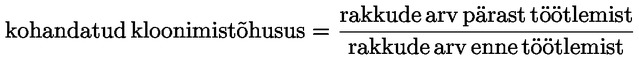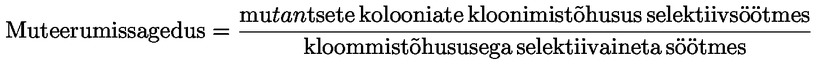|
8)
|
B osasse lisatakse järgmised peatükid:
„B.63. REPRODUKTIIV-/ARENGUTOKSILISUSE SÕELUURING
SISSEJUHATUS
|
1.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 421 (2016). Kemikaalide uurimist käsitlevaid OECD katsejuhendeid vaadatakse teaduse arengu arvessevõtmiseks korrapäraselt läbi. Algne sõeluuringut käsitlev katsejuhend nr 421 võeti vastu 1995. aastal ühe esialgse reproduktiivtoksilisuse sõeluuringu katse-eeskirja põhjal, mida arutati kahel ekspertide koosolekul, mis toimusid 1990. aastal Londonis (1) ja 1992. aastal Tokyos (2).
|
|
2.
|
Käesolevat katsemeetodit on ajakohastamise eesmärgil täiendatud endokriinfunktsiooni kahjustavate kemikaalide uurimiseks sobivate lõppnäitajatega, mis lisati järelmeetmena lähtuvalt prioriteetsest tegevussuunast, mille OECD käivitas 1998. aastal eesmärgiga vaadata läbi olemasolevad ja töötada välja uued katsejuhendid, et võimaldada sõeluuringute tegemist võimalike endokriinfunktsiooni kahjustavate kemikaalide tuvastamiseks ja selliste kemikaalide hindamist (3). Näiteks täiendati 2008. aastal OECD katsejuhendit nr 407 (käesoleva lisa peatükk B.7 „Suukaudse kordusdoosi toksilisuse 28-päevane uuring närilistel“) parameetritega, mis sobivad uuritavate kemikaalide endokriinse toime tuvastamiseks. Katsejuhendi nr 421 ajakohastamise eesmärk oli lisada sõeluuringut käsitlevasse juhendisse teatavad endokriinfunktsiooni kahjustavate kemikaalide uurimiseks sobivad lõppnäitajad, mida saab hinnata juhul, kui kokkupuuteperiood hõlmab mõnda tundlikku arenguperioodi (sünnieelset või varajast sünnijärgset perioodi).
|
|
3.
|
Peale selle lisati katsejuhendisse nr 421 täiendavad endokriinfunktsiooni kahjustavate kemikaalide uurimiseks sobivad lõppnäitajad, mida sisaldab ka katsejuhend nr 443 (käesoleva lisa peatükk B.56 „Laiendatud ühe põlvkonna reproduktiivtoksilisuse uuring“); need lõppnäitajad valiti lähtuvalt ühest teostatavusuuringust, milles käsitletakse asjaomaste lõppnäitajate lisamisega seotud teaduslikke ja tehnilisi küsimusi, ning võimalikust vajadusest katsemeetodit nende lisamiseks kohandada (4).
|
|
4.
|
Käesolev katsemeetod on kavandatud piiratud hulga andmete saamiseks uuritava kemikaali mõju kohta isas- ja emasloomade paljunemisvõimele, näiteks sugunäärmete talitlusele, paaritumiskäitumisele, eostumisele, sügoodi arengule ja sünnitamisele. See ei ole ette nähtud kasutamiseks olemasolevate katsemeetodite B.31, B.34, B35 või B.56 alternatiivina ega nende asendamiseks.
|
LÄHTEKAALUTLUSED
|
5.
|
Käesolevat sõeluuringumeetodit võib kasutada paljunemisvõimele ja/või arengule avalduva võimaliku mõju kohta esialgse teabe saamiseks kas kemikaalide toksikoloogiliste omaduste hindamise varajases etapis või probleemsete kemikaalide uurimisel. Seda võib kasutada ka osana esialgsete sõeluuringute seeriast, mille abil hinnatakse olemasolevaid kemikaale, mille kohta on vähe või ei ole üldse toksikoloogilisi andmeid, samuti ulatuslikumate paljunemisvõimet/arengut käsitlevate uuringute jaoks annusevahemiku kindlaksmääramiseks ning muudel asjakohaseks peetavatel juhtudel. Uuringu tegemisel tuleks järgida juhtpõhimõtteid ja kaalutlusi, mis on esitatud OECD juhenddokumendis nr 19, milles käsitletakse ohutuse hindamiseks kasutatavatel katseloomadel avalduvate kliiniliste tunnuste kindlakstegemist, hindamist ja kasutamist humaanse sekkumise vajadusele viitavate näitajatena (5).
|
|
6.
|
Käesolev katsemeetod ei võimalda saada ammendavat teavet kõikide paljunemisvõime ja arenguga seotud aspektide kohta. Eelkõige lubab see tuvastada sünnieelse kokkupuute tagajärjel sünnijärgselt avalduvaid ilminguid ja sünnijärgsest kokkupuutest tulenevat mõju üksnes piiratud ulatuses. Käesoleva meetodiga ei saa koguda tõendeid, mis lubaksid lõplikult kinnitada mõju puudumist; see on muu hulgas tingitud loomade suhteliselt väikesest arvust annuserühmades, lõppnäitajate selektiivsusest ja uuringu lühikesest kestusest. Täheldatavaid positiivseid tulemusi aga on võimalik muudest reproduktiiv-/arengutoksilisuse uuringutest saadavate andmete puudumisel kasutada esialgseks ohuhindamiseks ning need aitavad teha otsuseid täiendavate uuringute tegemise vajaduse ja selliste uuringute ajastuse kohta.
|
|
7.
|
Sisesekretsioonisüsteemiga seotud näitajaid käsitlevaid tulemusi tuleks vaadelda endokriinfunktsiooni kahjustavate kemikaalide uurimise ja hindamise OECD kontseptuaalse raamistiku (6) kontekstis. Nimetatud kontseptuaalses raamistikus on OECD täiendatud katsejuhendi nr 421 kohast meetodit käsitatud 4. taseme in vivo meetodina, mis võimaldab saada andmeid sisesekretsioonisüsteemiga seotud lõppnäitajate kaudu tuvastatava kahjuliku mõju kohta. Sisesekretsioonisüsteemist sõltuvat signaali ei pruugita eraldi võetuna siiski pidada piisavaks tõendiks selle kohta, et uuritava kemikaali puhul on tegemist endokriinfunktsiooni kahjustava kemikaaliga.
|
|
8.
|
Käesoleva katsemeetodi puhul eeldatakse, et uuritavat kemikaali manustatakse suu kaudu. Muu manustamisviisi kasutamise korral võib olla vaja teha meetodis muudatusi.
|
|
9.
|
Kui soovitakse saada kavandatud regulatiivsel eesmärgil andmeid segu kohta, tuleks enne katsemeetodi kasutamist kaaluda, kas ja kuidas see võimaldab saada kõnealusele eesmärgile vastavaid asjakohaseid tulemusi. Selline kaalumine ei ole vajalik, kui asjaomase segu hindamine on regulatiivse nõudega ette nähtud.
|
|
10.
|
Kasutatud mõisted on määratletud 1. liites.
|
KATSE PÕHIMÕTE
|
11.
|
Uuritavat kemikaali manustatakse astmeliselt suurenevates annustes mitme rühma isas- ja emasloomadele. Isasloomade puhul peaks manustamine toimuma vähemalt nelja nädala kestel kuni kavandatud surmamispäevale eelneva päevani, sealhulgas surmamiseelsel päeval (see hõlmab vähemalt kahte paaritumiseelset nädalat, paaritumisperioodi ja umbes kahte paaritumisjärgset nädalat). Kuna isasloomadel on paaritumiseelne manustamisperiood piiratud kestusega, ei pruugi viljakus olla eriti tundlik munanditele avalduva mürgisuse näitaja. Seepärast on vaja teha munandite üksikasjalik histoloogiline analüüs. Kahe nädala pikkust paaritumiseelset manustamisperioodi koos järgneva paaritumise/viljakuse jälgimisega ning manustamisperioodi üldkestusega vähemalt neli nädalat, millele järgneb isaslooma sugunäärmete üksikasjalik histopatoloogiline analüüs, peetakse enamiku isasloomal täheldatavate viljakusele ja spermatogeneesile avalduva mõju ilmingute tuvastamiseks piisavaks.
|
|
12.
|
Emasloomade puhul peaks manustamine toimuma kogu uuringu kestel. See hõlmab kahte paaritumiseelset nädalat (eesmärk on jälgida vähemalt kahte tervet innatsüklit), eostumisele eelnevat varieeruva pikkusega ajavahemikku, tiinuse kestust ja vähemalt kolmeteistkümne päeva pikkust sünnitusjärgset perioodi, mis kestab kuni kavandatud surmamispäevale eelneva päevani ja hõlmab ka surmamiseelset päeva.
|
|
13.
|
Kohanemisperioodile ja innatsükli manustamiseelsele hindamisele järgneva uuringu kestus sõltub emasloomade seisundist ning on umbes 63 päeva (vähemalt 14 päeva pikkune paaritumiseelne periood, (kuni) 14 päeva pikkune paaritumisperiood, 22 päeva pikkune tiinusperiood ja 13 päeva pikkune imetamisperiood).
|
|
14.
|
Manustamisperioodi jooksul jälgitakse loomi iga päev hoolikalt mürgisusnähtude suhtes. Uuringu kestel surnud või surmatud loomad lahatakse ning uuringu lõpus surmatakse ja lahatakse ka ellujäänud loomad.
|
MEETODI KIRJELDUS
Loomaliigi valimine
|
15.
|
Käesolev katsemeetod on kavandatud kasutamiseks rotil. Kui käesolevas katsemeetodis kirjeldatud näitajaid hinnatakse mõne muu liigi närilistel, tuleks seda üksikasjalikult põhjendada. Endokriinfunktsiooni kahjustavate kemikaalide kindlakstegemise rahvusvahelises valideerimisprogrammis, mille puhul on lähtutud OECD katsejuhendist nr 407 (see vastab käesoleva lisa peatükile B.7), kasutati ainsa liigina rotte. Ei tohiks kasutada liine, kus sigivus on väike või kus teadaolevalt esineb sageli arenguhäireid. Tuleks kasutada terveid varem paaritamata loomi, keda ei ole varem katsetes kasutatud. Katseloomi tuleks iseloomustada liigi, liini, soo, kehamassi ja vanuse järgi. Uuringu alguses peaksid kasutatavate loomade kehamassi erinevused olema minimaalsed ning mitte üle 20 % kummastki soost loomade keskmisest kehamassist. Kui uuring viiakse läbi pikaajalise või tervet põlvkonda hõlmava uuringu eeluuringuna, tuleks mõlemas uuringus soovitatavalt kasutada samast liinist ja sama päritolu loomi.
|
Pidamis- ja söötmistingimused
|
16.
|
Kõik katsetoimingud peaksid vastama kohalikele laboriloomade hooldamise normidele. Katseloomade ruumi temperatuur peaks olema 22 °C (± 3 °C). Ehkki suhteline õhuniiskus peaks olema vähemalt 30 % ja soovitatavalt mitte üle 70 %, v.a ruumi koristamise ajal, on sobivaim vahemik 50–60 %. Tuleks kasutada tehisvalgustust valgusrežiimiga 12 tundi valgust ja 12 tundi pimedust. Söötmiseks võib kasutada tavapärast laborisööta ja piiramatus koguses joogivett. Söödavalikut võib mõjutada vajadus tagada sööda sobivus uuritava kemikaali lisamiseks, kui kemikaali manustatakse sellisel viisil.
|
|
17.
|
Loomade pidamisel tuleks kasutada väikesi samasooliste isendite rühmi; loomi võib pidada eraldi, kui see on teaduslikult põhjendatud. Rühmas pidamisel ei tohiks puuris olla rohkem kui viis looma. Paaritamine peaks toimuma selleks sobivas puuris. Tiined emasloomad tuleks paigutada igaüks eraldi puuri, kus on olemas pesamaterjal. Imetavad emasloomad paigutatakse igaüks eraldi puuri koos järglaskonnaga.
|
|
18.
|
Sööta tuleks korrapäraselt analüüsida saasteainete suhtes. Asjaomase sööda proov tuleks kuni katseprotokolli lõpliku valmimiseni alles hoida.
|
Loomade ettevalmistamine
|
19.
|
Terved noored täiskasvanud loomad jagatakse juhuvaliku alusel kontroll- ja katserühmadesse. Puurid paigutatakse nii, et puuri asukohast tingitud võimalik mõju oleks võimalikult väike. Loomad idenditakse üheselt ja neil lastakse enne uuringu algust puuris vähemalt viis päeva laboritingimustega kohaneda.
|
Annuste ettevalmistamine
|
20.
|
Uuritavat kemikaali on soovitatav manustada suu kaudu, välja arvatud juhul, kui mõnda muud manustamisviisi peetakse sobivamaks. Uuritava kemikaali suukaudseks manustamiseks kasutatakse tavaliselt toitmissondi; selle asemel võib siiski kasutada ka sööda või joogiveega manustamist.
|
|
21.
|
Vajaduse korral lahustatakse või suspendeeritakse uuritav kemikaal sobivas kandeaines. Kui vähegi võimalik, on soovitatav esmalt kaaluda vesilahuse või veepõhise suspensiooni ja seejärel õlilahuse/-emulsiooni (nt maisiõli) kasutamist ning pärast seda lahustamist mõnes muus kandeaines. Muu kandeaine kui vee puhul peaksid olema teada selle mürgisusnäitajad. Tuleks teha kindlaks uuritava kemikaali püsivus ja homogeensus kandeaines.
|
KATSE KÄIK
Loomade arv ja sugu
|
22.
|
Soovitatavalt peaks igas rühmas olema alguses vähemalt 10 isaslooma ja 12–13 emaslooma. Emasloomi hinnatakse kokkupuute eel innatsükli alusel ja loomad, kelle innatsükkel erineb tavapärasest 4–5 päeva pikkusest tsüklist, jäetakse uuringust välja; seepärast on soovitatav kasutada suuremat arvu emasloomi, et igas rühmas oleks lõpuks 10 emaslooma. Eeldatavalt tiinestub igas rühmas vähemalt 8 emaslooma – seda peetakse tavapäraselt minimaalseks vastuvõetavaks tiinestumismääraks –, välja arvatud juhul, kui ilmneb märkimisväärne mürgine mõju. Eesmärk on saada piisav arv tiineid emasloomi, et oleks võimalik usaldusväärselt hinnata uuritava kemikaali võimet mõjutada viljakust, tiinust, ema- ja imetamiskäitumist ning F1-põlvkonna järglaste kasvu ja arengut eostamisest kuni kolmeteistkümnenda sünnijärgse päevani.
|
Annused
|
23.
|
Üldjuhul tuleks kasutada vähemalt kolme katserühma ja ühte kontrollrühma. Annuste valimisel võib lähtuda ägeda mürgisuse hindamise katsetega saadud andmetest või korduvannustamisel põhinevate uuringute tulemustest. Kontrollrühma loomi tuleks kohelda täpselt samal viisil kui katserühma loomi; ainus erinevus seisneb uuritava kemikaali manustamata jätmises. Kui uuritava kemikaali manustamisel kasutatakse kandeainet, tuleks võrdlusrühma loomadele manustada kandeainet suurimas kasutatavas koguses.
|
|
24.
|
Annuste valimisel tuleks võtta arvesse kõiki kättesaadavaid mürgisusandmeid ja (toksiko)kineetilisi andmeid. Samuti tuleks arvesse võtta, et tiinete ja mittetiinete loomade tundlikkus võib olla erinev. Suurim annus tuleks valida nii, et kemikaal avaldaks mürgist mõju, kuid ei põhjustaks surma ega tõsiseid kannatusi. Seejärel tuleks valida rida järk-järgult vähenevaid annuseid, mis võimaldaksid tõendada mõju sõltuvust annusest ja teha väikseima annuse põhjal kindlaks täheldatava kahjuliku toime puudumise annuse (NOAEL). Sageli on optimaalne vähenevate annuste vaheline erinevus kahe- kuni neljakordne ning üksteisest väga palju (näiteks üle 10 korra) erinevate annuste kasutamise asemel on tihti soovitatav lisada neljas katserühm.
|
|
25.
|
Kui täheldatakse üldist mürgisust (nt kehamassi vähenemine, mõju maksale, südamele, kopsudele või neerudele jne) või muid muutusi, mis ei pruugi olla tingitud mürgisusest (nt väiksem söödatarbimine, maksa suurenemine), tuleks täheldatud mõju sisesekretsioonisüsteemiga seotud lõppnäitajatele tõlgendada ettevaatusega.
|
Piirsisalduskatse
|
26.
|
Kui käesoleva meetodi kohaselt läbi viidud suukaudse manustamise uuringus, milles kasutatakse ainsa annusena annust vähemalt 1 000 mg kehamassi kilogrammi kohta päevas või sööda või joogiveega manustamise korral sellega samaväärset protsentuaalset sisaldust söödas või joogivees, ei täheldata mürgist mõju ja kui mürgisus ei ole uuritava kemikaaliga struktuurilt sarnaste ainetega saadud andmete põhjal ootuspärane, ei pruugita pidada mitme eri annusega täismahus uuringu tegemist vajalikuks. Piirsisalduskatse tulemused on kohaldatavad, kui inimese kokkupuutega seotud andmed ei viita vajadusele kasutada suukaudsel manustamisel suuremat annusemäära. Muu manustamisviisi korral, näiteks sissehingamise teel või naha kaudu manustamisel võib uuritava kemikaali füüsikalis-keemilistest omadustest tulenevalt olla tihti vaja kasutada suurimat saavutatavat kontsentratsiooni.
|
Annuste manustamine
|
27.
|
Uuritavat kemikaali manustatakse loomadele seitsmel päeval nädalas. Kui manustamiseks kasutatakse toitmissondi, tuleks kemikaali loomadele manustada ühe annusena maosondi või sobiva intubeerimiskanüüli abil. Suurim korraga manustatav vedelikukogus sõltub katselooma suurusest. See kogus ei tohiks olla suurem kui 1 ml 100 g kehamassi kohta, välja arvatud vesilahuse puhul, mida võib manustada koguses 2 ml 100 g kehamassi kohta. Katses kasutatava vedelikukoguse varieeruvuse minimeerimiseks tuleks reguleerida uuritava kemikaali kontsentratsiooni nii, et lahuse maht oleks kõikide annuste puhul ühesugune; see ei ole nõutav ärritava või söövitava uuritava kemikaali puhul, mille mõju kontsentratsiooni suurenedes tavaliselt tugevneb.
|
|
28.
|
Sööda või joogiveega manustatava uuritava kemikaali puhul on oluline tagada, et seda kasutatakse koguses, mis ei muuda tavapärast toitumise või joogivee tarbimise tasakaalu. Kui uuritavat kemikaali manustatakse söödaga, võib kasutada kas muutumatut kontsentratsiooni söödas (ppm) või looma kehamassi suhtes muutumatut annusemäära; tuleks täpsustada, kumba võimalust kasutatakse. Toitmissondi kaudu manustatava uuritava kemikaali puhul peaks manustamine toimuma igal päeval sarnasel kellaajal ning annusemäära tuleks vähemalt kord nädalas kohandada, et see püsiks looma kehamassi suhtes muutumatuna.
|
Katse ajakava
|
29.
|
Annuste manustamist tuleks kummagi soo puhul alustada vähemalt kaks nädalat enne paaritamist pärast seda, kui loomadel on lastud vähemalt viis päeva kohaneda ja emasloomi on hinnatud lähtuvalt innatsükli tavapärasest kestusest (töötlemiseelse kahe nädala pikkuse perioodi vältel). Uuringu ajakava peaks olema selline, et innatsükli hindamist alustatakse varsti pärast seda, kui loomad on saanud täielikult suguküpseks. See aeg võib olla eri rotiliinidel ja eri laborites veidi erinev: näiteks Sprague Dawley rottidel saabub suguküpsus 10 nädala vanuses ja Wistari rottidel umbes 12 nädala vanuses. Järglastega emasloomad tuleks surmata 13. sünnitusjärgsel päeval või veidi hiljem. Sünnitamispäeva (päev, mil sünnitus lõpeb) loetakse sünnitusjärgseks 0-päevaks. Emasloomad, kellel ei täheldata paaritumise ilminguid, surmatakse 24–26 päeva pärast paaritumisperioodi viimast päeva. Paaritumisperioodi vältel jätkatakse kummastki soost loomadele kemikaali manustamist. Isasloomadele manustamist tuleks paaritumisperioodi järgselt jätkata vähemalt seni, kuni manustamisperioodi algusest on möödunud kokku minimaalselt 28 päeva. Seejärel loomad surmatakse või teise võimalusena jätkatakse neile kemikaali manustamist, et viia võimaluse korral läbi teine paaritamine, kui seda peetakse asjakohaseks.
|
|
30.
|
Tiinestunud emasloomade puhul tuleks igapäevast manustamist jätkata kogu tiinuse vältel ning vähemalt kuni 13. sünnitusjärgse päevani, sealhulgas nimetatud päeval, või kuni surmamiseelse päevani. Uuringute puhul, kus uuritavat kemikaali manustatakse sissehingamise teel või naha kaudu, tuleks manustamist jätkata vähemalt kuni 19. tiinuspäevani, sealhulgas nimetatud päeval, ning taasalustada seda võimalikult varakult ja mitte hiljem kui 4. sünnitusjärgsel päeval.
|
|
31.
|
Katse skemaatiline ajakava on esitatud 2. liites.
|
Paaritamine
|
32.
|
Käesoleva meetodi kohases uuringus tuleks üldjuhul kasutada paaritamisel suhet 1: 1 (üks isasloom ühe emaslooma kohta). Erandi võib teha juhul, kui isasloomad juhtuvad surema. Emaslooma tuleks hoida sama isaslooma juures seni, kuni täheldatakse paaritumise ilminguid või on möödunud kaks nädalat. Emasloomi tuleks igal hommikul uurida sperma või tupekorgi esinemise suhtes. Tiinuse 0-päevaks loetakse päeva, mil paaritumise toimumine leiab kinnitust (leitakse tupekork või spermat). Kui paaritamine ebaõnnestub, võib kaaluda emaslooma uuesti paaritamist sama rühma viljakaks osutunud isasloomaga.
|
Pesakonna suurus
|
33.
|
Neljandal sünnijärgsel päeval võib iga pesakonna suurust korrigeerida; selleks kõrvaldatakse juhuvaliku alusel üleliigsed järglased nii, et pesakonda jääks sõltuvalt kasutatava rotiliini puhul tavapärasest pesakonna suurusest võimalikult täpselt neli või viis järglast kummagi soo kohta. Üleliigsetest järglastest kahelt tuleks võtta vereproov, moodustada nendest koondproov ja kasutada seda seerumi T4-sisalduse määramiseks. Järglaste selektiivne kõrvaldamine, näiteks kehamassi või anogenitaalse vahemaa (AGD) alusel, ei ole sobiv. Kui emaste või isaste järglaste arv ei võimalda jätta pesakonda nelja või viit järglast kummastki soost, on vastuvõetav ka osaline korrigeerimine (nt kuus isast ja neli emast). Kui pesakond jääks kõrvaldamise tagajärjel väiksemaks kui kõrvaldamisega saavutatav soovitud sihtarv (8 või 10 järglast pesakonna kohta), ei kõrvaldata ühtki järglast. Kui kõrvaldamisega saavutatava soovitud sihtarvuga võrreldes on üle ainult üks järglane, kõrvaldataksegi vaid üks järglane, kellelt võetakse vereproov võimalikuks seerumi T4-sisalduse hindamiseks.
|
|
34.
|
Kui pesakonna suurust ei korrigeerita, surmatakse 4. sünnijärgsel päeval igast pesakonnast kaks järglast ning neilt võetakse vereproov seerumi kilpnäärmehormoonisisalduse mõõtmiseks. Võimaluse korral peaksid kõnealused kaks järglast igast pesakonnast olema emased, et võimaldada isaste järglaste kasutamist nisade esinemise hindamiseks; see soovitus ei kehti juhul, kui kõnealuse kahe järglase eemaldamisel ei jääks pesakonda ühtki emast, keda uuringu lõpus hinnata. Kui pesakonda jääks kõrvaldamise tagajärjel vähem kui 8 või 10 järglast (olenevalt kasutatava rotiliini puhul tavapärasest pesakonna suurusest), ei kõrvaldata ühtki järglast. Kui pesakonna tavapärase suurusega võrreldes on üle ainult üks järglane, kõrvaldataksegi vaid üks järglane, kellelt võetakse vereproov võimalikuks seerumi T4-sisalduse hindamiseks.
|
Elusloomadel tehtavad vaatlused
Kliinilised vaatlused
|
35.
|
Üldisi kliinilisi vaatlusi tuleks kogu uuringuperioodi vältel teha vähemalt kord päevas ja mürgisusnähtude ilmnemisel sagedamini. Vaatlusi tuleks soovitatavalt teha igal päeval samal ajal või samadel aegadel; seejuures tuleks võtta arvesse manustamisjärgse eeldatava mõju maksimaalse avaldumise aega. Tuleks registreerida asjakohased muutused käitumises, raskele või kauakestnud sünnitusele viitavad ilmingud ja kõik mürgisusnähud, sealhulgas suremus. Registreeritavad andmed peaksid hõlmama mürgisusnähtude esinemise algusaega, ulatust ja kestust.
|
Kehamass ja sööda/vee tarbimine
|
36.
|
Isas- ja emasloomi tuleks kaaluda esimesel manustamispäeval ning seejärel vähemalt kord nädalas, samuti surmamise ajal. Tiinuse ajal tuleks emasloomi kaaluda tiinuse 0-päeval, 7., 14. ja 20. päeval, samuti tuleks neid kaaluda 24 tunni jooksul pärast sünnitust (sünnitusjärgsel 0-päeval või 1. päeval) ning vähemalt 4. ja 13. sünnitusjärgsel päeval. Nimetatud vaatluste tulemused tuleks esitada eraldi iga täiskasvanud looma kohta.
|
|
37.
|
Paaritumiseelsel perioodil ning tiinuse ja imetamise ajal tuleks söödatarbimist mõõta vähemalt kord nädalas. Paaritumisperioodil ei ole söödatarbimise mõõtmine kohustuslik. Kui uuritavat kemikaali manustatakse joogiveega, tuleks eelnimetatud perioodidel mõõta ka veetarbimist.
|
Innatsükkel
|
38.
|
Enne töötlemise alustamist tuleks jälgida innatsüklit ning valida selle alusel uuringusse emasloomad, kelle tsükkel on korrapärane (vt punkt 22). Tupeäigeid tuleks igapäevaselt hinnata ka alates töötlemisperioodi algusest kuni paaritumise ilmingute tuvastamiseni. Kui on kahtlus, et innatsükkel võib pärast manustamise alustamist ägeda stressi mõjul muutuda, võib katseloomi laboris 2 nädala vältel uuritava kemikaaliga töödelda ning seejärel koguda paaritumiseelsel perioodil innatsükli jälgimiseks vähemalt kahe nädala jooksul iga päev tupeäigeid ja jätkata jälgimist ka paaritumisperioodil, kuni täheldatakse paaritumise ilminguid. Tupe-/emakakaelarakkude võtmisel tuleks olla ettevaatlik, et mitte kahjustada limaskesta, kuna see võib põhjustada pseudotiinuse teket (7, 8).
|
Järglastega seotud näitajad
|
39.
|
Tiinuse kestus tuleks registreerida; selle arvutamisel lähtutakse tiinuse 0-päevast. Iga pesakonda tuleks pärast sündi võimalikult varakult hinnata, et teha kindlaks järglaste arv ja sugu, surnultsünnid, elussünnid, kängunud järglaste (vastavatest kontrolljärglastest palju väiksemate järglaste) arv ja silmaga nähtavate kõrvalekallete esinemine.
|
|
40.
|
Elus järglased tuleks loendada ja määrata nende sugu ning pesakonda tuleks kaaluda 24 tunni jooksul pärast sünnitust (sünnitusjärgsel 0-päeval või 1. päeval) ning vähemalt 4. ja 13. sünnitusjärgsel päeval. Peale punktis 35 kirjeldatud vaatluste tulemuste tuleks registreerida ka kõik järglaste ebatavalise käitumise ilmingud.
|
|
41.
|
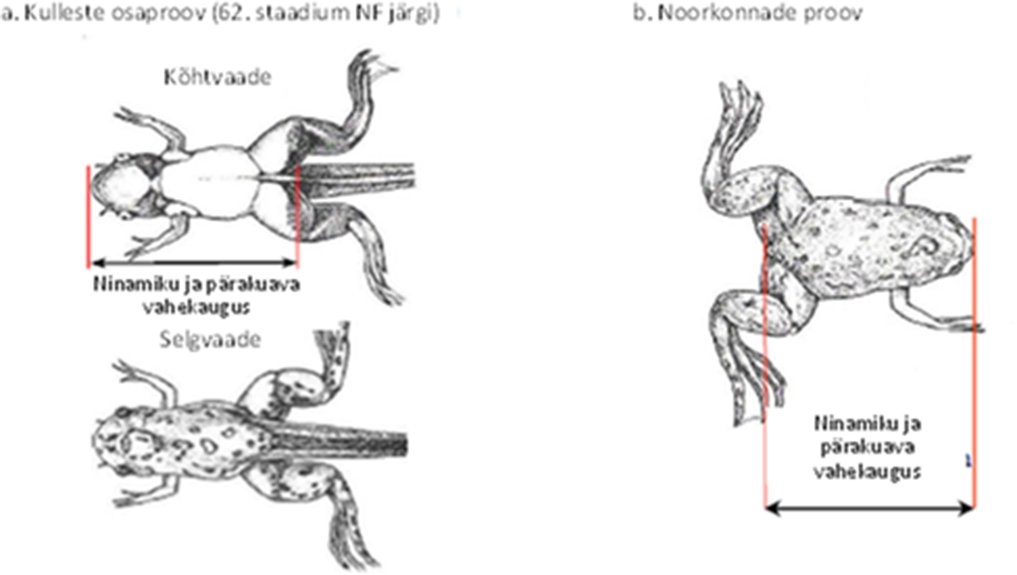
Kõikidel järglastel tuleks ajavahemikul sünnijärgsest 0-päevast kuni 4. sünnijärgse päevani mõõta ühel ja samal päeval AGD. Järglaste kehamassi andmed tuleks koguda AGD mõõtmise päeval ja AGD tuleks normaliseerida järglase suurust kajastava näitaja, soovitatavalt kehamassi kuupjuure suhtes (9). Isastel järglastel tuleks 12. või 13. sünnijärgsel päeval loendada nisade/areoolide arv, nagu on soovitatud OECD katsejuhendis nr 151 (10).
|
Kliiniline biokeemiline analüüs
|
42.
|
Vereproovid võetakse kindlaksmääratud kohast järgmise kava alusel:
|
—
|
igas pesakonnas vähemalt kahelt järglaselt 4. sünnijärgsel päeval, kui järglaste arv seda võimaldab (vt punktid 33–34),
|
|
—
|
kõikidelt järglastega emasloomadelt ja igas pesakonnas vähemalt kahelt järglaselt surmamise ajal 13. päeval ning
|
|
—
|
kõikidelt täiskasvanud isasloomadelt surmamise ajal.
|
|
Kõiki vereproove säilitatakse sobivates tingimustes. Järglastelt 13. päeval võetud vereproovides ja täiskasvanud isasloomade vereproovides analüüsitakse kilpnäärmehormoonide (T4) sisaldust seerumis. Kui see on asjakohane, hinnatakse T4 sisaldust ka järglastega emasloomade vereproovides ja järglastelt 4. päeval võetud vereproovides. Kui see on asjakohane, võib soovi korral mõõta ka muude hormoonide sisaldust. Järglaste vere võib kilpnäärmehormoonide analüüsimiseks pesakonnakaupa kokku koondada. Soovitatavalt tuleks mõõta kilpnäärmehormoonide (T4 ja TSH) üldsisaldust.
|
43.
|
Hormoonide määramisel võivad varieeruvust ja absoluutset kontsentratsiooni mõjutada järgmised tegurid:
|
—
|
surmamise aeg tulenevalt asjaolust, et hormoonide kontsentratsioon ööpäeva jooksul muutub;
|
|
—
|
surmamisviis – tuleks hoiduda tekitamast loomadele tarbetut stressi, mis võib mõjutada hormoonide kontsentratsiooni;
|
|
—
|
hormoonide määramise komplektid, milles kasutatavad standardkõverad võivad olla erinevad.
|
|
|
44.
|
Konkreetselt hormoonide määramiseks ette nähtud eri plasmaproovid tuleks koguda umbes samal kellaajal. Hormoonisisalduse analüüsimisel saadavad arvväärtused on müügilolevate eri katsekomplektide puhul erinevad.
|
Patoloogiline analüüs
Täielik lahkamine
|
45.
|
Surmatud või uuringu vältel surnud täiskasvanud loomi tuleks kohe pärast surma makroskoopiliselt uurida mis tahes kõrvalekallete või patoloogiliste muutuste suhtes. Erilist tähelepanu tuleks pöörata reproduktiivsüsteemi elunditele. Tuleks registreerida pesastumiskohtade arv. Lahkamispäeva hommikul tuleks analüüsida tupeäiet, et teha kindlaks innatsükli etapp ja võimaldada korreleerimist munasarja histopatoloogilise analüüsi tulemustega.
|
|
46.
|
Kõikide täiskasvanud isasloomade munandid ja munandimanused ning eesnääre ja seemnepõiekesed koos koagulatsiooninäärmetega tuleks vajaduse korral vabastada nende külge jäänud kudedest ja kuivamise ärahoidmiseks tuleks elundite märgmass määrata võimalikult kiiresti pärast nende väljalõikamist. Peale selle võib soovi korral kaaluda isasloomade puhul sellised elundid nagu pärakutõsturlihase ja sibulakäsnkeha-lihase kompleks, Cowperi näärmed ja sugutilukk ning emasloomade puhul munasarjade paar (märgmass) ja emakas (koos emakakaelaga); kui seda tehakse, peaks kaalumine toimuma võimalikult kiiresti pärast elundite väljalõikamist.
|
|
47.
|
Surnud järglasi ja 13. sünnijärgsel päeval või varsti pärast seda surmatud järglasi tuleks miinimumnõudena väliselt hoolikalt uurida silmaga nähtavate kõrvalekallete suhtes. Erilist tähelepanu tuleks pöörata välistele suguelunditele, mida tuleks uurida arenguhäirete ilmingute suhtes. Kolmeteistkümnendal sünnijärgsel päeval tuleks võtta säilitamiseks kilpnääre iga pesakonna ühelt isaselt ja ühelt emaselt järglaselt.
|
|
48.
|
Samuti tuleks säilitada kõikide täiskasvanud loomade munasarjad, munandid, suguelundid (emakas ja emakakael, munandimanused, eesnääre, seemnepõiekesed koos koagulatsiooninäärmetega), kilpnääre ja kõik makroskoopiliste kahjustustega elundid. Munandite ja munandimanuste tavapärasel analüüsil ei soovitata kasutada fikseerimist formaliiniga. Nende kudede puhul võib vastuvõetava meetodina kasutada Bouini fikseerimislahust või muudetud Davidsoni lahust (11). Fikseerimislahuse kiire sissetungimise võimaldamiseks võib elundi kummaski otsas valkjaskesta ettevaatlikult väikese sügavuseni nõelaga läbi torgata.
|
Histopatoloogiline analüüs
|
49.
|
Suurima annusega töödeldud rühma ja kontrollrühma loomade puhul tuleks viia läbi munasarjade, munandite ja munandimanuste üksikasjalik histoloogiline uuring (seejuures tuleks pöörata erilist tähelepanu spermatogeneesi etappidele ja munandite interstitsiaalrakustruktuuri histopatoloogilisele analüüsile). Vajaduse korral võib uurida ka teisi säilitatud elundeid, sealhulgas järglastelt ja täiskasvanud loomadelt saadud kilpnääret. Kilpnäärme massi võib määrata pärast fikseerimist. Liigsed koed tuleks samuti eemaldada väga ettevaatlikult alles pärast fikseerimist, et kudesid mitte kahjustada. Koekahjustus võib mõjutada histopatoloogilise analüüsi tulemusi. Kui suurima annusega töödeldud rühmas täheldatakse muutusi, peaks analüüs hõlmama ka teiste annuserühmade loomi. Täiendav üksikasjalik teave sisesekretsioonisüsteemi kudede väljalõikamise, fikseerimise, neist lõikude tegemise ja nende histopatoloogilise analüüsi kohta on esitatud histopatoloogilise analüüsi juhendis (11).
|
ANDMED JA ARUANDLUS
Andmed
|
50.
|
Andmed tuleks esitada iga looma kohta eraldi. Peale selle tuleks esitada kõik andmed kokkuvõtliku tabeli kujul, kus on iga katserühma puhul toodud ära loomade arv katse alguses, katse käigus surnud või humaansetel kaalutlustel surmatud loomade arv ning surma või humaanse surmamise aeg, viljakate loomade arv, tiinete emasloomade arv, mürgisusnähtudega loomade arv, mürgisusnähtude kirjeldus, sealhulgas mürgise mõju ilmnemisaeg, kestus ja tugevus, histopatoloogiliste muutuste liik ja kõik asjakohased andmed pesakonna kohta. Paljunemisele/arengule avalduva mõju hindamiseks väga kasulikuks osutunud aruandevorm kokkuvõtliku tabeli kujul on esitatud 3. liites.
|
|
51.
|
Uuringu piiratud ulatusest tulenevalt on tulemuste olulisuse tuvastamiseks tehtava statistilise analüüsi väärtus paljude lõppnäitajate, eelkõige paljunemisega seotud lõppnäitajate puhul piiratud. Statistilise analüüsi kasutamisel tuleks valida meetod, mis on uuritava muutuja väärtuste jaotuse analüüsimiseks sobiv, ning see valik tuleks teha enne uuringu alustamist. AGD ja nisade esinemise statistiline analüüs peaks põhinema iga üksiku järglase andmetel ning seejuures tuleks arvesse võtta pesakonnas ilmnevat mõju. Vajaduse korral käsitatakse analüüsiüksusena pesakonda. Järglaste kehamassi statistiline analüüs peaks põhinema iga üksiku järglase andmetel ning seejuures tuleks arvesse võtta pesakonna suurust. Ühtlasi võib rühmade väiksusest tulenevalt olla kasulik kasutada uuringutulemuste tõlgendamise hõlbustamiseks varasemaid kontrollidega saadud andmeid (nt pesakonna suuruse kohta), kui need on olemas.
|
Tulemuste hindamine
|
52.
|
Käesoleva meetodi kohase mürgisusuuringu tulemusi tuleks hinnata lähtuvalt täheldatud mõjust, lahkamistulemustest ja mikroskoopilistest leidudest. Hindamine hõlmab seose kindlakstegemist uuritava kemikaali annuse ja kõrvalekallete – sealhulgas silmaga nähtavate kahjustuste, tuvastatud sihtelunditele avalduva mõju, viljatuse, kliiniliste kõrvalekallete, paljunemisvõimele ja pesakonnale avalduva mõju, kehamassi muutuste, suremusele avalduva mõju ja mis tahes muu mürgise mõju – esinemise või puudumise, esinemissageduse ja raskusastme vahel.
|
|
53.
|
Isasloomade töötlemise lühiajalisuse tõttu tuleks isasloomade paljunemisele avalduva mõju hindamisel vaadelda munandite ja munandimanuste histopatoloogilise analüüsi tulemusi koos viljakusandmetega. Ühtlasi võib uuringutulemuste tõlgendamise hõlbustamiseks olla kasulik kasutada kontrollidega saadud varasemaid paljunemist/arengut käsitlevaid andmeid (nt pesakonna suuruse, AGD, nisade esinemise või seerumi T4-sisalduse kohta), kui need on olemas.
|
|
54.
|
Kvaliteedikontrolli eesmärgil on soovitatav koguda kontrolle käsitlevaid varasemaid andmeid ja arvutada arvandmete jaoks variatsioonikordajad, eriti näitajate puhul, mis on seotud endokriinfunktsiooni kahjustavate kemikaalide kindlakstegemisega. Neid andmeid võib kasutada võrdlusalusena käsiloleva uuringu tulemuste hindamiseks.
|
Katseprotokoll
|
55.
|
Katseprotokollis tuleks esitada järgmine teave.
|
|
Uuritav kemikaal:
|
—
|
allikas, partii number, aegumiskuupäev, kui on olemas;
|
|
—
|
uuritava kemikaali püsivus, kui see on teada;
|
|
|
|
ühest koostisosast koosnev aine:
|
—
|
füüsiline välimus, lahustuvus vees ja muud asjakohased füüsikalis-keemilised omadused;
|
|
—
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus, CASi number, SMILESi või InChI kood, struktuurivalem, puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
|
|
mitut koostisosa sisaldav aine, UVCB või segu:
|
—
|
võimalikult täpne omaduste kirjeldus lähtuvalt koostisosade keemilisest määratlusest (vt eespool), kvantitatiivsest sisaldusest ja asjakohastest füüsikalis-keemilistest omadustest.
|
|
|
|
Kandeaine (vajaduse korral):
|
—
|
kandeaine valimise põhjendus, kui kandeaine ei ole vesi.
|
|
|
|
Katseloomad:
|
—
|
loomade arv, vanus ja sugu;
|
|
—
|
päritolu, pidamistingimused, söötmisandmed jne;
|
|
—
|
iga looma kehamass katse alguses;
|
|
—
|
liigi valimise põhjendus, kui katseloom ei ole rott.
|
|
|
|
Katsetingimused:
|
—
|
annuste valimise põhjendus;
|
|
—
|
üksikasjad uuritava kemikaali valmistise / söödavalmistise, saavutatud kontsentratsioonide ning valmistise püsivuse ja homogeensuse kohta;
|
|
—
|
uuritava kemikaali manustamise üksikasjad;
|
|
—
|
vajaduse korral andmed söödas/joogivees sisalduva uuritava kemikaali kontsentratsiooni (ppm) teisendamiseks tegelikuks annuseks (mg kehamassi kilogrammi kohta päevas);
|
|
—
|
sööda ja vee kvaliteedi üksikasjad;
|
|
—
|
järglaste surmamise korral üksikasjalik kirjeldus surmatavate järglaste juhusliku valimise korra kohta.
|
|
|
|
Tulemused:
|
—
|
kehamass / kehamassi muutused;
|
|
—
|
söödatarbimine ja veetarbimise andmed, kui need on olemas;
|
|
—
|
mürgise mõju andmed kummagi soo ja iga annusemäära kohta, sealhulgas viljakusele ja tiinusele avalduva mõju ning mis tahes muude mürgisusnähtude kohta;
|
|
—
|
mürgisus või muu mõju paljunemisele, järglastele, sünnijärgsele kasvule jne;
|
|
—
|
kliiniliste ilmingute laad, raskusaste ja kestus (nähtude pöörduvus või pöördumatus);
|
|
—
|
normaalse või ebanormaalse innatsükliga täiskasvanud emasloomade arv ja tsükli kestus;
|
|
—
|
elussündide arv ja pesastumisjärgne kadu;
|
|
—
|
järglaste kehamassi andmed;
|
|
—
|
iga järglase AGD (ja kehamass AGD mõõtmise päeval);
|
|
—
|
nisade esinemine isastel järglastel;
|
|
—
|
kilpnäärmehormoonide sisaldus järglastelt 13. päeval võetud proovides ja täiskasvanud isasloomade proovides (ning järglastega emasloomade proovides ja järglastelt 4. päeval võetud proovides, kui seda on mõõdetud);
|
|
—
|
silmaga nähtavate kõrvalekalletega järglaste arv, väliste suguelundite makroskoopilise hindamise tulemused, kängunud järglaste arv;
|
|
—
|
katse vältel suremise aeg või teave selle kohta, et loomad olid kuni surmamiseni elus;
|
|
—
|
pesastunud embrüote arv, pesakonna suurus ja pesakonna kaal mõõtmishetkel;
|
|
—
|
andmed järglastega loomade kehamassi kohta surmamise hetkel ning selliste loomade elundite massi kohta;
|
|
—
|
histopatoloogiliste leidude üksikasjalik kirjeldus;
|
|
—
|
andmed absorbeerumise kohta (kui on olemas);
|
|
—
|
vajaduse korral tulemuste statistilise analüüsi andmed.
|
|
Tulemuste arutelu
Järeldused
|
Tulemuste tõlgendamine
|
56.
|
Kirjeldatud uuring võimaldab hinnata annuste korduval manustamisel täheldatavat paljunemisele/arengule avalduvat mürgist mõju (vt punktid 5 ja 6). Saadud tulemused võivad viidata vajadusele teha täiendavaid uuringuid ja hõlbustavad järgnevate uuringute kavandamist. Paljunemise ja arenguga seotud tulemuste hõlpsamaks tõlgendamiseks tuleks tutvuda OECD juhenddokumendiga nr 43 (12). Käesoleva katsemeetodi puhul võib olla kasu närilistega tehtavate sisesekretsioonisüsteemi ja paljunemisega seotud katsete tulemuste histoloogilist hindamist käsitlevas OECD juhenddokumendis nr 106 (11) esitatud teabest (sisesekretsiooni)elundite ja tupeäiete ettevalmistamise ja hindamise kohta.
|
KIRJANDUS
|
(1)
|
OECD (1990). Kemikaalide töörühma ja halduskomitee 14. ühiskoosoleku koosolekudokument nr 1. Taotluse korral kättesaadav Majanduskoostöö ja Arengu Organisatsioonist Pariisis.
|
|
(2)
|
OECD (1992). Ajutise eksperdirühma esimehe aruanne reproduktiivtoksilisust käsitlevate sõeluuringumeetodite alase koosoleku kohta. Tokyo, 27.–29. oktoober 1992. Taotluse korral kättesaadav Majanduskoostöö ja Arengu Organisatsioonist Pariisis.
|
|
(3)
|
OECD (1998). Endokriinfunktsiooni kahjustavate kemikaalide uurimise ja hindamise (EDTA) OECD rakkerühma esimese koosoleku aruanne. 10.–11. märts 1998. Taotluse korral kättesaadav Majanduskoostöö ja Arengu Organisatsioonist Pariisis.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment No. 217. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Series on Testing and Assessment No. 19. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment No. 150. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(7)
|
Goldman, J. M., Murr, A. S., ja Cooper, R. L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies. Birth Defects Res. B 80(2): 84–97.
|
|
(8)
|
Sadleir, R. M. F. S. (1979). Cycles and Seasons. Väljaandes: Reproduction in Mammals: I. Germ Cells and Fertilization. Auston, C. R., ja Short, R. V. (toim.), Cambridge, New York.
|
|
(9)
|
Gallavan, R. H. Jr., Holson, J. F., Stump, D. G., Knapp, J. F., ja Reynolds, V. L. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights. Reprod. Toxicol. 13: 383–390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment No. 151. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment No. 106. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment No. 43. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
1. liide
MÕISTED (VT KA OECD JUHENDDOKUMENT NR 150 (6))
Androgeensus – kemikaali võime toimida imetaja organismis samal viisil kui looduslik androgeen (nt testosteroon).
Annus – manustatav uuritava kemikaali kogus. Annust väljendatakse uuritava kemikaali kogusena massiühikutes katselooma kehamassi ühiku kohta päevas (näiteks milligrammides kehamassi kilogrammi kohta päevas) või kemikaali püsiva kontsentratsioonina söödas.
Annustamine – üldmõiste, mis hõlmab annust, selle manustamise sagedust ja annustamise kestust.
Antiandrogeensus – kemikaali võime pärssida imetaja organismis loodusliku androgeeni (nt testosterooni) toimet.
Antitüreoidne aktiivsus – kemikaali võime pärssida imetaja organismis loodusliku kilpnäärmehormooni (nt T3) toimet.
Antiöstrogeensus – kemikaali võime pärssida imetaja organismis loodusliku östrogeeni (nt 17β-östradiooli) toimet.
Arengutoksilisus – reproduktiivtoksilisuse ilming, mis avaldub järglaste sünnieelse, perinataalse või sünnijärgse struktuuri- või funktsioonihäirena.
Emale avalduv mürgisus – tiinele emasloomale avalduv kahjulik mõju, mis on spetsiifiline (otsene mõju) või mittespetsiifiline (kaudne mõju).
Ilmne mürgisus – üldmõiste, mille abil kirjeldatakse uuritava kemikaali manustamise järel täheldatavaid selgeid mürgisusnähte. Kõnealused nähud peaksid olema ohu hindamiseks piisavad ja sellised, et manustatava annuse suurendamisega kaasnevad eeldatavalt tõsised mürgisusnähud ja tõenäoliselt ka surm.
Kemikaal – aine või segu.
NOAEL – lühend, millega tähistatakse annusemäära, mille juures ei täheldata kahjulikku toimet. Tegemist on suurima annusega, mis ei põhjusta manustamisest tingitud kahjulikke ilminguid.
Reproduktiivtoksilisus – kahjulik mõju järglastele ja/või isas- või emasloomade paljunemisfunktsioonile või paljunemisvõimele.
Türeoidne aktiivsus – kemikaali võime toimida imetaja organismis samal viisil kui looduslik kilpnäärmehormoon (nt T3).
Uuritav kemikaal – iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
Valideerimine – teaduslik protsess, mille eesmärk on iseloomustada katsemeetodi rakendamisega seotud nõudeid ja piiranguid ning tõendada meetodi usaldusväärsust ja sobivust konkreetsel eesmärgil kasutamiseks.
Viljakuse vähenemine – isas- või emaslooma paljunemisfunktsiooni häire või paljunemisvõime langus.
Östrogeensus – kemikaali võime toimida imetaja organismis samal viisil kui looduslik östrogeen (nt 17β-östradiool).
2. liide
UURINGU MAKSIMUMKESTUSELE VASTAV TÄISPIKAL 14-PÄEVASEL PAARITUMISPERIOODIL PÕHINEV KATSE SKEMAATILINE AJAKAVA

3. liide
TABELI KUJUL KOKKUVÕTLIK ARUANNE PALJUNEMISELE/ARENGULE AVALDUVA MÕJU KOHTA
|
VAATLUSED
|
VÄÄRTUSED
|
|
|
|
Annus (ühik)
|
0 (kontroll)
|
…
|
…
|
…
|
…
|
|
Paaride arv katse alguses (N)
|
|
|
|
|
|
|
Innatsükkel (vähemalt keskmine pikkus ja ebaregulaarsete tsüklite esinemissagedus)
|
|
|
|
|
|
|
Emasloomad, kellel täheldatakse paaritumise ilminguid (N)
|
|
|
|
|
|
|
Tiinestunud emasloomad (N)
|
|
|
|
|
|
|
Eostunud 1.–5. päeval (N)
|
|
|
|
|
|
|
Eostunud 6.–… (21) päeval (N)
|
|
|
|
|
|
|
Tiinuse kestus ≤ 21 päeva (N)
|
|
|
|
|
|
|
Tiinuse kestus 22 päeva (N)
|
|
|
|
|
|
|
Tiinuse kestus ≥ 23 päeva (N)
|
|
|
|
|
|
|
Elusalt sündinud järglastega emasloomad (N)
|
|
|
|
|
|
|
4. sünnijärgse päevani elus püsinud järglastega emasloomad (N)
|
|
|
|
|
|
|
Pesastunud embrüote arv tiinestunud emaslooma kohta (keskmine)
|
|
|
|
|
|
|
Elussündide arv järglastega emaslooma kohta (keskmine)
|
|
|
|
|
|
|
4. päevani elus püsinud järglaste arv järglastega emaslooma kohta (keskmine)
|
|
|
|
|
|
|
Sugude vahekord (isased/emased) sünnihetkel (keskmine)
|
|
|
|
|
|
|
Sugude vahekord (isased/emased) 4. päeval (keskmine)
|
|
|
|
|
|
|
Pesakonna kaal sünnihetkel (keskmine)
|
|
|
|
|
|
|
Pesakonna kaal 4. päeval (keskmine)
|
|
|
|
|
|
|
Järglaste kehamass sünnihetkel (keskmine)
|
|
|
|
|
|
|
Järglaste kehamass AGD mõõtmise hetkel (isasloomade keskmine, emasloomade keskmine)
|
|
|
|
|
|
|
Järglaste AGD samal sünnijärgsel päeval ajavahemikul sündimispäevast kuni 4. päevani (isasloomade keskmine, emasloomade keskmine; märkida sünnijärgne mõõtmispäev)
|
|
|
|
|
|
|
Järglaste kehamass 4. päeval (keskmine)
|
|
|
|
|
|
|
Nisade esinemine isastel järglastel 13. päeval (keskmine)
|
|
|
|
|
|
|
Järglaste kehamass 13. päeval (keskmine)
|
|
|
|
|
|
|
|
|
KÕRVALEKALLETEGA JÄRGLASED
|
|
Järglastega emasloomad, kellel on 0 kõrvalekalletega järglast
|
|
|
|
|
|
|
Järglastega emasloomad, kellel on 1 kõrvalekalletega järglane
|
|
|
|
|
|
|
Järglastega emasloomad, kellel on ≥ 2 kõrvalekalletega järglast
|
|
|
|
|
|
|
|
|
JÄRGLASTE KADU
|
|
|
|
Sünnieelne/pesastumisjärgne (pesastunud embrüote arvu ja elussündide arvu vahe)
|
|
0 järglast kaotanud emasloomad
|
|
|
|
|
|
|
1 järglase kaotanud emasloomad
|
|
|
|
|
|
|
2 järglast kaotanud emasloomad
|
|
|
|
|
|
|
≥ 3 järglast kaotanud emasloomad
|
|
|
|
|
|
|
|
|
Sünnijärgne (elussündide arvu ja 13. sünnijärgse päevani elus püsinud järglaste arvu vahe)
|
|
0 järglast kaotanud emasloomad
|
|
|
|
|
|
|
1 järglase kaotanud emasloomad
|
|
|
|
|
|
|
2 järglast kaotanud emasloomad
|
|
|
|
|
|
|
≥ 3 järglast kaotanud emasloomad
|
|
|
|
|
|
B.64 REPRODUKTIIV-/ARENGUTOKSILISUSE SÕELUURINGUT HÕLMAV KORDUVANNUSTAMISEL PÕHINEV MÜRGISUSUURING
SISSEJUHATUS
|
1.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 422 (2016). Kemikaalide uurimist käsitlevaid OECD katsejuhendeid vaadatakse teaduse arengu arvessevõtmiseks korrapäraselt läbi. Algne sõeluuringut käsitlev katsejuhend nr 422 võeti vastu 1996. aastal ühe reproduktiiv-/arengutoksilisuse hindamist hõlmava korduvannustamisel põhineva sõeluuringu katse-eeskirja põhjal, mida arutati kahel ekspertide koosolekul, mis toimusid 1990. aastal Londonis (1) ja 1992. aastal Tokyos (2).
|
|
2.
|
Käesoleva katsemeetodi puhul on reproduktiiv-/arengutoksilisuse sõeluuring, mis põhineb liikmesriikides suures mahus toodetavate olemasolevate kemikaalide hindamiseks kasutatud algse meetodiga omandatud ja positiivseid kontrollaineid hõlmavatest ettevalmistavatest katsetest saadud kogemustel (3, 4), ühendatud korduvannustamisel avalduva mürgisuse uuringuga, mis on kooskõlas OECD katsejuhendiga nr 407 (sellele vastab käesoleva lisa peatükk B.7 „Suukaudse kordusdoosi toksilisuse 28-päevane uuring närilistel“).
|
|
3.
|
Käesolevat katsemeetodit on ajakohastamise eesmärgil täiendatud endokriinfunktsiooni kahjustavate kemikaalide uurimiseks sobivate lõppnäitajatega, mis lisati järelmeetmena lähtuvalt prioriteetsest tegevussuunast, mille OECD käivitas 1998. aastal eesmärgiga vaadata läbi olemasolevad ja töötada välja uued katsejuhendid, et võimaldada sõeluuringute tegemist võimalike endokriinfunktsiooni kahjustavate kemikaalide tuvastamiseks ja selliste kemikaalide hindamist (5). Selles kontekstis täiendati 2008. aastal katsejuhendit nr 407 (sellele vastab käesoleva lisa peatükk B.7) parameetritega, mis sobivad uuritavate kemikaalide endokriinse toime tuvastamiseks. Katsejuhendi nr 422 ajakohastamise eesmärk oli lisada sõeluuringut käsitlevasse juhendisse teatavad endokriinfunktsiooni kahjustavate kemikaalide uurimiseks sobivad lõppnäitajad, mida saab hinnata juhul, kui kokkupuuteperiood hõlmab mõnda tundlikku arenguperioodi (sünnieelset või varajast sünnijärgset perioodi).
|
|
4.
|
Peale selle lisati katsejuhendisse nr 422 täiendavad endokriinfunktsiooni kahjustavate kemikaalide uurimiseks sobivad lõppnäitajad, mida sisaldab ka katsejuhend nr 443 (sellele vastab käesoleva lisa peatükk B.56 „Laiendatud ühe põlvkonna reproduktiivtoksilisuse uuring“); need lõppnäitajad valiti lähtuvalt ühest teostatavusuuringust, milles käsitletakse asjaomaste lõppnäitajate lisamisega seotud teaduslikke ja tehnilisi küsimusi, ning võimalikust vajadusest katsemeetodit nende lisamiseks kohandada (6).
|
|
5.
|
Käesolev katsemeetod on kavandatud piiratud hulga andmete saamiseks uuritava kemikaali mõju kohta isas- ja emasloomade paljunemisvõimele, näiteks sugunäärmete talitlusele, paaritumiskäitumisele, eostumisele, sügoodi arengule ja sünnitamisele. See ei ole ette nähtud kasutamiseks olemasolevate katsemeetodite B.31, B.34, B35 või B.56 alternatiivina ega nende asendamiseks.
|
LÄHTEKAALUTLUSED
|
6.
|
Uuritava kemikaali mürgisust põhjustavate omaduste kindlakstegemiseks ja hindamiseks võib teha korduval suukaudsel manustamisel avalduva mürgisuse uuringu pärast seda, kui ägeda mürgisuse katsetest on saadud esialgsed andmed kemikaali mürgisuse kohta. Käesoleva meetodi kohane uuring võimaldab saada teavet võimalike terviseohtude kohta, mis avalduvad suhteliselt piiratud ajavahemiku jooksul toimuva korduva kokkupuute tagajärjel. Käesolev meetod hõlmab lihtsat korduvannustamisel põhinevat mürgisusuuringut, mida võib kasutada kemikaalide jaoks, mille puhul 90-päevase uuringu tegemine ei ole põhjendatud (näiteks kui tootmismaht ei ületa teatavat piiri), või eeluuringuna enne pikaajalise uuringu läbiviimist. Uuringu tegemisel tuleks järgida juhtpõhimõtteid ja kaalutlusi, mis on esitatud OECD juhenddokumendis nr 19, milles käsitletakse ohutuse hindamiseks kasutatavatel katseloomadel avalduvate kliiniliste tunnuste kindlakstegemist, hindamist ja kasutamist humaanse sekkumise vajadusele viitavate näitajatena (7).
|
|
7.
|
Peale selle hõlmab käesolev katsemeetod ka reproduktiiv-/arengutoksilisuse sõeluuringut ning seepärast saab seda ühtlasi kasutada uuritava kemikaali toksikoloogiliste omaduste hindamise varajases etapis või probleemse kemikaali uurimisel esialgse teabe saamiseks võimaliku mõju kohta isas- ja emasloomade paljunemisvõimele, näiteks sugunäärmete talitlusele, paaritumiskäitumisele, eostumisele, sügoodi arengule ja sünnitamisele. Käesolev katsemeetod ei võimalda saada ammendavat teavet kõikide paljunemisvõime ja arenguga seotud aspektide kohta. Eelkõige lubab see tuvastada sünnieelse kokkupuute tagajärjel sünnijärgselt avalduvaid ilminguid ja sünnijärgsest kokkupuutest tulenevat mõju üksnes piiratud ulatuses. Käesoleva meetodiga ei saa koguda tõendeid, mis lubaksid lõplikult kinnitada paljunemisele/arengule avalduva mõju puudumist; see on muu hulgas tingitud lõppnäitajate selektiivsusest ja uuringu lühikesest kestusest. Täheldatavaid positiivseid tulemusi aga on võimalik muudest reproduktiiv-/arengutoksilisuse uuringutest saadavate andmete puudumisel kasutada esialgseks ohuhindamiseks ning need aitavad teha otsuseid täiendavate uuringute tegemise vajaduse ja selliste uuringute ajastuse kohta.
|
|
8.
|
Sisesekretsioonisüsteemiga seotud näitajaid käsitlevaid tulemusi tuleks vaadelda endokriinfunktsiooni kahjustavate kemikaalide uurimise ja hindamise OECD kontseptuaalse raamistiku (8) kontekstis. Nimetatud kontseptuaalses raamistikus on OECD täiendatud katsejuhendi nr 422 kohast meetodit käsitatud 4. taseme in vivo meetodina, mis võimaldab saada andmeid sisesekretsioonisüsteemiga seotud lõppnäitajate kaudu tuvastatava kahjuliku mõju kohta. Sisesekretsioonisüsteemist sõltuvat signaali ei pruugita eraldi võetuna siiski pidada piisavaks tõendiks selle kohta, et uuritava kemikaali puhul on tegemist endokriinfunktsiooni kahjustava kemikaaliga.
|
|
9.
|
Käesoleva katsemeetodi puhul pööratakse ühtlasi suurt tähelepanu konkreetse lõppnäitajana vaadeldavale neuroloogilisele mõjule ja rõhutatakse loomade hoolika kliinilise jälgimise vajadust, et saada võimalikult palju teavet. Meetod peaks võimaldama teha kindlaks kemikaalide võimaliku neurotoksilisuse, mida võib olla vaja täiendavalt põhjalikumalt uurida. Peale selle võib käesoleva meetodiga saada esialgset teavet immunoloogilise mõju kohta.
|
|
10.
|
Täheldatavaid positiivseid tulemusi on võimalik muudest süsteemse mürgisuse, reproduktiiv-/arengutoksilisuse, neurotoksilisuse ja/või immunotoksilisuse uuringutest saadavate andmete puudumisel kasutada esialgseks ohuhindamiseks ning need aitavad teha otsuseid täiendavate uuringute tegemise vajaduse ja selliste uuringute ajastuse kohta. Katse tulemused võivad olla eriti kasulikud OECD sõeluuringute andmekogumi (SIDS) osana selliste olemasolevate kemikaalide hindamisel, mille kohta on vähe toksikoloogilist teavet või see puudub, ning sellist katset saab kasutada kahe eraldi katse asemel, milles hinnatakse vastavalt korduvannustamisel avalduvat mürgisust (OECD katsejuhend nr 407, mis vastab käesoleva lisa peatükile B.7) ja reproduktiiv-/arengutoksilisust (OECD katsejuhend nr 421, mis vastab käesoleva lisa peatükile B.63). Samuti võib seda kasutada ulatuslikumate paljunemisvõimet/arengut käsitlevate uuringute jaoks annusevahemiku kindlaksmääramiseks ning muudel asjakohaseks peetavatel juhtudel.
|
|
11.
|
Üldjuhul eeldatakse, et tiinete ja mittetiinete loomade tundlikkus on erinev. Seepärast võib kõnealuses ühendkatses kahe eraldi katse läbiviimisega võrreldes olla keerulisem teha kindlaks sobivad annusemäärad, mis võimaldaksid üheaegselt hinnata nii üldist süsteemset mürgisust kui ka konkreetselt reproduktiiv-/arengutoksilisust. Peale selle võib üldise süsteemse mürgisusega seotud katsetulemuste tõlgendamine olla raskem kui korduvannustamisel põhineva eraldi katse tegemisel, eriti juhul, kui seeruminäitajaid ja histopatoloogilisi näitajaid ei analüüsita uuringus üheaegselt. Nende tehniliste aspektide keerukusest tulenevalt nõuab kõnealuse ühendatud sõeluuringu läbiviimine ulatuslikke kogemusi mürgisuse hindamisel. Teisest küljest võimaldab selline ühendkatse peale väiksema arvu loomade kasutamise paremini eristada paljunemisele/arengule avalduvat otsest mõju mõjust, mis ilmneb muu (süsteemse) mõju tagajärjel.
|
|
12.
|
Käesoleva meetodi puhul on manustamisperiood pikem kui tavapärases 28-päevases korduvannustamisel põhinevas uuringus. Samal ajal on kummastki soost loomade arv igas rühmas väiksem kui olukorras, kus peale tavapärase 28-päevase korduvannustamisel põhineva uuringu tehakse veel ka reproduktiiv-/arengutoksilisuse sõeluuring.
|
|
13.
|
Käesoleva katsemeetodi puhul eeldatakse, et uuritavat kemikaali manustatakse suu kaudu. Muu manustamisviisi kasutamise korral võib olla vaja teha meetodis muudatusi.
|
|
14.
|
Kui soovitakse saada kavandatud regulatiivsel eesmärgil andmeid segu kohta, tuleks enne katsemeetodi kasutamist kaaluda, kas ja kuidas see võimaldab saada kõnealusele eesmärgile vastavaid asjakohaseid tulemusi. Selline kaalumine ei ole vajalik, kui asjaomase segu hindamine on regulatiivse nõudega ette nähtud.
|
|
15.
|
Kasutatud mõisted on määratletud 1. liites.
|
KATSE PÕHIMÕTE
|
16.
|
Uuritavat kemikaali manustatakse astmeliselt suurenevates annustes mitme rühma isas- ja emasloomadele. Isasloomade puhul peaks manustamine toimuma vähemalt nelja nädala kestel kuni kavandatud surmamispäevale eelneva päevani, sealhulgas surmamiseelsel päeval (see hõlmab vähemalt kahte paaritumiseelset nädalat, paaritumisperioodi ja umbes kahte paaritumisjärgset nädalat). Kuna isasloomadel on paaritumiseelne manustamisperiood piiratud kestusega, ei pruugi viljakus olla eriti tundlik munanditele avalduva mürgisuse näitaja. Seepärast on vajateha munandite üksikasjalik histoloogiline analüüs. Kahe nädala pikkust paaritumiseelset manustamisperioodi koos järgneva paaritumise/viljakuse jälgimisega ning manustamisperioodi üldkestusega vähemalt neli nädalat, millele järgneb isaslooma sugunäärmete üksikasjalik histopatoloogiline analüüs, peetakse enamiku isasloomal täheldatavate viljakusele ja spermatogeneesile avalduva mõju ilmingute tuvastamiseks piisavaks.
|
|
17.
|
Emasloomade puhul peaks manustamine toimuma kogu uuringu kestel. See hõlmab kahte paaritumiseelset nädalat (eesmärk on jälgida vähemalt kahte tervet innatsüklit), eostumisele eelnevat varieeruva pikkusega ajavahemikku, tiinuse kestust ja vähemalt kolmeteistkümne päeva pikkust sünnitusjärgset perioodi, mis kestab kuni kavandatud surmamispäevale eelneva päevani ja hõlmab ka surmamiseelset päeva.
|
|
18.
|
Kohanemisperioodile ja innatsükli manustamiseelsele hindamisele järgneva uuringu kestus sõltub emasloomade seisundist ning on umbes 63 päeva (vähemalt 14 päeva pikkune paaritumiseelne periood, (kuni) 14 päeva pikkune paaritumisperiood, 22 päeva pikkune tiinusperiood ja 13 päeva pikkune imetamisperiood).
|
|
19.
|
Manustamisperioodi jooksul jälgitakse loomi iga päev hoolikalt mürgisusnähtude suhtes. Uuringu kestel surnud või surmatud loomad lahatakse ning uuringu lõpus surmatakse ja lahatakse ka ellujäänud loomad.
|
MEETODI KIRJELDUS
Loomaliigi valimine
|
20.
|
Käesolev katsemeetod on kavandatud kasutamiseks rotil. Kui katsejuhendis nr 422 kirjeldatud näitajaid hinnatakse mõne muu liigi närilistel, tuleks seda üksikasjalikult põhjendada. Endokriinfunktsiooni kahjustavate kemikaalide kindlakstegemise rahvusvahelises valideerimisprogrammis, mille puhul on lähtutud katsejuhendist nr 407, kasutati ainsa liigina rotte. Ei tohiks kasutada liine, kus sigivus on väike või kus teadaolevalt esineb sageli arenguhäireid. Tuleks kasutada terveid varem paaritamata loomi, keda ei ole varem katsetes kasutatud. Katseloomi tuleks iseloomustada liigi, liini, soo, kehamassi ja vanuse järgi. Uuringu alguses peaksid kasutatavate loomade kehamassi erinevused olema minimaalsed ning mitte üle ± 20 % kummastki soost loomade keskmisest kehamassist. Kui uuring viiakse läbi pikaajalise või tervet põlvkonda hõlmava uuringu eeluuringuna, tuleks mõlemas uuringus soovitatavalt kasutada samast liinist ja sama päritolu loomi.
|
Pidamis- ja söötmistingimused
|
21.
|
Kõik katsetoimingud peaksid vastama kohalikele laboriloomade hooldamise normidele. Katseloomade ruumi temperatuur peaks olema 22 °C (± 3 °C). Suhteline õhuniiskus peaks olema vähemalt 30 % ja soovitatavalt mitte üle 70 %, v.a ruumi koristamise ajal. Tuleks kasutada tehisvalgustust valgusrežiimiga 12 tundi valgust ja 12 tundi pimedust. Söötmiseks võib kasutada tavapärast laborisööta ja piiramatus koguses joogivett. Söödavalikut võib mõjutada vajadus tagada sööda sobivus uuritava kemikaali lisamiseks, kui kemikaali manustatakse sellisel viisil.
|
|
22.
|
Loomade pidamisel tuleks kasutada väikesi samasooliste isendite rühmi; loomi võib pidada eraldi, kui see on teaduslikult põhjendatud. Rühmas pidamisel ei tohiks puuris olla rohkem kui viis looma. Paaritamine peaks toimuma selleks sobivas puuris. Tiined emasloomad tuleks paigutada igaüks eraldi puuri, kus on olemas pesamaterjal. Imetavad emasloomad paigutatakse igaüks eraldi puuri koos järglaskonnaga.
|
|
23.
|
Sööta tuleks korrapäraselt analüüsida saasteainete suhtes. Asjaomase sööda proov tuleks kuni katseprotokolli lõpliku valmimiseni alles hoida.
|
Loomade ettevalmistamine
|
24.
|
Terved noored täiskasvanud loomad jagatakse juhuvaliku alusel katserühmadesse ja puuridesse. Puurid paigutatakse nii, et puuri asukohast tingitud võimalik mõju oleks võimalikult väike. Loomad idenditakse üheselt ja neil lastakse enne uuringu algust puuris vähemalt viis päeva laboritingimustega kohaneda.
|
Annuste ettevalmistamine
|
25.
|
Uuritavat kemikaali on soovitatav manustada suu kaudu, välja arvatud juhul, kui mõnda muu manustamisviisi peetakse sobivamaks. Uuritava kemikaali suukaudseks manustamiseks kasutatakse tavaliselt toitmissondi; selle asemel võib siiski kasutada ka sööda või joogiveega manustamist.
|
|
26.
|
Vajaduse korral lahustatakse või suspendeeritakse uuritav kemikaal sobivas kandeaines. Kui vähegi võimalik, on soovitatav esmalt kaaluda vesilahuse või veepõhise suspensiooni ja seejärel õlilahuse või õlipõhise suspensiooni (nt maisiõli) kasutamist ning pärast seda lahustamist mõnes muus kandeaines. Muu kandeaine kui vee puhul peaksid olema teada selle mürgisusnäitajad. Tuleks teha kindlaks uuritava kemikaali püsivus ja homogeensus kandeaines.
|
KATSE KÄIK
Loomade arv ja sugu
|
27.
|
Soovitatavalt peaks igas rühmas olema alguses vähemalt 10 isaslooma ja 12–13 emaslooma. Emasloomi hinnatakse kokkupuute eel innatsükli alusel ja loomad, kelle innatsükkel erineb tavapärasest 4–5 päeva pikkusest tsüklist, jäetakse uuringust välja; seepärast on soovitatav kasutada suuremat arvu emasloomi, et igas rühmas oleks lõpuks 10 emaslooma. Eeldatavalt tiinestub igas rühmas vähemalt 8 emaslooma – seda peetakse tavapäraselt minimaalseks vastuvõetavaks tiinestumismääraks –, välja arvatud juhul, kui ilmneb märkimisväärne mürgine mõju. Eesmärk on saada piisav arv tiineid emasloomi, et oleks võimalik usaldusväärselt hinnata uuritava kemikaali võimet mõjutada viljakust, tiinust, ema- ja imetamiskäitumist ning F1-põlvkonna järglaste kasvu ja arengut eostamisest kuni kolmeteistkümnenda sünnijärgse päevani. Kui osa loomi on kavas surmata juba katse ajal, tuleks suurendada loomade arvu nende loomade arvu võrra, kes surmatakse plaanijärgselt enne katse lõppu. Tuleks kaaluda võimalust luua kontrollrühma ja suurima annusega töödeldava rühma juurde täiendrühm, millesse kuulub kummastki soost viis looma, et jälgida vähemalt 14 päeva jooksul pärast manustamise lõpetamist süsteemse mürgise mõju pöörduvust, püsivust või viivitusega avaldumist. Täiendrühma loomi ei paaritata ja neid ei kasutata seega reproduktiiv-/arengutoksilisuse hindamisel.
|
Annused
|
28.
|
Üldjuhul tuleks kasutada vähemalt kolme katserühma ja ühte kontrollrühma. Sobivate üldise mürgisuse andmete puudumisel võib kasutatavate annuste kindlaksmääramise hõlbustamiseks teha annusevahemiku kindlaksmääramise uuringu, kus kasutatakse samast liinist ja sama päritolu loomi. Kontrollrühma loomi tuleks kohelda täpselt samal viisil kui katserühma loomi; ainus erinevus seisneb uuritava kemikaali manustamata jätmises. Kui uuritava kemikaali manustamisel kasutatakse kandeainet, tuleks võrdlusrühma loomadele manustada kandeainet suurimas kasutatavas koguses.
|
|
29.
|
Annuste valimisel tuleks võtta arvesse kõiki kättesaadavaid mürgisusandmeid ja (toksiko)kineetilisi andmeid. Samuti tuleks arvesse võtta, et tiinete ja mittetiinete loomade tundlikkus võib olla erinev. Suurim annus tuleks valida nii, et kemikaal avaldaks mürgist mõju, kuid ei põhjustaks surma ega ilmseid kannatusi. Seejärel tuleks valida rida järk-järgult vähenevaid annuseid, mis võimaldaksid tõendada mõju sõltuvust annusest ja väikseima annuse puhul kahjuliku mõju puudumist. Sageli on eri annuste vaheline optimaalne erinevus kahe- kuni neljakordne ning üksteisest väga palju (näiteks üle 10 korra) erinevate annuste kasutamise asemel on tihti soovitatav lisada neljas katserühm.
|
|
30.
|
Kui täheldatakse üldist mürgisust (nt kehamassi vähenemine, mõju maksale, südamele, kopsudele või neerudele jne) või muid muutusi, mis ei pruugi olla tingitud mürgisusest (nt väiksem söödatarbimine, maksa suurenemine), tuleks täheldatud mõju sisesekretsioonisüsteemiga seotud lõppnäitajatele tõlgendada ettevaatusega.
|
Piirsisalduskatse
|
31.
|
Kui käesoleva meetodi kohaselt läbi viidud suukaudse manustamise uuringus, milles kasutatakse ainsa annusena annust vähemalt 1 000 mg kehamassi kilogrammi kohta päevas või sööda või joogiveega manustamise korral sellega samaväärset protsentuaalset sisaldust söödas või joogivees (see määratakse kindlaks kehamassi mõõtmise alusel), ei täheldata mürgist mõju ja kui mürgisus ei ole uuritava kemikaaliga struktuurilt sarnaste ainetega saadud andmete põhjal ootuspärane, ei pruugita pidada mitme eri annusega täismahus uuringu tegemist vajalikuks. Piirsisalduskatse tulemused on kohaldatavad, kui inimese kokkupuutega seotud andmed ei viita vajadusele kasutada suuremat annusemäära. Muu manustamisviisi korral, näiteks sissehingamise teel või naha kaudu manustamisel võib uuritava kemikaali füüsikalis-keemilistest omadustest tulenevalt olla tihti vaja kasutada suurimat saavutatavat kokkupuutemäära.
|
Annuste manustamine
|
32.
|
Uuritavat kemikaali manustatakse loomadele seitsmel päeval nädalas. Kui manustamiseks kasutatakse toitmissondi, tuleks kemikaali loomadele manustada ühe annusena maosondi või sobiva intubeerimiskanüüli abil. Suurim korraga manustatav vedelikukogus sõltub katselooma suurusest. See kogus ei tohiks olla suurem kui 1 ml 100 g kehamassi kohta, välja arvatud vesilahuse puhul, mida võib manustada koguses 2 ml 100 g kehamassi kohta. Katseskasutatava vedelikukoguse varieeruvuse minimeerimiseks tuleks reguleerida uuritava kemikaali kontsentratsiooni nii, et lahuse maht oleks kõikide annuste puhul ühesugune; see ei ole nõutav ärritava või söövitava uuritava kemikaali puhul, mille mõju kontsentratsiooni suurenedes tavaliselt tugevneb.
|
|
33.
|
Sööda või joogiveega manustatava uuritava kemikaali puhul on oluline tagada, et seda kasutatakse koguses, mis ei muuda tavapärast toitumise või joogivee tarbimise tasakaalu. Kui uuritavat kemikaali manustatakse söödaga, võib kasutada kas muutumatut kontsentratsiooni söödas (ppm) või looma kehamassi suhtes muutumatut annusemäära; tuleks täpsustada, kumba võimalust kasutatakse. Toitmissondi kaudu manustatava uuritava kemikaali puhul peaks manustamine toimuma igal päeval sarnasel kellaajal ning annusemäära tuleks vähemalt kord nädalas kohandada, et see püsiks looma kehamassi suhtes muutumatuna. Kui ühendkatse viiakse läbi pikaajalise või täismahus reproduktiivtoksilisuse uuringu eeluuringuna, tuleks mõlemas uuringus kasutada samasugust sööta.
|
Katse ajakava
|
34.
|
Annuste manustamist tuleks kummagi soo puhul alustada kaks nädalat enne paaritamist pärast seda, kui loomadel on lastud vähemalt viis päeva kohaneda ja emasloomi on hinnatud lähtuvalt innatsükli tavapärasest kestusest (töötlemiseelse kahe nädala pikkuse perioodi vältel). Uuringu ajakava peaks olema selline, et innatsükli hindamist alustatakse varsti pärast seda, kui loomad on saanud täielikult suguküpseks. See aeg võib olla eri rotiliinidel ja eri laborites veidi erinev: näiteks Sprague Dawley rottidel saabub suguküpsus 10 nädala vanuses ja Wistari rottidel umbes 12 nädala vanuses. Järglastega emasloomad tuleks surmata 13. sünnitusjärgsel päeval või veidi hiljem. Järglastega emasloomi ja järglasi ei pea tingimata surmama samal päeval, kui järglastega emasloomadel eelistatakse lasta enne vereproovi võtmist hommikuni paastuda. Sünnitamispäeva (päev, mil sünnitus lõpeb) loetakse sünnitusjärgseks 0-päevaks. Emasloomad, kellel ei täheldata paaritumise ilminguid, surmatakse 24–26 päeva pärast paaritumisperioodi viimast päeva. Paaritumisperioodi vältel jätkatakse kummastki soost loomadele kemikaali manustamist. Isasloomadele manustamist tuleks paaritumisperioodi järgselt jätkata vähemalt seni, kuni manustamisperioodi algusest on möödunud kokku minimaalselt 28 päeva. Seejärel loomad surmatakse või teise võimalusena jätkatakse neile kemikaali manustamist, et viia võimaluse korral läbi teine paaritamine, kui seda peetakse asjakohaseks.
|
|
35.
|
Tiinestunud emasloomade puhul tuleks igapäevast manustamist jätkata kogu tiinuse vältel ning vähemalt kuni 13. sünnitusjärgse päevani, sealhulgas nimetatud päeval, või kuni surmamiseelse päevani. Uuringute puhul, kus uuritavat kemikaali manustatakse sissehingamise teel või naha kaudu, tuleks manustamist jätkata vähemalt kuni 19. tiinuspäevani, sealhulgas nimetatud päeval, ning taasalustada seda võimalikult varakult ja mitte hiljem kui 4. sünnitusjärgsel päeval.
|
|
36.
|
Järelvaatluste jaoks ette nähtud täiendrühma kasutamisel jäetakse sellise rühma loomad paaritamata. Neid tuleks pärast järglastega emasloomade esimest plaanijärgset surmamist pidada veel vähemalt 14 päeva ja selle aja jooksul ei tohiks neile kemikaali manustada, et oleks võimalik hinnata mürgise mõju viivitusega avaldumist või selle püsivust või mürgisest mõjust taastumist.
|
|
37.
|
Katse skemaatiline ajakava on esitatud 2. liites.
|
Innatsükkel
|
38.
|
Enne töötlemise alustamist tuleks jälgida innatsüklit ning valida selle alusel uuringusse emasloomad, kelle tsükkel on korrapärane (vt punkt 27). Tupeäigeid tuleks igapäevaselt hinnata ka alates töötlemisperioodi algusest kuni paaritumise ilmingute tuvastamiseni. Kui on kahtlus, et innatsükkel võib pärast manustamise alustamist ägeda stressi mõjul muutuda, võib katseloomi laboris 2 nädala vältel uuritava kemikaaliga töödelda ning seejärel koguda paaritumiseelsel perioodil innatsükli jälgimiseks vähemalt kahe nädala jooksul iga päev tupeäigeid ja jätkata jälgimist ka paaritumisperioodil, kuni täheldatakse paaritumise ilminguid. Tupe-/emakakaelarakkude võtmisel tuleks olla ettevaatlik, et mitte kahjustada limaskesta, kuna see võib põhjustada pseudotiinuse teket (8, 9).
|
Paaritamine
|
39.
|
Käesoleva meetodi kohases uuringus tuleks üldjuhul kasutada paaritamisel suhet 1: 1 (üks isasloom ühe emaslooma kohta). Erandi võib teha juhul, kui isasloomad juhtuvad surema. Emaslooma tuleks hoida sama isaslooma juures seni, kuni täheldatakse paaritumise ilminguid või on möödunud kaks nädalat. Emasloomi tuleks igal hommikul uurida sperma või tupekorgi esinemise suhtes. Tiinuse 0-päevaks loetakse päeva, mil paaritumise toimumine leiab kinnitust (leitakse tupekork või spermat). Kui paaritamine ebaõnnestub, võib kaaluda emaslooma uuesti paaritamist sama rühma viljakaks osutunud isasloomaga.
|
Pesakonna suurus
|
40.
|
Neljandal sünnijärgsel päeval võib iga pesakonna suurust korrigeerida; selleks kõrvaldatakse juhuvaliku alusel üleliigsed järglased nii, et pesakonda jääks sõltuvalt kasutatava rotiliini puhul tavapärasest pesakonna suurusest võimalikult täpselt neli või viis järglast kummagi soo kohta. Üleliigsetest järglastest kahelt tuleks võtta vereproov, moodustada nendest koondproov ja kasutada seda seerumi T4-sisalduse määramiseks. Järglaste selektiivne kõrvaldamine, näiteks kehamassi või anogenitaalse vahemaa (AGD) alusel, ei ole sobiv. Kui emaste või isaste järglaste arv ei võimalda jätta pesakonda nelja või viit järglast kummastki soost, on vastuvõetav ka osaline korrigeerimine (nt kuus isast ja neli emast). Kui pesakond jääks kõrvaldamise tagajärjel väiksemaks kui kõrvaldamisega saavutatav soovitud sihtarv (8 või 10 järglast pesakonna kohta), ei kõrvaldata ühtki järglast. Kui kõrvaldamisega saavutatava soovitud sihtarvuga võrreldes on üle ainult üks järglane, kõrvaldataksegi vaid üks järglane, kellelt võetakse vereproov võimalikuks seerumi T4-sisalduse hindamiseks.
|
|
41.
|
Kui pesakonna suurust ei korrigeerita, surmatakse 4. sünnijärgsel päeval igast pesakonnast kaks järglast ning neilt võetakse vereproov seerumi kilpnäärmehormoonisisalduse mõõtmiseks. Võimaluse korral peaksid kõnealused kaks järglast igast pesakonnast olema emased, et võimaldada isaste järglaste kasutamist nisade esinemise hindamiseks; see soovitus ei kehti juhul, kui kõnealuse kahe järglase eemaldamisel ei jääks pesakonda ühtki emast, keda uuringu lõpus hinnata. Kui pesakonda jääks kõrvaldamise tagajärjel vähem kui 8 või 10 järglast (olenevalt kasutatava rotiliini puhul tavapärasest pesakonna suurusest), ei kõrvaldata ühtki järglast. Kui pesakonna tavapärase suurusega võrreldes on üle ainult üks järglane, kõrvaldataksegi vaid üks järglane, kellelt võetakse vereproov võimalikuks seerumi T4-sisalduse hindamiseks.
|
Vaatlused
|
42.
|
Üldisi kliinilisi vaatlusi tuleks teha vähemalt kord päevas, igal päeval soovitatavalt samal ajal või samadel aegadel; seejuures tuleks võtta arvesse manustamisjärgse eeldatava mõju maksimaalse avaldumise aega. Tuleks registreerida loomade tervislik seisund. Kõiki loomi kontrollitakse haigestumise ja surmajuhtude tuvastamiseks vähemalt kaks korda päevas.
|
|
43.
|
Kõikide paaritatavate loomade puhul tuleks läbi viia üksikasjalikud kliinilised vaatlused üks kord enne uuritava kemikaali manustamist (võimaldamaks sama loomaga saadud tulemuste võrdlemist) ning seejärel vähemalt kord nädalas. Need vaatlused tuleks teha standardalal väljaspool looma pidamiseks kasutatavat puuri ja igal vaatluste tegemise päeval soovitatavalt samal kellaajal. Tulemused tuleks hoolikalt registreerida; selleks tuleks soovitatavalt kasutada katset läbi viivas laboris selgelt kindlaks määratud hindamissüsteeme. Tuleks püüda tagada, et katsetingimused varieeruksid võimalikult vähe ja vaatlusi teeksid soovitatavalt isikud, kes ei ole asjaomasest töötlemisest teadlikud. Tuleks registreerida vähemalt järgmised ilmingud: nahal, karvastikus, silmades ja limaskestadel täheldatavad muutused, sekreetide ja eritiste esinemine ning autonoomse närvisüsteemiga seotud ilmingud (nt pisaravoolus, piloerektsioon, pupillide suuruse muutus, ebaharilik hingamisrütm). Peale selle tuleks registreerida muutused kõnnakus, kehahoius ja selles, kuidas loomad nendega ümberkäimisele reageerivad, samuti klooniliste ja tooniliste liigutuste esinemine, stereotüüpia (nt ülemäärane karvastiku hooldamine, ringiratast keerlemine), raske või kauakestnud sünnitus ja kummaline käitumine (nt enese vigastamine, tagurpidi kõndimine) (10).
|
|
44.
|
Uuringu kestel tuleks igast rühmast juhuvaliku alusel valitud viiel isasloomal ja viiel emasloomal hinnata ühel korral sensoorseid reaktsioone eri liiki ärritajatele (nt kuulmis-, nägemis- ja propriotseptiivsetele ärritajatele) (8, 9, 11), haardetugevust (12) ja motoorset tegevust (13). Asjaomaste võimalike meetodite täpsem kirjeldus on esitatud vastavate viidete kohastes allikates. Viidatud meetodite asemel võib siiski kasutada ka muid meetodeid. Isasloomadel tuleks kõnealused funktsionaalsed vaatlused läbi viia manustamisperioodi lõppjärgus veidi enne plaanijärgset surmamist, kuid enne hematoloogiliseks uuringuks või kliiniliseks biokeemiliseks analüüsiks vereproovi võtmist (vt punktid 53–56, sh joonealune märkus 1). Emasloomad peaksid nende funktsionaalsete vaatluste ajal olema sarnases füsioloogilises seisundis ja neid tuleks soovitatavalt vaadelda ühel korral viimasel imetamisnädalal (nt 6.–13. imetamispäeval) veidi enne plaanijärgset surmamist. Ajavahemik, mille vältel järglased on emast eraldatud, peaks olema võimalikult lühike.
|
|
45.
|
Uuringu lõppjärgus ühel korral tehtavad funktsionaalsed vaatlused võib ära jätta, kui uuring viiakse läbi järgnevalt tehtava subkroonilise mürgisuse 90-päevase uuringu või pikaajalise uuringu eeluuringuna. Sellisel juhul tuleks kõnealused funktsionaalsed vaatlused läbi viia vastava järeluuringu käigus. Teisest küljest aga võivad käesoleva meetodi kohase korduvannustamisel põhineva uuringu käigus tehtud funktsionaalsete vaatluste andmed hõlbustada järgnevalt tehtava subkroonilise mürgisuse uuringu või pikaajalise uuringu jaoks annusemäärade valimist.
|
|
46.
|
Funktsionaalsed vaatlused võib erandkorras ära jätta ka rühmade puhul, kus täheldatakse nii ulatuslikke mürgisusnähte, et see mõjutab märkimisväärselt funktsionaalse hindamise tulemuslikkust.
|
|
47.
|
Tiinuse kestus tuleks registreerida; selle arvutamisel lähtutakse tiinuse 0-päevast. Iga pesakonda tuleks pärast sündi võimalikult varakult hinnata, et teha kindlaks järglaste arv ja sugu, surnultsünnid, elussünnid, kängunud järglaste (vastavatest kontrolljärglastest palju väiksemate järglaste) arv ja silmaga nähtavate kõrvalekallete esinemine.
|
|
48.
|
Elus järglased tuleks loendada ja määrata nende sugu ning pesakonda tuleks kaaluda 24 tunni jooksul pärast sünnitust (sünnitusjärgsel 0-päeval või 1. päeval) ning vähemalt 4. ja 13. sünnitusjärgsel päeval. Peale paaritatavatel loomadel tehtavate vaatluste tulemuste (vt punktid 43–44) tuleks registreerida ka kõik järglaste ebatavalise käitumise ilmingud.
|
|
49.
|
Kõikidel järglastel tuleks ajavahemikul sünnijärgsest 0-päevast kuni 4. sünnijärgse päevani mõõta ühel ja samal päeval AGD. Järglaste kehamassi andmed tuleks koguda AGD mõõtmise päeval ja AGD tuleks normaliseerida järglase suurust kajastava näitaja, soovitatavalt kehamassi kuupjuure suhtes (14). Isastel järglastel tuleks 12. või 13. sünnijärgsel päeval loendada nisade/areoolide arv, nagu on soovitatud OECD katsejuhendis nr 151 (15).
|
Kehamass ja sööda/vee tarbimine
|
50.
|
Isas- ja emasloomi tuleks kaaluda esimesel manustamispäeval ning seejärel vähemalt kord nädalas, samuti surmamise ajal. Tiinuse ajal tuleks emasloomi kaaluda tiinuse 0-päeval, 7., 14. ja 20. päeval, samuti tuleks neid kaaluda 24 tunni jooksul pärast sünnitust (sünnitusjärgsel 0-päeval või 1. päeval) ning vähemalt 4. ja 13. sünnitusjärgsel päeval. Nimetatud vaatluste tulemused tuleks esitada eraldi iga täiskasvanud looma kohta.
|
|
51.
|
Paaritumiseelsel perioodil ning tiinuse ja imetamise ajal tuleks söödatarbimist mõõta vähemalt kord nädalas. Paaritumisperioodil ei ole söödatarbimise mõõtmine kohustuslik. Kui uuritavat kemikaali manustatakse veega, tuleks eelnimetatud perioodidel mõõta ka veetarbimist.
|
Hematoloogiline analüüs
|
52.
|
Uuringu vältel tuleks igast rühmast juhuvaliku alusel valitud viiel emasloomal ja viiel isasloomal viia ühel korral läbi hematoloogiline analüüs järgmiste näitajate määramiseks: hematokrit, hemoglobiinisisaldus, erütrotsüütide arv, retikulotsüütide arv, leukotsüütide üldarv ja eri tüüpi leukotsüütide arv, vereliistakute arv ja vere hüübimisaeg/hüübimisvõime. Kui uuritav kemikaal või mõni selle võimalikest metaboliitidest on või võib olla oksüdeerivate omadustega, tuleks teha muu hulgas kindlaks veel ka methemoglobiinisisaldus ja Heinzi kehakeste esinemine.
|
|
53.
|
Vereproov tuleks võtta kindlaksmääratud kohast. Emasloomad peaksid proovivõtu ajal olema sarnases füsioloogilises seisundis. Tiinuse algusaja varieeruvusega seotud praktilistest raskustest tulenevalt võib emasloomadelt vereproovi võtta juba paaritumiseelse perioodi lõpus, selmet võtta see loomade humaanse surmamise käigus või vahetult enne seda. Isasloomadelt tuleks vereproov võtta loomade humaanse surmamise käigus või vahetult enne seda. Teise võimalusena võib isasloomadelt vereproovi samuti võtta paaritumiseelse perioodi lõpus, kui emasloomade puhul eelistatakse seda proovivõtuaega.
|
|
54.
|
Vereproove tuleks säilitada sobivates tingimustes.
|
Kliiniline biokeemiline analüüs
|
55.
|
Kliinilise biokeemilise analüüsi jaoks, mille eesmärk on hinnata kudedes avalduvat olulist mürgist mõju ning konkreetset mõju neerudele ja maksale, tuleks kasutada kõnealuse igast rühmast valitud viie emaslooma ja viie isaslooma vereproove. Loomadel on soovitatav enne vereproovi võtmist hommikuni paastuda (22). Plasma või seerumi analüüs peaks hõlmama naatriumi, kaaliumi, glükoosi, kolesterooli üldsisalduse, karbamiidi, kreatiniini, valgu üldsisalduse ja albumiini, vähemalt kahe maksarakkudele avalduvale toimele osutava ensüümi (näiteks alaniini aminotransferaasi, aspartaadi aminotransferaasi ja sorbitooli dehüdrogenaasi) ning sapphapete määramist. Teatavatel juhtudel võib kasulikku teavet saada veel muude (maksas või mujal toodetud) ensüümide ja bilirubiini määramisest.
|
|
56.
|
Vereproovid võetakse kindlaksmääratud kohast järgmise kava alusel:
|
—
|
igas pesakonnas vähemalt kahelt järglaselt 4. sünnijärgsel päeval, kui järglaste arv seda võimaldab (vt punktid 40-41),
|
|
—
|
kõikidelt järglastega emasloomadelt ja igas pesakonnas vähemalt kahelt järglaselt surmamise ajal 13. päeval ning
|
|
—
|
kõikidelt täiskasvanud isasloomadelt surmamise ajal.
|
Kõiki vereproove säilitatakse sobivates tingimustes. Järglastelt 13. päeval võetud vereproovides ja täiskasvanud isasloomade vereproovides analüüsitakse kilpnäärmehormoonide (T4) sisaldust seerumis. Kui see on asjakohane, hinnatakse T4 sisaldust ka järglastega emasloomade vereproovides ja järglastelt 4. päeval võetud vereproovides. Kui see on asjakohane, võib soovi korral mõõta ka muude hormoonide sisaldust. Järglaste vere võib kilpnäärmehormoonide analüüsimiseks pesakonnakaupa kokku koondada. Soovitatavalt tuleks mõõta kilpnäärmehormoonide (T4 ja TSH) üldsisaldust.
|
|
57.
|
Uuringu viimasel nädalal võib igast rühmast juhuvaliku alusel valitud viielt isasloomalt koguda teatud kindla aja jooksul uriiniproovi ja määrata uriinianalüüsi käigus järgmised näitajad: välimus, ruumala, osmolaalsus või suhteline tihedus, pH, valgusisaldus, glükoosisisaldus ja veresisaldus / vererakkude sisaldus.
|
|
58.
|
Peale selle tuleks kaaluda üldistele koekahjustustele osutavate seerumimarkerite uurimist. Kui uuritav kemikaal on oma teadaolevatest omadustest tulenevalt võimeline mõjutama asjaomast ainevahetusprofiili või kui esineb sellekohane kahtlus, peaks määramine muu hulgas hõlmama veel ka kaltsiumi, fosfaati, paastumisjärgselt määratavaid triglütseriide ja glükoosi, teatavaid hormoone, methemoglobiini ja koliini esteraasi. Kõnealused näitajad tuleb igal konkreetsel juhul eraldi kindlaks määrata.
|
|
59.
|
Hormoonide määramisel võivad varieeruvust ja absoluutset kontsentratsiooni mõjutada järgmised tegurid:
|
—
|
surmamise aeg tulenevalt asjaolust, et hormoonide kontsentratsioon ööpäeva jooksul muutub;
|
|
—
|
surmamisviis – tuleks hoiduda tekitamast loomadele tarbetut stressi, mis võib mõjutada hormoonide kontsentratsiooni;
|
|
—
|
hormoonide määramise komplektid, milles kasutatavad standardkõverad võivad olla erinevad.
|
|
|
60.
|
Konkreetselt hormoonide määramiseks ette nähtud eri plasmaproovid tuleks koguda umbes samal kellaajal. Hormoonisisalduse analüüsimisel saadavad arvväärtused on müügilolevate eri katsekomplektide puhul erinevad.
|
|
61.
|
Kui varasemad võrdlusandmed ei ole piisavad, tuleks kaaluda hematoloogiliste ja kliiniliste biokeemiliste näitajate määramist enne manustamisperioodi algust või soovitatavalt loomadel, kes ei kuulu ühessegi katserühma. Emasloomade puhul tuleb andmete saamiseks kasutada imetavaid loomi.
|
PATOLOOGILINE ANALÜÜS
Täielik lahkamine
|
62.
|
Kõikidele uuringus kasutatud täiskasvanud loomadele tuleks teha täielik, põhjalik lahkamine, mille käigus uuritakse hoolikalt keha välispinda, kõiki avausi ning kolju-, rinna- ja kõhuõõnt ja nende sisu. Erilist tähelepanu tuleks pöörata reproduktiivsüsteemi elunditele. Tuleks registreerida pesastumiskohtade arv. Lahkamispäeval tuleks analüüsida tupeäiet, et teha kindlaks innatsükli etapp ja võimaldada korreleerimist emassuguelundite histopatoloogilise analüüsi tulemustega.
|
|
63.
|
Kõikide täiskasvanud isasloomade munandid ja munandimanused ning eesnääre ja seemnepõiekesed koos koagulatsiooninäärmetega tuleks vajaduse korral vabastada nende külge jäänud kudedest ja kuivamise ärahoidmiseks tuleks elundite märgmass määrata võimalikult kiiresti pärast nende väljalõikamist. Peale selle võib soovi korral kaaluda isasloomade puhul sellised elundid nagu pärakutõsturlihase ja sibulakäsnkeha-lihase kompleks, Cowperi näärmed ja sugutilukk ning emasloomade puhul munasarjade paar (märgmass) ja emakas (koos emakakaelaga); kui seda tehakse, peaks kaalumine toimuma võimalikult kiiresti pärast elundite väljalõikamist. Kõikide täiskasvanud loomade munasarjad, munandid, munandimanused, suguelundid ja kõik makroskoopiliste kahjustustega elundid tuleks säilitada.
|
|
64.
|
Kõikidelt täiskasvanud isas- ja emasloomadelt ning igast pesakonnast ühelt isaselt ja ühelt emaselt 13 päeva vanuselt järglaselt võetud kilpnääret tuleks säilitada fikseerimislahuses, mis on järgneva kavandatud histopatoloogilise analüüsi läbiviimiseks kõige sobivam. Kilpnäärme massi võib määrata pärast fikseerimist. Liigsed koed tuleks samuti eemaldada väga ettevaatlikult alles pärast fikseerimist, et kudesid mitte kahjustada. Koekahjustus võib mõjutada histopatoloogilise analüüsi tulemusi. Vereproovid tuleks võtta kindlaksmääratud kohast vahetult enne loomade humaanset surmamist või selle ajal ja neid tuleks säilitada sobivates tingimustes (vt punkt 56).
|
|
65.
|
Peale selle tuleks igast rühmast juhuvaliku alusel valitud vähemalt viielt täiskasvanud isas- ja emasloomalt (juhuvalik ei hõlma loomi, kes on leitud olevat surmaeelses seisundis ja/või on enne uuringu lõppu humaanselt surmatud) võtta maks, neerud, neerupealised, harknääre, põrn, aju ja süda, eemaldada vastavalt vajadusele nende külge jäänud koed ning määrata kuivamise ärahoidmiseks võimalikult kiiresti pärast väljalõikamist nende märgmass. Nii koetüübi kui ka kavandatud järgneva histopatoloogilise uuringu jaoks kõige sobivamas fikseerimislahuses tuleks säilitada järgmisi kudesid: kõik silmaga nähtavate kahjustustega koed, aju (representatiivsed piirkonnad, sealhulgas suuraju, väikeaju ja ajusild), seljaaju, silm, magu, peensool ja jämesool (sealhulgas Peyeri naastud), maks, neerud, neerupealised, põrn, süda, harknääre, hingetoru ja kopsud (säilitamiseks täidetakse need fikseerimislahusega ja sukeldatakseseejärel lahusesse), sugunäärmed (munandid ja munasarjad), suguelundid (emakas ja emakakael, munandimanused, eesnääre ja seemnepõiekesed koos koagulatsiooninäärmetega), tupp, kusepõis, lümfisõlmed (peale manustamiskohale kõige lähemal asuva lümfisõlme tuleks võtta labori kogemustest lähtuvalt veel teinegi lümfisõlm (16)), perifeerne närv (istmiku- või sääreluunärv), soovitatavalt lihase läheduses, skeletilihas ning luu koos luuüdiga (lõik või teise võimalusena värskelt aspireeritud luuüdi preparaat). Munandid on soovitatav fikseerida Bouini või muudetud Davidsoni fikseerimislahusesse sukeldamise teel (16, 17, 18); formaliinis fikseerimine ei ole nende kudede puhul soovitatav. Fikseerimislahuse kiire sissetungimise võimaldamiseks võib elundi kummaski otsas valkjaskesta ettevaatlikult väikese sügavuseni nõelaga läbi torgata. Kliinilised ja muud leiud võivad osutada vajadusele uurida ka muid kudesid. Tuleks säilitada ka kõik elundid, mida uuritava kemikaali teadaolevatest omadustest tulenevalt peetakse tõenäoliseks sihtelundiks.
|
|
66.
|
Sisesekretsioonisüsteemiga seotud mõju kohta võib saada väärtuslikku teavet järgmiste kudede uurimisel: sugunäärmed (munasarjad ja munandid), suguelundid (emakas koos emakakaelaga, munandimanused, seemnepõiekesed koos koagulatsiooninäärmetega, eesnäärme dorsolateraalne ja ventraalne osa), tupp, ajuripats, isaslooma rinnanäärmed ja neerupealised. Isaslooma rinnanäärmetes aset leidvad muutused on ebapiisavalt dokumenteeritud, kuid see näitaja võib olla väga tundlik östrogeense toimega ainete suhtes. Punktis 65 loetlemata elundite/kudede analüüsimine ei ole kohustuslik.
|
|
67.
|
Surnud järglasi ja 13. sünnijärgsel päeval või varsti pärast seda surmatud järglasi tuleks miinimumnõudena väliselt hoolikalt uurida silmaga nähtavate kõrvalekallete suhtes. Erilist tähelepanu tuleks pöörata välistele suguelunditele, mida tuleks uurida arenguhäirete ilmingute suhtes.
|
Histopatoloogiline analüüs
|
68.
|
Kontrollrühmast ja suurima annusega töödeldud rühmast valitud loomadelt pärit säilitatud elundite ja kudede puhul tuleks viia läbi täismahus histopatoloogiline uuring (seejuures tuleks pöörata erilist tähelepanu spermatogeneesi etappidele isaslooma sugunäärmetes ja munandite interstitsiaalrakustruktuuri histopatoloogilisele analüüsile). Vajaduse korral võib uurida ka järglastelt ja ülejäänud täiskasvanud loomadelt võetud kilpnääret. Kui suurima annusega töödeldud rühmas täheldatakse manustamisega seotud muutusi, peaks kõnealune analüüs hõlmama ka teiste annuserühmade loomi. Täiendav üksikasjalik teave sisesekretsioonisüsteemi kudede väljalõikamise, fikseerimise, neist lõikude tegemise ja nende histopatoloogilise analüüsi kohta on esitatud histopatoloogilise analüüsi juhendis (10).
|
|
69.
|
Tuleks uurida kõiki silmaga nähtavaid kahjustusi. NOAELi kindlakstegemise hõlbustamiseks tuleks analüüsida ka teiste annuserühmade loomade sihtelundeid, eelkõige sellistest rühmadest pärit sihtelundeid, mida iseloomustavate tulemuste põhjal saab väidetavalt määrata kindlaks NOAELi.
|
|
70.
|
Täiendrühma kasutamise korral tuleks histopatoloogilise analüüsi raames hinnata neid kudesid ja elundeid, kus katserühmade loomade puhul täheldati mõju avaldumist.
|
ANDMED JA ARUANDLUS
Andmed
|
71.
|
Andmed tuleks esitada iga looma kohta eraldi. Peale selle tuleks esitada kõik andmed kokkuvõtliku tabeli kujul, kus on iga katserühma puhul toodud ära loomade arv katse alguses, katse käigus surnud või humaansetel kaalutlustel surmatud loomade arv ning surma või humaanse surmamise aeg, viljakate loomade arv, tiinete emasloomade arv, mürgisusnähtudega loomade arv, mürgisusnähtude kirjeldus, sealhulgas mürgise mõju ilmnemisaeg, kestus ja tugevus, histopatoloogiliste muutuste liik ja kõik asjakohased andmed pesakonna kohta. Paljunemisele/arengule avalduva mõju hindamiseks väga kasulikuks osutunud aruandevorm kokkuvõtliku tabeli kujul on esitatud 3. liites.
|
|
72.
|
Numbrilisi tulemusi tuleks võimaluse korral hinnata asjakohase üldiselt tunnustatud statistilise meetodiga. Eri annuste puhul täheldatud mõju võrdlemisel tuleks hoiduda mitme t-testi kasutamisest. Kasutatavad statistilised meetodid tuleks valida uuringu kavandamise etapis. AGD ja nisade esinemise statistiline analüüs peaks põhinema iga üksiku järglase andmetel ning seejuures tuleks arvesse võtta pesakonnas ilmnevat mõju. Vajaduse korral käsitatakse analüüsiüksusena pesakonda. Järglaste kehamassi statistiline analüüs peaks põhinema iga üksiku järglase andmetel ning seejuures tuleks arvesse võtta pesakonna suurust. Uuringu piiratud ulatusest tulenevalt on tulemuste olulisuse tuvastamiseks tehtava statistilise analüüsi väärtus paljude lõppnäitajate, eelkõige paljunemisega seotud lõppnäitajate puhul piiratud. Mõned kõige sagedamini kasutatavatest meetoditest, eelkõige parameetrilised testid keskmise suundumuse näitajate analüüsimiseks, on ebasobivad. Statistilise analüüsi kasutamisel tuleks valida meetod, mis on uuritava muutuja väärtuste jaotuse analüüsimiseks sobiv, ning see valik tuleks teha enne uuringu alustamist.
|
Tulemuste hindamine
|
73.
|
Käesoleva meetodi kohase mürgisusuuringu tulemusi tuleks hinnata lähtuvalt täheldatud mõjust, lahkamistulemustest ja mikroskoopilistest leidudest. Hindamine hõlmab seose kindlakstegemist uuritava kemikaali annuse ja kõrvalekallete – sealhulgas silmaga nähtavate kahjustuste, tuvastatud sihtelunditele avalduva mõju, viljatuse, kliiniliste kõrvalekallete, paljunemisvõimele ja pesakonnale avalduva mõju, kehamassi muutuste, suremusele avalduva mõju ja mis tahes muu mürgise mõju – esinemise või puudumise, esinemissageduse ja raskusastme vahel.
|
|
74.
|
Isasloomade töötlemise lühiajalisuse tõttu tuleks isasloomade paljunemisele avalduva mõju hindamisel vaadelda munandite ja munandimanuste histopatoloogilise analüüsi tulemusi koos viljakusandmetega. Ühtlasi võib uuringutulemuste tõlgendamise hõlbustamiseks olla kasulik kasutada kontrollidega saadud varasemaid paljunemist/arengut käsitlevaid andmeid (nt pesakonna suuruse, AGD, nisade esinemise või seerumi T4-sisalduse kohta), kui need on olemas.
|
|
75.
|
Kvaliteedikontrolli eesmärgil on soovitatav koguda kontrolle käsitlevaid varasemaid andmeid ja arvutada arvandmete jaoks variatsioonikordajad, eriti näitajate puhul, mis on seotud endokriinfunktsiooni kahjustavate kemikaalide kindlakstegemisega. Neid andmeid võib kasutada võrdlusalusena käsiloleva uuringu tulemuste hindamiseks.
|
Katseprotokoll
|
76.
|
Katseprotokollis tuleks esitada järgmine teave.
|
|
Uuritav kemikaal:
|
—
|
allikas, partii number, aegumiskuupäev, kui on olemas;
|
|
—
|
uuritava kemikaali püsivus, kui see on teada;
|
|
|
|
ühest koostisosast koosnev aine:
|
—
|
füüsiline välimus, lahustuvus vees ja muud asjakohased füüsikalis-keemilised omadused;
|
|
—
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus, CASi number, SMILESi või InChI kood, struktuurivalem, puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
|
|
mitut koostisosa sisaldav aine, UVCB või segu:
|
—
|
võimalikult täpne omaduste kirjeldus lähtuvalt koostisosade keemilisest määratlusest (vt eespool), kvantitatiivsest sisaldusest ja asjakohastest füüsikalis-keemilistest omadustest.
|
|
|
|
Kandeaine (vajaduse korral):
|
—
|
kandeaine valimise põhjendus, kui kandeaine ei ole vesi.
|
|
|
|
Katseloomad:
|
—
|
loomade arv, vanus ja sugu;
|
|
—
|
päritolu, pidamistingimused, söötmisandmed jne;
|
|
—
|
iga looma kehamass katse alguses;
|
|
—
|
liigi valimise põhjendus, kui katseloom ei ole rott.
|
|
|
|
Katsetingimused:
|
—
|
annuste valimise põhjendus;
|
|
—
|
üksikasjad uuritava kemikaali valmistise / söödavalmistise, saavutatud kontsentratsioonide ning valmistise püsivuse ja homogeensuse kohta;
|
|
—
|
uuritava kemikaali manustamise üksikasjad;
|
|
—
|
vajaduse korral andmed söödas/joogivees sisalduva uuritava kemikaali kontsentratsiooni (ppm) teisendamiseks tegelikuks annuseks (mg kehamassi kilogrammi kohta päevas);
|
|
—
|
sööda ja vee kvaliteedi üksikasjad;
|
|
—
|
järglaste surmamise korral üksikasjalik kirjeldus surmatavate järglaste juhusliku valimise korra kohta.
|
|
|
|
Tulemused:
|
—
|
kehamass / kehamassi muutused;
|
|
—
|
vajaduse korral söödatarbimine ja veetarbimine;
|
|
—
|
mürgise mõju andmed kummagi soo ja iga annusemäära kohta, sealhulgas viljakusele ja tiinusele avalduva mõju ning mis tahes muude mürgisusnähtude kohta;
|
|
—
|
mürgisus või muu mõju paljunemisele, järglastele, sünnijärgsele kasvule jne;
|
|
—
|
kliiniliste ilmingute laad, raskusaste ja kestus (nähtude pöörduvus või pöördumatus);
|
|
—
|
sensoorse aktiivsuse, haardetugevuse ja motoorse tegevuse hindamise tulemused;
|
|
—
|
hematoloogilise analüüsi tulemused koos asjaomaste võrdlusväärtustega;
|
|
—
|
kliinilise biokeemilise analüüsi tulemused koos asjaomaste võrdlusväärtustega;
|
|
—
|
normaalse või ebanormaalse innatsükliga täiskasvanud emasloomade arv ja tsükli kestus;
|
|
—
|
elussündide arv ja pesastumisjärgne kadu;
|
|
—
|
silmaga nähtavate kõrvalekalletega järglaste arv; väliste suguelundite makroskoopilise hindamise tulemused, kängunud järglaste arv;
|
|
—
|
katse vältel suremise aeg või teave selle kohta, et loomad olid kuni surmamiseni elus;
|
|
—
|
pesastunud embrüote arv, pesakonna suurus ja pesakonna kaal mõõtmishetkel;
|
|
—
|
järglaste kehamassi andmed;
|
|
—
|
iga järglase AGD (ja kehamass AGD mõõtmise päeval);
|
|
—
|
nisade esinemine isastel järglastel;
|
|
—
|
kilpnäärmehormoonide sisaldus järglastelt 13. päeval võetud proovides ja täiskasvanud isasloomade proovides (ning järglastega emasloomade proovides ja järglastelt 4. päeval võetud proovides, kui seda on mõõdetud);
|
|
—
|
andmed järglastega loomade kehamassi kohta surmamise hetkel ning selliste loomade elundite massi kohta;
|
|
—
|
histopatoloogiliste leidude üksikasjalik kirjeldus;
|
|
—
|
andmed absorbeerumise kohta (kui on olemas);
|
|
—
|
vajaduse korral tulemuste statistilise analüüsi andmed.
|
|
|
Tulemuste tõlgendamine
|
77.
|
Kirjeldatud uuring võimaldab hinnata annuste korduval manustamisel täheldatavat paljunemisele/arengule avalduvat mürgist mõju. Kuna rõhk on seatud nii üldise mürgisuse kui ka reproduktiiv-/arengutoksilisuse lõppnäitajate hindamisele, võimaldavad uuringu tulemused eelkõige eristada üldise mürgisuse puudumisel paljunemisele/arengule avalduvat mõju sellisest mõjust paljunemisele/arengule, mis avaldub üksnes kontsentratsioonil, mille juures kemikaal on mürgine ka järglaste vanematele (vt punktid 7–11). Saadud tulemused võivad viidata vajadusele teha täiendavaid uuringuid ja võivad hõlbustada järgnevate uuringute kavandamist. Paljunemise ja arenguga seotud tulemuste hõlpsamaks tõlgendamiseks tuleks tutvuda OECD juhenddokumendiga nr 43 (19). Käesoleva katsemeetodi puhul võib olla kasu närilistega tehtavate sisesekretsioonisüsteemi ja paljunemisega seotud katsete tulemuste histoloogilist hindamist käsitlevas OECD juhenddokumendis nr 106 (16) esitatud teabest (sisesekretsiooni)elundite ja tupeäiete ettevalmistamise ja hindamise kohta.
|
KIRJANDUS
|
(1)
|
OECD (1990). Kemikaalide töörühma ja halduskomitee 14. ühiskoosoleku koosolekudokument nr 1. Taotluse korral kättesaadav Majanduskoostöö ja Arengu Organisatsioonist Pariisis.
|
|
(2)
|
OECD (1992). Ajutise eksperdirühma esimehe aruanne reproduktiivtoksilisust käsitlevate sõeluuringumeetodite alase koosoleku kohta. Tokyo, 27.–29. oktoober 1992. Taotluse korral kättesaadav Majanduskoostöö ja Arengu Organisatsioonist Pariisis.
|
|
(3)
|
Mitsumori, K., Kodama, Y., Uchida, O., Takada, K., Saito, M., Naito, K., Tanaka, S., Kurokawa, Y., Usami, M., Kawashima, K., Yasuhara, K., Toyoda, K., Onodera, H., Furukawa, F., Takahashi, M., ja Hayashi, Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol. Sci. 19: 141–149.
|
|
(4)
|
Tanaka, S., Kawashima, K., Naito, K., Usami, M., Nakadate, M., Imaida, K., Takahashi, M., Hayashi, Y., Kurokawa, Y., ja Tobe, M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol. 18: 89–95.
|
|
(5)
|
OECD (1998). Endokriinfunktsiooni kahjustavate kemikaalide uurimise ja hindamise (EDTA) OECD rakkerühma esimese koosoleku aruanne. 10.–11. märts 1998. Taotluse korral kättesaadav Majanduskoostöö ja Arengu Organisatsioonist Pariisis.
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment No. 217. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environment, Health and Safety Publications, Series on Testing and Assessment No. 19. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(8)
|
Goldman, J. M., Murr, A. S., ja Cooper, R. L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies. Birth Defects Res. B 80(2): 84–97.
|
|
(9)
|
Sadleir, R. M. F. S. (1979). Cycles and Seasons. Väljaandes: Reproduction in Mammals: I. Germ Cells and Fertilization. Auston, C. R., ja Short, R. V. (toim.), Cambridge, New York.
|
|
(10)
|
Rahvusvaheline kemikaaliohutuse programm (IPCS) (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document No. 60.
|
|
(11)
|
Moser, V. C., McDaniel, K. M., ja Phillips, P. M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol. 108: 267–283.
|
|
(12)
|
Meyer, O. A., Tilson, H. A., Byrd, W. C., ja Riley, M. T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol. 1: 233–236.
|
|
(13)
|
Crofton, K. M., Howard, J. L., Moser, V. C., Gill, M. W., Reiter, L. W., Tilson, H. A., ja MacPhail, R. C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13: 599–609.
|
|
(14)
|
Gallavan, R. H. Jr., Holson, J. F., Stump, D. G., Knapp, J. F., ja Reynolds, V. L. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights. Reprod. Toxicol. 13: 383–390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment No. 151. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(16)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment No. 106. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(17)
|
Hess, R. A., ja Moore, B. J. (1993). Histological Methods for the Evaluation of the Testis. Väljaandes: Methods in Reproductive Toxicology. Chapin, R. E., ja Heindel, J. J. (toim.), Academic Press, San Diego, CA. Lk 52–85.
|
|
(18)
|
Latendresse, J. R., Warbrittion, A. R., Jonassen, H., ja Creasy, D. M. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s Fluid. Toxicol. Pathol. 30: 524–533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment No. 43. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(20)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Series on Testing and Assessment No. 150. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
1. liide
MÕISTED (VT KA OECD JUHENDDOKUMENT NR 150 (20))
Androgeensus kemikaali võime toimida imetaja organismis samal viisil kui looduslik androgeen (nt testosteroon).
Annus manustatav uuritava kemikaali kogus. Annust väljendatakse uuritava kemikaali kogusena massiühikutes katselooma kehamassi ühiku kohta päevas (näiteks milligrammides kehamassi kilogrammi kohta päevas) või kemikaali püsiva kontsentratsioonina söödas.
Annustamine üldmõiste, mis hõlmab annust, selle manustamise sagedust ja annustamise kestust.
Antiandrogeensus kemikaali võime pärssida imetaja organismis loodusliku androgeeni (nt testosterooni) toimet.
Antitüreoidne aktiivsus kemikaali võime pärssida imetaja organismis loodusliku kilpnäärmehormooni (nt T3) toimet.
Antiöstrogeensus kemikaali võime pärssida imetaja organismis loodusliku östrogeeni (nt 17β-östradiooli) toimet.
Arengutoksilisus reproduktiivtoksilisuse ilming, mis avaldub järglaste sünnieelse, perinataalse või sünnijärgse struktuuri- või funktsioonihäirena.
Emale avalduv mürgisus tiinele emasloomale avalduv kahjulik mõju, mis on spetsiifiline (otsene mõju) või mittespetsiifiline (kaudne mõju) ning on seotud tiinusega.
Ilmne mürgisus üldmõiste, mille abil kirjeldatakse uuritava kemikaali manustamise järel täheldatavaid selgeid mürgisusnähte. Kõnealused nähud peaksid olema ohu hindamiseks piisavad ja sellised, et manustatava annuse suurendamisega kaasnevad eeldatavalt tõsised mürgisusnähud ja tõenäoliselt ka surm.
Kemikaal aine või segu.
NOAEL lühend, millega tähistatakse annusemäära, mille juures ei täheldata kahjulikku toimet. Tegemist on suurima annusega, mis ei põhjusta manustamisest tingitud kahjulikke ilminguid.
Reproduktiivtoksilisus kahjulik mõju järglastele ja/või isas- või emasloomade paljunemisfunktsioonile või paljunemisvõimele.
Türeoidne aktiivsus kemikaali võime toimida imetaja organismis samal viisil kui looduslik kilpnäärmehormoon (nt T3).
Uuritav kemikaal iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
Valideerimine teaduslik protsess, mille eesmärk on iseloomustada katsemeetodi rakendamisega seotud nõudeid ja piiranguid ning tõendada meetodi usaldusväärsust ja sobivust konkreetsel eesmärgil kasutamiseks.
Viljakuse vähenemine isas- või emaslooma paljunemisfunktsiooni häire või paljunemisvõime langus.
Östrogeensus – kemikaali võime toimida imetaja organismis samal viisil kui looduslik östrogeen (nt 17β-östradiool).
2. liide
UURINGU MAKSIMUMKESTUSELE VASTAV TÄISPIKAL 14-PÄEVASEL PAARITUMISPERIOODIL PÕHINEV KATSE SKEMAATILINE AJAKAVA


3. liide
TABELI KUJUL KOKKUVÕTLIK ARUANNE PALJUNEMISELE/ARENGULE AVALDUVA MÕJU KOHTA
|
VAATLUSED
|
VÄÄRTUSED
|
|
Annus (ühik)
|
0 (kontroll)
|
…
|
…
|
…
|
…
|
|
Paaride arv katse alguses (N)
|
|
|
|
|
|
|
Innatsükkel (vähemalt keskmine pikkus ja ebaregulaarsete tsüklite esinemissagedus)
|
|
|
|
|
|
|
Emasloomad, kellel täheldatakse paaritumise ilminguid (N)
|
|
|
|
|
|
|
Tiinestunud emasloomad (N)
|
|
|
|
|
|
|
Eostunud 1.–5. päeval (N)
|
|
|
|
|
|
|
Eostunud 6.–…. (23) päeval (N)
|
|
|
|
|
|
|
Tiinuse kestus ≤ 21 päeva (N)
|
|
|
|
|
|
|
Tiinuse kestus 22 päeva (N)
|
|
|
|
|
|
|
Tiinuse kestus ≥ 23 päeva (N)
|
|
|
|
|
|
|
Elusalt sündinud järglastega emasloomad (N)
|
|
|
|
|
|
|
4. sünnijärgse päevani elus püsinud järglastega emasloomad (N)
|
|
|
|
|
|
|
Pesastunud embrüote arv tiinestunud emaslooma kohta (keskmine)
|
|
|
|
|
|
|
Elussündide arv järglastega emaslooma kohta (keskmine)
|
|
|
|
|
|
|
4. päevani elus püsinud järglaste arv järglastega emaslooma kohta (keskmine)
|
|
|
|
|
|
|
Sugude vahekord (isased/emased) sünnihetkel (keskmine)
|
|
|
|
|
|
|
Sugude vahekord (isased/emased) 4. päeval (keskmine)
|
|
|
|
|
|
|
Pesakonna kaal sünnihetkel (keskmine)
|
|
|
|
|
|
|
Pesakonna kaal 4. päeval (keskmine)
|
|
|
|
|
|
|
Järglaste kehamass sünnihetkel (keskmine)
|
|
|
|
|
|
|
Järglaste kehamass AGD mõõtmise hetkel (isasloomade keskmine, emasloomade keskmine)
|
|
|
|
|
|
|
Järglaste AGD samal sünnijärgsel päeval ajavahemikul sündimispäevast kuni 4. päevani (isasloomade keskmine, emasloomade keskmine; märkida sünnijärgne mõõtmispäev)
|
|
|
|
|
|
|
Järglaste kehamass 4. päeval (keskmine)
|
|
|
|
|
|
|
Järglaste kehamass 13. päeval (keskmine)
|
|
|
|
|
|
|
Nisade esinemine isastel järglastel 13. päeval (keskmine)
|
|
|
|
|
|
|
KÕRVALEKALLETEGA JÄRGLASED
|
|
Järglastega emasloomad, kellel on 0 kõrvalekalletega järglast
|
|
|
|
|
|
|
Järglastega emasloomad, kellel on 1 kõrvalekalletega järglane
|
|
|
|
|
|
|
Järglastega emasloomad, kellel on ≥ 2 kõrvalekalletega järglast
|
|
|
|
|
|
|
JÄRGLASTE KADU
|
|
Sünnieelne (pesastunud embrüote arvu ja elussündide arvu vahe)
|
|
0 järglast kaotanud emasloomad
|
|
|
|
|
|
|
1 järglase kaotanud emasloomad
|
|
|
|
|
|
|
2 järglast kaotanud emasloomad
|
|
|
|
|
|
|
≥ 3 järglast kaotanud emasloomad
|
|
|
|
|
|
|
Sünnijärgne (elussündide arvu ja 13. sünnijärgse päevani elus püsinud järglaste arvu vahe)
|
|
0 järglast kaotanud emasloomad
|
|
|
|
|
|
|
1 järglase kaotanud emasloomad
|
|
|
|
|
|
|
2 järglast kaotanud emasloomad
|
|
|
|
|
|
|
≥ 3 järglast kaotanud emasloomad
|
|
|
|
|
|
B.65
IN VITRO MEMBRAANIBARJÄÄRI KATSEMEETOD NAHASÖÖVITUSE UURIMISEKS
SISSEJUHATUS
|
1.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 435 (2015). Nahasöövitus on Ühinenud Rahvaste Organisatsiooni (ÜRO) ühtse ülemaailmse kemikaalide klassifitseerimise ja märgistamise süsteemis (GHS (Globally Harmonized System) (1) ning Euroopa Liidu määruses (EÜ) nr 1272/2008, mis käsitleb ainete ja segude klassifitseerimist, märgistamist ja pakendamist (CLP-määrus), (24) esitatud määratluse kohaselt pöördumatu nahakahjustuse, st marrasknahast pärisnahani ulatuva nähtava nekroosi tekkimine uuritava kemikaali pealekandmise tagajärjel. Käesolev katsemeetod, mis on samaväärne ajakohastatud OECD katsejuhendiga nr 435, on in vitro membraanibarjääri katsemeetod, mis saab kasutada söövitavate kemikaalide kindlakstegemiseks. Katsemeetodis kasutatakse tehismembraani, mis on kavandatud söövitavatele kemikaalidele reageerima sarnaselt loomanahale in situ.
|
|
2.
|
Nahka söövitavat toimet on tavapäraselt hinnatud, kandes uuritavat kemikaali elusloomade nahale ja hinnates nahakahjustuse ulatust pärast kindla perioodi möödumist (2). Peale käesoleva meetodi on söövitavate kemikaalide kindlakstegemiseks kasutatava standardse küüliku in vivo nahakatse (käesoleva lisa peatükk B.4, samaväärne OECD katsejuhendiga nr 404) (2) alternatiividena vastu võetud mitu muud in vitro katsemeetodit (3, 4). ÜRO GHSi astmelise katsetamise ja hindamise strateegia nahka söövitava toime hindamiseks ja klassifitseerimiseks ning OECD juhenddokumendis nahaärrituse/-söövitusega seotud katsete tegemist ja hindamist käsitleva ühtse lähenemisviisi kohta soovitatakse kasutada valideeritud ja heaks kiidetud in vitro katsemeetodeid moodulite 3 ja 4 kohaselt (1, 5). Kõnealuse katsete tegemist ja hindamist käsitleva ühtse lähenemisviisi raames kirjeldatakse mitut moodulit, milles teabeallikad ja analüüsivahendid on rühmitatud, ning i) antakse suuniseid, kuidas lõimida ja kasutada olemasolevaid katse- ja muid andmeid kemikaalide võimaliku nahka ärritava ja nahka söövitava toime hindamiseks ja ii) pakutakse välja lähenemisviis juhuks, kui on vaja teha täiendavaid katseid, kaasa arvatud juhuks, kui saadakse negatiivsed tulemused (5). Selle modulaarse lähenemisviisi korral saab kasutada in vitro katsemeetoditega saadud positiivseid tulemusi kemikaali klassifitseerimiseks söövitavana, ilma et oleks vaja täiendavaid loomkatseid; seega vähendatakse ja täiustatakse loomade kasutamist ja välditakse nende kasutamisega kaasneda võivaid valu ja kannatusi.
|
|
3.
|
Valideerimisuuringud viidi läbi in vitro membraanibarjääri mudeliga, mis on müügil nimega Corrositex® (6, 7, 8) ja millega 163 ainest ja segust koosneva andmebaasi järgi oli kogutäpsus nahka söövitava toime prognoosimisel 79 % (128/163), tundlikkus 85 % (76/89) ning spetsiifilisus 70 % (52/74) (7). Tunnustatud nõuetekohasuse alusel on seda valideeritud võrdlusmeetod (VRM) soovitatav kasutada astmelise katsetamise strateegia osana kemikaalide võimalik nahka söövitava toime hindamisel (5, 7). Enne kui in vitro membraanibarjääri mudeliga katsemeetodit nahasöövituse uurimiseks võib kasutada regulatiivsel eesmärgil, tuleb kooskõlas eelnevalt kindlaksmääratud toimivusnõuetega (10) kindlaks teha selle usaldusväärsus, asjakohasus (täpsus) ja selle kavandatud kasutusega seotud piirangud, et tagada selle sarnasus valideeritud võrdlusmeetodiga (9). Andmete vastastikune tunnustamine vastavalt OECD kokkuleppele tagatakse ainult pärast seda, kui mis tahes uus või ajakohastatud kavandatud meetod on toimivusnõudeid järgides läbi vaadatud ja lisatud samaväärsesse OECD katsejuhendisse. Praegu on OECD katsejuhendiga nr 435 hõlmatud ainult üks in vitro ja käesoleva katsemeetod müügil oleva Corrositex®i mudeliga.
|
|
4.
|
Teised katsemeetodid nahka söövitava toime uurimiseks põhinevad rekonstrueeritud inimnaha (OECD katsejuhend nr 431) (3) ja isoleeritud rotinaha kasutamisel (OECD katsejuhend nr 430) (4). Käesoleva katsesuunise järgi saab söövitavaid kemikaali jagada kolme ÜRO GHSi kohasesse söövitavuse alamkategooriatesse ja söövitusohu järgi kolme ÜRO veopakendi rühma. Käesolev katsejuhend võeti esmakordselt vastu 2006. aastal ja seda ajakohastati 2015. aastal, et viidata katsete tegemist ja hindamist käsitleva ühtse lähenemisviisi juhenddokumendile ning ajakohastada pädevuse tõendamise ainete loendit.
|
MÕISTED
|
5.
|
Kasutatud mõisted on esitatud liites.
|
LÄHTEKAALUTLUSED JA PIIRANGUD
|
6.
|
Selles katsemeetodis kirjeldatud katse-eeskiri võimaldab kvalitatiivselt analüüsida söövitavaid uuritavaid kemikaale ning jagada need ÜRO GHSi / CLP-määruse kohastesse alamkategooriatesse (tabel 1). Peale selle võib sellist katsemeetodit kasutada otsustamaks konkreetsete kemikaaliklasside, nt orgaaniliste ja anorgaaniliste hapete, happederivaatide (25) ja aluste söövitavate ja mittesöövitavate omaduste üle teatavate veokatsete puhul (7, 11, 12). Käesolevas katsemeetodis kirjeldatakse valideeritud võrdlusmeetodile (7) sarnanevat üldmetoodikat. See katsemeetod ei anna asjakohast teavet nahaärrituse kohta, seega tuleb märkida, et katsemeetodiga B.46 (samaväärne OECDkatsejuhendiga nr 439) uuritakse spetsiaalselt in vitro sellist tervisemõju nagu nahaärritus (13). Nahale pärast ühekordset kokkupuudet avalduva paikse toime täielikuks hindamiseks tuleb juhinduda OECD juhenddokumendist katsete tegemist ja hindamist käsitleva ühtse lähenemisviisi kohta (5).
Tabel 1
ÜRO GHSi kohane nahka söövitava aine kategooria ja alamkategooriad
(26)
|
Söövituskategooria (1. kategooria) (ametiasutustele, kes alamkategooriaid ei kasuta)
|
Võimalikud söövituse alamkategooriad (26) (ametiasutustele, kes kasutavad alamkategooriaid, sealhulgas CLP-määrust)
|
Söövitav vähemalt ühe looma puhul kolmest
|
|
Kokkupuuteaeg
|
Vaatlus
|
|
Söövitav
|
Söövitav, alamkategooria 1A
|
≤ 3 minutit
|
≤ 1 tund
|
|
Söövitav, alamkategooria 1B
|
> 3 minutit / ≤ 1 tund
|
≤ 14 päeva
|
|
Söövitav, alamkategooria 1C
|
> 1 tund / ≤ 4 tundi
|
≤ 14 päeva
|
|
|
7.
|
Valideeritud võrdlusmeetodi (7) puuduseks on asjaolu, et paljud mittesöövitavad kemikaalid ja mõned söövitavad kemikaalid ei tarvitse esialgse sobivuskatse (vt punkt 13 alusel) uurimiseks sobida. Sageli ei sobi uurimiseks kemikaalide vesilahused, mille pH on vahemikus 4,5…8,5; 85 % kõnealuse pH-vahemikuga uuritud kemikaalidest osutusid loomkatsetel mittesöövitavaks (7). In vitro membraanibarjääri meetodit võib kasutada tahkete ainete (vees lahustuvad või mittelahustuvad), vedelike (vesilahused või mitte) ja emulsioonide uurimiseks. Kuid uuritavaid kemikaale, mis sobivuskatsel tuvastatavat muutust (nt värvuse muutust valideeritud võrdlusmeetodi kemikaalide detekteerimissüsteemis) esile ei kutsu, ei saa membraanibarjääri meetodiga uurida ning nende uurimiseks tuleks kasutada muid katsemeetodeid.
|
KATSE PÕHIMÕTE
|
8.
|
Katsesüsteem koosneb kahest osast: sünteetilisest makromolekulaarsest biobarjäärist ja kemikaalide detekteerimissüsteemist; selle katsemeetodiga tuvastatakse kemikaalide detekteerimissüsteemi abil membraanibarjääri kahjustus, mille on põhjustanud söövitav uuritav kemikaal pärast sünteetilise makromolekulaarse membraanibarjääri pinnale kandmist (7) eeldatavast sama(de) söövitusmehhanismi(de) järgi mis toimivad elusorganismi nahal.
|
|
9.
|
Membraanibarjääri läbimist saab mõõta mitme toiminguga või kemikaali detekteerimissüsteemiga, sealhulgas pH indikaatori värvuse muutuse või mõne muu indikaatorlahuse omaduse muutusega barjääri all.
|
|
10.
|
Membraanibarjäär peab olema ettenähtud kasutamiseks nõuetekohane, st asjakohane ja usaldusväärne. Muu hulgas peavad olema tagatud preparaatide ühtsed barjääriomadused, st barjääri säilimine mittesöövitavate kemikaalide suhtes ja võimalus liigitada kemikaalid söövitusomaduste järgi erinevatesse ÜRO GHSi söövitavuse alamkategooriatesse (1). Klassifitseerimine põhineb ajal, mis kulub kemikaali läbitungimiseks membraanibarjäärist ja indikaatorlahuseni jõudmiseks.
|
PÄDEVUSE TÕENDAMINE
|
11.
|
Enne käesoleval meetodil põhinevat in vitro membraanibarjääri meetodi rutiinset kasutamist tuleb laboril tõendada oma tehnilist pädevust, klassifitseerides õigesti tabelis 2 soovitatud kaksteist pädevuse kontrolli ainet. Olukorras, kus loetletud aine ei ole kättesaadav, või kui see on põhjendatud, võib kasutada muud ainet (nt võrdluskemikaalide loetelust (10)), mille puhul on kättesaadavad piisavad võrdlusandmed in vivo ja in vitro katsete kohta, eeldusel, et järgitakse samu valikukriteeriumeid nagu esitatud tabelis 1.
Tabel 2
Pädevuse kontrolli ained
(27)
|
Aine (28)
|
CASi nr
|
Aineklass
|
In vivo ÜRO GHSi kohane alamkategooria (29)
|
In vitro ÜRO GHSi kohane alamkategooria (29)
|
|
Boortrifluoriiddihüdraat
|
13319-75-0
|
Anorgaanilised happed
|
1A
|
1A
|
|
Lämmastikhape
|
7697-37-2
|
Anorgaanilised happed
|
1A
|
1A
|
|
Fosforpentakloriid
|
10026-13-8
|
Anorgaaniliste hapete prekursorid
|
1A
|
1A
|
|
Valerüülkloriid
|
638-29-9
|
Hapete kloriidid
|
1B
|
1B
|
|
Naatriumhüdroksiid
|
1310-73-2
|
Anorgaanilised alused
|
1B
|
1B
|
|
1-(2-aminoetüül)piperasiin
|
140-31-8
|
Alifaatsed amiinid
|
1B
|
1B
|
|
Benseensulfonüülkloriid
|
98-09-9
|
Hapete kloriidid
|
1C
|
1C
|
|
N,N-dimetüülbensüülamiin
|
103-83-3
|
Aniliinid
|
1C
|
1C
|
|
Tetraetüleenpentamiin
|
112-57-2
|
Alifaatsed amiinid
|
1C
|
1C
|
|
Eugenool
|
97-53-0
|
Fenoolid
|
MS
|
MS
|
|
Nonüülakrülaat
|
2664-55-3
|
Akrülaadid/metakrülaadid
|
MS
|
MS
|
|
Naatriumvesinikkarbonaat
|
144-55-8
|
Anorgaanilised soolad
|
MS
|
MS
|
|
METOODIKA
|
12.
|
Järgmistes punktides kirjeldatakse söövitavate omaduste hindamiseks kasutatava tehismembraanibarjääri katsemeetodi (7, 15) osi ja metoodikaid, mis põhinevad praegusel valideeritud võrdlusmeetodil, st müügil oleval Corrositex®-iga kasutaval meetodil. Membraanibarjääri, sobivuskatsel kasutatavad ja indikaatorlahused ning lahused, mis võimaldavad klassifitseerimist, võib sünteesida, valmistada või osta, nagu valideeritud võrdlusmeetodi puhul ostetakse Corrositex®. Saadaval on valideeritud katsemeetodi katse-eeskirja näidis (7). Katsed tuleb läbi viia toatemperatuuril (17–25 °C) ja nende osad peavad vastama järgmistele tingimustele.
|
Uuritava kemikaali sobivuskatse
|
13.
|
Enne membraanibarjääri katse tegemist tehakse sobivuskatse, et selgitada välja, kas kemikaali detekteerimissüsteem suudab uuritavat kemikaali avastada. Kui kemikaali detekteerimissüsteem uuritavat kemikaali avastada ei suuda, ei sobi membraanibarjääri katsemeetod selle konkreetse uuritava kemikaali võimalike söövitavate omaduste hindamiseks ja selleks tuleks kasutada teistsugust katsemeetodit. Sobivuskatses kasutatav kemikaali detekteerimissüsteem ja valitsevad kokkupuutetingimused peavad imiteerima järgnevas membraanibarjäärikatses toimuvat kokkupuudet.
|
Uuritava kemikaali ajakategooria katse
|
14.
|
Kui see on katsemeetodi juures asjakohane, peab sobivuskatse läbinud uuritav kemikaal läbima ka ajakategooria katse, st sõelkatse, millega eristatakse üksteisest nõrgad ja tugevad happed ja alused. Näiteks valideeritud võrdlusmeetodis kasutatakse ajakategooria katset selleks, et teha kindlaks, millist ajavahemikku kahest võimalikust kasutada, võttes aluseks leitud puhvermahtuvuse suuruse. Olenevalt uuritava kemikaali puhvermahtuvusest tuleks söövitavate omaduste ja ÜRO GHSi nahasöövituse alamkategooriate kindlaksmääramisel kasutada kaht erinevat läbimisaja vahemikku.
|
MEMBRAANIBARJÄÄRI KATSEMEETODI OSAD
Membraanibarjäär
|
15.
|
Membraanibarjäär koosneb kahest osast: Makromolekulaarne veepõhine valgugeel ja läbitav tugimembraan. Valgugeel peab olema vedelikele ja tahketele ainetele läbitungimatu, kuid see võib muutuda läbitavaks söövitamise tulemusena. Valmis konstrueeritud membraanibarjääri tuleb hoida eelnevalt kindlaksmääratud tingimustes, mis välistavad geeli riknemise, nt kuivamise, mikroobide kasvu, kihistumise, pragunemise vms, mis halvendaks selle toimivust. Tuleb kindlaks määrata vastuvõetav säilitusaeg ja pärast selle möödumist membraanbarjääri preparaate mitte kasutada.
|
|
16.
|
Läbitav tugimembraan pakub valgugeelile mehaanilist tuge geelistumise ja uuritava kemikaaliga kokkupuute ajal. Tugimembraan peaks takistama geeli kortsumist ja nihkumist ning laskma hästi läbi kõiki uuritavaid kemikaale.
|
|
17.
|
Valgugeel, mis koosneb näiteks keratiinist, kollageenist või valkude segust, mis moodustavad geelimaatriksi, on uuritava kemikaali sihtobjekt. Valgumaterjal kantakse tugimembraani pinnale ja lastakse enne membraanibarjääri indikaatorlahuse kohale seadmist geelistuda. Valgugeel peab olema läbivalt ühtlase paksuse ja tihedusega, ilma õhumullide ja muude defektideta, mis võiksid mõjutada selle funktsionaalset terviklikkust.
|
Kemikaalide detekteerimissüsteem
|
18.
|
Indikaatorlahus, mis on sama mis sobivuskatses kasutatud, peab reageerima uuritava kemikaali olemasolule. Kasutada tuleb värvainest või värvainete segust pH-indikaatorit, nt kresoolpunast või metüüloranži, mis uuritava kemikaali olemasolule reageerides värvi muudavad. Mõõtesüsteem võib olla visuaalne või elektrooniline.
|
|
19.
|
Detekteerimissüsteeme, mis on välja töötatud tuvastama uuritava kemikaali läbiminekut membraanibarjäärist, tuleb hinnata asjakohasuse ja usaldusväärsuse seisukohast, et teha kindlaks avastatavate kemikaalide hulk ja kvantitatiivne avastamispiir.
|
KATSE KÄIK
Katsemeetodi osade kokkupanek
|
20.
|
Membraanibarjäär paigutatakse indikaatorlahust sisaldavasse viaali (või katseklaasi), nii et tugimembraan on indikaatorlahusega täielikus kontaktis ja õhumulle ei esine. Tuleb olla ettevaatlik, et barjääri terviklikkus säiliks.
|
Uuritava kemikaaliga töötlemine
|
21.
|
Membraanibarjääri pealmisele pinnale kantakse hoolikalt ühe kihina sobiv kogus (nt 500 μl vedelikku või 500 mg pulbristatud tahket ainet (7)) uuritavat kemikaali ja aetakse ühtlaselt laiali. Iga uuritava kemikaaliga tehakse sobiv hulk paralleelproove, nt neli (7), samuti tehakse paralleelsed kontrollproovid (vt punktid 23–25). Registreeritakse uuritava kemikaali membraanibarjäärile kandmise aeg. Et tagada lühikese söövitusaja täpne registreerimine, ei kanta uuritavat kemikaali paralleelproovidega viaalidesse samaaegselt.
|
Membraanibarjääri läbimise mõõtmine
|
22.
|
Iga viaali jälgitakse ning registreeritakse ajahetk, mil indikaatorlahus esmakordselt muutub, st barjäär läbitakse, samuti tehakse kindlaks kemikaali pealekandmisest membraanibarjääri läbimiseni kulunud aeg.
|
Kontrollproovid
|
23.
|
Katsetes, kus koos uuritava kemikaaliga kasutatakse kandeainet või lahustit, peab kandeaine või lahusti membraanibarjäärisüsteemiga sobima, st see ei tohi muuta membraanibarjääri süsteemi terviklikkust ega uuritava kemikaali söövitavaid omadusi. Vajaduse korral tuleb uuritava kemikaaliga paralleelselt teha kontrollproov lahusti (või kandeainega), et veenduda lahusti sobivuses membraanibarjäärisüsteemiga.
|
|
24.
|
Selleks, et hinnata katsesüsteemi vastuvõetavat toimivust, tehakse uuritava kemikaaliga paralleelselt katse positiivse (söövitava) kontrollprooviga, mille söövitavad omadused on keskmised, nt 110 ± 15 mg naatriumhüdroksiidiga (ÜRO GHSi söövitavate ainete alamkategooria 1B) (7). Söövitava uuritava kemikaali suhtelise söövituspotentsiaali hindamiseks võib kasutada teist positiivset kontrollproovi ainest, mis kuulub uuritava kemikaaliga samasse aineklassi. Positiivsetesse kontrollproovidesse tuleb valida ained, mille söövitavad omadused on keskmise tugevusega (nt ÜRO GHSi alamkategooriasse 1B kuuluvad ained), et avastada muutusi läbitungimisajas, mis võib olla lubamatult pikem või lühem kui kindlaksmääratud võrdlusväärtus, näidates seega, et katsesüsteem ei toimi nõuetekohaselt. Sellisel juhul on vähe kasu äärmiselt söövitavatest (ÜRO GHSi alamkategooria 1A) ja mittesöövitavatest kemikaalidest. Söövitav kemikaal, mis kuulub ÜRO GHSi alamkategooriast 1B, võimaldaks avastada liiga pikka või liiga lühikest läbitungimisaega. Nõrgalt söövitavat ainet (ÜRO GHSi alamkategooria 1C) võib kasutada positiivses kontrollproovis, et mõõta, kui järjepidevalt on katsemeetodiga võimalik eristada nõrgalt söövitavaid ja mittesöövitavaid kemikaale. Olenemata lähenemisviisist tuleks positiivse kontrollproovidega saadud vastuvõetavate tulemuste vahemik välja töötada laboris varem kasutatud positiivsete kontrollproovidega saadud läbitungimisaegade vahemiku (nt keskväärtus ± 2–3 standardhälvet) alusel. Iga uuringu käigus tuleb määrata positiivse kontrollproovi täpne läbitungimisaeg, et tuvastada vastuvõetavast vahemikust väljapoole langevad kõrvalekalded.
|
|
25.
|
Paralleelselt uuritava kemikaaliga tuleb teha katse ka negatiivse (mittesöövitava) kontrollprooviga (nt 10 % sidrunhape, 6 % propioonhape (7)), mis on veel üks kvaliteedikontrolli meede membraanibarjääri funktsionaalse terviklikkuse tõendamiseks.
|
Uuringu nõuetekohasuse kriteeriumid
|
26.
|
Vastavalt iga ÜRO GHSi söövitavate omaduste alamkategooria kohta kindlaksmääratud ajaparameetritele kasutatakse uuritava kemikaali söövitavate omaduste prognoosimiseks uuritava kemikaali membraanibarjäärile kandmisest barjääri läbimiseni kulunud aega (minutites). Et uuringut saaks lugeda nõuetekohaseks, peab paralleelne positiivne kontrollproov andma eeldatava läbitungimisaja (st 8–16 minutit, kui positiivse kontrollainena kasutatakse naatriumhüdroksiidi), paralleelne negatiivne kontrollproov ei tohi olla söövitav ja lahusti kontrollproov, kui seda uuritakse, et tohi olla söövitav ega muuta uuritava kemikaali söövituspotentsiaali. Enne käesoleval katsemeetodil põhineva meetodi rutiinset kasutamist peaksid laborid tõendama oma tehnilist pädevust, kasutades tabelis 2 soovitatud kahtteistkümmet ainet. Niisuguste käesoleva katsemeetodi alusel välja töötatud samalaadsete meetodite puhul, mis on valideeritud võrdlusmeetodiga (14) struktuurilt ja funktsioonilt sarnased, tuleks kasutada eelnevalt kindlaksmääratud toimivusnõudeid, et tõendada uue meetodi usaldusväärsust ja täpsust enne selle kasutamist regulatiivsel eesmärgil (10).
|
Tulemuste tõlgendamine ja uuritavate kemikaalide klassifitseerimine söövitava toime järgi
|
27.
|
Uuritava kemikaali membraanibarjäärile kandmisest barjäärist läbitungimiseni kulunud aja (minutites) järgi klassifitseeritakse uuritav kemikaal ÜRO GHSi söövitavate ainete alamkategooriasse (1) ja ÜRO pakendirühma (16), kui see on asjakohane. Iga soovitatava katsemeetodi jaoks tehakse kindlaks läbitungimisaja läviväärtused kõigi kolme söövitavate omaduste alamkategooria puhul. Läbitungimisaja läviväärtuste kohta lõpliku otsuse tegemisel tuleb arvestada vajadust minimeerida söövitusohu alahindamist ja madalamasse kategoorias klassifitseerimist (st valenegatiivseid tulemusi). Praeguse katsejuhendi kohaselt tuleks kasutada Corrositex®iga kehtivaid läbitungimisaja läviväärtusi, mis on esitatud tabelis 3, sest see on ainus katsemeetod, mis hetkel katsejuhendile vastab (7).
Tabel 3
Corrositex®i prognoosimismudel
|
Keskmine läbitungimisaeg (min)
|
Prognoositav ÜRO GHSi kategooria (32)
|
|
1. kategooria uuritav kemikaal (30) (määratud meetodi kategoriseerimiskatse alusel)
|
2. kategooria uuritav kemikaal (31) (määratud meetodi kategoriseerimiskatse alusel)
|
|
0–3 min
|
0–3 min
|
Söövitav võimalik alamkategooria 1A
|
|
> 3 kuni 60 min
|
> 3 kuni 30 min
|
Söövitav võimalik alamkategooria 1B
|
|
> 60 kuni 240 min
|
> 30 kuni 60 min
|
Söövitav võimalik alamkategooria 1C
|
|
> 240 min
|
> 60 min
|
Mittesöövitav
|
|
ANDMED JA KATSEPROTOKOLLI KOOSTAMINE
Andmed
|
28.
|
Uuritava kemikaaliga ja positiivse(te) kontrollproovi(de)ga saadud aeg (minutites), mis kulus pealekandmisest barjääri läbimiseni, tuleb esitada tabelina, kus on näidatud nii eraldi andmed iga paralleelproovi kohta kui ka iga katse keskmine tulemus ± standardhälve.
|
Katseprotokoll
|
29.
|
Katseprotokollis tuleks esitada järgmine teave.
|
|
Uuritav kemikaal ja kontrollained
|
—
|
Ühest koostisosast koosnev aine: kemikaali identimisandmed, näiteks IUPACi või CASi nimetus, CASi number, SMILESi või InChI kood, struktuurivalem, puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
—
|
mitme koostisosaga aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal ja segu: võimalikult täpne omaduste kirjeldus lähtuvalt koostisosade keemilisest määratlusest (vt eespool), kvantitatiivsest sisaldusest ja asjakohastest füüsikalis-keemilistest omadustest;
|
|
—
|
füüsiline välimus, lahustuvus vees ja muud asjakohased füüsikalis-keemilised omadused;
|
|
—
|
tarnija ja partii number, kui see on olemas;
|
|
—
|
vajaduse korral teave uuritava kemikaali / kontrollaine töötlemise kohta enne katset (näiteks soojendamine, peenestamine);
|
|
—
|
uuritava kemikaali püsivus ja aegumiskuupäev või kordusanalüüsi tegemise tähtpäev, kui see on teada;
|
|
|
|
Kandeaine
|
—
|
Identimisandmed, kontsentratsioon (vajaduse korral), kasutatud ruumala;
|
|
—
|
kandeaine valimise põhjendus.
|
|
|
|
Kasutatud in vitro membraanibarjäär ja metoodika, sealhulgas tõendatud täpsus ja usaldusväärsus
|
|
|
Katsetingimused:
|
—
|
kasutatud aparatuur ja preparaadi ettevalmistusmeetodid;
|
|
—
|
kasutatud in vitro membraanibarjääri tarnija ja koostis;
|
|
—
|
indikaatorlahuse koostis ja omadused;
|
|
—
|
uuritava kemikaali ja kontrollainete kogused;
|
|
—
|
ajakategooria katse kirjeldus ja selle valimise põhjendus;
|
|
—
|
kasutatud hindamis- ja klassifitseerimiskriteeriumide kirjeldus;
|
|
—
|
Katsemeetodi kasutamise pädevuse tõendamine, analüüsides asjaomaseid kemikaale enne meetodi rutiinset kasutamist.
|
|
|
|
Tulemused
|
—
|
Iga uuritava ja kontrollprooviga igal paralleelkatsel saadud andmed tabelina;
|
|
—
|
muude täheldatud toimete kirjeldus;
|
|
—
|
tuletatud klassifikatsioon(id) viitega kasutatud prognoosimudelile ja/või otsustamiskriteeriumidele.
|
|
Tulemuste arutelu
Järeldused
|
KIRJANDUS
|
(1)
|
Ühinenud Rahvaste Organisatsioon (ÜRO), Globally Harmonized System of Classification and Labelling of Chemicals (GHS), 5. täiendatud väljaanne, United Nations, New York ja Genf, 2013. Kättesaadav aadressil http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Käesoleva lisa peatükk B.4, „Äge nahaärritus, söövitus“.
|
|
(3)
|
Käesoleva lisa peatükk B.40bis, „Nahasöövituskatse in vitro: rekonstrueeritud inimese epidermist (RHE) kasutav katsemeetod“.
|
|
(4)
|
Käesoleva lisa peatükk B.40, „Nahasöövituskatse in vitro: transkutaanse elektritakistuse (TER) mõõtmine“.
|
|
(5)
|
OECD (2015), „Guidance Document on an Integrated Approach to Testing and Assessment (IATA) for Skin Corrosion and Irritation“, Environmental, Health and Safety Publications, Series on Testing and Assessment (nr 203), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. ja Liebsch, M. (1998), „The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and Evaluation by the Management Team“, Toxicology In Vitro, nr 12, lk 483–524.
|
|
(7)
|
ICCVAM (1999), „Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM“, NIEHS, NIH Publication (nr 99–4495).
|
|
(8)
|
Gordon V.C., Harvell J.D. ja Maibach H.I. (1994), „Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization“, Alternative Methods in Toxicology, nr 10, lk 37–45.
|
|
(9)
|
OECD (2005), „Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment“, Environmental, Health and Safety Publications, Series on testing and Assessment (nr 34).
|
|
(10)
|
OECD (2014), „Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435“, Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001), „Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing“, ECVAMi teadusliku nõuandekomitee 15. istungjärk, ISPRA; Itaalia. ATLA nr 29, lk 96–97.
|
|
(12)
|
USA Transpordiministeerium (2002), Exemption DOT-E-10904 (5. täiendatud väljaanne), (20. september 2002), Washington, D.C., U.S. DOT.
|
|
(13)
|
Käesoleva lisa peatükk B.46, „Nahasöövituskatse in vitro: rekonstrueeritud inimese epidermist (RHE) kasutav katsemeetod“, ICCVAM (2004), „ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion“, NIEHS, NIH Publication (nr 04–4510). Kättesaadav aadressil http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
USA Keskkonnakaitseamet (1996), „Method 1120, Dermal Corrosion“. Kättesaadav aadressil http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
Ühinenud Rahvaste Organisatsioon (ÜRO) (2013), UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18. täiendatud väljaanne (peatükk 2.8), UN, 2013. Kättesaadav aadressil http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Liide
MÕISTED
Täpsus
: katsemeetodiga saadud tulemuste ja nõuetekohaste võrdlusväärtuste kokkulangevuse määr. Täpsus näitab katsemeetodi tulemuslikkust ja on üks olulistest aspektidest. Seda terminit kasutatakse ka vastavuse tähenduses ja see näitab, kui sageli annab katsemeetod õige tulemuse (9).
Kemikaal
: aine või segu.
Kemikaalide detekteerimissüsteem
: visuaalne või elektrooniline mõõtesüsteem indikaatorlahusega, mis reageerib uuritava kemikaali või muude kemikaalide olemasolule või elektrokeemilise reaktsiooni toimumisele nt pH-indikaatorvärvaine või värvainete segu värvuse muutusega.
Vastavus
: katsemeetodi tulemuslikkuse näitaja katsemeetodite puhul, millega saadakse kategooriline tulemus, on üks asjakohasuse aspekte. Terminit kasutatakse mõnikord ka täpsuse tähenduses ja määratletakse kõigi nende uuritavate kemikaalide osakaaluna, mis saadud positiivse või negatiivse tulemuse alusel klassifitseeritakse õigesti. Vastavus on tugevas sõltuvuses positiivset tulemust andvate kemikaalide esinemissagedusest uuritavate kemikaalide hulgas (9).
GHS (Globally Harmonized System of Classification and Labelling of Chemicals – kemikaalide ühtne ülemaailmne klassifitseerimis- ja märgistamissüsteem)
: süsteem, milles kemikaalid (ained ja segud) on klassifitseeritud vastavalt nende füüsilise ohtlikkuse ning kahjuliku tervise- ja keskkonnamõju standarditud tüübile ja -tasemele ning mis hõlmab asjaomaseid teavitustähiseid, nagu piktogrammid, märksõnad, ohulaused, hoiatuslaused ja ohutuskaardid, et anda edasi inimeste (sealhulgas tööandjate, töötajate, vedajate, tarbijate ja päästetöötajate) ja keskkonna kaitsmiseks vajalikku teavet kõnealuste kemikaalide kahjuliku mõju kohta (1).
IATA
: Integrated Approach on Testing and Assessment – ühtne lähenemisviis katsete tegemisele ja hindamisele.
Segu
: kahest või enamast ainest koosnev segu või lahus.
Ühest koostisosast koosnev aine
: aine, mis on määratletud oma kvantitatiivse koostisega, milles ühe peamise koostisosa sisaldus on vähemalt 80 massiprotsenti.
Mitme koostisosaga aine
: aine, mis on määratletud oma kvantitatiivse koostisega, milles rohkem kui ühe peamise koostisosa kontsentratsioon on ≥ 10 % ja < 80 % (massiprotsendid). Mitme koostisosaga aine saadakse tootmisprotsessi käigus. Erinevus segu ja mitme koostisosaga aine vahel seisneb selles, et segu saadakse kahe või enama aine kokku segamisel, ilma et toimuks keemilist reaktsiooni. Mitme koostisosaga aine saadakse keemilise reaktsiooni tulemusel.
MS
: mittesöövitav
Toimivusnõuded
: valideeritud katsemeetodil põhinevad nõuded, mille alusel hinnatakse, kuivõrd saab esitatud katsemeetodit võrrelda teostuslikult ja tööpõhimõttelt samalaadse meetodiga. See hõlmab järgmist: i) olulisemad katsemeetodi osad; ii) valideeritud katsemeetodi nõuetekohase toimivuse tõendamiseks kasutatud kemikaalide hulgast valitud võrdluskemikaalide miinimumloend ja iii) sarnased usaldusväärsuse ja täpsuse tasemed, mis põhinevad valideeritud katsemeetodiga saadud tulemustel ja mis tuleks kavandatud katsemeetodi hindamisel saavutada võrdluskemikaalide miinimumloendi kasutamisel (9).
Asjakohasus
: mõiste, millega väljendatakse seda, kas katsemeetod võimaldab uurida huvipakkuvat mõju, kas meetod on mõttekas ja sobib konkreetse eesmärgi jaoks. See näitab, mil määral saab katsemeetodiga õigesti mõõta või prognoosida huvipakkuvat bioloogilist mõju. Asjakohasus hõlmab ka katsemeetodi täpsuse (vastavuse) aspekti (9).
Usaldusväärsus
: iseloomustab katsemeetodi korratavust, kui meetodit rakendatakse ühe ja sama katse-eeskirja alusel ühes laboris ja eri laborites pikema aja jooksul. Usaldusväärsuse hindamiseks arvutatakse laborisisene ja laboritevaheline korratavus (9).
Tundlikkus
: kõigi positiivsete/reaktsioonivõimeliste kemikaalide osakaal, mis katsemeetodi alusel klassifitseeritakse õigesti. Tundlikkusega iseloomustatakse katsemeetodi täpsust, kui saadud tulemusi saab kategoriseerida, ning see on oluline aspekt katsemeetodi asjakohasuse hindamisel (9).
Nahasöövitus in vivo
: naha pöördumatu kahjustuse tekitamine, läbi epidermise pärisnahka (dermasse) ulatuv nähtav nekroos, mis tekib kuni neljatunnisel kokkupuutel katseainega. Tüüpilised söövitusnähud on haavandid, verejooks, verised kärnad ning 14-päevase vaatlusperioodi lõpuks ilmnev värvimuutus naha valastumise tõttu, täielik alopeetsia ja armistumine. Ebaselgete kahjustuste hindamiseks tuleks kaaluda histopatoloogilise analüüsi tegemist.
Spetsiifilisus
: kõigi niisuguste negatiivsete/mittereageerivate kemikaalide osakaal, mis katsemeetodiga klassifitseeritakse õigesti. Spetsiifilisusega iseloomustatakse katsemeetodi täpsust, kui saadud tulemusi saab kategoriseerida, ning see on oluline aspekt katsemeetodi asjakohasuse hindamisel (9).
Aine
: looduslik või tootmisprotsessi teel saadud keemiline element ja selle ühendid, kaasa arvatud stabiilsuse säilitamiseks vajalikud lisaained ja tootmisprotsessist tingitud lisandid, välja arvatud mis tahes lahusti, mida on võimalik aine stabiilsust vähendamata ja selle koostist muutmata eraldada.
Uuritav kemikaal
: iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
UVCB
: tundmatu või muutuva koostisega ained, komplekssed reaktsioonisaadused või bioloogilist päritolu materjalid.
B.66 STABIILSE TRANSFEKTSIOONIGA TRANSAKTIVATSIOONI IN VITRO KATSEMEETODID, MILLEGA AVASTATAKSE ÖSTROGEENIRETSEPTORITE AGONISTE JA ANTAGONISTE
ÜLDINE SISSEJUHATUS
OECD toimivusel põhinev katsejuhend
|
1.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 455 (2016). Katsejuhend nr 455 on toimivusel põhinev katsejuhend, milles kirjeldatakse stabiilse transfektsiooniga transaktivatsiooni in vitro katsemeetodeid östrogeeniretseptorite agonistide ja antagonistide tuvastamiseks (ER TA katsed). See hõlmab mitut teostuslikult ja tööpõhimõttelt samalaadset katsemeetodit östrogeeniretseptorite (st ERα, ja/või ERβ) agonistide ja antagonistide kvalitatiivseks määramiseks ning peaks hõlbustama uute samaväärsete või modifitseeritud katsemeetodite väljatöötamist kooskõlas valideerimispõhimõtetega, mis on esitatud OECD juhenddokumendis uute ja ajakohastatud ohu hindamise katsemeetodite valideerimise ja rahvusvahelise tunnustamise kohta (1). Täielikult valideeritud võrdlusmeetodid (liited 2 ja 3), mis on toimivuse põhineva katsejuhendi aluseks, on järgmised:
|
—
|
stabiilse transfektsiooniga transaktivatsiooni katsemeetod (2), milles kasutatakse rakuliini (h)ERα-HeLa-9903 ning
|
|
—
|
VM7Luc ER TA katsemeetod (3), milles kasutatakse rakuliini VM7Luc4E2, (33) mis ekspresseerib peamiselt hERα-d ja mõningal määral hERβ-d (4, 5).
|
Sama ohunäitaja analüüsimiseks kasutatavate samaväärsete katsemeetodite väljatöötamiseks ja valideerimiseks on olemas toimivusnõuded (6, 7) ja neid tuleks kasutada. Need võimaldavad toimivusel põhinevat katsejuhendit nr 455 õigeaegselt muuta, nii et ajakohastatud katsejuhendisse saaks lisada uued samalaadsed katsemeetodid, kuid need lisatakse üksnes pärast seda, kui OECD on need läbi vaadanud ja nõustunud, et toimivusnõuded on täidetud. Selleks, et täita OECD liikmesriikide nõuded östrogeeniretseptorite transaktivatsioonikatse tulemustele, võib kasutada suvalist meetodit katsejuhendiga nr 455 hõlmatud katsemeetoditest, kasutades ühtlasi andmete vastastikust tunnustamist vastavalt OECD kokkuleppele.
|
Käesoleva katsemeetodiga hõlmatud meetodite taust ja põhimõtted
|
2.
|
OECD algatas 1998. aastal prioriteetse tegevussuunana olemasolevate katsejuhendite läbivaatamise ja uute väljatöötamise, et sõeluuringutega selgitada välja endokriinfunktsiooni potentsiaalselt kahjustava kemikaalid ja neid katsetada. OECD kontseptuaalne raamistik endokriinfunktsiooni potentsiaalselt kahjustavate kemikaalide uurimiseks ja hindamiseks vaadati läbi 2012. aastal. Esialgne ja läbivaadatud kontseptuaalne raamistik on esitatud endokriinseid häireid põhjustavate kemikaalide hindamist käsitlevate standarditud katsejuhendite kohta koostatud OECD juhenddokumendi (8) lisades. Kontseptuaalne raamistik koosneb viiest tasemest, millest igaüks vastab erinevale bioloogilise keerukuse tasemele. Käesolevas katsemeetodis kirjeldatud ERi transaktivatsiooni katsed on 2. tasemel, mis hõlmab „in vitro katseid, millega saadakse andmeid teatud kindla(te) endokriinse(te) mehhanismi(de) või raja/radade kohta.“ See katsemeetod kirjeldab in vitro transaktivatsiooni (TA) katseid, mis on välja töötatud östrogeeniretseptorite (ER) agonistide ja antagonistide määramiseks.
|
|
3.
|
Östrogeenide vastastiktoime östrogeeniretseptoritega võib mõjutada östrogeenide reguleeritavate geenide transkriptsiooni, mis võib põhjustada rakuprotsesside induktsiooni või pärssimist, kaasa arvatud rakkude paljunemiseks, loote normaalseks arenguks ja reproduktiivfunktsiooni tööks vajalikud protsessid (9, 10, 11). Normaalse östrogeensõltuva süsteemi häirimine võib olla kahjuliku toimega normaalsele arengule (ontogeneesile), reproduktiivtervisele ja reproduktiivsüsteemi terviklikkusele.
|
|
4.
|
In vitro transaktivatsiooni katsemeetodid põhinevad ainete otsese või kaudse vastastikmõjul konkreetse retseptoriga, mis reguleerib reportergeeni produkti transkriptsiooni. Selliseid katsemeetodeid on laialdaselt kasutatud spetsiifiliste tuumaretseptorite, nt ERide poolt reguleeritud geeniekspressiooni hindamiseks (12, 13, 14, 15, 16). Neid on soovitatud kasutada ERi reguleeritava östrogeense transaktivatsiooni tuvastamiseks (17, 18, 19). Tuuma ERidel on olemas vähemalt kaks peamist alatüüpi, α ja β, mida kodeerivad erinevad geenid. Vastavatel valkudel on erinevad bioloogilised funktsioonid, samuti erinev jaotumine kudedes ja erinev ligandisidumisvõime (20, 21, 22, 23, 24, 25, 26). Tuuma ERα vahendab klassikalist reaktsiooni östrogeenile (27, 28, 29, 30) ning seetõttu on enamik ERi aktiveerimise või inhibeerimise mõõtmiseks hetkel välja töötatavaid mudelid ERα-spetsiifilised. Katsemeetodeid kasutatakse selleks, et välja selgitada kemikaalid, mis aktiveerivad (või pärsivad) ERi pärast ligandi sidumist, misjärel retseptori-ligandi kompleks seondub DNA spetsiifilise vastuselemendiga ja transaktiveerib reportergeeni, mille tulemuseks on markervalgu suurenenud rakusisene ekspressioon. Nendes katsemeetodites võib kasutada erinevate reporterite reaktsioone. Lutsiferaasil põhinevates süsteemides transformeerib ensüüm lutsiferaas substraadilutsiferiini helendavaks bioloogiliseks saaduseks, mida saab kvantitatiivselt mõõta luminomeetriga. Muud näited levinud reporteritest on fluorestseeruv valk ja lacZ-geen, mis kodeerib β-galaktosidaasi, ensüümi, mis transformeerib värvitu substraadi X-gal-i (5-bromo-4-kloro-indolüülgalaktopüranosiid) siniseks saaduseks, mida saab kvantitatiivselt mõõta spektrofotomeetriga. Neid reportereid saab kiiresti ja odavalt hinnata müügilolevate katsekomplektidega.
|
|
5.
|
STTA ja VM7Luc TA katsemeetodite valideerimisuuringutest nähtub, et need on ettenähtud eesmärgil kasutades asjakohased ja usaldusväärsed (3, 4, 5, 30). Rinna rakuliine kasutavate ja luminestsentsil põhinevate ERi transaktivatsiooni katsemeetodite toimivusnõuded on esitatud ICCVAMi katsemeetodi hindamisaruandes „ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (VM7Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals“ (3). Neid toimivusnõudeid on muudetud, et neid saaks kohaldada nii STTA kui ka VM7Luc TA katsemeetoditele (2).
|
|
6.
|
Käesolevas katsemeetodis kasutatud mõisted ja lühendid on esitatud 1. liites.
|
Transaktivatsiooni katsemeetodite rakendusala ja piirangud
|
7.
|
Käesolevaid katsemeetodeid on soovitatav kasutada sõelumise ja prioriseerimise eesmärgil, kuid need võivad anda ka niisugust teavet mehhanismide kohta, mida saab kasutada tõendite kaalukust arvestava lähenemisviisi korral. Need katsed uurivad ERiga seondunud kemikaali esilekutsutud transaktsiooni in vitro süsteemis. Seega ei tohiks tulemust otse ekstrapoleerida terve in vivo endokriinsüsteemi keerukale signaliseerimis- ja reguleerimismehhanismidele.
|
|
8.
|
ERide vahendatud transaktsiooni peetakse üheks peamiseks endokriinse häire põhjustamise mehhanismiks, kuigi endokriinne häire võib ilmneda ka muude mehhanismide kaudu, sealhulgas i) vastasmõjud endokriinsüsteemi teiste retseptorite ja ensüümisüsteemidega, ii) hormoonide süntees, iii) hormoonide metaboolne aktiveerimine ja/või inaktiveerimine, iv) hormoonide jaotumine sihtkudedes ja v) hormoonide väljumine organismist. Neid toimemehhanisme ei käsitle ükski käesoleva katsemeetodiga hõlmatud meetod.
|
|
9.
|
See katsemeetod uurib kemikaalide võimet ER-sõltuvat transkriptsiooni aktiveerida (st toimida agonistina) ja pärssida (st toimida antagonistina). Mõned kemikaalid võivad olenevalt rakutüübist toimida nii agonisti kui ka antagonistina, olles tuntud kui selektiivsed östrogeeniretseptori modulaatorid (SERMid). Nende katsemeetoditega negatiivse tulemuse andnud kemikaale võiks enne järeldamist, et kemikaal ei seo retseptorit, hinnata ERi sidumise katse abil. Peale selle annavad katsed tõenäoliselt teavet üksnes lähtemolekuli toime kohta, võttes arvesse, et in vitro rakusüsteemide metaboliseerimisvõime on piiratud. Arvestades, et valideerimisel kasutati üksnes puhtaid aineid, ei ole rakendatavust uuritavate segude puhul kontrollitud. See katsemeetod on siiski teoreetiliselt kohaldatav mitme koostisosaga ainete, tundmatu või muutuva koostisega ainete ning segude uurimiseks. Kui soovitakse saada kavandatud regulatiivsel eesmärgil andmeid mitme koostisosaga aine, tundmatu või muutuva koostisega aine või segu kohta, tuleks enne katsemeetodi kasutamist kaaluda, kas ja kuidas see võimaldab saada kõnealusele eesmärgile vastavaid asjakohaseid tulemusi. Selline kaalumine ei ole vajalik, kui asjaomase segu hindamine on regulatiivse nõudega ette nähtud.
|
|
10.
|
Teavitamise eesmärgil on tabelis 1 esitatud agonistikatse tulemused 34 aine kohta, mille uurimisel kasutati mõlemat käesolevas katsemeetodis kirjeldatud täielikult valideeritud võrdlusmeetodit. Nendest ainetest 26 on liigitatud kindlateks ERi agonistideks ja 8 negatiivseteks, võttes aluseks avaldatud aruanded, sealhulgas in vitro ERi sidumise ja transaktivatsiooni katsemeetodite ja/või uterotroofse katsemeetodi kohta (2, 3, 18, 31, 32, 33, 34). Tabelis 2 on esitatud antagonistikatse tulemused 15 aine kohta, mille uurimisel kasutati mõlemat käesolevas katsemeetodis kirjeldatud täielikult valideeritud võrdlusmeetodit. Nendest ainetest 4 on liigitatud kindlateks/eeldatavateks ERi antagonistideks ja 10 negatiivseteks, võttes aluseks avaldatud aruanded, sealhulgas in vitro ERi sidumise ja transaktivatsiooni katsemeetodite kohta (2, 3, 18, 31). Tabelites 1 ja 2 esitatud andmete kohaselt langesid kahe võrdlusmeetodiga saadud klassifitseerimise aluseks olevad tulemused 100 % kokku kõigi ainete puhul, välja arvatud ühe aine (mifepristoon) antagonistikatse tulemus, ning kõik ained klassifitseeriti korrektselt ERi agonistiks/antagonistiks või negatiivseks. Lisateave selle kemikaalirühma kohta, samuti muud kemikaalid, mida valideerimisuuringute käigus STTA ja VM7Luc ER TA katsemeetoditega uuriti, on esitatud 2. liites ER TA toimivusnõuetes (tabelid 1, 2 ja 3) (6, 7).
|
Tabel 1
STTA ja VM7Luc ER TA katsemeetodite tulemuste ülevaade ainete kohta, mida uuriti mõlema agonistikatsega ja mis klassifitseeriti kas ERi agonistideks (POS) või negatiivseteks (NEG)
|
|
Aine
|
CASi nr
|
STTA katsemeetod (34)
|
VM7Luc ER TA katsemeetod (35)
|
Klassifitseerimise andmete allikas (37)
|
|
ER TA toime
|
PC10 väärtus (M)
|
PC50 väärtus (2) (M)
|
ER TA toime
|
EC50 väärtus (2), (36) (M)
|
Muud ER TAd (3)
|
ERi siduv
|
Uterotroofne
|
|
1
|
17ß-östradiool (1)
|
50-28-2
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
5,63 × 10-12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-östradiool (1)
|
57-91-0
|
POS
|
7,24 × 10-11
|
6,44 × 10-10
|
POS
|
1,40 × 10-9
|
POS (11/11)
|
POS
|
POS
|
|
3
|
17α-etinüülöstradiool (1)
|
57-63-6
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
7,31 × 10-12
|
POS (22/22)
|
POS
|
POS
|
|
4
|
17β-trenboloon
|
10161-33-8
|
POS
|
1,78 × 10-8
|
2,73 × 10-7
|
POS
|
4,20 × 10-8
|
POS (2/2)
|
NT
|
NT
|
|
5
|
19-nortestosteroon (1)
|
434-22-0
|
POS
|
9,64 × 10-9
|
2,71 × 10-7
|
POS
|
1,80 × 10-6
|
POS (4/4)
|
POS
|
POS
|
|
6
|
4-kumüülfenool (1)
|
599-64-4
|
POS
|
1,49 × 10-7
|
1,60 × 10-6
|
POS
|
3,20 × 10-7
|
POS (5/5)
|
POS
|
NT
|
|
7
|
4-tert-oktüülfenool (1)
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
POS
|
3,19 × 10-8
|
POS (21/24)
|
POS
|
POS
|
|
8
|
Apigeniin (1)
|
520-36-5
|
POS
|
1,31 × 10-7
|
5,71 × 10-7
|
POS
|
1,60 × 10-6
|
POS (26/26)
|
POS
|
NT
|
|
9
|
Atrasiin (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Bisfenool A (1)
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
POS
|
5,33 × 10-7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisfenool B (1)
|
77-40-7
|
POS
|
2,36 × 10-8
|
2,11 × 10-7
|
POS
|
1,95 × 10-7
|
POS (6/6)
|
POS
|
POS
|
|
12
|
Butüülbensüülftalaat (1)
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
POS
|
1,98 × 10-6
|
POS (12/14)
|
POS
|
NEG
|
|
13
|
Kortikosteroon (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (6/6)
|
NEG
|
NT
|
|
14
|
Kumestrool (1)
|
479-13-0
|
POS
|
1,23 × 10-9
|
2,00 × 10-8
|
POS
|
1,32 × 10-7
|
POS (30/30)
|
POS
|
NT
|
|
15
|
Daidseiin (1)
|
486-66-8
|
POS
|
1,76 × 10-8
|
1,51 × 10-7
|
POS
|
7,95 × 10-7
|
POS (39/39)
|
POS
|
POS
|
|
16
|
Dietüülstilböstrool (1)
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POS
|
3,34 × 10-11
|
POS (42/42)
|
POS
|
NT
|
|
17
|
Di-n-butüülftalaat
|
84-74-2
|
POS
|
4,09 × 10-6
|
|
POS
|
4,09 × 10-6
|
POS (6/11)
|
POS
|
NEG
|
|
18
|
Etüülparabeen
|
120-47-8
|
POS
|
5,00 × 10-6
|
(PC50 puudub)
|
POS
|
2,48 × 10-5
|
POS
|
|
NT
|
|
19
|
Östroon (1)
|
53-16-7
|
POS
|
3,02 × 10-11
|
5,88 × 10-10
|
POS
|
2,34 × 10-10
|
POS (26/28)
|
POS
|
POS
|
|
20
|
Genisteiin (1)
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
POS
|
2,71 × 10-7
|
POS (100/102)
|
POS
|
POS
|
|
21
|
Haloperidool
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kamferool (1)
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
POS
|
3,99 × 10-6
|
POS (23/23)
|
POS
|
NT
|
|
23
|
Kepoon (1)
|
143-50-0
|
POS
|
7,11 × 10-7
|
7,68 × 10-6
|
POS
|
4,91 × 10-7
|
POS (14/18)
|
POS
|
NT
|
|
24
|
Ketokonasool
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Linuroon (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
meso-heksöstrool (1)
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POS
|
1,65 × 10-11
|
POS (4/4)
|
POS
|
NT
|
|
27
|
Metüültestosteroon (1)
|
58-18-4
|
POS
|
1,73 × 10-7
|
4,11 × 10-6
|
POS
|
2,68 × 10-6
|
POS (5/6)
|
POS
|
NT
|
|
28
|
Moriin
|
480-16-0
|
POS
|
5,43 × 10-7
|
4,16 × 10-6
|
POS
|
2,37 × 10-6
|
POS (2/2)
|
POS
|
NT
|
|
29
|
Noretünodreel (1)
|
68-23-5
|
POS
|
1,11 × 10-11
|
1,50 × 10-9
|
POS
|
9,39 × 10-10
|
POS (5/5)
|
POS
|
NT
|
|
30
|
p,p’-metoksükloor (1)
|
72-43-5
|
POS
|
1,23 × 10-6
|
(PC50 puudub) (2)
|
POS
|
1,92 × 10-6
|
POS (24/27)
|
POS
|
POS
|
|
31
|
Fenobarbitaal (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
32
|
Reserpiin
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
33
|
Spironolaktoon (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
34
|
Testosteroon
|
58-22-0
|
POS
|
2,82 × 10-8
|
9,78 × 10-6
|
POS
|
1,75 × 10-5
|
POS (5/10)
|
POS
|
NT
|
|
Lühendid: CASi nr – Chemical Abstracts Service’i number; M = molaarsus; EC50 = 50 % uuritava kemikaali suurima toime avaldumise kontsentratsioonist; NEG = negatiivne; POS = positiivne; NT = ei uuritud; PC10 (ja PC50) = uuritava aine kontsentratsioon, mille puhul toime on 10 % (või PC50 korral 50 %) võrreldes positiivse kontrollaine (E2, 1nM) toimega igal plaadil.
|
Tabel 2
STTA ja VM7Luc ER TA katsemeetodite tulemuste võrdlus ainete kohta, mida uuriti mõlema antagonistikatsega ja mis klassifitseeriti kas ERi antagonistideks (POS) või negatiivseteks (NEG)
|
|
Aine (4)
|
CASi nr
|
ER STTA katsemeetod (38)
|
VM7Luc ER TA katsemeetod (39)
|
ER STTA oletatav toime (41)
|
ICCVAMi (42) konsensusklassifikatsioon
|
MeSHi (43) aineklass
|
Tooteklass (44)
|
|
ER TA toime
|
IC50 väärtus (5) (M)
|
ER TA toime
|
IC50 väärtus (5), (40) (M)
|
|
1
|
4-hüdroksütamoksüfeen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
POS
|
2,08 × 10-7
|
Mõõdukalt POS
|
POS
|
Süsivesinik (tsükliline)
|
Ravim
|
|
2
|
Dibenso[a,h]antratseen
|
53-70-3
|
POS
|
IC50 puudub
|
POS
|
IC50 puudub
|
POS
|
PP
|
Polütsükliline ühend
|
Laborikemikaal, looduslik toode
|
|
3
|
Mifepristoon
|
84371-65-3
|
POS
|
5,61 × 10-6
|
NEG
|
—
|
Nõrgalt POS
|
NEG
|
Steroid
|
Ravim
|
|
4
|
Raloksüfeenvesinikkloriid
|
82640-04-8
|
POS
|
7,86 × 10-10
|
POS
|
1,19 × 10-9
|
Mõõdukalt POS
|
POS
|
Süsivesinik (tsükliline)
|
Ravim
|
|
5
|
Tamoksüfeen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
POS
|
8,17 × 10-7
|
POS
|
POS
|
Süsivesinik (tsükliline)
|
Ravim
|
|
6
|
17β-östradiool
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Steroid
|
Ravim, veterinaarravim
|
|
7
|
Apigeniin
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Heterotsükliline ühend
|
Värvaine, looduslik toode, ravimite vaheaine
|
|
8
|
Atrasiin
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Heterotsükliline ühend
|
Herbitsiid
|
|
9
|
Di-n-butüülftalaat
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Ester, ftaalhape
|
Kosmeetikatoodete koostisaine, tööstuskemikaal, plastifikaator
|
|
10
|
Fenarimool
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
pole katsetatud
|
PN
|
Heterotsükliline ühend, pürimidiin
|
Fungitsiid
|
|
11
|
Flavoon
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Flavonoid, heterotsükliline ühend
|
Looduslik toode, ravim
|
|
12
|
Flutamiid
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Amiid
|
Ravim, veterinaarravim
|
|
13
|
Genisteiin
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Flavonoid, heterotsükliline ühend
|
Looduslik toode, ravim
|
|
14
|
p-n-nonüülfenool
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
pole katsetatud
|
NEG
|
Fenool
|
Keemiline vaheühend
|
|
15
|
Resveratrool
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Süsivesinik (tsükliline)
|
Looduslik toode
|
|
Lühendid: CASi nr – Chemical Abstracts Service’i number; M = molaarsus; IC50 = 50 % uuritava kemikaali suurima inhibeeriva toime avaldumise kontsentratsioonist; NEG = negatiivne; PN = eeldatavasti negatiivne; POS = positiivne; PP = eeldatavasti positiivne.
|
ER TA KATSEMEETODI OSAD
Katsemeetodi olulised osad
|
11.
|
Käesolev katsemeetod kirjeldab katseid, milles kasutatakse stabiilse transfektsiooniga või endogeenseid ERα retseptori ja stabiilse transfektsiooniga reportergeeni konstrukti, mida reguleerib üks või mitu östrogeeni vastuselementi; leiduda võib siiski ka muid retseptoreid, nt ERβ. Need on katsemeetodi olulised osad.
|
Kontrollproovid
|
12.
|
Kirjeldada tuleb iga agonisti- ja antagonistikatse paralleelsetes kontrollproovides kasutatavate etalonainete soovituste alust. Asjakohaste paralleelsete kontrollproovide (negatiivne, lahusti ja positiivne) eesmärk on näidata, et katsemeetod katsetingimustes toimib, ning olla eksperimentide omavahelise võrdlemise aluseks; tavaliselt on paralleelsed kontrollproovid osa konkreetse eksperimendi nõuetekohasuse kriteeriumidest (1).
|
Kvaliteedikontrolli standardkord
|
13.
|
Kvaliteedikontrolli standardkorda tuleb kirjeldatud viisil järgida iga katse puhul tagamaks, et rakuliin on püsivalt stabiilne mitme passaaži jooksul, ei sisalda mükoplasmat (st ei ole bakteriaalselt saastunud) ja säilitab võime anda aja jooksul oodatavaid ERi vahendatud vastuseid. Lisaks tuleb kontrollida rakuliinide õiget identiteeti, samuti teiste saasteainete (nt seened, pärmseened ja viirused) sisaldust.
|
Labori pädevuse tõendamine
|
14.
|
Enne tundmatute kemikaalide uurimist mis tahes selle katsemeetodiga hõlmatud meetodi abil peab iga labor tõendama oma pädevust kõnealuse katsemeetodi kasutamiseks. Pädevuse tõendamiseks peab iga labor viima läbi katsed tabelis 3 loetletud 14 pädevuse tõendamise ainega agonistikatse puhul ja tabelis 4 loetletud 10 pädevuse tõendamise ainega antagonistikatse puhul. Pädevuskatsed kinnitavad ühtlasi katsesüsteemi reageerimisvõimet. Pädevuse tõendamise ainete loetelu on osa võrdlusainete loetelust, mis on esitatud ER TA katsemeetodite toimivusnõuetes (6). Need ained on müügil, esindavad niisuguseid kemikaaliklasse, mida üldiselt seostatakse ER agonistliku või antagonistliku toimega, avaldavad ER agonisti või antagonisti puhul eeldatavat toimet sobivas vahemikus (st on tugevad või nõrgad) ja hõlmavad negatiive. Pädevuse tõendamise ainetega tuleb teha vähemalt kaks korduskatset eri päevadel. Pädevust tõendab iga pädevuse tõendamise aine õige klassifitseerimine (positiivne/negatiivne). Pädevuskatset peaks katsemeetodite õppimise ajal korduvalt tegema iga laboritöötaja. Olenevalt rakutüübist võivad mõned neist pädevuse tõendamise ainetest käituda SERMidena ja toimida nii agonisti kui ka antagonistina. Tabelites 3 ja 4 on pädevuse tõendamise ained siiski klassifitseeritud nende teadaoleva peamise toime järgi, mida tuleks pädevuse tõendamisel arvesse võtta.
|
|
15.
|
Tulemuslikkuse tõendamiseks ja kvaliteedi kontrolli eesmärgil peaks iga labor võrdlema aja jooksul agonistide ja antagonistide kohta koostatud andmebaase andmetega etalonaine (nt 17β-östradiool ja tamoksüfeen), positiivses ja negatiivses kontrollproovis kasutatud kemikaalide ning lahusti (nt DMSO) kontrollproovi kohta. Kõigepealt tuleks luua andmebaas vähemalt 10 sõltumatu agonisti (nt 17β-östradiool) ja 10 sõltumatu antagonisti (nt tamoksüfeen) katse andmete põhjal. Kõnealuste etalonainete ja lahusti kontrollproovidega edaspidi saadud tulemused tuleks andmebaasi laiendamiseks sinna lisada; nii tagatakse laboris biokatsemeetodi järjepidevus ja tulemuslikkus läbi aja.
|
Tabel 3
Pädevuse tõendamise ainete (14) loetelu agonistikatse jaoks
(52)
|
Nr (51)
|
Aine
|
CASi nr
|
Eeldatav vastus (45)
|
STTA katsemeetod
|
VM7Luc ER TA katsemeetod
|
MeSHi aineklass (49)
|
Tooteklass (50)
|
|
PC10 väärtus (M) (46)
|
PC50 väärtus (M) (46)
|
Katsel kasutatud kontsentratsioonivahemik (M)
|
VM7Luc EC50 väärtus (M) (47)
|
Suurim kontsentratsioon vahemikuotsingus (M) (48)
|
|
14
|
Dietüülstilböstrool
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Süsivesinik (tsükliline)
|
Ravim, veterinaarravim
|
|
12
|
17α-östradiool
|
57-91-0
|
POS
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steroid
|
Ravim, veterinaarravim
|
|
15
|
meso-heksöstrool
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Süsivesinik (tsükliline), fenool
|
Ravim, veterinaarravim
|
|
11
|
4-tert-oktüülfenool
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenool
|
Keemiline vaheühend
|
|
9
|
Genisteiin
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoid, heterotsükliline ühend
|
Looduslik toode, ravim
|
|
6
|
Bisfenool A
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenool
|
Keemiline vaheühend
|
|
2
|
Kamferool
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoid, heterotsükliline ühend
|
Looduslik toode
|
|
3
|
Butüülbensüülftalaat
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Karboksüülhape, ester, ftaalhape
|
Plastifikaator, tööstuskemikaal
|
|
4
|
p,p’-metoksükloor
|
72-43-5
|
POS
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Süsivesinik (halogeenitud)
|
Pestitsiid, veterinaarravim
|
|
1
|
Etüülparabeen
|
120-47-8
|
POS
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Karboksüülhape, fenool
|
Ravim, konservant
|
|
17
|
Atrasiin
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Heterotsükliline ühend
|
Herbitsiid
|
|
20
|
Spironolaktoon
|
52-01-7
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Laktoon, steroid
|
Ravim
|
|
21
|
Ketokonasool
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Heterotsükliline ühend
|
Ravim
|
|
22
|
Reserpiin
|
50-55-5
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Heterotsükliline ühend, indool
|
Ravim, veterinaarravim
|
|
Lühendid: CASi nr – Chemical Abstracts Service’i number; EC50 = 50 % uuritava kemikaali suurima toime avaldumise kontsentratsioonist; NEG = negatiivne; POS = positiivne; PC10 (ja PC50) = uuritava aine kontsentratsioon, mille puhul toime on 10 % (või PC50 korral 50 %) võrreldes positiivse kontrollaine (E2, 1nM) toimega igal plaadil.
|
Tabel 4
Pädevuse tõendamise ainete (10) loetelu antagonistikatse jaoks
|
|
Aine (6)
|
CASi nr
|
ER STTA katsemeetod (53)
|
VM7Luc ER TA katsemeetod (54)
|
ER STTA (53) oletatav mõju
|
ICCVAMi (57) konsensusklassifikatsioon
|
MeSHi (58) aineklass
|
Tooteklass (59)
|
|
ER TA toime
|
IC50 (M)
|
Katsel kasutatud kontsentratsioonivahemik (M)
|
ER TA toime
|
IC50
(55) (M)
|
Suurim kontsentratsioon vahemikuotsingus (M) (56)
|
|
1
|
4-hüdroksütamoksüfeen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
10-12 – 10-7
|
POS
|
2,08 × 10-7
|
2,58 × 10-4
|
Mõõdukalt POS
|
POS
|
Süsivesinik (tsükliline)
|
Ravim
|
|
2
|
Raloksüfeenvesinikkloriid
|
82640-04-8
|
POS
|
7,86 × 10-10
|
10-12 – 10-7
|
POS
|
1,19 × 10-9
|
1,96 × 10-4
|
Mõõdukalt POS
|
POS
|
Süsivesinik (tsükliline)
|
Ravim
|
|
3
|
Tamoksüfeen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
10-10 – 10-5
|
POS
|
8,17 × 10-7
|
2,69 × 10-4
|
POS
|
POS
|
Süsivesinik (tsükliline)
|
Ravim
|
|
4
|
17β-östradiool
|
50-28-2
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,67 × 10-3
|
eeldatavalt negatiivne (*4)
|
PN
|
Steroid
|
Ravim, veterinaarravim
|
|
5
|
Apigeniin
|
520-36-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Heterotsükliline ühend
|
Värvaine, looduslik toode, ravimite vaheaine
|
|
6
|
Di-n-butüülftalaat
|
84-74-2
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Ester, ftaalhape
|
Kosmeetikatoodete koostisaine, tööstuskemikaal, plastifikaator
|
|
7
|
Flavoon
|
525-82-6
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,50 × 10-4
|
eeldatavalt negatiivne (*4)
|
PN
|
Flavonoid, heterotsükliline ühend
|
Looduslik toode, ravim
|
|
8
|
Genisteiin
|
446-72-0
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
eeldatavalt negatiivne (*4)
|
NEG
|
Flavonoid, heterotsükliline ühend
|
Looduslik toode, ravim
|
|
9
|
p-n-nonüülfenool
|
104-40-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
4,54 × 10-4
|
pole katsetatud
|
NEG
|
Fenool
|
Keemiline vaheühend
|
|
10
|
Resveratrool
|
501-36-0
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,38 × 10-4
|
eeldatavalt negatiivne (*4)
|
NEG
|
Süsivesinik (tsükliline)
|
Looduslik toode
|
|
Lühendid: CASi nr – Chemical Abstracts Service’i number; M = molaarsus; IC50 = 50 % uuritava kemikaali suurima inhibeeriva toime avaldumise kontsentratsioonist; NEG = negatiivne; PN = eeldatavasti negatiivne; POS = positiivne.
|
Katse nõuetekohasuse kriteeriumid
|
16.
|
Katse nõuetekohaseks tunnistamise või tagasilükkamise aluseks on iga katse tegemisel kasutatud etalonaine ja kontrollproovidega saadud tulemuste hinnang. Etalonainete PC50 (EC50) või IC50 väärtused peaksid vastama valitud katsemeetodi (vt 2. liide STTA kohta ja 3. liide VM7Luc ER TA kohta) nõuetekohasuse kriteeriumidele ning kõik positiivsed/negatiivsed kontrollproovid peaksid igas nõuetekohaseks tunnistatud katses olema õigesti klassifitseeritud. Katse järjepideva läbiviimise võimet tuleb tõendada aja jooksul etalonainete ja kontrollproovide kohta kogutud andmeid sisaldava andmebaasi arendamise ja hooldamisega (vt punkt 15). Laborisisese korratavuse mõõduna võib kasutada mitme katsega saadud etalonainete kõverate ja parameetrite sobitamise tulemusel leitud keskväärtuste standardhälbeid (SD) või variatsioonikordajaid (CV). Peale selle peavad seoses nõuetekohasuse kriteeriumidega olema täidetud järgmised tingimused:
|
—
|
andmed peavad olema ERi aktiveerimise (agonistikatse) või pärssimise (antagonistikatse) (st tõhususe ja toime) kvantitatiivseks hindamiseks piisavad;
|
|
—
|
piisava tundlikkuse tagamiseks peab reporteri keskmine aktiivsus võrdlusöstrogeeni võrdluskontsentratsiooni puhul olema võrdne vähemalt kandeaine (lahusti) kontrollproovi analüüsil määratud miinimumiga. STTA ja VM7Luc ER TA katsemeetodite korral on see igal plaadil neljakordne kandeaine kontrollprooviga saadud keskmine;
|
|
—
|
uuritavad kontsentratsioonid peaksid jääma uuritavate kemikaalide lahustuvusvahemikku ega tohiks olla tsütotoksilised.
|
|
Andmeanalüüs
|
17.
|
Positiivse ja negatiivse vastuse klassifitseerimiseks tuleks iga katsemeetodi puhul järgida kindlaksmääratud andmete tõlgendamise eeskirja.
|
|
18.
|
Nõuetekohasuse kriteeriumide (punkt 16) täitmine näitab, et katsemeetod toimib korrektselt, kuid see ei taga täpsete andmete saamist mis tahes konkreetsel katsel. Esimese katse tulemuste kordumine on parim märk täpsete andmete saamisest. Kui kahe katsega saadakse korratavad tulemused (st mõlema katse tulemused näitavad, et uuritav kemikaal on positiivne), ei ole kolmanda katse tegemine vajalik.
|
|
19.
|
Kui kahe katsega saadud tulemused ei ole korratavad (nt uuritav kemikaal on ühe katse põhjal positiivne ja teise põhjal negatiivne) või kõnealuse katsemeetodi tulemuste puhul on nõutav suurem paikapidavuse aste, tuleks läbi viia vähemalt kolm sõltumatut katset. Sellisel juhul võetakse klassifitseerimisel aluseks kaks kokkulangevat tulemust kolmest.
|
Andmete tõlgendamise üldkriteeriumid
|
20.
|
Praegu ei ole olemas üldtunnustatud meetodit ER TA andmete tõlgendamiseks. Kuid nii ERi vahendatud toime kvalitatiivne (nt positiivne/negatiivne) kui ka kvantitatiivne (nt EC50, PC50, IC50) hindamine peab põhinema empiiriliste andmetel ja põhjendatud teaduslikel otsustel. Positiivseid tulemusi tuleks võimaluse korral iseloomustada nii toime suurusjärgu poolest kandeaine (lahusti) kontrollproovi või etalonöstrogeeniga võrreldes kui ka toime avaldumise kontsentratsiooni (nt EC50, PC50, RPCmax, IC50 jms) poolest.
|
Katseprotokoll
|
21.
|
Katseprotokollis tuleks esitada järgmine teave.
|
|
Katsemeetod:
|
—
|
kontrollaine, etalonaine, uuritav kemikaal;
|
|
—
|
tarnija, partii number, aegumise kuupäev, kui olemas;
|
|
—
|
uuritava kemikaali püsivus, kui see on teada;
|
|
—
|
uuritava kemikaali lahustuvus ja stabiilsus lahustis, kui teada;
|
|
—
|
vastavalt vajadusele mõõtmistulemused söötme pH ja osmolaalsuse ning sademe tekke kohta pärast uuritava kemikaali lisamist.
|
|
|
|
Ühest koostisosast koosnev aine:
|
—
|
füüsiline välimus, lahustuvus vees ja muud asjakohased füüsikalis-keemilised omadused;
|
|
—
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus, CASi number, SMILESi või InChI kood, struktuurivalem, puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne.
|
|
|
|
Mitme koostisosaga aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal ja segu:
|
—
|
võimalikult täpne omaduste kirjeldus lähtuvalt koostisosade keemilisest määratlusest (vt eespool), kvantitatiivsest sisaldusest ja asjakohastest füüsikalis-keemilistest omadustest.
|
|
|
|
Lahusti/kandeaine:
|
—
|
iseloomustus (omadused, tarnija ja partii);
|
|
—
|
lahusti/kandeaine valiku põhjendus;
|
|
—
|
uuritava kemikaali lahustuvus ja püsivus lahustis/kandeaines, kui teada.
|
|
|
|
Rakud:
|
—
|
rakkude tüüp ja päritolu:
|
—
|
kas ERi ekspresseeritakse endogeenselt? Kui ei, siis milline/millised retseptor(id) transfekteeriti?
|
|
—
|
kasutatud reporterikonstrukt(id) (sealhulgas päritoluliigid);
|
|
—
|
stabiilse transfektsiooni säilitamiseks kasutatud selekteerimismeetod (kui asjakohane);
|
|
—
|
kas transfektsioonimeetod on stabiilsete liinide puhul asjakohane?
|
|
|
—
|
rakupassaažide arv (sulatamisest alates);
|
|
—
|
rakupassaažide arv sulatamisel;
|
|
—
|
rakukultuuride säilitamise meetodid.
|
|
|
|
Katsetingimused:
|
—
|
lahustuvusest tingitud piirangud;
|
|
—
|
eluvõimelisuse hindamiseks kasutatud meetodite kirjeldus;
|
|
—
|
söötme koostis, CO2 kontsentratsioon;
|
|
—
|
uuritava kemikaali kontsentratsioonid;
|
|
—
|
kandeaine ja lisatud uuritava kemikaali kogus;
|
|
—
|
inkubeerimise temperatuur ja niiskus;
|
|
—
|
rakutiheduse töötluse alguses ja selle ajal;
|
|
—
|
positiivsed ja negatiivsed etalonained;
|
|
—
|
reporterreagendid (toote nimetus, tarnija ja partii);
|
|
—
|
kriteeriumid, mille alusel katsetulemused liigitatakse positiivseteks, negatiivseteks või ebaselgeteks.
|
|
|
|
Nõuetekohasuse kontroll:
|
—
|
kordne induktsioon iga analüüsitava plaadi puhul ja kas need vastavad katses nõutavatele miinimumnõuetele, mille aluseks on aja jooksul kontrollproovidega saadud tulemused;
|
|
—
|
nõuetekohasuse kriteeriumide, nt log10EC50, log10PC50, logIC50 tegelikud väärtused ja Hilli kõvera tõusu väärtused paralleelselt uuritud positiivsete kontrollainete / etalonainete korral.
|
|
|
|
Tulemused
|
—
|
toor- ja normaliseeritud andmed;
|
|
—
|
maksimaalne kordse induktsiooni tase;
|
|
—
|
tsütotoksilisuse andmed;
|
|
—
|
vähim toimet avaldav kontsentratsioon, kui see on olemas;
|
|
—
|
RPCMax, PCmax, PC50, IC50 ja/või EC50 väärtused, vajaduse korral;
|
|
—
|
võimaluse korral andmed vastuse sõltuvuse kohta kontsentratsioonist;
|
|
—
|
statistiline analüüs, kui see on olemas, koos mõõtevea ja usalduspiiridega (nt aritmeetilise keskmise standardviga, standardhälve, variatsioonikordaja või usaldusvahemik usaldusnivool 95 %) ning kirjeldus selle kohta, kuidas need väärtused saadi.
|
|
|
KIRJANDUS
|
(1)
|
OECD (2005), „Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment“, Environment, Health and Safety Publications, Series on Testing and Assessment (nr 34), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(2)
|
OECD (2015), „Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity“, Environment, Health and Safety Publications, Series on Testing and Assessment (nr 225), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(3)
|
ICCVAM (2011), „ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists“, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol, P. et al. (1998), „Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis“, Cancer. Res., nr 58(23), lk 5367–73.
|
|
(5)
|
Rogers, J. M. ja Denison, M. S. (2000), „Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals“, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, nr 13(1), lk 67–82.
|
|
(6)
|
OECD (2012), „Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455)“, Environment, Health and Safety Publications, Series on Testing and Assessment (nr 173), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(7)
|
OECD (2015), „Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists“ Environment, Health and Safety Publications, Series on Testing and Assessment (nr 174), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(8)
|
OECD (2012), „Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption“, Environment, Health and Safety Publications, Series on Testing and Assessment (nr 150), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(9)
|
Cavailles, V. (2002), „Estrogens and Receptors: an Evolving Concept“, Climacteric, nr 5, 2 lisa, lk 20–6.
|
|
(10)
|
Welboren, W. J. et al. (2009), „Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated?“, Endocr. Relat. Cancer, nr 16(4), lk 1073–89.
|
|
(11)
|
Younes, M. ja Honma, N. (2011), „Estrogen Receptor Beta“, Arch. Pathol. Lab. Med., nr 135(1), lk 63–6.
|
|
(12)
|
Jefferson, W.N. et al. (2002), „Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses“, Journal of Chromatography B, nr 777(1–2), lk 179–189.
|
|
(13)
|
Sonneveld, E. et al. (2006), „Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities“, Toxicol. Sci., nr 89(1), lk 173–187.
|
|
(14)
|
Takeyoshi, M. et al. (2002), „The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions“, Toxicology Letters, nr 126(2), lk 91–98.
|
|
(15)
|
Combes, R. D. (2000), „Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans“, ATLA Alternatives to Laboratory Animals, nr 28(1), lk 81–118.
|
|
(16)
|
Escande, A. et al. (2006), „Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta“, Biochem. Pharmacol., nr 71(10), lk 1459–69.
|
|
(17)
|
Gray, L.E. Jr. (1998), „Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens“, Toxicol. Lett., nr 102–103, lk 677–680.
|
|
(18)
|
EDSTAC (1998), „Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report“.
|
|
(19)
|
ICCVAM (2003), „ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays“.
|
|
(20)
|
Gustafsson, J. Ö. (1999), „Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action“, Journal of Endocrinology, nr 163(3), lk 379–383.
|
|
(21)
|
Ogawa, S. et al. (1998), „The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERαIn Vivo and In Vitro“, Biochemical and Biophysical Research Communications, nr 243 (1), lk 122–126.
|
|
(22)
|
Enmark, E. et al. (1997), „Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern“, Journal of Clinical Endocrinology and Metabolism, nr 82(12), lk 4258–4265.
|
|
(23)
|
Ball, L. J. et al. (2009), „Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements“, Molecular and Cellular Endocrinology, nr 299(2), lk 204–211.
|
|
(24)
|
Barkhem, T. et al. (1998), „Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists“, Mol. Pharmacol., nr 54(1), lk 105–12.
|
|
(25)
|
Deroo, B. J. ja Buensuceso, A. V. (2010), „Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies“, Molecular Endocrinology, nr 24(9), lk 1703–1714.
|
|
(26)
|
Harris, D. M. et al. (2005), „Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells“, Experimental Biology and Medicine, nr 230(8), lk 558–568.
|
|
(27)
|
Anderson, J. N., Clark, J. H. ja Peck, E. J. Jr. (1972), „The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses“, Biochemical and Biophysical Research Communications, nr 48(6), lk 1460–1468.
|
|
(28)
|
Toft, D. (1972), „The Interaction of Uterine Estrogen Receptors with DNA“, Journal of Steroid Biochemistry, nr 3(3): lk 515–522.
|
|
(29)
|
Gorski, J. et al. (1968), „Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus“, Recent Progress in Hormone Research, nr 24, lk 45–80.
|
|
(30)
|
Jensen, E. V. et al. (1967), „Estrogen-Receptor Interactions in Target Tissues“, Archives d’Anatomie Microscopique et de Morphologie Experimentale, nr 56(3), lk 547–569.
|
|
(31)
|
ICCVAM (2002), „Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay“, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno, J. et al. (2001), „The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1“, Environ. Health Persp., nr 109, lk 785–94.
|
|
(33)
|
Kanno, J. et al. (2003), „The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies“, Environ. Health Persp., nr 111, lk 1530–1549.
|
|
(34)
|
Kanno, J. et al. (2003), „The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies“, Environ. Health Persp., nr 111, lk 1550–1558.
|
|
(35)
|
Geisinger et al. (1989), „Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors“, Cancer, nr 63, lk 280–288.
|
|
(36)
|
Baldwin et al. (1998), „BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro“, In Vitro Cell. Dev. Biol. – Animal, nr 34, lk 649–654.
|
|
(37)
|
Li, Y. et al. (2014), „Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant“, Mol. Endo., nr 28, lk 2072–2081.
|
|
(38)
|
Rogers, J. M. ja Denison, M. S. (2000), „Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals“, In Vitro & Molec. Toxicol. nr 13, lk 67–82.
|
1. liide
MÕISTED JA LÜHENDID
Katse nõuetekohasuse kriteeriumid
: katse kontrollproovide ja etalonainete toimivuse miinimumnõuded. Selleks, et katse loetaks kehtivaks, peavad kõik nõuetekohasuse kriteeriumid olema täidetud.
Täpsus (vastavus)
: katsemeetodiga saadud tulemuste ja nõuetekohaste võrdlusväärtuste kokkulangevuse määr. Täpsus näitab katsemeetodi tulemuslikkust ja on üks olulistest aspektidest. Seda terminit kasutatakse ka vastavuse tähenduses ja see näitab, kui sageli annab katsemeetod õige tulemuse (1).
Agonist
: aine, mis konkreetse retseptoriga seondudes põhjustab vastuse, nt transkriptsiooni.
Antagonist
: retseptori ligandi või kemikaali liik, mis retseptoriga seondudes ei kutsu esile bioloogilist vastust, vaid blokeerib või summutab agonisti vahendatud vastuseid.
Antiöstrogeenne toime
: kemikaali võime pärssida 17β-östradiooli toimet, mida vahendatakse östrogeeniretseptorite kaudu.
Raku morfoloogia
: koekultuuri plaadi süvendis monokihis kasvanud rakkude kuju ja välimus. Surevate rakkude morfoloogia on sageli ebatavaline.
CF
: OECD kontseptuaalne raamistik endokriinfunktsiooni kahjustavate kemikaalide uurimiseks ja hindamiseks.
Töötlemine aktiivsöe/dekstraaniga
: rakukultuuris kasutatud seerumi töötlemine. Aktiivsöe/dekstraaniga töötlemise teel eemaldatakse endogeensed hormoonid ja hormoone siduvad valgud.
Kemikaal
: aine või segu.
Tsütotoksilisus
: kahjulik mõju raku struktuurile või funktsioonile, mis lõpuks võib põhjustada raku surma ning mida kajastab pärast kokkupuuteperioodi lõppemist süvendis leiduvate rakkude arvu vähenemine või rakufunktsiooni mõõtnäitajate vähenemine paralleelselt uuritud kandeaine kontrollprooviga võrreldes.
CV
: variatsioonikordaja.
DCC-FBS
: dekstraaniga kaetud aktiivsöega töödeldud veiselooteseerum.
DMEM
: Dulbecco modifitseeritud Eagle'i sööde.
DMSO
: dimetüülsulfoksiid.
E2
: 17β-östradiool.
EC50
: 50 % uuritava kemikaali suurima toime avaldumise kontsentratsioonist.
ED
: endokriinsed häired.
hERα
: inimese östrogeeni alfa-retseptor.
hERß
: inimese östrogeeni beeta-retseptor.
EFM
: östrogeenivaba sööde. Dulbecco modifitseeritud Eagle'i minimaalsööde (DMEM), millele on lisatud 4,5 % aktiivsöe/dekstraaniga töödeldud veiselooteseerumit, 1,9 % L-glutamiini ja 0,9 % Pen-Strepi.
ER
: östrogeeniretseptor.
ERE
: östrogeeni vastuselement.
Östrogeenne toime
: kemikaali võime matkida 17β-östradiooli, sidudes östrogeeniretseptoreid ja neid aktiveerides. hERα vahendatud östrogeenset toimet saab tuvastada käesoleva katsemeetodiga.
ERTA
: östrogeeniretseptori transaktivatsioon.
FBS
: veiselooteseerum.
HeLa
: inimese emakakaelarakkude surematu liin.
HeLa9903
: HeLa raku subkloon, millesse on stabiilselt transfekteeritud hERαα ja lutsiferaasi reportergeen.
IC
50
: 50 % uuritava pärssiva kemikaali suurima toime avaldumise kontsentratsioonist.
ICCVAM
: The Interagency Coordinating Committee on the Validation of Alternative Methods.
Laboritevaheline korratavus
: suurus, mis näitab, millisel määral suudavad erinevad kvalifitseeritud laborid, kasutades sama katse-eeskirja ja uurides samu aineid, saada kvalitatiivselt ja kvantitatiivselt sarnaseid tulemusi. Laboritevaheline reprodutseeritavus määratakse eelvalideerimise ja valideerimise käigus ning see näitab, millisel määral võib uurimismeetodit edukalt üle viia ühest laborist teise (1).
Laborisisene korratavus
: suurus, mis näitab, millisel määral suudavad sama labori kvalifitseeritud töötajad konkreetset katse-eeskirja kasutades saada eri aegadel samasuguseid tulemusi (1).
LEC
: vähim toimet avaldav kontsentratsioon on uuritava kemikaali vähim kontsentratsioon, mis kutsub esile vastuse (st uuritava kemikaali vähim kontsentratsioon, mille juures kordne induktsioon on paralleelselt uuritud kandeaine kontrollproovi omaga võrreldes statistiliselt erinev).
Samalaadne katsemeetod
: katsemeetod, mis on struktuurilt ja funktsioonilt sarnane valideeritud ja tunnustatud võrdlusmeetodiga. Nimetatakse ka samaväärseks katsemeetodiks.
MT
: metallotioneiin.
MMTV
: hiire piimanäärmekasvaja viirus.
OHT
: 4-hüdroksütamoksüfeen.
PBTG
: toimivusel põhinev katsejuhend.
PC (positiivne kontrollproov)
: väga aktiivne aine, eelistatavalt 17ß-östradiool, mida uuritakse kõigis katsetes, et tagada katsemeetodi nõuetekohane toimivus.
PC
10
: uuritava kemikaali kontsentratsioon, mille juures agonistikatses mõõdetud toime on 10 % maksimaalsest toimest, mida positiivne kontrollproov (STTA katsemeetodi korral E2 kontsentratsiooniga 1 nM) igal plaadil esile kutsub.
PC
50
: uuritava kemikaali kontsentratsioon, mille juures agonistikatses mõõdetud toime on 50 % maksimaalsest toimest, mida positiivne kontrollproov (E2 katsemeetodis täpsustatud võrdluskontsentratsioonis) igal plaadil esile kutsub.
PC
max
: uuritava kemikaali kontsentratsioon, mis kutsub esile maksimaalse vastuse.
Toimivusnõuded
: valideeritud katsemeetodil põhinevad nõuded, mille alusel hinnatakse, kuivõrd saab esitatud katsemeetodit võrrelda teostuslikult ja tööpõhimõttelt samalaadse meetodiga. Sealhulgas on 1) katsemeetodi olulised osad, 2) valideeritud katsemeetodi vastuvõetava tulemuslikkuse tõendamiseks kasutatud kemikaalide hulgast valitud võrdluskemikaalide miinimumloetelu ja 3) võrreldavat usaldusväärsuse ja täpsuse taset, mille puhul on lähtutud valideeritud katsemeetodiga saadud tulemustest ja mis tuleks saavutada kavandatud katsemeetodi hindamisel võrdluskemikaalide miinimumloetelu kasutamise teel (1).
Pädevuse tõendamise ained
: toimivusnõuetes sisalduvas alamloetelus esitatud võrdlusained, mida laborid võivad kasutada, et tõendada oma tehnilist pädevust standardse katsemeetodi kasutamiseks. Nende ainete tüüpilised valikukriteeriumid on, et need esindaks laia vastusevahemikku, oleksid müügil ja et nende kohta oleksid kättesaadavad kvaliteetsed võrdlusandmed.
Pädevus
: enne tundmatute ainete uurimist tõendatud võime katsemeetodit korrektselt kasutada.
Võrdlusöstrogeen (positiivne kontrollaine)
: 17β-östradiool (E2, CAS 50-28-2).
Etalonaine
: võrdlusaine, mida kasutatakse katsemeetodi sobivuse tõendamiseks. STTA ja VM7Luc ER TA katsemeetodite puhul on etalonaineks 17β-östradiool.
Valideeritud katsemeetodid
: katsemeetodid, millel põhineb toimivusel põhinev katsejuhend nr 455.
Asjakohasus
: mõiste, millega väljendatakse seda, kas katsemeetod võimaldab uurida huvipakkuvat mõju, kas meetod on mõttekas ja sobib konkreetse eesmärgi jaoks. See näitab, mil määral saab katsemeetodiga õigesti mõõta või prognoosida huvipakkuvat bioloogilist mõju. Asjakohasus hõlmab ka katsemeetodi täpsuse (vastavuse) aspekti (1).
Usaldusväärsus
: iseloomustab katsemeetodi korratavust, kui meetodit rakendatakse ühe ja sama katse-eeskirja alusel ühes laboris ja eri laborites pikema aja jooksul. Usaldusväärsuse hindamisel arvutatakse laborisisene ja laboritevaheline korratavus.
RLU
: suhteline valgusühik.
RNA
: ribonukleiinhape.
RPCmax
: uuritava kemikaali indutseeritud maksimaalne vastus, väljendatuna protsendina 1 nM E2 indutseeritud vastusest samal plaadil.
RPMI
: sööde RPMI-1 640, millele on lisatud 0,9 % Pen-Strepi ja 8,0 % veiselooteseerumit (FBS).
Katse
: üksikeksperiment, millega hinnatakse kemikaali mõju bioloogilise analüüsi tulemustele. Iga katse on tervikeksperiment, mis viiakse läbi paralleelsüvendites rakkudega, mis on külvatud ühisest rakukogumist ühel ja samal ajal.
Sõltumatu katse
: eraldiseisev sõltumatu eksperiment, millega hinnatakse kemikaali mõju bioloogilise analüüsi tulemustele, kasutades teisest rakukogumist pärit rakke ja värskelt valmistatud kemikaalilahuseid; katse võib läbi viia erineval päeval võiv võib seda samal päeval teha teine töötaja.
SD
: standardhälve.
Tundlikkus
: kõigi niisuguste positiivsete/aktiivsete ainete osakaal, mis katse tulemusel klassifitseeritakse õigesti. Tundlikkusega iseloomustatakse katsemeetodi täpsust, kui saadud tulemusi saab kategoriseerida, ning see on oluline aspekt katsemeetodi asjakohasuse hindamisel (1).
Spetsiifilisus
: kõigi niisuguste negatiivsete/mitteaktiivsete ainete osakaal, mis katse tulemusel klassifitseeritakse õigesti. Spetsiifilisusega iseloomustatakse katsemeetodi täpsust, kui saadud tulemusi saab kategoriseerida, ning see on oluline aspekt katsemeetodi asjakohasuse hindamisel (1).
Stabiilne transfektsioon
: kui DNA transfekteeritakse kultiveeritud rakkudesse nii, et see integreerub stabiilselt rakkude genoomi, mille tulemusena toimub transfekteeritud geenide stabiilne ekspressioon. Stabiilselt transfekteeritud rakkude kloonid valitakse välja stabiilsete markerite järgi (nt G418-resistentsus).
STTA katsemeetod
: stabiilse transfektsiooniga transaktivatsiooni katse, ERα transkriptsiooni aktivatsiooni katse, milles kasutatakse rakuliini HeLa 9 903.
Uuring
: kogu katsetega seotud töö, mis on tehtud, et hinnata üht konkreetset ainet, kasutades konkreetset katsemeetodit. Uuring koosneb etappidest, mis hõlmavad kasutatavas uuritava aine söötmes lahustuvuse uurimist, eelkatseid vahemiku leidmiseks, kõiki vajalikke tervikkatseid kokku, andmeanalüüsi, kvaliteedi tagamist, tsütotoksilisuse hindamist jms. Uuringu lõpuleviimine võimaldab klassifitseerida uuritava kemikaali toimet (st, kas on aktiivne, mitteaktiivne või ei anna ühest tulemust) uuritava toksilise omaduse seisukohast, mida kasutava katsemeetodi abil hinnatakse, ja hinnata mõju tugevust positiivse võrdluskemikaaliga võrreldes.
Aine
: REACH-määruses (60) on aine määratletud kui looduslik või tootmisprotsessi teel saadud keemiline element või selle ühendid koos stabiilsuse säilitamiseks vajalike ja tootmisprotsessist johtuvate lisanditega, välja arvatud lahustid, mida on võimalik ainest eraldada, mõjutamata aine stabiilsust või muutmata selle koostist. ÜRO GHSis (1) on kasutatud väga sarnast määratlust.
TA (transaktivatsioon): mRNA sünteesi alustamine vastusena keemilisele signaalile, näiteks östrogeeni seondumisele östrogeeniretseptoriga.
Katsemeetod
: siinses kontekstis on katsemeetod üks metoodikatest, mis on tunnistatud valiidseks, sest see vastab väljatöötatud toimivuskriteeriumidele. Katsemeetodi osad hõlmavad näiteks konkreetset rakuliini koos sellega seotud kasvutingimustega, konkreetset söödet, milles katse läbi viiakse, plaadi ettevalmistamise tingimusi, uuritavate kemikaalide valimist ja lahjendamist, samuti mis tahes muid nõutavaid kvaliteedikontrolli meetmeid ja meetodiga seotud andmehindamisetappe.
Uuritav kemikaal
: iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
Transkriptsioon
: mRNA süntees.
UVCB
: tundmatu või muutuva koostisega kemikaal, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal.
Valideeritud katsemeetod
: katsemeetod, mille valideerimisuuringud on lõpule viidud, st on määratud selle asjakohasus (sealhulgas täpsus) ja usaldusväärsus konkreetse eesmärgi puhul. Oluline on märkida, et valideeritud katsemeetod ei tarvitse olla piisavalt kasutuskõlblik täpsuse ja usaldusväärsuse osas, et seda saaks lugeda vastuvõetavaks konkreetse eesmärgi puhul (1).
Valideerimine
: protsess, millega tehakse kindlaks konkreetse lähenemisviisi, meetodi, analüüsi, protsessi või hindamisviisi usaldusväärsus ja asjakohasus seoses määratud eesmärgiga (1).
VC (kandeaine kontrollproov)
: uuritavate ja kontrollkemikaalide lahustamiseks kasutatavat lahustit uuritakse eraldi kui kandeainet, mis ei sisalda lahustatud kemikaali.
VM7
: immortaliseeritud adenokartsinoomirakk, mis ekspresseerib endogeenselt östrogeeniretseptorit.
VM7Luc4E2
: rakuliin VM7Luc4E2 aretati inimese immortaliseeritud VM7 adenokartsinoomirakkudest, mis ekspresseerivad endogeenselt mõlemat liiki östrogeeniretseptorit (ERα ja ERβ) ning millesse on stabiilselt transfekteeritud plasmiid pGudLuc7.ERE. Kõnealune plasmiid sisaldab nelja sünteetilise oligonukleotiidi koopiat, milles östrogeeni vastuselement jääb ülespoole hiire piimanäärmekasvaja viiruse promootorist ja jaanimardika lutsiferaasi geenist.
Nõrgalt positiivne kontrollaine
: võrdluskemikaalide loetelust valitud nõrgalt aktiivne aine, mida uuritakse kõigis katsetes, et tagada katsemeetodi nõuetekohane toimivus.
2. liide
STABIILSE TRANSFEKTSIOONIGA INIMESE ÖSTROGEENI Α-RETSEPTORI TRANSAKTIVATSIOONI (TA) KATSEMEETOD, MILLES UURITAKSE KEMIKAALIDE TOIMET ÖSTROGEENI AGONISTI VÕI ANTAGONISTINA, KASUTADES RAKULIINI HERΑ-HELA-9903
LÄHTEKAALUTLUSED JA PIIRANGUD (VT KA ÜLDIST SISSEJUHATUST)
|
1.
|
Transaktivatsiooni (TA) katsemeetodis kasutatakse rakuliini hERα-HeLa-9 903, et tuvastada toime östrogeeni agonistina, mida vahendab inimese östrogeeni α-retseptor (hERα). Stabiilse transfektsiooniga transaktivatsiooni (STTA) katsemeetodi valideerimisuuring, mille tegi Jaapani kemikaalide hindamise ja uurimise instituut (CERI), kasutades inimese östrogeeni α-retseptori vahendatava östrogeeni agonistliku ja antagonistliku toime tuvastamiseks rakuliini hERα-HeLa-9 903, tõendas katsemeetodi asjakohasust ja usaldusväärsust ettenähtud eesmärgil kasutades (1).
|
|
2.
|
See katsemeetod on välja töötatud spetsiaalselt hERα-vahendatud transaktivatsiooni tuvastamiseks, kasutades mõõdetava parameetrina kemoluminestsentsi. Kuid üle 1 μM fütoöstrogeeni kontsentratsioonide juures on teadaolevalt saadud luminestsentssignaali, mida ei vahenda retseptorid, vaid mida põhjustab lutsiferaasi reportergeeni üleaktiveerimine (2, 3). Kuigi annuse-vastuse kõver näitab, et ERi süsteemi aktiveerumine toimub väiksematel kontsentratsioonidel, tuleb stabiilse transfektsiooniga ER TA katsesüsteemides hoolitalt uurida suurtel kontsentratsioonidel ilmnevat lutsiferaasi ekspressiooni, mida põhjustavad fütoöstrogeenid või samalaadsed ühendid, mis sarnaselt fütoöstrogeenile tekitavad lutsiferaasi reportergeeni üleaktivatsiooni (1. liide).
|
|
3.
|
Enne käesoleva katsemeetodi regulatiivsel eesmärgil kasutamist tuleks lugeda peatükke „Üldine sissejuhatus“ ja „ER TA katsemeetodi osad“. Käesolevas katsejuhendis kasutatud mõisted ja lühendid on esitatud liites 2.1.
|
KATSEMEETODI PÕHIMÕTE (VT KA ÜLDIST SISSEJUHATUST)
|
4.
|
Katsemeetodit kasutatakse selleks, et tuvastada östrogeeniretseptori seondumist ligandiga. Ligandi seondumise järel translokeerub retseptori-ligandi kompleks tuuma, kus seondub DNA spetsiifiliste vastuselementidega ja transaktiveerib jaanimardika lutsiferaasi reportergeeni, mille tulemusena suureneb ensüüm lutsiferaasi ekspressioon rakus. Lutsiferiin on substraat, mille ensüüm lutsiferaas transformeerib bioluminestsentsiga, mida saab luminomeetriga kvantitatiivselt mõõta. Lutsiferaasi aktiivsust saab kiiresti ja odavalt hinnata paljude müügilolevate katsekomplektidega.
|
|
5.
|
Katsesüsteemi kasutatakse rakuliini hERα-HeLa-9 903, mis on saadud inimese emakakaelakasvajast ja millele on stabiilselt lisatud kaks konstrukti: i) hERαα ekspressiooni konstrukt (kodeerib inimese täispikkusega retseptorit) ja ii) jaanimardika lutsiferaasi reporterkonstrukt, milles on viis vitellogeniini östrogeeni vastuselemendi (ERE) tandemkordust, mida juhib hiire metallotioneiini (MT) promootori TATA-element. Hiire MT TATA geenikonstrukt on osutunud kõige paremini toimivaks ja seetõttu seda enamasti kasutatakse. Sellest tulenevalt saab selle hERα-HeLa-9 903 rakuliiniga mõõta uuritava kemikaali võimet indutseerida lutsiferaasi geeni ekspressiooni hERα-vahendatud transaktivatsiooni.
|
|
6.
|
ERi agonistikatse korral oleneb andmete tõlgendamine sellest, kas uuritava kemikaali indutseeritud maksimaalse vastuse tase on suurem või võrdne 10 %-ga positiivse kontrollaine (PC) 17β-östradiooli (E2) maksimaalselt indutseeriva kontsentratsiooniga (1 nM) indutseeritud vastuse tasemest (st PC10-ga). ERi antagonistikatse korral oleneb andmete tõlgendamine sellest, kas vastuse toime on vähemalt 30 % väiksem kui vastus, mille indutseeris mittetsütotoksiline kontrollproov, kuhu oli lisatud võrdlusainet (25 pM E2). Andmete analüüsimist ja tõlgendamist on üksikasjalikult kirjeldatud punktides 34–48.
|
METOODIKA
Rakuliinid
|
7.
|
Katsemeetodiga tuleks kasutada stabiilse transfektsiooniga hERα-HeLa-9 903 rakuliini. Seda rakuliini saab osta Jaapani teadusbioressursside kogu (Japanese Collection of Research Bioresources, JCRB) rakupangast (61) materjali üleandmise lepingu (Material Transfer Agreement, MTA) allakirjutamisel.
|
|
8.
|
Katsete tegemisel tohib kasutada üksnes mükoplasmavabu rakke. Mükoplasmainfektsiooni tuvastamiseks on sobiv tundlik meetod RT-PCR-meetod (polümeraasiahela reaktsioon reaalajas) (4, 5, 6).
|
Rakuliini stabiilsus
|
9.
|
Rakuliini stabiilsuse jälgimiseks tuleks agonistikatse korral kasutada etalonainetena E2, 17α-östradiooli, 17α-metüültestosterooni ja kortikosterooni ning kontrollida kogu kontsentratsiooni-vastuse kõverat tabelis 1 esitatud uuritavate kontsentratsioonide vahemikus vähemalt üks kord iga kord, kui katsemeetodit kasutatakse; tulemused peavad langema kokku tabelis 1 esitatud tulemustega.
|
|
10.
|
Antagonistikatse korral tuleks iga katsega paralleelselt kontrollida kahe etalonaine, tamoksüfeeni ja flutamiidi täielikke kontsentratsioonikõveraid. Jälgida tuleb nende kahe kemikaali korrektset positiivseks või negatiivseks klassifitseerimist.
|
Rakkude kasvatamine ja plaadile kandmise tingimused
|
11.
|
Rakke tuleb hoida ilma fenoolpunaseta Eagle'i essentsiaalses minimaalsöötmes (EMEM), millele on lisatud 60 mg/l of antibiootikumi kanamütsiini ja 10 % dekstraankattega aktiivsöega töödeldud veiselooteseerumit (DCC-FBS), in a CO2-inkubaatoris (5 % CO2) temperatuuril 37±1 °C. 75–90 % konfluentsuse saavutamisel võib rakud ümber külvata, lisades 100 mm Petri tassi 10 ml rakukultuuri tihedusega 0,4 × 105 – 1 × 105 rakku/ml. Rakud suspendeeritakse 10 % FBS-EMEMiga (sama mis DCC-FBSiga EMEM) ning külvatakse mikroplaadi süvenditesse tihedusega 1 × 104 rakku/(100 μl × süvend). Järgmiseks inkubeeritakse rakke 5 % CO2 inkubaatoris temperatuuril 37±1 °C kolme tunni jooksul enne kemikaaliga kokku puutumist. Plasttarvikud ei tohi ilmutada östrogeenset toimet.
|
|
12.
|
Vastuse usaldusväärsuse säilitamiseks tuleks rakke kasvatada külmutatud varudest konditsioneeritud söötmes rohkem kui ühe passaaži võrra, kuid mitte üle 40 passaaži. hERα-HeLa-9 903 rakuliini puhul kestab see alla kolme kuu. Rakkude toimivus võib väheneda, kui neid kasvatatakse ebasobivates kasvutingimustes.
|
|
13.
|
DCC-FBSi võib valmistada liites 2.2 esitatud kirjelduse järgi või hankida kaubandusest.
|
Katse nõuetekohasuse kriteeriumid
ERi agonistikatses kasutatavad positiivsed ja negatiivsed etalonained
|
14.
|
Enne uuringut ja selle ajal tuleb kontrollida katsesüsteemi reageerimisvõimet, kasutades asjakohastes kontsentratsioonides tugevat östrogeeni (E2), nõrka östrogeeni (17α-östradiooli), väga nõrka agonisti (17α-metüültestosterooni) ja negatiivset ainet (kortikosterooni). Valideerimisuuringus (1) leitud vastuvõetava vahemiku väärtused on esitatud tabelis 1. Nende nelja etalonaine paralleelproovid tuleb lisada igasse katsesse ning saadud tulemused peavad jääma antud vastuvõetavatesse piiridesse. Kui see nii ei ole, siis tuleb kindlaks teha nõuetekohasuse kriteeriumidele mittevastavuse põhjus (kontrollida nt rakkude käitlemist, seerumi ja antibiootikumi kvaliteeti ja kontsentratsiooni) ning analüüsi korrata. Kui nõuetekohasuse kriteeriumid on täidetud, on äärmiselt oluline rakkude kasvatamise materjalide ühtlane kasutamine, et tagada EC50, PC50 ja PC10 väärtuste minimaalne varieeruvus. Nelja etalonaine paralleelproovidega,mis lisatakse igasse katsesse (uuritakse samades tingimustes, sealhulgas samade materjalide, rakupassaažide arvu ja tehnikutega), saab kontrollida analüüsi tundlikkust, sest kolme positiivse etalonaine PC10 väärtused peaksid jääma vastuvõetavatesse piiridesse, samuti ka PC50 ja EC50 väärtused, kui neid on võimalik arvutada (vt tabelit 1).
|
Tabel 1
ERi agonistikatses kasutatava nelja etalonaine vastuvõetava vahemiku väärtused
|
Nimetus
|
logPC50
|
logPC10
|
logPC50
|
Hilli kõvera tõus
|
Uuritav vahemik
|
|
17β-östradiool (E2) CASi nr: 50-28-2
|
–11,4 ~ –10,1
|
< –11
|
–11,3 ~ –10,1
|
0,7 ~ 1,5
|
10–14 ~ 10–8 M
|
|
17α-östradiool CASi nr: 57-91-0
|
–9,6 ~ –8,1
|
–10,7 ~ –9,3
|
–9,6 ~ –8,4
|
0,9 ~ 2,0
|
10–12 ~ 10–6 M
|
|
Kortikosteroon CASi nr: 50-22-6
|
—
|
—
|
—
|
—
|
10–10 ~ 10–4 M
|
|
17α-metüültestosteroon CASi nr: 58-18-4
|
–6,0 ~ –5,1
|
–8,0 ± –6,2
|
—
|
—
|
10–11 ~ 10–5 M
|
ERi antagonistikatses kasutatavad positiivsed ja negatiivsed etalonained
|
15.
|
Enne uuringut ja selle ajal tuleb kontrollida katsesüsteemi reageerimisvõimet, kasutades asjakohastes kontsentratsioonides positiivset ainet (tamoksüfeeni) ja negatiivset ainet (flutamiidi). Valideerimisuuringus (1) leitud vastuvõetava vahemiku väärtused on esitatud tabelis 2. Nende kahe etalonaine paralleelproovid tuleb lisada igasse katsesse ning saadud tulemused hinnata õigeks vastavalt kriteeriumidele. Kui seda teha ei saa, siis tuleb kindlaks teha kriteeriumidele mittevastavuse põhjus (kontrollida nt rakkude käitlemist, seerumi ja antibiootikumi kvaliteeti ja kontsentratsiooni) ning analüüsi korrata. Peale selle tuleb arvutada positiivse aine (tamoksüfeeni) IC50 väärtused ning tulemused peavad jääma esitatud vastuvõetavatesse piiridesse. Kui nõuetekohasuse kriteeriumid on täidetud, on äärmiselt oluline rakkude kasvatamise materjalide ühtlane kasutamine, et tagada IC50 väärtuste minimaalne varieeruvus. Kahe etalonaine paralleelproovidega, mis lisatakse igasse katsesse (uuritakse samades tingimustes, sealhulgas samade materjalide, rakupassaažide arvu ja tehnikutega), saab kontrollida analüüsi tundlikkust (vt tabelit 2).
|
Tabel 2
ERi antagonistikatses kasutatava kahe etalonaine kriteeriumid ja vastuvõetava vahemiku väärtused
|
Nimetus
|
Kriteeriumid
|
LogIC50
|
Uuritav vahemik
|
|
Tamoksüfeen CASi nr: 10 540 -29-1
|
Positiivne: arvutada tuleb IC50
|
–5,942 ~ –7,596
|
10–10 ~ 10–5 M
|
|
Flutamiid CASi nr: 13 311 -84-7
|
Negatiivne: IC30 ei pea arvutama
|
—
|
10–10 ~ 10–5 M
|
Positiivne kontrollproov ja kandeaine kontrollproov
|
16.
|
ERi agonistikatses kasutatavast positiivsest kontrollainest (1 nM E2) ja ERi antagonistikatses kasutatavast positiivsest kontrollainest (10 μM TAM) tuleb igale plaadile lisada vähemalt kolm paralleelproovi. Uuritava kemikaali lahustamiseks kasutatavast kandeainest tuleb igale plaadile lisada vähemalt kolm paralleelproovi. Kui positiivse kontrollaine lahustamiseks kasutatakse muud kandeainet, kui seda, milles lahustatakse uuritav kemikaal, tuleb positiivsete kontrollproovidega samale plaadile lisada kolm paralleelproovi ka teisest kandeainest.
|
ERi agonistikatse kvaliteedi kriteeriumid
|
17.
|
Positiivse kontrollainega (1 nM E2) saavutatud lutsiferaasi keskmine aktiivsuse peab olema vähemalt neljakordne igal plaadil kandeaine kontrollprooviga saavutatud keskmisega võrreldes. See kriteerium on kindlaks määratud valideerimisuuringus leitud näitajate väärtustega (aja jooksul mõõdetud 4- kuni 30-kordsed).
|
|
18.
|
Katsemeetodi kvaliteedikontrolli seisukohast peaks positiivse kontrollproovi (1 nM E2) PC10 väärtusele vastav kordne induktsioon olema suurem kui 1+2 SD kandeaine kontrollproovi kordse induktsiooni väärtusest (= 1). Prioriseerimise eesmärgil võib PC10 väärtus olla kasulik andmete vajalik analüüsimise hõlbustamiseks statistilise analüüsiga võrreldes. Kuigi statistiline analüüs annab teavet olulisuse kohta, ei ole niisugune analüüs kvantitatiivne parameeter kontsentratsioonil põhineva võimaliku toime puhul ega ole seega prioriseerimise seisukohast nii kasulik.
|
ERi antagonistikatse kvaliteedi kriteeriumid
|
19.
|
Kontrollproovi lisatud ainega (25 pM E2) saavutatud lutsiferaasi keskmine aktiivsuse peab olema vähemalt neljakordne igal plaadil kandeaine kontrollprooviga saavutatud keskmisega võrreldes. See kriteerium on kindlaks määratud valideerimisuuringus leitud näitajate väärtustega.
|
|
20.
|
Katsemeetodi kvaliteedikontrolli seisukohast peaks 1 nM E2 suhteline transkriptsiooni aktivatsioon (RTA) olema üle 100 %, 1 μM 4-hüdroksütamoksüfeeni (OHT) RTA alla 40,6 % ja 100 μM digitoniini (Dig) RTA alla 0 %.
Labori pädevuse tõendamine (vt käesoleva katsemeetodi jaotise „ER TA katsemeetodi osad“ punkti 14 ning tabeleid 3 ja 4).
|
Kandeaine
|
21.
|
Kandeaine kontrollprooviks tuleks kasutada dimetüülsulfoksiidi (DMSO) või asjakohast lahustit samas kontsentratsiooni, nagu seda kasutatakse erinevates positiivsetes ja negatiivsetes kontrollproovides ja uuritavate kemikaalide proovides. Uuritavad kemikaalid tuleb lahustada lahustis, milles nad lahustuvad ja mis on võimeline segunema rakkude söötmega. Sobivad kandeained on vesi, etanool (puhtusega 95–100 %) ja DMSO. Kui kasutatakse DMSOd, ei tohi selle kontsentratsioon ületada 0,1 mahuprotsenti. Mis tahes kandeaine korral tuleb tõendada, et maksimaalne kasutatav kogus ei ole tsütotoksiline ega sega analüüsimeetodi toimivust.
|
Uuritavate kemikaalide ettevalmistamine
|
22.
|
Üldiselt tuleks kemikaalid lahustada DMSOs või muus sobivas lahustis ning seejärel lahjendada sama lahustiga tavapärases vahekorras 1: 10, et saada lahused söötmega lahjendamiseks.
|
Lahustuvus ja tsütotoksilisus: vahemiku leidmise kaalutlused
|
23.
|
Uuritava kemikaali sobiva kontsentratsioonivahemiku määramiseks ning kontrollimiseks, kas uuritava kemikaali puhul võib tekkida probleeme lahustuvuse või tsütotoksilisusega, tuleb teha eelkatse. Kõigepealt uuritakse kemikaale maksimaalse kontsentratsiooniga kuni 1 μl/ml, 1 mg/ml või 1 mM olenevalt sellest, milline on väikseim. Olenevalt eelkatse täheldatud tsütotoksilisuse ulatusest või lahustuvuse puudumisest uuritakse esimeses põhikatses kemikaali 10-kordsete lahjenduste seerias alates maksimaalsest vastuvõetavast kontsentratsioonist (nt 1 mM, 100 μM, 10 μM jne) ning dokumenteeritakse täheldatav hägusus, sade või tsütotoksilisus. Teises ja vajaduse korral ka kolmandas katses tuleks kontsentratsioone sobivalt kohandada, nii et oleks võimalik paremini iseloomustada kontsentratsiooni-vastuse kõverat ning vältida kontsentratsioone, mille juures lahustuvus puudus või mis indutseerisid liigset tsütotoksilisust.
|
|
24.
|
ERi agonistide ja antagonistide korral võib suurenenud tsütotoksilisus tüüpilist sigmoidset vastust oluliselt moonutada või summutada ning seda tuleks andmete tõlgendamisel arvesse võtta. Kasutada tuleks tsütotoksilisuse uurimismeetodeid, millega on võimalik saada teavet 80 % rakkude eluvõimelisuse kohta, kasutades labori kogemuste põhinevat asjakohast katset.
|
|
25.
|
Kui tsütotoksilisuse katse tulemus näitab, et uuritava kemikaali kontsentratsioon on rakkude arvu vähendanud 20 % või rohkem, tuleb see kontsentratsioon lugeda tsütotoksiliseks ning tsütotoksilise kontsentratsiooni lähedased ja sellest kõrgemad kontsentratsioonid tuleb hindamisest välja jätta.
|
Kemikaaliga kokkupuude ja analüüsiplaadi jaotus
|
26.
|
Kemikaali lahjendamise (1. ja 2. etapp) ning rakkudega kokkupuute (3. etapp) saab läbi viia järgmiselt.
|
1. etapp: igast uuritavast kemikaalist tehakse DMSO või sobiva lahustiga lahjenduste seeria ning lisatakse need mikroplaadi süvenditesse, et saada vahemiku leidmise eelkatses määratud lõplik lahjenduste seeria (tavaliselt on seerias nt kontsentratsioonid 1 mM, 100 μM, 10 μM, 1 μM, 100 nM, 10 nM, 1 nM, 100 pM ja 10 pM (10–3 – 10–11 M)) kolme paralleelprooviga.
2. etapp: kemikaali lahjendamine: kõigepealt lahjendada 1,5 μl uuritavat kemikaali lahust nii, et saadakse 500 μl söödet.
3. etapp: rakkude kokkupuude kemikaaliga: lisada 50 μl söötmes lahust (valmistati 2. etapis) analüüsisüvendis, mis sisaldab 104 rakku/100 μl/süvend.
Igas süvendis on söötme soovitatav lõplik ruumala 150 μl. Uuritavad proovid ja etalonained võib paigutada nii, nagu näidatud tabelites 3 ja 4.
Tabel 3
Etalonainete eri kontsentratsioonidega proovide mikroplaadile paigutamise näide ERi agonistikatse puhul.
|
Rida
|
17α-metüültestosteroon
|
Kortikosteroon
|
17α-östradiool
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
Konts. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
Konts. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
Konts. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
Konts. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
Konts. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
Konts. 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
Konts. 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: kandeaine kontrollproov (0,1 % DMSO); PC: positiivne kontrollproov (1 nM E2)
|
|
27.
|
Etalonaineid (E2, 17α-östradiool, 17α-metüültestosteroon ja kortikosteroon) tuleb analüüsida iga katse käigus (tabel 3). Igal uuritaval plaadil peavad olema süvendid positiivse kontrollprooviga (1 nM E2), mis on võimeline esile kutsuma E2 maksimaalse vastuse, ja kandeaine kontrollprooviga (DMSO või sobiv lahusti). Kui sama katse käigus kasutatakse eri allikatest pärit rakke (nt erineva passaažide arvuga, eri partiidest pärit vms), tuleb etalonaineid analüüsida iga päritoluga rakkudega.
|
Tabel 4
Eri kontsentratsioonidega uuritava kemikaali ja kontrollproovide mikroplaadile paigutamise näide ERi agonistikatse puhul.
|
Rida
|
Uuritav kemikaal 1
|
Uuritav kemikaal 2
|
Uuritav kemikaal 3
|
Uuritav kemikaal 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
Konts. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
Konts. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
Konts. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
Konts. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
Konts. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
Konts. 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
Konts. 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: kandeaine kontrollproov (0,1 % DMSO); PC: positiivne kontrollproov (1 nM E2)
|
Tabel 5
Etalonainete eri kontsentratsioonidega proovide mikroplaadile paigutamise näide ERi antagonistikatse puhul

|
28.
|
Selleks, et hinnata kemikaalide antagonistlikku toimet, tuleb ridades A kuni G olevatesse süvenditesse lisada 25 pM E2. Etalonaineid (tamoksüfeen ja flutamiid) tuleb analüüsida iga katse käigus. Igal katses analüüsitaval plaadil peaks olema positiivsete kontrollproovidega süvendid (töödeldud 1 nM E2ga, millega saab kontrollida hERα-HeLa-9 903 rakuliini kvaliteeti), kandeaine kontrollproovidega süvendid (töödeldud DMSO või sobiva lahustiga), 0,1 % DMSOga süvendid, kuhu lisatakse veel E2 (rikastatud kontrollproov), lõplikus kontsentratsioonis 1 μM OHTga töödeldud süvendid ja 100 μM digitoniiniga töödeldud süvendid (tabel 5). Järgmisel analüüsitaval plaadil peaks olema sama paigutus ilma etalonaineteta (tabel 6). Kui sama katse käigus kasutatakse eri allikatest pärit rakke (nt erineva passaažide arvuga, eri partiidest pärit vms), tuleb etalonaineid analüüsida iga päritoluga rakkudega.
|
Tabel 6
Eri kontsentratsioonidega uuritava kemikaali ja kontrollproovide mikroplaadile paigutamise näide ERi antagonistikatse puhul

|
29.
|
Vajadust mööda tuleb kinnitada ääreefektide puudumist, kuid kui on ääreefektide kahtlus, tuleks plaadi paigutust muuta nii, et neid efekte saaks vältida. Näiteks võib proovide paigutamisel plaadile äärmised süvendid kasutamata jätta.
|
|
30.
|
Pärast kemikaalide lisamist inkubeeritakse analüüsitavaid plaate 5 % CO2 inkubaatoris temperatuuril 37±1 °C 20–24 tunni jooksul, et indutseerida reportergeeni produkte.
|
|
31.
|
Väga lenduvate ühendite korral tuleb rakendada eritingimusi. Sellisel juhul võivad lähedalasuvate süvendites kontrollproovid anda valepositiivseid tulemusi, mida tuleb eeldatavate ja aja jooksul kogutud kontrollväärtuse puhul arvesse võtta. Mõnel juhul, kui lenduvus võib olla probleemiks, võib kasutada kilekatteid, mis tõhusalt eraldavad üksikud süvendid katse läbiviimise ajaks ja mille kasutamine on seetõttu sellistel juhtudel soovitatav.
|
|
32.
|
Sõltumatuse tagamiseks tuleb sama kemikaaliga korduskatsed läbi viia eri päevadel.
|
Lutsiferaasi mõõtmine
|
33.
|
Kui nõuetekohasuse kriteeriumid on täidetud, võib mõõtmiseks kasutada müügilolevat lutsiferaasi analüüsireagenti [nt Steady-Glo® Luciferase Assay System (Promega, E2510, või samaväärne)] või standardset lutsiferaasi analüüsisüsteemi (nt Promega, E1500 või samaväärne). Analüüsireagendid tuleb valida kasutatava luminomeetri tundlikkusest lähtuvalt. Standardset lutsiferaasi analüüsisüsteemi kasutades tuleks enne substraadi lisamist kasutada rakukultuuri lüüsimise reagenti (nt Promega, E1531 või samaväärne). Lutsiferaasi analüüsireagenti tuleb kasutada vastavalt tootja juhistele.
|
ANDMEANALÜÜS
ERi agonistikatse
|
34.
|
ERi agonistikatse korral tuleb positiivse kontrollaine (1 nM E2) suhtelise transkriptsiooni aktiivsuse leidmiseks analüüsida sama plaadi luminestsentssignaale, järgides järgmisi etappe (ka muud matemaatilised võtted on lubatud):
|
1. etapp. Arvutada kandeaine kontrollproovi keskväärtus.
2. etapp. Andmete normaliseerimiseks lahutada kandeaine kontrollproovi keskväärtus iga süvendi puhul saadud väärtusest.
3. etapp. Arvutada normaliseeritud positiivse kontrollproovi keskväärtus.
4. etapp. Jagada plaadi iga süvendi normaliseeritud väärtus normaliseeritud positiivse kontrollproovi keskväärtusega (PC = 100 %).
Viimane iga süvendi kohta saadu väärtus on kõnealuse süvendi suhteline transkriptsiooni aktiivsus positiivse kontrollproovi vastusega võrreldes.
5. etapp. Arvutada uuritava kemikaali iga kontsentratsioonirühma suhteliste transkriptsiooni aktiivsuste keskväärtus. Vastusega on seotud kaks mõõdet: keskmine transkriptsiooni aktiivsus (vastus) ja kontsentratsioon, mille juures vastus ilmneb (vt järgmist jaotist).
Kaalutlused EC50, PC50 ja PC10 induktsiooni kohta
|
35.
|
EC50 arvutamiseks on nõutav täielik kontsentratsiooni-vastuse kõver, kuid alati ei ole seda võimalik koostada või ei ole see praktiline uuritavast kontsentratsioonivahemikust tulenevate piirangute tõttu (nt tsütotoksilisus või probleemid lahustuvusega). EC50 ja maksimaalne induktsioonitase (mis vastab Hilli võrrandi suurimale väärtusele) on informatiivsed parameetrid, mida võimaluse korral tuleks dokumenteerida. EC50 ja maksimaalse induktsioonitaseme arvutamiseks tuleks kasutada asjakohast statistikatarkvara (nt Graphpad Prism). Kui Hilli logistiline võrrand on kontsentratsiooni-vastuse andmetele rakendatav, tuleks EC50 arvutada järgmise valemi alusel (7):
Y = alumine + (ülemine – alumine) / (1+10 exp ((log EC50 –X) × Hilli tõus)), kus:
X on logaritm kontsentratsioonist ning
Y on vastus, mis algab sigmoidse kõvera alumiselt platoolt ja liigub ülemise platooni. Alumiseks platooks on Hilli logistilises võrrandis võetud null.
|
|
36.
|
Iga uuritava kemikaali kohta peavad olema esitatud järgmised andmed:
RPCmax, mis on uuritava kemikaali indutseeritud maksimaalne vastus, väljendatuna protsendina 1 nM E2 indutseeritud vastusest samal plaadil, samuti PCmax (RPCmax-ga seotud kontsentratsioon) ning
positiivsete kemikaalide korral indutseerivad kontsentratsioonid PC10 ja, kui see on asjakohane, siis PC50.
|
|
37.
|
PCx väärtuse saab arvutada, interpoleerides kahe punkti x- ja y-koordinaadi vahel, kusjuures üks neist punktidest on vahetult all- ja teine vahetult ülalpool PCx väärtust. Vahetult all- ja ülalpool PCx väärtust olevate andmepunktide koordinaatide (vastavalt (a,b) ja (c,d)) alusel saab järgmise valemi abil arvutada PCx väärtuse:
log[PCx] = log[c]+(x–d)/(d–b)
|
|
38.
|
PC väärtusi kirjeldatakse allpool joonisel 1.
Joonis 1
Näide PC väärtuste arvutamisest. PC (1 nM E2) on lisatud igale analüüsitavale plaadile.

|
ERi antagonistikatse
|
39.
|
ERi antagonistikatse korral tuleb positiivse kontrollaine (25 nM E2) suhtelise transkriptsiooni aktiivsuse (rikastatud kontrollproovi suhtes, 25 pM E2) leidmiseks analüüsida sama plaadi luminestsentssignaale, järgides järgmisi etappe (ka muud matemaatilised võtted on lubatud):
1. etapp. Arvutada kandeaine kontrollproovi keskväärtus.
2. etapp. Andmete normaliseerimiseks lahutada kandeaine kontrollproovi keskväärtus iga süvendi puhul saadud väärtusest. 3. etapp. Arvutada normaliseeritud rikastatud kontrollproovi keskväärtus.
4. etapp. Jagada plaadi iga süvendi normaliseeritud väärtus normaliseeritud rikastatud kontrollproovi keskväärtusega (rikastatud kontrollproov = 100 %).
Viimane iga süvendi kohta saadu väärtus on kõnealuse süvendi suhteline transkriptsiooni aktiivsus rikastatud kontrollproovi vastusega võrreldes.
5. etapp. Arvutada iga töötluse kohta suhteliste transkriptsiooni aktiivsuste keskväärtus.
|
Kaalutlused IC30 ja IC50 induktsiooni kohta
|
40.
|
Positiivsete kemikaalide korral tuleb esitada indutseerivad kontsentratsioonid IC30 ja, kui see on asjakohane, siis IC50.
|
|
41.
|
ICx väärtuse saab arvutada, interpoleerides kahe punkti x- ja y-koordinaadi vahel, kusjuures üks neist punktidest on vahetult all- ja teine vahetult ülalpool ICx väärtust. Vahetult all- ja ülalpool ICx väärtust olevate andmepunktide koordinaatide (vastavalt (a,b) ja (c,d)) alusel saab järgmise valemi abil arvutada PCx väärtuse:
lin ICx = a–(b–(100–x)) (a–c) /(b–d)
Joonis 2
Näide IC väärtuste arvutamisest. Rikastatud kontrollproov (25 pM E2) on lisatud igale analüüsitavale plaadile

RTA: suhteline transkriptsiooni aktiivsus.
|
|
42.
|
Tulemused peavad põhinema kahel (või kolmel) sõltumatul katsel. Kui kaks katset annavad võrreldavad ja seega korratavad tulemused, ei ole kolmandat katset vaja teha. Selleks, et olla vastuvõetavad, peavad tulemused:
|
—
|
vastama nõuetekohasuse kriteeriumidele (vt nõuetekohasuse kriteeriumid punktides 14–20),
|
|
Andmete tõlgendamise kriteeriumid
Tabel 7
Positiivseks ja negatiivseks klassifitseerimise otsuse kriteeriumid ERi agonistikatse korral
|
Positiivne
|
Kui leitud RPCmax on suurem kui 10 % positiivse kontrollprooviga vähemalt kahel katsel kahest või kolmest saadud vastusest või sellega võrdne.
|
|
Negatiivne
|
Kui leitud RPCmax ei ole vähemalt 10 % positiivse kontrollprooviga vähemalt kahel katsel kahest või kolmest saadud vastusest.
|
Tabel 8
Positiivseks ja negatiivseks klassifitseerimise otsuse kriteeriumid ERi antagonistikatse korral
|
Positiivne
|
Kui vähemalt kahel katsel kahest või kolmest on võimalik arvutada IC30.
|
|
Negatiivne
|
Kui vähemalt kahel katsel kahest või kolmest ei ole võimalik IC30 arvutada.
|
|
43.
|
Andmete tõlgendamise kriteeriumid on esitatud tabelites 7 ja 8. Positiivset tulemust iseloomustatakse nii toime suurusjärgu kui ka kontsentratsiooniga, mille juures toime avaldub. Selleks võib tulemusi väljendada kontsentratsioonina, mille juures agonistikatses saadakse 50 % (PC50) või 10 % (PC10) positiivse kontrollproovi väärtustest ning antagonistikatses 50 % (IC50) või 30 % (IC30) rikastatud kontrollproovi väärtustest. Uuritav kemikaal määratakse siiski positiivseks, kui tema maksimaalne indutseeritud vastus (RPCmax) on suurem kui 10 % positiivse kontrollprooviga vähemalt kahel katsel kahest või kolmest saadud vastusest või sellega võrdne, ning uuritav kemikaal loetakse negatiivseks, kui RPCmax jääb vähemalt kahel katsel kahest või kolmest alla 10 % positiivse kontrollprooviga saadud vastusest.
|
|
44.
|
ERi agonistikatse PC10, PC50 ja PCmax ning ERi antagonistikatse IC30 ja IC50 arvutused võib teha OECD avalikul veebisaidil (62) katsejuhendi juures leitava arvutustabeli abil.
|
|
45.
|
On piisav, kui PC10 või PC50 ja IC30 või IC50 väärtused saadakse vähemalt kaks korda. Kuid kui sama kontsentratsioonivahemiku andmete baasjoon on varieeruv ning vastuvõetamatult suure variatsioonikordajaga (CV, %), võidakse andmed lugeda ebausaldusväärseks ning suure varieeruvuse põhjus tuleb kindlaks teha. PC10 arvutamiseks kasutatavate andmepunktide andmed, mida saadakse kolmest paralleelkatsest (st luminestsentsi intensiivsuse andmed), peavad olema variatsioonikordajaga alla 20 %.
|
|
46.
|
Nõuetekohasuse kriteeriumide täitmine näitab, et katsesüsteem toimib korrektselt, kuid see ei taga täpsete andmete saamist mis tahes konkreetsel katsel. Esimese katse tulemuste kordamine on parim võimalus tagada täpsete andmete saamine.
|
|
47.
|
Kui tegemist on ERi agonistikatsega, kus lisaks sõelumise ja prioriseerimise eesmärgile on nõutav täiendav teave, on positiivsete uuritavate kemikaalide puhul, eriti nende puhul, mille PC väärtus on 10–49 %, samuti kemikaalide puhul, mida kahtlustatakse olevat lutsiferaasi ülestimuleerijad, võimalik ERα antagonisti kasutades kinnitada, et tähendatud lutsiferaasi aktiivsus on üksnes ERα-spetsiifiline vastus (vt liide 2.1).
|
KATSEPROTOKOLL
|
48.
|
Vt punkti 20 jaotises „ER TA katsemeetodi osad“.
|
KIRJANDUS
|
(1)
|
OECD (2015), „Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity“, Environment, Health and Safety Publications, Series on Testing and Assessment (nr 225), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(2)
|
Escande, A. et al. (2006), „Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta“, Biochem. Pharmacol., nr 71, lk 1 459–1 469.
|
|
(3)
|
Kuiper, G. G., et al. (1998), „Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta“, Endocrinol., nr 139, 4 252–4 263.
|
|
(4)
|
Spaepen, M., et al. (1992), „Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction“, FEMS Microbiol. Lett., nr 78(1), lk 89–94.
|
|
(5)
|
Kobayashi, H., et al. (1995), „Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products“, J. Vet. Med. Sci., nr 57(4), lk 769–71.
|
|
(6)
|
Dussurget, O. ja Roulland-Dussoix, D. (1994), „Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera“, Appl. Environ. Microbiol., nr 60(3), lk 953–9.
|
|
(7)
|
De Lean, A., Munson, P. J. ja Rodbard, D. (1978), „Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves“, Am. J. Physiol., nr 235, lk E97–E102.
|
Liide 2.1
VALEPOSITIIVSED TULEMUSED: NENDE LUMINESTSENTSSIGNAALIDE HINDAMINE, MIDA RETSEPTORID EI VAHENDA
|
1.
|
ERi agonistikatse valepositiivsed tulemused võivad olla põhjustatud lutsiferaasigeeni aktivatsioonist, mida ei vahenda ER, või geeniprodukti otsesest aktivatsioonist või sõltumatust fluorestsentsist. Selliseid mõjusid kajastab mittetäielik või ebahariliku kujuga annuse-vastuse kõver. Kui esineb selliste mõjude kahtlus, tuleks uurida ERi antagonisti (nt 4-hüdroksütamoksüfeen (OHT) mittetoksilises kontsentratsioonis) mõju vastusele. Puhas antagonist ICI 1827 80 ei pruugi selleks otstarbeks sobida, sest piisavas kontsentratsioonis võib ICI 1827 80 kandeaine kontrollprooviga saadud väärtust vähendada, mis omakorda mõjutab andmete analüüsi.
|
|
2.
|
Selle lähenemisviisi nõuetekohasuse tagamiseks tuleb samal plaadil analüüsida järgmist:
|
—
|
tundmatu kemikaali agonistlik toime koos 10 μM OHTga ja ilma selleta;
|
|
—
|
kandeaine kontrollproov (kolm proovi);
|
|
—
|
1 nM E2 (kolm proovi), agonisti positiivne kontrollproov;
|
|
—
|
1 nM E2 + OHT (kolm proovi).
|
|
Andmete tõlgendamise kriteeriumid
Märkus: Kõigis süvendites olevaid proove tuleb töödelda samas kontsentratsioonis kandeainega.
|
—
|
Kui ERi antagonistiga töötlemine ei mõjuta tundmatu kemikaali agonistlikku toimet, klassifitseeritakse tundmatu kemikaal negatiivseks.
|
|
—
|
Kui tundmatu kemikaali agonistlik toime täielikult pärsitakse, rakendada otsustuskriteeriume.
|
|
—
|
Kui madalaima kontsentratsiooni juures on agonistlik toime suurem kui PC10 saadud vastus või sellega võrdne, siis on tundmatu kemikaal inhibeeritud PC10 saadud vastusega võrdselt või rohkem. Arvutatakse ERiga töödeldud ja töötlemata süvendites saadud vastuste vahe, mida käsitletakse tõese vastusena ja kasutatakse asjakohaste parameetrite arvutamisel, mis võimaldavad teha otsuse klassifitseerimise kohta.
|
Andmete analüüs
Kontrollida toimivusnõudeid.
Kontrollida samades tingimustes töödeldud süvendites saadud tulemuste variatsioonikordajaid.
|
1.
|
Arvutada kandeaine kontrollproovi keskväärtus.
|
|
2.
|
Lahutada kandeaine kontrollproovi keskväärtus igas niisuguses süvendis saadud väärtusest, mida ei ole OHTga töödeldud.
|
|
3.
|
Arvutada OHTga saadud tulemuste keskväärtus.
|
|
4.
|
Lahutada kandeaine kontrollproovi keskväärtus igas OHTga töödeldud süvendis saadud väärtusest.
|
|
5.
|
Arvutada positiivse kontrollproovi keskväärtus.
|
|
6.
|
Arvutada kõigi teiste süvendite suhteline transkriptsiooni aktiivsus positiivse kontrollprooviga võrreldes.
|
Liide 2.2
DEKSTRAANIGA KAETUD AKTIIVSÖEGA (DCC) TÖÖDELDUD SEERUMI VALMISTAMINE
|
1.
|
Seerumi töötlemine dekstraaniga kaetud aktiivsöega (DCC) on levinud meetod rakkudes söötmesse lisatavast seerumist östrogeensete komponentide eemaldamiseks, et vältida vastuse nihet seoses seerumis sisalduvate jääköstrogeenidega. Seda metoodikat järgides saab töödelda 500 ml veiselooteseerumit (FBS).
|
Osad
|
2.
|
Vaja läheb järgmisi materjale ja seadmeid.
|
Materjalid
|
Magneesiumkloriidheksahüdraat, (MgCl2· 6H2O)
|
|
1 M HEPES-puhverlahus (pH 7,4)
|
|
Filtrisüsteemist saadud ülipuhas vesi
|
Seadmed
Autoklaavitud klaasanum (suurus valitakse vajaduse järgi) Harilik laboritsentrifuug (mille temperatuuriks saab seada 4 °C)
Töö käik
|
3.
|
Allpool kirjeldatud toimingud sobivad kasutamiseks 50 ml tsentrifuugianumatega.
[1. päev] Valmistada dekstraankattega aktiivsöe suspensioon 1 l ülipuhta veega, mis sisaldab 1,5 mM MgCl2, 0,25 M sahharoosi, 2,5 g aktiivsütt, 0,25 g dekstraani ja 5 mM HEPES-puhvrit, ning segada seda 4 °C juures üleöö.
[2. päev] Jaotada suspensioon 50 ml tsentrifuugianumatesse ja tsentrifuugida kiirusel 10 000 p/min 4 °C juures 10 minutit. Eemaldada supernatant ja säilitada pool söesadet 4 °C juures 3. päeval kasutamiseks. Teine pool söest suspendeerida FBSis, mis on sadenemise vältimiseks aeglaselt sulatatud ning 30 minuti jooksul 56 °C juures inaktiveeritud, seejärel viia suspensioon autoklaavitud klaasnõusse, näiteks koonilisse kolbi. Segada suspensiooni aeglaselt 4 °C juures üleöö.
[3. päev] Jaotada FBSis valmistatud suspensioon tsentrifuugianumatesse ja tsentrifuugida kiirusel 10 000 p/min 4 °C juures 10 minutit. Koguda FBS kokku ja viia uude söesademesse, mis valmistati 2. päeval. Suspendeerida söesade ja segada saadud suspensiooni aeglaselt autoklaavitud klaasnõus 4 °C juures üleöö.
[4. päev] Jaotada suspensiooni tsentrifuugianumatesse ja tsentrifuugida kiirusel 10 000 p/min 4 °C juures 10 minutit, seejärel steriliseerida supernatant läbi 0,2 μm steriilse filtri filtrimise teel. Seda DCCga töödeldud FBSi tuleb säilitada -20 °C juures ja see tuleb ära kasutada ühe aasta jooksul.
|
3. Liide
VM7LUC ÖSTROGEENIRETSEPTORITE TRANSAKTIVATSIOONI KATSEMEETOD, MILLEGA TEHAKSE KINDLAKS ÖSTROGEENIRETSEPTORI AGONISTID JA ANTAGONISTID
LÄHTEKAALUTLUSED JA PIIRANGUD (VT KA ÜLDIST SISSEJUHATUST)
|
1.
|
Selle katsemeetodiga kasutatakse rakuliini VM7Luc4E2 (63). Selle on valideerinud Ameerika Ühendriikide riikliku toksikoloogiaprogrammi alternatiivsete toksikoloogiameetodite ametitevaheline keskus (National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, NICEATM) ja alternatiivmeetodite valideerimise ametitevaheline koordineerimiskomitee (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) (1). VM7Luc-rakuliinid ekspresseerivad valdavalt endogeenset ERα-d ja vähesel määral endogeenset ERβ-d (2, 3, 4).
|
|
2.
|
See katsemeetod sobib paljude ainete uurimiseks eeldusel, et neid saab lahustada dimetüülsulfoksiidis (DMSO, CASi nr 67-68-5), need ei reageeri DMSOga ega rakkude söötmega ega ole uuritavate kontsentratsioonide juures tsütotoksilised. Kui DMSOd ei saa kasutada, võib kasutada muud kandeainet, näiteks etanooli või vett (vt punkt 12). VM7Luc ER TA (ant)agonistikatse tõendatud toimivus lubab oletada, et käesoleva katsemeetodiga saadud andmed võivad anda teavet ERi vahendatud toimemehhanismide kohta ning tuleks kaaluda nende kasutamist ainete prioriseerimiseks edasise uurimise eesmärgil.
|
|
3.
|
See katsemeetod on välja töötatud spetsiaalselt hERα ja hERß vahendatud transaktivatsiooni tuvastamiseks, kasutades mõõdetava parameetrina kemoluminestsentsi. Kemoluminestsentsi kasutamine biokatsetes on laialt levinud, sest luminestsentsil on suur signaali-müra suhe. Jaanimardika lutsiferaasi aktiivsust võivad rakupõhistes katsetes segada ained, mis inhibeerivad lutsiferaasi ensüümi, põhjustades kas näilist inhibitsiooni või suurenenud luminestsentsi valgu stabiliseerimise tõttu. Peale selle on mõnede lutsiferaasipõhiste ERi reportergeeni uurivate katsemeetoditega üle 1 μM fütoöstrogeeni kontsentratsioonide juures teadaolevalt saadud luminestsentssignaali, mida ei vahenda retseptorid, vaid mida põhjustab lutsiferaasi reportergeeni üleaktiveerimine. Kuigi annuse-vastuse kõver näitab, et ERi süsteemi aktiveerumine toimub väiksematel kontsentratsioonidel, tuleb stabiilse transfektsiooniga ER TA katsesüsteemides hoolitalt uurida suurtel kontsentratsioonidel ilmnevat lutsiferaasi ekspressiooni, mida põhjustavad fütoöstrogeenid või samalaadsed ühendid, mis sarnaselt fütoöstrogeenile tekitavad lutsiferaasi reportergeeni üleaktivatsiooni.
|
|
4.
|
Enne käesoleva katsemeetodi regulatiivsel eesmärgil kasutamist tuleks lugeda peatükke „Üldine sissejuhatus“ ja „ER TA katsemeetodi osad“. Käesolevas katsemeetodis kasutatud mõisted ja lühendid on esitatud 1. liites.
|
KATSEMEETODI PÕHIMÕTE (VT KA ÜLDIST SISSEJUHATUST)
|
5.
|
Katsemeetodit kasutatakse ERi ligandi sidumise kindlakstegemiseks, millele järgneb retseptori-ligandi kompleksi translokatsioon tuuma. Tuumas seondub retseptori-ligandi kompleks spetsiifilise DNA-vastuselemendiga ja transaktiveerib reportergeeni (luc), mille tulemusena tekib lutsiferaas ja valguse eraldumine, mida saab luminomeetriga kvantitatiivselt mõõta. Lutsiferaasi aktiivsust saab kiiresti ja odavalt hinnata paljude müügilolevate komplektidega. VM7Luc ER TA katsemeetodis kasutatakse in vitro ERi agonisti või antagonistina toimivate ainete kindlakstegemiseks inimese rinna adenokartsinoomi ER-tundlikku rakuliini VM7, millele on stabiilselt transfekteeritud jaanimardika luc-reporterkonstrukt, mida kontrollivad neli hiire piimanäärmekasvaja promootorist (MMTV) ülespoole jäävad östrogeeni vastuselementi. MMTV promootor annab ainult vähesel määral ristreaktsioone teiste steroidsete jamittesteroidsete hormoonidega (8). Andmete tõlgendamise kriteeriumeid on üksikasjalikult kirjeldatud punktis 41. Lühidalt öeldes tehakse positiivne vastus kindlaks kontsentratsiooni-vastuse kõvera abil, millel on vähemalt kolm punkti, mille veavahemikud (keskväärtus ± SD) ei kattu, samuti amplituudi muutusena (normeeritud suhtelisi valgusühikuid (RLU) vähemalt 20 % etalonainega (agonistikatse korral 17β-östradiool (E2, CASi nr 50-28-2)), antagonistikatse korral raloksüfeen HCl (Ral, CASi nr 84449-90-1) / E2 saadud maksimumväärtusest).
|
METOODIKA
Rakuliin
|
6.
|
Katsemeetodiga tuleks kasutada stabiilse transfektsiooniga VM7Luc4E2 rakuliini. See rakuliin on praegu saadaval ainult tehnilise litsentsilepingu alusel California Ülikoolist asukohaga Davis, California, USA (64) ja ettevõttest Xenobiotic Detection Systems Inc., asukohaga Durham, North Carolina, USA (65).
|
Rakuliini stabiilsus
|
7.
|
Rakuliini stabiilsuse ja terviklikkuse säilitamiseks tuleks rakke kasvatada külmutatud varudest kultiveerimissöötmes rohkem kui ühe passaaži võrra (vt punkt 9). Rakke ei tohiks kasvatada rohkem kui 30 passaaži. VM7Luc4E2 rakuliini puhul kestavad 30 passaaži kokku ligikaudu 3 kuud.
|
Rakkude kasvatamine ja plaadile kandmise tingimused
|
8.
|
Järgida tuleb rakkude kasvatamise heas tavas (5, 6) esitatud eeskirju, et tagada kõigi materjalide ja meetodite kvaliteet ning säilitada mis tahes tehtava töö terviklikkus, nõuetekohasus ja korratavus.
|
|
9.
|
VM7Luc4E2 rakke hoitakse RPMI 1 640 söötmes, millele on lisatud 0,9 % Pen-Strepi ja 8,0 % veiselooteseerumit (FBS), spetsiaalses koekultuuride inkubaatoris temperatuuril 37 ± 1oC ja 90 ± 5 % niiskuse juures keskkonnas, kus õhk sisaldab 5,0 ± 1 % CO2.
|
|
10.
|
Ligikaudu 80 % konfluentsuse saavutamisel külvatakse VM7Luc4E2 rakud ümber ja lastakse kohaneda östrogeenivaba keskkonnaga 48 tunni jooksul enne nende kandmist 96 süvendiga plaatidele, et neid uuritavate kemikaalidega töödelda ning analüüsida östrogeenist sõltuvat lutsiferaasi aktiivsuse induktsiooni. Östrogeenivaba sööde (EFM) sisaldab Dulbecco modifitseeritud fenoolpunasevaba Eagle’i minimaalsöödet (DMEM), millele on lisatud 4,5 % aktiivsöe/dekstraaniga töödeldud veiselooteseerumit, 1,9 % L-glutamiini ja 0,9 % Pen-Strepi. Ükski plasttarvik ei tohi ilmutada östrogeenset toimet (vt üksikasjalikku juhendit (7))
|
Katse nõuetekohasuse kriteeriumid
|
11.
|
Katse nõuetekohaseks tunnistamise või tagasilükkamise aluseks on kõigi 96 süvendiga plaadil tehtud katsete puhul kasutatud etalonaine ja kontrollproovidega saadud tulemuste hinnang. Iga etalonainet uuritakse mitme eri kontsentratsiooni juures ning iga etalonaine ja kontrollaine kontsentratsiooniga tehakse mitu paralleelproovi. Tulemusi võrreldakse nende kvaliteedikontrolli proovide (QC) parameetritega, mis saadi agonistide ja antagonistide kohtaaja jooksul kogutud andmeid sisaldavatest andmebaasidest, mille iga labor on koostanud pädevuse tõendamise käigus. Kogutud andmetega andmebaase ajakohastatakse pidevalt etalonaine ja kontrollprooviga saadud väärtustega. Varustuse või laboritingimuste muutused võivad tingida vajaduse ajakohastatud andmebaasi loomiseks.
|
Agonistikatse
Vahemiku leidmise katse
|
•
|
Induktsioon: induktsiooni mõõtmiseks plaadil jagatakse etalonaine E2 suurima kontsentratsiooni juures saadud suhteliste valgusühikute (RLU) keskväärtus DMSO kontrollprooviga saadud RLU keskväärtusega. Tavaliselt saavutatakse viiekordne induktsioon ning nõuetekohaseks tunnistamiseks peab induktsioon olema vähemalt neljakordne.
|
|
•
|
DMSO kontrollprooviga saadud tulemused: lahusti kontrollprooviga saadud RLU väärtused ei tohi aja jooksul kogutud lahusti kontrollproovide tulemuste keskmisest RLU väärtusest erineda rohkem kui 2,5 standardhälbe võrra.
|
|
•
|
Katse, mille puhul ei ole täidetud ükskõik kumb nõuetekohasuse kriteerium, tuleb tühistada ja uuesti läbi viia.
|
Tervikkatse
See hõlmab agonisti vahemiku leidmise katse nõuetekohasuse kriteeriume ning järgmist.
|
•
|
Etalonainega saadud tulemused: etalonaine E2 kontsentratsiooni-vastuse kõver peab olema sigmoidne ning vähemalt kolm väärtust peavad paiknema kontsentratsiooni-vastuse kõvera lineaarvahemikus.
|
|
•
|
Positiivse kontrolliga saadud tulemused: metoksükloori kontrollprooviga saadud RLU väärtused peavad olema suuremad kui DMSOga saadud keskväärtus pluss kolmekordne DMSO keskväärtuse standardhälve.
|
|
•
|
Katse, mille puhul ei ole täidetud ükskõik milline nõuetekohasuse kriteerium, tuleb tühistada ja uuesti läbi viia.
|
Antagonistikatse
Vahemiku leidmise katse
|
•
|
Vähenemine: vähenemise mõõtmiseks jagatakse etalonaine Ral/E2 suurima kontsentratsiooni juures saadud RLU keskväärtus DMSO kontrollprooviga saadud RLU keskväärtusega. Tavaliselt saavutatakse viiekordne vähenemine ning nõuetekohaseks tunnistamiseks peab vähenemine olema vähemalt kolmekordne.
|
|
•
|
E2 kontrollproovidega saadud tulemused: E2 kontrollprooviga saadud RLU väärtused ei tohi aja jooksul kogutud E2 kontrollproovide tulemuste keskmisest RLU väärtusest erineda rohkem kui 2,5 standardhälbe võrra.
|
|
•
|
DMSO kontrollprooviga saadud tulemused: DMSO kontrollprooviga saadud RLU väärtused ei tohi aja jooksul kogutud lahusti kontrollproovide tulemuste keskmisest RLU väärtusest erineda rohkem kui 2,5 standardhälbe võrra.
|
|
•
|
Katse, mille puhul ei ole täidetud ükskõik milline nõuetekohasuse kriteerium, tühistatakse ja tehakse uuesti.
|
Tervikkatse
See hõlmab antagonisti vahemiku leidmise katse nõuetekohasuse kriteeriume ning järgmist.
|
•
|
Etalonainega saadud tulemused: etalonaine Ral/E2 kontsentratsiooni-vastuse kõver peab olema sigmoidne ning vähemalt kolm väärtust peavad paiknema kontsentratsiooni-vastuse kõvera lineaarvahemikus.
|
|
•
|
Positiivse kontrolliga saadud tulemused: tamoksüfeeni/E2 kontrollprooviga saadud RLU väärtused peavad olema väiksemad kui E2 kontrollprooviga saadud keskväärtus miinus kolmekordne E2 kontrollproovi keskväärtuse standardhälve.
|
|
•
|
Katse, mille puhul ei ole täidetud ükskõik milline nõuetekohasuse kriteerium, tühistatakse ja tehakse uuesti.
|
Etalonained, positiivsed ja lahusti kontrollproovid
Lahusti kontrollproov (agonisti- ja antagonistikatsed)
|
12.
|
Uuritava kemikaali lahustamiseks kasutatavat kandeainet tuleb katseliselt kontrollida kandeaine kontrollproovina. VM7Luc ER TA katsemeetodi valideerimisel kasutatud kandeaine oli 1 % (mahuprotsent) dimetüülsulfoksiid (DMSO, CASi nr 67-68-5) (vt punkt 24). Kui DMSO asemel kasutatakse muud kandeainet, tuleb katsed kõigi etalonainete, kontrollproovide ja uuritavate kemikaalidega vajaduse korral läbi viia samas kandeaines.
|
Etalonaine (agonisti vahemiku leidmine)
|
13.
|
Etalonaine on E2 (CASi nr 50-28-2). Vahemiku leidmiskatseks tehakse etalonainest järjestikused lahjendused, nii et saadakse neljas kontsentratsioonis E2 (1,84 × 10-10, 4,59 × 10-11, 1,15 × 10-11 ja 2,87 × 10-12 M), ning iga kontsentratsiooniga tehakse katse kahes süvendis.
|
Etalonaine (agonisti tervikkatse)
|
14.
|
Tervikkatses koosnevad E2 proovid järjestikustest 1 : 2 lahjendustest, mille tulemusena saadakse 11 eri kontsentratsiooniga (3,67 × 10-10 kuni 3,59 × 10-13 M) E2 proovi, millest igaüht analüüsitakse paralleelselt kahes süvendis.
|
Etalonaine (antagonisti vahemiku leidmine)
|
15.
|
Etalonaineks on Rali (CASi nr 84449-90-1) ja E2 (CASi nr 50-28-2) segu. Vahemiku leidmise katses valmistatakse Ral/E2 järjestikused lahjendused nii, et saadakse kolmes kontsentratsioonis Ral (3,06 × 10-9, 7,67 × 10-10 ja 1,92 × 10-10M), millest igaühele on lisatud muutmatus kontsentratsioonis (9,18 × 10-11 M) E2, analüüsitakse paralleelselt kahes süvendis.
|
Etalonaine (antagonisti tervikkatse)
|
16.
|
Tervikkatses valmistatakse Ral/E2 järjestikused 1 : 2 lahjendused nii, et saadakse üheksas kontsentratsioonis Ral (2,45× 10-8 kuni 9,57 × 10-11M), millest igaühele on lisatud muutmatus kontsentratsioonis (9,18 × 10-11 M) E2, analüüsitakse paralleelselt kahes süvendis.
|
Nõrgalt positiivne kontrollaine (agonist)
|
17.
|
Nõrgalt positiivne kontrollaine on 9,06 × 10-6 M p,p’-metoksükloor (metoksükloor; CASi nr 72-43-5) EFMis.
|
Nõrgalt positiivne kontrollaine (antagonist)
|
18.
|
Nõrgalt positiivne kontrollaine on 3,36 × 10-6 M tamoksüfeen (CASi nr 10540-29-1) EFMis, millele on lisatud 9,18 × 10-11 M E2.
|
E2 kontrollproov (ainult agonistikatse)
|
19.
|
E2 kontrollproov koosneb 9,18 × 10-11 M E2-st EFMis ja seda kasutatakse negatiivse kontrollproovi baasjoone leidmiseks.
|
Kordne induktsioon (agonist)
|
20.
|
Lutsiferaasi aktiivsuse indutseerimine etalonaine (E2) mõjul leitakse, jagades etalonaine E2 suurima kontsentratsiooniga saadud RLU keskväärtuse DMSO kontrollprooviga saadud RLU keskväärtusega ning tulemus peab olema rohkem kui neljakordne.
|
Kordne vähenemine (antagonist)
|
21.
|
Etalonaine (Ral/E2) lutsiferaasi keskmine aktiivsus leitakse, jagades etalonaine Ral/E2 suurima kontsentratsiooniga saadud RLU keskväärtuse DMSO kontrollprooviga saadud RLU keskväärtusega ning tulemus peab olema rohkem kui kolmekordne.
Labori pädevuse tõendamine (vt käesoleva katsemeetodi jaotise „ER TA katsemeetodi osad“ punkti 14 ning tabeleid 3 ja 4).
|
Kandeaine
|
22.
|
Uuritavad kemikaalid tuleb lahustada lahustis, milles nad lahustuvad ja mis on võimeline segunema rakkude söötmega. Sobivad kandeained on vesi, etanool (puhtusega 95–100 %) ja DMSO. Kui kasutatakse DMSOd, ei tohi selle kontsentratsioon ületada 1 mahuprotsenti. Mis tahes kandeaine korral tuleb tõendada, et maksimaalne kasutatav kogus ei ole tsütotoksiline ega sega analüüsimeetodi toimivust. Etalonaine ja kontrollained lahustatakse 100 % lahustis ning lahjendatakse seejärel EFMiga sobivate kontsentratsioonideni.
|
Uuritavate kemikaalide ettevalmistamine
|
23.
|
Uuritavad kemikaalid lahustatakse 100 % DMSOs (või sobivas lahustis) ning lahjendatakse seejärel EFMiga sobivate kontsentratsioonideni. Enne lahustamist ja lahjendamist tuleb kõigil uuritavatel kemikaalidel lasta saavutada toatemperatuur. Uuritavate kemikaalide lahused tuleb iga katse jaoks värskelt valmistada. Lahustes ei tohi olla nähtavat sadet ega hägu. Etalonaine ja kontrollainetega võib valmistada suurema koguse põhilahust, kuid etalonaine, kontrollaine ja uuritavate kemikaalide lõplikud lahused tuleb valmistada värskelt enne igat katset ning kasutada 24 tunni jooksul valmistamisest alates.
|
Lahustuvus ja tsütotoksilisus: vahemiku leidmise kaalutlused
|
24.
|
Vahemiku leidmise katse koosneb seitsme punkti leidmisest järjestikuste 1 : 10 lahjendustega, millest igaühest tehakse kaks paralleelproovi. Kõigepealt tehakse uuritavate kemikaalidega katseid kuni maksimaalse kontsentratsioonini 1 mg/ml (~1 mM) agonistikatse korral ja 20 μg/ml (~10 μM) antagonistikatse korral. Vahemiku leidmise katsete abil määratakse järgmised parameetrid:
|
|
—
|
uuritava kemikaali algkontsentratsioon tervikkatses kasutamiseks;
|
|
—
|
uuritava kemikaali lahjendused (1 : 2 või 1 : 5) tervikkatses kasutamiseks.
|
|
25.
|
Agonisti- ja antagonistikatse eeskirjad hõlmavad raku eluvõimelisuse / tsütotoksilisuse hindamist ning see kuulub ka vahemiku leidmise ja tervikkatse juurde. VM7Luc ER TA valideerimisel (1) rakkude eluvõimelisuse hindamiseks kasutatud tsütotoksilisuse määramise meetod oli skaleeritud kvalitatiivne visuaalne jälgimismeetod, kuid tsütotoksilisuse määramiseks võib kasutada ka kvantitatiivset meetodit (vt eeskirja (7)). Nende uuritava kemikaali kontsentratsioonidega saadud andmeid, mis põhjustavad eluvõimelisuse rohkem kui 20 % vähenemise, ei saa kasutada.
|
Kemikaaliga kokkupuude ja analüüsiplaadi jaotus
|
26.
|
Rakud loendatakse ja külvatakse 96 süvendiga koekultuuriplaadile (2 × 105 rakku süvendi kohta), kus neid inkubeeritakse EFMis 24 tundi, et võimaldada rakkudel plaadile kinnituda. EFM eemaldatakse ning asendatakse EFMis lahustatud uuritavate ja võrdluskemikaalidega, plaati inkubeeritakse 19–24 tundi. Suure lenduvusega ainete korral on vaja erimeetmeid, sest kontrollproovid lähedal paiknevates süvendites võivad anda valepositiivseid tulemusi. Sellisel juhul on soovitatav kasutada kilekatteid, mis tõhusalt eraldavad üksikud süvendid katse läbiviimise ajaks.
|
Vahemiku leidmise katsed
|
27.
|
Vahemiku leidmise katsetes kasutatakse kõiki 96 süvendiga plaadi süvendeid, et katsetada kuni kuut uuritavat kemikaali seitsmes järjestikuste 1 : 10 lahjendustega saadud kontsentratsioonis kahe paralleelproovina (vt jooniseid 1 ja 2).
|
|
—
|
Agonisti vahemiku leidmise katses kasutatakse etalonainena E2 nelja kontsentratsiooni kahes paralleelproovis ning neli süvendit kulub DMSO kontrolli paralleelproovidele.
|
|
—
|
Antagonisti vahemiku leidmise katses kasutatakse etalonainena Ral/E2, kus Ral on kolmes eri kontsentratsioonis, millele on lisatud 9,18 × 10-11 M E2; seda lahust analüüsitakse kahes paralleelproovis ning nii E2 kontrollproovile kui ka DMSO-kontrollproovile kulub kolm paralleelsüvendit.
|
Joonis 1.
Agonisti vahemiku leidmise katse 96 süvendiga plaadi paigutus

Lühendid: E2-1 kuni E2-4 = etalonaine E2 kontsentratsioonid (suuremast väiksemani); TC1-1 kuni TC1-7 = 1. uuritava kemikaali (TC1) kontsentratsioonid (suuremast väiksemani); TC2-1 kuni TC2-7 = 2. uuritava kemikaali (TC2) kontsentratsioonid (suuremast väiksemani); TC3-1 kuni TC3-7 = 3. uuritava kemikaali (TC3) kontsentratsioonid (suuremast väiksemani); TC4-1 kuni TC4-7 = 4. uuritava kemikaali (TC4) kontsentratsioonid (suuremast väiksemani); TC5-1 kuni TC5-7 = 5. uuritava kemikaali (TC5) kontsentratsioonid (suuremast väiksemani); TC6-1 kuni TC6-7 = 6. uuritava kemikaali (TC6) kontsentratsioonid (suuremast väiksemani); VC = kandeaine kontrollproov (DMSO [1 % (mahuprotsent) EFM]).
Joonis 2.
Antagonisti vahemiku leidmise katse 96 süvendiga plaadi paigutus

Lühendid: E2 = E2 kontrollproov; Ral-1 kuni Ral-3 = etalonaine raloksüfeeni/E2 kontsentratsioonid (suuremasti väiksemani); TC1-1 kuni TC1-7 = 1. uuritava kemikaali (TC1) kontsentratsioonid (suuremast väiksemani); TC2-1 kuni TC2-7 = 2. uuritava kemikaali (TC2) kontsentratsioonid (suuremast väiksemani); TC3-1 kuni TC3-7 = 3. uuritava kemikaali (TC3) kontsentratsioonid (suuremast väiksemani); TC4-1 kuni TC4-7 = 4. uuritava kemikaali (TC4) kontsentratsioonid (suuremast väiksemani); TC5-1 kuni TC5-7 = 5. uuritava kemikaali (TC5) kontsentratsioonid (suuremast väiksemani); TC6-1 kuni TC6-7 = 6. uuritava kemikaali (TC6) kontsentratsioonid (suuremast väiksemani); VC = kandeaine kontrollproov (DMSO [1 % (mahuprotsent) EFM]).
Märkus: kõigi uuritavate kemikaalidega viiakse katse läbi 9,18 × 10-11 M E2 juuresolekul.
|
28.
|
Igas süvendis on söötme soovitatav lõplik ruumala 200 μl. Kasutada ainult neid analüüsitavaid plaate, milles rakkude eluvõimelisus kõigis süvendites on vähemalt 80 %.
|
|
29.
|
Agonisti
tervikkatse jaoks lähtekontsentratsioonide määramist kirjeldatakse põhjalikult agonistikatse eeskirjas (7). Lühidalt, kasutatakse järgmisi kriteeriume.
|
|
—
|
Kui uuritava kemikaali kontsentratsioonikõveral ei ole punkte, mis on suuremad kui DMSO kontrollprooviga saadud keskväärtus pluss kolmekordne standardhälve, viiakse tervikkatse läbi 11 järjestikuste 1 : 2 lahjendustega saadud kontsentratsioonis, alustades maksimaalsest lahustuvast kontsentratsioonist.
|
|
—
|
Kui uuritava kemikaali kontsentratsioonikõveral on punkte, mis on suuremad kui DMSO kontrollprooviga saadud keskväärtus pluss kolmekordne standardhälve, peab 11 lahjenduse lähtekontsentratsioon tervikkatses olema 1 log suurem kui kontsentratsioon, millega vahemiku leidmise katses saadi suurim kohandatud RLU väärtus. 11 kontsentratsiooniga lahjendusskeem põhineb kas 1 : 2 või 1 : 5 lahjendustel vastavalt järgmistele kriteeriumidele:
|
11 kontsentratsiooniga järjestikusi 1 : 2 lahjendusi tuleks kasutada, kui vahemiku leidmise katses saadud kontsentratsiooni-vastuse kõveral põhjal kajastab saadav kontsentratsioonide vahemik kogu vastuste vahemikku. Muul juhul tuleks lahjendada vahekorras 1 : 5.
|
|
|
—
|
Kui uuritava kemikaali vahemiku leidmise katses saadud kontsentratsiooni-vastuse kõver on kahefaasiline, peab ka tervikkatse kajastama mõlemat faasi.
|
|
30.
|
Antagonisti
tervikkatse jaoks lähtekontsentratsioonide määramist kirjeldatakse põhjalikult antagonistikatse eeskirjas (7). Lühidalt, kasutatakse järgmisi kriteeriume.
|
|
—
|
Kui uuritava kemikaali kontsentratsioonikõveral ei ole punkte, mis on väiksemad kui E2 kontrollprooviga saadud keskväärtus miinus kolmekordne standardhälve, viiakse tervikkatse läbi 11 järjestikuste 1 : 2 lahjendustega saadud kontsentratsioonis, alustades maksimaalsest lahustuvast kontsentratsioonist.
|
|
—
|
Kui uuritava kemikaali kontsentratsioonikõveral on punkte, mis on väiksemad kui E2 kontrollprooviga saadud keskväärtus miinus kolmekordne standardhälve, peab 11 lahjenduse lähtekontsentratsioon tervikkatses olema üks järgmistest:
|
—
|
kontsentratsioon, mis vahemiku leidmise katses annab vähima kohandatud RLU väärtuse;
|
|
—
|
maksimaalne lahustuv kontsentratsioon (vt agonistikatse eeskiri (7), joonis 14-2);
|
|
—
|
vähim tsütotoksiline kontsentratsioon (vt näiteks antagonistikatse eeskiri (7), joonis 14-3).
|
|
|
—
|
11 kontsentratsiooniga lahjendusskeem põhineb kas järjestikustel 1 : 2 või 1 : 5 lahjendustel vastavalt järgmistele kriteeriumidele:
|
11 kontsentratsiooniga järjestikusi 1 : 2 lahjendusi tuleks kasutada, kui vahemiku leidmise katses saadud kontsentratsiooni-vastuse kõveral põhjal kajastab saadav kontsentratsioonide vahemik kogu vastuste vahemikku. Muul juhul tuleks lahjendada vahekorras 1 : 5.
|
|
Tervikkatsed
|
31.
|
Tervikkatse koosneb 11 kontsentratsiooniga järjestikustest lahjendustest (järjestikused lahjendused vahekorras kas 1 : 2 või 1 : 5 olenevalt tervikkatse kriteeriumide kohasest lähtekontsentratsioonist), kusjuures iga kontsentratsiooniga tehakse paralleelkatse 96 süvendiga plaadi kolmes süvendis (vt joonised 3 ja 4).
|
|
—
|
Agonisti tervikkatses kasutatakse etalonainena E2 11 kontsentratsioonis kahes paralleelproovis. Igal plaadil tehakse neljas süvendis DMSO kontrollproovi paralleelkatsed ja veel neljas metoksükloori kontrollproovi (9,06 × 10-6 M) paralleelkatsed.
|
|
—
|
Antagonisti tervikkatses kasutatakse etalonainena Ral/E2 lahust Rali üheksas kontsentratsioonis, millele on lisatud muutumatult 9,18 × 10-11 M E2, neljas süvendis on 9,18 × 10-11 M E2 paralleelkontrollproovid, neljas DMSO kontrollproovid ja neljas 3,36 × 10-6M tamoksüfeeni paralleelproovid.
|
Joonis 3.
Agonisti tervikkatse 96 süvendiga plaadi paigutus
Lühendid: TC1-1 kuni TC1-11 = 1. uuritava kemikaali kontsentratsioonid (suuremast väiksemani); TC2-1 kuni TC2-11 = 2. uuritava kemikaali kontsentratsioonid (suuremast väiksemani); E2-1 kuni E2-11 = etalonaine E2 kontsentratsioonid (suuremast väiksemani); Meth = p,p’-metoksükloori nõrgalt positiivne kontrollproov; VC = DMSO (1 mahuprotsent) EFM kandeaine kontrollproov
Joonis 4.
Antagonisti tervikkatse 96 süvendiga plaadi paigutus

Lühendid: E2 = E2 kontrollproov; Ral-1 kuni Ral-9 = etalonaine raloksüfeeni/E2 kontsentratsioonid (suuremasti väiksemani); Tam = tamoksüfeeni/E2 nõrgalt positiivne kontrollproov; TC1-1 kuni TC1-11 = 1. uuritava kemikaali (TC1) kontsentratsioonid (suuremast väiksemani); TC2-1 kuni TC2-11 = 2. uuritava kemikaali (TC2) kontsentratsioonid (suuremast väiksemani); VC = kandeaine kontrollproov (DMSO [1 % (mahuprotsent) EFM]).
Märkus: nagu märgitud, sisaldavad kõik süvendid muutumatus kontsentratsioonis E2 (9,18 × 10-11 M).
|
32.
|
Sõltumatuse tagamiseks tuleb sama kemikaaliga tervikkatseid korrata eri päevadel. Kokku tuleks teha vähemalt kaks tervikkatset. Kui katsete tulemused on vasturääkivad (nt üks positiivne, teine negatiivne) või kui üks katsetest ei ole nõuetekohane, tuleb täiendavalt teha kolmas katse.
|
Luminestsentsi mõõtmine
|
33.
|
Luminestsentsi mõõdetakse lainepikkuste vahemikus 300 kuni 650 nm injektoriga luminomeetriga ning tarkvaraga, mis reguleerib süstitavat kogust ja mõõteintervalli (7). Igast süvendist emiteeritud valgust väljendatakse RLUdes süvendi kohta.
|
ANDMEANALÜÜS
EC50/IC50 määramine
|
34.
|
EC50 väärtus (50 % uuritava kemikaali suurima toime avaldumise kontsentratsioonist (agonisti puhul)) ja IC50 väärtus (50 % uuritava kemikaali suurima inhibeeriva toime avaldumise kontsentratsioonist (antagonisti puhul)) määratakse kontsentratsiooni-vastuse andmete järgi. Ühe või mitme kontsentratsiooni juures positiivse uuritava kemikaali puhul arvutatakse uuritava kemikaali kontsentratsioon, mis on 50 % suurima toime avaldumise kontsentratsioonist (IC50 või EC50), Hilli funktsiooni analüüsi või asjakohase alternatiivi abil. Hilli funktsioon on nelja parameetriga matemaatiline logistiline mudel, mis esitab uuritava kemikaali kontsentratsiooni seose vastusega (tavaliselt sigmoidse kõvera järgi) allpool toodud võrrandi kohaselt,
|
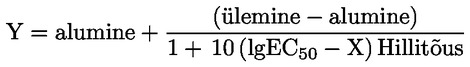
kus:
Y= vastus (st RLUd);
X= logaritm kontsentratsioonist;
alumine (platoo)= minimaalne vastus;
ülemine (platoo)= maksimaalne vastus;
lg EC50 (või lg IC50)= logaritm X-ist, vastusest, mis jääb ülemise ja alumise platoo vahele;
Hilli tõus= kõvera tõus.
Mudeliga arvutatakse parim sobiv joon ülemise ja alumise platoo ning tõusu järgi ning parameetrite IC50 ja EC50. EC50 ja IC50 väärtuste arvutamiseks tuleks kasutada asjakohast statistikatarkvara (nt Graphpad PrismR).
Võõrväärtuste määramine
|
35.
|
Head statistilist hindamist võib hõlbustada Q-testi jms (vt eeskirjad agonisti ja antagonisti puhul (7)) kasutamine „kasutute“ süvendite leidmiseks, millega saadud tulemused tuleb andmete analüüsimisel kõrvale jätta.
|
|
36.
|
E2 etalonaine paralleelproovide (kaks tk) korral loetakse paralleelproovi igasugune kohandatud RLU väärtus antud E2 kontsentratsiooni juures võõrväärtuseks, kui selle on rohkem kui 20 % suurem või väiksem kui aja jooksul kogutud andmete andmebaasis olev kohandatud RLU väärtus kõnealuse kontsentratsiooni jaoks.
|
Luminomeetriliste andmete kogumine ja kohandamine vahemiku leidmise katseks
|
37.
|
Luminomeetrist saadud töötlemata andmed tuleb kanda katse jaoks välja töötatud arvutustabelisse. Tuleb kindlaks teha, kas on olemas võõrväärtusi, mis tuleks eemaldada. (Vt katse nõuetekohasuse kriteeriume nende parameetrite osas, mida analüüsides määratakse.) Teha tuleb järgmised arvutused.
|
Agonist
|
1. etapp
|
Arvutada kandeaine (DMSO) kontrollproovi (VC) keskväärtus.
|
|
2. etapp
|
Andmete normaliseerimiseks lahutada kandeaine (DMSO) kontrollproovi (VC) keskväärtus iga süvendi puhul saadud väärtusest.
|
|
3. etapp
|
Arvutada etalonaine (E2) kordse induktsiooni keskväärtus.
|
|
4. etapp
|
Arvutada uuritavate kemikaalide EC50 keskväärtused.
|
Antagonist
|
1. etapp
|
Arvutada kandeaine DMSO kontrollproovi (VC) keskväärtus.
|
|
2. etapp
|
Andmete normaliseerimiseks lahutada kandeaine (DMSO) kontrollproovi (VC) keskväärtus iga süvendi puhul saadud väärtusest.
|
|
3. etapp
|
Arvutada etalonaine (Ral/E2) põhjustatud kordse vähenemise keskväärtus.
|
|
4. etapp
|
Arvutada etalonaine E2 kontrollproovide keskväärtus.
|
|
5. etapp
|
Arvutada uuritavate kemikaalide IC50 keskväärtused.
|
Luminomeetriliste andmete kogumine ja kohandamine tervikkatseks
|
38.
|
Luminomeetrist saadud töötlemata andmed tuleb kanda katse jaoks välja töötatud arvutustabelisse. Tuleb kindlaks teha, kas on olemas võõrväärtusi, mis tuleks eemaldada. (Vt katse nõuetekohasuse kriteeriume nende parameetrite osas, mida analüüsides määratakse.) Tehakse järgmised arvutused.
|
Agonist
|
1. etapp
|
Arvutada kandeaine DMSO kontrollproovi (VC) keskväärtus.
|
|
2. etapp
|
Andmete normaliseerimiseks lahutada kandeaine (DMSO) kontrollproovi (VC) keskväärtus iga süvendi puhul saadud väärtusest.
|
|
3. etapp
|
Arvutada etalonaine (E2) kordse induktsiooni keskväärtus.
|
|
4. etapp
|
Arvutada E2 ja uuritavate kemikaalide EC50 keskväärtused.
|
|
5. etapp
|
Arvutada metoksükloori kohandatud RLU keskväärtus.
|
Antagonist
|
1. etapp
|
Arvutada kandeaine DMSO kontrollproovi (VC) keskväärtus.
|
|
2. etapp
|
Andmete normaliseerimiseks lahutada kandeaine (DMSO) kontrollproovi (VC) keskväärtus iga süvendi puhul saadud väärtusest.
|
|
3. etapp
|
Arvutada etalonaine (Ral/E2) põhjustatud kordse induktsiooni keskväärtus.
|
|
4. etapp
|
Arvutada Ral/E2 ja uuritavate kemikaalide IC50 keskväärtused.
|
|
5. etapp
|
Arvutada tamoksüfeeni kohandatud RLU keskväärtus.
|
|
6. etapp
|
Arvutada etalonaine E2 kontrollproovide keskväärtus.
|
Andmete tõlgendamise kriteeriumid
|
39.
|
VM7Luc ER TA katsemeetod on kavandatud osana tõendite kaalukust arvestavast lähenemisviisist, et aidata aineid prioriseerida endokriinse häire põhjustamise in vivo uurimiseks. Üks kõnealuse prioriseerimise osadest on uuritava kemikaali klassifitseerimine kas positiivseks või negatiivseks ERi suhtes agonistliku või antagonistliku toime seisukohast. VM7Luc ER TA valideerimisuuringus kasutatud positiivseks ja negatiivseks klassifitseerimise otsustuskriteeriumid on esitatud tabelis 1.
|
Tabel 1.
Positiivseks ja negatiivseks klassifitseerimise otsustuskriteeriumid
|
|
|
AGONISTLIK TOIME
|
|
Positiivne
|
|
—
|
Kõigil uuritavatel kemikaalidel, mis on ERi agonistliku toime järgi klassifitseeritud positiivseks, pean kontsentratsiooni-vastuse kõver sisaldama baasjoont, millele järgneb positiivne tõus, mis lõpeb platoo või tipuga. Mõnel juhul on võimalik kindlaks määrata üksnes kaks neist osadest (alumine platoo ja tõus või tõus ja ülemine platoo).
|
|
—
|
Positiivse tõusu joonel peab olema vähemalt kolm punkti, mille veavahemikud (keskväärtus ± SD) ei kattu. Alumist platood moodustavad punktid jäetakse välja, kuid kõvera lineaarvahemikku moodustavad punktid võivad sisaldada tipupunkti või platoo esimest punkti.
|
|
—
|
Positiivseks klassifitseerimiseks peab vastuse amplituud, st alumise ja ülemise platoo vaheline erinevus olema vähemalt 20 % etalonainega E2 saadud maksimaalsest väärtusest (st vähemalt 2000 RLUd, kui etalonaine (E2) vastuse maksimaalseks väärtuseks on seatud 10 000 ) RLUd).
|
|
—
|
Võimaluse korral arvutatakse iga positiivse uuritava kemikaali EC50 väärtus.
|
|
|
Negatiivne
|
Konkreetse kontsentratsiooni keskmine kohandatud RLU on väiksem või võrdne DMSO kontrollproovi RLU keskväärtusega, millele on liidetud DMSO kontrollproovi RLU väärtuse kolmekordne standardhälve.
|
|
Ebapiisav
|
Ebapiisavaks loetakse andmed, mida suurte kvalitatiivsete või kvantitatiivsete puuduste tõttu ei saa tõlgendada usaldusväärsetena toime olemasolu või selle puudumise kajastamisel, ning neid andmeid ei saa kasutada määramaks, kas uuritav kemikaal on positiivne või negatiivne. Kemikaalidega tuleks teha uued katsed.
|
|
|
|
ANTAGONISTLIK TOIME
|
|
Positiivne
|
|
—
|
Uuritava kemikaalide andmetest saab koostada kontsentratsiooni-vastuse kõvera, mis koosneb baasjoonest, millele järgneb negatiivne tõus.
|
|
—
|
Negatiivse tõusu joonel peab olema vähemalt kolm punkti, mille veavahemikud ei kattu. Baasjoont moodustavad punktid jäetakse välja, kuid kõvera lineaarvahemikku moodustavad punktid võivad sisaldada platoo esimest punkti.
|
|
—
|
Toime peaks olema vähemalt 20 % väiksem etalonainega Ral/E2 saadud maksimaalsest väärtusest (st 8 000 RLUd või vähem, kui etalonaine (Ral/E2) vastuse maksimaalseks väärtuseks on seatud 10 000 RLUd).
|
|
—
|
Uuritava kemikaali suurim mittetsütotoksiline kontsentratsioon peab olema 1 × 10-5 M või alla selle.
|
|
—
|
Võimaluse korral arvutatakse iga positiivse uuritava kemikaali IC50 väärtus.
|
|
|
Negatiivne
|
Kõik andmepunktid on suuremad kui EC80 väärtus (80 % E2-ga saadud vastusest või 8 000 RLUd) kontsentratsioonil alla 1,0 × 10-5 M.
|
|
Ebapiisav
|
Ebapiisavaks loetakse andmed, mida suurte kvalitatiivsete või kvantitatiivsete puuduste tõttu ei saa tõlgendada usaldusväärsetena toime olemasolu või selle puudumise kajastamisel, ning neid andmeid ei saa kasutada määramaks, kas uuritav kemikaal on positiivne või negatiivne. Kemikaaliga tuleks teha uued katsed.
|
|
40.
|
Positiivset tulemust iseloomustatakse nii toime suurusjärgu kui ka võimaluse korral kontsentratsiooniga, mille juures toime avaldub. Näited positiivsetest, negatiivsetest ja ebapiisavatest andmetest on esitatud joonistel 5 ja 6.
|
Joonis 5.
Näited agonisti kohta: positiivsed, negatiivsed ja ebapiisavad andmed
Punktiirjoon tähistab 20 % E2-ga saadud vastusest, 2000 kohandatud ja normaliseeritud RLUd.
Joonis 6.
Näited antagonisti kohta: positiivsed, negatiivsed ja ebapiisavad andmed

Punktiirjoon tähistab 80 % Ral/E2-ga saadud vastusest, 8 000 kohandatud ja normaliseeritud RLUd.
Pidevjoon tähistab kontsentratsiooni 1,00 × 10-5 M. Selleks, et vastus loetakse positiivseks, peab see kontsentratsioonidel alla 1,00 × 10-5M jääma allapoole 8 000 RLU joont.
Meso-heksestrooli kõveral tärniga tähistatud kontsentratsioonidel juures on eluvõimelisus 2 või suurem.
Meso-heksestrooliga saadud katsetulemused loetakse ebapiisavateks andmetest, sest ainus vastus, mis jääb alla 8 000 RLU, ilmneb kontsentratsioonil 1,00 × 10-5 M.
|
41.
|
EC50 ja IC50 saab arvutada nelja parameetriga Hilli funktsiooni abil (täpsem teave on agonistikatse ja antagonistikatse eeskirjades (7)). Nõuetekohasuse kriteeriumide täitmine näitab, et süsteem toimib korrektselt, kuid see ei taga täpsete andmete saamist mis tahes konkreetsel katsel. Esimese katse tulemuste kordamine on parim võimalus tagada täpsete andmete saamine (vt punkti 19 jaotises „ER TA katsemeetodi osad“).
|
KATSEPROTOKOLL
|
42.
|
Vt punkti 20 jaotises „ER TA katsemeetodi osad“.
|
KIRJANDUS
|
(1)
|
ICCVAM (2011), „ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists“, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5367-5373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5941.
|
|
(5)
|
Balls, M. et al. (2006), „The Importance of Good Cell Culture Practice (GCCP)“, ALTEX, nr 23 (Lisa), lk 270–273.
|
|
(6)
|
Coecke, S. et al. (2005), „Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice“, Alternatives to Laboratory Animals, nr 33, lk 261–287.
|
|
(7)
|
ICCVAM (2011), „ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals“, NIH Publication nr 11–7850.
|
|
(8)
|
Rogers, J.M., Denison, M.S. (2000), „Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals“, In Vitro Mol. Toxicol., nr 13(1), lk 67–82.
|
|
(9)
|
Escande, A. et al. (2006), „Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta“, Biochem. Pharmacol., nr 71(10), lk 1459–69.
|
|
(10)
|
Thorne, N., Inglese, J., Auld, D. S. (2010), „Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology“, Chemistry and Biology nr 17(6), lk 646–57.
|
|
(11)
|
Kuiper, G. G. et al. (1998), „Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta“, Endocrinology, nr 139(10), lk 4252–63.
|
|
(12)
|
Geisinger et al. (1989), „Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors“, Cancer, nr 63, lk 280–288.
|
|
(13)
|
Baldwin et al. (1998), „BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro“, In Vitro Cell. Dev. Biol. – Animal, nr 34, lk 649–654.
|
|
(14)
|
Li, Y. et al. (2014), „Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant“, Mol. Endo., nr 28, lk 2072–2081.
|
|
(15)
|
Rogers, J. M. ja Denison, M. S. (2000), „Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals“, In Vitro & Molec. Toxicol., nr 13, lk 67–82.
|
4. Liide
STABIILSE TRANSFEKTSIOONIGA INIMESE ÖSTROGEENI Α-RETSEPTORI TRANSAKTIVATSIOONI (TA) KATSEMEETOD, MILLES UURITAKSE KEMIKAALIDE TOIMET ÖSTROGEENI AGONISTI VÕI ANTAGONISTINA, KASUTADES RAKULIINI ERΑ CALUX
LÄHTEKAALUTLUSED JA PIIRANGUD (VT KA ÜLDIST SISSEJUHATUST)
|
1.
|
ERα CALUX transaktivatsiooni katsemeetodis kasutatakse inimese rakuliini U2OS, et tuvastada toime östrogeeni agonistina või antagonistina, mida vahendab inimese östrogeeni α-retseptor (hERα). Stabiilse transfektsiooniga ERα CALUX biokatsemeetodi valideerimisuuring, mille tegi BioDetection Systems BV (Amsterdam, Madalmaad), tõendas katsemeetodi asjakohasust ja usaldusväärsust ettenähtud eesmärgil kasutades (1). Rakuliin ERα CALUX ekspresseerib ainult stabiilse transfektsiooniga inimese ERα-d (2) (3).
|
|
2.
|
See katsemeetod on välja töötatud spetsiaalselt hERα-vahendatud transaktivatsiooni tuvastamiseks, kasutades mõõdetava parameetrina bioluminestsentsi. Bioluminestsentsi kasutamine on suure signaali-müra suhte tõttu biokatsetes üldlevinud (4).
|
|
3.
|
Üle 1 μM fütoöstrogeeni kontsentratsioonid teadaolevalt üleaktiveerivad lutsiferaasi reportergeeni, mille tulemusena toimub luminestsents, mida ei vahenda retseptorid (5, 6, 7). Seetõttu tuleb stabiilse transfektsiooniga ERi transaktivatsiooni katsesüsteemides hoolikalt uurida fütoöstrogeenide või muude sarnaste ühendite suuri kontsentratsioone, mis võivad lutsiferaasi ekspressiooni üleaktiveerida (vt 2. liide).
|
|
4.
|
Enne käesoleva katsemeetodi regulatiivsel eesmärgil kasutamist tuleks lugeda peatükke „Üldine sissejuhatus“ ja „ER TA katsemeetodi osad“. Käesolevas katsemeetodis kasutatud mõisted ja lühendid on esitatud 1. liites.
|
KATSEMEETODI PÕHIMÕTE (VT KA ÜLDIST SISSEJUHATUST)
|
5.
|
Biokatsemeetodit kasutatakse ERi ligandi sidumise ja sellele järgneva retseptori-ligandi kompleksi tuuma suunatud translokatsiooni hindamiseks. Tuumas seondub retseptori-ligandi kompleks DNA spetsiifiliste vastuselementidega ja transaktiveerib jaanimardika lutsiferaasi reportergeeni, mille tulemusena suureneb ensüüm lutsiferaasi ekspressioon rakus. Pärast lutsiferaasi substraadi lutsiferiini lisamist muundatakse lutsiferiin bioluminestseeruvaks saaduseks. Eralduvat valgust on luminomeetri abil lihtne tuvastada ja kvantitatiivselt mõõta.
|
|
6.
|
Katsesüsteemis kasutatakse stabiilse transfektsiooniga ERα CALUX rakke. ERα CALUX rakud on pärit inimese osteoblastilise osteosarkoomi rakuliinist U2OS. Inimese U2OS rakud transfekteeriti stabiilselt 3xHRE-TATA-Luci ja pSG5-neo-hERα-ga, kasutades kaltsiumfosfaadiga kaassadestamise meetodit. Rakuliin U2OS valiti välja kui parim östrogeenile (ja teistele steroidhormoonidele) reageeriv reporterrakuliin, mis põhineb tähelepanekul, et rakuliinil U2OS on retseptori endogeenne aktiivsus väike või puudub hoopis. Endogeensete retseptorite puudumist hinnati ainult lutsiferaasi reporterplasmiidide abil, mis ei ilmutanud retseptorligandide lisamisel mingit aktiivsust. Peale selle toetas nimetatud rakuliin tugevat hormoonvahendatud vastust, kui sellesse viidi ajutiselt sugulasretseptoreid (2, 3, 8).
|
|
7.
|
Kemikaalide östrogeense või antiöstrogeense toime uurimine rakuliini ERα CALUX abil hõlmab eelnevat sõelkatset ja tervikkatseid. Eelneva sõelkatse käigus määratakse uuritavate kemikaalide lahustuvus, tsütotoksilisus ja täpsustatud, tervikkatses uuritavate kontsentratsioonide vahemik. Tervikkatsete käigus analüüsitakse uuritavaid kemikaale täpsustatud kontsentratsioonide vahemikus ERα CALUX biokatsemeetodi abil, millele järgneb uuritavate kemikaalide klassifitseerimine agonistideks ja antagonistideks.
|
|
8.
|
Andmete tõlgendamise kriteeriumeid on üksikasjalikult kirjeldatud punktis 59. Lühidalt loetakse uuritav kemikaal antagonismi seisukohast positiivseks, kui vähemalt kaks uuritava kemikaali järjestikust kontsentratsiooni annavad vastuse, mis on suurem või võrdne 10 %ga etalonaine 17β-östradiooliga saadud maksimaalsest vastusest (PC10). Uuritav kemikaal loetakse antagonismi seisukohast positiivseks, kui vähemalt kaks uuritava kemikaali järjestikust kontsentratsiooni annavad vastuse, mis on väiksem või võrdne 80 %ga etalonaine tamoksüfeeniga saadud maksimaalsest vastusest (PC80).
|
METOODIKA
Rakuliinid
|
9.
|
Katsemeetodiga tuleks kasutada stabiilse transfektsiooniga rakuliini U2OS ERα CALUX. Rakuliin on tehnilise litsentsilepingu alusel saadaval ettevõttest BioDetection Systems BV, asukohaga Amsterdam, Madalmaad.
|
|
10.
|
Kasutada tuleks ainult mükoplasmavabu rakukultuure. Rakupartiidel peab olema sertifikaat, mis tõendab, et need ei ole mükoplasmaga saastunud, või tuleb enne kasutamist mükoplasma olemasolu katseliselt kontrollida. Mükoplasmainfektsiooni tuvastamiseks tuleks kasutada tundlikku RT-PCR-meetodit (polümeraasiahela reaktsioon reaalajas) (9).
|
Rakuliini stabiilsus
|
11.
|
CALUX-rakkude stabiilsuse ja terviklikkuse säilitamiseks tuleb rakke hoida vedelas lämmastikus (–80 °C). Pärast rakkude sulatamist uue kultuuri külvamiseks tuleks rakke vähemalt kaks korda ümber külvata, enne kui neid kasutatakse kemikaalide agonistliku ja antagonistliku toime hindamiseks östrogeeni suhtes. Rakke ei tohiks ümber külvata rohkem kui 30 passaaži.
|
|
12.
|
Selleks, et jälgida rakuliini stabiilsust ajas, tuleb kontrollida agonistliku ja antagonistliku toime katsesüsteemi reageerimisvõimet, hinnates etalonainega saadud parameetri EC50 või IC50 väärtusi. Peale selle tuleb kontrollida positiivse kontrollprooviga (PC) ja negatiivse kontrollprooviga (NC) saadud suhtelist induktsiooni. Tulemused peavad olema kooskõlas agonistliku toime (tabel 3C) või antagonistliku toime (tabel 4C) hindamiseks tehtava ERα CALUXi biokatse nõuetekohasuse kriteeriumidega. Etalonained ning positiivsed ja negatiivsed kontrollained on agonistliku ja antagonistliku toime hindamiseks esitatud vastavalt tabelites 1 ja 2.
|
Rakkude kasvatamine ja külvamistingimused
|
13.
|
U2OS rakke kasvatatakse söötmes (sööde DMEM/F12 (1 : 1), mis sisaldab indikaatorina fenoolpunast ja millele on lisatud veiselooteseerumit (7,5 %), asendatavaid aminohappeid (1 %), 10 ühikut/ml penitsilliini, streptomütsiini ja selektiivainena genetitsiini (G-418)). Rakud pannakse CO2-inkubaatorisse (5 % CO2), kus temperatuur on 37 °C ja õhuniiskus 100 %. Kui rakud saavutavad 85–95 % konfluentsuse, külvatakse need ümber või valmistatakse ette külvamiseks 96 süvendiga mikrotiiterplaadile. Viimasel juhul suspendeeritakse rakud uuesti kontsentratsiooniga 1 × 105 rakku/ml katsesöötmes (DMEM/F12 (1 : 1), mis ei sisalda fenoolpunast ja millele on lisatud dekstraankattega aktiivsöega töödeldud veiselooteseerumit (5 mahuprotsenti), asendatavaid aminohappeid (1 mahuprotsent), 10 ühikut/ml penitsilliini ja streptomütsiini) ja külvatakse 96 süvendiga mikrotiiterplaadi süvenditesse (100 μl homogeniseeritud rakususpensiooni). Enne uuritava ainega töötlemist inkubeeritakse rakke 24 tundi CO2-inkubaatoris (5 % CO2, 37 °C, õhuniiskus 100 %) Plasttarvikud peavad olema östrogeenivabad.
|
Katse nõuetekohasuse kriteeriumid
|
14.
|
Uuritava(te) kemikaali(de) agonistlikku ja antagonistlikku toimet uuritakse katseseeriate abil. Katseseeria koosneb maksimaalselt kuuest mikrotiiterplaadist. Igas katseseerias on vähemalt üks täisseeria etalonaine lahjendustest, positiivne kontrollproov, negatiivne kontrollproov ja lahusti kontrollproovid. Joonistel 1 ja 2 on esitatud proovide paigutus plaadil agonisti ja antagonisti katseseeriate korral.
|
|
15.
|
Iga etalonaine lahjendust, uuritavat kemikaali, lahusti kontrollproovi ning positiivset ja negatiivset kontrollproovi analüüsitakse kolme paralleelproovina. Kõik need kolm paralleelset analüüsi peavad vastama tabelis 3A ja 4A esitatud nõuetele.
|
|
16.
|
Iga katseseeria esimesel plaadil mõõdetakse etalonaine (agonismi korral 17β-östradiool, antagonismi korral tamoksüfeen) täielikku lahjenduste seeriat. Selleks, et oleks võimalik võrrelda ülejäänud viie mikrotiiterplaadi analüüsi tulemusi etalonaine täieliku kontsentratsiooni-vastuse kõvera punkte sisaldava esimese mikrotiiterplaadi omadega, peavad kõik plaadid sisaldama kolme kontrollproovi: lahusti kontrollproov, uuritava etalonaine suurim kontsentratsioon ning etalonaine ligikaudne kontsentratsioon EC50 (agonismi korral) või IC50 (antagonismi korral). Esimese plaadi ja ülejäänud viie plaadi kontrollproovide keskmine suhtarv peab vastama tabelis 3C (agonismi korral) või 4C (antagonismi korral) esitatud nõuetele.
|
|
17.
|
Iga katseseeria osaks oleva mikrotiiterplaadi kohta arvutatakse z-tegur (10). Z-teguri arvutamiseks kasutatakse etalonaine suurima ja vähima kontsentratsiooniga saadud vastuste abil. Mikrotiiterplaat loetakse nõuetekohaseks, kui see vastab tabelis 3C (agonismi korral) või 4C (antagonismi korral) esitatud nõuetele.
|
|
18.
|
Etalonainega saadud annuse-vastuse kõver peab olema sigmoidne. Etalonaine lahjenduste seeriaga saadud vastustest tuletatud EC50 või IC50 peab vastama tabelis 3C (agonismi korral) või 4C (antagonismi korral) esitatud nõuetele.
|
|
19.
|
Igas katseseerias peab olema positiivne ja negatiivne kontrollproov. Nii positiivse kui ka negatiivse kontrollproovi kohta arvutatud suhteline induktsioon peab vastama tabelis 3C (agonismi korral) või 4C (antagonismi korral) esitatud nõuetele.
|
|
20.
|
Kõigi mõõtmiste käigus tuleb mõõta etalonaine suurima kontsentratsiooni induktsioonitegur, jagades selleks etalonaine 17β-östradiooli suurima kontsentratsiooniga saadud vastuse (suhtelistes valgusühikutes, RLU) keskmise väärtuse lahusti kontrollprooviga saadud vastuse (RLU) keskmise väärtusega. See induktsioonitegur peab vastama kordse induktsiooni miinimumnõuetele, mis on esitatud tabelis 3C (agonismi korral) või 4C (antagonismi korral).
|
|
21.
|
Nõuetekohaseks loetakse ainult need mikrotiiterplaadid, mis vastavad kõigile eespool nimetatud kriteeriumidele; sel juhul võib neid kasutada uuritavate kemikaalide esile kutsutud vastuste hindamiseks.
|
|
22.
|
Nõuetekohasuse kriteeriume kohaldatakse nii eelneva sõelkatse kui ka tervikkatsete suhtes.
|
Tabel 1.
Etalonaine, positiivse kontrollaine (PC) ja negatiivse kontrollaine (NC) kontsentratsioonid agonistliku toime uurimiseks CALUXiga biokatsemeetodiga
|
|
Aine
|
CASi nr
|
Uuritav vahemik (M)
|
|
Etalonaine
|
17β-östradiool
|
50-28-2
|
1 × 10-13 – 1 × 10-10
|
|
Positiivne kontrollaine (PC)
|
17α-metüültestosteroon
|
58-18-4
|
3 × 10-6
|
|
Negatiivne kontrollaine (NC)
|
Kortikosteroon
|
50-22-6
|
1 × 10-8
|
Tabel 2.
Etalonaine, positiivse kontrollaine (PC) ja negatiivse kontrollaine (NC) kontsentratsioonid antagonistliku toime uurimiseks CALUX biokatsemeetodiga
|
|
Aine
|
CASi nr
|
Uuritav vahemik (M)
|
|
Etalonaine
|
Tamoksüfeen
|
10540-29-1
|
3 × 10-9 – 1 × 10-5
|
|
Positiivne kontrollaine (PC)
|
4-hüdroksütamoksüfeen
|
68047-06-3
|
1 × 10-9
|
|
Negatiivne kontrollaine (NC)
|
Resveratrool
|
501-36-0
|
1 × 10-5
|
Tabel 3.
Nõuetekohasuse kriteeriumid agonistliku toime uurimiseks ERα CALUX biokatsemeetodiga
|
A – üksikproove plaadil
|
Kriteerium
|
|
1
|
Kolmes süvendis paralleelproovide (negatiivne kontrollproov, positiivne kontrollproov, kõik uuritava kemikaali ja etalonaine lahjendused, välja arvatud C0) maksimaalne %SD
|
< 15 %
|
|
2
|
Kolmes süvendis paralleelproovide (etalonaine ja uuritava kemikaali lahusti kontrollproovid (C0, SC)) maksimaalne %SD
|
< 30 %
|
|
3
|
Maksimaalne LDH teke tsütotoksilisuse mõõduna.
|
< 120 %
|
|
B – ühe mikrotiiterplaadi ulatuses
|
|
|
4
|
Etalonaine lahusti kontrollproovi (C0, plaat 1) ja uuritava kemikaali lahusti kontrollproovi (SC; plaadid 2–x) suhe
|
0,5 – 2,0
|
|
5
|
Plaadil 1 oleva etalonaine ligikaudse EC50 ja suurima kontsentratsiooni ning plaatidel 2–x olevate etalonaine ligikaudsete EC50 ja suurimate kontsentratsioonide (C4, C8) suhe
|
0,70 – 1,30
|
|
6
|
Iga plaadi z-tegur
|
> 0,6
|
|
C – ühe analüüsiseeria ulatuses (kõik ühe analüüsiseeria plaadid)
|
|
|
7
|
Etalonaine sigmoidne kõver
|
Jah (17ß-östradiool)
|
|
8
|
Etalonaine 17ß-östradiooli EC50 vahemik
|
4 × 10-12 – 4 × 10-11 M
|
|
9
|
17ß-östradiooli suurima kontsentratsiooni vähim kordne induktsioon võrrelduna etalonaine lahusti kontrollprooviga.
|
5
|
|
10
|
Positiivse kontrollproovi suhteline induktsioon (%)
|
> 30 %
|
|
11
|
Negatiivse kontrollproovi suhteline induktsioon (%)
|
< 10 %
|
PC: positiivne kontrollproov; NC: negatiivne kontrollproov; SC: uuritava kemikaali lahusti kontrollproov; C0: etalonaine lahusti kontrollproov; SD: standardhälve; LDH: laktaadi dehüdrogenaas
Tabel 4.
Nõuetekohasuse kriteeriumid antagonistliku toime uurimiseks ERα CALUX biokatsemeetodiga
|
A – üksikproove plaadil
|
Kriteerium
|
|
1
|
Kolmes süvendis paralleelproovide (negatiivne kontrollproov, positiivne kontrollproov, kõik uuritava kemikaali ja etalonaine lahjendused, lahusti kontrollproov C0) maksimaalne %SD
|
< 15 %
|
|
2
|
Kolmes süvendis paralleelproovide (kandeaine kontrollproov (VC) ja etalonaine suurim kontsentratsioon (C8)) maksimaalne %SD
|
< 30 %
|
|
3
|
Maksimaalne LDH teke tsütotoksilisuse mõõduna.
|
< 120 %
|
|
B – ühe mikrotiiterplaadi ulatuses
|
|
|
4
|
Etalonaine lahusti kontrollproovi (C0, plaat 1) ja uuritava kemikaali lahusti kontrollproovi (SC; plaadid 2–x) suhe
|
0,70 – 1,30
|
|
5
|
Plaadil 1 oleva etalonaine ligikaudse IC50 kontsentratsiooni ning plaatidel 2–x olevate etalonaine ligikaudsete IC50 kontsentratsioonide (C4) suhe
|
0,70 – 1,30
|
|
6
|
Plaadil 1 oleva etalonaine suurima kontsentratsiooni ning plaatidel 2–x olevate etalonaine suurimate kontsentratsioonide (C8) suhe
|
0,50 – 2,0
|
|
7
|
Iga plaadi z-tegur
|
> 0,6
|
|
C – ühe analüüsiseeria ulatuses (kõik ühe analüüsiseeria plaadid)
|
|
|
8
|
Etalonaine sigmoidne kõver
|
Jah (tamoksüfeen)
|
|
9
|
Etalonaine (tamoksüfeen) IC50 vahemik
|
1 × 10-8 – 1 × 10-7 M
|
|
10
|
Etalonaine lahusti kontrollproovi vähim kordne induktsioon võrrelduna tamoksüfeeni suurima kontsentratsiooniga.
|
2,5
|
|
11
|
Positiivse kontrollproovi suhteline induktsioon (%)
|
< 70 %
|
|
12
|
Negatiivse kontrollproovi suhteline induktsioon (%)
|
< 85 %
|
PC: positiivne kontrollproov; NC: negatiivne kontrollproov; VC: kandeaine kontrollproov (lahusti kontroll ilma agonisti etalonaine määratud kontsentratsioonita); SC: uuritava kemikaali lahusti kontrollproov; C0: etalonaine lahusti kontrollproov; SD: standardhälve; LDH: laktaadi dehüdrogenaas
Lahusti/kandeaine kontrollproov, etalonained, positiivsed kontrollproovid, negatiivsed kontrollproovid
|
23.
|
Eelnevas sõelkatses ja tervikkatsetes kasutatakse sama lahusti/kandeaine kontrollproovi, samu etalonaineid, positiivseid ja negatiivsed kontrollproove. Lisaks peab etalonainete, positiivsete ja negatiivsete kontrollproovide kontsentratsioon olema sama.
|
Lahusti kontrollproov
|
24.
|
Uuritava kemikaali lahustamiseks kasutatavat lahustit tuleb katseliselt kontrollida lahusti kontrollproovina. ERα CALUX biokatsemeetodi valideerimisel kasutati kandeainena dimetüülsulfoksiidi (DMSO, 1 % (mahuprotsent), CASi nr 67-68-5). Kui DMSO asemel kasutatakse muud lahustit, tuleb katsed kõigi etalonainete, kontrollproovide ja uuritavate kemikaalidega läbi viia samas kandeaines. Antagonistlike omaduste uurimise korral sisaldab lahusti kontrollproov kindlas kontsentratsioonis agonistlikku etalonainet 17β-östradiooli (ligikaudne EC50 kontsentratsioon). Antagonistlike omaduste uurimisel kasutatava lahusti kontrollimiseks valmistatakse kandeaine kontrollproov ja analüüsitakse seda.
|
Kandeaine kontrollproov (antagonism)
|
25.
|
Antagonistlike omaduste uurimiseks lisatakse katsesöötmesse kindlas kontsentratsioonis agonistlikku etalonainet 17β-östradiooli (ligikaudne EC50 kontsentratsioon). Antagonistlike omaduste suhtes uuritavate kemikaalide lahustamiseks kasutatava lahusti katseliseks kontrollimiseks valmistatakse katsesööde ilma kindlas kontsentratsioonis lisatud agonistliku etalonaineta (17β-östradiool). Seda kontrollproovi nimetatakse kandeaine kontrollprooviks. ERα CALUX biokatsemeetodi valideerimisel kasutati kandeainena dimetüülsulfoksiidi (DMSO, 1 % (mahuprotsent), CASi nr 67-68-5). Kui DMSO asemel kasutatakse muud lahustit, tuleb katsed kõigi etalonainete, kontrollproovide ja uuritavate kemikaalidega läbi viia samas kandeaines.
|
Etalonained
|
26.
|
Agonistlik etalonaine on 17β-östradiool (tabel 1). Etalonaineteks on 17β-östradiooli lahjenduste seeria, mis hõlmab kaheksat kontsentratsiooni (1 × 10-13, 3 × 10-13, 1 × 10-12, 3 × 10-12, 6 × 10-12, 1 × 10-11, 3 × 10-11, 1 × 10-10 M).
|
|
27.
|
Antagonistlik etalonaine on tamoksüfeen (tabel 2). Etalonaineteks on tamoksüfeeni lahjenduste seeria, mis hõlmab kaheksat kontsentratsiooni (3 × 10-9, 1 × 10-8, 3 × 10-8, 1 × 10-7, 3 × 10-7, 1 × 10-6, 3 × 10-6, 1 × 10-5 M). Antagonistliku etalonaine kõigis kontsentratsioonides lahjendusi inkubeeritakse koos kindlas kontsentratsioonis lisatud agonistliku etalonainega (17β-östradiool, 3 × 10-12 M)
|
Positiivne kontrollproov
|
28.
|
Agonistlike omaduste uurimise korral on positiivseks kontrollaineks 17α-metüültestosteroon (tabel 1).
|
|
29.
|
Antagonistlike omaduste uurimise korral on positiivseks kontrollaineks 4-hüdroksütamoksüfeen (tabel 2). Antagonistlikku positiivset kontrollproovi inkubeeritakse koos kindlas kontsentratsioonis lisatud agonistliku etalonainega (17β-östradiool, 3 × 10-12 M)
|
Negatiivne kontrollproov
|
30.
|
Agonistlike omaduste uurimise korral on negatiivseks kontrollaineks kortikosteroon (tabel 1).
|
|
31.
|
Antagonistlike omaduste uurimise korral on negatiivseks kontrollaineks resveratrool (tabel 2). Antagonistlikku negatiivset kontrollproovi inkubeeritakse koos kindlas kontsentratsioonis lisatud agonistliku etalonainega (17β-östradiool, 3 × 10-12 M)
|
Labori pädevuse tõendamine (vt käesoleva katsemeetodi jaotise „ER TA katsemeetodi osad“ punkti 14 ning tabeleid 3 ja 4).
Kandeaine
|
32.
|
Uuritava kemikaali lahustamiseks kasutatav lahusti peab uuritava kemikaali täielikult lahustama ja segunema rakkude kasvusöötmega. Sobivad kandeained on DMSO, vesi ja etanool (puhtusega 95–100 %). Kui lahustina kasutatakse DMSOd, ei tohi DMSO maksimaalne kontsentratsioon inkubeerimise ajal ületada 1 mahuprotsenti. Enne kasutamist tuleb katseliselt kontrollides veenduda, et lahusti ei ole tsütotoksiline ega häiri katsemeetodi toimivust.
|
Etalonainete, positiivsete ja negatiivsete kontrollproovide ning uuritavate kemikaalide ettevalmistamine
|
33.
|
Etalonained, positiivsed ja negatiivsed kontrollained ning uuritavad kemikaalid lahustatakse 100 % DMSOs (või sobivas lahustis). Seejärel valmistatakse samas lahustis asjakohased lahjendused (seeriana). Enne lahustamist lastakse kõigil ainetele saavutada toatemperatuur. Etalonainete, positiivsete ja negatiivsete kontrollainete ning uuritava kemikaalide värskelt valmistatud põhilahustes ei tohi olla nähtavat sadet ega hägu. Etalonaine ja kontrollainetega võib valmistada suurema koguse põhilahust. Uuritavate kemikaalide põhilahused valmistatakse värskelt enne igat katset. Etalonainete, positiivsete ja negatiivsete kontrollainete ja uuritavate kemikaalide lõplikud lahjendused tuleb valmistada värskelt enne igat katset ning kasutada 24 tunni jooksul valmistamisest alates.
|
Lahustuvus, tsütotoksilisus ja vahemiku leidmine
|
34.
|
Eelneva sõelkatsega määratakse uuritava kemikaali lahustuvus valitud lahustis. Valmistatakse põhilahus maksimaalse kontsentratsiooniga 0,1 M. Kui selle kontsentratsiooni juures tekib probleeme lahustuvusega, valmistatakse lahjemaid põhilahuseid, kuni uuritavad kemikaalid täielikult lahustuvad. Eelneva sõelkatse käigus analüüsitakse uuritava kemikaali lahjenduste seeriat, mis valmistatakse vahekorras 1 : 10. Maksimaalne analüüsitav kontsentratsioon agonistlike ja antagonistlike omaduste uurimisel on 1 mM. Pärast eelnevat sõelkatset tuletatakse täpsem sobivate kontsentratsioonide vahemik, mida tervikkatsete käigus analüüsida. Tervikkatsetes kasutatavad lahjendused peaksid olema 1-, 3-, 10-, 30-, 100-, 300-, 1000- ja 3000-kordsed.
|
|
35.
|
Agonisti- ja antagonistikatse eeskirjad (11) hõlmavad tsütotoksilisuse uurimist. Tsütotoksilisuse katseline kontrollimine on ka eelneva sõelkatse ja tervikkatsete osa. ERα CALUXi biokatsemeetodi valideerimisel tsütotoksilisuse hindamiseks kasutatud meetod oli laktaadi dehüdrogenaasi (LDH) eraldumise katse koos rakkude kvalitatiivse visuaalse kontrolliga (vt liide 4.1) pärast uuritava kemikaaliga töötlemist. Tsütotoksilisuse määramiseks võib kasutada ka muid kvantitatiivseid meetodeid (nt kolorimeetriline (MTT) meetod tetrasooliumiga, või CALUXi tsütotoksilisuse biokatse). Üldiselt loetakse tsütotoksiliseks uuritava kemikaali kontsentratsioon, mille juures rakkude eluvõimelisus väheneb rohkem kui 20 % ning mida seetõttu ei saa andmete hindamisel kasutada. LDH eraldumise katse korral loetakse uuritava kemikaali kontsentratsioon tsütotoksiliseks, kui LDH eraldumine on üle 120 %.
|
Uuritava kemikaaliga kokkupuude ja analüüsiplaadi jaotus
|
36.
|
Pärast kolvitäie konfluentse rakukultuuri trüpsineerimist suspendeeritakse rakud uuesti kontsentratsiooniga 1 × 105 rakku/ml östrogeenivabas katsesöötmes. 100 μl uuesti suspendeeritud rakke külvatakse 96 süvendiga mikrotiiterplaadi seesmistesse süvenditesse. Välimised süvendid täidetakse 200 μl fosfaadiga puhverdatud füsioloogilise lahusega (PBS) (vt joonised 1 ja 2). Plaadile külvatud rakke inkubeeritakse 24 tundi CO2-inkubaatoris (5 % CO2, 37 °C, õhuniiskus 100 %).
|
|
37.
|
Pärast inkubeerimist kontrollitakse plaate visuaalselt tsütotoksilisuse (vt liide 4.1), saastumise ja konfluentsuse suhtes. Katse läbiviimiseks kasutatakse ainult neid plaate, millel ei ole märke tsütotoksilisusest ega saastumisest ja mille konfluentsus on vähemalt 85 %. Seesmistest süvenditest eemaldatakse hoolikalt sööde ning asendatakse see 200 μl östrogeenivaba katsesöötmega, mis sisaldab asjakohases seeriana saadud lahjendusastmes etalonaineid, uuritavaid kemikaale, positiivseid ja negatiivseid kontrollaineid ning lahusti kontrollproovi (tabel 5: agonistlike omaduste uuring; tabel 6: antagonistlike omaduste uuring). Kõiki etalonaineid, uuritavaid kemikaale, positiivseid janegatiivseid kontrollproove ning lahusti kontrollproove analüüsitakse kolme paralleelproovina. Joonisel 1 on esitatud plaadi paigutus agonistikatse korral. Joonisel 2 on esitatud plaadi paigutus antagonistikatse korral. Eelneva sõelkatse ja tervikkatsete korral on plaadi paigutus samasugune. Antagonistikatse korral sisaldavad kõik seesmised süvendid peale kandeaine kontrollproovidega süvendite (VC) ka kindlas kontsentratsioonis agonistlikku etalonainet 17β-östradiooli (3 × 10-12 M). Etalonained C8 ja C4 tuleb lisada igale uuritava kemikaali proove sisaldavale plaadile.
|
|
38.
|
Pärast rakkude töötlemist kõigi kemikaalidega inkubeeritakse 96 süvendiga mikrotiiterplaati veel 24 tundi CO2-inkubaatoris (5 % CO2, 37 °C, õhuniiskus 100 %).
|
Joonis 1.
Proovide paigutus 96 süvendiga mikrotiiterplaadil eelneva sõelkatse ja agonistliku toime hindamise korral.
C0 = etalonaine lahusti.
C(1–8) = etalonaine lahjenduste seeria (1–8, vähimast kontsentratsioonist suurimani).
PC = positiivne kontrollproov.
NC = negatiivne kontrollproov.
TCx-(1–8) = uuritava kemikaali lahjendused (1–8, vähimast kontsentratsioonist suurimani) eelnevaks sõelkatseks ja uuritava kemikaali × agonistliku toime hindamiseks.
SC = uuritava kemikaali lahusti kontrollproov (optimaalne on sama lahusti, mis proovis C0, kuid see võib olla teisest põhilahusest).
Hallid väljad = välimised süvendid, mis täidetakse 200 μl PBSiga.
Joonis 2.
Proovide paigutus 96 süvendiga mikrotiiterplaadil antagonistliku toime uuringule eelneva sõelkatse ja antagonistliku toime hindamise korral.

C0 = etalonaine lahusti.
C(1–8) = etalonaine lahjenduste seeria (1–8, vähimast kontsentratsioonist suurimani).
NC = negatiivne kontrollproov.
PC = positiivne kontrollproov.
TCx-(1–8) = uuritava kemikaali lahjendused (1–8, vähimast kontsentratsioonist suurimani) eelnevaks sõelkatseks ja uuritava kemikaali × agonistliku toime hindamiseks.
SC = uuritava kemikaali lahusti kontrollproov (optimaalne on sama lahusti, mis proovis C0, kuid see võib olla teisest põhilahusest).
VC = kandeaine kontrollproov (lahusti kontroll ilma agonisti etalonaine 17β-östradiooli määratud kontsentratsioonita).
Hallid väljad = välimised süvendid, mis täidetakse 200 μl PBSiga.
Märkus: kõik seesmised süvendid peale kandeaine kontrollproovidega süvendite (VC) sisaldavad ka kindlas kontsentratsioonis agonistlikku etalonainet 17β-östradiooli (3,0 × 10-12 M).
Luminestsentsi mõõtmine
|
39.
|
Luminestsentsi mõõtmist on üksikasjalikult kirjeldatud agonisti- ja antagonistikatse eeskirjades (10). Süvenditest eemaldatakse sööde ja rakud lüüsitakse, millele järgneb 24 tundi inkubeerimist, et rakumembraan avaneks ja lutsiferaasi aktiivsust oleks võimalik mõõta.
|
|
40.
|
Luminestsentsi mõõtmiseks on vaja kahe injektoriga luminomeetrit. Lutsiferaasi reaktsioon käivitatakse substraadi lutsiferiini süstimisega. Reaktsioon peatatakse 0,2 M NaOH lisamisega. Reaktsiooni peatatakse, et vältida luminestsentsi ülekandumist ühest süvendist teise.
|
|
41.
|
Igast süvendist eraldunud valgus väljendatakse suhtelistes valgusühikutes (RLU) süvendi kohta.
|
Eelnev sõelkatse
|
42.
|
Eelnevas sõelkatses saadud analüüsitulemusi kasutatakse uuritavate kemikaalide täpsema kontsentratsioonivahemiku määramiseks tervikkatsete jaoks. Eelnevas sõelkatses saadud analüüsitulemuste hindamist ja uuritavate kemikaalide täpsema kontsentratsioonivahemiku määramist tervikkatsete jaoks on süvitsi kirjeldatud agonisti- ja antagonistikatse eeskirjades (10). Siin on esitatud lühikokkuvõte eeskirjadest, mida järgides määratakse uuritavate kemikaalide kontsentratsioonivahemik agonisti- ja antagonistikatse jaoks. Lahjendusteseeria ülesehituse juhised on esitatud tabelites 5 ja 6.
|
Kontsentratsioonide valimine agonistliku toime hindamiseks
|
43.
|
Eelnevas sõelkatses analüüsitakse uuritavaid kemikaale, kasutades tabelites 5 (agonism) ja 6 (antagonism) esitatud lahjendusteseeriaid. Kõiki kontsentratsioone analüüsitakse paralleelproovidena kolmes süvendis vastavalt joonisel 1 (agonism) või 2 (antagonism) esitatud proovide paigutusele plaadil.
|
|
44.
|
Nõuetekohaseks loetakse ainult need analüüsitulemused, mis vastavad nõuetekohasuse kriteeriumidele (tabel 3); sel juhul võib neid kasutada uuritavate kemikaalide esile kutsutud vastuste hindamiseks. Kui üks või mitu mikrotiiterplaati analüüsiseerias ei vasta nõuetekohasuse kriteeriumidele, tuleb vastavaid mikrotiiterplaate uuesti analüüsida. Kui nõuetekohasuse kriteeriumidele ei vasta esimene plaat, mis sisaldab etalonaine lahjenduste täielikku seeriat, tuleb uuesti analüüsida kogu katseseeria (6 plaati).
|
|
45.
|
Uuritavate kemikaalide kontsentratsioonide vahemikku tuleb kohandada ja eelnevat sõelkatset korrata, kui:
|
—
|
ilmneb tsütotoksilisus. Eelnevalt sõelkatset tuleb korrata uuritava kemikaali väiksema kontsentratsiooniga, mis ei ole tsütotoksiline;
|
|
—
|
uuritava kemikaali eelnevas sõelkatses ei saa täielikku doosi-vastuse kõverat, sest uuritavad kontsentratsioonid annavad maksimaalse induktsiooni. Eelnevalt sõelkatset tuleb korrata uuritava kemikaali väiksema kontsentratsiooniga.
|
|
|
46.
|
Kui leitakse nõuetekohane doosi-vastuse seos, tuleb valida (vähim) kontsentratsioon, mille juures saadakse maksimaalne induktsioon ega esine tsütotoksilisust. Tervikkatsetes analüüsitav uuritava kemikaali suurim kontsentratsioon peaks olema kõnealusest valitud kontsentratsioonist 3 korda suurem.
|
|
47.
|
Uuritava kemikaali täielik täpsem lahjenduste seeria valmistatakse tabelis 5 esitatud lahjendusetappide kohaselt, alustatud eespool määratud suurimast kontsentratsioonist.
|
|
48.
|
Uuritavat kemikaali, mille puhul ei ilmne mingit agonistlikku toimet, tuleks tervikkatsetes analüüsida, alustades eelnevas sõelkatses määratud suurima kontsentratsiooniga, mis ei ole tsütotoksiline.
|
Kontsentratsioonide valimine antagonistliku toime hindamiseks
|
49.
|
Nõuetekohaseks loetakse ainult need analüüsitulemused, mis vastavad nõuetekohasuse kriteeriumidele (tabel 4); sel juhul võib neid kasutada uuritavate kemikaalide esile kutsutud vastuste hindamiseks. Kui üks või mitu mikrotiiterplaati analüüsiseerias ei vasta nõuetekohasuse kriteeriumidele, tuleb vastavaid mikrotiiterplaate uuesti analüüsida. Kui nõuetekohasuse kriteeriumidele ei vasta esimene plaat, mis sisaldab etalonaine lahjenduste täielikku seeriat, tuleb uuesti analüüsida kogu katseseeria (6 plaati).
|
|
50.
|
Uuritavate kemikaalide kontsentratsioonide vahemikku tuleb kohandada ja eelnevat sõelkatset korrata, kui:
|
—
|
ilmneb tsütotoksilisus. Eelnevalt sõelkatset tuleb korrata uuritava kemikaali väiksema kontsentratsiooniga, mis ei ole tsütotoksiline;
|
|
—
|
uuritava kemikaali eelnevas sõelkatses ei saa täielikku doosi-vastuse kõverat, sest uuritavad kontsentratsioonid annavad maksimaalse inhibitsiooni. Eelnevalt sõelkatset tuleb korrata uuritava kemikaali väiksema kontsentratsiooniga.
|
|
|
51.
|
Kui leitakse nõuetekohane doosi-vastuse seos, tuleb valida (vähim) kontsentratsioon, mille juures saadakse maksimaalne inhibitsioon ega esine tsütotoksilisust. Tervikkatsetes analüüsitav uuritava kemikaali suurim kontsentratsioon peaks olema kõnealusest valitud kontsentratsioonist 3 korda suurem.
|
|
52.
|
Uuritava kemikaali täielik täpsem lahjenduste seeria valmistatakse tabelis 6 esitatud lahjendusetappide kohaselt, alustatud eespool määratud suurimast kontsentratsioonist.
|
|
53.
|
Uuritavat kemikaali, mille puhul ei ilmne mingit antagonistlikku toimet, tuleks tervikkatsetes analüüsida, alustades eelnevas sõelkatses uuritud suurima kontsentratsiooniga, mis ei ole tsütotoksiline.
|
Tervikkatsed
|
54.
|
Pärast täpsema kontsentratsioonivahemiku valimist analüüsitakse uuritavaid kemikaale tervikkatsetes, kasutades tabelites 5 (agonism) ja 6 (antagonism) esitatud lahjendusteseeriaid. Kõiki kontsentratsioone analüüsitakse paralleelproovidena kolmes süvendis vastavalt joonisel 1 (agonism) või 2 (antagonism) esitatud proovide paigutusele plaadil.
|
|
55.
|
Nõuetekohaseks loetakse ainult need analüüsitulemused, mis vastavad nõuetekohasuse kriteeriumidele (tabelid 3 ja 4); sel juhul võib neid kasutada uuritavate kemikaalide esile kutsutud vastuste hindamiseks. Kui üks või mitu mikrotiiterplaati analüüsiseerias ei vasta nõuetekohasuse kriteeriumidele, tuleb vastavaid mikrotiiterplaate uuesti analüüsida. Kui nõuetekohasuse kriteeriumidele ei vasta esimene plaat, mis sisaldab etalonaine lahjenduste täielikku seeriat, tuleb uuesti analüüsida kogu katseseeria (6 plaati).
|
Tabel 5.
Etalonainete, kontrollainete ja uuritavate kemikaalide kontsentratsioonid ja lahjendused agonistikatse jaoks
|
Etalonaine 17β-östradiool
|
TCx – eelkatse
|
TCx – tervikkatse
|
Kontrollproovide
|
|
konts. (M)
|
lahjendus
|
Lahjendus
|
konts. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 ×
|
TCx-1
|
3 000 ×
|
PC
|
3 × 1006
|
|
C1
|
1 × 10-13
|
TCx-2
|
1 000 000 ×
|
TCx-2
|
1 000 ×
|
NC
|
1 × 10-8
|
|
C2
|
3 × 10-13
|
TCx-3
|
100 000 ×
|
TCx-3
|
300 ×
|
C0
|
0
|
|
C3
|
1 × 10-12
|
TCx-4
|
10 000 ×
|
TCx-4
|
100 ×
|
SC
|
0
|
|
C4
|
3 × 10-12
|
TCx-5
|
1 000 ×
|
TCx-5
|
30 ×
|
|
|
|
C5
|
6 × 10-12
|
TCx-6
|
100 ×
|
TCx-6
|
10 ×
|
|
|
|
C6
|
1 × 10-11
|
TCx-7
|
10 ×
|
TCx-7
|
3 ×
|
|
|
|
C7
|
3 × 10-11
|
TCx-8
|
1 ×
|
TCx-8
|
1 ×
|
|
|
|
C8
|
1 × 10-10
|
|
|
|
|
|
|
|
TCx – uuritav kemikaal x
PC – positiivne kontrollproov (17α-metüültestosteroon)
NC – negatiivne kontrollproov (kortikosteroon)
C0 – etalonaine lahusti kontrollproov
SC – uuritava kemikaali lahusti kontrollproov
|
Tabel 6.
Etalonainete, kontrollainete ja uuritavate kemikaalide kontsentratsioonid ja lahjendused antagonistikatse jaoks
|
Etalonaine tamoksüfeen
|
TCx – eelkatse
|
TCx – tervikkatse
|
Kontrollproovide
|
|
konts. (M)
|
lahjendus
|
lahjendus
|
konts. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 ×
|
TCx-1
|
3 000 ×
|
PC
|
1 × 10-9
|
|
C1
|
3 × 10-09
|
TCx-2
|
1 000 000 ×
|
TCx-2
|
1 000 ×
|
NC
|
1 × 10-5
|
|
C2
|
1 × 10-08
|
TCx-3
|
100 000 ×
|
TCx-3
|
300 ×
|
C0
|
0
|
|
C3
|
3 × 10-08
|
TCx-4
|
10 000 ×
|
TCx-4
|
100 ×
|
SC
|
0
|
|
C4
|
1 × 10-07
|
TCx-5
|
1 000 ×
|
TCx-5
|
30 ×
|
|
|
|
C5
|
3 × 10-07
|
TCx-6
|
100 ×
|
TCx-6
|
10 ×
|
Lisatud agonisti
|
|
C6
|
1 × 10-06
|
TCx-7
|
10 ×
|
TCx-7
|
3 ×
|
konts. (M)
|
|
C7
|
3 × 10-06
|
TCx-8
|
1 ×
|
TCx-8
|
1 ×
|
17β-östradiool
|
3 × 10-12
|
|
C8
|
1 × 10-05
|
|
|
|
|
|
|
|
TCx – uuritav kemikaal x
PC – positiivne kontrollproov (4-hüdroksütamoksüfeen)
NC – negatiivne kontrollproov (resveratrool)
C0 – etalonaine lahusti kontrollproov
SC – uuritava kemikaali lahusti kontrollproov
VC – kandeaine kontrollproov (ei sisalda kindlas kontsentratsioonis lisatud agonistlikku etalonainet (17β-östradiool, 3,0 × 10-12 M)
|
Andmete kogumine ja analüüs
|
56.
|
Pärast eelnevat sõelkatset ja tervikkatseid määratakse agonistikatse korral uuritava kemikaali EC10, EC50, PC10, PC50 ning maksimaalset induktsiooni esile kutsuv kontsentratsioon (TCxmax). Antagonistikatse korral arvutatakse IC20, IC50, PC80, PC50 ja minimaalset induktsiooni esile kutsuv kontsentratsioon (TCxmin). Joonistel 3 (agonism) ja 4 (antagonism) on need parameetrid graafiliselt esitatud. Nõutavad parameetrid arvutatakse iga uuritava kemikaali suhtelise induktsiooni (võrreldes etalonaine maksimaalse induktsiooniga (= 100 %)) järgi. Andmete hindamiseks kasutatakse mittelineaarset regressiooni (muutuv tõus, 4 parameetrit) vastavalt järgmisele võrrandile:

kus:
X = doosi või kontsentratsiooni logaritm
Y = vastus (suhteline induktsioon (%))
Ülemine (platoo) = maksimaalne induktsioon (%)
Alumine (platoo) = minimaalne induktsioon (%)
LogEC50 = logaritm kontsentratsioonist, mille juures täheldati 50 % maksimaalsest vastusest
HillSlope = Hilli tõus (Hilli koefitsient)
|
|
57.
|
Luminomeetrist saadud töötlemata andmed tuleb kanda eelneva sõelkatse ja tervikkatsete jaoks välja töötatud arvutustabelisse. Töötlemata andmed peavad vastama nõuetekohasuse kriteeriumidele, mis on esitatud tabelites 3A ja 3B (antagonism) või 4A ja 4B (antagonism). Kui töötlemata andmed vastavad nõuetekohasuse kriteeriumidele, määratakse nõutavad parameetrid järgmisi arvutusetappi järgides.
|
Agonism
|
—
|
Lahutada etalonaine lahusti kontrollproovi keskmine RLU väärtus etalonainete töötlemata analüüsitulemusest.
|
|
—
|
Lahutada uuritava kemikaali lahusti kontrollproovi keskmine RLU väärtus uuritava kemikaali proovide töötlemata analüüsitulemusest.
|
|
—
|
Arvutada etalonaine iga kontsentratsiooni jaoks suhteline induktsioon. Võtta 100 % induktsiooniks etalonaine suurima kontsentratsiooniga saadud induktsioon.
|
|
—
|
Arvutada uuritava kemikaali iga kontsentratsiooniga seotud suhteline induktsioon võrreldes etalonaine suurima kontsentratsiooniga saadud induktsiooniga (100 %).
|
|
—
|
Hinnata analüüsitulemust mittelineaarse regressiooni (muutuv tõus, 4 parameetrit) alusel.
|
|
—
|
Leida etalonaine EC50 ja EC10.
|
|
—
|
Leida uuritavate kemikaalide EC50 ja EC10.
|
|
—
|
Leida uuritava kemikaali maksimaalne suhteline induktsioon (TCmax).
|
|
—
|
Leida uuritavate kemikaalide PC10 ja PC50.
|
Uuritavate kemikaalide puhul ei ole näiteks tsütotoksilisuse või lahustuvusprobleemide tõttu võimalik alati täielikku doosi-vastuse kõverat leida. Sel juhul ei saa EC50, EC10 ja PC50 leida. Siis saab leida üksnes PC10 ja TCmax.
Antagonism
|
—
|
Lahutada etalonaine suurima kontsentratsiooniga saadud keskmine RLU väärtus etalonainete töötlemata analüüsitulemusest.
|
|
—
|
Lahutada etalonaine suurima kontsentratsiooniga saadud keskmine RLU väärtus uuritavate kemikaalide töötlemata analüüsitulemusest.
|
|
—
|
Arvutada etalonaine iga kontsentratsiooni jaoks suhteline induktsioon. Võtta 100 % induktsiooniks etalonaine vähima kontsentratsiooniga saadud induktsioon.
|
|
—
|
Arvutada uuritava kemikaali iga kontsentratsiooniga seotud suhteline induktsioon võrreldes etalonaine vähima kontsentratsiooniga saadud induktsiooniga (100 %).
|
|
—
|
Hinnata analüüsitulemust mittelineaarse regressiooni (varieeruv tõus, 4 parameetrit) alusel.
|
|
—
|
Leida etalonaine IC50 ja IC20.
|
|
—
|
Leida uuritavate kemikaalide IC50 ja IC20.
|
|
—
|
Leida uuritava kemikaali minimaalne suhteline induktsioon (TCmin).
|
|
—
|
Leida uuritavate kemikaalide PC80 ja PC50.
|
Joonis 3.
Agonistikatsega leitud parameetrite ülevaade

EC10 = aine kontsentratsioon, mille juures täheldati 10 % selle maksimaalsest vastusest.
EC50 = aine kontsentratsioon, mille juures täheldati 50 % selle maksimaalsest vastusest.
PC10 = uuritava kemikaali kontsentratsioon, mille juures vastus on võrdne etalonaine EC10 väärtusega.
PC50 = uuritava kemikaali kontsentratsioon, mille juures vastus on võrdne etalonaine EC50 väärtusega.
TCxmax = uuritava kemikaali maksimaalne suhteline induktsioon.
Joonis 4.
Antagonistikatsega leitud parameetrite ülevaade

IC20 = aine kontsentratsioon, mille juures täheldati 80 % selle maksimaalsest vastusest (20 % inhibeerimine).
IC50 = aine kontsentratsioon, mille juures täheldati 50 % selle maksimaalsest vastusest (50 % inhibeerimine).
PC80 = uuritava kemikaali kontsentratsioon, mille juures vastus on võrdne etalonaine IC20 väärtusega.
PC50 = uuritava kemikaali kontsentratsioon, mille juures vastus on võrdne etalonaine IC50 väärtusega.
TCxmin = uuritava kemikaali minimaalne suhteline induktsioon.
Uuritavate kemikaalide puhul ei ole näiteks tsütotoksilisuse või lahustuvusprobleemide tõttu võimalik alati täielikku doosi-vastuse kõverat leida. Sel juhul ei saa IC50, IC20 ja PC50 leida. Siis saab leida üksnes PC20 ja TCmin.
|
58.
|
Tulemused peavad põhinema kahel (või kolmel) sõltumatul katsel. Kui kaks katset annavad võrreldavad ja seega korratavad tulemused, ei ole kolmandat katset vaja teha. Selleks, et olla vastuvõetavad, peavad tulemused:
|
—
|
vastama nõuetekohasuse kriteeriumidele (vt nõuetekohasuse kriteeriumid punktides 14-22),
|
|
Andmete tõlgendamise kriteeriumid
|
59.
|
Andmete tõlgendamiseks ja otsustamiseks, kas uuritav kemikaal lugeda positiivseks või negatiivseks, kasutatakse järgmisi kriteeriume.
|
Iga tervikkatse korral loetakse uuritav kemikaal positiivseks, kui:
|
1.
|
TCmax on suurem kui 10 % etalonainega (REF10) saadud maksimaalsest vastusest või sellega võrdne.
|
|
2.
|
Vähemalt kaks uuritava kemikaali kontsentratsiooniga saadud tulemust on suuremad kui REF10 või sellega võrdsed.
|
|
|
Iga tervikkatse korral loetakse uuritav kemikaal negatiivseks, kui:
|
1.
|
TCmax ei ületa 10 % etalonainega (REF10) saadud maksimaalsest vastusest.
|
|
2.
|
Vähem kui kahe uuritava kemikaali kontsentratsiooniga saadud tulemused on suuremad kui REF10 või sellega võrdsed.
|
|
|
Iga tervikkatse korral loetakse uuritav kemikaal positiivseks, kui:
|
1.
|
TCmin on väiksem kui 80 % etalonainega (REF80 = 20 % inhibeerimine) saadud maksimaalsest vastusest või sellega võrdne.
|
|
2.
|
Vähemalt kaks uuritava kemikaali kontsentratsiooniga saadud tulemust on väiksemad kui REF80 või sellega võrdsed.
|
|
|
Iga tervikkatse korral loetakse uuritav kemikaal negatiivseks, kui:
|
1.
|
TCmin on suurem kui 80 % etalonainega (REF80 = 20 % inhibeerimine) saadud maksimaalsest vastusest.
|
|
2.
|
Vähem kui kahe uuritava kemikaali kontsentratsiooniga saadud tulemused on väiksemad kui REF80 või sellega võrdsed.
|
|
|
|
60.
|
Selleks, et iseloomustada uuritava kemikaali võimet kutsuda esile positiivset vastust, tuleb katseprotokolli kanda toime suurusjärk (agonism: TCmax; antagonism: TCmin) ning kontsentratsioon, mille juures toime avaldub (agonism: EC10, EC50, PC10, PC50; antagonism: IC20, IC50, PC80, PC50).
|
KATSEPROTOKOLL
|
61.
|
Vt punkti 20 jaotises „ER TA katsemeetodi osad“.
|
KIRJANDUS
|
(1)
|
OECD (2016), „Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential“, Environmental, Health and Safety Publications, Series on Testing and Assessment (No 240), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(2)
|
Sonneveld, E., Jansen, H. J., Riteco, J. A., Brouwer, A., van der Burg, B. (2005), „Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays“, Toxicol Sci., nr 83(1), lk 136–148.
|
|
(3)
|
Quaedackers, M. E., van den Brink, C. E., Wissink, S., Schreurs, R. H. M. M., Gustafsson, J. A., van der Saag, P. T. ja van der Burg, B. (2001), „4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ“, Endocrinology, nr 142(3), lk 1156–1166.
|
|
(4)
|
Thorne, N., Inglese, J. ja Auld, D. S. (2010), „Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology“, Chemistry and Biology, nr 17(6), lk 646–57.
|
|
(5)
|
Escande, A., Pillon, A., Servant, N., Cravedi, J. P., Larrea, F., Muhn, P., Nicolas, J. C., Cavaillès, V. ja Balaguer, P.(2006), „Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta“, Biochem. Pharmacol., nr 71, lk 1459–1469.
|
|
(6)
|
Kuiper, G. G., Lemmen, J. G., Carlsson, B., Corton, J. C., Safe, S. H., van der Saag, P. T., van der Burg, B. ja Gustafsson, J. A. (1998), „Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta“, Endocrinol., nr 139, lk 4252–4263.
|
|
(7)
|
Sotoca, A. M., Bovee, T. F. H., Brand, W., Velikova, N., Boeren, S., Murk, A.J., Vervoort, J., Rietjens, I. M. C. M. (2010), „Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism“, J. Steroid. Biochem. Mol. Biol., nr 122, lk 204–211.
|
|
(8)
|
Sonneveld, E., Riteco, J. A. C., Jansen, H. J., Pieterse, B., Brouwer, A., Schoonen, W. G. ja van der Burg, B. (2006), „Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities“, Toxicol. Sci., nr 89(1), lk 173–187.
|
|
(9)
|
Kobayashi, H., Yamamoto, K., Eguchi, M., Kubo, M., Nakagami, S., Wakisaka, S., Kaizuka, M. ja Ishii, H. (1995), „Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products“, J. Vet. Med. Sci., nr 57(4), lk 769–771.
|
|
(10)
|
Zhang, J.-H., Chung, T. D. Y. ja Oldenburg, K. R. (1999), „A simple statistical parameter for use in evaluation and validation of high throuphut screening assays“, J. Biomol. Scr., nr 4, lk 67–73.
|
|
(11)
|
Besselink, H., Middelhof, I. ja Felzel, E. (2014), „Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells“. BioDetection Systems BV (BDS), Amsterdam, Madalmaad.
|
Liide 4.1
RAKKUDE ELUVÕIMELISUSE VISUAALNE KONTROLL
B.67 TÜMIDIINI KINAASI GEENIL PÕHINEVAD IN VITRO GEENIMUTATSIOONIKATSED IMETAJARAKKUDEGA
SISSEJUHATUS
|
1.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 490 (2016). Katsemeetodeid vaadatakse korrapäraselt läbi ja täiendatakse, et võtta arvesse teaduse arengut, regulatiivseid vajadusi ja loomade heaolu. Hiire lümfoomi katse (MLA) ja TK6-katse, milles kasutatakse tümidiini kinaasi (TK) lookust, olid varem hõlmatud katsemeetodiga B.17. Genotoksilisuse uuringute rahvusvahelise seminari (IWGT) MLA ekspertide töörühm on välja töötanud rahvusvaheliselt ühtlustatud soovitused katsemeetodi nõuetekohasuse kriteeriumide ja andmete tõlgendamise kohta MLA korral (1, 2, 3, 4, 5) ja need soovitused on hõlmatud käesoleva uue katsemeetodiga B.67. Käesoleva katsemeetod on kirjutatud MLA jaoks, samuti TK6-katse jaoks, kuna viimases kasutatakse samuti TK lookust. Kuigi MLAd on regulatiivsel eesmärgil laialdaselt kasutatud, on TK6 kasutus palju harvem. Näitajate sarnasusest hoolimata ei ole kaks rakuliini üksteisega asendatavad ning regulatiivsetes kavades võidakse õiguspäraselt väljendada konkreetse regulatiivse eesmärgiga seoses eelistust ühe või teise katse suhtes. Näiteks on MLA valideerimine tõendanud selle sobivust mitte üksnes geenimutatsioonide tuvastamiseks, vaid ka selle kindlakstegemiseks, kas uuritav kemikaal võib indutseerida struktuurse kromosoomiaberratsiooni. Käesolev katsemeetod kuulub geneetilise toksikoloogia katsemeetodite seeriasse. OECD on välja töötanud dokumendi, milles on esitatud kokkuvõtlik teave geneetilise toksikoloogia valdkonna katsete kohta ning ülevaade genotoksilisust käsitlevates OECD katsejuhendites tehtud viimastest muudatustest (6).
|
|
2.
|
Käesoleva meetodi kohaste imetajarakkudega läbi viidavate in vitro geenimutatsioonikatsete eesmärk on tuvastada kemikaalide põhjustatud geenimutatsioone. Kõnealustes katsetes kasutatakse rakuliine, mis võimaldavad tuvastada otsemutatsioone reportergeenides, täpsemalt endogeenses tümidiini kinaasi geenis (inimese rakkudes TK ja näriliserakkudes Tk, käesolevas katsemeetodis viidatakse neile koos kui geenile TK). Käesolev katsemeetod on ette nähtud kasutamiseks kahe rakuliiniga: hiire lümfoomi rakuliin the L5178Y TK+/--3.7.2C (üldise nimetusega L5178Y) ja inimese lümfoblastoidi rakuliin TK6 (üldise nimetusega TK6). Kuigi kahe rakuliini päritolu, rakkude kasvamine, p53-staatus jms varieerub, saab TK-geenimutatsioonikatsed mõlema rakutüübiga läbi viia sarnasel viisil, nagu käesolevas katsemeetodis kirjeldatud.
|
|
3.
|
Tümidiini kinaasi geeni autosoomsus ja heterosügootsus võimaldavad tuvastada eluvõimelisi kolooniaid, mille rakkudel esineb ensüümi tümidiini kinaasi defitsiit mutatsiooni TK+/-
–> TK-/-
tagajärjel. Kõnealune defitsiit võib olla TK-geeni mõjutavate sündmuste tagajärg, kusjuures sündmusteks võivad olla nii geenimutatsioonid (punktmutatsioonid, raaminihkemutatsioonid, väikesed deletsioonid jms) kui ka kromosoomidega toimuvad sündmused (suured deletsioonid, kromosoommutatsioonid ja mitootiline rekombinatsioon). Viimased sündmused väljenduvad heterosügootsuse kaoga, mis on tuumorsupressorgeeni tavaline geneetiline muutus inimesel kasvaja tekke puhul. Teoreetiliselt saab MLAga tuvastada mitoosikäävi kahjustusest ja/või mitootilisest mittelahknemisest põhjustatud terve TK-geeni kandva kromosoomi kadu. Kombineeritud tsütogeneetiline ja molekulaaranalüüs näitavad tõepoolest selgesti, et MLA TK-mutandid on mittelahknemise tagajärg. Kuid tõendite kaalukusest nähtub, et TK-geenimutatsioonikatsetega ei ole võimalik usaldusväärselt tuvastada aneugeene, kui kohaldatakse standardseid tsütotoksilisuse kriteeriume (nagu käesolevas katsemeetodis kirjeldatud), ning seetõttu ei sobi nimetatud katsed aneugeenide tuvastamiseks (7, 8, 9).
|
|
4.
|
TK-geenimutatsioonikatsetes luuakse kaks erinevat TK-mutantide fenotüübiklassi; normaalse kasvuga mutandid, mis kasvavad sama kiirusega nagu TK heterosügootsed rakud, ning aeglase kasvuga mutandid, mille kasvamisel on populatsiooni kahekordistumisaeg pikenenud. Normaalse kasvuga ja aeglase kasvuga mutante nimetatakse MLA korral suurte kolooniate ja väikeste kolooniatega mutantideks ning TK6 korral vara tekkivate kolooniate ja hilja tekkivate kolooniatega mutantideks. Nii suurte kui ka väikeste kolooniatega MLA mutantide molekulaarset ja tsütogeneetilist olemust on uuritud täpsemalt (8, 10, 11, 12, 13). Vara tekkivate kolooniate ja hilja tekkivate kolooniatega TK6 mutantide molekulaarset ja tsütogeneetilist olemust on samuti täpsemalt uuritud (14, 15, 16, 17). Mõlemat tüüpi rakkude aeglase kasvuga mutandid on saanud geneetilise kahjustuse, mis hõlmab oletatavat/oletatavaid kasvu regulaatorgeeni või -geene TK-lookuse lähedal, mille tulemusena kahekordistumise aeg pikeneb ja kolooniad moodustuvad hiljem või on väikesed (18). Aeglase kasvuga mutantide tekkimist on seostatud kemikaalidega, mis põhjustavad suuri struktuurimuutusi kromosoomi tasandil. Rakud, mille kahjustus ei hõlma oletatavat/oletatavaid kasvu regulaatorgeeni või -geene TK-lookuse lähedal, kasvavad vanemrakkudele sarnase kiirusega ja neist saavad normaalse kasvuga mutandid. Peamiselt normaalse kasvuga mutantide tekkimist on seostatud kemikaalidega, mis toimivad peamiselt punktmutageenidena. Sellest tulenevalt on oluline loendada nii aeglase kui ka normaalse kasvuga mutante, et leida kõik mutandid ja saada ülevaade uuritava kemikaali põhjustatud kahjustuste liigist või liikidest (mutageenid vs. klastogeenid) (10, 12, 18, 19).
|
|
5.
|
Katsemeetod on koostatud nii, et see annaks üldist teavet nii MLA kui ka TK6 kohta ning spetsiifilisi suuniseid kummagi katse kohta eraldi.
|
|
6.
|
Kasutatud mõisted on määratletud 1. liites.
|
LÄHTEKAALUTLUSED JA PIIRANGUD
|
7.
|
In vitro katsete puhul tuleb metabolismi aktiveerimiseks üldjuhul kasutada eksogeenset allikat. Eksogeenne metabolismi aktiveerimise süsteem ei võimalda täielikult matkida in vivo tingimusi.
|
|
8.
|
Tuleks hoiduda kasutamast tingimusi, mille puhul võib saada valepositiivseid tulemusi, mis ei ole põhjustatud uuritava kemikaali interaktsioonist raku geneetilise materjaliga (st võib esineda vastastikmõju katsesüsteemiga); sellised tingimused hõlmavad pH või osmolaalsuse muutusi, söötme koostisosadega reageerimist (20, 21) või liiga suurt tsütotoksilisust (22, 23, 24). MLA ja TK6-katse puhul loetakse liiga suureks tsütotoksilisuseks punktis 28 määratletud soovitatavat suurimat tsütotoksilisuse määra ületavat tsütotoksilisust. Lisaks tuleb tähelepanu pöörata sellele, et uuritavad kemikaalid, mis on tümidiini analoogid või käituvad sellisena, võivad raku töötlemise ajal suurendada muteerumissagedust muude spontaansete mutantide selektiivse kasvu teel ning nende nõuetekohaseks hindamiseks tuleb kasutada täiendavaid katsemeetodeid (25).
|
|
9.
|
Tehislike nanomaterjalide uurimiseks võib olla vaja käesolevat katsemeetodit kohandada, kuid neid kohandusi ei ole käesolevas katsemeetodis kirjeldatud.
|
|
10.
|
Enne kui kasutada käesolevat meetodit segu korral, et saada andmeid kavandatud regulatiivse eesmärgi jaoks, tuleb kaaluda, kas see katse annab selle eesmärgi jaoks piisavaid tulemusi ja kui annab, siis miks. Selline kaalumine ei ole vajalik, kui asjaomase segu hindamine on regulatiivse nõudega ette nähtud.
|
|
11.
|
Muteerunud rakud, millel TK
+/- —> TK
-/--mutatsiooni tulemusel puudub ensüümi tümidiini kinaasi aktiivsus, on resistentsed pürimidiini analoogi trifluorotümidiini (TFT) tsütotoksiliste mõjude suhtes. Rakud, kus on olemas TK, on tundlikud TFT suhtes, mis pärsib rakkude ainevahetust ja peatab rakkude edasise jagunemise. Seega on muteerunud rakud võimelised TFT juuresolekul paljunema ja nähtavaid kolooniaid moodustama, kuid ensüümi TK sisaldavad rakud seda ei suuda.
|
KATSE PÕHIMÕTE
|
12.
|
Rakususpensioon viiakse sobivaks ajavahemikuks (vt punkt 33) kokkupuutesse uuritava kemikaaliga – nii koos metabolismi aktiveeriva eksogeense allikaga kui ka ilma selleta (vt punkt 19) – ning seejärel külvatakse rakud ümber, et hinnata tsütotoksilisust ja võimaldada fenotüübi avaldumist enne mutantide selekteerimist. Tsütotoksilisuse hindamiseks kasutatakse MLA korral suhtelist kogukasvu (vt punkt 25) ja TK6 korral suhtelist ellujäämust (vt punkt 26). Töödeldud kultuure hoitakse söötmes piisava aja jooksul, mis on iseloomulik igale rakutüübile (vt punkt 37), võimaldamaks saavutatud mutatsioonide fenotüüpide avaldumist võimalikult lähedal optimaalsele tasemele. Fenotüübi avaldumise järgselt külvatakse teadaolev arv rakke muteerumissageduse määramiseks nii söötmesse, mis sisaldab muteerunud kolooniate tuvastamist võimaldavat selektiivainet, kui ka selektiivaineta söötmesse, et teha kindlaks kloonimistõhusus (eluvõimelisus). Sobiva inkubatsiooniperioodi lõppedes kolooniad loendatakse. Muteerumissagedus arvutatakse muteerunud kolooniate arvu põhjal, mida on korrigeeritud lähtuvalt kloonimistõhususest mutantide selekteerimise ajal.
|
MEETODI KIRJELDUS
Ettevalmistused
Rakud
|
13.
|
MLA korral: kuna MLA on välja töötatud L5178Y rakkude alaliini TK
+/- -3.7.2C kasutades ning selle kasutamine seda katsemeetodit iseloomustab, tuleb MLA korral kasutada seda konkreetset alaliini. Rakuliin L5178Y on saadud DBA-2 hiire tüümuse lümfoomist, mille on esile kutsunud metüülkolantreen (26). Clive ja kaastöötajad töötlesid L5178Y rakke (Clive'i tähistus TK+/+ -3) etüülmetaansulfonaadiga ja eraldasid TK-/- (tähistus TK-/- -3.7) klooni, kasutades selektiivainena bromodeoksüuridiini. TK-/- kloonist eraldati spontaanne TK+/- kloon (tähistus TK+/- -3.7.2.) ja alamkloon (tähistus TK+/--3.7.2C) ning iseloomustati neid MLAs kasutamiseks sobivana (27). Rakuliini karüotüüp on avaldatud (28, 29, 30, 31). Modaalne kromosoomiarv on 40. Leidub üks metatsentrilinekromosoom (t12;13), mis tuleb loendada ühe kromosoomina. Hiire TK lookus asub kromosoomi 11 distaalses otsas. Rakuliinil L5178Y TK
+/- -3.7.2C on mutatsioonid mõlemas p53 alleelis ning see toodab muteerunud p53 valku (32, 33). TK+/--3.7.2C rakuliini p53 seisund mõjutab tõenäoliselt seda, kuidas on katsega võimalik tuvastada ulatuslikke kahjustusi (17).
|
|
14.
|
TK6 korral: TK6 on inimese lümfoblastoidi rakuliin. Vanemrakuliin on Epstein-Barri viiruse toimel transformeerunud rakuliin WI-L2, mis algselt saadi päriliku sferotsütoosiga 5aastaselt meesisikult. Esimene isoleeritud kloon HH4 mutageniseeriti ICR191ga ja saadi TK heterosügootne rakuliin TK6 (34). TK6 rakud on peaaegu diploidsed ja esindav karüotüüp on 47, XY, 13+, t(14; 20), t(3; 21) (35). Inimese TK lookus asub kromosoomi 17 pikas õlas. TK6 on p53 sisaldav rakuliin, sest selle mõlemas alleelis on metsiktüüpi p53 järjestus ning see ekspresseerib üksnes metsiktüüpi p53 valku (36).
|
|
15.
|
Nii MLA kui ka TK6 puhul on soovitatav, et katselabor rakkude tüvikultuuri luues või uuendades veenduks Mycoplasma puudumises, kontrolliks rakkude karüotüüpi või värviks TK lookusega kromosoomid ning kontrolliks populatsiooni kahekordistumise aegu. Katselaboris kasutatavate rakkude puhul tuleb teha kindlaks normaalne rakutsükliks kuluv aeg ja see peab vastama rakkude kohta avaldatud iseloomulikele näitajatele (16, 19, 37). Tüvikultuuri säilitatakse –150 °C juures või madalamal temperatuuril ning kasutatakse kõigi rakkude töökultuuride valmistamiseks.
|
|
16.
|
Enne suure hulga krüosäilitatud töökultuuride valmistamist või otse enne katses kasutamist võib olla vajalik puhastada kultuur eelnevalt leiduvatest muteerunud rakkudest (kui lahusti kontrollproovi muteerumissagedus (MF) jääb juba lubatavatesse piiridesse – vt tabel 2 MLA kohta). Selleks kasutatakse metotreksaati (aminopteriini), et selekteerida rakud, mis ei ole TK defitsiidiga, ning lisatakse kultuurile tümidiini, hüpoksantiini ja glütsiini (L5178Y) või 2’-deoksütsütidiini (TK6), et tagada TK sisaldusega rakkude optimaalne kasv (19, 38, 39) (ja TK6 kohta (40)). Üldised nõuanded rakukultuuride säilitamise hea tava kohta ning konkreetsed nõuanded L5178Y ja TK6 rakkude kohta on esitatud allikates (19, 31, 37, 39, 41). Laboritele, kus on vaja tüvikultuure MLA või TK6 alustamiseks või uusi tüvikultuure, on ligipääsetav hästi iseloomustatud rakkudega rakupank (37).
|
Söötmed ja kultiveerimistingimused
|
17.
|
Mõlema katsemeetodi korral tuleks rakukultuuride kasvatamisel kasutada sobivat söödet ja sobivaid inkubeerimistingimusi (nt kasvunõud, 5 % CO2-sisaldusega niiske atmosfäär, inkubeerimistemperatuur 37 °C). Kultiveerimisel tuleks alati järgida tingimusi, millega tagatakse rakkude püsimine logaritmilises kasvufaasis. On väga oluline valida söötmed ja kasvutingimused nii, et oleks tagatud rakkude optimaalne kasv ekspressiooniperioodil ning optimaalne kloonimine nii muteerunud kui ka muteerumata rakkude puhul. MLA ja TK6 korral on oluline ka see, et kultiveerimistingimused tagaksid nii suurte / vara tekkivate kolooniatega kui ka väikeste / hilja tekkivate kolooniatega Tk mutantide optimaalse kasvu. Rohkem üksikasju kultiveerimise, kaasa arvatud vajaduse kohta hobuseseerum soojusega korralikult inaktiveerida, kui kasutatakse RPMI söödet, on leitavad allikatest (19, 31, 38, 39, 40, 42).
|
Rakukultuuride ettevalmistamine
|
18.
|
Rakkude paljundamiseks tüvikultuurist külvatakse need söötmesse sellise tihedusega, et rakkude eksponentsiaalne kasv suspensioonis jätkuks kogu töötlemis- ja ekspressiooniperioodi vältel.
|
Metabolismi aktiveerimine
|
19.
|
L5178Y jaTK6 rakkude kasutamise korral tuleks kasutada eksogeenseid metabolismisüsteeme, sest nende rakkude endogeenne metaboliseerimisvõime on ebapiisav. Kui ei ole teisiti põhjendatud, soovitatakse vaikevalikuna kõige sagedamini kasutatavat süsteemi, milleks on näriliste (tavaliselt roti) maksast valmistatud postmitokondriline fraktsioon S9, millele on lisatud kofaktor ning mida on töödeldud ensüümide induktoritega, nagu Aroclor 1254 (43, 44, 45) või fenobarbitaali ja β-naftoflavooni kombinatsiooniga (46, 47, 48, 49, 50, 51). Viimati nimetatud kombinatsiooni kasutamine ei ole vastuolus püsivate orgaaniliste saasteainete Stockholmi konventsiooniga (52) ning seeon osutunud segafunktsiooniga oksüdaaside induktorina sama tõhusaks kui Aroclor 1254 (45, 46, 47, 48, 49). S9-fraktsiooni kasutatakse tavaliselt kontsentratsioonivahemikus 1–2 % (mahuprotsent), kuid lõplikus katsesöötmes võib kontsentratsiooni suurendada kuni 10 %-ni (mahuprotsent). Kasutatava eksogeense metabolismi aktiveerimise süsteemi või ensüümiinduktori liigi ja kontsentratsiooni valikut võib mõjutada uuritava kemikaali klass.
|
Uuritava kemikaali ettevalmistamine
|
20.
|
Enne rakkude töötlemist tuleks tahke uuritav kemikaal lahustada sobivas lahustis ja vajaduse korral tuleks saadud lahust lahjendada (vt punkt 21). Vedelat uuritavat kemikaali võib lisada otse katsesüsteemi või seda enne lisamist lahjendada. Gaasilise või lenduva kemikaali kasutamisel tuleks standardmeetodit asjakohaselt muuta, näiteks viia kemikaaliga töötlemine läbi suletud kultiveerimisnõus (53, 54, 55). Uuritav kemikaal tuleks katseks ette valmistada vahetult enne kasutamist, välja arvatud juhul, kui püsivuse andmetest nähtub, et kemikaali säilitamine on vastuvõetav.
|
KATSETINGIMUSED
Lahustid
|
21.
|
Lahusti tuleks valida nii, et oleks tagatud uuritava kemikaali optimaalne lahustumine, kuid lahusti ei mõjutaks samal ajal negatiivselt katse käiku näiteks rakkude kasvukiiruse või uuritava kemikaali omaduste muutmise, kultiveerimisnõuga reageerimise või metabolismi aktiveerimise süsteemi kahjustamise teel. Esmajärjekorras soovitatakse võimaluse korral kaaluda veepõhise lahusti (või söötme) kasutamist. Tavalised lahustid on näiteks vesi või dimetüülsulfoksiid. Üldjuhul ei tohiks töötlusetapis kasutatavas lõppsöötmes orgaanilise lahusti sisaldus ületada 1 % (mahuprotsent) ja veepõhise lahusti (füsioloogiline lahus või vesi) sisaldus ei tohiks ületada 10 % (mahuprotsent). Kui kasutatakse muid kui tavaliselt kasutatavaid lahusteid (nt etanooli või atsetooni), tuleb nende kasutamiseks esitada andmed, millest nähtub nende kokkusobivus uuritava kemikaali ja katsesüsteemiga ning genotoksilise toime puudumine kasutatava kontsentratsiooni juures. Selliste tõendavate andmete puudumisel on oluline lisada katsesse kemikaaliga töötlemata kontrollproovid (vt 1. liide, määratlused), et tõendada valitud lahustist tuleneva kahjuliku või mutageense toime puudumist.
|
TSÜTOTOKSILISUSE MÕÕTMINE JA TÖÖTLEMISKONTSENTRATSIOONIDE VALIMINE
|
22.
|
Uuritava kemikaali suurima kontsentratsiooni valimisel tuleks hoiduda valimast kontsentratsiooni, mille puhul on võimalik saada valepositiivseid tulemusi, näiteks tulenevalt liiga suurest tsütotoksilisusest (vt punkt 28), söötmes sadenemisest (vt punkt 29) või pH või osmolaalsuse olulisest muutumisest (vt punkt 8). Kui uuritav kemikaal muudab lisamise hetkel oluliselt söötme pH-d, võib pH reguleerimiseks lisada töötlemiseks kasutatavasse lõppsöötmesse puhvrit, et hoida ära valepositiivsete tulemuste saamist ja säilitada sobivad kultiveerimistingimused.
|
|
23.
|
Kontsentratsioonide valimisel lähtutakse tsütotoksilisusest ja muudest kaalutlustest (vt punktid 27–30). Ehkki tsütotoksilisuse hindamine eelkatses võib põhikatses kasutatavate kontsentratsioonide täpsemaks kindlakstegemiseks kasulikuks osutuda, ei ole eelkatse tegemine kohustuslik. Põhikatses on tsütotoksilisuse mõõtmine igas kultuuris vajalik ka juhul, kui tsütotoksilisust on eelnevalt juba hinnatud. Kui tehakse vahemiku leidmise katse, peab see hõlmama suurt kontsentratsioonide vahemikku ning selle võib lõpetada kas 1. päeval pärast töötlemist või pikendada seda ekspresseerimiseni ja mutantide selekteerimiseni 2. päeval (kui selgub, et kasutatavad kontsentratsioonid on selleks sobivad).
|
|
24.
|
Tsütotoksilisust tuleks määrata iga üksiku katsekultuuri ja kontrollkultuuri puhul: MLA (2) ja TK6 (15) korral kasutatavad meetodid on määratletud rahvusvaheliselt kokkulepitud tavaga.
|
|
25.
|
Nii MLA agari- kui ka mikrotiiterplaadi variandi korral: tsütotoksilisuse hindamiseks tuleks kasutada suhtelist kogukasvu, mille määratlesid esmakordselt Clive ja Spector 1975. aastal (2). See näitaja hõlmab suhtelist suspensioonikasvu (katsekultuur vs. lahusti kontrollproov) rakkude töötlemise ajal, ekspresseerimisaega ja kloonimise suhtelist tõhusust (katsekultuur vs. lahusti kontrollproov) mutantide selekteerimise ajal (2). Tuleks võtta arvesse, et suhteline suspensioonikasv hõlmab igasugust rakkude arvu võimalikku vähenemist, mis töötlemise ajal katsekultuuris tekib (vt valemid 2. liites).
|
|
26.
|
TK6 korral: tsütotoksilisuse hindamiseks tuleks kasutada suhtelist ellujäämust, st võrrelda vahetult pärast töötlemist välja külvatud rakkude kloonimise tõhusust, mida on kohandatud rakkude arvu võimaliku vähenemise suhtes töötlemise ajal, negatiivsete kontrollproovidega, mille ellujäämuseks loetakse 100 % (vt valem 2. liites).
|
|
27.
|
Kemikaali tuleks hinnata vähemalt neljal nõuetekohasuse kriteeriumidele (sobiv tsütotoksilisuse määr, rakkude arv jne) vastaval katsekontsentratsioonil (see ei hõlma lahusti kontrollproovi ega positiivseid kontrollproove). Katse läbiviimisel on soovitatav kasutada iga katsekontsentratsiooni puhul kahte paralleelkultuuri, ent võib kasutada ka suuremat arvu paralleelkultuure või ühteainsat kultuuri. Iga kontsentratsiooni puhul tuleks paralleelkultuuridega saadud tulemused esitada eraldi, kuid andmeanalüüsi jaoks võib need koondada (55). Uuritavate kemikaalide puhul, mille tsütotoksilisus on väike või millel see puudub, sobivad tavaliselt kontsentratsioonid, mis erinevad üksteisest umbes kaks kuni kolm korda. Tsütotoksilisuse avaldumise korral, tuleb kontsentratsioonid valida nii, et need kataksid tsütotoksilisuse vahemiku alates kontsentratsioonist, mille juures avaldub tsütotoksilisus (nagu on kirjeldatud punktis 28), ja hõlmama kontsentratsioonid, mille juures esineb mõõdukas ja väike (või olematu) tsütotoksilisus. Paljudel uuritavatel kemikaalidel on kontsentratsiooni-vastuse kõver järsu tõusuga ning tsütotoksilisuse täieliku vahemiku katmiseks või kontsentratsioonist-vastuse sõltuvuse üksikasjalikuks uurimiseks võib olla vaja kasutada üksteisest vähem erinevaid kontsentratsioone ja enam kui nelja kontsentratsiooni, eriti olukorras, kus on vaja teha korduskatse (vt punkt 70). Enam kui nelja kontsentratsiooni kasutamine võib osutuda eriti oluliseks, kui kasutatakse ühteainsat kultuuri.
|
|
28.
|
Kui suurima kontsentratsiooni valimisel lähtutakse tsütotoksilisusest, tuleks suurim kontsentratsioon valida nii, et MLA puhul saavutataks suhteline kogukasv 10–20 % ning TK6 puhul suhteline ellujäämus 10–20 % (punkt 67).
|
|
29.
|
Raskesti lahustuva uuritava kemikaali puhul, mis ei ole tsütotoksiline kontsentratsioonidel, mis on väiksemad kui vähim kontsentratsioon, mille juures kemikaal ei lahustu, peaks suurimal analüüsitaval kontsentratsioonil olema pärast uuritava kemikaaliga töötlemise lõppemist täheldatav hägusus või silma või invertmikroskoobiga nähtav sade. Isegi kui tsütotoksilisus avaldub suuremal kontsentratsioonil kui vähim kontsentratsioon, mille juures kemikaal ei lahustu, on soovitatav teha katse ainult ühe sellise kontsentratsiooniga, mille juures tekib hägusus või nähtav sade, sest sade võib toimet moonutada. Kuna MLA ja TK6 korral kasutatakse suspendeeritud kultuure, tuleb eriti hoolikalt veenduda, et sade ei mõjuta katse käiku. Enne katse alustamist võib olla kasulik määrata ka kemikaali lahustuvus söötmes.
|
|
30.
|
Kui ei täheldata ei sadet ega liigset tsütotoksilisust, peaks suurim katsekontsentratsioon olema vähim järgmistest kontsentratsioonidest: 10 mM, 2 mg/ml või 2 μl/ml (57, 58). Kui uuritava kemikaali koostis ei ole määratav, näiteks kui see on tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal (UVCB), keskkonnast saadud ekstrakt vms, võib juhul, kui ei täheldata kemikaali piisavat tsütotoksilisust, olla vaja selle iga koostisosa kontsentratsiooni suurendamiseks kasutada suuremat maksimumkontsentratsiooni (nt 5 mg/ml). Tuleks siiski märkida, et inimravimite puhul võivad need nõuded olla teistsugused (59).
|
Kontrollproovid
|
31.
|
Iga katserežiimi puhul tuleks kasutada paralleelseid negatiivseid kontrollproove (vt punkt 21), mis koosnevad üksnes töötlemiseks kasutatavale söötmele lisatud lahustist ja mida käideldakse samamoodi kui töödeldud kultuure.
|
|
32.
|
Paralleelseid positiivseid kontrollproove on vaja, et tõendada labori pädevust kasutatud katsemetoodika tingimustel määrata mutageene ja eksogeense metaboolse aktivatsiooni süsteemi tõhusust (kui asjakohane) ning tõendada suutlikkust nii väikeste / hilja tekkivate kui ka suure / vara tekkivate TK mutantide piisaval tuvastamisel. Positiivsete kontrollainete näited on esitatud allpool tabelis 1. Kui see on põhjendatud, võib positiivse kontrollainena kasutada mõnda muud ainet. Kuna in vitro genotoksilisuse katsed imetajarakkudega on piisavalt standarditud lühiajaliste (3–4 tundi) töötluste puhul, mis tehakse paralleelkatsetena metaboolse aktivatsiooniga või ilma selleta, kasutades sama kestusega töötlust, võib positiivse kontrollproovi osas piirduda metaboolset aktiveerimist vajava mutageeniga. Sel juhul võimaldab üheainsa kõnealuse positiivse kontrollprooviga saadud tulemus tõendada nii metabolismi aktiveerimise süsteemi toimimist kui ka katsesüsteemi reageerimisvõimet. Pikaajalise töötluse jaoks (st 24 tundi ilma S9-fraktsioonita), kui seda kasutatakse, peab siiski olema oma positiivne kontrollproov, sest töötluse kestus erineb sellest, mida kasutatakse metaboolse aktivatsiooniga katses. Iga positiivset kontrollainet puhul tuleks kasutada ühes või mitmes kontsentratsioonis, mille juures võib eeldada, et saadakse korratavad ja tuvastatavad, tausttaset ületavad tulemused, mis võimaldavad tõendada katsesüsteemi tundlikkust, ning samal ajal ei tohiks tulemused olla mõjutatud käesolevas katsemeetodis sätestatud piirmäära ületavast tsütotoksilisusest (vt punkt 28).
|
Tabel 1
Labori pädevuse hindamiseks ja positiivsete kontrollainete valimiseks soovitatavad etalonained
|
Kategooria
|
Aine
|
CASi nr
|
|
1. Metaboolse aktivatsioonita toimivad mutageenid
|
|
Metüülmetaansulfonaat
|
66-27-3
|
|
Mitomütsiin C
|
50-07-7
|
|
4-nitrokinoliin-N-oksiid
|
56-57-5
|
|
2. Metaboolset aktivatsiooni vajavad mutageenid
|
|
Benso[a]püree
|
50-32-8
|
|
Tsüklofosfamiid (monohüdraat)
|
50-18-0 (6055-19-2)
|
|
7,12-dimetüülbensantratseen
|
57-97-6
|
|
3-metüülkolantreen
|
56-49-5
|
METOODIKA
Uuritava kemikaaliga töötlemine
|
33.
|
Paljunevaid rakke töödeldakse uuritava kemikaaliga nii metabolismi aktiveerimise süsteemi juuresolekul kui ka ilma selleta. Kokkupuuteperiood peab olema sobiva pikkusega (tavaliselt piisab 3–4 tunnist). Tuleks siiski märkida, et inimravimite puhul võivad need nõuded olla teistsugused (59). Kui MLA korral annab lühiajaline töötlemine negatiivsed tulemused ja leidub teavet, mille kohaselt oleks vaja pikemat töötlemist [nt nukleosiidi analoogid, halvasti lahustuvad kemikaalid (5, 59)], tuleks kaaluda katse tegemist pikema, nt 24-tunnise töötlemisega ilma S9-fraktsioonita.
|
|
34.
|
Rakkude minimaalse arvu valimisel iga katsekultuuri (nii kontroll- kui ka töödeldud kultuuride) ja iga katseetapi jaoks tuleks lähtuda spontaanse muteerumise sagedusest. Üldsuunisena tuleks igas katsekultuuris töödelda ja ümber külvata piisav hulk rakke, et tagada igas katseetapis (töötlemine, fenotüübi ekspressioon ja mutantide selektsioon) vähemalt 10, kuid ideaaljuhul 100 mutandi spontaanne teke (56).
|
|
35.
|
MLA puhul on soovituslik vastuvõetav spontaanse muteerumise sagedus vahemikus 35–140 × 10-6 (agarivariant) ja 50–170 × 10-6 (mikrotiiterplaadi variant) (vt tabel 2). Selleks, et saada igas katsekultuuris vähemalt 10, kuid ideaaljuhul 100 töötluses ellujäänud, spontaanselt tekkinud mutanti, on vaja töödelda vähemalt 6 × 106 rakku. Sellise arvu rakkude töötlemine ja rakkude piisava arvu tagamine ekspresseerumise ning mutantide selekteerimiseks vajaliku kloonimise etappides tagab katse igas etapis piisava arvu (10 või rohkem) spontaanselt tekkinud mutante ka juhul, kui kultuure töödeldakse kontsentratsioonidel, mis on 90 % ulatuses tsütotoksilised (mõõdetuna 10 % suhtelise kogukasvuna) (19, 38, 39).
|
|
36.
|
TK6 korral jääb spontaanse muteerumise sagedus üldjuhul vahemikku 2–20 × 10-6. Selleks, et saada igas kultuuris vähemalt 10 töötluses ellujäänud, spontaanselt tekkinud mutanti, on vaja töödelda vähemalt 20 × 106 rakku. Sellise arvu rakkude töötlemine tagab piisava arvu (10 või rohkem) spontaanselt tekkinud mutante ka juhul, kui kultuure töödeldakse kontsentratsioonidel, mis on 90 % ulatuses tsütotoksilised (suhteline ellujäämus 10 %). Peale selle tuleb ekspressiooniperioodi vältel kultiveerida ja mutantide selekteerimiseks välja külvata piisaval arvul rakke (60).
|
Fenotüübi avaldumise aeg ning tsütotoksilisuse ja muteerumissageduse mõõtmine
|
37.
|
Töötlemisperioodi lõppedes kasvatatakse rakke teatava ajavahemiku jooksul, et võimaldada äsja tekkinud mutantide fenotüübi optimaalset või sellelähedast avaldumist; ajavahemik oleneb kasutavavast rakuliinist. MLA korral on fenotüübi avaldumise periood 2 päeva. TK6 korral on fenotüübi avaldumise periood 3–4 päeva. Kui kasutatakse 24-tunnist töötlemist, algab avaldumisperiood pärast töötlemise lõppu.
|
|
38.
|
Fenotüübi avaldumise perioodi jooksul loendatakse rakke iga päev. MLA korral kasutatakse päevaseid rakkude arve päevase suspensioonikasvu arvutamiseks. Pärast 2-päevast avaldumisperioodi suspendeeritakse rakud nii selektiivainet sisaldavas kui ka ilma selektiivaineta söötmes, et teha kindlaks mutantide arv (selekteerimisplaadid) ja kloonimistõhusus (eluvõimelisuse plaadid). MLA korral on kaks võrdselt tunnustatud meetodit mutantide selektiivseks kloonimiseks: üks pehmes agaris ja teine vedelsöötmes 96 süvendiga plaatidel (19, 38, 39). TK6 korral kloonitakse vedelsöötmetes 96 süvendiga plaatidel (16).
|
|
39.
|
TK mutantide puhul on ainus soovitatav selektiivaine trifluorotümidiin (TFT) (61).
|
|
40.
|
MLA korral loendatakse agariplaate ja mikrotiiterplaate pärast 10–12-päevast inkubeerimist. TK6 korral loendatakse mikrotiiterplaatidel vara tekkivaid mutante 10–14 päeva möödudes. Aeglase kasvuga (hilja tekkivate) TK6 mutantide avastamiseks on vaja rakkudele pärast vara tekkivate mutantide loendamist lisada söödet ja TFT-d ning inkubeerida plaate veel 7–10 päeva (62). Punktides 42 ja 44 on arutletud aeglase ja normaalse kasvuga TK mutantide loendamise üle.
|
|
41.
|
MLA puhul sobivad, mõlemat meetodit (agar ja mikrotiiterplaat) hõlmavad arvutused on esitatud 2. liites. Muteerumissageduse arvutamiseks MLA agarimeetodi korral loendatakse kolooniad ja kohandatakse mutantide kolooniate arvu kloonimistõhususe järgi. MLA ja TK6 mikrotiiterplaadimeetodi korral tehakse kloonimistõhusus nii selekteerimis- kui ka kloonimistõhususe plaatidel kindlaks vastavalt Poissoni jaotusele (63). Muteerumissagedus arvutatakse nende kahe kloonimistõhususe alusel.
|
Mutantide kolooniate iseloomustamine
|
42.
|
Kui MLA meetodil uuritav kemikaal on positiivne (vt punktid 62–63), tuleks kolooniaid iseloomustada nende arvu või kasvukiirusega vähemalt ühes uuritavas kultuuris (tavaliselt suurim vastuvõetav positiivne kontsentratsioon) ning negatiivsetes ja positiivsetes kontrollproovides. Kui uuritav kemikaal on negatiivne (vt punkt 64), tuleks mutantide kolooniaid iseloomustada nende arvuga negatiivsetes ja positiivsetes kontrollproovides. MLA mikrotiiterplaadimeetodi korral määratletakse väikese kolooniaga mutandid kui mutandid, mis katavad alla 25 % süvendi diameetrist, ja suure kolooniaga mutandid kui mutandid, mis katavad üle 25 % süvendi diameetrist. Agarimeetodi korral kasutatakse mutantide kolooniate loendamiseks ja suuruse määramiseks kolooniate automaatloendurit. Üksikasjalikud lähenemisviisid kolooniate suuruse määramisele on esitatud kirjandusallikates (19, 38, 40). Kolooniate iseloomustamine negatiivsetes ja positiivsete kontrollproovides on vajalik, et tõendada uuringute asjakohast läbiviimist.
|
|
43.
|
Uuritavat kemikaali ei saa lugeda negatiivseks, kui positiivses kontrollproovis ei tuvastata piisavalt ei suure ega väikese kolooniaga mutante. Kolooniate iseloomustust võib kasutada üldise teabe saamiseks uuritava kemikaali võime kohta põhjustada punktmutatsioone ja/või kromosoomidega toimuvaid sündmusi (punkt 4).
|
|
44.
|
TK6: Normaalse ja aeglase kasvuga mutandid eristatakse üksteisest erinevate inkubatsiooniaegade abil (vt punkt 40). TK6 puhul loendatakse üldiselt kõigis kultuurides, kaasa arvatud negatiivsetes ja positiivsete kontrollproovides nii vara kui ka hilja tekkivad mutandid. Kolooniate iseloomustamine negatiivsetes ja positiivsete kontrollproovides on vajalik, et tõendada uuringute asjakohast läbiviimist. Uuritavat kemikaali ei saa lugeda negatiivseks, kui positiivses kontrollproovis ei tuvastata piisavalt ei vara ega hilja tekkivaid mutante. Kolooniate iseloomustust võib kasutada üldise teabe saamiseks uuritava kemikaali võime kohta põhjustada punktmutatsioone ja/või kromosoomidega toimuvaid sündmusi (punkt 4).
|
Labori pädevus
|
45.
|
Laboris peaks enne katsemeetodi rutiinset kasutamist olema katse läbiviimiseks piisava kogemuse tõendamiseks tehtud katseseeria positiivse kontrollainena kasutatud etalonainetega, millel on eri toimemehhanismid (tabelis 1 loetletud ainete hulgast valitud ained, millest vähemalt üks vajab toimimiseks metabolismi aktiveerimist ja vähemalt üks seda ei vaja), ning mitmesuguste negatiivsete kontrollainetega (sealhulgas töötlemata kultuurid ja mitmesugused lahustid/kandeained). Selliste positiivsete ja negatiivsete kontrollainetega saadud tulemused peaksid olema kooskõlas kirjanduse andmetega. Seda nõuet ei kohaldata kogemusega laborite puhul, kui on olemas varasemate tulemuste andmebaas, nagu on määratletud punktides 47–50. MLA korral peavad nii positiivsete kui ka negatiivsete kontrollainetega saadud tulemused olema kooskõlas IWGT soovitustega (vt tabel 2).
|
|
46.
|
Valitud positiivseid kontrollaineid (vt tabel 1) tuleks uurida nii lühiajalise kui ka pikaajalise töötlusega (kui pikaajalist töötlust kasutatakse) aktiveerimata metabolismi tingimustes ning lühiajalise töötlusega aktiveeritud metabolismi tingimustes, et tõendada mutageensete kemikaalide tuvastamise ja metabolismi aktiveerimise süsteemi tõhususe kindlakstegemise võimekust, samuti rakkude kasvutingimuste sobivust töötlemise, fenotüübi avaldumise ja mutantide selekteerimise ajal ning kasutatavate hindamismeetodite asjakohasust. Kõnealuste valitud ainete puhul tuleb katsesüsteemi tundlikkuse ja dünaamilise kasutusulatuse sobivuse tõendamiseks valida kontsentratsioonivahemikud nii, et oleks võimalik saada korratavaid, tausttaset ületavaid ja kontsentratsioonist sõltuvaid tulemusi.
|
Varasemad andmed kontrollproovide kohta
|
47.
|
Laboris tuleks kindlaks teha:
|
—
|
positiivsete kontrollainetega saadud varasemate tulemuste vahemik ja jaotus,
|
|
—
|
negatiivsete kontrollainetega (töötlemata rakud, lahusti kontrollproov) saadud varasemate tulemuste vahemik ja jaotus.
|
|
|
48.
|
Kui hangitakse algandmeid varasemate negatiivsete kontrollproovide tulemuste jaotuse määramiseks, peavad paralleelsete negatiivsete kontrollproovidega saadud andmed olema kooskõlas kontrollproovide kohta avaldatud andmetega. Kontrollproovidega saadud tulemuste jaotuse lähteandmetele üha uute katseandmete lisandumisel peaksid paralleelsete negatiivsete kontrollproovidega saadavad tulemused jääma ideaaljuhul selle jaotuse usaldusvahemikku usaldusnivool 95 % (64, 65).
|
|
49.
|
Laboris negatiivsete kontrollproovidega saadud varasemate tulemuste andmebaas peaks algselt hõlmama vähemalt 10 katse andmeid, kuid soovitatavalt peaks see sisaldama vähemalt 20 võrreldavates tingimustes tehtud katse andmeid. Laboris tuleks kasutada kvaliteedikontrolli meetodeid, näiteks kontrollkaarte (nt vastavuse kontrollkaardid või X-tüüpi kontrollkaardid (65)), et teha kindlaks, kui suures ulatuses positiivsete ja negatiivsete kontrollproovide andmed varieeruvad, ja tõendada, et laboris kasutatav metoodika on usaldusväärne (66). Üksikasju ja soovitusi aja jooksul andmete kogumise ja kasutamise kohta võib leida kirjandusest (64).
|
|
50.
|
Negatiivsete kontrollproovidega saadud andmed peaksid hõlmama muteerumissagedust ühes või soovitatavalt mitmes paralleelses kultuuris, nagu on kirjeldatud punktis 27. Paralleelsete negatiivsete kontrollproovide tulemused peaksid ideaaljuhul jääma labori varasemate kontrollproovi tulemuste jaotuse usaldusvahemikku usaldusnivool 95 %. Kui negatiivsete kontrollproovidega saadud tulemused jäävad väljapoole kõnealust usaldusvahemikku usaldusnivool 95 %, võivad need olla kontrollproovidega saadud varasemate tulemuste jaotuses arvessevõtmiseks vastuvõetavad juhul, kui tegemist ei ole äärmuslike võõrväärtustega ning on tõendatud, et katsesüsteem on usaldusväärne (vt punkt 49), ja puuduvad tõendid selle kohta, et tegemist on tehnilise vea või inimliku eksimusega.
|
|
51.
|
Katsemetoodika mis tahes muudatuse puhul tuleks kaaluda, kas see mõjutab katseandmete vastavust laboris kontrollproovidega varem saadud olemasolevatele andmetele. Iga olulise mittevastavuse korral tuleks luua uus kontrollproovidega saadud varasemate tulemuste andmebaas.
|
ANDMED JA KATSEPROTOKOLLI KOOSTAMINE
Tulemuste esitamine
|
52.
|
Nii MLA kui ka TK6 korral kuuluvad esitatavate andmete hulka nii töötlemata kui ka kontrollkultuuridega saadud andmed, mis on vajalik tsütotoksilisuse (vastavalt suhteline kogukasv või suhteline ellujäämus) ja muteerumissageduse arvutamiseks allpool kirjeldatud viisil.
|
|
53.
|
MLA korral tuleb iga üksiku kultuuri kohta esitada suhteline suspensioonikasv, suhteline kogukasv, kloonimistõhusus mutantide selekteerimise ajal ning mutantide kolooniate arv (agarivariandi korral) või tühjade süvendite arv (mikrotiiterplaadi variandi korral). Muteerumissagedust väljendatakse mutantrakkude arvuga miljoni ellujäänud raku kohta. Kui vastus on positiivne, esitatakse väikese ja suure koloonia muteerumissagedused (ja/või protsentuaalne osakaal muteerumise üldsagedusest), mis on leitud uuritava kemikaali vähemalt ühe kontsentratsiooniga (tavaliselt suurim positiivne kontsentratsioon), negatiivse kontrollprooviga ja positiivse kontrollprooviga. Negatiivse vastuse korral tuleb esitada negatiivse kontrollproovi ja positiivse kontrollprooviga saadud väikese ja suure koloonia muteerumissagedused.
|
|
54.
|
TK6 korral tuleb iga üksiku kultuuri kohta esitada suhteline ellujäämus, kloonimistõhusus mutantide selekteerimise ajal ning tühjade süvendite arv vara ja hilja tekkivate mutantide puhul. Muteerumissagedust väljendatakse mutantrakkude arvuga ellujäänud rakkude arvu kohta ning see peaks hõlmama muteerumise üldsagedust ning vara ja hilja tekkivate mutantide muteerumissagedust (ja/või protsentuaalselt osakaalu muteerumise üldsagedusest).
|
Katse nõuetekohasuse kriteeriumid
|
55.
|
Nii MLA kui ka TK6 korral peavad enne konkreetse uuritava kemikaali üldiste tulemuste määramist olema täidetud järgmised kriteeriumid:
|
—
|
katse on tehtud kahes eri tingimuses (lühiajaline töötlus metabolismi aktiveerimisega ja ilma selleta, vt punkt 33), välja arvatud juhul, kui neist ühe puhul on saadud positiivsed tulemused;
|
|
—
|
analüüsitud rakkude ja kontsentratsioonide arv on piisav (vt punktid 27 ja 34–36);
|
|
—
|
suurima kontsentratsiooni valimise kriteeriumid on kooskõlas kriteeriumidega, mis on kirjeldatud punktides 28–30.
|
|
Negatiivsete ja positiivsete kontrollproovide nõuetekohasuse kriteeriumid
|
56.
|
IWGT MLA ekspertide töörühma tehtud analüüs suure hulga MLA andmete kohta andis tulemuseks rahvusvahelise konsensuse MLA konkreetsete nõuetekohasuse kriteeriumide osas (1, 2, 3, 4, 5). Seega annab käesolev katsemeetod konkreetseid soovitusi negatiivsete ja positiivsete kontrollproovide vastuvõetavuse kindlakstegemiseks ning MLAga saadud üksikainete tulemuste hindamiseks. TK6 andmebaas on palju väiksem ja töörühm ei ole seda hinnanud.
|
|
57.
|
MLA korral tuleb iga katse juures hinnata, kas töötlemata/lahusti kontrollproov vastab IWGT MLA töörühma koostatud nõuetekohasuse kriteeriumidele ((4) ja tabel 2 allpool) järgmiste näitajate poolest: 1) muteerumissagedus (tuleb arvestada, et IWGT nõuetekohased muteerumissagedused on MLA agari- ja mikrotiiterplaadi variandi puhul erinevad); 2) kloonimistõhusus mutantide selekteerimise ajal ja 3) suspensioonikasv lahusti kontrollproovis (valemid on leitavad 2. liites).
Tabel 2
MLA nõuetekohasuse kriteeriumid
|
Näitaja
|
Pehme agari meetod
|
Mikrotiiterplaadi meetod
|
|
Muteerumissagedus:
|
35–140 × 10-6
|
50–170 × 10-6
|
|
Kloonimistõhusus
|
65–120 %
|
65–120 %
|
|
Suspensioonikasv
|
8–32-kordne (3–4-tunnine töötlus) 32–180-kordne (24-tunnine töötlus, kui seda kasutatakse)
|
8–32-kordne (3–4-tunnine töötlus) 32–180-kordne (24-tunnine töötlus, kui seda kasutatakse)
|
|
|
58.
|
Samuti tuleb MLA korral iga katse juures hinnata, kas positiivne kontrollkatse või positiivsed kontrollkatse vastavad vähemalt ühele järgmisest kahest nõuetekohasuse kriteeriumist, mille on välja töötanud IWGT töörühm:
|
—
|
positiivses kontrollproovis peab ilmnema muteerumise üldsageduse absoluutne suurenemine, st suurenemine spontaansest muteerumisest tulenevast taustsagedusest (indutseeritud muteerumissagedus) ülespoole vähemalt 300 × 10-6. Vähemalt 40 % indutseeritud muteerumissagedusest peab kajastuma väikese koloonia muteerumissageduses;
|
|
—
|
positiivses kontrollproovis peab väikese koloonia muteerumissagedus olema vähemalt 150 × 10-6 võrra suurem töötlemata/lahusti kontrollproovis esinevast (väikese koloonia indutseeritud muteerumissagedus 150 × 10-6).
|
|
|
59.
|
TK6 korral on katse nõuetekohane, kui paralleelsete negatiivsete kontrollproovide tulemused on vastuvõetavad, et lisada need labori varasemate negatiivsete kontrollproovidega saadud tulemuste andmebaasi, nagu on kirjeldatud punktides 48–49. Peale selle peavad paralleelsete positiivsete kontrollproovidega (vt punkt 32) saadud tulemused olema võrreldavad positiivsete kontrollproovidega varem saadud tulemustega ning saadud väärtused peaksid paralleelset negatiivset kontrollproovi iseloomustavate väärtustega võrrelduna olema statistiliselt oluliselt suuremad.
|
|
60.
|
Mõlema katse korral peab positiivses kontrollkultuuris leitud tsütotoksilisus olema sama mis katsekultuurides. See tähendab, et suhteline kogukasv või suhteline ellujäämus ei tohi jääda alla 10 % Positiivse kontrollproovi vastuvõetavuse kriteeriumidele vastavuse tõendamiseks piisab ühe kontsentratsiooni kasutamisest (või kui kasutatakse rohkem kontsentratsioone, siis ühe positiivse kontrollkultuuriga kasutatava kontsentratsiooni kasutamisest). Peale selle peab positiivse kontrollproovi muteerumissagedus jääma labori jaoks määratud vastuvõetavasse vahemikku.
|
Tulemuste hindamine ja tõlgendamine
|
61.
|
MLA kohta on IWGT hiire lümfoomi ekspertide töörühm ära teinud olulise töö positiivse vastuse bioloogilise olulisuse ja kriteeriumidega seoses (4). Seega annab käesolev katsemeetod konkreetseid soovitusi uuritava kemikaali MLA-katsega saadud tulemuste tõlgendamiseks (vt punktid 62–64). TK6 andmebaas on palju väiksem ja töörühm ei ole seda hinnanud. Seepärast on TK6 puhul esitatud soovitused andmete tõlgendamiseks üldisemad (vt punktid 65–66). Täiendavad soovitused kehtivad mõlema katsemeetodi kohta (vt punktid 67–71).
|
MLA
|
62.
|
Positiivsete ja negatiivsete vastuste määratlemiseks on soovitatav kasutada lähenemisviisi, mis tagab, et suurenenud muteerumissagedus on bioloogiliselt oluline. Muude katsete puhul tavaliselt kasutatava statistilise analüüsi asemel põhineb see lähenemisviis eelnevalt määratletud indutseeritud muteerumise sagedusel (st muteerumissageduse suurenemisel ülespoole paralleelse kontrollprooviga saadud väärtusest), mis on nimetatud globaalseks hindamisteguriks (GEF) ja mille aluseks on osalevate laborite negatiivse kontrollproovi muteerumissageduse andmete jaotuse analüüs (4). MLA agariversiooni korral on GEF 90 × 10-6 ja MLA mikrotiiterplaadi variandi korral 126 × 10-6.
|
|
63.
|
Tingimusel, et kõik nõuetekohasuse kriteeriumid on täidetud, loetakse uuritav kemikaal selgelt positiivseks, kui ükskõik millistel uuritud katsetingimustel (vt punkt 33) ületab muteerumissageduse kasv paralleelprooviga mõõdetud taustasageduse rohkem kui GEF võrra ning suurenemine on sõltuv kontsentratsioonist (nt kasutades trenditesti meetodit). Sel juhul käsitatakse uuritavat kemikaali võimelisena põhjustama selles katsesüsteemis mutatsiooni.
|
|
64.
|
Tingimusel, et kõik nõuetekohasuse kriteeriumid on täidetud, loetakse uuritav kemikaal selgelt negatiivseks, kui kõigis uuritud katsetingimustes (vt punkt 33) puudub kontsentratsioonist sõltuv vastus või kui muteerumissagedus on suurenenud, siis see ei ületa GEFi. Sel juhul käsitatakse uuritavat kemikaali võimetuna põhjustama selles katsesüsteemis mutatsioone.
|
TK6
|
65.
|
Tingimusel, et kõik nõuetekohasuse kriteeriumid on täidetud, loetakse uuritav kemikaal selgelt positiivseks, kui mis tahes uuritud katsetingimustes (vt punkt 33):
|
—
|
vähemalt ühe katsekontsentratsiooni juures täheldatakse saadud väärtuse statistiliselt olulist suurenemist paralleelse negatiivse kontrollprooviga võrreldes;
|
|
—
|
sobiva trenditestiga hindamisest nähtub, et see suurenemine on kontsentratsioonist sõltuv (vt punkt 33);
|
|
—
|
ükski nendest tulemustest ei ole väljaspool labori varasemate negatiivse kontrollprooviga saadud tulemuste jaotust (nt väljaspool Poissoni jaotuse usaldusvahemikku usaldusnivool 95 %; vt punkt 48).
|
Kui kõik need kriteeriumid on täidetud, käsitatakse uuritavat kemikaali võimelisena põhjustama selles katsesüsteemis mutatsiooni. Soovitused kõige asjakohasemate statistiliste meetodite kohta on esitatud kirjanduses (66, 67).
|
|
66.
|
Tingimusel, et kõik nõuetekohasuse kriteeriumid on täidetud, loetakse uuritav kemikaal selgelt negatiivseks, kui kõikides uuritud katsetingimustes (vt punkt 33):
|
—
|
ei täheldata ühegi katsekontsentratsiooni juures saadud väärtuse statistiliselt olulist suurenemist paralleelse negatiivse kontrollprooviga võrreldes;
|
|
—
|
sobiva trenditestiga hindamisest nähtub, et kontsentratsioonist sõltuvat väärtuse suurenemist ei esine;
|
|
—
|
kõik need tulemused on labori varasemate negatiivse kontrollprooviga saadud tulemuste jaotuse vahemikus (nt Poissoni jaotuse usaldusvahemikus usaldusnivool 95 %; vt punkt 48).
|
|
|
67.
|
Sel juhul käsitatakse uuritavat kemikaali võimetuna põhjustama selles katsesüsteemis mutatsioone.
|
MLA ja TK6
|
68.
|
Kui suurima kontsentratsiooni valimisel lähtutakse tsütotoksilisusest, tuleks suurim kontsentratsioon valida nii, et saavutataks suhteline kogukasv / suhteline ellujäämus 10–20 %. Ühine seisukoht on, et tuleb olla hoolikas niisuguste positiivsete tulemuste tõlgendamisel, mis on leitud suhtelise kogukasvu / suhtelise ellujäämuse vahemikus 10–20 %, ning et tulemust ei loeta positiivseks, kui muteerumissageduse suurenemine ilmnes üksnes 10 % või väiksema suhtelise kogukasvu / suhtelise ellujäämuse juures (kui seda hinnati) (2, 59).
|
|
69.
|
Leidub asjaolusid, mille puhul täiendav teave võib olla abiks otsustamisel, et uuritav kemikaal ei ole mutageenne, kui ühegi kultuuriga ei saada suhtelise kogukasvu / suhtelise ellujäämuse väärtust vahemikus 10–20 %. Niisugused olukorrad on järgmised: 1) tõendid mutageensuse kohta puuduvad (st puudub doosi-vastuse seos, puuduvad paralleelse negatiivse kontrollprooviga saadud muteerumissagedusest või aja jooksul kogutud andmetest saadud taustväärtusest suuremad muteerumissagedused) andmepunktide seerias, mis on suhtelise kogukasvu / suhtelise ellujäämuse vahemikus 20–100 %, ning vähemalt üks andmepunkt on suhtelise kogukasvu / suhtelise ellujäämuse vahemikus 20–25 %; 2) tõendid mutageensuse kohta puuduvad (st puudub doosi-vastuse seos, puuduvad paralleelse negatiivse kontrollprooviga saadud muteerumissagedusest või aja jooksul kogutud andmetest saadud taustväärtusest suuremad muteerumissagedused) andmepunktide seerias, mis on suhtelise kogukasvu / suhtelise ellujäämuse vahemikus 25–100 %, ning olemas on ka negatiivne andmepunkt, mis jääb veidi alla 10 % suhtelise kogukasvu / suhtelise ellujäämuse. Mõlemal juhul võib uuritava kemikaali lugeda negatiivseks.
|
|
70.
|
Selgelt positiivset või negatiivset vastust ei ole vaja üle kontrollida.
|
|
71.
|
Juhul, kui vastus ei ole eespool kirjeldatu kohaselt selgelt negatiivne ega selgelt positiivne ja/või kui eesmärk on aidata kindlaks teha tulemuse bioloogiline olulisus, tuleks andmete hindamiseks kasutada eksperdihinnangut ja/või täiendavaid uuringuid. Sellises olukorras võib olla kasulik teha korduskatse ja muuta vajaduse korral katsetingimusi (nt kontsentratsiooniintervalli, et suurendada andmepunktide saamise tõenäosust suhtelise kogukasvu / suhtelise ellujäämuse vahemikus 10–20 %, või metabolismi aktiveerimise tingimusi (st S9 kontsentratsiooni või S9 päritolu) ja töötlemise kestust).
|
|
72.
|
Harvadel juhtudel ei võimalda andmed isegi pärast täiendavaid uuringuid teha järeldust selle kohta, kas tulemus on positiivne või negatiivne. Seepärast tuleks sel juhul lugeda uuritava kemikaaliga saadud vastus ebaselgeks (seda tõlgendatakse positiivse ja negatiivse tulemuse võrdvõimalikkusena).
|
KATSEPROTOKOLL
|
73.
|
Katseprotokollis tuleks esitada järgmine teave.
|
|
Uuritav kemikaal:
|
—
|
allikas, partii number, aegumiskuupäev, kui on olemas;
|
|
—
|
uuritava kemikaali püsivus, kui see on teada;
|
|
—
|
uuritava kemikaali lahustuvus ja püsivus lahustis, kui see on teada;
|
|
—
|
vastavalt vajadusele mõõtmistulemused söötme pH ja osmolaalsuse ning sademe tekke kohta pärast uuritava kemikaali lisamist.
|
|
|
Ühest koostisosast koosnev aine:
|
—
|
füüsiline välimus, lahustuvus vees ja muud asjakohased füüsikalis-keemilised omadused;
|
|
—
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus, CASi number, SMILESi või InChI kood, struktuurivalem, puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne.
|
|
|
|
Mitme koostisosaga aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal ja segu:
|
—
|
võimalikult täpne omaduste kirjeldus lähtuvalt koostisosade keemilisest määratlusest (vt eespool), kvantitatiivsest sisaldusest ja asjakohastest füüsikalis-keemilistest omadustest.
|
|
|
|
|
Lahusti:
|
—
|
lahusti valimise põhjendus;
|
|
—
|
lahusti protsentuaalne sisaldus lõppsöötmes.
|
|
|
|
Rakud:
|
|
labori tüvikultuuride puhul:
|
—
|
rakkude tüüp ja päritolu ning nendega katselaboris tehtud toimingud;
|
|
—
|
karüotüübi andmed ja/või kromosoomide modaalarv;
|
|
—
|
rakukultuuride säilitamise meetodid;
|
|
—
|
teave mükoplasma puudumise kohta;
|
|
—
|
rakkude arvu kahekordistumise aeg.
|
|
|
|
|
Katsetingimused:
|
—
|
kontsentratsioonide ja rakukultuuride arvu valiku põhjendus, sealhulgas näiteks andmed tsütotoksilisuse ja lahustuvuspiirangute kohta;
|
|
—
|
söötme koostis, CO2 kontsentratsioon, niiskusesisaldus;
|
|
—
|
uuritava kemikaali kontsentratsioon, väljendatuna lõppkontsentratsioonina söötmes (nt μg/ml või mg/ml või mM);
|
|
—
|
söötmele lisatud lahusti ja uuritava kemikaali kontsentratsioon (ja/või ruumala);
|
|
—
|
inkubeerimistemperatuur;
|
|
—
|
rakutihedus töötluse ajal;
|
|
—
|
metabolismi aktiveerimise süsteemi liik ja koostis (fraktsiooni S9 allikas, S9 segu valmistamise meetod, S9 segu ja S9 kontsentratsioon või ruumala lõppsöötmes, S9 kvaliteedikontrolli andmed);
|
|
—
|
positiivsed ja negatiivsed kontrollained ja nende lõppkontsentratsioonid iga töötlemistingimuse puhul;
|
|
—
|
ekspressiooniperioodi pikkus (samuti väljakülvatud rakkude arv ning vajaduse korral teave ümberkülvide ja söötmevahetusrežiimi kohta);
|
|
—
|
selektiivaine identimisandmed ja kontsentratsioon;
|
|
—
|
MLA korral tuleb näidata kasutatud variant (agar või mikrotiiterplaat);
|
|
—
|
katse nõuetekohasuse kriteeriumid;
|
|
—
|
eluvõimeliste ja muteerunud rakkude loendamiseks kasutatud meetodid;
|
|
—
|
tsütotoksilisuse mõõtmiseks kasutatud meetodid;
|
|
—
|
muu täiendav asjakohane teave tsütotoksilisuse ja kasutatud meetodi kohta;
|
|
—
|
väljakülvamisele järgneva inkubeerimise kestus;
|
|
—
|
kolooniate määratlus, milles on arvesse võetud kolooniate arvu ja tüüpi (sh „väikeste“ ja „suurte“ kolooniate kriteeriumid, vajaduse korral);
|
|
—
|
uuringutulemuse positiivseks, negatiivseks või ebaselgeks liigitamise kriteeriumid;
|
|
—
|
pH, osmolaalsuse (kui määratakse) ja sademe määramiseks kasutatud meetodid, kui on asjakohane.
|
|
|
|
Tulemused:
|
—
|
töödeldud rakkude arv ja ümberkülvatud rakkude arv igas kultuuris;
|
|
—
|
toksilisuse näitajad (MLA korral suhteline kogukasv ja TK6 korral suhteline ellujäämus);
|
|
—
|
sadenemise ilmingud ja nende täheldamise aeg;
|
|
—
|
selektiivainega ja selektiivaineta söötmesse külvatud rakkude arv;
|
|
—
|
kolooniate arv selektiivaineta söötmes ja resistentsete kolooniate arv selektiivainega söötmes ning vastav muteerumissagedus;
|
|
—
|
kolooniate suurus negatiivsetes ja positiivsete kontrollproovides ning kui uuritav kemikaal on positiivne, siis vähemalt ühe kontsentratsiooni juures, samuti vastavad muteerumissagedused;
|
|
—
|
võimaluse korral andmed vastuse sõltuvuse kohta kontsentratsioonist;
|
|
—
|
andmed paralleelsete negatiivsete (lahusti) kontrollproovide ja positiivsete kontrollproovide kohta (kontsentratsioonid ja lahustid);
|
|
—
|
varasemate negatiivsete (lahusti) ja positiivsete kontrollproovide andmed (kontsentratsioonid ja lahustid), sh vahemikud, keskväärtused ja standardhälbed; kontrollkatsete arv, mille alusel varasemad andmed on saadud;
|
|
—
|
statistilise analüüsi tulemused (iga kultuuri kohta eraldi ja vajaduse korral paralleelkultuuride koondandmed) ja vajaduse korral p-väärtused ning MLA korral GEF-hinnang.
|
|
|
KIRJANDUS
|
(1)
|
Moore, M. M., Honma, M., Clements, J. (koostaja), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. ja Stankowski, L.F. Jr. (2000), „Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report“, Environ. Mol. Mutagen., nr 35 (3), lk 185–190.
|
|
(2)
|
Moore, M. M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. ja Stankowski, L.F. Jr. (2002), „Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (Aprill 2000)“, Environ. Mol. Mutagen., nr 40 (4), lk 292–299.
|
|
(3)
|
Moore, M. M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. ja Yoshimura, I. (2003), „Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report“, Mutation Res., nr 540, lk 127–140.
|
|
(4)
|
Moore, M. M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. ja Yoshimura, I. (2006), „Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation“, Environ. Mol. Mutagen., nr 47 (1), lk 1–5.
|
|
(5)
|
Moore, M. M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. ja Van Goethem, F. (2007), „Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment“, Mutation. Res., nr 627 (1), lk 36–40.
|
|
(6)
|
OECD (2016), Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014–2015. Environment, Health and Safety Publications, Series on Testing and Assessment No. 238. OECD, Pariis.
|
|
(7)
|
Fellows M. D., Luker, T., Cooper, A. ja O'Donovan, M.R. (2012), „Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors“, Mutation, Res., nr 746 (1), lk 21–28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. ja Hayashi, M. (2001), „Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction“, Mutation Res., nr 493 (1–2), lk 101–114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. ja Moore, M. M. (2009), „The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy“, Toxicol. Sci., nr 109 (1), lk 96–105.
|
|
(10)
|
Applegate, M. L., Moore, M. M., Broder, C. B., Burrell, A. ja Hozier, J. C. (1990), „Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells“, Proc. National. Academy. Sci. USA, nr 87 (1), lk 51–55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. ja Clive, D. (1981), „Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System“, Mutation Res., nr 84 (1), lk 169–181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. ja Moore, M. M. (1985), „Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History“, Mutation Res., nr 147 (5), lk 237–242.
|
|
(13)
|
Moore, M. M., Clive, D., Hozier, J. C., Howard, B. E., Batson, A. G., Turner, N. T. ja Sawyer, J. (1985), „Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells“, Mutation Res., nr 151 (1), lk 161–174.
|
|
(14)
|
Liber, H. L., Call, K. M. ja Little, J. B. (1987), „Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells“, Mutation Res., nr 178 (1), lk 143–153.
|
|
(15)
|
Li, C. Y., Yandell, D. W. ja Little, J. B. (1992), „Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro“, Somat. Cell Mol. Genet., nr 18 (1), lk 77–87.
|
|
(16)
|
Honma, M., Hayashi, M. ja Sofuni, T. (1997), „Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells“, Mutation. Res., nr 374 (1), lk 89–98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. ja Hayashi, M. (2000), „Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity“, Mol. Carcinogen., nr 28 (4), lk 203–14.
|
|
(18)
|
Amundson, S. A. ja Liber, H. L. (1992), „A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants“, Mutation Res., nr 267 (1), lk 89–95.
|
|
(19)
|
Schisler, M. R., Moore, M. M. ja Gollapudi, B. B. (2013), „In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay“, Protocols in Genotoxicity Assessment, toim. A. Dhawan ja M. Bajpayee, Springer Protocols, Humana Press, lk 27–50.
|
|
(20)
|
Long, L. H., Kirkland, D., Whitwell, J. ja Halliwell, B. (2007), „Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium“, Mutation Res., nr 634 (1–2), lk 177–183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. ja Marzin, D. (2008), „Characterization of the Genotoxicity of Nitrilotriacetic Acid“, Environ. Mol. Mutagen., nr 49 (6), lk 439–452.
|
|
(22)
|
Brusick, D. (1986), „Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations“, Environ. Mutagen., nr 8 (6), lk 879–886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. ja Okumura, K. (1992), „Clastogenicity of Low pH to Various Cultured Mammalian Cells“, Mutation Res., nr 268 (2), lk 297–305.
|
|
(24)
|
Scott, D., Galloway, S. M., Marshall, R. R., Ishidate, M. Jr, Brusick, D., Ashby, J. ja Myhr, B. C. (1991), „Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9“, Mutation Res., nr 257, lk 147–204.
|
|
(25)
|
Wang, J., Heflich, R. H. ja Moore, M. M. (2007), „A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay“, Mutation Res., nr 626 (1–2), lk 185–190.
|
|
(26)
|
Fischer, G. A. (1958), „Studies on the Culture of Leukemic Cells In Vitro“, Ann. N.Y. Academy Sci., nr 76, lk 673–680.
|
|
(27)
|
Clive, D., Johnson, K. O., Spector, J. F. S., Batson, A. G. ja Brown, M. M. M. (1979), „Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System“, Mutation Res., nr 59(1), lk 61–108.
|
|
(28)
|
Sawyer, J., Moore, M. M., Clive, D. ja Hozier, J. (1985), „Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line“, Mutation Res., nr 147 (5), lk 243–253.
|
|
(29)
|
Sawyer J. R., Moore M. M. ja Hozier J. C. (1989), „High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line“, Mutation Res., nr 214 (2), lk 181–193.
|
|
(30)
|
Sawyer, J. R., Binz, R. L., Wang, J. ja Moore, M. M. (2006), „Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line“, Environ. Mol. Mutagen., nr 47 (2), lk 127–131.
|
|
(31)
|
Fellows, M. D., McDermott, A., Clare, K. R., Doherty, A. ja Aardema, M. J. (2014), „The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay“, Environ. Mol. Mutagen., nr 55 (1), lk 35–42.
|
|
(32)
|
Storer, R. D., Jraynak, A. R., McKelvey, T. W., Elia, M. C., Goodrow, T.L. ja DeLuca, J.G. (1997), „The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene“, Mutation. Res., nr 373 (2), lk 157–165.
|
|
(33)
|
Clark L. S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S. H. ja Moore, M. M. (2004), „Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells“, Mutagen., nr 19 (4), lk 263–268.
|
|
(34)
|
Skopek, T. R., Liber, H. L., Penman, B. W. ja Thilly, W. G. (1978), „Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay“, Biochem. Biophys. Res. Commun., nr 84 (2), lk 411–416.
|
|
(35)
|
Honma, M. (2005), „Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells“, Environ. Mol. Mutagen., nr 45 (2–3): lk 162–176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y. H., Tsang, N. M., Yandell, D. W., Kelsey, K. T. ja Liber, H. L. (1995), „Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines“, Cancer. Res., nr 55 (1), lk 12–15.
|
|
(37)
|
Lorge, E., Moore, M., Clements, J., O'Donovan, M., Honma, M., Kohara, A., van Benthem, J., Galloway, S., Armstrong, M. J., Thybaud, V., Gollapudi, B., Aardema, M., Kim, J., Sutter, A., Kirkland, D. J. (2015), „Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing.“ (Pooleliolev käsikiri.)
|
|
(38)
|
Lloyd, M. ja Kidd, D. (2012), „The Mouse Lymphoma Assay“, Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, toim. Parry ja Parry, Humana Press, ISBN 978-1-61779-420-9, lk 35–54.
|
|
(39)
|
Mei, N., Guo, X. ja Moore M. M. (2014), „Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity“ väljaandes Optimization in Drug Discover: In Vitro Methods, toim. Yan, Z. ja Caldwell, 2. trükk, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber, H. L. ja Thilly, W. G. (1982), „Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts“, Mutation Res., nr 94 (2), lk 467–485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, O. W., Price, A., Schechtman, L., Stacey, G. ja Stokes, W. (2005), „Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice“, ATLA, nr 33 (3), lk 261–287.
|
|
(42)
|
Moore, M. M. ja Howard, B. E. (1982), „Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium“, Mutation Res., nr 104 (4–5), lk 287–294.
|
|
(43)
|
Ames, B. N., McCann, J. ja Yamasaki, E. (1975), „Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test“, Mutation Res., nr 31 (6), lk 347–364.
|
|
(44)
|
Maron, D. M. ja Ames, B. N. (1983), „Revised Methods for the Salmonella Mutagenicity Test“, Mutation Res., nr 113 (3–4), lk 173–215.
|
|
(45)
|
Natarajan, A. T., Tates, A. D, Van Buul, P. P. W., Meijers, M. ja de Vogel, N. (1976), „Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes“, Mutation Res., nr 37 (1), lk 83–90.
|
|
(46)
|
Matsuoka, A., Hayashi, M. ja Ishidate, M. Jr. (1979), „Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro“, Mutation Res., nr 66 (3), lk 277–290.
|
|
(47)
|
Ong, T. M. et al. (1980), „Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver“, J. Environ. Pathol. Toxicol., nr 4 (1), lk 55–65.
|
|
(48)
|
Elliott, B. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. ja Wolf, R. C. (1992), „Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays“, Mutagen., nr 7 (3), lk 175–177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. ja Sugimura, T. (1976), „A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems“ väljaandes In Vitro Metabolic Activation in Mutagenesis Testing, toim. de Serres F. J. et al. , Elsevier, North-Holland, lk 85–88.
|
|
(50)
|
Galloway, S. M. et al. (1994), „Report from Working Group on In Vitro Tests for Chromosomal Aberrations“, Mutation Res., nr 312 (3), lk 241–261.
|
|
(51)
|
Johnson, T. E., Umbenhauer, D. R. ja Galloway, S. M. (1996), „Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9“, Environ. Mol. Mutagen., nr 28 (1), lk 51–59.
|
|
(52)
|
UNEP (2001), Püsivate orgaaniliste saasteainete Stockholmi konventsioon. Ühinenud Rahvaste Organisatsiooni Keskkonnaprogramm (UNEP).
|
|
(53)
|
Krahn, D. F., Barsky, F. C. ja McCooey, K. T. (1982), „CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids“ väljaandes Genotoxic Effects of Airborne Agents, toim. Tice, R. R., Costa, D. L. ja Schaich, K. M., New York, Plenum, lk 91–103.
|
|
(54)
|
Zamora, P. O., Benson, J. M., Li, A. P. ja Brooks, A. L. (1983), „Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay“, Environ. Mutagen., nr 5 (6), lk 795–801.
|
|
(55)
|
Asakura, M., Sasaki, T., Sugiyama, T., Arito, H., Fukushima, S. ja Matsushima, T. (2008), „An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay“, Mutation Res., nr 652 (2), lk 122–130.
|
|
(56)
|
Arlett, C. F. et al. (1989), „Mammalian Cell Gene Mutation Assays Based upon Colony Formation“ väljaandes Statistical Evaluation of Mutagenicity Test Data, toim. Kirkland, D. J., Cambridge University Press, lk 66–101.
|
|
(57)
|
Morita, T., Honma, M. ja Morikawa, K. (2012), „Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity“, Mutation Res., nr 741 (1–2), lk 32–56.
|
|
(58)
|
Brookmire, L., Chen, J. J. ja Levy, D. D. (2013), „Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay“, Environ. Mol. Mutagen., nr 54 (1), lk 36–43.
|
|
(59)
|
USA Toidu- ja Ravimiamet (2012), International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Kättesaadav aadressil [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma, M. ja Hayashi, M. (2011), „Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells“, Environ. Mol. Mutagen., nr 52 (5), lk 373–384.
|
|
(61)
|
Moore-Brown, M. M., Clive, D., Howard, B. E., Batson, A. G. ja Johnson, K. O. (1981), „The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells“, Mutation Res., nr 85 (5), lk 363–378.
|
|
(62)
|
Liber, H. L., Yandell, D. W. ja Little, J. B. (1989), „A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus“, Mutation Res., nr 216 (1), lk 9–17.
|
|
(63)
|
Furth, E. E., Thilly, W. G., Penman, B. W., Liber, H. L. ja Rand, W. M. (1981), „Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates“, Anal. Biochem., nr 110 (1), lk 1–8.
|
|
(64)
|
Hayashi, M., Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. ja Thybaud, V. (2011), „Compilation and Use of Genetic Toxicity Historical Control Data“, Mutation Res., nr 723 (2), lk 87–90.
|
|
(65)
|
Ryan, T. P. (2000), Statistical Methods for Quality Improvement. 2. trükk. John Wiley & Sons, New York.
|
|
(66)
|
OECD (2014), Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (nr 199), Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(67)
|
Fleiss, J. L., Levin, B. ja Paik, M. C. (2003), Statistical Methods for Rates and Proportions. 3. trükk. John Wiley & Sons, New York.
|
1. liide
MÕISTED
Aneugeen
: iga kemikaal või protsess, mis põhjustab interaktsiooni kaudu mitootilise või meiootilise rakujagunemise tsükli komponentidega rakkude või organismide aneuploidsust.
Aneuploidsus
: iga kõrvalekalle normaalsest diploidsest (või haploidsest) kromosoomiarvust ühe või enama kromosoomi võrra, kuid mitte terve(te) kromosoomikomplekti(de) võrra (polüploidsus).
Aluspaarivahetust põhjustav mutageen
: kemikaal, mis kutsub esile aluspaari vahetuse DNAs.
Kemikaal
: aine või segu.
Kloonimistõhusus
: väikesel tihedusel välja külvatud rakkude hulgas esinevate selliste rakkude protsentuaalne osakaal, mis on võimelised moodustama loendatava koloonia.
Klastogeen
: iga kemikaal, mis põhjustab rakkude või organismide populatsioonis struktuurseid kromosoomiaberratsioone.
Tsütotoksilisus
: käesoleva katsemeetodi kohaste katsete puhul on tsütotoksilisus määratletud kui suhtelise kogukasvu (MLA korral) või suhtelise ellujäämuse (TK6 korral) vähenemine.
Otsemutatsioon
: geenimutatsioon, mille puhul algne geen võtab mutantse vormi ja mille tulemusena muutub või kaob asjaomase kodeeritud valgu ensümaatiline aktiivsus või funktsioon.
Raaminihet põhjustav mutageen
: kemikaal, mille mõjul lisatakse DNA molekuli või eemaldatakse sealt üks või mitu aluspaari.
Genotoksilisus
: üldmõiste, mis hõlmab DNA ja kromosoomide igat liiki kahjustusi, sealhulgas DNA-ahela katkemisi, adukte, ümberkorraldusi, mutatsioone, kromosoomiaberratsioone ja aneuploidsust põhjustavaid omadusi. Genotoksiline mõju ei väljendu alati mutatsiooni või püsiva kromosoomikahjustusena.
Mitootiline rekombinatsioon
: mitoosi ajal homoloogsete kromatiidide vahel toimuv rekombinatsioon, millega võib kaasneda DNA kahe ahela katkemine või heterosügootsuse kadu.
Mutageensus
: omadus, mis põhjustab pärilikke muutusi geenide DNA aluspaaride järjestuses või kromosoomide struktuuris (kromosoomiaberratsioonid).
Muteerumissagedus (MF)
: vaadeldud mutantrakkude arv, mis on jagatud elujõuliste rakkude arvuga.
Fenotüübi ekspressiooniaeg
: töötlusele järgnev aeg, mille vältel geneetiline muutus kinnistub genoomis ja mis tahes varem esinenud geeniproduktide sisaldus väheneb nii palju, et asjaomane fenotüübiline tunnus muutub.
Suhteline ellujäämus (RS)
: näitaja, mille kaudu TK6 korral väljendatakse töötlusest tulenevat tsütotoksilisust. See on vahetult pärast töötlemist välja külvatud rakkude kloonimise tõhusus (CE), mida on kohandatud rakkude arvu võimaliku vähenemise suhtes töötlemise ajal, võrrelduna negatiivse kontrollprooviga leitud kloonimistõhususega.
Suhteline suspensioonikasv (RSG)
: MLA korral katsekultuuri kogu suhteline suspensioonikasv kahe päeva jooksul, võrrelduna negatiivse/lahusti kontrollprooviga leitud kogu suspensioonikasvuga kahe päeva jooksul (Clive ja Spector, 1975). RSG peaks hõlmama katsekultuuri suhtelist kasvu võrrelduna negatiivse/lahusti kontrollprooviga töötlemisperioodi jooksul.
Suhteline kogukasv (RTG)
: näitaja, mille kaudu MLA korral väljendatakse töötlusest tulenevat tsütotoksilisust. Sellega väljendatakse katsekultuuride suhtelist (kandeaine kontrollprooviga võrreldud) kasvu katse töötlemis-, kahepäevases avaldumis- ja mutantide selektiivse kloonimise faasis. Iga katsekultuuri RSG korrutatakse katsekultuuri suhtelise kloonimistõhususega mutantide selekteerimise ajal ning väljendatakse negatiivse/kandeaine kontrollprooviga leitud kloonimistõhususe suhtes (Clive ja Spector, 1975).
Maksafraktsioon S9
: maksa homogenaadi supernatant, mis on eraldatud pärast tsentrifuugimist 9 000g juures, st maksa toorekstrakt.
S9 segu
: maksafraktsiooni S9 ja metaboolsete ensüümide aktiivsuse tagamiseks vajalike kofaktorite segu.
Suspensioonikasv (SG)
: rakkude arvu kordne suurenemine MLA töötlemis- ja avaldumisfaasi jooksul. SG arvutamiseks korrutatakse lühiajalise töötlemise (3 või 4 tundi) korral kordne kasv 1. päeval kordse kasvuga 2. päeval. Kui kasutati 24-tunnist töötlemist, on SG 24 tunnise töötlemisaja kordse kasvu korrutis 1. ja 2. avaldumispäeva kordsete kasvudega.
Lahusti kontrollproov
: üldmõiste, millega tähistatakse kontrollkultuuri, kuhu on lisatud üksnes uuritava kemikaali lahustamiseks kasutatud lahustit.
Uuritav kemikaal
: iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
Töötlemata kontrollproov
: töötlemata kontrollproovid on rakukultuurid, mida ei ole töödeldud (st ei uuritava kemikaali ega lahustiga), kuid mida on käideldud samaaegselt samal viisil kui uuritava kemikaaliga töödeldud rakukultuure.
2. liide
VALEMID
Tsütotoksilisus
Nii MLA agari- kui ka mikrotiiterplaadi variandi korral
Tsütotoksilisus on määratletud suhtelise kogukasvuna (RTG), mis hõlmab suhtelise kogukasvu (RSG) 2-päevase avaldumisperioodi jooksul ja mutantide selekteerimise ajal leitud suhtelise kloonimistõhususe (RCE). Nii RTG, RSG kui ka RCE väljendatakse protsentides.
RSG arvutamine: suspensioonikasv SG1 on 0-päeva ja 1. päeva vaheline kasvumäär (rakkude kontsentratsiooni 1. päeval / rakkude kontsentratsioon 0-päeval) ja suspensioonikasv SG2 on 1. päeva ja 2. päeva vaheline kasvumäär (rakkude kontsentratsioon 2. päeval / rakkude kontsentratsioon 1. päeval). RSG on töödeldud kultuuri kogu SG (SG1 × SG2) võrrelduna töötlemata/lahusti kontrollproovi omaga. Seega: RSG = [SG1(uuritav) × SG2(uuritav)] / [SG1(kontrollproov) × SG2(kontrollproov)]. SG1 arvutamisel tuleb lähtuda rakkude algsest kontsentratsioonist, mida kasutati rakkude töötlemise alguses. Seega võetakse arvesse rakkude töötlemise ajal katsekultuuris ilmnevaid erinevaid tsütotoksilisi toimeid.
RCE on katsekultuuri suhteline kloonimistõhusus võrreldes töötlemata/lahusti kontrollproovi suhtelise kloonimistõhususega mutantide selekteerimise ajal.
Suhteline kogukasv (RTG):
RTG=RSG × RCE
TK6
Suhteline Ellujäämus (RS):
tsütotoksilisust hinnatakse suhtelise ellujäämusega, st vahetult pärast töötlemist välja külvatud rakkude kloonimise tõhususega, kohandatuna rakkude arvu võimaliku vähenemise suhtes töötlemise käigus ja võrrelduna kloonimistõhususega negatiivsete kontrollproovide puhul, mille ellujäämuseks loetakse 100 %. Kohandusarvutus, mis võtab arvesse rakkude arvu vähenemist töötlemise ajal, on järgmine:
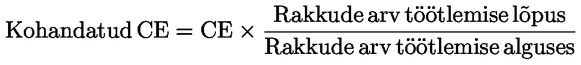
Uuritava kemikaaliga töödeldud kultuuri RS arvutatakse järgmiselt:

Muteerumissagedus MLA ja TK6 Korral
Muteerumissagedus (MF) on mutantsete kolooniate kloonimistõhusus selektiivsöötmes (CEM), mis on kohandatud selekteerimisaegse kloonimistõhususega selektiivaineta söötmes (CEV). Seega MF = CEM/CEV. Nende kahe kloonimistõhususe arvutuskäigud on esitatud allpool kloonimise agari- ja mikrotiiterplaadimeetodi jaoks.
MLA agarivariant: MLA pehme agari variandi korral saadakse kolooniate arv mutantide selekteerimisplaadil (CM) ja kolooniate arv selektiivaineta või kloonimistõhususe (eluvõimeliste rakkude arv) leidmise plaadil (CV) vahetult kolooniaid loendades. Kui 600 rakku külvatakse kloonimistõhususe (CE) määramiseks mutantide selekteerimisplaatidele (CEM) ja selektiivaineta või kloonimistõhususe (eluvõimeliste rakkude arv) määramise plaatidele (CEV) ja mutantide selekteerimiseks kasutatakse 3 × 106 rakku, siis
CEM = CM / (3 × 106) = (CM / 3) × 10-6
CEV = CV / 600
MLA ja TK6 mikrotiiterplaadi variant:
MLA ja TK6 mikrotiiterplaadi variandi korral arvutatakse CM ja CV korrutisena mikrotiiterplaadi süvendite koguarvust (TW) ja tõenäolisest kolooniate arvust süvendi kohta (P).
CM = PM × TWM
CV = PV × TWV
Lähtudes Poissoni jaotuse kujust, kui argument on null (Furth et al., 1981), saab P avaldada järgmiselt:
P = – ln (EW / TW),
kus EW on tühjade süvendite arv ja TW on süvendite koguarv. Niisiis:
CEM = CM / TM = (PM × TWM) / TM
CEV = CV / TV = (PV × TWV) / TV
MLA mikrotiiterplaadi variandi korral arvutatakse väikeste ja suurte kolooniate muteerumissagedused samal viisil, kasutades vastavalt väikeste ja suurte kolooniatega seotud tühjade süvendite arvu.
TK6 korral on väikeste ja suurte kolooniate muteerumissageduste aluseks vara tekkivad ja hilja tekkivad mutandid.
B.68 LÜHIAJALISE KOKKUPUUTE IN VITRO KATSEMEETOD SELLISTE KEMIKAALIDE KINDLAKSTEGEMISEKS, I) MIS TEKITAVAD RASKEID SILMAKAHJUSTUSI, JA II) MIDA EI OLE VAJA KLASSIFITSEERIDA SEOSES SILMADE ÄRRITUSE VÕI RASKE SILMAKAHJUSTUSEGA
SISSEJUHATUS
|
1.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 491 (2017). Lühiajalise kokkupuute katsemeetod on in vitro meetod, mida võib teatavatel asjaoludel ja teatavate piirangutega kasutada, et klassifitseerida ja märgistada kemikaale (aineid ja segusid), mis põhjustavad rasket silmakahjustust või silmade ärritust, nagu need on määratletud Ühinenud Rahvaste Organisatsiooni ühtses ülemaailmses kemikaalide klassifitseerimise ja märgistamise süsteemis (GHS) (1) ning Euroopa Liidu määruses (EÜ) nr 1272/2008, mis käsitleb ainete ja segude klassifitseerimist, märgistamist ja pakendamist (CLP-määrus) (66).
|
|
2.
|
Paljude aastate jooksul on kemikaalidest tingitud silmakahjustuse ohu hindamiseks kasutatud peamiselt in vivo silmakatset küülikul (katsemeetod B.5 (8), samaväärne OECD katsejuhendiga nr 405). Üldiselt ollakse arvamusel, et lähitulevikus ei ole võimalik rasket silmakahjustust tekitava / silmi ärritava toime kogu ulatuse prognoosimisel eri kemikaaliklasside puhul ühegi alternatiivse in vitro katsega täielikult asendada in vivo silmakatset küülikul. Kuid astmelise katsetamise strateegia osana kasutatava mitme alternatiivse katsemeetodi strateegilise kombineerimisega võib siiski olla võimalik küüliku silmakatset (2) täielikult asendada. Ülalt-alla lähenemisviisi tuleks kasutada katsetes kemikaalidega, mille puhul olemasoleva teabe põhjal võib eeldada, et asjaomane kemikaal võib silma tugevasti ärritada või põhjustada raske silmakahjustuse. Alt-üles lähenemisviisi tuleks kasutada vastupidiselt katsetes kemikaalidega, millel puhul olemasoleva teabe põhjal võib eeldada, et asjaomane kemikaal ei põhjusta klassifitseerimiseks piisavat silmade ärritust. Kuigi ei peeta võimalikuks, et lühiajalise kokkupuute katsemeetod asendaks täielikult in vivo silmakatset küülikuga, sobib see kasutamiseks astmelise katsetamise strateegia osana õigusliku klassifitseerimise ja märgistamise jaoks näiteks ülalt-alla ja alt-üles lähenemisviisi puhul, et teha täiendava katsetamiseta kindlaks i) kemikaalid, mis põhjustavad raske silmakahjustuse (ÜRO GHSi / CLP-määruse kohane 1. kategooria) ja ii) kemikaalid (välja arvatud väga lenduvad ained ja kõik tahked kemikaalid peale pindaktiivsete ainete), mida ei ole vaja klassifitseerida seoses silmade ärrituse või raske silmakahjustusega (ÜRO GHSi / CLP-määruse kohane kategooria puudub) (1, 2). Kuid niisugune kemikaal, mille puhul lühiajalise kokkupuute katsemeetodiga ei prognoosita raske silmakahjustuse põhjustamist (ÜRO GHSi / CLP-määruse kohane 1. kategooria) ega ka ÜRO GHSi / CLP-määruse kohase kategooria puudumist (ei põhjusta rasket silmakahjustust ega silmade ärritust), vajab siiski täiendavat uurimist, et saaks määrata lõpliku klassifikatsiooni. Peale selle tuleks konsulteerida asjakohaste reguleerivate asutustega, enne kui lühiajalise kokkupuute katset hakatakse muu kui ÜRO GHSi / CLP-määruse klassifitseerimisskeemi korral kasutama alt-üles lähenemisviisi rakendamiseks. Kõige asjakohasema katsemeetodi valimist ja selle kasutamist tuleks vaadelda raske silmakahjustuse ja silmade ärrituse põhjustamise uurimiseks ja hindamiseks kasutatavaid kompleksseid lähenemisviise käsitleva OECD juhenddokumendi (14) kontekstis.
|
|
3.
|
Käesoleva katsemeetodi eesmärk on kirjeldada eeskirju, mida kasutatakse, et hinnata uuritava kemikaali võimalikku ohtlikkust silmale selle võime põhjal kutsuda esile tsütotoksilisust lühiajalise kokkupuute meetodi kasutamisel. Kemikaalide tsütotoksiline mõju sarvkesta epiteelirakkudele on oluline toimemehhanism, mille tulemusena tekib sarvkesta epiteeli kahjustus ja silmade ärritus. Lühiajalise kokkupuute katsemeetodiga mõõdetakse rakkude eluvõimelisust vitaalvärvaine MTT (3-(4,5-dimetüültiasool-2-üül)-2,5-difenüültetrasooliumbromiid, tiasolüülsinine) ensümaatilise muundamise järgi siniseks formasaansoolaks, mis pärast rakkudest ekstraheerimist määratakse kvantitatiivselt (3). Saadud rakkude eluvõimelisust võrreldakse lahusti kontrollprooviga (suhteline eluvõimelisus) ja kasutatakse uuritavast kemikaalist tingitud võimaliku silmakahjustuse ohu hindamiseks. Uuritav kemikaal klassifitseeritakse ÜRO GHSi / CLP-määruse kohasesse 1. kategooriasse, kui nii 5 % kui ka 0,05 % kontsentratsiooni juures on rakkude eluvõimelisus alla 70 % või sellega võrdne. Kui rakkude eluvõimelisus nii 5 % kui ka 0,05 % kontsentratsiooni juures on üle 70 %, viidatakse kemikaalile kui ÜRO GHSi / CLP-määruse kohase kategooriata kemikaalile.
|
|
4.
|
Käesolevas katsemeetodis kasutatakse terminit „uuritav kemikaal“ uurimisobjektile viitava terminina ja see ei ole seotud lühiajalise kokkupuute katsemeetodi kohaldatavusega ainete ja/või segude uurimiseks. Mõisted on esitatud liites.
|
LÄHTEKAALUTLUSED JA PIIRANGUD
|
5.
|
Käesolev katsemeetod põhineb Kao Corporationi välja töötatud metoodikal (4), millega viidi läbi kaks eraldi valideerimisuuringut: ühe tegi Jaapani loomkatsete alternatiivide ühing (JSAAE) (5) ning teise Jaapanialternatiivmeetodite valideerimiskeskus (JaCVAM) (6). Vastastikuse eksperdihinnangu tegid Ameerika Ühendriikide riikliku toksikoloogiaprogrammi (NTP) alternatiivsete toksikoloogiameetodite ametitevaheline keskus (National Toxicology Program (NTP) Interagency Center for the Evaluation of Alternative Toxicological Methods, NICEATM) ja alternatiivmeetodite valideerimise ametitevaheline koordineerimiskomitee (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) (7) valideerimisuuringute aruannete ning katsemeetodi taustaülevaatedokumentide põhjal.
|
|
6.
|
Kui lühiajalise kokkupuute katsemeetodit kasutatakse selliste kemikaalide (ainete ja segude) kindlakstegemiseks, mis tekitavad rasket silmakahjustust (ÜRO GHSi / CLP-määruse kohane 1. kategooria) (1), on lühiajalise kokkupuute katsemeetodil 125 kemikaali (nii ained kui ka segud) kohta saadud andmete kogutäpsus 83 % (104/125), valepositiivsete tulemuste määr on 1 % (1/86) ja valenegatiivsete tulemuste määr 51 % (20/39), võrreldes küüliku in vivo silmakatsega (7). Saadud valenegatiivsete tulemuste määr ei ole selles kontekstis kriitiline, sest kõiki uuritavaid kemikaale, mille puhul rakkude eluvõimelisus 5 % kontsentratsiooni juures on ≤ 70 % ja 0,05 % kontsentratsiooni juures > 70 %, uuritakse järgmiseks muude asjakohaselt valideeritud in vitro katsemeetoditega või viimase võimalusena, olenevalt regulatiivsetest nõuetest, küüliku in vivo silmakatsetes kooskõlas järjestikuse katsete tegemise strateegia ning hetkel soovitatava tõendite kaalukuse lähenemisviisiga (1, 8). Põhiliselt uuriti ühe koostisosaga aineid, kuigi piiratud kogus andmeid on olemas ka katsetest segudega. See katsemeetod on siiski tehniliselt kohaldatav mitme koostisosaga ainete ja segude uurimiseks. Enne kui kasutada seda katsemeetodit segu korral, et saada andmeid kavandatud regulatiivse eesmärgi jaoks, tuleb siiski kaaluda, kas see katse annab selle eesmärgi jaoks piisavaid tulemusi, ja kui annab, siis miks. Sellist kaalumist ei ole vaja, kui regulatiivne nõue eeldabki segu katsetamist. Lühiajalise kokkupuute katsemeetodil ei ole spetsiifilisi puudusi, kui seda kasutatakse nende uuritavate kemikaalide kindlakstegemisel, mis kuuluvad ÜRO GHSi / CLP-määruse kohasesse 1. kategooriasse. Teadlased võiksid kaaluda uuritavate kemikaalide puhul kõnealuse katsemeetodi kasutamist, kusjuures rakkude eluvõimelisus ≤ 70 % nii 5 % kui ka 0,05 % kontsentratsiooni juures tuleks lugeda tõendiks, et kemikaal põhjustab rasket silmakahjustust ja tuleks liigitada ÜRO GHSi / CLP-määruse kohasesse 1. kategooriasse ilma edasise katsete tegemiseta.
|
|
7.
|
Kui lühiajalise kokkupuute katsemeetodit kasutatakse selliste kemikaalide (ainete ja segude) kindlakstegemiseks, mida ei ole vaja klassifitseerida seoses silmade ärrituse ja raske silmakahjustusega (ÜRO GHSi / CLP-määruse kohane kategooria puudub), on lühiajalise kokkupuute katsemeetodil 130 kemikaali (nii ained kui ka segud) kohta saadud andmete kogutäpsus 85 % (110/130), valenegatiivsete tulemuste määr on 12 % (9/73) ja valepositiivsete tulemuste määr 19 % (11/57), võrreldes küüliku in vivo silmakatsega (7). Kui andmestikust jäetakse välja väga lenduvad ained ja tahked ained peale pindaktiivsete ainete, paraneb kogutäpsus 90 %-ni (92/102), valenegatiivsete tulemuste määra 2 %-ni (1/54) ja valepositiivsete tulemuste määr 19 %-ni (9/48) (7). Seega on lühiajalise kokkupuute katsemeetodi võimalik puudus nende kemikaalide kindlakstegemiseks kasutamise korral, mida ei ole vaja klassifitseerida seoses silmade ärrituse või raske silmakahjustusega (ÜRO GHSi / CLP-määruse kohane kategooria puudub), suur valepositiivsete tulemuste määr, kui tegemist on i) üle 6 kPa aururõhuga väga lenduvate ainetega ning ii) tahkete kemikaalidega (ained ja segud), välja arvatud pindaktiivsed ained ja ainult pindaktiivsetest ainetest koosnevad segud. Niisugused kemikaalid jäetakse lühiajalise kokkupuute katsemeetodi (7) kohaldamisvaldkonnast välja.
|
|
8.
|
Lisaks punktides 6 ja 7 nimetatud kemikaalidele sisaldab lühiajalise kokkupuute katsemeetodiga saadud andmestik ettevõttesiseseid andmeid 40 segu kohta ning võrreldes Draize'i in vivo silmakatsega on nende andmete täpsus 88 % (35/40), valepositiivsete tulemuste määr 50 % (5/10) ja valenegatiivsete tulemuste määr 0 % (0/30), kui prognoosida nende segude omadusi, mida ei ole vaja ÜRO GHSi / CLP-määruse klassifitseerimissüsteemide kohaselt klassifitseerida (9). Seega võib lühiajalise kokkupuute katsemeetodit kasutada ÜRO GHSi / CLP-määruse kohase kategooriata segude kindlakstegemiseks alt-üles lähenemisviisi järgides, välja arvatud muude tahkete segude korral kui ainult pindaktiivsetest ainetest koosnevad segud, kuna nendele laieneb tahkete ainetega seotud piirang. Peale selle tuleks segusid, mis sisaldavad üle 6 kPa aururõhuga aineid, hoolikalt hinnata, et vältida võimalikku alahindamist, ning see peab olema igal üksikjuhtumil põhjendatud.
|
|
9.
|
Lühiajalise kokkupuute katsemeetodit ei saa kasutada nende uuritavate kemikaalide kindlakstegemiseks, mida klassifitseerida ÜRO GHSi / CLP-määruse kohasesse 2. kategooriasse, ÜRO GHSi kohasesse 2A (silmade ärritus) või 2B kategooriasse (kerge silmade ärritus); selle põhjuseks on nende ÜRO GHSi / CLP-määruse kohase 1. kategooria kemikaalide suur arv, mis on alahinnatud 2., 2A või 2B kategooriasse kuuluvaks, samuti nende ÜRO GHSi / CLP-määruse kohase kategooriata kemikaalide suur arv, mis on ülehinnatud 2., 2A või 2B kategooriasse kuuluvaks (7). Selleks võib vaja minna täiendavaid uuringuid muu sobiva meetodiga.
|
|
10.
|
Lühiajalise kokkupuute meetod sobib uuritavate kemikaalide puhul, mis lahustuvad füsioloogilises lahuses, 5 % dimetüülsulfoksiidisisaldusega füsioloogilises lahuses või mineraalõlis või suspendeeruvad nendes ühtlaselt vähemalt viieks minutiks. Lühiajalise kokkupuute meetod ei sobi uuritavate kemikaalide puhul, mis füsioloogilises lahuses, 5 % dimetüülsulfoksiidisisaldusega füsioloogilises lahuses või mineraalõlis ei lahustu ega nendes vähemalt viieks minutiks ühtlaselt ei suspendeeru. Mineraalõli kasutamine lühiajalise kokkupuute meetodi korral on võimalik lühiajalise kokkupuute tõttu. Seega sobib lühiajalise kokkupuute meetod vees lahustumatute uuritavate kemikaalide (nt pikaahelalised rasvhapped või ketoonid) võimaliku silmale ohtlikkuse hindamiseks tingimusel, et kõnealused kemikaalid segunevad vähemalt ühega kolmest eespool soovitatud lahustist (4).
|
|
11.
|
Käesolevas katsemeetodis kasutatakse terminit „uuritav kemikaal“ uurimisobjektile viitava terminina (67) ja see ei ole seotud lühiajalise kokkupuute katsemeetodi kohaldatavusega ainete ja/või segude uurimiseks.
|
KATSE PÕHIMÕTE
|
12.
|
Lühiajalise kokkupuute katsemeetod on tsütotoksilisusel põhinev in vitro meetod, mis viiakse läbi Taani riikliku seerumiinstituudi küüliku sarvkesta (Statens Seruminstitut Rabbit Cornea – SIRC) rakkude konfluentse monokihiga, mis kasvatatakse 96 süvendiga, polükarbonaadist mikrotiiterplaadil (4). Pärast 5-minutist kokkupuudet uuritava kemikaaliga määratakse tsütotoksilisus kvantitatiivselt SIRC-rakkude suhtelise eluvõimelisusena MTT-katsel (4). Rakkude vähenenud eluvõimelisuse alusel prognoositakse silmakahjustusi põhjustavat võimalikku kahjulikku mõju.
|
|
13.
|
On teatatud, et 80 % küüliku silma tilgutatud lahusest väljutatakse sidekestakoti kaudu kolme kuni nelja minuti jooksul ning üle 80 % inimese silma tilgutatud lahusest väljutatakse ühe kuni kahe minuti jooksul (10). Lühiajalise kokkupuute katsemeetodiga on püütud neid kokkupuuteaegu ühtlustada ning kasutatud tsütotoksilisust näitajana, mille alusel hinnata pärast 5-minutist kokkupuudet uuritava kemikaaliga SIRC-rakkudele tekitatud kahjustuste ulatust.
|
PÄDEVUSE TÕENDAMINE
|
14.
|
Enne käesoleval katsemeetodil põhineva lühiajalise kokkupuute meetodi rutiinset kasutamist tuleb laboril tõendada oma tehnilist pädevust, klassifitseerides õigesti tabelis 1 soovitatud üksteist ainet. Need ained on valitud nii, et need esindavad rasket silmakahjustust või silmade ärritust tekitavate ainete põhjustatud reaktsioonide täielikku vahemikku, mis põhineb küüliku silma in vivo katse (katsemeetod 405) ja ÜRO GHSi / CLP-määruse kohase klassifitseerimissüsteemi tulemustel (1). Teisteks valikukriteeriumideks olid ainete kaubanduslik kättesaadavus, kvaliteetsete võrdlusandmete kättesaadavus in vivo katsete kohta ning kvaliteetsete võrdlusandmete kättesaadavus lühiajalise kokkupuute meetodil tehtud in vitro katsete kohta (3). Olukorras, kus loetletud aine ei ole kättesaadav, või kui see on põhjendatud, võib kasutada muud ainet, mille puhul on kättesaadavad piisavad võrdlusandmed in vivo ja in vitro katsete kohta, eeldusel, et järgitakse eespool kirjeldatud kriteeriumeid.
Tabel 1
Pädevuse tõendamise ainete loend
|
Aine
|
CASi nr
|
Aineklass (68)
|
Füüsikaline olek
|
In vivo katsel määratud ÜRO GHSi /CLP-määruse kohane kategooria (69)
|
Lühiajalise kokkupuute katsel kasutatud lahusti
|
Lühiajalise kokkupuute katsel määratud ÜRO GHSi /CLP-määruse kohane kategooria
|
|
Bensalkooniumkloriid (10 % vesilahus)
|
8001-54-5
|
Ooniumühend
|
Vedel
|
1. kategooria
|
Füsioloogiline lahus
|
1. kategooria
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Eeter
|
Vedel
|
1. kategooria
|
Füsioloogiline lahus
|
1. kategooria
|
|
Happeline punane 92
|
18472-87-2
|
Heterotsükliline ühend; broomiühend; klooriühend
|
Tahke
|
1. kategooria
|
Füsioloogiline lahus
|
1. kategooria
|
|
Naatriumhüdroksiid
|
1310-73-2
|
Leelis; anorgaaniline aine
|
Tahke
|
1. kategooria (70)
|
Füsioloogiline lahus
|
1. kategooria
|
|
Butürolaktoon
|
96-48-0
|
Laktoon; heterotsükliline ühend
|
Vedel
|
2A kategooria (CLP-määruse kohane 2. kategooria)
|
Füsioloogiline lahus
|
Prognoosida ei ole võimalik
|
|
1-oktanool
|
111-87-5
|
Alkohol
|
Vedel
|
2A/B kategooria (71)(CLP-määruse kohane 2. kategooria)
|
Mineraalõli
|
Prognoosida ei ole võimalik
|
|
Tsüklopentanool
|
96-41-3
|
Alkohol; süsivesinik (tsükliline)
|
Vedel
|
2A/B kategooria (72) (CLP-määruse kohane 2. kategooria)
|
Füsioloogiline lahus
|
Prognoosida ei ole võimalik
|
|
2-etoksüetüülatsetaat
|
111-15-9
|
Alkohol; eeter
|
Vedel
|
Kategooriata
|
Füsioloogiline lahus
|
Kategooriata
|
|
Dodekaan
|
112-40-3
|
Süsivesinik (atsükliline)
|
Vedel
|
Kategooriata
|
Mineraalõli
|
Kategooriata
|
|
Metüülisobutüülketoon
|
108-10-1
|
Ketoon
|
Vedel
|
Kategooriata
|
Mineraalõli
|
Kategooriata
|
|
1,1-dimetüülguanidiinsulfaat
|
598-65-2
|
Amidiin Väävliühend
|
Tahke
|
Kategooriata
|
Füsioloogiline lahus
|
Kategooriata
|
|
Lühendid: CASi nr = Chemical Abstracts Service’i registrinumber
|
|
KATSE KÄIK
Rakkude monokihi valmistamine
|
15.
|
Lühiajalise kokkupuute katsemeetodi rakendamiseks tuleks kasutada küüliku sarvkesta rakkude SIRC-liini. Soovitatav on soetada SIRC-rakud usaldusväärsest rakupangast, näiteks USA tüüpkultuuride kollektsioonist (American Type Culture Collection), tootekood CCL60.
|
|
16.
|
SIRC-rakke kasvatatakse 37 °C juures reguleeritud õhuniiskusega 5 % CO2-sisaldusega atmosfääris kultuurikolvis, mis sisaldab kasvusöödet, mis koosneb Eagle'i essentsiaalsest minimaalsöötmest (MEM), millele on lisatud 10 % veise loote seerumit (FBS), 2 mM L-glutamiini, 50–100 ühikut/ml penitsilliini ja 50–100 μg/ml streptomütsiini. Rakud, mis on saavutanud kultuurikolvis konfluentsuse, eraldatakse trüpsiini etüleendiamiintetraäädikhappe lahusega, kasutada võib rakukaabitsat. Enne rutiinanalüüsis kasutamist kasvatatakse rakke (nt 2–3 passaaži) kultuurikolvis ning need ei tohiks pärast sulatamist läbida üle 25 passaaži.
|
|
17.
|
Lühiajalise kokkupuute katses kasutamiseks valmis rakud külvatakse sobiva tihedusega 96 süvendiga plaatidesse. Kui kultuuri ruumala on 200 μl, on rakkude soovitatav külvitihedus 6,0 × 103 rakku süvendis, kui rakke kasutatakse neli päeva pärast külvamist, või 3,0 × 103 rakku süvendis, kui rakke kasutatakse viis päeva pärast külvamist. Lühiajalise kokkupuute katses kasutatavad rakud, mis külvatakse kasvusöötmesse sobiva tihedusega, saavutavad katse läbiviimise ajaks, st 4. või 5. külvijärgseks päevaks üle 80 % konfluentsuse.
|
Uuritavate kemikaalide ja kontrollainete kasutamine
|
18.
|
Uuritavate kemikaalide lahustamiseks või suspendeerimiseks tuleks esmajoones valida füsioloogiline lahus. Kui uuritavad kemikaalid lahustuvad füsioloogilises lahuses halvasti või neid ei saa selles vähemalt viieks minutiks lahustada või ühtlaselt suspendeerida, tuleks teise lahustina valida 5 % DMSO (CASi nr 67-68-5) sisaldusega füsioloogiline lahus. Nende uuritavate kemikaalide jaoks, mida ei saa vähemalt viieks minutiks lahustada või ühtlaselt suspendeerida ei füsioloogilises lahuses ega 5 % DMSO-sisaldusega füsioloogilises lahuses, kasutatakse kolmanda valikuna mineraalõli (CASi nr 8042-47-5).
|
|
19.
|
Uuritavad kemikaalid lahustatakse või suspendeeritakse valitud lahustis ühtlaselt 5 % kontsentratsiooni (massiprotsent) juures ning valmistatakse järjestikuste 10-kordsete lahjendamiste teel 0,5 % ja 0,05 % kontsentratsiooniga lahused. Iga uuritava kemikaaliga viiakse katse läbi nii 5 % kui ka 0,05 % kontsentratsiooni juures. 96 süvendiga plaadis kasvatatud rakud viiakse toatemperatuuril viieks minutiks kokkupuutesse uuritava kemikaali lahuse või suspensiooniga, mille kontsentratsioon on kas 5 % või 0,05 % ja mida lisatakse 200 μl süvendi kohta. Uuritavaid kemikaale (ühe või mitme koostisainega ained või segud) käsitletakse nende puhtusastet arvestamata puhaste ainetena ning need lahustatakse või suspendeeritakse vastavalt meetodis esitatud juhistele.
|
|
20.
|
Punktis 16 kirjeldatud kasvusöödet kasutatakse söötmega kontrolliks iga korduskatse igas plaadis. Peale selle tuleb rakud iga korduskatse igas plaadis viia kokkupuutesse ka lahusti kontrollproovidega. On kindlaks tehtud, et punktis 18 loetletud lahustitel ei ole kahjulikku mõju SIRC-rakkude eluvõimelisusele.
|
|
21.
|
Lühiajalise kokkupuute katsemeetodi korral kasutatakse iga korduskatse igas plaadis positiivseks kontrolliks 0,01 % naatriumlaurüülsulfaadisisaldusega füsioloogilist lahust. Positiivse kontrolli rakkude eluvõimelisuse arvutamiseks peab iga korduskatse iga plaat hõlmama ka lahusti kontrollproovi füsioloogilise lahusega.
|
|
22.
|
Tühiproov on vajalik optilise tiheduse kompensatsiooni määramiseks ja seda tuleks kontrollida süvendites, mis sisaldavad ainult fosfaadiga puhverdatud füsioloogilist lahust (PBS) ega sisalda kaltsiumi, magneesiumi ega rakke.
|
|
23.
|
Igat proovi (uuritava kemikaali 5 % ja 0,05 % lahus, lahusti kontrollproov, söötme kontrollproov ja positiivne kontroll) tuleb igas korduskatses analüüsida kolme paralleelproovina, viies rakud toatemperatuuril viieks minutiks kokku 200 μl asjaomase uuritava või kontrollkemikaaliga.
|
|
24.
|
Võrdlusained võimaldavad hinnata teatavasse kemikaali- või tooteklassi kuuluva uurimata kemikaali omadust põhjustada silmade ärritust või hinnata teatava silmi ärritava aine mõju tugevust teatavas ärritava toime vahemikus.
|
Raku eluvõimelisuse mõõtmine
|
25.
|
Pärast kokkupuudet pestakse rakke kaks korda 200 μl PBSiga ja lisatakse 200 μl MTT lahust (0,5 mg MTTd 1 ml kasvusöötme kohta). Pärast kahetunnise reaktsiooniaja möödumist inkubaatoris (37 °C, 5 % CO2) MTT lahus dekanteeritakse ja lisatakse MTT formasaani ekstraheerimiseks 200 μl 0,04 N vesinikkloriidhappe lahust isopropanoolis, lastakse lahusel 60 minutit pimedas toatemperatuuril seista ning mõõdetakse plaadilugeriga neelduvus MTT formasaani lahuses lainepikkusel 570 nm. Uuritavate kemikaalide segav mõju MTT-katses (värvainete või MTT otseste redutseerijate puhul) esineb ainult juhul, kui uuritavasse süsteemi jääb pärast kokkupuutejärgset pesu märkimisväärne kogus uuritavat kemikaali. Seda juhtub inimese kolmemõõtmelise rekonstrueeritud sarvkesta või inimese kolmemõõtmelise rekonstrueeritud epidermise kudede puhul, kuid lühiajalise kokkupuute katsemeetodis kasutatud kahemõõtmeliste rakukultuuride puhul on see tähtsusetu.
|
Tulemuste tõlgendamine ja prognoosimismudel
|
26.
|
Iga uuritava kemikaali kohta saadud optilise tiheduse (OD) väärtusi kasutatakse selleks, et arvutada rakkude eluvõimelisus lahusti kontrollproovi suhtes, mille puhul eluvõimelisuseks loetakse 100 %. Rakkude suhtelist eluvõimelisust väljendatakse protsentides ning see saadakse, jagades uuritava kemikaali optilise tiheduse väärtuse lahusti kontrollproovi omaga, kusjuures eelnevalt on neist mõlemast lahutatud tühiproovi optilise tiheduse väärtus.

Samamoodi väljendatakse protsentides rakkude suhtelist eluvõimelisust igas lahusti kontrollproovis ning see saadakse, jagades iga lahusti kontrollproovi optilise tiheduse väärtuse söötme kontrollproovi omaga, kusjuures eelnevalt on neist mõlemast lahutatud tühiproovi optilise tiheduse väärtus.
|
|
27.
|
Läbi tuleb viia kolm sõltumatut korduskatset, millest igaüks toimub paralleelselt kolme süvendiga (st n=9). Rakkude suhtelise eluvõimelisuse aritmeetilise keskmise arvutamiseks kasutatakse sõltumatutel korduskatsetel iga uuritava kemikaali ja lahustiga kontrollprooviga kolmes süvendis saadud tulemuste aritmeetilist keskmist. Rakkude eluvõimelisuse lõplik aritmeetiline keskmine arvutatakse kolme sõltumatu korduskatse põhjal.
|
|
28.
|
Rakkude eluvõimelisuse läviväärtused, mille järgi uuritavaid kemikaale, mis põhjustavad rasket silmakahjustust (ÜRO GHSi / CLP-määruse kohane 1. kategooria), eristatakse uuritavatest kemikaalidest, mida ei ole vaja klassifitseerida seoses silmade ärrituse või raske silmakahjustusega (ÜRO GHSi / CLP-määruse kohase kategooriata kemikaal), on esitatud allpool.
|
Tabel 2
Lühiajalise kokkupuute katsemeetodi prognoosimismudel
|
Rakkude eluvõimelisus
|
ÜRO GHSi / CLP-määruse kohane klassifikatsioon
|
Kohaldamine
|
|
5 % korral
|
0,05 % korral
|
|
> 70 %
|
> 70 %
|
Kategooriata
|
Ained ja segud, välja arvatud: i) väga lenduvad ained aururõhuga üle 6 kPa (73) ja ii) tahked kemikaalid (ained ja segud) peale pindaktiivsete ainete ja ainul pindaktiivsetest ainetest koosnevate segude
|
|
≤ 70 %
|
> 70 %
|
Prognoosida ei ole võimalik
|
Ei kohaldata
|
|
≤ 70 %
|
≤ 70 %
|
1. kategooria
|
Ained ja segud (74)
|
Nõuetekohasuse kriteeriumid
|
29.
|
Katsetulemused loetakse nõuetekohaseks, kui on täidetud kõik järgmised kriteeriumid.
|
a)
|
Söötme kontrollproovi (kasvusööde, mida kokkupuutel kasutati) optiline tihedus peab pärast tühiproovi optilise tiheduse väärtuse lahutamist olema vähemalt 0,3;
|
|
b)
|
Eluvõimelisus lahusti kontrollproovis peab olema vähemalt 80 %, võrreldes eluvõimelisusega söötme kontrollproovis. Kui igal korduskatsel kasutatakse mitut lahusti kontrollproovi, peab neist igaühes rakkude eluvõimelisus olema üle 80 %, et nende lahustitega analüüsitud uuritavate kemikaalide omadusi oleks võimalik nõuetekohaselt hinnata.
|
|
c)
|
Positiivse kontrollprooviga (0,01 % naatriumlaurüülsulfaat) saadud rakkude eluvõimelisus võib varasemate uuringute keskväärtusest erineda kahe standardhälbe ulatuses. Positiivse kontrollproovi nõuetekohasuse ülem- ja alampiiri tuleb sageli, st iga kolme kuu möödudes ajakohastada, või tuleb laborites, kus katseid tehakse harva (st vähem kui kord kuus), iga kord teha ka nõuetekohane katse. Kui laboris ei viida läbi piisaval arvul eksperimente, et saavutada positiivse kontrollproovi tulemuste statistiliselt robustne jaotus, on lubatud kasutada meetodi väljatöötaja antud nõuetekohasuse ülem- ja alampiiri, st vahemikku 21,1–62,3 % vastavalt laboris aja jooksul kogutud andmetele, samas kui laborisisene jaotus tehakse kindlaks esimeste rutiinsete katsetega.
|
|
d)
|
Kolmest sõltumatust korduskatsest saadud rakkude lõpliku eluvõimelisuse standardhälve peab nii uuritava kemikaali 5 % kui ka 0,05 % kontsentratsiooni juures jääma alla 15 %.
Kui üks või mitu nendest kriteeriumidest ei ole täidetud, tuleb tulemused kehtetuks lugeda ja läbi viia veel kolm sõltumatut korduskatset.
|
|
ANDMED JA ARUANDLUS
Andmed
|
30.
|
Esitada tuleb iga korduskatse iga üksiksüvendi kohta saadud andmed (nt rakkude eluvõimelisuse väärtused) ning üldine keskväärtus, standardhälve ja klassifikatsioon.
|
Katseprotokoll
|
31.
|
Katseprotokollis tuleks esitada järgmine teave.
|
|
Uuritav kemikaal ja kontrollained
|
—
|
Ühest koostisosast koosnev aine: kemikaali identifitseerimisandmed, nagu IUPACi või CASi nimetus(ed) ja CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muud identifikaatorid;
|
|
—
|
mitme koostisosaga aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal ja segu: võimalikult täpne omaduste kirjeldus lähtuvalt koostisosade keemilisest määratlusest (vt eespool), kvantitatiivsest sisaldusest ja asjakohastest füüsikalis-keemilistest omadustest (vt eespool), kättesaadavas ulatuses;
|
|
—
|
füüsikaline olek, lenduvus, pH, log P, molekulmass, aineklass ja muud uuringu tegemisega seotud asjakohased füüsikalis-keemilised omadused, kättesaadavas ulatuses;
|
|
—
|
puhtus, lisandite keemiline määratlus, kui see on asjakohane ja praktiliselt teostatav jne;
|
|
—
|
töötlemine enne katset, kui asjakohane (nt soojendamine, peenestamine);
|
|
—
|
säilitamise tingimused ja stabiilsus säilitamisel, kättesaadavas ulatuses.
|
|
|
|
Katsetingimused ja -eeskirjad
|
—
|
Sponsori, katselabori ja uuringu juhi nimi ja aadress;
|
|
—
|
kasutatud katsemeetodi kirjeldus;
|
|
—
|
kasutatud rakuliin, selle päritolu, passaažide arv ja katses kasutatud rakkude konfluentsus;
|
|
—
|
kasutatud katse-eeskirja üksikasjad;
|
|
—
|
kordus- ja paralleelproovide arv;
|
|
—
|
uuritava kemikaali kasutatud kontsentratsioonid (kui erinevad soovitatust);
|
|
—
|
iga uuritava kemikaali puhul lahusti valimise põhjendus;
|
|
—
|
uuritava kemikaaliga kokkupuute kestus (kui erineb soovitatust);
|
|
—
|
kõikide katse-eeskirjas tehtud muudatuste kirjeldus;
|
|
—
|
kasutatud hindamis- ja otsustamiskriteeriumide kirjeldus;
|
|
—
|
viide aja jooksul kogutud positiivsete kontrollproovide tulemuste keskväärtusele ja standardhälbele;
|
|
—
|
– labori kasutatud meetod, millega tõendatakse katsemeetodi kasutamise pädevust (nt pädevuse kontrolli ainetega katsetamine) või meetod, millega tõendatakse katsemeetodi korratavat toimivust ajas.
|
|
|
|
Tulemused
|
—
|
Iga uuritava kemikaali ja kontrollaine ning iga uuritava kontsentratsiooni kohta tuleb tabeli kujul esitada optiline tihedus igas paralleelkatse süvendis, iga sõltumatu korduskatse optilise tiheduse väärtuste aritmeetiline keskmine, iga sõltumatu korduskatse rakkude eluvõimelisus protsentides ning kokku kolmes korduskatses saadud rakkude eluvõimelisuse (%) lõplik aritmeetiline keskmine ja standardhälve;
|
|
—
|
söötme kontrollproovi, lahusti kontrollproovi ja positiivse kontrollproovi tulemused, mis tõendavad vastavust uuringu nõuetekohasuse kriteeriumidele;
|
|
—
|
muude täheldatud toimete kirjeldus;
|
|
—
|
tuletatud üldine/üldised klassifikatsioon(id) viitega kasutatud prognoosimudelile ja/või otsustamiskriteeriumidele.
|
|
|
KIRJANDUS
|
(1)
|
Ühinenud Rahvaste Organisatsioon (ÜRO), Globally Harmonized System of Classification and Labelling of Chemicals (GHS). 5. täiendatud väljaanne, United Nations Publications, New York & Genf, 2013. ISBN: 978-92-1-117006-1. Kättesaadav aadressil http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott, L. et al. (2010), „A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-up and Top-down Approaches“, Toxicol. in Vitro, nr 24, lk 1–9.
|
|
(3)
|
Mosmann, T. (1983). „Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays“, J. Immunol. Methods, nr 65, lk 55–63.
|
|
(4)
|
Takahashi, Y. et al. (2008), „Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells“, Toxicol. In Vitro, nr 22, lk 760–770.
|
|
(5)
|
Sakaguchi, H. et al. (2011), „Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals“, Toxicol. In Vitro, nr 25, lk 796–809.
|
|
(6)
|
Kojima, H. et al. (2013), „Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals“, Toxicol. In Vitro, nr 27, lk 1855–1869.
|
|
(7)
|
ICCVAM (2013), Short Time Exposure (STE) Test Method Summary Review Document, NIH. Kättesaadav aadressil [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Käesoleva lisa peatükk B.5 „Äge mürgisus: silmade ärritus/söövitus“.
|
|
(9)
|
Saito, K. et al (2015), „Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures“,
|
|
(10)
|
Mikkelson, T.J., Chrai, S.S. ja Robinson, J.R. (1973), „Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction“, J. Pharm. Sci., lk 1 648–1 653.
|
|
(11)
|
ECETOC (1998), Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brüssel, Belgia.
|
|
(12)
|
Gautheron, P. et al. (1992). „Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy“, Fundam. Appl. Toxicol., nr 18, lk 442–449.
|
|
(13)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(14)
|
OECD (2017), Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
Liide
MÕISTED
Täpsus
: katsemeetodiga saadud tulemuste ja nõuetekohaste võrdlusväärtuste kokkulangevuse määr. Täpsus näitab katsemeetodi tulemuslikkust ja on üks olulistest aspektidest. Seda terminit kasutatakse ka vastavuse tähenduses ja see näitab katsemeetodiga saadud õigete tulemuste osakaalu (13).
Võrdlusaine
: aine, mida kasutatakse standardina võrdlemiseks uuritava kemikaaliga. Võrdlusainel peaksid olema järgmised omadused: i) pidev ja usaldusväärne tarnija; ii) struktuuriline ja funktsionaalne sarnasus uuritava ainega; iii) teadaolevad füüsikalised ja keemilised omadused; iv) teadaolevat toimet tõendavad andmed ja v) teadaolev mõjusus soovitavas reaktsioonitugevuse vahemikus.
Alt-üles lähenemisviis
: etapiviisiline lähenemisviis, mida kasutatakse niisuguste uuritavate kemikaalide korral, mille puhul on põhjust arvata, et need ei vaja klassifitseerimist silmade ärrituse või raske silmakahjustusega seoses; nimetatud lähenemisviisi puhul eristatakse kõigepealt kemikaalid, mis ei vaja klassifitseerimist (tulemus on negatiivne), muudest kemikaalidest (tulemus on positiivne).
Kemikaal
: aine või segu.
Silmade ärritus
: muutuse tekkimine silmas pärast uuritava kemikaali silma eespinnale manustamist, seejuures on muutus 21 päeva jooksul pärast manustamist täielikult taandunud. Samatähenduslik mõistetega „pöörduv mõju silmale“ ja „ÜRO GHSi / CLP-määruse kohane 2. kategooria“.
Valenegatiivsete tulemuste määr
: kõikide selliste kemikaalide osakaal, mis peaksid andma positiivse tulemuse, kuid on katsemeetodit järgides andnud negatiivse tulemuse. See on üks katsemeetodi tulemuslikkuse näitaja.
Valepositiivsete tulemuste määr
: kõikide selliste kemikaalide osakaal, mis peaksid andma negatiivse tulemuse, kuid on katsemeetodit järgides andnud positiivse tulemuse. See on üks katsemeetodi tulemuslikkuse näitaja.
Oht
: niisuguse mõjuri või olukorra loomupärane omadus, mis võib avaldada kahjulikku toimet, kui organism, süsteem või (ala)populatsioon selle mõjuriga kokku puutub.
Söötme kontrollproov
: töötlemata paralleelproov, mis sisaldab kõiki katsesüsteemi komponente. Seda proovi analüüsitakse koos uuritava kemikaaliga töödeldud proovide ja teiste kontrollproovidega, et teha kindlaks, kas lahusti mõjutab katsesüsteemi.
Segu
: kahest või enamast ainest koosnev segu või lahus.
Ühe koostisosaga aine
: aine, mis on määratletud oma kvantitatiivse koostisega, milles ühe peamise koostisosa sisaldus on vähemalt 80 massiprotsenti.
MTT
: 3-(4,5-dimetüültiasool-2-üül)-2,5-difenüültetrasooliumbromiid, tiasolüülsinine.
Mitme koostisosaga aine
: aine, mis on määratletud oma kvantitatiivse koostisega, milles rohkem kui ühe peamise koostisosa kontsentratsioon on ≥ 10 % ja < 80 % (massiprotsendid). Mitme koostisosaga aine saadakse tootmisprotsessi käigus. Erinevus segu ja mitme koostisosaga aine vahel seisneb selles, et segu saadakse kahe või enama aine kokku segamisel, ilma et toimuks keemilist reaktsiooni. Mitme koostisosaga aine saadakse keemilise reaktsiooni tulemusel.
OD
: optiline tihedus.
Positiivne kontrollproov
: paralleelproov, mis sisaldab kõiki katsesüsteemide komponente ja mida on töödeldud ainega, mis teadaolevalt kutsub esile positiivse reaktsiooni. Selleks, et oleks võimalik hinnata positiivse kontrollproovi esilekutsutud reaktsiooni varieeruvust ajas, ei tohi positiivne reaktsioon olla ülemäära tugev.
Asjakohasus
: mõiste, millega väljendatakse seda, kas katsemeetod võimaldab uurida huvipakkuvat mõju, kas meetod on mõttekas ja sobib konkreetse eesmärgi jaoks. See näitab, mil määral saab katsemeetodiga õigesti mõõta või prognoosida huvipakkuvat bioloogilist mõju. Asjakohasus hõlmab ka katsemeetodi täpsuse (vastavuse) aspekti (10).
Usaldusväärsus
: iseloomustab katsemeetodi korratavust, kui meetodit rakendatakse ühe ja sama katse-eeskirja alusel ühes laboris ja eri laborites pikema aja jooksul. Usaldusväärsuse hindamiseks arvutatakse laborisisene ja laboritevaheline korratavus ning laborisisene korduvus (13).
Tundlikkus
: kõigi niisuguste positiivsete/reaktsioonivõimeliste kemikaalide osakaal, mis katse tulemusel klassifitseeritakse õigesti. Tundlikkusega iseloomustatakse katsemeetodi täpsust, kui saadud tulemusi saab kategoriseerida, ning see on oluline aspekt katsemeetodi asjakohasuse hindamisel (10).
Raske silmakahjustus
: sellise koekahjustuse tekkimine silmas või selline tugev nägemisteravuse langus pärast seda, kui uuritavat kemikaali on manustatud silma eespinnale, mis ei ole 21 päeva jooksul pärast manustamist täielikult pöörduv. Samatähenduslik mõistetega „pöördumatu mõju silmale“ ja „ÜRO GHSi / CLP-määruse kohane 1. kategooria“.
Lahusti/kandeaine kontrollproov
: töötlemata proov, mis sisaldab kõiki katsesüsteemi komponente, sealhulgas lahustit või kandeainet, ning mida analüüsitakse koos uuritava kemikaaliga töödeldud proovide ja muude kontrollproovidega, et määrata kindlaks samas lahustis või kandeaines lahustatud uuritava kemikaaliga töödeldud proovide nulljoonetase. Kui seda proovi analüüsitakse samaaegselt söötme kontrollprooviga, näitab see samuti, kas lahusti või kandeaine mõjutab katsesüsteemi.
Spetsiifilisus
: kõigi niisuguste negatiivsete/mittereageerivate kemikaalide osakaal, mis katse tulemusel klassifitseeritakse õigesti. Spetsiifilisusega iseloomustatakse katsemeetodi täpsust, kui saadud tulemusi saab kategoriseerida, ning see on oluline aspekt katsemeetodi asjakohasuse hindamisel (13).
Aine
: looduslik või tootmisprotsessi teel saadud keemiline element ja selle ühendid, kaasa arvatud stabiilsuse säilitamiseks vajalikud lisaained ja tootmisprotsessist tingitud lisandid, välja arvatud mis tahes lahusti, mida on võimalik aine stabiilsust vähendamata ja selle koostist muutmata eraldada.
Pindaktiivne aine
: kemikaal, näiteks pesuaine, mis vähendab vedeliku pindpinevust ning muudab selle vahutavaks või tahket materjali läbivaks, seda nimetatakse ka märgavaks aineks.
Uuritav kemikaal
: iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
Astmelise katsetamise strateegia
: etapiviisi katsete tegemise strateegia, milles enne järgmise etapiga alustamist vaadatakse kindlaksmääratud korras läbi kogu uuritava kemikaali kohta olemasolev teave, kasutades igas etapis tõendite kaalukuse menetlust, et otsustada, kas teave on ohuklassi määramiseks piisav. Kui olemasoleva teabe põhjal saab otsustada uuritava kemikaali ärritava toime üle, ei ole täiendavaid katseid vaja teha. Kui olemasoleva teabe põhjal ei saa otsustada uuritava kemikaali ärritava toime üle, jätkatakse järjestikuste loomkatsete etapiti tegemist, kuni kemikaali saab üheselt klassifitseerida.
Ülalt-alla lähenemisviis
: etapiviisiline lähenemisviis, mida kasutatakse niisuguste uuritavate kemikaalide korral, mille puhul on põhjust arvata, et need põhjustavad rasket silmakahjustust; nimetatud lähenemisviisi puhul eristatakse kõigepealt rasket silmakahjustust tekitavad kemikaalid (tulemus on positiivne) muudest kemikaalidest (tulemus on negatiivne).
Ühinenud Rahvaste Organisatsiooni ühtne ülemaailmne kemikaalide klassifitseerimise ja märgistamise süsteem (ÜRO GHS (Globally Harmonized System))
: süsteem, milles kemikaalid (ained ja segud) on klassifitseeritud vastavalt nende füüsilise ohtlikkuse ning kahjuliku tervise- ja keskkonnamõju standarditud tüübile ja -tasemele ning mis hõlmab asjaomaseid teavitustähiseid, nagu piktogrammid, märksõnad, ohulaused, hoiatuslaused ja ohutuskaardid, et anda edasi inimeste (sealhulgas tööandjate, töötajate, vedajate, tarbijate ja päästetöötajate) ja keskkonna kaitsmiseks vajalikku teavet kõnealuste kemikaalide kahjuliku mõju kohta (1).
ÜRO GHSi / CLP-määruse kohane 1. kategooria
: vt „Raske silmakahjustus“.
ÜRO GHSi / CLP-määruse kohane 2. kategooria
: vt „Silmade ärritus“.
ÜRO GHSi / CLP-määruse kohase kategooriata
: kemikaalid, mis ei ole klassifitseeritud ÜRO GHSi / CLP-määruse kohasesse 1. ega 2. kategooriasse (ega ÜRO GHSi 2A või 2B kategooriasse).
UVCB
: tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal.
B.69 REKONSTRUEERITUD INIMESE SARVKESTA SARNASE EPITEELI (RhCE) KATSEMEETOD NENDE KEMIKAALIDE KINDLAKSTEGEMISEKS, MIDA EI OLE VAJA KLASSIFITSEERIDA EGA MÄRGISTADA SEOSES SILMADE ÄRRITUSE VÕI RASKE SILMAKAHJUSTUSEGA
SISSEJUHATUS
|
1.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 492 (2017). Raske silmakahjustus on Ühinenud Rahvaste Organisatsiooni ühtse ülemaailmse kemikaalide klassifitseerimise ja märgistamise süsteemis (ÜRO GHS (Globally Harmonized System) (1) ning Euroopa Liidu määruses (EÜ) nr 1272/2008, mis käsitleb ainete ja segude klassifitseerimist, märgistamist ja pakendamist (CLP-määrus) (75), esitatud määratluse kohaselt sellise koekahjustuse tekkimine silmas või selline tugev nägemisteravuse langus pärast seda, kui uuritavat kemikaali on manustatud silma eespinnale, mis ei ole 21 päeva jooksul pärast manustamist täielikult pöörduv. Samuti on ÜRO GHSi ja CLP-määruse kohaselt silmade ärritus muutuste tekkimine silmas pärast uuritava kemikaali silma eespinnale manustamist, seejuures on muutus 21 päeva jooksul pärast manustamist täielikult taandunud. Raske silmakahjustust põhjustavad uuritavad kemikaalid klassifitseeritakse ÜRO GHSi ja CLP-määruse kohasesse 1. kategooriasse ning silmade ärritust põhjustavad uuritavad kemikaalid ÜRO GHSi ja CLP-määruse kohasesse 2. kategooriasse. Kemikaal, mida ei klassifitseerita seoses silmade ärrituse või raske silmakahjustusega, on määratletud kui kemikaal, mis ei vasta ÜRO GHSi 1. või 2. (2A või 2B) kategooriasse klassifitseerimise nõuetele, see tähendab, et sellele viidatakse kui ÜRO GHSi kategooriata kemikaalile.
|
|
2.
|
Rasket silmakahjustust / silmade ärritust tekitava toime hindamisel on tavaliselt kasutatud laboriloomi (katsemeetod B.5 (2)). Kõige asjakohasema katsemeetodi valimist ja selle kasutamist tuleks vaadelda raske silmakahjustuse ja silmade ärrituse põhjustamise uurimiseks ja hindamiseks kasutatavaid kompleksseid lähenemisviise käsitleva OECD juhenddokumendi (39) kontekstis.
|
|
3.
|
Käesolevas katsemeetodis kirjeldatakse in vitro meetodit, mis võimaldab kindlaks teha kemikaalid (ained ja segud), mida ei ole vaja seoses silmade ärrituse või raske silmakahjustusega ÜRO GHSi ja CLP-määruse kohaselt klassifitseerida ega märgistada. Meetodis kasutatakse rekonstrueeritud inimese sarvkesta sarnast epiteeli, mille histoloogilised, morfoloogilised, biokeemilised ja füsioloogilised omadused on ülimalt sarnased inimese sarvkesta epiteeli omadele. Valideeritud, teaduslikult põhjendatuiks arvatud ning katsemeetoditena B.47 (3), B.48 (4), B.61 (5) ja B.68 (6) vastu võetud on veel neli in vitro katsemeetodit, mis uurivad rasket silmakahjustust / silmade ärritust inimtervisega seotud näitajana.
|
|
4.
|
Käesolev katsemeetod hõlmab kaht valideeritud katset, mille puhul kasutatakse müügil olevaid RhCE-mudeleid. Valideerimisuuringud silmade ärrituse / raske silmakahjustuse hindamiseks viidi läbi (7, 8, 9, 10, 11, 12, 13) EpiOcular™i silmaärrituse katse ja SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katse põhjal. Mõlemas katses kasutatakse katsesüsteemina müügil olevaid RhCE-koemudeleid, millele edaspidises tekstis viidatakse kui valideeritud võrdlusmeetoditele VRM1 ja VRM2 (VRM – Validated Reference Methods). Nende valideerimisuuringute ja seotud sõltumatu vastastikuse eksperdihinnangu (9, 12) põhjal tehti järeldus, et EpiOcular™i silmaärrituse katse ja SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse sobivad nende kemikaalide (nii ainete kui ka segude) korrektseks kindlakstegemiseks, mida ei ole vaja seoses silmade ärrituse ega raske silmakahjustusega ÜRO GHSi kohaselt klassifitseerida ja märgistada, ning soovitati kõnealuseid meetodeid kui selleks eesmärgiks sobivaid teaduslikult põhjendatud meetodeid (13).
|
|
5.
|
Praegu ollakse üldiselt arvamusel, et lähitulevikus ei ole võimalik rasket silmakahjustust tekitava / silmi ärritava toime kogu ulatuse prognoosimisel eri kemikaaliklasside puhul ühegi in vitro katsemeetodiga täielikult asendada Draize’i in vivo silmakatset (2, 14). Kuid mitme alternatiivse katsemeetodi kombineerimisega (astmelise) katsetamise strateegiat (nt alt-üles või ülalt-alla lähenemisviisi) rakendades võib siiski olla võimalik Draize’i silmakatset (15) täielikult asendada. Alt-üles lähenemisviisi (15) tuleks kasutada siis, kui olemasoleva teabe põhjal võib eeldada, et kemikaal ei põhjusta piisavalt silma ärritust, et seda oleks vaja klassifitseerida; ülalt-alla lähenemisviisi (15) tuleks kasutada siis, kui olemasoleva teabe põhjal võib eeldada, et kemikaal põhjustab raske silmakahjustuse. EpiOcular™i silmaärrituse katset ja SkinEthic™i inimese sarvkesta epiteeli kasutavat silmaärrituse katset soovitatakse kasutada selleks, et teha täiendava katsetamiseta kindlaks need kemikaalid, mida ei ole vaja seoses silmade ärrituse või raske silmakahjustusega ÜRO GHSi / CLP-määruse kohaselt klassifitseerida (kategooria puudub), kasutades neid näiteks Scott et al. soovitatud alt-üles / ülalt-alla lähenemisviisidele sarnastes katsestrateegiates, nt esimese etapina alt-üles lähenemisviisipuhul ja ühena viimastest etappidest ülalt-alla lähenemisviisi puhul (15). EpiOcular™i silmaärrituse katse ja SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse ei ole siiski ette nähtud ÜRO GHSi / CLP-määruse kohase 1. kategooria (raske silmakahjustus) ja 2. kategooria (silmade ärritus) eristamiseks. Selliseks eristamiseks peab katsestrateegias olema mõni muu etapp (15). Kui EpiOcular™i silmaärrituse katse või SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katsega on kindlaks tehtud, et uuritavat kemikaali on vaja seoses silmade ärrituse või raske silmakahjustusega klassifitseerida, on lõpliku järelduse (kas tegemist on ÜRO GHSi / CLP-määruse kohase kategooriata, 1. kategooria või 2. kategooria kemikaaliga) tegemiseks vaja täiendavaid katseid (in vitro ja/või in vivo), kasutades näiteks katsemeetodit B.47, B.48, B.61 või B.68.
|
|
6.
|
Käesoleva katsemeetodi eesmärk on kirjeldada eeskirja, mida kasutatakse, et hinnata uuritava kemikaali võimalikku ohtlikkust silmale selle võime põhjal kutsuda esile tsütotoksilisust RhCE-koemudelis; kõnealust võimet mõõdetakse MTT-katsega (16) (vt punkt 21). RhCE-koe eluvõimelisuse määramiseks pärast uuritava kemikaaliga kokkupuudet võrreldakse seda kudedega, mida on töödeldud negatiivse kontrollainega (saadakse eluvõimelisuse %), ning saadud tulemust kasutatakse, et prognoosida uuritava kemikaali võimalikku ohtlikkust silmale.
|
|
7.
|
Kättesaadavad on toimivusnõuded (17), mis hõlbustavad EpiOcular™i silmaärrituse katsega või SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katsega sarnaste uute või modifitseeritud RhCE-põhiste in vitro katsemeetodite valideerimist kooskõlas OECD juhenddokumendis nr 34 (18) esitatud põhimõtetega, ning võimaldavad OECD katsejuhendit nr 492 õigeaegselt muuta, lisades sinna kõnealused meetodid. Andmete vastastikune tunnustamine vastavalt OECD kokkuleppele tagatakse vaid nende katsemeetodite puhul, mis on valideeritud kooskõlas toimivusnõuetega, kui need katsemeetodid on läbi vaadatud ja lisatud OECD poolt vastavasse juhendisse.
|
MÕISTED
|
8.
|
Mõisted on esitatud 1. liites.
|
LÄHTEKAALUTLUSED JA PIIRANGUD
|
9.
|
Käesolevas katsemeetodis kasutatakse kaubanduslikult saadaolevaid kolmemõõtmelisi RhCE-koemudeleid, mis on valmistatud kas inimese epidermise esmastest keratinotsüütidest (nt EpiOcular™ OCL-200) või inimese sarvkesta immortaliseeritud epiteelirakkudest (nt SkinEthic™ HCE/S). RhCE-koemudelid EpiOcular™ OCL-200 ja SkinEthic™ HCE/S sarnanevad in vivo sarvkesta kolmemõõtmelise struktuuriga epiteelile ning nende valmistamiseks kasutatakse huvipakkuva liigi rakke (19, 20). Peale selle mõõdetakse nende katsetega otseselt tsütotoksilisust, mis ilmneb kemikaali tungimisel läbi sarvkesta ning raku- ja koekahjustuste tekkides; tsütotoksilisusreaktsioon on aluseks üldisele in vivo katsel saadud tulemusele raske silmakahjustuse / silmade ärrituse kohta. Rakukahjustuse tekkimise mehhanismid võivad olla erinevad (vt punkt 20), kuid tsütotoksilisusel on koekahjustust põhjustanud füüsikalis-keemilistest protsessidest olenemata oluline või isegi peamine mehhanistlik roll kemikaali põhjustatud üldise reaktsiooni (raske silmakahjustus või silmade ärritus) määramisel, reaktsioon avaldub in vivo peamiselt sarvkesta läbipaistmatuse, vikerkestapõletiku, sidekesta punetuse ja/või sidekesta tursena.
|
|
10.
|
Käesoleva katsemeetodi valideerimisuuringus uuriti katseliselt paljusid kemikaale, mis kuulusid mitmesse eri liiki ja kemikaaliklassi ning millel oli erinev molekulmass, log P, keemiline struktuur jne. EpiOcular™i silmaärrituse katse valideerimise andmebaas sisaldas kokku andmeid 113 kemikaali kohta, millel olid OECD rakendusega QSAR Toolbox tehtud analüüsi (8) kohaselt esindatud 95 erinevat funktsionaalrühma. Enamik neist kemikaalidest olid ühe koostisosaga ained, kuid uuringusse oli võetud ka mitu mitme koostisosaga ainet (sealhulgas 3 homopolümeeri, 5 kopolümeeri ja 10 kvaasipolümeeri). Füüsikalise oleku ja ÜRO GHSi / CLP-määruse kohase kategooria järgi jagunesid 113 uuritud kemikaali järgmiselt: 13 vedelikku 1. kategooriast, 15 tahket ainet 1. kategooriast, 6 vedelikku 2A kategooriast, 10 tahket ainet 2A kategooriast, 7 vedelikku 2B kategooriast, 7 tahket ainet 2B kategooriast, 27 kategooriata vedelikku ja 28 kategooriata tahket ainet (8). SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katse valideerimise andmebaas sisaldas kokku andmeid 200 kemikaali kohta, milles esindatud 165 erinevat funktsionaalrühma (8, 10, 11). Enamik neist kemikaalidest olid ühe koostisosaga ained, kuid uuringusse oli võetud ka mitu mitme koostisosaga ainet (sealhulgas 10 polümeeri). Füüsikalise oleku ja ÜRO GHSi / CLP-määruse kohase kategooria järgi jagunesid 200 uuritud kemikaali järgmiselt: 27 vedelikku 1. kategooriast, 24 tahket ainet 1. kategooriast, 19 vedelikku 2A kategooriast, 10 tahket ainet 2A kategooriast, 9 vedelikku 2B kategooriast, 8 tahket ainet 2B kategooriast, 50 kategooriata vedelikku ja 53 kategooriata tahket ainet (10, 11).
|
|
11.
|
Seda katsemeetodit saab kasutada ainete ja segude uurimiseks tahkete ainete, vedelike, pooltahkete ainete ja vahade puhul. Vedelikud võivad olla vesilahused või muud vedelikud; tahked ained võivad olla vees lahustuvad või lahustumatud. Tahked ained tuleks enne pealekandmist võimaluse korral peenestada peeneks pulbriks; proovi muud eeltöötlust ei ole vaja. Gaase ja aerosoole ei ole valideerimisuuringuga veel hinnatud. Kuigi on mõeldav, et neidki saab uurida RhCE-koemudeli abil, ei võimalda praegune katsemeetod gaaside ega aerosoolide uurimist.
|
|
12.
|
Uuritavad kemikaalid, mis (iseloomulikult või pärast töötlemist) neelavad valgust MTT formasaaniga samas lainepikkuste vahemikus või suudavad vitaalvärvaine MTT-d otse redutseerida (MTT formasaaniks), võivad koe eluvõimelisuse mõõtmist segada ning siis on vaja kasutada kohandatud kontrollproove paranduste tegemiseks. Vajalike kohandatud kontrollproovide liik varieerub olenevalt uuritava kemikaali põhjustatud häire iseloomust ja MTT formasaani kvantitatiivse mõõtmise metoodikast (vt punkte 36–42).
|
|
13.
|
Eelvalideerimisel (21, 22) ja täielikus valideerimisuuringus (8, 10, 11) saadud tulemused näitasid, et nii EpiOcular™i silmaärrituse katse kui ka SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse on rakendatavad laboris, millel ei ole nimetatud analüüside tegemise kogemust, samuti on need korratavad nii laborisiseselt kui ka laboritevaheliselt. Kõnealuste uuringute kohaselt on 113 kemikaali kohta saadud andmete põhjal EpiOcular™i silmaärrituse katse eeldatav korratavuse suurusjärk prognooside kokkulangevuse järgi laborisisese korratavuse puhul 95 % ja laboritevahelise korratavuse puhul 93 %. 120 kemikaali kohta saadud andmete põhjal on SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katse eeldatav korratavuse suurusjärk prognooside kokkulangevuse järgi laborisisese korratavuse puhul 92 % ja laboritevahelise korratavuse puhul 95 %.
|
|
14.
|
EpiOcular™i silmaärrituse katset saab kasutada niisuguste kemikaalide kindlakstegemiseks, mida ei ole vaja seoses silmade ärrituse või raske silmakahjustusega ÜRO GHSi / CLP-määruse klassifitseerimissüsteemi järgi klassifitseerida. Võttes arvesse valideerimisuuringus saadud andmeid (8), on EpiOcular™i silmaärrituse katse kogutäpsus 80 % (112 kemikaali põhjal), tundlikkus 96 % (57 kemikaali põhjal), valenegatiivsete tulemuste määr 4 % (57 kemikaali põhjal), spetsiifilisus 63 % (55 kemikaali põhjal) ja valepositiivsete tulemuste määr 37 % (55 kemikaali põhjal), võrreldes võrdluskatsena kasutataval küüliku in vivo silmakatsel (katsemeetod B.5) (2, 14) saadud andmetega, mille põhjal loetletud tooted olid klassifitseeritud ÜRO GHSi / CLP-määruse klassifikatsioonisüsteemi kohaselt. 97 vedel agrokeemiatoote uuringus EpiOcular™i silmaärrituse katsega leiti et katsemeetodi toimivus seda liiki segude puhul sarnaneb valideerimisuuringus leitud toimivusega (23). Nimetatud 97 toodet jagunesid järgmiselt: 21 toodet 1. kategooriast, 19 toodet 2A kategooriast, 14 toodet 2B kategooriast ja 43 kategooriata toodet, mis olid klassifitseeritud ÜRO GHSi klassifitseerimissüsteemi järgi võrdluskatsena kasutataval küüliku in vivo silmakatsel (katsemeetod B.5) (2, 14) saadud andmete alusel. Leiti, et kogutäpsus on 82 % (97 toote põhjal), tundlikkus 91 % (54 toote põhjal), valenegatiivsete tulemuste määr 9 % (54 toote põhjal), spetsiifilisus 72 % (43 toote põhjal) ja valepositiivsete tulemuste määr 28 % (43 toote põhjal) (23).
|
|
15.
|
SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katset saab kasutada niisuguste kemikaalide kindlakstegemiseks, mida ei ole vaja seoses silmade ärrituse või raske silmakahjustusega ÜRO GHSi / CLP-määruse klassifitseerimissüsteemi järgi klassifitseerida. Võttes arvesse valideerimisuuringus saadud andmeid (10, 11), on SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katse kogutäpsus 84 % (200 kemikaali põhjal), tundlikkus 95 % (97 kemikaali põhjal), valenegatiivsete tulemuste määr 5 % (97 kemikaali põhjal), spetsiifilisus 72 % (103 kemikaali põhjal) ja valepositiivsete tulemuste määr 28 % (103 kemikaali põhjal), võrreldes võrdluskatsena kasutataval küüliku in vivo silmakatsel (katsemeetod B.5) (2, 14) saadud andmetega, mille põhjal loetletud tooted olid klassifitseeritud ÜRO GHSi / CLP-määruse klassifikatsioonisüsteemi kohaselt.
|
|
16.
|
Mõlema RhCE-põhise katsemeetodiga nii ainete kui ka segude puhul korduvatel katsetel saadud valenegatiivsete tulemuste määrade alusel on üldine tõenäosus 12 %, et kemikaalid klassifitseeritakse Draize’i in vivo silmakatse tulemusena kas ÜRO GHSi ja CLP-määruse kohasesse 2. kategooriasse või neil ÜRO GHSi ja CLP-määrusekohane kategooria puudub. seda põhjustab meetodile iseloomulik katsesisene hajuvus (24). Mõlema RcHE-põhise katsemeetodiga nii ainete kui ka segude puhul saadud valepositiivsete tulemuste määrad ei ole selle katsemeetodi kontekstis kriitilised, sest kõiki uuritavaid kemikaale, mille puhul koe eluvõimelisus on võrdne kindlaksmääratud läviväärtusega (vt punkt 44) või sellest väiksem, tuleb järgmiseks uurida muude in vitro katsemeetoditega või viimase võimalusena, olenevalt regulatiivsetest nõuetest, katsetes küülikutega kooskõlas järjestikuse katsete tegemise strateegia ning tõendite kaalukuse lähenemisviisiga. Neid katsemeetodeid võib kasutada iga tüüpi kemikaalide puhul, kusjuures negatiivse tulemuse võib aktsepteerida kui tõendi, et kemikaali ei ole vaja seoses silmaärrituse või raske silmakahjustusega klassifitseerida (ÜRO GHSi ja CLP-määruse kohane kategooria puudub). Kui EpiOcular™i silmaärrituse katset ja SkinEthic™i inimese sarvkesta epiteeli kasutavat silmaärrituse katset hakatakse kasutama muu kui ÜRO GHSi / CLP-määruse klassifitseerimisskeemiga seoses, tuleks eelnevalt konsulteerida asjakohaste reguleerivate asutustega.
|
|
17.
|
Selle katsemeetodi kasutamist piirab asjaolu, et see ei võimalda vahet teha silmade ärritusel / pöörduval mõjul silmale (2. kategooria) ja tõsisel silmakahjustusel / pöördumatul mõjul silmale (1. kategooria), nagu need on määratletud ÜRO GHSis ja CLP-määruses, ega ka silma ärritaval (valikuline 2A kategooria) ja silma kergelt ärritaval (valikuline 2B kategooria) kemikaalil, nagu need on määratletud ÜRO GHSis (1). Selleks on vajalik täiendav uuring muude in vitro katsemeetoditega.
|
|
18.
|
Käesolevas katsemeetodis kasutatakse terminit „uuritav kemikaal“ uurimisobjektile viitava terminina (76) ja see ei ole seotud RhCE-põhise katsemeetodi kohaldatavusega ainete ja/või segude uurimiseks.
|
KATSE PÕHIMÕTE
|
19.
|
Uuritav kemikaal kantakse vähemalt kahele kolmemõõtmelisele RhCE-koemudelile ning pärast kokkupuute ja töötlemise järgset ajavahemikku mõõdetakse koe eluvõimelisus. RhCE-koed rekonstrueeritakse inimese epidermise esmastest keratinotsüütidest või inimese sarvkesta immortaliseeritud epiteelirakkudest, mida on mitme päeva jooksul kasvatatud, et saada mitmekihiline, ülimalt diferentseerunud lameepiteel, mis morfoloogiliselt sarnaneb inimese sarvkestal leiduvale. EpiOcular™i RhCE-koemudel koosneb vähemalt kolmest eluvõimelisest rakukihist ja sarvestumata pinnast, mille struktuur on sarvkesta sarnane ja analoogne in vivo esinevale. SkinEthic™i inimese sarvkesta epiteeli RhCE-koemudel koosneb vähemalt neljast eluvõimelisest rakukihist, sealhulgas kolumnaarsed basaalrakud, sarvkesta pealispinnaalused üleminekurakud ja pindmised lamerakud, ning sarnaneb tavalisele inimese sarvkesta epiteelile (20, 26).
|
|
20.
|
Kemikaalist põhjustatud raske silmakahjustus / silmade ärritus, mis avaldub in vivo peamiselt sarvkesta läbipaistmatuse, vikerkestapõletiku, sidekesta punetuse ja/või sidekesta tursena, on selliste järjestikuste muutuste tagajärg, mis algavad kemikaali tungimisega läbi sarvkesta ja/või sidekesta ja rakukahjustuse tekitamisega. Rakukahjustuse tekkimise mehhanismid võivad olla erinevad, sealhulgas rakumembraani lüüs (nt pindaktiivsete ainete, orgaaniliste lahustite toimel), makromolekulide (eelkõige valkude) koaguleerumine (nt pindaktiivsete ainete, orgaaniliste lahustite, leeliste ja hapete toimel), lipiidide seebistumine (nt leeliste toimel) ning alküülimine või muud kovalentsed vastastikmõjud makromolekulidega (nt pleegitite, peroksiidide ja alküülijate toimel) (15, 27, 28). Siis on näidatud, et tsütotoksilisusel on koekahjustust põhjustanud füüsikalis-keemilistest protsessidest olenemata oluline või isegi peamine mehhanistlik roll kemikaali põhjustatud üldise reaktsiooni (raske silmakahjustus või silmade ärritus) määramisel (29, 30). Peale selle määratakse kemikaali võime põhjustada rasket silmakahjustust / silmade ärritust põhimõtteliselt esialgse vigastuse ulatuse järgi (31), mis on vastavuses rakkude hävimise (29) ning sellest tulenevate reaktsioonide ja lõpptulemuse ulatusega (32). Seega mõjutavad silma kergelt ärritavad kemikaalid üldiselt üksnes sarvkesta pindmist epiteeli, kergelt ja mõõdukalt ärritavad kemikaalid kahjustavad põhimõtteliselt epiteeli ja pindmist stroomat ning tugevalt ärritavad kemikaalid kahjustavad epiteeli, strooma süvakihte ja kohati sarvkesta endoteeli (30, 33). RhCE-koemudeli eluvõimelisuse mõõtmine pärast pindmist kokkupuudet uuritava kemikaaliga, et teha kindlaks kemikaalid, mida ei ole vaja seoses raske silmakahjustuse / silmade ärritusega klassifitseerida (ÜRO GHSi ja CLP-määruse kohane kategooria puudub), põhineb eeldusel, et kõik kemikaalid, mis põhjustavad raske silmakahjustuse või silmade ärrituse, põhjustavad sarvkesta epiteelis ja/või sidekestas tsütotoksilisust.
|
|
21.
|
Tavapäraselt mõõdetakse RhCE-koe eluvõimelisust vitaalvärvaine MTT [3-(4,5-dimetüültiasool-2-üül)-2,5-difenüültetrasooliumbromiid, tiasolüülsinine; CASi number 298-93-1] ensümaatilise muundamise järgi siniseks MTT formasaansoolaks, mis pärast koest ekstraheerimist määratakse kvantitatiivselt (16). Kemikaalid, mida ei ole vaja ÜRO GHSi / CLP-määruse kohaselt klassifitseerida ja märgistada (kategooria puudub), on kemikaalid, mis uurimisel ei vähenda koe eluvõimelisust allapoole määratud läviväärtust (st EpiOcular™i silmaärrituse katsel ja SkinEthic™i inimese sarvkesta epiteeli kasutavat silmaärrituse katsel EITL (77) koe eluvõimelisus > 60 % või SkinEthic™i inimese sarvkesta epiteeli kasutavat silmaärrituse katsel EITS (78) > 50 %) (vt punkt 44).
|
PÄDEVUSE TÕENDAMINE
|
22.
|
Enne RhCE-koemudeliga tehtavate katsete rutiinset kasutamist regulatiivsel eesmärgil tuleb laboril tõendada oma tehnilist pädevust, prognoosides õigesti tabelis 1 loetletud pädevuse tõendamise kemikaalide omadused. Need kemikaalid valiti välja valideeritud võrdlusmeetodite valideerimisuuringutes kasutatud kemikaalide seast (8, 10, 11). Valikus on võimaluste piires esindatud järgmiste omadustega kemikaalid: i) neil erinevad füüsikalised olekud; ii) nad esindavad in vivo raske silmakahjustuse / silmade ärrituse reaktsioonide täit vahemikku võrdluskatsena kasutataval küüliku in vivo silmakatsel (katsemeetod B.5) (2, 14) saadud kvaliteetsete andmete ning ÜRO GHSi klassifitseerimissüsteemi (st 1. kategooria, 2A, 2B kategooria või klassifitseerimata) ja CLP-määruse kohase klassifitseerimissüsteemi (st 1. või 2. kategooria või kategooria puudub) alusel; iii) nad esindavad mitmesuguseid aluseid in vivo klassifitseerimiseks (24, 25); iv) kuuluvad samadesse kemikaaliklassidesse, mida kasutati valideerimisuuringus (8, 10, 11); v) neis on hästi esindatud mitmesugused funktsionaalrühmad (8, 10, 11); vi) nende keemiline ehitus on hästi teada (8, 10, 11); vii) nad on värvilised ja/või MTT otsesed redutseerijad; viii) nad andsid valideerimisel RhCE-koemudelitega katsemeetodeid kasutades korratavad tulemused; ix) nende omadused prognoositi valideerimisuuringutes RhCE-koemudeleid kasutavate katsemeetoditega õigesti; x) nad esindavad RhCE-koemudelit kasutava katsemeetodiga saadud kvaliteetsete andmete kohaselt in vitro reaktsioonide täit vahemikku (eluvõimelisus 0 kuni 100 %) xi) nad on müügil ja xii) nendega ei kaasne ülemääraseid soetamis- ja/või kõrvaldamiskulusid. Kui mõnda loetletud kemikaali ei ole saada või seda ei saa muul õigustatud põhjusel kasutada, võib kasutada muud eespool kirjeldatud kriteeriumidele vastavat kemikaali näiteks valideeritud võrdlusmeetodi valideerimisel kasutatud kemikaalide hulgast. Sellised kõrvalekalded peavad siiski olema põhjendatud.
Tabel 1
Pädevuse tõendamise kemikaalide loend
|
Keemiline nimetus
|
CASi nr
|
Funktsionaalrühm (79)
|
Füüsikaline olek
|
Eluvõimelisus meetodiga VRM1 (%) (80)
|
Eluvõimelisus meetodiga VRM2 (%) (81)
|
Prognoos VRM-meetodiga
|
MTT redutseerija
|
Segava värvusega
|
|
In vivo 1. kategooria (82)
|
|
|
Metüültioglükolaat
|
2365-48-2
|
Karboksüülhappeester, tioalkohol
|
Vedel
|
10,9 ± 6,4
|
5,5 ± 7,4
|
Prognoosida ei ole võimalik
|
Jah (tugev)
|
Ei
|
|
Hüdroksüetüülakrülaat
|
818-61-1
|
Akrülaat, alkohol
|
Vedel
|
7,5 ± 4,7 (83)
|
1,6 ± 1,0
|
Prognoosida ei ole võimalik
|
Ei
|
Ei
|
|
2,5-dimetüül-2,5-heksaandiool
|
110-03-2
|
Alkohol
|
Tahke
|
2,3 ± 0,2
|
0,2 ± 0,1
|
Prognoosida ei ole võimalik
|
Ei
|
Ei
|
|
Naatriumoksalaat
|
62-76-0
|
Oksokarboksüülhape
|
Tahke
|
29,0 ± 1,2
|
5,3 ± 4,1
|
Prognoosida ei ole võimalik
|
Ei
|
Ei
|
|
In vivo 2A kategooria (82)
|
|
|
N,N″-bis(4-klorofenüül)-3,12-diimino-2,4,11,13-tetraasatetradekaandiimiidamiidi di-D-glükonaat (20 % vesilahus) (84)
|
18472-51-0
|
Aromaatne heterotsükliline haliid, arüülhaliid, dihüdroksüülrühm, guanidiin
|
Vedel
|
4,0 ± 1,1
|
1,3 ± 0,6
|
Prognoosida ei ole võimalik
|
Ei
|
Jah (nõrk)
|
|
Naatriumbensoaat
|
532-32-1
|
Arüül, karboksüülhape
|
Tahke
|
3,5 ± 2,6
|
0,6 ± 0,1
|
Prognoosida ei ole võimalik
|
Ei
|
Ei
|
|
In vivo 2B kategooria (82)
|
|
|
Dietüültoluamiid
|
134-62-3
|
Bensamiid
|
Vedel
|
15,6 ± 6,3
|
2,8 ± 0,9
|
Prognoosida ei ole võimalik
|
Ei
|
Ei
|
|
2,2-dimetüül-3-metüleenbitsüklo[2.2.1]heptaan
|
79-92-5
|
Alkaan, hargnev, tertsiaarse süsinikuga; alkeen; bitsükloheptaan; sildtsüklilised ühendid; tsükloalkaan
|
Tahke
|
4,7 ± 1,5
|
15,8 ± 1,1
|
Prognoosida ei ole võimalik
|
Ei
|
Ei
|
|
In vivo kategooria puudub (82)
|
|
|
1-etüül-3-metüülimidasooliumetüülsulfaat
|
342573-75-5
|
Alkoksürühm; ammooniumsool; arüülrühm; imidasool; sulfaat
|
Vedel
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Kategooria puudub
|
Ei
|
Ei
|
|
Dikaprülüüleeter
|
629-82-3
|
Alkoksürühm; eeter
|
Vedel
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Kategooria puudub
|
Ei
|
Ei
|
|
Piperonüülbutoksiid
|
51-03-6
|
Alkoksürühm; bensodioksool; bensüülrühm; eeter
|
Vedel
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Kategooria puudub
|
Ei
|
Ei
|
|
Hüdrogeenitud kastoorõli polüetüleenglükoolester (PEG-40)
|
61788-85-0
|
Atsülaalrühm, alkohol; allüülrühm, eeter
|
Viskoosne
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Kategooria puudub
|
Ei
|
Ei
|
|
1-(4-klorofenüül)-3-(3,4-diklorofenüül)karbamiid
|
101-20-2
|
Aromaatne heterotsükliline haliid, arüülhaliid, Karbamiidi derivaadid
|
Tahke
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Kategooria puudub
|
Ei
|
Ei
|
|
2,2ʹ-metüleen-bis-(6-(2H-bensotriasool-2-üül)-4-(1,1,3,3-tetrametüülbutüül)fenool
|
103597-45-1
|
Alkaan, hargnev, kvaternaarse süsinikuga; kondenseerunud tsüklitega aromaatne ühend; kondenseerunud küllastunud heterotsüklid; kinoidühendite prekursor; tert-butüülrühm
|
Tahke
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Kategooria puudub
|
Ei
|
Ei
|
|
Kaaliumtetrafluoroboraat
|
14075-53-7
|
Anorgaaniline sool
|
Tahke
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Kategooria puudub
|
Ei
|
Ei
|
|
Lühendid:
CASi nr – Chemical Abstracts Service’i number; ÜRO GHS – Ühinenud Rahvaste Organisatsiooni ühtne ülemaailmne kemikaalide klassifitseerimise ja märgistamise süsteem (1); VRM1 – valideeritud võrdlusmeetod (Validated Reference Method), EpiOcular™i silmaärrituse katse; VRM2 – valideeritud võrdlusmeetod (Validated Reference Method), SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse; segava värvusega – ühendi värvus segab MTT formasaani neelduvuse (optilise tiheduse) mõõtmist.
|
|
|
23.
|
Pädevuse tõendamise katsete osana soovitatakse kasutajal pärast kudede saamist kontrollida nende kaitsekihiomaduste vastavust RhCE-koemudeli tootja kirjeldusele (vt punktid 25, 27 ja 30). See on eriti oluline pärast kudede pikaajalist või kaugvedu. Kui katsemeetod on edukalt omandatud ning oskus seda kasutada on saavutatud ja tõendatud, ei ole katse rutiinse kasutamise puhul selline kontrollimine igapäevaselt vajalik. Katse rutiinse kasutamise puhul soovitatakse siiski jätkata kaitsekihiomaduste kontrollimist korrapäraste ajavahemike järel.
|
KATSE KÄIK
|
24.
|
Selle katsemeetodiga praegu hõlmatud katsed on teaduslikult põhjendatud EpiOcular™i silmaärrituse katse ja SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse, millele viidatakse kui valideeritud võrdlusmeetoditele (vastavalt VRM1 ja VRM2). RhCE-koemudelit kasutava katsemeetodi standardeeskiri on kättesaadav ja seda tuleb järgida katsemeetodi juurutamisel ja kasutamisel laboris (34, 35). Järgmistes punktides ja 2. liites kirjeldatakse RhCE-koemudelit kasutavate katsete peamisi osi ja eeskirju.
|
RHCE-KOEMUDELIT KASUTAVA KATSEMEETODI OSAD
Üldtingimused
|
25.
|
Sarvkesta epiteelile sarnaneva kolmemõõtmelise koe rekonstrueerimiseks tuleb kasutada asjakohaseid inimese rakke; kude peaks koosnema progressiivselt kihistunud, kuid mitte sarvestunud rakkudest. RhCE-koemudel valmistatakse plaatidel, millel on sünteetiline poorne membraan, millest toitained läbi liiguvad ja rakkudesse suunduvad. Rekonstrueeritud sarvkesta epiteelile sarnanevas koes peab olema mitu kihti eluvõimelisi sarvestumata epiteelirakke. RhCE-koemudeli epiteelipind peaks olema otseses kokkupuutes õhuga, et võimaldada otsest pindmist kokkupuudet uuritavate kemikaalidega sarnaselt sellele, kuidas sarvkesta epiteeli kokkupuude toimuks in vivo. RhCE-koemudel peab moodustama funktsionaalse kaitsekihi, mis on piisavalt tugev, et takistada tsütotoksiliste võrdlusainete, nt Triton X-100 või naatriumdodetsüülsulfaadi kiiret läbitungimist. Nimetatud kaitsekihina toimimist tuleks tõendada ja seda saab hinnata kas sellise kokkupuuteaja määramisega, mille jooksul võrdlusaine teatava kindla kontsentratsiooni (nt 100 μl 0,3-mahuprotsendilist Triton X-100) juures vähendab koe eluvõimelisust 50 % võrra (ET50), või sellise kontsentratsiooni määramisega, mille juures võrdlusaine vähendab kindla kokkupuuteaja (nt 30-minutine töötlemine 50 μl naatriumdodetsüülsulfaadiga) jooksul koe eluvõimelisust 50 % võrra (IC50). RhCE-koemudel peab olema selline, et uuritav kemikaal ei saaks tungida eluskudedesse ümber serva, vastasel korral ei oleks tegemist sarvkesta ja uuritava aine kokkupuute õige mudeliga. RhCE-koemudeli valmistamiseks kasutatavad inimese rakud ei tohi olla saastunud bakterite, viiruste, mükoplasma ega seentega. Koemudeli steriilsust peaks kontrollima tarnija, kes jälgib, et mudel ei oleks saastatud seente ega bakteritega.
|
Funktsionaalsed tingimused
Eluvõimelisus
|
26.
|
Koe eluvõimelisuse määramiseks kasutatakse MTT-katset (16). RhCE-koemudeli eluvõimelised rakud redutseerivad vitaalvärvaine MTT siniseks MTT formasaaniks, mis sadeneb ning ekstraheeritakse seejärel koestisopropanooliga (või sarnase lahustiga). Ekstraheeritud MTT formasaani saab määrata kvantitatiivselt kas neelduvuse (optilise tiheduse) mõõtmisega või HPLC-/UPLC-spektromeetrilise ühendmeetodiga (36). Puhta ekstraheerimislahusti optiline tihedus peab olema piisavalt madal, väiksem kui 0,1. RhCE-koemudeli kasutajad peaksid tagama, et iga kasutatav RhCE-koemudeli partii vastab kindlaksmääratud negatiivse kontrollproovi kriteeriumidele. Negatiivse kontrollproovi optilise tiheduse lubatavad vahemikud valideeritud võrdlusmeetodite puhul on esitatud tabelis 2. HPLC-/UPLC-spektromeetrilise ühendmeetodi kasutaja peaks negatiivse kontrollproovi nõuetekohasuse kriteeriumina kasutama tabelis 2 esitatud negatiivse kontrollproovi optilise tiheduse vahemikke. Katseprotokollis peaks olema dokumenteeritud, et negatiivse võrdlusainega töödeldud koed on kultuuris stabiilsed (annavad sarnaseid koe eluvõimelisuse mõõtmistulemusi) katses kasutatava kokkupuuteaja jooksul. Samasugust eeskirja peaks koepartii kvaliteedikontrolli osana järgima koemudeli tootja, kuid sellisel juhul võivad kehtida teised nõuetekohasuse kriteeriumid kui tabelis 2 esitatud. RhCE-koemudeli väljatöötaja/tarnija peaks kindlaks määrama negatiivse kontrollproovi optilise tiheduse väärtuste (kvaliteedikontrolli katsetingimuste korral) nõuetekohasuse vahemiku (ülem- ja alampiiri).
|
Tabel 2
Negatiivse kontrollproovi optilise tiheduse väärtuste nõuetekohasuse vahemik (katse läbiviijatele)
|
Katse
|
Vastuvõetav alampiir
|
Vastuvõetav ülempiir
|
|
EpiOcular™i silmaärrituse katse (OCL-200) – VRM1 (nii vedelate kui ka tahkete ainetega töötlemisel)
|
> 0,8 (85)
|
> 2,5
|
|
SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse (HCE/S) – VRM2 (nii vedelate kui ka tahkete ainetega töötlemisel)
|
> 1,0
|
≤ 2,5
|
Toimimine kaitsekihina
|
27.
|
RhCE-koemudel peab olema piisavalt paks ja tugev, et takistada tsütotoksiliste võrdlusainete kiiret läbitungimist, seda hinnatakse ET50 (Triton X-100) või IC50 (naatriumdodetsüülsulfaat) abil (tabel 3). RhCE-koemudeli väljatöötaja/müüja peab lõppkasutajale kudesid tarnides tõendama iga RhCE-koemudeli partii toimivust kaitsekihina (vt punkt 30).
|
Morfoloogia
|
28.
|
RhCE-koemudeli histoloogilisel uuringul peab see osutuma olevaks inimese sarvkesta epiteeli sarnase struktuuriga (sealhulgas vähemalt kolm eluvõimeliste epiteelirakkude kihti ja sarvestumata pind). VRMide puhul on sobiv morfoloogiline struktuur väljatöötaja/tarnija poolt kindlaks määratud ning seetõttu ei pea katsemeetodi kasutaja selle omadusi iga kasutatava koepartii juures uuesti tõendama.
|
Korratavus
|
29.
|
Katsemeetodiga peavad positiivsete ja negatiivsete kontrollproovide tulemused olema ajas korratavad.
|
Kvaliteedikontroll
|
30.
|
RhCE-koemudelit tuleks kasutada üksnes siis, kui selle väljatöötaja/tarnija tõendab, et kõik kasutatavad RhCE-koemudelipartiid vastavad toote kindlaksmääratud kvaliteedikriteeriumidele, mille seas kõige olulisemad on eluvõimelisuse (punkt 26) ja kaitsekihina toimivuse (vt punkti 27) kriteeriumid. RhCE-koemudeli väljatöötaja/tarnija peaks kindlaks määrama ET50- või IC50-meetodiga (vt punktid 25 ja 26) mõõdetava kaitsekihina toimivuse nõuetekohasuse vahemiku (ülem- ja alampiiri). ET50 ja IC50 nõuetekohasuse vahemikud, mida RhCE-koemudelite (kasutatavad VRMides) väljatöötaja/tarnija kasutab partii kvaliteedikontrolli kriteeriumina, on esitatud tabelis 3. RhCE-koemudeli väljatöötaja/tarnija peab esitama katsemeetodi kasutajale andmed, mis tõendavad vastavust kõigile toote kvaliteedikriteeriumidele, nii et meetodi kasutaja saaks selle teabe katseprotokolli lisada. Ainult kõigile toote kvaliteedikriteeriumidele vastavate kudedega saadud tulemused sobivad nende kemikaalide kohta usaldusväärsete prognooside tegemiseks, mida ei ole vaja seoses silmade ärrituse või raske silmakahjustusega ÜRO GHSi / CLP-määruse kohaselt klassifitseerida ega märgistada.
|
Tabel 3
Partii kvaliteedi kontrollimise kriteerium
|
Katse
|
Vastuvõetav alampiir
|
Vastuvõetav ülempiir
|
|
EpiOcular™i silmaärrituse katse (OCL-200) – VRM1 (100 μl 0,3-mahuprotsendilist Triton X-100)
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse (HCE/S) – VRM2 (30-minutine töötlemine 50 μl naatriumdodetsüülsulfaadiga)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Uuritavate kemikaalide ja kontrollainete kasutamine
|
31.
|
Iga uuritava kemikaali ja iga kontrollainega tuleks iga kord kasutada vähemalt kaht koe paralleelproovi. Kasutatakse kaht erinevat töötlemismeetodit, üht vedelate ja teist tahkete uuritavate kemikaalide puhul (34, 35). Mõlema meetodi ja töötlemismeetodi korral tuleb koemudeli pinda enne uuritava kemikaaliga töötlemist niisutada kaltsiumi- ja magneesiumvaba Dulbecco fosfaadiga puhverdatud füsioloogilise lahusega (Ca2+/Mg2+-vaba DPBS), et jäljendada inimsilma niiskustingimusi. Kudede töötlemine algab kokkupuutesse viimisega uuritava(te) kemikaali(de) ja kontrollainetega. Mõlemas valideeritud võrdlusmeetodis tuleb mõlema töötlemismeetodi puhul kanda piisav kogus uuritavat kemikaali või kontrollainet ühtlaselt epiteeli pinnale, vältides lõpmatut annust (vt punktid 32 ja 33) (liide 2).
|
|
32.
|
Uuritavaid kemikaale, mida 37 °C juures või madalamal temperatuuril saab pipeteerida (vajaduse korral kolbpipetti kasutades), käsitletakse valideeritud võrdlusmeetodites vedelikena, muul juhul tuleb neid käsitleda tahkete ainetena (vt punkt 33). Valideeritud võrdlusmeetodites levitatakse vedelad uuritavad kemikaalid ühtlaselt koe pinnale (st kantakse peale vähemalt 60 μl pinna cm2 kohta) (vt liidet 2) (33, 34). Plaadile kantavate väikeste ruumalade tõttu ilmneda võivat kapillaarjõudu (pindpinevusjõudu) tuleks võimalikult palju vältida, et tagada õige annustamine koele. Vedela uuritava kemikaaliga töödeldud kudesid inkubeeritakse 30 minutit rakkude kasvatamise standardtingimustes (37 ± 2 °C, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %). Kokkupuuteaja lõppedes tuleb vedel uuritav kemikaal ja kontrollained koe pinnalt hoolikalt eemaldada, loputades ohtra Ca2+/Mg2+-vaba DPBSiga toatemperatuuril. Loputusetapile järgneb kokkupuutejärgne sukeldamine värskesse söötmesse toatemperatuuril (et eemaldada igasugune koesse imendunud uuritav kemikaal) eelnevalt kindlaks määratud ajavahemikuks, mis varieerub olenevalt kasutatavast valideeritud võrdlusmeetodist. Kokkupuutejärgset inkubeerimist värskes söötmes rakkude kasvatamise standardtingimustes enne MTT-katse tegemist rakendatakse üksnes VRM1 korral (vt liide 2) (34, 35).
|
|
33.
|
Uuritavaid kemikaale, mida temperatuuril kuni 37 °C pipeteerida ei saa, käsitletakse valideeritud võrdlusmeetodites tahkete ainetena. Pealekantava uuritava kemikaali kogus peaks olema piisav, et katta kogu koepind, st peale tuleb kanda vähemalt 60 mg ainet pinna cm2 kohta (liide 2). Võimaluse korral tuleks tahket ainet alati katsetada peene pulbri kujul. Tahke uuritava kemikaaliga töödeldud kudesid inkubeeritakse eelnevalt kindlaks määratud ajavahemiku (oleneb kasutatavast valideeritud võrdlusmeetodist) jooksul rakkude kasvatamise standardtingimustes (vt liide 2) (34, 35). Kokkupuuteaja lõppedes tuleb tahke uuritav kemikaal ja kontrollained koe pinnalt hoolikalt eemaldada, loputades ohtra Ca2+/Mg2+-vaba DPBSiga toatemperatuuril. Loputusetapile järgneb kokkupuutejärgne sukeldamine värskesse söötmesse toatemperatuuril (et eemaldada igasugune koesse imendunud uuritav kemikaal) eelnevalt kindlaks määratud ajavahemikuks, mis varieerub olenevalt kasutatavast valideeritud võrdlusmeetodist, ning kokkupuutejärgne inkubeerimine värskes söötmes rakkude kasvatamise standardtingimustes enne MTT-katse tegemist (vt liide 2) (34, 35).
|
|
34.
|
Iga analüüsitsükkel peaks sisaldama ühtlasi ka negatiivset ja positiivset kontrollproovi, millega tõendatakse, et kudede eluvõimelisus (määratakse negatiivse kontrollprooviga) ja tundlikkus (määratakse positiivse kontrollprooviga) jäävad aja jooksul kogutud andmete põhjal kindlaks määratud nõuetekohasesse vahemikku. Samaaegselt analüüsitav negatiivne kontrollproov annab samuti lähtetaseme (koe eluvõimelisus 100 %) uuritava kemikaaliga töödeldud kudede suhtelise eluvõimelisuse arvutamiseks protsentides (%eluvõimelisustuuritav). Valideeritud võrdlusmeetoditega kasutamiseks soovitatav positiivne kontrollaine on puhas metüülatsetaat (CASi nr 79-20-9, saab osta nt Sigma-Aldrichist, katalooginr 45997, vedelik). Valideeritud võrdlusmeetoditega VRM1 ja VRM2 kasutamiseks soovitatavad negatiivsed kontrollained on vastavalt ülipuhas H2O ja Ca2+/Mg2+-vaba DPBS. Neid kasutati kontrollainetena valideeritud võrdlusmeetodite valideerimisuuringutes ja nende kohta on olemas kõige rohkem aja jooksul kogutud andmeid. Sobivate alternatiivsete positiivsete ja negatiivsete kontrollainete kasutamine peab olema teaduslikult ja piisavalt põhjendatud. Negatiivsete ja positiivsete kontrollainetega tuleb katse teha sama(de) katse-eeskirja(de) järgi, mida kasutatakse analüüsitsüklis olevate uuritavate kemikaalide puhul (nt vedelike ja tahkete ainete puhul). Sellele peaks järgnema töötlemine kokkupuutesse viimise teel, loputamine, kokkupuutejärgne sukeldamine ja vajaduse korral kokkupuutejärgne inkubeerimine, nagu kirjeldatud vedelate uuritavate kemikaalidega paralleelselt samas analüüsitsüklis kasutatavate kontrollainete puhul (vt punkt 32) ja tahkete uuritavate kemikaalidega paralleelselt samas analüüsitsüklis kasutatavate kontrollainete puhul (vt punkt 33), enne kui tehakse MTT-katse (vt punkt 35) (34, 35). Ühest negatiivsete ja positiivsete kontrollproovide komplektist piisab kõigi samas analüüsitsüklis uuritavate samas olekus (vedelad või tahked) kemikaalide jaoks.
|
Koe eluvõimelisuse mõõtmine
|
35.
|
MTT-katse on standarditud kvantitatiivne meetod (16), mida tuleks käesoleva katsemeetodi puhul kasutada koe eluvõimelisuse mõõtmiseks. Meetodit saab kasutada kolmemõõtmelises koemudelis. MTT-katse tehakse vahetult pärast kokkupuutejärgse inkubatsiooniperioodi lõppemist. Valideeritud võrdlusmeetodite puhul asetatakse RhCE-koemudeli proov rakkude kasvatamise standardtingimustes 180±15 minutiks 0,3 ml MTT-lahusesse, mille kontsentratsioon on 1 mg/ml. RhCE-koemudeli eluvõimelised rakud redutseerivad vitaalvärvaine MTT siniseks sadenevaks MTT formasaaniks. Seejärel ekstraheeritakse sadenenud sinine MTT formasaan koest sobiva koguse isopropanooliga (või sarnase lahustiga) (34, 35). Vedelate kemikaalide uurimiseks kasutatud kudede puhul tuleks ekstraheerida nii koe pealt kui ka alt, kuid tahkete kemikaalide ja värviliste vedelike uurimiseks kasutatud kudede puhul tuleks ekstraheerida ainult koe alt (et minimeerida ekstraheerimiseks kasutatava isopropanoolilahuse võimalikku saastumist igasuguse uuritava kemikaaliga, mis võib olla koesse jäänud). Niisuguste vedelate kemikaalide uurimiseks kasutatud kudede puhul, mida ei saa hõlpsasti maha pesta, võib samuti ekstraheerida üksnes koe alt. Samaaegselt uuritud negatiivse ja positiivse kontrollaine proove tuleb töödelda sarnaselt uuritava kemikaali proovidega. Ekstraheeritud MTT formasaani võib kvantitatiivselt määrata kas neelduvuse (optilise tiheduse) mõõtmisega lainepikkusel 570 nm, kasutades spektririba laiust kuni ± 30 nm, või HPLC-/UPLC-spektrofotomeetrilisel ühendmeetodil (vt punkt 42) (11, 36).
|
|
36.
|
Uuritava kemikaali optilised omadused või selle keemiline reaktsioon MTTga võib MTT formasaani mõõtmist segada ja põhjustada vea koe eluvõimelisuse hindamisel. Uuritava kemikaalid võivad MTT formasaani mõõtmist segada, redutseerides MTT siniseks MTT formasaaniks ja/või olles segava värvusega, st uuritav kemikaal neelab tavaolekus või töötlemisprotsessi tõttu samas lainepikkuste vahemikus kui MTT formasaan (st 570 nm ümbruses). Enne katse läbiviimist tuleks teha eelkontroll, et teha kindlaks võimalikud MTT otsesed redutseerijad ja/või segava värvusega kemikaalid, samuti tuleks kasutada täiendavaid kontrollproove, et leida niisuguste kemikaalide põhjustatud võimalikud häired ja neid korrigeerida (vt punktid 37–41). See on eriti oluline, kui konkreetsest uuritavat kemikaali ei saa loputades RhCE-koemudelist täielikult eemaldada või kui see läbib sarvkesta epiteeliga sarnase koe ja esineb seetõttu RhCE-koemudelis MTT-katse tegemise ajal. Nende MTT formasaaniga samas lainepikkuste vahemikus valgust neelavate uuritavate kemikaalide puhul, mis ei võimalda tugeva segava mõju tõttu (st tugeva neelduvuse tõttu lainepikkusel 570±30 nm) MTT-formasaani neelduvuse (optilise tiheduse) mõõtmist, võib MTT formasaani sisalduse mõõtmiseks kasutada HPLC-/UPLC-spektromeetrilist ühendmeetodit (vt punktid 41 ja 42) (11, 36). Üksikasjalik kirjeldus, kuidas tuvastada MTT otsene redutseerimine ja värviliste ainete segav mõju ning teha parandusi nende arvestamiseks, on esitatud valideeritud võrdlusmeetodite standardeeskirjades (34, 35). Samuti on III ja IV liites esitatud vastavalt VRM1- ja VRM2-meetodi puhul illustreerivad vooskeemid, millest juhinduda MTT otseste redutseerijate ja/või segava värvusega kemikaalide kindlakstegemisel ja nendega tegelemisel.
|
|
37.
|
MTT formasaaniga samas lainepikkuste vahemikus valgust neelavate (loomulikus olekus või pärast töötlemist) uuritavate kemikaalide võimaliku segava mõju kindlakstegemiseks ning täiendavate kontrollproovide vajalikkuse üle otsustamiseks lisatakse uuritavat kemikaali vette ja/või isopropanooli ning inkubeeritakse sobiva aja jooksul toatemperatuuril (vt liide 2) (34, 35). Kui VRM1-meetodi puhul neelab vees ja/või etanoolis lahustatud uuritav kemikaal küllaldaselt valgust vahemikus 570±20 nm (vt liide 3) või VRM2-meetodi puhul saadakse uuritava kemikaali segamisel veega värviline lahus (vt liide 4), segab uuritav kemikaal eeldatavalt MTT formasaani neelduvuse (optilise tiheduse) mõõtmist ning teha tuleks täiendavaid kontrollkatseid värvainetega või tuleks kasutada HPLC-/UPLC-spektromeetrilist ühendmeetodit – sel juhul ei ole nimetatud kontrollkatsete tegemine vajalik (vt punkte 41 ja 41 ning III ja IV liidet) (34, 35). Neelavuse (optilise tiheduse) mõõtmise korral tuleb iga uuritavat kemikaali kanda vähemalt kahele elujõulise koe paralleelproovile, millega tehakse läbi kogu katse käik, kuid MTTga inkubeerimise etapis inkubeeritakse neid MTT asemel söötmega, et saada kontrollproov, mille eluskudedes on mittespetsiifiline värvus (NSCelus) (34, 35). Katse NSCelus-kontrollprooviga tuleb läbi viia paralleelselt uuritava värvilise kemikaali prooviga ning korduvate katsete korral tuleb eluskudede bioloogilise varieeruvuse tõttu koos iga tehtava katsega (igas analüüsitsüklis) analüüsida ka eraldi NSCelus-kontrollproovi. Koe tegelik eluvõimelisus arvutatakse järgmiselt: segava uuritava kemikaaliga kokku puutunud ja MTT-lahusega inkubeeritud eluskudedega saadud eluvõimelisuse protsent (%eluvõimelisustuuritav) miinus segava uuritava kemikaaliga kokku puutunud ning söötmega ja ilma MTTta inkubeeritud eluskudedega saadud mittespetsiifiline värvus (%NSCelus), mis saadakse paralleelselt selle katse läbiviimisega, mille tulemused vajavad korrigeerimist, st koe tegelik eluvõimelisus = [%eluvõimelisustuuritav] – [%NSCelus].
|
|
38.
|
MTT otseste redutseerijate kindlakstegemiseks tuleb iga uuritavat kemikaali lisada värskelt valmistatud MTT-lahusele. MTT-lahusele lisatakse sobiv kogus uuritavalt kemikaali ning segu inkubeeritakse rakkude kasvatamise standardtingimustes ligikaudu 3 tundi (vt III ja IV liide) (34, 35). Kui uuritavat kemikaali sisaldav MTT-segu (või mittelahustuvate kemikaalide korral suspensioon) muutub siniseks/lillaks, võib eeldada, et uuritav kemikaal redutseerib otseselt MTTd ning olenemata sellest, kas kasutatakse neelavuse (optilise tiheduse) mõõtmist või HPLC-/UPLC-spektromeetrilist ühendmeetodit, tuleb mitteeluvõimelise RhCE-koemudeliga toimimist täiendavalt kontrollida. Täiendav toimivuskontroll tehakse surnud kudedega, millel on üksnes metaboolne jääkaktiivsus, kuid mis imavad ja seovad uuritavat kemikaali sarnaselt eluvõimeliste kudedega. VRM1-meetodi puhul saadakse surnud koed madalal temperatuuril hoides (edaspidi „külmutades saadud surnud koed“). VRM2-meetodi puhul saadakse surnud koed pikaajalisel (vähemalt 24±1 tundi) hoidmisel vees, millele järgneb säilitamine madalal temperatuuril (edaspidi „vees saadud surnud koed“). Iga MTT-d redutseeriv uuritav kemikaal kantakse vähemalt kahele surnud loe paralleelproovile, mis läbivad kogu katse käigu, et saada kontrollproov, milles MTT redutseerimine on mittespetsiifiline (NSMTT) (34, 35). Piisab ühest NSMTT-kontrollproovist iga uuritava kemikaali kohta, olenemata sellest, kui palju üksikkatseid/katsetsükleid tehakse. Koe tegelik eluvõimelisus arvutatakse järgmiselt: MTT redutseerijaga kokku puutunud eluskudedega saadud eluvõimelisuse protsent (%eluvõimelisustuuritav) miinus sama MTT redutseerijaga kokku puutunud surnud kudedega saadud MTT redutseerimine, mis arvutatakse negatiivse kontrollproovi suhtes, mida analüüsitakse paralleelselt korrigeerimist vajavate tulemustega katse läbiviimisega, st koe tegelik eluvõimelisus = [%eluvõimelisustuuritav] – [%NSMTT].
|
|
39.
|
Kui on selgunud, et uuritav kemikaal on nii segava värvusega (vt punkt 37) kui ka MTT otsene redutseerija (vt punkt 38), on neelduvuse (optilise tiheduse) mõõtmise korral peale eelmistes punktides kirjeldatud NSMTT- ja NSCelus-kontrollproovide vajalik ka kolmas kontrollproovide komplekt. See on tavaline tumeda värvusega uuritavate kemikaalide puhul, mis neelavad valgust lainepikkuste vahemikus 570±30 nm (nt sinine, lilla, must), sest nende loomulik värv takistab nende MTT otsese redutseerimise võime hindamist vastavalt punktis 38 kirjeldatule. Selline olukord nõuab automaatselt nii NSMTT- kui ka NSCelus-kontrollproovide kasutamist. Uuritavaid kemikaale, millega viiakse läbi nii NSMTT- kui ka NSCelus-kontrollproovid, võivad absorbeerida ja siduda nii elus kui ka surnud loed. Seega võib juhtuda, et NSMTT-kontrollproov ei aita parandustes arvestada mitte ainult uuritava kemikaali MTT otsese redutseerimise võimet, vaid ka uuritava kemikaali surnud kudedesse absorbeerumisel ja seal püsimisel tekkivat segavat värvust. See võib põhjustada segavast värvusest tingitud vigade topeltparandusi, sest uuritava kemikaali elusas koes absorbeerumisest ja seal püsimisest tingitud värvuse häirivat mõju korrigeeritakse juba NSCelus-kontrollproovi järgi. Et vältida segavast värvusest tingitud vigade topeltparandusi, on vaja teha kolmas kontrollkatse, millega analüüsitakse mittespetsiifilist värvust surnud kudedes (NSCsurnud) (vt III ja IV liide) (34, 35). Uuritavat kemikaali kantakse vähemalt kahele surnud koe paralleelproovile, millega tehakse läbi kogu katse käik, kuid MTTga inkubeerimise etapis inkubeeritakse neid MTT lahuse asemel söötmega. Piisab ühest NSCsurnud-kontrollproovist iga uuritava kemikaalikohta, olenemata sellest, kui palju üksikkatseid/katsetsükleid tehakse, kuid kõnealust kontrollproovi tuleks uurida paralleelselt NSMTT-kontrollprooviga ning kasutada mõlemas sama koepartiid. Koe tegelik eluvõimelisus arvutatakse järgmiselt: uuritava kemikaaliga kokku puutunud eluskudedega saadud eluvõimelisuse protsent (%eluvõimelisustuuritav) miinus %NSMTT miinus %NSCelus pluss segava värvusega uuritava kemikaaliga kokku puutunud ning söötmega ja ilma MTTta inkubeeritud surnud kudedega saadud mittespetsiifilise värvuse %, mis arvutatakse negatiivse kontrollproovi suhtes, mida analüüsitakse paralleelselt korrigeerimist vajavate tulemustega katse läbiviimisega, st koe tegelik eluvõimelisus = [%eluvõimelisustuuritav] – [%NSMTT] – [%NSCelus] + [%NSCsurnud].
|
|
40.
|
Oluline on arvestada, et mittespetsiifiline MTT redutseerimine ja mittespetsiifilise värvuse häiriv mõju võivad suurendada koeekstrakti optilist tihedust (kui mõõdetakse neelduvust) nii, et see jääb ülespoole spektrofotomeetri lineaarsusvahemikku, ning et mittespetsiifiline MTT redutseerimine võib suurendada koeekstraktis sisalduva MTT formasaani piigi pindala (kui mõõdetakse HPLC-/UPLC-spektromeetrilise ühendmeetodiga) nii, et see jääb ülespoole spektrofotomeetri lineaarsusvahemikku. Seetõttu on oluline et iga labor määraks näiteks MTT formasaaniga (CASi nr 57360-69-7, saab osta nt Sigma-Aldrichist, katalooginr M2003) kindlaks oma spektrofotomeetri optilise tiheduse / piigi pindala lineaarsusvahemiku, enne kui alustab uuritavate kemikaalide analüüsimist regulatiivsel eesmärgil.
|
|
41.
|
Neelduvuse (optilise tiheduse) mõõtmine spektrofotomeetriga on asjakohane MTT otseste redutseerijate ning segava värvusega uuritavate kemikaalide hindamisel, kui MTT formasaaniga mõõtmisel täheldatud segav mõju ei ole väga tugev (st uuritava kemikaaliga saadud koeekstraktide optilised tihedused ilma MTT otsest redutseerimist ja/või värvuse segavat mõju arvestavate parandusteta on spektrofotomeetri lineaarsusvahemikus). Sellegipoolest tuleb ettevaatlikult suhtuda nende uuritavate kemikaalide puhul saadud tulemustesse, mille negatiivse kontrollproovi tulemus on %NSMTT ja/või %NSCelus ≥ 60 % (VRM1, ja VRM2 vedelike analüüsimise metoodika) või 50 % (VRM2 tahkete ainete analüüsimise metoodika), sest need on kindlaksmääratud läviväärtused, mille järgi valideeritud võrdlusmeetodites eristatakse klassifitseeritud ja klassifitseerimata kemikaale (vt punkt 44). Neelduvust (optilist tihedust) ei saa siiski mõõta, kui MTT formasaani sisalduse mõõtmist segav mõju on liiga tugev (st põhjustab uuritavate koeekstraktide korrigeerimata optiliste tiheduste sattumist väljapoole spektrofotomeetri lineaarsusvahemikku). Neid uuritavaid kemikaale, mis on värvilised või muutuvad värviliseks kokkupuutel vee või isopropanooliga ja segavad liiga tugevalt MTT formasaani neelduvuse (optilise tiheduse) mõõtmist, saab siiski analüüsida HPLC-/UPLC-spektromeetrilist ühendmeetodit kasutades (vt III ja IV liidet). HPLC-/UPLC-aparatuur võimaldab MTT formasaani enne selle kvantitatiivset määramist kemikaalist eraldada (36). Sel põhjusel ei ole HPLC-/UPLC-spektromeetrist ühendmeetodit kasutades vaja analüüsida NSCelus- või NSCsurnud-kontrollproove olenemata sellest, missugust kemikaali uuritakse. Siiski tuleks uurida NSMTT-kontrollproove, kui on kahtlus, et uuritav kemikaal redutseerib otseselt MTTd (toimides punktis 38 kirjeldatu kohaselt). Samuti tuleks NSMTT-kontrollproove kasutada värviliste (iseloomuliku või veega kokkupuutel tekkiva värvusega) uuritavate kemikaalide puhul, kui värvus takistab hinnata nende võimet otseselt MTTd redutseerida (nimetatud kontrollproovide kasutamist on kirjeldatud punktis 38). Kui MTT formasaani sisalduse mõõtmiseks kasutatakse HPLC-/UPLC-spektromeetrilist ühendmeetodit, arvutatakse koe eluvõimelisus (protsentides) uuritava kemikaaliga kokku puutunud elusa koega saadud MTT formasaani piigi pindala protsendina paralleelse negatiivse kontrollprooviga saadud MTT formasaani piigi suhtes. Nende uuritavate kemikaalide puhul, mis on võimelised otseselt MTT redutseerima, arvutatakse koe tegelik eluvõimelisus järgmiselt: %Eluvõimelisustuuritav miinus %NSMTT, nagu on kirjeldatud punkti 38 viimases lauses. Lõpuks tuleb märkida, et neid otseseid MTT redutseerijaid (mis võivad olla ka segava värvusega), mis jäävad kudedesse pärast töötlemist püsima ja redutseerivad MTTd nii tugevalt, et see põhjustab uuritavate koeekstraktide optiliste tiheduste (kui mõõdetakse optilist tihedust) või piigi pindalade (kui kasutatakse HPLC-/UPLC-spektromeetrilist ühendmeetodit) sattumist väljapoole spektrofotomeetri lineaarsusvahemikku, ei saa analüüsida RhCE-katsemeetoditega, kuigi neid esineb eeldatavast väga harva.
|
|
42.
|
HPLC-/UPLC-spektromeetrilist ühendmeetodit võib kasutada MTT formasaani sisalduse mõõtmiseks igat tüüpi uuritava kemikaali korral (värvilised, värvitud, MTT redutseerijad ja mitteredutseerijad) (11, 36). Kuna HPLC-/UPLC-spektrofotomeetria aparatuurid on erinevad, ei ole igal kasutajal lihtne luua süsteemis täpselt samasugused tingimused. Enne HPLC-/UPLC-spektromeetria aparatuuri kasutamist koeekstraktidest pärit MTT formasaani kvantitatiivseks mõõtmiseks tuleb tõendada aparatuuri nõuetekohasust; selleks peavad olema täidetud mitme standardse kvalifikatsiooninäitajaga seotud nõuetekohasuse kriteeriumid, mille aluseks on näitajad ja kriteeriumid, mida on kirjeldatud USA Toidu- ja Ravimiameti juhendis bioanalüütilise meetodi valideerimise kohta tööstuses (36, 38). Need põhinäitajad ja nende nõuetekohasuse kriteeriumid on esitatud 5. liites. Kui 5. liites määratletu nõuetekohasuse kriteeriumid on täidetud, loetakse HPLC-/UPLC-fotomeetriaaparatuur nõuetekohaseks ning see on valmis MTT formasaani sisalduse mõõtmiseks käesolevas katsemeetodis kirjeldatud katsetingimuste juures.
|
Nõuetekohasuse kriteeriumid
|
43.
|
Igas analüüsitsüklis, kus kasutakse kvaliteedikontrolli (vt punkt 30) läbinud RhCE-koepartiisid, peab negatiivse kontrollainega töödeldud kudede optiline tihedus kajastama kudede veo-, vastuvõtu- ja katse-eeskirjakohaste töötluste järgset kvaliteeti ega tohi jääda väljapoole varem kindlaksmääratud piire, mis on esitatud tabelis 2 (vt punkt 26). Samuti peab positiivse kontrollainega, nt metüülatsetaadiga töödeldud kudede keskmine eluvõimelisus negatiivse kontrollprooviga võrreldes olema < 50 %, kui järgitakse VRM1-meetodi vedelike või tahkete ainete katse-eeskirja, ja ≤ 30 % (vedelikel) või ≤ 20 % (tahketel ainetel), kui kasutatakse VRM2-meetodit; see näitaja kirjeldab kudede võimet reageerida ärritavale uuritavale kemikaalile katsemeetodiga ettenähtud tingimustes (34, 35). Uuritava kemikaalide ja kontrollainetega töödeldud paralleelsete koeproovidega saadud tulemuste varieeruvus peab jääma vastuvõetavatesse piiridesse (st kahe paralleelse koeproovi eluvõimelisuse erinevus peab olema alla 20 % või ei tohi kolme paralleelse koeprooviga saadud tulemuste standardhälve ületada 18 %). Kui analüüsitsüklisse võetud negatiivse või positiivse kontrollprooviga saadud tulemus jääb väljapoole vastuvõetavaid piire, loetakse analüüsitsükkel ebakvaliteetseks ja seda tuleb korrata. Kui uuritava kemikaalige töödeldud paralleelsete koeproovidega saadud tulemuste varieeruvus jääb väljapoole vastuvõetavaid piire, loetakse katse ebakvaliteetseks ja katset uuritava kemikaaliga tuleb korrata.
|
Tulemuste tõlgendamine ja prognoosimismudel
|
44.
|
Iga uuritava kemikaaliga töödeldud paralleelsete koeproovide ekstraktidega saadud optilise tiheduse väärtuste / piikide pindalade järgi arvutatakse protsentides koe keskmine eluvõimelisus (paralleelsete koeproovidega saadud tulemuste keskväärtus) normaliseerituna negatiivse kontrollproovi järgi, mille väärtuseks võetakse 100 %. Koe eluvõimelisuse (protsentides) läviväärtused nende uuritavate kemikaalide kindlakstegemiseks, mida ei ole vaja klassifitseerida seoses silmade ärrituse või raske silmakahjustusega (ÜRO GHSi / CLP-määruse kohane kategooria puudub), on esitatud tabelis 4. Tulemusi tuleb seega tõlgendada järgmiselt:
|
—
|
uuritav kemikaal määratletakse ÜRO GHSi ja CLP-määruse kohast klassifitseerimist ja märgistamist mittevajava kemikaalina (kategooria puudub), kui koe keskmine eluvõimelisus (protsentides) pärast kemikaaliga kokkupuudet ja kokkupuutejärgset inkubeerimist on suurem kui (>) koe eluvõimelisuse (protsentides) kindlaksmääratud läviväärtus, mis on esitatud tabelis 4. Sellisel juhul ei ole vaja täiendavaid katseid teiste katsemeetoditega;
|
|
—
|
kui koe keskmine eluvõimelisus (protsentides) pärast kemikaaliga kokkupuudet ja kokkupuutejärgset inkubeerimist on väiksem kui koe eluvõimelisuse (protsentides) kindlaksmääratud läviväärtus või sellega võrdne (≤), ei ole võimalik prognoose teha, nagu näidatud tabelis 4. Sellisel juhul on vaja täiendavaid kaitset teiste katsemeetoditega, sest RhCEd kasutavad katsemeetodid annavad teatava hulga valepositiivseid tulemusi (vt punktid 14–15) ega suuda eristada URO GHSi ja CLP-määruse kohastesse kategooriatesse 1 ja 2 kuuluvaid kemikaale (vt punkt 17)
|
Tabel 4
Prognoosimismudelid ÜRO GHSi ja CLP-määruse klassifikatsiooni järgi
|
VRM
|
Kategooriata
|
Prognoosida ei ole võimalik
|
|
VRM1 – EpiOcular™i silmaärrituse katse (mõlemad katse-eeskirjad)
|
Koe keskmine eluvõimelisus > 60 %
|
Koe keskmine eluvõimelisus ≤ 60 %
|
|
VRM2 – SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse (katse-eeskiri vedelike korral)
|
Koe keskmine eluvõimelisus > 60 %
|
Koe keskmine eluvõimelisus ≤ 60 %
|
|
VRM2 – SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse (katse-eeskiri tahkete ainete korral)
|
Koe keskmine eluvõimelisus > 50 %
|
Koe keskmine eluvõimelisus ≤ 50 %
|
|
|
45.
|
Üksainus katse vähemalt kahe paralleelse koeprooviga uuritava kemikaali kohta peaks olema piisav, kui tulemus on ühene. Piiripealse tulemuse puhul, nagu lahkuminevad tulemused paralleelproovide mõõtmisel ja/või keskmine eluvõimelisuse protsent 60 ± 5 % (VRM1 ja VRM2 vedelike korral) või 50 ± 5 % (VRM2 tahkete ainete korral), tuleks kaaluda teise ja isegi kolmanda katse läbiviimist, kui kaks esimest katset annavad vastuolulise tulemuse.
|
|
46.
|
Kui see on asjakohane ja õigustatud, võib teatavat liiki segude puhul kaaluda teistsuguste koe eluvõimelisuse (protsentides) läviväärtuste kasutamist, millega eristatakse klassifitseeritud ja klassifitseerimata uuritavad kemikaalid, et suurendada katsemeetodi üldist toimivust seda liiki segude korral (vt punkt 14). Võrdluskemikaalid võivad aidata hinnata tundmatu uuritava kemikaali või tooteklassi võimet põhjustada rasket silmakahjustust / silmade ärritust või hinnata klassifitseeritud kemikaali võimalikku suhtelist toksilisust silmade suhtes teatavas positiivsete reaktsioonide vahemikus.
|
ANDMED JA ARUANDLUS
Andmed
|
47.
|
Iga analüüsitsüklis oleva paralleelse koeprooviga saadud andmed (nt uuritavate kemikaalide ja kontrollainete optilise tiheduse väärtused / piikide pindalad ja arvutatud koe eluvõimelisuse (protsentides) andmed ning RhCE-katsemeetodi kohane lõplik prognoos) esitatakse tabelina iga uuritava kemikaali kohta, samuti tuleb vajaduse korral esitada korduskatsetel saadud andmed. Peale selle tuleb esitada kudede keskmine eluvõimelisus (protsentides) ja kahe paralleelse koeproovi eluvõimelisuse erinevus (kui n = 2 paralleelset koeproovi) või iga uuritava kemikaali ja kontrollainega saadud tulemuste standardhälve (kui n ≥ 3 paralleelset koeproovi). Kui uuritud kemikaali puhul täheldati segavat mõju MTT formasaani sisalduse mõõtmisele MTT otsese redutseerimise tõttu ja/või segavat värvust, tuleb iga niisuguse kemikaali puhul esitada sellekohased andmed.
|
Katseprotokoll
|
48.
|
Katseprotokollis tuleks esitada järgmine teave.
Uuritav kemikaal
|
|
Ühest koostisosast koosnev aine:
|
—
|
kemikaali identifitseerimisandmed, nagu IUPACi või CASi nimetus(ed) ja CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muud identifikaatorid;
|
|
—
|
füüsikaline olek, lenduvus, pH, log P, molekulmass, aineklass ja muud uuringu tegemisega seotud asjakohased füüsikalis-keemilised omadused, kättesaadavas ulatuses;
|
|
—
|
puhtus, lisandite keemiline määratlus, kui see on asjakohane ja praktiliselt teostatav jne;
|
|
—
|
töötlemine enne katset, kui asjakohane (nt soojendamine, peenestamine);
|
|
—
|
säilitamise tingimused ja stabiilsus säilitamisel, kättesaadavas ulatuses.
|
|
|
|
Mitme koostisosaga aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal ja segu:
|
—
|
võimalikult täpne omaduste kirjeldus lähtuvalt koostisosade keemilisest määratlusest (vt eespool), kvantitatiivsest sisaldusest ja asjakohastest füüsikalis-keemilistest omadustest (vt eespool), kättesaadavas ulatuses;
|
|
—
|
füüsikaline olek ja muud uuringu tegemisega seotud asjakohased füüsikalis-keemilised omadused, kättesaadavas ulatuses;
|
|
—
|
puhtus, lisandite keemiline määratlus, kui see on asjakohane ja praktiliselt teostatav jne;
|
|
—
|
töötlemine enne katset, kui asjakohane (nt soojendamine, peenestamine);
|
|
—
|
säilitamise tingimused ja stabiilsus säilitamisel, kättesaadavas ulatuses.
|
|
Positiivsed ja negatiivsed kontrollained
|
—
|
Kemikaali identifitseerimisandmed, nagu IUPACi või CASi nimetus(ed) ja CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muud identifikaatorid;
|
|
—
|
füüsikaline olek, lenduvus, molekulmass, aineklass ja muud uuringu tegemisega seotud asjakohased füüsikalis-keemilised omadused, kättesaadavas ulatuses;
|
|
—
|
puhtus, lisandite keemiline määratlus, kui see on asjakohane ja praktiliselt teostatav jne;
|
|
—
|
töötlemine enne katset, kui asjakohane (nt soojendamine, peenestamine);
|
|
—
|
säilitamise tingimused ja stabiilsus säilitamisel, kättesaadavas ulatuses;
|
|
—
|
muu negatiivse kontrollaine kui ülipuhta H2O või Ca2+/Mg2+-vaba DPBSi kasutamise põhjendus, kui see on asjakohane;
|
|
—
|
muu positiivse kontrollaine kui puhta metüülatsetaadi kasutamise põhjendus, kui see on asjakohane;
|
|
—
|
viide varasematele positiivse ja negatiivse kontrollproovi tulemustele, mis tõendavad sobivaid analüüsitsükli nõuetekohasuse kriteeriume.
|
Teave sponsori ja katselabori kohta
|
—
|
Sponsori, katselabori ja uuringu juhi nimi ja aadress.
|
|
—
|
Kasutatud RhCE-koemudel ja katse-eeskiri (koos valikute põhjendustega, kui see on asjakohane)
|
Katsemeetodi tingimused
|
—
|
Kasutatud RhCE-koemudel, sealhulgas partii number;
|
|
—
|
MTT formasaani kvantitatiivseks määramiseks kasutatud lainepikkus ja spektririba laius (kui asjakohane) ning mõõteseadme (nt spektrofotomeetri) lineaarsusvahemik;
|
|
—
|
MTT formasaani kvantitatiivse määramise meetodi kirjeldus;
|
|
—
|
kasutatud HPLC-/UPLC-fotomeetriaaparatuuri kirjeldus, kui asjakohane;
|
|
—
|
kogu täiendav teave kasutatud konkreetse RhE-koemudeli ja selle toimivuse kohta. See hõlmab vähemalt järgmist:
|
i)
|
eluvõimelisus kvaliteedi kontrollil (tarnija andmed);
|
|
ii)
|
eluvõimelisus katsemeetodiga nõutavates tingimustes (kasutaja andmed);
|
|
iii)
|
kaitsekihina toimivuse kontroll;
|
|
iv)
|
morfoloogiaandmed, kui on olemas;
|
|
v)
|
reprodutseeritavus ja prognoosivõime;
|
|
vi)
|
muude RhCE-koemudeli kvaliteedi kontrollide andmed, kui on olemas;
|
|
|
—
|
viide varasematele andmetele RhCE-koemudeli kohta. See hõlmab vähemalt järgmist: kvaliteedikontrolli andmete nõuetekohasus, arvestades varasemaid partii andmeid;
|
|
—
|
kinnitus, et katselabor on enne katsemeetodi rutiinset kasutamist tõendanud oma pädevust asjaomaseid kemikaale analüüsides.
|
Analüüsiseeria ja katse nõuetekohasuse kriteeriumid
|
—
|
Positiivsete ja negatiivsete kontrollproovidega saadud tulemuste keskväärtused ja nõuetekohased vahemikud, mille aluseks on aja jooksul kogutud andmed;
|
|
—
|
positiivsel ja negatiivsel kontrollkatsel paralleelsete koeproovidega saadud tulemuste lubatav varieeruvus;
|
|
—
|
uuritava kemikaaliga paralleelsete koeproovidega saadud tulemuste lubatav varieeruvus.
|
Katse käik
|
—
|
Kasutatud katse-eeskirja üksikasjad;
|
|
—
|
uuritava kemikaali ja kontrollainete kasutatud doosid;
|
|
—
|
kokkupuute, kokkupuutejärgse sukeldatuse ja inkubeerimise kestus ja temperatuur nende ajal (kui asjakohane);
|
|
—
|
kõikide katse-eeskirjas tehtud muudatuste kirjeldus;
|
|
—
|
otseste MTT redutseerijate ja/või värviliste uuritavate kemikaalide puhul kasutatud kontrollproovid, kui asjakohane;
|
|
—
|
kasutatud paralleelsete koeproovide arv uuritava kemikaali kohta ja kontrollproovid (positiivne kontrollproov, negatiivse kontrollproov, NSMTT, NSCelus ja NSCsurnud, kui asjakohane).
|
Tulemused
|
—
|
Tabeli kujul esitatud andmed igas analüüsitsüklis ja paralleelmõõtmises olnud iga uuritava kemikaali ja kontrollaine kohta (kaasa arvatud korduskatsete andmed, kui see on asjakohane), sealhulgas optilise tiheduse väärtus või MTT formasaani piigi pindala, koe eluvõimelisus (protsentides), koe keskmine eluvõimelisus (protsentides), paralleelsete koeproovidega saadud tulemuste erinevus või standardhälve ning lõplik prognoos;
|
|
—
|
kui see on asjakohane, siis otseselt MTT redutseerivate ja/või värviliste uuritavate kemikaalide puhul kasutatud kontrollproovide tulemused, sealhulgas optilise tiheduse väärtus või MTT formasaani piigi pindala, %NSMTT, %NSCelus, %NSCsurnud, paralleelsete koeproovidega saadud tulemuste erinevus või standardhälve, lõplik korrigeeritud koe eluvõimelisus (protsentides) ja lõplik prognoos;
|
|
—
|
uuritava(te) kemikaali(de)ga ja kontrollainetega saadud tulemuste võrdlus analüüsitsükli ja katsete nõuetekohasuse kriteeriumidega;
|
|
—
|
muude täheldatud mõjude (nt kudede värvumine värvilise uuritava kemikaali tõttu) kirjeldus.
|
Tulemuste arutelu
Järeldused
|
KIRJANDUS
|
(1)
|
Ühinenud Rahvaste Organisatsioon (ÜRO), „United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS)“, ST/SG/AC.10/30/Rev 6, 6., täiendatud väljaanne, United Nations, New York ja Genf, 2015. Kättesaadav aadressil http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Käesoleva lisa peatükk B.5 „Äge mürgisus: silmade ärritus/söövitus“.
|
|
(3)
|
Käesoleva lisa peatükk B.47, „Veise sarvkesta hägususe ja läbilaskvuse katsemeetod selliste kemikaalide kindlakstegemiseks, i) mis tekitavad raskeid silmakahjustusi, ja ii) mida ei ole vaja klassifitseerida seoses silmade ärrituse või raske silmakahjustusega“.
|
|
(4)
|
Käesoleva lisa peatükk B.48, „Isoleeritud kanasilma katsemeetod selliste kemikaalide kindlakstegemiseks, i) mis tekitavad raskeid silmakahjustusi, ja ii) mida ei ole vaja klassifitseerida“.
|
|
(5)
|
Käesoleva lisa peatükk B.61, „Fluorestseiini lekke katsemeetod silmi söövitavate ja tugevalt ärritavate kemikaalide kindlakstegemiseks“.
|
|
(6)
|
Käesoleva lisa peatükk B.68, „Lühiajalise kokkupuute in vitro katsemeetod selliste kemikaalide kindlakstegemiseks, i) mis tekitavad raskeid silmakahjustusi, ja ii) mida ei ole vaja klassifitseerida seoses silmade ärrituse või raske silmakahjustusega“.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010), „Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing“, ALTEX, nr 27, eriväljaanne 2010, lk 261–266.
|
|
(8)
|
EC EURL ECVAM (2014), „The EURL ECVAM – Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report.“ EUR 28 125 EN; doi:10.2787/41680. Kättesaadav aadressil http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAMi teaduslik nõuandekomitee (2014), „ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan.“ ESACi arvamus nr 2014-03, 17. november 2014; EUR 28 173 EN; doi: 10.2787/043697. Kättesaadav aadressil http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016), „Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals“, Toxicol. In Vitro, nr 31, lk 43–53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016), „Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing“, Toxicol. In Vitro, nr 34, lk 55–70.
|
|
(12)
|
EURL ECVAMi teaduslik nõuandekomitee (2016), „ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT)“, ESACi arvamus nr 2016-02, 24. juuni 2016; EUR 28 175 EN; doi: 10.2787/390390. Kättesaadav aadressil http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016), „Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS“ (pooleliolev käsikiri).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944), „Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes“, Journal of Pharmacol. and Exp. Therapeutics, nr 82, lk 377–390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010), „A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches“, Toxicol. In Vitro, nr 24, lk 1–9.
|
|
(16)
|
Mosmann, T. (1983), „Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays“, J. Immunol. Methods, nr 65, lk 55–63.
|
|
(17)
|
OECD (2016), „Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492“, Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(18)
|
OECD (2005), „Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment“, Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011), „Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation“, Altern. Lab. Anim., nr 39, lk 339–364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003), „Three-dimensional construct of the human corneal epithelium for in vitro toxicology“, Väljaandes: Salem, H., Katz, S.A. (toim.), Alternative Toxicological Methods, CRC Press, lk 147–159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013), „Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation“, Toxicol. In Vitro, nr 27, lk 619–626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013), „Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation“, Toxicol. In Vitro, nr 27, lk 1 476–1 488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015), „The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications“, Altern. Lab. Anim., nr 43, lk 1–18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014), „Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods“, Arch. Toxicol., nr 88, lk 701–723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017), „Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD)“, Arch. Toxicol., nr 91, lk 521–547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011), „Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium“, Molecular Vision, nr 17, lk 113–126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991), „Eye Irritation“, väljaandes Advances in Modern Toxicology: Dermatoxicology Marzulli F.N. ja Maibach H.I. (toim.), 4th trükk, lk 749–815, Hemisphere Publishing Corporation, Washington, DC, USA.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008), „Toxic Responses of the Ocular and Visual System,“ Väljaandes „Casarett & Doull’s Toxicology: The Basic Science of Poisons“, Klaassen C.D. (toim.), 7. trükk, lk 665–697, McGraw-Hill Ryerson, Withby, ON, Kanada.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998), „Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death“, Invest. Ophthalmol. Vis. Sci., nr 39, lk 922–936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002), „Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays“, Reg. Tox. Pharmacol., nr 36, lk 106–117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001), „Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests“, Toxicol. In Vitro, nr 15, lk 115–130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998), „Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses“, Invest. Ophthalmol. Vis. Sci., nr 39, lk 2 610–2 625.
|
|
(33)
|
Jester, J.V. (2006), „Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test“, Cutan. Ocul. Toxicol., nr 25, lk 41–54.
|
|
(34)
|
EpiOcular™i silmaärrituse katse standardeeskiri, 8. versioon (5. märts 2013), „EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals“, kättesaadav aadressil https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index
|
|
(35)
|
SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katse standardeeskiri, 1. versioon (20. juuli 2015), „SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals“, kättesaadav aadressil https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015), „Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals“, Toxicol. In Vitro, nr 29, lk 741–761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015), „EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times“, Altern. Lab. Anim., nr 43, lk 101–127.
|
|
(38)
|
USA Toidu- ja Ravimiamet (2001), „Guidance for Industry: Bioanalytical Method Validation“, U.S. Department of Health and Human Services, Food and Drug Administration, mai 2001. Kättesaadav aadressil http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017), „Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263“, ENV Publications, Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
1. liide
MÕISTED
Täpsus
: katsemeetodiga saadud tulemuste ja nõuetekohaste võrdlusväärtuste kokkulangevuse määr. Täpsus näitab katsemeetodi tulemuslikkust ja on üks olulistest aspektidest. Seda terminit kasutatakse ka vastavuse tähenduses ja see näitab katsemeetodiga saadud õigete tulemuste osakaalu (18).
Võrdluskemikaal
: kemikaal, mida kasutatakse standardina võrdlemiseks uuritava kemikaaliga. Võrdluskemikaalil peaksid olema järgmised omadused: i) pidev ja usaldusväärne tarnija, kes kemikaali identifitseerib ja seda kirjeldab; ii) struktuuriline ja funktsionaalne sarnasus uuritava(te) kemikaali(de)ga ja/või sellega/nendega sarnane kemikaali-/tooteklass; iii) teadaolevad füüsikalis-keemilised omadused; iv) teadaolevat toimet tõendavad andmed ja v) teadaolev mõjusus soovitavas reaktsioonitugevuse vahemikus.
Alt-üles lähenemisviis
: etapiviisiline lähenemisviis, mida kasutatakse niisuguste uuritavate kemikaalide korral, mille puhul on põhjust arvata, et need ei vaja klassifitseerimist ega märgistamist silmade ärrituse või raske silmakahjustusega seoses; nimetatud lähenemisviisi puhul eristatakse kõigepealt kemikaalid, mis ei vaja klassifitseerimist ega märgistamist (tulemus on negatiivne), muudest kemikaalidest (tulemus on positiivne).
Kemikaal
: aine või segu.
Vastavus
: vt „Täpsus“.
Sarvkest
: silmamuna esikülje läbipaistev osa, mis katab vikerkesta ja pupilli ning laseb valgusel tungida silma sisse.
CV
: variatsioonikordaja.
Dev
: hälve.
EIT
: silmaärrituse katse.
EURL ECVAM
: loomkatsete alternatiivide Euroopa Liidu referentlabor.
Silmade ärritus
: muutuse tekkimine silmas pärast uuritava kemikaali silma eespinnale manustamist, seejuures on muutus 21 päeva jooksul pärast manustamist täielikult taandunud. Samatähenduslik mõistetega „pöörduv mõju silmale“ ja „ÜRO GHSi / CLP-määruse kohane 2. kategooria“.
ET50
: kokkupuuteaeg, mille jooksul pealekantud, teatava kindla kontsentratsiooniga võrdluskemikaal vähendab koe eluvõimelisust 50 % võrra.
Valenegatiivsete tulemuste määr
: kõikide selliste ainete osakaal, mis peaksid andma positiivse tulemuse, kuid on katsemeetodit järgides andnud negatiivse tulemuse. See on üks katsemeetodi tulemuslikkuse näitaja.
Valepositiivsete tulemuste määr
: kõikide selliste ainete osakaal, mis peaksid andma negatiivse tulemuse, kuid on katsemeetodit järgides andnud positiivse tulemuse. See on üks katsemeetodi tulemuslikkuse näitaja.
Oht
: niisuguse mõjuri või olukorra loomupärane omadus, mis võib avaldada kahjulikku toimet, kui organism, süsteem või (ala)populatsioon selle mõjuriga kokku puutub.
HCE
: SkinEthic™i inimese sarvkesta epiteel.
HPLC
: kõrgefektiivne vedelikkromatograafia.
IC50
: kontsentratsioon, mille juures võrdluskemikaal vähendab kindla kokkupuuteaja (nt 30-minutine töötlemine naatriumdodetsüülsulfaadiga) jooksul koe eluvõimelisust 50 % võrra.
Lõpmatu annus
: RhCE-koemudelile kantud uuritava kemikaali kogus, mis on suurem epiteeli pinna täielikuks ja ühtlaseks katmiseks vajalikust kogusest.
Pöördumatu mõju silmale
: vt „Raske silmakahjustus“.
LLOQ
: alumine määramispiir.
LogP
: oktanool-vesi jaotuskoefitsiendi logaritm.
Segu
: kahest või enamast ainest koosnev segu või lahus.
Ühe koostisosaga aine
: aine, mis on määratletud oma kvantitatiivse koostisega, milles ühe peamise koostisosa sisaldus on vähemalt 80 massiprotsenti.
Mitme koostisosaga aine
: aine, mis on määratletud oma kvantitatiivse koostisega, milles rohkem kui ühe peamise koostisosa kontsentratsioon on ≥ 10 % ja < 80 % (massiprotsendid). Mitme koostisosaga aine saadakse tootmisprotsessi käigus. Erinevus segu ja mitme koostisosaga aine vahel seisneb selles, et segu saadakse kahe või enama aine kokku segamisel, ilma et toimuks keemilist reaktsiooni. Mitme koostisosaga aine saadakse keemilise reaktsiooni tulemusel.
MTT
: 3-(4,5-dimetüültiasool-2-üül)-2,5-difenüültetrasooliumbromiid, tiasolüülsinine.
Negatiivne kontrollproov
: proov, mis sisaldab kõiki katsesüsteemi komponente ja mida on töödeldud ainega, mis teadaolevalt kutsub katsesüsteemis esile positiivse reaktsiooni. Seda proovi analüüsitakse koos uuritava kemikaaliga töödeldud proovide ja teiste kontrollproovidega, et määrata kindlaks 100 % koe eluvõimelisus.
Klassifitseerimata
: kemikaalid, mida ei ole klassifitseeritud seoses silmade ärrituse (ÜRO GHSi / CLP-määruse kohane 2. kategooria, ÜRO GHSi kategooria 2A või 2B) ega raske silmakahjustusega (ÜRO GHSi / CLP-määruse kohane 1. kategooria). Samatähenduslik mõistega „ÜRO GHSi /CLP-määruse kohane kategooria puudub“.
NSCsurnud
: mittespetsiifiline värvus surnud koes.
NSCelus
: mittespetsiifiline värvus elavas koes.
NSMTT
: mittespetsiifiline MTT redutseerimine.
OD
: optiline tihedus.
Toimivusnõuded
: teaduslikult põhjendatuks arvatud valideeritud katsemeetodil põhinevad nõuded, mille alusel hinnatakse, kuivõrd saab kavandatud katsemeetodit võrrelda teostuslikult ja tööpõhimõttelt samalaadse meetodiga. Hõlmab järgmist: i) katsemeetodi olulised osad; ii) valideeritud katsemeetodi nõuetekohase toimivuse tõendamiseks kasutatud kemikaalide hulgast valitud võrdluskemikaalide miinimumloend ja iii) võrreldavad täpsuse ja usaldusväärsuse tasemed, mis põhinevad valideeritud katsemeetodiga saadud tulemustel ja mis tuleks kavandatud katsemeetodi hindamisel saavutada võrdluskemikaalide miinimumloendi kasutamisel (18).
Positiivne kontrollproov
: proov, mis sisaldab kõiki katsesüsteemi komponente ja mida on töödeldud ainega, mis teadaolevalt kutsub katsesüsteemis esile positiivse reaktsiooni. Seda proovi analüüsitakse koos uuritava kemikaaliga töödeldud proovide ja teiste kontrollproovidega. Selleks, et oleks võimalik hinnata positiivse kontrollproovi esilekutsutud reaktsiooni varieeruvust ajas, ei tohi positiivne reaktsioon olla ülemäära tugev.
Asjakohasus
: mõiste, millega väljendatakse seda, kas katsemeetod võimaldab uurida huvipakkuvat mõju, kas meetod on mõttekas ja sobib konkreetse eesmärgi jaoks. See näitab, mil määral saab katsemeetodiga õigesti mõõta või prognoosida huvipakkuvat bioloogilist mõju. Asjakohasus hõlmab ka katsemeetodi täpsuse (vastavuse) aspekti (18).
Usaldusväärsus
: iseloomustab katsemeetodi korratavust, kui meetodit rakendatakse ühe ja sama katse-eeskirja alusel ühes laboris ja eri laborites pikema aja jooksul. Usaldusväärsuse hindamiseks arvutatakse laborisisene ja laboritevaheline korratavus ning laborisisene korduvus (18).
Asendav meetod
: katsemeetod, mis on kavandatud rutiinses kasutuses oleva kinnitatud meetodi asendamiseks ohtude kindlakstegemise ja/või riskide hindamise meetodina ning mille puhul on kindlaks tehtud, et see tagab kas inimeste või loomade tervise või keskkonna samaväärse või parema kaitse kui kinnitatud katsemeetod kõikide võimalike määramisolukordade ja kemikaalide puhul (18).
Korratavus
: sama uuritava kemikaali ja sama katsemetoodikaga korduvatel katsetel saadud tulemuste kokkulangevus (18).
Pöörduv mõju silmale
: vt „Silmade ärritus“.
RhCE
: rekonstrueeritud inimese sarvkesta sarnane epiteel.
Analüüsitsükkel
: analüüsitsükkel hõlmab ühe või mitme uuritava kemikaali analüüsi koos negatiivse ja positiivse kontrollprooviga.
SD
: standardhälve.
Tundlikkus
: kõigi niisuguste positiivsete/reaktsioonivõimeliste uuritavate kemikaalide osakaal, mis katse tulemusel klassifitseeritakse õigesti. Tundlikkusega iseloomustatakse katsemeetodi täpsust, kui saadud tulemusi saab kategoriseerida, ning see on oluline aspekt katsemeetodi asjakohasuse hindamisel (18).
Raske silmakahjustus
: sellise koekahjustuse tekkimine silmas või selline tugev nägemisteravuse langus pärast seda, kui uuritavat ainet on manustatud silma eespinnale, mis ei ole 21 päeva jooksul pärast manustamist täielikult pöörduv. Samatähenduslik mõistetega „pöördumatu mõju silmale“ ja „ÜRO GHSi ja CLP-määruse kohane 1. kategooria“.
Standardeeskiri
: kirjalik formaalne eeskiri, mis kirjeldab üksikasjalikult, kuidas viia läbi rutiinseid ja katsega seotud laboritoiminguid. See on nõutav hea laboritava kohaselt.
Spetsiifilisus
: kõigi niisuguste negatiivsete/mittereageerivate uuritavate kemikaalide osakaal, mis katse tulemusel klassifitseeritakse õigesti. Spetsiifilisusega iseloomustatakse katsemeetodi täpsust, kui saadud tulemusi saab kategoriseerida, ning see on oluline aspekt katsemeetodi asjakohasuse hindamisel (18).
Aine
: looduslik või tootmisprotsessi teel saadud keemiline element ja selle ühendid, kaasa arvatud stabiilsuse säilitamiseks vajalikud lisaained ja tootmisprotsessist tingitud lisandid, välja arvatud mis tahes lahusti, mida on võimalik aine stabiilsust vähendamata ja selle koostist muutmata eraldada.
Katse
: ühe uuritava kemikaali analüüsimine vähemalt kahe paralleelse koeprooviga, nagu määratletud vastavas standardeeskirjas.
Koe eluvõimelisus
: rekonstrueeritud koe rakupopulatsiooni üldist aktiivsust (nt rakkude võimet redutseerida vitaalvärvainet MTT) väljendav parameeter, mis olenevalt mõõdetavast näitajast ja katseplaanist on vastavuses elusrakkude koguarvu ja/või elujõulisusega.
Ülalt-alla lähenemisviis
: etapiviisiline lähenemisviis, mida kasutatakse niisuguste kemikaalide korral, mille puhul on põhjust arvata, et need põhjustavad rasket silmakahjustust; nimetatud lähenemisviisi puhul eristatakse kõigepealt rasket silmakahjustust tekitavad kemikaalid (tulemus on positiivne) muudest kemikaalidest (tulemus on negatiivne).
Uuritav kemikaal
: iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
Astmelise katsetamise strateegia
: Etapiviisi katsete tegemise strateegia, milles kasutatakse järjestikku eri katsemeetodeid. Enne strateegia järgmise etapiga alustamist vaadatakse igas etapis läbi kogu uuritava kemikaali kohta olemasolev teave, kasutades tõendite kaalukuse menetlust, et otsustada, kas teave on ohuklassi määramiseks piisav. Kui olemasoleva teabe põhjal saab antud etapis otsustada uuritava kemikaali võimaliku ohtlikkuse üle, ei ole täiendavaid katseid vaja teha (18).
ULOQ
: ülemine määramispiir.
Ühinenud Rahvaste Organisatsiooni ühtne ülemaailmne kemikaalide klassifitseerimise ja märgistamise süsteem (ÜRO GHS (Globally Harmonized System))
: süsteem, milles kemikaalid (ained ja segud) on klassifitseeritud vastavalt nende füüsilise ohtlikkuse ning kahjuliku tervise- ja keskkonnamõju standarditud tüübile ja -tasemele ning mis hõlmab asjaomaseid teavitustähiseid, nagu piktogrammid, märksõnad, ohulaused, hoiatuslaused ja ohutuskaardid, et anda edasi inimeste (sealhulgas tööandjate, töötajate, vedajate, tarbijate ja päästetöötajate) ja keskkonna kaitsmiseks vajalikku teavet kõnealuste kemikaalide kahjuliku mõju kohta (1).
ÜRO GHSi ja CLP-määruse kohane 1. kategooria
: vt „Raske silmakahjustus“.
ÜRO GHSi ja CLP-määruse kohane 2. kategooria
: vt „Silmade ärritus“.
ÜRO GHSi ja CLP-määruse kohase kategooriata
: kemikaalid, mis ei vasta ÜRO GHSi / CLP-määruse kohasesse 1. ega 2. kategooriasse (ega ÜRO GHSi 2A või 2B kategooriasse) klassifitseerimise nõuetele. Samatähenduslik mõistega „Klassifitseerimata“.
UPLC
: ultraefektiivne vedelikkromatograafia.
UVCB
: tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal.
Teaduslikult põhjendatud katsemeetod
: katsemeetod, mille asjakohasust ja usaldusväärsust konkreetse eesmärgi puhul käsitatakse piisavana ning mis põhineb teaduslikult tõendatud põhimõtetel. Katsemeetod ei ole kunagi teaduslikult põhjendatud absoluutses tähenduses, vaid ainult seoses määratud eesmärgiga (18).
Valideeritud katsemeetod
: katsemeetod, mille valideerimisuuringud on lõpule viidud, st on määratud selle asjakohasus (sealhulgas täpsus) ja usaldusväärsus konkreetse eesmärgi puhul. Oluline on märkida, et valideeritud katsemeetod ei tarvitse olla piisavalt kasutuskõlblik täpsuse ja usaldusväärsuse osas, et seda saaks lugeda vastuvõetavaks konkreetse eesmärgi puhul (18).
VRM
: valideeritud võrdlusmeetod.
VRM1
: EpiOcular™i silmaärrituse katsele viidatakse kui 1. valideeritud võrdlusmeetodile.
VRM2
: SkinEthic™i inimese sarvkesta epiteeli kasutava silmaärrituse katsele viidatakse kui 2. valideeritud võrdlusmeetodile.
Tõendusmaterjali kaalukuse hindamine
: protsess, milles kaalutakse mitmesuguste andmete tugevaid ja nõrku külgi, et jõuda otsuseni uuritava aine võimaliku ohtlikkuse kohta ja toetada sellist otsust.
2. liide
KATSEMEETODI PEAMISED OSAD – RHCE-KOEMUDELIT KASUTAV KATSEMEETOD, MIS ON VALIDEERITUD NENDE KEMIKAALIDE KINDLAKSTEGEMISEKS, MIDA EI OLE VAJA KLASSIFITSEERIDA EGA MÄRGISTADA SEOSES SILMADE ÄRRITUSE VÕI RASKE SILMAKAHJUSTUSEGA
|
Katse osad
|
EpiOcular™i silmaärrituse katse
(VRM 1)
|
SkinEthic™i inimese sarvkesta epiteeli kasutav silmaärrituse katse
(VRM 2)
|
|
Metoodikad
|
Vedelikud
(pipeteeritavad 37 ± 1 °C juures või madalamal temperatuuril 15 minuti jooksul)
|
Tahked ained
(mittepipeteeritavad)
|
Vedelikud ja viskoossed ained
(pipeteeritavad)
|
Tahked ained
(mittepipeteeritavad)
|
|
Mudeli pindala
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Paralleelsete koeproovide arv
|
Vähemalt 2
|
Vähemalt 2
|
Vähemalt 2
|
Vähemalt 2
|
|
Segava värvuse eelkontroll
|
50 μl + 1 ml H2O 60 minuti jooksul temperatuuril 37 ± 2oC, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 % (värvuseta uuritavad kemikaalid) või 50 μl + 2 ml isopropanooli, segada toatemperatuuril 2–3 tundi (värvilised uuritavad kemikaalid)
|
 kui uuritava kemikaali optiline tihedus lainepikkusel 570 ± 20 nm pärast isopropanooli või vee optilise tiheduse lahutamist on > 0,08 (mis on ligikaudu 5 % negatiivse kontrollproovi keskmisest optilisest tihedusest), tuleks teha kohandatud kontrollkatsed eluskoega.
kui uuritava kemikaali optiline tihedus lainepikkusel 570 ± 20 nm pärast isopropanooli või vee optilise tiheduse lahutamist on > 0,08 (mis on ligikaudu 5 % negatiivse kontrollproovi keskmisest optilisest tihedusest), tuleks teha kohandatud kontrollkatsed eluskoega.
|
|
50 mg + 1 ml H2O 60 minuti jooksul temperatuuril 37 ± 2oC, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
(värvuseta uuritavad kemikaalid) või
50 mg + 2 ml isopropanooli, segada toatemperatuuril 2–3 tundi (värvilised ja värvuseta uuritavad kemikaalid)
|
 kui uuritava kemikaali optiline tihedus lainepikkusel 570 ± 20 nm pärast isopropanooli või vee optilise tiheduse lahutamist on > 0,08 (mis on ligikaudu 5 % negatiivse kontrollproovi keskmisest optilisest tihedusest), tuleks teha kohandatud kontrollkatsed eluskoega.
kui uuritava kemikaali optiline tihedus lainepikkusel 570 ± 20 nm pärast isopropanooli või vee optilise tiheduse lahutamist on > 0,08 (mis on ligikaudu 5 % negatiivse kontrollproovi keskmisest optilisest tihedusest), tuleks teha kohandatud kontrollkatsed eluskoega.
|
|
10 μl + 90 μl H2O, segada 30 ± 2 minutit toatemperatuuril (18–28oC)
|
 kui uuritav kemikaal on värviline, tuleks teha kohandatud kontrollkatsed eluskoega.
kui uuritav kemikaal on värviline, tuleks teha kohandatud kontrollkatsed eluskoega.
|
|
10 mg + 90 μl H2O, segada 30 ± 2 minutit toatemperatuuril
|
 kui uuritav kemikaal on värviline, tuleks teha kohandatud kontrollkatsed eluskoega.
kui uuritav kemikaal on värviline, tuleks teha kohandatud kontrollkatsed eluskoega.
|
|
|
MTT otsese redutseerimise eelkontroll
|
50 μl + 1 ml MTT 1 mg/ml lahust, 180 ± 15 min 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
 kui lahus muutub siniseks/lillaks, tuleks teha kohandatud kontrollkatsed külmutades saadud surnud kudedega
kui lahus muutub siniseks/lillaks, tuleks teha kohandatud kontrollkatsed külmutades saadud surnud kudedega
(negatiivse kontrollainena kasutatakse 50 μl steriilset deioniseeritud vett MTT lahuses)
|
|
50 μg + 1 ml MTT 1 mg/ml lahust, 180 ± 15 min 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
 kui lahus muutub siniseks/lillaks, tuleks teha kohandatud kontrollkatsed külmutades saadud surnud kudedega
kui lahus muutub siniseks/lillaks, tuleks teha kohandatud kontrollkatsed külmutades saadud surnud kudedega
(negatiivse kontrollainena kasutatakse 50 μl steriilset deioniseeritud vett MTT lahuses)
|
|
30 μl + 300 μl MTT 1 mg/ml lahust, 180 ± 15 min 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
 kui lahus muutub siniseks/lillaks, tuleks teha kohandatud kontrollkatsed vees saadud surnud kudedega
kui lahus muutub siniseks/lillaks, tuleks teha kohandatud kontrollkatsed vees saadud surnud kudedega
(negatiivse kontrollainena kasutatakse 30 μl steriilset deioniseeritud vett MTT lahuses)
|
|
30 mg + 300 μl MTT 1 mg/ml lahust, 180 ± 15 min 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
 kui lahus muutub siniseks/lillaks, tuleks teha kohandatud kontrollkatsed vees saadud surnud kudedega
kui lahus muutub siniseks/lillaks, tuleks teha kohandatud kontrollkatsed vees saadud surnud kudedega
(negatiivse kontrollainena kasutatakse 30 μl steriilset deioniseeritud vett MTT lahuses)
|
|
|
Eeltöötlus
|
20 μl Ca2+/Mg2+-vaba DPBS,
30 ± 2 min 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %, valguse eest kaitstud.
|
20 μl Ca2+/Mg2+-vaba DPBS,
30 ± 2 min 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %, valguse eest kaitstud.
|
–
|
–
|
|
Töötlemiskogused ja pealekandmine
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) kalibreeritud vahendiga (nt tasandatud lusikatäis lusikaga, mis on kalibreeritud 50 g naatriumkloriidi mõõtmiseks).
|
10 μl Ca2+/Mg2+-vaba DPBS, + 30 ± 2 μl (60 μl/cm2)
Viskoossete vedelike korral kasutada nailonvõrku
|
30 μl Ca2+/Mg2+-vaba DPBS, + 30 ± 2 mg (60 mg/cm2)
|
|
Kokkupuuteaeg ja -temperatuur
|
30 min (± 2 min)
söötmes
37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
6 tundi (± 0,25 tundi)
söötmes
37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
30 min (± 2 min)
söötmes
37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
4 tundi (± 0,1 tundi)
söötmes
37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
|
Loputamine toatemperatuuril
|
3 korda 100 ml Ca2+/Mg2+-vaba DPBSiga
|
3 korda 100 ml Ca2+/Mg2+-vaba DPBSiga
|
20 ml Ca2+/Mg2+-vaba DPBSiga
|
25 ml Ca2+/Mg2+-vaba DPBSiga
|
|
Kokkupuutejärgne sukeldamine
|
12 min (± 2 min) toetemperatuuril söötmes
|
25 min (± 2 min) toetemperatuuril söötmes
|
30 min (± 2 min) 37oC juures, 5 % CO2, suhteline õhuniiskus 95 %, söötmes
|
30 min (± 2 min) toatemperatuuril söötmes
|
|
Kokkupuutejärgne inkubeerimine
|
120 min (± 15 min) söötmes 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
18 tundi (± 0,25 tundi) söötmes 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
puudub
|
18 tundi (± 0,5 tundi) söötmes 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
|
Negatiivne kontrollproov
|
50 μl H2O
Analüüsitakse paralleelselt
|
50 μl H2O
Analüüsitakse paralleelselt
|
30 ± 2 μl Ca2+/Mg2+-vaba DPBSi
Analüüsitakse paralleelselt
|
30 ± 2 μl Ca2+/Mg2+-vaba DPBSi
Analüüsitakse paralleelselt
|
|
Positiivne kontrollproov
|
50 μl metüülatsetaati
Analüüsitakse paralleelselt
|
50 μl metüülatsetaati
Analüüsitakse paralleelselt
|
30 ± 2μl metüülatsetaati
Analüüsitakse paralleelselt
|
30 ± 2μl metüülatsetaati
Analüüsitakse paralleelselt
|
|
MTT lahus
|
300 μl, 1 mg/ml
|
300 μl, 1 mg/ml
|
300 μl, 1 mg/ml
|
300 μl, 1 mg/ml
|
|
MTT inkubeerimisaeg ja -temperatuur
|
180 min (± 15 min) 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
180 min (± 15 min) 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
180 min (± 15 min) 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
180 min (± 15 min) 37 ± 2oC juures, 5 ± 1 % CO2, suhteline õhuniiskus ≥ 95 %
|
|
Ekstraheerimislahusti
|
2 ml isopropanooli
(ekstraheerimine plaadi pealt ja alt, kude läbi torgates)
|
2 ml isopropanooli
(ekstraheerimine plaadi alt, kude läbi torgates)
|
1,5 ml isopropanooli
(ekstraheerimine plaadi pealt ja alt)
|
1,5 ml isopropanooli
(ekstraheerimine plaadi alt)
|
|
Ekstraheerimisaeg ja -temperatuur
|
2–3 tundi loksutades (~120 pööret minutis) toatemperatuuril või üle öö temperatuuril 4–10 °C
|
2–3 tundi loksutades (~120 pööret minutis) toatemperatuuril või üle öö temperatuuril 4–10 °C
|
4 tundi loksutades (~120 pööret minutis) toatemperatuuril või vähemalt üle öö ilma loksutamata temperatuuril 4–10 °C
|
Vähemalt 2 tundi loksutades (~120 pööret minutis) toatemperatuuril
|
|
Optilise tiheduse mõõtmine
|
570 nm (550–590 nm)
ilma võrdlusfiltrita
|
570 nm (550–590 nm)
ilma võrdlusfiltrita
|
570 nm (540–600 nm)
ilma võrdlusfiltrita
|
570 nm (540–600 nm)
ilma võrdlusfiltrita
|
|
Koe kvaliteedi kontroll
|
Töötlemine 100 μl 0,3-mahuprotsendilise Triton X-100-ga
12,2 min ≤ ET50 ≤ 37,5 min
|
Töötlemine 100 μl 0,3-mahuprotsendilise Triton X-100-ga
12,2 min ≤ ET50 ≤ 37,5 min
|
30-minutine töötlemine naatriumdodetsüülsulfaadiga (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
30-minutine töötlemine naatriumdodetsüülsulfaadiga (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Nõuetekohasuse kriteeriumid
|
|
1.
|
Negatiivse kontrollainega töödeldud paralleelsete koeproovide keskmine optiline tihedus peab olema > 0,8 ja < 2,5
|
|
2.
|
30 minutit positiivse kontrollainega kokku puutunud paralleelsete koeproovide keskmine eluvõimelisus, väljendatuna protsendina negatiivse kontrollainega saadud tulemusest, peab olema < 50 %
|
|
3.
|
Kahe paralleelse koeproovi eluvõimelisuse erinevus ei tohi ületada 20 %
|
|
|
1.
|
Negatiivse kontrollainega töödeldud paralleelsete koeproovide keskmine optiline tihedus peab olema > 0,8 ja < 2,5
|
|
2.
|
6 tundi positiivse kontrollainega kokku puutunud paralleelsete koeproovide keskmine eluvõimelisus, väljendatuna protsendina negatiivse kontrollainega saadud tulemusest, peab olema < 50 %
|
|
3.
|
Kahe paralleelse koeproovi eluvõimelisuse erinevus ei tohi ületada 20 %
|
|
|
1.
|
Negatiivse kontrollainega töödeldud paralleelsete koeproovide keskmine optiline tihedus peab olema > 1,0 ja ≤ 2,5
|
|
2.
|
30 minutit positiivse kontrollainega kokku puutunud paralleelsete koeproovide keskmine eluvõimelisus, väljendatuna protsendina negatiivse kontrollainega saadud tulemusest, peab olema ≤ 30 %
|
|
3.
|
Kahe paralleelse koeproovi eluvõimelisuse erinevus ei tohi ületada 20 %
|
|
|
1.
|
Negatiivse kontrollainega töödeldud paralleelsete koeproovide keskmine optiline tihedus peab olema > 1,0 ja ≤ 2,5
|
|
2.
|
4 tundi positiivse kontrollainega kokku puutunud paralleelsete koeproovide keskmine eluvõimelisus, väljendatuna protsendina negatiivse kontrollainega saadud tulemusest, peab olema ≤ 20 %
|
|
3.
|
Kahe paralleelse koeproovi eluvõimelisuse erinevus ei tohi ületada 20 %
|
|
3. liide
VRM1 STANDARDEESKIRJAL PÕHINEV ILLUSTREERIV VOOSKEEM, MILLEST JUHINDUDA MTT OTSESTE REDITSEERIJATE JA/VÕI SEGAVA VÄRVUSEGA KEMIKAALIDE KINDLAKSTEGEMISEL JA NENDEGA TEGELEMISEL

4. liide
VRM2 STANDARDEESKIRJAL PÕHINEV ILLUSTREERIV VOOSKEEM, MILLEST JUHINDUDA MTT OTSESTE REDITSEERIJATE JA/VÕI SEGAVA VÄRVUSEGA KEMIKAALIDE KINDLAKSTEGEMISEL JA NENDEGA TEGELEMISEL

5. liide
HPLC-/UPLC-SPEKTROMEETRIA APARATUURI PEAMISED KVALIFIKATSIOONINÄITAJAD JA NENDE NÕUETEKOHASUSE KRITEERIUMID SEOSES RHCE-KOEMUDELIST EKSTRAHEERITUD MTT FORMASAANI MÕÕTMISEGA
|
Näitaja
|
USA Toidu- ja Ravimiameti juhendist (36, 38) tuletatud eeskiri
|
Nõuetekohasuse kriteeriumid
|
|
Selektiivsus
|
Isopropanooliga ekstraheeritud eluskoe tühiproovi (töötlemata elusa RhCE-koemudeli ekstrakt isopropanooliga), surnud koe tühiproovi (töötlemata surnud RhCE-koemudeli ekstrakt isopropanooliga) ja värvaine (nt metüleensinine) analüüs
|
Pindalasegav ≤ 20 % pindalastLLOQ
(86)
|
|
Kordustäpsus
|
Kvaliteedi kontrollimise katsed (st MTT formasaan kontsentratsiooniga 1,6 μg/ml, 16 μg/ml ja 160 μg/ml) isopropanooliga (n = 5)
|
CV ≤ 15 % või alumise määramispiiri korral ≤ 20 %
|
|
Täpsus
|
Kvaliteedi kontrollimise katsed isopropanooliga (n = 5)
|
%Dev ≤ 15 % või alumise määramispiiri korral ≤ 20 %
|
|
Maatriksefekt
|
Kvaliteedi kontrollimise katsed elusa koega tühiprooviga (n = 5)
|
85 % ≤ %maatriksefekt ≤ 115 %
|
|
Proovimälu
|
Isopropanooli analüüs pärast ULOQ (87) standardit
|
Pindalasegav ≤ 20 % pindalastLLOQ
|
|
Korratavus (samal päeval)
|
3 sõltumatut kaliibrimisgraafikut (MTT formasaani lahus isopropanoolis, 6 järjestikust lahjendust 1/3 võrra alates ülemisest määramispiirist, st 200 μg/ml)
Kvaliteedi kontrollimise katsed isopropanooliga (n = 5)
|
Kaliibrimiskõverad: %Dev ≤ 15 % või alumise määramispiiri korral ≤ 20 %
Kvaliteedi kontrollimise katsed: %Dev ≤ 15 % ja CV ≤ 15 %
|
|
Korratavus (eri päevadel)
|
1. päev: 1 kaliibrimiskõver ja kvaliteedi kontrollimise katsed isopropanooliga (n=3)
2. päev: 1 kaliibrimiskõver ja kvaliteedi kontrollimise katsed isopropanooliga (n=3)
3. päev: 1 kaliibrimiskõver ja kvaliteedi kontrollimise katsed isopropanooliga (n=3)
|
|
MTT formasaani lühiajaline stabiilsus RhCE-koe ekstraktis
|
Kvaliteedi kontrollimise katsed elusa koega tühiproovidega (n = 3), analüüsitud valmistamise päeval ja pärast 24 tundi toatemperatuuril seismist
|
%Dev ≤ 15 %
|
|
MTT formasaani pikaajaline stabiilsus RhCE-koe ekstraktis, kui see on nõutav
|
Kvaliteedi kontrollimise katsed elusa koega tühiproovidega (n = 3), analüüsitud valmistamise päeval ja pärast mitu päeva –20 °C juures seismist
|
%Dev ≤ 15 %
|
B.70 REKOMBINANTSEL INIMESE ÖSTROGEENIRETSEPTORIL (hrER) PÕHINEVAD IN VITRO ANALÜÜSIMEETODID ÖSTROGEENIRETSEPTORIGA SEONDUVATE KEMIKAALIDE TUVASTAMISEKS
ÜLDINE SISSEJUHATUS
Tulemusnäitajatel põhinev Majanduskoostöö ja Arengu Organisatsiooni (OECD) katsejuhend
|
1.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 493 (2015). Katsejuhend nr 493 on tulemusnäitajatel põhinev katsejuhend, milles kirjeldatakse in vitro analüüsimeetodeid inimese rekombinantse östrogeeniretseptoriga (ER) seonduvate ainete tuvastamiseks (hrERiga seondumise katsed). See hõlmab kahte vaadeldava mehhanismi ja funktsionaalsuse poolest sarnast analüüsimeetodit östrogeeniretseptoriga (st α-ERiga) seonduvate kemikaalide tuvastamiseks ja peaks hõlbustama uute sarnaste või modifitseeritud analüüsimeetodite väljatöötamist vastavalt valideerimispõhimõtetele, mis on esitatud ohuhindamiseks kasutatavate uute või ajakohastatud katsemeetodite valideerimist ja rahvusvahelist tunnustamist käsitlevas OECD juhenddokumendis (1). Kõnealuse tulemusnäitajatel põhineva katsejuhendi aluseks on järgmised täielikult valideeritud võrdlusmeetodid (2. ja 3. liide):
|
—
|
täispikal rekombinantsel inimese α-ERil põhinev Freybergeri-Wilsoni (FW) in vitro meetod östrogeeniretseptoriga seondumise analüüsimiseks (2) ja
|
|
—
|
ligandiga seonduval rekombinantsel inimese valgudomeenil põhinev Jaapani Kemikaalide Hindamise ja Uurimise Instituudi (CERI) in vitro meetod östrogeeniretseptoriga seondumise analüüsimiseks (2).
|
On kehtestatud tulemuslikkuse standardid (3), mis hõlbustavad sama ohunäitaja hindamiseks kasutatavate sarnaste katsemeetodite väljatöötamist ja valideerimist ning võimaldavad tulemusnäitajatel põhinevat katsejuhendit nr 493 õigeaegselt muuta, et täiendada seda ajakohastamise eesmärgil uute sarnaste analüüsimeetoditega. Siiski lisatakse sarnased katsemeetodid juhendisse üksnes pärast seda, kui OECD on need läbi vaadanud ja kinnitanud nende vastavust tulemuslikkuse standarditele. Östrogeeniretseptoriga seondumise katsete tulemusi käsitlevatele OECD liikmesriikide nõuetele vastavuse saavutamiseks võib kasutada ükskõik millist katsejuhendis nr 493 esitatud analüüsimeetodit ning seejuures saadakse kasu OECD otsuse kohasest andmete vastastikusest tunnustamisest.
|
Käesoleva katsemeetodiga hõlmatud analüüsimeetodite taust ja põhimõtted
|
2.
|
OECD algatas 1998. aastal prioriteetse tegevussuunana olemasolevate katsejuhendite läbivaatamise ja uute väljatöötamise, et võimaldada sõeluuringute tegemist võimalike endokriinfunktsiooni kahjustavate kemikaalide tuvastamiseks ja selliste kemikaalide hindamist. OECD kontseptuaalset raamistikku võimalike endokriinfunktsiooni kahjustavate kemikaalide analüüsimiseks ja hindamiseks muudeti 2012. aastal. Nii algne kui ka muudetud kontseptuaalne raamistik on esitatud kemikaalide võimaliku endokriinfunktsiooni kahjustava toime hindamise standardiseeritud katsejuhendeid käsitleva juhenddokumendi lisadena (4). Kõnealune kontseptuaalne raamistik hõlmab viit taset; iga tase vastab bioloogilise keerukuse eri tasemele. Käesolevas katsemeetodis kirjeldatud ERiga seondumise katsed kuuluvad 2. tasemele, mis hõlmab „in vitro katseid valitud endokriinse(te) mehhanismi(de) või raja/radade kohta andmete saamiseks“. Käesolev katsemeetod on ette nähtud retseptoriga seondumise in vitro katsete tegemiseks eesmärgiga tuvastada inimese östrogeeniretseptori α-vormi (α-ER) ligandid.
|
|
3.
|
ERiga tehtavate in vitro seondumiskatsete asjakohasus bioloogiliste funktsioonide hindamisel on selgelt tõendatud. ERiga tehtavad seondumiskatsed on ette nähtud selliste kemikaalide tuvastamiseks, millel võib olla kahjulik mõju östrogeenirajale, ning neid on viimase kahe kümnendi jooksul laialdaselt kasutatud, et iseloomustada ERi jaotust kudedes ja identifitseerida ERi agoniste/antagoniste. Need katsed põhinevad ligandi ja retseptori vahelisel interaktsioonil, mis on esimene samm östrogeeni signaalirajas ja on hädavajalik kõigi selgroogsete paljunemisvõime tagamiseks.
|
|
4.
|
Östrogeenide interaktsioon ERidega võib mõjutada östrogeeni kontrollitavate geenide transkriptsiooni ja avaldada mittegenoomset mõju, mille tulemusena võidakse käivitada või pärssida muu hulgas selliseid rakuprotsesse, mis on vajalikud rakkude paljunemiseks, loote normaalseks arenguks ja paljunemisvõime säilitamiseks (5, 6, 7). Normaalsete östrogeensete süsteemide häiretel võib olla kahjulik mõju normaalsele arengule (ontogenees), reproduktiivtervisele ja reproduktiivsüsteemi terviklikkusele. Ebasobiv ERist lähtuv signaaliülekanne võib näiteks suurendada hormoonsõltuva vähi tekke riski, vähendada viljakust ja põhjustada muutusi loote kasvus ja arengus (8).
|
|
5.
|
In vitro seondumiskatsed põhinevad aine otsesel interaktsioonil retseptori spetsiifilise ligandiseondumiskohaga, mille kaudu reguleeritakse geenitranskriptsiooni. Inimese rekombinantse α-östrogeeniretseptoriga (hr-α-ER) seondumise katses mõõdetakse põhinäitajana radiomärgistatud ligandi ([3H]17β-östradiool) võimet seonduda ERiga üha suuremas kontsentratsioonis esineva uuritava kemikaali (st konkureeriva ligandi) juuresolekul. Uuritavad kemikaalid, millel on ERi suhtes suur afiinsus, konkureerivad radiomärgistatud ligandiga väiksemal kontsentratsioonil kui kemikaalid, mille afiinsus retseptori suhtes on väiksem. Kõnealune meetod hõlmab kahte põhielementi: küllastava seondumise katset, millega iseloomustatakse retseptori ja ligandi vahelise interaktsiooni parameetreid ja dokumenteeritakse spetsiifilisus ERi suhtes, ning sellele järgnevat konkureeriva seondumise katset, millega iseloomustatakse uuritava kemikaali ja radiomärgistatud ligandi vahelist konkurentsi ERile seondumisel.
|
|
6.
|
CERI ja FW seondumiskatsete asjakohasus ja usaldusväärsus sihtotstarbelisel kasutamisel on valideerimisuuringutega tõendatud (2).
|
|
7.
|
Käesolevas katsemeetodis kasutatud mõisted ja lühendid on esitatud 1. liites.
|
Retseptoriga seondumise katsete rakendusala ja piirangud
|
8.
|
Kõnealused katsed on ette nähtud sõeluuringu tegemiseks ja prioriseerimiseks, kuid nende käigus võib ühtlasi saada teavet molekulaarse lähtesündmuse kohta, mida saab kasutada tõendite kaalukusel põhineva lähenemisviisi puhul. Katsetes vaadeldakse kemikaali seondumist α-ERi ligandi siduvale domeenile in vitro süsteemis. Seega tuleks hoiduda saadud tulemuste otsesest ekstrapoleerimisest tervikliku endokriinsüsteemi keerukale signaaliülekande- ja reguleerimismehhanismile in vivo.
|
|
9.
|
Loodusliku ligandi 17β-östradiooli seondumine on esimene samm selliste molekulaarsete sündmuste jadas, mille abil aktiveeritakse sihtgeenide transkriptsioon ja mille lõpptulemuseks on füsioloogiline muutus (9). Seega on seondumine α-ERi ligandi siduvale domeenile üks peamisi ERi vahendatavate endokriinsüsteemi häirete tekkemehhanisme, kuigi kõnealuseid häireid võivad põhjustada ka muud mehhanismid, sealhulgas i) interaktsioon α-ERi teiste piirkondadega peale ligandisidumistasku, ii) interaktsioon östrogeeni signaalirajaga seotud muude retseptoritega, α-ERiga ja G-valguga seotud östrogeeniretseptoriga ning teiste endokriinsüsteemi retseptorite ja ensüümisüsteemidega, iii) hormoonide süntees, iv) hormoonide metaboolne aktiveerimine ja/või inaktiveerimine, v) hormoonide jaotumine sihtkudedes ja vi) hormoonide väljutamine kehast. Selliseid toimemehhanisme ei vaadelda üheski käesoleva katsemeetodi kohases katses.
|
|
10.
|
Käesoleva katsemeetodi puhul uuritakse ainete võimet seonduda inimese α-ERiga ja see ei võimalda eristada α-ERi agoniste α-ERi antagonistidest. Kõnealustes katsetes ei vaadelda ka seondumisele järgnevaid sündmusi, näiteks geenitranskriptsiooni ega füsioloogilisi muutusi. Kuna valideerimisel kasutati ühekaupa aineid, mis koosnevad ainult ühest koostisosast, ei ole uuritud selle meetodi kohaldatavust segude analüüsimiseks. Teoreetiliselt on kõnealustes katsetes siiski võimalik analüüsida ka mitut koostisosa sisaldavaid aineid ja segusid. Kui soovitakse saada kavandatud regulatiivsel eesmärgil andmeid segu kohta, tuleks enne katsemeetodi kasutamist kaaluda, kas ja kuidas see võimaldab saada kõnealusele eesmärgile vastavaid asjakohaseid tulemusi. Selline kaalumine ei ole vajalik, kui asjaomase segu hindamine on regulatiivse nõudega ette nähtud.
|
|
11.
|
Rakuvabadel retseptorisüsteemidel puudub metaboliseerimisvõime ja neid ei ole valideeritud kombinatsioonis metaboolsete ensüümisüsteemidega. Siiski võib olla võimalik lisada uuringumudelisse metaboliseerimisvõimet tagavad elemendid, kuid see nõuab täiendavat valideerimist.
|
|
12.
|
Kemikaale, mis võivad valku (st retseptorvalku) denatureerida – näiteks pindaktiivseid aineid ja analüüsipuhvri pH-d muuta võivaid kemikaale –, ei tohi analüüsida või võib analüüsida üksnes kontsentratsioonidel, mille juures selline mõju puudub. Muudel juhtudel võib uuritava kemikaali analüüsimiseks kasutada kontsentratsioonivahemikku, mille puhul kemikaal on analüüsipuhvris lahustuv.
|
|
13.
|
Tabelis 1 on teavitamise eesmärgil esitatud katsetulemused 24 aine kohta, mida on analüüsitud mõlema käesolevas katsemeetodis kirjeldatud täielikult valideeritud analüüsimeetodiga. Avaldatud aruannete kohaselt, mis muu hulgas hõlmavad in vitro katseid ERi-seoselise transkriptsiooni aktiveerimise tuvastamiseks ja uterotroofse mõju analüüsi, liigitati nendest ainetest 17 ERiga seonduvateks aineteks ja kuus mitteseonduvateks aineteks (9, 10, 11, 12, 13, 14, 15). Vastavalt tabelis 1 esitatud kokkuvõtlikele andmetele on kahe analüüsimeetodiga saadud liigitamistulemused kõigi ainete puhul kuni kontsentratsioonini 10-4 M peaaegu täielikult kattuvad ja iga aine on õigesti liigitatud ERiga seonduvaks või ERiga mitteseonduvaks. Lisateave selle ainete rühma ja valideerimisuuringute raames tehtud ERiga seondumise katsetes analüüsitud täiendavate ainete kohta on esitatud hrERiga seondumise katsete tulemuslikkuse standardite (3) 2. lisas (tabelid 1, 2 ja 3).
Tabel 1
Ainete liigitamine ERiga seonduvaks või mitteseonduvaks nende analüüsimisel FW ja CERI meetodite kohastes hrERiga seondumise katsetes ning võrdlus eeldatava tulemusega
|
|
Aine nimetus
|
CASi nr
|
Eeldatav tulemus
|
FW meetod
|
CERI meetod
|
MeSHi kemikaaliklass
|
Tootekategooria
|
|
|
Kontsentratsioonivahemik (M)
|
Liigitus
|
Kontsentratsioonivahemik (M)
|
Liigitus
|
|
1
|
17β-östradiool
|
50-28-2
|
Seonduv
|
1 × 10-11 – 1 × 10-6
|
Seonduv
|
1 × 10-11 – 1 × 10-6
|
Seonduv
|
Steroid
|
Ravim, veterinaarravim
|
|
2
|
Noretünodreel
|
68-23-5
|
Seonduv
|
3 × 10-9 – 30 × 10-4
|
Seonduv
|
3 × 10-9 – 30 × 10-4
|
Seonduv
|
Steroid
|
Ravim, veterinaarravim
|
|
3
|
Noretindroon
|
68-22-4
|
Seonduv
|
3 × 10-9 – 30 × 10-4
|
Seonduv
|
3 × 10-9 – 30 × 10-4
|
Seonduv
|
Steroid
|
Ravim, veterinaarravim
|
|
4
|
Di-n-butüülftalaat
|
84-74-2
|
Mitteseonduv (*5)
|
1 × 10-10 – 1 × 10-4
|
Mitteseonduv (*6)
(1)
|
1 × 10-10 – 1 × 10-4
|
Mitteseonduv (*6)
(1)
|
Süsivesinik (tsükliline), ester
|
Plastifikaator, keemiline vaheühend
|
|
5
|
DES
|
56-53-1
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Süsivesinik (tsükliline), fenool
|
Ravim, veterinaarravim
|
|
6
|
17α-etünüülöstradiool
|
57-63-6
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Steroid
|
Ravim, veterinaarravim
|
|
7
|
Mesoheksöstrool
|
84-16-2
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Süsivesinik (tsükliline), fenool
|
Ravim, veterinaarravim
|
|
8
|
Genisteiin
|
446-72-0
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Süsivesinik (heterotsükliline), flavonoid
|
Looduslik ühend
|
|
9
|
Ekvool
|
531-95-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Fütoöstrogeeni metaboliit
|
Looduslik ühend
|
|
10
|
Butüülparabeen (n-butüül-4-hüdroksübensoaat)
|
94-26-8
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Parabeen
|
Säilitusaine
|
|
11
|
Nonüülfenool (segu)
|
84852-15-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Alküülfenool
|
Vaheühend
|
|
12
|
o,p’-DDT
|
789-02-6
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Kloororgaaniline ühend
|
Insektitsiid
|
|
13
|
Kortikosteroon
|
50-22-6
|
Mitteseonduv (*5)
|
1 × 10-10 – 1 × 10-4
|
Mitteseonduv
|
1 × 10-10 – 1 × 10-4
|
Mitteseonduv
|
Steroid
|
Looduslik ühend
|
|
14
|
Zearalenoon
|
17924-92-4
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Süsivesinik (heterotsükliline), laktoon
|
Looduslik ühend
|
|
15
|
Tamoksifeen
|
10540-29-1
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Süsivesinik (tsükliline)
|
Ravim, veterinaarravim
|
|
16
|
5α-dihüdrotestosteroon
|
521-18-6
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Steroid, mittefenoolne
|
Looduslik ühend
|
|
17
|
Bisfenool A
|
80-05-7
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Fenool
|
Keemiline vaheühend
|
|
18
|
4-n-heptüülfenool
|
1987-50-4
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Ebaselge (7)
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Alküülfenool
|
Vaheühend
|
|
19
|
Kepoon (kloordekoon)
|
143-50-0
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Süsivesinik (halogeenitud)
|
Pestitsiid
|
|
20
|
Bens[a]antratseen
|
56-55-3
|
Mitteseonduv
|
1 × 10-10 – 1 × 10-3
|
Mitteseonduv (8)
|
1 × 10-10 – 1 × 10-3
|
Mitteseonduv (8)
|
Aromaatne süsivesinik
|
Vaheühend
|
|
21
|
Enterolaktoon
|
78473-71-9
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Seonduv
|
Fütoöstrogeen
|
Looduslik ühend
|
|
22
|
Progesteroon
|
57-83-0
|
Mitteseonduv (*5)
|
1 × 10-10 – 1 × 10-4
|
Mitteseonduv
|
1 × 10-10 – 1 × 10-4
|
Mitteseonduv
|
Steroid
|
Looduslik ühend
|
|
23
|
Oktüültrietoksüsilaan
|
2943-75-1
|
Mitteseonduv
|
1 × 10-10 – 1 × 10-3
|
Mitteseonduv
|
1 × 10-10 – 1 × 10-3
|
Mitteseonduv
|
Silaan
|
Pinnamodifikaator
|
|
24
|
Atrasiin
|
1912-24-9
|
Mitteseonduv (*5)
|
1 × 10-10 – 1 × 10-4
|
Mitteseonduv
|
1 × 10-10 – 1 × 10-4
|
Mitteseonduv
|
Heterotsükliline ühend
|
Herbitsiid
|
|
hrER-GA SEONDUMISE KATSE ELEMENDID
Katse põhielemendid
|
14.
|
Käesolevat katsemeetodit kohaldatakse katsete puhul, kus kasutatakse östrogeeniretseptorit ja sellega tugevalt seonduvat sobivat ligandi, mida on võimalik kasutada markeri/märgistusainena ja mida saab uuritava kemikaali kontsentratsiooni suurendamise teel välja tõrjuda. Seondumiskatsed hõlmavad kahte järgmist põhielementi: 1) küllastav seondumine ja 2) konkureeriv seondumine. Küllastava seondumise katset kasutatakse retseptoripreparaadi spetsiifilisuse ja aktiivsuse kontrollimiseks, konkureeriva seondumise katset aga selleks, et hinnata uuritava kemikaali võimet seonduda hrERiga.
|
Kontrollid
|
15.
|
Kavandatud paralleelsete kontrollide ja võrdlusöstrogeeni valikut tuleks põhjendada. Paralleelseid kontrolle (lahusti (kandeaine), positiivsed kontrollid (ERiga tugevalt seonduv aine ja nõrgalt seonduv aine), negatiivne kontroll (mitteseonduv aine)) kasutatakse vastavalt vajadusele meetodi töökindluse kontrollimiseks asjaomastes katsetingimustes ja nende alusel saab katseid omavahel võrrelda; need kuuluvad tavaliselt katse nõuetekohasuse kriteeriumide hulka (1). Igas analüüsitsüklis tuleks ühel plaadil kasutada võrdlusöstrogeeni ja kontrolle (st nõrgalt seonduv aine ja mitteseonduv aine) täieliku kontsentratsioonikõvera ulatuses. Kõik ülejäänud plaadid peaksid sisaldama: 1) nii E2 kui ka nõrgalt seonduvat ainet suures (umbes radiomärgistatud ligandi täielikuks väljatõrjumiseks vajalikus) ja keskmises (umbes IC50-le vastavas) kontsentratsioonis kolmes paralleelis; 2) lahustiga kontrolli ja mittespetsiifilise seondumise kontrolli, kumbagi kolmes paralleelis.
|
Standardne kvaliteedikontrolli kord
|
16.
|
Standardset kvaliteedikontrolli korda tuleks kohaldada vastavalt iga analüüsimeetodi kohta esitatud kirjeldusele, et tagada retseptorite aktiivsus, õigete kemikaalikontsentratsioonide kasutamine, hälbepiirides püsimine mitme korduskatse vältel ja ERiga seondumist käsitlevate ootuspäraste tulemuste saamise võimekus pikema aja vältel.
|
Labori pädevuse tõendamine
|
17.
|
Enne tundmatute kemikaalide analüüsimist käesoleva katsemeetodi kohases katses tuleks igas laboris tõendada asjaomase meetodi kasutamise pädevust; selleks viiakse läbi küllastava seondumise katsed, et kontrollida ERi preparaadi spetsiifilisust ja aktiivsust, ning konkureeriva seondumise katsed võrdlusöstrogeeni ja kontrollidega (nõrgalt seonduv aine ja mitteseonduv aine). Laboris tuleks luua varasemate tulemuste andmebaas, mis sisaldab võrdlusöstrogeeni ja kontrollidega saadud tulemusi eri päevadel läbi viidud 3–5 sõltumatust katsest. Nende katsetega luuakse laboris varasemate võrdlusöstrogeeni ja kontrolle hõlmavate tulemuste näol võrdlusalus, mida kasutatakse järgnevate katsete nõuetekohasuse hindamise osana.
|
|
18.
|
Katsesüsteemi reageerimisvõime kontrollimiseks analüüsitakse ka tabelis 2 loetletud pädevusaineid. Sellesse loetellu kantud pädevusained on valitud võrdlusainete hulgast, mida on kirjeldatud ERiga seondumise katsete tulemuslikkuse standardites (3). Kõnealused ained on turul kättesaadavad, kuuluvad kemikaaliklassidesse, mida tavaliselt seostatakse ERiga seondumise võimega, nende afiinsus ERi suhtes jääb sobivasse vahemikku (st tugevast kuni nõrgani) ja nende hulgas on ka mitteseonduvaid aineid (st negatiivsed kontrollid). Iga pädevusaine puhul kasutatavad kontsentratsioonid peaksid hõlmama tabelis 2 esitatud vahemikku. Iga ainega tuleks läbi viia vähemalt kolm katset ning nende katsete tulemused peaksid vastama aine eeldatavale aktiivsusele. Iga katse tuleks läbi viia sõltumatult (st kasutada retseptori, kemikaalide ja reaktiivi värskelt valmistatud lahjendusi); seejuures tuleks iga kontsentratsiooni puhul kasutada kolme paralleelproovi. Pädevuse tõendamiseks tuleb iga pädevusaine liigitada õigesti positiivseks või negatiivseks. Pädevuse tõendamise katsed peaks läbi viima iga asjaomaseid analüüsimeetodeid omandav laborant.
Tabel 2
hrERiga tehtavas konkureeriva seondumise katses kasutatavate kontrollide ja pädevusainete loetelu
(88)
|
Nr
|
Aine nimetus
|
CASi nr (89)
|
Eeldatav tulemus (90)
(91)
|
Uuritav kontsentratsioonivahemik (M)
|
MeSHi kemikaaliklass (92)
|
Tootekategooria (93)
|
|
Kontrollid (võrdlusöstrogeen, nõrgalt seonduv aine, mitteseonduv aine)
|
|
1
|
17β-östradiool
|
50-28-2
|
Seonduv
|
1 × 10-11 – 1 × 10-6
|
Steroid
|
Ravim, veterinaarravim
|
|
2
|
Noretünodreel (või) Noretindroon
|
68-23-5 (või) 68-22-4
|
Seonduv
|
3 × 10-9 – 30 × 10-6
|
Steroid
|
Ravim, veterinaarravim
|
|
3
|
Oktüültrietoksüsilaan
|
2943-75-1
|
Mitteseonduv
|
1 × 10-10 – 1 × 10-3
|
Silaan
|
Pinnamodifikaator
|
|
Pädevusained (93)
|
|
4
|
Dietüülstilböstrool
|
56-53-1
|
Seonduv
|
1 × 10-11 – 1 × 10-6
|
Süsivesinik (tsükliline), fenool
|
Ravim, veterinaarravim
|
|
5
|
17α-etünüülöstradiool
|
57-63-6
|
Seonduv
|
1 × 10-11 – 1 × 10-6
|
Steroid
|
Ravim, veterinaarravim
|
|
6
|
Mesoheksöstrool
|
84-16-2
|
Seonduv
|
1 × 10-11 – 1 × 10-6
|
Süsivesinik (tsükliline), fenool
|
Ravim, veterinaarravim
|
|
7
|
Tamoksifeen
|
10540-29-1
|
Seonduv
|
1 × 10-11 – 1 × 10-6
|
Süsivesinik (tsükliline)
|
Ravim, veterinaarravim
|
|
8
|
Genisteiin
|
446-72-0
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Heterotsükliline ühend, flavonoid
|
Looduslik ühend
|
|
9
|
Bisfenool A
|
80-05-7
|
Seonduv
|
1 × 10-10 – 1 × 10-3
|
Fenool
|
Keemiline vaheühend
|
|
10
|
Zearalenoon
|
17924-92-4
|
Seonduv
|
1 × 10-11 – 1 × 10-3
|
Heterotsükliline ühend, laktoon
|
Looduslik ühend
|
|
11
|
Butüülparabeen
|
94-26-8
|
Seonduv
|
1 × 10-11 – 1 × 10-3
|
Karboksüülhape, fenool
|
Säilitusaine
|
|
12
|
Atrasiin
|
1912-24-9
|
Mitteseonduv
|
1 × 10-11 – 1 × 10-6
|
Heterotsükliline ühend
|
Herbitsiid
|
|
13
|
Di-n-butüülftalaat (DBP) (94)
|
84-74-2
|
Mitteseonduv (95)
|
1 × 10-10 – 1 × 10-4
|
Süsivesinik (tsükliline), ester
|
Plastifikaator, keemiline vaheühend
|
|
14
|
Kortikosteroon
|
50-22-6
|
Mitteseonduv
|
1 × 10-11 – 1 × 10-4
|
Steroid
|
Looduslik ühend
|
|
Uuritava kemikaali lahustuvuse analüüsimine ja kontsentratsioonivahemiku kindlakstegemine
|
19.
|
Iga uuritava kemikaali puhul tuleks teha eelkatse, et määrata lahustuvuspiir ja teha kindlaks katse läbiviimiseks sobiv kontsentratsioonivahemik. Iga uuritava kemikaali lahustuvuspiir tuleb algselt määrata lahustis ja seejärel tuleb kontrollida seda katsetingimustes. Katses kasutatav lõppkontsentratsioon ei tohiks olla suurem kui 1 mM. Kontsentratsioonivahemiku kindlakstegemise katses kasutatakse lahustiga kontrolli koos kaheksa järjestikuse üksteisest log-ühiku võrra erineva lahjendusega, mille puhul alustatakse maksimaalsest vastuvõetavast kontsentratsioonist (nt 1 mM või väiksem, olenevalt lahustuvuspiirist), ja registreeritakse hägu või sademe tekkimine. Teises ja kolmandas katses tuleks kontsentratsioone vajaduse korral kohandada, et kontsentratsioonist sõltuvuse kõverat oleks võimalik paremini iseloomustada.
|
Katse nõuetekohasuse kriteeriumid
|
20.
|
Katse tunnistamine nõuetekohaseks või nõuetele mittevastavaks põhineb igas analüüsitsüklis kasutatud võrdlusöstrogeeni ja kontrollidega saadud tulemuste hindamisel. Esiteks peaks 1. plaadil olevate võrdluskontrollide täielikud kontsentratsioonikõverad igas analüüsitsüklis vastama lähenduskõvera parameetritega (nt IC50 ja Hilli tõus) seotud tulemusnäitajatele, mis põhinevad vastavates CERI ja FW meetodite kohastes katse-eeskirjades käsitletud tulemustel (2. ja 3. liide) ja katset läbi viivas laboris kontrollidega varem saadud andmetel. Kõik kontrollid (võrdlusöstrogeen, nõrgalt seonduv aine ja mitteseonduv aine) peaksid igas analüüsitsüklis olema õigesti liigitatud. Teiseks tuleb hinnata kõigil ülejäänud plaatidel olevate kontrollide andmete kooskõla 1. plaadi puhul saadud tulemustega. Tuleks kasutada piisavalt suurt uuritava kemikaali kontsentratsioonide vahemikku, et konkureeriva seondumise kõvera ülemine osa selgelt välja joonistuks. Paralleelproovide vaheline varieeruvus igal uuritava kemikaali kontsentratsioonil ja kolme sõltumatu analüüsitsükli vaheline varieeruvus peaks olema mõõdukas ja teaduslikult põhjendatav. Katse järjepideva läbiviimise võimekuse tõendamiseks tuleks luua võrdlusöstrogeeni ja kontrolle iseloomustavate varasemate tulemuste andmebaas ja seda hallata. Laborisisese reprodutseeritavuse hindamiseks võib kasutada mitmest eri katsest pärinevate võrdlusöstrogeenile ja nõrgalt seonduvale kontrollainele vastavate lähenduskõverate parameetrite keskväärtuste standardhälbeid (SD) ja variatsioonikordajaid (CV). Plaatidel olevate kontrollidega saadud tulemuste hindamine igas analüüsitsüklis ja iga uuritava kemikaali puhul peaks põhinema eksperdihinnangul.
Lisaks tuleks juhinduda järgmistest katse nõuetekohasuse kriteeriumidega seotud põhimõtetest:
|
—
|
andmed peaksid olema piisavad ERiga seondumise kvantitatiivseks hindamiseks;
|
|
—
|
kasutatud kontsentratsioonid peaksid jääma uuritava kemikaali lahustuvusvahemikku.
|
|
Andmeanalüüs
|
21.
|
Küllastava ja konkureeriva seondumise andmete analüüsimise kindlaksmääratud korra puhul tuleks järgida retseptor ja ligandi vaheliste interaktsioonide iseloomustamise aluspõhimõtteid. Tavaliselt kasutatakse küllastava seondumise andmete analüüsimiseks mittelineaarse regressiooni mudelit, milles võetakse arvesse summaarset ja mittespetsiifilist seondumist. Bmax-i ja Kd määramisel võib olla vaja korrigeerida andmeid ligandi väljatõrjumise suhtes (nt Swillens, 1995 (19)). Konkureeriva seondumise katsete puhul kasutatakse tavaliselt transformeeritud andmeid (nt spetsiifiline seondumine protsentides ja uuritava kemikaali kontsentratsioon (log M)). Iga uuritava kemikaali puhul tuleks IC50 hinnangulise logaritmitud väärtuse kindlakstegemiseks kasutada sobivat mittelineaarse kõvera lähendamise tarkvara, milles rakendatakse neljaparameetrilist Hilli võrrandit. Pärast esmast analüüsi tuleks määrata lähenduskõvera parameetrid ja visuaalselt hinnata, kui hästi sobituvad seondumisandmed saadud konkureeriva seondumise kõveraga. Mõnel juhul võib olla vaja viia läbi täiendav analüüs, et leida parim lähenduskõver (nt kõvera ülemise ja/või alumise osa piiritlemine, 10 % reegli kasutamine; vt 4. liide ja allikas 2 (jaotis III.A.2)).
|
|
22.
|
Katsete vastavus nõuetekohasuse kriteeriumidele (punkt 20) viitab sellele, et katsesüsteem töötab nõuetekohaselt, kuid sellega ei ole tagatud, et igas konkreetses katses saadavad andmed on täpsed. Esimese katse nõuetekohaste tulemuste korduvus on parim viide sellele, et saadud andmed on täpsed.
|
Üldised andmete tõlgendamise kriteeriumid
|
23.
|
Praegu puudub ühtne kokkulepitud meetod ERiga seondumise andmete tõlgendamiseks. Siiski tuleks hrERi vahendatava mõju kvalitatiivsel (nt seonduv/mitteseonduv) ja/või kvantitatiivsel (nt log IC50, suhteline seondumisafiinsus (RBA) jne) analüüsimisel lähtuda empiirilistest andmetest ja usaldusväärsest teaduslikust hinnangust.
|
Katseprotokoll
|
24.
|
Katseprotokollis tuleks esitada järgmine teave.
|
|
Analüüsimeetod:
|
—
|
kasutatud analüüsimeetod.
|
|
|
|
Kontrollkemikaal / võrdluskemikaal / uuritav kemikaal:
|
—
|
allikas, partii number, aegumiskuupäev, kui on teada;
|
|
—
|
uuritava kemikaali püsivus, kui on teada;
|
|
—
|
uuritava kemikaali lahustuvus ja püsivus lahustis, kui on teada;
|
|
—
|
lisatud uuritava kemikaaliga söötme pH ja osmolaalsus ning sademe teke selles (vastavalt vajadusele);
|
|
|
|
ühest koostisosast koosnev aine:
|
—
|
füüsiline välimus, lahustuvus vees ja muud asjakohased füüsikalis-keemilised omadused;
|
|
—
|
kemikaali identifitseerimisandmed, näiteks IUPACi või CASi nimetus, CASi number, SMILESi või InChI kood, struktuurivalem, puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
|
|
mitut koostisosa sisaldav aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal (UVCB) või segu:
|
—
|
võimalikult täpne omaduste kirjeldus lähtuvalt koostisosade keemilisest määratlusest (vt eespool), kvantitatiivsest sisaldusest ja asjakohastest füüsikalis-keemilistest omadustest.
|
|
|
|
Lahusti/kandeaine:
|
—
|
kirjeldus (aine liik, tarnija ja partii);
|
|
—
|
lahusti/kandeaine valiku põhjendus;
|
|
—
|
uuritava kemikaali lahustuvus ja püsivus lahustis/kandeaines, kui see on teada.
|
|
|
|
Retseptorid:
|
—
|
retseptorite päritolu (tarnija, katalooginumber, partii, retseptori liik, retseptori aktiivne kontsentratsioon tarnija andmetel, sertifikaat tarnijalt)
|
|
—
|
retseptorite kirjeldus (sh küllastava seondumise tulemused): Kd, Bmax;
|
|
—
|
retseptorite säilitamine.
|
|
—
|
Radiomärgistatud ligand:
|
|
—
|
tarnija, katalooginumber, partii, eriaktiivsus.
|
|
|
|
Katsetingimused:
|
—
|
lahustuvuspiir katsetingimustes;
|
|
—
|
seondumispuhvri koostis;
|
|
—
|
retseptori kontsentratsioon;
|
|
—
|
märgistusaine (st radiomärgistatud ligandi) kontsentratsioon;
|
|
—
|
uuritava kemikaali kontsentratsioonid;
|
|
—
|
kandeaine protsentuaalne sisaldus lõppkatses;
|
|
—
|
inkubeerimise temperatuur ja kestus;
|
|
—
|
meetod vaba aine eraldamiseks seondunud ainest;
|
|
—
|
positiivsed ja negatiivsed kontrollid/võrdlusained;
|
|
—
|
kriteeriumid, mille alusel katsetulemus liigitatakse positiivseks, negatiivseks või ebaselgeks.
|
|
|
|
Nõuetekohasuse kontroll:
|
—
|
paralleelsete positiivsete kontrollidega/võrdlusainetega saadud tegelikud IC50 ja Hilli tõusu väärtused.
|
|
|
|
Tulemused:
|
—
|
algandmed ja andmed seondunud/vaba aine kohta;
|
|
—
|
vajaduse korral denatureerumise kontrollimise tulemused;
|
|
—
|
väikseim efektiivne kontsentratsioon (LEC), kui see on olemas;
|
|
—
|
vastavalt vajadusele RBA ja/või IC50 väärtused;
|
|
—
|
võimaluse korral mõju sõltuvus kontsentratsioonist;
|
|
—
|
statistilise analüüsi tulemused, kui need on olemas, koos veamäära ja usaldusnivooga (nt keskväärtuse standardviga (SEM), SD, CV või usaldusvahemik usaldusnivool 95 %) ning kirjeldus selle kohta, kuidas need väärtused saadi.
|
|
|
|
Tulemuste arutelu:
|
—
|
10 % reegli rakendamine.
|
|
Järeldused
|
KIRJANDUS
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment No. 34. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα). Health and Safety Publications, Series on Testing and Assessment No. 226. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα). Health and Safety Publications, Series on Testing and Assessment No. 222. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(4)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment No. 150. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(5)
|
Cavailles, V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric 5 Suppl. 2: 20–26.
|
|
(6)
|
Welboren, W. J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer 16(4): 1073–1089.
|
|
(7)
|
Younes, M., ja Honma, N. (2011). Estrogen Receptor Beta. Arch. Pathol. Lab. Med. 135(1): 63–66.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Scientific Statement. Endocr. Rev. 30(4): 293–342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. NIH Publication No 03-4504. National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori, Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals. Toxicol. In Vitro 22(1): 225–231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment No. 67. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity – The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line. Jaapani Kemikaalide Hindamise ja Uurimise Instituut (CERI), Jaapan. Lk 1–188.
|
|
(15)
|
Yamasaki, K., Noda, S., Imatanaka, N., ja Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity. Toxicol. Lett. 146: 111–120.
|
|
(16)
|
Kummer, V., Maskova, J., Zraly, Z., Neca, J., Simeckova, P., Vondracek, J., ja Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Lett. 180: 213–221.
|
|
(17)
|
Gozgit, J. M., Nestor, K. M., Fasco, M. J., Pentecost, B. T., ja Arcaro, K. F. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. Appl. Pharmacol. 196: 58–67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere 34: 835–848.
|
|
(19)
|
Swillens, S. (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis. Mol. Pharmacol. 47(6): 1197–1203.
|
1. liide
MÕISTED JA LÜHENDID
10 % reegel– võimalus jätta analüüsist välja andmepunktid, kus [3H]17β-östradiooli spetsiifilist seondumist iseloomustav paralleelproovide andmete keskväärtus on vähemalt 10 % suurem kui vastav keskväärtus mõnel väiksemal kontsentratsioonil (vt 4. liide).
Asjakohasus– näitaja, mille abil kirjeldatakse, kuivõrd katsemeetod võimaldab uurida huvipakkuvat toimet, kas selle kasutamine on mõttekas ja kas see on konkreetse eesmärgi jaoks sobiv. Asjakohasusest nähtub, mil määral saab katsemeetodiga õigesti mõõta või ennustada huvipakkuvat bioloogilist toimet. Asjakohasuse puhul võetakse arvesse ka katsemeetodiga saavutatavat täpsust (vastavust) (1).
CV– variatsioonikordaja.
E2– 17β-östradiool.
ER– östrogeeniretseptor.
hr-α-ER– rekombinantne inimese α-östrogeeniretseptor.
IC50– kontsentratsioon, mille juures pärssiva uuritava kemikaali mõju on poole väiksem maksimaalsel efektiivsel kontsentratsioonil avalduvast mõjust.
ICCVAM– alternatiivsete meetodite valideerimise ametitevaheline koordineerimiskomitee.
Kemikaal– aine või segu.
Kontseptuaalne raamistik– OECD kontseptuaalne raamistik võimalike endokriinfunktsiooni kahjustavate kemikaalide analüüsimiseks ja hindamiseks.
Laborisisene reprodutseeritavus– näitaja, mis iseloomustab seda, millisel määral suudavad sama labori kvalifitseeritud töötajad saada konkreetset katse-eeskirja kasutades eri aegadel ühesuguseid tulemusi (1).
Laboritevaheline reprodutseeritavus– näitaja, mis iseloomustab seda, millisel määral suudavad eri pädevad laborid sama katse-eeskirja alusel samu aineid uurides saada kvalitatiivselt ja kvantitatiivselt sarnaseid tulemusi. Laboritevaheline reprodutseeritavus määratakse valideerimiseelse ja valideerimisega seotud tegevuse käigus ja see näitab, millisel määral on katsemeetod eri laborites edukalt kasutatav (1).
LEC– väikseim efektiivne kontsentratsioon: uuritava kemikaali väikseim kontsentratsioon, mille juures kemikaal avaldab veel mõju (st uuritava kemikaali väikseim kontsentratsioon, mille juures mõju on statistiliselt erinev üksnes kandeainet sisaldava paralleelse kontrolli puhul täheldatavast mõjust).
Mina-ka-meetod– kõnekeelne väljend katsemeetodi kohta, mis on oma ülesehituselt ja funktsionaalsuse poolest sarnane valideeritud ja tunnustatud standardse katsemeetodiga. Seda väljendit kasutatakse sarnase katsemeetodi tähenduses.
Nõuetekohasuse kriteeriumid– katses kasutatavate kontrollide ja võrdlusstandardite minimaalsed tulemuslikkuse standardid. Katse nõuetekohaseks tunnistamiseks peaksid kõik nõuetekohasuse kriteeriumid olema täidetud.
Pädevus– enne tundmatute ainete analüüsimist tõendatud võimekus katsemeetodi kohaseid katseid nõuetekohaselt läbi viia.
Pädevusained– tulemuslikkuse standarditega hõlmatud alarühm võrdlusaineid, mida võib laboris kasutada standardiseeritud analüüsimeetodi abil tehnilise pädevuse tõendamiseks. Nende ainete valikukriteeriumide hulka kuulub tavaliselt nõue, et nende ainete mõju tugevus oleks erinev, need oleksid turul kättesaadavad ja nende kohta oleksid olemas kvaliteetsed võrdlusandmed.
RBA– suhteline seondumisafiinsus. Aine RBAd väljendatakse aine IC50 logaritmitud väärtuse protsentuaalse osakaaluna 17β-östradiooli IC50 logaritmitud väärtusest.
SD– standardhälve.
Standardsed katsemeetodid– analüüsimeetodid, millele tuginetakse tulemusnäitajatel põhinevas katsejuhendis nr 493.
Tulemuslikkuse standardid– valideeritud katsemeetodil põhinevad standardid, mille alusel hinnatakse vaadeldava mehhanismi ja funktsionaalsuse poolest sarnase kavandatud katsemeetodi võrreldavust. Need hõlmavad 1) katsemeetodi olulisi elemente, 2) valideeritud katsemeetodi vastuvõetava tulemuslikkuse tõendamiseks kasutatud kemikaalide hulgast valitud võrdluskemikaalide miinimumloetelu ja 3) võrreldavat usaldusväärsuse ja täpsuse taset, mille puhul on lähtutud valideeritud katsemeetodiga saadud tulemustest ja mis tuleks saavutada kavandatud katsemeetodi hindamisel võrdluskemikaalide miinimumloetelu kasutamise teel (1).
Täpsus (vastavus)– katsetulemuste ja vastuvõetavate võrdlusväärtuste kooskõla määr. Täpsus näitab analüüsimeetodi tulemuslikkust ja on üks asjakohastest aspektidest. Seda mõistet kasutatakse sageli vastavuse tähenduses, et väljendada õigete katsetulemuste osakaalu (1).
Usaldusväärsus– näitaja, mis iseloomustab katsemeetodiga saadud tulemuste reprodutseeritavust pikema aja jooksul samas laboris ja eri laborites, kui meetodit rakendatakse ühe ja sama katse-eeskirja alusel. Usaldusväärsuse hindamiseks arvutatakse laborisisene ja laboritevaheline reprodutseeritavus.
Uuritav kemikaal– iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
Valideerimine– tegevus, mille käigus määratakse teatud kindla lähenemisviisi, meetodi, katsemudeli, protsessi või hindamisviisi usaldusväärsus ja asjakohasus kindlaksmääratud eesmärgil kasutamisel (1).
Valideeritud katsemeetod– katsemeetod, mille puhul valideerimisuuringud on lõpule viidud, st on määratud selle asjakohasus (sealhulgas täpsus) ja usaldusväärsus konkreetsel eesmärgil kasutamisel. Oluline on märkida, et valideeritud katsemeetod ei pruugi olla täpsuse ja usaldusväärsuse poolest piisavalt tulemuslik, et seda saaks lugeda kavandatud eesmärgi puhul vastuvõetavaks (1).
Võrdlusöstrogeen– 17ß-östradiool (E2, CASi nr: 50-28-2).
Östrogeenne toime– kemikaali võime jäljendada 17β-östradiooli võimet seonduda östrogeeniretseptoriga. Käesolev katsemeetod võimaldab tuvastada seondumist inimese α-ERiga.
2. liide
ÖSTROGEENIRETSEPTORIL (A-ER) PÕHINEVAD FREYBERGERI-WILSONI IN VITRO MEETODID KÜLLASTAVA JA KONKUREERIVA SEONDUMISE ANALÜÜSIMISEKS TÄISPIKA REKOMBINANTSE A-ER-I ABIL
LÄHTEKAALUTLUSED JA PIIRANGUD (VT KA ÜLDINE SISSEJUHATUS)
|
1.
|
Käesoleva östrogeeniretseptoril (α-ER) põhineva, küllastava ja konkureeriva seondumise analüüsimist võimaldava in vitro meetodi puhul kasutatakse täispikka inimese α-östrogeeniretseptorit (hr-α-ER), mille tootmiseks ja eraldamiseks on kasutatud bakuloviirusega nakatatud putukarakke. Freybergeri ja Wilsoni välja töötatud katse-eeskirja on hinnatud rahvusvahelises mitme labori osalusel läbi viidud valideerimisuuringus (2), mille käigus tõendati, et see analüüsimeetod on kavandatud kasutuseesmärgi puhul asjakohane ja usaldusväärne.
|
|
2.
|
Käesolev meetod on rakendatav sõeluuringu jaoks, mille eesmärk on tuvastada aineid, mis on võimelised seonduma täispika hr-α-ERiga. Seda kasutatakse selleks, et hinnata uuritava kemikaali võimet konkureerida hr-α-ERile seondumisel 17β-östradiooliga. Kvantitatiivsed katsetulemused võivad hõlmata uuritava kemikaali IC50 (uuritava kemikaali kontsentratsioon, mis on vajalik poole [3H]17β-östradiooli väljatõrjumiseks hr-α-ERilt) ja suhtelist seondumisafiinsust hr-α-ERi suhtes võrreldes 17β-östradiooliga. Kemikaalide sõeluuringu puhul võivad vastuvõetavad kvalitatiivsed katsetulemused hõlmata uuritava kemikaali liigitamist hr-α-ERiga seonduvaks aineks, mitteseonduvaks aineks või ebaselge mõjuga aineks lähtuvalt seondumiskõverate suhtes kohaldatavatest kriteeriumidest.
|
|
3.
|
Käesoleva analüüsimeetodi puhul kasutatakse radioaktiivset ligandi, mille jaoks peab laboril olema radioaktiivsete materjalide käitlemise luba. Kõikide radioisotoopide ja ohtlike kemikaalidega tehtavate toimingute puhul tuleks järgida siseriiklikes õigusaktides kirjeldatud eeskirju ja korda.
|
|
4.
|
Enne käesoleva meetodi kasutamist regulatiivsel eesmärgil tuleks lugeda peatükke „ÜLDINE SISSEJUHATUS“ ja „hrER-GA SEONDUMISE KATSE ELEMENDID“. Käesolevas katsemeetodis kasutatud mõisted ja lühendid on esitatud 1. liites.
|
MEETODI PÕHIMÕTE (VT KA ÜLDINE SISSEJUHATUS)
|
5.
|
Inimese rekombinantse α-östrogeeniretseptoriga (hr-α-ER) seondumise analüüsimisel mõõdetakse radiomärgistatud ligandi ([3H]17β-östradiool) võimet seonduda ERiga üha suuremas kontsentratsioonis esineva uuritava kemikaali (st konkureeriva ligandi) juuresolekul. Uuritavad kemikaalid, millel on ERi suhtes suur afiinsus, konkureerivad radiomärgistatud ligandiga väiksemal kontsentratsioonil kui kemikaalid, mille afiinsus retseptori suhtes on väiksem.
|
|
6.
|
Kõnealune meetod hõlmab kahte põhielementi: küllastava seondumise katset, millega iseloomustatakse retseptori ja ligandi vahelise interaktsiooni parameetreid, ning sellele järgnevat konkureeriva seondumise katset, millega iseloomustatakse uuritava kemikaali ja radiomärgistatud ligandi vahelist konkurentsi ERile seondumisel.
|
|
7.
|
Küllastava seondumise katse eesmärk on teha konkureeriva seondumise katse eel kindlaks konkreetse partii retseptorite seondumisafiinsus ja arv. Küllastava seondumise katses mõõdetakse tasakaalutingimustes teatud kindlas kontsentratsioonis kasutatava östrogeeniretseptori afiinsust loodusliku ligandi suhtes (seda väljendab dissotsiatsioonikonstant Kd) ja retseptori aktiivsete seondumiskohtade kontsentratsiooni (Bmax).
|
|
8.
|
Konkureeriva seondumise katses mõõdetakse aine afiinsuse hindamiseks selle võimet konkureerida ERile seondumisel [3H]17β-östradiooliga. Afiinsust väljendatakse uuritava kemikaali sellise kontsentratsiooni kaudu, mille juures kemikaal vähendab tasakaalutingimustes [3H]17β-östradiooli spetsiifilist seondumist 50 % võrra (sellele vastab mõiste „50 % ulatuses inhibeeriv kontsentratsioon“ ehk IC50). Afiinsust on võimalik hinnata ka suhtelise seondumisafiinsuse (RBA) kaudu, mis määratakse samas analüüsitsüklis eraldi mõõdetud östradiooli IC50 suhtes. Konkureeriva seondumise katses mõõdetakse teatud kindlas kontsentratsioonis kasutatava [3H]17β-östradiooli seondumist laia vahemikku (kaheksat suurusjärku) hõlmavates kontsentratsioonides esineva uuritava kemikaali juuresolekul. Seejärel kohaldatakse saadud andmete suhtes võimaluse korral teatavat liiki Hilli võrrandit (Hill, 1910), millega kirjeldatakse radioligandi väljatõrjumist konkureeriva seonduva ainega, millel on üks seondumiskoht. Radiomärgistatud östradiooli väljatõrjumise ulatust tasakaalutingimustes kasutatakse uuritava kemikaali liigitamiseks seonduvaks aineks, mitteseonduvaks aineks või ebaselge mõjuga aineks.
|
KATSE KÄIK
hr-α-ERi vastuvõetava funktsionaalsuse tõendamine
|
9.
|
Enne küllastava seondumise ja konkureeriva seondumise katsete regulaarset kasutamist tuleks iga uue hr-α-ERi partii puhul tõendada selle nõuetekohast funktsionaalsust laboris, kus seda kasutama hakatakse. Funktsionaalsuse tõendamine peaks hõlmama kahte etappi. Need etapid on järgmised.
|
—
|
Viiakse läbi [3H]17β-östradiooli küllastava seondumise katse, et tõendada hr-α-ERi spetsiifilisust ja küllastumist. Nende andmete analüüsimisel mittelineaarse regressiooni teel (nt BioSoft; McPherson, 1985; Motulsky, 1995) ja järgnevalt saadud Scatchardi graafiku alusel tuleks iga hr-α-ERi partii puhul dokumenteerida [3H]17β-östradiooli seondumisafiinsus hr-α-ERi suhtes (Kd) ja retseptorite arv (Bmax).
|
|
—
|
Viiakse läbi konkureeriva seondumise katse kontrollainetega (võrdlusöstrogeen (17β-östradiool)), nõrgalt seonduv aine (nt noretünodreel või noretindroon) ja mitteseonduv aine (oktüültrietoksüsilaan (OTES)). Igas laboris tuleks luua varasemate tulemuste andmebaas, et võimaldada dokumenteerida võrdlusöstrogeeni ja nõrgalt seonduva aine IC50 ja muude asjakohaste näitajate väärtuste kooskõla määra eri katsete lõikes ja hr-α-ERi eri partiide puhul. Kontrollaineid iseloomustavate konkureeriva seondumise kõverate parameetrid peaksid jääma usaldusnivoole 95 % vastavasse usaldusvahemikku (vt tabel 1), mis tehti kindlaks kõnealuse analüüsimeetodi valideerimise uuringus osalenud laborites saadud andmete põhjal (2).
|
|
Tabel 1.
FW meetodi kohases hrERiga seondumise katses võrdlusöstrogeeni ja nõrgalt seonduva aine puhul kohaldatavad tulemuslikkuse kriteeriumid
|
Aine
|
Näitaja
|
Keskmine (9)
|
Standardhälve (n)
|
Usaldusvahemik usaldusnivool 95 % (10)
|
|
Alampiir
|
Ülempiir
|
|
17β-östradiool
|
Maksimumväärtus (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Miinimumväärtus (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Hilli tõus
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
Log IC50 (M)
|
-8,92 (11)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretünodreel
|
Maksimumväärtus (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Miinimumväärtus (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Hilli tõus
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
Log IC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Noretindroon (11)
|
Maksimumväärtus (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Miinimumväärtus (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Hilli tõus
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
Log IC50 (M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Labori pädevuse tõendamine
|
10.
|
Tutvuge käesoleva katsemeetodi punktidega 17 ja 18 ning tabeliga 2 peatükis „hrER-GA SEONDUMISE KATSE ELEMENDID“. Iga katse (nii küllastava kui ka konkureeriva seondumise puhul) peaks koosnema kolmest eri päevadel läbi viidavast sõltumatust analüüsitsüklist (st kasutatakse retseptori, kemikaalide ja reaktiivide värskelt valmistatud lahjendusi) ja igas tsüklis tuleks kasutada kolme paralleelproovi.
|
Retseptori (hr-α-ER) kontsentratsiooni määramine
|
11.
|
Aktiivse retseptori kontsentratsioon varieerub pisut eri partiide ja säilitustingimuste puhul. Seepärast tuleks määrata tarnijalt saadud aktiivse retseptori kontsentratsioon. See võimaldab kasutada katses sobivat aktiivse retseptori kontsentratsiooni.
|
|
12.
|
Retseptorit tuleks konkureerivale seondumisele vastavates tingimustes (st 1 nM [3H]östradiool) inkubeerida nominaalsel kontsentratsioonil 0,25, 0,5, 0,75 ja 1 nM ilma märgistamata östradioolita (summaarne seondumine) ja koos 1 μM märgistamata östradiooliga (mittespetsiifiline seondumine). Spetsiifiline seondumine, mis arvutatakse summaarse ja mittespetsiifilise seondumise vahena, esitatakse graafikul vastavuses retseptori nominaalse kontsentratsiooniga. Retseptori kontsentratsioon, mille puhul spetsiifilise seondumise määr vastab 20 protsendile lisatud radiomärgise kogusest, on seotud retseptori vastava nominaalse kontsentratsiooniga ja seda retseptori kontsentratsiooni tuleks kasutada küllastava ja konkureeriva seondumise katsetes. Sageli vastab nendele tingimustele hrERi lõppkontsentratsioon 0,5 nM.
|
|
13.
|
Kui 20 % kriteeriumi täitmine korduvalt ebaõnnestub, tuleks kontrollida katseplaani võimalike vigade suhtes. Suutmatus saavutada vastavust 20 % kriteeriumile võib viidata sellele, et rekombinantse valgu partiis on väga vähe aktiivset retseptorit, ning sel juhul tuleks kaaluda mõne muu retseptoripartii kasutamist.
|
Küllastava seondumise katse
|
14.
|
[3H]17β-östradiooli tuleks hindamiseks kasutada kaheksas eri kontsentratsioonis, iga kontsentratsiooni puhul kolmes paralleelis, ning kohaldada seejuures järgmist kolme tingimust (vt tabel 2).
|
—
|
Seondumine märgistamata 17β-östradiooli puudumisel ja ERi juuresolekul. Nii määratakse summaarne seondumine, mida väljendab radioaktiivsus kannudes, kus on ainult [3H]17β-östradiool.
|
|
—
|
Seondumine märgistamata 17β-östradiooli juuresolekul 1000-kordses liias, võrreldes radiomärgistatud 17β-östradiooliga, ning ERi juuresolekul. Nende tingimuste puhul on eesmärk küllastada aktiivsed seondumiskohad märgistamata 17β-östradiooliga ja määrata kannudes radioaktiivsuse mõõtmise teel mittespetsiifiline seondumine. Radioaktiivse östradiooli mis tahes kogus, mis on veel võimeline retseptoriga seonduma, loetakse olevat seondunud mittespetsiifiliselt, sest märgistamata östradiooli kontsentratsioon peaks olema nii suur, et kõik saadaolevad spetsiifilise seondumise kohad retseptoril on hõivatud.
|
|
—
|
Seondumine märgistamata 17β-östradiooli puudumisel ja ERi puudumisel (summaarse radioaktiivsuse määramine).
|
|
[3H]17β-östradiooli ja märgistamata 17β-östradiooli lahuste valmistamine
|
15.
|
[3H]17β-östradiooli lahjenduste saamiseks tuleks 12 nM [3H]17β-östradiooli põhilahusele analüüsipuhvri lisamise teel valmistada lahused, kus kontsentratsioon jääb algselt vahemikku 0,12 nM kuni 12 nM. Pärast 40 μl sellise lahuse lisamist 96-kannulise mikrotiiterplaadi vastavasse kannu (saadav lõppruumala on 160 μl) saadakse lahused lõpliku katsekontsentratsiooniga 0,03 kuni 3,0 nM. Analüüsipuhvri, [3H]17β-östradiooli põhilahuse ja lahjenduste valmistamist ning kontsentratsiooni määramist on põhjalikult kirjeldatud FW meetodi kohases katse-eeskirjas (2).
|
|
16.
|
Etanoolis lahustatud 17β-östradiooli lahjenduste saamiseks tuleks analüüsipuhvri lisamise teel valmistada kaheksa eri kontsentratsioonidega lahust, mille kontsentratsioon jääb algselt vahemikku 0,06 μM kuni 6 μM. Pärast 80 μl sellise lahuse lisamist 96-kanulise mikrotiiterplaadi vastavasse kannu (saadav lõppruumala on 160 μl) saadakse lahused lõpliku katsekontsentratsiooniga 0,03 μM kuni 3 μM. Märgistamata 17β-östradiooli lõppkontsentratsioon igas mittespetsiifilise seondumise mõõtmiseks kasutatavas kannus peaks olema 1000 korda suurem kui märgistatud [3H]17β-östradiooli kontsentratsioon. Märgistamata 17β-östradiooli lahjenduste valmistamist on põhjalikult kirjeldatud FW meetodi kohases katse-eeskirjas (2).
|
|
17.
|
Tuleks kasutada retseptori sellist nominaalset kontsentratsiooni, mille puhul spetsiifiline seondumine on 20 ± 5 % (vt punktid 12–13). hr-α-ERi lahus tuleks valmistada vahetult enne kasutamist.
|
|
18.
|
96-kannulised mikrotiiterplaadid valmistatakse ette vastavalt tabelis 2 esitatud skeemile, kus iga kontsentratsiooni puhul kasutatakse kolme paralleelproovi. Näide plaadil kasutatavate [3H]17β-östradiooli, märgistamata 17β-östradiooli, puhvri ja retseptori kontsentratsioonide ja koguste kohta on esitatud liites 2.2.
|
Tabel 2.
Küllastava seondumise katses kasutatav mikrotiiterplaadi skeem
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H]E2 + ER
|
0,06 nM [3H]E2 + ER
|
0,08 nM [3H]E2 + ER
|
0,10 nM [3H]E2 + ER
|
Summaarne seondumine(ainult lahusti)
|
|
B
|
0,30 nM [3H]E2 + ER
|
0,60 nM [3H]E2 + ER
|
1,0 nM [3H]E2 + ER
|
3,0 nM [3H]E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H]E2 + ER + 0,03 μM E2
|
0,06 nM [3H]E2 + ER + 0,06 μM E2
|
0,08 nM [3H]E2 + ER + 0,08 μM E2
|
0,10 nM [3H]E2 + ER + 0,10 μM E2
|
Mittespetsiifiline seondumine
|
|
E
|
0,30 nM [3H]E2 + ER + 0,30 μM E2
|
0,60 nM [3H]E2 + ER + 0,60 μM E2
|
1,0 nM [3H]E2 + ER + 1,0 μM E2
|
3,0 nM [3H]E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
|
[3H]E2– [3H]17β-östradiool
ER– östrogeeniretseptor
E2– märgistamata 17β-östradiool
|
|
19.
|
Katses kasutatavaid mikrotiiterplaate tuleks inkubeerida pöördloksutil 2–8 °C juures 16 kuni 20 tundi.
|
hr-α-ERiga seondunud [3H]17β-östradiooli koguse mõõtmine
|
20.
|
hr-α-ERiga seondunud [3H]17β-östradiooli eraldamiseks vabast [3H]17β-östradioolist tuleks lisada igasse kannu 80 μl külma DCC suspensiooni, loksutada mikrotiiterplaate 10 minutit ja tsentrifuugida neid 10 minutit kiirusel umbes 2500 pööret minutis. Et vähendada seondunud [3H]17β-östradiooli dissotsieerumist hr-α-ERilt eraldamisprotsessi käigus, on väga oluline hoida puhvreid ja analüüsitavaid kanne temperatuuril 2–8 °C ning viia kõik etapid läbi kiirelt. Mikrotiiterplaadiloksuti on vajalik, et plaate tõhusalt ja kiiresti töödelda.
|
|
21.
|
Seejärel tuleks DCC puudutamisest tuleneda võiva kannude saastumise ärahoidmiseks väga ettevaatlikult võtta 50 μl supernatanti, mis sisaldab hr-α-ERiga seondunud [3H]17β-östradiooli, ning kanda see teisele mikrotiiterplaadile.
|
|
22.
|
Seejärel lisatakse igasse kannu (A1–B12 ja D1–E12) 200 μl stsintillatsioonivedelikku, mis on võimeline muutma tuumakiirguse kineetilise energia valgusenergiaks. Mõõteplaadi kannudesse G1–H12 (kasutatakse summaarse radioaktiivsuse mõõtmiseks) tuleks otse stsintillatsioonivedelikku lisada [3H]17β-östradiooli lahused (40 μl) järjestikuste lahjendustena, nagu on näidatud tabelis 3; need kannud sisaldavad seega üksnes 200 μl stsintillatsioonivedelikku ja vastava lahjendusastmega [3H]17β-östradiooli lahust. Nendest kannudest saadud andmetest nähtub, kui palju [3H]17β-östradiooli (mõõdetuna dpm-des) lisati iga rühma kannudesse summaarse seondumise ja mittespetsiifilise seondumise hindamiseks.
|
Tabel 3.
Küllastava seondumise katses radioaktiivsuse mõõtmisel kasutatav mikrotiiterplaadi skeem
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H]E2 + ER
|
0,06 nM [3H]E2 + ER
|
0,08 nM [3H]E2 + ER
|
0,10 nM [3H]E2 + ER
|
Summaarne seondumine(ainult lahusti)
|
|
B
|
0,30 nM [3H]E2 + ER
|
0,60 nM [3H]E2 + ER
|
1,0 nM [3H]E2 + ER
|
3,0 nM [3H]E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H]E2 + ER + 0,03 μM E2
|
0,06 nM [3H]E2 + ER + 0,06 μM E2
|
0,08 nM [3H]E2 + ER + 0,08 μM E2
|
0,10 nM [3H]E2 + ER + 0,10 μM E2
|
Mittespetsiifiline seondumine
|
|
E
|
0,30 nM [3H]E2 + ER + 0,30 μM E2
|
0,60 nM [3H]E2 + ER + 0,60 μM E2
|
1,0 nM [3H]E2 + ER + 1,0 μM E2
|
3,0 nM [3H]E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H]E2 (summaarne radioaktiivsus dpm-des)
|
0,06 nM [3H]E2
|
0,08 nM [3H]E2
|
0,10 nM [3H]E2
|
Summaarne radioaktiivsus dpm-des (*7)
|
|
H
|
0,30 nM [3H]E2
|
0,60 nM [3H]E2
|
1,0 nM [3H]E2
|
3,0 nM [3H]E2
|
|
[3H]E2– [3H]17β-östradiool
ER– östrogeeniretseptor
E2– märgistamata 17β-östradiool
dpm– lagunemiste arv minutis
|
|
23.
|
Mõõtmist tuleks alustada pärast vähemalt kahe tunni pikkust viivitust ja loendamisaeg peaks olema 40 minutit kannu kohta. Radioaktiivsuse mõõtmiseks dpm-des kannu kohta tuleks kasutada mikrotiiterplaadi stsintillatsiooniloendurit ja võtta seejuures arvesse sumbumisparandit. Kui mikrotiiterplaadi stsintillatsiooniloendur ei ole kättesaadav, võib proovide analüüsimiseks kasutada tavalist loendurit. Sellisel juhul võib kaaluda loendamisaja lühendamist.
|
Konkureeriva seondumise katse
|
24.
|
Konkureeriva seondumise katse käigus mõõdetakse ühel kontsentratsioonil kasutatava [3H]17β-östradiooli seondumist üha suuremas kontsentratsioonis esineva uuritava kemikaali juuresolekul. Igas analüüsitsüklis tuleks iga kontsentratsiooni puhul kasutada üheaegselt kolme paralleelproovi. Lisaks tuleks iga uuritava kemikaaliga läbi viia kolm järjestikust analüüsitsüklit. Katse läbiviimiseks tuleks kasutada ühte või mitut 96-kannulist mikrotiiterplaati.
|
Kontrollid
|
25.
|
Katse läbiviimisel tuleks igas analüüsitsüklis kasutada paralleelset lahustiga kontrolli ja teisi kontrolle (st võrdlusöstrogeeni, nõrgalt seonduvat ainet ja mitteseonduvat ainet). Igas analüüsitsüklis tuleks ühel plaadil kasutada võrdlusöstrogeeni ja kontrollaineid (st nõrgalt seonduvat ainet ja mitteseonduvat ainet) täieliku kontsentratsioonikõvera ulatuses. Kõikidele ülejäänud plaatidele tuleks kanda i) E2 ja nõrgalt seonduv aine suures (maksimaalsele väljatõrjumisele vastavas) ja keskmises (umbes IC50-le vastavas) kontsentratsioonis kolmes paralleelis ning ii) lahustiga kontroll ja mittespetsiifilise seondumise kontroll, kumbki vähemalt kolmes paralleelis. Analüüsipuhvri, kontrollide, [3H]17β-östradiooli, hr-α-ERi ja uuritava kemikaali lahuste valmistamist on kirjeldatud allikas 2 (K lisa; vt FW meetodi kohane katse-eeskiri).
|
Lahustiga kontroll
|
26.
|
Lahustiga kontroll võimaldab tõendada, et lahusti ei mõjuta katsesüsteemi, ja ühtlasi mõõta summaarset seondumist (SS). Eelistatud lahusti on etanool. Kui uuritav kemikaal on suurimal kontsentratsioonil etanoolis mittelahustuv, võib lahustina kasutada DMSOd. Etanooli või DMSO lõppkontsentratsioon katsekannudes on 1,5 %; see ei tohi olla suurem kui 2 %.
|
Puhvrikontroll
|
27.
|
Puhvrikontroll (PK) ei tohiks sisaldada lahustit ega uuritavat kemikaali, kuid peaks sisaldama kõiki teisi analüüsikomponente. Puhvrikontrolliga saadud tulemusi võrreldakse lahustiga kontrolliga saadud tulemustega, tõendamaks, et kasutatud lahusti ei mõjuta analüüsisüsteemi.
|
Tugevalt seonduv aine (võrdlusöstrogeen)
|
28.
|
17β-östradiool (CASi nr: 50-28-2) on ERi endogeenne ligand ja seondub retseptori α-vormiga suure afiinsusega. Igas hr-α-ERiga tehtavas konkureeriva seondumise katses tuleks kasutada märgistamata 17β-östradiooli standardkõverat, et oleks võimalik hinnata katsetulemuste varieeruvust samas laboris pikema aja jooksul. Märgistamata 17β-östradioolist tuleks katsekannudes kasutamiseks valmistada etanoolis kaheksa lahust kontsentratsioonivahemikus 100 nM kuni 10 pM (-7 [log M] kuni -11 [log M]); lahuste kontsentratsioonid on järgmised: -7 [log M], -8 [log M], -8,5 [log M], -9 [log M], - 9,5 [log M], -10 [log M], -11 [log M]. Märgistamata 17β-östradiooli suurimat kontsentratsiooni (1 μM) kasutatakse ka mittespetsiifilise seondumise hindamiseks. See kontsentratsioon on tabelis 4 tähistatud märgisega „MSS“, kuigi see on samuti osa standardkõverast.
|
Nõrgalt seonduv aine
|
29.
|
Igas analüüsitsüklis tuleks tundlikkuse tõendamiseks ja pikema aja jooksul saadud tulemuste varieeruvuse hindamiseks kasutada ka nõrgalt seonduvat ainet (noretünodreeli (CASi nr: 68-23-5) või noretindrooni (CASi nr: 68-22-4)). Nõrgalt seonduvast ainest tuleks katsekannudes kasutamiseks valmistada etanoolis kaheksa lahust kontsentratsioonvahemikus 3 nM kuni 30 μM (-8,5 [log M] kuni -4,5 [log M]); lahuste kontsentratsioonid on järgmised: -4,5 [log M], -5 [log M], -5,5 [log M], -6 [log M], -6,5 [log M], -7 [log M], -7,5 [log M], -8,5 [log M].
|
Mitteseonduv aine
|
30.
|
Negatiivse kontrollina (mitteseonduva ainena) tuleks kasutada oktüültrietoksüsilaani (OTES; CASi nr: 2943-75-1). Sellega tagatakse, et katse käigus on võimalik tuvastada hr-α-ERiga mitteseonduvaid uuritavaid kemikaale. Mitteseonduvast ainest tuleks katsekannudes kasutamiseks valmistada etanoolis kaheksa lahust log-ühiku kaupa suurenevate kontsentratsioonide vahemikus 0,1 nM kuni 1000 μM (-10 [log M] kuni -3 [log M]). Alternatiivse mitteseonduva ainena võib kasutada di-n-butüülftalaati (DBP). Selle maksimaalne lahustuvus on teadaolevalt -4 [log M].
|
hr-α-ERi kontsentratsioon
|
31.
|
Tuleks kasutada sellist retseptori kogust, mille puhul spetsiifiline seondumine radioligandi kontsentratsioonil 1 nM on 20 ± 5 % (vt 2. liite punktid 12–13). hr-α-ERi lahus tuleks valmistada vahetult enne kasutamist.
|
[3H]17β-östradiool
|
32.
|
[3H]17β-östradiooli kontsentratsioon katsekannudes peaks olema 1,0 nM.
|
Uuritavad kemikaalid
|
33.
|
Iga uuritava kemikaali puhul on esmalt vaja läbi viia lahustuvuskatse, et määrata lahustuvuspiir ja teha kindlaks katse-eeskirja järgimiseks sobiv kontsentratsioonivahemik. Iga uuritava kemikaali lahustuvuspiir tuleb algselt määrata lahustis ja seejärel kontrollida seda katsetingimustes. Katses kasutatav lõppkontsentratsioon ei tohiks olla suurem kui 1 mM. Kontsentratsioonivahemiku leidmise katses kasutatakse lahustiga kontrolli koos kaheksa järjestikuse üksteisest log-ühiku võrra erineva lahjendusega, mille puhul alustatakse maksimaalsest vastuvõetavast kontsentratsioonist (nt 1 mM või väiksem, olenevalt lahustuvuspiirist), ja registreeritakse nähtava hägu või sademe tekkimine (vt ka punkt 35). Uuritava kemikaali analüüsimiseks tuleks kasutada eelnevas kontsentratsioonivahemiku leidmise katses kindlaks tehtud kaheksal järjestikusel üksteisest log-ühiku võrra erineval kontsentratsioonil põhinevat kõverat. Teises ja kolmandas katses tuleks kontsentratsioone vajaduse korral kohandada, et kontsentratsioonist sõltuvuse kõverat oleks võimalik paremini iseloomustada.
|
|
34.
|
Uuritava kemikaali lahjendused tuleks valmistada sobivas lahustis (vt 2. liite punkt 26). Kui uuritav kemikaal ei lahustu suurimal kontsentratsioonil etanoolis ega DMSO-s ja suurema koguse lahusti lisamisel tõuseks lahusti sisaldus lõpplahuses üle lubatud piirmäära, võib suurima kontsentratsioonina kasutada lahjenduste reas järgmist väiksemat kontsentratsiooni. Sellisel juhul võib lisada kontsentratsioonirea lahjemasse otsa ühe lisalahjenduse. Teised kontsentratsioonid lahjenduste reas tuleks jätta muutmata.
|
|
35.
|
Uuritava kemikaali lahuseid tuleks pärast katsekannu lisamist hoolikalt jälgida, sest uuritav kemikaal võib kannu lisamisel välja sadeneda. Kõigi sadet sisaldavate kannude andmed tuleks kõvera lähendamisel välja jätta ja registreerida andmete väljajätmise põhjus.
|
|
36.
|
Kui eelnevalt on olemas muudest allikatest pärinev teave uuritava kemikaali log IC50 kohta, võib olla asjakohane kasutada lahjenduste geomeetrilist jaotust eeldatava log IC50 ümber (nt sammuga 0,5 log-ühikut). Lõpptulemuses peaks kajastuma piisav kontsentratsioonide ulatus kummalgi pool log IC50 väärtust, sealhulgas kõvera ülemine ja alumine osa, nii et seondumiskõverat oleks võimalik piisavalt iseloomustada.
|
Katseplaatide kasutusskeemid
|
37.
|
Mikrotiiterplaatide ettevalmistamisel tuleks need tähistada koodidega, mis vastavad lahustiga kontrollile, võrdlusöstrogeeni suurimale kontsentratsioonile – selle abil hinnatakse ühtlasi mittespetsiifilist seondumist (MSS) – ja puhvrikontrollile, mida kõiki kasutatakse kuues paralleelis, ning igale kontsentratsioonile kaheksast mitteseonduva kontrollaine (oktüültrietoksüsilaani) kontsentratsioonist, võrdlusöstrogeeni seitsmest väiksemast kontsentratsioonist, nõrgalt seonduva aine kaheksast kontsentratsioonist ja iga uuritava kemikaali (UK) kaheksast kontsentratsioonist, mida kõiki kasutatakse kolmes paralleelis. Võrdlusöstrogeeni ja kontrollide täielikke kontsentratsioonikõveraid hõlmav plaadi näidisskeem on esitatud tabelis 4. Uuritavate kemikaalide analüüsimiseks kasutatakse eraldi mikrotiiterplaate, mis peaksid sisaldama ka järgmisi plaadikontrolle: 1) E2 ja nõrgalt seonduvat ainet suures (maksimaalsele väljatõrjumisele vastavas) ja keskmises (umbes IC50-le vastavas) kontsentratsioonis, kõikidel juhtudel kolmes paralleelis; 2) lahustiga kontrolli ja mittespetsiifilise seondumise kontrolli, kummalgi juhul kuues paralleelis (tabel 5). Konkureeriva seondumise katses kasutatavale mikrotiiterplaadi skeemile vastav näidistööleht kolme tundmatu uuritava kemikaali analüüsimiseks on esitatud liites 2.3. Tabelites 4 ja 5 on esitatud katses kasutatavad lõppkontsentratsioonid. E2 maksimaalne kontsentratsioon peaks olema 1 × 10-7 M ja nõrgalt seonduva aine puhul tuleks kasutada 1. plaadil kasutatud suurimat kontsentratsiooni. IC50 tuleb laboris määrata kontrollidega varem saadud tulemuste andmebaasi alusel. Eeldatavalt on see väärtus sarnane valideerimisuuringutes täheldatud väärtusega (vt tabel 1).
|
Tabel 4.
Konkureeriva seondumise katses kasutatav mikrotiiterplaadi skeem, mis hõlmab võrdlusöstrogeeni ja kontrollide täielikke kontsentratsioonikõveraid (1. plaat)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
SS (ainult lahusti)
|
SS (ainult lahusti)
|
MSS
|
MSS
|
|
B
|
E2 (1 × 10-7)
|
E2 (1 × 10-8)
|
E2 (1 × 10-8,5)
|
E2 (1 × 10-9)
|
|
C
|
E2 (1 × 10-9,5)
|
E2 (1 × 10-10)
|
E2 (1 × 10-11)
|
Tühi kann (*8)
|
|
D
|
NE (1 × 10-4,5)
|
NE (1 × 10-5)
|
NE (1 × 10-5,5)
|
NE (1 × 10-6)
|
|
E
|
NE (1 × 10-6,5)
|
NE (1 × 10-7)
|
NE (1 × 10-7,5)
|
NE (1 × 10-8,5)
|
|
F
|
OTES (1 × 10-3)
|
OTES (1 × 10-4)
|
OTES (1 × 10-5)
|
OTES (1 × 10-6)
|
|
G
|
OTES (1 × 10-7)
|
OTES (1 × 10-8)
|
OTES (1 × 10-9)
|
OTES (1 × 10-10)
|
|
H
|
Tühiproov (radiomärgise jaoks) (*9)
|
Tühiproov (radiomärgise jaoks) (*9)
|
Puhvrikontroll
|
Puhvrikontroll
|
|
Selles näites on nõrgalt seonduv aine noretinodreel (NE).
|
Tabel 5.
Konkureeriva seondumise katses kasutatav mikrotiiterplaadi skeem, mis hõlmab uuritavate kemikaalide täielikke kontsentratsioonikõveraid ja plaadikontrolle
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
SS (ainult lahusti)
|
SS (ainult lahusti)
|
MSS
|
MSS
|
|
B
|
UK1 (1 × 10-3)
|
UK1 (1 × 10-4)
|
UK1 (1 × 10-5)
|
UK1 (1 × 10-6)
|
|
C
|
UK1 (1 × 10-7)
|
UK1 (1 × 10-8)
|
UK1 (1 × 10-9)
|
UK1 (1 × 10-10)
|
|
D
|
UK2 (1 × 10-3)
|
UK2 (1 × 10-4)
|
UK2 (1 × 10-5)
|
UK2 (1 × 10-6)
|
|
E
|
UK2 (1 × 10-7)
|
UK2 (1 × 10-8)
|
UK2 (1 × 10-9)
|
UK2 (1 × 10-10)
|
|
F
|
UK3 (1 × 10-3)
|
UK3 (1 × 10-4)
|
UK3 (1 × 10-5)
|
UK3 (1 × 10-6)
|
|
G
|
UK3 (1 × 10-7)
|
UK3 (1 × 10-8)
|
UK3 (1 × 10-9)
|
UK3 (1 × 10-10)
|
|
H
|
NE (IC50)
|
NE (1 × 10-4,5)
|
E2 (IC50)
|
E2 (1 × 10-7)
|
|
Selles näites on nõrgalt seonduv aine noretinodreel (NE).
|
Konkureeriva seondumise katse lõpetamine
|
38.
|
Nagu on näidatud tabelis 6, tuleks kannudesse lisada 80 μl lahustiga kontrolli, puhvrikontrolli, võrdlusöstrogeeni, nõrgalt seonduvat ainet, mitteseonduvat ainet ja uuritavaid kemikaale, mille lahused on valmistatud analüüsipuhvris. Seejärel lisatakse igasse kannu 40 μl 4 nM [3H]17β-östradiooli lahust. Pärast 10 kuni 15 minuti pikkust tasast loksutamist temperatuuril 2–8 °C tuleks lisada 40 μl hr-α-ERi lahust. Katses kasutatavaid mikrotiiterplaate tuleks inkubeerida pöördloksutil 2–8 °C juures 16 kuni 20 tundi.
|
Tabel 6.
hrERil põhineva konkureeriva seondumise katse komponentide kogused mikrotiiterplaadil
|
Kogus (μl)
|
Koostisosa
|
|
80
|
Märgistamata 17β-östradiool, noretünodreel, OTES, uuritav kemikaal, lahusti või puhver
|
|
40
|
4 nM [3H]17β-östradiooli lahus
|
|
40
|
Määratud kontsentratsiooniga hr-α-ERi lahus
|
|
160
|
Lõppruumala igas katsekannus
|
|
39.
|
Seejärel tuleks viia läbi hr-α-ERiga seondunud [3H]17β-östradiooli kvantifitseerimine pärast seda, kui hr-α-ERiga seondunud [3H]17β-östradiool on eraldatud vabast [3H]17β-östradioolist igasse kannu 80 μl külma DCC suspensiooni lisamise teel, nagu on kirjeldatud küllastava seondumise katset käsitlevates punktides 20–23.
|
|
40.
|
Kannusid H1–H6 (tabelis 4 tähistatud kui tühiproovid (radiomärgise jaoks)) kasutatakse 40 μl [3H]-ga märgistatud östradiooli radioaktiivsuse määramiseks dpm-des. 40 μl suurused alikvoodid tuleks lisada otse kannudes H1–H6 olevasse stsintillatsioonivedelikku.
|
Katse nõuetekohasuse kriteeriumid
Küllastava seondumise katse
|
41.
|
Spetsiifilise seondumise kõver peaks jõudma [3H]17β-östradiooli kontsentratsiooni suurenedes platoole; see viitab hr-α-ERi küllastumisele ligandiga.
|
|
42.
|
Spetsiifiline seondumine [3H]17β-östradiooli kontsentratsioonil 1 nM peaks jääma vastuvõetavasse vahemikku 15–25 % eri analüüsitsüklites mõõdetud, lisatud radiomärgise kogusele vastava summaarse radioaktiivsuse näitajate keskväärtusest. Aeg-ajalt esinevad väikesed kõrvalekalded sellest vahemikust on lubatud, ent kui analüüsitsüklites saadud tulemused on pidevalt sellest vahemikust väljaspool või mõnes konkreetses analüüsitsüklis saadud väärtus jääb sellest vahemikust väga kaugele, tuleks valgu kontsentratsiooni kohandada ja küllastamiskatset korrata.
|
|
43.
|
Andmed peaksid võimaldama saada lineaarse Scatchardi graafiku.
|
|
44.
|
Mittespetsiifiline seondumine ei tohi olla liiga ulatuslik. Mittespetsiifilise seondumise määr on tavaliselt < 35 % summaarse seondumise määrast. See suhtarv võib siiski olla aeg-ajalt suurem, kui mõõdetakse väga väikest dpm-des väljendatavat radioaktiivsust radiomärgistatud 17β-östradiooli väikseimal katses kasutataval kontsentratsioonil.
|
Konkureeriva seondumise katse
|
45.
|
Märgistamata 17β-östradiool peaks kontsentratsiooni suurenedes tõrjuma [3H]17β-östradiooli retseptorilt välja viisil, mis vastab konkureerivale seondumisele ühes seondumiskohas.
|
|
46.
|
Võrdlusöstrogeeni (st 17β-östradiooli) IC50 väärtus peaks olema ligikaudselt võrdne [3H]17β-östradiooli molaarse kontsentratsiooni ja küllastava seondumise katses määratud Kd summaga.
|
|
47.
|
Spetsiifiline seondumine peaks järjepidevalt olema vastuvõetavas vahemikus 20 ± 5 %, kui igas kannus täheldatavale summaarsele radioaktiivsusele vastav lisatud radiomärgise keskmine mõõdetud kontsentratsioon on eri analüüsitsüklite lõikes 1 nM. Aeg-ajalt esinevad väikesed kõrvalekalded sellest vahemikust on lubatud, ent kui analüüsitsüklites saadud tulemused on pidevalt sellest vahemikust väljaspool või mõnes konkreetses analüüsitsüklis saadud väärtus jääb sellest vahemikust väga kaugele, tuleks valgu kontsentratsiooni kohandada.
|
|
48.
|
Lahusti ei tohiks mõjutada analüüsimeetodi tundlikkust ega tulemuste reprodutseeritavust. Lahustiga kontrolliga (SSi kannudega) saadud tulemusi võrreldakse puhvrikontrolliga saadud tulemustega, tõendamaks, et kasutatud lahusti ei mõjuta katsesüsteemi. Kui lahustil puudub mõju katsetulemustele, peaksid SSi proove ja puhvrikontrolli iseloomustavad tulemused olema võrreldavad.
|
|
49.
|
Mitteseonduv aine ei tohiks tõrjuda hr-α-ERilt välja rohkem kui 25 % [3H]17β-östradioolist, kui sellist ainet analüüsitakse kontsentratsioonil kuni 10-3 M (OTES) või 10-4 M (DBP).
|
|
50.
|
Tulemuslikkuse kriteeriumide väljatöötamiseks võrdlusöstrogeeni ja kahe nõrgalt seonduva aine (nt noretünodreeli ja noretindrooni) jaoks on kasutatud hrERiga seondumist analüüsida võimaldava FW meetodi valideerimise uuringu andmeid (allikas 2, N lisa). Valideerimisuuringus osalenud laborites tehtud kõikide kontrollkatsete tulemuste puhul on keskväärtuse (n) ± SD kõrval esitatud ka usaldusvahemik usaldusnivool 95 %. Usaldusvahemik usaldusnivool 95 % on arvutatud võrdlusöstrogeeni ja nõrgalt seonduvaid aineid iseloomustavate lähenduskõvera parameetrite (st maksimumi, miinimumi, Hilli tõusu, log IC50) jaoks ning võrdlusöstrogeeni suhtes väljendatava nõrgalt seonduvate ainete log10 RBA jaoks ning seda näitajat kasutatakse positiivsete kontrollide puhul tulemuslikkuse kriteeriumina. Tabelis 1 on esitatud tulemuslikkuse kriteeriumidena kasutatavate lähenduskõvera parameetrite eeldatavad väärtusevahemikud. Praktikas võib IC50 väärtuste vahemik retseptoripreparaadi Kd-st ja ligandi kontsentratsioonist sõltuvalt pisut varieeruda.
|
|
51.
|
Uuritavate kemikaalide jaoks ei ole lähenduskõvera parameetreid käsitlevaid tulemuslikkuse kriteeriume kindlaks määratud, kuna olemasolevate võimalike uuritavate kemikaalide hulk on lai ning nende võimalik afiinsus ja saadavad tulemused varieeruvad (nt täielik kõver, osaline kõver, sobiva kõvera puudumine). Uuritava kemikaaliga igas analüüsitsüklis saadud tulemuste hindamisel tuleks siiski lähtuda eksperdihinnangust. Tuleks kasutada piisavalt suurt uuritava kemikaali kontsentratsioonide vahemikku, et konkureeriva seondumise kõvera ülemine osa (nt osa, kus seondumine on 90–100 %) selgelt välja joonistuks. Paralleelproovide vaheline varieeruvus igal uuritava kemikaali kontsentratsioonil ja kolme järjestikuse analüüsitsükli vaheline varieeruvus peaks olema mõõdukas ja teaduslikult põhjendatav. Uuritava kemikaali hindamisel peaksid kontrollidega saadud tulemused igas analüüsitsüklis ligilähedaselt vastama FW meetodit iseloomustavatele tulemusnäitajatele ja olema kooskõlas asjaomases laboris kontrollidega varem saadud tulemustega.
|
ANDMEANALÜÜS
Küllastava seondumise katse
|
52.
|
Mõõdetakse nii summaarset kui ka mittespetsiifilist seondumist. Nende väärtuste põhjal arvutatakse üha suuremas kontsentratsioonis esineva [3H]17β-östradiooli spetsiifilise seondumise näitaja tasakaalutingimustes; selleks lahutatakse summaarse seondumise näitajast mittespetsiifilise seondumise näitaja. [3H]17β-östradiooli kontsentratsioonist sõltuva spetsiifilise seondumise kõver peaks jõudma platoole, kus [3H]17β-östradiooli spetsiifiline seondumine on hr-α-ERi küllastumisest tulenevalt maksimaalne. Lisaks peaks andmeanalüüsist nähtuma [3H]17β-östradiooli seondumine ühteainsasse suure afiinsusega seondumiskohta. Küllastava seondumise graafikul peaks olema näidatud mittespetsiifiline, summaarne ja spetsiifiline seondumine. Järgnevas andmeanalüüsis tuleks kasutada mittelineaarset regressiooni (nt BioSoft; McPherson, 1985; Motulsky, 1995) ja andmed tuleks lõplikul kujul esitada Scatchardi graafikul.
|
|
53.
|
Andmeanalüüsi käigus tuleks määrata Bmax ja Kd üksnes summaarse seondumise andmete põhjal ning lähtuda seejuures eeldusest, et mittespetsiifiline seondumine on lineaarne, v.a juhul, kui esitatakse põhjendus teistsuguse meetodi kasutamise kohta. Lisaks tuleks parima lähenduskõvera leidmiseks kasutada robustset regressioonanalüüsi, v.a juhul, kui esitatakse põhjendus muu lähenemisviisi kasutamise kohta. Tuleks märkida, millist robustse regressioonanalüüsi meetodit kasutatakse. Küllastava seondumise andmete alusel Bmax-i ja Kd määramisel tuleks andmeid alati korrigeerida ligandi väljatõrjumise suhtes (nt meetodi abil, mida on kirjeldanud Swillens (1995)).
|
Konkureeriva seondumise katse
|
54.
|
Konkureeriva seondumise kõver esitatakse graafikul nii, et ühel teljel on [3H]17β-östradiooli spetsiifiline seondumine ja teisel konkureeriva aine kontsentratsioon (log10-ühikutes). Uuritava kemikaali kontsentratsioon, mille juures [3H]17β-östradiooli spetsiifiline seondumine on 50 % maksimummäärast, on IC50 väärtus.
|
|
55.
|
Positiivsete kontrollide (nt võrdlusöstrogeeni ja nõrgalt seonduva aine) log IC50 hinnanguliste väärtuste kindlakstegemiseks tuleks kasutada sobivat mittelineaarse kõvera lähendamise tarkvara, milles rakendatakse neljaparameetrilist Hilli võrrandit (nt BioSoft; McPherson, 1985; Motulsky, 1995). Kõnealuste kõverate lähendamisel tuleks kõvera ülemine ja alumine osa, tõus ja log IC50 üldjuhul jätta piiritlemata. Parima lähenduskõvera leidmiseks tuleks kasutada robustset regressioonanalüüsi, v.a juhul, kui esitatakse põhjendus muu lähenemisviisi kasutamise kohta. Andmeid ei tohiks korrigeerida ligandi väljatõrjumise suhtes. Pärast esmast analüüsi tuleks iga seondumiskõver üle vaadata, et veenduda mudeli ja andmete omavahelises sobivuses. Nõrgalt seonduva aine suhtelise seondumisafiinsuse (RBA) leidmiseks tuleks arvutada nõrgalt seonduva aine log IC50 väärtuse protsentuaalne osakaal 17β-östradiooli log IC50 väärtusest. Positiivsete kontrollidega ja mitteseonduval ainel põhineva kontrolliga saadud tulemusi tuleks hinnata lähtuvalt käesoleva 2. liite punktide 45–50 kohastest katsemeetodi tulemuslikkuse näitajatest.
|
|
56.
|
Kõigi uuritavate kemikaalide andmete analüüsimisel tuleks kasutada astmelist lähenemist, et tagada andmete asjakohane analüüs ja kõigi konkureeriva seondumise kõverate nõuetekohane liigitamine. Soovitatavalt tuleks uuritava kemikaaliga läbi viidud iga analüüsitsükli puhul esmalt rakendada täpselt sama standardiseeritud andmeanalüüsi, mida kasutatakse võrdlusöstrogeenil ja nõrgalt seonduval ainel põhinevate kontrollide puhul (vt eespool punkt 55). Pärast esmast analüüsi tuleks iga analüüsitsükli puhul teha lähenduskõvera parameetrite tehniline kontroll ja visuaalselt hinnata, kui hästi sobituvad seondumisandmed saadud konkureeriva seondumise kõveraga. Tehnilise kontrolli käigus täheldatav kontsentratsioonist sõltuv [3H]17β-östradiooli spetsiifilise seondumise protsentuaalse määra vähenemine, tehniliste paralleelproovide andmete vaheline väike varieeruvus kemikaali kõigil kontsentratsioonidel ja lähenduskõvera parameetrite sarnased väärtused kolmes analüüsitsüklis viitavad sellele, et katse ja andmeanalüüs on läbi viidud nõuetekohaselt.
|
Andmete tõlgendamine
|
57.
|
Kui kõik nõuetekohasuse kriteeriumid on täidetud, loetakse uuritav kemikaal hr-α-ERiga seonduvaks, kui seondumiskõver sobitub andmetega ja katseandmete vahemikku jäävale sõltuvuskõvera madalaimale punktile vastav väärtus on väiksem kui 50 % (joonis 1).
|
|
58.
|
Kui kõik nõuetekohasuse kriteeriumid on täidetud, loetakse uuritav kemikaal hr-α-ERiga mitteseonduvaks, kui:
|
—
|
seondumiskõver sobitub andmetega ja katseandmete vahemikku jäävale lähendatud sõltuvuskõvera madalaimale punktile vastav väärtus on suurem kui 75 % või
|
|
—
|
sobiv seondumiskõver puudub ja väikseim silumata keskmine seondumisprotsent eri kontsentratsioonidel saadud andmete hulgas on suurem kui 75 %.
|
|
|
59.
|
Kui ükski eespool kirjeldatud tingimus ei ole täidetud (nt lähendatud sõltuvuskõvera madalaimale punktile vastav väärtus on vahemikus 75–50 %), liigitatakse uuritav kemikaal ebaselgeks.
|
Tabel 7.
Uuritava kemikaali konkureeriva seondumise kõveral põhinevad liigitamiskriteeriumid
|
Liigitus
|
Kriteeriumid
|
|
Seonduva
|
Seondumiskõver sobitub andmetega.
Katseandmete vahemikku jäävale sõltuvuskõvera madalaimale punktile vastav väärtus on väiksem kui 50 %.
|
|
Mitteseonduvb
|
Seondumiskõvera sobitumisel andmetega
on katseandmete vahemikku jäävale lähendatud sõltuvuskõvera madalaimale punktile vastav väärtus suurem kui 75 %.
Sobiva seondumiskõvera puudumisel
on väikseim silumata keskmine seondumisprotsent eri kontsentratsioonidel saadud andmete hulgas suurem kui 75 %.
|
|
Ebaselgec
|
Ükskõik millises nõuetekohases analüüsitsüklis saadud tulemus, mille alusel ei saa uuritavat kemikaali liigitada seonduvaks aineks ega mitteseonduvaks aineks
(näiteks vastab lähendatud sõltuvuskõvera madalaim punkt väärtusele 75–50 %).
|
Joonis 1
Näited uuritava kemikaali liigitamise kohta konkureeriva seondumise kõvera alusel

|
60.
|
Samas laboris uuritava kemikaaliga läbi viidud mitme analüüsitsükli tulemuste kokkuvõtmiseks omistatakse iga analüüsitsükli tulemusele arvväärtus ja leitakse nende arvväärtuste keskmine, nagu on kirjeldatud tabelis 8. Igas laboris analüüsitsüklite tulemuste koondamisel saadud tulemust võrreldakse iga uuritava kemikaali puhul eeldatava liigitamistulemusega.
|
Tabel 8.
Meetod uuritava kemikaali liigitamiseks samas laboris läbi viidud mitme analüüsitsükli alusel
|
Iga analüüsitsükli tulemusele väärtuse omistamine
|
|
|
Liigitus
|
Arvväärtus
|
|
Seonduv
|
2
|
|
Ebaselge
|
1
|
|
Mitteseonduv
|
0
|
|
Liigitamine eri analüüsitsüklites saadud arvväärtuste keskmise alusel
|
|
|
Liigitus
|
Arvväärtus
|
|
Seonduv
|
Keskmine ≥ 1,5
|
|
Ebaselge
|
0,5 ≤ keskmine < 1,5
|
|
Mitteseonduv
|
Keskmine < 0,5
|
KATSEPROTOKOLL
|
61.
|
Vt käesoleva katsemeetodi punkt 24 peatükis „hrER-GA SEONDUMISE KATSE ELEMENDID“.
|
Liide 2.1
MÕISTETE LOETELU
[3H]E2– triitiumiga radiomärgistatud 17β-östradiool.
Analüüsipuhver– 10 mM Tris, veise seerumi albumiin kontsentratsioonis 10 mg/ml, 2 mM DTT, 10 % glütserooli, 0,2 mM leupeptiin, pH 7,5.
Analüüsitsükkel– mikrotiiterplaadi katsekannude täieliku komplekti üheaegne analüüs, mille tulemusena saadakse kogu vajalik teave uuritava kemikaali ja hr-α-ERi omavahelise seondumise iseloomustamiseks (nimelt katsekannu lisatud [3H]17β-östradiooli üldkogus, [3H]17β-östradiooli maksimaalne seondumismäär hr-α-ERiga seondumisel, mittespetsiifilise seondumise määr ja summaarse seondumise määr uuritava kemikaali eri kontsentratsioonidel). Analüüsitsüklis võidakse igal kontsentratsioonil kasutada ka ainult ühte katsekannu (st paralleeli), ent kuna käesolevas katse-eeskirjas nõutakse kolme paralleelproovi kasutamist, analüüsitakse käesoleval juhul ühes analüüsitsüklis iga kontsentratsiooni puhul kolme katsekannu. Peale selle viiakse käesoleva katse-eeskirja kohaselt iga kemikaaliga läbi kolm iseseisvat (st järjestikust) analüüsitsüklit.
DCC– dekstraaniga kaetud süsi.
E2– märgistamata 17β-östradiool (inertne).
hr-α-ER– rekombinantne inimese α-östrogeeniretseptor.
Paralleelproov– üks mitmest kannust, mis sisaldavad samu koostisaineid samas kontsentratsioonis ning mida analüüsitakse üheaegselt samas analüüsitsüklis. Käesoleva katse-eeskirja puhul analüüsitakse uuritavat kemikaali igal kontsentratsioonil kolmes paralleelis, st igal uuritava kemikaali kontsentratsioonil analüüsitakse üheaegselt kolme paralleelproovi.
Liide 2.2
TAVAPÄRANE [3H]17β-ÖSTRADIOOLIGA KÜLLASTAMISE KATSE KOLME PARALLEELKANNUGA
|
Tavapärane [3H]17β-östradiooliga küllastamise katse kolme paralleelkannuga
|
|
Asukoht
|
Paralleelproov
|
Kannuliigi kood
|
Radioaktiivse E2 algkontsentratsioon (nM)
|
Radioaktiivse E2 kogus (μl)
|
Radioaktiivse E2 lõppkontsentratsioon (nM)
|
Märgistamata E2 algkontsentratsioon (μM)
|
Märgistamata E2 kogus (μl)
|
Märgistamata E2 lõppkontsentratsioon (μM)
|
Puhvri kogus (μl)
|
Retseptori kogus (μl)
|
Üldkogus kannus (μl)
|
|
A1
|
1
|
R
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
R
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
R
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
R
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
R
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
R
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
R
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
R
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
R
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
R
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
R
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
R
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
R
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
R
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
R
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
R
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
R
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
R
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
R
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
R
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
R
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
R
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
R
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
R
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
RM
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
RM
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
RM
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
RM
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
RM
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
RM
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
RM
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
RM
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
RM
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
RM
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
RM
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
RM
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
RM
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
RM
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
RM
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
RM
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
RM
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
RM
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
RM
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
RM
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
RM
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
RM
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
RM
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
RM
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Radiomärgis
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Radiomärgis
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Radiomärgis
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Radiomärgis
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Radiomärgis
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Radiomärgis
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Radiomärgis
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Radiomärgis
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Radiomärgis
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Radiomärgis
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Radiomärgis
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Radiomärgis
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Radiomärgis
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Radiomärgis
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Radiomärgis
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Radiomärgis
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Radiomärgis
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Radiomärgis
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Radiomärgis
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Radiomärgis
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Radiomärgis
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Radiomärgis
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Radiomärgis
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Radiomärgis
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Tuleb silmas pidada, et kannud tähisega „Radiomärgis“ on inkubeerimise ajal tühjad. Ettenähtud 40 μl lisatakse ainult stsintillatsioonide loendamiseks.
|
Liide 2.3
KONKUREERIVA SEONDUMISE KATSES KASUTATAV MIKROTIITERPLAADI SKEEM
|
Plaat
|
Asukoht
|
Paralleelproov
|
Kannu liik
|
Kannu kood
|
Kontsentratsiooni kood
|
Konkureeriva aine algkontsentratsioon (M)
|
hrERi põhilahus (μl)
|
Puhvri kogus (μl)
|
Märgise (radioaktiivse E2) kogus (μl)
|
Lahjendusplaadilt lisatav kogus (μl)
|
Lõppruumala (μl)
|
Konkureeriva aine lõppkontsentratsioon (M)
|
|
S
|
A1
|
1
|
Summaarne seondumine
|
SS
|
SS1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
Summaarne seondumine
|
SS
|
SS2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
Summaarne seondumine
|
SS
|
SS3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
Summaarne seondumine
|
SS
|
SS4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
Summaarne seondumine
|
SS
|
SS5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
Summaarne seondumine
|
SS
|
SS6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A8
|
2
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A9
|
3
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A10
|
1
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A11
|
2
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A12
|
3
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
B1
|
1
|
Märgistamata E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B2
|
2
|
Märgistamata E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B3
|
3
|
Märgistamata E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B4
|
1
|
Märgistamata E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B5
|
2
|
Märgistamata E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B6
|
3
|
Märgistamata E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B7
|
1
|
Märgistamata E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B8
|
2
|
Märgistamata E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B9
|
3
|
Märgistamata E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B10
|
1
|
Märgistamata E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B11
|
2
|
Märgistamata E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B12
|
3
|
Märgistamata E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
C1
|
1
|
Märgistamata E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
Märgistamata E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
Märgistamata E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C4
|
1
|
Märgistamata E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
Märgistamata E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
Märgistamata E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
Märgistamata E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
Märgistamata E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
Märgistamata E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
Tühiproov
|
Tühiproov
|
T1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
Tühiproov
|
Tühiproov
|
T2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
Tühiproov
|
Tühiproov
|
T3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
Noretünodreel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D2
|
1
|
Noretünodreel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D3
|
1
|
Noretünodreel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D4
|
1
|
Noretünodreel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D5
|
1
|
Noretünodreel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D6
|
1
|
Noretünodreel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D7
|
1
|
Noretünodreel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D8
|
1
|
Noretünodreel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D9
|
1
|
Noretünodreel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D10
|
1
|
Noretünodreel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D11
|
1
|
Noretünodreel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D12
|
1
|
Noretünodreel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
E1
|
1
|
Noretünodreel
|
NE
|
WP
|
6,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E2
|
2
|
Noretünodreel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E3
|
3
|
Noretünodreel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E4
|
1
|
Noretünodreel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E5
|
2
|
Noretünodreel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E6
|
3
|
Noretünodreel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E7
|
1
|
Noretünodreel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E8
|
2
|
Noretünodreel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E9
|
3
|
Noretünodreel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E10
|
1
|
Noretünodreel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E11
|
2
|
Noretünodreel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E12
|
3
|
Noretünodreel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
Radiomärgis
|
R
|
R
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
Radiomärgis
|
R
|
R
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
Radiomärgis
|
R
|
R
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
Radiomärgis
|
R
|
R
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
Radiomärgis
|
R
|
R
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
Radiomärgis
|
R
|
R
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
Puhvrikontroll
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
Puhvrikontroll
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
Puhvrikontroll
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
Puhvrikontroll
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
Puhvrikontroll
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
Puhvrikontroll
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Tuleb silmas pidada, et kannud tähisega „Radiomärgis“ on inkubeerimise ajal tühjad. Ettenähtud 40 μl lisatakse ainult stsintillatsioonide loendamiseks.
|
Konkureeriva seondumise katses kasutatav mikrotiiterplaadi skeem
|
|
Plaat
|
Asukoht
|
Paralleelproov
|
Kannu liik
|
Kannu kood
|
Kontsentratsiooni kood
|
Konkureeriva aine algkontsentratsioon (M)
|
hrERi põhilahus (μl)
|
Puhvri kogus (μl)
|
Märgise (radioaktiivse E2) kogus (μl)
|
Lahjendusplaadilt lisatav kogus (μl)
|
Lõppruumala (μl)
|
Konkureeriva aine lõppkontsentratsioon (M)
|
|
P1
|
A1
|
1
|
Summaarne seondumine
|
SS
|
SS1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
Summaarne seondumine
|
SS
|
SS2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
Summaarne seondumine
|
SS
|
SS3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
Summaarne seondumine
|
SS
|
SS4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
Summaarne seondumine
|
SS
|
SS5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
Summaarne seondumine
|
SS
|
SS6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A8
|
2
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A9
|
3
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A10
|
1
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A11
|
2
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A12
|
3
|
Märgistamata E2 (suur)
|
MSS
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B1
|
1
|
Uuritav kemikaal nr 1
|
UK1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B2
|
2
|
Uuritav kemikaal nr 1
|
UK1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B3
|
3
|
Uuritav kemikaal nr 1
|
UK1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B4
|
1
|
Uuritav kemikaal nr 1
|
UK1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B5
|
2
|
Uuritav kemikaal nr 1
|
UK1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B6
|
3
|
Uuritav kemikaal nr 1
|
UK1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B7
|
1
|
Uuritav kemikaal nr 1
|
UK1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B8
|
2
|
Uuritav kemikaal nr 1
|
UK1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B9
|
3
|
Uuritav kemikaal nr 1
|
UK1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B10
|
1
|
Uuritav kemikaal nr 1
|
UK1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B11
|
2
|
Uuritav kemikaal nr 1
|
UK1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B12
|
3
|
Uuritav kemikaal nr 1
|
UK1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
C1
|
1
|
Uuritav kemikaal nr 1
|
UK1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C2
|
2
|
Uuritav kemikaal nr 1
|
UK1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C3
|
3
|
Uuritav kemikaal nr 1
|
UK1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C4
|
1
|
Uuritav kemikaal nr 1
|
UK1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C5
|
2
|
Uuritav kemikaal nr 1
|
UK1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C6
|
3
|
Uuritav kemikaal nr 1
|
UK1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C7
|
1
|
Uuritav kemikaal nr 1
|
UK1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C8
|
2
|
Uuritav kemikaal nr 1
|
UK1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C9
|
3
|
Uuritav kemikaal nr 1
|
UK1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C10
|
1
|
Uuritav kemikaal nr 1
|
UK1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
Uuritav kemikaal nr 1
|
UK1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
Uuritav kemikaal nr 1
|
UK1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
Uuritav kemikaal nr 2
|
UK2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D2
|
2
|
Uuritav kemikaal nr 2
|
UK2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D3
|
3
|
Uuritav kemikaal nr 2
|
UK2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D4
|
1
|
Uuritav kemikaal nr 2
|
UK2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D5
|
2
|
Uuritav kemikaal nr 2
|
UK2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D6
|
3
|
Uuritav kemikaal nr 2
|
UK2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D7
|
1
|
Uuritav kemikaal nr 2
|
UK2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D8
|
2
|
Uuritav kemikaal nr 2
|
UK2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D9
|
3
|
Uuritav kemikaal nr 2
|
UK2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D10
|
1
|
Uuritav kemikaal nr 2
|
UK2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D11
|
2
|
Uuritav kemikaal nr 2
|
UK2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D12
|
3
|
Uuritav kemikaal nr 2
|
UK2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
E1
|
1
|
Uuritav kemikaal nr 2
|
UK2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E2
|
2
|
Uuritav kemikaal nr 2
|
UK2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E3
|
3
|
Uuritav kemikaal nr 2
|
UK2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E4
|
1
|
Uuritav kemikaal nr 2
|
UK2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E5
|
2
|
Uuritav kemikaal nr 2
|
UK2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E6
|
3
|
Uuritav kemikaal nr 2
|
UK2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E7
|
1
|
Uuritav kemikaal nr 2
|
UK2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E8
|
2
|
Uuritav kemikaal nr 2
|
UK2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E9
|
3
|
Uuritav kemikaal nr 2
|
UK2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E10
|
1
|
Uuritav kemikaal nr 2
|
UK2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
Uuritav kemikaal nr 2
|
UK2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
Uuritav kemikaal nr 2
|
UK2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
Uuritav kemikaal nr 3
|
UK3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F2
|
2
|
Uuritav kemikaal nr 3
|
UK3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F3
|
3
|
Uuritav kemikaal nr 3
|
UK3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F4
|
1
|
Uuritav kemikaal nr 3
|
UK3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F5
|
2
|
Uuritav kemikaal nr 3
|
UK3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F6
|
3
|
Uuritav kemikaal nr 3
|
UK3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F7
|
1
|
Uuritav kemikaal nr 3
|
UK3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F8
|
2
|
Uuritav kemikaal nr 3
|
UK3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F9
|
3
|
Uuritav kemikaal nr 3
|
UK3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F10
|
1
|
Uuritav kemikaal nr 3
|
UK3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F11
|
2
|
Uuritav kemikaal nr 3
|
UK3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F12
|
3
|
Uuritav kemikaal nr 3
|
UK3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
G1
|
1
|
Uuritav kemikaal nr 3
|
UK3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G2
|
2
|
Uuritav kemikaal nr 3
|
UK3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G3
|
3
|
Uuritav kemikaal nr 3
|
UK3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G4
|
1
|
Uuritav kemikaal nr 3
|
UK3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G5
|
2
|
Uuritav kemikaal nr 3
|
UK3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G6
|
3
|
Uuritav kemikaal nr 3
|
UK3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G7
|
1
|
Uuritav kemikaal nr 3
|
UK3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G8
|
2
|
Uuritav kemikaal nr 3
|
UK3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G9
|
3
|
Uuritav kemikaal nr 3
|
UK3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G10
|
1
|
Uuritav kemikaal nr 3
|
UK3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
Uuritav kemikaal nr 3
|
UK3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
Uuritav kemikaal nr 3
|
UK3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
Noretünodreel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
Noretünodreel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
Noretünodreel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
Noretünodreel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
Noretünodreel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
Noretünodreel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
Märgistamata E2
|
S
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
Märgistamata E2
|
S
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
Märgistamata E2
|
S
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
Märgistamata E2
|
S
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
Märgistamata E2
|
S
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
Märgistamata E2
|
S
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
3. liide
LIGANDIGA SEONDUVAL REKOMBINANTSEL INIMESE α-ER-I VALGUDOMEENIL PÕHINEV JAAPANI KEMIKAALIDE HINDAMISE JA UURIMISE INSTITUUDI (CERI) IN VITRO MEETOD ÖSTROGEENIRETSEPTORIGA SEONDUMISE ANALÜÜSIMISEKS
LÄHTEKAALUTLUSED JA PIIRANGUD (VT KA ÜLDINE SISSEJUHATUS)
|
1.
|
Käesoleva östrogeeniretseptoril (α-ER) põhineva, küllastava ja konkureeriva seondumise analüüsimist võimaldava in vitro meetodi puhul kasutatakse inimese α-ERi (hr-α-ER) ligandiga seonduvat domeeni (LBD). See valgukonstrukt on toodetud Jaapani Kemikaalide Hindamise ja Uurimise Instituudis (CERI) ja seda ekspresseeritakse glutatiooni S-transferaasiga (GST) ühendatud liitvalguna bakteris E. coli. CERI katse-eeskirja on hinnatud rahvusvahelises mitme labori osalusel läbi viidud valideerimisuuringus (2), mille käigus tõendati, et see analüüsimeetod on kavandatud kasutuseesmärgi puhul asjakohane ja usaldusväärne.
|
|
2.
|
Käesolev meetod on rakendatav sõeluuringu jaoks, mille eesmärk on tuvastada aineid, mis on võimelised seonduma hr-α-ERiga. Seda kasutatakse selleks, et hinnata uuritava kemikaali võimet konkureerida hr-α-ERi LBD-le seondumisel 17β-östradiooliga. Kvantitatiivsed katsetulemused võivad hõlmata uuritava kemikaali IC50 (uuritava kemikaali kontsentratsioon, mis on vajalik poole [3H]17β-östradiooli väljatõrjumiseks hr-α-ERilt) ja suhtelist seondumisafiinsust hr-α-ERi suhtes võrreldes 17β-östradiooliga. Kemikaalide sõeluuringu puhul võivad vastuvõetavad kvalitatiivsed katsetulemused hõlmata uuritavate kemikaalide liigitamist hr-α-ERiga seonduvaks aineks, mitteseonduvaks aineks või ebaselge mõjuga aineks lähtuvalt seondumiskõverate suhtes kohaldatavatest kriteeriumidest.
|
|
3.
|
Käesoleva analüüsimeetodi puhul kasutatakse radioaktiivset ligandi, mille jaoks peab laboril olema radioaktiivsete materjalide käitlemise luba. Kõikide radioisotoopide ja ohtlike kemikaalidega tehtavate toimingute puhul tuleks järgida siseriiklikes õigusaktides kirjeldatud eeskirju ja korda.
|
|
4.
|
Enne käesoleva katsemeetodi kasutamist regulatiivsel eesmärgil tuleks lugeda peatükke „ÜLDINE SISSEJUHATUS“ ja „hrER-GA SEONDUMISE KATSE ELEMENDID“. Käesolevas katsemeetodis kasutatud mõisted ja lühendid on esitatud 1. liites.
|
MEETODI PÕHIMÕTE (VT KA ÜLDINE SISSEJUHATUS)
|
5.
|
Inimese rekombinantse α-östrogeeniretseptoriga (hr-α-ER) seondumise analüüsimisel mõõdetakse radiomärgistatud ligandi ([3H]17β-östradiool) võimet seonduda ERiga üha suuremas kontsentratsioonis esineva uuritava kemikaali (st konkureeriva ligandi) juuresolekul. Uuritavad kemikaalid, millel on ERi suhtes suur afiinsus, konkureerivad radiomärgistatud ligandiga väiksemal kontsentratsioonil kui kemikaalid, mille afiinsus retseptori suhtes on väiksem.
|
|
6.
|
Kõnealune meetod hõlmab kahte põhielementi: küllastava seondumise katset, millega iseloomustatakse retseptor ja ligandi vahelise interaktsiooni parameetreid, ning sellele järgnevat konkureeriva seondumise katset, millega iseloomustatakse uuritava kemikaali ja radiomärgistatud ligandi vahelist konkurentsi ERile seondumisel.
|
|
7.
|
Küllastava seondumise katse eesmärk on teha konkureeriva seondumise katse eel kindlaks konkreetse partii retseptorite seondumisafiinsus ja arv. Küllastava seondumise katses mõõdetakse tasakaalutingimustes teatud kindlas kontsentratsioonis kasutatava östrogeeniretseptori afiinsust loodusliku ligandi suhtes (seda väljendab dissotsiatsioonikonstant Kd) ja retseptori aktiivsete seondumiskohtade kontsentratsiooni (Bmax).
|
|
8.
|
Konkureeriva seondumise katses mõõdetakse aine afiinsuse hindamiseks selle võimet konkureerida ERile seondumisel [3H]17β-östradiooliga. Afiinsust väljendatakse uuritava kemikaali sellise kontsentratsiooni kaudu, mille juures kemikaal vähendab tasakaalutingimustes [3H]17β-östradiooli spetsiifilist seondumist 50 % võrra (sellele vastab mõiste „50 % ulatuses inhibeeriv kontsentratsioon“ ehk IC50). Afiinsust on võimalik hinnata ka suhtelise seondumisafiinsuse (RBA) kaudu, mis määratakse samas analüüsitsüklis eraldi mõõdetud östradiooli IC50 suhtes. Konkureeriva seondumise katses mõõdetakse teatud kindlas kontsentratsioonis kasutatava [3H]17β-östradiooli seondumist laia vahemikku (kaheksat suurusjärku) hõlmavates kontsentratsioonides esineva uuritava kemikaali juuresolekul. Seejärel kohaldatakse saadud andmete suhtes võimaluse korral teatavat liiki Hilli võrrandit (Hill, 1910), millega kirjeldatakse radioligandi väljatõrjumist konkureeriva seonduva ainega, millel on üks seondumiskoht. Radiomärgistatud östradiooli väljatõrjumise ulatust tasakaalutingimustes kasutatakse uuritava kemikaali liigitamiseks seonduvaks aineks, mitteseonduvaks aineks või ebaselge mõjuga aineks.
|
KATSE KÄIK
hr-α-ERi vastuvõetava funktsionaalsuse tõendamine
|
9.
|
Enne küllastava seondumise ja konkureeriva seondumise katsete regulaarset kasutamist tuleks iga uue hr-α-ERi partii puhul tõendada selle nõuetekohast funktsionaalsust laboris, kus seda kasutama hakatakse. Funktsionaalsuse tõendamine peaks hõlmama kahte etappi. Need etapid on järgmised.
|
—
|
Viiakse läbi [3H]17β-östradiooli küllastava seondumise katse, et tõendada hr-α-ERi spetsiifilisust ja küllastumist. Nende andmete analüüsimisel mittelineaarse regressiooni teel (nt BioSoft; McPherson, 1985; Motulsky, 1995) ja järgnevalt saadud Scatchardi graafiku alusel tuleks iga hr-α-ERi partii puhul dokumenteerida [3H]17β-östradiooli seondumisafiinsus hr-α-ERi suhtes (Kd) ja retseptorite arv (Bmax).
|
|
—
|
Viiakse läbi konkureeriva seondumise katse kontrollainetega (võrdlusöstrogeen (17β-östradiool), nõrgalt seonduv aine (nt noretünodreel või noretindroon) ja mitteseonduv aine (oktüültrietoksüsilaan (OTES)). Igas laboris tuleks luua varasemate tulemuste andmebaas, et võimaldada dokumenteerida võrdlusöstrogeeni ja nõrgalt seonduva aine IC50 ja muude asjakohaste näitajate väärtuste kooskõla määra eri katsete lõikes ja hr-α-ERi eri partiide puhul. Kontrollaineid iseloomustavate konkureeriva seondumise kõverate parameetrid peaksid jääma usaldusnivoole 95 % vastavasse usaldusvahemikku (vt tabel 1), mis tehti kindlaks kõnealuse analüüsimeetodi valideerimise uuringus osalenud laborites saadud andmete põhjal (2).
|
Tabel 1
CERI meetodi kohases hrERiga seondumise katses võrdlusöstrogeeni ja nõrgalt seonduva aine puhul kohaldatavad tulemuslikkuse kriteeriumid
|
Aine
|
Näitaja
|
Keskmine (12)
|
Standardhälve (n)
|
Usaldusvahemik usaldusnivool 95 % (13)
|
|
Alampiir
|
Ülempiir
|
|
17β-östradiool
|
Maksimumväärtus
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Miinimumväärtus
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Hilli tõus
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
Log IC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretünodreel
|
Maksimumväärtus
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Miinimumväärtus
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Hilli tõus
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
Log IC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Noretindroon (14)
|
Maksimumväärtus
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Miinimumväärtus
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Hilli tõus
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
Log IC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Labori pädevuse tõendamine
|
10.
|
Tutvuge käesoleva katsemeetodi punktidega 17 ja 18 ning tabeliga 2 peatükis „hrER–GA SEONDUMISE KATSE ELEMENDID“. Iga katse (nii küllastava kui ka konkureeriva seondumise puhul) peaks koosnema kolmest eri päevadel läbi viidavast sõltumatust analüüsitsüklist (st kasutatakse retseptori, kemikaalide ja reaktiivide värskelt valmistatud lahjendusi) ja igas tsüklis tuleks kasutada kolme paralleelproovi.
|
Retseptori (hr-α-ER) kontsentratsiooni määramine
|
11.
|
Aktiivse retseptori kontsentratsioon varieerub pisut eri partiide ja säilitustingimuste puhul. Seepärast tuleks määrata tarnijalt saadud aktiivse retseptori kontsentratsioon. See võimaldab kasutada katses sobivat aktiivse retseptori kontsentratsiooni.
|
|
12.
|
Retseptorit tuleks konkureerivale seonduvale vastavates tingimustes (st 0,5 nM [3H]östradiool) inkubeerida nominaalsel kontsentratsioonil 0,1, 0,2, 0,4 ja 0,6 nM ilma märgistamata östradioolita (summaarne seondumine) ja koos 1 μM märgistamata östradiooliga (mittespetsiifiline seondumine). Spetsiifiline seondumine, mis arvutatakse summaarse ja mittespetsiifilise seondumise vahena, esitatakse graafikul vastavuses retseptori nominaalse kontsentratsiooniga. Retseptori kontsentratsioon, mille puhul spetsiifilise seondumise määr vastab 40 protsendile lisatud radiomärgise kogusest, on seotud retseptori vastava nominaalse kontsentratsiooniga ja seda retseptori kontsentratsiooni tuleks kasutada küllastava ja konkureeriva seondumise katsetes. Sageli vastab neile tingimustele hrERi lõppkontsentratsioon 0,2 nM.
|
|
13.
|
Kui 40 % kriteeriumi täitmine korduvalt ebaõnnestub, tuleks kontrollida katseplaani võimalike vigade suhtes. Suutmatus saavutada vastavust 40 % kriteeriumile võib viidata sellele, et rekombinantse valgu partiis on väga vähe aktiivset retseptorit, ning sel juhul tuleks kaaluda mõne muu retseptoripartii kasutamist.
|
Küllastava seondumise katse
|
14.
|
[3H]17β-östradiooli tuleks hindamiseks kasutada kaheksas eri kontsentratsioonis, iga kontsentratsiooni puhul kolmes paralleelis, ning kohaldada seejuures järgmist kolme tingimust (vt tabel 2).
|
a.
|
Seondumine märgistamata 17β-östradiooli puudumisel ja ERi juuresolekul. Nii määratakse summaarne seondumine, mida väljendab radioaktiivsus kannudes, kus on ainult [3H]17β-östradiool.
|
|
b.
|
Seondumine märgistamata 17β-östradiooli juuresolekul 2000-kordses liias, võrreldes radiomärgistatud 17β-östradiooliga, ning ERi juuresolekuga. Nende tingimuse puhul on eesmärk küllastada aktiivsed seondumiskohad märgistamata 17β-östradiooliga ja määrata kannudes radioaktiivsuse mõõtmise teel mittespetsiifiline seondumine. Radioaktiivse östradiooli mis tahes kogus, mis on veel võimeline retseptoriga seonduma, loetakse olevat seondunud mittespetsiifiliselt, sest märgistamata östradiooli kontsentratsioon peaks olema nii suur, et kõik saadaolevad spetsiifilise seondumise kohad retseptoril on hõivatud.
|
|
c.
|
Seondumine märgistamata 17β-östradiooli puudumisel ja ERi puudumisel (summaarse radioaktiivsuse määramine).
|
[3H]17β-östradiooli, märgistamata 17β-östradiooli ja hr-α-ERi lahuste valmistamine
|
15.
|
[3H]17β-östradiooli 1 μM põhilahusest tuleks valmistada [3H]17β-östradiooli 40 nM lahus DMSOs; selleks lisatakse toatemperatuuril DMSOd (et saada 200 nM lahus) ja analüüsipuhvrit (et saada 40 nM lahus). Saadud 40 nM lahusest valmistatakse toatemperatuuril analüüsipuhvri lisamise teel rida [3H]17β-östradiooli lahjendusi kontsentratsiooniga 0,313 nM kuni 40 nM (nagu on kirjeldatud tabeli 2 kaheteistkümnendas veerus). Lõplik katsekontsentratsioon vahemikus 0,0313–4,0 nM saavutatakse 10 μl sellise lahuse lisamisega 96-kannulise mikrotiiterplaadi vastavasse katsekannu (vt tabelid 2 ja 3). Analüüsipuhvri valmistamist, eriaktiivsusel põhinevat [3H]17β-östradiooli algse põhilahuse kontsentratsiooni arvutamist, lahjenduste valmistamist ning kontsentratsiooni määramist on põhjalikult kirjeldatud CERI katse-eeskirjas (2).
|
|
16.
|
Märgistamata 17β-östradiooli lahuse lahjenduste valmistamiseks tuleks kasutada 17β-östradiooli 1 nM põhilahust, millele lisatakse analüüsipuhvrit, et saada kaheksa eri kontsentratsioonidega lahust, mille kontsentratsioon jääb algselt vahemikku 0,625 μM kuni 80 μM. Lõplik katsekontsentratsioon vahemikus 0,0625–8 μM saavutatakse 10 μl sellise lahuse lisamisega 96-kannulise mikrotiiterplaadi vastavasse katsekannu, mis on ette nähtud mittespetsiifilise seondumise mõõtmiseks (vt tabelid 2 ja 3). Märgistamata 17β-östradiooli lahjenduste valmistamist on põhjalikult kirjeldatud CERI katse-eeskirjas (2).
|
|
17.
|
Tuleks kasutada retseptori sellist kontsentratsiooni, mille puhul spetsiifiline seondumine on 40 ± 10 % (vt punktid 12–13). hr-α-ERi lahus tuleks valmistada jääkülma analüüsipuhvri lisamise teel vahetult enne kasutamist, st pärast seda, kui kõik summaarse seondumise, mittespetsiifilise seondumise ja ainult radioaktiivse ligandi määramiseks ette nähtud kannud on katseks ette valmistatud.
|
|
18.
|
96-kannulised mikrotiiterplaadid valmistatakse ette vastavalt tabelis 2 esitatud skeemile, kus [3H]17β-östradiooli iga kontsentratsiooni puhul kasutatakse kolme paralleelproovi. Kasutatavad [3H]17β-östradiooli, märgistamata 17β-östradiooli, puhvri ja retseptori kogused on esitatud tabelis 3.
Tabel 2
Küllastava seondumise katses kasutatav mikrotiiterplaadi skeem
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
SSi mõõtmiseks
|
MSSi mõõtmiseks
|
Ainult radioaktiivse ligandi määramiseks
|
|
Märgistamata E2 lahjendused plaadi 4.–6. tulba jaoks
|
[3H]E2 lahjendused plaadi 1.–9. tulba jaoks
|
|
A
|
0,0313 nM [3H]E2+ ER
|
0,0313 nM [3H]E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H]E2+ ER
|
0,0625 nM [3H]E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H]E2+ ER
|
0,125 nM [3H]E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H]E2+ ER
|
0,250 nM [3H]E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [3H]E2+ ER
|
0,50 nM [3H]E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H]E2+ ER
|
1,00 nM [3H]E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H]E2+ ER
|
2,00 nM [3H]E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H]E2+ ER
|
4,00 nM [3H]E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
SS
|
-
|
summaarne seondumine
|
|
MSS
|
-
|
mittespetsiifiline seondumine
|
|
[3H]E2
|
-
|
[3H]17β-östradiool
|
|
E2
|
-
|
märgistamata 17β-östradiool
|
|
Tabel 3
Reaktiivide kogused küllastamiskatses kasutataval mikrotiiterplaadil
|
Tulba number
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Katseetapid
|
SSi kannud
|
MSSi kannud
|
Ainult radioaktiivne ligand
|
|
Eespool kirjeldatud reaktsioonikannudesse lisatavate komponentide kogused ja lisamise järjekord
|
Puhver
|
60 μl
|
50 μl
|
90 μl
|
|
Märgistamata E2 tabeli 2 üheteistkümnendast veerust
|
–
|
10 μl
|
–
|
|
[3H]E2 tabeli 2 kaheteistkümnendast veerust
|
10 μl
|
10 μl
|
10 μl
|
|
hr-α-ER
|
30 μl
|
30 μl
|
—
|
|
Reaktsiooni koguruumala
|
100 μl
|
100 μl
|
100 μl
|
|
Inkubeerimine
|
PÄRAST 2 TUNNI PIKKUST INKUBEERIMIST
|
Radioaktiivsuse kvantifitseerimine kohe pärast lahuse valmistamist; inkubeerimist ei toimu
|
|
Töötlus 0,4 % DCCga
|
Jah
|
Jah
|
Ei
|
|
0,4 % DCC kogus
|
100 μl
|
100 μl
|
–
|
|
Filtrimine
|
Jah
|
Jah
|
Ei
|
|
RADIOAKTIIVSUSE MÕÕTMINE DPM-DES
|
|
Stsintillatsioonikokteilile kvantifitseerimiseks lisatav kogus
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Summaarse seondumise ja mittespetsiifilise seondumise määramiseks kasutatavaid mikrotiiterplaate tuleks inkubeerida toatemperatuuril (22–28 °C) kaks tundi.
|
|
hr-α-ERiga seondunud [3H]17β-östradiooli mõõtmine
|
20.
|
Pärast kahetunnist inkubatsiooniperioodi tuleks hr-α-ERiga seondunud [3H]17β-östradiooli eraldamiseks vabast [3H]17β-östradioolist lisada kannudesse 100 μl jääkülma 0,4 % DCC suspensiooni. Seejärel tuleks plaadid asetada 10 minutiks jääle ning DCC eraldamiseks tuleks reaktsioonisegu ja DCC suspensioon kanda filtrimiseks mikrotiiterplaadifiltrile. Järgnevalt tuleks 100 μl filtraati lisada vedelikstsintillatsiooniloenduri viaalis olevale stsintillatsioonivedelikule, et määrata vedelikstsintillatsiooniloenduri abil igas viaalis lagunemiste arv minutis (dpm).
|
|
21.
|
Kui mikrotiiterplaadifilter ei ole kättesaadav, võib DCC eemaldamiseks kasutada tsentrifuugimist. Seejärel tuleks DCC puudutamisest tuleneda võiva kannude saastumise ärahoidmiseks väga ettevaatlikult võtta 50 μl supernatanti, mis sisaldab hr-α-ERiga seondunud [3H]17β-östradiooli, ning kasutada seda stsintillatsioonide loendamiseks.
|
|
22.
|
Ainult radioaktiivset ligandi sisaldavaid proove kasutatakse selleks, et määrata katsekannudesse lisatud [3H]17β-östradioolis toimuvate lagunemiste arv minutis (dpm). Radioaktiivsus tuleks kvantifitseerida vahetult pärast proovide valmistamist. Kõnealuseid kanne ei tohiks inkubeerida ega töödelda DCC suspensiooniga, vaid nende sisu tuleks lisada otse stsintillatsioonivedelikku. Saadavatest mõõtmistulemustest nähtub, kui palju [3H]17β-östradiooli lisati dpm-des iga rühma kannudesse summaarse seondumise ja mittespetsiifilise seondumise hindamiseks.
|
Konkureeriva seondumise katse
|
23.
|
Konkureeriva seondumise katse käigus mõõdetakse ühel kontsentratsioonil kasutatava [3H]17β-östradiooli seondumist üha suuremas kontsentratsioonis esineva uuritava kemikaali juuresolekul. Igas analüüsitsüklis tuleks iga kontsentratsiooni puhul kasutada üheaegselt kolme paralleelproovi. Lisaks tuleks iga uuritava kemikaaliga läbi viia kolm järjestikust analüüsitsüklit. Katse läbiviimiseks tuleks kasutada ühte või mitut 96-kannulist mikrotiiterplaati.
|
Kontrollid
|
24.
|
Katse läbiviimisel tuleks igas analüüsitsüklis kasutada paralleelset lahustiga kontrolli ja teisi kontrolle (st võrdlusöstrogeeni, nõrgalt seonduvat ainet ja mitteseonduvat ainet). Igas analüüsitsüklis tuleks ühel plaadil kasutada võrdlusöstrogeeni ja kontrollaineid (st nõrgalt seonduvat ainet ja mitteseonduvat ainet) täieliku kontsentratsioonikõvera ulatuses. Kõikidele ülejäänud plaatidele tuleks kanda i) E2 ja nõrgalt seonduv aine suures (maksimaalsele väljatõrjumisele vastavas, st radiomärgistatud ligandi praktiliselt täielikuks väljatõrjumiseks vajalikus) ja keskmises (umbes IC50-le vastavas) kontsentratsioonis kolmes paralleelis ning ii) lahustiga kontroll ja mittespetsiifilise seondumise kontroll, kumbki kolmes paralleelis. Analüüsipuhvri, [3H]17β-östradiooli, hr-α-ERi ja uuritava kemikaali lahuste valmistamist on põhjalikult kirjeldatud CERI katse-eeskirjas (2).
|
Lahustiga kontroll
|
25.
|
Lahustiga kontroll võimaldab tõendada, et lahusti ei mõjuta katsesüsteemi, ja ühtlasi mõõta summaarset seondumist (SS). Eelistatud lahusti on DMSO. Kui uuritav kemikaal on suurimal kontsentratsioonil DMSOs mittelahustuv, võib lahustina kasutada etanooli. DMSO lõppkontsentratsioon katsekannudes peaks olema 2,05 %; kui uuritav kemikaal on vähelahustuv, võib DMSO sisaldust suurendada kuni 2,5 protsendini. DMSOd ei tohiks kasutada kontsentratsioonis üle 2,5 %, sest suuremal kontsentratsioonil hakkab lahusti mõjutama katsetulemusi. Kui uuritav kemikaal ei ole DMSOs lahustuv, kuid lahustub etanoolis, võib katses kasutada etanooli maksimaalses kontsentratsioonis 2 %, ilma et see mõjutaks katsetulemusi.
|
Puhvrikontroll
|
26.
|
Puhvrikontroll (PK) ei tohiks sisaldada lahustit ega uuritavat kemikaali, kuid peaks sisaldama kõiki teisi analüüsikomponente. Puhvrikontrolliga saadud tulemusi võrreldakse lahustiga kontrolliga saadud tulemustega, tõendamaks, et kasutatud lahusti ei mõjuta analüüsisüsteemi.
|
Tugevalt seonduv aine (võrdlusöstrogeen)
|
27.
|
17β-östradiool (CASi nr: 50-28-2) on ERi endogeenne ligand ja seondub retseptori α-vormiga suure afiinsusega. Igas hr-α-ERiga tehtavas konkureeriva seondumise katses tuleks kasutada märgistamata 17β-östradiooli standardkõverat, et oleks võimalik hinnata katsetulemuste varieeruvust samas laboris pikema aja jooksul. DMSOs ja analüüsipuhvris tuleks valmistada kaheksa märgistamata 17β-östradiooli lahust; lõppkontsentratsioonid standardkõverate saamiseks kasutatavates katsekannudes on järgmised: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. Märgistamata 17β-östradiooli suurimat kontsentratsiooni (1 μM) kasutatakse ka mittespetsiifilise seondumise hindamiseks. See kontsentratsioon on tabelis 4 tähistatud märgisega „MSS“, kuigi see on samuti osa standardkõverast.
|
Nõrgalt seonduv aine
|
28.
|
Igas analüüsitsüklis tuleks tundlikkuse tõendamiseks ja pikema aja jooksul saadud tulemuste varieeruvuse hindamiseks kasutada ka nõrgalt seonduvat ainet (noretünodreeli (CASi nr: 68-23-5) või noretindrooni (CASi nr: 68-22-4)). Nõrgalt seonduvast ainest tuleks valmistada DMSOs ja analüüsipuhvris kaheksa lahust; katsekannudes kasutatavad lõppkontsentratsioonid on järgmised: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 ja 10–9 M.
|
Mitteseonduv aine
|
29.
|
Negatiivse kontrollina (mitteseonduva ainena) tuleks kasutada oktüültrietoksüsilaani (OTES; CASi nr: 2943-75-1). Sellega tagatakse, et katse käigus on võimalik tuvastada hr-α-ERiga mitteseonduvaid uuritavaid kemikaale. Mitteseonduvast ainest tuleks valmistada DMSOs ja analüüsipuhvris kaheksa lahust; katsekannudes kasutatavad lõppkontsentratsioonid on järgmised: 10–3, 10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Alternatiivse mitteseonduva ainena võib kasutada di-n-butüülftalaati (DBP; CASi nr: 84-72-2), ent seda võib analüüsida üksnes kontsentratsioonini 10–4 M. DBP maksimaalne lahustuvus katsetingimustes on teadaolevalt 10–4 M.
|
hr-α-ERi kontsentratsioon
|
30.
|
Tuleks kasutada sellist retseptori kogust, mille puhul spetsiifiline seondumine on 40 ± 10 % (vt 3. liite punktid 12–13). hr-α-ERi lahus, mis saadakse funktsionaalse hr-α-ERi lahjendamisel jääkülmas analüüsipuhvris, tuleks valmistada vahetult enne kasutamist.
|
[3H]17β-östradiool
|
31.
|
[3H]17β-östradiooli lõppkontsentratsioon katsekannudes peaks olema 0,5 nM.
|
Uuritavad kemikaalid
|
32.
|
Iga uuritava kemikaali puhul on esmalt vaja läbi viia lahustuvuskatse, et määrata lahustuvuspiir ja teha kindlaks katse-eeskirja järgimiseks sobiv kontsentratsioonivahemik. Iga uuritava kemikaali lahustuvuspiir tuleb algselt määrata lahustis ja seejärel kontrollida seda katsetingimustes. Katses kasutatav lõppkontsentratsioon ei tohiks olla suurem kui 1 mM. Kontsentratsioonivahemiku leidmise katses kasutatakse lahustiga kontrolli koos vähemalt kaheksa järjestikuse üksteisest log-ühiku võrra erineva lahjendusega, mille puhul alustatakse maksimaalsest vastuvõetavast kontsentratsioonist (nt 1 mM või väiksem, olenevalt lahustuvuspiirist), ja registreeritakse nähtava hägu või sademe tekkimine (vt ka 3. liite punkt 35). Kui katse jaoks sobiv kontsentratsioonivahemik on kindlaks tehtud, tuleks uuritavat kemikaali analüüsida eelnevas kontsentratsioonivahemiku leidmise katses kindlaks tehtud kaheksal üksteisest log-ühiku võrra erineval kontsentratsioonil. Teises ja kolmandas katses tuleks kontsentratsioone vajaduse korral kohandada, et kontsentratsioonist sõltuvuse kõverat oleks võimalik paremini iseloomustada.
|
|
33.
|
Uuritava kemikaali lahjendused tuleks valmistada sobivas lahustis (vt 3. liite punkt 25). Kui uuritava kemikaal ei lahustu suurimal kontsentratsioonil DMSOs ega etanoolis ja suurema koguse lahusti lisamisel tõuseks lahusti sisaldus lõpplahuses üle lubatud piirmäära, võib suurima kontsentratsioonina kasutada lahjenduste reas järgmist väiksemat kontsentratsiooni. Sellisel juhul võib lisada kontsentratsioonirea lahjemasse otsa ühe lisalahjenduse. Teised kontsentratsioonid lahjenduste reas tuleks jätta muutmata.
|
|
34.
|
Uuritava kemikaali lahuseid tuleks pärast katsekannu lisamist hoolikalt jälgida, sest uuritav kemikaal võib kannu lisamisel välja sadeneda. Kõigi sadet sisaldavate kannude andmed tuleks kõvera lähendamisel välja jätta ja registreerida andmete väljajätmise põhjus.
|
|
35.
|
Kui eelnevalt on olemas muudest allikatest pärinev teave uuritava kemikaali log IC50 kohta, võib olla asjakohane kasutada lahjenduste geomeetrilist väiksema sammuga jaotust eeldatava log IC50 ümber (nt sammuga 0,5 log-ühikut). Lõpptulemuses peaks kajastuma piisav kontsentratsioonide ulatus kummalgi pool log IC50 väärtust, sealhulgas kõvera ülemine ja alumine osa, nii et seondumiskõverat oleks võimalik piisavalt iseloomustada.
|
Katseplaatide kasutusskeemid
|
36.
|
Tähistatud mikrotiiterplaatide ettevalmistamisel tuleks lähtuda sellest, et katses kasutatakse lahustiga kontrolli, võrdlusöstrogeeni (E2) suurimat kontsentratsiooni – selle abil hinnatakse ühtlasi mittespetsiifilist seondumist (MSS) – ja puhvrikontrolli kuues paralleelis ning mitteseonduva kontrollaine (oktüültrietoksüsilaani) kaheksat kontsentratsiooni, võrdlusöstrogeeni (E2) seitset väiksemat kontsentratsiooni, nõrgalt seonduva aine (noretünodreeli või noretindrooni) kaheksat kontsentratsiooni ja iga uuritava kemikaali (UK) kaheksat kontsentratsiooni kolmes paralleelis. Võrdlusöstrogeeni ja kontrollide täielikke kontsentratsioonikõveraid hõlmav plaadi näidisskeem on esitatud tabelis 4. Uuritavate kemikaalide analüüsimiseks kasutatakse eraldi mikrotiiterplaate, mis peaksid sisaldama ka järgmisi plaadikontrolle: i) E2 ja nõrgalt seonduvat ainet suures (maksimaalsele väljatõrjumisele vastavas) ja keskmises (umbes IC50-le vastavas) kontsentratsioonis, kõikidel juhtudel kolmes paralleelis; ii) lahustiga kontrolli (summaarse seondumise hindamiseks) ja mittespetsiifilise seondumise kontrolli, kummalgi juhul kuues paralleelis (tabel 5). Konkureeriva seondumise katses kasutatavale mikrotiiterplaadi skeemile vastav näidistööleht kolme tundmatu uuritava kemikaali analüüsimiseks on esitatud liites 3.3. Töölehel ning tabelites 4 ja 5 on esitatud igas katsekannus kasutatavad lõppkontsentratsioonid. E2 maksimaalne kontsentratsioon peaks olema 1 × 10–7 M ja nõrgalt seonduva aine puhul tuleks kasutada 1. plaadil kasutatud suurimat kontsentratsiooni. IC50 tuleb laboris määrata kontrollidega varem saadud tulemuste andmebaasi alusel. Eeldatavalt on see väärtus sarnane valideerimisuuringutes täheldatud väärtusega (vt tabel 1).
|
Tabel 4
Konkureeriva seondumise katses kasutatav mikrotiiterplaadi skeem (96), (97), mis hõlmab võrdlusöstrogeeni ja kontrollide täielikke kontsentratsioonikõveraid (1. plaat)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Puhvrikontroll ja positiivne kontroll (E2)
|
Nõrgalt positiivne aine (noretünodreel)
|
Negatiivne kontroll (OTES)
|
SS ja MSS
|
|
A
|
Tühi kann (*14)
|
1 × 10–9 M
|
1 × 10–10 M
|
SS (lahustiga kontroll) (2,05 % DMSO)
|
|
B
|
E2 (1 × 10–11 M)
|
1 × 10–8 M
|
1 × 10–9 M
|
|
C
|
E2 (1 × 10–10 M)
|
1 × 10–7,5 M
|
1 × 10–8 M
|
MSS (10–6 M E2)
|
|
D
|
E2 (1 × 10–9,5 M)
|
1 × 10–7 M
|
1 × 10–7 M
|
|
E
|
E2 (1 × 10–9 M)
|
1 × 10–6,5 M
|
1 × 10–6 M
|
Puhvrikontroll
|
|
F
|
E2 (1 × 10–8,5 M)
|
1 × 10–6 M
|
1 × 10–5 M
|
|
G
|
E2 (1 × 10–8 M)
|
1 × 10–5,5 M
|
1 × 10–4 M
|
Tühiproov (radiomärgise jaoks) (*15)
|
|
H
|
E2 (1 × 10–7 M)
|
1 × 10–4,5 M
|
1 × 10–3 M
|
Tabel 5
Konkureeriva seondumise katses kasutatav mikrotiiterplaadi skeem uuritavaid kemikaale (UK) ja plaadikontrolle sisaldavate ülejäänud plaatide jaoks
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Uuritav kemikaal nr 1 (UK-1)
|
Uuritav kemikaal nr 2 (UK-2)
|
Uuritav kemikaal nr 3 (UK-3)
|
Kontrollid
|
|
A
|
UK-1 (1 × 10–10 M)
|
UK-2 (1 × 10–10 M)
|
UK-3 (1 × 10–10 M)
|
E2 (1 × 10
-7
M)
|
|
B
|
UK-1 (1 × 10–9 M)
|
UK-2 (1 × 10–9 M)
|
UK-3 (1 × 10–9 M)
|
E2 (IC50)
|
|
C
|
UK-1 (1 × 10–8 M)
|
UK-2 (1 × 10–8 M)
|
UK-3 (1 × 10–8 M)
|
NE (1 × 10
-4,5
M)
|
|
D
|
UK-1 (1 × 10–7 M)
|
UK-2 (1 × 10–7 M)
|
UK-3 (1 × 10–7 M)
|
NE (IC50)
|
|
E
|
UK-1 (1 × 10–6 M)
|
UK-2 (1 × 10–6 M)
|
UK-3 (1 × 10–6 M)
|
MSS (10–6 M E2)
|
|
F
|
UK-1 (1 × 10–5 M)
|
UK-2 (1 × 10–5 M)
|
UK-3 (1 × 10–5 M)
|
|
G
|
UK-1 (1 × 10–4 M)
|
UK-2 (1 × 10–4 M)
|
UK-3 (1 × 10–4 M)
|
SS (lahustiga kontroll)
|
|
H
|
UK-1 (1 × 10–3 M)
|
UK-2 (1 × 10–3 M)
|
UK-3 (1 × 10–3 M)
|
|
Selles näites on nõrgalt seonduv aine noretinodreel (NE).
|
Konkureeriva seondumise katse lõpetamine
|
37.
|
Nagu on näidatud tabelis 6, tuleks igasse kannu – välja arvatud summaarse seondumise ja tühiproovide (radiomärgise) analüüsimiseks ette nähtud kannud – lisada 50 μl analüüsipuhvrit ja seejärel 10 μl lahustiga kontrolli, võrdlusöstrogeeni (E2), nõrgalt seonduvat ainet, mitteseonduvat ainet või uuritavat kemikaali ning 10 μl 5 nM [3H]17β-östradiooli lahust. Seejärel lisatakse igasse kannu 30 μl jääkülma retseptorilahust ja segatakse saadud lahust ettevaatlikult. hr-α-ERi lahus tuleks lisada viimasena. Katses kasutatavaid mikrotiiterplaate inkubeeritakse toatemperatuuril (22–28 °C) kaks tundi.
Tabel 6
hrERil põhineva konkureeriva seondumise katse komponentide kogused mikrotiiterplaadil
|
Ettevalmistusetapid
|
Muud kannud peale SSi kannude
|
SSi kannud
|
Tühiproov (radiomärgise jaoks)
|
|
Eespool kirjeldatud reaktsioonikannudesse lisatavate komponentide kogused ja lisamise järjekord
|
Toatemperatuuril analüüsipuhver
|
50 μl
|
60 μl
|
90 μl
|
|
Märgistamata E2, nõrgalt seonduv aine, mitteseonduv aine, lahusti või uuritav kemikaal (*16)
|
10 μl
|
—
|
—
|
|
[3H]17β-östradiool kontsentratsioonis, mille puhul saadav lõppkontsentratsioon on 0,5 nM (st 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
Määratud kontsentratsiooniga hr-α-ERi lahus (vt punktid 12–13)
|
30 μl
|
30 μl
|
—
|
|
Lõppruumala igas katsekannus
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Seejärel tuleks viia läbi hr-α-ERiga seondunud [3H]17β-östradiooli kvantifitseerimine pärast seda, kui hr-α-ERiga seondunud [3H]17β-östradiool on eraldatud vabast [3H]17β-östradioolist igasse kannu 100 μl jääkülma DCC suspensiooni lisamise teel, nagu on kirjeldatud küllastava seondumise katset käsitlevates 3. liite punktides 21–23.
|
|
39.
|
Kannusid G10–G12 ja H10–H12 (tabelis 4 tähistatud kui tühiproovid (radiomärgise jaoks)) kasutatakse 10 μl [3H]-ga märgistatud östradiooli radioaktiivsuse määramiseks dpm-des. 10 μl suurused alikvoodid tuleks lisada otse stsintillatsioonivedelikku.
|
Katse nõuetekohasuse kriteeriumid
Küllastava seondumise katse
|
40.
|
Spetsiifilise seondumise kõver peaks jõudma [3H]17β-östradiooli kontsentratsiooni suurenedes platoole; see viitab hr-α-ERi küllastumisele ligandiga.
|
|
41.
|
Spetsiifiline seondumine [3H]17β-östradiooli kontsentratsioonil 0,5 nM peaks jääma vastuvõetavasse vahemikku 30–50 % eri analüüsitsüklites mõõdetud, lisatud radiomärgise kogusele vastava summaarse radioaktiivsuse näitajate keskväärtusest. Aeg-ajalt esinevad väikesed kõrvalekalded sellest vahemikust on lubatud, ent kui analüüsitsüklites saadud tulemused on pidevalt sellest vahemikust väljaspool või mõnes konkreetses analüüsitsüklis saadud väärtus jääb sellest vahemikust väga kaugele, tuleks valgu kontsentratsiooni kohandada ja küllastamiskatset korrata.
|
|
42.
|
Andmed peaksid võimaldama saada lineaarse Scatchardi graafiku.
|
|
43.
|
Mittespetsiifiline seondumine ei tohi olla liiga ulatuslik. Mittespetsiifilise seondumise määr on tavaliselt < 35 % summaarse seondumise määrast. See suhtarv võib siiski olla aeg-ajalt suurem, kui mõõdetakse väga väikest dpm-des väljendatavat radioaktiivsust radiomärgistatud 17β-östradiooli väikseimal katses kasutataval kontsentratsioonil.
|
Konkureeriva seondumise katse
|
44.
|
Märgistamata 17β-östradiool peaks kontsentratsiooni suurenedes tõrjuma [3H]17β-östradiooli retseptorilt välja viisil, mis vastab konkureerivale seondumisele ühes seondumiskohas.
|
|
45.
|
Võrdlusöstrogeeni (st 17β-östradiooli) IC50 väärtus peaks olema ligikaudselt võrdne [3H]17β-östradiooli molaarse kontsentratsiooni ja küllastava seondumise katses määratud Kd summaga.
|
|
46.
|
Spetsiifiline seondumine peaks järjepidevalt olema vastuvõetavas vahemikus 40 ± 10 %, kui igas kannus täheldatavale summaarsele radioaktiivsusele vastav lisatud radiomärgise keskmine mõõdetud kontsentratsioon on eri analüüsitsüklite lõikes 0,5 nM. Aeg-ajalt esinevad väikesed kõrvalekalded sellest vahemikust on lubatud, ent kui analüüsitsüklites saadud tulemused on pidevalt sellest vahemikust väljaspool või mõnes konkreetses analüüsitsüklis saadud väärtus jääb sellest vahemikust väga kaugele, tuleks valgu kontsentratsiooni kohandada.
|
|
47.
|
Lahusti ei tohiks mõjutada analüüsimeetodi tundlikkust ega tulemuste reprodutseeritavust. Lahustiga kontrolliga (SSi kannudega) saadud tulemusi võrreldakse puhvrikontrolliga saadud tulemustega, tõendamaks, et kasutatud lahusti ei mõjuta katsesüsteemi. Kui lahustil puudub mõju katsetulemustele, peaksid SSi proove ja puhvrikontrolli iseloomustavad tulemused olema võrreldavad.
|
|
48.
|
Mitteseonduv aine ei tohiks tõrjuda hr-α-ERilt välja rohkem kui 25 % [3H]17β-östradioolist, kui sellist ainet analüüsitakse kontsentratsioonil kuni 10–3 M (OTES) või 10–4 M (DBP).
|
|
49.
|
Tulemuslikkuse kriteeriumide väljatöötamiseks võrdlusöstrogeeni ja kahe nõrgalt seonduva aine (nt noretünodreeli ja noretindrooni) jaoks on kasutatud hrERiga seondumist analüüsida võimaldava CERI meetodi valideerimise uuringu andmeid (allikas 2, N lisa). Valideerimisuuringus osalenud neljas laboris tehtud kõikide kontrollkatsete tulemuste puhul on keskväärtuse ± SD (n) kõrval esitatud ka usaldusvahemik usaldusnivool 95 %. Usaldusvahemik usaldusnivool 95 % on arvutatud võrdlusöstrogeeni ja nõrgalt seonduvaid aineid iseloomustavate lähenduskõvera parameetrite (st maksimumi, miinimumi, Hilli tõusu, log IC50) jaoks ning võrdlusöstrogeeni suhtes väljendatava nõrgalt seonduvate ainete log10 RBA jaoks. Tabelis 1 on esitatud tulemuslikkuse kriteeriumidena kasutatavate lähenduskõvera parameetrite eeldatavad väärtusevahemikud. Praktikas võib IC50 väärtuste vahemik retseptoripreparaadi katseliselt määratud Kd-st ja katses kasutatavast ligandi kontsentratsioonist sõltuvalt pisut varieeruda.
|
|
50.
|
Uuritavate kemikaalide jaoks ei ole lähenduskõvera parameetreid käsitlevaid tulemuslikkuse kriteeriume kindlaks määratud, kuna olemasolevate võimalike uuritavate kemikaalide hulk on lai ning nende võimalik afiinsus ja saadavad tulemused varieeruvad (nt täielik kõver, osaline kõver, sobiva kõvera puudumine). Uuritava kemikaaliga igas analüüsitsüklis saadud tulemuste hindamisel tuleks siiski lähtuda eksperdihinnangust. Tuleks kasutada piisavalt suurt uuritava kemikaali kontsentratsioonide vahemikku, et konkureeriva seondumise kõvera ülemine osa (nt osa, kus seondumine on 90–100 %) selgelt välja joonistuks. Paralleelproovide vaheline varieeruvus igal uuritava kemikaali kontsentratsioonil ja kolme järjestikuse analüüsitsükli vaheline varieeruvus peaks olema mõõdukas ja teaduslikult põhjendatav. Uuritava kemikaali hindamisel peaksid kontrollidega saadud tulemused igas analüüsitsüklis ligilähedaselt vastama CERI meetodit iseloomustavatele tulemusnäitajatele ja olema kooskõlas asjaomases laboris kontrollidega varem saadud tulemustega.
|
ANDMEANALÜÜS
Küllastava seondumise katse
|
51.
|
Mõõdetakse nii summaarset kui ka mittespetsiifilist seondumist. Nende väärtuste põhjal arvutatakse üha suuremas kontsentratsioonis esineva [3H]17β-östradiooli spetsiifilise seondumise näitaja tasakaalutingimustes; selleks lahutatakse summaarse seondumise näitajast mittespetsiifilise seondumise näitaja. [3H]17β-östradiooli kontsentratsioonist sõltuva spetsiifilise seondumise kõver peaks jõudma platoole, kus [3H]17β-östradiooli spetsiifiline seondumine on hr-α-ERi küllastumisest tulenevalt maksimaalne. Lisaks peaks andmeanalüüsist nähtuma [3H]17β-östradiooli seondumine ühteainsasse suure afiinsusega seondumiskohta. Küllastava seondumise graafikul peaks olema näidatud mittespetsiifiline, summaarne ja spetsiifiline seondumine. Järgnevas andmeanalüüsis tuleks kasutada mittelineaarset regressiooni (nt BioSoft; McPherson, 1985; Motulsky, 1995) ja andmed tuleks lõplikul kujul esitada Scatchardi graafikul.
|
|
52.
|
Andmeanalüüsi käigus tuleks määrata Bmax ja Kd üksnes summaarse seondumise andmete põhjal ning lähtuda seejuures eeldusest, et mittespetsiifiline seondumine on lineaarne, v.a juhul, kui esitatakse põhjendus teistsuguse meetodi kasutamise kohta. Lisaks tuleks parima lähenduskõvera leidmiseks kasutada robustset regressioonanalüüsi, v.a juhul, kui esitatakse põhjendus muu lähenemisviisi kasutamise kohta. Tuleks märkida, millist robustse regressioonanalüüsi meetodit kasutatakse. Küllastava seondumise andmete alusel Bmax-i ja Kd määramisel tuleks andmeid alati korrigeerida ligandi väljatõrjumise suhtes (nt meetodi abil, mida on kirjeldanud Swillens (1995)).
|
Konkureeriva seondumise katse
|
53.
|
Konkureeriva seondumise kõver esitatakse graafikul nii, et ühel teljel on [3H]17β-östradiooli spetsiifiline seondumine ja teisel konkureeriva aine kontsentratsioon (log10-ühikutes). Uuritava kemikaali kontsentratsioon, mille juures [3H]17β-östradiooli spetsiifiline seondumine on 50 % maksimummäärast, on IC50 väärtus.
|
|
54.
|
Positiivsete kontrollide (nt võrdlusöstrogeeni ja nõrgalt seonduva aine) log IC50 hinnanguliste väärtuste kindlakstegemiseks tuleks kasutada sobivat mittelineaarse kõvera lähendamise tarkvara, milles rakendatakse neljaparameetrilist Hilli võrrandit (nt BioSoft; McPherson, 1985; Motulsky, 1995). Kõnealuste kõverate lähendamisel tuleks kõvera ülemine ja alumine osa, tõus ja log IC50 üldjuhul jätta piiritlemata. Parima lähenduskõvera leidmiseks tuleks kasutada robustset regressioonanalüüsi, v.a juhul, kui esitatakse põhjendus muu lähenemisviisi kasutamise kohta. Andmeid ei tohiks korrigeerida ligandi väljatõrjumise suhtes. Pärast esmast analüüsi tuleks iga seondumiskõver üle vaadata, et veenduda mudeli ja andmete omavahelises sobivuses. Nõrgalt seonduva aine suhtelise seondumisafiinsuse (RBA) leidmiseks tuleks arvutada nõrgalt seonduva aine log IC50 väärtuse protsentuaalne osakaal 17β-östradiooli log IC50 väärtusest. Positiivsete kontrollidega ja mitteseonduval ainel põhineva kontrolliga saadud tulemusi tuleks hinnata lähtuvalt käesoleva 3. liite punktide 44–49 kohastest katsemeetodi tulemuslikkuse näitajatest.
|
|
55.
|
Kõigi uuritavate kemikaalide andmete analüüsimisel tuleks kasutada astmelist lähenemist, et tagada andmete asjakohane analüüs ja kõigi konkureeriva seondumise kõverate nõuetekohane liigitamine. Soovitatavalt tuleks uuritava kemikaaliga läbi viidud iga analüüsitsükli puhul esmalt rakendada täpselt sama standardiseeritud andmeanalüüsi, mida kasutatakse võrdlusöstrogeenil ja nõrgalt seonduval ainel põhinevate kontrollide puhul (vt käesoleva 3. liite punkt 54). Pärast esmast analüüsi tuleks iga analüüsitsükli puhul teha lähenduskõvera parameetrite tehniline kontroll ja visuaalselt hinnata, kui hästi sobituvad seondumisandmed saadud konkureeriva seondumise kõveraga. Tehnilise kontrolli käigus täheldatav kontsentratsioonist sõltuv [3H]17β-östradiooli spetsiifilise seondumise protsentuaalse määra vähenemine, tehniliste paralleelproovide andmete vaheline väike varieeruvus kemikaali kõigil kontsentratsioonidel ja lähenduskõvera parameetrite sarnased väärtused kolmes analüüsitsüklis viitavad sellele, et katse ja andmeanalüüs on läbi viidud nõuetekohaselt.
|
Andmete tõlgendamine
|
56.
|
Kui kõik nõuetekohasuse kriteeriumid on täidetud, loetakse uuritav kemikaal hr-α-ERiga seonduvaks, kui seondumiskυver sobitub andmetega ja katseandmete vahemikku jδδvale sυltuvuskυvera madalaimale punktile vastav vδδrtus on vδiksem kui 50 % (joonis 1).
|
|
57.
|
Kui kυik nυuetekohasuse kriteeriumid on tδidetud, loetakse uuritav kemikaal hr-α-ERiga mitteseonduvaks, kui:
|
—
|
seondumiskõver sobitub andmetega ja katseandmete vahemikku jäävale lähendatud sõltuvuskõvera madalaimale punktile vastav väärtus on suurem kui 75 % või
|
|
—
|
sobiv seondumiskõver puudub ja väikseim silumata keskmine seondumisprotsent eri kontsentratsioonidel saadud andmete hulgas on suurem kui 75 %.
|
|
|
58.
|
Kui ükski eespool kirjeldatud tingimus ei ole täidetud (nt lähendatud sõltuvuskõvera madalaimale punktile vastav väärtus on vahemikus 75–50 %), liigitatakse uuritav kemikaal ebaselgeks.
|
Tabel 7
Uuritava kemikaali konkureeriva seondumise kõveral põhinevad liigitamiskriteeriumid
|
Liigitus
|
Kriteeriumid
|
|
Seonduva
|
Seondumiskõver sobitub andmetega.
Katseandmete vahemikku jäävale sõltuvuskõvera madalaimale punktile vastav väärtus on väiksem kui 50 %.
|
|
Mitteseonduvb
|
Seondumiskõvera sobitumisel andmetega
on katseandmete vahemikku jäävale lähendatud sõltuvuskõvera madalaimale punktile vastav väärtus suurem kui 75 %.
Sobiva seondumiskõvera puudumisel
on väikseim silumata keskmine seondumisprotsent eri kontsentratsioonidel saadud andmete hulgas suurem kui 75 %.
|
|
Ebaselgec
|
Ükskõik millises nõuetekohases analüüsitsüklis saadud tulemus, mille alusel ei saa uuritavat kemikaali liigitada seonduvaks aineks ega mitteseonduvaks aineks
(näiteks vastab lähendatud sõltuvuskõvera madalaim punkt väärtusele 75–50 %).
|
Joonis 1.
Näited uuritava kemikaali liigitamise kohta konkureeriva seondumise kõvera alusel

|
59.
|
Samas laboris uuritava kemikaaliga läbi viidud mitme analüüsitsükli tulemuste kokkuvõtmiseks omistatakse iga analüüsitsükli tulemusele arvväärtus ja leitakse nende arvväärtuste keskmine, nagu on kirjeldatud tabelis 8. Igas laboris analüüsitsüklite tulemuste koondamisel saadud tulemust võrreldakse iga uuritava kemikaali puhul eeldatava liigitamistulemusega.
|
Tabel 8
Meetod uuritava kemikaali liigitamiseks samas laboris läbi viidud mitme analüüsitsükli alusel
|
Iga analüüsitsükli tulemusele väärtuse omistamine
|
|
Liigitus
|
Arvväärtus
|
|
Seonduv
|
2
|
|
Ebaselge
|
1
|
|
Mitteseonduv
|
0
|
|
Liigitamine eri analüüsitsüklites saadud arvväärtuste keskmise alusel
|
|
Liigitus
|
Arvväärtus
|
|
Seonduv
|
Keskmine ≥ 1,5
|
|
Ebaselge
|
0,5 ≤ keskmine < 1,5
|
|
Mitteseonduv
|
Keskmine < 0,5
|
KATSEPROTOKOLL
|
60.
|
Vt käesoleva katsemeetodi punkt 24 peatükis „hrER-GA SEONDUMISE KATSE ELEMENDID“.
|
Liide 3.1
MÕISTETE LOETELU
[3H]E2
– triitiumiga radiomärgistatud 17β-östradiool.
Analüüsipuhver
– 10 mM Tris-HCl, pH 7,4, 1 mM EDTA, 1 mM EGTA, 1 mM NaVO3, 10 % glütserooli, 0,2 mM leupeptiin, 1 mM ditiotreitool ja veise seerumi albumiin kontsentratsioonis 10 mg/ml.
Analüüsitsükkel
– mikrotiiterplaadi katsekannude täieliku komplekti üheaegne analüüs, mille tulemusena saadakse kogu vajalik teave uuritava kemikaali ja hr-α-ERi omavahelise seondumise iseloomustamiseks (nimelt katsekannu lisatud [3H]17β-östradiooli üldkogus, [3H]17β-östradiooli maksimaalne seondumismäär hr-α-ERiga seondumisel, mittespetsiifilise seondumise määr ja summaarse seondumise määr uuritava kemikaali eri kontsentratsioonidel). Analüüsitsüklis võidakse igal kontsentratsioonil kasutada ka ainult ühte katsekannu (st paralleeli), ent kuna käesolevas katse-eeskirjas nõutakse kolme paralleelproovi kasutamist, analüüsitakse käesoleval juhul ühes analüüsitsüklis iga kontsentratsiooni puhul kolme katsekannu. Peale selle viiakse käesoleva katse-eeskirja kohaselt iga kemikaaliga läbi kolm iseseisvat (st järjestikust) analüüsitsüklit.
DCC
– dekstraaniga kaetud süsi.
E2
– märgistamata 17β-östradiool (inertne).
hr-α-ER
– rekombinantne inimese α-östrogeeniretseptor (ligandiga seonduv domeen).
Paralleelproov
– Üks mitmest kannust, mis sisaldavad samu koostisaineid samas kontsentratsioonis ning mida analüüsitakse üheaegselt samas analüüsitsüklis. Käesoleva katse-eeskirja puhul analüüsitakse uuritavat kemikaali igal kontsentratsioonil kolmes paralleelis, st igal uuritava kemikaali kontsentratsioonil analüüsitakse üheaegselt kolme paralleelproovi.
Liide 3.2
KONKUREERIVA SEONDUMISE KATSES KASUTATAV MIKROTIITERPLAADI SKEEM
|
Plaat
|
Asukoht
|
Paralleelproov
|
Kannu liik
|
Kannu kood
|
Kontsentratsiooni kood
|
Konkureeriva aine algkontsentratsioon (M)
|
hrERi põhilahus (μl)
|
Puhvri kogus (μl)
|
Märgise (radioaktiivse E2) kogus (μl)
|
Lahjendusplaadilt lisatav kogus (μl)
|
Lõppruumala (μl)
|
Konkureeriva aine lõppkontsentratsioon (M)
|
|
S
|
A1
|
1
|
Tühi kann
|
TK
|
TK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Tühi kann
|
TK
|
TK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Tühi kann
|
TK
|
TK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
Märgistamata E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B2
|
2
|
Märgistamata E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B3
|
3
|
Märgistamata E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
C1
|
1
|
Märgistamata E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C2
|
2
|
Märgistamata E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C3
|
3
|
Märgistamata E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
D1
|
1
|
Märgistamata E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D2
|
2
|
Märgistamata E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D3
|
3
|
Märgistamata E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
E1
|
1
|
Märgistamata E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E2
|
2
|
Märgistamata E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E3
|
3
|
Märgistamata E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
F1
|
1
|
Märgistamata E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F2
|
2
|
Märgistamata E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F3
|
3
|
Märgistamata E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
G1
|
1
|
Märgistamata E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G2
|
2
|
Märgistamata E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G3
|
3
|
Märgistamata E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
H1
|
1
|
Märgistamata E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H2
|
2
|
Märgistamata E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H3
|
3
|
Märgistamata E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
A4
|
1
|
Noretünodreel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A5
|
2
|
Noretünodreel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A6
|
3
|
Noretünodreel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B4
|
1
|
Noretünodreel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B5
|
2
|
Noretünodreel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B6
|
3
|
Noretünodreel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C4
|
1
|
Noretünodreel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C5
|
2
|
Noretünodreel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C6
|
3
|
Noretünodreel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
D4
|
1
|
Noretünodreel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D5
|
2
|
Noretünodreel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D6
|
3
|
Noretünodreel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E4
|
1
|
Noretünodreel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E5
|
2
|
Noretünodreel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E6
|
3
|
Noretünodreel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
F4
|
1
|
Noretünodreel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F5
|
2
|
Noretünodreel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F6
|
3
|
Noretünodreel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
G4
|
1
|
Noretünodreel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G5
|
2
|
Noretünodreel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G6
|
3
|
Noretünodreel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
H4
|
1
|
Noretünodreel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H5
|
2
|
Noretünodreel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H6
|
3
|
Noretünodreel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
A10
|
1
|
Summaarne seondumine
|
SS
|
SS1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
Summaarne seondumine
|
SS
|
SS2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
Summaarne seondumine
|
SS
|
SS3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
Summaarne seondumine
|
SS
|
SS4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B11
|
5
|
Summaarne seondumine
|
SS
|
SS5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
Summaarne seondumine
|
SS
|
SS6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
Märgistamata E2 (suur)
|
MSS
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C11
|
2
|
Märgistamata E2 (suur)
|
MSS
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C12
|
3
|
Märgistamata E2 (suur)
|
MSS
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D10
|
4
|
Märgistamata E2 (suur)
|
MSS
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D11
|
5
|
Märgistamata E2 (suur)
|
MSS
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D12
|
6
|
Märgistamata E2 (suur)
|
MSS
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E10
|
1
|
Puhvrikontroll
|
PK
|
PK1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
Puhvrikontroll
|
PK
|
PK2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
Puhvrikontroll
|
PK
|
PK3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
Puhvrikontroll
|
PK
|
PK4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
Puhvrikontroll
|
PK
|
PK5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F12
|
6
|
Puhvrikontroll
|
PK
|
PK6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
Tühiproov (radiomärgise jaoks)
|
Radiomärgis
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G11 (*17)
|
2
|
Tühiproov (radiomärgise jaoks)
|
Radiomärgis
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
Tühiproov (radiomärgise jaoks)
|
Radiomärgis
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
Tühiproov (radiomärgise jaoks)
|
Radiomärgis
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
Tühiproov (radiomärgise jaoks)
|
Radiomärgis
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12*
|
6
|
Tühiproov (radiomärgise jaoks)
|
Radiomärgis
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Tundmatu nr 1
|
T1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
Tundmatu nr 1
|
T1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
Tundmatu nr 1
|
T1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
Tundmatu nr 1
|
T1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B2
|
2
|
Tundmatu nr 1
|
T1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B3
|
3
|
Tundmatu nr 1
|
T1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C1
|
1
|
Tundmatu nr 1
|
T1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C2
|
2
|
Tundmatu nr 1
|
T1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C3
|
3
|
Tundmatu nr 1
|
T1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D1
|
1
|
Tundmatu nr 1
|
T1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D2
|
2
|
Tundmatu nr 1
|
T1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D3
|
3
|
Tundmatu nr 1
|
T1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E1
|
1
|
Tundmatu nr 1
|
T1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E2
|
2
|
Tundmatu nr 1
|
T1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E3
|
3
|
Tundmatu nr 1
|
T1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F1
|
1
|
Tundmatu nr 1
|
T1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F2
|
2
|
Tundmatu nr 1
|
T1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F3
|
3
|
Tundmatu nr 1
|
T1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G1
|
1
|
Tundmatu nr 1
|
T1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G2
|
2
|
Tundmatu nr 1
|
T1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G3
|
3
|
Tundmatu nr 1
|
T1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H1
|
1
|
Tundmatu nr 1
|
T1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H2
|
2
|
Tundmatu nr 1
|
T1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H3
|
3
|
Tundmatu nr 1
|
T1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A4
|
1
|
Tundmatu nr 2
|
T2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
Tundmatu nr 2
|
T2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
Tundmatu nr 2
|
T2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
Tundmatu nr 2
|
T2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B5
|
2
|
Tundmatu nr 2
|
T2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B6
|
3
|
Tundmatu nr 2
|
T2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C4
|
1
|
Tundmatu nr 2
|
T2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C5
|
2
|
Tundmatu nr 2
|
T2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C6
|
3
|
Tundmatu nr 2
|
T2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D4
|
1
|
Tundmatu nr 2
|
T2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D5
|
2
|
Tundmatu nr 2
|
T2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D6
|
3
|
Tundmatu nr 2
|
T2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E4
|
1
|
Tundmatu nr 2
|
T2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E5
|
2
|
Tundmatu nr 2
|
T2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E6
|
3
|
Tundmatu nr 2
|
T2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F4
|
1
|
Tundmatu nr 2
|
T2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F5
|
2
|
Tundmatu nr 2
|
T2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F6
|
3
|
Tundmatu nr 2
|
T2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G4
|
1
|
Tundmatu nr 2
|
T2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G5
|
2
|
Tundmatu nr 2
|
T2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G6
|
3
|
Tundmatu nr 2
|
T2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H4
|
1
|
Tundmatu nr 2
|
T2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H5
|
2
|
Tundmatu nr 2
|
T2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H6
|
3
|
Tundmatu nr 2
|
T2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A7
|
1
|
Tundmatu nr 3
|
T3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
Tundmatu nr 3
|
T3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
Tundmatu nr 3
|
T3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
Tundmatu nr 3
|
T3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B8
|
2
|
Tundmatu nr 3
|
T3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B9
|
3
|
Tundmatu nr 3
|
T3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C7
|
1
|
Tundmatu nr 3
|
T3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C8
|
2
|
Tundmatu nr 3
|
T3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C9
|
3
|
Tundmatu nr 3
|
T3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D7
|
1
|
Tundmatu nr 3
|
T3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D8
|
2
|
Tundmatu nr 3
|
T3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D9
|
3
|
Tundmatu nr 3
|
T3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E7
|
1
|
Tundmatu nr 3
|
T3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E8
|
2
|
Tundmatu nr 3
|
T3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E9
|
3
|
Tundmatu nr 3
|
T3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F7
|
1
|
Tundmatu nr 3
|
T3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F8
|
2
|
Tundmatu nr 3
|
T3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F9
|
3
|
Tundmatu nr 3
|
T3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G7
|
1
|
Tundmatu nr 3
|
T3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G8
|
2
|
Tundmatu nr 3
|
T3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G9
|
3
|
Tundmatu nr 3
|
T3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H7
|
1
|
Tundmatu nr 3
|
T3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H8
|
2
|
Tundmatu nr 3
|
T3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H9
|
3
|
Tundmatu nr 3
|
T3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A10
|
1
|
Kontroll: E2 (maks.)
|
S
|
E2maks1
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A11
|
2
|
Kontroll: E2 (maks.)
|
S
|
E2maks2
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A12
|
3
|
Kontroll: E2 (maks.)
|
S
|
E2maks3
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
B10
|
1
|
Kontroll: E2 (IC50)
|
S
|
E2IC501
|
E2 IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2 IC50
|
|
P1
|
B11
|
2
|
Kontroll: E2 (IC50)
|
S
|
E2IC502
|
E2 IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2 IC50
|
|
P1
|
B12
|
3
|
Kontroll: E2 (IC50)
|
S
|
E2IC503
|
E2 IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2 IC50
|
|
P1
|
C10
|
1
|
Kontroll: NE (maks.)
|
S
|
NEmaks1
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C11
|
2
|
Kontroll: NE (maks.)
|
S
|
NEmaks2
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C12
|
3
|
Kontroll: NE (maks.)
|
S
|
NEmaks3
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
D10
|
1
|
Kontroll: NE (IC50)
|
S
|
NEIC501
|
NE IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NE IC50
|
|
P1
|
D11
|
2
|
Kontroll: NE (IC50)
|
S
|
NEIC502
|
NE IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NE IC50
|
|
P1
|
D12
|
3
|
Kontroll: NE (IC50)
|
S
|
NEIC503
|
NE IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NE IC50
|
|
P1
|
E10
|
1
|
Märgistamata E2 (suur)
|
MSS
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E11
|
2
|
Märgistamata E2 (suur)
|
MSS
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E12
|
3
|
Märgistamata E2 (suur)
|
MSS
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F10
|
4
|
Märgistamata E2 (suur)
|
MSS
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F11
|
5
|
Märgistamata E2 (suur)
|
MSS
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F12
|
6
|
Märgistamata E2 (suur)
|
MSS
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
G10
|
1
|
Summaarne seondumine
|
SS
|
SS1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
Summaarne seondumine
|
SS
|
SS2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
Summaarne seondumine
|
SS
|
SS3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
Summaarne seondumine
|
SS
|
SS4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
Summaarne seondumine
|
SS
|
SS5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
Summaarne seondumine
|
SS
|
SS6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
4. liide
hrER-IL PÕHINEVA KONKUREERIVA SEONDUMISE KATSE ANDMETE ANALÜÜSIMISE KAALUTLUSED
|
1.
|
hr-α-ERil põhineva konkureeriva seondumise katse käigus mõõdetakse ühel kontsentratsioonil kasutatava [3H]17β-östradiooli seondumist üha suuremas kontsentratsioonis esineva uuritava kemikaali juuresolekul. Konkureeriva seondumise kõver esitatakse graafikul nii, et ühel teljel on [3H]17β-östradiooli spetsiifiline seondumine ja teisel konkureeriva aine kontsentratsioon (log10-ühikutes). Uuritava kemikaali kontsentratsioon, mille juures [3H]17β-östradiooli spetsiifiline seondumine on 50 % maksimummäärast, on IC50 väärtus.
|
Võrdlusöstrogeeni ja nõrgalt seonduva aine andmete analüüs (1)
|
2.
|
Kontrollidega saadud andmete edasiseks analüüsimiseks need transformeeritakse (st [3H]17β-östradiooli spetsiifilist seondumist väljendatakse protsentides ja kontrollkemikaali kontsentratsiooni logaritmituna). Positiivsete kontrollide (nt võrdlusöstrogeeni ja nõrgalt seonduva aine) log IC50 hinnangulise väärtuse kindlakstegemiseks tuleks kasutada sobivat mittelineaarse kõvera lähendamise tarkvara, milles rakendatakse neljaparameetrilist Hilli võrrandit (nt BioSoft; GraphPad Prism) (2). Kõnealuste kõverate lähendamisel tuleks kõvera ülemine ja alumine osa, tõus ja log IC50 üldjuhul jätta piiritlemata. Parima lähenduskõvera leidmiseks tuleks kasutada robustset regressioonanalüüsi, v.a juhul, kui esitatakse põhjendus muu lähenemisviisi kasutamise kohta. Tuleks märkida, millist robustse regressioonanalüüsi meetodit kasutatakse. Andmete korrigeerimine ligandi väljatõrjumise suhtes ei ole hrERil põhinevate FW ja CERI meetodite puhul vajalik, kuid seda võib vajaduse korral kaaluda. Pärast esmast analüüsi tuleks iga seondumiskõver üle vaadata, et veenduda mudeli ja andmete omavahelises sobivuses. Nõrgalt seonduva aine suhtelise seondumisafiinsuse (RBA) leidmiseks võib arvutada nõrgalt seonduva aine log IC50 väärtuse protsentuaalne osakaal 17β-östradiooli log IC50 väärtusest. Positiivsete kontrollidega ja mitteseonduval ainel põhineva kontrolliga saadud tulemusi tuleks hinnata lähtuvalt katsemeetodi tulemuslikkuse näitajatest ja nõuetekohasuse kriteeriumidest, mida on kirjeldatud käesolevas katsemeetodis (punkt 20), 2. liites (FW analüüsimeetod, punktid 41–51) ja 3. liites (CERI analüüsimeetod, punktid 41–51). Võrdlusöstrogeeni ja nõrgalt seonduva ainega läbi viidud kolme analüüsitsükli tulemusi hõlmavad näited on esitatud joonisel 1.
|
Joonis 1.
Võrdlusöstrogeeni ja nõrgalt seonduvat kontrollainet iseloomustavate konkureeriva seondumise kõverate näited

Uuritavate kemikaalide andmete analüüs
|
3.
|
Kõigi uuritavate kemikaalide andmete analüüsimisel tuleks kasutada astmelist lähenemist, et tagada andmete asjakohane analüüs ja kõigi konkureeriva seondumise kõverate nõuetekohane liigitamine. Uuritava kemikaaliga läbi viidud iga analüüsitsükli puhul tuleks esmalt rakendada täpselt sama standardiseeritud andmeanalüüsi, mida kasutatakse võrdlusöstrogeenil ja nõrgalt seonduval ainel põhinevate kontrollide puhul. Pärast esmast analüüsi tuleks iga analüüsitsükli puhul teha lähenduskõvera parameetrite tehniline kontroll ja visuaalselt hinnata, kui hästi sobituvad seondumisandmed saadud konkureeriva seondumise kõveraga. Tehnilise kontrolli käigus täheldatav kontsentratsioonist sõltuv [3H]17β-östradiooli spetsiifilise seondumise protsentuaalse määra vähenemine, tehniliste paralleelproovide andmete vaheline väike varieeruvus kemikaali kõigil kontsentratsioonidel ja lähenduskõvera parameetrite sarnased väärtused kolmes analüüsitsüklis viitavad sellele, et katse ja andmeanalüüs on läbi viidud nõuetekohaselt. Uuritava kemikaaliga igas analüüsitsüklis saadud andmete hindamisel tuleks lähtuda eksperdihinnangust ja tulemused, mille alusel iga uuritav kemikaal liigitatakse seonduvaks või mitteseonduvaks, peaksid olema teaduslikult põhjendatavad.
|
|
4.
|
Aeg-ajalt võib esineda andmeid, mis vajavad täiendavat tähelepanu, et hrERiga seondumise tulemusi oleks võimalik nõuetekohaselt analüüsida ja tõlgendada. Varasemates uuringutes on täheldatud juhtumeid, kus retseptoripõhise konkureeriva seondumise andmete analüüsi võib muuta keeruliseks asjaolu, et spetsiifilise seondumise protsentuaalne määr kemikaali suurimatel kontsentratsioonidel suureneb (joonis 2). See on tuntud probleem, millega on kokku puututud mitme retseptoripõhist konkureerivat seondumist käsitleva katse-eeskirja puhul (3). Sellistel juhtudel täheldatakse väiksematel kontsentratsioonidel kontsentratsioonist sõltuvust, kuid kui uuritava kemikaali kontsentratsioon läheneb lahustuvuspiirile, ei suurene [3H]17β-östradiooli väljatõrjumine enam. Sellisel juhul nähtub suurematel kontsentratsioonidel saadud andmetest, et on jõutud analüüsimeetodi bioloogilise kasutuspiirini. Näiteks on seda nähtust paljudel juhtudel seostatud kemikaali mittelahustuvusega ja väljasadenemisega suurel kontsentratsioonil, kuid see võib olla põhjustatud ka asjaolust, et dekstraaniga kaetud süsi ei ole kemikaali suurimatel kontsentratsioonidel võimeline siduma eraldamisetapis enam rohkem radiomärgistatud vaba ligandi. Selliste andmepunktide sissejätmine sigmoidkõvera lähendamisel konkureeriva seondumise katse andmetele võib vahel põhjustada uuritava kemikaali valesti liigitamist ERiga seondumise võime alusel (joonis 2). Sellise olukorra ärahoidmiseks on hrERiga seondumisel põhinevate FW ja CERI meetodite katse-eeskirjas nähtud ette võimalus jätta analüüsist välja andmepunktid, kus [3H]17β-östradiooli spetsiifilist seondumist iseloomustav paralleelproovide andmete keskväärtus on vähemalt 10 % suurem kui vastav keskväärtus mõnel väiksemal kontsentratsioonil (sellele viidatakse tavaliselt kui 10 % reeglile). Seda reeglit võib iga kõvera puhul kasutada ainult ühe korra ja seejuures peavad allesjäänud andmed hõlmama vähemalt kuut kontsentratsiooni, et kemikaali oleks võimalik kõvera alusel õigesti liigitada.
|
Joonis 2.
Näide konkureeriva seondumise kõvera kohta enne ja pärast 10 % reegli kasutamist

|
5.
|
Asjaomaste kõverate korrigeerimisel tuleks 10 % reegli asjakohast kasutamist hoolikalt kaaluda ja kasutada seda üksnes juhul, kui esineb selgeid viiteid selle kohta, et tegemist on hrERiga seonduva ainega. hrERiga seondumisel põhineva FW meetodi kohaste katsete tegemisel valideerimisuuringu raames täheldati, et 10 % reegli kasutamisel on mõnikord soovimatu ja ettenägematu tagajärg. Kemikaalide puhul, mis retseptoriga ei interakteeru (st tõelised mitteseonduvad ained), täheldati tasemel, kus radioligandi seondumine oli umbes 100 %, kasutatud kontsentratsioonide lõikes sageli varieeruvust, mis oli suurem kui 10 %. Kui väikseimale täheldatud väärtusele vastas juhtumisi väike kontsentratsioon, olnuks võimalik kõik sellest suuremal kontsentratsioonil saadud andmed 10 % reegli alusel analüüsist välja jätta, isegi kui kõnealustest andmetest võinuks olla kasu kemikaali liigitamisel mitteseonduvaks. Joonisel 3 on esitatud näited, mille puhul 10 % reegli kasutamine ei ole asjakohane.
|
Joonis 3.
Näited konkureeriva seondumise andmete kohta, mille puhul 10 % reegli kasutamine ei ole asjakohane

Viited
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα). Health and Safety Publications, Series on Testing and Assessment No. 226. Majanduskoostöö ja Arengu Organisatsioon, Pariis.
|
|
(2)
|
Motulsky, H., ja Christopoulos, A. (2003). The law of mass action. Väljaandes: Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA. Lk 187–191. www.graphpad.com/manuals/Prism4/RegressionBook.pdf.
|
|
(3)
|
Laws, S. C., Yavanhxay, S., Cooper, R. L., ja Eldridge, J. C. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicol. Sci. 94(1): 46–56.
|
B.71 NAHA SENSIBILISEERIMISE IN VITRO KATSED, MILLEGA UURITAKSE DENDRIITRAKKUDE AKTIVEERIMISEGA SEOTUD OLULIST ETAPPI NAHA SENSIBILISEERIMISES SEISNEVA KAHJULIKU TOIME RAJAS
ÜLDINE SISSEJUHATUS
Dendriitrakkude aktiveerimisega seotud olulisel etapil põhinev katsemeetod
|
1.
|
Naha sensibilisaator on aine, mis põhjustab nahaga kokkupuutel allergilise reaktsiooni, nagu on määratletud ÜRO ühtses ülemaailmses kemikaalide klassifitseerimise ja märgistamise süsteemis (ÜRO GHS) (1) ja Euroopa Liidu määruses (EL) 1272/2008, milles käsitletakse ainete ja segude klassifitseerimist, märgistamist ja pakendamist (CLP-määrus) (98). Naha sensibiliseerimise oluliste bioloogiliste etappide suhtes on olemas ühisarusaam. Praegused teadmised naha sensibiliseerimisega seotud keemiliste ja bioloogiliste mehhanismide kohta on kokku võetud kahjuliku toime radu käsitleva OECD programmiga hõlmatud asjaomase kahjuliku toime raja käsitluses (2), milles kirjeldatakse nii protsessi käivitavat molekulaarset sündmust, järgnevaid vaheetappe kui ka kahjuliku toime, st allergilise kontaktdermatiidi avaldumist. Kõnealusel juhul on käivitav molekulaarne sündmus (st esimene oluline etapp) elektrofiilsete ainete kovalentne seondumine naha valkude nukleofiilsete tsentritega. Kahjuliku toime raja teine oluline etapp toimub keratinotsüütides ning hõlmab nii põletikureaktsioone kui ka geeniekspressiooni muutusi, mis on seotud selliste konkreetsete signaaliülekanderadadega nagu antioksüdatiivse/elektrofiiliseoselise reaktsiooni elemendist (ARE) sõltuvad rajad. Kolmas oluline etapp on dendriitrakkude aktiveerimine, mida hinnatakse tavaliselt spetsiifiliste raku pinnamarkerite, kemokiinide ja tsütokiinide ekspressiooni alusel. Neljas oluline etapp on T-rakkude aktiveerimine ja paljunemine, mida hinnatakse kaudselt hiire lokaalsete lümfisõlmede katses (LLNA) (3).
|
|
2.
|
Käesolev katsemeetod on samaväärne OECD katsejuhendiga nr 442E (2017). Selles kirjeldatakse in vitro katseid, millega uuritakse naha sensibiliseerimises seisneva kahjuliku toime rajas dendriitrakkude aktiveerimisega seotud olulise etapi puhul kirjeldatud mehhanisme (2). Katsemeetodit kasutatakse selleks, et aidata kooskõlas ÜRO GHSi ja CLP-määrusega eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri.
|
|
Käesolevas katsemeetodis kirjeldatakse järgmisi katseid:
|
—
|
inimese rakuliini aktiveerimise katse (h-CLAT);
|
|
—
|
rakuliini U937 aktiveerimise katse (U-SENS™);
|
|
—
|
interleukiin 8 reportergeeni katse (IL-8 lutsiferaasikatse).
|
|
|
|
3.
|
Käesolev katsemeetod ja OECD vastav katsejuhend võivad erineda andmete ja mõõdetavate näitajate saamiseks kasutatavate menetluste poolest, kuid OECD otsuse kohase andmete vastastikuse tunnustamise raames võib kasutada ükskõik kumba meetodit, et saada asjaomases riigis kehtivate nõuete kohaseid katsetulemusi naha sensibiliseerimises seisneva kahjuliku toime rajas dendriitrakkude aktiveerimisega seotud olulise etapi kohta.
|
Olulisel etapil põhineva katsemeetodi kohaste katsete taust ja põhimõtted
|
4.
|
Nahka sensibiliseeriva toime hindamiseks on tavaliselt kasutatud laboriloomi. Merisigade kasutamisel põhinevate klassikaliste meetoditega – merisigadega tehtava Magnussoni ja Kligmani maksimeerimiskatse ja Buehleri katsega (katsemeetod B.6) (4) – uuritakse nii naha sensibiliseerimise induktsiooni- kui ka esilekutsumisfaasi. Hiirtega läbi viidavate katsetega – LLNA (katsemeetod B.42) (3) ja selle kahe radioaktiivsest märgisest loobumist võimaldava modifikatsiooniga (LLNA: DA (katsemeetod B.50) (5) ja LLNA: BrdU-ELISA (katsemeetod B.51) (6)) – hinnatakse ainult indutseerivat toimet ja need on samuti heaks kiidetud, sest need on merisigadega tehtavate katsetega võrreldes eelistatud nii loomade heaolu seisukohast kui ka seepärast, et nendega saab naha sensibiliseerimise induktsioonifaasi objektiivselt mõõta.
|
|
5.
|
Viimasel ajal on kemikaalidest tingitud naha sensibiliseerimise ohu hindamiseks kasutusele võetud mehhanistlikul käsitlusel põhinevad in chemico ja in vitro katsemeetodid, millega uuritakse naha sensibiliseerimises seisneva kahjuliku toime raja esimest olulist etappi (katsemeetod B.59: otsene reaktsioonivõime katse peptiididega (7)) ja teist olulist etappi (katsemeetod B.60: ARE-Nrf2 lutsiferaasikatse (8)).
|
|
6.
|
Käesoleva meetodi kohaste katsetega tehakse kvantitatiivselt kindlaks raku pinnamarkeri(te) (nt CD54, CD86) ekspressiooni muutus monotsüütide ja dendriitrakkude aktiveerimisel pärast kokkupuudet sensibilisaatoritega või dendriitrakkude aktiveerimisega seotud tsütokiini IL-8 ekspressiooni muutus. Naha sensibilisaatorite kohta on teatatud, et need indutseerivad selliste rakumembraani markerite nagu CD40, CD54, CD80, CD83 ja CD86 ekspressiooni, lisaks kutsuvad need esile dendriitrakkude aktiveerimisega seotud (2), põletikku soodustavate tsütokiinide, näiteks IL-1β ja TNF-α, ning mitme kemokiini, näiteks IL-8 (CXCL8) ja CCL3 ekspressiooni (9, 10, 11, 12).
|
|
7.
|
Ent kuna dendriitrakkude aktiveerimine on ainult üks oluline etapp naha sensibiliseerimises seisneva kahjuliku toime rajas (2, 13), ei pruugi üksnes dendriitrakkude aktiveerimise markerite mõõtmisel põhinevate katsetega kogutud teave olla piisav, et teha järeldusi kemikaali nahka sensibiliseeriva toime olemasolu või puudumise kohta. Seepärast ollakse seisukohal, et käesoleva meetodiga saadud andmed võimaldavad toetada naha sensibilisaatorite (st ÜRO GHSi / CLP-määruse kohaselt 1. kategooria kemikaalide) eristamist nahka mitte sensibiliseerivatest kemikaalidest, kui neid andmeid kasutatakse katsete tegemist ja hindamist käsitleva ühtse lähenemisviisi (edaspidi „ühtne lähenemisviis“) raames koos asjakohase täiendava teabega, näiteks teabega, mis on saadud naha sensibiliseerimises seisneva kahjuliku toime raja teisi olulisi etappe hõlmavate in vitro katsetega ning muude mittekatseliste meetoditega, sealhulgas võrdlemisel kemikaali analoogidega (13). On avaldatud näiteid kõnealuste katsetega saadud andmete kasutamisest vastavalt kindlaksmääratud metoodikale, mille puhul on standarditud nii kasutatavate teabeallikate kogum kui ka andmete töötlemise viis prognooside tegemiseks (13), ning neid saab rakendada ühtse lähenemisviisi kasulike osadena.
|
|
8.
|
Käesoleva katsemeetodi kohaseid katseid ei saa kasutada eraldiseisvana selleks, et liigitada naha sensibilisaatoreid ÜRO GHSis / CLP-määruses määratletud alamkategooriatesse 1A või 1B ametiasutuste jaoks, kes neid mittekohustuslikke alamkategooriaid kasutavad, ega ka selleks, et prognoosida toime tugevust ohutushindamisega seotud otsuste tegemiseks. Sõltuvalt õigusraamistikust võib kõnealuste meetoditega saadud positiivseid tulemusi siiski kasutada eraldiseisvana kemikaali klassifitseerimiseks ÜRO GHSi / CLP-määruse kohasesse 1. kategooriasse.
|
|
9.
|
Käesolevas katsemeetodis kasutatakse mõistet „uuritav kemikaal“ uurimisobjektile viitava mõistena (99) ning see ei ole seotud meetodi sobivusega ühest või mitmest koostisosast koosnevate ainete ja/või segude uurimiseks. Praegu on vähe andmeid meetodi kohaldatavuse kohta mitmest koostisosast koosnevate ainete ja segude puhul (14, 15). Kõnealuste katsete kasutamine mitmest koostisosast koosnevate ainete ja segude uurimiseks on tehniliselt võimalik. Enne katsemeetodi kasutamist selleks, et saada kavandatud regulatiivsel eesmärgil andmeid segu kohta, tuleks siiski kaaluda, kas ja kuidas see meetod võimaldab saada taotletava eesmärgi jaoks vastuvõetavaid tulemusi. (100) Seda ei ole vaja kaaluda, kui asjaomase segu hindamine on regulatiivse nõudega ette nähtud. Peale selle tuleks mitmest koostisosast koosnevate ainete ja segude uurimisel võtta arvesse tsütotoksiliste koostisosade võimalikku segavat mõju vaadeldavatele toimetele.
|
KIRJANDUS
|
(1)
|
Ühinenud Rahvaste Organisatsioon (ÜRO) (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Kuues, täiendatud väljaanne. New York ja Genf: United Nations Publications. ISBN: 978-92-1-117006-1. Kättesaadav aadressil https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Kättesaadav aadressil http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Käesoleva lisa peatükk B.42 „Naha sensibiliseerimine: lokaalsete lümfisõlmede katse“.
|
|
(4)
|
Käesoleva lisa peatükk B.6 „Naha sensibiliseerimine“.
|
|
(5)
|
Käesoleva lisa peatükk B.50 „Naha sensibiliseerimine: lokaalsete lümfisõlmede katse: DA“.
|
|
(6)
|
Käesoleva lisa peatükk B.51 „Naha sensibiliseerimine: lokaalsete lümfisõlmede katse: BrdU-ELISA“.
|
|
(7)
|
Käesoleva lisa peatükk B.59 „Naha sensibiliseerimine in chemico: otsene reaktsioonivõime katse peptiididega (direct peptide reactivity assay, DPRA)“.
|
|
(8)
|
Käesoleva lisa peatükk B.60 „Naha sensibiliseerimine in vitro: ARE-Nrf2 lutsiferaasi katsemeetod“.
|
|
(9)
|
Steinman, R. M. (1991). The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 9: 271–296.
|
|
(10)
|
Caux, C., Vanbervliet, B., Massacrier, C., Azuma, M., Okumura, K., Lanier, L. L., ja Banchereau, J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J. Exp. Med. 180: 1841–1847.
|
|
(11)
|
Aiba, S., Terunuma, A., Manome, H., ja Tagami, H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur. J. Immunol. 27: 3031–3038.
|
|
(12)
|
Aiba, S., Manome, H., Nakagawa, S., Mollah, Z. U., Mizuashi, M., Ohtani, T., Yoshino, Y., ja Tagami, H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J. Invest. Dermatol. 120: 390–398.
|
|
(13)
|
OECD (2016). Series on Testing and Assessment No 256. Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga, T., Sakaguchi, H., Sono, S., Kosaka, N., Ishikawa, M., Nukada, Y., Miyazawa, M., Ito, Y., Nishiyama, N., ja Itagaki, H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38: 275–284.
|
|
(15)
|
Piroird, C., Ovigne, J. M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., ja Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29: 901–916.
|
1. liide
NAHA SENSIBILISEERIMINE IN VITRO: INIMESE RAKULIINI AKTIVEERIMISE KATSE (H-CLAT)
LÄHTEKAALUTLUSED JA PIIRANGUD
|
1.
|
Inimese rakuliini aktiveerimise katses tehakse inimese monotsüütse leukeemia rakuliinis THP-1 kindlaks monotsüütide ja dendriitrakkude aktiveerimisega seotud raku pinnamarkerite (CD86 ja CD54) ekspressiooni kvantitatiivsed muutused pärast kokkupuudet sensibilisaatoriga (1, 2). Seejärel kasutatakse raku pinnamarkerite CD86 ja CD54 ekspressiooni mõõdetud taset toetava teabena, mis aitab eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri.
|
|
2.
|
h-CLATd on hinnatud loomkatsete alternatiivide Euroopa Liidu referentlabori (EURL ECVAM) koordineeritud valideerimisuuringus ning seejärel on selle sõltumatult läbi vaadanud EURL ECVAMi teaduslik nõuandekomitee. Võttes arvesse kõiki kättesaadavaid andmeid ning reguleerivate asutuste ja sidusrühmade seisukohti, soovitas EURL ECVAM kasutada h-CLATd (3) ühtse lähenemisviisi osana, et aidata ohu alusel klassifitseerimise ja märgistamise eesmärgil eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri. Näited h-CLATga saadud andmete kasutamisest muu teabega kombineerituna on avaldatud kirjanduses (4, 5, 6, 7, 8, 9, 10, 11).
|
|
3.
|
h-CLAT on osutunud rakendatavaks laborites, kus on olemas rakukultuurimeetodite kasutamise ja läbivoolutsütomeetrilise analüüsi kogemus. Selle meetodi puhul eeldatav prognooside reprodutseeritavuse määr on nii laborisisese kui ka laboritevahelise reprodutseeritavuse puhul ligikaudu 80 % (3, 12). Valideerimisuuringust (13) ja muudest avaldatud uuringutulemustest (14) nähtub üldiselt, et LLNA tulemustega võrreldes on selles katses naha sensibilisaatorite (st ÜRO GHSi / CLP-määruse kohaselt 1. kategooria kemikaalide) mittesensibiliseerivatest kemikaalidest eristamise täpsus 85 % (N = 142), tundlikkus 93 % (94/101) ja spetsiifilisus 66 % (27/41) (vastavalt EURL ECVAMi kordusanalüüsile (12), milles võeti arvesse kõiki olemasolevaid andmeid ning vastavalt punktis 4 esitatud kirjeldusele jäeti arvestamata negatiivsed tulemused selliste kemikaalide puhul, mille log Kow on suurem kui 3,5). h-CLAT kohaste valenegatiivsete prognooside tõenäosus on suurem vähesel määral kuni mõõdukalt nahka sensibiliseerivate (st ÜRO GHSi / CLP-määruse kohaselt alamkategooria 1B) kemikaalide puhul ja väiksem siis, kui tegemist on tugevalt nahka sensibiliseeriva (st ÜRO GHSi / CLP-määruse kohaselt alamkategooria 1A) kemikaaliga (4, 13, 15). Kokkuvõttes nähtub sellest teabest, et h-CLAT on kasulik meetod, mis aitab kindlaks teha naha sensibiliseerimise ohtu. Ainsa meetodina kasutatava h-CLATga saavutatav eespool nimetatud täpsus on siiski üksnes orienteeriv, sest katseandmeid tuleks vaadelda ühtse lähenemisviisi kontekstis koos muudest allikatest pärit teabega ja kooskõlas üldise sissejuhatuse punktide 7 ja 8 sätetega. Kui hinnatakse naha sensibiliseerimise katsemeetodeid, mille puhul ei kasutata katseloomi, tuleb lisaks arvesse võtta, et LLNA ja muude loomkatsete tulemused ei pruugi täielikult kajastada kemikaali toimet inimesele.
|
|
4.
|
Praeguse olemasoleva teabe põhjal on tõendatud, et h-CLAT on kohaldatav paljude uuritavate kemikaalide puhul, mis erinevad üksteisest orgaanilise funktsionaalrühma, toimemehhanismi, in vivo katsetega määratud nahka sensibiliseeriva toime tugevuse ja füüsikalis-keemiliste omaduste poolest (3, 14, 15). h-CLAT on kohaldatav uuritavate kemikaalide puhul, mis on sobivas lahustis/kandeaines lahustuvad või moodustavad selles püsiva dispersiooni (st kolloidlahuse või suspensiooni, milles uuritav kemikaal ei sadestu ega eraldu lahustist/kandeainest eri faasi) (vt punkt 14). Kui uuritava kemikaali log Kow on suurem kui 3,5, saadakse sageli valenegatiivsed tulemused (14). Seepärast tuleks uuritavate kemikaalide puhul, mille log Kow on suurem kui 3,5, jätta negatiivsed tulemused arvesse võtmata. Kui uuritava kemikaali log Kow on üle 3,5, võib saadud positiivseid tulemusi sellegipoolest kasutada uuritava kemikaali naha sensibilisaatoriks liigitamise toetamiseks. Peale selle võidakse kasutatud rakuliini piiratud metaboliseerimisvõime (16) või katsetingimuste tõttu saada h-CLATga negatiivseid tulemusi ka prohapteenide (st ained, mis vajavad ensümaatilist aktiveerimist, nt P450 ensüümide abil) ja prehapteenide (st ained, mis aktiveeruvad oksüdeerumisel) puhul, eelkõige aeglaselt oksüdeeruvad ainete puhul (15). h-CLAT võimaldab hinnata ka fluorestseeruvaid uuritavaid kemikaale (17), kuid tugevalt fluorestseeruvad uuritavad kemikaalid, mille kiiratava kiirguse lainepikkus on sama kui fluorestseiinisotiotsüanaadil (FITC) või propiidiumjodiidil (PI), segavad läbivoolutsütomeetrilist analüüsi ning neid ei ole võimalik FITCga konjugeeritud antikehade või PI abil õigesti hinnata. Sellisel juhul võib kasutada vastavalt muu fluorokroomiga märgistatud antikehasid või muud tsütotoksilisuse markerit, kui näiteks katsetega, mis tehakse liites 1.2 loetletud pädevusainetega, on võimalik tõendada, et sellise fluorokroomi või markeriga saadakse samasugused tulemused kui FITCga märgistatud antikehade (vt punkt 24) või PIga (vt punkt 18). Eespool kirjeldatust lähtuvalt tuleks negatiivseid tulemusi tõlgendada kõnealuste piirangute kontekstis ja koos ühtse lähenemisviisi raames muudest allikatest saadud teabega. Kui leidub andmeid, mille kohaselt h-CLATd ei saa kasutada mõne muu konkreetse uuritavate kemikaalide rühma puhul, ei tohiks seda asjaomase konkreetse kemikaalirühma puhul kasutada.
|
|
5.
|
Nagu eespool kirjeldatud, aitab h-CLAT eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri. Ent kui seda kasutatakse näiteks katsete tegemist ja hindamist käsitleva ühtse lähenemisviisi raames, võib see aidata hinnata ka sensibiliseeriva toime tugevust (4, 5, 9). Võimalust kasutada h-CLATga saadud tulemusi toime tugevuse hindamisel on siiski vaja täiendavalt uurida, soovitatavalt inimuuringutest saadud andmete alusel.
|
|
6.
|
Mõisted on esitatud liites 1.1.
|
KATSE PÕHIMÕTE
|
7.
|
h-CLAT on in vitro meetod, millega määratakse kvantitatiivselt raku pinnamarkerite (CD86 ja CD54) ekspressiooni muutusi inimese monotsüütse leukeemia rakuliini THP-1 rakkudes pärast 24-tunnist kokkupuudet uuritava kemikaaliga. Kõnealused pinnamolekulid on monotsüütse THP-1 tüüpilised markerid ja võivad võimaldada jäljendada dendriitrakkude aktiveerimist, millel on väga oluline roll T-rakkude aktivatsioonis. Pinnamarkerite ekspressiooni muutusi mõõdetakse läbivoolutsütomeetriliselt pärast rakkude värvimist fluorokroomiga märgistatud antikehadega. Samal ajal mõõdetakse ka tsütotoksilisust, et hinnata, kas pinnamarkerite ekspressiooni suurenemine toimub kontsentratsioonil, mille juures ei täheldata tsütotoksilisust. Arvutatakse pinnamarkerite fluorestsentssignaali suhteline tugevus lahustiga/kandeainega kontrolli näitajatega võrrelduna ja seda tulemust kasutatakse prognoosimudelis (vt punkt 26), et aidata eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri.
|
PÄDEVUSE TÕENDAMINE
|
8.
|
Enne katsemeetodi B.71 juurde kuuluvas käesolevas liites kirjeldatud meetodi rutiinset kasutamist tuleks tõendada labori tehnilist pädevust ning kasutada selleks liites 1.2 loetletud kümmet pädevusainet. Lisaks peaksid meetodi kasutajad haldama reageerimisvõime kontrollimise käigus kogutud andmeid (vt punkt 11) ning positiivsete ja lahustiga/kandeainega kontrollidega saadud varasemaid andmeid (vt punktid 20–22) sisaldavat andmebaasi ning kontrollima nende andmete põhjal, kas katsetulemused on laboris aja möödudes jätkuvalt reprodutseeritavad.
|
KATSE KÄIK
|
9.
|
Käesolev meetod põhineb loomkatsetele alternatiivsete meetodite andmebaasi (DataBase on ALternative Methods, DB-ALM) teenusena kättesaadaval h-CLATd käsitleval katse-eeskirjal nr 158 (18), mis vastab EURL ECVAMi koordineeritud valideerimisuuringus kasutatud katse-eeskirjale. Seda katse-eeskirja soovitatakse kasutada laboris h-CLAT juurutamisel ja rakendamisel. Allpool kirjeldatakse h-CLAT põhikomponente ja katse käiku, mis koosneb kahest etapist: annuse kindlaksmääramise katsest ja CD86/CD54 ekspressiooni mõõtmisest.
|
Rakkude ettevalmistamine
|
10.
|
h-CLAT puhul tuleks kasutada inimese monotsüütse leukeemia rakuliini THP-1. Soovitatavalt tuleks rakud (TIB-202™) soetada usaldusväärsest rakupangast, näiteks Ameerika Tüüpkultuuride Kollektsioonist (American Type Culture Collection).
|
|
11.
|
THP-1 rakke kasvatatakse 37oC juures reguleeritud õhuniiskusega 5-protsendilise CO2-sisaldusega atmosfääris söötmes RPMI-1640, mis sisaldab 10 % veiselooteseerumit, 2-merkaptoetanooli kontsentratsioonis 0,05 mM, 100 ühikut penitsilliini milliliitri kohta ja streptomütsiini kontsentratsioonis 100 μg/ml. Penitsilliini ja streptomütsiini kasutamisest söötmes võib loobuda. Sel juhul peavad kasutajad siiski kontrollima, et antibiootikumide puudumine söötmest ei mõjuta tulemusi. Näiteks võib selleks teha katseid liites 1.2 loetletud pädevusainetega. Igal juhul tuleb saastumisohu vähendamiseks järgida rakukultuuridega töötamise head tava olenemata sellest, kas rakukultuuri sööde sisaldab antibiootikume või mitte. THP-1 rakke külvatakse regulaarselt iga 2–3 päeva järel ümber algtihedusega 0,1–0,2 × 106 rakku/ml. Kultuure tuleb säilitada tihedusvahemikus 0,1–1,0 × 106 rakku/ml. Enne rakkude kasutamist katses tuleb teha reageerimisvõime kontroll, et veenduda rakkude sobivuses. Rakkude reageerimisvõime kontroll tuleks teha kaks nädalat pärast sulatamist ning kasutada selleks positiivse kontrollina 2,4-dinitroklorobenseeni (DNCB) (CASi nr: 97-00-7; puhtus ≥ 99 %) ja nikkelsulfaati (NiSO4) (CASi nr: 10101-97-0; puhtus ≥ 99 %) ning negatiivse kontrollina piimhapet (CASi nr: 50-21-5; puhtus ≥ 85 %). Nii DNCB kui ka NiSO4 peaksid esile kutsuma raku pinnamarkerite CD86 ja CD54 alusel tuvastatava positiivse reaktsiooni, piimhape aga negatiivse reaktsiooni. Katses võib kasutada ainult reageerimisvõime kontrolli läbinud rakke. Rakke võib paljundada kuni kahe kuu jooksul pärast sulatamist. Passaažide arv ei tohiks ületada 30. Reageerimisvõime kontroll tuleb teha vastavalt punktides 20–24 esitatud kirjeldusele.
|
|
12.
|
Katses kasutamiseks külvatakse THP-1 rakud välja tihedusega 0,1 × 106 rakku/ml või 0,2 × 106 rakku/ml ning eelkasvatatakse neid rakukultuuripudelis vastavalt 72 või 48 tundi. Rakkude tihedus rakukultuuripudelis peaks vahetult pärast eelkasvatamise perioodi lõppu olema igas katses võimalikult sarnane (selle saavutamiseks kasutatakse ühte kahest eespool kirjeldatud eelkasvatamise tingimusest), sest rakkude tihedus pudelis vahetult pärast eelkasvatamist võib mõjutada allergeenide indutseeritavat CD86/CD54 ekspressiooni (19). Katse päeval eemaldatakse rakud rakukultuuripudelist ja suspendeeritakse uuesti värskes söötmes tihedusel 2 × 106 rakku/ml. Seejärel jaotatakse rakud 500 μl kaupa 24-kannulise lamedapõhjalise plaadi kannudesse (1 × 106 rakku kannu kohta) või 80 μl kaupa 96-kannulise lamedapõhjalise plaadi kannudesse (1,6 × 105 rakku kannu kohta).
|
Annuse kindlaksmääramise katse
|
13.
|
Annuse kindlaksmääramise katse eesmärk on leida CV75 väärtus ehk uuritava kemikaali kontsentratsioon, mille juures rakkude eluvõimelisus on 75 % lahustiga/kandeainega kontrolli puhul täheldatud väärtusest. CV75 väärtuse põhjal tehakse kindlaks CD86/CD54 ekspressiooni mõõtmisel (vt punktid 20–24) kasutatav uuritava kemikaali kontsentratsioon.
|
Uuritavate kemikaalide ja kontrollainete ettevalmistamine
|
14.
|
Uuritavad kemikaalid ja kontrollained valmistatakse ette katse tegemise päeval. h-CLAT puhul tehakse uuritavatest kemikaalidest lahus või püsiv dispersioon (vt ka punkt 4); lahustina/kandeainena kasutatakse eelistatavalt füsioloogilist lahust või söödet või kui uuritav kemikaal ei ole nendes lahustuv või ei moodusta neis püsivat dispersiooni, siis dimetüülsulfoksiidi (DMSO, puhtusega ≥ 99 %). Kemikaali lõppkontsentratsioon peab olema 100 mg/ml (füsioloogilises lahuses või söötmes) või 500 mg/ml (DMSOs). Kui see on teaduslikult piisavalt põhjendatud, võib kasutada ka eespool nimetatutest erinevat lahustit/kandeainet. Arvesse tuleks võtta uuritava kemikaali püsivust lahustis/kandeaines lõppkontsentratsioonil.
|
|
15.
|
Uuritava kemikaali põhilahust kontsentratsiooniga 100 mg/ml (füsioloogilises lahuses või söötmes) või 500 mg/ml (DMSOs) lahjendatakse järgmiselt.
|
—
|
Lahustina/kandeainena füsioloogilise lahuse või söötme kasutamisel: asjaomase lahustiga/kandeainega valmistatakse kahekordsete lahjenduste reana kaheksa eri kontsentratsiooniga põhilahust. Seejärel lahjendatakse neid põhilahuseid söötmes veel 50 korda (töölahused). Kui plaadil kasutataval suurimal lõppkontsentratsioonil 1 000 μg/ml ei täheldata mürgisust, tuleks maksimaalne kontsentratsioon uue tsütotoksilisuse katsega uuesti kindlaks määrata. Füsioloogilises lahuses või söötmes lahustatud või püsiva dispersiooni moodustanud uuritava kemikaali lõppkontsentratsioon plaadil ei tohiks ületada 5 000 μg/ml.
|
|
—
|
Lahustina/kandeainena DMSO kasutamisel: asjaomase lahustiga/kandeainega valmistatakse kahekordsete lahjenduste reana kaheksa eri kontsentratsiooniga põhilahust. Seejärel lahjendatakse neid põhilahuseid söötmes veel 250 korda (töölahused). Lõppkontsentratsioon plaadil ei tohiks ületada 1 000 μg/ml, isegi kui sellel kontsentratsioonil ei täheldata mürgisust.
|
Seejärel kasutatakse töölahuseid kokkupuutekatses: plaadil olevale THP-1 rakkude suspensioonile lisatakse võrdne kogus töölahust (vt ka punkt 17), et saavutada täiendav kahekordne lahjendus (tavaliselt on lõppkontsentratsioonide vahemik plaadil 7,81–1 000 μg/ml).
|
|
16.
|
h-CLAT puhul kontrollina kasutatav lahusti/kandeaine on sööde (kui uuritavast kemikaalist tehakse lahus või püsiv dispersioon (vt punkt 4) söötmes või füsioloogilises lahuses) või DMSO (kui uuritavast kemikaalist tehakse lahus või püsiv dispersioon DMSOs), mida kasutatakse plaadil ühes lõppkontsentratsioonis 0,2 %. Seda lahjendatakse samal viisil, nagu on kirjeldatud töölahuste saamist käsitlevas punktis 15.
|
Uuritavate kemikaalide ja kontrollainetega töötlemine
|
17.
|
Punktides 15 ja 16 kirjeldatud sööde ja töölahused segatakse 24- või 96-kannulisele lamedapõhjalisele plaadile kantud rakususpensiooniga (vt punkt 12) mahuvahekorras 1:1. Seejärel inkubeeritakse töödeldud plaate 37oC juures 5-protsendilise CO2-sisaldusega atmosfääris 24 ± 0,5 tundi. Tuleb jälgida, et lenduvad uuritavad kemikaalid ei aurustuks, ning hoida ära kannudevahelist uuritava kemikaaliga ristsaastumist. Selleks võib plaadi enne uuritava kemikaaliga inkubeerimist näiteks tihedalt kinni katta (20).
|
Propiidiumjodiidiga (PI) värvimine
|
18.
|
Pärast 24 ± 0,5 tundi kestnud kokkupuudet viiakse rakud katsutitesse ja eraldatakse tsentrifuugimise teel. Supernatant visatakse ära ja allesjäänud rakud suspendeeritakse uuesti 200 (96-kannulise plaadi puhul) või 600 (24-kannulise plaadi puhul) mikroliitris fosfaadiga puhverdatud soolalahuses, mis sisaldab 0,1 % veise seerumi albumiini (värvimispuhver). 200 μl rakususpensiooni viiakse üle 96-kannulisele ümarapõhjalisele plaadile (96-kannulise plaadi puhul) või mikrokatsutisse (24-kannulise plaadi puhul) ning rakke pestakse kaks korda 200 μl (96-kannulise plaadi puhul) või 600 μl (24-kannulise plaadi puhul) värvimispuhvriga. Lõpuks suspendeeritakse rakud uuesti värvimispuhvris (nt 400 μl) ning lisatakse PI lahus (nt 20 μl; PI lõppkontsentratsioon võib olla näiteks 0,625 μg/ml). Võib kasutada ka muid tsütotoksilisuse markereid, näiteks 7-aminoaktinomütsiin D-d (7-AAD) või trüpaansinist, kui näiteks katsetega, mis tehakse liites 1.2 loetletud pädevusainetega, on võimalik tõendada, et asjaomase muu värvainega ja PIga saadavad tulemused on sarnased.
|
Tsütotoksilisuse mõõtmine läbivoolutsütomeetriga ja CV75 väärtuse hindamine
|
19.
|
PI sisenemist rakkudesse analüüsitakse läbivoolutsütomeetriga; seejuures kasutatakse registreerimiskanalit FL3. Kokku registreeritakse 10 000 elusat (PIga värvumata) rakku. Tsütomeetri analüüsiprogrammi abil rakkude eluvõimelisuse arvutamiseks võib kasutada allpool esitatud valemit. Kui rakkude eluvõimelisus on väike, tuleks registreerida koos surnud rakkudega kokku kuni 30 000 rakku. Teine võimalus on registreerida andmeid ühe minuti jooksul alates analüüsi alustamise hetkest.

CV75 väärtus (vt punkt 13) ehk kontsentratsioon, mille juures elusate THP-1 rakkude osakaal on 75 % (tsütotoksilisus on 25 %), arvutatakse log-lineaarse interpolatsiooni teel järgmise valemiga:,
kus
a on rakkude eluvõimelisuse vähim väärtus, mis ületab 75 %,
c on rakkude eluvõimelisuse suurim väärtus, mis jääb alla 75 %, ning
b ja d on kontsentratsioonid, mille juures rakkude eluvõimelisus on vastavalt a või c.

CV75 leidmiseks on lubatud kasutada ka muid meetodeid, kui on tõendatud (näiteks katsetega, mis on tehtud pädevusainetega), et see ei mõjuta tulemusi.
|
CD86/CD54 ekspressiooni mõõtmine
Uuritavate kemikaalide ja kontrollainete ettevalmistamine
|
20.
|
Uuritava kemikaali lahuse või püsiva dispersiooni valmistamiseks kasutatakse sobivat lahustit/kandeainet (füsioloogiline lahus, sööde või DMSO; vt punkt 14). Kõigepealt lahjendatakse uuritavat kemikaali kontsentratsioonini, mis on 100 korda (füsioloogilise lahuse või söötme puhul) või 500 korda (DMSO puhul) suurem kui 1,2 × CV75, mis on kindlaks tehtud annuse kindlaksmääramise katsega (vt punkt 19). Kui CV75 väärtust ei ole võimalik kindlaks teha (nt annuse kindlaksmääramise katses ei täheldata piisavat tsütotoksilisust), tuleks lähtekontsentratsioonina kasutada suurimat kontsentratsiooni, mille juures uuritav kemikaal on asjaomases lahustis/kandeaines veel lahustuv või moodustab selles püsiva dispersiooni. Tuleb silmas pidada, et lõppkontsentratsioon plaadil ei tohi olla suurem kui 5 000 μg/ml (füsioloogilise lahuse või söötme puhul) või 1 000 μg/ml (DMSO puhul). Seejärel tehakse vastava lahustiga/kandeainega 1,2-kordsete lahjenduste rida, et saada h-CLAT puhul kasutatavad põhilahused (kaheksa kontsentratsiooni vahemikus 100 × 1,2 × CV75 kuni 100 × 0,335 × CV75 (füsioloogilise lahuse või söötme puhul) või vahemikus 500 × 1,2 × CV75 kuni 500 × 0,335 × CV75 (DMSO puhul); annuste kasutamise näide on esitatud DB-ALMi katse-eeskirjas nr 158). Seejärel lahjendatakse põhilahuseid söötmes veel 50 korda (füsioloogilise lahuse või söötme puhul) või 250 korda (DMSO puhul) (töölahused). Neid töölahuseid kasutatakse lõpuks kokkupuutekatses, kus neist tehakse plaadil veel viimane kahekordne lahjendus. Kui rakkude eluvõimelisuse hindamise tulemused ei vasta punktides 29 ja 30 kirjeldatud nõuetekohasuse kriteeriumitele, võib CV75 väärtuse täpsemaks kindlakstegemiseks annuse kindlaksmääramise katset korrata. CD86/CD54 ekspressiooni mõõtmiseks saab kasutada ainult 24-kannulisi plaate.
|
|
21.
|
Lahustiga/kandeainega kontroll valmistatakse ette vastavalt punktis 16 esitatud kirjeldusele. h-CLAT puhul kasutatav positiivne kontroll on DNCB (vt punkt 11), millest tehakse DMSOs põhilahused, mida lahjendatakse vastavalt punktis 20 esitatud põhilahuste lahjendamise juhistele. DNCBd kasutatakse CD86/CD54 ekspressiooni mõõtmisel positiivse kontrollina plaadil ühes lõppkontsentratsioonis (tavaliselt 4,0 μg/ml). DNCB kasutamiseks plaadil kontsentratsioonis 4,0 μg/ml valmistatakse DNCBst DMSOs põhilahus kontsentratsiooniga 2 mg/ml ning lahjendatakse seda seejärel söötmes 250 korda, et saada töölahus kontsentratsiooniga 8 μg/ml. Teise variandina võib positiivse kontrolli kontsentratsioonina kasutada DNCBga saadud CV75 väärtust, mis tehakse kindlaks igas katselaboris. Võib kasutada ka mõnda muud sobivat positiivset kontrolli, kui on olemas varasemad andmed, mis võimaldavad kindlaks määrata võrreldavad analüüsitsükli nõuetekohasuse kriteeriumid. Positiivse kontrolli lõppkontsentratsioon plaadil ei tohiks olla suurem kui 5 000 μg/ml (füsioloogilise lahuse või söötme puhul) või 1 000 μg/ml (DMSO puhul). Analüüsitsükli nõuetekohasuse kriteeriumid on samad kui uuritava kemikaali puhul (vt punkt 29), välja arvatud viimane nõuetekohasuse kriteerium, sest positiivset kontrolli hinnatakse vaid ühel kontsentratsioonil.
|
Uuritavate kemikaalide ja kontrollainetega töötlemine
|
22.
|
Iga uuritava kemikaali ja kontrollaine puhul on prognoosi saamiseks vaja ühte katset. Iga katse koosneb vähemalt kahest sõltumatust analüüsitsüklist CD86/CD54 ekspressiooni mõõtmiseks (vt punktid 26–28). Iga sõltumatu analüüsitsükkel viiakse läbi eri päeval; samal päeval võib läbi viia mitu tsüklit juhul, kui iga analüüsitsükli, jaoks: a) valmistatakse sõltumatult värsked uuritava kemikaali põhilahused ja töölahused ning antikehade lahused; b) kasutatakse sõltumatult kogutud rakke (st rakud kogutakse erinevatest rakukultuuripudelitest); rakud võivad olla siiski samast passaažist. Uuritavate kemikaalide ja kontrollainete töölahused (500 μl) segatakse 500 μl rakususpensiooniga (1 × 106 rakku) vahekorras 1:1 ja rakke inkubeeritakse 24 ± 0,5 tundi, nagu on kirjeldatud punktides 20 ja 21. Igas tsüklis piisab ühest proovist uuritava kemikaali ja kontrollaine iga kontsentratsiooni kohta, kuna prognoosi saamiseks on vaja vähemalt kahte sõltumatut tsüklit.
|
Rakkude värvimine ja analüüs
|
23.
|
Pärast 24 ± 0,5 tundi kestnud kokkupuudet viiakse rakud 24-kannuliselt plaadilt katsutisse, kogutakse tsentrifuugimise teel ja seejärel pestakse kaks korda 1 ml värvimispuhvriga (vajaduse korral võib pesemisetappe lisada). Pärast värvimist blokeeritakse rakud 600 μl blokeerimislahusega (värvimispuhver, mis sisaldab 0,01 % (massiühikutes mahuühiku kohta) globuliini (inimese globuliini Cohni fraktsioonid II, III (SIGMA, #G2388-10G) või samaväärne)) ja inkubeeritakse neid 4oC juures 15 min. Pärast blokeerimist jaotatakse rakud kolme 180 μl suuruse alikvoodina 96-kannulisele ümarapõhjalisele plaadile või mikrokatsutitesse.
|
|
24.
|
Pärast tsentrifuugimist värvitakse rakke 4oC juures 30 minuti vältel 50 μl CD86- või CD54-vastase antikehaga või hiire IgG1-ga (isotüübikontroll), mis on märgistatud FITCga. Tuleks kasutada DB-ALMi h-CLATi katse-eeskirjas nr 158 (18) kirjeldatud antikehasid, mis lahjendatakse värvimispuhvris mahuvahekorras 3: 25 (CD86 puhul (BD-PharMingen, #5556 57, kloon Fun-1)) või 3: 50 (CD54 puhul (DAKO, #F7143, kloon 6.5B5) ja IgG1 puhul (DAKO, #X0927)). Katsemeetodi väljatöötajate andmetel saavutatakse nimetatud lahjendusteguritega kõige paremsignaali ja müra suhe. Katsemeetodi väljatöötajate kogemuste põhjal on eri partiide antikehade fluorestsentssignaali tugevus tavaliselt sarnane. Kasutajad võivad siiski kaaluda antikehade tiitrimist oma labori tingimustes, et teha kindlaks kõige paremad kasutamiseks sobivad kontsentratsioonid. Mõne muu fluorokroomiga märgistatud CD86-vastaseid ja/või CD54-vastaseid antikehasid võib kasutada juhul, kui näiteks katsetega, mis tehakse liites 1.2 loetletud pädevusainetega, on võimalik tõendada, et asjaomaste antikehade ja FITCga konjugeeritud antikehade puhul saadavad tulemused on sarnased. Tuleks arvesse võtta, et DB-ALMi h-CLATi katse-eeskirjas nr 158 (18) kirjeldatud antikehade klooni või tarnija vahetamine võib tulemusi mõjutada. Pärast vähemalt kaks korda 150 μl värvimispuhvriga pesemist suspendeeritakse rakud uuesti värvimispuhvris (nt 400 μl), millele lisatakse PI lahust (nt 20 μl lõppkontsentratsiooni 0,625 μg/ml saamiseks) või muu tsütotoksilisuse markeri lahust (vt punkt 18). CD86 ja CD54 ekspressiooni taset ning rakkude eluvõimelisust analüüsitakse läbivoolutsütomeetriga.
|
ANDMED JA ARUANDLUS
Andmete hindamine
|
25.
|
CD86 ja CD54 ekspressiooni analüüsitakse läbivoolutsütomeetriga; seejuures kasutatakse registreerimiskanalit FL1. Fluorestsentssignaali tugevuse geomeetrilise keskväärtuse (mean fluorescence intensity, MFI) põhjal arvutatakse positiivse kontrolli rakkude CD86 ja CD54 puhul täheldatav fluorestsentssignaali suhteline tugevus (relative fluorescence intensity, RFI) vastavalt järgmisele valemile:.

Lisaks arvutatakse punktis 19 esitatud valemi järgi IgG1-ga värvitud isotüübikontrolli rakkude eluvõimelisus.
|
Prognoosimudel
|
26.
|
CD86/CD54 ekspressiooni mõõtmisel hinnatakse iga uuritavat kemikaali vähemalt kahes sõltumatus analüüsitsüklis, mille põhjal tehakse üks prognoosijäreldus (POSITIIVNE või NEGATIIVNE). h-CLAT kohast prognoosi käsitatakse POSITIIVSENA, kui vähemalt kahes kolmest sõltumatust analüüsitsüklist on täidetud vähemalt üks järgmistest tingimustest; vastasel korral tunnistatakse h-CLAT kohane prognoos NEGATIIVSEKS (joonis 1):
|
—
|
CD86 puhul on RFI mis tahes kasutatud kontsentratsioonil 150 % või suurem (ja rakkude eluvõimelisus ≥ 50 %);
|
|
—
|
CD54 puhul on RFI mis tahes kasutatud kontsentratsioonil 200 % või suurem (ja rakkude eluvõimelisus ≥ 50 %).
|
|
|
27.
|
Seega kui kahes esimeses analüüsitsüklis saadakse CD86 ja/või CD54 puhul positiivne tulemus, tunnistatakse h-CLAT kohane prognoos POSITIIVSEKS ja kolmandat analüüsitsüklit ei ole vaja läbi viia. Kui kahes esimeses analüüsitsüklis saadakse kummagi markeri puhul negatiivne tulemus, tunnistatakse h-CLAT kohane prognoos NEGATIIVSEKS (seejuures võetakse nõuetekohaselt arvesse punkti 30 sätteid) ja kolmandat tsüklit ei ole vaja. Kui aga kahe esimese tsükli tulemused ei lange vähemalt ühe markeri (CD54 või CD86) puhul kokku, on vaja kolmandat tsüklit ja lõplik prognoos tehakse kolme tsükli lõikes suurema osakaaluga tulemuse põhjal (st kaks kolmest). Seoses sellega tuleb silmas pidada, et kui viiakse läbi kaks sõltumatut tsüklit, millest ühes saadakse positiivne tulemus ainult CD86 puhul (edaspidi P1) ja teises positiivne tulemus ainult CD54 puhul (edaspidi P2), tuleb läbi viia kolmas tsükkel. Kui kolmandas tsüklis saadakse kummagi markeri puhul negatiivne tulemus (edaspidi N), tunnistatakse h-CLAT kohane prognoos NEGATIIVSEKS. Ent kui kolmandas tsüklis saadakse ükskõik kumma markeri puhul positiivne tulemus (P1 või P2) või mõlema markeri puhul positiivne tulemus (edaspidi P12), tunnistatakse h-CLAT kohane prognoos POSITIIVSEKS.

Joonis 1. h-CLAT puhul kasutatav prognoosimudel; h-CLAT kohast prognoosi tuleb arvesse võtta ühtse lähenemisviisi raamistikus ning kooskõlas üldise sissejuhatuse punktide 7 ja 8 sätetega
P1 – analüüsitsükkel, kus ainult CD86 on positiivne; P2 – analüüsitsükkel, kus ainult CD54 on positiivne; P12 – analüüsitsükkel, kus nii CD86 kui ka CD54 on positiivsed; N – analüüsitsükkel, kus ei CD86 ega CD54 ei ole positiivne.
* Kastides on esitatud kahe esimese analüüsitsükli tulemuste kombinatsioonid järjekorras, mis ei pruugi vastata nende saamise järjekorrale.
# Kastides on esitatud kolme analüüsitsükli tulemuste kombinatsioonid lähtuvalt ülemises kastis esitatud kahe esimese tsükli tulemustest, kuid esitatu ei kajasta tulemuste saamise järjekorda.
|
|
28.
|
h-CLAT kohase POSITIIVSE prognoosi saanud uuritava kemikaali puhul võib soovi korral määrata kaks toime avaldumise kontsentratsiooni: CD86 puhul EC150 ja CD54 puhul EC200. Need vastavad kontsentratsioonile, mille juures uuritava kemikaali põhjustatud RFI on vastavalt 150 või 200. Kui neid EC väärtusi kasutatakse näiteks katsete tegemist ja hindamist käsitleva ühtse lähenemisviisi raames (4, 5, 6, 7, 8), võivad need aidata hinnata sensibiliseeriva toime tugevust (9). Neid saab arvutada järgmiste valemitega: , ,
EC 150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
kus
Aconc on väikseim kontsentratsioon (μg/ml), mille juures RFI > 150 (CD86) või 200 (CD54),
Bconc on suurim kontsentratsioon (μg/ml), mille juures RFI < 150 (CD86) või 200 (CD54),
ARFI on RFI väärtus väikseimal kontsentratsioonil, mille juures RFI > 150 (CD86) või 200 (CD54), ning
BRFI on RFI väärtus suurimal kontsentratsioonil, mille juures RFI < 150 (CD86) või 200 (CD54).
EC150 ja EC200 väärtuste täpsemaks määramiseks võib vaja minna kolme sõltumatut CD86/CD54 ekspressiooni mõõtmist. Lõplikud EC150 ja EC200 väärtused arvutatakse seejärel kolmes sõltumatus analüüsitsüklis saadud EC väärtuste mediaanina. Kui positiivsuse tingimused (vt punktid 26–27) on täidetud ainult kahes sõltumatus analüüsitsüklis kolmest, kasutatakse kahest asjaomasest arvutatud EC150 või EC200 väärtusest suuremat.
|
Nõuetekohasuse kriteeriumid
|
29.
|
h-CLAT kasutamisel peaksid olema täidetud järgmised katse nõuetekohasuse kriteeriumid (22, 27).
|
—
|
Söötmega ja lahustiga/kandeainega kontrollide puhul peaks rakkude eluvõimelisus olema suurem kui 90 %.
|
|
—
|
Lahustiga/kandeainega kontrollis ei tohiks RFI väärtus CD86 ega CD54 puhul ületada positiivsuse kriteeriumi kohast väärtust (CD86 puhul RFI ≥ 150 % ja CD54 puhul RFI ≥ 200 %). Lahustiga/kandeainega kontrolli puhul arvutatakse RFI väärtus punktis 25 esitatud valemiga ("kemikaaliga" tuleks asendada muutujaga "lahustiga/kandeainega" ning "lahustiga/kandeainega töödeldud" tuleks asendada muutujaga "(söötmega) kontrolli").
|
|
—
|
Söötmega ja lahustiga/kandeainega kontrollide puhul peaks nii CD86 kui ka CD54 puhul olema vastava MFI väärtuse ja isotüübikontrolliga saadud MFI väärtuse suhtarv > 105 %.
|
|
—
|
Positiivse kontrolli (DNCB) puhul peaks RFI väärtus nii CD86 kui ka CD54 puhul vastama positiivsuse kriteeriumile (CD86 puhul RFI ≥ 150 % ja CD54 puhul RFI ≥ 200 %) ning rakkude eluvõimelisus peaks olema üle 50 %.
|
|
—
|
Uuritava kemikaali puhul peaks rakkude eluvõimelisus olema igas tsüklis vähemalt neljal kasutatud kontsentratsioonil suurem kui 50 %.
|
|
|
30.
|
Negatiivsed tulemused võetakse arvesse üksnes juhul, kui uuritava kemikaali puhul on rakkude eluvõimelisus suurimal katses kasutatud kontsentratsioonil (st 1,2 × CV75 vastavalt punktis 20 kirjeldatud järjestikuse lahjendamise skeemile) väiksem kui 90 %. Kui rakkude eluvõimelisus 1,2 × CV75 juures on 90 % või suurem, tuleks negatiivne tulemus kõrvale jätta. Sellisel juhul soovitatakse kasutatava annuse täpsustamiseks korrata CV75 määramist. Tuleb märkida, et kui uuritava kemikaali suurim katsekontsentratsioon on füsioloogilises lahuses (või söötmes või muus lahustis/kandeaines) 5 000 μg/ml või DMSOs 1 000 μg/ml või suurim kontsentratsioon, mille juures kemikaal veel lahustub, võetakse negatiivne tulemus arvesse ka siis, kui rakkude eluvõimelisus on üle 90 %.
|
Katseprotokoll
|
31.
|
Katseprotokoll peaks sisaldama järgmist teavet.
|
Uuritav kemikaal
|
Ühest koostisosast koosnev aine:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed), CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muu identimisteave;
|
|
füüsiline välimus, log Kow, lahustuvus vees, lahustuvus DMSOs, molekulmass ja muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti/kandeaine valimise põhjendus.
|
|
|
Mitmest koostisosast koosnev aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal või segu:
|
võimalikult täpne omaduste kirjeldus lähtuvalt näiteks koostisosade keemilisest määratlusest (vt eespool), puhtusest, kvantitatiivsest sisaldusest ja asjakohaseid füüsikalis-keemilisi omadusi käsitlevatest kättesaadavatest andmetest (vt eespool);
|
|
füüsiline välimus, lahustuvus vees, lahustuvus DMSOs ja muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
teadaoleva koostisega segu/polümeeri puhul molekulmass või näiline molekulmass või muu uuringu tegemiseks asjakohane teave;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti/kandeaine valimise põhjendus.
|
|
|
|
Kontrollid
|
Positiivne kontroll:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed), CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muu identimisteave;
|
|
füüsiline välimus, log Kow, lahustuvus vees, lahustuvus DMSOs, molekulmass ja vajaduse korral muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
vajaduse korral viide varasematele positiivse kontrolliga saadud tulemustele, millest nähtub vastavus asjakohastele analüüsitsükli nõuetekohasuse kriteeriumidele.
|
|
|
|
Negatiivne ja lahustiga/kandeainega kontroll:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed), CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muu identimisteave;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
asjaomases katsejuhendis nimetamata lahusti/kandeaine kasutamisel kontrollina kättesaadav teave selle füüsilise välimuse, molekulmassi ja muude asjakohaste füüsikalis-keemiliste omaduste kohta;
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti/kandeaine valimise põhjendus.
|
|
|
Katsetingimused:
|
sponsori, katselabori ja uuringu juhi nimi ja aadress;
|
|
kasutatud katsemeetodi kirjeldus;
|
|
kasutatud rakuliin, selle säilitamise tingimused ning päritolu (nt asutus, kellelt rakud saadi);
|
|
kasutatud läbivoolutsütomeeter (nt mudel) ja selle seaded, kasutatud globuliin, antikehad ja tsütotoksilisuse marker;
|
|
pädevusainete analüüsimise meetod, mida kasutati labori pädevuse tõendamiseks, ning katsemeetodiga saadud tulemuste pikaajalise reprodutseeritavuse tõendamise viis, näiteks laboris kontrollidega ja/või reageerimisvõime kontrollimisel varem saadud andmed.
|
|
|
Katse nõuetekohasuse kriteeriumid:
|
lahustiga/kandeainega kontrolli puhul saadud rakkude eluvõimelisuse, MFI ja RFI väärtused võrrelduna vastavate nõuetekohasust kajastavate vahemikega;
|
|
positiivse kontrolliga saadud rakkude eluvõimelisuse ja RFI väärtused võrrelduna vastavate nõuetekohasust kajastavate vahemikega;
|
|
rakkude eluvõimelisus uuritud kemikaali igal katses kasutatud kontsentratsioonil.
|
|
|
Katse käik:
|
uuritava kemikaali kontsentratsioonid, lisamisviis ja kasutatud kokkupuuteaeg (kui erineb soovitatust);
|
|
kasutatud hindamis- ja otsustamiskriteeriumide kirjeldus;
|
|
kõikide katse käigus tehtud muudatuste kirjeldus.
|
|
|
Tulemused:
|
tabeli kujul andmed uuritava kemikaali ja positiivse kontrolli kohta igas analüüsitsüklis, sh CV75 (vajaduse korral), eraldi MFI, RFI ja rakkude eluvõimelisuse väärtused, EC150/EC200 väärtused (vajaduse korral) ning prognoosimudelil põhinev hinnang uuritava kemikaali kohta;
|
|
vajaduse korral mis tahes muude asjakohaste vaatlusandmete kirjeldus.
|
|
|
Tulemuste arutelu:
|
h-CLATga saadud tulemuste arutelu;
|
|
katse tulemuste hindamine ühtse lähenemisviisi kontekstis, kui muu asjakohane teave on kättesaadav.
|
|
|
KIRJANDUS
|
(1)
|
Ashikaga, T., Yoshida, Y., Hirota, M., Yoneyama, K., Itagaki, H., Sakaguchi, H., Miyazawa, M., Ito, Y., Suzuki, H., ja Toyoda, H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20: 767–773.
|
|
(2)
|
Miyazawa, M., Ito, Y., Yoshida, Y., Sakaguchi, H., ja Suzuki, H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21: 428–437.
|
|
(3)
|
EURL ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Kättesaadav aadressil https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations.
|
|
(4)
|
Takenouchi, O., Fukui, S., Okamoto, K., Kurotani, S., Imai, N., Fujishiro, M., Kyotani, D., Kato, Y., Kasahara, T., Fujita, M., Toyoda, A., Sekiya, D., Watanabe, S., Seto, H., Hirota, M., Ashikaga, T., ja Miyazawa, M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J. Appl. Toxicol. 35: 1318–1332.
|
|
(5)
|
Hirota, M., Fukui, S., Okamoto, K., Kurotani, S., Imai, N., Fujishiro, M., Kyotani, D., Kato, Y., Kasahara, T., Fujita, M., Toyoda, A., Sekiya, D., Watanabe, S., Seto, H., Takenouchi, O., Ashikaga, T., ja Miyazawa, M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J. Appl. Toxicol. 35: 1333–1347.
|
|
(6)
|
Bauch, C., Kolle, S. N., Ramirez, T., Fabian, E., Mehling, A., Teubner, W., van Ravenzwaay, B., ja Landsiedel, R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul. Toxicol. Parmacol. 63: 489–504.
|
|
(7)
|
Van der Veen, J. W., Rorije, E., Emter, R., Natch, A., van Loveren, H., ja Ezendam, J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul. Toxicol. Pharmacol. 69: 371–379.
|
|
(8)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P. S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., ja Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Parmacol. 71: 337–351.
|
|
(9)
|
Jaworska, J. S., Natsch, A., Ryan, C., Strickland, J., Ashikaga, T., ja Miyazawa, M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch. Toxicol. 89: 2355–2383.
|
|
(10)
|
Strickland, J., Zang, Q., Kleinstreuer, N., Paris, M., Lehmann, D. M., Choksi, N., Matheson, J., Jacobs, A., Lowit, A., Allen, D., ja Casey, W. (2016). Integrated decision strategies for skin sensitization hazard. J. Appl. Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada, Y., Ashikaga, T., Miyazawa, M., Hirota, M., Sakaguchi, H., Sasa, H., ja Nishiyama, N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26: 1150–60.
|
|
(12)
|
EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Kättesaadav aadressil https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing.
|
|
(13)
|
EURL ECVAM (2012). Human Cell Line Activation Test (h-CLAT) Validation Study Report. Kättesaadav aadressil https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations.
|
|
(14)
|
Takenouchi, O., Miyazawa, M., Saito, K., Ashikaga, T., ja Sakaguchi, H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38: 599–609.
|
|
(15)
|
Ashikaga, T., Sakaguchi, H., Sono, S., Kosaka, N., Ishikawa, M., Nukada, Y., Miyazawa, M., Ito, Y., Nishiyama, N., ja Itagaki, H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38: 275–284.
|
|
(16)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., ja Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87: 1683–1969.
|
|
(17)
|
Okamoto, K., Kato, Y., Kosaka, N., Mizuno, M., Inaba, H., Sono, S., Ashikaga, T., Nakamura, T., Okamoto, Y., Sakaguchi, H., Kishi, M., Kuwahara, H., ja Ohno, Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15: 81–88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol No. 158: human Cell Line Activation Test (h-CLAT). 23 lk. Kättesaadav aadressil http://ecvam-dbalm.jrc.ec.europa.eu/.
|
|
(19)
|
Mizuno, M., Yoshida, M., Kodama, T., Kosaka, N., Okamato, K., Sono, S., Yamada, T., Hasegawa, S., Ashikaga, T., Kuwahara, H., Sakaguchi, H., Sato, J., Ota, N., Okamoto, Y., ja Ohno, Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13: 70–82.
|
|
(20)
|
Sono, S., Mizuno, M., Kosaka, N., Okamoto, K., Kato, Y., Inaba, H., Nakamura, T., Kishi, M., Kuwahara, H., Sakaguchi, H., Okamoto, Y., Ashikaga, T., ja Ohno, Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15: 89–96.
|
|
(21)
|
OECD (2005). Guidance Document No. 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Majanduskoostöö ja Arengu Organisatsioon, Pariis, Prantsusmaa. 96 lk.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Kättesaadav aadressil http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(23)
|
Ühinenud Rahvaste Organisatsioon (ÜRO) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Viies, täiendatud väljaanne. United Nations Publications, New York ja Genf. ISBN: 978-92-1-117006-1. Kättesaadav aadressil http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(24)
|
Euroopa Kemikaalide Ökotoksikoloogia ja Toksikoloogia Keskus (ECETOC) (2003). Contact Sensitization: Classification According to Potency. Technical Report No. 87. Euroopa Kemikaalide Ökotoksikoloogia ja Toksikoloogia Keskus.
|
|
(25)
|
Ashikaga, T., Sakaguchi, H., Okamoto, K., Mizuno, M., Sato, J., Yamada, T., Yoshida, M., Ota, N., Hasegawa, S., Kodama, T., Okamoto, Y., Kuwahara, H., Kosaka, N., Sono, S., ja Ohno, Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13: 27–35.
|
Liide 1.1
MÕISTED
Aine
: looduslik või mis tahes tootmisprotsessi tulemusena saadud keemiline element või selle ühend koos püsivuse tagamiseks vajalike lisaainete ja tootmisprotsessi käigus tekkinud lisanditega, mis ei hõlma siiski lahusteid, mida on võimalik aine püsivust mõjutamata või selle koostist muutmata ainest eraldada.
Analüüsitsükkel
: ühe või mitme uuritava kemikaali analüüsimine lahustiga/kandeainega kontrolli ja positiivse kontrolliga paralleelselt.
Asjakohasus
: näitaja, mille abil kirjeldatakse, kuivõrd katsemeetod võimaldab uurida huvipakkuvat toimet, kas selle kasutamine on mõttekas ja kas see on konkreetse eesmärgi jaoks sobiv. Asjakohasusest nähtub, mil määral saab katsemeetodiga õigesti mõõta või ennustada huvipakkuvat bioloogilist toimet. Asjakohasuse puhul võetakse arvesse ka katsemeetodiga saavutatavat täpsust (vastavust) (21).
CV75
: hinnanguline kontsentratsioon, mille juures rakkude eluvõimelisus on 75 %.
EC150
: kontsentratsioon, mille juures CD86 ekspressiooni kajastav RFI väärtus on 150.
EC200
: kontsentratsioon, mille juures CD54 ekspressiooni kajastav RFI väärtus on 200.
Fluorestsentssignaali suhteline tugevus (RFI)
: kemikaaliga töödeldud rakkude fluorestsentssignaali tugevuse väärtuste geomeetrilise keskmise suhteline väärtus võrrelduna lahustiga/kandeainega töödeldud rakkude fluorestsentssignaali tugevuse väärtuste geomeetrilise keskmisega.
Kahjuliku toime rada
: sündmuste ahel alates sihtkemikaali või rühma sarnaste kemikaalide keemilisest struktuurist sõltuvast käivitavast molekulaarsest sündmusest kuni vaadeldava toime avaldumiseni in vivo (22).
Katsete tegemist ja hindamist käsitlev ühtne lähenemisviis
: struktureeritud lähenemisviis, mida kasutatakse kemikaali või rühma kemikaalide ohtlikkuse tuvastamiseks (võimalik toime), ohu kirjeldamiseks (toime tugevus) ja/või ohutuse hindamiseks (võimalik toime / toime tugevus ja kokkupuude) ning mille raames lõimitakse strateegiliselt ja kaalutakse läbi kõik asjakohased andmed võimalikku ohtu ja/või riski ja/või täiendavate sihipäraste ja seega minimaalses ulatuses katsete tegemise vajadust käsitleva regulatiivse otsuse tegemiseks.
Kemikaal
: aine või segu.
Lahustiga/kandeainega kontroll
: töötlemata proov, mis sisaldab kõiki katsesüsteemi komponente peale uuritava kemikaali, sealhulgas lahustit/kandeainet. Selle abil tehakse kindlaks lähtetase samas lahustis/kandeaines lahustatud või püsiva dispersiooni moodustanud uuritava kemikaaliga töödeldud proovide jaoks. Kui sellist töötlemata proovi hinnatakse paralleelselt söötmega kontrolliga, võimaldab see ühtlasi kindlaks teha, kas lahusti/kandeaine mõjutab katsesüsteemi.
Läbivoolutsütomeetria
: tsütomeetriline meetod, mille puhul vedelikus suspendeeritud rakud liiguvad üksteise järel läbi ergastava valguse fookuse ning sellest tingitud valguse hajumise viis kajastab rakkude ja nende koostisosade omadusi; sageli märgistatakse rakud fluorestseeruvate markeritega, mille puhul valgus kõigepealt neeldub ja seejärel kiirgub muutunud sagedusel.
Mitmest koostisosast koosnev aine
: aine, mis on määratletud kvantitatiivse koostise alusel ja milles enam kui ühe põhikoostisosa sisaldus on vähemalt 10, kuid väiksem kui 80 massiprotsenti. Mitmest koostisosast koosnev aine saadakse tootmisprotsessi tulemusena. Erinevus segu ja mitmest koostisosast koosneva aine vahel seisneb selles, et segu saadakse kahe või enama aine kokku segamisel, ilma et toimuks keemilist reaktsiooni. Mitmest koostisosast koosnev aine saadakse keemilise reaktsiooni tulemusel.
Nõuetekohane katsemeetod
: katsemeetod, mille asjakohasust ja usaldusväärsust konkreetse eesmärgi puhul peetakse piisavaks ning mis rajaneb teaduslikult usaldusväärsetel põhimõtetel. Katsemeetod ei ole kunagi nõuetekohane absoluutses tähenduses, vaid üksnes seoses teatud kindla eesmärgiga (21).
Ohtlikkus
: mõjurit või olukorda iseloomustav omadus, millest võib tuleneda kahjulik toime, kui organism, süsteem või (alam)populatsioon selle mõjuriga kokku puutub.
Positiivne kontroll
: paralleelproov, mis sisaldab kõiki katsesüsteemi komponente ja mida on töödeldud ainega, millega teadaolevalt saadakse positiivne tulemus. Positiivse tulemuse väärtus ei tohiks olla liiga suur, et võimaldada hinnata positiivse kontrolliga saadud tulemuste varieeruvust ajas.
Prehapteenid
: kemikaalid, mis muutuvad sensibilisaatoriks abiootilise muundumise teel.
Prohapteenid
: kemikaalid, mis vajavad nahka sensibiliseeriva toime tekkimiseks ensümaatilist aktiveerimist.
Segu
: kahest või enamast ainest koosnev segu või lahus.
Spetsiifilisus
: katsemeetodiga õigesti negatiivseks/reaktsioonivõimetuks liigitatud uuritavate kemikaalide osakaal kõikide negatiivsete kemikaalide hulgas. Spetsiifilisus kajastab sellise katsemeetodi täpsust, millega saadakse kategoriseeritav tulemus, ning see on oluline näitaja katsemeetodi asjakohasuse hindamiseks (21).
Söötmega kontroll
: töötlemata paralleelproov, mis sisaldab kõiki katsesüsteemi komponente. Sellist proovi käideldakse koos uuritava kemikaaliga töödeldud proovidega ja muude kontrollproovidega, et teha kindlaks, kas lahusti/kandeaine mõjutab katsesüsteemi.
Tundlikkus
: katsemeetodiga õigesti positiivseks/reaktsioonivõimeliseks liigitatud uuritavate kemikaalide osakaal kõikide positiivsete kemikaalide hulgas. Tundlikkus kajastab sellise katsemeetodi täpsust, millega saadakse kategoriseeritav tulemus, ning see on oluline näitaja katsemeetodi asjakohasuse hindamiseks (21).
Täpsus
: katsetulemuste ja vastuvõetavate võrdlusväärtuste kooskõla määr. Täpsus näitab katsemeetodi tulemuslikkust ja on üks asjakohastest aspektidest. Seda mõistet kasutatakse sageli vastavuse tähenduses, et väljendada katsemeetodiga saadud õigete tulemuste osakaalu (21).
Usaldusväärsus
: näitaja, mis iseloomustab katsemeetodiga saadud tulemuste reprodutseeritavust pikema aja jooksul samas laboris ja eri laborites, kui meetodit rakendatakse ühe ja sama katse-eeskirja alusel. Usaldusväärsuse hindamiseks arvutatakse laborisisene ja laboritevaheline reprodutseeritavus ning laborisisene korratavus (21).
Uuritav kemikaal
: iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
UVCB
: tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal.
Värvimispuhver
: fosfaadiga puhverdatud soolalahus, mis sisaldab 0,1 % veise seerumi albumiini.
Ühest koostisosast koosnev aine
: aine, mis on määratletud kvantitatiivse koostise alusel ja mille ühe põhikoostisosa sisaldus on vähemalt 80 massiprotsenti.
Ühinenud Rahvaste Organisatsiooni ühtne ülemaailmne kemikaalide klassifitseerimise ja märgistamise süsteem (ÜRO GHS)
: süsteem, millega nähakse ette kemikaalide (ainete ja segude) klassifitseerimine vastavalt nende füüsilise ohtlikkuse ning kahjuliku tervise- ja keskkonnamõju standarditud liigile ja tasemele ning mis hõlmab selliseid asjaomaseid teavitustähiseid nagu piktogrammid, märksõnad, ohulaused, hoiatuslaused ja ohutuskaardid, et anda inimeste (sealhulgas tööandjate, töötajate, vedajate, tarbijate ja päästetöötajate) ning keskkonna kaitsmiseks vajalikku teavet kõnealuste kemikaalide kahjuliku toime kohta (23).
Liide 1.2
PÄDEVUSAINED
Enne katsemeetodi B.71 juurde kuuluvas käesolevas liites kirjeldatud meetodi rutiinset kasutamist tuleks tõendada labori tehnilist pädevust. Selleks tuleks h-CLAT kasutamisel saada tabelis 1 esitatud kümne soovitatava aine puhul eeldatavale tulemusele vastav prognoos ning CV75, EC150 ja EC200 väärtused, mis jäävad kümnest pädevusainest vähemalt kaheksa puhul asjaomasesse võrdlusvahemikku. Pädevusained on valitud nii, et nende puhul oleksid esindatud naha sensibiliseerimise ohuga seotud eri reaktsioonid. Peale selle on valikukriteeriumidena võetud arvesse ainete kaubanduslikku kättesaadavust, kvaliteetsete in vivo võrdlusandmete olemasolu ja h-CLATga saadud kvaliteetsete in vitro andmete olemasolu. Lisaks on h-CLAT kohta olemas avaldatud võrdlusandmed (3, 14).
Tabel 1.
h-CLAT kasutamise tehnilise pädevuse tõendamiseks soovitatavad ained
|
Pädevusained
|
CASi nr
|
Füüsikaline olek
|
In vivo toime prognoos (101)
|
CV75 võrdlusvahemik, μg/ml (102)
|
h-CLAT tulemus CD86 puhul (EC150 võrdlusvahemik, μg/ml) (102)
|
h-CLAT tulemus CD54 puhul (EC200 võrdlusvahemik, μg/ml) (102)
|
|
2,4-dinitroklorobenseen
|
97-00-7
|
Tahkis
|
Sensibilisaator (äärmiselt tugev)
|
2–12
|
Positiivne (0,5–10)
|
Positiivne (0,5–15)
|
|
4-fenüleendiamiin
|
106-50-3
|
Tahkis
|
Sensibilisaator (tugev)
|
5–95
|
Positiivne (< 40)
|
Negatiivne (> 1,5) (103)
|
|
Nikkelsulfaat
|
10101-97-0
|
Tahkis
|
Sensibilisaator (mõõdukas)
|
30–500
|
Positiivne (< 100)
|
Positiivne (10–100)
|
|
2-merkaptobensotiasool
|
149-30-4
|
Tahkis
|
Sensibilisaator (mõõdukas)
|
30–400
|
Negatiivne (> 10) (103)
|
Positiivne (10–140)
|
|
R(+)-limoneen
|
5989-27-5
|
Vedelik
|
Sensibilisaator (nõrk)
|
> 20
|
Negatiivne (> 5) (103)
|
Positiivne (< 250)
|
|
Imidasolidinüülkarbamiid
|
39236-46-9
|
Tahkis
|
Sensibilisaator (nõrk)
|
25–100
|
Positiivne (20–90)
|
Positiivne (20–75)
|
|
Isopropanool
|
67-63-0
|
Vedelik
|
Sensibiliseeriva toimeta
|
> 5 000
|
Negatiivne (> 5 000 )
|
Negatiivne (> 5 000 )
|
|
Glütserool
|
56-81-5
|
Vedelik
|
Sensibiliseeriva toimeta
|
> 5 000
|
Negatiivne (> 5 000 )
|
Negatiivne (> 5 000 )
|
|
Piimhape
|
50-21-5
|
Vedelik
|
Sensibiliseeriva toimeta
|
1 500 –5 000
|
Negatiivne (> 5 000 )
|
Negatiivne (> 5 000 )
|
|
4-aminobensoehape
|
150-13-0
|
Tahkis
|
Sensibiliseeriva toimeta
|
> 1 000
|
Negatiivne (> 1 000 )
|
Negatiivne (> 1 000 )
|
|
Lühendid: CASi nr – Chemical Abstracts Service’i registrinumber.
|
2. liide
NAHA SENSIBILISEERIMINE IN VITRO: RAKULIINI U937 AKTIVEERIMISE KATSE (U-SENS™)
LÄHTEKAALUTLUSED JA PIIRANGUD
|
1.
|
Katsemeetodiga U-SENS™ tehakse inimese histiotsüütse lümfoomi rakuliinis U937 kindlaks monotsüütide ja dendriitrakkude aktiveerimisega seotud raku pinnamarkeri (CD86) ekspressiooni kvantitatiivsed muutused pärast kokkupuudet sensibilisaatoriga (1). Seejärel kasutatakse rakuliinis U937 mõõdetud raku pinnamarkeri CD86 ekspressioonitaset toetava teabena, mis aitab eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri.
|
|
2.
|
Katsemeetodit U-SENS™ on hinnatud L’Oreali koordineeritud valideerimisuuringus (2) ning seejärel on selle sõltumatult läbi vaadanud loomkatsete alternatiivide Euroopa Liidu referentlabori (EURL ECVAM) teaduslik nõuandekomitee (3). Võttes arvesse kõiki kättesaadavaid andmeid ning reguleerivate asutuste ja sidusrühmade seisukohti, soovitas EURL ECVAM kasutada katsemeetodit U-SENS™ (4) ühtse lähenemisviisi osana, et aidata ohu alusel klassifitseerimise ja märgistamise eesmärgil eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri. Oma juhenddokumendis, milles käsitletakse naha sensibiliseerimisega seotud ühtse lähenemisviisi puhul kasutatavaid andmete lõimimise struktureeritud metoodikaid ja teabeallikaid, vaatleb OECD mitut juhtumiuuringut, milles kirjeldatakse eri katsestrateegiaid ja prognoosimudeleid. Üks kõnealustest kindlaksmääratud metoodikatest põhineb katsemeetodil U-SENS (5). Näited meetodiga U-SENS™ saadud andmete kasutamisest kombineerituna muu teabega, sealhulgas varasemate katseandmete ja nõuetekohastest inimuuringutest saadud andmetega (6) on avaldatud kirjanduses (4, 5, 7).
|
|
3.
|
U-SENS™ on osutunud rakendatavaks laborites, kus on olemas rakukultuurimeetodite kasutamise ja läbivoolutsütomeetrilise analüüsi kogemus. Selle meetodi puhul eeldatav prognooside reprodutseeritavuse ligikaudne määr on laborisisese ja laboritevahelise reprodutseeritavuse puhul vastavalt 90 % ja 84 % (8). Valideerimisuuringust (8) ja muudest avaldatud uuringutulemustest (1) nähtub üldiselt, et LLNA tulemustega võrreldes on selles katses naha sensibilisaatorite (st ÜRO GHSi / CLP-määruse kohaselt 1. kategooria kemikaalide) mittesensibiliseerivatest kemikaalidest eristamise täpsus 86 % (N = 166), tundlikkus 91 % (118/129) ja spetsiifilisus 65 % (24/37). Inimuuringute tulemustega võrreldes on naha sensibilisaatorite (st ÜRO GHSi / CLP-määruse kohaselt 1. kategooria kemikaalide) mittesensibiliseerivatest kemikaalidest eristamise täpsus 77 % (N = 101), tundlikkus 100 % (58/58) ja spetsiifilisus 47 % (20/43). LLNA tulemustega võrreldes on meetodi U-SENS™ kohaste valenegatiivsete prognooside tõenäosus suurem vähesel määral kuni mõõdukalt nahka sensibiliseerivate (st ÜRO GHSi / CLP-määruse kohaselt alamkategooria 1B) kemikaalide puhul ja väiksem siis, kui tegemist on tugevalt nahka sensibiliseeriva (st ÜRO GHSi / CLP-määruse kohaselt alamkategooria 1A) kemikaaliga (1, 8, 9). Kokkuvõttes nähtub sellest teabest, et U-SENS™ on kasulik meetod, mis aitab kindlaks teha naha sensibiliseerimise ohtu. Ainsa meetodina kasutatava U-SENS™-iga saavutatav eespool nimetatud täpsus on siiski üksnes orienteeriv, sest katseandmeid tuleks vaadelda ühtse lähenemisviisi kontekstis koos muudest allikatest pärit teabega ja kooskõlas üldise sissejuhatuse punktide 7 ja 8 sätetega. Kui hinnatakse naha sensibiliseerimise katsemeetodeid, mille puhul ei kasutata katseloomi, tuleb lisaks arvesse võtta, et LLNA ja muude loomkatsete tulemused ei pruugi täielikult kajastada kemikaali toimet inimesele.
|
|
4.
|
Praeguse olemasoleva teabe põhjal on tõendatud, et U-SENS™ on kohaldatav paljude uuritavate kemikaalide (sealhulgas kosmeetikatoodete koostisainete, nt säilitusainete, pindaktiivsete ainete, toimeainete, värvainete) puhul, mis erinevad üksteisest orgaanilise funktsionaalrühma, füüsikalis-keemiliste omaduste, in vivo katsetega määratud nahka sensibiliseeriva toime tugevuse ja teadaolevalt naha sensibiliseerimisega seotud reaktsioonimehhanismide (Michaeli aktseptor, Schiffi aluse teke, atsüülrühma ülekande reaktiiv, nukleofiilne bimolekulaarne asendusreaktsioon [SN2] või nukleofiilne aromaatne asendusreaktsioon [SNAr]) poolest (1, 8, 9, 10). U-SENS™ on kohaldatav uuritavate kemikaalide puhul, mis on sobivas lahustis/kandeaines lahustuvad või moodustavad selles püsiva dispersiooni (st kolloidlahuse või suspensiooni, milles uuritav kemikaal ei sadestu ega eraldu lahustist/kandeainest eri faasi) (vt punkt 13). Andmestikus esindatud kemikaalidega, mis on teadaolevalt prehapteenid (oksüdeerumisel aktiveeruvad ained) või prohapteenid (ained, mis vajavad ensümaatilist aktiveerimist, nt P450 ensüümide abil), saadi meetodi U-SENS™ kasutamisel õige prognoositulemus (1, 10). Membraani lõhkuvate ainete puhul võidakse CD86 ekspressiooni mittespetsiifilise suurenemise tõttu saada valepositiivsed tulemused, nagu nähtub asjaolust, et in vivo võrdlusliigitusest lähtuvalt saadud seitsmest valepositiivsest tulemusest kolm olid seotud pindaktiivsete ainetega (1). Seetõttu tuleks pindaktiivsete ainetega saadud positiivsetesse tulemustesse suhtuda ettevaatusega, aga pindaktiivsete ainetega saadudnegatiivseid tulemusi võib arvesse võtta uuritava kemikaali liigitamisel kemikaaliks, mis nahka ei sensibiliseeri. U-SENS™ võimaldab hinnata ka fluorestseeruvaid uuritavaid kemikaale (1), kuid tugevalt fluorestseeruvad uuritavad kemikaalid, mille kiiratava kiirguse lainepikkus on sama kui fluorestseiinisotiotsüanaadil (FITC) või propiidiumjodiidil (PI), segavad läbivoolutsütomeetrilist analüüsi ning neid ei ole võimalik FITCga konjugeeritud antikehade või PI abil õigesti hinnata (esimese puhul võidakse saada valepositiivsed tulemused ja teise puhul ei saa mõõta eluvõimelisust). Sellisel juhul võib kasutada vastavalt muu fluorokroomiga märgistatud antikehasid või muud tsütotoksilisuse markerit, kui näiteks katsetega, mis tehakse liites 2.2 loetletud pädevusainetega, on võimalik tõendada, et sellise fluorokroomi või markeriga saadakse samasugused tulemused kui FITCga märgistatud antikehade või PIga (vt punkt 18). Eespool kirjeldatust lähtuvalt tuleks pindaktiivsete ainetega saadud positiivseid tulemusi ja tugevalt fluorestseeruvate uuritavate kemikaalidega saadud negatiivseid tulemusi tõlgendada kõnealuste piirangute kontekstis ja koos ühtse lähenemisviisi raames muudest allikatest saadud teabega. Kui leidub andmeid, mille kohaselt U-SENS™ ei sobi kasutamiseks mõne muu konkreetse uuritavate kemikaalide rühma puhul, ei tohiks seda asjaomase konkreetse kemikaalirühma puhul kasutada.
|
|
5.
|
Nagu eespool kirjeldatud, aitab U-SENS™ eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri. Ent kui seda kasutatakse näiteks katsete tegemist ja hindamist käsitleva ühtse lähenemisviisi raames, võib see aidata hinnata ka sensibiliseeriva toime tugevust. Võimalust kasutada meetodiga U-SENS™ saadud tulemusi toime tugevuse hindamisel on siiski vaja täiendavalt uurida, soovitatavalt inimuuringutest saadud andmete alusel.
|
|
6.
|
Mõisted on esitatud liites 2.1.
|
KATSE PÕHIMÕTE
|
7.
|
U-SENS™ on in vitro meetod, millega määratakse kvantitatiivselt raku pinnamarkeri CD86 ekspressiooni muutusi inimese histiotsüütse lümfoomi rakuliini U937 rakkudes pärast 45 ± 3 tunni pikkust kokkupuudet uuritava kemikaaliga. Pinnamarker CD86 on üks U937 aktiveerimise tüüpiline marker. Teadaolevalt on CD86 kostimulatoorne molekul, mis võib võimaldada jäljendada monotsüütide aktiveerimist, millel on väga oluline roll T-rakkude aktivatsioonis. Pinnamarkeri CD86 ekspressiooni muutusi mõõdetakse läbivoolutsütomeetriliselt pärast rakkude värvimist, milleks tavaliselt kasutatakse fluorestseiinisotiotsüanaadiga (FITC) märgistatud antikehasid. Samal ajal mõõdetakse ka tsütotoksilisust (nt PIga), et hinnata, kas pinnamarkeri CD86 ekspressiooni suurenemine toimub kontsentratsioonil, mille juures ei täheldata tsütotoksilisust. Arvutatakse pinnamarkeri CD86 stimulatsiooniindeks (SI) lahustiga/kandeainega kontrolli näitajaga võrrelduna ja seda tulemust kasutatakse prognoosimudelis (vt punkt 19), et aidata eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri.
|
PÄDEVUSE TÕENDAMINE
|
8.
|
Enne katsemeetodi B.71 juurde kuuluva käesolevas liites kirjeldatud meetodi rutiinset kasutamist tuleks tõendada labori tehnilist pädevust ning kasutada selleks liites 2.2 loetletud kümmet pädevusainet kooskõlas in vitro meetodeid käsitleva hea tavaga (11). Lisaks peaksid meetodi kasutajad haldama reageerimisvõime kontrollimise käigus kogutud andmeid (vt punkt 11) ning positiivsete ja lahustiga/kandeainega kontrollidega saadud varasemaid andmeid (vt punktid 15–16) sisaldavat andmebaasi ning kontrollima nende andmete põhjal, kas katsetulemused on laboris aja möödudes jätkuvalt reprodutseeritavad.
|
KATSE KÄIK
|
9.
|
Käesolev meetod põhineb loomkatsetele alternatiivsete meetodite andmebaasi (DataBase on ALternative Methods, DB-ALM) teenusena kättesaadaval U-SENS™-i käsitleval katse-eeskirjal nr 183 (12). Meetodi U-SENS™ laboris juurutamisel ja rakendamisel tuleks kasutada standardset töökorda. U-SENS™-i rakendamiseks võib kasutada automaatsüsteemi, kui näiteks katsetega, mis tehakse liites 2.2 loetletud pädevusainetega, on võimalik tõendada, et sellise süsteemi kasutamisel saadakse sarnased tulemused. Allpool on kirjeldatud U-SENS™-i põhielemente ja katse käiku.
|
Rakkude ettevalmistamine
|
10.
|
U-SENS™-i puhul tuleks kasutada inimese histiotsüütse lümfoomi rakuliini U937 (13). Soovitatavalt tuleks rakud (kloon CRL1593.2) soetada usaldusväärsest rakupangast, näiteks Ameerika Tüüpkultuuride Kollektsioonist (American Type Culture Collection).
|
|
11.
|
U937 rakke kasvatatakse 37 °C juures reguleeritud õhuniiskusega 5-protsendilise CO2-sisaldusega atmosfääris söötmes RPMI-1 640, mis sisaldab 10 % veiselooteseerumit, 2 mM L-glutamiini, 100 ühikut penitsilliini milliliitri kohta ja streptomütsiini kontsentratsioonis 100 μg/ml (täissööde). U937 rakke külvatakse regulaarselt iga 2–3 päeva järel ümber algtihedusega vastavalt 1,5 või 3 × 105 rakku/ml. Rakkude tihedus ei tohiks olla suurem kui 2 × 106 rakku/ml ja trüpaansinisega mittevärvumise alusel mõõdetud rakkude eluvõimelisus peaks olema ≥ 90 % (ei kohaldata esimesel sulatamisjärgsel väljakülvamisel). Enne katses kasutamist tuleks iga rakkude, veiselooteseerumi ja antikehade partii puhul teha reageerimisvõime kontroll, et veenduda nende sobivuses. Rakkude reageerimisvõime kontroll tuleks teha vähemalt üks nädal pärast sulatamist ning kasutada selleks positiivse kontrollina pikrüülsulfoonhapet (2,4,6-trinitrobenseensulfoonhape, TNBS) (CASi nr: 2 508-19-2; puhtus ≥ 99 %) ja negatiivse kontrollina piimhapet (CASi nr: 50-21-5, puhtus ≥ 85 %). Reageerimisvõime kontrollimisel tuleks kummagi kontrollkemikaali puhul kasutada kuut lõppkontsentratsiooni (TNBS puhul 1, 12,5, 25, 50, 75 ja 100 μg/ml ning piimhappe puhul 1, 10, 20, 50, 100 ja 200 μg/ml). Täissöötmes lahustatud TNBS peaks esile kutsuma CD86 alusel tuvastatava positiivse kontsentratsioonist sõltuva reaktsiooni (nt kui kontsentratsioonile, mille puhul reaktsioon on positiivne, st CD86 SI ≥ 150, järgneb kontsentratsioon, mille juures CD86 SI kasvab) ning täissöötmes lahustatud piimhappe puhul peaks CD86 alusel saadav tulemus olema negatiivne (vt punkt 21). Katses tuleks kasutada üksnes reageerimisvõime kontrolli kaks korda läbinud rakke. Rakke võib paljundada kuni seitsme nädala jooksul pärast sulatamist. Passaažide arv ei tohiks ületada 21. Reageerimisvõime kontroll tuleks teha vastavalt punktides 18–22 esitatud kirjeldusele.
|
|
12.
|
Katses kasutamiseks külvatakse U937 rakud välja tihedusega 3 × 105 rakku/ml või 6 × 105 rakku/ml ning eelkasvatatakse neid rakukultuuripudelis vastavalt kaks päeva või üks päev. Eespool kirjeldatust erinevaid eelkasvatamise tingimusi võib kasutada juhul, kui see on teaduslikult piisavalt põhjendatud ja näiteks katsetega, mis tehakse liites 2.2. loetletud pädevusainetega, on võimalik tõendada, et saadavad tulemused on sarnased. Katse päeval eemaldatakse rakud rakukultuuripudelist ja suspendeeritakse uuesti värskes söötmes tihedusel 5 × 105 rakku/ml. Seejärel jaotatakse rakud 100 μl kaupa 96-kannulise lamedapõhjalise plaadi kannudesse (rakkude lõpptihedus on 0,5 × 105 rakku kannu kohta).
|
Uuritavate kemikaalide ja kontrollainete ettevalmistamine
|
13.
|
Enne katse tegemist hinnatakse lahustuvust. Selleks tehakse uuritavast kemikaalist lahus või püsiv dispersioon kontsentratsiooniga 50 mg/ml; lahustina/kandeainena kasutatakse eelistatavalt täissöödet või kui uuritav kemikaal ei ole täissöötmes lahustuv, siis dimetüülsulfoksiidi (DMSO, puhtusega ≥ 99 %). Katse tegemiseks lahustatakse uuritav kemikaal täissöötmes lõppkontsentratsioonini 0,4 mg/ml, kui kemikaal on kõnealuses lahustis/kandeaines lahustuv. Kui kemikaal lahustub üksnes DMSOs, lahustatakse kemikaal selles kontsentratsioonil 50 mg/ml. Kui see on teaduslikult piisavalt põhjendatud, võib kasutada ka eespool nimetatutest erinevat lahustit/kandeainet. Tuleks võtta arvesse uuritava kemikaali püsivust lahustit/kandeainet sisaldavas lõpplahuses.
|
|
14.
|
Uuritavad kemikaalid ja kontrollained valmistatakse ette katse tegemise päeval. Kuna annuse kindlaksmääramise katset ei tehta, kasutatakse esimeses analüüsitsüklis kuut lõppkontsentratsiooni (1, 10, 20, 50, 100 ja 200 μg/ml) vastavas lahustis/kandeaines, milleks on kas täissööde või 0,4 % DMSOd sisaldav sööde. Järgnevate analüüsitsüklite jaoks valmistatakse uuritavast kemikaalist vastava lahusti/kandeaine abil vähemalt neli töölahust (st vähemalt nelja eri kontsentratsiooniga); seejuures alustatakse täissöötmes lahustamisel kontsentratsioonist 0,4 mg/ml ja DMSOs lahustamisel kontsentratsioonist 50 mg/ml. Lõpuks lisatakse töölahustega töötlemiseks plaadil olevale töölahusele võrdne kogus U937 rakkude suspensiooni (vt punkt 11), et saavutada täiendav kahekordne lahjendus (12). Võimalikes täiendavates analüüsitsüklites kasutatavad kontsentratsioonid (vähemalt neli) valitakse kõigi eelnenud analüüsitsüklite tulemuste põhjal (8). Kasutamiseks sobivad lõppkontsentratsioonid on 1, 2, 3, 4, 5, 7,5, 10, 12,5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 ja 200 μg/ml. Suurim lubatud lõppkontsentratsioon on 200 μg/ml. Kui kontsentratsioonil 1 μg/ml täheldatakse CD86 puhul positiivset väärtust, hinnatakse kontsentratsiooni 0,1 μg/ml, et leida uuritava kemikaali selline kontsentratsioon, mille juures CD86-põhist positiivsuse läviväärtust ei ületata. Igas analüüsitsüklis, kus täheldatakse CD86-põhist kontsentratsioonist sõltuvat positiivset reaktsiooni, arvutatakse EC150 (kemikaali kontsentratsioon, mille juures saavutatakse CD86-põhine positiivsuse läviväärtus 150 %; vt punkt 19). Kui uuritav kemikaal kutsub esile kontsentratsioonist sõltumatu positiivse CD86-põhisereaktsiooni, ei pruugi EC150 arvutamine olla asjakohane, nagu on kirjeldatud U-SENS™-i käsitlevas DB-ALMi katse-eeskirjas nr 183 (12). Igas analüüsitsüklis arvutatakse võimaluse korral alati CV70 (kontsentratsioon, mille juures kemikaali tsütotoksilisus saavutab läviväärtuse 70 %; vt punkt 19) (12). Et uurida CD86 taseme tõusu sõltuvust kontsentratsioonist, tuleks kasutamiseks sobivate kontsentratsioonide hulgast valida sellised kontsentratsioonid, mis on ühtlaselt jaotunud vahemikus EC150-st (või suurimast mittetsütotoksilisest kontsentratsioonist, mille juures CD86-põhine tulemus on negatiivne) kuni CV70-ni (või suurima lubatud kontsentratsioonini 200 μg/ml). Igas analüüsitsüklis tuleks kasutada vähemalt nelja kontsentratsiooni, millest kaks peaksid võrdlemise võimaldamiseks olema samad kui eelnenud analüüsitsükli(te)s.
|
|
15.
|
U-SENS™-i puhul kontrollina kasutatav lahusti/kandeaine on täissööde (kui uuritavast kemikaalist tehakse lahus või püsiv dispersioon söötmes) (vt punkt 4) või 0,4 % DMSO täissöötmes (kui uuritavast kemikaalist tehakse lahus või püsiv dispersioon DMSOs).
|
|
16.
|
U-SENS™-i puhul kasutatav positiivne kontroll on TNBS (vt punkt 11), mille ettevalmistamiseks kasutatakse täissöödet. TNBSi tuleks CD86 ekspressiooni mõõtmisel kasutada plaadil positiivse kontrollina ühes lõppkontsentratsioonis (50 μg/ml), mille puhul rakkude eluvõimelisus on > 70 %. TNBSi kasutamiseks plaadil kontsentratsioonis 50 μg/ml valmistatakse TNBSist täissöötmes 1 M põhilahus (st kontsentratsiooniga 293 mg/ml) ning lahjendatakse seda seejärel täissöötmes 2 930 korda, et saada töölahus kontsentratsiooniga 100 μg/ml. Negatiivse kontrollina tuleks kasutada piimhapet (CASi nr: 50-21-5), mille põhilahusest kontsentratsiooniga 0,4 mg/ml valmistatakse täissöötmes lahus kontsentratsiooniga 200 μg/ml. Igas analüüsitsüklis valmistatakse igal plaadil ette kolm täissöödet sisaldava töötlemata kontrolli, lahustiga/kandeainega kontrolli ning negatiivse ja positiivse kontrolli paralleelproovi (12). Võib kasutada ka mõnda muud sobivat positiivset kontrolli, kui on olemas varasemad andmed, mis võimaldavad kindlaks määrata võrreldavad analüüsitsükli nõuetekohasuse kriteeriumid. Kõnealused analüüsitsükli nõuetekohasuse kriteeriumid on samad kui uuritava kemikaali puhul (vt punkt 12).
|
Uuritavate kemikaalide ja kontrollainetega töötlemine
|
17.
|
Punktides 14–16 kirjeldatud lahustiga/kandeainega kontrollproovid ja töölahused segatakse 96-kannulisele lamedapõhjalisele plaadile kantud rakususpensiooniga (vt punkt 12) mahuvahekorras 1: 1. Seejärel inkubeeritakse töödeldavaid plaate 37 °C juures 5-protsendilise CO2-sisaldusega atmosfääris 45 ± 3 tundi. Enne inkubeerimist kaetakse plaadid poolläbilaskva membraaniga, et takistada lenduvate uuritavate kemikaalide aurustumist ja uuritavate kemikaalidega töödeldud rakkude ristsaastumist (12).
|
Rakkude värvimine
|
18.
|
Pärast 45 ± 3 tundi kestnud kokkupuudet viiakse rakud V-kujuliste kannudega mikrotiiterplaadile ja kogutakse tsentrifuugimise teel. Lahustuvushäire korral tekivad kristallid või tilgad, mis on mikroskoobi all vaadeldavad pärast 45 ± 3 tunni pikkust töötlemist (enne rakkude värvimist). Supernatant visatakse ära ja allesjäänud rakke pestakse üks kord 100 μl jääkülmas fosfaadiga puhverdatud soolalahuses (PBS), mis sisaldab 5 % veiselooteseerumit (värvimispuhver). Pärast tsentrifuugimist suspendeeritakse rakud uuesti 100 μl värvimispuhvris ja neid värvitakse 4 °C juures 30 minuti vältel valguse eest kaitstult 5 μl (nt 0,25 μg) CD86-vastase antikehaga või hiire IgG1ga (isotüübikontroll), mis on märgistatud FITCga. Tuleks kasutada U-SENS™-i käsitlevas DB-ALMi katse-eeskirjas nr 183 (12) kirjeldatud antikehasid (CD86 puhul: BD-PharMingen, katalooginumber 5556 57, kloon Fun-1, või Caltag/Invitrogen, katalooginumber MHCD8601, kloon BU63; IgG1 puhul: BD-PharMingen, katalooginumber 5557 48, või Caltag/Invitrogen, katalooginumber GM4992). Katsemeetodi väljatöötajate kogemuste põhjal on eri partiide antikehade fluorestsentssignaali tugevus tavaliselt sarnane. Katses võib kasutada muid kloone või muu tarnija antikehi, millega on edukalt viidud läbi reageerimisvõime kontroll (vt punkt 11). Kasutajad võivad siiski kaaluda antikehade tiitrimist oma labori tingimustes, et teha kindlaks kõige paremad kasutamiseks sobivad kontsentratsioonid. Mõnda muud tuvastamissüsteemi, näiteks muu fluorokroomiga märgistatud CD86-vastaseid antikehasid võib kasutada juhul, kui näiteks katsetega, mis tehakse liites 2.2 loetletud pädevusainetega, on võimalik tõendada, et asjaomaste antikehadeja FITCga konjugeeritud antikehade puhul saadavad tulemused on sarnased. Pärast vähemalt kaks korda 100 μl värvimispuhvriga ja üks kord 100 μl jääkülma PBSiga pesemist suspendeeritakse rakud uuesti jääkülmas PBSis (nt katsuti kaupa käsitsi analüüsitavate proovide puhul 125 mikroliitris või proovide automaatsisestamiseks ette nähtud plaadi kasutamisel 50 mikroliitris) ning lisatakse PI lahus (lõppkontsentratsioon 3 μg/ml). Võib kasutada ka muid tsütotoksilisuse markereid, näiteks 7-aminoaktinomütsiin D-d (7-AAD) või trüpaansinist, kui näiteks katsetega, mis tehakse liites 2.2 loetletud pädevusainetega, on võimalik tõendada, et asjaomase muu värvainega ja PIga saadavad tulemused on sarnased.
|
Läbivoolutsütomeetriga analüüsimine
|
19.
|
CD86 ekspressioonitaset ja rakkude eluvõimelisust analüüsitakse läbivoolutsütomeetriga. Esimeses väravas R1 täheldatava rakupopulatsiooni selgeks tuvastamiseks ja rakuprügi elimineerimiseks kujutatakse rakke suuruse (FSC) ja granulaarsuse (SSC) punktdiagrammil, mis on esitatud logaritmskaalal. Iga kannu kohta registreeritakse väravas R1 kokku 10 000 rakku. Sama värava R1 rakke kujutatakse FL3 või FL4 ja SSC punktdiagrammil. Eluvõimeliste rakkude eristamiseks kasutatakse teist väravat R2, mille abil valitakse välja propiidiumjodiidi suhtes negatiivsete rakkude populatsioon (kanal FL3 või FL4). Tsütomeetri analüüsiprogrammi abil rakkude eluvõimelisuse arvutamiseks võib kasutada allpool esitatud valemit. Kui rakkude eluvõimelisus on väike, võib registreerida koos surnud rakkudega kokku kuni 20 000 rakku. Teine võimalus on registreerida andmeid ühe minuti jooksul alates analüüsi alustamise hetkest.

Seejärel mõõdetakse väravas R2 (R1 sees) olevate eluvõimeliste rakkude seas kanalis FL1 positiivsena registreeritud rakkude protsentuaalne osakaal. CD86 ekspressiooni rakkude pinnal analüüsitakse eluvõimelisi rakke (R2) kujutaval FL1 ja SSC punktdiagrammil.
Täissöötmega/IgG1ga kannude puhul seadistatakse analüüsimarker põhipopulatsioonile lähedale selliselt, et IgG1 signaaliga rakkude osakaal täissöötmega kontrollides jääks vahemikku 0,6–0,9 %.
Värvusest tulenev segav mõju avaldub nihkena FITCga märgistatud IgG1 punktdiagrammil (kanalis FL1 registreeritud IgG1 signaalile vastava SI geomeetriline keskmine on ≥ 150 %).
Töötlemata või 0,4 % DMSOd sisaldavate kontrollproovide rakkude ja kemikaaliga töödeldud rakkude CD86 stimulatsiooniindeks arvutatakse vastavalt järgmisele valemile:.

IgG1+-rakkude % töötlemata kontrollis – töötlemata eluvõimeliste rakkude seas analüüsimarkeriga kindlaks määratud, kanalis FL1 registreeritud positiivsete IgG1 signaaliga rakkude protsentuaalne osakaal (lubatav vahemik on ≥ 0,6 % ja < 1,5 %; vt punkt 22).
IgG1+/CD86+-rakkude % kontrollis/töödeldud proovis – kanalis FL1 registreeritud positiivsete IgG1/CD86 signaaliga rakkude protsentuaalne osakaal, mille mõõtmisel ei ole analüüsimarkerit eluvõimeliste kontrollrakkude / töödeldud rakkude suhtes nihutatud.
|
ANDMED JA ARUANDLUS
Andmete hindamine
|
20.
|
U-SENS™-i puhul arvutatakse järgmised näitajad: CV70 väärtus ehk kontsentratsioon, mille juures elusate U937 rakkude osakaal on 70 % (tsütotoksilisus on 30 %), ja EC150 väärtus ehk kontsentratsioon, mille juures uuritava kemikaali põhjustatud CD86 stimulatsiooniindeks (SI) on 150 %.
CV70 arvutatakse log-lineaarse interpolatsiooni teel järgmise valemiga:
CV70 = C1 + [(V1 – 70) / (V1 – V2) * (C2 – C1)],
kus
V1 on rakkude eluvõimelisuse vähim väärtus, mis ületab 70 %,
V2 on rakkude eluvõimelisuse suurim väärtus, mis jääb alla 70 %, ning
C1 ja C2 on kontsentratsioonid, mille juures rakkude eluvõimelisus on vastavalt V1 või V2.

CV70 leidmiseks on lubatud kasutada ka muid meetodeid, kui on tõendatud (nt katsetega, mis on tehtud pädevusainetega), et see ei mõjuta tulemusi.
EC150 arvutatakse log-lineaarse interpolatsiooni teel järgmise valemiga:
EC150 = C1 + [(150 – SI1) / (SI2 – SI1) * (C2 – C1)],
kus
C1 on suurim kontsentratsioon (μg/ml), mille juures CD86 SI < 150 % (SI1), ja
C2 on vähim kontsentratsioon (μg/ml), mille juures CD86 SI ≥ 150 % (SI2).
EC150 ja CV70 väärtused arvutatakse järgmiselt:
|
—
|
iga analüüsitsükli puhul saadud konkreetseid EC150 ja CV70 väärtusi kasutatakse selleks, et uurida CD86 taseme tõusu sõltuvust kontsentratsioonist (vt punkt 14);
|
|
—
|
keskmiste eluvõimelisuse väärtuste põhjal tehakse kindlaks üldine CV70 (12);
|
|
—
|
keskmiste CD86 SI väärtuste põhjal tehakse U-SENS™-i kohase prognoosi järgi POSITIIVSEKS loetud uuritava kemikaali puhul (vt punkt 21) kindlaks üldine EC150 (12).
|
|
Prognoosimudel
|
21.
|
CD86 ekspressiooni mõõtmisel hinnatakse iga uuritavat kemikaali vähemalt neljal kontsentratsioonil ja vähemalt kahes sõltumatus eri päevadel läbi viidavas analüüsitsüklis, mille põhjal tehakse üks prognoosijäreldus (NEGATIIVNE või POSITIIVNE).
|
—
|
U-SENS™-i iga üksiku analüüsitsükli kohast prognoosi käsitatakse negatiivsena (N), kui CD86 SI on kõigil mittetsütotoksilistel kontsentratsioonidel (rakkude eluvõimelisus on ≥ 70 %) alla 150 % ja ei täheldata segavat mõju (seoses tsütotoksilisusega, lahustuvusega (vt punkt 18) või värvusega (vt punkt 19), olenemata mittetsütotoksilisest kontsentratsioonist, mille juures segav mõju ilmneb). Kõigil muudel juhtudel – kui CD86 SI on 150 % või suurem ja/või täheldatakse segavat mõju – käsitatakse U-SENS™-i iga üksiku analüüsitsükli kohast prognoosi positiivsena (P).
|
|
—
|
U-SENS™-i kohast prognoosi käsitatakse NEGATIIVSENA, kui vähemalt kahe sõltumatu analüüsitsükli kohased prognoosid on negatiivsed (N) (joonis 1). Kui kahes esimeses analüüsitsüklis saadakse negatiivne (N) tulemus, siis tunnistatakse U-SENS™-i kohane prognoos NEGATIIVSEKS ja kolmandat analüüsitsüklit ei ole vaja läbi viia.
|
|
—
|
U-SENS™-i kohast prognoosi käsitatakse POSITIIVSENA, kui vähemalt kahe sõltumatu analüüsitsükli kohased prognoosid on positiivsed (P) (joonis 1). Kui kahes esimeses analüüsitsüklis saadakse positiivne (P) tulemus, siis tunnistatakse U-SENS™-i kohane prognoos POSITIIVSEKS ja kolmandat analüüsitsüklit ei ole vaja läbi viia.
|
|
—
|
Kuna annuse kindlaksmääramise katset ei tehta, käsitatakse erandina olukorda, kus esimeses analüüsitsüklis on CD86 SI 150 % või suurem üksnes suurimal mittetsütotoksilisel kontsentratsioonil. Sel juhul käsitatakse analüüsitsüklis saadud tulemust EBASELGENA (ES) ning tuleks läbi viia täiendavad analüüsitsüklid, kus kasutatakse ka muid kontsentratsioone (mis jäävad suurima mittetsütotoksilise ja vähima tsütotoksilise kontsentratsiooni vahele; vt punkt 20). Kui analüüsitsükli tulemus on ES, tuleks läbi viia veel vähemalt kaks analüüsitsüklit, millele lisandub neljas analüüsitsükkel juhul, kui teise ja kolmanda tsükli tulemused (teineteisest sõltumatult N ja/või P) ei lange kokku (joonis 1). Täiendavate analüüsitsüklite tulemust käsitatakse positiivsena isegi juhul, kui CD86 SI väärtust 150 % või enam täheldatakse üksnes ühel mittetsütotoksilisel kontsentratsioonil, sest kontsentratsioone on kohandatud vastavalt konkreetsele uuritavale kemikaalile. Lõplik prognoos tehakse selle põhjal, millise tulemuse osakaal on kolme või nelja eraldi analüüsitsükli lõikes suurem (st kaks kolmest või kaks neljast) (joonis 1).
|
Joonis 1. U-SENS™-i puhul kasutatav prognoosimudel; U-SENS™-i kohast prognoosi tuleks vaadelda ühtse lähenemisviisi raames ning kooskõlas punkti 4 ja üldise sissejuhatuse punktide 7, 8 ja 9 nõuetega
N – analüüsitsükkel, kus CD86 ei ole positiivne ja segavat mõju ei täheldata;
P – analüüsitsükkel, kus CD86 on positiivne ja/või täheldatakse segavat mõju;
ES – ebaselge; ebaselge tulemusega esimene analüüsitsükkel, kus CD86 on positiivne üksnes suurima mittetsütotoksilise kontsentratsiooni juures;
# – üksnes esimese analüüsitsükli põhjal ebaselgeks (ES) loetud üksiktulemus tingib automaatselt vajaduse viia läbi kolmas analüüsitsükkel, et saavutada positiivsete (P) või negatiivsete (N) tulemuste ülekaal vähemalt kolmest sõltumatust analüüsitsüklist vähemalt kahes;
$ – kastides on esitatud kolme analüüsitsükli tulemuste kombinatsioonid lähtuvalt ülemises kastis esitatud kahe esimese tsükli tulemustest;
° – kastides on esitatud nelja analüüsitsükli tulemuste kombinatsioonid lähtuvalt ülemises kastis esitatud kolme esimese tsükli tulemustest.
|
Nõuetekohasuse kriteeriumid
|
22.
|
Meetodi U-SENS™ kasutamisel peaksid olema täidetud järgmised katse nõuetekohasuse kriteeriumid (12).
|
—
|
U937 rakkude keskmine eluvõimelisus kolmes töötlemata paralleelproovis peab 45 ± 3 tundi kestnud kokkupuute lõpus olema > 90 % ja CD86 ekspressioonis ei tohi esineda triivi. Töötlemata U937 rakkude CD86 ekspressiooni baastasemele vastav väärtus peab jääma vahemikku ≥ 2 % ja ≤ 25 %.
|
|
—
|
DMSO kasutamisel lahustina arvutatakse DMSO juuresolekul saadud SI võrrelduna töötlemata rakkudega saadud SIga, et hinnata kandeainena DMSOd sisaldava kontrolli nõuetekohasust; seejuures peab kolme paralleelproovi rakkude keskmine eluvõimelisus olema > 90 %. Kandeainena DMSOd sisaldav kontroll on nõuetekohane, kui selle kolme paralleelprooviga saadud CD86 SI keskväärtus on väiksem kui 250 % töötlemata U937 rakkude kolme paralleelprooviga saadud CD86 SI keskväärtusest.
|
|
—
|
Analüüsitsükkel tunnistatakse nõuetekohaseks, kui töötlemata U937 rakkude puhul jääb vähemalt kaks kolmest IgG1ga saadud signaali väärtusest vahemikku ≥ 0,6 % ja < 1,5 %.
|
|
—
|
Samaaegselt hinnatav negatiivne kontroll (piimhappega) tunnistatakse nõuetekohaseks, kui kolmest paralleelproovist vähemalt kaks on negatiivsed (CD86 SI on < 150 %) ja neis ei täheldata tsütotoksilisust (rakkude eluvõimelisus on ≥ 70 %).
|
|
—
|
Positiivne kontroll (TNBSiga) tunnistatakse nõuetekohaseks, kui kolmest paralleelproovist vähemalt kaks on positiivsed (CD86 SI on ≥ 150 %) ja neis ei täheldata tsütotoksilisust (rakkude eluvõimelisus on ≥ 70 %).
|
|
Katseprotokoll
|
23.
|
Katseprotokoll peaks sisaldama järgmist teavet.
|
Uuritav kemikaal
Ühest koostisosast koosnev aine:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed) ja CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muu identimisteave;
|
|
füüsiline välimus, lahustuvus täissöötmes, lahustuvus DMSOs, molekulmass ja muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti/kandeaine valimise põhjendus.
|
Mitmest koostisosast koosnev aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal või segu:
|
võimalikult täpne omaduste kirjeldus lähtuvalt näiteks koostisosade keemilisest määratlusest (vt eespool), puhtusest, kvantitatiivsest sisaldusest ja asjakohaseid füüsikalis-keemilisi omadusi käsitlevatest kättesaadavatest andmetest (vt eespool);
|
|
füüsiline välimus, lahustuvus täissöötmes, lahustuvus DMSOs ja muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
teadaoleva koostisega segu/polümeeri puhul molekulmass või näiline molekulmass või muu uuringu tegemiseks asjakohane teave;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti/kandeaine valimise põhjendus.
|
|
|
Kontrollid
Positiivne kontroll:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed) ja CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muu identimisteave;
|
|
füüsiline välimus, lahustuvus DMSOs, molekulmass ja vajaduse korral muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
vajaduse korral viide varasematele positiivse kontrolliga saadud tulemustele, millest nähtub vastavus asjakohastele analüüsitsükli nõuetekohasuse kriteeriumidele.
|
Negatiivne ja lahustiga/kandeainega kontroll:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed) ja CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muu identimisteave;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
asjaomases katsejuhendis nimetamata lahusti/kandeaine kasutamisel kontrollina kättesaadav teave selle füüsilise välimuse, molekulmassi ja muude asjakohaste füüsikalis-keemiliste omaduste kohta;
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti/kandeaine valimise põhjendus.
|
|
|
Katsetingimused:
|
sponsori, katselabori ja uuringu juhi nimi ja aadress;
|
|
kasutatud katsemeetodi kirjeldus;
|
|
kasutatud rakuliin, selle säilitamistingimused ning päritolu (nt asutus, kellelt rakud saadi);
|
|
kasutatud läbivoolutsütomeeter (nt mudel) ja selle seaded, kasutatud globuliin, antikehad ja tsütotoksilisuse marker;
|
|
pädevusainete analüüsimise meetod, mida kasutati labori pädevuse tõendamiseks, ning katsemeetodiga saadud tulemuste pikaajalise reprodutseeritavuse tõendamise viis, näiteks laboris kontrollidega ja/või reageerimisvõime kontrollimisel varem saadud andmed.
|
|
|
Katse nõuetekohasuse kriteeriumid:
|
lahustiga/kandeainega kontrolli puhul saadud rakkude eluvõimelisuse ja CD86 SI väärtused võrrelduna vastavate nõuetekohasust kajastavate vahemikega;
|
|
positiivse kontrolliga saadud rakkude eluvõimelisuse ja SI väärtused võrrelduna vastavate nõuetekohasust kajastavate vahemikega;
|
|
rakkude eluvõimelisus uuritud kemikaali igal katses kasutatud kontsentratsioonil.
|
|
|
Katse käik:
|
uuritava kemikaali kontsentratsioonid, lisamisviis ja kasutatud kokkupuuteaeg (kui erineb soovitatust);
|
|
kasutatud hindamis- ja otsustamiskriteeriumide kirjeldus;
|
|
kõikide katse käigus tehtud muudatuste kirjeldus.
|
|
|
Tulemused:
|
tabeli kujul andmed uuritava kemikaali ja positiivse kontrolli kohta igas analüüsitsüklis, sh CV70 (vajaduse korral), SI, rakkude eluvõimelisuse väärtused, EC150 väärtused (vajaduse korral) ja prognoosimudelil põhinev hinnang uuritava kemikaali kohta;
|
|
vajaduse korral mis tahes muude asjakohaste vaatlusandmete kirjeldus.
|
|
|
Tulemuste arutelu:
|
U-SENS™-iga saadud tulemuste arutelu;
|
|
katse tulemuste hindamine ühtse lähenemisviisi kontekstis, kui muu asjakohane teave on kättesaadav.
|
|
Järeldused
|
KIRJANDUS
|
(1)
|
Piroird, C., Ovigne, J. M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., ja Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29: 901–916.
|
|
(2)
|
ECVAM (2017). The U-SENS™ test method Validation Study Report. Kättesaadav aadressil http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations.
|
|
(3)
|
ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2787/815737. Kättesaadav aadressil [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2760/588955. Kättesaadav aadressil https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014. Cosmetics 3: 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation. Series on Testing and Assessment No. 256. ENV/JM/MONO(2016)29. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil [http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P. S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., ja Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71: 337–351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., ja Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol. In Vitro 30: 373–382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., ja Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29: 259–270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., ja Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87: 1 683–1 696.
|
|
(11)
|
OECD. (2018). Draft Guidance Document on Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil http://www.oecd.org/env/ehs/testing/OECDFinal Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no. 183: Myeloid U937 Skin Sensitization Test (U-SENS™). 33 lk. Kättesaadav aadressil http://ecvam-dbalm.jrc.ec.europa.eu/.
|
|
(13)
|
Sundström, C., ja Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17: 565–577.
|
|
(14)
|
OECD (2005). Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Series on Testing and Assessment No. 34. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
Ühinenud Rahvaste Organisatsioon (ÜRO) (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6. Kuues, täiendatud väljaanne. United Nations Publications, New York ja Genf. Kättesaadav aadressil http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Contact Sensitization: Classification According to Potency. Technical Report No. 87. Euroopa Kemikaalide Ökotoksikoloogia ja Toksikoloogia Keskus, Brüssel. Kättesaadav aadressil https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Liide 2.1
MÕISTED
Aine
– looduslik või mis tahes tootmisprotsessi tulemusena saadud keemiline element või selle ühend koos püsivuse tagamiseks vajalike lisaainete ja tootmisprotsessi käigus tekkinud lisanditega, mis ei hõlma siiski lahusteid, mida on võimalik aine püsivust mõjutamata või selle koostist muutmata ainest eraldada.
Analüüsitsükkel
– ühe või mitme uuritava kemikaali analüüsimine lahustiga/kandeainega kontrolli ja positiivse kontrolliga paralleelselt.
Asjakohasus
– näitaja, mille abil kirjeldatakse, kuivõrd katsemeetod võimaldab uurida huvipakkuvat toimet, kas selle kasutamine on mõttekas ja kas see on konkreetse eesmärgi jaoks sobiv. Asjakohasusest nähtub, mil määral saab katsemeetodiga õigesti mõõta või ennustada huvipakkuvat bioloogilist toimet. Asjakohasuse puhul võetakse arvesse ka katsemeetodiga saavutatavat täpsust (vastavust) (14).
CD86
– le avalduv kontsentratsioonist sõltuv mõju sõltuvus kontsentratsioonist esineb juhul, kui positiivse tulemuse (CD86 SI ≥ 150) saamiseks vajalikule kontsentratsioonile järgneval kontsentratsioonil täheldatakse CD86 SI suurenemist.
CV70
– hinnanguline kontsentratsioon, mille juures rakkude eluvõimelisus on 70 %.
EC150
– hinnanguline kontsentratsioon, mille puhul CD86 ekspressiooni kajastav SI on 150 %.
Kahjuliku toime rada
– sündmuste ahel alates sihtkemikaali või rühma sarnaste kemikaalide keemilisest struktuurist sõltuvast käivitavast molekulaarsest sündmusest kuni vaadeldava toime avaldumiseni in vivo (15).
Katsete tegemist ja hindamist käsitlev ühtne lähenemisviis
– struktureeritud lähenemisviis, mida kasutatakse kemikaali või rühma kemikaalide ohtlikkuse tuvastamiseks (võimalik toime), ohu kirjeldamiseks (toime tugevus) ja/või ohutuse hindamiseks (võimalik toime / toime tugevus ja kokkupuude) ning mille raames lõimitakse strateegiliselt ja kaalutakse läbi kõik asjakohased andmed võimalikku ohtu ja/või riski ja/või täiendavate sihipäraste ja seega minimaalses ulatuses katsete tegemise vajadust käsitleva regulatiivse otsuse tegemiseks.
Kemikaal
– aine või segu.
Lahustiga/kandeainega kontroll
– töötlemata proov, mis sisaldab kõiki katsesüsteemi komponente peale uuritava kemikaali, sealhulgas lahustit/kandeainet. Selle abil tehakse kindlaks lähtetase samas lahustis/kandeaines lahustatud või püsiva dispersiooni moodustanud uuritava kemikaaliga töödeldud proovide jaoks. Kui sellist töötlemata proovi hinnatakse paralleelselt söötmega kontrolliga, võimaldab see ühtlasi kindlaks teha, kas lahusti/kandeaine mõjutab katsesüsteemi.
Läbivoolutsütomeetria
– tsütomeetriline meetod, mille puhul vedelikus suspendeeritud rakud liiguvad üksteise järel läbi ergastava valguse fookuse ning sellest tingitud valguse hajumise viis kajastab rakkude ja nende koostisosade omadusi; sageli märgistatakse rakud fluorestseeruvate markeritega, mille puhul valgus kõigepealt neeldub ja seejärel kiirgub muutunud sagedusel.
Mitmest koostisosast koosnev aine
– aine, mis on määratletud kvantitatiivse koostise alusel ja milles enam kui ühe põhikoostisosa sisaldus on vähemalt 10, kuid väiksem kui 80 massiprotsenti. Mitmest koostisosast koosnev aine saadakse tootmisprotsessi tulemusena. Erinevus segu ja mitmest koostisosast koosneva aine vahel seisneb selles, et segu saadakse kahe või enama aine kokku segamisel, ilma et toimuks keemilist reaktsiooni. Mitmest koostisosast koosnev aine saadakse keemilise reaktsiooni tulemusel.
Nõuetekohane katsemeetod
– katsemeetod, mille asjakohasust ja usaldusväärsust konkreetse eesmärgi puhul peetakse piisavaks ning mis rajaneb teaduslikult usaldusväärsetel põhimõtetel. Katsemeetod ei ole kunagi nõuetekohane absoluutses tähenduses, vaid üksnes seoses teatud kindla eesmärgiga (14).
Ohtlikkus
– mõjurit või olukorda iseloomustav omadus, millest võib tuleneda kahjulik toime, kui organism, süsteem või (alam)populatsioon selle mõjuriga kokku puutub.
Positiivne kontroll
– paralleelproov, mis sisaldab kõiki katsesüsteemi komponente ja mida on töödeldud ainega, millega teadaolevalt saadakse positiivne tulemus. Positiivse tulemuse väärtus ei tohiks olla liiga suur, et võimaldada hinnata positiivse kontrolliga saadud tulemuste varieeruvust ajas.
Prehapteenid
– kemikaalid, mis muutuvad sensibilisaatoriks abiootilise muundumise, nt oksüdeerumise teel.
Prohapteenid
– kemikaalid, mis vajavad nahka sensibiliseeriva toime tekkimiseks ensümaatilist aktiveerimist.
Segu
– kahest või enamast ainest koosnev segu või lahus.
SI
– stimulatsiooniindeks. Kemikaaliga töödeldud rakkude fluorestsentssignaali tugevuse väärtuste geomeetrilise keskmise suhteline väärtus võrrelduna lahustiga töödeldud rakkude fluorestsentssignaali tugevuse väärtuste geomeetrilise keskmisega.
Spetsiifilisus
– katsemeetodiga õigesti negatiivseks/reaktsioonivõimetuks liigitatud uuritavate kemikaalide osakaal kõikide negatiivsete kemikaalide hulgas. Spetsiifilisus kajastab sellise katsemeetodi täpsust, millega saadakse kategoriseeritav tulemus, ning see on oluline näitaja katsemeetodi asjakohasuse hindamiseks (14).
Triiv
– triiv esineb juhul, kui i) kolmanda töötlemata kontrollprooviga saadud korrigeeritud CD86+(%) väärtus on väiksem kui 50 % esimese ja teise töötlemata kontrollprooviga saadud korrigeeritud CD86+(%) väärtuste keskmisest ning ii) negatiivse kontrolli kolmanda prooviga saadud korrigeeritud CD86+(%) väärtus on väiksem kui 50 % negatiivse kontrolli esimese ja teise prooviga saadud korrigeeritud CD86+(%) väärtuste keskmisest.
Tundlikkus
– katsemeetodiga õigesti positiivseks/reaktsioonivõimeliseks liigitatud uuritavate kemikaalide osakaal kõikide positiivsete kemikaalide hulgas. Tundlikkus kajastab sellise katsemeetodi täpsust, millega saadakse kategoriseeritav tulemus, ning see on oluline näitaja katsemeetodi asjakohasuse hindamiseks (14).
Täpsus
– katsetulemuste ja vastuvõetavate võrdlusväärtuste kooskõla määr. Täpsus näitab katsemeetodi tulemuslikkust ja on üks asjakohastest aspektidest. Seda mõistet kasutatakse sageli vastavuse tähenduses, et väljendada katsemeetodiga saadud õigete tulemuste osakaalu (14).
Usaldusväärsus
– näitaja, mis iseloomustab katsemeetodiga saadud tulemuste reprodutseeritavust pikema aja jooksul samas laboris ja eri laborites, kui meetodit rakendatakse ühe ja sama katse-eeskirja alusel. Usaldusväärsuse hindamiseks arvutatakse laborisisene ja laboritevaheline reprodutseeritavus ning laborisisene korratavus (14).
Uuritav kemikaal
– iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
UVCB
– tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal.
Värvimispuhver
– fosfaadiga puhverdatud soolalahus, mis sisaldab 5 % veiselooteseerumit.
Ühest koostisosast koosnev aine
– aine, mis on määratletud kvantitatiivse koostise alusel ja mille ühe põhikoostisosa sisaldus on vähemalt 80 massiprotsenti.
Ühinenud Rahvaste Organisatsiooni ühtne ülemaailmne kemikaalide klassifitseerimise ja märgistamise süsteem (ÜRO GHS)
– süsteem, millega nähakse ette kemikaalide (ainete ja segude) klassifitseerimine vastavalt nende füüsilise ohtlikkuse ning kahjuliku tervise- ja keskkonnamõju standarditud liigile ja tasemele ning mis hõlmab selliseid asjaomaseid teavitustähiseid nagu piktogrammid, märksõnad, ohulaused, hoiatuslaused ja ohutuskaardid, et anda inimeste (sealhulgas tööandjate, töötajate, vedajate, tarbijate ja päästetöötajate) ning keskkonna kaitsmiseks vajalikku teavet kõnealuste kemikaalide kahjuliku toime kohta (16).
Liide 2.2
PÄDEVUSAINED
Enne katsemeetodi B.71 juurde kuuluvas käesolevas liites kirjeldatud meetodi rutiinset kasutamist tuleks tõendada labori tehnilist pädevust. Selleks tuleks U-SENS™-i kasutamisel saada tabelis 1 esitatud soovitatava kümne aine puhul eeldatavale tulemusele vastav prognoos ning CV70 ja EC150 väärtused, mis jäävad kümnest pädevusainest vähemalt kaheksa puhul asjaomasesse võrdlusvahemikku. Pädevusained on valitud nii, et nende puhul oleksid esindatud naha sensibiliseerimise ohuga seotud eri reaktsioonid. Peale selle on valikukriteeriumidena võetud arvesse ainete kaubanduslikku kättesaadavust, kvaliteetsete in vivo võrdlusandmete olemasolu ja U-SENS™-iga saadud kvaliteetsete in vitro andmete olemasolu. Lisaks on U-SENS™-i kohta olemas avaldatud võrdlusandmed (1, 8).
Tabel 1. 1
U-SENS™-i kasutamise tehnilise pädevuse tõendamiseks soovitatavad ained
|
Pädevusained
|
CASi nr
|
Füüsikaline olek
|
In vivo toime prognoos (104)
|
U-SENS Lahusti/kandeaine
|
U-SENS CV70 võrdlusvahemik, μg/ml (105)
|
U-SENS EC150 võrdlusvahemik, μg/ml (105)
|
|
4-fenüleendiamiin
|
106-50-3
|
Tahkis
|
Sensibilisaator (tugev)
|
Täissööde (106)
|
< 30
|
Positiivne (≤ 10)
|
|
Pikrüülsulfoonhape
|
2508-19-2
|
Vedelik
|
Sensibilisaator (tugev)
|
Täissööde
|
> 50
|
Positiivne (≤ 50)
|
|
Dietüülmaleaat
|
141-05-9
|
Vedelik
|
Sensibilisaator (mõõdukas)
|
DMSO
|
10–100
|
Positiivne (≤ 20)
|
|
Resortsinool
|
108-46-3
|
Tahkis
|
Sensibilisaator (mõõdukas)
|
Täissööde
|
> 100
|
Positiivne (≤ 50)
|
|
Kaneelalkohol
|
104-54-1
|
Tahkis
|
Sensibilisaator (nõrk)
|
DMSO
|
> 100
|
Positiivne (10–100)
|
|
4-allüülanisool
|
140-67-0
|
Vedelik
|
Sensibilisaator (nõrk)
|
DMSO
|
> 100
|
Positiivne (< 200)
|
|
Sahhariin
|
81-07-2
|
Tahkis
|
Sensibiliseeriva toimeta
|
DMSO
|
> 200
|
Negatiivne (> 200)
|
|
Glütserool
|
56-81-5
|
Vedelik
|
Sensibiliseeriva toimeta
|
Täissööde
|
> 200
|
Negatiivne (> 200)
|
|
Piimhape
|
50-21-5
|
Vedelik
|
Sensibiliseeriva toimeta
|
Täissööde
|
> 200
|
Negatiivne (> 200)
|
|
Salitsüülhape
|
69-72-7
|
Tahkis
|
Sensibiliseeriva toimeta
|
DMSO
|
> 200
|
Negatiivne (> 200)
|
|
Lühendid: CASi nr – Chemical Abstracts Service’i registrinumber.
|
3. liide
NAHA SENSIBILISEERIMINE IN VITRO: IL-8 LUTSIFERAASIKATSE
LÄHTEKAALUTLUSED JA PIIRANGUD
|
1.
|
Erinevalt raku pinnamarkerite ekspressiooni analüüsimiseks ette nähtud katsetest mõõdetakse IL-8 lutsiferaasikatsega muutusi IL-8 ekspressioonis. IL-8 on tsütokiin, mis on seotud dendriitrakkude aktiveerimisega. IL-8 ekspressiooni mõõdetakse THP-1 baasil loodud IL-8 reporterrakuliinis (THP-G8, mis on saadud inimese ägeda monotsüütse leukeemia rakuliinist THP-1) pärast rakkude kokkupuudet sensibilisaatoriga (1). Selle meetodi puhul hinnatakse lutsiferaasi ekspressiooni, et aidata eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri.
|
|
2.
|
IL-8 lutsiferaasikatse meetodit on hinnatud Alternatiivsete Meetodite Valideerimise Jaapani Keskuse (JaCVAM), Jaapani majandus-, kaubandus- ja tööstusministeeriumi ning Loomkatsete Alternatiivide Jaapani Ühingu (JSAAE) läbi viidud valideerimisuuringus (2) ning seejärel on see vastastikuse eksperdihinnangu raames sõltumatult läbi vaadatud (3) JaCVAMi ning Jaapani tervishoiu-, töö- ja sotsiaalministeeriumi egiidi all ning alternatiivsete meetodite alase rahvusvahelise koostöö foorumi (ICATM) toetusel. Kõikidest kättesaadavatest andmetest ning reguleerivate asutuste ja sidusrühmade seisukohtadest lähtuvalt peetakse IL-8 lutsiferaasikatset ühtse lähenemisviisi kasulikuks osaks, mis võimaldab aidata ohu alusel klassifitseerimise ja märgistamise eesmärgil eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri. Näited IL-8 lutsiferaasikatsega saadud andmete kasutamisest muu teabega kombineerituna on avaldatud kirjanduses (4, 5, 6).
|
|
3.
|
IL-8 lutsiferaasikatse meetod on osutunud rakendatavaks laborites, kus on olemas rakukultuuride kasutamise ja lutsiferaasi aktiivsuse mõõtmise kogemus. Laborisisese ja laboritevahelise reprodutseeritavuse määr on vastavalt 87,7 % ja 87,5 % (2). Valideerimisuuringu (2) ja muude avaldatud tööde (1, 6) andmetest nähtub, et LLNA tulemustega võrreldes liigitati IL-8 lutsiferaasikatsega 143 kemikaalist 118 positiivseks või negatiivseks ja 25 kemikaali puhul oli tulemus ebaselge ning IL-8 lutsiferaasikatses oli naha sensibilisaatorite (ÜRO GHSi / CLP-määruse kohaselt 1. kategooria kemikaalide) mittesensibiliseerivatest kemikaalidest (ÜRO GHSi /CLP-määruse kohaselt kategoriseerimata kemikaalidest) eristamise täpsus 86 % (101/118), tundlikkus 96 % (92/96) ja spetsiifilisus 41 % (9/22). Kui allpool kirjeldatud kohaldamisalast (punkt 5) välja jäävad ained kõrvale jätta, liigitati IL-8 lutsiferaasikatsega 136 kemikaalist 113 positiivseks või negatiivseks ja 23 kemikaali puhul oli tulemus ebaselge ning IL-8 lutsiferaasikatse meetodi täpsus oli 89 % (101/113), tundlikkus 96 % (92/96) ja spetsiifilisus 53 % (9/17). Inimuuringute andmete alusel, millele on osutanud Urbisch et al. (7), liigitati IL-8 lutsiferaasikatsega 90 kemikaalist 76 positiivseks või negatiivseks ja 14 kemikaali puhul oli tulemus ebaselge ning meetodi täpsus oli 80 % (61/76), tundlikkus 93 % (54/58) ja spetsiifilisus 39 % (7/18). Kui kohaldamisalast välja jäävad ained kõrvale jätta, liigitati IL-8 lutsiferaasikatsega 84 kemikaalist 71 positiivseks või negatiivseks ja 13 kemikaali puhul oli tulemus ebaselge ning meetodi täpsus oli 86 % (61/71), tundlikkus 93 % (54/58) ja spetsiifilisus 54 % (7/13). IL-8 lutsiferaasikatses valenegatiivsete prognooside saamise tõenäosus on suurem vähesel määral kuni mõõdukalt nahka sensibiliseerivate (ÜRO GHSi / CLP-määruse kohaselt alamkategooria 1B) kemikaalide puhul ja väiksem siis, kui tegemist on tugevalt nahka sensibiliseeriva (ÜRO GHSi / CLP-määruse kohaselt alamkategooria 1A) kemikaaliga (6). Kokkuvõttes nähtub sellest teabest, et IL-8 lutsiferaasikatset saab kasutada naha sensibiliseerimise ohu kindlakstegemisel. Ainsa meetodina kasutatava IL-8 lutsiferaasikatsega saavutatav eespool nimetatud täpsus on siiski üksnes orienteeriv, sest katseandmeid tuleks vaadelda ühtse lähenemisviisi kontekstis koos muudest allikatest pärit teabega ja kooskõlas üldise sissejuhatuse punktide 8 ja 7 sätetega. Kui hinnatakse naha sensibiliseerimise katsemeetodeid, mille puhul ei kasutata katseloomi, tuleb lisaks meeles pidada, et LLNA ja muude loomkatsete tulemused ei pruugi täielikult kajastada kemikaali toimet inimesele.
|
|
4.
|
Praeguse olemasoleva teabe põhjal on tõendatud, et IL-8 lutsiferaasikatse meetod on kohaldatav paljude uuritavate kemikaalide puhul, mis erinevad üksteisest orgaanilise funktsionaalrühma, toimemehhanismi, in vivo katsetega määratud nahka sensibiliseeriva toime tugevuse ja füüsikalis-keemiliste omaduste poolest (2, 6).
|
|
5.
|
Kuigi kõnealuses IL-8 lutsiferaasikatses kasutatav lahusti on X-VIVOTM 15, võimaldab see katse anda õige hinnangu kemikaalide kohta, mille log Kow > 3,5 ja vees lahustuvus on programmiga EPI SuiteTM tehtud arvutuste kohaselt ligikaudu 100 μg/ml, ning tuvastada vees halvasti lahustuvaid nahka sensibiliseerivaid kemikaale tõhusamalt kui IL-8 lutsiferaasikatse, milles kasutatakse lahustina dimetüülsulfoksiidi (DMSO) (2). Ent kontsentratsioonil 20 mg/ml mittelahustuvate kemikaalide puhul saadavad negatiivsed tulemused võivad olla valenegatiivsed, kuna sellised kemikaalid ei lahustu X-VIVOTM 15-s. Seepärast tuleks kõnealuste kemikaalide puhul jätta negatiivsed tulemused arvesse võtmata. Valideerimisuuringus täheldati suurt valenegatiivsete tulemuste määra anhüdriidide puhul. Peale selle võidakse kasutatava rakuliini piiratud metaboliseerimisvõime (8) või katsetingimuste tõttu saada katses negatiivseid tulemusi ka prohapteenidega (ained, mis vajavad metaboolset aktiveerimist) ja prehapteenidega (ained, mis aktiveeruvad õhus oksüdeerumisel). Ehkki võimaliku pre- või prohapteeni puhul saadavate negatiivsete tulemuste tõlgendamisel tuleks olla ettevaatlik, oli IL-8 lutsiferaasikatset käsitlevas andmestikus esindatud ainete kohta tehtud õigete järelduste määr prehapteenide puhul 11/11, prohapteenide puhul 6/6 ja pre-/prohapteenide puhul 6/8 (2). Lähtuvalt pre- ja prohapteenide tuvastamiseks kasutatava kolme loomadega mitteseotud katsemeetodi (DPRA, KeratinoSens™ ja h-CLAT) hiljutisest põhjalikust analüüsist (9) ja tulenevalt asjaolust, et IL-8 lutsiferaasikatses kasutatav rakuliin THP-G8 on saadud h-CLATs kasutatavast rakuliinist THP-1, võib IL-8 lutsiferaasikatse meetod muude meetoditega kombineerituna aidata suurendada muudel kui loomkatsetel põhinevate meetodite tundlikkust pre- ja prohapteenide tuvastamisel. Seni uuritud pindaktiivsete ainetega on saadud (vale)positiivsed tulemused, seda olenemata nende liigist (nt katioonsed, anioonsed või mitteioonsed). Lisaks võivad lutsiferaasile segavat mõju avaldavad kemikaalid moonutada selle aktiivsust või mõõtmistulemusi ning seeläbi põhjustada näilist inhibitsiooni või tugevamat luminestsentsi (10). Näiteks on muude lutsiferaasipõhiste reportergeenikatsete puhul täheldatud, et fütoöstrogeenid, mis esinevad suuremas kontsentratsioonis kui 1 μM, mõjutavad luminestsentsisignaale lutsiferaasi reportergeeni üleaktiveerimise teel. Seepärast tuleb lutsiferaasi ekspressioonitaset, mis on mõõdetud suures kontsentratsioonis esineva fütoöstrogeeni või sellise ühendi juuresolekul, mille puhul kahtlustatakse fütoöstrogeeniga sarnast lutsiferaasi reportergeeni aktiveerivat toimet, hoolikalt analüüsida (11). Eespool kirjeldatust tulenevalt ei kuulu pindaktiivsed ained, anhüdriidid ja lutsiferaasile segavat mõju avaldavad kemikaalid käesoleva katsemeetodi kohaldamisalasse. Kui leidub andmeid, mille kohaselt IL-8 lutsiferaasikatset ei saa kasutada mõne muu konkreetse uuritavate kemikaalide rühma puhul, ei tohiks seda asjaomase konkreetse kemikaalirühma puhul kasutada.
|
|
6.
|
Nagu eespool kirjeldatud, aitab IL-8 lutsiferaasikatse eristada naha sensibilisaatoreid kemikaalidest, mis nahka ei sensibiliseeri. Täiendavalt on vaja uurida seda, kas IL-8 lutsiferaasikatse tulemusi saab muudest allikatest pärit teabega kombineerituna kasutada toime tugevuse hindamisel; seda tuleks soovitatavalt teha inimuuringutest saadud andmete alusel.
|
|
7.
|
Mõisted on esitatud liites 3.1.
|
KATSE PÕHIMÕTE
|
8.
|
IL-8 lutsiferaasikatses kasutatakse inimese monotsüütse leukeemia rakuliini THP-1, mis on pärit Ameerika Tüüpkultuuride Kollektsioonist (Manassas, VA, Ameerika Ühendriigid). Selle rakuliini baasil on Tohoku Ülikooli arstiteaduskonna dermatoloogiaosakonnas loodud IL-8 reporterrakuliin THP-G8, mis sisaldab oranži valgust tekitava püsiva lutsiferaasi (SLO) ja punast valgust tekitava püsiva lutsiferaasi (SLR) geene, mis on vastavalt IL-8 ja glütseeraldehüüd-3-fosfaadi dehüdrogenaasi (GAPDH) promootori kontrolli all (1). See võimaldab pärast kokkupuudet sensibiliseeriva kemikaaliga luminestsentskiirguse tuvastamise teel kvantitatiivselt mõõta lutsiferaasi geeni aktiveerumist IL-8 ja GAPDH aktiivsuse näitajana; selleks otstarbeks kasutatakse üldkasutatavaid valgust tekitavaid lutsiferaasi substraate.
|
|
9.
|
Kahe värvuse tekitamist võimaldav katsesüsteem koosneb oranži valgust tekitavast lutsiferaasist (SLO; λmaks = 580 nm) (12), mis võimaldab mõõta IL-8 promootorist sõltuvat geeniekspressiooni, ja punast valgust tekitavast lutsiferaasist (SLR; λmaks = 630 nm) (13), mis võimaldab mõõta sisemise kontrollina kasutatavat GAPDH promootorist sõltuvat geeniekspressiooni. Jaanimardika D-lutsiferiin kiirgab nende kahe lutsiferaasiga reageerides eri värvi valgust ning tekkivat luminestsentskiirgust mõõdetakse üheaegselt üheetapilises reaktsioonis: selleks kasutatakse katsesegust lähtuva valguskiirguse lahutamist optilise filtri abil (14) (liide 3.2).
|
|
10.
|
THP-G8 rakke töödeldakse 16 tunni jooksul uuritava kemikaaliga ning seejärel mõõdetakse lutsiferaasi SLO aktiivsus (SLO-LA), mis kajastab IL-8 promootori aktiivsust, ja lutsiferaasi SLR aktiivsus (SLR-LA), mis kajastab GAPDH promootori aktiivsust. Parema arusaadavuse huvides kasutatakse lühendite SLO-LA ja SLR-LA asemel vastavalt tähiseid IL8LA ja GAPLA. Tabelis 1 on esitatud ülevaade IL-8 lutsiferaasikatse puhul kasutatavatest lutsiferaasi aktiivsusega seotud mõistetest. Mõõdetud väärtuste põhjal arvutatakse järgmised näitajad: normaliseeritud IL8LA (nIL8LA), mida väljendatakse IL8LA ja GAPLA suhtarvuna; nIL8LA suurenemine (Ind-IL8LA), mida väljendatakse neljas uuritava kemikaaliga töödeldud THP-G8 rakkude paralleelproovis mõõdetud nIL8LA väärtuste aritmeetilise keskmise ja neljas töötlemata THP-G8 rakkude paralleelproovis mõõdetud nIL8LA väärtuste aritmeetilise keskmise suhtarvuna; GAPLA inhibeerimise määr (Inh-GAPLA), mida väljendatakse neljas uuritava kemikaaliga töödeldud THP-G8 rakkude paralleelproovis mõõdetud GAPLA väärtuste aritmeetilise keskmise ja neljas töötlemata THP-G8 rakkude paralleelproovis mõõdetud GAPLA väärtuste aritmeetilise keskmise suhtarvuna ning mida kasutatakse tsütotoksilisuse näitajana.
Tabel 1.
IL-8 lutsiferaasikatse puhul kasutatavad lutsiferaasi aktiivsusega seotud mõisted
|
Lühendid
|
Määratlus
|
|
GAPLA
|
Lutsiferaasi SLR aktiivsus, mis kajastab GAPDH promootori aktiivsust
|
|
IL8LA
|
Lutsiferaasi SLO aktiivsus, mis kajastab IL-8 promootori aktiivsust
|
|
nIL8LA
|
IL8LA: GAPLA
|
|
Ind-IL8LA
|
nIL8LA kemikaaliga töödeldud THP-G8 rakkudes: nIL8LA töötlemata rakkudes
|
|
Inh-GAPLA
|
GAPLA kemikaaliga töödeldud THP-G8 rakkudes: GAPLA töötlemata rakkudes
|
|
CV05
|
Kemikaali vähim kontsentratsioon, mille juures Inh-GAPLA on < 0,05
|
|
|
11.
|
Siin kirjeldatud IL-8 lutsiferaasikatse meetodiga sarnaste muudetud in vitro IL-8 lutsiferaasikatse meetodite valideerimise hõlbustamiseks on olemas tulemuslikkuse standardid (15), mis võimaldavad OECD katsejuhendit 442E selliste meetodite lisamiseks õigeaegselt muuta. OECD otsuse kohane andmete vastastikune tunnustamine on tagatud vaid sellise katsemeetodi puhul, mis on valideeritud kooskõlas tulemuslikkuse standarditega ning mille OECD on läbi vaadanud ja katsejuhendisse 442E lisanud (16).
|
PÄDEVUSE TÕENDAMINE
|
12.
|
Enne katsemeetodi B.71 juurde kuuluvas käesolevas liites kirjeldatud meetodi rutiinset kasutamist tuleks tõendada labori tehnilist pädevust ning kasutada selleks liites 3.3 loetletud kümmet pädevusainet kooskõlas in vitro meetodeid käsitleva hea tavaga (17). Lisaks peaksid meetodi kasutajad haldama reageerimisvõime kontrollimise käigus kogutud andmeid (vt punkt 15) ning positiivsete ja lahustiga/kandeainega kontrollidega saadud varasemaid andmeid (vt punktid 21–24) sisaldavat andmebaasi ning kontrollima nende andmete põhjal, kas katsetulemused on laboris aja möödudes jätkuvalt reprodutseeritavad.
|
KATSE KÄIK
|
13.
|
IL-8 lutsiferaasikatse jaoks on olemas standardne töökord, mida tuleks katsemeetodi rakendamisel kasutada (18). Katset läbi viia soovivad laborid saavad soetada rekombinantse THP-G8 rakuliini Jaapanis Tottoris asuvastettevõttest GPC Lab. Co. Ltd., kui nad allkirjastavad OECD vormil esitatud tingimustele vastava materjali üleandmise lepingu. Järgmistes punktides on kirjeldatud katsemeetodi põhielemente ja katse käiku.
|
Rakkude ettevalmistamine
|
14.
|
IL-8 lutsiferaasikatse tegemiseks tuleks kasutada THP-G8 rakuliini, mis on soetatud Jaapanis Tottoris asuvast ettevõttest GPC Lab. Co. Ltd. (vt punktid 8 ja 13). Pärast rakkude kättesaamist paljundatakse neid (2–4 passaaži) ja säilitatakse need külmutatult homogeense tüvikultuurina. Kõnealuse tüvikultuuri rakkude paljundamiseks võib neid kuni 12 korda ümber külvata või kasvatada neid kuni kuue nädala jooksul. Paljundamiseks kasutatakse söödet RPMI-1640, mis sisaldab 10 % veiselooteseerumit, antibiootikume / seentevastast ainet (0,85 % füsioloogilises lahuses 100 ühikut penitsilliin G-d milliliitri kohta, streptomütsiini kontsentratsioonis 100 μg/ml ja amfoteritsiin B-d kontsentratsioonis 0,25 μg/ml) (nt GIBCO, katalooginumber 15240-062), puromütsiini (nt CASi numbriga 58-58-2) kontsentratsioonis 0,15 μg/ml ja G418 (nt CASi numbriga 108321-42-2) kontsentratsioonis 300 μg/ml.
|
|
15.
|
Enne rakkude kasutamist katses tuleks teha reageerimisvõime kontroll, et veenduda rakkude sobivuses. Kõnealune kontroll tuleks teha 1–2 nädalat pärast sulatamist või pärast 2–4 korda ümber külvamist ning kasutada selleks positiivse kontrollina 4-nitrobensüülbromiidi (4-NBB) (CASi nr: 100-11-8; puhtus ≥ 99 %) ja negatiivse kontrollina piimhapet (CASi nr: 50-21-5; puhtus ≥ 85 %). 4-NBB puhul peaks Ind-IL8LA alusel tuvastatav reaktsioon olema positiivne (Ind-IL8LA ≥ 1,4) ja piimhappe puhul negatiivne (Ind-IL8LA < 1,4). Katses kasutatakse üksnes reageerimisvõime kontrolli läbinud rakke. Reageerimisvõime kontroll tuleb teha vastavalt punktides 22–24 esitatud kirjeldusele.
|
|
16.
|
Katses kasutamiseks külvatakse THP-G8 rakud välja tihedusega 2–5 × 105 rakku/ml ning eelkasvatatakse neid rakukultuuripudelis 48–96 tundi. Katse päeval pestakse rakukultuuripudelist eemaldatud rakke söötmega RPMI-1640, mis sisaldab 10 % veiselooteseerumit, aga ei sisalda antibiootikume, ja rakud suspendeeritakse uuesti 10 % veiselooteseerumit sisaldavas antibiootikumideta söötmes RPMI-1640 tihedusel 1 × 106 rakku/ml. Seejärel jaotatakse rakud 50 μl kaupa 96-kannulise lamedapõhjalise plaadi kannudesse (nt Costar, katalooginumber 3603) (5 × 104 rakku kannu kohta).
|
Uuritava kemikaali ja kontrollainete ettevalmistamine
|
17.
|
Uuritav kemikaal ja kontrollained valmistatakse ette katse tegemise päeval. IL-8 lutsiferaasikatse puhul lahustatakse uuritav kemikaal turul kättesaadavas seerumita söötmes X-VIVOTM 15 (Lonza, 04-418Q) lõppkontsentratsioonini 20 mg/ml. X-VIVOTM 15 lisatakse mikrotsentrifuugi katsutis olevale 20 mg uuritavale kemikaalile (olenemata kemikaali lahustuvusest) kuni koguseni 1 ml. Seejärel segatakse lahust ägedalt keerissegistil ja loksutatakse 30 minutit rootori maksimumkiirusel 8 pööret minutis ümbritseva õhu temperatuuril umbes 20oC juures. Kui katsutisse jääb veel lahustumata tahket kemikaali, töödeldakse seda ultraheliga, kuni kemikaal on täielikult lahustunud või moodustab püsiva dispersiooni. Kui uuritav kemikaal on X-VIVOTM 15-s lahustuv, siis lahjendatakse lahust viis korda X-VIVOTM 15-ga ja kasutatakse saadud lahust X-VIVOTM 15-s lahustatud uuritava kemikaali põhilahusena (4 mg/ml). Kui uuritav kemikaal ei ole X-VIVOTM 15-s lahustuv, loksutatakse segu pöördloksutil veel vähemalt 30 minutit ning tsentrifuugitakse seejärel 5 minutit kiirusel 15000 pööret minutis (≈ 20 000 g); saadud supernatanti kasutatakse X-VIVOTM 15-s lahustatud uuritava kemikaali põhilahusena. Muu lahusti, näiteks DMSO, vee või söötme kasutamisel tuleks esitada sellekohane teaduslik põhjendus. Kemikaalide lahustamise korda on üksikasjalikult kirjeldatud liites 3.5. Punktides 18–23 kirjeldatud X-VIVOTM 15 lahused segatakse 96-kannulisele lamedapõhjalisele mustale plaadile kantud rakususpensiooniga (vt punkt 16) mahuvahekorras 1: 1.
|
|
18.
|
Esimese analüüsitsükli puhul on eesmärk teha kindlaks tsütotoksilisuse avaldumise kontsentratsioon ja uurida kemikaali võimet nahka sensibiliseerida. X-VIVOTM 15-s lahustatud uuritava kemikaali põhilahusest tehakse 96-kannulisel katseplaadil (nt Costar, katalooginumber EW-01729-03) X-VIVOTM 15 abil rida kahekordseid lahjendusi (vt liide 3.5). Seejärel lisatakse 96-kannulisele lamedapõhjalisele mustale plaadile kantud 50 μl rakususpensioonile 50 μl lahjendatud lahust kannu kohta. Nii saadakse X-VIVOTM 15-s lahustuva uuritava kemikaali lõppkontsentratsioonid vahemikus 0,002–2 mg/ml (liide 3.5). Selliste uuritavate kemikaalide puhul, mis X-VIVOTM 15-s kontsentratsioonil 20 mg/ml ei lahustu, tehakse kindlaks ainult lahjendustegurid vahemikus 2–210, kuid uuritava kemikaali tegelikud lõppkontsentratsioonid ei ole teada ja sõltuvad uuritava kemikaali küllastuskontsentratsioonist X-VIVOTM 15-s valmistatud põhilahuses.
|
|
19.
|
Järgnevates analüüsitsüklites (st teises, kolmandas ja neljandas tsüklis) valmistatakse põhilahus X-VIVOTM 15-s neli korda suurema kontsentratsiooniga, kui oli esimeses tsüklis kasutatud kontsentratsioon, mille juures rakkude eluvõimelisus oli läviväärtusest väiksem (CV05 – väiksem kontsentratsioon, mille juures Inh-GAPLA on < 0,05). Kui Inh-GAPLA ei lange alla 0,05 ka kõige suuremal esimeses analüüsitsüklis kasutatud kontsentratsioonil, valmistatakse põhilahus X-VIVOTM 15-s esimeses analüüsitsüklis kasutatud suurima kontsentratsiooniga. CV05-le vastava kontsentratsiooni arvutamiseks jagatakse esimeses analüüsitsüklis kasutatud põhilahuse kontsentratsioon CV05 puhul kasutatud lahendusteguriga X (lahjendustegur CV05 (X) – lahjendustegur, mida tuleb kasutada põhilahuse lahjendamiseks kontsentratsioonini CV05) (vt liide 3.5). Selliste uuritavate ainete puhul, mis kontsentratsioonil 20 mg/ml X-VIVOs ei lahustu, tehakse CV05 kindlaks nii, et põhilahuse kontsentratsioon korrutatakse teguriga 1/X. 2.–4. analüüsitsükli jaoks valmistatakse teine põhilahus kontsentratsiooniga 4 × CV05 (liide 3.5).
|
|
20.
|
Teisest põhilahusest valmistatakse X-VIVOTM 15-ga 96-kannulisel katseplaadil rida 1,5-kordseid lahjendusi. Seejärel lisatakse 96-kannulise lamedapõhjalise musta plaadi kannudesse kantud 50 μl rakususpensioonile 50 μl lahjendatud lahust kannu kohta. Iga uuritavat kemikaali tuleks igal kontsentratsioonil analüüsida neljas kannus. Järgnevalt segatakse proovid plaadiloksutil ja neid inkubeeritakse 37oC juures 5-protsendilise CO2-sisaldusega atmosfääris 16 tundi. Seejärel mõõdetakse lutsiferaasi aktiivsus allpool kirjeldatud viisil.
|
|
21.
|
Lahustiga kontrollina kasutatakse segu, mis sisaldab kannu kohta 50 μl X-VIVOTM 15 ja 50 μl rakususpensiooni 10 % veiselooteseerumit sisaldavas RPMI-1640-s.
|
|
22.
|
Soovitatav positiivse kontrolli aine on 4-NBB. 1,5 ml mikrotsentrifuugi katsutisse viiakse 20 mg 4-NBBd ning lisatakse X-VIVOTM 15 kuni koguseni 1 ml. Lahust segatakse ägedalt keerissegistil ja loksutatakse vähemalt 30 minutit jooksul rootori maksimumkiirusel 8 pööret minutis. Pärast 5 minutit tsentrifuugimist kiirendusega 20000 g lahjendatakse supernatanti X-VIVOTM 15-ga neli korda ning 500 μl lahjendatud supernatanti kantakse 96-kannulise katseplaadi ühte kannu. Lahjendatud supernatanti lahjendatakse X-VIVOTM 15-ga uuesti kaks ja neli korda ning igast saadud lahusest lisatakse 50 μl 96-kannulise lamedapõhjalise musta plaadi kannudesse, mis sisaldavad 50 μl THP-G8 rakkude suspensiooni (liide 3.6). Positiivset kontrolli tuleks igal kontsentratsioonil analüüsida neljas kannus. Plaati loksutatakse plaadiloksutil ja inkubeeritakse CO2 inkubaatoris 37oC juures 5-protsendilise CO2-sisaldusega atmosfääris 16 tundi. Seejärel mõõdetakse punktis 29 kirjeldatud viisil lutsiferaasi aktiivsus.
|
|
23.
|
Soovitatav negatiivse kontrolli aine on piimhape. 1,5 ml mikrotsentrifuugi katsutisse viiakse 20 mg piimhapet ning lisatakse X-VIVOTM 15 kuni koguseni 1 ml (20 mg/ml). Piimhappelahust kontsentratsiooniga 20 mg/ml lahjendatakse X-VIVOTM 15-ga veel viis korda (4 mg/ml); 500 μl saadud piimhappelahusest kontsentratsiooniga 4 mg/ml kantakse 96-kannulise katseplaadi ühte kannu. Seda lahust lahjendatakse X-VIVOTM 15-ga kaks korda ja seejärel uuesti kaks korda, et saada lahused kontsentratsiooniga 2 mg/ml ja 1 mg/ml. Igast saadud kolmest lahusest ja kandeainega kontrollist (X-VIVOTM 15) lisatakse 50 μl 96-kannulise lamedapõhjalise musta plaadi kannudesse, mis sisaldavad 50 μl THP-G8 rakkude suspensiooni. Negatiivset kontrolli analüüsitakse igal kontsentratsioonil neljas kannus. Plaati loksutatakse plaadiloksutil ja inkubeeritakse CO2 inkubaatoris 37oC juures 5-protsendilise CO2-sisaldusega atmosfääris 16 tundi. Seejärel mõõdetakse punktis 29 kirjeldatud viisil lutsiferaasi aktiivsus.
|
|
24.
|
Võib kasutada ka mõnda muud sobivat positiivset või negatiivset kontrolli, kui on olemas varasemad andmed, mis võimaldavad kindlaks määrata võrreldavad analüüsitsükli nõuetekohasuse kriteeriumid.
|
|
25.
|
Tuleks jälgida, et lenduvad uuritavad kemikaalid ei aurustuks, ning hoida ära kannudevahelist uuritava kemikaaliga ristsaastumist. Selleks võib plaadi enne uuritava kemikaaliga inkubeerimist näiteks tihedalt kinni katta.
|
|
26.
|
Uuritavate kemikaalide ja lahustiga kontrolli puhul on positiivse või negatiivse prognoosi saamiseks vaja läbi viia 2–4 analüüsitsüklit (vt tabel 2). Iga analüüsitsükkel viiakse läbi eri päeval; igas tsüklis kasutatakse X-VIVOTM 15-s lahustatud uuritava kemikaali värsket põhilahust ja sõltumatult kogutud rakke. Rakud võivad pärineda samast passaažist.
|
Lutsiferaasi aktiivsuse mõõtmine
|
27.
|
Luminestsentskiirgust mõõdetakse 96-kannulise mikroplaadi jaoks ette nähtud luminomeetriga, mis on varustatud optiliste filtritega – nt seadmega Phelios (ATTO, Tokyo, Jaapan) või Tristan 941 (Berthold, Bad Wildbad, Saksamaa) või ARVO-seeria seadmega (PerkinElmer, Waltham, MA, Ameerika Ühendriigid). Reprodutseeritavuse tagamiseks tuleb luminomeeter iga katse eel kalibreerida (19). Kalibreerimiseks on saadaval oranži või punast valgust tekitavad rekombinantsed lutsiferaasid.
|
|
28.
|
Igasse kemikaaliga töödeldud või töötlemata rakususpensiooni sisaldavasse plaadi kannu lisatakse 100 μl lutsiferaasikatse eelsoojendatud reaktiivi Tripluc® (Tripluc). Plaati loksutatakse 10 minutit ümbritseva õhu temperatuuril umbes 20oC juures. Lutsiferaasi aktiivsuse mõõtmiseks asetatakse plaat luminomeetrisse. Bioluminestsentsi mõõdetakse 3 sekundit ilma optilise filtrita (F0) ja 3 sekundit optilise filtriga (F1). Kui kasutatakse muid seadeid, mis võivad sõltuda näiteks kasutatava luminomeetri mudelist, tuleks seda põhjendada.
|
|
29.
|
Mõõdetud väärtuste põhjal arvutatakse soovitud näitajad iga kontsentratsiooni puhul, nt IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, IL8LA keskväärtus ± standardhälve, GAPLA keskväärtus ± standardhälve, nIL8LA keskväärtus ± standardhälve, Ind-IL8LA keskväärtus ± standardhälve, Inh-GAPLA keskväärtus ± standardhälve ja Ind-IL8LA usaldusvahemik usaldusnivool 95 %. Käesolevas punktis nimetatud näitajate määratlused on esitatud liidetes 3.1 ja 3.4.
|
|
30.
|
Mitme värvuse tuvastamist võimaldavate reporterkatsete puhul kasutatakse mõõtmise eel eri värvuste lahutamiseks üldjuhul selliseid detektorseadmeid (luminomeeter ja plaadilugemisseade), mis on varustatud optiliste filtritega, nt teravapiiriliste madal- või kõrgpääsufiltrite või ribapääsufiltritega. Enne katset tuleks määrata filtrite läbilaskvustegurid iga bioluminestsentssignaali värvuse jaoks vastavalt liitele 3.2.
|
ANDMED JA ARUANDLUS
Andmete hindamine
|
31.
|
Positiivseks/negatiivseks liigitamise otsus tehakse igas analüüsitsüklis järgmiste kriteeriumide alusel:
|
—
|
IL-8 lutsiferaasikatsel põhinev prognoos tunnistatakse positiivseks, kui uuritava kemikaaliga saadud Ind-IL8LA väärtus on ≥ 1,4 ja Ind-IL8LAd iseloomustava usaldusnivoole 95 % vastava usaldusvahemiku alampiir on ≥ 1,0;
|
|
—
|
IL-8 lutsiferaasikatsel põhinev prognoos tunnistatakse negatiivseks, kui uuritava kemikaaliga saadud Ind-IL8LA väärtus on < 1,4 ja/või Ind-IL8LAd iseloomustava usaldusnivoole 95 % vastava usaldusvahemiku alampiir on < 1,0.
|
|
Prognoosimudel
|
32.
|
Uuritav kemikaal, mille puhul on esimese, teise, kolmanda ja neljanda analüüsitsükli lõikes saadud kaks positiivset tulemust, tunnistatakse positiivseks; uuritav kemikaal, mille puhul on esimese, teise, kolmanda ja neljanda analüüsitsükli lõikes saadud kolm negatiivset tulemust, tunnistatakse arvatavalt negatiivseks (tabel 2). Arvatavalt negatiivsete kemikaalide seas tunnistatakse negatiivseks sellised kemikaalid, mis lahustuvad X-VIVOTM 15-s kontsentratsioonil 20 mg/ml; kemikaalid, mis X-VIVOTM 15-s kontsentratsioonil 20 mg/ml ei lahustu, tuleks kõrvale jätta (joonis 1).
Tabel 2.
Positiivsete ja arvatavalt negatiivsete kemikaalide kindlakstegemise kriteeriumid
|
1. tsükkel
|
2. tsükkel
|
3. tsükkel
|
4. tsükkel
|
Lõplik prognoos
|
|
Positiivne
|
Positiivne
|
—
|
—
|
Positiivne
|
|
Negatiivne
|
Positiivne
|
—
|
Positiivne
|
|
Negatiivne
|
Positiivne
|
Positiivne
|
|
Negatiivne
|
Arvatavalt negatiivne
|
|
Negatiivne
|
Positiivne
|
Positiivne
|
—
|
Positiivne
|
|
Negatiivne
|
Positiivne
|
Positiivne
|
|
Negatiivne
|
Arvatavalt negatiivne
|
|
Negatiivne
|
Positiivne
|
Positiivne
|
Positiivne
|
|
Negatiivne
|
Arvatavalt negatiivne
|
|
Negatiivne
|
—
|
Arvatavalt negatiivne
|
|
Joonis 1.
Prognoosimudel lõppotsuse tegemiseks

Nõuetekohasuse kriteeriumid
|
33.
|
IL-8 lutsiferaasikatse puhul peaksid olema täidetud järgmised nõuetekohasuse kriteeriumid.
|
—
|
Ind-IL8LA peaks olema igas analüüsitsüklis vähemalt ühel positiivse kontrolli (4-NBB) kontsentratsioonil suurem kui 5,0.
|
|
—
|
Ind-IL8LA peaks olema igas analüüsitsüklis kõikidel negatiivse kontrolli (piimhape) kontsentratsioonidel väiksem kui 1,4.
|
|
—
|
Andmed, mis on saadud plaatidelt, kus rakke ja Triplucit sisaldavates ilma kemikaalita kontrollkannudes täheldatud GAPLA väärtus on alla viie korra suurem kui GAPLA väärtus kannus, mis sisaldab ainult katsesöödet (kannu kohta 50 μl söödet RPMI-1640, mis sisaldab 10 % veiselooteseerumit, ja 50 μl X-VIVOTM 15), tuleks jätta arvesse võtmata.
|
|
—
|
Andmed, mis on saadud plaatidelt, kus uuritava või kontrollkemikaaliga saadud Inh-GAPLA väärtus on kõigil kontsentratsioonidel väiksem kui 0,05, tuleks jätta arvesse võtmata. Sellisel juhul tuleks esimest katset korrata nii, et korduskatses kasutatakse suurima lõppkontsentratsioonina eelmises katses kasutatud vähimat lõppkontsentratsiooni.
|
|
Katseprotokoll
|
34.
|
Katseprotokoll peaks sisaldama järgmist teavet.
|
Uuritavad kemikaalid
Ühest koostisosast koosnev aine:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed) ja CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muu identimisteave;
|
|
füüsiline välimus, lahustuvus vees, molekulmass ja muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
lahustuvus X-VIVOTM 15-s. X-VIVOTM 15-s mittelahustuvate kemikaalide puhul teave selle kohta, kas pärast tsentrifuugimist on näha sadet või hõljumit;
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti/kandeaine valimise põhjendus juhul, kui ei kasutatud X-VIVOTM 15.
|
Mitmest koostisosast koosnev aine, tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal või segu:
|
võimalikult täpne omaduste kirjeldus lähtuvalt näiteks koostisosade keemilisest määratlusest (vt eespool), puhtusest, kvantitatiivsest sisaldusest ja asjakohaseid füüsikalis-keemilisi omadusi käsitlevatest kättesaadavatest andmetest (vt eespool);
|
|
füüsiline välimus, lahustuvus vees ja muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
teadaoleva koostisega segu/polümeeri puhul molekulmass või näiline molekulmass või muu uuringu tegemiseks asjakohane teave;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
lahustuvus X-VIVOTM 15-s. X-VIVOTM 15-s mittelahustuvate kemikaalide puhul teave selle kohta, kas pärast tsentrifuugimist on näha sadet või hõljumit;
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti/kandeaine valimise põhjendus juhul, kui ei kasutatud X-VIVOTM 15.
|
|
|
Kontrollid
Positiivne kontroll:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed) ja CASi number/numbrid, SMILESi või InChI kood, struktuurivalem ja/või muu identimisteave;
|
|
füüsiline välimus, lahustuvus vees, molekulmass ja muud asjakohased kättesaadavad andmed füüsikalis-keemiliste omaduste kohta;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
vajaduse korral teave töötlemise kohta enne katset (nt soojendamine, peenestamine);
|
|
kasutatud kontsentratsioon(id);
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
vajaduse korral viide varasematele positiivse kontrolliga saadud tulemustele, millest nähtub vastavus asjakohastele nõuetekohasuse kriteeriumidele.
|
Negatiivne kontroll:
|
kemikaali identimisandmed, näiteks IUPACi või CASi nimetus(ed) ja CASi number/numbrid ja/või muu identimisteave;
|
|
puhtus, võimaluse ja vajaduse korral lisandite keemiline määratlus jne;
|
|
asjaomases katsejuhendis nimetamata negatiivsete kontrolli aine kasutamisel kättesaadav teave selle füüsilise välimuse, molekulmassi ja muude asjakohaste füüsikalis-keemiliste omaduste kohta;
|
|
kättesaadav teave säilitamistingimuste ja püsivuse kohta;
|
|
iga uuritava kemikaali puhul lahusti valimise põhjendus.
|
|
|
Katsetingimused:
|
sponsori, katselabori ja uuringu juhi nimi ja aadress;
|
|
kasutatud katsemeetodi kirjeldus;
|
|
kasutatud rakuliin, selle säilitamistingimused ning päritolu (nt asutus, kellelt rakud saadi);
|
|
veiselooteseerumi partiinumber ja päritolu, tarnija nimi, 96-kannulise lamedapõhjalise musta plaadi partiinumber ja reaktiivi Tripluc partiinumber;
|
|
passaaži number ja rakkude tihedus katses kasutamisel;
|
|
enne katset rakkude külvamisel kasutatud rakuloendusmeetod ning võetud meetmed, millega tagati rakkude homogeenne jaotus;
|
|
kasutatud luminomeeter (nt mudel), sealhulgas seadme seaded, kasutatud lutsiferaasisubstraat ja liites 3.2 kirjeldatud kontrollkatsel põhinev tõendus asjakohaste luminestsentskiirguse mõõtmiste läbiviimise kohta;
|
|
laboris katse tegemiseks vajaliku pädevuse tõendamiseks (nt pädevusainetega katsete tegemiseks) kasutatud meetod või katsemeetodiga saadud tulemuste pikaajalise reprodutseeritavuse tõendamiseks kasutatud meetod.
|
|
|
Katse käik:
|
proovide ja analüüsitsüklite arv;
|
|
uuritava kemikaali kontsentratsioonid, lisamisviis ja kasutatud kokkupuuteaeg (kui erineb soovitatust);
|
|
kasutatud hindamis- ja otsustamiskriteeriumide kirjeldus;
|
|
kasutatud katse nõuetekohasuse kriteeriumide kirjeldus;
|
|
kõikide katse käigus tehtud muudatuste kirjeldus.
|
|
|
Tulemused:
|
mõõdetud IL8LA ja GAPLA väärtused;
|
|
arvutatud nIL8LA, Ind-IL8LA ja Inh-GAPLA väärtused;
|
|
Ind-IL8LA usaldusvahemik usaldusnivool 95 %;
|
|
graafik, millel on kujutatud lutsiferaasi aktiivsuse ja rakkude eluvõimelisuse kontsentratsioonist sõltuvuse kõverad;
|
|
vajaduse korral mis tahes muude asjakohaste vaatlusandmete kirjeldus.
|
|
|
Tulemuste arutelu:
|
IL-8 lutsiferaasikatsega saadud tulemuste arutelu;
|
|
katse tulemuste hindamine ühtse lähenemisviisi kontekstis, kui muu asjakohane teave on kättesaadav.
|
|
Järeldused
|
KIRJANDUS
|
(1)
|
Takahashi, T., Kimura, Y., Saito, R., Nakajima, Y., Ohmiya, Y., Yamasaki, K., ja Aiba, S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol. Sci. 124: 359–369.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No. 267. ENV/JM/MONO(2017)19. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No. 258. ENV/JM/MONO(2017)20. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016). Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation. Series on Testing and Assessment No. 256. ENV/JM/MONO(2016)29. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen, J. W., Rorije, E., Emter, R., Natsch, A., van Loveren, H., ja Ezendam, J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul. Toxicol. Pharmacol. 69: 371–379.
|
|
(6)
|
Kimura, Y., Fujimura, C., Ito, Y., Takahashi, T., Nakajima, Y., Ohmiya, Y., ja Aiba, S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol. In Vitro 29: 1816–1830.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P. S., Gerberick, F., et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71: 337–351.
|
|
(8)
|
Ashikaga, T., Sakaguchi, H., Sono, S., Kosaka, N., Ishikawa, M., Nukada, Y., Miyazawa, M., Ito, Y., Nishiyama, N., ja Itagaki, H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). ATLA 38: 275–284.
|
|
(9)
|
Patlewicz, G., Casati, S., Basketter, D. A., Asturiol, D., Roberts, D. W., Lepoittevin, J.-P., Worth, A., ja Aschberger, K. (2016). Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul. Toxicol. Pharmacol. 82: 147–155.
|
|
(10)
|
Thorne, N., Inglese, J., ja Auld, D. S. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem. Biol. 17: 646–657.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists. OECD Publishing, Pariis. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani, V., Uchida, A., Suenaga, N., Ryufuku, M., ja Ohmiya, Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem. Biophys. Res. Commun. 280:1286–1291.
|
|
(13)
|
Viviani, V. R., Bechara, E. J., ja Ohmiya, Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38: 8271–8279.
|
|
(14)
|
Nakajima, Y., Kimura, T., Sugata, K., Enomoto, T., Asakawa, A., Kubota, H., Ikeda, M., ja Ohmiya, Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38: 891–894.
|
|
(15)
|
OECD (2017). Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. Environment, Health and Safety Publications, Series on Testing and Assessment. Avaldamisel. OECD, Pariis, Prantsusmaa.
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment No. 34. OECD, Pariis, Prantsusmaa.
|
|
(17)
|
OECD (2018). Draft Guidance Document on Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Majanduskoostöö ja Arengu Organisatsioon, Pariis. Kättesaadav aadressil http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol. Kättesaadav aadressil http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa, K., Ichino, Y., Kumata, S., Nakajima, Y., Hiraishi, Y., Kato, D., Viviani, V. R., ja Ohmiya, Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem. Photobiol. 86: 1046–1049.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Environment, Health and Safety Publications, Series on Testing and Assessment No. 168. OECD, Pariis, Prantsusmaa.
|
|
(21)
|
Ühinenud Rahvaste Organisatsioon (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Kuues, täiendatud väljaanne. United Nations Publications, New York ja Genf. ISBN: 978-92-1-117006-1. Kättesaadav aadressil http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Liide 3.1
MÕISTED
Aine
– looduslik või mis tahes tootmisprotsessi tulemusena saadud keemiline element või selle ühend koos püsivuse tagamiseks vajalike lisaainete ja tootmisprotsessi käigus tekkinud lisanditega, mis ei hõlma siiski lahusteid, mida on võimalik aine püsivust mõjutamata või selle koostist muutmata ainest eraldada.
Analüüsitsükkel
– ühe või mitme uuritava kemikaali analüüsimine lahustiga/kandeainega kontrolli ja positiivse kontrolliga paralleelselt.
Asjakohasus
– näitaja, mille abil kirjeldatakse, kuivõrd katsemeetod võimaldab uurida huvipakkuvat toimet, kas selle kasutamine on mõttekas ja kas see on konkreetse eesmärgi jaoks sobiv. Asjakohasusest nähtub, mil määral saab katsemeetodiga õigesti mõõta või ennustada huvipakkuvat bioloogilist toimet. Asjakohasuse puhul võetakse arvesse ka katsemeetodiga saavutatavat täpsust (vastavust) (16).
CV05
– rakkude eluvõimelisu se tasemes kajastuv näitaja ehk vähim kontsentratsioon, mille juures kemikaali toimel avalduv Inh-GAPLA on väiksem kui 0,05.
FInSLO-LA
– IL-8 lutsiferaasikatset käsitlevas valideerimisaruandes ja varasemates väljaannetes kasutatud lühend, millega osutatakse näitajale Ind-IL8LA. Mõiste Ind-IL8LA määratlus on esitatud allpool.
GAPLA
– punast valgust tekitava püsiva lutsiferaasi (SLR) (λmaks = 630 nm) aktiivsus, mida reguleerib GAPDH promootor ning mis kajastab rakkude eluvõimelisust ja eluvõimeliste rakkude arvu.
II-SLR-LA
– IL-8 lutsiferaasikatset käsitlevas valideerimisaruandes ja varasemates väljaannetes kasutatud lühend, millega osutatakse näitajale Inh-GAPLA. Mõiste Inh-GAPLA määratlus on esitatud allpool.
IL-8 (interleukiin 8)
– endoteelirakkude, fibroblastide, keratinotsüütide, makrofaagide ja monotsüütide toodetav tsütokiin, mis põhjustab neutrofiilide ja T-lümfotsüütide kemotaksist.
IL8LA
– oranži valgust tekitava püsiva lutsiferaasi (SLO) (λmaks = 580 nm) aktiivsus, mida reguleerib IL-8 promootor.
Ind-IL8LA
– nIL8LA suurenemine kordades. Selle arvutamiseks jagatakse kemikaaliga töödeldud THP-G8 rakkude puhul täheldatud nIL8LA väärtus töötlemata THP-G8 rakkude puhul täheldatud nIL8LA väärtusega ning see näitab, mil määral kemikaal suurendab IL-8 promootori aktiivsust.
Induktsiooni alampiir (MIT)
– väiksem kontsentratsioon, mille juures kemikaal vastab positiivsuse kriteeriumidele.
Inh-GAPLA
– GAPLA inhibeerimise määr. Selle arvutamiseks jagatakse kemikaaliga töödeldud THP-G8 rakkude puhul täheldatud GAPLA väärtus töötlemata THP-G8 rakkude puhul täheldatud GAPLA väärtusega ning see näitab kemikaali tsütotoksilisust.
Kahjuliku toime rada
– sündmuste ahel alates sihtkemikaali või rühma sarnaste kemikaalide keemilisest struktuurist sõltuvast käivitavast molekulaarsest sündmusest kuni vaadeldava toime avaldumiseni in vivo (20).
Katsete tegemist ja hindamist käsitlev ühtne lähenemisviis
– struktureeritud lähenemisviis, mida kasutatakse kemikaali või rühma kemikaalide ohtlikkuse tuvastamiseks (võimalik toime), ohu kirjeldamiseks (toime tugevus) ja/või ohutuse hindamiseks (võimalik toime / toime tugevus ja kokkupuude) ning mille raames lõimitakse strateegiliselt ja kaalutakse läbi kõik asjakohased andmed võimalikku ohtu ja/või riski ja/või täiendavate sihipäraste ja seega minimaalses ulatuses katsete tegemise vajadust käsitleva regulatiivse otsuse tegemiseks.
Kemikaal
– aine või segu.
Lahustiga/kandeainega kontroll
– töötlemata proov, mis sisaldab kõiki katsesüsteemi komponente peale uuritava kemikaali, sealhulgas lahustit/kandeainet. Selle abil tehakse kindlaks lähtetase samas lahustis/kandeaines lahustatud või püsiva dispersiooni moodustanud uuritava kemikaaliga töödeldud proovide jaoks. Kui sellist töötlemata proovi hinnatakse paralleelselt söötmega kontrolliga, võimaldab see ühtlasi kindlaks teha, kas lahusti/kandeaine mõjutab katsesüsteemi.
Mitmest koostisosast koosnev aine
– aine, mis on määratletud kvantitatiivse koostise alusel ja milles enam kui ühe põhikoostisosa sisaldus on vähemalt 10, kuid väiksem kui 80 massiprotsenti. Mitmest koostisosast koosnev aine saadakse tootmisprotsessi tulemusena. Erinevus segu ja mitmest koostisosast koosneva aine vahel seisneb selles, et segu saadakse kahe või enama aine kokku segamisel, ilma et toimuks keemilist reaktsiooni. Mitmest koostisosast koosnev aine saadakse keemilise reaktsiooni tulemusel.
nIL8LA
– IL-8 promootori aktiivsust kajastav lutsiferaasi SLO aktiivsus (IL8LA), mis on normaliseeritud GAPDH promootori aktiivsust kajastava lutsiferaasi SLR aktiivsuse (GAPLA) suhtes. Sellega väljendatakse IL-8 promootori aktiivsust pärast rakkude eluvõimelisuse või rakkude arvu arvessevõtmist.
nSLO-LA
– IL-8 lutsiferaasikatset käsitlevas valideerimisaruandes ja varasemates väljaannetes kasutatud lühend, millega osutatakse näitajale nIL8LA. Mõiste nIL8LA määratlus on esitatud eespool.
Nõuetekohane katsemeetod
– katsemeetod, mille asjakohasust ja usaldusväärsust konkreetse eesmärgi puhul peetakse piisavaks ning mis rajaneb teaduslikult usaldusväärsetel põhimõtetel. Katsemeetod ei ole kunagi nõuetekohane absoluutses tähenduses, vaid üksnes seoses teatud kindla eesmärgiga (29).
Ohtlikkus
– mõjurit või olukorda iseloomustav omadus, millest võib tuleneda kahjulik toime, kui organism, süsteem või (alam)populatsioon selle mõjuriga kokku puutub.
Pindaktiivne aine
– detergent või muu aine, mis võib vähendada vedeliku pindpinevust ja võimaldab sellel vahutada või tungida tahkesse ainesse; sama mis märgav aine (katsejuhend nr 437).
Positiivne kontroll
– paralleelproov, mis sisaldab kõiki katsesüsteemi komponente ja mida on töödeldud ainega, millega teadaolevalt saadakse positiivne tulemus. Positiivse tulemuse väärtus ei tohiks olla liiga suur, et võimaldada hinnata positiivse kontrolliga saadud tulemuste varieeruvust ajas.
Prehapteenid
– kemikaalid, mis muutuvad sensibilisaatoriks abiootilise muundumise teel.
Prohapteenid
– kemikaalid, mis vajavad nahka sensibiliseeriva toime tekkimiseks ensümaatilist aktiveerimist.
Segu
– kahest või enamast ainest koosnev segu või lahus.
SLO-LA
– IL-8 lutsiferaasikatset käsitlevas valideerimisaruandes ja varasemates väljaannetes kasutatud lühend, millega osutatakse näitajale IL8LA. Mõiste IL8LA määratlus on esitatud eespool.
SLR-LA
– IL-8 lutsiferaasikatset käsitlevas valideerimisaruandes ja varasemates väljaannetes kasutatud lühend, millega osutatakse näitajale GAPLA. Mõiste GAPLA määratlus on esitatud eespool.
Spetsiifilisus
– katsemeetodiga õigesti negatiivseks/reaktsioonivõimetuks liigitatud uuritavate kemikaalide osakaal kõikide negatiivsete kemikaalide hulgas. Spetsiifilisus kajastab sellise katsemeetodi täpsust, millega saadakse kategoriseeritav tulemus, ning see on oluline näitaja katsemeetodi asjakohasuse hindamiseks (16).
THP-G8
– IL-8 lutsiferaasikatses kasutatav IL-8 reporterrakuliin. Inimese makrofaagilaadsete rakkude liin THP-1, millesse on transfekteeritud lutsiferaaside SLO ja SLR geenid, mis on vastavalt IL-8 ja GAPDH promootori kontrolli all.
Tundlikkus
– katsemeetodiga õigesti positiivseks/reaktsioonivõimeliseks liigitatud uuritavate kemikaalide osakaal kõikide positiivsete kemikaalide hulgas. Tundlikkus kajastab sellise katsemeetodi täpsust, millega saadakse kategoriseeritav tulemus, ning see on oluline näitaja katsemeetodi asjakohasuse hindamiseks (16).
Täpsus
– katsetulemuste ja vastuvõetavate võrdlusväärtuste kooskõla määr. Täpsus näitab katsemeetodi tulemuslikkust ja on üks asjakohastest aspektidest. Seda mõistet kasutatakse sageli vastavuse tähenduses, et väljendada katsemeetodiga saadud õigete tulemuste osakaalu (16).
Usaldusväärsus
– näitaja, mis iseloomustab katsemeetodiga saadud tulemuste reprodutseeritavust pikema aja jooksul samas laboris ja eri laborites, kui meetodit rakendatakse ühe ja sama katse-eeskirja alusel. Usaldusväärsuse hindamiseks arvutatakse laborisisene ja laboritevaheline reprodutseeritavus ning laborisisene korratavus (16).
Uuritav kemikaal
– iga aine või segu, mida uuritakse käesoleva katsemeetodi abil.
UVCB
– tundmatu või muutuva koostisega aine, kompleksne reaktsioonisaadus või bioloogilist päritolu materjal.
Ühest koostisosast koosnev aine
– aine, mis on määratletud kvantitatiivse koostise alusel ja mille ühe põhikoostisosa sisaldus on vähemalt 80 massiprotsenti.
Ühinenud Rahvaste Organisatsiooni ühtne ülemaailmne kemikaalide klassifitseerimise ja märgistamise süsteem (ÜRO GHS)
– süsteem, millega nähakse ette kemikaalide (ainete ja segude) klassifitseerimine vastavalt nende füüsilise ohtlikkuse ning kahjuliku tervise- ja keskkonnamõju standarditud liigile ja tasemele ning mis hõlmab selliseid asjaomaseid teavitustähiseid nagu piktogrammid, märksõnad, ohulaused, hoiatuslaused ja ohutuskaardid, et anda inimeste (sealhulgas tööandjate, töötajate, vedajate, tarbijate ja päästetöötajate) ning keskkonna kaitsmiseks vajalikku teavet kõnealuste kemikaalide kahjuliku toime kohta (21).
Liide 3.2
LUTSIFERAASI AKTIIVSUSE MÕÕTMISE PÕHIMÕTE NING SLO JA SLR-I PUHUL KOHALDATAVATE OPTILISE FILTRI LÄBILASKVUSTEGURITE KINDLAKSTEGEMINE
Mitme reporteriga katsesüsteemi Tripluc on võimalik kasutada mitme värvuse tuvastamist võimaldava mikroplaatide jaoks ette nähtud luminomeetriga, mille saab varustada optilise filtriga (nt Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). Mõõtmiseks kasutatav optiline filter on madal- või kõrgpääsufilter piirlainepikkusega 600–620 nm või ribapääsufilter lainepikkusele 600–700 nm.
Lutsiferaaside tekitatava kahe värvuse mõõtmine optilise filtri abil
Selles näites kasutatakse luminomeetrit Phelios AB-2350 (ATTO). See luminomeeter on varustatud 600 nm madalpääsufiltriga (R60, HOYA Co., filter 1), mis võimaldab eristada SLO (λmaks = 580 nm) ja SLRi (λmaks = 630 nm) põhjustatud luminestsentsi.
600 nm madalpääsufiltri läbilaskvustegurite kindlakstegemiseks kasutatakse puhastatud lutsiferaase SLO ja SLR ning i) mõõdetakse SLO ja SLRi põhjustatud bioluminestsentsi tugevus ilma filtrita (F0), ii) mõõdetakse SLO ja SLRi põhjustatud, 600 nm madalpääsufiltri (filter 1) läbinud bioluminestsentskiirguse tugevus ja iii) arvutatakse allpool loetletud 600 nm madalpääsufiltri läbilaskvustegurid SLO ja SLRi puhul.
|
Läbilaskvustegurid
|
Lühend
|
Määratlus
|
|
SLO
|
Filter 1: läbilaskvustegur
|
=κOR60
|
Filtri läbilaskvustegur SLO puhul
|
|
SLR
|
Filter 1: läbilaskvustegur
|
κRR60
|
Filtri läbilaskvustegur SLRi puhul
|
Kui valida katseproovis SLO ja SLRi põhjustatud luminestsentsi tugevuse tähisteks vastavalt O ja R, siis on i) filtri puudumisel täheldatav kogu optilist spektrit hõlmava valguse tugevus F0 ja ii) 600 nm madalpääsufiltri (filter 1) läbinud valguse tugevus F1 kirjeldatavad järgmiste valemitega.
F0 = O + R
F1 = κOR60 × O + κRR60 × R
Need valemid võib esitada ka järgmisel kujul:

Seejärel saab arvutatud läbilaskvustegurite (κOR60 ja κRR60) ning mõõdetud F0 ja F1 väärtuste abil leida O ja Ri väärtused järgmise valemiga:.

Läbilaskvusteguri kindlakstegemiseks kasutatavad materjalid ja meetodid
|
1)
|
Reaktiivid
Puhastatud eraldi lutsiferaasid:
|
lüofiliseeritud puhastatud SLO
|
|
lüofiliseeritud puhastatud SLR
|
|
(valideerimisuuringu jaoks hangiti need koos rakuliiniga THP-G8 Jaapanis Tottoris asuvast ettevõttest GPC Lab. Co. Ltd.)
|
Katsereaktiiv:
|
lutsiferaasikatse reaktiiv Tripluc® (nt ettevõttest TOYOBO; katalooginumber MRA-301)
|
Sööde: lutsiferaasikatse sööde (30 ml, säilitatakse temperatuuril 2–8 °C)
|
|
Reaktiiv
|
Konts.
|
Lõppkonts. söötmes
|
Vajalik kogus
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
Veiselooteseerum
|
—
|
10 %
|
3 ml
|
|
2)
|
Ensüümilahuse valmistamine
Lüofiliseeritud puhastatud lutsiferaasi lahustamiseks katsutis lisatakse sellele 200 μl 10–100 mM Tris/HCl-i või Hepes/HCl-i (pH 7,5–8,0), mis sisaldab 10 % glütserooli massiühikutes ruumalaühiku kohta. Ensüümilahus jaotatakse 10 μl alikvootidena ühekordsetesse 1,5 ml katsutitesse ja säilitatakse sügavkülmikus temperatuuril -80 °C. Külmutatud ensüümilahus on kasutatav kuni kuue kuu jooksul. Kasutamisel lisatakse igasse ensüümilahust (lahjendatud ensüümilahust) sisaldavasse katsutisse 1 ml lutsiferaasikatse söödet (10 % veiselooteseerumit sisaldav RPMI-1640) ja inaktiveerumise takistamiseks hoitakse katsuteid jääl.
|
|
3)
|
Bioluminestsentsi mõõtmine
Lutsiferaasikatse reaktiiv Tripluc® sulatatakse ja seda hoitakse toatemperatuuril veevannis või ümbritseva õhu temperatuuril. Luminomeeter lülitatakse sisse 30 minutit enne mõõtmise algust, et fotokordisti jõuaks stabiliseeruda. 100 μl lahjendatud ensüümilahust kantakse 96-kannulisele mustale lamedapõhjalisele plaadile (SLO proovid kannudesse B1, B2 ja B3, SLRi proovid kannudesse D1, D2, ja D3). Seejärel kantakse igasse ensüümilahust sisaldavasse plaadi kannu pipetiga 100 μl eelsoojendatud Triplucit. Plaati loksutatakse plaadiloksutil toatemperatuuril (umbes 25 °C) 10 minutit. Vajaduse korral kõrvaldatakse kannudes olevast lahusest mullid. Lutsiferaasi aktiivsuse mõõtmiseks asetatakse plaat luminomeetrisse. Bioluminestsentsi mõõdetakse 3 sekundit ilma optilise filtrita (F0) ja 3 sekundit optilise filtriga (F1).
Optilise filtri läbilaskvustegur arvutatakse vastavalt järgmistele valemitele.
Läbilaskvustegur (SLO (κOR60)) = (B1/F1 + B2/F1 + B3/F1): (B1/F0 + B2/F0 + B3/F0)
Läbilaskvustegur (SLR (κRR60)) = (D1/F1 + D2/F1 + D3/F1): (D1/F0 + D2/F0 + D3/F0)
Arvutatud läbilaskvustegureid kasutatakse kõigi sama luminomeetriga tehtavate mõõtmiste puhul.
|
Seadmete kvaliteedi kontrollimine
Tuleks kasutada IL-8 lutsiferaasikatset käsitlevas katse-eeskirjas kirjeldatud korda (18).
Liide 3.3
PÄDEVUSAINED
Enne katsemeetodi B.71 juurde kuuluvas käesolevas liites kirjeldatud meetodi rutiinset kasutamist tuleks tõendada labori tehnilist pädevust. Selleks tuleks IL-8 lutsiferaasikatses saada tabelis 1 esitatud soovitatava üheksa aine puhul eeldatavale tulemusele vastav prognoos ning üheksast pädevusainest vähemalt kaheksa puhul väärtused, mis jäävad asjaomasesse võrdlusvahemikku (pädevusained on valitud nii, et nende puhul oleksid esindatud naha sensibiliseerimise ohuga seotud eri reaktsioonid). Peale selle on valikukriteeriumidena võetud arvesse ainete kaubanduslikku kättesaadavust, kvaliteetsete in vivo võrdlusandmete olemasolu ja IL-8 lutsiferaasikatsega saadud kvaliteetsete in vitro andmete olemasolu. Lisaks on IL-8 lutsiferaasikatse kohta olemas avaldatud võrdlusandmed (1, 6).
Tabel 1.
IL-8 lutsiferaasikatse puhul tehnilise pädevuse tõendamiseks soovitatavad ained
|
Pädevusained
|
CASi nr
|
Olek
|
Lahustuvus X-VIVO15-s kontsentratsioonil 20 mg/ml
|
In vivo toime prognoos (107)
|
IL-8 lutsiferaasikatse kohane prognoos (108)
|
Võrdlusvahemik (μg/ml) (109)
|
|
|
CV05
(110)
|
IL-8 lutsiferaasikatse kohane MIT (111)
|
|
2,4-dinitroklorobenseen
|
97-00-7
|
Tahkis
|
Ei lahustu
|
Sensibilisaator (äärmiselt tugev)
|
Positiivne
|
2,3–3,9
|
0,5–2,3
|
|
Formaldehüüd
|
50-00-0
|
Vedelik
|
Lahustub
|
Sensibilisaator (tugev)
|
Positiivne
|
9–30
|
4–9
|
|
2-merkaptobensotiasool
|
149-30-4
|
Tahkis
|
Ei lahustu
|
Sensibilisaator (mõõdukas)
|
Positiivne
|
250–290
|
60–250
|
|
Etüleendiamiin
|
107-15-3
|
Vedelik
|
Lahustub
|
Sensibilisaator (mõõdukas)
|
Positiivne
|
500–700
|
0,1–0,4
|
|
Etüleenglükooldimetakrülaat
|
97-90-5
|
Vedelik
|
Ei lahustu
|
Sensibilisaator (nõrk)
|
Positiivne
|
> 2000
|
0,04–0,1
|
|
4-allüülanisool (estragool)
|
140-67-0
|
Vedelik
|
Ei lahustu
|
Sensibilisaator (nõrk)
|
Positiivne
|
> 2000
|
0,01–0,07
|
|
Streptomütsiinsulfaat
|
3810-74-0
|
Tahkis
|
Lahustub
|
Sensibiliseeriva toimeta
|
Negatiivne
|
> 2000
|
> 2000
|
|
Glütserool
|
56-81-5
|
Vedelik
|
Lahustub
|
Sensibiliseeriva toimeta
|
Negatiivne
|
> 2000
|
> 2000
|
|
Isopropanool
|
67-63-0
|
Vedelik
|
Lahustub
|
Sensibiliseeriva toimeta
|
Negatiivne
|
> 2000
|
> 2000
|
|
Lühendid: CASi nr – Chemical Abstracts Service’i registrinumber.
|
Liide 3.4
INDEKSID JA HINDAMISKRITEERIUMID
nIL8LA (nSLO-LA)
IL8LA (SLO-LA) ja GAPLA (SLR-LA) puhul tehakse mõõtmisi j-ndas analüüsitsüklis (j = 1–4) i-ndal kontsentratsioonil (i = 0–11). Normaliseeritud IL8LA tähis on nIL8LA (nSLO-LA) ja selle määratlus on järgmine: .
nIL8LAij = IL8LAij/GAPLAij
See on käesoleva katsemeetodi puhul kasutatav lähtesuurus.
Ind-IL8LA (FInSLO-LA)
Ind-IL8LA on käesoleva katsemeetodi puhul peamine näitaja, mis väljendab nIL8LA (nSLO-LA) keskmise väärtuse suurenemist kordades konkreetses analüüsitsüklis i-ndal kontsentratsioonil võrrelduna sama näitaja väärtusega nullkontsentratsioonil. Kõnealune suhe on esitatav järgmise valemiga:

Juhtlabori ettepaneku kohaselt vastab uuritava kemikaali puhul positiivsele tulemusele väärtus 1,4. Nimetatud väärtus põhineb juhtlabori varasemate katseandmete analüüsil. Andmehalduse töörühm on kasutanud seda väärtust valideerimisuuringu kõigis etappides. Nagu valemist nähtub, on katse esmane tulemusnäitaja Ind-IL8LA kahe aritmeetilise keskmise suhtarv.
Usaldusvahemik usaldusnivool 95 % (CI95 %)
Kõnealuse esmase mõõdetava tulemusnäitaja kordustäpsuse hindamiseks saab leida nimetatud suhtarvul põhineva usaldusnivoole 95 % vastava usaldusvahemiku (CI95 %). Kui CI95 % alampiir on ≥ 1, siis see näitab, et kontsentratsioonil nr i on nIL8LA oluliselt suurem kui lahustiga kontrolli puhul. CI95 % leidmiseks on mitu võimalust. Valideerimisuuringus kasutatud meetod põhineb Fielleri teoreemil. Selle teoreemi järgi leitakse usaldusvahemik usaldusnivool 95 % valemiga

kus .


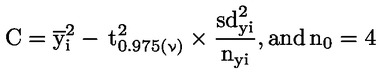


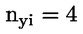


on keskse t-jaotuse 97,5-protsentiil vabadusastmete arvu ν puhul, kus

Inh-GAPLA (II-SLR-LA)
Inh-GAPLA on konkreetses analüüsitsüklis i-ndal kontsentratsioonil täheldatava GAPLA (SLR-LA) keskväärtuse ja lahustiga kontrolli puhul täheldatava sama näitaja keskväärtuse suhtarv ning see on esitatav järgmisel kujul:

Kuna GAPLA asub nIL8LA arvutamise valemi nimetajas, põhjustab see väga väikestel väärtustel suurt varieeruvust nIL8LA väärtustes. Seepärast võib Ind-IL8LA väärtusi, mis on saadud Inh-GAPLA väga väikeste väärtuste puhul (alla 0,05), pidada ebatäpseks.
Liide 3.5
IL-8 LUTSIFERAASIKATSES KASUTATAV KEMIKAALIDE LAHUSTAMISE SKEEM
|
a)
|
X-VIVOTM 15-s kontsentratsioonil 20 mg/ml lahustuvad kemikaalid
|
|
b)
|
X-VIVOTM 15-s kontsentratsioonil 20 mg/ml lahustumatud kemikaalid

|
Liide 3.6
IL-8 LUTSIFERAASIKATSES POSITIIVSE KONTROLLINA KASUTATAVA 4-NBB LAHUSTAMISE SKEEM

“ |