ISSN 1725-2512
Diario Oficial
de la Unión Europea
L 54

Edición en lengua española
Legislación
52o año
26 de febrero de 2009
|
ISSN 1725-2512 |
||
|
Diario Oficial de la Unión Europea |
L 54 |
|
|
|
||
|
Edición en lengua española |
Legislación |
52o año |
|
Sumario |
|
I Actos adoptados en aplicación de los Tratados CE/Euratom cuya publicación es obligatoria |
Página |
|
|
|
REGLAMENTOS |
|
|
|
* |
Reglamento (CE) no 152/2009 de la Comisión, de 27 de enero de 2009, por el que se establecen los métodos de muestreo y análisis para el control oficial de los piensos ( 1 ) |
|
|
|
||
|
|
* |
|
|
|
|
|
(1) Texto pertinente a efectos del EEE |
|
ES |
Los actos cuyos títulos van impresos en caracteres finos son actos de gestión corriente, adoptados en el marco de la política agraria, y que tienen generalmente un período de validez limitado. Los actos cuyos títulos van impresos en caracteres gruesos y precedidos de un asterisco son todos los demás actos. |
I Actos adoptados en aplicación de los Tratados CE/Euratom cuya publicación es obligatoria
REGLAMENTOS
|
26.2.2009 |
ES |
Diario Oficial de la Unión Europea |
L 54/1 |
REGLAMENTO (CE) no152/2009 DE LA COMISIÓN
de 27 de enero de 2009.
por el que se establecen los métodos de muestreo y análisis para el control oficial de los piensos
(Texto pertinente a efectos del EEE)
LA COMISIÓN DE LAS COMUNIDADES EUROPEAS,
Visto el Tratado constitutivo de la Comunidad Europea,
Visto el Reglamento (CE) no 882/2004 del Parlamento Europeo y del Consejo, de 29 de abril de 2004, sobre los controles oficiales efectuados para garantizar la verificación del cumplimiento de la legislación en materia de piensos y alimentos y la normativa sobre salud animal y bienestar de los animales (1), y, en particular, su artículo 11, apartado 4, letras a), b) y c),
Considerando lo siguiente:
|
(1) |
Los actos que se citan a continuación se adoptaron para implementar la Directiva 70/373/CEE y siguen estando en vigor según el artículo 62, apartado 2, del Reglamento (CE) no 882/2004:
|
|
(2) |
Dado que la Directiva 70/373/CEE fue sustituida por el Reglamento (CE) no 882/2004, conviene reemplazar los actos de desarrollo de dicha Directiva por un único reglamento. Al mismo tiempo, es conveniente adaptar los métodos a los nuevos conocimientos científicos y tecnológicos. Los métodos que han dejado de ser válidos para el fin al que están destinados deben suprimirse. Aunque está previsto actualizar a su debido tiempo las disposiciones relativas al muestreo, a fin de tener en cuenta los últimos avances en las formas de producción, almacenamiento, transporte y comercialización de los piensos, conviene mantener por el momento las disposiciones existentes a este respecto. |
|
(3) |
Deben, pues, derogarse las Directivas 71/250/CEE, 71/393/CEE, 72/199/CEE, 73/46/CEE, 76/371/CEE, 76/372/CEE, 78/633/CEE, 81/715/CEE, 84/425/CEE, 86/174/CEE, 93/70/CEE, 93/117/CE, 98/64/CE, 1999/27/CE, 1999/76/CE, 2000/45/CE, 2002/70/CE y 2003/126/CE. |
|
(4) |
Las medidas previstas en el presente Reglamento se ajustan al dictamen del Comité permanente de la cadena alimentaria y de sanidad animal. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
El muestreo para el control oficial de los piensos en lo que se refiere a la determinación de los componentes, los aditivos y las sustancias indeseables, con excepción de los residuos de plaguicidas y los microorganismos, se llevará a cabo de acuerdo con los métodos expuestos en el anexo I.
Artículo 2
La preparación de las muestras para el análisis y la expresión de los resultados se efectuarán conforme a los métodos expuestos en el anexo II.
Artículo 3
Los análisis para el control oficial de los piensos se realizarán aplicando los métodos expuestos en el anexo III (Métodos de análisis para el control de la composición de los materiales para piensos y los piensos compuestos), el anexo IV (Métodos de análisis para el control del nivel de aditivos autorizados en los piensos), el anexo V (Métodos de análisis para el control de sustancias indeseables en los piensos) y el anexo VI (Métodos de análisis para la determinación de componentes de origen animal con fines de control oficial de los piensos).
Artículo 4
El valor energético de los piensos compuestos para aves de corral se calculará conforme al anexo VII.
Artículo 5
Los métodos de análisis expuestos en el anexo VIII, utilizados para controlar la presencia ilícita de aditivos que ya no están autorizados en los piensos, se emplearán con fines de confirmación.
Artículo 6
Quedan derogadas las Directivas 71/250/CEE, 71/393/CEE, 72/199/CEE, 73/46/CEE, 76/371/CEE, 76/372/CEE, 78/633/CEE, 81/715/CEE, 84/425/CEE, 86/174/CEE, 93/70/CEE, 93/117/CE, 98/64/CE, 1999/27/CE, 1999/76/CE, 2000/45/CE, 2002/70/CE y 2003/126/CE.
Las referencias a las Directivas derogadas se entenderán hechas al presente Reglamento y se leerán con arreglo a la tabla de correspondencias que figura en el anexo IX.
Artículo 7
El presente Reglamento entrará en vigor el vigésimo día siguiente al de su publicación en el Diario Oficial de la Unión Europea.
Será aplicable a partir del 26 de agosto de 2009
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 27 de enero de 2009.
Por la Comisión
Androulla VASSILIOU
Miembro de la Comisión
(1) DO L 165 de 30.4.2004, p. 1. Versión corregida en el DO L 191 de 28.5.2004, p. 1.
(2) DO L 155 de 12.7.1971, p. 13.
(3) DO L 279 de 20.12.1971, p. 7.
(4) DO L 123 de 29.5.1972, p. 6.
(5) DO L 83 de 30.3.1973, p. 21.
(6) DO L 102 de 15.4.1976, p. 1.
(7) DO L 102 de 15.4.1976, p. 8.
(8) DO L 206 de 29.7.1978, p. 43.
(9) DO L 257 de 10.9.1981, p. 38.
(10) DO L 238 de 6.9.1984, p. 34.
(11) DO L 130 de 16.5.1986, p. 53.
(12) DO L 234 de 17.9.1993, p. 17.
(13) DO L 329 de 30.12.1993, p. 54.
(14) DO L 257 de 19.9.1998, p. 14.
(15) DO L 118 de 6.5.1999, p. 36.
(16) DO L 207 de 6.8.1999, p. 13.
(17) DO L 174 de 13.7.2000, p. 32.
(18) DO L 209 de 6.8.2002, p. 15.
(19) DO L 339 de 24.12.2003, p. 78.
ANEXO I
MÉTODOS DE MUESTREO
1. OBJETO Y ÁMBITO DE APLICACIÓN
Las muestras destinadas al control oficial de los piensos se tomarán siguiendo los métodos que se describen a continuación. Las muestras así obtenidas se considerarán representativas de las porciones objeto de muestreo.
2. PERSONAL ENCARGADO DEL MUESTREO
La toma de muestras correrá a cargo de personas autorizadas al efecto por los Estados miembros.
3. DEFINICIONES
Lote objeto de muestreo: una cantidad de producto que constituye una unidad y tiene características presuntamente uniformes.
Muestra elemental: cantidad tomada en un punto del lote objeto de muestreo.
Muestra global: suma de las muestras elementales tomadas del mismo lote objeto de muestreo.
Muestra reducida: parte representativa de la muestra global, obtenida por reducción de esta.
Muestra final: parte de la muestra reducida o de la muestra global homogeneizada.
4. INSTRUMENTAL
|
4.1. |
El instrumental de muestreo debe estar hecho de materiales que no puedan contaminar los productos de los se hayan de tomar muestras. Este instrumental puede ser aprobado oficialmente por los Estados miembros. |
4.2. Instrumental recomendado para el muestreo de piensos sólidos
4.2.1. Muestreo manual
|
4.2.1.1. |
Pala de fondo plano y bordes verticales |
|
4.2.1.2. |
Sonda con hendidura larga o compartimentos. Las dimensiones de la sonda deben ajustarse a las características del lote objeto de muestreo (profundidad del recipiente, dimensiones del saco, etc.) y al tamaño de las partículas del pienso. |
4.2.2. Muestreo mecánico
Podrán utilizarse aparatos mecánicos homologados para el muestreo de piensos en movimiento.
4.2.3. Divisor
Para tomar muestras elementales y preparar muestras reducidas y muestras finales podrán utilizarse aparatos diseñados para dividir la muestra en partes aproximadamente iguales.
5. REQUISITOS CUANTITATIVOS
|
5.A. |
En relación con el control de sustancias o productos repartidos de manera uniforme en el pienso |
|
|
5.A.1. |
Lote objeto de muestreo El lote objeto de muestreo debe tener un tamaño que permita tomar muestras de todas las partes que lo compongan. |
|
|
5.A.2. |
Muestras elementales |
|
|
5.A.2.1. |
Piensos a granel |
Número mínimo de muestras elementales |
|
5.A.2.1.1. |
Lotes objeto de muestreo que no superen las 2,5 toneladas |
Siete |
|
5.A.2.1.2. |
Lotes objeto de muestreo que superen las 2,5 toneladas |
√ Veinte veces el número de toneladas que constituyen el lote objeto de muestreo (1), hasta un máximo de 40 muestras elementales |
|
5.A.2.2. |
Piensos envasados |
Número mínimo de envases de los que deben tomarse muestras (2) |
|
5.A.2.2.1. |
Envases con un contenido superior a 1 kg |
|
|
5.A.2.2.1.1. |
Lotes objeto de muestreo compuestos de uno a cuatro envases |
Todos los envases |
|
5.A.2.2.1.2. |
Lotes objeto de muestreo compuestos de cinco a 16 envases |
Cuatro |
|
5.A.2.2.1.3. |
Lotes objeto de muestreo compuestos de más de 16 envases |
√ Número de envases que componen el lote objeto de muestreo (1), hasta un máximo de 20 envases |
|
5.A.2.2.2. |
Envases de no más de 1 kg |
Cuatro |
|
5.A.2.3. |
Piensos líquidos o semilíquidos |
Número mínimo de recipientes de los que deben tomarse muestras (2) |
|
5.A.2.3.1. |
Recipientes de más de 1 l |
|
|
5.A.2.3.1.1. |
Lotes objeto de muestreo compuestos de uno a cuatro recipientes |
Todos los recipientes |
|
5.A.2.3.1.2. |
Lotes objeto de muestreo compuestos de cinco a 16 recipientes |
Cuatro |
|
5.A.2.3.1.3. |
Lotes objeto de muestreo compuestos de más de 16 recipientes |
√ Número de recipientes que componen el lote objeto de muestreo (1), hasta un máximo de 20 recipientes |
|
5.A.2.3.2. |
Recipientes de no más de 1 l |
Cuatro |
|
5.A.2.4. |
Piensos en bloques y piedras para lamer de sales minerales |
Número mínimo de bloques o piedras para lamer de los que deben tomarse muestras (2): un bloque o una piedra para lamer por lote objeto de muestreo compuesto de 25 unidades, hasta un máximo de cuatro bloques o piedras. |
|
5.A.3. |
Muestra global Se requiere una sola muestra global por cada lote objeto de muestreo. La cantidad total de las muestras elementales que constituyen la muestra global no será inferior a las cantidades siguientes: |
|
|
5.A.3.1. |
Piensos a granel |
4 kg |
|
5.A.3.2. |
Piensos envasados |
|
|
5.A.3.2.1. |
Envases de más de 1 kg |
4 kg |
|
5.A.3.2.2. |
Envases de no más de 1 kg |
Peso del contenido de cuatro envases originales |
|
5.A.3.3. |
Piensos líquidos o semilíquidos: |
|
|
5.A.3.3.1. |
Recipientes de más de 1 l |
4 l |
|
5.A.3.3.2. |
Recipientes de no más de 1 l |
Volumen del contenido de cuatro envases originales |
|
5.A.3.4. |
Piensos en bloques o piedras para lamer |
|
|
5.A.3.4.1. |
De un peso superior a 1 kg cada uno |
4 kg |
|
5.A.3.4.2. |
De un peso no superior a 1 kg cada uno |
El peso de cuatro bloques o piedras para lamer |
|
5.A.4. |
Muestras finales La muestra global da lugar, una vez reducida si es necesario, a las muestras finales. Se requiere el análisis de, por lo menos, una muestra final. La cantidad de muestra final destinada al análisis no será inferior a lo que se indica a continuación: |
|
|
|
Piensos sólidos |
500 g |
|
|
Piensos líquidos o semilíquidos |
500 ml |
|
5.B. |
En relación con el control de sustancias o productos indeseables que pueden estar repartidos de manera no uniforme en el pienso, como son las aflatoxinas, el cornezuelo del centeno, el ricino y las crotalarias en los materiales para piensos (3) |
|
|
5.B.1. |
Lote objeto de muestreo: véase 5.A.1. |
|
|
5.B.2. |
Muestras elementales |
|
|
5.B.2.1. |
Piensos a granel: véase 5.A.2.1. |
|
|
5.B.2.2. |
Piensos envasados |
Número mínimo de envases de los que deben tomarse muestras |
|
5.B.2.2.1. |
Lotes objeto de muestreo compuestos de uno a cuatro envases |
Todos los envases |
|
5.B.2.2.2. |
Lotes objeto de muestreo compuestos de cinco a 16 envases |
Cuatro |
|
5.B.2.2.3. |
Lotes objeto de muestreo compuestos de más de 16 envases |
√ Número de envases que componen el lote objeto de muestreo (1), hasta un máximo de 40 envases |
|
5.B.3. |
Muestras globales El número de muestras globales variará en función del tamaño del lote objeto de muestreo. A continuación se indica el número mínimo de muestras globales por cada lote objeto de muestreo. El peso total de las muestras elementales que componen cada muestra global no será inferior a 4 kg. |
|
|
5.B.3.1. |
Piensos a granel |
|
|
|
Peso del lote objeto de muestreo en toneladas |
Número mínimo de muestras globales por cada lote objeto de muestreo |
|
|
hasta 1 |
1 |
|
|
más de 1 y hasta 10 |
2 |
|
|
más de 10 y hasta 40 |
3 |
|
|
más de 40 |
4 |
|
5.B.3.2. |
Piensos envasados |
|
|
|
Tamaño del lote objeto de muestreo en número de envases |
Número mínimo de muestras globales por cada lote objeto de muestreo |
|
|
1 a 16 |
1 |
|
|
17 a 200 |
2 |
|
|
201 a 800 |
3 |
|
|
más de 800 |
4 |
|
5.B.4. |
Muestras finales Cada muestra global da lugar, una vez reducida, a las muestras finales. Se requiere el análisis de por lo menos una muestra final por cada muestra global. El peso de la muestra final destinada al análisis no puede ser inferior a 500 g. |
|
6. INSTRUCCIONES PARA LA TOMA, LA PREPARACIÓN Y EL ENVASADO DE LAS MUESTRAS
6.1. Generalidades
Las muestras deben tomarse y prepararse lo más rápidamente posible, tomando las precauciones necesarias para evitar que el producto se altere o contamine. Los instrumentos, así como las superficies y los recipientes destinados a recibir las muestras, deben estar limpios y secos.
6.2. Muestras elementales
6.2.A. En relación con el control de sustancias o productos repartidos de manera uniforme en el pienso
Las muestras elementales deben tomarse al azar en todo el lote objeto de muestreo y tener aproximadamente el mismo tamaño.
6.2.A.1.
El lote objeto de muestreo deberá dividirse simbólicamente en varias partes aproximadamente iguales. Deberá seleccionarse al azar un número de partes que se corresponda con el número de muestras elementales requeridas en el punto 5.A.2, y de cada una de esas partes deberá tomarse al menos una muestra.
En su caso, el muestreo puede realizarse mientras el lote objeto de muestreo está en movimiento (carga o descarga).
6.2.A.2.
Una vez seleccionado el número requerido de envases para muestreo según se indica en el punto 5.A.2, deberá tomarse parte del contenido de cada envase con una sonda o una pala. Si es necesario, las muestras se tomarán después de haber vaciado por separado los envases. Los grumos se desharán, si es necesario, apartándolos y devolviéndolos a la muestra, en cada muestra global por separado.
6.2.A.3.
Una vez seleccionado el número requerido de recipientes para muestreo según se indica en el punto 5.A.2, deberá homogeneizarse el contenido, si es necesario, y tomarse una cantidad de cada recipiente.
Las muestras elementales pueden tomarse mientras se vacía el contenido.
6.2.A.4.
Una vez seleccionado el número requerido de recipientes para muestreo según se indica en el punto 5.A.2, se tomarán muestras en diferentes niveles.
También pueden tomarse muestras mientras se vacía el contenido, pero, en ese caso, deberán desecharse las primeras fracciones.
En cualquier caso, el volumen total recogido no debe ser inferior a 10 l.
6.2.A.5.
Una vez seleccionado el número requerido de bloques o piedras para muestreo según se indica en el punto 5.A.2, se tomará una parte de cada bloque o piedra para lamer.
6.2.B. En relación con el control de sustancias o productos indeseables que pueden estar repartidos de manera no uniforme en el pienso, como son las aflatoxinas, el cornezuelo del centeno, el ricino y las crotalarias en los materiales para piensos
El lote objeto de muestreo deberá dividirse simbólicamente en un número de partes aproximadamente iguales correspondiente al número de muestras globales que se establece en el punto 5.B.3. Si este número es mayor de uno, el número total de muestras elementales establecido en el punto 5.B.2 se distribuirá de manera aproximadamente igual en las diferentes partes. A continuación se tomarán muestras de aproximadamente el mismo tamaño (4), de manera que la cantidad total en las muestras de cada parte no sea inferior al mínimo de 4 kg requerido para cada muestra global. No se unirán muestras elementales tomadas de partes diferentes.
6.3. Preparación de muestras globales
6.3.A. En relación con el control de sustancias o productos repartidos de manera uniforme en el pienso
Las muestras elementales se mezclarán para formar una sola muestra global.
6.3.B. En relación con el control de sustancias o productos indeseables que pueden estar repartidos de manera no uniforme en el pienso, como son las aflatoxinas, el cornezuelo del centeno, el ricino y las crotalarias en los materiales para piensos
Se mezclarán las muestras elementales de cada parte del lote objeto de muestreo y se constituirán el número de muestras globales que se establece en el punto 5.B.3, teniendo cuidado de anotar el origen de cada muestra global.
6.4. Preparación de muestras finales
El material de cada muestra global deberá mezclarse cuidadosamente para obtener una mezcla homogénea (5). Si fuera necesario, la muestra global deberá reducirse hasta un mínimo de 2 kg o 2 l (muestra reducida), bien con ayuda de un divisor mecánico o automático, bien con el método de cuarteo.
Deberán prepararse a continuación, como mínimo, tres muestras finales que tengan aproximadamente la misma cantidad y se ajusten a las exigencias cuantitativas del punto 5.A.4 o 5.B.4. Cada muestra deberá introducirse en un recipiente apropiado. Deberán tomarse todas las precauciones necesarias para evitar cualquier alteración en la composición de la muestra o cualquier contaminación o adulteración que pudiera sobrevenir durante el transporte o el almacenamiento.
6.5. Envasado de muestras finales
Los recipientes o los envases deberán precintarse y etiquetarse (la etiqueta debe estar totalmente incorporada en el precinto) de manera que sea imposible abrirlos sin deteriorar el precinto.
7. ACTA DE MUESTREO
De cada muestreo deberá levantarse un acta que permita identificar sin ambigüedad el lote objeto de muestreo.
8. DESTINO DE LA MUESTRAS
De cada muestra global deberá enviarse cuanto antes al laboratorio de análisis autorizado, como mínimo, una muestra final, con la información necesaria para el analista.
(1) Cuando la cifra obtenida sea un quebrado, deberá redondearse al siguiente número entero.
(2) En el caso de envases o recipientes cuyo contenido no exceda de 1 kg o 1 l, y en el de los bloques o piedras para lamer que no pesen más de 1 kg cada uno, la muestra elemental estará formada por el contenido de un envase o recipiente original, o por un bloque o una piedra para lamer.
(3) Los métodos presentados en 5.A se emplean para el control de las aflatoxinas, el cornezuelo del centeno, el ricino y las crotalarias en piensos completos y complementarios.
(4) En el caso de piensos envasados, parte del contenido de los envases que vayan a someterse a muestreo se extraerá con una sonda o una pala, después de haber vaciado los envases por separado, si es necesario.
(5) Los grumos se desharán (si es necesario, apartándolos y devolviéndolos a la muestra) en cada muestra global por separado.
ANEXO II
DISPOSICIONES GENERALES SOBRE MÉTODOS DE ANÁLISIS PARA PIENSOS
A. PREPARACIÓN DE LAS MUESTRAS PARA EL ANÁLISIS
1. Objeto
Los procedimientos descritos a continuación se refieren a la preparación para el análisis de muestras finales enviadas a los laboratorios de control tras ser tomadas conforme a lo dispuesto en el anexo I.
Estas muestras deben prepararse de manera que las cantidades pesadas según disponen los métodos de análisis sean homogéneas y representativas de las muestras finales.
2. Precauciones que deben tomarse
El procedimiento que debe seguirse para preparar las muestras depende de los métodos de análisis empleados. Por tanto, es muy importante que dicho procedimiento se adecúe al método de análisis aplicado.
Todas las operaciones necesarias deben realizarse de modo que se eviten en lo posible la contaminación de la muestra y los cambios en su composición.
La molienda, la mezcla y el tamizado deberán efectuarse lo más rápidamente posible, a fin de minimizar la exposición de la muestra al aire y a la luz. No se emplearán molinos ni moledoras que puedan calentar perceptiblemente la muestra.
Para los piensos especialmente sensibles al calor se recomienda la molienda manual. Deberá cuidarse también de que el propio instrumental no sea fuente de contaminación de los oligoelementos.
Si la muestra no puede prepararse sin que su contenido de humedad sufra cambios significativos, debe determinarse dicho contenido antes y después de prepararla, de acuerdo con el método establecido en la parte A del anexo III.
3. Procedimiento
Dividir la muestra en las submuestras adecuadas para ser analizadas y servir de referencia, empleando técnicas divisorias apropiadas como la formación de montones alternativos con pala o la subdivisión con divisores mecánicos estacionarios o rotatorios. No se recomienda la técnica de conos y cuarteo, pues las submuestras resultantes pueden presentar un elevado error de división. Guardar la muestra de referencia en un recipiente adecuado, limpio y seco, con tapa hermética, y preparar las submuestras para el análisis de al menos 100 g, según se indica más adelante.
3.1. Piensos que pueden molerse tal como se presentan
Salvo que se especifique lo contrario en los métodos de análisis, tamizar la muestra completa por un tamiz con una luz de malla de 1 mm (conforme a la Recomendación ISO R565) tras molerla, si es necesario. No moler en exceso.
Mezclar la muestra tamizada y recogerla en un recipiente limpio y seco adecuado, provisto de tapón hermético. Volver a mezclar inmediatamente antes de pesar la cantidad para análisis.
3.2. Piensos que pueden molerse tras secarse
Salvo que se especifique lo contrario en los métodos de análisis, secar la muestra hasta que su contenido de humedad disminuya a un nivel del 8 % al 12 %, de acuerdo con el procedimiento preliminar de secado descrito en el punto 4.3 del método de determinación de la humedad mencionado en la parte A del anexo III. Proceder a continuación como se indica en el punto 3.1.
3.3. Piensos líquidos o semilíquidos
Colocar la muestra en un recipiente limpio y seco adecuado, provisto de tapón hermético. Mezclar bien inmediatamente antes de pesar la cantidad para análisis.
3.4. Otros piensos
Las muestras que no puedan prepararse conforme a uno de los procedimientos anteriores deberán someterse a cualquier otro procedimiento que garantice que las cantidades pesadas para el análisis son homogéneas y representativas de las muestras finales.
4. Almacenamiento de las muestras
Las muestras deben almacenarse a una temperatura que no altere su composición. Las destinadas al análisis de vitaminas o sustancias especialmente fotosensibles deberán guardarse en recipientes de vidrio marrón.
B. DISPOSICIONES RELATIVAS A LOS REACTIVOS Y EL INSTRUMENTAL EMPLEADOS EN LOS MÉTODOS DE ANÁLISIS
|
1. |
Salvo que se especifique lo contrario en el método de análisis, todos los reactivos deben ser analíticamente puros (a.p.). Si se analizan oligoelementos, debe comprobarse la pureza de los reactivos por medio de un ensayo en blanco. Dependiendo de los resultados que se obtengan, quizá sea necesaria una mayor purificación de los reactivos. |
|
2. |
Siempre que en los métodos de análisis se mencionen operaciones que impliquen preparación de soluciones, dilución, enjuague o lavado sin indicar la naturaleza del disolvente o el diluyente, debe utilizarse agua. Por regla general, el agua deberá desmineralizarse o destilarse. En casos particulares, indicados en los métodos de análisis, debe someterse a procedimientos especiales de purificación. |
|
3. |
Habida cuenta del equipamiento que se encuentra normalmente en los laboratorios de control, en los métodos de análisis solo se hace referencia a los instrumentos y aparatos especiales o que requieren un uso específico. Deben estar limpios, sobre todo cuando hayan de determinarse cantidades muy pequeñas de sustancias. |
C. APLICACIÓN DE LOS MÉTODOS DE ANÁLISIS Y EXPRESIÓN DE LOS RESULTADOS
1. Procedimiento de extracción
Varios métodos establecen un procedimiento de extracción específico. Como regla general, puede aplicarse un procedimiento de extracción distinto al mencionado en el método si se ha demostrado que su eficacia de extracción es equivalente para la matriz analizada.
2. Procedimiento de limpieza (cleanup)
Varios métodos establecen un procedimiento de limpieza específico. Como regla general, puede aplicarse un procedimiento de limpieza distinto al mencionado en el método si se ha demostrado que sus resultados analíticos son equivalentes para la matriz analizada.
3. Comunicación del método de análisis empleado
En general se establece un único método de análisis para determinar cada una de las sustancias del pienso. Si se ofrecen varios métodos, en el informe de análisis debe indicarse el método particular empleado por el laboratorio de control.
4. Número de determinaciones
El resultado indicado en el informe de análisis deberá ser el valor medio obtenido de, como mínimo, dos determinaciones, efectuadas en porciones de la muestra distintas y de repetibilidad satisfactoria.
Sin embargo, en el caso de análisis de sustancias indeseables, si el resultado de la primera determinación es significativamente (> 50 %) inferior a la especificación que ha de controlarse, no serán necesarias más determinaciones, a condición de que se apliquen los procedimientos de calidad adecuados.
Si se controla el contenido declarado de una sustancia o un ingrediente y el resultado de la primera determinación confirma dicho contenido, es decir, que el resultado analítico entra en el intervalo de variación aceptable del contenido declarado, no será necesaria una segunda determinación, siempre que se apliquen los procedimientos de calidad apropiados.
En algunos casos, este intervalo de variación aceptable está definido en la legislación, por ejemplo en la Directiva 79/373/CEE de la Comisión (1).
5. Comunicación de los resultados analíticos
El resultado analítico se expresará según establezca el método de análisis, con el número adecuado de cifras significativas, y se corregirá, si es necesario, con respecto al contenido de humedad de la muestra final antes de la preparación.
6. Incertidumbre de medida y tasa de recuperación en caso de análisis de sustancias indeseables
Por lo que se refiere a las sustancias indeseables según se definen en la Directiva 2002/32/CE, en especial las dioxinas y los PCB similares a las dioxinas, se considerará que un producto destinado a la alimentación animal no cumple los requisitos relativos al contenido máximo establecido si se estima que el resultado analítico excede del contenido máximo, teniendo en cuenta la incertidumbre de medida expandida y la corrección en función de la recuperación. Para evaluar el cumplimiento se emplea la concentración analizada, una vez corregida en función de la recuperación y tras deducirse la incertidumbre de medida expandida. Este procedimiento solo es aplicable en los casos en que el método de análisis permite estimar la incertidumbre de medida expandida y la corrección en función de la recuperación (no es posible, por ejemplo, en caso de análisis microscópico).
El resultado analítico se comunicará como sigue (en la medida en que el método de análisis utilizado permita estimar la incertidumbre de medida y la tasa de recuperación):
|
a) |
corregido en función de la recuperación, indicando el nivel de la misma; dicha corrección no será necesaria si la tasa de recuperación es del 90 % al 110 %; |
|
b) |
como «x +/- U», donde x es el resultado analítico y U la incertidumbre de medida expandida, utilizando un factor de cobertura de 2, que da un nivel de confianza del 95 % aproximadamente. |
Sin embargo, si el resultado del análisis fuera notablemente inferior (> 50 %) a la especificación que ha de controlarse, podría comunicarse sin corrección en función de la recuperación, y la tasa de recuperación y la incertidumbre de medida podrían omitirse, a condición de que se aplicaran los procedimientos de calidad apropiados y de que el análisis sirviera exclusivamente para comprobar el cumplimiento de las disposiciones legales.
ANEXO III
MÉTODOS DE ANÁLISIS PARA EL CONTROL DE LA COMPOSICIÓN DE LOS MATERIALES PARA PIENSOS Y LOS PIENSOS COMPUESTOS
A. DETERMINACIÓN DE LA HUMEDAD
1. Objeto y ámbito de aplicación
Este método permite determinar el contenido de humedad de los piensos. Si el pienso contiene sustancias volátiles, como ácidos orgánicos, debe tenerse presente que, junto con el contenido de humedad, también se determina una cantidad importante de esas sustancias.
No incluye el análisis de productos lácteos como materiales para piensos, el análisis de sustancias minerales y mezclas compuestas predominantemente de sustancias minerales, el análisis de grasas y aceites animales y vegetales, ni el análisis de semillas y frutos oleaginosos.
2. Principio
La muestra se deseca en las condiciones especificadas, que varían en función de la naturaleza del pienso. La pérdida de peso se determina por pesada. Cuando se trata de piensos sólidos con un elevado contenido de humedad, es necesario efectuar un secado preliminar.
3. Instrumental
|
3.1. |
Trituradora de material que no absorba la humedad, fácil de limpiar, que permita un triturado rápido y uniforme sin provocar un calentamiento sensible, evite al máximo el contacto con el aire exterior y cumpla los requisitos indicados en los puntos 4.1.1 y 4.1.2 (por ejemplo, microtrituradoras de martillos o de enfriamiento por agua, molinos de conos plegables, trituradoras de movimiento lento o de discos dentados). |
|
3.2. |
Balanza analítica, con una exactitud de 1 mg |
|
3.3. |
Recipientes secos de metal no corrosible o de vidrio, con tapas que garanticen un cierre hermético; la superficie de trabajo debe permitir que la muestra de ensayo se esparza a razón de 0,3 g/cm2 aproximadamente. |
|
3.4. |
Estufa isotérmica de calentamiento eléctrico (± 2 oC), adecuadamente ventilada, que permita una rápida regulación de la temperatura (1). |
|
3.5. |
Estufa de vacío regulable de calentamiento eléctrico, provista de una bomba de aceite y un mecanismo para introducir, o bien aire desecado caliente, o bien un agente desecante (por ejemplo, óxido de calcio) |
|
3.6. |
Desecador con una placa gruesa perforada de metal o porcelana, que contenga un agente desecante eficaz. |
4. Procedimiento
|
Nota |
: |
Las operaciones que se describen en esta sección deben realizarse inmediatamente después de abrir los paquetes de muestras. Los análisis deben efectuarse, como mínimo, por duplicado. |
4.1. Preparación
4.1.1.
Tomar, como mínimo, 50 g de la muestra. Si es necesario, triturar o dividir de manera que no se produzca variación alguna en el contenido de humedad (véase el punto 6).
4.1.2.
Tomar, como mínimo, 50 g de la muestra. Moler en partículas de las que al menos el 50 % pasen por un tamiz con una luz de malla de 0,5 mm y no dejen más de un 10 % de desecho en un tamiz con una luz de malla redonda de 1 mm.
4.1.3.
Tomar unos 25 g de la muestra, pesar con una precisión de 10 mg, añadir una cantidad apropiada de arena anhidra pesada con una precisión de 10 mg y mezclar hasta obtener un producto homogéneo.
4.2. Desecación
4.2.1.
Tarar un recipiente (3.3) con su tapa, con una precisión de 1 mg. En el recipiente tarado, pesar, con una precisión de 1 mg, unos 5 g de la muestra y esparcirla uniformemente. Colocar el recipiente, sin su tapa, en la estufa precalentada a 103 oC. Para impedir que la estufa se enfríe en exceso, introducir el recipiente lo más rápidamente posible. Dejar secar durante cuatro horas, contadas a partir del momento en que la estufa haya alcanzado de nuevo una temperatura de 103 oC. Volver a colocar la tapa sobre el recipiente, retirarlo de la estufa, dejarlo enfriar durante 30 a 45 minutos en el desecador (3.6) y pesar con una precisión de 1 mg.
En el caso de piensos compuestos predominantemente de aceites y grasas, secar en la estufa durante otros 30 minutos a 130 oC. La diferencia entre las dos pesadas no debe superar el 0,1 % de humedad.
4.2.2.
Tarar un recipiente (3.3) con su tapa, con una precisión de 0,5 mg. En el recipiente tarado, pesar, con una precisión de 1 mg, unos 5 g de la muestra triturada y esparcirla uniformemente. Colocar el recipiente, sin su tapa, en la estufa precalentada a 130 oC. Para impedir que la estufa se enfríe en exceso, introducir el recipiente lo más rápidamente posible. Dejar secar durante dos horas, contadas a partir del momento en que la estufa haya alcanzado de nuevo una temperatura de 130 oC. Volver a colocar la tapa sobre el recipiente, retirarlo de la estufa, dejarlo enfriar durante 30 a 45 minutos en el desecador (3.6) y pesar con una precisión de 1 mg.
|
4.2.3. |
Piensos compuestos con más de un 4 % de sacarosa o lactosa: materiales para piensos como algarrobas, productos cerealísticos hidrolizados, semillas de malta, lonchas de remolacha desecadas, pescado y azúcares solubles; piensos compuestos con más de un 25 % de sales minerales que contengan agua de cristalización. Tarar un recipiente (3.3) con su tapa, con una precisión de 0,5 mg. En el recipiente tarado, pesar, con una precisión de 1 mg, unos 5 g de la muestra y esparcirla uniformemente. Colocar el recipiente, sin su tapa, en la estufa de vacío (3.5) precalentada a una temperatura comprendida entre 80 oC y 85 oC. Para impedir que la estufa se enfríe en exceso, introducir el recipiente lo más rápidamente posible. Elevar la presión a 100 Torr y dejar secar durante cuatro horas a esa presión, bien en una corriente de aire seco caliente, bien mediante un agente desecante (unos 300 g para 20 muestras). En este último caso, desconectar la bomba de vacío cuando se haya alcanzado la presión prescrita. Calcular el tiempo de secado a partir del momento en que la estufa haya alcanzado de nuevo una temperatura de 80 oC a 85 oC. A continuación, restablecer con precaución la presión atmosférica en la estufa. Abrirla, tapar inmediatamente el recipiente, retirarlo de la estufa, dejarlo enfriar durante 30 a 45 minutos en el desecador (3.6) y pesar con una precisión de 1 mg. Proceder a una desecación complementaria de 30 minutos en la estufa de vacío a una temperatura de 80 oC a 85 oC y volver a pesar. La diferencia entre las dos pesadas no debe superar el 0,1 % de humedad. |
4.3. Predesecación
4.3.1.
Los piensos sólidos con un contenido de humedad elevado que dificulte su trituración deben someterse a una predesecación, del siguiente modo:
Pesar, con una precisión de 10 mg, unos 50 g de la muestra no triturada (los piensos comprimidos o aglomerados pueden dividirse someramente, si es necesario) en un recipiente apropiado (por ejemplo, una placa de aluminio de 20 × 12 cm con un borde de 0,5 cm). Dejar secar en una estufa a una temperatura de 60 oC a 70 oC, hasta que el contenido de humedad se haya reducido a un valor comprendido entre el 8 % y el 12 %. Retirar de la estufa, dejar enfriar al descubierto en el laboratorio durante una hora y pesar con una precisión de 10 mg. Triturar inmediatamente después como se indica en el punto 4.1.1 y efectuar la desecación como se indica en el punto 4.2.1 o 4.2.3, según la naturaleza del pienso.
4.3.2.
Los granos con un índice de humedad superior al 17 % deben someterse a una desecación preliminar del siguiente modo:
Pesar, con una precisión de 10 mg, unos 50 g de grano sin moler en un recipiente apropiado (por ejemplo, una placa de aluminio de 20 × 12 cm con un borde de 0,5 cm). Dejar secar en una estufa durante cinco a siete minutos a una temperatura de 130 oC. Retirar de la estufa, dejar enfriar al descubierto en el laboratorio durante dos horas y pesar con una precisión de 10 mg. Moler inmediatamente después como se indica en el punto 4.1.2 y efectuar la desecación como se indica en el punto 4.2.2.
5. Cálculo de los resultados
El contenido de humedad (X), en porcentaje de la muestra, se calcula con las fórmulas siguientes:
5.1. Desecación sin predesecación
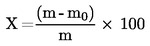
donde:
|
m |
= |
peso inicial, en gramos, de la muestra de ensayo; |
|
m0 |
= |
peso, en gramos, de la muestra de ensayo seca. |
5.2. Desecación con predesecación

donde:
|
m |
= |
peso inicial, en gramos, de la muestra de ensayo; |
|
m1 |
= |
peso, en gramos, de la muestra de ensayo tras la predesecación; |
|
m2 |
= |
peso, en gramos, de la muestra de ensayo una vez triturada o molida; |
|
m0 |
= |
peso, en gramos, de la muestra de ensayo seca. |
5.3. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no excederá del 0,2 % del valor absoluto de humedad.
6. Observación
Cuando resulte necesario efectuar una trituración y se considere que esta modifica el contenido de humedad del producto, los resultados del análisis de los componentes del pienso deben corregirse atendiendo al contenido de humedad de la muestra en su estado inicial.
B. DETERMINACIÓN DE LA HUMEDAD EN GRASAS Y ACEITES ANIMALES Y VEGETALES
1. Objeto y ámbito de aplicación
Este método permite determinar el contenido de agua y sustancias volátiles de las grasas y los aceites animales y vegetales.
2. Principio
Se deseca la muestra a 103 oC con un peso constante (la pérdida de peso entre dos pesadas sucesivas debe ser inferior o igual a 1 mg). La pérdida de peso se determina mediante pesada.
3. Instrumental
|
3.1. |
Plato de fondo plano de material resistente a la corrosión, de 8 cm a 9 cm de diámetro y de aproximadamente 3 cm de alto |
|
3.2. |
Termómetro de bulbo reforzado con tubo capilar en el extremo superior, graduado entre aproximadamente 80 oC y, como mínimo, 110 oC, de unos 10 cm de longitud |
|
3.3. |
Baño de arena o placa calefactora eléctrica |
|
3.4. |
Desecador con un agente desecante eficaz |
|
3.5. |
Balanza analítica |
4. Procedimiento
Pesar, con una precisión de 1 mg, unos 20 g de la muestra homogeneizada en el plato tarado seco (3.1) que contiene el termómetro (3.2). Calentar en el baño de arena o la placa calefactora (3.3), removiendo continuamente con el termómetro, de manera que la temperatura alcance 90 oC en unos siete minutos.
Reducir el calor, observando con qué frecuencia salen burbujas del fondo del plato. La temperatura no debe sobrepasar los 105 oC. Seguir removiendo, raspando el fondo del plato, hasta que dejen de formarse burbujas.
Para eliminar completamente la humedad, calentar varias veces a 103 oC ± 2 oC, enfriando a 93 oC entre los sucesivos calentamientos. A continuación, dejar enfriar a temperatura ambiente en el desecador (3.4) y pesar. Repetir esta operación hasta que la pérdida de peso entre dos pesadas sucesivas deje de sobrepasar los 2 mg.
|
Nota |
: |
El incremento del peso de la muestra tras varios calentamientos es indicio de una oxidación de la grasa, en cuyo caso debe calcularse el resultado a partir de la pesada efectuada inmediatamente antes de que empezara a producirse ese incremento. |
5. Cálculo de los resultados
El contenido de humedad (X), como porcentaje de la muestra, viene dado por la fórmula siguiente:

donde:
|
m |
= |
peso, en gramos, de la muestra de ensayo; |
|
m1 |
= |
peso, en gramos, del plato con su contenido, antes del calentamiento; |
|
m2 |
= |
peso, en gramos, del plato con su contenido, tras el calentamiento. |
Los resultados inferiores al 0,05 % deben registrarse como «inferior al 0,05 %».
Repetibilidad
La diferencia de humedad entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe exceder del 0,05 %, en valor absoluto.
C. DETERMINACIÓN DEL CONTENIDO DE PROTEÍNA BRUTA
1. Objeto y ámbito de aplicación
Este método permite determinar el contenido de proteína bruta del pienso sobre la base del contenido de nitrógeno, determinado por el método Kjeldahl.
2. Principio
La muestra se digiere con ácido sulfúrico en presencia de un catalizador. La solución ácida se alcaliniza son una solución de hidróxido de sodio. El amoniaco se destila y se recoge en una cantidad medida de ácido sulfúrico, cuyo exceso se titula con una solución patrón de hidróxido de sodio.
Alternativamente, el amoniaco desprendido se destila en un exceso de solución de ácido bórico, tras lo cual se efectúa la titulación con una solución de ácido clorhídrico o ácido sulfúrico.
3. Reactivos
|
3.1. |
Sulfato de potasio |
|
3.2. |
Catalizador: óxido de cobre (II), CuO, o sulfato de cobre (II) pentahidratado, CuSO4 5H2 O |
|
3.3. |
Cinc granulado |
|
3.4. |
Acido sulfúrico, ρ20 = 1,84 g/ml |
|
3.5. |
Ácido sulfúrico, solución volumétrica patrón, c(H2SO4) = 0,25 mol/l |
|
3.6. |
Ácido sulfúrico, solución volumétrica patrón, c(H2SO4) = 0,10 mol/l |
|
3.7. |
Ácido sulfúrico, solución volumétrica patrón, c(H2SO4) = 0,05 mol/l |
|
3.8. |
Indicador de rojo de metilo; disolver 300 mg de rojo de metilo en 100 ml de etanol, σ = 95-96 % (v/v). |
|
3.9. |
Solución de hidróxido de sodio (puede utilizarse la calidad técnica), β = 40 g/100 ml (m/v: 40 %) |
|
3.10. |
Hidróxido de sodio, solución volumétrica patrón, c(NaOH) = 0,25 mol/l |
|
3.11. |
Hidróxido de sodio, solución volumétrica patrón, c(NaOH) = 0,10 mol/l |
|
3.12. |
Piedra pómez granulada, lavada en ácido clorhídrico y calcinada |
|
3.13. |
Acetanilida (punto de fusión = 114 oC, contenido de N = 10,36 %) |
|
3.14. |
Sacarosa (exenta de nitrógeno) |
|
3.15. |
Ácido bórico (H3BO3) |
|
3.16. |
Solución de indicador de rojo de metilo: disolver 100 mg de rojo de metilo en 100 ml de etanol o metanol. |
|
3.17. |
Solución de verde de bromocresol: disolver 100 mg de verde de bromocresol en 100 ml de etanol o metanol. |
|
3.18. |
Solución de ácido bórico (10 g/l a 40 g/l, en función del instrumental empleado) Si se aplica la detección colorimétrica del punto final, debe añadirse rojo de metilo y verde de bromocresol a las soluciones de ácido bórico. Si se prepara 1 l de solución de ácido bórico, antes de ajustar el volumen deberán añadirse 7 ml de solución de indicador de rojo de metilo (3.16) y 10 ml de solución de verde de bromocresol (3.17). Dependiendo del agua utilizada, el pH de la solución de ácido bórico podría variar de un lote a otro. A menudo es necesario un ajuste con un pequeño volumen de álcali para obtener un blanco positivo.
|
|
3.19 |
Ácido clorhídrico, solución volumétrica patrón, c(HCl) = 0,10 mol/l
|
4. Instrumental
El adecuado para efectuar la digestión, la destilación y la titulación conforme al procedimiento Kjeldahl.
5. Procedimiento
5.1. Digestión
Pesar, con una precisión de 0,001 g, 1 g de la muestra, y pasar esta al matraz del aparato de digestión. Añadir 15 g de sulfato de potasio (3.1.), una cantidad apropiada de catalizador (3.2) (de 0,3 g a 0,4 g de óxido de cobre [II] o de 0,9 g a 1,2 g de sulfato de cobre [II] pentahidratado), 25 ml de ácido sulfúrico (3.4) y, si es necesario, unos pocos gránulos de piedra pómez (3.12), y mezclar.
Calentar el matraz, moderadamente al principio, agitándolo en círculos de vez en cuando, si es necesario, hasta que la masa se haya carbonizado y la espuma haya desaparecido; a continuación, calentar más intensamente hasta que el líquido hierva de manera constante. El calentamiento es el adecuado si el ácido en ebullición se condensa en la pared del matraz. Evitar que los lados se sobrecalienten y que se adhieran a ellos partículas orgánicas.
Cuando la solución se aclare y adopte un color verde claro, seguir hirviendo durante otras dos horas, y dejar enfriar a continuación.
5.2. Destilación
Añadir con cuidado suficiente agua para que los sulfatos se disuelvan por completo. Dejar enfriar y, a continuación, si es necesario, añadir unos pocos gránulos de cinc (3.3). Proceder conforme al punto 5.2.1 o 5.2.2.
5.2.1.
Verter en el matraz receptor del aparato de destilación exactamente 25 ml de ácido sulfúrico (3.5) o (3.7), en función del supuesto contenido de nitrógeno. Añadir unas pocas gotas del indicador de rojo de metilo (3.8).
Conectar el matraz de digestión al refrigerante del aparato de destilación y sumergir el extremo del refrigerante, como mínimo, 1 cm en el líquido contenido en el matraz receptor (véase la observación del punto 8.3). Verter lentamente 100 ml de solución de hidróxido de sodio (3.9) en el matraz de digestión sin pérdida de amoniaco (véase la observación del punto 8.1). Calentar el matraz hasta que el amoniaco se haya destilado.
5.2.2.
Si la titulación del contenido de amoniaco del destilado se efectúa manualmente, se aplica el procedimiento mencionado a continuación. Si la unidad de destilación está completamente automatizada e incluye la titulación del contenido de amoniaco del destilado, seguir las instrucciones de empleo del fabricante.
Colocar un matraz receptor que contenga de 25 ml a 30 ml de la solución de ácido bórico (3.18) bajo la salida del refrigerante, de manera que el tubo de descarga quede bajo la superficie del exceso de solución de ácido bórico. Ajustar la unidad de destilación para que dispense 50 ml de solución de hidróxido de sodio (3.9). Poner en funcionamiento la unidad de destilación siguiendo las instrucciones del fabricante y destilar el amoniaco desprendido por la adición de la solución de hidróxido de sodio. Recoger el destilado en la solución receptora de ácido bórico. La cantidad de destilado (tiempo de destilación de vapor) depende de la cantidad de nitrógeno que contiene la muestra. Seguir las instrucciones del fabricante.
|
Nota |
: |
En una unidad de destilación semiautomática, la adición de exceso de hidróxido de sodio y la destilación de vapor se realizan de forma automática. |
5.3. Titulación
Proceder conforme al punto 5.3.1 o 5.3.2.
5.3.1.
Titular el exceso de ácido sulfúrico en el matraz receptor con solución de hidróxido de sodio (3.10 o 3.11), dependiendo de la concentración del ácido sulfúrico empleado, hasta alcanzar el punto final.
5.3.2.
Titular, empleando una bureta, el contenido del matraz receptor con la solución volumétrica patrón de ácido clorhídrico (3.19) o con la solución volumétrica patrón de ácido sulfúrico (3.6), y leer la cantidad de titulante utilizado.
Si se aplica la detección colorimétrica del punto final, este se alcanza cuando aparece el primer rastro de coloración rosa en el contenido. Leer la bureta con una precisión de 0,05 ml. Una placa agitadora magnética iluminada o un detector fotométrico pueden ayudar a visualizar el punto final.
También puede hacerse automáticamente utilizando un destilador de vapor con titulación automática.
Utilizar el destilador o el destilador/titulador siguiendo las correspondientes instrucciones del fabricante.
|
Nota |
: |
Si se utiliza un sistema de titulación automática, la titulación empieza inmediatamente después de que comience la destilación, y se emplea la solución de ácido bórico al 1 % (3.18). Si se utiliza una unidad de destilación completamente automática, la titulación automática del amoniaco puede también llevarse a cabo con detección del punto final mediante un sistema de pH potenciométrico. En este caso se emplea un titulador automático con pehachímetro. El pehachímetro deberá calibrarse adecuadamente en el intervalo de pH 4 a pH 7, siguiendo los procedimientos normales de laboratorio para la calibración del pH. El punto final del pH de la titulación se alcanza con un pH 4,6, que es el punto álgido de la curva de titulación (punto de inflexión). |
5.4. Ensayo en blanco
Para confirmar que los reactivos no tienen nitrógeno, efectuar un ensayo en blanco (digestión, destilación y titulación) con un 1 g de sacarosa (3.14) en lugar de la muestra.
6. Cálculo de los resultados
Los cálculos se realizan conforme al punto 6.1 o 6.2.
6.1. Cálculo para la titulación según el punto 5.3.1
El contenido de proteína bruta, expresado en porcentaje en peso, se calcula según la fórmula siguiente:

donde:
|
V0 |
= |
es el volumen (ml) de NaOH (3.10 o 3.11) empleado en el ensayo en blanco; |
|
V1 |
= |
es el volumen (ml) de NaOH (3.10 o 3.11) empleado en la titulación de la muestra; |
|
c |
= |
es la concentración (mol/l) de hidróxido de sodio (3.10 o 3.11); |
|
m |
= |
es el peso (g) de la muestra. |
6.2 Cálculo para la titulación según el punto 5.3.2
6.2.1.
El contenido de proteína bruta, expresado en porcentaje en peso, se calcula según la fórmula siguiente:

donde:
|
m |
= |
es el peso (g) de la porción de ensayo; |
|
c |
= |
es la concentración (mol/l) de solución volumétrica patrón de ácido clorhídrico (3.19); |
|
V0 |
= |
es el volumen (en mililitros) de ácido clorhídrico empleado en el ensayo en blanco; |
|
V1 |
= |
es el volumen (en mililitros) de ácido clorhídrico empleado para la porción de ensayo. |
6.2.2.
El contenido de proteína bruta, expresado en porcentaje en peso, se calcula según la fórmula siguiente:
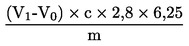
donde:
|
m |
= |
es el peso (g) de la porción de ensayo; |
|
c |
= |
es la concentración (mol/l) de solución volumétrica patrón de ácido sulfúrico (3.6); |
|
V0 |
= |
es el volumen (en mililitros) de ácido sulfúrico (3.6) empleado para el ensayo en blanco; |
|
V1 |
= |
es el volumen (en mililitros) de ácido sulfúrico (3.6) empleado para la porción de ensayo. |
7. Verificación del método
7.1. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe exceder del:
|
— |
0,2 % en valor absoluto, en el caso de contenidos de proteína bruta inferiores al 20 %, |
|
— |
1,0 % del valor superior, en el caso de contenidos de proteína bruta del 20 % a 40 %, |
|
— |
0,4 % en valor absoluto, en el caso de contenidos de proteína bruta superiores al 40 %. |
7.2. Exactitud
Efectuar el análisis (digestión, destilación y titulación) con 1,5 g a 2,0 g de acetanilida (3.13) en presencia de 1 g de sacarosa (3.14); 1 g de acetanilida consume 14,80 ml de ácido sulfúrico (3.5). La recuperación debe ser de al menos el 99 %.
8. Observaciones
|
8.1. |
El instrumental puede ser de tipo manual, semiautomático o automático. Si requiere un transvase entre la digestión y la destilación, este debe tener lugar sin pérdidas. Si el matraz del aparato de destilación no va provisto de un embudo de decantación, añadir el hidróxido de sodio inmediatamente antes de conectarlo al refrigerante, vertiendo el líquido de forma que caiga lentamente por la pared del matraz. |
|
8.2. |
Si el producto de la digestión se solidifica, recomenzar la determinación con una cantidad de ácido sulfúrico (3.4) mayor que la especificada anteriormente. |
|
8.3. |
En el caso de productos con un bajo contenido de nitrógeno, el volumen de ácido sulfúrico (3.7) que se pone en el matraz receptor puede reducirse, si es necesario, a 10 ml o 15 ml, enrasando a continuación con agua hasta los 25 ml. |
|
8.4. |
Aunque para los análisis ordinarios pueden emplearse métodos de determinación de la proteína bruta alternativos, el método Kjeldahl descrito en esta parte C es el método de referencia. La equivalencia de los resultados obtenidos con el método alternativo (por ejemplo, DUMAS) respecto de los obtenidos con el método de referencia debe demostrarse para cada una de las matrices. Puesto que los resultados obtenidos con un método alternativo, aun habiéndose verificado la equivalencia, pueden desviarse ligeramente de los obtenidos con el método de referencia, es necesario mencionar en el informe analítico el método de análisis empleado para la determinación de la proteína bruta. |
D. DETERMINACIÓN DE LA UREA
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de urea en los piensos.
2. Principio
La muestra se suspende en agua con un agente clarificante. A continuación se filtra la suspensión. El contenido de urea del filtrado se determina tras añadir 4-dimetilaminobenzaldehído (4-DMAB) midiendo la densidad óptica a una longitud de onda de 420 nm.
3. Reactivos
|
3.1. |
Solución de 4-dimetilaminobenzaldehído: disolver 1,6 g de 4-DMAB en 100 ml de etanol al 96 % y añadir 10 ml de ácido clorhídrico (ρ201,19 g/ml). Este reactivo se conserva como máximo dos semanas. |
|
3.2. |
Solución de Carrez I: disolver en agua 21,9 g de acetato de cinc, Zn(CH3COO)2 · 2H2O, y 3 g de ácido acético glacial. Enrasar con agua a 100 ml. |
|
3.3. |
Solución de Carrez II: disolver en agua 10,6 g de ferrocianuro de potasio, K4Fe(CN)6 · 3H2O. Enrasar con agua a 100 ml. |
|
3.4. |
Carbón activo que no absorba urea (debe comprobarse). |
|
3.5. |
Solución de urea al 0,1 % (p/v) |
4. Instrumental
|
4.1. |
Mezclador (tambor): de 35 a 40 revoluciones por minuto aproximadamente |
|
4.2. |
Tubos de ensayo: 160 × 16 mm con tapones esmerilados |
|
4.3. |
Espectrofotómetro |
5. Procedimiento
5.1. Análisis de la muestra
Pesar, con una precisión de 1 mg, 2 g de la muestra e introducirlos con 1 g de carbón activo (3.4) en un matraz aforado de 500 ml. Añadir 400 ml de agua y 5 ml de solución de Carrez I (3.2), mezclar durante aproximadamente 30 segundos y añadir 5 ml de solución de Carrez II (3.3). Mezclar durante 30 minutos en el tambor. Enrasar con agua, agitar y filtrar.
Retirar 5 ml de los filtrados incoloros transparentes, colocarlos en tubos de ensayo con tapones esmerilados, añadir 5 ml de solución de 4-DMAB (3.1) y mezclar. Colocar los tubos en una baño maría a 20 oC (+/- 4 oC). Transcurridos 15 minutos, medir la densidad óptica de la solución de muestra con el espectrofotómetro a 420 nm. Comparar con la solución de ensayo en blanco de los reactivos.
5.2. Curva de calibración
Retirar de la solución de urea (3.5) unos volúmenes de 1, 2, 4, 5 y 10 ml, verterlos en matraces aforados de 100 ml y enrasar con agua. Retirar 5 ml de cada solución, añadir a cada una de ellas 5 ml de solución de 4-DMAB (3.1), homogeneizar y medir la densidad óptica, como se ha mostrado anteriormente, comparándola con una solución de control que contenga 5 ml de 4-DMAB y 5 ml de agua sin urea. Trazar la curva de calibración.
6. Cálculo de los resultados
Determinar la cantidad de urea de la muestra empleando la curva de calibración.
Expresar el resultado en porcentaje de la muestra.
7. Observaciones
|
7.1. |
Si el contenido de urea excede del 3 %, reducir la muestra a 1 g o diluir la solución inicial de manera que en 500 ml no haya más de 50 mg de urea. |
|
7.2. |
Si los contenidos de urea son bajos, incrementar la muestra, siempre que el filtrado siga siendo transparente e incoloro. |
|
7.3. |
Si la muestra contiene compuestos nitrogenados simples, como aminoácidos, la densidad óptica deberá medirse a 435 nm. |
E. DETERMINACIÓN DE LAS BASES NITROGENADAS VOLÁTILES
I. POR MICRODIFUSIÓN
1. Objeto y ámbito de aplicación
Este método permite determinar el contenido de bases nitrogenadas volátiles de los piensos, expresadas en amoniaco.
2. Principio
La muestra se extrae con agua y la solución se clarifica y se filtra. Las bases nitrogenadas volátiles se desplazan por microdifusión con una solución de carbonato de potasio, se recogen en una solución de ácido bórico y se titulan con ácido sulfúrico.
3. Reactivos
|
3.1. |
Ácido tricloroacético, solución al 20 % (p/v) |
|
3.2. |
Indicador: disolver 33 mg de verde de bromocresol y 65 mg de rojo de metilo en 100 ml de etanol al 95-96 % (v/v). |
|
3.3. |
Solución de ácido bórico: en un matraz aforado de 1 l, disolver 10 g de ácido bórico en 200 ml de etanol al 95-96 % (v/v) y 700 ml de agua. Añadir 10 ml de indicador (3.2). Mezclar y, si es necesario, ajustar la coloración de la solución al rojo claro añadiendo una solución de hidróxido de sodio. Con 1 ml de esta solución se fijarán, como máximo, 300 μg de NH3. |
|
3.4. |
Solución saturada de carbonato de potasio: disolver 100 g de carbonato de potasio en 100 ml de agua en ebullición. Dejar enfriar y filtrar. |
|
3.5. |
Acido sulfúrico de 0,01 mol/l |
4. Instrumental
|
4.1. |
Mezclador (tambor): de 35 a 40 revoluciones por minuto aproximadamente |
|
4.2. |
Células de Conway de vidrio o de plástico (véase el diagrama) |
|
4.3. |
Microburetas graduadas a 1/100 ml |
5. Procedimiento
Pesar, con una precisión de 1 mg, 10 g de la muestra e introducirlos con 100 ml de agua en un matraz aforado de 200 ml. Mezclar o remover en el tambor durante 30 minutos. Añadir 50 ml de solución de ácido tricloracético (3.1), enrasar con agua, agitar enérgicamente y filtrar por un filtro de pliegues.
Empleando una pipeta, verter 1 ml de solución de ácido bórico (3.3) en la parte central de la célula de Conway y 1 ml del filtrado de la muestra en la corona de la célula. Cubrir parcialmente con la tapa engrasada. Dejar caer rápidamente en la corona 1 ml de solución saturada de carbonato de potasio (3.4) y cerrar la tapa de manera que la célula quede herméticamente cerrada. Girar con precaución la célula haciéndola rotar en un plano horizontal, de forma que se mezclen los dos reactivos. Dejar incubar, bien durante al menos cuatro horas a temperatura ambiente, bien durante una hora a 40 oC.
Con ayuda de una microbureta (4.3), titular las bases volátiles en la solución de ácido bórico con ácido sulfúrico (3.5).
Efectuar un ensayo en blanco siguiendo el mismo procedimiento, pero sin la muestra que debe analizarse.
6. Cálculo de los resultados
1 ml de una solución de 0,01 mol/l de H2SO4 corresponde a 0,34 mg de amoniaco.
Expresar el resultado en porcentaje de la muestra.
Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no excederá del:
|
— |
10 % en valor relativo, en el caso de contenidos de amoniaco inferiores al 1,0 %, |
|
— |
0,1 % en valor absoluto, en el caso de contenidos de amoniaco iguales o superiores al 1,0 %. |
7. Observación
Si el contenido de amoniaco de la muestra es superior al 0,6 %, diluir el filtrado inicial.
CONWAY CELL
Scale 1/1

II. POR DESTILACIÓN
1. Objeto y ámbito de aplicación
Este método permite determinar el contenido de bases nitrogenadas volátiles, expresadas en amoniaco, de la harina de pescado que no contenga prácticamente urea. Únicamente es aplicable para contenidos de amoniaco inferiores al 0,25 %.
2. Principio
La muestra se extrae con agua y la solución se clarifica y se filtra. Las bases nitrogenadas volátiles se desplazan en el punto de ebullición añadiendo óxido de magnesio y se recogen en una cantidad determinada de ácido sulfúrico, cuyo exceso se titula por retroceso con una solución de hidróxido de sodio.
3. Reactivos
|
3.1. |
Ácido tricloroacético, solución al 20 % (p/v) |
|
3.2. |
Óxido de magnesio |
|
3.3. |
Emulsión antiespumante (por ejemplo, silicona) |
|
3.4. |
Acido sulfúrico de 0,05 mol/l |
|
3.5. |
Solución de hidróxido de sodio de 0,1 mol/l |
|
3.6. |
Solución de rojo de metilo al 0,3 % en etanol al 95 %-96 % (v/v) |
4. Instrumental
|
4.1. |
Mezclador (tambor): de 35 a 40 revoluciones por minuto aproximadamente |
|
4.2. |
Aparato de destilación de tipo Kjeldahl |
5. Procedimiento
Pesar, con una precisión de 1 mg, 10 g de la muestra e introducirlos con 100 ml de agua en un matraz aforado de 200 ml. Mezclar o remover en el tambor durante 30 minutos. Añadir 50 ml de solución de ácido tricloracético (3.1), enrasar con agua, agitar enérgicamente y filtrar por un filtro de pliegues.
Tomar una cantidad de filtrado limpio que sea adecuada para el contenido supuesto de bases nitrogenadas volátiles (en general, basta con 100 ml). Diluir hasta 200 ml y añadir 2 g de óxido de magnesio (3.2) y unas pocas gotas de emulsión antiespumante (3.3). La solución debe ser alcalina en el papel de tornasol; si no lo es, añadir un poco de óxido de magnesio (3.2). Proceder conforme a los puntos 5.2 y 5.3 del método de análisis para la determinación del contenido de proteína bruta (parte C del presente anexo).
Efectuar un ensayo en blanco siguiendo el mismo procedimiento, pero sin la muestra que debe analizarse.
6. Cálculo de los resultados
1 ml de solución de 0,05 mol/l de H2SO4 corresponde a 1,7 mg de amoniaco.
Expresar el resultado en porcentaje de la muestra.
Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no excederá del 10 % de amoniaco, en valor relativo.
F. DETERMINACIÓN DE LOS AMINOÁCIDOS (EXCEPTO TRIPTÓFANO)
1. Objeto y ámbito de aplicación
Este método permite determinar la presencia en los piensos de aminoácidos libres (sintéticos y naturales) y totales (unidos en péptidos y libres), utilizando un analizador de aminoácidos. Es aplicable a los siguientes aminoácidos: cistina y cisteína, metionina, lisina, treonina, alanina, arginina, ácido aspártico, ácido glutámico, glicina, histidina, isoleucina, leucina, fenilalanina, prolina, serina, tirosina y valina.
El método no distingue entre las sales de los aminoácidos, y tampoco permite diferenciar entre sus formas D y L. No es válido para determinar el triptófano ni los análogos hidroxilados de los aminoácidos.
2. Principio
2.1. Aminoácidos libres
Los aminoácidos libres se extraen con ácido clorhídrico diluido. Las macromoléculas nitrogenadas extraídas a la vez se precipitan con ácido sulfosalicílico y se eliminan por filtración. El pH de la solución filtrada se ajusta a 2,20. Los aminoácidos se separan por cromatografía de intercambio iónico y se determinan por reacción con ninhidrina mediante detección fotométrica a 570 nm.
2.2. Aminoácidos totales
El procedimiento elegido depende de los aminoácidos estudiados. La cistina, la cisteína y la metionina deben oxidarse a ácido cisteico y metionina sulfona, respectivamente, antes de la hidrólisis. La tirosina debe determinarse en hidrolizados de muestras no oxidadas. Todos los demás aminoácidos citados en el punto 1 pueden determinarse tanto en muestras oxidadas como no oxidadas.
La oxidación se realiza a 0 oC con una mezcla de ácido perfórmico y fenol. El exceso de reactivo oxidante se descompone con disulfito de sodio. La muestra oxidada o no oxidada se hidroliza con ácido clorhídrico (3.20) durante 23 horas. El pH del hidrolizado se ajusta a 2,20. Los aminoácidos se separan por cromatografía de intercambio iónico y se determinan por reacción con ninhidrina mediante detección fotométrica a 570 nm (440 nm en el caso de la prolina).
3. Reactivos
Debe utilizarse agua bidestilada o agua de calidad equivalente (conductividad < 10 μS).
|
3.1. |
Peróxido de hidrógeno, p (p/p) = 30 % |
|
3.2. |
Ácido fórmico, p (p/p) = 98-100 % |
|
3.3. |
Fenol |
|
3.4. |
Disulfito de sodio |
|
3.5. |
Hidróxido de sodio |
|
3.6. |
Ácido 5-sulfosalicílico dihidratado |
|
3.7. |
Ácido clorhídrico, con una densidad aproximada de 1,18 g/ml |
|
3.8. |
Citrato trisódico dihidratado |
|
3.9. |
2,2′-Tiodietanol (tiodiglicol) |
|
3.10. |
Cloruro de sodio |
|
3.11. |
Ninhidrina |
|
3.12. |
Éter de petróleo, con un intervalo de ebullición de 40-60 oC |
|
3.13. |
Norleucina, u otro compuesto adecuado para ser utilizado como patrón interno |
|
3.14. |
Gas nitrógeno (< 10 ppm de oxígeno) |
|
3.15. |
1-Octanol |
|
3.16. |
Aminoácidos |
|
3.16.1. |
Sustancias patrón enumeradas en el punto 1. Compuestos puros sin agua de cristalización. Desecar en vacío sobre P2O5 o H2SO4 durante una semana antes de su utilización. |
|
3.16.2. |
Ácido cisteico |
|
3.16.3. |
Metionina sulfona |
|
3.17. |
Solución de hidróxido de sodio, c = 7,5 mol/l: Disolver 300 g de NaOH (3.5) en agua y enrasar a 1 l. |
|
3.18. |
Solución de hidróxido de sodio, c = 1 mol/l: Disolver 40 g de NaOH (3.5) en agua y enrasar a 1 l. |
|
3.19. |
Solución de ácido fórmico y fenol: Mezclar 889 g de ácido fórmico (3.2) con 111 g de agua y añadir 4,73 g de fenol (3.3). |
|
3.20. |
Mezcla de hidrólisis, c = 6 mol de HCl/l, con 1 g de fenol/l: Añadir 1 g de fenol (3.3) a 492 ml de HCl (3.7) y enrasar a 1 l con agua. |
|
3.21. |
Mezcla de extracción, c = 0,1 mol de HCl/l, con un 2 % de tiodiglicol: tomar 8,2 ml de HCl (3.7), diluir con unos 900 ml de agua, añadir 20 ml de tiodiglicol (3.9) y enrasar a 1 l con agua (no mezclar 3.7 y 3.9 directamente). |
|
3.22. |
Ácido 5-sulfosalicílico, ß = 6 %: Disolver 60 g de ácido 5-sulfosalicílico (3.6) en agua y enrasar a 1 l con agua. |
|
3.23. |
Mezcla de oxidación (ácido perfórmico y fenol): Mezclar 0,5 ml de peróxido de hidrógeno (3.1) con 4,5 ml de solución de ácido fórmico y fenol (3.19) en un pequeño vaso de precipitado. Incubar a 20-30 oC durante una hora a fin de que se forme ácido perfórmico y, a continuación, enfriar sobre baño de hielo y agua (15 minutos) antes de añadir a la muestra. Precaución: evitar el contacto con la piel y llevar vestimenta de protección. |
|
3.24. |
Solución reguladora de citrato, c = 0,2 mol de Na+/l, pH = 2,20: Disolver 19,61 g de citrato de sodio (3.8), 5 ml de tiodiglicol (3.9), 1 g de fenol (3.3) y 16,50 ml de HCl (3.7) en unos 800 ml de agua. Ajustar el pH a 2,20. Enrasar a 1 l con agua. |
|
3.25. |
Soluciones reguladoras de elución, preparadas según las condiciones del analizador utilizado (4.9) |
|
3.26. |
Reactivo de ninhidrina, preparado según las condiciones del analizador utilizado (4.9) |
|
3.27. |
Soluciones patrón de aminoácidos. Estas soluciones deberán conservarse a una temperatura inferior a 5 oC. |
|
3.27.1. |
Solución patrón madre de aminoácidos (3.16.1) c = 2,5 μmol/ml de cada uno en ácido clorhídrico Disponibles en los comercios |
|
3.27.2. |
Solución patrón madre de ácido cisteico y metionina sulfona, c = 1,25 μmol/ml Disolver 0,2115 g de ácido cisteico (3.16.2) y 0,2265 g de metionina sulfona (3.16.3) en solución reguladora de citrato (3.24) en un matraz aforado de 1 l y enrasar con solución reguladora de citrato. Conservar a menos de 5 oC durante un período máximo de 12 meses. Esta solución no se utiliza si la solución patrón madre (3.27.1) contiene ácido cisteico y metionina sulfona. |
|
3.27.3. |
Solución patrón madre del patrón interno, por ejemplo, norleucina, c = 20 μmol/ml Disolver 0,6560 g de norleucina (3.13) en solución reguladora de citrato (3.24) en un matraz aforado y enrasar a 250 ml con solución reguladora de citrato. Conservar a menos de 5 oC durante un período máximo de seis meses. |
|
3.27.4. |
Solución de calibración de los aminoácidos patrón para utilizar con los hidrolizados, c = 5 nmol/50 μl de ácido cisteico y metionina sulfona y c = 10 nmol/50 μl de los demás aminoácidos. Disolver 2,2 g de cloruro de sodio (3.10) en un vaso de precipitado de 100 ml con 30 ml de solución reguladora de citrato (3.24). Añadir 4,00 ml de solución patrón madre de aminoácidos (3.27.1), 4,00 ml de solución patrón madre de ácido cisteico y metionina sulfona (3.27.2) y 0,50 ml de solución patrón madre de patrón interno (3.27.3), si se utiliza. Ajustar el pH a 2,20 con hidróxido de sodio (3.18). Transvasar cuantitativamente a un matraz aforado de 50 ml, enrasar con solución reguladora de citrato (3.24) y mezclar. Conservar a menos de 5 oC durante un período máximo de tres meses. Véanse también las observaciones del punto 9.1. |
|
3.27.5. |
Solución de calibración de los aminoácidos patrón para utilizar con los hidrolizados preparados según el punto 5.3.3.1 y con los extractos (5.2). La solución de calibración se prepara con arreglo al punto 3.27.4, pero sin cloruro de sodio. Conservar a menos de 5 oC durante un período máximo de tres meses. |
4. Instrumental
|
4.1. |
Matraz de fondo redondo de 100 ml o 250 ml, provisto de refrigerante de reflujo |
|
4.2. |
Frasco de vidrio borosilicato de 100 ml con tapón de rosca provisto de junta de goma/teflón (por ejemplo Duran, Schott), para uso en estufa |
|
4.3. |
Estufa con ventilación forzada y con un regulador de la temperatura de exactitud superior a ± 2 oC |
|
4.4. |
Pehachímetro (lectura con tres decimales) |
|
4.5. |
Filtro de membrana (0,22 μm) |
|
4.6. |
Centrífuga |
|
4.7. |
Evaporador rotativo de vacío |
|
4.8. |
Agitador mecánico o magnético |
|
4.9. |
Analizador de aminoácidos o equipo de cromatografía de líquidos de alta resolución (CLAR) con columna de intercambio iónico, dispositivo para ninhidrina, derivatización postcolumna y detector fotométrico La columna se llena con resinas de poliestireno sulfonadas capaces de separar unos aminoácidos de otros y los aminoácidos de otros materiales que reaccionan con la ninhidrina. El flujo en las conducciones de solución reguladora y de ninhidrina se regula mediante bombas con una estabilidad de flujo de ±0,5 % en el período que abarca tanto la fase de calibración del patrón como el análisis de la muestra. Con algunos analizadores de aminoácidos pueden utilizarse procedimientos de hidrólisis en los que el hidrolizado presenta una concentración de sodio de c = 0,8 mol/l y contiene todo el ácido fórmico residual de la fase de oxidación. Otros no proporcionan una separación satisfactoria de determinados aminoácidos si el hidrolizado contiene ácido fórmico en exceso o elevadas concentraciones de ión sodio. En este caso, el volumen de ácido se reduce por evaporación hasta aproximadamente 5 ml después de la hidrólisis y antes de ajustar el pH. La evaporación deberá realizarse en vacío a 40 oC como máximo. |
5. Procedimiento
5.1. Preparación de la muestra
La muestra se muele hasta que pase por un tamiz de 0,5 mm. Las muestras con humedad elevada deben secarse al aire a una temperatura no superior a 50 oC, o bien liofilizarse antes de la molienda. Las muestras con un elevado contenido de grasa deberán someterse a extracción con éter de petróleo (3.12) antes de la molienda.
5.2. Determinación de los aminoácidos libres en piensos y premezclas
Pesar en un Erlenmeyer, con una precisión de 0,2 mg, una cantidad adecuada (1-5 g) de la muestra preparada (5.1), y añadir 100,0 ml de mezcla de extracción (3.21). Agitar la mezcla durante 60 minutos utilizando un agitador mecánico o magnético (4.8). Dejar que sedimente y pipetear 10,0 ml de la solución sobrenadante a un vaso de precipitado de 100 ml.
Añadir removiendo 5,0 ml de solución de ácido sulfosalicílico (3.22) y seguir removiendo con ayuda de un agitador magnético durante cinco minutos. Filtrar o centrifugar el sobrenadante para eliminar el posible precipitado. Verter 10,0 ml de la solución resultante en un vaso de precipitado de 100 ml y ajustar el pH a 2,20 con solución de hidróxido de sodio (3.18), transvasar a un matraz aforado de volumen adecuado con solución reguladora de citrato (3.24) y enrasar con esta misma solución reguladora.
Si se utiliza un patrón interno, añadir 1,00 ml de patrón interno (3.27.3) por cada 100 ml de solución final y enrasar con la solución reguladora (3.24).
Pasar a la fase de cromatografía según el punto 5.4.
Si los extractos no se examinan el mismo día, deben conservarse a menos de 5 oC.
5.3. Determinación de los aminoácidos totales
5.3.1.
Pesar, con una precisión de 0,2 mg, entre 0,1 g y 1 g de la muestra preparada (5.1), en:
|
— |
un matraz de fondo redondo de 100 ml (4.1) para hidrólisis abierta (5.3.2.3), o |
|
— |
un matraz de fondo redondo de 250 ml (4.1), si se requiere una baja concentración de sodio (5.3.3.1), o |
|
— |
un frasco de 100 ml con tapón de rosca (4.2) para hidrólisis cerrada (5.3.2.4). |
La porción de muestra pesada debe tener un contenido de nitrógeno de unos 10 mg y un contenido de humedad no superior a 100 mg.
Colocar el matraz o el frasco en un baño de hielo y agua y enfriarlo a 0 oC, añadir 5 ml de mezcla de oxidación (3.23) y mezclar con una espátula de vidrio de punta curvada. Cerrar el matraz o el frasco, con la espátula dentro, por medio de una película impermeable al aire, colocar el baño de hielo y agua con el recipiente cerrado en un frigorífico a 0 oC y dejar durante 16 horas. Transcurridas esas 16 horas, sacar del frigorífico y descomponer el exceso de reactivo de oxidación añadiendo 0,84 g de disulfito de sodio (3.4).
Pasar al punto 5.3.2.1.
5.3.2.
5.3.2.1.
Añadir 25 ml de mezcla de hidrólisis (3.20) a la muestra oxidada preparada según el punto 5.3.1, procurando arrastrar cualquier residuo de muestra que hubiera quedado adherido a las paredes del recipiente y a la espátula.
Según el procedimiento de hidrólisis utilizado, proceder según el punto 5.3.2.3 o 5.3.2.4.
5.3.2.2.
Pesar en un matraz de fondo redondo de 100 ml o 250 ml (4.1), o en un frasco de 100 ml con tapón de rosca (4.2), con una precisión de 0,2 mg, entre 0,1 g y 1 g de la muestra preparada (5.1). La porción de muestra pesada debe tener un contenido de nitrógeno de unos 10 mg. Añadir cuidadosamente 25 ml de mezcla de hidrólisis (3.20) y mezclar con la muestra. Proceder según el punto 5.3.2.3 o 5.3.2.4.
5.3.2.3.
Añadir 3 perlas de vidrio a la mezcla del matraz (preparada según el punto 5.3.2.1 o 5.3.2.2) y hervir con borboteo continuo y reflujo durante 23 horas. Al terminar la hidrólisis, lavar el refrigerante con 5 ml de solución reguladora de citrato (3.24). Desconectar el matraz y enfriarlo en un baño de hielo.
Proceder según el punto 5.3.3.
5.3.2.4.
Colocar el frasco con la mezcla preparada según el punto 5.3.2.1 o 5.3.2.2 en una estufa (4.3) a 110 oC. Durante la primera hora, para prevenir la formación de presión (debido a la producción de sustancias gaseosas) y evitar el peligro de explosión, poner el tapón de rosca encima del recipiente, pero sin cerrarlo. Al cabo de una hora, cerrar el recipiente con el tapón y dejarlo en la estufa (4.3) durante 23 horas. Una vez terminada la hidrólisis, sacar el frasco de la estufa, desenroscar cuidadosamente el tapón y colocar el frasco en un baño de hielo y agua. Dejar enfriar.
En función del método de ajuste del pH (5.3.3), transvasar cuantitativamente el contenido del frasco a un vaso de precipitado de 250 ml o a un matraz de fondo redondo de 250 ml, utilizando solución reguladora de citrato (3.24).
Proceder según el punto 5.3.3.
5.3.3.
En función de la tolerancia al sodio del analizador de aminoácidos (4.9), proceder según el punto 5.3.3.1 o 5.3.3.2 para ajustar el pH.
5.3.3.1.
Es recomendable utilizar una solución patrón madre de patrón interno (3.27.3) si se utilizan analizadores de aminoácidos que requieran una baja concentración de sodio (cuando haya que reducir el volumen de ácido).
En este caso, añadir al hidrolizado 2,00 ml de la solución patrón madre (3.27.3) interna antes de la evaporación.
Añadir 2 gotas de 1-octanol (3.15) al hidrolizado obtenido según el punto 5.3.2.3 o 5.3.2.4.
Por medio de un evaporador rotativo (4.7), reducir el volumen a 5-10 ml en vacío a 40 oC. Si el volumen se reduce accidentalmente a menos de 5 ml, debe desecharse el hidrolizado y recomenzarse el análisis.
Ajustar el pH a 2,20 con solución de hidróxido de sodio (3.18) y pasar al punto 5.3.4.
5.3.3.2.
Tomar los hidrolizados obtenidos de acuerdo con el punto 5.3.2.3 o 5.3.2.4 y neutralizarlos parcialmente añadiendo, mientras se remueve, 17 ml de solución de hidróxido de sodio (3.17), cuidando de que la temperatura se mantenga por debajo de 40 oC.
Ajustar el pH a 2,20 a temperatura ambiente con solución de hidróxido de sodio (3.17) y, finalmente, con solución de hidróxido de sodio (3.18). Pasar al punto 5.3.4.
5.3.4.
Transvasar cuantitativamente el hidrolizado de pH ajustado (5.3.3.1 o 5.3.3.2) con solución reguladora de citrato (3.24) a un matraz aforado de 200 ml y enrasar con solución reguladora (3.24).
Si aún no se ha utilizado un patrón interno, añadir 2,00 ml de patrón interno (3.27.3) y enrasar con solución reguladora de citrato (3.24). Mezclar perfectamente.
Pasar a la fase de cromatografía (5.4).
Si las soluciones de muestra no se van a examinar el mismo día, deben conservarse a menos de 5 oC.
5.4. Cromatografía
Antes de realizar la cromatografía, llevar el extracto (5.2) o el hidrolizado (5.3.4) a temperatura ambiente. Agitar la mezcla y filtrar una cantidad adecuada a través de un filtro de membrana de 0,22 μm (4.5). Se obtiene una solución clara que se somete a cromatografía de intercambio iónico, utilizando un analizador de aminoácidos (4.9).
La inyección puede realizarse de forma manual o automática. Es importante que siempre se añada a la columna la misma cantidad de solución ±0,5 % para el análisis de patrones y muestras, excepto cuando se utilice un patrón interno, y que las relaciones sodio/aminoácidos de las soluciones patrón y de muestra sean lo más parecidas posible.
En general, la frecuencia de las calibraciones depende de la estabilidad del reactivo de ninhidrina y del sistema analítico. El patrón o la muestra se diluyen con solución reguladora de citrato (3.24) para conseguir un área de pico del patrón equivalente al 30-200 % del área de pico de los aminoácidos de la muestra.
La cromatografía de aminoácidos variará ligeramente según el tipo de analizador empleado y la resina utilizada. El sistema elegido debe ser capaz de separar unos aminoácidos de otros y los aminoácidos de otros materiales que reaccionan con la ninhidrina. En el intervalo de funcionamiento, el sistema cromatográfico debe proporcionar una respuesta lineal a los cambios en las cantidades de aminoácidos que se añadan a la columna.
Durante la fase de cromatografía, cuando se analice una solución equimolar (de los aminoácidos determinados), se aplicarán las relaciones altura de valle/altura de pico que se mencionan más adelante. Esta solución equimolar debe contener al menos el 30 % de la carga máxima de cada aminoácido que puede medirse con exactitud con el sistema de análisis de aminoácidos (4.9).
Para separar la treonina de la serina, la relación altura de valle/altura de pico del más bajo de los dos aminoácidos que se solapan en el cromatograma no debe pasar de 2/10 (si solo se determinan cistina, cisteína, metionina, treonina y lisina, una separación insuficiente entre picos adyacentes afectará negativamente a la determinación). Respecto a los demás aminoácidos, la separación debe ser mejor que 1/10.
El sistema debe garantizar que la lisina se separe de los «artefactos de lisina» y de la ornitina.
6. Cálculo de los resultados
Las áreas de los picos de la muestra y el patrón se miden para cada aminoácido y la cantidad correspondiente (X) se calcula en gramos de aminoácidos por kilogramo de muestra.

Si se utiliza un patrón interno, debe multiplicarse por: 
|
A |
= |
área de pico, hidrolizado o extracto |
|
B |
= |
área de pico, solución patrón de calibración |
|
C |
= |
área de pico, patrón interno en el hidrolizado o el extracto |
|
D |
= |
área de pico, patrón interno, solución patrón de calibración |
|
M |
= |
peso molecular del aminoácido que se está determinando |
|
c |
= |
concentración del patrón en μmol/ml |
|
m |
= |
peso de la muestra (g) (corregido para obtener el peso original si se ha desecado o desengrasado) |
|
V |
= |
mililitros de hidrolizado total (5.3.4) o mililitros de volumen de dilución total calculado del extracto (6.1). |
Tanto la cistina como la cisteína se determinan como ácido cisteico en hidrolizados de la muestra oxidada, pero se calculan como cistina (C6H12N2O4S2, M 240,30 g/mol) utilizando un M de 120,15 g/mol (= 0,5 x 240,30 g/mol).
La metionina se determina como metionina sulfona en hidrolizados de muestra oxidada, pero se calcula como metionina utilizando el M de la metionina: 149,21 g/mol.
La metionina libre añadida se determina tras extracción como metionina, utilizando para el cálculo el mismo M.
|
6.1. |
El volumen total de dilución de los extractos (F) para la determinación de los aminoácidos libres (5.2) se calcula como sigue: 
|
7. Evaluación del método
El método se puso a prueba en un estudio intercomparativo realizado a nivel internacional en 1990 con cuatro piensos diferentes (pienso mezclado para cerdos, pienso compuesto para pollos de engorde, concentrado de proteínas y premezcla). Los resultados, tras descartar los valores atípicos, de la media y de la desviación típica se indican en los cuadros que figuran en el presente punto:
Medias en g/kg
|
Material de referencia |
Aminoácido |
||||||
|
Treonina |
Cistina y cisteína |
Metionina |
Lisina |
||||
|
Pienso mezclado para cerdos |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|||
|
Pienso compuesto para pollos de engorde |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|||
|
Concentrado de proteínas |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|||
|
Premezcla |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|||
|
|||||||
7.1. Repetibilidad
La repetibilidad del citado estudio intercomparativo, expresada como «desviación típica intralaboratorio», se indica en los cuadros siguientes:
Desviación típica intralaboratorio (Sr) en g/kg
|
Material de referencia |
Aminoácido |
||||||
|
Treonina |
Cistina y cisteína |
Metionina |
Lisina |
||||
|
Pienso mezclado para cerdos |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|||
|
Pienso compuesto para pollos de engorde |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|||
|
Concentrado de proteínas |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|||
|
Premezcla |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|||
|
|||||||
Coeficiente de variación (%) de la desviación típica intralaboratorio (Sr)
|
Material de referencia |
Aminoácido |
||||||
|
Treonina |
Cistina y cisteína |
Metionina |
Lisina |
||||
|
Pienso mezclado para cerdos |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|||
|
Pienso compuesto para pollos de engorde |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|||
|
Concentrado de proteínas |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|||
|
Premezcla |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|||
|
|||||||
7.2 Reproducibilidad
En el cuadro siguiente se indican los resultados de la desviación típica interlaboratorios obtenida en el citado estudio intercomparativo:
Desviación típica interlaboratorios (SR) en g/kg
|
Material de referencia |
Aminoácido |
||||||
|
Treonina |
Cistina y cisteína |
Metionina |
Lisina |
||||
|
Pienso mezclado para cerdos |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|||
|
Pienso compuesto para pollos de engorde |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|||
|
Concentrado de proteínas |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|||
|
Premezcla |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|||
|
|||||||
Coeficiente de variación (%) de la desviación típica interlaboratorios (SR)
|
Material de referencia |
Aminoácido |
||||||
|
Treonina |
Cistina y cisteína |
Metionina |
Lisina |
||||
|
Pienso mezclado para cerdos |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|||
|
Pienso compuesto para pollos de engorde |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|||
|
Concentrado de proteínas |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|||
|
Premezcla |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|||
|
|||||||
8. Uso de materiales de referencia
La correcta aplicación del método se verificará haciendo mediciones reiteradas de materiales de referencia certificados, cuando estén disponibles. Se recomienda la calibración con solución de calibración de aminoácidos certificada.
9. Observaciones
|
9.1. |
Debido a las diferencias entre los analizadores de aminoácidos, las concentraciones finales de las soluciones de calibración de los aminoácidos patrón (véanse los puntos 3.27.4 y 3.27.5) y del hidrolizado (véase el punto 5.3.4) deberán entenderse como orientativas. El intervalo de respuesta lineal del aparato debe comprobarse con todos los aminoácidos. La solución patrón se diluye con una solución reguladora de citrato para obtener áreas de pico en el centro del intervalo. |
|
9.2. |
Si se usa un equipo de cromatografía de líquidos de alta resolución para analizar los hidrolizados, deben optimizarse las condiciones experimentales siguiendo las recomendaciones del fabricante. |
|
9.3. |
Si se aplica este método a piensos que contengan más de un 1 % de cloruro (concentrados, piensos minerales, piensos complementarios), es posible que se subestimen los valores de metionina, por lo que debe aplicarse un tratamiento especial. |
G. DETERMINACIÓN DEL TRIPTÓFANO
1. Objeto y ámbito de aplicación
Este método permite determinar el triptófano total y libre en los piensos. No distingue entre formas L y D.
2. Principio
Para la determinación del triptófano total, la muestra se hidroliza en condiciones alcalinas con solución saturada de hidróxido de bario y se calienta a 110 oC durante veinte horas. Tras la hidrólisis se añade patrón interno.
Para la determinación del triptófano libre, la muestra se somete a extracción en condiciones de acidez suave en presencia de patrón interno.
El triptófano y el patrón interno presentes en el hidrolizado o en el extracto se determinan mediante CLAR con detección por fluorescencia.
3. Reactivos
|
3.1. |
Debe utilizarse agua bidestilada o agua de calidad equivalente (conductividad < 10 μS/cm). |
|
3.2. |
Sustancia patrón: triptófano (pureza/contenido ≥ 99 %), desecado al vacío sobre pentóxido de fósforo |
|
3.3. |
Patrón interno: α-metiltriptófano (pureza/contenido ≥ 99 %), desecado al vacío sobre pentóxido de fósforo |
|
3.4. |
Hidróxido de bario octahidratado (deberá procurarse no exponer excesivamente el Ba(OH)2 · 8 H2O al aire a fin de evitar la formación de BaCO3, que podría interferir en la determinación) (véase la observación del punto 9.3) |
|
3.5. |
Hidróxido de sodio |
|
3.6. |
Ácido ortofosfórico, p (p/p) = 85 % |
|
3.7. |
Ácido clorhídrico, ρ20 = 1,19 g/ml |
|
3.8. |
Metanol, equivalente al de calidad CLAR |
|
3.9. |
Éter de petróleo, con un intervalo de ebullición de 40-60 oC |
|
3.10. |
Solución de hidróxido de sodio, c = 1 mol/l: Disolver 40,0 g de NaOH (3.5) en agua y enrasar a 1 l con agua (3.1). |
|
3.11. |
Ácido clorhídrico, c = 6 mol/l: Tomar 492 ml de HCl (3.7) y enrasar a 1 l con agua. |
|
3.12. |
Ácido clorhídrico, c = 1 mol/l: Tomar 82 ml de HCl (3.7) y enrasar a 1 l con agua. |
|
3.13. |
Ácido clorhídrico, c = 0,1 mol/l: Tomar 8,2 ml de HCl (3.7) y enrasar a 1 l con agua. |
|
3.14. |
Ácido ortofosfórico, c = 0,5 mol/l: Tomar 34 ml de ácido ortofosfórico (3.6) y enrasar a 1 l con agua (3.1). |
|
3.15. |
Solución concentrada de triptófano (3.2), c = 2,50 μmol/ml: En un matraz aforado de 500 ml, disolver 0,2553 g de triptófano (3.2) en ácido clorhídrico (3.13) y enrasar con ácido clorhídrico (3.13). Conservar a - 18 oC durante cuatro semanas como máximo. |
|
3.16. |
Solución concentrada de patrón interno, c = 2,50 μmol/ml: En un matraz aforado de 500 ml, disolver 0,2728 g de α-metiltriptófano (3.3) en ácido clorhídrico (3.13) y enrasar con ácido clorhídrico (3.13). Conservar a - 18 oC durante cuatro semanas como máximo. |
|
3.17. |
Solución patrón de calibración de triptófano y patrón interno: Tomar 2,00 ml de solución concentrada de triptófano (3.15) y 2,00 ml de solución concentrada de patrón interno (α-metiltriptófano) (3.16). Diluir con agua (3.1) y metanol (3.8) hasta conseguir aproximadamente el mismo volumen y la misma concentración de metanol (10-30 %) que en el hidrolizado acabado. Esta solución debe prepararse poco antes de utilizarse. Proteger de la luz solar directa mientras se prepara. |
|
3.18. |
Ácido acético |
|
3.19. |
1,1,1-tricloro-2-metil-2-propanol |
|
3.20. |
Etanolamina p (p/p) > 98 % |
|
3.21. |
Solución de 1 g de 1,1,1-tricloro-2-metil-2-propanol (3.19) en 100 ml de metanol (3.8) |
|
3.22. |
Fase móvil para la CLAR: 3,00 g de ácido acético (3.18) + 900 ml de agua (3.1) + 50,0 ml de solución (3.21) de 1,1,1-tricloro-2-metil-2-propanol (3.19) en metanol (3.8) (1 g/100 ml). Ajustar el pH a 5,00 con etanolamina (3.20). Enrasar a 1 000 ml con agua (3.1). |
4. Instrumental
|
4.1. |
Equipo de CLAR con detector por espectrofluorometría |
|
4.2. |
Columna cromatográfica de líquidos, de 125 mm x 4 mm, C18, relleno de 3 μm, o equivalente |
|
4.3. |
pH-metro |
|
4.4. |
Matraz de polipropileno, de 125 ml de capacidad, cuello ancho y tapón de rosca |
|
4.5. |
Filtro de membrana de 0,45 μm |
|
4.6. |
Autoclave, 110 (± 2) oC, 1,4 (±0,1) bar |
|
4.7. |
Agitador mecánico o magnético |
|
4.8. |
Agitador vortex |
5. Procedimiento
5.1. Preparación de las muestras
La muestra se muele hasta que pase por un tamiz de 0,5 mm. Las muestras con humedad elevada deben secarse al aire a una temperatura no superior a 50 oC, o bien liofilizarse antes de la molienda. Las muestras con un elevado contenido de grasa deberán someterse a extracción con éter de petróleo (3.9) antes de la molienda.
5.2. Determinación del triptófano libre (extracto)
Pesar en un Erlenmeyer, con una precisión de 1 mg, una cantidad adecuada (1-5 g) de la muestra preparada (5.1). Añadir 100,0 ml de ácido clorhídrico (3.13) y 5,00 ml de solución concentrada de patrón interno (3.16). Agitar o mezclar durante 60 minutos utilizando un agitador mecánico o magnético (4.7). Dejar sedimentar y pipetear 10,0 ml de la solución sobrenadante a un vaso de precipitado. Añadir 5 ml de ácido ortofosfórico (3.14). Ajustar el pH a 3 utilizando hidróxido de sodio (3.10). Añadir suficiente metanol (3.8) para obtener en el volumen final una concentración de metanol del 10 % al 30 %. Transvasar a un matraz aforado de capacidad adecuada y diluir con agua hasta conseguir el volumen necesario para la cromatografía (aproximadamente el mismo volumen que la solución patrón de calibración [3.17]).
Filtrar unos pocos mililitros de la solución por un filtro de membrana de 0,45 μm (4.5) antes de inyectarla en la columna de CLAR. Pasar a la fase de cromatografía según el punto 5.4.
Proteger la solución patrón y los extractos de la luz solar directa. Si no es posible analizar los extractos el mismo día, pueden conservarse a 5 oC durante tres días como máximo.
5.3. Determinación del triptófano total (hidrolizado)
Pesar en el matraz de polipropileno (4.4), con una precisión de 0,2 mg, entre 0,1 g y 1 g de la muestra preparada (5.1). La porción de muestra pesada deberá tener un contenido de nitrógeno de unos 10 mg. Añadir 8,4 g de hidróxido de bario octahidratado (3.4) y 10 ml de agua. Mezclar con un agitador vortex (4.8) o un agitador magnético (4.7). Dejar dentro de la mezcla el imán recubierto de teflón. Lavar las paredes del recipiente con 4 ml de agua. Poner el tapón de rosca y cerrar el matraz sin apretar. Pasar al autoclave (4.6) con agua hirviendo y dejar al vapor durante 30 a 60 minutos. Cerrar el autoclave y dejar a 110 (± 2) oC durante 20 horas.
Antes de abrir el autoclave, reducir la temperatura hasta justo por debajo de 100 oC. Para evitar la cristalización del Ba(OH)2 · 8 H2O, añadir a la mezcla caliente 30 ml de agua que esté a temperatura ambiente. Agitar o remover suavemente. Añadir 2,00 ml de solución concentrada de patrón interno (α-metiltriptófano) (3.16). Enfriar el recipiente en baño de agua y hielo durante 15 minutos.
A continuación, añadir 5 ml de ácido ortofosfórico (3.14). Mantener el recipiente en el baño frío y neutralizar con HCl (3.11), al tiempo que se remueve; ajustar el pH a 3,0 con HCl (3.12). Añadir suficiente metanol para obtener en el volumen final una concentración de metanol del 10 % al 30 %. Pasar a un matraz aforado de capacidad adecuada y diluir con agua hasta conseguir el volumen definido necesario para la cromatografía (por ejemplo, 100 ml). La adición de metanol no deberá producir precipitación.
Filtrar unos pocos mililitros de la solución por un filtro de membrana de 0,45 μm (4.5) antes de inyectarla en la columna de CLAR. Pasar a la fase de cromatografía según el punto 5.4.
Proteger la solución patrón y los hidrolizados de la luz solar directa. Si no es posible analizar los hidrolizados el mismo día, pueden conservarse a 5 oC durante tres días, como máximo.
5.4. Determinación mediante CLAR
Las siguientes condiciones de elución isocrática tienen carácter orientativo; pueden aplicarse otras, siempre que ofrezcan resultados equivalentes (véanse también las observaciones de los puntos 9.1 y 9.2).
|
Columna cromatográfica de líquidos: |
125 mm x 4 mm, C18, relleno de 3 μm, o equivalente |
|
Temperatura de la columna: |
Temperatura ambiente |
|
Fase móvil (3.22): |
3,00 g de ácido acético (3.18) + 900 ml de agua (3.1) +50,0 ml de solución (3.21) de 1,1,1-tricloro-2-metil-2-propanol (3.19) en metanol (3.8) (1 g/100 ml). Ajustar el pH a 5,00 con etanolamina (3.20). Enrasar a 1 000 ml con agua (3.1). |
|
Caudal: |
1 ml/min. |
|
Tiempo total del ciclo: |
Aproximadamente 34 minutos |
|
Longitud de onda de detección: |
Excitación: 280 nm, emisión: 356 nm. |
|
Volumen de inyección: |
20 μl |
6. Cálculo de los resultados
Se calcula la cantidad de triptófano (X) en gramos por cada 100 g de muestra.

|
A |
= |
área de pico del patrón interno, solución patrón de calibración (3.17) |
|
B |
= |
área de pico del triptófano, extracto (5.2) o hidrolizado (5.3) |
|
V1 |
= |
volumen en mililitros (2 ml) de la solución concentrada de triptófano (3.15) añadida a la solución de calibración (3.17) |
|
c |
= |
concentración en μmol/ml (= 2,50) de la solución concentrada de triptófano (3.15) añadida a la solución de calibración (3.17) |
|
V2 |
= |
volumen en mililitros de la solución concentrada de patrón interno (3.16) añadida al extracto (5.2) (= 5,00 ml) o al hidrolizado (5.3) (= 2,00 ml) |
|
C |
= |
área de pico del patrón interno, extracto (5.2) o hidrolizado (5.3) |
|
D |
= |
área de pico del triptófano, solución patrón de calibración (3.17) |
|
V3 |
= |
volumen en mililitros (= 2,00 ml) de la solución concentrada de patrón interno (3.16) añadida a la solución patrón de calibración (3.17) |
|
m |
= |
peso de la muestra en gramos (corregido al peso original si se ha desecado o desengrasado) |
|
M |
= |
peso molecular del triptófano (= 204,23 g/mol) |
7. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe rebasar el 10 % del resultado más elevado.
8. Resultados de un estudio colaborativo
Se organizó un estudio colaborativo de la Comunidad Europea (cuarta comparación interlaboratorios), en el cual hasta 12 laboratorios analizaron tres muestras para certificar el método de hidrólisis. Con cada muestra se repitieron varios análisis (5). Los resultados se recogen en el cuadro siguiente:
|
|
Muestra 1 Pienso para cerdos |
Muestra 2 Pienso para cerdos con complemento de L-triptófano |
Muestra 3 Pienso concentrado para cerdos |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Media [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [ %] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [ %] |
6,3 |
6,0 |
2,2 |
|
L |
= |
número de laboratorios que presentaron resultados |
|
n |
= |
número de resultados individuales aceptados tras eliminar los valores atípicos (identificados mediante las pruebas de Cochran y Dixon) |
|
sr |
= |
desviación típica de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
r |
= |
repetibilidad |
|
R |
= |
reproducibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad, en tanto por ciento |
|
CVR |
= |
coeficiente de variación de la reproducibilidad, en tanto por ciento |
En otro estudio colaborativo de la Comunidad Europea (tercera comparación interlaboratorios), hasta trece laboratorios analizaron dos muestras para certificar el método de extracción del triptófano libre. Con cada muestra se repitieron varios análisis (5). Los resultados se recogen en el cuadro siguiente:
|
|
Muestra 4 Mezcla de trigo y soja |
Muestra 5 Mezcla de trigo y soja (= muestra 4) con triptófano añadido (0,457 g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Media [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [ %] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [ %] |
4,71 |
5,11 |
|
L |
= |
número de laboratorios que presentaron resultados |
|
n |
= |
número de resultados individuales aceptados tras eliminar los valores atípicos (identificados mediante las pruebas de Cochran y Dixon) |
|
sr |
= |
desviación típica de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
r |
= |
repetibilidad |
|
R |
= |
reproducibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad, en tanto por ciento |
|
CVR |
= |
coeficiente de variación de la reproducibilidad, en tanto por ciento |
En otro estudio intercomparativo de la Comunidad Europea, hasta siete laboratorios analizaron cuatro muestras para certificar el método de hidrólisis de triptófano. A continuación figuran los resultados del estudio. Con cada muestra se repitieron varios análisis (5).
|
|
Muestra 1 Pienso mezclado para cerdos (CRM 117) |
Muestra 2 Harina de pescado con poca grasa (CRM 118) |
Muestra 3 Sémola de soja (CRM 119) |
Muestra 4 Leche desnatada en polvo (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Media [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [ %] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [ %] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
número de laboratorios que presentaron resultados |
|
n |
= |
número de resultados individuales aceptados tras eliminar los valores atípicos (identificados mediante las pruebas de Cochran y Dixon) |
|
sr |
= |
desviación típica de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
r |
= |
repetibilidad |
|
R |
= |
reproducibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad, en tanto por ciento |
|
CVR |
= |
coeficiente de variación de la reproducibilidad, en tanto por ciento |
9. Observaciones
|
9.1. |
Las siguientes condiciones cromatográficas pueden ofrecer una mejor separación entre el triptófano y el α-metiltriptófano. Elución isocrática, seguida de lavado de la columna de gradiente:
|
||||||||||||||||||||||||||||||||||||||||||||
|
9.2. |
La cromatografía variará en función del tipo de CLAR y del material de relleno de la columna. El sistema elegido debe proporcionar una separación en la línea base entre el triptófano y el patrón interno. Asimismo, es importante que los productos de degradación se separen bien del triptófano y del patrón interno. Deberán pasarse hidrolizados sin patrón interno para comprobar las impurezas de la línea base bajo el patrón interno. También es importante que el ciclo dure lo suficiente para que se eluyan todos los productos de degradación, pues, de lo contrario, los picos de elución tardía pueden interferir en los ciclos cromatográficos posteriores. En el intervalo de funcionamiento, el sistema cromatográfico deberá dar una respuesta lineal. Esta deberá medirse con una concentración constante (la normal) del patrón interno y con concentraciones variables de triptófano. Es importante que el tamaño de los picos tanto de triptófano como de patrón interno estén dentro del intervalo lineal del sistema de CLAR/fluorescencia. Si los picos de triptófano o de patrón interno son demasiado pequeños o demasiado grandes, deberá repetirse el análisis con otro tamaño de muestra o con un volumen final distinto. |
|
9.3. |
Hidróxido de bario
Con el tiempo, el hidróxido de bario es más difícil de disolver. Esto hace que la solución para la determinación por CLAR esté turbia, lo que puede dar unos resultados bajos de triptófano. |
H. DETERMINACIÓN DE LOS ACEITES Y LAS GRASAS BRUTOS
1. Objeto y ámbito de aplicación
Este método sirve para determinar el contenido de aceites y grasas brutos de los piensos. No abarca el análisis de semillas y frutos oleaginosos.
La aplicación de uno u otro de los dos procedimientos que se describen a continuación dependerá de la naturaleza y la composición del pienso y de la razón por la que se lleve a cabo el análisis.
1.1. Procedimiento A. Aceites y grasas brutos directamente extraíbles
Este método es aplicable a los materiales para piensos de origen vegetal, excepto los incluidos en el ámbito de aplicación del procedimiento B.
1.2. Procedimiento B. Aceites y grasas brutos totales
Este método es aplicable a los materiales para piensos de origen animal y a todos los piensos compuestos. Se ha de utilizar con todos los materiales de los que los aceites y las grasas no puedan extraerse completamente sin hidrólisis previa (por ejemplo, los glútenes, las levaduras, las proteínas de patata y los productos sujetos a procesos como la extrusión, la floculación y el calentamiento).
1.3. Interpretación de los resultados
En todos los casos en que el resultado obtenido con el procedimiento B sea mayor que el obtenido con el procedimiento A, el resultado obtenido con el procedimiento B se aceptará como el valor real.
2. Principio
2.1. Procedimiento A
La muestra se somete a extracción con éter de petróleo. El disolvente se extrae por destilación y el residuo se seca y se pesa.
2.2. Procedimiento B
La muestra se trata en caliente con ácido clorhídrico. La mezcla se enfría y se filtra. Una vez lavado y secado, el residuo se somete a la determinación según el procedimiento A.
3. Reactivos
|
3.1. |
Éter de petróleo con un intervalo de ebullición de 40 oC a 60 oC. El índice de bromo debe ser inferior a 1 y el residuo en evaporación inferior a 2 mg/100 ml. |
|
3.2. |
Sulfato de sodio, anhidro |
|
3.3. |
Ácido clorhídrico, c = 3 mol/l |
|
3.4. |
Coadyuvante de filtración, como, por ejemplo, Kieselgur o Hyflo Supercel |
4. Instrumental
|
4.1. |
Aparato de extracción. Si el aparato está provisto de un sifón (aparato Soxhtel), el caudal de reflujo deberá regularse de forma que se obtengan como mínimo diez ciclos por hora; si se trata de un aparato sin sifón, el caudal de reflujo deberá ser de alrededor de 10 ml por minuto. |
|
4.2. |
Cartuchos de extracción, exentos de sustancias solubles en el éter de petróleo y de porosidad compatible con las exigencias del punto 4.1 |
|
4.3. |
Estufa de secado, bien de vacío a 75 oC ± 3 oC, bien de aire a 100 oC ± 3 oC |
5. Procedimiento
5.1. Procedimiento A (véase el punto 8.1)
Pesar, con una precisión de 1 mg, 5 g de la muestra, pasarlos a un cartucho de extracción (4.2) y cubrirlos con un poco de algodón hidrófilo exento de grasa.
Colocar el cartucho en un extractor (4.1) y extraer durante seis horas con éter de petróleo (3.1). Recoger el extracto de éter de petróleo en un matraz seco tarado que contenga fragmentos de piedra pómez (2).
Extraer el disolvente por destilación. Secar el residuo dejando el matraz hora y media en la estufa de secado (4.3). Dejar enfriar en un desecador y pesar. Volver a secar durante 30 minutos para que el peso de los aceites y las grasas se mantenga constante (la pérdida de peso entre dos pesadas sucesivas debe ser inferior o igual a 1 mg).
5.2. Procedimiento B
Pesar, con una precisión de 1 mg, 2,5 g de la muestra (véase el punto 8.2), introducirlos en un vaso de precipitado de 400 ml o en un Erlenmeyer de 300 ml y añadir 100 ml de ácido clorhídrico (3.3), junto con algunos fragmentos de piedra pómez. Cubrir el vaso de precipitado con un vidrio de reloj o dotar al Erlenmeyer de un refrigerante de reflujo. Llevar la mezcla a ebullición suave sobre una llama pequeña o una placa calefactora eléctrica y mantenerla así durante una hora. No dejar que el producto se adhiera a las paredes del recipiente.
Enfriar y añadir una cantidad de coadyuvante de filtración (3.4) suficiente para evitar que se pierda aceite o grasa durante la filtración. Filtrar sobre un papel de filtro doble humedecido exento de grasa. Lavar el residuo con agua fría hasta obtener un filtrado neutro. Comprobar que el filtrado no contiene aceites ni grasas. Si los contiene, la muestra debe someterse a extracción con éter de petróleo, según el procedimiento A, antes de la hidrólisis.
Colocar el papel de filtro doble que contiene el residuo sobre un vidrio de reloj y secar durante hora y media en la estufa de aire (4.3) a 100 oC ± 3 oC.
Introducir el papel de filtro doble que contiene el residuo seco en un cartucho de extracción (4.2) y cubrirlo con un poco de algodón hidrófilo exento de grasa. Colocar el cartucho en un extractor (4.1) y proceder según se indica en los párrafos segundo y tercero del punto 5.1.
6. Expresión de los resultados
Expresar el peso del residuo en porcentaje de la muestra.
7. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra por el mismo analista no deberá superar:
|
— |
el 0,2 % en valor absoluto, en el caso de contenidos de aceites y grasas brutos inferiores al 5 %, |
|
— |
el 4,0 % del resultado más elevado, en relación con contenidos del 5 % al 10 %, |
|
— |
el 0,4 % en valor absoluto, en el caso de contenidos superiores al 10 %. |
8. Observaciones
|
8.1. |
Con los productos que tienen un alto contenido de aceites y grasas y son difíciles de triturar o inapropiados para obtener una muestra de ensayo reducida homogénea, proceder del modo siguiente: Pesar, con una precisión de 1 mg, 20 g de la muestra y mezclarlos con 10 g o más de sulfato de sodio anhidro (3.2). Extraer con éter de petróleo (3.1) según se indica en el punto 5.1. Enrasar el extracto obtenido a 500 ml con éter de petróleo (3.1) y mezclar. Tomar 50 ml de la solución y ponerlos en un matraz pequeño, seco y tarado con fragmentos de piedra pómez. Extraer el disolvente por destilación, secar y proceder según se indica en el último párrafo del punto 5.1. Eliminar el disolvente del residuo de extracción que haya quedado en el cartucho, triturar el residuo a una finura de 1 mm, colocarlo de nuevo en el cartucho de extracción (no añadir sulfato de sodio) y proceder según se indica en los párrafos segundo y tercero del punto 5.1. Calcular el contenido de aceites y grasas en porcentaje de la muestra aplicando la siguiente fórmula: (10 m1 + m2) × 5 donde:
|
|
8.2. |
En el caso de productos pobres en aceites y grasas, la muestra de ensayo puede aumentarse a 5 g. |
|
8.3. |
Es posible que los piensos para animales de compañía con un alto contenido de agua deban mezclarse con sulfato de sodio anhidro antes de la hidrólisis y la extracción conforme al procedimiento B. |
|
8.4. |
En el punto 5.2 puede resultar más eficaz utilizar agua caliente en lugar de agua fría para lavar el residuo tras la filtración. |
|
8.5. |
Con algunos piensos quizá sea necesario aumentar el tiempo de secado de hora y media. Se evitará un secado excesivo, ya que puede producir resultados bajos. También puede utilizarse un horno microondas. |
|
8.6. |
Si el contenido de aceites o grasas brutos es superior al 15 %, se recomienda la preextracción por el procedimiento A antes de la hidrólisis y la reextracción por el procedimiento B. Hasta cierto punto, esto depende de la naturaleza del pienso y de la naturaleza del aceite o la grasa que contenga. |
I. DETERMINACIÓN DE LA FIBRA BRUTA
1. Objeto y ámbito de aplicación
Este método permite determinar las sustancias orgánicas de los piensos exentas de grasa, que son insolubles en medios ácidos y alcalinos y se designan convencionalmente como fibra bruta.
2. Principio
La muestra, si es necesario desengrasada, se trata sucesivamente con soluciones en ebullición de ácido sulfúrico e hidróxido de potasio de concentraciones determinadas. El residuo se separa por filtración en un filtro de vidrio sinterizado, se lava, se deseca, se pesa y se calcina en un intervalo de 475 oC a 500 oC. La pérdida de peso resultante de la calcinación corresponde a la fibra bruta presente en la muestra de ensayo.
3. Reactivos
|
3.1. |
Acido sulfúrico, c = 0,13 mol/l |
|
3.2. |
Agente antiespumante (por ejemplo, n-octanol) |
|
3.3. |
Coadyuvante de filtración (Celite 545 o equivalente), calentado a 500 oC durante cuatro horas (8.6) |
|
3.4. |
Acetona |
|
3.5. |
Éter de petróleo, con un intervalo de ebullición de 40 oC a 60 oC |
|
3.6. |
Ácido clorhídrico, c = 0,5 mol/l |
|
3.7. |
Solución de hidróxido de sodio, c = 0,23 mol/l |
4. Instrumental
|
4.1. |
Unidad de calentamiento para digestión con ácido sulfúrico y solución de hidróxido de potasio, provista de un soporte para el crisol filtrante (4.2) y de un tubo de salida con una llave conectada a la salida de líquidos y al vacío, posiblemente con aire comprimido. Antes de usarla, precalentar la unidad todos los días con agua hirviendo durante cinco minutos. |
|
4.2. |
Crisol de vidrio filtrante, con placa filtrante de vidrio sinterizado fundido de porosidad comprendida entre 40 μm y 90 μm. Antes de utilizarlo por primera vez, calentarlo a 500 oC durante unos minutos y enfriarlo (8.6). |
|
4.3. |
Probeta apta para ebullición, de 270 ml como mínimo, con refrigerante de reflujo |
|
4.4. |
Horno de secado con termostato |
|
4.5. |
Horno de mufla con termostato |
|
4.6. |
Unidad de extracción consistente en una placa soporte para el crisol filtrante (4.2), con un tubo de descarga provisto de llave conectada al vacío y a la salida de líquidos |
|
4.7. |
Aros de conexión para unir la unidad de calentamiento (4.1), el crisol (4.2) y la probeta (4.3) y conectar la unidad de extracción en frío (4.6) y el crisol |
5. Procedimiento
Pesar, con una precisión de 1 mg, 1 g de la muestra, ponerlo en el crisol (4.2) (véanse las observaciones 8.1, 8.2 y 8.3) y añadir 1 g de coadyuvante de filtración (3.3).
Tras ensamblar la unidad de calentamiento (4.1) y el crisol filtrante (4.2), unir a este último la probeta (4.3). Verter 150 ml de ácido sulfúrico (3.1) en ebullición en la probeta y el crisol ensamblados y, si es necesario, añadir unas pocas gotas de agente antiespumante (3.2).
Llevar el líquido a ebullición en 5 ± 2 minutos y dejar hervir con fuerza durante 30 minutos exactos.
Abrir la llave del tubo de descarga (4.1), filtrar al vacío el ácido sulfúrico a través del crisol filtrante y lavar tres veces consecutivas con 30 ml de agua hirviendo cada vez, cuidando de que el residuo se filtre seco después de cada lavado.
Cerrar la llave de salida y verter 150 ml de solución de hidróxido de potasio (3.7) hirviendo en la probeta y el crisol ensamblados, añadiendo después unas pocas gotas de agente antiespumante (3.2). Llevar el líquido al punto de ebullición en 5 ± 2 minutos y dejar hervir con fuerza durante 30 minutos exactos. Filtrar y repetir el proceso de lavado empleado en la fase de ácido sulfúrico.
Después del lavado y secado finales, desconectar el crisol con su contenido y volverlo a conectar a la unidad de extracción en frío (4.6). Aplicar el vacío y lavar el residuo en el crisol tres veces consecutivas con 25 ml de acetona (3.4) cada vez, cuidando de que el residuo se filtre seco después de cada lavado.
Secar el crisol en la estufa a 130 oC hasta alcanzar un peso constante. Después de cada secado, enfriar en el desecador y pesar rápidamente. Colocar el crisol en un horno de mufla y calcinar, hasta alcanzar un peso constante (la pérdida de peso entre dos pesadas sucesivas debe ser inferior o igual a 2 mg), a 475 oC durante 30 minutos como mínimo.
Después de cada calentamiento, enfriar, primero en el horno y después en el desecador, antes de pesar.
Realizar una prueba en blanco sin la muestra. La pérdida de peso debida a la calcinación no debe exceder de 4 mg.
6. Cálculo de los resultados
El contenido de fibra bruta en porcentaje de la muestra viene dado por la fórmula:

donde:
|
m |
= |
peso de la muestra, en gramos; |
|
m0 |
= |
pérdida de peso tras la calcinación durante la determinación, en gramos; |
|
m1 |
= |
pérdida de peso tras la calcinación durante el ensayo en blanco, en gramos. |
7. Repetibilidad
La diferencia entre dos determinaciones paralelas efectuadas con la misma muestra no debe exceder:
|
— |
del 0,6 % en valor absoluto, en el caso de contenidos de fibra bruta inferiores al 10 %, |
|
— |
del 6 % del resultado superior, en el caso de contenidos de fibra bruta iguales o superiores al 10 %. |
8. Observaciones
|
8.1. |
Los piensos con más de un 10 % de grasa bruta deben desengrasarse antes de efectuar el análisis con éter de petróleo (3.5). Conectar el crisol filtrante (4.2), con su contenido, a la unidad de extracción en frío (4.6), aplicar vacío y lavar tres veces consecutivas con 30 ml de éter de petróleo cada vez, cuidando de que el residuo esté seco. Conectar el crisol, con su contenido, a la unidad de calentamiento (4.1) y continuar según se describe en el punto 5. |
|
8.2. |
Los piensos que contengan grasas que no puedan extraerse directamente con éter de petróleo (3.5) deben desengrasarse con arreglo al punto 8.1 y someterse a un nuevo desengrasado después de haber sido hervidos con ácido. Tras el hervor con ácido y el subsiguiente lavado, unir el crisol, con su contenido, a la unidad de extracción en frío (4.6) y lavar tres veces con 30 ml de acetona y otras tres veces con 30 ml de éter de petróleo cada vez. Filtrar en vacío hasta sequedad y continuar el análisis como se describe en el punto 5, comenzando por el tratamiento con hidróxido de potasio. |
|
8.3. |
Si los piensos contienen más de un 5 % de carbonatos, expresados en carbonato de calcio, conectar el crisol (4.2), con la muestra pesada, a la unidad de calentamiento (4.1). Lavar la muestra tres veces con 30 ml de ácido clorhídrico (3.6). Después de cada adición, dejar reposar la muestra durante un minuto aproximadamente antes de filtrar. Lavar una vez con 30 ml de agua y proseguir como se describe en el punto 5. |
|
8.4. |
Si se utiliza una batería de aparatos (varios crisoles unidos a la misma unidad de calentamiento), no pueden realizarse dos determinaciones distintas de la misma muestra en la misma serie. |
|
8.5. |
Si, tras el hervor, resulta difícil filtrar las soluciones ácidas y alcalinas, introducir aire comprimido por el tubo de descarga de la unidad de calentamiento y seguir filtrando a continuación. |
|
8.6. |
Con objeto de alargar la duración de los crisoles filtrantes de vidrio, la temperatura de calcinación no superará los 500 oC. Asimismo, deben evitarse los cambios bruscos de temperatura en los ciclos de calentamiento y enfriamiento. |
J. DETERMINACIÓN DEL AZÚCAR
1. Objeto y ámbito de aplicación
Este método permite determinar la cantidad de azúcares reductores y azúcares totales tras la inversión, expresados en glucosa o, si procede, en sacarosa, por conversión mediante un factor de 0,95. Es aplicable a los piensos compuestos. Para otros piensos se establecen métodos especiales. Si es necesario, la lactosa deberá medirse por separado y tenerse en cuenta al calcular los resultados.
2. Principio
Los azúcares se extraen en etanol diluido; la solución se clarifica con las soluciones de Carrez I y II. Tras eliminar el etanol se determinan las cantidades antes y después de la inversión, siguiendo el método de Luff-Schoorl.
3. Reactivos
|
3.1. |
Solución de etanol al 40 % (v/v), de 0,948 g/ml de densidad a 20 oC, neutralizada frente a la fenolftaleína. |
|
3.2. |
Solución de Carrez I: disolver en agua 21,9 g de acetato de cinc, Zn(CH3COO)2 · 2H2O, y 3 g de ácido acético glacial. Enrasar con agua a 100 ml. |
|
3.3. |
Solución de Carrez II: disolver en agua 10,6 g de ferrocianuro de potasio, K4Fe(CN)6 · 3H2O. Enrasar con agua a 100 ml. |
|
3.4. |
Naranja de metilo, solución al 0,1 % (p/v). |
|
3.5. |
Acido clorhídrico de 4 mol/l. |
|
3.6. |
Acido clorhídrico de 0,1 mol/l. |
|
3.7. |
Solución de hidróxido de sodio de 0,1 mol/l. |
|
3.8. |
Reactivo de Luff-Schoorl: Verter la solución de ácido cítrico (3.8.2) en la solución de carbonato de sodio (3.8.3), removiendo con cuidado. Añadir la solución de sulfato de cobre (3.8.1) y enrasar a 1 l con agua. Dejar reposar una noche y filtrar. Comprobar la concentración del reactivo así obtenido (0,05 mol de Cu/l; 1 mol de Na2 CO3/l), véase el punto 5.4, último párrafo. El pH de la solución deberá ser de 9,4 aproximadamente. |
|
3.8.1. |
Solución de sulfato de cobre: disolver 25 g de sulfato de cobre, Cu SO4 · 5H2O, exento de hierro, en 100 ml de agua. |
|
3.8.2. |
Solución de ácido cítrico: disolver 50 g de ácido cítrico, C6H8O7· H2O, en 50 ml de agua. |
|
3.8.3. |
Solución de carbonato de sodio: disolver 143,8 g de carbonato de sodio anhidro en 300 ml aproximadamente de agua caliente. Dejar enfriar. |
|
3.9. |
Solución de tiosulfato de sodio de 0,1 mol/l |
|
3.10. |
Solución de almidón: añadir una mezcla de 5 g de almidón soluble en 30 ml de agua a 1 l de agua hirviendo. Hervir durante tres minutos, dejar enfriar y, si es necesario, añadir 10 mg de yoduro de mercurio como conservante. |
|
3.11. |
Acido sulfúrico de 3 mol/l |
|
3.12. |
Yoduro de potasio, solución al 30 % (p/v) |
|
3.13. |
Piedra pómez granulada, hervida en ácido clorhídrico, lavada en agua y desecada |
|
3.14. |
3-metilbutan-l-ol |
4. Instrumental
Mezclador (tambor): de 35 a 40 revoluciones por minuto aproximadamente
5. Procedimiento
5.1. Extracción de la muestra
Pesar, con una precisión de 1 mg, 2,5 g de la muestra e introducirlos en un matraz aforado de 250 ml. Añadir 200 ml de etanol (3.1) y mezclar en el tambor durante una hora. Añadir 5 ml de solución de Carrez I (3.2) y remover durante 30 segundos aproximadamente. Añadir 5 ml de solución de Carrez II (3.3) y volver a remover durante un minuto. Enrasar con etanol (3.1), homogeneizar y filtrar. Tomar 200 ml del filtrado y evaporar alrededor de la mitad del volumen, con el fin de eliminar la mayor parte del etanol. Transvasar cuantitativamente el residuo de evaporación, por medio de agua caliente, a un matraz aforado de 200 ml, enfriar, enrasar con agua, homogeneizar y filtrar, si es necesario. Esta solución se empleará para determinar la cantidad de azúcares reductores y, tras la inversión, la cantidad de azúcares totales.
5.2. Determinación de los azúcares reductores
Pipetear no más de 25 ml de la solución que contenga menos de 60 mg de azúcares reductores, expresados en glucosa. Si es necesario, enrasar a 25 ml con agua destilada y determinar el contenido de azúcares reductores siguiendo el método de Luff-Schoorl. El resultado se expresará en porcentaje de glucosa de la muestra.
5.3. Determinación de los azúcares totales tras la inversión
Pipetear 50 ml de solución y transvasarlos a un matraz aforado de 100 ml. Añadir a continuación unas pocas gotas de solución de naranja de metilo (3.4), con cuidado y sin parar de remover, y añadir ácido clorhídrico (3.5) hasta que el líquido se vuelva definitivamente rojo. Añadir 15 ml de ácido clorhídrico (3.6), sumergir el matraz en un baño maría en fuerte ebullición y mantenerlo allí durante 30 minutos. Enfriar rápidamente a unos 20 oC y añadir 15 ml de solución de hidróxido de sodio (3.7). Enrasar a 100 ml con agua y homogeneizar. Retirar no más de 25 ml con un contenido de azúcares reductores inferior a 60 mg, expresados en glucosa. Si es necesario, enrasar a 25 ml con agua destilada y determinar el contenido de azúcares reductores siguiendo el método de Luff-Schoorl. El resultado se expresa en porcentaje de glucosa o, si procede, de sacarosa, multiplicando por un factor de 0,95.
5.4. Titulación por el método de Luff-Schoorl
Pipetear 25 ml del reactivo de Luff-Schoorl (3.8) y transvasarlos a un Erlenmeyer de 300 ml; añadir exactamente 25 ml de la solución de azúcares clarificada. Añadir dos gránulos de piedra pómez (3.13), calentar, removiendo manualmente, sobre una llama desnuda de altura media y llevar el líquido a ebullición en dos minutos aproximadamente. Colocar inmediatamente el Erlenmeyer sobre una tela metálica revestida de amianto con un orificio de unos 6 cm de diámetro, bajo el cual se habrá encendido una llama. La llama se regulará de forma que solo se caliente el fondo del Erlenmeyer. Ajustar a este último un refrigerante de reflujo. Hervir durante diez minutos exactos. Enfriar inmediatamente en agua fría y, transcurridos unos cinco minutos, titular como se indica a continuación:
Añadir 10 ml de solución de yoduro de potasio (3.12), e inmediatamente después (con cuidado, ya que puede formarse mucha espuma) 25 ml de ácido sulfúrico (3.11). Titular con solución de tiosulfato de sodio (3.9) hasta que aparezca una coloración amarilla mate, añadir el indicador de almidón (3.10) y completar la titulación.
Efectuar la misma titulación en una mezcla exactamente medida de 25 ml de reactivo de Luff-Schoorl (3.8) y 25 ml de agua, después de añadir 10 ml de solución de yoduro de potasio (3.12) y 25 ml de ácido sulfúrico (3.11) sin hervir.
6. Cálculo de los resultados
Establecer, con ayuda de la tabla, la cantidad de glucosa en miligramos que corresponde a la diferencia entre los valores de las dos titulaciones, expresados en miligramos de tiosulfato de sodio de 0,1 mol/l. Expresar el resultado en porcentaje de la muestra.
7. Procedimientos especiales
|
7.1. |
En el caso de piensos ricos en melaza y otros piensos no muy homogéneos, pesar 20 g e introducirlos con 500 ml de agua en un matraz aforado de 1 l. Mezclar durante una hora en el tambor. Clarificar con los reactivos de Carrez I (3.2) y II (3.3) según se describe en el punto 5.1, empleando esta vez una cantidad cuatro veces superior de cada reactivo. Enrasar con etanol al 80 % (v/v). Homogeneizar y filtrar. Eliminar el etanol como se describe en el punto 5.1. En ausencia de almidón dextrinado, enrasar con agua destilada. |
|
7.2. |
En el caso de melazas y materiales para piensos que sean ricos en azúcares y no tengan prácticamente almidón (algarrobos, peladuras desecadas de remolachas, etc.), pesar 5 g e introducirlos en un matraz aforado de 250 ml, añadir 200 ml de agua destilada y mezclar en el tambor durante una hora o más, si es necesario. Clarificar con los reactivos de Carrez I (3.2) y II (3.3) según se describe en el punto 5.1. Enrasar con agua fría, homogeneizar y filtrar. Para determinar la cantidad de azúcares totales, proseguir como se describe en el punto 5.3. |
8. Observaciones
|
8.1. |
Es recomendable añadir (sea cual sea el volumen) aproximadamente 1 ml de 3-metilbutan-l-ol (3.14) antes de hervir con el reactivo de Luff-Schoorl, a fin de evitar la formación de espuma. |
|
8.2. |
La diferencia entre el contenido de azúcares totales después de la inversión, expresados en glucosa, y el contenido de azúcares reductores, expresados en glucosa, multiplicada por 0,95, da el porcentaje de sacarosa. |
|
8.3. |
Para determinar el contenido de azúcares reductores, excluida la lactosa, pueden adoptarse dos métodos: |
|
8.3.1. |
Para un cálculo aproximado, multiplicar por 0,675 el contenido de lactosa establecido mediante un método de análisis diferente y restar el resultado obtenido al contenido de azúcares reductores. |
|
8.3.2. |
Para un cálculo exacto de los azúcares reductores, excluida la lactosa, debe emplearse la misma muestra en las dos determinaciones finales. Uno de los análisis se efectúa con una parte de la solución obtenida según el punto 5.1, y el otro con una parte de la solución obtenida al determinar la lactosa conforme al método establecido al efecto (previa fermentación de los otros tipos de azúcares y clarificación). En ambos casos, la cantidad de azúcar presente se determina según el método de Luff-Schoorl y se calcula en miligramos de glucosa. Los dos valores se restan y la diferencia se expresa en porcentaje de la muestra. Ejemplo Los dos volúmenes tomados corresponden, para cada determinación, a una muestra de 250 mg. En el primer caso se consumen 17 ml de solución de tiosulfato de sodio de 0,1 mol/l, lo que corresponde a 44,2 mg de glucosa; en el segundo, 11 ml, que corresponden a 27,6 mg de glucosa. La diferencia es de 16,6 mg de glucosa. El contenido de azúcares reductores (excluida la lactosa), calculado en glucosa, es pues de:
|
Tabla de valores correspondientes a 25 ml de reactivo de Luff-Schoorl
mililitros de Na2 S2 O3 de 0,1 mol/l, calentamiento de dos minutos, hervor de diez minutos
|
Na2 S2 O3 0,1 mol/l |
Glucosa, fructosa y azúcares invertidos C6 H12 O6 |
Lactosa C12 H22 O11 |
Maltosa C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
diferencia |
mg |
diferencia |
mg |
diferencia |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. DETERMINACIÓN DE LA LACTOSA
1. Objeto y ámbito de aplicación
Este método permite determinar el contenido de lactosa de los piensos que contienen más de un 0,5 % de este azúcar.
2. Principio
Los azúcares se disuelven en agua. La solución se somete a fermentación por la levadura Saccharomyces cerevisiae, que deja intacta la lactosa. Tras clarificación y filtración se determina el contenido de lactosa por el método de Luff-Schoorl.
3. Reactivos
|
3.1. |
Suspensión de Saccharomyces cerevisiae: suspender 25 g de levadura fresca en 100 ml de agua. En el frigorífico, el período máximo de conservación de la suspensión es de una semana. |
|
3.2. |
Solución de Carrez I: disolver en agua 21,9 g de acetato de cinc, Zn(CH3COO)2 · 2H2O, y 3 g de ácido acético glacial. Enrasar con agua a 100 ml. |
|
3.3. |
Solución de Carrez II: disolver en agua 10,6 g de ferrocianuro de potasio, K4Fe(CN)6 · 3H2O. Enrasar con agua a 100 ml. |
|
3.4. |
Reactivo de Luff-Schoorl: Verter la solución de ácido cítrico (3.4.2) en la solución de carbonato de sodio (3.4.3), removiendo con cuidado. Añadir la solución de sulfato de cobre (3.4.1) y enrasar a 1 l con agua. Dejar reposar una noche y filtrar. Comprobar la concentración del reactivo así obtenido (0,05 mol de Cu/l; 1 mol de Na2 CO3/l). El pH de la solución deberá ser de 9,4 aproximadamente. |
|
3.4.1. |
Solución de sulfato de cobre: disolver 25 g de sulfato de cobre, Cu SO4 · 5H2O, exento de hierro, en 100 ml de agua. |
|
3.4.2. |
Solución de ácido cítrico: disolver 50 g de ácido cítrico, C6H8O7· H2O, en 50 ml de agua. |
|
3.4.3. |
Solución de carbonato de sodio: disolver 143,8 g de carbonato de sodio anhidro en 300 ml aproximadamente de agua caliente. Dejar enfriar. |
|
3.5. |
Piedra pómez granulada, hervida en ácido clorhídrico, lavada en agua y desecada. |
|
3.6. |
Yoduro de potasio, solución al 30 % (p/v). |
|
3.7. |
Acido sulfúrico de 3 mol/l. |
|
3.8. |
Solución de tiosulfato de sodio de 0,1 mol/l. |
|
3.9. |
Solución de almidón: añadir una mezcla de 5 g de almidón soluble en 30 ml de agua a 1 l de agua hirviendo. Hervir durante tres minutos, dejar enfriar y, si es necesario, añadir 10 mg de yoduro de mercurio como conservante. |
4. Instrumental
Baño maría con termostato, regulado a 38-40 oC
5. Procedimiento
Pesar, con una precisión de 1 mg, 1 g de la muestra e introducirlo en un matraz aforado de 100 ml. Añadir de 25 ml a 30 ml de agua. Colocar el matraz durante 30 minutos en un baño maría hirviendo y, a continuación, enfriar a 35 oC aproximadamente. Añadir 5 ml de suspensión de levadura (3.1) y homogeneizar. Dejar reposar el matraz durante dos horas en un baño maría, a una temperatura de 38-40 oC. Enfriar a 20 oC aproximadamente.
Añadir 2,5 ml de solución de Carrez I (3.2) y remover durante 30 segundos, añadiendo a continuación 2,5 ml de solución de Carrez II (3.3) y volviendo a remover durante otros 30 segundos. Enrasar a 100 ml con agua, mezclar y filtrar. Pipetear no más de 25 ml de filtrado que contenga preferiblemente de 40 mg a 80 mg de lactosa y transvasarlos a un Erlenmeyer de 300 ml. Si es necesario, enrasar a 25 ml con agua.
Efectuar de la misma manera un ensayo en blanco con 5 ml de suspensión de levadura (3.1). Determinar, como se indica a continuación, el contenido de lactosa siguiendo el método de Luff-Schoorl: añadir exactamente 25 ml de reactivo de Luff-Schoorl (3.4) y dos gránulos de piedra pómez (3.5). Remover manualmente sobre una llama desnuda de media altura y llevar el líquido a ebullición en dos minutos aproximadamente. Colocar inmediatamente el Erlenmeyer sobre una tela metálica revestida de amianto con un orificio de unos 6 cm de diámetro, bajo el cual se habrá encendido una llama. La llama se regulará de forma que solo se caliente el fondo del Erlenmeyer. Ajustar a este último un refrigerante de reflujo. Hervir durante diez minutos exactos. Enfriar inmediatamente en agua fría y, transcurridos unos cinco minutos, titular como se indica a continuación:
Añadir 10 ml de solución de yoduro de potasio (3.6), e inmediatamente después (con cuidado, ya que puede formarse mucha espuma) 25 ml de ácido sulfúrico (3.7). Titular con solución de tiosulfato de sodio (3.8) hasta que aparezca una coloración amarilla mate, añadir el indicador de almidón (3.9) y completar la titulación.
Efectuar la misma titulación en una mezcla exactamente medida de 25 ml de reactivo de Luff-Schoorl (3.4) y 25 ml de agua, después de añadir 10 ml de solución de yoduro de potasio (3.6) y 25 ml de ácido sulfúrico (3.7) sin hervir.
6. Cálculo de los resultados
Establecer, con ayuda de la tabla aneja, la cantidad de lactosa en miligramos que corresponde a la diferencia entre los resultados de las dos titulaciones, expresados en mililitros de tiosulfato de sodio de 0,1 mol/l.
Expresar el resultado de lactosa anhidra como porcentaje de la muestra.
7. Observación
Para productos que contengan más de un 40 % de azúcares fermentables, emplear más de 5 ml de suspensión de levadura (3.1).
Tabla de valores correspondientes a 25 ml de reactivo de Luff-Schoorl
mililitros de Na2 S2 O3 de 0,1 mol/l, calentamiento de dos minutos, hervor de diez minutos
|
Na2 S2 O3 0,1 mol/l |
Glucosa, fructosa y azúcares invertidos C6 H12 O6 |
Lactosa C12 H22 O11 |
Maltosa C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
diferencia |
mg |
diferencia |
mg |
diferencia |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. DETERMINACIÓN DEL ALMIDÓN
MÉTODO POLARIMÉTRICO
1. Objeto y ámbito de aplicación
Este método permite determinar los niveles de almidón y de productos de la degradación del almidón de elevado peso molecular presentes en los piensos, a fin de comprobar que se cumple el valor energético declarado (disposiciones del anexo VII) y lo dispuesto en la Directiva 96/25/CE del Consejo (3).
2. Principio
El método comprende dos determinaciones. En la primera, la muestra se trata con ácido clorhídrico diluido. Tras clarificación y filtración, se mide la rotación óptica de la solución por polarimetría.
En la segunda, la muestra se extrae con etanol al 40 %. Tras acidificación del filtrado con ácido clorhídrico, clarificación y filtración, se mide la rotación óptica como en la primera determinación.
La diferencia entre las dos mediciones, multiplicada por un factor conocido, da el contenido de almidón de la muestra.
3. Reactivos
|
3.1. |
Ácido clorhídrico, solución al 25 % (p/p), con una densidad de 1,126 g/ml |
|
3.2. |
Ácido clorhídrico, solución al 1,13 % (p/v) La concentración debe comprobarse por titulación con una solución de hidróxido de sodio de 0,1 mol/l en presencia de rojo de metilo al 0,1 % (p/v) en etanol al 94 % (v/v). Para la neutralización de 10 ml se necesitan 30,94 ml de NaOH de 0,1 ml/l. |
|
3.3. |
Solución de Carrez I: disolver en agua 21,9 g de acetato de cinc, Zn(CH3COO)2 · 2H2O, y 3 g de ácido acético glacial. Enrasar con agua a 100 ml. |
|
3.4. |
Solución de Carrez II: disolver en agua 10,6 g de ferrocianuro de potasio, K4Fe(CN)6 · 3H2O. Enrasar con agua a 100 ml. |
|
3.5. |
Etanol, solución al 40 % (v/v), con una densidad de 0,948 g/ml a 20 oC |
4. Instrumental
|
4.1. |
Erlenmeyer de 250 ml con junta esmerilada estándar y refrigerante de reflujo. |
|
4.2. |
Polarímetro o sacarímetro. |
5. Procedimiento
5.1. Preparación de la muestra
Triturar la muestra hasta que sea lo suficientemente fina para pasar en su totalidad por un tamiz con una luz de malla redonda de 0,5 mm.
5.2. Determinación de la rotación óptica total (P o S) (véase la observación del punto 7.1)
Pesar, con una precisión de 1 mg , 2,5 g de la muestra triturada e introducirlos en un matraz aforado de 100 ml. Añadir 25 ml de ácido clorhídrico (3.2), agitar para obtener una distribución uniforme de la muestra de ensayo y añadir otros 25 ml de ácido clorhídrico (3.2). Sumergir el matraz en un baño maría hirviendo y, durante los tres primeros minutos, agitar enérgica y constantemente para evitar la formación de aglomerados. La cantidad de agua del baño maría debe ser suficiente para que pueda mantenerse en ebullición cuando se introduzca en él el matraz. Este no debe retirarse del baño mientras se agita. Transcurridos exactamente 15 minutos, sacarlo del baño, añadir 30 ml de agua fría y enfriar inmediatamente a 20 oC.
Añadir 5 ml de solución de Carrez I (3.3) y agitar durante 30 segundos aproximadamente. Añadir 5 ml de solución de Carrez I (3.4) y agitar durante 30 segundos aproximadamente. Enrasar con agua, mezclar y filtrar. Si el filtrado no está perfectamente claro (lo cual es raro), repetir la determinación con una cantidad mayor de soluciones de Carrez I y II, por ejemplo 10 ml.
Medir la rotación óptica de la solución en un tubo de 200 mm con el polarímetro o el sacarímetro.
5.3. Determinación de la rotación óptica (P' o S') de sustancias solubles en etanol al 40 %
Pesar, con una precisión de 1 mg, 5 g de la muestra, introducirlos en un matraz aforado de 100 ml y añadir unos 80 ml de etanol (3.5) (véase la observación 7.2). Dejar reposar el matraz durante una hora a temperatura ambiente; durante este tiempo, agitar enérgicamente seis veces de manera que la muestra se mezcle completamente con el etanol. Enrasar con etanol (3.5), mezclar y filtrar.
Pipetear 50 ml del filtrado (correspondientes a 2,5 g de la muestra) y transvasarlos a un Erlenmeyer de 250 ml, añadiendo a continuación 2,1 ml de ácido clorhídrico (3.1) y agitando enérgicamente. Ajustar un refrigerante de reflujo al Erlenmeyer y sumergir este último en un baño maría hirviendo. Transcurridos exactamente 15 minutos, retirar el Erlenmeyer del baño, transvasar su contenido a un matraz aforado de 100 ml, enjuagando con un poco de agua fría, y enfriar a 20 oC.
Clarificar con soluciones de Carrez I (3.3) y II (3.4), enrasar con agua, mezclar, filtrar y medir la rotación óptica como se indica en los párrafos segundo y tercero del punto 5.2.
6. Cálculo de los resultados
El contenido de almidón (%) se calcula como sigue:
6.1. Medición con polarímetro
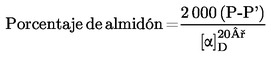
|
P |
= |
rotación óptica total en grados de ángulo |
||||||||||||||
|
P' |
= |
rotación óptica en grados de ángulo de las sustancias solubles en etanol al 40 % (V/V) |
||||||||||||||
|
|
= |
rotación óptica específica del almidón puro. Los valores numéricos D aceptados convencionalmente para este factor son los siguientes:
|
6.2. Medición con sacarímetro

|
S |
= |
rotación óptica total, en grados de sacarímetro |
|
S' |
= |
rotación óptica, en grados de sacarímetro, de las sustancias solubles en etanol al 40 % (v/v) |
|
N |
= |
peso (g) de la sacarosa en 100 ml de agua que da una rotación óptica de 100 grados de sacarímetro cuando se mide con un tubo de 200 mm 16,29 g para sacarímetros franceses 26,00 g para sacarímetros alemanes 20,00 g para sacarímetros mixtos |
|
|
= |
rotación óptica específica del almidón puro (véase el punto 6.1) |
6.3. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe exceder de 0,4 en valor absoluto, en el caso de un contenido de almidón inferior al 40 %, ni del 1 % en el caso de contenidos de almidón iguales o superiores al 40 %.
7. Observaciones
|
7.1. |
Si la muestra contiene más de un 6 % de carbonatos, calculados en carbonato de calcio, deben destruirse mediante un tratamiento con la cantidad exacta apropiada de ácido sulfúrico diluido antes de proceder a la determinación de la rotación óptica total. |
|
7.2. |
En el caso de productos con un elevado contenido de lactosa, como el suero de leche en polvo o la leche desnatada en polvo, proceder como sigue tras añadir 80 ml de etanol (3.5). Ajustar un refrigerante de reflujo al Erlenmeyer y sumergir este último en un baño maría a 50 oC durante 30 minutos. Dejar enfriar y seguir con el análisis como se indica en el punto 5.3. |
|
7.3. |
Cuando están presentes en cantidades importantes en los piensos, los siguientes materiales producen interferencias en la determinación del contenido de almidón por el método polarimétrico, lo que podría dar lugar a resultados incorrectos:
|
M. DETERMINACIÓN DE LA CENIZA BRUTA
1. Objeto y ámbito de aplicación
Este método permite determinar el contenido de ceniza bruta de los piensos.
2. Principio
La muestra se incinera a 550 oC; el residuo se pesa.
3. Reactivos
Nitrato de amonio, solución al 20 % (p/v).
4. Instrumental
|
4.1. |
Placa calefactora. |
|
4.2. |
Horno eléctrico de mufla con termostato. |
|
4.3. |
Crisoles de incineración de sílice, porcelana o platino, bien rectangulares (60 x 40 x 25 mm, aproximadamente), bien redondos (60 mm a 75 mm de diámetro y 20 mm a 40 mm de altura). |
5. Procedimiento
Pesar, con una precisión de 1 mg, 5 g aproximadamente de la muestra (2,5 g en el caso de productos con tendencia a hincharse) e introducirlos en un crisol de incineración previamente calentado a 550 oC, enfriado y tarado. Colocar el crisol sobre la placa calefactora y calentar gradualmente hasta que se carbonice la sustancia. Incinerar conforme al punto 5.1 o 5.2.
|
5.1. |
Introducir el crisol en el horno de mufla calibrado, regulado a 550 oC. Mantener a esta temperatura hasta obtener una ceniza blanca, gris claro o rojiza aparentemente exenta de partículas carbonosas. Colocar el crisol en un desecador, dejar enfriar y pesar inmediatamente. |
|
5.2. |
Introducir el crisol en el horno de mufla calibrado, regulado a 550 oC. Incinerar durante tres horas. Colocar el crisol en un desecador, dejar enfriar y pesar inmediatamente. Volver a incinerar durante 30 minutos para que el peso de la ceniza se mantenga constante (la pérdida de peso entre dos pesadas sucesivas debe ser inferior o igual a 1 mg). |
6. Cálculo de los resultados
Calcular el peso del residuo deduciendo la tara.
Expresar el resultado en porcentaje de la muestra.
7. Observaciones
|
7.1. |
La ceniza de sustancias difíciles de incinerar debe someterse a una primera incineración de, como mínimo, tres horas y después enfriarse, para a continuación añadirle unas pocas gotas de una solución de nitrato de amonio al 20 % o agua (con cuidado de que no se disperse la ceniza ni se formen grumos). Continuar la calcinación después de desecar en la estufa. Repetir la operación las veces necesarias hasta que la incineración sea completa. |
|
7.2. |
En el caso de sustancias resistentes al tratamiento descrito en el punto 7.1, proceder como sigue: tras incinerar durante tres horas, poner la ceniza en agua caliente y filtrar por un filtro pequeño sin cenizas. Incinerar el filtro y su contenido en el crisol inicial. Colocar el filtrado en el crisol enfriado, evaporar hasta que esté seco, incinerar y pesar. |
|
7.3. |
En el caso de aceites y grasas, pesar con exactitud una muestra de 25 g en un crisol de tamaño adecuado. Carbonizar inflamando la sustancia con una tira de papel de filtro sin cenizas. Tras la combustión, humedecer con el mínimo estrictamente necesario de agua. Secar e incinerar como se describe en el punto 5. |
N. DETERMINACIÓN DE LA CENIZA INSOLUBLE EN ÁCIDO CLORHÍDRICO
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de sustancias minerales de los piensos que son insolubles en ácido clorhídrico. Pueden seguirse dos métodos, en función de la naturaleza de la muestra.
|
1.1. |
Método A: aplicable a los materiales para piensos orgánicos y a la mayor parte de los piensos compuestos. |
|
1.2. |
Método B: aplicable a los compuestos y mezclas minerales y a los piensos compuestos cuyo contenido de sustancias insolubles en ácido clorhídrico, determinado por el método A, sea superior al 1 %. |
2. Principio
|
2.1. |
Método A: la muestra se incinera, la ceniza se hierve en ácido clorhídrico y el residuo insoluble se filtra y se pesa. |
|
2.2. |
Método B: la mezcla se trata con ácido clorhídrico. La solución se filtra, el residuo se incinera y la ceniza así obtenida se trata conforme al método A. |
3. Reactivos
|
3.1. |
Acido clorhídrico de 3 mol/l. |
|
3.2. |
Ácido tricloroacético, solución al 20 % (p/v). |
|
3.3. |
Ácido tricloroacético, solución al 1 % (p/v). |
4. Instrumental
|
4.1. |
Placa calefactora. |
|
4.2. |
Horno eléctrico de mufla con termostato. |
|
4.3. |
Crisoles de incineración de sílice, porcelana o platino, bien rectangulares (60 x 40 x 25 mm, aproximadamente), bien redondos (60 mm a 75 mm de diámetro y 20 mm a 40 mm de altura). |
5. Procedimiento
5.1. Método A:
Incinerar la muestra según el método descrito para la determinación de la ceniza bruta. También puede emplearse la ceniza obtenida en ese análisis.
Introducir la ceniza en un vaso de precipitado de 250 ml a 400 ml, empleando 75 ml de ácido clorhídrico (3.1). Llevar lentamente a ebullición y hervir suavemente durante 15 minutos. Filtrar la solución caliente por un papel de filtro sin cenizas y lavar el residuo con agua caliente hasta que deje de ser visible la reacción ácida. Secar el filtro que contiene el residuo e incinerarlo en un crisol tarado a una temperatura no inferior a 550 oC ni superior a 700 oC. Enfriar en un desecador y pesar.
5.2. Método B
Pesar, con una precisión de 1 mg , 5 g de la muestra e introducirlos en un vaso de precipitado de 250 ml a 400 ml. Añadir sucesivamente 25 ml de agua y 25 ml de ácido clorhídrico (3.1), mezclar y esperar a que cese la efervescencia. Añadir otros 50 ml de ácido clorhídrico (3.1). Esperar a que cese cualquier posible desprendimiento de gas, colocar a continuación el vaso de precipitado en un baño maría hirviendo y mantenerlo allí durante 30 minutos o más, si fuera necesario, con el fin de hidrolizar completamente el almidón que pueda estar presente. Filtrar en caliente por un filtro sin cenizas y lavar el filtro en 50 ml de agua caliente (véase la observación del punto 7). Colocar el filtro que contiene el residuo en un crisol de incineración, secar e incinerar a una temperatura no inferior a 550 oC ni superior a 700 oC. Introducir la ceniza en un vaso de precipitado de 250 ml a 400 ml, empleando 75 ml de ácido clorhídrico (3.1); continuar como se describe en el punto 5.1, párrafo segundo.
6. Cálculo de los resultados
Calcular el peso del residuo deduciendo la tara. Expresar el resultado en porcentaje de la muestra.
7. Observación
Si la filtración resultara difícil, recomenzar el análisis sustituyendo los 50 ml de ácido clorhídrico (3.1) por 50 ml de ácido tricloracético al 20 % (3.2) y lavando el filtro en una solución caliente de ácido tricloracético al 1 % (3.3).
O. DETERMINACIÓN DE LOS CARBONATOS
1. Objeto y ámbito de aplicación
Este método permite determinar la cantidad de carbonatos, convencionalmente expresados en carbonato de calcio, en la mayoría de los piensos.
Sin embargo, en determinados casos (carbonato de hierro, por ejemplo), es necesario emplear un método especial.
2. Principio
Los carbonatos se descomponen en ácido clorhídrico; el dióxido de carbono desprendido se recoge en un tubo graduado y su volumen se compara con el desprendido, en las mismas condiciones, por una cantidad conocida de carbonato de calcio.
3. Reactivos
|
3.1. |
Ácido clorhídrico de 1,10 g/ml de densidad. |
|
3.2. |
Carbonato de calcio. |
|
3.3. |
Ácido sulfúrico, de aproximadamente 0,05 mol/l, coloreado con rojo de metilo. |
4. Instrumental
Aparato de Scheibler-Dietrich (véase el diagrama) o equivalente.
5. Procedimiento
Según el contenido de carbonatos de la muestra, pesar una porción de muestra según se indica a continuación:
|
— |
0,5 g en el caso de productos que contengan del 50 % al 100 % de carbonatos, expresados en carbonato de calcio, |
|
— |
1 g en el caso de productos que contengan del 40 % al 50 % de carbonatos, expresados en carbonato de calcio, |
|
— |
2 g a 3 g en los demás casos. |
Introducir la porción de muestra en el matraz especial (4) del aparato, provisto de un pequeño tubo de material irrompible con 10 ml de ácido clorhídrico (3.1), y conectar el matraz al aparato. Girar el grifo de tres vías (5) de forma que el tubo (1) se conecte con el exterior. Con ayuda del tubo móvil (2), que está lleno de ácido sulfúrico coloreado (3.3) y conectado al tubo graduado (1), llevar el nivel del líquido a la marca cero. Girar el grifo (5) para conectar los tubos (1) y (2) y comprobar que el nivel está en cero.
Dejar correr lentamente el ácido clorhídrico (3.1) sobre la porción de muestra, inclinando el matraz (4). Igualar la presión bajando el tubo (2). Agitar el matraz (4) hasta que deje de desprenderse dióxido de carbono por completo.
Restablecer la presión llevando el líquido al mismo nivel en los tubos (1) y (2). Hacer la lectura cuando hayan transcurrido unos pocos minutos y el volumen de gas se haya hecho constante.
Efectuar un ensayo de control en las mismas condiciones con 0,5 g de carbonato de calcio (3.2).
6. Cálculo de los resultados
El contenido de carbonatos, expresados en carbonato de calcio, se calcula con la siguiente fórmula:

donde:
|
X |
= |
porcentaje (p/p) de carbonatos de la muestra, expresados en carbonato de calcio; |
|
V |
= |
mililitros de CO2 desprendidos por la porción de muestra; |
|
V1 |
= |
mililitros de CO2 desprendidos por 0,5 g de CaCO3; |
|
m |
= |
peso en gramos de la porción de muestra. |
7. Observaciones
|
7.1. |
Cuando la porción de muestra pese más de 2 g, introducir previamente en el matraz (4) 15 ml de agua destilada y mezclar antes de comenzar el ensayo. Emplear el mismo volumen de agua para el ensayo de control. |
|
7.2. |
Si el aparato utilizado tiene un volumen diferente al del aparato de Scheibler-Dietrich, deben adaptarse en consecuencia tanto las porciones de muestra y de sustancia de control como el cálculo de los resultados.
|
P. DETERMINACIÓN DEL FÓSFORO TOTAL
MÉTODO FOTOMÉTRICO
1. Objeto y ámbito de aplicación
Este método permite determinar el contenido de fósforo total de los piensos. Está indicado, en particular, para el análisis de los productos pobres en fósforo. En determinados casos (productos ricos en fósforo), puede aplicarse un método gravimétrico.
2. Principio
La muestra se mineraliza, bien por combustión seca (en el caso de piensos orgánicos), bien por digestión ácida (en el caso de compuestos minerales y piensos líquidos), y se pone en una solución ácida. La solución se trata con el reactivo de molibdovanadato. La densidad óptica de la solución amarilla así formada se mide en el espectrofotómetro a 430 nm.
3. Reactivos
|
3.1. |
Carbonato de calcio. |
|
3.2. |
Ácido clorhídrico, ρ20 = 1,10 g/ml (aproximadamente 6 mol/l). |
|
3.3. |
Ácido nítrico, ρ20 = 1,045 g/ml. |
|
3.4. |
Ácido nítrico, ρ20 = 1,38 a 1,42 g/ml. |
|
3.5. |
Ácido sulfúrico, ρ20 = 1,84 g/ml. |
|
3.6. |
Reactivo de molibdovanadato: mezclar 200 ml de solución de heptamolibdato de amonio (3.6.1), 200 ml de solución de monovanadato de amonio (3.6.2) y 134 ml de ácido nítrico (3.4) en un matraz aforado de 1 l. Enrasar con agua. |
|
3.6.1. |
Solución de heptamolibdato de amonio: disolver en agua caliente 100 g de heptamolibdato de amonio, (NH4) 6Mo7O24 · 4H2O. Añadir 10 ml de amoniaco (densidad 0,91 g/ml) y enrasar a 1 l con agua. |
|
3.6.2. |
Solución de monovanadato de amonio: disolver 2,35 g de monovanadato de amonio, NH4VO3, en 400 ml de agua caliente. Añadir lentamente y sin parar de remover 20 ml de ácido nítrico diluido (7 ml de HNO3 (3.4) + 13 ml de H2O) y enrasar a 1 l con agua. |
|
3.7. |
Solución patrón de 1 mg de fósforo por mililitro: disolver en agua 4,387 g de dihidrogenofosfato de potasio, KH2PO4. Enrasar a 1 l con agua. |
4. Instrumental
|
4.1. |
Crisoles de incineración de sílice, porcelana o platino. |
|
4.2. |
Horno eléctrico de mufla con termostato, regulado a 550 oC. |
|
4.3. |
Matraz Kjeldahl de 250 ml. |
|
4.4. |
Matraces aforados y pipetas de precisión. |
|
4.5. |
Espectrofotómetro. |
|
4.6. |
Tubos de ensayo de 16 mm de diámetro aproximadamente, con tapones rebajados a un diámetro de 14,5 mm y con una capacidad de 25 ml a 30 ml. |
5. Procedimiento
5.1. Preparación de la solución
Según la naturaleza de la muestra, preparar una solución como se indica en el punto 5.1.1 o 5.1.2.
5.1.1.
Pesar, con una precisión de 1 mg, 1 g o más de la muestra. Introducirla en un matraz Kjeldahl, añadir 20 ml de ácido sulfúrico (3.5), agitar para impregnar completamente la sustancia de ácido y evitar que se adhiera a las paredes del matraz, calentar y mantener durante diez minutos en el punto de ebullición. Dejar enfriar ligeramente, añadir 2 ml de ácido nítrico (3.4), calentar suavemente, dejar enfriar ligeramente, añadir un poco más de ácido nítrico (3.4) y llevar de nuevo al punto de ebullición. Repetir este procedimiento hasta obtener una solución incolora. Enfriar, añadir un poco de agua y decantar el líquido en un matraz aforado de 500 ml, enjuagando el matraz Kjeldahl con agua caliente. Dejar enfriar, enrasar con agua, homogeneizar y filtrar.
5.1.2.
Pesar, con una precisión de 1 mg, 2,5 g aproximadamente de la muestra en un crisol de incineración. Mezclar la muestra de ensayo hasta su completa fusión con 1 g de carbonato de calcio (3.1). Incinerar en el horno a 550 oC hasta obtener una ceniza blanca o gris (no importa que quede algo de carbón). Transferir la ceniza a un vaso de precipitado de 250 ml. Añadir 20 ml de agua y de ácido clorhídrico (3.2) hasta que cese la efervescencia. Añadir otros 10 ml de ácido clorhídrico (3.2). Poner el vaso de precipitado sobre un baño de arena y evaporar hasta sequedad para insolubilizar la sílice. Volver a disolver el residuo en 10 ml de ácido nítrico (3.3) y hervir durante cinco minutos sobre el baño de arena o la placa calefactora, sin evaporación, hasta sequedad. Decantar el líquido en un matraz aforado de 500 ml, enjuagando el vaso de precipitado varias veces con agua caliente. Dejar enfriar, enrasar con agua, homogeneizar y filtrar.
5.2. Desarrollo de la coloración y medición de la densidad óptica
Diluir una parte alícuota del filtrado obtenido con el procedimiento del punto 5.1.1 o 5.1.2 para obtener una concentración de fósforo no superior a 40 μg/ml. Introducir 10 ml de esta solución en un tubo de ensayo (4.6) y añadir 10 ml del reactivo de molibdovanadato (3.6). Homogeneizar y dejar reposar un mínimo de diez minutos a 20 oC. Medir la densidad óptica en un espectrofotómetro a 430 nm por comparación con una solución obtenida añadiendo 10 ml del reactivo de molibdovanadato (3.6) a 10 ml de agua.
5.3. Curva de calibración
A partir de la solución patrón (3.7), preparar soluciones que contengan, respectivamente, 5, 10, 20, 30 y 40 μg de fósforo por mililitro. Tomar 10 ml de cada una de estas soluciones y añadirles 10 ml del reactivo de molibdovanadato (3.6). Homogeneizar y dejar reposar un mínimo de diez minutos a 20 oC. Medir la densidad óptica como se indica en el punto 5.2. Trazar la curva de calibración relacionando las densidades ópticas con las correspondientes cantidades de fósforo. La curva será lineal para concentraciones comprendidas entre 0 μg/ml y 40 μg/ml.
6. Cálculo de los resultados
Determinar la cantidad de fósforo de la muestra de ensayo por medio de la curva de calibración.
Expresar el resultado en porcentaje de la muestra.
Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no excederá del:
|
— |
3 % del resultado superior, en el caso de contenidos de fósforo inferiores al 5 %, |
|
— |
0,15 % en valor absoluto, en el caso de contenidos de fósforo iguales o superiores al 5 %. |
Q. DETERMINACIÓN DEL CLORO DE LOS CLORUROS
1. Objeto y ámbito de aplicación
Este método permite determinar la cantidad de cloro de los cloruros solubles en agua, expresada convencionalmente en cloruro de sodio. Es aplicable a todos los piensos.
2. Principio
Los cloruros se disuelven en agua. Si el producto contiene materia orgánica, se procede a una clarificación. La solución se acidifica ligeramente con ácido nítrico y los cloruros se precipitan en forma de cloruro de plata por medio de una solución de nitrato de plata. El exceso de nitrato de plata se titula con una solución de tiocianato de amonio por el método de Volhard.
3. Reactivos
|
3.1. |
Solución de tiocianato de amonio de 0,1 mol/l. |
|
3.2. |
Solución de nitrato de plata de 0,1 mol/l. |
|
3.3. |
Solución saturada de sulfato férrico de amonio (NH4)Fe(SO4)2. |
|
3.4. |
Ácido nítrico, de 1,38 g/ml de densidad. |
|
3.5. |
Éter dietílico. |
|
3.6. |
Acetona. |
|
3.7. |
Solución de Carrez I: disolver en agua 21,9 g de acetato de cinc, Zn(CH3COO)2 · 2H2O, y 3 g de ácido acético glacial. Enrasar con agua a 100 ml. |
|
3.8. |
Solución de Carrez II: disolver en agua 10,6 g de ferrocianuro de potasio, K4Fe(CN)6 · 3H2O. Enrasar con agua a 100 ml. |
|
3.9. |
Carbón activo, exento de cloruros y que no los absorba. |
4. Instrumental
Mezclador (tambor): de 35 a 40 revoluciones por minuto aproximadamente.
5. Procedimiento
5.1. Preparación de la solución
Según la naturaleza de la muestra, preparar una solución como se muestra en el punto 5.1.1, 5.1.2 o 5.1.3.
Al mismo tiempo, efectuar un ensayo en blanco sin la muestra que debe analizarse.
5.1.1.
Pesar, con una precisión de 1 mg, una muestra de no más de 10 g que no contenga más de 3 g de cloro en forma de cloruros. Introducirla con 400 ml de agua en un matraz aforado de 500 ml a unos 20 oC. Mezclar durante 30 minutos en el tambor, enrasar, homogeneizar y filtrar.
5.1.2.
Pesar, con una precisión de 1 mg, 5 g aproximadamente de la muestra e introducirlos con 1 g de carbón activo en un matraz aforado de 500 ml. Añadir 400 ml de agua a unos 20 oC y 5 ml de solución de Carrez I (3.7), remover durante 30 segundos y, a continuación, añadir 5 ml de solución de Carrez II (3.8). Mezclar durante 30 minutos en el tambor, enrasar, homogeneizar y filtrar.
5.1.3.
Preparar la solución como se describe en el punto 5.1.2, pero no filtrar. Decantar (si fuera necesario, centrifugar), retirar 100 ml del líquido sobrenadante y transvasar a un matraz aforado de 200 ml. Mezclar con acetona (3.6) y enrasar con este disolvente, homogeneizar y filtrar.
5.2. Titulación
Transvasar con una pipeta al Erlenmeyer de 25 ml a 100 ml del filtrado (según el contenido supuesto de cloro) obtenido según se describe en el punto 5.1.1, 5.1.2 o 5.1.3. La parte alícuota no debe contener más de 150 mg de cloro (Cl). Diluir, si es necesario, a no menos de 50 ml con agua, añadir 5 ml de ácido nítrico (3.4), 20 ml de solución saturada de sulfato férrico de amonio (3.3) y dos gotas de solución de triocianato de amonio (3.1) transvasadas mediante una bureta llena hasta la marca cero. Transvasar con una bureta la solución de nitrato de plata (3.2) de forma que se obtenga un exceso de 5 ml. Añadir 5 ml de éter dietílico (3.5) y agitar fuertemente para coagular el precipitado. Titular el exceso de nitrato de plata con la solución de tiocianato de amonio (3.1) hasta que la tinción marrón rojiza haya persistido un minuto.
6. Cálculo de los resultados
El contenido de cloro, expresado en porcentaje de cloruro de sodio, se calcula con la fórmula siguiente:

donde:
|
V1 |
= |
mililitros de solución de nitrato de plata de 0,1 mol/l añadidos; |
|
V2 |
= |
mililitros de solución de tiocianato de amonio de 0,1 mol/l empleados para la titulación; |
|
m |
= |
peso de la muestra. |
Si el ensayo en blanco indica un consumo de solución de nitrato de plata de 0,1 mol/l, deducir este valor del volumen (V1-V2).
7. Observaciones
|
7.1. |
La titulación también puede hacerse por potenciometría. |
|
7.2. |
En el caso de productos muy ricos en aceites y grasas, proceder primero a un desengrasado con éter dietílico o éter de petróleo. |
|
7.3. |
En el caso de las harinas de pescado, la titulación puede efectuarse por el método Mohr. |
(1) Para el secado de cereales, harina, grañones y sémola, la estufa debe tener una capacidad térmica tal que, precalentada a 131 oC, recupere esa temperatura en menos de 45 minutos una vez puestas a secar simultáneamente en su interior el máximo número de muestras de ensayo. La ventilación debe ser tal que, tras dos horas secando el máximo número de muestras de trigo candeal que pueda contener, los resultados difieran en menos de un 0,15 % de los obtenidos tras cuatro horas de secado.
(2) Sustituir los fragmentos de piedra pómez por perlas de vidrio cuando el aceite o la grasa deban someterse a exámenes cualitativos ulteriores.
ANEXO IV
MÉTODOS DE ANÁLISIS PARA EL CONTROL DEL NIVEL DE ADITIVOS AUTORIZADOS EN LOS PIENSOS
A. DETERMINACIÓN DE LA VITAMINA A
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de vitamina A (retinol) en piensos y premezclas. La vitamina A incluye el alcohol todo-trans-retinilo y sus isómeros cis, que se determinan con este método. El contenido de vitamina A se expresa en unidades internacionales (UI) por kilogramo. Una UI corresponde a la actividad de 0,300 μg de alcohol todo-trans-retinilo, 0,344 μg de acetato de todo-trans-retinilo o 0,550 μg de palmitato de todo-trans-retinilo.
El límite de cuantificación es de 2 000 UI de vitamina A por kilogramo.
2. Principio
La muestra se hidroliza con solución etanólica de hidróxido de potasio y la vitamina A se extrae en éter de petróleo. El disolvente se elimina por evaporación y el residuo se disuelve en metanol y, en caso necesario, se diluye hasta conseguir la concentración necesaria. El contenido de vitamina A se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa con detector de UV o de fluorescencia. Los parámetros cromatográficos se seleccionan de forma que no haya separación entre el alcohol todo-trans-retinilo y sus isómeros cis.
3. Reactivos
|
3.1. |
Etanol, σ = 96 %. |
|
3.2. |
Éter de petróleo, con un intervalo de ebullición de 40 oC-60 oC. |
|
3.3. |
Metanol. |
|
3.4. |
Solución de hidróxido de potasio, c = 50 g/100 ml. |
|
3.5. |
Solución de ascorbato de sodio, c = 10 g/100 ml (véanse las observaciones del punto 7.7). |
|
3.6. |
Sulfuro de sodio, Na2S· x H2O (x = 7 — 9). |
|
3.6.1. |
Solución de sulfuro de sodio, c = 0,5 mol/l en glicerol, ß = 120 g/l (para x = 9) (véanse las observaciones del punto 7.8). |
|
3.7. |
Solución de fenolftaleína, c = 2 g/100 ml en etanol (3.1). |
|
3.8. |
2-Propanol. |
|
3.9. |
Fase móvil para la CLAR: mezcla de metanol (3,3) y agua, por ejemplo 980 + 20 (v+v). La proporción exacta se determinará en función de las características de la columna empleada. |
|
3.10. |
Nitrógeno, exento de oxígeno. |
|
3.11. |
Acetato de todo-trans-retinilo, extra puro, de actividad certificada, por ejemplo 2,80 x 106 UI/g. |
|
3.11.1. |
Solución madre de acetato de todo-trans-retinilo: pesar, con una precisión de 0,1 mg, 50 mg de acetato de vitamina A (3.11) en un matraz aforado de 100 ml. Disolver en 2-propanol (3.8) y enrasar con el mismo disolvente. La concentración nominal de esta solución es de 1 400 UI de vitamina A por mililitro. El contenido exacto ha de determinarse según el punto 5.6.3.1. |
|
3.12. |
Palmitato de todo-trans-retinilo, extra puro, de actividad certificada, por ejemplo 1,80 x 106 UI/g. |
|
3.12.1. |
Solución madre de palmitato de todo-trans-retinilo: pesar, con una precisión de 0,1 mg, 80 mg de palmitato de vitamina A (3.12) en un matraz aforado de 100 ml. Disolver en 2-propanol (3.8) y enrasar con el mismo disolvente. La concentración nominal de esta solución es de 1 400 UI de vitamina A por mililitro. El contenido exacto ha de determinarse según el punto 5.6.3.2. |
|
3.13. |
2,6-Di-tert-butil-4-metilfenol (BHT) (véanse las observaciones del punto 7.5). |
4. Instrumental
|
4.1. |
Evaporador rotativo de vacío. |
|
4.2. |
Material de vidrio ámbar. |
|
4.2.1. |
Matraces Erlenmeyer o de fondo plano, de 500 ml, con boca esmerilada. |
|
4.2.2. |
Matraces aforados con tapón esmerilado, de cuello estrecho, de 10, 25, 100 y 500 ml. |
|
4.2.3. |
Embudos cónicos de decantación, de 1 000 ml, con tapón esmerilado. |
|
4.2.4. |
Matraces piriformes, de 250 ml, con boca esmerilada. |
|
4.3. |
Condensador de Allihn, con camisa de 300 mm de longitud, junta esmerilada y adaptador para tubo de alimentación de gas. |
|
4.4. |
Papel de filtro de pliegues para separación de fases, de 185 mm de diámetro (por ejemplo, Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
Equipo de CLAR con sistema de inyección. |
|
4.5.1. |
Columna cromatográfica de líquidos, de 250 mm x 4 mm, C18, relleno de 5 μm o 10 μm, o equivalente (criterio de funcionamiento: un único pico para todos los isómeros de retinol en las condiciones de CLAR). |
|
4.5.2. |
Detector de UV o de fluorescencia, con ajuste de longitud de onda variable. |
|
4.6. |
Espectrofotómetro con cubetas de cuarzo de 10 mm. |
|
4.7. |
Baño maría con agitador magnético. |
|
4.8. |
Aparato de extracción (véase la figura 1) con los siguientes elementos: |
|
4.8.1. |
Probeta de 1 l de capacidad con cuello y tapón esmerilados. |
|
4.8.2. |
Pieza esmerilada provista de una oliva lateral y de un tubo ajustable que la atraviesa por el centro. El extremo inferior del tubo ajustable deberá tener forma de U, mientras que el superior debe ser una tobera, de forma que pueda transvasarse la fase superior de líquido de la probeta a un embudo de decantación. |
5. Procedimiento
|
Nota |
: |
La vitamina A es sensible a la luz (UV) y a la oxidación. Todas las operaciones deberán realizarse en ausencia de luz (utilizando material de vidrio ámbar o protegido con papel de aluminio) y de oxígeno (chorro de nitrógeno). Durante la extracción, el aire que se encuentre por encima del líquido deberá sustituirse por nitrógeno (evitar un exceso de presión aflojando el tapón de vez en cuando). |
5.1. Preparación de la muestra
Triturar la muestra de modo que pase por un tamiz con una luz de malla de 1 mm, cuidando de que no se produzca calor. La trituración debe hacerse inmediatamente antes de la pesada y la saponificación, pues, de lo contrario, puede haber pérdidas de vitamina A.
5.2. Saponificación
En función del contenido de vitamina A, pesar, con una precisión de 1 mg, entre 2 g y 25 g de la muestra en un matraz Erlenmeyer o de fondo plano de 500 ml (4.2.1). Añadir sucesivamente, agitando en círculos, 130 ml de etanol (3.1), unos 100 mg de BHT (3.13), 2 ml de solución de ascorbato de sodio (3.5) y 2 ml de solución de sulfuro de sodio (3.6). Ajustar un refrigerante (4.3) al matraz y sumergir este en un baño maría con agitador magnético (4.7). Calentar hasta ebullición y dejar refluir durante cinco minutos. Añadir entonces 25 ml de solución de hidróxido de potasio (3.4) a través del refrigerante (4.3) y dejar refluir durante otros 25 minutos, removiendo bajo una corriente lenta de nitrógeno. Enjuagar después el refrigerante con unos 20 ml de agua y enfriar el contenido del matraz hasta la temperatura ambiente.
5.3. Extracción
Transvasar cuantitativamente por decantación la solución de saponificación a un embudo de decantación de 1 000 ml (4.2.3) o al aparato de extracción (4.8), enjuagando con un volumen total de 250 ml de agua. Enjuagar el matraz de saponificación sucesivamente con 25 ml de etanol (3.1) y 100 ml de éter de petróleo (3.2) y transvasar los líquidos de enjuague al embudo de decantación o al aparato de extracción. La proporción de agua y etanol en las soluciones combinadas debe ser aproximadamente de 2/1. Agitar enérgicamente durante dos minutos y dejar reposar durante otros dos minutos.
5.3.1.
Cuando se hayan separado las fases (véase la observación del punto 7.3), transvasar la fase de éter de petróleo a otro embudo de decantación (4.2.3). Repetir esta extracción dos veces con 100 ml de éter de petróleo (3.2) y dos veces con 50 ml de éter de petróleo (3.2).
Lavar dos veces los extractos combinados en el embudo de decantación, agitando suavemente en círculos (para evitar la formación de emulsiones), con sendas porciones de 100 ml de agua y después, agitando repetidas veces, con más porciones de 100 ml de agua hasta que el agua no se coloree al añadir solución de fenolftaleína (3.7) (normalmente es suficiente con lavar cuatro veces). Pasar el extracto lavado a un matraz aforado de 500 ml (4.2.2) a través de un filtro de pliegues seco para separación de fases (4.4), a fin de eliminar el agua que pudiera quedar en suspensión. Enjuagar el embudo de decantación y el filtro con 50 ml de éter de petróleo (3.2), enrasar con éter de petróleo (3.2) y mezclar bien.
5.3.2.
Cuando se hayan separado las fases (véase la observación del punto 7.3), sustituir el tapón de la probeta (4.8.1) por la pieza esmerilada (4.8.2) y colocar el extremo inferior con forma de U del tubo ajustable de manera que quede justo por encima del nivel de la interfase. Aplicando a la oliva la presión de un conducto de nitrógeno, transvasar la fase superior de éter de petróleo a un embudo de decantación de 1 000 ml (4.2.3). Añadir 100 ml de éter de petróleo (3.2) a la probeta, tapar y agitar bien. Dejar que se separen las fases y transvasar la fase superior al embudo de decantación, como antes. Repetir el procedimiento de extracción con otros 100 ml de éter de petróleo (3.2) y otras dos veces con sendas porciones de 50 ml de éter de petróleo (3.2), añadiendo a continuación las fases de éter de petróleo al embudo de decantación.
Lavar los extractos combinados de éter de petróleo como se describe en el punto 5.3.1 y seguir el procedimiento allí descrito.
5.4. Preparación de la solución de muestra para la CLAR
Pipetear una parte alícuota de la solución de éter de petróleo (de 5.3.1 o 5.3.2) a un matraz piriforme de 250 ml (4.2.4). Evaporar el disolvente casi hasta sequedad en el evaporador rotativo (4.1) a presión reducida y a una temperatura del baño no superior a 40 oC. Restaurar la presión atmosférica introduciendo nitrógeno (3.10) y sacar el matraz del evaporador rotativo. Eliminar el disolvente restante con una corriente de nitrógeno (3.10) y disolver inmediatamente el residuo en un volumen conocido (10-100 ml) de metanol (3.3) (la concentración de vitamina A debe quedar en un intervalo de 5 UI/ml a 30 UI/ml).
5.5. Determinación mediante CLAR
La vitamina A se separa en una columna de fase reversa de C18 (4.5.1) y su concentración se mide mediante un detector de UV (325 nm) o un detector de fluorescencia (excitación: 325 nm, emisión: 475 nm) (4.5.2).
Inyectar una parte alícuota (por ejemplo, 20 μl) de la solución metanólica obtenida según el punto 5.4 y eluir con la fase móvil (3.9). Calcular la altura (área) media de pico de varias inyecciones de la misma solución de muestra, así como la altura (área) media de pico de varias inyecciones de las soluciones de calibración (5.6.2).
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
|
Columna cromatográfica de líquidos (4.5.1): |
250 mm x 4 mm, C18, relleno de 5 μm o 10 μm, o equivalente |
|
Fase móvil (3.9): |
Mezcla de metanol (3.3) y agua, por ejemplo 980 + 20 (v+v) |
|
Caudal: |
1-2 ml/min. |
|
Detector (4.5.2): |
Detector de UV (325 nm) o detector de fluorescencia (excitación: 325 nm; emisión: 475 nm) |
5.6. Calibración
5.6.1.
Pipetear a un matraz Erlenmeyer o de fondo plano de 500 ml (4.2.1) 20 ml de la solución madre de acetato de vitamina A (3.11.1) o 20 ml de la solución madre de palmitato de vitamina A (3.12.1), e hidrolizar según se describe en el punto 5.2, pero sin añadir BHT. Extraer después con éter de petróleo (3.2) según el punto 5.3 y enrasar a 500 ml con éter de petróleo (3.2). Evaporar 100 ml de este extracto casi hasta sequedad en el evaporador rotativo (véase el punto 5.4), eliminar el disolvente restante con una corriente de nitrógeno (3,10) y volver a disolver el residuo en 10,0 ml de metanol (3.3). La concentración nominal de esta solución es de 560 UI de vitamina A por mililitro. El contenido exacto debe determinarse según el punto 5.6.3.3. La solución patrón de trabajo debe prepararse poco antes de utilizarse.
Pipetear 2,0 ml de esta solución patrón de trabajo a un matraz aforado de 20 ml, enrasar con metanol (3.3) y mezclar. La concentración nominal de esta solución patrón de trabajo diluida es de 56 UI de vitamina A por mililitro.
5.6.2.
Transvasar 1,0, 2,0, 5,0 y 10,0 ml de la solución patrón de trabajo diluida a una serie de matraces aforados de 20 ml, enrasar con metanol (3.3) y mezclar. Las concentraciones nominales de estas soluciones son de 2,8, 5,6, 14,0 y 28,0 UI de vitamina A por mililitro.
Inyectar varias veces 20 μl de cada solución de calibración y determinar las alturas (áreas) medias de pico. Utilizar estas últimas para trazar la curva de calibración teniendo en cuenta los resultados del control de UV (5.6.3.3).
5.6.3.
5.6.3.1.
Pipetear 2,0 ml de la solución madre de acetato de vitamina A (3.11.1) a un matraz aforado de 50 ml (4.2.2) y enrasar con 2-propanol (3.8). La concentración nominal de esta solución es de 56 UI de vitamina A por mililitro. Pipetear 3,0 ml de esta solución diluida de acetato de vitamina A a un matraz aforado de 25 ml y enrasar con 2-propanol (3.8). La concentración nominal de esta solución es de 6,72 UI de vitamina A por mililitro. Medir en el espectrofotómetro (4.6) a 300-400 nm el espectro de UV de esta solución frente al 2-propanol (3.8). El coeficiente máximo de extinción debe ser de 325 nm a 327 nm.
Cálculo del contenido de vitamina A:
UI de vitamina A/ml = E326 × 19,0
( para el acetato de vitamina A = 1 530 a 326 nm en 2-propanol)
para el acetato de vitamina A = 1 530 a 326 nm en 2-propanol)
5.6.3.2.
Pipetear 2,0 ml de la solución madre de palmitato de vitamina A (3.12.1) a un matraz aforado de 50 ml (4.2.2) y enrasar con 2-propanol (3.8). La concentración nominal de esta solución es de 56 UI de vitamina A por mililitro. Pipetear 3,0 ml de esta solución diluida de palmitato de vitamina A a un matraz aforado de 25 ml y enrasar con 2-propanol (3.8). La concentración nominal de esta solución es de 6,72 UI de vitamina A por mililitro. Medir en el espectrofotómetro (4.6) a 300-400 nm el espectro de UV de esta solución frente al 2-propanol (3.8). El coeficiente máximo de extinción debe ser de 325 nm a 327 nm.
Cálculo del contenido de vitamina A:
UI de vitamina A/ml = E326 × 19,0
( para el palmitato de vitamina A = 957 a 326 nm en 2-propanol)
para el palmitato de vitamina A = 957 a 326 nm en 2-propanol)
5.6.3.3.
Pipetear a un matraz aforado de 50 ml (4.2.2) 3,0 ml de la solución patrón de trabajo de vitamina A sin diluir, preparada de acuerdo con el punto 5.6.1, y enrasar con 2-propanol (3.8). Pipetear 5,0 ml de esta solución a un matraz aforado de 25 ml y enrasar con 2-propanol (3.8). La concentración nominal de esta solución es de 6,72 UI de vitamina A por mililitro. Medir en el espectrofotómetro (4.6) a 300-400 nm el espectro de UV de esta solución frente al 2-propanol (3.8). El coeficiente máximo de extinción debe ser de 325 nm a 327 nm.
Cálculo del contenido de vitamina A:
UI de vitamina A/ml = E326 × 18,3
( para el alcohol de vitamina A = 1 821 a 325 nm en 2-propanol)
para el alcohol de vitamina A = 1 821 a 325 nm en 2-propanol)
6. Cálculo de los resultados
A partir de la altura (área) media de los picos de vitamina A de la solución de muestra, determinar la concentración de esta última en UI/ml tomando como referencia la curva de calibración (5.6.2).
El contenido p de vitamina A de la muestra en UI/kg viene dado por la siguiente fórmula:
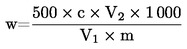 [UI/kg]
[UI/kg]
en la cual:
|
c |
= |
concentración de vitamina A de la solución de muestra (5.4), en UI/ml; |
|
V1 |
= |
volumen de la solución de muestra (5.4), en mililitros; |
|
V2 |
= |
volumen de la parte alícuota tomada en el punto 5.4, en mililitros; |
|
m |
= |
peso de la porción de ensayo, en gramos. |
7. Observaciones
|
7.1. |
En caso de muestras con baja concentración de vitamina A puede ser útil combinar los extractos de éter de petróleo de dos cargas de saponificación (cantidad pesada: 25 g) en una única solución de muestra para la determinación por CLAR. |
|
7.2. |
La muestra tomada para el análisis no contendrá en peso más de 2 g de grasa. |
|
7.3. |
Si no se produce la separación de las fases, añadir unos 10 ml de etanol (3.1) para romper la emulsión. |
|
7.4. |
Con aceite de hígado de bacalao y otras grasas puras, el tiempo de saponificación deberá prolongarse hasta durar de 45 a 60 minutos. |
|
7.5. |
Puede utilizarse hidroquinona en lugar de BHT. |
|
7.6. |
Con una columna de fase normal se pueden separar los isómeros del retinol. Pero, en ese caso, para hacer los cálculos deben sumarse las alturas (áreas) de todos los picos de los isómeros cis y trans. |
|
7.7. |
En lugar de la solución de ascorbato de sodio pueden utilizarse unos 150 mg de ácido ascórbico. |
|
7.8. |
En lugar de la solución de sulfuro de sodio pueden utilizarse unos 50 mg de EDTA. |
|
7.9. |
Cuando se analice la vitamina A de sustitutivos de la leche, debe prestarse una atención especial a:
Para comprobar si el método de análisis aplicado arroja resultados fiables para esta matriz concreta (sustitutivo de la leche), deberá efectuarse un ensayo de recuperación con una porción de ensayo adicional. Si el porcentaje de recuperación es inferior al 80 %, el resultado analítico debe corregirse para tener en cuenta la recuperación. |
8. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe rebasar el 15 % del resultado superior.
9. Resultados de un estudio colaborativo (1)
|
|
Premezcla |
Pienso de premezcla |
Concentrado de minerales |
Pienso proteínico |
Lechón |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
media [UI/kg] |
17,02 x 106 |
1,21 x 106 |
537 100 |
151 800 |
18 070 |
|
Sr [UI/kg] |
0,51 x 106 |
0,039 x 106 |
22 080 |
12 280 |
682 |
|
r [UI/kg] |
1,43 x 106 |
0,109 x 106 |
61 824 |
34 384 |
1 910 |
|
CVr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
SR [UI/kg] |
1,36 x 106 |
0,069 x 106 |
46 300 |
23 060 |
3 614 |
|
R [UI/kg] |
3,81 x 106 |
0,193 x 106 |
129 640 |
64 568 |
10 119 |
|
CVR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
número de laboratorios |
|
n |
= |
número de valores individuales |
|
Sr |
= |
desviación típica de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
r |
= |
repetibilidad |
|
R |
= |
reproducibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad. |
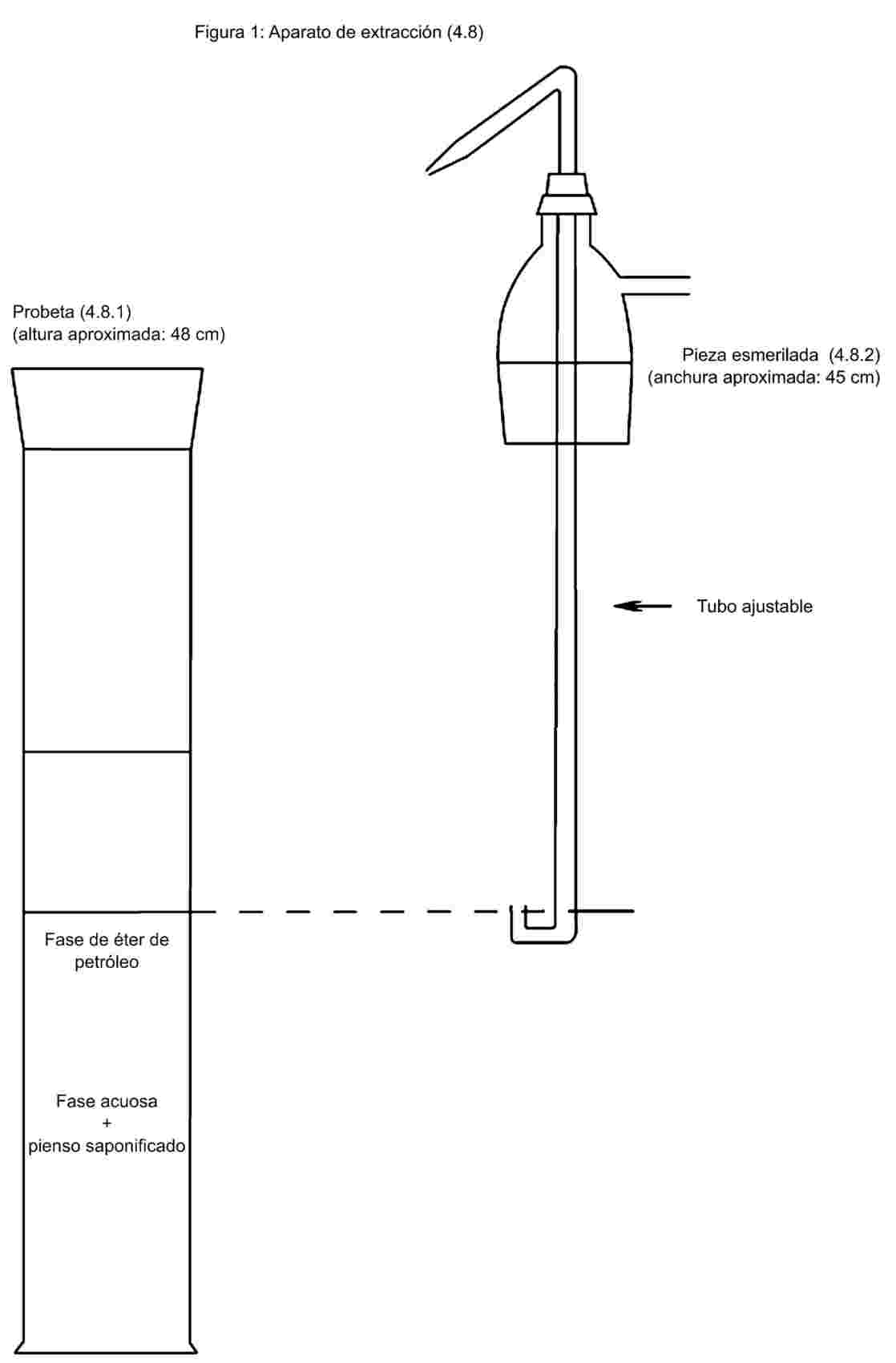
B. DETERMINACIÓN DE LA VITAMINA E
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de vitamina E en piensos y premezclas. El contenido de vitamina E se expresa en miligramos de acetato de DL-α-tocoferol por kilogramo. Un miligramo de acetato de DL-α-tocoferol corresponde a 0,91 mg de DL-α-tocoferol (vitamina E).
El límite de cuantificación es de 2 mg de vitamina E por kilogramo. Este límite de cuantificación solo puede alcanzarse con un detector de fluorescencia. Con un detector de UV, el límite de cuantificación es de 10 mg/kg.
2. Principio
La muestra se hidroliza con solución etanólica de hidróxido de potasio y la vitamina E se extrae en éter de petróleo. El disolvente se elimina por evaporación y el residuo se disuelve en metanol y, en caso necesario, se diluye hasta conseguir la concentración necesaria. El contenido de vitamina E se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa con detector de fluorescencia o de UV.
3. Reactivos
|
3.1. |
Etanol, σ = 96 %. |
|
3.2. |
Éter de petróleo con un intervalo de ebullición de 40 oC-60 oC. |
|
3.3. |
Metanol. |
|
3.4. |
Solución de hidróxido de potasio, c = 50 g/100 ml. |
|
3.5. |
Solución de ascorbato de sodio, c = 10 g/100 ml (véanse las observaciones del punto 7.7). |
|
3.6. |
Sulfuro de sodio, Na2S· x H2O (x = 7 — 9). |
|
3.6.1. |
Solución de sulfuro de sodio, c = 0,5 mol/l en glicerol, β = 120 g/l (para x = 9) (véanse las observaciones del punto 7.8). |
|
3.7. |
Solución de fenolftaleína, c = 2 g/100 ml en etanol (3.1). |
|
3.8. |
Fase móvil para la CLAR: mezcla de metanol (3.3) y agua, por ejemplo 980 + 20 (v+v). La proporción exacta estará en función de las características de la columna empleada. |
|
3.9. |
Nitrógeno, exento de oxígeno. |
|
3.10. |
Acetato de DL-α-tocoferol, extra puro, de actividad certificada. |
|
3.10.1. |
Solución madre de acetato de DL-α-tocoferol: pesar, con una precisión de 0,1 mg, 100 mg de acetato de DL-α-tocoferol (3.10) en un matraz aforado de 100 ml. Disolver en etanol (3.1) y enrasar con el mismo disolvente. Un mililitro de esta solución contiene 1 mg de acetato de DL-α-tocoferol. (Con respecto al control de UV, véase el punto 5.6.1.3; con respecto a la estabilización, véanse las observaciones del punto 7.4). |
|
3.11. |
Acetato de DL-α-tocoferol, extra puro, de actividad certificada. |
|
3.11.1. |
Solución madre de DL-α-tocoferol: pesar, con una precisión de 0,1 mg, 100 mg de DL-α-tocoferol (3.10) en un matraz aforado de 100 ml. Disolver en etanol (3.1) y enrasar con el mismo disolvente. Un mililitro de esta solución contiene 1 mg de DL-α-tocoferol. (Con respecto al control de UV, véase el punto 5.6.2.3; con respecto a la estabilización, véanse las observaciones del punto 7.4). |
|
3.12. |
2,6-Di-tert-butil-4-metilfenol (BHT) (véanse las observaciones del punto 7.5). |
4. Instrumental
|
4.1. |
Evaporador rotativo de película. |
|
4.2. |
Material de vidrio ámbar. |
|
4.2.1. |
Matraces Erlenmeyer o de fondo plano, de 500 ml, con boca esmerilada. |
|
4.2.2. |
Matraces aforados con tapón esmerilado, de cuello estrecho, de 10, 25, 100 y 500 ml. |
|
4.2.3. |
Embudos cónicos de decantación, de 1 000 ml, con tapón esmerilado. |
|
4.2.4. |
Matraces piriformes, de 250 ml, con boca esmerilada. |
|
4.3. |
Condensador de Allihn, con camisa de 300 mm de longitud, junta esmerilada y adaptador para tubo de alimentación de gas. |
|
4.4. |
Papel de filtro de pliegues para separación de fases, de 185 mm de diámetro (por ejemplo, Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
Equipo de CLAR con sistema de inyección. |
|
4.5.1. |
Columna cromatográfica de líquidos, de 250 mm x 4 mm, C18, relleno de 5 μm o 10 μm, o equivalente. |
|
4.5.2. |
Detector de fluorescencia o de UV, con ajuste de longitud de onda variable. |
|
4.6. |
Espectrofotómetro con cubetas de cuarzo de 10 mm. |
|
4.7. |
Baño maría con agitador magnético. |
|
4.8. |
Aparato de extracción (véase la figura 1) con los siguientes elementos: |
|
4.8.1. |
Probeta de 1 l de capacidad con cuello y tapón esmerilados. |
|
4.8.2. |
Pieza esmerilada provista de una oliva lateral y de un tubo ajustable que la atraviesa por el centro. El extremo inferior del tubo ajustable deberá tener forma de U, mientras que el superior debe ser una tobera, de forma que pueda pasarse la fase superior de líquido de la probeta a un embudo de decantación. |
5. Procedimiento
|
Nota |
: |
La vitamina E es sensible a la luz (UV) y a la oxidación. Todas las operaciones deberán realizarse en ausencia de luz (utilizando material de vidrio ámbar o protegido con papel de aluminio) y de oxígeno (chorro de nitrógeno). Durante la extracción, el aire que se encuentre por encima del líquido deberá sustituirse por nitrógeno (evitar un exceso de presión aflojando el tapón de vez en cuando). |
5.1. Preparación de la muestra.
Triturar la muestra de modo que pase por un tamiz con una luz de malla de 1 mm, cuidando de que no se produzca calor. La trituración debe hacerse inmediatamente antes de la pesada y la saponificación, pues, de lo contrario, puede haber pérdidas de vitamina E.
5.2. Saponificación.
En función del contenido de vitamina E, pesar, con una precisión de 0,01 g, entre 2 g y 25 g de la muestra en un matraz Erlenmeyer o de fondo plano de 500 ml (4.2.1). Añadir sucesivamente, agitando en círculos, 130 ml de etanol (3.1), unos 100 mg de BHT (3.12), 2 ml de solución de ascorbato de sodio (3.5) y 2 ml de solución de sulfuro de sodio (3.6). Ajustar el refrigerante (4.3) al matraz y sumergir este en un baño maría con agitador magnético (4.7). Calentar hasta ebullición y dejar refluir durante cinco minutos. Añadir entonces 25 ml de solución de hidróxido de potasio (3.4) a través del refrigerante (4.3) y dejar refluir durante otros 25 minutos, removiendo bajo una corriente lenta de nitrógeno. Enjuagar después el refrigerante con unos 20 ml de agua y enfriar el contenido del matraz hasta la temperatura ambiente.
5.3. Extracción.
Pasar cuantitativamente por decantación la solución de saponificación, enjuagando con un volumen total de 250 ml de agua, a un embudo de decantación de 1 000 ml (4.2.3) o al aparato de extracción (4.8). Enjuagar el matraz de saponificación sucesivamente con 25 ml de etanol (3.1) y 100 ml de éter de petróleo (3.2) y transvasar los líquidos de enjuague al embudo de decantación o al aparato de extracción. La proporción de agua y etanol en las soluciones combinadas debe ser aproximadamente de 2/1. Agitar enérgicamente durante dos minutos y dejar reposar durante otros dos minutos.
5.3.1.
Cuando se hayan separado las fases (véase la observación del punto 7.3), transvasar la fase de éter de petróleo a otro embudo de decantación (4.2.3). Repetir esta extracción dos veces con 100 ml de éter de petróleo (3.2) y dos veces con 50 ml de éter de petróleo (3.2).
Lavar dos veces los extractos combinados en el embudo de decantación, agitando en círculos suavemente (para evitar la formación de emulsiones), con sendas porciones de 100 ml de agua y después, agitando repetidas veces, con más porciones de 100 ml de agua hasta que el agua no se coloree al añadir solución de fenolftaleína (3.7) (normalmente es suficiente con lavar cuatro veces). Pasar el extracto lavado a un matraz aforado de 500 ml (4.2.2) a través de un filtro de pliegues seco para separación de fases (4.4), a fin de eliminar el agua que pudiera quedar en suspensión. Enjuagar el embudo de decantación y el filtro con 50 ml de éter de petróleo (3.2), enrasar con éter de petróleo (3.2) y mezclar bien.
5.3.2.
Cuando se hayan separado las fases (véase la observación del punto 7.3), sustituir el tapón de la probeta (4.8.1) por la pieza esmerilada (4.8.2) y colocar el extremo inferior con forma de U del tubo ajustable de manera que quede justo por encima del nivel de la interfase. Aplicando a la oliva la presión de un conducto de nitrógeno, transvasar la fase superior de éter de petróleo a un embudo de decantación de 1 000 ml (4.2.3). Añadir 100 ml de éter de petróleo (3.2) a la probeta, tapar y agitar bien. Dejar que se separen las fases y transvasar la fase superior al embudo de decantación, como antes. Repetir el procedimiento de extracción con otros 100 ml de éter de petróleo (3.2) y otras dos veces con sendas porciones de 50 ml de éter de petróleo (3.2), añadiendo a continuación las fases de éter de petróleo al embudo de decantación.
Lavar los extractos combinados de éter de petróleo como se describe en el punto 5.3.1 y seguir el procedimiento allí descrito.
5.4. Preparación de la solución de muestra para la CLAR
Pipetear una parte alícuota de la solución de éter de petróleo (de 5.3.1 o 5.3.2) a un matraz piriforme de 250 ml (4.2.4). Evaporar el disolvente casi hasta sequedad en el evaporador rotativo (4.1) a presión reducida y a una temperatura del baño no superior a 40 oC. Restaurar la presión atmosférica introduciendo nitrógeno (3.9) y sacar el matraz del evaporador rotativo. Eliminar el disolvente restante con una corriente de nitrógeno (3.9) y disolver inmediatamente el residuo en un volumen conocido (10-100 ml) de metanol (3.3) (la concentración de DL-α-tocoferol debe quedar en un intervalo de 5 μg/ml a 30 μg/ml).
5.5. Determinación mediante CLAR
La vitamina E se separa en una columna de fase reversa de C18 (4.5.1) y su concentración se mide mediante un detector de fluorescencia (excitación: 295 nm, emisión: 330 nm) o un detector de UV (292 nm) (4.5.2).
Inyectar una parte alícuota (por ejemplo, 20 μl) de la solución metanólica obtenida según el punto 5.4 y eluir con la fase móvil (3.8). Calcular las alturas (áreas) medias de pico de varias inyecciones de la misma solución de muestra, así como las alturas (áreas) medias de pico de varias inyecciones de las soluciones de calibración (5.6.2).
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
|
Columna cromatográfica de líquidos (4.5.1): |
250 mm x 4 mm, C18, relleno de 5 μm o 10 μm, o equivalente |
|
Fase móvil (3.8): |
Mezcla de metanol (3.3) y agua, por ejemplo 980 + 20 (v+v) |
|
Caudal: |
1-2 ml/min. |
|
Detector (4.5.2): |
Detector de fluorescencia (excitación: 295 nm/emisión: 330 nm) o detector de UV (292 nm) |
5.6. Calibración (acetato de DL-α-tocoferol o DL-α-tocoferol)
5.6.1.
5.6.1.1.
Pipetear a un matraz Erlenmeyer o de fondo plano de 500 ml (4.2.1) 25 ml de la solución madre de acetato de DL-α-tocoferol (3.10.1) e hidrolizar según se describe en el punto 5.2 Extraer después con éter de petróleo (3.2) según el punto 5.3 y enrasar a 500 ml con éter de petróleo. Evaporar 25 ml de este extracto casi hasta sequedad en el evaporador rotativo (véase el punto 5.4), eliminar el disolvente restante con una corriente de nitrógeno (3.9) y volver a disolver el residuo en 25,0 ml de metanol (3.3). La concentración nominal de esta solución es de 45,5 μg de DL-α-tocoferol por mililitro, equivalente a 50 μg de acetato de DL-α-tocoferol por mililitro. La solución patrón de trabajo debe prepararse poco antes de utilizarse.
5.6.1.2.
Transvasar 1,0, 2,0, 4,0 y 10,0 ml de la solución patrón de trabajo a una serie de matraces aforados de 20 ml, enrasar con metanol (3.3) y mezclar. Las concentraciones nominales de estas soluciones son de 2,5, 5,0, 10,0 y 25,0 μg/ml de acetato de DL-α-tocoferol, es decir, 2,28, 4,55, 9,10 y 22,8 μg/ml de DL-α-tocoferol.
Inyectar varias veces 20 μl de cada solución de calibración y determinar las alturas (áreas) medias de pico. Utilizar las alturas (áreas) medias de pico para trazar una curva de calibración.
5.6.1.3.
Diluir 5,0 ml de solución madre de acetato de DL-α-tocoferol (3.10.1) con etanol hasta un volumen de 25 ml y medir en el espectrofotómetro (4.6) a 250-320 nm el espectro de UV de esta solución frente al etanol (3.1).
El máximo de absorción a 284 nm deberá ser de:
 = 43,6 a 284 nm en etanol
= 43,6 a 284 nm en etanol
Con esta dilución debe obtenerse un valor de extinción de 0,84 a 0,88.
5.6.2.
5.6.2.1.
Pipetear 2 ml de la solución madre de DL-α-tocoferol (3.11.1) a un matraz aforado de 50 ml, disolver en metanol (3.3) y enrasar con metanol. La concentración nominal de esta solución es de 40 μg de DL-α-tocoferol por mililitro, equivalente a 44,0 μg de acetato de DL-α-tocoferol por mililitro. La solución patrón de trabajo debe prepararse poco antes de utilizarse.
5.6.2.2.
Transvasar 1,0, 2,0, 4,0 y 10,0 ml de la solución patrón de trabajo a una serie de matraces aforados de 20 ml, enrasar con metanol (3.3) y mezclar. Las concentraciones nominales de estas soluciones son de 2,0, 4,0, 8,0 y 20,0 μg/ml de DL-α-tocoferol, es decir, 2,20, 4,40, 8,79 y 22,0 μg/ml de acetato de DL-α-tocoferol.
Inyectar varias veces 20 μl de cada solución de calibración y determinar las alturas (áreas) medias de pico. Utilizar las alturas (áreas) medias de pico para trazar una curva de calibración.
5.6.2.3.
Diluir 2,0 ml de solución madre de DL-α-tocoferol (3.11.1) con etanol hasta un volumen de 25 ml y medir en el espectrofotómetro (4.6) a 250-320 nm el espectro de UV de esta solución frente al etanol (3.1). El máximo de absorción a 292 nm deberá ser de:
 = 75,8 a 292 nm en etanol
= 75,8 a 292 nm en etanol
Con esta dilución debe obtenerse un valor de extinción de 0,6.
6. Cálculo de los resultados
A partir de la altura (área) media de los picos de vitamina E de la solución de muestra, determinar la concentración de esta última en microgramos por mililitro (calculada en acetato de α-tocoferol) tomando como referencia la curva de calibración (5.6.1.2 o 5.6.2.2).
El contenido p de vitamina E de la muestra, en miligramos por kilogramo, viene dado por la siguiente fórmula:
 [mg/kg]
[mg/kg]
en la cual:
|
c |
= |
concentración de vitamina E (en acetato de α-tocoferol) de la solución de muestra (5.4), en microgramos por mililitro; |
|
V1 |
= |
volumen de la solución de muestra (5.4), en mililitros; |
|
V2 |
= |
volumen de la parte alícuota tomada en el punto 5.4, en mililitros; |
|
m |
= |
peso de la porción de ensayo, en gramos. |
7. Observaciones
|
7.1. |
En caso de muestras con baja concentración de vitamina E puede ser útil combinar los extractos de éter de petróleo de dos cargas de saponificación (cantidad pesada: 25 g) en una única solución de muestra para la determinación por CLAR. |
|
7.2. |
La muestra tomada para el análisis no contendrá en peso más de 2 g de grasa. |
|
7.3. |
Si no se produce la separación de las fases, añadir unos 10 ml de etanol (3.1) para romper la emulsión. |
|
7.4. |
Tras la medición espectrofotométrica de la solución de acetato de DL-α-tocoferol o de la solución de DL-α-tocoferol conforme al punto 5.6.1.3 o 5.6.2.3, respectivamente, añadir unos 10 mg de BHT (3.12) a la solución (3.10.1 o 3.10.2) y guardarla en el frigorífico (período máximo de conservación: cuatro semanas). |
|
7.5. |
Puede utilizarse hidroquinona en lugar de BHT. |
|
7.6. |
Con una columna de fase normal se pueden separar el α-tocoferol, el β-tocoferol, el γ-tocoferol y el δ-tocoferol. |
|
7.7. |
En lugar de la solución de ascorbato de sodio pueden utilizarse unos 150 mg de ácido ascórbico. |
|
7.8. |
En lugar de la solución de sulfuro de sodio pueden utilizarse unos 50 mg de EDTA. |
|
7.9. |
El acetato de vitamina E se hidroliza muy deprisa en condiciones alcalinas y, por tanto, es muy sensible a la oxidación, especialmente en presencia de oligoelementos como el hierro o el cobre. La vitamina E puede degradarse si se determina en premezclas a niveles superiores a 5 000 mg/kg. Por consiguiente, se recomienda confirmar los resultados mediante una CLAR que incluya la digestión enzimática de la formulación de vitamina E, sin la fase de saponificación alcalina. |
8. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe rebasar el 15 % del resultado superior.
9. Resultados de un estudio colaborativo (2)
|
|
Premezcla |
Pienso de premezcla |
Concentrado de minerales |
Pienso proteínico |
Lechón |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
media [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
Sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [ %] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
SR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [ %] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
número de laboratorios |
|
n |
= |
número de valores individuales |
|
S |
= |
desviación típica de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
r |
= |
repetibilidad |
|
R |
= |
reproducibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad. |

C. DETERMINACIÓN DE LOS OLIGOELEMENTOS HIERRO, COBRE, MANGANESO Y CINC
1. Objeto y ámbito de aplicación
Este método permite determinar los oligoelementos hierro, cobre, manganeso y cinc en los piensos. Los límites de cuantificación son:
|
— |
hierro (Fe): 20 mg/kg, |
|
— |
cobre (Cu): 10 mg/kg, |
|
— |
manganeso (Mn): 20 mg/kg, |
|
— |
cinc (Zn): 20 mg/kg. |
2. Principio
Tras destruir las posibles materias orgánicas, la muestra se disuelve en ácido clorhídrico. Los oligoelementos hierro, cobre, manganeso y cinc se determinan, tras la dilución apropiada, por espectrometría de absorción atómica.
3. Reactivos
Observaciones preliminares
Para preparar los reactivos y las soluciones analíticas, utilizar agua exenta de los cationes por determinar, obtenida bien por destilación doble en un frasco de vidrio borosilicato o un alambique de cuarzo, bien por tratamiento doble en resina de intercambio iónico.
Los reactivos deben ser al menos de calidad analítica. La ausencia del elemento por determinar debe comprobarse en un ensayo en blanco. Si es necesario, los reactivos deben someterse a una purificación más profunda.
Las soluciones patrón descritas a continuación pueden sustituirse por soluciones patrón comerciales, siempre que estén garantizadas y se hayan comprobado antes de ser utilizadas.
|
3.1. |
Ácido clorhídrico (d: 1,19 g/ml). |
|
3.2. |
Acido clorhídrico (6 mol/l). |
|
3.3. |
Acido clorhídrico (0,5 mol/l). |
|
3.4. |
Ácido fluorhídrico al 38-40 % (v/v), con un contenido de hierro (Fe) inferior a 1 mg/l y un residuo de evaporación inferior a 10 mg (en sulfatos)/l. |
|
3.5. |
Ácido sulfúrico (d: 1,84 g/ml). |
|
3.6. |
Peróxido de hidrógeno (aproximadamente 100 volúmenes de oxígeno [30 % en peso]). |
|
3.7. |
Solución patrón de hierro (1 000 μg Fe/ml), preparada como sigue, o solución comercial equivalente: disolver 1 g de alambre de hierro en 200 ml de ácido clorhídrico de 6 mol/l (3.2), añadir 16 ml de peróxido de hidrógeno (3.6) y enrasar a 1 l con agua. |
|
3.7.1. |
Solución patrón de hierro de trabajo (100 μg Fe/ml), preparada diluyendo una parte de solución patrón (3.7) con nueve partes de agua. |
|
3.8. |
Solución patrón de cobre (1 000 μg Cu/ml), preparada como sigue, o solución comercial equivalente:
|
|
3.8.1. |
Solución patrón de cobre de trabajo (10 μg Cu/ml), preparada diluyendo una parte de solución patrón (3.8) con nueve partes de agua, y diluyendo a continuación una parte de la solución resultante con nueve partes de agua. |
|
3.9. |
Solución patrón de manganeso (1 000 μg Mn/ml), preparada como sigue, o solución comercial equivalente:
|
|
3.9.1. |
Solución patrón de manganeso de trabajo (10 μg Mn/ml), preparada diluyendo una parte de solución patrón (3.9) con nueve partes de agua, y diluyendo a continuación una parte de la solución resultante con nueve partes de agua. |
|
3.10. |
Solución patrón de cinc (1 000 μg Zn/ml), preparada como sigue, o solución comercial equivalente:
|
|
3.10.1. |
Solución patrón de cinc de trabajo (10 μg Zn/ml), preparada diluyendo una parte de solución patrón (3.10) con nueve partes de agua, y diluyendo a continuación una parte de la solución resultante con nueve partes de agua. |
|
3.11. |
Solución de cloruro de lantano: disolver 12 g de óxido de lantano en 150 ml de agua, añadir 100 ml de ácido clorhídrico de 6 mol/l (3.2) y enrasar a 1 l con agua. |
4. Instrumental
|
4.1. |
Horno de mufla de temperatura regulable y, preferiblemente, con registrador. |
|
4.2. |
El material de vidrio debe ser de borosilicato resistente, y se recomienda utilizar un instrumental que esté reservado exclusivamente a la determinación de oligoelementos. |
|
4.3. |
Espectrofotómetro de absorción atómica que responda a las exigencias del método en cuanto a sensibilidad y precisión en el intervalo requerido. |
5. Procedimiento (3)
5.1. Muestras que contienen materia orgánica
5.1.1. (4)
|
5.1.1.1. |
Introducir de 5 g a 10 g de la muestra, pesados con una precisión de 0,2 mg, en un crisol de cuarzo o de platino [véase la nota b)], secar en una estufa a 105 oC e introducir el crisol en el horno de mufla (4.1) frío. Cerrar el horno [véase la nota c)] y elevar progresivamente la temperatura hasta alcanzar de 450 oC a 475 oC en 90 minutos aproximadamente. Mantener dicha temperatura durante cuatro a 16 horas (por ejemplo, durante la noche) para eliminar el material carbonoso, abrir a continuación el horno y dejar enfriar [véase la nota d)]. Humedecer las cenizas con agua e introducirlas en un vaso de precipitado de 250 ml. Lavar el crisol con no más de 5 ml de ácido clorhídrico (3.1) y añadir este último lentamente y con precaución al vaso de precipitado (puede producirse una fuerte reacción debido a la formación de CO2). Añadir gota a gota, agitando, ácido clorhídrico (3.1) hasta que cese la efervescencia. Evaporar hasta sequedad, removiendo de vez en cuando con una varilla de vidrio. A continuación, añadir al residuo 15 ml de ácido clorhídrico de 6 mol/l (3.2), y luego unos 120 ml de agua. Remover con la varilla de vidrio, que deberá dejarse en el vaso de precipitado, y cubrir este con un vidrio de reloj. Llevar suavemente a ebullición y mantener en el punto de ebullición hasta que aparentemente ya no se disuelvan las cenizas. Filtrar en un papel de filtro exento de cenizas y recoger el filtrado en un matraz aforado de 250 ml. Lavar el vaso de precipitado y el filtro con 5 ml de ácido clorhídrico de 6 mol/l (3.2) caliente y dos veces con agua hirviendo. Enrasar el matraz aforado con agua (concentración de HCl: 0,5 mol/l aproximadamente). |
|
5.1.1.2. |
Si el residuo que queda en el filtro es negro (carbón), volver a introducirlo en el horno y a incinerarlo a una temperatura de 450 oC a 475 oC. Esta incineración, que solo requiere unas horas (de tres a cinco horas, aproximadamente), se termina cuando la ceniza tiene un aspecto blanco o casi blanco. Disolver el residuo con unos 2 ml de ácido clorhídrico (3.1), evaporar hasta sequedad y añadir 5 ml de ácido clorhídrico de 6 mol/l (3.2). Calentar, filtrar la solución en el matraz aforado y enrasar con agua (concentración de HCl: 0,5 mol/l, aproximadamente). Notas:
|
5.1.2.
5.1.2.1.
Preparar, para cada elemento por determinar, una gama de soluciones de calibración a partir de las soluciones patrón de trabajo de los puntos 3.7.1, 3.8.1, 3.9.1 y 3.10.1, de forma que cada solución de calibración tenga una concentración de HCl de 0,5 mol/l aproximadamente y (en el caso del hierro, del manganeso y del cinc) una concentración de cloruro de lantano equivalente a 0,1 % La (p/v).
Las concentraciones escogidas de oligoelementos deben encontrarse en el intervalo de sensibilidad del espectrofotómetro utilizado. Los cuadros siguientes muestran, a título de ejemplo, las composiciones de gamas típicas de soluciones de calibración; sin embargo, en función del tipo y la sensibilidad del espectrofotómetro utilizado, puede ser necesario escoger otras concentraciones.
Hierro
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml de solución patrón de trabajo (3.7.1) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
añadir 10 ml de solución de cloruro de lantano (3.11) y enrasar a 100 ml con agua |
|||||||
Cobre
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml de solución patrón de trabajo (3.8.1) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
Manganeso
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml de solución patrón de trabajo (3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
añadir 10 ml de solución de cloruro de lantano (3.11) y enrasar a 100 ml con agua |
|||||||
Cinc
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml de solución patrón de trabajo (3.10.1) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
añadir 10 ml de solución de cloruro de lantano (3.11) y enrasar a 100 ml con agua |
|||||||
5.1.2.2.
Para la determinación del cobre, la solución preparada según el punto 5.1.1 puede, en general, utilizarse directamente. Si es necesario ajustar su concentración a la gama de las soluciones de calibración, puede pipetearse una parte alícuota a un matraz aforado de 100 ml, enrasando a continuación con ácido clorhídrico de 0,5 mol/l (3.3).
Para la determinación del hierro, del manganeso y del cinc, pipetear una parte alícuota de la solución preparada según el punto 5.1.1 a un matraz aforado de 100 ml, añadir 10 ml de solución de cloruro de lantano (3.11) y enrasar con ácido clorhídrico de 0,5 mol/l (3.3) (véase también la observación del punto 8).
5.1.2.3.
El experimento en blanco debe incluir todas las etapas prescritas del procedimiento, pero omitiendo el material de muestra. La solución de calibración «0» no debe utilizarse como blanco.
5.1.2.4.
Medir la absorción atómica de las soluciones de calibración y de la solución objeto de análisis utilizando una llama oxidante aire-acetileno con las siguientes longitudes de onda:
|
|
Fe: 248,3 nm |
|
|
Cu: 324,8 nm |
|
|
Mn: 279,5 nm |
|
|
Zn: 213,8 nm |
Realizar cuatro veces cada medición.
5.2. Piensos minerales
Si la muestra no contiene materia orgánica, no es necesaria la incineración previa. Proceder como se describe en el punto 5.1.1.1, comenzando a partir del párrafo segundo. Puede omitirse la evaporación con ácido fluorhídrico.
6. Cálculo de los resultados
Por medio de una curva de calibración, calcular la concentración de oligoelementos de la solución objeto de análisis y expresar el resultado en miligramos de oligoelemento por kilogramo de muestra (ppm).
7. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra por el mismo analista no deberá superar:
|
— |
5 mg/kg en valor absoluto, en el caso de que el contenido del oligoelemento en cuestión no supere los 50 mg/kg, |
|
— |
el 10 % del resultado superior, en el caso de que el contenido del oligoelemento en cuestión sea de 50-100 mg/kg, |
|
— |
10 mg/kg en valor absoluto, en el caso de que el contenido del oligoelemento en cuestión sea de 100-200 mg/kg, |
|
— |
el 5 % del resultado superior, en el caso de que el contenido del oligoelemento en cuestión supere los 200 mg/kg. |
8. Observación
La presencia de grandes cantidades de fosfatos puede interferir en la determinación del hierro, del manganeso y del cinc. Tal interferencia debe corregirse añadiendo solución de cloruro de lantano (3.11). Si, de todas formas, la relación de peso Ca + Mg/P de la muestra es > 2, no es necesario añadir solución de cloruro de lantano (3.11) a la solución objeto de análisis ni a las soluciones de calibración.
D. DETERMINACIÓN DE LA HALOFUGINONA
Bromhidrato de DL-trans-7-bromo-6-cloro-3-[3-(3-hidroxi-2-piperidil)acetonil]-quinazolin-4-(3H)-ona
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de halofuginona en los piensos. El límite de cuantificación es de 1 mg/kg.
2. Principio
Tras tratamiento con agua caliente, la halofuginona se extrae como base libre en acetato de etilo y posteriormente se somete a separación como clorhidrato en una solución ácida acuosa. El extracto se purifica mediante cromatografía de intercambio iónico. El contenido de halofuginona se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa, empleando un detector de UV.
3. Reactivos
|
3.1. |
Acetonitrilo, equivalente al de calidad CLAR. |
|
3.2. |
Resina Amberlite XAD-2. |
|
3.3. |
Acetato de amonio. |
|
3.4. |
Acetato de etilo. |
|
3.5. |
Ácido acético glacial. |
|
3.6. |
Halofuginona patrón (hidrobromuro de DL-trans-7-bromo-6-cloro-3-[3-(3-hidroxi-2-piperidil)acetonil]-quinazolin-4-(3H)-ona, E 764). |
|
3.6.1. |
Pesar, con una precisión de 0,1 mg, 50 mg de halofuginona (3.6) en un matraz aforado de 500 ml, disolver en solución reguladora de acetato de amonio (3.18), enrasar con la solución reguladora y mezclar. Esta solución se mantiene estable durante tres semanas a 5 oC, si se guarda al abrigo de la luz. |
|
3.6.2. |
Transvasar 1,0, 2,0, 3,0, 4,0 y 6,0 ml de la solución patrón madre (3.6.1) a una serie de matraces aforados de 100 ml. Enrasar con fase móvil (3.21) y mezclar. Estas soluciones tienen concentraciones de halofuginona de 1,0, 2,0, 3,0, 4,0 y 6,0 μg/ml, respectivamente. Deben prepararse poco antes de utilizarse. |
|
3.7. |
Ácido clorhídrico (ρ20 = 1,16 g/ml aproximadamente). |
|
3.8. |
Metanol. |
|
3.9. |
Nitrato de plata. |
|
3.10. |
Ascorbato de sodio. |
|
3.11. |
Carbonato de sodio. |
|
3.12. |
Cloruro de sodio. |
|
3.13. |
EDTA (ácido etilendiaminotetracético, sal disódica). |
|
3.14. |
Agua, equivalente a la de calidad CLAR. |
|
3.15. |
Solución de carbonato de sodio, c = 10 g/100 ml. |
|
3.16. |
Solución de carbonato de sodio saturada de cloruro de sodio, c = 5 g/100 ml. Disolver 50 g de carbonato de sodio (3.11) en agua, diluir hasta 1 l y añadir cloruro de sodio (3.12) hasta que la solución esté saturada. |
|
3.17. |
Ácido clorhídrico de aproximadamente 0,1 mol/l. Diluir 10 ml de HCl (3.7) con agua, hasta 1 l. |
|
3.18. |
Solución reguladora de acetato de amonio de aproximadamente 0,25 mol/l. Disolver 19,3 g de acetato de amonio (3.3) y 30 ml de ácido acético (3.5) en agua (3.14) y diluir hasta 1 l. |
|
3.19. |
Preparado de resina Amberlite XAD-2. Lavar una cantidad adecuada de resina Amberlite (3.2) con agua hasta que se hayan eliminado todos los iones de cloruro, lo cual se comprobará mediante una prueba con nitrato de plata (3.20) en la fase acuosa desechada. A continuación, lavar la resina con 50 ml de metanol (3.8), desechar el metanol y guardar la resina bajo metanol nuevo. |
|
3.20. |
Solución de nitrato de plata de aproximadamente 0,1 mol/l. Disolver 0,17 g de nitrato de plata (3.9) en 10 ml de agua. |
|
3.21. |
Fase móvil para la CLAR. Mezclar 500 ml de acetonitrilo (3.1) con 300 ml de solución reguladora de acetato de amonio (3.18) y 1 200 ml de agua (3.14). Ajustar el pH a 4,3 empleando ácido acético (3.5). Filtrar por un filtro de 0,22 μm (4.8) y desgasificar la solución (por ejemplo, aplicando ultrasonidos durante diez minutos). Esta solución, almacenada al abrigo de la luz y en un recipiente cerrado, es estable durante un mes. |
4. Instrumental
|
4.1. |
Baño ultrasónico. |
|
4.2. |
Evaporador rotativo de película. |
|
4.3. |
Centrífuga. |
|
4.4. |
Equipo para CLAR con detector ultravioleta de longitud de onda variable o detector de red de diodos. |
|
4.4.1. |
Columna cromatográfica de líquidos, de 300 mm x 4 mm, C18, relleno de 10 μm, o equivalente. |
|
4.5. |
Columna de vidrio (300 mm x 10 mm) provista de filtro de vidrio sinterizado y llave de cierre. |
|
4.6. |
Filtros de fibra de vidrio de un diámetro de 150 mm. |
|
4.7. |
Filtros de membrana de 0,45 μm. |
|
4.8. |
Filtros de membrana de 0,22 μm. |
5. Procedimiento
|
Nota |
: |
La halofuginona como base libre es inestable en soluciones alcalinas y de acetato de etilo. No deberá permanecer en acetato de etilo durante más de 30 minutos. |
5.1. Generalidades
|
5.1.1. |
Deberá analizarse un pienso en blanco para comprobar la ausencia de halofuginona y de sustancias interferentes. |
|
5.1.2. |
Asimismo, deberá realizarse un ensayo de recuperación analizando el pienso en blanco, que se habrá enriquecido añadiendo una cantidad de halofuginona similar a la presente en la muestra. Para enriquecer a un nivel de 3 mg/kg, añadir 300 μl de la solución patrón madre (3.6.1) a 10 g del pienso en blanco, mezclar y esperar diez minutos antes de proceder a la extracción (5.2).
|
5.2. Extracción
Pesar, con una precisión de 0,1 g, 10 g de la muestra preparada en un tubo de centrífuga de 200 ml, añadir 0,5 g de ascorbato de sodio (3.10), 0,5 g de EDTA (3.13) y 20 ml de agua, y mezclar. Poner el tubo en un baño maría a (80 oC) durante cinco minutos. Tras enfriar a temperatura ambiente, añadir 20 ml de solución de carbonato de sodio (3.15) y mezclar. Añadir inmediatamente 100 ml de acetato de etilo (3.4) y agitar enérgicamente a mano durante 15 segundos. Colocar después el tubo en el baño ultrasónico (4.1) durante tres minutos y aflojar el tapón. Centrifugar durante dos minutos y decantar la fase de acetato de etilo en un embudo de decantación de 500 ml a través de un filtro de fibra de vidrio (4.6). Repetir la extracción de la muestra con una segunda porción de 100 ml de acetato de etilo. Lavar los extractos combinados durante un minuto con 50 ml de solución de carbonato de sodio saturada de cloruro de sodio (3.16) y desechar la fase acuosa.
Extraer la fase orgánica durante un minuto con 50 ml de ácido clorhídrico (3.17). Pasar la fase ácida inferior a un embudo de decantación de 250 ml. Extraer de nuevo la fase orgánica durante minuto y medio con otros 50 ml de ácido clorhídrico y combinar con el primer extracto. Lavar los extractos ácidos combinados agitando en círculos durante aproximadamente diez segundos con 10 ml de acetato de etilo (3.4).
Transvasar cuantitativamente la fase acuosa a un matraz de fondo redondo de 250 ml y desechar la fase orgánica. Evaporar todo el acetato de etilo que quede en la solución ácida empleando un evaporador rotativo de película (4.2). La temperatura del baño maría no debe superar los 40 oC. A un vacío de aproximadamente 25 mbar, todo el acetato de etilo residual se eliminará en cinco minutos a 38 oC.
5.3. Limpieza (cleanup)
5.3.1.
Preparar una columna de XAD-2 para cada extracto de muestra. Pasar 10 g de resina Amberlite preparada (3.19) a una columna de vidrio (4.5) con metanol (3.8). Poner un trozo pequeño de lana de vidrio encima del lecho de resina. Dejar salir el metanol de la columna y lavar la resina con 100 ml de agua, cortando el flujo cuando el líquido alcance la parte superior del lecho de resina. Antes de utilizarla, dejar que la columna se equilibre durante diez minutos. No dejar nunca que se seque.
5.3.2.
Transferir el extracto (5.2) cuantitativamente a la parte superior de la columna de resina Amberlite preparada (5.31) y eluir, desechando el eluido. La velocidad de elución no debe exceder de 20 ml/min. Enjuagar el matraz de fondo redondo con 20 ml de ácido clorhídrico (3.17) y emplear este líquido para lavar la columna de resina. Eliminar todo resto de solución ácida con un chorro de aire. Desechar los líquidos de lavado. Añadir 100 ml de metanol (3.8) a la columna y dejar eluir de 5 ml a 10 ml, recogiendo el eluido en un matraz de fondo redondo de 250 ml. Dejar que el metanol restante se equilibre durante diez minutos con la resina y continuar con la elución a una velocidad que no supere los 20 ml/min., recogiendo el eluido en el mismo matraz de fondo redondo. Evaporar el metanol en el evaporador rotativo de película (4.2) sin que la temperatura del baño maría sobrepase los 40 oC. Transferir cuantitativamente el residuo a un matraz aforado de 10 ml empleando la fase móvil (3.21). Enrasar con fase móvil y mezclar. Filtrar una parte alícuota por un filtro de membrana (4.7). Reservar esta solución para la determinación mediante CLAR (5.4).
5.4. Determinación mediante CLAR
5.4.1.
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
Columna cromatográfica de líquidos (4.4.1).
Fase móvil para la CLAR (3.21).
Caudal: 1,5-2 ml/min.
Longitud de onda de detección: 243 nm.
Volumen de inyección: 40-100 μl.
Comprobar la estabilidad del sistema cromatográfico inyectando varias veces la solución de calibración (3.6.2) de 3,0 μg/ml, hasta que se alcancen alturas (o áreas) de pico y tiempos de retención constantes.
5.4.2.
Inyectar varias veces cada solución de calibración (3.6.2) y medir las alturas (áreas) de pico de cada concentración. Trazar una curva de calibración empleando las alturas o áreas de pico medias de las soluciones de calibración como ordenadas y las concentraciones correspondientes, en microgramos por mililitro, como abscisas.
5.4.3.
Inyectar varias veces el extracto de muestra (5.3.2) empleando el mismo volumen que para las soluciones de calibración, y determinar la altura (área) media de los picos de halofuginona.
6. Cálculo de los resultados
Determinar la concentración de la solución de muestra, en microgramos por mililitro, a partir de la altura (área) media de sus picos de halofuginona, tomando como referencia la curva de calibración (5.4.2).
El contenido de halofuginona p (mg/kg) de la muestra viene dado por la siguiente fórmula:

en la cual:
|
c |
= |
concentración de halofuginona de la solución de muestra, en microgramos por mililitro; |
|
m |
= |
peso de la porción de ensayo, en gramos. |
7. Validación de los resultados
7.1. Identidad
La identidad del analito puede confirmarse mediante cocromatografía, o utilizando un detector de red de diodos con el cual se comparan los espectros del extracto de muestra y de la solución de calibración (3.6.2) de 6,0 μg/ml.
7.1.1.
Enriquecer un extracto de muestra añadiéndole una cantidad apropiada de solución de calibración (3.6.2). La cantidad de halofuginona añadida debe ser similar a la cantidad estimada de halofuginona hallada en el extracto de muestra.
Solo la altura del pico de halofuginona deberá aumentar en función de la cantidad añadida y de la dilución del extracto. La anchura del pico, a la mitad de su altura máxima, debe ser igual ± 10 % a la anchura original.
7.1.2.
Los resultados se evalúan con arreglo a los siguientes criterios:
|
a) |
la longitud de onda de absorción máxima de los espectros de la muestra y del patrón, registrados en el vértice del pico del cromatograma, debe ser la misma dentro de un margen determinado por el poder de resolución del sistema de detección; en el caso de la detección por red de diodos, el margen típico se sitúa en ± 2 nm; |
|
b) |
entre 225 nm y 300 nm, los espectros de la muestra y del patrón registrados en el vértice del pico del cromatograma no deben ser diferentes por lo que respecta a aquellas partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los dos espectros excede del 15 % de la absorbancia del analito patrón; |
|
c) |
entre 225 nm y 300 nm, los espectros de la pendiente ascendente, del vértice y de la pendiente descendente del pico producido por el extracto de muestra no deben diferir entre sí por lo que respecta a las partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los espectros excede del 15 % de la absorbancia del espectro del vértice. |
Si no se cumple alguno de estos criterios, la presencia del analito no queda confirmada.
7.2. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas llevadas a cabo con la misma muestra no debe superar los 0,5 mg/kg, en relación con contenidos de halofuginona de hasta 3 mg/kg.
7.3. Recuperación
La recuperación de la muestra en blanco enriquecida deberá ser al menos del 80 %.
8. Resultados de un estudio colaborativo
Se organizó un estudio colaborativo (5) en el que ocho laboratorios analizaron tres muestras.
Resultados
|
|
Muestra A (blanco) A la recepción |
Muestra B (sémola) |
Muestra C (gránulos) |
||
|
|
|
A la recepción |
Tras dos meses |
A la recepción |
Tras dos meses |
|
Media [mg/kg] |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [ %] |
— |
16 |
18 |
14 |
17 |
|
Rec. [ %] |
|
86 |
74 |
88 |
75 |
|
ND |
= |
no detectada |
|
SR |
= |
desviación típica de la reproducibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad (%) |
|
Rec. |
= |
recuperación (%) |
E. DETERMINACIÓN DE LA ROBENIDINA
Clorhidrato de 1,3-bis [(4-clorobencilideno)amino]guanidina
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de robenidina en los piensos. El límite de cuantificación es de 5 mg/kg.
2. Principio
La muestra se extrae con metanol acidificado. El extracto se deseca y una parte alícuota se limpia en una columna de óxido de aluminio. La robenidina se eluye de la columna con metanol, se concentra y se enrasa a un volumen adecuado con fase móvil. El contenido de robenidina se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa, empleando un detector de UV.
3. Reactivos
|
3.1. |
Metanol |
|
3.2. |
Metanol acidificado Transvasar 4,0 ml de ácido clorhídrico (ρ20 = 1,18 g/ml) a un matraz aforado de 500 ml, enrasar con metanol (3.1) y mezclar. Esta solución deberá prepararse poco antes de utilizarse. |
|
3.3. |
Acetonitrilo, equivalente al de calidad CLAR |
|
3.4. |
Tamiz molecular Tipo 3A, perlas de 8-12 mallas (perlas de 1,6 - 2,5 mm, aluminosilicato cristalino, poros de 0,3 mm de diámetro). |
|
3.5. |
Óxido de aluminio de actividad ácida de grado I para cromatografía de columna Transferir 100 g de óxido de aluminio a un recipiente apropiado y añadir 2,0 ml de agua. Tapar y agitar durante 20 minutos aproximadamente. Almacenar en un recipiente bien cerrado. |
|
3.6. |
Solución de dihidrogenofosfato de potasio, c = 0,025 mol/l Disolver 3,40 g de dihidrogenofosfato de potasio en agua (de calidad CLAR) en un matraz aforado de 1 000 ml, enrasar y mezclar. |
|
3.7. |
Solución de hidrogenofosfato de disodio, c = 0,025 mol/l Disolver 3,55 g de hidrogenofosfato de disodio anhidro (o 4,45 g de dihidrato u 8,95 g de dodecahidrato) en agua (equivalente a la de calidad CLAR) en un matraz aforado de 1 l, enrasar y mezclar. |
|
3.8. |
Fase móvil para la CLAR Mezclar los reactivos siguientes:
Filtrar por un filtro de 0,22 μm (4.6) y desgasificar la solución (por ejemplo, aplicando ultrasonidos durante diez minutos). |
|
3.9. |
Sustancia patrón Robenidina pura: clorhidrato de 1,3-bis [(4-clorobencilideno)amino]guanidina |
|
3.9.1. |
Pesar, con una precisión de 0,1 mg, 30 mg de sustancia patrón de robenidina (3.9). Disolver en metanol acidificado (3.2) en un matraz aforado de 100 ml, enrasar con el mismo disolvente y mezclar. Envolver el matraz con papel de aluminio y guardar al abrigo de la luz. |
|
3.9.2. |
Transvasar 10,0 ml de la solución patrón madre (3.9.1) a un matraz aforado de 250 ml, enrasar con la fase móvil (3.8) y mezclar. Envolver el matraz con papel de aluminio y guardar al abrigo de la luz. |
|
3.9.3. |
Transvasar 5,0, 10,0, 15,0, 20,0 y 25,0 ml de la solución patrón intermedia (3.9.2) a una serie de matraces aforados de 50 ml. Enrasar con fase móvil (3.8) y mezclar. Estas soluciones corresponden, respectivamente, a 1,2, 2,4, 3,6, 4,8 y 6,0 μg/ml de robenidina. Deben prepararse poco antes de utilizarse. |
|
3.10. |
Agua, equivalente a la de calidad CLAR |
4. Instrumental
|
4.1. |
Columna de vidrio Columna de vidrio ámbar provista de llave de cierre y de un depósito de aproximadamente 150 ml de capacidad, con un diámetro interior de 10-15 mm y una longitud de 250 mm. |
|
4.2. |
Agitador mecánico o magnético |
|
4.3. |
Evaporador rotativo de película |
|
4.4. |
Equipo para CLAR con detector ultravioleta de longitud de onda variable o detector de red de diodos que funcione en el intervalo de 250-400 nm |
|
4.4.1. |
Columna cromatográfica de líquidos: 300 mm x 4 mm, C18, relleno de 10 μm, o equivalente. |
|
4.5. |
Papel de filtro de fibra de vidrio (Whatman GF/A o equivalente) |
|
4.6. |
Filtros de membrana de 0,22 μm |
|
4.7. |
Filtros de membrana de 0,45 μm |
5. Procedimiento
|
Nota |
: |
La robenidina es sensible a la luz. Deberá utilizarse material de vidrio ámbar en todas las operaciones. |
5.1. Generalidades
|
5.1.1. |
Deberá analizarse un pienso en blanco para comprobar la ausencia de robenidina y de sustancias interferentes. |
|
5.1.2. |
Asimismo, deberá realizarse un ensayo de recuperación analizando el pienso en blanco (5.1.1), que se habrá enriquecido añadiendo una cantidad de robenidina similar a la presente en la muestra. Para enriquecer a un nivel de 60 mg/kg, transvasar 3,0 ml de la solución patrón madre (3.9.1) a un Erlenmeyer de 250 ml. Evaporar la solución en una corriente de nitrógeno hasta que resten 0,5 ml, aproximadamente. Añadir 15 g del pienso en blanco, mezclar y esperar diez minutos antes de proceder a la extracción (5.2).
|
5.2. Extracción
Pesar, con una precisión de 0,01 g, 15 g aproximadamente de la muestra preparada. Transferirlos a un Erlenmeyer de 250 ml y añadir 100,0 ml de metanol acidificado (3.2), tapar y agitar durante una hora en el agitador (4.2). Filtrar la solución por un papel de filtro de fibra de vidrio (4.5) y recoger todo el filtrado en un Erlenmeyer de 150 ml. Añadir 7,5 g de tamiz molecular (3.4), tapar y agitar durante cinco minutos. Filtrar inmediatamente por un papel de filtro de fibra de vidrio. Conservar esta solución para la fase de purificación (5.3).
5.3. Purificación
5.3.1.
Introducir un pequeño tapón de lana de vidrio en el extremo inferior de la columna de vidrio (4.1) y atacarlo con una varilla de vidrio. Pesar 11,0 g del óxido de aluminio preparado (3.5) y transferirlos a la columna. Durante esta fase deberá procurarse minimizar la exposición a la atmósfera. Golpear suavemente el extremo inferior de la columna cargada a fin de que se sedimente el óxido de aluminio.
5.3.2.
Pipetear a la columna 5,0 ml del extracto de muestra preparado según el punto 5.2, colocando la punta de la pipeta cerca de la pared de la columna y dejando que la solución se absorba en el óxido de aluminio. Eluir la robenidina de la columna con 100 ml de metanol (3.1), manteniendo un caudal de 2-3 ml/min., y recoger el eluido en un matraz de fondo redondo de 250 ml. Evaporar hasta sequedad la solución de metanol a presión reducida y a una temperatura de 40 oC, utilizando un evaporador rotativo de película (4.3). Disolver nuevamente el residuo en 3-4 ml de fase móvil (3.8) y transvasar cuantitativamente a un matraz aforado de 10 ml. Enjuagar el matraz con varias porciones de 1-2 ml de fase móvil y transvasar estos líquidos de enjuague al matraz aforado. Enrasar con el mismo disolvente y mezclar. Filtrar una parte alícuota por un filtro de membrana de 0,45 μm (4.7). Reservar esta solución para la determinación mediante CLAR (5.4).
5.4. Determinación mediante CLAR.
5.4.1.
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
Columna cromatográfica de líquidos (4.4.1).
Fase móvil para la CLAR (3.8).
Caudal: 1,5-2 ml/min.
Longitud de onda del detector: 317 nm.
Volumen de inyección: 20-50 μl.
Comprobar la estabilidad del sistema cromatográfico inyectando varias veces la solución de calibración (3.9.3) de 3,6 μg/ml, hasta que se alcancen alturas de pico y tiempos de retención constantes.
5.4.2.
Inyectar varias veces cada solución de calibración (3.9.3) y medir las alturas (áreas) de pico de cada concentración. Trazar una curva de calibración empleando las alturas o áreas de pico medias de las soluciones de calibración como ordenadas y las concentraciones correspondientes, en microgramos por mililitro, como abscisas.
5.4.3.
Inyectar varias veces el extracto de muestra (5.3.2) empleando el mismo volumen que para las soluciones de calibración, y determinar la altura (área) media de los picos de robenidina.
6. Cálculo de los resultados
Determinar la concentración de la solución de muestra, en microgramos por mililitro, a partir de la altura (área) media de sus picos de robenidina, tomando como referencia la curva de calibración (5.4.2).
El contenido de robenidina p (mg/kg) de la muestra viene dado por la siguiente fórmula:
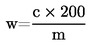
en la cual:
|
c |
= |
concentración de robenidina de la solución de muestra, en microgramos por mililitro; |
|
m |
= |
peso de la porción de ensayo, en gramos. |
7. Validación de los resultados
7.1. Identidad.
La identidad del analito puede confirmarse mediante cocromatografía, o utilizando un detector de red de diodos con el cual se comparan los espectros del extracto de muestra y de la solución de calibración (3.9.3) de 6 μg/ml.
7.1.1.
Enriquecer un extracto de muestra añadiéndole una cantidad apropiada de solución de calibración (3.9.3). La cantidad de robenidina añadida debe ser similar a la cantidad estimada de robenidina hallada en el extracto de muestra.
Solo la altura del pico de robenidina deberá aumentar en función de la cantidad añadida y de la dilución del extracto. La anchura del pico, a la mitad de su altura máxima, debe ser igual ± 10 % a la anchura original.
7.1.2.
Los resultados se evalúan con arreglo a los siguientes criterios:
|
a) |
la longitud de onda de absorción máxima de los espectros de la muestra y del patrón, registrados en el vértice del pico del cromatograma, debe ser la misma dentro de un margen determinado por el poder de resolución del sistema de detección; en el caso de la detección por red de diodos, el margen típico se sitúa en aproximadamente 2 nm; |
|
b) |
entre 250 nm y 400 nm, los espectros de la muestra y del patrón registrados en el vértice del pico del cromatograma no deben ser diferentes por lo que respecta a aquellas partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los dos espectros excede del 15 % de la absorbancia del analito patrón; |
|
c) |
entre 250 nm y 400 nm, los espectros de la pendiente ascendente, del vértice y de la pendiente descendente del pico producido por el extracto de muestra no deben diferir entre sí por lo que respecta a las partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los espectros excede del 15 % de la absorbancia del espectro del vértice. |
Si no se cumple alguno de estos criterios, la presencia del analito no queda confirmada.
7.2. Repetibilidad.
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe rebasar el 10 % del resultado superior en el caso de contenidos de robenidina mayores de 15 mg/kg.
7.3. Recuperación.
La recuperación de la muestra en blanco enriquecida deberá ser al menos del 85 %.
8. Resultados de un estudio colaborativo
Se organizó un estudio colaborativo de la Comunidad Europea en el que 12 laboratorios analizaron cuatro muestras de piensos para aves de corral y conejos, en forma de sémola o de gránulos. De cada muestra se hicieron análisis por duplicado. Los resultados se recogen en el cuadro siguiente:
|
|
Aves de corral |
Conejos |
||
|
|
Sémola |
Gránulos |
Sémola |
Gránulos |
|
Media [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [ %] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [ %] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Recuperación [ %] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
desviación típica de la repetibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad, en tanto por ciento |
|
SR |
= |
desviación típica de la reproducibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad, en tanto por ciento |
F) DETERMINACIÓN DEL DICLAZURILO
(+)-4-clorofenil [2,6-dicloro-4-(2,3,4,5-tetrahidro-3,5-dioxo-1,2,4-triazin-2-il)fenil]acetonitrilo
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de diclazurilo en piensos y premezclas. El límite de detección es de 0,1 mg/kg; el de cuantificación, de 0,5 mg/kg.
2. Principio
Tras añadir un patrón interno, la muestra se extrae con metanol acidificado. En el caso de los piensos, se purifica una parte alícuota del extracto en un cartucho de extracción en fase sólida de C18. El diclazurilo se eluye del cartucho con una mezcla de metanol acidificado y agua. Previa evaporación, el residuo se disuelve en DMF/agua. En el caso de las premezclas, el extracto se evapora y el residuo se disuelve en DMF/agua. El contenido de diclazurilo se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa y gradiente ternario, empleando un detector de UV.
3. Reactivos
|
3.1. |
Agua, equivalente a la de calidad CLAR. |
|
3.2. |
Acetato de amonio. |
|
3.3. |
Hidrogenosulfato de tetrabutilamonio. |
|
3.4. |
Acetonitrilo, equivalente al de calidad CLAR. |
|
3.5. |
Metanol, equivalente al de calidad CLAR. |
|
3.6. |
N, N-dimetilformamida (DMF). |
|
3.7. |
Ácido clorhídrico, ρ20 = 1,19 g/ml. |
|
3.8. |
Sustancia patrón: diclazurilo II-24, es decir, (+)-4-clorofenil [2,6-dicloro-4-(2,3,4,5-tetrahidro-3,5-dioxo-1,2,4-triazin-2-il)fenil]acetonitrilo de pureza garantizada, E 771. |
|
3.8.1. |
Pesar, con una precisión de 0,1 mg, 25 mg de sustancia patrón de diclazurilo (3.8) en un matraz aforado de 50 ml. Disolver en DMF (3.6), enrasar con DMF (3.6) y mezclar. Envolver el matraz con papel de aluminio, o utilizar un matraz ámbar, y guardarlo en el frigorífico. A una temperatura de ≤ 4 oC, la solución es estable durante un mes. |
|
3.8.2. |
Transvasar 5,00 ml de la solución patrón madre (3.8.1) a un matraz aforado de 50 ml, enrasar con DMF (3.6) y mezclar. Envolver el matraz con papel de aluminio, o utilizar un matraz ámbar, y guardarlo en el frigorífico. A una temperatura de ≤ 4 oC, la solución es estable durante un mes. |
|
3.9. |
Patrón interno: 2,6-dicloro-α-(4-clorofenil)-4-(4,5-dihidro-3,5-dioxo-1,2,4-triazina-2 (3H)-il)-α-metilbenceno-acetonitrilo. |
|
3.9.1. |
Pesar, con una precisión de 0,1 mg, 25 mg de patrón interno (3.9) en un matraz aforado de 50 ml. Disolver en DMF (3.6), enrasar con DMF (3.6) y mezclar. Envolver el matraz con papel de aluminio, o utilizar un matraz ámbar, y guardarlo en el frigorífico. A una temperatura de ≤ 4 oC, la solución es estable durante un mes. |
|
3.9.2. |
Transvasar 5,00 ml de la solución patrón madre de patrón interno (3.9.1) a un matraz aforado de 50 ml, enrasar con DMF (3.6) y mezclar. Envolver el matraz con papel de aluminio, o utilizar un matraz ámbar, y guardarlo en el frigorífico. A una temperatura de ≤ 4 oC, la solución es estable durante un mes. |
|
3.9.3. |
(p = contenido nominal de diclazurilo en la premezcla, en miligramos por kilogramo). Pesar, con una precisión de 0,1 mg, p/10 mg de patrón interno en un matraz aforado de 100 ml, disolver en DMF (3.6) en un baño ultrasónico (4.6), enrasar con DMF y mezclar. Envolver el matraz con papel de aluminio, o utilizar un matraz ámbar, y guardarlo en el frigorífico. A una temperatura de ≤ 4 oC, la solución es estable durante un mes. |
|
3.10. |
Solución de calibración de 2 μg/ml. Pipetear 2,00 ml de la solución patrón de diclazurilo (3.8.2) y 2,00 ml de la solución de patrón interno (3.9.2) a un matraz aforado de 50 ml. Añadir 16 ml de DMF (3.6), enrasar con agua y mezclar. Esta solución debe prepararse poco antes de utilizarse. |
|
3.11. |
Cartucho de extracción en fase sólida C18, por ejemplo Bond Elut; tamaño: 1 cc; peso del sorbente: 100 mg. |
|
3.12. |
Disolvente de extracción: metanol acidificado. Pipetear 5,0 ml de ácido clorhídrico (3.7) a 1 000 ml de metanol (3.5) y mezclar. |
|
3.13. |
Fase móvil para la CLAR. |
|
3.13.1. |
Eluyente A: solución de acetato de amonio e hidrogenosulfato de tetrabutilamonio. Disolver 5 g de acetato de amonio (3.2) y 3,4 g de hidrogenosulfato de tetrabutilamonio (3.3) en 1 000 ml de agua (3.1) y mezclar. |
|
3.13.2. |
Eluyente B: acetonitrilo (3.4). |
|
3.13.3. |
Eluyente C: metanol (3.5). |
4. Instrumental
|
4.1. |
Agitador mecánico. |
|
4.2. |
Equipo para CLAR de gradiente ternario. |
|
4.2.1. |
Columna cromatográfica de líquidos, Hypersil ODS, relleno de 3 μm, de 100 mm x 4,6 mm, o equivalente. |
|
4.2.2. |
Detector de UV de longitud de onda variable, o detector de red de diodos. |
|
4.3. |
Evaporador rotativo de película. |
|
4.4. |
Filtro de membrana de 0,45 μm. |
|
4.5. |
Colector de vacío. |
|
4.6. |
Baño ultrasónico. |
5. Procedimiento
5.1. Generalidades.
5.1.1.
Deberá analizarse un pienso en blanco para comprobar la ausencia de diclazurilo y de sustancias interferentes. El pienso en blanco deberá ser de tipo similar al de la muestra y en su análisis no deberá detectarse diclazurilo ni sustancias interferentes.
5.1.2.
Deberá realizarse un ensayo de recuperación analizando el pienso en blanco, que se habrá enriquecido añadiendo una cantidad de diclazurilo similar a la presente en la muestra. Para enriquecer a un nivel de 1 mg/kg, añadir 0,1 ml de la solución patrón madre (3.8.1) a 50 g del pienso en blanco, mezclar completamente y esperar diez minutos, volviendo a mezclar varias veces antes de proceder a la extracción (5.2).
Si no se dispone de un pienso en blanco de tipo similar al de la muestra (véase el punto 5.1.1), también puede realizarse un ensayo de recuperación por el método de adición de patrón. En este caso, la muestra que debe analizarse se enriquece con una cantidad de diclazurilo similar a la que ya esté presente en ella. Esta muestra se analiza junto con la muestra sin enriquecer, y la recuperación puede calcularse por sustracción.
5.2. Extracción.
5.2.1.
Pesar, con una precisión de 0,01 g, unos 50 g de la muestra. Pasarlos a un Erlenmeyer de 500 ml, añadir 1,00 ml de solución de patrón interno (3.9.2) y 200 ml de disolvente de extracción (3.12) y tapar el matraz. Agitar la mezcla en el agitador (4.1) hasta el día siguiente. Dejar reposar durante diez minutos. Transvasar una parte alícuota de 20 ml del sobrenadante a un recipiente de vidrio adecuado y diluir con 20 ml de agua. Transvasar esta solución a un cartucho de extracción (3.11) y pasarla a su través aplicando vacío (4.5). Lavar el cartucho con 25 ml de una mezcla de disolvente de extracción (3.12) y agua, 65 + 35 (V + V). Desechar las fracciones recogidas y eluir los compuestos con 25 ml de una mezcla de disolvente de extracción (3.12) y agua, 80 + 20 (V + V). Evaporar esta fracción hasta llegar justo a la sequedad mediante el evaporador rotativo (4.3) a 60 oC. Disolver el residuo en 1,0 ml de DMF (3.6), añadir 1,5 ml de agua (3.1) y mezclar. Filtrar a través de un filtro de membrana (4.4). Proceder a la determinación mediante CLAR (5.3).
5.2.2.
Pesar, con una precisión de 0,001 g, aproximadamente 1 g de la muestra. Pasarlo a un Erlenmeyer de 500 ml, añadir 1,00 ml de solución de patrón interno (3.9.3) y 200 ml de disolvente de extracción (3.12) y tapar el matraz. Agitar la mezcla en el agitador (4.1) hasta el día siguiente. Dejar reposar durante diez minutos. Pasar una parte alícuota de 10 000/p ml (p = contenido nominal de diclazurilo en la premezcla, en miligramos por kilogramo) del sobrenadante a un matraz de fondo redondo de tamaño adecuado. Evaporar hasta llegar justo a la sequedad a presión reducida y a 60 oC, por medio del evaporador rotativo (4.3). Volver a disolver el residuo en 10,0 ml de DMF (3.6), añadir 15,0 ml de agua (3.1) y mezclar. Proceder a la determinación mediante CLAR (5.3).
5.3. Determinación mediante CLAR.
5.3.1.
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
|
Columna cromatográfica de líquidos (4.2.1.): |
100 mm × 4,6 mm, Hypersil ODS, relleno de 3 μm, o equivalente |
|
||||||
|
Fase móvil: |
Eluyente A (3.13.1): |
Solución acuosa de acetato de amonio e hidrogenosulfato de tetrabutilamonio |
||||||
|
|
Eluyente B (3.13.2): |
Acetonitrilo |
||||||
|
|
Eluyente C (3.13.3): |
Metanol |
||||||
|
Modo de elución: |
Chorro de B durante diez minutos |
|||||||
|
Caudal: |
1,5-2 ml/min. |
|
||||||
|
Volumen de inyección: |
20 μl |
|
||||||
|
Longitud de onda del detector: |
280 nm |
|
||||||
Comprobar la estabilidad del sistema cromatográfico inyectando varias veces la solución de calibración (3.10) de 2 μg/ml, hasta que se alcancen alturas de pico y tiempos de retención constantes.
5.3.2.
Inyectar varias veces 20 μl de la solución de calibración (3.10) y determinar la altura (área) media de los picos de diclazurilo y patrón interno.
5.3.3.
Inyectar varias veces 20 μl de la solución de muestra (5.2.1 o 5.2.2) y determinar la altura (área) media de los picos de diclazurilo y patrón interno.
6. Cálculo de los resultados
6.1. Piensos.
El contenido p (mg/kg) de diclazurilo de la muestra viene dado por la siguiente fórmula:
 [mg/kg]
[mg/kg]
donde:
|
hd, s |
= |
altura (área) del pico de diclazurilo en la solución de muestra (5.2.1); |
|
hi, s |
= |
altura (área) del pico de patrón interno en la solución de muestra (5.2.1); |
|
hd, c |
= |
altura (área) del pico de diclazurilo en la solución de calibración (3.10); |
|
hi, c |
= |
altura (área) del pico de patrón interno en la solución de calibración (3.10); |
|
cd, c |
= |
concentración de diclazurilo en la solución de calibración (3.10), en microgramos por mililitro; |
|
m |
= |
peso de la porción de ensayo, en gramos; |
|
V |
= |
volumen del extracto de muestra según el punto 5.2.1 (es decir, 2,5 ml). |
6.2. Premezclas.
El contenido p (mg/kg) de diclazurilo de la muestra viene dado por la siguiente fórmula:
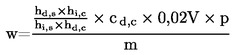 [mg/kg]
[mg/kg]
donde:
|
hd, c |
= |
altura (área) del pico de diclazurilo en la solución de calibración (3.10); |
|
hi, c |
= |
altura (área) del pico de patrón interno en la solución de calibración (3.10); |
|
hd, s |
= |
altura (área) del pico de diclazurilo en la solución de muestra (5.2.2); |
|
hi, s |
= |
altura (área) del pico de patrón interno en la solución de muestra (5.2.2); |
|
cd, c |
= |
concentración de diclazurilo en la solución de calibración (3.10), en microgramos por mililitro; |
|
m |
= |
peso de la porción de ensayo, en gramos; |
|
V |
= |
volumen del extracto de muestra según el punto 5.2.2 (es decir, 25 ml); |
|
p |
= |
contenido nominal de diclazurilo de la premezcla, en miligramos por kilogramo. |
7. Validación de los resultados
7.1. Identidad.
La identidad del analito puede confirmarse mediante cocromatografía, o utilizando un detector de red de diodos con el cual se comparan los espectros del extracto de muestra (5.2.1 o 5.2.2) y de la solución de calibración (3.10).
7.1.1.
Enriquecer un extracto de muestra (5.2.1 o 5.2.2) añadiéndole una cantidad apropiada de solución de calibración (3.10). La cantidad de diclazurilo añadida debe ser similar a la hallada en el extracto de muestra.
Solo la altura del pico de diclazurilo y del pico de patrón interno deberá aumentar en función de la cantidad añadida y de la dilución del extracto. La anchura del pico, a la mitad de su altura, debe ser igual ± 10 % a la anchura original del pico de diclazurilo o el pico de patrón interno del extracto de muestra sin enriquecer.
7.1.2.
Los resultados se evalúan con arreglo a los siguientes criterios:
|
a) |
la longitud de onda de absorción máxima de los espectros de la muestra y del patrón, registrados en el vértice del pico del cromatograma, debe ser la misma dentro de un margen determinado por el poder de resolución del sistema de detección; en el caso de la detección por red de diodos, el margen típico se sitúa en ± 2 nm; |
|
b) |
entre 230 nm y 320 nm, los espectros de la muestra y del patrón registrados en el vértice del pico del cromatograma no deben ser diferentes por lo que respecta a aquellas partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los dos espectros excede del 15 % de la absorbancia del analito patrón; |
|
c) |
entre 230 nm y 320 nm, los espectros de la pendiente ascendente, del vértice y de la pendiente descendente del pico producido por el extracto de muestra no deben diferir entre sí por lo que respecta a las partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los espectros excede del 15 % de la absorbancia del espectro del vértice del pico. |
Si no se cumple alguno de estos criterios, la presencia del analito no queda confirmada.
7.2. Repetibilidad.
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe exceder de:
|
— |
el 30 % del valor superior, en el caso de contenidos de diclazurilo de 0,5 mg/kg a 2,5 mg/kg, |
|
— |
0,75 mg/kg, en el caso de contenidos de diclazurilo de 2,5 mg/kg a 5 mg/kg, |
|
— |
el 15 % del valor superior, en el caso de contenidos de diclazurilo superiores a 5 mg/kg. |
7.3. Recuperación.
La recuperación de la muestra (en blanco) enriquecida deberá ser al menos del 80 %.
8. Resultados de un estudio colaborativo
Se organizó un estudio colaborativo en el que 11 laboratorios analizaron cinco muestras. Estas muestras consistieron en dos premezclas; una se mezcló con una matriz orgánica (O 100) y otra con una matriz inorgánica (A 100). El contenido teórico de diclazurilo fue de 100 mg por kilogramo. Los tres piensos mezclados para aves de corral fueron preparados por tres fabricantes diferentes (NL) (L1/Z1/K1). El contenido teórico de diclazurilo fue de 1 mg por kilogramo. Se dijo a los laboratorios que analizaran cada muestra una vez o por duplicado. (Puede encontrarse información más detallada sobre este estudio colaborativo en Journal of AOAC International, volumen 77, no 6, 1994, pp. 1359-1361). Los resultados se recogen en el cuadro siguiente:
|
|
Muestra 1 A 100 |
Muestra 2 O 100 |
Muestra 3 L1 |
Muestra 4 Z1 |
Muestra 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Media |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr (%) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR (%) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Contenido nominal (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
número de laboratorios |
|
n |
= |
número de valores individuales |
|
Sr |
= |
desviación típica de la repetibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad |
9. Observaciones
Previamente debe haberse demostrado que la respuesta del diclazurilo es lineal en todo el intervalo de concentraciones sometidas a medición.
G) DETERMINACIÓN DEL LASALOCID SÓDICO
Sal sódica de un poliéter de ácido monocarboxílico producido por Streptomyces lasaliensis
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de lasalocid sódico en piensos y premezclas. El límite de detección es de 5 mg/kg; el de cuantificación, de 10 mg/kg.
2. Principio
El lasalocid sódico se extrae de la muestra en metanol acidificado y se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa, empleando un detector espectrofluorométrico.
3. Reactivos
|
3.1. |
Dihidrogenofosfato de potasio (KH2PO4). |
|
3.2. |
Ácido ortofosfórico, p (p/p) = 85 %. |
|
3.3. |
Solución de ácido ortofosfórico, c = 20 %. Diluir 23,5 ml de ácido ortofosfórico (3.2) con agua hasta 100 ml. |
|
3.4. |
6-metil-2-heptilamina (1,5-dimetilhexilamina), p (p/p) = 99 %. |
|
3.5. |
Metanol, equivalente al de calidad CLAR. |
|
3.6. |
Ácido clorhídrico, de 1,19 g/ml de densidad. |
|
3.7. |
Solución reguladora de fosfato, c = 0,01 mol/l. Disolver 1,36 g de KH2PO4 (3.1) en 500 ml de agua (3.11), añadir 3,5 ml de ácido ortofosfórico (3.2) y 10,0 ml de 6-metil-2-heptilamina (3.4). Ajustar el pH a 4,0 con solución de ácido ortofosfórico (3.3) y diluir con agua (3.11) hasta 1 000 ml. |
|
3.8. |
Metanol acidificado. Transvasar 5,0 ml de ácido clorhídrico (3.6) a un matraz aforado de 1 000 ml, enrasar con metanol (3.5) y mezclar. Esta solución debe prepararse poco antes de utilizarse. |
|
3.9. |
Fase móvil para la CLAR: solución reguladora de fosfato y metanol, 5 + 95 (V + V). Mezclar 5 ml de solución reguladora de fosfato (3.7) con 95 ml de metanol (3.5). |
|
3.10. |
Sustancia patrón de lasalocid sódico de pureza garantizada, C34H53O8Na (sal sódica de un poliéter de ácido monocarboxílico producido por Streptomyces lasaliensis), E 763. |
|
3.10.1. |
Pesar, con una precisión de 0,1 mg, 50 mg de lasalocid sódico (3.10) en un matraz aforado de 100 ml, disolver con metanol acidificado (3.8), enrasar con este mismo disolvente y mezclar. Esta solución debe prepararse poco antes de utilizarse. |
|
3.10.2. |
Pipetear 10,0 ml de solución patrón madre (3.10.1) a un matraz aforado de 100 ml, enrasar con metanol acidificado (3.8) y mezclar. Esta solución debe prepararse poco antes de utilizarse. |
|
3.10.3. |
Transvasar 1,0, 2,0, 4,0, 5,0 y 10,0 ml de la solución patrón intermedia (3.10.2) a una serie de matraces aforados de 50 ml. Enrasar con metanol acidificado (3.8) y mezclar. Estas soluciones corresponden, respectivamente, a 1,0, 2,0, 4,0, 5,0 y 10,0 μg de lasalocid sódico por mililitro. Deben prepararse poco antes de utilizarse. |
|
3.11. |
Agua, equivalente a la de calidad CLAR. |
4. Instrumental
|
4.1. |
Baño ultrasónico (o baño de agua con agitación), con control de temperatura. |
|
4.2. |
Filtros de membrana de 0,45 μm. |
|
4.3. |
Equipo para CLAR con un sistema de inyección que permita inyectar volúmenes de 20 μl. |
|
4.3.1. |
Columna cromatográfica de líquidos, de 125 mm x 4 mm, fase reversa, C18, relleno de 5 μm, o equivalente. |
|
4.3.2. |
Espectrofluorímetro con ajuste variable de las longitudes de onda de excitación y emisión. |
5. Procedimiento
5.1. Generalidades.
5.1.1.
Para el ensayo de recuperación (5.1.2) deberá analizarse un pienso en blanco, a fin de comprobar la ausencia de lasalocid sódico y de sustancias interferentes. El pienso en blanco deberá ser de tipo similar al de la muestra y en su análisis no deberá detectarse lasalocid sódico ni sustancias interferentes.
5.1.2.
Deberá realizarse un ensayo de recuperación analizando el pienso en blanco, que se habrá enriquecido añadiendo una cantidad de lasalocid sódico similar a la presente en la muestra. Para enriquecer a un nivel de 100 mg/kg, transvasar 10,0 ml de la solución patrón madre (3.10.1) a un Erlenmeyer de 250 ml y evaporar la solución a unos 0,5 ml. Añadir 50 g del pienso en blanco, mezclar completamente y esperar diez minutos, mezclando varias veces más antes de proceder a la extracción (5.2).
Si no se dispone de un pienso en blanco de tipo similar al de la muestra (véase el punto 5.1.1), también puede realizarse un ensayo de recuperación por el método de adición de patrón. En este caso, la muestra que debe analizarse se enriquece con una cantidad de lasalocid sódico similar a la que ya esté presente en ella. Esta muestra se analiza junto con la muestra sin enriquecer, y la recuperación se calcula por sustracción.
5.2. Extracción.
5.2.1.
Pesar, con una precisión de 0,01 g, de 5 g a 10 g de la muestra en un Erlenmeyer de 250 ml con tapón. Añadir con la pipeta 100,0 ml de metanol acidificado (3.8). Tapar sin apretar y agitar en círculos para dispersar. Colocar el matraz en un baño ultrasónico (4.1) a unos 40 oC durante 20 minutos, remover a continuación y enfriar a temperatura ambiente. Dejar reposar durante aproximadamente una hora hasta que la materia en suspensión se haya sedimentado; a continuación, verter una parte alícuota en un recipiente adecuado filtrándola por un filtro de membrana de 0,45 μm. Proceder a la determinación mediante CLAR (5.3).
5.2.2.
Pesar, con una precisión de 0,001 g, unos 2 g de premezcla sin moler en un matraz aforado de 250 ml. Añadir 100,0 ml de metanol acidificado (3.8) y agitar en círculos para dispersar. Colocar el matraz con su contenido en un baño ultrasónico (4.1) a unos 40 oC durante 20 minutos, remover a continuación y enfriar a temperatura ambiente. Diluir hasta la marca con metanol acidificado (3.8) y mezclar completamente. Dejar reposar durante una hora hasta que la materia en suspensión se haya sedimentado; a continuación, filtrar una parte alícuota por un filtro de membrana de 0,45 μm (4.2). Diluir un volumen apropiado del filtrado limpio con metanol acidificado (3.8) para producir una solución final de ensayo que contenga en torno a 4 μm/ml de lasalocid sódico. Proceder a la determinación mediante CLAR (5.3).
5.3. Determinación mediante CLAR.
5.3.1.
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
|
Columna cromatográfica de líquidos (4.3.1): |
125 mm × 4 mm, fase reversa C18, relleno de 5 μm, o equivalente |
|
Fase móvil (3.9): |
Mezcla de solución reguladora de fosfato(3.7) y metanol (3.5), 5+95 (V+V) |
|
Caudal: |
1,2 ml/min. |
|
Longitudes de onda de detección: |
|
|
Excitación: |
10 nm |
|
Emisión: |
419 nm |
|
Volumen de inyección: |
20 μl |
Comprobar la estabilidad del sistema cromatográfico inyectando varias veces la solución de calibración (3.10.3) de 4,0 μg/ml, hasta que se alcancen alturas (o áreas) de pico y tiempos de retención constantes.
5.3.2.
Inyectar varias veces cada solución de calibración (3.10.3) y determinar las alturas (áreas) de pico medias de cada concentración. Trazar una curva de calibración empleando las alturas (áreas) de pico medias como ordenadas y las concentraciones correspondientes, en microgramos por mililitro, como abscisas.
5.3.3.
Inyectar varias veces los extractos de muestra obtenidos en el punto 5.2.1 o 5.2.2 empleando el mismo volumen que para la solución de calibración, y determinar las alturas (áreas) medias de los picos de lasalocid sódico.
6. Cálculo de los resultados
Determinar la concentración de lasalocid sódico (μg/ml) a partir de la altura (área) de pico media producida por la inyección de la solución de muestra (5.3.3), tomando como referencia la curva de calibración.
6.1. Piensos.
El contenido p (mg/kg) de lasalocid sódico de la muestra viene dado por la siguiente fórmula:
 [mg/kg]
[mg/kg]
donde:
|
c |
= |
concentración de lasalocid sódico de la solución de muestra (5.2.1), en microgramos por mililitro; |
|
V1 |
= |
volumen del extracto de muestra según el punto 5.2.1, en mililitros (es decir, 100); |
|
m |
= |
peso de la porción de ensayo, en gramos. |
6.2. Premezclas.
El contenido p (mg/kg) de lasalocid sódico de la muestra viene dado por la siguiente fórmula:
 [mg/kg]
[mg/kg]
donde:
|
c |
= |
concentración de lasalocid sódico de la solución de muestra (5.2.2), en microgramos por mililitro; |
|
V2 |
= |
volumen del extracto de muestra según el punto 5.2.2, en mililitros (es decir, 250); |
|
f |
= |
factor de dilución según el punto 5.2.2; |
|
m |
= |
peso de la porción de ensayo, en gramos. |
7. Validación de los resultados
7.1. Identidad.
Los métodos basados en la espectrofluorometría están menos sujetos a interferencias que aquellos en los que se emplea la detección de UV. La identidad del analito puede confirmarse por cocromatografía.
7.1.1.
Enriquecer un extracto de muestra (5.2.1 o 5.2.2) añadiéndole una cantidad apropiada de solución de calibración (3.10.3). La cantidad de lasalocid sódico añadida debe ser similar a la hallada en el extracto de muestra. Solo la altura del pico de lasalocid sódico deberá aumentar en función de la cantidad de lasalocid sódico añadida y de la dilución del extracto. La anchura del pico, a la mitad de su altura, debe ser igual ± 10 % a la anchura del pico original producida por el extracto de muestra sin enriquecer.
7.2. Repetibilidad.
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe exceder de:
|
— |
el 15 % del valor superior, en el caso de contenidos de lasalocid sódico de 30 mg/kg a 100 mg/kg, |
|
— |
15 mg/kg, en el caso de contenidos de lasalocid sódico de 100 mg/kg a 200 mg/kg, |
|
— |
el 7,5 % del valor superior, en el caso de contenidos de lasalocid sódico superiores a 200 mg/kg. |
7.3. Recuperación.
La recuperación de la muestra (en blanco) de pienso enriquecida deberá ser al menos del 80 %. En el caso de las muestras de premezclas enriquecidas, la recuperación deberá ser al menos del 90 %.
8. Resultados de un estudio colaborativo
Se organizó un estudio colaborativo (6) en el que 12 laboratorios analizaron dos premezclas (muestras 1 y 2) y cinco piensos (muestras 3 a 7). De cada muestra se hicieron análisis por duplicado. Los resultados se recogen en el cuadro siguiente:
|
|
Muestra 1 Premezcla para pollos |
Muestra 2 Premezcla para pavos |
Muestra 3 Gránulos para pavos |
Muestra 4 Migajas para pollos |
Muestra 5 Pienso para pavos |
Muestra 6 Pienso A para pollos |
Muestra 7 Pienso B para pollos |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Media [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
Sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
SR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Contenido nominal [mg/kg] |
5 000 (7) |
16 000 (7) |
80 (7) |
105 (7) |
120 (7) |
50 (8) |
35 (8) |
|
L |
= |
número de laboratorios |
|
n |
= |
número de resultados individuales |
|
Sr |
= |
desviación típica de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad, en tanto por ciento |
|
CVR |
= |
coeficiente de variación de la reproducibilidad, en tanto por ciento |
(1) Realizado por el Grupo de Trabajo sobre Piensos de la Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(2) Realizado por el Grupo de Trabajo sobre Piensos de la Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(3) Pueden emplearse otros métodos de digestión, siempre que se haya demostrado que ofrecen resultados similares (por ejemplo, digestión a presión por microondas).
(4) El forraje verde (fresco o desecado) puede contener grandes cantidades de sílice vegetal, que puede retener oligolementos y debe eliminarse. Por tanto, con las muestras de estos piensos debe aplicarse el procedimiento modificado que sigue. Efectuar la operación del punto 5.1.1.1, hasta la filtración. Lavar el papel de filtro que contiene el residuo insoluble dos veces con agua hirviendo e introducirlo en un crisol de cuarzo o platino. Calcinar en el horno de mufla (4.1) a una temperatura por debajo de 550 oC hasta que haya desaparecido por completo todo el material carbonoso. Dejar enfriar, añadir unas pocas gotas de agua y, a continuación, de 10 ml a 15 ml de ácido fluorhídrico (3.4), evaporando después hasta sequedad a unos 150 oC. Si el residuo sigue conteniendo sílice, volver a disolverlo en unos pocos mililitros de ácido fluorhídrico (3.4) y evaporar hasta sequedad. Añadir cinco gotas de ácido sulfúrico (3.5) y calentar hasta que dejen de desprenderse vapores. Tras añadir 5 ml de ácido clorhídrico de 6 mol/l (3.2) y unos 30 ml de agua, calentar, filtrar la solución en el matraz aforado de 250 ml y enrasar con agua (concentración de HCl: 0,5 mol/l aproximadamente). Proseguir entonces con la determinación a partir del punto 5.1.2.
(5) The Analyst 108, 1983, pp. 1252-1256.
(6) The Analyst, 1995, 120, pp. 2175-2180
(7) contenido declarado por el fabricante
(8) pienso preparado en el laboratorio
ANEXO V
MÉTODOS DE ANÁLISIS PARA EL CONTROL DE SUSTANCIAS INDESEABLES EN LOS PIENSOS
A. DETERMINACIÓN DEL GOSIPOL LIBRE Y TOTAL
1. Objeto y ámbito de aplicación
Este método permite determinar el gosipol libre, el gosipol total y las sustancias químicamente relacionadas presentes en las semillas de algodón y en las sémolas y tortas de semillas de algodón, así como en los piensos compuestos que contengan estos materiales para piensos, cuando los contenidos de gosipol libre, gosipol total y sustancias químicamente relacionadas superen los 20 mg/kg.
2. Principio
El gosipol se extrae en presencia de 3-aminopropan-1-ol, ya sea mediante una mezcla de propan-2-ol y hexano, para la determinación del gosipol libre, ya con dimetilformamida, para la determinación del gosipol total. El gosipol se transforma mediante anilina en gosipol-dianilina, cuya densidad óptica se mide a 440 nm.
3. Reactivos
|
3.1. |
Mezcla de propan-2-ol y hexano: mezclar 60 partes en volumen de propan-2-ol con 40 partes en volumen de n-hexano. |
|
3.2. |
Disolvente A: verter en un matraz aforado de 1 l unos 500 ml de la mezcla de propan-2-ol y hexano (3.1), 2 ml de 3-aminopropan-1-ol, 8 ml de ácido acético glacial y 50 ml de agua. Enrasar con la mezcla de propan-2-ol y hexano (3.1). Este reactivo se mantiene estable durante una semana. |
|
3.3. |
Disolvente B: pipetear 2 ml de 3-aminopropan-1-ol y 10 ml de ácido acético glacial a un matraz aforado de 100 ml. Enfriar a temperatura ambiente y enrasar con N, N-dimetilformamida. Este reactivo se mantiene estable durante una semana. |
|
3.4. |
Anilina: si la densidad óptica del ensayo en blanco excede de 0,022, destilar la anilina sobre polvo de cinc, desechando la primera y la última fracción del destilado, cada una de ellas equivalente a un 10 % del mismo. Este reactivo se conserva varios meses si está refrigerado y se guarda en un matraz de vidrio oscuro tapado. |
|
3.5. |
Solución patrón de gosipol A: introducir 27,9 mg de acetato de gosipol en un matraz aforado de 250 ml. Disolver y enrasar con el disolvente A (3.2). Pipetear 50 ml de esta solución a un matraz aforado de 250 ml y enrasar con el disolvente A. La concentración de gosipol de esta solución es de 0,02 mg/ml. Dejarla reposar durante una hora a temperatura ambiente antes de utilizarla. |
|
3.6. |
Solución patrón de gosipol B: introducir 27,9 mg de acetato de gosipol en un matraz aforado de 50 ml. Disolver y enrasar con el disolvente B (3.3). La concentración de gosipol de esta solución es de 0,5 mg/ml. Las soluciones patrón de gosipol A y B se mantendrán estables durante veinticuatro horas si están al abrigo de la luz. |
4. Instrumental
|
4.1. |
Mezclador (tambor): aproximadamente 35 revoluciones por minuto. |
|
4.2. |
Espectrofotómetro. |
5. Procedimiento
5.1. Muestra de ensayo
La cantidad de muestra de ensayo empleada depende del contenido supuesto de gosipol de la muestra. Es preferible trabajar con una muestra de ensayo pequeña y una parte alícuota del filtrado relativamente grande, de forma que se obtenga una cantidad de gosipol suficiente para poder efectuar una medición fotométrica precisa. Para la determinación del gosipol libre en las semillas de algodón y en las sémolas y tortas de semillas de algodón, la muestra de ensayo no excederá de 1 g; en el caso de los piensos compuestos, puede llegar a 5 g. En la mayoría de los casos, una parte alícuota de 10 ml de filtrado es adecuada; deberá contener de 50 μg a 100 μg de gosipol. Para la determinación del gosipol total, la muestra de ensayo deberá ser de 0,5 g a 5 g, de modo que una parte alícuota de 2 ml de filtrado contenga de 40 μg a 200 μg de gosipol.
El análisis deberá efectuarse a una temperatura ambiente en torno a los 20 oC.
5.2. Determinación del gosipol libre
Introducir la muestra en un matraz de cuello esmerilado de 250 ml, cuyo fondo esté cubierto de vidrio triturado. Añadir con la pipeta 50 ml de disolvente A (3.2), tapar el matraz y mezclar durante una hora en el mezclador. Filtrar por un filtro seco y recoger el filtrado en un matraz pequeño de cuello esmerilado. Durante la filtración, cubrir el embudo con un vidrio de reloj.
Pipetear a dos matraces aforados de 25 ml (A y B) partes alícuotas idénticas de filtrado que contengan de 50 μg a 100 μg de gosipol. Si es necesario, enrasar a 10 ml con disolvente A (3.2). A continuación, enrasar el matraz (A) con la mezcla de propan-2-ol y hexano (3.1). Esta solución servirá de referencia para medir la solución de muestra.
Pipetear a otros dos matraces aforados de 25 ml (C y D), respectivamente, 10 ml de disolvente A (3.2). Enrasar el matraz (C) con la mezcla de propan-2-ol y hexano (3.1). Esta solución servirá de referencia para medir la solución de ensayo en blanco.
Añadir 2 ml de anilina (3.4) al matraz (D) y al matraz (B). Calentar durante 30 minutos sobre un baño maría en ebullición para colorar. Enfriar a temperatura ambiente, enrasar con la mezcla de propan-2-ol y hexano (3.1), homogeneizar y dejar reposar durante una hora.
Determinar las densidades ópticas de la solución de ensayo en blanco (D) y de la solución de muestra (B) comparándolas, respectivamente, con la solución de referencia (C) y la solución de referencia (A) en el espectrofotómetro a 440 nm, empleando cubetas de vidrio de 1 cm.
Restar la densidad óptica de la solución de ensayo en blanco a la de la solución de muestra (= densidad óptica corregida). Partiendo de este valor, calcular el contenido de gosipol libre como se indica en el punto 6.
5.3. Determinación del gosipol total
Introducir una muestra de ensayo que contenga de 1 mg a 5 mg de gosipol en un matraz aforado de 50 ml y añadir 10 ml de disolvente B (3.3). Preparar simultáneamente un ensayo en blanco vertiendo 10 ml de disolvente B (3.3) en otro matraz aforado de 50 ml. Calentar ambos matraces durante 30 minutos sobre un baño maría en ebullición. Enfriar a temperatura ambiente y enrasar los dos matraces con la mezcla de propan-2-ol y hexano (3.1). Homogeneizar y dejar reposar durante diez a 15 minutos, filtrando a continuación y recogiendo los filtrados en matraces de cuello esmerilado.
Pipetear a dos matraces aforados de 25 ml, respectivamente, 2 ml del filtrado de muestra, y a otros dos matraces de 25 ml, respectivamente, 2 ml del filtrado de ensayo en blanco. Enrasar a 25 ml un matraz de cada serie con la mezcla de propan-2-ol y hexano (3.1). Estas soluciones se emplearán como soluciones de referencia.
Añadir 2 ml de anilina (3.4) a cada uno de los otros dos matraces. Calentar durante 30 minutos sobre un baño maría en ebullición para colorar. Enfriar a temperatura ambiente, enrasar a 25 ml con la mezcla de propan-2-ol y hexano (3.1), homogeneizar y dejar reposar durante una hora.
Determinar la densidad óptica como se indica en el punto 5.2 para el gosipol libre. Partiendo de este valor, calcular el contenido de gosipol total como se indica en el punto 6.
6. Cálculo de los resultados
Los resultados pueden calcularse, o bien a partir de la densidad óptica específica (6.1), o bien tomando como referencia una curva de calibración (6.2).
6.1. A partir de la densidad óptica específica
Las densidades ópticas específicas, en las condiciones descritas, serán las siguientes:
|
Gosipol libre |
|
|
Gosipol total |
|
El contenido de gosipol libre o total de la muestra se calcula por medio de la siguiente fórmula:

donde
|
E |
= |
densidad óptica corregida, determinada según se indica en el punto 5.2; |
|
p |
= |
muestra de ensayo, en gramos; |
|
a |
= |
parte alícuota del filtrado, en mililitros. |
6.2. A partir de una curva de calibración
6.2.1.
Preparar dos series de cinco matraces aforados de 25 ml. Pipetear partes alícuotas de 2,0, 4,0, 6,0, 8,0 y 10,0 ml de la solución patrón de gosipol A (3.5) en cada serie de matraces. Enrasar a 10 ml con el disolvente A (3.2). Completar cada serie con un matraz aforado de 25 ml que contenga únicamente 10 ml del disolvente A (3.2) (ensayo en blanco).
Enrasar a 25 ml los matraces de la primera serie (incluido el matraz para el ensayo en blanco) con la mezcla de propan-2-ol y hexano (3.1) (serie de referencia).
Añadir 2 ml de anilina (3.4) a cada uno de los matraces de la segunda serie (incluido el matraz para el ensayo en blanco). Calentar durante 30 minutos sobre un baño maría en ebullición para colorar. Enfriar a temperatura ambiente, enrasar con la mezcla de propan-2-ol y hexano (3.1), homogeneizar y dejar reposar durante una hora (serie de referencia).
Determinar la densidad óptica de las soluciones de la serie patrón como se indica en el punto 5.2, comparándola con las correspondientes soluciones de la serie de referencia. Trazar la curva de calibración relacionando las densidades ópticas con las cantidades de gosipol (en microgramos).
6.2.2.
Preparar seis matraces aforados de 50 ml. Verter 10 ml del disolvente B (3.3) en el primer matraz y, en el resto, 2,0, 4,0, 6,0, 8,0 y 10,0 ml, respectivamente, de solución patrón de gosipol B (3.6). Enrasar cada matraz a 10 ml con disolvente B (3.3). Calentar durante 30 minutos sobre un baño maría en ebullición. Enfriar a temperatura ambiente, enrasar con la mezcla de propan-2-ol y hexano (3.1) y homogeneizar.
Verter respectivamente 2,0 ml de estas soluciones en dos series de seis matraces aforados de 25 ml. Enrasar los matraces de la primera serie a 25 ml con la mezcla de propan-2-ol y hexano (3.1) (serie de referencia).
Añadir 2 ml de anilina (3.4) a cada matraz de la segunda serie. Calentar durante 30 minutos sobre un baño maría en ebullición. Enfriar a temperatura ambiente, enrasar con la mezcla de propan-2-ol y hexano (3.1), homogeneizar y dejar reposar durante una hora (serie de referencia).
Determinar la densidad óptica de las soluciones de la serie patrón como se indica en el punto 5.2, comparándola con las correspondientes soluciones de la serie de referencia. Trazar la curva de calibración relacionando las densidades ópticas con las cantidades de gosipol (en microgramos).
6.3. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe exceder de:
|
— |
el 15 % del valor superior, en el caso de contenidos de gosipol inferiores a 500 ppm, |
|
— |
75 ppm en valor absoluto, en el caso de contenidos no inferiores a 500 ppm ni superiores a 750 ppm, |
|
— |
el 10 % del valor superior, en el caso de contenidos superiores a 750 ppm. |
B. DETERMINACIÓN DE LOS NIVELES DE DIOXINAS (PCDD/PCDF) Y PCB SIMILARES A LAS DIOXINAS
I. MÉTODOS DE MUESTREO E INTERPRETACIÓN DE LOS RESULTADOS ANALÍTICOS
1. Objeto y ámbito de aplicación
Las muestras destinadas al control oficial de los niveles de dioxinas (dibenzo-p-dioxinas policloradas [PCDD] y dibenzofuranos policlorados [PCDF]) y bifenilos policlorados similares a las dioxinas (PCB) (1) en los piensos se tomarán conforme a las disposiciones del anexo I. Son de aplicación los requisitos cuantitativos relacionados con el control de las sustancias o los productos distribuidos uniformemente por el pienso que se establecen en el punto 5.A del anexo I. Las muestras globales así obtenidas se considerarán representativas de los lotes o sublotes de los que se tomen. El cumplimiento de los niveles máximos establecidos en la Directiva 2002/32/CE del Parlamento Europeo y del Consejo (2) se establecerá sobre la base de los niveles determinados en las muestras de laboratorio.
2. Conformidad del lote o sublote con la especificación
El lote es aceptado si el resultado analítico de un análisis único no supera el nivel máximo correspondiente fijado en la Directiva 2002/32/CE, teniendo en cuenta la incertidumbre de medida.
Se considera que el lote incumple el nivel máximo establecido en la Directiva 2002/32/CE si el resultado analítico del límite superior (3), confirmado por el análisis por duplicado (4), supera el nivel máximo más allá de cualquier duda razonable, teniendo en cuenta la incertidumbre de medida.
La incertidumbre de medida puede tenerse en cuenta con arreglo a uno de los siguientes métodos:
|
— |
calculando la incertidumbre expandida con un factor de cobertura de 2, que ofrece un nivel de confianza del 95 % aproximadamente; un lote no es conforme si el valor medido menos U está por encima del nivel máximo; en caso de que se determinen por separado las dioxinas y los PCB similares a las dioxinas, para la suma de ambos debe emplearse la suma de la incertidumbre expandida calculada correspondiente a los resultados analíticos obtenidos por separado para las dioxinas y los PCB similares a las dioxinas, |
|
— |
estableciendo el límite de decisión (CCα) con arreglo a la Decisión 2002/657/CE de la Comisión (5) (punto 3.1.2.5 del anexo, en el caso de sustancias para las que se ha establecido un límite permitido); un lote no es conforme si el valor medido es igual o superior al CCα. |
Las presentes normas de interpretación se aplican al resultado analítico obtenido con la muestra para control oficial. No afectan al derecho de los Estados miembros de aplicar normas nacionales a los análisis con fines de defensa o arbitraje.
II. PREPARACIÓN DE LAS MUESTRAS Y REQUISITOS APLICABLES A LOS MÉTODOS DE ANÁLISIS UTILIZADOS EN EL CONTROL OFICIAL DE LOS NIVELES DE DIOXINAS (PCDD/PCDF) Y PCB SIMILARES A LAS DIOXINAS
1. Objeto y ámbito de aplicación
Estos requisitos deberán aplicarse en el análisis de piensos y materiales para piensos realizado para la determinación de dioxinas [dibenzo-p-dioxinas policloradas (PCDD) y dibenzofuranos policlorados (PCDF)] y policlorobifenilos similares a las dioxinas (PCB).
El control de la presencia de dioxinas en los piensos puede efectuarse mediante una estrategia que incluya un método de cribado para seleccionar aquellas muestras cuyo nivel de dioxinas y PCB similares a las dioxinas sea menos de un 25 % inferior al nivel considerado o exceda de dicho nivel. La concentración de dioxinas en esas muestras con niveles significativos debe determinarse o confirmarse mediante un método de confirmación.
Los métodos de cribado son los que se utilizan para detectar la presencia de dioxinas y PCB similares a las dioxinas con el nivel considerado. Estos métodos se caracterizan por un elevado rendimiento de muestras y se utilizan para cribar grandes cantidades de muestras en busca de posibles positivos. Están específicamente diseñados para evitar los falsos negativos.
Son métodos de confirmación los que proporcionan una información completa o complementaria que permite la identificación y cuantificación inequívoca de las dioxinas y los PCB similares a las dioxinas con el nivel considerado.
2. Antecedentes
Ya que las muestras ambientales y biológicas (incluidas las muestras de piensos o materiales para piensos) contienen, por lo general, mezclas complejas de diferentes congéneres de dioxinas, se ha desarrollado el concepto de factores de equivalencia tóxica (FET) para facilitar la evaluación de los riesgos. Estos FET se han establecido para expresar concentraciones de mezclas de PCDD y PCDF sustituidos en las posiciones 2, 3, 7 y 8 y de algunos PCB no-ortosustituidos y mono-ortosustituidos con actividad similar a las dioxinas en equivalentes tóxicos (EQT) de 2,3,7,8-TCDD. Las concentraciones de cada sustancia en una muestra dada se multiplican por sus respectivos FET y se suman a continuación para obtener la concentración total de compuestos similares a las dioxinas, expresada en EQT.
Únicamente a los efectos del presente Reglamento, el límite de cuantificación específico aceptado de un congénere individual es la concentración de un analito en el extracto de una muestra que produce una respuesta instrumental a dos iones diferentes que debe controlarse con una relación señal/ruido (S/R) de 3/1 para la señal menos sensible, y que cumple requisitos básicos tales como, por ejemplo, el tiempo de retención y la relación isotópica con arreglo al procedimiento de determinación descrito en el método EPA 1613, revisión B.
3. Requisitos de aseguramiento de la calidad que han de cumplirse en la preparación de las muestras
Son de aplicación las disposiciones generales sobre preparación de las muestras para análisis que se establecen en el anexo II.
Deben cumplirse, además, los siguientes requisitos:
|
— |
las muestras deben almacenarse y transportarse en recipientes de vidrio, aluminio, polipropileno o polietileno. Deben eliminarse del recipiente que contiene la muestra los restos de polvo de papel. Los recipientes de vidrio deberán enjuagarse con disolventes previamente sometidos a un control de detección de dioxinas, |
|
— |
debe efectuarse un análisis en blanco realizando todo el procedimiento analítico, únicamente sin la muestra, |
|
— |
el peso de la muestra utilizada para la extracción debe ser suficiente para que se cumplan los requisitos relativos a la sensibilidad. |
4. Requisitos que deben cumplir los laboratorios
|
— |
Los laboratorios deberán demostrar la eficacia de un método en el intervalo del nivel considerado, por ejemplo 0,5, una y dos veces el nivel considerado, con un coeficiente de variación aceptable para análisis repetidos. Por lo que se refiere a los criterios de aceptación, véase el punto 5. |
|
— |
El límite de cuantificación en un método de confirmación deberá situarse en un intervalo de aproximadamente un quinto del nivel considerado, a fin de garantizar coeficientes de variación aceptables en el intervalo de dicho nivel. |
|
— |
Como medidas internas de control de calidad, deberán realizarse regularmente controles en blanco y experimentos con muestras enriquecidas o análisis de muestras de control (de preferencia, si existe, material de referencia certificado). |
|
— |
La participación con éxito en estudios interlaboratorios, que evalúan la aptitud del laboratorio, es la mejor manera de demostrar su competencia para efectuar análisis específicos. No obstante, la participación con éxito en estudios interlaboratorios con muestras, por ejemplo, de suelos o de aguas residuales, no demuestra necesariamente la competencia en relación con muestras de alimentos o de piensos, que presentan niveles de contaminación más bajos. Por tanto, es obligatoria la participación continua en estudios interlaboratorios para la determinación de dioxinas y PCB similares a las dioxinas en las matrices pertinentes de piensos o alimentos. |
|
— |
Los laboratorios deberán estar acreditados por un organismo reconocido que opere de conformidad con la Guía ISO 58, para asegurarse de que llevan a cabo el aseguramiento de la calidad analítica. Dicha acreditación deberá efectuarse conforme a la norma ISO/IEC/17025. |
5. Requisitos aplicables a los procedimientos analíticos para dioxinas y PCB similares a las dioxinas
Requisitos básicos de aceptación de los procedimientos analíticos:
|
— |
Sensibilidad elevada y límites de detección bajos. En el caso de las PCDD y los PCDF, los umbrales de detección deben situarse en el intervalo de picogramos de EQT (10-12 g), habida cuenta de la extrema toxicidad de algunos de estos compuestos. Se sabe que los PCB se presentan en cantidades más elevadas que las PCDD y los PCDF. Para la mayoría de los congéneres de PCB, es suficiente una sensibilidad en el intervalo de nanogramos (10-9 g). No obstante, para medir los congéneres de PCB similares a las dioxinas más tóxicos (en particular, los congéneres no-ortosustituidos), es preciso conseguir la misma sensibilidad que para las PCDD y los PCDF. |
|
— |
Selectividad elevada (especificidad). Es necesario distinguir las PCDD, los PCDF y los PCB similares a las dioxinas de una multitud de otros compuestos extraídos simultáneamente de la muestra, que posiblemente interfieran y que están presentes en concentraciones de hasta varios órdenes de magnitud superiores a las de los analitos considerados. Por lo que respecta a los métodos de cromatografía de gases/espectrometría de masas (CG/EM), es necesario distinguir entre varios congéneres, en particular entre los tóxicos (por ejemplo, los 17 PCDD y PCDF sustituidos en las posiciones 2, 3, 7 y 8 y los PCB similares a las dioxinas) y otros. Los bioensayos deben permitir una determinación selectiva de los valores EQT como suma de PCDD, PCDF y PCB similares a las dioxinas. |
|
— |
Exactitud elevada (veracidad y precisión). La determinación deberá proporcionar una estimación válida y fiable de la concentración real en una muestra. A fin de evitar que el resultado del análisis de una muestra sea rechazado debido a la escasa fiabilidad de la estimación de EQT, es necesario lograr un alto grado de exactitud (exactitud de la medición: grado de concordancia entre el resultado de la medición y el valor real o atribuido del mensurando). La exactitud se expresa como veracidad (diferencia entre el valor medio medido de un analito en un material certificado y su valor certificado, expresado como porcentaje de este valor) y precisión (desviación típica relativa, RSDR, calculada a partir de los resultados obtenidos en condiciones de reproducibilidad). |
Los métodos de cribado pueden incluir bioensayos y métodos CG/EM; los métodos de confirmación son métodos de cromatografía de gases de alta resolución/espectrometría de masas de alta resolución (CGAR/EMAR).
Deben cumplirse los siguientes criterios con respecto al valor de EQT total:
|
|
Métodos de cribado |
Métodos de confirmación |
|
Porcentaje de falsos negativos |
< 1 % |
|
|
Veracidad |
|
20 % a + 20 % |
|
Precisión RSDR |
< 30 % |
< 15 % |
6. Requisitos específicos que deben cumplir los métodos cg/em utilizados con fines de cribado o de confirmación
|
— |
A fin de validar el procedimiento analítico, deben añadirse, desde el mismo comienzo del método analítico —por ejemplo, antes de la extracción—, patrones internos de PCDD/F clorosustituidos en las posiciones 2, 3, 7 y 8 y marcados con 13C, así como patrones internos de PCB similares a las dioxinas marcados con 13C. Debe añadirse al menos un congénere por cada grupo homólogo tetra a octoclorado de PCDD/F y al menos un congénere por cada grupo homólogo de PCB similares a las dioxinas (otra posibilidad es añadir al menos un congénere por cada función de registro de iones seleccionada por espectrometría de masas y utilizada para el control de PCDD/F y PCB similares a las dioxinas). Se utilizará preferiblemente, y por supuesto en los métodos de confirmación, el conjunto de los diecisiete patrones internos de PCDD/F sustituidos en las posiciones 2, 3, 7 y 8 y marcados con 13C, así como los 12 patrones internos de PCB similares a las dioxinas marcados con 13C. |
|
— |
Deberán determinarse asimismo factores de respuesta relativos en el caso de los congéneres para los que no se añade ningún análogo marcado con 13C, empleando las soluciones de calibración apropiadas. |
|
— |
Para los piensos de origen vegetal y de origen animal con un contenido de grasa inferior al 10 %, es obligatorio añadir patrones internos antes de proceder a la extracción. Por lo que respecta a los piensos de origen animal con un contenido de grasa superior al 10 %, los patrones internos pueden añadirse antes de la extracción o después de la extracción de grasas. Deberá validarse adecuadamente la eficacia de la extracción, en función de la fase en la que se introduzcan los patrones internos y de si los resultados notificados se refieren al producto o a las grasas. |
|
— |
Con anterioridad al análisis mediante CG/EM, deben añadirse uno o dos patrones de recuperación (sustitutos). |
|
— |
Es preciso realizar un control de la recuperación. En los métodos de confirmación, los porcentajes de recuperación de cada patrón interno deben situarse en un intervalo del 60 % al 120 %. Se pueden aceptar porcentajes de recuperación inferiores o superiores para congéneres concretos, en particular en relación con algunas dibenzodioxinas y algunos dibenzofuranos hepta y octoclorados, siempre y cuando su contribución al valor de EQT no supere el 10 % del valor total (basado en la suma de PCDD/F y PCB similares a las dioxinas). En los métodos de cribado, los porcentajes de recuperación deben situarse en un intervalo del 30 % al 140 %. |
|
— |
Las dioxinas deberán separarse de los compuestos clorados interferentes, como son los PCB no similares a las dioxinas y los éteres difenílicos clorados, mediante técnicas de cromatografía adecuadas (de preferencia con una columna de florisil, alúmina o carbono, o de varias de estas sustancias). |
|
— |
La separación de los isómeros por cromatografía de gases será suficiente (< 25 % de pico a pico entre 1,2,3,4,7,8-HxCDF y 1,2,3,6,7,8-HxCDF). |
|
— |
La determinación deberá realizarse con arreglo al método EPA 1613, revisión B: Tetra- through octa-chlorinated dioxins and furans by isotope dilution HRGC/HRMS (Dioxinas y furanos tetra a octoclorados por dilución isotópica con CGAR/EMAR), u otro método con criterios de rendimiento equivalentes. |
|
— |
La diferencia entre el límite superior y el límite inferior no debe exceder del 20 % en los piensos cuya contaminación por dioxinas esté próxima o sea superior al nivel máximo. En el caso de los piensos con niveles de contaminación muy inferiores al nivel máximo, la diferencia puede ser del 25 % al 40 %. |
7. Métodos analíticos de cribado
7.1. Introducción
En un método de cribado es posible aplicar distintos enfoques analíticos: un enfoque puramente de cribado y un enfoque cuantitativo.
La respuesta de las muestras se compara con la de una muestra de referencia en el nivel considerado. Las muestras cuya respuesta es inferior a la de la muestra de referencia se declaran negativas, mientras que las muestras cuya respuesta es superior se consideran supuestamente positivas. Requisitos:
|
— |
en cada serie de ensayos deben utilizarse una muestra en blanco y una o varias muestras de referencia, extraídas y analizadas al mismo tiempo y en condiciones idénticas. La respuesta de la muestra de referencia debe ser claramente superior a la del blanco, |
|
— |
deberán incluirse otras muestras de referencia con una respuesta 0,5 y dos veces mayor que el nivel considerado, a fin de demostrar la eficacia del ensayo en el intervalo de interés para el control del nivel considerado, |
|
— |
cuando se analicen otras matrices, debe demostrarse la validez de las muestras de referencia, de preferencia utilizando muestras cuyo nivel de EQT, determinado mediante CGAR/EMAR, sea similar, aproximadamente, al de la muestra de referencia o, de lo contrario, un blanco enriquecido hasta ese nivel, |
|
— |
puesto que en los bioensayos no pueden utilizarse patrones internos, los ensayos de repetibilidad son muy importantes para obtener información sobre la desviación típica en una serie de ensayos. El coeficiente de variación debe ser inferior al 30 %, |
|
— |
en el caso de los bioensayos, deberán definirse los compuestos de interés, las posibles interferencias y los niveles máximos tolerables de blanco. |
El enfoque cuantitativo exige series de diluciones patrón, procesos de limpieza y medición dobles o triples, así como controles en blanco y de recuperación. El resultado puede expresarse en EQT, suponiendo en tal caso que los compuestos responsables de la señal cumplen el principio de EQT. Para ello, puede utilizarse la TCDD (o una mezcla patrón de dioxinas/furanos/PCB similares a las dioxinas) a fin de producir una curva de calibración que permita calcular el nivel de EQT en el extracto y, por tanto, en la muestra. Posteriormente, este resultado se corrige con respecto al nivel de EQT calculado para una muestra en blanco (a fin de tener en cuenta las impurezas procedentes de los disolventes o productos químicos utilizados) y una recuperación (calculada a partir del nivel de EQT en una muestra de control de calidad próxima al límite considerado). Es fundamental tener en cuenta que parte de la pérdida de recuperación aparente puede deberse a efectos matriciales o a las diferencias entre los valores de FET de los bioensayos y los valores oficiales de FET fijados por la OMS.
7.2. Requisitos aplicables a los métodos analíticos de cribado
|
— |
El cribado puede realizarse utilizando métodos analíticos de CG/EM o bioensayos. Para los métodos de CG/EM deben aplicarse los criterios establecidos en el punto 6. Por lo que se refiere a los bioensayos celulares y los bioensayos realizados con kits, los requisitos específicos aplicables figuran, respectivamente, en los puntos 7.3 y 7.4. |
|
— |
Es necesario proporcionar información sobre el número de resultados falsos positivos y falsos negativos obtenidos en una amplia serie de muestras por debajo y por encima del nivel máximo o umbral de intervención, en comparación con el contenido de EQT determinado mediante un método analítico de confirmación. Los porcentajes reales de falsos negativos deben ser inferiores al 1 %. La tasa de falsas muestras positivas deberá ser lo suficientemente baja para que la herramienta de cribado resulte eficaz. |
|
— |
Los resultados positivos deben confirmarse siempre mediante un método analítico de confirmación (CGAR/EMAR). Además, las muestras de una gama amplia de EQT deberán ser confirmadas por CGAR/EMAR (aproximadamente del 2 % al 10 % de las muestras negativas). Deberá facilitarse información sobre la correspondencia entre los resultados de los bioensayos y los de la CGAR/EMAR. |
7.3. Requisitos específicos aplicables a los bioensayos celulares
|
— |
Al efectuar un bioensayo, debe utilizarse en cada prueba una serie de concentraciones de referencia de TCDD o una mezcla de dioxinas y furanos (curva de respuesta con un R2 > 0,95 para una dosis completa). Sin embargo, a efectos del cribado, en el análisis de las muestras de baja concentración podría utilizarse una curva detallada en los niveles bajos. |
|
— |
Para los resultados del bioensayo en un intervalo de tiempo constante, deberá utilizarse una concentración de referencia de la TCDD (aproximadamente tres veces el límite de cuantificación) en una ficha de control de calidad. Otra posibilidad sería utilizar la respuesta relativa de una muestra de referencia comparada con la línea de calibración de la TCDD, ya que la respuesta de las células puede depender de múltiples factores. |
|
— |
Deberán registrarse y comprobarse los gráficos de control de calidad correspondientes a cada tipo de material de referencia, para asegurarse de que el resultado es conforme con las directrices establecidas. |
|
— |
La inducción de la dilución de la muestra utilizada debe situarse en la parte lineal de la curva de respuesta, en particular para los cálculos cuantitativos. Las muestras situadas por encima de la parte lineal de la curva de respuesta deben diluirse y analizarse de nuevo. Por tanto, se recomienda analizar tres o más diluciones a la vez. |
|
— |
La desviación típica porcentual no deberá ser superior al 15 % en una determinación por triplicado de cada dilución de la muestra, ni superior al 30 % entre tres experimentos independientes. |
|
— |
El límite de detección puede fijarse en tres veces la desviación típica del blanco de disolvente o de la respuesta de fondo. Otro enfoque consiste en aplicar una respuesta superior a la de fondo (factor de inducción cinco veces superior al blanco de disolvente) calculada a partir de la curva de calibración del día. El límite de cuantificación puede fijarse en cinco a seis veces la desviación típica del blanco de disolvente o de la respuesta de fondo, o bien puede aplicarse una respuesta muy superior a la de fondo (factor de inducción diez veces superior al blanco de disolvente) calculada a partir de la curva de calibración del día. |
7.4. Requisitos específicos aplicables a los bioensayos realizados con kits
|
— |
Deberá estar garantizado que los bioensayos realizados con kits tienen la suficiente sensibilidad y son lo bastante fiables para ser empleados con piensos. |
|
— |
Deben respetarse las instrucciones del fabricante por lo que respecta a la preparación de las muestras y los análisis. |
|
— |
Los kits no se utilizarán después de la fecha de caducidad. |
|
— |
No se utilizarán materiales o componentes previstos para otros kits. |
|
— |
Los kits de ensayo deberán conservarse y utilizarse a las temperaturas de almacenamiento y empleo indicadas. |
|
— |
El límite de detección para los inmunoensayos viene determinado por la suma de la media más un valor igual a tres veces la desviación típica, para una serie de diez análisis repetidos del blanco, cuyo resultado ha de dividirse por el valor de la pendiente de la ecuación de regresión lineal. |
|
— |
Deberán utilizarse patrones de referencia para los análisis de laboratorio, a fin de garantizar que el grado de respuesta al patrón se sitúa en un intervalo aceptable. |
8. Comunicación de los resultados
En la medida en que el procedimiento analítico seguido lo permita, los resultados del análisis deberán incluir los niveles de los congéneres individuales de PCDD/F y PCB e indicarse como límite inferior, límite superior y límite intermedio, a fin de aportar el máximo de información posible en la comunicación de los resultados, permitiendo así interpretarlos en función de los requisitos específicos.
El informe deberá indicar, asimismo, el contenido en lípidos de la muestra, así como el método utilizado para su extracción.
Deben indicarse los porcentajes de recuperación de cada patrón interno en caso de que estén fuera del intervalo mencionado en el punto 6 o de que se supere el nivel máximo, y en otros casos en que se solicite.
Puesto que, al decidir sobre la conformidad de una muestra, se ha de tener en cuenta la incertidumbre de medida, también deberá indicarse este parámetro. Así pues, los resultados analíticos deberán expresarse como x +/- U, donde x es el resultado analítico y U la incertidumbre de medida expandida, aplicando un factor de cobertura de 2, que ofrece un nivel de confianza aproximado del 95 %. En caso de que se determinen por separado las dioxinas y los PCB similares a las dioxinas, para la suma de ambos debe emplearse la suma de la incertidumbre expandida calculada correspondiente a los resultados analíticos obtenidos por separado para las dioxinas y los PCB similares a las dioxinas.
Si la incertidumbre de medida se tuviera en cuenta aplicando un CCα (según se describe en el punto I.2 de esta parte B), deberá indicarse este parámetro.
(1) Tabla de FET (= factores de equivalencia tóxica) correspondientes a dioxinas, furanos y PCB similares a las dioxinas
|
Congénere |
Valor FET |
Congénere |
Valor FET |
|
Dibenzo-p-dioxinas («PCDD») |
|
PCB similares a las dioxinas: |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
PCB no-orto |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0001 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,01 |
|
OCDD |
0,0001 |
PCB mono-orto |
|
|
|
|
PCB 105 |
0,0001 |
|
Dibenzofuranos («PCDF») |
|
PCB 114 |
0,0005 |
|
2,3,7,8-TCDF |
0,1 |
PCB 118 |
0,0001 |
|
1,2,3,7,8-PeCDF |
0,05 |
PCB 123 |
0,0001 |
|
2,3,4,7,8-PeCDF |
0,5 |
PCB 156 |
0,0005 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 157 |
0,0005 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00001 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 189 |
0,0001 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
|
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
|
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0001 |
|
|
|
Abreviaturas utilizadas: «T» = tetra; «Pe» = penta; «Hx» = hexa; «Hp» = hepta; «O» = octo; «CDD» = clorodibenzo-p-dioxina; «CDF» = clorodibenzofurano; «CB» = clorobifenilo. |
|||
(2) DO L 140 de 30.5.2002, p. 10.
(3) El concepto de «límite superior» exige suponer que la contribución de cada congénere no cuantificado al equivalente tóxico (EQT) es igual al límite de cuantificación.
El concepto de «límite inferior» exige suponer que la contribución de cada congénere no cuantificado al EQT es igual a cero.
El concepto de «límite intermedio» exige suponer que la contribución de cada congénere no cuantificado al EQT es igual a la mitad del límite de cuantificación.
(4) El análisis por duplicado es necesario para descartar la posibilidad de contaminación cruzada interna o de combinación accidental de muestras. El primer análisis, teniendo en cuenta la incertidumbre de medida, se realiza para verificar el cumplimiento.
Si el análisis se lleva a cabo en el marco de un incidente de contaminación con dioxinas, puede prescindirse de la confirmación mediante un análisis por duplicado si la trazabilidad pone de manifiesto que las muestras seleccionadas para el análisis están relacionadas con dicho incidente.
ANEXO VI
MÉTODOS DE ANÁLISIS PARA LA DETERMINACIÓN DE COMPONENTES DE ORIGEN ANIMAL CON FINES DE CONTROL OFICIAL DE LOS PIENSOS
Condiciones para la detección, identificación o estimación microscópica de los componentes de origen animal presentes en los piensos
1. Objeto y ámbito de aplicación
Las presentes condiciones se aplicarán cuando la detección de componentes de origen animal (definidos como productos derivados del procesamiento de cuerpos y partes de cuerpos de mamíferos, aves de corral y pescado) en los piensos se realice mediante examen microscópico en el marco del programa de inspección coordinado en el ámbito de la alimentación animal, conforme al Reglamento (CE) no 882/2004 del Parlamento Europeo y del Consejo (1). Si en todos los exámenes oficiales se utilizan los métodos del presente anexo, podrá también efectuarse un segundo examen utilizando variantes de los métodos o métodos alternativos, a fin de mejorar la detección de determinados tipos de componentes animales o precisar más el origen de estos componentes. Por otra parte, en el examen de determinados componentes animales específicos, como son el plasma o los huesos presentes en el sebo (véase el punto 9), podrá utilizarse una variante del protocolo, a condición de que los correspondientes análisis se realicen como complemento de los previstos en el programa de inspección coordinado.
2. Sensibilidad
En función de su naturaleza, pueden detectarse en los piensos cantidades muy pequeñas de componentes de origen animal (< 0,1 %).
3. Principio
Para la identificación se emplea una muestra representativa tomada conforme a lo dispuesto en el anexo I y preparada convenientemente. El siguiente protocolo es apto para la manipulación de piensos con bajo contenido de humedad. Los piensos con un contenido de humedad superior al 14 % deberán secarse (condensarse) antes de ser manipulados. Determinados piensos o materiales para piensos (por ejemplo, grasas y aceites) requieren un tratamiento específico (véase el punto 9). Los componentes de origen animal se identifican sobre la base de unas características típicas microscópicamente identificables (por ejemplo, fibras musculares y otras partículas de carne, cartílago, huesos, cuerno, pelo, cerdas, sangre, plumas, cáscaras de huevo y espinas o escamas de pescado). La identificación debe realizarse tanto en la fracción de tamiz (6.1) como en el sedimento concentrado (6.2) de la muestra.
4. Reactivos
4.1. Agente de inclusión
|
4.1.1. |
Hidrato de cloral (acuoso, 60 % p/v). |
|
4.1.2. |
Lejía (NaOH al 2,5 % p/v, o KOH al 2,5 % p/v) para fracciones de tamiz. |
|
4.1.3. |
Aceite de parafina o glicerol (viscosidad: 68-81) para las observaciones microscópicas en el sedimento. |
4.2. Agentes de lavado
|
4.2.1. |
Alcohol al 96 %. |
|
4.2.2. |
Acetona. |
4.3. Agente de concentración
|
4.3.1. |
Tetracloroetileno (densidad 1,62). |
4.4. Reactivos de tinción
|
4.4.1. |
Solución de yodo y yoduro de potasio (disolver 2 g de yoduro de potasio en 100 ml de agua y añadir 1 g de yodo, agitando con frecuencia). |
|
4.4.2. |
Rojo de alizarina (diluir 2,5 ml de ácido clorhídrico 1M en 100 ml de agua y añadir a esta solución 200 mg de rojo de alizarina). |
|
4.4.3. |
Reactivo de cistina (2 g de acetato de plomo, 10 g de NaOH/100 ml de H2O). |
|
4.4.4. |
Solución de yodo y yoduro de potasio (disuelta en etanol al 70 %). |
4.5. Reactivo decolorante
|
4.5.1 |
Solución comercial de hipoclorito de sodio (9,6 % de cloro activo). |
5. Equipo y accesorios
|
5.1. |
Balanza analítica (exactitud de 0,01 g, salvo para el sedimento concentrado: 0,001 g). |
|
5.2. |
Material de trituración (molino o mortero, especialmente para piensos que contengan > 15 % de grasa en análisis). |
|
5.3. |
Tamiz con luces de malla cuadradas de 0,50 mm de lado como máximo. |
|
5.4. |
Embudo de decantación o vaso de precipitado de fondo cónico. |
|
5.5. |
Lupa binocular (mínimo de 40 aumentos). |
|
5.6. |
Microscopio compuesto (mínimo de cuatrocientos aumentos) de luz transmitida o polarizada. |
|
5.7. |
Material de vidrio habitual de laboratorio. Todo el equipo se limpiará a fondo. Los embudos de decantación y el material de vidrio deben lavarse en máquina lavadora. Los tamices deben limpiarse con un cepillo de cerdas rígidas. |
6. Procedimiento
Los piensos granulados pueden tamizarse previamente si ambas fracciones se analizan como muestras aparte.
Se tratarán al menos 50 g de muestra (cuidadosamente molidos con el equipo adecuado (5.2), si es necesario para lograr una estructura apropiada). Se tomarán dos porciones representativas del material molido, una para la fracción de tamiz (5 g como mínimo) (6.1) y otra para el sedimento concentrado (5 g como mínimo) (6.2). Para facilitar la identificación, pueden utilizarse también reactivos de tinción (6.3).
Para indicar la naturaleza de las proteínas animales y el origen de las partículas, puede utilizarse un sistema de apoyo a la decisión como ARIES y pueden documentarse muestras de referencia.
6.1. Identificación de componentes de origen animal en las fracciones de tamiz
Se pasan por el tamiz (5.3) un mínimo de 5 g de la muestra en dos fracciones.
La fracción o fracciones de tamiz con partículas grandes (o una parte representativa de la fracción) se aplican en una capa fina a un portaobjetos adecuado y se observan sistemáticamente a la lupa binocular (5.5) a diversos aumentos, para comprobar la presencia de componentes de origen animal.
Las extensiones hechas con la fracción o las fracciones de tamiz con partículas finas se observan sistemáticamente al microscopio compuesto (5.6) a diversos aumentos, para comprobar la presencia de componentes de origen animal.
6.2. Determinación de componentes de origen animal del sedimento concentrado
Se transferirán un mínimo de 5 g (con una exactitud de 0,01 g) de la muestra a un embudo de decantación o un vaso de precipitado de fondo cónico y se tratarán con al menos 50 ml de tetracloroetileno (4.3.1). La mezcla se agitará o removerá varias veces.
|
— |
Si se utiliza un embudo de decantación cerrado, deberá dejarse reposar el sedimento durante tiempo suficiente (al menos tres minutos) antes de separarlo. Después volverá a agitarse y a dejarse reposar el sedimento durante al menos tres minutos. Seguidamente, el sedimento volverá a separarse. |
|
— |
Si se utiliza un vaso de precipitado abierto, el sedimento se dejará reposar durante al menos cinco minutos antes de separarlo. |
El sedimento total se secará y posteriormente se pesará (con una exactitud de 0,001 g). La pesada solo será necesaria si se requiere una estimación. Si el sedimento se compone de muchas partículas grandes, podrá pasarse por un tamiz (5.3) en dos fracciones. El sedimento seco se examinará a la lupa binocular (5.5) y al microscopio compuesto (5.6) para buscar componentes óseos.
6.3. Uso de agentes de inclusión y de reactivos de tinción
La identificación microscópica de los componentes de origen animal puede facilitarse utilizando agentes de inclusión y reactivos de tinción especiales.
Hidrato de cloral (4.1.1): al calentarse cuidadosamente, las estructuras celulares pueden apreciarse con más claridad, pues los granos de almidón se gelatinizan y el contenido celular no deseado se elimina.
Lejía (4.1.2.): tanto el hidróxido de sodio como el hidróxido de potasio clarifican las materias constitutivas de los piensos, lo que ayuda a detectar fibras musculares, pelos y otras estructuras queratínicas.
Aceite de parafina o glicerol (4.1.3): los componentes óseos pueden identificarse bien en este agente de inclusión, ya que la mayor parte de las lagunas se mantienen llenas de aire y aparecen como agujeros negros de 5-15 μm.
Solución de yodo y yoduro de potasio (4.4.1): se utiliza para la detección de almidón (color entre azul y violeta) y proteínas (color entre amarillo y naranja). Si es necesario, las soluciones pueden diluirse.
Solución de rojo de alizarina (4.4.2): coloración roja/rosada de huesos, espinas y escamas. Antes de secar el sedimento (véase el punto 6.2), el sedimento total se transferirá a un tubo de ensayo de vidrio y se enjuagará dos veces con unos 5 ml de alcohol (4.2.1) (en ambas ocasiones deberá utilizarse un vortex y deberá dejarse reposar el disolvente aproximadamente un minuto antes de decantarlo). Antes de utilizar este reactivo de tinción, el sedimento se decolorará añadiendo al menos 1 ml de solución de hipoclorito de sodio (4.5.1). Se dejará que la reacción continúe durante diez minutos. El tubo se llenará de agua y se dejará sedimentar durante dos a tres minutos, tras lo cual se decantarán el agua y las partículas suspendidas. El sedimento se enjuagará dos veces más con unos 10 ml de agua (utilizar un vortex, dejar reposar y decantar el agua cada vez). Se añadirán de dos a diez gotas o más (dependiendo de la cantidad de residuo) de la solución de rojo de alizarina. Se agitará la mezcla y se dejará reaccionar unos segundos. El sedimento coloreado se enjuagará dos veces con aproximadamente 5 ml de alcohol (4.2.1) y seguidamente una vez con acetona (4.2.2) (en cada ocasión deberá utilizarse un vortex y deberá dejarse reposar el disolvente alrededor de un minuto antes de decantarlo). El sedimento estará entonces listo para secar.
Reactivo de cistina (4.4.3): calentados cuidadosamente, los componentes que contienen cistina (pelo, plumas, etc.) adquieren un color entre negro y marrón.
6.4. Examen de piensos que posiblemente contienen harina de pescado
Se examinará al microscopio compuesto al menos una extensión de la fracción fina de tamiz y de la fracción fina del sedimento (véanse los puntos 6.1 y 6.2).
Cuando la etiqueta indique que los ingredientes incluyen harina de pescado, o si se sospecha o se ha detectado su presencia en el examen inicial, se examinarán al menos dos extensiones adicionales de la fracción fina de tamiz de la muestra original y de la fracción de sedimento total.
7. Cálculo y evaluación
Los Estados miembros velarán por que se apliquen los procedimientos descritos en este punto cuando se realice un análisis oficial para estimar la cantidad (y no sólo la presencia) de componentes animales.
El cálculo solo puede hacerse si los componentes de origen animal contienen fragmentos óseos.
En el portaobjetos, los fragmentos óseos de especies terrestres de sangre caliente (es decir, mamíferos y aves) pueden distinguirse de los diversos tipos de espinas de pescado gracias a sus típicas lagunas. La proporción de componentes de origen animal en el material de muestra se estima tomando en consideración:
|
— |
la proporción estimada (porcentaje en peso) de fragmentos óseos en el sedimento concentrado, y |
|
— |
la proporción (porcentaje en peso) de hueso presente en los componentes de origen animal. |
La estimación debe basarse en al menos tres (si es posible) extensiones y al menos cinco campos por extensión. En los piensos compuestos, el sedimento concentrado contiene, por regla general, no solo fragmentos de huesos de animales terrestres y espinas de pescado, sino también otras partículas de peso específico elevado como, por ejemplo, minerales, arena, fragmentos vegetales lignificados y similares.
7.1. Valor estimado del porcentaje de fragmentos óseos
Porcentaje de fragmentos de huesos de animales terrestres = (S × c)/P.
Porcentaje de fragmentos de espinas y escamas de pescados = (S × d)/P.
|
(S = |
peso del sedimento [miligramos]; c = factor de corrección [tanto por ciento] para la porción estimada de huesos de animales terrestres del sedimento; d = factor de corrección [tanto por ciento] para la porción estimada de fragmentos de espinas y escamas de pescado del sedimento, P = peso del material de muestra para la sedimentación [miligramos]). |
7.2. Valor estimado de los componentes de origen animal
La proporción de componentes óseos en productos animales puede variar considerablemente. (El porcentaje óseo en el caso de las harinas de huesos es del orden del 50-60 %, y en el de las harinas de carne del orden del 20-30 %; en el caso de las harinas de pescado, el contenido de espinas y escamas varía según la categoría y el origen de la harina, pero normalmente es del orden del 10-20 %).
Si se conoce el tipo de harina animal presente en la muestra, pueden hacerse la siguientes estimaciones del contenido:
contenido estimado de componentes de productos de animales terrestres (en tanto por ciento) = (S × c)/(P × f) × 100,
contenido estimado de componentes de productos de pescado (en tanto por ciento) = (S × d)/(P × f) × 100.
|
(S = |
peso del sedimento [miligramos]; c = factor de corrección [tanto por ciento] para la porción estimada de componentes de huesos de animales terrestres del sedimento; d = factor de corrección [tanto por ciento] para la porción estimada de fragmentos de espinas y escamas de pescado del sedimento; f = factor de corrección para la proporción ósea de los componentes de origen animal de la muestra examinada; P = peso del material de muestra para la sedimentación [miligramos]). |
8. Expresión del resultado del examen
El informe contendrá, como mínimo, información sobre la presencia de componentes derivados de animales terrestres y de harina de pescado. Los diversos casos se comunicarán de la siguiente forma:
|
8.1. |
En cuanto a la presencia de componentes derivados de animales terrestres:
|
|
8.2. |
Y, en cuanto a la presencia de harina de pescado:
En caso de que se detecten componentes derivados de pescado o de animales terrestres, el informe del resultado del examen puede incluir también, si es necesario, una estimación de la cantidad de componentes detectada (x %, < 0,1 %, 0,1-0,5 %, 0,5-5 % o > 5 %) y especificar el tipo de animal terrestre, si es posible, así como los componentes animales identificados (fibras musculares, cartílago, huesos, cuerno, pelo, cerdas, plumas, sangre, cáscaras de huevo y espinas o escamas de pescado). Si se hace una estimación de la cantidad de ingredientes animales, se indicará el factor de corrección, f, utilizado. Si se identifican componentes óseos de animales terrestres, el informe deberá incluir la frase siguiente: «No puede descartarse la posibilidad de que los componentes mencionados procedan de mamíferos». No será necesario incluir esta frase cuando se haya especificado que los fragmentos óseos de animales terrestres proceden de aves de corral o de mamíferos. |
9. Protocolo opcional para el análisis de grasas o aceites
En el análisis de grasas o aceites podrá utilizarse el siguiente protocolo:
|
— |
Si la grasa es sólida, se calienta, por ejemplo, en un microondas hasta su licuefacción. |
|
— |
A continuación, se pipetean 40 ml de grasa del fondo de la muestra a un tubo de centrifugación. |
|
— |
Se centrifuga durante diez minutos a 4 000 revoluciones por minuto. |
|
— |
Si, tras la centrifugación, la grasa se ha solidificado, se vuelve a calentar en una estufa hasta su licuefacción. Se repite la centrifugación durante cinco minutos a 4 000 revoluciones por minuto. |
|
— |
Por medio de una cucharilla o una espátula se transfiere la mitad de las impurezas decantadas a una pequeña placa de Petri o a un portaobjetos, para determinar al microscopio la posible presencia de componentes de origen animal (fibras de carne, plumas, fragmentos óseos, etc.). Como agente de inclusión para la microscopia se recomienda aceite de parafina o glicerol. |
|
— |
Las impurezas restantes se utilizan para la sedimentación descrita en el punto 6.2. |
(1) DO L 165 de 30.4.2004, p. 1. Versión corregida en el DO L 191 de 28.5.2004, p. 1.
ANEXO VII
MÉTODO DE CÁLCULO DEL VALOR ENERGÉTICO DE LOS PIENSOS PARA AVES DE CORRAL
1. Método de cálculo y expresión del valor energético
El valor energético de los piensos compuestos para aves de corral debe calcularse según la fórmula que figura más abajo, sobre la base de los porcentajes de determinados componentes analíticos de los piensos. Este valor ha de expresarse en megajulios (MJ) de energía metabolizable (EM), corregida en nitrógeno, por kilogramo de pienso compuesto:
MJ/kg de EM = 0,1551 × % proteína bruta +0,3431 × % grasa bruta +0,1669 × % almidón +0,1301 × % azúcar total (expresada en sacarosa).
2. Tolerancias aplicables a los valores declarados
Si la inspección oficial pone de manifiesto una discrepancia (valor energético del pienso aumentado o reducido) entre el resultado de la inspección y el valor energético declarado, se aplicará una tolerancia mínima de 0,4 MJ/kg de EM.
3. Expresión del resultado
El resultado obtenido tras aplicar la fórmula anteriormente indicada se expresará con un decimal.
4. Métodos de muestreo y análisis
El muestreo del pienso compuesto y la determinación del contenido de los compuestos analíticos indicados en el método de cálculo deben realizarse de conformidad, respectivamente, con los métodos comunitarios de muestreo y los métodos comunitarios de análisis para el control oficial de los piensos.
Han de aplicarse:
|
— |
para la determinación del contenido de grasa bruta: el procedimiento B del método para la determinación de los aceites y las grasas brutos, establecido en la parte H del anexo III, |
|
— |
para la determinación del contenido de almidón: el método polarimétrico establecido en la parte L del anexo III. |
ANEXO VIII
MÉTODOS DE ANÁLISIS PARA EL CONTROL DE LA PRESENCIA ILEGAL DE ADITIVOS YA NO AUTORIZADOS EN LOS PIENSOS
Nota importante:Para detectar la presencia ilegal de aditivos ya no autorizados en los piensos pueden emplearse métodos de análisis más sensibles que los mencionados en el presente anexo.Los métodos de análisis expuestos en este anexo se utilizarán con fines de confirmación.
A) DETERMINACIÓN DEL METILBENZOCUATO
7-benziloxi-6-butil-3-metoxicarbonil-4-quinolona
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de metilbenzocuato en los piensos. El límite de cuantificación es de 1 mg/kg.
2. Principio
El metilbenzocuato se extrae de la muestra con solución metanólica de ácido metanosulfónico. El extracto se purifica con diclorometano, por cromatografía de intercambio iónico y, a continuación, nuevamente con diclorometano. El contenido de metilbenzocuato se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa con un detector de UV.
3. Reactivos
|
3.1. |
Diclorometano |
|
3.2. |
Metanol, equivalente al de calidad CLAR |
|
3.3. |
Fase móvil para la CLAR Mezcla de metanol (3.2) y agua (equivalente a la de calidad CLAR) 75 + 25 (v + v). Filtrar por un filtro de 0,22 μm (4.5) y desgasificar la solución (por ejemplo, aplicando ultrasonidos durante diez minutos). |
|
3.4. |
Solución de ácido metanosulfónico, c = 2 % Diluir 20,0 ml de ácido metanosulfónico a 1 000 ml con metanol (3.2). |
|
3.5. |
Solución de ácido clorhídrico, c = 10 % Diluir 100 ml de ácido clorhídrico (ρ201,18 g/ml) a 1 000 ml con agua. |
|
3.6. |
Resina de intercambio catiónico Amberlite CG-120 (Na), de 100 a 200 mallas La resina se trata previamente antes de utilizarla. Preparar una lechada con 100 g de resina y 500 ml de solución de ácido clorhídrico (3.5) y llevar a ebullición en una placa calefactora sin parar de remover. Dejar enfriar y decantar el ácido. Filtrar en vacío a través de un papel de filtro. Lavar la resina dos veces con porciones de agua de 500 ml y, a continuación, con 250 ml de metanol (3.2). Enjuagar la resina con otra porción de 250 ml de metanol y secar haciendo pasar aire a través de la torta de filtro. Guardar la resina desecada en una botella tapada. |
|
3.7. |
Sustancia patrón: metilbenzocuato puro (7-benziloxi-6-butil-3-metoxicarbonil-4-quinolona). |
|
3.7.1. |
Pesar, con una precisión de 0,1 mg, 50 mg de la sustancia patrón (3.7), disolver en solución de ácido metanosulfónico (3.4) en un matraz aforado de 100 ml, enrasar y mezclar. |
|
3.7.2. |
Transvasar 5,0 ml de la solución patrón madre de metilbenzocuato (3.7.1) a un matraz aforado de 50 ml, enrasar con metanol (3.2) y mezclar. |
|
3.7.3. |
Transvasar 1,0, 2,0, 3,0, 4,0 y 5,0 ml de la solución patrón intermedia de metilbenzocuato (3.7.2) a una serie de matraces aforados de 25 ml. Enrasar con la fase móvil (3.3) y mezclar. Estas soluciones tienen concentraciones de metilbenzocuato de 2,0, 4,0, 6,0, 8,0 y 10,0 μg/ml, respectivamente. Deben prepararse poco antes de utilizarse. |
4. Instrumental
|
4.1. |
Agitador de laboratorio. |
|
4.2. |
Evaporador rotativo de película. |
|
4.3. |
Columna de vidrio (250 mm × 15 mm) provista de una llave de cierre y de un depósito de 200 ml de capacidad, aproximadamente. |
|
4.4. |
Equipo para CLAR con detector ultravioleta de longitud de onda variable o detector de red de diodos. |
|
4.4.1. |
Columna cromatográfica de líquidos: 300 mm x 4 mm, C18, relleno de 10 μm, o equivalente. |
|
4.5. |
Filtros de membrana de 0,22 μm. |
|
4.6. |
Filtros de membrana de 0,45 μm. |
5. Procedimiento
5.1. Generalidades
|
5.1.1. |
Deberá analizarse un pienso en blanco para comprobar la ausencia de metilbenzocuato y de sustancias interferentes. |
|
5.1.2. |
Asimismo, deberá realizarse un ensayo de recuperación analizando el pienso en blanco, que se habrá enriquecido añadiendo una cantidad de metilbenzocuato similar a la presente en la muestra. Para enriquecer a un nivel de 15 mg/kg, añadir 600 μl de la solución patrón madre (3.7.1) a 20 g del pienso en blanco, mezclar y esperar diez minutos antes de proceder a la extracción (5.2). Debe tenerse en cuenta que, a los efectos de este método, el pienso en blanco deberá ser de tipo similar al de la muestra y en su análisis no debe detectarse metilbenzocuato. |
5.2. Extracción
Pesar, con una precisión de 0,01 g, unos 20 g de la muestra preparada y pasarlos a un Erlenmeyer de 250 ml. Añadir 100,0 ml de la solución de ácido metanosulfónico (3.4) y agitar mecánicamente (4.1) durante 30 minutos. Filtrar la solución por un papel de filtro y conservar el filtrado para la fase de separación líquido-líquido (5.3).
5.3. Separación líquido-líquido
Transvasar 25,0 ml del filtrado obtenido en el punto 5.2 a un embudo de decantación de 500 ml que contenga 100 ml de solución de ácido clorhídrico (3.5). Añadir 100 ml de diclorometano (3.1) al embudo y agitar durante un minuto. Dejar que se separen las fases y decantar la fase inferior (diclorometano) en un matraz de fondo redondo de 500 ml. Repetir la extracción de la fase acuosa con otras dos porciones de 40 ml de diclorometano y combinarlas con el primer extracto en el matraz de fondo redondo. Evaporar el extracto de diclorometano hasta sequedad en el evaporador rotativo (4.2) a 40 oC y a presión reducida. Disolver el residuo en 20-25 ml de metanol (3.2), tapar el matraz y guardar todo el extracto para la cromatografía de intercambio iónico (5.4).
5.4. Cromatografía de intercambio iónico
5.4.1.
Introducir un tapón de lana de vidrio en el extremo inferior de una columna de vidrio (4.3). Preparar una lechada con 5,0 g de la resina de intercambio catiónico tratada (3.6) y 50 ml de ácido clorhídrico (3.5), verterla en la columna de vidrio y dejarla reposar. Eliminar el ácido sobrante hasta justo por encima de la superficie de resina y lavar la columna con agua hasta que el líquido efluyente dé neutro al tornasol. Transvasar 50 ml de metanol (3.2) a la columna y dejar evacuar hasta la superficie de la resina.
5.4.2.
Pipetear cuidadosamente a la columna el extracto obtenido en el punto 5.3. Enjuagar el matraz de fondo redondo con dos porciones de 5 ml a 10 ml de metanol (3.2) y transvasar estos líquidos de lavado a la columna. Dejar correr el extracto hasta la superficie de resina y lavar la columna con 50 ml de metanol, cuidando de que el caudal no sea superior a 5 ml por minuto. Desechar el líquido efluyente. Eluir el metilbenzocuato de la columna utilizando 150 ml de solución de ácido metanosulfónico (3.4) y recoger el eluido de la columna en un Erlenmeyer de 250 ml.
5.5. Separación líquido-líquido
Transferir el eluido obtenido en el punto 5.4.2 a un embudo de decantación de 1 l. Enjuagar el Erlenmeyer con 5 ml a 10 ml de metanol (3.2) y combinar los líquidos de lavado con el contenido del embudo de decantación. Añadir 300 ml de una solución de ácido clorhídrico (3.5) y 130 ml de diclorometano (3.1). Agitar durante un minuto y dejar que las fases se separen. Decantar la fase inferior (diclorometano) en un matraz de fondo redondo de 500 ml. Repetir la extracción de la fase acuosa con otras dos porciones de 70 ml de diclorometano y combinar estos extractos con el primero en el matraz de fondo redondo.
Evaporar el extracto de diclorometano hasta sequedad en el evaporador rotativo (4.2) a 40 oC y a presión reducida. Disolver el residuo en el matraz con unos 5 ml de metanol (3.2) y transvasar cuantitativamente esta solución a un matraz aforado de 10 ml. Enjuagar el matraz de fondo redondo con otras dos porciones de 1 ml a 2 ml de metanol y transvasarlas al matraz aforado. Enrasar con metanol y mezclar. Filtrar una parte alícuota por un filtro de membrana (4.6). Reservar esta solución para la determinación mediante CLAR (5.6).
5.6. Determinación mediante CLAR
5.6.1.
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes:
|
— |
columna cromatográfica de líquidos (4.4.1), |
|
— |
fase móvil para la CLAR: mezcla de metanol y agua (3.3), |
|
— |
caudal: 1-1,5 ml/min, |
|
— |
longitud de onda de detección: 265 nm, |
|
— |
volumen de inyección: 20-50 μl. |
Comprobar la estabilidad del sistema cromatográfico inyectando varias veces la solución de calibración (3.7.3) de 4 μg/ml, hasta que se alcancen alturas (o áreas) de pico y tiempos de retención constantes.
5.6.2.
Inyectar varias veces cada solución de calibración (3.7.3) y medir las alturas (áreas) de pico de cada concentración. Trazar una curva de calibración empleando las alturas o áreas de pico medias de las soluciones de calibración como ordenadas y las concentraciones correspondientes, en microgramos por mililitro, como abscisas.
5.6.3.
Inyectar varias veces el extracto de muestra (5.5) empleando el mismo volumen que para las soluciones de calibración, y determinar la altura (área) media de los picos de metilbenzocuato.
6. Cálculo de los resultados
A partir de la altura (área) media de los picos de metilbenzocuato de la solución de muestra, determinar la concentración de esta última, en microgramos por mililitro, tomando como referencia la curva de calibración (5.6.2).
El contenido de metilbenzocuato p (miligramos por kilogramo) de la muestra viene dado por la siguiente fórmula:

en la cual:
|
c |
= |
concentración de metilbenzocuato de la solución de muestra, en microgramos por mililitro; |
|
m |
= |
peso de la porción de ensayo, en gramos. |
7. Validación de los resultados
7.1. Identidad
La identidad del analito puede confirmarse mediante cocromatografía, o utilizando un detector de red de diodos con el cual se comparan los espectros del extracto de muestra y de la solución de calibración (3.7.3) de 10 μg/ml.
7.1.1.
Enriquecer un extracto de muestra añadiéndole una cantidad apropiada de la solución patrón intermedia (3.7.2). La cantidad de metilbenzocuato añadida debe ser similar a la cantidad estimada de metilbenzocuato en el extracto de muestra.
Solo la altura del pico de metilbenzocuato deberá aumentar en función de la cantidad añadida y de la dilución del extracto. La anchura del pico, a la mitad de su altura máxima, debe ser igual ± 10 % a la anchura original.
7.1.2.
Los resultados se evalúan con arreglo a los siguientes criterios:
|
a) |
la longitud de onda de absorción máxima de los espectros de la muestra y del patrón, registrados en el vértice del pico del cromatograma, debe ser la misma dentro de un margen determinado por el poder de resolución del sistema de detección; en el caso de la detección por red de diodos, el margen típico se sitúa en aproximadamente 2 nm; |
|
b) |
entre 220 nm y 350 nm, los espectros de la muestra y del patrón registrados en el vértice del pico del cromatograma no deben ser diferentes por lo que respecta a aquellas partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los dos espectros excede del 15 % de la absorbancia del analito patrón; |
|
c) |
entre 220 nm y 350 nm, los espectros de la pendiente ascendente, del vértice y de la pendiente descendente del pico producido por el extracto de muestra no deben diferir entre sí por lo que respecta a las partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los espectros excede del 15 % de la absorbancia del espectro del vértice. |
Si no se cumple alguno de estos criterios, la presencia del analito no queda confirmada.
7.2. Repetibilidad
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe exceder del 10 % del resultado superior, en el caso de contenidos de metilbenzocuato de 4-20 mg/kg.
7.3. Recuperación
La recuperación de la muestra en blanco enriquecida deberá ser al menos del 90 %.
8. Resultados de un estudio colaborativo
Se analizaron cinco muestras en diez laboratorios. De cada muestra se hicieron análisis por duplicado.
|
|
Blanco |
Sémola 1 |
Gránulo 1 |
Sémola 2 |
Gránulo 2 |
|
Media [mg/kg] |
ND |
4,50 |
4,50 |
8,90 |
8,70 |
|
Sr [mg/kg] |
—— |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [ %] |
—— |
6,70 |
4,40 |
6,70 |
5,70 |
|
SR [mg/kg] |
—— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [ %] |
—— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Recuperación [ %] |
—— |
92,00 |
93,00 |
92,00 |
89,00 |
|
ND |
= |
no detectado |
|
Sr |
= |
desviación típica de la repetibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad, en tanto por ciento |
|
SR |
= |
desviación típica de la reproducibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad, en tanto por ciento |
B) DETERMINACIÓN DEL OLAQUINDOX
N 1 ,N 4 -dióxido de 2-[N-2'-(hidroxietil)carbamoil]-3-metilquinoxalina
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de olaquindox en los piensos. El límite de cuantificación es de 5 mg/kg.
2. Principio
La muestra se extrae mediante una mezcla de agua y metanol. El contenido de olaquindox se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa, empleando un detector de UV.
3. Reactivos
|
3.1. |
Metanol. |
|
3.2. |
Metanol, equivalente al de calidad CLAR. |
|
3.3. |
Agua, equivalente a la de calidad CLAR. |
|
3.4. |
Fase móvil para la CLAR. Mezcla de agua (3.3) y metanol (3.2), 900 + 100 (V + V). |
|
3.5. |
Sustancia patrón: olaquindox puro, N1,N4-dióxido de 2-[N-2'-(hidroxietil)carbamoil]-3-metilquinoxalina, E 851. |
|
3.5.1. |
Pesar, con una precisión de 0,1 mg, 50 mg de olaquindox (3.5) en un matraz aforado de 200 ml y añadir unos 190 ml de agua. A continuación, introducir el matraz durante 20 minutos en un baño ultrasónico (4.1). Después del tratamiento de ultrasonidos, llevar la solución a temperatura ambiente, enrasar con agua y mezclar. Envolver el matraz con papel de aluminio y guardarlo en el frigorífico. Esta solución debe prepararse de nuevo cada mes. |
|
3.5.2. |
Transvasar 10,0 ml de la solución patrón madre (3.5.1) a un matraz aforado de 100 ml, enrasar con la fase móvil (3.4) y mezclar. Envolver el matraz con papel de aluminio y guardarlo en el frigorífico. Esta solución debe prepararse de nuevo cada día. |
|
3.5.3. |
Transvasar 1,0, 2,0, 5,0, 10,0, 15,0 y 20,0 ml de la solución patrón intermedia (3.5.2) a una serie de matraces aforados de 50 ml. Enrasar con la fase móvil (3.4) y mezclar. Envolver los matraces con papel de aluminio. Estas soluciones corresponden, respectivamente, a 0,5, 1,0, 2,5, 5,0, 7,5 y 10,0 μg/ml de olaquindox. Deben prepararse de nuevo cada día. |
4. Instrumental
|
4.1. |
Baño ultrasónico. |
|
4.2. |
Agitador mecánico. |
|
4.3. |
Equipo para CLAR con detector ultravioleta de longitud de onda variable o detector de red de diodos. |
|
4.3.1. |
Columna cromatográfica de líquidos, de 250 mm x 4 mm, C18, relleno de 10 μm, o equivalente. |
|
4.4. |
Filtros de membrana de 0,45 μm. |
5. Procedimiento
|
Nota: |
El olaquindox es sensible a la luz. Realizar todas las operaciones con luz tenue o con material de vidrio ámbar. |
5.1. Generalidades.
|
5.1.1. |
Deberá analizarse un pienso en blanco para comprobar la ausencia de olaquindox y de sustancias interferentes. |
|
5.1.2. |
Asimismo, deberá realizarse un ensayo de recuperación analizando el pienso en blanco, que se habrá enriquecido añadiendo una cantidad de olaquindox similar a la presente en la muestra. Para enriquecer a un nivel de 50 mg/kg, transvasar 10,0 ml de la solución patrón madre (3.5.1) a un Erlenmeyer de 250 ml y evaporar la solución a unos 0,5 ml. Añadir 50 g del pienso en blanco, mezclar completamente y esperar diez minutos, mezclando varias veces más antes de proceder a la extracción (5.2).
|
5.2. Extracción.
Pesar, con una precisión de 0,01 g, aproximadamente 50 g de la muestra. Pasarlos a un Erlenmeyer de 1 000 ml, añadir 100 ml de metanol (3.1) e introducir el matraz durante cinco minutos en el baño ultrasónico (4.1). Añadir 410 ml de agua y dejar en el baño ultrasónico durante otros 15 minutos. Retirar el matraz del baño ultrasónico, agitar durante 30 minutos en el agitador (4.2) y filtrar por un filtro plegado. Transvasar 10,0 ml del filtrado a un matraz aforado de 20 ml, enrasar con agua y mezclar. Filtrar una parte alícuota por un filtro de membrana (4.4) (véase la observación del punto 9). Proceder a la determinación mediante CLAR (5.3).
5.3. Determinación mediante CLAR.
5.3.1.
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
|
Columna analítica (4.3.1) |
|
|
Fase móvil (3.4): |
Mezcla de metanol (3.2) y agua (3.3), 900 + 100 (V + V) |
|
Caudal: |
1,5-2 ml/min. |
|
Longitud de onda de detección: |
380 nm |
|
Volumen de inyección: |
20-100 μl |
Comprobar la estabilidad del sistema cromatográfico inyectando varias veces la solución de calibración (3.5.3) de 2,5 μg/ml, hasta que se alcancen alturas de pico y tiempos de retención constantes.
5.3.2.
Inyectar varias veces cada solución de calibración (3.5.3) y determinar las alturas (áreas) de pico medias de cada concentración. Trazar una curva de calibración empleando las alturas (áreas) de pico medias de las soluciones de calibración como ordenadas y las concentraciones correspondientes, en microgramos por mililitro, como abscisas.
5.3.3.
Inyectar varias veces el extracto de muestra (5.2) empleando el mismo volumen que para las soluciones de calibración, y determinar la altura (área) media de los picos de olaquindox.
6. Cálculo de los resultados
Determinar la concentración de la solución de muestra, en microgramos por mililitro, a partir de la altura (área) media de sus picos de olaquindox, tomando como referencia la curva de calibración (5.3.2).
El contenido p de olaquindox de la muestra, en miligramos por kilogramo, viene dado por la siguiente fórmula:
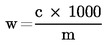
en la cual:
|
c |
= |
concentración de olaquindox del extracto de muestra (5.2), en microgramos por mililitro; |
|
m |
= |
peso de la porción de ensayo, en gramos por mililitro (5.2). |
7. Validación de los resultados
7.1. Identidad.
La identidad del analito puede confirmarse mediante cocromatografía, o utilizando un detector de red de diodos con el cual se comparan los espectros del extracto de muestra (5.2) y de la solución de calibración (3.5.3) de 5,0 μg/ml.
7.1.1.
Enriquecer un extracto de muestra (5.2) añadiéndole una cantidad apropiada de solución de calibración (3.5.3). La cantidad de olaquindox añadida debe ser similar a la hallada en el extracto de muestra.
Solo la altura del pico de olaquindox deberá aumentar en función de la cantidad añadida y de la dilución del extracto. La anchura del pico, a la mitad de su altura, debe ser igual ± 10 % a la anchura original del pico de olaquindox del extracto de muestra sin enriquecer.
7.1.2.
Los resultados se evalúan con arreglo a los siguientes criterios:
|
a) |
la longitud de onda de absorción máxima de los espectros de la muestra y del patrón, registrados en el vértice del pico del cromatograma, debe ser la misma dentro de un margen determinado por el poder de resolución del sistema de detección; en el caso de la detección por red de diodos, el margen típico se sitúa en ± 2 nm; |
|
b) |
entre 220 nm y 400 nm, los espectros de la muestra y del patrón registrados en el vértice del pico del cromatograma no deben ser diferentes por lo que respecta a aquellas partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los dos espectros excede del 15 % de la absorbancia del analito patrón; |
|
c) |
entre 220 nm y 400 nm, los espectros de la pendiente ascendente, del vértice y de la pendiente descendente del pico producido por el extracto de muestra no deben diferir entre sí por lo que respecta a las partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los espectros excede del 15 % de la absorbancia del espectro del vértice del pico. |
Si no se cumple alguno de estos criterios, la presencia del analito no queda confirmada.
7.2. Repetibilidad.
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe rebasar el 15 % del resultado superior, en el caso de contenidos de olaquindox de 10-200 mg/kg.
7.3. Recuperación.
La recuperación de la muestra en blanco enriquecida deberá ser al menos del 90 %.
8. Resultados de un estudio colaborativo
Se organizó un estudio colaborativo comunitario en el que hasta trece laboratorios analizaron cuatro muestras de piensos para lechones, incluido un pienso en blanco. A continuación figuran los resultados del estudio.
|
|
Muestra 1 |
Muestra 2 |
Muestra 3 |
Muestra 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
media [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
Contenido nominal |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
recuperación % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
número de laboratorios |
|
n |
= |
número de valores individuales |
|
Sr |
= |
desviación típica de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad |
9. Observación
Aunque el método no ha sido validado para piensos que contengan más de 100 mg/kg de olaquindox, quizá puedan obtenerse resultados satisfactorios tomando una muestra de menor peso y/o diluyendo el extracto (5.2) hasta alcanzar una concentración que se encuentre en el intervalo de la curva de calibración (5.3.2).
C) DETERMINACIÓN DEL AMPROLIO
Clorhidrato de cloruro de 1-[(4-amino-2-propilpirimidin-5-il)metil]-2-metil-piridinio
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de amprolio en piensos y premezclas. El límite de detección es de 1 mg/kg; el de cuantificación, de 5 mg/kg.
2. Principio
La muestra se extrae con una mezcla de agua y metanol. Tras dilución con la fase móvil y filtración por un filtro de membrana, el contenido de amprolio se determina mediante cromatografía de líquidos de alta resolución (CLAR) de intercambio catiónico, empleando un detector de UV.
3. Reactivos
|
3.1. |
Metanol. |
|
3.2. |
Acetonitrilo, equivalente al de calidad CLAR. |
|
3.3. |
Agua, equivalente a la de calidad CLAR. |
|
3.4. |
Solución de dihidrogenofosfato de sodio, c = 0,1 mol/l. Disolver 13,80 g de dihidrogenofosfato de sodio monohidrato en agua (3.3) en un matraz aforado de 1 000 ml, enrasar con agua (3.3) y mezclar. |
|
3.5. |
Solución de perclorato de sodio, c = 1,6 mol/l. Disolver 224,74 g de perclorato de sodio monohidrato en agua (3.3) en un matraz aforado de 1 000 ml, enrasar con agua (3.3) y mezclar. |
|
3.6. |
Fase móvil para la CLAR (véase la observación 9.1). Mezcla de acetonitrilo (3.2), solución de dihidrogenofosfato de sodio (3.4) y solución de perclorato de sodio (3.5), 450 + 450 + 100 (v + v + v). Antes de utilizarla, filtrar la solución por un filtro de membrana de 0,22 μm (4.3) y desgasificarla (por ejemplo, en el baño ultrasónico [4.4] durante, como mínimo, 15 minutos). |
|
3.7. |
Sustancia patrón: amprolio puro, clorhidrato de cloruro de 1-[(4-amino-2-propilpirimidin-5-il)metil]-2-metil-piridinio, E 750 (véase el punto 9.2). |
|
3.7.1. |
Pesar, con una precisión de 0,1 mg, 50 mg de amprolio (3.7) en un matraz aforado de 100 ml, disolver en 80 ml de metanol (3.1) y poner el matraz en un baño ultrasónico (4.4) durante diez minutos. Después del tratamiento de ultrasonidos, llevar la solución a temperatura ambiente, enrasar con agua y mezclar. A una temperatura de ≤ 4 oC, la solución es estable durante un mes. |
|
3.7.2. |
Pipetear 5,0 ml de la solución patrón madre (3.7.1) a un matraz aforado de 50 ml, enrasar con el disolvente de extracción (3.8) y mezclar. A una temperatura de ≤ 4 oC, la solución es estable durante un mes. |
|
3.7.3. |
Transvasar 0,5, 1,0 y 2,0 ml de la solución patrón intermedia (3.7.2) a una serie de matraces aforados de 50 ml. Enrasar con la fase móvil (3.6) y mezclar. Estas soluciones corresponden, respectivamente, a 0,5, 1,0 y 2,0 μg/ml de amprolio. Deben prepararse poco antes de utilizarse. |
|
3.8. |
Disolvente de extracción. Mezcla de metanol (3.1) y agua 2 + 1 (v + v). |
4. Instrumental
|
4.1. |
Equipo para CLAR con un sistema de inyección que permita inyectar volúmenes de 100 μl. |
|
4.1.1. |
Columna cromatográfica de líquidos, de 125 mm x 4 mm, Nucleosil 10 SA de intercambio catiónico, relleno de 5 μm o 10 μm, o equivalente. |
|
4.1.2. |
Detector de UV de longitud de onda variable, o detector de red de diodos. |
|
4.2. |
Filtro de membrana de politetrafluoretileno de 0,45 μm. |
|
4.3. |
Filtro de membrana de 0,22 μm. |
|
4.4. |
Baño ultrasónico. |
|
4.5. |
Agitador mecánico o magnético. |
5. Procedimiento
5.1. Generalidades
5.1.1.
Para el ensayo de recuperación (5.1.2) deberá analizarse un pienso en blanco, a fin de comprobar la ausencia de amprolio y de sustancias interferentes. El pienso en blanco deberá ser de tipo similar al de la muestra y en su análisis no debe detectarse amprolio ni sustancias interferentes.
5.1.2
Deberá realizarse un ensayo de recuperación analizando el pienso en blanco, que se habrá enriquecido añadiendo una cantidad de amprolio similar a la presente en la muestra. Para enriquecer a un nivel de 100 mg/kg, transvasar 10,0 ml de la solución patrón madre (3.7.1) a un Erlenmeyer de 250 ml y evaporar la solución a unos 0,5 ml. Añadir 50 g del pienso en blanco, mezclar completamente y esperar diez minutos, mezclando varias veces más antes de proceder a la extracción (5.2).
Si no se dispone de un pienso en blanco de tipo similar al de la muestra (véase el punto 5.1.1), también puede realizarse un ensayo de recuperación por el método de adición de patrón. En este caso, la muestra que debe analizarse se enriquece con una cantidad de amprolio similar a la que ya esté presente en ella. Esta muestra se analiza junto con la muestra sin enriquecer, y la recuperación puede calcularse por sustracción.
5.2. Extracción
5.2.1.
En función del contenido de amprolio, pesar, con una precisión de 0,01 g, entre 5 g y 40 g de la muestra en un Erlenmeyer de 500 ml y añadir 200 ml de disolvente de extracción (3.8). Poner el matraz en el baño ultrasónico (4.4) y dejarlo 15 minutos. Retirar el matraz del baño ultrasónico, agitar durante una hora en el agitador o remover en el agitador magnético (4.5). Diluir una parte alícuota del extracto con la fase móvil (3.6) hasta obtener un contenido de amprolio de 0,5-2 μg/ml y mezclar (véase la observación del punto 9.3). Filtrar de 5 ml a 10 ml de esta solución diluida por un filtro de membrana (4.2). Proceder a la determinación mediante CLAR (5.3).
5.2.2.
En función del contenido de amprolio, pesar, con una precisión de 0,001 g, entre 1 g y 4 g de la premezcla en un Erlenmeyer de 500 ml y añadir 200 ml de disolvente de extracción (3.8). Poner el matraz en el baño ultrasónico (4.4) y dejarlo 15 minutos. Retirar el matraz del baño ultrasónico, agitar durante una hora en el agitador o remover en el agitador magnético (4.5). Diluir una parte alícuota del extracto con la fase móvil (3.6) hasta obtener un contenido de amprolio de 0,5-2 μg/ml y mezclar. Filtrar de 5 ml a 10 ml de esta solución diluida por un filtro de membrana (4.2). Proceder a la determinación mediante CLAR (5.3).
5.3. Determinación mediante CLAR
5.3.1.
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
|
Columna cromatográfica |
|
|
de líquidos (4.1.1): |
125 mm × 4 mm, Nucleosil 10 SA de intercambio catiónico, relleno de 5 μm o 10 μm, o equivalente |
|
Fase móvil (3.6): |
Mezcla de acetonitrilo (3.2), solución de dihidrogenofosfato de sodio (3.4) y solución de perclorato de sodio (3.5), 450 + 450 + 100 (v + v + v). |
|
Caudal: |
0,7-1 ml/min. |
|
Longitud de onda de detección: |
264 nm |
|
Volumen de inyección: |
100 μl |
Comprobar la estabilidad del sistema cromatográfico inyectando varias veces la solución de calibración (3.7.3) de 1,0 μg/ml, hasta que se alcancen alturas de pico y tiempos de retención constantes.
5.3.2.
Inyectar varias veces cada solución de calibración (3.7.3) y determinar las alturas (áreas) de pico medias de cada concentración. Trazar una curva de calibración empleando las alturas (áreas) de pico medias de las soluciones de calibración como ordenadas y las concentraciones correspondientes, en microgramos por mililitro, como abscisas.
5.3.3.
Inyectar varias veces el extracto de muestra (5.2) empleando el mismo volumen que para las soluciones de calibración, y determinar la altura (área) media de los picos de amprolio.
6. Cálculo de los resultados
Determinar la concentración de la solución de muestra, en microgramos por mililitro, a partir de la altura (área) media de sus picos de amprolio, tomando como referencia la curva de calibración (5.3.2).
El contenido p de amprolio de la muestra, en miligramos por kilogramo, viene dado por la siguiente fórmula:
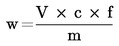 [mg/kg]
[mg/kg]
en la cual:
|
V |
= |
volumen del disolvente de extracción (3.8) según el punto 5.2, en mililitros (es decir, 200 ml); |
|
c |
= |
concentración de amprolio del extracto de muestra (5.2), en microgramos por mililitro; |
|
f |
= |
factor de dilución según el punto 5.2; |
|
m |
= |
peso de la porción de ensayo, en gramos. |
7. Validación de los resultados
7.1. Identidad.
La identidad del analito puede confirmarse mediante cocromatografía, o utilizando un detector de red de diodos con el cual se comparan los espectros del extracto de muestra (5.2) y de la solución de calibración (3.7.3) de 2,0 μg/ml.
7.1.1.
Enriquecer un extracto de muestra (5.2) añadiéndole una cantidad apropiada de solución de calibración (3.7.3). La cantidad de amprolio añadida debe ser similar a la hallada en el extracto de muestra.
Solo la altura del pico de amprolio deberá aumentar en función de la cantidad añadida y de la dilución del extracto. La anchura del pico, a la mitad de su altura, debe ser igual ± 10 % a la anchura original del pico de amprolio del extracto de muestra sin enriquecer.
7.1.2.
Los resultados se evalúan con arreglo a los siguientes criterios:
|
a) |
la longitud de onda de absorción máxima de los espectros de la muestra y del patrón, registrados en el vértice del pico del cromatograma, debe ser la misma dentro de un margen determinado por el poder de resolución del sistema de detección; en el caso de la detección por red de diodos, el margen típico se sitúa en ± 2 nm; |
|
b) |
entre 210 nm y 320 nm, los espectros de la muestra y del patrón registrados en el vértice del pico del cromatograma no deben ser diferentes por lo que respecta a aquellas partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los dos espectros excede del 15 % de la absorbancia del analito patrón; |
|
c) |
entre 210 nm y 320 nm, los espectros de la pendiente ascendente, del vértice y de la pendiente descendente del pico producido por el extracto de muestra no deben diferir entre sí por lo que respecta a las partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los espectros excede del 15 % de la absorbancia del espectro del vértice del pico. |
Si no se cumple alguno de estos criterios, la presencia del analito no queda confirmada.
7.2. Repetibilidad.
La diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe exceder de:
|
— |
el 15 % del valor superior, en el caso de contenidos de amprolio de 25 mg/kg a 500 mg/kg, |
|
— |
75 mg/kg, en el caso de contenidos de amprolio de 500 mg/kg a 1 000 mg/kg, |
|
— |
el 7,5 % del valor superior, en el caso de contenidos de amprolio superiores a 1 000 mg/kg. |
7.3. Recuperación.
La recuperación de la muestra (en blanco) enriquecida deberá ser al menos del 90 %.
8. Resultados de un estudio colaborativo
Se organizó un estudio colaborativo en el que se analizaron tres piensos para aves de corral (muestras 1 a 3), un pienso mineral (muestra 4) y una premezcla (muestra 5). Los resultados se recogen en el cuadro siguiente:
|
|
Muestra 1 (pienso en blanco) |
Muestra 2 |
Muestra 3 |
Muestra 4 |
Muestra 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
media [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Contenido nominal [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
número de laboratorios |
|
n |
= |
número de valores individuales |
|
sr |
= |
desviación típica de la repetibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad |
|
sR |
= |
desviación típica de la reproducibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad |
9. Observaciones
|
9.1. |
Si la muestra contiene tiamina, su pico aparece en el cromatograma poco antes que el de amprolio. Según este método, el amprolio y la tiamina deben separarse. Si la columna (4.1.1) utilizada en este método no separa el amprolio de la tiamina, sustituir hasta el 50 % de la porción de acetonitrilo de la fase móvil (3.6) por metanol. |
|
9.2. |
De acuerdo con la British Pharmacopoeia, el espectro de una solución de amprolio (c = 0,02 mol/l) en ácido clorhídrico (c = 0,1 mol/l) presenta máximos a 246 nm y 262 nm. La absorbancia será de 0,84 a 246 nm y de 0,80 a 262 nm. |
|
9.3. |
El extracto debe estar siempre diluido con la fase móvil, ya que, de lo contrario, el tiempo de retención del pico de amprolio puede variar significativamente, debido a cambios en la fuerza iónica. |
D) DETERMINACIÓN DEL CARBADOX
N1,N4-dióxido de metil 3-(2-quinoxalinilmetileno)carbazato
1. Objeto y ámbito de aplicación
Este método permite determinar el nivel de carbadox en piensos, premezclas y preparados. El límite de detección es de 1 mg/kg; el de cuantificación, de 5 mg/kg.
2. Principio
La muestra se equilibra con agua y se extrae con una mezcla de metanol y acetonitrilo. En el caso de los piensos, una parte alícuota del extracto filtrado se limpia en una columna de óxido de aluminio. En el caso de las premezclas y los preparados, una parte alícuota del extracto filtrado se diluye con agua, metanol y acetonitrilo hasta obtener una concentración apropiada. El contenido de carbadox se determina mediante cromatografía de líquidos de alta resolución (CLAR) de fase reversa, empleando un detector de UV.
3. Reactivos
|
3.1. |
Metanol. |
|
3.2. |
Acetonitrilo, equivalente al de calidad CLAR. |
|
3.3. |
Ácido acético, p = 100 %. |
|
3.4. |
Óxido de aluminio: neutro, con grado de actividad I. |
|
3.5. |
Mezcla de metanol y acetonitrilo 1 + 1 (v + v). Mezclar 500 ml de de metanol (3.1) con 500 ml de acetonitrilo (3.2). |
|
3.6. |
Ácido acético, σ = 10 %. Diluir 10 ml de ácido acético (3.3) a 100 ml con agua. |
|
3.7. |
Acetato de sodio. |
|
3.8. |
Agua, equivalente a la de calidad CLAR. |
|
3.9. |
Solución reguladora de acetato, c = 0,01 mol/l, pH = 6,0. Disolver 0,82 g de acetato de sodio (3.7) en 700 ml de agua (3.8) y ajustar el pH a 6,0 con ácido acético (3.6). Transvasar a un matraz aforado de 1 000 ml, enrasar con agua (3.8) y mezclar. |
|
3.10. |
Fase móvil para la CLAR. Mezclar 825 ml de solución reguladora de acetato (3.9) con 175 ml de acetonitrilo (3.2). Filtrar por un filtro de 0,22 μm (4.5) y desgasificar la solución (por ejemplo, aplicando ultrasonidos durante diez minutos). |
|
3.11. |
Sustancia patrón. Carbadox puro: N1,N4-dióxido de metil 3-(2-quinoxalinilmetileno)carbazato, E 850. |
|
3.11.1. |
Pesar, con una precisión de 0,1 mg, 25 mg de sustancia patrón de carbadox (3.11) en un matraz aforado de 250 ml. Disolver en una mezcla de metanol y acetonitrilo (3.5) por medio de ultrasonidos (4.7). Después del tratamiento de ultrasonidos, llevar la solución a temperatura ambiente, enrasar con la mezcla de metanol y acetonitrilo (3.5) y mezclar. Envolver el matraz con papel de aluminio, o utilizar material de vidrio ámbar, y guardarlo en el frigorífico. A una temperatura de ≤ 4 oC, la solución es estable durante un mes. |
|
3.11.2. |
Transvasar 2,0, 5,0, 10,0 y 20,0 ml de la solución patrón madre (3.11.1) a una serie de matraces aforados de 100 ml. Añadir 30 ml de agua, enrasar con la mezcla de metanol y acetonitrilo (3.5) y mezclar. Envolver los matraces con papel de aluminio. Estas soluciones corresponden, respectivamente, a 2,0, 5,0, 10,0 y 20,0 μg/ml de carbadox. Las soluciones de calibración deben prepararse poco antes de utilizarse.
|
|
3.12. |
Mezcla de agua y metanol-acetonitrilo (3.5), 300 + 700 (v + v). Mezclar 300 ml de agua con 700 ml de la mezcla de metanol y acetonitrilo (3.5). |
4. Instrumental
|
4.1. |
Agitador de laboratorio o agitador magnético. |
|
4.2. |
Papel de filtro de fibra de vidrio (Whatman GF/A o equivalente). |
|
4.3. |
Columna de vidrio (de 300 mm a 400 mm de longitud y 10 mm aproximadamente de diámetro interior) con frita de vidrio sinterizado y válvula de extracción.
|
|
4.4. |
Equipo para CLAR con un sistema de inyección que permita inyectar volúmenes de 20 μl. |
|
4.4.1. |
Columna cromatográfica de líquidos: 300 mm x 4 mm, C18, relleno de 10 μm, o equivalente. |
|
4.4.2. |
Detector de UV de longitud de onda ajustable, o detector de red de diodos que funcione en el intervalo de 225-400 nm. |
|
4.5. |
Filtro de membrana de 0,22 μm. |
|
4.6. |
Filtro de membrana de 0,45 μm. |
|
4.7. |
Baño ultrasónico. |
5. Procedimiento
|
Nota: |
El carbadox es sensible a la luz. Realizar todas las operaciones con luz tenue o con material de vidrio ámbar o envuelto con papel de aluminio. |
5.1. Generalidades.
5.1.1.
Para el ensayo de recuperación (5.1.2) deberá analizarse un pienso en blanco, a fin de comprobar la ausencia de carbadox y de sustancias interferentes. El pienso en blanco deberá ser de tipo similar al de la muestra y en su análisis no debe detectarse carbadox ni sustancias interferentes.
5.1.2.
Deberá realizarse un ensayo de recuperación analizando el pienso en blanco (5.1.1), que se habrá enriquecido añadiendo una cantidad de carbadox similar a la presente en la muestra. Para enriquecer a un nivel de 50 mg/kg, transvasar 5,0 ml de la solución patrón madre (3.11.1) a un Erlenmeyer de 200 ml. Evaporar la solución en una corriente de nitrógeno hasta que resten 0,5 ml, aproximadamente. Añadir 10 g del pienso en blanco, mezclar y esperar diez minutos antes de proceder a la extracción (5.2).
Si no se dispone de un pienso en blanco de tipo similar al de la muestra (véase el punto 5.1.1), también puede realizarse un ensayo de recuperación por el método de adición de patrón. En este caso, la muestra se enriquece con una cantidad de carbadox similar a la que ya esté presente en ella. Esta muestra se analiza junto con la muestra sin enriquecer, y la recuperación puede calcularse por sustracción.
5.2. Extracción.
5.2.1.
Pesar, con una precisión de 0,01 g, 10 g de la muestra preparada y pasarlos a un Erlenmeyer de 200 ml. Añadir 15,0 ml de agua, mezclar y equilibrar durante cinco minutos. Añadir 35,0 ml de la mezcla de metanol y acetonitrilo (3.5), tapar y agitar durante 30 minutos en el agitador o remover en el agitador magnético (4.1). Filtrar la solución por un papel de filtro de fibra de vidrio (4.2). Conservar esta solución para la fase de purificación (5.3).
5.2.2.
Pesar, con una precisión de 0,001 g, 1 g de la muestra sin moler y pasarlo a un Erlenmeyer de 200 ml. Añadir 15,0 ml de agua, mezclar y equilibrar durante cinco minutos. Añadir 35,0 ml de la mezcla de metanol y acetonitrilo (3.5), tapar y agitar durante 30 minutos en el agitador o remover en el agitador magnético (4.1). Filtrar la solución por un papel de filtro de fibra de vidrio (4.2).
Pipetear una parte alícuota de filtrado a un matraz aforado de 50 ml. Añadir 15,0 ml de agua, enrasar con la mezcla de metanol y acetonitrilo (3.5) y mezclar. La concentración de carbadox de la solución final deberá ser de aproximadamente 10 μg/ml. Filtrar una parte alícuota por un filtro de 0,45 μm (4.6).
Proceder a la determinación mediante CLAR (5.4).
5.2.3.
Pesar, con una precisión de 0,001 g, 0,2 g de la muestra sin moler y pasarlos a un Erlenmeyer de 250 ml. Añadir 45,0 ml de agua, mezclar y equilibrar durante cinco minutos. Añadir 105,0 ml de mezcla de metanol y acetonitrilo (3.5), tapar y homogeneizar. Aplicar ultrasonidos (4.7) a la muestra durante 15 minutos y, a continuación, agitar o remover durante otros 15 minutos (4.1). Filtrar la solución por un papel de filtro de fibra de vidrio (4.2).
Diluir una parte alícuota de filtrado con una mezcla de agua, metanol y acetonitrilo (3.12) hasta obtener una concentración de carbadox de 10-15 μg/ml (para un preparado al 10 %, el factor de dilución es 10). Filtrar una parte alícuota por un filtro de 0,45 μm (4.6).
Proceder a la determinación mediante CLAR (5.4).
5.3. Purificación.
5.3.1.
Pesar 4 g de óxido de aluminio (3.4) y transferirlos a la columna de vidrio (4.3).
5.3.2.
Pasar 15 ml del extracto filtrado (5.2.1) a la columna de óxido de aluminio y desechar los primeros 2 ml de eluido. Recoger los 5 ml siguientes y filtrar una parte alícuota por un filtro de 0,45 μm (4.6).
Proceder a la determinación mediante CLAR (5.4).
5.4. Determinación mediante CLAR.
5.4.1.
Las siguientes condiciones se ofrecen con carácter orientativo; pueden aplicarse otras si arrojan resultados equivalentes.
|
Columna cromatográfica |
|
|
de líquidos (4.4.1): |
300 mm × 4 mm, C18, relleno de 10 μm, o equivalente |
|
Fase móvil (3.10): |
Mezcla de solución reguladora de acetato (3.9) y acetonitrilo (3.2), 825 + 175 (v + v) |
|
Caudal: |
1,5-2 ml/min. |
|
Longitud de onda de detección: |
365 nm |
|
Volumen de inyección: |
20 μl |
Comprobar la estabilidad del sistema cromatográfico inyectando varias veces la solución de calibración (3.11.2) de 5,0 μg/ml, hasta que se alcancen alturas (áreas) de pico y tiempos de retención constantes.
5.4.2.
Inyectar varias veces cada solución de calibración (3.11.2) y medir las alturas (áreas) de pico de cada concentración. Trazar una curva de calibración empleando las alturas o áreas de pico medias de las soluciones de calibración como ordenadas y las concentraciones correspondientes, en microgramos por mililitro, como abscisas.
5.4.3.
Inyectar varias veces el extracto de muestra ([5.3.2] para los piensos, [5.2.2] para las premezclas y [5.2.3] para los preparados) y determinar la altura (área) media de los picos de carbadox.
6. Cálculo de los resultados
Determinar la concentración de carbadox de la solución de muestra, en microgramos por mililitro, a partir de la altura (área) media de sus picos de carbadox, tomando como referencia la curva de calibración (5.4.2).
6.1. Piensos.
El contenido de carbadox p (mg/kg) de la muestra viene dado por la siguiente fórmula:
 [mg/kg]
[mg/kg]
en la cual:
|
c |
= |
concentración de carbadox del extracto de muestra (5.3.2), en microgramos por mililitro; |
|
V1 |
= |
volumen de extracción, en mililitros (es decir, 50); |
|
m |
= |
peso de la porción de ensayo, en gramos. |
6.2. Premezclas y preparados.
El contenido de carbadox p (mg/kg) de la muestra viene dado por la siguiente fórmula:
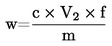 [mg/kg]
[mg/kg]
en la cual:
|
c |
= |
concentración de carbadox del extracto de muestra (5.2.2 o 5.2.3), en microgramos por mililitro; |
|
V2 |
= |
volumen de extracción, en mililitros (es decir, 50 para las premezclas y 150 para los preparados); |
|
f |
= |
factor de dilución según el punto 5.2.2 (premezclas) o 5.2.3 (preparados); |
|
m |
= |
peso de la porción de ensayo, en gramos. |
7. Validación de los resultados
7.1. Identidad.
La identidad del analito puede confirmarse mediante cocromatografía, o utilizando un detector de red de diodos con el cual se comparan los espectros del extracto de muestra y de la solución de calibración (3.11.2) de 10,0 μg/ml.
7.1.1.
Enriquecer un extracto de muestra añadiéndole una cantidad apropiada de solución de calibración (3.11.2). La cantidad de carbadox añadida debe ser similar a la cantidad estimada de carbadox hallada en el extracto de muestra.
Solo la altura del pico de carbadox deberá aumentar en función de la cantidad añadida y de la dilución del extracto. La anchura del pico, a la mitad de su altura máxima, debe ser igual ± 10 % a la anchura original.
7.1.2.
Los resultados se evalúan con arreglo a los siguientes criterios:
|
a) |
la longitud de onda de absorción máxima de los espectros de la muestra y del patrón, registrados en el vértice del pico del cromatograma, debe ser la misma dentro de un margen determinado por el poder de resolución del sistema de detección; en el caso de la detección por red de diodos, el margen típico se sitúa en ± 2 nm; |
|
b) |
entre 225 nm y 400 nm, los espectros de la muestra y del patrón registrados en el vértice del pico del cromatograma no deben ser diferentes por lo que respecta a aquellas partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los dos espectros excede del 15 % de la absorbancia del analito patrón; |
|
c) |
entre 225 nm y 400 nm, los espectros de la pendiente ascendente, del vértice y de la pendiente descendente del pico producido por el extracto de muestra no deben diferir entre sí por lo que respecta a las partes del espectro que se encuentran en un intervalo del 10 % al 100 % de la absorbancia relativa; se cumple este criterio cuando están presentes los mismos máximos y en ningún punto observado la desviación entre los espectros excede del 15 % de la absorbancia del espectro del vértice. |
Si no se cumple alguno de estos criterios, la presencia del analito no queda confirmada.
7.2. Repetibilidad.
Tratándose de contenidos iguales o superiores a 10 mg/kg, la diferencia entre los resultados de dos determinaciones paralelas efectuadas con la misma muestra no debe rebasar el 15 % del resultado superior.
7.3. Recuperación.
La recuperación de la muestra (en blanco) enriquecida deberá ser al menos del 90 %.
8. Resultados de un estudio colaborativo
Se organizó un estudio colaborativo en el que ocho laboratorios analizaron seis piensos, cuatro premezclas y tres preparados. De cada muestra se hicieron análisis por duplicado. (Puede encontrarse información más detallada sobre este estudio colaborativo en Journal of AOAC International, volumen 71, 1988, pp. 484-490). A continuación figuran los resultados del estudio (excluidos los valores atípicos).
Cuadro 1
Resultados del estudio colaborativo correspondientes a los piensos
|
|
Muestra 1 |
Muestra 2 |
Muestra 3 |
Muestra 4 |
Muestra 5 |
Muestra 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Media (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr ( %) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR ( %) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Contenido nominal (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Cuadro 2
Resultados del estudio colaborativo correspondientes a las premezclas y los preparados
|
|
Premezclas |
Preparados |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Media (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr ( %) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR ( %) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Contenido nominal (mg/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
número de laboratorios |
|
n |
= |
número de valores individuales |
|
Sr |
= |
desviación típica de la repetibilidad |
|
CVr |
= |
coeficiente de variación de la repetibilidad |
|
SR |
= |
desviación típica de la reproducibilidad |
|
CVR |
= |
coeficiente de variación de la reproducibilidad |
ANEXO IX
TABLAS DE CORRESPONDENCIAS A LAS QUE SE REFIERE EL ARTÍCULO 6
1. Directiva 71/250/CEE
|
Directiva 71/250/CEE |
Presente Reglamento |
|
Artículo 1, párrafo primero |
Artículo 3 |
|
Artículo 1, párrafo segundo |
Artículo 2 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo, parte 1 |
Anexo II |
|
Anexo, parte 2 |
—— |
|
Anexo, parte 3 |
—— |
|
Anexo, parte 4 |
Anexo III, parte O |
|
Anexo, parte 5 |
Anexo III, parte M |
|
Anexo, parte 6 |
Anexo III, parte N |
|
Anexo, parte 7 |
Anexo III, parte Q |
|
Anexo, parte 9 |
Anexo III, parte K |
|
Anexo, parte 10 |
—— |
|
Anexo, parte 11 |
—— |
|
Anexo, parte 12 |
Anexo III, parte J |
|
Anexo, parte 14 |
Anexo III, parte D |
|
Anexo, parte 16 |
—— |
2. Directiva 71/393/CEE
|
Directiva 71/393/CEE |
Presente Reglamento |
|
Artículo 1 |
Artículo 3 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo, parte I |
Anexo III, parte A |
|
Anexo, parte II |
Anexo III, parte E |
|
Anexo, parte III |
Anexo III, parte P |
|
Anexo, parte IV |
Anexo III, parte H |
3. Directiva 72/199/CEE
|
Directiva 72/199/CEE |
El presente Reglamento |
|
Artículo 1 |
Artículo 3 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Artículo 4 |
—— |
|
Anexo I, parte 1 |
Anexo III, parte L |
|
Anexo I, parte 2 |
Anexo III, parte C |
|
Anexo I, parte 3 |
—— |
|
Anexo I, parte 4 |
—— |
|
Anexo I, parte 5 |
Anexo V, parte A |
|
Anexo II |
—— |
4. Directiva 73/46/CEE
|
Directiva 73/46/CEE |
El presente Reglamento |
|
Artículo 1 |
Artículo 3 |
|
Artículo 3 |
—— |
|
Artículo 4 |
—— |
|
Anexo I, parte 1 |
Anexo III, parte B |
|
Anexo I, parte 2 |
—— |
|
Anexo I, parte 3 |
Anexo III, parte I |
5. Directiva 76/371/CEE
|
Directiva 76/371/CEE |
Presente Reglamento |
|
Artículo 1 |
Artículo 1 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo |
Anexo I |
6. Directiva 76/372/CEE
|
Directiva 76/372/CEE |
Presente Reglamento |
|
Artículo 1 |
—— |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo |
—— |
7. Directiva 78/633/CEE
|
Directiva 78/633/CEE |
Presente Reglamento |
|
Artículo 1 |
Artículo 3 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo, parte 1 |
—— |
|
Anexo, parte 2 |
—— |
|
Anexo, parte 3 |
Anexo IV, parte C |
8. Directiva 81/715/CEE
|
Directiva 81/715/CEE |
Presente Reglamento |
|
Artículo 1 |
—— |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo |
—— |
9. Directiva 84/425/CEE
|
Directiva 84/425/CEE |
Presente Reglamento |
|
Artículo 1 |
—— |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo |
—— |
10. Directiva 86/174/CEE
|
Directiva 86/174/CEE |
Presente Reglamento |
|
Artículo 1 |
Artículo 4 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo |
Anexo VII |
11. Directiva 93/70/CEE
|
Directiva 93/70/CEE |
Presente Reglamento |
|
Artículo 1 |
Artículo 3 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo |
Anexo IV, parte D |
12. Directiva 93/117/CE
|
Directiva 93/117/CE |
Presente Reglamento |
|
Artículo 1 |
Artículos 3 y 5 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Anexo, parte 1 |
Anexo IV, parte E |
|
Anexo, parte 2 |
Anexo VIII, parte A |
13. Directiva 98/64/CE
|
Directiva 98/64/CE |
Presente Reglamento |
|
Artículo 1 |
Artículos 3 y 5 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Artículo 4 |
—— |
|
Anexo, parte A |
Anexo III, parte F |
|
Anexo, parte C |
Anexo VIII, parte B |
14. Directiva 1999/27/CE
|
Directiva 1999/27/CE |
Presente Reglamento |
|
Artículo 1 |
Artículos 3 y 5 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Artículo 4 |
—— |
|
Artículo 5 |
—— |
|
Artículo 6 |
—— |
|
Artículo 7 |
—— |
|
Anexo, parte A |
Anexo VIII, parte C |
|
Anexo, parte B |
Anexo IV, parte F |
|
Anexo, parte C |
Anexo VIII, parte D |
15. Directiva 1999/76/CE
|
Directiva 1999/76/CE |
Presente Reglamento |
|
Artículo 1 |
Artículo 3 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Artículo 4 |
—— |
|
Anexo |
Anexo IV, parte G |
16. Directiva 2000/45/CE
|
Directiva 2000/45/CE |
Presente Reglamento |
|
Artículo 1 |
Artículo 3 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Artículo 4 |
—— |
|
Anexo, parte A |
Anexo IV, parte A |
|
Anexo, parte B |
Anexo IV, parte B |
|
Anexo, parte C |
Anexo III, parte G |
17. Directiva 2002/70/CE
|
Directiva 2002/70/CE |
Presente Reglamento |
|
Artículo 1 |
Artículo 1 |
|
Artículo 2 |
Artículos 2 y 3 |
|
Artículo 3 |
—— |
|
Artículo 4 |
—— |
|
Artículo 5 |
—— |
|
Anexo I |
Anexo I y anexo V, parte B (I) |
|
Anexo II |
Anexo II y anexo V, parte B (II) |
18. Directiva 2003/126/CE
|
Directiva 2003/126/CE |
Presente Reglamento |
|
Artículo 1 |
Artículo 3 |
|
Artículo 2 |
—— |
|
Artículo 3 |
—— |
|
Artículo 4 |
—— |
|
Artículo 5 |
—— |
|
Artículo 6 |
—— |
|
Anexo |
Anexo VI |
|
26.2.2009 |
ES |
Diario Oficial de la Unión Europea |
L 54/s3 |
NOTA AL LECTORLas instituciones han decidido no mencionar en sus textos la última modificación de los actos citados.Salvo indicación en contrario, se entenderá que los actos a los que se hace referencia en los textos aquí publicados son los actos en su versión actualmente en vigor.