III LISA
RASKETEST KÕRVALTOIMETEST TEATAMINE


|
25.10.2006 |
ET |
Euroopa Liidu Teataja |
L 294/32 |
KOMISJONI DIREKTIIV 2006/86/EÜ,
24. oktoober 2006,
millega rakendatakse Euroopa Parlamendi ja nõukogu direktiivi 2004/23/EÜ jälgitavusnõuete, rasketest kõrvaltoimetest ja tõsistest kõrvalekalletest teatamise ning inimkudede ja -rakkude kodeerimist, töötlemist, säilitamist, ladustamist ja jaotamist käsitlevate teatavate tehniliste nõuete osas
(EMPs kohaldatav tekst)
EUROOPA ÜHENDUSTE KOMISJON,
võttes arvesse Euroopa Ühenduse asutamislepingut,
võttes arvesse Euroopa Parlamendi ja nõukogu 31. märtsi 2004. aasta direktiivi 2004/23/EÜ inimkudede ja -rakkude annetamise, hankimise, uurimise, töötlemise, säilitamise, ladustamise ja jaotamise kvaliteedi- ja ohutusstandardite kehtestamise kohta, (1) eriti selle artiklit 8, artikli 11 lõiget 4 ja artikli 28 punkte a, c, g ja h,
ning arvestades järgmist:
|
(1) |
Direktiivis 2004/23/EÜ on sätestatud kvaliteedi- ja ohutusstandardid inimkasutuseks ette nähtud inimkudede ja -rakkude ning nendest valmistatud ja inimkasutuseks ette nähtud toodete annetamise, hankimise, uurimise, töötlemise, säilitamise, ladustamise ja jaotamise kohta, et tagada inimeste tervise kõrgetasemeline kaitse. |
|
(2) |
Et vältida haiguste edasikandumist inimkasutuseks ette nähtud inimkudede ja -rakkude kaudu ning tagada kvaliteedi ja ohutuse võrdväärne tase, on direktiiviga 2004/23/EÜ kehtestatud tehnilised nõuded kõikidele inimkudede ja -rakkude kasutamisprotsessi etappidele, sealhulgas standardid ja spetsifikatsioonid, mis on seotud koepankade kvaliteedisüsteemiga. |
|
(3) |
Vastavalt direktiivile 2004/23/EÜ tuleks liikmesriikides kehtestada koepankade ning koepankades tehtavate ettevalmistusprotsesside akrediteerimise, määramise, lubamise või litsentsimise süsteem, et tagada inimeste tervise kõrgetasemeline kaitse. Selle süsteemi jaoks on vaja kehtestada tehnilised nõuded. |
|
(4) |
Koepankade akrediteerimise, määramise, lubamise või litsentsimise nõuded peaksid hõlmama organisatsiooni ja selle juhtimist, personali, seadmeid ja materjale, tööruume, dokumentatsiooni, andmeid ja kvaliteedikontrolli. Akrediteeritud, määratud, loa saanud või litsentsitud koepangad peaksid täitma ka nende konkreetse tegevusega seotud lisanõudeid. |
|
(5) |
Oluliseks teguriks, mis võib suurendada kudede ja rakkude saastumise ohtu, on õhu kvaliteet kudede ja rakkude töötlemise ajal. Üldiselt nõutakse, et osakeste ja mikroorganismide kolooniate sisaldus õhus vastaks Euroopa hea tootmistava suuniste 1. lisas määratletud A-klassi standardile ja komisjoni direktiivile 2003/94/EÜ. (2) Teatavates olukordades ei ole siiski nõutud, et osakeste ja mikroorganismide kolooniate sisaldus õhus vastaks A-klassi standardile. Sel juhul tuleks näidata ja dokumenteerida, et valitud keskkond vastab asjaomase koe- ja rakuliigi, protsessi ja kasutusvaldkonna suhtes kehtestatud kvaliteedi- ja ohutusnõuetele. |
|
(6) |
Käesoleva direktiivi reguleerimisala hõlmab inimkudede ja -rakkude kvaliteeti ja ohutust kodeerimisel, töötlemisel, säilitamisel, ladustamisel ja jaotamisel tervishoiuasutustele, kus neid kasutatakse inimkehas. See ei peaks siiski laienema kudede ja rakkude inimkasutusele (nt implantaatide paigaldamine, perfusioon, viljastamine või embrüote siirdamine). Käesoleva direktiivi sätteid, mis käsitlevad jälgitavust ning rasketest kõrvaltoimetest ja tõsistest kõrvalekalletest teatamist, kohaldatakse ka inimkudede ja -rakkude annetamise, hankimise ja uurimise suhtes, mida reguleeritakse direktiiviga 2006/17/EÜ. (3) |
|
(7) |
Inimkasutuseks ette nähtud kudede ja rakkude kasutamisega kaasneb haiguste edasikandumise ning muude ebasoovitavate kõrvaltoimete oht. Selliste kõrvaltoimete ohu jälgimiseks ja vähendamiseks tuleks kehtestada jälgitavusnõuded ning ühenduse menetlus rasketest kõrvaltoimetest ja tõsistest kõrvalekalletest teatamiseks. |
|
(8) |
Rasketest kõrvaltoimetest nii doonoril kui ka retsipiendil ja tõsistest kõrvalekalletest, mis võivad mõjutada kudede ja rakkude kvaliteeti ja ohutust ja mis on tingitud inimkudede ja -rakkude hankimisest (sealhulgas doonorite hindamine ja valik), uurimisest, töötlemisest, säilitamisest, ladustamisest ja jaotamisest, tuleks viivitamata teatada pädevale asutusele. |
|
(9) |
Raskeid kõrvaltoimeid võib esineda elusdoonoritel rakkude ja kudede hankimise ajal või pärast seda, samuti kudede või rakkude inimkasutuse ajal või pärast seda. Neist tuleks teatada vastavasse koepanka, et see juhtumit uuriks ja teataks sellest pädevale asutusele. See ei peaks takistama hankimise või inimkasutuse eest vastutavat organisatsiooni soovi korral pädevat asutust ka otse teavitamast. Direktiiviga tuleks kindlaks määrata miinimumandmed, mida on vaja pädeva asutuse teavitamiseks, ilma et see mõjutaks liikmesriikide võimalust säilitada või kehtestada oma territooriumil asutamislepingu nõuetele vastavad rangemad kaitsemeetmed. |
|
(10) |
Teavitamiskulude minimeerimiseks, kattumiste vältimiseks ja haldusliku tõhususe suurendamiseks tuleks teabe edastamisel ja töötlemisel kasutada kaasaegset tehnoloogiat ja e-valitsuse lahendusi. See tehnoloogia peaks põhinema teabevahetuse standardvormil ja selleks tuleks kasutada andmete haldamiseks sobivat süsteemi. |
|
(11) |
Jälgitavuse parandamiseks ning kudede ja rakkude põhiomadusi käsitleva teabevahetuse edendamiseks on vaja kindlaks määrata ühises Euroopa koodis sisalduvad põhiandmed. |
|
(12) |
Käesolevas direktiivis austatakse põhiõigusi ja järgitakse iseäranis Euroopa Liidu põhiõiguste hartas tunnustatud põhimõtteid. |
|
(13) |
Käesoleva direktiiviga ette nähtud meetmed on kooskõlas direktiivi 2004/23/EÜ artikli 29 kohaselt asutatud komitee arvamusega, |
ON VASTU VÕTNUD KÄESOLEVA DIREKTIIVI:
Artikkel 1
Kohaldamisala
1. Käesolevat direktiivi kohaldatakse alljärgneva materjali kodeerimise, töötlemise, säilitamise, ladustamise ja jaotamise suhtes:
|
a) |
inimkasutuseks ette nähtud inimkoed ja -rakud; |
|
b) |
inimkudedest ja -rakkudest valmistatud ja inimkasutuseks ette nähtud tooted, mis ei kuulu teiste direktiivide reguleerimisalasse. |
2. Käesoleva direktiivi artikleid 5–9, mis käsitlevad jälgitavust ning rasketest kõrvaltoimetest ja tõsistest kõrvalekalletest teatamist, kohaldatakse ka inimkudede ja -rakkude annetamise, hankimise ja uurimise suhtes.
Artikkel 2
Mõisted
Käesolevas direktiivis kasutatakse järgmisi mõisteid:
a) sugurakud– koed ja rakud, mis on ette nähtud kasutamiseks kunstlikul viljastamisel;
b) partneri annetus– sugurakkude annetamine mehe ja naise vahel, kes kinnitavad, et neil on intiimsuhe;
c) kvaliteedisüsteem– kvaliteedijuhtimisega seotud organisatsiooniline struktuur, kindlaksmääratud kohustused, menetlused, protsessid ja vahendid; mõiste hõlmab kõiki otseselt või kaudselt kvaliteeti parandavaid tegevusi;
d) kvaliteedijuhtimine– kooskõlastatud tegevus organisatsiooni kvaliteedi parandamiseks ja kontrolliks;
e) standardne töökord– kirjalikud juhised, milles kirjeldatakse teatava protsessi etappe, sealhulgas kasutatavaid materjale ja meetodeid ning eeldatavat lõpptoodet;
f) valideerimine (seadmete või keskkondade puhul kvalifitseerimine)– dokumenteeritud tõendusmaterjali loomine, millega tagatakse kindlus, et konkreetse protsessi, seadme või keskkonna abil toodetakse järjekindlalt toodet, mis vastab eelnevalt kindlaks määratud nõuetele ja kvaliteedistandarditele; protsess valideeritakse selleks, et hinnata süsteemi toimimise tõhusust, lähtudes kasutuseesmärgist;
g) jälgitavus– võime kude/rakk identifitseerida ja selle asukoht kindlaks määrata protsessi igas etapis alates koe/raku hankimisest, töötlemisest, uurimisest ja ladustamisest ning lõpetades selle üleandmisega retsipiendile või hävitamisega, sealhulgas võime teha kindlaks doonor ja kudesid/rakke vastu võttev, töötlev või säilitav koepank või muu asutus ning võime teha kudesid/rakke kasutava(te)s meditsiiniasutus(t)es kindlaks retsipient (retsipiendid); jälgitavus hõlmab ka võimet teha kindlaks kõik asjakohased andmed kõnealuste kudede/rakkudega kokku puutuvate toodete ja materjalide kohta;
h) kriitiline– potentsiaalselt rakkude ja kudede kvaliteeti ja/või ohutust mõjutav või nendega kokku puutuv;
i) hankimisasutus– tervishoiuasutus või haiglaüksus või muu asutus, mis tegeleb inimkudede ja -rakkude hankimisega ning mida ei ole akrediteeritud ega määratud tegutsema koepangana ja millele ei ole antud vastavat luba ega litsentsi;
j) inimkasutuse eest vastutav asutus– tervishoiuasutus või haiglaüksus või muu asutus, mis tegeleb kudede ja -rakkude inimkasutusega.
Artikkel 3
Nõuded koepankade akrediteerimiseks, määramiseks ja neile loa või litsentsi andmiseks
Koepank peab vastama I lisas sätestatud nõuetele.
Artikkel 4
Nõuded kudede ja rakkude ettevalmistusprotsesside akrediteerimiseks, määramiseks või nende jaoks loa või litsentsi andmiseks
Koepankade ettevalmistusprotsessid peavad vastama II lisas sätestatud nõuetele.
Artikkel 5
Rasketest kõrvaltoimetest teatamine
1. Liikmesriigid tagavad, et
|
a) |
hankimisasutustes on kehtestatud menetlused, mille kohaselt säilitatakse andmed hangitud kudede ja rakkude kohta ning teavitatakse koepankasid viivitamata elusdoonoril esinevatest rasketest kõrvaltoimetest, mis võivad mõjutada kudede ja rakkude kvaliteeti ja ohutust; |
|
b) |
inimkasutuse eest vastutavates asutustes on kehtestatud menetlused, mille kohaselt säilitatakse andmed kasutatud kudede ja rakkude kohta ning teavitatakse koepankasid viivitamata kliinilise kasutamise ajal või pärast seda täheldatud rasketest kõrvaltoimetest, millel võib olla seos kudede ja rakkude kvaliteedi ja ohutusega; |
|
c) |
koepangad, kes jaotavad kudesid ja rakke inimkasutuseks, annavad kudede ja rakkude inimkasutuse eest vastutavale asutusele teada, kuidas kõnealune asutus peab punktis b nimetatud rasketest kõrvaltoimetest teatama. |
2. Liikmesriigid tagavad, et koepangad
|
a) |
on kehtestanud menetluse, mis võimaldab lõike 1 punktides a ja b nimetatud raske kõrvaltoime kahtluse korral pädevale asutusele viivitamata edastada kogu vajaliku teabe; |
|
b) |
on kehtestanud menetluse, mis võimaldab pädevat asutust viivitamata teavitada kõrvaltoime põhjuste ja tagajärgede analüüsi tulemustest. |
3. Liikmesriigid tagavad, et
|
a) |
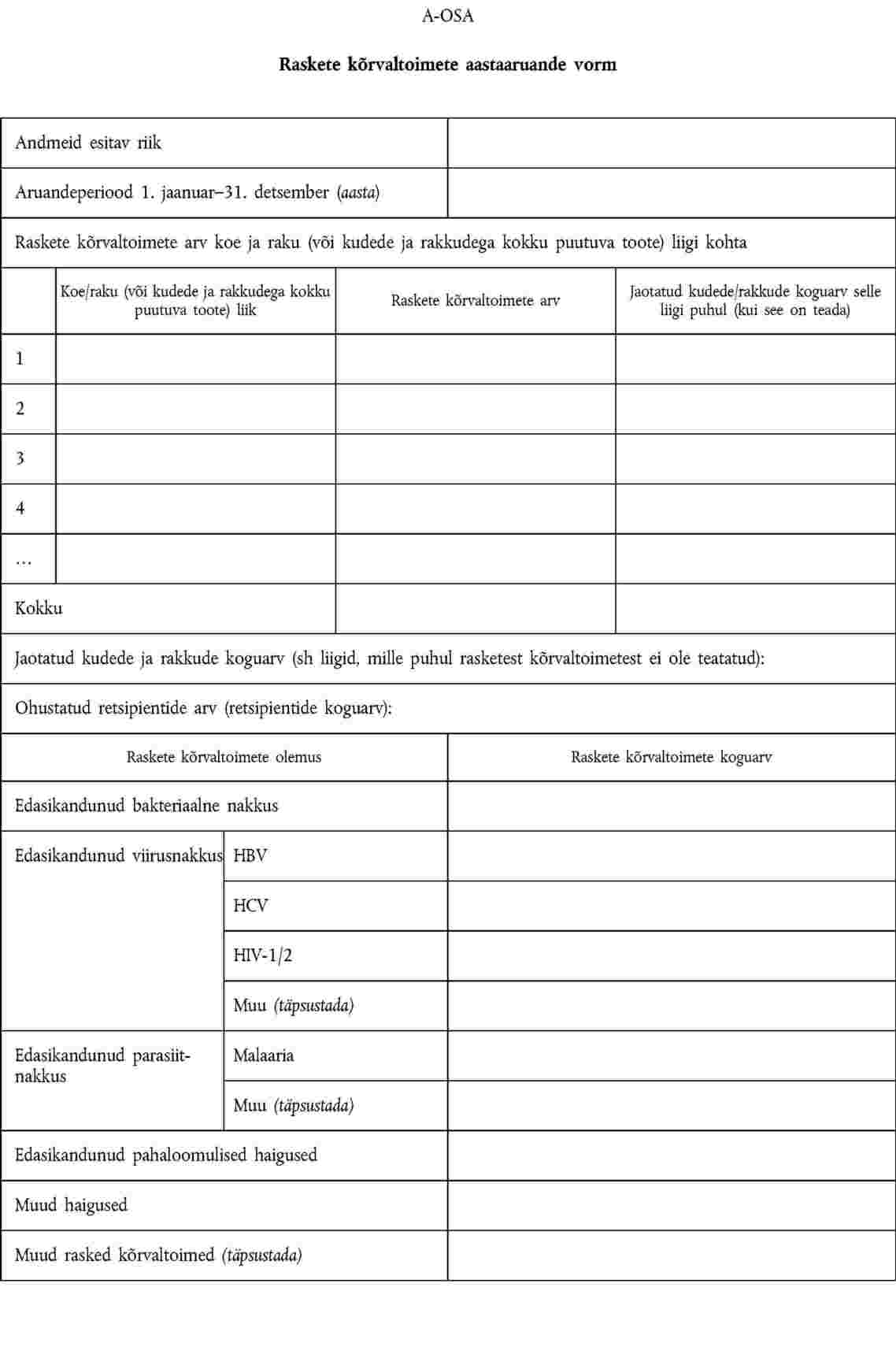
direktiivi 2004/23/EÜ artiklis 17 osutatud vastutav isik edastab pädevale asutusele III lisa A-osas kirjeldatud teabe; |
|
b) |
koepangad teavitavad pädevaid asutusi teiste inimkasutuseks ette nähtud ja jaotatud ning kahtluse all olevate kudede ja rakkude suhtes võetud meetmetest; |
|
c) |
koepangad teavitavad pädevat asutust uurimise tulemustest, esitades vähemalt III lisa B-osas kirjeldatud teabe. |
Artikkel 6
Tõsistest kõrvalekalletest teatamine
1. Liikmesriigid tagavad, et
|
a) |
hankimisasutused ja koepangad on kehtestanud menetluse koepankade viivitamatuks teavitamiseks hankimise ajal esinenud mis tahes tõsistest kõrvalekalletest, mis võivad mõjutada inimkudede ja -rakkude kvaliteeti ja/või ohutust, ja selliseid kõrvalekaldeid käsitlevate dokumentide säilitamiseks; |
|
b) |
kudede ja rakkude inimkasutuse eest vastutavad asutused on kehtestanud menetluse koepankade viivitamatuks teavitamiseks mis tahes tõsistest kõrvalekalletest, mis võivad mõjutada kudede ja rakkude kvaliteeti ja ohutust; |
|
c) |
koepangad annavad inimkasutuse eest vastutavale asutusele teada, kuidas kõnealune asutus peab neile teatama tõsistest kõrvalekalletest, mis võivad mõjutada kudede ja rakkude kvaliteeti ja ohutust. |
2. Kunstliku viljastamise puhul peetakse tõsiseks kõrvalekaldeks sugurakkude või embrüote mis tahes väärtuvastamist või segiajamist. Kõik isikud või hankimisasutused või inimkasutuse eest vastutavad/kunstlikku viljastamist tegevad asutused teatavad sellistest kõrvalekalletest vastavatele koepankadele, et need juhtumit uuriks ja teataks sellest pädevale asutusele.
3. Liikmesriigid tagavad, et koepangad
|
a) |
on kehtestanud menetluse, mis võimaldab lõike 1 punktides a ja b nimetatud tõsise kõrvalekalde kahtluse korral pädevale asutusele viivitamata edastada kogu vajaliku teabe; |
|
b) |
on kehtestanud menetluse, mis võimaldab pädevat asutust viivitamatult teavitada kõrvalekalde põhjuste ja tagajärgede analüüsi tulemustest. |
4. Liikmesriigid tagavad, et
|
a) |
direktiivi 2004/23/EÜ artiklis 17 osutatud vastutav isik edastab pädevale asutusele IV lisa A-osas kirjeldatud teabe; |
|
b) |
koepangad hindavad tõsiseid kõrvalekaldeid, et selgitada välja nende põhjusi, mida saaks ennetada; |
|
c) |
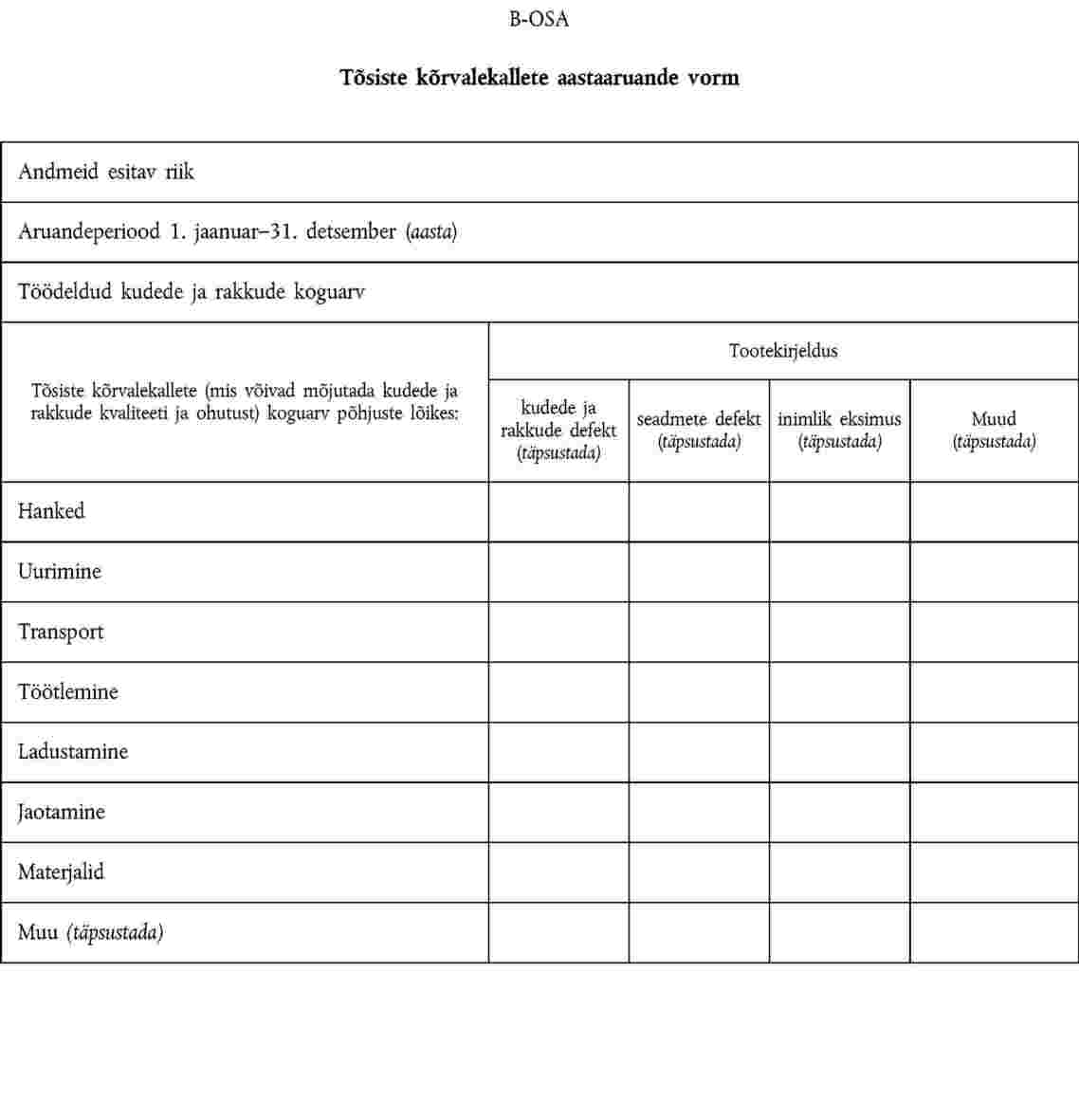
koepangad teavitavad pädevat asutust uurimise tulemustest, esitades vähemalt IV lisa B-osas kirjeldatud teabe. |
Artikkel 7
Aastaaruanded
1. Liikmesriigid esitavad komisjonile järgneva aasta 30. juuniks aastaaruande pädevale asutusele teatatud raskete kõrvaltoimete ja tõsiste kõrvalekallete kohta. Komisjon esitab liikmesriikide pädevatele asutustele saadud aruannetest kokkuvõtte. Pädev asutus tagab, et nimetatud aruanne oleks kättesaadav ka koepankadele.
2. Andmeid edastatakse V lisa A- ja B-osas esitatud vormide alusel ning need peavad sisaldama teavet, mida on vaja saatja identifitseerimiseks.
Artikkel 8
Pädevate asutuste vaheline teabevahetus ja teabe edastamine komisjonile
Liikmesriigid tagavad, et nende pädevad asutused edastavad üksteisele ja komisjonile vajaliku teabe seoses raskete kõrvaltoimete ja tõsiste kõrvalekalletega, et saaks võtta piisavaid meetmeid.
Artikkel 9
Jälgitavus
1. Koepangad kehtestavad tõhusad ja täpsed süsteemid vastuvõetud ja jaotatud rakkude/kudede üheseks identifitseerimiseks ja märgistamiseks.
2. Koepangad ja inimkasutuse eest vastutavad asutused säilitavad VI lisas nimetatud andmeid vähemalt 30 aastat nõuetekohasel ja loetaval kujul.
Artikkel 10
Euroopa kodeerimissüsteem
1. Koepangas antakse kogu annetatud materjalile üleeuroopaline identifitseerimiskood, mis võimaldab täpselt kindlaks teha doonori, tagab kogu annetatud materjali jälgitavuse ning sisaldab teavet kudede ja rakkude põhiomaduste kohta. Kood sisaldab vähemalt VII lisas ette nähtud andmeid.
2. Lõiget 1 ei kohaldata sugurakkude suhtes, kui tegemist on partneri annetusega.
Artikkel 11
Ülevõtmine
1. Liikmesriigid jõustavad käesoleva direktiivi järgimiseks vajalikud õigusnormid hiljemalt 1. septembriks 2007. Nad edastavad kõnealuste normide teksti ning kõnealuste normide ja käesoleva direktiivi vahelise vastavustabeli viivitamata komisjonile.
Liikmesriigid jõustavad käesoleva direktiivi artikli 10 järgimiseks vajalikud normid 1. septembriks 2008.
Kui liikmesriigid võtavad need õigusnormid vastu, lisavad nad normidesse või normide ametliku avaldamise korral nende juurde viite käesolevale direktiivile. Sellise viitamise viisi näevad ette liikmesriigid.
2. Liikmesriigid edastavad komisjonile käesoleva direktiiviga reguleeritavas valdkonnas nende poolt vastu võetud põhiliste riiklike õigusnormide teksti.
Artikkel 12
Jõustumine
Käesolev direktiiv jõustub kahekümnendal päeval pärast selle avaldamist Euroopa Liidu Teatajas.
Artikkel 13
Adressaadid
Käesolev direktiiv on adresseeritud liikmesriikidele.
Brüssel, 24. oktoober 2006
Komisjoni nimel
komisjoni liige
Markos KYPRIANOU
(1) ELT L 102, 7.4.2004, lk 48.
(2) http://pharmacos.eudra.org/F2/eudralex/vol-4/home.htm ja ELT L 262, 14.10.2003, lk 22.
(3) ELT L 38, 9.2.2006, lk 40.
I LISA
Artiklis 3 viidatud nõuded koepankade akrediteerimiseks, määramiseks või neile loa või litsentsi andmiseks
A. ORGANISATSIOON JA JUHTIMINE
|
1. |
Tuleb määrata vastutav isik, kelle kvalifikatsioon ja ülesanded on sätestatud direktiivi 2004/23/EÜ artiklis 17. |
|
2. |
Koepangal peab olema organisatsiooniline struktuur ja töökord, mis on kohased tegevusele, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse; organisatsiooni skeemil peavad olema selgelt näidatud vastutusalad ja aruandlussuhted. |
|
3. |
Igal koepangal peab olema võimalus konsulteerida selleks määratud registreeritud arstiga, kes nõustab ja kontrollib meditsiinilist tegevust, nagu doonorite valik, kasutatud kudede ja rakkude kliiniline kontroll või suhtlemine kliiniliste kasutajatega. |
|
4. |
Tegevuse puhul, millele akrediteerimist, määramist, luba või litsentsi taotletakse, peab olema kehtestatud dokumenteeritud kvaliteedijuhtimissüsteem, mis vastab käesolevas direktiivis sätestatud standarditele. |
|
5. |
Kudede ja rakkude kvaliteedi ja ohutuse huvides tuleb tagada, et bioloogilise materjali kasutamise ja käitlemisega seotud ohud tehakse kindlaks ja minimeeritakse. Need ohud hõlmavad ka koepanga menetluste, keskkonna ja töötajate tervisliku seisundiga seotud riske. |
|
6. |
Koepankade ja kolmandate isikute vahelised lepingud peavad vastama direktiivi 2004/23/EÜ artiklile 24. Kolmandate isikutega sõlmitud lepingutes tuleb täpsustada lepingulise suhte aluseks olevad tingimused ja lepinguosaliste vastutus, samuti protokollid, mida tuleb järgida, et tegevus vastaks nõuetele. |
|
7. |
Peab olema kehtestatud dokumenteeritud süsteem, mis allub vastutava isiku järelevalvele ja mille alusel kinnitatakse, et koed ja/või rakud vastavad ohutus- ja kvaliteedistandarditele ja neid võib kasutusse lubada. |
|
8. |
Tegevuse lõpetamise korral hõlmavad direktiivi 2004/23/EÜ artikli 21 lõike 5 kohaselt sõlmitud lepingud ja kehtestatud menetlused jälgitavusandmeid ning rakkude ja kudede kvaliteedi ja ohutusega seotud materjali. |
|
9. |
Peab olema kehtestatud dokumenteeritud süsteem, millega tagatakse iga koe- või rakuüksuse identifitseerimine kõigil selle tegevuse etappidel, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse. |
B. TÖÖTAJAD
|
1. |
Koepankades peab olema piisaval hulgal vajaliku kvalifikatsiooniga töötajaid. Töötajate pädevust tuleb hinnata kvaliteedisüsteemis kindlaks määratud ajavahemike tagant. |
|
2. |
Kõigi töötajate jaoks tuleb koostada selged ja ajakohastatud kirjalikud ametikirjeldused. Tööülesanded, vastutusala ja aruandekohustus tuleb selgelt dokumenteerida ja need peavad olema üheselt mõistetavad. |
|
3. |
Töötajatele tuleb pakkuda baasväljaõpet ja täiendõpet seoses menetluste muutumise ja arenevate teadmistega, samuti piisavaid võimalusi ametialaseks arenguks. Koolitusprogrammi raames tuleb tagada ja dokumenteerida, et
|
C. SEADMED JA MATERJALID
|
1. |
Kõik seadmed ja materjalid tuleb kavandada ja neid tuleb hooldada vastavalt kasutamise eesmärgile ning nii, et retsipiendid ja/või töötajad oleksid võimalikult vähe ohustatud. |
|
2. |
Kõik kriitilised ja tehnilised seadmed tuleb märgistada ja valideerida, neid tuleb korrapäraselt kontrollida ja ennetavalt hooldada vastavalt tootja juhistele. Kui seadmed või materjalid mõjutavad töötlemise või säilitamisega seotud kriitilisi parameetreid (nt temperatuur, rõhk, osakeste sisaldus, mikroobne saastumine), tuleb need märgistada ja varustada häiresüsteemiga ning neid tuleb kontrollida ja võtta vajaduse korral korrektiivmeetmeid, et avastada rikkeid ja defekte ning tagada kriitiliste parameetrite hoidmine ettenähtud piirides. Kõik seadmed, mida kasutatakse kriitilisteks mõõtmisteks, tuleb kalibreerida, võimaluse korral jälgitava standardi järgi. |
|
3. |
Uusi ja remonditud seadmeid tuleb paigaldamise ajal kontrollida ja need tuleb enne kasutamist valideerida. Kontrolli tulemused tuleb dokumenteerida. |
|
4. |
Kriitilisi seadmeid tuleb regulaarselt hooldada, puhastada ja desinfitseerida ning säilitada vastav dokumentatsioon. |
|
5. |
Iga kriitilise seadme jaoks peab olema koostatud põhjalik töökirjeldus, milles antakse juhiseid ka tegevuse kohta seadme rikke või tõrke korral. |
|
6. |
Sellise tegevuse kirjelduses, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse, tuleb anda põhjalik ülevaade kõigi kriitiliste materjalide ja reaktiivide omadustest. Eelkõige tuleb kirjeldada lisaainete (nt lahuste) ja pakkematerjalide omadusi. Kriitilised reaktiivid ja materjalid peavad vastama dokumenteeritud nõuetele ja spetsifikaatidele ning vajaduse korral nõukogu 14. juuni 1993. aasta direktiivi 93/42/EMÜ (meditsiiniseadmete kohta) (1) ja Euroopa Parlamendi ja nõukogu 27. oktoobri 1998. aasta direktiivi 98/79/EÜ (meditsiiniliste in vitro diagnostikavahendite kohta) (2) nõuetele. |
D. RUUMID
|
1. |
Tegevuse jaoks, millele akrediteerimist, määramist, luba või litsentsi taotletakse, peavad koepangal olema sobivad ruumid, mis vastavad käesolevas direktiivis sätestatud standarditele. |
|
2. |
Kui see tegevus hõlmab kudede ja rakkude töötlemist nii, et need puutuvad kokku väliskeskkonnaga, peavad õhu kvaliteet ja keskkonna puhtus vastama kindlaksmääratud normidele, et minimeerida saastumise (sh annetustevahelise ristsaastumise) ohtu. Nende meetmete tõhusust tuleb valideerida ja kontrollida. |
|
3. |
Kui punktis 4 ei ole sätestatud teisiti, nõutakse juhul, kui koed või rakud puutuvad töötlemise ajal kokku väliskeskkonnaga ja pärast töötlemist ei toimu mikroobset inaktiveerimist, et osakeste ja mikroorganismide kolooniate sisaldus õhus vastaks A-klassi standardile, mis on kindlaks määratud praegustes Euroopa hea tootmistava suuniste 1. lisas ja direktiivis 2003/94/EÜ, taustkeskkonna näitajad peavad olema asjaomase koe/raku töötlemiseks sobivad, kusjuures osakeste ja mikroobide sisaldus peab olema samaväärne vähemalt hea tootmistava suuniste D-klassi näitajatega. |
|
4. |
Normid võivad olla punktis 3 sätestatust leebemad, kui
|
|
5. |
Punkti 4 alapunktides a–d tuleb täpsustada, mis keskkonnaga on tegemist. Tuleb näidata ja dokumenteerida, et valitud keskkond vastab vajalikul määral kvaliteedi- ja ohutusnõuetele, võttes arvesse kavandatud eesmärki, kasutusviisi ning retsipiendi immuunseisundit. Koepanga igas asjaomases osakonnas peavad olema sobivad rõivad ning kaitse- ja hügieenivahendid koos kirjalike hügieeni- ja riietumisjuhistega. |
|
6. |
Kui tegevus, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse, hõlmab kudede ja rakkude ladustamist, tuleb kindlaks määrata kudede ja rakkude omaduste säilimiseks vajalikud tingimused, sealhulgas sellised parameetrid nagu temperatuur, niiskus ja õhu kvaliteet. |
|
7. |
Kriitilisi parameetreid (nt temperatuur, niiskus, õhu kvaliteet) tuleb kontrollida, jälgida ja registreerida, et näidata nende vastavust kindlaksmääratud ladustamistingimustele. |
|
8. |
Ruumid, kus kudesid ja rakke hoitakse, peavad olema sellised, et kudesid ja rakke, mille kasutamiseks pole veel luba antud/mis on karantiinis, saab hoida selgelt eraldi nendest kudedest ja rakkudest, mille kasutamiseks on luba antud, ja nendest, mille puhul on loa andmisest keeldutud, et vältida kudede ja rakkude segiminekut ja ristsaastumist. Nii karantiinis kui ka kasutamiseks lubatud materjali ladustamiskohtades peavad olema füüsiliselt eraldi asuvad alad või säilitusvahendid või kindlat eristamist võimaldavad säilitusvahendid teatavate erikriteeriumide alusel kogutud kudede ja rakkude hoidmiseks. |
|
9. |
Koepangal peavad olema kirjalikud eeskirjad ja menetlused kontrollitud juurdepääsuks, puhastamiseks ja hooldamiseks, jäätmete kõrvaldamiseks ja teenuste ümberkorraldamiseks eriolukorras. |
E. DOKUMENDID JA ANDMETE SÄILITAMINE
|
1. |
Koepankades peab olema kehtestatud süsteem, mis võimaldab tegevuse kohta, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse, säilitada selgeid ja informatiivseid dokumente, täpseid andmeid ja registreid ning kinnitatud standardse töökorra. Dokumendid tuleb korrapäraselt läbi vaadata ning need peavad vastama käesoleva direktiiviga kehtestatud nõuetele. Süsteem peab tagama, et tehtav töö oleks standardiseeritud ning et kõik etapid (kodeerimine, doonorite kõlblikkus, hankimine, töötlemine, säilitamine, ladustamine, transport, jaotamine või hävitamine ning kvaliteedikontroll) oleksid jälgitavad. |
|
2. |
Iga kriitilise tegevuse puhul tuleb kindlaks teha ja dokumenteerida kasutatud materjalid ja seadmed ning sellega seotud töötajad. |
|
3. |
Koepankades peavad selleks volitatud töötajad kõik dokumentide muudatused läbi vaatama, kuupäevastama, heaks kiitma, dokumenteerima ja neid viivitamata rakendama. |
|
4. |
Koepankades peab olema kehtestatud dokumentide kontrolli menetlus, mis võimaldab anda ülevaate dokumentide läbivaatamistest ja muudatustest ning tagada, et kasutatakse ainult dokumentide ajakohastatud versioone. |
|
5. |
Tuleb näidata, et andmed on usaldusväärsed ja kajastavad tulemusi tõeselt. |
|
6. |
Andmed peavad olema loetavad ja kustumatud ning need võivad olla kirjutatud käsitsi või registreeritud mõne teise valideeritud süsteemi abil, nagu arvuti või mikrofilm. |
|
7. |
Ilma et see piiraks artikli 9 lõike 2 kohaldamist, hoitakse kõiki andmeid, sealhulgas algandmeid, mis on kudede ja rakkude ohutuse ja kvaliteedi jaoks kriitilise tähtsusega, nii, et neile oleks tagatud juurdepääs vähemalt kümne aasta jooksul pärast kudede ja rakkude aegumiskuupäeva, kliinilist kasutamist või likvideerimist. |
|
8. |
Andmed peavad vastama direktiivi 2004/23/EÜ artiklis 14 kehtestatud konfidentsiaalsusnõuetele. Juurdepääs registritele ja andmetele peab olema võimaldatud ainult isikutele, kellel on selleks vastutava isiku volitus, ning pädevale asutusele inspekteerimise ja kontrollimise eesmärgil. |
F. KVALITEEDIKONTROLL
|
1. |
Tegevuse suhtes, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse, peab olema kehtestatud auditeerimissüsteem. Vähemalt iga kahe aasta järel peavad vastava koolituse läbinud pädevad isikud korraldama sõltumatu auditi, mis võimaldab kontrollida tegevuse vastavust kinnitatud protokollidele ja õiguslikele nõuetele. Tulemused ja parandusmeetmed tuleb dokumenteerida. |
|
2. |
Kui nõutud kvaliteedi- ja ohutusstandarditest kõrvale kaldutakse, tuleb korraldada dokumenteeritud uurimine, millega kaasneb otsus võimalike parandus- ja ennetusmeetmete kohta. Kudede ja rakkude osas, mis ei vasta nõuetele, tuleb vastutava isiku juhtimisel vastu võtta otsus, lähtudes kirjalikust menetlusest, ning kõnealune otsus dokumenteerida. Kõik asjaomased koed ja rakud tuleb kindlaks teha ja nende üle peetakse arvestust. |
|
3. |
Parandusmeetmed tuleb dokumenteerida ning need tuleb kasutusele võtta ja lõpule viia õigeaegselt ja tõhusalt. Pärast ennetus- ja parandusmeetmete rakendamist tuleb hinnata nende tõhusust. |
|
4. |
Koepank peab kehtestama protsessid kvaliteedijuhtimissüsteemide läbivaatamiseks, et neid pidevalt ja süstemaatiliselt täiendada. |
(1) EÜT L 169, 12.7.1993, lk 1. Direktiivi on viimati muudetud Euroopa Parlamendi ja nõukogu määrusega (EÜ) nr 1882/2003 (ELT L 284, 31.10.2003, lk 1).
(2) EÜT L 331, 7.12.1998, lk 1. Direktiivi on muudetud määrusega (EÜ) nr 1882/2003.
II LISA
Artiklis 4 osutatud nõuded kudede ja rakkude ettevalmistusprotsesside kohta loa andmiseks
Pädev asutus annab kudede ja rakkude ettevalmistamisprotsessile loa pärast seda, kui on hinnanud doonorite valiku kriteeriume ja hankemenetlust, protsessi iga etapi kohta koostatud protokolle, kvaliteedijuhtimise kriteeriume ning rakkude ja kudede lõplikke kvantitatiivseid ja kvalitatiivseid kriteeriume. Hindamine peab vastama vähemalt käesolevas lisas sätestatud nõuetele.
A. VASTUVÕTMINE KOEPANGAS
Kudede ja rakkude vastuvõtmisel koepangas peavad need vastama direktiivis 2006/17/EÜ sätestatud nõuetele.
B. TÖÖTLEMINE
Kui tegevus, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse, hõlmab kudede ja rakkude töötlemist, peavad koepanga menetlused vastama järgmistele kriteeriumidele.
|
1. |
Kriitilised töötlemismenetlused tuleb valideerida ning nende tulemusel ei tohi koed või rakud kaotada oma kliinilist toimet või retsipienti kahjustada. Valideerimine peab põhinema uuringutel, mida teeb koepank ise, või avaldatud uurimuste andmetel või varem kasutatud töötlemismenetluste puhul koepanga väljastatud kudede kliiniliste tulemuste retrospektiivsel hinnangul. |
|
2. |
Tuleb näidata, et koepanga töötajad suudavad valideeritud protsessi järjepidevalt ja tõhusalt läbi viia. |
|
3. |
Menetlused tuleb dokumenteerida standardses töökorras, mis peab vastama valideeritud meetodile ning käesolevas direktiivis sätestatud nõuetele vastavalt I lisa E-osa punktidele 1–4. |
|
4. |
Tuleb tagada, et kõik protsessid oleksid vastavuses kinnitatud standardse töökorraga. |
|
5. |
Kui kudede ja rakkude suhtes kasutatakse mikroobset inaktiveerimist, tuleb seda protsessi täpselt kirjeldada, dokumenteerida ja valideerida. |
|
6. |
Enne töötlemise märkimisväärset muutmist tuleb muudetav protsess valideerida ja dokumenteerida. |
|
7. |
Töötlemismenetlusi peab korrapäraselt kriitiliselt hindama, et nende abil oleks jätkuvalt võimalik saavutada soovitud tulemusi. |
|
8. |
Kudede ja rakkude hävitamisel tuleb vältida teiste annetuste ja toodete, töötlemiskeskkonna ja töötajate kahjustamist. Menetlused peavad vastama riikide õigusaktidele. |
C. TOODETE LADUSTAMINE JA KASUTUSSE LUBAMINE
Kui tegevus, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse, hõlmab kudede ja rakkude ladustamist ja kasutusse lubamist, peavad koepanga menetlused vastama järgmistele kriteeriumidele.
|
1. |
Tuleb määrata ladustamistingimustest sõltuv maksimaalne ladustamisaeg. Aja valikul tuleb muu hulgas arvesse võtta kudede ja rakkude omaduste võimalikku halvenemist. |
|
2. |
Peab olema kehtestatud kudede ja rakkude hoidmise süsteem, mis aitab tagada, et kudesid ja rakke ei lubataks kasutusse enne, kui kõik käesoleva direktiiviga sätestatud nõudmised on täidetud. Standardses töökorras tuleb üksikasjalikult kirjeldada kudede ja rakkude kasutusse lubamise tingimusi ja menetlusi ning sellega seotud tööülesandeid. |
|
3. |
Peab olema kehtestatud süsteem, mis võimaldaks kudesid ja rakke igal töötlemisetapil identifitseerida ja millega eristataks selgelt kasutusse lubatud koed ja rakud neist, mida pole veel kasutusse lubatud (mis on karantiinis) või mille puhul on loa andmisest keeldutud. |
|
4. |
Dokumentides tuleb näidata, et enne, kui koed ja rakud kasutusse lubatakse, on kõik nõuded täidetud ning isik, kelle direktiivi 2004/23/EÜ artiklis 17 osutatud vastutav isik on selleks volitanud, on vastavalt kirjalikule menetlusele kontrollinud kõiki kehtivaid deklaratsioone, asjakohaseid meditsiinilisi andmeid, töötlemisandmeid ja uurimistulemusi. Kui tulemused saadetakse laborist välja arvuti teel, tuleb kontrolljäljes näidata, kes nende väljasaatmise eest vastutab. |
|
5. |
Pärast uute doonorite valiku kriteeriumi või uue kontrollikriteeriumi kasutuselevõttu või mõne töötlemisetapi olulist muudatust, mille eesmärk on suurendada ohutust ja parandada kvaliteeti, tuleb läbi viia dokumenteeritud riskihindamine, et teha kindlaks, mida teha kõigi ladustatud kudede ja rakkudega; riskihindamise tulemused kinnitab direktiivi 2004/23/EÜ artiklis 17 määratletud vastutav isik. |
D. JAOTAMINE JA TAGASIVÕTMINE
Kui tegevus, mille jaoks akrediteerimist, määramist, luba või litsentsi taotletakse, hõlmab kudede ja rakkude jaotamist, peavad koepanga menetlused vastama järgmistele kriteeriumidele.
|
1. |
Tuleb kindlaks määrata kriitilised transporditingimused, nt temperatuur ja ajapiirang, mis on vajalikud kudede ja rakkude omaduste säilimiseks. |
|
2. |
Mahuti/pakend peab olema turvaline ja tagama, et koed ja rakud säilivad kindlaksmääratud tingimustes. Kõik mahutid ja pakendid tuleb valideerida. |
|
3. |
Kui jaotamisega tegeleb lepingulisel alusel kolmas isik, tuleb sõlmida kirjalik leping, et tagada nõutud tingimuste säilimine. |
|
4. |
Koepangas peab olema töötajaid, keda on volitatud hindama tagasivõtmise vajadust ning kes peavad selleks vajaliku tegevuse algatama ja seda koordineerima. |
|
5. |
Koepangas peab olema kehtestatud tõhus tagasivõtmise menetlus, milles kirjeldatakse ka kohustusi ja tehtavaid toiminguid. Menetlus peab hõlmama ka pädeva asutuse teavitamist. |
|
6. |
Tagasivõtmine peab toimuma kindlaksmääratud ajavahemiku jooksul ning see hõlmab kõikide asjaomaste kudede ja rakkude jälgitavust ja vajaduse korral tagasijälgitavust. Uurimise eesmärk on identifitseerida doonor, kes võis põhjustada retsipiendi reaktsiooni, ning kõrvaldada sellelt doonorilt saadud koed ja rakud, samuti teavitada samalt doonorilt saadud kudede ja rakkude vastuvõtjaid ja retsipiente juhul, kui nad on võinud ohtu sattuda. |
|
7. |
Koepangas peab olema kehtestatud menetlus kudede ja rakkude tellimuste haldamiseks. Kudede ja rakkude jaotamiseks teatavatele patsientidele või tervishoiuasutustele peavad olema kirjalikud eeskirjad, mis tuleb nimetatud isikutele nende soovi korral esitada. |
|
8. |
Tagasivõetud toodete haldamiseks peab olema kehtestatud dokumenteeritud süsteem, sh ka kriteeriumid nende loendisse kandmiseks. |
E. LÕPLIK JAOTAMISEELNE MÄRGISTAMINE
|
1. |
Kudede/rakkude esmasele pakendile tuleb märkida järgmised andmed:
Kui punktides d ja e nõutud teavet ei ole võimalik esmase pakendi etiketile kanda, tuleb see esitada koos pakendiga eraldi lehel. Leht tuleb esmasele pakendile lisada nii, et see pakendist ei eralduks. |
|
2. |
Järgmine teave tuleb esitada kas etiketil või pakendile lisatud dokumentides:
|
F. VEOKONTEINERI MÄRGISTAMINE
Transpordiks tuleb esmane pakend asetada veokonteinerisse, millel peab olema vähemalt järgmine teave:
|
a) |
vastava koepanga identifitseerimisandmed, sh aadress ja telefoninumber; |
|
b) |
inimkasutuse eest vastutava organisatsiooni identifitseerimisandmed, sh aadress ja telefoninumber; |
|
c) |
teave selle kohta, et pakend sisaldab inimkudesid/-rakke, ja märge “ETTEVAATUST KÄSITSEMISEL”; |
|
d) |
kui siirdamiseks on vaja elusrakke, nt tüvirakke, sugurakke või embrüosid, tuleb lisada keeld “MITTE KIIRITADA”; |
|
e) |
soovitatavad transporditingimused (nt hoida jahedas, püstises asendis jne); |
|
f) |
ohutusjuhised/jahutusmeetod (vajaduse korral). |
III LISA
RASKETEST KÕRVALTOIMETEST TEATAMINE
IV LISA
TÕSISTEST KÕRVALEKALLETEST TEATAMINE

V LISA
AASTAARUANDE VORM
VI LISA
Artiklis 9 nõutud minimaalne teave, mis tuleb säilitada doonorite/retsipientide kohta
A. KOEPANKADES
Doonori identifitseerimisandmed
Annetuse identifitseerimisandmed, sh vähemalt
|
— |
hankiva asutuse või koepanga identifitseerimisandmed, |
|
— |
annetuse identifitseerimisnumber, |
|
— |
hankimise kuupäev, |
|
— |
hankimise koht, |
|
— |
annetuse liik (nt üks või mitu kude; autoloogne või allogeenne; elusdoonor või surnud doonor). |
Toote identifitseerimisandmed, sh vähemalt
|
— |
koepanga identifitseerimistunnus, |
|
— |
koe või raku/toote liik (põhinomenklatuur), |
|
— |
partii number (kui see on olemas), |
|
— |
osapartii number (kui see on olemas), |
|
— |
säilivusaeg, |
|
— |
koe/raku seisund (st karantiinis, sobib kasutamiseks jne), |
|
— |
toodete kirjeldus ja päritolu, läbitud töötlemise etapid, kudede ja rakkudega kokku puutunud materjalid ja lisaained, mis mõjutavad nende kvaliteeti ja/või ohutust, |
|
— |
lõpliku etiketi väljastanud asutuse identifitseerimistunnus. |
Inimkasutuse identifitseerimisandmed, sh vähemalt
|
— |
jaotamise/hävitamise kuupäev, |
|
— |
arsti või lõppkasutaja/asutuse identifitseerimisandmed. |
B. INIMKASUTUSE EEST VASTUTAVAS ORGANISATSIOONIS
|
a) |
koepanga identifitseerimisandmed; |
|
b) |
arsti või lõppkasutaja/asutuse identifitseerimisandmed; |
|
c) |
kudede ja rakkude liik; |
|
d) |
toote identifitseerimisandmed; |
|
e) |
retsipiendi identifitseerimisandmed; |
|
f) |
kasutamise kuupäev. |
VII LISA
Euroopa kodeerimissüsteemis nõutav teave
|
a) |
Annetuse identifitseerimisandmed:
|
|
b) |
Toote identifitseerimisandmed:
|