ISSN 1977-0685
Diario Oficial
de la Unión Europea
L 180

Edición en lengua española
Legislación
64.° año
21 de mayo de 2021
|
ISSN 1977-0685 |
||
|
Diario Oficial de la Unión Europea |
L 180 |
|
|
|
||
|
Edición en lengua española |
Legislación |
64.° año |
|
|
|
|
|
(1) Texto pertinente a efectos del EEE. |
|
ES |
Los actos cuyos títulos van impresos en caracteres finos son actos de gestión corriente, adoptados en el marco de la política agraria, y que tienen generalmente un período de validez limitado. Los actos cuyos títulos van impresos en caracteres gruesos y precedidos de un asterisco son todos los demás actos. |
II Actos no legislativos
REGLAMENTOS
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/1 |
REGLAMENTO DE EJECUCIÓN (UE) 2021/804 DEL CONSEJO
de 20 de mayo de 2021
por el que se aplica el artículo 15, apartado 3, del Reglamento (UE) n.o 747/2014 relativo a medidas restrictivas habida cuenta de la situación en Sudán
EL CONSEJO DE LA UNIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (UE) n.o 747/2014 del Consejo, de 10 de julio de 2014, relativo a medidas restrictivas habida cuenta de la situación en Sudán, y por el que se derogan los Reglamentos (CE) n.o 131/2004 y (CE) n.o 1184/2005 (1), y en particular su artículo 15, apartado 3,
Vista la propuesta del Alto Representante de la Unión para Asuntos Exteriores y Política de Seguridad,
Considerando lo siguiente:
|
(1) |
El 10 de julio de 2014, el Consejo adoptó el Reglamento (UE) n.o 747/2014. |
|
(2) |
El 5 de marzo de 2021, el Comité del Consejo de Seguridad de las Naciones Unidas, creado en virtud de la Resolución 1591 (2005) de dicho Consejo de Seguridad, decidió suprimir el nombre de una persona de la lista de personas y entidades sujetas a medidas restrictivas. |
|
(3) |
Por lo tanto, procede modificar el anexo I del Reglamento (UE) n.o 747/2014 en consecuencia. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
El anexo I del Reglamento (UE) n.o 747/2014 se modifica como se establece en el anexo del presente Reglamento.
Artículo 2
El presente Reglamento entrará en vigor el día de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 20 de mayo de 2021.
Por el Consejo
El Presidente
A. SANTOS SILVA
ANEXO
En la lista que figura en el anexo I del Reglamento (UE) n.o 747/2014, se suprime la entrada correspondiente a la siguiente persona:
|
3. |
SHAREIF, Adam. |
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/3 |
REGLAMENTO DELEGADO (UE) 2021/805 DE LA COMISIÓN
de 8 de marzo de 2021
por el que se modifica el anexo II del Reglamento (UE) 2019/6 del Parlamento Europeo y del Consejo
(Texto pertinente a efectos del EEE)
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (UE) 2019/6 del Parlamento Europeo y del Consejo, de 11 de diciembre de 2018, sobre medicamentos veterinarios y por el que se deroga la Directiva 2001/82/CE (1), y en particular su artículo 146, apartado 2,
Considerando lo siguiente:
|
(1) |
Procede actualizar sustancialmente los requisitos establecidos en el anexo II del Reglamento (UE) 2019/6, en el que se retomaron los requisitos aplicables a los expedientes establecidos en el anexo I de la Directiva 2001/82/CE del Parlamento Europeo y del Consejo (2), ya que dicho Reglamento no actualizó tales requisitos al derogar la Directiva. Los requisitos aplicables a los expedientes establecidos en el anexo I de la Directiva 2001/82/CE se actualizaron por última vez en 2009. Por consiguiente, procede modificar el anexo II para tener en cuenta el progreso y los avances científicos desde 2009, como las directrices internacionales de la Conferencia Internacional sobre Armonización de los Requisitos Técnicos para el Registro de los Medicamentos de Uso Veterinario (VICH), la Organización Mundial de la Salud (OMS) y las normas de la Organización de Cooperación y Desarrollo Económicos (OCDE). |
|
(2) |
También procede establecer requisitos para los medicamentos veterinarios biológicos y los medicamentos veterinarios para nuevas terapias introducidos como categorías nuevas de medicamentos veterinarios por el Reglamento (UE) 2019/6. Es preciso definir para estos productos los requisitos técnicos específicos que deben cumplirse al solicitar una autorización de comercialización. |
|
(3) |
Reconociendo que la resistencia a los antimicrobianos provocada por los medicamentos es un problema de salud creciente en la Unión y en todo el mundo, el Reglamento (UE) 2019/6 introdujo disposiciones jurídicas específicas destinadas a limitar el riesgo de desarrollo de resistencia a los antimicrobianos. Por tanto, procede introducir requisitos técnicos específicos para los medicamentos veterinarios antimicrobianos. |
|
(4) |
El presente Reglamento debe aplicarse a partir del 28 de enero de 2022, de conformidad con el artículo 153, apartado 3, del Reglamento (UE) 2019/6. |
|
(5) |
Procede, por tanto, modificar el Reglamento (UE) 2019/6 en consecuencia. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
El anexo II del Reglamento (UE) 2019/6 se sustituye por el texto que figura en el anexo del presente Reglamento.
Artículo 2
El presente Reglamento entrará en vigor a los veinte días de su publicación en el Diario Oficial de la Unión Europea.
Será aplicable a partir del 28 de enero de 2022.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 8 de marzo de 2021.
Por la Comisión
La Presidenta
Ursula VON DER LEYEN
(1) DO L 4 de 7.1.2019, p. 43.
(2) Directiva 2001/82/CE del Parlamento Europeo y del Consejo, de 6 de noviembre de 2001, por la que se establece un código comunitario sobre medicamentos veterinarios (DO L 311 de 28.11.2001, p. 1).
ANEXO
«ANEXO II
REQUISITOS CONTEMPLADOS EN EL ARTÍCULO 8, APARTADO 1, LETRA B)
Índice
|
SECCIÓN I |
PRINCIPIOS Y REQUISITOS GENERALES | 11 |
|
I.1. |
Principios generales | 11 |
|
I.2. |
Requisitos aplicables a la composición de los expedientes | 11 |
|
I.2.1. |
Parte 1: Resumen del expediente | 11 |
|
I.2.2. |
Parte 2: Documentación sobre calidad (información fisicoquímica, biológica o microbiológica) | 12 |
|
I.2.3. |
Parte 3: Documentación relativa a la seguridad (pruebas de seguridad y de residuos) | 13 |
|
I.2.4. |
Parte 4: Documentación sobre la eficacia (estudios preclínicos y ensayos clínicos) | 13 |
|
I.2.5. |
Requisitos detallados para distintos tipos de medicamentos veterinarios o de expedientes de solicitud de autorización de comercialización | 14 |
|
SECCIÓN II |
REQUISITOS RELATIVOS A LOS MEDICAMENTOS VETERINARIOS NO BIOLÓGICOS | 14 |
|
II.1. |
PARTE 1: Resumen del expediente | 14 |
|
II.2. |
PARTE 2: Documentación sobre calidad (información fisicoquímica, biológica o microbiológica) | 14 |
|
II.2A. |
Descripción del producto | 14 |
|
II.2A1. |
Composición cualitativa y cuantitativa | 14 |
|
II.2A2. |
Desarrollo farmacéutico | 16 |
|
II.2B. |
Descripción del método de fabricación | 16 |
|
II.2C. |
Producción y control de los materiales de partida | 16 |
|
II.2C1. |
Principios activos | 17 |
|
II.2C1.1. |
Principios activos descritos en las farmacopeas | 18 |
|
II.2C1.2. |
Principios activos no descritos en una farmacopea | 18 |
|
II.2C1.3. |
Características fisicoquímicas que pueden modificar la biodisponibilidad | 18 |
|
II.2C2. |
Excipientes | 19 |
|
II.2C3. |
Envases (recipientes y sistemas de cierre) | 19 |
|
II.2C3.1. |
Principio activo | 19 |
|
II.2C3.2. |
Producto terminado | 19 |
|
II.2C4. |
Sustancias de origen biológico | 20 |
|
II.2D. |
Pruebas de control efectuadas en sustancias intermedias aisladas durante el proceso de fabricación | 20 |
|
II.2E. |
Pruebas de control del producto terminado | 20 |
|
II.2E1. |
Características generales del producto terminado | 21 |
|
II.2E2. |
Identificación y determinación de los principios activos | 21 |
|
II.2E3. |
Identificación y determinación de los excipientes | 21 |
|
II.2E4. |
Controles microbiológicos | 21 |
|
II.2E5. |
Constancia entre lotes | 21 |
|
II.2E6. |
Otros controles | 22 |
|
II.2F. |
Estudio de estabilidad | 22 |
|
II.2F1. |
Principios activos | 22 |
|
II.2F2. |
Producto terminado | 22 |
|
II.2G. |
Otra información | 23 |
|
II.3 |
PARTE 3: Documentación relativa a la seguridad (pruebas de seguridad y de residuos) | 23 |
|
II.3A. |
Pruebas de seguridad | 23 |
|
II.3A1. |
Identificación exacta del medicamento y sus principios activos | 24 |
|
II.3A2. |
Farmacología | 24 |
|
II.3A2.1. |
Farmacodinamia | 24 |
|
II.3A2.2. |
Farmacocinética | 25 |
|
II.3A3. |
Toxicología | 25 |
|
II.3A4. |
Otros requisitos | 26 |
|
II.3A.4.1. |
Estudios especiales | 26 |
|
II.3A.4.2. |
Observaciones sobre el uso terapéutico en personas | 26 |
|
II.3A.4.3. |
Aparición de resistencia y riesgo para las personas | 27 |
|
II.3A5. |
Seguridad para el usuario | 27 |
|
II.3A6. |
Evaluación del riesgo medioambiental | 27 |
|
II.3B. |
Estudio de los residuos | 28 |
|
II.3B1. |
Identificación del medicamento | 28 |
|
II.3B2. |
Eliminación de los residuos (metabolismo y cinética de los residuos) | 28 |
|
II.3B3. |
Método de análisis de residuos | 29 |
|
II.4. |
PARTE 4: Documentación sobre la eficacia (estudios preclínicos y ensayos clínicos) | 29 |
|
II.4A. |
Estudios preclínicos | 29 |
|
II.4A1. |
Farmacología | 29 |
|
II.4A.1.1. |
Farmacodinamia | 29 |
|
II.4A.1.2. |
Farmacocinética | 29 |
|
II.4A2. |
Aparición de resistencia y riesgo para los animales | 30 |
|
II.4A3. |
Determinación y confirmación de la dosis | 30 |
|
II.4A4. |
Tolerancia en la especie animal de destino | 30 |
|
II.4B. |
Ensayos clínicos | 31 |
|
II.4B1. |
Principios generales | 31 |
|
II.4B2. |
Documentación | 31 |
|
II.4AB2.1. |
Resultado de los estudios preclínicos | 31 |
|
II.4AB2.2. |
Resultado de los ensayos clínicos | 32 |
|
SECCIÓN III |
REQUISITOS RELATIVOS A LOS MEDICAMENTOS VETERINARIOS BIOLÓGICOS | 32 |
|
SECCIÓN IIIa |
REQUISITOS RELATIVOS A LOS MEDICAMENTOS VETERINARIOS BIOLÓGICOS NO INMUNOLÓGICOS | 33 |
|
IIIa.1. |
PARTE 1: Resumen del expediente | 33 |
|
IIIa.2. |
PARTE 2: Documentación sobre calidad (información fisicoquímica, biológica o microbiológica) | 33 |
|
IIIa.2A. |
Descripción del producto | 33 |
|
IIIa.2A1. |
Composición cualitativa y cuantitativa | 33 |
|
IIIa.2A2. |
Desarrollo farmacéutico | 34 |
|
IIIa.2A3. |
Caracterización | 34 |
|
IIIa.2A3.1. |
Elucidación de la estructura y otras características | 34 |
|
IIIa.2A3.2. |
Impurezas | 35 |
|
IIIa.2B. |
Descripción del método de fabricación | 35 |
|
IIIa.2C. |
Producción y control de los materiales de partida | 35 |
|
IIIa.2C1. |
Materiales de partida descritos en las farmacopeas | 36 |
|
IIIa.2C2. |
Materiales de partida no descritos en una farmacopea | 36 |
|
IIIa.2C2.1. |
Materiales de partida de origen biológico | 36 |
|
IIIa.2C2.2. |
Materiales de partida de origen no biológico | 37 |
|
IIIa.2D. |
Pruebas de control efectuadas durante el proceso de fabricación | 37 |
|
IIIa.2E. |
Pruebas de control del producto terminado | 38 |
|
IIIa.2E1 |
Especificaciones del producto terminado | 38 |
|
IIIa.2E2 |
Descripciones del método y validación de pruebas de liberación | 38 |
|
IIIa.2E3. |
Normas o materiales de referencia | 39 |
|
IIIa.2F. |
Constancia entre lotes | 39 |
|
IIIa.2F1. |
Principio activo | 39 |
|
IIIa.2F2. |
Producto terminado | 39 |
|
IIIa.2G. |
Estudios de estabilidad | 39 |
|
IIIa.2H. |
Otra información | 40 |
|
IIIa.3. |
PARTE 3: Documentación relativa a la seguridad (pruebas de seguridad y de residuos) | 40 |
|
IIIa.3A. |
Pruebas de seguridad | 41 |
|
IIIa.3A1. |
Identificación exacta del medicamento y sus principios activos | 41 |
|
IIIa.3A2. |
Farmacología | 41 |
|
IIIa.3A2.1. |
Farmacodinamia | 42 |
|
IIIa.3A2.2. |
Farmacocinética | 42 |
|
IIIa.3A3. |
Toxicología | 42 |
|
IIIa.3A3.1. |
Toxicidad por dosis única | 42 |
|
IIIa.3A3.2. |
Toxicidad por administración repetida | 42 |
|
IIIa.3A3.3. |
Tolerancia en la especie de destino | 43 |
|
IIIa.3A3.4. |
Efectos tóxicos en la función reproductora, incluida la teratogenicidad | 43 |
|
IIIa.3A3.5. |
Genotoxicidad | 43 |
|
IIIa.3A3.6. |
Carcinogenicidad | 43 |
|
IIIa.3A3.7. |
Excepciones | 43 |
|
IIIa.3A4. |
Otros requisitos | 44 |
|
IIIa.3A4.1. |
Estudios especiales | 44 |
|
IIIa.3A4.2. |
Observaciones sobre el uso terapéutico en personas | 44 |
|
IIIa.3A4.3. |
Aparición de resistencia y riesgo para las personas | 44 |
|
IIIa.3A5. |
Seguridad para el usuario | 45 |
|
IIIa.3A6. |
Evaluación del riesgo medioambiental | 45 |
|
IIIa.3A6.1. |
Evaluación del riesgo medioambiental de medicamentos veterinarios que no contienen o no consisten en organismos modificados genéticamente | 45 |
|
IIIa.3A6.2. |
Evaluación del riesgo medioambiental de medicamentos veterinarios que contienen o consisten en organismos modificados genéticamente | 45 |
|
IIIa.3B. |
Evaluación del riesgo medioambiental de medicamentos veterinarios que contienen o consisten en organismos modificados genéticamentePruebas de residuos | 46 |
|
IIIa.3B1. |
Identificación del medicamento | 46 |
|
IIIa.3B2. |
Eliminación de los residuos | 46 |
|
IIIa.3B3. |
Método de análisis de residuos | 46 |
|
IIIa.4. |
PARTE 4: Documentación sobre la eficacia (estudios preclínicos y ensayos clínicos) | 47 |
|
IIIa.4A. |
Estudios preclínicos | 47 |
|
IIIa.4A1. |
Farmacología | 47 |
|
IIIa.4A1.1. |
Farmacodinamia | 47 |
|
IIIa.4A1.2. |
Farmacocinética | 47 |
|
IIIa.4A2. |
Aparición de resistencia y riesgo para los animales | 48 |
|
IIIa.4A3. |
Determinación y confirmación de la dosis | 48 |
|
IIIa.4A4. |
Tolerancia en la especie animal de destino | 48 |
|
IIIa.4B. |
Ensayos clínicos | 48 |
|
IIIa.4B1. |
Principios generales | 48 |
|
IIIa.4B2. |
Documentación | 49 |
|
IIIa.4B2.1. |
Resultado de los estudios preclínicos | 49 |
|
IIIa.4B2.2. |
Resultado de los ensayos clínicos | 49 |
|
SECCIÓN IIIb |
REQUISITOS RELATIVOS A LOS MEDICAMENTOS VETERINARIOS INMUNOLÓGICOS | 50 |
|
IIIb.1. |
PARTE 1: Resumen del expediente | 50 |
|
IIIb.2. |
PARTE 2: Documentación sobre calidad (información fisicoquímica, biológica y microbiológica) | 50 |
|
IIIb.2.A. |
Descripción del producto | 50 |
|
IIIb.2A1. |
Composición cualitativa y cuantitativa | 50 |
|
IIIb.2A2. |
Desarrollo farmacéutico | 51 |
|
IIIb.2B. |
Descripción del método de fabricación | 52 |
|
IIIb.2C. |
Producción y control de los materiales de partida | 52 |
|
IIIb.2C1. |
Materiales de partida descritos en las farmacopeas | 53 |
|
IIIb.2C2. |
Materiales de partida no descritos en una farmacopea | 53 |
|
IIIb.2C2.1. |
Materiales de partida de origen biológico | 53 |
|
IIIb.2C2.2. |
Materiales de partida de origen no biológico | 54 |
|
IIIb.2D. |
Pruebas de control efectuadas durante el proceso de fabricación | 54 |
|
IIIb.2E. |
Pruebas de control del producto terminado | 55 |
|
IIIb.2F. |
Constancia entre lotes | 56 |
|
IIIb.2G. |
Estudios de estabilidad | 56 |
|
IIIb.2H. |
Otra información | 57 |
|
IIIb.3. |
PARTE 3: Documentación relativa a la seguridad (pruebas de seguridad y de residuos) | 57 |
|
IIIb.3A. |
Requisitos generales | 57 |
|
IIIb.3B. |
Estudios preclínicos | 58 |
|
IIIb.3C. |
Ensayos clínicos | 60 |
|
IIIb.3D. |
Evaluación del riesgo medioambiental | 60 |
|
IIIb.3E. |
Evaluación de los medicamentos veterinarios que contienen o consisten en organismos modificados genéticamente | 61 |
|
IIIb.3F. |
Pruebas de residuos que deben incluirse en los estudios preclínicos | 61 |
|
IIIb.4. |
PARTE 4: Documentación sobre la eficacia (estudios preclínicos y ensayos clínicos) | 61 |
|
IIIb.4A. |
Requisitos generales | 61 |
|
IIIb.4B. |
Estudios preclínicos | 62 |
|
IIIb.4C. |
Ensayos clínicos | 63 |
|
SECCIÓN IV |
REQUISITOS RELATIVOS A SOLICITUDES ESPECÍFICAS DE AUTORIZACIÓN DE COMERCIALIZACIÓN | 64 |
|
IV.1. |
Solicitudes para medicamentos veterinarios genéricos | 64 |
|
IV.2. |
Solicitudes para medicamentos veterinarios híbridos | 65 |
|
IV.3. |
Solicitudes para medicamentos veterinarios basados en la combinación de principios activos | 66 |
|
IV.4. |
Solicitudes basadas en el consentimiento informado | 66 |
|
IV.5. |
Solicitudes basadas en datos bibliográficos | 66 |
|
IV.6. |
Solicitudes para mercados limitados | 68 |
|
IV.7. |
Solicitudes en circunstancias excepcionales | 68 |
|
SECCIÓN V |
REQUISITOS RELATIVOS A SOLICITUDES DE AUTORIZACIÓN DE COMERCIALIZACIÓN DE DETERMINADOS MEDICAMENTOS VETERINARIOS | 68 |
|
V.1. |
Medicamentos veterinarios para nuevas terapias | 68 |
|
V.1.1. |
Requisitos generales | 68 |
|
V.1.2. |
Requisitos de calidad | 69 |
|
V.1.3. |
Requisitos de seguridad | 70 |
|
V.1.4. |
Requisitos de eficacia | 70 |
|
V.1.5. |
Requisitos de datos específicos aplicables a determinados tipos de medicamentos para nuevas terapias | 70 |
|
V.1.5.1. |
Principios | 70 |
|
V.1.5.2. |
Medicamentos veterinarios para terapia génica | 70 |
|
V.1.5.3. |
Medicamentos veterinarios de medicina regenerativa, ingeniería tisular y terapia celular | 71 |
|
V.1.5.4. |
Medicamentos veterinarios diseñados específicamente para terapia de fagos | 72 |
|
V.1.5.5. |
Medicamentos veterinarios generados a partir de nanotecnologías | 72 |
|
V.1.5.6. |
Medicamentos de terapia de ARN de antisentido y de terapia de interferencia por ARN | 73 |
|
V.2. |
Archivo maestro de antígenos vacunales | 74 |
|
V.3. |
Expediente multicepas | 75 |
|
V.4. |
Tecnología de plataforma vacunal | 75 |
|
V.5. |
Medicamentos veterinarios homeopáticos autorizados | 76 |
SECCIÓN I
PRINCIPIOS Y REQUISITOS GENERALES
I.1. Principios generales
|
I.1.1. |
La documentación que acompañe a una solicitud de autorización de comercialización con arreglo a los artículos 8 y 18 a 25 se presentará de conformidad con los requisitos establecidos en el presente anexo y tendrá en cuenta los documentos de orientación publicados por la Comisión y los requisitos relativos al modelo de presentación por vía electrónica publicados por la Agencia. |
|
I.1.2. |
Al constituir el expediente de solicitud de autorización de comercialización, los solicitantes deberán tener asimismo en cuenta los conocimientos veterinarios más actualizados y las directrices científicas sobre la calidad, la seguridad y la eficacia de los medicamentos veterinarios publicadas por la Agencia. |
|
I.1.3. |
A las partes correspondientes de los expedientes de los medicamentos veterinarios se aplican todas las monografías pertinentes de la Farmacopea Europea, incluidas las monografías generales y los capítulos generales. |
|
I.1.4. |
Los procesos de fabricación de los principios activos y los productos terminados deberán cumplir las prácticas correctas de fabricación. |
|
I.1.5. |
En la solicitud deberá incluirse toda la información útil para la evaluación del medicamento veterinario en cuestión, tanto si resulta favorable para el producto como si no. En particular, se proporcionarán todos los detalles pertinentes sobre todo ensayo o estudio incompleto o abandonado en relación con el medicamento veterinario. |
|
I.1.6. |
Se llevarán a cabo estudios farmacológicos, toxicológicos, de residuos y preclínicos de conformidad con las disposiciones sobre buenas prácticas de laboratorio establecidas en las Directivas 2004/10/CE (1) y 2004/9/CE (2) del Parlamento Europeo y del Consejo. |
|
I.1.7. |
Todos los experimentos con animales deberán llevarse a cabo teniendo en consideración los principios establecidos en la Directiva 2010/63/UE, independientemente del lugar donde se efectúen. |
|
I.1.8. |
El expediente incluirá un documento separado con la evaluación del riesgo medioambiental relacionado con la aprobación de medicamentos veterinarios que contengan o consistan en organismos modificados genéticamente en el sentido del artículo 2 de la Directiva 2001/18/CE. La información se presentará de conformidad con las disposiciones de la Directiva 2001/18/CE, teniendo en cuenta los documentos de orientación publicados por la Comisión. |
|
I.1.9. |
En la parte 1 del expediente de una solicitud de autorización de comercialización, el solicitante confirmará que ninguno de los datos presentados relativos a la calidad, la seguridad y la eficacia del medicamento veterinario, incluidos los que estén a disposición pública, está sujeto a protección de la documentación técnica. |
I.2. Requisitos aplicables a la composición de los expedientes
Todo expediente relativo a una solicitud de autorización de comercialización de un medicamento veterinario constará de las partes siguientes:
I.2.1. Parte 1: Resumen del expediente
La parte 1 incluirá la información administrativa indicada en el anexo I, a saber:
|
a) |
parte 1A: los puntos 1 a 4, y 6.1 a 6.4; |
|
b) |
parte 1B: el punto 5; |
|
c) |
parte 1C: el punto 6.5. |
Por lo que respecta a la parte 1B, punto 5.1, en conexión con el artículo 35, apartado 1, letra l), una solicitud en la que se proponga la clasificación de un medicamento veterinario como «no sujeto a prescripción veterinaria» incluirá una revisión crítica de las características del medicamento para justificar la adecuación de esa clasificación teniendo en cuenta la seguridad para la especie animal de destino y otras que no sean de destino, la salud pública y la seguridad medioambiental, como se indica en los criterios que figuran en el artículo 34, apartado 3, letras a) a g).
Cada informe pericial crítico se elaborará teniendo en cuenta el estado del conocimiento científico en el momento de la presentación de la solicitud. Contendrá una evaluación de los diversos ensayos y pruebas que constituyen el expediente de solicitud de autorización de comercialización, y abordará todos los aspectos pertinentes para evaluar la calidad, la seguridad y la eficacia del medicamento veterinario. Dará resultados detallados de las pruebas y de los ensayos realizados y referencias bibliográficas exactas. Se proporcionarán copias de las referencias bibliográficas citadas.
Los informes periciales críticos irán firmados y fechados por su autor correspondiente y contendrán información sobre la educación, la formación académica y la experiencia profesional de esa persona. Se hará constar la relación profesional del autor con el solicitante.
Estos informes periciales críticos y sus apéndices remitirán con precisión y claridad a la información contenida en la documentación técnica.
Cuando la parte 2 se presente con el formato de documento técnico común (DTC), el resumen global de la calidad se usará para el informe pericial crítico sobre calidad.
En el caso de las partes 3 y 4, el informe pericial crítico incluirá también un resumen tabulado de toda la documentación técnica y todos los datos pertinentes que se hayan presentado.
I.2.2. Parte 2: Documentación sobre calidad (información fisicoquímica, biológica o microbiológica)
|
1) |
Figurarán como información farmacéutica sobre calidad (fisicoquímica, biológica o microbiológica) del principio activo y del medicamento veterinario terminado datos del proceso de fabricación, la caracterización y las propiedades, los procedimientos y requisitos de control de calidad, la estabilidad, así como una descripción de la composición, el desarrollo y la presentación del medicamento veterinario. |
|
2) |
Serán aplicables todas las monografías, incluidas las específicas, las generales y los capítulos generales de la Farmacopea Europea. En el caso de los medicamentos veterinarios inmunológicos, serán aplicables todas las monografías, incluidas las específicas, las generales y los capítulos generales de la Farmacopea Europea, salvo justificación. Cuando no exista ninguna monografía de la Farmacopea Europea, podrá aplicarse la monografía de la farmacopea de un Estado miembro. Cuando una sustancia no esté descrita en la Farmacopea Europea ni en la farmacopea de un Estado miembro, es admisible que se siga la monografía de la farmacopea de un tercer país, si queda demostrado que es adecuada; en estos casos, el solicitante presentará un ejemplar de la monografía acompañada, cuando sea necesario, de una traducción. Se presentarán los datos probatorios de que la monografía permite controlar adecuadamente la calidad de la sustancia. |
|
3) |
Si se utilizan pruebas distintas de las mencionadas en la farmacopea, dicha utilización se justificará demostrando que los materiales, en caso de someterlos a prueba conforme a la farmacopea, cumplirían los requisitos de calidad establecidos en la monografía de la farmacopea en cuestión. |
|
4) |
Todos los procedimientos para el análisis y el control de la calidad tendrán en cuenta las directrices y los requisitos establecidos. Se proporcionarán los resultados de los estudios de validación. Todos los procedimientos analíticos deberán describirse de forma suficientemente detallada, con objeto de que puedan reproducirse en las pruebas de control que se efectúen a solicitud de las autoridades competentes, para que estas puedan evaluarlos adecuadamente. Deberá asimismo describirse de manera adecuada todo instrumento o equipo especial que pueda utilizarse, incluyendo esquemas, si resulta pertinente. La composición de los reactivos de laboratorio deberá completarse, en su caso, con el método de preparación. En el caso de procedimientos analíticos incluidos en la Farmacopea Europea o en la farmacopea de un Estado miembro, esta descripción podrá sustituirse por una referencia precisa a la farmacopea en cuestión. |
|
5) |
Cuando exista, se utilizará material de referencia químico y biológico de la Farmacopea Europea. Si se utilizan otras preparaciones y normas de referencia, se identificarán y se describirán detalladamente. |
|
6) |
La información farmacéutica sobre calidad (fisicoquímica, biológica o microbiológica) del principio activo o del medicamento terminado podrá incluirse en el expediente en formato de documento técnico común (DTC). |
|
7) |
En el caso de los medicamentos veterinarios biológicos, entre ellos los inmunológicos, se incluirá en el expediente la información sobre los disolventes necesarios para hacer la preparación final. Un medicamento veterinario biológico se considerará como un solo producto aunque necesite más de un disolvente, de modo que puedan obtenerse diversas preparaciones del producto final, que podrá administrarse por diversas vías o mediante métodos diferentes. Los disolventes suministrados con medicamentos veterinarios biológicos podrán envasarse junto con los viales del principio activo o por separado. |
|
8) |
De conformidad con la Directiva 2010/63/UE y con el Convenio Europeo sobre Protección de los Animales Vertebrados Utilizados con Fines Experimentales y Otros Fines Científicos, las pruebas se realizarán de forma que se utilice el menor número de animales y se cause el menor dolor, sufrimiento, angustia o daño duradero. Se utilizarán pruebas alternativas in vitro, si las hay, cuando puedan sustituir o disminuir el uso de animales o reducir su sufrimiento. |
I.2.3. Parte 3: Documentación relativa a la seguridad (pruebas de seguridad y de residuos)
|
1) |
El expediente sobre los estudios de seguridad incluirá:
|
|
2) |
El expediente incluirá:
|
I.2.4. Parte 4: Documentación sobre la eficacia (estudios preclínicos y ensayos clínicos)
|
1) |
En el expediente sobre la eficacia figurará toda la documentación preclínica y clínica, ya sea favorable o desfavorable para el medicamento veterinario, que permita una evaluación general objetiva de la relación beneficio-riesgo del medicamento. |
|
2) |
El expediente sobre los estudios de eficacia incluirá:
|
|
3) |
El expediente incluirá:
|
|
4) |
El objetivo de los ensayos descritos en esta parte es demostrar la eficacia del medicamento veterinario. Todas las declaraciones realizadas por el solicitante en relación con las propiedades, los efectos y la utilización del medicamento deberán estar plenamente justificadas por los resultados de ensayos específicos incluidos en la solicitud de autorización de comercialización. |
|
5) |
Todos los ensayos de eficacia se realizarán con arreglo a un protocolo pormenorizado que deberá consignarse por escrito antes de iniciar el ensayo. Durante la elaboración de todo protocolo de ensayo y a lo largo de la realización de este, se tendrá en cuenta en todo momento el bienestar de los animales de experimentación, aspecto que será objeto de supervisión veterinaria. |
|
6) |
Salvo debida justificación, los ensayos clínicos (ensayos de campo) se llevarán a cabo de conformidad con los principios establecidos de buenas prácticas clínicas. |
|
7) |
Antes de empezar cualquier ensayo de campo, será necesario obtener y acreditar documentalmente el consentimiento informado del propietario de los animales que se vayan a utilizar en el ensayo. En particular, se informará por escrito al propietario de los animales acerca de las consecuencias de la participación en el ensayo por lo que se refiere al destino posterior de los animales tratados o a la obtención de productos alimenticios a partir de estos. |
I.2.5. Requisitos detallados para distintos tipos de medicamentos veterinarios o de expedientes de solicitud de autorización de comercialización
|
1) |
En las secciones siguientes del presente anexo se indican los requisitos detallados para distintos tipos de medicamentos veterinarios o para tipos específicos de expedientes de solicitud de autorización de comercialización:
|
SECCIÓN II
REQUISITOS RELATIVOS A LOS MEDICAMENTOS VETERINARIOS NO BIOLÓGICOS
Los siguientes requisitos detallados se aplicarán a los medicamentos veterinarios no biológicos, salvo indicación contraria en la sección IV.
II.1. Parte 1: Resumen del expediente
Véase la sección I.
II.2. Parte 2: Documentación sobre calidad (información fisicoquímica, biológica o microbiológica)
II.2A. Descripción del producto
II.2A1. Composición cualitativa y cuantitativa
|
1) |
Se entenderá por «composición cualitativa» de todos los componentes del medicamento la designación o descripción de:
|
|
2) |
Sin perjuicio de lo dispuesto en el artículo 8, se entenderá por «terminología usual» para la designación de los componentes de los medicamentos veterinarios:
|
|
3) |
Para proporcionar la composición cuantitativa de todos los principios activos y excipientes del medicamento veterinario, será preciso, según la forma farmacéutica, especificar la masa o el número de unidades de actividad biológica, bien sea por dosis o por unidad de masa o de volumen, de cada principio activo y excipiente. |
|
4) |
Las unidades de actividad biológica se emplearán en las sustancias que no pueden definirse en términos químicos. Cuando se haya definido una unidad internacional de actividad biológica, se utilizará. En los casos en los que no se haya definido una unidad internacional, las unidades de actividad biológica se expresarán de forma que proporcionen información inequívoca sobre la actividad de la sustancia, utilizando, cuando proceda, las unidades de la Farmacopea Europea. |
|
5) |
La composición cuantitativa se completará con lo siguiente:
|
|
6) |
Los principios activos presentes en forma de compuestos o derivados se designarán cuantitativamente por su masa total y, si es necesario o procedente, por la masa de las fracciones activas de la molécula. |
|
7) |
En el caso de los medicamentos veterinarios que contengan un principio activo cuya autorización de comercialización se haya solicitado en la Unión por primera vez, la declaración cuantitativa de un principio activo que sea una sal o un hidrato se expresará sistemáticamente en términos de masa de las fracciones activas de la molécula. La composición cuantitativa se expresará de la misma manera para el mismo principio activo en todas las autorizaciones ulteriores de medicamentos veterinarios que se concedan en los Estados miembros. |
II.2A2. Desarrollo farmacéutico
|
1) |
Se explicará la elección de la composición, los componentes, el acondicionamiento, el embalaje, la función prevista de los excipientes en el producto terminado y el método de fabricación, con una justificación de la selección de dicho método y detalles de los procesos de esterilización o asépticos utilizados en el producto terminado. Esta explicación se justificará con datos científicos sobre el desarrollo farmacéutico. Deberá indicarse y justificarse toda posible sobredosificación. Las características microbiológicas (pureza microbiológica y actividad antimicrobiana) y las instrucciones de uso tendrán que ser adecuadas al uso previsto del medicamento veterinario según lo especificado en el expediente de solicitud de autorización de comercialización. |
|
2) |
Se presentará un estudio sobre la interacción del medicamento terminado y el acondicionamiento primario en los casos en que el riesgo de dicha interacción se considere posible, especialmente cuando se trate de preparados inyectables. |
|
3) |
Los tamaños propuestos para el envase se justificarán en relación con la vía de administración, la posología y las especies de destino propuestas, en particular para las sustancias o los principios activos antimicrobianos. |
|
4) |
Cuando el producto terminado se suministre con un dispositivo para su dosificación, se demostrará la exactitud de las dosis. |
|
5) |
Cuando se recomiende utilizar una prueba junto con el producto terminado (por ejemplo, un estudio de diagnóstico), se proporcionará la información pertinente sobre dicha prueba. |
|
6) |
En el caso de medicamentos veterinarios que se incorporarán en piensos, se dará información sobre tasas de incorporación, instrucciones de incorporación, homogeneidad en el pienso y compatibilidad o adecuación de los piensos. |
II.2B. Descripción del método de fabricación
|
1) |
La descripción del método de fabricación que, conforme a lo establecido en el artículo 8, debe acompañar a la solicitud de autorización de comercialización se redactará de forma que ofrezca una idea clara del carácter de las operaciones efectuadas. |
|
2) |
Con este fin, deberá incluir, como mínimo:
|
II.2C. Producción y control de los materiales de partida
|
1) |
A los efectos del presente punto, se entenderá por «materiales de partida» los principios activos, los excipientes y el envase (el acondicionamiento primario con su sistema de cierre y, si procede, el embalaje exterior, junto con cualquier dispositivo para su dosificación suministrado con el medicamento veterinario). |
|
2) |
En el expediente figurará información técnica detallada sobre las pruebas que se llevarán a cabo para el control de calidad de todos los lotes de materiales de partida. |
|
3) |
Las pruebas sistemáticas que se realicen en los materiales de partida deberán llevarse a cabo tal como estén descritas en el expediente. |
|
4) |
Cuando la Dirección Europea de Calidad del Medicamento y la Asistencia Sanitaria haya emitido un certificado de conformidad de un material de partida, un principio activo o un excipiente, dicho certificado constituirá la referencia a la correspondiente monografía de la Farmacopea Europea. |
|
5) |
Cuando se haga referencia a un certificado de conformidad, el fabricante dará por escrito al solicitante garantías de que el proceso de fabricación no se ha modificado desde la emisión del certificado de conformidad por la Dirección Europea de Calidad del Medicamento y la Asistencia Sanitaria. Cuando el recuadro sobre «declaración de acceso» del certificado esté cumplimentado y firmado, ese requisito se considerará cumplido y no será preciso dar garantías adicionales. |
|
6) |
Se presentarán certificados de análisis de los materiales de partida para demostrar el cumplimiento de las características técnicas definidas. |
II.2C1. Principios activos
|
1) |
Los datos necesarios se presentarán en una de las tres formas descritas en los puntos 2 a 4 siguientes. |
|
2) |
Se presentará la información siguiente:
|
|
3) |
Archivo maestro del principio activo
En el caso de un principio activo no biológico, el solicitante puede concertar la presentación directa por el fabricante a las autoridades competentes de la información sobre dicho principio activo indicada en el punto 2 anterior en forma de archivo maestro del principio activo. En este caso, el fabricante del principio activo proporcionará al solicitante todos los datos (la parte del archivo maestro del principio activo correspondiente al solicitante) que sean necesarios para que este último pueda asumir la responsabilidad del medicamento veterinario. En el expediente del medicamento se incluirá una copia de los datos proporcionados por el fabricante del principio activo al solicitante. El fabricante del principio activo deberá comprometerse por escrito ante el solicitante a garantizar la constancia de los lotes y a no modificar el proceso de fabricación o las especificaciones sin haberle informado. |
|
4) |
Certificado de conformidad emitido por la Dirección Europea de Calidad del Medicamento y la Asistencia Sanitaria
Se proporcionará el certificado de conformidad junto con cualquier dato adicional relacionado con la presentación que no figure en este. |
II.2C1.1. Principios activos descritos en las farmacopeas
|
1) |
A efectos del cumplimiento de lo dispuesto en el artículo 8 será suficiente la conformidad de los principios activos respecto a las prescripciones de la Farmacopea Europea o, cuando no exista ninguna monografía de la Farmacopea Europea, de la farmacopea de uno de los Estados miembros. En este caso, la descripción de los métodos y procesos analíticos será reemplazada en cada sección pertinente por la correspondiente referencia a la farmacopea en cuestión. |
|
2) |
Cuando la especificación de una monografía de la Farmacopea Europea o de la farmacopea nacional de un Estado miembro no sea suficiente para garantizar la calidad de la sustancia, las autoridades competentes podrán exigir especificaciones más apropiadas al solicitante, incluidos criterios de aceptación de impurezas específicas con procedimientos analíticos validados. |
|
3) |
Las autoridades competentes informarán de ello a las autoridades responsables de la farmacopea de que se trate. El titular de la autorización de comercialización comunicará a las autoridades responsables de dicha farmacopea los pormenores de la pretendida insuficiencia y las especificaciones adicionales que se hayan aportado. |
II.2C1.2. Principios activos no descritos en una farmacopea
|
1) |
Los principios activos que no estén descritos en ninguna farmacopea serán objeto de una monografía que haga referencia a cada uno de los puntos siguientes:
|
|
2) |
Esos datos demostrarán que el conjunto propuesto de procedimientos analíticos es suficiente para verificar la calidad del principio activo de la fuente definida. |
II.2C1.3. Características fisicoquímicas que pueden modificar la biodisponibilidad
Deberán proporcionarse los datos que figuran a continuación como elementos de la descripción general de los principios activos cuando condicionen la biodisponibilidad del medicamento veterinario:
|
a) |
forma cristalina y solubilidad; |
|
b) |
tamaño de partículas; |
|
c) |
estado de hidratación; |
|
d) |
coeficiente de reparto aceite/agua; |
|
e) |
valores de pK/pH. |
Las letras a) a c) no se aplicarán a las sustancias que se utilicen únicamente en solución.
II.2C2. Excipientes
|
1) |
A efectos del cumplimiento de lo dispuesto en el artículo 8, será suficiente la conformidad de los excipientes respecto a las prescripciones de la Farmacopea Europea o, cuando no exista ninguna monografía de la Farmacopea Europea, de la farmacopea de uno de los Estados miembros. En ese caso, la descripción de los métodos y procesos analíticos será reemplazada en cada sección pertinente por la correspondiente referencia a la farmacopea en cuestión. En su caso, los requisitos de la monografía se completarán con pruebas adicionales para controlar parámetros tales como el tamaño de las partículas, la esterilidad o los disolventes residuales. |
|
2) |
Cuando no exista una monografía de una farmacopea, se propondrá y justificará una especificación. Se seguirán los requisitos establecidos para los principios activos en la parte II.2C1.2., punto 1, letras a) a e). Se presentarán los métodos propuestos y los datos de validación que los apoyan. |
|
3) |
Se presentará una declaración en la que se confirme que los colorantes que vayan a formar parte de medicamentos veterinarios cumplen los requisitos de la Directiva 2009/35/CE del Parlamento Europeo y del Consejo (3), salvo si la solicitud de autorización de comercialización se refiere a determinados medicamentos veterinarios de uso tópico, como collares medicinales y marcas crotales. |
|
4) |
Se presentará una declaración en la que se confirme que los colorantes utilizados cumplen los criterios de pureza establecidos en el Reglamento (UE) n.o 231/2012 de la Comisión (4). |
|
5) |
En el caso de excipientes nuevos, es decir, que se usan por primera vez en la Unión en un medicamento veterinario o por una nueva vía de administración, se darán los detalles de fabricación, caracterización y controles, con referencias en apoyo de los datos de seguridad tanto clínicos como no clínicos. En el caso de los colorantes, se considerarán suficientes las declaraciones de cumplimiento indicadas en los puntos 3 y 4 anteriores. |
II.2C3. Envases (recipientes y sistemas de cierre)
II.2C3.1. Principio activo
|
1) |
Se dará información sobre el recipiente y su sistema de cierre para el principio activo, que incluirá la identidad del material del acondicionamiento primario y sus especificaciones. El grado de información requerido vendrá determinado por el estado físico (sólido o líquido) del principio activo. |
|
2) |
Cuando se presente un certificado de conformidad del principio activo de la fuente propuesta en el que se especifique un recipiente y su sistema de cierre, podrá sustituirse la información detallada sobre ambos para el principio activo de esa fuente por una referencia al certificado de conformidad válido. |
|
3) |
Cuando se presente un archivo maestro del principio activo de la fuente propuesta en el que se especifique un recipiente y su sistema de cierre, podrá sustituirse la información detallada sobre ambos para el principio activo de esa fuente por una referencia al archivo maestro del principio activo. |
II.2C3.2. Producto terminado
|
1) |
Se dará información sobre el recipiente y su sistema de cierre y cualquier dispositivo del producto terminado, que incluirá la identidad del material del acondicionamiento primario y sus especificaciones. El grado de información requerido vendrá determinado por la vía de administración del medicamento veterinario y el estado físico (sólido o líquido) de la presentación. |
|
2) |
Cuando no exista una monografía de una farmacopea, se propondrá y justificará una especificación del material de envasado. |
|
3) |
Se presentará información sobre la composición, fabricación y seguridad de los materiales de envasado que se usen por primera vez en la Unión y que estén en contacto con el medicamento. |
II.2C4. Sustancias de origen biológico
|
1) |
Se proporcionará información sobre la fuente, el procesamiento, la caracterización y el control de todos los materiales de origen biológico (humano, animal, vegetal o de microorganismos) utilizados en la fabricación de los medicamentos veterinarios, incluidos los datos sobre seguridad vírica, de conformidad con las directrices pertinentes. |
|
2) |
Se suministrará documentación para demostrar que los materiales procedentes de especies animales por las que puedan transmitirse las encefalopatías espongiformes transmisibles (EET) se ajustan a la Nota explicativa sobre cómo minimizar los riesgos de transmisión de los agentes de las encefalopatías espongiformes animales a través de los medicamentos humanos y veterinarios, así como a la correspondiente monografía de la Farmacopea Europea. Como justificante de cumplimiento podrán utilizarse los certificados de conformidad emitidos por la Dirección Europea de Calidad del Medicamento y la Asistencia Sanitaria, con referencia a la correspondiente monografía de la Farmacopea Europea. |
II.2D. Pruebas de control efectuadas en sustancias intermedias aisladas durante el proceso de fabricación
|
1) |
A los efectos de la presente sección, se entenderá por «sustancia intermedia aislada» un material procesado parcialmente que puede ser almacenado durante un período de tiempo definido y que se someterá a nuevas fases de procesamiento antes de convertirse en un producto terminado. |
|
2) |
Se establecerá una especificación para cada sustancia intermedia y se describirán y validarán los métodos analíticos, si procede. |
|
3) |
Se proporcionará la información impresa en el acondicionamiento primario de la sustancia intermedia si difiere de la relativa al producto terminado. |
|
4) |
El período de validez y las condiciones de almacenamiento para la sustancia intermedia se definirán con arreglo a los datos obtenidos de los estudios de estabilidad. |
II.2E. Pruebas de control del producto terminado
|
1) |
A efectos de control del producto terminado, se entenderá por «lote» de un producto terminado el conjunto de unidades de una forma farmacéutica que provengan de una misma cantidad inicial de material y hayan sido sometidas a la misma serie de operaciones de fabricación o esterilización. En caso de un proceso de fabricación continua, el tamaño del lote podrá expresarse por período de tiempo o por cantidad de producto, y especificarse por rangos. |
|
2) |
Figurará una lista de las pruebas que se realicen en el producto terminado. Se justificará la especificación propuesta. Se hará constar y se justificará la frecuencia de las pruebas que no se lleven a cabo de forma sistemática. Se indicarán también los criterios de aceptación para la aprobación. |
|
3) |
Se adjuntarán al expediente datos sobre las pruebas de control realizadas en el producto terminado en el momento de la aprobación y sobre la validación de esas pruebas. Dichos controles se presentarán con arreglo a los requisitos siguientes. |
|
4) |
Si se emplean procedimientos analíticos y criterios de aceptación distintos de los mencionados en las monografías correspondientes y en los capítulos generales de la Farmacopea Europea o, en su defecto, de la farmacopea de un Estado miembro, se justificarán demostrando que el producto terminado, en caso de someterlo a prueba conforme a esas monografías, cumpliría los requisitos cualitativos de esa farmacopea aplicables a la forma farmacéutica de que se trate. |
II.2E1. Características generales del producto terminado
|
1) |
Entre los controles de un producto terminado figurarán siempre determinados controles de sus características generales. Estas pruebas de control se referirán, siempre que sea procedente, a la determinación de las masas o los volúmenes medios y las desviaciones máximas, estudios mecánicos o físicos, la apariencia visual, características físicas como el pH o el tamaño de las partículas. El solicitante deberá definir, para cada una de estas características, las normas y los criterios de aceptación. |
|
2) |
Las condiciones de los ensayos y, en su caso, el aparato o equipo utilizado y las normas se describirán con suficientes detalles cuando no figuren en la Farmacopea Europea o las farmacopeas de los Estados miembros, o cuando no sean aplicables los métodos prescritos por dichas farmacopeas. |
II.2E2. Identificación y determinación de los principios activos
|
1) |
La identificación y la determinación de los principios activos se efectuarán bien con una muestra representativa del lote de fabricación, bien con un determinado número de dosis analizadas aisladamente. |
|
2) |
Salvo debida justificación, la desviación máxima tolerable del contenido del principio activo en el producto terminado no podrá ser superior a ± 5 % en el momento de la fabricación. |
|
3) |
En ciertos casos excepcionales de mezclas particularmente complejas, en las que la determinación de los principios activos, muy numerosos o presentes solo en pequeñas proporciones, requiera investigaciones complejas y difícilmente aplicables a cada lote de fabricación, podrá omitirse la determinación de uno o varios principios activos en el producto terminado, con la condición expresa de que estas determinaciones se efectúen en fases intermedias del proceso de producción. Esta técnica simplificada no podrá extenderse a la caracterización de las sustancias afectadas y deberá completarse con un método de evaluación cuantitativa que permita a las autoridades competentes verificar la conformidad del medicamento con sus características técnicas después de su comercialización. |
|
4) |
Si los métodos fisicoquímicos no bastan para proporcionar suficiente información sobre la calidad del producto, será obligatorio un bioanálisis in vivo o in vitro. Siempre que sea posible, en este bioanálisis deberán emplearse materiales de referencia y un análisis estadístico que permitan calcular los límites de confianza. Cuando esos controles no puedan realizarse en el producto terminado, será admisible que se lleven a cabo en una fase intermedia, lo más cerca posible del fin del proceso de fabricación. |
|
5) |
Deberán indicarse los niveles máximos aceptables, individuales y totales, de los productos de degradación inmediatamente después de la fabricación. Se justificará la inclusión o exclusión de los productos de degradación en la especificación. |
II.2E3. Identificación y determinación de los excipientes
Se presentará obligatoriamente una prueba de identificación y una prueba de límite superior e inferior de cada conservante antimicrobiano y cada excipiente que pueda afectar a la biodisponibilidad del principio activo, a menos que esta quede garantizada por otras pruebas apropiadas. Se establecerá obligatoriamente en el momento de la aprobación una prueba de identificación y una prueba de límite superior de cualquier antioxidante o excipiente que pueda afectar adversamente a las funciones fisiológicas, y también una prueba de límite inferior de los antioxidantes.
II.2E4. Controles microbiológicos
Deberán figurar en el expediente analítico los pormenores de las pruebas microbiológicas, como las de esterilidad y de endotoxinas bacterianas, cuando deban efectuarse sistemáticamente para comprobar la calidad del producto.
II.2E5. Constancia entre lotes
Con el fin de garantizar que la calidad del medicamento es constante entre lotes y demostrar su conformidad con las especificaciones, se presentarán datos por lotes con los resultados de todas las pruebas realizadas en general sobre [tres] lotes fabricados en los lugares de fabricación propuestos según el proceso de producción descrito.
II.2E6. Otros controles
Se someterá a control cualquier otra prueba que se considere necesaria para confirmar la calidad del medicamento.
II.2F. Estudio de estabilidad
II.2F1. Principios activos
|
1) |
Se especificarán las condiciones de almacenamiento y el período de reanálisis del principio activo, excepto cuando el fabricante del producto terminado someta el principio activo a una nueva prueba completa inmediatamente antes de usarlo para la fabricación del producto terminado. |
|
2) |
Se presentarán datos de estabilidad para aportar pruebas sobre la variación de la calidad de un principio activo en el tiempo por la influencia de diversos factores medioambientales y en apoyo del período de reanálisis y de las condiciones de almacenamiento definidos, si procede. Se describirá el tipo de estudios de estabilidad realizados, los protocolos y procedimientos analíticos utilizados y su validación, junto con sus resultados detallados. |
|
3) |
Cuando exista un certificado de conformidad del principio activo de la fuente propuesta que especifique las condiciones de almacenamiento y el período de reanálisis, podrán sustituirse los datos de estabilidad del principio activo de dicha fuente por una referencia al certificado de conformidad válido. |
|
4) |
Cuando se presente un archivo maestro del principio activo de la fuente propuesta en el que se especifiquen los datos de estabilidad, podrá sustituirse la información detallada sobre la estabilidad para el principio activo de esa fuente por una referencia al archivo maestro del principio activo. |
II.2F2. Producto terminado
|
1) |
Deberán describirse las investigaciones que hayan permitido determinar el período de validez, las condiciones de almacenamiento que se recomiendan y las especificaciones al final del período de validez que propone el solicitante. |
|
2) |
Se describirá el tipo de estudios de estabilidad realizados, los protocolos y procedimientos analíticos utilizados y su validación, junto con sus resultados detallados. |
|
3) |
Si un producto terminado tiene que ser reconstituido o diluido previamente a su administración, se especificarán los pormenores relativos al período de validez y a las especificaciones que se proponen para el medicamento reconstituido o diluido, junto con los datos pertinentes sobre su estabilidad. |
|
4) |
Cuando se trate de envases multidosis, se presentarán los datos relativos a la estabilidad para justificar el período de validez del medicamento después de su primera apertura o utilización, y se definirán las especificaciones una vez abierto. |
|
5) |
Cuando un producto terminado pueda dar lugar a productos de degradación, el solicitante declarará dichos productos, indicando los métodos de identificación y los procedimientos analíticos utilizados. |
|
6) |
Cuando los datos de estabilidad muestren que la determinación del principio activo disminuye en almacenamiento, la descripción de los métodos de control del producto terminado incluirá, en su caso, el estudio químico y, si fuera necesario, el estudio toxicofarmacológico de la alteración sufrida por este principio activo, con caracterización o determinación, si ha lugar, de los productos de degradación. |
|
7) |
Deberá indicarse y justificarse el nivel máximo aceptable, individual y total, de los productos de degradación al expirar el período de validez. |
|
8) |
Sobre la base de los resultados del estudio de estabilidad, se enumerarán y justificarán las pruebas, junto con sus criterios de aceptación, que se lleven a cabo en el producto terminado durante su período de validez. |
|
9) |
En las conclusiones figurarán los resultados de los análisis y se justificará el período de validez propuesto y, si procede, el período de validez una vez abierto, en las condiciones de almacenamiento recomendadas. |
|
10) |
Además, en el caso de medicamentos veterinarios que se incorporarán en piensos, se dará información sobre la estabilidad y el período de validez propuesto después de su incorporación en los piensos. Asimismo se indicarán las especificaciones de los piensos medicamentosos fabricados con esos medicamentos veterinarios de conformidad con las instrucciones recomendadas para su uso. |
II.2G. Otra información
El expediente podrá contener en este punto información sobre la calidad del medicamento veterinario que no haya quedado cubierta en otros puntos de esta parte.
II.3 Parte 3: Documentación relativa a la seguridad (pruebas de seguridad y de residuos)
|
1) |
Cada informe de estudio incluirá:
|
|
2) |
Podrán aceptarse estudios publicados si contienen una cantidad de datos y detalles suficientes para permitir una evaluación independiente. Las técnicas experimentales se describirán de tal forma que puedan reproducirse, y quien realice la investigación demostrará su validez. No se aceptarán como documentación válida resúmenes de estudios sobre los cuales no se disponga de informes detallados. Cuando la sustancia se haya evaluado con anterioridad para establecer su límite máximo de residuos (LMR) a fin de cumplir ciertos requisitos de seguridad, podrá hacerse referencia a los informes públicos europeos de evaluación de los LMR (EPMAR, por sus siglas en inglés). Si se hace referencia a un EPMAR, no será necesario presentar estudios ya evaluados durante la evaluación del LMR; solamente se facilitarán estudios nuevos que no estuvieran disponibles para la evaluación del LMR. Si la vía de exposición (por ejemplo, para el usuario) no es idéntica a la vía utilizada de conformidad con el Reglamento (UE) 2018/782 de la Comisión (5), podrían necesitarse nuevos estudios. |
II.3A. Pruebas de seguridad
|
1) |
La documentación relativa a la seguridad será adecuada para evaluar lo siguiente:
|
|
2) |
En algunos casos podrá ser necesario someter a prueba los metabolitos del compuesto original, cuando constituyan los residuos en cuestión. |
|
3) |
Un excipiente que se utilice por primera vez en un medicamento veterinario o con una vía de administración nueva deberá considerarse como un principio activo. |
II.3A1. Identificación exacta del medicamento y sus principios activos
|
a) |
denominación común internacional (DCI); |
|
b) |
nombre de la Unión Internacional de Química Pura y Aplicada (UIQPA); |
|
c) |
número CAS (Chemical Abstract Service); |
|
d) |
clasificación terapéutica, farmacológica y química; |
|
e) |
sinónimos y abreviaturas; |
|
f) |
fórmula estructural; |
|
g) |
fórmula molecular; |
|
h) |
peso molecular; |
|
i) |
grado de pureza; |
|
j) |
composición cualitativa y cuantitativa de las impurezas; |
|
k) |
descripción de las propiedades físicas:
|
|
l) |
formulación del producto. |
II.3A2. Farmacología
|
1) |
Los estudios farmacológicos resultan fundamentales para poner en evidencia los mecanismos por los que el medicamento veterinario produce sus efectos terapéuticos y, por consiguiente, se incluirán los estudios farmacológicos realizados con especies animales de destino y de experimentación. Se hará referencia, si procede, a estudios presentados en la parte 4 del expediente. |
|
2) |
Si un medicamento veterinario produce efectos farmacológicos exentos de toxicidad, o bien los efectos se logran con dosis inferiores a las tóxicas, habrá que tener en cuenta dichos efectos farmacológicos al evaluar la seguridad del medicamento veterinario para el usuario. |
|
3) |
Los pormenores de los estudios farmacológicos practicados con animales de laboratorio, así como la información pertinente relativa a los estudios clínicos realizados con el animal de destino, deberán preceder siempre a la documentación sobre la seguridad. |
II.3A2.1 Farmacodinamia
Se dará información sobre el mecanismo de acción de los principios activos, y sobre los efectos farmacodinámicos primarios y secundarios, que ayude a comprender cualquier efecto adverso que se produzca en los estudios con animales. En la parte 4A del expediente se declarará información detallada sobre las propiedades farmacodinámicas relacionadas con el efecto terapéutico.
II.3A2.2 Farmacocinética
Se proporcionarán datos sobre el destino del principio activo y sus metabolitos en animales de laboratorio, entre los que figurarán la absorción, la distribución, el metabolismo y la excreción. Para determinar la exposición adecuada, los datos se cotejarán con los resultados de la relación dosis-efecto que figuran en los estudios farmacológicos y toxicológicos.
II.3A3. Toxicología
|
1) |
La documentación toxicológica se ajustará a las directrices publicadas por la Agencia en cuanto al planteamiento general de la prueba y a los estudios particulares. En general, los estudios de toxicidad se llevarán a cabo con el principio activo, no con el medicamento formulado, a menos que se exija específicamente lo contrario. |
|
2) |
Los estudios con animales se realizarán en cepas establecidas de animales de laboratorio para las cuales se disponga de datos históricos (preferiblemente). |
|
3) |
Toxicidad por dosis única
El estudio de la toxicidad por dosis única puede ser útil para prever:
El estudio de la toxicidad por dosis única pondrá en evidencia los efectos tóxicos agudos de la sustancia y el tiempo que tardan en aparecer y remitir. Los estudios que deban llevarse a cabo se seleccionarán de manera que den información sobre la seguridad para el usuario; por ejemplo, si se prevé una exposición sustancial del usuario del medicamento veterinario por inhalación o contacto cutáneo, se estudiarán estas vías de exposición. |
|
4) |
Toxicidad por administración repetida
Los estudios de toxicidad por administración repetida tendrán como objeto revelar las alteraciones fisiológicas o patológicas subsiguientes a la administración repetida del principio activo o de la asociación de principios activos que se estén estudiando, y establecer la relación de dichas alteraciones con la posología. Normalmente será suficiente el estudio de la toxicidad por administración repetida en una especie animal de experimentación. Este estudio podrá sustituirse por uno realizado en la especie animal de destino. La frecuencia y la vía de administración, así como la duración del estudio, se determinarán teniendo en cuenta las condiciones previstas de uso clínico o exposición del usuario. El solicitante justificará la envergadura y la duración de los estudios, así como las dosis escogidas. |
|
5) |
Tolerancia en la especie de destino
Se presentará un resumen de los signos de intolerancia que se hayan observado durante los estudios realizados con la especie de destino, normalmente con el producto terminado, de conformidad con los requisitos de la parte II.4A4 (Tolerancia en la especie animal de destino). Se indicarán los estudios correspondientes, la dosis a la que se manifiesta la intolerancia, así como la especie y la raza de que se trate. Se detallará también cualquier alteración fisiológica imprevista. Los informes detallados de esos estudios se incluirán en la parte 4 del expediente. |
|
6) |
Efectos tóxicos en la función reproductora, incluida la teratogenicidad
Estudio de los efectos en la reproducción En el caso de medicamentos que vayan a administrarse a animales destinados a la cría, se proporcionarán estudios sobre la seguridad para la función reproductora según las directrices VICH GL43. No se prevé la presentación de estudios sobre los efectos tóxicos en la función reproductora en animales de laboratorio para la evaluación de los efectos en el usuario. |
|
7) |
Estudio de la teratogenicidad
En el caso de la evaluación de los efectos en la especie animal de destino, no se requieren estudios de la teratogenicidad para los medicamentos que vayan a administrarse exclusivamente a animales no destinados a la cría. Para otros medicamentos, se realizará un estudio de la teratogenicidad como mínimo en una especie, que podrá ser la especie de destino. Si el estudio se realiza con la especie de destino, aquí se facilitará un resumen, y el informe completo del estudio se incluirá en la parte 4 del expediente. Para la evaluación de la seguridad del usuario, en todos los casos en que pueda preverse una exposición significativa del usuario se realizarán estudios estándar de teratogenicidad de conformidad con las pruebas normalizadas según las directrices establecidas (como VICH GL32 y de la OCDE). |
|
8) |
Genotoxicidad
Se llevarán a cabo pruebas del potencial genotóxico que pongan de manifiesto los cambios que una sustancia puede causar en el material genético de las células. Se evaluarán las propiedades genotóxicas de toda sustancia que vaya a utilizarse por primera vez en un medicamento veterinario. Los principios activos se presentarán normalmente a una serie estándar de pruebas de genotoxicidad de conformidad con las pruebas normalizadas según las directrices establecidas (como VICH GL23 y de la OCDE). |
|
9) |
Carcinogenicidad
Para decidir si es necesario realizar pruebas de carcinogenicidad se tendrán en cuenta los resultados de las pruebas de genotoxicidad, las relaciones entre estructura y actividad, y los resultados de las pruebas de toxicidad por administración repetida que puedan demostrar el potencial de alteraciones hiperplásicas o neoplásicas. Se tendrá en cuenta cualquier especificidad de especie que se conozca del mecanismo de toxicidad, y cualquier diferencia del metabolismo entre las especies de laboratorio, las de destino y las personas. Las pruebas de carcinogenicidad se realizarán de conformidad con las pruebas normalizadas según las directrices establecidas (como VICH GL28 y de la OCDE). |
|
10) |
Excepciones
Se deberá estudiar la absorción sistémica de los medicamentos veterinarios de uso tópico en la especie animal de destino. Si se demuestra que dicha absorción es desdeñable, se podrán omitir las pruebas de toxicidad por administración repetida, las de toxicidad sobre la función reproductora y de teratogenicidad y las de carcinogenicidad, a menos que:
|
II.3A4. Otros requisitos
II.3A.4.1 Estudios especiales
En caso de determinados grupos de sustancias, o si los efectos observados en estudios de administración repetida a los animales apuntan a variaciones de la inmunotoxicidad, neurotoxicidad o a disfunciones endocrinas, por ejemplo, se requerirán otras pruebas, como estudios de sensibilización o ensayos de neurotoxicidad diferida. Según la naturaleza del medicamento, podrán necesitarse estudios adicionales para evaluar el mecanismo subyacente del efecto tóxico o del potencial de irritación.
En caso de medicamentos con los que pueda darse una exposición cutánea u ocular, se proporcionarán estudios de irritación y sensibilización. Esos estudios se llevarán a cabo con el producto terminado.
Al diseñar estos estudios y valorar sus resultados se tendrán en cuenta el estado de los conocimientos científicos más recientes y las directrices establecidas.
II.3A.4.2. Observaciones sobre el uso terapéutico en personas
Se facilitará información que demuestre si los principios farmacológicamente activos del medicamento veterinario tienen un uso terapéutico humano o no. En caso afirmativo, se recopilarán todos los efectos observados en las personas (incluidas las reacciones adversas) y sus causas, en la medida en que puedan resultar importantes para valorar la seguridad del medicamento veterinario, y, cuando proceda, se aportarán los resultados de estudios publicados. Si los componentes del medicamento veterinario no tienen o han dejado de tener un uso terapéutico humano, se harán constar los motivos, cuando sean de acceso público.
II.3A.4.3. Aparición de resistencia y riesgo para las personas
Los requisitos de datos descritos en este punto se relacionan con sustancias antibacterianas y pueden no ser plenamente aplicables a otros tipos de antimicrobianos (a saber, antivirales, antimicóticos y antiprotozoarios) aunque, en principio, podrán seguirse tales requisitos cuando corresponda.
Para esos productos, son necesarios los datos sobre la posible aparición de bacterias resistentes o determinantes de resistencia de interés para la salud humana y que estén vinculados con el uso de medicamentos veterinarios. Especial importancia al respecto reviste el mecanismo de aparición y selección de dicha resistencia. Cuando sea necesario, el solicitante propondrá medidas para limitar la aparición de resistencia con la utilización prevista del medicamento veterinario.
Los datos de resistencia de interés para el uso clínico del medicamento en animales de destino se abordarán de conformidad con la parte II.4A2. Cuando proceda, se hará referencia a los datos establecidos en la parte II.4A2.
|
1) |
En el caso de animales productores de alimentos, la evaluación del riesgo abordará:
|
|
2) |
En el caso de animales de compañía, el examen del riesgo para la salud humana o para la salud pública abordará:
|
|
3) |
Se abordará la resistencia en el medio ambiente. |
II.3A5. Seguridad para el usuario
En esta sección se evaluarán los efectos encontrados desde la parte II.3A hasta la parte II.3A4, que se relacionarán con el tipo y grado de exposición humana al medicamento con objeto de formular las correspondientes advertencias para el usuario y demás medidas de gestión del riesgo.
La seguridad para el usuario se abordará de conformidad con las directrices del Comité de Medicamentos de Uso Veterinario.
II.3A6. Evaluación del riesgo medioambiental
|
1) |
Se evaluará el riesgo medioambiental en busca de los posibles efectos nocivos del uso del medicamento veterinario y para determinar el riesgo de tales efectos. En la evaluación se especificará asimismo cualquier medida preventiva que pueda ser necesaria para reducir tal riesgo. |
|
2) |
Esta evaluación consta de dos fases. La primera fase de la evaluación se realizará en todos los casos. Los detalles de la evaluación se presentarán según las directrices publicadas por la Agencia. Se indicará la posible exposición del medio ambiente al medicamento y el riesgo asociado con tal exposición, especialmente teniendo en cuenta los elementos siguientes:
|
|
3) |
En la segunda fase se investigarán de manera específica el destino y los efectos del medicamento en ecosistemas particulares, según las directrices publicadas por la Agencia. Se tendrán en cuenta la amplitud de la exposición del medio ambiente al medicamento y la información disponible sobre las propiedades fisicoquímicas, farmacológicas o toxicológicas de las sustancias en cuestión, incluidos los metabolitos en caso de riesgo detectado, que se haya obtenido durante la realización de las demás pruebas y los demás ensayos exigidos en virtud del presente Reglamento. |
|
4) |
En el caso de los medicamentos destinados a especies productoras de alimentos, las sustancias persistentes, bioacumulables y tóxicas (PBT) o muy persistentes y muy bioacumulables (mPmB) se clasificarán según los criterios establecidos en el anexo XIII del Reglamento (CE) n.o 1907/2006 del Parlamento Europeo y del Consejo (6) (Reglamento REACH) y se evaluarán según las directrices para la evaluación de sustancias PBT y mPmB en medicamentos veterinarios publicadas por la Agencia. |
II.3B. Estudio de los residuos
|
1) |
A efectos del presente punto, se aplicarán las definiciones del Reglamento (CE) n.o 470/2009. |
|
2) |
El propósito de estudiar la eliminación de residuos de los tejidos comestibles o de los huevos, la leche y la miel (o la cera, si procede) procedentes de animales tratados es determinar en qué condiciones y hasta qué punto pueden persistir residuos en productos alimenticios procedentes de esos animales. Además, estos estudios permitirán determinar el tiempo de espera. |
|
3) |
Cuando se trate de medicamentos veterinarios para animales productores de alimentos, la documentación relativa a los residuos deberá poner de manifiesto:
|
II.3B1. Identificación del medicamento
Los medicamentos veterinarios utilizados en el estudio se identificarán mediante los siguientes datos:
|
a) |
composición; |
|
b) |
resultados de pruebas físicas y químicas (potencia y pureza) de los lotes en cuestión; |
|
c) |
identificación del lote. |
II.3B2. Eliminación de los residuos (metabolismo y cinética de los residuos)
|
1) |
Estos estudios, que consisten en medir la velocidad de eliminación de los residuos en la especie animal de destino después de la última administración del medicamento veterinario, tienen por objeto permitir la determinación de los tiempos de espera necesarios para garantizar que en los productos alimenticios procedentes de animales tratados no queden residuos que puedan poner en peligro a los consumidores. |
|
2) |
Se notificará la situación actual del LMR para los componentes del medicamento veterinario en la especie de destino pertinente. |
|
3) |
Los niveles de residuos presentes se determinarán en un número suficiente de momentos después de la última administración del medicamento veterinario a los animales de experimentación. Los estudios en mamíferos y aves se realizarán según las directrices VICH GL48 y otras directrices aplicables. Los estudios de residuos en la miel se realizarán según las directrices VICH GL56 y los de eliminación de residuos en especies acuáticas seguirán las directrices VICH GL57. |
|
4) |
Con arreglo a la evaluación, se justificará el tiempo de espera propuesto. |
II.3B3. Método de análisis de residuos
Los estudios de eliminación de residuos, los métodos analíticos y su validación se realizarán de conformidad con las directrices VICH GL49.
El método analítico propuesto tendrá en cuenta el estado de los conocimientos científicos y técnicos en el momento en que se presente la solicitud.
II.4. Parte 4: Documentación sobre la eficacia (estudios preclínicos y ensayos clínicos)
II.4A. Estudios preclínicos
Los estudios preclínicos tienen por objeto investigar la seguridad para el animal de destino y la eficacia del medicamento, y son necesarios para determinar la actividad farmacológica, las propiedades farmacocinéticas, la dosis, el intervalo de administración, la resistencia (si procede) y la tolerancia en el animal de destino del medicamento.
II.4A1. Farmacología
II.4A.1.1. Farmacodinamia
|
1) |
Se caracterizarán los efectos farmacodinámicos de los principios activos del medicamento veterinario. |
|
2) |
Se describirán de manera adecuada el mecanismo de acción y los efectos farmacológicos, incluidos los efectos secundarios (en su caso), en que se basa la aplicación recomendada. Por lo general se examinarán los efectos en las principales funciones corporales. Los resultados se expresarán de forma cuantitativa (por ejemplo, mediante curvas dosis-efecto o tiempo-efecto) y, en la medida de lo posible, comparándolos con los de una sustancia cuya actividad se conozca bien (cuando se afirme que su actividad es mayor que la de la sustancia conocida, se demostrará que la diferencia existe y es estadísticamente significativa). |
|
3) |
Se estudiará cualquier efecto de las demás características de los medicamentos (como la vía de administración o la formulación) en la actividad farmacológica del principio activo. |
|
4) |
Las técnicas experimentales, a menos que sean procedimientos normalizados, se describirán de forma tal que permitan su reproducción y que se pueda demostrar su validez. Los resultados de los métodos experimentales se expresarán claramente y se presentará el resultado de las comparaciones estadísticas. |
|
5) |
Deberá investigarse igualmente, salvo justificación apropiada, toda modificación cuantitativa de los efectos que resulte de la administración repetida del producto. |
II.4A.1.2. Farmacocinética
|
1) |
Se necesitan los datos farmacocinéticos básicos del principio activo en el contexto de la evaluación de la seguridad y la eficacia del medicamento veterinario para la especie de destino, en particular si se trata de un principio o una formulación nuevos. |
|
2) |
Los objetivos de estudios farmacocinéticos en las especies animales de destino pueden dividirse en cuatro ámbitos principales:
|
|
3) |
En las especies animales de destino, los estudios farmacocinéticos son necesarios, por regla general, como complemento a los estudios farmacodinámicos para contribuir a establecer pautas posológicas seguras y efectivas (vía y lugar de la administración, dosis, intervalo de administración, número de administraciones, etc.). Pueden ser precisos otros estudios farmacocinéticos para establecer pautas posológicas según determinadas variables de población. |
|
4) |
Cuando se hayan presentado estudios farmacocinéticos en la parte 3 del expediente, podrá hacerse referencia a ellos. En el caso de combinaciones fijas, véase la sección IV. |
II.4A2. Aparición de resistencia y riesgo para los animales
|
1) |
Para los medicamentos veterinarios pertinentes (por ejemplo, antimicrobianos o antiparasitarios), se proporcionará información sobre la resistencia actual (si procede) y sobre la posible aparición de resistencia de interés clínico para la indicación alegada en la especie animal de destino. Si es posible, se presentará información sobre los mecanismos de resistencia, la base genética molecular de la resistencia y la tasa de transmisión de los determinantes de resistencia. Cuando proceda, se presentará información sobre corresistencia y resistencia cruzada. El solicitante propondrá medidas para limitar la aparición en los organismos de resistencia de interés clínico para la utilización prevista del medicamento veterinario. |
|
2) |
Los datos de resistencia relacionados con el riesgo para las personas se abordarán de conformidad con la parte II.3A.4.3. Cuando proceda, se hará referencia a los datos establecidos en la parte II.3A.4.3. |
II.4A3. Determinación y confirmación de la dosis
Se facilitarán los datos apropiados para justificar las propuestas relativas a la dosis, el intervalo de administración, la duración del tratamiento y posibles repeticiones del tratamiento.
En el caso de los estudios realizados en condiciones de campo, se facilitará la información correspondiente indicada en la parte II.4B, salvo debida justificación.
II.4A4. Tolerancia en la especie animal de destino
Se estudiará la tolerancia local y sistémica del medicamento veterinario en la especie animal de destino. La finalidad de estos estudios de seguridad para la especie animal de destino es caracterizar los signos de intolerancia y establecer un margen adecuado de seguridad para las vías de administración recomendadas. Esto puede lograrse aumentando la dosis o la duración del tratamiento. Los informes sobre los estudios contendrán detalles de todos los efectos farmacológicos previsibles y de todas las reacciones adversas. Estos estudios de seguridad para la especie animal de destino se realizarán de conformidad con las directrices internacionales de la Conferencia Internacional sobre Armonización de los Requisitos Técnicos para el registro de Medicamentos Veterinarios (VICH) y las directrices pertinentes publicadas por la Agencia. También se puede proporcionar información sobre la seguridad en las especies de destino mediante otros estudios preclínicos, entre ellos los incluidos en la parte 3, y ensayos clínicos, además de en la bibliografía existente. Se incluirán aquí los estudios de teratogenicidad realizados en las especies animales de destino, y en la parte 3 del expediente se incluirá un resumen.
II.4B. Ensayos clínicos
II.4B1. Principios generales
|
1) |
Los ensayos clínicos se diseñarán, realizarán y comunicarán teniendo debidamente en cuenta las directrices internacionales de buenas prácticas clínicas de la VICH y las directrices pertinentes publicadas por la Agencia. Los datos procedentes de ensayos clínicos realizados fuera de la Unión podrán ser tomados en consideración al evaluar una solicitud de autorización de comercialización únicamente si los datos son suficientemente representativos de la situación en la Unión. |
|
2) |
Salvo debida justificación, los datos experimentales, como los de ensayos exploratorios o piloto, o los resultados de planteamientos no experimentales deberán confirmarse mediante ensayos clínicos. |
|
3) |
La finalidad de los ensayos clínicos es examinar, en condiciones de campo, la seguridad para el animal de destino y la eficacia de un medicamento veterinario en condiciones normales de cría de animales o como parte de prácticas veterinarias normales. Con ellos se demostrará el efecto del medicamento veterinario tras la administración a la especie de destino siguiendo las pautas posológicas propuestas y por las vías de administración recomendadas. El diseño de los ensayos tendrá por objeto apoyar las indicaciones y tener en cuenta las posibles contraindicaciones en función de la especie, la edad, la raza y el sexo, las instrucciones de uso del medicamento veterinario y cualquier reacción adversa que pueda darse. |
|
4) |
Todos los ensayos clínicos veterinarios se llevarán a cabo de conformidad con un protocolo de ensayo pormenorizado. |
|
5) |
En el caso de las formulaciones destinadas a su uso en ensayos clínicos veterinarios en la Unión, en el etiquetado deberá figurar de forma visible e indeleble la expresión «uso exclusivo en ensayos clínicos veterinarios». |
|
6) |
Salvo debida justificación, los ensayos clínicos se realizarán con animales testigo (ensayos clínicos comparativos). Los resultados de eficacia obtenidos con el nuevo medicamento se compararán con los de las especies animales de destino a las que se haya administrado un medicamento veterinario autorizado en la Unión que haya demostrado un nivel aceptable de eficacia y que haya sido aprobado para las indicaciones propuestas y para uso en las mismas especies animales de destino, o un placebo o ningún tratamiento. Se informará de todos los resultados obtenidos, ya sean positivos o negativos. |
|
7) |
Salvo debida justificación, en el diseño, el análisis y la evaluación del protocolo de ensayos clínicos se utilizarán principios estadísticos establecidos de conformidad con las directrices pertinentes publicadas por la Agencia. |
II.4B2. Documentación
II.4B2.1. Resultado de los estudios preclínicos
Cuando sea posible, se aportarán datos sobre los resultados de:
|
a) |
los estudios que pongan de manifiesto la actividad farmacológica, incluidos los que evidencien los mecanismos farmacodinámicos responsables del efecto terapéutico y los que demuestren el principal perfil farmacocinético; |
|
b) |
las pruebas e investigaciones sobre la resistencia, si procede; |
|
c) |
los estudios de seguridad para la especie animal de destino; |
|
d) |
los estudios que demuestren y confirmen la dosis (incluidos el intervalo de administración, la duración del tratamiento y posibles repeticiones del tratamiento). |
Si durante la realización de las pruebas aparecen resultados inesperados, se describirán detalladamente. Deberá justificarse la omisión de cualquiera de esos datos. En todos los informes sobre estudios preclínicos se incluirán los elementos siguientes:
|
a) |
un resumen; |
|
b) |
un protocolo del estudio; |
|
c) |
una descripción detallada de los objetivos, el diseño y la realización que incluya los métodos, instrumentos y materiales utilizados, la identificación de los animales, su especie, edad, peso, sexo, número y raza o linaje, la posología, vía y pauta de administración; |
|
d) |
un análisis estadístico de los resultados, si procede; |
|
e) |
una presentación objetiva de los resultados obtenidos, de la que se deduzcan conclusiones sobre la eficacia y la seguridad para el animal de destino del medicamento veterinario. |
II.4B2.2. Resultado de los ensayos clínicos
Todos los investigadores deberán proporcionar la información en fichas individuales cuando el tratamiento sea individual y en fichas colectivas cuando el tratamiento sea colectivo.
El titular de la autorización de comercialización del medicamento veterinario velará por que la documentación original, que constituye la base de la información facilitada, se conserve durante cinco años, como mínimo, desde el momento en que se retire la autorización.
Respecto de las observaciones de cada ensayo clínico, se hará un resumen sinóptico de los ensayos y sus resultados, que incluirá:
|
a) |
el número de animales testigo y de experimentación, tratados individual o colectivamente, desglosados por especie, raza o linaje, edad y sexo; |
|
b) |
el número de animales cuyo estudio haya sido interrumpido antes del final, y los motivos de dicha interrupción; |
|
c) |
para los animales testigo, la información siguiente:
|
|
d) |
la frecuencia de las reacciones adversas observadas; |
|
e) |
observaciones relativas al efecto en la producción de los animales, si procede; |
|
f) |
los datos sobre los animales de experimentación que puedan presentar mayor riesgo en razón de su edad, modo de cría, alimentación o destino, o cuyo estado fisiológico o patológico requiera una atención especial; |
|
g) |
una evaluación estadística de los resultados. |
La persona responsable de la investigación extraerá conclusiones generales sobre la eficacia y la seguridad para el animal de destino del medicamento veterinario en las condiciones de uso propuestas y, en particular, toda información relativa a las indicaciones y contraindicaciones, la posología y la duración media del tratamiento y, en su caso, las interacciones observadas con otros medicamentos veterinarios o aditivos de los piensos, así como las precauciones particulares que deban tomarse durante el tratamiento y los signos clínicos de sobredosis, cuando los haya.
SECCIÓN III
REQUISITOS RELATIVOS A LOS MEDICAMENTOS VETERINARIOS BIOLÓGICOS
Sin perjuicio de los requisitos concretos establecidos por la legislación de la Unión para el control y la erradicación de enfermedades animales infecciosas específicas, los requisitos siguientes se aplicarán a los medicamentos veterinarios biológicos, excepto cuando estén destinados al uso en determinadas especies o con indicaciones específicas, según lo definido en las secciones IV y V, y en las directrices pertinentes.
SECCIÓN IIIa
REQUISITOS RELATIVOS A LOS MEDICAMENTOS VETERINARIOS BIOLÓGICOS NO INMUNOLÓGICOS
Los requisitos siguientes se aplicarán a los medicamentos veterinarios biológicos definidos en el artículo 4, punto 6, con excepción de los medicamentos definidos en el artículo 4, punto 5, o salvo indicación contraria en la sección IV.
Se permite cierta flexibilidad en relación con el cumplimiento de los requisitos especificados en la presente sección, pero toda variación respecto de los requisitos del presente anexo se justificará científicamente y estará basada en propiedades específicas del medicamento biológico. Para ciertas sustancias, según la naturaleza del medicamento, tal vez sean necesarios datos sobre seguridad además de los requisitos indicados en la presente sección.
IIIa.1. Parte 1: Resumen del expediente
Véase la sección I.
IIIa.2. Parte 2: Documentación sobre calidad (información fisicoquímica, biológica o microbiológica)
IIIa.2A. Descripción del producto
IIIa.2A1. Composición cualitativa y cuantitativa
|
1) |
Se hará constar la composición cualitativa y cuantitativa del medicamento veterinario biológico. En esta sección se incluirá información relativa a:
|
|
2) |
Para proporcionar la composición cuantitativa de todos los principios activos y excipientes del medicamento veterinario, será preciso, según la forma farmacéutica, especificar la masa o el número de unidades de actividad biológica, bien sea por dosis o por unidad de masa o de volumen, de cada principio activo y excipiente. |
|
3) |
Cuando sea posible, se indicará la actividad biológica por unidad de masa o volumen. Salvo debida justificación, cuando se haya definido una unidad internacional de actividad biológica, se utilizará. En los casos en los que no se haya definido una unidad internacional, las unidades de actividad biológica se expresarán de forma que proporcionen información inequívoca sobre la actividad de la sustancia utilizando, cuando proceda, las unidades de la Farmacopea Europea. |
|
4) |
Sin perjuicio de lo dispuesto en el artículo 8, se entenderá por «terminología usual» para la designación de los componentes de los medicamentos veterinarios biológicos:
|
IIIa.2A2. Desarrollo farmacéutico
Se explicarán los siguientes aspectos, entre otros:
|
a) |
la elección de la composición y la elección de los componentes, especialmente en relación con la función prevista y la concentración de cada uno; |
|
b) |
se justificará la inclusión de conservantes en la composición; |
|
c) |
el acondicionamiento primario y la adecuación del recipiente y su sistema de cierre utilizados para el almacenamiento y la utilización del medicamento terminado. Se presentará un estudio sobre la interacción del medicamento terminado y el acondicionamiento primario en los casos en que el riesgo de dicha interacción se considere posible, especialmente cuando se trate de preparados inyectables; |
|
d) |
las características microbiológicas (pureza microbiológica y actividad antimicrobiana) y las instrucciones de uso; |
|
e) |
otros materiales de envasado o embalaje exterior, si procede; |
|
f) |
los tamaños propuestos para el envase en relación con la vía de administración, la posología y las especies de destino propuestas; |
|
g) |
toda sobredosificación en la formulación para garantizar una potencia mínima al final del período de validez, con su justificación; |
|
h) |
la selección del proceso de fabricación del principio activo y el producto terminado; |
|
i) |
se comentarán las diferencias entre los procesos de fabricación utilizados para producir los lotes destinados a ensayos clínicos y los procesos descritos en la solicitud de autorización de comercialización; |
|
j) |
cuando el producto terminado se suministre con un dispositivo para su dosificación, se demostrará la exactitud de las dosis; |
|
k) |
cuando se recomiende utilizar una prueba junto con el producto terminado (por ejemplo, un estudio de diagnóstico), se proporcionará la información pertinente sobre dicha prueba. |
|
l) |
Esta explicación se justificará con datos científicos sobre el desarrollo farmacéutico. |
IIIa.2A3. Caracterización
IIIa.2A3.1. Elucidación de la estructura y otras características
|
1) |
La caracterización de una sustancia biotecnológica o biológica (lo que incluye determinar las propiedades fisicoquímicas, la actividad biológica, las propiedades inmunoquímicas, la pureza y las impurezas) mediante las técnicas apropiadas es necesaria para permitir una definición adecuada de las especificaciones. No es aceptable hacer referencia exclusivamente a datos de la bibliografía, salvo que esto se justifique por conocimientos anteriores de moléculas similares para modificaciones cuando no haya ningún problema de seguridad. La caracterización adecuada se realizará durante la fase de desarrollo y, cuando proceda, tras cambios significativos en el proceso. |
|
2) |
Se facilitará toda la información pertinente disponible sobre la estructura primaria, secundaria y de orden superior, incluidas las modificaciones postraduccionales (por ejemplo, glicoformas) y otras modificaciones del principio activo. |
|
3) |
Se incluirán detalles sobre la actividad biológica (es decir, la capacidad o habilidad específica de un medicamento para alcanzar un efecto biológico definido). Normalmente, la actividad biológica se determinará o se evaluará utilizando un método apropiado, fiable y cualificado. Se justificará la ausencia de tal determinación. Se reconoce que la amplitud de los datos de caracterización aumentará durante el desarrollo. |
|
4) |
Se explicará el motivo por el que se han seleccionado los métodos utilizados para la caracterización y se justificará su adecuación. |
IIIa.2A3.2. Impurezas
|
1) |
Se informará sobre las impurezas relacionadas con el proceso (por ejemplo, proteínas de células hospedadoras, ADN de células hospedadoras, residuos de los medios o lixiviables de columna) y relacionadas con el medicamento (por ejemplo, precursores, formas escindidas, productos de degradación o agregados). La información sobre las impurezas se proporcionará en forma cuantitativa e incluirá la cantidad máxima para la mayor dosis. En el caso de ciertas impurezas relacionadas con el proceso (por ejemplo, antiespumantes), podrá justificarse una estimación del aclaramiento. |
|
2) |
Si se proporcionan solamente datos cualitativos para ciertas impurezas, deberá justificarse. |
IIIa.2B. Descripción del método de fabricación
|
1) |
La descripción del método de fabricación que, conforme a lo establecido en el artículo 8, debe acompañar a la solicitud de autorización de comercialización se redactará de forma que ofrezca una idea clara del carácter de las operaciones efectuadas. |
|
2) |
Se harán constar los nombres, las direcciones y las responsabilidades de cada fabricante, incluidos los contratistas, junto con cada lugar de fabricación propuesto o instalación que interviene en la fabricación, las pruebas y la liberación de lotes. |
|
3) |
La descripción del proceso de fabricación incluirá, como mínimo:
|
|
4) |
Se facilitará una descripción, documentación y los resultados de los estudios de validación o evaluación para los pasos cruciales o las determinaciones críticas utilizados en el proceso de fabricación (por ejemplo, validación del proceso de esterilización o el procesamiento aséptico o el llenado) y se demostrará la validación del proceso de producción en su conjunto aportando los resultados de tres lotes consecutivos producidos con el método descrito. |
IIIa.2C. Producción y control de los materiales de partida
|
1) |
A los efectos del presente punto, se entenderá por «materiales de partida» todos los componentes, incluidos los principios activos, utilizados en la producción del medicamento veterinario biológico. Los medios de cultivo utilizados para producir los principios activos se considerarán un solo material de partida. |
|
2) |
Se presentará la composición cualitativa y cuantitativa siempre que las autoridades consideren que esta información es pertinente para la calidad del producto terminado y para cualquier riesgo que pudiera surgir. |
|
3) |
Si se utilizan materiales de origen animal para preparar estos medios de cultivo, tendrá que indicarse la especie animal y el tejido utilizados, y se demostrará el cumplimiento de las monografías pertinentes, incluidas las generales, y de los capítulos generales de la Farmacopea Europea. |
|
4) |
El solicitante suministrará documentación para demostrar que los materiales de partida, incluidos los materiales de siembra, las líneas celulares, los lotes de suero y demás materiales procedentes de especies animales por las que puedan transmitirse las EET, y la fabricación del medicamento veterinario se ajustan a los requisitos de la Nota explicativa sobre cómo minimizar los riesgos de transmisión de los agentes de las encefalopatías espongiformes animales a través de los medicamentos humanos y veterinarios, así como a los requisitos de la correspondiente monografía de la Farmacopea Europea. |
|
5) |
Como justificante de cumplimiento podrán utilizarse los certificados de conformidad emitidos por la Dirección Europea de Calidad del Medicamento y la Asistencia Sanitaria, con referencia a la correspondiente monografía de la Farmacopea Europea. |
|
6) |
El expediente contendrá las especificaciones, información sobre los estudios que se realizarán para el control de calidad de todos los lotes de materiales de partida y los resultados de un lote de todos los componentes utilizados, y se presentará de conformidad con las siguientes disposiciones. |
|
7) |
Se presentarán certificados de análisis de los materiales de partida para demostrar el cumplimiento de las características técnicas definidas. |
|
8) |
En todos los casos, las materias colorantes deberán reunir los requisitos que se establecen en la Directiva 2009/35/CE. |
|
9) |
La utilización de antibióticos durante la producción, así como la de conservantes, cumplirán la Farmacopea Europea. |
|
10) |
En el caso de excipientes nuevos, es decir, que se usan por primera vez en la Unión en un medicamento veterinario o por una nueva vía de administración, se darán los detalles de fabricación, caracterización y controles, con referencias en apoyo de los datos de seguridad tanto clínicos como no clínicos. En el caso de los colorantes, se considerarán suficientes las declaraciones de cumplimiento indicadas en la parte II.2C2, puntos 3 y 4. |
IIIa.2C1. Materiales de partida descritos en las farmacopeas
|
1) |
Las monografías de la Farmacopea Europea serán aplicables a todos los materiales de partida que figuren en ella, salvo justificación adecuada. |
|
2) |
Para las restantes sustancias, cada Estado miembro podrá exigir el cumplimiento de su farmacopea nacional en relación con los productos que se fabriquen en su territorio. |
|
3) |
La descripción de los métodos de análisis podrá sustituirse por una referencia detallada a la farmacopea de que se trate. |
|
4) |
Los estudios sistemáticos que se realicen en cada lote de materiales de partida se llevarán a cabo tal y como estén descritos en la solicitud de autorización de comercialización. Si se utilizan estudios distintos de los mencionados en una farmacopea, deberá demostrarse que los materiales de partida corresponden a las exigencias de calidad de dicha farmacopea. |
|
5) |
Cuando las especificaciones u otras disposiciones incluidas en una monografía de la Farmacopea Europea o en la farmacopea de un Estado miembro puedan no ser suficientes para garantizar la calidad de la sustancia, las autoridades competentes podrán exigir al solicitante de la autorización de comercialización que presente especificaciones más apropiadas. Se informará de la presunta insuficiencia a las autoridades responsables de la farmacopea en cuestión. |
IIIa.2C2. Materiales de partida no descritos en una farmacopea
IIIa.2C2.1. Materiales de partida de origen biológico
|
1) |
Cuando en la fabricación del medicamento veterinario se utilicen materiales de partida biológicos como microorganismos, tejidos de origen vegetal o animal, células o líquidos (incluida la sangre) de origen humano o animal, así como construcciones celulares biotecnológicas, se describirán y documentarán el origen, incluida la región geográfica, y los antecedentes de los materiales de partida. Se indicarán el origen, el estado de salud general y la situación inmunitaria de los animales utilizados para la producción, y se utilizarán conjuntos identificados de materiales de partida biológicos. |
|
2) |
Se demostrará la ausencia de agentes extraños (bacterias, micoplasma, hongos y virus) con arreglo a la Farmacopea Europea para los materiales de siembra, incluidas las líneas celulares y los conjuntos de sueros, así como para los materiales de partida biológicos de los que proceden, cuando sea posible. |
|
3) |
Se dará información sobre todas las sustancias de origen biológico utilizadas en cualquier fase del proceso de fabricación. Esa información incluirá la estrategia de fabricación, los procedimientos de purificación e inactivación, junto con su validación, y todos los procedimientos de control durante el proceso cuya finalidad sea asegurar la calidad, seguridad y constancia de los lotes del producto terminado, además de datos de las pruebas de control de contaminación realizadas con cada lote de la sustancia. Se indicará cualquier precaución especial que pueda ser necesaria durante el almacenamiento del material de partida y, en su caso, su período de validez. |
|
4) |
Cuando se utilicen materiales de partida de origen animal o humano, se describirán las medidas que se han tomado para garantizar la ausencia de agentes extraños. Si se detecta o sospecha la presencia de agentes extraños, el material correspondiente se eliminará o se procesará con un tratamiento validado para reducir el riesgo de dicha presencia. Si, después del tratamiento, se detecta o sospecha tal presencia, el material correspondiente se utilizará únicamente cuando un procesamiento posterior del producto garantice la eliminación o inactivación de esos agentes; esta eliminación o inactivación de los agentes extraños deberá demostrarse. |
|
5) |
Cuando se utilicen líneas celulares, deberá demostrarse que las características celulares se han mantenido inalteradas hasta el pase más alto utilizado en la producción. |
|
6) |
En caso de materiales de partida procedentes de ingeniería genética, esta información incluirá aspectos como la descripción del origen de las células o cepas, la construcción del vector de expresión (denominación, origen, función del replicón, estimulador del promotor y otros elementos reguladores), control de la secuencia de ADN o ARN insertada correctamente, secuencias de oligonucleótidos del vector plasmídico en las células, plásmido utilizado para la cotransfección, genes adicionados o suprimidos, propiedades biológicas de la construcción final y los genes expresados, número de copias y estabilidad genética. |
|
7) |
Cuando un medicamento veterinario contenga o consista en organismos modificados genéticamente, se adjuntarán también a la parte de la solicitud dedicada a la calidad los documentos exigidos de conformidad con la Directiva 2001/18/CE. |
|
8) |
En caso necesario, deberán proporcionarse a las autoridades competentes muestras del material biológico de partida o de los reactivos utilizados en los métodos de ensayo, a fin de que puedan disponer la realización de los estudios necesarios de comprobación. |
IIIa.2C2.2. Materiales de partida de origen no biológico
|
1) |
La descripción se presentará en forma de monografía con los siguientes encabezamientos:
|
IIIa.2D. Pruebas de control efectuadas durante el proceso de fabricación
|
1) |
El expediente incluirá detalles de las pruebas de control realizadas durante la producción, que se efectúan en fases intermedias de la fabricación, para verificar la constancia del proceso de fabricación y del producto final. Se establecerán especificaciones para cada prueba de control y se describirán los métodos analíticos. Se proporcionará la validación de las pruebas de control, salvo debida justificación. |
|
2) |
En las especificaciones para los lotes del principio activo se definirán criterios de aceptación junto con las pruebas utilizadas para lograr un control suficiente de la calidad de dicho principio activo. Salvo debida justificación, se incluirá una prueba de actividad biológica. Se establecerán límites superiores para las impurezas, teniendo en cuenta las condiciones de seguridad. Se especificará la calidad microbiológica para el principio activo. Se demostrará la ausencia de agentes extraños (bacterias, micoplasma, hongos y virus) con arreglo a la Farmacopea Europea. |
|
3) |
De conformidad con la Directiva 2010/63/UE y con el Convenio Europeo sobre Protección de los Animales Vertebrados Utilizados con Fines Experimentales y Otros Fines Científicos, las pruebas se realizarán de forma que se utilice el menor número de animales y se cause el menor dolor, sufrimiento, angustia o daño duradero. Se utilizarán pruebas alternativas in vitro, si las hay, cuando puedan sustituir o disminuir el uso de animales o reducir su sufrimiento. |
IIIa.2E. Pruebas de control del producto terminado
IIIa.2E1 Especificaciones del producto terminado
En todas las pruebas deberán describirse de forma detallada las técnicas de análisis del producto terminado, de modo que sea posible la evaluación de calidad.
Cuando haya monografías adecuadas, si se utilizan procedimientos analíticos y límites distintos de los mencionados en las monografías de la Farmacopea Europea o, en su defecto, de la farmacopea de un Estado miembro, deberá demostrarse que el producto terminado, si se sometiera a control con arreglo a estas monografías, cumpliría los requisitos de calidad establecidos en dicha farmacopea para la forma farmacéutica correspondiente. La solicitud de autorización de comercialización presentará una relación de estas pruebas, que se realizarán con muestras representativas de cada lote del producto terminado. Se hará constar la frecuencia de las pruebas realizadas en el producto final a granel, en lugar de en el lote o los lotes preparados, si procede. Se justificará la frecuencia de las pruebas que no se lleven a cabo de forma sistemática. Se indicarán y justificarán los criterios de aceptación para la aprobación. Se proporcionará la validación de las pruebas de control que se realicen en el producto terminado.
Se establecerán límites superiores para las impurezas, teniendo en cuenta las condiciones de seguridad.
IIIa.2E2 Descripciones del método y validación de pruebas de liberación
|
1) |
Características generales
Los controles de las características generales se referirán, siempre que sea procedente, a la apariencia del producto terminado y a estudios físicos o químicos como el pH, la osmolalidad, etc. El solicitante deberá definir en cada caso las especificaciones de cada una de esas características con límites de confianza apropiados. |
|
2) |
Identificación y prueba de potencia
En caso necesario, se realizará una prueba específica de identificación del principio activo. Cuando proceda, la prueba de identificación podrá combinarse con la prueba de potencia. Se realizará una prueba de actividad o una prueba para la cuantificación del principio activo o una prueba para medir cuantitativamente la funcionalidad (actividad biológica o efecto funcional) que esté vinculada a las propiedades biológicas correspondientes, a fin de demostrar que cada lote contiene la potencia adecuada para garantizar su seguridad y eficacia. Si los métodos fisicoquímicos no proporcionan suficiente información sobre la calidad del producto, será obligatorio un bioanálisis. Siempre que sea posible, en este bioanálisis deberán emplearse materiales de referencia y un análisis estadístico que permitan calcular los límites de confianza. Cuando esos controles no puedan realizarse en el producto terminado, será admisible que se lleven a cabo en una fase intermedia, lo más cerca posible del fin del proceso de fabricación. Cuando se produzca degradación durante la fabricación del producto terminado, se indicarán las tasas aceptables máximas de degradación de cada producto y las totales inmediatamente después de la fabricación. |
|
3) |
Identificación y determinación de los excipientes
En la medida de lo necesario, el excipiente o excipientes serán objeto, como mínimo, de pruebas de identificación. Los conservantes se presentarán obligatoriamente a una prueba de límite superior e inferior. Cualquier otro componente del excipiente que pueda producir una reacción adversa se presentará obligatoriamente a una prueba de límite superior. Si procede, deberá verificarse en el producto terminado la cantidad y la naturaleza del adyuvante y sus componentes, salvo justificación. |
|
4) |
Pruebas de esterilidad y pureza
Se demostrará la ausencia de agentes extraños (bacterias, micoplasma, hongos y endotoxinas bacterianas cuando proceda) con arreglo a la Farmacopea Europea. Deberán realizarse pruebas adecuadas para demostrar la ausencia de contaminación por otras sustancias, según la naturaleza del medicamento veterinario biológico, el método y las condiciones de fabricación. Si con cada lote se realiza sistemáticamente un número menor de pruebas que las requeridas por la correspondiente monografía de la Farmacopea Europea, las pruebas que se lleven a cabo serán vitales para determinar el cumplimiento de los requisitos. Se demostrará que el medicamento veterinario biológico cumpliría los requisitos si se sometiera al conjunto de las pruebas de la monografía. |
|
5) |
Humedad residual
Cada lote de producto liofilizado o en comprimido se presentará a una prueba de determinación de la humedad residual. |
|
6) |
Volumen de llenado
Se llevarán a cabo pruebas adecuadas para demostrar el volumen de llenado correcto. |
IIIa.2E3. Normas o materiales de referencia
Se proporcionará información sobre el proceso de fabricación utilizado para establecer el material de referencia. Si se han utilizado dos o más normas de referencia para una prueba concreta durante el desarrollo del producto, se facilitará un historial de cualificación en el que se describa cómo se mantuvo la relación entre las distintas normas.
Si se utilizan preparaciones y normas de referencia distintas de las indicadas en la Farmacopea Europea, se identificarán y se describirán detalladamente.
IIIa.2F. Constancia entre lotes
IIIa.2F1. Principio activo
Con el fin de garantizar que la calidad del principio activo es constante entre lotes y demostrar su conformidad con las especificaciones, se presentarán datos de lotes representativos.
IIIa.2F2. Producto terminado
Con el fin de garantizar que la calidad del medicamento es constante entre lotes y demostrar su conformidad con las especificaciones, se presentará un protocolo completo de tres lotes consecutivos representativos de la producción sistemática.
IIIa.2G. Estudios de estabilidad
|
1) |
Los estudios de estabilidad se refieren a la estabilidad del principio activo y del producto terminado, incluidos los disolventes, si procede. Si los principios activos se almacenan, se definirán las condiciones y la duración del almacenamiento sobre la base de los datos de estabilidad; estos pueden obtenerse bien mediante pruebas en los principios activos propiamente dichos o bien mediante las pruebas adecuadas en el medicamento terminado. |
|
2) |
Deberán describirse las pruebas realizadas para determinar el período de validez, las condiciones recomendadas de almacenamiento y las especificaciones al final del período de validez que propone el solicitante. Estos estudios serán siempre en tiempo real; se llevarán a cabo en un mínimo de tres lotes representativos fabricados según el proceso de producción descrito y en productos almacenados en su recipiente final; incluirán estudios de estabilidad biológica y fisicoquímica, realizados a intervalos periódicos, del producto terminado hasta el final del período de validez alegado. |
|
3) |
En las conclusiones figurarán los resultados de los análisis y se justificará el período de validez propuesto en todas las condiciones propuestas de almacenamiento. Los resultados obtenidos durante el estudio de estabilidad se tendrán en cuenta al definir las especificaciones apropiadas de formulación y liberación, a fin de garantizar la conformidad del medicamento con el período de validez alegado. |
|
4) |
Cuando se trate de medicamentos que se administren con piensos, también se incluirá la información necesaria sobre el período de validez del producto en las diferentes fases de mezcla, cuando se mezcle con arreglo a las instrucciones de uso recomendadas. |
|
5) |
Si un producto terminado tiene que ser reconstituido previamente a su administración, o se administra con el agua de bebida, se especificará el período de validez que se propone para el medicamento reconstituido según lo recomendado. Deberán presentarse datos que justifiquen el período de validez propuesto para el producto reconstituido. |
|
6) |
Cuando se trate de envases multidosis, si procede, se presentarán los datos relativos a la estabilidad para justificar el período de validez del medicamento después de su primera apertura o utilización, y se definirán las especificaciones una vez abierto. |
|
7) |
Cuando un producto terminado pueda dar lugar a productos de degradación, el solicitante declarará dichos productos, indicando los métodos de identificación y los procedimientos analíticos utilizados. |
|
8) |
Los datos de estabilidad de medicamentos combinados podrán utilizarse, cuando se justifique debidamente, para los productos derivados que contengan uno o más de los mismos componentes. |
|
9) |
Deberá demostrarse la eficacia del sistema de conservación utilizado. Para ello podrá ser suficiente la información sobre la eficacia de los conservantes en otros medicamentos veterinarios biológicos similares del mismo fabricante. |
IIIa.2H. Otra información
El expediente podrá contener información sobre la calidad del medicamento veterinario biológico que no haya quedado cubierta en las partes IIIa.2 a IIIa2G.
IIIa.3. Parte 3: Documentación relativa a la seguridad (pruebas de seguridad y de residuos)
|
1) |
Cada informe de estudio incluirá:
|
|
2) |
Podrán aceptarse estudios publicados si contienen una cantidad de datos y detalles suficientes para permitir una evaluación independiente. Las técnicas experimentales se describirán de tal forma que puedan reproducirse, y quien realice la investigación demostrará su validez. No se aceptarán como documentación válida resúmenes de estudios sobre los cuales no se disponga de informes detallados. Cuando la sustancia se haya evaluado con anterioridad para establecer su límite máximo de residuos (LMR) a fin de cumplir ciertos requisitos de seguridad, podrá hacerse referencia a los informes públicos europeos de evaluación de los LMR (EPMAR, por sus siglas en inglés). Si se hace referencia a los EPMAR, no será necesario presentar estudios ya evaluados durante la evaluación del LMR; solamente se facilitarán estudios nuevos que no estuvieran disponibles para la evaluación del LMR. Si la vía de exposición (por ejemplo, para el usuario) no es idéntica a la vía utilizada de conformidad con el Reglamento (UE) 2018/78, podrían necesitarse nuevos estudios. |
IIIa.3A. Pruebas de seguridad
|
1) |
La documentación relativa a la seguridad será adecuada para evaluar lo siguiente:
|
|
2) |
En algunos casos podrá ser necesario someter a prueba los metabolitos del compuesto original, cuando constituyan los residuos en cuestión. |
|
3) |
Un excipiente que se utilice por primera vez en un medicamento veterinario o con una vía de administración nueva deberá considerarse como un principio activo. |
|
4) |
Se abordarán todas las secciones indicadas en la parte IIIa.3A. Según la naturaleza del medicamento, tal vez ciertas secciones no sean pertinentes y puedan omitirse estudios, cuando esté justificado. |
IIIa.3A1. Identificación exacta del medicamento y sus principios activos
|
a) |
denominación común internacional (DCI); |
|
b) |
nombre de la Unión Internacional de Química Pura y Aplicada (UIQPA); |
|
c) |
número CAS (Chemical Abstract Service); |
|
d) |
clasificación terapéutica, farmacológica y química; |
|
e) |
sinónimos y abreviaturas; |
|
f) |
fórmula estructural; |
|
g) |
fórmula molecular; |
|
h) |
peso molecular; |
|
i) |
grado de impureza; |
|
j) |
composición cualitativa y cuantitativa de las impurezas; |
|
k) |
descripción de las propiedades físicas; |
|
l) |
solubilidad en el agua y en los disolventes orgánicos expresada en g/l, con indicación de la temperatura; |
|
m) |
refracción de la luz, rotación óptica, etc.; |
|
n) |
formulación del producto. |
IIIa.3A2. Farmacología
|
1) |
Los estudios farmacológicos resultan fundamentales para poner en evidencia los mecanismos por los que el medicamento veterinario produce sus efectos terapéuticos y, por consiguiente, se incluirán los estudios farmacológicos realizados con las especies animales de destino y, cuando proceda, con otras especies que no sean de destino. Se hará referencia, si procede, a estudios presentados en la parte 4 del expediente. |
|
2) |
Los estudios farmacológicos también pueden ser de utilidad para el análisis de los fenómenos toxicológicos. Si un medicamento veterinario produce efectos farmacológicos exentos de toxicidad, o bien los efectos se logran con dosis inferiores a las tóxicas, habrá que tener en cuenta dichos efectos farmacológicos al evaluar la seguridad del medicamento veterinario. |
|
3) |
Los pormenores de los estudios farmacológicos practicados con animales de laboratorio, así como la información pertinente relativa a los estudios clínicos realizados con el animal de destino, deberán preceder siempre a la documentación sobre la seguridad. |
IIIa.3A2.1. Farmacodinamia
Se dará información sobre el mecanismo de acción de los principios activos, y sobre los efectos farmacodinámicos primarios y secundarios, que ayude a comprender cualquier efecto adverso que se produzca en los estudios con animales. En la parte 4A del expediente se declarará información detallada sobre las propiedades farmacodinámicas relacionadas con el efecto terapéutico.
IIIa.3A2.2. Farmacocinética
Se proporcionarán datos sobre el destino del principio activo y sus metabolitos en animales de laboratorio, entre los que figurarán la absorción, la distribución, el metabolismo y la excreción. Para determinar la exposición adecuada, los datos se cotejarán con los resultados de la relación dosis-efecto que figuran en los estudios farmacológicos y toxicológicos.
IIIa.3A3. Toxicología
|
1) |
La documentación toxicológica se ajustará a las directrices publicadas por la Agencia en cuanto al planteamiento general de la prueba y a los estudios particulares. Estas directrices incluyen los datos toxicológicos necesarios para establecer la seguridad para el usuario, y la evaluación de efectos adversos para los animales de destino y el medio ambiente. |
|
2) |
Los estudios de toxicidad se llevarán a cabo con el principio activo, no con el medicamento formulado, a menos que se exija específicamente lo contrario. |
|
3) |
Los estudios con animales se realizarán en cepas establecidas de animales de laboratorio para las cuales se disponga de datos históricos (preferiblemente). |
IIIa.3A3.1. Toxicidad por dosis única
El estudio de la toxicidad por dosis única puede ser útil para prever:
|
a) |
los posibles efectos en la especie animal de destino tras una sobredosis aguda; |
|
b) |
los posibles efectos tras una administración accidental a personas; |
|
c) |
las dosis convenientes para el estudio de la administración repetida. |
El estudio de la toxicidad por dosis única pondrá en evidencia los efectos tóxicos agudos de la sustancia y el tiempo que tardan en aparecer y remitir.
Los estudios que deban llevarse a cabo se seleccionarán de manera que den información sobre la seguridad para el usuario; por ejemplo, si se prevé una exposición sustancial del usuario del medicamento veterinario por inhalación o contacto cutáneo, se estudiarán estas vías de exposición.
IIIa.3A3.2. Toxicidad por administración repetida
Los estudios de toxicidad por administración repetida tendrán como objeto revelar las alteraciones fisiológicas o patológicas subsiguientes a la administración repetida del principio activo o de la asociación de principios activos que se estén estudiando, y establecer la relación de dichas alteraciones con la posología.
Normalmente será suficiente el estudio de la toxicidad por administración repetida en una especie animal de experimentación. Este estudio podrá sustituirse por uno realizado en la especie animal de destino. La frecuencia y la vía de administración, así como la duración del estudio, se determinarán teniendo en cuenta las condiciones previstas de uso clínico o exposición del usuario. El solicitante justificará la envergadura y la duración de los estudios, así como las dosis escogidas.
IIIa.3A3.3. Tolerancia en la especie de destino
Se presentará un resumen de los signos de intolerancia que se hayan observado durante los estudios realizados con la especie de destino, normalmente con el producto terminado, de conformidad con los requisitos de la parte IIIa.4A4 (seguridad para la especie animal de destino). Se indicarán los estudios correspondientes, la dosis a la que se manifiesta la intolerancia, así como la especie y la raza de que se trate. Se detallará también cualquier alteración fisiológica imprevista. Los informes detallados de esos estudios se incluirán en la parte 4 del expediente.
IIIa.3A3.4. Efectos tóxicos en la función reproductora, incluida la teratogenicidad
|
1) |
Estudio de los efectos en la reproducción
En el caso de medicamentos que vayan a administrarse a animales destinados a la cría, se proporcionarán estudios sobre la seguridad para la función reproductora según las directrices VICH GL43. No se prevé la presentación de estudios sobre los efectos tóxicos en la función reproductora en animales de laboratorio para la evaluación de los efectos en el usuario. |
|
2) |
Estudio de la teratogenicidad
En el caso de la evaluación de los efectos en la especie animal de destino, no se requieren estudios de la teratogenicidad para los medicamentos que vayan a administrarse exclusivamente a animales no destinados a la cría. Para otros medicamentos, se realizará un estudio de la teratogenicidad como mínimo en una especie, que podrá ser la especie de destino. Para la evaluación de la seguridad del usuario, en todos los casos en que pueda preverse una exposición significativa del usuario se realizarán estudios estándar de teratogenicidad de conformidad con las pruebas normalizadas según las directrices establecidas (como VICH GL32 y de la OCDE). |
IIIa.3A3.5. Genotoxicidad
Salvo debida justificación, se llevarán a cabo pruebas del potencial genotóxico que pongan de manifiesto los cambios que una sustancia puede causar en el material genético de las células. Se evaluarán las propiedades genotóxicas de toda sustancia que vaya a utilizarse por primera vez en un medicamento veterinario.
Los principios activos se presentarán normalmente a una serie estándar de pruebas de genotoxicidad de conformidad con las pruebas normalizadas según las directrices establecidas (como VICH GL23 y de la OCDE).
IIIa.3A3.6. Carcinogenicidad
Para decidir si es necesario realizar pruebas de carcinogenicidad se tendrán en cuenta los resultados de las pruebas de genotoxicidad, las relaciones entre estructura y actividad, y los resultados de las pruebas de toxicidad por administración repetida que puedan demostrar el potencial de alteraciones hiperplásicas o neoplásicas.
Se tendrá en cuenta cualquier especificidad de especie que se conozca del mecanismo de toxicidad, y cualquier diferencia del metabolismo entre las especies de laboratorio, las de destino y las personas.
Las pruebas de carcinogenicidad se realizarán de conformidad con las pruebas normalizadas según las directrices establecidas (como VICH GL28 y de la OCDE).
IIIa.3A3.7. Excepciones
Se deberá estudiar la absorción sistémica de los medicamentos veterinarios de uso tópico en la especie animal de destino. Si se demuestra que dicha absorción es desdeñable, se podrán omitir las pruebas de toxicidad por administración repetida, las de teratogenicidad y las de carcinogenicidad, a menos que:
|
a) |
se prevea la ingestión del medicamento veterinario por parte del animal en las condiciones de uso establecidas, o |
|
b) |
se prevea la exposición del usuario del medicamento veterinario por vía oral en las condiciones de uso establecidas. |
IIIa.3A4. Otros requisitos
IIIa.3A4.1. Estudios especiales
En caso de determinados grupos de sustancias, o si los efectos observados en estudios de administración repetida a los animales apuntan a variaciones de la inmunogenicidad, inmunotoxicidad, neurotoxicidad o a disfunciones endocrinas, por ejemplo, se requerirán otras pruebas, como estudios de sensibilización o ensayos de neurotoxicidad diferida. Según la naturaleza del medicamento, podrán necesitarse estudios adicionales para evaluar el mecanismo subyacente del efecto tóxico o del potencial de irritación.
En caso de medicamentos con los que pueda darse una exposición cutánea u ocular, se proporcionarán estudios de irritación y sensibilización. En general, esos estudios se llevarán a cabo con el producto terminado.
Al diseñar estos estudios y valorar sus resultados se tendrán en cuenta el estado de los conocimientos científicos y las directrices establecidas.
IIIa.3A4.2. Observaciones sobre el uso terapéutico en personas
Se facilitará información sobre si los principios farmacológicamente activos del medicamento veterinario tienen un uso terapéutico humano o no. En caso afirmativo, se recopilarán a partir de los estudios publicados todos los efectos observados en las personas (incluidas las reacciones adversas) y sus causas, en la medida en que puedan resultar importantes para valorar la seguridad del medicamento veterinario; si los componentes del medicamento veterinario no tienen o han dejado de tener un uso terapéutico humano por motivos de seguridad, se harán constar esos motivos cuando sean de acceso público.
IIIa.3A4.3. Aparición de resistencia y riesgo para las personas
Los requisitos de datos descritos en este punto se relacionan con sustancias antibacterianas y tal vez no sean plenamente aplicables a otros tipos de antimicrobianos (a saber, antivirales, antimicóticos y antiprotozoarios); en el caso de sustancias distintas de las antibacterianas para las cuales esté bien determinada la existencia de resistencia a los antimicrobianos, podrán seguirse los mismos requisitos, cuando corresponda.
Son necesarios los datos sobre la posible aparición de bacterias resistentes o determinantes de resistencia de interés para la salud humana que estén vinculados con el uso de medicamentos veterinarios. Especial importancia al respecto reviste el mecanismo de aparición y selección de dicha resistencia. En caso necesario, se propondrán medidas para limitar la aparición de resistencia con la utilización prevista del medicamento veterinario.
Los datos de resistencia de interés para el uso clínico del medicamento en animales de destino se abordarán de conformidad con la parte IIIa.4A2. Cuando proceda, se hará referencia a los datos establecidos en la parte IIIa.4A2.
|
1) |
En el caso de animales productores de alimentos, la evaluación del riesgo abordará:
|
|
2) |
En el caso de animales de compañía, el examen del riesgo para la salud humana o para la salud pública abordará:
|
|
3) |
Se abordará la resistencia en el medio ambiente. |
IIIa.3A5. Seguridad para el usuario
En esta sección sobre seguridad para el usuario se evaluarán los efectos encontrados desde la parte IIIa.3A hasta la parte IIIa.3A4, que se relacionarán con el tipo y grado de exposición humana al medicamento con objeto de formular las correspondientes advertencias para el usuario y demás medidas de gestión del riesgo.
La seguridad para el usuario se abordará de conformidad con las directrices del Comité de Medicamentos de Uso Veterinario.
IIIa.3A6. Evaluación del riesgo medioambiental
IIIa.3A6.1. Evaluación del riesgo medioambiental de medicamentos veterinarios que no contienen o no consisten en organismos modificados genéticamente
|
1) |
Se evaluará el riesgo medioambiental en busca de los posibles efectos nocivos del uso del medicamento veterinario y para determinar el riesgo de tales efectos. En la evaluación se especificará asimismo cualquier medida preventiva que pueda ser necesaria para reducir tal riesgo. |
|
2) |
Esta evaluación consta de dos fases. La primera fase de la evaluación se realizará en todos los casos. Los detalles de la evaluación se presentarán según las directrices publicadas por la Agencia. Se indicará la posible exposición del medio ambiente al medicamento y el riesgo asociado con tal exposición, teniendo especialmente en cuenta los puntos siguientes:
|
|
3) |
En la segunda fase se investigarán de manera específica el destino y los efectos del medicamento en ecosistemas particulares, según las directrices publicadas por la Agencia. Se tendrán en cuenta la amplitud de la exposición del medio ambiente al medicamento y la información disponible sobre las propiedades fisicoquímicas, farmacológicas o toxicológicas de las sustancias en cuestión, incluidos los metabolitos en caso de riesgo detectado, que se haya obtenido durante la realización de las demás pruebas y los demás ensayos exigidos en virtud del presente Reglamento.
En el caso de los medicamentos destinados a especies productoras de alimentos, las sustancias persistentes, bioacumulables y tóxicas (PBT) o muy persistentes y muy bioacumulables (mPmB) se clasificarán según los criterios establecidos en el anexo XIII del Reglamento REACH y se evaluarán según las directrices para la evaluación de sustancias PBT y mPmB en medicamentos veterinarios publicadas por la Agencia. |
IIIa.3A6.2. Evaluación del riesgo medioambiental de medicamentos veterinarios que contienen o consisten en organismos modificados genéticamente
|
1) |
Cuando un medicamento veterinario contenga o consista en organismos modificados genéticamente, se adjuntarán también a la solicitud los documentos establecidos en el artículo 2 y en la parte C de la Directiva 2001/18/CE. |
|
2) |
Los posibles efectos adversos para la salud humana y el medio ambiente que puedan producirse mediante la transferencia de genes de organismos modificados genéticamente a otros organismos o que surjan de modificaciones genéticas se evaluarán con precisión caso por caso. Esa evaluación del riesgo medioambiental tiene como objetivo determinar y valorar posibles efectos adversos directos e indirectos, inmediatos o no, del organismo modificado genéticamente para la salud humana y el medio ambiente (incluidos vegetales y animales), y se llevará a cabo de conformidad con los principios del anexo II de la Directiva 2001/18/CE. |
IIIa.3B. Pruebas de residuos
|
1) |
A efectos del presente punto, se aplicarán las definiciones del Reglamento (CE) n.o 470/2009. |
|
2) |
El propósito de estudiar la eliminación de residuos de los tejidos comestibles o de los huevos, la leche y la miel (o la cera, si procede) procedentes de animales tratados es determinar en qué condiciones y hasta qué punto pueden persistir residuos en productos alimenticios procedentes de esos animales. Además, estos estudios permitirán determinar el tiempo de espera. |
|
3) |
Cuando se trate de medicamentos veterinarios para animales productores de alimentos, la documentación relativa a los residuos deberá poner de manifiesto:
|
IIIa.3B1. Identificación del medicamento
Los medicamentos veterinarios utilizados en el estudio se identificarán mediante los siguientes datos:
|
a) |
composición; |
|
b) |
resultados de pruebas físicas y químicas (potencia y pureza) de los lotes en cuestión; |
|
c) |
identificación del lote. |
IIIa.3B2. Eliminación de los residuos
|
1) |
Estos estudios, que consisten en medir la velocidad de eliminación de los residuos en la especie animal de destino después de la última administración del medicamento veterinario, tienen por objeto permitir la determinación de los tiempos de espera necesarios para garantizar que en los productos alimenticios procedentes de animales tratados no queden residuos que puedan poner en peligro a los consumidores. |
|
2) |
Se notificará la situación actual de los límites máximos de residuos para los componentes del medicamento veterinario en la especie de destino pertinente. |
|
3) |
Los niveles de residuos presentes se determinarán en un número suficiente de momentos después de la última administración del medicamento veterinario a los animales de experimentación. Los estudios en mamíferos y aves se realizarán según las directrices VICH GL48 y otras directrices aplicables. Los estudios de residuos en la miel se realizarán según las directrices VICH GL56 y los de eliminación de residuos en especies acuáticas seguirán las directrices VICH GL57. |
|
4) |
Con arreglo a la evaluación, se justificará el tiempo de espera propuesto. |
IIIa.3B3. Método de análisis de residuos
|
1) |
Los estudios de eliminación de residuos, los métodos analíticos y su validación se realizarán de conformidad con las directrices VICH GL49. |
|
2) |
La idoneidad del método analítico propuesto se valorará a la luz del estado de los conocimientos científicos y técnicos en el momento en que se presente la solicitud. |
IIIa.4. Parte 4: Documentación sobre la eficacia (estudios preclínicos y ensayos clínicos)
IIIa.4A. Estudios preclínicos
Los estudios preclínicos tienen por objeto investigar la seguridad para el animal de destino y la eficacia del medicamento, y son necesarios para determinar la actividad farmacológica, las propiedades farmacocinéticas, la dosis, el intervalo de administración, la resistencia (si procede) y la tolerancia en el animal de destino del medicamento.
IIIa.4A1. Farmacología
IIIa.4A1.1. Farmacodinamia
|
1) |
Se caracterizarán los efectos farmacodinámicos de los principios activos del medicamento veterinario. |
|
2) |
Se describirán de manera adecuada el mecanismo de acción y los efectos farmacológicos, incluidos los efectos secundarios (en su caso), en que se basa la aplicación recomendada. Por lo general se examinarán los efectos en las principales funciones corporales. Los resultados se expresarán de forma cuantitativa (por ejemplo, mediante curvas dosis-efecto o tiempo-efecto) y, en la medida de lo posible, comparándolos con los de una sustancia cuya actividad se conozca. Cuando se alegue una actividad mayor para un principio activo, se demostrará que la diferencia existe y es estadísticamente significativa. |
|
3) |
Se estudiará cualquier efecto de las demás características de los medicamentos (como la vía de administración o la formulación) en la actividad farmacológica del principio activo. |
|
4) |
Las técnicas experimentales, a menos que sean procedimientos normalizados, se describirán de forma tal que permitan su reproducción y que se pueda demostrar su validez. Los resultados de los métodos experimentales se expresarán claramente y se presentará el resultado de las comparaciones estadísticas. |
|
5) |
Deberá investigarse igualmente, salvo justificación apropiada, toda modificación cuantitativa de los efectos que resulte de la administración repetida del producto. |
IIIa.4A1.2. Farmacocinética
|
1) |
Se necesitan los datos farmacocinéticos básicos del principio activo en el contexto de la evaluación de la seguridad y la eficacia del medicamento veterinario para la especie de destino, en particular si se trata de un principio o una formulación nuevos. |
|
2) |
Los objetivos de estudios farmacocinéticos en las especies animales de destino pueden dividirse en cuatro ámbitos principales:
|
|
3) |
En las especies animales de destino, los estudios farmacocinéticos son necesarios, por regla general, como complemento a los estudios farmacodinámicos para contribuir a establecer pautas posológicas efectivas (vía y lugar de la administración, dosis, intervalo de administración, número de administraciones, etc.). Pueden ser precisos otros estudios farmacocinéticos para establecer pautas posológicas según determinadas variables de población. |
|
4) |
Cuando se hayan presentado estudios farmacocinéticos en la parte 3 del expediente, podrá hacerse referencia a ellos. |
|
5) |
En el caso de combinaciones fijas, véase la sección IV. |
IIIa.4A2. Aparición de resistencia y riesgo para los animales
|
1) |
Para los medicamentos veterinarios biológicos pertinentes (por ejemplo, sustancias con actividad antimicrobiana o antiparasitaria), se proporcionará información sobre la resistencia actual (si procede) y sobre la posible aparición de resistencia de interés clínico para la indicación alegada en la especie animal de destino. Si es posible, se presentará información sobre los mecanismos de resistencia, la base genética molecular de la resistencia y la tasa de transmisión de los determinantes de resistencia. Cuando proceda, se presentará información sobre corresistencia y resistencia cruzada. El solicitante propondrá medidas para limitar la aparición en los organismos de resistencia de interés clínico para la utilización prevista del medicamento veterinario. |
|
2) |
Los datos de resistencia relacionados con el riesgo para las personas se abordarán en la parte 3 del expediente. Cuando proceda, se hará referencia a los datos establecidos en la parte 3 del expediente. |
IIIa.4A3. Determinación y confirmación de la dosis
|
1) |
Se facilitarán los datos apropiados para justificar las propuestas relativas a la dosis, el intervalo de administración, la duración del tratamiento y posibles repeticiones del tratamiento. |
|
2) |
En el caso de los estudios realizados en condiciones de campo, se facilitará la información correspondiente indicada en los estudios clínicos. |
IIIa.4A4. Tolerancia en la especie animal de destino
|
1) |
Se estudiará la tolerancia local y sistémica del medicamento veterinario en la especie animal de destino. La finalidad de estos estudios de seguridad para la especie animal de destino es caracterizar los signos de intolerancia y establecer un margen adecuado de seguridad para las vías de administración recomendadas. Esto puede lograrse aumentando la dosis o la duración del tratamiento. |
|
2) |
Los informes sobre los estudios contendrán detalles de todos los efectos farmacológicos previsibles y de todas las reacciones adversas. Estos estudios de seguridad para la especie animal de destino se realizarán de conformidad con las directrices de la VICH y las directrices pertinentes publicadas por la Agencia. También se puede obtener información sobre la seguridad en las especies de destino en otros estudios preclínicos y ensayos clínicos, además de en la bibliografía existente. |
IIIa.4B. Ensayos clínicos
IIIa.4B1. Principios generales
|
1) |
Los ensayos clínicos se diseñarán, realizarán y comunicarán teniendo en cuenta las directrices de la VICH y las directrices pertinentes publicadas por la Agencia. Los datos procedentes de ensayos clínicos realizados fuera de la Unión podrán ser tomados en consideración al evaluar una solicitud de autorización de comercialización únicamente si los datos son suficientemente representativos de la situación en la Unión. |
|
2) |
Salvo debida justificación, los datos experimentales, como los de ensayos exploratorios o piloto, o los resultados de planteamientos no experimentales deberán confirmarse mediante datos obtenidos en condiciones de campo normales. |
|
3) |
La finalidad de los ensayos clínicos es examinar, en condiciones de campo, la seguridad para el animal de destino y la eficacia de un medicamento veterinario en condiciones normales de cría de animales o como parte de prácticas veterinarias normales. Con ellos se demostrará el efecto del medicamento veterinario tras la administración a la especie de destino siguiendo las pautas posológicas propuestas y por las vías de administración recomendadas. El diseño de los ensayos tendrá por objeto apoyar las indicaciones y tener en cuenta las posibles contraindicaciones en función de la especie, la edad, la raza y el sexo, las instrucciones de uso del medicamento veterinario y cualquier reacción adversa que pueda darse. |
|
4) |
Todos los ensayos clínicos veterinarios se llevarán a cabo de conformidad con un protocolo de ensayo pormenorizado. En el caso de las formulaciones destinadas a su uso en ensayos clínicos veterinarios en la Unión, en el etiquetado deberá figurar de forma visible e indeleble la expresión «uso exclusivo en ensayos clínicos veterinarios». |
|
5) |
Salvo debida justificación, los ensayos clínicos se realizarán con animales testigo (ensayos clínicos comparativos). Los resultados de eficacia obtenidos con el nuevo medicamento se compararán con los de las especies animales de destino a las que se haya administrado un medicamento veterinario autorizado en la Unión que haya demostrado un nivel aceptable de eficacia y que haya sido aprobado para las indicaciones propuestas y para uso en las mismas especies animales de destino, o un placebo o ningún tratamiento. Se informará de todos los resultados obtenidos, ya sean positivos o negativos. |
|
6) |
Salvo debida justificación, en el diseño, el análisis y la evaluación del protocolo de ensayos clínicos se utilizarán principios estadísticos establecidos de conformidad con las directrices pertinentes publicadas por la Agencia. |
IIIa.4B2. Documentación
En el expediente sobre la eficacia figurará toda la documentación preclínica y clínica, ya sea favorable o desfavorable para el medicamento veterinario, que permita una evaluación general objetiva de la relación beneficio-riesgo del medicamento.
IIIa.4B2.1. Resultado de los estudios preclínicos
Cuando sea posible, se aportarán datos sobre los resultados de:
|
a) |
los estudios que pongan de manifiesto la actividad farmacológica; |
|
b) |
los estudios que evidencien los mecanismos farmacodinámicos responsables del efecto terapéutico; |
|
c) |
los estudios que demuestren el principal perfil farmacocinético; |
|
d) |
los estudios de seguridad para la especie animal de destino; |
|
e) |
los estudios que demuestren y confirmen la dosis (incluidos el intervalo de administración, la duración del tratamiento y posibles repeticiones del tratamiento); |
|
f) |
las pruebas e investigaciones sobre la resistencia, si procede. |
Si durante la realización de las pruebas aparecen resultados inesperados, se describirán detalladamente. Además, en todos los informes sobre estudios preclínicos se incluirán los elementos siguientes:
|
a) |
un resumen; |
|
b) |
un protocolo del estudio; |
|
c) |
una descripción detallada de los objetivos, el diseño y la realización que incluya los métodos, instrumentos y materiales utilizados, la identificación de los animales, su especie, edad, peso, sexo, número y raza o linaje, la posología, vía y pauta de administración; |
|
d) |
un análisis estadístico de los resultados; |
|
e) |
una presentación objetiva de los resultados obtenidos, de la que se deduzcan conclusiones sobre la eficacia y la seguridad para el animal de destino del medicamento veterinario. |
Deberá justificarse la omisión de cualquiera de esos datos.
IIIa.4B2.2. Resultado de los ensayos clínicos
Todos los investigadores deberán proporcionar la información en fichas individuales cuando el tratamiento sea individual y en fichas colectivas cuando el tratamiento sea colectivo.
El titular de la autorización de comercialización del medicamento veterinario velará por que la documentación original, que constituye la base de la información facilitada, se conserve durante cinco años, como mínimo, desde el momento en que se retire la autorización.
Respecto de las observaciones de cada ensayo clínico, se hará un resumen sinóptico de los ensayos y sus resultados, que incluirá:
|
a) |
el número de animales testigo y de experimentación, tratados individual o colectivamente, desglosados por especie, raza o linaje, edad y sexo; |
|
b) |
el número de animales cuyo estudio haya sido interrumpido antes del final, y los motivos de dicha interrupción; |
|
c) |
para los animales testigo, la información siguiente:
|
|
d) |
la frecuencia de las reacciones adversas observadas; |
|
e) |
observaciones relativas al efecto en la producción de los animales, si procede; |
|
f) |
los datos sobre los animales de experimentación que puedan presentar mayor riesgo en razón de su edad, modo de cría, alimentación o destino, o cuyo estado fisiológico o patológico requiera una atención especial; |
|
g) |
una evaluación estadística de los resultados. |
La persona responsable de la investigación extraerá conclusiones generales sobre la eficacia y la seguridad para el animal de destino del medicamento veterinario en las condiciones de uso propuestas y, en particular, toda información relativa a las indicaciones y contraindicaciones, la posología y la duración media del tratamiento y, en su caso, las interacciones observadas con otros medicamentos veterinarios o aditivos de los piensos, así como las precauciones particulares que deban tomarse durante el tratamiento, y los signos clínicos de sobredosificación, cuando los haya.
SECCIÓN IIIb
REQUISITOS RELATIVOS A LOS MEDICAMENTOS VETERINARIOS INMUNOLÓGICOS
Los requisitos siguientes se aplicarán a los medicamentos veterinarios inmunológicos definidos en el artículo 4, punto 5, salvo indicación contraria en la sección IV.
IIIb.1. Parte 1: Resumen del expediente
Véase la sección I.
IIIb.2. Parte 2: Documentación sobre calidad (información fisicoquímica, biológica y microbiológica)
IIIb.2.A. Descripción del producto
IIIb.2A1. Composición cualitativa y cuantitativa
|
1) |
Se entenderá por «composición cualitativa» de todos los componentes del medicamento veterinario inmunológico la designación o descripción de:
|
|
2) |
Los datos indicados en el punto 1 irán acompañados de toda información útil sobre el acondicionamiento primario y, si procede, sobre el embalaje exterior y, en su caso, el tipo de cierre, junto con detalles sobre los dispositivos que se empleen para utilizar o administrar el medicamento veterinario inmunológico y que se suministren con él. Cuando el dispositivo no se suministre junto con el medicamento veterinario inmunológico, se dará sobre el mismo la información que sea precisa para evaluar el medicamento. |
|
3) |
Sin perjuicio de lo dispuesto en el artículo 8, se entenderá por «terminología usual» para la designación de los componentes de los medicamentos veterinarios inmunológicos:
|
|
4) |
Para proporcionar la composición cuantitativa de los principios activos de un medicamento veterinario inmunológico, será preciso especificar siempre que sea posible el número de organismos, su contenido proteico específico, la masa, el número de unidades internacionales (UI) o unidades de actividad biológica, bien sea por dosis o por unidad de volumen, y, por lo que respecta al adyuvante y a los componentes de los excipientes, la masa o el volumen de cada uno de ellos, teniendo debidamente en cuenta lo especificado en la parte IIb.2B. |
|
5) |
Cuando se haya definido una unidad internacional de actividad biológica, se utilizará. |
|
6) |
Las unidades de actividad biológica de las que no existan datos publicados se expresarán de forma que se proporcione información inequívoca sobre la actividad de los ingredientes, por ejemplo haciendo constar la cantidad determinada por titrimetría o mediante pruebas de potencia del medicamento final. |
|
7) |
La composición se hará constar en términos de cantidades mínimas y, si procede, con cantidades máximas. |
IIIb.2A2. Desarrollo farmacéutico
|
1) |
Se explicarán los siguientes aspectos, entre otros:
|
|
2) |
Esta explicación se justificará con datos científicos sobre el desarrollo farmacéutico. |
IIIb.2B. Descripción del método de fabricación
|
1) |
La descripción del método de fabricación que, conforme a lo establecido en el artículo 8, debe acompañar a la solicitud de autorización de comercialización se redactará de forma que ofrezca una idea clara del carácter de las operaciones efectuadas, incluida la determinación de las etapas clave del proceso de producción. |
|
2) |
La descripción del proceso de fabricación incluirá, como mínimo:
|
|
3) |
Se describirá y documentará la validación de todos los métodos de control utilizados en el proceso de fabricación, y se proporcionarán los resultados, salvo justificación. Se demostrarán la validación de etapas clave del proceso de producción y la validación del proceso de producción en su conjunto, aportando los resultados de tres lotes consecutivos producidos con el método descrito. |
IIIb.2C. Producción y control de los materiales de partida
|
1) |
A los efectos de la presente parte, se entenderá por «materiales de partida» todos los componentes utilizados en la producción del medicamento veterinario inmunológico. |
|
2) |
Los sistemas de adyuvantes preparados para usar disponibles en el mercado designados con una marca, así como los medios de cultivo utilizados para producir los principios activos y que consten de varios componentes, se considerarán un solo material de partida. No obstante, se presentará la composición cualitativa y cuantitativa siempre que las autoridades consideren que esta información es pertinente para la calidad del producto terminado y para cualquier riesgo que pudiera surgir. |
|
3) |
Si se utilizan materiales de origen animal para preparar estos medios de cultivo o sistemas de adyuvantes, tendrá que indicarse la especie animal y el tejido utilizados, y se demostrará el cumplimiento de las monografías pertinentes, incluidas las generales, y de los capítulos generales de la Farmacopea Europea. |
|
4) |
El solicitante suministrará documentación para demostrar que los materiales de partida, incluidos los materiales de siembra, las líneas celulares, los lotes de suero y demás materiales procedentes de especies animales por las que puedan transmitirse las EET, y la fabricación del medicamento veterinario se ajustan a los requisitos de la Nota explicativa sobre cómo minimizar los riesgos de transmisión de los agentes de las encefalopatías espongiformes animales a través de los medicamentos humanos y veterinarios, así como a los requisitos de la correspondiente monografía de la Farmacopea Europea. Como justificante de cumplimiento podrán utilizarse los certificados de conformidad emitidos por la Dirección Europea de Calidad del Medicamento y la Asistencia Sanitaria, con referencia a la correspondiente monografía de la Farmacopea Europea. |
|
5) |
El expediente contendrá las especificaciones, información sobre los estudios que se realizarán para el control de calidad de todos los lotes de materiales de partida y los resultados de un lote de todos los componentes utilizados, y se presentará de conformidad con los requisitos indicados en la presente parte. |
|
6) |
Se presentarán certificados de análisis de los materiales de partida para demostrar el cumplimiento de las características técnicas definidas. |
|
7) |
En todos los casos, las materias colorantes deberán reunir los requisitos que se establecen en la Directiva 2009/35/CE. |
|
8) |
La utilización de antibióticos durante la producción y la inclusión de conservantes en la composición del producto terminado deberán justificarse y cumplir la Farmacopea Europea. |
|
9) |
En el caso de excipientes nuevos, es decir, que se usan por primera vez en la Unión en un medicamento veterinario o por una nueva vía de administración, se darán los detalles de fabricación, caracterización y controles, con referencias en apoyo de los datos de seguridad tanto clínicos como no clínicos. En el caso de los colorantes, se considerarán suficientes las declaraciones de cumplimiento indicadas en la parte II.2C2, puntos 3 y 4. |
IIIb.2C1. Materiales de partida descritos en las farmacopeas
|
1) |
Las monografías de la Farmacopea Europea serán aplicables a todos los materiales de partida que figuren en ella, salvo justificación adecuada. |
|
2) |
Para las restantes sustancias, cada Estado miembro podrá exigir el cumplimiento de su farmacopea nacional en relación con los productos que se fabriquen en su territorio. |
|
3) |
La descripción de los métodos de análisis podrá sustituirse por una referencia detallada a la farmacopea de que se trate. |
|
4) |
Los estudios sistemáticos que se realicen en cada lote de materiales de partida se llevarán a cabo tal y como estén descritos en la solicitud de autorización de comercialización. Si se utilizan estudios distintos de los mencionados en una farmacopea, deberá demostrarse que los materiales de partida corresponden a las exigencias de calidad de dicha farmacopea. |
|
5) |
Cuando las especificaciones u otras disposiciones incluidas en una monografía de la Farmacopea Europea o en la farmacopea de un Estado miembro puedan no ser suficientes para garantizar la calidad de la sustancia, las autoridades competentes podrán exigir al solicitante de la autorización de comercialización que presente especificaciones más apropiadas. Se informará de la presunta insuficiencia a las autoridades responsables de la farmacopea en cuestión. |
IIIb.2C2. Materiales de partida no descritos en una farmacopea
IIIb.2C2.1. Materiales de partida de origen biológico
|
1) |
La descripción se presentará en forma de monografía. |
|
2) |
La producción de vacunas se basará en un sistema de lotes de siembra y en líneas celulares bien establecidas, siempre que sea posible. Para la producción de medicamentos veterinarios inmunológicos consistentes en sueros, se indicarán el origen, el estado de salud general y la situación inmunitaria de los animales donantes y se utilizarán conjuntos identificados de materiales de partida biológicos. |
|
3) |
Deberán describirse y documentarse el origen, incluida la región geográfica, y los antecedentes de los materiales de partida. |
|
4) |
En caso de materiales de partida procedentes de ingeniería genética, esta información incluirá aspectos como la descripción del origen de las células o cepas, la construcción del vector de expresión (denominación, origen, función del replicón, estimulador del promotor y otros elementos reguladores), control de la secuencia de ADN o ARN insertada correctamente, secuencias de oligonucleótidos del vector plasmídico en las células, plásmido utilizado para la cotransfección, genes adicionados o suprimidos, propiedades biológicas de la construcción final y los genes expresados, número de copias y estabilidad genética. |
|
5) |
Cuando un medicamento veterinario contenga o consista en organismos modificados genéticamente, se adjuntarán también a la parte de la solicitud dedicada a la calidad los documentos establecidos de conformidad con la Directiva 2001/18/CE. |
|
6) |
Se comprobará la identidad de los materiales de siembra, las líneas celulares y el suero de partida para producción de antisueros, y se demostrará la ausencia de agentes extraños según la Farmacopea Europea. |
|
7) |
Se dará información sobre todas las sustancias de origen biológico utilizadas en cualquier fase del proceso de fabricación. Esta información incluirá:
|
|
8) |
Si se detecta o sospecha la presencia de agentes extraños, el material correspondiente se eliminará o se procesará con un tratamiento validado para reducir el riesgo de dicha presencia. Si, después del tratamiento, se detecta o sospecha tal presencia, el material correspondiente se utilizará únicamente cuando un procesamiento posterior del producto garantice la eliminación o inactivación de esos agentes; esta eliminación o inactivación de los agentes extraños deberá demostrarse. |
|
9) |
Cuando se utilicen líneas celulares, deberá demostrarse que las características celulares se han mantenido inalteradas hasta el pase más alto utilizado en la producción. |
|
10) |
En el caso de vacunas atenuadas, deberá proporcionarse una confirmación de la estabilidad de las características de atenuación de la siembra. A menos que una característica específica esté asociada a la atenuación (por ejemplo, marcador genético o estabilidad térmica), esto se consigue normalmente mediante la ausencia de reversión a la virulencia en la especie animal de destino. |
|
11) |
En caso necesario, deberán proporcionarse a las autoridades competentes muestras del material biológico de partida o de los reactivos utilizados en los métodos de ensayo, a fin de que puedan disponer la realización de los estudios necesarios de comprobación. |
IIIb.2C2.2. Materiales de partida de origen no biológico
La descripción se presentará en forma de monografía con los siguientes encabezamientos:
|
a) |
la denominación del material de partida que cumpla los requisitos de la parte IIIb.2A1, punto 3, irá acompañada por sus sinónimos comerciales o científicos; |
|
b) |
la descripción del material de partida, establecida de forma similar a la utilizada en las descripciones de la Farmacopea Europea; |
|
c) |
la función del material de partida; |
|
d) |
métodos de identificación; |
|
e) |
cualquier precaución que pueda ser necesaria durante el almacenamiento del material de partida y, en su caso, su período de validez. |
IIIb.2D. Pruebas de control efectuadas durante el proceso de fabricación
|
1) |
El expediente incluirá detalles de las pruebas de control, realizadas en fases intermedias de la fabricación, para verificar la constancia del proceso de fabricación y del producto final. Se establecerán especificaciones para cada prueba de control y se describirán los métodos analíticos. Se proporcionará la validación de las pruebas de control en relación con los parámetros considerados críticos para el proceso de fabricación, salvo debida justificación. |
|
2) |
Para las vacunas inactivadas o detoxificadas, se comprobarán durante cada ciclo de producción la inactivación o detoxificación lo antes posible una vez finalizadas y, en su caso, tras la neutralización, pero siempre antes de la siguiente fase de producción. |
|
3) |
De conformidad con lo dispuesto en la Directiva 2010/63/UE y con el Convenio Europeo sobre Protección de los Animales Vertebrados Utilizados con Fines Experimentales y Otros Fines Científicos, las pruebas se realizarán de forma que se utilice el menor número de animales y se cause el menor dolor, sufrimiento, angustia o daño duradero. Se utilizarán pruebas alternativas in vitro, si las hay, cuando puedan sustituir o disminuir el uso de animales o reducir su sufrimiento. |
IIIb.2E. Pruebas de control del producto terminado
|
1) |
En todas las pruebas deberán describirse de forma detallada las técnicas de análisis del producto terminado, de modo que sea posible la evaluación de calidad. |
|
2) |
Cuando haya monografías adecuadas, si se utilizan procedimientos analíticos y límites distintos de los mencionados en las monografías de la Farmacopea Europea o, en su defecto, de la farmacopea de un Estado miembro, deberá demostrarse que el producto terminado, si se sometiera a control con arreglo a estas monografías, cumpliría los requisitos de calidad establecidos en dicha farmacopea para la forma farmacéutica correspondiente. La solicitud de autorización de comercialización presentará una relación de estas pruebas, que se realizarán con muestras representativas de cada lote del producto terminado. Se hará constar la frecuencia de las pruebas realizadas en la vacuna final a granel, en lugar de en el lote o los lotes preparados. Se indicarán y justificarán los límites de liberación. Se proporcionará la validación de las pruebas de control que se realicen en el producto terminado. |
|
3) |
Se facilitará información sobre el establecimiento y la sustitución de material de referencia. Si se han utilizado dos o más normas de referencia, se facilitará un historial de cualificación en el que se describa cómo se mantuvo la relación entre las distintas normas. |
|
4) |
Cuando exista, se utilizará material de referencia químico y biológico de la Farmacopea Europea. Si se utilizan otras preparaciones y normas de referencia, se identificarán y se describirán detalladamente. |
|
5) |
De conformidad con lo dispuesto en la Directiva 2010/63/UE y con el Convenio Europeo sobre Protección de los Animales Vertebrados Utilizados con Fines Experimentales y Otros Fines Científicos, las pruebas se realizarán de forma que se utilice el menor número de animales y se cause el menor dolor, sufrimiento, angustia o daño duradero. Se utilizarán pruebas alternativas in vitro, si las hay, cuando puedan sustituir o disminuir el uso de animales o reducir su sufrimiento. |
|
6) |
Características generales del producto terminado
Los controles de las características generales se referirán, siempre que sea procedente, a la apariencia y a estudios físicos o químicos como la conductividad, el pH, la viscosidad, etc. El solicitante deberá definir las especificaciones de cada una de esas características, con límites de aceptación apropiados. |
|
7) |
Identificación de los principios activos
En caso necesario, se realizará una prueba específica de identificación. Cuando proceda, la prueba de identificación podrá combinarse con la prueba de título o potencia del lote. |
|
8) |
Título o potencia de lote
El principio activo se cuantificará en cada lote para confirmar que cada uno de ellos contiene la potencia o el título apropiados que garanticen su seguridad y su eficacia. |
|
9) |
Identificación y determinación de los adyuvantes
Deberán verificarse en el producto terminado la cantidad y la naturaleza del adyuvante y sus componentes, salvo justificación. |
|
10) |
Identificación y determinación de los excipientes
En la medida de lo necesario, el excipiente o excipientes serán objeto, como mínimo, de pruebas de identificación. Los conservantes se presentarán obligatoriamente a una prueba de límite superior e inferior. Cualquier otro componente del excipiente que pueda producir una reacción adversa se presentará obligatoriamente a una prueba de límite superior. |
|
11) |
Pruebas de esterilidad y pureza
Se demostrará la ausencia de agentes extraños (bacterias, micoplasma, hongos y endotoxinas bacterianas, cuando proceda) para los productos administrados por vía parenteral con arreglo a la Farmacopea Europea. Para los productos que no sean líquidos administrados por vía no parenteral, cuando se justifique adecuadamente, podrá aceptarse el cumplimiento de un límite máximo de carga biológica en lugar de la prueba de esterilidad. Deberán realizarse pruebas adecuadas para demostrar la ausencia de contaminación por agentes extraños u otras sustancias, según la naturaleza del medicamento veterinario inmunológico, el método y las condiciones de fabricación. Para demostrar la ausencia de agentes extraños se utilizará un planteamiento basado en el riesgo descrito en la Farmacopea Europea. |
|
12) |
Humedad residual
Cada lote de producto liofilizado se presentará a una prueba de determinación de la humedad residual. |
|
13) |
Volumen de llenado
Se llevarán a cabo pruebas adecuadas para demostrar el volumen de llenado correcto. |
IIIb.2F. Constancia entre lotes
Con el fin de garantizar que la calidad del medicamento es constante entre lotes y demostrar su conformidad con las especificaciones, se presentará un protocolo completo de tres lotes consecutivos representativos de la producción sistemática en el que figurarán los resultados de todas las pruebas realizadas durante la producción y con el producto terminado. Los datos de constancia de medicamentos combinados podrán utilizarse para los productos derivados que contengan uno o más de los mismos componentes.
IIIb.2G. Estudios de estabilidad
|
1) |
Los estudios de estabilidad se refieren a la estabilidad del principio activo y del producto terminado, incluidos los disolventes, si procede. |
|
2) |
Deberán describirse las pruebas realizadas para determinar el período de validez, las condiciones recomendadas de almacenamiento y las especificaciones al final del período de validez que se hayan propuesto para el principio activo y el producto terminado. Estos estudios serán siempre en tiempo real.
Si los productos intermedios obtenidos en diversas fases del proceso de fabricación se almacenan, se justificarán adecuadamente las condiciones y la duración previstas del almacenamiento sobre la base de los datos de estabilidad disponibles. |
|
3) |
Los estudios de estabilidad del producto terminado se llevarán a cabo en un mínimo de tres lotes representativos fabricados según el proceso de producción descrito y en productos almacenados en su recipiente final; incluirán estudios de estabilidad biológica y fisicoquímica, realizados a intervalos periódicos, del producto terminado hasta tres meses después del final del período de validez alegado. |
|
4) |
En las conclusiones figurarán los resultados de los análisis y se justificará el período de validez propuesto en todas las condiciones propuestas de almacenamiento. Los resultados obtenidos durante el estudio de estabilidad se tendrán en cuenta al definir las especificaciones apropiadas de formulación y liberación, a fin de garantizar la conformidad del medicamento con el período de validez alegado. |
|
5) |
Cuando se trate de medicamentos que se administren con piensos, también se incluirá la información necesaria sobre el período de validez del producto en las diferentes fases de mezcla, cuando se mezcle con arreglo a las instrucciones de uso recomendadas. |
|
6) |
Si un producto terminado tiene que ser reconstituido previamente a su administración, o se administra con el agua de bebida, se especificará el período de validez que se propone para el medicamento reconstituido según lo recomendado. Deberán presentarse datos que justifiquen el período de validez propuesto para el producto reconstituido. |
|
7) |
Los datos de estabilidad de medicamentos combinados podrán utilizarse, cuando se justifique debidamente, para los productos derivados que contengan uno o más de los mismos componentes. |
|
8) |
Cuando se trate de envases multidosis, se presentarán los datos relativos a la estabilidad para justificar el período de validez del medicamento después de su primera apertura o utilización, y se definirán las especificaciones sobre el período de validez una vez abierto. |
|
9) |
Deberá demostrarse la eficacia del sistema de conservación utilizado. |
|
10) |
Para ello podrá ser suficiente la información sobre la eficacia de los conservantes en otros medicamentos veterinarios inmunológicos similares del mismo fabricante. |
|
11) |
Si los principios activos se almacenan, se definirán las condiciones y la duración del almacenamiento sobre la base de los datos de estabilidad. Estos datos pueden obtenerse bien mediante pruebas en los principios activos propiamente dichos o bien mediante las pruebas adecuadas en el medicamento terminado. |
IIIb.2H. Otra información
El expediente podrá contener información sobre la calidad del medicamento veterinario inmunológico que no haya quedado cubierta en la presente sección.
IIIb.3. Parte 3: Documentación relativa a la seguridad (pruebas de seguridad y de residuos)
IIIb.3A. Requisitos generales
|
1) |
La documentación relativa a la seguridad será adecuada para evaluar lo siguiente:
|
|
2) |
Los estudios preclínicos se llevarán a cabo de conformidad con los requisitos de las buenas prácticas de laboratorio.
Los estudios que no cumplan las buenas prácticas de laboratorio podrán aceptarse si se trata de estudios sobre especies que no son las de destino o estudios en los que se evalúen las propiedades inmunológicas, biológicas o genéticas de las cepas vacunales, en condiciones adecuadamente controladas. Los demás casos deberán justificarse. |
|
3) |
Todos los ensayos de seguridad se realizarán con arreglo a un protocolo pormenorizado que deberá consignarse por escrito antes de iniciar el ensayo. Durante la elaboración de todo protocolo de ensayo y a lo largo de la realización de este, se tendrá en cuenta en todo momento el bienestar de los animales de experimentación, aspecto que será objeto de supervisión veterinaria. |
|
4) |
Se exigirán sistemáticamente procedimientos escritos preestablecidos referentes a la organización, realización, recopilación de datos, documentación y comprobación de los ensayos de seguridad. |
|
5) |
Los ensayos clínicos (ensayos de campo) se llevarán a cabo de conformidad con los principios establecidos de buenas prácticas clínicas. Toda desviación deberá justificarse. |
|
6) |
Los estudios de seguridad cumplirán los requisitos pertinentes de la Farmacopea Europea. Toda desviación deberá justificarse. |
|
7) |
Los estudios de seguridad se realizarán en la especie de destino. La dosis que debe utilizarse será la cantidad del producto que debe recomendarse para el uso, y el lote utilizado para la prueba de seguridad se tomará de un lote o lotes producidos según el proceso de fabricación descrito en la parte 2 de la solicitud. |
|
8) |
Para las pruebas de laboratorio descritas en las secciones B.1, B.2 y B.3, la dosis del medicamento veterinario será la que contenga el nivel máximo del título, el contenido de antígeno o la potencia. En caso necesario podrá ajustarse la concentración del antígeno para lograr la dosis requerida. |
|
9) |
La seguridad de un medicamento veterinario inmunológico deberá demostrarse para cada categoría de especie animal de destino para la que se recomiende su uso, mediante cada vía y modo de administración recomendados y siguiendo la pauta de administración propuesta. Podrá utilizarse el caso más desfavorable posible para la vía y el modo de administración si existe justificación científica para ello. |
|
10) |
En el caso de medicamentos veterinarios inmunológicos que contengan organismos vivos, en la sección B.6 se incluyen requisitos especiales. |
|
11) |
Los datos y documentos que acompañen a una solicitud de autorización de comercialización se presentarán conforme a los requisitos para los estudios preclínicos y los ensayos clínicos descritos en la parte IIIb.4B, punto 4, y en la parte IIIb.4C, punto 3. |
IIIb.3B. Estudios preclínicos
|
1) |
Seguridad de la administración de una sola dosis
El medicamento veterinario inmunológico se administrará a la dosis recomendada, y por cada vía y modo de administración recomendados, a animales de cada especie y categoría en las que se prevea utilizarlo (por ejemplo, animales de la edad mínima o hembras gestantes, según corresponda). Los animales se observarán y examinarán diariamente para detectar posibles signos de reacciones generalizadas y locales hasta que ya no pueda preverse reacción alguna pero, en todos los casos, durante un período mínimo de catorce días a partir de la última administración. Cuando corresponda, esos estudios incluirán exámenes detallados microscópicos y macroscópicos detallados del lugar de la inyección, realizados tras la muerte del animal. Se registrarán otros criterios objetivos, como la temperatura rectal y mediciones de la producción. Este estudio puede formar parte del estudio de administración repetida de una dosis establecido en el punto 3, o bien omitirse si el estudio de sobredosis establecido en el punto 2 no pone de manifiesto signos importante de reacción generalizada o local. De omitirse, las reacciones generalizadas o locales observadas en el estudio de sobredosis se tomarán como base para describir la seguridad del producto en el resumen de las características del medicamento. |
|
2) |
Seguridad de una sola administración de una sobredosis
Solamente los medicamentos veterinarios inmunológicos que contengan microorganismos vivos han de someterse a la prueba de la sobredosis. Se administrará una sobredosis del medicamento veterinario inmunológico, compuesta normalmente por diez dosis, por cada vía y cada modo recomendados de administración a animales de las categorías más sensibles de las especies de destino, a menos que esté justificado seleccionar la vía más sensible de varias similares. En el caso de los medicamentos veterinarios inmunológicos administrados por inyección, se elegirán las dosis, las vías y los modos de administración para tener en cuenta el volumen máximo que puede administrarse en cualquier punto único de inyección. Los animales se observarán y examinarán diariamente durante un período mínimo de catorce días a partir de la última administración para detectar posibles signos de reacciones generalizadas y locales. Se registrarán otros criterios objetivos, como la temperatura rectal y mediciones de la producción. Cuando corresponda, esos estudios incluirán exámenes detallados microscópicos y macroscópicos del lugar de la inyección, realizados tras la muerte del animal, si no se han hecho para el punto 1. |
|
3) |
Seguridad de la administración repetida de una sola dosis
En el caso de los medicamentos veterinarios inmunológicos que deben administrarse más de una vez, como parte del sistema básico de administración, será preciso estudiar la administración repetida de una sola dosis para poner de manifiesto los posibles efectos adversos inducidos por dicha administración. La prueba se llevará a cabo en las categorías más sensibles de las especies de destino (como determinadas razas o categorías de edad), utilizando cada vía y cada modo de administración recomendados. El número de administraciones no será inferior al número máximo recomendado; en el caso de las vacunas, se tendrá en cuenta el número de administraciones para la primovacunación y la primera revacunación. El intervalo entre administraciones puede ser más breve que el indicado en el resumen de las características del medicamento. Se justificará el intervalo elegido en relación con las condiciones de uso propuestas. Los animales se observarán y examinarán diariamente durante un período mínimo de catorce días a partir de la última administración para detectar posibles signos de reacciones generalizadas y locales. Se registrarán otros criterios objetivos, como la temperatura rectal y mediciones de la producción. |
|
4) |
Examen de la función reproductora
Se tendrá en cuenta un examen de la función reproductora cuando el medicamento veterinario inmunológico vaya a administrarse o pueda usarse en hembras gestantes o aves de puesta y cuando haya datos que sugieran que el material de partida del que deriva el producto pueda ser un factor de riesgo. Se estudiará la función reproductora de machos y de hembras gestantes y no gestantes con la dosis recomendada y por la vía y el modo de administración más sensibles. En el caso de medicamentos veterinarios inmunológicos que estén recomendados para hembras gestantes, el examen de la función reproductora abordará la seguridad de la administración durante todo el período de gestación o durante el período de gestación específico que refleje el uso al que esté destinado el producto. El período de observación se ampliará hasta el parto a fin de investigar posibles efectos nocivos en la progenie, incluidos los efectos teratogénicos y abortivos. Esos estudios podrán formar parte de los estudios de seguridad descritos en los puntos 1, 2 y 3 o de los ensayos de campo previstos en la parte IIIb.3C. |
|
5) |
Examen de la función inmunitaria
Cuando el medicamento veterinario inmunológico pueda afectar negativamente a la respuesta inmunitaria del animal vacunado o su progenie, deberán realizarse estudios adecuados de la función inmunitaria. |
|
6) |
Requisitos especiales para las vacunas atenuadas |
1) Transmisión de la cepa vacunal
Se investigará la transmisión de la cepa vacunal desde los animales de destino vacunados hacia los no vacunados, por la vía de administración recomendada que con más probabilidad pueda dar lugar a la transmisión. Además, podrá ser necesario investigar la transmisión a especies animales no destinatarias que puedan ser muy sensibles a la cepa de la vacuna atenuada. Se proporcionará una evaluación del número de pases entre animales susceptibles de ocurrir en condiciones normales de uso y sus posibles consecuencias.
2) Distribución en el animal vacunado
Se investigará la presencia del microorganismo en las heces, orina, leche, huevos y secreciones orales, nasales y de otro tipo, según proceda. Además, podrá ser necesario realizar estudios sobre la distribución de la cepa vacunal en el cuerpo, prestando especial atención a los lugares favoritos de replicación del microorganismo. En el caso de las vacunas atenuadas contra zoonosis, en el sentido de la Directiva 2003/99/CE del Parlamento Europeo y del Consejo, destinadas a animales productores de alimentos, dichos estudios tendrán particularmente en cuenta la persistencia del microorganismo en el lugar de inyección.
3) Aumento de la virulencia
El aumento de la virulencia o la reversión a la virulencia se investigará con la semilla primaria. Si no se dispone de semilla primaria en cantidad suficiente, se investigará con material procedente del pase menos atenuado utilizado para la producción. Se justificará el uso de otro pase. La vacunación inicial se realizará por la vía y el modo de administración que puedan producir con mayor probabilidad un aumento de la virulencia que apunte a la reversión a la virulencia. Se llevarán a cabo pases seriados en cinco grupos de animales de la especie de destino, salvo que esté justificado hacer más pases o que el microorganismo desaparezca antes de los animales de experimentación. Si el microorganismo no se replica adecuadamente, se llevarán a cabo tantos pases como sea posible en la especie de destino.
4) Propiedades biológicas de la cepa vacunal
Puede ser necesario hacer otras pruebas para determinar con la mayor precisión posible las propiedades biológicas intrínsecas de la cepa vacunal (por ejemplo, el neurotropismo).
En el caso de vacunas que contengan organismos modificados genéticamente atenuados, cuando el producto de un gen foráneo se incorpore en la cepa en forma de proteína estructural, se abordará el riesgo de alteración del tropismo o la virulencia de la cepa y, si es necesario, se realizarán pruebas específicas.
5) Recombinación o redistribución genómica de las cepas
Se evaluará la probabilidad de recombinación o redistribución genómica con cepas silvestres o de otro tipo y se comentarán las consecuencias de tales eventos.
|
7) |
Seguridad para el usuario
En esta sección se comentarán los efectos encontrados desde la parte IIIb.3A hasta la parte IIIb.3B, que se relacionarán con el tipo y grado de exposición humana al medicamento con objeto de formular las correspondientes advertencias para el usuario y demás medidas de gestión del riesgo. La seguridad para el usuario se abordará de conformidad con las directrices pertinentes publicadas por la Agencia. |
|
8) |
Interacciones
Si en el resumen de las características del medicamento hay una declaración de compatibilidad con otros medicamentos veterinarios, se investigará la seguridad de la asociación. Se describirán otras interacciones conocidas con medicamentos veterinarios. |
IIIb.3C. Ensayos clínicos
Salvo debida justificación, los resultados de estudios preclínicos se completarán con datos de ensayos clínicos realizados con lotes representativos del proceso de fabricación descrito en la solicitud de autorización de comercialización. En los mismos ensayos clínicos pueden investigarse tanto la seguridad como la eficacia.
IIIb.3D. Evaluación del riesgo medioambiental
|
1) |
Se evaluará el riesgo medioambiental en busca de los posibles efectos nocivos del uso del medicamento veterinario y para determinar el riesgo de tales efectos. En la evaluación se especificará asimismo cualquier medida preventiva que pueda ser necesaria para reducir tal riesgo. |
|
2) |
Esta evaluación consta de dos fases. La primera fase de la evaluación se realizará en todos los casos. Los detalles de la evaluación se presentarán según las directrices publicadas por la Agencia. Se indicará la posible exposición del medio ambiente al medicamento y el riesgo asociado con tal exposición, teniendo especialmente en cuenta los puntos siguientes:
|
|
3) |
En el caso de cepas de vacunas atenuadas posiblemente zoonóticas, se evaluará el riesgo para las personas. |
|
4) |
Cuando las conclusiones de la primera fase apunten a un posible riesgo del medicamento para el medio ambiente, el solicitante pasará a la segunda fase e investigará el riesgo que podría constituir el medicamento veterinario para el medio ambiente. Cuando sea necesario, se realizarán estudios complementarios sobre las repercusiones del medicamento (en el suelo, las aguas, el aire, los sistemas acuáticos o los organismos que no sean de destino). |
|
5) |
En el caso de las vacunas de ADN, un problema de seguridad específico es el riesgo de migración del ADN a tejido gonadal y la potencial transferencia del ADN a la línea germinal de animales machos y hembras vacunados, con la posibilidad subsiguiente de transmisión a la descendencia. El solicitante evaluará y explicará los posibles riesgos que esos medicamentos veterinarios inmunológicos puedan plantear para la salud humana y el medio ambiente (incluidos vegetales y animales). Si se detectan posibles riesgos, se llevarán a cabo investigaciones sobre las repercusiones de la vacuna según se utilice en animales de compañía o en animales productores de alimentos, a fin de proporcionar información al respecto. |
IIIb.3E. Evaluación de los medicamentos veterinarios que contienen o consisten en organismos modificados genéticamente
|
1) |
Cuando un medicamento veterinario contenga o consista en organismos modificados genéticamente, se adjuntarán también a la solicitud los documentos establecidos en el artículo 2 y en la parte C de la Directiva 2001/18/CE y en las directrices específicas sobre organismos modificados genéticamente. |
|
2) |
Los posibles efectos adversos para la salud humana y el medio ambiente que puedan producirse mediante la transferencia de genes de organismos modificados genéticamente a otros organismos o que surjan de modificaciones genéticas se evaluarán con precisión caso por caso. Esa evaluación del riesgo medioambiental tiene como objetivo determinar y valorar posibles efectos adversos directos e indirectos, inmediatos o no, del organismo modificado genéticamente para la salud humana y el medio ambiente (incluidos vegetales y animales), y se llevará a cabo de conformidad con los principios del anexo II de la Directiva 2001/18/CE. |
IIIb.3F. Pruebas de residuos que deben incluirse en los estudios preclínicos
|
1) |
En el caso de medicamentos veterinarios inmunológicos, normalmente no será necesario realizar un estudio de residuos. |
|
2) |
Cuando los antibióticos, adyuvantes, conservantes o cualquier otro excipiente se utilicen en la fabricación de medicamentos veterinarios inmunológicos destinados a especies productoras de alimentos o se incluyan en el producto terminado, se tendrá en cuenta la posibilidad de exposición de los consumidores a residuos en los productos alimenticios procedentes de animales tratados, así como el cumplimiento de la legislación sobre los límites máximos de residuos. Se abordarán las implicaciones que su posible presencia en el medicamento terminado tenga para la seguridad de los consumidores. |
|
3) |
En el caso de vacunas atenuadas para enfermedades zoonóticas bien establecidas, además de los estudios de distribución tal vez sea necesario determinar la presencia de microorganismos residuales en el lugar de la inyección. En caso necesario, habrá que investigar los efectos de tales residuos. |
|
4) |
Se presentará una propuesta de tiempo de espera y se comentará su validez en relación con los estudios de residuos que se hayan realizado. |
IIIb.4. Parte 4: Documentación sobre la eficacia (estudios preclínicos y ensayos clínicos)
IIIb.4A. Requisitos generales
|
1) |
Se respetarán los siguientes requisitos generales:
|
|
2) |
En general, los estudios preclínicos se confirmarán con ensayos realizados en condiciones de campo.
Cuando los estudios preclínicos confirmen plenamente las alegaciones formuladas en el resumen de las características del medicamento, no será necesario realizar ensayos en condiciones de campo. Salvo debida justificación, los resultados de estudios preclínicos se completarán con datos de ensayos clínicos realizados con lotes representativos del proceso de fabricación descrito en la solicitud de autorización de comercialización. En los mismos ensayos clínicos pueden investigarse tanto la seguridad como la eficacia. |
|
3) |
Todos los ensayos se describirán de manera pormenorizada para que las autoridades competentes puedan evaluarlos adecuadamente. Se demostrará la validez de todas las técnicas utilizadas en el ensayo. |
|
4) |
Se indicarán todos los resultados obtenidos, tanto si son favorables como si no:
|
IIIb.4B. Estudios preclínicos
|
1) |
En principio, la demostración de la eficacia deberá realizarse en condiciones de laboratorio perfectamente controladas mediante una prueba de provocación tras la administración del medicamento veterinario inmunológico al animal de destino en las condiciones de uso recomendadas. Siempre que sea posible, las condiciones en que se realiza la prueba de provocación reflejarán las condiciones naturales de la infección. Se dará información detallada de la cepa utilizada en la provocación y su pertinencia. |
|
2) |
En el caso de las vacunas atenuadas, el medicamento utilizado para el estudio de eficacia se tomará de un lote o lotes que contengan el título o la potencia mínimos. En los demás casos, se utilizará un medicamento tomado de lotes con el contenido activo o la potencia mínimos previstos al final del período de validez, salvo justificación. |
|
3) |
Si es posible, se especificará y documentará el mecanismo inmunitario (humoral o mediado por células, inmunoglobulinas generales o locales) que se desencadena tras la administración del medicamento veterinario inmunológico a la especie de destino por la vía de administración recomendada. |
|
4) |
En cada estudio preclínico se hará constar lo siguiente:
|
IIIb.4C. Ensayos clínicos
|
1) |
Salvo debida justificación, los resultados de estudios preclínicos se completarán con datos de ensayos de campo realizados con lotes representativos del proceso de fabricación descrito en la solicitud de autorización de comercialización. En un mismo ensayo de campo pueden investigarse tanto la seguridad como la eficacia. |
|
2) |
Si los estudios preclínicos no pueden demostrar la eficacia, podrá aceptarse que únicamente se realicen ensayos de campo. |
|
3) |
Los datos relativos a los ensayos de campo serán lo suficientemente detallados como para que pueda hacerse un juicio objetivo. Incluirán los siguientes aspectos:
|
SECCIÓN IV
REQUISITOS RELATIVOS A SOLICITUDES ESPECÍFICAS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
IV.1. Solicitudes para medicamentos veterinarios genéricos
|
IV.1.1. |
Las solicitudes basadas en el artículo 18 (medicamentos veterinarios genéricos) contendrán los datos mencionados en las partes 1 y 2 de la sección II del presente anexo. De ser necesario según el artículo 18, apartado 7, incluirán una evaluación del riesgo medioambiental. Además, el expediente contendrá datos que demuestren que el medicamento tiene la misma composición cualitativa y cuantitativa en principios activos y la misma forma farmacéutica que el medicamento de referencia, junto con datos que demuestren la bioequivalencia con el medicamento de referencia o la justificación de por qué no se han realizado estudios al respecto, con referencia a directrices establecidas. Todas las formas farmacéuticas orales de liberación inmediata se considerarán una misma forma farmacéutica.
El planteamiento normal para los medicamentos veterinarios genéricos, en principio, no se considera apropiado en el caso de los medicamentos veterinarios biológicos (incluidos los inmunológicos), para los cuales se seguirá un planteamiento híbrido (véase la parte IV.2). |
|
IV.1.2. |
Los informes periciales críticos de la seguridad y la eficacia de los medicamentos veterinarios genéricos se centrarán particularmente en los siguientes elementos:
|
|
IV.1.3. |
En las solicitudes para medicamentos veterinarios genéricos que contengan sustancias antimicrobianas se incluirá información sobre el nivel de resistencia, obtenida a partir de datos bibliográficos. |
|
IV.1.4. |
En el caso de medicamentos veterinarios genéricos que contengan sustancias antiparasitarias se incluirá información sobre el nivel de resistencia, obtenida a partir de datos bibliográficos. |
|
IV.1.5. |
Se dará la siguiente información adicional de los medicamentos veterinarios genéricos que vayan a administrarse por vía intramuscular, subcutánea o transdérmica:
|
IV.2. Solicitudes para medicamentos veterinarios híbridos
|
IV.2.1. |
Las solicitudes basadas en el artículo 19 (medicamentos veterinarios híbridos) se refieren a medicamentos veterinarios que son similares a un medicamento veterinario de referencia pero que no cumplen las condiciones de la definición de medicamentos genéricos. |
|
IV.2.2. |
En esas solicitudes se incluirá la información siguiente:
|
|
IV.2.3. |
En el caso de los medicamentos veterinarios biológicos (incluidos los inmunológicos), se proporcionará una revisión exhaustiva de comparabilidad, que abordará la parte sobre la calidad, la seguridad y la eficacia. |
|
IV.2.4. |
Cuando se haga referencia a datos procedentes de otro medicamento veterinario autorizado, se justificará el uso y la pertinencia de esos datos para el nuevo producto. |
|
IV.2.5. |
La amplitud de los datos nuevos necesarios para confirmar la seguridad y la eficacia dependerá de las características específicas del medicamento nuevo en concreto, así como de sus diferencias con el medicamento veterinario de referencia, y se determinará caso por caso. Se presentarán nuevos datos preclínicos y clínicos para el producto nuevo relativos a todos los aspectos no confirmados por el medicamento veterinario de referencia. |
|
IV.2.6. |
Si se realizan estudios nuevos con lotes de un medicamento veterinario de referencia autorizado en un tercer país, el solicitante demostrará que ese medicamento de referencia ha sido autorizado de conformidad con requisitos equivalentes a los establecidos en la Unión y que ambos medicamentos son tan similares que pueden sustituirse mutuamente en estudios preclínicos o ensayos clínicos. |
IV.3. Solicitudes para medicamentos veterinarios basados en la combinación de principios activos
|
IV.3.1. |
Las solicitudes relativas a medicamentos de combinación fija con principios activos individuales que ya sean objeto de una autorización de comercialización para un medicamento veterinario en el Espacio Económico Europeo se presentarán en virtud del artículo 20.
Las relativas a medicamentos de combinación fija que contengan al menos un principio activo nuevo aún no autorizado para un medicamento veterinario en el Espacio Económico Europeo se presentarán en virtud del artículo 8. |
|
IV.3.2. |
Para las solicitudes presentadas en virtud del artículo 20, se presentará un expediente completo que contenga las partes 1, 2, 3 y 4. |
|
IV.3.3. |
Se incluirá una justificación científica sólida basada en principios terapéuticos válidos para la combinación de principios activos, junto con datos clínicos, con la que se demostrará la necesidad y la contribución de todos los principios activos en el momento del tratamiento. |
|
IV.3.4. |
En general, se presentarán todos los datos sobre la seguridad y la eficacia para el medicamento de combinación fija y no serán necesarios datos sobre la seguridad y la eficacia para cada uno de los principios activos, excepto con objeto de aclarar sus propiedades farmacológicas individuales. |
|
IV.3.5. |
Si el solicitante dispone de datos sobre la seguridad y la eficacia de un principio activo conocido con suficiente detalle, podrían facilitarse esos datos para evitar la necesidad de realizar algunos estudios con la combinación fija o de aportar información pertinente. En ese caso, también se investigará la posible interacción entre principios activos. |
|
IV.3.6. |
La evaluación de la seguridad para el usuario, la evaluación del riesgo medioambiental, los estudios de eliminación de residuos y los estudios clínicos sí se llevarán a cabo con el medicamento de combinación fija. |
|
IV.3.7. |
Salvo que la omisión esté justificada, se presentará un estudio de seguridad para la especie animal de destino con el producto terminado. |
IV.4. Solicitudes basadas en el consentimiento informado
|
IV.4.1. |
Las solicitudes basadas en el artículo 21 se refieren a productos con una composición, una forma farmacéutica y un proceso de fabricación (incluidas las materias primas y los materiales de partida, los parámetros del proceso y los lugares de fabricación) que son idénticos a los de medicamentos veterinarios ya autorizados. |
|
IV.4.2. |
El expediente de esas solicitudes solo incluirá datos en las partes 1A y 1B, como se describe en el anexo I (puntos 1 a 6.4), siempre que el titular de la autorización de comercialización del medicamento veterinario ya autorizado haya dado al solicitante su consentimiento por escrito para que haga referencia al contenido de las partes 1C, 2, 3 y 4 del expediente de dicho medicamento. En ese caso, tampoco hay necesidad de presentar informes periciales críticos de la calidad, la seguridad ni la eficacia. El solicitante presentará pruebas del consentimiento escrito junto con la solicitud. |
IV.5. Solicitudes basadas en datos bibliográficos
|
IV.5.1. |
Se aplicarán las siguientes normas específicas a los medicamentos veterinarios cuyos principios activos tengan o hayan tenido un uso veterinario bien establecido en el sentido del artículo 22 y presenten una eficacia reconocida y una seguridad aceptable. |
|
IV.5.2. |
Se presentará un expediente completo (que contenga las partes 1, 2, 3 y 4). El solicitante presentará las partes 1 y 2 tal como se especifica en el presente anexo. En lo que respecta a las partes 3 y 4, para abordar la seguridad y la eficacia se presentará una bibliografía científica detallada, junto con información que demuestre la relación adecuada entre las referencias bibliográficas y el medicamento veterinario. Los datos bibliográficos tal vez deban completarse con documentación específica del medicamento, por ejemplo, evaluaciones de la seguridad para el usuario y del riesgo medioambiental, o con datos de estudios de residuos, para justificar las propuestas de tiempo de espera. |
|
IV.5.3. |
Se aplicarán las normas específicas establecidas en las partes IV.5.3.1 a IV.5.3.12 para demostrar un uso veterinario bien establecido. |
|
IV.5.3.1. |
Para demostrar un uso veterinario bien establecido de los componentes de medicamentos veterinarios, se tendrán en cuenta los factores siguientes:
|
|
IV.5.3.2. |
Pueden ser necesarios lapsos de tiempo diferentes para declarar un uso veterinario bien establecido de diversos principios activos. De todos modos, el lapso necesario para declarar que un componente de un medicamento tiene un uso veterinario bien establecido no será inferior a diez años, contados a partir de su primera utilización sistemática y documentada como medicamento veterinario en la Unión. |
|
IV.5.3.3. |
El uso veterinario no significa exclusivamente una utilización como medicamento veterinario autorizado. El uso veterinario bien establecido se refiere al uso con un fin terapéutico específico en la especie de destino. |
|
IV.5.3.4. |
Si se propone una sustancia con un uso bien establecido para indicaciones terapéuticas completamente nuevas, no será posible declarar únicamente un uso veterinario bien establecido. Se facilitarán datos adicionales sobre la nueva indicación terapéutica, junto con los estudios adecuados de seguridad y de residuos y los datos preclínicos y clínicos y, en tal caso, no será posible presentar una solicitud basada en el artículo 21. |
|
IV.5.3.5. |
La documentación publicada presentada por el solicitante será de libre acceso público y estará publicada por una fuente con buena reputación, preferiblemente sometida a revisión por pares. |
|
IV.5.3.6. |
La documentación contendrá información suficiente para permitir una evaluación independiente. |
|
IV.5.3.7. |
La documentación cubrirá todos los aspectos de la evaluación de la seguridad o de la eficacia del medicamento para la indicación propuesta en la especie de destino utilizando la vía de administración y la pauta posológica propuestas. Incluirá o hará referencia a un estudio de la bibliografía al respecto, como estudios previos y posteriores a la comercialización y bibliografía científica relativa a la experiencia, en forma de estudios epidemiológicos, en particular comparativos. |
|
IV.5.3.8. |
Deberán comunicarse todos los documentos existentes, tanto favorables como desfavorables. Respecto a las disposiciones sobre un uso veterinario bien establecido, es particularmente necesario aclarar que podrá considerarse prueba válida de la seguridad y la eficacia de un medicamento la referencia bibliográfica a otras fuentes (estudios posteriores a la comercialización, estudios epidemiológicos, etc.), y no solo los datos relacionados con pruebas y ensayos, si el solicitante explica y justifica satisfactoriamente la utilización de tales fuentes. |
|
IV.5.3.9. |
No puede considerarse que los informes públicos de evaluación o los resúmenes de libertad de información proporcionen detalles suficientes, con excepción del informe de evaluación publicado por la Agencia tras evaluar una solicitud de establecimiento de límites máximos de residuos, que puede utilizarse adecuadamente como bibliografía, en particular para las pruebas de seguridad. |
|
IV.5.3.10. |
Se prestará especial atención a los datos que falten y se justificará por qué puede afirmarse la existencia de un nivel aceptable de seguridad o eficacia pese a la ausencia de información. |
|
IV.5.3.11. |
Los informes periciales críticos relativos a la seguridad y a la eficacia explicarán la pertinencia de cualquier dato presentado referido a un medicamento distinto del que se pretende comercializar. Se deberá apreciar si el producto estudiado en la bibliografía puede relacionarse de forma satisfactoria o científica con el medicamento cuya autorización de comercialización se ha solicitado, a pesar de las diferencias existentes. |
|
IV.5.3.12. |
La experiencia posterior a la comercialización de otros productos que contengan los mismos componentes es muy importante, y los solicitantes deberán insistir especialmente en este aspecto. |
IV.6. Solicitudes para mercados limitados
|
IV.6.1. |
Podrá concederse una autorización de comercialización para un mercado limitado a falta de datos exhaustivos sobre la seguridad o la eficacia cuando, con arreglo a lo dispuesto en el artículo 23, el solicitante demuestre que el medicamento se destina a un mercado limitado y que el beneficio de la disponibilidad del nuevo medicamento supera el riesgo asociado a la omisión de algunos de los datos sobre seguridad o eficacia exigidos en el presente anexo. |
|
IV.6.2. |
En estas solicitudes, el solicitante presentará las partes 1 y 2 tal como se especifica en el presente anexo. |
|
IV.6.3. |
Para las partes 3 y 4 podrán omitirse algunos de los datos de seguridad o eficacia exigidos por el presente anexo. Por lo que se refiere a la amplitud de los datos sobre seguridad y eficacia que puedan omitirse, se tendrán en cuenta las directrices pertinentes publicadas por la Agencia. |
IV.7. Solicitudes en circunstancias excepcionales
|
IV.7.1. |
En circunstancias excepcionales relacionadas con la sanidad animal o la salud pública, podrá concederse una autorización de comercialización de un medicamento veterinario con arreglo al artículo 25 si se cumplen determinadas obligaciones, condiciones o restricciones específicas. |
|
IV.7.2. |
Para esas solicitudes, el solicitante presentará la parte 1 como se especifica en el presente anexo, junto con una justificación de por qué el beneficio de la disponibilidad inmediata en el mercado del medicamento veterinario de que se trate supera el riesgo inherente al hecho de que no se haya facilitado determinada documentación sobre calidad, seguridad o eficacia. |
|
IV.7.3. |
En las partes 2, 3 y 4, podrán omitirse determinados datos sobre calidad, seguridad o eficacia exigidos en el presente anexo si el solicitante justifica que dichos datos no pueden facilitarse en el momento de la presentación. Con objeto de determinar los requisitos esenciales para todas esas solicitudes, se tendrán en cuenta las directrices pertinentes publicadas por la Agencia. |
|
IV.7.4. |
Podrán solicitarse estudios posteriores a la autorización como una de las condiciones para la autorización de comercialización; estos se diseñarán, llevarán a cabo, analizarán y presentarán con arreglo a los principios generales para los estudios de calidad, seguridad y eficacia establecidos en el presente anexo, y a los documentos de orientación pertinentes, según proceda en función de la cuestión que se trate en cada estudio. |
SECCIÓN V
REQUISITOS RELATIVOS A SOLICITUDES DE AUTORIZACIÓN DE COMERCIALIZACIÓN DE DETERMINADOS MEDICAMENTOS VETERINARIOS
En esta sección se establecen requisitos específicos para determinados medicamentos veterinarios, relacionados con la naturaleza de los principios activos que contienen.
V.1. Mdicamentos veterinarios para nuevas terapias
V.1.1. Requisitos generales
|
V.1.1.1. |
Dependiendo del principio activo y del mecanismo de acción, un medicamento veterinario para nuevas terapias podría entrar en cualquiera de las tres categorías de productos:
|
|
V.1.1.2. |
En general, las solicitudes de autorización de comercialización de medicamentos veterinarios para nuevas terapias, tal como se definen en el artículo 4, punto 43, cumplirán los requisitos relativos al formato y a los datos que se indican en las secciones II o III del presente anexo, según cómo se clasifique la nueva terapia. Normalmente se proporcionará un expediente completo que contenga las partes 1, 2, 3 y 4 de conformidad con los requisitos indicados en las secciones II o III y con las directrices pertinentes publicadas por la Agencia. Podrán producirse desviaciones respecto de los requisitos del presente anexo cuando esté justificado. Cuando proceda, y teniendo en cuenta las características específicas de los medicamentos para nuevas terapias, tal vez sean pertinentes requisitos adicionales para determinados tipos de medicamentos. |
|
V.1.1.3. |
Los procesos de fabricación de los medicamentos veterinarios para nuevas terapias respetarán los principios de las prácticas correctas de fabricación adaptados, cuando sea necesario, para reflejar la naturaleza específica de esos medicamentos. Se elaborarán directrices específicas para los medicamentos veterinarios para nuevas terapias a fin de reflejar adecuadamente las particularidades de su proceso de fabricación. |
|
V.1.1.4. |
En función de la naturaleza específica del medicamento para nuevas terapias, su uso podría estar asociado a riesgos específicos. Dichos riesgos se determinarán aplicando una metodología de elaboración de perfiles de riesgo que permita detectar los riesgos inherentes al medicamento específico y los factores de riesgo que contribuyen a ellos. En este contexto, se consideran riesgos los posibles efectos desfavorables atribuibles al uso del medicamento para nuevas terapias que afecten a la población de destino o al usuario, el consumidor o el medio ambiente. El análisis del riesgo puede cubrir todo el proceso. Entre los factores de riesgo que pueden tenerse en cuenta figuran el origen del material de partida (células, etc.), el mecanismo de acción en el animal (proliferación, inicio de la respuesta inmunitaria, permanencia en el organismo, etc.), el nivel de manipulación celular (por ejemplo, el proceso de fabricación), la combinación del principio activo con moléculas bioactivas o materiales estructurales, el grado de capacidad replicativa de los virus o microorganismos utilizados in vivo, el nivel de integración de los genes o las secuencias de ácidos nucleicos en el genoma, la funcionalidad a largo plazo, el riesgo de carcinogenicidad, los efectos colaterales y el modo de administración o de uso. |
|
V.1.1.5. |
Tras evaluar la información sobre los riesgos y los factores de riesgo detectados, se establecerá un perfil específico de cada riesgo individual asociado a un medicamento concreto, perfil que podrá utilizarse para determinar y justificar que el conjunto de datos facilitado ofrece las garantías necesarias de calidad, seguridad y eficacia y es adecuado para apoyar una solicitud de autorización de comercialización, especialmente en aquellos aspectos de los medicamentos para nuevas terapias que superan los conocimientos actuales. |
|
V.1.1.6. |
Con objeto de superar las carencias de datos o las incertidumbres en el momento de la autorización del producto, podrá estudiarse caso por caso la aplicación de medidas o estudios posteriores a la autorización. Con el fin de detectar señales tempranas o diferidas de reacciones adversas, prevenir las consecuencias clínicas de dichas reacciones y garantizar su tratamiento oportuno, y de obtener información sobre la seguridad y eficacia a largo plazo de los medicamentos veterinarios para nuevas terapias, un plan de gestión de riesgos detallará las medidas previstas para garantizar el seguimiento. |
|
V.1.1.7. |
En el caso de cualquier medicamento para nuevas terapias, en particular las consideradas como un campo emergente de la medicina veterinaria, se recomienda solicitar oportunamente el asesoramiento de la Agencia antes de presentar el expediente de autorización de comercialización para clasificar el medicamento, determinar la estructura del expediente aplicable y recibir la información pertinente sobre el conjunto de datos adicionales que pueda ser necesario para contribuir a la calidad, la seguridad y la eficacia. |
V.1.2. Requisitos de calidad
|
V.1.2.1. |
En general, se presentará una descripción de la composición, el método de fabricación, la constancia de la producción, los controles de los materiales de partida, los controles aplicados durante el proceso de fabricación, los estudios del medicamento terminado, incluida la realización de una prueba de actividad o una cuantificación del principio activo, y los datos sobre la estabilidad. |
|
V.1.2.2. |
Los requisitos en materia de datos para la fabricación y las pruebas de medicamentos veterinarios para nuevas terapias de origen biológico y clasificados como medicamentos biológicos o inmunológicos coincidirán, en general, con los de los medicamentos biológicos o inmunológicos (tal como se describen en la sección III del presente anexo), incluida la necesidad de efectuar una prueba de potencia pertinente. En ciertos casos tal vez se apliquen requisitos adicionales, por ejemplo, en construcciones de vector génico y celulares. |
|
V.1.2.3. |
En el caso de los medicamentos veterinarios para nuevas terapias construidos mediante síntesis química, en general se aplican los mismos requisitos de datos que para los medicamentos veterinarios no biológicos (descritos en la sección II del presente anexo). En ciertos casos tal vez se apliquen requisitos adicionales, por ejemplo, la necesidad de efectuar una prueba de potencia pertinente. |
V.1.3. Requisitos de seguridad
|
V.1.3.1. |
Dependiendo de la naturaleza del medicamento y de su uso previsto, podrían ser pertinentes datos adicionales a fin de evaluar la seguridad para el animal de destino, el usuario, el consumidor o el medio ambiente, según se determine mediante un análisis del riesgo en cada caso. |
|
V.1.3.2. |
Se tendrán en cuenta los requisitos de la Directiva 2001/18/CE cuando el propio animal tratado pueda convertirse en un organismo modificado genéticamente. Si bien la Directiva 2001/18/CE se aplica a los medicamentos terminados que contienen organismos modificados genéticamente, sigue siendo la mejor guía técnica disponible actualmente para indicar los datos necesarios. En particular, una cuestión importante es la tasa de integración del ADN en las células germinales (por lo tanto, transmisible a la descendencia) o la posible transmisión de las células modificadas genéticamente a la descendencia. También cabe señalar que este problema no es exactamente el mismo en relación con los animales de compañía y con los animales productores de alimentos (consumo humano de productos que contengan organismos modificados genéticamente). |
|
V.1.3.3. |
En el caso de las sustancias destinadas a su integración en el genoma o a la edición del genoma, se realizarán los estudios adecuados para evaluar el riesgo de modificaciones no intencionadas o de mutagénesis por inserción. |
V.1.4. Requisitos de eficacia
|
V.1.4.1. |
Los requisitos en materia de datos de eficacia difieren principalmente en función de las indicaciones de uso previstas en las especies de destino. Dependiendo de la categoría del medicamento para nuevas terapias y del uso previsto en las especies de destino, podrán serle de aplicación los requisitos de eficacia establecidos en las secciones II o III. |
|
V.1.4.2. |
Las indicaciones alegadas deberán confirmarse mediante datos adecuados en las especies de destino. |
V.1.5. Requisitos de datos específicos aplicables a determinados tipos de medicamentos para nuevas terapias
V.1.5.1. Principios
|
V.1.5.1.1. |
Teniendo en cuenta las especificidades de los medicamentos para nuevas terapias, puede ser adecuado aplicar determinados requisitos además de los habituales para la evaluación de la calidad, la seguridad y la eficacia. |
|
V.1.5.1.2. |
En las secciones siguientes se destacan los requisitos específicos que deben tenerse en cuenta para determinados tipos de medicamentos para nuevas terapias. Esta es una lista no exhaustiva de requisitos específicos que tal vez sea necesario adaptar al medicamento para nuevas terapias de que se trate caso por caso y sobre la base de un análisis del riesgo. |
|
V.1.5.13. |
En todos los casos, y especialmente en el caso de nuevas terapias que se consideran emergentes en el ámbito de la medicina veterinaria, los solicitantes deberán tener en cuenta el estado actual de los conocimientos veterinarios y las directrices científicas publicadas por la Agencia y por la Comisión, de conformidad con la sección I del presente anexo. |
V.1.5.2. Medicamentos veterinarios para terapia génica
|
V.1.5.2.1. |
Los medicamentos para terapia génica son medicamentos veterinarios biológicos que utilizan un principio activo que contiene o consiste en un ácido nucleico recombinante utilizado en animales o administrado a estos con el fin de regular, reparar, sustituir, añadir o suprimir una secuencia genética. Su efecto terapéutico, profiláctico o diagnóstico depende directamente de la secuencia del ácido nucleico recombinante que contengan, o del producto de la expresión genética de dicha secuencia. |
|
V.1.5.2.2. |
Además de los requisitos de datos establecidos en las secciones II o III, se aplicarán los requisitos siguientes:
|
V.1.5.3. Medicamentos veterinarios de medicina regenerativa, ingeniería tisular y terapia celular
|
V.1.5.3.1. |
Se considera que la medicina regenerativa abarca una amplia gama de medicamentos y terapias cuyo propósito general es restaurar las funciones. Una de ellas es la terapia celular, en la que se incluyen los productos de ingeniería tisular. |
|
V.1.5.3.2. |
Los medicamentos veterinarios de terapia celular son medicamentos veterinarios biológicos que contienen o consisten en células o tejidos que han sido objeto de manipulación sustancial en su naturaleza o función, de modo que se han alterado características biológicas, funciones fisiológicas o propiedades estructurales pertinentes para el uso clínico previsto, o que contienen o consisten en células o tejidos que no están destinados a utilizarse para las mismas funciones esenciales en el receptor que en el donante. Se presentan como poseedores de propiedades para tratar, prevenir o diagnosticar una enfermedad a través de la acción farmacológica, inmunológica o metabólica de sus células o tejidos, o para regenerar, reparar o sustituir un tejido, o bien se utilizan en animales o se les administran con esos fines. |
|
V.1.5.3.3. |
Además de los requisitos de datos establecidos en las secciones II o III, se aplicarán los requisitos siguientes:
|
V.1.5.4. Medicamentos veterinarios diseñados específicamente para terapia de fagos
|
V.1.5.4.1. |
Los bacteriófagos, o fagos, son virus que dependen de bacterias hospedadoras para su proliferación y actúan de manera muy específica en determinadas cepas bacterianas. La terapia de fagos, o fagoterapia, puede utilizarse, por ejemplo, como alternativa a los antibióticos. Generalmente, los bacteriófagos consisten en un genoma, compuesto de ADN o ARN monocatenario o bicatenario, encapsulado por una cápside proteica. Debido a la diversidad de especies de destino para el tratamiento y a la especificidad de los bacteriófagos, será necesario elegir, caso por caso, la cepa de bacteriófago adecuada para la cepa bacteriana causante de cada brote de la enfermedad. |
|
V.1.5.4.2 |
La calidad y la cantidad de bacteriófagos que vayan a utilizarse en el medicamento terminado suelen ser variables. Por lo tanto, será inhabitual contar con una composición cualitativa y cuantitativa fija de bacteriófagos, ya que los fagos deben adaptarse de forma continua. Sobre esta base, es necesario establecer y mantener una reserva de cepas bacteriófagas (lo que es comparable con el planteamiento multicepas). |
|
V.1.5.4.3. |
Los bacteriófagos y los bancos de bacterias hospedadoras o de células primarias para la fabricación se producirán preferiblemente a partir de un sistema de cepas primarias. Se confirmará que el bacteriófago utilizado es lítico. |
|
V.1.5.4.4. |
La ausencia de genes de resistencia y la ausencia de genes codificadores para los factores de virulencia se mostrarán en todas las cepas primarias. |
|
V.1.5.4.5. |
La indicación será para tratamiento profiláctico, metafiláctico o terapéutico de una o varias infecciones o enfermedades infecciosas específicas. La eficacia del tratamiento está vinculada a la actividad lítica de los fagos que confiere actividad bactericida a los bacteriófagos con especificidad para la cepa bacteriana en cuestión. |
|
V.1.5.4.6. |
En el caso de los fagos modificados genéticamente, se describirá la modificación genética. |
V.1.5.5. Medicamentos veterinarios generados a partir de nanotecnologías
|
V.1.5.5.1. |
Las nanotecnologías se consideran principalmente tecnologías para generar vectores de sustancias de síntesis química, pero también pueden ser vectores de sustancias biológicas. El uso de nanopartículas puede ser una forma de controlar la administración de sustancias con baja solubilidad o compuestos tóxicos. |
|
V.1.5.5.2. |
Por «nanotecnología» se entiende el diseño, la caracterización y la producción de nanomateriales controlando su forma y tamaño a nanoescala (hasta unos 100 nm). |
|
V.1.5.5.3. |
Se considera que las «nanopartículas» tienen dos o más dimensiones a nanoescala. |
|
V.1.5.5.4. |
En el ámbito veterinario, las nanopartículas para el sistema de administración de medicamentos son importantes en calidad de «medicamentos generados a partir de nanotecnologías»: las nanopartículas se conjugan con sustancias para cambiar sus propiedades farmacocinéticas o farmacodinámicas. Es preferible que los medicamentos de ARN mensajero (ARNm) estén encapsulados en sistema de administración por nanopartículas. |
|
V.1.5.5.5. |
Además de los requisitos de datos de calidad establecidos en las secciones II o III, se aplicarán los requisitos siguientes:
|
|
V.1.5.5.6. |
Por lo que se refiere a la seguridad, el uso de nanopartículas para la administración de medicamentos puede introducir peligros distintos de los convencionales provocados por las sustancias químicas en las matrices clásicas de administración. Por lo tanto, se tendrán en cuenta los aspectos siguientes en relación con la seguridad:
|
V.1.5.6. Medicamentos de terapia de ARN de antisentido y de terapia de interferencia por ARN
|
V.1.5.6.1. |
Los medicamentos de terapia de antisentido y de terapia de interferencia pueden generarse mediante síntesis o mediante técnicas recombinantes. |
|
V.1.5.6.2. |
El ARN de antisentido es un ARN monocatenario que es complementario a un ARN mensajero codificante de proteínas con el que se hibrida, bloqueando así su traducción en proteína. |
|
V.1.5.6.3. |
La interferencia por ARN (iARN) es un proceso biológico en el que las moléculas de ARN inhiben la expresión o la traducción de un gen mediante la neutralización de las moléculas de ARNm diana. |
|
V.1.5.6.4. |
Además de los requisitos de datos establecidos en las secciones II o III, se aplicarán los requisitos siguientes:
|
V.2. Archivo maestro de antígenos vacunales
No obstante lo dispuesto en la sección IIIb, parte 2, para determinados medicamentos veterinarios inmunológicos se introduce el concepto de archivo maestro de antígenos vacunales.
|
V.2.1. |
Principios |
|
V.2.1.1. |
A efectos del presente anexo, se entiende por «archivo maestro de antígenos vacunales» una parte independiente del expediente de solicitud de autorización de comercialización de una vacuna, en la que figura toda la información pertinente sobre la calidad de cada uno de los principios activos que forman parte del medicamento veterinario. La parte independiente podrá ser común a una o varias vacunas monovalentes o combinadas presentadas por el mismo solicitante o titular de autorización de comercialización. |
|
V.2.1.2. |
El uso de los archivos maestros de antígenos vacunales es opcional. En el caso de las vacunas combinadas, se especificará el antígeno o los antígenos vacunales que se deben incluir en el archivo maestro y se exigirá un archivo maestro para cada vacuna. |
|
V.2.1.3. |
La presentación y aprobación de un archivo maestro de antígenos vacunales se ajustará a las directrices pertinentes publicadas por la Agencia. |
|
V.2.2. |
Contenido
El expediente del archivo maestro de antígenos vacunales contendrá la información de las partes V.2.2.1 a V.2.3.3 extraída de las secciones pertinentes de la parte 1 (Resumen del expediente) y la parte 2 (Documentación sobre calidad), tal como se establece en la sección IIIb del presente anexo: |
|
V.2.2.1. |
Resumen del expediente (parte 1)
Se indicarán el nombre y la dirección del fabricante o fabricantes y los lugares que intervienen en las diferentes fases de fabricación y control del principio activo, y se adjuntarán copias de las autorizaciones de fabricación correspondientes. |
|
V.2.2.2. |
Datos cualitativos y cuantitativos de los componentes (parte 2.A)
Se indicará la denominación completa y exacta del principio activo (por ejemplo, cepa vírica o bacteriana, antígeno), tal como se mencione en los productos terminados. Se facilitará información sobre el desarrollo farmacéutico pertinente para el principio activo. |
|
V.2.2.3. |
Descripción del método de fabricación (parte 2.B)
Se facilitará la descripción del método de fabricación del principio activo, incluida la validación de las etapas clave del proceso de producción, y se justificará, si procede, el almacenamiento intermedio propuesto. En el caso de vacunas inactivadas, se facilitarán los datos relativos a la inactivación del principio activo, incluida la validación del proceso de inactivación. |
|
V.2.2.4. |
Producción y control de los materiales de partida (parte 2.C) |
|
V.2.2.4.1. |
Se aplicarán los requisitos estándar descritos en la sección IIIb.2C que sean pertinentes para el principio activo. |
|
V.2.2.4.2. |
Se facilitará información sobre el principio activo (por ejemplo, cepa vírica o bacteriana), los sustratos (células, medio de cultivo) y todas las materias primas (incluidas o no en la farmacopea, biológicas o no) que se hayan utilizado en la producción del principio activo. |
|
V.2.2.4.3. |
El expediente incluirá las especificaciones, información sobre los procesos realizados y sobre los estudios que se realizarán para el control de calidad de todos los lotes de materiales de partida y los resultados en un lote para todos los componentes utilizados. |
|
V.2.2.4.4. |
Cuando proceda, se facilitará una evaluación del riesgo de encefalopatías espongiformes transmisibles (EET) y de agentes extraños (EA). Cabe señalar que, en la evaluación del riesgo de EET y EA, se tendrán en cuenta las especies de destino indicadas para los productos terminados en referencia al archivo maestro de antígenos vacunales. Las advertencias o restricciones de uso pueden introducirse en el archivo maestro de antígenos vacunales en función de la información presentada y pueden atenuarse durante el análisis del riesgo del producto terminado. |
|
V.2.2.4.5. |
Si el principio activo se obtiene mediante técnicas recombinantes, se facilitarán todos los datos correspondientes sobre los virus o bacterias modificados genéticamente. |
|
V.2.2.5. |
Pruebas de control efectuadas durante el proceso de fabricación (parte 2.D)
Se aplicarán los requisitos estándar descritos en la sección IIIb.2D a las pruebas de control efectuadas durante el proceso de fabricación del principio activo, incluida la validación de las pruebas de control claves y, si procede, del almacenamiento intermedio propuesto (antes de la mezcla). |
|
V.2.2.6. |
Constancia entre lotes (parte 2.F)
Se aplicarán los requisitos estándar descritos en la sección IIIb.2F para demostrar la constancia en la fabricación del antígeno. |
|
V.2.2.7. |
Estabilidad (parte 2.G)
Se aplicarán los requisitos estándar descritos en la sección IIIb.2G para demostrar la estabilidad del antígeno y, cuando proceda, del almacenamiento intermedio. |
|
V.2.3. |
Evaluación y certificación |
|
V.2.3.1. |
En el caso de las vacunas que contengan nuevos antígenos vacunales para los cuales no exista ya un archivo maestro de antígenos vacunales, el solicitante presentará a la Agencia un expediente completo de solicitud de autorización de comercialización que incluya todos los archivos maestros correspondientes a cada uno de los antígenos vacunales para los que esté previsto utilizar el archivo maestro. La Agencia realizará una evaluación científica y técnica de cada archivo maestro de antígenos vacunales. La evaluación positiva supondrá la expedición de un certificado de cumplimiento de la legislación de la Unión relativo a cada archivo maestro de antígenos vacunales, que irá acompañado del informe de evaluación. El certificado tendrá validez en toda la Unión. |
|
V.2.3.2. |
La parte V.2.3.1 se aplicará también a todas las vacunas que consistan en una nueva combinación de antígenos vacunales, independientemente de si uno o varios de esos antígenos forman parte de vacunas ya autorizadas en la Unión. |
|
V.2.3.3. |
Toda alteración en el contenido de un archivo maestro de antígenos vacunales para una vacuna autorizada en la Unión estará sujeta a una evaluación científica y técnica llevada a cabo por la Agencia. En caso de evaluación positiva, la Agencia expedirá un certificado de cumplimiento de la legislación de la Unión relativo a ese archivo maestro de antígenos vacunales. El certificado tendrá validez en toda la Unión. |
V.3. Expediente multicepas
|
V.3.1. |
No obstante lo dispuesto en la sección IIIb, parte 2, para ciertos medicamentos veterinarios inmunológicos se introduce el concepto de presentación de un expediente multicepas. |
|
V.3.2. |
Un expediente multicepas es un solo expediente que contiene los datos pertinentes para una evaluación científica única y exhaustiva de las diferentes opciones de cepas o combinaciones de cepas, evaluación que permitirá autorizar vacunas inactivadas contra virus o bacterias con variabilidad antigénica para las cuales, a fin de garantizar su eficacia respecto a la situación epidemiológica sobre el terreno, sea necesario efectuar cambios rápidos o frecuentes en la composición de las formulaciones vacunales. En función de la situación epidemiológica en la cual se prevea utilizar la vacuna, podrían seleccionarse varias cepas de entre las incluidas en el expediente para formular un producto final. |
|
V.3.3. |
Cada expediente multicepas solo es aplicable a una especie vírica, un género de bacterias o un vector para una enfermedad dada; en el contexto de un expediente multicepas no podrán aprobarse mezclas de diversos virus pertenecientes a distintas familias, géneros o especies, ni de bacterias pertenecientes a diferentes familias o géneros. |
|
V.3.4. |
En el caso de nuevas solicitudes de autorización de comercialización con expediente multicepas cuando no exista aún ninguna vacuna multicepas autorizada para un virus, una bacteria o una enfermedad determinados, la Agencia deberá confirmar que se cumplen los requisitos para el planteamiento del expediente multicepas antes de que se presente la solicitud. |
|
V.3.5. |
La presentación de expedientes multicepas se ajustará a las directrices pertinentes publicadas por la Agencia. |
V.4. Tecnología de plataforma vacunal
|
V.4.1. |
Principios |
|
V.4.1.1. |
La tecnología de plataforma vacunal es una colección de tecnologías que tienen en común el uso de un vector o transportador «troncal» que se modifica con un antígeno o un conjunto de antígenos diferentes para cada vacuna derivada de la plataforma. Esto incluye, entre otras cosas, plataformas basadas en proteínas (partículas similares a virus), plataformas de vacunas de ADN, plataformas basadas en el ARNm, replicones (ARN autorreplicante) y vacunas con vector vírico y bacteriano. |
|
V.4.1.2. |
Se considera que las solicitudes de autorización de comercialización de medicamentos veterinarios inmunológicos fabricados con tecnologías de plataforma vacunal pueden cumplir los requisitos para la presentación de un conjunto reducido de datos. Se exige un expediente completo para el primer medicamento de un fabricante que esté basado en una tecnología de plataforma concreta para una especie de destino concreta. En el momento de la presentación del primer expediente (completo) basado en esa tecnología de plataforma, el solicitante podrá presentar paralelamente un «archivo maestro de tecnología de plataforma» que incluya todos los datos relativos a la plataforma sobre los que se tenga una certeza científica razonable de que permanecerán inalterados, independientemente de los antígenos o los genes de interés que se añadan a la plataforma. La naturaleza de los datos que se incluyan en el archivo maestro de tecnología de plataforma dependerá del tipo de plataforma. |
|
V.4.1.3. |
Una vez certificado un archivo maestro de tecnología de plataforma, dicho certificado podrá utilizarse como demostración del cumplimiento de los requisitos de datos pertinentes en solicitudes de autorización de comercialización posteriores basadas en la misma plataforma y destinadas a la misma especie de destino. |
V.4.2. Evaluación y certificación
|
V.4.2.1. |
La presentación de archivos maestros de tecnología de plataforma se ajustará a las directrices pertinentes publicadas por la Agencia. La Agencia realizará una evaluación científica y técnica del archivo maestro de tecnología de plataforma. La evaluación positiva supondrá la expedición de un certificado de cumplimiento de la legislación de la Unión relativo a cada archivo maestro de tecnología de plataforma, que irá acompañado del informe de evaluación. El certificado tendrá validez en toda la Unión. |
|
V.4.2.2. |
Toda alteración en el contenido de un archivo maestro de tecnología de plataforma para una vacuna autorizada en la Unión estará sujeta a una evaluación científica y técnica llevada a cabo por la Agencia. |
|
V.4.2.3. |
En caso de evaluación positiva, la Agencia expedirá un certificado de cumplimiento de la legislación de la Unión relativo a ese archivo maestro de tecnología de plataforma. |
V.5. Medicamentos veterinarios homeopáticos autorizados
|
V.5.1 |
Calidad (parte 2)
Lo dispuesto en la sección II.2., parte 2, se aplicará a los documentos presentados para la autorización de los medicamentos veterinarios homeopáticos contemplados en el artículo 85, apartado 2, con las modificaciones siguientes. |
|
V.5.2 |
Terminología
La denominación latina de la cepa homeopática descrita en el expediente de solicitud de autorización de comercialización deberá ser acorde con la denominación latina de la Farmacopea Europea o, en su ausencia, de una farmacopea oficial de un Estado miembro. Se indicarán, en su caso, los nombres tradicionales usados en cada Estado miembro. |
|
V.5.3 |
Control de los materiales de partida
Los datos y documentos sobre todos los materiales de partida utilizados, incluidas todas las materias primas y los productos intermedios hasta la dilución final que haya de incorporarse al medicamento veterinario homeopático terminado autorizado, que se adjunten a la solicitud se completarán con datos adicionales sobre la cepa homeopática. Los requisitos generales de calidad se aplicarán a todos los materiales de partida y materias primas, así como a los pasos intermedios del proceso de fabricación hasta la dilución final que haya de incorporarse al medicamento homeopático terminado. Cuando haya un componente tóxico, su presencia en la dilución final debe controlarse, si es posible. Cuando esto no sea posible por lo elevado de la dilución, el componente tóxico deberá controlarse en una fase previa. Se describirá pormenorizadamente cada paso del proceso de fabricación, desde los materiales de partida hasta la dilución final que haya de incorporarse al producto terminado. Cuando se realicen diluciones, estos pasos de dilución se harán de acuerdo con los métodos de fabricación homeopática establecidos en la correspondiente monografía de la Farmacopea Europea o, en su defecto, de una farmacopea oficial de un Estado miembro. |
|
V.5.4 |
Pruebas de control del medicamento terminado
Los requisitos generales de calidad se aplicarán a los medicamentos veterinarios homeopáticos terminados. Cualquier excepción habrá de ser debidamente justificada por el solicitante. Se efectuará la identificación y determinación cuantitativa de todos los componentes pertinentes desde un punto de vista toxicológico. Si se justifica que no es posible la identificación o determinación de todos los componentes pertinentes desde un punto de vista toxicológico (por ejemplo, dada su dilución en el medicamento terminado), la calidad se demostrará mediante la validación completa del proceso de fabricación y dilución. |
|
V.5.5 |
Estudios de estabilidad
Tendrá que demostrarse la estabilidad del medicamento terminado. Los datos de estabilidad de las cepas homeopáticas son generalmente transferibles a sus diluciones o potenciaciones. Si no es posible la identificación o determinación cuantitativa del principio activo dado el grado de dilución, podrán considerarse los datos de estabilidad de la forma farmacéutica. |
|
V.5.6 |
Documentación relativa a la seguridad (parte 3)
La parte 3 se aplicará a los medicamentos veterinarios homeopáticos mencionados en el artículo 4, apartado 10, del presente Reglamento con la especificación siguiente, sin perjuicio de lo dispuesto en el Reglamento (UE) n.o 37/2010 de la Comisión (7) relativo a las sustancias farmacológicamente activas y su clasificación por lo que se refiere a los límites máximos de residuos en los productos alimenticios de origen animal. Se justificará la ausencia de cualquier dato, por ejemplo, se indicará cómo se demuestra un grado aceptable de seguridad pese a la ausencia de determinados estudios. |
(1) Directiva 2004/10/CE del Parlamento Europeo y del Consejo, de 11 de febrero de 2004, sobre la aproximación de las disposiciones legales, reglamentarias y administrativas relativas a la aplicación de los principios de buenas prácticas de laboratorio y al control de su aplicación para las pruebas sobre las sustancias químicas (DO L 50 de 20.2.2004, p. 44).
(2) Directiva 2004/9/CE del Parlamento Europeo y del Consejo, de 11 de febrero de 2004, relativa a la inspección y verificación de las buenas prácticas de laboratorio (BPL) (DO L 50 de 20.2.2004, p. 28).
(3) Directiva 2009/35/CE del Parlamento Europeo y del Consejo, de 23 de abril de 2009, relativa a las materias que pueden añadirse a los medicamentos para su coloración (DO L 109 de 30.4.2009, p. 10).
(4) Reglamento (UE) n.o 231/2012 de la Comisión, de 9 de marzo de 2012, por el que se establecen especificaciones para los aditivos alimentarios que figuran en los anexos II y III del Reglamento (CE) n.o 1333/2008 del Parlamento Europeo y del Consejo (DO L 83 de 22.3.2012, p. 1).
(5) Reglamento (UE) 2018/782 de la Comisión, de 29 de mayo de 2018, por el que se establecen los principios metodológicos aplicables a la evaluación de los riesgos y a las recomendaciones para la gestión de los riesgos a que se refiere el Reglamento (CE) n.o 470/2009 (DO L 132 de 30.5.2018, p. 5).
(6) Reglamento (CE) n.o 1907/2006 del Parlamento Europeo y del Consejo, de 18 de diciembre de 2006, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH), por el que se crea la Agencia Europea de Sustancias y Mezclas Químicas, se modifica la Directiva 1999/45/CE y se derogan el Reglamento (CEE) n.o 793/93 del Consejo y el Reglamento (CE) n.o 1488/94 de la Comisión así como la Directiva 76/769/CEE del Consejo y las Directivas 91/155/CEE, 93/67/CEE, 93/105/CE y 2000/21/CE de la Comisión (DO L 396 de 30.12.2006, p. 1).
(7) Reglamento (UE) n.o 37/2010 de la Comisión, de 22 de diciembre de 2009, relativo a las sustancias farmacológicamente activas y su clasificación por lo que se refiere a los límites máximos de residuos en los productos alimenticios de origen animal (DO L 15 de 20.1.2010, p. 1).
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/78 |
REGLAMENTO DELEGADO (UE) 2021/806 DE LA COMISIÓN
de 10 de marzo de 2021
por el que se modifica el Reglamento (UE) n.o 528/2012 del Parlamento Europeo y del Consejo para incluir el dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión como sustancia activa en su anexo I
(Texto pertinente a efectos del EEE)
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (UE) n.o 528/2012 del Parlamento Europeo y del Consejo, de 22 de mayo de 2012, relativo a la comercialización y el uso de los biocidas (1), y en particular su artículo 28, apartado 1,
Considerando lo siguiente:
|
(1) |
El Reglamento Delegado (UE) n.o 1062/2014 de la Comisión (2) establece una lista de sustancias activas existentes que deben evaluarse para su posible aprobación para su uso en biocidas. En ella figura el dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión. |
|
(2) |
El dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión ha sido evaluado para su uso en biocidas del tipo 19 (repelentes y atrayentes), descrito en el anexo V del Reglamento (UE) n.o 528/2012. |
|
(3) |
Francia fue designada Estado miembro ponente, y su autoridad competente evaluadora presentó el informe de evaluación, junto con sus conclusiones, a la Agencia Europea de Sustancias y Mezclas Químicas («Agencia») el 18 de septiembre de 2019. |
|
(4) |
De conformidad con lo dispuesto en el artículo 7, apartado 2, del Reglamento Delegado (UE) n.o 1062/2014, el Comité de Biocidas adoptó el dictamen de la Agencia el 16 de junio de 2020 (3), teniendo en cuenta las conclusiones de la autoridad competente evaluadora. |
|
(5) |
Según dicho dictamen, cabe esperar que los biocidas del tipo 19 que utilicen dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión cumplan los requisitos del artículo 19, apartado 1, letra b), del Reglamento (UE) n.o 528/2012. En su dictamen, la Agencia confirmó asimismo que esta sustancia activa no se considera de posible riesgo y puede incluirse en el anexo I del Reglamento (UE) n.o 528/2012. |
|
(6) |
Teniendo en cuenta el dictamen de la Agencia, procede, por tanto, incluir el dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión en el anexo I del Reglamento (UE) n.o 528/2012. Dado que el dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión ha sido evaluado conforme a un expediente de sustancia activa que cumple los requisitos establecidos en el artículo 11, apartado 1, de la Directiva 98/8/CE del Parlamento Europeo y del Consejo (4), conviene incluirlo en la categoría 6 del anexo I del Reglamento (UE) n.o 528/2012 («Sustancias para las que un Estado miembro ha validado un expediente de sustancia activa de conformidad con el artículo 7, apartado 3, del presente Reglamento o lo ha aceptado de conformidad con el artículo 11, apartado 1, de la Directiva 98/8/CE»). |
|
(7) |
El artículo 89, apartado 3, del Reglamento (UE) n.o 528/2012 contempla medidas transitorias cuando una sustancia activa existente incluida en el programa de trabajo para el examen sistemático de sustancias activas existentes es aprobada de conformidad con dicho Reglamento. Por lo que respecta al dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión para el tipo de biocida 19, la fecha de aprobación a efectos del artículo 89, apartado 3, del Reglamento sobre biocidas debe fijarse en el 1 de julio de 2022, con el fin de dar suficiente tiempo para la presentación de solicitudes de autorización con arreglo al artículo 89, apartado 3, párrafo segundo, de dicho Reglamento. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
El anexo I del Reglamento (UE) n.o 528/2012 se modifica de conformidad con el anexo del presente Reglamento.
Artículo 2
A efectos del artículo 89, apartado 3, del Reglamento (UE) n.o 528/2012, la fecha de aprobación del dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión para el tipo de biocida 19 será el 1 de julio de 2022.
Artículo 3
El presente Reglamento entrará en vigor a los veinte días de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 10 de marzo de 2021.
Por la Comisión
La Presidenta
Ursula VON DER LEYEN
(1) DO L 167 de 27.6.2012, p. 1.
(2) Reglamento Delegado (UE) n.o 1062/2014 de la Comisión, de 4 de agosto de 2014, relativo al programa de trabajo para el examen sistemático de todas las sustancias activas existentes contenidas en los biocidas que se mencionan en el Reglamento (UE) n.o 528/2012 del Parlamento Europeo y del Consejo (DO L 294 de 10.10.2014, p. 1).
(3) «Biocidal Products Committee Opinion on the application for approval of the active substance: Carbon dioxide generated from propane, butane or a mixture of both by combustion, Product type: 19» [«Dictamen del Comité de Biocidas sobre la solicitud de aprobación del dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión como sustancia activa para el tipo de biocida 19», documento en inglés], ECHA/BPC/249/2020, adoptado el 16 de junio de 2020.
(4) Directiva 98/8/CE del Parlamento Europeo y del Consejo, de 16 de febrero de 1998, relativa a la comercialización de biocidas (DO L 123 de 24.4.1998, p. 1).
ANEXO
En el anexo I del Reglamento (UE) n.o 528/2012, en la categoría 6 de la lista de sustancias activas contempladas en el artículo 25, letra a), se añade la entrada siguiente:
|
Número CE |
Nombre/Grupo |
Restricción |
Comentario |
|
«204-696-9 |
Dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión (*1) |
|
N.o CAS 124-38-9 |
(*1) La fecha de aprobación del dióxido de carbono generado a partir de propano, butano o una mezcla de ambos por combustión para el tipo de biocida 19 a efectos del artículo 89, apartado 3, será el 1 de julio de 2022.».
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/81 |
REGLAMENTO DELEGADO (UE) 2021/807 DE LA COMISIÓN
de 10 de marzo de 2021
por el que se modifica el Reglamento (UE) n.o 528/2012 del Parlamento Europeo y del Consejo para incluir el sorbato de potasio como sustancia activa en su anexo I
(Texto pertinente a efectos del EEE)
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (UE) n.o 528/2012 del Parlamento Europeo y del Consejo, de 22 de mayo de 2012, relativo a la comercialización y el uso de los biocidas (1), y en particular su artículo 28, apartado 1,
Considerando lo siguiente:
|
(1) |
El (E,E)-Hexa-2,4-dienoato de potasio (sorbato de potasio) se ha evaluado como sustancia activa existente incluida en el programa de trabajo para el examen sistemático de todas las sustancias activas existentes, contemplado en el artículo 89, apartado 1, del Reglamento (UE) n.o 528/2012 y realizado de conformidad con el Reglamento Delegado (UE) n.o 1062/2014 de la Comisión (2). |
|
(2) |
De conformidad con el artículo 7, apartado 2, del Reglamento Delegado (UE) n.o 1062/2014, el 4 de diciembre de 2014 el Comité de Biocidas adoptó el dictamen de la Agencia Europea de Sustancias y Mezclas Químicas («Agencia») (3), teniendo en cuenta las conclusiones de la autoridad competente evaluadora. Dado que la evaluación de la autoridad competente finalizó el 20 de octubre de 2010, la solicitud de aprobación del sorbato de potasio se examinó de acuerdo con la Directiva 98/8/CE del Parlamento Europeo y del Consejo (4), conforme a lo dispuesto en el artículo 90, apartado 2, del Reglamento (UE) n.o 528/2012, y la Agencia concluyó en su dictamen que cabe esperar que los biocidas del tipo de producto 8 que contienen sorbato de potasio cumplan los requisitos del artículo 5 de la Directiva 98/8/CE. |
|
(3) |
Por consiguiente, el sorbato de potasio fue aprobado como sustancia activa para su uso en biocidas del tipo de producto 8 mediante el Reglamento de Ejecución (UE) 2015/1729 de la Comisión (5). |
|
(4) |
El sorbato de potasio está todavía incluido en el programa de trabajo para el examen sistemático de todas las sustancias activas existentes, para su uso en biocidas del tipo de producto 6. |
|
(5) |
En su dictamen de 4 de diciembre de 2014, la Agencia concluyó asimismo que el sorbato de potasio cumple los criterios para su inclusión en el anexo I del Reglamento (UE) n.o 528/2012. |
|
(6) |
Teniendo en cuenta el dictamen de la Agencia, procede incluir el sorbato de potasio en el anexo I del Reglamento (UE) n.o 528/2012. Dado que el sorbato de potasio se ha evaluado a partir de un expediente de sustancia activa que ha sido aceptado con arreglo al artículo 11, apartado 1, de la Directiva 98/8/CE, debe incluirse en la categoría 6 del anexo I del Reglamento (UE) n.o 528/2012. |
|
(7) |
El artículo 89, apartado 3, del Reglamento (UE) n.o 528/2012 contempla medidas transitorias cuando una sustancia activa existente incluida en el programa de trabajo para el examen sistemático de sustancias activas existentes es aprobada de conformidad con dicho Reglamento. Por lo que respecta al sorbato de potasio como tipo de producto 6, la fecha de aprobación a efectos del artículo 89, apartado 3, del Reglamento sobre biocidas debe fijarse en el 1 de febrero de 2023, con el fin de dar suficiente tiempo para la presentación de solicitudes de autorización con arreglo al artículo 89, apartado 3, párrafo segundo, de dicho Reglamento. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
El anexo I del Reglamento (UE) n.o 528/2012 se modifica de conformidad con el anexo del presente Reglamento.
Artículo 2
A efectos del artículo 89, apartado 3, del Reglamento (UE) n.o 528/2012, la fecha de aprobación del sorbato de potasio como tipo de producto 6 será el 1 de febrero de 2023.
Artículo 3
El presente Reglamento entrará en vigor a los veinte días de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 10 de marzo de 2021.
Por la Comisión
La Presidenta
Ursula VON DER LEYEN
(1) DO L 167 de 27.6.2012, p. 1.
(2) Reglamento Delegado (UE) n.o 1062/2014 de la Comisión, de 4 de agosto de 2014, relativo al programa de trabajo para el examen sistemático de todas las sustancias activas existentes contenidas en los biocidas que se mencionan en el Reglamento (UE) n.o 528/2012 del Parlamento Europeo y del Consejo (DO L 294 de 10.10.2014, p. 1).
(3) Dictamen del Comité de Biocidas sobre la solicitud de aprobación de la sustancia activa: sorbato de potasio, tipo de producto 8, ECHA/BPC/37/2014, adoptado el 4 de diciembre de 2014.
(4) Directiva 98/8/CE del Parlamento Europeo y del Consejo, de 16 de febrero de 1998, relativa a la comercialización de biocidas (DO L 123 de 24.4.1998, p. 1).
(5) Reglamento de Ejecución (UE) 2015/1729 de la Comisión, de 28 de septiembre de 2015, por el que se aprueba el uso del sorbato de potasio como sustancia activa existente en biocidas del tipo de producto 8 (DO L 252 de 29.9.2015, p. 24).
ANEXO
En el anexo I del Reglamento (UE) n.o 528/2012, en la categoría 6 de la lista de sustancias activas contempladas en el artículo 25, letra a), se añade la entrada siguiente:
|
Número CE |
Nombre/Grupo |
Restricción |
Observación |
|
«246-376-1 |
(E,E)-Hexa-2,4-dienoato de potasio (sorbato de potasio) (*1) |
Grado de pureza mínimo de la sustancia activa (*2): 990 g/kg |
N.o CAS: 24634-61-5 |
(*1) La fecha de aprobación del sorbato de potasio como tipo de producto 6 a efectos del artículo 89, apartado 3, será el 1 de febrero de 2023.
(*2) La pureza indicada en esta columna es el grado de pureza mínimo de la sustancia activa evaluada. La sustancia activa presente en el producto comercializado puede tener una pureza igual o diferente, si se ha demostrado que es técnicamente equivalente a la sustancia activa evaluada.».
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/84 |
REGLAMENTO DE EJECUCIÓN (UE) 2021/808 DE LA COMISIÓN
de 22 de marzo de 2021
relativo al funcionamiento de los métodos analíticos para los residuos de sustancias farmacológicamente activas utilizadas en animales productores de alimentos y a la interpretación de resultados, así como a los métodos que deben utilizarse para el muestreo, y por el que se derogan las Decisiones 2002/657/CE y 98/179/CE
(Texto pertinente a efectos del EEE)
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (UE) 2017/625 del Parlamento Europeo y del Consejo, de 15 de marzo de 2017, relativo a los controles y otras actividades oficiales realizados para garantizar la aplicación de la legislación sobre alimentos y piensos, y de las normas sobre salud y bienestar de los animales, sanidad vegetal y productos fitosanitarios, y por el que se modifican los Reglamentos (CE) n.o 999/2001, (CE) n.o 396/2005, (CE) n.o 1069/2009, (CE) n.o 1107/2009, (UE) n.o 1151/2012, (UE) n.o 652/2014, (UE) 2016/429 y (UE) 2016/2031 del Parlamento Europeo y del Consejo, los Reglamentos (CE) n.o 1/2005 y (CE) n.o 1099/2009 del Consejo, y las Directivas 98/58/CE, 1999/74/CE, 2007/43/CE, 2008/119/CE y 2008/120/CE del Consejo, y por el que se derogan los Reglamentos (CE) n.o 854/2004 y (CE) n.o 882/2004 del Parlamento Europeo y del Consejo, las Directivas 89/608/CEE, 89/662/CEE, 90/425/CEE, 91/496/CEE, 96/23/CE, 96/93/CE y 97/78/CE del Consejo y la Decisión 92/438/CEE del Consejo (Reglamento sobre controles oficiales) (1), y en particular su artículo 34, apartado 6,
Considerando lo siguiente:
|
(1) |
El Reglamento (UE) 2017/625 establece normas para la realización de los controles oficiales y otras actividades oficiales por parte de las autoridades competentes de los Estados miembros, a fin de verificar el cumplimiento de la legislación de la Unión, entre otros ámbitos, en el de la seguridad alimentaria en todas las fases del proceso de producción, transformación y distribución. Establece normas específicas sobre los controles oficiales relativos a sustancias cuyo uso pueda generar residuos en alimentos y piensos, así como requisitos generales para los métodos que deben utilizarse para el muestreo, los análisis y los ensayos de laboratorio durante los controles oficiales y otras actividades oficiales. |
|
(2) |
La Decisión 2002/657/CE de la Comisión (2) establece los requisitos para el funcionamiento de los métodos analíticos y la interpretación de los resultados de los análisis de determinadas sustancias y sus residuos en animales vivos y productos de origen animal, y la Decisión 98/179/CE de la Comisión (3) establece normas específicas relativas a la toma de muestras oficiales para el control de determinadas sustancias y sus residuos en animales vivos y productos de origen animal. Ambas decisiones fueron adoptadas con arreglo a la Directiva 96/23/CE del Consejo (4), derogada por el Reglamento (UE) 2017/625. En vista de los nuevos avances científicos, estas normas deben actualizarse e integrarse en el marco de los controles oficiales establecido en el Reglamento (UE) 2017/625. |
|
(3) |
Según lo dispuesto en el artículo 1, párrafo segundo, de la Decisión 2002/657/CE, dicha Decisión no será aplicable a sustancias sujetas a normas más específicas de la legislación de la Unión, a saber, micotoxinas en los productos alimenticios, dioxinas y policlorobifenilos (PCB) similares a las dioxinas en productos alimenticios, y plomo, cadmio, mercurio y benzo(a)pireno en los productos alimenticios. Las micotoxinas presentes en los productos alimenticios deben cumplir los requisitos establecidos en el Reglamento (CE) n.o 401/2006 de la Comisión (5), por el que se establecen los métodos de muestreo y de análisis para el control oficial del contenido de micotoxinas en los productos alimenticios. En el caso de las dioxinas y los PCB similares a las dioxinas, se aplica el Reglamento (UE) 2017/644 de la Comisión (6), por el que se establecen métodos de muestreo y de análisis para el control de los niveles de dioxinas, PCB similares a las dioxinas y PCB no similares a las dioxinas en determinados productos alimenticios. El Reglamento (CE) n.o 333/2007 de la Comisión (7) establece las disposiciones relativas al muestreo y el análisis para los controles oficiales de plomo, cadmio, mercurio y benzo(a)pireno en los productos alimenticios. |
|
(4) |
Por motivos de claridad y seguridad jurídica, conviene fusionar las disposiciones aplicables al muestreo y análisis de las sustancias farmacológicamente activas en un único acto jurídico, como en el caso de las micotoxinas, las dioxinas, los PCB similares a las dioxinas, el plomo, el cadmio, el mercurio y el benzo(a)pireno en los productos alimenticios. |
|
(5) |
Por tanto, deben derogarse las Decisiones 98/179/CE y 2002/657/CE y sustituirse por el presente Reglamento. |
|
(6) |
Según lo dispuesto en el Reglamento (CE) n.o 1831/2003 del Parlamento Europeo y del Consejo (8), los coccidiostáticos e histomonóstatos pueden ser utilizados como aditivos para piensos, por lo que el Reglamento (CE) n.o 152/2009 de la Comisión (9), que establece los métodos de muestreo y análisis para el control oficial de los piensos, se aplica a los análisis del contenido de estas sustancias en los piensos. No obstante, dicho Reglamento debe aplicarse cuando el análisis de los piensos se realiza como parte de las medidas de seguimiento durante las investigaciones de la fuente de muestras no conformes en casos de incumplimiento, supuesto o demostrado, de las normas de la Unión aplicables al uso de sustancias farmacológicamente activas autorizadas, o sus residuos, en medicamentos veterinarios o como aditivos de piensos, o de las normas de la Unión aplicables al uso de sustancias farmacológicamente activas no autorizadas o prohibidas, o sus residuos. |
|
(7) |
A fin de garantizar la continuidad de la ejecución de los controles oficiales y otras actividades oficiales en residuos de sustancias farmacológicamente activas, y con el fin de evitar que todos los métodos deban ser revalidados al mismo tiempo, los métodos que hayan sido validados antes de la fecha de entrada en vigor del presente Reglamento podrán seguir utilizándose durante un período de tiempo limitado, siempre que se cumplan los requisitos de los puntos 2 y 3 del anexo I de la Decisión 2002/657/CE. Procede, por tanto, conceder a los Estados miembros tiempo suficiente para aplicar los requisitos establecidos en el presente Reglamento a todos los métodos analíticos. |
|
(8) |
Las medidas previstas en el presente Reglamento se ajustan al dictamen del Comité Permanente de Vegetales, Animales, Alimentos y Piensos. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
Objeto y ámbito de aplicación
El presente Reglamento establece las normas sobre los métodos analíticos utilizados para el muestreo y los análisis de laboratorio relativos a residuos de sustancias farmacológicamente activas en animales vivos productores de alimentos, partes y fluidos de su cuerpo, excrementos, tejidos, productos de origen animal, subproductos animales, piensos y agua. Establece asimismo las normas para la interpretación de los resultados de dichos análisis de laboratorio.
El presente Reglamento se aplica a los controles oficiales destinados a verificar el cumplimiento de los requisitos relativos a la presencia de residuos de sustancias farmacológicamente activas.
Artículo 2
Definiciones
A efectos del presente Reglamento, se aplicarán las definiciones del artículo 2 del Reglamento Delegado (UE) 2019/2090 de la Comisión (10), del Reglamento (UE) 2019/1871 de la Comisión (11), del artículo 2 del Reglamento (CE) n.o 470/2009 del Parlamento Europeo y del Consejo (12) y del Reglamento (CEE) n.o 315/93 del Consejo (13).
También se aplicarán las siguientes definiciones:
|
1) |
«recuperación absoluta»: cantidad de analito recuperado en la fase final de un proceso analítico dividida entre la cantidad de analito de la muestra original, expresada como porcentaje; |
|
2) |
«exactitud»: grado de acuerdo entre un resultado de ensayo y el valor de referencia verdadero aceptado, determinada estimando la veracidad y la precisión (14); |
|
3) |
«error alfa (α)»: probabilidad de que la muestra analizada sea conforme, aunque se haya obtenido un resultado de medición no conforme; |
|
4) |
«analito»: componente de un sistema que debe analizarse; |
|
5) |
«sustancia autorizada»: sustancia farmacológicamente activa autorizada para el uso en animales productores de alimentos de conformidad con la Directiva 2001/82/CE del Parlamento Europeo y del Consejo (15); |
|
6) |
«error beta (β)»: probabilidad de que la muestra analizada sea en realidad no conforme, aunque se haya obtenido un resultado de medición conforme; |
|
7) |
«sesgo»: diferencia entre el valor estimado del resultado del ensayo y un valor de referencia aceptado; |
|
8) |
«patrón de calibración»: referencia trazable para las mediciones que representa la cantidad de sustancia de interés de una manera que vincula su valor a una base de referencia; |
|
9) |
«material de referencia certificado (MRC)»: material de referencia acompañado por la documentación emitida por un organismo autorizado, que proporciona uno o varios valores de propiedades especificadas, con incertidumbres y trazabilidades asociadas, empleando procedimientos válidos (16); |
|
10) |
«cocromatografía»: técnica en la que se aplica a un soporte cromatográfico una sustancia desconocida junto con uno o varios compuestos conocidos, con la expectativa de que el comportamiento relativo de las sustancias desconocidas y conocidas ayude a identificar la sustancia desconocida; |
|
11) |
«estudio colaborativo»: análisis de la misma muestra o muestras utilizando el mismo método para determinar las características de funcionamiento del método en diferentes laboratorios. Permite calcular el error aleatorio de medición y el sesgo de laboratorio del método utilizado; |
|
12) |
«método de confirmación»: método que proporciona información completa o complementaria que permite identificar y, de ser necesario, cuantificar de manera inequívoca la sustancia de alguna de las siguientes maneras:
|
|
13) |
«factor de cobertura (k)»: número que expresa el nivel de confianza deseado y que está asociado a la incertidumbre expandida; |
|
14) |
«límite de decisión para confirmación (CCα)»: límite en el cual y a partir del cual se puede concluir con una probabilidad de error de α que una muestra no es conforme, y el valor 1 – α significa certeza estadística en porcentaje de que se ha superado el límite permitido; |
|
15) |
«capacidad de detección del cribado (CCβ)»: contenido mínimo de analito que puede ser detectado o cuantificado en una muestra con una probabilidad de error de β:
|
|
16) |
«material de muestra enriquecido»: muestra enriquecida con una cantidad conocida del analito que debe detectarse o cuantificarse; |
|
17) |
«estudio interlaboratorios»: organización, realización y evaluación de ensayos de la misma muestra o muestras por dos laboratorios o más, con arreglo a condiciones predeterminadas, para evaluar el funcionamiento de los ensayos, ya sea en forma de estudio colaborativo o de ensayo de aptitud; |
|
18) |
«patrón interno»: sustancia, no contenida en la muestra, que tiene propiedades fisicoquímicas lo más parecidas posible a las del analito que debe identificarse o cuantificarse; |
|
19) |
«nivel de interés»: concentración de una sustancia o un analito en una muestra que es significativa para determinar su conformidad con la legislación en lo relativo a:
|
|
20) |
«nivel mínimo calibrado (NMC)»: concentración mínima a la que se ha calibrado el sistema de medición; |
|
21) |
«matriz»: material del que se toma una muestra; |
|
22) |
«efecto matriz»: diferencia de respuesta analítica entre un patrón disuelto en el disolvente y un patrón similar a la matriz, con o sin corrección mediante patrón interno; |
|
23) |
«patrón similar a la matriz»: matriz en blanco (libre de analito) a la que se añade analito en un rango de concentraciones tras el tratamiento de la muestra; |
|
24) |
«patrón de matriz enriquecida»: matriz en blanco (libre de analito) a la que, antes de la extracción con disolventes y del tratamiento de la muestra, se ha añadido el analito en un rango de concentraciones; |
|
25) |
«mensurando»: cantidad particular que se somete a medición; |
|
26) |
«incertidumbre de medida»: parámetro no negativo, asociado con el resultado de la medición, que caracteriza la dispersión de los valores que pueden atribuirse razonablemente al mensurando, a partir de la información utilizada; |
|
27) |
«criterios de funcionamiento»: requisitos aplicables a una característica de funcionamiento en función de los cuales se puede determinar que un método analítico es adecuado para el uso previsto y ofrece resultados fiables; |
|
28) |
«precisión»: grado de acuerdo entre los resultados independientes de ensayos obtenidos en condiciones prescritas. Se expresa como la desviación estándar o el coeficiente de variación de los resultados del ensayo; |
|
29) |
«método cualitativo»: método analítico que detecta o identifica una sustancia o grupo de sustancias basándose en sus propiedades químicas, biológicas o físicas; |
|
30) |
«método cuantitativo»: método analítico que determina la cantidad o la fracción de la masa de una sustancia de forma que pueda expresarse como valor numérico de unidades apropiadas; |
|
31) |
«recuperación»: cantidad correcta recuperada de un analito dividida entre la cantidad enriquecida del analito en la muestra matriz, expresada como porcentaje; |
|
32) |
«corrección de la recuperación»: uso de patrones internos, de una curva de calibración de matriz y de un factor de corrección de la recuperación, y también una combinación de estos métodos; |
|
33) |
«material de referencia»: material, suficientemente homogéneo y estable con respecto a una o más propiedades especificadas, que se ha establecido que es idóneo para el uso previsto en un proceso de medición o en un examen de las propiedades nominales (19); |
|
34) |
«efecto matriz relativo»: diferencia de respuesta analítica entre un patrón disuelto en el disolvente y un patrón similar a la matriz con corrección mediante patrón interno; |
|
35) |
«repetibilidad»: precisión bajo condiciones en las que se obtienen resultados independientes, con el mismo método, sobre idénticas muestras, en el mismo laboratorio, por el mismo operador, y utilizando los mismos equipos de medición, durante un corto intervalo de tiempo; |
|
36) |
«reproducibilidad»: precisión bajo condiciones en las que los resultados se obtienen con el mismo método, sobre muestras idénticas, en laboratorios diferentes, con operadores distintos y utilizando equipos diferentes (20); |
|
37) |
«robustez»: vulnerabilidad de un método analítico frente a cambios de las condiciones experimentales en las que puede aplicarse el método tal cual o con determinadas modificaciones menores; |
|
38) |
«método de cribado»: método utilizado para el cribado de una sustancia o clase de sustancia en el nivel de interés; |
|
39) |
«concentración de cribado establecida»: concentración igual o inferior a la CCβ en la que una medición de cribado clasifica la muestra como «cribado positivo» potencialmente no conforme y que da lugar a que se realice un ensayo de confirmación; |
|
40) |
«selectividad»: capacidad de un método de distinguir entre el analito que se está midiendo y otras sustancias; |
|
41) |
«estudio intralaboratorio» o «validación interna»: estudio analítico realizado por un solo laboratorio que utiliza el mismo método para proceder a análisis, separados por largos intervalos de tiempo justificados, de idénticos o diferentes materiales de ensayo en condiciones distintas; |
|
42) |
«adición de patrón»: procedimiento en el que una parte de la muestra se analiza tal cual, y a las demás porciones de ensayo se les añade, antes del análisis, cantidades conocidas del analito patrón; |
|
43) |
«analito patrón»: analito de contenido y pureza conocidos y certificados que se utiliza como referencia en el análisis; |
|
44) |
«sustancia»: materia de composición constante caracterizada por las entidades que la componen y por determinadas propiedades físicas; |
|
45) |
«porción de ensayo»: cantidad de material extraído de la muestra en la que se realiza el análisis o la observación; |
|
46) |
«veracidad»: grado de concordancia existente entre el valor medio obtenido de una gran serie de resultados y un valor de referencia aceptado; |
|
47) |
«unidades»: unidades descritas en la norma ISO 80000 (21) y la Directiva 80/181/CEE del Consejo (22); |
|
48) |
«validación»: demostración mediante examen y puesta a disposición de pruebas efectivas de que se cumplen los requisitos particulares de un uso específico previsto (23), gracias a un estudio intralaboratorio o un estudio colaborativo; |
|
49) |
«reproducibilidad intralaboratorio» o «reproducibilidad de precisión intermedia/interna»: precisión de la medición en determinadas condiciones intralaboratorio en un laboratorio específico. |
Artículo 3
Métodos de análisis
Los Estados miembros garantizarán que las muestras tomadas de conformidad con el artículo 34 del Reglamento (UE) 2017/625 se analicen utilizando métodos que cumplan los siguientes requisitos:
|
1) |
estar documentados en instrucciones de ensayo, de preferencia con arreglo a los anexos de la norma ISO 78-2:1999 Química. Diseños para normas. Parte 2: Métodos de análisis químico (24); |
|
2) |
cumplir los criterios de funcionamiento y otros requisitos aplicables a los métodos analíticos establecidos en el capítulo 1 del anexo I del presente Reglamento; |
|
3) |
haber sido validados de conformidad con los requisitos establecidos en los capítulos 2 y 4 del anexo I del presente Reglamento; |
|
4) |
permitir el cumplimiento de los valores de referencia a efectos de intervención establecidos en el Reglamento (UE) 2019/1871, la detección de sustancias prohibidas y no autorizadas, así como el cumplimiento de los contenidos máximos (CM) fijados con arreglo al Reglamento (CEE) n.o 315/93 y al Reglamento (CE) n.o 124/2009 y los límites máximos de residuos (LMR) fijados con arreglo a los Reglamentos (CE) n.o 1831/2003 y n.o 470/2009. |
Artículo 4
Control de calidad
Los Estados miembros garantizarán la calidad de los resultados de los análisis efectuados de conformidad con el Reglamento (UE) 2017/625, en particular controlando los resultados de los ensayos o de la calibración con arreglo a la norma ISO/IEC 17025:2017 Requisitos generales para la competencia de los laboratorios de ensayo y calibración, y de conformidad con los requisitos de control de calidad durante análisis de rutina establecidos en el capítulo 3 del anexo I del presente Reglamento.
Artículo 5
Interpretación de los resultados
1) El resultado de un análisis se considerará no conforme cuando sea igual o superior al límite de decisión para confirmación(CCα).
2) En el caso de sustancias autorizadas para las que se haya establecido un LMR o un CM, el límite de decisión para confirmación (CCα) será la concentración a partir de la cual pueda decidirse, con certeza estadística de valor numérico 1 – α, que se ha superado el límite permitido.
3) En el caso de sustancias no autorizadas o prohibidas para las que no se haya establecido un LMR o un CM en una especie o producto determinados, el límite de decisión para confirmación (CCα) será el nivel de concentración más bajo en el que pueda decidirse, con certeza estadística de valor numérico 1 – α, que el analito se encuentra presente.
4) En el caso de sustancias farmacológicamente activas no autorizadas o prohibidas, el error α será igual o inferior al 1 %. Para todas las demás sustancias, el error α será igual o inferior al 5 %.
Artículo 6
Métodos de muestreo
Los Estados miembros garantizarán que las muestras se tomen, manipulen y etiqueten de conformidad con los métodos detallados de muestreo establecidos en el anexo II del presente Reglamento.
Artículo 7
Derogaciones y medidas transitorias
Quedan derogadas las Decisiones 2002/657/CE y 98/179/CE a partir de la fecha de entrada en vigor del presente Reglamento.
No obstante, hasta el 10 de junio de 2026, los requisitos establecidos en los puntos 2 y 3 del anexo I de la Decisión 2002/657/CE seguirán aplicándose a los métodos que hayan sido validados antes de la fecha de entrada en vigor del presente Reglamento.
Artículo 8
Entrada en vigor
El presente Reglamento entrará en vigor a los veinte días de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 22 de marzo de 2021.
Por la Comisión
La Presidenta
Ursula VON DER LEYEN
(1) DO L 95 de 7.4.2017, p. 1.
(2) Decisión 2002/657/CE de la Comisión, de 14 de agosto de 2002, por la que se aplica la Directiva 96/23/CE del Consejo en cuanto al funcionamiento de los métodos analíticos y la interpretación de los resultados (DO L 221 de 17.8.2002, p. 8).
(3) Decisión 98/179/CE de la Comisión, de 23 de febrero de 1998, por la que se fijan normas específicas relativas a la toma de muestras oficiales para el control de determinadas sustancias y sus residuos en los animales vivos y sus productos (DO L 65 de 5.3.1998, p. 31).
(4) Directiva 96/23/CE del Consejo, de 29 de abril de 1996, relativa las medidas de control aplicables respecto de determinadas sustancias y sus residuos en los animales vivos y sus productos y por la que se derogan las Directivas 85/358/CEE y 86/469/CEE y las Decisiones 89/187/CEE y 91/664/CEE (DO L 125 de 23.5.1996, p. 10).
(5) Reglamento (CE) n.o 401/2006 de la Comisión, de 23 de febrero de 2006, por el que se establecen los métodos de muestreo y de análisis para el control oficial del contenido de micotoxinas en los productos alimenticios (DO L 070 de 9.3.2006, p. 12).
(6) Reglamento (UE) 2017/644 de la Comisión, de 5 de abril de 2017, por el que se establecen métodos de muestreo y de análisis para el control de los niveles de dioxinas, PCB similares a las dioxinas y PCB no similares a las dioxinas en determinados productos alimenticios y por el que se deroga el Reglamento (UE) n.o 589/2014 (DO L 92 de 6.4.2017, p. 9).
(7) Reglamento (CE) n.o 333/2007 de la Comisión, de 28 de marzo de 2007, por el que se establecen los métodos de muestreo y análisis para el control de los niveles de elementos traza y de los contaminantes de proceso en los productos alimenticios (DO L 088 de 29.3.2007, p. 29).
(8) Reglamento (CE) n.o 1831/2003 del Parlamento Europeo y del Consejo, de 22 de septiembre de 2003, sobre los aditivos en la alimentación animal (DO L 268 de 18.10.2003, p. 29).
(9) Reglamento (CE) n.o 152/2009 de la Comisión, de 27 de enero de 2009, por el que se establecen los métodos de muestreo y análisis para el control oficial de los piensos (DO L 54 de 26.2.2009, p. 1).
(10) Reglamento Delegado (UE) 2019/2090 de la Comisión, de 19 de junio de 2019, que complementa al Reglamento (UE) 2017/625 del Parlamento Europeo y del Consejo en lo que respecta a los casos de sospecha o constatación de incumplimiento de las normas de la Unión aplicables al uso de sustancias farmacológicamente activas autorizadas o sus residuos en medicamentos veterinarios o como aditivos de piensos o de las normas de la Unión aplicables al uso de sustancias farmacológicamente activas no autorizadas o prohibidas o sus residuos (DO L 317 de 9.12.2019, p. 28).
(11) Reglamento (UE) 2019/1871 de la Comisión, de 7 de noviembre de 2019, relativo a los valores de referencia para las sustancias farmacológicamente activas no autorizadas presentes en los alimentos de origen animal y por el que se deroga la Decisión 2005/34/CE (DO L 289 de 8.11.2019, p. 41).
(12) Reglamento (CE) n.o 470/2009 del Parlamento Europeo y del Consejo, de 6 de mayo de 2009, por el que se establecen procedimientos comunitarios para la fijación de los límites de residuos de las sustancias farmacológicamente activas en los alimentos de origen animal, se deroga el Reglamento (CEE) n.o 2377/90 del Consejo y se modifican la Directiva 2001/82/CE del Parlamento Europeo y del Consejo y el Reglamento (CE) n.o 726/2004 del Parlamento Europeo y del Consejo (DO L 152 de 16.6.2009, p. 11).
(13) Reglamento (CEE) n.o 315/93 del Consejo, de 8 de febrero de 1993, por el que se establecen procedimientos comunitarios en relación con los contaminantes presentes en los productos alimenticios (DO L 037 de 13.2.1993, p. 1).
(14) ISO 3534-1: 2006. Estadística. Vocabulario y símbolos. Parte 1: Términos estadísticos generales y términos empleados en el cálculo de probabilidades (capítulo 1).
(15) Directiva 2001/82/CE del Parlamento Europeo y del Consejo, de 6 de noviembre de 2001, por la que se establece un código comunitario sobre medicamentos veterinarios (DO L 311 de 28.11.2001, p. 1).
(16) JCGM 200:2008, Vocabulario Internacional de Metrología. Conceptos fundamentales y generales y términos asociados (VIM) (capítulo 5. Patrones de medida). Tercera edición (2008): https://www.iso.org/sites/JCGM/VIM-JCGM200.htm.
(17) Reglamento (CE) n.o 124/2009 de la Comisión, de 10 de febrero de 2009, que establece los contenidos máximos de coccidiostáticos e histomonóstatos presentes en los alimentos como resultado de la transferencia inevitable de estas sustancias en los piensos a los que no están destinadas (DO L 040 de 11.2.2009, p. 7).
(18) Reglamento (UE) n.o 37/2010 de la Comisión, de 22 de diciembre de 2009, relativo a las sustancias farmacológicamente activas y su clasificación por lo que se refiere a los límites máximos de residuos en los productos alimenticios de origen animal (DO L 015 de 20.1.2010, p. 1).
(19) «Directrices sobre la terminología analítica» (CAC/GL 72-2009) de la Comisión del Codex Alimentarius (Organización de las naciones Unidas para la Alimentación y la Agricultura/Organización Mundial de la Salud).
(20) ISO 5725-1:1994. Exactitud (veracidad y precisión) de resultados y métodos de medición. Parte 1: Principios generales y definiciones (capítulo 3).
(21) ISO 80000-1:2009. Magnitudes y unidades. Parte 1: Generalidades.
(22) Directiva 80/181/CEE del Consejo, de 20 de diciembre de 1979, relativa a la aproximación de las legislaciones de los Estados miembros sobre las unidades de medida y por la que se deroga la Directiva 71/354/CEE (DO L 39 de 15.2.1980, p. 40).
(23) ISO/IEC 17025:2017. Requisitos generales para la competencia de los laboratorios de ensayo y calibración (capítulo 3).
(24) ISO 78-2: 1999 Química. Diseños para normas. Parte 2: Métodos de análisis químico (anexos).
ANEXO I
CAPÍTULO 1
CRITERIOS DE FUNCIONAMIENTO Y OTROS REQUISITOS DE LOS MÉTODOS ANALÍTICOS
1.1. Requisitos aplicables a los métodos de cribado
1.1.1. Categorías de métodos adecuados de cribado
Como métodos adecuados de cribado se utilizarán métodos cualitativos, semicuantitativos o cuantitativos.
1.1.2. Requisitos aplicables a los métodos de cribado biológicos, bioquímicos o físicoquímicos
La CCβ de las sustancias prohibidas o no autorizadas será la más baja posible de alcanzar razonablemente y, en cualquier caso, inferior al valor de referencia a efectos de intervención (VRI) de las sustancias para las que se haya establecido dicho valor en virtud del Reglamento (UE) 2019/1871.
En el caso de las sustancias farmacológicamente activas autorizadas, la CCβ será inferior al límite máximo de residuos (LMR) o al contenido máximo (CM).
Solo se utilizarán para el cribado aquellos métodos analíticos de los que se pueda demostrar de manera documentada que están validados y que tienen un porcentaje de falsos conformes (error β) igual o inferior al 5 %. Si se sospecha que un resultado es no conforme, dicho resultado se confirmará mediante un método de confirmación.
Los métodos cuantitativos de cribado utilizados tanto para el cribado como para la confirmación cumplirán los mismos requisitos de exactitud, rango y precisión que los descritos en los puntos 1.2.2.1 y 1.2.2.2.
1.2. Requisitos aplicables a los métodos de confirmación
1.2.1. Requisitos generales para métodos de confirmación
El CCα de las sustancias prohibidas o no autorizadas será el más bajo posible de alcanzar razonablemente. El CCα de las sustancias prohibidas o no autorizadas para las que se haya establecido un VRI en virtud del Reglamento (UE) 2019/1871 será igual o inferior al VRI.
El CCα de las sustancias autorizadas será superior, pero lo más cercano posible, al LMR o al CM.
Para la confirmación, solo se utilizarán aquellos métodos analíticos de los que se pueda demostrar de manera documentada que están validados y que tienen un porcentaje de falsos no conformes (error α) igual o inferior al 1 % para las sustancias prohibidas o no autorizadas, o igual o inferior al 5 % para las sustancias autorizadas.
Los métodos de confirmación proporcionarán información sobre la composición química estructural del analito. En consecuencia, los métodos de confirmación basados exclusivamente en análisis cromatográficos sin recurrir a la detección por espectrometría de masas no son adecuados por sí solos como métodos de confirmación para sustancias farmacológicamente activas prohibidas o no autorizadas. En caso de que la espectrometría de masas no sea adecuada para sustancias autorizadas, pueden utilizarse otros métodos, como la HPLC-DAD y la HPLC-FLD o una combinación de ambas.
Cuando el método de confirmación así lo requiera, al comenzar el procedimiento de extracción se añadirá un patrón interno adecuado a la porción de ensayo. Se emplearán, según su disponibilidad, formas marcadas por isótopos estables del analito, especialmente adecuadas para la detección por espectrometría de masas, o compuestos análogos cuya estructura esté estrechamente relacionada con el analito. Cuando no pueda utilizarse un patrón interno adecuado, la identificación del analito se confirmará, de preferencia, mediante cocromatografía (1). En este caso, solo se obtendrá un pico, en el cual la altura (o superficie) máxima será equivalente a la cantidad de analito añadida. Si esto no es posible, se utilizarán patrones similares a la matriz o de matriz enriquecida.
1.2.2. Criterios generales de funcionamiento para métodos de confirmación
1.2.2.1. Veracidad por recuperación
En el caso de análisis repetidos de un material de referencia certificado, la desviación entre la fracción de masa media determinada experimentalmente, con corrección de recuperación, y el valor certificado cumplirá los rangos de veracidad mínima recogidos en el cuadro 1.
Cuadro 1
Veracidad mínima de los métodos cuantitativos
|
Fracción de masa |
Rango |
|
≤ 1 μg/kg |
– 50 % a +20 % |
|
> 1 μg/kg a 10 μg/kg |
– 30 % a +20 % |
|
≥ 10 μg/kg |
– 20 % a +20 % |
Cuando no se disponga de materiales de referencia certificados, es aceptable que la veracidad de las mediciones se evalúe de otras maneras como, por ejemplo, utilizando materiales con valores asignados a partir de estudios interlaboratorios o añadiendo cantidades conocidas del analito o analitos a una matriz en blanco.
1.2.2.2. Precisión
El coeficiente de variación (CV) para el análisis repetido de un material de referencia o enriquecido, en condiciones de reproducibilidad intralaboratorio, no superará el nivel calculado mediante la ecuación de Horwitz. La ecuación es la siguiente:
CV = 2(1 – 0.5 log C)
donde C es la fracción de masa expresada como potencia (exponente) de 10 (por ejemplo, 1 mg/g = 10-3). Para las fracciones de masa inferiores a 120 μg/kg, la aplicación de la ecuación de Horwitz arroja valores inaceptablemente altos. Por consiguiente, el coeficiente de variación máximo autorizado no deberá ser superior a los valores presentados en el cuadro 2.
Cuadro 2
Coeficiente de variación aceptable
|
Fracción de masa |
CV de reproducibilidad (%) |
|
> 1 000 μg/kg |
16 (adaptado a partir de la ecuación de Horwitz) |
|
> 120 μg/kg – 1 000 μg/kg |
22 (adaptado a partir de la ecuación de Horwitz) |
|
10 – 120 μg/kg |
25 (*) |
|
< 10 μg/kg |
30 (*) |
En el caso de los análisis efectuados en condiciones de repetibilidad, el coeficiente de variación en condiciones de repetibilidad será igual o inferior a dos tercios de los valores enumerados en el cuadro 2.
1.2.3. Requisitos aplicables a la separación cromatográfica
Para la cromatografía líquida (LC) o de gases GC), el tiempo de retención mínimo aceptable para los analitos examinados será el doble del tiempo de retención correspondiente al volumen vacío de la columna. El tiempo de retención del analito en el extracto corresponderá al del patrón de calibración, al de un patrón similar a la matriz o al de un patrón de matriz enriquecida, con una tolerancia de ± 0,1 minuto. Para la cromatografía rápida, en la que el tiempo de retención es inferior a 2 minutos, se aceptará una desviación inferior al 5 % del tiempo de retención. En caso de que se utilice un patrón interno, la relación entre el tiempo de retención cromatográfica del analito y el del patrón interno, es decir, el tiempo relativo de retención del analito, se corresponderá con el del patrón de calibración, el patrón similar a la matriz o el patrón de matriz enriquecida, con una desviación máxima del 0,5 % para la cromatografía de gases y del 1 % para la cromatografía líquida, en el caso de métodos validados a partir de la fecha de entrada en vigor del presente Reglamento.
1.2.4. Criterios específicos de funcionamiento para la espectrometría de masas
1.2.4.1. Detección por espectrometría de masas
La detección por espectrometría de masas se llevará a cabo mediante alguna de las opciones siguientes:
|
1. |
registro de espectros de masa de barrido completo (FS); |
|
2. |
registro selectivo de iones (SIM); |
|
3. |
técnicas de espectrometría de masas secuencial (MSn), como el registro selectivo de la reacción (SRM); |
|
4. |
una combinación de técnicas de espectrometría de masas (MS) o espectrometría de masas secuencial (MSn) con modos de ionización adecuados. |
Son adecuadas la espectrometría de masas de baja resolución (LRMS, a resolución de masa unitaria) y la espectrometría de masas de alta resolución (HRMS), incluidos, por ejemplo, los instrumentos de doble focalización, de tiempo de vuelo (TOF) y orbitrap.
Para confirmar la identidad de un analito en HRMS, la desviación de la masa de todos los iones de diagnóstico será inferior a 5 ppm (o en el caso de m/z < 200, inferior a 1 mDa). Sobre esta base, debe seleccionarse la resolución efectiva idónea para el fin previsto. La resolución será normalmente superior a 10 000 para todo el rango de masas a un valle del 10 %, o superior a 20 000 en anchura a media altura (FWHM).
Cuando la determinación mediante espectrometría de masas se lleve a cabo registrando espectros de barrido completo (tanto LRMS como HRMS), solo son adecuados los iones de diagnóstico con una intensidad relativa superior al 10 % en el espectro de referencia del patrón de calibración, patrón similar a la matriz o patrones de matriz enriquecida. Los iones de diagnóstico incluirán el ion molecular (si se encuentra presente en una intensidad de ≥ 10 % del pico base) y los iones fragmento o iones producto característicos.
Selección del ion precursor: cuando la determinación mediante espectrometría de masas se realiza por fragmentación tras la selección del ion precursor, la selección del ion precursor se realiza con una resolución de masa unitaria o mejor. El ion precursor seleccionado será el ion molecular, los aductos característicos del ion molecular, los iones producto característicos o uno de sus iones isotópicos. Si la selección del precursor tiene una ventana de selección de la masa de más de un dalton (por ejemplo, en caso de adquisición independiente de datos), la técnica se considerará un análisis confirmatorio de barrido completo.
Iones fragmento e iones producto: los iones producto o fragmento seleccionados serán iones de diagnóstico para el analito o producto medido. Siempre que sea posible, se omitirán las transiciones no selectivas (por ejemplo, el catión de tropilio o la pérdida de agua). La abundancia de iones de diagnóstico se determinará a partir del área o la altura del pico de los cromatogramas integrados de ion extraído. Esto también se aplica cuando se utilizan mediciones de barrido completo para la identificación. La relación señal-ruido de todos los iones de diagnóstico será igual o superior a tres a uno (3:1).
Intensidades relativas: las intensidades relativas de los iones de diagnóstico (relación de iones) se expresan como porcentaje de la intensidad del ion o de la transición más abundante. La relación de iones debe determinarse comparando espectros o integrando las señales de las trazas de masa de iones extraídos. La relación de iones del analito que debe confirmarse corresponderá a las de los patrones similares a la matriz, los patrones de matriz enriquecida o las disoluciones patrón en concentraciones comparables, medidas en las mismas condiciones, con una desviación relativa de ± 40 %.
Para todos los análisis mediante espectrometría de masas, se determinará al menos una relación de iones. Se trata preferentemente de iones obtenidos en un único barrido, pero los iones también pueden proceder de distintos barridos en la misma inyección (es decir, barrido completo y barrido de fragmentación).
1.2.4.2. Identificación
Se utilizará un sistema de puntos de identificación para seleccionar los modos de adquisición y los criterios de evaluación adecuados. Para confirmar la identidad de sustancias para las que se ha establecido un LMR (uso autorizado) presentes en una matriz, se requiere un mínimo de 4 puntos de identificación. En el caso de las sustancias no autorizadas o prohibidas, se requieren 5 puntos de identificación. Un punto puede proceder de la separación cromatográfica. El cuadro 3 muestra el número de puntos de identificación que aporta cada una de las técnicas. Para alcanzar el número de puntos de identificación necesario para la confirmación, pueden añadirse puntos de identificación obtenidos a partir de diferentes técnicas.
|
1. |
Todos los análisis mediante espectrometría de masas deberán combinarse con una técnica de separación que muestre una potencia de separación y una selectividad suficientes para la aplicación específica. Entre las técnicas de separación adecuadas se encuentran la cromatografía líquida y de gases, la electroforesis capilar (CE) y la cromatografía de fluidos supercríticos (SFC). En los casos en que el analito presente cualquier compuesto isobárico o isomérico, la aceptabilidad del tiempo de retención (a saber, ± 0.5 % en GC y ± 1 % en LC y SFC) es obligatoria para confirmar su identidad |
|
2. |
Se combinará un máximo de tres técnicas distintas para alcanzar el número mínimo de puntos de identificación. |
|
3. |
Los diferentes modos de ionización (por ejemplo, la ionización por impacto electrónico y la ionización química) se consideran técnicas diferentes. Cuadro 3 Puntos de identificación por técnica
Cuadro 4 Ejemplos de número de puntos de identificación, técnicas específicas y combinaciones de técnicas (n = un número entero)
|
1.2.5. Criterios de funcionamiento específicos para la determinación de un analito mediante cromatografía líquida con técnicas de detección distintas de la espectrometría de masas
Solo en el caso de sustancias autorizadas, podrán utilizarse las técnicas siguientes como alternativa a los métodos basados en la espectrometría de masas, siempre que se cumplan los criterios pertinentes para estas técnicas:
|
1. |
espectrofotometría con detección por diodos en serie (DAD) mediante barrido completo en caso de utilizarse con HPLC; |
|
2. |
espectrofotometría de detección por fluorescencia (FLD) en caso de utilizarse con HPLC. |
Por sí sola, la cromatografía líquida con detección de UV/VIS (longitud de onda única) no es apropiada como método de confirmación.
1.2.5.1. Criterios de funcionamiento de la espectrofotometría con detección por diodos en serie mediante barrido completo
Deberán cumplirse los criterios de funcionamiento de la separación cromatográfica incluidos en el capítulo 1.2.3.
Los máximos de absorción en el espectro UV del analito presentarán las mismas longitudes de onda que las del patrón de calibración en matriz, dentro de un margen máximo determinado por la resolución del sistema de detección. Para la detección por diodos en serie, este margen máximo suele ser de ± 2 nm. El espectro del analito por encima de los 220 nm, para aquellas partes de ambos espectros con una absorbancia relativa superior o igual al 10 %, no deberán diferir visualmente del espectro del patrón de calibración. Este criterio se satisface, en primer lugar, cuando se presentan máximos iguales y, en segundo lugar, cuando en ninguno de los puntos la diferencia entre ambos espectros es superior al 10 % de la absorbancia del patrón de calibración. Si se recurre a la búsqueda bibliográfica asistida por ordenador y a la concordancia, la comparación de los datos del espectro en las muestras oficiales con los de la disolución de calibración debe superar un factor crítico de correspondencia. Este factor se determinará durante el proceso de validación para cada analito, sobre la base de espectros para los que se cumplen los criterios descritos anteriormente. Se controlará la variabilidad debida a la matriz de la muestra y el funcionamiento del detector.
1.2.5.2. Criterios de funcionamiento para la espectrofotometría de detección por fluorescencia
Deberán cumplirse los criterios de funcionamiento de la separación cromatográfica incluidos en el capítulo 1.2.3.
La selección de las longitudes de onda de excitación y de emisión en combinación con las condiciones cromatográficas se harán de tal manera que se minimicen los efectos de componentes de interferencia en extractos de muestras en blanco. Debe haber un mínimo de 50 nanómetros entre la longitud de onda de excitación y la de emisión.
El máximo del pico más próximo en el cromatograma estará separado del pico del analito designado por al menos una anchura del pico, medida al 10 % de la altura máxima del pico del analito.
Ello se aplica a las moléculas que presentan fluorescencia natural y a las que la presentan después de transformación o derivatización.
CAPÍTULO 2
VALIDACIÓN
2.1. Características de funcionamiento que deben determinarse para los métodos analíticos
Mediante la validación del método, se demostrará que el método analítico cumple los criterios relativos a las características de funcionamiento aplicables. Controles con objetivos diferentes requieren distintas categorías de métodos. El cuadro 5 determina qué característica de funcionamiento debe verificarse para qué tipo de método. En el presente capítulo se ofrece una explicación más detallada de cada parámetro.
Cuadro 5
Clasificación de los métodos analíticos en función de las características de funcionamiento que deben determinarse
|
Método |
Confirmación |
Cribado |
|||
|
Cualitativo |
Cuantitativo |
Cualitativo |
Semicuantitativo |
Cuantitativo |
|
|
Sustancias |
A |
A, B |
A, B |
A, B |
A, B |
|
Identificación con arreglo a 1.2 |
x |
x |
|
|
|
|
CCα |
x |
x |
|
|
|
|
CCβ |
- |
|
x |
x |
x |
|
Veracidad |
x |
|
|
x |
|
|
Precisión |
x |
|
(x) |
x |
|
|
Efecto matriz relativo/recuperación absoluta (*) |
x |
|
|
x |
|
|
Selectividad/especificidad |
x |
x |
x |
x |
|
|
Estabilidad (#) |
x |
x |
x |
x |
|
|
Robustez |
x |
x |
x |
x |
|
|
x: Deberá demostrarse mediante la validación que se cumplen los requisitos de la característica de funcionamiento. x) Los métodos de cribado semicuantitativo no necesitan cumplir los requisitos de precisión del capítulo 1.2.2.2. No obstante, se determinará la precisión con el fin de demostrar la idoneidad del método para evitar resultados analíticos que den falsos conformes. A: sustancias prohibidas o no autorizadas B: sustancias autorizadas |
|||||
2.2. Veracidad, repetibilidad y reproducibilidad intralaboratorio
En este capítulo se ofrecen ejemplos y referencias de procedimientos de validación. Pueden utilizarse otros enfoques para demostrar que el método cumple los criterios de funcionamiento, a condición de que alcancen la misma cantidad y calidad de información.
2.2.1. Validación convencional
El cálculo de los parámetros por métodos convencionales exige la realización de diversos experimentos aislados. Debe determinarse cada característica de funcionamiento para cada cambio importante (véase la sección 2.4). Con los métodos multianalito pueden analizarse varios analitos simultáneamente, siempre que se hayan descartado posibles interferencias de importancia. De modo similar pueden determinarse varias características de funcionamiento. Así pues, para minimizar la carga de trabajo es conveniente combinar los experimentos cuando sea posible (por ejemplo, la repetibilidad y la reproducibilidad intralaboratorio con la especificidad, el análisis de muestras en blanco para determinar el límite decisión para confirmación y las pruebas de especificidad).
2.2.1.1. Veracidad mediante material de referencia certificado
Es preferible determinar la veracidad de un método analítico mediante material de referencia certificado (MRC). El procedimiento para ello se describe en la norma ISO 5725-4:1994 (2).
Presentamos un ejemplo a continuación:
|
1. |
analice seis muestras idénticas del MRC siguiendo las instrucciones de ensayo del método; |
|
2. |
determine la concentración del analito en cada una de dichas muestras; |
|
3. |
calcule la media, la desviación estándar y el coeficiente de variación (%) para estas seis muestras; |
|
4. |
calcule la veracidad dividiendo la concentración media detectada entre el valor certificado (medido como concentración) y multiplíquelo por 100, para expresar el resultado en porcentaje. |
Veracidad (%) = (concentración media detectada tras introducción del factor corrector de recuperación) × 100/valor certificado.
2.2.1.2. Veracidad mediante muestras enriquecidas
Si no se dispone de material de referencia certificado, la veracidad del método se determinará mediante experimentos con una matriz en blanco enriquecida, como mínimo de acuerdo con el sistema siguiente:
|
1. |
Para los métodos validados a partir de la fecha de entrada en vigor del presente Reglamento, seleccione material en blanco y enriquezca en una concentración de:
|
|
2. |
En cada nivel deberá procederse al análisis con un mínimo de seis muestras idénticas. |
|
3. |
Analice las muestras. |
|
4. |
Calcule la concentración detectada en cada muestra. |
|
5. |
Calcule la veracidad de cada muestra utilizando la ecuación que figura a continuación y, seguidamente, calcule la veracidad media y el coeficiente de variación medio de los seis resultados en cada nivel de concentración. |
Veracidad (%) = (concentración media detectada tras introducción del factor corrector de recuperación) × 100/nivel de enriquecimiento
En el caso de los métodos para sustancias autorizadas validados antes de la fecha de aplicación del presente Reglamento, será suficiente una determinación de la veracidad del método utilizando seis alícuotas enriquecidas a 0,5, 1,0 y 1,5 veces el LMR o el CM.
2.2.1.3. Repetibilidad
|
1. |
Para los métodos validados a partir de la fecha de entrada en vigor del presente Reglamento, se preparará un conjunto de muestras de matrices en blanco idénticas de la misma especie. Se enriquecerán con el analito para obtener concentraciones equivalentes a:
|
|
2. |
En cada nivel deberá procederse al análisis con un mínimo de seis muestras idénticas. |
|
3. |
Analice las muestras. |
|
4. |
Calcule la concentración detectada en cada muestra. |
|
5. |
Calcule la concentración media, la desviación estándar y el coeficiente de variación (%) de las muestras enriquecidas. |
|
6. |
Repita estos pasos al menos otras dos veces. |
|
7. |
Calcule las medias generales de las concentraciones, de las desviaciones estándar (haciendo la media de las desviaciones estándar al cuadrado de las distintas ocasiones y calculando la raíz cuadrada del resultado obtenido) y de los coeficientes de variación de las muestras enriquecidas.
En el caso de los métodos para sustancias autorizadas validados antes de la fecha de aplicación del presente Reglamento, será suficiente una determinación de la repetibilidad con matrices enriquecidas en concentraciones a 0,5, 1,0 y 1,5 veces el LMR o el CM. Otra posibilidad es calcular la repetibilidad con arreglo a la norma ISO 5725-2: 2019 (7). |
2.2.1.4. Reproducibilidad intralaboratorio
|
1. |
Para las validaciones efectuadas tras la entrada en vigor del presente Reglamento, prepare un conjunto de muestras de materiales de ensayo especificados (de matrices idénticas o diferentes), enriquecidas con el analito o analitos para dar concentraciones equivalentes a:
|
|
2. |
Realice el análisis de cada nivel de concentración con al menos seis muestras idénticas de material en blanco. |
|
3. |
Analice las muestras. |
|
4. |
Calcule la concentración detectada en cada muestra. |
|
5. |
Repita estos pasos al menos otras dos veces con diferentes lotes de material en blanco, diferentes operadores y tantas condiciones ambientales diferentes como sea posible, por ejemplo lotes diferentes de reactivos y disolventes, temperaturas ambiente diferentes, distintos instrumentos o una variación de otros parámetros. |
|
6. |
Determine la concentración media, la desviación estándar y el coeficiente de variación (%) de las muestras enriquecidas.
En el caso de los métodos para sustancias autorizadas validados antes de la fecha de aplicación del presente Reglamento, será suficiente una determinación de la reproducibilidad intralaboratorio con matrices enriquecidas en concentraciones a 0,5, 1,0 y 1,5 veces el LMR o el CM. Otra posibilidad es calcular la reproducibilidad intralaboratorio/precisión intermedia de acuerdo con las normas ISO 5725-2: 2019, ISO 11843-1: 1997 (8) o Codex CAC/GL 59-2006 (9). |
2.2.2. Validación mediante modelos alternativos
El cálculo de los parámetros mediante modelos alternativos exige la realización de un plan experimental, que deberá elaborarse en función del número de especies y de factores diferentes que se pretenda estudiar. Por ello, en el primer paso de todo el proceso de validación se estudiarán las poblaciones de muestras que se analizarán en el futuro en el laboratorio para determinar las especies más importantes y los factores que pueden influir en los resultados de las mediciones. El enfoque factorial permite evaluar la incertidumbre de medida de los resultados de ensayos obtenidos en un laboratorio determinado en diversas condiciones de ensayo, como diferentes analistas, instrumentos, lotes de reactivos, matrices, duraciones de ensayo y temperaturas de ensayo. Posteriormente, se debe elegir el rango de concentraciones adaptado a la finalidad en función del LMR o del CM de sustancias autorizadas, o del VRI o del NMC de sustancias prohibidas o no autorizadas.
El enfoque factorial tiene por objeto establecer datos de precisión y de medición fiables mediante una variación controlada simultánea de los factores seleccionados. Permite evaluar el impacto combinado de los efectos factoriales y los efectos aleatorios. El diseño experimental también permite la investigación de la robustez (10) del método analítico y la determinación de la desviación estándar de la reproducibilidad interna con las distintas matrices.
Se presenta a continuación un ejemplo de planteamiento alternativo mediante un plan de diseño experimental ortogonal.
Pueden examinarse hasta siete factores (factores de ruido). El estudio está diseñado de tal manera que la precisión, la veracidad (basada en muestras enriquecidas), la sensibilidad, la incertidumbre de medida y las concentraciones críticas puedan determinarse simultáneamente mediante la aplicación del plan experimental.
Cuadro 6
Ejemplo de plan de diseño experimental ortogonal con siete factores (I-VII) variados en dos niveles (A/B) en un estudio de validación con ocho tandas (combinación de nivel de factores)
|
Factor |
I |
II |
III |
IV |
V |
VI |
VII |
|
Tanda 01 |
A |
A |
A |
A |
A |
A |
A |
|
Tanda 02 |
A |
A |
B |
A |
B |
B |
B |
|
Tanda 03 |
A |
B |
A |
B |
A |
B |
B |
|
Tanda 04 |
A |
B |
B |
B |
B |
A |
A |
|
Tanda 05 |
B |
A |
A |
B |
B |
A |
B |
|
Tanda 06 |
B |
A |
B |
B |
A |
B |
A |
|
Tanda 07 |
B |
B |
A |
A |
B |
B |
A |
|
Tanda 08 |
B |
B |
B |
A |
A |
A |
B |
El cálculo de las características del método se efectuará según lo descrito por Jülicher et al (11).
2.2.3. Otros enfoques de validación
Pueden utilizarse otros enfoques para demostrar que el método cumple los criterios de funcionamiento para las características de funcionamiento, a condición de que alcancen la misma cantidad y calidad de información. La validación también puede realizarse a través de estudios interlaboratorios, como se establece en el Codex Alimentarius, las normas ISO o la IUPAC (12), o de acuerdo con métodos alternativos tales como estudios en un solo laboratorio o la validación interna (13). Si se aplican procedimientos alternativos de validación, se indicará en el protocolo de validación el modelo y la estrategia, con sus respectivos requisitos previos, hipótesis y fórmulas, o, cuando menos, sus referencias.
2.3. Selectividad/especificidad
La capacidad de discriminación entre el analito y las sustancias estrechamente relacionadas se determinará al máximo posible. Se determinará la interferencia de homólogos, isómeros, productos de degradación, sustancias endógenas, análogos, productos metabólicos del residuo de interés, de compuestos de la matriz o de cualquier otra sustancia que pueda interferir y, en caso necesario, se modificará el método para evitar las interferencias detectadas. Para determinar la especificidad del método, se utilizará el siguiente planteamiento:
|
1. |
Seleccione una gama de compuestos químicamente relacionados u otras sustancias que puedan encontrarse en las muestras con el compuesto de interés, y compruebe si podrían interferir en el análisis de los analitos. |
|
2. |
Analice un número apropiado de muestras en blanco representativas como, por ejemplo, diferentes lotes o lotes de diferentes especies animales (n ≥ 20) y verifique posibles interferencias de señales, picos o indicios de iones en la región de interés en la que cabe esperar la elución del analito. |
|
3. |
Enriquezca muestras en blanco representativas hasta una concentración adecuada con sustancias que pueden interferir con la identificación o la cuantificación del analito e investigue si la sustancia añadida:
|
2.4. Robustez
El método analítico se someterá a ensayo para comprobar su funcionamiento continuado en diferentes condiciones experimentales, que incluyen, por ejemplo, condiciones de muestreo diferentes y cambios menores que pueden producirse en los ensayos rutinarios. Para comprobar la robustez del método, los cambios introducidos en las condiciones experimentales deben ser menores. Se evaluará la importancia de estos cambios. Se determinará cada característica de funcionamiento para todos los cambios menores que hayan tenido un efecto demostrado significativo en el funcionamiento del análisis.
2.5. Estabilidad
Se determinará la estabilidad del patrón de calibración, el patrón similar a la raíz y los patrones de matriz enriquecida, así como la estabilidad de los componentes del analito o la matriz en la muestra, durante el almacenamiento o el análisis, ya que las inestabilidades pueden influir en el resultado del ensayo.
Normalmente, la estabilidad del analito está debidamente caracterizada para diversas condiciones de almacenamiento. Los experimentos realizados para supervisar las condiciones de almacenamiento de los patrones y las muestras, que se llevan a cabo como parte del sistema normal de acreditación de laboratorio y control de calidad, pueden proporcionar la información requerida. Si los datos de estabilidad de los analitos en la matriz están disponibles (por ejemplo, a partir de información de los laboratorios de referencia de la UE, datos publicados, etc.), no es necesario que cada laboratorio determine dichos datos. Sin embargo, referirse a los datos disponibles sobre la estabilidad de los analitos en la disolución y en la matriz solo es aceptable si se aplican condiciones idénticas.
En caso de que no se disponga de los datos de estabilidad requeridos, deberán utilizarse los métodos descritos a continuación.
2.5.1. Determinación de la estabilidad del analito en la solución
|
1. |
Prepare nuevas soluciones madre del analito y dilúyalas, siguiendo las instrucciones de ensayo, para obtener el número suficiente de alícuotas (por ejemplo, 40) de cada concentración seleccionada. Se prepararán muestras de:
|
|
2. |
Mida el contenido de analito en la solución recién preparada, siguiendo las instrucciones de ensayo. |
|
3. |
Vierta los volúmenes adecuados en recipientes apropiados, etiquételos y almacénelos de acuerdo con las condiciones de luz y temperatura del esquema del cuadro 7. El tiempo de almacenamiento será el elegido teniendo en cuenta la práctica analítica aplicada, idealmente hasta que se observen los primeros fenómenos de degradación durante la identificación o la cuantificación. Si no se observa degradación durante el estudio de estabilidad, la duración de almacenamiento del estudio de estabilidad será igual a la duración del período máximo de almacenamiento de la solución. |
|
4. |
Calcule la concentración del analito (o los analitos) en cada alícuota en comparación con la concentración del analito en la solución recién preparada, siguiendo la siguiente fórmula:
analito restante (%) = Ci × 100/Crecién preparada Ci = concentración en el momento i Crecién preparada = concentración de la solución recién preparada El valor medio de cinco soluciones idénticas que se hayan almacenado no diferirá en más de un 15 % del valor medio de cinco soluciones idénticas recién preparadas. El valor medio de las cinco soluciones recién preparadas se utilizará como base para calcular la diferencia porcentual. Cuadro 7 Procedimiento para la determinación de la estabilidad del analito en la solución
|
2.5.2. Determinación de la estabilidad del analito (o los analitos) en la matriz
|
1. |
Utilice muestras reales siempre que sea posible. Cuando no disponga de una matriz real, utilice una matriz en blanco enriquecida con el analito. |
|
2. |
Si dispone de matriz real, determine la concentración en la matriz mientras aún está fresca. Almacene otras alícuotas de la matriz real homogeneizado a-20 °C, o menos si es necesario, y determine las concentraciones del analito mientras la muestra se conserve en el laboratorio. |
|
3. |
Si no dispone de matriz real, tome una matriz en blanco y homogenéicela. Divida la matriz en cinco alícuotas. Enriquezca cada alícuota con el analito, preparado de preferencia en una pequeña cantidad de solución acuosa. Analice una alícuota inmediatamente. Almacene las alícuotas restantes a una temperatura mínima de-20 °C, o inferior si es necesario, y analícelas tras un almacenamiento a corto, medio y largo plazo, teniendo en cuenta los métodos analíticos aplicados. |
|
4. |
Registre el tiempo máximo aceptable y las condiciones óptimas de almacenamiento.
El valor medio de cinco soluciones idénticas que se hayan almacenado no diferirá en más de la reproducibilidad intralaboratorio del método con respecto al valor medio de cinco soluciones idénticas recién preparadas. El valor medio de las cinco soluciones recién preparadas se utilizará como base para calcular la diferencia porcentual. |
2.6. Límite de decisión para confirmación (CCα)
El CCα se determinará para los métodos de confirmación. El CCα se establecerá en condiciones que cumplan los requisitos de identificación o identificación más cuantificación definidos en los «Criterios de funcionamiento y otros requisitos de los métodos analíticos» establecidos en el capítulo 1.
Para el control de conformidad de las muestras, la incertidumbre estándar combinada de medida ya ha sido tenida en cuenta en el valor del CCα (límite de decisión para confirmación).
|
1. |
En el caso de sustancias farmacológicamente activas no autorizadas o prohibidas, el CCα se calculará como sigue:
El método 2 para el cálculo del CCα solo podrá utilizarse hasta el 1 de enero de 2026 en el caso de métodos validados antes de la fecha de entrada en vigor del presente Reglamento. Para los métodos validados después de la entrada en vigor del presente Reglamento, solo se utilizarán los métodos 1 o 3. |
|
2. |
En el caso de sustancias autorizadas, el CCα se calculará como sigue:
|
2.7. Capacidad de detección del cribado (CCβ)
Se determinará la CCβ para los métodos de cribado. La CCβ se establecerá tal como se define en los «Criterios de funcionamiento y otros requisitos de los métodos analíticos» establecidos en el capítulo 1 del presente anexo, y de conformidad con los requisitos establecidos en el cuadro 5. Sin embargo, no es necesario aplicar a los métodos de cribado todos los requisitos de identificación (véanse los puntos 1.2.3, 1.2.4 y 1.2.5).
|
1. |
En el caso de sustancias farmacológicamente activas no autorizadas o prohibidas, se garantizará un error β máximo del 5 %. La CCβ se calculará de la manera siguiente:
|
|
2. |
En el caso de sustancias autorizadas, se garantizará un error β máximo del 5 %. La CCβ se calculará de la manera siguiente:
En el caso de las sustancias farmacológicamente activas para las que se haya establecido un LMR de la suma de diferentes sustancias, la CCβ de la sustancia con la concentración más alta de la muestra se utilizará como CCβ para evaluar la suma de las sustancias de la muestra medida. |
2.8. Curvas de calibración
Cuando se utilizan curvas de calibración para la cuantificación es preciso:
|
1) |
emplear al menos cinco niveles preferiblemente equidistantes (incluido el nivel cero) en la construcción de la curva; |
|
2) |
describir el intervalo de trabajo de la curva; |
|
3) |
describir la fórmula matemática de la curva y la bondad del ajuste de los datos (coeficiente de determinación R2) a la curva; |
|
4) |
describir los márgenes de aceptabilidad de los parámetros de la curva. |
Para las curvas de calibración basadas en una disolución patrón, en patrones similares a la matriz o en patrones de matriz enriquecida, se indicarán los márgenes aceptables para los parámetros de la curva de calibración, que pueden variar de una serie a otra.
2.9. Recuperación absoluta
La recuperación absoluta del método se determinará cuando no se utilice ningún patrón interno o ninguna calibración de matriz enriquecida.
Cuando se cumplan los requisitos de veracidad establecidos en el cuadro 1, podrá utilizarse un factor de corrección fijo. En caso contrario, se aplicará el factor de recuperación obtenido para ese lote específico. Como alternativa, se utilizará el procedimiento de adición de patrón (16) o un patrón interno, en lugar de utilizar un factor de corrección de recuperación.
La recuperación absoluta se calculará para al menos seis lotes representativos de matriz.
Una alícuota de matriz en blanco se enriquecerá con el analito antes de la extracción, y una segunda alícuota de matriz en blanco se enriquecerá tras la preparación de la muestra a un nivel de concentración adecuado, y se determinará la concentración del analito.
La recuperación se calculará de la manera siguiente:
Rec (analito) = (patrón de matriz enriquecida)/(patrón similar a la matriz) × 100
2.10. Efecto matriz relativo
El efecto matriz relativo se determinará en todos los casos. Esto puede hacerse como parte de la validación o en experimentos separados. Se efectuará el cálculo del efecto matriz relativo para al menos 20 lotes en blanco diferentes (matrices/especies), según el ámbito de aplicación del método, como, por ejemplo, diferentes especies que deben cubrirse.
Tras la extracción, la matriz en blanco debe enriquecerse con el analito al VRI, el LMR o el CM, y debe analizarse junto con una disolución pura del analito.
El efecto matriz relativo o factor matriz (FM) se calcula como sigue:


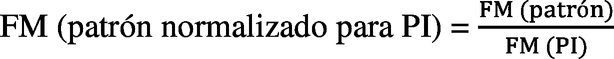
PI: patrón interno
PSM: patrón similar a la matriz
El coeficiente de variación no superará el 20 % para el FM (patrón normalizado para PI).
CAPÍTULO 3
CONTROL DE CALIDAD DURANTE ANÁLISIS DE RUTINA. VERIFICACIÓN CONTINUA DEL FUNCIONAMIENTO DEL MÉTODO
Deberán cumplirse los requisitos para garantizar la calidad de los resultados analíticos del capítulo 7.7 de la norma ISO/IEC 17025:2017 (17).
Durante los análisis de rutina, el análisis de los materiales de referencia certificados (MRC) es la opción preferible para aportar pruebas del funcionamiento del método. Dado que rara vez se dispone de MRC que contengan los analitos pertinentes en los niveles de concentración requeridos, también pueden utilizarse como alternativa materiales de referencia suministrados y caracterizados por los laboratorios de referencia de la Unión Europea o por laboratorios que posean una acreditación ISO/IEC 17043: 2010 (18). Otra posibilidad es utilizar materiales de referencia internos que se controlen periódicamente.
La verificación continua del funcionamiento del método durante análisis de rutina debe llevarse a cabo en la fase de cribado y en la fase de confirmación.
|
1. |
Fase de cribado: Para cada serie (tanda) de análisis realizados, se analizará simultáneamente un conjunto de las siguientes muestras de control de calidad:
|
|
2. |
Fase de confirmación: Para cada serie (tanda) de análisis realizados, se analizará simultáneamente un conjunto de las siguientes muestras de control de calidad:
|
Se recomienda el siguiente orden para las muestras de control de calidad: muestra de control de la adecuación del instrumento al sistema, muestra de control conforme, muestras pendientes de confirmación, muestra de control conforme de nuevo y muestra de control de calidad enriquecida (muestras de control no conformes).
En el caso de los métodos cuantitativos, con cada tanda de muestras oficiales se analizará y medirá una curva de calibración antes o después de las muestras enumeradas en el párrafo anterior.
Cuando sea posible, se evaluará la veracidad (a partir de muestras enriquecidas) de todos los analitos de las muestras de control no conformes, mediante gráficos de control de calidad de conformidad con el capítulo 7.7 de la norma ISO/IEC 17025: 2017. Si ello requiere un número desproporcionado de determinaciones de veracidad, el número de analitos podrá reducirse a varios analitos representativos.
CAPÍTULO 4
AMPLIACIÓN DEL ÁMBITO DE APLICACIÓN VALIDADO DE UN MÉTODO VALIDADO CON ANTERIORIDAD
A veces es necesario ampliar el ámbito de aplicación de un método validado globalmente con anterioridad. En estos casos, la ampliación del ámbito de aplicación debe llevarse a cabo de manera eficiente y sólida desde el punto de vista analítico. Esto puede lograrse realizando una validación en un número reducido de muestras (por ejemplo, la mitad de muestras), en comparación con una validación completa.
No obstante, el tipo y el número de modificaciones que deben validarse en un único sistema de validación reducido siempre se basará en conocimientos especializados y experiencias anteriores. Por ejemplo, un cambio en la técnica de detección requeriría una validación completa en cualquier caso.
En general, para garantizar que el método siga siendo válido, su funcionamiento será objeto de un seguimiento continuo y se comparará con los parámetros de validación obtenidos inicialmente. Idealmente, este control continuo del funcionamiento del método está diseñado de manera que los datos que faltan para una validación completa puedan recogerse con el tiempo (por ejemplo, con algunos puntos de datos procedentes de muestras de control de calidad en cada serie analítica).
4.1. Ampliaciones de los métodos relativas al rango de concentraciones
Debido a cambios en los LMR, los CM y los VRI, puede resultar necesario ajustar el rango de concentraciones para el que está validado un método. En tal caso, es aceptable la aplicación de un sistema de validación reducido.
Las curvas de calibración del intervalo modificado deben prepararse según el procedimiento validado. Deben analizarse diferentes lotes enriquecidos a diferentes niveles de concentración (véanse los puntos 2.2.1 y 2.2.2). La veracidad, la repetibilidad y la reproducibilidad intralaboratorio/precisión intermedia deben situarse dentro de un intervalo aceptable en comparación con los del método validado originalmente. Cuando proceda, debe volver a calcularse la CCβ (métodos de cribado) y el CCα (métodos de confirmación).
4.2. Ampliaciones de los métodos relativas a sustancias adicionales
En general, ampliar el método a otros compuestos solo es posible si se trata de analitos con estructura y características similares a las de los analitos que ya están incluidos en el método analítico. En tal caso, es aceptable la aplicación de un sistema de validación reducido. De la misma manera, no se permiten divergencias con respecto a la descripción del método.
Las curvas de calibración de las nuevas sustancias deben prepararse según el procedimiento validado. Deben analizarse diferentes tandas de materiales matriz enriquecidos a diferentes niveles de concentración (véanse los puntos 2.2.1 y 2.2.2). La veracidad, la repetibilidad y la reproducibilidad intralaboratorio/precisión intermedia deben situarse dentro de un intervalo comparable a los de los demás analitos del método validado originalmente, y deben ajustarse a los requisitos establecidos en el punto 1.2.2. Deben calcularse la CCβ (métodos de cribado) y el CCα (métodos de confirmación) para los nuevos analitos.
4.3. Ampliaciones de los métodos relativas a matrices o especies
La decisión de incluir nuevas matrices o especies en un método analítico ya validado se tomará siempre caso por caso, basándose en el conocimiento y la experiencia adquiridos hasta el momento con el método y en experimentos preliminares que evalúen posibles efectos matriz e interferencias. En general, esto solo será posible en el caso de matrices que presenten propiedades similares y de analitos no críticos (estabilidad, detectabilidad).
Las curvas de calibración (patrón o matriz) deben prepararse según el procedimiento validado. Deben analizarse diferentes tandas de material matriz enriquecido a diferentes niveles de concentración (véanse los puntos 2.2.1 y 2.2.2). La veracidad, la repetibilidad y la reproducibilidad intralaboratorio/precisión intermedia deben situarse dentro de un intervalo aceptable en comparación con las del método validado originalmente, y deben ajustarse a los requisitos establecidos en el punto 1.2.2. En función del método de validación, puede ser necesario volver a calcular la CCβ (métodos de cribado) o el CCα (métodos de confirmación).
Si los resultados no se encuentran dentro de un intervalo aceptable en comparación con los valores de la matriz original, será necesaria una nueva validación completa para determinar los parámetros específicos de funcionamiento de la matriz o especie.
En aquellos casos en los que el LMR de una sustancia específica difiera en determinadas matrices, probablemente resultará muy difícil adaptar el ámbito de aplicación a la nueva matriz o especie y a la nueva concentración, ya que, en este caso, deben tenerse en cuenta dos modificaciones. En estos casos se recomienda realizar una validación completa.
(1) La cocromatografía es un procedimiento en el que se divide el extracto muestra en dos partes antes de las etapas cromatográficas. Una parte se cromatografía tal cual. La otra parte se mezcla con el analito patrón que debe medirse y se cromatografía también esta mezcla. La cantidad de analito patrón añadido debe ser similar a la cantidad de analito estimada en el extracto. La cocromatografía se usa para mejorar la identificación de un analito por métodos cromatográficos, especialmente cuando no puede utilizarse un patrón interno adecuado.
(*) * El CV (%) presentado es una orientación y debe ser tan bajo como sea razonablemente posible.
(a) a No se obtiene ningún punto de identificación adicional para la selección del ion precursor, si este es el mismo ion (o un aducto o isótopo) que el ion HRMS registrado en barrido completo.
(#) Si se dispone de datos de estabilidad de los analitos de una matriz procedentes de publicaciones científicas o de otro laboratorio, no es necesario que el laboratorio en cuestión determine de nuevo estos datos. Sin embargo, las referencia a los datos disponibles sobre la estabilidad de los analitos en disolución solo son aceptables si se aplican condiciones idénticas.
(*) Pertinentes para que los métodos de MS demuestren mediante la validación que cumplen los requisitos aplicables a las características de funcionamiento. El efecto matriz relativo del método se determinará cuando dicho efecto no se haya evaluado durante el proceso de validación. La recuperación absoluta del método se determinará cuando no se utilice ningún patrón interno o ninguna calibración de matriz enriquecida.
(2) ISO 5725-4:2020 Exactitud (veracidad y precisión) de resultados y métodos de medición. Parte 4: Métodos básicos para la determinación de la veracidad de un método de medición normalizado (artículo 3).
(3) Cuando, para una sustancia farmacológicamente activa no autorizada, la validación de una concentración de 0,5 veces el VRI no sea razonablemente posible, la concentración de 0,5 veces el VRI puede sustituirse por la concentración más baja que pueda alcanzarse razonablemente entre 0,5 veces y 1,0 veces el VRI.
(4) Cuando, para una determinada sustancia farmacológicamente activa, la validación de una concentración de 0,1 veces el LMR no sea razonablemente posible, la concentración de 0,1 veces el LRM puede sustituirse por la concentración más baja que pueda alcanzarse razonablemente entre 0,1 veces y 0,5 veces el LMR.
(5) Cuando, para una sustancia farmacológicamente activa no autorizada, la validación de una concentración de 0,5 veces el VRI no sea razonablemente posible, la concentración de 0,5 veces el VRI puede sustituirse por la concentración más baja que pueda alcanzarse razonablemente entre 0,5 veces y 1,0 veces el VRI.
(6) Cuando, para una determinada sustancia farmacológicamente activa, la validación de una concentración de 0,1 veces el LMR no sea razonablemente posible, la concentración de 0,1 veces el LRM puede sustituirse por la concentración más baja que pueda alcanzarse razonablemente entre 0,1 veces y 0,5 veces el LMR.
(7) ISO 5725-2:2019 Exactitud (veracidad y precisión) de resultados y métodos de medición. Parte 2: Método básico para la determinación de la repetibilidad y la reproducibilidad de un método de medición normal (artículo 3).
(8) ISO 11843-1:1997 Capacidad de detección. Parte 1: Términos y definiciones
(9) «Directrices sobre la estimación de la incertidumbre de los resultados» (CAC/GL 59-2006) de la Comisión del Codex Alimentarius (Organización de las naciones Unidas para la Alimentación y la Agricultura/Organización Mundial de la Salud).
(10) Los cambios de las condiciones experimentales a que se hace referencia pueden consistir en cambios en los materiales de la muestra, los analitos, las condiciones de almacenamiento o las condiciones ambientales o de preparación de la muestra. Deberá indicarse cualquier variación de las condiciones experimentales susceptibles de fluctuación en la práctica (por ejemplo, estabilidad de los reactivos, composición de la muestra, pH o temperatura) que puedan afectar a los resultados analíticos.
(11) Jülicher, B., Gowik, P., Uhlig, S. (1998). Assessment of detection methods in trace analysis by means of a statistically based in-house validation concept. Analyst, 123, 173-179.
(12) IUPAC (1995). Protocol for the design, conduct and interpretation of method-performance studies. Pure & Applied Chem, 67(2), 331-343.
(13) Gowik, P., Jülicher, B., Uhlig, S. (1998). Multi-residue method for non-steroidal anti-inflammatory drugs in plasma using high performance liquid chromatography-photodiode-array detection. Method description and comprehensive in-house validation. J. Chromatogr., 716(1-2), 221-232.
(14) ISO 11843-1:1997 Capacidad de detección. Parte 1: Términos y definiciones
(15) Reglamento de Ejecución (UE) 2018/470 de la Comisión, de 21 de marzo de 2018, que establece normas detalladas sobre el límite máximo de residuos a tener en cuenta a efectos de control de alimentos derivados de animales tratados en la UE según lo dispuesto en el artículo 11 de la Directiva 2001/82/CE (DO L 79 de 22.3.2018, p. 16).
(16) La cantidad de analito patrón añadida puede ser, por ejemplo, entre dos y cinco veces superior al contenido estimado de analito en la muestra. Este procedimiento está diseñado para determinar el contenido de un analito en una muestra, teniendo en cuenta la recuperación del procedimiento analítico.
(17) Norma ISO/IEC 17025: 2017 Requisitos generales para la competencia de los laboratorios de ensayo y calibración (capítulo 7.7).
(18) ISO/IEC 17043:2010 Evaluación de la conformidad. Requisitos generales para los ensayos de aptitud.
ANEXO II
PROCEDIMIENTOS DE MUESTREO Y TRATAMIENTO DE LAS MUESTRAS OFICIALES
1. Cantidad de muestras
Las cantidades mínimas de muestras se definirán en el programa nacional de control de residuos. Dichas cantidades serán suficientes para que los laboratorios autorizados puedan efectuar los procedimientos analíticos necesarios para completar el cribado y los análisis de confirmación. Concretamente en el caso de las aves de corral, la acuicultura, los conejos, la caza de cría, los reptiles y los insectos, las muestras se compondrán de uno o varios animales, dependiendo de los requisitos de los métodos analíticos. En el caso de los huevos, el tamaño de la muestra será de al menos 12 huevos, según los métodos analíticos utilizados. Si deben analizarse varias categorías de sustancias en una única muestra con diferentes métodos analíticos, el tamaño de la muestra se incrementará en consecuencia.
2. División en submuestras
Cada muestra se dividirá al menos en dos submuestras equivalentes, de forma que sea posible realizar el procedimiento analítico completo de cada una, excepto si es técnicamente imposible o no lo exige la normativa nacional. La subdivisión podrá efectuarse en el lugar donde se recojan las muestras o en el laboratorio.
3. Trazabilidad
Cada muestra se tomará de forma que sea siempre posible obtener su trazabilidad hasta la explotación de origen y el animal o lote de animales, cuando proceda. En particular, las muestras de leche pueden tomarse, según decidan los Estados miembros, en cualquiera de los lugares siguientes:
|
1. |
en la explotación, en las cisternas colectoras; |
|
2. |
en la industria del sector lácteo, antes de descargar la leche. |
4. Recipientes para muestras
Las muestras se recogerán en recipientes adecuados para mantener la integridad y la trazabilidad de las muestras. En particular, los recipientes deberán evitar la sustitución, la contaminación cruzada y la degradación, y deberán estar provistos de un precinto oficial.
5. Informe de muestreo
Después de cada muestreo se elaborará un informe.
El inspector incluirá en el informe de muestreo al menos los datos siguientes:
|
1. |
dirección de las autoridades competentes; |
|
2. |
nombre y apellidos del inspector o código de identificación; |
|
3. |
número de código oficial de la muestra; |
|
4. |
fecha del muestreo; |
|
5. |
nombre, apellidos y dirección del propietario o de la persona responsable de los animales o de los productos de origen animal; |
|
6. |
nombre y dirección de la explotación de origen del animal (en caso de que el muestreo se haya realizado en la explotación); |
|
7. |
número de registro del establecimiento o número del matadero; |
|
8. |
identificación del animal o del producto; |
|
9. |
especie animal; |
|
10. |
matriz de la muestra; |
|
11. |
cuando proceda, medicación administrada en las cuatro semanas anteriores al muestreo (cuando el muestreo se haya realizado en la explotación); |
|
12. |
sustancia o grupos de sustancias objeto de examen; |
|
13. |
observaciones particulares. |
En función del procedimiento de muestreo, se facilitarán copias impresas o electrónicas del informe. El informe de muestreo y sus copias se cumplimentarán de forma que se garantice su autenticidad y validez jurídica, lo que puede requerir que estos documentos estén firmados por el inspector. Cuando el muestreo se realice en la explotación, podrá invitarse al ganadero o a su suplente a firmar el informe de muestreo original.
El original del informe de muestreo quedará en poder de la autoridad competente, que impedirá que puedan acceder a él personas no autorizadas.
En caso necesario, el ganadero o propietario de la explotación podrá ser informado de la toma de muestras.
6. Informe de muestreo para el laboratorio
El informe de muestreo para el laboratorio establecido por las autoridades competentes cumplirá los requisitos establecidos en el capítulo 7 de la norma ISO/IEC 17025:2017 (1) y contendrá al menos la información siguiente:
|
1. |
dirección de las autoridades competentes o de los organismos designados; |
|
2. |
nombre y apellidos del inspector o código de identificación, |
|
3. |
número de código oficial de la muestra; |
|
4. |
fecha del muestreo; |
|
5. |
especie animal; |
|
6. |
matriz de la muestra; |
|
7. |
sustancia o grupo de sustancias objeto de examen; |
|
8. |
observaciones particulares. |
El informe de muestreo para el laboratorio acompañará a la muestra cuando se envíe al laboratorio.
7. Transporte y almacenamiento
Los programas de control de residuos especificarán las condiciones adecuadas de almacenamiento y transporte de cada combinación de analito/matriz para garantizar la estabilidad del analito y la integridad de la muestra. El transporte durará lo menos posible y la temperatura durante el transporte deberá ser la adecuada para garantizar la estabilidad del analito.
Se prestará una atención especial a las cajas de transporte, la temperatura y el plazo de entrega al laboratorio responsable.
En caso de que se incumpla cualquier requisito del programa de control, el laboratorio informará de ello inmediatamente a la autoridad competente.
(1) Norma ISO/IEC 17025: 2017 Requisitos generales para la competencia de los laboratorios de ensayo y calibración (capítulo 7.7).
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/110 |
REGLAMENTO DE EJECUCIÓN (UE) 2021/809 DE LA COMISIÓN
de 20 de mayo de 2021
sobre la no aprobación del extracto fermentado de hojas de Symphytum officinale L. (consuelda) como sustancia básica de conformidad con el Reglamento (CE) n.o 1107/2009 del Parlamento Europeo y del Consejo, relativo a la comercialización de productos fitosanitarios
(Texto pertinente a efectos del EEE)
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (CE) n.o 1107/2009 del Parlamento Europeo y del Consejo, de 21 de octubre de 2009, relativo a la comercialización de productos fitosanitarios y por el que se derogan las Directivas 79/117/CEE y 91/414/CEE del Consejo (1), y en particular su artículo 13, apartado 2, en relación con su artículo 23, apartado 5,
Considerando lo siguiente:
|
(1) |
El 22 de enero de 2015 la Comisión recibió una solicitud de la empresa Greenprotech para la aprobación de la «maceración de consuelda» como sustancia básica para su uso como repelente de insectos y elicitor vegetal en árboles frutales, hierba y hortalizas. Dicha solicitud iba acompañada de la información que se exige en el artículo 23, apartado 3, párrafo segundo, del Reglamento (CE) n.o 1107/2009. |
|
(2) |
La Comisión pidió asistencia científica a la Autoridad Europea de Seguridad Alimentaria («la Autoridad»). El 28 de noviembre de 2019 la Autoridad presentó a la Comisión un informe técnico sobre la «maceración de consuelda» (2). Sobre la base de dicho informe técnico y de la documentación facilitada por el solicitante, procede definir que el ámbito de aplicación de la solicitud abarca la sustancia activa «extracto fermentado de hojas de Symphytum officinale L. (consuelda)». |
|
(3) |
La información facilitada por el solicitante sobre el extracto fermentado de hojas de Symphytum officinale L. (consuelda) no era suficiente para demostrar que esta sustancia cumple los criterios para ser considerada un producto alimenticio tal como se define en el artículo 2 del Reglamento (CE) n.o 178/2002 del Parlamento Europeo y del Consejo (3). |
|
(4) |
La Autoridad llegó a la conclusión de que la especificación del extracto fermentado de hojas de Symphytum officinale L. (consuelda) no está bien definida. La Autoridad también indicó que se sabe que la consuelda contiene componentes genotóxicos y carcinógenos, y no se disponía de información sobre la concentración de compuestos genotóxicos y carcinógenos en el extracto fermentado de hojas de Symphytum officinale L. Por consiguiente, no fue posible llegar a la conclusión de que el extracto fermentado de hojas de Symphytum officinale L. no deba considerarse una sustancia preocupante con arreglo a lo dispuesto en el artículo 23, apartado 1, letra a), del Reglamento (CE) n.o 1107/2009. |
|
(5) |
Además, la información disponible sobre el extracto fermentado de hojas de Symphytum officinale L. no permitió a la Autoridad finalizar una evaluación del riesgo de exposición no alimentaria ni la evaluación del riesgo para los consumidores. Por añadidura, no se disponía de suficiente información en relación con la exposición ambiental y los riesgos para los organismos no objetivo. |
|
(6) |
Tampoco se disponía de ninguna evaluación pertinente realizada de conformidad con otras normas de la Unión, tal como se menciona en el artículo 23, apartado 2, del Reglamento (CE) n.o 1107/2009. |
|
(7) |
La Comisión presentó el informe de revisión (4) y un proyecto de Reglamento al Comité Permanente de Vegetales, Animales, Alimentos y Piensos el 19 de mayo de 2020 y el 26 de enero de 2021, respectivamente, y los finalizó para la reunión de dicho Comité de 24 de marzo de 2021. |
|
(8) |
La Comisión invitó al solicitante a presentar observaciones sobre el informe técnico de la Autoridad y sobre el proyecto de informe de revisión de la Comisión. El solicitante presentó sus observaciones, que se examinaron con detenimiento. |
|
(9) |
Sin embargo, pese a los argumentos presentados por el solicitante, no pudieron descartarse los problemas relacionados con la sustancia. |
|
(10) |
Por consiguiente, no se ha demostrado el cumplimiento de los requisitos establecidos en el artículo 23 del Reglamento (CE) n.o 1107/2009. Por tanto, procede no aprobar el extracto fermentado de hojas de Symphytum officinale L. (consuelda) como sustancia básica. |
|
(11) |
El presente Reglamento no excluye la presentación de una nueva solicitud de aprobación del extracto fermentado de hojas de Symphytum officinale L. (consuelda) como sustancia básica de conformidad con el artículo 23, apartado 3, del Reglamento (CE) n.o 1107/2009, ni de una solicitud de aprobación como sustancia activa de conformidad con el artículo 7 de dicho Reglamento. |
|
(12) |
Las medidas previstas en el presente Reglamento se ajustan al dictamen del Comité Permanente de Vegetales, Animales, Alimentos y Piensos. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
No se aprueba como sustancia básica la sustancia extracto fermentado de hojas de Symphytum officinale L. (consuelda).
Artículo 2
El presente Reglamento entrará en vigor a los veinte días de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 20 de mayo de 2021.
Por la Comisión
La Presidenta
Ursula VON DER LEYEN
(1) DO L 309 de 24.11.2009, p. 1.
(2) EFSA (Autoridad Europea de Seguridad Alimentaria), 2019. Technical report on the outcome of the consultation with Member States and EFSA on the basic substance application for approval of Comfrey steeping to be used in plant protection as an insect repellent and plant elicitor in fruit trees, grass and vegetables (Informe técnico sobre el resultado de la consulta con los Estados miembros y la EFSA sobre la solicitud de aprobación de la maceración de consuelda para su uso fitosanitario como repelente de insectos y elicitor vegetal en árboles frutales, hierba y hortalizas). Publicación de referencia de la EFSA 2019:EN-1753. 64 páginas, doi:10.2903/sp.efsa.2019.EN-1753.
(3) Reglamento (CE) n.o 178/2002 del Parlamento Europeo y del Consejo, de 28 de enero de 2002, por el que se establecen los principios y los requisitos generales de la legislación alimentaria, se crea la Autoridad Europea de Seguridad Alimentaria y se fijan procedimientos relativos a la seguridad alimentaria (DO L 31 de 1.2.2002, p. 1).
(4) https://ec.europa.eu/food/plant/pesticides/eu-pesticides-db_en
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/112 |
REGLAMENTO DE EJECUCIÓN (UE) 2021/810 DE LA COMISIÓN
de 20 de mayo de 2021
por el que se modifica el Reglamento de Ejecución (UE) 2021/2021/808 en lo que respecta a las disposiciones transitorias para determinadas sustancias que figuran en el anexo II de la Decisión 2002/657/CE
(Texto pertinente a efectos del EEE)
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (UE) 2017/625 del Parlamento Europeo y del Consejo, de 15 de marzo de 2017, relativo a los controles y otras actividades oficiales realizados para garantizar la aplicación de la legislación sobre alimentos y piensos, y de las normas sobre salud y bienestar de los animales, sanidad vegetal y productos fitosanitarios, y por el que se modifican los Reglamentos (CE) n.o 999/2001, (CE) n.o 396/2005, (CE) n.o 1069/2009, (CE) n.o 1107/2009, (UE) n.o 1151/2012, (UE) n.o 652/2014, (UE) 2016/429 y (UE) 2016/2031 del Parlamento Europeo y del Consejo, los Reglamentos (CE) n.o 1/2005 y (CE) n.o 1099/2009 del Consejo, y las Directivas 98/58/CE, 1999/74/CE, 2007/43/CE, 2008/119/CE y 2008/120/CE del Consejo, y por el que se derogan los Reglamentos (CE) n.o 854/2004 y (CE) n.o 882/2004 del Parlamento Europeo y del Consejo, las Directivas 89/608/CEE, 89/662/CEE, 90/425/CEE, 91/496/CEE, 96/23/CE, 96/93/CE y 97/78/CE del Consejo y la Decisión 92/438/CEE del Consejo (Reglamento sobre controles oficiales) (1), y en particular su artículo 34, apartado 6,
Considerando lo siguiente:
|
(1) |
El Reglamento de Ejecución (UE) 2021/2021/808 de la Comisión (2) deroga, entre otros actos, la Decisión 2002/657/CE de la Comisión (3). El artículo 4 de dicha Decisión, en relación con su anexo II, establece los límites mínimos de funcionamiento exigidos para el cloranfenicol, los metabolitos de nitrofuranos, el acetato de medroxiprogesterona y el verde de malaquita, que son sustancias farmacológicamente activas, en determinadas matrices. |
|
(2) |
El artículo 8 del Reglamento (UE) 2019/1871 de la Comisión (4) establece las disposiciones transitorias relativas a los valores de referencia para las sustancias farmacológicamente activas que están prohibidas. Los límites mínimos de funcionamiento exigidos para el cloranfenicol, los metabolitos de nitrofuranos y la suma de verde de malaquita y de verde de leucomalaquita que figuran en el anexo II de la Decisión 2002/657/CE deben ser aplicables como valores de referencia para los alimentos de origen animal importados de terceros países y los alimentos de origen animal producidos en la Unión hasta el 27 de noviembre de 2022. |
|
(3) |
Por tanto, a efectos de lo dispuesto en el artículo 8 del Reglamento (UE) 2019/1871, el anexo II de la Decisión 2002/657/CE debe seguir siendo aplicable hasta el 27 de noviembre de 2022. |
|
(4) |
A fin de mantener la continuidad, el presente Reglamento debe aplicarse a partir de la misma fecha que el Reglamento de Ejecución (UE) 2021/2021/808. |
|
(5) |
Las medidas previstas en el presente Reglamento se ajustan al dictamen del Comité Permanente de Vegetales, Animales, Alimentos y Piensos. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
El artículo 7 del Reglamento de Ejecución (UE) 2021/2021/808 se sustituye por el texto siguiente:
«Artículo 7
Derogaciones y medidas transitorias
Quedan derogadas las Decisiones 2002/657/CE y 98/179/CE a partir de la fecha de entrada en vigor del presente Reglamento.
No obstante, hasta el 10 de junio de 2026, los requisitos establecidos en los puntos 2 y 3 del anexo I de la Decisión 2002/657/CE seguirán aplicándose a los métodos que hayan sido validados antes de la fecha de entrada en vigor del presente Reglamento.
A efectos de lo dispuesto en el artículo 8, párrafo segundo, del Reglamento (UE) 2019/1871, el anexo II de la Decisión 2002/657/CE seguirá siendo aplicable hasta el 27 de noviembre de 2022.».
Artículo 2
El presente Reglamento entrará en vigor a los veinte días de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 20 de mayo de 2021.
Por la Comisión
La Presidenta
Ursula VON DER LEYEN
(1) DO L 95 de 7.4.2017, p. 1.
(2) Reglamento de Ejecución (UE) 2021/2021/808 de la Comisión, de 22 de marzo de 2021, relativo al funcionamiento de los métodos analíticos para los residuos de sustancias farmacológicamente activas utilizadas en animales productores de alimentos y a la interpretación de resultados, así como a los métodos que deben utilizarse para el muestreo, y por el que se derogan las Decisiones 2002/657/CE y 98/179/CE (véase la página 84 del presente Diario Oficial).
(3) Decisión 2002/657/CE de la Comisión, de 14 de agosto de 2002, por la que se aplica la Directiva 96/23/CE del Consejo en cuanto al funcionamiento de los métodos analíticos y la interpretación de los resultados (DO L 221 de 17.8.2002, p. 8).
(4) Reglamento (UE) 2019/1871 de la Comisión, de 7 de noviembre de 2019, relativo a los valores de referencia para las sustancias farmacológicamente activas no autorizadas presentes en los alimentos de origen animal y por el que se deroga la Decisión 2005/34/CE (DO L 289 de 8.11.2019, p. 41).
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/114 |
REGLAMENTO DE EJECUCIÓN (UE) 2021/811 DE LA COMISIÓN
de 20 de mayo de 2021
que modifica el anexo I del Reglamento de Ejecución (UE) 2021/605, por el que se establecen medidas especiales de control de la peste porcina africana
(Texto pertinente a efectos del EEE)
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (UE) 2016/429 del Parlamento Europeo y del Consejo, de 9 de marzo de 2016, relativo a las enfermedades transmisibles de los animales y por el que se modifican o derogan algunos actos en materia de sanidad animal («Legislación sobre sanidad animal») (1), y en particular su artículo 71, apartado 3,
Considerando lo siguiente:
|
(1) |
La peste porcina africana es una enfermedad vírica infecciosa que afecta a los porcinos silvestres y en cautividad y puede tener graves repercusiones en la población animal afectada y en la rentabilidad de la ganadería, perturbando los desplazamientos de las partidas de esos animales y sus productos dentro de la Unión y las exportaciones a terceros países. |
|
(2) |
El Reglamento de Ejecución (UE) 2021/605 de la Comisión (2) se adoptó en el marco del Reglamento (UE) 2016/429, y en él se establecen medidas especiales de control de la peste porcina africana que los Estados miembros que figuran en su anexo I deben aplicar durante un período de tiempo limitado en las zonas restringidas enumeradas en dicho anexo. Las zonas enumeradas como zonas restringidas I, II y III en el anexo I del Reglamento de Ejecución (UE) 2021/605 se basan en la situación epidemiológica de la peste porcina africana en la Unión. El anexo I del Reglamento de Ejecución (UE) 2021/605 se modificó mediante el Reglamento de Ejecución (UE) 2021/687 de la Comisión (3), a fin de asegurar la continuidad y la coherencia de las medidas especiales de control de la peste porcina africana en la Unión a raíz de la expiración de la Decisión de Ejecución 2014/709/UE de la Comisión (4), y el inicio, el 21 de abril de 2021, de la aplicación del Reglamento de Ejecución (UE) 2021/605. |
|
(3) |
Cualquier modificación de las zonas restringidas I, II y III del anexo I del Reglamento de Ejecución (UE) 2021/605 debe basarse en la situación epidemiológica por lo que se refiere a la peste porcina africana en las zonas afectadas por esta enfermedad y en la situación epidemiológica general de la peste porcina africana en los Estados miembros afectados; en el nivel de riesgo de propagación de la enfermedad; en principios y criterios científicos para definir geográficamente la zonificación debida a la peste porcina africana; y en las directrices de la Unión acordadas con los Estados miembros y el Comité Permanente de Vegetales, Animales, Alimentos y Piensos y publicadas en el sitio web de la Comisión (5). Dichas modificaciones también deben tener en cuenta las normas internacionales, como el Código Sanitario para los Animales Terrestres (6) de la Organización Mundial de Sanidad Animal (Código de la OIE) y las justificaciones de zonificación facilitadas por las autoridades competentes de los Estados miembros afectados. |
|
(4) |
Desde la fecha de adopción del Reglamento de Ejecución (UE) 2021/687, ha habido nuevos brotes de peste porcina africana en porcinos silvestres en Eslovaquia y Polonia. |
|
(5) |
En abril de 2021 se detectó un caso de peste porcina africana en un porcino silvestre en la provincia eslovaca de Rimavska Sobota, en una zona que figura actualmente como zona restringida I en el anexo I del Reglamento de Ejecución (UE) 2021/605. Este nuevo brote de peste porcina africana en un porcino silvestre supone un aumento del nivel de riesgo que debe reflejarse en dicho anexo. Por consiguiente, esta zona de Eslovaquia que figura actualmente como zona restringida I en el mencionado anexo, afectada por el reciente brote de peste porcina africana mencionado, debe figurar ahora en dicho anexo como zona restringida II en vez de como zona restringida I. |
|
(6) |
En mayo de 2021 se detectó un caso de peste porcina africana en un porcino silvestre en la provincia polaca de szamotulski, en una zona que figura actualmente como zona restringida I en el anexo I del Reglamento de Ejecución (UE) 2021/605. Este nuevo brote de peste porcina africana en un porcino silvestre supone un aumento del nivel de riesgo que debe reflejarse en dicho anexo. Por consiguiente, esta zona de Polonia que figura actualmente como zona restringida I en el mencionado anexo, afectada por el reciente brote de peste porcina africana mencionado, debe figurar ahora en dicho anexo como zona restringida II en vez de como zona restringida I. |
|
(7) |
A raíz de estos brotes recientes de peste porcina africana en porcinos silvestres en Eslovaquia y Polonia, y habida cuenta de la actual situación epidemiológica respecto de la peste porcina africana en la Unión, la zonificación en dichos Estados miembros se ha evaluado de nuevo y se ha actualizado. Asimismo, también se han evaluado de nuevo y se han actualizado las medidas vigentes de gestión de riesgos. Estos cambios deben reflejarse en el anexo I del Reglamento de Ejecución (UE) 2021/605. |
|
(8) |
A fin de tener en cuenta la reciente evolución de la situación epidemiológica respecto a la peste porcina africana en la Unión y para combatir los riesgos asociados a la propagación de esta enfermedad de manera proactiva, deben delimitarse nuevas zonas restringidas de un tamaño suficiente en Eslovaquia y Polonia e incluirse convenientemente en las listas del anexo I del Reglamento de Ejecución (UE) 2021/605 como zona restringida II. Puesto que la situación con respecto a la peste porcina africana es muy dinámica en la Unión, al delimitar estas nuevas zonas restringidas se ha tenido en cuenta la situación en las zonas circundantes. |
|
(9) |
Dada la urgencia de la situación epidemiológica en la Unión por lo que respecta a la propagación de la peste porcina africana, es importante que las modificaciones que se introduzcan en el anexo I del Reglamento de Ejecución (UE) 2021/605 mediante el presente Reglamento de Ejecución surtan efecto lo antes posible. |
|
(10) |
Las medidas previstas en el presente Reglamento se ajustan al dictamen del Comité Permanente de Vegetales, Animales, Alimentos y Piensos. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
El anexo I del Reglamento de Ejecución (UE) 2021/605 se sustituye por el texto que figura en el anexo del presente Reglamento.
Artículo 2
El presente Reglamento entrará en vigor el día siguiente al de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 20 de mayo de 2021.
Por la Comisión
La Presidenta
Ursula VON DER LEYEN
(1) DO L 84 de 31.3.2016, p. 1.
(2) Reglamento de Ejecución (UE) 2021/605 de la Comisión, de 7 de abril de 2021, por el que se establecen medidas especiales de control de la peste porcina africana (DO L 129 de 15.4.2021, p. 1).
(3) Reglamento de Ejecución (UE) 2021/687 de la Comisión, de 26 de abril de 2021, que modifica el anexo I del Reglamento de Ejecución (UE) 2021/605 por el que se establecen medidas especiales de control de la peste porcina africana (DO L 143 de 27.4.2021, p. 11).
(4) Decisión de Ejecución 2014/709/UE de la Comisión, de 9 de octubre de 2014, sobre medidas de control zoosanitarias relativas a la peste porcina africana en determinados Estados miembros y por la que se deroga la Decisión de Ejecución 2014/178/UE (DO L 295 de 11.10.2014, p. 63).
(5) Documento de trabajo SANTE/7112/2015/Rev. 3: Principles and criteria for geographically defining ASF regionalisation [«Principios y criterios para definir geográficamente la regionalización de la peste porcina africana», documento en inglés]. https://ec.europa.eu/food/animals/animal-diseases/control-measures/asf_en
(6) Código Sanitario para los Animales Terrestres de la OIE, 28.a Edición, 2019. ISBN del volumen I: 978-92-95108-85-1; ISBN del volumen II: 978-92-95108-86-8. https://www.oie.int/standard-setting/terrestrial-code/access-online/
ANEXO
El anexo I del Reglamento de Ejecución (UE) 2021/605 se sustituye por el texto siguiente:
«ANEXO I
ZONAS RESTRINGIDAS
PARTE I
1. Alemania
Las siguientes zonas restringidas I de Alemania:
Bundesland Brandenburg:
|
— |
Landkreis Dahme-Spreewald:
|
|
— |
Landkreis Märkisch-Oderland:
|
|
— |
Landkreis Oder-Spree:
|
|
— |
Landkreis Spree-Neiße:
|
Bundesland Sachsen:
|
— |
Landkreis Bautzen
|
|
— |
Landkreis Görlitz:
|
2. Estonia
Las siguientes zonas restringidas I de Estonia:
|
— |
Hiiu maakond. |
3. Grecia
Las siguientes zonas restringidas I de Grecia:
|
— |
in the regional unit of Drama:
|
|
— |
in the regional unit of Xanthi:
|
|
— |
in the regional unit of Rodopi:
|
|
— |
in the regional unit of Evros:
|
|
— |
in the regional unit of Serres:
|
4. Letonia
Las siguientes zonas restringidas I de Letonia:
|
— |
Pāvilostas novada Vērgales pagasts, |
|
— |
Stopiņu novada daļa, kas atrodas uz rietumiem no autoceļa V36, P4 un P5, Acones ielas, Dauguļupes ielas un Dauguļupītes, |
|
— |
Grobiņas novada Medzes, Grobiņas un Gaviezes pagasts. Grobiņas pilsēta, |
|
— |
Rucavas novada Rucavas pagasts, |
|
— |
Nīcas novads. |
5. Lituania
Las siguientes zonas restringidas I de Lituania:
|
— |
Klaipėdos rajono savivaldybė: Agluonėnų, Dovilų, Gargždų, Priekulės, Vėžaičių, Kretingalės ir Dauparų-Kvietinių seniūnijos, |
|
— |
Palangos miesto savivaldybė. |
6. Hungría
Las siguientes zonas restringidas I de Hungría:
|
— |
Békés megye 950950, 950960, 950970, 951950, 952050, 952750, 952850, 952950, 953050, 953150, 953650, 953660, 953750, 953850, 953960, 954250, 954260, 954350, 954450, 954550, 954650, 954750, 954850, 954860, 954950, 955050, 955150, 955250, 955260, 955270, 955350, 955450, 955510, 955650, 955750, 955760, 955850, 955950, 956050, 956060, 956150 és 956160 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Bács-Kiskun megye 600150, 600850, 601550, 601650, 601660, 601750, 601850, 601950, 602050, 603250, 603750 és 603850 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Budapest 1 kódszámú, vadgazdálkodási tevékenységre nem alkalmas területe, |
|
— |
Csongrád-Csanád megye 800150, 800160, 800250, 802220, 802260, 802310 és 802450 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Fejér megye 400150, 400250, 400351, 400352, 400450, 400550, 401150, 401250, 401350, 402050, 402350, 402360, 402850, 402950, 403050, 403250, 403350, 403450, 403550, 403650, 403750, 403950, 403960, 403970, 404570, 404650, 404750, 404850, 404950, 404960, 405050, 405750, 405850, 405950, |
|
— |
406050, 406150, 406550, 406650 és 406750 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Jász-Nagykun-Szolnok megye 750150, 750160, 750260, 750350, 750450, 750460, 754450, 754550, 754560, 754570, 754650, 754750, 754950, 755050, 755150, 755250, 755350 és 755450 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Komárom-Esztergom megye 250150, 250250, 250350, 250450, 250460, 250550, 250650, 250750, 250850, 250950, 251050, 251150, 251250, 251350, 251360, 251450, 251550, 251650, 251750, 251850, 252150 és 252250, kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Pest megye 571550, 572150, 572250, 572350, 572550, 572650, 572750, 572850, 572950, 573150, 573250, 573260, 573350, 573360, 573450, 573850, 573950, 573960, 574050, 574150, 574350, 574360, 574550, 574650, 574750, 574850, 574860, 574950, 575050, 575150, 575250, 575350, 575550, 575650, 575750, 575850, 575950, 576050, 576150, 576250, 576350, 576450, 576650, 576750, 576850, 576950, 577050, 577150, 577350, 577450, 577650, 577850, 577950, 578050, 578150, 578250, 578350, 578360, 578450, 578550, 578560, 578650, 578850, 578950, 579050, 579150, 579250, 579350, 579450, 579460, 579550, 579650, 579750, 580250 és 580450 kódszámú vadgazdálkodási egységeinek teljes területe. |
7. Polonia
Las siguientes zonas restringidas I de Polonia:
w województwie warmińsko-mazurskim:
|
— |
gminy Wielbark i Rozogi w powiecie szczycieńskim, |
|
— |
gminy Janowiec Kościelny, Janowo i część gminy Kozłowo położona na południe od linii wyznaczonej przez linię kolejową w powiecie nidzickim, |
|
— |
gminy Iłowo – Osada, Lidzbark, Płośnica, miasto Działdowo, część gminy Rybno położona na południe od linii wyznaczonej przez drogę kolejową, część gminy wiejskiej Działdowo położona na południe od linii wyznaczonej przez linie kolejowe biegnące od wschodniej do zachodniej granicy gminy w powiecie działdowskim, |
|
— |
gminy Kisielice, Susz i część gminy wiejskiej Iława położona na zachód od linii wyznaczonej przez drogę nr 521 biegnącą od zachodniej granicy gminy do skrzyżowania z drogą łączącą miejscowości Szymbark - Ząbrowo - Segnowy – Laseczno – Gulb, a następnie na zachód od linii wyznaczonej przez drogę łączącą miejscowości Szymbark - Ząbrowo - Segnowy – Laseczno - Gulb biegnącą do południowej granicy gminy w powiecie iławskim, |
|
— |
gminy Biskupiec, Kurzętnik, część gminy wiejskiej Nowe Miasto Lubawskie położona na południe od linii wyznaczonej przez drogę biegnącą od zachodniej granicy gminy do miejscowości Lekarty, a następnie na południowy - zachód od linii wyznaczonej przez drogę łączącą miejscowości Lekarty – Nowy Dwór Bratiański biegnącą do północnej granicy gminy miejskiej Nowe Miasto Lubawskie oraz na południe od linii wyznaczonej przez drogę nr 538, część gminy Grodziczno położona na południe od linii wyznaczonej przez drogę nr 538 w powiecie nowomiejskim. |
w województwie podlaskim:
|
— |
gminy Wysokie Mazowieckie z miastem Wysokie Mazowieckie, Czyżew i część gminy Kulesze Kościelne położona na południe od linii wyznaczonej przez linię koleją w powiecie wysokomazowieckim, |
|
— |
gminy Miastkowo, Nowogród, Śniadowo i Zbójna w powiecie łomżyńskim, |
|
— |
gminy Szumowo, Zambrów z miastem Zambrów i część gminy Kołaki Kościelne położona na południe od linii wyznaczonej przez linię kolejową w powiecie zambrowskim, |
|
— |
gminy Grabowo, Kolno i miasto Kolno, Turośl w powiecie kolneńskim, |
w województwie mazowieckim:
|
— |
powiat ostrołęcki, |
|
— |
powiat miejski Ostrołęka, |
|
— |
gminy Bielsk, Brudzeń Duży, Bulkowo, Drobin, Gąbin, Łąck, Nowy Duninów, Radzanowo, Słupno, Staroźreby i Stara Biała w powiecie płockim, |
|
— |
powiat miejski Płock, |
|
— |
powiat ciechanowski, |
|
— |
gminy Baboszewo, Dzierzążnia, Joniec, Nowe Miasto, Płońsk i miasto Płońsk, Raciąż i miasto Raciąż, Sochocin w powiecie płońskim, |
|
— |
powiat sierpecki, |
|
— |
powiat żuromiński, |
|
— |
gminy Andrzejewo, Brok, Stary Lubotyń, Szulborze Wielkie, Wąsewo, Ostrów Mazowiecka z miastem Ostrów Mazowiecka, część gminy Małkinia Górna położona na północ od rzeki Brok w powiecie ostrowskim, |
|
— |
powiat mławski, |
|
— |
powiat przasnyski, |
|
— |
powiat makowski, |
|
— |
powiat pułtuski, |
|
— |
powiat wyszkowski, |
|
— |
powiat węgrowski, |
|
— |
gminy Dąbrówka, Jadów, Klembów, Poświętne, Radzymin, Strachówka Wołomin i Tłuszcz w powiecie wołomińskim, |
|
— |
gminy Mokobody i Suchożebry w powiecie siedleckim, |
|
— |
gminy Dobre, Jakubów, Kałuszyn, Stanisławów w powiecie mińskim, |
|
— |
gminy Bielany i gmina wiejska Sokołów Podlaski w powiecie sokołowskim, |
|
— |
gminy Kowala, Wierzbica, część gminy Wolanów położona na południe od linii wyznaczonej przez drogę nr 12 w powiecie radomskim, |
|
— |
powiat miejski Radom, |
|
— |
gminy Jastrząb, Mirów, Orońsko w powiecie szydłowieckim, |
|
— |
powiat gostyniński, |
w województwie podkarpackim:
|
— |
gminy Pruchnik, Rokietnica, Roźwienica, w powiecie jarosławskim, |
|
— |
gminy Fredropol, Krasiczyn, Krzywcza, Medyka, Orły, Żurawica, Przemyśl w powiecie przemyskim, |
|
— |
powiat miejski Przemyśl, |
|
— |
gminy Gać, Jawornik Polski, Kańczuga, część gminy Zarzecze położona na południe od linii wyznaczonej przez rzekę Mleczka w powiecie przeworskim, |
|
— |
powiat łańcucki, |
|
— |
gminy Trzebownisko, Głogów Małopolski i część gminy Sokołów Małopolski położona na południe od linii wyznaczonej przez drogę nr 875 w powiecie rzeszowskim, |
|
— |
gminy Dzikowiec, Kolbuszowa, Niwiska i Raniżów w powiecie kolbuszowskim, |
|
— |
gminy Borowa, Czermin, Gawłuszowice, Mielec z miastem Mielec, Padew Narodowa, Przecław, Tuszów Narodowy w powiecie mieleckim, |
w województwie świętokrzyskim:
|
— |
powiat opatowski, |
|
— |
powiat sandomierski, |
|
— |
gminy Bogoria, Łubnice, Oleśnica, Osiek, Połaniec, Rytwiany i Staszów w powiecie staszowskim, |
|
— |
gminy Bliżyn, Skarżysko – Kamienna, Suchedniów i Skarżysko Kościelne w powiecie skarżyskim, |
|
— |
gmina Wąchock, część gminy Brody położona na zachód od linii wyznaczonej przez drogę nr 9 oraz na południowy - zachód od linii wyznaczonej przez drogi: nr 0618T biegnącą od północnej granicy gminy do skrzyżowania w miejscowości Lipie, drogę biegnącą od miejscowości Lipie do wschodniej granicy gminy oraz na północ od drogi nr 42 i część gminy Mirzec położona na zachód od linii wyznaczonej przez drogę nr 744 biegnącą od południowej granicy gminy do miejscowości Tychów Stary a następnie przez drogę nr 0566T biegnącą od miejscowości Tychów Stary w kierunku północno - wschodnim do granicy gminy w powiecie starachowickim, |
|
— |
powiat ostrowiecki, |
|
— |
gminy Fałków, Ruda Maleniecka, Radoszyce, Smyków, część gminy Końskie położona na zachód od linii kolejowej, część gminy Stąporków położona na południe od linii kolejowej w powiecie koneckim, |
|
— |
gminy Mniów i Zagnańsk w powiecie kieleckim, |
w województwie łódzkim:
|
— |
gminy Łyszkowice, Kocierzew Południowy, Kiernozia, Chąśno, Nieborów, część gminy wiejskiej Łowicz położona na północ od linii wyznaczonej przez drogę nr 92 biegnącej od granicy miasta Łowicz do zachodniej granicy gminy oraz część gminy wiejskiej Łowicz położona na wschód od granicy miasta Łowicz i na północ od granicy gminy Nieborów w powiecie łowickim, |
|
— |
gminy Cielądz, Rawa Mazowiecka z miastem Rawa Mazowiecka w powiecie rawskim, |
|
— |
gminy Bolimów, Głuchów, Godzianów, Lipce Reymontowskie, Maków, Nowy Kawęczyn, Skierniewice, Słupia w powiecie skierniewickim, |
|
— |
powiat miejski Skierniewice, |
|
— |
gminy Mniszków, Paradyż, Sławno i Żarnów w powiecie opoczyńskim, |
|
— |
gminy Czerniewice, Inowłódz, Lubochnia, Rzeczyca, Tomaszów Mazowiecki z miastem Tomaszów Mazowiecki i Żelechlinek w powiecie tomaszowskim, |
|
— |
gmina Aleksandrów w powiecie piotrkowskim, |
w województwie pomorskim:
|
— |
gminy Ostaszewo, miasto Krynica Morska oraz część gminy Nowy Dwór Gdański położona na południowy - zachód od linii wyznaczonej przez drogę nr 55 biegnącą od południowej granicy gminy do skrzyżowania z drogą nr 7, następnie przez drogę nr 7 i S7 biegnącą do zachodniej granicy gminy w powiecie nowodworskim, |
|
— |
gminy Lichnowy, Miłoradz, Nowy Staw, Malbork z miastem Malbork w powiecie malborskim, |
|
— |
gminy Mikołajki Pomorskie, Stary Targ i Sztum w powiecie sztumskim, |
|
— |
powiat gdański, |
|
— |
Miasto Gdańsk, |
|
— |
powiat tczewski, |
|
— |
powiat kwidzyński, |
w województwie lubuskim:
|
— |
gminy Przytoczna, Pszczew, Skwierzyna i część gminy Trzciel położona na północ od linii wyznaczonej przez drogę nr 92 w powiecie międzyrzeckim, |
|
— |
gminy Lubniewice i Krzeszyce w powiecie sulęcińskim, |
|
— |
gminy Bogdaniec, Deszczno, Lubiszyn i część gminy Witnica położona na północny - wschód od drogi biegnącej od zachodniej granicy gminy od miejscowości Krześnica, przez miejscowości Kamień Wielki - Mościce -Witnica - Kłopotowo do południowej granicy gminy w powiecie gorzowskim, |
w województwie dolnośląskim:
|
— |
gminy Bolesławiec z miastem Bolesławiec, Gromadka i Osiecznica w powiecie bolesławieckim, |
|
— |
gmina Węgliniec w powiecie zgorzeleckim, |
|
— |
gmina Chocianów i część gminy Przemków położona na południe od linii wyznaczonej przez drogę nr 12 w powiecie polkowickim, |
|
— |
gmina Góra , Wąsosz, część gminy Niechlów położona na północny – wschód od linii wyznaczonej przez rzekę Barycz i część gminy Jemielno położona na wschód od linii wyznaczonej przez drogę nr 323 w powiecie górowskim, |
|
— |
gmina Wińsko w powiecie wołowskim, |
|
— |
gminy Ścinawa i Lubin z miastem Lubin w powiecie lubińskim, |
w województwie wielkopolskim:
|
— |
gminy Krzemieniewo, Osieczna, Rydzyna, część gminy Lipno położona na wschód od linii wyznaczonej przez drogę nr S5, część gminy Święciechowa położona na południe od linii wyznaczonej przez drogę nr 12 oraz na wschód od linii wyznaczonej przez drogę nr S5 w powiecie leszczyńskim, |
|
— |
powiat miejski Leszno, |
|
— |
gminy Chrzypsko Wielkie, Międzychód, część gminy Sieraków położona na zachód od linii wyznaczonej przez drogę nr 186 biegnącą od południowej granicy gminy do miejscowości Lutomek, a następnie na zachód od linii wyznaczonej przez drogę biegnącą od skrzyżowania z drogą nr 186 w miejscowości Lutomek biegnącą do skrzyżowania z ul. Leśną w miejscowości Lutom i dalej na zachód od ul. Leśnej biegnącej do wschodniej granicy gminy, część gminy Kwilcz położona na zachód linii wyznaczonej przez drogę nr 186 biegnącą od północnej granicy gminy do skrzyżowania z drogą nr 24, następnie na południe od linii wyznaczonej przez drogę nr 24 biegnącą od skrzyżowania z drogą nr 186 do skrzyżowania z drogą w miejscowości Pólko, i dalej na zachód od linii wyznaczonej przez drogę biegnącą od miejscowości Pólko przez miejscowość Wituchowo do południowej granicy gminy, w powiecie międzychodzkim, |
|
— |
gminy Lwówek, Kuślin, Opalenica, część gminy Miedzichowo położona na północ od linii wyznaczonej przez drogę nr 92, część gminy Nowy Tomyśl położona na wschód od linii wyznaczonej przez drogę nr 305 w powiecie nowotomyskim, |
|
— |
gminy Granowo, Grodzisk Wielkopolski i część gminy Kamieniec położona na wschód od linii wyznaczonej przez drogę nr 308 w powiecie grodziskim, |
|
— |
gminy Czempiń, Kościan i miasto Kościan, Krzywiń, część gminy Śmigiel położona na wschód od linii wyznaczonej przez drogę nr S5 w powiecie kościańskim, |
|
— |
powiat miejski Poznań, |
|
— |
gminy Buk, Dopiewo, Komorniki, Tarnowo Podgórne, Stęszew, Swarzędz, Pobiedziska, Czerwonak, Mosina, miasto Luboń, miasto Puszczykowo i część gminy Kórnik położona na zachód od linii wyznaczonych przez drogi: nr S11 biegnącą od północnej granicy gminy do skrzyżowania z drogą nr 434 i drogę nr 434 biegnącą od tego skrzyżowania do południowej granicy gminy, część gminy Rokietnica położona na południowy zachód od linii kolejowej biegnącej od północnej granicy gminy w miejscowości Krzyszkowo do południowej granicy gminy w miejscowości Kiekrz oraz część gminy wiejskiej Murowana Goślina położona na południe od linii kolejowej biegnącej od północnej granicy miasta Murowana Goślina do północno-wschodniej granicy gminy w powiecie poznańskim, |
|
— |
gmina Kiszkowo i część gminy Kłecko położona na zachód od rzeki Mała Wełna w powiecie gnieźnieńskim, |
|
— |
gminy Lubasz, Czarnków z miastem Czarnków, część gminy Połajewo na położona na północ od drogi łączącej miejscowości Chraplewo, Tarnówko-Boruszyn, Krosin, Jakubowo, Połajewo - ul. Ryczywolska do północno-wschodniej granicy gminy oraz część gminy Wieleń położona na południe od linii kolejowej biegnącej od wschodniej granicy gminy przez miasto Wieleń i miejscowość Herburtowo do zachodniej granicy gminy w powiecie czarnkowsko-trzcianeckim, |
|
— |
gmina Kaźmierz część gminy Duszniki położona na południowy – wschód od linii wyznaczonej przez drogę nr 306 biegnącą od północnej granicy gminy do miejscowości Duszniki, a następnie na południe od linii wyznaczonej przez ul. Niewierską oraz drogę biegnącą przez miejscowość Niewierz do zachodniej granicy gminy, część gminy Ostroróg, położona na wschód od linii wyznaczonej przez drogę nr 186 i 184 biegnące od granicy gminy do miejscowości Ostroróg, a następnie od miejscowości Ostroróg przez miejscowości Piaskowo – Rudki do południowej granicy gminy, część gminy Wronki położona na północ od linii wyznaczonej przez drogi nr 182 i 186, miasto Szamotuły i część gminy Szamotuły położona na wschód od linii wyznaczonej przez drogę nr 306 do linii wyznaczonej przez wschodnią granicę miasta Szamotuły i na południe od linii kolejowej biegnącej od południowej granicy miasta Szamotuły, do południowo-wschodniej granicy gminy oraz część gminy Obrzycko położona na zachód od drogi nr 185 łączącej miejscowości Gaj Mały, Słopanowo i Obrzycko do północnej granicy miasta Obrzycko, a następnie na zachód od drogi przebiegającej przez miejscowość Chraplewo w powiecie szamotulskim, |
|
— |
gmina Budzyń w powiecie chodzieskim, |
|
— |
gminy Mieścisko, Skoki i Wągrowiec z miastem Wągrowiec w powiecie wągrowieckim, |
|
— |
powiat pleszewski, |
|
— |
gmina Zagórów w powiecie słupeckim, |
|
— |
gmina Pyzdry w powiecie wrzesińskim, |
|
— |
gminy Kotlin, Żerków i część gminy Jarocin położona na wschód od linii wyznaczonej przez drogi nr S11 i 15 w powiecie jarocińskim, |
|
— |
gmina Rozdrażew, część gminy Koźmin Wielkopolski położona na wschód od linii wyznaczonej przez drogę nr 15, część gminy Krotoszyn położona na wschód od linii wyznaczonej przez drogę nr 15 oraz na wschód od granic miasta Krotoszyn w powiecie krotoszyńskim, |
|
— |
gminy Nowe Skalmierzyce, Raszków, Ostrów Wielkopolski z miastem Ostrów Wielkopolski w powiecie ostrowskim, |
|
— |
powiat miejski Kalisz, |
|
— |
gminy Blizanów, Stawiszyn, Żelazków, Ceków – Kolonia, Godziesze Wielkie, Koźminek, Lisków, Mycielin, Opatówek, Szczytniki w powiecie kaliskim, |
|
— |
gmina Malanów i część gminy Tuliszków położona na zachód od linii wyznaczonej przez drogę nr 72 w powiecie tureckim, |
|
— |
gminy Rychwał, Rzgów, Grodziec, część gminy Stare Miasto położona na południe od linii wyznaczonej przez autostradę nr A2 w powiecie konińskim, |
w województwie zachodniopomorskim:
|
— |
część gminy Dębno położona na wschód od linii wyznaczonej przez drogę nr 126 biegnącą od zachodniej granicy gminy do skrzyżowania z drogą nr 23 w miejscowości Dębno, następnie na wschód od linii wyznaczonej przez drogę nr 23 do skrzyżowania z ul. Jana Pawła II w miejscowości Cychry, następnie na północ od ul. Jana Pawła II do skrzyżowania z ul. Ogrodową i dalej na północ od linii wyznaczonej przez ul. Ogrodową, której przedłużenie biegnie do wschodniej granicy gminy w powiecie myśliborskim, |
|
— |
gminy Chojna, Trzcińsko - Zdrój oraz część gminy Cedynia położona na północ od linii wyznaczonej przez drogę nr 124 biegnącą od zachodniej granicy gminy do miasta Cedynia, a następnie na północ od linii wyznaczonej przez drogę nr 125 biegnącą od miasta Cedynia do wschodniej granicy gminy w powiecie gryfińskim. |
8. Eslovaquia
Las siguientes zonas restringidas I de Eslovaquia:
|
— |
the whole district of Vranov nad Topľou, except municipalities included in part II, |
|
— |
the whole district of Humenné, except municipalities included in part II, |
|
— |
the whole district of Snina, |
|
— |
the whole district of Medzilaborce |
|
— |
the whole district of Stropkov |
|
— |
the whole district of Svidník, except municipalities included in part II, |
|
— |
the whole district of Stará Ľubovňa, except municipalities included in part II, |
|
— |
the whole district of whole Kežmarok, |
|
— |
the whole district of Poprad, |
|
— |
the whole district of Veľký Krtíš, except municipalities included in part II, |
|
— |
in the whole district of Zvolen, except municipalities included in part II, |
|
— |
the whole district of Detva, except municipalities included in part II, |
|
— |
the whole district of Krupina, except municipalities included in part II, |
|
— |
the whole district of Brezno. |
PARTE II
1. Bulgaria
Las siguientes zonas restringidas II de Bulgaria:
|
— |
the whole region of Haskovo, |
|
— |
the whole region of Yambol, |
|
— |
the whole region of Stara Zagora, |
|
— |
the whole region of Pernik, |
|
— |
the whole region of Kyustendil, |
|
— |
the whole region of Plovdiv, |
|
— |
the whole region of Pazardzhik, |
|
— |
the whole region of Smolyan, |
|
— |
the whole region of Dobrich, |
|
— |
the whole region of Sofia city, |
|
— |
the whole region of Sofia Province, |
|
— |
the whole region of Blagoevgrad, |
|
— |
the whole region of Razgrad, |
|
— |
the whole region of Kardzhali, |
|
— |
the whole region of Burgas excluding the areas in Part III, |
|
— |
the whole region of Varna excluding the areas in Part III, |
|
— |
the whole region of Silistra, excluding the areas in Part III, |
|
— |
the whole region of Ruse, excluding the areas in Part III, |
|
— |
the whole region of Veliko Tarnovo, excluding the areas in Part III, |
|
— |
the whole region of Pleven, excluding the areas in Part III, |
|
— |
the whole region of Targovishte, excluding the areas in Part III, |
|
— |
the whole region of Shumen, excluding the areas in Part III, |
|
— |
the whole region of Sliven, excluding the areas in Part III, |
|
— |
the whole region of Vidin, excluding the areas in Part III. |
2. Alemania
Las siguientes zonas restringidas II de Alemania:
Bundesland Brandenburg:
|
— |
Landkreis Oder-Spree:
|
|
— |
Landkreis Dahme-Spreewald:
|
|
— |
Landkreis Spree-Neiße:
|
|
— |
Landkreis Märkisch-Oderland:
|
|
— |
kreisfreie Stadt Frankfurt (Oder), |
Bundesland Sachsen:
|
— |
Landkreis Görlitz:
|
3. Estonia
Las siguientes zonas restringidas II de Estonia:
|
— |
Eesti Vabariik (välja arvatud Hiiu maakond). |
4. Letonia
Las siguientes zonas restringidas II de Letonia:
|
— |
Ādažu novads, |
|
— |
Aizputes novada Aizputes, Cīravas un Lažas pagasts, Kalvenes pagasta daļa uz rietumiem no ceļa pie Vārtājas upes līdz autoceļam A9, uz dienvidiem no autoceļa A9, uz rietumiem no autoceļa V1200, Kazdangas pagasta daļa uz rietumiem no ceļa V1200, P115, P117, V1296, Aizputes pilsēta, |
|
— |
Aglonas novads, |
|
— |
Aizkraukles novads, |
|
— |
Aknīstes novads, |
|
— |
Alojas novads, |
|
— |
Alsungas novads, |
|
— |
Alūksnes novads, |
|
— |
Amatas novads, |
|
— |
Apes novads, |
|
— |
Auces novads, |
|
— |
Babītes novads, |
|
— |
Baldones novads, |
|
— |
Baltinavas novads, |
|
— |
Balvu novads, |
|
— |
Bauskas novads, |
|
— |
Beverīnas novads, |
|
— |
Brocēnu novads, |
|
— |
Burtnieku novads, |
|
— |
Carnikavas novads, |
|
— |
Cēsu novads |
|
— |
Cesvaines novads, |
|
— |
Ciblas novads, |
|
— |
Dagdas novads, |
|
— |
Daugavpils novads, |
|
— |
Dobeles novads, |
|
— |
Dundagas novads, |
|
— |
Durbes novads, |
|
— |
Engures novads, |
|
— |
Ērgļu novads, |
|
— |
Garkalnes novads, |
|
— |
Grobiņas novada Bārtas pagasts, |
|
— |
Gulbenes novads, |
|
— |
Iecavas novads, |
|
— |
Ikšķiles novads, |
|
— |
Ilūkstes novads, |
|
— |
Inčukalna novads, |
|
— |
Jaunjelgavas novads, |
|
— |
Jaunpiebalgas novads, |
|
— |
Jaunpils novads, |
|
— |
Jēkabpils novads, |
|
— |
Jelgavas novads, |
|
— |
Kandavas novads, |
|
— |
Kārsavas novads, |
|
— |
Ķeguma novads, |
|
— |
Ķekavas novads, |
|
— |
Kocēnu novads, |
|
— |
Kokneses novads, |
|
— |
Krāslavas novads, |
|
— |
Krimuldas novads, |
|
— |
Krustpils novads, |
|
— |
Kuldīgas novada, Laidu pagasta daļa uz ziemeļiem no autoceļa V1296, Padures, Rumbas, Rendas, Kabiles, Vārmes, Pelču, Ēdoles, Īvandes, Kurmāles, Turlavas, Gudenieku un Snēpeles pagasts, Kuldīgas pilsēta, |
|
— |
Lielvārdes novads, |
|
— |
Līgatnes novads, |
|
— |
Limbažu novads, |
|
— |
Līvānu novads, |
|
— |
Lubānas novads, |
|
— |
Ludzas novads, |
|
— |
Madonas novads, |
|
— |
Mālpils novads, |
|
— |
Mārupes novads, |
|
— |
Mazsalacas novads, |
|
— |
Mērsraga novads, |
|
— |
Naukšēnu novads, |
|
— |
Neretas novads, |
|
— |
Ogres novads, |
|
— |
Olaines novads, |
|
— |
Ozolnieku novads, |
|
— |
Pārgaujas novads, |
|
— |
Pāvilostas novada Sakas pagasts, Pāvilostas pilsēta, |
|
— |
Pļaviņu novads, |
|
— |
Preiļu novads, |
|
— |
Priekules novads, |
|
— |
Priekuļu novads, |
|
— |
Raunas novads, |
|
— |
republikas pilsēta Daugavpils, |
|
— |
republikas pilsēta Jelgava, |
|
— |
republikas pilsēta Jēkabpils, |
|
— |
republikas pilsēta Jūrmala, |
|
— |
republikas pilsēta Rēzekne, |
|
— |
republikas pilsēta Valmiera, |
|
— |
Rēzeknes novads, |
|
— |
Riebiņu novads, |
|
— |
Rojas novads, |
|
— |
Ropažu novads, |
|
— |
Rucavas novada Dunikas pagasts, |
|
— |
Rugāju novads, |
|
— |
Rundāles novads, |
|
— |
Rūjienas novads, |
|
— |
Salacgrīvas novads, |
|
— |
Salas novads, |
|
— |
Salaspils novads, |
|
— |
Saldus novads, |
|
— |
Saulkrastu novads, |
|
— |
Sējas novads, |
|
— |
Siguldas novads, |
|
— |
Skrīveru novads, |
|
— |
Skrundas novada Raņķu pagasta daļa uz ziemeļiem no autoceļa V1272 līdz robežai ar Ventas upi, Skrundas pagasta daļa no Skrundas uz ziemeļiem no autoceļa A9 un austrumiem no Ventas upes, |
|
— |
Smiltenes novads, |
|
— |
Stopiņu novada daļa, kas atrodas uz austrumiem no autoceļa V36, P4 un P5, Acones ielas, Dauguļupes ielas un Dauguļupītes, |
|
— |
Strenču novads, |
|
— |
Talsu novads, |
|
— |
Tērvetes novads, |
|
— |
Tukuma novads, |
|
— |
Vaiņodes novada Vaiņodes pagasts un Embūtes pagasta daļa uz dienvidiem autoceļa P116, P106, |
|
— |
Valkas novads, |
|
— |
Varakļānu novads, |
|
— |
Vārkavas novads, |
|
— |
Vecpiebalgas novads, |
|
— |
Vecumnieku novads, |
|
— |
Ventspils novads, |
|
— |
Viesītes novads, |
|
— |
Viļakas novads, |
|
— |
Viļānu novads, |
|
— |
Zilupes novads. |
5. Lituania
Las siguientes zonas restringidas II de Lituania:
|
— |
Alytaus miesto savivaldybė, |
|
— |
Alytaus rajono savivaldybė, |
|
— |
Anykščių rajono savivaldybė, |
|
— |
Akmenės rajono savivaldybė, |
|
— |
Birštono savivaldybė, |
|
— |
Biržų miesto savivaldybė, |
|
— |
Biržų rajono savivaldybė, |
|
— |
Druskininkų savivaldybė, |
|
— |
Elektrėnų savivaldybė, |
|
— |
Ignalinos rajono savivaldybė, |
|
— |
Jonavos rajono savivaldybė, |
|
— |
Joniškio rajono savivaldybė, |
|
— |
Jurbarko rajono savivaldybė: Eržvilko, Girdžių, Jurbarko miesto, Jurbarkų, Raudonės, Šimkaičių, Skirsnemunės, Smalininkų, Veliuonos ir Viešvilės seniūnijos, |
|
— |
Kaišiadorių rajono savivaldybė, |
|
— |
Kalvarijos savivaldybė, |
|
— |
Kauno miesto savivaldybė, |
|
— |
Kauno rajono savivaldybė: Akademijos, Alšėnų, Batniavos, Ežerėlio, Domeikavos, Garliavos, Garliavos apylinkių, Karmėlavos, Kulautuvos, Lapių, Linksmakalnio, Neveronių, Raudondvario, Ringaudų, Rokų, Samylų, Taurakiemio, Vandžiogalos, Užliedžių, Vilkijos, ir Zapyškio seniūnijos, Babtų seniūnijos dalis į rytus nuo kelio A1, ir Vilkijos apylinkių seniūnijos dalis į vakarus nuo kelio Nr. 1907, |
|
— |
Kazlų rūdos savivaldybė, |
|
— |
Kelmės rajono savivaldybė, |
|
— |
Kėdainių rajono savivaldybė: Dotnuvos, Gudžiūnų, Kėdainių miesto, Krakių, Pelėdnagių, Surviliškio, Šėtos, Truskavos, Vilainių ir Josvainių seniūnijos dalis į šiaurę ir rytus nuo kelio Nr. 229 ir Nr. 2032, |
|
— |
Klaipėdos rajono savivaldybė: Judrėnų, Endriejavo ir Veiviržėnų seniūnijos, |
|
— |
Kupiškio rajono savivaldybė, |
|
— |
Kretingos rajono savivaldybė, |
|
— |
Lazdijų rajono savivaldybė, |
|
— |
Marijampolės savivaldybė, |
|
— |
Mažeikių rajono savivaldybė, |
|
— |
Molėtų rajono savivaldybė, |
|
— |
Pagėgių savivaldybė, |
|
— |
Pakruojo rajono savivaldybė, |
|
— |
Panevėžio rajono savivaldybė, |
|
— |
Panevėžio miesto savivaldybė, |
|
— |
Pasvalio rajono savivaldybė, |
|
— |
Radviliškio rajono savivaldybė, |
|
— |
Rietavo savivaldybė, |
|
— |
Prienų rajono savivaldybė, |
|
— |
Plungės rajono savivaldybė: Žlibinų, Stalgėnų, Nausodžio, Plungės miesto, Šateikių ir Kulių seniūnijos, |
|
— |
Raseinių rajono savivaldybė: Betygalos, Girkalnio, Kalnujų, Nemakščių, Pagojukų, Paliepių, Raseinių miesto, Raseinių, Šiluvos, Viduklės seniūnijos, |
|
— |
Rokiškio rajono savivaldybė, |
|
— |
Skuodo rajono savivaldybės: Aleksandrijos, Ylakių, Lenkimų, Mosėdžio, Skuodo ir Skuodo miesto seniūnijos, |
|
— |
Šakių rajono savivaldybė, |
|
— |
Šalčininkų rajono savivaldybė, |
|
— |
Šiaulių miesto savivaldybė, |
|
— |
Šiaulių rajono savivaldybė, |
|
— |
Šilutės rajono savivaldybė, |
|
— |
Širvintų rajono savivaldybė, |
|
— |
Šilalės rajono savivaldybė, |
|
— |
Švenčionių rajono savivaldybė, |
|
— |
Tauragės rajono savivaldybė, |
|
— |
Telšių rajono savivaldybė, |
|
— |
Trakų rajono savivaldybė, |
|
— |
Ukmergės rajono savivaldybė, |
|
— |
Utenos rajono savivaldybė, |
|
— |
Varėnos rajono savivaldybė, |
|
— |
Vilniaus miesto savivaldybė, |
|
— |
Vilniaus rajono savivaldybė, |
|
— |
Vilkaviškio rajono savivaldybė, |
|
— |
Visagino savivaldybė, |
|
— |
Zarasų rajono savivaldybė. |
6. Hungría
Las siguientes zonas restringidas II de Hungría:
|
— |
Békés megye 950150, 950250, 950350, 950450, 950550, 950650, 950660, 950750, 950850, 950860, 951050, 951150, 951250, 951260, 951350, 951450, 951460, 951550, 951650, 951750, 952150, 952250, 952350, 952450, 952550, 952650, 953250, 953260, 953270, 953350, 953450, 953550, 953560, 953950, 954050, 954060, 954150, 956250, 956350, 956450, 956550, 956650 és 956750 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Borsod-Abaúj-Zemplén megye valamennyi vadgazdálkodási egységének teljes területe, |
|
— |
Fejér megye 403150, 403160, 403260, 404250, 404550, 404560, 405450, 405550, 405650, 406450 és 407050 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Hajdú-Bihar megye valamennyi vadgazdálkodási egységének teljes területe, |
|
— |
Heves megye valamennyi vadgazdálkodási egységének teljes területe, |
|
— |
Jász-Nagykun-Szolnok megye 750250, 750550, 750650, 750750, 750850, 750970, 750980, 751050, 751150, 751160, 751250, 751260, 751350, 751360, 751450, 751460, 751470, 751550, 751650, 751750, 751850, 751950, 752150, 752250, 752350, 752450, 752460, 752550, 752560, 752650, 752750, 752850, 752950, 753060, 753070, 753150, 753250, 753310, 753450, 753550, 753650, 753660, 753750, 753850, 753950, 753960, 754050, 754150, 754250, 754360, 754370, 754850, 755550, 755650 és 755750 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Komárom-Esztergom megye: 251950, 252050, 252350, 252450, 252460, 252550, 252650, 252750, 252850, 252860, 252950, 252960, 253050, 253150, 253250, 253350, 253450 és 253550 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Nógrád megye valamennyi vadgazdálkodási egységeinek teljes területe, |
|
— |
Pest megye 570150, 570250, 570350, 570450, 570550, 570650, 570750, 570850, 570950, 571050, 571150, 571250, 571350, 571650, 571750, 571760, 571850, 571950, 572050, 573550, 573650, 574250, 577250, 580050 és 580150 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Szabolcs-Szatmár-Bereg megye valamennyi vadgazdálkodási egységének teljes területe. |
7. Polonia
Las siguientes zonas restringidas II de Polonia:
w województwie warmińsko-mazurskim:
|
— |
gminy Kalinowo, Stare Juchy, Prostki oraz gmina wiejska Ełk w powiecie ełckim, |
|
— |
powiat elbląski, |
|
— |
powiat miejski Elbląg, |
|
— |
powiat gołdapski, |
|
— |
powiat piski, |
|
— |
powiat bartoszycki, |
|
— |
gminy Biskupiec, Jeziorany, Kolno, część gminy Olsztynek położona na południe od linii wyznaczonej przez drogę nr S51 biegnącą od wschodniej granicy gminy do miejscowości Ameryka oraz na zachód od linii wyznaczonej przez drogę biegnącą od skrzyżowania z drogą S51 do północnej granicy gminy, łączącej miejscowości Mańki – Mycyny – Ameryka w powiecie olsztyńskim, |
|
— |
powiat ostródzki, |
|
— |
powiat olecki, |
|
— |
powiat giżycki, |
|
— |
powiat braniewski, |
|
— |
powiat kętrzyński, |
|
— |
gminy Lubomino i Orneta w powiecie lidzbarskim, |
|
— |
gmina Nidzica i część gminy Kozłowo położona na północ od linii wyznaczonej przez linię kolejową w powiecie nidzickim, |
|
— |
gminy Dźwierzuty, Jedwabno, Pasym, Szczytno i miasto Szczytno i Świętajno w powiecie szczycieńskim, |
|
— |
powiat mrągowski, |
|
— |
gminy Lubawa, miasto Lubawa, Zalewo, miasto Iława i część gminy wiejskiej Iława położona na wschód od linii wyznaczonej przez drogę nr 521 biegnącą od zachodniej granicy gminy do skrzyżowania z drogą łączącą miejscowości Szymbark - Ząbrowo - Segnowy – Laseczno – Gulb, a następnie na wschód od linii wyznaczonej przez drogę łączącą miejscowości Szymbark - Ząbrowo - Segnowy – Laseczno - Gulb biegnącą do południowej granicy gminy w powiecie iławskim, |
|
— |
część gminy wiejskiej Nowe Miasto Lubawskie położona na północ od linii wyznaczonej przez drogę biegnącą od zachodniej granicy gminy do miejscowości Lekarty, a następnie na północny -wschód od linii wyznaczonej przez drogę łączącą miejscowości Lekarty – Nowy Dwór Bratiański biegnącą do północnej granicy gminy miejskiej Nowe Miasto Lubawskie oraz na północ od linii wyznaczonej przez drogę nr 538, część gminy Grodziczno położona na północ od linii wyznaczonej przez drogę nr 538 w powiecie nowomiejskim, |
|
— |
powiat węgorzewski, |
|
— |
część gminy Rybno położona na północ od linii kolejowej, część gminy wiejskiej Działdowo położona na północ od linii wyznaczonej przez linie kolejowe biegnące od wschodniej do zachodniej granicy gminy w powiecie działdowskim, |
w województwie podlaskim:
|
— |
powiat bielski, |
|
— |
powiat grajewski, |
|
— |
powiat moniecki, |
|
— |
powiat sejneński, |
|
— |
gminy Łomża, Piątnica, Jedwabne, Przytuły i Wizna w powiecie łomżyńskim, |
|
— |
powiat miejski Łomża, |
|
— |
powiat siemiatycki, |
|
— |
powiat hajnowski, |
|
— |
gminy Ciechanowiec, Klukowo, Szepietowo, Kobylin-Borzymy, Nowe Piekuty, Sokoły i część gminy Kulesze Kościelne położona na północ od linii wyznaczonej przez linię kolejową w powiecie wysokomazowieckim, |
|
— |
gmina Rutki i część gminy Kołaki Kościelne położona na północ od linii wyznaczonej przez linię kolejową w powiecie zambrowskim, |
|
— |
gminy Mały Potok i Stawiski w powiecie kolneńskim, |
|
— |
powiat białostocki, |
|
— |
powiat suwalski, |
|
— |
powiat miejski Suwałki, |
|
— |
powiat augustowski, |
|
— |
powiat sokólski, |
|
— |
powiat miejski Białystok, |
w województwie mazowieckim:
|
— |
gminy Domanice, Korczew, Kotuń, Mordy, Paprotnia, Przesmyki, Siedlce, Skórzec, Wiśniew, Wodynie, Zbuczyn w powiecie siedleckim, |
|
— |
powiat miejski Siedlce, |
|
— |
gminy Ceranów, Jabłonna Lacka, Kosów Lacki, Repki, Sabnie, Sterdyń w powiecie sokołowskim, |
|
— |
powiat łosicki, |
|
— |
powiat sochaczewski, |
|
— |
gminy Policzna, Przyłęk, Tczów i Zwoleń w powiecie zwoleńskim, |
|
— |
powiat kozienicki, |
|
— |
gminy Chotcza i Solec nad Wisłą w powiecie lipskim, |
|
— |
gminy Gózd, Jastrzębia, Jedlnia Letnisko, Pionki z miastem Pionki, Skaryszew, Jedlińsk, Przytyk, Zakrzew, część gminy Iłża położona na zachód od linii wyznaczonej przez drogę nr 9, część gminy Wolanów położona na północ od drogi nr 12 w powiecie radomskim, |
|
— |
gminy Bodzanów, Słubice, Wyszogród i Mała Wieś w powiecie płockim, |
|
— |
powiat nowodworski, |
|
— |
gminy Czerwińsk nad Wisłą, Naruszewo, Załuski w powiecie płońskim, |
|
— |
gminy: miasto Kobyłka, miasto Marki, miasto Ząbki, miasto Zielonka w powiecie wołomińskim, |
|
— |
gminy Borowie, Garwolin z miastem Garwolin, Miastków Kościelny, Parysów, Pilawa, część gminy Wilga położona na północ od linii wyznaczonej przez rzekę Wilga biegnącą od wschodniej granicy gminy do ujścia do rzeki Wisły, część gminy Górzno położona na północ od linii wyznaczonej przez drogę łączącą miejscowości Łąki i Górzno biegnącą od wschodniej granicy gminy, następnie od miejscowości Górzno na północ od drogi nr 1328W biegnącej do drogi nr 17, a następnie na północ od linii wyznaczonej przez drogę biegnącą |
|
— |
od drogi nr 17 do zachodniej granicy gminy przez miejscowości Józefów i Kobyla Wola w powiecie garwolińskim, |
|
— |
gminy Boguty – Pianki, Zaręby Kościelne, Nur i część gminy Małkinia Górna położona na południe od rzeki Brok w powiecie ostrowskim, |
|
— |
gminy Chlewiska i Szydłowiec w powiecie szydłowieckim, |
|
— |
gminy Cegłów, Dębe Wielkie, Halinów, Latowicz, Mińsk Mazowiecki i miasto Mińsk Mazowiecki, Mrozy, Siennica, miasto Sulejówek w powiecie mińskim, |
|
— |
powiat otwocki, |
|
— |
powiat warszawski zachodni, |
|
— |
powiat legionowski, |
|
— |
powiat piaseczyński, |
|
— |
powiat pruszkowski, |
|
— |
powiat grójecki, |
|
— |
powiat grodziski, |
|
— |
powiat żyrardowski, |
|
— |
powiat białobrzeski, |
|
— |
powiat przysuski, |
|
— |
powiat miejski Warszawa, |
w województwie lubelskim:
|
— |
powiat bialski, |
|
— |
powiat miejski Biała Podlaska, |
|
— |
gminy Batorz, Godziszów, Janów Lubelski, Modliborzyce i Potok Wielki w powiecie janowskim, |
|
— |
gminy Janowiec, Kazimierz Dolny, Końskowola, Kurów, Markuszów, Nałęczów, Puławy z miastem Puławy, Wąwolnica i Żyrzyn w powiecie puławskim, |
|
— |
gminy Nowodwór, miasto Dęblin i część gminy Ryki położona na południe od linii wyznaczonej przez linię kolejową powiecie ryckim, |
|
— |
gminy Adamów, Krzywda, Stoczek Łukowski z miastem Stoczek Łukowski, Wola Mysłowska, Trzebieszów, Stanin, Wojcieszków, gmina wiejska Łuków i miasto Łuków w powiecie łukowskim, |
|
— |
powiat lubelski, |
|
— |
powiat miejski Lublin, |
|
— |
gminy Niedźwiada, Ostrówek, Ostrów Lubelski, Serniki, Uścimów i Lubartów z miastem Lubartów w powiecie lubartowskim, |
|
— |
powiat łęczyński, |
|
— |
powiat świdnicki, |
|
— |
gminy Fajsławice, Gorzków, Izbica, Krasnystaw z miastem Krasnystaw, Kraśniczyn, Łopiennik Górny, Siennica Różana i część gminy Żółkiewka położona na północ od linii wyznaczonej przez drogę nr 842 w powiecie krasnostawskim, |
|
— |
gminy Chełm, Ruda – Huta, Sawin, Rejowiec, Rejowiec Fabryczny z miastem Rejowiec Fabryczny, Siedliszcze, Wierzbica, Żmudź, Dorohusk, Dubienka, Kamień, Leśniowice, Wojsławice w powiecie chełmskim, |
|
— |
powiat miejski Chełm, |
|
— |
powiat kraśnicki, |
|
— |
powiat opolski, |
|
— |
powiat parczewski, |
|
— |
powiat włodawski, |
|
— |
powiat radzyński, |
|
— |
powiat miejski Zamość, |
|
— |
gminy Sitno, Skierbieszów, Stary Zamość, Zamość w powiecie zamojskim |
w województwie podkarpackim:
|
— |
powiat stalowowolski, |
|
— |
gminy Oleszyce, Lubaczów z miastem Lubaczów, Wielkie Oczy w powiecie lubaczowskim, |
|
— |
część gminy Kamień położona na zachód od linii wyznaczonej przez drogę nr 19, część gminy Sokołów Małopolski położona na północ od linii wyznaczonej przez drogę nr 875 w powiecie rzeszowskim, |
|
— |
gminy Cmolas i Majdan Królewski w powiecie kolbuszowskim, |
|
— |
gminy Grodzisko Dolne, część gminy wiejskiej Leżajsk położona na południe od miasta Leżajsk oraz na zachód od linii wyznaczonej przez rzekę San, w powiecie leżajskim, |
|
— |
gmina Jarocin, część gminy Harasiuki położona na północ od linii wyznaczona przez drogę nr 1048 R, część gminy Ulanów położona na północ od linii wyznaczonej przez rzekę Tanew, część gminy Nisko położona na zachód od linii wyznaczonej przez drogę nr 19 oraz na północ od linii wyznaczonej przez linię kolejową biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 19, część gminy Jeżowe położona na zachód od linii wyznaczonej przez drogę nr 19 w powiecie niżańskim, |
|
— |
powiat tarnobrzeski, |
|
— |
część gminy wiejskiej Przeworsk położona na zachód od miasta Przeworsk i na zachód od linii wyznaczonej przez autostradę A4 biegnącą od granicy z gminą Tryńcza do granicy miasta Przeworsk, część gminy Zarzecze położona na zachód od linii wyznaczonej przez drogę nr 1594R biegnącą od północnej granicy gminy do miejscowości Zarzecze oraz na południe od linii wyznaczonej przez drogi nr 1617R oraz 1619R biegnącą do południowej granicy gminy oraz na północ od linii wyznaczonej przez rzekę Mleczka w powiecie przeworskim, |
w województwie pomorskim:
|
— |
gminy Dzierzgoń i Stary Dzierzgoń w powiecie sztumskim, |
|
— |
gmina Stare Pole w powiecie malborskim, |
|
— |
gminy Stegny, Sztutowo i część gminy Nowy Dwór Gdański położona na północny - wschód od linii wyznaczonej przez drogę nr 55 biegnącą od |
|
— |
południowej granicy gminy do skrzyżowania z drogą nr 7, następnie przez drogę nr 7 i S7 biegnącą do zachodniej granicy gminy w powiecie nowodworskim, |
w województwie świętokrzyskim:
|
— |
gmina Tarłów i część gminy Ożarów położona na północ od linii wyznaczonej przez drogę nr 74 w powiecie opatowskim, |
|
— |
część gminy Brody położona na zachód od linii kolejowej biegnącej od miejscowości Marcule i od północnej granicy gminy przez miejscowości Klepacze i Karczma Kunowska do południowej granicy gminy oraz na wschód od linii wyznaczonej przez drogę nr 9 i na północny - wschód od linii wyznaczonej przez drogę nr 0618T biegnącą od północnej granicy gminy do skrzyżowania w miejscowości Lipie oraz przez drogę biegnącą od miejscowości Lipie do wschodniej granicy gminy i część gminy Mirzec położona na wschód od linii wyznaczonej przez drogę nr 744 biegnącą od południowej granicy gminy do miejscowości Tychów Stary a następnie przez drogę nr 0566T biegnącą od miejscowości Tychów Stary w kierunku północno – wschodnim do granicy gminy w powiecie starachowickim, |
|
— |
gmina Gowarczów, część gminy Końskie położona na wschód od linii kolejowej, część gminy Stąporków położona na północ od linii kolejowej w powiecie koneckim, |
w województwie lubuskim:
|
— |
powiat wschowski, |
|
— |
gmina Kostrzyn nad Odrą i część gminy Witnica położona na południowy zachód od drogi biegnącej od zachodniej granicy gminy od miejscowości Krześnica, przez miejscowości Kamień Wielki - Mościce - Witnica - Kłopotowo do południowej granicy gminy w powiecie gorzowskim, |
|
— |
gminy Gubin z miastem Gubin, Maszewo i część gminy Bytnica położona na zachód od linii wyznaczonej przez drogę nr 1157F w powiecie krośnieńskim, |
|
— |
powiat słubicki, |
|
— |
gminy Słońsk, Sulęcin i Torzym w powiecie sulęcińskim, |
|
— |
gminy Bledzew i Międzyrzecz w powiecie międzyrzeckim, |
|
— |
gminy Kolsko, część gminy Kożuchów położona na południe od linii wyznaczonej przez drogę 283 biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 290 i na południe od linii wyznaczonej przez drogę nr 290 biegnącej od miasta Mirocin Dolny do zachodniej granicy gminy, część gminy Bytom Odrzański położona na północny zachód od linii wyznaczonej przez drogi nr 293 i 326, część gminy Nowe Miasteczko położona na zachód od linii wyznaczonych przez drogi 293 i 328, część gminy Siedlisko położona na północny zachód od linii wyznaczonej przez drogę biegnącą od rzeki Odry przy południowe granicy gminy do drogi nr 326 łączącej się z drogą nr 325 biegnącą w kierunku miejscowości Różanówka do skrzyżowania z drogą nr 321 biegnącą od tego skrzyżowania w kierunku miejscowości Bielawy, a następnie przedłużoną przez drogę przeciwpożarową biegnącą od drogi nr 321 w miejscowości Bielawy do granicy gminy w powiecie nowosolskim, |
|
— |
gminy Nowogród Bobrzański, Trzebiechów, część gminy Bojadła położona na północ od linii wyznaczonej przez drogę nr 278 biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 282 i na północ od linii |
|
— |
wyznaczonej przez drogę nr 282 biegnącej od miasta Bojadła do zachodniej granicy gminy, część gminy Sulechów położona na wschód od linii wyznaczonej przez drogę nr S3 oraz na południe od linii wyznaczonej przez drogę łączącą miejscowości Kępsko - Buków biegnącą od zachodniej granicy gminy do miejscowości Buków, a następnie na wschód od linii wyznaczonej przez drogę łączącą miejscowości Buków – Miłkowo biegnącą od miejscowości Buków do północnej granicy gminy w powiecie zielonogórskim, |
|
— |
powiat żarski, |
|
— |
gminy Brzeźnica, Iłowa, Małomice, Szprotawa, Wymiarki, Żagań, miasto Żagań, miasto Gozdnica, część gminy Niegosławice położona na zachód od linii wyznaczonej przez drogę nr 328 w powiecie żagańskim, |
|
— |
gmina Łagów, część gminy Lubrza położona na północ od linii wyznaczonej przez autostradę A2 i część gminy Świebodzin położona na północ od linii wyznaczonej przez autostradę A2w powiecie świebodzińskim, |
w województwie dolnośląskim:
|
— |
gmina Pęcław, część gminy Kotla położona na północ od linii wyznaczonej przez rzekę Krzycki Rów, część gminy wiejskiej Głogów położona na wschód od linii wyznaczonej przez drogi nr 12, 319 oraz 329, część miasta Głogów położona na wschód od linii wyznaczonej przez drogę nr 12 w powiecie głogowskim, |
|
— |
gminy Grębocice i Polkowice w powiecie polkowickim, |
|
— |
gmina Rudna w powiecie lubińskim, |
|
— |
część gminy Niechlów położona na południowy – zachód od linii wyznaczonej przez rzekę Barycz, część gminy Jemielno położona na zachód od linii wyznaczonej przez drogę nr 323 w powiecie górowskim, |
w województwie wielkopolskim:
|
— |
gminy Przemęt i Wolsztyn w powiecie wolsztyńskim, |
|
— |
gmina Wielichowo część gminy Kamieniec położona na zachód od linii wyznaczonej przez drogę nr 308 i część gminy Rakoniewice położona na zachód od linii wyznaczonej przez drogę nr 305 w powiecie grodziskim, |
|
— |
gminy Wijewo, Włoszakowice, część gminy Lipno położona na zachód od linii wyznaczonej przez drogę nr S5 i część gminy Święciechowa położona na północ od linii wyznaczonej przez drogę nr 12 oraz na zachód od linii wyznaczonej przez drogę nr S5 w powiecie leszczyńskim, |
|
— |
część gminy Śmigiel położona na zachód od linii wyznaczonej przez drogę nr S5, w powiecie kościańskim, |
|
— |
powiat obornicki, |
|
— |
część gminy Połajewo na położona na południe od drogi łączącej miejscowości Chraplewo, Tarnówko-Boruszyn, Krosin, Jakubowo, Połajewo - ul. Ryczywolska do północno-wschodniej granicy gminy w powiecie czarnkowsko-trzcianeckim, |
|
— |
gmina Suchy Las, część gminy wiejskiej Murowana Goślina położona na północ od linii kolejowej biegnącej od północnej granicy miasta Murowana Goślina do północno-wschodniej granicy gminy oraz część gminy Rokietnica położona na północ i na wschód od linii kolejowej biegnącej od północnej |
|
— |
granicy gminy w miejscowości Krzyszkowo do południowej granicy gminy w miejscowości Kiekrz w powiecie poznańskim, |
|
— |
gmina Pniewy, część gminy Duszniki położona na północny – zachód od linii wyznaczonej przez drogę nr 306 biegnącą od północnej granicy gminy do miejscowości Duszniki, a następnie na północ od linii wyznaczonej przez ul. Niewierską oraz drogę biegnącą przez miejscowość Niewierz do zachodniej granicy gminy, część gminy Ostroróg położona na zachód od linii wyznaczonej przez drogę nr 186 i 184 biegnące od granicy gminy do miejscowości Ostroróg, a następnie od miejscowości Ostroróg przez miejscowości Piaskowo – Rudki do południowej granicy gminy, część gminy Wronki położona na południe od linii wyznaczonej przez drogi nr 182 i 186, część gminy Szamotuły położona na zachód od linii wyznaczonej przez drogę nr 306 oraz na wschód od wschodniej granicy miasta Szamotuły i na północ od linii kolejowej biegnącej od południowej granicy miasta Szamotuły do południowo-wschodniej granicy gminy oraz część gminy Obrzycko położona na wschód od drogi nr 185 łączącej miejscowości Gaj Mały, Słopanowo i Obrzycko do północnej granicy miasta Obrzycko, a następnie na wschód od drogi przebiegającej przez miejscowość Chraplewo w powiecie szamotulskim, |
|
— |
część gminy Sieraków położona na wschód od linii wyznaczonej przez drogę nr 186 biegnącą od południowej granicy gminy do miejscowości Lutomek, a następnie na wschód od linii wyznaczonej przez drogę biegnącą od skrzyżowania z drogą nr 186 w miejscowości Lutomek biegnącą do skrzyżowania z ul. Leśną w miejscowości Lutom i dalej na wschód od ul. Leśnej biegnącej do wschodniej granicy gminy, część gminy Kwilcz położona na wschód od linii wyznaczonej przez drogę nr 186 biegnącą od północnej granicy gminy do skrzyżowania z drogą nr 24, następnie na północ od linii wyznaczonej przez drogę nr 24 biegnącą od skrzyżowania z drogą nr 186 do skrzyżowania z drogą w miejscowości Pólko, i dalej na wschód od linii wyznaczonej przez drogę biegnącą od miejscowości Pólko przez miejscowość Wituchowo do południowej granicy gminy w powiecie międzychodzkim |
w województwie łódzkim:
|
— |
gminy Białaczów, Drzewica, Opoczno i Poświętne w powiecie opoczyńskim, |
|
— |
gminy Biała Rawska, Regnów i Sadkowice w powiecie rawskim, |
|
— |
gmina Kowiesy w powiecie skierniewickim, |
w województwie zachodniopomorskim:
|
— |
gmina Boleszkowice i część gminy Dębno położona na zachód od linii wyznaczonej przez drogę nr 126 biegnącą od zachodniej granicy gminy do skrzyżowania z drogą nr 23 w miejscowości Dębno, następnie na zachód od linii wyznaczonej przez drogę nr 23 do skrzyżowania z ul. Jana Pawła II w miejscowości Cychry, następnie na południe od ul. Jana Pawła II do skrzyżowania z ul. Ogrodową i dalej na południe od linii wyznaczonej przez ul. Ogrodową, której przedłużenie biegnie do wschodniej granicy gminy w powiecie myśliborskim, |
|
— |
gminy Mieszkowice, Moryń, część gminy Cedynia położona na południe od linii wyznaczonej przez drogę nr 124 biegnącą od zachodniej granicy gminy do miasta Cedynia, a następnie na południe od linii wyznaczonej przez drogę nr 125 biegnącą od miasta Cedynia do wschodniej granicy gminy w powiecie gryfińskim. |
8. Eslovaquia
Las siguientes zonas restringidas II de Eslovaquia:
|
— |
the whole district of Gelnica, |
|
— |
the whole district of Spišská Nová Ves, |
|
— |
the whole district of Levoča, |
|
— |
in the whole district of Michalovce, |
|
— |
the whole district of Košice-okolie, |
|
— |
the whole district of Rožnava, |
|
— |
the whole city of Košice, |
|
— |
the whole district of Sobrance, |
|
— |
in the district of Vranov nad Topľou, the whole municipalities of Zámutov, Rudlov, Jusková Voľa, Banské, Cabov, Davidov, Kamenná Poruba, Vechec, Čaklov, Soľ, Komárany, Čičava, Nižný Kručov, Vranov nad Topľou, Sačurov, Sečovská Polianka, Dlhé Klčovo, Nižný Hrušov, Poša, Nižný Hrabovec, Hencovce, Kučín, Majerovce, Sedliská, Kladzany and Tovarnianska Polianka, Herrmanovce nad Topľou, Petrovce, Pavlovce, Hanušovce nad Topľou, Medzianky, Radvanovce, Babie, Vlača, Ďurďoš, Prosačov, Remeniny, |
|
— |
Skrabské, Bystré, Petkovce, Michalok, Vyšný Žipov, Čierne nad Topľou, Zlatník, Hlinné, Jastrabie nad Topľou, Merník, Ondavské Maťašovce, Tovarné, |
|
— |
in the district of Humenné the whole municipalities of Hudcovce, Brekov, Jasenov, Ptičie, Chlmec, Porúbka, |
|
— |
the whole district of Prešov, |
|
— |
in the whole district of Sabinov, |
|
— |
in the district of Svidník, the whole municipalities of Dukovce, Želmanovce, Kuková, Kalnište, Lužany pri Ondave, Lúčka, Giraltovce, Kračúnovce, Železník, Kobylince, Mičakovce, |
|
— |
the whole district of Bardejov, |
|
— |
in the district of Stará Ľubovňa, the whole municipalities of Kyjov, Pusté Pole, Šarišské Jastrabie, Čirč, Ruská Voľa nad Popradom, Obručné, Vislanka, Ďurková, Plaveč, Ľubotín, Orlov, |
|
— |
the whole district of Revúca, |
|
— |
the whole district of Rimavská Sobota, |
|
— |
in the district of Veľký Krtíš, the whole municipalities of Ľuboriečka, Muľa, Dolná Strehová, Závada, Pravica, Chrťany, Senné, Brusník, Horná Strehová, Slovenské Kľačany, Vieska, Veľký Lom, Suché Brezovo, Horné Strháre, Dolné Strháre, Modrý Kameň,Veľký Krtíš, Veľké Zlievce, Malé Zlievce, Veľké Stračiny, Malé Stračiny, Bušince, Čeláre, Gabušovce, Zombor, Olováry, Malý Krtíš, Nová Ves, Šuľa, Červeňany, Sucháň, Dačov Lom, |
|
— |
the whole district of Lučenec, |
|
— |
the whole district of Poltár |
|
— |
in the district of Zvolen, the whole municipalities Lešť, Pliešovce |
|
— |
in the district of Detva, the whole municipalities of Stará Huta, Vígľašská Huta,- Kalinka, Slatinské Lazy, Stožok, Klokoč, Vígľaš, Detva, |
|
— |
in the district of Krupina the whole municipalities of Senohrad, Horné Mladonice, Dolné Mladonice, Čekovce, Lackov. |
PARTE III
1. Bulgaria
Las siguientes zonas restringidas III de Bulgaria:
|
— |
the whole region of Gabrovo, |
|
— |
the whole region of Lovech, |
|
— |
the whole region of Montana, |
|
— |
the Pleven region:
|
|
— |
the Ruse region:
|
|
— |
the Shumen region:
|
|
— |
the Silistra region:
|
|
— |
the Sliven region:
|
|
— |
the Targovishte region:
|
|
— |
the Vidin region,
|
|
— |
the Veliko Tarnovo region:
|
|
— |
the whole region of Vratza, |
|
— |
in Varna region:
|
|
— |
in Burgas region:
|
2. Italia
Las siguientes zonas restringidas III de Italia:
|
— |
tutto il territorio della Sardegna. |
3. Letonia
Las siguientes zonas restringidas III de Letonia:
|
— |
Aizputes novada Kalvenes pagasta daļa uz austrumiem no ceļa pie Vārtājas upes līdz autoceļam A9, uz ziemeļiem no autoceļa A9, uz austrumiem no autoceļa V1200, Kazdangas pagasta daļa uz austrumiem no ceļa V1200, P115, P117, V1296, |
|
— |
Kuldīgas novada, Laidu pagasta daļa uz dienvidiem no autoceļa V1296, |
|
— |
Skrundas novada Rudbāržu, Nīkrāces pagasts, Raņķu pagasta daļa uz dienvidiem no autoceļa V1272 līdz robežai ar Ventas upi, Skrundas pagasts (izņemot pagasta daļa no Skrundas uz ziemeļiem no autoceļa A9 un austrumiem no Ventas upes), Skrundas pilsēta, |
|
— |
Vaiņodes novada Embūtes pagasta daļa uz ziemeļiem autoceļa P116, P106. |
4. Lituania
Las siguientes zonas restringidas III de Lituania:
|
— |
Jurbarko rajono savivaldybė: Seredžiaus ir Juodaičių seniūnijos, |
|
— |
Kauno rajono savivaldybė: Čekiškės seniūnija, Babtų seniūnijos dalis į vakarus nuo kelio A1ir Vilkijos apylinkių seniūnijos dalis į rytus nuo kelio Nr. 1907, |
|
— |
Kėdainių rajono savivaldybė: Pernaravos seniūnija ir Josvainių seniūnijos pietvakarinė dalis tarp kelio Nr. 229 ir Nr. 2032, |
|
— |
Plungės rajono savivaldybė: Alsėdžių, Babrungo, Paukštakių, Platelių ir Žemaičių Kalvarijos seniūnijos, |
|
— |
Raseinių rajono savivaldybė: Ariogalos ir Ariogalos miesto seniūnijos, |
|
— |
Skuodo rajono savivaldybės: Barstyčių, Notėnų ir Šačių seniūnijos. |
5. Polonia
Las siguientes zonas restringidas III de Polonia:
w województwie warmińsko-mazurskim:
|
— |
gminy Kiwity i Lidzbark Warmiński z miastem Lidzbark Warmiński w powiecie lidzbarskim, |
|
— |
gminy Barczewo, Gietrzwałd, Jonkowo, Dywity, Dobre Miasto, Purda, Stawiguda, Świątki, część gminy Olsztynek położona na północ od linii wyznaczonej przez drogę nr S51 biegnącą od wschodniej granicy gminy do miejscowości Ameryka oraz na wschód od linii wyznaczonej przez drogę biegnącą od skrzyżowania z drogą S51 do północnej granicy gminy, łączącej miejscowości Mańki – Mycyny – Ameryka w powiecie olsztyńskim, |
|
— |
powiat miejski Olsztyn, |
w województwie mazowieckim:
|
— |
gminy Łaskarzew z miastem Łaskarzew, Maciejowice, Sobolew, Trojanów, Żelechów, część gminy Wilga położona na południe od linii wyznaczonej przez rzekę Wilga biegnącą od wschodniej granicy gminy do ujścia do rzeki Wisły, część gminy Górzno położona na południe od linii wyznaczonej przez drogę łączącą miejscowości Łąki i Górzno biegnącą od wschodniej granicy gminy, następnie od miejscowości Górzno na południe od drogi nr 1328W biegnącej do drogi nr 17, a następnie na południe od linii wyznaczonej przez drogę biegnącą od drogi nr 17 do zachodniej granicy gminy przez miejscowości Józefów i Kobyla Wola w powiecie garwolińskim, |
|
— |
część gminy Iłża położona na wschód od linii wyznaczonej przez drogę nr 9 w powiecie radomskim, |
|
— |
gmina Kazanów w powiecie zwoleńskim, |
|
— |
gminy Ciepielów, Lipsko, Rzeczniów i Sienno w powiecie lipskim, |
w województwie lubelskim:
|
— |
powiat tomaszowski, |
|
— |
gmina Białopole w powiecie chełmskim, |
|
— |
gmina Rudnik i część gminy Żółkiewka położona na południe od linii wyznaczonej przez drogę nr 842 w powiecie krasnostawskim, |
|
— |
gminy Adamów, Grabowiec, Komarów – Osada, Krasnobród, Łabunie, Miączyn, Nielisz, Radecznica, Sułów, Szczebrzeszyn, Zwierzyniec w powiecie zamojskim, |
|
— |
powiat biłgorajski, |
|
— |
powiat hrubieszowski, |
|
— |
gminy Dzwola i Chrzanów w powiecie janowskim, |
|
— |
gmina Serokomla w powiecie łukowskim, |
|
— |
gminy Abramów, Kamionka, Michów, Firlej, Jeziorzany, Kock w powiecie lubartowskim, |
|
— |
gminy Kłoczew, Stężyca, Ułęż i część gminy Ryki położona na północ od linii wyznaczonej przez linię kolejową w powiecie ryckim, |
|
— |
gmina Baranów w powiecie puławskim, |
w województwie podkarpackim:
|
— |
gminy Cieszanów, Horyniec – Zdrój, Narol i Stary Dzików w powiecie lubaczowskim, |
|
— |
gminy Kuryłówka, Nowa Sarzyna, miasto Leżajsk, część gminy wiejskiej Leżajsk położona na północ od miasta Leżajsk oraz część gminy wiejskiej Leżajsk położona na wschód od linii wyznaczonej przez rzekę San, w powiecie leżajskim, |
|
— |
gminy Krzeszów, Rudnik nad Sanem, część gminy Harasiuki położona na południe od linii wyznaczona przez drogę nr 1048 R, część gminy Ulanów położona na południe od linii wyznaczonej przez rzekę Tanew, część gminy Nisko położona na wschód od linii wyznaczonej przez drogę nr 19 oraz na południe od linii wyznaczonej przez linię kolejową biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 19, część gminy Jeżowe położona na wschód od linii wyznaczonej przez drogę nr 19 w powiecie niżańskim, |
|
— |
gminy Chłopice, Jarosław z miastem Jarosław, Laszki, Wiązownica, Pawłosiów, Radymno z miastem Radymno, w powiecie jarosławskim, |
|
— |
gmina Stubno w powiecie przemyskim, |
|
— |
część gminy Kamień położona na wschód od linii wyznaczonej przez drogę nr 19 w powiecie rzeszowskim, |
|
— |
gminy Adamówka, Sieniawa, Tryńcza, miasto Przeworsk, część gminy wiejskiej Przeworsk położona na wschód od miasta Przeworsk i na wschód od linii wyznaczonej przez autostradę A4 biegnącą od granicy z gminą Tryńcza do granicy miasta Przeworsk, część gminy Zarzecze położona na wschód od linii wyznaczonej przez drogę nr 1594R biegnącą od północnej granicy gminy do miejscowości Zarzecze oraz na północ od linii wyznaczonej przez drogi nr 1617R oraz 1619R biegnącą do południowej granicy gminy w powiecie przeworskim, |
w województwie lubuskim:
|
— |
gminy Nowa Sól i miasto Nowa Sól, Otyń oraz część gminy Kożuchów położona na północ od linii wyznaczonej przez drogę nr 283 biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 290 i na północ od linii wyznaczonej przez drogę nr 290 biegnącej od miasta Mirocin Dolny do zachodniej granicy gminy, część gminy Bytom Odrzański położona na południowy wschód od linii wyznaczonej przez drogi nr 293 i 326, część gminy Nowe Miasteczko położona na wschód od linii wyznaczonych przez drogi 293 i 328, część gminy Siedlisko położona na południowy wschód od linii wyznaczonej przez drogę biegnącą od rzeki Odry przy południowe granicy gminy do drogi nr 326 łączącej się z drogą nr 325 biegnącą w kierunku miejscowości Różanówka do skrzyżowania z drogą nr 321 biegnącą od tego skrzyżowania w kierunku miejscowości Bielawy, a następnie przedłużoną przez drogę przeciwpożarową biegnącą od drogi nr 321 w miejscowości Bielawy do granicy gminy w powiecie nowosolskim, |
|
— |
gminy Babimost, Czerwieńsk, Kargowa, Świdnica, Zabór, część gminy Bojadła położona na południe od linii wyznaczonej przez drogę nr 278 biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 282 i na południe od linii wyznaczonej przez drogę nr 282 biegnącej od miasta Bojadła do zachodniej granicy gminy i część gminy Sulechów położona na zachód od linii wyznaczonej przez drogę nr S3 oraz na północ od linii wyznaczonej przez |
|
— |
drogę łączącą miejscowości Kępsko - Buków biegnącą od zachodniej granicy gminy do miejscowości Buków, a następnie na zachód od linii wyznaczonej przez drogę łączącą miejscowości Buków – Miłkowo biegnącą od miejscowości Buków do północnej granicy gminy w powiecie zielonogórskim, |
|
— |
część gminy Niegosławice położona na wschód od linii wyznaczonej przez drogę nr 328 w powiecie żagańskim, |
|
— |
powiat miejski Zielona Góra, |
|
— |
gminy Skąpe, Szczaniec, Zbąszynek , część gminy Lubrza położona na południe od linii wyznaczonej przez autostradę A2 i część gminy Świebodzin położona na południe od linii wyznaczonej przez autostradę A2 w powiecie świebodzińskim, |
|
— |
gminy Bobrowice, Dąbie, Krosno Odrzańskie i część gminy Bytnica położona na wschód od linii wyznaczonej przez drogę nr 1157F w powiecie krośnieńskim, |
|
— |
część gminy Trzciel położona na południe od linii wyznaczonej przez drogę nr 92 w powiecie międzyrzeckim, |
w województwie wielkopolskim:
|
— |
gmina Zbąszyń, część gminy Miedzichowo położona na południe od linii wyznaczonej przez drogę nr 92, część gminy Nowy Tomyśl położona na zachód od linii wyznaczonej przez drogę nr 305 w powiecie nowotomyskim, |
|
— |
gmina Siedlec w powiecie wolsztyńskim, |
|
— |
część gminy Rakoniewice położona na wschód od linii wyznaczonej przez drogę nr 305 w powiecie grodziskim, |
w województwie dolnośląskim:
|
— |
gminy Jerzmanowa, Żukowice, część gminy Kotla położona na południe od linii wyznaczonej przez rzekę Krzycki Rów, część gminy wiejskiej Głogów położona na zachód od linii wyznaczonej przez drogi nr 12, 319 oraz 329, część miasta Głogów położona na zachód od linii wyznaczonej przez drogę nr 12 w powiecie głogowskim, |
|
— |
gminy Gaworzyce, Radwanice i część gminy Przemków położona na północ od linii wyznaczonej prze drogę nr 12 w powiecie polkowickim, |
w województwie świętokrzyskim:
|
— |
część gminy Brody położona na wschód od linii kolejowej biegnącej od miejscowości Marcule i od północnej granicy gminy przez miejscowości Klepacze i Karczma Kunowska do południowej granicy gminy w powiecie starachowickim. |
6. Rumanía
Las siguientes zonas restringidas III de Rumanía:
|
— |
Zona orașului București, |
|
— |
Județul Constanța, |
|
— |
Județul Satu Mare, |
|
— |
Județul Tulcea, |
|
— |
Județul Bacău, |
|
— |
Județul Bihor, |
|
— |
Județul Bistrița Năsăud, |
|
— |
Județul Brăila, |
|
— |
Județul Buzău, |
|
— |
Județul Călărași, |
|
— |
Județul Dâmbovița, |
|
— |
Județul Galați, |
|
— |
Județul Giurgiu, |
|
— |
Județul Ialomița, |
|
— |
Județul Ilfov, |
|
— |
Județul Prahova, |
|
— |
Județul Sălaj, |
|
— |
Județul Suceava |
|
— |
Județul Vaslui, |
|
— |
Județul Vrancea, |
|
— |
Județul Teleorman, |
|
— |
Judeţul Mehedinţi, |
|
— |
Județul Gorj, |
|
— |
Județul Argeș, |
|
— |
Judeţul Olt, |
|
— |
Judeţul Dolj, |
|
— |
Județul Arad, |
|
— |
Județul Timiș, |
|
— |
Județul Covasna, |
|
— |
Județul Brașov, |
|
— |
Județul Botoșani, |
|
— |
Județul Vâlcea, |
|
— |
Județul Iași, |
|
— |
Județul Hunedoara, |
|
— |
Județul Alba, |
|
— |
Județul Sibiu, |
|
— |
Județul Caraș-Severin, |
|
— |
Județul Neamț, |
|
— |
Județul Harghita, |
|
— |
Județul Mureș, |
|
— |
Județul Cluj, |
|
— |
Județul Maramureş. |
7. Eslovaquia
Las siguientes zonas restringidas III de Eslovaquia:
|
— |
the whole district of Trebišov. |
DECISIONES
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/147 |
DECISIÓN (UE) 2021/812 DEL CONSEJO
de 10 de mayo de 2021
relativa a la posición que debe adoptarse en nombre de la Unión Europea en el seno del Comité de Asociación, en su configuración de Comercio, y en el Consejo de Asociación establecido por el Acuerdo de Asociación entre la Unión Europea y la Comunidad Europea de la Energía Atómica y sus Estados miembros, por una parte, y Georgia, por otra, en lo que respecta a un dictamen favorable sobre el plan de trabajo detallado aprobado por el Gobierno de Georgia para la aplicación de la legislación relativa a la contratación pública, y por el que se reconoce la finalización de la fase 1 del anexo XVI-B de dicho Acuerdo de Asociación
EL CONSEJO DE LA UNIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea, y en particular su artículo 207, apartado 4, párrafo primero, en relación con su artículo 218, apartado 9,
Vista la propuesta de la Comisión Europea,
Considerando lo siguiente:
|
(1) |
El Acuerdo de Asociación entre la Unión Europea y la Comunidad Europea de la Energía Atómica y sus Estados miembros, por una parte, y Georgia, por otra (1) (en lo sucesivo, «Acuerdo»), fue celebrado por la Unión mediante la Decisión (UE) 2016/838 del Consejo (2), y entró en vigor el 1 de julio de 2016. |
|
(2) |
En el artículo 145, apartado 1, del Acuerdo se establece que Georgia debe presentar al Comité de Asociación, en su configuración de Comercio, un plan de trabajo detallado para la aplicación de la legislación sobre contratación pública de Georgia, con un calendario y plazos, que debe incluir todas las reformas en términos de aproximación legislativa al acervo de la Unión. |
|
(3) |
En aplicación del artículo 145, apartado 2, del Acuerdo, el dictamen favorable del Comité de Asociación, en su configuración de Comercio, es necesario para que el plan de trabajo detallado se convierta en el documento de referencia para ejecutar la aproximación de la legislación sobre contratación pública de Georgia al acervo de la Unión sobre ese tema. |
|
(4) |
De conformidad con el artículo 146, apartado 2, del Acuerdo, la aproximación al acervo de la Unión debe llevarse a cabo en fases consecutivas, establecidas en el calendario que figura en el anexo XVI-B del Acuerdo. La ejecución de cada fase debe ser evaluada por el Comité de Asociación, en su configuración de Comercio, de conformidad con el artículo 408, apartado 4, del Acuerdo y, previa evaluación positiva de dicho Comité, debe vincularse a la concesión recíproca de acceso al mercado prevista en el anexo XVI-B del Acuerdo. |
|
(5) |
El Comité de Asociación, en su configuración de Comercio, con arreglo al artículo 11, apartado 2, de su reglamento interno, que figura en el anexo II de la Decisión n.o 1/2014 del Consejo de Asociación UE-Georgia (3), debe adoptar una decisión con un dictamen sobre el plan de trabajo detallado aprobado por el Gobierno de Georgia, así como una evaluación de la aproximación de la legislación de Georgia al acervo de la Unión realizada con vistas a la conclusión de la fase 1 prevista en el anexo XVI-B del Acuerdo. Dicho plan de trabajo fue aprobado por el Gobierno de Georgia mediante su Decreto n.o 536, de 31 de marzo de 2016, «relativo a los cambios previstos en el ámbito de la contratación pública en cumplimiento de las obligaciones entre Georgia y la UE en el ámbito de aplicación del Acuerdo sobre la zona de libre comercio de alcance amplio y profundo (ZLCAP)», modificado por los Decretos n.o 154, de 22 de enero de 2018, y n.o 974, de 12 de junio de 2020, del Gobierno de Georgia. |
|
(6) |
Tras el reconocimiento de la finalización de la fase 1 prevista en el anexo XVI-B del Acuerdo, el Consejo de Asociación, de conformidad con el artículo 11, apartado 2, de su reglamento interno, que figura en el anexo I de la Decisión n.o 1/2014 del Consejo de Asociación UE-Georgia, debe adoptar una decisión en cuanto a la concesión de acceso recíproco al mercado de suministros para las autoridades del gobierno central, de conformidad con el anexo XVI-B del Acuerdo. |
|
(7) |
Procede establecer la posición que debe adoptarse en nombre de la Unión en el Comité de Asociación, en su configuración de Comercio, y en el Consejo de Asociación, dado que las decisiones previstas serán vinculantes para la Unión. |
HA ADOPTADO LA PRESENTE DECISIÓN:
Artículo 1
La posición que debe adoptarse en nombre de la Unión en el Comité de Asociación, en su configuración de Comercio, por lo que respecta al plan de trabajo detallado aprobado por el Gobierno de Georgia, y la finalización de la fase 1 prevista en el anexo XVI-B del Acuerdo, se basará en el proyecto de Decisión del Comité de Asociación, en su configuración de Comercio (4).
Artículo 2
La posición que debe adoptarse en nombre de la Unión en el Consejo de Asociación en lo que respecta a la concesión de acceso recíproco al mercado de conformidad con el anexo XVI-B del Acuerdo se basará en el proyecto de Decisión del Consejo de Asociación (4).
Artículo 3
La presente Decisión entrará en vigor el día de su adopción.
Hecho en Bruselas, el 10 de mayo de 2021.
Por el Consejo
El Presidente
J. BORRELL FONTELLES
(1) DO L 261 de 30.8.2014, p. 4.
(2) Decisión (UE) 2016/838 del Consejo, de 23 de mayo de 2016, relativa a la celebración, en nombre de la Unión Europea, del Acuerdo de Asociación entre la Unión Europea y la Comunidad Europea de la Energía Atómica y sus Estados miembros, por una parte, y Georgia, por otra (DO L 141 de 28.5.2016, p. 26).
(3) Decisión n.o 1/2014 del Consejo de Asociación UE-Georgia, de 17 de noviembre de 2014, por la que se adopta su reglamento interno y el reglamento interno del Comité de Asociación y los subcomités [2015/2261] (DO L 321 de 5.12.2015, p. 60).
(4) Véase el documento ST 7791/21 en http://register.consilium.europa.eu.
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/149 |
DECISIÓN (PESC) 2021/813 DEL CONSEJO
de 20 de mayo de 2021
por la que se modifica la Decisión 2014/486/PESC relativa a la Misión asesora de la Unión Europea para la reforma del sector de la seguridad civil en Ucrania (EUAM Ucrania)
EL CONSEJO DE LA UNIÓN EUROPEA,
Visto el Tratado de la Unión Europea, y en particular su artículo 42, apartado 4, y su artículo 43, apartado 2,
Vista la propuesta del Alto Representante de la Unión para Asuntos Exteriores y Política de Seguridad,
Considerando lo siguiente:
|
(1) |
El 22 de julio de 2014, el Consejo adoptó la Decisión 2014/486/PESC relativa a la Misión asesora de la Unión Europea para la reforma del sector de la seguridad civil en Ucrania (EUAM Ucrania) (1). |
|
(2) |
El 13 de mayo de 2019, el Consejo adoptó la Decisión (PESC) 2019/761 (2), que prorrogaba el mandato de la EUAM Ucrania hasta el 31 de mayo de 2021. |
|
(3) |
En el contexto de una revisión estratégica de la EUAM Ucrania, el Comité Político y de Seguridad convino en que la EUAM Ucrania debía prorrogarse hasta el 31 de mayo de 2024 y en que, pasados dos años, se procedería a una evaluación estratégica centrada en la evolución de la dimensión política. |
|
(4) |
Por lo tanto, procede prorrogar la Decisión 2014/486/PESC hasta el 31 de mayo de 2024. |
|
(5) |
La EUAM Ucrania se llevará a cabo en el contexto de una situación que puede deteriorarse y que podría obstaculizar el logro de los objetivos de la acción exterior de la Unión que se recogen en el artículo 21 del Tratado. |
HA ADOPTADO LA PRESENTE DECISIÓN:
Artículo 1
La Decisión 2014/486/PESC se modifica como sigue:
|
1) |
En el artículo 14, apartado 1, se añade el párrafo siguiente: «El importe de referencia financiera destinado a cubrir los gastos asociados a la EUAM Ucrania para el período comprendido entre el 1 de junio de 2021 y el 31 de mayo de 2024 ascenderá a 88 500 000 EUR.». |
|
2) |
El artículo 18 se sustituye por el texto siguiente: «Artículo 18 Revisión estratégica Después del 31 de mayo de 2023 se procederá a una evaluación estratégica de la EUAM Ucrania centrada en la evolución de la dimensión política.». |
|
3) |
En el artículo 19, el párrafo segundo se sustituye por el texto siguiente: «Será aplicable hasta el 31 de mayo de 2024.». |
Artículo 2
La presente Decisión entrará en vigor el día de su adopción.
Hecho en Bruselas, el 20 de mayo de 2021.
Por el Consejo
El Presidente
A. SANTOS SILVA
(1) DO L 217 de 23.7.2014, p. 42.
(2) Decisión (PESC) 2019/761 del Consejo, de 13 de mayo de 2019, por la que se modifica la Decisión 2014/486/PESC relativa a la Misión asesora de la Unión Europea para la reforma del sector de la seguridad civil en Ucrania (EUAM Ucrania) (DO L 125 de 14.5.2019, p. 16).
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/151 |
DECISIÓN (PESC) 2021/814 DEL CONSEJO
de 20 de mayo de 2021
por la que se modifica la Decisión (PESC) 2017/915 sobre las actividades de sensibilización de la Unión en apoyo de la aplicación del Tratado sobre el Comercio de Armas
EL CONSEJO DE LA UNIÓN EUROPEA,
Visto el Tratado de la Unión Europea, y en particular su artículo 28, apartado 1, y su artículo 31, apartado 1,
Vista la propuesta del Alto Representante de la Unión para Asuntos Exteriores y Política de Seguridad,
Considerando lo siguiente:
|
(1) |
El 29 de mayo de 2017, el Consejo adoptó la Decisión (PESC) 2017/915 (1). |
|
(2) |
El 30 de julio de 2020, el Consejo adoptó la Decisión (PESC) 2020/1134 (2), por la que se modificaba la Decisión (PESC) 2017/915 y se ampliaba hasta el 30 de junio de 2021 el período de ejecución para las actividades a que se refiere su artículo 1. |
|
(3) |
El 31 de marzo de 2021, el Bundesamt für Wirtschaft und Ausfuhrkontrolle (Oficina federal alemana de economía y control de las exportaciones), y el 6 de abril de 2021, Expertise France, en su calidad de agencias de ejecución, solicitaron autorización a la Unión para prorrogar por segunda vez la aplicación de la Decisión (PESC) 2017/915 hasta el 31 de enero de 2022, debido a los desafíos derivados de la persistente pandemia de COVID-19. |
|
(4) |
La continuación de las actividades contempladas en el artículo 1 de la Decisión (PESC) 2017/915 no tiene repercusión alguna en materia de recursos financieros hasta el 31 de enero de 2022. |
HA ADOPTADO LA PRESENTE DECISIÓN:
Artículo 1
El artículo 5 de la Decisión (PESC) 2017/915 se sustituye por el texto siguiente:
«Artículo 5
La presente Decisión entrará en vigor el día de su adopción.
Expirará el 31 de enero de 2022.».
Artículo 2
La presente Decisión entrará en vigor el día de su adopción.
Hecho en Bruselas, el 20 de mayo de 2021.
Por el Consejo
El Presidente
A. SANTOS SILVA
(1) Decisión (PESC) 2017/915 del Consejo, de 29 de mayo de 2017, sobre las actividades de sensibilización de la Unión en apoyo de la aplicación del Tratado sobre el Comercio de Armas (DO L 139 de 30.5.2017, p. 38).
(2) Decisión (PESC) 2020/1134 del Consejo, de 30 de julio de 2020, por la que se modifica la Decisión (PESC) 2017/915 sobre las actividades de sensibilización de la Unión en apoyo de la aplicación del Tratado sobre el Comercio de Armas (DO L 247 de 31.7.2020, p. 24).
|
21.5.2021 |
ES |
Diario Oficial de la Unión Europea |
L 180/152 |
DECISIÓN DE EJECUCIÓN (PESC) 2021/815 DEL CONSEJO
de 20 de mayo de 2021
por la que se aplica la Decisión 2014/450/PESC relativa a medidas restrictivas habida cuenta de la situación en Sudán
EL CONSEJO DE LA UNIÓN EUROPEA,
Visto el Tratado de la Unión Europea, y en particular su artículo 31, apartado 2,
Vista la Decisión 2014/450/PESC del Consejo, de 10 de julio de 2014, relativa a medidas restrictivas habida cuenta de la situación en Sudán y por la que se deroga la Decisión 2011/423/PESC (1), y en particular su artículo 6,
Vista la propuesta del Alto Representante de la Unión para Asuntos Exteriores y Política de Seguridad,
Considerando lo siguiente:
|
(1) |
El 10 de julio de 2014, el Consejo adoptó la Decisión 2014/450/PESC. |
|
(2) |
El 5 de marzo de 2021, el Comité del Consejo de Seguridad de las Naciones Unidas, creado en virtud de la Resolución 1591 (2005) de dicho Consejo de Seguridad, decidió suprimir el nombre de una persona de la lista de personas y entidades sujetas a medidas restrictivas. |
|
(3) |
Por lo tanto, procede modificar el anexo de la Decisión 2014/450/PESC en consecuencia. |
HA ADOPTADO LA PRESENTE DECISIÓN:
Artículo 1
El anexo de la Decisión 2014/450/PESC se modifica como se establece en el anexo de la presente Decisión.
Artículo 2
La presente Decisión entrará en vigor el día de su publicación en el Diario Oficial de la Unión Europea.
Hecho en Bruselas, el 20 de mayo de 2021.
Por el Consejo
El Presidente
A. SANTOS SILVA
ANEXO
En la lista que figura en el anexo de la Decisión 2014/450/PESC, se suprime la entrada correspondiente a la siguiente persona:
|
3. |
SHAREIF, Adam. |