraktiot heitetään pois ja määritettävät aineet eluoidaan 25 ml:lla uuttoliuoksen (3.12) ja veden seosta, 80 + 20 (V + V). Tämä fraktio haihdutetaan pyöröhaihduttimella (4.3) 60 oC:ssa niin, että se on juuri ja juuri kuivunut. Jäännös liuotetaan 1,0 ml:aan DMF:ää (3.6), lisätään 1,5 ml vettä (3.1) ja sekoitetaan. Suodatetaan kalvosuodattimella (4.4). Tehdään HPLC-määritys (5.3).
5.2.2
Punnitaan noin 1 g näytettä 0,001 gramman tarkkuudella. Siirretään 500 ml:n erlenmeyerkolviin, lisätään 1,00 ml sisäistä standardiliuosta (3.9.3), 200 ml uuttoliuosta (3.12) ja suljetaan kolvi tulpalla. Seosta ravistellaan yli yön ravistelijalla (4.1). Annetaan laskeutua 10 minuuttia. Siirretään 10 000/p ml:n (p = diklatsuriilin nimellispitoisuus esiseoksessa mg/kg) erä supernatanttia sopivankokoiseen keittokolviin. Haihdutetaan alipaineessa pyöröhaihduttimella (4.3) 60 oC:ssa niin, että se on juuri ja juuri kuivunut. Jäännös liuotetaan uudelleen 10,0 ml:aan DMF:ää (3.6), lisätään 15,0 ml vettä (3.1) ja sekoitetaan. Tehdään HPLC-määritys (5.3).
5.3 HPLC-määritys
5.3.1
Seuraavat ajo-olosuhteet ovat ohjeellisia ja niitä voidaan muuttaa, mikäli ne antavat vastaavanlaiset tulokset.
|
Nestekromatografiakolonni (4.2.1.): |
100 mm × 4,6 mm, Hypersil ODS, hiukkaskoko 3 μm, tai vastaava |
|
||||||
|
Liikkuva faasi: |
Eluentti A (3.13.1): |
ammoniumasetaatin ja tetrabutyyliammoniumvetysulfaatin vesiliuos |
||||||
|
|
Eluentti B (3.13.2): |
asetonitriili |
||||||
|
|
Eluentti C (3.13.3): |
metanoli |
||||||
|
Eluointi: |
Huuhdotaan B:llä 10 min. |
|||||||
|
Virtausnopeus: |
1,5–2 ml/min |
|
||||||
|
Injektoitava määrä: |
20 μl |
|
||||||
|
Detektioaallonpituus: |
280 nm |
|
||||||
Kromatografiajärjestelmän stabiilisuus tarkistetaan injektoimalla useita kertoja kalibrointiliuosta (3.10), jonka konsentraatio on 2 μl/ml, kunnes piikkien korkeudet ja retentioajat ovat vakiot.
5.3.2
Injektoidaan 20 μl kalibrointiliuosta (3.10) useita kertoja ja määritetään diklatsuriilin ja sisäisen standardin piikkien korkeuksien (pinta-alojen) keskiarvot.
5.3.3
Injektoidaan 20 μl näyteliuosta (5.2.1 tai 5.2.2) useita kertoja ja määritetään diklatsuriilin ja sisäisen standardin piikkien korkeuksien (pinta-alojen) keskiarvot.
6. Tulosten laskeminen
6.1 Rehut
Näytteen diklatsuriilipitoisuus w (mg/kg) saadaan seuraavasta kaavasta:
 [mg/kg]
[mg/kg]
jossa:
|
hd, s |
= |
näyteliuoksen (5.2.1) diklatsuriilin piikin korkeus (pinta-ala), |
|
hi, s |
= |
näyteliuoksen (5.2.1) sisäisen standardin piikin korkeus (pinta-ala), |
|
hd, c |
= |
kalibrointiliuoksen (3.10) diklatsuriilin piikin korkeus (pinta-ala), |
|
hi, c |
= |
kalibrointiliuoksen (3.10) sisäisen standardin piikin korkeus (pinta-ala), |
|
cd, c |
= |
kalibrointiliuoksen (3.10) diklatsuriilipitoisuus, μg/ml, |
|
m |
= |
näyte-erän paino (g), |
|
V |
= |
5.2.1 kohdan mukaisesti valmistetun näyteuutteen tilavuus (2,5 ml). |
6.2 Esiseokset
Näytteen diklatsuriilipitoisuus w (mg/kg) saadaan seuraavasta kaavasta:
 [mg/kg]
[mg/kg]
jossa:
|
hd, c |
= |
kalibrointiliuoksen (3.10) diklatsuriilin piikin korkeus (pinta-ala), |
|
hi, c |
= |
kalibrointiliuoksen (3.10) sisäisen standardin piikin korkeus (pinta-ala), |
|
hd, s |
= |
näyteliuoksen (5.2.2) diklatsuriilin piikin korkeus (pinta-ala), |
|
hi, s |
= |
näyteliuoksen (5.2.2) sisäisen standardin piikin korkeus (pinta-ala), |
|
cd, c |
= |
kalibrointiliuoksen (3.10) diklatsuriilipitoisuus, μg/ml, |
|
m |
= |
näyte-erän paino (g), |
|
V |
= |
5.2.2 kohdan mukaisesti valmistetun näyteuutteen tilavuus (25 ml), |
|
p |
= |
diklatsuriilin nimellispitoisuus esiseoksessa, mg/kg. |
7. Tulosten validointi
7.1 Tunnistaminen
Analyytin tunnistaminen voidaan varmentaa yhteiskromatografialla tai käyttämällä diodirividetektoria, jolla verrataan näyteuutteen (5.2.1 tai 5.2.2) ja kalibrointiliuoksen (3.10) spektrejä.
7.1.1
Näyteuutteeseen (5.2.1 tai 5.2.2) lisätään sopiva määrä kalibrointiliuosta (3.10). Lisätyn diklatsuriilin määrän on oltava samaa suuruusluokkaa kuin näytteessä todettu diklatsuriilin määrä.
Ainoastaan diklatsuriilin ja sisäisen standardin piikit saavat kasvaa, kun otetaan huomioon sekä lisätty määrä että uutteen laimentaminen. Piikin leveys puolivälissä piikin korkeutta on oltava ± 10 % sellaisen näyteuutteen alkuperäisen diklatsuriilipiikin tai sisäisen standardin piikin leveydestä, johon ei ole tehty lisäystä.
7.1.2
Tulokset arvioidaan seuraavien arviointiperusteiden mukaisesti:
|
a) |
Kromatogrammin piikkien huipusta mitattujen näyte- ja standardispektrien maksimiabsorption aallonpituuksien on oltava samat ilmaisinjärjestelmän erotuskyvyn mukaan määräytyvissä rajoissa. Diodirividetektorilla tämä on tyypillisesti ± 2 nm. |
|
b) |
Välillä 230–320 nm kromatogrammin piikkien huipun kohdalla ajetut näyte- ja standardispektrit eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat eikä spektrien välinen ero ole missään kohdassa suurempi kuin 15 % standardianalyytin absorbanssista. |
|
c) |
Välillä 230–320 nm näyteuutteesta ajetut spektrit piikin nousun, huipun ja laskun kohdalla eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat eikä spektrien välinen ero ole missään kohdassa suurempi kuin 15 % piikin huipun spektrin absorbanssista. |
Jos jokin näistä edellytyksistä ei täyty, tutkittavan aineen esiintymistä ei ole vahvistettu.
7.2 Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla suurempi kuin
|
— |
30 % suuremmasta rinnakkaistuloksesta, kun diklatsuriilipitoisuus on 0,5–2,5 mg/kg |
|
— |
0,75 mg/kg, kun diklatsuriilipitoisuus on 2,5–5 mg/kg |
|
— |
15 % suuremmasta rinnakkaistuloksesta, kun diklatsuriilipitoisuus on yli 5 mg/kg. |
7.3 Saanto
Sellaisen nollanäytteen saannon, johon on lisätty mitattavaa ainetta, on oltava vähintään 80 %.
8. Laboratorioiden välisen vertailun tulokset
Laboratorioiden välisessä tutkimuksessa analysoitiin viisi näytettä 11 laboratoriossa. Näytteistä kaksi oli esiseoksia. Toiseen sekoitettiin orgaanista materiaalia (O 100) ja toiseen epäorgaanista materiaalia (A 100). Teoreettinen pitoisuus on 100 mg diklatsuriilia kilogrammassa. Näitä kolmea siipikarjan rehuseosta valmisti kolme eri valmistajaa (L1/Z1/K1) (NL). Teoreettinen pitoisuus on 1 mg diklatsuriilia kilogrammassa. Laboratorioita pyydettiin analysoimaan kukin näyte kerran tai kaksoismäärityksinä. (Tarkempia tietoja tästä laboratorioiden välisestä tutkimuksesta saa julkaisusta Journal of AOAC International, Volume 77, n:o 6, 1994, s. 1 359–1 361.) Tulokset esitetään seuraavassa taulukossa.
|
|
Näyte 1 A 100 |
Näyte 2 O 100 |
Näyte 3 L1 |
Näyte 4 Z1 |
Näyte 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Keskiarvo |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr ( %) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR ( %) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Nimellispitoisuus mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
yksittäisten arvojen lukumäärä |
|
Sr |
= |
toistettavuuden keskihajonta |
|
CVr |
= |
toistettavuuden variaatiokerroin |
|
SR |
= |
uusittavuuden keskihajonta |
|
CVR |
= |
uusittavuuden variaatiokerroin. |
9. Huomautukset
Diklatsuriilivasteen lineaarisuus on osoitettava etukäteen ao. konsentraatioalueilla.
G. LASALOSID-NATRIUMIN MÄÄRITTÄMINEN
Streptomyces lasaliensis -bakteerin tuottama polyeetterimonokarboksyylihapon natriumsuola
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää lasalosid-natriumin pitoisuus rehuissa ja esiseoksissa. Toteamisraja on 5 mg/kg ja määritysraja 10 mg/kg.
2. Periaate
Lasalosid-natrium uutetaan näytteestä happamoituun metanoliin ja määritetään korkean erotuskyvyn käänteisfaasinestekromatografialla (HPLC) käyttäen spektrofluorimetridetektoria.
3. Reagenssit
|
3.1 |
Kaliumdivetyfosfaatti (KH2PO4). |
|
3.2 |
Ortofosforihappo, w (w/w) = 85 %. |
|
3.3 |
Ortofosforihappoliuos, c = 20 %. Laimennetaan 23,5 ml ortofosforihappoa (3.2) vedellä 100 ml:aan. |
|
3.4 |
6-metyyli-2-heptyyliamiini (1,5-dimetyyliheksyyliamiini), w (w/w) = 99 %. |
|
3.5 |
Metanoli, HPLC-laatua vastaava. |
|
3.6 |
Kloorivetyhappo, tiheys = 1,19 g/ml. |
|
3.7 |
Fosfaattipuskuriliuos, c = 0,01 mol/1. Liuotetaan 1,36 g kaliumdivetyfosfaattia (3.1) 500 ml:aan vettä (3.11), lisätään 3,5 ml ortofosforihappoa (3.2) ja 10,0 ml 6-metyyli-2-heptyyliamiinia (3.4). Säädetään pH ortofosforihappoliuoksen avulla arvoon 4,0 ja laimennetaan vedellä (3.11) 1 000 ml:aan. |
|
3.8 |
Happamoitu metanoli. Pipetoidaan 5,0 ml suolahappoa (3.6) 1 000 ml:n mittapulloon, täytetään merkkiin asti metanolilla (3.5) ja sekoitetaan. Tämä liuos on valmistettava juuri ennen käyttöä. |
|
3.9 |
HPLC:n liikkuva faasi, fosfaattipuskuri-metanoliliuos 5 + 95 (V + V). Sekoitetaan 5 ml fosfaattipuskuriliuosta (3.7) ja 95 ml metanolia (3.5). |
|
3.10 |
Lasalosid-natriumstandardi, jonka puhtaus on taattu, C34H53O8Na (Streptomyces lasaliensis -bakteerin tuottama polyeetterimonokarboksyylihapon natriumsuola), E763. |
|
3.10.1 |
Punnitaan 50 mg lasalosid-natriumia (3.10) 0,1 mg:n tarkkuudella 100 ml:n mittapulloon, liuotetaan happamoituun metanoliin (3.8), täytetään merkkiin asti samalla liuottimella ja sekoitetaan. Tämä liuos on valmistettava välittömästi ennen käyttöä. |
|
3.10.2 |
Pipetoidaan 10,0 ml standardin kantaliuosta (3.10.1) 100 ml:n mittapulloon, täytetään merkkiin asti happamoidulla metanolilla (3.8) ja sekoitetaan. Tämä liuos on valmistettava juuri ennen käyttöä. |
|
3.10.3 |
Pipetoidaan 1,0, 2,0, 4,0, 5,0 ja 10,0 ml standardin välimuotoliuosta (3.10.2) 50 ml:n mittapulloihin. Täytetään merkkiin asti happamoidulla metanolilla (3.8) ja sekoitetaan. Nämä liuokset vastaavat konsentraatioita 1,0, 2,0, 4,0, 5,0 ja 10,0 μg/ml lasalosid-natriumia. Nämä liuokset on valmistettava aina juuri ennen käyttöä. |
|
3.11. |
Vesi, HPLC-laatua vastaava. |
4. Välineistö
|
4.1 |
Ultraäänihaude (tai ravisteleva vesihaude), jossa on lämpötilansäätö. |
|
4.2 |
Kalvosuodattimia, 0,45 μm. |
|
4.3 |
HPLC-laitteisto, jossa voidaan injektoida 20 μl:n näytteitä. |
|
4.3.1 |
Nestekromatografiakolonni, 125 mm × 4 mm, käänteisfaasi C18, hiukkaskoko 5 μm, tai vastaava. |
|
4.3.2 |
Spektrofluorimetri, jossa voidaan säätää eksitaatio- ja emissioaallonpituudet. |
5. Menettely
5.1 Yleistä
5.1.1
Saantokokeen (5.1.2) suorittamista varten analysoidaan nollanäyte, jotta varmistetaan, ettei se sisällä lasalosid-natriumia tai mittausta häiritseviä aineita. Nollanäytteen on oltava näytteen kaltainen, eikä siinä saa olla havaittavia määriä lasalosid-natriumia tai häiritseviä aineita.
5.1.2
Saanto on määritettävä analysoimalla nollanäyte, johon on lisätty näytteen lasalosid-natriumia vastaava määrä lasalosid-natriumia. Jotta saadaan liuos, jossa on 100 mg/kg lasalosid-natriumia vastaava lisäys, siirretään 10,0 ml standardin kantaliuosta (3.10.1) 250 ml:n erlenmeyerkolviin ja haihdutetaan noin 0,5 ml:aan. Lisätään 50 g nollanäytettä, sekoitetaan huolellisesti, annetaan tasoittua 10 minuuttia sekoittaen useita kertoja ennen uuttovaiheeseen siirtymistä (5.2).
Jos saatavilla ei ole näytteen kaltaista nollanäytettä (katso 5.1.1), voidaan saantokoe suorittaa vaihtoehtoisesti standardinlisäysmenetelmällä. Tällöin analysoitavaan näytteeseen lisätään näytteiden jo sisältämää määrää vastaava määrä lasalosid-natriumia. Tämä näyte analysoidaan yhdessä varsinaisen näytteen kanssa, ja saanto voidaan laskea vähennyslaskulla.
5.2 Uutto
5.2.1
Punnitaan 5–10 g näytettä 0,01 gramman tarkkuudella 250 ml:n tulpalliseen erlenmeyerkolviin. Lisätään pipetillä 100,0 ml happamoitua metanolia (3.8). Suljetaan löyhästi tulpalla ja sekoitetaan kunnes näyte on liuennut. Asetetaan pullo noin 40-asteiseen ultraäänihauteeseen (4.1) 20 minuutiksi, otetaan pois ja jäähdytetään huoneenlämpöiseksi. Annetaan laskeutua noin tunnin ajan ja suodatetaan määräosa 0,45 μm:n kalvosuodattimen (4.2) läpi sopivaan astiaan. Tehdään HPLC-määritys (5.3).
5.2.2
Punnitaan 2 g jauhamatonta esiseosta 0,001 gramman tarkkuudella 250 ml:n mittapulloon. Lisätään 100,0 ml happamoitua metanolia (3.8) ja sekoitetaan kunnes esiseos on liuennut. Laitetaan pullo sisältöineen noin 40-asteiseen ultraäänihauteeseen (4.1) 20 minuutiksi, otetaan pois ja jäähdytetään huoneenlämpöiseksi. Täytetään merkkiin asti happamoidulla metanolilla (3.8) ja sekoitetaan hyvin. Annetaan laskeutua tunnin ajan ja suodatetaan määräosa 0,45 μm:n kalvosuodattimen (4.2) läpi sopivaan astiaan. Laimennetaan sopiva määrä kirkasta suodosta happamoidulla metanolilla (3.8) siten, että lopullisessa koeliuoksessa on lasalosid-natriumia noin 4 μg/ml. Tehdään HPLC-määritys (5.3).
5.3 HPLC-määritys
5.3.1
Seuraavat ajo-olosuhteet ovat ohjeellisia; ajo voidaan suorittaa muissa olosuhteissa, jos niillä saadaan samat tulokset:
|
Nestekromatografia-kolonni (4.3.1): |
125 mm × 4 mm, käänteisfaasi C18, hiukkaskoko 5 μm, tai vastaava |
|
Liikkuva faasi (3.9): |
Fosfaattipuskuriliuoksen (3.7) ja metanolin (3.5) seos, 5 + 95 (V+V) |
|
Virtausnopeus: |
1,2 ml/min |
|
Mittausaallonpituudet: |
|
|
Eksitaatio: |
310 nm |
|
Emissio: |
419 nm |
|
Injektoitava määrä: |
20 μl. |
Kromatografiajärjestelmän stabiilisuus tarkistetaan injektoimalla useita kertoja 4,0 μg/ml:n kalibrointiliuosta (3.10.3), kunnes piikkien korkeudet (tai pinta-alat) ja retentioajat pysyvät vakioina.
5.3.2
Injektoidaan kutakin kalibrointiliuosta (3.10.3) useita kertoja ja lasketaan kutakin konsentraatiota vastaavat piikkien korkeuksien (pinta-alojen) keskiarvot. Piirretään kalibrointikäyrä merkiten piikkien korkeuksien (pinta-alojen) keskiarvot y-akselille ja vastaavat konsentraatiot (μg/ml) x-akselille.
5.3.3
Injektoidaan 5.2.1 tai 5.2.2 kohdassa saatuja näyteuutteita useita kertoja käyttäen samaa tilavuutta kuin kalibrointiliuoksille ja määritetään lasalosid-natriumpiikkien korkeuksien (pinta-alojen) keskiarvo.
6. Tulosten laskeminen
Näyteliuosta (5.3.3) injektoimalla saatujen piikkien korkeuksien (pinta-alojen) keskiarvosta määritetään lasalosid-natriumkonsentraatio (μg/ml) kalibrointikäyrän avulla.
6.1 Rehut
Näytteen lasalosid-natriumpitoisuus w (mg/kg) lasketaan seuraavalla kaavalla:
 [mg/kg]
[mg/kg]
jossa:
|
c |
= |
näyteliuoksen (5.2.1) lasalosid-natriumkonsentraatio (μg/ml), |
|
V1 |
= |
5.2.1 kohdan mukaisesti valmistetun näyteuutteen tilavuus (ts. 100 ml), |
|
m |
= |
näyte-erän paino (g). |
6.2 Esiseokset
Näytteen lasalosid-natriumpitoisuus w (mg/kg) lasketaan seuraavalla kaavalla:
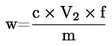 [mg/kg]
[mg/kg]
jossa:
|
c |
= |
näyteliuoksen (5.2.2) lasalosid-natriumkonsentraatio (μg/ml), |
|
V2 |
= |
5.2.2 kohdan mukaisesti valmistetun näyteuutteen tilavuus (ts. 250 ml), |
|
f |
= |
5.2.2 kohdan mukainen laimennuskerroin, |
|
m |
= |
näyte-erän paino (g). |
7. Tulosten validointi
7.1 Tunnistaminen
Spektrofluorimetriaan perustuvissa menetelmissä on vähemmän häiriömahdollisuuksia kuin UV-mittauksissa. Tutkittavan aineen tunnistaminen voidaan varmentaa yhteiskromatografialla.
7.1.1
Näyteuutteeseen (5.2.1 tai 5.2.2) lisätään sopiva määrä kalibrointiliuosta (3.10.3). Lisätyn lasalosid-natriummäärän on oltava suunnilleen sama kuin näyteliuoksesta määritetty lasalosid-natriummäärä. Ainoastaan lasalosid-natriumpiikin korkeus saa kasvaa, kun otetaan huomioon sekä lisätty määrä lasalosid-natriumia että näyteliuoksen laimeneminen. Piikin leveyden tulisi olla puolikorkeudessa ± 10 % sellaisen näyteliuoksen piikin leveydestä, johon ei ole lisätty lasalosid-natriumia.
7.2 Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla suurempi kuin
|
— |
15 % lasalosid-natriumpitoisuuden korkeammasta arvosta välillä 30–100 mg/kg, |
|
— |
15 mg/kg lasalosid-natriumpitoisuuden ollessa 100–200 mg/kg, |
|
— |
7,5 % lasalosid-natriumpitoisuuden korkeammasta arvosta pitoisuuksien ollessa yli 200 mg/kg. |
7.3 Saanto
Sellaisen nollanäytteen saannon, johon on lisätty määritettävää ainetta, on oltava vähintään 80 %. Sellaisen esiseosnäytteen saannon, johon on lisätty määritettävää ainetta, saannon on oltava vähintään 90 %.
8. Laboratorioiden välisen vertailun tulokset
Laboratorioiden välinen vertailu (6) järjestettiin siten, että 12 laboratoriossa tutkittiin 2 esiseosta (näytteet 1 ja 2) ja 5 rehua (näytteet 3–7). Kukin näyte määritettiin kaksi kertaa. Tulokset on esitetty seuraavassa taulukossa:
|
|
Näyte 1 Kananrehun esiseos |
Näyte 2 Kalkkunanrehun esiseos |
Näyte 3 Kalkkunanrehupelletit |
Näyte 4 Kananrehurakeet |
Näyte 5 Kalkkunanrehu |
Näyte 6 Siipikarjanrehu A |
Näyte 7 Siipikarjanrehu B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Keskiarvo [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
sR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Nimellispitoisuus [mg/kg] |
5 000 (7) |
16 000 (7) |
80 (7) |
105 (7) |
120 (7) |
50 (8) |
35 (8) |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
yksittäisten tulosten lukumäärä |
|
sr |
= |
toistettavuuden keskihajonta |
|
sR |
= |
uusittavuuden keskihajonta |
|
CVr |
= |
toistettavuuden variaatiokerroin, % |
|
CVR |
= |
uusittavuuden variaatiokerroin, %. |
(1) Vertailun järjestäjä: Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA), rehutyöryhmä.
(2) Vertailun järjestäjä: Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA), rehutyöryhmä.
(3) Myös muita hajotusmenetelmiä (kuten mikroaaltouunihajotusta paineen alaisena) voidaan käyttää sillä edellytyksellä, että niiden on osoitettu antavan samanlaiset tulokset.
(4) Vihreisiin rehuihin (tuoreisiin tai kuivattuihin) oletetaan sisältyvän suuria määriä kasvisperäisiä piiyhdisteitä, jotka saattavat sitoa mikromineraaleja, ja jotka on siksi poistettava. Sellaisten rehujen tarkastuksessa on siksi sovellettava seuraavaa muutettua menettelyä. Menetellään 5.1.1.1 alakohdassa esitetyllä tavalla aina suodattamisvaiheeseen asti. Pestään suodatinpaperi, jossa on liukenemattomat jäämät, kahteen kertaan kiehuvalla vedellä ja siirretään ne kvartsi- tai platinaupokkaaseen. Poltetaan tuhkaksi muhveliuunissa (4.1) alle 550 oC:ssa kunnes kaikki hiilipitoinen aines on hävinnyt kokonaan. Annetaan jäähtyä, lisätään muutama tippa vettä ja sen jälkeen 10–15 ml fluorivetyhappoa (3.4) ja haihdutetaan kuivaksi noin 150 oC:ssa. Jos jäännökseen jää piiyhdisteitä, se liuotetaan uudelleen muutamaan millilitraan fluorivetyhappoa (3.4) ja haihdutetaan kuivaksi. Lisätään viisi tippaa rikkihappoa (3.5) ja kuumennetaan kunnes valkoinen savu on haihtunut. Lisätään viisi tippaa kloorivetyhappoa, 6 mol/l (3.2), ja noin 30 ml vettä, kuumennetaan ja suodatetaan liuos 250 ml:n vetoiseen mittapulloon ja täytetään merkkiin asti vedellä (HCl-pitoisuus on noin 0,5 mol/l). Sen jälkeen jatketaan menettelyä 5.1.2 kohdasta alkaen.
(5) The Analyst 108, 1983, s. 1 252–1 256.
(6) Analyst, 1995, 120, 2175–2180.
(7) Valmistajan ilmoittama pitoisuus
(8) Laboratoriossa valmistettu rehu.
LIITE V
REHUJEN SISÄLTÄMIEN HAITALLISTEN AINEIDEN VALVONNASSA KÄYTETTÄVÄT MÄÄRITYSMENETELMÄT
A. VAPAAN GOSSYPOLIN JA KOKONAISGOSSYPOLIN MÄÄRITYS
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää vapaan gossypolin, kokonaisgossypolin ja niitä kemiallisesti lähellä olevien aineiden pitoisuus puuvillansiemenissä, puuvillansiemenjauhossa ja puuvillansiemenkakussa sekä näitä rehuaineita sisältävissä rehuseoksissa; vapaan gossypolin, kokonaisgossypolin ja niitä kemiallisesti lähellä olevien aineiden määrityksen alaraja on 20 mg/kg.
2. Periaate
Gossypoli uutetaan 3-amino-1-propanolin läsnä ollessa joko isopropanolin ja heksaanin seoksella vapaan gossypolin määrittämiseksi tai dimetyyliformamidilla kokonaisgossypolin määrittämiseksi. Gossypoli muutetaan aniliinilla gossypolidianiliiniksi, jonka optinen tiheys mitataan 440 nm:ssä.
3. Reagenssit
|
3.1 |
Isopropanoli-heksaaniseos: sekoitetaan 60 tilavuusosaa isopropanolia ja 40 tilavuusosaa n-heksaania. |
|
3.2 |
Liuotinseos A: Lisätään noin 500 ml isopropanoli-heksaani-seosta (3.1), 2 ml 3-amino-1-propanolia, 8 ml jääetikkaa ja 50 ml vettä 1 litran mittapulloon. Mittapullo täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1). Säilyy yhden viikon ajan. |
|
3.3 |
Liuotinseos B: Pipetoidaan 2 ml 3-amino-1-propanolia ja 10 ml jääetikkaa 100 ml:n mittapulloon. Annetaan jäähtyä huoneenlämpöön ja täytetään merkkiin asti N, N-dimetyyliformamidilla. Säilyy yhden viikon ajan. |
|
3.4 |
Aniliini: Jos nollakokeessa saatu optinen tiheys on yli 0,022, aniliini on tislattava sinkkijauheen päältä siten, että tisleestä jätetään pois 10 % sekä alusta että lopusta. Suljetussa ruskeassa lasipullossa kylmässä säilytettynä reagenssiliuos säilyy useiden kuukausien ajan. |
|
3.5 |
Gossypolin standardiliuos A: Punnitaan 27,9 g gossypoliasetaattia 250 ml:n mittapulloon. Liuotetaan liuotinseokseen A, täytetään merkkiin asti samalla liuotinseoksella (3.2). Pipetoidaan 50 ml tätä liuosta 250 ml:n mittapulloon ja täytetään merkkiin asti liuotinseoksella A. Tämän liuoksen gossypolikonsentraatio on 0,02 mg/ml. Annetaan seistä yhden tunnin ajan huoneenlämmössä ennen käyttöä. |
|
3.6 |
Gossypolin standardiliuos B: Punnitaan 27,9 g gossypoliasetaattia 50 ml:n mittapulloon. Liuotetaan liuotinseokseen B, täytetään merkkiin asti samalla liuotinseoksella (3.3). Tämän liuoksen gossypolikonsentraatio on 0,5 mg/ml. Gossypolin standardiliuokset A ja B säilyvät 24 tuntia valolta suojattuina. |
4. Välineistö
|
4.1 |
Sekoittaja: noin 35 kierr./min. |
|
4.2 |
Spektrofotometri. |
5. Menettely
5.1 Näyte
Punnitusmäärä riippuu näytteen oletetusta gossypolipitoisuudesta. On suositeltavaa käyttää pientä punnitusmäärää ja suhteellisen suurta suodosmäärää, jotta gossypolin määrä saataisiin riittävän suureksi tarkan spektrofotometrisen mittauksen suorittamiseen. Näyte ei saa olla suurempi kuin 1 g määritettäessä vapaan gossypolin pitoisuutta puuvillansiemenistä, puuvillansiemenjauhosta tai puuvillansiemenkakusta; jos määritys tapahtuu rehuseoksesta, näytekoko voi olla jopa 5 g. Useimmissa tapauksissa riittää 10 ml suodosta; suodoksen pitää sisältää 50–100 μg gossypolia. Kokonaisgossypolin määrittämiseen punnitusmäärän pitää olla 0,5–5 g siten, että 2 ml:n määrä suodosta sisältää 40–200 μg gossypolia.
Määritys on suoritettava noin 20 oC:n huoneenlämmössä.
5.2 Vapaan gossypolin määritys
Sopiva määrä näytettä punnitaan hiokselliseen, lasitulpalla suljettavaan 250 ml:n keittokolviin, jonka pohjalla on lasimurskaa. Lisätään pipetillä kolviin 50 ml liuotinseosta A (3.2), suljetaan kolvi tulpalla ja sekoitetaan yhden tunnin ajan sekoittajassa. Suodatetaan kuivan suodattimen läpi pieneen, hiokselliseen kolviin. Suodatinsuppilo peitetään kellonlasilla suodatuksen ajaksi.
Pipetoidaan kahteen 25 ml:n mittapulloon (A ja B) sama määrä suodosta, joka sisältää 50–100 μg gossypolia. Tarvittaessa täytetään 10 ml:aan liuotinseoksella A (3.2). Tämän jälkeen mittapullo (A) täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1). Tätä liuosta käytetään referenssiliuoksena näyteliuoksen mittauksessa.
Pipetoidaan 10 ml liuotinseosta A (3.2) kahteen 25 ml:n mittapulloon (C ja D). Toinen mittapullo (C) täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1). Tätä liuosta käytetään referenssiliuoksena nollakoeliuoksen mittauksessa.
Pipetoidaan mittapulloihin D ja B 2 ml aniliiniliuosta (3.4). Lämmitetään 30 minuutin ajan kiehuvalla vesihauteella värin kehittymiseksi. Annetaan jäähtyä huoneenlämpöön, täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1), homogenoidaan ja annetaan seistä yhden tunnin ajan.
Mitataan nollakoeliuoksen (D) optinen tiheys spektrometrillä 440 nm:ssä 1 cm:n lasikyvetissä vertailuliuosta (C) vastaan, ja näyteliuoksen (B) optinen tiheys vertailuliuosta (A) vastaan.
Nollakoeliuoksen optinen tiheys vähennetään näyteliuoksen optisesta tiheydestä (= korjattu optinen tiheys). Tästä arvosta lasketaan vapaan gossypolin pitoisuus 6 kohdassa kuvatulla tavalla.
5.3 Kokonaisgossypolin määritys
Näytemäärä, joka sisältää 1–5 mg gossypolia, punnitaan 50 ml:n mittapulloon ja siihen lisätään 10 ml liuotinseosta B (3.3). Samanaikaisesti valmistetaan nollakoeliuos pipetoimalla 10 ml liuotinseosta B (3.3) toiseen 50 ml:n mittapulloon. Molemmat mittapullot asetetaan kiehuvalle vesihauteelle 30 minuutin ajaksi. Annetaan jäähtyä huoneenlämpöön ja täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1). Homogenoidaan ja annetaan seistä 10–15 minuuttia, suodatetaan hioksellisiin pulloihin.
Sekä suodatettua näyteliuosta että suodatettua nollakoeliuosta pipetoidaan 2 ml 25 ml:n mittapulloihin. Toinen näyteliuosta ja toinen nollakoeliuosta sisältävä mittapullo täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1). Näitä liuoksia käytetään vertailuliuoksina.
Jäljelle jääneisiin kahteen mittapulloon lisätään 2 ml aniliiniliuosta (3.4). Lämmitetään 30 minuutin ajan kiehuvalla vesihauteella värin kehittymiseksi. Annetaan jäähtyä huoneenlämpöön, täytetään 25 ml:aan isopropanoli-heksaaniseoksella (3.1), homogenoidaan ja annetaan seistä yhden tunnin ajan.
Optinen tiheys mitataan 5.2 kohdan mukaisesti kuten vapaan gossypolin optinen tiheys. Tästä arvosta lasketaan kokonaisgossypolipitoisuus 6 kohdassa esitetyllä tavalla.
6. Tulosten laskeminen
Tulokset voidaan laskea joko optista ominaistiheyttä (6.1) tai kalibrointikäyrää (6.2) käyttäen.
6.1 Optinen ominaistiheys
Optinen ominaistiheys on annetuissa olosuhteissa seuraava:
|
Vapaa gossypoli: |
|
|
Kokonaisgossypoli: |
|
Näytteen vapaan gossypolin tai kokonaisgossypolin pitoisuus lasketaan seuraavalla kaavalla:

jossa:
|
E |
= |
korjattu optinen tiheys määritettynä 5.2 kohdan mukaisesti, |
|
p |
= |
punnittu näytemäärä grammoina, |
|
a |
= |
suodosmäärä millilitroina. |
6.2 Kalibrointikäyrän avulla
6.2.1
Valmistetaan kaksi viiden 25 ml:n mittapullon sarjaa. Molempiin mittapullosarjoihin pipetoidaan 2,0, 4,0, 6,0, 8,0 ja 10,0 ml gossypolistandardiliuosta A (3.5). Täytetään 10 ml:aan liuotinseoksella A (3.2). Molempiin sarjoihin otetaan mukaan 25 ml:n mittapullo, joka sisältää vain 10 ml liuotinseosta A (3.2) (nollakoe).
Toinen sarja mittapulloja (myös nollakokeessa käytettävä pullo) täytetään 25 ml:aan isopropanoli-heksaaniseoksella (3.1) (vertailusarja).
Toiseen mittapullosarjaan (myös nollakokeeseen) lisätään 2 ml aniliiniliuosta (3.4). Lämmitetään 30 minuutin ajan kiehuvalla vesihauteella värin kehittymiseksi. Annetaan jäähtyä huoneenlämpöön, täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1), homogenoidaan ja annetaan seistä yhden tunnin ajan (standardisarja).
Mitataan 5.2 kohdassa mainituissa olosuhteissa standardisarjan liuosten optinen tiheys vertailusarjan vastaavia liuoksia vastaan. Kalibrointikäyrä laaditaan piirtämällä optisia tiheyksiä ja näitä vastaavia gossypolimääriä (mikrogrammoina) esittävä kuvaaja.
6.2.2
Valmistetaan kuuden 50 ml:n mittapullon sarja. Ensimmäiseen mittapulloon pipetoidaan 10 ml liuotinseosta B (3.3) ja muihin 2,0; 4,0; 6,0; 8,0 ja 10,0 ml gossypolistandardiliuosta B (3.6). Pullojen tilavuus säädetään 10 ml:ksi liuotinseoksella B (3.3). Lämmitetään kiehuvalla vesihauteella 30 minuuttia. Annetaan jäähtyä huoneenlämpöön, täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1) ja homogenoidaan.
Kuhunkin kahden sarjan kuuteen 25 ml:n mittapulloon pipetoidaan 2,0 ml tätä liuosta. Ensimmäisen sarjan pullot täytetään 25 ml:ksi isopropanoli-heksaaniseoksella (3.1) (vertailusarja).
Kuhunkin toisen sarjan mittapulloon lisätään 2 ml aniliiniliuosta (3.4). Lämmitetään kiehuvalla vesihauteella 30 minuuttia. Annetaan jäähtyä huoneenlämpöön, täytetään merkkiin asti isopropanoli-heksaaniseoksella (3.1), homogenoidaan ja annetaan seistä yhden tunnin ajan (standardisarja).
Mitataan 5.2 kohdassa mainituissa olosuhteissa standardisarjan liuosten optinen tiheys vertailusarjan vastaavia liuoksia vastaan. Kalibrointikäyrä laaditaan piirtämällä optisia tiheyksiä ja näitä vastaavia gossypolimääriä (mikrogrammoina) esittävä kuvaaja.
6.3 Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla suurempi kuin
|
— |
15 %, suhteessa korkeampaan tasoon, kun gossypolipitoisuudet ovat alle 500 mg/kg, |
|
— |
75 mg/kg, absoluuttisena arvona ilmoitettuna, kun gossypolipitoisuudet ovat 500–750 mg/kg, |
|
— |
10 %, suhteessa korkeampaan arvoon, kun gossypolipitoisuudet ovat yli 750 mg/kg. |
B. DIOKSIINIEN (PCDD/PCDF) JA DIOKSIININ KALTAISTEN PCB-YHDISTEIDEN PITOISUUKSIEN MÄÄRITTÄMINEN
I. NÄYTTEENOTTOMENETELMÄT JA MÄÄRITYSTULOSTEN TULKINTA
1. Tarkoitus ja soveltamisala
Rehun dioksiinipitoisuuksien (polykloorattujen dibentso-p-dioksiinien (PCDD) ja polykloorattujen dibentsofuraanien (PCDF) pitoisuuksien) ja dioksiinin kaltaisten polykloorattujen bifenyylien (PCB-yhdisteiden) (1) pitoisuuksien viralliseen valvontaan tarkoitetut näytteet on otettava liitteen I säännösten mukaisesti. Rehuun tasaisesti jakautuneiden aineiden tai tuotteiden valvontaan on sovellettava liitteessä I olevassa 5.A kohdassa säädettyjä määrää koskevia vaatimuksia. Tällä tavoin saatujen kokoomanäytteiden katsotaan edustavan eriä tai osaeriä, joista ne on otettu. Laboratorionäytteistä määritettyjen pitoisuuksien perusteella arvioidaan, ovatko tutkittavat erät Euroopan parlamentin ja neuvoston direktiivissä 2002/32/EY (2) vahvistettujen enimmäismääriä koskevien vaatimusten mukaisia.
2. Erän tai osaerän säännöstenmukaisuus
Erä hyväksytään, jos yhden määrityksen tulos ei ylitä direktiivissä 2002/32/EY vahvistettua enimmäismäärää, kun otetaan huomioon mittausepävarmuus.
Katsotaan, että erä ei ole asetuksessa (EY) N:o 2002/32/EY vahvistettujen enimmäismääriä koskevien vaatimusten mukainen, jos kaksoismäärityksellä (3) vahvistettu suurempi määritystulos (4) ylittää enimmäismäärän selvästi, kun otetaan huomioon mittaukseen liittyvä epävarmuus.
Mittausepävarmuus voidaan ottaa huomioon jollakin seuraavista tavoista:
|
— |
laskemalla laajennettu epävarmuus käyttäen kattavuuskerrointa 2, jolloin luotettavuustaso on noin 95 %. Erä ei ole säännösten mukainen, jos mitattu arvo, josta on vähennetty mittausepävarmuus U, ylittää enimmäismäärän. Jos dioksiinipitoisuudet ja dioksiinin kaltaisten PCB-yhdisteiden pitoisuudet määritetään erikseen, on dioksiinipitoisuuksien ja dioksiinin kaltaisten PCB-yhdisteiden pitoisuuksien yhteydessä käytettävä dioksiinipitoisuuksien ja dioksiinin kaltaisten PCB-yhdisteiden erillisten määritystulosten yhteenlaskettua, arvioitua ja laajennettua epävarmuutta. |
|
— |
vahvistamalla päätöksen tekoon vaadittava raja-arvo (päätösraja) (CCα) komission päätöksen 2002/657/EY (5) mukaisesti (liitteen 3.1.2.5 kohta – aineet, joille on vahvistettu sallittu raja). Erä ei ole säännösten mukainen, jos mitattu arvo on yhtä suuri tai suurempi kuin CCα. |
Näitä tulkintasääntöjä sovelletaan virallista tarkastusta varten otettujen näytteiden määritystuloksiin. Ne eivät vaikuta jäsenvaltioiden oikeuteen soveltaa kansallisia sääntöjä oikeustoimia ja kiistanratkaisumenettelyjä varten tehtyihin määrityksiin.
II. NÄYTTEIDEN VALMISTUS SEKÄ DIOKSIINIEN (PCDD/PCDF) JA DIOKSIININ KALTAISTEN PCB-YHDISTEIDEN VIRALLISESSA VALVONNASSA KÄYTETTÄVIÄ MÄÄRITYSMENETELMIÄ KOSKEVAT VAATIMUKSET
1. Tarkoitus ja soveltamisala
Näitä vaatimuksia on sovellettava analysoitaessa rehuaineita ja rehuja dioksiinin (polykloorattujen dibentso-p-dioksiinien (PCDD-yhdisteet) ja polykloorattujen dibentsofuraanien (PCDF-yhdisteet)) ja dioksiinin kaltaisten polykloorattujen bifenyylien (PCB-yhdisteiden) määrittämiseksi.
Dioksiinien esiintymistä rehuissa voidaan valvoa strategialla, johon kuuluu seulontamenetelmä niiden näytteiden valitsemiseksi, joissa on vähemmän kuin 25 prosentilla tietyn tason alittavia tai sen ylittäviä määriä dioksiineja tai dioksiinin kaltaisia PCB-yhdisteitä. Merkittäviä dioksiinimääriä sisältävien näytteiden dioksiinipitoisuudet on määritettävä tai vahvistettava varmistavan menetelmän avulla.
Seulontamenetelmät ovat menetelmiä, joita käytetään osoittamaan dioksiinien tai dioksiinin kaltaisten PCB-yhdisteiden esiintyminen tietyllä tasolla. Menetelmien näytteenkäsittelykapasiteetti on suuri, ja niillä seulotaan suuria määriä näytteitä mahdollisesti positiivisten näytteiden havaitsemiseksi. Menetelmät on erityisesti suunniteltu välttämään väärät negatiiviset tulokset.
Varmistusmenetelmät ovat menetelmiä, joilla saadaan täydelliset tiedot tai lisätietoja, joiden avulla dioksiinit ja dioksiinin kaltaiset PCB-yhdisteet voidaan tunnistaa ja kvantifioida yksiselitteisesti tietyllä tasolla.
2. Taustaa
Koska ympäristönäytteet ja biologiset näytteet (rehuaineista ja rehuista otetut näytteet mukaan luettuina) sisältävät yleensä eri dioksiiniyhdisteiden monenlaisia seoksia, riskinarvioinnin helpottamiseksi on kehitetty toksisuusekvivalenssikertoimen (Toxic Equivalency Factor, TEF) käsite. Toksisuusekvivalenssikertoimet on kehitetty sitä varten, että niillä voidaan esittää 2,3,7,8-substituoitujen PCDD-yhdisteiden ja PCDF-yhdisteiden seoksien ja myös joidenkin muuhun kuin orto-asemaan substituoitujen tai toisaalta mono-orto-asemaan substituoitujen PCB-yhdisteiden — joilla on dioksiinin kaltaista aktiivisuutta — konsentraatioita 2,3,7,8-TCDD:n (2,3,7,8-tetraklooridibentso-p-dioksiinin) toksisuusekvivalentteina (TEQ). Tietyssä erässä olevien yksittäisten aineiden konsentraatiot kerrotaan kunkin aineen toksisuusekvivalenssikertoimella, ja saadut määrät lasketaan yhteen, jolloin tulokseksi saadaan dioksiinin kaltaisten yhdisteiden kokonaiskonsentraatio toksisuusekvivalentteina ilmaistuna.
Tässä asetuksessa yksittäisen yhdisteen hyväksytty määritysraja on se näytteessä olevan analyytin pitoisuus, joka tuottaa mitattaville kahdelle ionille mittauslaitteessa vasteen, jossa vähemmän herkän signaalin signaali-kohinasuhde on 3:1, ja joka täyttää EPA-menetelmän 1613 versiossa B kuvatun määritysmenetelmän vaatimukset, jotka koskevat esimerkiksi retentioaikaa ja isotooppisuhdetta.
3. Näytteen valmistamista koskevat laadunvarmistusvaatimukset
Tässä yhteydessä sovelletaan liitteessä II vahvistettuja yleisiä säännöksiä näytteiden valmistamisesta määritystä varten.
Lisäksi on noudatettava seuraavia vaatimuksia:
|
— |
Näytteet on säilytettävä ja kuljetettava lasista, alumiinista, polypropyleenistä tai polyetyleenistä valmistetuissa säiliöissä. Paperipölyjäämät on poistettava säiliöistä. Lasitavarat on huuhdeltava liuottimella, josta on ennalta tarkastettu, ettei niissä ole dioksiinijäämiä. |
|
— |
On tehtävä ilman näytettä nolla-analyysi, jossa tehdään kaikki analyysin vaiheet. |
|
— |
Uuttamisessa käytettävän näytteen määrän on oltava riittävä herkkyysvaatimusten täyttämiseksi. |
4. Laboratorioita koskevat vaatimukset
|
— |
Laboratorioiden on osoitettava menetelmän suorituskyky tarkoituksenmukaisella mittausalueella, esimerkiksi 0,5 ×, 1 × ja 2 × tärkeäksi katsottu taso, ja toistettujen mittausten hajonnan on oltava hyväksyttävä. Ks. hyväksymisvaatimuksia koskevat tiedot 5 kohdasta. |
|
— |
Varmistusmenetelmän määritysrajan on oltava noin yksi viidesosa tärkeäksi katsotusta tasosta, jotta varmistetaan, että vaihtelukertoimet ovat hyväksyttävät kyseisellä mittausalueella. |
|
— |
Sisäistä laadunvalvontaa varten on mitattava säännöllisesti nollanäytteitä tai näytteitä, joihin on lisätty analyyttiä, tai erityisiä kontrollinäytteitä mieluiten sertifioiduilla vertailuaineilla. |
|
— |
Paras tapa osoittaa pätevyys analyysien tekemisessä on osallistua menestyksekkäästi laboratorioiden välisiin laboratorioiden pätevyyttä arvioiviin tutkimuksiin. Menestyksekäs osallistuminen esimerkiksi maaperä- tai viemärinäytteitä koskeviin laboratorioiden välisiin tutkimuksiin ei kuitenkaan välttämättä osoita laboratorion pätevyyttä myös elintarvike- ja rehunäytteiden analysoinnin alalla, koska kyseisten näytteiden kontaminaatiotaso on alempi. Jatkuva osallistuminen laboratorioiden välisiin tutkimuksiin, jotka koskevat dioksiinien ja dioksiinin kaltaisten PCB-yhdisteiden määrittämistä rehu- tai elintarvikematriiseissa, on siksi välttämätöntä. |
|
— |
Laboratorioiden on oltava ISO-oppaan 58 mukaisesti toimivan tunnustetun laitoksen hyväksymiä, millä varmistetaan, että laboratoriot soveltavat analyyttistä laadunvarmistusta. Laboratorioiden hyväksyntä on tehtävä ISO/IEC/17025-standardin mukaisesti. |
5. Dioksiinien ja dioksiinin kaltaisten pcb-yhdisteiden määritysmenettelyjä koskevat vaatimukset
Määritysmenettelyjä koskevat perusvaatimukset:
|
— |
Hyvä herkkyys ja matalat toteamisrajat. PCDD- ja PCDF-yhdisteet on pystyttävä havaitsemaan jo määrinä, joiden suuruus on pikogrammoja TEQ (pikogramma = 10-12 g), sillä jotkin näistä yhdisteistä ovat erittäin myrkyllisiä. PCB-yhdisteiden tiedetään esiintyvän suurempina pitoisuuksina kuin PCDD- ja PCDF-yhdisteiden. Useimpien PCB-yhdisteiden osalta herkkyydeksi riittää jo nanogrammoina (10-9 g) ilmaistava alue. Niitä toksisempien dioksiinin kaltaisten PCB-yhdisteiden (erityisesti ei-orto-yhdisteiden) mittauksessa herkkyyden on oltava samaa luokkaa kuin PCDD- ja PCDF-yhdisteiden kohdalla. |
|
— |
Hyvä selektiivisyys (spesifisyys). PCDD- ja PCDF-yhdisteet sekä dioksiinin kaltaiset PCB:t on voitava erottaa lukuisista muista uuttamisessa mukana tulevista ja mahdollisesti häiriöitä aiheuttavista yhdisteistä, joiden pitoisuudet voivat olla moninkertaisia verrattuna näiden yhdisteiden pitoisuuksiin. Kaasukromatografia-massaspektrometriamenetelmissä (GC-MS) on pystyttävä tarvittaessa erottelemaan eri yhdisteet, kuten esimerkiksi 17 toksista 2,3,7,8-substituoitua PCDD- ja PCDF-yhdistettä ja dioksiinin kaltaista PCB-yhdistettä, ja muut näiden aineiden sukulaisaineet. Biotestien pitää pystyä määrittämään TEQ-arvot valikoiden PCDD- ja PCDF-yhdisteiden ja dioksiinin kaltaisten PCB-yhdisteiden summana. |
|
— |
Hyvä tarkkuus (oikeellisuus ja täsmällisyys). Määrityksessä pitää pystyä antamaan pätevä ja luotettava arvio aineen todellisesta pitoisuudesta näytteessä. Hyvä tarkkuus (mittauksen tarkkuus: mittauksen tuloksen ja mittaussuureen todellisen tai annetun arvon läheisyys) on välttämätöntä, jotta voidaan välttää näytteen määritystuloksen hylkääminen TEQ-arvion heikon luotettavuuden perusteella. Tarkkuus ilmaistaan oikeellisuutena (sertifioidusta materiaalista mitatun tutkittavan aineen määrän keskiarvon ja sertifioidun arvon erotus prosentteina tästä arvosta) ja täsmällisyytenä (RSDR on uusittavissa olosuhteissa saaduista tuloksista laskettu suhteellinen standardipoikkeama). |
Seulontamenetelmiin voi kuulua biotestejä ja GC-MS-menetelmiä; varmistusmenetelmät ovat korkean erotuskyvyn kaasukromatografia-massaspektrometriamenetelmiä (HRGC-HRMS-menetelmiä).
TEQ-arvon kokonaismäärän osalta on noudatettava seuraavia vaatimuksia:
|
|
Seulontamenetelmät |
Varmistusmenetelmät |
|
Väärien negatiivisten tulosten osuus |
< 1 % |
|
|
Oikeellisuus |
|
- 20 % – + 20 % |
|
Tarkkuus RSDR |
< 30 % |
< 15 % |
6. Seulonnassa tai varmistuksessa käytettäviä gc-ms-menetelmiä koskevat erityisvaatimukset
|
— |
Määritysmenetelmän validoimiseksi on aivan menetelmän alussa eli esimerkiksi ennen uuttamista lisättävä 13C-merkittyjä 2,3,7,8-kloorisubstituoituja sisäisiä PCDD/F-standardeja ja 13C-merkittyjä dioksiinin kaltaisten PCB-yhdisteiden sisäisiä standardeja. Vähintään yhtä näistä yhdisteistä on lisättävä kutakin tetra–oktakloorattua PCDD/F-homologiryhmää kohden ja vähintään yhtä näistä yhdisteistä kutakin dioksiinin kaltaisten PCB-yhdisteiden homologiryhmää kohden (tai vaihtoehtoisesti vähintään yhtä näistä yhdisteistä kutakin PCDD/F:ien ja dioksiinin kaltaisten PCB-yhdisteiden valvonnassa käytettyä massaspektrillä määritettyä ionia kohden.) Ainakin varmistusmenetelmien yhteydessä on käytettävä kaikkia 17:ää 13C-merkittyä 2,3,7,8-kloorisubstituoitua sisäistä PCDD/F-standardia ja kaikkia 12:ta 13C-merkittyä dioksiinin kaltaisten PCB-yhdisteiden sisäistä standardia. |
|
— |
Suhteelliset vastekertoimet olisi asianmukaista kalibrointiliuosta käyttäen määritettävä myös niille aineille, joiden määrityksessä ei lisätä 13C-merkittyä analogia. |
|
— |
Kasviperäisistä rehuista ja alle 10 % rasvaa sisältävistä eläinperäisistä rehuista otettuihin näytteisiin on sisäiset standardit on lisättävä ennen uuttamista. Yli 10 % rasvaa sisältävistä eläinperäisistä rehuista otettuihin näytteisiin ne voidaan lisätä joko ennen uuttamista tai rasvojen uuttamisen jälkeen. Uuttamisen tehokkuus on validoitava asianmukaisesti sen mukaan, missä vaiheessa sisäiset standardit lisätään, ja sen mukaan, ilmoitetaanko tulokset tuotteessa vai rasvassa olevan pitoisuuden perusteella. |
|
— |
Ennen GC-MS-analyysia on lisättävä 1 tai 2 saantostandardia. |
|
— |
Saantojen valvonta on välttämätöntä. Varmistusmenetelmissä sisäisten standardien saantojen pitää olla 60–120 %. Pienemmät tai suuremmat saantoarvot voidaan hyväksyä erityisesti hepta- ja oktakloorattujen dibentsodioksiinien ja dibentsofuraanien osalta, kunhan niiden vaikutus TEQ-arvoon on enintään 10 % TEQ-arvon kokonaismäärästä (perustana PCDD/F:ien ja dioksiinin kaltaisten PCB-yhdisteiden summa). Seulontamenetelmissä saantojen on oltava 30–140 %. |
|
— |
Dioksiinit on erotettava häiriöitä aiheuttavista klooratuista yhdisteistä, esimerkiksi muista kuin dioksiinin kaltaisista PCB-yhdisteistä ja klooratuista bifenyylieettereistä, soveltuvilla kromatografiatekniikoilla (mieluiten florisil-, alumiinioksidi- ja/tai hiilikolonneilla). |
|
— |
Isomeerien erotuksen kaasukromatografian avulla pitää olla riittävä (1,2,3,4,7,8-HxCDF:n ja 1,2,3,6,7,8-HxCDF:n välisen laakson korkeus ei saa olla yli 25 %:a). |
|
— |
Dioksiinien määrittämiseen on käytettävä EPA-menetelmän 1613 versiota B (Tetra- through octa-chlorinated dioxins and furans by isotope dilution HRGC/HRMS) tai vastaavat tehokkuuskriteerit täyttävää menetelmää. |
|
— |
Suurimman ja pienimmän arvon välinen ero saa olla enintään 20 % rehuilla, joiden dioksiinipitoisuus on enimmäismäärän rajoissa tai ylittää sen. Sellaisten rehujen osalta, joiden dioksiinipitoisuudet alittavat selvästi enimmäismäärät, ero voi olla 25–40 %. |
7. Seulovat analyysimenetelmät
7.1 Johdanto
Seulontaa voidaan käyttää erilaisiin analyyseihin: puhtaaseen seulontaan tai kvantitatiiviseen analyysiin.
Näytteiden vastetta verrataan vertailunäytteen vasteeseen kyseessä olevalla mittausalueella. Vertailunäytettä vähäisemmän vasteen antavia näytteitä pidetään negatiivisina, suuremman vasteen antaneita mahdollisesti positiivisina. Vaatimukset:
|
— |
Kussakin koesarjassa on käytettävä nollanäytettä ja vertailunäytettä (-näytteitä), jotka uutetaan ja testataan samaan aikaan identtisissä oloissa. Vertailunäytteen vasteen on oltava selvästi kohonnut verrattuna nollanäytteeseen. |
|
— |
Lisäksi on käytettävä mitattavan tason 0,5- ja 2-kertaista arvoa edustavia vertailunäytteitä, joilla osoitetaan testin toimivuus halutulla mittausalueella mitattavan tason kontrollia varten. |
|
— |
Muita matriiseja testattaessa on osoitettava vertailunäytteiden sopivuus mieluiten ottamalla mukaan näytteitä, joiden TEQ-arvoksi on korkean resoluution kaasukromatografia-massaspektrometrialla määritetty vertailunäytteen arvoa lähellä oleva arvo, taikka käyttämällä arvoltaan vastaavaksi säädettyä nollanäytettä. |
|
— |
Koska sisäisiä standardeja ei voida käyttää biotesteissä, on erittäin tärkeää tehdä toistettavuustestejä, jotta saadaan tietoja yksittäisen koesarjan keskihajonnasta. Vaihtelukertoimen on oltava pienempi kuin 30 %. |
|
— |
Biotestien osalta on määriteltävä kohdeyhdisteet, mahdolliset häiriöt sekä suurimmat hyväksyttävät nollatasot. |
Kvantitatiivisessa analyysissa tarvitaan standardilaimennussarja, kaksin- tai kolminkertainen puhdistus sekä mittaus-, nolla- ja saantokontrollit. Tulos voidaan ilmoittaa TEQ:na, jolloin oletetaan signaalista vastaavien yhdisteiden olevan TEQ-periaatteen mukaisia. Tämä voidaan tehdä tuottamalla TCDD:n (tai dioksiinin, furaanin tai dioksiinin kaltaisten PCP-yhdisteiden standardiseoksen) avulla kalibrointikuvaaja, jonka perusteella lasketaan TEQ-taso uutteessa ja sitä kautta näytteessä. Arvo korjataan nollanäytteelle (jotta otetaan huomioon epäpuhtaudet käytetyistä liuottimista ja kemikaaleista) lasketun TEQ-arvon ja saannon (lasketaan pitoisuudeltaan noin kyseessä olevaa tasoa edustavan laadunvalvontanäytteen TEQ-arvosta) perusteella. On huomattava, että osa ilmeisestä saantohäviöstä voi johtua matriisivaikutuksista ja/tai biotestien TEF-arvojen ja WHO:n virallisten TEF-arvojen eroista.
7.2 Seulontaan käytettäviä analyysimenetelmiä koskevat vaatimukset
|
— |
Seulontaan voidaan käyttää GC-MS-menetelmiä ja biotestimenetelmiä. GC-MS-menetelmien vaatimukset annetaan 6 kohdassa. Solupohjaisten biotestien erityisvaatimukset esitetään 7.3 kohdassa ja testisarjapohjaisten biotestien erityisvaatimukset 7.4 kohdassa. |
|
— |
On myös ilmoitettava väärien positiivisten ja väärien negatiivisten tulosten määrä suuressa määrässä näytteitä, joiden pitoisuudet ovat enimmäismäärien tai toimintarajojen ala- ja yläpuolella, ja niitä on verrattava varmistavalla analyysimenetelmällä määritettyyn TEQ-arvoon. Todellisten väärien negatiivisten tulosten osuuden pitää olla alle 1 %. Väärien positiivisten tulosten määrän pitää olla niin pieni, että seulontavälineen käytöstä on hyötyä. |
|
— |
Positiiviset tulokset on aina vahvistettava varmistavalla analyysimenetelmällä (HRGC/HRMS). Lisäksi laajaa TEQ-vaihtelualuetta edustavat näytteet on vahvistettava HRGC/HRMS:llä (noin 2–10 % negatiivisista näytteistä). Tiedot biotestin ja HRGC/HRMS:n tulosten vastaavuudesta on ilmoitettava. |
7.3 Solupohjaisia biotestejä koskevat erityisvaatimukset
|
— |
Biotestiä suoritettaessa on kussakin testauksessa käytettävä TCDD-yhdisteiden tai dioksiini-furaaniseoksen viitepitoisuussarjaa (kattava annos-vastekuvaaja, jossa R2 > 0,95). Seulonnassa voidaan kuitenkin käyttää matalien pitoisuuksien näytteitten analysointiin laajennettua matalien pitoisuuksien kuvaajaa. |
|
— |
Biotestin tulosten valvomiseen vakiomittaisella ajanjaksolla on käytettävä laadunvalvontalomakkeessa TCDD-viitepitoisuutta (noin 3 kertaa määritysraja). Vaihtoehtoisesti voidaan käyttää vertailunäytteen suhteellista vastetta verrattuna TCDD-kalibrointikäyrään, sillä solukohtainen vaste voi riippua useista eri tekijöistä. |
|
— |
Kullekin vertailumateriaalille on luotava laadunvalvontakortti, joka on tarkastettava; näin varmistetaan, että tulokset vastaavat ohjeita. |
|
— |
Erityisesti kvantitatiivisissa laskelmissa on käytetyn näytelaimennuksen oltava vastekäyrän lineaarisella osuudella. Pitoisuudeltaan vastekäyrän lineaarisen osuuden ylittävät näytteet on laimennettava ja testattava uudelleen. Onkin suositeltavaa testata kerralla vähintään kolme laimennosta. |
|
— |
Prosentuaalinen keskihajonta saa olla enintään 15 % kunkin näytelaimennoksen kolminkertaisessa määrityksessä ja enintään 30 % kolmen erillisen kokeen välillä. |
|
— |
Toteamisrajan arvoksi voidaan asettaa kolme kertaa liuotinnollan tai taustavasteen keskihajonnan arvo. Vaihtoehtoisesti voidaan käyttää tausta-arvoa suurempaa vastetta (5 kertaa liuotinnollan vaste) laskettuna kyseisen päivän kalibrointikäyrältä. Määritysrajaksi voidaan asettaa 5–6 kertaa liuotinnollan tai tausta-arvon keskihajonta, tai sitten voidaan käyttää selvästi tausta-arvoa suurempaa vastetta (10 kertaa liuotinnollan vaste) laskettuna kyseisen päivän kalibrointikäyrältä. |
7.4 Testisarjapohjaisia biotestejä koskevat erityisvaatimukset
|
— |
On varmistettava, että testisarjapohjaiset biotestit ovat riittävän herkkiä ja luotettavia rehujen testaukseen. |
|
— |
Valmistajan ohjeita näytteen valmistamiseen ja analyyseihin on noudatettava. |
|
— |
Viimeisen käyttöpäivän ohittaneita testisarjoja ei pidä käyttää. |
|
— |
Muiden testisarjojen yhteydessä käytettäviksi tarkoitettuja tarvikkeita tai komponentteja ei pidä käyttää. |
|
— |
Testisarjoja on säilytettävä annetuissa säilytyslämpötilarajoissa ja käytettävä annetuissa käyttölämpötiloissa. |
|
— |
Toteamisraja on immunomäärityksissä summa, joka saadaan laskemalla yhteen keskiarvo sekä 3 kertaa keskihajonta, joka on määritetty 10:llä nollanäytteen toistoanalyysillä, ja joka jaetaan lineaarisen regressiokuvaajan kulmakertoimella. |
|
— |
Laboratoriotesteissä on käytettävä vertailustandardeja, jotta taataan, että vaste standardiin on hyväksyttävissä rajoissa. |
8. Tulosten ilmoittaminen
Mikäli analyyttinen menetelmä sallii, analyysin tuloksissa on ilmoitettava yksittäisten PCDD/F:ien ja PCB-yhdisteiden pitoisuudet, ja analyysin tulokset on ilmoitettava pienimpinä, suurimpina ja väliarvoina, jotta tulosten raportointiin saadaan mukaan mahdollisimman paljon tietoja, ja tuloksia pystytään tulkitsemaan erityisten vaatimusten mukaisesti.
Raportissa on ilmoitettava myös näytteen rasvapitoisuus sekä rasvan uuttamiseen käytetty menetelmä.
Yksittäisten sisäisten standardien saantotiedot on toimitettava, jos saantojen arvo on kohdassa 6 mainitun alueen ulkopuolella tai jos enimmäismäärä ylittyy; muissa tapauksissa ne on toimitettava pyydettäessä.
Koska mittausepävarmuus on otettava huomioon päätettäessä näytteen säännöstenmukaisuudesta, kyseisen muuttujan on myös oltava saatavissa. Siksi määritystulos on raportoitava muodossa x +/- U, jossa x on määritystulos ja U on mittaukseen liittyvä laajennettu epävarmuustekijä, jossa käytetään kattavuuskerrointa 2, jolloin luotettavuustaso on noin 95 %. Jos dioksiinipitoisuudet ja dioksiinin kaltaisten PCB-yhdisteiden pitoisuudet määritetään erikseen, dioksiinien ja dioksiinin kaltaisten PCB-yhdisteiden erillisten määritystulosten yhteenlaskettua, arvioitua ja laajennettua epävarmuutta on käytettävä dioksiinipitoisuuksien ja dioksiinin kaltaisten PCB-yhdisteiden pitoisuuksien yhteydessä.
Jos mittausten epävarmuus otetaan huomioon soveltamalla päätösrajaa CCα (tämän B osan I luvun 2 kohdassa kuvatulla tavalla), tiedot tästä muuttujasta on ilmoitettava.
(1) Dioksiinien, furaanien ja dioksiin kaltaisten PCB-yhdisteiden toksisuusekvivalenssikertoimia (Toxic Equivalency Factor, TEF) koskeva taulukko
|
Yhdiste |
TEF-arvo |
Yhdiste |
TEF-arvo |
|
Dibentso-para-dioksiinit (PCDD:t) |
|
Dioksiinin kaltaiset PCB:t: |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
Ei-orto-PCB-yhdisteet |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0001 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,01 |
|
OCDD |
0,0001 |
Mono-orto-PCB-yhdisteet |
|
|
|
|
PCB 105 |
0,0001 |
|
Dibentsofuraanit (PCDF:t) |
|
PCB 114 |
0,0005 |
|
2,3,7,8-TCDF |
0,1 |
PCB 118 |
0,0001 |
|
1,2,3,7,8-PeCDF |
0,05 |
PCB 123 |
0,0001 |
|
2,3,4,7,8-PeCDF |
0,5 |
PCB 156 |
0,0005 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 157 |
0,0005 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00001 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 189 |
0,0001 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
|
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
|
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0001 |
|
|
|
Käytetyt lyhenteet: T = tetra, Pe = penta, Hx = heksa, Hp = hepta, O = okta, CDD = klooridibentso-p-dioksiini, CDF = klooridibentsofuraani, CB = klooribifenyyli. |
|||
(2) EYVL L 140, 30.5.2002, s. 10.
(3) Kaksoismääritys on tarpeen, jotta voidaan sulkea pois mahdollinen sisäinen ristikontaminaatio tai näytteiden sekoittuminen vahingossa. Säännöstenmukaisuus varmistetaan ensimmäisen määrityksen perusteella ja huomioon otetaan mittausepävarmuus.
Jos määritys suoritetaan dioksiinikontaminaatiotapauksen yhteydessä, varmennus kaksoismäärityksellä voidaan jättää tekemättä, mikäli määritykseen valitut näytteet liittyvät dioksiinikontaminaatiotapaukseen jäljitettävyyden perusteella.
(4) Suurimmat arvot: toksisuusekvivalenttia TEQ laskettaessa oletetaan kunkin määrittämättä jääneen yhdisteen arvoksi määritysrajaa vastaava arvo. Pienimmät arvot: TEQ:ta laskettaessa oletetaan kunkin määrittämättä jääneen yhdisteen arvoksi nolla.
Väliarvot: TEQ:ta laskettaessa oletetaan kunkin määrittämättä jääneen yhdisteen arvoksi puolet määritysrajaa vastaavasta arvosta.
LIITE VI
REHUJEN SISÄLTÄMIEN ELÄINPERÄISTEN AINESOSIEN MÄÄRITYSMENETELMÄT REHUJEN VIRALLISTA VALVONTAA VARTEN
Rehujen eläinperäisten ainesosien mikroskooppiseen toteamiseen, tunnistamiseen tai arviointiin sovellettavat vaatimukset
1. Tarkoitus ja soveltamisala
Näitä vaatimuksia on sovellettava, kun eläinperäisiä ainesosia (jotka on määritelty nisäkkäiden, siipikarjan ja kalojen ruhojen tai ruhon osien käsittelystä saataviksi tuotteiksi) tutkitaan rehuista mikroskooppisesti Euroopan parlamentin ja neuvoston asetuksen (EY) N:o 882/2004 (1) mukaisen rehujen yhteensovitetun tarkastusohjelman yhteydessä. Mikäli tässä liitteessä kuvattuja menetelmiä käytetään kaikissa virallisissa testeissä, voidaan tehdä myös toinen testi jotain menetelmän muunnosta tai vaihtoehtoista menetelmää käyttäen, jotta voitaisiin parantaa tiettyjen eläinperäisten ainesosien havaitsemista tai täsmentää eläinperäisten ainesosien alkuperää. Muunnettua menetelmää voidaan lisäksi käyttää tutkittaessa erityisiä eläinperäisiä ainesosia, esimerkiksi talissa olevaa plasmaa tai luita (ks. myös 9 kohta), kunhan nämä analyysit tehdään yhteensovitetussa tarkastusohjelmassa määrättyjen analyysien lisäksi.
2. Herkkyys
Eläinperäisten ainesosien luonteesta riippuen niitä voidaan havaita rehuista hyvinkin pieniä määriä (< 0,1 %).
3. Periaate
Tunnistukseen käytetään asianmukaisesti valmistettua, edustavaa näytettä, joka on otettu liitteen I säännösten mukaisesti. Seuraavassa esitettävä analyysimenetelmä soveltuu rehunäytteille, joiden kosteuspitoisuus on alhainen. Jos rehun kosteuspitoisuus on yli 14 %, se on ennen käsittelyä kuivattava (kondensoitava). Eräitä rehuja tai rehuaineita (esim. rasvoja ja öljyjä) varten tarvitaan erityiskäsittelyä (ks. 9 kohta). Eläinperäiset ainesosat tunnistetaan tyypillisten mikroskooppisesti tunnistettavissa olevien ominaisuuksien (lihassäikeiden ja muiden lihan palojen, ruston, luiden, sarvien, karvojen, harjasten, veren, höyhenten, munankuorten, kalanluiden ja suomujen) perusteella. Tutkimus on tehtävä sekä näytteen siiviläfraktiosta (6.1) että konsentroidusta sedimentistä (6.2).
4. Reagenssit
4.1 Mikroskopointireagenssit
|
4.1.1 |
Kloraalihydraatti (60-prosenttinen vesiliuos, w/v). |
|
4.1.2 |
Lipeä (NaOH 2,5 % w/v tai KOH 2,5 % w/v) siiviläfraktiota varten. |
|
4.1.3 |
Parafiiniöljy tai glyseroli (viskositeetti 68–81) sedimentistä tehtäviä mikroskopointia varten. |
4.2 Pesureagenssit
|
4.2.1 |
Alkoholi, 96 %. |
|
4.2.2 |
Asetoni. |
4.3 Konsentrointireagenssi
|
4.3.1 |
Tetrakloorietyleeni (tiheys 1,62). |
4.4 Värjäysreagenssit
|
4.4.1 |
Jodi-kaliumjodidiliuos (2 g kaliumjodidia liuotetaan 100 ml:aan vettä ja lisätään 1 g jodia, ja sekoitetaan useita kertoja). |
|
4.4.2 |
Alitsariinipunainen (2,5 ml 1 M kloorivetyhappoa laimennetaan 100 ml:aan vettä ja liuokseen lisätään 200 mg alitsariinipunaista). |
|
4.4.3 |
Kystiinireagenssi (2 g lyijyasetaattia ja 10 g NaOH:ta liuotetaan 100 ml:aan vettä). |
|
4.4.4 |
Jodi-kaliumjodidiliuos (liuotettu 70-prosenttiseen etanoliin). |
4.5. Värinpoistoreagenssi
|
4.5.1 |
Kaupallista natriumhypokloriittiliuosta (9,6 % aktiivista klooria). |
5. Laitteet ja tarvikkeet
|
5.1 |
Analyysivaaka (tarkkuus 0,01 g, paitsi konsentroitua sedimenttiä varten 0,001 g). |
|
5.2 |
Jauhamisvälineet (mylly tai huhmare, erityisesti rehulle, jonka rasvapitoisuus analyysissa on yli 15 %). |
|
5.3 |
Seula, jossa on neliönmuotoiset reiät, silmäkoko enintään 0,50 mm. |
|
5.4 |
Erotussuppilo tai kartiopohjainen selkeytysastia. |
|
5.5 |
Stereomikroskooppi (vähintään 40-kertainen suurennus). |
|
5.6 |
Valomikroskooppi (vähintään 400-kertainen suurennus), jossa on läpivalaisu- tai polarisaatio-optiikka. |
|
5.7 |
Tavanomaiset laboratorion lasiastiat. Kaikki välineet on puhdistettava huolellisesti. Erotussuppilot ja lasiastiat on pestävä pesukoneessa. Seulat on puhdistettava jäykkäharjaksisella harjalla. |
6. Menettely
Pelletöidyt rehut voidaan esiseuloa, jos molemmat fraktiot analysoidaan erillisinä näytteinä.
Näytettä käsitellään vähintään 50 g (tarvittaessa se jauhetaan varovasti sopivilla jauhamisvälineillä (5.2) sopivan rakenteen aikaansaamiseksi). Jauhetusta materiaalista otetaan kaksi edustavaa osanäytettä, yksi seulafraktiota (vähintään 5 g, ks. 6.1 kohta) ja yksi konsentroitua sedimenttiä varten (vähintään 5 g, ks. 6.2 kohta). Lisäksi voidaan käyttää värjäysreagenssia (6.3) tunnistamista varten.
Eläinproteiinien luonteen ja partikkelien alkuperän osoittamiseksi voidaan käyttää päätöksentekoa tukevaa järjestelmää (kuten ARIES) ja vertailunäytteet voidaan dokumentoida.
6.1 Eläinperäisten ainesosien osoittaminen seulafraktioista
Vähintään 5 g näytettä seulotaan (seula, ks. 5.3 kohta) kahteen fraktioon.
Karkea(t) seulafraktio(t) (tai edustava osa sitä) levitetään ohuena kerroksena sopivalle alustalle ja tutkitaan järjestelmällisesti stereomikroskoopilla (5.5) eri suurennuksilla eläinperäisten ainesosien osoittamiseksi.
Hienosta seulafraktiosta valmistetaan objektilasinäyte, joka tutkitaan järjestelmällisesti valomikroskoopissa (5.6) eri suurennuksilla eläinperäisten ainesosien osoittamiseksi.
6.2 Eläinperäisten ainesosien osoittaminen konsentroidusta sedimentistä
Vähintään 5 g näytettä punnitaan 0,01 g:n tarkkuudella erotussuppiloon tai kartiopohjaiseen selkeytysastiaan ja käsitellään vähintään 50 ml:lla tetrakloorietyleeniä (4.3.1). Seosta ravistetaan tai sekoitetaan toistuvasti.
|
— |
Käytettäessä suljettua erotussuppiloa sedimentin annetaan laskeutua astian pohjalle riittävän kauan (vähintään 3 minuuttia), ennen kuin se erotetaan. Suppiloa ravistetaan uudelleen, ja sedimentin annetaan jälleen laskeutua vähintään 3 minuuttia. Sedimentti erotetaan jälleen. |
|
— |
Käytettäessä avointa selkeytysastiaa sedimentin annetaan laskeutua astian pohjalle vähintään 5 minuuttia, minkä jälkeen se erotetaan. |
Koko sedimentti kuivataan ja punnitaan 0,001 g:n tarkkuudella. Punnitus on tarpeen vain, jos eläinperäisten ainesosien osuus on arvioitava. Jos sedimentti koostuu useista suurista partikkeleista, se voidaan seuloa (seulasta ks. 5.3 kohta) kahteen fraktioon. Kuivattu sedimentti tutkitaan stereomikroskoopilla (5.5) tai valomikroskoopilla (5.6) luuaineksen toteamiseksi.
6.3 Petaus- ja värjäysreagenssien käyttö
Eläinperäisten ainesosien mikroskooppisessa tunnistuksessa voidaan käyttää erityisiä petaus- ja värjäysreagensseja.
Kloraalihydraatti (4.1.1): Varovainen kuumennus helpottaa solurakenteiden tarkastelua, koska tärkkelysjyväset gelatinoituvat ja ei-toivottu solun sisältö häviää.
Lipeä (4.1.2): Natrium- tai kaliumhydroksidi kirkastaa rehumateriaalia, mikä helpottaa lihassäikeiden, karvojen ja muiden keratiinirakenteiden havaitsemista.
Parafiiniöljy ja glyseroli (4.1.3): Luuaines voidaan tunnistaa tässä petausaineessa helposti, koska useimmat luun ontelot (lakuunat) jäävät täyteen ilmaa ja näkyvät mustina noin 5–15 mikrometrin kokoisina reikinä.
Jodi-kaliumjodidiliuos (4.4.1): Liuosta käytetään tärkkelyksen (sinivioletti väri) ja valkuaisen (keltaoranssi väri) osoittamiseen. Liuosta voidaan tarvittaessa laimentaa.
Alitsariinipunaliuos (4.4.2): Liuos värjää luut, ruodot ja suomut punaisiksi tai vaaleanpunaisiksi. Ennen sedimentin kuivaamista (ks. 6.2 kohta) koko sedimentti siirretään lasiseen koeputkeen ja pestään kahdesti noin 5 ml:lla etanolia (4.2.1) (kummallakin kerralla käytetään koeputkisekoitinta ja annetaan liuottimen selkeytyä noin minuutin ajan ennen sen dekantointia). Ennen alitsariinipunaliuoksen käyttöä sedimentti käsitellään värinpoistoreagenssilla lisäämällä sedimenttiin vähintään 1 ml natriumhypokloriittiliuosta (4.5.1). Reaktion annetaan jatkua 10 minuutin ajan. Putki täytetään vedellä, sedimentin annetaan laskeutua 2–3minuuttia, ja sen jälkeen vesi ja suspendoituneet partikkelit dekantoidaan. Sedimentti pestään vielä kahdesti noin 10 ml:lla vettä (kummallakin kerralla käytetään koeputkisekoitinta, sedimentin annetaan laskeutua putken pohjaan ja vesi dekantoidaan). Lisätään alitsariinipunaliuosta vähintään kahdesta kymmeneen tippaa (sedimentin määrän mukaan). Seosta ravistetaan ja sedimentin annetaan reagoida liuoksen kanssa muutaman sekunnin ajan. Värjäytynyt sedimentti pestään kahdesti noin 5 ml:lla alkoholia (4.2.1) ja sen jälkeen kerran asetonilla (4.2.2) (joka kerta käytetään koeputkisekoitinta ja sedimentin annetaan laskeutua noin minuutin ajan, minkä jälkeen liuos dekantoidaan). Tämän jälkeen sedimentti on valmista kuivattavaksi.
Kystiinireagenssi (4.4.3): Varovainen kuumennus muuttaa kystiiniä sisältävät ainesosat (karvat, höyhenet jne.) mustanruskeiksi.
6.4 Kalajauhoa mahdollisesti sisältävän rehun tutkiminen
Vähintään yksi objektilasi hienosta seulafraktiosta ja hienosta sedimenttifraktiosta tutkitaan valomikroskoopilla (ks. kohdat 6.1 ja 6.2).
Jos rehu pakkausmerkintöjen mukaan sisältää kalajauhoa tai jos kalajauhon esiintymistä epäillään tai sitä havaitaan alustavassa tutkimuksessa, mikroskopoidaan lisäksi vähintään kaksi uutta objektilasia alkuperäisen näytteen hienosta seulafraktiosta ja koko sedimenttifraktiosta.
7. Laskutoimitukset ja arviointi
Kun virallisella analyysilla arvioidaan eläinperäisten ainesosien määrää (eikä vain niiden esiintymistä), jäsenvaltioiden on varmistettava, että käytetään tässä kohdassa kuvattuja menettelyjä.
Laskutoimitukset voidaan tehdä vain, jos eläinperäiset ainesosat sisältävät luupartikkeleita.
Lämminveristen maalla elävien lajien (ts. nisäkkäiden ja lintujen) luupartikkelit voidaan erottaa erityyppisistä kalanluista mikroskooppisesti tyypillisten ontelomuodostelmien (lakuunoiden) perusteella. Näytteen sisältämän eläinperäisen aineksen osuus arvioidaan ottamalla huomioon
|
— |
luupartikkelien arvioitu osuus (painoprosenttia) konsentroidussa sedimentissä ja |
|
— |
luun osuus (painoprosenttia) eläinperäisissä ainesosissa. |
Arvion on perustuttava vähintään kolmeen (jos mahdollista) objektilasinäytteeseen ja vähintään viiteen näkökenttään objektilasia kohti. Rehuseoksista saatu konsentroitu sedimentti sisältää yleensä maaeläinten luupartikkelien ja kalanluupartikkelien lisäksi muita raskaita partikkeleita kuten mineraaleja, hiekkaa, ligniiniä sisältäviä kasvinosia yms.
7.1 Luupartikkelien osuuden arvioitu määrä
prosenttia maaeläinten luupartikkeleita = (S × c) / W
prosenttia kalanluu- ja suomupartikkeleita = (S × d) / W
|
(S = |
sedimentin paino (mg), c = korjauskerroin (%) sedimentin sisältämien maaeläimistä peräisin olevien luiden arvioitua osuutta varten, d = korjauskerroin (%) sedimentin sisältämien kalanruotojen ja suomujen arvioitua osuutta varten ja W = sedimentointia varten punnittu näyte (mg)). |
7.2 Eläinperäisten ainesosien arvioitu osuus
Luun osuus eläinkunnan tuotteissa voi vaihdella paljon. (Luujauho sisältää luuta tavallisesti 50–60 % ja lihajauho 20–30 %; kalajauhossa luiden ja suomujen osuus vaihtelee kalajauhon tyypin ja alkuperän mukaan ja on tavallisesti 10–20 %.)
Jos näytteen sisältämän eläinperäisen jauhon tyyppi tiedetään, sen osuus näytteestä voidaan arvioida:
Maaeläinperäisten ainesosien arvioitu osuus (%) = (S × c) / (W × f) × 100
Kalatuotteiden ainesosien arvioitu osuus (%) = (S × d) / (W × f) × 100
|
(S = |
sedimentin paino (mg), c = korjauskerroin (%) sedimentin sisältämien maaeläimistä peräisin olevien luiden arvioitua osuutta varten, d = korjauskerroin (%) sedimentin sisältämien kalanruotojen ja suomujen arvioitua osuutta varten, f = korjauskerroin näytteen eläinperäisten ainesosien sisältämien luiden osuutta varten ja W = sedimentointia varten punnittu näyte (mg)). |
8. Tutkimuksen tulosten esittäminen
Raportissa on vähintään esitettävä tieto maaeläimistä ja kalajauhosta peräisin olevien ainesosien esiintymisestä. Tulokset esitetään seuraavasti:
|
8.1 |
Maaeläimistä peräisin olevien ainesosien esiintyminen:
|
|
8.2 |
Kalajauhon esiintyminen:
Jos kaloista tai maaeläimistä peräisin olevia ainesosia todetaan, tutkimusraportissa voidaan tarvittaessa esittää arvio havaittujen ainesosien määrästä (x %, < 0,1 %, 0,1–0,5 %, 0,5–5 % tai > 5 %), ja täsmentää — jos mahdollista — kyseessä olevan maaeläimen tyyppi ja havaitut eläinperäiset ainesosat (lihassäikeet, rusto, luu, sarvet, karvat, harjakset, höyhenet, veri, munankuoret, kalanluut, suomut). Esitettäessä arvio eläinperäisten ainesosien määrästä käytetty korjauskerroin f on mainittava. Kun tunnistetaan maaeläinten luuainesta, raporttiin on lisättävä seuraava maininta: ”Ainesosat voivat olla peräisin nisäkkäistä.” Tämä maininta ei ole tarpeen, jos maaeläinten luupartikkelit on täsmennetty siipikarjan tai nisäkkäiden luupartikkeleiksi. |
9. Vaihtoehtoinen menettely rasvan tai öljyn analysointiin
Seuraavaa menettelyä voidaan käyttää rasvan tai öljyn analysoinnissa:
|
— |
Jos rasva on kiinteää, sitä lämmitetään esimerkiksi mikroaaltouunissa, kunnes se muuttuu nestemäiseksi. |
|
— |
40 ml rasvaa pipetoidaan näytteen pohjaosasta sentrifugiputkeen. |
|
— |
Sentrifugoidaan 4 000 kierrosta minuutissa (rpm) 10 minuuttia. |
|
— |
Jos rasva on jähmettynyt sentrifugoinnin aikana, sitä lämmitetään taas mikroaaltouunissa, kunnes se muuttuu nestemäiseksi. Sentrifugoidaan uudestaan 4 000 kierrosta minuutissa (rpm) 5 minuuttia. |
|
— |
Puolet dekantoiduista epäpuhtauksista siirretään pienellä lusikalla tai spaattelilla petrimaljaan tai objektilasille mahdollisten eläinperäisten ainesosien (lihassäikeiden, höyhenten, luunpalasten jne.) mikroskooppista tutkimusta varten. Mikroskopoinnissa käytettäväksi petausaineeksi suositellaan parafiiniöljyä tai glyserolia. |
|
— |
Loput epäpuhtauksista käytetään sedimentointiin 6.2 kohdassa kuvatulla tavalla. |
(1) EUVL L 165, 30.4.2004, s. 1, oikaisu EUVL L 191, 28.5.2004, s. 1.
LIITE VII
SIIPIKARJAN REHUJEN ENERGIA-ARVON LASKENTAMENETELMÄ
1. Energia-arvon laskentamenetelmä ja ilmoittaminen
Siipikarjan rehuseosten energia-arvo on laskettava jäljempänä olevan kaavan mukaisesti tiettyjen rehun ravintoaineiden prosenttiosuuksien perusteella. Energia-arvo ilmoitetaan typen suhteen korjattuna muuntokelpoisena energiana (ME) käyttäen yksikköä megajoule (MJ) rehuseoskiloa kohti:
MJ/kg ME:tä = 0,1551 × raakavalkuaisprosentti +0,3431 × raakarasvaprosentti +0,1669 × tärkkelysprosentti +0,1301 × kokonaissokeriprosentti (sakkaroosina ilmaistuna).
2. Sallitut poikkeamat ilmoitetuista energia-arvoista
Jos virallisessa tarkastuksessa käy ilmi, että tarkastuksen perusteella saatu ja ilmoitettu energia-arvo poikkeavat toisistaan (pienempi tai suurempi kuin ilmoitettu energia-arvo), sallitaan ME:ssä 0,4 MJ/kg:n suuruinen enimmäispoikkeama.
3. Tuloksen ilmoittaminen
Edellä olevan laskukaavan mukaisesti saatu energia-arvo ilmoitetaan yhden desimaalin tarkkuudella.
4. Näytteenotto- ja määritysmenetelmät
Näytteenotto rehuseoksesta ja laskentakaavassa esiintyvien ravintoainepitoisuuksien määrittäminen on suoritettava yhteisön näytteenottomenetelmien ja virallisen rehuvalvonnan määritysmenetelmien mukaisesti.
Seuraavia menetelmiä on käytettävä:
|
— |
raakarasvapitoisuuden määrittäminen: raakarasvan ja -öljyn määrityksessä käytettävä menettelytapa B sellaisena kuin se on vahvistettu liitteessä III olevassa H osassa, |
|
— |
tärkkelyspitoisuuden määrittäminen: polarimetrinen menetelmä, joka on vahvistettu liitteessä III olevassa L osassa. |
LIITE VIII
MÄÄRITYSMENETELMÄT SELLAISTEN LISÄAINEIDEN OSOITTAMISEKSI, JOTKA EIVÄT ENÄÄ OLE SALLITTUJA REHUISSA
Tärkeä huomautus:Sellaisten lisäaineiden osoittamiseksi, jotka eivät enää ole sallittuja rehuissa, voidaan käyttää tässä liitteessä vahvistettuja määritysmenetelmiä herkempiä menetelmiä.Tässä liitteessä esitettyjä määritysmenetelmiä on käytettävä varmistustarkoituksiin
A. METYYLIBENTSOKVAATTIPITOISUUDEN MÄÄRITYS
7-Bentsyloksi-6-butyyli-3-metoksikarbonyyli-4-kinoloni
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehujen metyylibentsokvaattipitoisuus. Määritysraja on 1 mg/kg.
2. Periaate
Metyylibentsokvaatti uutetaan näytteestä metanolisella metaanisulfonihappoliuoksella. Uute puhdistetaan dikloorimetaanilla, sitten ioninvaihtokromatografialla ja sen jälkeen uudelleen dikloorimetaanilla. Metyylibentsokvaattipitoisuus määritetään korkean erotuskyvyn käänteisfaasinestekromatografialla (HPLC) ja UV-detektorilla.
3. Reagenssit
|
3.1 |
Dikloorimetaani. |
|
3.2 |
Metanoli, HPLC-laatua vastaava. |
|
3.3 |
HPLC:n liikkuva faasi. Metanolin (3.2) ja veden (HPLC-laatua vastaava) seos 75 + 25 (v + v). Suodatetaan 0,22 μm:n suodattimen (4.5) läpi ja poistetaan liuoksesta kaasut (esimerkiksi 10 minuutin ultraäänikäsittelyllä). |
|
3.4 |
Metaanisulfonihappoliuos, c = 2 %. Laimennetaan 20,0 ml metaanisulfonihappoa 1 000 ml:ksi metanolilla (3.2). |
|
3.5 |
Kloorivetyhappoliuos, c = 10 %. Laimennetaan 100 ml kloorivetyhappoa (ρ20 noin 1,18 g/ml) 1 000 ml:ksi vedellä. |
|
3.6 |
Kationinvaihtohartsi Amberlite CG-120 (Na), 100–200 mesh. Hartsi esikäsitellään ennen käyttöä: sekoitetaan 100 g hartsia 500 ml:aan kloorivetyhappoliuosta (3.5) ja saatetaan seos kiehuvaksi lämpölevyllä koko ajan sekoittaen. Annetaan jäähtyä ja dekantoidaan happo pois. Suodatetaan tyhjiössä suodatinpaperin läpi. Pestään hartsi kahdesti 500 ml:n annoksella vettä ja sen jälkeen 250 ml:lla metanolia (3.2). Huuhdellaan hartsi uudelleen 250 ml:lla metanolia (3.2) ja kuivataan johtamalla ilmavirta suodatuskakun läpi. Kuivattu hartsi säilytetään suljetussa pullossa. |
|
3.7 |
Standardiyhdiste: puhdas metyylibentsokvaatti (7-bentsyloksi-6-butyyli-3-metoksikarbonyyli-4-kinoloni). |
|
3.7.1 |
Punnitaan 50 mg standardiyhdistettä (3.7) 0,1 mg:n tarkkuudella, liuotetaan metaanisulfonihappoliuokseen (3.4) 100 ml:n mittapullossa, täytetään merkkiin asti ja sekoitetaan. |
|
3.7.2 |
Siirretään 5,0 ml standardin kantaliuosta (3.7.1) 50 ml:n mittapulloon, täytetään merkkiin asti metanolilla (3.2) ja sekoitetaan. |
|
3.7.3 |
Siirretään 25 ml:n mittapulloihin 1,0, 2,0, 3,0, 4,0 ja 5,0 ml standardin välimuotoliuosta (3.7.2). Täytetään merkkiin asti liikkuvalla faasilla (3.3) ja sekoitetaan. Näiden liuosten metyylibentsokvaattipitoisuudet ovat vastaavasti 2,0, 4,0, 6,0, 8,0 ja 10,0 μg/ml. Liuokset on valmistettava juuri ennen käyttöä. |
4. Välineistö
|
4.1 |
Laboratorioravistelija. |
|
4.2 |
Pyörivä kalvohaihdutin. |
|
4.3 |
Lasikolonni (250 mm × 15 mm), jossa on hana ja noin 200 ml:n säiliö. |
|
4.4 |
HPLC-laitteisto, jossa on vaihtuva-aallonpituuksinen UV-detektori tai diodirividetektori. |
|
4.4.1 |
Nestekromatografiakolonni: 300 mm x 4 mm, C18, hiukkaskoko 10 μm, tai vastaava. |
|
4.5 |
Kalvosuodattimia, 0,22 μm. |
|
4.6 |
Kalvosuodattimia, 0,45 μm. |
5. Menettely
5.1 Yleistä
|
5.1.1 |
Nollarehu on analysoitava, jotta varmistetaan, ettei se sisällä metyylibentsokvaattia tai mittausta häiritseviä aineita. |
|
5.1.2 |
Saantokoe on suoritettava analysoimalla nollarehu, johon on tehty näytteen metyylibentsokvaattipitoisuutta vastaava lisäys. Pitoisuuden 15 mg/kg aikaan saamiseksi lisätään 600 μl standardin kantaliuosta (3.7.1) 20 grammaan nollarehua, sekoitetaan ja annetaan seistä 10 minuuttia ennen uuttamista (5.2). Huomautus: Tätä menetelmää käytettäessä nollarehun on vastattava tyypiltään näytettä, ja metyylibentsokvaattia ei saa havaita määrityksessä. |
5.2 Uutto
Punnitaan 0,01 g:n tarkkuudella noin 20 g valmistettua näytettä ja siirretään 250 ml:n erlenmeyerkolviin. Lisätään 100,0 ml metaanisulfonihappoa (3.4) ja ravistellaan mekaanisesti (4.1) 30 minuutin ajan. Suodatetaan liuos suodatinpaperin läpi ja suodos säilytetään neste-neste-erotusta (5.3) varten.
5.3 Neste-neste-erotus
Siirretään 25 ml suodosta (5.2) 500 ml:n erotussuppiloon, joka sisältää 100 ml kloorivetyhappoliuosta (3.5). Lisätään 100 ml dikloorimetaania (3.1) suppiloon ja ravistellaan yhden minuutin ajan. Faasien erottumisen jälkeen valutetaan alempi faasi (dikloorimetaani) 500 ml:n pyörökolviin. Toistetaan vesifaasin uutto vielä kahdella 40 ml:n annoksella dikloorimetaania ja yhdistetään nämä ensimmäiseen uutteeseen pyörökolvissa. Dikloorimetaaniuute haihdutetaan täysin kuivaksi pyöröhaihduttimessa (4.2), joka toimii 40 oC:n lämpötilassa ja alennetussa paineessa. Liuotetaan jäännös 20–25 ml:aan metanolia (3.2), suljetaan kolvi tulpalla ja säilytetään koko uute ioninvaihtokromatografiaa (5.4) varten.
5.4 Ioninvaihtokromatografia
5.4.1
Laitetaan pieni tuppo lasivillaa lasikolonnin (4.3) alapäähän. Valmistetaan 5 grammasta käsiteltyä kationinvaihtohartsia (3.6) ja 50 ml:sta vetykloridihappoa (3.5) seos, joka kaadetaan kolonniin; annetaan asettua. Annetaan ylimääräisen hapon valua kolonnista siten, että sen pinta jää juuri ja juuri hartsipinnan yläpuolelle ja pestään kolonni vedellä, kunnes ulos virtaava liuos on lakmuspaperilla mitattuna neutraalia. Siirretään 50 ml metanolia (3.2) kolonniin ja annetaan valua, kunnes se saavuttaa hartsipinnan.
5.4.2
Pipetoidaan saatua uutetta (5.3) varovasti kolonniin. Huuhdellaan pyörökolvi kahdella 5–10 ml:n annoksella metanolia (3.2) ja siirretään nämä pesuliuokset kolonniin. Annetaan uutteen valua hartsipintaan ja pestään kolonni 50 ml:lla metanolia valvoen, ettei virtausnopeus ylitä 5 ml:aa minuutissa. Heitetään ulos valunut liuos pois. Metyylibentsokvaatti eluoidaan kolonnista käyttäen 150 ml:aa metaanisulfonihappoliuosta (3.4) ja kerätään eluaatti kolonnista 250 ml:n erlenmeyerkolviin.
5.5 Neste-neste-erotus
Siirretään saatu eluaatti (5.4.2) 1 litran erotussuppiloon. Huuhdellaan erlenmeyerkolvi 5–10 ml:lla metanolia (3.2) ja yhdistetään pesuliuos erotussuppilon sisällön kanssa. Lisätään 300 ml kloorivetyhappoliuosta (3.5) ja 130 ml dikloorimetaania (3.1). Ravistellaan yhden minuutin ajan ja annetaan faasien erottua. Valutetaan alempi faasi (dikloorimetaani) 500 ml:n pyörökolviin. Toistetaan vesifaasin uutto kahdella uudella 70 ml:n annoksella dikloorimetaania ja yhdistetään nämä uutteet ensimmäisen, pyörökolvissa olevan uutteen kanssa.
Dikloorimetaaniuute haihdutetaan täysin kuivaksi pyöröhaihduttimessa (4.2), joka toimii 40 oC:n lämpötilassa ja alennetussa paineessa. Liuotetaan kolvin jäännös noin 50 ml:aan metanolia (3.2) ja siirretään tämä liuos kvantitatiivisesti 10 ml:n mittapulloon. Pyörökolvi huuhdellaan kahdella uudella 1–2 ml:n annoksella metanolia ja siirretään liuos mittapulloon. Täytetään merkkiin asti metanolilla ja sekoitetaan. Suodatetaan määräosa kalvosuodattimen (4.6) läpi. Tämä liuos säilytetään HPLC-määritystä (5.6) varten.
5.6 HPLC-määritys
5.6.1
Seuraavat olosuhteet annetaan viitteeksi; muita olosuhteita voidaan käyttää, jos niillä saadaan vastaavat tulokset.
|
— |
nestekromatografiakolonni (4.4.1), |
|
— |
HPLC:n liikkuva faasi: metanoli-vesiseos (3.3), |
|
— |
virtausnopeus: 1–1,5 ml/minuutti, |
|
— |
detektioaallonpituus: 265 nm, |
|
— |
injektiotilavuus: 20–50 μl. |
Kromatografiajärjestelmän stabiilisuus on tarkastettava injektoimalla useita kertoja kalibrointiliuosta (3.7.3), jonka pitoisuus on 4 μg/ml, kunnes saadaan piikkejä, joiden korkeudet (pinta-alat) ja retentioajat pysyvät vakioina.
5.6.2
Injektoidaan useita kertoja kutakin kalibrointiliuosta (3.7.3) ja mitataan kunkin pitoisuuden piikkien korkeudet (pinta-alat). Piirretään kalibrointikäyrä siten, että y-akselilla ovat kalibrointiliuosten piikkien korkeuksien (pinta alojen) keskiarvot ja x-akselilla vastaavat konsentraatiot (μg/ml).
5.6.3
Injektoidaan useita kertoja näyteuutetta (5.5) käyttäen samaa tilavuutta kuin kalibrointiliuoksille, ja määritetään metyylibentsokvaattipiikkien korkeuden (pinta-alan) keskiarvo.
6. Tulosten laskeminen
Näyteliuoksen metyylibentsokvaattipiikkien korkeuden (pinta-alan) keskiarvosta määritetään näyteliuoksen pitoisuus μg/ml:na käyttäen kalibrointikäyrää (5.6.2).
Näytteessä olevan metyylibentsokvaatin määrä w (mg/kg) lasketaan seuraavalla kaavalla:
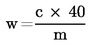
jossa:
|
c |
= |
näyteliuoksen metyylibentsokvaattipitoisuus μg/ml:na, |
|
m |
= |
näyte-erän paino grammoina. |
7. Tulosten validointi
7.1 Tunnistaminen
Tutkittavan aineen tunnistaminen voidaan varmentaa yhteiskromatografialla tai diodirividetektorilla, joilla näyteuutteen ja 10 μg/ml metyylibentsokvaattia sisältävän kalibrointiliuoksen (3.7.3) spektrejä voidaan verrata.
7.1.1
Näyteuutteeseen lisätään sopiva määrä standardin välimuotoliuosta (3.7.2). Lisätyn metyylibentsokvaatin määrän on vastattava näyteuutteessa olevan metyylibentsokvaatin arvioitua määrää.
Ainoastaan metyylibentsokvaatin piikin korkeus saa kasvaa, kun otetaan huomioon sekä lisätty määrä että uutteen laimeneminen. Piikin leveyden on oltava sen puolikorkeudella noin 10 % sen alkuperäisestä leveydestä.
7.1.2
Tulokset arvioidaan seuraavien arviointiperusteiden mukaisesti:
|
a) |
kromatogrammin piikin huipun kohdalla näytteen ja standardin spektrien maksimiabsorptioaallonpituuksien on oltava samat ilmaisinjärjestelmän erotuskyvyn mukaan määräytyvissä rajoissa. Diodirividetektorimäärityksessä tämä on yleensä noin ± 2 nm; |
|
b) |
välillä 220–350 nm kromatogrammin piikin huipun kohdalla ajetut näyte- ja standardispektrit eivät saa erota niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat ja näiden kahden spektrin välillä havaittu poikkeama ei ole missään kohdassa yli 15 %:a standardina olevan tutkittavan aineen absorbanssista; |
|
c) |
välillä 220–350 nm näyteliuoksesta ajetut spektrit piikin nousun, huipun ja laskun kohdalla eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun samat maksimit esiintyvät ja kun kaikissa havaittavissa kohdissa spektrien välinen poikkeama ei ole suurempi kuin 15 % huipun spektrin absorbanssista. |
Jos jokin näistä edellytyksistä ei täyty, tutkittavan aineen esiintymistä ei ole vahvistettu.
7.2 Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ero ei saa olla suurempi kuin 10 % suuremmasta tuloksesta, kun metyylibentsokvaattipitoisuudet ovat välillä 4–20 mg/kg.
7.3 Saanto
Sellaisen nollanäytteen saannon, johon on lisätty määritettävää ainetta, on oltava vähintään 90 %.
8. Laboratorioiden välisen vertailun tulokset
Laboratorioiden välisessä tutkimuksessa analysoitiin viisi näytettä 10 laboratoriossa. Kukin näyte määritettiin kaksi kertaa.
|
|
Nollanäyte |
Jauho 1 |
Rae 1 |
Jauho 2 |
Rae 2 |
|
Keskiarvo [mg/kg] |
Ei hav. |
4,50 |
4,50 |
8,90 |
8,70 |
|
sr [mg/kg] |
- |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [ %] |
- |
6,70 |
4,40 |
6,70 |
5,70 |
|
sR [mg/kg] |
- |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [ %] |
- |
8,90 |
11,10 |
10,10 |
11,50 |
|
Saanto [ %] |
- |
92,00 |
93,00 |
92,00 |
89,00 |
|
Ei hav. |
= |
Ei havaittu |
|
sr |
= |
toistettavuuden keskihajonta |
|
CVr |
= |
toistettavuuden variaatiokerroin, % |
|
sR |
= |
uusittavuuden keskihajonta |
|
CVR |
= |
uusittavuuden variaatiokerroin, %. |
B. OLAKVINDOKSIN MÄÄRITYS
(2-[N-2′-(hydroksietyyli)karbamoyyli]-3-metyylikinoksaliini-N 1 ,N 4 -dioksidi
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää rehujen olakvindoksipitoisuus. Määritysraja on 5 mg/kg.
2. Periaate
Näyte uutetaan vesi-metanoliseoksella. Olakvindoksipitoisuus määritetään korkean erotuskyvyn käänteisfaasinestekromatografialla (HPLC) UV-detektoria käyttäen.
3. Reagenssit
|
3.1 |
Metanoli. |
|
3.2 |
Metanoli, HPLC-laatua vastaava. |
|
3.3 |
Vesi, HPLC-laatua vastaava. |
|
3.4 |
HPLC:ssä käytettävä liikkuva faasi. Vesi (3.3)-metanoli (3.2)-seos, 900 + 100 (V + V). |
|
3.5 |
Standardiyhdiste: puhdas olakvindoksi, 2-[N-2′-(hydroksietyyli)karbamoyyli]-3-metyylikinoksaliini-N1,N4-dioksidi, E 851. |
|
3.5.1 |
Punnitaan 50 mg olakvindoksia (3.5) 0,1 mg:n tarkkuudella 200 ml:n mittapulloon ja lisätään noin 190 ml vettä. Asetetaan pullo 20 minuutiksi ultraäänihauteeseen (4.1). Ultraäänikäsittelyn jälkeen liuoksen annetaan jäähtyä huoneenlämpötilaan, täytetään merkkiin asti vedellä ja sekoitetaan. Kääritään pullo alumiinifolioon ja säilytetään jääkaapissa. Liuos on valmistettava kuukausittain. |
|
3.5.2 |
Pipetoidaan 10,0 ml kantaliuosta (3.5.1) 100 ml:n mittapulloon, täytetään merkkiin asti liikkuvalla faasilla (3.4) ja sekoitetaan. Kääritään pullo alumiinifolioon ja säilytetään jääkaapissa. Liuos on valmistettava päivittäin. |
|
3.5.3 |
Pipetoidaan 50 ml:n mittapulloihin 1,0, 2,0, 5,0, 10,0, 15,0 ja 20,0 ml standardin välimuotoliuosta (3.5.2). Täytetään merkkiin asti liikkuvalla faasilla (3.4) ja sekoitetaan. Kääritään pullot alumiinifolioon. Nämä liuokset sisältävät vastaavasti 0,5, 1,0, 2,5, 5,0, 7,5 ja 10,0 μg/ml olakvindoksia. Liuokset on valmistettava päivittäin. |
4. Välineistö
|
4.1 |
Ultraäänihaude. |
|
4.2 |
Mekaaninen ravistelija. |
|
4.3 |
HPLC-laitteisto, jossa on vaihtuva-aallonpituuksinen UV-detektori tai diodirividetektori. |
|
4.3.1 |
Nestekromatografiakolonni, 250 mm x 4 mm, C18, hiukkaskoko 10 μm, tai vastaava. |
|
4.4 |
Kalvosuodattimia, 0,45 μm. |
5. Menettely
|
Huomautus: |
Olakvindoksi on valoherkkää. Kaikki toimenpiteet on tehtävä himmennetyssä valaistuksessa tai ruskeita lasiastioita käyttäen. |
5.1 Yleistä
|
5.1.1 |
Nollanäyte on analysoitava, jotta varmistetaan, ettei se sisällä olakvindoksia tai mittausta häiritseviä aineita. |
|
5.1.2 |
Saantokoe suoritetaan analysoimalla nollanäyte, johon on lisätty näytteessä esiintyvää määrää vastaava määrä olakvindoksia. Lisäyksen saamiseksi tasolle 50 mg/kg pipetoidaan 10,0 ml standardin kantaliuosta (3.5.1) 250 ml:n erlenmeyerkolviin ja haihdutetaan liuos noin 0,5 ml:ksi. Lisätään 50 g nollanäytettä, sekoitetaan huolellisesti ja annetaan tasoittua 10 min sekoittaen useita kertoja ennen uuttovaiheeseen siirtymistä (5.2).
|
5.2 Uutto
Punnitaan noin 50 g näytettä 0,01 g:n tarkkuudella. Siirretään 1 000 ml:n erlenmeyerkolviin, lisätään 100 ml metanolia (3.1) ja asetetaan kolvi 5 minuutiksi ultraäänihauteeseen (4.1). Lisätään 410 ml vettä ja annetaan olla ultraäänihauteessa vielä 15 minuuttia. Otetaan kolvi ultraäänihauteesta, ravistellaan 30 minuutin ajan ravistelijassa (4.2) ja suodatetaan taitellun suodatinpaperin läpi. Pipetoidaan 10,0 ml suodosta 20 ml:n mittapulloon, täytetään merkkiin asti vedellä ja sekoitetaan. Suodatetaan näyte kalvosuodattimen (4.4) läpi (ks. huomautus 9). Jatketaan HPLC-määrityksellä (5.3).
5.3 HPLC-määritys
5.3.1
Seuraavat ajo-olosuhteet ovat ohjeellisia ja niitä voidaan muuttaa, mikäli ne antavat vastaavanlaiset tulokset.
|
Analyysikolonni (4.3.1) |
|
|
Liikkuva faasi (3.4): |
vesi (3.3) — metanoli (3.2.) seos, 900 + 100 (V + V) |
|
Virtausnopeus: |
1,5–2 ml/min |
|
Detektioaallonpituus: |
380 nm |
|
Injektiotilavuus: |
20–100 μl. |
Kromatografiajärjestelmän stabiilisuus tarkistetaan injektoimalla useita kertoja 2,5 μg/ml olakvindoksia sisältävää kalibrointiliuosta (3.5.3), kunnes piikkien korkeudet ja retentioajat pysyvät vakioina.
5.3.2
Injektoidaan kutakin kalibrointiliuosta (3.5.3) useita kertoja ja lasketaan kutakin konsentraatiota vastaavat piikkien korkeuksien (pinta-alojen) keskiarvot. Piirretään kalibrointikäyrä siten, että y-akselilla ovat kalibrointiliuosten piikkien korkeuksien (pinta alojen) keskiarvot ja x-akselilla vastaavat konsentraatiot (μg/ml).
5.3.3
Injektoidaan useita kertoja näyteliuosta (5.2) sama määrä kuin kalibrointiliuoksia ja lasketaan olakvindoksipiikkien korkeuden (pinta-alan) keskiarvo.
6. Tulosten laskeminen
Näyteliuoksen olakvindoksipitoisuus (μg/ml) lasketaan olakvindoksin piikkien korkeuden (pinta-alan) keskiarvosta kalibrointikäyrän (5.3.2) avulla.
Näytteen olakvindoksipitoisuus w (mg/kg) saadaan seuraavasta kaavasta:
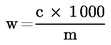
jossa:
|
c |
= |
näyteliuoksen (5.2) olakvindoksipitoisuus (μg/ml), |
|
m |
= |
näyte-erän paino grammoina (5.2). |
7. Tulosten validointi
7.1 Tutkittavan aineen tunnistaminen
Tutkittavan aineen tunnistaminen voidaan varmentaa yhteiskromatografialla tai diodirividetektorilla vertaamalla näyteliuoksen (5.2) ja 5,0 μg/ml olakvindoksia sisältävän kalibrointiliuoksen (3.5.3) spektrejä.
7.1.1
Näyteliuokseen (5.2) lisätään sopiva määrä kalibrointiliuosta (3.5.3). Lisätyn olakvindoksimäärän on oltava samaa luokkaa kuin näyteliuoksesta analysoitu olakvindoksin määrä.
Vain olakvindoksipiikin korkeus saa kasvaa, kun otetaan huomioon sekä lisätty määrä että olakvindoksin näyteliuoksen laimeneminen. Piikin leveyden puolivälissä sen korkeutta on oltava ± 10 prosenttia alkuperäisen näyteliuoksen olakvindoksipiikin leveydestä.
7.1.2
Tulokset arvioidaan seuraavien arviointiperusteiden mukaisesti:
|
a) |
Näytteen ja standardin spektrien maksimiabsorption aallonpituuden, mitattuna kromatogrammin piikin kärjestä, on oltava sama detektorin erotuskyvyn määräämissä rajoissa. Diodirividetektorilla tämä on tyypillisesti ± 2 nm. |
|
b) |
Välillä 220–400 nm näytteen ja standardin spektrit, kromatogrammin piikin kärjestä mitattuna, eivät saa erota niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat eikä spektrien välinen ero ole missään kohdassa suurempi kuin 15 % standardianalyytin absorbanssista. |
|
c) |
Välillä 220–400 nm näyteuutteesta ajetut spektrit piikin nousun, huipun ja laskun kohdalla eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat eikä spektrien välinen ero ole missään kohdassa suurempi kuin 15 % piikin huipun spektrin absorbanssista. |
Jos jokin näistä edellytyksistä ei täyty, tutkittavan aineen esiintymistä ei ole vahvistettu.
7.2 Toistettavuus
Kun olakvindoksipitoisuus on 10–200 mg/kg, näytteen kahden rinnakkaisen määrityksen välinen ero ei saa olla suurempi kuin 15 % suuremmasta rinnakkaistuloksesta.
7.3 Saanto
Sellaisen nollanäytteen saannon, johon on lisätty määritettävää ainetta, on oltava vähintään 90 %.
8. Laboratorioiden välisen vertailun tulokset
EY:n laboratorioiden välisessä menetelmävertailussa 13 laboratoriossa analysoitiin neljä porsaanrehunäytettä, joista yksi oli nollanäyte. Tulokset olivat seuraavat:
|
|
Näyte 1 |
Näyte 2 |
Näyte 3 |
Näyte 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
keskiarvo [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
nimellispitoisuus |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
saanto % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
yksittäisten arvojen lukumäärä |
|
Sr |
= |
toistettavuuden keskihajonta |
|
SR |
= |
uusittavuuden keskihajonta |
|
CVr |
= |
toistettavuuden variaatiokerroin |
|
CVR |
= |
uusittavuuden variaatiokerroin. |
9. Huomautus
Vaikka menetelmää ei ole validoitu rehuille, joissa on yli 100 mg/kg olakvindoksia, sillä on mahdollista saada tyydyttäviä tuloksia käyttämällä pienempää näytemäärää ja/tai laimentamalla näyteliuosta (5.2), jotta päästäisiin kalibrointikäyrän pitoisuusalueelle (5.3.2).
C. AMPROLIUMIN MÄÄRITYS
1 [(4-amino-2-propyylipyrimidin-5-yyli)metyyli]-2-metyylipyridiniumkloridihydrokloridi
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää amproliumin pitoisuus rehuissa ja esiseoksissa. Toteamisraja on 1 mg/kg ja määritysraja 5 mg/kg.
2. Periaate
Näyte uutetaan metanoli-vesiseoksella. Uute laimennetaan liikkuvalla faasilla, suodatetaan kalvosuodattimella, ja amprolium määritetään korkean erotuskyvyn kationinvaihtonestekromatografialla (HPLC) ja UV-detektorilla.
3. Reagenssit
|
3.1 |
Metanoli. |
|
3.2 |
Asetonitriili, HPLC-laatua vastaava. |
|
3.3 |
Vesi, HPLC-laatua vastaava. |
|
3.4 |
Natriumdivetyfosfaattiliuos, c = 0,1 mol/l. Liuotetaan 13,80 g natriumdivetyfosfaattimonohydraattia veteen (3.3) 1 000 ml:n mittapullossa, täytetään merkkiin asti vedellä (3.3) ja sekoitetaan. |
|
3.5 |
Natriumperkloraattiliuos, c = 1,6 mol/l. Liuotetaan 224,74 g natriumperkloraattimonohydraattia veteen (3.3) 1 000 ml:n mittapullossa, täytetään merkkiin asti vedellä (3.3) ja sekoitetaan. |
|
3.6 |
HPLC:ssä käytettävä liikkuva faasi (katso huomautus 9.1). Asetonitriilin (3.2), natriumdivetyfosfaattiliuoksen (3.4) ja natriumperkloraattiliuoksen (3.5) seos, 450 + 450 + 100 (v + v + v). Suodatetaan ennen käyttöä 0,22 μm:n kalvosuodattimella (4.3) ja poistetaan kaasut (esim. ultraäänihauteessa (4.4) vähintään 15 min). |
|
3.7 |
Standardiyhdiste: puhdas amproliumi, 1-[(4-amino-2-propyylipyrimidin-5-yyli)-metyyli]-2-metyylipyridi-niumkloridihydrokloridi, E 750 (katso kohta 9.2). |
|
3.7.1 |
Punnitaan 0,1 mg:n tarkkuudella 50 mg amproliumia (3.7) 100 ml:n mittapulloon, liuotetaan 80 ml:aan metanolia (3.1), ja pullo pannaan 10 minuutiksi ultraäänihauteeseen (4.4). Ultraäänikäsittelyn jälkeen liuoksen annetaan jäähtyä huoneenlämpötilaan, täytetään merkkiin asti vedellä ja sekoitetaan. Liuos säilyy ≤ 4 oC:n lämpötilassa yhden kuukauden. |
|
3.7.2 |
Pipetoidaan 5,0 ml standardin kantaliuosta (3.7.1) 50 ml:n mittapulloon, täytetään merkkiin asti uuttoliuoksella (3.8) ja sekoitetaan. Liuos säilyy ≤ 4 oC:n lämpötilassa yhden kuukauden. |
|
3.7.3 |
Siirretään 0,5, 1,0 ja 2,0 ml standardin välimuotoliuosta (3.7.2) 50 ml:n mittapulloihin. Täytetään merkkiin asti liikkuvalla faasilla (3.6) ja sekoitetaan. Nämä liuokset vastaavat 0,5, 1,0 ja 2,0 μg/ml amproliumia. Liuokset on valmistettava juuri ennen käyttöä. |
|
3.8 |
Uuttoliuos. Metanoli (3.1) -vesiseos 2 + 1 (v + v). |
4. Välineistö
|
4.1 |
HPLC-laitteisto, jossa voidaan injektoida 100 μl:n näytteitä. |
|
4.1.1 |
Nestekromatografiakolonni, 125 mm × 4 mm, kationinvaihto Nucleosil 10 SA, hiukkaskoko 5 μm tai 10 μm, tai vastaava. |
|
4.1.2 |
Vaihtuva-aallonpituuksinen UV-detektori tai diodirividetektori. |
|
4.2 |
Kalvosuodatin, PTFE:tä, 0,45 μm. |
|
4.3 |
Kalvosuodatin, 0,22 μm. |
|
4.4 |
Ultraäänihaude. |
|
4.5 |
Mekaaninen ravistelija tai magneettisekoitin. |
5. Menettely
5.1 Yleistä
5.1.1
Saantokokeen (5.1.2) toimivuuden määrittämiseksi on analysoitava nollarehu (nollanäyte), jotta varmistetaan, ettei se sisällä amproliumia tai mittausta häiritseviä aineita. Nollanäytteen on oltava näytteen kaltainen, eikä siinä saa olla havaittavia määriä amproliumia tai häiritseviä aineita.
5.1.2
Saanto on määritettävä analysoimalla nollanäyte, johon on lisätty näytteen amproliumia näytteen amproliumpitoisuutta vastaava määrä. Jotta saadaan 100 mg/kg amproliumia vastaava näyteliuos, siirretään 10,0 ml standardin kantaliuosta (3.7.1) 250 ml:n erlenmeyerkolviin, ja liuos haihdutetaan noin 0,5 ml:ksi. Lisätään 50 g nollanäytettä, sekoitetaan huolellisesti ja annetaan tasoittua 10 min sekoittaen useita kertoja ennen uuttovaiheeseen siirtymistä (5.2).
Jos saatavilla ei ole näytteen kaltaista nollanäytettä (katso 5.1.1), voidaan saantokoe suorittaa vaihtoehtoisesti standardinlisäysmenetelmällä. Tällöin analysoitavaan näytteeseen lisätään suunnilleen näytteiden jo sisältämä määrä amproliumia. Tämä näyte analysoidaan yhdessä varsinaisen näytteen kanssa, ja saanto voidaan laskea vähennyslaskulla.
5.2 Uutto
5.2.1
Punnitaan 0,01 gramman tarkkuudella 5–40 g näytettä, riippuen sen amproliumpitoisuudesta, 500 ml:n erlenmeyerkolviin ja lisätään 200 ml uuttoliuosta (3.8). Kolvi pannaan ultraäänihauteeseen (4.4) 15 minuutiksi. Otetaan kolvi ultraäänihauteesta, ravistellaan 1 minuutin ajan ravistelijassa tai sekoitetaan magneettisekoittimella (4.5). Uutteesta otettu erä laimennetaan liikkuvalla faasilla (3.6) siten, että sen amproliumpitoisuus on 0,5–2 μg/ml, ja sekoitetaan (katso 9.3). Suodatetaan 5–10 ml tätä laimennettua liuosta kalvosuodattimella (4.2). Tehdään HPLC-määritys (5.3).
5.2.2
Punnitaan 0,01 gramman tarkkuudella 1–4 g esiseosta, riippuen sen amproliumpitoisuudesta, 500 ml:n erlenmeyerkolviin ja lisätään 200 ml uuttoliuosta (3.8). Kolvi pannaan ultraäänihauteeseen (4.4) 15 minuutiksi. Otetaan kolvi ultraäänihauteesta, ravistellaan 1 minuutin ajan ravistelijassa tai sekoitetaan magneettisekoittimella (4.5). Uutteesta otettu erä laimennetaan liikkuvalla faasilla (3.6) siten, että sen amproliumpitoisuus on 0,5–2 μg/ml, ja sekoitetaan. Suodatetaan 5–10 ml tätä laimennettua liuosta kalvosuodattimella (4.2). Tehdään HPLC-määritys (5.3).
5.3 HPLC-määritys
5.3.1
Seuraavat ajo-olosuhteet ovat ohjeellisia ja niitä voidaan muuttaa, mikäli ne antavat vastaavanlaiset tulokset.
|
Nestekromatografiakolonni (4.1.1): |
125 mm × 4 mm, kationinvaihtaja Nucleosil 10 SA, hiukkaskoko 5 tai 10 μm, tai vastaava |
|
Liikkuva faasi (3.6): |
Asetonitriilin (3.2), natriumdivetyfosfaattiliuoksen (3.4) ja natriumperkloraattiliuoksen (3.5) seos, 450 + 450 + 100 (v + v + v). |
|
Virtausnopeus: |
0,7–1 ml/min |
|
Detektioaallonpituus: |
264 nm |
|
Injektiotilavuus: |
100 μl |
Kromatografiajärjestelmän stabiilisuus tarkistetaan injektoimalla useita kertoja kalibrointiliuosta (3.7.3), jonka konsentraatio on 1,0 μg/ml, kunnes piikkien korkeudet ja retentioajat pysyvät vakioina.
5.3.2
Injektoidaan kutakin kalibrointiliuosta (3.7.3) useita kertoja ja lasketaan kutakin konsentraatiota vastaavat piikkien korkeuksien (pinta-alojen) keskiarvot. Piirretään kalibrointikäyrä siten, että y-akselilla ovat kalibrointiliuosten piikkien korkeuksien (pinta alojen) keskiarvot ja x-akselilla vastaavat konsentraatiot (μg/ml).
5.3.3
Injektoidaan näyteliuosta (5.2) useita kertoja käyttäen samaa injektiotilavuutta kuin kalibrointiliuoksille ja määritetään amproliumpiikkien korkeuksien (pinta-alojen) keskiarvo.
6. Tulosten laskeminen
Näyteliuoksen amproliumkonsentraatio (μg/ml) lasketaan piikkien korkeuksien (pinta-alan) keskiarvosta kalibrointikäyrän (5.3.2) avulla.
Näytteen amproliumpitoisuus w (mg/kg) lasketaan seuraavan kaavan avulla:
 [mg/kg]
[mg/kg]
jossa:
|
V |
= |
uuttoliuoksen (3.8) tilavuus (ml) 5.2 kohdan mukaisesti (ts. 200 ml), |
|
c |
= |
näyteliuoksen (5.2) amproliumpitoisuus (μg/ml), |
|
f |
= |
5.2 kohdan mukainen laimennuskerroin, |
|
m |
= |
näyte-erän paino (g). |
7. Tulosten validointi
7.1 Tunnistaminen
Tutkittavan aineen tunnistaminen voidaan varmentaa yhteiskromatografialla tai käyttämällä diodirividetektoria, jolla verrataan näyteuutteen (5.2) ja 2,0 μg/ml sisältävän kalibrointiliuoksen (3.7.3) spektrejä.
7.1.1
Näyteuutteeseen (5.2) lisätään sopiva määrä kalibrointiliuosta (3.7.3). Lisätyn amproliumin määrän on oltava samaa suuruusluokkaa kuin näyteuutteessa todettu amproliumin määrä.
Ainoastaan amproliumin piikki saa kasvaa, kun otetaan huomioon sekä lisätty määrä että uutteen laimennus. Piikin leveyden puolivälissä sen korkeutta on oltava ± 10 % alkuperäisen näyteuutteen olakvindoksipiikin leveydestä.
7.1.2
Tulokset arvioidaan seuraavien arviointiperusteiden mukaisesti:
|
a) |
Kromatogrammin piikkien huipusta mitattujen näyte- ja standardispektrien maksimiabsorption aallonpituuksien on oltava samat ilmaisinjärjestelmän erotuskyvyn mukaan määräytyvissä rajoissa. Diodirividetektorilla tämä on tyypillisesti ± 2 nm. |
|
b) |
Välillä 210–320 nm kromatogrammin piikkien huipun kohdalla saadut näyte- ja standardispektrit eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat eikä spektrien välinen ero ole missään kohdassa suurempi kuin 15 % standardianalyytin absorbanssista. |
|
c) |
Välillä 210–320 nm näyteuutteesta saadut spektrit piikin nousun, huipun ja laskun kohdalla eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat eikä spektrien välinen ero ole missään kohdassa suurempi kuin 15 % piikin huipun spektrin absorbanssista. |
Jos jokin näistä edellytyksistä ei täyty, tutkittavan aineen esiintymistä ei ole vahvistettu.
7.2 Toistettavuus
Kahden rinnakkaismäärityksen välinen ero samasta näytteestä ei saa olla suurempi kuin
|
— |
15 % suuremmasta rinnakkaistuloksesta, kun amproliumpitoisuus on 25–500 mg/kg, |
|
— |
75 mg/kg, kun amproliumpitoisuus on 500–1 000 mg/kg, |
|
— |
75 % suuremmasta rinnakkaistuloksesta, kun amproliumpitoisuus on yli 1 000 mg/kg. |
7.3 Saanto
Sellaisen nollanäytteen saannon, johon on lisätty määritettävää ainetta, on oltava vähintään 90 %.
8. Laboratorioiden välisen vertailun tulokset
Eri laboratorioissa analysoitiin kolme siipikarjan rehua (näytteet 1–3), yksi kivennäisrehu (näyte 4) ja yksi esiseos (näyte 5). Tulokset esitetään seuraavassa taulukossa.
|
|
Näyte 1 (nollarehu) |
Näyte 2 |
Näyte 3 |
Näyte 4 |
Näyte 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
keskiarvo [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
nimellispitoisuus [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
yksittäisten arvojen lukumäärä |
|
sr |
= |
toistettavuuden keskihajonta |
|
CVr |
= |
toistettavuuden variaatiokerroin |
|
sR |
= |
uusittavuuden keskihajonta |
|
CVR |
= |
uusittavuuden variaatiokerroin. |
9. Huomautukset
|
9.1 |
Jos näytteessä on tiamiinia, tiamiinipiikki näkyy kromatogrammissa hiukan ennen amproliumpiikkiä. Tässä menetelmässä amproliumin ja tiamiinin on erotuttava toisistaan. Jos ne eivät erotu tässä menetelmässä käytetyllä kolonnilla (4.1.1), korvataan enintään 50 % liikkuvan faasin (3.6) asetonitriiliosuudesta metanolilla. |
|
9.2 |
British Pharmacopoeian mukaan kloorivetyhappoon (c = 0,1 mol/l) tehdyn amproliumliuoksen (c = 0,02 mol/l) spektrissä on maksimit aallonpituuksilla 246 nm ja 262 nm. Absorbanssin on saavutettava arvo 0,84 aallonpituudella 246 nm ja 0,80 aallonpituudella 262 nm. |
|
9.3 |
Uute on aina laimennettava liikkuvalla faasilla, koska muuten amproliumpiikin retentioaika voi muuttua merkittävästi ionivahvuuden muuttumisen johdosta. |
D. KARBADOKSIN MÄÄRITYS
Metyyli-3-(2-kinoksalinyylimetyleeni)-karbatsaatti-N 1 ,N 4 -dioksidi
1. Tarkoitus ja soveltamisala
Menetelmällä voidaan määrittää karbadoksin pitoisuus rehuissa ja esiseoksissa. Toteamisraja on 1 mg/kg. Määritysraja on 5 mg/kg.
2. Periaate
Näytteeseen lisätään vettä ja se uutetaan metanoli-asetonitriilillä. Rehuseosnäytteiden suodatetusta uutteesta puhdistetaan tietty tilavuusosa alumiinioksikolonnilla. Esiseosnäytteiden ja rehun lisäainevalmistenäytteiden suodatetusta uutteesta tietty tilavuusosa laimennetaan sopivalle pitoisuusalueelle vedellä, metanolilla ja asetonitriilillä. Karbadoksipitoisuus määritetään korkean erotuskyvyn käänteisfaasinestekromatografialla (HPLC) UV-detektoria käyttäen.
3. Reagenssit
|
3.1 |
Metanoli. |
|
3.2 |
Asetonitriili, HPLC-laatua vastaava. |
|
3.3 |
Etikkahappo, w = 100 %. |
|
3.4 |
Alumiinioksidi: neutraali, aktiivisuusaste I. |
|
3.5 |
Metanoli-asetonitriili 1 + 1 (v + v). Sekoitetaan 500 ml metanolia (3.1) ja 500 ml asetonitriiliä (3.2). |
|
3.6 |
Etikkahappo, σ = 10 %. Laimennetaan 10 ml etikkahappoa (3.3) 100 ml:aan asti vedellä. |
|
3.7 |
Natriumasetaatti. |
|
3.8 |
Vesi, HPLC-laatua vastaava. |
|
3.9 |
Asetaattipuskuriliuos, c = 0,01 mol/l, pH = 6,0. Liuotetaan 0,82 g natriumasetaattia (3.7) 700 ml:aan vettä (3.8) ja pH säädetään arvoon 6,0 etikkahapolla (3.6). Siirretään 1 000 ml:n mittapulloon, täytetään merkkiin asti vedellä (3.8) ja sekoitetaan. |
|
3.10 |
HPLC:ssä käytettävä liikkuva faasi. Sekoitetaan 825 ml asetaattipuskuriliuosta (3.9) ja 175 ml asetonitriiliä (3.2). Suodatetaan 0,22 μm:n suodattimen (4.5) läpi ja poistetaan liuoksesta kaasut (esimerkiksi 10 minuutin ultraäänikäsittelyllä). |
|
3.11 |
Standardiyhdiste. Puhdas karbadoksi: Metyyli-3-(2-kinoksalinyylimetyleeni)-karbatsaatti-N1,N4-dioksidi, E 850. |
|
3.11.1 |
Punnitaan 25 mg karbadoksia (3.11) 0,1 mg:n tarkkuudella 250 ml:n mittapulloon. Liuotetaan metanoli-asetonitriiliin (3.5) käsittelemällä ultraäänihauteessa (4.7). Ultraäänikäsittelyn jälkeen liuos jäähdytetään huoneen lämpötilaan, täytetään merkkiin asti metanoli-asetonitriilillä (3.5) ja sekoitetaan. Pullo kääritään alumiinifolioon tai käytetään tummaa pulloa ja säilytetään jääkaapissa. Liuos säilyy ≤ 4 oC:n lämpötilassa yhden kuukauden. |
|
3.11.2 |
Pipetoidaan 2,0, 5,0, 10,0 ja 20,0 ml standardin kantaliuosta (3.11.1) 100 ml:n mittapulloihin. Lisätään 30 ml vettä, täytetään merkkiin asti metanoli-asetonitriilillä (3.5) ja sekoitetaan. Kääritään pullot alumiinifolioon. Nämä liuokset sisältävät vastaavasti 2,0, 5,0, 10,0 ja 20,0 μg/ml karbadoksia. Kalibrointiliuokset on valmistettava juuri ennen käyttöä.
|
|
3.12 |
Vesi-[metanoli-asetonitriili] (3.5) -seos, 300 + 700 (v + v) Sekoitetaan 300 ml vettä ja 700 ml metanoli-asetonitriiliseosta (3.5). |
4. Välineistö
|
4.1 |
Laboratorioravistelija tai magneettisekoitin. |
|
4.2 |
Lasikuitusuodatinpaperia (Whatman GF/A tai vastaava). |
|
4.3 |
Lasikolonni (pituus 300–400 mm, sisähalkaisija noin 10 mm), jossa on sintterilasifritti ja poistohana.
|
|
4.4 |
HPLC-laitteisto, jossa voidaan injektoida 20 μl:n näytteitä. |
|
4.4.1 |
Nestekromatografiakolonni: 300 mm x 4 mm, C18, hiukkaskoko 10 μm, tai vastaava. |
|
4.4.2 |
Vaihtuva-aallonpituuksinen UV-detektori tai diodirividetektori, mittausaallonpituusalue 225–400 nm. |
|
4.5 |
Kalvosuodatin, 0,22 μm. |
|
4.6 |
Kalvosuodatin, 0,45 μm. |
|
4.7 |
Ultraäänihaude. |
5. Menettely
|
Huomautus: |
Karbadoksi on valonarkaa. Kaikki toimenpiteet on suoritettava himmennetyssä valaistuksessa, tai on käytettävä ruskeita tai alumiinifolioon käärittyjä lasitavaroita. |
5.1 Yleistä
5.1.1
Saantokokeen (5.1.2) suorittamista varten analysoidaan nollarehu, jotta varmistetaan, ettei se sisällä karbadoksia tai häiritseviä aineita. Nollarehun on oltava näytteen kaltainen, eikä siinä saa olla havaittavia määriä karbadoksia tai häiritseviä aineita.
5.1.2
Saantokoe suoritetaan analysoimalla nollarehu (5.1.1), johon on lisätty näytteessä esiintyvää määrää vastaava määrä karbadoksia. Lisäyksen saamiseksi tasolle 50 mg/kg karbadoksia pipetoidaan 5,0 ml standardin kantaliuosta (3.11.1) 200 ml:n erlenmeyerkolviin. Liuos haihdutetaan noin 0,5 ml:ksi typpivirrassa. Lisätään 10 g nollarehua, sekoitetaan ja odotetaan 10 minuuttia ennen siirtymistä uuttovaiheeseen (5.2).
Jos saatavilla ei ole näytteen kaltaista nollanäytettä (katso 5.1.1), voidaan saantokoe suorittaa vaihtoehtoisesti standardinlisäysmenetelmällä. Tällöin analysoitavaan näytteeseen lisätään suunnilleen näytteiden ennestään sisältämä määrä karbadoksia. Tämä näyte analysoidaan yhdessä varsinaisen näytteen kanssa, ja saanto voidaan laskea vähennyslaskulla.
5.2 Uutto
5.2.1
Punnitaan 10 g näytettä 0,01 gramman tarkkuudella ja siirretään 200 ml:n erlenmeyerkolviin. Lisätään 15,0 ml vettä, sekoitetaan ja annetaan seistä 5 minuuttia. Lisätään 35,0 ml metanoli-asetonitriiliseosta (3.5), suljetaan tulpalla ja ravistellaan 30 minuuttia ravistelijalla tai sekoitetaan magneettisekoittimella (4.1). Suodatetaan liuos lasikuitusuodatinpaperin läpi (4.2). Tämä liuos säilytetään puhdistusta (5.3) varten.
5.2.2
Punnitaan 1 g jauhamatonta näytettä 0,001 gramman tarkkuudella ja siirretään 200 ml:n erlenmeyerkolviin. Lisätään 15,0 ml vettä, sekoitetaan ja annetaan seistä 5 minuuttia. Lisätään 35,0 ml metanoli-asetonitriiliseosta (3.5), suljetaan tulpalla ja ravistellaan 30 minuuttia ravistelijalla tai sekoitetaan magneettisekoittimella (4.1). Suodatetaan liuos lasikuitusuodatinpaperin läpi (4.2).
Pipetoidaan määräosa suodoksesta 50 ml:n mittapulloon. Lisätään 15,0 ml vettä, täytetään merkkiin asti metanoli-asetonitriilillä (3.5) ja sekoitetaan. Lopullisen liuoksen karbadoksipitoisuuden pitää olla noin 10 μg/ml. Suodatetaan osa 0,45 μm:n kalvosuodattimen läpi (4.6).
Tehdään HPLC-määritys (5.4).
5.2.3
Punnitaan 0,2 g jauhamatonta näytettä 0,001 gramman tarkkuudella ja siirretään 250 ml:n erlenmeyerkolviin. Lisätään 45,0 ml vettä, sekoitetaan ja annetaan seistä 5 minuuttia. Lisätään 105,0 ml metanoli-asetonitriiliseosta (3.5), suljetaan tulpalla ja homogenoidaan. Pidetään näytettä ultraäänihauteessa (4.7) 15 minuuttia, minkä jälkeen liuosta ravistellaan tai sekoitetaan 15 minuuttia (4.1). Suodatetaan liuos lasikuitusuodatinpaperin läpi (4.2).
Laimennetaan määräosa suodosta vesi-metanoli-asetonitriiliseoksella (3.12) siten, että lopullisen liuoksen karbadoksipitoisuus on 10–15 μg/ml (10-prosenttisen lisäainevalmisteen laimennuskerroin on 10). Suodatetaan osa 0,45 μm:n kalvosuodattimen läpi (4.6).
Tehdään HPLC-määritys (5.4).
5.3 Puhdistus
5.3.1
Punnitaan 4 g alumiinioksidia (3.4) ja siirretään se lasikolonniin (4.3).
5.3.2
Lisätään 15 ml suodatettua uutetta (5.2.1) alumiinioksidikolonniin; ensimmäiset 2 ml eluaattia heitetään pois. Seuraavat 5 ml otetaan talteen ja suodatetaan osa 0,45 μm:n kalvosuodattimen läpi (4.6).
Tehdään HPLC-määritys (5.4).
5.4 HPLC-määritys
5.4.1
Seuraavat olosuhteet ovat ohjeellisia, ja myös muita muuttujia voidaan käyttää, jos niillä saadaan vastaavat tulokset:
|
Nestekromatografiakolonni (4.4.1): |
300 mm × 4 mm, C18, hiukkaskoko 10 μm, tai vastaava |
|
Liikkuva faasi (3.10): |
Asetaattipuskuriliuoksen (3.9) ja asetonitriilin (3.2) seos, 825 + 175 (v + v) |
|
Virtausnopeus: |
1,5–2 ml/min |
|
Detektioaallonpituus: |
365 nm |
|
Injektiotilavuus: |
20 μl. |
Kromatografiajärjestelmän stabiilisuus tarkistetaan injektoimalla useita kertoja 5,0 μg/ml karbadoksia sisältävää kalibrointiliuosta (3.11.2), kunnes piikkien korkeudet (pinta-alat) ja retentioajat pysyvät vakioina.
5.4.2
Injektoidaan jokaista kalibrointiliuosta (3.11.2) useita kertoja ja mitataan kutakin konsentraatiota vastaavat karbadoksipiikkien korkeudet (pinta-alat). Kalibrointikäyrä piirretään siten, että y-akselilla ovat kalibrointiliuosten piikkien korkeuksien (pinta alojen) keskiarvot ja x-akselilla vastaavat konsentraatiot μg/ml.
5.4.3
Injektoidaan näyteuutetta (rehuseosten (5.3.2), esiseosten (5.2.2) ja lisäainevalmisteiden (5.2.3)) useita kertoja ja määritetään karbadoksipiikkien korkeuksien (pinta-alojen) keskiarvo.
6. Tulosten laskeminen
Näyteliuoksen karbadoksipitoisuus μg/ml lasketaan karbadoksipiikkien korkeuksien (pinta-alan) keskiarvosta kalibrointikäyrän (5.4.2) avulla.
6.1 Rehut
Näytteen karbadoksipitoisuus w (mg/kg) lasketaan seuraavan kaavan avulla:
 [mg/kg]
[mg/kg]
jossa:
|
c |
= |
näyteuutteen (5.3.2) karbadoksipitoisuus (μg/ml), |
|
V1 |
= |
uuttoliuoksen tilavuus (ml) (ts. 50), |
|
m |
= |
näyte-erän paino (g). |
6.2 Esiseokset ja lisäainevalmisteet
Näytteen karbadoksipitoisuus w (mg/kg) lasketaan seuraavan kaavan avulla:
 [mg/kg]
[mg/kg]
jossa:
|
c |
= |
näyteuutteen (5.2.2 tai 5.2.3) karbadoksipitoisuus (μg/ml), |
|
V2 |
= |
uuttoliuoksen tilavuus (ml) (ts. 50 esiseoksille, 150 lisäainevalmisteille), |
|
f |
= |
laimennuskerroin 5.2.2 kohdan (esiseokset) tai 5.2.3 kohdan (lisäainevalmisteet) mukaisesti, |
|
m |
= |
näyte-erän paino (g). |
7. Tulosten validointi
7.1 Tutkittavan aineen tunnistaminen
Tutkittavan aineen tunnistaminen voidaan varmentaa yhteiskromatografialla tai diodirividetektorilla, mikä mahdollistaa näytteen uuttoliuoksen ja 10,0 μl/ml karbadoksia sisältävän kalibrointiliuoksen (3.11.2) spektrien vertailun.
7.1.1
Näytteen uuttoliuokseen lisätään sopiva määrä kalibrointiliuosta (3.11.2). Lisätyn karbadoksimäärän on oltava suunnilleen sama kuin näyteliuoksen karbadoksin määrä.
Ainoastaan karbadoksipiikin korkeus saa kasvaa, kun otetaan huomioon sekä lisätty määrä karbadoksia että näyteliuoksen laimeneminen. Piikin leveyden on oltava sen puolikorkeudella noin 10 % sen alkuperäisestä leveydestä.
7.1.2
Tulokset arvioidaan seuraavien arviontiperusteiden mukaisesti:
|
a) |
kromatogrammin piikkien huipusta mitattujen näyte- ja standardispektrien maksimiabsorption aallonpituuksien on oltava samat ilmaisinjärjestelmän erotuskyvyn mukaan määräytyvissä rajoissa. Diodirividetektorilla se on yleensä ± 2 nm; |
|
b) |
välillä 225–400 nm kromatogrammin piikin huipun kohdalla ajetut näyte- ja standardispektrit eivät saa erota niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun maksimit ovat samat ja näiden kahden spektrin välillä havaittu poikkeama ei ole missään kohdassa yli 15 %:a standardina olevan tutkittavan aineen absorbanssista; |
|
c) |
välillä 225–400 nm näyteliuoksesta ajetut spektrit piikin nousun, huipun ja laskun kohdalla eivät saa erota toisistaan niiltä spektrin osilta, joiden suhteellinen absorbanssi on 10–100 %. Tämä edellytys täyttyy, kun samat maksimit esiintyvät ja kun kaikissa havaittavissa kohdissa spektrien välinen poikkeama ei ole suurempi kuin 15 % huipun spektrin absorbanssista. |
Jos jokin näistä edellytyksistä ei täyty, tutkittavan aineen esiintymistä ei ole vahvistettu.
7.2 Toistettavuus
Kun karbadoksipitoisuus on 10 mg/kg tai suurempi, kahden rinnakkaisen samasta näytteestä tehdyn määrityksen tulosten ero ei saa olla suurempi kuin 15 % suuremmasta rinnakkaistuloksesta.
7.3 Saanto
Sellaisen nollanäytteen saannon, johon on lisätty määritettävää ainetta, on oltava vähintään 90 %.
8. Laboratorioiden välisen vertailun tulokset
Laboratorioiden välisessä tutkimuksessa analysoitiin 6 rehuseosta, 4 esiseosta ja 3 lisäainevalmistetta kahdeksassa laboratoriossa. Kukin näyte määritettiin kaksi kertaa. (Tarkempia tietoja tästä laboratorioiden välisestä tutkimuksesta löytyy julkaisusta Journal of AOAC International, Volume 71, 1988, s. 484–490.) Tulokset (lukuun ottamatta poikkeavia arvoja) olivat seuraavat:
Taulukko 1
Laboratorioiden välisen vertailun tulokset: rehut
|
|
Näyte 1 |
Näyte 2 |
Näyte 3 |
Näyte 4 |
Näyte 5 |
Näyte 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Keskiarvo (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr ( %) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR ( %) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Nimellispitoisuus (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Taulukko 2
Laboratorioiden välisen vertailun tulokset: esiseokset ja lisäainevalmisteet
|
|
Esiseokset |
Lisäainevalmisteet |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Keskiarvo (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr ( %) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR ( %) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Nimellispitoisuus (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
laboratorioiden lukumäärä |
|
n |
= |
yksittäisten arvojen lukumäärä |
|
Sr |
= |
toistettavuuden keskihajonta |
|
CVr |
= |
toistettavuuden variaatiokerroin |
|
SR |
= |
uusittavuuden keskihajonta |
|
CVR |
= |
uusittavuuden variaatiokerroin. |
LIITE IX
6 ARTIKLASSA TARKOITETUT VASTAAVUUSTAULUKOT
1. Direktiivi 71/250/ETY
|
Direktiivi 71/250/ETY |
Tämä asetus |
|
1 artiklan ensimmäinen alakohta |
3 artikla |
|
1 artiklan toinen alakohta |
2 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liitteessä oleva 1 osa |
Liite II |
|
Liitteessä oleva 2 osa |
— |
|
Liitteessä oleva 3 osa |
— |
|
Liitteessä oleva 4 osa |
Liitteessä III oleva O osa |
|
Liitteessä oleva 5 osa |
Liitteessä III oleva M osa |
|
Liitteessä oleva 6 osa |
Liitteessä III oleva N osa |
|
Liitteessä oleva 7 osa |
Liitteessä III oleva Q osa |
|
Liitteessä oleva 9 osa |
Liitteessä III oleva K osa |
|
Liitteessä oleva 10 osa |
— |
|
Liitteessä oleva 11 osa |
— |
|
Liitteessä oleva 12 osa |
Liitteessä III oleva J osa |
|
Liitteessä oleva 14 osa |
Liitteessä III oleva D osa |
|
Liitteessä oleva 16 osa |
— |
2. Direktiivi 71/393/ETY
|
Direktiivi 71/393/ETY |
Tämä asetus |
|
1 artikla |
3 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liitteessä oleva I osa |
Liitteessä III oleva A osa |
|
Liitteessä oleva II osa |
Liitteessä III oleva E osa |
|
Liitteessä oleva III osa |
Liitteessä III oleva P osa |
|
Liitteessä oleva IV osa |
Liitteessä III oleva H osa |
3. Direktiivi 72/199/ETY
|
Direktiivi 72/199/ETY |
Tämä asetus |
|
1 artikla |
3 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
4 artikla |
— |
|
Liitteessä I oleva 1 osa |
Liitteessä III oleva L osa |
|
Liitteessä I oleva 2 osa |
Liitteessä III oleva C osa |
|
Liitteessä I oleva 3 osa |
— |
|
Liitteessä I oleva 4 osa |
— |
|
Liitteessä I oleva 5 osa |
Liitteessä V oleva A osa |
|
Liite II |
— |
4. Direktiivi 73/46/ETY
|
Direktiivi 73/46/ETY |
Tämä asetus |
|
1 artikla |
3 artikla |
|
3 artikla |
— |
|
4 artikla |
— |
|
Liitteessä I oleva 1 osa |
Liitteessä III oleva B osa |
|
Liitteessä I oleva 2 osa |
— |
|
Liitteessä I oleva 3 osa |
Liitteessä III oleva I osa |
5. Direktiivi 76/371/ETY
|
Direktiivi 76/371/ETY |
Tämä asetus |
|
1 artikla |
1 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liite |
Liite I |
6. Direktiivi 76/372/ETY
|
Direktiivi 76/372/ETY |
Tämä asetus |
|
1 artikla |
— |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liite |
— |
7. Direktiivi 78/633/ETY
|
Direktiivi 78/633/ETY |
Tämä asetus |
|
1 artikla |
3 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liitteessä oleva 1 osa |
— |
|
Liitteessä oleva 2 osa |
— |
|
Liitteessä oleva 3 osa |
Liitteessä IV oleva C osa |
8. Direktiivi 81/715/ETY
|
Direktiivi 81/715/ETY |
Tämä asetus |
|
1 artikla |
— |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liite |
— |
9. Direktiivi 84/425/ETY
|
Direktiivi 84/425/ETY |
Tämä asetus |
|
1 artikla |
— |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liite |
— |
10. Direktiivi 86/174/ETY
|
Direktiivi 86/174/ETY |
Tämä asetus |
|
1 artikla |
4 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liite |
Liite VII |
11. Direktiivi 93/70/ETY
|
Direktiivi 93/70/ETY |
Tämä asetus |
|
1 artikla |
3 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liite |
Liitteessä IV oleva D osa |
12. Direktiivi 93/117/EY
|
Direktiivi 93/117/EY |
Tämä asetus |
|
1 artikla |
3 ja 5 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
Liitteessä oleva 1 osa |
Liitteessä IV oleva E osa |
|
Liitteessä oleva 2 osa |
Liitteessä VIII oleva A osa |
13. Direktiivi 98/64/EY
|
Direktiivi 98/64/EY |
Tämä asetus |
|
1 artikla |
3 ja 5 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
4 artikla |
— |
|
Liitteessä oleva A osa |
Liitteessä III oleva F osa |
|
Liitteessä oleva C osa |
Liitteessä VIII oleva B osa |
14. Direktiivi 1999/27/EY
|
Direktiivi 1999/27/EY |
Tämä asetus |
|
1 artikla |
3 ja 5 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
4 artikla |
— |
|
5 artikla |
— |
|
6 artikla |
— |
|
7 artikla |
— |
|
Liitteessä oleva A osa |
Liitteessä VIII oleva C osa |
|
Liitteessä oleva B osa |
Liitteessä IV oleva F osa |
|
Liitteessä oleva C osa |
Liitteessä VIII oleva D osa |
15. Direktiivi 1999/76/EY
|
Direktiivi 1999/76/EY |
Tämä asetus |
|
1 artikla |
3 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
4 artikla |
— |
|
Liite |
Liitteessä IV oleva G osa |
16. Direktiivi 2000/45/EY
|
Direktiivi 2000/45/EY |
Tämä asetus |
|
1 artikla |
3 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
4 artikla |
— |
|
Liitteessä oleva A osa |
Liitteessä IV oleva A osa |
|
Liitteessä oleva B osa |
Liitteessä IV oleva B osa |
|
Liitteessä oleva C osa |
Liitteessä III oleva G osa |
17. Direktiivi 2002/70/EY
|
Direktiivi 2002/70/EY |
Tämä asetus |
|
1 artikla |
1 artikla |
|
2 artikla |
2 ja 3 artikla |
|
3 artikla |
— |
|
4 artikla |
— |
|
5 artikla |
— |
|
Liite I |
Liite I ja liitteessä V olevan B osan I luku |
|
Liite II |
Liite II ja liitteessä V olevan B osan II luku |
18. Direktiivi 2003/126/EY
|
Direktiivi 2003/126/EY |
Tämä asetus |
|
1 artikla |
3 artikla |
|
2 artikla |
— |
|
3 artikla |
— |
|
4 artikla |
— |
|
5 artikla |
— |
|
6 artikla |
— |
|
Liite |
Liite VI |
|
26.2.2009 |
FI |
Euroopan unionin virallinen lehti |
L 54/s3 |
HUOMAUTUS LUKIJALLEToimielimet ovat päättäneet, ettei niiden säädöksissä enää viitata niissä mainittujen säädösten viimeisimpään muutokseen.Ellei toisin mainita, julkaistuissa teksteissä mainituilla säädöksillä tarkoitetaan säädöksiä niiden tällä hetkellä voimassa olevassa muodossa.
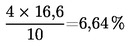

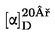 = puhtaan tärkkelyksen ominaiskiertokyky (ks. 6.1).
= puhtaan tärkkelyksen ominaiskiertokyky (ks. 6.1).