1991R2568 — ET — 11.10.2016 — 030.001
Käesolev tekst on üksnes dokumenteerimisvahend ning sel ei ole mingit õiguslikku mõju. Liidu institutsioonid ei vastuta selle teksti sisu eest. Asjakohaste õigusaktide autentsed versioonid, sealhulgas nende preambulid, on avaldatud Euroopa Liidu Teatajas ning on kättesaadavad EUR-Lexi veebisaidil. Need ametlikud tekstid on vahetult kättesaadavad käesolevasse dokumenti lisatud linkide kaudu
|
KOMISJONI MÄÄRUS (EMÜ) nr 2568/91, 11. juuli 1991, oliiviõlide ja pressimisjääkide omaduste ja asjakohaste analüüsimeetodite kohta (ELT L 248 5.9.1991, lk 1) |
Muudetud:
|
|
|
Euroopa Liidu Teataja |
||
|
nr |
lehekülg |
kuupäev |
||
|
L 349 |
36 |
18.12.1991 |
||
|
L 150 |
17 |
2.6.1992 |
||
|
L 176 |
27 |
30.6.1992 |
||
|
L 199 |
18 |
18.7.1992 |
||
|
L 327 |
28 |
13.11.1992 |
||
|
L 22 |
58 |
30.1.1993 |
||
|
L 87 |
6 |
7.4.1993 |
||
|
L 66 |
29 |
18.3.1993 |
||
|
L 24 |
33 |
29.1.1994 |
||
|
COMMISSION REGULATION (EC) No 2632/94 of 28 October 1994 (*) |
L 280 |
43 |
29.10.1994 |
|
|
L 69 |
1 |
29.3.1995 |
||
|
COMMISSION REGULATION (EC) No 2527/95 of 27 October 1995 (*) |
L 258 |
49 |
28.10.1995 |
|
|
COMMISSION REGULATION (EC) No 2472/97 of 11 December 1997 (*) |
L 341 |
25 |
12.12.1997 |
|
|
L 28 |
5 |
4.2.1998 |
||
|
COMMISSION REGULATION (EC) No 2248/98 of 19 October 1998 (*) |
L 282 |
55 |
20.10.1998 |
|
|
L 46 |
15 |
20.2.1999 |
||
|
L 65 |
9 |
7.3.2001 |
||
|
COMMISSION REGULATION (EC) No 2042/2001 of 18 October 2001 (*) |
L 276 |
8 |
19.10.2001 |
|
|
L 128 |
8 |
15.5.2002 |
||
|
L 295 |
57 |
13.11.2003 |
||
|
L 161 |
11 |
22.6.2007 |
||
|
L 178 |
11 |
5.7.2008 |
||
|
L 23 |
1 |
27.1.2011 |
||
|
L 192 |
3 |
20.7.2012 |
||
|
L 90 |
52 |
28.3.2013 |
||
|
KOMISJONI RAKENDUSMÄÄRUS (EL) nr 1348/2013, 16. detsember 2013, |
L 338 |
31 |
17.12.2013 |
|
|
KOMISJONI DELEGEERITUD MÄÄRUS (EL) 2015/1830, 8. juuli 2015, |
L 266 |
9 |
13.10.2015 |
|
|
L 266 |
29 |
13.10.2015 |
||
|
L 202 |
7 |
28.7.2016 |
||
|
KOMISJONI RAKENDUSMÄÄRUS (EL) 2016/1784, 30. september 2016, |
L 273 |
5 |
8.10.2016 |
|
|
(*) |
Käesolevat akti ei ole eesti keeles avaldatud. |
KOMISJONI MÄÄRUS (EMÜ) nr 2568/91,
11. juuli 1991,
oliiviõlide ja pressimisjääkide omaduste ja asjakohaste analüüsimeetodite kohta
Artikkel 1
1. Õli, mille omadused vastavad käesoleva määruse I lisa punktides 1 ja 2 sätestatutele, käsitatakse neitsioliiviõlina määruse nr 136/66/EMÜ lisa punkti 1 alapunktide a ja b tähenduses.
2. Õli, mille omadused vastavad käesoleva määruse I lisa punktis 3 sätestatutele, käsitatakse lambiõlina määruse nr 136/66/EMÜ lisa punkti 1 alapunkti c tähenduses.
3. Õli, mille omadused vastavad käesoleva määruse I lisa punktis 4 sätestatutele, käsitatakse rafineeritud oliiviõlina määruse nr 136/66/EMÜ lisa punkti 2 tähenduses.
4. Õli, mille omadused vastavad käesoleva määruse I lisa punktis 5 sätestatutele, käsitatakse rafineeritud oliiviõlist ja neitsioliiviõlist koosnevaks oliiviõliks määruse nr 136/66/EMÜ lisa punkti 3 tähenduses.
5. Õli, mille omadused vastavad käesoleva määruse I lisa punktis 6 sätestatutele, käsitatakse töötlemata oliivijääkõlina määruse nr 136/66/EMÜ lisa punkti 4 tähenduses.
6. Õli, mille omadused vastavad käesoleva määruse I lisa punktis 7 sätestatutele, käsitatakse rafineeritud oliivijääk- õlina määruse nr 136/66/EMÜ lisa punkti 5 tähenduses.
7. Õli, mille omadused vastavad käesoleva määruse I lisa punktis 8 sätestatutele, käsitatakse oliivijääkõlina määruse nr 136/66/EMÜ lisa punkti 6 tähenduses.
Artikkel 2
1. I lisas sätestatud õlide omadused määratakse kindlaks järgmiste analüüsimeetoditega:
a) oleiinhappe protsendina väljendatud vabad rasvhapped määratakse II lisas sätestatud meetodiga;
b) peroksiidarv määratakse kindlaks III lisas sätestatud meetodiga;
c) vahade sisaldus määratakse IV lisas sätestatud meetodiga;
d) steroolide ja triterpeendialkoholide koostis ja sisaldus määratakse gaasikromatograafiliselt kapillaarkolonniga, kasutades V lisas sätestatud meetodit;
e) 2-glütserüülmonopalmitaadi protsendiline sisaldus määratakse VII lisas sätestatud meetodiga;
f) spektrofotomeetriline analüüs tehakse IX lisas sätestatud meetodiga;
g) rasvhapete koostis määratakse X lisas sätestatud meetodiga;
h) halogeenitud lenduvad lahustid määratakse XI lisas sätestatud meetodiga;
i) neitsioliiviõli organoleptilisi omadusi hinnatakse XII lisas sätestatud meetodiga;
j) stigmastadieene määratakse XVII lisas sätestatud meetodiga;
k) ECN42-triglütseriidide sisaldus määratakse XVIII lisas sätestatud meetodiga;
l) alifaatsete alkoholide ja triterpeenalkoholide sisaldus määratakse XIX lisas sätestatud meetodiga;
m) vahade, rasvhapete metüülestrite ja etüülestrite sisaldus määratakse XX lisas sätestatud meetodiga.
▼M28 —————
2. Liikmesriikide ametiasutused või nende esindajad kontrollivad neitsioliiviõlide organoleptilisi omadusi liikmesriikide määratud degusteerimiskomisjonide kaudu.
Õli organoleptilised omadused, millele on osutatud esimeses lõigus, loetakse deklareeritud kategooriale vastavaks, kui liikmesriigi määratud degusteerimiskomisjon kinnitab klassifikatsiooni.
Kui degusteerimiskomisjon organoleptiliste omaduste põhjal ei kinnita deklareeritud kategooriat, korraldavad ametiasutused või nende esindajad huvitatud isiku palvel kaks kordushindamist muudes heakskiidetud degusteerimiskomisjonides, millest vähemalt ühe on heaks kiitnud asjaomase tootja liikmesriik. Asjaomaseid omadusi peetakse vastavaks deklareeritud kategooriale, kui vähemalt kahel kontrollhindamisel kinnitatakse deklareeritud kategooria. Vastupidisel juhul kannab huvitatud osaline kontrollhindamise kulud.
3. Kui riigi ametiasutused või nende esindajad kontrollivad õli omadusi, nagu on osutatud lõikes 1, võetakse proovid vastavalt rahvusvahelisele standardile EN ISO 661, mis käsitleb uuritavate proovide ettevalmistamist, ja EN ISO 5555, mis käsitleb proovide võtmist. Ent standardi EN ISO 5555 punktist 6.8 olenemata võetakse proov selliste esmapakendis õlipartiide korral vastavalt käesoleva määruse Ia lisale. Pakendamata õlide puhul, kui proove ei saa võtta vastavalt standardile EN ISO 5555, toimub proovide võtmine vastavalt liikmesriigi pädeva asutuse juhenditele.
Ilma, et see piiraks standardi EN ISO 5555 ja standardi EN ISO 661 6. peatüki kohaldamist, asetatakse võetud proovid võimalikult kiiresti pimedasse kohta ja eemale soojusallikatest ning saadetakse laborisse analüüsimiseks hiljemalt viiendal tööpäeval pärast võtmist; vastupidisel juhul hoitakse proove nii, et need vedamisel ega säilitamisel ei halveneks ega saaks kahjustatud enne laborisse saatmist.
4. Lõikes 3 osutatud kontrollimiste läbiviimisel tehakse pakendatud toodete puhul II, III, IX, XII ja XX lisas osutatud analüüsid ning vajaduse korral kõik siseriiklike seadustega nõutavad kontrollanalüüsid enne minimaalse säilimisaja tähtpäeva. Lahtiste õlide proovide puhul tehakse need analüüsid hiljemalt kuuendal kuul pärast proovi võtmist.
Muude käesolevas määruses sätestatud analüüside puhul ei kohaldata tähtaegu.
Kui analüüside tulemused ei vasta oliiviõli või oliivijääkõli deklareeritud kategooria omadustele, teatatakse huvitatud isikule sellest hiljemalt üks kuu enne esimeses lõigus sätestatud tähtaja lõppu, välja arvatud juhul, kui proov on võetud vähem kui kaks kuud enne minimaalse säilimisaja lõppu.
5. Oliiviõlide omaduste määramisel lõikes 1 ettenähtud meetoditega võrreldakse analüüsi tulemusi otse käesolevas määruses sätestatud piirväärtustega.
Artikkel 2a
1. Käesolevas artiklis tähendab asjaomase liikmesriigi „turustatav oliiviõli” oliiviõli ja oliivijääkõli summaarset kogust, mis tarbitakse kõnealuses liikmesriigis või eksporditakse kõnealusest liikmesriigist.
2. Liikmesriik tagab, et vastavuskontrolle tehakse valikuliselt, toetudes riskianalüüsile, ja piisavalt sageli tagamaks, et turustatav oliiviõli vastab deklareeritud kategooriale.
3. Riski hindamise kriteeriumid võivad olla muu hulgas:
a) õli kategooria, tootmise ajavahemik, õli hind muude taimeõlidega võrreldes, segamis- ja pakkimistoimingud, hoidmisvõimalused ja -tingimused, päritoluriik, sihtkohariik, transpordivahend või partii maht;
b) käitaja asukoht turustamisahelas, tema turustatava õli kogus ja/või väärtus, tema turustatavate õlikategooriate sortiment, äritegevuse tüüp, nagu pressimine, säilitamine, rafineerimine, segamine, pakkimine või jaemüük;
c) eelmiste kontrollide tulemused, sealhulgas leitud vigade arv ja liik, turustatavate õlide tavapärane kvaliteet, kasutatavat tehnilist varustust iseloomustavad näitajad;
d) käitajate kasutatava turustamisstandarditele vastavuse alase kvaliteeditagamise süsteemi või enesekontrolli süsteemi usaldusväärsus;
e) kontrolli teostamise koht, eriti kui see on Euroopa Liitu esmase sisenemise koht, Euroopa Liidust väljumise viimane punkt või koht, kus õlisid toodetakse, pakitakse, laaditakse või müüakse lõpptarbijale;
f) mis tahes muu teave, mis võib viidata standarditele mittevastavuse ohule.
4. Liikmesriigid sätestavad eelnevalt järgmist:
a) partiide standarditele mittevastavuse ohu hindamise kriteeriumid;
b) iga riskikategooria riskianalüüsi põhjal minimaalne arv ettevõtjaid või partiisid ja/või koguseid, mille vastavust tuleb kontrollida.
Liikmesriigis turustatava oliiviõli iga tuhande tonni kohta tuleb teha vähemalt üks vastavuse kontroll aastas.
5. Liikmesriik kontrollib vastavust järgmiselt:
a) viies selleks ükskõik millises järjekorras läbi I lisas sätestatud analüüsid või
b) Ib lisa otsustamisskeemil sätestatud järjekorras, kuni saavutatakse mõni otsustamisskeemil nimetatud otsustest.
▼M19 —————
Artikkel 3
Kui selgub, et õli ei vasta oma kategooria kirjeldusele, kohaldab asjaomane liikmesriik tõhusaid, proportsionaalseid ja hoiatavaid rahalisi karistusi, mis määratakse kindlaks eeskirjade eiramise raskusastet arvesse võttes, ilma et see piiraks muude karistuste kohaldamist.
Kui kontrolli tulemusel ilmnevad märkimisväärsed eeskirjade eiramised, suurendab liikmesriik turustamisstaadiumi, õli kategooria, päritolu või muude kriteeriumide alusel kontrollimise sagedust.
Artikkel 4
1. The Member States may approve assessment panels so that national authorities or their representatives can assess and verify organoleptic characteristics.
The terms of approval shall be set by Member States and ensure that:
— the requirements of Annex XII.4 are met,
— the panel head is given training recognised for this purpose by the Member State,
— continued approval depends on performance in annual checks arranged by the Member State.
Member States shall notify to the Commission a list of approved panels and the action taken under this paragraph.
2. Kui liikmesriikidel on raskusi degusteerimiskomisjonide loomisega, võivad nad kasutada mõnes teises liikmesriigis heakskiidetud komisjoni.
3. Iga liikmesriik koostab kutseorganisatsioonide või kutsealadevaheliste organisatsioonide loodud degusteerimiskomisjonide loetelu vastavalt lõikes 1 sätestatud tingimustele ning tagab kõnealuste tingimuste järgimise.
▼M19 —————
Artikkel 6
1. Oliiviõli ekstraheerimisest tulenev õlikoogi ja muude jääkide õlisisaldus (CN-koodid 2306 90 11 ja 2306 90 19 ) määratakse kindlaks XV lisas sätestatud meetodil.
2. Lõikes 1 osutatud õlisisaldus väljendatakse õli massiprotsendina kuivaine massist.
Artikkel 7
Kohaldatakse ühenduse sätteid, mis käsitlevad saasteainete olemasolu.
Halogeenitud lahustite puhul on kõikide oliiviõli kategooriate puhul piirmäärad järgmised:
— iga tuvastatud halogeenitud lahusti maksimaalne sisaldus: 0,1 mg/kg,
— iga tuvastatud halogeenitud lahusti maksimaalne üldsisaldus: 0,2 mg/kg.
Artikkel 7a
Füüsilised või juriidilised isikud või isikute rühmad, kes omavad oliiviõli või oliivijääkõli alates eraldamisest pressimiskäitises kuni villimisetapini (kaasa arvatud) mis tahes kutseliseks või äriliseks otstarbeks, on kohustatud pidama iga sellise kategooria õli saamise ja väljaandmise registrit.
Liikmesriik tagab, et esimeses lõigus sätestatud kohustust korralikult täidetaks.
Artikkel 8
1. Liikmesriik teatab komisjonile käesoleva määruse rakendamiseks võetud meetmed. Ta teavitab komisjoni kõigist hilisematest muudatustest.
2. Hiljemalt iga aasta 31. mail edastab liikmesriik komisjonile aruande käesoleva määruse rakendamise kohta eelmisel kalendriaastal. Aruanne sisaldab vähemalt oliiviõlide vastavuskontrolli tulemusi vastavalt XXI lisas sätestatud vormidele.
3. Käesolevas määruses osutatud teatised edastatakse vastavalt komisjoni määrusele (EÜ) nr 792/2009 ( 1 ).
Artikkel 9
Määrus (EMÜ) nr 1058/77 tunnistatakse kehtetuks.
Artikkel 10
1. Käesolev määrus jõustub kolmandal päeval pärast selle avaldamist Euroopa Ühenduste Teatajas.
XII lisas sätestatud meetodit kohaldatakse siiski ►M1 1. november 1992 ◄ , välja arvatud sekkumissüsteemiga seotud toimingute puhul.
Kõnealust meetodit ei kohaldata enne 1. novembrit 1992 pakendatud neitsioliiviõli suhtes.
2. Käesolevat määrust ei kohaldata enne käesoleva määruse jõustumist pakendatud ja kuni 31. oktoobrini 1992 turustatava oliiviõli ja oliivijääkõli suhtes.
Käesolev määrus on tervikuna siduv ja vahetult kohaldatav kõikides liikmesriikides.
LISAD
Kokkuvõte
|
I. lisa: |
Oliiviõlide omadused |
|
I A lisa: |
Proovide võtmine oliiviõlist või oliivijääkõlist, mis on tarnitud esmapakendis |
|
I B lisa: |
Otsustamisskeem, mille järgi kontrollitakse, kas oliiviõli proov on kooskõlas deklareeritud kategooriaga |
|
II lisa: |
Vabade rasvhapete määramine külmmeetodil |
|
III lisa: |
Peroksiidiarvu määramine |
|
IV lisa: |
Vahade sisalduse määramine kapillaargaasikromatograafia abil |
|
V lisa: |
Steroolide ja triterpeendialkoholide koostise ja sisalduse gaasikromatograafiline määramine kapillaarkolonniga |
|
VII lisa: |
►M21 2-glütserüülmonopalmitaadi protsendilise sisalduse määramine ◄ |
|
▼M20 ————— |
|
|
IX lisa: |
Spektrofotomeetriline analüüs ultraviolettpiirkonnas |
|
X lisa: |
Metüülestrite rasvhapete gaasikromatograafiline määramine |
|
XI lisa: |
Oliiviõli halogeenitud lenduvate lahustite määramine |
|
XII lisa: |
Rahvusvahelise oliiviõlinõukogu meetod neitsioliiviõlide organoleptiliseks hindamiseks |
|
▼M20 ————— |
|
|
▼M19 ————— |
|
|
XV lisa: |
Oliiviõli pressimisjääkide õlisisaldus |
|
XVI lisa: |
Joodiarvu määramine |
|
XVII lisa: |
Meetod stigmastadieenide määramiseks taimeõlides |
|
XVIII lisa: |
ECN 42-Triatsüülglütseroolide tegeliku ja teoreetiliselt arvutatud sisalduse vahe määramine |
|
Annex XIX: |
►M28 Alifaatsete alkoholide ja triterpeenalkoholide sisalduse kapillaarkolonnkromatograafiline määramine ◄ |
|
XX lisa: |
Meetod vahade, rasvhapete metüülestrite ja etüülestrite sisalduse määramiseks kapillaargaasikromatograafiaga |
|
▼M28 ————— |
|
|
ХХI lisa: |
Oliiviõlide vastavuse artikli 8 lõikes 2 osutatud kontrollimise tulemused |
I LISA
OLIIVIÕLI OMADUSED
|
Kategooria |
Rasvhapete etüülestrid (FAEEd) (*) |
Happesus (%) (*) |
Peroksiidiarv mekv O2/kg (*) |
Vahad mg/kg (**) |
2-glütserüül-monopalmitaat (%) |
Stigmastadieenid mg/kg (1) |
Vahe: ECN42 (HPLC) ja ECN42 (teoreetiline arvutus) |
K232 (*) |
K268 või K270 (*) |
Δ-K (*) |
Organoleptiline hinnang: puuduse mediaan (Md) (*) |
Organoleptiline hinnang: puuviljalisuse mediaan (Mf) (*) |
|
1. Ekstra-neitsioliiviõli |
FAEEd ≤ 40 mg/kg (viljelusaasta 2013–2014 (2)) FAEEd ≤ 35 mg/kg (viljelusaasta 2014–2016) FAEEd ≤ 30 mg/kg (viljelusaastad pärast 2016. a) |
≤ 0,8 |
≤ 20 |
C42 + C44 + C46 ≤ 150 |
≤ 0,9, kui palmitiinhappe üldsisaldus ≤ 14 % |
≤ 0,05 |
≤ |0,2| |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0 |
Mf > 0 |
|
≤ 1,0, kui palmitiinhappe üldsisaldus > 14 % |
||||||||||||
|
2. Neitsioliiviõli |
— |
≤ 2,0 |
≤ 20 |
C42 + C44 + C46 ≤ 150 |
≤ 0,9, kui palmitiinhappe üldsisaldus ≤ 14 % |
≤ 0,05 |
≤ |0,2| |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0 |
|
≤ 1,0, kui palmitiinhappe üldsisaldus > 14 % |
||||||||||||
|
3. Lambiõli |
— |
> 2,0 |
— |
C40 + C42 + C44 + C46 ≤ 300 (3) |
≤ 0,9, kui palmitiinhappe üldsisaldus ≤ 14 % |
≤ 0,50 |
≤ |0,3| |
— |
— |
— |
Md > 3,5 (4) |
— |
|
≤ 1,1, kui palmitiinhappe üldsisaldus > 14 % |
||||||||||||
|
4. Rafineeritud oliiviõli |
— |
≤ 0,3 |
≤ 5 |
C40 + C42 + C44 + C46 ≤ 350 |
≤ 0,9, kui palmitiinhappe üldsisaldus ≤ 14 % |
— |
≤ |0,3| |
— |
≤ 1,10 |
≤ 0,16 |
— |
— |
|
≤ 1,1, kui palmitiinhappe üldsisaldus > 14 % |
||||||||||||
|
5. Rafineeritud ja neitsioliiviõlist koosnev oliiviõli |
— |
≤ 1,0 |
≤ 15 |
C40 + C42 + C44 + C46 ≤ 350 |
≤ 0,9, kui palmitiinhappe üldsisaldus ≤ 14 % |
— |
≤ |0,3| |
— |
≤ 0,90 |
≤ 0,15 |
— |
— |
|
≤ 1,0, kui palmitiinhappe üldsisaldus > 14 % |
||||||||||||
|
6. Töötlemata oliivijääkõli |
— |
— |
— |
C40 + C42+ C44 + C46 > 350 (5) |
≤ 1,4 |
— |
≤ |0,6| |
— |
— |
— |
— |
— |
|
7. Rafineeritud oliivijääkõli |
— |
≤ 0,3 |
≤ 5 |
C40 + C42 + C44 + C46 > 350 |
≤ 1,4 |
— |
≤ |0,5| |
— |
≤ 2,00 |
≤ 0,20 |
— |
— |
|
8. Oliivijääkõli |
— |
≤ 1,0 |
≤ 15 |
C40 + C42 + C44 + C46 > 350 |
≤ 1,2 |
— |
≤ |0,5| |
— |
≤ 1,70 |
≤ 0,18 |
— |
— |
|
(1) Kokku isomeere, mis õnnestus (mida ei õnnestunud) lahutada kapillaarkolonniga. (2) Piirmäära kohaldatakse oliiviõlidele, mis on toodetud alates 1. märtsist 2014. (3) Õlisid vahasisaldusega 300–350 mg/kg käsitatakse lambiõlidena, kui alifaatsete alkoholide üldsisaldus on kuni 350 mg/kg või kui erütrodiooli ja uvaooli sisaldus on kuni 3,5 %. (4) Puuduse mediaan võib olla kuni 3,5 ning puuviljalisuse mediaan 0. (5) Õlisid vahasisaldusega 300–350 mg/kg käsitatakse töötlemata oliivijääkõlidena, kui alifaatsete alkoholide üldsisaldus on üle 350 mg/kg ning kui erütrodiooli ja uvaooli sisaldus on suurem kui 3,5 %. |
||||||||||||
|
Kategooria |
Rasvhappeline koostis (1) |
Oleiinhappe trans-isomeeride kogusumma (%) |
Linool- ja linoleenhappe trans-isomeeride summa (%) |
Steroolne koostis |
Steroolid kokku (mg/kg) |
Erütrodiool ja uvaool (%) (**) |
||||||||||
|
Müristiinhape (%) |
Linoleenhape (%) |
Arahhiinhape (%) |
Eikoseenhape (%) |
Beheenhape (%) |
Lignotseriinhape (%) |
Kolesterool (%) |
Brassikasterool (%) |
Kampesterool (2) (%) |
Stigmasterool (%) |
App. βsitosterool (3) (%) (**) |
Δ-7-stigmastenool (2) (%) |
|||||
|
1. Ekstra-neitsioliiviõli |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
2. Neitsioliiviõli |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
3. Lambiõli |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,10 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (4) |
|
4. Rafineeritud oliiviõli |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
5. Rafineeritud ja neitsioliiviõlist koosnev oliiviõli |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
6. Töötlemata oliivijääkõli |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (5) |
|
7. Rafineeritud oliivijääkõli |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
|
8. Oliivijääkõli |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
|
(1) Muude rasvhapete sisaldus (%): palmitiinhape: 7,50–20,00; palmitoleiinhape: 0,30–3,50; heptadekaanhape: ≤ 0,30; heptadetseenhape: ≤ 0,30; steariinhape: 0,50–5,00; oleiinhape: 55,00–83,00; linoolhape: 2,50–21,00. (2) Vt käesoleva lisa liide. (3) App. β-sitosterool: Δ-5,23-stigmastadienool + klerosterool + β-sitosterool + sitostanool + Δ-5-avenasterool + Δ-5,24-stigmastadienool. (4) Õlisid vahasisaldusega 300–350 mg/kg käsitatakse lambiõlidena, kui alifaatsete alkoholide üldsisaldus on kuni 350 mg/kg või kui erütrodiooli ja uvaooli sisaldus on kuni 3,5 %. (5) Õlisid vahasisaldusega 300–350 mg/kg käsitatakse töötlemata oliivijääkõlidena, kui alifaatsete alkoholide üldsisaldus on üle 350 mg/kg või kui erütrodiooli ja uvaooli sisaldus on suurem kui 3,5 %. |
||||||||||||||||
Märkused
a) Analüüside tulemused peavad olema väljendatud sama arvu kümnendkohtadega, kui iga omaduse puhul on ette nähtud. Viimast numbrit tuleb suurendada ühe ühiku võrra, kui järgmine number on suurem kui 4.
b) Õli kategooriat võib muuta või deklareerida õli käesoleva määruse kohaselt mittepuhtaks, kui ainult üks omadus ei vasta märgitud väärtusele.
c) Kui omadus on märgistatud tärniga (*), tähendab see õli kvaliteedi puhul järgmist: – lambiõli puhul võivad mõlemad asjakohased piirväärtused samaaegselt erineda deklareeritud väärtustest; – neitsioliiviõlide puhul muudetakse kategooriat, kui kas või ainult üks nendest väärtustest erineb deklareeritud väärtusest, kuigi sellise õli võib ikka veel klassifitseerida ühte neitsioliiviõli kategooriatest.
d) Kui omadus on tähistatud kahe tärniga (**), osutab see, et kõikide oliivijääkõli tüüpide puhul võivad mõlemad asjaomased näitajad korraga erineda märgitud piirmääradest.
Liide
OTSUSTAMISSKEEM
Kampesterooli otsustamisskeem neitsioliiviõli ja ekstra neitsioliiviõli kohta:
Muud näitajad peavad vastama käesoleva määrusega kehtestatud normidele.
Δ-7-stigmastenooli otsustamisskeem:
— Ekstra-neitsioliiviõlid ja neitsioliiviõlid
—

Muud näitajad peavad vastama käesoleva määrusega kehtestatud normidele.
— Oliivijääkõlid (toor- ja rafineeritud)
—

Ia LISA
PROOVIDE VÕTMINE OLIIVIÕLIST VÕI OLIIVIJÄÄKÕLIST, MIS ON TARNITUD ESMAPAKENDIS
Seda proovivõtumeetodit kohaldatakse esmapakendisse pakendatud oliiviõli või oliivijääkõli partiidele. Kasutatakse eri meetodeid sõltuvalt sellest, kas esmapakendi maht ületab 5 liitrit või mitte.
Partii on hulk kaubaüksuseid, mis toodetakse, valmistatakse ja pakitakse sellistes tingimustes, et igas kaubaüksuses olev õli on kõikide analüütiliste omaduste osas homogeenne. Partii tähistus peab vastama Euroopa Parlamendi ja nõukogu direktiivile 2011/91/EL ( 2 ).
„Osaproov” – õli kogus, mis on esmapakendis ja mis on võetud partii juhuslikult valitud punktist.
1. ÜKSIKPROOVI SISALDUS
1.1. Esmapakend ei ole suurem kui 5 liitrit
„Üksikproov” esmapakendi puhul, mis ei ole suurem kui 5 liitrit – partiist vastavalt tabelile 1võetud osaproovide arv.
Tabel 1
Üksikproovi maht peab olema vähemalt järgmine
|
Kui esmapakendi maht on |
Üksikprooviks tuleb võtta õli |
|
a) 1 liiter või rohkem |
a) 1 kontaktpakendist; |
|
b) vähem kui 1 liiter |
b) väikseimast arvust pakenditest, mille kogumaht on vähemalt 1,0 liiter |
Tabelis 1 osutatud pakendite arvu, mis moodustavad üksikproovi, võib iga liikmesriik suurendada vastavalt oma vajadustele (nt organoleptiline hindamine muus laboris kui keemilised analüüsid, kontrollanalüüsid jne).
1.2. Esmapakend on suurem kui 5 liitrit
„Üksikproov” esmapakendi puhul, mis on suurem kui 5 liitrit – kogutud osaproove esindav osa, mis on saadud vähendamise meetodiga ja vastavalt tabelile 2. Üksikproov peab koosnema eri näidistest.
„Näidis” – üksikproovi näidis on iga pakkeüksus nendest, mis kokku moodustavad üksikproovi.
Tabel 2
Valitavate osaproovide miinimumarv
|
Pakendite arv partiis |
Valitavate osaproovide miinimumarv |
|
kuni 10 |
1 |
|
alates … 11 kuni 150 |
2 |
|
alates … 151 kuni 500 |
3 |
|
alates … 501 kuni 1 500 |
4 |
|
alates … 1 501 kuni 2 500 |
5 |
|
kui > 2 500 , siis iga 1 000 pakkeüksuse kohta |
1 täiendav osaproov |
Selleks et vähendada esmapakendite võtmisega saadud mahtu, ühtlustatakse üksikproovi valmistamiseks võetud osaproovide sisu. Eri osaproovide õlikogused valatakse mahutisse, et ühtlustada proov segamisega; seejuures kaitstakse proovi nii hästi kui võimalik kokkupuute eest õhuga.
Üksikproovi sisu tuleb valada pakenditesse mahuga vähemalt 1,0 liiter; igas sellises pakendis on üksikproovi näidis.
Üksikproovi näidiste arvu võib iga liikmesriik suurendada vastavalt oma vajadustele (nt organoleptiline hindamine muus laboris kui keemilised analüüsid, kontrollanalüüsid jne).
Iga pakend peab olema täidetud nii, et õhku oleks selles võimalikult vähe, seejärel sobivalt suletud ja plommitud, et tagada toote võltsimiskindlus.
Need näidised peavad olema märgistatud, et tagada õige tuvastamine.
2. ANALÜÜSID JA TULEMUSED
2.1. Iga üksikproov tuleb jagada laboriproovideks vastavalt standardi EN ISO 5555 punktile 2.5 ja analüüsida vastavalt Ib lisa otsustamisskeemis näidatud järjekorrale või muus juhuslikus järjekorras.
2.2. Kui kõik analüüside tulemused vastavad deklareeritud õlikategooria omadustele, loetakse kogu asjaomane partii vastavaks.
Kui kas või üks analüüsitulemus ei vasta deklareeritud õlikategooria omadustele, loetakse kogu asjaomane partii mitte vastavaks.
3. PARTII KATEGOORIA KONTROLLIMINE
3.1. Partii kategooria kontrollimiseks võib pädev asutus suurendada partii eri kohtadest võetavate üksikproovide arvu vastavalt järgmisele tabelile:
Tabel 3
Partii suurusega määratud üksikproovide arv
|
Partii suurus (liitrites) |
Üksikproovide arv |
|
alla 7 500 |
2 |
|
7 500 kuni vähem kui 25 000 |
3 |
|
25 000 kuni vähem kui 75 000 |
4 |
|
75 000 kuni vähem kui 125 000 |
5 |
|
Alates 125 000 st |
6 + 1 iga täiendava 50 000 liitri kohta |
Iga osaproov, millest koosneb üksikproov, tuleb võtta partii jätkuvast kohast; on vaja märkida iga üksikproovi koht ja tähistada see üheselt mõistetavalt.
Iga üksikproovi moodustamine toimub punktides 1.1 ja 1.2 osutatud korras.
Iga üksikprooviga tehakse seejärel artikli 2 lõikes 1 osutatud analüüsid.
3.2. Kui artikli 2 lõikes 1 osutatud analüüside tulemuste poolest vähemalt üks üksikproov ei vasta õli deklareeritud kategooria omadustele, tunnistatakse kogu proovivõtupartii nõuetele mittevastavaks.
Ib LISA
OTSUSTAMISSKEEM, MILLE JÄRGI KONTROLLITAKSE, KAS OLIIVIÕLI PROOV ON KOOSKÕLAS DEKLAREERITUD KATEGOORIAGA
Tabel 1

Tabel 2

Tabel 3

1. liide
Käesoleva määruse lisade ja otsustamisskeemis osutatud analüüside vastavuse tabel
|
— Happesus |
II lisa |
Vabade rasvhapete määramine külmmeetodil |
|
— Peroksiidiarv |
III lisa |
Peroksiidiarvu määramine |
|
— UV-spektromeetria |
IX lisa |
Spektrofotomeetriline analüüs |
|
— Organoleptiline hindamine |
XII lisa |
Neitsioliiviõli organoleptiline hindamine |
|
— Etüülestrid |
XX lisa |
Meetod vahade, rasvhapete metüülestrite ja etüülestrite sisalduse määramiseks kapillaargaasikromatograafiaga |
|
— 3,5-stigmastadieenid |
XVII lisa |
Meetod stigmastadieenide määramiseks taimeõlides |
|
— Rasvhapete trans-isomeerid |
X lisa |
Rasvhapete metüülestrite gaasikromatograafiline määramine |
|
— Rasvhapete sisaldus |
X lisa |
Rasvhapete metüülestrite gaasikromatograafiline määramine |
|
— ΔECN42 |
XVIII lisa |
HPLC-meetodiga triglütseriidide koostise määramine ECN42 abil (erinevus HPLC-meetodiga määratud sisalduse ja teoreetilise sisalduse vahel) |
|
— Steroolide koostis ja üldsisaldus — Erütrodiool ja uvaool |
V lisa |
Steroolide ja triterpeendialkoholide koostise ja sisalduse gaasikromatograafiline määramine kapillaarkolonniga |
|
— Vahad |
IV lisa |
Vahade sisalduse määramine kapillaargaasikromatograafia abil |
|
— Alifaatsed alkoholid ja triterpeenalkoholid |
XIX lisa |
Alifaatsete alkoholide ja triterpeenalkoholide sisalduse kapillaarkolonnkromatograafiline määramine |
|
— Küllastunud rasvhapped 2. positsioonis |
VII lisa |
2-glütserüülmonopalmitaadi protsendilise sisalduse määramine |
II LISA
VABADE RASVHAPETE MÄÄRAMINE KÜLMMEETODIL
1. RAKENDATAVUS JA RAKENDUSALA
Kõnealuse meetodi puhul kirjeldatakse rasvhapete kindlaksmääramist oliiviõlides ja oliivijääkõli. Vabade rasvhapete sisaldust väljendatakse oleiinhappe protsendina arvutatud happesusena.
2. PÕHIMÕTE
Proov lahustatakse lahustite segus ja vabad rasvhapped tiitritakse kaaliumhüdroksiidi või naatriumhüdroksiidi lahuses.
3. REAGENDID
Kõik reagendid peavad olema tunnustatud analüütilise kvaliteediga ja vesi peab olema kas destilleeritud või samaväärse puhtusega.
|
3.1. |
Dietüüleeter; 95 % etanool (v/v), segu võrdsetes osades Segu neutraliseeritakse kasutamise hetkel kaaliumhüdroksiidi lahusega (3.2), lisades 100 ml segu kohta 0,3 ml fenoolftaleiini lahust (3.3). Märkus 1. Dietüüleeter on väga kergesti süttiv ning võib moodustada plahvatusohtlikke peroksiide. Selle kasutamisel tuleb olla ettevaatlik. Märkus 2. Kui dietüüleetrit ei ole võimalik kasutada, võib kasutada etanooli ja tolueeni sisaldavate lahustite segu. Vajaduse korral võib etanooli asendada 2-propanool. |
|
3.2. |
Kaaliumhüdroksiid või naatriumhüdroksiid, tiitritud etanoolilahusena, c(KOH) [või c(NaOH)] 0,1 mol/l või vajaduse korral c(KOH) [või c(NaOH)] ligikaudu 0,5 mol/l. Saadaval on kaubanduslikud lahused. Kaaliumhüdroksiidi lahuse (või naatriumhüdroksiidi lahuse) täpne kontsentratsioon peab olema teada ning seda tuleb enne kasutamist kontrollida. Kasutatakse lahust, mis on valmistatud vähemalt viis päeva enne kasutamist, ning see dekanteeritakse kummist punniga tumedasse klaaspudelisse. Lahus peab olema värvitu või helekollase värvusega. Kui kaaliumhüdroksiidi (või naatriumhüdroksiidi) vesilahuse kasutamisel võib täheldada faaside eraldumist, võib vesilahuse asendada etanoolilahusega. Märkus 3. Kaaliumhüdroksiidi (või naatriumhüdroksiidi) stabiilset värvusetut lahust saab valmistada järgmiselt. 1 000 ml etanooli või vett lastakse koos 8 g kaaliumhüdroksiidi (või naatriumhüdroksiidi) ja 0,5 g alumiiniumilaastudega keema ning keedetakse püstjahutiga kolvis tund aega. Destilleeritakse kohe. Destillaadis lahustatakse vajalik kogus kaaliumhüdroksiidi (või naatriumhüdroksiidi). Lahus jäetakse mitmeks päevaks seisma ja kaaliumkarbonaadi (või naatriumkarbonaadi) sademest dekanteeritakse selge supernatant. Lahuse võib valmistada ilma destilleerimiseta järgmiselt: 1 000 ml etanoolile (või veele) lisatakse 4 ml alumiiniumbutülaati ja segu jäetakse mitmeks päevaks seisma. Supernatant dekanteeritakse ja selles lahustatakse vajalik kogus kaaliumhüdroksiidi (või naatriumhüdroksiidi). Lahus on kasutamisvalmis. |
|
3.3. |
Fenoolftaleiin, 10 g/l lahus 95–96 % etanoolis (v/v) või alkalisinises 6B või tümoolftaleiinis, 20 g/l lahus 95–96 % etanoolis (v/v). Väga tugeva värviga õlide puhul tuleks kasutada alkalisinist või tümoolftaleiini. |
4. SEADMED
Tavalised laboriseadmed, sealhulgas:
4.1. Analüütiline kaal,
4.2. 250 ml kooniline kolb,
4.3. 10 ml bürett, klassist A, mõõtskaala ühikutega 0,05 ml, või samaväärne automaatbürett.
5. MÄÄRAMISE KÄIK
5.1. Katseproovi ettevalmistamine
Kui proov on hägune, tuleb see filtreerida.
5.2. Katsekogus
Proov võetakse eeldatava happesuse alusel vastavalt järgmisele tabelile.
|
Eeldatav happesus (oleiinhape g / 100 g) |
Proovi mass (g) |
Kaalumistäpsus (g) |
|
0–2 |
10 |
0,02 |
|
> 2–7,5 |
2,5 |
0,01 |
|
> 7,5 |
0,5 |
0,001 |
Proov kaalutakse koonilises kolvis (4.2).
5.3. Kindlaksmääramine
Proov (5.2) lahustatakse 50–100 ml dietüüleetri ja etanooli (3.1) varem neutraliseeritud segus.
Lahus tiitritakse, segades sellesse kaaliumhüdroksiidi (või naatriumhüdroksiidi) 0,1 mol/l lahust (3.2) (vt märkus 4), kuni indikaator muutub (värvunud indikaatori värvus püsib vähemalt 10 sekundit).
Märkus 4. Kui kaaliumhüdroksiidi (või naatriumhüdroksiidi) 0,1 mol/l lahuse nõutav kogus ületab 10 ml, kasutatakse 0,5 mol/l lahust või asendatakse proovi mass vastavalt eeldatavale vabade hapete sisaldusele ja tabelis pakutule.
Märkus 5. Kui lahus muutub tiitrimise ajal häguseks, tuleb lisada nii palju lahustit (3.1), kui on vaja selge lahuse saamiseks.
Teine määramine tehakse üksnes juhul, kui esimene tulemus on kõrgem kui vastava õlikategooria puhul kehtestatud piirmäär.
6. TULEMUSTE VÄLJENDAMINE
Happesus oleiinhappe massiprotsendina arvutatakse järgmiselt:
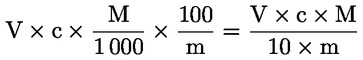
kus:
|
V |
= |
kasutatud tiitritud kaaliumhüdroksiidi (või naatriumoksiidi) maht milliliitrites; |
|
c |
= |
täpne kontsentratsioon moolides kasutatud kaaliumhüdroksiidi (või naatriumoksiidi) tiitritud lahuse liitri kohta; |
|
M |
= |
282 g/mol, molaarmass grammides oleiinhappe mooli kohta; |
|
m |
= |
proovi mass grammides. |
Oleiinhapet väljendatakse järgmiselt:
a) 0–1 puhul (viimane kaasa arvatud) kahe kümnendkoha täpsusega;
b) 1–100 puhul (viimane kaasa arvatud) ühe kümnendkoha täpsusega.
III LISA
PEROKSIIDIARVU MÄÄRAMINE
1. Kohaldamisala
Käesolevas lisas kirjeldatakse loomse ja taimse õli ja rasva peroksiidiarvu määramise meetodit.
2. Määratlus
Peroksiidiarv on nende proovis olevate ainete kogus aktiivse hapniku milliekvivalendina kilogrammi kohta, mis kirjeldatud töötamistingimustel kaaliumjodiidi oksüdeerivad.
3. Põhimõte
Katsekogust töödeldakse äädikhappe ja kloroformi lahuses kaaliumjodiidi lahusega. Eraldunud jood tiitritakse naatriumtiosulfaadi standardlahusega.
4. Seadmed
Seadmetel ei tohi olla redutseerivaid või oksüdeerivaid aineid.
Märkus 1. Lihvitud pindu ei tohi määrdeainega määrida.
4.1. 3 ml klaasist kühvel.
4.2. Lihvitud kaela ja korgiga 250 ml mahuga kolvid, enne kuivatatud ja täidetud puhta kuiva väärisgaasiga (lämmastik või soovitatavalt süsinikdioksiid).
4.3. 5 ml, 10 ml või 25 ml bürett mõõtskaala ühikutega vähemalt 0,05 ml, soovitavalt automaatse nullimisnupuga, või samaväärne automaatbürett.
4.4. Analüütilised kaalud.
5. Reagendid
5.1. Analüütiliselt puhta reaktiivi kvaliteediga kloroform, millest hapnik on vabastatud puhta, kuiva inertgaasi barboteerimisel.
5.2. Analüütiliselt puhta kvaliteediga jää-äädikas, millest hapnik on vabastatud puhta, kuiva inertgaasi barboteerimisel.
5.3. Kaaliumjodiidi küllastunud vesilahus, värskelt valmistatud ning joodi ja jodaatideta. Lahustada ligikaudu 14 g kaaliumjodiidi 10 ml toatemperatuuril vees.
5.4. Naatriumtiosulfaat, 0,01 mol/l (0,01 N), vahetult enne kasutamist täpselt standarditud vesilahus.
Valmistada iga päev 0,1 mol/l naatriumtiosulfaadi standardlahusest vahetult enne kasutamist 0,01 mol/l naatriumtiosulfaadi lahus, või määrata kindlaks täpne molaarsus. Kogemused näitavad, et lahuse stabiilsus on piiratud ning sõltub pH-väärtusest ja vaba süsinikdioksiidi sisaldusest. Lahjendamiseks kasutada ainult värskelt keedetud vett, mis on võimaluse korral lämmastikuga läbi puhutud.
Naatriumtiosulfaadi lahuse täpse molaarsuse kindlaksmääramiseks soovitatakse kasutada järgmist menetlust.
Kaaluda mõõtekolbi (250 ml või 500 ml) 0,001 g täpsusega 0,27–0,33 g kaaliumjodaati (mKIO3) ja lahjendada märgini, kasutades värskelt keedetud vett (V2), mis on jahutatud toatemperatuurini. Viia 5 ml või 10 ml eespool kirjeldatud kaaliumjodaadi lahust (V1) pipeti abil 250 ml Erlenmeyeri kolbi. Lisada 60 ml värskelt keedetud vett, 5 ml 4 mol/l vesinkkloriidhapet ja 25–50 mg kaaliumjodiidi või 0,5 ml küllastunud kaaliumjodiidi lahust. Tiitrida see lahus naatriumtiosulfaadi lahusega (V3), et määrata kindlaks naatriumtiosulfaadi lahuse täpne molaarsus.

kus
mKIO3 on kaaliumjodaadi mass grammides;
V1 on kaaliumjodaadi lahuse maht milliliitrites (5 ml või 10 ml);
V2 on kaaliumjodaadi lahuse kogumaht milliliitrites (250 ml või 500 ml);
V3 on naatriumtiosulfaadi lahuse maht milliliitrites;
wKIO3 on kaaliumjodaadi puhtus (g/100 g);
MKIO3 on kaaliumjodaadi molekulmass (214 g/mol);
T on kasutatud naatriumtiosulfaadi lahuse täpne molaarsus (mol/l).
5.5. Tärkliselahus, 10 g/l vesidispersioon, värskelt looduslikust lahustuvast tärklisest valmistatud. Kasutada võib ka samaväärseid reaktiive.
6. Proov
Tagatakse proovi võtmine ja selle säilitamine varjus, jahedas ja täielikult täidetud klaasmahutites, mis on lihvkorgi või korgist punniga hermeetiliselt suletud.
7. Menetlus
Katsed tuleb teha hämara päevavalguse või kunstliku valgustusega. Klaasist kühvlisse (4.1) või selle puudumise korral kolbi (4.2) kaalutakse 0,001 g täpsusega järgmisele tabelile vastava massiga proov, mille aluseks on eeldatav peroksiidiarv:
|
Hinnanguline peroksiidiarv (meq) |
Katsekoguse mass (g) |
|
0–12 |
5,0–2,0 |
|
12–20 |
2,0–1,2 |
|
20–30 |
1,2–0,8 |
|
30–50 |
0,8–0,5 |
|
50–90 |
0,5–0,3 |
Kolvilt (4.2) eemaldatakse kork ning sellesse pannakse klaasist kühvel, millel on katsekogus. Lisatakse 10 ml kloroformi (5.1). Katsekogus lahustatakse kiiresti segamise teel. Lisatakse 15 ml äädikhapet (5.2) ja seejärel 1 ml kaaliumjodiidi lahust (5.3). Kolvile pannakse kiiresti kork, loksutatakse üks minut ja jäetakse siis viieks minutiks temperatuurile 15–25 °C varju seisma.
Lisatakse umbes 75 ml destilleeritud vett. Vabastatud jood tiitritakse kõvasti loksutades naatriumtiosulfaadi lahusega (5.4), kasutades indikaatorina tärkliselahust (5.5).
Ühe ja sama prooviga tehakse 2 määramist.
Samal ajal tuleb teha ka pimekatse. Kui pimekatse tulemus on suurem kui 0,05 ml 0,01 N naatriumtiosulfaadi lahust (5.4), asendatakse puhastamata reaktiivid.
8. Tulemuste esitamine
Peroksiidiarv aktiivse hapniku milliekvivalentidena kilogrammi kohta saadakse valemiga

kus:
V on katses kasutatud naatriumtiosulfaadi standardlahuse (5.4) milliliitrite arv, mida on pimekatse arvessevõtmiseks parandatud;
T on kasutatud naatriumtiosulfaadi lahuse (5.4) täpne molaarsus (mol/l);
m on katsekoguse mass grammides.
Tulemus on kahe määramise aritmeetiline keskmine.
Määramise tulemus esitatakse ühe kümnendkoha täpsusega.
IV LISA
VAHADE SISALDUSE MÄÄRAMINE KAPILLAARGAASIKROMATOGRAAFIA ABIL
1. EESMÄRK
Käesoleva meetodiga määratakse vahade sisaldust oliiviõlides. Vahade eraldamine toimub süsinikuaatomite arvu alusel. Meetodit võib eeskätt kasutada pressimisega saadud oliiviõlide eristamiseks ekstraheerimisega saadud õlidest (oliivijääkõlidest).
2. PÕHIMÕTE
Rasvainele lisatakse sobivat sisestandardit, seejärel fraktsioneeritakse kromatograafiliselt, kasutades hüdrateeritud silikageelkolonni; katse tingimustes esimesena elueeritav fraktsioon ainetega, mis on triglütseriididest vähem polaarsed, eraldatakse ning seda analüüsitakse kapillaargaasikromatograafia abil.
3. SEADMED
|
3.1. |
25milliliitrine Erlenmeyeri kolb. |
|
3.2. |
Klaasist gaasikromatograafiakolonn sisediameeteriga 15,0 mm ja pikkusega 30–40 cm, mis on varustatud kraaniga. |
|
3.3. |
Sobiv kapillaarkolonniga gaasikromatograaf, mis on varustatud süsteemiga, mis võimaldab otse kolonni süstimist ja koosneb järgmistest osadest:
|
|
3.4. |
10mikroliitrine mikrosüstal otse kolonni süstimiseks, mille nõelal on tugevdatud pinnakiht. |
|
3.5. |
Elektriline vibraator. |
|
3.6. |
Pöördaurusti. |
|
3.7. |
Muhvelahi. |
|
3.8. |
Analüütiline kaal, kaalumistäpsus ± 0,1 mg. |
|
3.9. |
Tavalised klaasist laborinõud. |
4. REAKTIIVID
|
4.1. |
Silikageel, tera suurus 60–200 μm. Geel pannakse vähemalt neljaks tunniks ahju temperatuuriga 500 °C. Lastakse jahtuda, seejärel lisatakse vett koguses, mis vastab 2 %le silikageeli massist. Segatakse korralikult ühtlase massi saamiseni. Hoitakse pimedas vähemalt 12 tundi enne kasutamist. |
|
4.2. |
n-heksaan, kromatograafias kasutamiseks. |
|
4.3. |
Etüüleeter, kromatograafias kasutamiseks. |
|
4.4. |
n-heptaan, kromatograafias kasutamiseks. |
|
4.5. |
Laurüülarahidaadi 0,1 % (m/V) standardlahus heksaanis (sisestandard) (võib kasutada ka palmitüülpalmitaati või müristüülstearaati).
|
|
4.6. |
Kandegaas: puhas vesinik või heelium, gaasikromatograafias kasutamiseks. |
|
4.7. |
Abigaasid: — puhas vesinik, gaasikromatograafias kasutamiseks; — puhas õhk, gaasikromatograafias kasutamiseks. |
5. TÖÖ KÄIK
5.1. Kromatograafiakolonni valmistamine
n-heksaanis (4.2) suspendeeritakse 15 g silikageeli (4.1) ning viiakse kolonni (3.2). Lastakse settida. Homogeensema kromatograafilise kolonni saamiseks töödeldakse settinud kolonni elektrilise vibraatoriga (3.5) ja lisandite eemaldamiseks voolutatakse kolonni 30 ml n-heksaaniga. 25 milliliitrisesse Erlenmeyeri kolbi (3.1) kaalutakse täpselt kaalu (3.8) abil 500 mg proovi ning lisatakse sobiv kogus sisestandardit (4.5), arvestades eeldatavat vahasisaldust; nt oliiviõli puhul lisatakse 0,1 mg ja oliivijääkõli puhul 0,25–0,5 mg laurüülarahidaati. Eelkirjeldatud viisil ettevalmistatud proov kantakse kromatograafiakolonni, kasutades kahte n-heksaani (4.2) kogust mahuga à 2 ml.
Lahustil lastakse voolata, kuni absorbendi ülemise pinna kohale jääb 1 mm vedelikku, seejärel voolutatakse kolonni veel 70 ml n-heksaaniga, et eemaldada proovis tavaliselt sisalduvad n-alkaanid. Seejärel alustatakse voolutamist; kogutakse 180 ml n-heksaani-etüüleetri segu (vahekorras 99:1) voolukiirusel umbes 15 tilka 10 sekundi jooksul. Proovi elueerimine toimub toatemperatuuril 22 ± 4 °C.
NB:
— n-heksaani-dietüüleetri segu (99:1) tuleb valmistada igal päeval uuesti.
— Vahade nõuetekohase elueerimise visuaalseks kontrollimiseks võib proovi lahusele lisada 100 μl 1 % sudaani lahust elueerimissegus. Värvaine retentsiooniaeg on vahade ja triglütseriidide retentsiooniaegade vahel; kui värvaine jõuab kromatograafiakolonni põhjani, tuleb elueerimine peatada, sest kõik vahad on elueeritud.
Niimoodi saadud fraktsiooni aurustatakse pöördaurustis (3.6), kuni peaaegu kogu lahusti on eemaldatud. Viimased 2 ml lahustit eemaldatakse nõrga lämmastikuvoolu abil, seejärel lisatakse 2–4 ml n-heptaani.
5.2. Gaasikromatograafiline analüüs
5.2.1. Ettevalmistustööd
Kolonn pannakse gaasikromatograafi (3.3); sisselaskekoht ühendatakse on-column-süsteemiga ja väljalaskekoht detektoriga. Kontrollitakse, et gaasikromatograafiaseade oleks töökorras (gaasijuhtmete lekkekindlus, detektori tundlikkus, meerik jne).
Kui kolonni kasutatakse esimest korda, on soovitatav see tasakaalustada. Läbi kolonni hakatakse nõrga joana juhtima gaasi, seejärel lülitatakse gaasikromatograafiaseade sisse. Järk-järgult, 4 tunni jooksul, tõstetakse temperatuur 350 °C-ni. Sellist temperatuuri hoitakse vähemalt kaks tundi, seejärel reguleeritakse seade töörežiimile (reguleeritakse gaasivoolu kiirust, leegi süüdet, ühendatakse elektrilise meerikuga (3.3.4), reguleeritakse kolonni termostaatkapi temperatuuri, detektorit jne). Registreeritakse signaal vähemalt kaks korda suuremal tundlikkusel, kui on vaja analüüsi läbiviimiseks. Saadav nulljoon peaks olema sirge, ilma igasuguste piikideta, ning see ei tohiks kummalegi poole triivida.
Negatiivne sirge triiv näitab, et kolonn ei ole korralikult ühendatud; positiivne triiv osutab kolonni puudulikule tasakaalustamisele.
5.2.2. Töötingimuste valimine
Töötingimused on reeglina järgmised:
— kolonni temperatuur:
—
|
|
20 °C/min |
|
5 °C/min |
|
20 °C/min |
|
|
algul 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— detektori temperatuur: 350 °C;
— sisestatava aine kogus: 1 μl lahust n-heptaanis (2–4 ml);
— kandegaas: heelium või vesinik asjaomase gaasi optimaalsel voolukiirusel (vt liide);
— seadme tundlikkus peab vastama järgmistele tingimustele:
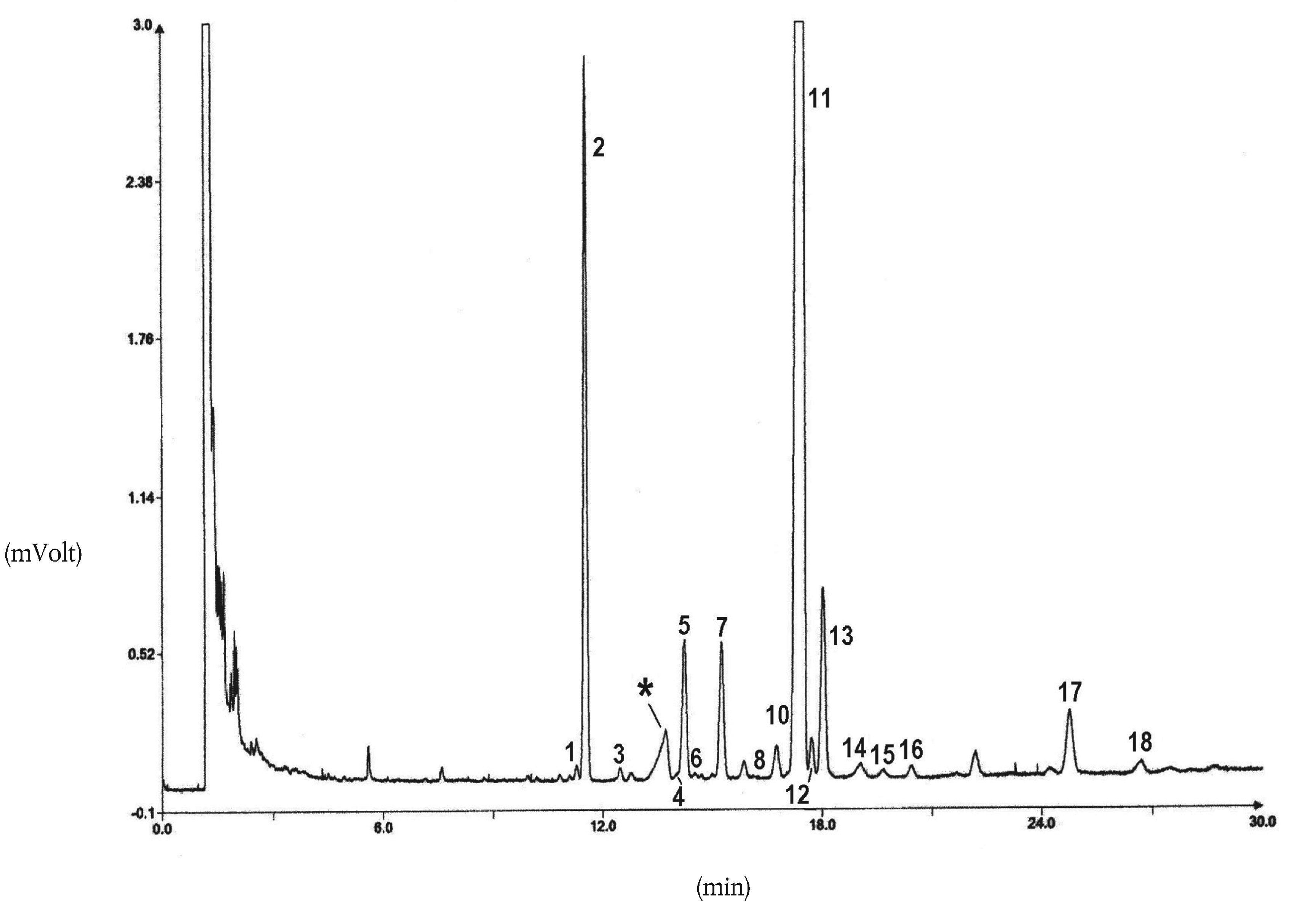
Olenevalt kolonni ja gaasikromatograafi omadustest võib tingimusi muuta, et saavutada kõikide vahade eraldumine, piikide rahuldav lahutus (vt joonis) ja et C32 sisestandardi retentsiooniaeg oleks 18 ± 3 minutit. Vaha kõige tüüpilisem piik peab olema vähemalt 60 % täisskaalast.
Piigi integreerimisparameetrid määratakse kindlaks nii, et uuritavate piikide pindalasid oleks võimalik õigesti hinnata.
NB:Arvesse võttes kõrget lõpptemperatuuri, on lubatav positiivne triiv, mis ei tohi ületada 10 % skaala ulatusest.
5.3. Analüüsi läbiviimine
10mikroliitrisesse mikrosüstlasse võetakse 1 μl lahust; kolb tõmmatakse tagasi, kuni nõel on tühi. Nõel viiakse injektsioonisüsteemi ning 1–2 sekundi pärast tehakse kiire süst. Umbes 5 sekundi pärast tõmmatakse nõel ettevaatlikult välja.
Detektori signaali registreeritakse seni, kuni vahad on täielikult elueeritud.
Nulljoon peab kogu aeg vastama nõuetele.
5.4. Piikide määramine
Piigid määratakse retentsiooniaja põhjal, võrdlusest samadel tingimustel analüüsitud teadaoleva retentsiooniajaga vahade segudega.
Joonisel on esitatud neitsioliiviõli vahade kromatogramm.
5.5. Kvantitatiivne hindamine
Sisestandardile ja alifaatsetele estritele C40–C46vastavate piikide pindalad arvutatakse integraatori abil.
Iga estri vahasisaldus milligrammides rasvaine kilogrammi kohta arvutatakse järgmiselt:

kus:
|
Ax |
= |
iga estri piigi pindala ruutmillimeetrites; |
|
As |
= |
sisestandardi piigi pindala ruutmillimeetrites; |
|
ms |
= |
lisatud sisestandardi mass milligrammides; |
|
m |
= |
määramisel kasutatud proovi mass grammides. |
6. TULEMUSTE VÄLJENDAMINE
Märgitakse erinevate C40- kuni C46-vahade sisalduste summa milligrammides rasva kilogrammi kohta (ppm).
NB. Määratavate koostisosade puhul osutatakse vahade piikidele, mis paiknevad paarisarvulise süsinikuaatomite arvuga estrite C40 ja C46 vahel, vastavalt järgneval joonisel esitatud oliiviõli vahade kromatogrammile. Kui ester C46 esineb kahekordsena, on soovitatav selle identifitseerimiseks analüüsida pressimisjääkide õli vahafraktsiooni, kus C46 piiki on kerge ära tunda, sest see on oluliselt suurem.
Tulemused väljendatakse ühe kümnendkoha täpsusega.
Joonis
Oliiviõli vahade kromatogramm ( 3 )
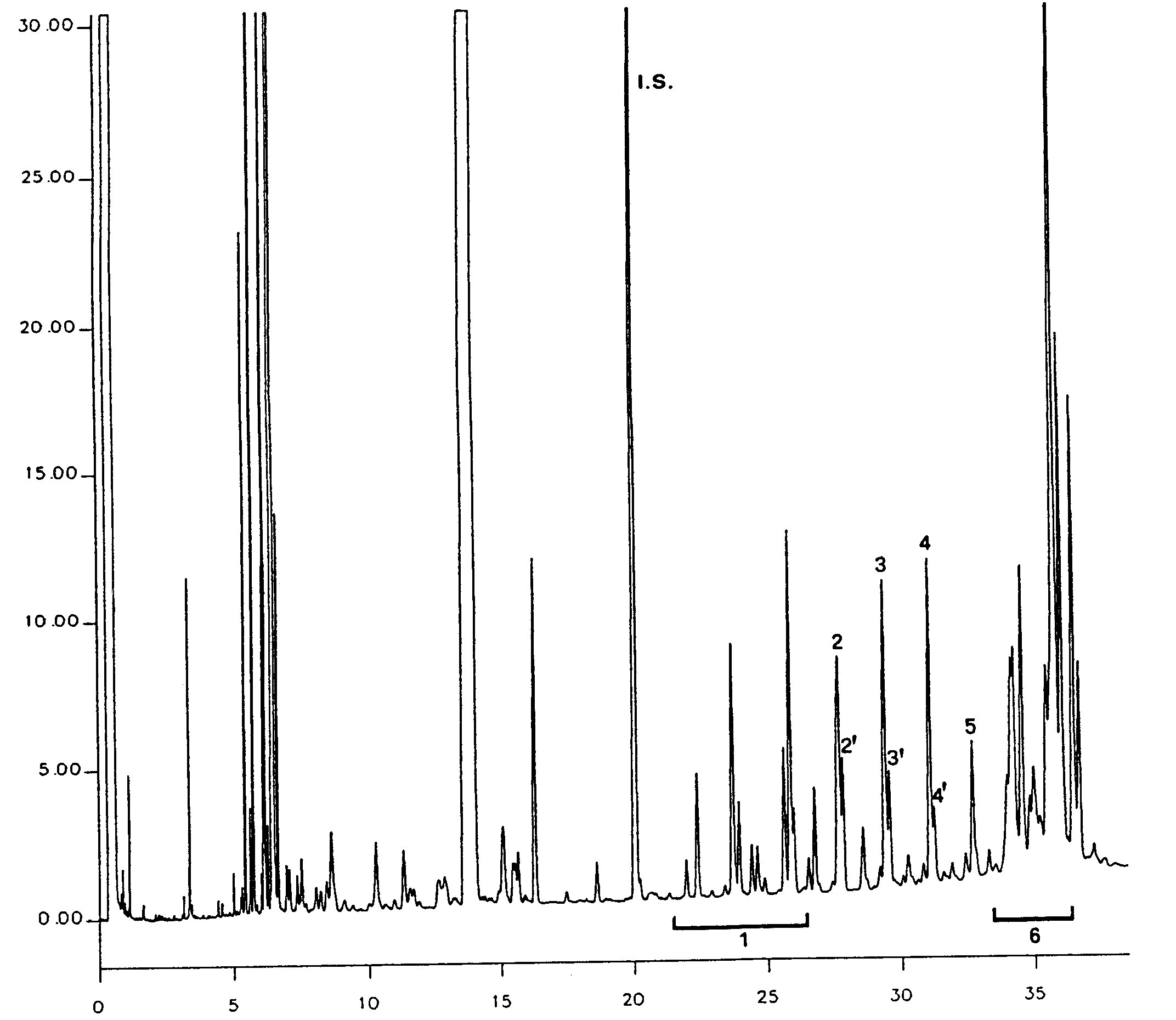
Tähistused:
|
I.S. |
= |
laurüülarahidaat |
|
1. |
= |
diterpeenestrid |
|
2 + 2′ |
= |
estrid C40 |
|
3 + 3′ |
= |
estrid C42 |
|
4 + 4′ |
= |
estrid C44 |
|
5. |
= |
estrid C46 |
|
6. |
= |
steroolestrid ja triterpeenalkoholid. |
Liide
Gaasi voolukiiruse määramine
Kui gaasikromatograaf on viidud tavapärasesse töörežiimi, süstitakse kolonni 1–3 μl metaani või propaani ning mõõdetakse stopperiga aeg süstimise hetkest kuni piigi alguseni (tM); selle ajaga läks metaan või propaan läbi kolonni.
Voolukiirus (cm/s) määratakse valemiga L/tM, kus L on kolonni pikkus sentimeetrites ja tM on stopperiga mõõdetud aeg sekundites.
V LISA
STEROOLIDE JA TRITERPEENDIALKOHOLIDE KOOSTISE JA SISALDUSE GAASIKROMATOGRAAFILINE MÄÄRAMINE KAPILLAARKOLONNIGA
1. KASUTAMISALA
Meetodis kirjeldatakse üksiksteroolide ja -triterpeendialkoholide ning steroolide ja triterpeendialkoholide üldsisalduse määramist oliiviõlides ja oliivijääkõlides.
2. PÕHIMÕTE
Õli, millele on sisestandardina lisatud α-kolestanooli, seebistatakse kaaliumhüdroksiidiga etanoolilahuses ja seebistumatud komponendid ekstraheeritakse dietüüleetriga.
Steroolide ja triterpeendialkoholide fraktsioon eraldatakse seebistamatust ainest õhukese kihi kromatograafiaga aluselisel silikageelplaadil. Silikageelist saadud fraktsioonid viiakse üle trimetüülsilüüleetriteks ja analüüsitakse gaasikromatograafiliselt kapillaarkolonniga.
3. SEADMED
Kasutatakse tavalisi laboriseadmeid, eelkõige järgmisi:
3.1. Püstjahutiga varustatud lihvühendustega 250 ml kolb.
3.2. 500 ml jaotuslehter.
3.3. 250 ml kolvid.
3.4. Kõik seadmed õhukese kihi kromatograafia tegemiseks 20 × 20 cm klaasplaatidel.
3.5. Ultraviolettlamp lainepikkusega 254 või 366 nm.
3.6. 100 μl ja 500 μl mikrosüstlad.
3.7. Silindriline poorse G3-filtriga (poori läbimõõt 15–40 μm) filterlehter, läbimõõduga ligikaudu 2 cm ja sügavusega 5 cm, mis sobib vaakumfiltrimiseks ja millel on lihvkern.
3.8. 50 ml kooniline vaakuumkolb klaaslihvmuhviga, mille saab ühendada filterlehtriga (punkt 3.7).
3.9. 10 ml katseklaas koonusekujulise põhja ja tihedalt sulguva klaaskorgiga.
3.10. Gaasikromatograaf, millel saab töötada kapillaarkolonniga ja millel on jaotatud vooluga injektorseade, mis koosneb järgmistest seadistest:
3.10.1. termostateeritav kolonnikamber, mis hoiab nõutavat temperatuuri täpsusega ± 1 °C;
3.10.2. reguleeritava temperatuuriga injektorseade persilaniseeritud klaasist aurustiga ja jaotatud voolu süsteemiga;
3.10.3. leekionisatsioondetektor;
3.10.4. Andmekogumissüsteem, mis sobib kasutamiseks koos leekionisatsioondetektoriga (punkt 3.10.3) ja mille abil saab käsitsi integreerida.
3.11. Sulatatud ränidioksiidist kapillaarkolonn, mille pikkus on 20–30 m, sisediameeter 0,25–0,32 mm, üleni kaetud ühtlase 0,10–0,30 μm paksuse 5 % difenüül- ja 95 % dimetüülpolüsiloksaani kihiga (statsionaarne faas SE-52 või SE-54 või samaväärne).
3.12. Mikrosüstal, maht 10 μml, gaasikromatograafia jaoks, liimitud nõelaga, mis sobib jaotatud voolu süstideks.
3.13. Kaltsiumdikloriidiga eksikaator
4. REAGENDID
4.1. Kaaliumhüdroksiid, miinimumtiiter 85 %.
4.2. Kaaliumhüdroksiidi umbes 2 N etanoollahus.
130 g kaaliumhüdroksiidi (punkt 4.1) lahustatakse samaaegsel jahutamisel 200 ml destilleeritud vees ning täiendatakse etanooliga (punkt 4.10) kuni 1 liitri märgini. Lahust hoitakse õhukindlalt suletud tumedast klaasist pudelites ja säilitatakse kuni kaks päeva.
4.3. Etüüleeter, analüütiliselt puhas.
4.4. Kaaliumhüdroksiidi umbes 0,2 N etanoollahus.
13 g kaaliumhüdroksiidi (punkt 4.1) lahustatakse samaaegsel jahutamisel 20 ml destilleeritud vees ning täiendatakse etanooliga (punkt 4.10) kuni 1 liitri märgini.
4.5. Veevaba naatriumsulfaat, analüütiliselt puhas.
4.6. Silikageeliga kaetud klaasplaadid (20 × 20 cm), fluorestsentsindikaatorita, paksus 0,25 mm (müügil kasutusvalmina).
4.7. Tolueen, kromatograafia jaoks.
4.8. Atsetoon, kromatograafia jaoks.
4.9. n-heksaan, kromatograafia jaoks.
4.10. Etüüleeter, kromatograafia jaoks.
4.11. Etanool, analüütiliselt puhas.
4.12. Etüülatsetaat, analüütiliselt puhas.
4.13. Võrdluslahus õhukese kihi kromatograafia jaoks: kolesterooli või fütosteroolide ja erütrodiooli 5 % lahus etüülatsetaadis (punkt 4.11).
4.14. 2,7-diklorofluorestseiin, 0,2 % etanoolilahus. Lahus muudetakse kergelt aluseliseks mõne tilga 2 N alkohoolse kaaliumhüdroksiidilahuse (punkt 4.2) lisamisega.
4.15. Veevaba püridiin, kromatograafia jaoks (vt märkus 5).
4.16. Heksametüüldisilasaan, analüütiliselt puhas.
4.17. Trimetüülklorosilaan, analüütiliselt puhas.
4.18. Sterooltrimetüülsilüüleetrite võrdluslahused.
Valmistatakse kasutamise ajal steroolidest ja erütrodioolist, mis on saadud neid sisaldavatest õlidest.
4.19. α-kolestanool, puhtus üle 99 % (puhtust tuleb kontrollida gaasikromatograafilise analüüsiga).
4.20. α-kolestanooli sisestandardi lahus, 0,2 % lahus (mass/ruumala) etüülatsetaadis (punkt 4.11).
4.21. Fenoolftaleiini lahus etanoolis (punkt 4.10), 10 g/l.
4.22. Kandegaas: lämmastik või heelium, gaasikromatograafiliselt puhas.
4.23. Abigaasid: vesinik, heelium, lämmastik ja õhk, gaasikromatograafiliselt puhtad.
4.24. n-heksaani (4.9) / dietüüleetri (punkt 4.10) segu 65:35 (maht/maht).
4.25. Silüülimisreaktiiv, mis kujutab endast püridiini-heksametüüldisilasaani-trimetüülklorosilaani segu vahekorras 9:3:1 (maht/maht/maht).
5. MÄÄRAMISE KÄIK
|
5.1. |
Seebistumatu aine valmistamine.
|
|
5.2. |
Steroolide ja triterpeendialkoholide (erütrodiool + uvaool) eraldamine
|
|
5.3. |
Trimetüülsilüüleetrite valmistamine.
|
|
5.4. |
Gaasikromatograafiline analüüs.
|
6. TULEMUSTE VÄLJENDAMINE
6.1. Esitatakse iga sterooli kontsentratsioon milligrammides kg rasvaine kohta ning nende summa (steroolide üldsisaldus).
Iga sterooli, erütrodiooli ja uvaooli sisaldus väljendatakse ühe kümnendkoha täpsusega.
Steroolide summaarne sisaldus väljendatakse ilma komakohtadeta.
6.2. Iga sterooli protsent arvutatakse vastava piigi pindala ja steroolide piikide kogupindala suhtena:
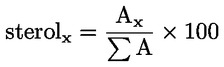
kus:
|
Ax |
= |
x'i piigi pindala; |
|
ΣA |
= |
steroolide piikide kogupindala. |
6.3. Näiline β-sitosterool: Δ5-23-stigmastadienool + klerosterool + β-sitosterool + sitostanool + Δ5-avenasterool + Δ5-24-stigmastadienool.
6.4. Arvutatakse erütrodiooli ja uvaooli protsendiline sisaldus:
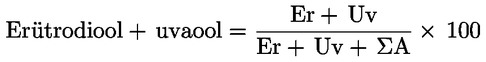
kus:
|
ΣA |
= |
steroolide piikide kogupindala arvutamissüsteemi ühikutes; |
|
Er |
= |
erütrodiooli piigi pindala arvutamissüsteemi ühikutes; |
|
Uv |
= |
uvaooli piigi pindala arvutamissüsteemi ühikutes. |
Liide
Gaasi lineaarse voolukiiruse määramine
Normaalsetel töötingimustel töötavasse gaasikromatograafi süstitakse 1–3 μl metaani (või propaani) ja mõõdetakse aeg, mis kulub gaasi voolamiseks läbi kolonni alates süstimisest kuni piigi ilmumiseni (tM).
Lineaarne voolukiirus cm/s on esitatud valemiga L/tM, kus L on kolonni pikkus sentimeetrites ja tM on mõõdetud aeg sekundites.
Tabel 1
Steroolide suhtelised retentsiooniajad
|
Piik |
Identifitseerimisandmed |
Suhteline retentsiooni-aeg |
||
|
SE 54 kolonn |
SE 52 kolonn |
|||
|
1 |
Kolesterool |
Δ-5-kolesteen-3ß-ool |
0,67 |
0,63 |
|
2 |
Kolestanool |
5α-kolestaan-3ß-ool |
0,68 |
0,64 |
|
3 |
Brassikasterool |
[24S]-24-metüül-Δ-5,22-kolestadieen-3ß-ool |
0,73 |
0,71 |
|
* |
Ergosterool |
[24S]-24-metüül-Δ5-7-22 kolestatrieen-3β-ool |
0,78 |
0,76 |
|
4 |
24-metüleen-kolesterool |
24-metüleen-Δ-5,24-kolestadieen-3ß-oo1 |
0,82 |
0,80 |
|
5 |
Kampesterool |
(24R)-24-metüül-Δ-5-kolesteen-3ß-ool |
0,83 |
0,81 |
|
6 |
Kampestanool |
(24R)-24-metüül-kolestaan-3ß-ool |
0,85 |
0,82 |
|
7 |
Stigmasterool |
(24S)-24-etüül-Δ-5,22-kolestadieen-3ß-ool |
0,88 |
0,87 |
|
8 |
Δ-7-kampesterool |
(24R)-24-metüül-Δ-7-kolesteen-3ß-ool |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadienool |
(24R,S)-24-etüül-Δ-5,23-koIestadieen-3ß-ool |
0,95 |
0,95 |
|
10 |
Klerosterool |
(24S)-24-etüül-Δ-5,25-kolestadieen-3ß-ool |
0,96 |
0,96 |
|
11 |
ß-sitosterool |
(24R)-24-etüül-Δ-5-kolesteen-3ß-ool |
1,00 |
1,00 |
|
12 |
Sitostanool |
24-etüül-kolestaan-3ß-ool |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterool |
(24Z)-24-etülideen-Δ-kolesteen-3ß-ool |
1,03 |
1,03 |
|
14 |
Δ-5-24-stigmastadienool |
(24R,S)-24-etüül-Δ-5,24-kolestadieen-3ß-ool |
1,08 |
1,08 |
|
15 |
Δ-7-stigmastenool |
(24R,S)-24-etüül-Δ-7-kolesteen-3ß-ool |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterool |
(24Z)-24-etülideen-Δ-7-kolesteen-3ß-ool |
1,16 |
1,16 |
|
17 |
Erütrodiool |
5α-oleaan-12een-3β28-diool |
1,41 |
1,41 |
|
18 |
Uvaool |
Δ12-urseen-3β28-diool |
1,52 |
1,52 |
Joonis 1
Lambiõli steroolide ja triterpeendialkoholide fraktsiooni gaasikromatogramm (lisatud on sisestandard)
Joonis 2
Rafineeritud oliiviõli steroolide ja triterpeendialkoholide fraktsiooni gaasikromatogramm (lisatud on sisestandard)

Joonis 3
Oliivijääkõli õhukese kihi kromatograafia plaat vööndiga, mis tuleb maha kraapida steroolide ja triterpeendialkoholide määramiseks

1 – skvaleen
2 – triterpeen ja alifaatsed alkoholid
3 – steroolid ja triterpeendialkoholid
4 – stardijoon ja vabad rasvhapped
▼M26 —————
VII LISA
2-GLÜTSERÜÜLMONOPALMITAADI PROTSENDILISE SISALDUSE MÄÄRAMINE
1. EESMÄRK JA RAKENDAMISALA
Käesoleva meetodiga määratakse triglütseriidide 2. positsioonis oleva palmitiinhappe protsendilist sisaldust 2-glütserüülmonopalmitaadi hindamise abil.
Seda meetodit saab kasutada toatemperatuuril (20 °C) vedelate taimeõlide puhul.
2. PÕHIMÕTE
Pärast õliproovi ettevalmistamist lastakse proovil reageerida pankrease lipaasiga – toimub triglütseriidi molekuli spetsiifiline osaline hüdrolüüs 1. ja 3. positsioonis, mille tulemusena tekivad 2-monoglütseriidid. 2-glütserüülmonopalmitaadi sisaldus monoglütseriidi fraktsioonis määratakse pärast silaanimist kapillaarkolonn-gaasikromatograafia abil.
3. KASUTATAVAD SEADMED JA MATERJALID
|
3.1. |
25milliliitrine Erlenmeyeri kolb |
|
3.2. |
100-, 250- ja 300milliliitrised keeduklaasid |
|
3.3. |
Klaasist kromatograafiakolonn sisediameetriga 21–23 mm ja pikkusega 400 mm, mis on varustatud klaasfilterketta ja kraaniga |
|
3.4. |
Gradueeritud 10-, 50-, 100- ja 200milliliitrised mõõtesilindrid |
|
3.5. |
100- ja 250milliliitrised ümarkolvid |
|
3.6. |
Pöördaurusti |
|
3.7. |
10milliliitrised lihvkorgiga tsentrifuugitopsid, millel on kooniline põhi |
|
3.8. |
Tsentrifuug tööks 10- ja 100milliliitriste tsentrifuugitopsidega |
|
3.9. |
Termostaat, mis võimaldab hoida temperatuuri 40 ± 0,5 °C |
|
3.10. |
Gradueeritud 1- ja 2milliliitrised pipetid |
|
3.11. |
1milliliitrine hüpodermiline süstal |
|
3.12. |
100mikroliitrine mikrosüstal |
|
3.13. |
1 000 milliliitrine jaotuslehter |
|
3.14. |
Kapillaarkolonniga gaasikromatograaf, mis on varustatud süsteemiga, mis võimaldab proovi otse külmalt kolonni süstimist, ja ahjuga, mis hoiab soovitud temperatuuri 1 °C täpsusega |
|
3.15. |
Külminjektor proovi viimiseks otse kolonni |
|
3.16. |
Leekionisatsioondetektor ja elektromeeter |
|
3.17. |
Koos elektromeetriga kasutatav integraatormeerik, mille reageerimisaeg on kuni 1 sekund ning mille paberi kiirust saab muuta |
|
3.18. |
Klaasist või sulatatud ränidioksiidist kapillaarkolonn pikkusega 8–12 m ja sisediameetriga 0,25–0,32 mm, mis sobib tööks kuni 370 °C juures ja mis on seestpoolt kaetud 5 % metüülpolüsiloksaani või fenüül-metüülpolüsiloksaani kihiga, mille paksus on 0,10–0,30 μm |
|
3.19. |
Vähemalt 7,5 cm pikkune 10mikroliitrine mikrosüstal otse kolonni süstimiseks, mille nõelal on tugevdatud pinnakiht |
4. REAKTIIVID
|
4.1. |
Silikageel tera suurusega 0,063–0,200 mm (sõelaava 70–280), mis on ette valmistatud järgmisel viisil. Silikageel pannakse portselankaussi, kuivatatakse kuivatuskapis 160 °C juures 4 tundi, lastakse eksikaatoris jahtuda toatemperatuurini. Lisatakse vett koguses, mis vastab 5 % le silikageeli massist, tehes seda järgmisel viisil: 500milliliitrisesse Erlenmeyeri kolbi kaalutakse 152 g silikageeli ja lisatakse 8 g destilleeritud vett, suletakse korgiga ja segatakse ettevaatlikult, et tagada vee ühtlane jaotumine. Enne kasutamist lastakse seista vähemalt 12 tundi. |
|
4.2. |
n-heksaan, kromatograafias kasutamiseks |
|
4.3. |
Isopropanool |
|
4.4. |
Isopropanooli vesilahus (mahusuhe 1:1) |
|
4.5. |
Pankrease lipaas. Kasutatava lipaasi aktiivsus peab olema vahemikus 2,0–10 lipaasiühikut mg kohta (müügilolevate lipaaside aktiivsus on 2–10 ühikut ensüümi mg kohta) |
|
4.6. |
Puhverlahus: 1 M tris-hüdroksümetüülaminometaani lahuse pH reguleeritakse potentsiomeetriga kontrollides väärtusele 8, lisades selleks kontsentreeritud soolhapet (mahusuhe 1:1) |
|
4.7. |
Naatriumkolaat, ensüümikvaliteediga, 0,1 % vesilahus (lahus tuleb ära kasutada 15 päeva jooksul pärast valmistamist) |
|
4.8. |
Kaltsiumkloriid, 22 % vesilahus |
|
4.9. |
Dietüüleeter, kromatograafias kasutamiseks |
|
4.10. |
Elueerimislahus: n-heksaani ja dietüüleetri segu (mahusuhe 87:13) |
|
4.11. |
Naatriumhüdroksiid, 12massiprotsendiline lahus |
|
4.12. |
Fenoolftaleiin, 1 % lahus etanoolis |
|
4.13. |
Kandegaas: vesinik või heelium gaasikromatograafias kasutamiseks |
|
4.14. |
Abigaasid: kuiv vesinik, vähemalt 99 % puhtusega, ei tohi sisaldada orgaanilisi aineid; õhk kromatograafias kasutamiseks, sama puhtusastmega |
|
4.15. |
Silaanimisreaktiiv: püridiini, heksametüüldisilasaani ja trimetüülklorosilaani segu (mahusuhe 9:3:1). (Müügil on vastavad valmissegud. Kasutada võib ka muid silaanimisreaktiive, näiteks bistrimetüülsilüültrifluoroatsetamiid + 1 % trimetüülklorosilaani, mis on lahustatud samas koguses veevabas püridiinis.) |
|
4.16. |
Standardproovid: puhtad monoglütseriidid või proovile sarnaste mahusuhetega monoglütseriidide segud, mille teadaolevad mahusuhted on samad kui proovil. |
5. TÖÖ KÄIK
5.1. Proovi ettevalmistamine
|
5.1.1. |
Õlisid, milles vabade hapete sisaldus on alla 3 %, ei ole vaja neutraliseerida enne kromatografeerimist silikageelkolonnis. Õlid, mille vabade hapete sisaldus on suurem kui 3 %, tuleb neutraliseerida vastavalt punktile 5.1.1.1.
|
|
5.1.2. |
1,0 g eespool kirjeldatud viisil ettevalmistatud õli pannakse 25milliliitrisesse Erlenmeyeri kolbi (3.1) ja lahustatakse 10 ml elueerimislahuses (4.10). Enne kromatografeerimist silikageelkolonnis lastakse lahusel vähemalt 15 minutit seista. Kui lahus on hägune, tsentrifuugitakse see, et luua optimaalsed tingimused kromatograafia jaoks. (On lubatud kasutada müügilolevaid valmis SPE silikageelipadruneid (500 mg)). |
|
5.1.3. |
Kromatograafiakolonni valmistamine Kolonni (3.3) kallatakse ligikaudu 30 ml elueerimislahust (4.10), kolonni alumisse otsa viiakse klaaspulga abil vatitükk ja pressitakse seda õhu eemaldamiseks. Keeduklaasis valmistatakse suspensioon 25 g silikageelist (4.1) ja ligikaudu 80 ml elueerimislahusest; suspensioon kallatakse lehtri abil kolonni. Kontrollitakse, et kogu silikageel oleks viidud kolonni, loputatakse seda elueerimislahusega (4.10), avatakse kraan ja lastakse vedelikul voolata, kuni silikageeli kohale jääb ligikaudu 2 mm vedelikku. |
|
5.1.4. |
Kolonnkromatograafia 25 milliliitrisesse Erlenmeyeri kolbi (3.1) kaalutakse täpselt 1,0 g punkti 5.1 kohaselt ettevalmistatud proovi. Proov lahustatakse 10 ml elueerimislahuses (4.10). Lahus kantakse üle punkti 5.1.3 kohaselt valmistatud kromatograafiakolonni. Tuleb vältida kolonnitäidise pinna liigutamist. Avatakse kraan ja proovi lahusel lastakse välja voolata seni, kuni tase jõuab silikageeli tasemeni. Elueeritakse 150 ml elueerimislahusega. Elueerimislahust lisatakse kiirusega 2 ml/min (150 ml lisamine kolonni võtab aega ligikaudu 60–70 min). Eluaat kogutakse eelnevalt kaalutud 250milliliitrisesse ümarkolbi. Lahusti aurustatakse vaakumis, viimased lahusti jäljed eemaldatakse lämmastikuvoolus. Ümarkolb kaalutakse ja arvutatakse eraldatud aine kogus. (Juhul kui kasutatakse müügilolevaid valmis SPE silikageelipadruneid, toimitakse järgmiselt: 1 ml lahust (5.1.2) viiakse padrunitesse, mis on eelnevalt ette valmistatud 3 ml n-heksaaniga. Pärast lahuse nõrgumist kolonni elueeritakse 4 ml n-heksaani-dietüüleetri lahusega (mahusuhe 9:1). Eluaat kogutakse 10milliliitrisesse katseklaasi ja aurustatakse lämmastikuvoolus kuivaks. Kuivaine hüdrolüüsitakse pankrease lipaasiga (5.2). Oluline on kontrollida rasvainete sisaldust enne ja pärast SPE-padruni kasutamist). |
5.2. Hüdrolüüs pankrease lipaasiga
|
5.2.1. |
Tsentrifuugitopsi kaalutakse 0,1 g punkti 5.1 kohaselt ettevalmistatud proovi. Lisatakse 2 ml puhverlahust (4.6), 0,5 ml naatriumkolaadi lahust (4.7) ja 0,2 ml kaltsiumkloriidi lahust; pärast iga lahuse lisamist segatakse hoolikalt. Tops suletakse lihvkorgiga ja asetatakse termostaati temperatuuriga 40 + 0,5 °C. |
|
5.2.2. |
Lisatakse 20 mg lipaasi, segatakse hoolikalt (vältides korgi kokkupuutumist lahusega) ning asetatakse tops täpselt 2 minutiks termostaati, seejärel võetakse see termostaadist välja, loksutatakse tugevasti täpselt ühe minuti jooksul ning lastakse jahtuda. |
|
5.2.3. |
Lisatakse 1 ml dietüüleetrit, suletakse tops korgiga ja loksutatakse tugevasti, seejärel segu tsentrifuugitakse ning eetrilahus kantakse mikrosüstla abil puhtasse ja kuiva topsi. |
5.3. Silaanitud ühendite valmistamine ja gaasikromatograafia
|
5.3.1. |
Mikrosüstla abil viiakse 100 μl lahust (5.2.3) 10milliliitrisesse koonilise põhjaga topsi. |
|
5.3.2. |
Lahusti eemaldatakse nõrga lämmastikuvoolu all, lisatakse 200 μl silaanimisreaktiivi (4.15), tops suletakse korgiga ja lastakse seista 20 minuti jooksul. |
|
5.3.3. |
20 minuti pärast lisatakse 1–5 ml n-heksaani (sõltuvalt kromatograafia läbiviimise tingimustest) – saadav lahus on valmis gaasikromatograafia läbiviimiseks. |
5.4. Gaasikromatograafia
Töö põhitingimused on järgmised:
— injektori (on-column-injektor) temperatuur on madalam lahusti keemistemperatuurist (68 °C);
— detektori temperatuur: 350 °C;
— kolonni temperatuur: ahju temperatuuri muudetakse järgmise programmi kohaselt: 60 °C 1 minuti jooksul, temperatuuri tõstetakse 15 °C minutis kuni temperatuurini 180 °C, seejärel 5 °C minutis kuni temperatuurini 340 °C, 340 °C 13 minuti jooksul;
— kandegaas: vesinik või heelium, mille lineaarkiirust reguleeritakse nii, et saada joonisel 1 kujutatud lahutus; triglütseriidi C54 retentsiooniaeg peab olema 40 + 5 minutit (vt joonis 2); (Need töö põhitingimused on ligikaudsed. Töö teostaja peab neid soovitud tulemuste saamiseks kohandama. 2-glütserüülmonopalmitaadi piigi kõrgus peab olema vähemalt 10 % registreerimisseadme skaala ulatusest.);
— kolonni süstitava aine kogus on 0,5–1 μl n-heksaani lahust (5 ml) (5.3.3).
5.4.1. Piikide määramine
Piigid määratakse retentsiooniaja põhjal, võrdlusest samadel tingimusel analüüsitud monoglütseriidide standardsegude puhul saadud tulemustega.
5.4.2. Kvantitatiivne hindamine
Elektroonilise integraatori abil leitakse iga piigi pindala.
6. TULEMUSTE VÄLJENDAMINE
Glütserüülmonopalmitaadi protsendiline sisaldus arvutatakse vastava piigi pindala ja monoglütseriidide piikide kogupindala suhtena (vt joonis 2) vastavalt järgmisele valemile:
Gycéril monopalmitate (%):
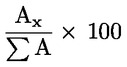
kus:
|
Ax |
= |
glütserüülmonopalmitaadi piigi pindala; |
|
ΣA |
= |
monoglütseriidide piikide kogupindala. |
Tulemus esitatakse ühe kümnendkoha täpsusega.
7. ANALÜÜSI PROTOKOLL
Analüüsi protokollis tuleb esitada järgmine teave:
— viide käesolevale meetodile;
— kogu teave, mis on vajalik proovi täielikuks identifitseerimiseks;
— analüüsi tulemus;
— kõik kõrvalekaldumised käesolevast meetodist, kas asjaomaste poolte kokkuleppe tõttu või muudel põhjustel;
— andmed laboratooriumi kohta, analüüsi läbiviimise kuupäev ja analüüsi läbiviimise eest vastutavate isikute allkirjad.
Joonis 1
Silaanimisreaktsiooni saaduste kromatogramm (rafineeritud oliiviõlile on lisatud 20 % täielikult esterdatud õli, segu on töödeldud lipaasiga ja reaktsioonisaadused on silaanitud)

Kirjad joonisel: Acides gras libres = vabad rasvhapped Huile d’olive raffinée + 20 % huile estérifiée = rafineeritud oliiviõli + 20 % esterdatud õli 1–2 monopalmitoléine = 1–2 monopalmitoleiin 1–2 monopalmitine = 1–2 monopalmitiin 1–2 monoC18 insat. = 1–2 monoC18 küllastamata Squalene = skvaleen]
Joonis 2
Kromatogramm:
|
A) |
esterdamata oliiviõli pärast lipaasiga töötlemist; pärast silüülimist; kõnealuste tingimuste juures (kapillaarkolonni pikkus 8–12 m) elueeritakse vahafraktsioon samaaegselt diglütseriidi fraktsiooniga või veidi hiljem. Pärast lipaasiga töötlemist ei tohi triglütseriidide sisaldus ületada 15 %. 
Tähistused:
|
Kromatogramm:
|
B) |
esterdatud õli pärast lipaasiga töötlemist; pärast silaanimist; kõnealuste tingimuste juures (kapillaarkolonni pikkus 8–12 m) elueeritakse vahafraktsioon samaaegselt diglütseriidi fraktsiooniga või veidi hiljem. Pärast lipaasiga töötlemist ei tohi triglütseriidide sisaldus ületada 15 %. 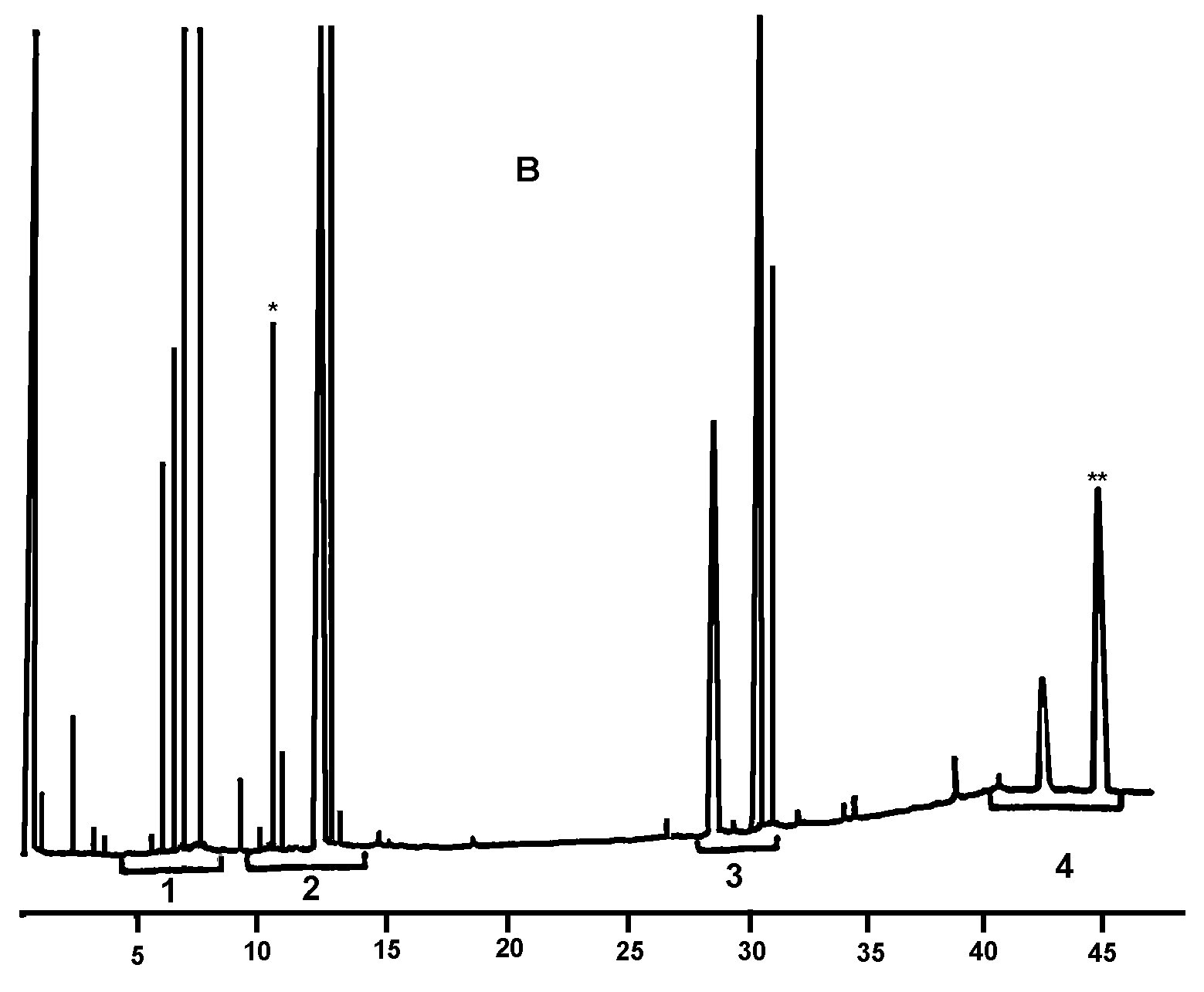
Tähistused:
|
8. MÄRKUSED
LIPAASI VALMISTAMINE
Müügil on rahuldava aktiivsusega lipaase. Lipaasi saab valmistada ka laboris järgmisel viisil:
5 kg värsket seapankreast jahutatakse temperatuurini 0 °C. Eemaldatakse tahke rasvkude ja seda ümbritsev sidekude ning pankreas peenestatakse lõikenugadega peenestajas kuni ühtlase vedela massi saamiseni. Saadud massi segatakse 4–6 tunni jooksul 2,5 liitri veevaba atsetooniga, seejärel tsentrifuugitakse. Saadud jääki ekstraheeritakse veel kolm korda sama koguse veevaba atsetooniga, seejärel kaks korda sama koguse atsetooni/dietüüleetri seguga (mahusuhe 1:1) ning veel kaks korda dietüüleetriga.
Saadud jääki kuivatatakse 48 tunni jooksul vaakumis; tulemusena saadakse stabiilne pulber, mida on võimalik külmkapis kuivas pikemaajaliselt hoida.
LIPAASI AKTIIVSUSE KONTROLLIMINE
Valmistatakse oliiviõli emulsioon järgmisel viisil:
Mikseris segatakse 10 minuti jooksul järgmist segu: 165 ml kummiaraabiku lahust kontsentratsiooniga 100 g/l, 15 g purustatud jääd ja 20 ml eelnevalt neutraliseeritud oliiviõli.
50milliliitrisesse keeduklaasi viiakse 10 ml saadud emulsiooni, seejärel 0,3 ml naatriumkolaadi lahust kontsentratsiooniga 0,2 g/ml ja 20 ml destilleeritud vett.
Keeduklaas asetatakse termostaati temperatuuriga 37 °C, lahusesse viiakse pH-meetri elektroodid ja spiraalsegaja.
Büreti abil lisatakse tilkhaaval naatriumhüdroksiidi 0,1 N lahust, kuni pH saavutab väärtuse 8,3.
Lisatakse teatav kogus lipaasi suspensiooni vees (0,1 g/ml lipaasi). Kui pH-meeter näitab pH väärtust 8,3, käivitatakse stopper ja hakatakse tilkhaaval lisama naatriumhüdroksiidi lahust sellise kiirusega, et hoida pH-d väärtusel 8,3. Iga minuti möödudes märgitakse üles selleks kulunud lahuse kogus.
Andmed kantakse graafikule, võttes abstsissteljeks ajatelje ja kandes ordinaatteljele 0,1 N leelise lahuse kogused milliliitrites, mis on kulunud pH väärtuse hoidmiseks konstantsena. Graafik peab olema lineaarne.
Lipaasi aktiivsus, mida väljendatakse lipaasi ühikutes mg kohta, arvutatakse järgmise valemi abil:
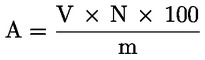
kus:
|
A |
aktiivsus lipaasi ühikutes mg kohta; |
|
V |
0,1 N naatriumhüdroksiidi lahuse kulu milliliitrites minuti kohta (arvutatud graafiku alusel); |
|
N |
naatriumhüdroksiidi lahuse normaalsus; |
|
m |
prooviks võetud lipaasi mass, mg. |
Lipaasi ühik on määratletud kui ensüümi kogus, mis vabastab 10 mikroekvivalenti hapet ühe minuti jooksul.
▼M20 —————
IX LISA
SPEKTROFOTOMEETRILINE ANALÜÜS ULTRAVIOLETTPIIRKONNAS
EESSÕNA
Spektrofotomeetriline analüüs ultraviolettpiirkonnas võib anda teavet rasva kvaliteedi, säilivusseisundi ja tehnoloogilistest protsessidest tulenenud muutuste kohta. Käesolevas meetodis osutatud lainepikkustel esinev neeldumine on tingitud konjugeeritud dieen- ja trieensüsteemide olemasolust olenevalt oksüdeerumisprotsessist ja/või rafineerimisviisist. Valguse neeldumine väljendatakse eriekstinktsioonina
![]()
(1-protsendilise (w/v) rasvalahuse ekstinktsioon teatavas lahustis 10 mm läbimõõduga küvetis), mida tavaliselt tähistatakse K-ga ja nimetatakse ka ekstinktsioonikoefitsiendiks.
1. KOHALDAMISALA
Käesolevas lisas kirjeldatakse oliiviõli spektrofotomeetrilist analüüsi ultraviolettpiirkonnas.
2. MEETODI PÕHIMÕTE
Proov lahustatakse ettenähtud lahustis ja lahuse neeldumist mõõdetakse teatavatel lainepikkustel, kasutades võrdluslahusena puhast lahustit.
Arvutatakse eriekstinktsioon 232 nm ja 268 nm juures isooktaanis või 232 nm ja 270 nm juures tsükloheksaanis kontsentratsioonil 1 % (mass/maht) küvetis läbimõõduga 10 mm.
3. SEADMED
|
3.1. |
Spektrofotomeeter, mis sobib mõõtmiseks ultraviolettkiirguse lainepikkustel (220–360 nm) ja mis võimaldab tulemusi lugeda ühe nanomeetri täpsusega. Neeldumise ja lainepikkuse skaala täpsust ja korratavust ning hajuvat valgust soovitatakse korrapäraselt kontrollida.
|
|
3.2. |
10 mm optilise teepikkusega risttahukakujulised korgiga kvartsküvetid, mis sobivad mõõtmiseks ultraviolettkiirguse lainepikkusel (220–360 nm). Vee või mõne muu sobiva lahustiga täidetuna ei tohiks küvettide tulemuste erinevus olla suurem kui 0,01 ekstinktsiooniühikut. |
|
3.3. |
Sama kaubamärgi alla kuuluvad A-klassi mõõtekolvid mahuga 25 ml. |
|
3.4. |
Analüütiline kaal, millelt saab andmeid lugeda täpsusega 0,0001 g. |
4. REAGENDID
Kui ei ole öeldud teisiti, kasutatakse analüüsi käigus ainult tunnustatud analüütiliselt puhtaid reagente ja demineraliseeritud või destilleeritud või samaväärse puhtusastmega vett.
Lahusti: isooktaan (2,2,4-trimetüülpentaan) mõõtmiseks 232 nm ja 268 nm juures või tsükloheksaan mõõtmiseks 232 nm ja 270 nm juures, mille neelduvus 10 mm küvetis 232 nm juures on väiksem kui 0,12 ja neelduvus 270 nm juures destilleeritud vee puhul mõõtmisel on väiksem kui 0,05.
5. TÖÖ KÄIK
|
5.1. |
Proov peab olema täiesti homogeenne ja hõljuvate lisanditeta. Kui see nii ei ole, tuleb proov filtreerida läbi paberi temperatuuril ligikaudu 30 °C. |
|
5.2. |
25 ml mõõtekolbi kaalutakse ligikaudu 0,25 g (täpsusega 1 mg) eespool kirjeldatud viisil ettevalmistatud proovi, täidetakse ettenähtud lahustiga kuni märgini ning homogeenitakse. Tekkiv lahus peab olema täiesti selge. Opalestsentsi või hägususe korral tuleb proov kiiresti läbi paberi filtreerida. MÄRKUS: üldiselt on kaal 0,25–0,30 g piisav neitsioliiviõli ja ekstra-neitsioliiviõli neelduvuse mõõtmiseks 268 nm ja 270 nm juures. Mõõtmiseks 232 nm juures on proovi nõutav kaal üldiselt 0,05 g, seega valmistatakse tavaliselt ette kaks eri lahust. Oliivijääkõli, rafineeritud oliiviõli ja võltsitud oliiviõli neelduvuse mõõtmiseks on nende suurema neeldumise tõttu vaja üldjuhul väiksemat proovi (kaaluga 0,1 g). |
|
5.3. |
Vajaduse korral korrigeeritakse nulljoont (220–290 nm) lahustiga mõlemas kvartsküvetis (proov ja võrdlusmaterjal), seejärel täidetakse proovi kvartsküvett testlahusega ja mõõdetakse ekstinktsioonid vahemikus 232, 268 või 270 nm, võttes võrdluseks kasutatud lahusti. Registreeritud ekstinktsiooniväärtused peavad jääma vahemikku 0,1–0,8 või spektrofotomeetri lineaarsusvahemikku, mida tuleks kontrollida. Vastasel korral tuleb mõõtmisi korrata ja kasutada vajaduse korral vähem või rohkem lahjendatud lahuseid. |
|
5.4. |
Pärast neelduvuse mõõtmist lainepikkusel 268 või 270 nm mõõdetakse neelduvus λmax, λmax + 4 ja λmax – 4. Neelduvuse väärtusi kasutatakse selleks, et määrata kindlaks eriekstinktsiooni variatsioon (ΔΚ). MÄRKUS: lahustina kasutatava isooktaani neelduvus (λmax) peab olema 268 nm ja tsükloheksaani neelduvus 270 nm. |
6. TULEMUSTE VÄLJENDAMINE
|
6.1. |
Registreeritakse eriekstinktsioonid (ekstinktsioonikoefitsiendid) eri lainepikkustel, mis arvutatakse järgmise võrrandi alusel:
kus: Kλ on eriekstinktsioon lainepikkusel λ; Eλ on ekstinktsioon lainepikkusel λ; c on lahuse kontsentratsioon g/100 ml; s on kvartsküveti teepikkus sentimeetrites; väljendatud kahe kümnendkoha täpsusega. |
|
6.2. |
Eriekstinktsiooni (ΔΚ) variatsioon Ekstinktsiooni (ΔΚ) absoluutväärtuse variatsioon on esitatud olukorras,
kus Km on eriekstinktsioon maksimaalse neeldumise lainepikkusel 270 nm ja 268 nm, sõltuvalt kasutatud lahustist, väljendatud kahe kümnendkoha täpsusega. |
X LISA
RASVHAPETE METÜÜLESTRITE GAASIKROMATOGRAAFILINE MÄÄRAMINE
1. KOHALDAMISALA
Käesolevas lisas antakse juhised, kuidas määrata gaasikromatograafiliselt vabu ja seotud rasvhappeid taimses rasvas ja õlis pärast nende teisendamist rasvhapete metüülestriteks (FAME).
Triatsüülglütseroolide (TAG) seotud rasvhapped ning sõltuvalt esterdusmeetodist vabad rasvhapped (FFA) teisendatakse rasvhapete metüülestriteks, mis määratakse kapillaarkolonnkromatograafiaga.
Käesolevas lisas kirjeldatud meetod võimaldab määrata rasvhapete metüülestreid vahemikus C12–C24, sealhulgas küllastunud, cis- ja trans-monoküllastumata ning cis- ja trans-polüküllastumata rasvhapete metüülestrid.
2. PÕHIMÕTE
Gaasikromatograafiat kasutatakse rasvhapete metüülestrite kvantitatiivseks analüüsiks. Rasvhapete metüülestrid valmistatakse vastavalt A osale. Seejärel süstitakse need gaasikromatograafi ja aurustatakse injektoris. Rasvhapete metüülestrid eraldatakse konkreetse polaarsuse ja pikkusega analüüsikolonnide abil. Rasvhapete metüülestrite kindlaksmääramiseks kasutatakse leekionisatsioonidetektorit. Analüüsi tingimused on esitatud B osas.
Rasvhapete metüülestrite kindlaksmääramisel leekionisatsioonidetektoris kasutatav kandegaas (liikuv faas) võib olla vesinik või heelium. Vesinik kiirendab eraldamist ja annab teravamad piigid. Statsionaarne faas on mikroskoopiliselt õhuke kiht tihedat vedelikku kvartsklaasist inertsel tahkel pinnal.
Kapillaarkolonni läbimisel analüüsitakse lenduvate ühendite ja kolonni sisepinda katva statsionaarse faasi omavahelist suhet. Eri ühendite erineva koosmõju tõttu on nende elueerumisaeg erinev ning seda nimetatakse ühendi peetumisajaks konkreetsetel analüüsiparameetritel. Eri ühendid identifitseeritakse peetumisaegade võrdlemise teel.
A OSA
RASVHAPETE METÜÜLESTRITE VALMISTAMINE OLIIVIÕLIST JA OLIIVIJÄÄKÕLIST
1. RAKENDUSALA
Käesolevas osas esitatakse üksikasjalikult rasvhapete metüülestrite valmistamisviis. Osa sisaldab meetodeid rasvhapete metüülestrite valmistamiseks oliiviõlist ja oliivijääkõlist.
2. KOHALDAMISALA
Rasvhapete metüülestrite valmistamiseks oliiviõlist ja oliivijääkõlist ümberesterdatakse kaaliumhüdroksiid metanoolilahusega toatemperatuuril. Vajadus proovi enne ümberesterdamist puhastada sõltub proovi vabade rasvhapete sisaldusest ja kindlaksmääratavatest analüütilistest parameetritest, vajaduse üle võib otsustada vastavalt järgmisele tabelile:
|
Õli kategooria |
Meetod |
|
Neitsioliiviõli happesusega ≤ 2,0 % |
1. Rasvhapped 2. Trans-rasvhapped 3. ΔECN42 (pärast puhastamist tahkefaasekstraktsiooni meetodil SPE silikageeliga) |
|
Rafineeritud oliiviõli |
|
|
Rafineeritud õlist ja neitsioliiviõlist koosnev oliiviõli |
|
|
Rafineeritud oliivijääkõli |
|
|
Oliivijääkõli |
|
|
Neitsioliiviõli happesusega > 2,0 % Töötlemata oliivijääkõli |
1. Rasvhapped (pärast puhastamist SPE silikageeliga) 2. Rasvhapped (pärast puhastamist SPE silikageeliga) 3. ΔECN42 (pärast puhastamist SPE silikageeliga) |
3. METOODIKA
3.1. Ümberesterdus kaaliumhüdroksiidi metanoolilahusega toatemperatuuril
3.1.1. Põhimõte
Metüülestrid tekivad ümberesterdusel kaaliumhüdroksiidi lahusega metanoolis, estrite teke on vaheetapp enne seebistumist.
3.1.2. Reagendid
|
3.1.2.1. |
Metanool, mille veesisaldus on kuni 0,5 massiprotsenti. |
|
3.1.2.2. |
Heksaan, kromatograafias kasutamiseks. |
|
3.1.2.3. |
Heptaan, kromatograafias kasutamiseks. |
|
3.1.2.4. |
Dietüüleeter, analüüsiks stabiliseeritud. |
|
3.1.2.5. |
Atsetoon, kromatograafias kasutamiseks. |
|
3.1.2.6. |
Elueerimislahusti õli puhastamiseks kolonnkromatograafia/SPE-kromatograafia abil: heksaani ja etüüleetri segu komponentide suhtega 87/13 (v/v). |
|
3.1.2.7. |
Kaaliumhüdroksiid, ligikaudu 2M metanoolilahus: 100 ml metanoolis lahustatakse 11,2 g kaaliumhüdroksiidi. |
|
3.1.2.8. |
Silikageelpadrunid, 1 g (6 ml), tahkefaasekstraktsiooni jaoks. |
3.1.3. Seadmed
|
3.1.3.1. |
Keeratava korgiga 5 ml katseklaasid, mille korgid on varustatud teflontihendiga. |
|
3.1.3.2. |
Mõõtpipetid või automaatpipetid, 2 ml ja 0,2 ml. |
3.1.4. Õliproovide puhastamine
Vajaduse korral puhastatakse proove õli juhtimisega läbi tahkefaasiekstraktsiooni silikageeli padruni. Silikageeli padrun (3.1.2.8) pannakse vaakumvoolutusseadmesse ja pestakse 6 ml heksaaniga (3.1.2.2); pesemine toimub ilma vaakumita. Seejärel kantakse õlilahus (ligikaudu 0,12 g) 0,5 ml heksaanis (3.1.2.2) kolonnile. Lahus imetakse kolonni sisse ja eludeeritakse siis vaakumit kasutades 10 ml heksaani-dietüüleetri seguga vahekorras 87:13 mahuosa (3.1.2.6). Ühendatud eluaadid homogeniseeritakse ja jagatakse võrdselt kaheks. Üks alikvoot aurutatakse rotaatoril vaakumis toatemperatuuril kuivaks. Jääk lahustatakse 1 ml heptaanis ja saadakse valmis lahus rasvhapete määramiseks gaasikromatograafia meetodil. Teine alikvoot aurutatakse kuivaks ja jääk lahustatakse 1 ml atsetoonis triglütseriidide määramiseks HPLC abil, kui see on vajalik.
3.1.5. Töö käik
5 ml keeratava korgiga katseklaasis (3.1.3.1) kaalutakse umbes 0,1 g õliproovi. Lisatakse 2 ml heptaani (3.1.2.2) ja loksutatakse. Lisatakse 0,2 ml kaaliumhüdroksiidi lahust metanoolis (3.1.2.7), suletakse teflontihendiga varustatud keeratava korgiga, keeratakse kork tugevasti kinni ja loksutatakse tugevalt 30 sekundit. Jäetakse kihistuma, kuni ülemine lahus muutub selgeks. Ülemine, metüülestreid sisaldav kiht kallatakse pealt ära. Heptaanilahus on valmis gaasikromatograafi süstimiseks. Kuni gaasikromatograafilise analüüsi tegemiseni soovitatakse lahust hoida külmkapis. Lahust ei ole soovitatav hoida kauem kui 12 tundi.
B OSA
RASVHAPETE METÜÜLESTRITE GAASIKROMATOGRAAFILINE ANALÜÜS
1. RAKENDUSALA
Käesolevas osas antakse üldjuhised kapillaarkolonnkromatograafia kohaldamiseks A osas kehtestatud meetodil saadud rasvhapete metüülestrite segu kvalitatiivse ja kvantitatiivse koostise kindlakstegemisel.
Käesolevat osa ei kohaldata polümeriseeritud rasvhapete suhtes.
2. REAGENDID
2.1. Kandegaas
Inertgaas (heelium või vesinik), põhjalikult kuivatatud ja hapnikusisaldusega kuni 10 mg/kg.
Märkus 1: vesinik võib analüüsi kiirust kahekordistada, kuid on ohtlik. Saadaval on ohutusseadmed.
2.2. Abigaasid
|
2.2.1. |
Vesinik (puhtus ≥ 99,9 %), orgaaniliste lisanditeta. |
|
2.2.2. |
Õhk või hapnik, orgaaniliste lisanditeta. |
|
2.2.3. |
Lämmastik (puhtus > 99 %). |
2.3. Võrdlusstandard
Puhaste rasvhapete metüülestrite segu või teadaoleva, eelistatult analüüsitava rasvainega samaväärse koostisega rasva metüülestrid. Oktadekeenhappe, oktadekadieenhappe ja oktadekatrieenhappe metüülestrite cis- ja trans-isomeerid aitavad kindlaks teha küllastumata hapete trans-isomeere.
Tuleks tagada, et polüküllastumata rasvhapped ei oksüdeeruks.
3. SEADMED
Käesolevad juhendid hõlmavad tavapäraseid gaasikromatograafia seadmeid, kus kasutatakse kapillaarkolonne ja leekionisatsioonidetektorit.
3.1. Gaasikromatograaf
Gaasikromatograaf koosneb järgmistest osadest.
3.1.1. Injektorseade
Kasutada tuleb kapillaarkolonnidega injektorseadet, mis peab olema spetsiaalselt ette nähtud selliste kolonnide kasutamiseks. See võib olla jaotatud või jaotamata vooluga injektor.
3.1.2. Ahi
Kapillaarkolonni peab olema võimalik ahjus kuumutada vähemalt temperatuurini 260 °C ning ahi peab soovitud temperatuuri hoidma 0,1 °C täpsusega. Viimane nõue on eriti oluline juhul, kui kasutatakse kvartsklaasist kolonni.
Temperatuuriregulaatoriga kuumutamine on kõikidel juhtudel soovitav, eelkõige rasvhapete puhul, millel on vähem kui 16 süsinikuaatomit.
3.1.3. Kapillaarkolonn
|
3.1.3.1. |
Katseklaas, mis on valmistatud analüüsitava ainega inertsest materjalist (tavaliselt klaas või kvartsklaas). Sisediameeter on 0,20–0,32 mm. Sisekülge tuleb enne statsionaarse faasi kihiga katmist nõuetekohaselt töödelda (nt pinna ettevalmistamine, inaktiveerimine). Pikkus 60 m on rasvhapete ja rasvhapete trans-isomeeride jaoks piisav. |
|
3.1.3.2. |
Statsionaarne faas, sobivad polaar-polüsiloksaani (tsüanopropüülsilikoon) seotud (võrkstruktuuriga) kolonnid. Märkus 2: polaar-polüsiloksaanid võivad raskendada linoleenhappe ja C20 hapete identifitseerimist ja eraldamist. Kiht peab olema õhuke, st 0,1–0,2 μm. |
|
3.1.3.3. |
Kolonni monteerimine ja tasakaalustamine Järgitakse kapillaarkolonnide tavapäraseid ettevaatusabinõusid: kolonni asetus ahjus (tugirajatised), liitekohtade valik ja paigaldus (lekkekindlus), kolonni otste asetus injektoris ja detektoris (võimalikult vähe tühja ruumi). Läbi kolonni juhitakse kandegaas (nt 0,3 baari (30 kPa) läbi 25 m pikkuse ja 0,3 mm sisediameetriga kolonni). Kolonn tasakaalustatakse ahju reguleerides selliselt, et temperatuur tõuseb ümbritseva õhu temperatuurilt 3 °C minutis, saavutades sellise temperatuuri, mis on 10 °C madalam kui statsionaarse faasi hajumistemperatuur. Ahju hoitakse sellisel temperatuuril tund aega, kuni nulljoon stabiliseerub. Temperatuuri alandatakse taas tagasi 180 °C-le ja katset jätkatakse isotermilistel tingimustel. Märkus 3: nõuetekohaselt tasakaalustatud kolonnid on kaubandusvõrgus müügil. |
3.1.4. Leekionisatsioondetektor ja muundurvõimendi
3.2. Süstal
Süstla ülemine mõõtepiir on 10 μl ning mõõtskaala on jagatud 0,1-mikroliitristeks ühikuteks.
3.3. Andmekogumissüsteem
Andmekogumissüsteem on internetipõhiselt ühendatud detektoritega ja seda kasutatakse piikide integreerimiseks ja normaliseerimiseks sobiva tarkvaraprogrammiga.
4. TÖÖ KÄIK
Punktides 4.1–4.3 kirjeldatud toimingud eeldavad leekionisatsioonidetektori kasutamist.
4.1. Katsetingimused
4.1.1. Kapillaarkolonni jaoks vajalike optimaalsete töötingimuste valik
Kapillaarkolonni tõhusus- ja täituvusomadused tingivad selle, et koostisosade eraldamine ja analüüsi kestus sõltuvad suuresti kandegaasi voolukiirusest kolonnis. Seetõttu on vaja töötingimusi optimeerida, muutes seda parameetrit (ehk lihtsalt kolonnipead vähendades) sõltuvalt sellest, kas soovitakse tõhustada eraldamist või kiirendada analüüsi.
Rasvhapete metüülestrite (C4–C26) eraldamiseks on osutunud sobivaks järgmised tingimused. Kromatogrammide näidised on esitatud B liites.
|
Injektori temperatuur: |
250 °C |
|
Detektori temperatuur: |
250 °C |
|
Ahju temperatuur: |
165 °C (8 min) kuni 210 °C temperatuuri tõusuga 2 °C/min |
|
Kandegaas vesinik: |
kolonnipea rõhk: 179 kPa |
|
Koguvool: |
154,0 ml/min; |
|
Jaotatud voolu suhe: |
1:100 |
|
Sissesüstitav ruumala: |
1 μl |
4.1.2. Resolutsiooni määramine (vt A liide)
Kahe külgneva piigi (I ja II) resolutsioon (R) arvutatakse järgmise valemi abil:
R = 2 × ((dr ( II ) – d r(I))/(ω(I) + ω(II))) või R = 2 × ((tr ( II ) – t r(I))/(ω(I) + ω(II))) (USP) (USA farmakopöa)
või
R = 1,18 × ((tr ( II ) – tr(I))/(ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB), (JP (Jaapani farmakopöa), EP (Euroopa farmakopöa), BP (Briti farmakopöa))
kus:
|
d r(I) |
I piigi peetumisdistants; |
|
d r(II) |
II piigi peetumisdistants; |
|
t r(I) |
I piigi peetumisaeg; |
|
t r(II) |
II piigi peetumisaeg; |
|
ω(I) |
I piigi laius nulljoonel; |
|
ω(II) |
II piigi laius nulljoonel; |
|
ω0,5 |
konkreetse ühendi piigi laius, piigi poolel kõrgusel; |
kui ω(I) ≈ ω(II), arvutatakse R järgmiste valemite abil:
R = (dr ( II ) – d r(I))/ω = (dr ( II ) – d r(I))/4σ
kus:
σ on standardhälve (vt A liide joonis 1).
Kui kahe piigi vaheline distants d r(II) – d r(I) võrdub 4σ, siis resolutsioonfaktor R = 1.
Kui kaks piiki ei ole täiesti eraldi, lõikuvad kahe piigi käänupunkti tõmmatud puutujad punktis C. Selleks et kahte piiki täielikult eraldada, peab kahe piigi vaheline kaugus olema järgmine:
d r(II) – d r(I) = 6 σ millest R = 1,5 (vt A liide joonis 3).
5. TULEMUSTE VÄLJENDAMINE
5.1. Kvalitatiivne analüüs
B liite joonise 1 kohase kromatogrammiga saadud proovi metüülestripiigid määratakse vajaduse korral interpolatsiooni abil või nende võrdlemise teel punktis 2.3 osutatud metüülestrite võrdlussegude piikidega.
5.2. Kvantitatiivne analüüs
5.2.1. Koostise määramine
Arvutada järgmisel viisil üksikute rasvhapete metüülestrite massiosa wi väljendatuna metüülestrite massiprotsendina.
5.2.2. Arvutusmeetod
5.2.2.1. Üldiselt
Teatava komponendi i sisaldus väljendatuna metüülestrite massiprotsendina arvutatakse selliselt, et määratakse piigipindala suhteline osa kõikide piikide pindalast järgmise valemi alusel.
wi = (Ai/ΣA) × 100
kus:
Ai on üksikute rasvhapete metüülestrite i piigipindala;
ΣA on kõikide üksikute rasvhapete metüülestrite piigipindalade summa.
Tulemused väljendatakse kahe kümnendkoha täpsusega.
Märkus 4: rasvade ja õlide puhul võrdub rasvhapete metüülestrite massiosa triatsüülglütseroolide massiosaga grammides 100 g kohta. Kui seda üldistust ei saa kohaldada, vt punkti 5.2.2.2.
5.2.2.2. Parandustegurite kasutamine
Teatavatel juhtudel, näiteks kui rasvhapetes on vähem kui kaheksa süsinikuaatomit või kui happes on sekundaarsed rühmad, tuleks piigipindalad korrigeerida kindla parandusteguriga (Fci). Parandustegur tuleb iga üksikjuhu puhul kindlaks määrata. Selleks tuleks kasutada sobivaid võrdlusmaterjale, mille vastavate vahemike rasvhapete koostis on sertifitseeritud.
Märkus 5: need parandustegurid ei ole samad mis teoreetilised leekionisatsioonidetektori parandustegurid, mis on esitatud A liites, vaid hõlmavad ka injektorseadme toimimist jne. Suuremate erinevuste esinemisel tuleb siiski kontrollida kogu süsteemi toimimist.
Selle võrdlussegu puhul määratakse rasvhapete metüülestrite massiprotsent i järgmise valemiga:
wi = (mi /Σm) × 100
kus:
m i on rasvhapete metüülestrite mass i võrdlussegus;
Σm on erinevate komponentide üldmass kui rasvhapete metüülestrite võrdlussegu.
Võrdlussegu kromatogrammi alusel arvutatakse rasvhapete metüülestrite pindala protsent i järgmiselt:
wi = (Ai/ΣA) × 100
kus:
Ai on rasvhapete metüülestrite pindala i võrdlussegus;
ΣA on kõikide rasvhapete metüülestrite pindalade summa võrdlussegus.
Parandustegur Fc on seega
Fc = (mi × ΣA)/(Ai/Σm)
Proovi iga rasvhappe metüülestri massiprotsent i on:
wi = (Fi × Ai)/Σ (Fi × Ai)
Tulemused väljendatakse kahe kümnendkoha täpsusega.
Märkus 6: arvutatud väärtus vastab iga üksiku rasvhappe massiprotsendile, mis on arvutatud kui triatsüülglütseroolid 100 grammi rasva kohta.
5.2.2.3. Sisestandardi kasutamine
Teatavate analüüside puhul (nt kui kõikide rasvhapete määr ei ole teada, juhul kui nelja ja kuue süsinikuaatomiga hapete kõrval esinevad ka 16 ja 18 süsinikuaatomiga happed, või kui on vaja määrata proovis olevate rasvhapete absoluutsumma) on vaja kasutada sisestandardit. Sageli kasutatakse 5, 15 või 17 süsinikuaatomiga rasvhappeid. Tuleks kindlaks määrata sisestandardi parandustegur (juhul kui see on vajalik).
Komponendi i massiprotsent väljendatuna metüülestritena arvutatakse valemiga:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
kus:
A i on rasvhapete metüülestrite pindala i;
A IS on sisestandardi pindala;
F i on rasvhapete parandustegur i, väljendatud rasvhapete metüülestritena;
F IS on sisestandardi parandustegur;
m on katsekoguse mass milligrammides;
m IS sisestandardi mass milligrammides.
Tulemused väljendatakse kahe kümnendkoha täpsusega.
6. KATSEARUANNE
Katsearuandesse märgitakse metüülestrite valmistamisel ja gaasikromatograafilisel analüüsimisel kasutatud meetodid. Selles märgitakse samuti kõik üksikasjad, mida ei ole nimetatud käesolevas standardmeetodis või mis on valikulised, samuti üksikasjad tulemust mõjutada võinud asjaolude kohta.
Katsearuandes peavad olema kõik proovi täielikuks tuvastamiseks vajalikud andmeid.
7. TÄPSUS
7.1. Laboritevahelise katse tulemused
Üksikasjad laboritevahelise katse meetodi täpsuse kohta on esitatud standardi IOC/T.20/Doc. nr 33 C lisas. Laboritevahelise katse põhjal saadud näitajaid tohib kohaldada üksnes esitatud kontsentratsioonivahemike ja põhiainete suhtes.
7.2. Korduvus
Absoluutne erinevus kahe sõltumatu üksikkatse (teostatud lühikese ajavahemiku jooksul samal meetodil, identse katsematerjaliga, samas laboris, sama teostaja poolt, samu katseseadmeid kasutades) tulemuste vahel võib üksnes 5 % juhtudest olla suurem kui r, mis on esitatud standardi IOC/T.20/Doc. nr 33 C lisa tabelites 1–14.
7.3. Korratavus
Absoluutne erinevus kahe üksikkatse (teostatud samal meetodil, identse katsematerjaliga, eri laborites, eri teostaja poolt, eri katseseadmeid kasutades) tulemuste vahe võib üksnes 5 % juhtudest olla suurem kui R, mis on esitatud standardi IOC/T.20/Doc. nr 33 C lisa tabelites 1–14.
A liide
Joonis 1

ω0,5 on laius kolmnurga (ABC) poolel kõrgusel ja b laius kolmnurga (NPM) poolel kõrgusel.
|
Joonis 2 |
Joonis 3 |
|
|
|
B liide
Joonis 1
Oliivijääkõli koostise gaasikromatograafiline määramine külmmetüleerimise meetodi abil

Kromatograafilised piigid vastavad metüül- ja etüülestritele, kui ei ole öeldud teisiti.
XI LISA
OLIIVIÕLI HALOGEENITUD LENDUVATE LAHUSTITE MÄÄRAMINE
1. MEETOD
Gaasikromatograafiline aurufaasi analüüs.
2. VAHENDID
|
2.1. |
Gaasikromatograafia seade, milles on elektronihaarde detektor (EHD). |
|
2.2. |
Aurufaasi analüüsimise seade. |
|
2.3. |
Klaasist gaasikromatograafia kolonn, pikkus 2 m ja läbimõõt 2 mm, statsionaarne faas. OV101 10 % või samaväärne, kaltsineeritud, happega pestud ja silaanitud kobediatomiiti immutatud, osakeste suurus 80–100 mesh’i. |
|
2.4. |
Kande- ja abigaas: lämmastik gaasikromatograafia jaoks, sobiv elektronihaardedetektorile. |
|
2.5. |
10- kuni 15-milliliitrised klaaspudelid, teflonpinna ja alumiiniumpunniga, milles on koht nõela sisseviimise jaoks. |
|
2.6. |
Klambrid hermeetiliseks sulgemiseks. |
|
2.7. |
Gaasisüstal, 0,5- kuni 2-milliliitrine. |
3. REAKTIIVID
Standard: halogeenitud lahustid, mille puhtusaste on gaasikromatograafia jaoks sobiv.
4. TÖÖ KÄIK
|
4.1. |
Klaaskolbi (ühekordselt kasutatav) kaalutakse umbes 3 g õli; suletakse hermeetiliselt. Pannakse termostaati temperatuurile 70 oC üheks tunniks. Süstlaga eemaldatakse ettevaatlikult 0,2–0,5 ml aurufaasi. See injekteeritakse järgmiselt reguleeritud gaasikromatograafiaseadme kolonni: — injektori temperatuur: 150 oC, — kolonni temperatuur: 70–80 oC, — detektori temperatuur: 200–250 oC. Kasutada võib ka teisi temperatuure tingimusel, et tulemused jäävad samaks. |
|
4.2. |
Võrdluslahused: valmistatakse ette standardlahused, kasutades rafineeritud oliiviõli, milles ei ole 0,05–1 ppm (mg/kg) kontsentratsiooniga lahusteid, ja mis vastab proovi eeldatavale sisaldusele. Halogeenitud lahustid võib pentaaniga lahjendada. |
|
4.3. |
Kvantitatiivne hindamine: võrreldakse proovi ja eeldatavalt kõige sarnasema kontsentratsiooniga standardlahuse piikide pindalasid või kõrgusi. Kui kõrvalekalle on suurem kui 10 %, tuleb analüüsi korrata mõne teise standardlahusega võrreldes, kuni kõrvalekalle jääb 10 % piiresse. Sisaldus määratakse kindlaks üksikute injekteerimiste keskmise põhjal. |
|
4.4. |
Tulemuste esitamine: ppm (mg/kg). Meetodi avastamispiir on 0,01 mg/kg. |
XII LISA
RAHVUSVAHELISE OLIIVIÕLINÕUKOGU MEETOD NEITSIOLIIVIÕLIDE ORGANOLEPTILISEKS HINDAMISEKS
1. EESMÄRK JA KOHALDAMISALA
Käesolevas lisas kirjeldatud rahvusvahelise meetodi eesmärk on kehtestada kord, kuidas hinnata neitsioliiviõlide organoleptilisi omadusi Euroopa Parlamendi ja nõukogu määruse (EL) nr 1308/2013 ( 4 ) VII lisa VIII osa punkti 1 tähenduses ning määrata kindlaks metoodika, mille järgi oliiviõlisid klassifitseeritakse osutatud omaduste põhjal. Metoodika hõlmab ühtlasi juhiseid valikuliseks märgistamiseks.
Kirjeldatud meetodit kohaldatakse ainult neitsioliiviõlide puhul, nende klassifitseerimisel või märgistamisel, võttes arvesse täheldatud puuduste intensiivsust, samuti puuviljalisust, mille on kindlaks määranud valitud, koolitatud ja testitud degustaatorite rühm (hindamiskomisjon).
Käesolevas lisas nimetatud rahvusvahelise oliivinõukogu standardite puhul kasutatakse nende viimast versiooni.
2. ORGANOLEPTILISEKS HINDAMISEKS VAJALIK ÜLDINE PÕHISÕNAVARA
Vt standard IOC/T.20/Doc. No 4 „Sensory Analysis: General Basic Vocabulary” (Organoleptiline analüüs: üldine põhisõnavara).
3. ERIALANE SÕNAVARA
3.1. Negatiivsed tunnused
Kopitanud/settene Iseloomulik maitse ja lõhn õlide puhul, mis on saadud kuhjas või ebakohastes tingimustes hoitud anaeroobse käärimise protsessis olevatest oliividest, või õlide puhul, mis on jäänud maa-alustes säilitusmahutites ja paakides settega seisma ja milles samuti on toimunud anaeroobne käärimine.
Hallitanud-niiske-mullane Iseloomulik maitse ja lõhn õlide puhul, mis on saadud oliividest, milles on arenenud palju hallitusseeni ja pärmseeni, kuna vilju on hoitud niisketes tingimustes mitu päeva, või oliividest, mis on kogutud mullase või mudasena ja mida ei ole pestud.
Veinine-äädikane/hapu-kibehapu Teatavatele õlidele iseloomulik maitse ja lõhn, mis meenutab veini või äädikat. Tingitud eelkõige oliivide või korralikult pesemata pressimismattide külge jäänud oliivimassi aeroobsest käärimisest, mille tulemusel tekivad äädikhape, etüülatsetaat ja etanool.
Räästunud Iseloomulik maitse ja lõhn õlide puhul, milles on toimunud intensiivne oksüdatsioon.
Külmavõetud oliivid (niiske puune) Puu otsas külma saanud oliividest valmistatud õlidele iseloomulik maitse ja lõhn.
3.1.1. Muud negatiivsed tunnused
|
Kuumutatud või kõrbenud |
Iseloomulik maitse ja lõhn, mille põhjuseks on ülemäärane ja/või pikaajaline kuumutamine tootmise ajal, eelkõige pasta kuumsegamine ebasobival temperatuuril. |
|
Heinane-puune |
Iseloomulik maitse ja lõhn kuivatatud oliividest toodetud õlide puhul. |
|
Tihke |
Teatavate vanade õlide puhul suus tekkiv paks, pastat meenutav puuteaisting. |
|
Määrdeainene |
Diislikütust, määret või mineraalõli meenutav maitse ja lõhn. |
|
Taim mahlane |
Iseloomulik maitse õlide puhul, mis on olnud kaua kokkupuutes viljadest pressimisel eraldunud käärinud taimemahlaga. |
|
Soolveene |
Soolvees säilitatud oliividest toodetud õlide maitse ja lõhn. |
|
Metalline |
Maitse, mis meenutab metalli. See on iseloomulik õlidele, mis on olnud purustamise, segamise, pressimise või säilitamise ajal kaua kokkupuutes metallpindadega. |
|
Espartone |
Iseloomulik maitse õli puhul, mis on saadud uutes espartomattides pressitud oliividest. Maitse võib erineda sõltuvalt sellest, kas matid on valmistatud haljast või kuivatatud espartost. |
|
Tõugumaitse |
Iseloomulik maitse õli puhul, mis on saadud oliivikärbse (Bactrocera oleae) tõukude poolt kõvasti kahjustatud oliividest. |
|
Kurgimaitse |
Maitse, mis tekib siis, kui õli on liiga kaua olnud hermeetiliselt pakendatud, eelkõige plekkpurkides, seda maitset seostatakse 2,6-nonadienaali tekkimisega. |
3.2. Positiivsed tunnused
|
Puuviljaline |
Õlile iseloomulik haistmisaistingute kogum, mis sõltub õli sordist ja tekib, kui oliivid on olnud veatud ja värsked, kas küpsed või toored. Seda tuntakse otse ja/või retronasaalselt. |
|
Mõru |
Iseloomulik esmane maitse õlide puhul, mis on valmistatud rohelistest oliividest või parajasti värvi muutvatest oliividest. Seda tuntakse keele V-piirkonnas asuvate keelenäsade kaudu. |
|
Terav |
Torkavusaisting, mis on iseloomulik viljelusaasta alguses peamiselt veel rohelistest oliividest toodetud õlidele. Seda võib tunda kogu suus ja eriti kurgus. |
3.3. Märgistamiseks vajalik valikuline terminoloogia
Nõudmise korral võib hindamiskomisjoni esimees tõendada, et hinnatud õlid vastavad mõistetele ja väljendites esitatud vahemikele ning tunnuste intensiivsuste ja nende tajumist märkivatele omadussõnadele.
Positiivsed tunnused (puuviljaline, mõru ja terav): Vastavalt tajumise intensiivsusele:
— tugev, kui tunnuse mediaan on suurem kui 6;
— keskmine, kui tunnuse mediaan on 3–6;
— õrn, kui tunnuse mediaan on väiksem kui 3.
|
Puuviljalisus |
Õlile iseloomulik haistmisaistingute kogum, mis sõltub oliivisordist ja tekib, kui oliivid on olnud veatud ja värsked, ja milles ei domineeri ei roheline ega küps puuviljalisus. Seda tuntakse otse ja/või retronasaalselt. |
|
Roheline puuviljalisus |
Õlile iseloomulik haistmisaistingute kogum, meenutab rohelisi puuvilju, sõltub oliivisordist ja tekib, kui oliivid on olnud rohelised, veatud ja värsked. Seda tuntakse otse ja/või retronasaalselt. |
|
Küps puuviljalisus |
Õlile iseloomulik haistmisaistingute kogum, meenutab küpseid puuvilju, sõltub oliivisordist ja tekib, kui oliivid on olnud veatud ja värsked. Seda tuntakse otse ja/või retronasaalselt. |
|
Hästi tasakaalus |
Õli, mis ei ole tasakaalust väljas; selle all mõistetakse haistmis-, maitse- ja puuteaistingut, mille puhul mõru tunnuse mediaan ja terava tunnuse mediaan ei ole kõrgemad kui kaks punkti üle puuviljalisuse mediaani. |
|
Mahe õli |
Õli, mille puhul mõru ja terava tunnuse mediaan on kuni 2. |
Omaduste loetelu vastavalt tajumise intensiivsusele:
|
Omadused, mille kohta antakse välja organoleptilise määramise sertifikaat. |
Tunnuse mediaan |
|
Puuviljalisus |
— |
|
Küps puuviljalisus |
— |
|
Roheline puuviljalisus |
— |
|
Õrn puuviljalisus |
Alla 3 |
|
Keskmine puuviljalisus |
Vahemikus 3–6 |
|
Tugev puuviljalisus |
Üle 6 |
|
Õrn küps puuviljalisus |
Alla 3 |
|
Keskmine küps puuviljalisus |
Vahemikus 3–6 |
|
Tugev küps puuviljalisus |
Üle 6 |
|
Õrn roheline puuviljalisus |
Alla 3 |
|
Keskmine roheline puuviljalisus |
Vahemikus 3–6 |
|
Tugev roheline puuviljalisus |
Üle 6 |
|
Õrn mõrudus |
Alla 3 |
|
Keskmine mõrudus |
Vahemikus 3–6 |
|
Tugev mõrudus |
Üle 6 |
|
Õrn teravus |
Alla 3 |
|
Keskmine teravus |
Vahemikus 3–6 |
|
Tugev teravus |
Üle 6 |
|
Hästi tasakaalus olev õli |
Mõruduse tunnuse mediaan ja teravuse tunnuse mediaan ei ole kõrgemad kui kaks punkti üle puuviljalisuse mediaani. |
|
Mahe õli |
Mõruduse tunnuse mediaan ja teravuse tunnuse mediaan ei ole kõrgemad kui kaks |
4. ÕLI DEGUSTEERIMISE KLAAS
Vt standard IOC/T.20/Doc. No. 5, „Glass for Oil Tasting” (Õli degusteerimise klaas).
5. DEGUSTEERIMISRUUM
Vt standard IOC/T.20/Doc. No. 6, „Guide for the Installation of a Test Room” (Degusteerimisruumi sisustamise juhised).
6. TÖÖVAHENDID
Selleks et degusteerija saaks oma ülesandeid nõuetekohaselt täita, peaksid igas boksis olema kergesti kättesaadavad järgmised töövahendid:
— proove sisaldavad (standarditud) klaasid, tähistatud numbrilise koodiga, kaetud kellaklaasiga, mida hoitakse temperatuuril 28 ± 2 °C;
— profiilileht (vt joonis 1) paberil; võib olla ka elektrooniline, kui profiililehe tingimused on täidetud ning juures on kasutusjuhend, kui see on vajalik;
— sulepea või kustumatu tint;
— kandikud õunaviilude ja/või veega, gaseeritud veega ja/või kuivikutega;
— klaas toatemperatuuril oleva veega;
— paberileht, millel on esitatud punktides 8.4 ja 9.1.1 loetletud üldreeglid;
— süljekausid.
7. HINDAMISKOMISJONI JUHATAJA JA DEGUSTEERIJAD
7.1. Hindamiskomisjoni juhataja
Hindamiskomisjoni juhataja peab olema piisava koolitusega ja ekspertteadmistega isik, kes tunneb väga hästi õlisid, millega ta töö käigus kokku puutuma hakkab. Ta on hindamiskomisjoni töös võtmeisik ja ta vastutab komisjoni töö korraldamise ja juhtimise eest.
Hindamiskomisjoni juhataja töö nõuab sensoorse analüüsi vahendite alast baaskoolitust, organoleptilise analüüsi vilumust, täpsust degusteerimiskatsete ettevalmistamisel, korraldamisel ja läbiviimisel ning vilumust ja kannatlikkust, et kavandada ja läbi viia katsed teaduslikul viisil.
Ta vastutab ainuisikuliselt degusteerijate valimise, koolitamise ja degustaatori võimekuse kontrolli eest. Seega vastutab ta degustaatorite heakskiitmise eest; degustaatorid peavad olema alati objektiivsed ja töötama selleks välja konkreetse korra, mis põhineb katsetel ja kindlatel heakskiitmise või tagasilükkamise kriteeriumidel. Vt standard IOC/t.20/doc. No. 14, „Guide for the selection, training and monitoring of skilled virgin olive oil tasters (Neitsioliiviõli vilunud degusteerijate valimise, koolitamise ja kontrolli juhised).
Komisjoni juhataja vastutab komisjoni töö ja seega ka hindamistegevuse eest, mille kohta ta peab esitama usaldusväärsed, objektiivsed tõendid. Igal juhul peab ta tõendama, et meetod ja degusteerijad on kontrolli all. Hindamiskomisjoni soovitatakse perioodiliselt kaliibrida (IOC/T.20/Doc. No 14, § 5).
Komisjoni juhatajal on lõplik vastutus komisjoni töö protokollimise eest. Komisjoni dokumendid peavad alati olema jälgitavad. Dokumendid peavad vastama kvaliteedi- ja selle tagamise nõuetele, mis on esitatud rahvusvahelistes organoleptilise analüüsi standardites, ja alati tagama proovide anonüümsuse.
Komisjoni juhataja vastutab seadmete ja varustuse üle arvestuse pidamise eest, samuti käesoleva meetodi nõuete täitmiseks vajalike seadmete ja varustuse nõuetekohase puhastamise ja töökorras hoidmise eest ning säilitab selle ja ka vajalike degusteerimistingimuste tagamise kohta kirjalikud tõendid.
Juhataja vastutab laborisse proovide saabumisel nende vastuvõtmise ja hoidmise eest, samuti nende hoidmise eest pärast degusteerimist. Seda tehes tagab ta alati proovide anonüümsuse ja vajaliku hoidmise ning koostab selleks kirjaliku korra, millega on tagatud kogu protsessi jälgitavus ja nõuete täitmise garanteeritus.
Lisaks sellele vastutab ta proovide ettevalmistamise, kodeerimise ja degusteerijatele esitamise eest vastavalt asjakohasele degusteerimise korraldusele, mis vastab eelnevalt koostatud töökirjeldusele, samuti degusteerijate saadud andmete kogumise ja statistilise töötlemise eest.
Juhataja vastutab kõigi muude töökorralduste ja menetluste koostamise eest, mis võivad olla vajalikud käesoleva standardi täiendamiseks ja hindamiskomisjoni nõuetekohase töö tagamiseks.
Juhataja peab otsima võimalusi hindamiskomisjoni tulemuste võrdlemiseks muude neitsioliiviõli hindamise komisjonide tulemustega, et kontrollida, kas komisjon toimib nõuetekohaselt.
Hindamiskomisjoni juhataja peab komisjoni liikmeid motiveerima, et suurendada nende huvi degusteerimise vastu, uudishimu ja konkurentsivaimu. Selleks soovitatakse tungivalt, et ta tagaks sujuva kahesuunalise teabevahetuse komisjoni liikmetega, hoiaks neid kursis teabega kõigi ülesannete kohta, mida nad täidavad, ja saadud tulemustega. Lisaks peab ta tagama, et tema enda arvamus ei oleks teada ning et võimalikud liidrid oma arvamust teistele degusteerijatele peale ei suruks.
Ta kutsub degusteerijad kohale piisavalt aegsasti ja vastab kõigile nende küsimustele katsete läbiviimise kohta, kuid ei ütle neile mingeid arvamusi proovi kohta.
7.1.1. Hindamiskomisjoni juhataja asetäitja
Hindamiskomisjoni juhatajat võib põhjendatud juhtudel asendada hindamiskomisjoni juhataja asetäitja, kes võib täita katsete tegemisega seotud kohustusi. Asetäitjal peavad olema kõik vajalikud oskused, mis on nõutavad hindamiskomisjoni juhataja puhul.
7.2. Degusteerijad
Oliiviõli organoleptilises analüüsis osalevad degusteerijad peavad tegema seda vabatahtlikult. Seepärast on soovitatav, et kandidaadid esitaksid kirjaliku avalduse. Hindamiskomisjoni juhataja valib kandidaadid, koolitab neid ja jälgib nende oskust eristada sarnaseid proove; tuleb arvestada, et eristamise täpsus suureneb koolitamisega.
Degusteerijad peavad tegutsema nagu tõelised organoleptilised vaatlejad, jätma kõrvale isiklikud maitse-eelistused ja teatama üksnes, milliseid aistinguid nad tajuvad. Selleks peavad nad alati töötama vaikuses, pingevabas õhkkonnas, kiirustamata, suunates kogu oma tähelepanu proovile, mida nad degusteerivad.
Katse tegemiseks on vaja 8–12 degusteerijat, kuid võimalike puudumiste puhuks oleks hea, kui on ka paar varudegusteerijat.
8. DEGUSTEERIMISE TINGIMUSED
8.1. Proovi esitamine degusteerimiseks
Õli esitatakse analüüsiks standarditud degusteerimisklaasides, mis vastavad standardile IOC/t.20/doc. No. 5, „Glass for oil tasting” (Õli degusteerimise klaas).
Klaas sisaldab 14–16 ml õli või 12,8–14,6 g õli, kui proovid tuleb kaaluda, ning on kaetud kellaklaasiga.
Igale klaasile on märgitud kood, mis koosneb numbritest või numbrite ja tähtede kombinatsioonist ja on valitud juhuslikult. Kood kirjutatakse klaasile meetodiga, mis ei jäta lõhna.
8.2. Degusteerimistemperatuur ja proovi temperatuur
Õli degusteerimiseks ette nähtud proovid peavad klaasides olema 28 °C ± 2 °C kogu katse vältel. See temperatuur on valitud sellepärast, et organoleptilisi erinevusi on kergem tajuda sellel temperatuuril kui toatemperatuuril; madalamal temperatuuril lenduvad õlidele omased aromaatsed ühendid halvemini, samas kui kõrgem temperatuur võib põhjustada kuumutatud õlile omaste lenduvate ühendite ilmumist. Vt standard IOC/t.20/doc. Nr 5 „Glass for Oil Tasting” (Õli degusteerimise klaas), kuidas tuleb soojendada klaasi kallatud proove.
Degusteerimisruumis peab temperatuur olema vahemikus 20o and 25o C (vt IOC/T.20/Doc. No 6).
8.3. Degusteerimise aeg
Parim aeg õli degusteerimiseks on hommik. On teada, et maitse- ja lõhnaomaduste tajumiseks on olemas optimaalselt sobivamad ajad. Maitsmis- ja/või haistmistaju on tugevam enne söömist ning nõrgem pärast söömist.
Selle kriteeriumi rakendamisega ei tohi siiski liiale minna, kuna nälg võib hajutada degusteerijate tähelepanu ja nõrgendada sellega nende eristamisvõimet, seepärast soovitatakse korraldada degusteerimisseansse hommikupoolikul kella 10 ja 12 vahel.
8.4. Üldised tegutsemiseeskirjad
Degusteerijatel tuleks oma töös järgida järgmisi soovitusi.
Kui hindamiskomisjoni juhataja kutsub degusteerijad osalema organoleptilisel määramisel, peab degusteerijal olema võimalik ettenähtud ajal kohale tulla ning ta peab silmas pidama järgmist:
— vähemalt 30 minutit enne katse toimumise aega ei tohi ta suitsetada ega kohvi juua;
— ta ei tohi olla kasutanud lõhnaõli, kosmeetikatoodet ega seepi, mille lõhn võib püsida katse tegemise ajani. Ta tohib käte pesemisel kasutada ainult lõhnastamata seepi ning ta peab käsi nii kaua loputama ja kuivatama, kuni kaob igasugune lõhn;
— ta ei tohi vähemalt 1 tund enne katse tegemist süüa;
— kui ta tunneb end füüsiliselt halvasti, eelkõige kui tema lõhna- või maitsetaju on halvenenud või kui mõni psühholoogiline mõju ei lase tal keskenduda tööle, peab degusteerija maitsmisest loobuma ja teatama hindamiskomisjoni juhatajale oma loobumisest;
— kui degusteerija puhul on eelpool kirjeldatud nõuded täidetud, võtab ta rahulikult ja vaikselt sisse endale määratud koha boksis;
— degusteerija loeb hoolikalt läbi hindamislehel olevad instruktsioonid ega alusta proovi analüüsimist enne, kui on täielikult ette valmistunud, et täita oma ülesanne rahulikult ja kiirustamata. Iga kahtluse korral peaks ta pidama eraviisiliselt nõu hindamiskomisjoni juhatajaga;
— oma ülesannete täitmisel degusteerijad vaikivad;
— degusteerija mobiiltelefon peab olema välja lülitatud, et mitte segada kolleegide keskendumist ja tööd.
9. NEITSIOLIIVIÕLI ORGANOLEPTILISE HINDAMISE JA KLASSIFITSEERIMISE KORD
9.1. Degusteerimise meetod
9.1.1. Degusteerija tõstab klaasi, hoides selle kellaklaasiga suletuna, ja kallutab seda ettevaatlikult; seejärel pöörab ta klaasi täisringi selles asendis, nii et katta siseseinast võimalikult suur osa õliga. Kui see etapp on lõpule viidud, võtab ta kellaklaasi ära ja õli hindamiseks nuusutab proovi ühtlaste sügavate hingetõmmetega. Proovi ei ole vaja nuusutada kauem kui 30 sekundit. Kui degusteerija ei jõua näitaja kohta selle aja jooksul arvamust kujundada, puhkab ta enne uut proovimist.
Kui nuusutamiskatse on lõpetatud, hindavad degusteerijad suutunnet (üldine retronasaalne haistmis-, maitsmis- ja puuteaisting). Selleks võtab ta väikse suutäie (umbes 3 ml) õli. On väga oluline, et õli kataks ühtlaselt kogu suuõõne, suu eesosast, keelest ja külgedest kuni tagaosa, suulae ja kurguni, kuna on teada, et nelja peamist maitset (magus, soolane, hapu ja mõru) ning puuteaistinguid tajutakse keele ja suulae eri osades erineva tugevusega.
Oluline on see, et piisava õlikogusega kaetaks väga aeglaselt keele pealmine pind kuni suulaega ühinemise koha ja kurguni samal ajal, kui degustaator keskendub sellele, millises järjekorras mõru ja terav maitse ilmnevad. Kui seda mitte teha, võivad mõlemad aistingud jääda mõne õli puhul märkamata või mõru aisting võib varjata terava aistingu.
Õhu läbi suu sissetõmbamine lühikeste järjestikuste hingetõmmetega võimaldab degusteerijal katta prooviga kogu suu pinna ning tajuda lenduvaid aromaatseid komponente ka nina tagaosa kaudu, kuna sellega ta sunnib end kasutama ka seda kanalit.
N.B. Kui degusteerija ei tunne proovis puuviljalisust ja klassifitseeriva negatiivse tunnuse intensiivsus on 3,5 või vähem, võib hindamiskomisjoni juhataja otsustada, et degusteerijad analüüsivad proovi uuesti toatemperatuuril (COI/T.20/Doc. No 6/Rev. 1, september 2007, punkt 3 „Üldsätted degusteerimisruumi seadmete kohta”, milles on esitatud ümbritseva keskkonnaga seotud tingimused ja toatemperatuuri mõiste.) Kui proov on saavutanud toatemperatuuri, peavad degusteerijad hindama uuesti üksnes seda, kas puuviljalisus on tuntav. Kui on, märgivad nad skaalal selle intensiivsuse.
Jälgida tuleks teravuse puuteaistingut. Selleks soovitatakse õli alla neelata.
9.1.2. Neitsioliiviõli organoleptilisel hindamisel on soovitatav, et igal hindamissessioonil hinnataks kuni nelja proovi ja et päevas võib olla kuni kolm sellist sessiooni, et vältida kontrastiefekti, mis võib tekkida, kui järjest maitstakse muid proove.
Kuna järjestikune hindamine võib eelmiste proovide kaudu tekitada väsimust või põhjustada tundlikkuse kadumist, on vaja kasutada toodet, mis kõrvaldab eelmisest degusteerimisest suus oleva õli jäägid.
Selleks soovitatakse kasutada väikest õunaviilu, mille võib pärast närimist süljekaussi sülitada. Seejärel loputatakse suud väikse koguse toatemperatuuril oleva veega. Ühe degusteerimissessiooni lõpu ja teise alguse vahel peab olema vähemalt 15 minutit.
9.2. Profiililehe kasutamine degusteerija poolt
Degusteerijale kasutamiseks ettenähtud profiilileht on esitatud käesoleva lisa joonisel 1.
Hindamiskomisjoni iga degusteerija nuusutab ja seejärel maitseb ( 5 ) hinnatavat õli. Seejärel kannab ta profiililehele iga negatiivse ja iga positiivse omaduse tema poolt tajutud intensiivsuse 10 cm skaalas, mis on näidatud esitatud profiililehel.
Kui degusteerija tajub negatiivseid omadusi, mida ei ole loetletud punktis 4, kannab ta need pealkirja „Muud” alla, kasutades termineid, mis kõige täpsemini iseloomustavad neid omadusi.
9.3. Andmete kasutamine hindamiskomisjoni juhataja poolt
Hindamiskomisjoni juhataja kogub kokku kõigi degusteerijate täidetud profiililehed ja kontrollib eri tunnustele omistatud intensiivsusastmeid. Kui ta leiab midagi ebanormaalset, palub ta degusteerijal oma profiilileht üle vaadata ja hindamist korrata, kui see on vajalik.
Hindamiskomisjoni juhataja sisestab iga komisjoniliikme hindamisandmed arvutiprogrammi, mis vastab ettenähtud standardile IOC/t.20/Doc. nr 15, et tulemusi statistiliselt töödelda, mille aluseks on tulemuste mediaani arvutamine. Vt punkt 9.4 ja käesoleva lisa liide. Proovi andmed kantakse maatriksisse, millel on üheksa veergu, mis vastavad üheksale organoleptilisele tunnusele, ja n rida, mis vastavad komisjoni liikmete arvule n.
Kui on märgatud puudust ja vähemalt 50 % komisjoniliikmetest on kandnud selle pealkirja „Muud” alla, arvutab hindamiskomisjoni juhataja puuduse mediaani ja leiab selle alusel klassifikatsiooni.
Robustse variatsioonikordaja väärtus, mille alusel määratakse klassifikatsioon (suurima intensiivsusega puudus ja puuviljalisuse tunnus), ei tohi olla üle 20 %.
Vastupidisel juhul peab hindamiskomisjoni juhataja kordama konkreetsete proovide hindamist muul degusteerimissessioonil.
Kui selline olukord tekib sageli, soovitatakse hindamiskomisjoni juhatajal korraldada degusteerijatele täiendav koolitus selle kohta (IOC/T.20/Doc. nr 14, § 5) ning hinnata komisjoni tööd korratavusnäitaja ja kõrvalekaldenäitaja alusel (IOC/T.20/Doc. nr 14, § 6).
9.4. Õli klassifitseerimine
Õli klassifitseeritakse puuduste mediaani ja puuviljalisuse mediaani põhjal järgnevalt esitatud kategooriatesse. Puuduste mediaani käsitatakse suurima intensiivsusega tajutud puuduse mediaanina. Puuduste mediaan ja puuviljalisuse mediaan väljendatakse ühe kümnendkoha täpsusega.
Õli klassifitseerimisel võrreldakse puuduste mediaani ja puuviljalisuse mediaani väärtust järgmiste osutatud etalonvahemike suhtes. Kõnealuste vahemike piirid on kindlaks määratud meetodi viga arvesse võttes ning neid loetakse seepärast absoluutseteks. Arvutitarkvara võimaldab klassifikatsiooni esitada statistikaandmete tabelina või graafiliselt.
a) Ekstra-neitsioliiviõli: puuduste mediaan on 0 ja puuviljalisuse mediaan on suurem kui 0.
b) Neitsioliiviõli: puuduste mediaan on suurem kui 0, kuid mitte üle 3,5, ja puuviljalisuse mediaan on suurem kui 0.
c) Lambiõli: puuduste mediaan on üle 3,5 või puuduste mediaan on kuni 3,5 ning puuviljalisuse mediaan on 0.
Märkus 1. Kui mõruduse ja/või teravuse mediaan on suurem kui 5,0, märgib hindamiskomisjoni juhataja seda degusteerimise kohta antavas sertifikaadis.
Vastavuse kontrollimiseks tehtud hindamiste puhul tehakse proov. Vasturääkivustega hindamiste puhul tuleb analüüse hinnata kaks korda erinevate hindamisseansside jooksul. Korduvanalüüsi tulemused peavad olema statistiliselt homogeensed. (Vt punkt 9.5.) Kui see nii ei ole, tuleb proovi uuesti kaks korda analüüsida. Tunnuste mediaani lõplik väärtus arvutatakse mõlema mediaani keskmise alusel.
9.5. Korduvanalüüside heakskiitmise või tagasilükkamise kriteeriumid.
Allpool määratletud normeeritud viga kasutatakse selleks, et määrata kindlaks, kas korduvanalüüsi kaks tulemust on homogeensed või statistiliselt vastuvõetavad:
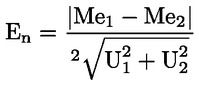
Kui Me1 ja Me2 on kahe korduvanalüüsi mediaanid (esimene ja teine analüüs) ning U1 ja U2 on liite kohaselt arvutatud kahe väärtuse laiendatud määramatused:
U
1 = c × s * and

Laiendatud määramatuse puhul c = 1,96; järelikult
U1 = 0,0196 × CVr × Me1
kus CVr robustse variatsioonikordaja väärtus.
Tuleb märkida, et kaks saadud väärtust ei ole statistiliselt erinevad, En peab olema väiksem kui 1,0 või sellega võrdne.
Liide
Mediaani ja usaldusvahemike arvutamise meetod
Mediaan
![]()
Mediaan on määratletud kui reaalarv Xm, mille puhul tõenäosus p, et jaotuse (X) väärtused on väiksemad kui Xm või sellega võrdsed, on väiksem kui 0,5 või sellega võrdne, ja samal ajal tõenäosus p, et jaotuse (X) väärtused on suuremad kui Xm või sellega võrdsed, on suurem kui 0,5 või sellega võrdne. Praktilisem määratlus on selline, et mediaan on suuruse järgi reastatud arvude 50-protsentiil. Lihtsamini öeldes on see paarituarvulise liikmete arvuga arvujada puhul jada keskmine arv ja paarisarvulise liikmete arvuga arvujada puhul kahe keskmise arvu keskmine.
Robustne standardhälve
Selleks, et usaldusväärselt hinnata varieeruvust keskväärtuse ümbruses, on vaja kasutada robustset standardhälvet, mis hinnatakse Stuarti ja Kendalli (4) järgi. Võrrandiga saab leida asümptootilise robustse standardhälbe, s.t vaadeldavate andmete varieeruvuse lihtsa hinnangulise väärtuse, milles N on vaatluste arv ja IQR on kvartiilhaare, mis hõlmab täpselt 50 % mingi tõenäosusliku jaotuse juhtudest:

Kvartiilhaarde leidmiseks arvutatakse 75-protsentiili ja 25-protsentiili vahe suurus.
![]()
.
Siin on protsentiil määratletud kui väärtus Xpc, mille puhul tõenäosus p, et jaotuse väärtused on väiksemad kui Xpc või sellega võrdsed, on mingi väiksem kui mingi konkreetne protsent või sellega võrdne, ja et samal ajal tõenäosus p, et jaotuse (X) väärtused on suuremad kui Xpc või sellega võrdsed, on suurem kui see protsent või sellega võrdne. Protsendimäär näitab jaotuse hõlmatud osa. Mediaani puhul on see võrdne 50/100-ga
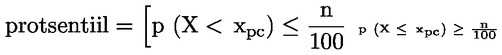
Praktikas on protsentiil jaotusväärtus, mis määrab kindlaks vastava suurusega osa jaotus-või tiheduskõveraga piiratud pindalast. Näiteks 25-protsentiil on jaotusväärtus, mis määrab kindlaks 0,25 või 25/100 sellest alast.
Selle meetodi puhul arvutatakse protsentiilid tegelike väärtuste alusel, mis on esitatud andmete maatriksis (protsentiilide arvutamine).
Robustne variatsioonikordaja (%)
CVr% on dimensioonita arv, mis näitab analüüsitava arvjada muutuvuse protsenti. Seepärast on kõnealune koefitsient väga kasulik hindamiskomisjoni liikmete usaldusväärsuse kontrollimiseks.

Mediaani 95 %-line usaldusvahemik
95 %-line usaldusvahemik (esimest liiki vea väärtus on 0,05 või 5 %) on vahemik, milles mediaan võiks varieeruda, kui katset oleks võimalik lõpmatult korrata. Tegelikkuses näitab see, millises vahemikus katse tulemus varieerub kindlaksmääratud katsetingimuste puhul, kui eeldada, et katset saab korrata palju kordi. Nagu CVr%, aitab ka see vahemik hinnata katse usaldusväärsust.


kus C = 1,96 usaldusvahemiku puhul 95 %-lise usaldusnivoo juures.
I lisas on esitatud näide, milline on arvutustabel vastavalt standardile IOC/t 20/doc. No 15.
Kasutatud kirjandus
(1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.
(2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
(3) Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam
(4) Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
(5) McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
(6) IOC/T.28/Doc. No 1 September 2007, Guidelines for the accreditation of sensory testing laboratories with particular reference to virgin olive oil according to standard ISO/IEC 17025:2005.
(7) IOC/T.20/Doc. No 14.
(8) IOC/T.20/Doc. No 15.
(9) ISO/IEC 17025:05.
▼M20 —————
▼M19 —————
XV LISA
1. OLIIVIÕLI PRESSIMISJÄÄKIDE ÕLISISALDUS
1.1. Seadmed
— nõuetekohane ekstraheerimisaparaat, milles on 200–250 ml ümarapõhjalised kolvid,
— elektriline vann (näiteks liivavann, veevann) või pliit,
— analüütilised kaalud,
— ahi, mida on võimalik reguleerida kuni 80 kraadini,
— elektriahi, milles on 103 ± 2 kraadini reguleeritav termostaat ja kuhu on võimalik suunata õhuvool või millega on võimalik töötada alandatud rõhu all,
— mehaaniline veski, mida on lihtne puhastada ja mis võimaldab oliiviõli pressimisjääke jahvatada ilma, et nende temperatuur tõuseks või nende niiskuse, lenduvate ainete või heksaaniga ekstraheeruvate ainete sisaldus märkimisväärselt muutuks,
— ekstraheerimishülss ja vatt või filterpaber, millest on heksaaniga ekstraheeruvad ained juba eemaldatud,
— eksikaator,
— sõel, mille avade diameeter on 1 mm,
— enne kuivatatud pimsskivi väikesed terakesed.
1.2. Reaktiiv
Tehniline n-heksaan, millest jääb täielikul aurustamisel 100 ml kohta kuni 0,002 g suurune jääk.
2. TÖÖ KÄIK
2.1. Uuritava proovi ettevalmistamine
Selleks et laboriproovi osakesed mahuksid täielikult läbi sõela, kasutatakse proovi jahvatamiseks vajaduse korral enne korralikult puhastatud mehaanilist veskit.
1/20 proovist kasutatakse veski puhastamiseks, jahvatamisel tekkiv aine visatakse ära, jahvatatakse ülejäänud osa proovist ning seejärel kogutakse see kokku, segatakse hoolikalt ning analüüsitakse kohe.
2.2. Katsekogus
Pärast jahvatamist kaalutakse katse tegemiseks 10 g proovi 0,01 g täpsusega.
2.3. Ekstraheerimishülsi ettevalmistamine
Katsekogus asetatakse hülssi ning suletakse vatitropiga. Filterpaberi kasutamise korral tuleb katsekogus sellesse keerata.
2.4. Eelnev kuivatamine
Kui oliiviõli pressimisjäägid on väga niisked (näiteks kui niiskusesisaldus ja lenduvate ainete sisaldus on üle 10 %), kuivatatakse nad enne, et niiskuse- ja lenduvate ainete sisaldust vähendada, milleks asetatakse laetud hülss (või filterpaber) ettenähtud aja jooksul kuni 80 oC kraadini kuumutatud ahju.
2.5. Ümarapõhjalise kolvi ettevalmistamine
Kolb, milles on üks või kaks ahjus 103 ± 2 oC juures enne kuivatatud pimsskivitera, kaalutakse 1 mg täpsusega ning seda jahutatakse eksikaatoris vähemalt üks tund.
2.6. Esimene ekstraheerimine
Ekstraheerimisaparaati pannakse hülss (või filterpaber), milles on katsekogus. Kolbi valatakse vajalik kogus heksaani. Kolb pannakse ekstraheerimisaparaati ning ekstraheerimisaparaat asetatakse elektriga kuumutatavasse vanni. Kuumutamise kiirust kohandatakse selliselt, et tagasivoolu kiirus oleks vähemalt kolm tilka sekundis (mõõdukas, mitte tugev keemine). Pärast neljatunnist ekstraheerimist lastakse jahtuda. Hülss võetakse ekstraheerimisaparaadist ning asetatakse õhuvoolu, selleks et kõrvaldada suurem osa immutuslahustist.
2.7. Teine ekstraheerimine
Hülsi sisu pannakse mikrojahvatamisaparaati ja see jahvatatakse võimalikult peeneks. Jahvatatud segu pannakse kohe hülssi tagasi ning hülss pannakse tagasi ekstraheerimisaparaati.
Ekstraheerimist jätkatakse täiendavalt kahe tunni jooksul, kasutades sedasama ümarapõhjalist kolbi, milles on esialgne ekstrakt.
Ekstraheerimiskolbis tekkiv lahus peab olema selge. Kui lahus ei ole selge, tuleb seda läbi filterpaberi filtreerida ning pesta algset kolbi ja filterpaberit mitu korda heksaaniga. Filtraat ja pesulahus kogutakse teise ümarapõhjalisse kolvi, mis on kuivatatud ning tareeritud 1 mg täpsusega.
2.8. Lahusti eemaldamine ja ekstrakti kaalumine
Suurem osa lahustist eemaldatakse elektrilisel vannil destilleerimisega. Viimased lahustijäägid eemaldatakse kolvi kuumutamisel 20 minutit ahjus temperatuuril 103 ± 2 oC. Eemaldamisel kasutatakse abivahendina teatud ajavahemike järel õhuvoolu, inertgaasi või alandatud rõhku.
Kolb jäetakse vähemalt üheks tunniks eksikaatorisse jahtuma ning kaalutakse 1 mg täpsusega.
Kuumutatakse uuesti 10 minutit samadel tingimustel, jahutatakse eksikaatoris ning kaalutakse uuesti.
Kahe kaalumistulemuse erinevus ei tohi olla suurem kui 10 mg. Kui erinevus on suurem, kuumutatakse kolbi 10 minutit, jahutatakse see ja kaalutakse ning seda korratakse seni, kuni erinevus on 10 mg või väiksem. Kolvi viimane kaal märgitakse üles.
Tehakse uuritava proovi dubleeriv määramine.
3. TULEMUSTE VÄLJENDAMINE
3.1. Arvutamismeetod ja valem
a) Ekstrakt saadud toote massiprotsendina on:
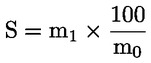
|
kus: |
S on saadud toote ekstrakti massiprotsent; mo on katsekoguse mass grammides; m1 on kuiva ekstrakti mass grammides. |
Tulemuseks loetakse kahe määramise aritmeetiline keskmine tingimusel, et korratavuse tingimused on täidetud.
Tulemus esitatakse ühe kümnendkoha täpsusega.
b) Ekstrakt on väljendatud kuivainena järgmise valemi alusel:

|
kus: |
: |
S = on saadud toote ekstrakti protsent (vt punkt a), U = on selle niiskuse- ja lenduvate ainete sisaldus. |
3.2. Korratavus
Ühel ja samal ajal või kohe üksteise järel sama analüüsija poolt tehtud kahe määramise tulemuste erinevus ei tohi olla suurem kui 0,2 g heksaaniekstrakti 100 g proovi kohta.
Kui see tingimus ei ole täidetud, korratakse analüüsi kahe teise katsekogusega. Kui ka nende puhul ületab erinevus 0,2 g, loetakse tulemuseks nelja määramise aritmeetiline keskmine.
XVI LISA
JOODIARVU MÄÄRAMINE
1. KOHALDAMISALA
Käesolev rahvusvaheline standard täpsustab loomsete ja taimsete rasvade ja õlide (edaspidi rasvade) joodiarvu määramise meetodi.
2. MÄÄRATLUS
Käesolevas rahvusvahelises standardis kasutatakse järgmist mõistet:
|
2.1. |
joodiarv. Joodiarv on proovis neeldunud joodi mass käesolevas rahvusvahelises standardis täpsustatud töötingimustel. Joodiarvu väljendatakse joodi grammides 100 g proovi kohta. |
3. PÕHIMÕTE
Katsekogus lahustatakse lahustis ja sellele lisatakse wijsi reaktiiv. pärast kindlaksmääratud aega lisatakse kaaliumjodiidi lahus ja vesi ning eraldunud jood tiitritakse naatriumtiosulfaadi lahusega.
4. REAKTIIVID
Kõik reaktiivid peavad olema analüütiliselt puhtad:
|
4.1. |
vesi, mis vastab 3. klassi nõuetele vastavalt ISO 3696 standardile. |
|
4.2. |
kaaliumjodiid, 100 g/l lahus, mis ei sisalda jodaati ega vaba joodi. |
|
4.3. |
tärklis, lahus. 5 g lahustuvat tärklist segatakse 30 ml vees, seejärel lisatakse see segu 1 000 ml keevale veele, keedetakse 3 minutit ning lastakse jahtuda. |
|
4.4. |
naatriumtiosulfaat, standardne tiitrimislahus c (Na2S2O3.5H2O) = 0,1 mol/l, standarditud kuni 7 päeva enne kasutamist. |
|
4.5. |
lahusti, valmistatud võrdses mahus tsükloheksaanist ja äädikhappest. |
|
4.6. |
Wijsi reaktiiv, mis sisaldab joodmonokloriidi äädikhappes. Kasutatakse müügilolevat Wijsi reaktiivi. |
5. SEADMED
Tavalised, eelkõige järgmised laboratooriumiseadmed:
|
5.1. |
klaasist kaalumiskühvlid, mis sobivad katsekoguse kaalumiseks ja kolbidesse (6.2) kandmiseks. |
|
5.2. |
500 ml Erlenmeyeri kolvid, lihvkorgiga ja täiesti kuivad . |
6. UURITAVA PROOVI ETTEVALMISTAMINE
Homogeniseeritud proov kuivatatakse naatriumsulfaadi kohal ja filtreeritakse.
7. TÖÖ KÄIK
|
7.1. |
Katsekogus Katsekoguse mass on tabelis 1 esitatud hinnanguliste joodiarvude puhul erinev.
Tabel 1
Katsekogus kaalutakse klaasist kaalumiskühvlis (5.1) 0,1 mg täpsusega. |
|
7.2. |
Määramine Katsekogus asetatakse 500milliliitrisesse kolbi (5.2). Rasva lahustamiseks lisatakse 20 ml lahustit (4.5). Lisatakse täpselt 25 ml Wijsi reaktiivi (4.6), suletakse punniga, segatakse ning seejärel asetatakse kolb pimedasse. Wijsi reaktiivi ei tohi pipeteerida suuga. Samal viisi valmistatakse lahusti ja reaktiiviga pimelahus, kuhu ei lisata katsekogust. Proovide puhul, mille joodiarv on alla 150, jäetakse kolvid pimedasse üheks tunniks; proovide puhul, mille joodiarv on üle 150, ja polümeriseeritud toodete või teatud määrani oksüdeeritud toodete puhul jäetakse kolvid pimedasse kaheks tunniks. Nimetatud aja möödudes lisatakse mõlemasse kolbi 20 ml kaaliumjodiidi lahust (4.2) ja 150 ml vett (4.1). Seejärel tiitritakse standardse naatriumtiosulfaadi tiitrimislahusega (4.4) kuni joodist tulenev kollane värv on peaaegu kadunud. Lisatakse paar tilka tärkliselahust (4.3) ja jätkatakse tiitrimist kuni sinine värv pärast väga tugevat loksutamist ära kaob. Märkus: Lubatud on lõpp-punkti potentsiomeetriline määramine. |
|
7.3. |
Määramiste arv Ühe ja sama prooviga tehakse 2 määramist. |
8. TULEMUSTE VÄLJENDAMINE
Joodiarv arvutatakse järgmise valemiga:

kus
|
c |
= |
on kasutatud standardse naatriumtiosulfaadi tiitrimislahuse (4.4) täpse kontsentratsiooni arvväärtus moolides liitri kohta; |
|
V1 |
= |
on pimekatses kasutatud standardse naatriumtiosulfaadi tiitrimislahuse (4.4) mahu arvväärtus milliliitrites; |
|
V2 |
= |
on määramisel kasutatud standardse naatriumtiosulfaadi tiitrimislahuse (4.4) mahu arvväärtus milliliitrites; |
|
m |
= |
on katsekoguse (7.1) massi arvväärtus grammides. |
Tulemuseks loetakse kahe määramise aritmeetiline keskmine tingimusel, et korratavuse nõue on täidetud.
XVII LISA
MEETOD STIGMASTADIEENIDE MÄÄRAMISEKS TAIMEÕLIDES
1. EESMÄRK
Stigmastadieenide määramine taimeõlides, mis sisaldavad neid süsivesinikke väikeses kontsentratsioonis, eeskätt esmases oliiviõlis ja oliivijääkidest saadud toorõlis.
2. RAKENDUSALA
Käesolevat standardmeetodit võib rakendada kõigi taimeõlide analüüsimisel, kuid mõõtmistulemused on usaldatavad ainult juhul, kui stigmastadieenide sisaldus on 0,01-4,0 mg/kg. Kuna rafineeritud õlid sisaldavad stigmastadieene, esmased õlid aga neid ei sisalda, sobib see meetod eriti hästi rafineeritud taimeõlide (oliiviõli, oliivijääkidest saadud õli, päevalilleõli, palmiõli jms) kindlakstegemiseks esmases oliiviõlis.
3. PÕHIMÕTE
Isoleeritakse mitteseebistuvad ained. Silikageel-kolonnkromatograafia abil eraldatakse steroidsete süsivesinike fraktsioon ning analüüsitakse seda kapillaargaasikromatograafiliselt.
4. SEADMED
|
4.1. |
250 ml kolvid, mida saab kasutada püstjahutiga. |
|
4.2. |
500 ml jaotuslehtrid. |
|
4.3. |
100 ml ümarkolvid. |
|
4.4. |
Pöördauruti. |
|
4.5. |
Klaasist kromatograafiakolonn (sisediameeter 1,5-2,0 cm; pikkus 50 cm), millel on teflonkraan ja põhjas klaasvillakork või klaasfilter. Silikageelkolonni ettevalmistamiseks valatakse kromatograafiakolonni heksaani umbes 5 cm kõrguseni ja seejärel täidetakse kolonn silikageeli suspensiooniga heksaanis (15 g silikageeli 40 ml heksaani kohta), kandes seda kolonni väikeste heksaaniportsjonite abil. Suspensioonil lastakse settida, rakendades settimise lõpus kerget vibratsiooni. Kolonni lisatakse umbes 0,5 cm paksune kiht veevaba naatriumsulfaati ja lastakse liigsel heksaanil välja voolata. |
|
4.6. |
Gaasikromatograaf, mis on varustatud leekionisatsioonidetektoriga, voolujaotusega või kolonnile kinnitatud külma injektoriga ning täpsusega ± 1 °C programmeeritava ahjuga. |
|
4.7. |
Sulatatud ränidioksiidist gaasikromatograafiline kapillaarkolonn (sisediameeter 0,25 või 0,32 mm, pikkus 25 m, kaetud 0,25 μm paksuse 5 % fenüülmetüülsilikoonfaasi kihiga). Märkus 1: Võib kasutada ka muid samasuguse või väiksema polaarsusega kolonne. |
|
4.8. |
Integreeriv isekirjuti, mis võimaldab integreerida miinimumist miinimumini. |
|
4.9. |
Liimitud nõelaga 5-10 μl gaasikromatograafiline mikrosüstal. |
|
4.10. |
Elektriline soojendussärk või termokamber. |
5. REAKTIIVID
Kui ei ole ette nähtud teisiti, peavad kõik reaktiivid olema analüüsipuhtad. Kasutatakse destilleeritud või vähemalt samaväärse puhtusastmega vett.
|
5.1. |
Heksaan või alkaanide segu keemisvahemikuga 65-70° C, destilleeritud läbi rektifitseerimiskolonni. Märkus 2: Solvent destilleeritakse lisandite eemaldamiseks. |
|
5.2. |
Etanool mahuprotsendiga 96. |
|
5.3. |
Veevaba naatriumsulfaat. |
|
5.4. |
10 % kaaliumhüdroksiidi lahus alkoholis. 50 g kaaliumhüdroksiidile lisatakse 10 ml vett, segatakse ja täiendatakse segu maht etanooliga 500 milliliitrini. Märkus 3: Kaaliumhüdroksiidi alkoholilahus muutub seismisel pruuniks. See valmistatakse iga päeva uuesti ja seda hoitakse hästi suletud tumedas pudelis. |
|
5.5. |
Gaasikromatograafiline silikageel 60, 70-230 mešši (Merck, Art. Nr. 7734 vms). Märkus 4: Tavaliselt võib pakendist võetud silikageeli kasutada eelneva töötlemiseta. Mõned silikageeli partiid on aga madala aktiivsusega, mis põhjustab halba kromatograafilist lahutamist. Sellist silikageeli tuleb töödelda järgmiselt. Silikageel aktiveeritakse, kuumutades vähemalt 4 tundi 550 °C juures. Seejärel jahutatakse silikageel eksikaatoris ja pannakse korgiga suletavasse kolbi. Lisatakse 2 % vett ja raputatakse, kuni klombid pudenevad laiali ja pulber voolab vabalt. Selline töötlemine on vajalik juhul, kui teatava silikageeli partii kasutamisel saadakse halvasti eraldunud piigid. Teine võimalus on kasutada eriti puhast silikageeli 60 (Merck, Art. Nr. 7754). |
|
5.6. |
Kolesta-3, 5-dieeni (Sigma, puhtusaste 99 %) põhilahus heksaanis kontsentratsiooniga 200 miljondikku (10 mg kolesta-3, 5-dieeni 50 ml-s lahuses). |
|
5.7. |
Kolesta-3, 5-dieeni standardlahus heksaanis kontsentratsiooniga 20 miljondikku; saadakse põhilahuse lahjendamisel. Märkus 5: Kui lahuseid 5.6 ja 5.7 säilitatakse madalamal temperatuuril kui 4 °C, püsivad need stabiilsena vähemalt 4 kuud. |
|
5.8. |
n-nonakosaani lahus heksaanis kontsentratsiooniga ligikaudu 100 miljondikku. |
|
5.9. |
Kromatograafiline kandegaas: heelium või vesinik, puhtusaste 99,9990 %. |
|
5.10. |
Leekionisatsioonidetektori töögaasid: 99,9990 % puhtusastmega vesinik ja puhastatud õhk. |
6. ANALÜÜSI KÄIK
6.1. Mitteseebistuva materjali ettevalmistamine
|
6.1.1. |
250 ml kolvis (4.1) kaalutakse 20 ± 0, 1 g õli, lisatakse 1 ml kolesta-3, 5-dieeni standardlahust (20 μg kolesta-3, 5-dieeni) ja 75 ml 10 % kaaliumhüdroksiidi alkoholilahust, kolb ühendatakse püstjahutiga ja keedetakse segu nõrgalt 30 minutit. Seejärel lastakse kolvil pisut jahtuda (palju jahutada ei tohi, sest proov võib sadeneda). Lisatakse 100 ml vett ja kantakse lahus 100 ml heksaani abil jaotuslehtrisse (4.2). Segu loksutatakse energiliselt 30 sekundit ja lastakse faasidel eralduda. Märkus 6: Kui tekib kaua püsiv emulsioon, lisatakse väike kogus etanooli. |
|
6.1.2. |
Alla kogunenud veefaas kantakse teise jaotuslehtrisse ja ekstraheeritakse veel kord 100 ml heksaaniga. Alumine faas lastakse uuesti välja, valatakse heksaaniekstraktid kokku eraldi jaotuslehtrisse ning pestakse kolm korda 100 ml etanooli ja vee seguga (1:1), nii et pH muutub neutraalseks. |
|
6.1.3. |
Heksaanilahus filtritakse läbi 50 g veevaba naatriumsulfaadi, pestakse 20 ml heksaaniga ja aurutatakse heksaan välja pöördaurutis alandatud rõhul 30 °C juures. |
6.2. Steroidsete süsivesinike fraktsiooni eraldamine
|
6.2.1. |
Jääk kantakse kahe ühemilliliitrise heksaaniportsjoni abil fraktsioneerimiskolonni, lastakse proovil voolata kolonni, kuni lahuse tase on alanenud naatriumsulfaadikihi ülemise servani, ja alustatakse kromatograafilist aeglast elueerimist heksaaniga kiirusega ligikaudu 1 ml/min. Esimesed 25-30 ml eluaati visatakse ära ja kogutakse seejärel 40 ml fraktsioon. See fraktsioon valatakse 100 ml ümarkolbi (4.3). Märkus 7: Esimene fraktsioon sisaldab küllastunud süsivesinikke (joonis 1a) ja teine steroidseid süsivesinikke. Edasisel elueerimisel väljuvad skvaleen ja selle analoogid. Küllastunud süsivesinike ja steroidsete süsivesinike hea lahknemise saavutamiseks on vaja optimeerida fraktsioonide mahud. Selleks tuleb esimese fraktsiooni maht valida nii, et teise fraktsiooni analüüsimisel osutuksid küllastunud süsivesinikele vastavad piigid madalaks (vt joonis 1c); kui need piigid ei ilmne üldse ja standardpiik on madal, tuleb esimese fraktsiooni mahtu vähendada. Esimese ja teise fraktsiooni komponendid ei pea olema siiski täielikult lahknenud, sest kui gaasikromatograafilise analüüsi tingimused on välja reguleeritud 6.3.1 kohaselt, siis gaasikromatograafilise analüüsi piigid ei kattu. Teise fraktsiooni mahu optimeerimine ei ole üldiselt vajalik, sest lahknemine hiljem väljuvatest komponentidest on hea. Kui siiski ilmub suur piik, mille peetumisaeg on ligikaudu 1,5 minutit väiksem kui standardi peetumisaeg, näitab see halba lahutamist. |
|
6.2.2. |
Teisest fraktsioonist aurutatakse heksaan välja pöördaurutis alandatud rõhul 30 °C juures ja jääk lahustatakse kohe 0,2 ml heksaanis. Lahust hoitakse kuni analüüsini külmikus. Märkus 8: Jääke 6.1.3 ja 6.2.2 ei tohi hoida toatemperatuuril kuivalt. Niipea kui need jäägid saadakse, tuleb neile lisada solvent ning säilitada lahust külmikus. |
6.3. Gaasikromatograafia
|
6.3.1. |
Töötingimused voolujaotusega injektsiooni korral: — injektori temperatuur: 300 °C, — detektori temperatuur: 320 °C, — integreeriv isekirjuti: integreerimise parameetrid tuleb kindlaks määrata nii, et pindalade hindamine annaks õigeid tulemusi. Soovitav on kasutada miinimumist miinimumini integreerimise meetodit, — tundlikkus: 16-kordne miinimumkustumine, — süstitav kogus: 1 μl, — ahju temperatuuri programmeerimine: esimese 6 minuti jooksul 235 °C, seejärel tõstetakse kiirusega 2 °C/min väärtuseni 285 °C, — injektor: voolujaotusega 1:15, — kandegaas: heelium või vesinik umbes 120 kPa rõhul. Neid tingimusi võib reguleerida vastavalt kromatograafi ja kolonni karakteristikutele, nii et kromatogrammid vastaksid järgmistele tingimustele: sisestandardi piigi peetumisaeg peab vastama punktis 6.3.2 märgitud ajale umbes 5 minuti täpsusega; sisestandardi piigi kõrgus peab olema vähemalt 80 % kogu skaala kõrgusest. Gaasikromatograafilise süsteemi kontrollimiseks süstitakse kolestadieeni (5.6) ja n-nonakosaani (5.8) põhilahuste segu. Kolesta-3, 5-dieeni piik peab ilmuma enne n-nonakosaani piiki (joonis 1c); kui see ei toimu, võib kasutada kahte abinõu: alandada ahju temperatuuri ja/või kasutada vähem polaarset kolonni. |
|
6.3.2. |
Piikide identifitseerimine Sisestandardi piik ilmub umbes 19. minutil; 3,5-stigmastadieeni suhteline peetumisaeg on umbes 1,29 (vt joonis 1b). 3,5-stigmastadieen esineb koos väikese isomeerilisandiga ja need elueeruvad tavaliselt koos, andes ühe kromatograafilise piigi. Juhul kui kolonn on liiga polaarne või kõrge lahutusvõimega, võib isomeer siiski väljuda väikese piigina enne stigmasta-3, 5-dieeni selle lähedal (vt joonis 2). Selle tagamiseks, et stigmastadieenid elueeruksid ühe piigina, on soovitav kasutada kas väiksema polaarsusega või suurema sisediameetriga kolonni. Märkus 9: Võrdlemiseks võib stigmastadieenide piigi saada rafineeritud taimeõli analüüsi teel, kasutades väiksemat proovi kogust (1-2 g). Stigmastadieenid annavad silmatorkava kergesti äratuntava piigi. |
|
6.3.3. |
Kvantitatiivne analüüs Stigmastadieenide sisaldus määratakse järgmise valemi abil: stigmastadieenid (mg/kg) =
Meetodi piirtundlikkus: ligikaudu 0,01 mg/kg. |
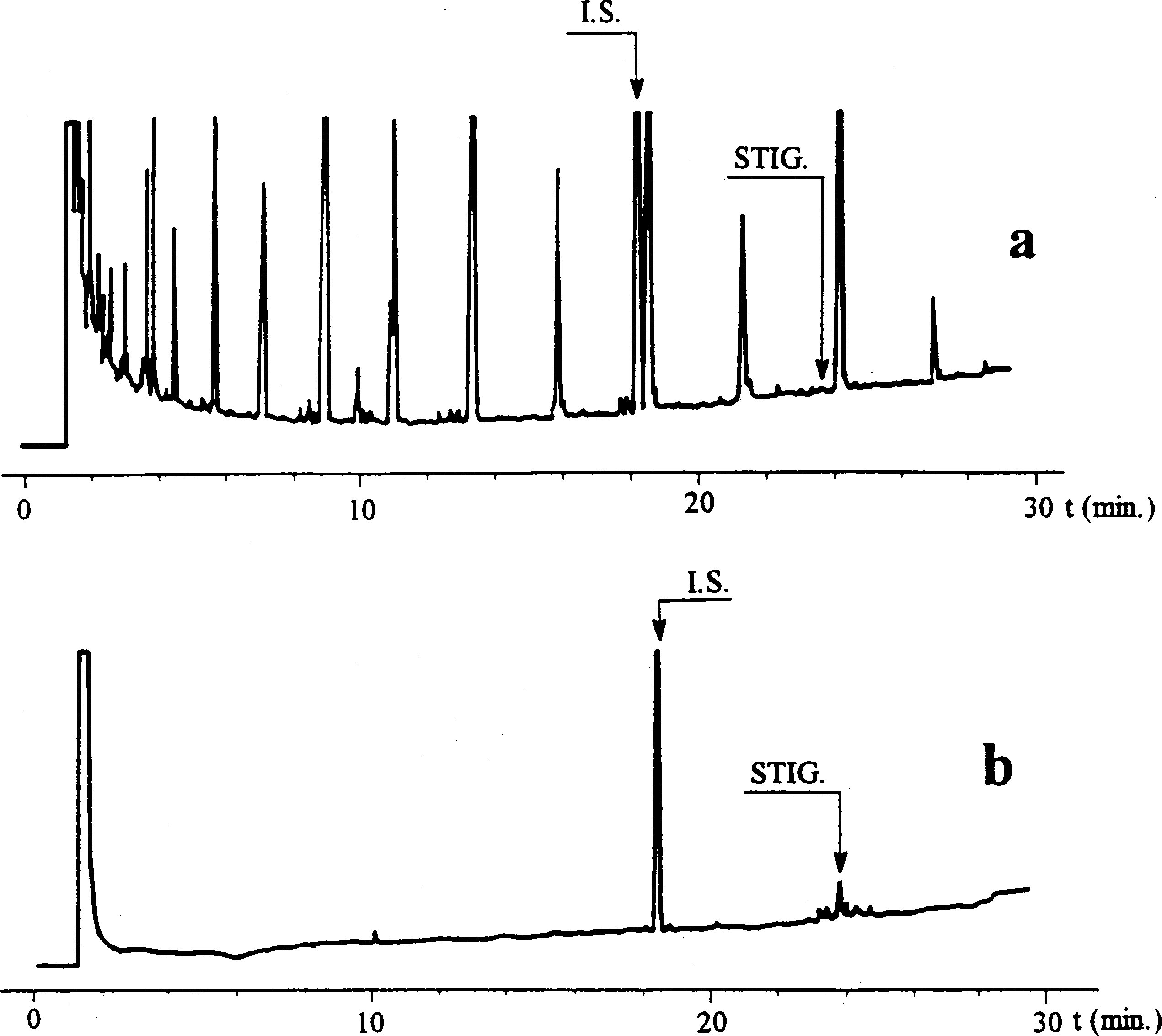
 Joonis 1.
Joonis 1.
Gaasikromatogrammid, mis on saadud katsetes oliiviõli proovidega sulatatud ränidioksiidist kapillaarkolonnis (sisediameeter 0,25 mm, pikkus 25 m, kaetud 0,25 μm paksuse 5 % fenüülmetüülsilikooni kihiga).
a) Esmase õli esimene fraktsioon (30 ml), millele on lisatud standardit.
b) Oliiviõli teine fraktsioon (40 ml), mis sisaldab 0,10 mg/kg stigmastadieene.
c) Teine fraktsioon (40 ml), mis sisaldab väikese koguse esimest fraktsiooni.
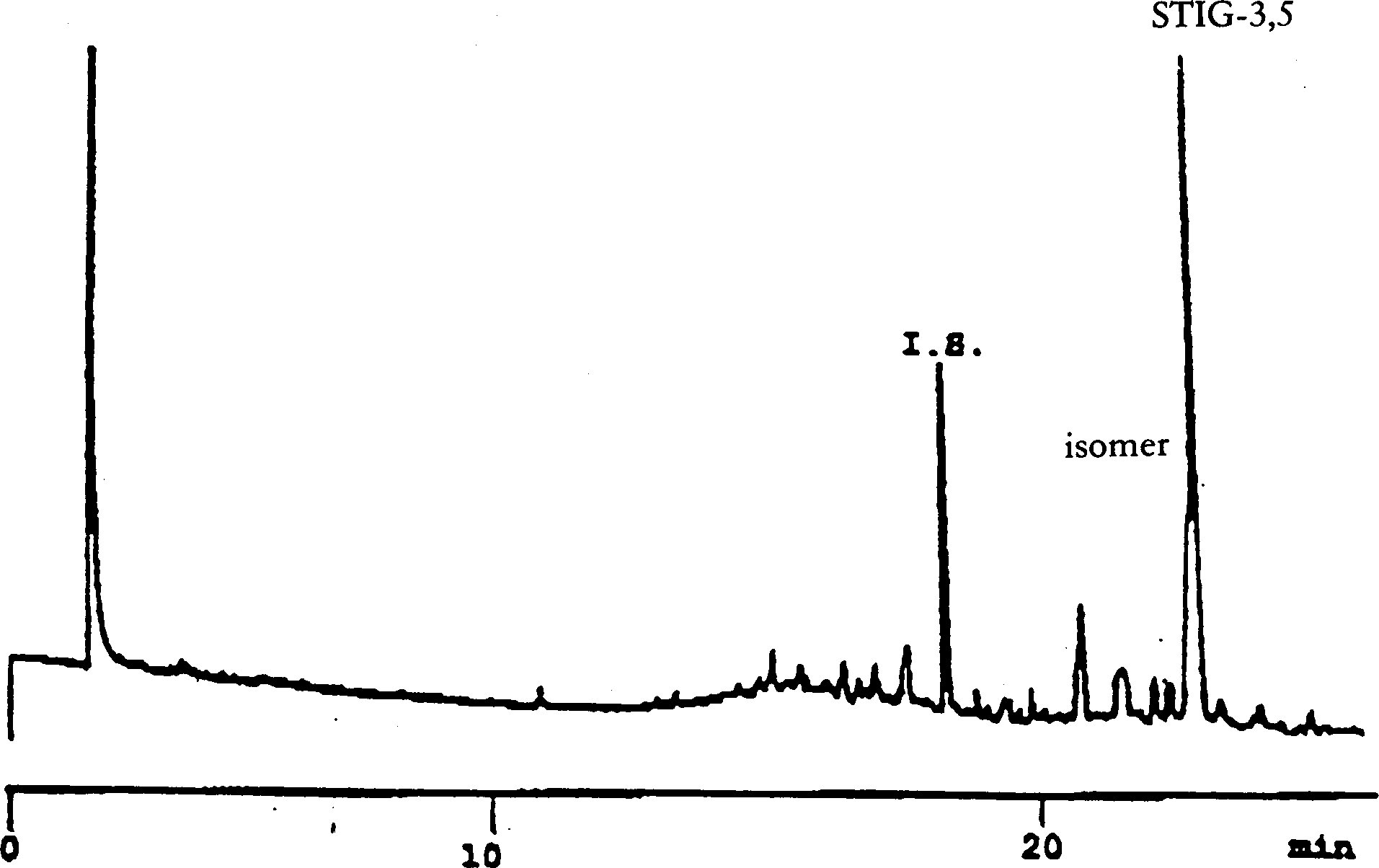 Joonis 2.
Joonis 2.
DB-5-kolonni abil analüüsitud rafineeritud oliiviõliproovi kromatogramm, millel on näha 3,5-stigmastadieeni isomeer.
XVIII LISA
ECN 42-TRIATSÜÜLGLÜTSEROOLIDE TEGELIKU JA TEOREETILISELT ARVUTATUD SISALDUSE VAHE MÄÄRAMINE
1. KASUTUSALA
Kõrgefektiivse vedelikkromatograafiaga (HPLC) määratud ekvivalentse süsinikuarvuga 42 (ECN42HPLC) triatsüülglütseroolide eksperimentaalsete väärtuste ja rasvhappekoostisest arvutatud ekvivalentse süsinikuarvuga 42 (ECN42teoreetiline) triatsüülglütseroolide teoreetiliste väärtuste absoluutarvulise vahe määramine.
2. KOHALDAMISALA
Käesolevat normi kohaldatakse oliiviõlidele. Meetodit võib kasutada (linoolhapperikka) seemneõli väikeste koguste avastamiseks iga kategooria oliiviõlides.
3. PÕHIMÕTE
HPLC-analüüsiga määratud ECN42-triatsüülglütseroolide sisaldus ja ECN42-triatsüülglütseroolide teoreetiline sisaldus (mis on arvutatud gaas-vedelik-kromatograafiaga määratud rasvhappekoostise põhjal) on ehtsate oliiviõlide puhul omavahel teataval määral kooskõlas. Nende vahe, mis on suurem iga õlitüübi jaoks heakskiidetud väärtusest, osutab sellele, et õli sisaldab seemneõlisid.
4. MEETOD
ECN42-triatsüülglütseroolide teoreetilise sisalduse arvutamine ja HPLC-andmetega võrdlemisest vahe leidmine toimub peamiselt muude meetoditega saadud analüüsiandmetega kooskõlastamise kaudu. Selles on võimalik eristada kolme etappi: rasvhappekoostise määramine kapillaargaasikromatograafiaga, ECN42-triatsüülglütseroolide teoreetilise sisalduse arvutamine, ECN42-triatsüülglütseroolide määramine HPLC meetodiga.
4.1. Seadmed
|
4.1.1. |
250 ml ja 500 ml ümarapõhjalised kolvid. |
|
4.1.2. |
100 ml keeduklaasid. |
|
4.1.3. |
Klaasist kromatograafiakolonn sisediameetriga 21 mm ja pikkusega 450 mm, varustatud kraaniga ja ülaosas normaallihvmuhviga. |
|
4.1.4. |
250 ml jaotuslehtrid, normaallihvkerniga alumises osas, mis sobib ühendamiseks kolonni ülemise otsaga. |
|
4.1.5. |
600 mm pikkune klaaspulk. |
|
4.1.6. |
Klaaslehter läbimõõduga 80 mm. |
|
4.1.7. |
50 ml mõõtkolvid. |
|
4.1.8. |
20 ml mõõtkolvid. |
|
4.1.9. |
Pöördauruti. |
|
4.1.10. |
Kõrgefektiivne vedelikkromatograaf, mille kolonni temperatuuri on võimalik hoida etteantud väärtuse juures. |
|
4.1.11. |
Sissesüstimissüsteem, millesse on võimalik süstida 10 μl proove. |
|
4.1.12. |
Detektorina kasutatakse diferentsiaalrefraktomeetrit. Kogu mõõtepiirkonna ulatuses peaks tundlikkus olema vähemalt 10–4 murdumisnäitaja ühikut. |
|
4.1.13. |
Kolonn: roostevabast terasest toru pikkusega 250 mm ja sisediameetriga 4,5 mm, täidiseks on ränidioksiidiosakesed läbimõõduga 5 μm; täidis sisaldab 22–23 % süsinikku oktadetsüülsilaani kujul. |
|
4.1.14. |
Andmetöötlustarkvara. |
|
4.1.15. |
Umbes 2 ml ruumalaga viaalid, millel on keeratavad korgid ja teflontihendid. |
4.2. Reaktiivid
Kõik reaktiivid peaksid olema puhtusastmega „analüütiliselt puhas”. Elueerimislahustid peaksid olema gaasivabad ja neid võib korduvalt kasutada, ilma et see mõjutaks lahutust.
|
4.2.1. |
Kromatograafiliselt puhas petrooleeter keemistemperatuuriga 40–60 °C või heksaan. |
|
4.2.2. |
Värskelt destilleeritud peroksiidivaba dietüüleeter. |
|
4.2.3. |
Elueerimislahusti õli puhastamiseks kolonnkromatograafia abil: petrooleetri ja etüüleetri segu komponentide suhtega 87/13 (v/v). |
|
4.2.4. |
Silikageel, mešš 70–230, tüüp Merck 7734, veesisaldus standarditud 5 massiprotsendile. |
|
4.2.5. |
Klaasvatt. |
|
4.2.6. |
Atsetoon HPLC jaoks. |
|
4.2.7. |
Atsetonitriil või propionitriil HPLC jaoks. |
|
4.2.8. |
HPLC-elueerimislahusti: atsetonitriil + atsetoon (vahekorda tuleb soovitud lahutuse saamiseks kohandada; alustada tuleks seguga 50:50) või propionitriil. |
|
4.2.9. |
Solubiliseerimislahusti: atsetoon. |
|
4.2.10. |
Võrdlustriglütseriidid: võib kasutada müügil olevaid triglütseriide (tripalmitiin, trioleiin, jne); nende retentsiooniajad kantakse graafikule funktsioonina ekvivalentsest süsinikuaatomite arvust; samuti võib kasutada võrdluskromatogramme, mis on saadud sojaõliga, seguga sojaõli-oliiviõli 30:70 ja puhta oliiviõliga (vt märkused 1 ja 2 ning joonised 1–4). |
|
4.2.11. |
Tahke faasi ekstraktsiooni kolonn mahuga 6 ml, sisaldab 1 g ränidioksiidi. |
4.3. Proovi ettevalmistamine
Kuna mitmed segavad ained võivad anda valepositiivseid tulemusi, tuleb proov alati puhastada vastavalt IUPACi meetodile 2.507, mida kasutatakse polaarsete ühendite määramiseks praadimisõlides.
4.3.1. Kromatograafilise kolonni ettevalmistamine.
Kolonni (4.1.3) valatakse umbes 30 ml elueerimislahustit (4.2.3), seejärel pannakse kolonni veidi klaasvatti (4.2.5) ja lükatakse see klaaspulga (4.1.5) abil kolonni põhja.
100 ml keeduklaasis suspendeeritakse 25 g silikageeli (4.2.4) 80 ml elueerimissegus (4.2.3), seejärel kallatakse suspensioon klaaslehtri (4.1.6) abil kolonni.
Silikageeli täielikuks ülekandmiseks kolonni loputatakse keeduklaasi elueerimisseguga ja kantakse ka loputamiseks kasutatud vedelikukogused kolonni.
Avatakse kraan ja lastakse lahustil kolonnist läbi voolata, kuni lahusti nivoo jääb umbes 1 cm kõrgusele silikageeli pinnast.
4.3.2. Kolonnkromatograafia
50 ml mõõtkolbi (4.1.7) kaalutakse täpsusega 0,001 g 2,5 ± 0,1 g õli, mis on eelnevalt filtritud, homogeenitud ja vajaduse korral muudetud veevabaks.
Õli lahustatakse umbes 20 ml elueerimislahustis (4.2.3). Vajaduse korral soojendatakse veidi, et hõlbustada lahustumist. Jahutatakse toatemperatuurini ja täiendatakse elueerimislahustiga märgini.
Mõõtpipetiga viiakse 20 ml lahust punkti 4.3.1 kohaselt ettevalmistatud kolonni, avatakse kraan ja lastakse lahustil voolata, kuni nivoo kolonnis jõuab silikageelikihi pinnani.
Seejärel elueeritakse 150 ml elueerimislahustiga (4.2.3), seades lahusti voolukiiruseks umbes 2 ml/min (150 ml lahusti voolamiseks läbi kolonni kulub umbes 60–70 minutit).
Eluaat kogutakse 250 ml ümarkolbi (4.1.1), mis on eelnevalt ahjus kuumutatud konstantse kaaluni ja täpselt kaalutud. Lahus aurutatakse pöördaurutil (4.1.9) vaakumis lahustist kuivaks ja kaalutakse jääk, mida kasutatakse HPLC-analüüsi ja metüülestrite sünteesimiseks kasutatavate lahuste valmistamiseks.
Proovist peab pärast kolonni olema võimalik taastuvastada vähemalt 90 % ekstraneitsiõli, neitsiõli, tavalise rafineeritud oliiviõli ja oliiviõli eri kategooriate puhul ning vähemalt 80 % lambiõli ja oliivijääkõli puhul.
4.3.3. Puhastamine tahke faasi ekstraktsiooniga
Ränidioksiidi sisaldav tahke faasi ekstraktsiooni kolonn aktiveeritakse, milleks sellest lastakse vaakumis läbi 6 ml heksaani (4.2.3), kuid välditakse kolonni kuivaks jäämist.
Kaalutakse 0,12 g täpsusega 0,001 g 2 ml viaali (4.1.15) ja lahustatakse 0,5 ml heksaanis (4.2.3).
Lahus kantakse tahke faasi ekstraktsiooni kolonni ja voolutatakse siis vaakumit kasutades 10 ml heksaani-dietüüleetri seguga vahekorras 87:13 mahuosa (4.2.3).
Kogutud fraktsioon aurutatakse rotaatoril (4.1.9) vaakumis toatemperatuuril kuivaks. Jääk lahustatakse 2 ml atsetoonis (4.2.6) triatsüülglütseroolide analüüsiks.
4.4. HPLC-analüüs
4.4.1. Proovide ettevalmistamine kromatograafilise analüüsi jaoks
Valmistatakse uuritava proovi 5 % lahus, milleks kaalutakse 10 ml mõõtkolbi 0,5 ± 0,001 g proovi ja katseklaas täidetakse solubiliseerimislahustiga (4.2.9) kuni 10 ml märgini.
4.4.2. Analüüsi käik
Koostatakse kromatograafiasüsteem. Kogu süsteemi läbipesemiseks pumbatakse sellest läbi elueerimislahustit (4.2.8) kiirusega 1,5 ml/min. Oodatakse, kuni tekib stabiilne nulljoon.
Kolonni süstitakse 10 μl proovi, mis on ette valmistatud vastavalt punktile 4.3.
4.4.3. Arvutuskäik ja tulemuste esitamine
Kasutatakse pindalade normeerimise meetodit, mille puhul eeldatakse, et ekvivalentse süsinikuaatomite arvuga 42 kuni 52 triatsüülglütseroolide piikide pindalade summa on 100 %.
Iga triglütserooli suhteline protsendiline sisaldus arvutatakse järgmise võrrandiga:
![]()
.
Tulemused esitatakse vähemalt kahe kümnendikkoha täpsusega.
Vt joonealused märkused 1–4.
4.5. Triatsüülglütseroolse koostise (moolprotsent) arvutamine rasvhappekoostise andmetest (pindalaprotsent)
4.5.1. Rasvhappekoostise määramine
Rasvhappekoostis määratakse meetodiga ISO 5508 kapillaarkolonni abil. Metüülestrid valmistatakse vastavalt dokumendis COI/T.20/Doc. No 24 esitatud eeskirjale.
4.5.2. Arvutustes kasutatavad rasvhapped
Glütseroolid rühmitatakse nende ekvivalentse süsinikuaatomite arvu (ECN) järgi, võttes arvesse ECNi ja rasvhapete järgmisi ekvivalentsusi. Arvestatakse üksnes rasvhappeid süsinikuaatomite arvuga 16 ja 18, kuna üksnes nimetatud rasvhapped on oliiviõli puhul olulised. Rasvhapped tuleks normeerida 100 %-le.
|
Rasvhape |
Lühend |
Molekulmass (MW) |
ECN |
|
Palmitiinhape |
P |
256,4 |
16 |
|
Palmitoleiinhape |
Po |
254,4 |
14 |
|
Steariinhape |
S |
284,5 |
18 |
|
Oleiinhape |
O |
282,5 |
16 |
|
Linoolhape |
L |
280,4 |
14 |
|
Linoleenhape |
Ln |
278,4 |
12 |
4.5.3. Pindalaprotsentide (area %) teisendamine kõikide rasvhapete moolideks (1) (area % X on rasvhappe X pindalaprotsent ja MW X on rasvhappe X molekulmass)
|
|
|
|
|
|
|
|
4.5.4. Rasvhapete moolide normeerimine 100 %-le (2)
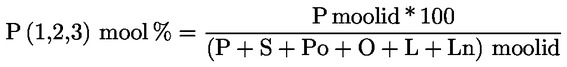





Tulemuseks saadakse iga rasvhappe moolprotsendiline osa 1,2,3-asendatud triatsüülglütseroolides.
Seejärel arvutatakse küllastunud rasvhapete (saturated fatty acids, SFA) P ja S ning küllastumata rasvhapete (unsaturated fatty acids, UFA) Po, O, L ja Ln summad (3):
![]()
![]()
4.5.5. Triatsüülglütseroolide 2- ja 1,3-asendis olevate rasvhapete koostise arvutamine
Rasvhapped jagatakse järgmisel viisil kolme rühma: üks rühm, milles rasvhape on 2-asendis, ning kaks identset rühma, milles rasvhape on 1- ja 3-asendis, kusjuures küllastunud (P ja S) ning küllastumata (Po, O, L ja Ln) hapete koefitsiendid on erinevad.
4.5.5.1. Küllastunud rasvhapped 2-asendis [P(2) ja S(2)] (4)
![]()
![]()
4.5.5.2. Küllastumata rasvhapped 2-asendis [Po(2), O(2), L(2) ja Ln(2)] (5):



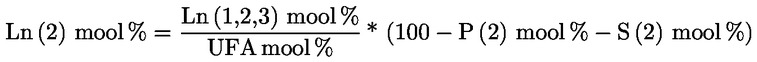
4.5.5.3. Rasvhapped 1,3-asendis [P(1,3), S(1,3), Po(1,3) O(1,3), L(1,3) ja Ln(1,3)] (6)

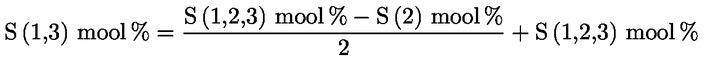

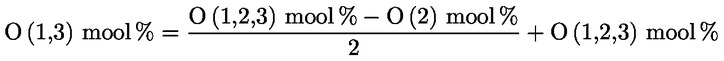
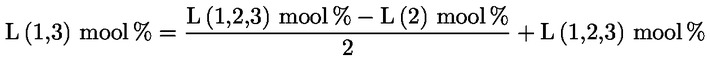

4.5.6. Triatsüülglütseroolide arvutamine
4.5.6.1. Ühe rasvhappe triatsüülglütseroolid (tähis AAA, siin: LLL, PoPoPo) (7)
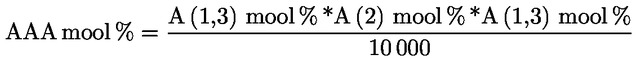
4.5.6.2. Kahe rasvhappe triatsüülglütseroolid (tähis AAB, siin: PoPoL, PoLL) (8)
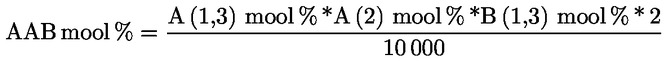

4.5.6.3. Kolme erineva rasvhappe triatsüülglütseroolid (tähis ABC, siin: OLLn, PLLn, PoOLn, PPoLn) (9)

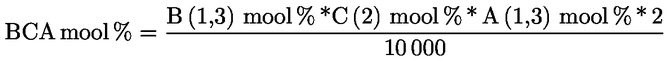
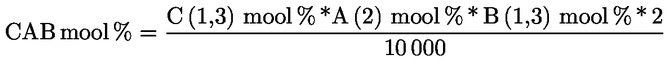
4.5.6.4. Triatsüülglütseroolid ekvivalentse süsinikuaatomite arvuga 42 (ECN42)
ECN42-triatsüülglütseroolid arvutatakse võrranditega 7, 8 ja 9 ning need on esitatud vastavalt nende oodatavale elueerumisele HPLCga määramisel (tavaliselt ainult kolm piiki).
LLL
PoLL ja asendiisomeer LPoL
OLLn ning asendiisomeerid OLnL ja LnOL
PoPoL ja asendiisomeer PoLPo
PoOLn ning asendiisomeerid OPoLn ja OLnPo
PLLn ning asendiisomeerid LLnP ja LnPL
PoPoPo
SLnLn ja asendiisomeer LnSLn
PPoLn ning asendiisomeerid PLnPo ja PoPLn
ECN42-triatsüülglütseroolid esitatakse üheksa triatsüülglütserooli ja nende asendiisomeeride summana. Tulemused esitatakse vähemalt kahe kümnendikkoha täpsusega.
5. TULEMUSTE HINDAMINE
Arvutusega määratud teoreetilist sisaldust võrreldakse HPLC-analüüsi abil määratud sisaldusega. Kui HPLC-andmetest määratud sisalduse ja teoreetiliselt arvutatud sisalduse vahe absoluutväärtus on suurem väärtusest, mis on asjaomase õlikategooria jaoks esitatud standardis, sisaldab proov seemneõli.
Tulemused esitatakse kahe kümnendikkoha täpsusega.
6. NÄIDE (NUMBRITEGA ON VIIDATUD MEETODI KIRJELDUSE PUNKTIDELE)
— 4.5.1. Rasvhapete moolprotsendi arvutamine gaas-vedelik-kromatograafia andmetest (normeeritud pindalaprotsent)
Gaas-vedelik-kromatograafia abil on rasvhappekoostise kohta saadud järgmised andmed:
|
FA |
P |
S |
Po |
O |
L |
Ln |
|
MW |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
Pindalaprotsent |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3. Pindalaprotsentide teisendamine kõikide rasvhapete moolideks (vt võrrand 1)
|
P moole |
= |
|
|
S moole |
= |
|
|
Po moole |
= |
|
|
O moole |
= |
|
|
L moole |
= |
|
|
Ln moole |
= |
|
|
Kokku |
= |
0,35821 mooli triatsüülglütseroole |
— 4.5.4. Rasvhapete moolide normeerimine 100 %-le (vt võrrand 2)
|
P(1,2,3) mool % |
= |
|
|
S(1,2,3) mool % |
= |
|
|
Po(1,2,3) mool % |
= |
|
|
O(1,2,3) mool % |
= |
|
|
L(1,2,3) mool % |
= |
|
|
Ln(1,2,3) mool % |
= |
|
|
Kokku moolprotsente |
= |
100 % |
Triatsüülglütseroolide 1,2,3-asendis olevate küllastunud ja küllastumata rasvhapete summa (vt võrrand 3):
![]()
![]()
— 4.5.5. Triatsüülglütseroolide 2- ja 1,3-asendis olevate rasvhapete koostise arvutamine
— 4.5.5.1. Küllastunud rasvhapped 2-asendis [P(2) ja S(2)] (vt võrrand 4)
![]()
![]()
— 4.5.5.2. Küllastumata rasvhapped 2-asendis [Po(1,3), O(1,3), L(1,3) ja Ln(1,3)] (vt võrrand 5)

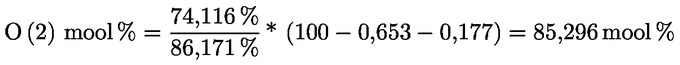

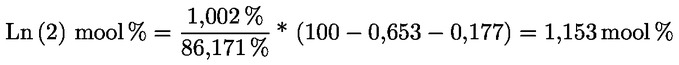
— 4.5.5.3. Rasvhapped 1,3-asendis [P(1,3), S(1,3), Po(1,3) O(1,3), L(1,3) ja Ln(1,3)] (vt võrrand 6)
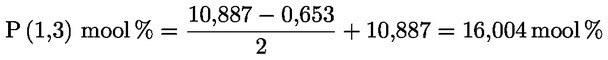





— 4.5.6. Triatsüülglütseroolide arvutamine
Arvutatud stereokeemilise numeratsiooni kohaselt 2- ja 1,3-asendis olevate rasvhapete koostisest:
|
Rasvhape |
1,3-asendis |
2-asendis |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Summa |
100,0 % |
100,0 % |
arvutatakse järgmised triatsüülglütseroolid:
LLL
PoPoPo
PoLL ühe asendiisomeeriga
SLnLn ühe asendiisomeeriga
PoPoL ühe asendiisomeeriga
PPoLn kahe asendiisomeeriga
OLLn kahe asendiisomeeriga
PLLn kahe asendiisomeeriga
PoOLn kahe asendiisomeeriga
— 4.5.6.1. Ühe rasvhappega triatsüülglütseroolid (LLL, Po,Po,Po) (vt võrrand 7)
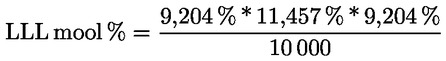 = 0,09706 LLL mooli
= 0,09706 LLL mooli

— 4.5.6.2. Kahe rasvhappega triatsüülglütseroolid (PoLL, SLnLn, PoPoL) (vt võrrand 8)


0,03210 PoLL mooli


0,00094 SLnLn mooli


0,00354 PoPoL mooli
— 4.5.6.3. Kolme erineva rasvhappe triatsüülglütseroolid (PoPLn, OLLn, PLLn, PoOLn) (vt võrrand 9)

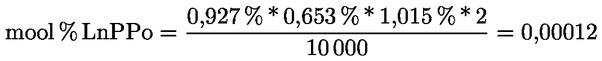

0,00761 PPoLn mooli

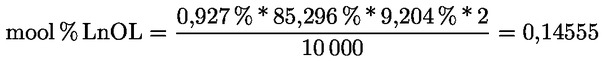
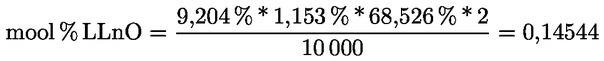
0,43655 OLLn mooli
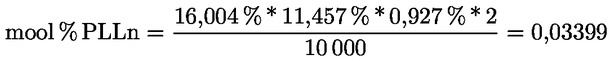

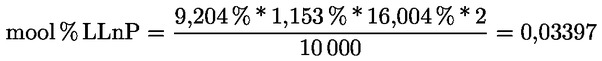
0,06907 PLLn mooli


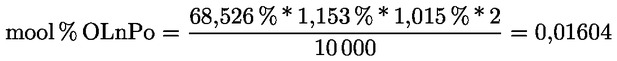
0,04812 PoOLn mooli
ECN42-triatsüülglütseroole = 0,69512 mooli
Märkus 1: elueerumisjärjekorda võib määrata ekvivalentse süsinikuarvu arvutamise teel, mida väljendab sageli seos ![]() , kus CN on süsinikuaatomite arv ja n on kaksiksidemete arv; kaksiksideme päritolu arvesse võttes on seda võimalik täpsemalt arvutada. Kui no, nl ja nln on vastavalt oleiinhappele, linoolhappele ja linoleenhappele omistatud kaksiksidemete arvud, on ekvivalentset süsinikuaatomite arvu võimalik arvutada järgmise valemiga:
, kus CN on süsinikuaatomite arv ja n on kaksiksidemete arv; kaksiksideme päritolu arvesse võttes on seda võimalik täpsemalt arvutada. Kui no, nl ja nln on vastavalt oleiinhappele, linoolhappele ja linoleenhappele omistatud kaksiksidemete arvud, on ekvivalentset süsinikuaatomite arvu võimalik arvutada järgmise valemiga:
![]()
,
milles koefitsiente do, d1 ja d1n on võimalik arvutada võrdlustriglütseriidide andmetest. Käesolevas meetodis täpsustatud tingimustel on võrrand ligikaudu järgmine:
![]()
Märkus 2: mitme võrdlustriglütseriidi abil on võimalik arvutada trioleiini lahutus:
![]()
trioleiinkasutades taandatud retentsiooniaega
![]()
.
log α ja kaksiksidemete arvu f vahelise sõltuvuse graafiku järgi saab määrata kindlaks kõikide võrdlustrigütseriidides sisalduvate rasvhapete triglütseriidide retentsiooniajad (vt joonis 1).
Märkus 3: kolonni lahutusvõime peab võimaldama trilinoleiini piigi selget eristumist lähedase retentsiooniajaga triglütseriidide piikidest. Voolutamist jätkatakse kuni ECN52 piigini.
Märkus 4: kõikide kõnealuse analüüsi seisukohast huvipakkuvate piikide pindalade õige määramine on tagatud juhul, kui ECN50-le vastava teise piigi kõrgus on 50 % meeriku täisskaalast.
Joonis 1
Graafik: log α sõltuvus kaksiksidemete arvust f

Kaksiksidemete arv
La: lauriinhape; My: müristiinhape; P: palmitiinhape; S: steariinhape; O: oleiinhape; L: linoolhape; Ln: linoleenhape.
Joonis 2
Vähese linoolhappesisaldusega oliiviõli
a)
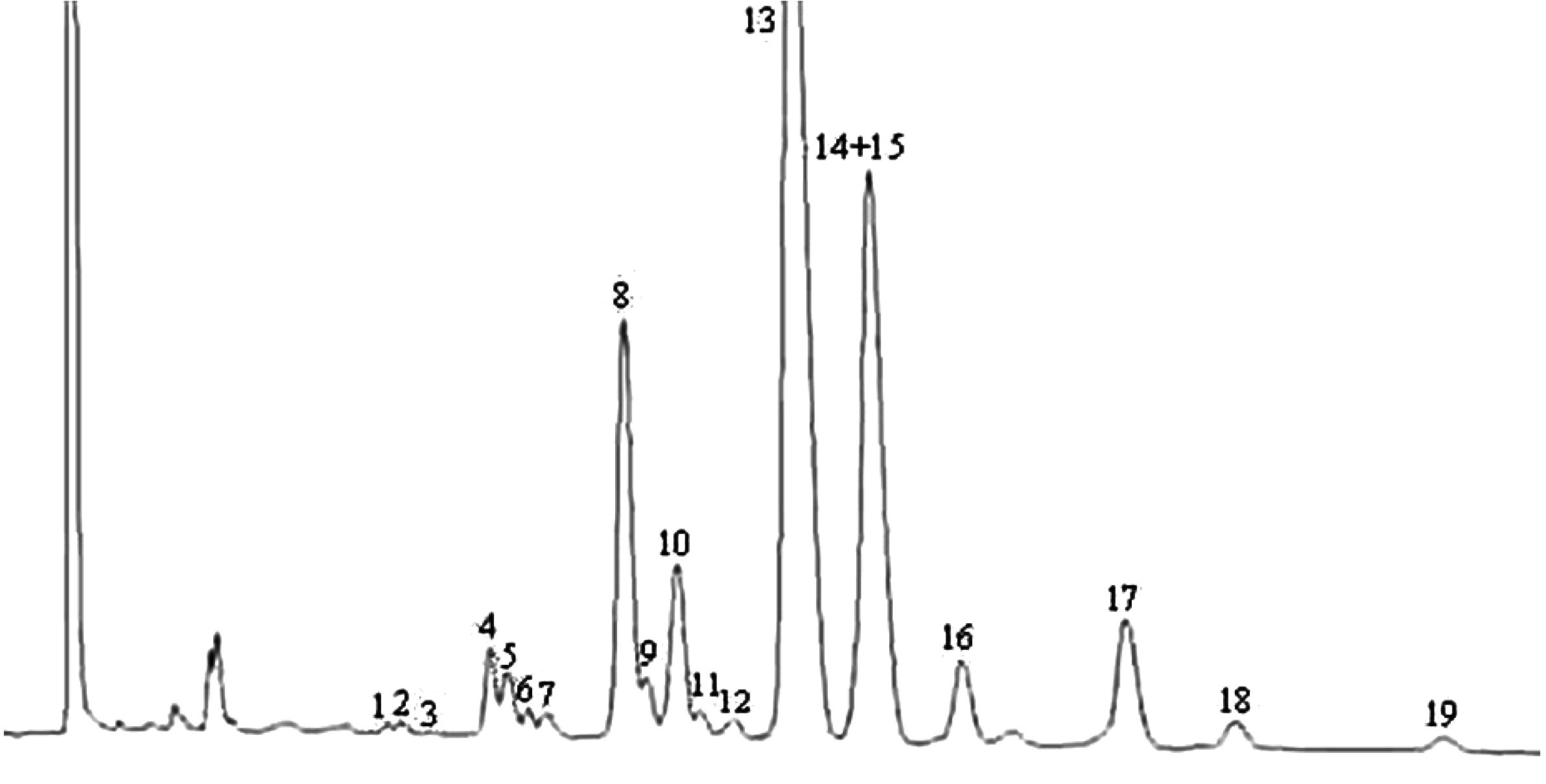
Lahusti: atsetoon/atsetonitriil.
Kromatogramm a: kromatograafiliste piikide peamised komponendid: ECN42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
b)

Lahusti: propionitriil
Kromatogramm b: kromatograafiliste piikide peamised komponendid: ECN42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
Joonis 3
Suure linoolhappesisaldusega oliiviõli
a)

Lahusti: atsetoon/atsetonitriil (50:50).
Kromatogramm a: kromatograafiliste piikide peamised komponendid: ECN42: (1’) LLL + PoLL; (2’) OLLn + PoOLn; (3’) PLLn; ECN44: (4’) OLL + PoOL; (5’) OOLn + PLL; (6’) POLn + PPoPo; ECN46: (7’) OOL + PoOO; (8’) PLO + SLL + PoOP; (9’) PLP + PoPP; ECN48: (10’) OOO; (11’) POO + SLL + PPoO; (12’) POP + PLS; ECN50: (13’) SOO; (14’) POS + SLS.
b)
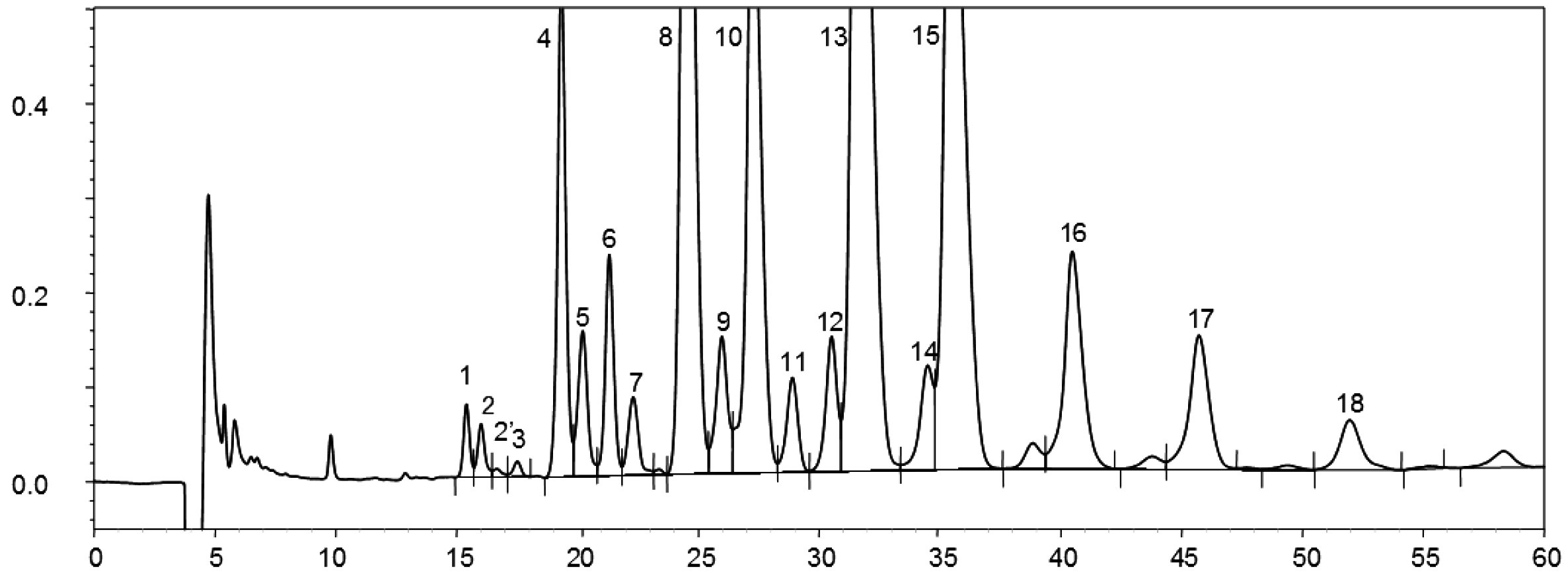
Lahusti: propionitriil.
Kromatogramm b: kromatograafiliste piikide peamised komponendid: ECN42: (1) LLL; (2 + 2’) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS; ECN52: (19) AOO.
ANNEX XIX
Alifaatsete alkoholide ja triterpeenalkoholide sisalduse kapillaarkolonnkromatograafiline määramine
1. SISU
Käesolevas lisas kirjeldatakse alifaatsete alkoholide ja triterpeenalkoholide sisalduse määramist õlides ja rasvades.
2. PRINCIPLE OF THE METHOD
The fatty substance, with 1-eicosanol added as internal standard, is saponified with ethanolic potassium hydroxide and then the unsaponifiable matter extracted with ethyl ether. The alcoholic fraction is separated from the unsaponifiable matter by chromatography on a basic silica gel plate; the alcohols recovered from the silica gel are transformed into trimethylsilyl ethers and analysed by capillary gas chromatography.
3. EQUIPMENT
3.1. 250 ml round-bottomed flask fitted with a reflux condenser having ground-glass joints.
3.2. 500 ml separating funnel.
3.3. 250 ml round-bottomed flasks.
3.4. Chromatographic tank for thin-layer chromatographic analysis, for glass plates of dimensions 20 x 20 cm.
3.5. Ultraviolet lamp having a wavelength of 366 or 254 nm.
3.6. 100 μl and 500 μl microsyringes.
3.7. A cylindrical filter funnel with a G3 porous septum (porosity 15 to 40 μm) of diameter approximately 2 cm and a depth of some 5 cm, with an attachment suitable for filtration under vacuum and a 12/21 male ground glass joint.
3.8. 50 ml vacuum conical flask with a 12/21 ground-glass female joint which can be fitted to the filter funnel (3.7).
3.9. A 10 ml test tube with a tapering bottom and a sealing stopper.
3.10. Gas chromatograph for use with a capillary column, and provided with a splitting system composed of:
3.10.1. Thermostatic chamber for columns (column oven) to hold the temperature desired with a precision of ± 1 °C.
3.10.2. A temperature-adjustable injection unit with a persilanised glass vaporising element.
3.10.3. A flame ionisation detector and converter-amplifier.
3.10.4. Recorder-integrator for operation with the converter-amplifier (3.10.3), with response time not exceeding one second and with variable paper-speed.
3.11. Glass or fused silica capillary column, of length 20 to 30 m, internal diameter 0,25 to 0,32 mm, with SE-52 or SE-54 liquid phase or equivalent, with a film thickness between 0,10 and 0,30 μm.
3.12. Microsyringe for gas chromatography, of 10 μl capacity with hardened needle.
3.13. Analytical balance sensitive to 1 mg (with 0,1 mg display).
4. REAGENTS
4.1. Potassium hydroxide, approximately 2 N ethanolic solution: 130 g potassium hydroxide (minimum concentration 85 %) is dissolved, with cooling, in 200 ml distilled water and then made up to one litre with ethanol. The solution should be stored in a well-stoppered opaque glass bottle.
4.2. Ethyl ether, pure for analysis.
4.3. Anhydrous sodium sulphate, analytical purity.
4.4. Glass plates coated with silica gel, without fluorescence indicator, thickness 0,25 mm (commercially available ready for use).
4.5. Potassium hydroxide, approximately 0,2 N ethanolic solution; 13 g of potassium hydroxide are dissolved in 20 ml of distilled water and made up to one litre with ethanol.
4.6. Benzene, for chromatography (see 5.2.2).
4.7. Acetone, for chromatography (See 5.2.2).
4.8. Hexane, for chromatography (see 5.2.2).
4.9. Ethyl ether, for chromatography (see 5.2.2).
4.10. Chloroform, for chromatography.
4.11. Võrdluslahus õhukese kihi kromatograafia jaoks: C20–C28 alkoholid 0,5 % kloroformis, või alkoholide fraktsioon, mis on saadud punkti 5.2 kohaselt oliivijääkõli seebistumatust ainest.
4.12. 0,2 % solution of 2', 7'-dichlorofluorescein in ethanol. Make slightly basic by adding a few drops of 2 N alcoholic potassium hydroxide solution.
4.13. Anhydrous pyridine, for chromatography.
4.14. Hexamethyl disilazane.
4.15. Trimethylchlorosilane.
4.16. Standard solutions of trimethylsilyl ethers of aliphatic alcohols from C20 to C28. They may be prepared from mixtures of pure alcohols at the time they are required for use.
4.17. A 0,1 % (m/v) solution of 1-eicosanol in chloroform (internal standard).
4.18. Carrier gas: hydrogen or helium, gas-chromatographic purity.
4.19. Auxiliary gas: nitrogen, gas-chromatographic purity.
5. PROCEDURE
5.1. Preparation of the unsaponifiables
|
5.1.1. |
Using a 500 μl microsyringe place, into a 250 ml round-bottom flask, a volume of 0,1 % 1-eicosanol solution in chloroform (4.17) containing a quantity of 1-eicosanol approximately equal to 10 % of the aliphatic alcohols content in that portion of sample to be taken for analysis. For example, to 5 g of sample add 250 μl of the 0,1 % 1-eicosanol solution if olive oil and 1 500 μl if olive pomace oil. Evaporate to dryness in current of nitrogen and then weigh accurately 5 g of the dry filtered sample into the same flask. |
|
5.1.2. |
Add 50 ml of 2 N potassium hydroxide ethanolic solution, fit the reflux condenser and heat the apparatus to slight boiling on a steam bath, stirring continuously throughout the heating process until saponification has taken place (the solution becomes clear). Continue heating for a further 20 minutes and then add 50 ml of distilled water through the condenser. The condenser is then disconnected and the flask cooled to approximately 30 °C. |
|
5.1.3. |
The contents of the flask are quantitatively transferred to a separating funnel of 500 ml capacity by adding distilled water several times, using a total of around 50 ml distilled water. Add approximately 80 ml of ethyl ether, shake vigorously for approximately 30 seconds and allow to settle (Note 1). Separate off the lower aqueous phase collecting it in a second separating funnel. Two further extractions are effected on the aqueous phase, in the same manner, using each time 60 to 70 ml ethyl ether. Note 1: Emulsions may be eliminated by adding, using as a spray, small quantities of ethyl alcohol or methyl alcohol. |
|
5.1.4. |
The ethyl ether extracts are combined in a separating funnel and washed with distilled water (50 ml at a time) until the washing water gives a neutral reaction. Discard the washing water, dry with anhydrous sodium sulphate and filter, into a flask of 250 ml capacity which has been weighed beforehand, the funnel and filter being washed with small quantities of ethyl ether which are added to the total. |
|
5.1.5. |
Distil the ether down to a few ml, then bring to dryness under a slight vacuum or in a current of nitrogen, completing drying in an oven at 100 °C for approximately a quarter of an hour, and then weigh after cooling in a desiccator. |
5.2. Separation of alcoholic fractions
|
5.2.1. |
Preparation of basic TLC plates: the silica gel plates (4.4) are immersed completely, in 0,2 N potassium hydroxide solution (4.5) for 10 seconds, and then left to dry for two hours under an extractor hood and finally placed in an oven at 100 °C for one hour. Remove from the oven and keep in a calcium chloride desiccator until required for use (plates treated in this way must be used within 15 days). Note 2: When basic silica gel plates are used to separate the alcoholic fraction there is no need to treat the unsaponifiables with alumina. It follows that all acid compounds (fatty acids and others) are retained at the origin thereby obtaining both aliphatic alcohol and terpenic alcohol bands which are both separated distinctly from the sterol band. |
|
5.2.2. |
Place a 65/35 by volume hexane/ethyl ether mixture in the plate-developing chamber to a depth of approximately 1 cm ( 6 ). Close the chamber using an appropriate cover and leave for half an hour to allow equilibration between vapour and liquid. Strips of filter paper dipping into the eluent may be affixed to the inside surfaces of the tank to reduce the development time by approximately one third and obtain more uniform, regular elution of the components. Note 3: The developing solution must be replaced for each analysis in order to obtain reproducible developing conditions. |
|
5.2.3. |
An approximately 5 % solution of unsaponifiable matter (5.1.5) in chloroform is prepared and 0,3 ml of the solution is streaked as a uniform strip of minimum thickness, using the 100 μl microsyringe, on a TLC plate at approximately 2 cm from the bottom of the TLC plate. Aligned with the origin, 2 to 3 μl of the aliphatic alcohols reference solution (4.11) are spotted for the identification of the aliphatic alcohols band after development has been completed. |
|
5.2.4. |
Place the plate inside the development tank as stated in 5.2.2. The ambient temperature should be maintained between 15 and 20 °C. Immediately close the chamber with the cover and allow to elute until the solvent front reaches approximately 1 cm from the upper edge of the plate. The plate is then removed from the development chamber and the solvent evaporated under a hot air current or the plate is left for a while under the extractor hood. |
|
5.2.5. |
Plaadile pihustatakse ettevaatlikult ja ühtlaselt 2′,7′-diklorofluorestseiini lahust, kui plaati vaadeldakse ultraviolettlambi all. Alifaatsete alkoholide vööndid võib määrata vastavalt nende asendile, mida võrreldakse võrdluslahuse lahutamisel tekkinud vöönditega: vööndite piirjooned joonistatakse üle musta pliiatsiga, kusjuures alifaatsete alkoholide vöönd ja otse selle kohal asuv terpeenalkoholide vöönd arvestatakse üheks vööndiks (märkus 4). Märkus 4: alifaatsete alkoholide vöönd ja terpeenalkoholide vöönd tuleb arvestada kokku, kuna mõned alifaatsed alkoholid võivad olla migreerunud triterpeenalkoholide vööndisse. Õhukese kihi kromatograafia abil eraldamise näide on esitatud liite joonises 1. |
|
5.2.6. |
Metallspaatliga kraabitakse märgistatud alalt maha silikageel. Kraapimisega saadud peenestatud materjal pannakse filterlehtrisse (3.7). Lisatakse 10 ml kuuma kloroformi, segatakse hoolikalt metallist spaatliga ning filtritakse vaakumiga; filtraat kogutakse filterlehtriga ühendatud koonilisse kolbi (3.8). Silikageeli pestakse klaasfilterlehtris kolm korda dietüüleetriga (iga kord umbes 10 ml) ning filtraat kogutakse sama lehtriga ühendatud kolbi. Filtraat aurustatakse kokku mahuni 4–5 ml ning jääklahus valatakse eelnevalt kaalutud 10 ml katseklaasi (3.9), seejärel aurustatakse nõrga lämmastikuvoo läbijuhtimisel ettevaatlikult kuumutades kuivaks, lahustatakse uuesti paari tilga atsetooniga, aurustatakse taas kuivaks ning asetatakse umbes kümneks minutiks kuivatuskappi temperatuurile 105 °C, lastakse eksikaatoris jahtuda ja kaalutakse. Katseklaasis olev jääk koosneb alkoholifraktsioonist. |
5.3. Preparation of the trimethylsilyl ethers
|
5.3.1. |
The reagent for silylation, consisting of a mixture of 9:3:1 by volume (Note 5) of pyridine-hexamethyldisilazane-trimethylchlorosilane in the proportion of 50 μl for each milligram of aliphatic alcohols, is added to the test tube containing the alcoholic fraction, avoiding all absorption of moisture (Note 6). Note 5: Solutions which are ready for use are available commercially. Other silanising reagents such as, for example, bis-trimethylsilyl, trifluor acetamide + 1 % trimethyl chlorosilane, which has to be diluted with an equal volume of anhydrous pyridine, are also available. Note 6: The slight opalescence which may form is normal and does not cause any interference. The formation of a white floc or the appearance of a pink colour are indicative of the presence of moisture or deterioration of the reagent. If these occur the test must be repeated. |
|
5.3.2. |
Stopper the test tube, shake carefully (without overturning) until the aliphatic alcohols are completely dissolved. Stand for at least 15 minutes at ambient temperature and then centrifuge for a few minutes. The clear solution is ready for gas chromatographic analysis. |
5.4. Gas chromatography analysis
5.4.1. Preliminary operations, column packing
|
5.4.1.1. |
Fit the column in the gas chromatograph, attaching the inlet end to the injector connected to the splitting system and the outlet end to the detector. Carry out a general check of the gas chromatography assembly (tightness of gas fittings, efficiency of the detector, efficiency of the splitting system and of the recording system, etc.). |
|
5.4.1.2. |
If the column is being used for the first time it is recommended that it should be subjected to conditioning. A little carrier gas is passed through the capillary column and then the gas chromatography assembly is switched on and gradually heated until a temperature not less than 20 °C above the operating temperature (see Note 7) is attained. That temperature is held for not less than two hours and then the assembly is brought to the operating conditions (regulation of gas flow, split flame ignition, connection to the electronic recorder, adjustment of the temperature of the capillary column oven, the detector and the injector, etc.) and the signal is adjusted to a sensitivity not less than twice the highest level contemplated for the execution of the analysis. The course of the base line must be linear, without peaks of any kind, and must not drift. A negative straight-line drift indicates leakage from the column connections; a positive drift indicates inadequate conditioning of the column. Note 7: The conditioning temperature shall be at least 20 °C less than the maximum temperature contemplated for the liquid phase employed. |
5.4.2. Choice of operating conditions
5.4.2.1. The guideline operating conditions are as follows:
— column temperature: the initial isotherm is set at 180 °C for eight minutes and then programmed at 5 °C/minute to 260 °C and a further 15 minutes at 260 °C,
— temperature of evaporator: 280 °C,
— temperature of detector: 290 °C,
— linear velocity of carrier gas: helium 20 to 35 cm/s, hydrogen 30 to 50 cm/s,
— splitting ratio: 1:50 to 1:100,
— sensitivity of instrument: 4 to 16 times the minimum attenuation,
— sensitivity of recording: 1 to 2 mV fs,
— paper speed: 30 to 60 cm/h,
— quantity of substance injected: 0,5 to 1 μl of TMSE solution.
The above conditions may be modified according to the characteristics of the column and of the gas chromatograph to obtain chromatograms satisfying the following conditions:
— alcohol C26 retention time shall be 18 ± 5 minutes,
— the alcohol C22 peak shall be 80 ± 20 % of the full scale value for olive oil and 40 ± 20 % of the full scale value for seed oil.
5.4.2.2. The above requirements are checked by repeated injection of the standard TMSE mixture of alcohols and the operating conditions are adjusted to yield the best possible results.
5.4.2.3. The parameters for the integration of peaks shall be set so that a correct appraisal of the areas of the peaks considered is obtained.
5.4.3. Analytical procedure
5.4.3.1. Using the microsyringe of 10 μl capacity draw in 1 μl of hexane followed by 0,5 μl of air and subsequently 0,5 to 1 μl of the sample solution; raise the plunger of the syringe further so the needle is emptied. Push the needle through the membrane of the injection unit and after one to two seconds inject rapidly, then slowly remove the needle after some five seconds.
5.4.3.2. Recording is effected until the TMSE of the aliphatic alcohols present have been eluted completely. The base line shall always correspond to the requirements of 5.4.1.2.
5.4.4. Piigi määramine
Üksikpiigid määratakse peetumisaja alusel, võrreldes neid samadel tingimustel analüüsitud alifaatsete alkoholide trimetüülsilüüleetrite segu kromatogrammiga.
Rafineeritud oliiviõli alkoholifraktsiooni kromatogrammi näited on esitatud liite joonistel 2 ja 3.
5.4.5. Quantitative evaluation
5.4.5.1. The peak areas of 1-eicosanol and of the aliphatic alcohols C22, C24, C26 and C28 are calculated by electronic integration.
5.4.5.2. The contents of each aliphatic alcohol, expressed in mg/1 000 g fatty substance, are calculated as follows:
![]()
where:
|
Ax |
= |
area of the alcohol peak x |
|
As |
= |
area of 1-eicosanol |
|
ms |
= |
mass of 1-eicosanol in milligrams |
|
m |
= |
mass of sample drawn for determination, in grams. |
6. EXPRESSION OF THE RESULTS
The contents of the individual aliphatic alcohols in mg/1 000 g of fatty substance and the sum of the „total aliphatic alcohols” are reported.
Liide
Õhukese kihi kromatograafia abil eraldamise näide ja kromatogrammi näited
Joonis 1
Heksaani/etüüleetriga (65/35) eludeeritud oliiviõli seebistamatu fraktsiooni õhukese kihi kromatograafia plaat

|
1 |
Alkohol C26 |
A |
Steroolid |
|
2 |
Alkohol C30 |
B |
Alifaatsed alkoholid |
|
3 |
Alkohol C20 |
C |
Triterpeenalkoholid |
|
4 |
Alkoholisegu C20-22-26-30 |
D |
Skvaleen |
|
5 |
Ekstra-neitsioliiviõli seebistumatus |
|
|
Joonis 2
Rafineeritud oliiviõli alkoholifraktsiooni kromatogramm

|
1 = Fütool |
5 = Alkohol C24 |
9 = 24-Metüültsükloartenool |
|
2 = Geranüülgerinool |
6 = Alkohol C26 |
10 = Tsitrostadienool |
|
3 = Alkohol C20 (IS) |
7 = Alkohol C28 |
11 = Tsüklobranool |
|
4 = Alkohol C22 |
8 = Tsükloartenool |
|
Joonis 3
Rafineeritud oliiviõli ja teistkordselt tsentrifuugitud oliiviõli alifaatsed alkoholid ja triterpeenalkoholid

|
1 = Fütool |
5 = Alkohol C24 |
9 = 24-Metüültsükloartenool (24MeCA) |
|
2 = Geranüülgerinool (CX) |
6 = Alkohol C26 |
10 = Tsitrostadienool |
|
3 = Alkohol C20 |
7 = Alkohol C28 |
11 = Tsüklobranool |
|
4 = Alkohol C22 |
8 = Tsükloartenool (CA) |
|
XX LISA
Meetod vahade, rasvhapete metüülestrite ja etüülestrite sisalduse määramiseks kapillaargaasikromatograafiaga
1. EESMÄRK
Käesolev meetod on ette nähtud vahade ning rasvhapete metüül- ja etüülestrite määramiseks oliiviõlides. Üksikud vahad ja alküülestrid eraldatakse üksteisest vastavalt süsinikuaatomite arvule. Meetodit soovitatakse vahendina, mis võimaldab eristada oliiviõli ja oliivijääkõli, samuti on see meetod ekstra neitsioliiviõli kvaliteedinäitaja, mis võimaldab kindlaks teha võltsinguid, milles ekstra neitsioliiviõlisid on segatud madalamakvaliteediliste õlidega – neitsiõlide, lambiõlide või ka mõne lõhnatustatud õliga.
2. PÕHIMÕTE
Rasvainele lisatakse sobivaid sisestandardeid ja seejärel fraktsioneeritakse rasvaine kromatograafiliselt, kasutades hüdrateeritud silikageelkolonni. Katsetingimustel elueeritud fraktsioon, mille polaarsus on väiksem kui triatsüülglütseriididel, eraldatakse ning seda analüüsitakse kapillaargaasikromatograafia abil.
3. VAHENDID
|
3.1. |
25-milliliitrine Erlenmeyeri kolb |
|
3.2. |
Klaasist vedelikkromatograafiakolonn sisediameeteriga 15 mm ja pikkusega 30–40 cm, mis on varustatud sobiva kraaniga |
|
3.3. |
Sobiv kapillaarkolonniga gaasikromatograaf, mis on varustatud süsteemiga, mis võimaldab otse kolonni süstimist ja koosneb järgmistest osadest:
|
|
3.4. |
Mikrosüstal, 10 μl, otse kolonni süstimiseks, mille nõelal on tugevdatud pinnakiht |
|
3.5. |
Elektriline vibraator |
|
3.6. |
Pöördauruti |
|
3.7. |
Muhvelahi |
|
3.8. |
Analüütiline kaal, mis võimaldab kaaluda täpsusega ± 0,1 mg |
|
3.9. |
Harilikud laboratoorsed klaasnõud |
4. REAKTIIVID
|
4.1. |
Silikageel, terasuurus 60–200 μm. Silikageel pannakse vähemalt neljaks tunniks muhvelahju temperatuuriga 500 °C. Lastakse jahtuda, seejärel lisatakse vett koguses, mis vastab 2 %-le silikageeli massist. Loksutatakse korralikult ühtlase suspensiooni saamiseks ja hoitakse enne kasutamist eksikaatoris vähemalt 12 tundi. |
|
4.2. |
n-heksaan, kromatograafia jaoks või jäägivaba (puhtust on vaja kontrollida) ETTEVAATUST! Aur on süttimisohtlik! Hoidke eemal soojusallikatest, sädemetest ja lahtisest leegist. Jälgige, et pudel oleks alati kindlalt suletud. Kasutamise ajal peab ruumis olema korralik ventilatsioon. Vältige aurude kogunemist ja eemaldage kõik tuleohu allikad, nagu soojendusriistad või elektriaparaadid, mis ei ole süttimisohutust materjalist. Sissehingamisel kahjulik, kuna võib kahjustada närvirakke. Vältige aurude sissehingamist. Vajaduse korral kasutage sobivat respiraatorit. Vältige sattumist nahale või silma. |
|
4.3. |
Dietüüleeter kromatograafia jaoks
ETTEVAATUST! Väga tuleohtlik ja mõõdukalt mürgine. Ärritab nahka. Sissehingamisel kahjulik. Võib kahjustada silmi. Toime võib avalduda alles hiljem. Võib moodustada plahvatusohtlikke peroksiide. Aur on süttimisohtlik. Hoidke eemal soojusallikatest, sädemetest ja lahtisest leegist. Jälgige, et pudel oleks alati kindlalt suletud. Kasutamise ajal peab ruumis olema korralik ventilatsioon. Vältige aurude kogunemist ja eemaldage kõik tuleohu allikad, nagu soojendusriistad või elektriaparaadid, mis ei ole süttimisohutust materjalist. Ärge aurutage kuivaks või peaaegu kuivaks. Vee või sobiva taandava reagendi lisamine võib vähendada peroksiidide moodustumist. Mitte juua. Vältige aurude sissehingamist. Vältige kestvat või korduvat kokkupuudet nahaga. |
|
4.4. |
n-heptaan kromatograafias kasutamiseks või isooktaan ETTEVAATUST! Tuleohtlik. Sissehingamisel kahjulik. Hoidke eemal soojusallikatest, sädemetest ja lahtisest leegist. Jälgige, et pudel oleks alati kindlalt suletud. Kasutamise ajal peab ruumis olema korralik ventilatsioon. Vältige aurude sissehingamist. Vältige kestvat või korduvat kokkupuudet nahaga. |
|
4.5. |
Laurüülarahidaadi (vt märkus 3) standardlahus 0,05 % (m/V) heptaanis (vahade sisestandard) Märkus 3. Võib kasutada ka palmitüülpalmitaati, müristüülstearaati või arahidüüllauraati. |
|
4.6. |
Metüülheptadekanaadi standardlahus 0,02 % (m/V) heptaanis (metüül- ja etüülestrite sisestandard) |
|
4.7. |
Sudaan 1 (1-fenüülaso-2-naftool) |
|
4.8. |
Kandegaas: vesinik või heelium gaasikromatograafias kasutamiseks HOIATUS Vesinik. Väga tuleohtlik, kokkusurutud gaas. Hoidke eemal soojusallikatest, sädemetest, lahtisest leegist ja elektriaparaatidest, mis ei ole süttimisohutust materjalist. Jälgige, et balloonikraan oleks suletud, kui vesinikku ei kasutata. Kasutage alati rõhureduktorit. Enne balloonikraani avamist vabastage reduktori vedru pinge alt. Ärge seiske ballooniava ees kraani avamise ajal. Kasutamise ajal peab ruumis olema korralik ventilatsioon. Ärge kandke vesinikku üle ühest balloonist teise. Ärge segage gaase balloonis. Kinnitage balloon, et seda ei saaks ümber ajada. Hoidke balloonid eemal päiksevalgusest ja soojusallikatest. Hoidke balloone keskkonnas, kus need ei puutu kokku söövitavate ainetega. Ärge kasutage kahjustatud või etiketita balloone. Heelium. Kokkusurutud gaas kõrge rõhu all. Heelium vähendab sissehingamisel kättesaadava hapniku hulka. Jälgige, et balloon oleks suletud. Kasutamise ajal peab ruumis olema korralik ventilatsioon. Ärge sisenege gaasiballoonide hoidmise ruumi, kui seal ei ole korralikku ventilatsiooni. Kasutage alati rõhureduktorit. Enne balloonikraani avamist vabastage reduktori vedru pinge alt. Ärge kandke gaasi üle ühest balloonist teise. Kinnitage balloon, et seda ei saaks ümber ajada. Ärge seiske ballooniava ees kraani avamise ajal. Hoidke balloonid eemal päiksevalgusest ja soojusallikatest. Hoidke balloone keskkonnas, kus need ei puutu kokku söövitavate ainetega. Ärge kasutage kahjustatud või etiketita balloone. Ärge hingake gaasi sisse. Kasutage ainult tehniliseks otstarbeks. |
|
4.9. |
Abigaasid: — vesinik, puhas, gaasikromatograafias kasutamiseks; — õhk, puhas, gaasikromatograafias kasutamiseks. HOIATUS Õhk. Kokkusurutud gaas kõrge rõhu all. Olge ettevaatlik, kui kasutate põlevate ainete juuresolekul, kuna enamiku orgaaniliste ainete isesüttimistemperatuur õhus on kõrgel rõhul oluliselt madalam. Jälgige, et balloonikraan oleks suletud, kui ballooni ei kasutata. Kasutage alati rõhureduktorit. Enne balloonikraani avamist vabastage reduktori vedru pinge alt. Ärge seiske ballooniava ees kraani avamise ajal. Ärge kandke gaasi üle ühest balloonist teise. Ärge segage gaase balloonis. Kinnitage balloon, et seda ei saaks ümber ajada. Hoidke balloonid eemal päiksevalgusest ja soojusallikatest. Hoidke balloone keskkonnas, kus need ei puutu kokku söövitavate ainetega. Ärge kasutage kahjustatud või etiketita balloone. Tehniliseks otstarbeks ettenähtud õhku ei kohi kasutada sissehingamiseks või hingamisaparaatides. |
5. MÄÄRAMISE KÄIK
5.1. Kromatograafiakolonni valmistamine
n-heksaanis (4.2) suspendeeritakse 15 g silikageeli (4.1) ning viiakse kolonni (3.2). Lastakse settida. Ühtlasema kromatograafiakolonni saamiseks töödeldakse kolonni pärast spontaanse settimise lõppu elektrilise vibraatoriga. Lisandite eemaldamiseks voolutatakse kolonni 30 ml n-heksaaniga. Analüütilise kaaluga (3.8) kaalutakse 25 ml kolbi (3.1) täpselt ligikaudu 500 mg proovi ning lisatakse sobiv kogus sisestandardit (4.5), olenevalt eeldatavast vahasisaldustest, nt oliiviõli puhul lisatakse 0,1 mg ja oliivijääkõli puhul 0,25–0,5 mg laurüülarahidaati ja oliiviõlide puhul 0,05 mg metüülheptadekanaati (4.6).
Ettevalmistatud proov kantakse kahe 2 ml n-heksaanikoguse (4.2) abil kromatograafiakolonni.
Lahustil lastakse voolata, kuni absorbendi pinna kohale jääb 1 mm kiht vedelikku. Kolonni voolutatakse täiendavalt n-heksaani-dietüüleetri seguga (99:1) kiirusel umbes 15 tilka 10 sekundi kohta ja kogutakse 220 ml lahust. (See fraktsioon sisaldab metüül- ja etüülestreid ning vahasid). (Vt märkused 4 ja 5.)
Märkus 4. n-heksaani-dietüüleetri segu (99:1) tuleb valmistada igal päeval uuesti.
Märkus 5. Vahade elueerimise täielikkuse visuaalseks kontrollimiseks võib proovi lahusele lisada 100 μl värvaine sudaan I 1-protsendilist lahust elueerimissegus.
Sudaan I retentsiooniaeg on vahade ja triatsüülglütseriidide vahel. Kui värvaine jõuab kolonni põhjani, tuleb elueerimine peatada, kuna selleks ajaks on kõik vahad kolonnist väljunud.
Tekkinud fraktsioone aurustatakse vaakumpöördaurutis, kuni peaaegu kogu lahusti on eemaldatud. Viimased 2 ml eemaldatakse nõrga lämmastikuvoolu läbijuhtimisel. Metüül- ja etüülestreid sisaldav fraktsioon viiakse lahusesse 2–4 ml n-heptaani või isooktaaniga.
5.2. Gaasikromatograafiline analüüs
5.2.1. Ettevalmistus
Kolonn pannakse gaasikromatograafi (3.3), sisselaskekoht ühendatakse on-column-süsteemiga ja väljalaskekoht detektoriga. Kontrollitakse gaasikromatograafiaseadet (gaasijuhtmete tööd, detektori ja meeriku toimimist jne).
Kui kolonni kasutatakse esimest korda, on soovitav see tasakaalustada. Kolonni juhitakse nõrk gaasivool, seejärel lülitatakse gaasikromatograafiaseade sisse. Umbes 4 tunniga tõstetakse temperatuur järk-järgult kuni 350 °C-ni.
Sellist temperatuuri hoitakse vähemalt 2 tundi, seejärel reguleeritakse seadet, kuni jõutakse töötingimusteni (reguleeritakse gaasi juurdevoolu, süüdatakse leek, ühendatakse elektrilise meerikuga (3.3.4), reguleeritakse kolonniahju temperatuuri, detektorit jne). Registreeritakse signaal vähemalt kaks korda suuremal tundlikkusel, kui on analüüsi läbiviimiseks nõutud. Tekkiv nulljoon peaks olema lineaarne, ilma piikideta ega tohi triivida.
Sirgjooneline negatiivne triiv näitab, et kolonni ühendused ei ole korras, samas kui positiivne triiv näitab, et kolonn ei ole korralikult töökorda viidud.
5.2.2. Töötingimuste valimine vahade ning metüül- ja etüülestrite määramiseks (märkus 6)
Töötingimused on reeglina järgmised:
|
— |
kolonni temperatuur : 20 °C/min 5 °C/min alguses 80 °C (1’) |
|
— |
detektori temperatuur : 350 °C; |
|
— |
süstitav ainekogus : 1 μl lahust n-heptaanis (2–4 ml); |
|
— |
kandegaas : heelium või vesinik asjaomase gaasi optimaalsel voolukiirusel (vt liide A); |
|
— |
seadme tundlikkus : piisav eespoolnimetatud tingimuste täitmiseks. |
Märkus 6. Kõrge lõpptemperatuuri tõttu on lubatud positiivne triiv, kuid see ei tohi olla suurem kui 10 % täisskaalast.
Neid tingimusi võib muuta kolonni ja gaasikromatograafi omaduste arvessevõtmiseks, et eraldada kõik vahad ja rasvhapete metüül- ja etüülestrid ning saavutada rahuldav piikide lahutus (vt joonised 2, 3 ja 4) ning sisestandardi laurüülarahidaadi retentsiooniaeg 18 ± 3 minutit. Kõige suurem vahade piik peab olema üle 60 % koguskaalast ning metüül- ja etüülestrite sisestandardi metüülheptadekanaadi piik peab ulatuma skaala lõpuni.
Piigi integreerimisparameetrid tuleks määrata kindlaks nii, et uuritavaid piigipindalasid oleks võimalik õigesti hinnata.
5.3. Analüüsi läbiviimine
Võetakse 10 μl mikrosüstlaga kuni 10 μl lahust ja tõmmatakse kolb tagasi, kuni süstla nõel on tühi. Nõel viiakse injektsioonisüsteemi ning 1–2 sekundi pärast süstitakse kiiresti. Umbes 5 sekundi pärast tõmmatakse nõel ettevaatlikult välja.
Tulemusi registreeritakse seni, kuni vahad või stigmastadieenid on täielikult elueeritud, olenevalt analüüsitavast fraktsioonist.
Nulljoon peab kogu aeg vastama nõuetele.
5.4. Piikide määramine
Piigid määratakse retsentsiooniaegade põhjal, võrreldes samadel tingimusel analüüsitud vahasegudega, mille retentsiooniajad on teada. Alküülestrid määratakse oliiviõlides esinevate peamiste rasvhapete (palmitiin- ja oleiinhape) metüül- ja etüülestrite segust.
Joonisel 1 on esitatud neitsioliiviõli vahade kromatogramm. Joonistel 2 ja 3 on näidatud kahe jaemüügis oleva ekstra neitsioliiviõli kromatogrammid, neist üks on metüül- ja etüülestritega ja teine ilma. Joonisel 4 on väga kvaliteetse ekstra neitsioliiviõli kromatogramm ja sellise segu kromatogramm, milles samale õlile on lisatud 20 % lõhnatustatud õli.
5.5. Vahade kvantitatiivne määramine
Sisestandardile laurüülarahidaadile ja alifaatsetele estritele C40–C46 vastavate piikide pindala arvutatakse integraatori abil.
Kõigi üksikute vahade sisalduse (mg rasva kg kohta) liitmisega määratakse vahade summaarne sisaldus järgmise valemiga:

kus:
|
Ax |
= |
igale üksikule estrile vastava piigi pindala, leitud arvutiga; |
|
As |
= |
sisestandardile laurüülarahidaadile vastava piigi pindala, leitud arvutiga; |
|
ms |
= |
lisatud sisestandardi laurüülarahidaadi mass milligrammides; |
|
m |
= |
määramisel kasutatud proovi mass (g). |
5.5.1. Metüül- ja etüülestrite kvantitatiivne määramine
Integraatori abil määratakse sisestandardile metüülheptadekanaadile vastava piigi ning C16- ja C18-rasvhapete metüül- ja etüülestritele vastavate piikide pindalad.
Iga alküülestri sisaldus (mg rasva kg kohta) määratakse järgmise valemiga:

kus:
|
Ax |
= |
igale üksikule C16- ja C18-estrile vastava piigi pindala, leitud arvutiga; |
|
As |
= |
sisestandardile metüülheptadekanaadile vastava piigi pindala, leitud arvutiga; |
|
ms |
= |
lisatud sisestandardi metüülheptadekanaadi mass milligrammides; |
|
m |
= |
määramisel kasutatud proovi mass (g). |
6. TULEMUSTE VÄLJENDAMINE
Esitatakse eri vahade (C40–C46 (vt märkus 7)) sisalduste summa milligrammides rasva kilogrammi kohta.
Esitatakse metüül- ja etüülestrite (C16–C18) sisalduste summa ja kahe kõnealuse väärtuse kogusumma.
Tulemused ühikutes mg/kg ümardatakse lähima täisarvuni.
Märkus 7. Koguseliselt määratavate komponentide sisaldus leitakse paarisarvulise süsinikuarvuga C40- kuni C46-estrite piikidest, vastavalt juurdelisatud joonisel esitatud oliiviõli vahade näidiskromatogrammile. Kui C46-estri piik on lõhestunud, soovitatakse komponentide kindlakstegemiseks analüüsida oliivijääkõli vahade fraktsiooni, milles C46 piik on eristatav, kuna see on selgelt suurem.
Esitatakse etüülestrite ja metüülestrite suhtarv.
Joonis 1
Oliiviõli vahafraktsiooni gaasikromatogrammi näide ( 7 )

Rasvhapete metüül- ja etüülestrite piigid retentsiooniajaga 5–8 minutit:
Selgitus:
|
I.S. |
= |
laurüülarahidaat (sisestandard) |
|
1 |
= |
diterpeenestrid |
|
2+2′ |
= |
C40-estrid |
|
3+3′ |
= |
C42-estrid |
|
4+4′ |
= |
C44-estrid |
|
5 |
= |
C46-estrid |
|
6 |
= |
steroolestrid ja triterpeenalkoholid |
Joonis 2
Neitsioliiviõli metüülestrid, etüülestrid ja vahad

Selgitus:
|
1 |
– |
metüül-C16 |
|
2 |
– |
etüül-C16 |
|
3 |
– |
metüülheptadekanaat (sisestandard) |
|
4 |
– |
metüül-C18 |
|
5 |
– |
etüül-C18 |
|
6 |
– |
skvaleen |
|
7 |
– |
laurüülarahidaat (sisestandard) |
|
A |
– |
diterpeenestrid |
|
B |
– |
vahad |
|
C |
– |
steroolestrid ja triterpeenestrid |
Joonis 3
Ekstra neitsioliiviõli metüülestrid, etüülestrid ja vahad
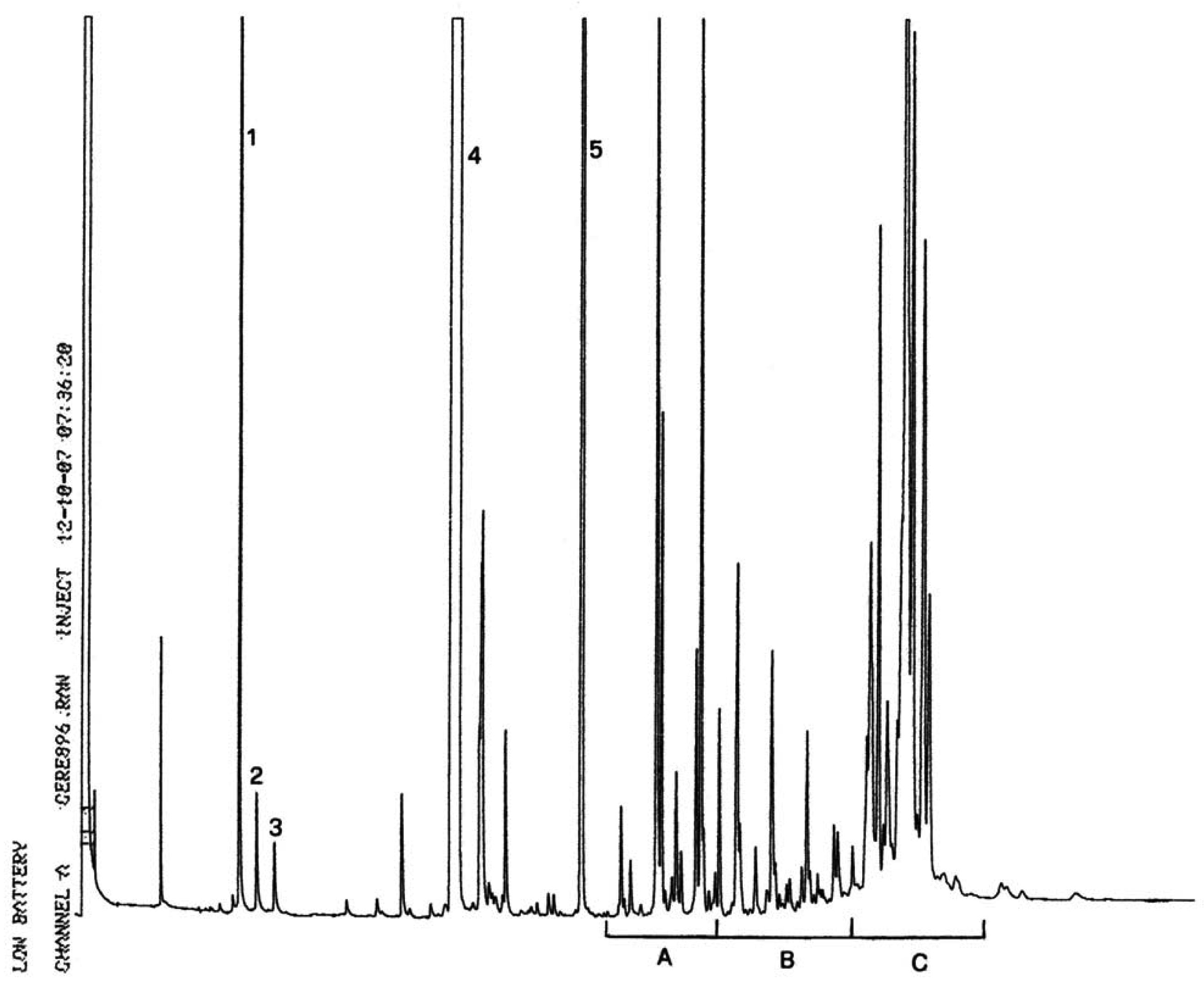
Selgitus:
|
1 |
– |
metüülheptadekanaat (sisestandard) |
|
2 |
– |
metüül-C18 |
|
3 |
– |
etüül-C18 |
|
4 |
– |
skvaleen |
|
5 |
– |
laurüülarahidaat (sisestandard) |
|
A |
– |
diterpeenestrid |
|
B |
– |
vahad |
|
C |
– |
steroolestrid ja triterpeenestrid |
Joonis 4
Osa ekstra neitsioliiviõli kromatogrammist ja sellise segu kromatogrammist, milles samale õlile on lisatud lõhnatustatud õli

Selgitus:
|
1 |
– |
metüülmüristaat (sisestandard) |
|
2 |
– |
metüülpalmitaat |
|
3 |
– |
etüülpalmitaat |
|
4 |
– |
metüülheptadekanaat (sisestandard) |
|
5 |
– |
metüüllinoleaat |
|
6 |
– |
metüüloleaat |
|
7 |
– |
metüülstearaat |
|
8 |
– |
etüüllinoleaat |
|
9 |
– |
etüüloleaat |
|
10 |
– |
etüülstearaat |
Liide A
Gaasi voolukiiruse määramine
Pärast gaasikromatograafi viimist tavapärastele töötingimustele süstitakse sellesse 1–3 μl metaani (või propaani). Mõõdetakse aeg, mis kulub gaasi voolamiseks läbi kolonni alates süstimisest kuni piigi ilmumiseni (tM).
Voolukiirus cm/s avaldub valemiga L/tM, kus L on kolonni pikkus sentimeetrites ja tM on mõõdetud aeg sekundites.
▼M28 —————
XXI LISA
Oliiviõlide vastavuse artikli 8 lõikes 2 osutatud kontrollimise tulemused
|
|
Märgistamine |
Keemilised näitajad |
Organoleptilised omadused (4) |
Lõppjäreldus |
|||||||||||||
|
Proov |
Kategooria |
Päritoluriik |
Kontrollimise koht (1) |
Ametlik nimi |
Päritolu-nimetus |
Säilitustingimused |
Vigane teave |
Loetavus |
V/EV (3) |
Näitajate kõrvalekalded on üle piiri Jah/Ei |
Kui „jah”, siis millised (2) |
V/EV (3) |
Puuduse mediaan |
Puuviljasuse mediaan |
V/EV (3) |
Vajalik meede |
Karistused |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Siseturg (tootmiskäitis, villimiskäitis, jaemüügietapp), eksport, import. (2) Igale I lisas esitatud oliiviõli näitajale on omistatud kood. (3) Vastab / Ei vasta. (4) Ei nõuta oliiviõli ja oliivijääkõli puhul. |
|||||||||||||||||
( 1 ) ELT L 228, 1.9.2009, lk 3.
( 2 ) Euroopa Parlamendi ja nõukogu direktiiv 2011/91/EL, 13. detsember 2011, toidupartiide tähistamise ja märgistamise kohta (ELT L 334, 16.12.2011, lk 1).
( 3 ) Pärast steroolestrite väljumist ei tohi kromatogrammil enam ilmuda märkimisväärseid piike (triglütseriide).
( 4 ) Euroopa Parlamendi ja nõukogu määrus (EL) nr 1308/2013, 17. detsember 2013, millega kehtestatakse põllumajandustoodete ühine turukorraldus ning millega tunnistatakse kehtetuks nõukogu määrused (EMÜ) nr 922/72, (EMÜ) nr 234/79, (EÜ) nr 1037/2001 ja (EÜ) nr 1234/2007 (ELT L 347, 20.12.2013, lk 671).
( 5 ) Degusteerija võib õli maitsmisest loobuda, kui ta tajub otsese haistmismeelega äärmiselt intensiivset negatiivset omadust; sel juhul märgib ta kõnealuse erandliku asjaolu profiililehele.
( 6 ) In these cases in particular, a 95/5 by volume benzene/acetone eluent mixture must be used to obtain distinct band separation.
( 7 ) Pärast steroolestrite väljumist ei tohi kromatogrammil enam ilmuda märkimisväärseid piike (triatsüülglütseriide).