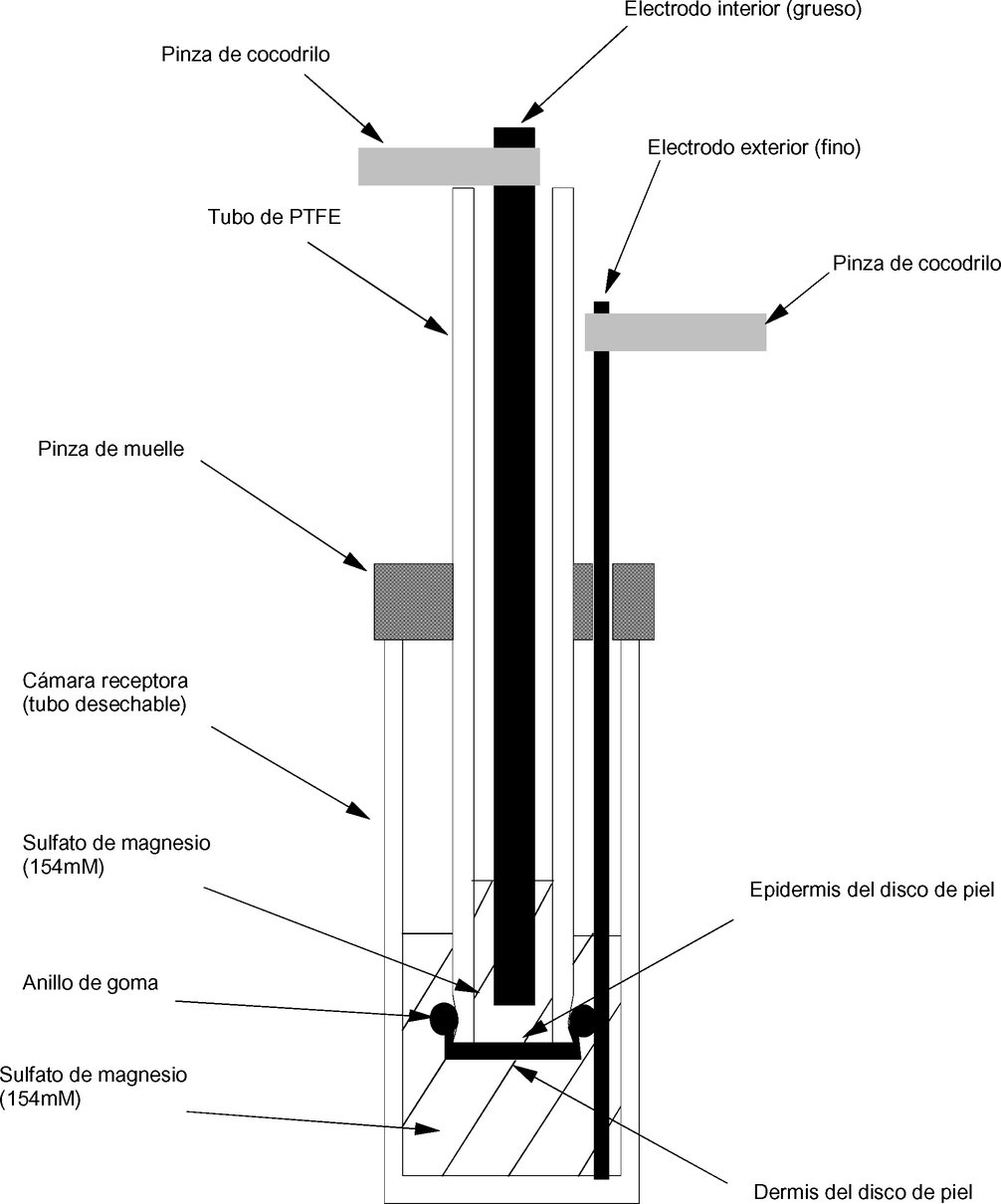
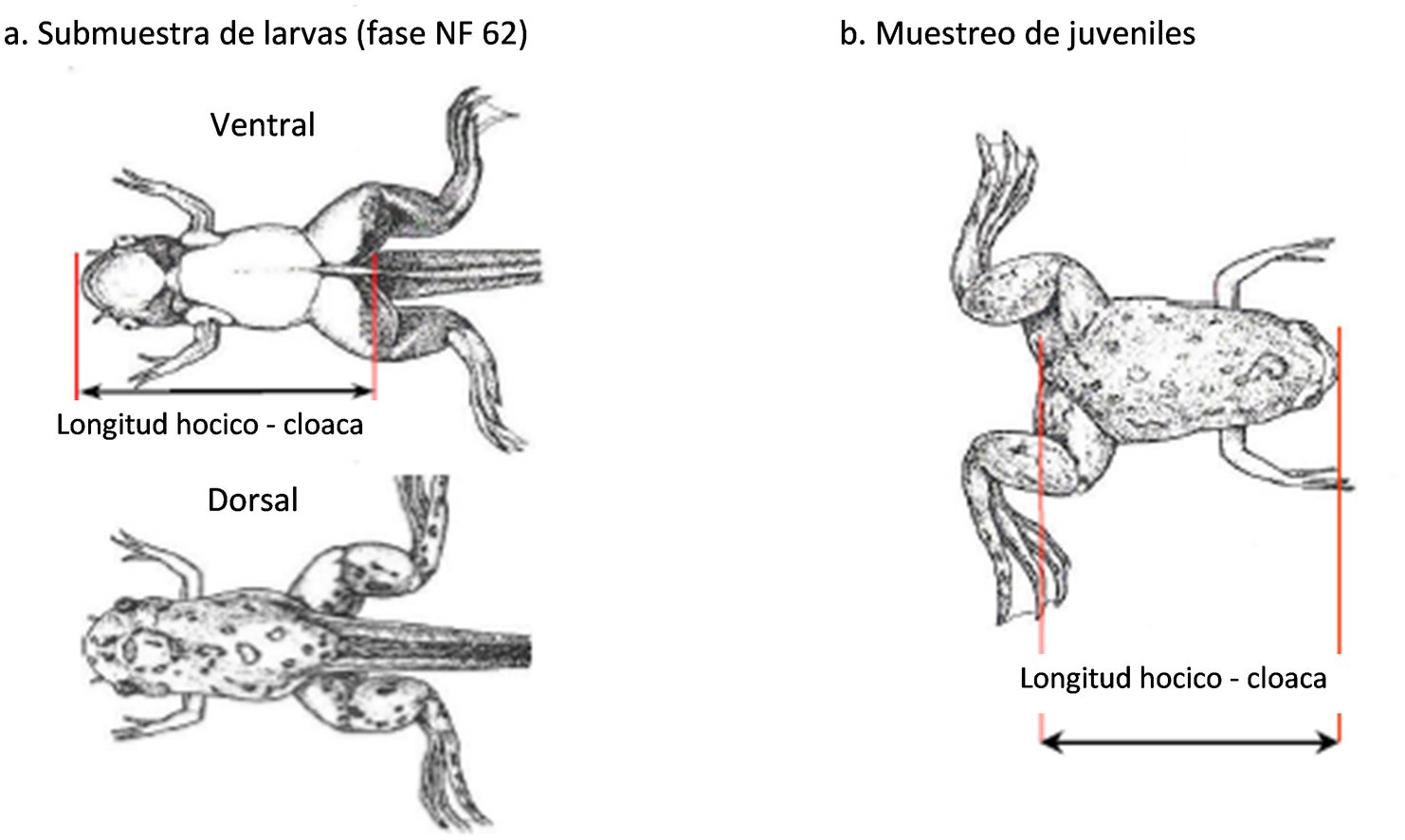
|
8)
|
En la parte B se añaden los capítulos siguientes:
«B.63 ENSAYO DE CRIBADO DE TOXICIDAD PARA LA REPRODUCCIÓN O EL DESARROLLO
INTRODUCCIÓN
|
1.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 421 de la OCDE (2016). Las directrices de la OCDE sobre los ensayos de productos se revisan periódicamente a la luz del progreso científico,. Las directrices de ensayo de cribado TG 421 originales se adoptaron en 1995 sobre la base de un protocolo de un «ensayo de cribado preliminar de toxicidad para la reproducción», debatido en dos reuniones de expertos, celebradas en Londres en 1990 (1) y en Tokio en 1992 (2).
|
|
2.
|
Este método de ensayo se ha actualizado con parámetros pertinentes de alteración endocrina, como continuación de la actividad prioritaria iniciada en 1998 en la OCDE para revisar las directrices de ensayo vigentes y elaborar nuevas directrices de ensayo para el cribado y el ensayo de los alteradores endocrinos potenciales (3). Por ejemplo, las directrices de ensayo TG 407 de la OCDE [Toxicidad oral por administración continuada (28 días) en roedores, capítulo B.7 del presente anexo] se reforzaron en 2008 con parámetros adecuados para detectar la actividad endocrina de los productos problema. El objetivo de la actualización de las directrices de ensayo TG 421 era incluir algunos parámetros relevantes de alteración endocrina en las directrices de ensayo de cribado, en las que los períodos de exposición cubren algunos de los períodos sensibles durante el desarrollo (periodos prenatal y posnatal inicial).
|
|
3.
|
En las directrices de ensayo TG 421 se incluyeron los parámetros adicionales pertinentes de alteración endocrina seleccionados, que también forman parte de las directrices TG 443 (Estudio ampliado de toxicidad para la reproducción en una generación, capítulo B.56 del presente anexo), sobre la base de un estudio de viabilidad sobre cuestiones científicas y técnicas relacionadas con su inclusión, así como las posibles adaptaciones del diseño del ensayo necesarias para su inclusión (4).
|
|
4.
|
El presente método de ensayo está diseñado para generar información limitada sobre los efectos de un producto problema en el comportamiento reproductor de los machos y de las hembras, tales como la actividad gonadal, el comportamiento relativo al apareamiento, la concepción, el desarrollo del embrión y el parto. No es una alternativa ni sustituye a los métodos de ensayo existentes B.31, B.34, B.35 o B.56.
|
CONSIDERACIONES INICIALES
|
5.
|
Este método de ensayo de cribado puede utilizarse para proporcionar información inicial en cuanto a los posibles efectos sobre la reproducción o el desarrollo, en una fase temprana de la evaluación de las propiedades toxicológicas de los productos, o en cuanto a productos preocupantes. También puede utilizarse como parte de una serie de ensayos iniciales de cribado con sustancias existentes sobre las que se dispone de escasa o nula información toxicológica, como estudio de determinación del intervalo de dosis para estudios más amplios sobre la reproducción o el desarrollo, o en cualquier otro caso que se considere pertinente. Al realizar el estudio, deben aplicarse los principios y consideraciones básicos recogidos en el documento de orientación de la OCDE n.o 19 sobre el reconocimiento, evaluación y utilización de los signos clínicos como parámetros de tratamiento compasivo para los animales de experimentación empleados en las evaluaciones de la seguridad (5).
|
|
6.
|
El presente método de ensayo no proporciona información completa sobre todos los aspectos de la reproducción y el desarrollo. En particular, solo ofrece medios limitados para detectar las manifestaciones postnatales de una exposición prenatal, o los efectos que puedan inducirse durante la exposición posnatal. Debido (entre otras razones) al número relativamente reducido de animales en los grupos tratados, la selectividad de los criterios de valoración y la corta duración del estudio, este método no aporta pruebas para hacer alegaciones definitivas de efectos nulos. Por otra parte, a falta de datos de otros ensayos de toxicidad para la reproducción o el desarrollo, los resultados positivos son útiles para la evaluación inicial del peligro y contribuyen a la adopción de decisiones con respecto a la necesidad y el calendario de ensayos adicionales.
|
|
7.
|
Los resultados obtenidos por medio de parámetros endocrinos deben contemplarse en el contexto del «Marco conceptual para el estudio y la evaluación de los alteradores endocrinos» (Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals) de la OCDE (6). En este Marco conceptual, las directrices TG 421 mejoradas de la OCDE figuran en el nivel 4 como ensayo in vivo que proporciona datos sobre los efectos adversos relativos a los parámetros endocrinos pertinentes. Sin embargo, una señal endocrina podría no considerarse prueba suficiente por sí misma de que el producto problema es un alterador endocrino.
|
|
8.
|
En este método de ensayo se parte de la administración oral del producto problema. Es posible que se requieran modificaciones si se utilizan otras vías de exposición.
|
|
9.
|
Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines normativos, debe considerarse si podría proporcionar resultados adecuados a tales fines y, en caso afirmativo, por qué. Dichas consideraciones no son necesarias cuando los requisitos normativos estipulan que la mezcla debe someterse a ensayo.
|
|
10.
|
En el apéndice 1 se dan las definiciones utilizadas.
|
PRINCIPIO DEL ENSAYO
|
11.
|
Se administra el producto problema en dosis escalonadas a varios grupos de machos y hembras. Los machos deben recibir el producto durante un mínimo de cuatro semanas y hasta el día anterior al sacrificio programado (incluido dicho día) (lo que comprende un mínimo de dos semanas antes del apareamiento, el período de apareamiento y, aproximadamente, dos semanas después del apareamiento). Teniendo en cuenta lo limitado del período de administración a los machos antes del apareamiento, la fertilidad puede no ser un indicador específico sensible de la toxicidad testicular. Por lo tanto, es esencial un examen histológico detallado de los testículos. Para permitir la detección de la mayor parte de los efectos sobre la fertilidad masculina y la espermatogénesis, se considera suficiente la combinación de un período de administración antes del apareamiento de dos semanas y, posteriormente, observaciones sobre el apareamiento y la fertilidad, con un período de administración total de al menos cuatro semanas, seguido de un examen histopatológico detallado de las gónadas masculinas.
|
|
12.
|
Las hembras deben recibir el producto problema a lo largo de todo el estudio. Se incluyen aquí dos semanas antes del apareamiento (con el objetivo de abarcar por lo menos dos ciclos estrales completos), el período de tiempo variable hasta la concepción, la duración de la gestación y al menos trece días después del parto, hasta el día anterior al sacrifico programado, inclusive.
|
|
13.
|
La duración del estudio, tras la aclimatación y la evaluación del ciclo estral antes de la administración del producto problema, depende del comportamiento de las hembras y es de aproximadamente 63 días, [14 días como mínimo antes del apareamiento, (hasta) 14 días de apareamiento, 22 días de gestación y 13 días de lactancia].
|
|
14.
|
A lo largo del período de administración, se observa a los animales todos los días atentamente con el fin de descubrir la posible aparición de signos de toxicidad. Se practica la autopsia a los animales que mueran o sean sacrificados durante el período del ensayo, y al final del mismo se sacrifican y someten a autopsia los animales supervivientes.
|
DESCRIPCIÓN DEL MÉTODO
Selección de la especie animal
|
15.
|
Este método de ensayo está diseñado para su uso con la rata. Si los parámetros especificados en el presente método de ensayo se investigan en otra especie de roedor, habrá que justificarlo minuciosamente. En el programa internacional de validación de la detección de alteradores endocrinos en las directrices de ensayo TG 407 de la OCDE (correspondientes al capítulo B.7 del presente anexo), la única especie utilizada fue la rata. Debe evitarse la utilización de cepas que presenten un bajo índice de fertilidad o una incidencia notoriamente elevada de anomalías del desarrollo. Han de utilizarse animales vírgenes sanos, no sometidos a experimentación previa. Deben especificarse la especie, la cepa, el sexo, el peso y la edad de los animales utilizados en el ensayo. Al empezar el estudio, la variación del peso de los animales utilizados ha de ser mínima y no exceder del ± 20 % del peso medio de cada sexo. Cuando el estudio se realice como fase preliminar de un estudio a largo plazo o de una generación completa, es preferible que en ambos estudios se utilicen animales de la misma cepa y de la misma fuente.
|
Alojamiento y alimentación de los animales
|
16.
|
Todos los procedimientos serán conformes a las normas habituales de cuidado de los animales de laboratorio. El local de los animales de experimentación ha de estar a una temperatura de 22 °C (± 3 °C). Aunque la humedad relativa debe ser del 30 % como mínimo y preferiblemente no superar el 70 %, excepto durante la limpieza del animalario, el objetivo debe ser el 50-60 %. La iluminación debe ser artificial, con un fotoperíodo de 12 horas de luz y 12 horas de oscuridad. Para la alimentación se podrán utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber. La elección de la dieta puede verse influida por la necesidad de garantizar una mezcla conveniente de la sustancia problema si se administra por este método.
|
|
17.
|
Los animales deben alojarse en pequeños grupos del mismo sexo; pueden alojarse de forma individual si está científicamente justificado. En caso de que se utilicen jaulas colectivas, no habrá más de cinco animales en cada jaula. El apareamiento debe llevarse a cabo en jaulas adecuadas al efecto. Las hembras gestantes deben enjaularse de forma individual y disponer de materiales de nidificación. Las hembras en período de lactancia deben enjaularse de forma individual con sus crías.
|
|
18.
|
Se debe analizar regularmente la comida para detectar posibles contaminantes. Se conservará una muestra de la dieta hasta el momento de concluir el informe.
|
Preparación de los animales
|
19.
|
Se reparten al azar animales adultos jóvenes y sanos entre los grupos tratados y los grupos testigo. Las jaulas se deben disponer de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Se identifica a los animales de forma unívoca y se los mantiene en las jaulas al menos cinco días antes de empezar el estudio para que se acostumbren a las condiciones de laboratorio.
|
Preparación de las dosis
|
20.
|
Se recomienda administrar el producto problema oralmente, a menos que se consideren más adecuadas otras vías de administración. Cuando se selecciona la vía oral, el producto problema suele administrarse por sonda; sin embargo, los productos problema pueden administrarse alternativamente con el alimento o el agua de bebida.
|
|
21.
|
En caso necesario, el producto problema se disuelve o suspende en un vehículo adecuado. Se recomienda considerar en primer lugar, siempre que sea posible, el uso de una solución o suspensión acuosa, después el uso de una solución o emulsión oleosa (por ejemplo, en aceite de maíz) y, por último, la posible disolución en otros vehículos. Si se emplean vehículos distintos del agua, deben conocerse sus características tóxicas. Debe determinarse la estabilidad y homogeneidad del producto problema en el vehículo.
|
PROCEDIMIENTO
Número y sexo de los animales
|
22.
|
Se recomienda que cada grupo comience con un mínimo de 10 machos y entre 12 y 13 hembras. Las hembras se evaluarán antes de la exposición en relación con el ciclo estral y no se incluirán en el estudio los animales que no presenten ciclos típicos de 4 a 5 días; por lo tanto, se recomienda añadir más hembras para conseguir 10 hembras por grupo. Excepto en el caso de efectos tóxicos marcados, se espera que de esta forma se consigan al menos 8 hembras gestantes por grupo, que normalmente es el número mínimo aceptable de hembras gestantes por grupo. El objetivo es conseguir suficientes gestaciones y descendientes para garantizar una evaluación significativa del potencial del producto problema para afectar a la fertilidad, gestación, comportamiento materno y lactante, y al crecimiento y desarrollo de los descendientes F1, desde la concepción hasta 13 días después del parto.
|
Posología
|
23.
|
Deben utilizarse generalmente como mínimo tres grupos de ensayo y un grupo testigo. Las dosis pueden basarse en la información procedente de ensayos de toxicidad aguda o en los resultados de estudios de administración continuada. A excepción de la administración del producto problema, los animales del grupo testigo deben tratarse de la misma manera que los de los grupos de ensayo. Si se utiliza un vehículo para administrar el producto problema, el grupo testigo debe recibir el mayor volumen utilizado de dicho vehículo.
|
|
24.
|
Las dosis deben seleccionarse teniendo en cuenta los eventuales datos existentes sobre toxicidad y (toxico)cinética disponibles. También debe tenerse en cuenta que puede haber diferencias de sensibilidad entre los animales gestantes y los no gestantes. La dosis más elevada debe seleccionarse con el propósito de inducir efectos tóxicos pero sin llegar a la muerte ni a un sufrimiento intenso. Posteriormente, debe seleccionarse una secuencia descendente de dosis destinadas a demostrar le eventual relación dosis-respuesta y la ausencia de efectos adversos observados (NOAEL) con la dosis inferior. Los intervalos del doble al cuádruple suelen ser óptimos para establecer los niveles descendentes y frecuentemente es preferible añadir un cuarto grupo de ensayo antes que utilizar intervalos muy amplios (por ejemplo con un factor superior a 10) entre dosis.
|
|
25.
|
Si se observan signos de toxicidad general (por ejemplo, peso corporal reducido, efectos hepáticos, cardíacos, pulmonares o renales, etc.) u otras alteraciones que no son necesariamente respuestas tóxicas (por ejemplo, ingesta de alimentos reducida, hiperplasia hepática), habrá que interpretar con prudencia los efectos observados sobre los parámetros sensibles endocrinos.
|
Ensayo límite
|
26.
|
Si en un estudio oral en el que se administra una sola dosis de al menos 1 000 mg/kg peso corporal/día o, en caso de administración en la comida o en el agua de bebida, en el que se utiliza un porcentaje equivalente en los alimentos o el agua de bebida, siguiendo los procedimientos descritos en este estudio, no se produce ningún efecto tóxico observable y si, a partir de datos de sustancias estructuralmente afines, no debiera esperarse la aparición de toxicidad, puede considerarse innecesario realizar un estudio completo con varias dosis diferentes. El ensayo límite es válido excepto cuando la exposición humana indique la necesidad de utilizar una dosis oral superior. Si se trata de otras vías de administración, como la inhalación o la aplicación cutánea, suelen ser las características fisicoquímicas del producto problema las que determinan la concentración máxima que puede alcanzarse.
|
Administración del producto problema
|
27.
|
El producto problema se administra diariamente a los animales durante 7 días a la semana. Si el producto problema se administra por sonda, debe hacerse en una dosis única y con una sonda gástrica o una cánula de intubación adecuada. El volumen máximo de líquido que puede administrarse en una sola vez depende del tamaño del animal utilizado. Dicho volumen no debe superar 1 ml/100 g de peso corporal, salvo en el caso de las soluciones acuosas, en que puede llegarse a 2 ml/100 g de peso corporal. Salvo en el caso de productos problema irritantes o corrosivos, que por lo general producen efectos exacerbados con concentraciones mayores, deberá reducirse al mínimo la variabilidad del volumen de ensayo ajustando la concentración de manera que el volumen sea el mismo con las diferentes dosis.
|
|
28.
|
En el caso de productos problema administrados con los alimentos o el agua de bebida, es importante cerciorarse de que las cantidades administradas de producto problema no interfieren con la nutrición normal ni con el equilibrio hídrico. Cuando el producto problema se administre con el alimento, se puede utilizar una concentración alimentaria constante (ppm) o bien una dosis constante por unidad de peso corporal de los animales; debe indicarse qué alternativa se ha seguido. Si el producto problema se administra por sonda, la dosis debe darse todos los días a una hora similar, y ajustarse al menos una vez por semana para mantener una dosis constante respecto al peso corporal del animal.
|
Calendario del experimento
|
29.
|
La administración del producto a ambos sexos debe comenzar al menos dos semanas antes del apareamiento, una vez que se han aclimatado durante al menos cinco días y las hembras han sido sometidas a cribado para confirmar que sus ciclos estrales son normales (en un período de dos semanas antes del tratamiento). El estudio debe programarse de tal manera que la evaluación del ciclo estral comience pronto después de que los animales hayan alcanzado la madurez sexual completa. Esto puede variar ligeramente según las diferentes cepas de ratas en distintos laboratorios; por ejemplo, en las ratas Sprague Dawley corresponde a 10 semanas de edad y en las ratas Wistar a unas 12 semanas de edad. Las madres con descendencia deben sacrificarse el día 13 después del parto, o poco después. El día del nacimiento (es decir, cuando se completa el parto) se define como el día 0 después del parto. Las hembras que no presentan signos de apareamiento se sacrifican entre 24 y 26 días después del último día del período de apareamiento. Se continúa la administración del producto problema a los dos sexos durante el período de apareamiento. Los machos deben seguir recibiendo el producto problema después del período de apareamiento, al menos hasta que se haya completado el período mínimo total de tratamiento de 28 días. A continuación se les da muerte o, alternativamente, se conservan y se les sigue administrando el producto para la posible realización de un segundo apareamiento si se considera adecuado.
|
|
30.
|
La administración diaria del producto problema a las hembras parentales debe mantenerse a lo largo de toda la gestación y, como mínimo, hasta el día 13 después del parto o el día anterior al sacrificio, inclusive. En los estudios en que el producto problema se administre por inhalación o por vía cutánea, esta administración debe continuar como mínimo hasta el día 19 de la gestación, inclusive, y debe volver a iniciarse lo antes posible y, a más tardar, el día posnatal (DPN) 4.
|
|
31.
|
En el apéndice 2 figura un diagrama del programa experimental.
|
Procedimiento del apareamiento
|
32.
|
Normalmente, en este estudio deben utilizarse apareamientos 1: 1 (un macho con una hembra). Pueden darse excepciones en caso de muertes ocasionales de machos. Se recomienda colocar a la hembra con el mismo macho hasta que se observen signos de apareamiento o hayan transcurrido dos semanas. Las hembras deben examinarse cada mañana para determinar la presencia de esperma o de tapón vaginal. El día 0 de la gestación se define como el día en que se confirman los signos de apareamiento (se observa la presencia de un tapón vaginal o de esperma). Si no tiene éxito el apareamiento, puede plantearse otro intento de aparear hembras con machos de comprobada capacidad reproductora del mismo grupo.
|
Tamaño de la camada
|
33.
|
El día 4 después del nacimiento, podrá ajustarse el tamaño de cada camada eliminando las crías excedentarias mediante selección aleatoria con el fin de obtener, en la medida de lo posible, cuatro o cinco crías por sexo y por camada en función del tamaño normal de la camada en la cepa de ratas utilizada. Se deben tomar muestras de sangre de dos de las crías excedentarias; tales muestras se mezclan y se utilizan para determinar los niveles de T4 en el suero. No es adecuado eliminar crías de forma selectiva, por ejemplo sobre la base del peso corporal o la distancia anogenital (DAG). Cuando el número de crías machos o hembras impida lograr que cada camada cuente con cuatro o cinco de cada sexo, es aceptable una adaptación parcial (por ejemplo, seis machos y cuatro hembras). No se eliminará ninguna cría cuando el tamaño de la camada descienda por debajo del objetivo de la eliminación (8 o 10 crías/camada). Si solo se dispone de una única cría por encima del objetivo de la eliminación, solo se eliminará una cría, que se utilizará para la extracción de sangre con el fin de efectuar la posible evaluación de T4 en el suero.
|
|
34.
|
Si no se ajusta el tamaño de la camada, se sacrifican dos crías por camada el día 4 después del nacimiento y se toman muestras de sangre para medir las concentraciones de la hormona tiroidea en el suero. Si es posible, las dos crías por camada deben ser hembras, a fin de reservar las crías macho para las evaluaciones del mantenimiento de pezones, salvo en caso de que la eliminación de estas crías no deje hembras suficientes para la evaluación en el momento del sacrificio. No se eliminarán las crías cuando el tamaño de la camada descienda por debajo de 8 o 10 crías/camada (dependiendo del tamaño normal de la camada en la cepa de ratas utilizada). Si solo se dispone de una única cría por encima del tamaño normal de la camada, solo se eliminará una cría, que se utilizará para la extracción de sangre con el fin de efectuar la posible evaluación de T4 en el suero.
|
OBSERVACIONES EN VIVO
Observaciones clínicas
|
35.
|
Durante todo el ensayo, deben hacerse observaciones clínicas generales al menos una vez al día, y con mayor frecuencia cuando se observen signos de toxicidad. Deben hacerse preferentemente a la misma hora u horas cada día y teniendo en cuenta el período más agudo de los efectos previstos tras la administración. Se deben registrar los cambios de conducta pertinentes, los signos de parto difícil o prolongado y todos los signos de toxicidad, incluida la mortalidad. Estos registros deben incluir el momento de aparición, el grado y la duración de los signos de toxicidad.
|
Peso corporal y consumo de alimento y agua
|
36.
|
Los machos y las hembras deben pesarse el primer día de la administración del producto problema, después al menos una vez por semana, y en el momento del sacrificio. Durante la gestación, las hembras deben pesarse en los días 0, 7, 14 y 20 y dentro de las 24 horas siguientes al parto (día 0 o 1 después del parto) y al menos en los días 4 y 13 después del parto. Estas observaciones se registran respecto a cada animal adulto por separado.
|
|
37.
|
Durante el período previo al apareamiento, la gestación y la lactancia, el consumo de alimentos debe medirse al menos semanalmente. La medición del consumo de alimentos durante el apareamiento es opcional. El consumo de agua durante estos períodos también debe medirse si el producto problema se administra a través del agua de bebida.
|
Ciclos estrales
|
38.
|
Deben supervisarse los ciclos estrales antes de que se inicie el tratamiento, a fin de seleccionar para el estudio las hembras con ciclos regulares (véase el punto 22). También hay que efectuar un seguimiento diario de los frotis vaginales desde el principio del período de tratamiento hasta que se observen signos de apareamiento. Si hay preocupación por que los efectos del estrés agudo puedan modificar los ciclos estrales con el inicio de la administración del producto, los laboratorios pueden exponer a los animales de ensayo durante 2 semanas, hacer a continuación frotis vaginales diarios para supervisar el ciclo estral durante un mínimo de dos semanas durante el período previo al apareamiento, y continuar la supervisión en el período de apareamiento hasta que haya signos de apareamiento. En el momento de la obtención de las células de la vagina o del cuello del útero, hay que tener cuidado para evitar la alteración de la mucosa, que podría provocar una pseudogestación (7) (8).
|
Parámetros de los descendientes
|
39.
|
Debe registrarse la duración de la gestación y calcularse a partir del día 0 de la gestación. Cada camada debe examinarse lo antes posible después del parto para determinar el número y sexo de las crías, los mortinatos, los nacidos vivos y los nacidos con retraso evidente (crías que sean significativamente menores que las crías testigo correspondientes) y la presencia de anomalías macroscópicas.
|
|
40.
|
Las crías vivas deben contarse y clasificarse por sexo, y las camadas pesarse dentro de las 24 horas siguientes al parto (día 0 o 1 después del parto) y al menos en los días 4 y 13 después del parto. Además de las observaciones descritas en el punto 35, debe registrarse cualquier eventual comportamiento anómalo de la descendencia.
|
|
41.
|
La distancia anogenital (DAG) de cada cría debe medirse el mismo día postnatal, entre el DPN 0 y el DPN 4. El peso corporal de las crías debe tomarse el día en que se mida la DAG y esta DAG se debe normalizar en función de una medida del tamaño de las crías, de preferencia la raíz cúbica del peso corporal (9). El número de pezones/areolas en las crías macho debe contarse el DPN 12 o 13, según lo recomendado en el documento de orientaciones GD 151 de la OCDE (10).
|
Bioquímica clínica
|
42.
|
Se toman muestras de sangre de un lugar definido sobre la base del siguiente calendario:
|
—
|
de al menos dos crías por camada el día 4 después del parto, si el número de crías lo permite (véanse los puntos 33-34),
|
|
—
|
de todas las madres y al menos dos crías por camada, en el momento del sacrificio el día 13, y
|
|
—
|
de todos los machos adultos, en el momento del sacrificio.
|
|
Todas las muestras de sangre se conservan en condiciones adecuadas. Las muestras de sangre de las crías del día 13 y las de los machos adultos se evalúan en cuanto a los niveles de hormonas tiroideas en suero (T4). Si procede, se lleva a cabo una evaluación adicional de la T4 en las muestras de sangre de las madres y de las crías del día 4. Como opción pueden medirse otras hormonas si son pertinentes. La sangre de las crías puede ponerse en común por camadas para efectuar los análisis de hormonas tiroideas. Las hormonas tiroideas (T4 y TSH) deben medirse preferentemente como «total».
|
43.
|
Los siguientes factores pueden influir en la variabilidad y en el valor absoluto de las concentraciones hormonales:
|
—
|
el momento del sacrificio, debido a la variación diurna de las concentraciones hormonales,
|
|
—
|
el método de sacrificio, que se escogerá para evitar a los animales un sufrimiento indebido que pueda influir en las concentraciones hormonales,
|
|
—
|
los equipos de ensayo para la determinación de las hormonas, que pueden presentar diferencias en sus curvas patrón.
|
|
|
44.
|
Las muestras de plasma destinadas específicamente a la determinación de las hormonas deben obtenerse a una hora comparable del día. Las cifras obtenidas al analizar las concentraciones de hormonas varían con los equipos comerciales de ensayo que se usan para el análisis.
|
Anatomía patológica
Autopsia macroscópica
|
45.
|
En el momento del sacrificio o de la muerte en el transcurso del estudio, los animales adultos deben someterse a un examen macroscópico para detectar los eventuales cambios patológicos o anomalías. Se debe prestar especial atención a los órganos sexuales. Debe registrarse el número de puntos de implantación. Deben examinarse frotis vaginales en la mañana del día de la autopsia para determinar la fase del ciclo estral y poder establecer una correlación con el examen histopatológico de los ovarios.
|
|
46.
|
Los testículos y epidídimos, así como la próstata y las vesículas seminales con las glándulas coagulantes en conjunto, de todos los machos adultos deben limpiarse de los tejidos adherentes, según proceda, y su peso húmedo debe tomarse lo antes posible tras la disección para evitar su desecación. Además, el peso de órganos opcionales podría incluir el complejo del músculo elevador del ano y el músculo bulbocavernoso, las glándulas de Cowper y el glande del pene en los machos y los ovarios emparejados (peso húmedo) y el útero (incluido el cuello) en las hembras; si se incluyen, estos pesos deben tomarse lo antes posible tras la disección.
|
|
47.
|
Las crías muertas y las crías sacrificadas el día 13 después del parto, o poco después, deben, al menos, ser examinadas externamente de forma detenida para detectar la eventual presencia de anomalías macroscópicas. Debe prestarse especial atención a los órganos genitales externos, en los que debe estudiarse la eventual presencia de signos de alteración en el desarrollo. El día 13 debe tomarse y conservarse la glándula tiroidea de 1 cría macho y 1 cría hembra por camada.
|
|
48.
|
Se deben conservar los ovarios, testículos, órganos sexuales secundarios (útero y cuello, epidídimos, próstata, vesículas seminales más glándulas coagulantes), glándula tiroidea y todos los órganos que muestren lesiones macroscópicas de todos los animales adultos. No se recomienda la fijación con formol para el examen sistemático de testículos y epidídimos. Un método aceptable es el uso del fijador de Bouin o el de Davidson modificado para estos tejidos (11). Con una aguja se puede puncionar suave y superficialmente la túnica albugínea por ambos polos del órgano para permitir la rápida penetración del fijador.
|
Examen histopatológíco
|
49.
|
Se debe realizar un examen histológico detallado de los ovarios, testículos y epidídimos (con especial énfasis en las fases de espermatogénesis e histopatología de la estructura celular testicular intersticial) de los animales del grupo de dosis más elevada y del grupo testigo. En caso necesario, podrán examinarse los demás órganos conservados, incluida la glándula tiroidea de las crías y de los animales adultos. El peso de la glándula tiroidea puede determinarse después de su fijación. Su disección debe hacerse también de manera muy cuidadosa y solo después de la fijación para evitar daños tisulares, ya que estos podrían comprometer el examen histopatológico. El examen debe ampliarse a los animales de otros grupos de tratamiento si se observan cambios en el grupo que ha recibido la dosis mayor. El documento de orientación sobre histopatología (11) proporciona más información acerca de la disección, fijación, sección e histopatología de los tejidos endocrinos.
|
DATOS E INFORME
Datos
|
50.
|
Los resultados de cada animal deben darse por separado. Además, todos los datos deben resumirse en forma de cuadro que recoja, respecto a cada grupo de ensayo, el número de animales al inicio del ensayo, el número de animales hallados muertos durante el mismo o sacrificados por razones compasivas, el momento de la muerte o del sacrificio compasivo, el número de animales fértiles, el número de hembras gestantes, el número de animales que presenten signos de toxicidad, una descripción de dichos signos (con inclusión del momento de su aparición, duración y gravedad), los tipos de cambios histopatológicos y todos los datos pertinentes sobre la camada. En el apéndice 3 figura un modelo de informe resumido en forma de cuadro que ha demostrado ser muy útil para la evaluación del efecto sobre la reproducción y el desarrollo.
|
|
51.
|
Debido a las dimensiones limitadas del estudio, los análisis estadísticos en forma de ensayos de “significación” tienen un valor limitado respecto a muchos parámetros, especialmente a los de la reproducción. Si se utilizan análisis estadísticos, entonces el método elegido debe ser apropiado para la distribución de la variable examinada, y ser seleccionado antes de que se inicie el estudio. El análisis estadístico de la DAG y del mantenimiento de pezones debe basarse en los datos de cada cría, teniendo en cuenta los efectos de camada. Cuando proceda, la camada es la unidad de análisis. El análisis estadístico del peso corporal de las crías debe basarse en los datos de cada cría, teniendo en cuenta el tamaño de la camada. Debido a lo reducido del tamaño del grupo, la utilización de datos históricos de control (por ejemplo, sobre el tamaño de la camada), en su caso, también puede ser útil como ayuda para la interpretación del estudio.
|
Evaluación de los resultados
|
52.
|
Los resultados de este estudio de toxicidad deben evaluarse en términos de efectos observados, autopsia y resultados microscópicos. La evaluación debe referirse a la relación entre la dosis de producto problema y la presencia o ausencia, incidencia y gravedad de las anomalías, incluyendo lesiones macroscópicas, órganos diana identificados, esterilidad, anomalías clínicas, alteración del comportamiento reproductor y con la prole, cambios del peso corporal, efectos sobre la mortalidad y cualquier otro efecto tóxico.
|
|
53.
|
Debido al breve período de tratamiento de los machos, el examen histopatológico de los testículos y epidídimos debe tenerse en cuenta junto con los datos de fertilidad, al evaluar los efectos para la reproducción masculina. También puede ser útil como ayuda para la interpretación del estudio el uso de datos de controles históricos sobre reproducción o desarrollo (por ejemplo, en cuanto al tamaño de la camada, DAG, mantenimiento de pezones, niveles séricos de T4), cuando se disponga de ellos.
|
|
54.
|
Por motivos de control de calidad, se propone recopilar los datos de los controles históricos y calcular los coeficientes de variación de los datos numéricos, especialmente en cuanto a los parámetros relacionados con la detección de alteradores endocrinos. Estos datos se pueden usar con fines de comparación al evaluar los estudios actuales.
|
Informe del ensayo
|
55.
|
El informe del ensayo debe incluir la información siguiente:
|
|
Producto problema:
|
—
|
origen, número de lote, fecha límite de utilización, si se dispone de ellos;
|
|
—
|
estabilidad del producto problema, si se conoce.
|
|
|
|
Sustancias de un solo componente:
|
—
|
aspecto físico, hidrosolubilidad y otras propiedades fisicoquímicas pertinentes;
|
|
—
|
identificación química, como nombre IUPAC o CAS, número CAS, notación SMILES o InChI, fórmula estructural, pureza, identidad química de las impurezas si procede y es viable en la práctica, etc.
|
|
|
|
Sustancias de componentes múltiples, UVCB y mezclas:
|
—
|
deben caracterizarse en la medida de lo posible por la identidad química (véase más arriba), la cantidad en que están presentes y las propiedades fisicoquímicas pertinentes de sus componentes.
|
|
|
|
Vehículo (si procede):
|
—
|
justificación de la elección del vehículo, si es distinto del agua.
|
|
|
|
Animales de ensayo:
|
—
|
especie y cepa empleada;
|
|
—
|
número, edad y sexo de los animales;
|
|
—
|
origen, condiciones de alojamiento, dieta, etc.;
|
|
—
|
peso de cada animal al inicio del ensayo;
|
|
—
|
justificación de la especie si es distinta de la rata.
|
|
|
|
Condiciones del ensayo:
|
—
|
justificación de la selección de las dosis;
|
|
—
|
datos sobre la formulación del producto problema o su preparación con los alimentos, concentraciones obtenidas, estabilidad y homogeneidad del preparado;
|
|
—
|
datos sobre la administración del producto problema;
|
|
—
|
factor de conversión de la concentración (ppm) del producto problema en los alimentos o en el agua de bebida a dosis reales (mg/kg peso corporal/día), si procede;
|
|
—
|
datos de la calidad de los alimentos y el agua;
|
|
—
|
descripción detallada del procedimiento de aleatorización de la selección de crías para el sacrificio, en su caso.
|
|
|
|
Resultados:
|
—
|
peso corporal y variaciones del mismo;
|
|
—
|
consumo de alimentos y de agua, si se ha medido;
|
|
—
|
datos de la respuesta tóxica por sexo y dosis, incluida la fertilidad, la gestación y cualquier otro signo de toxicidad;
|
|
—
|
duración de la gestación;
|
|
—
|
efectos tóxicos o de otro tipo sobre la reproducción, la descendencia, el crecimiento postnatal, etc.;
|
|
—
|
naturaleza, gravedad y duración de las observaciones clínicas (sean reversibles o no);
|
|
—
|
número de hembras adultas con ciclo estral normal y anormal y duración del ciclo;
|
|
—
|
número de nacidos vivos y de pérdidas postimplantatorias;
|
|
—
|
datos sobre el peso corporal de las crías;
|
|
—
|
DAG de todas las crías (y peso corporal el día de la medición de la DAG);
|
|
—
|
mantenimiento de pezones en las crías macho;
|
|
—
|
niveles de la hormona tiroidea, en machos adultos y crías el día 13 (y madres y crías el día 4 si se miden);
|
|
—
|
número de crías con anomalías visibles macroscópicamente, evaluación macroscópica de los genitales externos, número de nacidos con retraso evidente;
|
|
—
|
momento de la muerte durante el ensayo o indicación de que los animales han sobrevivido hasta el sacrificio;
|
|
—
|
número de implantes, tamaño de la camada y pesos de la camada en el momento del registro;
|
|
—
|
peso corporal en el momento del sacrificio y peso de los órganos de los animales parentales;
|
|
—
|
observaciones de la autopsia;
|
|
—
|
descripción detallada de los hallazgos histopatológicos;
|
|
—
|
datos relativos a la absorción (si procede);
|
|
—
|
tratamiento estadístico de los resultados, si procede.
|
|
Discusión de los resultados.
Conclusiones.
|
Interpretación de los resultados
|
56.
|
El estudio pretende evaluar la toxicidad para la reproducción o el desarrollo relacionada con la administración continuada de dosis (véanse los puntos 5 y 6). Podría dar una indicación de la necesidad de realizar nuevas investigaciones y proporciona orientaciones para el diseño de estudios posteriores. Debe consultarse el documento de orientación n.o 43 de la OCDE para obtener ayuda en la interpretación de los resultados de toxicidad para la reproducción y el desarrollo (12). El documento de orientación n.o 106 de la OCDE sobre la evaluación histológica de los ensayos endocrinos y de reproducción en roedores (11) proporciona información sobre la preparación y evaluación de los órganos (endocrinos) y los frotis vaginales que puede ser útil para las presentes directrices de ensayo.
|
BIBLIOGRAFÍA
|
(1)
|
OCDE (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Disponible previa solicitud a la Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(2)
|
OCDE (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Disponible previa solicitud a la Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(3)
|
OCDE (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Disponible previa solicitud a la Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(4)
|
OCDE (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(5)
|
OCDE (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organización para la Cooperación y el Desarrollo Económicos, París.
|
|
(6)
|
OCDE (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment(No 150), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, Nueva York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L.(1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OCDE (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(11)
|
OCDE (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No 106), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(12)
|
OCDE (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organización para la Cooperación y el Desarrollo Económico, París.
|
Apéndice 1
DEFINICIONES [VÉASE TAMBIÉN EL DOCUMENTO DE ORIENTACIONES GD 150 DE LA OCDE (6)]
Androgenicidad: Capacidad de un producto para actuar como una hormona androgénica natural (por ejemplo, testosterona) en el organismo de un mamífero.
Antiandrogenicidad: Capacidad de un producto para inhibir la acción de una hormona androgénica natural (por ejemplo, testosterona) en el organismo de un mamífero.
Antiestrogenicidad: Capacidad de un producto para inhibir la acción de una hormona estrogénica natural (por ejemplo, 17ß-estradiol) en el organismo de un mamífero.
Actividad antitiroidea: Capacidad de un producto para inhibir la acción de una hormona tiroidea natural (por ejemplo, T3) en el organismo de un mamífero.
Producto: Sustancia o mezcla.
Toxicidad para el desarrollo: Manifestación de la toxicidad para la reproducción que consiste en la presencia de trastornos pre-, peri- y posnatales, de tipo estructural o funcional, en la descendencia.
Posología: Término general que abarca la dosis administrada, y la frecuencia y duración de la administración.
Dosis: Cantidad de producto problema administrada. La dosis se expresa en peso del producto problema por unidad de peso corporal de animal de ensayo y día (p. ej., mg/kg peso corporal/día), o como concentración alimentaria constante.
Toxicidad evidente: Término general que describe signos claros de toxicidad tras la administración de un producto problema. Estos signos deben ser suficientes para la evaluación del peligro y tales que, ante un aumento de la dosis administrada, quepa esperar como resultado la aparición de signos tóxicos graves y una mortalidad probable.
Disminución de la fertilidad: Trastornos en la capacidad o las funciones reproductoras de las hembras o de los machos.
Toxicidad para la madre: Efectos adversos en las hembras gestantes, tanto si se producen de forma específica (efecto directo) como inespecífica (efecto indirecto).
NOAEL: Abreviatura en inglés de nivel sin efectos adversos observados (no-observed-adverse effect level). Se trata de la dosis más elevada a la que no se observa ningún efecto adverso debido al tratamiento.
Estrogenicidad: Capacidad de un producto para actuar como una hormona estrogénica natural (por ejemplo, 17ß-estradiol) en el organismo de un mamífero.
Toxicidad para la reproducción: Efectos nocivos para la descendencia y/o disminución de la capacidad o las funciones reproductoras de las hembras y de los machos.
Producto problema: Sustancia o mezcla estudiada con este método de ensayo.
Actividad tiroidea: Capacidad de un producto para actuar como una hormona tiroidea natural (por ejemplo, T3) en el organismo de un mamífero.
Validación: Procedimiento científico diseñado para caracterizar las exigencias y limitaciones operativas de un método de ensayo y demostrar su fiabilidad y pertinencia para un fin particular.
Apéndice 2
DIAGRAMA DEL PROGRAMA EXPERIMENTAL CON INDICACIÓN DE LA DURACIÓN MÁXIMA DEL ESTUDIO, SOBRE LA BASE DE UN PERÍODO DE APAREAMIENTO COMPLETO DE CATORCE DÍAS

Apéndice 3
INFORME RESUMIDO EN FORMA DE CUADRO SOBRE LOS EFECTOS PARA LA REPRODUCCIÓN O EL DESARROLLO
|
OBSERVACIONES
|
VALORES
|
|
|
|
Dosis (unidades)
|
0 (testigo)
|
…
|
…
|
…
|
…
|
|
Parejas al inicio (N)
|
|
|
|
|
|
|
Ciclo estral (por lo menos, longitud media y frecuencia de ciclos irregulares)
|
|
|
|
|
|
|
Hembras que presentan signos de cópula (N)
|
|
|
|
|
|
|
Hembras que llegan a la gestación (N)
|
|
|
|
|
|
|
Días de concepción 1-5 (N)
|
|
|
|
|
|
|
Días de concepción 6-...(
(21)
) (N)
|
|
|
|
|
|
|
Gestación ≤ 21 días (N)
|
|
|
|
|
|
|
Gestación = 22 días (N)
|
|
|
|
|
|
|
Gestación ≥ 23 días (N)
|
|
|
|
|
|
|
Madres con crías nacidas vivas (N)
|
|
|
|
|
|
|
Madres con crías vivas el día 4 después del parto (N)
|
|
|
|
|
|
|
Implantes/madre (media)
|
|
|
|
|
|
|
Crías vivas/madre en el momento del nacimiento (media)
|
|
|
|
|
|
|
Crías vivas/madre el día 4 (media)
|
|
|
|
|
|
|
Proporción de sexos (m/f) en el momento del nacimiento (media)
|
|
|
|
|
|
|
Proporción de sexos (m/f) el día 4 (media)
|
|
|
|
|
|
|
Peso de la camada al nacer (media)
|
|
|
|
|
|
|
Peso de la camada el día 4 (media)
|
|
|
|
|
|
|
Peso de las crías al nacer (media)
|
|
|
|
|
|
|
Peso de las crías en el momento de la medición de la DAG (media de los machos, media de las hembras)
|
|
|
|
|
|
|
DAG de las crías en el mismo día posnatal, nacimiento – día 4 (media de los machos, media de las hembras, indíquese el DPN)
|
|
|
|
|
|
|
Peso de las crías el día 4 (media)
|
|
|
|
|
|
|
Mantenimiento de pezones en las crías macho al día 13 (media)
|
|
|
|
|
|
|
Peso de las crías el día 13 (media)
|
|
|
|
|
|
|
|
|
CRÍAS ANÓMALAS
|
|
Madres con 0
|
|
|
|
|
|
|
Madres con 1
|
|
|
|
|
|
|
Madres con ≥ 2
|
|
|
|
|
|
|
|
|
PÉRDIDA DE DESCENDENCIA
|
|
|
|
Prenatal tras el implante (implantes menos nacimientos vivos)
|
|
Hembras con 0
|
|
|
|
|
|
|
Hembras con 1
|
|
|
|
|
|
|
Hembras con 2
|
|
|
|
|
|
|
Hembras con ≥ 3
|
|
|
|
|
|
|
|
|
Posnatal (nacidos vivos menos vivos el día 13 posnatal)
|
|
Hembras con 0
|
|
|
|
|
|
|
Hembras con 1
|
|
|
|
|
|
|
Hembras con 2
|
|
|
|
|
|
|
Hembras con ≥ 3
|
|
|
|
|
|
B.64. ESTUDIO DE TOXICIDAD POR ADMINISTRACIÓN CONTINUADA COMBINADO CON EL ENSAYO DE CRIBADO DE TOXICIDAD PARA LA REPRODUCCIÓN O EL DESARROLLO
INTRODUCCIÓN
|
1.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 422 de la OCDE (2016). Las directrices de la OCDE sobre los ensayos de productos se revisan periódicamente a la luz del progreso científico,. Las directrices de ensayo de cribado TG 422 originales se adoptaron en 1996 sobre la base de un protocolo de un «Estudio de toxicidad por administración continuada combinado con el ensayo de cribado de toxicidad para la reproducción o el desarrollo», debatido en dos reuniones de expertos, celebradas en Londres en 1990 (1) y en Tokio en 1992 (2).
|
|
2.
|
El presente método de ensayo combina una parte de cribado de la toxicidad para la reproducción o el desarrollo que se basa en la experiencia adquirida en los Estados miembros a partir de la utilización del método original con productos existentes de alto volumen de producción y en ensayos exploratorios con sustancias testigo positivo (3) (4), así como una parte de toxicidad por administración continuada, en concordancia con las directrices de ensayo TG 407 de la OCDE [estudio de toxicidad oral por administración continuada (28 días) en roedores, correspondiente al capítulo B.7 del presente anexo].
|
|
3.
|
Este método de ensayo se ha actualizado con parámetros pertinentes de alteración endocrina, como continuación de la actividad prioritaria iniciada en 1998 en la OCDE para revisar las directrices de ensayo vigentes y elaborar nuevas directrices de ensayo para el cribado y el ensayo de los alteradores endocrinos potenciales (5). En este contexto, las TG 407 (correspondiente al capítulo B.7 del presente anexo) se mejoraron en 2008 mediante parámetros adecuados para detectar la actividad endocrina de los productos problema. El objetivo de la actualización de las directrices de ensayo TG 422 era incluir algunos parámetros relevantes de alteración endocrina en las directrices de ensayo de cribado, en las que los períodos de exposición cubren algunos de los períodos sensibles durante el desarrollo (periodos prenatal y posnatal inicial).
|
|
4.
|
En las directrices de ensayo TG 422 se incluyeron los parámetros adicionales pertinentes de alteración endocrina seleccionados, que también forman parte de las directrices TG 443 (Estudio ampliado de toxicidad para la reproducción en una generación, correspondiente al capítulo B.56 del presente anexo), sobre la base de un estudio de viabilidad sobre cuestiones científicas y técnicas relacionadas con su inclusión, así como las posibles adaptaciones del diseño del ensayo necesarias para su inclusión (6).
|
|
5.
|
El presente método de ensayo está diseñado para generar información limitada sobre los efectos de un producto problema en el comportamiento reproductor de los machos y de las hembras, tales como la actividad gonadal, el comportamiento relativo al apareamiento, la concepción, el desarrollo del embrión y el parto. No es una alternativa ni sustituye a los métodos de ensayo existentes B.31, B.34, B.35 o B.56.
|
CONSIDERACIONES INICIALES
|
6.
|
Al determinar y evaluar las características tóxicas de un producto problema, el estudio de la toxicidad oral por administración continuada puede estar indicado cuando la información inicial relativa a la toxicidad se haya obtenido mediante ensayos de toxicidad aguda. Este estudio proporciona información sobre los posibles peligros para la salud que pueden derivarse de la exposición continuada durante un período de tiempo relativamente limitado. El método describe el estudio básico de toxicidad por administración continuada que puede aplicarse en el caso de productos para los que no está justificado un estudio de 90 días (por ejemplo, cuando el volumen de producción no supera ciertos límites), o bien como estudio preliminar de un estudio de toxicidad a largo plazo. Al realizar el estudio, deben aplicarse los principios y consideraciones básicos recogidos en el documento de orientación n.o 19 de la OCDE sobre el reconocimiento, evaluación y utilización de los signos clínicos como parámetros de tratamiento compasivo para los animales de experimentación empleados en las evaluaciones de la seguridad (7).
|
|
7.
|
Además, incluye un ensayo de cribado de la toxicidad para la reproducción y el desarrollo y, por tanto, también puede utilizarse para proporcionar información inicial sobre los posibles efectos en el comportamiento reproductivo de los machos y las hembras, tales como la función gonadal, el comportamiento relativo al apareamiento, la concepción, el desarrollo del embrión y el parto, sea en una fase temprana de la evaluación de las propiedades toxicológicas de los productos problema o sea con los productos problema preocupantes. El presente método de ensayo no proporciona información completa sobre todos los aspectos de la reproducción y el desarrollo. En particular, solo ofrece medios limitados para detectar las manifestaciones postnatales de una exposición prenatal, o los efectos que puedan inducirse durante la exposición posnatal. Debido (entre otras razones) a la selectividad de los criterios de valoración y a la corta duración del estudio, este método no aporta pruebas para hacer alegaciones definitivas de efectos nulos sobre la reproducción o el desarrollo. Por otra parte, a falta de datos de otros ensayos de toxicidad para la reproducción o el desarrollo, los resultados positivos son útiles para la evaluación inicial del peligro y contribuyen a la adopción de decisiones con respecto a la necesidad y el calendario de ensayos adicionales.
|
|
8.
|
Los resultados obtenidos por medio de parámetros endocrinos deben contemplarse en el contexto del «Marco conceptual para el estudio y la evaluación de los alteradores endocrinos» (Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals) de la OCDE (8). En este Marco conceptual, las directrices TG 422 mejoradas de la OCDE figuran en el nivel 4 como ensayo in vivo que proporciona datos sobre los efectos adversos relativos a los parámetros endocrinos pertinentes. Sin embargo, una señal endocrina podría no considerarse prueba suficiente por sí misma de que el producto problema es un alterador endocrino.
|
|
9.
|
El método de ensayo insiste más en los efectos neurológicos como parámetro específico, y destaca la necesidad de hacer observaciones clínicas atentas de los animales, a fin de obtener la máxima información posible. El método debe detectar los productos con potencial neurotóxico, y respecto a los que pueda justificarse la realización de investigaciones más profundas de este aspecto. Además, el método puede dar también una indicación básica de los efectos inmunológicos.
|
|
10.
|
A falta de datos de otros estudios de toxicidad sistémica, de toxicidad para la reproducción o el desarrollo, de neurotoxicidad o de inmunotoxicidad, los resultados positivos son útiles para la evaluación inicial del peligro y contribuyen a la adopción de decisiones con respecto a la necesidad y el calendario de ensayos adicionales. El ensayo puede ser especialmente útil como parte de la serie de datos de información de examen (SIDS, Screening Information Data Set) de la OCDE para la evaluación de los productos existentes respecto a los que se dispone de escasa o nula información toxicológica y pueden servir como alternativa a la realización de dos ensayos aparte, uno de la toxicidad por administración continuada (TG 407 de la OCDE, correspondientes al capítulo B.7 del presente anexo) y otro de la toxicidad para la reproducción o el desarrollo (TG 421 de la OCDE, correspondientes al capítulo B.63 del presente anexo). También puede utilizarse como estudio de determinación del intervalo de dosis para estudios más amplios sobre la reproducción o el desarrollo, o en cualquier otro caso que se considere pertinente.
|
|
11.
|
En general, se supone que existen diferencias de sensibilidad entre los animales gestantes y los no gestantes. Por consiguiente, puede ser más complicado determinar qué dosis son adecuadas para evaluar tanto la toxicidad general sistémica como la toxicidad específica para la reproducción o el desarrollo en esta combinación de ensayos que cuando se realizan por separado los distintos ensayos. Por otra parte, la interpretación de los resultados de los ensayos con respecto a la toxicidad sistémica general puede ser más difícil que cuando se realiza un estudio aparte de administración continuada, especialmente si los parámetros séricos y de histopatología no se evalúan al mismo tiempo en el estudio. Debido a estas complejidades técnicas, se requiere una considerable experiencia con los ensayos de toxicidad para la realización de este ensayo de cribado combinado. Por otra parte, aparte del menor número de animales implicados, el ensayo combinado puede ofrecer un medio mejor de discriminar los efectos directos sobre la reproducción o el desarrollo frente a los que son secundarios a otros efectos (sistémicos).
|
|
12.
|
En este ensayo, el período de administración es más largo que en un estudio de administración continuada de 28 días. Sin embargo, utiliza menos animales de cada sexo por grupo en comparación con la situación en la que se lleva a cabo un estudio clásico de administración continuada de de 28 días además de un ensayo de cribado de la toxicidad para la reproducción o el desarrollo.
|
|
13.
|
En este método de ensayo se parte de la administración oral del producto problema. Es posible que se requieran modificaciones si se utilizan otras vías de exposición.
|
|
14.
|
Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines normativos, debe considerarse si podría proporcionar resultados adecuados a tales fines y, en caso afirmativo, por qué. Dichas consideraciones no son necesarias cuando los requisitos normativos estipulan que la mezcla debe someterse a ensayo.
|
|
15.
|
En el apéndice 1 se dan las definiciones utilizadas.
|
PRINCIPIO DEL ENSAYO
|
16.
|
Se administra el producto problema en dosis escalonadas a varios grupos de machos y hembras. Los machos deben recibir el producto durante un mínimo de cuatro semanas, hasta el día anterior al sacrificio programado (incluido dicho día), lo que comprende un mínimo de dos semanas antes del apareamiento, el período de apareamiento y, aproximadamente, dos semanas después del apareamiento. Teniendo en cuenta lo limitado del período de administración a los machos antes del apareamiento, la fertilidad puede no ser un indicador particularmentesensible de la toxicidad testicular. Por lo tanto, es esencial un examen histológico detallado de los testículos. Para permitir la detección de la mayor parte de los efectos sobre la fertilidad masculina y la espermatogénesis, se considera suficiente la combinación de un período de administración antes del apareamiento de dos semanas y, posteriormente, observaciones sobre el apareamiento y la fertilidad, con un período de administración total de al menos cuatro semanas, seguido de un examen histopatológico detallado de las gónadas masculinas.
|
|
17.
|
Las hembras deben recibir el producto problema a lo largo de todo el estudio. Se incluyen aquí dos semanas antes del apareamiento (con el objetivo de abarcar por lo menos dos ciclos estrales completos), el período de tiempo variable hasta la concepción, la duración de la gestación y al menos trece días después del parto, hasta el día anterior al sacrifico programado, inclusive.
|
|
18.
|
La duración del estudio, tras la aclimatación y la evaluación del ciclo estral antes de la administración del producto problema, depende del comportamiento de las hembras y es de aproximadamente 63 días, [14 días como mínimo antes del apareamiento, (hasta) 14 días de apareamiento, 22 días de gestación y 13 días de lactancia].
|
|
19.
|
A lo largo del período de administración, se observa a los animales todos los días atentamente con el fin de descubrir la posible aparición de signos de toxicidad. Se practica la autopsia a los animales que mueran o sean sacrificados durante el ensayo y, al final del mismo, se sacrifican y someten a autopsia los animales supervivientes.
|
DESCRIPCIÓN DEL MÉTODO
Selección de la especie animal
|
20.
|
Este método de ensayo está diseñado para su uso con la rata. Si los parámetros especificados en estas directrices de ensayo TG 422 se investigan en otra especie de roedor, habrá que justificarlo minuciosamente. En el programa internacional de validación de la detección de alteradores endocrinos en relación con las directrices de ensayo TF 407, la única especie utilizada fue la rata. Debe evitarse la utilización de cepas que presenten un bajo índice de fertilidad o una incidencia notoriamente elevada de anomalías del desarrollo. Han de utilizarse animales vírgenes sanos, no sometidos a experimentación previa. Deben especificarse la especie, la cepa, el sexo, el peso y la edad de los animales utilizados en el ensayo. Al principio del experimento, la diferencia de peso entre los animales empleados ha de ser mínima y no superar el ± 20 % del peso medio de cada sexo. Cuando el estudio se realice como fase preliminar de un estudio a largo plazo o de una generación completa, es preferible que en ambos estudios se utilicen animales de la misma cepa y de la misma fuente.
|
Alojamiento y alimentación de los animales
|
21.
|
Todos los procedimientos serán conformes a las normas habituales del cuidado de los animales de laboratorio. El local de los animales de experimentación ha de estar a una temperatura de 22 °C (± 3 °C). La humedad relativa debe ser como mínimo del 30 % y preferentemente no superar el 70 %, salvo durante la limpieza del local. La iluminación debe ser artificial, con un fotoperíodo de 12 horas de luz y 12 horas de oscuridad. Para la alimentación se podrán utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber. La elección de la dieta puede verse influida por la necesidad de garantizar una mezcla conveniente de la sustancia problema si se administra por este método.
|
|
22.
|
Los animales deben alojarse en pequeños grupos del mismo sexo; pueden alojarse de forma individual si está científicamente justificado. En caso de que se utilicen jaulas colectivas, no habrá más de cinco animales en cada jaula. El apareamiento debe llevarse a cabo en jaulas adecuadas al efecto. Las hembras gestantes deben enjaularse de forma individual y disponer de materiales de nidificación. Las hembras en período de lactancia deben enjaularse de forma individual con sus crías.
|
|
23.
|
Se debe analizar regularmente la comida para detectar posibles contaminantes. Se conservará una muestra de la dieta hasta el momento de concluir el informe.
|
Preparación de los animales
|
24.
|
Se eligen al azar animales adultos jóvenes y sanos, y se asignan a las jaulas y grupos tratados. Las jaulas se deben disponer de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Se identifica a los animales de forma unívoca y se los mantiene en las jaulas al menos cinco días antes de empezar el estudio para que se acostumbren a las condiciones de laboratorio.
|
Preparación de las dosis
|
25.
|
Se recomienda administrar el producto problema oralmente a menos que se consideren más adecuadas otras vías de administración. Cuando se selecciona la vía oral, el producto problema suele administrarse por sonda; sin embargo, los productos problema también pueden administrarse alternativamente con el alimento o el agua de bebida.
|
|
26.
|
En caso necesario, el producto problema se disuelve o suspende en un vehículo adecuado. Se recomienda considerar en primer lugar, siempre que sea posible, el uso de una solución o suspensión acuosa, después el uso de una solución o suspensión oleosa (por ejemplo, en aceite de maíz) y, por último, la posible disolución en otros vehículos. En caso de vehículos no acuosos, deben conocerse sus características tóxicas. Debe determinarse la estabilidad y homogeneidad del producto problema en el vehículo.
|
PROCEDIMIENTO
Número y sexo de los animales
|
27.
|
Se recomienda que cada grupo comience con un mínimo de 10 machos y entre 12 y 13 hembras. Las hembras se evaluarán antes de la exposición en relación con el ciclo estral y no se incluirán en el estudio los animales que no presenten ciclos típicos de 4 a 5 días; por lo tanto, se recomienda añadir más hembras para conseguir 10 hembras por grupo. Excepto en el caso de efectos tóxicos marcados, se espera que de esta forma se consigan al menos 8 hembras gestantes por grupo, que normalmente es el número mínimo aceptable de hembras gestantes por grupo. El objetivo es conseguir suficientes gestaciones y descendientes para garantizar una evaluación significativa del potencial del producto problema para afectar a la fertilidad, gestación, comportamiento materno y lactante, y al crecimiento y desarrollo de los descendientes F1, desde la concepción hasta 13 días después del parto. Si se van a sacrificar animales durante el experimento, habrá que añadir el número de animales que se haya previsto sacrificar antes de acabar el estudio. Debe estudiarse la conveniencia de utilizar un grupo satélite adicional de cinco animales por sexo en los grupos testigo y de dosis más elevada para observar la reversibilidad, la persistencia o la aparición retardada de efectos tóxicos sistémicos, al menos durante los 14 días siguientes al tratamiento. Los animales de los grupos satélite no se aparean y, por consiguiente, no se utilizan para la evaluación de la toxicidad para la reproducción o el desarrollo.
|
Posología
|
28.
|
Deben utilizarse generalmente como mínimo tres grupos de ensayo y un grupo testigo. Si no se dispone de datos generales apropiados sobre la toxicidad, puede realizarse un estudio (con animales de la misma cepa y procedencia) para ayudar a determinar las dosis que deben emplearse. A excepción de la administración del producto problema, los animales del grupo testigo deben tratarse de la misma manera que los de los grupos de ensayo. Si se utiliza un vehículo para administrar el producto problema, el grupo testigo debe recibir el mayor volumen utilizado de dicho vehículo.
|
|
29.
|
Las dosis deben seleccionarse teniendo en cuenta los eventuales datos existentes sobre toxicidad y (toxico)cinética disponibles. También debe tenerse en cuenta que puede haber diferencias de sensibilidad entre los animales gestantes y los no gestantes. La dosis más elevada debe seleccionarse con el propósito de inducir efectos tóxicos pero sin llegar a la muerte ni a un sufrimiento evidente. Posteriormente, debe seleccionarse una secuencia descendente de dosis destinadas a demostrar la eventual relación dosis-respuesta y la ausencia de efectos adversos con la dosis inferior. Los intervalos del doble al cuádruple suelen ser óptimos y a menudo es preferible añadir un cuarto grupo de ensayo en lugar de utilizar intervalos muy amplios (por ejemplo, con un factor superior a 10) entre dosis.
|
|
30.
|
Si se observan signos de toxicidad general (por ejemplo, peso corporal reducido, efectos hepáticos, cardíacos, pulmonares o renales, etc.) u otras alteraciones que no son necesariamente respuestas tóxicas (por ejemplo, ingesta de alimentos reducida, hiperplasia hepática), habrá que interpretar con prudencia los efectos observados sobre los parámetros sensibles endocrinos.
|
Ensayo límite
|
31.
|
Si un estudio oral con una sola dosis de, al menos, 1 000 mg/kg peso corporal/día o, en caso de administración con los alimentos, de un porcentaje equivalente en los alimentos o el agua de bebida (según las determinaciones del peso corporal), siguiendo los procedimientos descritos en el presente estudio, no se produce ningún efecto tóxico observable y si, a partir de datos de sustancias estructuralmente afines, no debiera esperarse la aparición de toxicidad, puede considerarse innecesario realizar un estudio completo con varias dosis. El ensayo límite es válido excepto cuando la exposición humana indique la necesidad de utilizar una dosis superior. Si se trata de otras vías de administración, como la inhalación o la aplicación cutánea, suelen ser las características fisicoquímicas del producto problema las que determinan la exposición máxima que puede alcanzarse.
|
Administración del producto problema
|
32.
|
El producto problema se administra diariamente a los animales durante 7 días a la semana. Si el producto problema se administra por sonda, debe hacerse en una dosis única y con una sonda gástrica o una cánula de intubación adecuada. El volumen máximo de líquido que puede administrarse en una sola vez depende del tamaño del animal utilizado. Dicho volumen no debe superar 1 ml/100 g de peso corporal, salvo en el caso de las solucionesacuosas, en que puede llegarse a 2 ml/100 g de peso corporal. Salvo en el caso de productos problema irritantes o corrosivos, que por lo general producen efectos exacerbados con concentraciones mayores, deberá reducirse al mínimo la variabilidad del volumen de ensayo ajustando la concentración de manera que el volumen sea el mismo con las diferentes dosis.
|
|
33.
|
En el caso de productos problema administrados con los alimentos o el agua de bebida, es importante cerciorarse de que las cantidades administradas de producto problema no interfieren con la nutrición normal ni con el equilibrio hídrico. Cuando el producto problema se administre con el alimento, se puede utilizar una concentración alimentaria constante (ppm) o bien una dosis constante por unidad de peso corporal de los animales; debe indicarse qué alternativa se ha seguido. Si el producto problema se administra por sonda, la dosis debe darse todos los días a una hora similar, y ajustarse al menos una vez por semana para mantener una dosis constante respecto al peso corporal del animal. Cuando el estudio combinado se utilice como fase preliminar de un ensayo de toxicidad a largo plazo o de un estudio completo de toxicidad para la reproducción, en ambos estudios deberá utilizarse una dieta similar.
|
Calendario del experimento
|
34.
|
La administración del producto a ambos sexos debe comenzar dos semanas antes del apareamiento, una vez que se han aclimatado durante al menos cinco días y las hembras han sido sometidas a cribado para confirmar que sus ciclos estrales son normales (en un período de dos semanas antes del tratamiento). El estudio debe programarse de tal manera que la evaluación del ciclo estral comience pronto después de que los animales hayan alcanzado la madurez sexual completa. Esto puede variar ligeramente según las diferentes cepas de ratas en distintos laboratorios; por ejemplo, en las ratas Sprague Dawley corresponde a 10 semanas de edad y en las ratas Wistar a unas 12 semanas de edad. Las madres con descendencia deben sacrificarse el día 13 después del parto, o poco después. A fin de permitir el ayuno desde la víspera de las madres antes de la recogida de sangre (si se prefiere esta opción), no es necesario dar muerte a las madres y a sus descendientes el mismo día. El día del nacimiento (es decir, cuando se completa el parto) se define como el día 0 después del parto. Las hembras que no presentan signos de apareamiento se sacrifican entre 24 y 26 días después del último día del período de apareamiento. Se continúa la administración del producto problema a los dos sexos durante el período de apareamiento. Los machos deben seguir recibiendo el producto problema después del período de apareamiento, al menos hasta que se haya completado el período mínimo total de tratamiento de 28 días. A continuación se les da muerte o, alternativamente, se conservan y se les sigue administrando el producto para la posible realización de un segundo apareamiento si se considera adecuado.
|
|
35.
|
La administración diaria del producto problema a las hembras parentales debe mantenerse a lo largo de toda la gestación y, como mínimo, hasta el día 13 después del parto o el día anterior al sacrificio, inclusive. En los estudios en que el producto problema se administre por inhalación o por vía cutánea, esta administración debe continuar como mínimo hasta el día 19 de la gestación, inclusive, y debe volver a iniciarse lo antes posible y, a más tardar, el día posnatal (DPN) 4.
|
|
36.
|
No se aparean los animales del eventual grupo satélite previsto para las observaciones de seguimiento. Deben mantenerse al menos 14 días después del primer sacrificio programado de las madres, sin tratamiento alguno, para detectar la aparición retardada, la persistencia o la recuperación de los efectos tóxicos.
|
|
37.
|
En el apéndice 2 figura un diagrama del programa experimental.
|
Ciclos estrales
|
38.
|
Deben supervisarse los ciclos estrales antes de que se inicie el tratamiento, a fin de seleccionar para el estudio las hembras con ciclos regulares (véase el punto 27). También hay que efectuar un seguimiento diario de los frotis vaginales desde el principio del período de tratamiento hasta que se observen signos de apareamiento. Si hay preocupación por que los efectos del estrés agudo puedan modificar los ciclos estrales con el inicio de la administración del producto, los laboratorios pueden exponer a los animales de ensayo durante 2 semanas, hacer a continuación frotis vaginales diarios para supervisar el ciclo estral durante un mínimo de dos semanas durante el período previo al apareamiento, y continuar la supervisión en el período de apareamiento hasta que haya signos de apareamiento. En el momento de la obtención de las células de la vagina o del cuello del útero, hay que tener cuidado para evitar la alteración de la mucosa, que podría provocar una pseudogestación (8) (9).
|
Procedimiento del apareamiento
|
39.
|
Normalmente, en este estudio deben utilizarse apareamientos 1: 1 (un macho con una hembra). Pueden darse excepciones en caso de muertes ocasionales de machos. Se recomienda colocar a la hembra con el mismo macho hasta que se observen signos de apareamiento o hayan transcurrido dos semanas. Las hembras deben examinarse cada mañana para determinar la presencia de esperma o de tapón vaginal. El día 0 de la gestación se define como el día en que se confirman los signos de apareamiento (se observa la presencia de un tapón vaginal o de esperma). Si no ha tenido éxito el apareamiento, puede plantearse otro intento de aparear hembras con machos de comprobada capacidad reproductora del mismo grupo.
|
Tamaño de la camada
|
40.
|
El día 4 después del nacimiento, podrá ajustarse el tamaño de cada camada eliminando las crías excedentarias mediante selección aleatoria con el fin de obtener, en la medida de lo posible, cuatro o cinco crías por sexo y por camada en función del tamaño normal de la camada en la cepa de ratas utilizada. Se deben tomar muestras de sangre de dos de la crías excedentarias; tales muestras se mezclan y se utilizan para determinar los niveles de T4 en el suero. No es adecuado eliminar crías de forma selectiva, por ejemplo sobre la base del peso corporal o la distancia anogenital (DAG). Cuando el número de crías machos o hembras impida lograr que cada camada cuente con cuatro o cinco de cada sexo, es aceptable una adaptación parcial (por ejemplo, seis machos y cuatro hembras). No se eliminará ninguna cría cuando el tamaño de la camada descienda por debajo del objetivo de la eliminación (8 o 10 crías/camada). Si solo se dispone de una única cría por encima del objetivo de la eliminación, solo se eliminará una cría, que se utilizará para la extracción de sangre con el fin de efectuar la posible evaluación de T4 en el suero.
|
|
41.
|
Si no se ajusta el tamaño de la camada, se sacrifican dos crías por camada el día 4 después del nacimiento y se toman muestras de sangre para medir las concentraciones de la hormona tiroidea en el suero. Si es posible, las dos crías por camada deben ser hembras, a fin de reservar las crías macho para las evaluaciones del mantenimiento de pezones, salvo en caso de que la eliminación de estas crías no deje hembras suficientes para la evaluación en el momento del sacrificio. No se eliminarán las crías cuando el tamaño de la camada descienda por debajo de 8 o 10 crías/camada (dependiendo del tamaño normal de la camada en la cepa de ratas utilizada). Si solo se dispone de una única cría por encima del tamaño normal de la camada, solo se eliminará una cría, que se utilizará para la extracción de sangre con el fin de efectuar la posible evaluación de T4 en el suero.
|
Observaciones
|
42.
|
Debe hacerse una observación clínica general al menos una vez al día, preferentemente a la misma hora cada día y teniendo en cuenta el período más agudo de los efectos previstos tras la administración. Se debe registrar el estado de salud de los animales. Se observan todos los animales al menos dos veces el día en relación con la morbilidad y la mortalidad.
|
|
43.
|
Todos los animales parentales deben someterse a una observación clínica exhaustiva antes de la primera exposición (para permitir realizar comparaciones con un mismo sujeto) y, después, al menos a una por semana. Estas observaciones deben hacerse fuera de la jaula de alojamiento en un ambiente normal y, de preferencia, a la misma hora cada día. Deben registrarse cuidadosamente, de preferencia con sistemas de puntuación definidos expresamente por el laboratorio de ensayo. Debe procurarse que las variaciones en las condiciones de ensayo sean mínimas y que las observaciones sean realizadas de preferencia por observadores ajenos al tratamiento. Los signos observados deben incluir, sin ánimo de exhaustividad, los cambios de la piel, pelo, ojos, mucosas, la presencia de secreciones y excreciones y la actividad neurovegetativa (por ejemplo, lagrimeo, piloerección, tamaño de la pupila, respiración anómala). Deben registrarse también los cambios en la marcha, postura y respuesta a la manipulación, así como la presencia de movimientos clónicos o tónicos, o estereotipados (por ejemplo, realización excesiva de movimientos de limpieza o recorridos circulares repetitivos), los partos prolongados o difíciles, o los comportamientos anómalos (automutilación, marcha hacia atrás, etc.) (10).
|
|
44.
|
En un momento del estudio, deben llevarse a cabo, en cinco machos y cinco hembras seleccionados aleatoriamente de cada grupo, una evaluación de la reactividad sensorial a estímulos de diferentes modalidades (p. ej., estímulos auditivos, visuales y propioceptivos) (8) (9) (11), una evaluación de la fuerza de prensión (12) y una evaluación de la actividad motriz (13). En las referencias respectivas se da más información sobre los procedimientos que pueden seguirse. Sin embargo, también pueden utilizarse procedimientos alternativos a los citados. En los machos, estas observaciones funcionales deben realizarse hacia el final del período de administración del producto, poco antes del sacrificio programado, pero antes del muestreo de sangre para hematología o bioquímica clínica (véanse los puntos 53-56, incluida la nota a pie de página 1). Las hembras deben estar en un estado fisiológico similar durante estas pruebas funcionales y deben someterse a ensayo preferentemente una vez durante la última semana de lactancia (p. ej. DL 6-13), poco antes del sacrificio programado. En la medida de lo posible, han de reducirse al mínimo los tiempos de separación de las madres y las crías.
|
|
45.
|
Las observaciones funcionales realizadas una vez hacia la finalización del estudio pueden omitirse cuando el estudio se realice como estudio preliminar de un estudio posterior de toxicidad subcrónica (90 días) o a largo plazo. En este caso, las observaciones funcionales deberán incluirse en este estudio de continuación. Por otra parte, la disponibilidad de datos sobre las observaciones funcionales en este estudio de administración continuada puede facilitar la selección de las dosis utilizadas en un estudio posterior sobre toxicidad subcrónica o a largo plazo.
|
|
46.
|
De forma excepcional, también pueden omitirse las observaciones funcionales en los grupos que muestren signos de toxicidad de otro tipo tales que interfieran significativamente con los resultados de las pruebas funcionales.
|
|
47.
|
Debe registrarse la duración de la gestación y calcularse a partir del día 0 de la gestación. Cada camada debe examinarse lo antes posible después del parto para determinar el número y sexo de las crías, los mortinatos, los nacidos vivos y los nacidos con retraso evidente (crías que sean significativamente menores que las crías testigo correspondientes), y la presencia de anomalías macroscópicas.
|
|
48.
|
Las crías vivas deben contarse y clasificarse por sexo, y las camadas pesarse dentro de las 24 horas siguientes al parto (día 0 o 1 después del parto) y al menos en los días 4 y 13 después del parto. Además de las observaciones sobre los animales parentales (véanse los puntos 43 y 44), debe registrarse cualquier eventual comportamiento anómalo de la descendencia.
|
|
49.
|
La distancia anogenital (DAG) de cada cría debe medirse el mismo día posnatal, entre el DPN 0 y el DPN 4. El peso corporal de las crías debe recogerse el día en que se mida la DAG y esta DAG se debe normalizar en función de una medida del tamaño de las crías, de preferencia la raíz cúbica del peso corporal (14). El número de pezones/areolas en las crías macho debe contarse el DPN 12 o 13, según lo recomendado en el documento de orientaciones GD 151 de la OCDE (15).
|
Peso corporal y consumo de alimento y agua
|
50.
|
Los machos y las hembras deben pesarse el primer día de la administración del producto problema, después al menos una vez por semana, y en el momento del sacrificio. Durante la gestación, las hembras deben pesarse en los días 0, 7, 14 y 20 y dentro de las 24 horas siguientes al parto (día 0 o 1 después del parto) y al menos en los días 4 y 13 después del parto. Estas observaciones se registran respecto a cada animal adulto por separado.
|
|
51.
|
Durante el período previo al apareamiento, la gestación y la lactancia, el consumo de alimentos debe medirse al menos semanalmente. La medición del consumo de alimentos durante el apareamiento es opcional. El consumo de agua durante estos períodos también debe medirse si el producto problema se administra a través del agua de bebida.
|
Hematología
|
52.
|
Una vez durante el estudio, deben practicarse los siguientes exámenes hematológicos con cinco machos y cinco hembras seleccionados aleatoriamente de cada grupo: hematocrito, concentraciones de hemoglobina, recuento de eritrocitos, reticulocitos, recuento de leucocitos y fórmula leucocitaria, recuento de plaquetas y medida del tiempo o capacidad de coagulación sanguínea. Si se sabe o sospecha que el producto problema o sus posibles metabolitos tienen propiedades oxidantes, habrá que determinar además la concentración de metahemoglobina y los corpúsculos de Heinz.
|
|
53.
|
Las muestras de sangre deben tomarse de un sitio designado. Durante el muestreo, las hembras deben estar en un estado fisiológicamente similar. A fin de evitar dificultades prácticas relacionadas con la variabilidad en cuanto al principio de la gestación, la recogida de sangre de las hembras puede hacerse al final del período anterior al apareamiento como alternativa al muestreo justo antes del procedimiento de sacrificio de los animales, o como parte de este. Las muestras de sangre de los machos deben tomarse preferentemente justo antes del procedimiento de sacrificio de los animales, o como parte de este. Otra posibilidad es recoger la sangre de los machos al final del período previo a la apareamiento cuando se haya preferido este punto para las hembras.
|
|
54.
|
Las muestras de sangre deben conservarse en condiciones adecuadas.
|
Bioquímica clínica
|
55.
|
Las determinaciones de bioquímica clínica para investigar los principales efectos tóxicos en los tejidos y, en concreto, en el riñón y el hígado, deben realizarse con muestras de sangre obtenidas de los cinco machos y cinco hembras seleccionados de cada grupo. Se recomienda que los animales estén en ayunas desde el día anterior a la toma de muestras (22). Los parámetros medidos en el plasma o del suero deben incluir las concentraciones de sodio, potasio, glucosa, colesterol total, urea, creatinina, proteínas totales y albúminas, al menos dos enzimas indicadoras de los efectos hepatocelulares (alanina-transaminasa, aspartato-transaminasa y sorbitol-deshidrogenasa) y ácidos biliares. La determinación de otras enzimas (de origen hepático o no) y de la bilirrubina puede proporcionar información útil en ciertas circunstancias.
|
|
56.
|
Se toman muestras de sangre de un lugar definido sobre la base del siguiente calendario:
|
—
|
de al menos dos crías por camada el día 4 después del parto, si el número de crías lo permite (véanse los puntos 40-41),
|
|
—
|
de todas las madres y al menos dos crías por camada en el momento del sacrificio el día 13, y
|
|
—
|
de todos los machos adultos, en el momento del sacrificio.
|
Todas las muestras de sangre se conservan en condiciones adecuadas. Las muestras de sangre de las crías del día 13 y las de los machos adultos se evalúan en cuanto a los niveles de hormonas tiroideas en suero (T4). Si procede, se lleva a cabo una evaluación adicional de la T4 en las muestras de sangre de las madres y de las crías del día 4. Como opción pueden medirse otras hormonas si son pertinentes. La sangre de las crías puede ponerse en común por camadas para efectuar los análisis de hormonas tiroideas. Las hormonas tiroideas (T4 y TSH) deben medirse preferentemente como «total».
|
|
57.
|
Pueden realizarse, con carácter opcional, las siguientes determinaciones en los análisis de orina de cinco machos seleccionados aleatoriamente de cada grupo durante la última semana del estudio utilizando la recogida programada de orina; aspecto, volumen, osmolalidad o densidad, pH, contenido en proteínas, glucosa, sangre y glóbulos sanguíneos.
|
|
58.
|
Además de ello, debe plantearse el estudio de los marcadores séricos de lesiones tisulares generales. Entre las otras determinaciones que deben realizarse si se sabe o sospecha que las propiedades conocidas del producto problema pueden afectar a las características metabólicas correspondientes se incluyen las concentraciones de calcio, fosfato, triglicéridos en ayunas y glucosa en ayunas, hormonas específicas, metahemoglobina y colinesterasa. Debe decidirse caso por caso si se procede a estas determinaciones.
|
|
59.
|
Los siguientes factores pueden influir en la variabilidad y en el valor absoluto de las concentraciones hormonales:
|
—
|
el momento del sacrificio, debido a la variación diurna de las concentraciones hormonales,
|
|
—
|
el método de sacrificio, que se escogerá para evitar a los animales un sufrimiento indebido que pueda influir en las concentraciones hormonales,
|
|
—
|
los equipos de ensayo para la determinación de las hormonas, que pueden presentar diferencias en sus curvas patrón.
|
|
|
60.
|
Las muestras de plasma destinadas específicamente a la determinación de las hormonas deben obtenerse a una hora comparable del día. Las cifras obtenidas al analizar las concentraciones de hormonas varían con los equipos comerciales de ensayo que se usan para el análisis.
|
|
61.
|
Si los datos de referencia previos son insuficientes, habrá que considerar la posible determinación de los parámetros hematológicos y de bioquímica clínica antes de iniciar la administración o, preferentemente, con un conjunto de animales no incluidos en los grupos experimentales. En el caso de las hembras, los datos deben ser de animales lactantes.
|
ANATOMÍA PATOLÓGICA
Autopsia macroscópica
|
62.
|
Debe practicarse una autopsia macroscópica completa y detallada a todos los animales adultos empleados en el estudio, que incluya un examen detenido de la superficie corporal externa, todos los orificios y las cavidades craneana, torácica y abdominal con su contenido. Se debe prestar especial atención a los órganos sexuales. Debe registrarse el número de puntos de implantación. Deben examinarse frotis vaginales en el día de la autopsia para determinar la fase del ciclo estral y poder establecer una correlación con el examen histopatológico de los sexuales femeninos.
|
|
63.
|
Los testículos y epidídimos, así como la próstata y las vesículas seminales con las glándulas coagulantes en conjunto, de todos los machos adultos deben limpiarse de los tejidos adherentes, según proceda, y su peso húmedo debe tomarse lo antes posible tras la disección para evitar su desecación. Además, el peso de órganos opcionales podría incluir el complejo de músculo elevador del ano y el músculo bulbocavernoso, las glándulas de Cowper y el glande del pene en los machos y los ovarios emparejados (peso húmedo) y el útero (incluido el cuello) en las hembras; si se incluyen, estos pesos deben recogerse lo antes posible tras la disección. Deben conservarse los ovarios, testículos, epidídimos, órganos sexuales secundarios y todos los órganos que muestren lesiones macroscópicas de todos los animales adultos.
|
|
64.
|
Deben conservarse en el medio de fijación más adecuado para el examen histopatológico posterior previsto las glándulas tiroideas de todos los adultos machos y hembras y de una cría macho y una cría hembra del día 13 de cada camada. El peso de la glándula tiroidea puede determinarse después de su fijación. Su disección debe hacerse también de manera muy cuidadosa y solo después de la fijación para evitar daños tisulares. Los daños tisulares podrían comprometer el examen histopatológico. Las muestras de sangre deben tomarse de un punto indicado, justo antes del procedimiento de sacrificio de los animales (o como parte de este procedimiento), y conservarse en condiciones adecuadas (véase el punto 56).
|
|
65.
|
Además, en el caso de un mínimo de cinco machos y hembras adultos, seleccionados aleatoriamente entre cada grupo (aparte de los moribundos o sacrificados antes de la terminación del estudio), el hígado, los riñones, las glándulas suprarrenales, el timo, el bazo, el encéfalo y el corazón deben limpiarse de los tejidos adherentes, según convenga, y debe medirse su peso húmedo lo antes posible tras la disección para evitar su desecación. Los tejidos que se enumeran a continuación deben conservarse en el medio de fijación más adecuado teniendo en cuenta tanto el tipo de tejido como el examen histopatológico posterior previsto: todas las lesiones macroscópicas, el encéfalo (regiones representativas, incluidos el cerebro, el cerebelo y la protuberancia), la médula espinal, los ojos, el estómago, el intestino delgado y grueso (incluidas las placas de Peyer), el hígado, los riñones, las glándulas suprarrenales, el bazo, el corazón, el timo, la tráquea y los pulmones (conservados mediante distensión pulmonar confijador y después inmersión), las gónadas (testículos y ovarios), los órganos sexuales secundarios (útero con el cuello, epidídimos, próstata, vesículas seminales más glándulas coagulantes), la vagina, la vejiga urinaria, los ganglios linfáticos [además del ganglio de drenaje más proximal, debe tomarse otro ganglio linfático según la experiencia del laboratorio (16)], un nervio periférico (ciático o tibial) preferentemente muy próximo al músculo, músculo esquelético y hueso, con médula ósea (una sección o, como alternativa, un aspirado de médula ósea recién montado). Se recomienda fijar los testículos mediante inmersión en fijador de Bouin o en fijador de Davidson modificado (16) (17) (18); no se recomienda para estos tejidos la fijación con formol. Con una aguja se puede puncionar suave y superficialmente la túnica albugínea por ambos polos del órgano para permitir la rápida penetración del fijador. Las observaciones clínicas y de otro tipo pueden indicar la necesidad de examinar otros tejidos. Se conservará también cualquier otro órgano que se considere diana teniendo en cuenta las propiedades conocidas de la sustancia problema.
|
|
66.
|
Los siguientes tejidos pueden proporcionar valiosas indicaciones sobre los efectos de carácter endocrino: gónadas (ovarios y testículos), órganos sexuales secundarios (útero con el cuello, epidídimos, vesículas seminales con glándulas coagulantes, próstata dorsolateral y ventral), vagina, hipófisis, glándula mamaria y glándula suprarrenal. Las alteraciones de las glándulas mamarias de machos no se han documentado suficientemente, pero este parámetro puede ser muy sensible a las sustancias dotadas de actividad estrogénica. La observación de órganos/tejidos que no se enumeran en el punto 65 es opcional.
|
|
67.
|
Las crías muertas y las crías sacrificadas el día 13 después del parto, o poco después, deben, al menos, ser examinadas externamente de forma detenida para detectar la eventual presencia de anomalías macroscópicas. Debe prestarse especial atención a los órganos genitales externos, en los que debe estudiarse la eventual presencia de signos de alteración en el desarrollo.
|
Examen histopatológíco
|
68.
|
Es preciso realizar un examen histopatológico completo de los órganos y tejidos conservados de los animales seleccionados en los grupos testigo y tratados con la dosis más elevada (haciendo especial hincapié en las fases de la espermatogénesis en las gónadas masculinas y en la histopatología de la estructura de las células intersticiales de los testículos). En caso necesario, podrán examinarse la glándula tiroidea de las crías y de los animales adultos restantes. En caso de que se hayan observado cambios relacionados con el tratamiento en el grupo con la dosis más elevada, estos exámenes se ampliarán a animales de otros grupos tratados. El documento de orientación sobre histopatología (10) proporciona más información acerca de la disección, fijación, sección e histopatología de los tejidos endocrinos.
|
|
69.
|
Deben examinarse todas las lesiones macroscópicas. Para contribuir a la determinación de los NOAEL, deben examinarse los órganos diana en otros grupos tratados, especialmente en los grupos de los que se alega que muestran un NOAEL.
|
|
70.
|
Si se utiliza un grupo satélite, debe hacerse un examen histopatológico de los órganos y tejidos señalados por presentar efectos en los grupos tratados.
|
DATOS E INFORME
Datos
|
71.
|
Los resultados de cada animal deben darse por separado. Además, todos los datos deben resumirse en forma de cuadro que recoja, respecto a cada grupo de ensayo, el número de animales al inicio del ensayo, el número de animales hallados muertos durante el mismo o sacrificados por razones compasivas, el momento de la muerte o del sacrificio compasivo, el número de animales fértiles, el número de hembras gestantes, el número de animales que presenten signos de toxicidad, una descripción de dichos signos (con inclusión del momento de su aparición, duración y gravedad), los tipos de cambios histopatológicos y todos los datos pertinentes sobre la camada. En el apéndice 3 figura un modelo de informe resumido en forma de cuadro, que ha demostrado ser muy útil para la evaluación de los efectos sobre la reproducción y el desarrollo.
|
|
72.
|
Siempre que sea posible, los resultados numéricos deberán evaluarse mediante un método estadístico adecuado y comúnmente aceptado. La comparación del efecto a lo largo de un intervalo de dosis debe evitar el uso de múltiples pruebas t. Los métodos estadísticos deben seleccionarse durante el diseño del estudio. El análisis estadístico de la DAG y del mantenimiento de pezones debe basarse en los datos de cada cría, teniendo en cuenta los efectos de camada. Cuando proceda, la camada es la unidad de análisis. El análisis estadístico del peso corporal de las crías debe basarse en los datos de cada cría, teniendo en cuenta el tamaño de la camada. Debido a las dimensiones limitadas del estudio, los análisis estadísticos en forma de ensayos de «significación» tienen un valor limitado respecto a muchos parámetros, especialmente a los de la reproducción. Son inadecuados algunos de los métodos más utilizados, especialmente los ensayos paramétricos con medidas de tendencia central. Si se utilizan análisis estadísticos, entonces el método elegido debe ser apropiado para la distribución de la variable examinada y ser seleccionado antes de que se inicie el estudio.
|
Evaluación de los resultados
|
73.
|
Los resultados de este estudio de toxicidad deben evaluarse en términos de efectos observados, autopsia y resultados microscópicos. La evaluación debe referirse a la relación entre la dosis de producto problema y la presencia o ausencia, incidencia y gravedad de las anomalías, incluyendo lesiones macroscópicas, órganos diana identificados, esterilidad, anomalías clínicas, alteración del comportamiento reproductor y con la prole, cambios del peso corporal, efectos sobre la mortalidad y cualquier otro efecto tóxico.
|
|
74.
|
Debido al breve período de tratamiento de los machos, el examen histopatológico de los testículos y epidídimos debe tenerse en cuenta junto con los datos de fertilidad, al evaluar los efectos para la reproducción masculina. También puede ser útil como ayuda para la interpretación del estudio el uso de datos de controles históricos sobre reproducción o desarrollo (por ejemplo, en cuanto al tamaño de la camada, DAG, mantenimiento de pezones, niveles séricos de T4), cuando se disponga de ellos.
|
|
75.
|
Por motivos de control de calidad, se propone recopilar datos de controles históricos y calcular coeficientes de variación de los datos numéricos, especialmente en cuanto a los parámetros relacionados con la detección de alteradores endocrinos. Estos datos se pueden usar con fines de comparación al evaluar los estudios actuales.
|
Informe del ensayo
|
76.
|
El informe del ensayo debe incluir la información siguiente:
|
|
Producto problema:
|
—
|
origen, número de lote, fecha límite de utilización, si se dispone de ellos;
|
|
—
|
estabilidad del producto problema, si se conoce.
|
|
|
|
Sustancias de un solo componente:
|
—
|
aspecto físico, hidrosolubilidad y otras propiedades fisicoquímicas pertinentes;
|
|
—
|
identificación química, como nombre IUPAC o CAS, número CAS, notación SMILES o InChI, fórmula estructural, pureza, identidad química de las impurezas si procede y es viable en la práctica, etc.
|
|
|
|
Sustancias de componentes múltiples, UVCB y mezclas:
|
—
|
deben caracterizarse en la medida de lo posible por la identidad química (véase más arriba), la cantidad en que están presentes y las propiedades fisicoquímicas pertinentes de sus componentes
|
. |
|
|
Vehículo (si procede):
|
—
|
Justificación de la elección del vehículo, si es distinto del agua.
|
|
|
|
Animales de ensayo:
|
—
|
especie y cepa empleada;
|
|
—
|
número, edad y sexo de los animales;
|
|
—
|
origen, condiciones de alojamiento, dieta, etc.;
|
|
—
|
peso de cada animal al inicio del ensayo;
|
|
—
|
justificación de la especie si es distinta de la rata.
|
|
|
|
Condiciones del ensayo:
|
—
|
justificación de la selección de las dosis;
|
|
—
|
datos sobre la formulación de la sustancia problema o su preparación con los alimentos, concentración obtenida, estabilidad y homogeneidad del preparado;
|
|
—
|
datos sobre la administración del producto problema;
|
|
—
|
factor de conversión de la concentración (ppm) del producto problema en los alimentos o en el agua de bebida a dosis reales (mg/kg peso corporal/día), si procede;
|
|
—
|
datos de la calidad de los alimentos y el agua;
|
|
—
|
descripción detallada del procedimiento de aleatorización de la selección de crías para el sacrificio, en su caso.
|
|
|
|
Resultados:
|
—
|
peso corporal y variaciones del mismo;
|
|
—
|
consumo de alimentos y de agua, si se ha medido;
|
|
—
|
datos de la respuesta tóxica por sexo y dosis, incluida la fertilidad, la gestación y cualquier otro signo de
|
|
—
|
duración de la gestación;
|
|
—
|
efectos tóxicos o de otro tipo sobre la reproducción, la descendencia, el crecimiento postnatal, etc.;
|
|
—
|
naturaleza, gravedad y duración de las observaciones clínicas (sean reversibles o no);
|
|
—
|
evaluación de la actividad sensorial, de la fuerza de prensión y de la actividad motriz;
|
|
—
|
pruebas hematológicas con los correspondientes valores de referencia;
|
|
—
|
pruebas bioquímicas con los correspondientes valores de referencia;
|
|
—
|
número de hembras adultas con ciclo estral normal y anormal y duración del ciclo;
|
|
—
|
número de nacidos vivos y de pérdidas postimplantatorias;
|
|
—
|
número de crías con anomalías visibles macroscópicamente; evaluación macroscópica de los genitales externos, número de nacidos con retraso evidente;
|
|
—
|
momento de la muerte durante el ensayo o indicación de que los animales han sobrevivido hasta el sacrificio;
|
|
—
|
número de implantes, tamaño de la camada y pesos de la camada en el momento del registro;
|
|
—
|
datos sobre el peso corporal de las crías; DAG de todas las crías (y peso corporal el día de la medición de la DAG);
|
|
—
|
mantenimiento de pezones en las crías macho;
|
|
—
|
niveles de la hormona tiroidea, en machos adultos y crías el día 13 (y madres y crías el día 4 si se miden);
|
|
—
|
peso corporal en el momento del sacrificio y peso de los órganos de los animales parentales;
|
|
—
|
observaciones de la autopsia;
|
|
—
|
descripción detallada de los hallazgos histopatológicos;
|
|
—
|
datos relativos a la absorción (si procede);
|
|
—
|
tratamiento estadístico de los resultados, si procede.
|
|
|
|
Discusión de los resultados.
|
|
Interpretación de los resultados
|
77.
|
El estudio pretende evaluar la toxicidad para la reproducción o el desarrollo relacionada con la administración continuada de dosis. En particular, dado que el estudio se centra en parámetros tanto de toxicidad general como de toxicidad para la reproducción y el desarrollo, los resultados deben permitir distinguir los efectos sobre la reproducción y el desarrollo que se producen en ausencia de toxicidad general, frente a los que se manifiestan únicamente a dosis que también son tóxicas para los animales parentales (véanse los puntos 7-11). Podría dar una indicación de la necesidad de realizar nuevas investigaciones y proporcionar orientaciones para el diseño de estudios posteriores. Debe consultarse el documento de orientación n.o 43 de la OCDE para obtener ayuda en la interpretación de los resultados de toxicidad para la reproducción y el desarrollo (19). El documento de orientación n.o 106 de la OCDE sobre la evaluación histológica de los ensayos endocrinos y de reproducción en roedores (16) proporciona información sobre la preparación y evaluación de los órganos (endocrinos) y los frotis vaginales que puede ser útil para el presente método de ensayo.
|
BIBLIOGRAFÍA
|
(1)
|
OCDE (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(2)
|
OCDE (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. and Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OCDE (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998, Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(6)
|
OCDE (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(7)
|
OCDE (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M.and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. and Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds. (1999). «Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights», Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OCDE (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(16)
|
OCDE (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No 106) Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(17)
|
Hess RA and Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (Eds.). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OCDE (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(20)
|
OCDE (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organización para la Cooperación y el Desarrollo Económico, París.
|
Apéndice 1
DEFINICIONES (VÉASE TAMBIÉN EL DOCUMENTO DE ORIENTACIONES GD 150 DE LA OCDE) (20)
Androgenicidad Capacidad de un producto para actuar como una hormona androgénica natural (por ejemplo, testosterona) en el organismo de un mamífero.
Antiandrogenicidad Capacidad de un producto para inhibir la acción de una hormona androgénica natural (por ejemplo, testosterona) en el organismo de un mamífero.
Antiestrogenicidad Capacidad de un producto para inhibir la acción de una hormona estrogénica natural (por ejemplo, 17ß-estradiol) en el organismo de un mamífero.
Actividad antitiroidea Capacidad de un producto para inhibir la acción de una hormona tiroidea natural (por ejemplo, T3) en el organismo de un mamífero.
Producto: Sustancia o mezcla.
Toxicidad para el desarrollo Manifestación de la toxicidad para la reproducción que consiste en la presencia de trastornos pre-, peri- y posnatales, de tipo estructural o funcional, en la descendencia.
Dosis Cantidad de producto problema administrada. La dosis se expresa en peso del producto problema por unidad de peso corporal de animal de ensayo y día (p. ej., mg/kg peso corporal/día), o como concentración alimentaria constante.
Posología Término general que abarca la dosis administrada, y la frecuencia y duración de la administración.
Toxicidad evidente Término general que describe signos claros de toxicidad tras la administración de un producto problema. Estos signos deben ser suficientes para la evaluación del peligro y tales que, ante un aumento de la dosis administrada, quepa esperar como resultado la aparición de signos tóxicos graves y una mortalidad probable.
Disminución de la fertilidad Trastornos en la capacidad o las funciones reproductoras de las hembras o de los machos.
Toxicidad para la madre Efectos adversos en las hembras gestantes, tanto si se producen de forma específica (efecto directo) como inespecífica (efecto indirecto) y relacionados con la gestación.
NOAEL Abreviatura en inglés de nivel sin efectos adversos observados (no-observed-adverse effect level). Se trata de la dosis más elevada a la que no se observa ningún efecto adverso debido al tratamiento.
Estrogenicidad Capacidad de un producto para actuar como una hormona estrogénica natural (por ejemplo, 17ß-estradiol) en el organismo de un mamífero.
Toxicidad para la reproducción Efectos nocivos para la descendencia y/o disminución de la capacidad o las funciones reproductoras de las hembras y de los machos.
Producto problema Sustancia o mezcla estudiada con este método de ensayo.
Actividad tiroidea Capacidad de un producto para actuar como una hormona tiroidea natural (por ejemplo, T3) en el organismo de un mamífero.
Validación Procedimiento científico diseñado para caracterizar las exigencias y limitaciones operativas de un método de ensayo y demostrar su fiabilidad y pertinencia para un fin particular.
Apéndice 2
DIAGRAMA DEL PROGRAMA EXPERIMENTAL CON INDICACIÓN DE LA DURACIÓN MÁXIMA DEL ESTUDIO, SOBRE LA BASE DE UN PERÍODO DE APAREAMIENTO COMPLETO DE CATORCE DÍAS


Apéndice 3
INFORME RESUMIDO EN FORMA DE CUADRO SOBRE LOS EFECTOS PARA LA REPRODUCCIÓN O EL DESARROLLO
|
OBSERVACIONES
|
VALORES
|
|
Dosis (unidades) ……
|
0 (testigo)
|
…
|
…
|
…
|
…
|
|
Parejas al inicio (N)
|
|
|
|
|
|
|
Ciclo estral (por lo menos, longitud media y frecuencia de los ciclos irregulares)
|
|
|
|
|
|
|
Hembras que presentan signos de cópula (N)
|
|
|
|
|
|
|
Hembras que llegan a la gestación (N)
|
|
|
|
|
|
|
Días de concepción 1-5 (N)
|
|
|
|
|
|
|
Días de concepción 6-. ..([1]
(23)
) (N)
|
|
|
|
|
|
|
Gestación ≤ 21 días (N)
|
|
|
|
|
|
|
Gestación = 22 días (N)
|
|
|
|
|
|
|
Gestación ≥ 23 días (N)
|
|
|
|
|
|
|
Madres con crías nacidas vivas (N)
|
|
|
|
|
|
|
Madres con crías vivas el día 4 después del parto (N)
|
|
|
|
|
|
|
Implantes/madre (media)
|
|
|
|
|
|
|
Crías vivas/madre en el momento del nacimiento (media)
|
|
|
|
|
|
|
Crías vivas/madre el día 4 (media)
|
|
|
|
|
|
|
Proporción de sexos (m/f) en el momento del nacimiento (media)
|
|
|
|
|
|
|
Proporción de sexos (m/f) el día 4 (media)
|
|
|
|
|
|
|
Peso de la camada al nacer (media)
|
|
|
|
|
|
|
Peso de la camada el día 4 (media)
|
|
|
|
|
|
|
Peso de las crías al nacer (media)
|
|
|
|
|
|
|
Peso de las crías en el momento de la medición de la DAG (media de los machos, media de las hembras)
|
|
|
|
|
|
|
DAG de las crías en el mismo día posnatal, nacimiento – día 4 (media de los machos, media de las hembras, indíquese el DPN)
|
|
|
|
|
|
|
Peso de las crías el día 4 (media)
|
|
|
|
|
|
|
Peso de las crías el día 13 (media)
|
|
|
|
|
|
|
Mantenimiento de pezones en las crías macho al día 13 (media)
|
|
|
|
|
|
|
CRÍAS ANÓMALAS
|
|
Madres con 0
|
|
|
|
|
|
|
Madres con 1
|
|
|
|
|
|
|
Madres con ≥ 2
|
|
|
|
|
|
|
PÉRDIDA DE DESCENDENCIA
|
|
Prenatal (implantes menos nacimientos vivos)
|
|
Hembras con 0
|
|
|
|
|
|
|
Hembras con 1
|
|
|
|
|
|
|
Hembras con 2
|
|
|
|
|
|
|
Hembras con ≥ 3
|
|
|
|
|
|
|
Posnatal (nacidos vivos menos vivos el día 13 posnatal)
|
|
Hembras con 0
|
|
|
|
|
|
|
Hembras con 1
|
|
|
|
|
|
|
Hembras con 2
|
|
|
|
|
|
|
Hembras con ≥ 3
|
|
|
|
|
|
B.65. MÉTODO DE ENSAYO IN VITRO CON BARRERA DE MEMBRANA PARA EVALUAR LA CORROSIÓN CUTÁNEA
INTRODUCCIÓN
|
1.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 435 de la OCDE (2015). La corrosión cutánea se refiere a la producción de una lesión irreversible en la piel, que se manifiesta como necrosis visible a través de la epidermis y que llega a la dermis, como consecuencia de la aplicación de un producto problema según la definición del Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA) de las Naciones Unidas (1) y del Reglamento (CE) n.o 1272/2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) de la Unión Europea (24). El presente método de ensayo, que es equivalente a las directrices de ensayo TG 435 actualizadas de la OCDE, proporciona un método de ensayo con barrera de membrana in vitro que puede utilizarse para identificar los productos corrosivos. El método de ensayo utiliza una membrana artificial diseñada para responder a productos corrosivos de forma similar a la piel animal in situ.
|
|
2.
|
Tradicionalmente, la corrosividad cutánea se ha evaluado aplicando el producto problema a la piel de animales vivos y evaluando el alcance de los daños tisulares tras un período de tiempo determinado (2). Además del presente método de ensayo, se han adoptado otros métodos de ensayo in vitro como alternativas (3) (4) al procedimiento normal de piel de conejo in vivo (capítulo B.4 del presente anexo, equivalente a las TG 404 de la OCDE) utilizado para identificar productos corrosivos (2). La estrategia escalonada de ensayo y valoración del SGA de las Naciones Unidas para la evaluación y clasificación de la corrosividad cutánea y el documento de orientación sobre los enfoques integrados de ensayos y evaluación (IATA, Integrated Approaches to Testing and Assessment) en relación con la irritación y la corrosión cutáneas recomiendan utilizar métodos de ensayo in vitro validados y aceptados con arreglo a los módulos 3 y 4 (1) (5). Los IATA describen varios módulos que agrupan las fuentes de información y herramientas de análisis, y i) aportan orientaciones sobre cómo integrar y utilizar los datos disponibles, tanto de ensayo como no experimentales, para la evaluación de la capacidad de irritación cutánea y de corrosión cutánea de los productos químicos, y ii) proponen un enfoque cuando se precisan más ensayos, incluido el caso de que se encuentren resultados negativos (5). En este enfoque modular, los resultados positivos de métodos de ensayo in vitro pueden utilizarse para clasificar un producto como corrosivo sin necesidad de realizar ensayos con animales, reduciendo así y afinando el uso de animales y evitando el dolor y la angustia que podrían producirse si se utilizaran animales para este fin.
|
|
3.
|
Se han realizado estudios de validación para el modelo de barrera de membrana in vitro, comercializado como Corrositex® (6) (7) (8), que muestra una exactitud general para predecir la corrosividad cutánea del 79 % (128/163), una sensibilidad del 85 % (76/89), y una especificidad del 70 % (52/74) con una base de datos de 163 sustancias y mezclas (7). Sobre la base de su validez reconocida, se ha recomendado la utilización de este método de referencia validado (MRV) como parte de una estrategia escalonada de ensayos para evaluar el peligro potencial de corrosión cutánea de los productos (5) (7). Antes de que pueda utilizarse con fines normativos un modelo de barrera de membrana in vitro de evaluación de la corrosión cutánea, deben determinarse su fiabilidad, pertinencia (exactitud) y limitaciones para el uso propuesto, a fin de garantizar su similitud con el MRV (9), de acuerdo con las normas de comportamiento predefinidas (10). La aceptación mutua de datos por la OCDE solo se garantizará después de que se hayan revisado e incluido en las directrices de ensayo equivalentes de la OCDE los eventuales métodos nuevos o actualizados propuestos que sigan las normas de comportamiento. En la actualidad, solo uno de los métodos in vitro está cubierto por las directrices de ensayo 435 de la OCDE y el presente método de ensayo, el modelo comercializado Corrositex®.
|
|
4.
|
Otros métodos de ensayo para la evaluación de la corrosividad cutánea se basan en la utilización de piel humana reconstituida (TG 431 de la OCDE) (3) y de piel de rata aislada (TG 430 de la OCDE) (4). Las presentes directrices de ensayo contemplan también la subcategorización de los productos corrosivos en las tres subcategorías de corrosividad del SGA de las Naciones Unidas y los tres grupos de embalaje para el transporte de las Naciones Unidas en cuanto al peligro de corrosividad. Las directrices de ensayo se adoptaron originalmente en 2006 y se actualizaron en 2015 para referirse al documento de orientación sobre los IATA y actualizar la lista de sustancias para la prueba de la competencia.
|
DEFINICIONES
|
5.
|
En el apéndice se dan las definiciones utilizadas.
|
CONSIDERACIONES INICIALES Y LIMITACIONES
|
6.
|
El ensayo descrito en este método de ensayo permite la identificación de productos problema corrosivos y permite la subcategorización de los productos problema corrosivos según el SGA de las Naciones Unidas y del CLP (cuadro 1). Además, un método de ensayo de este tipo puede utilizarse para tomar decisiones sobre la corrosividad y la no corrosividad de determinadas clases de productos como, por ejemplo, ácidos orgánicos e inorgánicos, derivados de ácidos (25) y bases, a efectos de determinados ensayos relacionados con el transporte (7) (11) (12). Elpresente método de ensayo describe un procedimiento genérico similar al método de ensayo de referencia validado (7). Aunque el presente método no proporciona información adecuada sobre la irritación cutánea, cabe señalar que el método de ensayo B.46 (equivalente a las directrices de ensayo TG 439 de la OCDE) aborda específicamente el aspecto de la irritación cutánea in vitro (13). Para una evaluación completa de los efectos cutáneos locales tras una exposición cutánea única, debe consultarse el documento de orientación de la OCDE sobre los enfoques integrados de ensayos y evaluación (5).
Cuadro 1
Categorías y subcategorías de corrosión cutánea del SGA de las Naciones Unidas (26)
|
Categoría de corrosivo (categoría 1) (en el caso de las autoridades que no utilizan subcategorías)
|
Posibles subcategorías de corrosivo (26) (en el caso de las autoridades que utilizan subcategorías, incluido el Reglamento CLP)
|
Corrosivo en ≥ 1 de 3 animales
|
|
Tiempo de exposición
|
Observación
|
|
Corrosivo
|
Corrosivo de subcategoría 1A
|
≤ 3 minutos
|
≤ 1 hora
|
|
Corrosivo de subcategoría 1B
|
> 3 minutos / ≤ 1 hora
|
≤ 14 días
|
|
Corrosivo de subcategoría 1C
|
> 1 hora / ≤ 4 horas
|
≤ 14 días
|
|
|
7.
|
Una limitación del método de referencia validado (7) es que muchos productos no corrosivos y algunos productos corrosivos no pueden someterse a ensayo, sobre la base de los resultados del ensayo de compatibilidad inicial (véase el punto 13). Los productos acuosos con un pH comprendido entre 4,5 y 8,5 a menudo no cumplen las condiciones para someterse a ensayo; sin embargo, el 85 % de los productos sometidos a ensayo en este intervalo de pH no eran corrosivos, en los ensayos con animales (7). El método con barrera de membrana in vitro puede utilizarse para el ensayo de sólidos (solubles o insolubles en agua), líquidos (acuosos o no acuosos) y emulsiones. Sin embargo, los productos problema que no provocan un cambio detectable en el ensayo de compatibilidad (es decir, cambio de color en el sistema de detección del producto que tiene el método de ensayo de referencia validado) no pueden someterse a ensayo utilizando el método con barrera de membrana y deben ensayarse utilizando otros métodos de ensayo.
|
PRINCIPIO DEL ENSAYO
|
8.
|
El sistema de ensayo consta de dos componentes: una biobarrera macromolecular sintética y un sistema de detección del producto (CDS); este método de ensayo detecta, a través del CDS, el daño causado en la barrera de membrana por los productos problema corrosivos tras la aplicación del producto problema en la superficie de la barrera de membrana macromolecular sintética (7), presumiblemente por el mismo mecanismo o mecanismos de corrosión que funcionan en la piel viva.
|
|
9.
|
La penetración a través de la barrera de membrana (o perforación) podría medirse por varios procedimientos o CDS, incluido un cambio en el color de un colorante indicador de pH o en alguna otra propiedad de la solución indicadora situada por debajo de la barrera.
|
|
10.
|
Debe confirmarse que la barrera de membrana es válida, es decir, que es pertinente y fiable para el uso previsto. Esto incluye garantizar que diferentes preparados son constantes con respecto a las propiedades de la barrera como, por ejemplo, que son capaces de mantener una barrera frente a los productos no corrosivos, para poder clasificar las propiedades corrosivas de los productos en las diversas subcategorías de corrosividad del SGA de las Naciones Unidas (1). La clasificación asignada se basa en el tiempo que tarda un producto en penetrar a través de la barrera de membrana hasta llegar a la solución indicadora.
|
DEMOSTRACIÓN DE LA COMPETENCIA
|
11.
|
Antes de proceder al uso sistemático del método con barrera de membrana in vitro, según el presente método de ensayo, los laboratorios deben demostrar su competencia técnica mediante la correcta clasificación de las doce sustancias para la prueba de la competencia recomendadas en el cuadro 2. Cuando una sustancia de la lista no esté disponible o en casos justificados, podrá utilizarse otra sustancia sobre la que se disponga de datos adecuados de referencia in vivo e in vitro [por ejemplo, de la lista de productos de referencia (10)] siempre que se apliquen los mismos criterios de selección que los descritos en el cuadro 1.
Cuadro 2
Sustancias para la prueba de la competencia
(27)
|
Sustancia (28)
|
N.o CAS
|
Clase química
|
Subcategoría del SGA de las Naciones Unidas in vivo
(29)
|
Subcategoría del SGA de las Naciones Unidas in vitro
(29)
|
|
Trifluoruro de boro dihidratado
|
13319-75-0
|
Ácidos inorgánicos
|
1A
|
1A
|
|
Ácido nítrico
|
7697-37-2
|
Ácidos inorgánicos
|
1A
|
1A
|
|
Pentacloruro de fósforo
|
10026-13-8
|
Precursores de ácidos inorgánicos
|
1A
|
1A
|
|
Cloruro de valerilo
|
638-29-9
|
Cloruros de ácidos
|
1B
|
1B
|
|
Hidróxido de sodio
|
1310-73-2
|
Bases inorgánicas
|
1B
|
1B
|
|
1-(2-Aminoetil)piperazina
|
140-31-8
|
Aminas alifáticas
|
1B
|
1B
|
|
Cloruro de bencenosulfonilo
|
98-09-9
|
Cloruros de ácidos
|
1C
|
1C
|
|
N,N-Dimetilbencilamina
|
103-83-3
|
Anilinas
|
1C
|
1C
|
|
Tetraetilen-pentaamina
|
112-57-2
|
Aminas alifáticas
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Fenoles
|
NC
|
NC
|
|
Acrilato de nonilo
|
2664-55-3
|
Acrilatos/metacrilatos
|
NC
|
NC
|
|
Bicarbonato de sodio
|
144-55-8
|
Sales inorgánicas
|
NC
|
NC
|
|
PROCEDIMIENTO
|
12.
|
En los puntos siguientes se describen los componentes y procedimientos de un método de ensayo con barrera de membrana artificial para la evaluación de la corrosividad (7) (15), basado en el MRV actual, es decir, el método Corrositex® disponible en el comercio. La barrera de membrana y la solución indicadora / de compatibilidad, así como la de categorización, pueden construirse, prepararse u obtenerse en el comercio, como sucede con el MRV, Corrositex®. Se dispone de un protocolo de método de ensayo de muestras para el método de ensayo de referencia validado (7). Los ensayos deben realizarse a temperatura ambiente (17-25 °C) y los componentes deben cumplir las siguientes condiciones.
|
Ensayo de compatibilidad con el producto problema
|
13.
|
Antes de realizar el ensayo con barrera de membrana, se realiza un ensayo de compatibilidad para determinar si el producto problema es detectable por el CDS. Si el CDS no detecta el producto problema, el método de ensayo con barrera de membrana no es adecuado para evaluar la posible corrosividad del producto problema y debe utilizarse otro método de ensayo diferente. El CDS y las condiciones de exposición utilizadas en el ensayo de compatibilidad deben reflejar la exposición en el subsiguiente ensayo con barrera de membrana.
|
Ensayo de categoría del producto problema según la escala temporal
|
14.
|
Si es apropiado para el método de ensayo, el producto problema que haya pasado con éxito el ensayo de compatibilidad debe someterse a un ensayo de categoría según la escala temporal, es decir, a una prueba de cribado para distinguir entre ácidos o bases débiles y fuertes. Por ejemplo, en el método de ensayo de referencia validado se utiliza un ensayo de categorización según la escala temporal para indicar cuál de las dos escalas temporales debe utilizarse basándose en si se detecta una reserva ácida o alcalina significativa. Deben utilizarse dos escalas temporales diferentes de la perforación para determinar la corrosividad y la subcategoría de corrosividad cutánea del SGA de las Naciones Unidas, sobre la base de la reserva ácida o alcalina del producto problema.
|
COMPONENTES DEL MÉTODO DE ENSAYO CON BARRERA DE MEMBRANA
Barrera de membrana
|
15.
|
La barrera de membrana consta de dos componentes: un gel acuoso macromolecular proteínico y una membrana permeable de soporte. El gel proteínico debe ser impermeable a los líquidos y sólidos, pero puede corroerse y hacerse permeable. La barrera de membrana totalmente construida debe conservarse en condiciones predeterminadas de las que se haya confirmado que impiden el deterioro del gel por fenómenos como, por ejemplo, secado, crecimiento microbiano, deslizamiento o cuarteado, que podrían afectar a su comportamiento. Debe determinarse el período de conservación aceptable, tras el que no se podrán utilizar los preparados de barrera de membrana.
|
|
16.
|
La membrana permeable de soporte proporciona apoyo mecánico al gel proteínico durante el proceso de gelificación y la exposición al producto problema. La membrana de soporte debe evitar que el gel se ablande o se deslice, y ha de ser fácilmente permeable a todos los productos problema.
|
|
17.
|
El gel proteínico, compuesto de proteínas como, por ejemplo, queratina, colágeno o mezclas de proteínas, que forman una matriz de gel, sirve como diana del producto problema. El material proteínico se coloca en la superficie de la membrana de soporte y se deja que gelifique antes de colocar la barrera de membrana encima de la solución indicadora. El gel proteínico debe tener por todos sitios el mismo espesor y la misma densidad, sin burbujas de aire ni defectos que puedan afectar a su integridad funcional.
|
Sistema de detección del producto (CDS)
|
18.
|
La solución indicadora, que es la misma solución utilizada en el ensayo de compatibilidad, debe responder a la presencia de una producto problema. Debe utilizarse un colorante (o una combinación de colorantes) indicador de pH como, por ejemplo, rojo del cresol y naranja de metilo, que muestre un cambio de color en respuesta a la presencia del producto problema. El sistema de medición puede ser visual o electrónico.
|
|
19.
|
Los sistemas de detección que se hayan elaborado para detectar el paso del producto problema a través de la membrana de barrera deben evaluarse en cuanto a su pertinencia y fiabilidad, a fin de demostrar la gama de productos que pueden detectarse y los límites cuantitativos de su detección.
|
REALIZACIÓN DEL ENSAYO
Conjunto de los componentes del método de ensayo
|
20.
|
La barrera de membrana se coloca en un frasco (o en un tubo) que contenga la solución indicadora, de forma que la membrana de soporte esté en pleno contacto con la solución indicadora y no haya presencia de burbujas de aire. Debe velarse por que se mantenga la integridad de la barrera.
|
Aplicación del producto problema
|
21.
|
Una cantidad adecuada del producto problema, por ejemplo 500 μl de un líquido o 500 mg de un sólido en polvo fino (7) se pone con cuidado en una capa por encima de la superficie superior de la barrera de membrana y se distribuye uniformemente. Se prepara un número adecuado de réplicas, por ejemplo cuatro (7), para cada producto problema y los testigos correspondientes (véanse los puntos 23 a 25). Se registra el momento de aplicación del producto problema a la barrera de membrana. Para garantizar que los tiempos de corrosión cortos se registran exactamente, se escalonan los tiempos de aplicación del producto problema a los frascos replicados.
|
Medición de la penetración en las barreras de las membranas
|
22.
|
Cada frasco se controla adecuadamente, y se registra el tiempo del primer cambio de la solución indicadora, es decir, la penetración a través de la barrera, y se determina el tiempo transcurrido entre la aplicación y la penetración a través de la barrera de membrana.
|
Testigos
|
23.
|
En los ensayos que impliquen el uso de un vehículo o disolvente con el producto problema, el vehículo o disolvente debe ser compatible con el sistema de barrera de membrana, es decir, no alterar la integridad del sistema de barrera de membrana, y no alterar la corrosividad del producto problema. Si procede, el control del disolvente (o del vehículo) debe someterse a ensayo en paralelo con el producto problema para demostrar la compatibilidad del disolvente con el sistema de barrera de membrana.
|
|
24.
|
Un testigo positivo (corrosivo) con una actividad de corrosividad intermedia, por ejemplo 110 ± 15 mg de hidróxido de sodio (corrosivo de la categoría 1B del SGA de las Naciones Unidas) (7), debe someterse a ensayo en paralelo con el producto problema para evaluar si el sistema de ensayo se comporta de manera aceptable. Un segundo testigo positivo que sea de la misma clase química que el producto problema puede resultar útil para evaluar el potencial relativo de corrosividad de un producto problema corrosivo. Deben seleccionarse uno o más testigos positivos que sean intermedios en su corrosividad (por ejemplo, de la subcategoría 1B del SGA de las Naciones Unidas) a fin de detectar eventuales cambios en el tiempo de penetración que puedan ser inaceptablemente superiores o inferiores al valor de referencia establecido, lo que indicaría que el sistema de ensayo no funciona correctamente. Con este fin, son de utilidad limitada los productos extremadamente corrosivos (de la subcategoría 1A del SGA de las Naciones Unidas) o no corrosivos. Un producto corrosivo de la subcategoría 1B del SGA de las Naciones Unidas permitiría detectar un tiempo de perforación demasiado corto o demasiado largo. Un producto ligeramente corrosivo (de la subcategoría 1C del SGA de las Naciones Unidas) podría utilizarse como testigo positivo a fin de medir la capacidad del método de ensayo para distinguir sistemáticamente entre productos débilmente corrosivos y no corrosivos. Independientemente del enfoque utilizado, debe elaborarse un intervalo de respuesta aceptable de los testigos positivos sobre la base del intervalo histórico de los tiempos de perforación de los testigos positivos empleados, como la media ± 2-3 desviaciones típicas. En cada estudio debe determinarse el tiempo exacto de perforación correspondiente al testigo positivo, de forma que puedan detectarse las desviaciones fuera del intervalo aceptable.
|
|
25.
|
También debe someterse a ensayo en paralelo con el producto problema un testigo negativo (no corrosivo) como, por ejemplo, ácido cítrico al 10 % o ácido propiónico al 6 % (7), como otra medida de control de la calidad para demostrar la integridad funcional de la barrera de membrana.
|
Criterios de aceptabilidad del estudio
|
26.
|
Según los parámetros temporales establecidos para cada una de las subcategorías de corrosividad del SGA de las Naciones Unidas, el tiempo (en minutos) transcurrido entre la aplicación de un producto problema a la barrera de membrana y la penetración a través de la barrera se utiliza para predecir la corrosividad del producto problema. Para que un estudio se considere aceptable, el testigo positivo en paralelo debe dar el tiempo previsto de respuesta a la penetración (por ejemplo, un tiempo de perforación de 8-16 minutos si se utiliza el hidróxido sódico como testigo positivo), el testigo negativo en paralelo no debe ser corrosivo y, cuando se incluye, el control del disolvente en paralelo no debe ser corrosivo ni alterar el potencial de corrosividad del producto problema. Antes de proceder al uso sistemático de un método que se ajuste al presente método de ensayo, los laboratorios deben demostrar su competencia técnica, utilizando las doce sustancias recomendadas en el cuadro 2. En el caso de los nuevos métodos «de imitación» elaborados en el marco del presente método de ensayo que son estructural y funcionalmente similares al método de referencia validado (14), deben utilizarse las normas de comportamiento predefinidas para demostrar la fiabilidad y exactitud del nuevo método antes de su uso en pruebas normativas (10).
|
Interpretación de los resultados y clasificación de la corrosividad de los productos problema
|
27.
|
El tiempo (en minutos) transcurrido entre la aplicación del producto problema a la barrera de membrana y la penetración a través de esta se utiliza para clasificar el producto problema en términos de subcategorías de corrosión del SGA de las Naciones Unidas (1) y, si procede, de grupos de embalaje de las Naciones Unidas (16). Se establecen para cada método de ensayo propuesto unos valores umbral de tiempo correspondientes a cada una de las tres subcategorías de corrosión. Las decisiones finales sobre los tiempos umbral deben considerar la necesidad de minimizar la subclasificación del peligro corrosivo (es decir, los falsos negativos). En las presentes directrices de ensayo, deben utilizarse los tiempos umbral de Corrositex®, descritos en el cuadro 3, ya que representa el único método de ensayo que actualmente se ajusta a las directrices de ensayo (7).
Cuadro 3
Modelo de predicción de Corrositex®
|
Tiempo medio de perforación (min.)
|
Predicción del SGA de las Naciones Unidas (32)
|
|
Productos problema de categoría 1 (30) (determinados por el ensayo de categorización del método)
|
Productos problema de categoría 2 (31) (determinados por el ensayo de categorización del método)
|
|
0-3 min.
|
0-3 min.
|
Corrosivo Subcategoría optativa 1A
|
|
> 3 a 60 min.
|
> 3 a 30 min.
|
Corrosivo Subcategoría optativa 1B
|
|
> 60 a 240 min.
|
> 30 a 60 min.
|
Corrosivo Subcategoría optativa 1C
|
|
> 240 min.
|
> 60 min.
|
No corrosivo
|
|
DATOS E INFORME
Datos
|
28.
|
El tiempo (en minutos) transcurrido entre la aplicación y la penetración a través de la barrera del producto problema y del testigo o testigos positivos debe comunicarse en forma de cuadro con los datos de las distintas réplicas, así como la media ± la desviación típica de cada ensayo.
|
Informe del ensayo
|
29.
|
El informe del ensayo debe incluir la información siguiente:
|
|
Producto problema y sustancias testigo:
|
—
|
Sustancias de un solo componente: identificación química, como nombre IUPAC o CAS, número CAS, notación SMILES o InChI, fórmula estructural, pureza, identidad química de las impurezas si procede y es viable en la práctica, etc.;
|
|
—
|
Sustancias de componentes múltiples, sustancias UVCB y mezclas: deben caracterizarse en la medida de lo posible por la identidad química (véase más arriba), la cantidad en que están presentes y las propiedades fisicoquímicas pertinentes de sus componentes;
|
|
—
|
Aspecto físico, hidrosolubilidad y otras propiedades fisicoquímicas pertinentes;
|
|
—
|
Origen; número de lote, si está disponible;
|
|
—
|
Tratamiento del producto problema / sustancia testigo antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
—
|
Estabilidad del producto problema, fecha límite de utilización, o fecha de nuevo análisis, si se conoce;
|
|
—
|
Condiciones de conservación.
|
|
|
|
Vehículo:
|
—
|
Identificación, concentración (en su caso), volumen utilizado;
|
|
—
|
Motivación de la elección del vehículo.
|
|
|
|
Modelo de barrera de membrana in vitro y protocolo utilizados, incluidas la exactitud y la fiabilidad demostradas
|
|
|
Condiciones de ensayo:
|
—
|
Descripción del equipo y de los procedimientos de preparación utilizados;
|
|
—
|
Origen y composición de la barrera de membrana in vitro utilizada;
|
|
—
|
Composición y propiedades de la solución indicadora;
|
|
—
|
Cantidades de producto problema y de sustancias testigo;
|
|
—
|
Descripción y justificación del ensayo de categorización de la escala temporal;
|
|
—
|
Tiempos de observación;
|
|
—
|
Descripción de los criterios de evaluación y clasificación aplicados;
|
|
—
|
Demostración de la competencia para la aplicación del método de ensayo antes de su uso sistemático mediante ensayo de las sustancias para la prueba de la competencia.
|
|
|
|
Resultados:
|
—
|
Cuadro de datos brutos individuales de las distintas muestras de producto problema y de testigo respecto a cada réplica;
|
|
—
|
Descripción de otros efectos observados;
|
|
—
|
Clasificación derivada con referencia al modelo de predicción o a los criterios de decisión utilizados.
|
|
Discusión de los resultados
Conclusiones
|
BIBLIOGRAFÍA
|
(1)
|
Naciones Unidas (2013). Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), quinta edición revisada, Naciones Unidas, Nueva York y Ginebra, 2013. Disponible en: http://www.unece.org/es/trans/danger/publi/ghs/ghs_rev05/05files_s.html
|
|
(2)
|
Capítulo B.4 del presente anexo, Toxicidad aguda: irritación/corrosión cutánea.
|
|
(3)
|
Capítulo B.40 bis del presente anexo, Corrosión cutánea in vitro: método de ensayo con epidermis humana reconstruida (EHR).
|
|
(4)
|
Capítulo B.40 del presente anexo, Corrosión cutánea in vitro: Resistencia eléctrica transcutánea (RET).
|
|
(5)
|
OCDE (2015): Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. and Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OCDE (2005): Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (No 34).
|
|
(10)
|
OCDE (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (September 20, 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Capítulo B.46 del presente anexo, Irritación cutánea in vitro: Método de ensayo con epidermis humana reconstruida. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Disponible en: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Disponible en: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
Naciones Unidas (2013). Recomendaciones de las Naciones Unidas relativas al transporte de mercancías peligrosas, Reglamentación modelo, 18.a edición revisada (parte, capítulo 2.8), Naciones Unidas, 2013. Disponible en: https://shop.un.org/es/series/recomendaciones-relativas-al-transporte-de-mercanc-peligrosas-reglamentaci-n-modelo.
|
Apéndice
DEFINICIONES
Exactitud
: Grado de coincidencia entre los resultados obtenidos con el método de ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del método de ensayo y es un aspecto de su pertinencia. Este término y el de “concordancia” se suelen usar indistintamente para indicar la proporción de resultados correctos de un método de ensayo (9).
Producto
: Sustancia o mezcla.
Sistema de detección del producto (CDS)
: Sistema de medición visual o electrónico con una solución indicadora que responde a la presencia de un producto problema, por ejemplo mediante un cambio en un colorante (o una combinación de colorantes) indicador de pH que muestre un cambio de color en respuesta a la presencia del producto problema o mediante otros tipos de reacciones químicas o electroquímicas.
Concordancia
: Se trata de una medida del comportamiento del método de ensayo que da un resultado categorial, y es un aspecto de su pertinencia. Este término y el de “exactitud” se pueden usar indistintamente, y se define como la proporción de todos los productos estudiados que se clasifican correctamente como positivos o negativos. La concordancia depende en gran medida de la prevalencia de positivos en los tipos de producto problema que se están examinando (9).
SGA (Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos)
: Sistema que propone la clasificación de los productos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (1).
IATA
: Enfoque integrado de pruebas y evaluación (Integrated Approach to Testing and Assessment).
Mezcla
: Mezcla o solución compuesta de dos o más sustancias.
Sustancia de un solo componente:
: Sustancia, definida por su composición cuantitativa, en la que un solo componente principal representa al menos el 80 % (p/p).
Sustancia de componentes múltiples
: Sustancia, definida por su composición cuantitativa, en la que hay varios componentes principales presentes a una concentración ≥ 10 % (p/p) y < 80 % (p/p). Una sustancia de componentes múltiples es el resultado de un proceso de fabricación. La diferencia entre mezcla y sustancia de componentes múltiples es que una mezcla se obtiene mezclando dos o más sustancias sin reacción química. Una sustancia de componentes múltiples es el resultado de una reacción química.
NC
: No corrosivo.
Normas de comportamiento
: Normas, basadas en un método de referencia validado, que proporcionan la base para evaluar la comparabilidad de un método de ensayo propuesto que es similar desde el punto de vista mecánico y funcional. Se incluyen aquí: i) los componentes fundamentales del método de ensayo; ii) una lista mínima de productos de referencia seleccionados de entre los productos utilizados para demostrar el comportamiento aceptable del método de ensayo validado; y iii) los niveles similares de fiabilidad y exactitud, basados en lo obtenido con el método de ensayo validado, que el método de ensayo propuesto debe demostrar cuando se evalúa con la lista mínima de productos de referencia (9).
Pertinencia
: Descripción de la relación del método de ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el método de ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un método de ensayo (9).
Fiabilidad
: Medida del grado en que un método de ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios (9).
Sensibilidad
: Proporción de todos los productos activos/positivos que se clasifican correctamente mediante el método de ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (9).
Corrosión cutánea in vivo
: Producción de una lesión irreversible de la piel; en particular, necrosis visible a través de la epidermis y que llega a la dermis, como consecuencia de la aplicación de un producto problema durante hasta cuatro horas. Las reacciones corrosivas se caracterizan por úlceras, sangrado, costras sanguinolentas y, hacia el final del período de observación de catorce días, por decoloración debida al blanqueo de la piel, zonas completas de alopecia y cicatrices. Debe considerarse la realización de un examen histopatológico para evaluar las lesiones dudosas.
Especificidad
: Proporción de todos los productos inactivos/negativos que se clasifican correctamente mediante el método de ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (9).
Sustancia
: Elemento químico y sus compuestos naturales u obtenidos mediante algún proceso de producción, incluidos los eventuales aditivos necesarios para mantener su estabilidad y las eventuales impurezas que se produzcan en el proceso, con exclusión de los eventuales disolventes que puedan separarse sin afectar a la estabilidad de la sustancia ni modificar su composición.
Producto problema
: Sustancia o mezcla estudiada con este método de ensayo.
UVCB
: Sustancias de composición desconocida o variable, productos complejos de reacción o materiales biológicos.
B.66. ENSAYOS IN VITRO DE TRANSACTIVACIÓN TRANSFECTADA ESTABLEMENTE PARA DETECTAR AGONISTAS Y ANTAGONISTAS DE LOS RECEPTORES ESTROGÉNICOS
INTRODUCCIÓN GENERAL
Directrices de ensayo de la OCDE sobre la base del comportamiento
|
1.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 455 de la OCDE (2016). Estas son unas directrices de ensayo basadas en el comportamiento (PBTG, performance-based test guideline), que describen la metodología de los ensayos in vitro de transactivación transfectada establemente para detectar agonistas y antagonistas de los receptores estrogénicos (ensayos ER TA, Estrogen Receptor Transactivation Assays). Comprenden varios métodos de ensayo similares desde el punto de vista mecánico y funcional para la identificación de los agonistas y antagonistas de los receptores estrogénicos (es decir, ERα, y/o ERβ) y deben facilitar la elaboración de nuevos métodos de ensayo similares o modificados, de conformidad con los principios de validación expuestos en el documento de orientación de la OCDE sobre la validación y la aceptación internacional de métodos de ensayo nuevos o actualizados para la evaluación de peligros (Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment) (1). Los métodos de ensayo de referencia plenamente validados (apéndice 2 y apéndice 3) que sirven de base a estas PBTG son los siguientes:
|
—
|
el ensayo de transactivación transfectada establemente (STTA, Stably Transfected TA) (2) con la línea celular (h) ERα-HeLa-9903; y
|
|
—
|
el ensayo VM7Luc ER TA (3) con la línea celular VM7Luc4E2 (33) que expresa predominantemente hERα con cierta contribución de hERβ (4) (5).
|
Para la elaboración y la validación de ensayos similares relativos al mismo parámetro de peligro, se dispone de normas de comportamiento (6) (7), que deben utilizarse. Permiten la oportuna modificación de las PBTG 455, para que puedan añadirse a unas PBTG actualizadas nuevos ensayos similares; sin embargo, esos ensayos similares solo se añadirán después de que la OCDE haya examinado si se cumplen las normas de comportamiento y haya manifestado que es así. Los ensayos incluidos en las TG 455 pueden utilizarse indistintamente para satisfacer los requisitos de los países miembros de la OCDE en cuanto a los resultados de los ensayos sobre la transactivación de receptores estrogénicos, y también se benefician de la aceptación mutua de datos de la OCDE.
|
Antecedentes y principios de los ensayos incluidos en este método de ensayo
|
2.
|
La OCDE lanzó en 1998 una actividad muy prioritaria para revisar las directrices existentes y elaborar nuevas directrices sobre los ensayos de cribado y finales de posibles alteradores endocrinos. El marco conceptual de la OCDE para los ensayos y la evaluación de posibles alteradores endocrinos se revisó en 2012. Los marcos conceptuales originales y revisados se incluyen como anexos en el documento de orientación de la OCDE sobre las directrices de ensayo normalizadas para la evaluación de los productos en cuanto a las alteraciones endocrinas (8). El marco conceptual se compone de cinco niveles, cada uno de los cuales corresponde a un nivel diferente de complejidad biológica. Los ensayos de transactivación de receptores estrogénicos (ER TA) descritos en este método de ensayo son de nivel 2, que incluye los ensayos in vitro que aportan datos sobre el mecanismo o mecanismos y rutas endocrinos seleccionados. Este método de ensayo se refiere a ensayos de transactivación (TA) in vitro diseñados para identificar a los agonistas y antagonistas de los receptores estrogénicos (ER).
|
|
3.
|
La interacción de los estrógenos con los ER puede afectar a la transcripción de genes controlados por estrógenos, lo cual puede dar lugar a la inducción o a la inhibición de procesos celulares, incluidos los necesarios para la proliferación celular, el desarrollo fetal normal y la función reproductora (9) (10) (11). La perturbación de los sistemas estrogénicos normales puede provocar efectos adversos sobre el desarrollo normal (ontogénesis), la salud reproductiva y la integridad del aparato reproductor.
|
|
4.
|
Los ensayos TA in vitro se basan en una interacción directa o indirecta de las sustancias con un receptor específico que regula la transcripción de un producto del gen marcador. Estos ensayos se han utilizado ampliamente para evaluar la expresión génica regulada por receptores nucleares específicos como, por ejemplo, los ER (12) (13) (14) (15) (16), y se han propuesto para la detección de la transactivación estrogénica regulada por los ER (17) (18) (19). Hay por lo menos dos subtipos importantes de ER nucleares, los α y los β, que están codificados por distintos genes. Las respectivas proteínas tienen funciones biológicas diferentes, así como diferentes distribuciones tisulares y afinidades para unirse a ligandos (20) (21) (22) (23) (24) (25) (26). El ERα nuclear media la respuesta estrogénica clásica (27) (28) (29) (30), por lo que la mayoría de los modelos que se están elaborando actualmente para medir la activación o inhibición del ER son específicos de ERα. Los ensayos se utilizan para identificar los productos que activan (o inhiben) el ER tras la unión al ligando, después de lo cual el complejo receptor-ligando se une a unos elementos específicos de respuesta de ADN y transactiva un gen marcador, lo que da lugar a mayor expresión celular de una proteína marcadora. En estos ensayos pueden utilizarse diferentes respuestasde marcadores. En los sistemas basados en la luciferasa, esta enzima transforma el sustrato de luciferina en un producto bioluminiscente que puede medirse cuantitativamente con un luminómetro. Otros ejemplos de marcadores comunes son el gen de la proteína fluorescente y el gen LacZ, que codifica la β-galactosidasa, enzima que puede transformar el sustrato incoloro X-gal (5-bromo-4-cloro-indolil-galactopiranósido) en un producto de color azul que puede cuantificarse con un espectrofotómetro. Estos marcadores pueden evaluarse de forma rápida y poco costosa con los equipos de pruebas disponibles en el comercio.
|
|
5.
|
Los estudios de validación de los ensayos STTA y VM7Luc TA han demostrado su pertinencia y fiabilidad con respecto a su finalidad prevista (3) (4) (5) (30). Las normas de comportamiento de los ensayos ER TA basados en la luminiscencia que utilizan líneas celulares de cáncer de mama se incluyen en el informe de evaluación de métodos de ensayo del ICCVAM relativo al método de ensayo LUMI-CELL® ER (VM7Luc ER TA): ensayo in vitro para identificar la actividad agonista y antagonista de los receptores estrogénicos humanos de los productos (3). Estas normas de comportamiento se han modificado para que sean aplicables tanto a los ensayos STTA como a los VM7Luc TA (2).
|
|
6.
|
Las definiciones y abreviaturas utilizadas en el presente método de ensayo se recogen en el apéndice 1.
|
Ámbito de aplicación y limitaciones relativas a los ensayos TA
|
7.
|
Estos ensayos se proponen para fines de cribado y establecimiento de prioridades, pero también pueden proporcionar información mecánica que pueda utilizarse con un enfoque de ponderación de los ensayos. Se refieren a la TA inducida por un producto que se une a los ER en un sistema in vitro. Así pues, los resultados no deben extrapolarse directamente a la compleja señalización y regulación del sistema endocrino intacto in vivo.
|
|
8.
|
La TA mediada por los ER se considera uno de los mecanismos clave de la alteración endocrina (AE), aunque existen otros mecanismos a través de los cuales se puede producir esta, entre ellos: i) interacciones con otros receptores y sistemas enzimáticos dentro del sistema endocrino, ii) síntesis de hormonas, iii) activación y/o inactivación metabólica de hormonas, iv) distribución de hormonas a los tejidos diana, y v) eliminación de hormonas del organismo. Ninguno de los ensayos con arreglo al presente método de ensayo aborda estos modos de acción.
|
|
9.
|
El presente método de ensayo aborda la capacidad de los productos para activar (es decir, actuar como agonistas) y también suprimir (es decir, actuar como antagonistas) la transcripción dependiente de los ER. Algunos productos pueden, en función del tipo de células, mostrar actividad tanto agonista como antagonista y se conocen como moduladores selectivos de receptores estrogénicos (SERM). Los productos que son negativos en estos ensayos pueden evaluarse en un ensayo de unión a ER antes de llegar a la conclusión de que el producto no se une al receptor. Además, solo es probable que los ensayos informen sobre la actividad de la molécula madre teniendo en cuenta la limitada capacidad de metabolización de los sistemas celulares in vitro. Teniendo en cuenta que durante la validación únicamente se utilizaron sustancias solas, no se ha abordado la aplicabilidad a mezclas problema. Sin embargo, el método de ensayo es teóricamente aplicable a los ensayos de sustancias de componentes múltiples, UVCB y mezclas. Antes de la utilización del método de ensayo con una sustancia de componentes múltiples, UVCB o mezcla para obtener datos con fines normativos, debe considerarse si podría proporcionar resultados adecuados a tales fines y, en caso afirmativo, por qué. Dichas consideraciones no son necesarias cuando los requisitos normativos estipulan que la mezcla debe someterse a ensayo.
|
|
10.
|
A efectos de información, en el cuadro 1 se presentan los resultados de los ensayos de actividad agonista correspondientes a las 34 sustancias que se sometieron a ensayo con los dos métodos de ensayo de referencia plenamente validados descritos en el presente método de ensayo. De estas sustancias, 26 se clasifican como agonistas ER definitivos y 8 son negativas sobre la base de los informes publicados, incluidos los ensayos in vitro de unión a ER y de TA, y/o el ensayo uterotrófico (2) (3) (18) (31) (32) (33) (34). En el cuadro 2 se presentan los resultados de los ensayos de actividad antagonista correspondientes a las 15 sustancias que se sometieron a ensayo con los dos métodos de ensayo de referencia plenamente validados descritos en el presente método de ensayo. De estas sustancias, 4 se clasifican como antagonistas ER definitivos/supuestos y 10 son negativas sobre la base de los informes publicados, incluidos los ensayos in vitro de unión a ER y de TA (2) (3) (18) (31). En referencia a los datos resumidos en los cuadros 1 y 2, había un 100 % de concordancia entre los dos métodos de ensayo de referencia en la clasificación de todas las sustancias, excepto una (mifepristona), en cuanto al ensayo de la actividad antagonista, y cada sustancia se clasificó correctamente como agonista/antagonista ER o negativa. En las normas de comportamiento aplicables a la ER TA (6) (7), apéndice 2 (cuadros 1, 2 y 3) se ofrece información suplementaria sobre este grupo de productos, así como sobre otros productos objeto de los ensayos de tipo STTA y VM7Luc ER durante los estudios de validación.
|
Cuadro 1
Resumen de los resultados de los ensayos STTA y VM7Luc ER AT con sustancias objeto de los dos ensayos de la actividad agonista y clasificadas como agonistas ER (POS) o negativas (NEG)
|
|
Sustancia
|
N.o CAS
|
Ensayo STTA (35)
|
Ensayo VM7Luc ER TA (36)
|
Procedencia de los datos para la clasificación (38)
|
|
Actividad ER TA
|
Valor CP10 (M)
|
Valor CP50
(2) (M)
|
Actividad ER TA
|
Valor CE50
(2), (37) (M)
|
Otras ER TA (34)
|
Unión ER
|
Uterotrófico
|
|
1
|
17ß-Estradiol (1)
|
50-28-2
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
5,63 × 10-12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-Estradiol (1)
|
57-91-0
|
POS
|
7,24 × 10-11
|
6,44 × 10-10
|
POS
|
1,40 × 10-9
|
POS (11/11)
|
POS
|
POS
|
|
3
|
17α-Etinil-estradiol (1)
|
57-63-6
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
7,31 × 10-12
|
POS (22/22)
|
POS
|
POS
|
|
4
|
17β-Trembolona
|
10161-33-8
|
POS
|
1,78 × 10-8
|
2,73 × 10-7
|
POS
|
4,20 × 10-8
|
POS (2/2)
|
NE
|
NE
|
|
5
|
19-Nortestosteron (1)
|
434-22-0
|
POS
|
9,64 × 10-9
|
2,71 × 10-7
|
POS
|
1,80 × 10-6
|
POS (4/4)
|
POS
|
POS
|
|
6
|
4-Cumilfenol (1)
|
599-64-4
|
POS
|
1,49 × 10-7
|
1,60 × 10-6
|
POS
|
3,20 × 10-7
|
POS (5/5)
|
POS
|
NE
|
|
7
|
4-terc-Octilfenol (1)
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
POS
|
3,19 × 10-8
|
POS (21/24)
|
POS
|
POS
|
|
8
|
Apigenin (1)
|
520-36-5
|
POS
|
1,31 × 10-7
|
5,71 × 10-7
|
POS
|
1,60 × 10-6
|
POS (26/26)
|
POS
|
NE
|
|
9
|
Atrazin (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NE
|
|
10
|
Bisfenol A (1)
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
POS
|
5,33 × 10-7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisfenol B (1)
|
77-40-7
|
POS
|
2,36 × 10-8
|
2,11 × 10-7
|
POS
|
1,95 × 10-7
|
POS (6/6)
|
POS
|
POS
|
|
12
|
Ftalato de butilbencilo (1)
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
POS
|
1,98 × 10-6
|
POS (12/14)
|
POS
|
NEG
|
|
13
|
Corticosteron (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (6/6)
|
NEG
|
NE
|
|
14
|
Cumestrol (1)
|
479-13-0
|
POS
|
1,23 × 10-9
|
2,00 × 10-8
|
POS
|
1,32 × 10-7
|
POS (30/30)
|
POS
|
NE
|
|
15
|
Daidzeín (1)
|
486-66-8
|
POS
|
1,76 × 10-8
|
1,51 × 10-7
|
POS
|
7,95 × 10-7
|
POS (39/39)
|
POS
|
POS
|
|
16
|
Dietilestilbestrol (1)
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POS
|
3,34 × 10-11
|
POS (42/42)
|
POS
|
NE
|
|
17
|
Ftalato de di-n-butilo
|
84-74-2
|
POS
|
4,09 × 10-6
|
|
POS
|
4,09 × 10-6
|
POS (6/11)
|
POS
|
NEG
|
|
18
|
Etilparabeno
|
120-47-8
|
POS
|
5,00 × 10-6
|
(sin CP50)
|
POS
|
2,48 × 10-5
|
POS
|
|
NE
|
|
19
|
Estron (1)
|
53-16-7
|
POS
|
3,02 × 10-11
|
5,88 × 10-10
|
POS
|
2,34 × 10-10
|
POS (26/28)
|
POS
|
POS
|
|
20
|
Genisteín (1)
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
POS
|
2,71 × 10-7
|
POS (100/102)
|
POS
|
POS
|
|
21
|
Haloperidol
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NE
|
|
22
|
Canferol (1)
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
POS
|
3,99 × 10-6
|
POS (23/23)
|
POS
|
NE
|
|
23
|
Kepone (1)
|
143-50-0
|
POS
|
7,11 × 10-7
|
7,68 × 10-6
|
POS
|
4,91 × 10-7
|
POS (14/18)
|
POS
|
NE
|
|
24
|
Ketoconazol
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NE
|
|
25
|
Linurón (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NE
|
|
26
|
meso-Hexestrol (1)
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POS
|
1,65 × 10-11
|
POS (4/4)
|
POS
|
NE
|
|
27
|
Metil-testosteron (1)
|
58-18-4
|
POS
|
1,73 × 10-7
|
4,11 × 10-6
|
POS
|
2,68 × 10-6
|
POS (5/6)
|
POS
|
NE
|
|
28
|
Morina
|
480-16-0
|
POS
|
5,43 × 10-7
|
4,16 × 10-6
|
POS
|
2,37 × 10-6
|
POS (2/2)
|
POS
|
NE
|
|
29
|
Noretinodrel (1)
|
68-23-5
|
POS
|
1,11 × 10-11
|
1,50 × 10-9
|
POS
|
9,39 × 10-10
|
POS (5/5)
|
POS
|
NE
|
|
30
|
p,p’-Metoxicloro (1)
|
72-43-5
|
POS
|
1,23 × 10-6
|
(sin CP50) (2)
|
POS
|
1,92 × 10-6
|
POS (24/27)
|
POS
|
POS
|
|
31
|
Fenobarbital (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NE
|
|
32
|
Reserpina
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NE
|
|
33
|
Espironolacton (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NE
|
|
34
|
Testosterona
|
58-22-0
|
POS
|
2,82 × 10-8
|
9,78 × 10-6
|
POS
|
1,75 × 10-5
|
POS (5/10)
|
POS
|
NE
|
|
Abreviaturas: N.o CAS = número de registro del Chemical Abstracts Service; M = molar; CE50 = concentración de la sustancia problema que produce la mitad del efecto máximo; NEG = negativo; POS = positivo; NE = No se ha ensayado; CP10 (y CP50) = concentración de una sustancia problema a la que la respuesta es del 10 % (o del 50 % en el caso de la CP50) de la respuesta inducida por el testigo positivo (E2, 1 nm) en cada placa.
|
Cuadro 2
Resumen de los resultados de los ensayos STTA y VM7Luc ER TA con sustancias objeto de los dos ensayos de la actividad antagonista y clasificadas como antagonistas ER (POS) o negativas (NEG)
|
|
Sustancia (3)
|
N.o CAS
|
Ensayo ER STTA (40)
|
Ensayo VM7Luc ER TA (41)
|
Efectos candidatos del ER STTA (43)
|
Clasificación de consenso del ICCVAM (44) Clase química
|
del MeSH (45)
|
Clase de producto (46)
|
|
Actividad ER TA
|
Valor de CI50
(39) (M)
|
Actividad ER TA
|
Valor de CI50
(39), (42) (M)
|
|
1
|
4-Hidroxitamoxifeno
|
68047-06-3
|
POS
|
3,97 × 10-9
|
POS
|
2,08 × 10-7
|
POS moderado
|
POS
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos
|
|
2
|
Dibenzo[a,h]antraceno
|
53-70-3
|
POS
|
Sin CI50
|
POS
|
Sin CI50
|
POS
|
SP
|
Compuestos policíclicos
|
Productos de laboratorio, productos naturales
|
|
3
|
Mifepristona
|
84371-65-3
|
POS
|
5,61 × 10-6
|
NEG
|
—
|
POS suave
|
NEG
|
Esteroides
|
Productos farmacéuticos
|
|
4
|
Raloxifeno HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
POS
|
1,19 × 10-9
|
POS moderado
|
POS
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos
|
|
5
|
Tamoxifeno
|
10540-29-1
|
POS
|
4,91 × 10-7
|
POS
|
8,17 × 10-7
|
POS
|
POS
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos
|
|
6
|
17β-Estradiol
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
SN
|
SN
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
7
|
Apigenina
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Compuestos heterocíclicos
|
Colorantes, productos naturales, productos intermedios farmacéuticos
|
|
8
|
Atrazina
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
SN
|
Compuestos heterocíclicos
|
Herbicidas
|
|
9
|
Ftalato de di-n-butilo
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Ésteres, ácido ftálico
|
Ingredientes de cosméticos, productos industriales, plastificantes
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
No se ha ensayado
|
SN
|
Compuestos heterocíclicos, pirimidina
|
Fungicidas
|
|
11
|
Flavona
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
SN
|
SN
|
Flavonoides, compuestos heterocíclicos
|
Productos naturales, productos farmacéuticos
|
|
12
|
Flutamida
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
SN
|
Amidas
|
Productos farmacéuticos, productos veterinarios
|
|
13
|
Genisteína
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
SN
|
NEG
|
Flavonoides, compuestos heterocíclicos
|
Productos naturales, productos farmacéuticos
|
|
14
|
p-n-Nonilfenol
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
No se ha ensayado
|
NEG
|
Fenoles
|
Productos intermedios
|
|
15
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
SN
|
NEG
|
Hidrocarburos (cíclicos)
|
Productos naturales
|
|
Abreviaturas: N.o CAS = número de registro del Chemical Abstracts Service; M = molar; CI50 = concentración de la sustancia problema que produce la mitad de la inhibición máxima; NEG = negativo; SP = supuesto negativo; POS = positivo; SP = supuesto positivo.
|
COMPONENTES DEL ENSAYO ER TA
Componentes esenciales del ensayo
|
11.
|
Este método de ensayo se aplica a los ensayos que utilizan un receptor ERα transfectado establemente o endógeno y una construcción de gen marcador transfectado establemente bajo el control de uno o más elementos de respuesta estrogénica; sin embargo, pueden estar presentes otros receptores, como el ERβ. Estos son los componentes esenciales del ensayo.
|
Testigos
|
12.
|
Debe describirse la base de los patrones de referencia en paralelo propuestos para cada ensayo de agonistas y de antagonistas. Los testigos en paralelo (negativos, de disolvente y positivos), según proceda, sirven para indicar que el ensayo funciona en las condiciones de ensayo y proporcionan la base para comparar unos experimentos con otros; generalmente forman parte de los criterios de aceptabilidad de un determinado experimento (1).
|
Procedimientos normales de control de calidad
|
13.
|
Deben llevarse a cabo procedimientos normales de control de calidad, tal como se describe para cada ensayo, a fin de garantizar que la línea celular se mantiene estable a lo largo de varios pases, exenta de micoplasmas (es decir, libre de contaminación bacteriana), y conserva a lo largo del tiempo la capacidad de proporcionar las respuestas esperadas mediadas por ER. Las líneas celulares deben someterse a un control adicional para comprobar su identidad correcta, así como para detectar la eventual presencia de otros contaminantes (por ejemplo, hongos, levaduras y virus).
|
Demostración de la competencia del laboratorio
|
14.
|
Antes de someter productos desconocidos a alguno de los ensayos con arreglo al presente método de ensayo, cada laboratorio debe demostrar su competencia para utilizar el ensayo. Para demostrar tal competencia, cada laboratorio debe probar las catorce sustancias para la prueba de la competencia que figuran en el cuadro 3 en relación con el ensayo de agonistas y las diez sustancias para la prueba de la competencia que figuran en el cuadro 4 en relación con el ensayo de antagonistas. Esta prueba de la competencia también confirmará la sensibilidad del sistema de ensayo. La lista de sustancias para la prueba de la competencia es un subconjunto de las sustancias de referencia que figuran en las normas de comportamiento relativas a los ensayos ER TA (6). Estas sustancias están disponibles en el mercado, representan las clases de productos normalmente asociadas con la actividad agonista o antagonista ER, presentan una gama adecuada de potencia esperada en cuanto a su actividad como agonistas/antagonistas ER (es decir, de fuerte a débil) e incluyen negativos. Los ensayos de las sustancias para la prueba de la competencia deben repetirse al menos dos veces, en diferentes días. La competencia se demuestra por la clasificación correcta (positiva/negativa) de cada sustancia para la prueba de la competencia. Cada técnico debe repetir la prueba de la competencia al aprender los ensayos. En función del tipo de células, algunas de estas sustancias para la prueba de la competencia pueden comportarse como SERM y mostrar actividad como agonistas y como antagonistas. Sin embargo, las sustancias para la prueba de la competencia se clasifican en los cuadros 3 y 4 por su actividad predominante conocida, que es la que debe utilizarse para la evaluación de la competencia.
|
|
15.
|
Para demostrar el comportamiento y con fines de control de la calidad, cada laboratorio debe compilar bases de datos históricos de agonistas y de antagonistas con datos sobre los patrones de referencia (por ejemplo, 17β-estradiol y tamoxifeno), las sustancias testigo positivo y negativo y los controles del disolvente (por ejemplo, DMSO). En primer lugar, la base de datos debe generarse a partir de al menos diez tandas independientes con agonistas (por ejemplo, 17β-estradiol) y diez tandas independientes con antagonistas (por ejemplo, tamoxifeno). Deben añadirse los resultados de futuros análisis de estos patrones de referencia y controles del disolvente para ampliar la base de datos a fin de garantizar la coherencia y el comportamiento del bioensayo realizado por el laboratorio a lo largo del tiempo.
|
Cuadro 3
Lista de catorce sustancias para la prueba de la competencia con el ensayo de agonistas
(47)
|
N.o
(54)
|
Sustancia
|
N.o CAS
|
Respuesta prevista (48)
|
Ensayo STTA
|
Ensayo VM7Luc ER TA
|
Clase química del sistema MeSH (52)
|
Clase de producto (53)
|
|
Valor de CP10 (M) (49)
|
Valor de CP50 (M) (49)
|
Intervalo conc. ensayo (M)
|
Valor de CE50 en VM7Luc (M) (50)
|
Conc. máxima en la determinación del intervalo (M) (51)
|
|
14
|
Dietilestilbestrol
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos, productos veterinarios
|
|
12
|
17α-Estradiol
|
57-91-0
|
POS
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
15
|
meso-Hexestrol
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Hidrocarburos (cíclicos), fenoles
|
Productos farmacéuticos, productos veterinarios
|
|
11
|
4-terc-Octilfenol
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenoles
|
Productos intermedios
|
|
9
|
Genisteína
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoides, compuestos heterocíclicos
|
Productos naturales, productos farmacéuticos
|
|
6
|
Bisfenol A
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenoles
|
Productos intermedios
|
|
2
|
Canferol
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoides, compuestos heterocíclicos
|
Productos naturales
|
|
3
|
Ftalato de butilbencilo
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Ácidos carboxílicos, ésteres, ácido ftalico
|
Plastificantes, productos industriales
|
|
4
|
p,p’-Metoxicloro
|
72-43-5
|
POS
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Hidrocarburos (halogenados)
|
Plaguicidas, productos veterinarios
|
|
1
|
Etilparabeno
|
120-47-8
|
POS
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Ácidos carboxílicos, ésteres
|
Productos farmacéuticos, conservantes
|
|
17
|
Atrazina
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Compuestos heterocíclicos
|
Herbicidas
|
|
20
|
Espironolactona
|
52-01-7
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Lactonas, esteroides
|
Productos farmacéuticos
|
|
21
|
Ketoconazol
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Compuestos heterocíclicos
|
Productos farmacéuticos
|
|
22
|
Reserpina
|
50-55-5
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Compuestos heterocíclicos, indoles
|
Productos farmacéuticos, productos veterinarios
|
|
Abreviaturas: N.o CAS = número de registro del Chemical Abstracts Service; CE50 = concentración de la sustancia problema que produce la mitad del efecto máximo; NEG = negativo; POS = positivo; CP10 (y CP50) = concentración de una sustancia problema a la que la respuesta es del 10 % (o del 50 % en el caso de la CP50) de la respuesta inducida por el testigo positivo (E2, 1 nm) en cada placa.
|
Cuadro 4
Lista de diez sustancias para la prueba de la competencia con el ensayo de antagonistas
|
|
Sustancia (4)
|
N.o CAS
|
Ensayo ER STTA (55)
|
Ensayo VM7Luc ER TA (56)
|
Efectos candidatos del ER STTA (55)
|
Clasificación de consenso del ICCVAM (59)
|
Clase química del MeSH (60)
|
Clase de producto (61)
|
|
Actividad ER TA
|
CI50 (M)
|
Intervalo conc. ensayo (M)
|
Actividad ER TA
|
CI50
(57) (M)
|
Conc. máxima en la determinación del intervalo (M) (58)
|
|
1
|
4-Hidroxitamoxifeno
|
68047-06-3
|
POS
|
3,97 × 10-9
|
10-12 – 10-7
|
POS
|
2,08 × 10-7
|
2,58 × 10-4
|
POS moderado
|
POS
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos
|
|
2
|
Raloxifeno HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
10-12 – 10-7
|
POS
|
1,19 × 10-9
|
1,96 × 10-4
|
POS moderado
|
POS
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos
|
|
3
|
Tamoxifeno
|
10540-29-1
|
POS
|
4,91 × 10-7
|
10-10 – 10-5
|
POS
|
8,17 × 10-7
|
2,69 × 10-4
|
POS
|
POS
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos
|
|
4
|
17β-Estradiol
|
50-28-2
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,67 × 10-3
|
Previsto que sea negativo (*4)
|
SN
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
5
|
Apigenina
|
520-36-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Compuestos heterocíclicos
|
Colorantes, productos naturales, productos intermedios farmacéuticos
|
|
6
|
Ftalato de di-n-butilo
|
84-74-2
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Ésteres, ácido ftálico
|
Ingredientes de cosméticos, productos industriales, plastificantes
|
|
7
|
Flavona
|
525-82-6
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,50 × 10-4
|
Previsto que sea negativo (*4)
|
SN
|
Flavonoides, compuestos heterocíclicos
|
Productos naturales, productos farmacéuticos
|
|
8
|
Genisteína
|
446-72-0
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
Previsto que sea negativo (*4)
|
NEG
|
Flavonoides, compuestos heterocíclicos
|
Productos naturales, productos farmacéuticos
|
|
9
|
p-n-Nonilfenol
|
104-40-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
4,54 × 10-4
|
No se ha ensayado
|
NEG
|
Fenoles
|
Productos intermedios
|
|
10
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,38 × 10-4
|
Previsto que sea negativo (*4)
|
NEG
|
Hidrocarburos (cíclicos)
|
Productos naturales
|
|
Abreviaturas: N.o CAS = número de registro del Chemical Abstracts Service; M = molar; CI50 = concentración de la sustancia problema que produce la mitad de la inhibición máxima; NEG = negativo; SP = supuesto negativo; POS = positivo;
|
Criterios de aceptabilidad de las tandas de ensayo
|
16.
|
La aceptación o el rechazo de una tanda de ensayo se basa en la evaluación de los resultados obtenidos con los patrones de referencia y los testigos utilizados para cada experimento. Los valores correspondientes a CP50 (CE50) o CI50 de los patrones de referencia deben cumplir los criterios de aceptabilidad establecidos para el ensayo seleccionado (para el STTA véase el apéndice 2, para el VM7Luc TA véase el apéndice 3), y todos los testigos positivos o negativos deben clasificarse correctamente con cada experimento aceptado. La capacidad de realizar el ensayo de forma coherente debe demostrarse mediante la elaboración y el mantenimiento de una base de datos históricos sobre los patrones de referencia y los testigos (véase el punto 15). Las desviaciones típicas (DT) o los coeficientes de variación (CV) de las medias de los parámetros de ajuste de la curva de los patrones de referencia a partir de experimentos múltiples pueden utilizarse como medida de la reproducibilidad intralaboratorios. Además, deben cumplirse los siguientes principios en relación con los criterios de aceptabilidad:
|
—
|
Los datos deben ser suficientes para una evaluación cuantitativa de la activación del ER (para el ensayo de agonistas) o de su supresión (para el ensayo de antagonistas) (es decir, eficacia y potencia).
|
|
—
|
La actividad media del marcador correspondiente a la concentración de referencia del estrógeno de referencia debe ser, al menos, la mínima especificada en los ensayos en relación con la del control del vehículo (disolvente) para garantizar una sensibilidad adecuada. En los ensayos STTA y VM7Luc ER TA, esto es cuatro veces el valor de la media del control del vehículo en cada placa.
|
|
—
|
Las concentraciones ensayadas deben mantenerse dentro del intervalo de solubilidad de los productos problema y no mostrar citotoxicidad.
|
|
Análisis de los datos
|
17.
|
Para clasificar una respuesta como positiva y negativa debe utilizarse el procedimiento definido de interpretación de datos correspondiente a cada ensayo.
|
|
18.
|
El cumplimiento de los criterios de aceptabilidad (punto 16) indica que el ensayo funciona correctamente, pero no garantiza que cualquier tanda de ensayo concreta produzca datos exactos. La reproducción de los resultados de la primera tanda es la mejor indicación de que se han producido datos exactos. Si dos tandas dan resultados reproducibles (por ejemplo, si los resultados de ambas tandas de ensayo indican que es positivo un producto problema), no es necesario realizar una tercera tanda.
|
|
19.
|
Si no se obtienen resultados reproducibles en dos tandas (por ejemplo, un producto problema es positivo en una sola tanda y negativo en la otra), o si es necesario un grado de certeza más elevado en relación con el resultado de este ensayo, deben realizarse al menos tres tandas independientes. En este caso, la clasificación se basa en los dos resultados concordantes de los tres.
|
Criterios generales de interpretación de los datos
|
20.
|
Actualmente no se dispone de un método aceptado universalmente para interpretar los datos del ensayo ER TA. Sin embargo, tanto las evaluaciones cualitativas (por ejemplo, positivo/negativo) como las cuantitativas (por ejemplo, CE50, CP50, CI50) de la actividad mediada por ER deben basarse en datos empíricos y en un sólido juicio científico. Cuando sea posible, los resultados positivos deben caracterizarse tanto por la magnitud del efecto en comparación con el control del vehículo (disolvente) o el estrógeno de referencia como por la concentración a la que se produce el efecto (por ejemplo, CE50, CP50, RCPMáx, CI50, etc.).
|
Informe del ensayo
|
21.
|
El informe del ensayo debe incluir la información siguiente:
|
|
Ensayo:
|
—
|
testigo/ patrón de referencia / producto problema;
|
|
—
|
origen, número de lote, fecha límite de utilización, si se dispone de ellos;
|
|
—
|
estabilidad del producto problema en sí, si se conoce;
|
|
—
|
solubilidad y estabilidad del producto problema en el disolvente, si se conocen;
|
|
—
|
medición del pH, osmolalidad y precipitado en el medio de cultivo al que se ha añadido el producto problema, según proceda.
|
|
|
|
Sustancias de un solo componente:
|
—
|
aspecto físico, hidrosolubilidad y otras propiedades fisicoquímicas pertinentes;
|
|
—
|
identificación química, como nombre IUPAC o CAS, número CAS, notación SMILES o InChI, fórmula estructural, pureza, identidad química de las impurezas si procede y es viable en la práctica, etc.
|
|
|
|
Sustancias de componentes múltiples, UVCB y mezclas:
|
—
|
deben caracterizarse en la medida de lo posible por la identidad química (véase más arriba), la cantidad en que están presentes y las propiedades fisicoquímicas pertinentes de sus componentes.
|
|
|
|
Disolvente o vehículo:
|
—
|
caracterización (naturaleza, proveedor y lote);
|
|
—
|
justificación de la elección del disolvente o vehículo;
|
|
—
|
solubilidad y estabilidad del producto problema en el disolvente o vehículo, si se conocen.
|
|
|
|
Células:
|
—
|
tipo y origen de las células;
|
—
|
¿Se expresa el ER de forma endógena? En caso contrario, ¿qué receptor o receptores se han transfectado?
|
|
—
|
Construcción o construcciones marcadoras utilizadas (incluyendo las especies origen);
|
|
—
|
Método de transfección;
|
|
—
|
Método de selección para mantener la transfección estable (cuando proceda);
|
|
—
|
¿Es pertinente para las líneas estables el método de transfección?
|
|
|
—
|
número de pases celulares (después de la descongelación);
|
|
—
|
número del pase de las células en el momento de la descongelación;
|
|
—
|
métodos de mantenimiento de los cultivos celulares.
|
|
|
|
Condiciones del ensayo:
|
—
|
limitaciones de la solubilidad;
|
|
—
|
descripción de los métodos de evaluación de la viabilidad aplicados;
|
|
—
|
composición de los medios, concentración de CO2;
|
|
—
|
concentración del producto problema;
|
|
—
|
volúmenes de vehículo y de producto problema añadidos;
|
|
—
|
temperatura y humedad de la incubación;
|
|
—
|
duración del tratamiento;
|
|
—
|
densidad celular al principio del tratamiento y durante este;
|
|
—
|
patrones de referencia positivos y negativos;
|
|
—
|
reactivos del marcador (nombre del producto, proveedor y lote);
|
|
—
|
criterios empleados para considerar que las tandas de ensayo son positivas, negativas o dudosas.
|
|
|
|
Comprobación de la aceptabilidad:
|
—
|
factor de inducción para cada placa de ensayo y si cumple el mínimo requerido por el ensayo sobre la base de los controles históricos;
|
|
—
|
valores reales para los criterios de aceptabilidad, por ejemplo valores de log10 CE50, log10 CP50, log CI50 y pendiente de Hill, correspondientes a los testigos positivos en paralelo y a los patrones de referencia.
|
|
|
|
Resultados:
|
—
|
datos en bruto y normalizados;
|
|
—
|
nivel máximo del factor de inducción;
|
|
—
|
datos de citotoxicidad;
|
|
—
|
si existe, concentración efectiva mínima (CEMín);
|
|
—
|
RCPMáx, CPMáx, CP50, CI50 y/o CE50, según corresponda;
|
|
—
|
relación concentración-respuesta, cuando sea posible;
|
|
—
|
análisis estadísticos, en su caso, junto con una medida del error y de la confianza (por ejemplo, SEM, DT, CV o IC del 95 %) y una descripción de cómo se han obtenido dichos valores.
|
|
|
|
Discusión de los resultados
|
|
BIBLIOGRAFÍA
|
(1)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(2)
|
OCDE (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): p. 5367-73.
|
|
(5)
|
Rogers J.M. and Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): p. 67-82.
|
|
(6)
|
OCDE (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(7)
|
OCDE (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(8)
|
OCDE (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: p. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): p. 1073-89.
|
|
(11)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): p. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): p. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): p. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): p. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol,71(10): p. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): p. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERαIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): p. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): p. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): p. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): p. 105-12.
|
|
(25)
|
Deroo B.J. and Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): p. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): p. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. and Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): p. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): p. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: p. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3):p. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. and Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Apéndice 1
DEFINICIONES Y ABREVIATURAS
Criterios de aceptabilidad
: Normas mínimas para la realización de controles experimentales y patrones de referencia. Para que un experimento se considere válido deben cumplirse todos los criterios de aceptabilidad.
Exactitud (concordancia)
: Grado de coincidencia entre los resultados de un ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del ensayo y es un aspecto de su pertinencia. Este término y el de “concordancia” se suelen usar indistintamente para indicar la proporción de resultados correctos de un ensayo (1).
Agonista
: Sustancia que produce una respuesta, por ejemplo transcripción, cuando se une a un receptor específico.
Antagonista
: Tipo de producto o de ligando de un receptor que no provoca una respuesta biológica en sí como consecuencia de su unión a un receptor, sino que bloquea o amortigua las respuestas mediadas por los agonistas.
Actividad antiestrogénica
: Capacidad de un producto de suprimir la acción del 17β-estradiol mediada a través de receptores estrogénicos.
Morfología celular
: Forma y aspecto de las células cultivadas en una monocapa en un solo pocillo de una placa de cultivo de tejidos. Las células que están muriendo suelen presentar una morfología celular anormal.
MC
: Marco conceptual de la OCDE para los ensayos y la evaluación de los alteradores endocrinos.
Tratamiento de carbón/dextrano
: Tratamiento del suero utilizado en el cultivo celular. El tratamiento con carbón/dextrano (a menudo denominado «separación») elimina las hormonas endógenas y las proteínas de unión de hormonas.
Producto
: Sustancia o mezcla.
Citotoxicidad
: Efectos nocivos para la estructura o la función celular que pueden finalmente causar la muerte de las células y puede reflejarse mediante una reducción del número de células presentes en el pocillo al final del período de exposición o mediante una reducción de la capacidad respecto a una medida de la función celular en comparación con el control en paralelo del vehículo.
CV
: Coeficiente de variación.
DCC-FBS
: Suero bovino fetal tratado con carbón recubierto de dextrano.
DMEM
: Modificación de Dulbecco del medio de Eagle.
DMSO
: Dimetilsulfóxido.
E2
: 17β-estradiol.
CE50
: Concentración del producto problema que produce la mitad del efecto máximo.
AE
: Alteración endocrina.
hERα
: Receptor estrogénico humano alfa.
hERß
: Receptor estrogénico humano beta.
EFM
: Medio libre de estrógenos. Modificación de Dulbecco del medio de Eagle (DMEM), complementado con 4,5 % de FBS tratado con carbón/dextrano, 1,9 % de L-glutamina y 0,9 % de Pen-Strep (penicilina-estreptomicina).
ER
: Receptor estrogénico.
ERE
: Elemento de respuesta a estrógenos.
Actividad estrogénica
: Capacidad de un producto para imitar al 17β-estradiol en su posibilidad de unirse a los receptores estrogénicos y activarlos. La actividad estrogénica mediada por hERα puede detectarse con este método de ensayo.
ERTA
: Transactivación de los receptores estrogénicos.
FBS
: Suero bovino fetal.
HeLa
: Línea celular de cuello de útero humana inmortal.
HeLa9903
: Un subclón de la línea celular HeLa al que se han transfectado de forma estable un gen marcador de la luciferasa y hERαα.
CI50
: Concentración del producto problema que produce la mitad de la inhibición máxima.
ICCVAM
: Comité de Coordinación Interagencias sobre la Validación de Métodos Alternativos de Estados Unidos (Interagency Coordinating Committee on the Validation of Alternative Methods).
Reproducibilidad interlaboratorios
: Medida del grado en que distintos laboratorios cualificados, utilizando el mismo protocolo y sometiendo a ensayo las mismas sustancias, pueden producir resultados cualitativa y cuantitativamente similares. La reproducibilidad interlaboratorios se determina durante los procesos de prevalidación y validación, e indica el grado en que un ensayo puede transferirse con éxito entre laboratorios; se denomina también reproducibilidad entre laboratorios (1).
Reproducibilidad intralaboratorios
: Determinación del grado en que personas cualificadas del mismo laboratorio pueden repetir con éxito los resultados en momentos diferentes, utilizando un protocolo especificado. También se denomina reproducibilidad dentro del laboratorio (1).
CEMín
: Concentración efectiva mínima; es la concentración más baja del producto problema que produce una respuesta (es decir, la concentración más baja del producto problema en la que el factor de inducción es estadísticamente diferente al del control del vehículo en paralelo).
Ensayos de imitación
: Expresión coloquial para denominar los ensayos que son estructural y funcionalmente similares a un método de ensayo de referencia validado y aceptado. También se pueden denominar métodos de ensayo similares.
MT
: Metalotioneína.
MMTV
: Virus de tumor mamario de ratón.
OHT
: 4-Hidroxitamoxifeno.
PBTG
: Directrices de ensayo basadas en el comportamiento.
TP(testigo positivo):: Sustancia muy activa, de preferencia el 17ß-estradiol, que se incluye en todos los ensayos para contribuir a garantizar el buen funcionamiento de estos.
CP10
: Concentración de un producto problema a la que la actividad medida en un ensayo de agonistas es el 10 % de la actividad máxima inducida por el TP (E2 1 nM en el ensayo STTA) en cada placa.
CP50
: Concentración de un producto problema a la que la actividad medida en un ensayo de agonistas es el 50 % de la actividad máxima inducida por el TP (E2 a la concentración de referencia especificada en el método de ensayo) en cada placa.
CPMáx
: Concentración a la que un producto problema induce la RCPMáx.
Normas de comportamiento
: Normas, basadas en un ensayo validado, que proporcionan la base para evaluar la comparabilidad de un ensayo propuesto que es similar desde el punto de vista mecánico y funcional. Se incluye lo siguiente: 1) los componentes esenciales del ensayo; 2) una lista mínima de productos de referencia seleccionados de entre los productos utilizados para demostrar el comportamiento aceptable del método de ensayo validado; y 3) los niveles similares de fiabilidad y exactitud, basados en lo obtenido con el método de ensayo validado, que el ensayo propuesto debe demostrar cuando se evalúa con la lista mínima de productos de referencia (1).
Sustancias para la prueba de la competencia
: Subconjunto de las sustancias de referencia incluidas en las normas de comportamiento que pueden ser utilizadas por los laboratorios para demostrar la competencia técnica con un método de ensayo normalizado. Los criterios de selección de estas sustancias suelen incluir que representen la gama de respuestas, que estén disponibles en el mercado y que se disponga sobre ellas de datos de referencia de alta calidad.
Competencia
: Capacidad demostrada de realizar correctamente un ensayo antes de someter a este sustancias desconocidas.
Estrógeno de referencia (testigo positivo, TC)
: 17β-estradiol (E2, CAS 50-28-2).
Patrón de referencia
: Sustancia de referencia utilizada para demostrar la adecuación de un ensayo. El 17β-estradiol es el patrón de referencia de los ensayos STTA y VM7Luc ER TA.
Métodos de ensayo de referencia
: Ensayos sobre los que se basan las PBTG 455.
Pertinencia
: Descripción de la relación de un ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un ensayo (1).
Fiabilidad
: Medida del grado en que un ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios.
ULR
: Unidades luminosas relativas.
ARN
: Ácido ribonucleico.
RCPMáx
: Nivel máximo de respuesta inducido por un producto problema, expresado como porcentaje de la respuesta inducida por la solución 1 nM de E2 en la misma placa.
RPMI
: Medio RPMI 1 640 complementado con 0,9 % de Pen-Strep y 8,0 % de suero bovino fetal (FBS).
Tanda
: Experimento individual que evalúa la acción del producto sobre los resultados biológicos del ensayo. Cada tanda es un experimento completo realizado en pocillos replicados de células cultivadas a partir de un conjunto común de células al mismo tiempo.
Tanda independiente
: Experimento aparte e independiente que evalúa la acción del producto sobre los resultados biológicos del ensayo, utilizando células procedentes de un conjunto diferente y productos recién diluidos, realizada en días distintos o el mismo día por personal diferente.
DT
: Desviación típica.
Sensibilidad
: Proporción de todas las sustancias activas/positivas que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un ensayo (1).
Especificidad
: Proporción de todas las sustancias inactivas/negativas que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un ensayo (1).
Transfección estable
: Cuando se transfecta ADN a células cultivadas de tal manera que se integra de forma estable en el genoma de las células, dando lugar a la expresión estable de genes transfectados. Los clones de células transfectadas de forma estable se seleccionan mediante marcadores estables (por ejemplo, resistencia al antibiótico G418).
Ensayo STTA
: Ensayo de transactivación transfectada establemente, que es el ensayo de activación transcripcional ERα en el que se utiliza la línea celular HeLa 9 903.
Estudio
: Todo el conjunto de trabajo experimental realizado para evaluar una única sustancia específica mediante un ensayo específico. El estudio comprende todas las fases, incluidos los ensayos de dilución de la sustancia problema en los medios de ensayo, las tandas de determinación preliminar del intervalo, todas las tandas completas necesarias, los análisis de datos, la garantía de calidad, las evaluaciones de la citotoxicidad, etc. La realización de un estudio permite clasificar la actividad del producto problema en relación con la diana de toxicidad (es decir, activo, inactivo o no concluyente) que se evalúa mediante el ensayo utilizado y una estimación de la potencia relativa al producto de referencia positivo.
Sustancia
: Según REACH (62), una sustancia se define como un elemento químico y sus compuestos naturales o los obtenidos mediante algún proceso industrial, incluidos los aditivos necesarios para conservar su estabilidad y las impurezas que inevitablemente se produzcan en el proceso, con exclusión de todos los disolventes que puedan separarse sin afectar a la estabilidad de la sustancia ni modificar su composición. En el contexto del SGA de las Naciones Unidas se utiliza una definición muy similar (1).
TA (transactivación)
: Inicio de la síntesis de ARNm en respuesta a una señal química específica, como la fijación de un estrógeno al receptor estrogénico.
Ensayo
: En el contexto de este método de ensayo, un ensayo es uno de los métodos aceptados como válido por cumplir los criterios de comportamiento establecidos. Los componentes del ensayo incluyen, por ejemplo, la línea celular específica con las condiciones de crecimiento asociadas, los medios específicos en los que se realiza el ensayo, las condiciones de configuración de las placas, la disposición y las diluciones de los productos problema, junto con cualquier otra medida requerida de control de la calidad y las correspondientes fases de evaluación de los datos.
Producto problema
: Sustancia o mezcla estudiada con este método de ensayo.
Transcripción
: Síntesis de ARNm.
UVCB
: Sustancias químicas de composición desconocida o variable, productos complejos de reacción y materiales biológicos.
Método de ensayo validado
: Ensayo sobre el cual se han completado estudios de validación para determinar su pertinencia (incluida su exactitud) y su fiabilidad con un fin específico. Es importante señalar que un método de ensayo validado podría no tener un comportamiento suficiente en términos de exactitud y fiabilidad como para considerarse aceptable a efectos del fin propuesto (1).
Validación
: Proceso por el que se establece para un fin determinado la fiabilidad y pertinencia de un enfoque, método, ensayo, proceso o evaluación concretos (1).
CVe (control del vehículo)
: El disolvente que se utiliza para disolver el producto problema y las sustancias testigo se somete a ensayo únicamente como vehículo, sin producto disuelto.
VM7
: Células de adenocarcinoma inmortalizado que expresan de forma endógena el receptor estrogénico.
VM7Luc4E2
: La línea celular VM7Luch4E2 se ha obtenido de células de adenocarcinoma inmortalizado de origen humano VM7 que expresan de forma endógena ambas formas del receptor estrogénico (ERα y ERβ) y se han transfectado establemente con el plásmido pGudLuc7.ERE. Este plásmido contiene cuatro copias de un oligonucleótido sintético que incluye el elemento de respuesta a estrógenos en dirección 5’ respecto al promotor vírico del tumor de mama del ratón (MMTV) y el gen de la luciferasa de la luciérnaga.
Testigo positivo débil
: Sustancia débilmente activa, seleccionada de la lista de productos de referencia y que se incluye en todos los ensayos para contribuir a garantizar el buen funcionamiento de estos.
Apéndice 2
ENSAYO DE TRANSACTIVACIÓN DEL RECEPTOR ESTROGÉNICO Α HUMANO TRANSFECTADO ESTABLEMENTE PARA LA DETECCIÓN DE LA ACTIVIDAD AGONISTA Y ANTAGONISTA DE LOS PRODUCTOS, UTILIZANDO LA LÍNEA CELULAR HERΑ-HELA-9903
CONSIDERACIONES Y LIMITACIONES INICIALES (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
1.
|
Este ensayo de transactivación (TA) utiliza la línea celular hERα-HeLa-9903 para detectar la actividad de agonistas estrogénicos mediada a través del receptor estrogénico humano alfa (hERα). El estudio de validación del ensayo de transactivación transfectada establemente (STTA), realizado por el Instituto Japonés de Evaluación e Investigación de Productos Químicos (CERI) utilizando la línea celular hERα-HeLa-9903 para detectar la actividad estrogénica agonista y antagonista mediada a través del receptor estrogénico humano alfa (hERα), demostró la pertinencia y fiabilidad del ensayo para su fin previsto (1).
|
|
2.
|
Este ensayo está diseñado específicamente para detectar la TA mediada a través del hERα midiendo la quimioluminiscencia como parámetro. Sin embargo, se han comunicado señales de luminiscencia no mediadas por el receptor a concentraciones de fitoestrógenos superiores a 1 μM como consecuencia de la sobreactivación del gen marcador de la luciferasa (2) (3). Si bien la curva dosis-respuesta indica que la activación real del sistema ER se produce a concentraciones más bajas, debe examinarse detenidamente la expresión de la luciferasa obtenida a altas concentraciones de fitoestrógenos o de compuestos similares sospechosos de producir una sobreactivación de tipo fitoestrogénico del gen marcador de la luciferasa, en los sistemas de ensayo ER TA transfectados de forma estable (apéndice 1).
|
|
3.
|
Las secciones “INTRODUCCIÓN GENERAL” y “COMPONENTES DEL ENSAYO ER TA” deben leerse antes de utilizar este ensayo con fines normativos. Las definiciones y abreviaturas utilizadas en las presentes directrices de ensayo se recogen en el apéndice 2.1.
|
PRINCIPIO DEL ENSAYO (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
4.
|
El ensayo se utiliza para detectar la unión del receptor estrogénico con un ligando. Tras la unión del ligando, el complejo receptor-ligando se traslada al núcleo, donde se une a determinados elementos específicos de respuesta del ADN y transactiva un gen marcador de la luciferasa de la luciérnaga, lo que provoca un aumento de la expresión celular de la enzima luciferasa. La luciferina constituye un sustrato que es transformado por la enzima luciferasa en un producto de bioluminiscencia que puede medirse cuantitativamente con un luminómetro. La actividad de la luciferasa puede evaluarse de forma rápida y poco costosa con varios equipos de pruebas disponibles en el comercio.
|
|
5.
|
El sistema de ensayo utiliza la línea celular de hERα-HeLa-9903, procedente de un tumor de cuello de útero humano, con dos construcciones introducidas de forma estable: i) la construcción de expresión de hERαα (que codifica el receptor humano completo), y ii) la construcción del marcador de la luciferasa de la luciérnaga, que lleva cinco repeticiones en tándem de un elemento sensible a los estrógenos (ERE, Estrogen-Responsive Element) de vitelogenina movilizado por un elemento TATA del promotor de metalotioneína (MT) del ratón. Se ha demostrado que la construcción génica MT TATA del ratón tiene el mejor comportamiento, por lo que se utiliza normalmente. Por consiguiente, esta línea celular hERα-HeLa-9903 puede medir la capacidad de un producto problema para inducir la transactivación mediada por hERα de la expresión del gen de la luciferasa.
|
|
6.
|
En el caso de un ensayo de agonistas ER, la interpretación de los datos se basa en si el nivel máximo de respuesta inducida por un producto problema es igual o mayor al de una respuesta de agonistas igual al 10 % de la inducida por una concentración inductora máxima (1 nM) del testigo positivo (TP) 17β-estradiol (E2) (es decir, la CP10). En el caso de un ensayo de antagonistas ER, la interpretación de los datos se basa en si la respuesta muestra al menos una reducción del 30 % de la actividad respecto a la respuesta inducida por el control con el testigo añadido (solución 25 pM de E2) sin citotoxicidad. El análisis y la interpretación de los datos se detallan en los puntos 34 a 48.
|
PROCEDIMIENTO
Líneas celulares
|
7.
|
Debe utilizarse para el ensayo la línea celular hERα-HeLa-9903, transfectada de forma estable. La línea celular puede obtenerse del banco de células de la Japanese Collection of Research Bioresources (JCRB) (63), previa firma de un acuerdo de transferencia de material (ATM).
|
|
8.
|
En los ensayos solo deben utilizarse células caracterizadas como libres de micoplasmas. La RCP-TR (reacción en cadena de la polimerasa en tiempo real) es el método preferido para la detección sensible de la infección por micoplasmas (4) (5) (6).
|
Estabilidad de la línea celular
|
9.
|
Para supervisar la estabilidad de la línea celular, deben utilizarse el E2, el 17α-estradiol, la 17α-metiltestosterona y la corticosterona como patrones de referencia para el ensayo de agonistas, y debe obtenerse una curva completa concentración-respuesta en el intervalo de concentraciones de ensayo que figura en el cuadro 1 al menos cada vez que se realice el ensayo, y los resultados deben estar de acuerdo con los resultados proporcionados en el cuadro 1.
|
|
10.
|
En el caso de un ensayo de antagonistas, deben medirse simultáneamente con cada tanda curvas completas de concentración correspondientes a dos patrones de referencia, tamoxifeno y flutamida. Debe supervisarse la clasificación cualitativa correcta de los dos productos como positivos o negativos.
|
Condiciones de cultivo y siembra de las células
|
11.
|
Las células deben mantenerse en medio esencial mínimo de Eagle (EMEM) sin rojo de fenol, complementado con 60 mg/l del antibiótico kanamicina y un 10 % de suero bovino fetal tratado con carbón recubierto de dextrano (DCC-FBS), en una incubadora con CO2 (5 % de CO2) a 37 ± 1oC. Tras alcanzar una confluencia del 75 -90 %, se hace un subcultivo de las células con 10 ml de 0,4 x 105 – 1 x 105 células/ml por placa de cultivo celular de 100 mm. Las células deben suspenderse con un 10 % de FBS-EMEM (que es lo mismo que EMEM con DCCB-FBS) y, a continuación, deben depositarse en pocillos de una microplaca en la proporción de 1 x 104 células (100 μl/pocillo). A continuación, las células deben preincubarse en una incubadora con 5 % de CO2 a una temperatura de 37 ± 1oC durante 3 horas antes de la exposición al producto. El material de plástico debe estar libre de actividad estrogénica.
|
|
12.
|
Para mantener la integridad de la respuesta, las células deben cultivarse durante más de un pase después del cultivo madre congelado en los medios acondicionados y no deben cultivarse durante más de cuarenta pases. En el caso de la línea celular hERα-HeLa-9903, esto significa menos de tres meses. Sin embargo, las características de las células pueden hacerse menos favorables si se cultivan en condiciones inadecuadas.
|
|
13.
|
El DCCB-FBS puede prepararse tal y como se describe en el apéndice 2.2 u obtenerse de fuentes comerciales.
|
Criterios de aceptabilidad
Patrones de referencia positivos y negativos para el ensayo de agonistas ER
|
14.
|
Antes del estudio y durante el mismo, debe verificarse la sensibilidad del sistema de ensayo utilizando las concentraciones adecuadas de un estrógeno fuerte (E2), un estrógeno débil (17α-estradiol), un agonista muy débil (17α-metiltestosterona), y una sustancia negativa (corticosterona). En el cuadro 1 figuran los valores del intervalo aceptable derivados del estudio de validación (1). Estos cuatro patrones de referencia en paralelo deben incluirse en cada experimento y los resultados deben quedar dentro de los límites aceptables establecidos. En caso contrario, debe determinarse la causa del incumplimiento de los criterios de aceptabilidad (por ejemplo, manipulación de las células, o calidad y concentración de suero y antibióticos) y repetirse el ensayo. Una vez cumplidos los criterios de aceptabilidad, para garantizar una variabilidad mínima de los valores de CE50, CP50 y CP10, es esencial la utilización uniforme de los materiales para el cultivo celular. Los cuatro patrones de referencia en paralelo, que deben incluirse en cada experimento (realizado en las mismas condiciones, incluidos los materiales, el nivel de pases de las células y el personal técnico), pueden garantizar la sensibilidad del ensayo, ya que los valores de CP10 de los tres patrones de referencia positivos deben quedar dentro del intervalo aceptable, como también los valores de CP50 y de CE50, cuando puedan calcularse (véase el cuadro 1).
|
Cuadro 1
Intervalo aceptable de valores de los cuatro patrones de referencia para el ensayo de agonistas ER
|
Nombre
|
log CP50
|
log CP10
|
log CE50
|
Pendiente de Hill
|
Intervalo de ensayo
|
|
17β-Estradiol (E2) N.o CAS: 50-28-2
|
-11,4 ~ -10,1
|
< -11
|
-11,3 ~ -10,1
|
0,7 ~ 1,5
|
10-14~10-8 M
|
|
17α-Estradiol N.o CAS: 57-91-0
|
-9,6 ~ -8,1
|
-10,7 ~ -9,3
|
-9,6 ~ -8,4
|
0,9 ~ 2,0
|
10-12~10-6 M
|
|
Corticosterona N.o CAS: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4 M
|
|
17α-Metiltestosterona N.o CAS: 58-18-4
|
-6,0 ~ -5,1
|
-8,0 ~ -6,2
|
—
|
—
|
10-11~10-5 M
|
Patrones de referencia positivos y negativos para el ensayo de antagonistas del ER
|
15.
|
Antes del estudio y durante el mismo, debe verificarse la sensibilidad del sistema de ensayo utilizando las concentraciones adecuadas de una sustancia positiva (tamoxifeno) y una sustancia negativa (flutamida). En el cuadro 2 figuran los valores del intervalo aceptable derivados del estudio de validación (1). Estos dos patrones de referencia en paralelo deben incluirse en cada experimento y los resultados deben considerarse correctos según se indica en los criterios. En caso contrario, debe determinarse la causa del incumplimiento de los criterios (por ejemplo, manipulación de las células, o calidad y concentración de suero y antibióticos) y repetirse el ensayo. Además, deben calcularse los valores de CI50 de una sustancia positiva (tamoxifeno) y los resultados deben quedar dentro de los límites aceptables establecidos. Una vez cumplidos los criterios de aceptabilidad, para garantizar una variabilidad mínima de los valores de CI50, es esencial la utilización uniforme de los materiales para el cultivo celular. Los dos patrones de referencia en paralelo, que deben incluirse en cada experimento (realizado en las mismas condiciones, incluidos los materiales, el nivel de pases de las células y el personal técnico), pueden garantizar la sensibilidad del ensayo (véase el cuadro 2).
|
Cuadro 2
Criterios e intervalo aceptable de valores de los dos patrones de referencia para el ensayo de antagonistas del ER.
|
Nombre
|
Criterios
|
Log CI50
|
Intervalo de ensayo
|
|
Tamoxifeno N.o CAS: 10540-29-1
|
Positivo: debe calcularse la CI50.
|
-5,942 ~ -7,596
|
10-10 ~ 10-5 M
|
|
Flutamida N.o CAS: 13311-84-7
|
Negativo: no debe calcularse la CI30.
|
—
|
10-10 ~ 10-5 M
|
Testigos positivos y control del vehículo
|
16.
|
El testigo positivo (TP) del ensayo de agonistas ER (1 nM de E2) y del ensayo de antagonistas ER (10 μM de tamoxifeno) se someterá a ensayo al menos por triplicado en cada placa. El vehículo que se utilice para disolver un producto problema debe someterse a ensayo como control del vehículo (CVe) al menos por triplicado en cada placa. Además de este CVe, si el TP utiliza un vehículo diferente al del producto problema, debe ensayarse otro CVe al menos por triplicado en la misma placa con el TP.
|
Criterios de calidad para el ensayo de agonistas ER
|
17.
|
La actividad media de la luciferasa del testigo positivo (E2 1 nM) debe ser al menos cuatro veces la media del CVe de cada placa. Este criterio se establece sobre la base de la fiabilidad de los valores de los parámetros obtenidos en el estudio de validación (históricamente, un factor multiplicador de entre cuatro y treinta).
|
|
18.
|
Con respecto al control de calidad del ensayo, el factor multiplicador de la inducción correspondiente al valor de CP10 del TP en paralelo (E2 1 nM) debe ser superior a 1 + 2 DT del valor del factor multiplicador de la inducción (= 1) del CVe en paralelo. A efectos de priorización, el valor de CP10 puede ser útil para simplificar el análisis de datos requerido en comparación con un análisis estadístico. Aunque un análisis estadístico proporciona información sobre la significación, dicho análisis no es un parámetro cuantitativo en relación con el potencial basado en la concentración, por lo que es menos útil a efectos de priorización.
|
Criterios de calidad para el ensayo de agonistas del ER
|
19.
|
La actividad media de la luciferasa del control con el testigo añadido (solución 25 pM de E2) debe ser al menos cuatro veces la media del CVe de cada placa. Este criterio se establece sobre la base de la fiabilidad de los valores de los parámetros obtenidos en el estudio de validación.
|
|
20.
|
Por lo que respecta al control de calidad del ensayo, la actividad transcripcional relativa (ATR) de la solución 1 nM de E2 debe ser superior al 100 %; la ATR de la solución 1 μM de 4-hidroxitamixofeno (OHT) debe ser inferior al 40,6 %, y la ATR de solución 100 μM de digitonina (Dig) debe ser inferior al 0 %.
Demostración de la competencia del laboratorio (véanse el punto 14 y los cuadros 3 y 4 de la sección “COMPONENTES DEL ENSAYO ER TA” del presente método de ensayo)
|
Vehículo
|
21.
|
Debe utilizarse como CVe en paralelo el dimetilsulfóxido (DMSO), o el disolvente adecuado, a la misma concentración utilizada con los diferentes testigos positivos y negativos y los productos problema. Los productos problema deben disolverse en un disolvente que los solubilice y sea miscible con el medio celular. Son vehículos adecuados el agua, el etanol (pureza del 95 % al 100 %) y el DMSO. Si se utiliza el DMSO, el nivel no debe superar el 0,1 % (v/v). Con cualquier vehículo, debe demostrarse que el volumen máximo utilizado no es citotóxico ni interfiere con el comportamiento del ensayo.
|
Preparación de los productos problema
|
22.
|
En general, los productos problema deben disolverse en DMSO o en otro disolvente adecuado, y diluirse en serie con el mismo disolvente a una proporción común de 1: 10, a fin de preparar soluciones para la dilución con los medios.
|
Solubilidad y citotoxicidad: Consideraciones para la determinación del intervalo de dosis
|
23.
|
Debe realizarse un ensayo preliminar para determinar el intervalo de concentraciones apropiadas del producto problema, y determinar si el producto problema puede tener dificultades en cuanto a su solubilidad y citotoxicidad. Inicialmente, los productos se someten a ensayo hasta la concentración máxima que suponga el valor más bajo de los siguientes: 1 μl/ml, 1 mg/ml, o 1 mM. Sobre la base del grado de citotoxicidad o falta de solubilidad observados en el ensayo preliminar, en la primera tanda definitiva debe someterse a ensayo el producto en diluciones logarítmicas empezando en la concentración máxima aceptable (p. ej., 1 mM, 100 μM, 10 μM, etc.) y debe anotarse la presencia de turbidez o precipitado o de citotoxicidad. Las concentraciones en la segunda y, si es necesaria, tercera tanda deben ajustarse según convenga para caracterizar mejor la curva concentración-respuesta y evitar concentraciones que resulten que son insolubles o inducen una citotoxicidad excesiva.
|
|
24.
|
En el caso de los agonistas y antagonistas ER, la presencia de niveles crecientes de citotoxicidad puede alterar significativamente o eliminar la respuesta sigmoidea típica y debe tenerse en cuenta al interpretar los datos. Se utilizarán métodos de ensayo de citotoxicidad que puedan proporcionar información sobre una viabilidad celular del 80 %, utilizando un ensayo adecuado basado en la experiencia de laboratorio.
|
|
25.
|
En caso de que los resultados del ensayo de citotoxicidad muestren que la concentración del producto problema ha reducido el número de células en un 20 % o más, esta concentración debe considerarse citotóxica, y las concentraciones iguales o superiores a la concentración citotóxica deben excluirse de la evaluación.
|
Exposición al producto y organización de la placa del ensayo
|
26.
|
El procedimiento correspondiente a las diluciones del producto (fases 1 y 2) y a la exposición de las células (fase 3) se puede llevar a cabo del siguiente modo:
|
Fase 1: Cada producto problema debe diluirse en serie en DMSO, o en otro disolvente adecuado, y añadirse a los pocillos de una placa de microvaloración a fin de alcanzar las concentraciones seriadas finales determinadas por el ensayo preliminar de determinación del intervalo de concentraciones [normalmente en una serie de, por ejemplo, 1 mM , 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM y 10 pM (10-3 - 10-11 M)] para ensayos por triplicado.
Fase 2: Dilución del producto: diluir en primer lugar 1,5 μl del producto problema en el disolvente en un volumen de 500 μl de medio.
Fase 3: Exposición de las células al producto: añadir 50 μl de dilución en el medio (preparada en la fase 2) a un pocillo de ensayo que contenga 104 células/100 μl/pocillo.
El volumen final recomendado de medio necesario para cada pocillo es de 150 μl. Pueden asignarse las muestras de ensayo y los patrones de referencia como se muestra en el cuadro 3 y en el cuadro 4.
Cuadro 3
Ejemplo de asignación de las concentraciones de los patrones de referencia en la placa de ensayo en el ensayo de agonistas ER
|
Fila
|
17α-Metiltestosterona
|
Corticosterona
|
17α-Estradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
Conc 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
Conc 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
Conc 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
Conc 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
Conc 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
Conc 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
Conc 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
CVe
|
→
|
→
|
→
|
→
|
→
|
TP
|
→
|
→
|
→
|
→
|
→
|
|
CVe: control del vehículo (DMSO al 0,1 %); TP: testigo positivo (E2 1 nM).
|
|
27.
|
Los patrones de referencia (E2, 17α-estradiol, Los patrones de referencia (E2, 17α-estradiol, 17 α-metil-testosterona y corticosterona) deben someterse a ensayo en cada tanda (cuadro 3). En cada placa del ensayo (cuadro 4) se deben incluir pocillos de TP tratados con solución 1 nM de E2 que puedan dar como resultado la máxima inducción de E2 y pocillos de CVe tratados únicamente con DMSO (o disolvente apropiado). Si en el mismo experimento se utilizan células procedentes de distintas fuentes (por ejemplo, con diferente número de pases, de un lote diferente, etc.), los patrones de referencia deben someterse a ensayo respecto a cada fuente de las células.
|
Cuadro 4
Ejemplo de asignación de las concentraciones de los productos problema y de los testigos de las placas en la placa de ensayo de agonistas ER
|
Fila
|
Producto problema 1
|
Producto problema 2
|
Producto problema 3
|
Producto problema 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
Conc 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
Conc 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
Conc 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
Conc 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
Conc 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
Conc 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
Conc 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
CVe
|
→
|
→
|
→
|
→
|
→
|
TP
|
→
|
→
|
→
|
→
|
→
|
|
CVe: control del vehículo (DMSO al 0,1 %); TP: testigo positivo (E2 1 nM).
|
Cuadro 5
Ejemplo de asignación de las concentraciones de los patrones de referencia en la placa de ensayo en el ensayo de antagonistas ER

|
28.
|
Para evaluar la actividad antagonista de los productos, a los pocillos de ensayo situados en las filas de A a G se les debe añadir solución 25 pM de E2. Los patrones de referencia (tamoxifeno y flutamida) deben someterse a ensayo en cada tanda. En cada placa del ensayo deben incluirse pocillos de TP tratados con solución 1 nM de E2 que pueden servir de control de la calidad de la línea celular hERα-HeLa-9903, pocillos de CVe tratados con DMSO (o disolvente apropiado), pocillos con solución al 0,1 % de DMSO tratados con adición de DMSO al E2 añadido correspondiente al “control con el testigo añadido”, pocillos tratados con la concentración final 1 μM de OHT y pocillos tratados con solución 100 μM de Dig (cuadro 5). La placa del ensayo posterior debe seguir la misma disposición de placa sin los pocillos de los patrones de referencia (cuadro 6). Si en el mismo experimento se utilizan células procedentes de distintas fuentes (por ejemplo, con diferente número de pases, de un lote diferente, etc.), los patrones de referencia deben someterse a ensayo respecto a cada fuente de las células.
|
Cuadro 6
Ejemplo de asignación de las concentraciones de los productos problema y de los testigos de las placas del ensayo de antagonistas ER

|
29.
|
Debe confirmarse la ausencia de efectos de borde, según proceda, y, si se sospecha que hay presencia de estos efectos, debe modificarse la disposición de la placa para evitarlos. Por ejemplo, se puede utilizar una disposición de placa en la que se excluyan los pocillos del borde.
|
|
30.
|
Después de añadir los productos, las placas de ensayo deben incubarse en una incubadora con 5 % de CO2 a 37 ± 1oC durante 20-24 horas para inducir la formación de los productos del gen marcador.
|
|
31.
|
Deben aplicarse consideraciones especiales a los compuestos que sean ligeramente volátiles. En tales casos, los pocillos testigo cercanos pueden generar falsos positivos, lo que debe tenerse en cuenta a la luz de los valores previstos e históricos de los testigos. En los pocos casos en los que la volatilidad puede ser preocupante, el uso de “selladores de placas” puede ayudar a aislar eficazmente los distintos pocillos durante el ensayo, por lo que se recomienda en tales casos.
|
|
32.
|
La repetición de los ensayos definitivos con el mismo producto debe hacerse en días distintos a fin de garantizar su independencia.
|
Ensayo de la luciferasa
|
33.
|
Para el ensayo puede utilizarse un reactivo comercial del ensayo de la luciferasa [p. ej., el sistema de ensayo de la luciferasa Steady-Glo® (Promega, E2510, o equivalente)] o un sistema de ensayo de la luciferasa normal (p. ej., Promega, E1500, o equivalente), siempre que se cumplan los criterios de aceptabilidad. Los reactivos del ensayo deben seleccionarse sobre la base de la sensibilidad del luminómetro que se vaya a usar. Cuando se utilice el sistema de ensayo de la luciferasa normal, antes de añadir el sustrato se debe utilizar el reactivo de lisis del cultivo celular (p. ej., Promega, E1531, o equivalente). El reactivo de la luciferasa debe aplicarse siguiendo las instrucciones del fabricante.
|
ANÁLISIS DE LOS DATOS
Ensayo de agonistas ER
|
34.
|
En el caso de un ensayo de agonistas ER, para obtener la actividad transcripcional relativa respecto al TP (solución 1 nM de E2), las señales de luminiscencia de la misma placa pueden analizarse con arreglo a las siguientes fases (también son aceptables otros procesos matemáticos equivalentes):
|
Fase 1. Calcular el valor medio del CVe.
Fase 2. Restar el valor medio del CVe del valor de cada pocillo para normalizar los datos.
Fase 3. Calcular la media del TP normalizado.
Fase 4. Dividir el valor normalizado de cada pocillo en la placa por el valor medio del TP normalizado (TP = 100 %).
El valor final de cada pocillo es la actividad transcripcional relativa de ese pocillo en comparación con la respuesta del TP.
Fase 5. Calcular el valor medio de la actividad transcripcional relativa de cada grupo de concentraciones del producto problema. La respuesta tiene dos dimensiones: la media de la actividad transcripcional (respuesta) y la concentración a la que se produce la respuesta (véase la sección siguiente).
Consideraciones de la inducción respecto a CE50, CP50 y CP10
|
35.
|
La curva completa concentración-respuesta es necesaria para el cálculo de la CE50, pero puede que esto no sea siempre factible o práctico debido a las limitaciones del intervalo de concentraciones de ensayo (a causa de, por ejemplo, problemas de solubilidad o de citotoxicidad). Sin embargo, dado que la CE50 y el nivel de inducción máximo (que corresponde al valor superior de la ecuación de Hill) son parámetros informativos, estos parámetros deben comunicarse cuando sea posible. Para el cálculo de la CE50 y del nivel de inducción máximo, deben utilizarse los programas estadísticos adecuados (p. ej., los programas informáticos estadísticos Prism). Si la ecuación logística de Hill es aplicable a los datos de respuesta a la concentración, la CE50 debe calcularse con la ecuación siguiente (7):
Y = valor inferior + (valor superior - valor inferior) / {1 + 10 exp [(log CE50 - X) x pendiente de Hill]}, donde:
X es el logaritmo de la concentración; e
Y es la respuesta; Y se inicia en el valor inferior y llega al valor superior siguiendo una curva sigmoidea. El valor inferior se fija en cero en la ecuación logística de Hill.
|
|
36.
|
Respecto a cada producto problema debe proporcionarse la siguiente información:
la RCPMáx, que es el nivel máximo de respuesta inducido por un producto problema, expresado como porcentaje de la respuesta inducida por una solución 1 nM de E2 en la misma placa, así como la CPMáx (concentración asociada a la RCPMáx); y
en el caso de productos positivos, las concentraciones que inducen la CP10 y, si procede, la CP50.
|
|
37.
|
El valor de la CPx puede calcularse interpolando entre dos puntos en las coordenadas X-Y, uno inmediatamente por encima y otro inmediatamente por debajo de un valor de CPx. Si los puntos de datos situados inmediatamente por encima y por debajo del valor de CPx tienen las coordenadas (a, b) y (c, d) respectivamente, entonces el valor de CPx podrá calcularse utilizando la siguiente ecuación:
log [CPx] = log [c] + (x-d)/(d-b)
|
|
38.
|
En la figura 1 se ofrecen descripciones de los valores de CP.
Figura 1
Ejemplo de cómo calcular los valores de CP. El TP (solución 1 nM de E2) se incluye en cada placa de ensayo.

|
Ensayo de antagonistas ER
|
39.
|
En el caso de un ensayo de antagonistas ER, para obtener la actividad transcripcional relativa (ATR) respecto al control con el testigo añadido (solución 25 pM de E2), las señales de luminiscencia de la misma placa pueden analizarse con arreglo a las siguientes fases (también son aceptables otros procesos matemáticos equivalentes):
Fase 1. Calcular el valor medio del CVe.
Fase 2. Restar el valor medio del CVe del valor de cada pocillo para normalizar los datos. Fase 3. Calcular la media del control con el testigo añadido normalizado.
Fase 4. Dividir el valor normalizado de cada pocillo en la placa por el valor medio del "control con el testigo añadido" normalizado (control con el testigo añadido = 100 %).
El valor final de cada pocillo es la actividad transcripcional relativa de ese pocillo en comparación con la respuesta del control con el testigo añadido.
Fase 5. Calcular el valor medio de la actividad transcripcional relativa de cada tratamiento.
|
Consideraciones de la inducción respecto a CI30 y CI50
|
40.
|
En el caso de los productos positivos, deben indicarse las concentraciones que inducen la CI30 y, si procede, la CI50.
|
|
41.
|
El valor de CIx puede calcularse interpolando entre dos puntos en las coordenadas X-Y, uno inmediatamente por encima y otro inmediatamente por debajo de un valor CIx. Si los puntos de datos situados inmediatamente por encima y por debajo del valor de CIx tienen las coordenadas (c, d) y (a, b) respectivamente, entonces el valor de CIx podrá calcularse utilizando la siguiente ecuación:
lin CIx = a - (b - (100 - x)) (a - c) /(b - d)
Figura 2
Ejemplo de cómo calcular los valores de CI. El control con el testigo añadido (solución 25 pM de E2) se incluye en cada placa de ensayo.

ATR: actividad transcripcional relativa.
|
|
42.
|
Los resultados deben basarse en dos (o tres) tandas independientes. Si dos tandas dan resultados comparables y, por lo tanto, reproducibles, no es necesario realizar una tercera tanda. Para ser aceptables, los resultados deben:
|
—
|
cumplir los criterios de aceptabilidad (véanse los criterios de aceptabilidad, puntos 14-20),
|
|
Criterios de interpretación de los datos
Cuadro 7
Criterios de decisión sobre el carácter positivo o negativo en el ensayo de agonistas ER.
|
Positivo
|
Si se obtiene una RCPMáx que es igual o superior al 10 % de la respuesta del testigo positivo en al menos dos de dos o dos de tres tandas.
|
|
Negativo
|
Si se obtiene una RCPMáx que es inferior al 10 % de la respuesta del testigo positivo en dos de dos o dos de tres tandas.
|
Cuadro 8
Criterios de decisión sobre el carácter positivo o negativo en el ensayo de antagonistas ER.
|
Positivo
|
Si se calcula la CI30 en al menos dos de dos o dos de tres tandas.
|
|
Negativo
|
Si no se calcula la CI30 en dos de dos o dos de tres tandas.
|
|
43.
|
Los criterios de interpretación de los datos figuran en los cuadros 7 y 8. Los resultados positivos se caracterizarán tanto por la magnitud del efecto como por la concentración a la que se produce el efecto. Ambos objetivos se alcanzan mediante la expresión de los resultados como la concentración a la que se alcanza un 50 % (CP50) o un 10 % (CP10) de los valores del TP en el ensayo de agonistas, y el 50 % (CI50) o el 30 % (CI30) del valor del control con el testigo añadido en el ensayo de antagonistas. Sin embargo, se determina que un producto problema es positivo si la respuesta máxima inducida por el producto problema es igual o superior al 10 % de la respuesta del TP en al menos dos de dos o dos de tres tandas, mientras que un producto problema se considera negativo si la RCPMáx no alcanza al menos el 10 % de la respuesta del testigo positivo en dos de dos o dos de tres tandas.
|
|
44.
|
Los cálculos de CP10, CP50 y CPMáx en el ensayo de agonistas ER, y CI30 y CI50 en el ensayo de antagonistas ER pueden efectuarse utilizando una hoja de cálculo disponible con las directrices de ensayo en el sitio web público de la OCDE (64).
|
|
45.
|
Debe ser suficiente con obtener los valores CP10 o CP50 y CI30 o CI50 al menos dos veces. Sin embargo, si los valores resultantes de referencia para los datos en el mismo intervalo de concentraciones presenta variabilidad con un coeficiente de variación inaceptablemente elevado (CV; %), los datos no se pueden considerar fiables y debe identificarse la fuente de esa elevada variabilidad. El CV de los datos brutos triplicados (es decir, datos de intensidad de la luminiscencia) de los puntos de datos que se utilizan para el cálculo del valor de CP10 debe ser inferior al 20 %.
|
|
46.
|
El cumplimiento de los criterios de aceptabilidad indica que el sistema de ensayo funciona correctamente, pero no garantiza que cualquier tanda de ensayo concreta produzca datos exactos. La duplicación de los resultados de la primera tanda es la mejor garantía de que se han producido datos exactos.
|
|
47.
|
En el caso de un ensayo de agonistas ER, cuando se requiere más información adicional a los objetivos de cribado y priorización de las presentes TG con respecto a los productos problema positivos, especialmente productos CP10 - CP49, así como productos sospechosos de sobreestimular la luciferasa; puede confirmarse que la actividad de luciferasa observada es únicamente una respuesta específica del ERα, utilizando un antagonista ERα (véase el apéndice 2.1).
|
INFORME DEL ENSAYO
|
48.
|
Véase el punto 20 de la sección “COMPONENTES DEL ENSAYO ER TA”.
|
BIBLIOGRAFÍA
|
(1)
|
OCDE (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1459-1469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4252-4263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769- 71.
|
|
(6)
|
Dussurget O. and Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. and Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Apéndice 2.1
FALSOS POSITIVOS: EVALUACIÓN DE LAS SEÑALES DE LUMINISCENCIA NO MEDIADAS POR EL RECEPTOR
|
1.
|
Los falsos positivos en el ensayo de agonistas ER pueden generarse debido a la activación del gen de la luciferasa no mediada por el ER, o bien debido a la activación directa del producto génico o a fluorescencia no relacionada. Estos efectos se indican mediante una curva dosis-respuesta incompleta o inusual. Si se sospechan tales efectos, debe examinarse el efecto que tiene sobre la respuesta un antagonista ER [p. ej., 4-hidroxitamoxifeno (OHT) a una concentración no tóxica]. El antagonista puro ICI 182780 puede no ser adecuado para este fin, ya que una concentración suficiente de ICI 182780 puede reducir el valor del CVe, lo que afectará al análisis de los datos.
|
|
2.
|
Para garantizar la validez de este enfoque, debe someterse a ensayo lo siguiente en la misma placa:
|
—
|
actividad agonista del producto desconocido sin/con solución 10 μM de OHT
|
|
—
|
solución 1 nM de E2 (por triplicado) como TP agonista
|
|
—
|
solución 1 nM de E2 + OHT (por triplicado).
|
|
Criterios de interpretación de los datos
Nota: Todos los pocillos deben tratarse con la misma concentración del vehículo.
|
—
|
Si la actividad agonista del producto desconocido NO se ve afectada por el tratamiento con antagonista ER, se clasifica como “negativo”.
|
|
—
|
Si se inhibe completamente la actividad agonista del producto desconocido, deben aplicarse los criterios de decisión.
|
|
—
|
Si la actividad agonista a la concentración más baja es igual o superior a la respuesta CP10, el producto desconocido se inhibe en una medida igual o superior a la respuesta CP10. Se calcula la diferencia en las respuestas entre los pocillos no tratados y los pocillos tratados con el antagonista ER, y esta diferencia debe considerarse como la respuesta verdadera y debe utilizarse en el cálculo de los parámetros adecuados para permitir la adopción de una decisión de clasificación.
|
Análisis de los datos
Comprobar la norma de comportamiento.
Comprobar el CV entre los pocillos tratados en las mismas condiciones.
|
1.
|
Calcular el valor medio del CVe.
|
|
2.
|
Restar la media del CVe de los valores de cada pocillo no tratado con OHT.
|
|
3.
|
Calcular el valor medio de OHT.
|
|
4.
|
Restar la media del CVe de los valores de cada pocillo tratado con OHT.
|
|
5.
|
Calcular el valor medio del TP.
|
|
6.
|
Calcular la actividad transcripcional relativa de todos los demás pocillos respecto al TP.
|
Apéndice 2.2
PREPARACIÓN DE SUERO TRATADO CON CARBÓN RECUBIERTO DE DEXTRANO (DCC)
|
1.
|
El tratamiento de suero con carbón recubierto de dextrano (DCC) es un método general para la eliminación de los compuestos estrogénicos del suero que se añade al medio celular, con el fin de excluir una respuesta sesgada asociada a la presencia residual de estrógenos en el suero. Pueden tratarse mediante este procedimiento 500 ml de suero bovino fetal (FBS).
|
Componentes
|
2.
|
Se necesitan los siguientes materiales y equipo:
|
Materiales
|
Cloruro de magnesio hexahidratado (MgCl2· 6 H2O)
|
|
Solución amortiguadora HEPES 1 M (pH 7,4)
|
|
Agua ultrapura producida a partir de un sistema de filtro
|
Equipo
Centrífuga general de laboratorio con recipientes de vidrio esterilizados en autoclave (el tamaño debe ajustarse según proceda), que pueda fijar la temperatura a 4 °C
Procedimiento
|
3.
|
Se ajusta el siguiente procedimiento para la utilización de tubos de centrífuga de 50 ml:
[Día-1] Preparar la suspensión de carbón recubierto de dextrano con 1 l de agua ultrapura con MgCl2 1,5 mM, sacarosa 0,25 M, 2,5 g de carbón vegetal, 0,25 g de dextrano y HEPES 5 mM, y agitar a 4 °C durante la noche.
[Día-2] Poner la suspensión en tubos de centrífuga de 50 ml y centrifugar a 10 000 rpm a 4 °C durante 10 minutos. Retirar el sobrenadante y conservar la mitad del sedimento de carbón vegetal a 4 °C para su uso el día-3. Suspender la otra mitad del carbón vegetal con FBS que se ha descongelado suavemente para evitar la precipitación y se ha inactivado a 56 °C durante 30 minutos; transferir después a un recipiente de vidrio esterilizado en autoclave, como por ejemplo un matraz Erlenmeyer. Agitar suavemente esta suspensión a 4 °C durante la noche.
[Día-3] Poner la suspensión con FBS en tubos de centrífuga de 50 ml y centrifugar a 10 000 rpm a 4 °C durante 10 minutos. Recoger el FBS y transferirlo al nuevo sedimento de carbón vegetal preparado y conservado el día-2. Suspender el sedimento de carbón vegetal y agitar suavemente esta suspensión en un recipiente de vidrio esterilizado en autoclave, a una temperatura de 4 °C durante la noche.
[Día-4] Disponer la suspensión para su centrifugación a 10 000 rpm a 4 °C durante 10 minutos y esterilizar el sobrenadante mediante filtración a través de un filtro estéril de 0,2 μm. Este DCC-FBS debe conservarse a -20 °C y se puede utilizar durante un máximo de un año.
|
Apéndice 3
ENSAYO DE TRANSACTIVACIÓN DEL RECEPTOR ESTROGÉNICO PARA IDENTIFICAR LOS AGONISTAS Y ANTAGONISTAS DEL RECEPTOR ESTROGÉNICO CON VM7LUC
CONSIDERACIONES Y LIMITACIONES INICIALES (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
1.
|
Este ensayo utiliza la línea celular VM7Luc4E2 (65). Ha sido validado por el Centro Interagencias del Programa Nacional de Toxicología para la Evaluación de Métodos Toxicológicos Alternativo (National Toxicology Center for the Evaluation of Alternative Toxicological Methods, NICEATM), y el Comité de Coordinación Interagencias sobre la Validación de Métodos Alternativos (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) (1). Las líneas celulares VM7Luc expresan predominantemente el receptor ERα endógeno y una pequeña cantidad de ERβ endógeno (2) (3) (4).
|
|
2.
|
Este ensayo es aplicable a una amplia gama de sustancias, a condición de que puedan disolverse en dimetilsulfóxido (DMSO; n.o CAS 67-68-5), no reaccionen con el DMSO ni con el medio de cultivo celular y no sean citotóxicas a las concentraciones utilizadas en el ensayo. Si el uso de DMSO no es posible, podrá emplearse otro vehículo, como etanol o agua (véase el punto 12). El comportamiento demostrado del ensayo de (ant)agonistas VM7Luc ER TA indica que los datos generados con este ensayo pueden dar información sobre los mecanismos de actuación mediados por el ER y podrían tenerse en cuenta a la hora de asignar prioridad a las sustancias para someterse a ensayos adicionales.
|
|
3.
|
Este ensayo está diseñado específicamente para detectar la TA mediada a través del hERα y el hERß midiendo la quimioluminiscencia como parámetro. La utilización de la quimioluminiscencia en bioensayos está extendida porque la luminiscencia presenta una alta relación de señal a fondo. Sin embargo, la actividad de la luciferasa de luciérnaga en ensayos celulares puede confundirse por la presencia de sustancias que inhiben la luciferasa, lo que provoca una inhibición aparente o un aumento de la luminiscencia debido a la estabilización de la proteína. Sin embargo, en algunos ensayos de gen marcador de ER a base de luciferasa se han comunicado señales de luminiscencia no mediadas por el receptor a concentraciones de fitoestrógenos superiores a 1 μM como consecuencia de la sobreactivación del gen marcador de la luciferasa (9) (11). Si bien la curva dosis-respuesta indica que la activación real del sistema ER se produce a concentraciones más bajas, debe examinarse detenidamente la expresión de la luciferasa obtenida a altas concentraciones de fitoestrógenos o de compuestos similares sospechosos de producir una sobreactivación de tipo fitoestrogénico del gen marcador de la luciferasa, en los sistemas de ensayo ER TA transfectados de forma estable (véase el apéndice 2).
|
|
4.
|
Las secciones “INTRODUCCIÓN GENERAL” y “COMPONENTES DEL ENSAYO ER TA” deben leerse antes de utilizar este ensayo con fines normativos. Las definiciones y abreviaturas utilizadas en el presente método de ensayo se recogen en el apéndice 1.
|
PRINCIPIO DEL ENSAYO (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
5.
|
El ensayo se utiliza para indicar la unión de un ligando al ER, seguida por la translocación del complejo receptor-ligando al núcleo. En el núcleo, el complejo receptor-ligando se une a elementos específicos de respuesta de ADN y transactiva el gen marcador (luc), lo que da lugar a la producción de luciferasa y a la consiguiente emisión de luz, que puede cuantificarse utilizando un luminómetro. La actividad de la luciferasa puede evaluarse de forma rápida y poco costosa con varios equipos de pruebas disponibles en el comercio. El VM7Luc ER TA utiliza una línea celular de adenocarcinoma de mama humana sensible al ER, la línea VM7, que se ha transfectado de forma estable con una construcción del marcador luc de la luciérnaga bajo el control de cuatro elementos de respuesta estrogénica situados en dirección 5’ respecto al promotor vírico del tumor de mama del ratón (MMTV), a fin de detectar sustanciascon actividad agonista o antagonista ER in vitro. El promotor MMTV muestra reactividad cruzada solo de pequeña intensidad con otras hormonas esteroideas y no esteroideas (8). Los criterios para la interpretación de los datos se describen con detalle en el punto 41. En síntesis, una respuesta positiva se identifica mediante una curva concentración-respuesta que contiene al menos tres puntos con barras de error no solapadas (media ± DT), así como un cambio en la amplitud [unidad luminosa relativa normalizada (ULR)] de al menos el 20 % del valor máximo del patrón de referencia [17β-estradiol (E2; n.o CAS 50-28-2)] en el caso del ensayo de agonistas, clorhidrato de raloxifeno [Ral; n.o CAS 84449-90 -1]/E2 en el caso del ensayo de antagonistas).
|
PROCEDIMIENTO
Línea celular
|
6.
|
Debe utilizarse para el ensayo la línea celular VM7Luc4E2, transfectada de forma estable. En la actualidad, solo se puede conseguir la línea celular mediante un acuerdo de licencia técnica en la Universidad de California, Davis, California, EE. UU. (66), y en Xenobiotic Detection Systems Inc., Durham, Carolina del Norte, EE. UU. (67).
|
Estabilidad de la línea celular
|
7.
|
Para mantener la estabilidad e integridad de la línea celular, las células deben cultivarse en medio de mantenimiento celular a lo largo de más de un pase a partir del cultivo madre congelado (véase el punto 9). Las células no se deben cultivar a lo largo de más de treinta pases. En el caso de la línea celular VM7Luc4E2, treinta pases serán aproximadamente tres meses.
|
Condiciones de cultivo y siembra de las células
|
8.
|
Deben seguirse los procedimientos especificados en las Orientaciones sobre buenas prácticas en materia de cultivo de células (5) (6) para garantizar la calidad de todos los materiales y métodos con el fin de mantener la integridad, validez y reproducibilidad de los trabajos realizados.
|
|
9.
|
Las células VM7Luc4E2 se mantienen en el medio RPMI 1 640 complementado con 0,9 % de Pen-Strep y 8,0 % de suero bovino fetal (FBS) en una incubadora de cultivos tisulares aparte, a una temperatura de 37oC ± 1oC, una humedad del 90 % ± 5 % y un contenido de CO2 en el aire del 5,0 % ± 1 %.
|
|
10.
|
Al llegar al ~ 80 % de confluencia, las células VM7Luc4E2 se subcultivan y se ponen en un medio libre de estrógenos durante 48 horas antes de sembrarlas en placas de 96 pocillos para exponerlas a los productos problema y analizar la inducción de la actividad de la luciferasa en función de los estrógenos. El medio libre de estrógenos (EFM) contiene la modificación de Dulbecco del medio de Eagle (DMEM), sin rojo de fenol, complementado con 4,5 % de FBS tratado con carbón/dextrano, 1,9 % de L-glutamina y 0,9 % de Pen-Strep. Todo el material de plástico debe estar libre de actividad estrogénica [véase el protocolo detallado (7)].
|
Criterios de aceptabilidad
|
11.
|
La aceptación o el rechazo de un ensayo se basa en la evaluación de los resultados obtenidos con los patrones de referencia y los testigos en cada experimento realizado con una placa de 96 pocillos. Cada patrón de referencia se somete a ensayo a diversas concentraciones y hay varias muestras de cada concentración de patrón de referencia yde testigo. Se comparan los resultados con los controles de calidad en relación con estos parámetros derivados de las bases de datos históricos de agonistas y de antagonistas generadas por cada laboratorio durante la demostración de la competencia. Las bases de datos históricos se actualizan continuamente con valores de los patrones de referencia y de los testigos. Los cambios en el equipo o en las condiciones de laboratorio pueden requerir la actualización de las bases de datos históricos.
|
Ensayo de agonistas
Ensayo de determinación del intervalo
|
•
|
Inducción: La inducción en la placa se mide dividiendo el valor medio más alto de unidades luminosas relativas (ULR) del patrón de referencia E2 por el valor medio de ULR del testigo de DMSO. Se alcanza por lo general un factor de inducción de cinco pero, a efectos de aceptación, dicho factor de inducción debe ser superior o igual a cuatro.
|
|
•
|
Resultados del control de DMSO: Los valores de ULR del control del disolvente deben situarse en el intervalo de 2,5 veces la desviación típica respecto al valor medio histórico de ULR del control del disolvente.
|
|
•
|
Se debe descartar y repetir todo experimento que incumpla cualquiera de estos criterios de aceptación.
|
Ensayo completo
Incluye los criterios de aceptabilidad del ensayo de determinación del intervalo de agonistas y los siguientes:
|
•
|
Resultados con el patrón de referencia: La curva concentración-respuesta del patrón de referencia E2 debe ser de forma sigmoidea y tener al menos tres valores dentro de la porción lineal de la curva concentración-respuesta.
|
|
•
|
Resultados del testigo positivo: Los valores de ULR del testigo de metoxicloro deben ser mayores que la media obtenida con el DMSO más el triple de la desviación típica respecto a dicha media.
|
|
•
|
Se debe descartar y repetir todo experimento que incumpla cualquiera de estos criterios de aceptación.
|
Ensayo de antagonistas
Ensayo de determinación del intervalo
|
•
|
Reducción: La reducción en la placa se mide dividiendo el valor medio más alto de ULR del patrón de referencia Ral/E2 por el valor medio de ULR del testigo de DMSO. Se alcanza por lo general un factor de reducción de cinco pero, a efectos de aceptación, dicho factor de reducción debe ser superior o igual a tres.
|
|
•
|
Resultados del testigo E2: Los valores de ULR del testigo de E2 deben situarse en el intervalo de 2,5 veces la desviación típica respecto al valor medio histórico de ULR del testigo de E2.
|
|
•
|
Resultados del control de DMSO: Los valores de ULR del control de DMSO deben situarse en el intervalo de 2,5 veces la desviación típica respecto al valor medio histórico de ULR del control del disolvente.
|
|
•
|
Se debe descartar y repetir todo experimento que incumpla cualquiera de estos criterios de aceptación.
|
Ensayo completo
Incluye los criterios de aceptación del ensayo de determinación del intervalo de antagonistas y los siguientes:
|
•
|
Resultados con el patrón de referencia: La curva concentración-respuesta del patrón de referencia Ral/E2 debe ser de forma sigmoidea y tener al menos tres valores dentro de la porción lineal de la curva concentración-respuesta.
|
|
•
|
Resultados del testigo positivo: Los valores de ULR del testigo de tamoxifeno/E2 deben ser menores que la media obtenida con el testigo de E2 menos el triple de la desviación típica respecto a dicha media.
|
|
•
|
Se debe descartar y repetir todo experimento que incumpla cualquiera de estos criterios de aceptación.
|
Patrones de referencia, testigos positivos y controles de los vehículos
Control de los vehículos (ensayo de agonistas y antagonistas)
|
12.
|
El vehículo que se utilice para disolver los productos problema debe someterse a ensayo como control del vehículo. El vehículo utilizado durante la validación del ensayo VM7Luc ER TA fue la solución al 1 % (v/v) de dimetilsulfóxido (DMSO, n.o CAS 67-68-5) (véase el punto 24). Si se utiliza un vehículo distinto del DMSO, todos los patrones de referencia, testigos y productos problema deben ensayarse en el mismo vehículo, si procede.
|
Patrón de referencia (determinación del intervalo de agonistas)
|
13.
|
El patrón de referencia es el E2 (n.o CAS 50-28-2). Para los ensayos de determinación del intervalo, el patrón de referencia consiste en una serie de diluciones de cuatro concentraciones de E2 (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 y 2,87 x 10-12 M) y cada concentración se somete al ensayo en pocillos duplicados.
|
Patrón de referencia (ensayo completo de agonistas)
|
14.
|
El E2 para el ensayo completo consiste en una serie de diluciones 1: 2, compuesta por once concentraciones (desde 3,67 x 10-10 hasta 3,59 x 10-13 M) de E2 en pocillos duplicados.
|
Patrón de referencia (determinación del intervalo de antagonistas)
|
15.
|
El patrón de referencia es una combinación de Ral (n.o CAS 84449-90-1) y E2 (n.o CAS 50-28-2). El Ral/E2 para los ensayos de determinación del intervalo consiste en una serie de diluciones de tres concentraciones de Ral (3,06 x 10-9, 7,67 x 10-10, y 1,92 x 10-10 M) más una concentración fija (9,18 × 10-11 M) de E2 en pocillos duplicados.
|
Patrón de referencia (ensayo completo de antagonistas)
|
16.
|
El Ral/E2 para los ensayos completos consiste en una serie de diluciones 1: 2 de Ral (desde 2,45x 10-8 hasta 9,57 x 10-11 M) más una concentración fija (9,18 × 10-11 M) de E2, dando nueve concentraciones de Ral/E2 que se someten al ensayo en pocillos duplicados.
|
Testigo positivo débil (agonistas):
|
17.
|
El testigo positivo débil es una disolución 9,06 x 10-6 M de p,p’-metoxicloro (metoxicloro; n.o CAS 72-43-5) en EFM.
|
Testigo positivo débil (antagonistas):
|
18.
|
El control positivo débil consiste en una disolución de tamoxifeno (n.o CAS 10540-29-1) 3,36 x 10-6 M con E2 9,18 × 10-11 M en EFM.
|
Testigo de E2 (ensayo de antagonistas solo)
|
19.
|
El testigo de E2 consiste en una solución de E2 9,18 × 10-11 M en EFM y se utiliza como testigo negativo de referencia.
|
Factor de inducción (agonistas)
|
20.
|
La inducción de la actividad de luciferasa del patrón de referencia (E2) se mide dividiendo el valor medio más alto de ULR del patrón de referencia E2 por el valor medio de ULR del control de DMSO, y el resultado debe ser mayor que cuatro.
|
Factor de reducción (antagonistas)
|
21.
|
La actividad media de luciferasa del patrón de referencia (Ral/E2) se mide dividiendo el valor medio más alto de ULR del patrón de referencia Ral/E2 por el valor medio de ULR del control de DMSO, y el resultado debe ser mayor que tres.
Demostración de la competencia del laboratorio (véanse el punto 14 y los cuadros 3 y 4 de la sección “COMPONENTES DEL ENSAYO ER TA” del presente método de ensayo)
|
Vehículo
|
22.
|
Los productos problema deben disolverse en un disolvente que solubilice el producto problema y sea miscible con el medio celular. Son vehículos adecuados el agua, el etanol (pureza del 95 % al 100 %) y el DMSO. Si se utiliza el DMSO, el nivel no debe superar el 1 % (v/v). Con cualquier vehículo, debe demostrarse que el volumen máximo utilizado no es citotóxico ni interfiere con el comportamiento del ensayo. Los patrones de referencia y los testigos se disuelven en disolvente del 100 % y, a continuación, se diluyen hasta las concentraciones adecuadas en EFM.
|
Preparación de los productos problema
|
23.
|
Los productos problema se disuelven en DMSO (o el disolvente adecuado) del 100 % y, a continuación, se diluyen hasta las concentraciones adecuadas en EFM. Debe dejarse que todos los productos problema se estabilicen a temperatura ambiente antes de disolverse y diluirse. Las soluciones de los productos problema deben prepararse de nuevo para cada experimento. Las soluciones no deben presentar ningún precipitado ni turbidez apreciables. Las soluciones madre de los patrones de referencia y de los testigos pueden prepararse en gran cantidad; sin embargo, las diluciones finales de los patrones de referencia y de los testigos, así como los productos problema deben prepararse de nuevo para cada experimento y utilizarse en las 24 horas siguientes a la preparación.
|
Solubilidad y citotoxicidad: consideraciones para la determinación del intervalo de las dosis
|
24.
|
El ensayo de determinación del intervalo de las dosis consiste en una tanda con siete diluciones en serie 1: 10 por duplicado. Al principio, los productos problema se ensayan hasta la concentración máxima de 1 mg/ml (~ 1 mM) en las pruebas de agonistas y de 20 μg/ml (~ 10 μM) en las pruebas de antagonistas. Los experimentos de determinación del intervalo de las dosis se utilizan para determinar lo siguiente:
|
|
—
|
Concentraciones iniciales del producto problema que deben utilizarse durante los ensayos completos,
|
|
—
|
Diluciones del producto problema (1: 2 or 1: 5) que deben utilizarse durante los ensayos completos.
|
|
25.
|
En los protocolos de ensayo de agonistas y antagonistas (7) se incluye una evaluación de la viabilidad celular / citotoxicidad, que se incorpora en los ensayos de determinación del intervalo y en los ensayos completos. El método de citotoxicidad que se utilizó para evaluar la viabilidad celular durante la validación del VM7Luc ER TA (1) era un método de observación visual cualitativa en una serie graduada; sin embargo, puede utilizarse un método cuantitativo para la determinación de la citotoxicidad [véase el protocolo (7)]. No pueden utilizarse los datos de las concentraciones de producto problema que provoquen una reducción de la viabilidad superior al 20 %.
|
Exposición al producto problema y organización de la placa del ensayo
|
26.
|
Se hace el recuento de las células y se siembran en placas de cultivo tisular de 96 pocillos (2 x 105 células por pocillo) en EFM y se incuban durante 24 horas para que las células puedan adherirse a la placa. Se retira el EFM; se ponen en su lugar los productos problema y de referencia en EFM, y se incuban durante 19-24 horas. Se debe prestar especial atención a las sustancias que son muy volátiles, ya que la proximidad de los pocillos de los testigos puede generar falsos resultados positivos. En tales casos, el uso de “selladores de placas” puede ayudar a aislar eficazmente los distintos pocillos durante el ensayo, por lo que resulta recomendable.
|
Ensayos de determinación del intervalo
|
27.
|
Los ensayos de determinación del intervalo utilizan todos los pocillos de la placa de 96 pocillos para estudiar hasta seis productos problema, en una serie de siete diluciones 1: 10 por duplicado (véanse las figuras 1 y 2).
|
|
—
|
Las pruebas de determinación del intervalo de agonistas utiliza cuatro concentraciones de E2 por duplicado como patrón de referencia y cuatro pocillos replicados para el control de DMSO.
|
|
—
|
Las pruebas de determinación del intervalo de antagonistas utiliza tres concentraciones de Ral/E2 con una concentración 9,18 × 10-11 M de E2 por duplicado como patrón de referencia, y tres pocillos replicados para el testigo de E2 y el control de DMSO.
|
Figura 1
Disposición de las placas de 96 pocillos para el ensayo de determinación del intervalo de agonistas

Abreviaturas: E2-1 a E2-4 = concentraciones del patrón de referencia E2 (de altas a bajas); TC1-1 a TC1-7 = concentraciones del producto problema 1 (TC1) (de altas a bajas); TC2-1 a TC2-7 = concentraciones del producto problema 2 (TC2) (de altas a bajas); TC3-1 a TC3-7 = concentraciones del producto problema 3 (TC3) (de altas a bajas); TC4-1 a TC4-7 = concentraciones del producto problema 4 (TC4) (de altas a bajas); TC5-1 a TC5-7 = concentraciones del producto problema 5 (TC5) (de altas a bajas); TC6-1 a TC6-7 = concentraciones del producto problema 6 (TC6) (de altas a bajas); CVe = control del vehículo [DMSO (1 % v/v en EFM)].
Figura 2
Disposición de las placas de 96 pocillos para el ensayo de determinación del intervalo de antagonistas

Abreviaturas: E2 = testigo E2; Ral-1 a Ral-3 = concentraciones del patrón de referencia raloxifeno/E2 (de altas a bajas); TC1-1 a TC1-7 = concentraciones del producto problema 1 (TC1) (de altas a bajas); TC2-1 a TC2-7 = concentraciones del producto problema 2 (TC2) (de altas a bajas); TC3-1 a TC3-7 = concentraciones del producto problema 3 (TC3) (de altas a bajas); TC4-1 a TC4-7 = concentraciones del producto problema 4 (TC4) (de altas a bajas); TC5-1 a TC5-7 = concentraciones del producto problema 5 (TC5) (de altas a bajas); TC6-1 a TC6-7 = concentraciones del producto problema 6 (TC6) (de altas a bajas); CVe = control del vehículo (DMSO [1 % v/v en EFM]).
Nota: Todos los productos problema se someten a ensayo en presencia de E2 a la concentración de 9,18 × 10-11 M.
|
28.
|
El volumen final recomendado de medio necesario para cada pocillo es de 200 μl. Deben utilizarse únicamente placas de ensayo en las que las células de todos los pocillos tengan una viabilidad igual o superior al 80 %.
|
|
29.
|
En el protocolo de agonistas (7) se describe la determinación de las concentraciones iniciales de los ensayos completos de
agonistas
. En resumen, se utilizan los siguientes criterios:
|
|
—
|
Si en la curva de concentración del producto problema no hay puntos que sean superiores a la media más tres veces la desviación típica del control de DMSO, se realizará un ensayo completo utilizando una dilución en serie 1: 2 con once puntos empezando a la concentración soluble máxima.
|
|
—
|
Si en la curva de concentración del producto problema hay puntos que sean superiores a la media más tres veces la desviación típica del control de DMSO, la concentración inicial que se utilizará para la serie de diluciones con once puntos en el ensayo completo debe ser una unidad logarítmica superior a la concentración que dé el mayor valor de ULR ajustado en la determinación del intervalo. La serie de diluciones con once puntos se debe basar en la relación 1: 2 o 1: 5, con arreglo a los criterios siguientes:
|
Se debe utilizar una serie de diluciones con once puntos en la relación 1: 2 en caso de que el intervalo de concentraciones resultante abarque toda la gama de respuestas según la curva concentración-respuesta generada en el ensayo de determinación del intervalo. En caso contrario, debe utilizarse una relación de dilución de 1: 5.
|
|
|
—
|
Si un producto problema presenta una curva de respuesta bifásica en el ensayo de determinación del intervalo, ambas fases deben también incluirse en el ensayo completo.
|
|
30.
|
En el protocolo de antagonistas (7) se describe la determinación de las concentraciones iniciales de los ensayos completos de
antagonistas
. En resumen, se utilizan los siguientes criterios:
|
|
—
|
Si en la curva de concentración del producto problema no hay puntos que sean inferiores a la media menos tres veces la desviación típica del testigo de E2, se realizará un ensayo completo utilizando una serie de diluciones con once puntos en la relación 1: 2 empezando a la concentración soluble máxima.
|
|
—
|
Si en la curva de concentración del producto problema hay puntos que sean inferiores a la media menos tres veces la desviación típica del testigo de E2, la concentración inicial que se utilizará para la serie de diluciones con once puntos en el ensayo completo debe ser una de las siguientes:
|
—
|
la concentración que proporciona el valor de ULR ajustado más bajo en el ensayo de determinación del intervalo,
|
|
—
|
la concentración soluble máxima [véase el protocolo de antagonistas (7), figura 14-2],
|
|
—
|
la concentración citotóxica mínima [véase un ejemplo relacionado en el protocolo de antagonistas (7), figura 14-3].
|
|
|
—
|
La serie de once diluciones se debe basar en la relación 1: 2 o 1: 5, con arreglo a los criterios siguientes:
|
Se debe utilizar una serie de diluciones con once puntos en la relación 1: 2 en caso de que el intervalo de concentraciones resultante abarque toda la gama de respuestas según la curva concentración-respuesta generada en el ensayo de determinación del intervalo. En caso contrario, debe utilizarse una relación de dilución de 1: 5.
|
|
Ensayos completos
|
31.
|
El ensayo completo se compone de una serie de once diluciones (en la relación 1: 2 o 1: 5 diluciones a partir de la concentración inicial según los criterios de ensayo completo), y cada concentración se somete a ensayo en pocillos triplicados de la placa de 96 pocillos (véanse las figuras 3 y 4).
|
|
—
|
El ensayo completo de agonistas utiliza once concentraciones de E2 por duplicado como patrón de referencia. Se incluyen en cada placa cuatro pocillos replicados del control de DMSO y cuatro pocillos replicados del control de metoxicloro (a la concentración de 9,06 x 10-6 M).
|
|
—
|
El ensayo completo de antagonistas utiliza nueve concentraciones de Ral/E2 con una concentración de E2 de 9,18 × 10-11 M por duplicado como patrón de referencia, con cuatro pocillos replicados para el testigo E2 a la concentración de 9,18 x 10-11 M, cuatro pocillos replicados para los controles de DMSO, y cuatro pocillos replicados para el tamoxifeno a la concentración de 3,36 x 10-6 M.
|
Figura 3
Disposición de las placas de 96 pocillos para el ensayo completo de agonistas
Abreviaturas: TC1-1 a TC1-11 = concentraciones del producto problema 1 (de altas a bajas); TC2-1 a TC2-11 = concentraciones del producto problema 2 (de altas a bajas); E2-1 a E2-11 = concentraciones del patrón de referencia E2 (de altas a bajas); Met = p,p’-metoxicloro, testigo positivo débil; CVe = disolución de DMSO en EFM al 1 % v/v, control del vehículo
Figura 4
Disposición de las placas de 96 pocillos para el ensayo completo de antagonistas

Abreviaturas: E2 = testigo E2; Ral-1 a Ral-9 = concentraciones del patrón de referencia raloxifeno/E2 (de altas a bajas); Tam = tamoxifeno/E2, control positivo débil; TC1-1 a TC1-11 = concentraciones del producto problema 1 (TC1) (de altas a bajas); TC2-1 a TC2-11 = concentraciones del producto problema 2 (TC2) (de altas a bajas); CVe = control del vehículo (DMSO [1 % v/v en EFM]).
Nota: Como se indica, todos los pocillos de referencia y de ensayo contienen una concentración fija de E2 (9,18 x 10-11M).
|
32.
|
La repetición de los ensayos completos con el mismo producto debe hacerse en días distintos, a fin de garantizar su independencia. Deben realizarse al menos dos ensayos completos. Si los resultados de los ensayos se contradicen (por ejemplo, un ensayo es positivo y el otro negativo), o si uno de ellos es inadecuado, se realizará un tercer ensayo adicional.
|
Medición de la luminiscencia
|
33.
|
Se mide la luminiscencia en el intervalo de 300 a 650 nm, utilizando un luminómetro de inyección y programas informáticos que controlen el volumen de inyección y el intervalo de medición (7). La emisión de luz de cada pocillo se expresa como ULR por pocillo.
|
ANÁLISIS DE LOS DATOS
Determinación de la CE50/CI50
|
34.
|
El valor de la CE50 [concentración de producto problema que produce la mitad del efecto máximo (agonistas)] y el valor de la CI50 [concentración del producto problema que produce la mitad de la inhibición máxima (antagonistas)] se determinan a partir de los datos de concentración-respuesta. En el caso de productos problema que sean positivos a una o más concentraciones, la concentración del producto problema que causa la mitad de la respuesta máxima (CI50 o CE50) se calcula utilizando un análisis de función de Hill o una alternativa adecuada. La función de Hill consiste en un modelo matemático logístico de cuatro parámetros que relaciona la concentración del producto problema con la respuesta (por lo general, según una curva sigmoidea) utilizando la siguiente ecuación:
|

donde:
Y= respuesta (es decir, ULR);
X= logaritmo de la concentración;
Valor inferior= respuesta mínima;
Valor superior= respuesta máxima;
lg CE50 (o lg CI50
= logaritmo de X como respuesta intermedia entre el valor superior y el valor inferior;
Pendiente de Hill= pendiente de la curva.
El modelo calcula el mejor ajuste de los parámetros valor superior, valor inferior, pendiente de Hill, y CI50 y CE50. Para el cálculo de los valores CI50 y CE50, deben utilizarse programas informáticos estadísticos adecuados (por ejemplo, los de Graphpad PrismR).
Determinación de los valores atípicos
|
35.
|
Podría facilitarse un buen juicio estadístico si se incluyera el test Q (pero sin limitarse a este) [véanse los protocolos de agonistas y de antagonistas (7) para determinar los pocillos “inutilizables” que se excluirán del análisis de los datos].
|
|
36.
|
En el caso de las réplicas del patrón de referencia E2 (tamaño de la muestra de dos), se considerará que un valor de ULR ajustado correspondiente a una réplica a una concentración dada de E2 es atípico si su valor es superior o inferior en más del 20 % al valor de ULR ajustado a esa concentración en la base de datos históricos.
|
Recogida y ajuste de los datos del luminómetro correspondientes al ensayo de determinación del intervalo
|
37.
|
Los datos brutos del luminómetro deben transferirse a una hoja de cálculo diseñada para el ensayo. Debe determinarse si hay puntos de datos atípicos que hayan de eliminarse. (Véanse los criterios de aceptación del ensayo en relación con los parámetros que se determinan en los análisis). Deben llevarse a cabo los siguientes cálculos:
|
Agonista
|
Fase 1
|
Calcular el valor medio del control del vehículo de DMSO (CVe).
|
|
Fase 2
|
Restar el valor medio del CVe de DMSO del valor de cada pocillo para normalizar los datos.
|
|
Fase 3
|
Calcular el factor medio de inducción del patrón de referencia (E2).
|
|
Fase 4
|
Calcular el valor medio de CE50 de los productos problema.
|
Antagonista
|
Fase 1
|
Calcular el valor medio del CVe de DMSO.
|
|
Fase 2
|
Restar el valor medio del CVe de DMSO del valor de cada pocillo para normalizar los datos.
|
|
Fase 3
|
Calcular el factor medio de reducción del patrón de referencia (Ral/E2).
|
|
Fase 4
|
Calcular el valor medio del patrón de referencia E2.
|
|
Fase 5
|
Calcular el valor medio de CI50 de los productos problema.
|
Recogida y ajuste de los datos del luminómetro correspondientes al ensayo completo
|
38.
|
Los datos brutos del luminómetro deben transferirse a una hoja de cálculo diseñada para el ensayo. Debe determinarse si hay puntos de datos atípicos que hayan de eliminarse. (Véanse los criterios de aceptación del ensayo en relación con los parámetros que se determinan en los análisis). Deben llevarse a cabo los siguientes cálculos:
|
Agonista
|
Fase 1
|
Calcular el valor medio del CVe de DMSO.
|
|
Fase 2
|
Restar el valor medio del CVe de DMSO del valor de cada pocillo para normalizar los datos.
|
|
Fase 3
|
Calcular el factor medio de inducción del patrón de referencia (E2).
|
|
Fase 4
|
Calcular el valor medio de CE50 de E2 y los productos problema.
|
|
Fase 5
|
Calcular el valor de ULR medio ajustado del metoxicloro.
|
Antagonista
|
Fase 1
|
Calcular el valor medio del CVe de DMSO.
|
|
Fase 2
|
Restar el valor medio del CVe de DMSO del valor de cada pocillo para normalizar los datos.
|
|
Fase 3
|
Calcular el factor medio de inducción del patrón de referencia (Ral/E2).
|
|
Fase 4
|
Calcular el valor medio de CI50 de Ral/E2 y los productos problema.
|
|
Fase 5
|
Calcular el valor de ULR medio ajustado del tamoxifeno.
|
|
Fase 6
|
Calcular el valor medio del patrón de referencia E2.
|
Criterios de interpretación de los datos
|
39.
|
El VM7Luc TE TA se entiende como parte de un enfoque de ponderación de las pruebas para ayudar a asignar prioridades a las sustancias para el ensayo de la alteración endocrina in vivo. Parte de este procedimiento de asignación de prioridades será la clasificación del producto problema como positivo o negativo en cuanto a la actividad como agonista o antagonista del ER. Los criterios de decisión como producto positivo o negativo utilizados en el estudio de validación VM7Luc ER TA se describen en el cuadro 1.
|
Cuadro 1
Criterios de decisión como producto positivo o negativo
|
|
|
ACTIVIDAD COMO AGONISTA
|
|
Positivo
|
|
—
|
Todos los productos problema clasificados como positivos en cuanto a la actividad como agonista del ER deben presentar una curva concentración-respuesta que consista en una línea de base, seguida de una pendiente positiva, y que se concluya en una meseta o pico. En algunos casos, pueden estar definidas solo dos de estas características (línea de base - pendiente, o pendiente - pico).
|
|
—
|
La línea que define la pendiente positiva debe contener al menos tres puntos con barras de error no solapadas (media ± DT). Se excluyen los puntos que forman la línea de base, pero la parte lineal de la curva puede incluir el pico o el primer punto de la meseta.
|
|
—
|
Una clasificación como producto positivo requiere una amplitud de respuesta (la diferencia entre la línea de base y el pico) de al menos el 20 % del valor máximo del patrón de referencia, E2 [es decir, 2 000 ULR o más cuando el valor de la respuesta máxima del patrón de referencia (E2) se ajusta a 10 000 ULR].
|
|
—
|
Si es posible, deberá calcularse un valor de la CE50 para cada producto problema positivo.
|
|
|
Negativo
|
El valor de ULR ajustado medio para una concentración dada es igual o inferior a valor de ULR medio del control de DMSO más el triple de la desviación típica del valor de ULR del DMSO.
|
|
Inadecuado
|
Se considera que los datos que no pueden interpretarse como válidos para demostrar la presencia o la ausencia de actividad por limitaciones cualitativas o cuantitativas importantes son inadecuados y no pueden utilizarse para determinar si el producto problema es positivo o negativo. Los productos deben someterse a ensayo de nuevo.
|
|
|
|
ACTIVIDAD COMO ANTAGONISTA
|
|
Positivo
|
|
—
|
Los datos del producto problema producen una curva concentración-respuesta que consiste en una línea de base, a la que sigue una pendiente negativa.
|
|
—
|
La línea que define la pendiente negativa debe contener al menos tres puntos con barras de error no solapadas; se excluyen los puntos que forman la línea de base, pero la parte lineal de la curva puede incluir el primer punto de la meseta.
|
|
—
|
Debe haber al menos una reducción del 20 % en la actividad respecto al valor máximo del patrón de referencia, Ral/E2 (es decir, 8 000 ULR o menos cuando el valor de la respuesta máxima del patrón de referencia (Ral/E2) se ajusta a 10 000 ULR).
|
|
—
|
Las concentraciones no citotóxicas más altas del producto problema deben ser inferiores o iguales a 1 x 10-5 M.
|
|
—
|
Si es posible, deberá calcularse un valor de la CI50 para cada producto problema positivo.
|
|
|
Negativo
|
Todos los puntos de datos se sitúan por encima del valor CE80 (80 % de la respuesta del E2 u 8 000 ULR), a concentraciones inferiores a 1,0 x 10-5 M.
|
|
Inadecuado
|
Se considera que los datos que no pueden interpretarse como válidos para demostrar la presencia o la ausencia de actividad por limitaciones cualitativas o cuantitativas importantes son inadecuados y no pueden utilizarse para determinar si el producto problema es positivo o negativo. El producto debe someterse a ensayo de nuevo.
|
|
40.
|
Los resultados positivos se caracterizarán tanto por la magnitud del efecto como por la concentración a la que se produce el efecto, cuando sea posible. En las figuras 5 y 6 se muestran ejemplos de datos positivos, negativos e inadecuados.
|
Figura 5
Ejemplos de agonistas: datos positivos, negativos e inadecuados

La línea discontinua indica el 20 % de la respuesta del E2, 2 000 ULR ajustadas y normalizadas.
Figura 6
Ejemplos de antagonistas: datos positivos, negativos e inadecuados

La línea discontinua indica el 80 % de la respuesta del Ral/E2, 8 000 ULR ajustadas y normalizadas.
La línea continua indica 1,00 x 10-5 M. Para que una respuesta se considere positiva, debe estar por debajo de la línea de 8 000 ULR y a concentraciones inferiores a 1,00 x 10-5 M.
Las concentraciones indicadas con asteriscos en la gráfica del meso-hexestrol indican unas puntuaciones de viabilidad iguales o superiores a “2”.
Se considera que los resultados de los ensayos del ensayo correspondientes al meso-hexestrol son inadecuados porque la única respuesta que se encuentra por debajo de 8 000 ULR se da a la concentración de 1,00 x 10-5 M.
|
41.
|
Los cálculos de CE50 y CI50 pueden hacerse utilizando una función de Hill de cuatro parámetros [véanse más detalles en el protocolo de agonistas y en el de antagonistas (7)]. El cumplimiento de los criterios de aceptabilidad indica que el sistema funciona correctamente, pero no garantiza que cualquier tanda de ensayo concreta produzca datos exactos. La duplicación de los resultados de la primera tanda es la mejor garantía de que se han producido datos exactos (véase el punto 19 de la sección “COMPONENTES DEL ENSAYO ER TA”.
|
INFORME DEL ENSAYO
|
42.
|
Véase el punto 20 de la sección “COMPONENTES DEL ENSAYO ER TA”.
|
BIBLIOGRAFÍA
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5367-5373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): p. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: p. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7 850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10):4252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. and Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Apéndice 4
ENSAYO DE TRANSACTIVACIÓN DEL RECEPTOR ESTROGÉNICO Α HUMANO TRANSFECTADO ESTABLEMENTE PARA LA DETECCIÓN DE LA ACTIVIDAD AGONISTA Y ANTAGONISTA DE LOS PRODUCTOS, UTILIZANDO LA LÍNEA CELULAR ERΑ CALUX
CONSIDERACIONES Y LIMITACIONES INICIALES (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
1.
|
Este ensayo de transactivación ERα CALUX utiliza la línea celular U2OS humana para detectar la actividad de agonistas y antagonistas estrogénicos mediada a través del receptor estrogénico humano alfa (hERα). El estudio de validación del bioensayo ERα CLUX con transfección estable realizado por BioDetection Systems BV (Amsterdam, Países Bajos) puso de manifiesto la pertinencia y la fiabilidad del ensayo para su finalidad prevista (1). La línea celular ERα CALUX expresa solo el ERα humano con transfección estable (2) (3).
|
|
2.
|
Este ensayo está diseñado específicamente para detectar la transactivación mediada a través del hERα midiendo la bioluminiscencia como parámetro. La bioluminiscencia se utiliza normalmente en los bioensayos debido a la alta relación señal-ruido (4).
|
|
3.
|
Se ha notificado que las concentraciones de fitoestrógenos superiores a 1 μM sobreactivan el gen marcador de la luciferasa, lo que da lugar a una luminiscencia no mediada por el receptor (5) (6) (7). Por tanto, en los ensayos de transactivación del ER con transfección estable hay que examinar cuidadosamente las concentraciones más elevadas de fitoestrógenos u otros compuestos similares que puedan sobreactivar la expresión de la luciferasa (véase el apéndice 2).
|
|
4.
|
Las secciones “INTRODUCCIÓN GENERAL” y “COMPONENTES DEL ENSAYO ER TA” deben leerse antes de utilizar este ensayo con fines normativos. Las definiciones y abreviaturas utilizadas en el presente método de ensayo se recogen en el apéndice 1.
|
PRINCIPIO DEL ENSAYO (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
5.
|
El bioensayo se utiliza para evaluar la unión de un ligando al ER, seguida por la translocación del complejo receptor-ligando al núcleo. En el núcleo, el complejo receptor-ligando se une a determinados elementos específicos de respuesta del ADN y transactiva un gen marcador de la luciferasa de la luciérnaga, lo que provoca un aumento de la expresión celular de la enzima luciferasa. Como consecuencia de la adición del sustrato de la luciferasa, que es la luciferina, esta se transforma en un producto bioluminiscente. La luz producida puede detectarse y cuantificarse fácilmente con un luminómetro.
|
|
6.
|
El sistema de ensayo utiliza células CALUX transfectadas de forma estable. Las células ERα CALUX proceden de la línea celular U2OS de osteosarcoma osteoblástico humano. Las células U2OS humanas se han transfectado de forma estable con 3 x HRE-TAT-Luc y pSG5-neo-hERα utilizando el método de coprecipitación con fosfato de calcio. Se ha seleccionado la línea celular U2OS como el mejor candidato para servir de línea celular marcadora sensible a los estrógenos (y a otras hormonas esteroideas), basándose en la observación de que la línea celular U2OS mostraba poca o ninguna actividad de receptores endógenos. La ausencia de receptores endógenos se evaluó utilizando únicamente plásmidos de marcador de luciferasa, sin que se pusiera de manifiesto actividad alguna cuando se añadían los ligandos de los receptores. Además, esta línea celular permitía fuertes respuestas medidas por hormonas, cuando se introducían con carácter transitorio receptores análogos (2) (3) (8).
|
|
7.
|
El ensayo de productos para detectar su actividad estrogénica o antiestrogénica utilizando la línea celular ERα CALUX incluye una tanda de precribado y unas tandas completas. Durante la tanda de precribado se determina la solubilidad, la citotoxicidad y el intervalo afinado de concentraciones para el ensayo completo. Durante las tandas completas, se someten a ensayo los intervalos afinados de concentración de los productos problema según el bioensayo ERα CALUX, seguido de la clasificación de los productos problema en cuanto a su agonismo o antagonismo.
|
|
8.
|
Los criterios para la interpretación de los datos se describen con detalle en el punto 59. En resumen, el producto problema se considera positivo como agonista en el caso de que al menos dos concentraciones consecutivas suyas muestren una respuesta igual o superior al 10 % de la respuesta máxima del patrón de referencia 17β-estradiol (CP10). El producto problema se considera positivo como antagonista en el caso de que al menos dos concentraciones consecutivas suyas muestren una respuesta igual o inferior al 80 % de la respuesta máxima del patrón de referencia tamoxifeno (CP80).
|
PROCEDIMIENTO
Línea celular
|
9.
|
Debe utilizarse para el ensayo la línea celular ERα CALUX de U2OS, transfectada de forma estable. La línea celular puede obtenerse de BioDetection Systems BV, Amsterdam, Países Bajos, mediante un acuerdo de licencia técnica.
|
|
10.
|
Solo deben utilizarse cultivos celulares libres de micoplasmas. Los lotes usados de células deben estar certificados como negativos en cuanto a la contaminación por micoplasmas, o bien antes de su uso debe llevarse a cabo un ensayo para excluir la presencia de estos. Debe utilizarse la RCP-TR (reacción en cadena de la polimerasa en tiempo real) para la detección sensible de la infección por micoplasmas (9).
|
Estabilidad de la línea celular
|
11.
|
Para mantener la estabilidad y la integridad de las células CALUX, deben conservarse en nitrógeno líquido (-80oC). Tras descongelarlas para iniciar un nuevo cultivo, las células deben subcultivarse al menos dos veces antes de utilizarse para evaluar los productos en cuanto a su actividad como agonista y antagonista estrogénico. Las células no se deben subcultivar a lo largo de más de treinta pases.
|
|
12.
|
Para supervisar la estabilidad de la línea celular a lo largo del tiempo, debe verificarse la sensibilidad del sistema de ensayo de agonistas y antagonistas mediante la evaluación del patrón de referencia (CE50 o CI50). Además, debe supervisarse la inducción relativa de la muestra de testigo positivo (TP) y la muestra de testigo negativo (TN). Los resultados deben estar de acuerdo con los criterios de aceptación para el bioensayo CELUX de agonistas (cuadro 3C) o antagonistas (cuadro 4C). Los patrones de referencia, testigos positivos y negativos, figuran en el cuadro 1 y en el cuadro 2, en relación con el modo agonista y el modo antagonista.
|
Condiciones de cultivo y siembra de las células
|
13.
|
Las células U2OS se deben cultivar en medio de cultivo [DMEM/F12 (1: 1) con rojo de fenol como indicador de pH, complementado con suero bovino fetal (7,5 %), aminoácidos no esenciales (1 %), 10 unidades/ml de penicilina, estreptomicina y geneticina (G-418) como marcador de selección]. Las células deben colocarse en una incubadora con CO2 (5 % de CO2) a 37,0oC y 100 % de humedad. Cuando las células alcancen una confluencia del 85-95 %, deben subcultivarse o prepararse para la siembra en placas de microvaloración de 96 pocillos. En este último caso, las células se deben resuspender a la concentración de 1 x 105 células/ml en medio de ensayo libre de estrógenos [DMEM/F12 (1: 1) sin rojo de fenol, complementado con suero bovino fetal tratado con carbón recubierto de dextrano (5 % v/v), aminoácidos no esenciales (1 % v/v), 10 unidades/ml de penicilina y estreptomicina] y ponerse en los pocillos de placas de microvaloración de 96 pocillos (100 μl de suspensión homogeneizada de células). Las células deben preincubarse en una incubadora de CO2 (5 % de CO2, 37,0oC, 100 % de humedad) durante 24 horas antes de la exposición. Los artículos de plástico deben estar libres de estrógenos.
|
Criterios de aceptabilidad
|
14.
|
Se somete a ensayo el producto o productos problema en cuanto a su actividad como agonista y antagonista. Una serie de ensayo consiste en un máximo de 6 placas de microvaloración. Cada serie de ensayo contiene al menos una serie completa de diluciones de un patrón de referencia, una muestra de testigo positivo, una muestra de testigo negativo y controles del disolvente. Las figuras 1 y 2 describen la disposición de las placas para las series de pruebas de agonistas y antagonistas.
|
|
15.
|
Debe analizarse por triplicado cada una de las diluciones de los patrones de referencia, de los productos problema, de todos los controles del disolvente, y de los testigos positivos y negativos. Cada uno de los análisis por triplicado deberá cumplir los requisitos que figuran en los cuadros 3A y 4A.
|
|
16.
|
En la primera placa de cada serie de ensayo se mide una serie completa de diluciones del patrón de referencia (17β-estradiol para los agonistas; tamoxifeno para los antagonistas). A fin de poder comparar los resultados del análisis de las otras cinco placas de microvaloración con la primera placa de microvaloración que contiene la curva completa concentración-respuesta del patrón de referencia, todas las placas deben contener 3 muestras de control: control del disolvente, concentración más alta del patrón de referencia sometida a ensayo, y la concentración aproximada CE50 (agonistas) o CI50 (antagonistas) del patrón de referencia. La relación entre las muestras de control medias en la primera placa y las otras cinco placas debe cumplir los requisitos indicados en el cuadro 3C (agonistas) o en el cuadro 4C (antagonistas).
|
|
17.
|
Se calcula el factor z de cada una de las placas de microvaloración dentro de una serie de ensayo (10). El factor z debe calcularse utilizando las respuestas obtenidas a las concentraciones más alta y más baja del patrón de referencia. Una placa de microvaloración se considera válida en caso de que cumpla los requisitos establecidos en el cuadro 3C (agonistas) o en el cuadro 4C (antagonistas).
|
|
18.
|
El patrón de referencia debe presentar una curva dosis-respuesta sigmoidea. Los valores de CE50 o CI50 derivados de la respuesta de la serie de diluciones del patrón de referencia deben cumplir los requisitos indicados en el cuadro 3C (agonistas) o en el cuadro 4C (antagonistas).
|
|
19.
|
Cada serie de ensayo debe contener una muestra de testigo positivo y una muestra de testigo negativo. La inducción relativa calculada con la muestra de testigo tanto positivo como negativo debe cumplir los requisitos indicados en el cuadro 3C (agonistas) o en el cuadro 4C (antagonistas).
|
|
20.
|
Durante todas las mediciones, el factor de inducción de la concentración más alta del patrón de referencia debe medirse dividiendo la media de la respuesta máxima en unidades luminosas relativas (ULR) del patrón de referencia, 17β-estradiol, por la media de la respuesta en ULR del control del disolvente de referencia. Este factor de inducción debe cumplir los requisitos mínimos aplicables al factor de inducción que se indican en el cuadro 3C (agonistas) o en el cuadro 4C (antagonistas).
|
|
21.
|
Solo las placas de microvaloración que cumplan todos los criterios de aceptación antes mencionados se consideran válidas y pueden utilizarse para evaluar la respuesta de los productos problema.
|
|
22.
|
Los criterios de aceptación son aplicables tanto a las tandas de precribado como a las tandas de los ensayos completos.
|
Cuadro 1
Concentraciones de patrón de referencia, testigo positivo (TP) y testigo negativo (TN) para el bioanálisis CALUX de agonistas
|
|
Sustancia
|
N.o CAS
|
Intervalo de ensayo (M)
|
|
Patrón de referencia
|
17β-Estradiol
|
50-28-2
|
1 * 10-13 - 1 * 10-10
|
|
Testigo positivo (TP)
|
17α-Metiltestosterona
|
58-18-4
|
3 * 10-6
|
|
Testigo negativo (TN)
|
Corticosterona
|
50-22-6
|
1 * 10-8
|
Cuadro 2
Concentraciones de patrón de referencia, testigo positivo (TP) y testigo negativo (TN) para el bioanálisis CALUX de antagonistas
|
|
Sustancia
|
N.o CAS
|
Intervalo de ensayo (M)
|
|
Patrón de referencia
|
Tamoxifeno
|
10540-29-1
|
3 * 10-9 - 1 * 10-5
|
|
Testigo positivo (TP)
|
4-Hidroxitamoxifeno
|
68047-06-3
|
1 * 10-9
|
|
Testigo negativo (TN)
|
Resveratrol
|
501-36-0
|
1 * 10-5
|
Cuadro 3
Criterios de aceptación para el bioensayo ERα CALUX de agonistas
|
A — Muestras individuales en una placa
|
Criterio
|
|
1
|
DT % máxima de los pocillos triplicados (de TN, TP, de cada dilución del producto problema y del patrón de referencia, excepto C0)
|
< 15 %
|
|
2
|
DT % máxima de los pocillos triplicados [del patrón de referencia y de los controles del disolvente del producto problema (C0, CD)]
|
< 30 %
|
|
3
|
Pérdida máxima de LDH como medida de la citotoxicidad
|
< 120 %
|
|
B — Dentro de una sola placa de microvaloración
|
|
|
4
|
Relación entre el control del disolvente del patrón de referencia (C0; placa 1) y el control del disolvente del producto problema (CD; placas 2 a x)
|
0,5 a 2,0
|
|
5
|
Relación de las concentraciones aprox. CE50 y máxima del patrón de referencia en la placa 1 y las concentraciones aprox. CE50 y máxima del patrón de referencia en las placas 2 a x (C4, C8)
|
0,70 a 1,30
|
|
6
|
Factor z de cada placa
|
> 0,6
|
|
C — Dentro de una sola serie de análisis (todas las placas de una serie)
|
|
|
7
|
Curva sigmoidea del patrón de referencia
|
Sí (17ß-estradiol)
|
|
8
|
Intervalo de CE50 del patrón de referencia 17ß-estradiol
|
4 * 10-12 - 4 * 10-11 M
|
|
9
|
Factor mínimo de inducción de la mayor concentración de 17ß-estradiol, respecto al control del disolvente del patrón de referencia
|
5
|
|
10
|
Inducción relativa (%) del TP
|
> 30 %
|
|
11
|
Inducción relativa (%) del TN
|
< 10 %
|
Aprox.: aproximado; TP: testigo positivo; TN: testigo negativo; CD: control del disolvente del producto problema; C0: control del disolvente del patrón de referencia; DT: desviación típica; LDH: lactato-deshidrogenasa.
Cuadro 4
Criterios de aceptación para el bioensayo ERα CALUX de antagonistas
|
A — Muestras individuales en una placa
|
Criterio
|
|
1
|
DT % máxima de los pocillos triplicados [de TN, TP, de cada dilución del producto problema y del patrón de referencia, control del disolvente (C0)]
|
< 15 %
|
|
2
|
DT % máxima de los pocillos triplicados [del control del vehículo (CVe) y de la concentración máxima del patrón de referencia (C8)]
|
< 30 %
|
|
3
|
Pérdida máxima de LDH como medida de la citotoxicidad
|
< 120 %
|
|
B — Dentro de una sola placa de microvaloración
|
|
|
4
|
Relación entre el control del disolvente del patrón de referencia (C0; placa 1) y el control del disolvente del producto problema (CD; placas 2 a x)
|
0,70 a 1,30
|
|
5
|
Relación de las concentraciones aprox. CI50 del patrón de referencia en la placa 1 y las concentraciones aprox. CI50 del patrón de referencia en las placas 2 a x (C4)
|
0,70 a 1,30
|
|
6
|
Relación de las concentraciones máximas del patrón de referencia en la placa 1 y las concentraciones máximas del patrón de referencia en las placas 2 a x (C8)
|
0,50 a 2,0
|
|
7
|
Factor z de cada placa
|
> 0,6
|
|
C — Dentro de una sola serie de análisis (todas las placas de una serie)
|
|
|
8
|
Curva sigmoidea del patrón de referencia
|
Sí (tamoxifeno)
|
|
9
|
Intervalo de CI50 del patrón de referencia (tamoxifeno)
|
1 * 10-8 - 1 * 10-7 M
|
|
10
|
Factor mínimo de inducción del control del disolvente del patrón de referencia, con respecto a la concentración más alta de tamoxifeno
|
2,5
|
|
11
|
Inducción relativa (%) del TP
|
< 70 %
|
|
12
|
Inducción relativa (%) del TN
|
> 85 %
|
Aprox.: aproximado; TP: testigo positivo; TN: testigo negativo; CVe: control del vehículo (control del disolvente sin concentración fija del patrón de referencia de agonista); CD: control del disolvente del producto problema; C0: control del disolvente del patrón de referencia; DT: desviación típica; LDH: lactato-deshidrogenasa.
Control del disolvente/vehículo, patrones de referencia, testigos positivos, testigos negativos
|
23.
|
Debe utilizarse el mismo control del disolvente/vehículo, patrones de referencia, testigos positivos y testigos negativos tanto en la tanda de precribado como en las tandas de los ensayos completos. Además, la concentración de los patrones de referencia, testigos positivos y testigos negativos debe ser la misma.
|
Control del disolvente
|
24.
|
El disolvente que se utilice para disolver los productos problema debe someterse a ensayo como control del disolvente. El dimetilsulfóxido [DMSO, 1 % (v/v); n.o CAS 67-68-5] se utilizó como vehículo durante la validación del bioensayo ERα CALUX. Si se utiliza un disolvente distinto del DMSO, todos los patrones de referencia, testigos y productos problema deben ensayarse con el mismo vehículo. Téngase en cuenta que el control del disolvente para los estudios de antagonistas contiene una concentración fija del patrón de referencia agonista, que es el 17β-estradiol (CE50 aproximada). Para comprobar el disolvente utilizado en los estudios de antagonistas, debe prepararse y ensayarse un control del vehículo.
|
Control del vehículo (antagonistas)
|
25.
|
Para los estudios de antagonistas, el medio de ensayo tiene el complemento de una concentración fija del patrón de referencia agonista, que es el 17β-estradiol (CE50 aproximada). A fin de comprobar el disolvente utilizado para disolver los productos problema del estudio de antagonistas, debe prepararse un medio de ensayo sin una concentración fija del patrón de referencia agonista, que es el 17β-estradiol. Esta muestra de control se indica como control del vehículo. El dimetilsulfóxido [DMSO, 1 % (v/v); n.o CAS 67-68-5] se utilizó como vehículo durante la validación del bioensayo ERα CALUX. Si se utiliza un disolvente distinto del DMSO, todos los patrones de referencia, testigos y productos problema deben ensayarse con el mismo vehículo.
|
Patrones de referencia
|
26.
|
El patrón de referencia de los agonistas es el 17β-estradiol (cuadro 1). Los patrones de referencia comprenden una serie de diluciones de ocho concentraciones de 17β-estradiol (1 * 10-13, 3 * 10-13, 1 * 10-12, 3 * 10-12, 6 * 10-12, 1 * 10-11, 3 * 10-11, 1 * 10-10 M).
|
|
27.
|
El patrón de referencia de los antagonistas es el tamoxifeno (cuadro 2). Los patrones de referencia comprenden una serie de diluciones de ocho concentraciones de tamoxifeno (3 * 10-9, 1 * 10-8, 3 * 10-8, 1 * 10-7, 3 * 10-7, 1 * 10-6, 3 * 10-6, 1 * 10-5 M). Cada una de las concentraciones del patrón de referencia de los antagonistas se incuba conjuntamente con una concentración fija del patrón de referencia de los agonistas, el 17β-estradiol (3 * 10-12 M).
|
Testigo positivo
|
28.
|
El testigo positivo de los estudios de agonistas es la 17α-metiltestosterona (cuadro 1).
|
|
29.
|
El testigo positivo de los estudios de antagonistas es el 4-hidroxitamoxifeno (cuadro 2). El testigo positivo de los antagonistas se incuba conjuntamente con una concentración fija del patrón de referencia de los agonistas, el 17β-estradiol (3 * 10-12 M).
|
Testigo negativo
|
30.
|
El testigo negativo de los estudios de agonistas es la corticosterona (cuadro 1).
|
|
31.
|
El testigo negativo de los estudios de antagonistas es el resveratrol (cuadro 2). El testigo negativo de los antagonistas se incuba conjuntamente con una concentración fija del patrón de referencia de los agonistas, el 17β-estradiol (3 * 10-12 M).
|
Demostración de la competencia del laboratorio (véanse el punto 14 y los cuadros 3 y 4 de la sección “COMPONENTES DEL ENSAYO ER TA” del presente método de ensayo)
Vehículo
|
32.
|
El disolvente utilizado para disolver los productos problema debe solubilizarlos completamente y ser miscible con el medio celular. Son disolventes adecuados el DMSO, el agua y el etanol (del 95 % al 100 % de pureza). En caso de que se emplee el DMSO como disolvente, su concentración máxima durante la incubación no debe superar el 1 % (v/v). Antes de su utilización, el disolvente debe someterse a ensayo para comprobar la ausencia de citotoxicidad y de interferencia con el comportamiento del ensayo.
|
Preparación de los patrones de referencia, testigos positivos, testigos negativos y productos problema
|
33.
|
Se disuelven en 100 % de DMSO (o un disolvente adecuado) los patrones de referencia, los testigos positivos, los testigos negativos y los productos problema. A continuación, deben prepararse diluciones (en serie) adecuadas en el mismo disolvente. Antes de su disolución, debe permitirse que todas las sustancias se equilibren a la temperatura ambiente. Las soluciones madre recién preparadas de los patrones de referencia, los testigos positivos, los testigos negativos y los productos problema no deben tener precipitado ni turbidez apreciables. Las soluciones madre de los patrones de referencia y de los testigos pueden prepararse en gran cantidad. Las soluciones madre de los productos problema deben prepararse de nuevo antes de cada experimento. Las diluciones finales de los patrones de referencia, testigos positivos, testigos negativos y productos problema deben prepararse de nuevo para cada experimento y utilizarse en las 24 horas siguientes a la preparación.
|
Solubilidad, citotoxicidad y determinación del intervalo
|
34.
|
Durante la tanda de precribado se determina la solubilidad de los productos problema en el disolvente elegido. Se prepara una solución madre a la concentración máxima de 0,1 M. En caso de que esta concentración muestre problemas de solubilidad, deben prepararse soluciones madre más diluidas hasta que los productos problema estén completamente solubilizados. Durante la tanda de precribado, se someten a ensayo diluciones de producto problema en serie 1: 10. La concentración máxima par el ensayo de agonistas o antagonistas es 1 mM. Tras realizar el precribado, se obtiene un intervalo afinado de concentraciones de los productos problema para utilizarlo en las tandas de los ensayos completos. Las diluciones utilizadas para los ensayos completos deben ser de 1 x, 3 x, 10 x, 30 x, 100 x, 300 x, 1000 x y 3000 x.
|
|
35.
|
El ensayo de citotoxicidad está incluido en el protocolo de ensayo de agonistas y antagonistas (11). El ensayo de citotoxicidad se incorpora tanto a la tanda de precribado como a las tandas de ensayos completos. El método utilizado para evaluar la citotoxicidad durante la validación del bioensayo del ERα CALUX es la prueba de pérdida de lactato-deshidrogenasa (LDH) combinada con la inspección visual cualitativa de las células (véase el apéndice 4.1) tras la exposición a los productos problema. No obstante, pueden utilizarse otros métodos cuantitativos para la determinación de la citotoxicidad [por ejemplo, el ensayo colorimétrico con tetrazolio (MTT) o el bioensayo CALUX de citotoxicidad]. En general, las concentraciones del producto problema que muestran una reducción de más del 20 % de de la viabilidad celular se consideran citotóxicas, por lo que no pueden utilizarse para la evaluación de los datos. Por lo que respecta al ensayo de pérdida de LDH, se considera que la concentración del producto problema es citotóxica cuando el porcentaje de pérdida de LDH es superior al 120 %.
|
Exposición al producto problema y organización de la placa del ensayo
|
36.
|
Tras la tripsinización de un matraz confluente de células cultivadas, las células se vuelven a suspender a la concentración de 1 x 105 células/ml en medio de ensayo libre de estrógenos. En los pocillos interiores de una placa de microvaloración de 96 pocillos se siembran 100 μl de células resuspendidas. Los pocillos exteriores se llenan con 200 μl de solución salina amortiguadora de fosfato (PBS) (véanse las figuras 1 y 2). Las placas se preincuban durante 24 horas en una incubadora de CO2 (5 % de CO2, 37oC, 100 % de humedad).
|
|
37.
|
Tras la preincubación, las placas se inspeccionan para comprobar visualmente la citotoxicidad (véase el apéndice 4.1), la contaminación y la confluencia. Solo se utilizan para el ensayo las placas que visualmente no muestren citotoxicidad ni contaminación y que presenten una confluencia del 85 % como mínimo. El medio de los pocillos interiores se retira cuidadosamente y se sustituye por 200 μl de medio de ensayo libre de estrógenos, con diluciones en serie apropiadas de los patrones de referencia, productos problema, testigos positivos, testigos negativosy controles del disolvente (cuadro 5: estudios de agonistas; cuadro 6: estudios de antagonistas). Todos los patrones de referencia, productos problema, testigos positivos, testigos negativos y controles de los disolventes se someten a ensayo por triplicado. En la figura 1 se indica la disposición de la placa para los ensayos de agonistas. En la figura 2 se indica la disposición de la placa para los ensayos de antagonistas. La disposición de la placa es la misma para las pruebas de precribado y para el ensayo completo. En los ensayos de antagonistas, todos los pocillos interiores, excepto los pocillos de control del vehículo (CVe), también contendrán una concentración fija del patrón de referencia agonista, el 17β-estradiol (3 * 10-12 M). Obsérvese que deben añadirse a cada placa de producto problema los patrones de referencia C8 y C4.
|
|
38.
|
Tras la exposición de las células a todos los productos, las placas de microvaloración de 96 pocillos deben incubarse durante otras 24 horas en una incubadora de CO2 (5 % CO2, 37oC, 100 % de humedad).
|
Figura 1
Disposición de las placas de microvaloración de 96 pocillos para el precribado y la valoración del efecto agonista.

C0 = disolvente del patrón de referencia;
C(1-8) = serie de diluciones (1-8, concentración de baja a elevada) del patrón de referencia.
TP = testigo positivo;
TN = testigo negativo;
TCx-(1-8) = diluciones (1-8, concentraciones de baja a elevada) del producto problema para la tanda de precribado y para la evaluación del efecto agonista del producto problema x.
CD = control del disolvente del producto problema (idealmente, el mismo disolvente que en C0, pero puede ser de otro lote).
Casillas grises = pocillos exteriores, llenados con 200 μl de PBS.
Figura 2
Disposición de las placas de microvaloración de 96 pocillos para el precribado de antagonistas y la valoración del efecto antagonista.

C0 = disolvente del patrón de referencia;
C(1-8) = serie de diluciones (1-8, concentración de baja a elevada) del patrón de referencia.
TN = testigo negativo;
TP = testigo positivo;
TCx-(1-8) = diluciones (1-8, concentraciones de baja a elevada) del producto problema para la tanda de precribado y para la evaluación del efecto agonista del producto problema x.
CD = control del disolvente del producto problema (idealmente, el mismo disolvente que en C0, pero puede ser de otro lote).
CVe = control del vehículo (control del disolvente sin concentración fija del patrón de referencia de agonista, 17β-estradiol).
Casillas grises = pocillos exteriores, llenados con 200 μl de PBS.
Nota: Todos los pocillos interiores, excepto los pocillos de control del vehículo (CVe), también contendrán una concentración fija del patrón de referencia agonista, el 17β-estradiol (3,0 * 10-12 M).
Medición de la luminiscencia
|
39.
|
La medición de la luminiscencia se describe con detalle en el protocolo del ensayo de agonistas y antagonistas (10). Debe retirarse el medio de los pocillos, y las células deben lisarse tras 24 horas de incubación, a fin de abrir la membrana celular y permitir la medición de la actividad de la luciferasa.
|
|
40.
|
Para medir la luminiscencia, este procedimiento requiere un luminómetro equipado con dos inyectores. La reacción de la luciferasa comienza al inyectarse el sustrato luciferina. La reacción se detiene mediante la adición de NaOH 0,2 M. La reacción se detiene para evitar que pase luminiscencia de un pocillo al otro.
|
|
41.
|
La luz emitida de cada pocillo se expresa como unidades luminosas relativas (ULR) por pocillo.
|
Tanda de precribado
|
42.
|
Los resultados del análisis de precribado se utilizan para determinar un intervalo afinado de concentraciones de los productos problema para los ensayos completos. En el protocolo de ensayo de agonistas y antagonistas (10) se describe en profundidad la evaluación de los resultados del análisis de precribado y la determinación del intervalo afinado de concentraciones de los productos problema para los ensayos completos. Aquí se ofrece un breve resumen de los procedimientos para determinar el intervalo de concentraciones de productos problema para los ensayos de agonistas y de antagonistas. Véanse en los cuadros 5 y 6 las orientaciones para el diseño de la serie de diluciones.
|
Selección de las concentraciones para la evaluación de los efectos agonistas
|
43.
|
Durante la tanda de precribado, los productos problema deben someterse a ensayo en la serie de diluciones como se indica en los cuadros 5 (agonistas) y 6 (antagonistas). Todas las concentraciones deben ensayarse en pocillos por triplicado, siguiendo la disposición de la placa indicada en la figura 1 (agonistas) o 2 (antagonistas).
|
|
44.
|
Solo los resultados de los análisis que cumplen los criterios de aceptación (cuadro 3) se consideran válidos y pueden utilizarse para evaluar la respuesta de los productos problema. En caso de que una o varias placas de microvaloración en una serie de análisis no cumplan los criterios de aceptación, deberán volver a analizarse las correspondientes placas de microvaloración. En caso de que la primera placa que contenga la serie completa de diluciones del patrón de referencia no supere los criterios de aceptación, habrá que volver a analizar la serie completa de ensayos (6 placas).
|
|
45.
|
Los intervalos iniciales de concentración de los productos problema deben ajustarse y la tanda de precribado debe repetirse en caso de que:
|
—
|
se observe citotoxicidad. El procedimiento de precribado debe repetirse con concentraciones menores no citotóxicas del producto problema.
|
|
—
|
el precribado del producto problema no muestre una curva completa de dosis-respuesta, porque las concentraciones ensayadas generan la inducción máxima. La tanda de precribado debe repetirse con concentraciones menores del producto problema.
|
|
|
46.
|
Cuando se observe una respuesta válida relacionada con la dosis, se debe seleccionar la concentración (más baja) a la que se haya observado la inducción máxima sin presencia de citotoxicidad. La concentración máxima del producto problema que se va a someter a las tandas del ensayo completo debe ser 3 veces esta concentración seleccionada.
|
|
47.
|
Debe prepararse una serie completa de diluciones afinadas del producto problema con las fases de dilución indicadas en el cuadro 5, empezando por la mayor concentración anteriormente determinada.
|
|
48.
|
Los productos problema que no provoquen ningún efecto agonista deben someterse a ensayo en las tandas completas empezando por la mayor concentración no citotóxica que se haya identificado durante el precribado.
|
Selección de las concentraciones para la evaluación de los efectos antagonistas
|
49.
|
Solo los resultados de los análisis que cumplen los criterios de aceptación (cuadro 4) se consideran válidos y pueden utilizarse para evaluar la respuesta de los productos problema. En caso de que una o varias placas de microvaloración en una serie de análisis no cumplan los criterios de aceptación, deberán volver a analizarse las correspondientes placas de microvaloración. En caso de que la primera placa que contenga la serie completa de diluciones del patrón de referencia no supere los criterios de aceptación, habrá que volver a analizar la serie completa de ensayos (6 placas).
|
|
50.
|
Los intervalos iniciales de concentración de los productos problema deben ajustarse y la tanda de precribado debe repetirse en caso de que:
|
—
|
se observe citotoxicidad. El procedimiento de precribado debe repetirse con concentraciones menores no citotóxicas del producto problema.
|
|
—
|
el precribado del producto problema no muestre una curva completa de dosis-respuesta, porque las concentraciones ensayadas generan la inhibición máxima. El precribado debe repetirse con concentraciones menores del producto problema.
|
|
|
51.
|
Cuando se encuentre una respuesta válida relacionada con la dosis, se debe seleccionar la concentración (más baja) a la que se haya observado la inhibición máxima sin presencia de citotoxicidad. La concentración máxima del producto problema que se va a someter a las tandas del ensayo completo debe ser 3 veces esta concentración seleccionada.
|
|
52.
|
Debe prepararse una serie completa de diluciones afinadas del producto problema con las fases de dilución indicadas en el cuadro 6, empezando por la mayor concentración anteriormente determinada.
|
|
53.
|
Los productos problema que no provoquen ningún efecto antagonista deben someterse a ensayo en las tandas completas empezando por la mayor concentración no citotóxica que se haya identificado durante el precribado.
|
Tandas completas
|
54.
|
Tras la selección de los intervalos afinados de concentraciones, los productos problema deben someterse al ensayo completo en la serie de diluciones que se indica en los cuadros 5 (agonistas) y 6 (antagonistas). Todas las concentraciones deben ensayarse en pocillos por triplicado, siguiendo la disposición de la placa indicada en la figura 1 (agonistas) o 2 (antagonistas).
|
|
55.
|
Solo los resultados de los análisis que cumplan los criterios de aceptación (cuadros 3 y 4) se consideran válidos y pueden utilizarse para evaluar la respuesta de los productos problema. En caso de que una o varias placas de microvaloración en una serie de análisis no cumplan los criterios de aceptación, deberán volver a analizarse las correspondientes placas de microvaloración. En caso de que la primera placa que contenga la serie completa de diluciones del patrón de referencia no supere los criterios de aceptación, habrá que volver a analizar la serie completa de ensayos (6 placas).
|
Cuadro 5
Concentración y diluciones de los patrones de referencia, testigos y productos problema utilizados para los ensayos de agonistas
|
Concentración (M) del
|
Dilución del TCx
|
Dilución del TCx
|
Concentración (M) de los
|
|
17β-estradiol de referencia
|
en la tanda de precribado
|
en el ensayo completo
|
testigos
|
|
C0
|
0
|
TCx-1
|
10000000 x
|
TCx-1
|
3000 x
|
TP
|
3 * 10-6
|
|
C1
|
1 * 10-13
|
TCx-2
|
1000000 x
|
TCx-2
|
1000 x
|
TN
|
1 * 10-8
|
|
C2
|
3 * 10-13
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1 * 10-12
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
CD
|
0
|
|
C4
|
3 * 10-12
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6 * 10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1 * 10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3 * 10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1 * 10-10
|
|
|
|
|
|
|
|
TCx: producto problema x.
TP: testigo positivo (17α-metiltestosterona).
TN: testigo negativo (corticosterona).
C0: control del disolvente del patrón de referencia.
CD: control del disolvente del producto problema.
|
Cuadro 6
Concentración y diluciones de los patrones de referencia, testigos y productos problema utilizados para los ensayos de antagonistas
|
Concentración (M) del
|
Dilución del TCx
|
Dilución del TCx
|
Concentración (M) de los
|
|
tamoxifeno de referencia
|
en la tanda de precribado
|
en el ensayo completo
|
testigos
|
|
C0
|
0
|
TCx-1
|
10000000 x
|
TCx-1
|
3000 x
|
TP
|
1 * 10-9
|
|
C1
|
3 * 10-9
|
TCx-2
|
1000000 x
|
TCx-2
|
1000 x
|
TN
|
1 * 10-5
|
|
C2
|
1 * 10-8
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3 * 10-8
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
CD
|
0
|
|
C4
|
1 * 10-7
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3 * 10-7
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Concentración (M) del
|
|
C6
|
1 * 10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
agonista complementado
|
|
C7
|
3 * 10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-Estradiol
|
3 * 10-12
|
|
C8
|
1 * 10-5
|
|
|
|
|
|
|
|
TCx: producto problema x.
TP: testigo positivo (4-hidroxitamoxifeno).
TN: testigo negativo (resveratrol).
C0: control del disolvente del patrón de referencia.
CD: control del disolvente del producto problema.
CVe: control del vehículo (no contiene una concentración fija del patrón de referencia de los agonistas, el 17β-estradiol (3,0 * 10-12 M).
|
Recogida y análisis de los datos
|
56.
|
Tras las tandas de precribado y del ensayo completo, en el ensayo de agonistas deben determinarse los valores de CE10, CE50, CP10, CP50 e inducción máxima (TCxmáx) de un producto problema. En el ensayo de antagonistas deben calcularse los valores de CI20, CI50, CP80, CP50 e inducción mínima (TCxmín). En las figuras 3 (agonistas) y 4 (antagonistas), se ofrece una representación gráfica de estos parámetros. Los parámetros requeridos se calculan sobre la base de la inducción relativa de cada producto problema [respecto a la inducción máxima del patrón de referencia (= 100 %)]. Para la evaluación de los datos, debe utilizarse una regresión no lineal (pendiente variable, cuatro parámetros) con arreglo a la ecuación siguiente:

donde:
X = log. de la dosis o de la concentración
Y = respuesta [inducción relativa (%)]
Valor superior = inducción máxima (%)
Valor inferior = inducción mínima (%)
Log CE50 = log. de la concentración a la que se observa el 50 % de la respuesta máxima
Pendiente de Hill = factor de pendiente o pendiente de Hill.
|
|
57.
|
Los datos brutos del luminómetro, expresados como unidades luminosas relativas (ULR), deben transferirse a la hoja de cálculo del análisis de los datos diseñada para las tandas de precribado y de ensayo completo. Los datos brutos deben cumplir los criterios de aceptación que se indican en los cuadros 3A y 3B (agonistas) o 4A y 4B (antagonistas). En el caso de que los datos brutos cumplan los criterios de aceptación, se efectuarán las siguientes etapas de cálculo para determinar los parámetros requeridos:
|
Agonistas
|
—
|
Restar, de cada uno de los datos brutos del análisis de los patrones de referencia, el valor medio de ULR del control del disolvente del patrón de referencia.
|
|
—
|
Restar, de cada uno de los datos brutos del análisis de los productos problema, el valor medio de ULR del control del disolvente del producto problema.
|
|
—
|
Calcular la inducción relativa de cada concentración del patrón de referencia. Fijar en el 100 % la inducción de la concentración máxima del patrón de referencia.
|
|
—
|
Calcular la inducción relativa de cada concentración de producto problema en comparación con la concentración máxima del patrón de referencia, que es el 100 %.
|
|
—
|
Evaluar los resultados del análisis tras una regresión no lineal (pendiente variable, cuatro parámetros).
|
|
—
|
Determinar los valores CE50 y CE10 del patrón de referencia.
|
|
—
|
Determinar los valores CE50 y CE10 de los productos problema.
|
|
—
|
Determinar la inducción relativa máxima del producto problema (TCmáx).
|
|
—
|
Determinar los valores de CP10 y CP50 de los productos problema.
|
En relación con los productos problema, puede que no siempre se consiga una curva dosis-respuesta completa debido, por ejemplo, a problemas de citotoxicidad o de solubilidad. Por tanto, no es posible determinar los valores CE50, CE10 y TP50. En tal caso, solo pueden determinarse los valores de CP10 y TCmáx
Antagonistas
|
—
|
Restar, de cada uno de los datos brutos del análisis de los patrones de referencia, el valor medio de ULR de la mayor concentración del patrón de referencia.
|
|
—
|
Restar, de cada uno de los datos brutos del análisis de los productos problema, el valor medio de ULR de la mayor concentración del patrón de referencia.
|
|
—
|
Calcular la inducción relativa de cada concentración del patrón de referencia. Fijar en el 100 % la inducción de la concentración mínima del patrón de referencia.
|
|
—
|
Calcular la inducción relativa de cada concentración de producto problema en comparación con la concentración mínima del patrón de referencia, que es el 100 %.
|
|
—
|
Evaluar los resultados del análisis tras una regresión no lineal (pendiente variable, cuatro parámetros).
|
|
—
|
Determinar los valores de CI50 y CI20 del patrón de referencia.
|
|
—
|
Determinar los valores de CI50 y CI20 de los productos problema.
|
|
—
|
Determinar la inducción relativa mínima del producto problema (TCmín).
|
|
—
|
Determinar los valores de CP80 y CP50 de los productos problema.
|
Figura 3
Visión general de los parámetros determinados en el ensayo de agonistas

CE10 = concentración de una sustancia a la que se observa el 10 % de su respuesta máxima.
CE50 = concentración de una sustancia a la que se observa el 50 % de su respuesta máxima.
CP10 = concentración de un producto problema a la que su respuesta es igual a la CE10 del patrón de referencia.
CP50 = concentración de un producto problema a la que su respuesta es igual a la CE50 del patrón de referencia.
TCxmáx = inducción relativa máxima del producto problema.
Figura 4
Visión general de los parámetros determinados en el ensayo de antagonistas

CI20 = concentración de una sustancia a la que se observa el 80 % de su respuesta máxima (20 % de inhibición).
CI50 = concentración de una sustancia a la que se observa el 50 % de su respuesta máxima (50 % de inhibición).
TP80 = concentración de un producto problema a la que su respuesta es igual a la CI20 del patrón de referencia.
TP50 = concentración de un producto problema a la que su respuesta es igual a la CI50 del patrón de referencia.
TCxmín = inducción relativa mínima del producto problema.
En relación con los productos problema, puede que no siempre se consiga una curva dosis-respuesta completa debido, por ejemplo, a problemas de citotoxicidad o de solubilidad. Por tanto, no es posible determinar los valores CI50, CI20 y TP50. En tal caso, solo pueden determinarse los valores TP20 y TCmín.
|
58.
|
Los resultados deben basarse en dos (o tres) tandas independientes. Si dos tandas dan resultados comparables y, por lo tanto, reproducibles, no es necesario realizar una tercera tanda. Para ser aceptables, los resultados deben:
|
—
|
cumplir los criterios de aceptabilidad (véanse los criterios de aceptabilidad, puntos 14-22),
|
|
Criterios de interpretación de los datos
|
59.
|
Para interpretar los datos y tomar la decisión de si un producto problema se considera positivo o negativo, deben utilizarse los criterios siguientes:
|
En cada tanda del ensayo completo, se considera que un producto problema es positivo cuando:
|
1.
|
El valor de TCmáx es igual o superior al 10 % de la respuesta máxima del patrón de referencia (REF10).
|
|
2.
|
Al menos dos concentraciones consecutivas del producto problema son iguales o superiores a REF10.
|
|
|
En cada tanda del ensayo completo, se considera que un producto problema es negativo cuando:
|
1.
|
El valor de TCmáx no supera el 10 % de la respuesta máxima del patrón de referencia (REF10).
|
|
2.
|
Son iguales o superiores a REF10 menos de dos concentraciones del producto problema.
|
|
|
En cada tanda del ensayo completo, se considera que un producto problema es positivo cuando:
|
1.
|
El valor de TCmín es igual o inferior al 80 % de la respuesta máxima del patrón de referencia (REF80 = 20 % de inhibición).
|
|
2.
|
Al menos dos concentraciones consecutivas del producto problema son iguales o inferiores a REF80.
|
|
|
En cada tanda del ensayo completo, se considera que un producto problema es negativo cuando:
|
1.
|
El valor de TCmín es superior al 80 % de la respuesta máxima del patrón de referencia (REF80 = 20 % de inhibición).
|
|
2.
|
Son iguales o inferiores a REF80 menos de dos concentraciones del producto problema.
|
|
|
|
60.
|
Para caracterizar la potencia de la respuesta positiva de un producto problema, deben indicarse la magnitud del efecto (agonistas: TCmáx; antagonistas: TCmín) y la concentración a la que se produce el efecto (agonistas: CE10, CE50, CP10, CP50; antagonistas: CI20, CI50, TP80, TP50).
|
INFORME DEL ENSAYO
|
61.
|
Véase el punto 20 de la sección “COMPONENTES DEL ENSAYO ER TA”.
|
BIBLIOGRAFÍA
|
(1)
|
OCDE (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, and van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V and Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M and Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, and Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I, and Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, Países Bajos.
|
Apéndice 4.1
INSPECCIÓN VISUAL DE LA VIABILIDAD CELULAR
B.67. ENSAYO DE MUTACIÓN GÉNICA DE CÉLULAS DE MAMÍFERO IN VITRO UTILIZANDO EL GEN DE LA TIMIDINA-CINASA
INTRODUCCIÓN
|
1.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 490 de la OCDE (2016). Los métodos de ensayo se examinan y se revisan periódicamente a la luz del progreso científico, los cambios en las necesidades normativas y el bienestar animal. El ensayo con células de linfoma de ratón (MLA) y el ensayo con células TK6 utilizando el locus de la timidina-cinasa (TC) estaban originalmente incluidos en el método de ensayo B.17. Posteriormente, el grupo de trabajo de expertos MLA del Taller Internacional sobre Ensayos de Genotoxicidad (International Workshop for Genotoxicity Testing, IWGT) ha elaborado recomendaciones internacionalmente armonizadas en relación con los criterios de aceptación del ensayo y la interpretación de los datos respecto al ensayo MLA (1) (2) (3) (4) (5), y estas recomendaciones se incorporan en el presente y nuevo método de ensayo B.67. Este método de ensayo está escrito para el ensayo MLA y, dado que también utiliza el locus TK, para el ensayo TK6. Mientras que el MLA se ha utilizado ampliamente con fines normativos, el TK6 se ha utilizado mucho menos a menudo. Hay que señalar que, a pesar de la similitud entre los parámetros, las dos líneas celulares no son intercambiables y los programas normativos pueden expresar válidamente una preferencia por uno sobre el otro con vistas a un uso normativo particular. Por ejemplo, la validación del punto MLA demostró su adecuación para detectar no solo la mutación génica, sino también la capacidad de un producto problema para inducir una lesión cromosómica estructural. El presente método forma parte de una serie de métodos de ensayo sobre toxicología genética. La OCDE ha elaborado un documento que aporta información sucinta sobre los ensayos de toxicología genética y una síntesis de los recientes cambios aportados a las directrices de ensayo de la OCDE sobre toxicidad genética (6).
|
|
2.
|
El objetivo de los ensayos de mutación génica con células de mamífero in vitro es detectar las mutaciones génicas inducidas por los productos. Las líneas celulares utilizadas en estos ensayos miden las mutaciones directas en genes marcadores y, en concreto, en el gen endógeno de la timidina-cinasa (TK en el caso de las células humanas y Tk en el caso de las células de roedores, denominadas conjuntamente TK en el presente método de ensayo). Este método de ensayo está diseñado para su uso con dos líneas celulares: la línea celular de linfoma de ratón L5178Y TK+/--3.7.2C (generalmente denominada L5178Y) y la línea celular linfoblastoide humana TK6 (generalmente denominada TK6). Aunque las dos líneas celulares varían debido a su origen, crecimiento celular, situación de p53, etc., los ensayos de mutación génica TK pueden realizarse de forma similar con ambos tipos de células, tal como se describe en el presente método de ensayo.
|
|
3.
|
La naturaleza autosómica y heterocigótica del gen de la timidina-cinasa permite detectar las colonias viables cuyas células sean deficientes en la enzima timidina-cinasa tras la mutación de TK+/-
a TK-/-
. Esta deficiencia puede deberse a fenómenos genéticos que afecten al gen TK, incluidas las mutaciones génicas (mutaciones puntuales, mutaciones de desplazamiento del marco de lectura, pequeñas supresiones, etc.) y los fenómenos cromosómicos (supresiones grandes, reorganizaciones cromosómicas y recombinación mitótica). Estos últimos fenómenos se expresan como pérdida de heterocigosis, que es un cambio genético común de los genes supresores de tumores en la oncogénesis humana. En teoría, con el MLA puede detectarse la pérdida de todo el cromosoma portador del gen TK que resulta de la alteración del huso o de la ausencia de disyunción mitótica. De hecho, una combinación de análisis citogenético y molecular muestra claramente que algunos mutantes TK según el MLA son el resultado de una ausencia de disyunción. Sin embargo, la ponderación de las pruebas muestra que los ensayos de mutación del gen TK no pueden detectar con fiabilidad los anéugenos cuando se aplican los criterios de citotoxicidad normales (tal como se describen en el presente método de ensayo) y, por tanto, no es adecuado utilizar estos ensayos para detectar anéugenos (7) (8) (9).
|
|
4.
|
En las pruebas de mutación del gen TK, se generan dos clases fenotípicas distintas de mutantes TK; los mutantes de crecimiento normal, que crecen al mismo ritmo que las células heterocigóticas TK, y los mutantes de crecimiento lento, que crecen con tiempos de generación prolongados. Los mutantes de crecimiento normal y de crecimiento lento se reconocen como mutantes de colonias grandes y de colonias pequeñas en el análisis MLA y como mutantes de colonias de aparición temprana y de aparición tardía en el ensayo TK6. Debe estudiarse detenidamente la naturaleza molecular y citogenética de los mutantes MLA tanto de colonias grandes como de colonias pequeñas (8) (10) (11) (12) (13). También debe estudiarse a fondo la naturaleza molecular y citogenética de los mutantes TK6 tanto de aparición temprana como de aparición tardía (14) (15) (16) (17). Los mutantes de crecimiento lento de ambos tipos de células han sufrido lesiones genéticas que afectan al posible gen o genes de regulación del crecimiento cerca del locus TK, lo que da lugar a tiempos de generación prolongados y a la formación de colonias pequeñas o de aparición tardía (18). La inducción de mutantes de crecimiento lento se ha asociado con productos que inducen cambios estructurales macroscópicos a nivel cromosómico. Las células cuyas lesiones no afectan al posible gen o genes de regulación del crecimiento cerca del locus TK crecen a ritmos similares a los de las células parentales y se convierten en mutantes de crecimiento normal. La inducción de mutantes de crecimiento básicamente normal se asocia con productos que actúan fundamentalmente como mutágenos puntuales. Por consiguiente, es esencial recontar tanto los mutantes de crecimiento lento como los mutantes de crecimiento normal, a fin de aislar todos los mutantes y hacerse una idea del tipo o tipos de lesiones (mutágenos frente a clastógenos) inducidas por el producto problema (10) (12) (18) (19).
|
|
5.
|
El método de ensayo se organiza de manera que proporcione información general aplicable tanto al ensayo MLA como al TK6, así como orientaciones especializadas para los distintos ensayos.
|
|
6.
|
En el apéndice 1 se dan las definiciones utilizadas.
|
CONSIDERACIONES INICIALES Y LIMITACIONES
|
7.
|
Los ensayos efectuados in vitro requieren generalmente el uso de una fuente exógena de activación metabólica. El sistema exógeno de activación metabólica no reproduce completamente las condiciones in vivo.
|
|
8.
|
Hay que procurar evitar las condiciones que puedan llevar a la producción de resultados positivos que sean artefactos (es decir, debidos a la posible interacción con el sistema de ensayo), no causados por la interacción entre el producto problema y el material genético de la célula; pueden mencionarse entre tales condiciones los cambios de pH o de osmolalidad, la interacción con los componentes del medio (20) (21), o unos niveles excesivos de citotoxicidad (22) (23) (24). Se considera excesiva para el ensayo MLA y el TK6 la citotoxicidad que supere los niveles máximos recomendados de citotoxicidad que se definen en el punto 28. Además, debe tenerse en cuenta que los productos problema que sean análogos de la timidina, o se comporten como tales, pueden aumentar la frecuencia de la aparición de mutantes mediante un crecimiento selectivo de los mutantes espontáneos de fondo durante el tratamiento celular y exigir métodos de ensayo adicionales para una evaluación adecuada (25).
|
|
9.
|
En caso de nanomateriales fabricados, puede ser necesario recurrir a adaptaciones específicas del presente método de ensayo, pero estas no se describen aquí.
|
|
10.
|
Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines normativos, debe considerarse si podría proporcionar resultados adecuados a tales fines y, en caso afirmativo, por qué. Dichas consideraciones no son necesarias cuando los requisitos normativos estipulan que la mezcla debe someterse a ensayo.
|
|
11.
|
Las células mutantes deficientes en actividad enzimática de timidina-cinasa a causa de una mutación TK
+/- a TK
-/- son resistentes a los efectos citostáticos de la trifluorotimidina (TFT), análogo de la pirimidina. Las células capaces de producir timidina-cinasa son sensibles a la TFT, lo que inhibe el metabolismo celular y detiene la división celular. Así pues, las células mutantes son capaces de proliferar en presencia de TFT y forman colonias visibles, al contrario que las células que contienen la enzima TK.
|
PRINCIPIO DEL ENSAYO
|
12.
|
Se exponen al producto problema células en suspensión, tanto en presencia como en ausencia de una fuente exógena de activación metabólica (véase el punto 19), durante un plazo adecuado (véase el punto 33), y después se subcultivan para determinar la citotoxicidad y permitir la expresión fenotípica antes de la selección de los mutantes. La citotoxicidad se determina mediante el crecimiento relativo total (CRT, véase el punto 25) en el MLA y mediante la supervivencia relativa (SR, véase el punto 26) en el TK6. Los cultivos tratados se mantienen en un medio de crecimiento durante un período de tiempo suficiente y específico para cada tipo celular (véase el punto 37), de manera que la expresión fenotípica de las mutaciones inducidas sea casi óptima. Tras la expresión fenotípica, la frecuencia de mutantes se determina sembrando un número conocido de células en un medio con el agente selectivo para detectar las colonias de mutantes, y en otro medio sin dicho agente para determinar la eficiencia de clonación (viabilidad). Tras un período de incubación adecuado, se cuentan las colonias. La frecuencia de mutantes se calcula en función del número de colonias de mutantes, corregido para tener en cuenta la eficiencia de clonación, en el momento de la selección de los mutantes.
|
DESCRIPCIÓN DEL MÉTODO
Preparaciones
Células
|
13.
|
En cuanto al ensayo MLA: Dado que el ensayo MLA se ha desarrollado y caracterizado a partir de la sublínea TK
+/- -3.7.2C de las células L5178Y, esta es la sublínea específica que debe utilizarse en el ensayo MLA. La línea celular L5178Y se ha obtenido a partir de un linfoma de timo inducido con metilcolantreno en un ratón DBA-2 (26). Clive y colaboradores trataron células L5178Y (designadas por Clive como TK+/+ -3) con sulfonato de etilmetanoy aislaron un clon TK-/- (designado como TK-/- -3.7) utilizando bromodesoxiuridina como agente selectivo. A partir del clon TK-/- se aislaron un clon TK+/- espontáneo (designado como TK+/- -3.7.2.) y un subclón (designado como TK+/--3.7.2), que se caracterizaron para utilizarse en el ensayo MLA (27). Se ha publicado el cariotipo de la línea celular (28) (29) (30) (31). El número modal de cromosomas es 40. Hay un cromosoma metacéntrico (t12;13) que debe contarse como un solo cromosoma. El locus TK del ratón se encuentra en el extremo distal del cromosoma 11. La línea celular L5178Y TK
+/- -3.7.2C tiene mutaciones en los dos alelos p53 y produce proteína p53 mutante (32) (33). Es probable que la situación de la línea celular TK+/--3.7.2C en cuanto a la proteína p53 sea responsable de la capacidad del ensayo para detectar lesones a gran escala (17).
|
|
14.
|
En cuanto al ensayo TK6: TK6 es una línea celular linfoblastoide humana. La línea celular original es una línea celular transformada con virus de Epstein-Barr, WI-L2, que inicialmente se obtuvo de un varón de 5 años con esferocitosis hereditaria. El primer clon aislado, el HH4, fue sometido a tratamiento mutagénico con CIR191 y se generó una línea celular TK heterocigótica, la TK6 (34). Las células TK6 son casi diploides y el cariotipo representativo es 47, XY, 13 +, t(14; 20), t(3; 21) (35). El locus TK del hombre se encuentra en el brazo largo del cromosoma 17. La línea celular TK6 es competente respecto a p53 porque tiene una secuencia p53 de tipo natural en ambos alelos y expresa únicamente la proteína p53 de tipo natural (36).
|
|
15.
|
Tanto en el ensayo MLA como en el TK6, cuando se establezca por primera vez o se reponga un cultivo madre, es aconsejable que el laboratorio de ensayo garantice la ausencia de contaminación por Mycoplasma, establezca el cariotipo de las células o marque los cromosomas que albergan el locus TK, y compruebe los tiempos de duplicación de la población. Debe determinarse la duración del ciclo celular normal de las células utilizadas en el laboratorio de ensayo, la cual debe ser coherente con las características celulares publicadas (16) (19) (37). Este cultivo madre debe conservarse a una temperatura igual o inferior a -150 °C y utilizarse para preparar todos los cultivos celulares de trabajo.
|
|
16.
|
Antes de establecer un gran número de cultivos de trabajo crioconservados o justo antes de su utilización en un experimento, puede ser necesario suprimir de los cultivos las células mutantes preexistentes [a menos que la frecuencia de los mutantes (FM) del control del disolvente esté ya dentro del intervalo aceptable (véase el cuadro 2 en relación con el MLA)]. Esto se consigue utilizando metotrexato (aminopterina) para excluir las células deficientes en cuanto al TK y añadiendo timidina, hipoxantina y glicina (L5178Y) o 2’-desoxicitidina (TK6) al cultivo para garantizar un crecimiento óptimo de las células competentes en cuanto al TK (19) (38) (39), y (40) respecto a las células TK6. En las referencias (19) (31) (37) (39) (41) se ofrece asesoramiento general sobre buenas prácticas para el mantenimiento de los cultivos celulares, así como asesoramiento específico sobre las células L5178Y y TK6. Para los laboratorios que necesiten cultivos celulares madre para iniciar el ensayo MLA o el TK6, o bien para obtener nuevos cultivos celulares madre, se dispone de un depósito celular de células bien caracterizadas (37).
|
Medios y condiciones de cultivo
|
17.
|
En ambos ensayos, para el mantenimiento de los cultivos deben utilizarse medios de cultivo y condiciones de incubación adecuados (es decir, recipientes de cultivo, atmósfera humidificada con 5 % de CO2, y temperatura de incubación de 37 °C). Los cultivos celulares deben mantenerse siempre en condiciones que garanticen que se encuentran en la fase logarítmica de crecimiento. Es especialmente importante elegir medios y condiciones de cultivo que garanticen un crecimiento celular óptimo durante el período de expresión y clonación de las células, tanto mutantes como no mutantes. En los ensayos MLA y TK6, también es importante que las condiciones de cultivo aseguren un crecimiento óptimo de los mutantes TK tanto de colonias grandes / aparición temprana como de colonias pequeñas / aparición tardía. En (19) (31) (38) (39) (40) (42) se pueden encontrar más precisiones sobre los cultivos, incluida la necesidad de calentar convenientemente el suero de caballo para inactivarlo, si se utiliza el medio RPMI durante la selección de mutantes.
|
Preparación de los cultivos
|
18.
|
Las células se propagan a partir de cultivos madre, y se siembran en medio de cultivo a una densidad tal que los cultivos en suspensión sigan creciendo exponencialmente a lo largo de los períodos de tratamiento y de expresión.
|
Activación metabólica
|
19.
|
Se debe recurrir a sistemas exógenos de metabolización cuando se utilicen células L5178Y y TK6 porque su capacidad metabólica endógena es inadecuada. El sistema utilizado con más frecuencia que se recomienda por defecto, salvo en casos justificados, es una fracción postmitocondrial (S9) a la que se añaden cofactores y que se obtiene a partir del hígado de roedores (generalmente ratas) tratados con inductores enzimáticos como el aroclor 1254 (43) (44) (45) o una combinación de fenobarbital y β-naftoflavona (46) (47) (48) (49) (50) (51). Esta última combinación no infringe el Convenio de Estocolmo sobre contaminantes orgánicos persistentes(52), y se ha visto que es tan efectiva como el aroclor 1254 para inducir oxidasas de función mixta (45) (46) (47) (48) (49). La fracción S9 se utiliza normalmente a concentraciones que varían entre el 1 y el 2 %, pero que pueden aumentarse hasta el 10 % (v/v) en el medio de ensayo final. La elección del tipo y de la concentración del sistema exógeno de activación metabólica o del inductor metabólico utilizado puede estar influida por la clase de los productos problema.
|
Preparación del producto problema
|
20.
|
Los productos problema sólidos deben prepararse en disolventes adecuados y, si es conveniente, diluirse antes de tratar las células (véase el punto 21). Los productos problema líquidos pueden añadirse directamente al sistema de ensayo o diluirse antes del tratamiento del sistema de ensayo. Los productos problema gaseosos o volátiles deben someterse a ensayo aplicando modificaciones adecuadas a los protocolos normales, tales como el tratamiento en recipientes de cultivo sellados (53) (54) (55). La preparación del producto problema debe hacerse justo antes del tratamiento, salvo que se cuente con datos de estabilidad que avalen la posibilidad de su conservación.
|
CONDICIONES DE ENSAYO
Disolventes
|
21.
|
El disolvente debe elegirse para optimizar la solubilidad del producto problema sin tener un impacto negativo en la realización del ensayo, por ejemplo por cambiar el crecimiento celular, afectar a la integridad del producto problema, reaccionar con los recipientes de cultivo, o interferir con el sistema de activación metabólica. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente (o medio de cultivo) acuoso. Son disolventes bien establecidos el agua o el dimetilsulfóxido. En general, los disolventes orgánicos no deben exceder del 1 % (v/v) y los disolventes acuosos (solución salina o agua) no deben superar el 10 % (v/v) en el medio de tratamiento final. Si se utilizan disolventes distintos de los que están bien establecidos (por ejemplo, etanol o acetona), debe disponerse de datos justificativos que indiquen su compatibilidad con el sistema de ensayo y los productos problema, y su ausencia de toxicidad genética a la concentración utilizada. En ausencia de estos datos justificativos, es importante utilizar testigos sin tratar (véanse las definiciones en el apéndice 1) para demostrar que el disolvente elegido no es nocivo ni induce efectos mutagénicos.
|
MEDICIÓN DE LA CITOTOXICIDAD Y SELECCIÓN DE LAS CONCENTRACIONES DE TRATAMIENTO
|
22.
|
Al determinar la concentración máxima de producto problema, debe evitarse llegar a concentraciones que puedan producir respuestas positivas que sean artefactos, tales como las que provocan una citotoxicidad excesiva (véase el punto 28), precipitación en el medio de cultivo (véase el punto 29), o cambios marcados del pH o de la osmolalidad (véase el punto 8). Si el producto problema provoca un cambio marcado en el pH del medio en el momento de su adición, el pH puede ajustarse amortiguando el medio de tratamiento final para evitar resultados positivos que sean artefactos y mantener unas condiciones de cultivo adecuadas.
|
|
23.
|
La selección de las concentraciones se basa en la citotoxicidad y en otras consideraciones (véanse los puntos 27-30). Si bien la evaluación de la citotoxicidad en un ensayo inicial puede ser útil para definir mejor las concentraciones que deben utilizarse en el experimento principal, no es obligatorio efectuar ese ensayo inicial. Incluso aunque se realice una evaluación inicial de la citotoxicidad, sigue siendo necesario medir la citotoxicidad en cada cultivo del experimento principal. Si se lleva a cabo un experimento de determinación del intervalo, debe abarcar un amplio intervalo de concentraciones y puede finalizarse el día 1 después del tratamiento o prolongarse durante los dos días de la expresión y hasta la selección de mutantes (si se ve que las concentraciones utilizadas son las adecuadas).
|
|
24.
|
La citotoxicidad se debe determinar en relación con cada cultivo de ensayo y con cada cultivo testigo: los métodos del ensayo MLA (2) y del TK6 (15) están definidos por prácticas acordadas a nivel internacional.
|
|
25.
|
En relación con las versiones del MLA tanto en agar como en micropocillos: la citotoxicidad debe evaluarse utilizando el crecimiento relativo total (CRT), definido originalmente por Clive y Spector en 1975 (2). Esta medida incluye el crecimiento relativo en la suspensión (CRS: cultivo de ensayo frente al control del disolvente) durante el tratamiento celular, el tiempo de expresión y la eficacia relativa de clonación (ERC: cultivo de ensayo frente al control del disolvente) en el momento en que se seleccionan los mutantes (2). Cabe señalar que el CRS incluye cualquier pérdida de células que se produzca en el cultivo de ensayo durante el tratamiento (véanse las fórmulas en el apéndice 2).
|
|
26.
|
En cuanto al ensayo TK6: la citotoxicidad debe evaluarse utilizando la supervivencia relativa (SR), es decir, la eficiencia de clonación de las células sembradas inmediatamente después del tratamiento, ajustada para tener en cuenta las eventuales pérdidas de células durante el tratamiento, sobre la base del recuento celular, frente a la del testigo negativo (al que se asigna una supervivencia del 100 %) (véase la fórmula en el apéndice 2).
|
|
27.
|
Deben evaluarse al menos cuatro concentraciones de ensayo (sin incluir el testigo positivo ni el control del disolvente) que cumplan los criterios de aceptabilidad (citotoxicidad apropiada, número de células, etc.). Si bien es aconsejable utilizar cultivos duplicados, a cada concentración estudiada podrán utilizarse cultivos tratados bien replicados o bien únicos. Los resultados obtenidos en los cultivos replicados a una concentración determinada deben comunicarse por separado, pero pueden agruparse para el análisis de datos (55). En el caso de productos problema que muestren escasa o nula citotoxicidad, normalmente serán adecuados los intervalos de concentración de aproximadamente el doble o el triple. Cuando se produce citotoxicidad, deben seleccionarse las concentraciones de ensayo para cubrir el intervalo de citotoxicidad a partir de la concentración que provoca citotoxicidad según se describe en el punto 28, con inclusión en particular de las concentraciones a las que existe citotoxicidad moderada y débil o nula. Muchos productos problema presentan curvas concentración-respuesta de elevada pendiente y, a fin de abarcar toda la gama de citotoxicidad o de estudiar la relación concentración-respuesta en detalle, puede ser necesario utilizar concentraciones más próximas entre sí y en un número superior a cuatro, en particular en situaciones en las que se requiera repetir un experimento (véase el punto 70). La utilización de más de cuatro concentraciones puede ser especialmente importante si se utilizan cultivos únicos.
|
|
28.
|
Si la concentración máxima se basa en la citotoxicidad, la concentración más alta debe aspirar a proporcionar entre el 20 y el 10 % de CRT en el caso del MGA, y entre el 20 y el 10 % de SR en el caso del TK6 (punto 67).
|
|
29.
|
Con productos problema poco solubles que no son citotóxicos a concentraciones inferiores a la concentración mínima insoluble, la mayor concentración analizada debe producir turbidez o precipitado visibles a simple vista o con ayuda de un microscopio invertido al final del tratamiento con el producto problema. Incluso si se produce citotoxicidad por encima del límite de solubilidad, es aconsejable hacer el ensayo a una única concentración que produzca turbidez o precipitado visible porque este puede provocar efectos falsos. Dado que los ensayos MLA y TK6 utilizan cultivos en suspensión, hay que tener especial cuidado para que el precipitado no interfiera con la realización del ensayo. Puede ser útil también determinar la solubilidad en el medio de cultivo antes de efectuar el experimento.
|
|
30.
|
Si no se observa precipitado ni citotoxicidad limitante, la concentración de ensayo más elevada debe corresponder a la más baja de las siguientes: 10 mM, 2 mg/ml o 2 μl/ml (57) (58). Cuando el producto problema no tenga una composición definida y se trate, p. ej., de una sustancia de composición desconocida o variable, de productos complejos de reacción o de materiales biológicos [es decir, sustancias de composición desconocida o variable (UVCB)], extractos medioambientales, etc., es posible que la concentración superior tenga que ser mayor (p. ej., 5 mg/ml), en ausencia de citotoxicidad suficiente, para aumentar la concentración de cada uno de los componentes. Conviene señalar, no obstante, que estos requisitos pueden variar en caso de medicamentos de uso humano (59).
|
Testigos
|
31.
|
Se deben incluir, con cada condición experimental, testigos negativos en paralelo (véase el punto 21), consistentes en el disolvente solo en el medio de tratamiento y manipulados de la misma manera que los cultivos tratados.
|
|
32.
|
Es necesario disponer de testigos positivos en paralelo a fin de demostrar la capacidad del laboratorio para detectar mutágenos en las condiciones establecidas en el protocolo de ensayo utilizado, la efectividad del sistema exógeno de activación metabólica (si procede), y la detección adecuada de los mutantes TK tanto grandes / de aparición temprana como pequeños / de aparición tardía. En el cuadro 1 a continuación se encuentran ejemplos de testigos positivos. Es posible utilizar como testigos positivos otras sustancias, si se justifica. Debido a que los ensayos de toxicidad genética con células de mamífero in vitro están suficientemente normalizados respecto a los tratamientos a corto plazo (3-4 horas) realizados en paralelo, con y sin activación metabólica, utilizando la misma duración del tratamiento, el uso de testigos positivos puede limitarse a un mutágeno que requiera activación metabólica. En este caso, una respuesta de este único testigo positivo demostrará tanto la actividad del sistema de activación metabólica como la sensibilidad del sistema de ensayo. Si se utilizan, los tratamientos a largo plazo (es decir, 24 horas sin fracción S9) deben tener, no obstante, su propio testigo positivo, ya que la duración del tratamiento será diferente de la del ensayo con activación metabólica. Cada testigo positivo debe utilizarse a una o varias concentraciones de las que se espere que den un aumento reproducible y detectable respecto al nivel de fondo, a fin de demostrar la sensibilidad del sistema de ensayo, y la respuesta no debe ponerse en peligro por una citotoxicidad que supere los límites especificados en el presente método de ensayo (véase el punto 28).
|
Cuadro 1
Sustancias de referencia recomendadas para evaluar la competencia del laboratorio, y para la selección de los testigos positivos
|
Categoría
|
Sustancia
|
N.o CAS
|
|
1. Mutágenos activos sin activación metabólica
|
|
Metanosulfonato de metilo
|
66-27-3
|
|
Mitomicina C
|
50-07-7
|
|
N-Óxido de 4-nitroquinolina
|
56-57-5
|
|
2. Mutágenos que necesitan activación metabólica
|
|
Benzo(a)pireno
|
50-32-8
|
|
Ciclofosfamida (monohidrato)
|
50-18-0 (6055-19-2)
|
|
7,12-Dimetilbenzoantraceno
|
57-97-6
|
|
3-Metilcolantreno
|
56-49-5
|
PROCEDIMIENTO
Tratamiento con el producto problema
|
33.
|
Se tratan células en crecimiento con el producto problema en presencia y en ausencia de un sistema de activación metabólica. La exposición debe durar un plazo de tiempo adecuado (generalmente de 3 a 4 horas). Conviene señalar, no obstante, que estos requisitos pueden variar en caso de medicamentos de uso humano (59). En relación con el ensayo MLA, en los casos en que el tratamiento a corto plazo dé resultados negativos, y se disponga de información que sugiera la necesidad de un tratamiento más prolongado [por ejemplo, análogos de nucleósidos, productos poco solubles (5) (59)], debe prestarse atención a la realización del ensayo con un tratamiento más largo, es decir, 24 horas sin S9.
|
|
34.
|
El número mínimo de células utilizadas para cada cultivo del ensayo (testigo y tratado) en cada fase del ensayo debe basarse en la frecuencia de mutantes espontáneos. Una orientación general consiste en tratar y resembrar suficientes células de cada cultivo experimental de forma que se mantengan al menos 10 mutaciones espontáneas, aunque lo ideal es que sean 100, en todas las fases del ensayo (tratamiento, expresión fenotípica y selección de mutantes) (56).
|
|
35.
|
Para el ensayo MLA, la frecuencia de mutantes espontáneos aceptable recomendada se sitúa entre 35 y 140 × 10-6 (versión en agar) y entre 50 y 170 × 10-6 (versión en micropocillos) (véase el cuadro 2). Para tener como mínimo 10, e idealmente 100, mutantes espontáneos que sobrevivan al tratamiento en cada cultivo de ensayo, es necesario tratar al menos 6 × 106 células. El tratamiento de este número de células, y el mantenimiento de células suficientes durante la expresión y la clonación para la selección de mutantes, proporciona un número suficiente de mutantes espontáneos (10 o más) en todas las fases del experimento, incluso en el caso de los cultivos tratados con concentraciones que dan lugar a una citotoxicidad del 90 % (medida por un CRT del 10 %) (19) (38) (39).
|
|
36.
|
En el caso del ensayo TK6, la frecuencia de mutantes espontáneos suele estar entre 2 y 10 × 10-6. Para tener como mínimo 10 mutantes espontáneos que sobrevivan al tratamiento en cada cultivo, es necesario tratar al menos 20 × 106 células. El tratamiento de este número de células ofrece un número suficiente de mutaciones espontáneas (10 o más), incluso en el caso de los cultivos tratados con concentraciones que provocan una citotoxicidad del 90 % durante el tratamiento (10 % de SR). Además, ha de disponerse de un número suficiente de células para cultivarlas durante el período de expresión y para sembrarlas a efectos de la selección de mutantes (60).
|
Período de expresión fenotípica y medición de la citotoxicidad y de la frecuencia de mutantes
|
37.
|
Al final del período de tratamiento, las células se cultivan durante un tiempo determinado para permitir una expresión fenotípica casi óptima de los mutantes recién inducidos; este tiempo es específico de cada línea celular. En el ensayo MLA, el período de expresión fenotípica es de 2 días. En el ensayo TK6, el período de expresión fenotípica es de 3-4 días. Si se utiliza un tratamiento de 24 horas, el período de expresión comienza una vez finalizado el tratamiento.
|
|
38.
|
Durante el período de expresión fenotípica, las células se enumeran diariamente. En el caso del MLA se utilizan los recuentos celulares diarios para calcular el crecimiento diario en la suspensión (CS). Tras el período de expresión de 2 días, las células se suspenden en medio con y sin agente selectivo para determinar el número de mutantes (placas de selección) y la eficiencia de clonación (placas de viabilidad), respectivamente. En el caso del ensayo MLA, existen dos métodos igualmente aceptables para la clonación de la selección de mutantes; uno con agar blando y el otro con medio líquido en placas de 96 pocillos (19) (38) (39). La clonación en el ensayo TK6 se realiza utilizando medios líquidos y placas de 96 pocillos (16).
|
|
39.
|
La trifluorotimidina (TFT) es el único agente selectivo recomendado para los mutantes TK (61).
|
|
40.
|
En el MLA, las placas de agar y las placas de micropocillos se cuentan después de 10-12 días de incubación. En el TK6, las colonias de las placas de micropocillos se examinan después de 10 o 14 días en cuanto a la presencia de mutantes de aparición temprana. Con el fin de recuperar los mutantes TK6 de crecimiento lento (aparición tardía), es necesario volver a aportar a las células medio de cultivo y TFT después de haber recontado los mutantes de aparición temprana y, a continuación, incubar las placas durante un período adicional de 7-10 días (62). Véase en los puntos 42 y 44 una discusión del recuento de los mutantes TK de crecimiento lento y normal.
|
|
41.
|
Los cálculos apropiados para los dos ensayos, incluidos los dos métodos (agar y micropocillos) del ensayo MLA, figuran en el apéndice 2. En el método con agar del MLA, se recuentan las colonias y se ajusta el número de colonias mutantes en función de la eficiencia de clonación para calcular la FM. En la versión con micropocillos del MLA y en el TK6, la eficiencia de clonación en las placas tanto de selección como de eficiencia de clonación se determina con arreglo a la distribución de Poisson (63). La FM se calcula a partir de estas dos eficiencias de clonación.
|
Caracterización de las colonias de mutantes
|
42.
|
En el caso del MLA, si el producto problema es positivo (véanse los puntos 62-63), debe realizarse la caracterización de las colonias en función de su tamaño o crecimiento al menos en uno de los cultivos de ensayo (en general, a la concentración positiva más alta aceptable) y en los testigos positivos y negativos. Si el producto problema es negativo (véase el punto 64), debe procederse a la caracterización de las colonias de mutantes en los testigos positivos y negativos. En el método de micropocillos del MLA se definen los mutantes de colonias pequeñas como aquellos que cubren menos del 25 % del diámetro del pocillo y los mutantes de colonias grandes como los que cubren más del 25 % del diámetro del pocillo. En el caso del método de agar, se utiliza un contador automático de colonias para enumerar las colonias mutantes y para determinar su tamaño. En la bibliografía se detallan los enfoques relativos al tamaño de las colonias (19) (38) (40). La caracterización de las colonias en los testigos positivos y negativos es necesaria para demostrar que los estudios se llevan a cabo adecuadamente.
|
|
43.
|
No puede determinarse que el producto problema sea negativo si no se detectan adecuadamente en el testigo positivo tanto las colonias grandes como las pequeñas. La caracterización de las colonias puede utilizarse para proporcionar información general sobre la capacidad del producto problema para causar mutaciones puntuales o fenómenos cromosómicos (punto 4).
|
|
44.
|
TK6: los mutantes de crecimiento normal y los de crecimiento lento se diferencian por una variación en el tiempo de incubación (véase el punto 40). En el caso del TK6, en general los mutantes de aparición temprana y tardía se puntúan en relación con todos los cultivos, incluidos los testigos positivos y negativos. La caracterización de las colonias en los testigos positivos y negativos es necesaria para demostrar que los estudios se llevan a cabo adecuadamente. No puede determinarse que el producto problema sea negativo si no se detectan adecuadamente en el testigo positivo tanto los mutantes de aparición temprana como los mutantes de aparición tardía. La caracterización de las colonias puede utilizarse para proporcionar información general sobre la capacidad del producto problema para causar mutaciones puntuales o fenómenos cromosómicos (punto 4).
|
Competencia del laboratorio
|
45.
|
Con el fin de demostrar la suficiente experiencia con el ensayo antes de utilizarlo para los ensayos sistemáticos, el laboratorio debe haber efectuado una serie de experimentos con sustancias positivas de referencia que actúen a través de mecanismos diferentes (como mínimo, una activa con activación metabólica y otra activa sin ella, seleccionadas de entre las sustancias enumeradas en el cuadro 1) y con diversos testigos negativos (incluyendo cultivos sin tratar y diferentes disolventes o vehículos). Las respuestas obtenidas con estos testigos positivos y negativos deben ser coherentes con la bibliografía. Este requisito no es aplicable a los laboratorios que tienen experiencia, esto es, que disponen de una base de datos históricos, según se define en los puntos 47-50. En el caso del ensayo MLA, los valores obtenidos con los testigos tanto positivos como negativos deben ser coherentes con las recomendaciones del IWGT (véase el cuadro 2).
|
|
46.
|
Debe investigarse una selección de sustancias testigo positivo (véase el cuadro 1) con tratamientos cortos y largos, en ausencia de activación metabólica, y también con tratamientos cortos en presencia de activación metabólica, para demostrar la capacidad de detectar productos mutágenos, para determinar la efectividad del sistema de activación metabólica y para demostrar la adecuación de las condiciones de crecimiento celular durante el tratamiento, la expresión fenotípica y la selección de mutantes, así como la de los procedimientos de examen. Debe elegirse una gama de concentraciones de las sustancias seleccionadas de forma que produzcan aumentos sobre el nivel de fondo relacionados con la concentración y reproducibles, para demostrar la sensibilidad y el intervalo dinámico del sistema de ensayo.
|
Datos sobre testigos históricos
|
47.
|
El laboratorio debe determinar:
|
—
|
un intervalo y una distribución de los testigos positivos históricos,
|
|
—
|
un intervalo y una distribución de los testigos negativos (sin tratar, disolventes) históricos.
|
|
|
48.
|
Cuando se obtengan datos por primera vez en relación con la distribución de testigos negativos históricos, los testigos negativos en paralelo deben ser coherentes con los datos publicados de los testigos negativos. Según se añadan más datos experimentales sobre la distribución de los testigos, los testigos negativos en paralelo deben situarse idealmente dentro de los límites de control del 95 % de dicha distribución (64) (65).
|
|
49.
|
La base de datos de testigos negativos históricos del laboratorio debe constituirse en un principio con un mínimo de 10 experimentos, pero preferiblemente con al menos 20 experimentos realizados en condiciones experimentales comparables. Los laboratorios deben utilizar métodos de control de calidad, como gráficos de control [por ejemplo, gráficos C o gráficos de medias (65)], con el fin de determinar la variabilidad de sus datos sobre los testigos positivos y negativos, y de demostrar que la metodología está “controlada” en su laboratorio (66). En la bibliografía se encuentran más datos y recomendaciones sobre cómo conseguir y utilizar los datos históricos (64).
|
|
50.
|
Los datos de los testigos negativos deben consistir en frecuencias de mutantes procedentes de cultivos únicos o preferiblemente replicados, tal como se describe en el punto 27. Lo ideal sería que los testigos negativos en paralelo estuvieran dentro de los límites de control del 95 % de la distribución de la base de datos de testigos negativos históricos del laboratorio. Cuando hay datos de los testigos negativos que quedan fuera del límite de control del 95 %, su inclusión en la distribución de testigos históricos puede ser aceptable en la medida en que dichos datos no sean valores atípicos extremos, y haya pruebas de que el sistema de ensayo está “controlado” (véase el punto 49) y de la ausencia de fallos técnicos o humanos.
|
|
51.
|
Cualquier cambio en el protocolo experimental debe considerarse en función de la coherencia de los datos con las bases de datos de testigos históricos existentes del laboratorio. Cualquier incoherencia importante debería dar lugar a la creación de una nueva base de datos de testigos históricos.
|
DATOS E INFORME
Presentación de los resultados
|
52.
|
La presentación de los datos correspondientes a los ensayos MLA y TK6 debe incluir, tanto respecto a los cultivos tratados como a los testigos, los datos necesarios para el cálculo de la citotoxicidad (CRT o SR, respectivamente) y las frecuencias de mutantes, según se describe a continuación.
|
|
53.
|
En el caso del ensayo MLA, deben facilitarse datos sobre los distintos cultivos en cuanto al CRS, el CRT, la eficiencia de clonación en el momento de la selección de mutantes y el número de colonias mutantes (en la versión con agar) o el número de pocillos vacíos (en la versión con micropocillos). La FM debe expresarse como número de células mutantes por millón de células supervivientes. Si la respuesta es positiva, deben darse las FM de las colonias pequeñas y grandes (y/o el porcentaje de la FM total) al menos a una concentración del producto problema (en general, la mayor concentración positiva) y con los testigos positivos y negativos. En caso de respuesta negativa, debe darse las FM de las colonias pequeñas y grandes en relación con el control negativo y el control positivo.
|
|
54.
|
En el ensayo TK6, deben facilitarse datos sobre los distintos cultivos en relación con la SR, la eficiencia de clonación en el momento de la selección de mutantes y el número de pocillos vacíos correspondientes a los mutantes de aparición temprana y a los de aparición tardía. La FM debe expresarse como número de células mutantes por número de células supervivientes, y debe incluir la FM total, así como la FM (y/o el porcentaje de la FM total) de los mutantes de aparición temprana y de aparición tardía.
|
Criterios de aceptabilidad
|
55.
|
Tanto en el ensayo MLA como en el TK6 deben cumplirse los siguientes criterios antes de determinar los resultados globales correspondientes a un producto problema específico:
|
—
|
Se han estudiado dos condiciones experimentales (tratamiento corto con y sin activación metabólica, véase el punto 33), salvo que se hayan obtenido resultados positivos en una de ellas.
|
|
—
|
Son analizables números y concentraciones apropiados de células (véanse los puntos 27 y 34-36).
|
|
—
|
Los criterios de selección de la concentración superior son coherentes con los descritos en los puntos 28-30.
|
|
Criterios de aceptabilidad de los testigos positivos y negativos
|
56.
|
El análisis, realizado por el grupo de trabajo de expertos MLA del IWGT, de una amplia cantidad de datos del MLA dio lugar a un consenso internacional en torno a los criterios de aceptabilidad específicos del MLA (1) (2) (3) (4) (5). Por lo tanto, este método de ensayo ofrece recomendaciones específicas para determinar la aceptabilidad de los testigos negativos y positivos y para evaluar los resultados de distintas sustancias con el ensayo MLA. El TK6 tiene una base de datos mucho más pequeña y no ha sido objeto de evaluación por parte de un grupo de trabajo.
|
|
57.
|
En el caso del ensayo MLA, se debe evaluar cada experimento para determinar si el testigo sin tratar / control del disolvente cumple los criterios de aceptación del grupo de trabajo de expertos MLA del IWGT [(4) y cuadro 2, más abajo] en relación con: 1) la FM (obsérvese que las FM aceptables para el IWGT son diferentes para la versión con agar y para la versión con micropocillos del MGA), 2) la eficiencia de clonación (EC) en el momento de la selección de los mutantes, y 3) el crecimiento en la suspensión (CS) del control del disolvente (véanse las fórmulas en el apéndice 2).
Cuadro 2
Criterios de aceptabilidad del MLA
|
Parámetro
|
Método de agar blando
|
Método de micropocillos
|
|
Frecuencia de mutantes
|
35 – 140 × 10-6
|
50 – 170 × 10-6
|
|
Eficiencia de clonación
|
65 – 120 %
|
65 – 120 %
|
|
Crecimiento en la suspensión
|
8 – 32 veces (tratamiento de 3 a 4 horas) 32 – 180 veces (tratamiento de 24 horas, si se lleva a cabo)
|
8 – 32 veces (tratamiento de 3 a 4 horas) 32 – 180 veces (tratamiento de 24 horas, si se lleva a cabo)
|
|
|
58.
|
En el caso del ensayo MLA, también debe evaluarse cada ensayo en cuanto a si el testigo o testigos positivos cumplen al menos uno de los dos criterios de aceptación siguientes, elaborados por el grupo de trabajo del IWGT:
|
—
|
El testigo positivo debe mostrar un aumento absoluto de la FM total, es decir, un aumento por encima de la FM de fondo espontánea [una FM inducida (FMI)], de al menos 300 × 10-6. Al menos el 40 % de la FMI debe reflejarse en la FM de las colonias pequeñas.
|
|
—
|
El testigo positivo tiene un aumento en la FM de las colonias pequeñas de al menos 150 × 10-6 por encima de la observada en el caso de un testigo sin tratar / control del disolvente (una FMI de las pequeñas colonias de 150 × 10-6).
|
|
|
59.
|
En el caso del TK6, el ensayo será aceptable si el testigo negativo en paralelo se considera aceptable para añadirse a la base de datos de testigos negativos históricos del laboratorio de acuerdo con lo descrito en los puntos 48-49. Además, los testigos positivos en paralelo (véase el punto 32) deben inducir respuestas compatibles con las obtenidas en la base de datos de testigos positivos históricos y producir un aumento estadísticamente significativo en comparación con el testigo negativo en paralelo.
|
|
60.
|
En ambos ensayos, el límite superior de citotoxicidad observado en el cultivo del testigo positivo debe ser el mismo que en los cultivos experimentales. Es decir, el valor de CRT o SR no debe ser inferior al 10 %. Basta con utilizar una sola concentración (o una de las concentraciones de los cultivos de los testigos positivos si se utiliza más de una concentración) para demostrar que se cumplen los criterios de aceptación del testigo positivo. Además, la FM del testigo positivo deberá encontrarse dentro del intervalo aceptable establecido para el laboratorio.
|
Evaluación e interpretación de los resultados
|
61.
|
En el caso del MLA, el grupo de trabajo de expertos en linfoma de ratón del IWGT ha llevado a cabo un trabajo significativo en relación con la pertinencia biológica y los criterios de respuesta positiva (4). Por lo tanto, este método de ensayo ofrece recomendaciones específicas para la interpretación de los resultados de los productos problema obtenidos con el ensayo MLA (véanse los puntos 62 a 64). El TK6 tiene una base de datos mucho más pequeña y no ha sido objeto de evaluación por parte de un grupo de trabajo. Por lo tanto, las recomendaciones para la interpretación de los datos del TK6 se dan en términos más generales (véanse los puntos 65-66). Se aplican recomendaciones adicionales a ambos ensayos (véanse los puntos 67-71).
|
MLA
|
62.
|
Se recomienda un planteamiento para definir las respuestas positivas y negativas a fin de garantizar que el aumento de la FM es biológicamente pertinente. En lugar del análisis estadístico utilizado generalmente con otros ensayos, este planteamiento se basa en el uso de una frecuencia de mutantes inducida predefinida (es decir, el aumento de la FM por encima del control en paralelo), designada como factor de evaluación global (FEG), el cual se basa en el análisis de la distribución de los datos de FM de los testigos negativos conseguidos en los laboratorios participantes (4). Para la versión del MLA con agar, el FEG es 90 × 10-6, y para la versión del MLA con micropocillos, el FEG es 126 × 10-6.
|
|
63.
|
Siempre que se cumplan todos los criterios de aceptabilidad, se considera que un producto problema es claramente positivo si, en alguna de las condiciones experimentales examinadas (véase el punto 33), el aumento de la FM por encima del fondo de referencia en paralelo excede del FEG y el aumento está relacionado con la concentración (por ejemplo, según una prueba de tendencia). El producto problema se considera entonces capaz de inducir mutaciones en este sistema de ensayo.
|
|
64.
|
Siempre que se cumplan todos los criterios de aceptabilidad, se considera que un producto problema es claramente negativo si, en todas las condiciones experimentales examinadas (véase el punto 33), no hay ninguna respuesta relacionada con la concentración o, si se produce un aumento de la FM, este no supera el FEG. El producto problema se considera entonces incapaz de inducir mutaciones en este sistema de ensayo.
|
TK6
|
65.
|
Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente positivo si, en alguna de las condiciones experimentales examinadas (véase el punto 33):
|
—
|
al menos una de las concentraciones de ensayo muestra un aumento estadísticamente significativo en comparación con el testigo negativo en paralelo,
|
|
—
|
el aumento está relacionado con la concentración cuando se evalúa con una prueba de tendencia adecuada (véase el punto 33),
|
|
—
|
alguno de estos resultados está fuera de la distribución de los datos históricos de los testigos negativos (por ejemplo, límite de control del 95 % según la distribución de Poisson; véase el punto 48).
|
Cuando se cumplen todos estos criterios, el producto problema se considera capaz de inducir mutaciones en este sistema de ensayo. En la bibliografía se encuentran recomendaciones sobre los métodos estadísticos más adecuados (66) (67).
|
|
66.
|
Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente negativo si, en todas las condiciones experimentales examinadas (véase el punto 33):
|
—
|
ninguna de las concentraciones de ensayo muestra un aumento estadísticamente significativo en comparación con el testigo negativo en paralelo,
|
|
—
|
no hay ningún aumento relacionado con la concentración cuando se evalúa con una prueba de tendencia adecuada,
|
|
—
|
todos los resultados están dentro de la distribución de los datos históricos de los testigos negativos (por ejemplo, límite de control del 95 % según la distribución de Poisson; véase el punto 48).
|
El producto problema se considera entonces incapaz de inducir mutaciones en este sistema de ensayo.
|
Tanto con MLA como con TK6:
|
67.
|
Si la concentración máxima se basa en la citotoxicidad, la concentración más elevada debe intentar conseguir un valor de CRT o SR entre el 20 y el 10 %. Hay consenso en cuanto a la necesidad de tener cuidado a la hora de interpretar los resultados positivos que solo se encuentren entre el 20 y el 10 % de CRT/SR, y en cuanto a no considerar positivo un resultado si el aumento de la FM se produce solo a un valor inferior o igual al 10 % de CRT/SR (si se evalúa) (2) (59).
|
|
68.
|
Hay algunas circunstancias en las que cierta información adicional puede ayudar a determinar que un producto problema no es mutágeno cuando no existe ningún cultivo que muestre un valor de CRT entre el 10 y el 20 % de CRT/SR. Estas situaciones se exponen a continuación: 1) No hay pruebas de mutagenicidad (por ejemplo, no hay respuesta en función de las dosis, no hay ninguna frecuencia de mutantes por encima de las registradas con los testigos negativos en paralelo o en los intervalos de fondo históricos, etc.) en una serie de puntos de datos de entre el 100 % y el 20 % de CRT/SR y hay por lo menos un punto de datos entre el 20 y el 25 % de CRT/SR. 2) No hay pruebas de mutagenicidad (por ejemplo, no hay respuesta en función de las dosis, no hay ninguna frecuencia de mutantes por encima de las registradas con los testigos negativos en paralelo o en los intervalos de fondo históricos, etc.) en una serie de puntos de datos de entre el 100 % y el 25 % de CRT/SR y hay también un punto de datos negativo ligeramente por debajo del 10 % de CRT/SR. En ambas situaciones puede concluirse que el producto problema es negativo.
|
|
69.
|
No se requiere ninguna verificación de una respuesta claramente positiva o negativa.
|
|
70.
|
En los casos en que la respuesta no sea ni claramente positiva ni claramente negativa como se describe más arriba, o a fin de ayudar a determinar la relevancia biológica de un resultado, los datos deben ser evaluados por expertos o mediante más investigaciones. Puede ser útil repetir el experimento modificando quizás las condiciones experimentales [por ejemplo, la separación entre concentraciones para aumentar la probabilidad de alcanzar puntos de datos dentro del intervalo del 10-20 % de CRT/SR, las condiciones de activación metabólica (por ejemplo, la concentración o el origen de la fracción S9) y la duración del tratamiento].
|
|
71.
|
En casos raros, incluso después de hacer más investigaciones, el conjunto de datos no permite que se extraiga una conclusión de resultado positivo o negativo. Por lo tanto, debe concluirse que la respuesta del producto problema es dudosa (lo que se interpreta como que resulta igualmente probable que sea positiva o negativa).
|
INFORME DEL ENSAYO
|
72.
|
El informe del ensayo debe incluir la información siguiente:
|
|
Producto problema:
|
—
|
origen, número de lote, fecha límite de utilización, si se conocen;
|
|
—
|
estabilidad del producto problema en sí, si se conoce;
|
|
—
|
solubilidad y estabilidad del producto problema en el disolvente, si se conocen;
|
|
—
|
medición del pH, osmolalidad y precipitado en el medio de cultivo al que se ha añadido el producto problema, según proceda.
|
|
|
Sustancias de un solo componente:
|
—
|
aspecto físico, hidrosolubilidad y otras propiedades fisicoquímicas pertinentes;
|
|
—
|
identificación química, como nombre IUPAC o CAS, número CAS, notación SMILES o InChI, fórmula estructural, pureza, identidad química de las impurezas si procede y es viable en la práctica, etc.
|
|
|
|
Sustancias de componentes múltiples, UVCB y mezclas:
|
—
|
deben caracterizarse en la medida de lo posible por la identidad química (véase más arriba), la cantidad en que están presentes y las propiedades fisicoquímicas pertinentes de sus componentes.
|
|
|
|
|
Disolvente:
|
—
|
justificación de la elección del disolvente;
|
|
—
|
porcentaje de disolvente en el medio de cultivo final.
|
|
|
|
Células:
|
|
En el caso de cultivos madre de laboratorio:
|
—
|
tipo y origen de las células, e historia en el laboratorio de ensayo;
|
|
—
|
características del cariotipo y/o número modal de los cromosomas;
|
|
—
|
métodos de mantenimiento de los cultivos celulares;
|
|
—
|
ausencia de micoplasmas;
|
|
—
|
tiempos de duplicación de las células.
|
|
|
|
|
Condiciones del ensayo:
|
—
|
fundamento de la selección de las concentraciones y del número de los cultivos celulares; incluidos, por ejemplo, datos relativos a la citotoxicidad y limitaciones de solubilidad;
|
|
—
|
composición de los medios, concentración de CO2 , nivel de humedad;
|
|
—
|
concentración del producto problema, expresada como concentración final en el medio de cultivo (por ejemplo, mM, o μg o mg/ml de medio de cultivo);
|
|
—
|
concentración (y/o volumen) del disolvente y del producto problema añadidos al medio de cultivo;
|
|
—
|
temperatura de incubación;
|
|
—
|
duración del tratamiento;
|
|
—
|
densidad celular durante el tratamiento;
|
|
—
|
tipo y composición del sistema de activación metabólica (origen de la fracción S9, método de preparación de la mezcla S9, concentración o volumen de la mezcla S9 y de la fracción S9 en el medio de cultivo final, controles de calidad de la fracción S9);
|
|
—
|
sustancias testigo positivo y negativo, concentraciones finales en cada una de las condiciones de tratamiento;
|
|
—
|
duración del período de expresión (con el número de células sembradas, subcultivos y pautas de nutrición, si procede);
|
|
—
|
identidad del agente selectivo y su concentración;
|
|
—
|
para el ensayo MLA debe indicarse la versión utilizada (agar o micropocillos);
|
|
—
|
criterios de aceptabilidad de los estudios;
|
|
—
|
métodos empleados para contar las células viables y las mutantes;
|
|
—
|
métodos utilizados para medir la citotoxicidad;
|
|
—
|
cualquier información adicional relativa a la citotoxicidad y método utilizado;
|
|
—
|
duración de la incubación después de la siembra;
|
|
—
|
definición de las colonias de las que se considera el tamaño y el tipo (incluidos, en su caso, los criterios de distinción de colonias “grandes” y “pequeñas”);
|
|
—
|
criterios empleados para considerar si los resultados de los estudios son positivos, negativos o dudosos;
|
|
—
|
métodos utilizados para determinar el pH y la osmolalidad, si se aplican, y la precipitación si es pertinente.
|
|
|
|
Resultados:
|
—
|
número de células tratadas y número de células subcultivadas por cada cultivo;
|
|
—
|
parámetros de toxicidad (CRT con el MLA y SR con el TK6);
|
|
—
|
signos de precipitación y momento de la determinación;
|
|
—
|
número de células sembradas en medio selectivo y no selectivo;
|
|
—
|
número de colonias en medio no selectivo y número de colonias resistentes en medio selectivo, y frecuencias de mutantes correspondientes;
|
|
—
|
determinación del tamaño de las colonias de los testigos positivos y negativos y si el producto problema es positivo, al menos a una concentración, y las frecuencias de mutantes correspondientes;
|
|
—
|
relación concentración-respuesta, cuando sea posible;
|
|
—
|
datos de los testigos negativos (disolvente) y positivos (concentraciones y disolventes) en paralelo;
|
|
—
|
datos de los testigos negativos (disolvente) y positivos (concentraciones y disolventes) históricos, con intervalos, medias y desviaciones típicas; número de ensayos en los que se basan los controles históricos;
|
|
—
|
análisis estadísticos (correspondientes a los cultivos individuales y a las réplicas combinadas, en su caso), y valores p, en su caso; y, en relación con el MLA, la evaluación del FEG.
|
|
|
|
Discusión de los resultados
|
|
BIBLIOGRAFÍA
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OCDE (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. and O'Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. and Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. and Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., and Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. and Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. and Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. and Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. and Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. and Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. and Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. and Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. and Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. and Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. and Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. and Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. and Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. and Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. and Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. and Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. and Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. and DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. and Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. and Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411–416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. and Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscript in preparation).
|
|
(38)
|
Lloyd M. and Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. and Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan Z and Caldwell(Eds) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. and Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. and Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. and Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. and De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. and Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. and Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
PNUMA (2001). Convenio de Estocolmo sobre Contaminantes Orgánicos Persistentes, Programa de las Naciones Unidas para el Medio Ambiente (PNUMA).
|
|
(53)
|
Krahn D.F., Barsky F.C. and McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, pp. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. and Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. and Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. and Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Disponible en: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. and Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. and Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. and Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. and Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OCDE (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 199), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(67)
|
Fleiss J.L., Levin B. and Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Apéndice 1
DEFINICIONES
Anéugeno
: Producto o proceso que, por interacción con los componentes del ciclo de división celular mitótica y meiótica, produce la aneuploidía de células u organismos.
Aneuploidía
: Desviación respecto al número diploide (o haploide) normal de cromosomas que afecta a uno o varios cromosomas, pero no a dotaciones completas de cromosomas (poliploidía).
Mutágenos por sustitución de pares de bases
: Productos que provocan la sustitución de pares de bases en el ADN.
Producto
: Sustancia o mezcla.
Eficiencia de clonación
: Porcentaje de células sembradas a baja densidad que pueden crecer para formar una colonia que puede contarse.
Clastógeno
: Producto o proceso que provoca aberraciones cromosómicas estructurales en poblaciones de células u organismos.
Citotoxicidad
: Para los ensayos incluidos en este método de ensayo, la citotoxicidad se identifica como una reducción del crecimiento relativo total (CRT) o de la supervivencia relativa (SR) respecto a los ensayos MLA y TK6, respectivamente.
Mutación directa
: Mutación génica del tipo parental a la forma mutante, que da lugar a una alteración o pérdida de la actividad enzimática o de la función de la proteína codificada.
Mutágenos por desplazamiento del marco de lectura
: Productos que provocan la adición o supresión de uno o varios pares de bases en la molécula de ADN.
Genotoxicidad
: Término general que engloba todos los tipos de lesión del ADN o del cromosoma, con inclusión de roturas de ADN, aductos, reorganizaciones, mutaciones, aberraciones cromosómicas, y aneuploidía. No todos los tipos de efectos genotóxicos resultan en mutaciones o en lesiones cromosómicas estables.
Recombinación mitótica
: Durante la mitosis, recombinación entre cromátidas homólogas que puede dar lugar a la inducción de roturas de la doble cadena de ADN, o a una pérdida de heterocigosis.
Mutágeno
: Agente que provoca un cambio hereditario en una o varias secuencias de pares de bases del ADN en los genes o en la estructura de los cromosomas (aberraciones cromosómicas).
Frecuencia de mutantes (FM)
: Número de células mutantes observadas dividido por el número de células viables.
Período de expresión fenotípica
: Tiempo después del tratamiento en el que la alteración genética se fija en el genoma y los eventuales productos génicos preexistentes se agotan hasta que se modifica el rasgo fenotípico.
Supervivencia relativa (SR)
: Se utiliza como medida de la citotoxicidad relacionada con el tratamiento en el ensayo TK6. Es la eficiencia de clonación relativa (EC) de las células sembradas inmediatamente después del tratamiento celular ajustada para tener en cuenta la pérdida de células durante el tratamiento en comparación con la eficiencia de clonación del testigo negativo.
Crecimiento relativo en la suspensión (CRS)
: Para el ensayo MLA, el crecimiento relativo total en la suspensión durante dos días del cultivo de ensayo, en comparación con el crecimiento total en la suspensión durante dos días del testigo negativo / control del disolvente (Clive y Spector, 1975). El CRS debe incluir el crecimiento relativo del cultivo de ensayo en comparación con el testigo negativo / control del disolvente durante el período de tratamiento.
Crecimiento relativo total (CRT)
: Se utiliza como medida de la citotoxicidad relacionada con el tratamiento en el ensayo MLA. Es una medida del crecimiento relativo (comparado con el control del vehículo) de los cultivos de ensayo durante el tratamiento, el periodo de expresión de dos días y la fase de clonación de la selección de mutantes del ensayo. El CRT de cada cultivo de ensayo se multiplica por la eficiencia relativa de clonación del cultivo de ensayo en el momento de la selección de los mutantes y se expresa en relación con la eficiencia de clonación del testigo negativo / control del disolvente (Clive y Spector, 1975).
Fracciones hepáticas S9
: Sobrenadante de homogeneizado de hígado después de centrifugación a 9 000 g, es decir, extracto de hígado crudo.
Mezcla S9
: Mezcla de la fracción hepática S9 y de cofactores necesarios para la actividad metabólica de las enzimas.
Crecimiento en la suspensión (CS)
: El factor multiplicador del número de células a lo largo de las fases de tratamiento y expresión del MLA. El CS se calcula multiplicando el factor multiplicador del día 1 por el factor multiplicador del día 2 en caso de un tratamiento corto (3 o 4 horas). Si se utiliza un tratamiento de 24 horas, el CS es el factor multiplicador durante el tratamiento de 24 horas, multiplicado por los factores multiplicadores de los días 1 y 2.
Control del disolvente
: Término general para definir los cultivos testigo que reciben solo el disolvente utilizado para disolver el producto problema.
Producto problema
: Sustancia o mezcla estudiada con este método de ensayo.
Testigos sin tratar
: Cultivos que no reciben tratamiento (es decir, ni producto problema ni disolvente), pero que se someten al mismo proceso que los cultivos que reciben el producto problema.
Apéndice 2
FÓRMULAS
Citotoxicidad
Para las dos versiones del MLA (con agar y con micropocillos)
La citotoxicidad se define como el crecimiento relativo total (CRT), que incluye el crecimiento relativo en la suspensión (CRS) durante el período de expresión de 2 días y la eficiencia relativa de clonación (ERC) obtenida en el momento de la selección de los mutantes. Los valores de CRT, CRS y ERC se expresan en porcentaje.
Cálculo del CRS: El crecimiento en la suspensión uno (CS1) es la tasa de crecimiento entre el día 0 y el día 1 (concentración celular el día 1 / concentración celular el día 0) y el crecimiento en la suspensión dos (CS2) es la tasa de crecimiento entre el día 1 y el día 2 (concentración celular el día 2 / concentración celular el día 1). El CRS es el CS total (CS1 x CS2) del cultivo tratado en comparación con el testigo sin tratar / control del disolvente. Esto es: CRS = [CS1(tratado) x CS2(tratado)] / [CS1(testigo) x CS2(testigo)] El CS1 debe calcularse a partir de la concentración celular inicial utilizada al principio del tratamiento de las células. Esto tiene en cuenta la evenutal citotoxicidad diferencial que se produzca en los cultivos de ensayo durante el tratamiento celular.
La ERC es la eficiencia relativa de clonación del cultivo tratado en comparación con la eficiencia relativa de clonación del testigo sin tratar / control del disolvente obtenida en el momento de la selección de los mutantes.
Crecimiento relativo total (CRT):
CRT = CRS x ERC
TK6
Supervivencia relativa (SR):
La citotoxicidad se evalúa mediante la supervivencia relativa, es decir, la eficiencia de clonación (EC) de las células sembradas inmediatamente después del tratamiento, ajustada para tener en cuenta la eventual pérdida de células durante el tratamiento en comparación con la eficiencia de clonación en los testigos negativos (a los que se asigna una supervivencia del 100 %). El ajuste para tener en cuenta la pérdida de células durante el tratamiento puede calcularse como sigue:
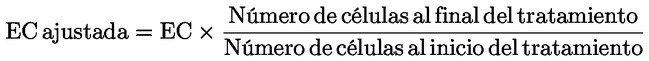
La SR correspondiente a un cultivo tratado con un producto problema se calcula de la siguiente manera:

Frecuencia de mutantes tanto con MLA como con TK6
La frecuencia de mutantes (FM) es la eficiencia de clonación de las colonias mutantes en el medio selectivo (ECM) dividida por la eficiencia de clonación en el medio no selectivo en relación en el momento de la selección (ECV). Es decir, FM = ECM/ECV. El cálculo de estas dos eficiencias de clonación se describe a continuación para los métodos de clonación con agar y con micropocillos.
Versión del MLA con agar
: En la versión del MLA con agar blando, el número de colonias en la placa de selección de mutantes (CM) y el número de colonias en la placa de eficiencia de clonación o sin selección (recuento de viables) (CV) se obtienen mediante el recuento directo de los clones. Para ver la eficiencia de clonación (EC) se utilizan las fórmulas siguientes cuando se siembran 600 células en las placas de selección de mutantes (ECM) y en las placas de eficiencia de la clonación o sin selección (recuento de viables) (ECV) y se utilizan 3 x 106 células para la selección de mutantes:
ECM = CM / (3 x 106) = (CM / 3) x 10-6
ECV = CV / 600
Versión del MLA y TK6 con micropocillos:
En la versión del MLA con micropocillos, los valores de CM y CV se determinan como el producto del número total de micropocillos (TW) y del número probable de colonias por pocillo (P) en las placas de micropocillos.
CM = PM x TWM
CV = PV x TWV
A partir del término cero de la distribución de Poisson (Furth et al., 1981), el valor de P viene dado por:
P = - ln (EW / TW)
donde EW es el número de pocillos vacíos y TW es el número total de pocillos. Por tanto,
ECM = CM / TM = (PM x TWM) / TM
ECV = CV / TV = (PV x TWV) / TV
En el caso de la versión con micropocillos del MLA, las frecuencias de mutantes de colonias pequeñas y grandes se calcularán de la misma manera, utilizando el número pertinente de pocillos vacíos correspondientes a las colonias pequeñas y grandes.
En el caso del TK6, las frecuencias de mutantes de colonias pequeñas y grandes se basan en los mutantes de aparición temprana y de aparición tardía.
B.68. MÉTODO DE ENSAYO IN VITRO CON EXPOSICIÓN DE BREVE DURACIÓN PARA DETECTAR: I) PRODUCTOS QUE PROVOCAN LESIONES OCULARES GRAVES Y II) PRODUCTOS QUE NO REQUIEREN CLASIFICACIÓN POR IRRITACIÓN OCULAR O LESIONES OCULARES GRAVES
INTRODUCCIÓN
|
1.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 491 de la OCDE (2017). El método de ensayo con exposición de breve duración (EBD) es un método in vitro que puede utilizarse en determinadas circunstancias y con limitaciones específicas para la clasificación de los peligros y el etiquetado de productos (sustancias y mezclas) que provocan lesiones oculares graves, así como de aquellos que no requieren clasificación por lesiones oculares graves ni por irritación ocular, según se definen en el Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA) de las Naciones Unidas (1) y el Reglamento (UE) n.o 1272/2008 sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) de la Unión Europea (68).
|
|
2.
|
Durante muchos años, el potencial de peligro para los ojos de los productos se ha evaluado principalmente mediante un ensayo in vivo con ojos de conejo [método B.5 (8), equivalente a las directrices TG 405 de la OCDE)]. Generalmente se acepta que, en un futuro próximo, ningún ensayo alternativo in vitro podrá sustituir completamente por sí solo el ensayo in vivo con ojos de conejo para evaluar toda la gama de respuestas de lesiones oculares graves o de irritación ocular que pueden causar diferentes clases de productos. Sin embargo, unas combinaciones estratégicas de métodos de ensayo alternativos dentro de una estrategia de ensayos (escalonados) sí podrán sustituir completamente al ensayo con ojos de conejo (2). El enfoque descendente está diseñado para ensayar productos de los que quepa esperar, sobre la base de la información existente, que tengan un alto potencial de irritación o provoquen lesiones oculares graves. Por el contrario, el enfoque ascendente está diseñado para ensayar productos de los que quepa esperar, sobre la base de la información existente, que no provoquen una irritación ocular suficiente para exigir una clasificación. Si bien el método de ensayo EBD no se considera una sustitución completa del ensayo con ojos de conejo in vivo, puede utilizarse como parte de una estrategia de ensayo escalonada para la clasificación y el etiquetado normativos, como es el enfoque descendente/ascendente, para detectar sin necesidad de realizar más ensayos i) los productos que provocan lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas y del CLP) y ii) los productos (salvo las sustancias muy volátiles y todos los productos sólidos distintos de los tensioactivos) que no requieren clasificación por irritación ocular o lesiones oculares graves (sin categoría del SGA de las Naciones Unidas y del CLP) (1) (2). Sin embargo, un producto del que no pueda decirse mediante el método de ensayo EBD que causa lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas y del CLP) ni que corresponde al grupo sin categoría del SGA de las Naciones Unidas y del CLP (no induce lesiones oculares graves ni irritación ocular), requeriría ensayos adicionales para establecer una clasificación definitiva. Además, hay que consultar a las autoridades reguladoras competentes antes de utilizar el ensayo EBD en un enfoque ascendente en relación con otros regímenes de clasificación distintos del SGA de las Naciones Unidas y del CLP. La elección del método de ensayo más adecuado y el uso de este método de ensayo deben considerarse en el contexto del documento de orientación de la OCDE sobre un enfoque integrado de ensayos y evaluación de las lesiones oculares graves y la irritación ocular (14).
|
|
3.
|
El objetivo del presente método de ensayo es describir los procedimientos utilizados para evaluar el potencial de peligro para los ojos de un producto problema en función de su capacidad de inducir citotoxicidad en el método de ensayo con exposición de breve duración. El efecto citotóxico de los productos en las células epiteliales de la córnea es un modo de acción importante (MDA) que lleva a lesiones epiteliales de la córnea e irritación ocular. La viabilidad celular en el método de ensayo EBD se evalúa mediante la medición cuantitativa, tras la extracción de las células, de la sal de formazano azul producida por las células vivas a través de la conversión enzimática del colorante vital MTT [bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio)], también conocido como bromuro de tetrazolio de azul de tiazolilo (3). La viabilidad celular obtenida se compara con el control del disolvente (viabilidad relativa) y se utiliza para estimar el potencial de peligro para los ojos que tiene el producto problema. Un producto problema se clasifica en la categoría 1 del SGA de las Naciones Unidas y del CLP cuando tanto la concentración del 5 % como la del 0,05 % dan lugar a una viabilidad celular menor o igual (≤) al 70 %. Por el contrario, un producto problema se asigna al grupo sin categoría del SGA de las Naciones Unidas y del CLP cuando tanto la concentración del 5 % como la del 0,05 % dan lugar a una viabilidad celular superior (>) al 70 %.
|
|
4.
|
El término “producto problema” se utiliza en el presente método de ensayo para referirse al objeto del ensayo y no está relacionado con la aplicabilidad del método de ensayo EBD al ensayo de sustancias o mezclas. En el apéndice se dan las definiciones utilizadas.
|
CONSIDERACIONES INICIALES Y LIMITACIONES
|
5.
|
Este método de ensayo se basa en un protocolo elaborado por Kao Corporation (4), que fue objeto de dos estudios de validación diferentes: uno por el Comité de Validación de la Sociedad Japonesa para la Alternativa a la Experimentación con Animales (Japanese Society for Alternative to Animal Experiments, JSAAE) (5) y otro por elCentro Japonés para la Validación de Métodos Alternativos (Japanese Center for the Validation of Alternative Methods, JaCVAM) (6). NICEATM/ICCVAM han efectuado una revisión por pares sobre la base de los informes de los estudios de validación y de los documentos de revisión de fondo del método de ensayo (7).
|
|
6.
|
Cuando se utiliza para detectar productos (sustancias y mezclas) que provocan lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas y del CLP) (1), los datos obtenidos con el método de ensayo EBD respecto a 125 productos (entre los que se incluyen tanto sustancias como mezclas) muestran una exactitud global del 83 % (104/125), una tasa de falsos positivos del 1 % (1/86) y una tasa de falsos negativos del 51 % (20/39) en comparación con el ensayo in vivo con ojos de conejo (7). La tasa obtenida de falsos negativos no es crítica en el contexto actual, ya que todos los productos problema que inducen una viabilidad celular ≤ 70 % a una concentración del 5 % y > 70 % a una concentración del 0,05 % serían analizadas posteriormente con otros métodos de ensayo in vitro debidamente validados o, como última opción, en el ensayo con ojos de conejo in vivo, en función de los requisitos normativos y de conformidad con la estrategia de evaluación secuencial y con los enfoques de ponderación de las pruebas recomendados actualmente (1) (8). Se sometieron a ensayo principalmente sustancias de un solo componente, aunque también se dispone de una cantidad limitada de datos sobre ensayos con mezclas. Sin embargo, el método de ensayo es técnicamente aplicable a los ensayos de sustancias de componentes múltiples y de mezclas. No obstante, antes de la utilización de este método de ensayo con una mezcla para obtener datos con fines normativos, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué. Tales consideraciones no son necesarias si la normativa impone el ensayo de la mezcla. El método de ensayo EBD no ha mostrado ninguna otra deficiencia específica cuando se utiliza para identificar productos problema como de categoría 1 del SGA de las Naciones Unidas y del CLP. Los investigadores podrían considerar la posibilidad de utilizar este método de ensayo con productos problema, de manera que una viabilidad celular ≤ 70 % a las concentraciones tanto del 5 % como del 0,05 % debería aceptarse como indicativa de una respuesta inductora de lesiones oculares graves que impondría la clasificación en la categoría 1 del SGA de las Naciones Unidas y del CLP sin más ensayos.
|
|
7.
|
Cuando se utiliza para detectar productos (sustancias y mezclas) que no requieren clasificación por irritación ocular o lesiones oculares graves (es decir, sin categoría del SGA de las Naciones Unidas y del CLP), los datos obtenidos con el método de ensayo EBD respecto a 130 productos (entre los que se incluyen tanto sustancias como mezclas) muestran una exactitud global del 85 % (110/130), una tasa de falsos negativos del 12 % (9/73) y una tasa de falsos positivos del 19 % (11/57) en comparación con el ensayo in vivo con ojos de conejo (7). Si se excluyen del conjunto de datos las sustancias muy volátiles y las sustancias sólidas distintas de los tensioactivos, la exactitud global mejora al 90 % (92 /102), la tasa de falsos negativos al 2 % (1/54), y la de falsos positivos al 19 % (9/48) (7). Como consecuencia de ello, las posibles deficiencias del método de ensayo EBD cuando se utiliza para identificar productos problema que no requieren clasificación por irritación ocular o lesiones oculares graves (sin categoría del SGA de las Naciones Unidas y del CLP) son una tasa elevada de falsos negativos para i) sustancias muy volátiles, con una presión de vapor de más de 6 kPa, y ii) productos sólidos (sustancias y mezclas) distintos de los tensioactivos y las mezclas compuestas únicamente de tensioactivos. Estos productos están excluidos del ámbito de aplicación del método de ensayo EBD (7).
|
|
8.
|
Además de los productos mencionados en los puntos 6 y 7, el conjunto de datos generados por el método de ensayo EBD contiene también datos internos sobre 40 mezclas, que, en comparación con el ensayo ocular de Draize in vivo, muestran una exactitud del 88 % (35/40), una tasa de falsos positivos del 50 % (5/10), y una tasa de falsos negativos del 0 % (0/30) para asignar mezclas que no requieren clasificación con arreglo a los sistemas de clasificación del SGA de las Naciones Unidas y del CLP (9). Por lo tanto, el método de ensayo EBD se puede aplicar para detectar las mezclas del grupo sin categoría del SGA de las Naciones Unidas y del CLP en un enfoque ascendente, salvo las mezclas sólidas distintas de las constituidas únicamente por tensioactivos como ampliación de su limitación respecto a las sustancias sólidas. Además, las mezclas que contengan sustancias con presión de vapor superior a 6 kPa deben evaluarse con cuidado para evitar posibles subasignaciones, y deben justificarse caso por caso.
|
|
9.
|
El método de ensayo EBD no puede utilizarse para la detección de los productos problema de la categoría 2 del SGA de las Naciones Unidas y del CLP, ni de la categoría 2A (irritación ocular) o 2B (irritación ocular leve) del SGA de las Naciones Unidas, debido al considerable número de productos de la categoría 1 del SGA de las Naciones Unidas que resultan subasignadas como de categoría 2, 2A o 2 B, y de productos del grupo sin categoría del SGA de las Naciones Unidas y del CLP que resultan sobreasignadas como de categoría 2, 2A o 2B (7). A tal fin, pueden resultar necesarias pruebas adicionales con otro método adecuado.
|
|
10.
|
El método de ensayo EBD es adecuado para los productos problema que se disuelven o suspenden uniformemente durante al menos 5 minutos en suero fisiológico, dimetilsulfóxido (DMSO) al 5 % en solución salina, o en aceite mineral. El método de ensayo EBD no es adecuado para los productos problema que son insolubles o que no se suspenden uniformemente durante al menos 5 minutos en suero fisiológico, dimetilsulfóxido (DMSO) al 5 % en solución salina, o en aceite mineral. El uso de aceite mineral en el método de ensayo EBD es posible a causa de la breve duración de la exposición. Por lo tanto, el método de ensayo EBD es adecuado para prever el potencial de peligro para los ojos que tienen los productos problema insolubles en agua (por ejemplo, cetonas o alcoholes grasos de cadena larga) siempre que sean miscibles en al menos uno de los tres disolventes propuestos (4).
|
|
11.
|
El término “producto problema” se utiliza en el presente método de ensayo para referirse al objeto del ensayo (69) y no está relacionado con la aplicabilidad del método de ensayo EBD al ensayo de sustancias o mezclas.
|
PRINCIPIO DEL ENSAYO
|
12.
|
El método de ensayo EBD consiste en un ensayo in vitro basado en la citotoxicidad, que se realiza con una monocapa confluente de células de córnea de conejo del Statens Seruminstitut (SIRC), cultivadas en una microplaca de policarbonato de 96 pocillos (4). Tras una exposición de cinco minutos a un producto problema, la citotoxicidad se mide cuantitativamente como la viabilidad relativa de las células SIRC utilizando el ensayo con MTT (4). La disminución de la viabilidad celular se utiliza para predecir los posibles efectos adversos que provocan lesiones oculares.
|
|
13.
|
Se ha informado de que el 80 % de una solución aplicada al ojo de un conejo se excreta a través del saco conjuntival en el plazo de tres o cuatro minutos, mientras que más del 80 % de una solución aplicada al ojo humano se excreta en el plazo de uno o dos minutos (10). El método de ensayo EBD intenta aproximar estos tiempos de exposición y utiliza la citotoxicidad como parámetro para evaluar el grado de las lesiones a las células SIRC tras una exposición de cinco minutos al producto problema.
|
DEMOSTRACIÓN DE LA COMPETENCIA
|
14.
|
Antes de proceder al uso sistemático del método de ensayo EBD aquí descrito, los laboratorios deben demostrar su competencia técnica mediante la correcta clasificación de las once sustancias recomendadas en el cuadro 1. Estas sustancias se seleccionaron para representar la gama completa de respuestas en cuanto a lesiones oculares graves o irritación ocular sobre la base de los resultados de los ensayos con ojos de conejo in vivo (TG 405) y el sistema de clasificación SGA de las Naciones Unidas y del CLP (1). Entre los otros criterios de selección figuran los siguientes: que las sustancias estén disponibles en el mercado, que se disponga de datos de referencia in vivo de alta calidad y que se disponga de datos in vitro de alta calidad procedentes del método de ensayo EBD (3). Cuando una sustancia de la lista no esté disponible o en casos justificados, podrá utilizarse otra sustancia sobre la que se disponga de datos adecuados de referencia in vivo e in vitro siempre que se apliquen los mismos criterios de selección que los aquí descritos.
Cuadro 1
Lista de sustancias para la prueba de la competencia
|
Sustancia
|
N.o CAS
|
Clase química (70)
|
Estado físico
|
Categoría del SGA de las Naciones Unidas y del CLP según el ensayo in vivo
(71)
|
Disolvente en el ensayo EBD
|
Categoría del SGA de las Naciones Unidas y del CLP según el ensayo EBD
|
|
Cloruro de benzalconio (solución acuosa al 10 %)
|
8001-54-5
|
Compuesto onio
|
Líquido
|
Categoría 1
|
Solución salina
|
Categoría 1
|
|
Tritón X-100 (al 100 %)
|
9002-93-1
|
Éter
|
Líquido
|
Categoría 1
|
Solución salina
|
Categoría 1
|
|
Acid Red 92
|
18472-87-2
|
Compuesto heterocíclico; compuesto bromado; compuesto clorado
|
Sólido
|
Categoría 1
|
Solución salina
|
Categoría 1
|
|
Hidróxido de sodio
|
1310-73-2
|
Álcali; producto inorgánico
|
Sólido
|
Categoría 1 (72)
|
Solución salina
|
Categoría 1
|
|
Butirolactona
|
96-48-0
|
Lactona; compuesto heterocíclico
|
Líquido
|
Categoría 2A (categoría 2 en CLP)
|
Solución salina
|
No puede hacerse ninguna asignación.
|
|
1-Octanol
|
111-87-5
|
Alcohol
|
Líquido
|
Categoría 2A/B (73) (categoría 2 en CLP)
|
Aceite mineral
|
No puede hacerse ninguna asignación.
|
|
Ciclopentanol
|
96-41-3
|
Alcohol; Hidrocarburo (cíclico)
|
Líquido
|
Categoría 2A/B (74) (categoría 2 en CLP)
|
Solución salina
|
No puede hacerse ninguna asignación.
|
|
Acetato de 2-etoxietilo
|
111-15-9
|
Alcohol; éter
|
Líquido
|
Sin categoría
|
Solución salina
|
Sin categoría
|
|
Dodecano
|
112-40-3
|
Hidrocarburo (acíclico)
|
Líquido
|
Sin categoría
|
Aceite mineral
|
Sin categoría
|
|
Metil-isobutil-cetona
|
108-10-1
|
Cetona
|
Líquido
|
Sin categoría
|
Aceite mineral
|
Sin categoría
|
|
Sulfato de 1,1-dimetilguanidina
|
598-65-2
|
Amidina; compuesto de azufre
|
Sólido
|
Sin categoría
|
Solución salina
|
Sin categoría
|
|
Abreviaturas: No CAS = número de registro del Chemical Abstracts Service.
|
|
PROCEDIMIENTO
Preparación de la monocapa celular
|
15.
|
Para llevar a cabo el método de ensayo EBD debe utilizarse la línea celular de córnea de conejo SIRC. Se recomienda que las células SIRC se obtengan a partir de un banco de células adecuado, por ejemplo la CCL60 de la American Type Culture Collection.
|
|
16.
|
Se cultivan las células SIRC a 37 °C en atmósfera humidificada y con un 5 % de CO2, en un matraz de cultivo con medio de cultivo que comprende el medio esencial mínimo (MEM) de Eagle, complementado con un 10 % de suero bovino fetal (FBS), L-glutamina 2 mM, 50-100 unidades/ml de penicilina y 50-100 μg/ml de estreptomicina. Las células que hayan llegado a ser confluyentes en el matraz de cultivo deben separarse por medio de la solución de tripsina con ácido etilendiaminotetraacético, eventualmente utilizando un raspador celular. Se propagan las células (por ejemplo, 2 o 3 pases) en un matraz de cultivo antes de emplearse en los ensayos sistemáticos y no deben ser objeto de más de 25 pases tras la descongelación.
|
|
17.
|
Las celdas listas para su uso en el ensayo EBD se preparan a continuación a la densidad adecuada y se siembran en placas de 96 pocillos. La densidad de siembra celular recomendada es de 6,0 × 103 células por pocillo cuando se utilizan las células cuatro días después de la siembra, o de 3,0 × 103 células por pocillo cuando se utilizan las células cinco días después de la siembra, con un volumen de cultivo de 200 μl. Las células utilizadas en el ensayo EBD sembradas en un medio de cultivo a la densidad apropiada alcanzarán una confluencia de más del 80 % en el momento del ensayo, es decir, cuatro o cinco días después de la siembra.
|
Aplicación de los productos problema y de las sustancias testigo
|
18.
|
La primera opción de disolvente para disolver o suspender los productos problema es el suero fisiológico. Si el producto problema presenta baja solubilidad o no puede disolverse o suspenderse de manera uniforme durante al menos cinco minutos en solución salina sola, se utiliza la solución salina con un 5 % de DMSO (n.o CAS 67-68-5) como segunda opción. En el caso de los productos problema que no puedan disolverse ni suspenderse de manera uniforme durante al menos cinco minutos en solución salina sola o con un 5 % de DMSO, el aceite mineral (n.o CAS 8042-47-5) se utiliza como disolvente de tercera opción.
|
|
19.
|
Los productos problema se disuelven o suspenden uniformemente en el disolvente seleccionado, a una concentración del 5 % (p/p), y se someten posteriormente a dicuciones decimales en serie hasta llegar a las concentraciones del 0,5 % y 0,05 %. Cada producto problema debe someterse a ensayo a las dos concentraciones del 5 % y del 0,05 %. Las células cultivadas en la placa de 96 pocillos se exponen a 200 μl/pocillo de la solución (o suspensión) del producto problema a la concentración del 5 % o del 0,05 % durante cinco minutos a temperatura ambiente. Los productos problema (sustancias de un solo componente o sustancias de componentes múltiples o mezclas) se consideran sustancias puras y se diluyen o se suspenden con arreglo al método, independientemente de su pureza.
|
|
20.
|
El medio de cultivo descrito en el punto 16 se utiliza como testigo del medio en cada placa de cada repetición. Además, las células han de exponerse también a muestras de control del disolvente en cada placa de cada repetición. Se ha confirmado que los disolventes enumerados en el punto 18 no tienen ningún efecto adverso sobre la viabilidad de las células SIRC.
|
|
21.
|
En el método de ensayo EBD, se debe utilizar como testigo positivo en cada placa de cada repetición solución salina con un 0,01 % de laurilsulfato sódico (LSS). Para calcular la viabilidad celular del testigo positivo, cada placa de cada repetición debe incluir también un control del disolvente salino.
|
|
22.
|
Es necesario un ensayo en blanco para determinar la compensación respecto a la densidad óptica y debe realizarse con pocillos que contengan únicamente solución salina amortiguadora de fosfato, pero sin calcio ni magnesio (PBS-) ni células.
|
|
23.
|
Cada muestra (producto problema al 5 % y 0,05 %, testigo del medio, control del disolvente y testigo positivo) debe ensayarse por triplicado en cada repetición, exponiendo las células a 200 μl del producto problema o testigo adecuado durante cinco minutos a temperatura ambiente.
|
|
24.
|
Es útil disponer de sustancias de referencia para evaluar el potencial de irritación ocular de sustancias desconocidas dentro de una clase específica de sustancias o productos, o para evaluar la capacidad de irritación relativa de un irritante ocular dentro de una gama específica de respuestas de irritación.
|
Medición de la viabilidad celular
|
25.
|
Tras la exposición, las células se lavan dos veces con 200 μl de PBS y se añaden 200 μl de solución de MTT (0,5 mg MTT/ml de medio de cultivo). Tras un tiempo de reacción de dos horas en una incubadora (37 °C, 5 % de CO2), se decanta la solución de MTT, se extrae el formazano de MTT mediante la adición de 200 μl de isopropanol-ácido clorhídrico 0,04 N durante 60 minutos en la oscuridad a temperatura ambiente, y la absorbancia del formazano de MTT se mide a 570 nm con un lector de placas. Se produce interferencia de los productos problema con el ensayo del MTT (en caso de colorantes o reductores directos del MTT) solo si se mantiene una cantidad significativa de producto problema en el sistema de ensayo después del lavado tras la exposición, lo que sucede con los tejidos de epidermis humana reconstruida o la córnea humana reconstruida en 3D, pero no es pertinente en el caso de los cultivos celulares de 2D utilizados para el método de ensayo EBD.
|
Interpretación de los resultados y modelo de asignación
|
26.
|
Los valores de densidad óptica (DO) obtenidos con cada producto problema se utilizan a continuación para calcular la viabilidad celular respecto a la del control del disolvente, que se fija arbitrariamente en el 100 %. La viabilidad celular relativa se expresa en porcentaje y se obtiene dividiendo la DO del producto problema por la DO del control del disolvente, después de restar de ambos valores la DO del ensayo en blanco.

Análogamente, la viabilidad celular relativa se expresa en porcentaje y se obtiene dividiendo la DO del producto problema por la DO del control del disolvente, después de restar de ambos valores la DO del ensayo en blanco.
|
|
27.
|
Deben efectuarse tres repeticiones independientes, cada una de ellas con tres pocillos replicados (es decir, n = 9). Para calcular la media aritmética de la viabilidad celular relativa se utiliza la media aritmética de los tres pocillos de cada producto problema y del control del disolvente en cada repetición independiente. La media aritmética final de la viabilidad celular se calcula a partir de las tres repeticiones independientes.
|
|
28.
|
A continuación figuran los valores de corte de la viabilidad celular para la identificación de productos problema que provocan lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas y del CLP) y de productos problema que no requieren clasificación por irritación ocular o lesiones oculares graves (sin categoría del SGA de las Naciones Unidas y del CLP).
|
Cuadro 2
Modelo de asignación del método de ensayo EBD
|
Viabilidad celular
|
Clasificación del SGA de las Naciones Unidas / CLP
|
Aplicabilidad
|
|
Al 5 %
|
Al 0,05 %
|
|
> 70 %
|
> 70 %
|
Sin categoría
|
Sustancias y mezclas, excepto: i) las sustancias muy volátiles, con una presión de vapor de más de 6 kPa (75) y ii) los productos sólidos (sustancias y mezclas) distintos de los tensioactivos y las mezclas que solo se componen de tensioactivos
|
|
≤ 70 %
|
> 70 %
|
No puede hacerse ninguna asignación.
|
No aplicable.
|
|
≤ 70 %
|
≤ 70 %
|
Categoría 1
|
Sustancias y mezclas (76)
|
Criterios de aceptación
|
29.
|
Los resultados de los ensayos se consideran aceptables cuando se cumplen los siguientes criterios:
|
a)
|
La densidad óptica del testigo del medio (exposición al medio de cultivo) debe ser igual o superior a 0,3 tras restar la densidad óptica del blanco.
|
|
b)
|
La viabilidad del control del disolvente debe ser igual o superior al 80 % respecto al valor del testigo del medio. Si se utilizan varios testigos del disolvente en cada repetición, cada uno de estos testigos debe mostrar una viabilidad celular superior al 80 % para poder evaluar los productos problema ensayados con esos disolventes.
|
|
c)
|
La viabilidad celular obtenida con el testigo positivo (LSS al 0,01 %) debe encontrarse dentro del intervalo de dos veces la desviación típica respecto a la media histórica. Los límites de aceptación superior e inferior para el testigo positivo deben actualizarse con frecuencia, es decir, cada tres meses, o cada vez que se realice un ensayo aceptable en los laboratorios en los que se realizan ensayos con poca frecuencia (es decir, menos de una vez al mes). Cuando un laboratorio no complete un número suficiente de experimentos para establecer una distribución de los testigos positivos que sea estadísticamente sólida, puede utilizar los límites de aceptación superior e inferior establecidos por el diseñador del método, es decir, entre el 21,1 % y el 62,3 % en función de sus datos históricos de laboratorio, mientras que durante los primeros ensayos sistemáticos se construye una distribución interna.
|
|
d)
|
La desviación típica de la viabilidad celular final derivada de tres repeticiones independientes debe ser inferior al 15 % a las concentraciones tanto del 5 % como del 0,05 % del producto problema.
Si no se cumplen uno o varios de estos criterios, se deben descartar los resultados y se deben realizar otras tres repeticiones independientes.
|
|
DATOS E INFORME
Datos
|
30.
|
Deben comunicarse los datos de cada pocillo (por ejemplo, valores de viabilidad celular) de cada repetición, así como la media global, la desviación típica y la clasificación.
|
Informe del ensayo
|
31.
|
El informe del ensayo debe incluir la información siguiente:
|
|
Producto problema y sustancias testigo
|
—
|
Sustancias de un solo componente: identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
—
|
Sustancias de componentes múltiples, sustancias UVCB y mezclas: caracterización, en la medida de lo posible, mediante, por ejemplo, la identidad química (véase más arriba), pureza, presencia cuantitativa y propiedades fisicoquímicas pertinentes de los componentes (véase más arriba), según su disponibilidad;
|
|
—
|
Estado físico, volatilidad, pH, log P, peso molecular, clase química y otras propiedades fisicoquímicas pertinentes para la realización del estudio, según su disponibilidad;
|
|
—
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
—
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
—
|
Condiciones de conservación y estabilidad en la medida de lo posible.
|
|
|
|
Condiciones y procedimientos del método de ensayo
|
—
|
Nombre y dirección del promotor, laboratorio y director del estudio;
|
|
—
|
Descripción del método de ensayo utilizado;
|
|
—
|
Línea celular utilizada, su origen, número de pases y confluencia de las células utilizadas para los ensayos;
|
|
—
|
Particularidades del procedimiento de ensayo empleado;
|
|
—
|
Número de repeticiones y réplicas utilizadas;
|
|
—
|
Concentraciones del producto problema utilizadas (si son distintas de las recomendadas);
|
|
—
|
Justificación de la elección del disolvente para cada producto problema;
|
|
—
|
Duración de la exposición al producto problema (si es diferente de la recomendada);
|
|
—
|
Descripción de las eventuales modificaciones del procedimiento del ensayo;
|
|
—
|
Descripción de los criterios de evaluación y decisión seguidos;
|
|
—
|
Referencia a la media y desviación típica (DT) de los testigos positivos históricos:
|
|
—
|
Demostración de la competencia del laboratorio para utilizar el método de ensayo (por ejemplo, sometiendo a ensayo las sustancias para la prueba de la competencia) o demostración de la utilización reproducible del método de ensayo a lo largo del tiempo.
|
|
|
|
Resultados
|
—
|
Respecto a cada producto problema y sustancia testigo, y cada concentración sometida a ensayo, deben indicarse en un cuadro cada uno de los valores de DO por cada pocillo replicado, la media aritmética de los valores de DO de cada repetición independiente, la viabilidad celular porcentual de cada repetición independiente y la media aritmética final de la viabilidad celular porcentual y la desviación típica de las tres repeticiones;
|
|
—
|
Resultados de los testigos de medio, de disolvente y positivo, que demuestren el cumplimiento de los criterios adecuados de aceptación del estudio;
|
|
—
|
Descripción de otros efectos observados;
|
|
—
|
Clasificación general derivada con referencia al modelo de asignación o a los criterios de decisión utilizados.
|
|
|
|
Discusión de los resultados
|
|
BIBLIOGRAFÍA
|
(1)
|
Naciones Unidas (2013). Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA). Quinta edición revisada. Nueva York y Ginebra: Publicaciones de las Naciones Unidas. ISBN: 978-92-1-117006-1. Disponible en: http://www.unece.org/es/trans/danger/publi/ghs/ghs_rev05/05files_s.html
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, pp.1855-1869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Disponible en: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Capítulo B.5 del presente anexo, Irritación/corrosión ocular aguda.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS and Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1648-1653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Bruselas, Bélgica.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442–449.
|
|
(13)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(14)
|
OCDE (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Organización para la Cooperación y el Desarrollo Económico, París.
|
Apéndice
DEFINICIONES
Exactitud
: Grado de coincidencia entre los resultados obtenidos con el método de ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del método de ensayo y es un aspecto de su pertinencia. Este término y el de “concordancia” se suelen usar indistintamente para indicar la proporción de resultados correctos de un método de ensayo (13).
Sustancias de referencia
: Sustancias utilizadas como patrón para comparar con un producto problema. Las sustancias de referencia deben presentar las siguientes propiedades: i) un origen u orígenes coherentes y fiables; ii) similitud estructural y funcional con la clase de sustancias objeto del ensayo; iii) características físicas y químicas conocidas; iv) datos de apoyo sobre los efectos conocidos, y v) potencia conocida en la banda de la respuesta deseada.
Enfoque ascendente
: Enfoque gradual utilizado para un producto problema del que se piensa que no requiere clasificación por irritación ocular o lesiones oculares graves, que comienza con la determinación de los productos que no requieren clasificación (resultado negativo) frente a otros productos (resultado positivo).
Producto
: Sustancia o mezcla.
Irritación ocular
: Producción de cambios en el ojo, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, que son totalmente reversibles en los 21 días siguientes a la aplicación. Intercambiable con “efectos reversibles en el ojo” y con la categoría 2 del SGA de las Naciones Unidas y del CLP.
Tasa de falsos negativos
: Proporción de todos los productos positivos identificados erróneamente como negativos por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo.
Tasa de falsos positivos
: Proporción de todos los productos negativos identificados erróneamente como positivos por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo.
Peligro
: Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente.
Testigo del medio
: Muestra replicada no tratada que contiene todos los componentes de un sistema de ensayo. Esta muestra se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras testigo para determinar si el disolvente interactúa con el sistema de ensayo.
Mezcla
: Mezcla o solución compuesta de dos o más sustancias.
Sustancia de un solo componente
: Sustancia, definida por su composición cuantitativa, en la que un solo componente principal representa al menos el 80 % (p/p).
MTT
: Bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio; bromuro de tetrazolio de azul de tiazolilo.
Sustancia de componentes múltiples
: Sustancia, definida por su composición cuantitativa, en la que hay varios componentes principales presentes a una concentración ≥ 10 % (p/p) y < 80 % (p/p). Una sustancia de componentes múltiples es el resultado de un proceso de fabricación. La diferencia entre mezcla y sustancia de componentes múltiples es que una mezcla se obtiene mezclando dos o más sustancias sin reacción química. Una sustancia de componentes múltiples es el resultado de una reacción química.
DO
: Densidad óptica.
Testigo positivo
: Réplica que contiene todos los componentes de un sistema de ensayo y que se trata con una sustancia de la que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta positiva.
Pertinencia
: Descripción de la relación del ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o clasifica correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un método de ensayo (10).
Fiabilidad
: Medida del grado en que un método de ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios (13).
Sensibilidad
: Proporción de todos los productos activos/positivos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (10).
Lesiones oculares graves
: Producción de lesiones tisulares en el ojo o una degradación física severa de la vista, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, y que no es totalmente reversible en los 21 días siguientes a la aplicación. Intercambiable con “efectos irreversibles en el ojo” y con la categoría 1 del SGA de las Naciones Unidas y del CLP.
Control del disolvente/vehículo
: Muestra no tratada que contiene todos los componentes de un sistema de ensayo, incluido el disolvente o vehículo, y que se somete al mismo proceso que las muestras tratadas con el producto problema y otras muestras testigo a fin de determinar la respuesta de base correspondiente a las muestras tratadas con el producto problema disuelto en el mismo disolvente o vehículo. Cuando se somete a ensayo con un testigo del medio en paralelo, esta muestra pone de manifiesto también si el disolvente o vehículo interactúa con el sistema de ensayo.
Especificidad
: Proporción de todos los productos inactivos/negativos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (13).
Sustancia
: Elemento químico y sus compuestos naturales u obtenidos mediante algún proceso de producción, incluidos los eventuales aditivos necesarios para mantener su estabilidad y las eventuales impurezas que se produzcan en el proceso, con exclusión de los eventuales disolventes que puedan separarse sin afectar a la estabilidad de la sustancia ni modificar su composición.
Tensioactivo
: También denominado agente tensioactivo, es un producto, como un detergente, que puede reducir la tensión superficial de un líquido y, de este modo, permitir que forme espuma o penetre en los sólidos. También se conoce como agente humectante.
Producto problema
: Toda sustancia o mezcla sometida a ensayo con este método de ensayo.
Estrategia de ensayos escalonados
: Estrategia de ensayo por fases, en la que se revisa toda la información existente sobre un producto problema, siguiendo un orden especificado, en un proceso de ponderación de las pruebas en cada escalón, a fin de determinar si se dispone de información suficiente para tomar una decisión sobre la clasificación de un peligro, antes de pasar al escalón siguiente. Si puede establecerse la capacidad de irritación de un producto problema con la información disponible, no hace falta efectuar más ensayos. Si no puede establecerse la capacidad de irritación de un producto problema con la información disponible, se aplica un procedimiento gradual de ensayos con animales por fases hasta que pueda efectuarse una clasificación clara.
Enfoque descendente
: Enfoque gradual utilizado para un producto problema del que se sospecha que causa lesiones oculares graves, que comienza con la determinación de los productos que inducen lesiones oculares graves (resultado positivo) frente a otros productos (resultado negativo).
Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de las Naciones Unidas)
: Sistema que propone la clasificación de los productos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de respuesta a emergencias) y al medio ambiente (1).
Categoría 1 del SGA de las Naciones Unidas y del CLP
: Véase “Lesiones oculares graves”.
Categoría 2 del SGA de las Naciones Unidas y del CLP
: Véase “Irritación ocular”.
Sin categoría del SGA de las Naciones Unidas y del CLP
: Productos no clasificados en las categorías 1 o 2 del SGA de las Naciones Unidas y del CLP (o en las categorías 2A o 2B del SGA de las Naciones Unidas).
UVCB
: Sustancias de composición desconocida o variable, productos de reacción compleja y materiales biológicos.
B.69. MÉTODO DE ENSAYO CON EPITELIO HUMANO RECONSTRUIDO SIMILAR A LA CÓRNEA (EHRSC) PARA IDENTIFICAR PRODUCTOS QUE NO REQUIEREN CLASIFICACIÓN Y ETIQUETADO POR IRRITACIÓN OCULAR O LESIONES OCULARES GRAVES
INTRODUCCIÓN
|
1.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 492 de la OCDE (2017). El término lesiones oculares graves se refiere a la producción de lesiones tisulares en el ojo o de una degradación física severa de la vista, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, y que no es totalmente reversible en los 21 días siguientes a la aplicación según la definición del Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA) de las Naciones Unidas (1) y del Reglamento (UE) n.o 1272/2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) (77) de la Unión Europea. También de acuerdo con el SGA de las Naciones Unidas y el CLP, el término irritación ocular se refiere a la producción de cambios en el ojo, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, que son totalmente reversibles en los 21 días siguientes a la aplicación. Los productos problema que provocan lesiones oculares graves se clasifican en la categoría 1 del SGA de las Naciones Unidas y del CLP, mientras que los que inducen irritación ocular se clasifican en la categoría 2 del SGA de las Naciones Unidas y del CLP. Los productos problema no clasificados por irritación ocular o lesiones oculares graves se definen como aquellos que no cumplen los requisitos para la clasificación en las categorías 1 o 2 (2A o 2B) del SGA de las Naciones Unidas y del CLP, es decir, se mencionan como «sin categoría» del SGA de las Naciones Unidas y del CLP.
|
|
2.
|
La evaluación de la irritación ocular / lesiones oculares graves ha supuesto normalmente la utilización de animales de laboratorio [método de ensayo B.5 (2)]. La elección del método de ensayo más adecuado y el uso de este método de ensayo deben considerarse en el contexto del documento de orientación de la OCDE sobre un enfoque integrado de ensayos y evaluación de las lesiones oculares graves y la irritación ocular (39).
|
|
3.
|
Este método de ensayo describe un procedimiento in vitro que permite la identificación de los productos (sustancias y mezclas) que no requieren clasificación y etiquetado por lesiones oculares graves o irritación ocular según el SGA de las Naciones Unidas y el CLP. Hace uso del epitelio humano reconstruido similar a la córnea (EHRSC), que imita bien las propiedades histológicas, morfológicas, bioquímicas y fisiológicas del epitelio de córnea humana. Otros cuatro métodos de ensayo in vitro se han validado, se han considerado científicamente válidos y se han adoptado como métodos de ensayo B.47 (3), B.48 (4), B.61 (5) y B.68 (6) para evaluar el parámetro de las lesiones oculares graves / irritación ocular en la salud humana.
|
|
4.
|
En este método de ensayo se incluyen dos ensayos validados que utilizan modelos de EHRSC disponibles comercialmente. Se han realizado estudios de validación para evaluar la irritación ocular / lesiones oculares graves (7) (8) (9) (10) (11) (12) (13) utilizando el ensayo de irritación ocular (EIO) EpiOcular™ y el EIO con epitelio de córnea humana (ECH) SkinEthic™. En cada uno de ellos se utilizan como sistema de ensayo construcciones tisulares de EHRSC disponibles en el mercado, y se nombran en lo sucesivo como los métodos de referencia validados — MRV1 y MRV2, respectivamente. A partir de estos estudios de validación y de su revisión independiente por pares (9) (12) se llegó a la conclusión de que el EIO EpiOcular™ y el EIO con ECH SkinEthic™ son capaces de identificar correctamente los productos (tanto sustancias como mezclas) que no requieren clasificación y etiquetado por irritación ocular o lesiones oculares graves, según el SGA de las Naciones Unidas, y se recomendaron los ensayos como científicamente válidos para ese propósito (13).
|
|
5.
|
Actualmente se acepta que, en un futuro próximo, ningún método de ensayo in vitro podrá sustituir completamente por sí solo al ensayo ocular de Draize in vivo (2) (14) para clasificar toda la gama de respuestas de lesiones oculares graves o de irritación ocular que pueden causar diferentes clases de productos. Sin embargo, unas combinaciones estratégicas de varios métodos de ensayo alternativos dentro de una estrategia de ensayos (escalonados), como es el enfoque descendente/ascendente, sí podrán sustituir completamente al ensayo ocular de Draize (15). El enfoque ascendente (15) está diseñado para ser utilizado cuando, sobre la base de la información existente, se espere que un producto no cause irritación ocular suficiente para exigir una clasificación, mientras que el enfoque descendente (15) está diseñado para ser utilizado cuando, sobre la base de la información existente, se espere que un producto cause lesiones oculares graves. Se recomiendan el EIO EpiOcular™ y el EIO con ECH SkinEthic™ para identificar los productos que no requieren clasificación por irritación ocular o lesiones oculares graves de acuerdo con el SGA delas Naciones Unidas y del CLP (sin categoría) sin más ensayos, en el marco de una estrategia de ensayo como el enfoque ascendente/descendente sugerido por Scott et al., por ejemplo como un primer paso de un enfoque ascendente o como uno de los últimos pasos de un enfoque descendente (15). Sin embargo, el EIO EpiOcular™ y el EIO con ECH SkinEthic™ no pretenden diferenciar entre la categoría 1 (lesiones oculares graves) y la categoría 2 (irritación ocular) del SGA de las Naciones Unidas y del CLP. Esta diferenciación deberá abordarse a otro escalón de la estrategia de ensayo (15). Por tanto, un producto problema del que se sepa que hay que adjudicarle una categoría de clasificación por irritación ocular o lesiones oculares graves con el EIO EpiOcular™ o el EIO con ECH SkinEthic™ requerirá ensayos adicionales (in vitro o in vivo) para llegar a una conclusión definitiva (sin categoría, categoría 2 o categoría 1 del SGA de las Naciones Unidas y del CLP) utilizando, por ejemplo, los métodos de ensayo B.47, B.48, B.61 o B.68.
|
|
6.
|
El objetivo del presente método de ensayo es describir el procedimiento utilizado a fin de evaluar el potencial de peligro para los ojos de un producto problema en función de su capacidad de inducir citotoxicidad en una construcción tisular de EHRSC, medida con el ensayo de MTT (16) (véase el punto 21). La viabilidad del tejido del EHRSC tras la exposición a un producto problema se determina comparándola con la de los tejidos tratados con la sustancia testigo negativo (% de viabilidad), y se utiliza a continuación para clasificar el potencial de peligro para los ojos del producto problema.
|
|
7.
|
Se dispone de normas de comportamiento (17) que facilitan la validación de ensayos in vitro nuevos o modificados basados en el EHRSC, similares al EIO EpiOcular™ o al EIO con ECH SkinEthic™, de conformidad con los principios del documento de orientación n.o 34 de la OCDE (18), y tienen en cuenta la modificación oportuna de las directrices de ensayo TG 492 de la OCDE para su inclusión. La mutua aceptación de datos (MAD) con arreglo al acuerdo de la OCDE solo estará garantizada respecto a los ensayos validados de conformidad con las normas de comportamiento, si estos ensayos han sido revisados e incluidos en las correspondientes directrices de ensayo por la OCDE.
|
DEFINICIONES
|
8.
|
En el apéndice 1 se dan las definiciones pertinentes.
|
CONSIDERACIONES INICIALES Y LIMITACIONES
|
9.
|
Este método de ensayo se basa en construcciones tisulares de EHRSC tridimensionales disponibles en el mercado que se producen bien utilizando queratinocitos epidérmicos humanos primarios (es decir, OCL-200 en el caso de EpiOcular™) o células epiteliales de córnea humana inmortalizadas (es decir, ECH/S en el caso de SkinEthic™). Las construcciones tisulares de EHRSC OCL-200 de EpiOcular™ y ECH/S de SkinEthic™ son similares a la estructura tridimensional del epitelio de córnea in vivo y se producen a partir de células procedentes de la especie de interés (19) (20). Además, los ensayos miden directamente la citotoxicidad resultante de la penetración del producto a través de la córnea y la producción de lesiones celulares y tisulares; la respuesta citotóxica determina entonces el resultado global de lesiones oculares graves o irritación ocular in vivo. Pueden producirse lesiones celulares a través de varios modos de acción (véase el punto 20), pero la citotoxicidad desempeña un papel importante, si no el principal, en cuanto al mecanismo de la determinación de la respuesta global a un producto en forma de irritación ocular o lesiones oculares graves, y se manifiesta in vivo principalmente mediante opacidad de la córnea, iritis, enrojecimiento conjuntival y/o quemosis conjuntival, independientemente de los procesos fisicoquímicos que causan las lesiones tisulares.
|
|
10.
|
En el estudio de validación en que se basa este método de ensayo se ha probado una amplia gama de productos, que abarcan una gran variedad de tipos químicos, clases químicas, pesos moleculares, log P, estructuras químicas, etc. La base de datos de validación del EIO EpiOcular™ contenía 113 productos en total, correspondientes a 95 grupos funcionales orgánicos diferentes según un análisis con la caja de herramientas QSAR (relación cuantitativa estructura-actividad) de la OCDE (8). La mayoría de estos productos representaban sustancias de un solo componente, pero también se incluyeron en el estudio diversas sustancias de componentes múltiples (incluidos 3 homopolímeros, 5 copolímeros y 10 cuasipolímeros). En términos de estado físico y categorías del SGA de la ONU y del CLP, los 113 productos sometidos a ensayo se distribuían como sigue: 13 líquidos de categoría 1, 15 sólidos de categoría 1, 6 líquidos de categoría 2A, 10 sólidos de categoría 2A, 7 líquidos de categoría 2B, 7 sólidos de categoría 2B, 27 líquidos sin categoría y 28 sólidos sin categoría (8). La base de datos de validación del EIO con ECH SkinEthic™ contenía 200 productos en total, correspondientes a 165 grupos funcionales orgánicos (8) (10) (11). La mayoría de estos productos representaban sustancias de un solo componente, pero también se incluyeron en el estudio diversas sustancias de componentes múltiples (incluidos 10 polímeros). En términos de estado físico y categorías del SGA de la ONU y del CLP, los 200 productos sometidos a ensayo se distribuían como sigue: 27 líquidos de categoría 1, 24 sólidos de categoría 1, 19 líquidos de categoría 2A, 10 sólidos de categoría 2A, 9 líquidos de categoría 2B, 8 sólidos de categoría 2B, 50 líquidos sin categoría y 53 sólidos sin categoría (10) (11).
|
|
11.
|
Este método de ensayo es aplicable a sustancias y mezclas, y a sólidos, líquidos, semisólidos y ceras. Los líquidos pueden ser acuosos o no acuosos; los sólidos pueden ser solubles o insolubles en agua. Siempre que sea posible, los sólidos deben molerse hasta convertirse en polvo fino antes de su aplicación; no se requiere ningún otro tratamiento previo de la muestra. No se ha efectuado ningún estudio de validación con gases y aerosoles. Aunque no hay que descartar que los gases y aerosoles puedan estudiarse utilizando la tecnología del EHRSC, el actual método de ensayo no permite hacerlo.
|
|
12.
|
Los productos problema que absorben la luz en el mismo intervalo que el formazano de MTT (de forma natural o previo tratamiento) y los productos problema capaces de reducir directamente el colorante vital MTT (a formazano de MTT) pueden interferir con las mediciones de la viabilidad tisular y hacen necesario el uso de testigos adaptados para efectuar las correcciones oportunas. El tipo de testigos adaptados que puede ser necesario varía en función del tipo de interferencia producida por el producto problema y del procedimiento utilizado para cuantificar el formazano de MTT (véanse los puntos 36-42).
|
|
13.
|
Los resultados obtenidos en los estudios de prevalidación (21) (22) y de validación completa (8) (10) (11) han demostrado que tanto el EIO EpiOcular™ como el EIO con ECH SkinEthic™ son transferibles a laboratorios sin experiencia previa con la realización de los ensayos y que también pueden ser reproducibles en un mismo laboratorio y en distintos laboratorios. Sobre la base de estos estudios, el nivel de reproducibilidad en términos de concordancia de las asignaciones que cabe esperar del EIO EpiOcular™ a partir de datos de 113 productos es del orden del 95 % en un mismo laboratorio y del 93 % en distintos laboratorios. El nivel de reproducibilidad en términos de concordancia de las asignaciones que cabe esperar del EIO con ECH SkinEthic™ a partir de datos de 120 productos es del orden del 92 % en un mismo laboratorio y del 95 % en distintos laboratorios.
|
|
14.
|
El EIO EpiOcular™ puede utilizarse para identificar productos que no requieren clasificación por irritación ocular o lesiones oculares graves con arreglo al sistema de clasificación del SGA de las Naciones Unidas y del CLP. Teniendo en cuenta los datos obtenidos en el estudio de validación (8), el EIO EpiOcular™ tiene una exactitud global del 80 % (sobre 112 productos), una sensibilidad del 96 % (sobre 57 productos), una tasa de falsos negativos del 4 % (sobre 57 productos), una especificidad del 63 % (sobre 55 productos) y una tasa de falsos positivos del 37 % (sobre 55 productos), en comparación con los datos del ensayo de referencia con ojos de conejo in vivo (método de ensayo B.5) (2) (14) clasificados según el sistema de clasificación del SGA de las Naciones Unidas y del CLP. Un estudio en el que se analizaron 97 formulaciones agroquímicas líquidas con el EIO EpiOcular™ demostró un comportamiento del método de ensayo con este tipo de mezclas similar al obtenido en el estudio de validación (23). Las 97 formulaciones se distribuían del siguiente modo: 21 de categoría 1, 19 de categoría 2A, 14 de categoría 2B y 43 sin categoría, clasificadas según el sistema de clasificación del SGA de las Naciones Unidas sobre la base de los datos del ensayo de referencia con ojos de conejo in vivo (método de ensayo B.5) (2) (14). Se obtuvo una exactitud global del 82 % (sobre 97 formulaciones), una sensibilidad del 91 % (sobre 54 formulaciones), una tasa de falsos negativos del 9 % sobre 54 formulaciones), una especificidad del 72 % (sobre 43 formulaciones) y una tasa de falsos positivos del 28 % (sobre 43 formulaciones) (23).
|
|
15.
|
El EIO con ECH SkinEthic™ puede utilizarse para identificar productos que no requieren clasificación por irritación ocular o lesiones oculares graves con arreglo al sistema de clasificación del SGA de las Naciones Unidas y del CLP. Teniendo en cuenta los datos obtenidos en el estudio de validación (10) (11), el EIO con ECH SkinEthic™ tiene una exactitud global del 84 % (sobre 200 productos), una sensibilidad del 95 % (sobre 97 productos), una tasa de falsos negativos del 5 % (sobre 97 productos), una especificidad del 72 % (sobre 103 productos) y una tasa de falsos positivos del 28 % (sobre 103 productos), en comparación con los datos del ensayo de referencia con ojos de conejo in vivo (método de ensayo B.5) (2) (14) clasificados según el sistema de clasificación del SGA de las Naciones Unidas y del CLP.
|
|
16.
|
Las tasas de falsos negativos obtenidas con ambos ensayos EHRSC, con sustancias o con mezclas, se sitúan dentro de la probabilidad global del 12 % de que se identifiquen los productos bien como de la categoría 2 del SGA de las Naciones Unidas y del CLP, bien como sin categoría del SGA de las Naciones Unidas y del CLP por el ensayo ocular de Draize in vivo, en ensayos repetidos; esto se debe a la variabilidad dentro del ensayo que presenta el métodode forma inherente (24). Las tasas de falsos positivos obtenidas con ambos ensayos EHRSC, con sustancias o con mezclas, no son críticas en el contexto del presente método, ya que todos los productos problema que inducen una viabilidad celular igual o inferior a los valores límite establecidos (véase el punto 44) requieren nuevos análisis con otros métodos de ensayo in vitro o, como última opción, con conejos, en función de los requisitos normativos y de conformidad con una estrategia de evaluación secuencial dentro de un enfoque de ponderación de las pruebas. Estos métodos de ensayo pueden utilizarse con todos los tipos de productos, y un resultado negativo con ellos debe aceptarse para no clasificar un producto por irritación ocular ni lesiones oculares graves (sin categoría del SGA de las Naciones Unidas y del CLP). Hay que consultar a las autoridades reguladoras competentes antes de utilizar el EIO EpiOcular™ y el EIO con ECH SkinEthic™ en un sistema de clasificación distinto del SGA de las Naciones Unidas y del CLP.
|
|
17.
|
Una limitación de este método de ensayo es que no permite discriminar entre irritación ocular / efectos reversibles en el ojo (categoría 2) y lesiones oculares graves / efectos irreversibles en el ojo (categoría 1) según lo definido por el SGA de las Naciones Unidas y el CLP, ni entre irritantes oculares (categoría optativa 2A) e irritantes oculares leves (categoría optativa 2B), definidos por el SGA de las Naciones Unidas (1). A estos efectos, es necesario realizar más pruebas con otros métodos de ensayo in vitro.
|
|
18.
|
El término «producto problema» se utiliza en el presente método de ensayo para referirse al objeto del ensayo (78) y no está relacionado con la aplicabilidad del método de ensayo EHRSC al ensayo de sustancias o mezclas.
|
PRINCIPIO DEL ENSAYO
|
19.
|
El producto problema se aplica tópicamente a un mínimo de dos construcciones tisulares tridimensionales de EHRSC y se mide la viabilidad de los tejidos después de la exposición y de un período de incubación tras el tratamiento. Los tejidos de EHRSC se reconstruyen a partir de queratinocitos epidérmicos humanos primarios o de células epiteliales de córnea humana inmortalizadas, que se han cultivado durante varios días para formar un epitelio escamoso estratificado y muy diferenciado, similar al encontrado en la córnea humana. La construcción tisular de EHRSC del ensayo EpiOcular™ consiste en al menos tres capas viables de células y una superficie no queratinizada, con una estructura similar a la córnea, análoga a la que se encuentra in vivo. La construcción tisular de EHRSC del ensayo ECH SkinEthic™ consiste en al menos cuatro capas viables de células, con inclusión de células basales cilíndricas, células de transición aladas y células superficiales escamosas, similares a las del epitelio de córnea humana normal (20) (26).
|
|
20.
|
La irritación ocular / lesiones oculares graves inducidas por un producto, que se manifiestan in vivo principalmente mediante opacidad de la córnea, iritis, enrojecimiento conjuntival y/o quemosis conjuntival, son el resultado de una cascada de sucesos que comienzan por la penetración del producto a través de la córnea y/o de la conjuntiva y la producción de lesiones a las células. Estas lesiones celulares pueden producirse a través de varios modos de acción, como los siguientes: lisis de la membrana celular (por ejemplo, por agentes tensioactivos, disolventes orgánicos); coagulación de macromoléculas (en particular, proteínas) (por ejemplo, por agentes tensioactivos, disolventes orgánicos, álcalis y ácidos); saponificación de lípidos (por ejemplo, por álcalis); y alquilación u otras interacciones covalentes con macromoléculas (por ejemplo, por hipocloritos, peróxidos y alquilantes) (15) (27) (28). Sin embargo, se ha demostrado que la citotoxicidad desempeña un papel importante, si no el principal, en cuanto al mecanismo de la determinación de la respuesta global a un producto en forma de irritación ocular o lesiones oculares graves, independientemente de los procesos fisicoquímicos que causan las lesiones tisulares (29) (30). Por otra parte, el potencial de lesiones oculares graves / irritación ocular de un producto se determina principalmente por la magnitud de la lesión inicial (31), que se corresponde con la magnitud de la muerte celular (29) y con la magnitud de las respuestas posteriores y los resultados finales (32). Así pues, los irritantes leves generalmente afectan solo al epitelio superficial de la córnea, los irritantes suaves y moderados dañan principalmente el epitelio y el estroma superficial, y los irritantes intensos dañan el epitelio, el estroma profundo y a veces el endotelio de la córnea (30) (33). La medición de la viabilidad de la construcción tisular de EHRSC después de la exposición tópica a un producto problema a fin de identificar los productos que no requieren clasificación por lesiones oculares graves o irritación ocular (sin categoría del SGA de las Naciones Unidas y del CLP) se basa en el supuesto de que todos los productos que provoquen lesiones oculares graves o irritación ocular provocarán citotoxicidad en el epitelio de la córnea y/o en la conjuntiva.
|
|
21.
|
La viabilidad tisular del EHRSC se mide clásicamente mediante la conversión enzimática del colorante vital MTT [bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio; bromuro de tetrazolio de azul de tiazolilo; número CAS 298-93-1] por las células viables del tejido, que lo transforman en una sal azul de formazano de MTT, la cual se mide cuantitativamente tras su extracción de los tejidos (16). Los productos que no requieren clasificación y etiquetado de acuerdo con el SGA de las Naciones Unidas y el CLP (sin categoría) se identifican como aquellos que no disminuyen la viabilidad tisular por debajo de un determinado umbral (es decir, viabilidad tisular > 60 %, en el EIO EpiOcular™ y en el EIOL con ECH SkinEthic™ (79), o > 50 % en el EIOS con ECH SkinEthic™ (80)) (véase el punto 44).
|
DEMOSTRACIÓN DE LA COMPETENCIA
|
22.
|
Antes de proceder al uso sistemático de los ensayos con EHRSC con fines normativos, los laboratorios deben demostrar su competencia técnica mediante la asignación correcta de los quince productos para la prueba de la competencia que figuran en el cuadro 1. Estos productos se seleccionaron a partir de entre los productos utilizados en los estudios de validación de los MRV (8) (10) (11). En la selección se incluyen, en la medida de lo posible, productos que: i) tienen estados físicos diferentes; ii) abarcan toda la gama de respuestas in vivo de lesiones oculares graves / irritación ocular basadas en resultados de alta calidad obtenidos en el ensayo de referencia con ojos de conejo in vivo (método de ensayo B.5) (2) (14) y utilizando el SGA de las Naciones Unidas (es decir, categorías 1, 2A, 2B, o sin categoría) (1) y el sistema de clasificación del CLP (es decir, categorías 1, 2 o sin categoría); iii) abarcan los diversos factores in vivo de la clasificación (24) (25); iv) son representativos de las clases químicas utilizadas en el estudio de validación (8) (10) (11); v) suponen una buena y amplia representación de grupos funcionales orgánicos (8) (10) (11); vi) tienen estructuras químicas bien definidas (8) (10) (11); vii) están coloreados o son reductores directos del MTT; viii) producen resultados reproducibles según los métodos de ensayo con EHRSC durante sus validaciones; ix) durante sus estudios de validación, los métodos de ensayo con EHRSC las han clasificado correctamente; x) abarcan toda la gama de respuestas in vitro sobre la base de los datos de alta calidad de los métodos de ensayo con EHRSC (de 0 a 100 % de viabilidad); xi) están disponibles en el comercio; y xii) no tienen asociados costes prohibitivos de adquisición ni de eliminación. En situaciones en las que un producto de la lista no esté disponible o no pueda utilizarse por otras razones justificadas, podrá utilizarse otro producto que cumpla los criterios descritos anteriormente, por ejemplo alguno de los productos utilizados en la validación del MRV. No obstante, tales desviaciones deben estar justificadas.
Cuadro 1
Lista de sustancias para la prueba de la competencia
|
Nombre químico
|
N.o CAS
|
Grupo funcional orgánico (81)
|
Estado físico
|
Viabilidad del MRV1 (%) (82)
|
Viabilidad del MRV2 (%) (83)
|
Asignación según el MRV
|
Reductor del MTT
|
Interf. color
|
|
Categoría 1 in vivo
(84)
|
|
|
Tioglicolato de metilo
|
2365-48-2
|
Ésteres del ácido carboxílico; tioalcoholes
|
L
|
10,9 ± 6,4
|
5,5 ± 7,4
|
No puede hacerse ninguna asignación.
|
Sí (fuerte)
|
No
|
|
Acrilato de hidroxietilo
|
818-61-1
|
Acrilatos; alcoholes
|
L
|
7,5 ± 4,7 (85)
|
1,6 ± 1,0
|
No puede hacerse ninguna asignación.
|
No
|
No
|
|
2,5-Dimetil-2,5-hexanodiol
|
110-03-2
|
Alcoholes
|
S
|
2,3 ± 0,2
|
0,2 ± 0,1
|
No puede hacerse ninguna asignación.
|
No
|
No
|
|
Oxalato sódico
|
62-76-0
|
Ácidos oxocarboxílicos
|
S
|
29,0 ± 1,2
|
5,3 ± 4,1
|
No puede hacerse ninguna asignación.
|
No
|
No
|
|
Categoría 2A in vivo (84)
|
|
|
Di-D-gluconato de N,N″-bis(4-clorofenil)-3,12-diimino-2,4,11,13-tetraazatetradecano-diimidamida (solución acuosa al 20 %) (86)
|
18472-51-0
|
Haluros heterocíclicos aromáticos; haluros de arilo; grupos dihidroxilos; guanidinas
|
L
|
4,0 ± 1,1
|
1,3 ± 0,6
|
No puede hacerse ninguna asignación.
|
No
|
Sí (débil)
|
|
Benzoato sódico
|
532-32-1
|
Arilos; ácidos carboxílicos
|
S
|
3,5 ± 2,6
|
0,6 ± 0,1
|
No puede hacerse ninguna asignación.
|
No
|
No
|
|
Categoría 2B in vivo
(84)
|
|
|
Dietil-toluamida
|
134-62-3
|
Benzamidas
|
L
|
15,6 ± 6,3
|
2,8 ± 0,9
|
No puede hacerse ninguna asignación.
|
No
|
No
|
|
2,2-Dimetil-3-metilen-biciclo[2.2.1]heptano
|
79-92-5
|
Alcanos ramificados con carbono terciario; alquenos; bicicloheptanos; carbociclos con puente; cicloalcanos
|
S
|
4,7 ± 1,5
|
15,8 ± 1,1
|
No puede hacerse ninguna asignación.
|
No
|
No
|
|
Sin categoría in vivo
(84)
|
|
|
Etilsulfato de 1-etil-3-metilimidazolio
|
342573-75-5
|
Alcóxidos; sales de amonio; arilos; imidazoles; sulfatos
|
L
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Sin categoría
|
No
|
No
|
|
Éter dicaprílico
|
629-82-3
|
Alcóxidos; éteres
|
L
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Sin categoría
|
No
|
No
|
|
Butoxido de piperonilo
|
51-03-6
|
Alcóxidos; benzodioxoles; bencilos; éteres
|
L
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Sin categoría
|
No
|
No
|
|
Aceite de ricino hidrogenado con polietilenglicol (PEG-40)
|
61788-85-0
|
Acilales; alcoholes; alilos; éteres
|
Viscoso
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Sin categoría
|
No
|
No
|
|
1-(4-Clorofenil)-3-(3,4-diclorofenil)urea
|
101-20-2
|
Haluros heterocíclicos aromáticos; haluros de arilo; derivados de la urea
|
S
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Sin categoría
|
No
|
No
|
|
2,2′-Metilen-bis-(6-(2H-benzotriazol-2-il)-4-(1,1,3,3-tetrametil-butil)fenol
|
103597-45-1
|
Alcanos ramificados con carbono cuaternario; carbociclos aromáticos fusionados; heterociclos saturados fusionados; compuestos quinonoides precursores; terc-butilos
|
S
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Sin categoría
|
No
|
No
|
|
Tetrafluoroborato potásico
|
14075-53-7
|
Sales inorgánicas
|
S
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Sin categoría
|
No
|
No
|
|
Abreviaturas:
N.o CAS = número de registro del Chemical Abstracts Service; SAG de las Naciones Unidas = Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (1); MRV1 = Método de referencia validado, EIO EpiOcular™; MRV2 = Método de referencia validado, EIO con ECH SkinEthic™; Interf. color = interferencia de color con la medición normal de la absorbancia [densidad óptica (DO)] del formazano de MTT.
|
|
|
23.
|
Como parte de la prueba de la competencia, se recomienda que el usuario verifique las propiedades de barrera de los tejidos tras su recepción, siguiendo las especificaciones del productor de la construcción tisular del EHRSC (véanse los puntos 25, 27 y 30). Este aspecto es particularmente importante si el envío de los tejidos implica grandes distancias o largos plazos. Una vez se haya establecido con éxito un método y se haya adquirido y demostrado la competencia para su uso, no será necesario efectuar esta verificación de forma sistemática. Sin embargo, cuando se utilice sistemáticamente un método, se recomienda seguir evaluando las propiedades de barrera a intervalos periódicos.
|
PROCEDIMIENTO
|
24.
|
Los ensayos que se acogen actualmente a este método de ensayo son el EIO EpiOcular™ y el EIO con ECH SkinEthic™ (9) (12) (13), mencionados como el método de referencia validado (MRV1 y MRV2, respectivamente). Se dispone de procedimientos normalizados de trabajo (PNT) para los métodos de ensayo con EHRSC y deben emplearse cuando se aplican y utilizan los métodos de ensayo en el laboratorio (34) (35). En los puntos siguientes y en el apéndice 2 se describen los principales componentes y procedimientos de los ensayos con EHRSC.
|
COMPONENTES DEL MÉTODO DE ENSAYO CON EHRSC
Condiciones generales
|
25.
|
Deben utilizarse células pertinentes de origen humano para reconstruir el tejido tridimensional de epitelio similar a la córnea, tejido que debe estar compuesto de células progresivamente estratificadas pero no queratinizadas. La construcción tisular de EHRSC se prepara en piezas con una membrana sintética porosa a través de la cual los nutrientes pueden pasar a las células. En el epitelio reconstruido similar a la córnea deben estar presentes varias capas de células epiteliales viables y no queratinizadas. La construcción tisular de EHRSC debe tener la superficie epitelial en contacto directo con el aire para permitir la exposición tópica directa de los productos problema de forma similar a la de exposición in vivo del epitelio corneal. La construcción tisular de EHRSC debe constituir una barrera funcional con suficiente solidez para resistir la rápida penetración de sustancias citotóxicas de referencia como, por ejemplo, Tritón X-100 o dodecilsulfato sódico (DSS). La función de barrera debe demostrarse y puede evaluarse determinando bien el tiempo de exposición necesario para reducir la viabilidad tisular en un 50 % (TE50) tras la aplicación de una sustancia de referencia a una concentración fija especificada [por ejemplo, 100 μl de Tritón X-100 al 0,3 % (v/v)], o bien la concentración a la que una sustancia de referencia reduce la viabilidad de los tejidos en un 50 % (CI50) tras un tiempo fijo de exposición (por ejemplo, tratamiento de 30 minutos con 50 μl de DSS) (véase el punto 30). Las propiedades de aislamiento de la construcción tisular de EHRSC deben evitar que pase producto problema por la periferia del tejido viable, lo que podría reducir la calidad del modelo en cuanto a la exposición de la córnea. Las células de origen humano utilizadas para la construcción tisular de EHRSC deben estar libres de contaminación por bacterias, virus, micoplasmas y hongos. El proveedor debe comprobar la esterilidad de la construcción tisular en cuanto a la ausencia de contaminación por hongos y bacterias.
|
Condiciones funcionales
Viabilidad
|
26.
|
El ensayo utilizado para cuantificar la viabilidad tisular es el ensayo de MTT (16). Las células viables del conjunto tisular de EHRSC reducen el colorante vital MTT a un precipitado azul de formazano de MTT, que se extrae a continuación del tejido utilizando isopropanol (o un disolvente similar). El formazano de MTT extraído puede cuantificarse utilizando una medición normal de la absorbancia (densidad óptica, DO) o un procedimiento de espectrofotometría con HPLC/UPLC (36). La DO del disolvente de extracción solo debe ser suficientemente reducida, es decir, DO < 0,1. Los usuarios de la construcción tisular de EHRSC deben asegurarse de que cada lote utilizado de esta construcción cumple los criterios definidos en relación con el testigo negativo. En el cuadro 2 se indican los intervalos de aceptabilidad de los valores de DO de los testigos negativos correspondientes a los MRV. Como criterio de aceptación del testigo negativo, los usuarios de la espectrofotometría con HPLC/UPLC deben utilizar los intervalos de DO del testigo negativo que figuran en el cuadro 2. Hay que demostrar documentalmente en el informe del ensayo que los tejidos tratados con la sustancia testigo negativo son estables en cultivo (proporcionan unas mediciones similares de la viabilidad tisular) durante todo el tiempo de exposición del ensayo. El productor de tejidos debe seguir un procedimiento similar como parte del control de calidad para la aprobación de los lotes de tejidos, pero en este caso pueden ser de aplicación criterios de aceptación distintos de los especificados en el cuadro 2. El diseñador o proveedor de la construcción tisular de EHRSC debe establecer un intervalo de aceptabilidad (límites superior e inferior) de los valores de DO de los testigos negativos (en las condiciones del método de ensayo del control de calidad).
|
Cuadro 2
Intervalos de aceptabilidad de los valores de DO de los testigos negativos (para los usuarios del ensayo)
|
Ensayo
|
Límite inferior de aceptación
|
Límite superior de aceptación
|
|
EIO EpiOcular™ (OCL-200) — MRV1 (para los protocolos tanto de líquidos como de sólidos)
|
> 0,8 (87)
|
< 2,5
|
|
EIO con ECH SkinEthic™ (ECH/S) — MRV2 (para los protocolos tanto de líquidos como de sólidos)
|
> 1,0
|
≤ 2,5
|
Función de barrera
|
27.
|
La construcción tisular de EHRSC debe tener el suficiente espesor y solidez como para resistir la penetración rápida de sustancias citotóxicas de referencia, según la estimación, por ejemplo, mediante el TE50 (tritón X-100) o mediante la CI50 (DSS) (cuadro 3). La función de barrera de cada lote de construcción tisular de EHRSC utilizado debe estar demostrada por el diseñador o vendedor de esta construcción en el momento del suministro de los tejidos al usuario final (véase el punto 30).
|
Morfología
|
28.
|
El examen histológico de la construcción tisular de EHRSC debe mostrar una estructura epitelial humana similar a la córnea (con al menos tres capas de células epiteliales viables y una superficie no queratinizada). Respecto a los MRV, la morfología adecuada ha sido establecida por el diseñador o proveedor y, por tanto, no tiene que ser demostrada de nuevo por el usuario del método de ensayo para cada lote de tejidos utilizado.
|
Reproducibilidad
|
29.
|
Los resultados de los testigos positivos y negativos del método de ensayo deben demostrar su reproducibilidad a lo largo del tiempo.
|
Control de calidad (CC)
|
30.
|
La construcción tisular de EHRSC solo debe utilizarse si el diseñador o proveedor demuestra que cada lote utilizado de esta construcción cumple los criterios definidos de aprobación de la producción, los más importantes de los cuales son los relativos a la viabilidad (punto 26) y a la función de barrera (punto 27). El diseñador o proveedor de la construcción tisular de EHRSC debe establecer un intervalo de aceptabilidad (límites superior e inferior) para las funciones de barrera medidas mediante el TE50 o la CI50 (véanse los puntos 25 y 26). En el cuadro 3 figura el intervalo de aceptabilidad del TE50 o de la CI50 utilizado como criterio de control de calidad para la aprobación de los lotes por el diseñador o proveedor de las construcciones tisulares de EHRSC (utilizadas en los MRV). El diseñador/proveedor de las construcciones tisulares de EHRSC debe facilitar a los usuarios de los métodos de ensayo los datos que demuestren el cumplimiento de todos los criterios para la aprobación de la producción, de manera que puedan incluir esta información en el informe de ensayo. Solo los resultados obtenidos con tejidos que cumplan todos estos criterios de aprobación de la producción pueden aceptarse para la asignación fiable de productos que no requieren clasificación y etiquetado por irritación ocular o lesiones oculares graves de conformidad con el SGA de la ONU y del CLP.
|
Cuadro 3
Criterio de control de calidad para la aprobación de los lotes
|
Ensayo
|
Límite inferior de aceptación
|
Límite superior de aceptación
|
|
EIO EpiOcular™ (OCL-200) — MRV1 [100 μl de tritón X-100 al 0,3 % (v/v)]
|
TE50 = 12,2 min
|
TE50 = 37,5 min
|
|
EIO con ECH SkinEthic™ (ECH/S) — MRV2 (tratamiento de 30 minutos con 50 μl de DSS)
|
CI50 = 1 mg/ml
|
CI50 = 3,2 mg/ml
|
Aplicación de los productos problema y de las sustancias testigo
|
31.
|
En cada tanda deben utilizarse al menos dos réplicas tisulares con cada producto problema y cada sustancia testigo. Se utilizan dos protocolos de tratamiento diferentes, uno para productos problema líquidos y otro para productos problema sólidos (34) (35). Con ambos métodos y protocolos, la superficie de la construcción tisular debe humedecerse utilizando solución salina amortiguadora con fosfato de Dulbecco libre de calcio y magnesio (DPBS libre de Ca2+/Mg2+) antes de la aplicación de los productos problema, a fin de imitar las condiciones húmedas del ojo humano. El tratamiento de los tejidos se inicia con la exposición al producto o productos problema y a las sustancias testigo. Según ambos protocolos de tratamiento de los dos MRV, debe aplicarse una cantidad suficiente de producto problema o de sustancia testigo para cubrir uniformemente la superficie epitelial, evitando una dosis excesiva (véanse los puntos 32 y 33) (apéndice 2).
|
|
32.
|
Los productos problema que pueden pipetearse a 37 °C o a temperaturas más bajas (utilizando una pipeta de desplazamiento positivo, en caso necesario) se tratan como líquidos en los MRV; de lo contrario deben tratarse como sólidos (véase el punto 33). En los MRV, el producto problema líquido se distribuye uniformemente sobre la superficie tisular (es decir, aplicación de un mínimo de 60 μl/cm2) [véase el apéndice 2, (33) (34)]. Para garantizar la dosificación correcta del tejido deben evitarse en la medida de lo posible los efectos capilares (efectos de tensión superficial) que pueden producirse debido a los bajos volúmenes aplicados a la pieza (en la superficie tisular). Los tejidos tratados con productos problema líquidos se incuban durante 30 minutos en condiciones de cultivo normales (37 ± 2 °C, 5 ± 1 % de CO2, ≥ 95 % de HR). Al final del período de exposición, el producto problema líquido y las sustancias testigo deben retirarse con cuidado de la superficie tisular mediante un aclarado exhaustivo con DPBS libre de Ca2+/Mg2+ a temperatura ambiente. Este paso de aclarado va seguido de una inmersión tras la exposición en medio fresco a temperatura ambiente (para eliminar cualquier producto problema absorbido en el tejido) durante un plazo predeterminado que varía en función del MRV utilizado. Solo en el caso del MRV1 se aplica una incubación en medio fresco tras la exposición, en condiciones normales de cultivo, antes de realizar el ensayo del MTT [véase el apéndice 2, (34) (35)].
|
|
33.
|
Los productos problema que no pueden pipetearse a temperaturas de hasta 37 °C se tratan como sólidas en los MRV. La cantidad de producto problema aplicado debe ser suficiente para cubrir toda la superficie tisular, es decir, debe utilizarse un mínimo de 60 mg/cm2 (apéndice 2). Siempre que sea posible, los sólidos deben someterse a ensayo en forma de polvo fino. Los tejidos tratados con productos problema sólidos se incuban durante un período de tiempo predeterminado (dependiendo del MRV utilizado) en condiciones normales de cultivo [véase el apéndice 2, (34) (35)]. Al final del período de exposición, el producto problema sólido y las sustancias testigo deben retirarse con cuidado de la superficie tisular mediante un aclarado exhaustivo con DPBS libre de Ca2+/Mg2+ a temperatura ambiente. Este paso de aclarado va seguido de una inmersión tras la exposición en medio fresco a temperatura ambiente (para eliminar cualquier producto problema absorbido en el tejido) durante un plazo predeterminado que varía en función del MRV utilizado, y de una incubación en medio fresco tras la exposición, en condiciones normales de cultivo, antes de realizar el ensayo del MTT [véase el apéndice 2, (34) (35)].
|
|
34.
|
En cada tanda deben incluirse testigos positivos y negativos en paralelo para demostrar que la viabilidad (determinada con el testigo negativo) y la sensibilidad (determinada con el testigo positivo) de los tejidos se encuentran dentro de intervalos de aceptación definidos sobre la base de datos históricos. El testigo negativo en paralelo también aporta la línea de base (100 % de viabilidad tisular) para calcular la viabilidad porcentual relativa de los tejidos tratados con el producto problema (% viabilidadensayo). La sustancia testigo positivo recomendada para los MRV es el acetato de metilo puro (n.o CAS 79-20-9, disponible en el comercio, por ejemplo, Sigma-Aldrich, n.o cat. 45997; líquido). Las sustancias testigo negativo recomendadas para el MRV1 y el MRV2 son H2O ultrapura y DPBS libre de Ca2+/Mg2+, respectivamente. Se trata de las sustancias testigo utilizadas en los estudios de validación de los MRV y son aquellas sobre las que se dispone de más datos históricos. El uso de otras sustancias testigo positivo o negativo adecuadas debe estar justificado científica y adecuadamente. Deben someterse a ensayo testigos positivos y negativos con el mismo protocolo o protocolos utilizados con los productos problema incluidos en la tanda (es decir, con líquidos y/o sólidos). Esta aplicación debe ir seguida de la exposición de tratamiento, el aclarado, una inmersión tras la exposición, y una incubación tras la exposición cuando proceda, tal como se describe para las tandas de los testigos en paralelo con los productos problema líquidos (véase el punto 32) o para las tandas de los testigos en paralelo con los productos problema sólidos (véase el punto 33), antes de realizar el ensayo de MTT (véase el punto 35) (34) (35). Un único conjunto de testigos negativos y positivos es suficiente para todos los productos problema del mismo estado físico (líquidos o sólidos) incluidos en la misma tanda.
|
Medición de la viabilidad celular
|
35.
|
El ensayo del MTT es un método cuantitativo normalizado (16) que debe utilizarse para medir la viabilidad tisular según el presente método de ensayo. Es compatible con el uso de una construcción tisular tridimensional. El ensayo del MTT se realiza inmediatamente después del período de incubación tras la exposición. En los MRV, la muestra de la construcción tisular de EHRSC se coloca en 0,3 ml de solución de MTT a 1 mg/ml durante 180 ± 15 minutos en condiciones normales de cultivo. El colorante vital MTT es reducido a un precipitado azul de formazano de MTT por las células viables de la construcción tisular de EHRSC. A continuación se extrae del tejido el precipitado azul de formazano de MTT, utilizando un volumen adecuado de isopropanol (o un disolvente similar) (34) (35). En caso de tejidos sometidos a ensayo con productos problema líquidos la extracción debe hacerse tanto de la parte superior como de la parte inferior de los tejidos, mientras que en caso de tejidos sometidos a ensayo con productos problema sólidos y con líquidos coloreados la extracción debe hacerse solo de la parte inferior de los tejidos (para minimizar cualquier posible contaminación de la solución isopropanólica de extracción con cualquier producto problema que pueda haber permanecido en el tejido). Los tejidos sometidos a ensayo con productos problema líquidos que no son fáciles de lavar también pueden extraerse únicamente de la parte inferior del tejido. Las sustancias testigo positivo y negativo ensayadas en paralelo deben tratarse de forma similar al producto problema. El formazano de MTT extraído se puede cuantificar mediante una medición normal de la absorbancia (DO) a 570 nm utilizando un paso de banda máximo de ± 30 nm o mediante un procedimiento de espectrofotometría con HPLC/UPL (véase el punto 42) (11) (36).
|
|
36.
|
Las propiedades ópticas del producto problema o su acción química sobre el MTT pueden interferir con la medición del formazano de MTT, lo que daría lugar a una estimación falsa de la viabilidad del tejido. Ciertos productos problema pueden interferir con la medición del formazano de MTT mediante una reducción directa del MTT a formazano de MTT azul, y/o mediante interferencia de color si el producto problema absorbe, de forma natural o debido a los procedimientos del tratamiento, en la misma gama de DO que el formazano de MTT (es decir, a alrededor de 570 ± 30 nm). Deben realizarse controles previos antes de proceder a los ensayos para permitir la identificación de posibles reductores del MTT y/o de productos que interfieran con el color, y deben utilizarse controles adicionales para detectar y corregir posibles interferencias de tales productos problema (véanse los puntos 37-41). Esto es especialmente importante cuando un producto problema específico no se elimina completamente de la construcción tisular de EHRSC por el lavado o cuando penetra en el epitelio similar a la córnea y está presente, por tanto, en dicha construcción cuando se realiza el ensayo del MTT. En el caso de los productos problema que absorben luz en el mismo intervalo que el formazano de MTT (naturalmente o después de un tratamiento), que no son compatibles con la medición normal de la absorbancia del formazano de MTT debido a una interferencia excesiva, es decir, una fuerte absorción a 570 ± 30 nm, puede emplearse un procedimiento de espectrofotometría con HPLC/UPLC para medir el formazano de MTT (véanse los puntos 41 y 42) (11) (36). En los PNT de los MRV puede encontrarse una descripción pormenorizada de la forma de detectar y corregir la reducción directa del MTT y las interferencias debidas a agentes colorantes (34) (35). En los apéndices III y IV se presentan, respectivamente, diagramas de flujo ilustrativos con orientaciones sobre cómo identificar y tratar los reductores directos del MTT y/o los productos que interfieren con el color en el caso del MRV1 y del MRV2, respectivamente.
|
|
37.
|
Para identificar la posible interferencia de los productos problema que absorben la luz en el mismo intervalo que el formazano de MTT (naturalmente o después de un tratamiento) y decidir sobre la necesidad de utilizar testigos adicionales, el producto problema se añade al agua y/o al isopropanol y se incuba durante un tiempo adecuado a temperatura ambiente [véase el apéndice 2, (34) (35)]. Si el producto problema en el agua y/o isopropanol absorbe suficiente luz en el intervalo de 570 ± 20 nm con el MRV1 (véase el apéndice 3), o si se obtiene una solución coloreada cuando se mezcla el producto problema con agua con el MRV2 (véase el apéndice 4), se supone que el producto problema interfiere con la medición normal de la absorbancia del formazano de MTT y deben aplicarse otros testigos de colorante o bien debe utilizarse un procedimiento de espectrofotometría con HPLC/UPLC, en cuyo caso no serían necesarios esos otros testigos (véanse los puntos 41 y 42 y los apéndices III y IV) (34) (35). Cuando se realiza la medición normal de la absorbancia (DO), cada producto problema que interfiere debe aplicarse en al menos dos réplicas de tejido viable, que se someten al procedimiento de ensayo completo, pero se incuban con medio en lugar de con la solución de MTT durante la fase de incubación del MTT para generar un testigo de color inespecífico en los tejidos vivos (NSCvivo) (34) (35). El control NSCvivo debe realizarse al mismo tiempo que el ensayo del producto problema coloreado y, en caso de ensayos múltiples, debe llevarse a cabo un control NSCvivo independiente con cada ensayo realizado (en cada tanda) debido a la variabilidad biológica inherente de los tejidos vivos. La viabilidad tisular real se calcula de la manera siguiente: el porcentaje de viabilidad tisular obtenido con los tejidos vivos expuestos al producto problema que interfiere e incubados con solución de MTT (% viabilidadensayo) menos el porcentaje de color inespecífico obtenido con los tejidos vivos expuestos al producto problema que interfiere e incubados con medio sin MTT, en paralelo con el ensayo que se está corrigiendo (% NSCvivo), es decir, viabilidad tisular real = [% viabilidadensayo] - [% NSCvivo].
|
|
38.
|
Para identificar los reductores directos del MTT, cada producto problema debe añadirse a una solución de MTT recién preparada. Se añade una cantidad adecuada de producto problema a una solución de MTT y la mezcla se incuba durante aproximadamente 3 horas en condiciones normales de cultivo (véanse los apéndices III y IV) (34) (35). Si la mezcla de MTT que contiene el producto problema (o la suspensión en caso de productos problema insolubles) se pone azul/violeta, se supone que el producto problema reduce directamente el MTT y debe aplicarse otro testigo funcional con construcciones tisulares de EHRSC inviables, con independencia de que se utilice la medición normal de la absorbancia (DO) o un procedimiento de espectrofotometría con HPLC/UPLC. Este control funcional adicional emplea tejidos muertos que solo poseen actividad metabólica residual, pero que absorben y retienen el producto problema de forma similar a los tejidos viables. Los tejidos muertos del MRV1 se preparan por exposición a bajas temperaturas («muertos por congelación»). Los tejidos muertos del MRV2 se preparan mediante incubación prolongada (por ejemplo, al menos 24 ± 1 horas) en agua seguida de su almacenamiento a baja temperatura («muertos en agua»). Cada producto problema reductor del MTT se aplica al menos a dos réplicas de tejido muerto que se someten a todo el procedimiento de ensayo, para generar una reducción inespecífica del MTT (NSMTT) (34) (35). Un solo control NSMTT es suficiente por producto problema, independientemente del número realizado de ensayos o tandas independientes. La viabilidad tisular real se calcula de la manera siguiente: el porcentaje de viabilidad tisular obtenido con los tejidos vivos sometidos al reductor del MTT (% viabilidadensayo) menos el porcentaje de reducción inespecífica del MTT obtenido con los tejidos muertos expuestos al mismo reductor del MTT, calculado en relación con el testigo negativo aplicado en paralelo con el ensayo que se está corrigiendo (% NSMTT); es decir, viabilidad tisular real = [% viabilidadensayo] - [% NSMTT].
|
|
39.
|
Los productos problema que se identifiquen como causantes de interferencia de color (véase el punto 37) y de reducción directa del MTT (véase el punto 38) también requerirán una tercera serie de testigos cuando se realice la medición normal de la absorbancia (DO), aparte de los controles NSMTT y NSCvivo que se describen en los puntos anteriores. Esto es generalmente así en el caso de los productos problema de color oscuro que absorben luz en el intervalo de 570 ± 30 nm (p. ej., azul, violeta, negro) porque su color intrínseco impide la evaluación de su capacidad para reducir directamente el MTT, tal como se describe en el punto 38. Esto obliga, por defecto, a utilizar controles NSMTT junto con controles NSCvivo. Los productos problema para los que se aplican controles tanto NSMTT como NSCvivo pueden ser absorbidos y retenidos por los tejidos vivos y muertos. Por tanto, el control NSMTT puede facilitar la corrección no solo de la posible reducción directa del MTT por el producto problema, sino también dela interferencia de color derivada de la absorción y retención del producto problema por los tejidos muertos. Esto podría dar lugar a una doble corrección de la interferencia de color, puesto que el control NSCvivo ya corrige la interferencia de color derivada de la absorción y retención del producto problema por los tejidos vivos. Para evitar una posible doble corrección de la interferencia de color, es necesario aplicar un tercer control relativo al color inespecífico con tejidos muertos (NSCmuerto) (véanse los apéndices III y IV) (35). En este control adicional, el producto problema se aplica en al menos dos réplicas de tejido muerto, que se someten a todo el procedimiento de ensayo, pero se incuban con medio en lugar de con una solución de MTT durante la fase de incubación del MTT. Un solo control NSCmuerto es suficiente por producto problema cualquiera que sea el número realizado de ensayos o tandas independientes, pero debe aplicarse al mismo tiempo que el control NSMTT y, en la medida de lo posible, con el mismo lote de tejidos. La viabilidad tisular real se calcula de la manera siguiente: el porcentaje de viabilidad tisular obtenido con los tejidos vivos expuestos al producto problema (% viabilidadensayo) menos el % NSMTT menos el % NSCvivo
más el porcentaje de color inespecífico obtenido con los tejidos muertos expuestos al producto problema que interfiere e incubados con medio sin MTT, calculados en relación con el testigo negativo aplicado en paralelo con el ensayo que se está corrigiendo (% NSCmuerto), es decir, viabilidad tisular real = [% viabilidadensayo] - [% NSMTT] - [% NSCvivo] + [% NSCmuerto].
|
|
40.
|
Es importante señalar que las interferencias de reducción inespecífica del MTT y de color inespecífico pueden aumentar la DO (cuando se realizan mediciones normales de absorbancia) del extracto tisular por encima del intervalo de linealidad del espectrofotómetro y que la reducción inespecífica del MTT puede también aumentar el área del pico de formazano de MTT (cuando se realizan mediciones mediante espectrofotometría con HPLC/UPLC) del extracto tisular por encima del intervalo de linealidad del espectrofotómetro. Sobre esta base, es importante que cada laboratorio determine el intervalo de linealidad de DO / área bajo el pico de su espectrofotómetro con, p. ej., formazano de MTT (n.o CAS 57360-69-7), procedente de una fuente comercial como Sigma-Aldrich (n.o cat. M2003), antes de iniciar el ensayo de los productos problema con fines normativos.
|
|
41.
|
La medición normal de la absorbancia (DO) utilizando un espectrofotómetro es adecuada para evaluar los reductores directos del MTT y los productos problema que interfieren con el color, cuando la interferencia observada con la medición de formazano de MTT no es demasiado fuerte (es decir, las DO de los extractos tisulares obtenidos con el producto problema sin corrección alguna de la reducción directa del MTT y/o de la interferencia de color se encuentran dentro del intervalo lineal del espectrofotómetro). No obstante, los resultados correspondientes a productos problema que muestran un % NSMTT o un % NSCvivo ≥ 60 % (MRV1 y MRV2 con el protocolo para líquidos) o 50 % (MRV2 con el protocolo para sólidos) del testigo negativo deben tomarse con precaución, ya que este valor es el umbral utilizado en los MRV para distinguir los productos clasificados de los no clasificados (véase el punto 44). Sin embargo, la absorbancia (DO) no puede medirse normalmente cuando la interferencia con la medición del formazano de MTT es demasiado fuerte (es decir, lleva a valores de DO de los extractos tisulares del ensayo sin corregir que quedan fuera del intervalo lineal del espectrofotómetro). Los productos problema de color o los productos problema que se colorean en contacto con el agua o el isopropanol que interfieren excesivamente con la medición normal de la absorbancia (DO) del formazano de MTT pueden evaluarse todavía utilizando la espectrofotometría con HPLC/UPLC (véanse los apéndices III y IV). Esto es así porque el sistema de HPLC/UPLC permite separar el formazano de MTT del producto problema antes de su cuantificación (36). Por esta razón, los controles NSCvivo o NSCmuerto no son necesarios nunca si se utiliza la espectrofotometría con HPLC/UPLC, independientemente del producto sometido a ensayo. No obstante, deben utilizarse controles NSMTT si se sospecha que el producto problema reduce directamente el MTT (siguiendo el procedimiento descrito en el punto 38). También deben utilizarse controles NSMTT en caso de productos problema con color (intrínseco o de aparición cuando se pone en el agua) que obstaculiza la evaluación de su capacidad de reducir directamente el MTT, como se describe en el punto 38. Cuando se utiliza la espectrofotometría con HPLC/UPLC para medir el formazano de MTT, el porcentaje de viabilidad tisular se calcula como porcentaje del área bajo el pico de formazano de MTT obtenido con tejidos vivos expuestos al producto problema en relación con el pico de formazano de MTT obtenido con el testigo negativo en paralelo. En el caso de productos problema capaces de reducir directamente el MTT, la viabilidad tisular real se calcula de la siguiente manera: (% viabilidadensayo) menos % NSMTT, según se describe en la última frase del apartado 38. Por último, hay que señalar que no pueden evaluarse mediante los métodos de ensayo con EHRSC los reductores directos de MTT o los reductores directos de MTT que también interfieren con el color, que se mantienen en los tejidos después del tratamiento y reducen el MTT de manera tan intensa que llevan a unas DO (si se utiliza la medición normal de la DO) o a unas áreas bajo el pico (si se utiliza la espectrofotometría con UPLC/HPLC) de los extractos tisulares analizados que quedan fuera del intervalo de linealidad del espectrofotómetro, aunque se espera que estos casos se produzcan solo en muy raras situaciones.
|
|
42.
|
La espectrofotometría con HPLC/UPLC puede utilizarse para medir el formazano de MTT con todos los tipos de productos problema (con color, sin color, reductores del MTT, no reductores del MTT) (11) (36). Debido a la diversidad de sistemas de espectrofotometría con HPLC/UPLC, no es posible que cada usuario consiga exactamente las mismas condiciones del sistema. Antes de utilizar un sistema de espectrofotometría con HPLC/UPLC para cuantificar el formazano de MTT en extractos tisulares, ha de demostrarse que en sí es apto a tal fin mediante el cumplimiento de los criterios de aceptación respecto a un conjunto de parámetros normales de aptitud sobre la base de los descritos en el documento de orientación de la Food and Drug Administration de EE. UU. para la industria sobre la validación de métodos bioanalíticos (36) (38). Estos parámetros clave y sus criterios de aceptación figuran en el apéndice 5. Una vez se hayan cumplido los criterios de aceptación definidos en el apéndice 5, se considera que el sistema de espectrofotometría con HPLC/UPLC es apto y está listo para medir el formazano de MTT en las condiciones experimentales descritas en el presente método de ensayo.
|
Criterios de aceptación
|
43.
|
Para cada tanda que utilice lotes de tejido de EHRSC que pasen el control de calidad (véase el punto 30), los tejidos tratados con la sustancia testigo negativo deben presentar una DO que refleje la calidad de los tejidos que hayan pasado por las fases de transporte y recepción y por todos los procesos del protocolo, y no deben estar fuera de los límites históricos establecidos en el cuadro 2 (véase el punto 26). Del mismo modo, los tejidos tratados con la sustancia testigo positivo, es decir, acetato de metilo, deben mostrar una viabilidad tisular media < 50 % en relación con el testigo negativo en el MRV1, en los protocolos de líquidos o de sólidos, y ≤ 30 % (protocolo de líquidos), o ≤ 20 % (protocolo de sólidos) en relación con el testigo negativo en el MRV2, lo que refleja la capacidad de los tejidos para responder a un producto problema irritante en las condiciones del método de ensayo (34) (35). La variabilidad entre réplicas tisulares de productos problema y sustancias testigo debe respetar los límites aceptados [es decir, la diferencia de viabilidad entre dos réplicas tisulares debe ser inferior al 20 %, o la desviación típica (DT) entre tres réplicas tisulares no debe exceder del 18 %]. Si el testigo negativo o el testigo positivo incluido en una tanda queda fuera de los intervalos aceptados, se considera que la tanda no es apta y debe repetirse. Si la variabilidad entre las réplicas tisulares de un producto problema está fuera del intervalo aceptado, debe considerarse que el ensayo no es apto y el producto problema debe someterse a ensayo de nuevo.
|
Interpretación de los resultados y modelo de asignación
|
44.
|
Deben utilizarse los valores de DO / áreas bajo el pico obtenidos con los extractos tisulares replicados para cada producto problema a fin de calcular la viabilidad tisular media porcentual (media entre las réplicas tisulares) normalizadas con referencia al testigo negativo, cuyo valor se fija en el 100 %. En el cuadro 4 se indica el valor porcentual de corte de la viabilidad tisular para identificar los productos problema que no requieren clasificación por irritación ocular o lesiones oculares graves (sin categoría del SGA de las Naciones Unidas y CLP). Así pues, los resultados deben interpretarse como sigue:
|
—
|
Se dice que el producto problema no requiere clasificación y etiquetado de acuerdo con el SGA de las Naciones Unidas y el CLP (sin categoría) si el valor porcentual medio de la viabilidad tisular después de la exposición y la incubación tras la exposición es superior (>) al valor porcentual de corte establecido de la viabilidad tisular, como se muestra en el cuadro 4. En este caso, no es necesario realizar más ensayos con otros métodos de ensayo.
|
|
—
|
Si el valor porcentual medio de la viabilidad tisular después de la exposición y la incubación tras la exposición es inferior o igual (≤) al valor porcentual de corte establecido de la viabilidad tisular, no se podrá hacer ninguna asignación, como se muestra en el cuadro 4. En este caso, será necesario llevar a cabo ensayos adicionales con otros métodos de ensayo, ya que los métodos de ensayo con EHRSC muestran un número determinado de resultados positivos falsos (véanse los puntos 14-15) y no pueden resolverse entre las categorías 1 y 2 del SGA de la ONU y del CLP (véase el punto 17).
|
Cuadro 4
Modelos de asignación según la clasificación del SGA de las Naciones Unidas y del CLP
|
MRV
|
Sin categoría
|
No puede hacerse ninguna asignación.
|
|
MRV1 — EIO EpiOcular™ (con ambos protocolos)
|
Viabilidad tisular media > 60 %
|
Viabilidad tisular media ≤ 60 %
|
|
MRV2 — EIO con ECH SkinEthic™ (con el protocolo de líquidos)
|
Viabilidad tisular media > 60 %
|
Viabilidad tisular media ≤ 60 %
|
|
MRV2 — EIO con ECH SkinEthic™ (con el protocolo de sólidos)
|
Viabilidad tisular media > 50 %
|
Viabilidad tisular media ≤ 50 %
|
|
|
45.
|
Un solo ensayo compuesto al menos por dos réplicas tisulares debe ser suficiente para un producto problema, si el resultado es claro. Sin embargo, en casos de resultados dudosos, como el de mediciones no concordantes de las réplicas y/o el de un porcentaje de viabilidad tisular media igual al 60 ± 5 % (MRV1 y MRV2 con el protocolo para líquidos) o 50 ± 5 % (MRV2 con el protocolo para sólidos), debería plantearse la conveniencia de efectuar un segundo ensayo, así como un tercero en caso de resultados discordantes entre los dos primeros.
|
|
46.
|
A fin de mejorar el comportamiento global del método de ensayo con determinados tipos de mezclas (véase el punto 14), en relación con estos tipos de mezclas podrán tenerse en cuenta diferentes valores porcentuales de corte de la viabilidad tisular que distingan entre productos problema clasificados y no clasificados, siempre que sea apropiado y justificable. Los productos de referencia pueden ser útiles para evaluar el potencial de irritación ocular o de lesiones oculares graves de productos problema desconocidos o clases de productos, o para evaluar el potencial relativo de toxicidad ocular de un producto clasificado en una gama específica de respuestas positivas.
|
DATOS E INFORME
Datos
|
47.
|
Los datos de los distintos tejidos replicados de una tanda (por ejemplo, valores de DO o de área bajo el pico de formazano de MTT y valores porcentuales calculados de la viabilidad tisular relativos al producto problema y a los testigos, y la asignación final del método de ensayo con EHRSC) deben presentarse en forma de tabla para cada producto problema, incluidos los datos de ensayos repetidos, según proceda. Además, debe indicarse el valor porcentual medio de la viabilidad tisular y la diferencia de viabilidad entre dos réplicas tisulares (si n = 2 tejidos replicados) o la DT (si n ≥ 3 tejidos replicados) para cada producto problema y testigo. Se deben notificar respecto a cada producto problema las eventuales interferencias observadas que provoque este con la medición del formazano de MTT a través de la reducción directa del MTT o por interferencias de color.
|
Informe del ensayo
|
48.
|
El informe del ensayo debe incluir la información siguiente:
Producto problema
|
|
Sustancias de un solo componente
|
—
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
—
|
Estado físico, volatilidad, pH, log P, peso molecular, clase química y otras propiedades fisicoquímicas pertinentes para la realización del estudio, según su disponibilidad;
|
|
—
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
—
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
—
|
Condiciones de conservación y estabilidad en la medida de lo posible.
|
|
|
|
Sustancias de componentes múltiples, sustancias UVCB y mezclas
|
—
|
Caracterización, en la medida de lo posible, mediante, por ejemplo, la identidad química (véase más arriba), pureza, presencia cuantitativa y propiedades fisicoquímicas pertinentes de los componentes (véase más arriba), en la medida de lo posible;
|
|
—
|
Estado físico y otras propiedades fisicoquímicas pertinentes para la realización del estudio, según su disponibilidad;
|
|
—
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
—
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
—
|
Condiciones de conservación y estabilidad en la medida de lo posible.
|
|
Sustancias testigo positivo y negativo
|
—
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
—
|
Estado físico, volatilidad, peso molecular, clase química y otras propiedades fisicoquímicas pertinentes para la realización del estudio, según su disponibilidad;
|
|
—
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
—
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
—
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
—
|
Justificación del uso de un testigo negativo distinto del H2O ultrapura o de la solución DPBS libre de Ca2+/Mg2+, si procede;
|
|
—
|
Justificación del uso de un testigo positivo distinto del acetato de metilo puro, si procede;
|
|
—
|
Referencia a los resultados de los testigos positivos y negativos históricos que demuestren unos criterios adecuados de aceptación de las tandas.
|
Información referente al promotor y al laboratorio
|
—
|
Nombre y dirección del promotor, laboratorio y director del estudio.
|
|
—
|
Construcción tisular de EHRSC y protocolo utilizados (justificación de las opciones elegidas, si procede)
|
Condiciones del método de ensayo
|
—
|
Construcción tisular de EHRSC utilizada, incluido el número de lote;
|
|
—
|
Longitud de onda y paso de banda (si procede) utilizados para cuantificar el formazano de MTT y el intervalo de linealidad del dispositivo de medición (p. ej., espectrofotómetro);
|
|
—
|
Descripción del método utilizado para cuantificar el formazano de MTT;
|
|
—
|
Descripción del sistema utilizado de espectrofotometría con HPLC/UPLC, si procede;
|
|
—
|
Información justificativa completa sobre la construcción tisular de EHRSC concreta utilizada, incluido su comportamiento. Aquí deben incluirse, entre otras cosas:
|
i)
|
Control de calidad de la viabilidad (proveedor);
|
|
ii)
|
Viabilidad en las condiciones del método de ensayo (usuario);
|
|
iii)
|
Control de calidad de la función de barrera;
|
|
iv)
|
Morfología, si se dispone de ella;
|
|
v)
|
Reproducibilidad y capacidad de asignación;
|
|
vi)
|
Otros controles de calidad (CC) de la construcción tisular de EHRSC, si se dispone de ellos;
|
|
|
—
|
Referencia a datos históricos de la construcción tisular de EHRSC. Aquí deben incluirse, entre otras cosas, la aceptabilidad de los datos de CC con referencia a datos anteriores del lote;
|
|
—
|
Declaración de que el laboratorio ha demostrado su competencia para el uso del método de ensayo, antes de su uso sistemático, mediante el ensayo de los productos utilizados para la prueba de la competencia.
|
Criterios de aceptación de las tandas y del ensayo
|
—
|
Valores medios e intervalos de aceptación de los testigos positivos y negativos a partir de datos históricos;
|
|
—
|
Variabilidad aceptable entre las réplicas tisulares con respecto a los testigos positivos y negativos;
|
|
—
|
Variabilidad aceptable entre las réplicas tisulares correspondientes a cada producto problema.
|
Procedimiento de ensayo
|
—
|
Particularidades del procedimiento de ensayo empleado;
|
|
—
|
Dosis utilizadas del producto problema y de las sustancias testigo;
|
|
—
|
Duración y temperatura de los períodos de exposición, inmersión tras la exposición e incubación tras la exposición (en su caso);
|
|
—
|
Descripción de las eventuales modificaciones del procedimiento del ensayo;
|
|
—
|
Indicación de los controles utilizados con productos problema que son reductores directos del MTT y/o colorantes, en su caso;
|
|
—
|
Número de réplicas tisulares utilizadas con cada producto problema y testigo (testigo positivo, testigo negativo, NSMTT, NSCvivo y NSCmuerto, en su caso);
|
Resultados
|
—
|
Cuadros de datos de cada producto problema y testigo, con las mediciones de cada tanda (includios los experimentos repetidos, en su caso) y cada réplica, incluido el valor de DO o el área bajo el pico de formazano de MTT, el porcentaje de viabilidad tisular, el porcentaje medio de viabilidad tisular, las diferencias entre las réplicas tisulares o las desviaciones típicas, y asignación final;
|
|
—
|
En su caso, los resultados de los controles utilizados en relación con los productos problema que son reductores directos del MTT o coloreados, incluido el valor de DO o el área bajo el pico de formazano de MTT, el % NSMTT, el % NSCvivo, el % NSCmuerto, las diferencias entre las réplicas tisulares o las desviaciones típicas, porcentaje final correcto de viabilidad tisular, y asignación final;
|
|
—
|
Resultados obtenidos con el producto o productos problema y sustancias testigo en relación con los criterios definidos de aceptación de las tandas y del ensayo;
|
|
—
|
Descripción de otros efectos observados como, por ejemplo, coloración de los tejidos por un producto problema coloreado;
|
Discusión de los resultados
Conclusión
|
BIBLIOGRAFÍA
|
(1)
|
Naciones Unidas (2015). Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA) ST/SG/AC.10/30/Rev. 6, sexta edición revisada, Nueva York y Ginebra: Naciones Unidas. Disponible en: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/Spanish/ST-SG-AC10-30-Rev6sp.pdf.
|
|
(2)
|
Capítulo B.5 del presente anexo, Irritación/corrosión ocular aguda.
|
|
(3)
|
Capítulo B.47 del presente anexo, Método de ensayo de la opacidad y permeabilidad de la córnea de bovino para identificar: i) productos que provocan lesiones oculares graves y ii) productos que no requieren clasificación por irritación ocular ni lesiones oculares graves.
|
|
(4)
|
Capítulo B.48 del presente anexo, Método de ensayo de ojo de pollo aislado para identificar: i) productos que provocan lesiones oculares graves y ii) productos que no requieren clasificación.
|
|
(5)
|
Capítulo B.61 del presente anexo, Método de ensayo de pérdida de fluoresceína para detectar agentes corrosivos e irritantes intensos para los ojos.
|
|
(6)
|
Capítulo B.68 del presente anexo, Método de ensayo in vitro con exposición de breve duración para detectar: i) productos que provocan lesiones oculares graves y ii) productos que no requieren clasificación por irritación ocular o lesiones oculares graves.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2010,261-266.
|
|
(8)
|
LRUE del CEVMA (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Disponible en: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
Comité Consultivo Científico del CEVMA (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28 173 EN; doi: 10.2787/043697. Disponible en: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
Comité Consultivo Científico del CEVMA (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28 175 EN; doi: 10.2787/390390. Disponible en: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
LRUE del CEVMA (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscript in Preparation).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OCDE (2016) Series on Testing and Assessment, No. 216. Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(18)
|
OCDE (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, pp. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922–936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115–130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2610–2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41–54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Disponible en: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Disponible en: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. May 2001. Disponible en: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OCDE (2017) Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications, Organización para la Cooperación y el Desarrollo Económico, París.
|
Apéndice 1
DEFINICIONES
Exactitud
: Grado de coincidencia entre los resultados obtenidos con el método de ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del método de ensayo y es un aspecto de su “pertinencia”. Este término y el de “concordancia” se suelen usar indistintamente para indicar la proporción de resultados correctos de un método de ensayo (18).
Producto de referencia
: Producto utilizado como patrón para comparar con un producto problema. Los productos de referencia deben presentar las siguientes propiedades: i) tener un origen u orígenes coherentes y fiables para su identificación y caracterización; ii) presentar similitud estructural, funcional o en cuanto a la clase química o de producto con respecto al producto o productos que se somete a ensayo; iii) características fisicoquímicas conocidas; iv) datos de apoyo sobre los efectos conocidos; y v) potencia conocida en la banda de la respuesta deseada.
Enfoque ascendente
: Enfoque gradual utilizado para un producto problema del que se sospecha que no requiere clasificación y etiquetado por irritación ocular o lesiones oculares graves, que comienza con la determinación de los productos que no requieren clasificación y etiquetado (resultado negativo) frente a otros productos (resultado positivo).
Producto
: Sustancia o mezcla.
Concordancia
: Véase “Exactitud”.
Córnea
: Parte transparente delantera del globo ocular que cubre el iris y la pupila y permite el paso de la luz al interior.
CV
: Coeficiente de variación.
Desv.
: Desviación.
EIO
: Ensayo de irritación ocular.
LRUE del CEVMA
: Laboratorio de referencia de la Unión Europea para los métodos alternativos a la experimentación con animales
Irritación ocular
: Producción de cambios en el ojo, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, que son totalmente reversibles en los 21 días siguientes a la aplicación. Intercambiable con “efectos reversibles en el ojo” y con la categoría 2 del SGA de las Naciones Unidas y del CLP.
TE50
: Tiempo de exposición necesario para reducir la viabilidad tisular en un 50 % tras la administración de un producto de referencia a una concentración fija determinada.
Tasa de falsos negativos
: Proporción de todas las sustancias positivas identificadas erróneamente como negativas por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo.
Tasa de falsos positivos
: Proporción de todas las sustancias negativas identificadas erróneamente como positivas por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo.
Peligro
: Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente.
ECH
: Epitelio de córnea humana SkinEthic™.
HPLC
: Cromatografía líquida de alta resolución.
CI50
: Concentración a la que una sustancia de referencia reduce la viabilidad tisular en un 50 % tras un tiempo fijo de exposición (por ejemplo, tratamiento de 30 minutos con DSS).
Dosis excesiva
: Cantidad de producto problema aplicada a la construcción tisular de EHRSC que supera a la cantidad necesaria para cubrir completa y uniformemente la superficie epitelial.
Efectos irreversibles en el ojo
: Véase “Lesiones oculares graves”.
LIC
: Límite inferior de cuantificación.
Log P
: Logaritmo del coeficiente de reparto octanol/agua.
Mezcla
: Mezcla o solución compuesta de dos o más sustancias.
Sustancia de un solo componente
: Sustancia, definida por su composición cuantitativa, en la que un solo componente principal representa al menos el 80 % (p/p).
Sustancia de componentes múltiples
: Sustancia, definida por su composición cuantitativa, en la que hay varios componentes principales presentes a una concentración ≥ 10 % (p/p) y < 80 % (p/p). Una sustancia de componentes múltiples es el resultado de un proceso de fabricación. La diferencia entre mezcla y sustancia de componentes múltiples es que una mezcla se obtiene mezclando dos o más sustancias sin reacción química. Una sustancia de componentes múltiples es el resultado de una reacción química.
MTT
: Bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio; bromuro de tetrazolio de azul de tiazolilo.
Testigo negativo
: Muestra que contiene todos los componentes de un sistema de ensayo y que se trata con una sustancia de la que se sabe que no induce una respuesta positiva en el sistema de ensayo. Esta muestra se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras testigo, y se utiliza para determinar el 10 % de viabilidad tisular.
No clasificados
: Productos no clasificados por irritación ocular (categoría 2 del SGA de las Naciones Unidas y del CLP, categorías 2A o 2B del del SGA de las Naciones Unidas) o por lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas y del CLP). Intercambiable con “sin categoría del SGA de las Naciones Unidas y del CLP”.
NSCmuerto
: Color inespecífico en tejidos muertos.
NSCvivo
: Color inespecífico en tejidos vivos.
NSMTT
: Reducción inespecífica del MTT.
DO
: Densidad óptica.
Normas de comportamiento
: Normas, basadas en un método de referencia validado que se ha considerado científicamente válido, que proporcionan la base para evaluar la comparabilidad de un método de ensayo propuesto que es similar desde el punto de vista mecánico y funcional. Se incluyen: i) los componentes fundamentales del método de ensayo; ii) una lista mínima de productos de referencia seleccionados de entre los productos utilizados para demostrar el comportamiento aceptable del método de ensayo validado; y iii) los niveles similares de fiabilidad y exactitud, basados en lo obtenido con el método de ensayo validado, que el método de ensayo propuesto debe demostrar cuando se evalúa con la lista mínima de productos de referencia (18).
Testigo positivo
: Muestra que contiene todos los componentes de un sistema de ensayo y que se trata con una sustancia de la que se sabe que induce una respuesta positiva en el sistema de ensayo. Esta muestra se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras testigo. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta positiva.
Pertinencia
: Descripción de la relación del ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un método de ensayo (18).
Fiabilidad
: Medida del grado en que un método de ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios (18).
Ensayo de sustitución
: Ensayo que se diseña para sustituir un ensayo utilizado de forma sistemática y aceptado para la identificación de peligros o la evaluación de riesgos, y del que se ha determinado que proporciona una protección equivalente o mejorada de la salud humana o del medio ambiente, según corresponda, respecto al ensayo aceptado, en relación con todas las situaciones de ensayo y todos los productos posibles (18).
Reproducibilidad
: Coincidencia entre los resultados obtenidos en el ensayo repetido del mismo producto problema utilizando el mismo protocolo de ensayo (véase “Fiabilidad”) (18).
Efectos reversibles en el ojo
: Véase “Irritación ocular”.
EHRSC
: Epitelio humano reconstruido similar a la córnea.
Tanda
: Una tanda consiste en el ensayo de uno o más productos problema al mismo tiempo que un testigo negativo y un testigo positivo.
DT
: Desviación típica.
Sensibilidad
: Proporción de todos los productos problema activos/positivos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (18).
Lesiones oculares graves
: Producción de lesiones tisulares en el ojo o una degradación física severa de la vista, como consecuencia de la aplicación de una sustancia problema en la superficie anterior del ojo, y que no es totalmente reversible en los 21 días siguientes a la aplicación. Intercambiable con “efectos irreversibles en el ojo” y con la categoría 1 del SGA de las Naciones Unidas y del CLP.
Procedimientos normalizados de trabajo (PNT)
: Procedimientos escritos oficiales que describen en detalle cómo deben llevarse a cabo las operaciones de laboratorio corrientes y las específicas de cada ensayo. Son exigidas por las BPL.
Especificidad
: Proporción de todos los productos negativos o inactivos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (18).
Sustancia
: Elemento químico y sus compuestos naturales u obtenidos mediante algún proceso de producción, incluidos los eventuales aditivos necesarios para mantener su estabilidad y las eventuales impurezas que se produzcan en el proceso, con exclusión de los eventuales disolventes que puedan separarse sin afectar a la estabilidad de la sustancia ni modificar su composición.
Ensayo
: Un solo producto problema sometido a ensayo al mismo tiempo con un mínimo de dos réplicas tisulares como se define en los correspondientes PNT.
Viabilidad tisular
: Parámetro que mide la actividad total de una población celular en un tejido reconstruido como su capacidad de reducir el colorante vital MTT que, según el parámetro que se mida y el diseño del ensayo utilizado, se corresponde con el número total y/o la vitalidad de las células vivas.
Enfoque descendente
: Enfoque gradual utilizado para un producto del que se sospecha que causa lesiones oculares graves, que comienza con la determinación de los productos que inducen lesiones oculares graves (resultado positivo) frente a otros productos (resultado negativo).
Producto problema
: Sustancia o mezcla estudiada con este método de ensayo.
Estrategia de ensayos escalonados
: Estrategia de ensayo por fases, que utiliza los métodos de ensayo de forma secuencial. En cada escalón se revisa toda la información existente sobre un producto problema, en un proceso de ponderación de las pruebas, a fin de determinar si se dispone de información suficiente para tomar una decisión sobre la clasificación de un peligro, antes de pasar al escalón siguiente de la estrategia. Si puede establecerse la potencia o el potencial de peligro de un producto problema sobre la base de la información disponible en un escalón dado, no hace falta efectuar más ensayos (18).
LSC
: Límite superior de cuantificación.
Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de las Naciones Unidas)
: Sistema que propone la clasificación de los productos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (1).
Categoría 1 del SGA de las Naciones Unidas y del CLP
: Véase “Lesiones oculares graves”.
Categoría 2 del SGA de las Naciones Unidas y del CLP
: Véase “Irritación ocular”.
Sin categoría del SGA de las Naciones Unidas y del CLP
: Productos que no cumplen los requisitos de clasificación en las categorías 1 o 2 del SGA de las Naciones Unidas y del CLP (o en las categorías 2A o 2B del SGA de las Naciones Unidas). Intercambiable con “no clasificados”.
UPLC
: Cromatografía líquida de ultraalta resolución.
UVCB
: Sustancias de composición desconocida o variable, productos de reacción compleja y materiales biológicos.
Método de ensayo válido
: Método de ensayo del que se considera que tiene suficiente pertinencia y fiabilidad con un fin específico y que se basa en principios sólidos desde el punto de vista científico. Un método de ensayo nunca es válido en un sentido absoluto, sino únicamente en relación con un fin determinado (18).
Método de ensayo validado
: Método de ensayo sobre el cual se han completado estudios de validación para determinar su pertinencia (incluida su exactitud) y su fiabilidad con un fin específico. Es importante señalar que un método de ensayo validado podría no tener un comportamiento suficiente en términos de exactitud y fiabilidad como para considerarse aceptable a efectos del fin propuesto (18).
MRV
: Método de referencia validado.
MRV1
: El EIO EpiOcular™ se conoce como el método de referencia validado 1.
MRV2
: El EIO con ECH SkinEthic™ se conoce como el método de referencia validado 2.
Ponderación de los datos
: Proceso de consideración de los aspectos favorables y desfavorables de los distintos elementos de información a efectos de alcanzar y confirmar una conclusión en cuanto al peligro potencial de una sustancia problema.
Apéndice 2
PRINCIPALES COMPONENTES DE LOS ENSAYOS CON EHRSC PARA IDENTIFICAR PRODUCTOS QUE NO REQUIEREN CLASIFICACIÓN Y ETIQUETADO POR IRRITACIÓN OCULAR O LESIONES OCULARES GRAVES
|
Componentes del ensayo
|
EIO EpiOcula™
(MRV1)
|
EIO con ECH SkinEthic™
(MRV2)
|
|
Protocolos
|
Líquidos
(pipeteables a 37 ± 1 °C o temperaturas más bajas durante 15 minutos)
|
Sólidos
(no pipeteables)
|
Líquidos y viscosos
(pipeteables)
|
Sólidos
(no pipeteables)
|
|
Superficie del modelo
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Número de réplicas tisulares
|
Al menos dos
|
Al menos dos
|
Al menos dos
|
Al menos dos
|
|
Control previo en relación con la interferencia de color
|
50 μl + 1 ml H2O durante 60 min a 37 ± 2 °C, 5 ± 1 % de CO2, ≥ 95 % HR (productos problema no coloreados), o 50 μl + 2 ml de isopropanol mezclado durante 2-3 h a temperatura ambiente (productos problema coloreados)
|
 Si la DO del producto problema a 570 ± 20 nm, tras la sustracción de la DO correspondiente al isopropanol o al agua es > 0,08 (lo que corresponde aproximadamente al 5 % de la DO media del testigo negativo), deben utilizarse testigos adaptados con tejido vivo. Si la DO del producto problema a 570 ± 20 nm, tras la sustracción de la DO correspondiente al isopropanol o al agua es > 0,08 (lo que corresponde aproximadamente al 5 % de la DO media del testigo negativo), deben utilizarse testigos adaptados con tejido vivo.
|
|
50 mg + 1 ml H2O durante 60 min a 37 ± 2 °C, 5 ± 1 % de CO2, ≥ 95 % HR (productos problema no coloreados)
y/o
50 mg + 2 ml de isopropanol mezclado durante 2-3 h a temperatura ambiente (productos problema coloreados y no coloreados)
|
 Si la DO del producto problema a 570 ± 20 nm, tras la sustracción de la DO correspondiente al isopropanol o al agua es > 0,08 (lo que corresponde aproximadamente al 5 % de la DO media del testigo negativo), deben utilizarse testigos adaptados con tejido vivo. Si la DO del producto problema a 570 ± 20 nm, tras la sustracción de la DO correspondiente al isopropanol o al agua es > 0,08 (lo que corresponde aproximadamente al 5 % de la DO media del testigo negativo), deben utilizarse testigos adaptados con tejido vivo.
|
|
10 μl + 90 μl H2O mezclada durante 30 ± 2 min, a la temperatura ambiente (18-28oC).
|
 Si el producto problema es de color, deben utilizarse testigos adaptados con tejido vivo. Si el producto problema es de color, deben utilizarse testigos adaptados con tejido vivo.
|
|
10 mg + 90 μl de H2O mezclados durante 30 ± 2 min a temperatura ambiente
|
 Si el producto problema es de color, deben utilizarse testigos adaptados con tejido vivo. Si el producto problema es de color, deben utilizarse testigos adaptados con tejido vivo.
|
|
|
Control previo en relación con la reducción directa del MTT
|
50 μl + 1 ml de solución MTT 1 mg/ml durante 180 ± 15 min. a 37 ± 2oC, 5 ± 1 % de CO2, ≥95 % de humedad relativa
|
 Si la solución se pone de color azul/morado, deben utilizarse testigos adaptados con tejido muerto por congelación Si la solución se pone de color azul/morado, deben utilizarse testigos adaptados con tejido muerto por congelación
(se utilizan como testigo negativo 50 μl de agua desionizada estéril en solución de MTT)
|
|
50 mg + 1 ml de solución MTT 1 mg/ml durante 180 ± 15 min a 37 ± 2oC, 5 ± 1 % de CO2, ≥95 % de humedad relativa
|
 Si la solución se pone de color azul/morado, deben utilizarse testigos adaptados con tejido muerto por congelación Si la solución se pone de color azul/morado, deben utilizarse testigos adaptados con tejido muerto por congelación
(se utilizan como testigo negativo 50 μl de agua desionizada estéril en solución de MTT)
|
|
30 μl + 300 μl de solución MTT 1 mg/ml durante 180 ± 15 min a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
 Si la solución se pone de color azul/morado, deben utilizarse testigos adaptados con tejido muerto en agua. Si la solución se pone de color azul/morado, deben utilizarse testigos adaptados con tejido muerto en agua.
(se utilizan como testigo negativo 30 μl de agua desionizada estéril en solución de MTT)
|
|
30 mg + 300 μl de solución MTT 1 mg/ml durante 180 ± 15 min a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
 Si la solución se pone de color azul/morado, deben utilizarse testigos adaptados con tejido muerto en agua. Si la solución se pone de color azul/morado, deben utilizarse testigos adaptados con tejido muerto en agua.
(se utilizan como testigo negativo 30 μl de agua desionizada estéril en solución de MTT)
|
|
|
Pretratamiento
|
20 μl de DPBS libre de Ca2+/Mg2+
durante 30 ± 2 min a 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa, protegidos de la luz.
|
20 μl de DPBS libre de Ca2+/Mg2+
durante 30 ± 2 min a 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa, protegidos de la luz.
|
-
|
-
|
|
Dosis de tratamiento y aplicación
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) utilizando una herramienta calibrada (por ejemplo, una cucharada rasa calibrada para tener 50 mg de cloruro de sodio).
|
10 μl de DPBS libre de Ca2+/Mg2+ + 30 ± 2 μl (60 μl/cm2)
En caso de productos viscosos, se utiliza una malla de nailon.
|
30 μl de DPBS libre de Ca2+/Mg2+ + 30 ± 2 mg (60 mg/cm2)
|
|
Tiempo y temperatura de la exposición
|
30 min (± 2 min)
en medio de cultivo
a 37 ± 2oC, 5 ± 1 % de CO2, ≥ 95 % de humedad relativa
|
6 horas (± 0,25 h)
en medio de cultivo
a 37 ± 2oC, 5 ± 1 % de CO2, ≥ 95 % de humedad relativa
|
30 min (± 2 min)
en medio de cultivo
a 37 ± 2oC, 5 ± 1 % de CO2, ≥ 95 % de humedad relativa
|
4 horas (± 0,1 h)
en medio de cultivo
a 37 ± 2oC, 5 ± 1 % de CO2, ≥ 95 % de humedad relativa
|
|
Lavado a temperatura ambiente
|
3 veces en 100 ml de DPBS libre de Ca2+/Mg2+
|
3 veces en 100 ml de DPBS libre de Ca2+/Mg2+
|
20 ml de DPBS libre de Ca2+/Mg2+
|
25 ml de DPBS libre de Ca2+/Mg2+
|
|
Inmersión tras la exposición
|
12 min (± 2 min) a temperatura ambiente en medio de cultivo
|
25 min (± 2 min) a temperatura ambiente en medio de cultivo
|
30 min (±2 min) a 37oC, 5 % CO2, 95 % de humedad relativa en medio de cultivo
|
30 min (± 2 min) a temperatura ambiente en medio de cultivo
|
|
Incubación tras la exposición
|
120 min (± 15 min) en medio de cultivo a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
18 h (± 0,25 h) en medio de cultivo a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
Ninguna
|
18 h (± 0,5 h) en medio de cultivo a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
|
Testigo negativo
|
50 μl de H2O
Sometido a ensayo al mismo tiempo
|
50 μl de H2O
Sometido a ensayo al mismo tiempo
|
30 ± 2 μl de DPBS libre de Ca2+/Mg2+
Sometido a ensayo al mismo tiempo
|
30 ± 2 μl de DPBS libre de Ca2+/Mg2+
Sometido a ensayo al mismo tiempo
|
|
Testigo positivo
|
50 μl de acetato de metilo
Sometido a ensayo al mismo tiempo
|
50 μl de acetato de metilo
Sometido a ensayo al mismo tiempo
|
30 ± 2 μl de acetato de metilo
Sometido a ensayo al mismo tiempo
|
30 ± 2 μl de acetato de metilo
Sometido a ensayo al mismo tiempo
|
|
Solución de MTT
|
300 μl con 1 mg/ml
|
300 μl con 1 mg/ml
|
300 μl con 1 mg/ml
|
300 μl con 1 mg/ml
|
|
Tiempo y temperatura de incubación del MTT
|
180 min (± 15 min) a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
180 min (± 15 min) a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
180 min (± 15 min) a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
180 min (± 15 min) a 37 ±2 °C, 5 ± 1 % CO2, ≥ 95 % de humedad relativa
|
|
Disolvente de extracción
|
2 ml de isopropanol
(extracción de la parte superior y de la parte inferior de la pieza perforando el tejido)
|
2 ml de isopropanol
(extracción de la parte inferior de la pieza perforando el tejido)
|
1,5 ml de isopropanol
(extracción de la parte superior y de la parte inferior de la pieza)
|
1,5 ml de isopropanol
(extracción de la parte inferior de la pieza)
|
|
Tiempo y temperatura de extracción
|
2-3 h con agitación (~ 120 rpm) a temperatura ambiente o una noche a 4-10 °C
|
2-3 h con agitación (~ 120 rpm) a temperatura ambiente o una noche a 4-10 °C
|
4 h con agitación (~ 120 rpm) a temperatura ambiente o una noche a 4-10 °C
|
Al menos 2 h con agitación (~ 120 rpm) a temperatura ambiente
|
|
Lectura de la DO
|
570 nm (550 - 590 nm)
sin filtro de referencia
|
570 nm (550 - -590 nm)
sin filtro de referencia
|
570 nm (540 - 600 nm)
sin filtro de referencia
|
570 nm (540 - 600 nm)
sin filtro de referencia
|
|
Control de calidad del tejido
|
Tratamiento con 100 μl de tritón X-100 al 0,3 % (v/v)
12,2 min ≤ TE50 ≤ 37,5 min
|
Tratamiento con 100 μl de tritón X-100 al 0,3 % (v/v)
12,2 min ≤ TE50 ≤ 37,5 min
|
30 min de tratamiento con DSS (50 μl)
1,0 mg/ml ≤ CI50 ≤ 3,5 mg/ml
|
30 min de tratamiento con DSS (50 μl)
1,0 mg/ml ≤ CI50 ≤ 3,2 mg/ml
|
|
Criterios de aceptación
|
|
1.
|
La DO media de las réplicas tisulares tratadas con el testigo negativo debe ser > 0,8 y < 2,5.
|
|
2.
|
La viabilidad media de las réplicas tisulares expuestas durante 30 min con el testigo positivo, expresada en % del testigo negativo, debe ser < 50 %.
|
|
3.
|
La diferencia de viabilidad entre dos réplicas tisulares debe ser inferior al 20 %.
|
|
|
1.
|
La DO media de las réplicas tisulares tratadas con el testigo negativo debe ser > 0,8 y < 2,5.
|
|
2.
|
La viabilidad media de las réplicas tisulares expuestas durante 6 horas con el testigo positivo, expresada en % del testigo negativo, debe ser < 50 %.
|
|
3.
|
La diferencia de viabilidad entre dos réplicas tisulares debe ser inferior al 20 %.
|
|
|
1.
|
La DO media de las réplicas tisulares tratadas con el testigo negativo debe ser > 1,0 y ≤ 2,5.
|
|
2.
|
La viabilidad media de las réplicas tisulares expuestas durante 30 min con el testigo positivo, expresada en % del testigo negativo, debe ser ≤ 30 %.
|
|
3.
|
La diferencia de viabilidad entre dos réplicas tisulares debe ser inferior al 20 %.
|
|
|
1.
|
La DO media de las réplicas tisulares tratadas con el testigo negativo debe ser > 1,0 y ≤ 2,5.
|
|
2.
|
La viabilidad media de las réplicas tisulares expuestas durante 4 horas con el testigo positivo, expresada en % del testigo negativo, debe ser ≤ 20 %.
|
|
3.
|
La diferencia de viabilidad entre dos réplicas tisulares debe ser inferior al 20 %.
|
|
Apéndice 3
DIAGRAMA DE FLUJO ILUSTRATIVO CON ORIENTACIONES SOBRE CÓMO IDENTIFICAR Y TRATAR LOS REDUCTORES DIRECTOS DEL MTT Y/O LOS PRODUCTOS QUE INTERFIEREN CON EL COLOR, SOBRE LA BASE DE LOS PNT DEL MRV1

Apéndice 4
DIAGRAMA DE FLUJO ILUSTRATIVO CON ORIENTACIONES SOBRE CÓMO IDENTIFICAR Y TRATAR LOS REDUCTORES DIRECTOS DEL MTT Y/O LOS PRODUCTOS QUE INTERFIEREN CON EL COLOR, SOBRE LA BASE DE LOS PNT DEL MRV2

Apéndice 5
PARÁMETROS CLAVE Y CRITERIOS DE ACEPTACIÓN PARA CONSIDERAR APTO UN SISTEMA DE ESPECTROFOTOMETRÍA CON HPLC/UPLC PARA LA MEDICIÓN DEL FORMAZANO DE MTT EXTRAÍDO DE CONSTRUCCIONES TISULARES DE EHRSC
|
Parámetro
|
Protocolo obtenido de las orientaciones de la FDA (36) (38)
|
Criterios de aceptación
|
|
Selectividad
|
Análisis de isopropanol, blanco vivo (extracto isopropanólico de construcciones tisulares de EHRSC vivo sin ningún tratamiento), blanco muerto (extracto isopropanólico de construcciones tisulares de EHRSC muerto sin ningún tratamiento)
|
Áreainterferencia ≤ 20 % del ÁreaLIC
(88)
|
|
Precisión
|
Controles de calidad (es decir, formazano de MTT a 1,6 μg/ml, 16 μg/ml y 160 μg/ml) en isopropanol (n = 5)
|
CV ≤ 15 % o ≤ 20 % respecto al LIC
|
|
Exactitud
|
Controles de calidad en isopropanol (n = 5)
|
% Desv ≤ 15 % o ≤ 20 % respecto al LIC
|
|
Efecto de matriz
|
Controles de calidad con blanco vivo (n = 5)
|
85 % ≤ % efecto de matriz % ≤ 115 %
|
|
Efecto residual
|
Análisis de isopropanol después de un patrón para el LSC (89)
|
Áreainterferencia ≤ 20 % del ÁreaLIC
|
|
Reproducibilidad (intradiaria)
|
Tres curvas de calibración independientes (sobre la base de 6 diluciones consecutivas a 1/3 de formazano de MTT en isopropanol, comenzando en el LSC, es decir, 200 μg/ml);
Controles de calidad en isopropanol (n = 5)
|
Curvas de calibración: % Desv ≤ 15 % o ≤ 20 % respecto al LIC
Controles de calidad: % Desv ≤ 15 % y CV ≤ 15 %
|
|
Reproducibilidad (interdiaria)
|
Día 1: Una curva de calibración y controles de calidad en isopropanol (n = 3)
Día 2: Una curva de calibración y controles de calidad en isopropanol (n = 3)
Día 3: Una curva de calibración y controles de calidad en isopropanol (n = 3)
|
|
Estabilidad a corto plazo del formazano de MTT en el extracto tisular de EHRSC
|
Controles de calidad en el blanco vivo (n = 3) analizado el día de la preparación y después de 24 horas de conservación a temperatura ambiente
|
% Desv ≤ 15 %
|
|
Estabilidad a largo plazo del formazano de MTT en el extracto tisular de EHRSC, en caso necesario
|
Controles de calidad en el blanco vivo (n = 3) analizado el día de la preparación y después de varios días de conservación a -20 °C
|
% Desv ≤ 15 %
|
B.70. ENSAYOS IN VITRO CON RECEPTOR ESTROGÉNICO RECOMBINANTE HUMANO (hrER) PARA DETECTAR PRODUCTOS CON AFINIDAD PARA UNIRSE A LOS RECEPTORES ESTROGÉNICOS
INTRODUCCIÓN GENERAL
Directrices de ensayo de la OCDE sobre la base del comportamiento
|
1.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 493 de la OCDE (2015). Las TG 493 son unas directrices de ensayo basadas en el comportamiento (performance-based test guideline, PBTG), en las que se describe la metodología de los ensayos in vitro con receptores recombinantes humanos para detectar sustancias con afinidad para unirse a los receptores estrogénicos (ensayos de unión al hrER). Comprenden dos ensayos similares desde el punto de vista mecánico y funcional para la identificación de los ligandos de los receptores estrogénicos (es decir, ERα) y deben facilitar la elaboración de nuevos métodos de ensayo similares o modificados, de conformidad con los principios de validación expuestos en el documento de orientación de la OCDE sobre la validación y la aceptación internacional de métodos de ensayo nuevos o actualizados para la evaluación de peligros (Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment) (1). Los métodos de ensayo de referencia plenamente validados (apéndice 2 y apéndice 3) que sirven de base a dichas PBTG son estos:
|
—
|
el ensayo de Freyberg-Wilson (FW) de unión a receptores estrogénicos in vitro utilizando un ERα recombinante humano completo (2), y
|
|
—
|
el ensayo in vitro del Instituto de Evaluación e Investigación de Productos Químicos (Chemical Evaluation and Research Institute, CERI) de unión al receptor estrogénico con utilización de una proteína con un dominio de unión al ligando recombinante humano (2).
|
Se dispone de normas de comportamiento (NC) (3) para facilitar el desarrollo y la validación de métodos de ensayo similares para el mismo parámetro de peligro, considerando la modificación a su debido tiempo de las PBTG 493, de manera que puedan añadirse nuevos ensayos similares a unas PBTG actualizadas. Sin embargo, la adición de ensayos similares solo se efectuará previa revisión y acuerdo por parte de la OCDE en el sentido de que se cumplen las normas de comportamiento. Los ensayos incluidos en las TG 493 pueden utilizarse indistintamente para satisfacer los requisitos de los países miembros de la OCDE en cuanto a los resultados de los ensayos sobre la unión a los receptores estrogénicos, y también se benefician de la aceptación mutua de datos de la OCDE.
|
Antecedentes y principios de los ensayos incluidos en este método de ensayo
|
2.
|
La OCDE lanzó en 1998 una actividad muy prioritaria para revisar las directrices existentes y elaborar nuevas directrices sobre los ensayos, tanto de cribado como finales, de posibles alteradores endocrinos. El marco conceptual de la OCDE para los ensayos y la evaluación de posibles alteradores endocrinos se revisó en 2012. Los marcos conceptuales originales y revisados se incluyen como anexos en el documento de orientación sobre las directrices de ensayo normalizadas para la evaluación de los productos en cuanto a las alteraciones endocrinas (4). El marco conceptual se compone de cinco niveles, cada uno de los cuales corresponde a un nivel diferente de complejidad biológica. Los ensayos de unión a receptores estrogénicos descritos en este método de ensayo son de nivel 2, que incluye los ensayos in vitro que aportan datos sobre los mecanismos y rutas endocrinos seleccionados. Este método se refiere a los ensayos in vitro de unión a receptores diseñados para identificar los ligandos del receptor estrogénico humano alfa (ERα).
|
|
3.
|
Se ha demostrado claramente la pertinencia del ensayo in vitro de unión a ER respecto a funciones biológicas. Los ensayos de unión a ER están diseñados para identificar productos que pueden alterar la ruta de las hormonas estrogénicas, y han sido utilizadas ampliamente durante las dos últimas décadas para caracterizar la distribución tisular de los ER, así como para identificar agonistas/antagonistas de los ER. Estos ensayos reflejan la interacción ligando-receptor, que es la fase inicial de la vía estrogénica de señalización y es esencial para la función reproductora en todos los vertebrados.
|
|
4.
|
La interacción de los estrógenos con los ER puede afectar a la transcripción de genes controlados por estrógenos e inducir efectos no genómicos, lo cual puede dar lugar a la inducción o a la inhibición de procesos celulares, incluidos los necesarios para la proliferación celular, el desarrollo fetal normal y la función reproductora (5) (6) (7). La perturbación de los sistemas estrogénicos normales puede provocar efectos adversos sobre el desarrollo normal (ontogénesis), la salud reproductiva y la integridad del aparato reproductor. Una señalización inadecuada de los ER puede producir efectos tales como un mayor riesgo de cáncer dependiente de hormonas, trastornos de la fertilidad y alteraciones del crecimiento y el desarrollo del feto (8).
|
|
5.
|
Los ensayos de unión de ligandos in vitro se basan en la interacción directa de una sustancia con el punto de unión de un ligando a un receptor específico que regula la transcripción de un gen. El componente clave del ensayo de unión al receptor estrogénico recombinante humano alfa (hrERα) mide la capacidad de un ligando radiomarcado ([3H]17β-estradiol) para unirse al receptor estrogénico en presencia de concentraciones cada vez mayores de un producto problema (es decir, un competidor). Los productos problema que presentan una elevada afinidad por el receptor estrogénico compiten con el ligando radiomarcado a una concentración menor en comparación con los productos que presentan menor afinidad por el receptor. Este ensayo consta de dos componentes principales: un experimento de unión de saturación para caracterizar los parámetros de la interacción receptor-ligando y documentar la especificidad del ER, seguido de un experimento de unión de competencia que caracteriza la competencia entre un producto problema y un ligando radiomarcado para unirse al ER.
|
|
6.
|
Los estudios de validación de los ensayos de unión del CERI y de FW han demostrado su pertinencia y fiabilidad con respecto a su finalidad prevista (2).
|
|
7.
|
Las definiciones y abreviaturas utilizadas en el presente método de ensayo se recogen en el apéndice 1.
|
Ámbito de aplicación y limitaciones relativas a los ensayos de unión a receptores
|
8.
|
Estos ensayos se proponen para fines de cribado y establecimiento de prioridades, pero también pueden proporcionar información sobre un acontecimiento de iniciación molecular que pueda utilizarse en un enfoque de ponderación de las pruebas. Abordan la unión del producto al dominio de unión del ERα con el ligando en un sistema in vitro. Así pues, los resultados no deben extrapolarse directamente a la compleja señalización y regulación del sistema endocrino intacto in vivo.
|
|
9.
|
La unión del ligando natural, el 17β-estradiol, es la etapa inicial de una serie de acontecimientos moleculares que activa la transcripción de los genes diana y, en última instancia, culmina con un cambio fisiológico (9). Por lo tanto, la unión al dominio de unión del ERα con el ligando se considera uno de los mecanismos clave de la alteración endocrina (AE) con mediación de ER, aunque existen otros mecanismos a través de los cuales puede producirse la AE, entre los que se incluyen los siguientes: i) interacciones con puntos del ERα distintos de la cavidad de unión con el ligando, ii) interacciones con otros receptores pertinentes para la señalización estrogénica, ERβ y receptores estrogénicos acoplados con la proteína G, otros receptores y sistemas enzimáticos dentro del sistema endocrino, iii) síntesis de hormonas, iv) activación y/o inactivación metabólica de hormonas, v) distribución de hormonas a los tejidos diana, y vi) eliminación de hormonas del organismo. Ninguno de los ensayos con arreglo al presente método de ensayo aborda estos modos de acción.
|
|
10.
|
Este método de ensayo aborda la capacidad de las sustancias de unirse al ERα humano y no distingue entre agonistas o antagonistas del ERα. Estos ensayos tampoco abordan otros acontecimientos posteriores, como la transcripción de genes o los cambios fisiológicos. Teniendo en cuenta que durante la validación únicamente se utilizaron sustancias de un solo componente, no se ha abordado la aplicabilidad a mezclas problema. Sin embargo, los ensayos son técnicamente aplicables a las sustancias de componentes múltiples y mezclas. Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines normativos, debe considerarse si podría proporcionar resultados adecuados a tales fines y, en caso afirmativo, por qué. Dichas consideraciones no son necesarias cuando los requisitos normativos estipulan que la mezcla debe someterse a ensayo.
|
|
11.
|
Los sistemas de receptores sin células no tienen capacidad metabólica intrínseca y no han sido validados en combinación con sistemas enzimáticos metabólicos. Sin embargo, se podría incorporar la actividad metabólica en el diseño de un estudio, pero ello requeriría más esfuerzos de validación.
|
|
12.
|
Los productos que pueden desnaturalizar la proteína (es decir, la proteína del receptor), como los tensioactivos o los productos que pueden cambiar el pH de la solución amortiguadora del ensayo, no pueden someterse a ensayo o solo a concentraciones que no presenten tales interacciones. Por lo demás, el intervalo de concentraciones que pueden utilizarse en los ensayos respecto a un producto problema está limitado por su solubilidad en la solución amortiguadora del ensayo.
|
|
13.
|
A efectos de información, en el cuadro 1 se presentan los resultados de los ensayos correspondientes a las 24 sustancias que se estudiaron con los dos ensayos plenamente validados descritos en el presente método de ensayo. De estas sustancias, 17 se clasifican como ligandos del ER y 8 como no ligandos, sobre la base de los informes publicados, incluidos los ensayos in vitro de activación transcripcional de ER y/o el ensayo uterotrófico (9) (10) (11) (12) (13) (14) (15). En referencia a los datos resumidos en el cuadro 1, había casi un 100 % de concordancia entre los dos ensayos en la clasificación de todas las sustancias hasta 10-4 M, y cada sustancia se clasificó correctamente como ligando / no ligando del ER. En las normas de comportamiento aplicables a los ensayos de unión al hrER se ofrece información suplementaria sobre este grupo de sustancias, así como sobre otras sustancias objeto de ensayos de unión al hrER durante los estudios de validación (3), apéndice 2 (cuadros 1, 2 y 3).
Cuadro 1
Clasificación de sustancias como ligandos o no ligandos del ER cuando se someten a los ensayos de unión al hrER de FW y del CERI con referencia a la respuesta prevista
|
|
Nombre de la sustancia
|
N.o CAS
|
Respuesta prevista
|
Ensayo de FW
|
Ensayo del CERI
|
Clase química del MeSH
|
Clase de producto
|
|
|
Intervalo de concentración (M)
|
Clasificación
|
Intervalo de concentración (M)
|
Clasificación
|
|
1
|
17β-Estradiol
|
50-28-2
|
Ligando
|
1 × 10-11 – 1 × 10-6
|
Ligando
|
1 × 10-11 – 1 × 10-6
|
Ligando
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
2
|
Noretinodrel
|
68-23-5
|
Ligando
|
3 × 10-9 – 30 × 10-4
|
Ligando
|
3 × 10-9 – 30 × 10-4
|
Ligando
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
3
|
Noretindrona
|
68-22-4
|
Ligando
|
3 × 10-9 – 30 × 10-4
|
Ligando
|
3 × 10-9 – 30 × 10-4
|
Ligando
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
4
|
Ftalato de di-n-butilo
|
84-74-2
|
No ligando (*5)
|
1 × 10-10 – 1 × 10-4
|
No ligando (*6)
(1)
|
1 × 10-10 – 1 × 10-4
|
No ligando (*6)
(1)
|
Hidrocarburos (cíclicos), ésteres
|
Plastificantes, productos intermedios
|
|
5
|
DES
|
56-53-1
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Hidrocarburos (cíclicos), fenoles
|
Productos farmacéuticos, productos veterinarios
|
|
6
|
17α-Etinilestradiol
|
57-63-6
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
7
|
meso-Hexestrol
|
84-16-2
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Hidrocarburos (cíclicos), fenoles
|
Productos farmacéuticos, productos veterinarios
|
|
8
|
Genisteína
|
446-72-0
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Hidrocarburos (heterocíclicos), flavonoides
|
Productos naturales
|
|
9
|
Ecuol
|
531-95-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Metabolitos fitoestrogénicos
|
Productos naturales
|
|
10
|
Butilparabeno (4-hidroxibenzoato de n-butilo)
|
94-26-8
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Parabenos
|
Conservantes
|
|
11
|
Nonilfenol (mezcla)
|
84852-15-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Alquilfenoles
|
Compuestos intermedios
|
|
12
|
o,p’-DDT
|
789-02-6
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Organoclorados
|
Insecticidas
|
|
13
|
Corticosterona
|
50-22-6
|
No ligando (*5)
|
1 × 10-10 – 1 × 10-4
|
No ligando
|
1 × 10-10 – 1 × 10-4
|
No ligando
|
Esteroides
|
Productos naturales
|
|
14
|
Zearalenona
|
17924-92-4
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Hidrocarburos (heterocíclicos), lactonas
|
Productos naturales
|
|
15
|
Tamoxifeno
|
10540-29-1
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos, productos veterinarios
|
|
16
|
5α-Dihidrotestosterona
|
521-18-6
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Esteroides, no fenólicos
|
Productos naturales
|
|
17
|
Bisfenol A
|
80-05-7
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Fenoles
|
Productos intermedios
|
|
18
|
4-n-Heptilfenol
|
1987-50-4
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Dudoso (5)
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Alquilfenoles
|
Productos intermediarios
|
|
19
|
Kepona (clordecona)
|
143-50-0
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Hidrocarburos (halogenados)
|
Plaguicidas
|
|
20
|
Benzo[a]antraceno
|
56-55-3
|
No ligando
|
1 × 10-10 – 1 × 10-3
|
No ligando (6)
|
1 × 10-10 – 1 × 10-3
|
No ligando (6)
|
Hidrocarburos aromáticos
|
Productos intermediarios
|
|
21
|
Enterolactona
|
78473-71-9
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Ligando
|
Fitoestrógenos
|
Productos naturales
|
|
22
|
Progesterona
|
57-83-0
|
No ligando (*5)
|
1 × 10-10 – 1 × 10-4
|
No ligando
|
1 × 10-10 – 1 × 10-4
|
No ligando
|
Esteroides
|
Productos naturales
|
|
23
|
Octiltrietoxisilano
|
2943-75-1
|
No ligando
|
1 × 10-10 – 1 × 10-3
|
No ligando
|
1 × 10-10 – 1 × 10-3
|
No ligando
|
Silanos
|
Modificadores de superficie
|
|
24
|
Atrazina
|
1912-24-9
|
No ligando (*5)
|
1 × 10-10 – 1 × 10-4
|
No ligando
|
1 × 10-10 – 1 × 10-4
|
No ligando
|
Compuestos heterocíclicos
|
Herbicidas
|
|
COMPONENTES DEL ENSAYO DE UNIÓN AL hrER
Componentes esenciales del ensayo
|
14.
|
Este método de ensayo se aplica a los ensayos con un receptor ER y un ligando con la fuerza adecuada para unirse al receptor que puede utilizarse como marcador para el ensayo y puede ser desplazado con concentraciones cada vez mayores de un producto problema. Los ensayos de unión contienen los dos componentes principales siguientes: 1) unión de saturación y 2) unión de competencia. El ensayo de unión de saturación se utiliza para confirmar la especificidad y la actividad de los preparados de receptores, mientras que el experimento de unión de competencia se utiliza para evaluar la capacidad de un producto problema para unirse al hrER.
|
Testigos
|
15.
|
Debe describirse la base del estrógeno de referencia y de los testigos en paralelo propuestos. Los testigos en paralelo [de disolvente (vehículo), positivos (ligando del ER; con afinidad fuerte y débil), negativos (no ligando)], según proceda, sirven para indicar que el ensayo funciona en las condiciones de ensayo y proporcionan la base para comparar unos experimentos con otros; generalmente forman parte de los criterios de aceptabilidad de un experimento dado (1). Deben incluirse en una placa de cada tanda todos los puntos que forman las curvas de concentración completas del estrógeno de referencia y de los testigos (es decir, ligando débil y no ligando). Todas las demás placas deben contener: 1) una concentración alta (con desplazamiento aproximadamente total del ligando radiomarcado) y media (aproximadamente la CI50) de E2 y ligando débil, cada uno por triplicado; 2) el testigo de disolvente y el ligando inespecífico, cada uno por triplicado.
|
Procedimientos normales de control de calidad
|
16.
|
Deben llevarse a cabo procedimientos normales de control de calidad, tal como se describe para cada ensayo, a fin de garantizar que los receptores activos, las concentraciones correctas de los productos y los límites de tolerancia se mantienen estables a lo largo de varios pases, y conservan a lo largo del tiempo la capacidad de proporcionar las respuestas esperadas de unión a los ER.
|
Demostración de la competencia del laboratorio
|
17.
|
Antes de someter productos desconocidos a alguno de los ensayos con arreglo al presente método, cada laboratorio debe demostrar su competencia para utilizar el ensayo realizando ensayos de saturación para confirmar la especificidad y la actividad de la preparación del ER, y ensayos de unión de competencia del estrógeno de referencia y de los testigos (es decir, ligando débil y no ligando). El laboratorio debe establecer una base de datos históricos con los resultados del estrógeno de referencia y de los testigos generados mediante 3-5 experimentos independientes llevados a cabo en diferentes días. Estos experimentos servirán de base para el estrógeno de referencia y los testigos históricos del laboratorio, y se utilizarán como evaluación parcial de la aceptabilidad del ensayo para futuras tandas.
|
|
18.
|
La sensibilidad del sistema de ensayo también será confirmada mediante el ensayo de las sustancias para la prueba de la competencia que figuran en el cuadro 2. La lista de sustancias para la prueba de la competencia es un subconjunto de las sustancias de referencia que figuran en las normas de comportamiento relativas a los ensayos de unión a los ER (3). Estas sustancias están disponibles en el mercado, representan las clases de productos normalmente asociadas con la actividad de unión a los ER, y presentan una gama adecuada de potencia esperada como ligandos (es decir, de fuerte a débil) y no ligandos (es decir, negativos). Respecto a cada sustancia para la prueba de la competencia, las concentraciones ensayadas deben cubrir el intervalo indicado en el cuadro 2. Deben realizarse al menos tres experimentos con cada sustancia y los resultados deben ser conformes a la actividad química esperada. Cada experimento debe realizarse de forma independiente (es decir, con diluciones nuevas del receptor, de los productos y del reactivo), con tres réplicas por cada concentración. La competencia se demuestra por la clasificación correcta (positiva/negativa) de cada sustancia para la prueba de la competencia. Cada técnico debe realizar la prueba de la competencia al aprender los ensayos.
Cuadro 2
Lista de testigos y sustancias para la prueba de la competencia en relación con los ensayos de unión de competencia al hrER
(90)
|
N.o
|
Nombre de la sustancia
|
N.o CAS (91)
|
Respuesta esperada (92)
(93)
|
Intervalo conc. ensayo (M)
|
Clase química según el sistema MeSH (94)
|
Clase de producto (95)
|
|
Testigos (estrógeno de referencia, ligando débil, no ligando)
|
|
1
|
17β-Estradiol
|
50-28-2
|
Ligando
|
1 × 10-11 – 1 × 10-6
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
2
|
Noretinodrel (o) noretindrona
|
68-23-5 (o) 68-22-4
|
Ligando
|
3 × 10-9 – 30 × 10-6
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
3
|
Octiltrietoxisilano
|
2943-75-1
|
No ligando
|
1 × 10-10 – 1 × 10-3
|
Silanos
|
Modificadores de superficie
|
|
Sustancias para la prueba de la competencia (95)
|
|
4
|
Dietilestilbestrol
|
56-53-1
|
Ligando
|
1 × 10-11 – 1 × 10-6
|
Hidrocarburos (cíclicos), fenoles
|
Productos farmacéuticos, productos veterinarios
|
|
5
|
17α-Etinilestradiol
|
57-63-6
|
Ligando
|
1 × 10-11 – 1 × 10-6
|
Esteroides
|
Productos farmacéuticos, productos veterinarios
|
|
6
|
meso-Hexestrol
|
84-16-2
|
Ligando
|
1 × 10-11 – 1 × 10-6
|
Hidrocarburos (cíclicos), fenoles
|
Productos farmacéuticos, productos veterinarios
|
|
7
|
Tamoxifeno
|
10540-29-1
|
Ligando
|
1 × 10-11 – 1 × 10-6
|
Hidrocarburos (cíclicos)
|
Productos farmacéuticos, productos veterinarios
|
|
8
|
Genisteína
|
446-72-0
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Compuestos heterocíclicos, flavonoides
|
Productos naturales
|
|
9
|
Bisfenol A
|
80-05-7
|
Ligando
|
1 × 10-10 – 1 × 10-3
|
Fenoles
|
Productos intermedios
|
|
10
|
Zearalonona
|
17924-92-4
|
Ligando
|
1 × 10-11 – 1 × 10-3
|
Compuestos heterocíclicos, lactonas
|
Productos naturales
|
|
11
|
Butilparabenos
|
94-26-8
|
Ligando
|
1 × 10-11 – 1 × 10-3
|
Ácidos carboxílicos, fenoles
|
Conservantes
|
|
12
|
Atrazina
|
1912-24-9
|
No ligando
|
1 × 10-11 – 1 × 10-6
|
Compuestos heterocíclicos
|
Herbicidas
|
|
13
|
Ftalato de di-n-butilo (DBP) (96)
|
84-74-2
|
No ligando (97)
|
1 × 10-10 – 1 × 10-4
|
Hidrocarburos (cíclicos), ésteres
|
Plastificantes, productos intermedios
|
|
14
|
Corticosterona
|
50-22-6
|
No ligando
|
1 × 10-11 – 1 × 10-4
|
Esteroides
|
Productos naturales
|
|
Ensayo de solubilidad y determinación del intervalo de concentraciones de los productos problema
|
19.
|
Debe realizarse un ensayo preliminar para determinar el límite de solubilidad de cada producto problema y para identificar el intervalo adecuado de concentraciones que se ha de utilizar al realizar el ensayo. El límite de solubilidad de cada producto problema se determina inicialmente en el disolvente y se confirma después en las condiciones del ensayo. La concentración final utilizada en el ensayo no deberá ser superior a 1 mM. La determinación del intervalo de concentraciones consiste en someter a ensayo un control del disolvente, junto con ocho diluciones logarítmicas en serie, a partir de la concentración máxima aceptable (por ejemplo, 1 mM o menos, sobre la base del límite de solubilidad), y anotar la presencia de turbidez o precipitado. Las concentraciones en los experimentos segundo y tercero deben ajustarse según convenga para caracterizar mejor la curva concentración-respuesta.
|
Criterios de aceptabilidad de las tandas de ensayo
|
20.
|
La aceptación o el rechazo de una tanda de ensayo se basa en la evaluación de los resultados obtenidos con el estrógeno de referencia y el testigo utilizados para cada experimento. En primer lugar, para la placa 1, las curvas completas de concentración de los testigos de referencia de cada experimento deben cumplir los criterios de comportamiento con parámetros de ajuste de las curvas (por ejemplo, CI50 y pendiente de Hill), sobre la base de los resultados comunicados en relación con los respectivos protocolos de los ensayos del CERI y de FW (apéndices 2 y 3), y de los datos de controles históricos del laboratorio que realice el ensayo. Todos los testigos (estrógeno de referencia, ligando débil y no ligando) deben clasificarse correctamente en cada experimento. En segundo lugar, los testigos de todas las placas posteriores deben evaluarse en cuanto a su coherencia con la placa 1. Debe utilizarse un intervalo suficiente de concentraciones del producto problema para definir claramente la parte superior de la curva de unión de competencia. La variabilidad entre las réplicas de cada concentración del producto problema, así como entre las tres tandas independientes, debe ser razonable y científicamente justificable. La capacidad de realizar el ensayo de forma coherente debe demostrarse mediante la elaboración y el mantenimiento de una base de datos históricos sobre el estrógeno de referencia y los testigos. Las desviaciones típicas (DT) o los coeficientes de variación (CV) de las medias de los parámetros de ajuste de las curvas del estrógeno de referencia y de los ligandos débiles testigo a partir de experimentos múltiples pueden utilizarse como medida de la reproducibilidad intralaboratorios. A la hora de revisar los resultados de los testigos de las placas de cada tanda, así como respecto a cada producto problema, debe aplicarse un juicio profesional.
Además, deben seguirse los siguientes principios en relación con los criterios de aceptabilidad:
|
—
|
Los datos deben ser suficientes para una evaluación cuantitativa de la unión a los ER;
|
|
—
|
Las concentraciones ensayadas deben mantenerse dentro del intervalo de solubilidad del producto problema.
|
|
Análisis de los datos
|
21.
|
El procedimiento definido de análisis de los datos relativos a la unión de saturación y de competencia deben respetar los principios clave para caracterizar las interacciones receptor-ligando. Normalmente, los datos relativos a la unión de saturación se analizan utilizando un modelo de regresión no lineal que tiene en cuenta la unión total y la inespecífica. Al determinar los parámetros Bmax y Kd, puede ser necesario efectuar una corrección para tener en cuenta el agotamiento de los ligandos [p. ej., Swillens, 1995 (19)]. Los datos procedentes de la unión de competencia suelen transformarse [por ejemplo, porcentaje de unión específica y concentración del producto problema (log M)]. Deben estimarse los valores de log CI50 de cada producto problema, utilizando un programa informático adecuado de ajuste de curvas no lineales para ajustarse a una ecuación de Hill de cuatro parámetros. Tras el análisis inicial, debe llevarse a cabo una revisión visual y con los parámetros de ajuste de la curva sobre el grado de calidad del ajuste de los datos de la unión con la curva de unión de competencia generada. En algunos casos, puede ser necesario realizar un análisis adicional para obtener el mejor ajuste de la curva (por ejemplo, limitar la parte superior y/o inferior de la curva, utilizar la regla del 10 %); véase el apéndice 4 y la referencia 2 (sección III.A.2).
|
|
22.
|
El cumplimiento de los criterios de aceptabilidad (punto 20) indica que el sistema de ensayo funciona correctamente, pero no garantiza que cualquier ensayo concreto produzca datos exactos. La reproducción de los resultados correctos del primer ensayo es la mejor indicación de que se han producido datos exactos.
|
Criterios generales de interpretación de los datos
|
23.
|
Actualmente no se dispone de un método aceptado universalmente para interpretar los datos de la unión al ER. Sin embargo, tanto las evaluaciones cualitativas (por ejemplo, ligando / no ligando) como las cuantitativas [por ejemplo, log CI50, afinidad relativa de unión (Relative Binding Affinity, RBA)] de la actividad mediada por el hrER deben basarse en datos empíricos y en un sólido juicio científico.
|
Informe del ensayo
|
24.
|
El informe del ensayo debe incluir la información siguiente:
|
|
Testigo/ producto de referencia /producto problema
|
—
|
origen, número de lote, fecha límite de utilización, si se dispone de ellos;
|
|
—
|
estabilidad del producto problema en sí, si se conoce;
|
|
—
|
solubilidad y estabilidad del producto problema en el disolvente, si se conocen;
|
|
—
|
medición del pH, osmolalidad y precipitado en el medio de cultivo al que se ha añadido el producto problema, según proceda.
|
|
|
|
Sustancias de un solo componente:
|
—
|
aspecto físico, hidrosolubilidad y otras propiedades fisicoquímicas pertinentes;
|
|
—
|
identificación química, como nombre IUPAC o CAS, número CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores; pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.
|
|
|
|
Sustancias de componentes múltiples, UVCB y mezclas:
|
—
|
deben caracterizarse en la medida de lo posible por la identidad química (véase más arriba), la cantidad en que están presentes y las propiedades fisicoquímicas pertinentes de sus componentes.
|
|
|
|
Disolvente o vehículo:
|
—
|
caracterización (naturaleza, proveedor y lote);
|
|
—
|
justificación de la elección del disolvente o vehículo;
|
|
—
|
solubilidad y estabilidad del producto problema en el disolvente o vehículo, si se conocen.
|
|
|
|
Receptores:
|
—
|
fuente de receptores (proveedor, número de catálogo, lote, especie del receptor, concentración del receptor activo facilitada por el proveedor, certificación del proveedor);
|
|
—
|
caracterización de los receptores (incluidos los resultados de la unión de saturación): Kd, Bmax;
|
|
—
|
conservación de los receptores;
|
|
—
|
ligandos radiomarcados:
|
|
—
|
proveedor, número de catálogo, lote, actividad específica.
|
|
|
|
Condiciones del ensayo:
|
—
|
limitaciones de la solubilidad en las condiciones del ensayo;
|
|
—
|
composición de la solución amortiguadora del ligando;
|
|
—
|
concentración del receptor;
|
|
—
|
concentración del marcador (por ejemplo, ligando radiomarcado);
|
|
—
|
concentraciones del producto problema;
|
|
—
|
porcentaje del vehículo en el ensayo final;
|
|
—
|
temperatura y duración de la incubación;
|
|
—
|
método de separación entre sustancia unida y libre;
|
|
—
|
testigos positivos y negativos / sustancias de referencia;
|
|
—
|
criterios empleados para considerar que los resultados de los ensayos son positivos, negativos o dudosos.
|
|
|
|
Comprobación de la aceptabilidad:
|
—
|
valores reales de CI50 y pendiente de Hill, correspondientes a los testigos positivos en paralelo y a las sustancias de referencia.
|
|
|
|
Resultados:
|
—
|
datos brutos y datos sobre la sustancia unida y libre;
|
|
—
|
comprobación de confirmación de la desnaturalización, si procede;
|
|
—
|
si existe, concentración efectiva mínima (CEMín);
|
|
—
|
valores de RBA y/o de CI50, según proceda;
|
|
—
|
relación concentración-respuesta, cuando sea posible;
|
|
—
|
análisis estadísticos, en su caso, junto con una medida del error y de la confianza (por ejemplo, SEM, DT, CV o IC del 95 %) y una descripción de cómo se han obtenido dichos valores.
|
|
|
|
Discusión de los resultados:
|
—
|
aplicación de la regla del 10 %.
|
|
Conclusión
|
BIBLIOGRAFÍA
|
(1)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(2)
|
OCDE (2015): Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, París.
|
|
(3)
|
OCDE (2015): Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(4)
|
OCDE (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 150), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: p. 20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OCDE (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Apéndice 1
DEFINICIONES Y ABREVIATURAS
Regla del 10 %
: Opción de excluir de los análisis los puntos de datos en los que la media de las réplicas tiene un valor porcentual de unión especifíca al [3H]17β-estradiol que es igual o superior al 10 % del valor medio a una concentración inferior (véase el apéndice 4).
Criterios de aceptabilidad
: Normas mínimas para la realización de controles experimentales y patrones de referencia. Para que un experimento se considere válido deben cumplirse todos los criterios de aceptabilidad.
Exactitud (concordancia)
: Grado de coincidencia entre los resultados de un ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del ensayo y es un aspecto de su pertinencia. Este término y el de “concordancia” se suelen usar indistintamente para indicar la proporción de resultados correctos de un ensayo (1).
MC
: Marco conceptual de la OCDE para los ensayos y la evaluación de los alteradores endocrinos.
Producto
: Sustancia o mezcla.
CV
: Coeficiente de variación.
E2
: 17β-Estradiol
AE
: Alteración endocrina.
hERα
: Receptor estrogénico humano alfa.
ER
: Receptor estrogénico.
Actividad estrogénica
: Capacidad de un producto para imitar al 17β-estradiol en su posibilidad de unirse a los receptores estrogénicos y activarlos. La unión al hERα puede detectarse con este método de ensayo.
CI50
: Concentración del producto problema que produce la mitad de la inhibición máxima.
ICCVAM
: Comité de Coordinación Interagencias sobre la Validación de Métodos Alternativos de Estados Unidos (Interagency Coordinating Committee on the Validation of Alternative Methods).
Reproducibilidad interlaboratorios
: Medida del grado en que distintos laboratorios cualificados, utilizando el mismo protocolo y sometiendo a ensayo las mismas sustancias, pueden producir resultados cualitativa y cuantitativamente similares. La reproducibilidad interlaboratorios se determina durante los procesos de prevalidación y validación, e indica el grado en que un ensayo puede transferirse con éxito entre laboratorios; se denomina también reproducibilidad entre laboratorios (1).
Reproducibilidad intralaboratorios
: Determinación del grado en que personas cualificadas del mismo laboratorio pueden repetir con éxito los resultados en momentos diferentes, utilizando un protocolo especificado. También se denomina reproducibilidad dentro del laboratorio (1).
CE:Mín
: Concentración efectiva mínima; es la concentración más baja del producto problema que produce una respuesta (es decir, la concentración más baja del producto problema en la que el factor de inducción es estadísticamente diferente al del control del vehículo en paralelo).
Ensayos de imitación
: Expresión coloquial para denominar los ensayos que son estructural y funcionalmente similares a un método de ensayo de referencia validado y aceptado. También se pueden denominar métodos de ensayo similares.
PBTG
: Directrices de ensayo basadas en el comportamiento (Performance-Based Test Guideline).
Normas de comportamiento
: Normas, basadas en un método de ensayo validado, que proporcionan la base para evaluar la comparabilidad de un ensayo propuesto que es similar desde el punto de vista mecánico y funcional. Se incluye lo siguiente: 1) los componentes esenciales del ensayo; 2) una lista mínima de productos de referencia seleccionados de entre los productos utilizados para demostrar el comportamiento aceptable del método de ensayo validado; y 3) los niveles similares de fiabilidad y exactitud, basados en lo obtenido con el método de ensayo validado, que el ensayo propuesto debe demostrar cuando se evalúa con la lista mínima de productos de referencia (1).
Sustancias para la prueba de la competencia
: Subconjunto de las sustancias de referencia incluidas en las normas de comportamiento que pueden ser utilizadas por los laboratorios para demostrar la competencia técnica con un ensayo normalizado. Los criterios de selección de estas sustancias suelen incluir que representen la gama de respuestas, que estén disponibles en el mercado y que se disponga sobre ellas de datos de referencia de alta calidad.
Competencia
: Capacidad demostrada de realizar correctamente un ensayo antes de someter a este sustancias desconocidas.
Estrógeno de referencia
: 17ß-estradiol (E2, CAS 50-28-2).
Métodos de ensayo de referencia
: Ensayos sobre los que se basan las PBTG 493.
RBA
: Afinidad relativa de unión (Relative Binding Affinity). La RBA de una sustancia se calcula como porcentaje del log CI50 de la sustancia respecto al log CI50 del 17β-estradiol.
Pertinencia
: Descripción de la relación de un ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un ensayo (1).
Fiabilidad
: Medida del grado en que un ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios.
DT
: Desviación típica.
Producto problema
: Sustancia o mezcla estudiada con este método de ensayo.
Método de ensayo validado
: Ensayo sobre el cual se han completado estudios de validación para determinar su pertinencia (incluida su exactitud) y su fiabilidad con un fin específico. Es importante señalar que un método de ensayo validado podría no tener un comportamiento suficiente en términos de exactitud y fiabilidad como para considerarse aceptable a efectos del fin propuesto (1).
Validación
: Proceso por el que se establece para un fin determinado la fiabilidad y pertinencia de un enfoque, método, ensayo, proceso o evaluación concretos (1).
Apéndice 2
ENSAYOS DE FREYBERGER-WILSON DE UNIÓN DE SATURACIÓN Y DE COMPETENCIA A RECEPTORES ESTROGÉNICOS (ERΑ) IN VITRO UTILIZANDO EL ERα RECOMBINANTE HUMANO COMPLETO
CONSIDERACIONES INICIALES Y LIMITACIONES (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
1.
|
Este ensayo de unión de saturación y de competencia a receptores estrogénicos (ERα) in vitro utiliza un receptor ERα humano completo (hrERα) que se produce en células de insectos infectadas con baculovirus, de las que después se aísla. El protocolo, elaborado por Freyberger y Wilson, fue sometido a un estudio de validación internacional multilaboratorios (2), que demostró su pertinencia y fiabilidad para la finalidad prevista del ensayo.
|
|
2.
|
Este ensayo es un procedimiento de cribado para identificar sustancias que pueden unirse al hrERα completo. Se utiliza para determinar la capacidad de un producto problema de competir con el 17β-estradiol para unirse al hrERα. Los resultados cuantitativos del ensayo pueden incluir la CI50 (medida de la concentración del producto problema necesaria para desplazar la mitad del [3H]17β-estradiol del hrERα) y las afinidades relativas de unión de los productos problema respecto al hrERα en comparación con el 17β-estradiol. A efectos de cribado de los productos problema, entre los resultados cualitativos aceptables de los ensayos pueden incluirse la clasificación de los productos problema como ligandos o no ligandos del hrERα, o bien si el resultado es dudoso sobre la base de los criterios descritos para las curvas de unión.
|
|
3.
|
El ensayo utiliza un ligando radiactivo, lo que impone al laboratorio la obligación de disponer de una licencia de materiales radiactivos. Todos los procedimientos con radioisótopos y productos peligrosos deben ajustarse a las normas y procedimientos descritos en la legislación nacional.
|
|
4.
|
Las secciones “INTRODUCCIÓN GENERAL” y “COMPONENTES DEL ENSAYO DE UNIÓN AL hrER” deben leerse antes de utilizar este ensayo con fines normativos. Las definiciones y abreviaturas utilizadas en las presentes directrices de ensayo se recogen en el apéndice 1.
|
PRINCIPIOS DEL ENSAYO (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
5.
|
El ensayo de unión al hrERα mide la capacidad de un ligando radiomarcado ([3H]17β-estradiol) para unirse al receptor estrogénico en presencia de concentraciones cada vez mayores de un producto problema (es decir, un competidor). Los productos problema que presentan una elevada afinidad por el receptor estrogénico compiten con el ligando radiomarcado a una concentración menor en comparación con los productos que presentan menor afinidad por el receptor.
|
|
6.
|
Este ensayo consta de dos componentes principales: un experimento de unión de saturación para caracterizar los parámetros de la interacción receptor-ligando, seguido de un experimento de unión de competencia que caracteriza la competencia entre un producto problema y un ligando radiomarcado para unirse al ER.
|
|
7.
|
El objetivo del experimento de unión de saturación es caracterizar un determinado lote de receptores en cuanto a su afinidad de unión y número para preparar el experimento de unión de competencia. El experimento de unión de saturación mide, en condiciones de equilibrio, la afinidad de una concentración fija del receptor estrogénico por su ligando natural (representada por la constante de disociación, Kd) y la concentración de puntos activos de unión de los receptores (Bmax).
|
|
8.
|
El experimento de unión de competencia mide la afinidad de una sustancia para competir con el [3H]17β-estradiol en cuanto a la unión al ER. La afinidad se cuantifica por la concentración del producto problema que, en equilibrio, inhibe el 50 % de la unión específica del [3H]17β-estradiol (denominada "concentración inhibitoria del 50 %" o CI50). También puede evaluarse utilizando la afinidad relativa de unión (RBA, en relación con la CI50 del estradiol medida aparte en la misma tanda). El experimento de unión de competencia mide la unión del [3H]17β-estradiol a una concentración fija en presencia de una amplia gama de concentraciones (ocho órdenes de magnitud) del producto problema. A continuación, se ajustan los datos, en la medida de lo posible, a una forma de la ecuación de Hill (Hill, 1910) que describe el desplazamiento del radioligando por un ligando competitivo de un solo punto de unión. La magnitud del desplazamiento del estradiol radiomarcado en equilibrio se utiliza para caracterizar el producto problema como ligando, no ligando, o generador de una respuesta dudosa.
|
PROCEDIMIENTO
Demostración del comportamiento aceptable de la proteína del hrERα
|
9.
|
Con anterioridad a la realización sistemática de los ensayos de unión de saturación y de competencia, se debe demostrar que cada nuevo lote de hrERα presenta un comportamiento correcto en el laboratorio en el que se va a utilizar. Para demostrar el comportamiento debe seguirse un proceso de dos fases. Se trata de las siguientes:
|
—
|
Realizar un ensayo de unión de [3H]17β-estradiol de saturación para demostrar la especificidad y saturación del hrERα. El análisis de regresión no lineal de estos datos (por ejemplo, BioSoft; McPherson, 1985; Motulsky, 1995) y la consiguiente gráfica de Scatchard deben documentar la afinidad de unión al hrERα del [3H]17β-estradiol (Kd) y el número de receptores (Bmax) respecto a cada lote del hrERα.
|
|
—
|
Llevar a cabo un ensayo de unión de competencia utilizando las sustancias testigo [estrógeno de referencia (17β-estradiol)], un ligando débil (por ejemplo, noretinodrel o noretindrona), y un no ligando (octiltrietoxisilano, OTES). Cada laboratorio debe establecer una base de datos históricos para documentar la coherencia de la CI50 y de otros valores pertinentes sobre el estrógeno de referencia y el ligando débil entre experimentos y diferentes lotes de hrERα. Los parámetros de las curvas de unión de competencia de las sustancias testigo deben estar dentro de los límites del intervalo de confianza del 95 % (véase el cuadro 1) que se han establecido utilizando datos de los laboratorios que participaron en el estudio de validación de este ensayo (2).
|
|
Cuadro 1
Criterios de comportamiento establecidos para el estrógeno de referencia y el ligando débil, ensayo de FW de unión al hrER.
|
Sustancia
|
Parámetro
|
Media (7)
|
Desviación típica (n)
|
Intervalos de confianza del 95 % (8)
|
|
Límite inferior
|
Límite superior
|
|
17β-Estradiol
|
Parte superior (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Parte inferior (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Pendiente de Hill
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
Log CI50 (M)
|
-8,92 (9)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretinodrel
|
Parte superior (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Parte inferior (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Pendiente de Hill
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
Log CI50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Noretindrona (9)
|
Parte superior (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Parte inferior (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Pendiente de Hill
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
Log CI50 (M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Demostración del comportamiento del laboratorio
|
10.
|
Véanse los puntos 17 y 18 y el cuadro 2 de la sección “COMPONENTES DEL ENSAYO DE UNIÓN AL hrER” de este método de ensayo. Cada ensayo (unión de saturación y de competencia) debe consistir en tres tandas independientes (es decir, con diluciones nuevas del receptor, de los productos y de los reactivos) en días diferentes, y cada tanda debe contener tres réplicas.
|
Determinación de la concentración del receptor (hrERα)
|
11.
|
La concentración del receptor activo varía ligeramente en función del lote y de las condiciones de conservación. Por este motivo, debe determinarse la concentración del receptor activo tal como se recibe del proveedor. Esto dará la concentración adecuada del receptor activo en el momento de realizar la tanda.
|
|
12.
|
En condiciones correspondientes a la unión de competencia (es decir, [3H]-estradiol 1 nM), deben incubarse concentraciones nominales del receptor de 0,25, 0,5, 0,75 y 1 nM en ausencia (unión total) y presencia (unión inespecífica) de estradiol no marcado 1 μM. La unión específica, calculada como la diferencia de unión total e inespecífica, se representa frente a la concentración nominal del receptor. La concentración del receptor que da valores de unión específica correspondientes al 20 % de la sustancia radiomarcada añadida está relacionada con la correspondiente concentración nominal del receptor, y esta concentración del receptor debe utilizarse para los experimentos de unión de saturación y de competencia. Con frecuencia, una concentración final del rhER de 0,5 nM cumplirá esta condición.
|
|
13.
|
Si el criterio del 20 % no puede cumplirse de forma reiterada, deberá comprobarse si no se encuentran posibles errores en la configuración experimental. Si no se cumple el criterio del 20 %, puede deberse a que en el lote del recombinante hay muy poco de receptor activo, y en tal caso debe considerarse el uso de otro lote del receptor.
|
Ensayo de saturación
|
14.
|
Deben evaluarse por triplicado ocho concentraciones cada vez mayores de [3H]17β-estradiol, en las tres condiciones siguientes (véase el cuadro 2):
|
—
|
En ausencia de 17β-estradiol no marcado y presencia de ER. Se trata de la determinación de la unión total por medida de la radiactividad en los pocillos que solo tienen [3H]17β-estradiol.
|
|
—
|
En presencia de una concentración de 17-β-estradiol no marcado que supera 1000 veces la concentración de 17β-estradiol marcado y en presencia del ER. El propósito de esta condición es saturar los puntos activos de unión con 17β-estradiol no marcado y, midiendo la radiactividad en los pocillos, determinar la unión inespecífica. Se considera que el eventual estradiol marcado restante que pueda unirse al receptor se une a un punto de unión inespecífica, ya que el estradiol no marcado debe estar a una concentración tan elevada que esté unido a todos los puntos específicos disponibles del receptor.
|
|
—
|
En ausencia de 17β-estradiol no marcado y ausencia de ER (determinación de la radiactividad total).
|
|
Preparación de soluciones de [3H]17β-estradiol y de 17β-estradiol no marcado
|
15.
|
Deben prepararse diluciones de [3H]17β-estradiol añadiendo solución amortiguadora de ensayo a una solución madre 12 nM de [3H]17β-estradiol para obtener concentraciones inicialmente en el intervalo de 0,12 nM a 12 nM. Mediante la adición de 40 μl de estas soluciones a los pocillos de ensayo correspondientes de una placa de microvaloración de 96 pocillos (en un volumen final de 160 μl), se obtendrán las concentraciones finales de ensayo, entre 0,03 y 3,0 nM. La preparación de la solución amortiguadora de ensayo y de la solución madre de [3H]17β-estradiol y sus diluciones, así como la determinación de las concentraciones, se describen con detalle en el protocolo FW (2).
|
|
16.
|
Las diluciones de las soluciones etanólicas de 17β-estradiol deben prepararse añadiendo solución amortiguadora de ensayo para conseguir ocho concentraciones cada vez mayores que inicialmente van de 0,06 μM a 6 μM. Mediante la adición de 80 μl de estas soluciones a los pocillos de ensayo correspondientes de una placa de microvaloración de 96 pocillos (en un volumen final de 160 μl), se obtendrán las concentraciones finales de ensayo, que van de 0,03 μM hasta 3 μM. La concentración final de 17β-estradiol no marcado en los distintos pocillos de unión inespecífica debe ser 1 000 veces superior a la concentración de [3H]17β-estradiol marcado. La preparación de diluciones de 17β-estradiol no marcado se describe en profundidad en el protocolo FW (2).
|
|
17.
|
Debe utilizarse la concentración nominal del receptor que da una unión específica del 20 ± 5 % (véanse los puntos 12-13). La solución de hrERα debe prepararse inmediatamente antes de su utilización.
|
|
18.
|
Las placas de microvaloración de 96 pocillos se preparan como se ilustra en el cuadro 2, con 3 réplicas por concentración. En el apéndice 2.2 figura un ejemplo de asignación a las placas de concentraciones y volúmenes de [3H]17β-estradiol, de 17β-estradiol no marcado, de solución amortiguadora y de receptor.
|
Cuadro 2
Disposición de las placas de microvaloración para el ensayo de unión de saturación
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H]E2 + ER
|
0,06 nM [3H]E2 + ER
|
0,08 nM [3H]E2 + ER
|
0,10 nM [3H]E2 + ER
|
Unión total (disolvente)
|
|
B
|
0,30 nM [3H]E2 + ER
|
0,60 nM [3H]E2 + ER
|
1,0 nM [3H]E2 + ER
|
3,0 nM [3H]E2 + ER
|
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H]E2 + ER + 0,03 μM E2
|
0,06 nM [3H]E2 + ER + 0,06 μM E2
|
0,08 nM [3H]E2 + ER + 0,08 μM E2
|
0,10 nM [3H]E2 + E2 0,10 μM
|
Unión inespecífica
|
|
E
|
0,30 nM [3H]E2 + ER + 0,30 μM E2
|
0,60 nM [3H]E2 + ER + 0,60 μM E2
|
1,0 nM [3 H]E2 + ER + 1,0 μM E2
|
3,0 nM [3H]E2 + ER + 3,0 μM E2
|
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
|
[3H]E2: [3H]17β-Estradiol.
ER: Receptor estrogénico.
E2: 17β-Estradiol no marcado.
|
|
19.
|
Las placas de microvaloración del ensayo deben incubarse a una temperatura de entre 2o y 8oC durante entre 16 y 20 horas y colocarse en un rotador durante el período de incubación.
|
Medición del [3H]17β-estradiol unido al hrERα
|
20.
|
El [3H]17β-estradiol unido al hrERα debe separarse del [3H]17β-estradiol libre mediante la adición a cada pocillo de 80 μl de suspensión fría de DCC, seguida de agitación de las placas de microvaloración durante 10 minutos y de centrifugación durante 10 minutos a unas 2 500 rpm. Para reducir al mínimo la disociación de [3H]17β-estradiol unido al hrERα durante este proceso, es sumamente importante que la temperatura de las soluciones amortiguadoras y de los pocillos del ensayo se mantenga entre 2 y 8oC, y que cada paso se lleve a cabo con rapidez. Para tratar las placas de forma eficiente y rápida se necesita un agitador de placas de microvaloración.
|
|
21.
|
A continuación, se deben tomar 50 μl del sobrenadante que contiene el [3H]17β-estradiol unido al hrERα con muchísimo cuidado para evitar cualquier contaminación de los pocillos por contacto con la suspensión de DCC, y deben colocarse en una segunda placa de microvaloración.
|
|
22.
|
A continuación, deben añadirse a cada pocillo (A1-B12 y D1 a E12) 200 μl del líquido de centelleo, capaces de convertir la energía cinética de las emisiones nucleares en energía luminosa. Los pocillos G1-H12 (identificados como dpm totales) contienen diluciones seriadas del [3H]17β-estradiol (40 μl) que deben ponerse directamente en el líquido de centelleo de los pocillos de la placa de medición, como se indica en el cuadro 3; es decir, estos pocillos contienen solo 200 μl del líquido de centelleo y la dilución adecuada del [3H]17β-estradiol. Estas medidas muestran cuánto [3H]17β-estradiol medido en dpm se había añadido a cada conjunto de pocillos en relación con la unión total y con la unión inespecífica.
|
Cuadro 3
Disposición de las placas de microvaloración para el ensayo de unión de saturación, medición de la radiactividad
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H]E2 + ER
|
0,06 nM [3H]E2 + ER
|
0,08 nM [3H]E2 + ER
|
0,10 nM [3H]E2 + ER
|
Unión total (disolvente)
|
|
B
|
0,30 nM [3H]E2 + ER
|
0,60 nM [3H]E2 + ER
|
1,0 nM [3H]E2 + ER
|
3,0 nM [3H]E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H]E2 + ER + 0,03 μM E2
|
0,06 nM [3H]E2 + ER + 0,06 μM E2
|
0,08 nM [3H]E2 + ER + 0,08 μM E2
|
0,10 nM [3H]E2 + ER + 0,10 μM E2
|
Unión inespecífica
|
|
E
|
0,30 nM [3H]E2 + ER + 0,30 μM E2
|
0,60 nM [3H]E2 + ER + 0,60 μM E2
|
1,0 nM [3H]E2 + ER + 1,0 μM E2
|
3,0 nM [3H]E2 + ER + + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H]E2 (dpm totales)
|
0,06 nM [3H]E2
|
0,08 nM [3H]E2
|
0,10 nM [3H]E2
|
dpm totales (*7)
|
|
H
|
0,30 nM [3H]E2
|
0,60 nM [3H]E2
|
1,0 nM [3H]E2
|
3,0 nM [3H]E2
|
|
[3H]E2: [3H]17β-estradiol.
ER: Receptor estrogénico.
E2: 17β-Estradiol no marcado.
dpm: Desintegraciones por minuto.
|
|
23.
|
La medición debe comenzar cuando hayan pasado al menos 2 horas y el tiempo de recuento debe ser de 40 minutos por pocillo. Para la determinación de las dpm/pocillo con corrección de la atenuación debe utilizarse un contador de centelleo para placas de microvaloración. Si no se dispone de este contador de centelleo para placas de microvaloración, se puede recurrir también a medir las muestras con un contador convencional. En estas condiciones, puede considerarse una reducción del tiempo de recuento.
|
Ensayo de unión de competencia
|
24.
|
El ensayo de unión de competencia mide la unión de una única concentración de [3H]17β-estradiol en presencia de concentraciones cada vez mayores de un producto problema. Deben utilizarse en paralelo tres réplicas de cada concentración en cada tanda. Además, deben realizarse tres tandas no simultáneas con cada producto problema. El ensayo debe configurarse en una o más placas de microvaloración con 96 pocillos.
|
Testigos
|
25.
|
Al realizar el ensayo, deben incluirse en cada experimento testigos y disolventes en paralelo (es decir, estrógeno de referencia, ligando débil y no ligando). Deben incluirse en una placa de cada tanda todos los puntos que forman las curvas de concentración completas del estrógeno de referencia y de los testigos (es decir, ligando débil y no ligando). Todas las demás placas deben contener: i) una concentración elevada (desplazamiento máximo) y una media (aproximadamente la CI50) tanto de E2 como del ligando débil, por triplicado; ii) el testigo de disolvente y el ligando inespecífico, cada uno por triplicado. En la referencia 2 (anexo K, protocolo del ensayo FW) se describen los procedimientos para la preparación de las soluciones amortiguadoras, testigo, de [3H]17β-estradiol, de hrERα y de producto problema.
|
Control del disolvente:
|
26.
|
El control del disolvente indica que este no interactúa con el sistema de ensayo y que también mide la unión total (total binding, TB). El etanol es el disolvente preferido. Si la concentración más alta del producto problema no es soluble en etanol, también se podrá utilizar DMSO. La concentración de etanol o DMSO, si se utiliza este, en los pocillos del ensayo final es del 1,5 % y no puede ser superior al 2 %.
|
Testigo de la solución amortiguadora:
|
27.
|
El testigo de la solución amortiguadora (buffer control, BC) no debe contener ni disolvente ni producto problema, pero sí todos los demás componentes del ensayo. Los resultados del testigo de la solución amortiguadora se comparan con el control del disolvente para verificar que el disolvente utilizado no afecta al sistema de ensayo.
|
Ligando fuerte (estrógeno de referencia)
|
28.
|
El 17β-estradiol (n.o CAS 50-28-2) es el ligando endógeno y se une con gran afinidad al ER, subtipo alfa. Debe prepararse una curva patrón con 17β-estradiol no marcado para cada uno de los ensayos de unión de competencia al hrERα, a fin de permitir una evaluación de la variabilidad cuando se realice el ensayo a lo largo del tiempo dentro del mismo laboratorio. Deben prepararse en etanol ocho soluciones de 17β-estradiol no marcado, con concentraciones en los pocillos del ensayo comprendidas entre 100 nM y 10 pM [-7 (log M) hasta -11 (log M)], separadas como sigue: -7 (log M), -8 (log M), -8,5 (log M), -9 (log M), -9,5 (log M), -10 (log M), -11 (logM). La concentración más elevada de 17β-estradiol no marcado (1 μM) también sirve de indicador de unión inespecífica. Esta concentración se distingue con el rótulo «NSB» (non-specific binding) en el cuadro 4, aunque también forma parte de la curva patrón.
|
Ligando débil
|
29.
|
Debe incluirse un ligando débil [noretinodrel (n.o CAS 68-23-5) o noretindrona (n.o CAS 68-22-4)] para demostrar la sensibilidad de cada experimento y permitir una evaluación de la variabilidad cuando se realice el ensayo a lo largo del tiempo. Deben prepararse en etanol ocho soluciones del ligando débil, con concentraciones en los pocillos del ensayo comprendidas entre 3 nM y 30 μM [desde -8,5 (log M) hasta -4,5 (log M)], separadas como sigue: [-4,5 (log M), -5 (log M), -5,5 (log M), -6 (log M), -9,5 (log M), -7,5 (log M), -8,5 (logM)].
|
No ligando
|
30.
|
Debe utilizarse como testigo negativo (no ligando) el octiltrietoxisilano (OTES, n.o CAS 2943-75-1). Aporta la garantía de que el ensayo, tal como se realiza, detecta si los productos problema no se unen al hrERα. Deben prepararse en etanol ocho soluciones del no ligando, con concentraciones en los pocillos del ensayo comprendidas entre 0,1 nM y 1 000 μM [desde -10 (log M) hasta -3 (log M)], con incrementos logarítmicos. Puede utilizarse también como no ligando testigo el ftalato de di-n-butilo (DBP). Se ha visto que su solubilidad máxima es de -4 (log M).
|
Concentración del hrERα
|
31.
|
Debe utilizarse la cantidad del receptor que da una unión específica del 20 ± 5 % de radioligando 1 nM (véanse los puntos 12-13 del apéndice 2). La solución de hrERα debe prepararse inmediatamente antes de su utilización.
|
[3H]17β-estradiol
|
32.
|
La concentración de [3H]17β-estradiol en los pocillos de ensayo debe ser de 1,0 nM.
|
Productos problema
|
33.
|
En primer lugar, es necesario realizar un ensayo de solubilidad para determinar el límite de solubilidad de cada producto problema y para identificar el intervalo adecuado de concentraciones que se ha de utilizar al aplicar el protocolo del ensayo. El límite de solubilidad de cada producto problema se determina inicialmente en el disolvente y se confirma después en las condiciones del ensayo. La concentración final utilizada en el ensayo no deberá ser superior a 1 mM. La determinación del intervalo de concentraciones consiste en someter a ensayo un testigo de disolvente, junto con ocho diluciones logarítmicas en serie, a partir de la concentración máxima aceptable (por ejemplo, 1 mM o menos, sobre la base del límite de solubilidad), y anotar la presencia de turbidez o precipitado (véase el punto 35). El producto problema debe ensayarse utilizando curvas con ocho concentraciones a intervalos logarítmicos, definidas en el ensayo previo de detección del intervalo de concentraciones. Las concentraciones en los experimentos segundo y tercero deben ajustarse según convenga para caracterizar mejor la curva concentración-respuesta.
|
|
34.
|
Las diluciones del producto problema deben prepararse en el disolvente adecuado (véase el punto 26 del apéndice 2). Si la concentración más elevada del producto problema no fuera soluble en etanol o DMSO, y la adición de más disolvente hiciera que la concentración de disolvente en el tubo final fuera mayor que el límite aceptable, la concentración más elevada podría reducirse a la concentración inmediatamente inferior. En este caso, podría añadirse una concentración adicional en el extremo inferior de la serie de concentraciones. Las demás concentraciones de la serie deben permanecer inalteradas.
|
|
35.
|
Las soluciones del producto problema deben ser objeto de un estrecho seguimiento cuando se añaden al pocillo del ensayo, ya que el producto problema puede provocar una precipitación cuando se añade al pocillo del ensayo. Los datos de todos los pocillos que contengan precipitado deben quedar excluidos del ajuste de la curva, y debe anotarse el motivo de la exclusión de los datos.
|
|
36.
|
Si hay información previa de otras fuentes que proporcionen el log CI50 de un producto problema, puede ser adecuado espaciar geométricamente las diluciones (es decir, 0,5 unidades logarítmicas alrededor del log CI50 previsto. El resultado final debe reflejar la suficiente amplitud de las concentraciones a ambos lados del log CI50, incluidos la “parte superior” y la “parte inferior”, de manera que la curva de la unión pueda caracterizarse adecuadamente.
|
Organización de las placas del ensayo
|
37.
|
Deben prepararse placas de microvaloración etiquetadas que tengan en cuenta las incubaciones séxtuples con códigos para el control del disolvente, la concentración más elevada del estrógeno de referencia, que también sirve de indicador de la unión inespecífica (NSB), y el testigo de la solución amortiguadora, y que tengan en cuenta las incubaciones por triplicado con códigos para cada una de las ocho concentraciones del testigo no ligando (octiltrietoxisilano), las siete concentraciones inferiores del estrógeno de referencia, las ocho concentraciones del ligando débil y las ocho concentraciones de cada producto problema (test chemical, TC). En el cuadro 4 a continuación figura un ejemplo de diagrama de la disposición de la placa para las curvas de concentración completas correspondientes al estrógeno de referencia y al testigo. Se utilizan placas de microvaloración adicionales para los productos problema y deben incluir testigos de las placas, es decir: 1) una concentración elevada (desplazamiento máximo) y media (aproximadamente la CI50) tanto del E2 como de un ligando débil, por triplicado; 2) el testigo de disolvente y el ligando inespecífico, cada uno por sextuplicado (cuadro 5). En el apéndice 2.3 se presenta un ejemplo de hoja de cálculo de la configuración de la placa de microvaloración para el ensayo de competencia con tres productos problema desconocidos. Las concentraciones indicadas en los cuadros 4 y 5 son las concentraciones finales del ensayo. La concentración máxima de E2 debe ser 1 × 10-7 M y la del ligando débil debe ser la concentración más elevada utilizada para este en la placa 1. La concentración CI50 debe ser determinada por el laboratorio a partir de su base de datos de testigos históricos. Se espera que este valor sea similar al observado en los estudios de validación (véase el cuadro 1).
|
Cuadro 4
Disposición de las placas de microvaloración para el ensayo de unión de competencia, curvas completas de concentración del estrógeno de referencia y de los testigos (placa 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (solo disolvente)
|
TB (solo disolvente)
|
NSB
|
NSB
|
|
B
|
E2 (1 × 10-7)
|
E2 (1 × 10-8)
|
E2 (1 × 10-8,5)
|
E2 (1 × 10-9)
|
|
C
|
E2 (1 × 10-9,5)
|
E2 (1 × 10-10)
|
E2 (1 × 10-11)
|
Blanco (*8)
|
|
D
|
NE (1 × 10-4,5)
|
NE (1 × 10-5)
|
NE (1 × 10-5,5)
|
NE (1 × 10-6)
|
|
E
|
NE (1 × 10-6,5)
|
NE (1 × 10-7)
|
NE (1 × 10-7,5)
|
NE (1 × 10-8,5)
|
|
F
|
OTES (1 × 10-3)
|
OTES (1 × 10-4)
|
OTES (1 × 10-5)
|
OTES (1 × 10-6)
|
|
G
|
OTES (1 × 10-7)
|
OTES (1 × 10-8)
|
OTES (1 × 10-9)
|
OTES (1 × 10-10)
|
|
H
|
Blanco (de marcado) (*9)
|
Blanco (de marcado) (*9)
|
Testigo de la solución amortiguadora
|
Testigo de la solución amortiguadora
|
|
En este ejemplo, el ligando débil es el noretinodrel (NE).
|
Cuadro 5
Disposición de las placas de microvaloración para el ensayo de unión de competencia, curvas completas de concentración de los productos problema y de los testigos de las placas.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (solo disolvente)
|
TB (solo disolvente)
|
NSB
|
NSB
|
|
B
|
TC1 (1 × 10-3)
|
TC1 (1 × 10-4)
|
TC1 (1 × 10-5)
|
TC1 (1 × 10-6)
|
|
C
|
TC1 (1 × 10-7)
|
TC1 (1 × 10-8)
|
TC1 (1 × 10-9)
|
TC1 (1 × 10-10)
|
|
D
|
TC2 (1 × 10-3)
|
TC2 (1 × 10-4)
|
TC2 (1 × 10-5)
|
TC2 (1 × 10-6)
|
|
E
|
TC2 (1 × 10-7)
|
TC2 (1 × 10-8)
|
TC2 (1 × 10-9)
|
TC2 (1 × 10-10)
|
|
F
|
TC3 (1 × 10-3)
|
TC3 (1 × 10-4)
|
TC3 (1 × 10-5)
|
TC3 (1 × 10-6)
|
|
G
|
TC3 (1 × 10-7)
|
TC3 (1 × 10-8)
|
TC3 (1 × 10-9)
|
TC3 (1 × 10-10)
|
|
H
|
NE (CI50)
|
NE (1 × 10-4,5)
|
E2 (CI50)
|
E2 (1 × 10-7)
|
|
En este ejemplo, el ligando débil es el noretinodrel (NE).
|
Terminación del ensayo de unión de competencia
|
38.
|
Como se muestra en el cuadro 6, deben añadirse a los pocillos 80 μl del testigo de disolvente, del testigo de la solución amortiguadora, del estrógeno de referencia, del ligando débil, del no ligando y de los productos problema preparados en la solución amortiguadora de ensayo. A continuación, deben añadirse a cada pocillo 40 μl de una solución 4 nM de [3H]17β-estradiol. Tras una rotación suave durante entre 10 y 15 minutos, a una temperatura de entre 2o y 8oC, deben añadirse 40 μl de solución de hrERα. Las placas de microvaloración del ensayo deben incubarse a una temperatura de entre 2o y 8oC durante entre 16 y 20 horas y colocarse en un rotador durante el período de incubación.
|
Cuadro 6
Volumen de los componentes del ensayo para las placas de un ensayo de unión de competencia, placas de microvaloración.
|
Volumen (μl)
|
Componente
|
|
80
|
17β-Estradiol no marcado, noretinodrel, OTES, productos problema, disolvente o solución amortiguadora
|
|
40
|
Solución 4 nM de [3H]17β-estradiol
|
|
40
|
Solución de hrERα, concentración según se haya determinado
|
|
160
|
Volumen total en cada pocillo del ensayo
|
|
39.
|
A continuación debe llevarse a cabo la cuantificación del [3H]17β-estradiol unido al hrERα, tras la separación del [3H]17β-estradiol unido al hrERα respecto al [3H]17β-estradiol libre mediante la adición de 80 μl de suspensión fría de DCC a cada pocillo, como se describe en los puntos 20 a 23, para el ensayo de unión de saturación.
|
|
40.
|
Los pocillos H1-6 [identificados como blanco (de marcado) en el cuadro 4] presentan las dpm del estradiol marcado con [3H] en 40 μl. La alícuota de 40 μl debe añadirse directamente al líquido de centelleo en los pocillos H1 - H6.
|
Criterios de aceptabilidad
Ensayo de unión de saturación
|
41.
|
La curva de unión específica debe alcanzar una meseta a medida que se utilizan concentraciones crecientes de [3H]17β-estradiol, lo que indica la saturación del hrERα con el ligando.
|
|
42.
|
La unión específica a la concentración 1 nM de [3H]17β-estradiol debe situarse en el intervalo aceptable del 15 % - 25 % de la radiactividad total medida media que se ha añadido en todas las tandas. Pueden aceptarse ligeras salidas fuera de este intervalo, pero si las tandas están sistemáticamente fuera del mismo o una tanda concreta se sitúa significativamente fuera de este intervalo, debe ajustarse la concentración de proteínas y repetirse el ensayo de saturación.
|
|
43.
|
Los datos deben corresponder a una gráfica lineal de Scatchard.
|
|
44.
|
La unión inespecífica no debe ser excesiva. El valor de la unión inespecífica debe ser típicamente < 35 % de la unión total. Sin embargo, la proporción puede superar ocasionalmente este límite cuando se miden valores muy bajos de dpm con la concentración más baja de 17β-estradiol radiomarcado.
|
Ensayo de unión de competencia
|
45.
|
El aumento de las concentraciones de 17β-estradiol no marcado debe desplazar el [3H]17β-estradiol del receptor de manera coherente con una unión de competencia a un solo punto.
|
|
46.
|
El valor de CI50 para el estrógeno de referencia (es decir, el 17β-estradiol) debe ser aproximadamente igual a la concentración molar del [3H]17β-estradiol más la Kd determinada a partir del ensayo de unión de saturación.
|
|
47.
|
La unión específica total debe estar sistemáticamente dentro del intervalo aceptable del 20 ± 5 % cuando la concentración medida media de la radiactividad total añadida a cada pocillo es de 1 nM en todas las tandas. Pueden aceptarse ligeras salidas fuera de este intervalo, pero si las tandas están sistemáticamente fuera del mismo o una tanda concreta se sitúa significativamente fuera de este intervalo, debe ajustarse la concentración de proteínas.
|
|
48.
|
El disolvente no debe alterar la sensibilidad ni la reproducibilidad del ensayo. Los resultados del control del disolvente (pocillos TB) se comparan con el testigo de la solución amortiguadora para verificar que el disolvente utilizado no afecta al sistema de ensayo. Los resultados del testigo de la TB y de la solución amortiguadora deben ser comparables si el disolvente no produce ningún efecto sobre el ensayo.
|
|
49.
|
El no ligando no debe desplazar más del 25 % del [3H]17β-estradiol del hrERα cuando se somete a ensayo hasta la concentración de 10-3 M (OTES) o 10-4 M (DBP).
|
|
50.
|
Se han establecido criterios de comportamiento para el estrógeno de referencia y dos ligandos débiles (por ejemplo, noretinodrel, noretindrona) utilizando los datos del estudio de validación del ensayo de FW de unión al hrER (anexo N de la referencia 2). Se aportan los intervalos de confianza del 95 % para la media (n) ± DT de todas las tandas de los testigos en todos los laboratorios participantes en el estudio de validación. Se calcularon intervalos de confianza del 95 % para los parámetros de ajuste de las curvas (es decir, parte superior, parte inferior, pendiente de Hill, log CI50) en relación con el estrógeno de referencia y los ligandos débiles, y para el log10 RBA de los ligandos débiles en relación con el estrógeno de referencia, y se proporcionan como criterios de comportamiento respecto a los testigos positivos. En el cuadro 1 se ofrecen los intervalos previstos para los parámetros de ajuste de las curvas, que pueden utilizarse como criterios de comportamiento. En la práctica, el intervalo de la CI50 puede variar ligeramente según la Kd del preparado del receptor y la concentración del ligando.
|
|
51.
|
No se han establecido criterios de comportamiento sobre los parámetros de ajuste de las curvas en relación con los productos problema debido a la amplia gama de productos problema que puede haber y a la variación de las posibles afinidades y resultados (por ejemplo, ajuste de la curva completa o de la curva parcial, o sin ajuste alguno). Sin embargo, a la hora de revisar los resultados de cada tanda correspondientes a un producto problema, debe aplicarse un juicio profesional. Debe utilizarse un intervalo suficiente de concentraciones del producto problema para definir claramente la parte superior (p. ej., 90 - 100 % de unión) de la curva de competencia. La variabilidad entre las réplicas de cada concentración del producto problema, así como entre las tres tandas en momentos diferentes, debe ser razonable y científicamente justificable. Los testigos de cada tanda de un producto problema deben aproximarse a las medidas de comportamiento notificadas para este ensayo de FW y deben ser coherentes con los datos históricos de los testigos de cada laboratorio respectivo.
|
ANÁLISIS DE LOS DATOS
Ensayo de unión de saturación
|
52.
|
Se mide tanto la unión total como la inespecífica. A partir de estos valores, se calcula la unión específica con concentraciones en aumento de [3H]17β-estradiol en condiciones de equilibrio, restando la inespecífica de la total. La gráfica de la unión específica frente a la concentración de [3H]17β-estradiol debe alcanzar una meseta para la unión específica máxima, lo cual indica la saturación del hrERα con el [3H]17β-estradiol. Por otra parte, el análisis de los datos debe documentar la unión del [3H]17β-estradiol a un solo punto de unión de elevada afinidad. En la curva de unión de saturación deben ponerse de manifiesto la unión inespecífica, la total y la específica. El posterior análisis de estos datos debe utilizar una regresión no lineal (por ejemplo, BioSoft; McPherson, 1985; Motulsky, 1995) con una representación final de los datos como gráfica de Scatchard.
|
|
53.
|
El análisis de los datos debe determinar los parámetros Bmax y Kd a partir solo de los datos de unión total, aceptando que la unión inespecífica es lineal, salvo que se justifique utilizar otro método. Además, a la hora de determinar el mejor ajuste debe utilizarse una regresión sólida, salvo que se justifique otra opción. Debe indicarse el método elegido para la regresión sólida. Al determinar los parámetros Bmax y Kd a partir de los datos de la unión de saturación, siempre debe aplicarse una corrección para tener en cuenta el agotamiento de los ligandos (p. ej., utilizando el método de Swillens, 1995).
|
Ensayo de unión de competencia
|
54.
|
La curva de la unión de competencia se representa gráficamente como la unión específica del [3H]17β-estradiol frente a la concentración (unidades log10) del competidor. La concentración del producto problema que inhibe el 50 % de la unión máxima específica del [3H]17β-estradiol es el valor de la CI50.
|
|
55.
|
Deben estimarse los valores de log CI50 de los testigos positivos (p. ej., el estrógeno de referencia y el ligando débil), utilizando un programa informático adecuado de ajuste de curvas no lineales para ajustarse a una ecuación de Hill de cuatro parámetros (p. ej., BioSoft; McPherson, 1985; Motulsky, 1995). La parte superior, la parte inferior, la pendiente y log CI50 deben dejarse, por lo general, sin restricciones cuando se ajustan estas curvas. A la hora de determinar el mejor ajuste debe utilizarse una regresión sólida, salvo que se justifique otra opción. No debe utilizarse ninguna corrección por el agotamiento de los ligandos. Tras el análisis inicial, debe revisarse cada curva de los ligandos para garantizar que se ajusta adecuadamente al modelo. La afinidad relativa de unión (RBA) del ligando débil debe calcularse como porcentaje del log CI50 del ligando débil respecto al log CI50 del 17β-estradiol. Los resultados de los testigos positivos y del testigo de no ligando deben evaluarse utilizando las medidas del comportamiento del ensayo de los puntos 45 - 50 del presente apéndice 2.
|
|
56.
|
Los datos de todos los productos problema deben analizarse utilizando un enfoque gradual para garantizar que los datos se analizan de forma adecuada y que cada curva de unión de competencia se clasifica correctamente. Se recomienda que cada tanda con el producto problema se someta inicialmente a un análisis de datos normalizado idéntico al utilizado con el estrógeno de referencia y los testigos de ligandos débiles (véase el punto 55 anterior). Tras este análisis inicial, debe llevarse a cabo una revisión técnica de los parámetros de ajuste de las curvas, así como un examen visual sobre el grado de calidad del ajuste de los datos con la curva de la unión de competencia generada en cada tanda. Durante esta revisión técnica, la observación de una disminución dependiente de la concentración en el porcentaje de [3H]17β-estradiol unido específicamente, la de una baja variabilidad entre las réplicas técnicas de cada concentración de producto, y la de la coherencia de los parámetros de ajuste entre las tres tandas son una buena indicación de que el ensayo y los análisis de datos se han llevado a cabo adecuadamente.
|
Interpretación de los datos
|
57.
|
Siempre que se cumplan todos los criterios de aceptabilidad, se considera que un producto problema es un ligando del hrERα si puede ajustarse una curva de unión y el punto más bajo de la curva de respuesta dentro del intervalo de los datos es inferior al 50 % (figura 1).
|
|
58.
|
Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es no ligando del hrERα si:
|
—
|
puede ajustarse una curva de unión y el punto más bajo de la curva de respuesta ajustada dentro del intervalo de datos es superior al 75 %, o
|
|
—
|
no puede ajustarse una curva de unión y la unión porcentual media sin suavizar más baja entre los grupos de concentración de los datos es superior al 75 %.
|
|
|
59.
|
Los productos problema se consideran dudosos si no se cumple ninguna de las condiciones anteriores (por ejemplo, el punto más bajo de la curva de respuesta ajustada se sitúa entre el 76 y el 51 %).
|
Cuadro 7
Criterios de asignación de la clasificación a un producto problema sobre la base de una curva de unión de competencia.
|
Clasificación
|
Criterios
|
|
Ligandoa
|
Puede ajustarse una curva de unión.
El punto más bajo de la curva de respuesta dentro del intervalo de datos es inferior al 50 %.
|
|
No ligandob
|
Si puede ajustarse una curva de unión,
el punto más bajo de la curva de respuesta ajustada dentro del intervalo de datos es superior al 75 %.
Si no puede ajustarse una curva de unión,
la unión porcentual media sin suavizar más baja entre los grupos de concentración de los datos es superior al 75 %.
|
|
Dudosoc
|
Cualquier producto sometido al ensayo que no sea ni ligando ni no ligando
(p. ej., el punto más bajo de la curva de respuesta ajustada se sitúa entre el 76 y el 51 %).
|
Figura 1
Ejemplos de clasificación de productos problema utilizando una curva de unión de competencia.
|
60.
|
Se combinan las diversas tandas realizadas con un producto problema dentro de un laboratorio, Puede ajustarse una curva de unión.asignando un valor numérico a cada tanda y promediando entre ellas, como se muestra en el cuadro 8. Los resultados de las tandas combinadas dentro de cada laboratorio se comparan con la clasificación prevista de cada producto problema.
|
Cuadro 8
Método de clasificación del producto problema mediante varias tandas dentro de un laboratorio
|
Asignar valor a cada tanda:
|
|
|
Clasificación
|
Valor numérico
|
|
Ligando
|
2
|
|
Dudoso
|
1
|
|
No ligando
|
0
|
|
Clasificar según la media de los valores numéricos de las tandas:
|
|
|
Clasificación
|
Valor numérico
|
|
Ligando
|
Media ≥ 1,5
|
|
Dudoso
|
0,5 ≤ Media < 1,5
|
|
No ligando
|
Media < 0,5
|
INFORME DEL ENSAYO
|
61.
|
Véase el punto 24 de la sección “COMPONENTES DEL ENSAYO DE UNIÓN AL hrER” de este método de ensayo.
|
Apéndice 2.1
RELACIÓN DE TÉRMINOS
[3H]E2
: 17β-Estradiol radiomarcado con tritio.
DCC
: Carbón recubierto de dextrano.
E2
: 17β-Estradiol no marcado (inerte).
Solución amortiguadora del ensayo
: Tris 10 mM, 10 mg de seroalbúmina bovina / ml, DTT 2 mM, glicerol al 10 %, leupeptina 0,2 mM, pH 7,5.
hrERα
: Receptor estrogénico recombinante humano alfa.
Réplica
: Cada uno de los diversos pocillos que tienen el mismo contenido a la misma concentración y se someten a ensayo simultáneamente dentro de una misma tanda. En este protocolo, cada concentración del producto problema se somete al ensayo por triplicado; es decir, hay tres réplicas que se someten simultáneamente al ensayo con cada concentración del producto problema.
Tanda
: Conjunto completo de pocillos de ensayo en placas de microvaloración sometidos a ensayo a la vez, que aporta toda la información necesaria para caracterizar la unión de un producto problema al hrERα (a saber, [3H]17β-estradiol total añadido al pocillo de ensayo, unión máxima del [3H]17β-estradiol al hrERα, unión inespecífica, y unión total a diversas concentraciones del producto problema). Una tanda podría consistir simplemente en un solo pocillo (es decir, una réplica) por concentración, pero, puesto que este protocolo requiere que los ensayos se realicen por triplicado, una tanda estará compuesta por tres pocillos de ensayo por cada concentración. Además, este protocolo exige la realización de tres tandas independientes (es decir, no simultáneas) por producto.
Apéndice 2.2
ENSAYO NORMAL DE SATURACIÓN CON [3H]17Β-ESTRADIOL CON TRES POCILLOS REPLICADOS
|
Ensayo normal de saturación con [3H]17β-estradiol con tres pocillos replicados
|
|
Posición
|
Réplica
|
Código de tipo de pocillo
|
Concentración inicial de E2 marcado (nM)
|
Volumen de E2 marcado (μl)
|
Concentración final de E2 marcado (nM)
|
Concentración inicial de E2 no marcado (μM)
|
Volumen de E2 no marcado (μl)
|
Concentración final de E2 no marcado (μM)
|
Volumen de solución amortiguadora (μl)
|
Volumen de receptor (μl)
|
Volumen total en los pocillos
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Marcado
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Marcado
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Marcado
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Marcado
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Marcado
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Marcado
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Marcado
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Marcado
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Marcado
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Marcado
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Marcado
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Marcado
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Marcado
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Marcado
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Marcado
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Marcado
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Marcado
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Marcado
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Marcado
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Marcado
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Marcado
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Marcado
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Marcado
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Marcado
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Obsérvese que los pocillos “marcados” están vacíos durante la incubación. Los 40 μl se añaden solo para el recuento de centelleo.
|
Apéndice 2.3
DISTRIBUCIÓN DE LOS POCILLOS PARA EL ENSAYO DE UNIÓN DE COMPETENCIA
|
Placa
|
Posición
|
Réplica
|
Tipo de pocillo
|
Código del pocillo
|
Código de la concentración
|
Concentración inicial del competidor (M)
|
Solución madre de hrER (μl)
|
Volumen de solución amortiguadora (μl)
|
Volumen del marcador (E2 marcado) (μl)
|
Volumen tomado de la placa de dilución (μL)
|
Volumen final (μl)
|
Concentración final del competidor (M)
|
|
S
|
A1
|
1
|
Unión total
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
Unión total
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
Unión total
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
Unión total
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
Unión total
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
Unión total
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A8
|
2
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A9
|
3
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A10
|
1
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A11
|
2
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A12
|
3
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
B1
|
1
|
E2 no marcado
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B2
|
2
|
E2 no marcado
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B3
|
3
|
E2 no marcado
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B4
|
1
|
E2 no marcado
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B5
|
2
|
E2 no marcado
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B6
|
3
|
E2 no marcado
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B7
|
1
|
E2 no marcado
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B8
|
2
|
E2 no marcado
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B9
|
3
|
E2 no marcado
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B10
|
1
|
E2 no marcado
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B11
|
2
|
E2 no marcado
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B12
|
3
|
E2 no marcado
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
C1
|
1
|
E2 no marcado
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
E2 no marcado
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
E2 no marcado
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C4
|
1
|
E2 no marcado
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
E2 no marcado
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
E2 no marcado
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
E2 no marcado
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
E2 no marcado
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
E2 no marcado
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
Blanco
|
Blanco
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
Blanco
|
Blanco
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
Blanco
|
Blanco
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D2
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D3
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D4
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D5
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D6
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D7
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D8
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D9
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D10
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D11
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D12
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
E1
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E2
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E3
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E4
|
1
|
Noretinodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E5
|
2
|
Noretinodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E6
|
3
|
Noretinodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E7
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E8
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E9
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E10
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E11
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E12
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
Marcado
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
Marcado
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
Marcado
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
Marcado
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
Marcado
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
Marcado
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
Testi sol. amortig.
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
Testi sol. amortig
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
Testi sol. amortig
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
Testi sol. amortig
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
Testi sol. amortig
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
Testi sol. amortig
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Obsérvese que los pocillos “marcados” están vacíos durante la incubación. Los 40 μl se añaden solo para el recuento de centelleo.
|
Distribución de los pocillos para el ensayo de unión de competencia
|
|
Placa
|
Posición
|
Réplica
|
Tipo de pocillo
|
Código del pocillo
|
Código de la concentración
|
Concentración inicial del competidor (M)
|
Solución madre de hrER (μl)
|
Volumen de solución amortiguadora (μl)
|
Volumen del marcador (E2 marcado) (μl)
|
Volumen tomado de la placa de dilución (μl)
|
Volumen final (μl)
|
Concentración final del competidor (M)
|
|
P1
|
A1
|
1
|
Unión total
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
Unión total
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
Unión total
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
Unión total
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
Unión total
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
Unión total
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A8
|
2
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A9
|
3
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A10
|
1
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A11
|
2
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A12
|
3
|
E2 no marcado (elevada)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B1
|
1
|
Producto problema 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B2
|
2
|
Producto problema 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B3
|
3
|
Producto problema 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B4
|
1
|
Producto problema 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B5
|
2
|
Producto problema 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B6
|
3
|
Producto problema 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B7
|
1
|
Producto problema 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B8
|
2
|
Producto problema 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B9
|
3
|
Producto problema 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B10
|
1
|
Producto problema 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B11
|
2
|
Producto problema 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B12
|
3
|
Producto problema 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
C1
|
1
|
Producto problema 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C2
|
2
|
Producto problema 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C3
|
3
|
Producto problema 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C4
|
1
|
Producto problema 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C5
|
2
|
Producto problema 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C6
|
3
|
Producto problema 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C7
|
1
|
Producto problema 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C8
|
2
|
Producto problema 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C9
|
3
|
Producto problema 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C10
|
1
|
Producto problema 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
Producto problema 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
Producto problema 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
Producto problema 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D2
|
2
|
Producto problema 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D3
|
3
|
Producto problema 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D4
|
1
|
Producto problema 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D5
|
2
|
Producto problema 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D6
|
3
|
Producto problema 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D7
|
1
|
Producto problema 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D8
|
2
|
Producto problema 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D9
|
3
|
Producto problema 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D10
|
1
|
Producto problema 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D11
|
2
|
Producto problema 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D12
|
3
|
Producto problema 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
E1
|
1
|
Producto problema 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E2
|
2
|
Producto problema 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E3
|
3
|
Producto problema 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E4
|
1
|
Producto problema 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E5
|
2
|
Producto problema 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E6
|
3
|
Producto problema 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E7
|
1
|
Producto problema 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E8
|
2
|
Producto problema 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E9
|
3
|
Producto problema 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E10
|
1
|
Producto problema 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
Producto problema 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
Producto problema 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
Producto problema 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F2
|
2
|
Producto problema 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F3
|
3
|
Producto problema 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F4
|
1
|
Producto problema 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F5
|
2
|
Producto problema 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F6
|
3
|
Producto problema 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F7
|
1
|
Producto problema 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F8
|
2
|
Producto problema 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F9
|
3
|
Producto problema 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F10
|
1
|
Producto problema 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F11
|
2
|
Producto problema 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F12
|
3
|
Producto problema 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
G1
|
1
|
Producto problema 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G2
|
2
|
Producto problema 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G3
|
3
|
Producto problema 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G4
|
1
|
Producto problema 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G5
|
2
|
Producto problema 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G6
|
3
|
Producto problema 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G7
|
1
|
Producto problema 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G8
|
2
|
Producto problema 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G9
|
3
|
Producto problema 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G10
|
1
|
Producto problema 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
Producto problema 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
Producto problema 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
Noretinodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
Noretinodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
Noretinodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
E2 no marcado S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
E2 no marcado S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
E2 no marcado S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
E2 no marcado S
|
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
E2 no marcado S
|
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
E2 no marcado S
|
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
Apéndice 3
ENSAYO IN VITRO DEL INSTITUTO DE EVALUACIÓN E INVESTIGACIÓN DE PRODUCTOS QUÍMICOS (CERI) DE UNIÓN AL RECEPTOR ESTROGÉNICO CON UTILIZACIÓN DE UNA PROTEÍNA CON UN DOMINIO DE UNIÓN AL LIGANDO DEL ERΑ RECOMBINANTE HUMANO
CONSIDERACIONES INICIALES Y LIMITACIONES (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
1.
|
Este ensayo de unión al receptor estrogénico (ERα) de saturación y de competencia in vitro utiliza un dominio de unión al ligando (ligand binding domain, LBD) del receptor ERα humano (hrERα). Esta construcción proteínica fue producida por el Instituto de Evaluación e Investigación de Productos Químicos de Japón (Chemicals Evaluation Research Institute, CERI), existe como proteína de fusión con glutatión-S-transferasa (GST) y se expresa en E. coli. El protocolo del CERI fue sometido a un estudio de validación internacional multilaboratorios (2), que demostró su pertinencia y fiabilidad para la finalidad prevista del ensayo.
|
|
2.
|
Este ensayo es un procedimiento de cribado para identificar sustancias que pueden unirse al hrERα. Se utiliza para determinar la capacidad de un producto problema de competir con el 17β-estradiol para unirse al LBD del hrERα. Los resultados cuantitativos del ensayo pueden incluir la CI50 (medida de la concentración del producto problema necesaria para desplazar la mitad del [3H]17β-estradiol del hrERα) y las afinidades relativas de unión de los productos problema respecto al hrERα en comparación con el 17β-estradiol. A efectos de cribado de los productos problema, entre los resultados cualitativos aceptables de los ensayos pueden incluirse la clasificación de los productos problema como ligandos o no ligandos del hrERα, o bien si el resultado es dudoso sobre la base de los criterios descritos para las curvas de unión.
|
|
3.
|
El ensayo utiliza un ligando radiactivo, lo que impone al laboratorio la obligación de disponer de una licencia de materiales radiactivos. Todos los procedimientos con radioisótopos y productos peligrosos deben ajustarse a las normas y procedimientos descritos en la legislación nacional.
|
|
4.
|
Las secciones “INTRODUCCIÓN GENERAL” y “COMPONENTES DEL ENSAYO DE UNIÓN AL hrER” deben leerse antes de utilizar este ensayo con fines normativos. Las definiciones y abreviaturas utilizadas en las presentes directrices de ensayo se recogen en el apéndice 1.
|
PRINCIPIOS DEL ENSAYO (VÉASE TAMBIÉN LA INTRODUCCIÓN GENERAL)
|
5.
|
El ensayo de unión al hrERα mide la capacidad de un ligando radiomarcado ([3H]17β-estradiol) para unirse al receptor estrogénico en presencia de concentraciones cada vez mayores de un producto problema (es decir, un competidor). Los productos problema que presentan una elevada afinidad por el receptor estrogénico compiten con el ligando radiomarcado a una concentración menor en comparación con los productos que presentan menor afinidad por el receptor.
|
|
6.
|
Este ensayo consta de dos componentes principales: un experimento de unión de saturación para caracterizar los parámetros de la interacción receptor-ligando, seguido de un experimento de unión de competencia que caracteriza la competencia entre un producto problema y un ligando radiomarcado para unirse al ER.
|
|
7.
|
El objetivo del experimento de unión de saturación es caracterizar un determinado lote de receptores en cuanto a su afinidad de unión y número para preparar el experimento de unión de competencia. El experimento de unión de saturación mide, en condiciones de equilibrio, la afinidad de una concentración fija del receptor estrogénico por su ligando natural (representada por la constante de disociación, Kd) y la concentración de puntos activos de unión de los receptores (Bmax).
|
|
8.
|
El experimento de unión de competencia mide la afinidad de una sustancia para competir con el [3H]17β-estradiol en cuanto a la unión al ER. La afinidad se cuantifica por la concentración del producto problema que, en equilibrio, inhibe el 50 % de la unión específica del [3H]17β-estradiol (denominada "concentración inhibitoria del 50 %" o CI50). También puede evaluarse utilizando la afinidad relativa de unión (RBA, en relación con la CI50 del estradiol medida aparte en la misma tanda). El experimento de unión de competencia mide la unión del [3H]17β-estradiol a una concentración fija en presencia de una amplia gama de concentraciones (ocho órdenes de magnitud) del producto problema. A continuación, se ajustan los datos, en la medida de lo posible, a una forma de la ecuación de Hill (Hill, 1910) que describe el desplazamiento del radioligando por un ligando competitivo de un solo punto de unión. La magnitud del desplazamiento del estradiol radiomarcado en equilibrio se utiliza para caracterizar el producto problema como ligando, no ligando, o generador de una respuesta dudosa.
|
PROCEDIMIENTO
Demostración del comportamiento aceptable de la proteína del hrERα
|
9.
|
Con anterioridad a la realización sistemática de los ensayos de unión de saturación y de competencia, se debe demostrar que cada nuevo lote de hrERα presenta un comportamiento correcto en el laboratorio en el que se va a utilizar. Para demostrar el comportamiento debe seguirse un proceso de dos fases. Se trata de las siguientes:
|
—
|
Realizar un ensayo de unión de [3H]17β-estradiol de saturación para demostrar la especificidad y saturación del hrERα. El análisis no lineal de regresión de estos datos (por ejemplo, BioSoft; McPherson, 1985; Motulsky, 1995) y la consiguiente gráfica de Scatchard deben documentar la afinidad de unión al hrERα del [3H]17β-estradiol (Kd) y el número de receptores (Bmax) respecto a cada lote del hrERα.
|
|
—
|
Llevar a cabo un ensayo de unión de competencia utilizando las sustancias testigo [estrógeno de referencia (17β-estradiol)], un ligando débil (por ejemplo, noretinodrel o noretindrona), y un no ligando (octiltrietoxisilano, OTES). Cada laboratorio debe establecer una base de datos históricos para documentar la coherencia de la CI50 y de los valores pertinentes sobre el estrógeno de referencia y el ligando débil entre experimentos y diferentes lotes de hrERα. Además, los parámetros de las curvas de unión de competencia de las sustancias testigo deben estar dentro de los límites del intervalo de confianza del 95 % (véase el cuadro 1) que se han establecido utilizando datos de los laboratorios que participaron en el estudio de validación de este ensayo (2).
|
Cuadro 1
Criterios de comportamiento establecidos para el estrógeno de referencia y el ligando débil, ensayo del CERI de unión al hrER.
|
Sustancia
|
Parámetro
|
Media (10)
|
Desviación típica (n)
|
Intervalos de confianza del 95 % (11)
|
|
Límite inferior
|
Límite superior
|
|
17β-Estradiol
|
Parte superior
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Parte inferior
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Pendiente de Hill
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
Log CI50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretinodrel
|
Parte superior
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Parte inferior
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Pendiente de Hill
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
Log CI50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Noretindrona (12)
|
Parte superior
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Parte inferior
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Pendiente de Hill
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
Log CI50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Demostración del comportamiento del laboratorio
|
10.
|
Véanse los puntos 17 y 18 y el cuadro 2 en la sección “COMPONENTES DEL ENSAYO DE UNIÓN AL hrER” de este método de ensayo. Cada ensayo (unión de saturación y de competencia) debe consistir en tres tandas independientes (es decir, con diluciones nuevas del receptor, de los productos y de los reactivos) en días diferentes, y cada tanda debe contener tres réplicas.
|
Determinación de la concentración del receptor (hrERα)
|
11.
|
La concentración del receptor activo varía ligeramente en función del lote y de las condiciones de conservación. Por este motivo, debe determinarse la concentración del receptor activo tal como se recibe del proveedor. Esto dará la concentración adecuada del receptor activo en el momento de la tanda.
|
|
12.
|
En condiciones correspondientes a la unión de competencia (es decir, [3H]-estradiol 0,5 nM), deben incubarse concentraciones nominales del receptor de 0,1, 0,2, 0,4 y 0,6 nM en ausencia (unión total) y presencia (unión inespecífica) de estradiol no marcado 1 μM. La unión específica, calculada como la diferencia de unión total e inespecífica, se representa frente a la concentración nominal del receptor. La concentración del receptor que da valores de unión específica correspondientes al 40 % de la sustancia radiomarcada añadida está relacionada con la correspondiente concentración del receptor, y esta concentración del receptor debe utilizarse para los experimentos de unión de saturación y de competencia. Con frecuencia, una concentración final del rhER de 0,2 nM cumplirá esta condición.
|
|
13.
|
Si el criterio del 40 % no puede cumplirse de forma reiterada, deberá comprobarse si no se encuentran posibles errores en la configuración experimental. Si no se cumple el criterio del 40 %, puede deberse a que en el lote del recombinante hay muy poco de receptor activo, y en tal caso debe considerarse el uso de otro lote del receptor.
|
Ensayo de saturación
|
14.
|
Deben evaluarse por triplicado ocho concentraciones cada vez mayores de [3H]17β-estradiol, en las tres condiciones siguientes (véase el cuadro 2):
|
a.
|
En ausencia de 17β-estradiol no marcado y presencia de ER. Se trata de la determinación de la unión total por medida de la radiactividad en los pocillos que solo tienen [3H]17β-estradiol.
|
|
b.
|
En presencia de una concentración de 17-β-estradiol no marcado que supera 2000 veces la concentración de 17β-estradiol marcado y en presencia del ER. El propósito de esta condición es saturar los puntos activos de unión con 17β-estradiol no marcado y, midiendo la radiactividad en los pocillos, determinar la unión inespecífica. Se considera que el eventual estradiol marcado restante que pueda unirse al receptor se une a un punto de unión inespecífica, ya que el estradiol no marcado debe estar a una concentración tan elevada que está unido a todos los puntos específicos disponibles del receptor.
|
|
c.
|
En ausencia de 17β-estradiol no marcado y ausencia de ER (determinación de la radiactividad total).
|
Preparación de las soluciones de [3H]17β-estradiol, de 17β-estradiol no marcado y de hrERα
|
15.
|
Debe prepararse una solución 40 nM de [3H]-17β-estradiol a partir de una solución madre 1 μM de [3H]17β-estradiol en DMSO, mediante la adición de DMSO (para preparar una solución 200 nM) y de solución amortiguadora de ensayo a temperatura ambiente (para preparar la solución 40 nM). Utilizando esta solución 40 nM, se prepara la serie de diluciones de [3H]17β-estradiol, con concentraciones que van desde 0,313 nM hasta 40 nM con solución amortiguadora de ensayo a temperatura ambiente (representada en la columna 12 del cuadro 2). Las concentraciones finales del ensayo, que van de 0,0313 a 4,0 nM, se obtendrán añadiendo 10 μl de estas soluciones a los pocillos de ensayo respectivos de una placa de microvaloración de 96 pocillos (véanse los cuadros 2 y 3). La preparación de la solución amortiguadora de ensayo, el cálculo de la solución madre original de [3H]17β-estradiol sobre la base de su actividad específica, la preparación de las diluciones y la determinación de las concentraciones se describen con detalle en el protocolo del CERI (2).
|
|
16.
|
Las diluciones de las soluciones de 17β-estradiol no marcado deben prepararse a partir de una solución madre 1 nM de 17β-estradiol añadiendo solución amortiguadora de ensayo para conseguir ocho concentraciones cada vez mayores que inicialmente van de 0,625 μM a 80 μM. Las concentraciones finales del ensayo, que van de 0,0625 a 8 μM, se obtendrán añadiendo 10 μl de estas soluciones a los pocillos de ensayo respectivos destinados a la medición de la unión inespecífica en una placa de microvaloración de 96 pocillos (véanse los cuadros 2 y 3). La preparación de diluciones de 17β-estradiol no marcado se describe con detalle en el protocolo del CERI (2).
|
|
17.
|
Debe utilizarse la concentración del receptor que da una unión específica del 40 ± 10 % (véanse los puntos 12-13). La solución de hrERα debe prepararse con solución amortiguadora enfriada con hielo, justo antes de su utilización, es decir, después de que se hayan preparado todos los pocillos relacionados con la unión total, la unión inespecífica y el ligando marcado solo.
|
|
18.
|
Las placas de microvaloración de 96 pocillos se preparan como se ilustra en el cuadro 2, con 3 réplicas por concentración de [3H]17β-estradiol. En el cuadro 3 se indica la asignación de los volúmenes de [3H]17β-estradiol, de 17β-estradiol no marcado, de solución amortiguadora y de receptor.
Cuadro 2
Disposición de las placas de microvaloración para el ensayo de unión de saturación
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
Para la medición de la TB
|
Para la medición de la NSB
|
Para la determinación del ligando marcado solo
|
|
Diluciones de E2 no marcado en las columnas 4-6 de la placa
|
Diluciones de [3H]E2 en las columnas 1-9 de la placa
|
|
A
|
[3H]E2 0,0313 nM+ ER
|
[3H]E2 0,0313 nM+ E2 0,0625 μM+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
[3H]E2 0,0625 nM+ ER
|
[3H]E2 0,0625 nM+ E2 0,125 μM+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
[3H]E2 0,125 nM+ ER
|
[3H]E2 0,125 nM+ E2 0,25 μM+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
[3H]E2 0,250 nM+ ER
|
[3H]E2 0,250 nM+ E2 0,5 μM+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
[3H]E2 0,50 nM+ ER
|
[3H]E2 0,50 nM+ E2 1 μM+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
[3H]E2 1,00 nM+ ER
|
[3H]E2 1,00 nM+ E2 2 μM+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
[3H]E2 2,00 nM+ ER
|
[3H]E2 2,00 nM+ E2 4 μM+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
[3H]E2 4,00 nM+ ER
|
[3H]E2 4,00 nM+ E2 8 μM+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
:
|
unión total.
|
|
NSB
|
:
|
unión inespecífica.
|
|
[3H]E2
|
:
|
[3H]17β-estradiol.
|
|
E2
|
:
|
17β-estradiol no marcado.
|
|
Cuadro 3
Volúmenes de reactivo para la placa de microvaloración de saturación
|
Número de columna
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Fases de la preparación
|
Pocillos TB
|
Pocillos NSB
|
Ligando marcado solo
|
|
Volumen de los componentes de los pocillos de reacción indicados arriba y orden de adición
|
Solución amortiguadora
|
60 μl
|
50 μl
|
90 μl
|
|
E2 no marcado de la columna 11 del cuadro 2
|
–
|
10 μl
|
–
|
|
[3H]E2 de la columna 12 del cuadro 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
–
|
|
Volumen total de reacción
|
100 μl
|
100 μl
|
100 μl
|
|
Incubación
|
TRAS 2 HORAS DE REACCIÓN EN LA INCUBACIÓN
|
Cuantificación de la radiactividad justo después de la preparación. Sin incubación.
|
|
Tratamiento con DCC al 0,4 %
|
Sí
|
Sí
|
No
|
|
Volumen de DCC al 0,4 %
|
100 μl
|
100 μl
|
–
|
|
Filtración
|
Sí
|
Sí
|
No
|
|
MEDICIÓN DE LAS DPM
|
|
Volumen de cuantificación añadido a la mezcla de centelleo
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Las placas de microvaloración del ensayo para la determinación de la unión total y de la unión inespecífica deben incubarse a temperatura ambiente (de 22o C a 28 oC) durante dos horas.
|
|
Medición del [3H]17β-estradiol unido al hrERα
|
20.
|
Tras el período de incubación de dos horas, el [3H]17β-estradiol unido al hrERα debe separarse del [3H]-17β-estradiol libre añadiendo a los pocillos 100 μl de suspensión de DCC al 0,4 % enfriada con hielo. A continuación, las placas deben colocarse en hielo durante 10 minutos y la mezcla de reacción y la suspensión de DCC se deben filtrar mediante transferencia a un filtro de placas de microvaloración para retirar el DCC. A continuación, deben añadirse 100 μl del filtrado al líquido de centelleo en frascos LSC para determinar el número de desintegraciones por minuto (dpm) por frasco mediante recuento de centelleo líquido.
|
|
21.
|
Como alternativa, si no se dispone de filtro de microplacas, puede conseguirse la eliminación del DCC mediante centrifugación. A continuación, se deben tomar 50 μl del sobrenadante que contiene el [3H]17β-estradiol unido al hrERα con muchísimo cuidado para evitar cualquier contaminación de los pocillos por contacto con la suspensión de DCC, y deben utilizarse para el recuento de centelleo.
|
|
22.
|
El ligando marcado solo se utiliza para determinar las desintegraciones por minuto (dpm) del [3H]17β-estradiol añadido a los pocillos de ensayo. La radiactividad debe cuantificarse justo después de la preparación. Estos pocillos no deben incubarse y no deben tratarse con suspensión de DCC, sino que su contenido debe añadirse directamente al líquido de centelleo. Estas medidas muestran cuánto [3H]17β-estradiol medido en dpm se había añadido a cada conjunto de pocillos en relación con la unión total y con la unión inespecífica.
|
Ensayo de unión de competencia
|
23.
|
El ensayo de unión de competencia mide la unión de una única concentración de [3H]17β-estradiol en presencia de concentraciones cada vez mayores de un producto problema. Deben utilizarse en paralelo tres réplicas de cada concentración en cada tanda. Además, deben realizarse tres tandas no simultáneas con cada producto problema. El ensayo debe configurarse en una o más placas de microvaloración con 96 pocillos.
|
Testigos
|
24.
|
Al realizar el ensayo, deben incluirse en cada experimento testigos y disolventes en paralelo (es decir, estrógeno de referencia, ligando débil y no ligando). Deben incluirse en una placa de cada tanda todos los puntos que forman las curvas de concentración completas del estrógeno de referencia y de los testigos (es decir, ligando débil y no ligando). Todas las demás placas deben contener: i) una concentración elevada (desplazamiento máximo, es decir, aproximadamente el desplazamiento completo del ligando radiomarcado) y una media (aproximadamente la CI50) tanto de E2 como del ligando débil, por triplicado; 2) el testigo de disolvente y la unión inespecífica, cada uno por triplicado. En el protocolo del CERI se describen con detalle los procedimientos para la preparación de la solución amortiguadora y de las soluciones de [3H]17β-estradiol, de hrERα y de producto problema (2).
|
Control del disolvente:
|
25.
|
El control del disolvente indica que este no interactúa con el sistema de ensayo y que también mide la unión total (TB). El DMSO es el disolvente preferido. Si la concentración más alta del producto problema no es soluble en DMSO, también se podrá utilizar etanol. La concentración de DMSO en los pocillos finales del ensayo debe ser del 2,05 % y podría aumentarse hasta el 2,5 % en caso de falta de solubilidad del producto problema. No deben utilizarse concentraciones de DMSO por encima del 2,5 % debido a la interferencia de las concentraciones más elevadas de disolvente con el ensayo. En el caso de productos problema que no sean solubles en DMSO pero que sí sean solubles en etanol, podrá utilizarse sin interferencias en el ensayo un máximo de etanol del 2 %.
|
Testigo de la solución amortiguadora:
|
26.
|
El testigo de la solución amortiguadora (buffer control, BC) no debe contener ni disolvente ni producto problema, pero sí todos los demás componentes del ensayo. Los resultados del testigo de la solución amortiguadora se comparan con el testigo de disolvente para verificar que el disolvente utilizado no afecta al sistema de ensayo.
|
Ligando fuerte (estrógeno de referencia)
|
27.
|
El 17β-estradiol (n.o CAS 50-28-2) es el ligando endógeno y se une con gran afinidad al ER, subtipo alfa. Debe prepararse una curva patrón con 17β-estradiol no marcado para cada uno de los ensayos de unión de competencia al hrERα, para permitir una evaluación de la variabilidad cuando se realice el ensayo a lo largo del tiempo dentro del mismo laboratorio. Deben prepararse ocho soluciones de 17β-estradiol no marcado en DMSO y en solución amortiguadora de ensayo, y las concentraciones finales en los pocillos de ensayo que se vayan a utilizar para la curva patrón deben espaciarse como se indica a continuación: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. La concentración más elevada de 17β-estradiol no marcado (1 μM) debe servir de indicador de unión inespecífica (non-specific binding, NSB). Esta concentración se distingue con el rótulo "NSB" en el cuadro 4, aunque también forma parte de la curva patrón.
|
Ligando débil
|
28.
|
Debe incluirse un ligando débil [noretinodrel (n.o CAS 68-23-5) o, como alternativa, noretindrona (n.o CAS 68-22-4)] para demostrar la sensibilidad de cada experimento y permitir una evaluación de la variabilidad cuando se realice el ensayo a lo largo del tiempo. Deben prepararse ocho soluciones de ligando débil en DMSO y en solución amortiguadora de ensayo, y las concentraciones finales en los pocillos de ensayo deben espaciarse como se indica a continuación: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 y 10–9 M.
|
No ligando
|
29.
|
Debe utilizarse como testigo negativo (no ligando) el octiltrietoxisilano (OTES, n.o CAS 2943-75-1). Aporta la garantía de que el ensayo, tal como se realiza, detecta los productos problema que no se unen al hrERα. Deben prepararse ocho soluciones de no ligando en DMSO y en solución amortiguadora de ensayo, y las concentraciones finales en los pocillos de ensayo deben espaciarse como se indica a continuación: 10–3, 10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Puede utilizarse como no ligando alternativo el ftalato de di-n-butilo (DBP, n.o CAS 84-72-2), pero solo hasta la concentración de 10–4 M. Se ha demostrado que la solubilidad máxima del DBP en el ensayo es de 10–4 M.
|
Concentración del hrERα
|
30.
|
Debe utilizarse la cantidad del receptor que da una unión específica del 40 ± 10 % (véanse los puntos 12-13 del apéndice 3). La solución de hrERα debe prepararse mediante dilución de hrERα funcional en solución amortiguadora enfriada con hielo, justo antes de su utilización.
|
[3H]17β-estradiol
|
31.
|
La concentración final de [3H]17β-estradiol en los pocillos de ensayo debe ser de 0,5 nM.
|
Productos problema
|
32.
|
En primer lugar, es necesario realizar un ensayo de solubilidad para determinar el límite de solubilidad de cada producto problema y para identificar el intervalo adecuado de concentraciones que se ha de utilizar al aplicar el protocolo del ensayo. El límite de solubilidad de cada producto problema se determina inicialmente en el disolvente y después se confirma en las condiciones del ensayo. La concentración final utilizada en el ensayo no debe ser superior a 1 mM. La determinación del intervalo de concentraciones consiste en someter a ensayo un control del disolvente, junto con un mínimo de ocho diluciones logarítmicas en serie, a partir de la concentración máxima aceptable (por ejemplo, 1 mM o menos, según el límite de solubilidad), y anotar la presencia de turbidez o precipitado (véase también el punto 35 del apéndice 3). Una vez determinado el intervalo de concentraciones para los ensayos, debe someterse a ensayo el producto problema con 8 concentraciones logarítmicas espaciadas de forma adecuada, según los resultados de la determinación del intervalo de concentraciones. Las concentraciones sometidas a ensayo en los experimentos segundo y tercero deben ajustarse más según convenga para caracterizar mejor la curva concentración-respuesta, en caso necesario.
|
|
33.
|
Las diluciones del producto problema deben prepararse en el disolvente adecuado (véase el punto 25 del apéndice 3). Si la concentración más elevada del producto problema no fuera soluble en DMSO o en etanol, y la adición de más disolvente hiciera que la concentración de disolvente en el tubo final fuera mayor que el límite aceptable, la concentración más elevada podría reducirse a la concentración inmediatamente inferior. En este caso, puede añadirse una concentración adicional en el extremo inferior de la serie de concentraciones. Las demás concentraciones de la serie deben permanecer inalteradas.
|
|
34.
|
Las soluciones del producto problema deben ser objeto de un estrecho seguimiento cuando se añaden al pocillo del ensayo, ya que el producto problema puede provocar una precipitación cuando se añade al pocillo del ensayo. Los datos de todos los pocillos que contengan precipitado deben quedar excluidos del ajuste de la curva, y debe anotarse el motivo de la exclusión de los datos.
|
|
35.
|
Si hay información previa de otras fuentes que proporcionen el log CI50 de un producto problema, puede ser adecuado espaciar geométricamente las diluciones más cerca alrededor del log CI50 previsto (es decir, 0,5 unidades logarítmicas). Los resultados finales deben reflejar la suficiente amplitud de las concentraciones a ambos lados del log CI50 , incluidos la “parte superior” y la “parte inferior”, de manera que la curva de unión pueda caracterizarse adecuadamente.
|
Organización de las placas del ensayo
|
36.
|
Deben prepararse placas de microvaloración etiquetadas utilizando incubaciones séxtuples del control del disolvente, la concentración más elevada del estrógeno de referencia (E2), que también sirve de indicador de la unión inespecífica (NSB), el testigo de la solución amortiguadora, e incubaciones triplicadas de cada una de las ocho concentraciones del testigo no ligando (octiltrietoxisilano), las siete concentraciones inferiores del estrógeno de referencia (E2), las ocho concentraciones del ligando débil (noretinodrel o noretindrona) y las ocho concentraciones de cada producto problema (TC). En el cuadro 4 a continuación figura un ejemplo de diagrama de la disposición de la placa para las curvas de concentración completas correspondientes al estrógeno de referencia y al testigo. Se utilizan placas de microvaloración adicionales para el producto problema y deben incluir testigos de las placas, es decir, 1) una concentración elevada (desplazamiento máximo) y media (aproximadamente la CI50) tanto del E2 como de un ligando débil, por triplicado; 2) el testigo de disolvente (como unión total) y un ligando inespecífico, cada uno por sextuplicado (cuadro 5). En el apéndice 3.3 se presenta un ejemplo de una hoja de cálculo de la configuración de la placa de microvaloración para el ensayo de competencia con tres productos problema desconocidos. Las concentraciones indicadas en la hoja de cálculo, así como en los cuadros 4 y 5, se refieren a las concentraciones finales utilizadas en cada pocillo de ensayo. La concentración máxima de E2 debe ser 1 × 10–7 M y la del ligando débil debe ser la concentración más elevada utilizada para este en la placa 1. La concentración CI50 debe ser determinada por el laboratorio a partir de su base de datos de testigos históricos. Se espera que este valor sea similar al observado en los estudios de validación (véase el cuadro 1).
|
Cuadro 4
Disposición de las placas de microvaloración para el ensayo de unión de competencia
(98)
(99)
, curvas completas de concentración del estrógeno de referencia y de los testigos (placa 1).
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Testigo de la solución amortiguadora y testigo positivo (E2)
|
Positivo débil (noretinodrel)
|
Testigo negativo (OTES)
|
TB y NSB
|
|
A
|
Blanco (*14)
|
1 × 10–9 M
|
1 × 10–10 M
|
TB (control del disolvente) (DMSO al 2,05 %)
|
|
B
|
E2 (1 × 10–11 M)
|
1 × 10–8 M
|
1 × 10–9 M
|
|
C
|
E2 (1 × 10–10 M)
|
1 × 10–7,5 M
|
1 × 10–8 M
|
NSB (E2 10–6 M)
|
|
D
|
E2 (1 × 10–9,5 M)
|
1 × 10–7 M
|
1 × 10–7 M
|
|
E
|
E2 (1 × 10–9 M)
|
1 × 10–6,5 M
|
1 × 10–6 M
|
Testigo de la solución amortiguadora
|
|
F
|
E2 (1 × 10–8,5 M)
|
1 × 10–6 M
|
1 × 10–5 M
|
|
G
|
E2 (1 × 10–8 M)
|
1 × 10–5,5 M
|
1 × 10–4 M
|
Blanco (de marcado) (*15)
|
|
H
|
E2 (1 × 10–7 M)
|
1 × 10–4,5 M
|
1 × 10–3 M
|
Cuadro 5
Disposición de las placas de microvaloración para el ensayo de unión de competencia, placas adicionales para los productos problema (TC) y los testigos de las placas.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Producto problema-1 (TC-1)
|
Producto problema-2 (TC-2)
|
Producto problema-3 (TC-3)
|
Testigos
|
|
A
|
TC-1 (1 × 10–10 M)
|
TC-2 (1 × 10–10 M)
|
TC-3 (1 × 10–10 M)
|
E2 (1 × 10
-7
M)
|
|
B
|
TC-1 (1 × 10–9 M)
|
TC-2 (1 × 10–9 M)
|
TC-3 (1 × 10–9 M)
|
E2 (CI50)
|
|
C
|
TC-1 (1 × 10–8 M)
|
TC-2 (1 × 10–8 M)
|
TC-3 (1 × 10–8 M)
|
NE (1 × 10–4,5)
|
|
D
|
TC-1 (1 × 10–7 M)
|
TC-2 (1 × 10–7 M)
|
TC-3 (1 × 10–7 M)
|
NE (CI50)
|
|
E
|
TC-1 (1 × 10–6 M)
|
TC-2 (1 × 10–6 M)
|
TC-3 (1 × 10–6 M)
|
NSB (E2 10–6 M)
|
|
F
|
TC-1 (1 × 10–5 M)
|
TC-2 (1 × 10–5 M)
|
TC-3 (1 × 10–5 M)
|
|
G
|
TC-1 (1 × 10–4 M)
|
TC-2 (1 × 10–4 M)
|
TC-3 (1 × 10–4 M)
|
TB (control del disolvente)
|
|
H
|
TC-1 (1 × 10–3 M)
|
TC-2 (1 × 10–3 M)
|
TC-3 (1 × 10–3 M)
|
|
En este ejemplo, el ligando débil es el noretinodrel (NE).
|
Terminación del ensayo de unión de competencia
|
37.
|
Salvo en los pocillos destinados a la unión total y los blancos (de marcado), como se indica en el cuadro 6, se deben colocar en cada pocillo 50 μl de la solución amortiguadora de ensayo, y se deben mezclar con 10 μl de control del disolvente, estrógeno de referencia (E2), ligando débil, no ligando y productos problema, o, según corresponda, con 10 μl de una solución 5 nM de [3H]17β-estradiol. A continuación, se deben añadir a cada placa 30 μl de solución de receptor enfriada con hielo y se mezcla suavemente. La solución de hrERα debe ser el último reactivo que se añada. Las placas de microvaloración del ensayo deben incubarse a temperatura ambiente (22o a 28oC) durante 2 horas.
Cuadro 6
Volumen de los componentes del ensayo para las placas de un ensayo de unión de competencia, placas de microvaloración.
|
Etapas de preparación
|
Excepto los pocillos TB
|
Pocillos TB
|
Blanco (de marcado)
|
|
Volumen de los componentes de los pocillos de reacción indicados arriba y orden de adición
|
Solución amortiguadora del ensayo a temperatura ambiente
|
50 μl
|
60 μl
|
90 μl
|
|
E2 no marcado, ligando débil, no ligando, disolvente y productos problema (*16)
|
10 μl
|
–
|
–
|
|
[3H]17β-estradiol para obtener una concentración final de 0,5 nM (es decir, 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
Concentración de hrERα según se haya determinado (véanse los puntos 12-13)
|
30 μl
|
30 μl
|
–
|
|
Volumen total en cada pocillo del ensayo
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
A continuación debe llevarse a cabo la cuantificación del [3H]17β-estradiol unido al hrERα, tras la separación del [3H]17β-estradiol unido al hrERα respecto al [3H]17β-estradiol libre mediante la adición a cada pocillo de 100 μl de suspensión de DCC enfriada con hielo, como se describe en los puntos 21 a 23 del apéndice 3 en relación con el ensayo de unión de saturación.
|
|
39.
|
Los pocillos G10-12 y H10-12 [identificados como blanco (de marcado) en el cuadro 4] presentan las dpm del estradiol marcado con [3H] en 10 μl. La alícuota de 10 μl debe añadirse directamente al líquido de centelleo.
|
Criterios de aceptabilidad
Ensayo de unión de saturación
|
40.
|
La curva de la unión específica debe alcanzar una meseta a medida que se utilizan concentraciones crecientes de [3H]17β-estradiol, lo que indica la saturación del hrERα con el ligando.
|
|
41.
|
La unión específica a la concentración de 0,5 nM de [3H]17β-estradiol debe situarse en el intervalo aceptable del 30 % - 50 % de la radiactividad total medida media que se ha añadido en todas las tandas. Pueden aceptarse ligeras salidas fuera de este intervalo, pero si las tandas están sistemáticamente fuera del mismo o una tanda concreta se sitúa significativamente fuera de este intervalo, debe ajustarse la concentración de proteínas y repetirse el ensayo de saturación.
|
|
42.
|
Los datos deben corresponder a una gráfica lineal de Scatchard.
|
|
43.
|
La unión inespecífica no debe ser excesiva. El valor de la unión inespecífica debe ser típicamente < 35 % de la unión total. Sin embargo, la proporción puede superar ocasionalmente este límite cuando se miden valores muy bajos de dpm con la concentración más baja de 17β-estradiol radiomarcado.
|
Ensayo de unión de competencia
|
44.
|
El aumento de las concentraciones de 17β-estradiol no marcado debe desplazar el [3H]17β-estradiol del receptor de manera coherente con una unión de competencia a un solo punto.
|
|
45.
|
El valor de CI50 para el estrógeno de referencia (es decir, el 17β-estradiol) debe ser aproximadamente igual a la concentración molar del [3H]17β-estradiol más la Kd determinada a partir del ensayo de unión de saturación.
|
|
46.
|
La unión específica total debe estar sistemáticamente dentro del intervalo aceptable del 40 ± 10 % cuando la concentración media medida de la radiactividad total añadida a cada pocillo es de 0,5 nM en todas las tandas. Pueden aceptarse ligeras salidas fuera de este intervalo, pero si las tandas están sistemáticamente fuera del mismo o una tanda concreta se sitúa significativamente fuera de este intervalo, debe ajustarse la concentración de proteínas.
|
|
47.
|
El disolvente no debe alterar la sensibilidad ni la reproducibilidad del ensayo. Los resultados del control del disolvente (pocillos TB) se comparan con el testigo de la solución amortiguadora para verificar que el disolvente utilizado no afecta al sistema de ensayo. Los resultados del testigo de la TB y de la solución amortiguadora deben ser comparables si el disolvente no produce ningún efecto sobre el ensayo.
|
|
48.
|
El no ligando no debe desplazar más del 25 % del [3H]17β-estradiol del hrERα cuando se somete a ensayo hasta la concentración de 10–3 M (OTES) o 10–4 M (DBP).
|
|
49.
|
Se han establecido criterios de comportamiento para el estrógeno de referencia y dos ligandos débiles (por ejemplo, noretinodrel, noretindrona) utilizando los datos del estudio de validación del ensayo del CERI de unión al hrER (anexo N de la referencia 2). Se aportan los intervalos de confianza del 95 % para la media (n) ± DT de todas las tandas de los testigos en cuatro laboratorios participantes en el estudio de validación. Se calcularon los intervalos de confianza del 95 % para los parámetros de ajuste de las curvas (es decir, parte superior, parte inferior, pendiente de Hill y log CI50) en relación con el estrógeno de referencia y los ligandos débiles, y para el log10 RBA de los ligandos débiles en relación con el estrógeno de referencia. En el cuadro 1 se ofrecen los intervalos previstos para los parámetros de ajuste de la curva que pueden utilizarse como criterios de comportamiento. En la práctica, el intervalo de la CI50 puede variar ligeramente según la Kd obtenida experimentalmente del preparado del receptor y la concentración del ligando.
|
|
50.
|
No se han establecido criterios de comportamiento sobre los parámetros de ajuste de la curva en relación con los productos problema debido a la amplia gama de productos problema que puede haber y a la variación de las posibles afinidades y resultados (por ejemplo, ajuste de la curva completa o de la curva parcial, o sin ajuste alguno). Sin embargo, a la hora de revisar los resultados de cada tanda correspondientes a un producto problema, debe aplicarse un juicio profesional. Debe utilizarse un intervalo suficiente de concentraciones del producto problema para definir claramente la parte superior (p. ej., 90 - 100 % de unión) de la curva de competencia. La variabilidad entre las réplicas de cada concentración del producto problema, así como entre las tres tandas en momentos diferentes, debe ser razonable y científicamente justificable. Los testigos de cada tanda de un producto problema deben aproximarse a las medidas de comportamiento notificadas para este ensayo del CERI y deben ser coherentes con los datos históricos de los testigos de cada laboratorio respectivo.
|
ANÁLISIS DE LOS DATOS
Ensayo de unión de saturación
|
51.
|
Se mide tanto la unión total como la inespecífica. A partir de estos valores, se calcula la unión específica con concentraciones en aumento de [3H]17β-estradiol en condiciones de equilibrio, restando la inespecífica de la total. La gráfica de la unión específica frente a la concentración de [3H]17β-estradiol debe alcanzar una meseta para la unión específica máxima, lo cual indica la saturación del hrERα con el [3H]17β-estradiol. Por otra parte, el análisis de los datos debe documentar la unión del [3H]17β-estradiol a un solo punto de unión de elevada afinidad. En la curva de unión de saturación deben ponerse de manifiesto la unión inespecífica, la total y la específica. El posterior análisis de estos datos debe utilizar una regresión no lineal (por ejemplo, BioSoft; McPherson, 1985; Motulsky, 1995) con una representación final de los datos como gráfica de Scatchard.
|
|
52.
|
El análisis de los datos debe determinar los parámetros Bmax y Kd a partir solo de los datos de unión total, aceptando que la unión inespecífica es lineal, salvo que se justifique utilizar otro método. Además, a la hora de determinar el mejor ajuste debe utilizarse una regresión sólida, salvo que se justifique otra opción. Debe indicarse el método elegido para la regresión sólida. Al determinar los parámetros Bmax y Kd a partir de los datos de la unión de saturación, siempre debe aplicarse una corrección para tener en cuenta el agotamiento de los ligandos (p. ej., utilizando el método de Swillens, 1995).
|
Ensayo de unión de competencia
|
53.
|
La curva de unión de competencia se representa gráficamente como la unión específica del [3H]17β-estradiol frente a la concentración (unidades log10) del competidor. La concentración del producto problema que inhibe el 50 % de la unión máxima específica del [3H]17β-estradiol es el valor de la CI50.
|
|
54.
|
Deben estimarse los valores de log CI50 de los testigos positivos (p. ej., el estrógeno de referencia y el ligando débil), utilizando un programa informático adecuado de ajuste de curvas no lineales para ajustarse a una ecuación de Hill de cuatro parámetros (p. ej., BioSoft; McPherson, 1985; Motulsky, 1995). La parte superior, la parte inferior, la pendiente y log CI50 deben dejarse, por lo general, sin restricciones cuando se ajustan estas curvas. A la hora de determinar el mejor ajuste debe utilizarse una regresión sólida, salvo que se justifique otra opción. No debe utilizarse ninguna corrección por el agotamiento de los ligandos. Tras el análisis inicial, debe revisarse cada curva de los ligandos para garantizar que se ajusta adecuadamente al modelo. La afinidad relativa de unión (RBA) del ligando débil debe calcularse como porcentaje del log CI50 del ligando débil respecto al log CI50 del 17β-estradiol. Los resultados de los testigos positivos y del testigo de no ligando deben evaluarse utilizando las medidas del comportamiento del ensayo de los puntos 44 - 49 del presente apéndice 3.
|
|
55.
|
Los datos de todos los productos problema deben analizarse utilizando un enfoque gradual para garantizar que los datos se analizan de forma adecuada y que cada curva de unión de competencia se clasifica correctamente. Se recomienda que cada tanda con el producto problema se someta inicialmente a un análisis de datos normalizado idéntico al utilizado con el estrógeno de referencia y los testigos de ligandos débiles (véase el punto 54 del presente apéndice 3). Tras este análisis inicial, debe llevarse a cabo una revisión técnica de los parámetros de ajuste de la curva, así como un examen visual sobre el grado de calidad del ajuste de los datos con la curva de la unión de competencia generada en cada tanda. Durante esta revisión técnica, la observación de una disminución dependiente de la concentración en el porcentaje de [3H]17β-estradiol unido específicamente, la de una baja variabilidad entre las réplicas técnicas de cada concentración de producto problema, y la de la coherencia de los parámetros de ajuste entre las tres tandas son una buena indicación de que el ensayo y los análisis de datos se han llevado a cabo adecuadamente.
|
Interpretación de los datos
|
56.
|
Siempre que se cumplan todos los criterios de aceptabilidad, se considera que un producto problema es un ligando del hrERα si puede ajustarse una curva de unión y el punto más bajo de la curva de respuesta dentro del intervalo de los datos es inferior al 50 % (figura 1).
|
|
57.
|
Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es no ligando del hrERα si:
|
—
|
puede ajustarse una curva de unión y el punto más bajo de la curva de respuesta ajustada dentro del intervalo de datos es superior al 75 %, o
|
|
—
|
no puede ajustarse una curva de unión y la unión porcentual media sin suavizar más baja entre los grupos de concentración de los datos es superior al 75 %.
|
|
|
58.
|
Los productos problema se consideran dudosos si no se cumple ninguna de las condiciones anteriores (por ejemplo, el punto más bajo de la curva de respuesta ajustada se sitúa entre el 76 y el 51 %).
|
Cuadro 7
Criterios de asignación de la clasificación a un producto problema sobre la base de una curva de unión de competencia
|
Clasificación
|
Criterios
|
|
Ligandoa
|
Puede ajustarse una curva de unión.
El punto más bajo de la curva de respuesta dentro del intervalo de datos es inferior al 50 %.
|
|
No ligandob
|
Si puede ajustarse una curva de unión,
el punto más bajo de la curva de respuesta ajustada dentro del intervalo de datos es superior al 75 %.
Si no puede ajustarse una curva de unión,
la unión porcentual media sin suavizar más baja entre los grupos de concentración de los datos es superior al 75 %.
|
|
Dudosoc
|
Cualquier producto sometido al ensayo que no sea ni ligando ni no ligando
(p. ej., el punto más bajo de la curva de respuesta ajustada se sitúa entre el 76 y el 51 %).
|
Figura 1
Ejemplos de clasificación de productos problema utilizando una curva de unión de competencia.

|
59.
|
Se combinan las diversas tandas realizadas con un producto problema dentro de un laboratorio, asignando un valor numérico a cada tanda y promediando entre ellas, como se muestra en el cuadro 8. Los resultados de las tandas combinadas dentro de cada laboratorio se comparan con la clasificación prevista de cada producto problema.
|
Cuadro 8
Método de clasificación del producto problema mediante varias tandas dentro de un laboratorio
|
Asignar valor a cada tanda:
|
|
Clasificación
|
Valor numérico
|
|
Ligando
|
2
|
|
Dudoso
|
1
|
|
No ligando
|
0
|
|
Clasificar según la media de los valores numéricos de las tandas:
|
|
Clasificación
|
Valor numérico
|
|
Ligando
|
Media ≥ 1,5
|
|
Dudoso
|
0,5 ≤ Media < 1,5
|
|
No ligando
|
Media < 0,5
|
INFORME DEL ENSAYO
|
60.
|
Véase el punto 24 de la sección “COMPONENTES DEL ENSAYO DE UNIÓN AL hrER” de este método de ensayo.
|
Apéndice 3.1
RELACIÓN DE TÉRMINOS
[3H]E2
: 17β-estradiol radiomarcado con tritio.
DCC
: Carbón recubierto de dextrano.
E2
: 17β-Estradiol no marcado (inerte).
Solución amortiguadora del ensayo
: Tris-HCl 10 mM, pH 7,4, con EDTA 1 mM, EGTA 1mM, NaVO3 1 mM, 10 % de glicerol, leupeptina 0,2 mM, ditiotreitol 1 mM y 10 mg/ml de seroalbúmina bovina.
hrERα
: Receptor estrogénico recombinante humano alfa (dominio de unión al ligando).
Réplica
: Cada uno de los diversos pocillos que tienen el mismo contenido a la misma concentración y se someten a ensayo simultáneamente dentro de una misma tanda. En este protocolo, cada concentración del producto problema se somete al ensayo por triplicado; es decir, hay tres réplicas que se someten simultáneamente al ensayo con cada concentración del producto problema.
Tanda
: Conjunto completo de pocillos de ensayo en placas de microvaloración sometidos a ensayo a la vez, que aporta toda la información necesaria para caracterizar la unión de un producto problema al hrERα (a saber, [3H]17β-estradiol total añadido al pocillo de ensayo, unión máxima del [3H]17β-estradiol al hrERα, unión inespecífica, y unión total a diversas concentraciones del producto problema). Una tanda podría consistir simplemente en un solo pocillo (es decir, una réplica) por concentración, pero, puesto que este protocolo requiere que los ensayos se realicen por triplicado, una tanda estará compuesta por tres pocillos de ensayo por cada concentración. Además, este protocolo exige la realización de tres tandas independientes (es decir, no simultáneas) por producto.
Apéndice 3.2
DISTRIBUCIÓN DE LOS POCILLOS PARA EL ENSAYO DE UNIÓN DE COMPETENCIA
|
Placa
|
Posición
|
Réplica
|
Tipo de pocillo
|
Código del pocillo
|
Código de la concentración
|
Concentración inicial del competidor (M)
|
Solución madre de hrER (μl)
|
Volumen de solución amortiguadora (μl)
|
Volumen del marcador (E2 marcado) (μl)
|
Volumen tomado de la placa de dilución (μL)
|
Volumen final (μl)
|
Concentración final del competidor (M)
|
|
S
|
A1
|
1
|
Blanco
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Blanco
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Blanco
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
E2 no marcado
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B2
|
2
|
E2 no marcado
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B3
|
3
|
E2 no marcado
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
C1
|
1
|
E2 no marcado
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C2
|
2
|
E2 no marcado
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C3
|
3
|
E2 no marcado
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
D1
|
1
|
E2 no marcado
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D2
|
2
|
E2 no marcado
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D3
|
3
|
E2 no marcado
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
E1
|
1
|
E2 no marcado
|
S
|
S4
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E2
|
2
|
E2 no marcado
|
S
|
S4
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E3
|
3
|
E2 no marcado
|
S
|
S4
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
F1
|
1
|
E2 no marcado
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F2
|
2
|
E2 no marcado
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F3
|
3
|
E2 no marcado
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
G1
|
1
|
E2 no marcado
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G2
|
2
|
E2 no marcado
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G3
|
3
|
E2 no marcado
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
H1
|
1
|
E2 no marcado
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H2
|
2
|
E2 no marcado
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H3
|
3
|
E2 no marcado
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
A4
|
1
|
Noretinodrel
|
NE
|
WP1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A5
|
2
|
Noretinodrel
|
NE
|
WP1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A6
|
3
|
Noretinodrel
|
NE
|
WP1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B4
|
1
|
Noretinodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B5
|
2
|
Noretinodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B6
|
3
|
Noretinodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C4
|
1
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C5
|
2
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C6
|
3
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
D4
|
1
|
Noretinodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D5
|
2
|
Noretinodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D6
|
3
|
Noretinodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E4
|
1
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E5
|
2
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E6
|
3
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
F4
|
1
|
Noretinodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F5
|
2
|
Noretinodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F6
|
3
|
Noretinodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
G4
|
1
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G5
|
2
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G6
|
3
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
H4
|
1
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H5
|
2
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H6
|
3
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
A10
|
1
|
Unión total
|
TB
|
TB1
|
|
30
|
60
|
10
|
—
|
100
|
|
|
S
|
A11
|
2
|
Unión total
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
|
100
|
|
|
S
|
A12
|
3
|
Unión total
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
|
100
|
|
|
S
|
B10
|
4
|
Unión total
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
|
100
|
|
|
S
|
B11
|
5
|
Unión total
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
|
100
|
|
|
S
|
B12
|
6
|
Unión total
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
|
100
|
|
|
S
|
C10
|
1
|
E2 no marcado (elevada)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C11
|
2
|
E2 no marcado (elevada)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C12
|
3
|
E2 no marcado (elevada)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D10
|
4
|
E2 no marcado (elevada)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D11
|
5
|
E2 no marcado (elevada)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D12
|
6
|
E2 no marcado (elevada)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E10
|
1
|
Testigo de la solución amortiguadora
|
BC
|
BC1
|
|
|
100
|
|
|
100
|
|
|
S
|
E11
|
2
|
Testigo de la solución amortiguadora
|
BC
|
BC2
|
|
|
100
|
|
|
100
|
|
|
S
|
E12
|
3
|
Testigo de la solución amortiguadora
|
BC
|
BC3
|
|
|
100
|
|
|
100
|
|
|
S
|
F10
|
4
|
Testigo de la solución amortiguadora
|
BC
|
BC4
|
|
|
100
|
|
|
100
|
|
|
S
|
F11
|
5
|
Testigo de la solución amortiguadora
|
BC
|
BC5
|
|
|
100
|
|
|
100
|
|
|
S
|
F12
|
6
|
Testigo de la solución amortiguadora
|
BC
|
BC6
|
|
|
100
|
|
|
100
|
|
|
S
|
G10 (*17)
|
1
|
Blanco (de marcado)
|
Marcado
|
H1
|
|
90
|
|
10
|
|
100
|
|
|
S
|
G11 (*17)
|
2
|
Blanco (de marcado)
|
Marcado
|
H2
|
|
90
|
|
10
|
|
100
|
|
|
S
|
G12 (*17)
|
3
|
Blanco (de marcado)
|
Marcado
|
H3
|
|
90
|
|
10
|
|
100
|
|
|
S
|
H10 (*17)
|
4
|
Blanco (de marcado)
|
Marcado
|
H4
|
|
90
|
|
10
|
|
100
|
|
|
S
|
H11 (*17)
|
5
|
Blanco (de marcado)
|
Marcado
|
H5
|
|
90
|
|
10
|
|
100
|
|
|
S
|
H12
|
6
|
Blanco (de marcado)
|
Marcado
|
H6
|
|
90
|
|
10
|
|
100
|
|
|
P1
|
A1
|
1
|
Desconocido 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
Desconocido 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
Desconocido 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
Desconocido 1
|
U1
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B2
|
2
|
Desconocido 1
|
U1
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B3
|
3
|
Desconocido 1
|
U1
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C1
|
1
|
Desconocido 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C2
|
2
|
Desconocido 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C3
|
3
|
Desconocido 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D1
|
1
|
Desconocido 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D2
|
2
|
Desconocido 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D3
|
3
|
Desconocido 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E1
|
1
|
Desconocido 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E2
|
2
|
Desconocido 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E3
|
3
|
Desconocido 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F1
|
1
|
Desconocido 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F2
|
2
|
Desconocido 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F3
|
3
|
Desconocido 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G1
|
1
|
Desconocido 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G2
|
2
|
Desconocido 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G3
|
3
|
Desconocido 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H1
|
1
|
Desconocido 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H2
|
2
|
Desconocido 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H3
|
3
|
Desconocido 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A4
|
1
|
Desconocido 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
Desconocido 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
Desconocido 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
Desconocido 2
|
U2
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B5
|
2
|
Desconocido 2
|
U2
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B6
|
3
|
Desconocido 2
|
U2
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C4
|
1
|
Desconocido 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C5
|
2
|
Desconocido 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C6
|
3
|
Desconocido 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D4
|
1
|
Desconocido 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D5
|
2
|
Desconocido 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D6
|
3
|
Desconocido 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E4
|
1
|
Desconocido 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E5
|
2
|
Desconocido 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E6
|
3
|
Desconocido 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F4
|
1
|
Desconocido 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F5
|
2
|
Desconocido 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F6
|
3
|
Desconocido 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G4
|
1
|
Desconocido 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G5
|
2
|
Desconocido 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G6
|
3
|
Desconocido 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H4
|
1
|
Desconocido 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H5
|
2
|
Desconocido 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H6
|
3
|
Desconocido 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A7
|
1
|
Desconocido 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
Desconocido 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
Desconocido 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
Desconocido 3
|
U3
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B8
|
2
|
Desconocido 3
|
U3
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B9
|
3
|
Desconocido 3
|
U3
|
2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C7
|
1
|
Desconocido 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C8
|
2
|
Desconocido 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C9
|
3
|
Desconocido 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D7
|
1
|
Desconocido 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D8
|
2
|
Desconocido 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D9
|
3
|
Desconocido 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E7
|
1
|
Desconocido 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E8
|
2
|
Desconocido 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E9
|
3
|
Desconocido 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F7
|
1
|
Desconocido 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F8
|
2
|
Desconocido 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F9
|
3
|
Desconocido 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G7
|
1
|
Desconocido 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G8
|
2
|
Desconocido 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G9
|
3
|
Desconocido 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H7
|
1
|
Desconocido 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H8
|
2
|
Desconocido 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H9
|
3
|
Desconocido 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A10
|
1
|
Testigo E2 (máx)
|
S
|
E2max1
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A11
|
2
|
Testigo E2 (máx)
|
S
|
E2max2
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A12
|
3
|
Testigo E2 (máx)
|
S
|
E2max3
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
B10
|
1
|
Testigo E2 (CI50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Testigo E2 (CI50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Testigo E2 (CI50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Testigo NE (máx)
|
S
|
Nemax1
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C11
|
2
|
Testigo NE (máx)
|
S
|
Nemax2
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C12
|
3
|
Testigo NE (máx)
|
S
|
Nemax3
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
D10
|
1
|
Testigo NE (CI50)
|
S
|
NE CI501
|
NE CI50 x10
|
30
|
50
|
10
|
10
|
100
|
NE CI50
|
|
P1
|
D11
|
2
|
Testigo NE (CI50)
|
S
|
NE CI502
|
NE CI50 x10
|
30
|
50
|
10
|
10
|
100
|
NE CI50
|
|
P1
|
D12
|
3
|
Testigo NE (CI50)
|
S
|
NE CI503
|
NE CI50 x10
|
30
|
50
|
10
|
10
|
100
|
NE CI50
|
|
P1
|
E10
|
1
|
E2 no marcado (elevada)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E11
|
2
|
E2 no marcado (elevada)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E12
|
3
|
E2 no marcado (elevada)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F10
|
4
|
E2 no marcado (elevada)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F11
|
5
|
E2 no marcado (elevada)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F12
|
6
|
E2 no marcado (elevada)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
G10
|
1
|
Unión total
|
TB
|
TB1
|
|
30
|
60
|
10
|
|
100
|
|
|
P1
|
G11
|
2
|
Unión total
|
TB
|
TB2
|
|
30
|
60
|
10
|
|
100
|
|
|
P1
|
G12
|
3
|
Unión total
|
TB
|
TB3
|
|
30
|
60
|
10
|
|
100
|
|
|
P1
|
H10
|
4
|
Unión total
|
TB
|
TB4
|
|
30
|
60
|
10
|
|
100
|
|
|
P1
|
H11
|
5
|
Unión total
|
TB
|
TB5
|
|
30
|
60
|
10
|
|
100
|
|
|
P1
|
H12
|
6
|
Unión total
|
TB
|
TB6
|
|
30
|
60
|
10
|
|
100
|
|
Apéndice 4
CONSIDERACIONES PARA EL ANÁLISIS DE LOS DATOS PROCEDENTES DEL ANÁLISIS DE LA UNIÓN DE COMPETENCIA AL HRER
|
1.
|
El ensayo de unión de competencia al hrERα mide la unión de una única concentración de [3H]17β-estradiol en presencia de concentraciones cada vez mayores de un producto problema. La curva de la unión de competencia se representa gráficamente como la unión específica del [3H]17β-estradiol frente a la concentración (unidades log10) del competidor. La concentración del producto problema que inhibe el 50 % de la unión máxima específica del [3H]17β-estradiol es la CI50.
|
Análisis de datos para el estrógeno de referencia y el ligando débil (1)
|
2.
|
Los datos de las tandas de testigos se transforman (es decir, el porcentaje de la unión específica del [3H]17β-estradiol y la concentración logarítmcia del producto testigo) para su posterior análisis. Deben estimarse los valores de log CI50 de los testigos positivos (p. ej., el estrógeno de referencia y el ligando débil), utilizando un programa informático adecuado de ajuste de curvas no lineales para ajustarse a una ecuación de Hill de cuatro parámetros (p. ej., BioSoft; GraphPad Prism) (2). La parte superior, la parte inferior, la pendiente y log CI50 pueden dejarse, por lo general, sin restricciones cuando se ajustan estas curvas. A la hora de determinar el mejor ajuste debe utilizarse una regresión sólida, salvo que se justifique otra opción. Debe indicarse el método elegido para la regresión sólida. No fue necesario utilizar ninguna corrección para tener en cuenta el agotamiento de los ligandos en los ensayos FW o CERI, pero puede considerarse en caso necesario. Tras el análisis inicial, debe revisarse cada curva de los ligandos para garantizar que se ajusta adecuadamente al modelo. La afinidad relativa de unión (RBA) del ligando débil puede calcularse como porcentaje del log CI50 del ligando débil respecto al log CI50 del 17β-estradiol. Los resultados de los testigos positivos y del testigo de no ligando deben evaluarse utilizando medidas de comportamiento del ensayo y criterios de aceptabilidad, tal como se describe en el presente método de ensayo (punto 20), en el apéndice 2 (ensayo de FW, antes citado, puntos 41 a 51) y en el apéndice 3 (ensayo del CERI, puntos 41 a 51). En la figura 1 se muestran ejemplos de tres tandas para el estrógeno de referencia y el ligando débil.
|
Figura 1
Ejemplos de curvas de la unión de competencia para el estrógeno de referencia y para el ligando débil testigo.

Análisis de datos de los productos problema
|
3.
|
Los datos de todos los productos problema deben analizarse utilizando un enfoque gradual para garantizar que los datos se analizan de forma adecuada y que cada curva de la unión de competencia se clasifica correctamente. Cada tanda con el producto problema debe someterse inicialmente a un análisis de datos normalizado idéntico al utilizado con el estrógeno de referencia y los testigos de ligandos débiles. Tras este análisis inicial, debe llevarse a cabo una revisión técnica de los parámetros de ajuste de la curva, así como un examen visual sobre el grado de calidad del ajuste de los datos con la curva de la unión de competencia generada en cada tanda. Durante esta revisión técnica, la observación de una disminución dependiente de la concentración en el porcentaje de [3H]17β-estradiol unido específicamente, la de una baja variabilidad entre las réplicas técnicas de cada concentración de producto, y la de la coherencia de los parámetros de ajuste entre las tres tandas son una buena indicación de que el ensayo y los análisis de datos se han llevado a cabo adecuadamente. A la hora de revisar los resultados de cada tanda correspondientes a un producto problema, debe aplicarse un juicio profesional, y los datos utilizados para clasificar cada producto problema como ligando o no ligando deben ser científicamente justificables.
|
|
4.
|
En ocasiones puede haber ejemplos de datos que requieran una atención adicional para analizar e interpretar adecuadamente los datos de unión al hrER. Los estudios anteriores habían mostrado casos en los que el análisis y la interpretación de los datos sobre la unión a receptores de competencia pueden complicarse debido a un aumento del porcentaje de la unión específica cuando se someten a ensayo productos a las concentraciones más altas (figura 2). Se trata de una cuestión muy conocida que se ha presentado al utilizar protocolos para una serie de ensayos de unión a receptores de competencia (3). En estos casos, se observa una respuesta dependiente de la concentración a concentraciones más bajas pero, al aproximarse la concentración del producto problema al límite de solubilidad, el desplazamiento del [3H]17β-estradiol deja de disminuir. En estos casos, los datos de las concentraciones más elevadas indican que se ha alcanzado el límite biológico del ensayo. Por ejemplo, este fenómeno se asocia muchas veces a la insolubilidad y la precipitación del producto a altas concentraciones, o también puede ser un reflejo de la superación de la capacidad que tiene el carbón recubierto de dextrano para atrapar el ligando radiomarcado libre durante el procedimiento de separación a las concentraciones más elevadas del producto. El incluir estos puntos de datos cuando se ajustan los datos de la unión de competencia a una curva sigmoidea puede dar lugar a veces a una clasificación errónea del potencial de unión al ER de un producto problema (figura 2). Para evitarlo, el protocolo de los ensayos de unión al hrER de FW y del CERI incluye una opción para excluir de los análisis los puntos de datos en los que la media de las réplicas del porcentaje de unión específica del [3H]17β-estradiol supera al menos en el 10 % a la media observada a una concentración inferior (esto es lo que suele denominarse la regla del 10 %). Esta regla solo puede utilizarse una vez con una curva determinada y deben seguir quedando datos correspondientes a un mínimo de seis concentraciones, de forma que la curva pueda clasificarse correctamente.
|
Figura 2
Ejemplos de curvas de unión de competencia con y sin aplicación de la regla del 10 %

|
5.
|
El uso adecuado de la regla del 10 % para corregir estas curvas debe considerarse cuidadosamente y reservarse para aquellos casos en los que haya una clara indicación de unión al hrER. Durante la realización de experimentos para el estudio de validación del ensayo de unión al hrER de FW, se observó que la regla del 10 % tenía a veces una consecuencia imprevista y no deseada. Los productos que no interactuaban con el receptor (es decir, los no ligandos verdaderos) a menudo presentaban una variabilidad alrededor del 100 % de unión del radioligando que era superior al 10 % en toda la gama de concentraciones ensayadas. Si resultaba que el valor más bajo se encontraba a una concentración baja, los datos de todas las concentraciones superiores podrían eliminarse del análisis utilizando la regla del 10 %, aunque tales concentraciones podrían ser útiles para establecer que el producto es un ligando. El gráfico 3 muestra ejemplos en los que el uso de la regla del 10 % no es adecuado.
|
Figura 3
Ejemplos de datos de unión de competencia en los que la utilización de la regla del 10 % no es adecuada


BIBLIOGRAFÍA
|
(1)
|
OCDE (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organización para la Cooperación y el Desarrollo Económico, París.
|
|
(2)
|
Motulsky H. and Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71. ESTUDIOS DE SENSIBILIZACIÓN CUTÁNEA IN VITRO RELATIVOS AL FENÓMENO CLAVE DE LA ACTIVACIÓN DE LAS CÉLULAS DENDRÍTICAS EN LA RUTA DE RESULTADOS ADVERSOS (AOP) DE LA SENSIBILIZACIÓN CUTÁNEA
INTRODUCCIÓN GENERAL
Método de ensayo basado en el fenómeno clave de la activación de las células dendríticas
|
1.
|
Un sensibilizante cutáneo es una sustancia que induce una respuesta alérgica por contacto con la piel, tal como se define en el Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de las Naciones Unidas) (1) y en el Reglamento (UE) n.o 1272/2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) de la Unión Europea (100). Existe un acuerdo general sobre los principales fenómenos biológicos subyacentes a la sensibilización cutánea. El conocimiento actual de los mecanismos químicos y biológicos asociados a la sensibilización cutánea se ha resumido como una ruta de resultados adversos (AOP, Adverse Outcome Pathway) en el marco del programa AOP de la OCDE (2), empezando por el fenómeno iniciador molecular y pasando por fenómenos intermedios hasta llegar al efecto adverso, a saber, la dermatitis alérgica de contacto. En este caso, el fenómeno iniciador molecular (es decir, el primer fenómeno clave) es la unión covalente de sustancias electrofílicas a los centros nucleofílicos de las proteínas de la piel. El segundo fenómeno clave en esta AOP tiene lugar en los queratinocitos e incluye respuestas inflamatorias, así como cambios en la expresión génica relacionada con rutas específicas de comunicación celular, tales como las rutas dependientes del elemento de respuesta antioxidante/electrófilo (ARE, antioxidant/electrophile response element). El tercer fenómeno clave es la activación de las células dendríticas (DC, dendritic cells), normalmente evaluada por la expresión de marcadores específicos de superficie celular, quimiocinas y citocinas. El cuarto fenómeno clave es la activación y proliferación de las células T, que se evalúa indirectamente en el ensayo con ganglios linfáticos locales (LLNA, Local Lymph Node Assay) de múridos (3).
|
|
2.
|
El presente método de ensayo es equivalente a las directrices de ensayo (TG) 442E de la OCDE (2017). Describe los ensayos in vitro que abordan los mecanismos descritos en el fenómeno clave de la activación de las células dendríticas en la AOP de la sensibilización cutánea (2). El método de ensayo comprende los ensayos que deben utilizarse para justificar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes de conformidad con el SGA de las Naciones Unidas y el CLP.
|
|
Los ensayos descritos en el presente método de ensayo son los siguientes:
|
—
|
Ensayo de activación de la línea celular humana (h-CLAT),
|
|
—
|
Ensayo de activación de la línea celular U937 (U-SENS™),
|
|
—
|
Ensayo del gen marcador de la interleucina-8 (ensayo IL-8 Luc).
|
|
|
|
3.
|
Los ensayos incluidos en el presente método de ensayo y las TG de la OCDE correspondientes pueden diferir en relación con el procedimiento utilizado para generar los datos y los resultados medidos, pero pueden utilizarse de forma indiscriminada para satisfacer los requisitos de los países en cuanto a los resultados de los ensayos relativos al fenómeno clave de la activación de las células dendríticas en la AOP de la sensibilización cutánea, beneficiándose al mismo tiempo de la aceptación mutua de datos de la OCDE.
|
Antecedentes y principios de los ensayos incluidos en el método de ensayo basado en el fenómeno clave
|
4.
|
Tradicionalmente, la evaluación de la sensibilización cutánea se hacía con animales de laboratorio. Los métodos clásicos con cobayas, como el ensayo de maximización con cobayas (GMPT, Guinea Pig Maximisation Test) de Magnusson y Kligman y el ensayo de Buehler (método de ensayo B.6) (4), estudian tanto la fase de inducción como la de desencadenamiento de la sensibilización cutánea. Los ensayos con múridos, el ensayo con ganglios linfáticos locales (LLNA, Local Lymph Node Assay) (método de ensayo B.42) (3) y sus dos modificaciones no radiactivas, el LLNA: DA (método de ensayo B.50) (5) y el LLNA: BrdU-ELISA (método de ensayo B.51) (6), evalúan todos la respuesta de inducción exclusivamente, y también han conseguido su reconocimiento, ya que presentan una ventaja en relación con los ensayos con cobayas en términos de bienestar animal, junto con una medición objetiva de la fase de inducción de la sensibilización cutánea.
|
|
5.
|
Recientemente se han adoptado para contribuir a la evaluación del potencial de peligro de sensibilización cutánea de los productos varios métodos de ensayo in chemico e in vitro de base farmacodinámica relativos al primer fenómeno clave (método de ensayo B.59; Ensayo directo de reactividad peptídica) (7) y el segundo fenómeno clave (método de ensayo B.60; Método de ensayo de la luciferasa ARE-Nrf2) (8) de la AOP de la sensibilización cutánea.
|
|
6.
|
Los ensayos descritos en el presente método de ensayo cuantifican el cambio en la expresión de los marcadores de superficie celular asociados al proceso de activación de los monocitos y DC tras la exposición a sensibilizantes (por ejemplo, CD54, CD86), o los cambios en la expresión de la IL-8, citocina asociada a la activación de las DC. Se ha comunicado que ciertos sensibilizantes cutáneos inducen la expresión de marcadores de membrana celular como, por ejemplo, CD40, CD54, CD80, CD83 y CD86, además de inducir la producción de citocinas proinflamatorias, como la IL-1β y la TNF-α, y de varias quimiocinas, como la IL-8 (CXCL8) y la CCL3 (9) (10) (11) (12), asociadas con la activación de las DC (2).
|
|
7.
|
Sin embargo, como la activación de las DC representa por sí sola un fenómeno clave de la AOP de la sensibilización cutánea (2) (13), la información generada con los ensayos que miden los marcadores de la activación de las DC solas puede no ser suficiente para concluir sobre la presencia o la ausencia de potencial de sensibilización cutánea de los productos. Por consiguiente, se proponen los datos generados con los ensayos descritos en el presente método de ensayo como ayuda para discriminar entre los sensibilizantes cutáneos (es decir, la categoría 1 del SGA de las Naciones Unidas y del CLP) y los no sensibilizantes, cuando se utilicen en los enfoques integrados de ensayos y evaluación (IATA, Integrated Approaches to Testing and Assessment), junto con otros datos complementarios pertinentes derivados, por ejemplo, de ensayos in vitro relativos a otros fenómenos clave de la AOP de la sensibilización cutánea, así como métodos que no sean de ensayo, incluida la extrapolación a partir de análogos químicos (13). Se han publicado ejemplos de la utilización de los datos obtenidos con estos ensayos en enfoques definidos, es decir, enfoques normalizados tanto en relación con el conjunto de fuentes de información utilizadas como en el procedimiento aplicado a los datos para obtener asignaciones (13), y pueden emplearse como elementos útiles en el marco de los IATA.
|
|
8.
|
Los ensayos descritos en este método de ensayo no pueden utilizarse solos, ni para clasificar los sensibilizantes cutáneos en las subcategorías 1A y 1B definidas por el SGA de las Naciones Unidas y el CLP, en el caso de las autoridades que apliquen estas dos subcategorías facultativas, ni para asignar la potencia a efectos de las decisiones de evaluación de la seguridad. Sin embargo, dependiendo del marco normativo, los resultados positivos generados con estos métodos pueden utilizarse por sí solos para clasificar un producto en la categoría 1 del SGA de las Naciones Unidas y del CLP.
|
|
9.
|
En este método de ensayo se utiliza el término «producto problema» para referirse a lo que se somete a ensayo (101), y no está relacionado con la aplicabilidad de los ensayos al estudio de sustancias de un solo componente, sustancias de componentes múltiples y/o mezclas. Actualmente, se dispone de información limitada sobre la aplicabilidad de los ensayos a las sustancias de componentes múltiples / mezclas (14) (15). Sin embargo, los ensayos son técnicamente aplicables a las sustancias de componentes múltiples y mezclas. No obstante, antes de la utilización de este método de ensayo con una mezcla para obtener datos con fines normativos, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué (102). Dichas consideraciones no son necesarias cuando los requisitos normativos estipulan que la mezcla debe someterse a ensayo. Por otra parte, al ensayar sustancias de componentes múltiples o mezclas, deben tenerse en cuenta las posibles interferencias de componentes citotóxicos con las respuestas obtenidas.
|
BIBLIOGRAFÍA
|
(1)
|
Naciones Unidas (2015). Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA). Sexta edición revisada. Nueva York & Ginebra: Publicaciones de las Naciones Unidas. ISBN: 978-92-1-117006-1. Disponible en: https://www.unece.org/es/trans/danger/publi/ghs/ghs_rev06/06files_s.html.
|
|
(2)
|
OCDE (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Disponible en: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Capítulo B.42 del presente anexo: Ensayo con ganglios linfáticos locales.
|
|
(4)
|
Capítulo B.6 del presente anexo: Sensibilización de la piel.
|
|
(5)
|
Capítulo B.50 del presente anexo: Sensibilización cutánea: Ensayo con ganglios linfáticos locales: DA.
|
|
(6)
|
Capítulo B.51 del presente anexo: Sensibilización cutánea: Ensayo con ganglios linfáticos locales: BrdU-ELISA.
|
|
(7)
|
Capítulo B.59 del presente anexo: Sensibilización cutánea in chemico: Ensayo directo de reactividad peptídica (DPRA).
|
|
(8)
|
Capítulo B.60 del presente anexo: Sensibilización cutánea in vitro: Método de ensayo de la luciferasa ARE-Nrf2.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H, and Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y, and Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OCDE (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Apéndice 1
SENSIBILIZACIÓN CUTÁNEA IN VITRO: ENSAYO DE ACTIVACIÓN DE LA LÍNEA CELULAR HUMANA (H-CLAT)
CONSIDERACIONES INICIALES Y LIMITACIONES
|
1.
|
El ensayo h-CLAT cuantifica los cambios en la expresión de los marcadores de superficie celular asociados al proceso de activación de los monocitos y de las células dendríticas (DC) (es decir, CD86 y CD54), en la línea celular de la leucemia monocítica humana (THP-1), tras la exposición a sensibilizantes (1) (2). Los niveles de expresión medidos en los marcadores de superficie celular CD86 y CD54 se utilizan a continuación para justificar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes.
|
|
2.
|
El ensayo h-CLAT se ha evaluado en un estudio de validación coordinado por el laboratorio de referencia de la Unión Europea para las alternativas a los ensayos con animales (EURL ECVAM), con posterior revisión por pares independientes en el Comité Científico Consultivo del ECVAM (ESAC). Teniendo en cuenta todas las pruebas disponibles y las aportaciones de los reguladores y de las partes interesadas, el EURL ECVAM recomendó que el h-CLAT se utilizara como parte de un enfoque IATA para justificar la discriminación entre sensibilizantes y no sensibilizantes a efectos de la clasificación de los peligros y el etiquetado (3). En la bibliografía se recogen ejemplos del uso de datos del h-CLAT en combinación con otra información (4) (5) (6) (7) (8) (9) (10) (11).
|
|
3.
|
Se ha demostrado que el h-CLAT puede transferirse a laboratorios con experiencia en técnicas de cultivo celular y análisis de citometría de flujo. El nivel de reproducibilidad de las asignaciones que cabe esperar del ensayo es del orden del 80 % dentro de un laboratorio y entre laboratorios (3) (12). Los resultados obtenidos en el estudio de validación (13) y otros estudios publicados (14) indican en general que, en comparación con los resultados del LLNA, la exactitud de la distinción de los sensibilizantes cutáneos (es decir, categoría 1 del SGA de las Naciones Unidas y del CLP) frente a los no sensibilizantes es del 85 % (N = 142), con una sensibilidad del 93 % (94/101) y una especificidad del 66 % (27/41) [sobre la base de un nuevo análisis por el EURL ECVAM (12), considerando todos los datos existentes y no teniendo en cuenta los resultados negativos correspondientes a productos con un valor de log Kow superior a 3,5, como se describe en el punto 4]. Es más probable que las asignaciones negativas falsas con el ensayo h-CLAT afecten a los productos que muestran una potencia de sensibilización cutánea baja o moderada (es decir, subcategoría 1B del SGA de las Naciones Unidas y del CLP) que a los productos que presentan una potencia de sensibilización cutánea elevada (es decir, subcategoría 1A del SGA de las Naciones Unidas y del CLP) (4) (13) (15). En conjunto, esta información indica la utilidad del método del h-CLAT para contribuir a la identificación del peligro de sensibilización cutánea. Sin embargo, los valores de la exactitud aquí indicados para el h-CLAT utilizado como ensayo único solo son indicativos, ya que el ensayo debe considerarse en combinación con otras fuentes de información en el contexto de un enfoque IATA y de conformidad con las disposiciones de los puntos 7 y 8 de la introducción general. Además, a la hora de evaluar los métodos de ensayo de la sensibilización cutánea sin animales, debe tenerse en cuenta que el ensayo LLNA y otros ensayos con animales pueden no reflejar plenamente la situación en los seres humanos.
|
|
4.
|
Sobre la base de los datos actualmente disponibles, se ha demostrado que el h-CLAT es aplicable a productos problema que abarcan diversos grupos funcionales orgánicos, mecanismos de reacción, potencias de sensibilización cutánea (determinada en estudios in vivo) y propiedades fisicoquímicas (3) (14) (15). El método h-CLAT es aplicable a los productos problema solubles o que forman una dispersión estable (es decir, un coloide o una suspensión en la que el producto problema no sedimenta ni se separa del disolvente o vehículo para formar distintas fases) en un disolvente o vehículo adecuado (véase el punto 14). Los productos problema con un valor de log Kow superior a 3,5 tienden a producir resultados falsos negativos (14). Por tanto, no deben tenerse en cuenta los resultados negativos con productos problema cuyo valor de log Kow es superior a 3,5. Sin embargo, los resultados positivos obtenidos con productos problema cuyo valor de log Kow es superior a 3,5 podrían seguir utilizándose para justificar la identificación del producto problema como sensibilizante cutáneo. Por otra parte, debido a la limitada capacidad metabólica de la línea celular utilizada (16) y debido a las condiciones experimentales, pueden obtenerse también resultados negativos en el h-CLAT (15) con los pro-haptenos (es decir, sustancias que requieren activación enzimática, por ejemplo mediante enzimas P450) y los pre-haptenos (es decir, productos activados por oxidación), en particular con una baja velocidad de oxidación. Los productos problema fluorescentes pueden evaluarse con el h-CLAT (17); sin embargo, los productos problema muy fluorescentes que emiten a la misma longitud de onda que el isotiocianato de fluoresceína (FITC) o que el yoduro de propidio (PI), pueden interferir con la detección por citometría de flujo y, por tanto, no pueden evaluarse correctamente utilizando anticuerpos conjugados con el FITC o PI. En tal caso, pueden utilizarse otros anticuerpos marcados con fluorocromos u otros marcadores de citotoxicidad, según corresponda, siempre que se demuestre que proporcionan resultados similares a los de los anticuerpos marcados con FITC (véase el punto 24) o PI (véase el punto 18), por ejemplo sometiendo a ensayo las sustancias utilizadas para la prueba de la competencia recogidas en el apéndice 1.2. A la luz de lo anterior, los resultados negativos deben interpretarse en el contexto de las limitaciones indicadas y junto con otras fuentes de información en el marco de los enfoques IATA. En los casos en que haya pruebas que demuestren la inaplicabilidad del método h-CLAT a otras categorías específicas de productos problema, no deberá utilizarse este método con tales categorías específicas.
|
|
5.
|
Tal como se ha descrito anteriormente, el método h-CLAT sirve para justificar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes. No obstante, puede contribuir también a la evaluación de la potencia sensibilizante (4) (5) (9) cuando se utiliza en enfoques integrados, como los enfoques IATA. Sin embargo, es necesario seguir trabajando, preferentemente a partir de datos con seres humanos, para determinar de qué modo los resultados del h-CLAT pueden informar sobre la evaluación de la potencia.
|
|
6.
|
En el apéndice 1.1 se dan las definiciones pertinentes.
|
PRINCIPIO DEL ENSAYO
|
7.
|
El método h-CLAT es un ensayo in vitro que cuantifica los cambios de la expresión de los marcadores de superficie celular (es decir, CD86 y CD54) de la línea celular THP-1 de la leucemia monocítica humana, tras una exposición de 24 horas al producto problema. Estas moléculas de superficie son marcadores típicos de la activación de las células THP-1 monocíticas y pueden imitar la activación de las DC, la cual desempeña un papel fundamental en la estimulación de las células T. Los cambios en la expresión de los marcadores de superficie se miden mediante citometría de flujo tras la tinción celular mediante anticuerpos marcados con fluorocromo. También se lleva a cabo simultáneamente una medición de la citotoxicidad con el fin de evaluar si a concentraciones subcitotóxicas se produce un incremento de la expresión de los marcadores de superficie. Se calcula la intensidad de la fluorescencia relativa de los marcadores de superficie en comparación con el control del disolvente o de vehículo y se utiliza en el modelo de asignación (véase el punto 26), a fin de justificar la discriminación entre sensibilizantes y no sensibilizantes.
|
DEMOSTRACIÓN DE LA COMPETENCIA
|
8.
|
Antes de proceder al uso sistemático del ensayo descrito en el presente apéndice del método de ensayo B.71, los laboratorios deben demostrar su competencia técnica, utilizando las diez sustancias recomendadas al efecto en el apéndice 1.2. Por otra parte, los usuarios del ensayo deben mantener una base de datos históricos que contenga los generados en los controles de reactividad (véase el punto 11) y con los testigos positivos y el control del disolvente/vehículo (véanse los puntos 20 a 22), y utilizar estos datos para confirmar que la reproducibilidad del ensayo en su laboratorio se mantiene a lo largo del tiempo.
|
PROCEDIMIENTO
|
9.
|
Este ensayo se basa en el protocolo n.o 158 de servicio de la base de datos de h-CLAT sobre métodos alternativos a la experimentación con animales (DB-ALM) (18) que constituye el protocolo utilizado para el estudio de validación coordinado por el EURL ECVAM. Se recomienda que se siga este protocolo cuando se aplique y utilice el método h-CLAT en el laboratorio. A continuación se recoge una descripción de los principales componentes y procedimientos del método h-CLAT, que comprende dos etapas: ensayo de determinación de la dosis y medición de la expresión de los marcadores CD86/CD54.
|
Preparación de las células
|
10.
|
Para llevar a cabo el método h-CLAT debe utilizarse la línea celular de la leucemia monocítica humana, THP-1. Se recomienda que las células (TIB-202™) se obtengan de un banco de células adecuado, como la American Type Culture Collection.
|
|
11.
|
Se cultivan células THP-1, a 37oC, en atmósfera humidificada y con un 5 % de CO2, en medio RPMI-1640, complementado con un 10 % de suero bovino fetal (FBS), 2-mercaptoetanol 0,05 mM, 100 unidades/ml de penicilina y 100 μg/ml de estreptomicina. Puede evitarse el uso de penicilina y de estreptomicina en el medio de cultivo. Sin embargo, en tal caso, los usuarios deben verificar que la ausencia de antibióticos en el medio de cultivo no influye en los resultados, por ejemplo sometiendo a ensayo las sustancias para demostrar la competencia que figuran en el apéndice 1.2. En cualquier caso, para minimizar el riesgo de contaminación, deben seguirse buenas prácticas de cultivo celular, independientemente de la presencia o no de antibióticos en el medio de cultivo celular. Las células THP-1 se siembran sistemáticamente cada 2-3 días a una densidad de 0,1 a 0,2 × 106 células/ml. Deben mantenerse a densidades de 0,1 a 1,0 × 106 células/ml. Antes de utilizarse para el ensayo, las células deben ser objeto de un control de reactividad, que ha de realizarse utilizando los testigos positivos, 2,4-dinitroclorobenceno (DNCB) (n.o CAS 97-00-7, pureza ≥ 99 %) y sulfato de níquel (NiSO4) (n.o CAS 10101-97-0, pureza ≥ 99 %) y el testigo negativo, ácido láctico (LA) (n.o CAS 50-21-5, pureza ≥ 85 %), dos semanas después de la descongelación. Tanto el DNCB como el NiSO4 deben producir una respuesta positiva de los marcadores de superficie celular CD86 y CD54, y el LA debe producir una respuesta negativa de ambos tipos de marcadores. Solo se pueden utilizar para el ensayo las células que hayan superado el control de reactividad. Las células pueden propagarse hasta dos meses después de la descongelación. El número de pases no debe ser superior a 30. El control de reactividad debe realizarse con arreglo a los procedimientos descritos en los puntos 20 a 24.
|
|
12.
|
Para el ensayo, se siembran células THP-1 a una densidad de 0,1 × 106 células/ml o 0,2 × 106 células/ml, y se precultivan en matraces de cultivo durante 72 horas o durante 48 horas, respectivamente. Es importante que la densidad celular en el matraz de cultivo justo después del período de precultivo sea lo más constante posible en cada experimento (utilizando una de las dos condiciones de precultivo descritas anteriormente), ya que la densidad celular en el matraz de cultivo justo después del precultivo podría afectar a la expresión de CD86/CD54 inducida por alérgenos (19). En el día del ensayo, se toman células del matraz de cultivo y se suspenden con medio de cultivo fresco a la concentración de 2 × 106 células/ml. A continuación, se distribuyen las células en una placa de fondo plano de 24 pocillos (500 μl, 1 × 106 células/pocillo) o una placa de fondo plano de 96 pocillos (80 μl, 1,6 × 105 células/pocillo).
|
Ensayo de determinación de la dosis
|
13.
|
Se lleva a cabo un ensayo de determinación de la dosis para determinar la CV75, que es la concentración del producto problema que da el 75 % de viabilidad celular (CV) en comparación con el control del disolvente/vehículo. El valor de CV75 se utiliza para determinar la concentración de productos problema utilizada en la medición de la expresión de CD86/CD54 (véanse los puntos 20-24).
|
Preparación de los productos problema y de las sustancias testigo
|
14.
|
Los productos problema y las sustancias testigo se preparan el día del ensayo. Para el método h-CLAT, los productos problema se disuelven o dispersan de forma estable (véase también el punto 4) en solución salina o medio, como primeras opciones de disolvente/vehículo, o en dimetilsulfóxido (DMSO, ≥ 99 % de pureza) como segunda opción de disolvente/vehículo si el producto problema no es soluble o no forma una dispersión estable en los dos disolventes/vehículos anteriores, hasta obtener las concentraciones finales de 100 mg/ml (en solución salina o medio) o 500 mg/ml (en DMSO). Podrán utilizarse otros disolventes/vehículos distintos de los descritos anteriormente si se presenta una justificación científica suficiente. Debe tenerse en cuenta la estabilidad del producto problema en el disolvente o vehículo final.
|
|
15.
|
A partir de las soluciones madre de 100 mg/ml (en solución salina o en medio) o 500 mg/ml (en DMSO) de los productos problema, deben efectuarse las siguientes diluciones:
|
—
|
Con solución salina o medio como disolvente/vehículo: Se preparan ocho soluciones madre (ocho concentraciones), mediante diluciones a la mitad en serie utilizando el disolvente o vehículo correspondiente. Estas soluciones madre se diluyen posteriormente 50 veces en medio de cultivo (soluciones de trabajo). Si la concentración final máxima en la placa de 1 000 μg/ml no es tóxica, debe volver a determinarse la concentración máxima realizando un nuevo ensayo de citotoxicidad. La concentración final en la placa no debe exceder de 5 000 μg/ml en el caso de los productos problema disueltos o dispersos de forma estable en la solución salina o salina.
|
|
—
|
Con DMSO como disolvente/vehículo: Se preparan ocho soluciones madre (ocho concentraciones), mediante diluciones a la mitad en serie utilizando el disolvente o vehículo correspondiente. Estas soluciones madre se diluyen posteriormente 250 veces en medio de cultivo (soluciones de trabajo). la concentración final en la placa no debe exceder de 1 000 μg/ml, incluso aunque esta concentración no sea tóxica.
|
Las soluciones de trabajo se utilizan finalmente para la exposición añadiendo un volumen igual de solución de trabajo al volumen de la suspensión de células THP-1 en la placa (véase también el punto 17) para lograr otra dilución a la mitad (normalmente, el intervalo final de concentraciones en la placa es de 7,81-1 000 μg/ml).
|
|
16.
|
El control del disolvente o vehículo utilizado en el método h-CLAT es el medio de cultivo [en el caso de los productos problema disueltos o dispersos de forma estable (véase el punto 4), con medio o con solución salina] o el DMSO (en el caso de los productos problema disueltos o dispersos de forma estable en DMSO), y se somete a ensayo a una única concentración final en la placa del 0,2 %. Se somete al mismo proceso de dilución que se describe en el punto 15 para las soluciones de trabajo.
|
Aplicación de los productos problema y de las sustancias testigo
|
17.
|
El medio de cultivo o las soluciones de trabajo descritas en los puntos 15 y 16 se mezclan 1: 1 (v/v) con las suspensiones celulares preparadas en la placa de fondo plano de 24 pocillos o de 96 pocillos (véase el punto 12). A continuación, las placas tratadas se incuban durante 24 ± 0,5 horas a 37oC con 5 % de CO2. Deben tomarse precauciones para evitar la evaporación de los productos problema volátiles y la contaminación cruzada entre pocillos por productos problema, por ejemplo tapando la placa antes de la incubación con los productos problema (20).
|
Tinción con yoduro de propidio (PI)
|
18.
|
Tras 24 ± 0,5 horas de exposición, las células se transfieren a tubos de muestreo y se recogen por centrifugación. Los sobrenadantes se desechan y las células restantes se vuelven a suspender con 200 μl (en caso de 96 pocillos) o 600 μl (en caso de 24 pocillos) de una solución salina amortiguadora de fosfato que contenga un 0,1 % de seroalbúmina bovina (solución amortiguadora de tinción). Se transfieren 200 μl de suspensión celular a una placa de fondo redondo de 96 pocillos (en caso de 96 pocillos) o a un microtubo (en el caso de 24 pocillos) y se lavan dos veces con 200 μl (en caso de 96 pocillos) o 600 μl (en caso de 24 pocillos) de solución amortiguadora de tinción. Por último, las células se vuelven a suspender en solución amortiguadora de tinción (p. ej., 400 μl) y se añade la solución de PI (p. ej., 20 μl) (p. ej., la concentración final de PI es de 0,625 μg/ml). Pueden utilizarse otros marcadores de citotoxicidad, tales como la 7-aminoactinomicina D (7-AAD), azul tripano u otros, si se puede demostrar que las tinciones alternativas ofrecen resultados similares a los del PI, por ejemplo sometiendo a ensayo las sustancias para demostrar la competencia que figuran en el apéndice 1.2.
|
Medición de la citotoxicidad por citometría de flujo y estimación del valor de CV75
|
19.
|
La absorción del PI se analiza mediante citometría de flujo con el canal de medición FL-3. Se miden en total 10 000 células vivas (negativas para el PI). La viabilidad celular puede calcularse con la siguiente ecuación por el programa de análisis del citómetro. Cuando la viabilidad celular es baja, deben medirse hasta 30 000 células, incluidas las células muertas. Como alternativa, también es posible medir los datos durante un minuto tras el inicio del análisis.
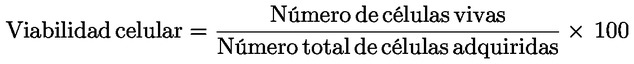
El valor de CV75 (véase el punto 13), es decir, la concentración que muestra el 75 % de supervivencia de las células THP-1 (25 % de citotoxicidad), se calcula mediante interpolación semilogarítmica con la siguiente ecuación:
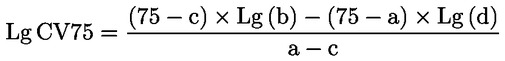
donde:
a es el valor mínimo de viabilidad celular superior al 75 %;
c es el valor máximo de viabilidad celular inferior al 75 %;
b y d son las concentraciones que muestran los valores de viabilidad celular a y c, respectivamente.

Pueden utilizarse otros métodos para obtener el valor de CV75 siempre que se demuestre que esto no afecta a los resultados (por ejemplo, sometiendo a ensayo las sustancias para demostrar la competencia).
|
Medición de la expresión de CD86/CD54
Preparación de los productos problema y de las sustancias testigo
|
20.
|
El disolvente o vehículo apropiado (solución salina, medio o DMSO; véase el punto 14). Los productos problema se diluyen primero a la concentración correspondiente a 100 veces (en caso de solución salina) o 500 veces (en caso de DMSO) la concentración 1,2 × CV75 determinada en el ensayo de determinación de la dosis (véase el punto 19). Si no puede determinarse la CV75 (es decir, si no se observa suficiente citotoxicidad en el ensayo de determinación de la dosis), debe utilizarse como concentración inicial la concentración más elevada soluble o dispersa de forma estable del producto problema preparado con cada disolvente o vehículo. Téngase en cuenta que la concentración final en la placa no debe superar los 5 000 μg/ml (en el caso de la solución salina o del medio) o los 1 000 μg/ml (en el caso del DMSO). A continuación, se utilizan diluciones en serie de factor 1,2 con el disolvente o vehículo correspondiente para obtener las soluciones madre [ocho concentraciones comprendidas entre 100 × 1,2 × CV75 y 100 × 0,335 × CV75 (con solución salina o medio) o entre 500 × 1,2 × CV75 y 500 × 0,335 × CV75 (con DMSO)] que deben someterse a ensayo con el método h-CLAT (véase en el protocolo DB-ALM, n.o 158 un ejemplo de sistema de dosificación). Las soluciones madre se diluyen posteriormente 50 veces (en caso de solución salina o medio) o 250 veces (en caso de DMSO) en el medio de cultivo (soluciones de trabajo). Estas soluciones de trabajo se utilizan finalmente para la exposición con otra dilución final a la mitad en la placa. Si los resultados no cumplen los criterios de aceptación descritos en los puntos 29 y 30 respecto a la viabilidad celular, podrá repetirse el ensayo de determinación de la dosis para determinar el valor de CV75 más precisamente. Téngase en cuenta que para la medición de la expresión de CD86/CD54 se pueden utilizar solo placas de 24 pocillos.
|
|
21.
|
El control del disolvente/vehículo se prepara como se describe en el punto 16. El testigo positivo utilizado en el método h-CLAT es el DNCB (véase el punto 11); se preparan las soluciones madre en DMSO y se diluyen como se describe en el punto 20 respecto a las soluciones madre. El DNCB debe utilizarse como testigo positivo para la medición de la expresión de CD86/CD54 en una única concentración final en la placa (por lo general, 4,0 μg/ml). Para obtener una concentración de 4,0 μg/ml de DNCB en la placa, se prepara una solución madre de 2 mg/ml de DNCB en DMSO, y se diluye después 250 veces con medio de cultivo hasta conseguir una solución de trabajo de 8 μg/ml. Alternativamente, la CV75 del DNCB, que se determina en cada laboratorio, también podría utilizarse como concentración de testigo positivo. Podrán utilizarse otros testigos positivos adecuados si se dispone de datos históricos para obtener criterios comparables de aceptación de tandas. Respecto a los testigos positivos, la concentración final única en la placa no debe superar los 5 000 μg/ml (en el caso de la solución salina o del medio) o los 1 000 μg/ml (en el caso del DMSO). Los criterios de aceptación de tandas son los mismos que los descritos para el producto problema (véase el punto 29), excepto en lo relativo al último criterio de aceptación, puesto que el testigo positivo se somete a ensayo a una sola concentración.
|
Aplicación de los productos problema y de las sustancias testigo
|
22.
|
Respecto a cada producto problema y sustancia testigo, se necesita un solo experimento para obtener una asignación. Cada experimento consiste en al menos dos tandas independientes para la medición de la expresión de CD86/CD54 (véanse los puntos 26-28). Cada tanda independiente se llevará a cabo en un día diferente o el mismo día, siempre que, respecto a cada una de las tandas: a) se preparen soluciones madre y soluciones de trabajo nuevas e independientes del producto problema y de los anticuerpos, y b) se utilicen células recolectadas de forma independiente (es decir, se recogen las células de distintos matraces de cultivo); no obstante, las células pueden proceder del mismo pase. Los productos problema y las sustancias testigo preparados como soluciones de trabajo (500 μl) se mezclan con 500 μl de células suspendidas (1 x 106 células) en la proporción 1: 1, y las células se incuban durante 24 ± 0,5 horas, como se describe en los puntos 20 y 21. En cada tanda, bastará una sola réplica de cada concentración del producto problema y de la sustancia testigo, ya que la clasificación se obtiene de al menos dos tandas independientes.
|
Tinción y análisis celular
|
23.
|
Después de 24 ± 0,5 horas de exposición, se transfieren las células de la placa de 24 pocillos a los tubos de muestreo, se recogen por centrifugación y, a continuación, se lavan dos veces con 1 ml de solución amortiguadora de tinción (en caso necesario, pueden aplicarse más etapas de lavado). Tras su lavado, las células se bloquean con 600 μl de solución de bloqueo [solución amortiguadora de tinción con 0,01 % (p/v) de globulina (fracciones II y III de Cohn, humanas; SIGMA, # G2388-10G o equivalente)] y se incuban a 4oC durante 15 min. Tras su bloqueo, las células se dividen en tres alícuotas de 180 μl en una placa de fondo redondo de 96 pocillos o microtubo.
|
|
24.
|
Tras su centrifugación, las células se tiñen con 50 μl de anticuerpos anti-CD86, anti-CD54 marcados con FITC o IgG1 (isotipo) de ratón a 4oC durante 30 min. Los anticuerpos descritos en el protocolo n.o 158 de la DB-ALM de h-CLAT (18) deben utilizarse mediante dilución 3: 25 v/v [para CD86 (BD-PharMingen, # 5556 57; Clone: Fun-1)] o 3: 50 v/v [para CD54 (DAKO, #F7143; Clone: 6.5B5) e IgG1 (DAKO, #X0927)] con solución amortiguadora de tinción. Los desarrolladores del ensayo definieron estos factores de dilución de los anticuerpos como los que proporcionan la mejor relación entre señal y ruido. Según la experiencia de los desarrolladores del ensayo, la intensidad de la fluorescencia de los anticuerpos suele ser coherente entre los diferentes lotes. No obstante, los usuarios pueden considerar la valoración de los anticuerpos en las condiciones de su propio laboratorio a fin de determinar las mejores concentraciones que han de utilizar. Podrán utilizarse otros anticuerpos marcados con fluorocromo antiCD86 y/o antiCD54 si se puede demostrar que proporcionan resultados similares a los de anticuerpos conjugados con FITC, por ejemplo sometiendo a ensayo las sustancias utilizadas para demostrar la competencia que se indican en el apéndice 1.2. Cabe señalar que la modificación del clon o del proveedor de los anticuerpos, tal como se recogen en el protocolo DB-ALM n.o 158 del h-CLAT (18), puede afectar a los resultados. Después de lavarlas dos veces o más con 150 μl de solución amortiguadora de tinción, las células se vuelven a suspender en esta solución amortiguadora de tinción (p. ej., 400 μl) y en la solución de PI (p. ej., 20 μl para obtener una concentración final de 0,625 μg/ml) o en la solución de otro marcador de citotoxicidad (véase el punto 18). Se analizan los niveles de expresión de CD86 y CD54, y la viabilidad celular, utilizando la citometría de flujo.
|
DATOS E INFORME
Evaluación de los datos
|
25.
|
La expresión de CD86 y CD54 se analiza mediante citometría de flujo con el canal de medida FL-1. Sobre la base de la media geométrica de la intensidad de la fluorescencia (MFI, mean fluorescence intensity), la intensidad de la fluorescencia relativa (RFI, relative fluorescence intensity) de CD86 y CD54 en el caso de las células testigo positivo (ctrl) y de las células tratadas con producto se calcula con arreglo a la ecuación siguiente:

La viabilidad celular de las células testigo de isotipo (ctrl) [que se tiñen con anticuerpos IgG1 (isotipo) de ratón] se calcula también con arreglo a la ecuación descrita en el punto 19.
|
Modelo de asignación
|
26.
|
Para la medición de la expresión de CD86/CD54, cada producto problema se somete al menos a dos tandas independientes a fin de obtener una única asignación (POSITIVA o NEGATIVA). La asignación del h-CLAT se considera POSITIVA si en 2 de 2, o en al menos 2 de 3, tandas independientes se cumple al menos una de las condiciones siguientes; de lo contrario, la asignación del h-CLAT se considera NEGATIVA (figura 1):
|
—
|
La RFI del CD86 es igual o superior al 150 % a cualquier concentración estudiada (con viabilidad celular ≥ 50 %);
|
|
—
|
La RFI del CD54 es igual o superior al 200 % a cualquier concentración estudiada (con viabilidad celular ≥ 50 %);
|
|
|
27.
|
Sobre la base de lo anterior, si las dos primeras tandas son positivas para el CD86 y/o son positivas para el CD54, la asignación del h-CLAT se considera POSITIVA y no es necesario realizar una tercera tanda. Del mismo modo, si las dos primeras tandas son negativas para ambos marcadores, la asignación del h-CLAT se considera NEGATIVA (teniendo debidamente en cuenta lo dispuesto en el punto 30) sin necesidad de una tercera tanda. Sin embargo, si las dos primeras tandas no son concordantes en relación al menos con uno de los marcadores (CD54 o CD86), se necesita una tercera tanda y la asignación final se basará en el resultado mayoritario de las tres tandas individuales (es decir, 2 de 3). A este respecto, hay que señalar que, si se llevan a cabo dos tandas independientes y una es solo positiva en el caso del CD86 (en lo sucesivo, P1) y la otro es solo positiva en el caso del CD54 (en lo sucesivo P2), se requiere una tercera tanda. Si esta tercera tanda es negativa para ambos marcadores (en lo sucesivo, N), la asignación del h-CLAT se considera NEGATIVA. Por otra parte, si la tercera tanda es positiva para uno de los dos marcadores (P1 o P2) o para ambos marcadores (en lo sucesivo, P12), la asignación del h-CLAT se considera POSITIVA.

Figura 1 Modelo de asignación utilizado en el método h-CLAT. Las asignaciones del h-CLAT deben interpretarse en el marco de un enfoque IATA y de conformidad con las disposiciones de los puntos 7 y 8 de la introducción general.
P1: tanda con solo CD86 positivo; P2: tanda con solo CD54 positivo; P12: tanda con CD86 y CD54 positivos; N: tanda sin CD86 ni CD54 positivos.
* Los recuadros muestran las combinaciones pertinentes de resultados de las dos primeras tandas, con independencia del orden en que puedan obtenerse.
# Los recuadros muestran las combinaciones pertinentes de resultados de las tres tandas sobre la base de los resultados obtenidos en las dos primeras, indicadas en el recuadro superior, pero no reflejan el orden en que puedan obtenerse.
|
|
28.
|
En el caso de los productos problema clasificados como POSITIVOS con el h-CLAT, opcionalmente se pueden determinar dos valores de concentraciones efectivas (CE), el valor de CE150 para el CD86 y el valor de CE200 para el CD54, es decir, la concentración a la que los productos problema inducen una RFI de 150 o de 200. Estos valores de CE podrían contribuir a la evaluación de la potencia sensibilizante (9) cuando se utilicen en enfoques integrados, como los enfoques IATA (4) (5) (6) (7) (8). Pueden calcularse con las siguientes ecuaciones:
EC 150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
donde:
Aconc es la concentración más baja, en μg/ml, con RFI > 150 (CD86) o 200 (CD54);
Bconc es la concentración más alta, en μg/ml, con RFI < 150 (CD86) o 200 (CD54);
ARFI es la RFI a la concentración más baja con RFI > 150 (CD86) o 200 (CD54);
BRFI es la RFI a la concentración más alta con RFI < 150 (CD86) o 200 (CD54).
Con el fin de obtener más precisamente los valores de CE150 y CE200, pueden ser necesarias tres tandas independientes para la medición de la expresión de CD86/CD54. Los valores finales de CE150 y CE200 se determinan a continuación como el valor mediano de las CE calculadas con las tres tandas independientes. Si solo dos de las tres tandas independientes cumplen los criterios de positividad (véanse los puntos 26 - 27), se adopta el valor de CE150 o CE200 más alto de los dos valores calculados.
|
Criterios de aceptación
|
29.
|
Cuando se use el método h-CLAT deben cumplirse los siguientes criterios de aceptación (22) (27).
|
—
|
Las viabilidades celulares de los controles de disolvente o vehículo y medio deben ser superiores al 90 %.
|
|
—
|
En el control del disolvente o vehículo, los valores de RFI tanto del CD86 como del CD54 no deben superar los criterios positivos (CD86 con RFI ≥ 150 % y CD54 con RFI ≥ 200 %). Los valores de RFI del control del disolvente o vehículo se calculan mediante la fórmula descrita en el punto 25 [MFI del producto debe sustituirse por MFI del disolvente/vehículo y MFI del disolvente/vehículo debe sustituirse por MFI del control (medio)].
|
|
—
|
Tanto en los controles de medio como en los de disolvente o vehículo, la proporción de la MFI tanto del CD86 como del CD54 respecto a la del testigo de isotipo deberá ser > 105 %.
|
|
—
|
En el testigo positivo (DNCB), los valores de RFI tanto del CD86 como del CD54 deben cumplir los criterios positivos (CD86 con RFI ≥ 150 y CD54 con RFI ≥ 200) y la viabilidad celular debe ser superior al 50 %.
|
|
—
|
En el caso del producto problema, la viabilidad celular debe ser superior al 50 % en al menos cuatro concentraciones sometidas a ensayo en cada tanda.
|
|
|
30.
|
Los resultados negativos son aceptables únicamente para los productos problema que presenten una viabilidad celular inferior al 90 % en la concentración más alta ensayada (es decir, 1,2 × CV75 según el sistema de diluciones en serie descrito en el punto 20). Si la viabilidad celular a 1,2 × CV75 es igual o superior al 90 %, debe descartarse el resultado negativo. En tal caso, se recomienda intentar afinar la selección de la dosis repitiendo la determinación de la CV75. Hay que señalar que, cuando se utiliza como concentración máxima de ensayo de un producto problema la concentración de 5 000 μg/ml en solución salina (o en medio o en otros disolventes/vehículos), la de 1 000 μg/ml en DMSO o la mayor concentración soluble, se puede aceptar un resultado negativo aunque la viabilidad celular sea superior al 90 %.
|
Informe del ensayo
|
31.
|
El informe del ensayo debe incluir la información siguiente:
|
Producto problema
|
Sustancias de un solo componente
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
Aspecto físico, log Kow, hidrosolubilidad, solubilidad en DMSO, peso molecular, y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Justificación de la elección del disolvente o vehículo para cada producto problema.
|
|
|
Sustancias de componentes múltiples, sustancias UVCB y mezclas
|
Caracterización, en la medida de lo posible, mediante, por ejemplo, la identidad química (véase más arriba), pureza, presencia cuantitativa y propiedades fisicoquímicas pertinentes de los componentes (véase más arriba), en la medida de lo posible;
|
|
Aspecto físico, hidrosolubilidad, solubilidad en DMSO y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible;
|
|
Peso molecular o peso molecular aparente en el caso de mezclas/polímeros de composición conocida u otra información pertinente para la realización del estudio;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Justificación de la elección del disolvente o vehículo para cada producto problema.
|
|
|
|
Testigos y controles
|
Testigo positivo
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
Aspecto físico, log Kow, hidrosolubilidad, solubilidad en DMSO, peso molecular, y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible y en los casos aplicables;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Referencia a los resultados de los testigos positivos históricos que demuestren unos criterios adecuados de aceptación de las tandas, si procede.
|
|
|
|
Testigo negativo y controles del disolvente o vehículo
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Aspecto físico, peso molecular, y propiedades fisicoquímicas pertinentes adicionales en el caso de que se utilicen unos controles de disolvente o vehículo distintos de los especificados en las directrices del ensayo y en la medida de lo posible;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Justificación de la elección del disolvente o vehículo para cada producto problema.
|
|
|
Condiciones del ensayo
|
Nombre y dirección del promotor, laboratorio y director del estudio;
|
|
Descripción del ensayo utilizado;
|
|
Línea celular utilizada, sus condiciones de conservación y su origen (por ejemplo, instalación en la que se ha obtenido);
|
|
Citometría de flujo utilizada (por ejemplo, modelo), incluidos los ajustes instrumentales, la globulina, los anticuerpos y el marcador de citotoxicidad utilizados;
|
|
Procedimiento utilizado para demostrar la competencia del laboratorio en la realización del ensayo mediante el ensayo de sustancias destinadas a tal fin, y procedimiento utilizado para demostrar que el ensayo tiene un comportamiento reproducible a lo largo del tiempo, por ejemplo datos de testigos históricos o datos históricos de controles de la reactividad.
|
|
|
Criterios de aceptación del ensayo
|
Valores de viabilidad celular, de MFI y de RFI obtenidos con el control del disolvente o vehículo en comparación con los intervalos de aceptación;
|
|
Valores de viabilidad celular y de RFI obtenidos con el testigo positivo en comparación con los intervalos de aceptación;
|
|
Viabilidad celular de todas las concentraciones sometidas a ensayo del producto problema.
|
|
|
Procedimiento de ensayo
|
Número de tandas utilizadas;
|
|
Concentraciones del producto problema, tiempo de aplicación y de exposición utilizado (si es diferente del recomendado);
|
|
Descripción de los criterios de evaluación y decisión seguidos;
|
|
Descripción de las eventuales modificaciones del procedimiento del ensayo.
|
|
|
Resultados
|
Tabulación de los datos, incluidos la CV75 (si procede), los distintos valores de la media geométrica de la intensidad de la fluorescencia, de la RFI y de la viabilidad celular, los valores de CE150/CE200 (si procede) obtenidos con el producto problema y con el testigo positivo en cada tanda, y una indicación de la calificación del producto problema según el modelo de asignación;
|
|
Descripción de cualesquiera otras observaciones pertinentes, si procede.
|
|
|
Discusión de los resultados
|
Discusión de los resultados obtenidos con el método h-CLAT;
|
|
Consideración de los resultados del ensayo en el contexto de un enfoque IATA, si se dispone de otra información pertinente.
|
|
|
BIBLIOGRAFÍA
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767-773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Puede consultarse en: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Puede consultarse en: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report Accessible at: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. Puede consultarse en: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OCDE (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organización para la Cooperación y el Desarrollo Económico, París (Francia), 2005, 96 pp.
|
|
(22)
|
OCDE (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment, No. 168. Disponible en: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
Naciones Unidas (2013). Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA). Quinta edición revisada. Nueva York & Ginebra: Publicaciones de las Naciones Unidas. ISBN: 978-92-1-117006-1. Disponible en: http://www.unece.org/es/trans/danger/publi/ghs/ghs_rev05/05files_s.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Apéndice 1.1
DEFINICIONES
Exactitud
: Grado de coincidencia entre los resultados del ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del ensayo y es un aspecto de su pertinencia. Este término y el de concordancia se suelen usar indistintamente para indicar la proporción de resultados correctos de un ensayo (21).
Ruta de resultados adversos (AOP, Adverse Outcome Pathway)
: Secuencia de fenómenos desde la estructura química de un producto diana o grupo de productos similares, pasando por el fenómeno molecular desencadenante, hasta un resultado in vivo de interés (22).
Producto
: Sustancia o mezcla.
CV75
: La concentración estimada que muestra una viabilidad celular del 75 %.
CE150
: Concentraciones que muestran un valor de RFI de 150 en la expresión del CD86.
CE200
: Concentraciones que muestran un valor de RFI de 200 en la expresión del CD54.
Citometría de flujo
: Técnica de citometría en la que las células suspendidas en un fluido fluyen una a una a través de un foco de luz excitadora, que se dispersa en patrones característicos según las células y sus componentes; las células están marcadas frecuentemente con marcadores fluorescentes, de modo que la luz se absorbe primero y, a continuación, se emite con una frecuencia modificada.
Peligro
: Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente.
IATA (Enfoque integrado de pruebas y evaluación, Integrated Approach to Testing and Assessment)
: Enfoque estructurado para la identificación del peligro (potencial), caracterización del peligro (potencia) o evaluación de la seguridad (potencial/potencia y exposición) de un producto o grupo de productos, que integra y pondera de forma estratégica todos los datos pertinentes para fundamentar una decisión normativa relativa a posibles peligros o riesgos o a la necesidad de realizar ensayos más específicos y, por tanto, mínimos.
Control del medio
: Réplica no tratada que contiene todos los componentes de un sistema de ensayo. Esta muestra se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras de control para determinar si el disolvente o vehículo interactúa con el sistema de ensayo.
Mezcla
: Mezcla o solución compuesta de dos o más sustancias.
Sustancias de un solo componente
: Sustancia, definida por su composición cuantitativa, en la que un solo componente principal representa al menos el 80 % (p/p).
Sustancia de componentes múltiples
: Sustancia, definida por su composición cuantitativa, en la que hay varios componentes principales presentes a una concentración ≥ 10 % (p/p) y < 80 % (p/p). Una sustancia de componentes múltiples es el resultado de un proceso de fabricación. La diferencia entre mezcla y sustancia de componentes múltiples es que una mezcla se obtiene mezclando dos o más sustancias sin reacción química. Una sustancia de componentes múltiples es el resultado de una reacción química.
Testigo positivo
: Réplica que contiene todos los componentes de un sistema de ensayo y que se trata con una sustancia de la que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta positiva.
Pre-haptenos
: Productos que pasan a ser sensibilizantes por una transformación abiótica.
Pro-haptenos
: Productos que requieren activación enzimática para ejercer su potencial de sensibilización cutánea.
Intensidad de fluorescencia relativa (RFI)
: Valores relativos de la media geométrica de la intensidad de la fluorescencia (MFI) de las células tratadas con el producto en comparación con la MFI de las células tratadas con disolvente o vehículo.
Pertinencia
: Descripción de la relación del ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un ensayo (21).
Fiabilidad
: Medida del grado en que un ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios (21).
Tanda
: Una tanda consta del ensayo de uno o más productos de forma simultánea con un control del disolvente o vehículo y con un testigo positivo.
Sensibilidad
: Proporción de todos los productos activos/positivos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un ensayo (21).
Solución amortiguadora de tinción
: Solución salina amortiguadora de fosfato con un 0,1 % de seroalbúmina bovina.
Control del disolvente o vehículo
: Muestra no tratada que contiene todos los componentes de un sistema de ensayo, excepto el producto problema, pero incluido el disolvente o vehículo utilizado. Se utiliza para establecer la respuesta de referencia para las muestras tratadas con el producto problema disuelto o disperso de forma estable en el mismo disolvente o vehículo. Cuando se somete a ensayo con un control del medio en paralelo, esta muestra pone de manifiesto también si el disolvente o vehículo interactúa con el sistema de ensayo.
Especificidad
: Proporción de todos los productos inactivos/negativos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un ensayo (21).
Sustancia
: Elemento químico y sus compuestos naturales u obtenidos mediante algún proceso de producción, incluidos los eventuales aditivos necesarios para mantener su estabilidad y las eventuales impurezas que se produzcan en el proceso, con exclusión de los eventuales disolventes que puedan separarse sin afectar a la estabilidad de la sustancia ni modificar su composición.
Producto problema
: Sustancia o mezcla estudiada con este método.
Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de las Naciones Unidas)
: Sistema que propone la clasificación de los productos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (23).
UVCB
: Sustancias de composición desconocida o variable, productos de reacción compleja o materiales biológicos.
Ensayo válido
: Ensayo del que se considera que tiene suficiente pertinencia y fiabilidad con un fin específico y que se basa en principios sólidos desde el punto de vista científico. Un ensayo nunca es válido en un sentido absoluto, sino únicamente en relación con un fin determinado (21).
Apéndice 1.2
SUSTANCIAS UTILIZADAS PARA DEMOSTRAR LA COMPETENCIA
Antes de proceder al uso sistemático del ensayo descrito en el presente apéndice del método B.71, los laboratorios deben demostrar su competencia técnica mediante la correcta obtención con el h.CLAT de la clasificación prevista de las diez sustancias recomendadas en el cuadro 1, y la obtención de valores de CV75, CE150 y CE200 que estén dentro del intervalo de referencia respectivo con al menos ocho de las diez sustancias utilizadas para demostrar la competencia. Estas sustancias de la prueba de la competencia se seleccionaron a fin de representar la gama de respuestas correspondientes a los peligros de sensibilización cutánea. Otros criterios de selección fueron que las sustancias estén disponibles en el mercado y que se disponga de datos de referencia in vivo de alta calidad, así como de datos in vitro de alta calidad obtenidos con el método h-CLAT. Asimismo, ha de disponerse de datos de referencia publicados sobre el método h-CLAT (3) (14).
Cuadro 1
Sustancias recomendadas para demostrar la competencia técnica con el método h-CLAT
|
Sustancias para la competencia
|
N.o CAS
|
Estado físico
|
Asignación in vivo
(103)
|
Intervalo de referencia de la CV75 en μg/ml (104)
|
Resultados con el h-CLAT sobre el CD86 (intervalo de referencia de la CE150 en μg/ml) (104)
|
Resultados con el h-CLAT sobre el CD54 (intervalo de referencia de la CE200 en μg/ml) (104)
|
|
2,4-Dinitroclorobenceno
|
97-00-7
|
Sólido
|
Sensibilizante (extremo)
|
2-12
|
Positivo (0,5-10)
|
Positivo (0,5-15)
|
|
4-Fenilendiamina
|
106-50-3
|
Sólido
|
Sensibilizante (fuerte)
|
5-95
|
Positivo (< 40)
|
Negativo (> 1,5) (105)
|
|
Sulfato de níquel
|
10101-97-0
|
Sólido
|
Sensibilizante (moderado)
|
30-500
|
Positivo (< 100)
|
Positivo (10-100)
|
|
2-Mercaptobenzotiazol
|
149-30-4
|
Sólido
|
Sensibilizante (moderado)
|
30-400
|
Negativo (> 10) (105)
|
Positivo (10-140)
|
|
R(+)-Limoneno
|
5989-27-5
|
Líquido
|
Sensibilizante (débil)
|
> 20
|
Negativo (> 5) (105)
|
Positivo (< 250)
|
|
Imidazolidinil-urea
|
39236-46-9
|
Sólido
|
Sensibilizante (débil)
|
25-100
|
Positivo (20-90)
|
Positivo (20-75)
|
|
Isopropanol
|
67-63-0
|
Líquido
|
No sensibilizante
|
> 5000
|
Negativo (> 5000)
|
Negativo (> 5000)
|
|
Glicerol
|
56-81-5
|
Líquido
|
No sensibilizante
|
> 5000
|
Negativo (> 5000)
|
Negativo (> 5000)
|
|
Ácido láctico
|
50-21-5
|
Líquido
|
No sensibilizante
|
1500-5000
|
Negativo (> 5000)
|
Negativo (> 5000)
|
|
Ácido 4-aminobenzoico
|
150-13-0
|
Sólido
|
No sensibilizante
|
> 1000
|
Negativo (> 1000)
|
Negativo (> 1000)
|
|
Abreviaturas: No CAS = número de registro del Chemical Abstracts Service.
|
Apéndice 2
SENSIBILIZACIÓN CUTÁNEA IN VITRO: ENSAYO DE ACTIVACIÓN DE LA LÍNEA CELULAR U937 (U-SENS™)
CONSIDERACIONES INICIALES Y LIMITACIONES
|
1.
|
El ensayo U-SENS™ cuantifica el cambio en la expresión de un marcador de superficie celular asociado al proceso de activación de los monocitos y de las células dendríticas (DC) (es decir, el CD86), en la línea celular del linfoma histiocítico humano U937, tras la exposición a sensibilizantes (1). Los niveles de expresión medidos del marcador de superficie celular CD86 en la línea celular U937 se utilizan a continuación para justificar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes.
|
|
2.
|
El ensayo U-SENS™ se ha evaluado en un estudio de validación (2) coordinado por L’Oréal y objeto posteriormente de revisión por pares independientes en el Comité Científico Consultivo (ESAC) del laboratorio de referencia de la Unión Europea para las alternativas a los ensayos con animales (EURL ECVAM) (3). Teniendo en cuenta todas las pruebas disponibles y las aportaciones de los reguladores y de las partes interesadas, el EURL ECVAM recomendó que el U-SENS™ se utilizara como parte de un enfoque IATA para justificar la discriminación entre sensibilizantes y no sensibilizantes a efectos de la clasificación de los peligros y el etiquetado (4). En su documento de orientación sobre la información de los enfoques estructurados relativos a la integración de datos y a las distintas fuentes de información utilizadas en los enfoques IATA en relación con la sensibilización cutánea, la OCDE examina actualmente una serie de estudios de casos que describen diferentes estrategias de ensayo y modelos de asignación. Uno de los diferentes enfoques definidos se basa en el ensayo U-SENS (5). En otras citas de la bibliografía (4) (5) (7) también se incluyen ejemplos de uso de los datos del ensayo U-SENS™ en combinación con otra información, incluidos datos históricos y datos humanos válidos ya existentes (6).
|
|
3.
|
Se ha demostrado que el ensayo U-SENS™ puede transferirse a laboratorios con experiencia en técnicas de cultivo celular y análisis de citometría de flujo. El nivel de reproducibilidad de las asignaciones que cabe esperar del ensayo es del orden del 90 % y del 84 % dentro de un laboratorio y entre laboratorios, respectivamente (8). Los resultados obtenidos en el estudio de validación (8) y otros estudios publicados (1) indican en general que, en comparación con los resultados del LLNA, la exactitud de la distinción de los sensibilizantes cutáneos (es decir, categoría 1 del SGA de las Naciones Unidas y del CLP) frente a los no sensibilizantes es del 86 % (N = 166), con una sensibilidad del 91 % (118/129) y una especificidad del 65 % (24/37). En comparación con los resultados obenidos con seres humanos, la exactitud de la distinción de los sensibilizantes cutáneos (es decir, categoría 1 del SGA de las Naciones Unidas y del CLP) frente a los no sensibilizantes es del 77 % (N = 101), con una sensibilidad del 100 % (58/58) y una especificidad del 47 % (20/43). Es más probable que las asignaciones negativas falsas obtenidas con el ensayo U-SENS™ en comparación con el LLNA afecten a los productos que muestran una potencia de sensibilización cutánea baja o moderada (es decir, subcategoría 1B del SGA de las Naciones Unidas y del CLP) que los productos que presentan una potencia de sensibilización cutánea elevada (es decir, subcategoría 1A del SGA de las Naciones Unidas y del CLP) (1) (8) (9). En conjunto, esta información indica la utilidad del ensayo U-SENS™ para contribuir a la identificación de los peligros de sensibilización cutánea. Sin embargo, los valores de la exactitud aquí indicados para el U-SENS™ utilizado como ensayo único solo son indicativos, ya que el ensayo debe considerarse en combinación con otras fuentes de información en el contexto de un enfoque IATA y de conformidad con las disposiciones de los puntos 7 y 8 de la introducción general. Además, a la hora de evaluar los métodos de ensayo de la sensibilización cutánea sin animales, debe tenerse en cuenta que el ensayo LLNA y otros ensayos con animales pueden no reflejar plenamente la situación en los seres humanos.
|
|
4.
|
Sobre la base de los datos actualmente disponibles, se ha demostrado que el ensayo U-SENS™ es aplicable a productos problema (incluidos los ingredientes de cosméticos, como los conservantes, tensioactivos, sustancias activas, colorantes, etc.) que abarcan una variedad de grupos funcionales orgánicos, de propiedades fisicoquímicas, de potencia de sensibilización cutánea (determinada en estudios in vivo) y el espectro de mecanismos de reacción de los que se sabe que están asociados con la sensibilización cutánea (como, por ejemplo, aceptor de Michael, formación de bases de Schiff, agentes de transferencia de grupos acilo, sustitución nucleofílica bi-molecular [SN2], o sustitución nucleofílica aromática [SNAr]) (1) (8) (9) (10). El ensayo U-SENS™ es aplicable a los productos problema que son solubles o que forman una dispersión estable (es decir, un coloide o una suspensión en la que el producto problema no sedimenta ni se separa del disolvente o vehículo para formar distintas fases) en un disolvente o vehículo adecuado (véase el punto 13). Los productos que en el conjunto de datos figuran como pre-haptenos (es decir, sustancias que se activan por oxidación) o pro-haptenos (es decir, sustancias que requieren activación enzimática, por ejemplo mediante enzimas P450) fueron clasificados correctamente por el ensayo U-SENS™ (1) (10). Las sustanciasque alteran la membrana pueden dar lugar a resultados positivos falsos debido a un aumento inespecífico de la expresión del CD86, ya que eran tensioactivos tres de los siete falsos positivos en relación con la clasificación de referencia in vivo (1). Tales resultados positivos con los tensioactivos deben considerarse con precaución, mientras que los resultados negativos con los tensioactivos podrían seguir utilizándose para apoyar la identificación del producto problema como no sensibilizante. Los productos problema fluorescentes pueden evaluarse con el ensayo U-SENS™ (1); sin embargo, los productos problema muy fluorescentes que emiten a la misma longitud de onda que el isotiocianato de fluoresceína (FITC) o que el yoduro de propidio (PI) pueden interferir con la detección por citometría de flujo y, por tanto, no pueden evaluarse correctamente utilizando anticuerpos conjugados con el FITC (posibles falsos negativos) ni con el PI (no puede medirse la viabilidad). En tal caso, pueden utilizarse otros anticuerpos marcados con fluorocromos u otros marcadores de citotoxicidad, según corresponda, siempre que se demuestre que proporcionan resultados similares a los de los anticuerpos marcados con FITC o PI (véase el punto 18), por ejemplo sometiendo a ensayo las sustancias utilizadas para la prueba de la competencia recogidas en el apéndice 2.2. A la luz de lo anterior, los resultados positivos obtenidos con tensioactivos y los resultados negativos obtenidos con fluorescentes fuertes deben interpretarse en el contexto de las limitaciones indicadas y junto con otras fuentes de información en el marco de los enfoques IATA. En los casos en que haya pruebas que demuestren la inaplicabilidad del ensayo U-SENS™ a otras categorías específicas de productos problema, no deberá utilizarse este ensayo con tales categorías específicas.
|
|
5.
|
Tal como se ha descrito anteriormente, el ensayo U-SENS™ sirve para justificar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes. No obstante, podrá contribuir también a la evaluación de la potencia sensibilizante cuando se utilice en enfoques integrados, como los enfoques IATA. Sin embargo, es necesario seguir trabajando, preferentemente a partir de datos con seres humanos, para determinar de qué modo los resultados del ensayo U-SENS™ pueden informar sobre la evaluación de la potencia.
|
|
6.
|
En el apéndice 2.1 se dan las definiciones pertinentes.
|
PRINCIPIO DEL ENSAYO
|
7.
|
El U-SENS™ es un ensayo in vitro que cuantifica los cambios de la expresión del marcador de superficie celular CD86 en las células U937 de la línea celular del linfoma histiocitico humano, tras una exposición de 45 ± 3 horas al producto problema. El marcador de superficie CD86 es un marcador típico de la activación de U937. Se sabe que el CD86 es una molécula coestimuladora que puede imitar la activación monocítica, la cual desempeña un papel fundamental en la estimulación de las células T. Los cambios en la expresión del marcador de superficie celular CD86 se miden recurriendo a la citometría de flujo tras la tinción celular normalmente mediante anticuerpos marcados con isotiocianato de fluoresceína (FITC). También se lleva a cabo simultáneamente una medición de la citotoxicidad (p. ej., utilizando PI) con el fin de evaluar si a concentraciones subcitotóxicas se produce un incremento de la expresión del marcador de superficie celular CD86. Se calcula el índice de estimulación (IE) del marcador de superficie celular CD86 en comparación con el control del disolvente o de vehículo y se utiliza en el modelo de asignación (véase el punto 19), a fin de justificar la discriminación entre sensibilizantes y no sensibilizantes.
|
DEMOSTRACIÓN DE LA COMPETENCIA
|
8.
|
Antes de proceder al uso sistemático del ensayo descrito en el presente apéndice del método de ensayo B.71, los laboratorios deben demostrar su competencia técnica, utilizando las diez sustancias para la prueba de la competencia que figuran en el apéndice 2.2, de conformidad con las buenas prácticas de los métodos in vitro (11). Por otra parte, los usuarios del ensayo deben mantener una base de datos históricos que contenga los generados en los controles de reactividad (véase el punto 11) y con los testigos positivos y los controles del disolvente o vehículo (véanse los puntos 15-16), y utilizar estos datos para confirmar que la reproducibilidad del ensayo en su laboratorio se mantiene a lo largo del tiempo.
|
PROCEDIMIENTO
|
9.
|
Este ensayo se basa en el protocolo n.o 183 de servicio de la base de datos de U-SENS™ sobre métodos alternativos a la experimentación con animales (DB-ALM) (12). Deben emplearse procedimientos normalizados de trabajo (PNT) a la hora de aplicar y utilizar el ensayo U-SENS™ en el laboratorio. Puede utilizarse un sistema automatizado para llevar a cabo el ensayo U-SENS™, si puede demostrarse que proporciona resultados similares, por ejemplo sometiendo a ensayo las sustancias utilizadas para la prueba de la competencia recogidas en el apéndice 2.2. A continuación se recoge una descripción de los principales componentes y procedimientos del ensayo U-SENS™.
|
Preparación de las células
|
10.
|
Para llevar a cabo el ensayo U-SENS™ debe utilizarse la línea celular del linfoma histiocítico humano, U937 (13). Las células (clon CRL1593.2) deben obtenerse de un banco de células adecuado, como la American Type Culture Collection.
|
|
11.
|
Se cultivan células U937 a 37 °C, en atmósfera humidificada y con un 5 % de CO2, en medio RPMI-1640 complementado con un 10 % de suero de ternera fetal (FCS), L-glutamina 2 mM, 100 unidades/ml de penicilina y 100 μg/ml de estreptomicina. Se hace un pase de las células U937 sistemáticamente cada 2-3 días a una densidad de 1,5 a 3 × 105 células/ml, respectivamente. La densidad celular no debe superar el valor de 2 × 106 células/ml y la viabilidad celular medida mediante la exclusión del azul tripano debe ser ≥ 90 % (no debe aplicarse al primer pase después de la descongelación). Antes de utilizarlos para el ensayo, cada lote de células, FCS o anticuerpos deben ser objeto de un control de reactividad. El control de reactividad de las células debe realizarse utilizando el testigo positivo, ácido picrilsulfónico (ácido 2,4,6-trinitro-bencenosulfónico, TNBS) (n.o CAS 2508-19-2, ≥ 99 % de pureza), y el testigo negativo, ácido láctico (LA) (n.o CAS 50-21-5, ≥ 85 % de pureza), al menos una semana después de la descongelación. En el control de reactividad, se deben someter a ensayo seis concentraciones finales de cada uno de los dos testigos (TNBS: 1, 12,5, 25, 50, 75, 100 μg/ml, y LA: 1, 10, 20, 50, 100, 200 μg/ml). El TNBS solubilizado en medio completo debe dar una respuesta del CD86 positiva y relacionada con la concentración (p. ej., cuando una concentración positiva, con IE de CD86 ≥ 150, va seguida de una concentración con un aumento del IE de CD86), y el LA solubilizado en medio completo debe dar una respuesta del CD86 negativa (véase el punto 21). Solo se puede utilizar para el ensayo el lote de células que haya superado dos veces el control de reactividad. Las células pueden propagarse hasta siete semanas después de la descongelación. El número de pases no debe ser superior a 21. El control de reactividad debe realizarse con arreglo a los procedimientos descritos en los puntos 18 a 22.
|
|
12.
|
Para el ensayo, se siembran células U937 a una densidad de 3 × 105 células/ml o 6 × 105 células/ml, y se precultivan en matraces de cultivo durante 2 días o durante 1 día, respectivamente. Podrán utilizarse otras condiciones de precultivo si se presenta una justificación científica suficiente y si puede demostrarse que proporcionan resultados similares, por ejemplo sometiendo a ensayo las sustancias para la prueba de la competencia recogidas en el apéndice 2.2. En el día del ensayo, se toman células del matraz de cultivo y se suspenden con medio de cultivo fresco a la concentración de 5 × 105 células/ml. A continuación, se distribuyen las células en una placa de fondo plano de 96 pocillos con 100 μl (densidad celular final de 0,5 × 105 células/pocillo).
|
Preparación de los productos problema y de las sustancias testigo
|
13.
|
La evaluación de la solubilidad se realiza antes del ensayo. Con este fin, los productos problema se disuelven o se dispersan de forma estable a una concentración de 50 mg/ml en medio completo como disolvente de primera opción o en dimetilsulfóxido (DMSO, ≥ 99 % de pureza) como vehículo/disolvente de segunda opción si el producto problema no es soluble en el disolvente o vehículo de medio completo. Para el ensayo, el producto problema se disuelve hasta una concentración final de 0,4 mg/ml en medio completo si el producto es soluble en este disolvente o vehículo. Si el producto es soluble únicamente en DMSO, se disuelve a una concentración de 50 mg/ml. Podrán utilizarse otros disolventes/vehículos distintos de los descritos anteriormente si se presenta una justificación científica suficiente. Debe tenerse en cuenta la estabilidad del producto problema en el disolvente o vehículo final.
|
|
14.
|
Los productos problema y las sustancias testigo se preparan el día del ensayo. Dado que no se realiza un ensayo de determinación de la dosis, para la primera tanda se deben ensayar seis concentraciones finales (1, 10, 20, 50, 100 y 200 μg/ml) en el disolvente o vehículo correspondiente, bien en medio completo o bien en DMSO al 0,4 % en medio. Para las tandas siguientes, a partir de las soluciones de los productos problema de 0,4 mg/ml en medio completo o de 50 mg/ml en DMSO, se preparan, al menos cuatro soluciones de trabajo (es decir, al menos cuatro concentraciones), utilizando el disolvente o vehículo correspondiente. Las soluciones de trabajo se utilizan finalmente para el tratamiento añadiendo un volumen igual de suspensión celular U937 (véase el punto 11) al volumen de la solución de trabajo en la placa para lograr una dilución adicional a la mitad (12). Las concentraciones (al menos cuatro concentraciones) para cualquier otra tanda se eligen sobre la base de los distintos resultados de todas las tandas anteriores (8). Las concentraciones finales utilizables son 1, 2, 3, 4, 5, 7,5, 10, 12,5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 y 200 μg/ml. La concentración máxima final es de 200 μg/ml. En caso de que se observe un valor positivo de CD86 a 1 μg/ml, se evalúa entonces a 0,1 μg/ml para encontrar la concentración del producto problema que no induce al CD86 por encima del umbral positivo. Con cada tanda, se calcula la CE150 (concentración a la que un producto alcanza el umbral positivo de CD86 del 150 %; véase elpunto 19) si se observa una relación positiva entre la respuesta del CD86 y la concentración. Cuando el producto problema induzca una respuesta positiva del CD86 no relacionada con la concentración, el cálculo de la CE150 podría no ser pertinente, como se describe en el protocolo n.o 183 de la DB-ALM de U-SENS™ (12). Con cada tanda, se calcula la CV70 (concentración a la que un producto alcanza el umbral de citotoxicidad del 70 %, véase el punto 19) siempre que sea posible (12). Para investigar el efecto de respuesta a la concentración como aumento de CD86, deben elegirse cualesquiera concentraciones de entre las utilizables, repartidas de forma uniforme entre la CE150 (o la mayor concentración no citotóxica negativa respecto al CD86) y la CV70 (o la mayor concentración permitida, es decir, 200 μg/ml). Se deben someter a ensayo como mínimo cuatro concentraciones por tanda, con un mínimo de dos concentraciones comunes con las de la tanda o tandas anteriores, con fines comparativos.
|
|
15.
|
El control del disolvente o vehículo utilizado en el ensayo U-SENS™ es el medio completo (en el caso de los productos problema solubilizados o dispersos de forma estable en medio completo) (véase el punto 4) o DMSO al 0,4 % en medio completo (en el caso de los productos problema solubilizados o dispersos de forma estable en DMSO).
|
|
16.
|
El testigo positivo utilizado en el ensayo U-SENS™ es el TNBS (véase el punto 11), preparado en el medio completo. El TNBS debe utilizarse como testigo positivo para la medición de la expresión de CD86 a una única concentración final en la placa (50 μg/ml) que da > 70 % de viabilidad celular. Para obtener una concentración de 50 μg/ml de TNBS en la placa, se prepara una solución madre de TNBS 1 M (es decir, 293 mg/ml) en medio completo, y se diluye después 2 930 veces con medio completo hasta conseguir una solución de trabajo de 100 μg/ml. El ácido láctico (LA, n.o CAS 50-21-5) debe utilizarse como testigo negativo a 200 μg/ml solubilizado en medio completo (a partir de una solución madre de 0,4 mg/ml). En cada placa de cada tanda se preparan tres réplicas del control de medio completo no tratado, del control del disolvente o del vehículo, y de los testigos positivos y negativos (12). Podrán utilizarse otros testigos positivos adecuados si se dispone de datos históricos para obtener criterios comparables de aceptación de tandas. Los criterios de aceptación de tandas son los mismos que los descritos para el producto problema (véase el punto 12).
|
Aplicación de los productos problema y de las sustancias testigo
|
17.
|
El control del disolvente o del vehículo o las soluciones de trabajo descritas en los puntos 14-16 se mezclan 1: 1 (v/v) con las suspensiones celulares preparadas en la placa de fondo plano de 96 pocillos (véase el punto 12). A continuación, las placas tratadas se incuban durante 45 ± 3 horas a 37oC con 5 % de CO2. Antes de la incubación, las placas se sellan con una membrana semipermeable, para evitar la evaporación de los productos problema volátiles y la contaminación cruzada entre células tratadas con productos problema (12).
|
Tinción celular
|
18.
|
Tras 45 ± 3 horas de exposición, las células se transfieren a una placa de microvaloración con forma de V y se recogen por centrifugación. La interferencia de la solubilidad se define como cristales o gotas observadas al microscopio a las 45 ± 3 horas después del tratamiento (antes de la tinción celular). Los sobrenadantes se desechan y las células restantes se lavan una vez con 100 μl de una solución salina amortiguadora de fosfato (PBS) enfriada con hielo y que contiene un 5 % de suero de ternera fetal (solución amortiguadora de tinción). Tras la centrifugación, se vuelven a suspender las células con 100 μl de solución amortiguadora de tinción y se tiñen con 5 μl (p. ej., 0,25 μg) de anticuerpos anti-CD86 marcados con FITC o IgG1 (isotipo) de ratón a 4oC durante 30 min protegidas de la luz. Deben utilizarse los anticuerpos descritos en el protocolo n.o 183 de la DB-ALM de U-SENS™ (12) (para CD86: BD-PharMingen, # 555 657 Clon: Fun-1, o Caltag/Invitrogen # MHCD8601 Clon: BU63; y para IgG1: BD-PharMingen #555 748, o Caltag/Invitrogen # GM4992). Según la experiencia de los desarrolladores del ensayo, la intensidad de la fluorescencia de los anticuerpos suele ser coherente entre los diferentes lotes. Para el ensayo podrán utilizarse otros clones o proveedores de anticuerpos que hayan superado el control de reactividad (véase el punto 11). No obstante, los usuarios pueden considerar la valoración de los anticuerpos en las condiciones de su propiolaboratorio a fin de determinar la mejor concentración que han de utilizar. Podrá utilizarse otro sistema de detección como, p. ej., anticuerpos marcados con fluorocromo antiCD86, si se puede demostrar que proporcionan resultados similares a los de anticuerpos conjugados con FITC, por ejemplo sometiendo a ensayo las sustancias utilizadas para demostrar la competencia que se indican en el apéndice 2.2. Después de lavar dos veces con 100 μl de solución amortiguadora de tinción y una vez con 100 μl de PBS enfriada con hielo, se vuelven a suspender las células en PBS enfriada con hielo (por ejemplo, 125 μl en caso de muestras analizadas manualmente tubo por tubo, o 50 μl si se utiliza una placa de auto-muestreador) y se añade una solución de PI (concentración final de 3 μg/ml). Pueden utilizarse otros marcadores de citotoxicidad, tales como la 7-aminoactinomicina D (7-AAD) o el azul tripano, si se puede demostrar que las tinciones alternativas ofrecen resultados similares a los del PI, por ejemplo sometiendo a ensayo las sustancias para demostrar la competencia que figuran en el apéndice 2.2.
|
Análisis por citometría de flujo
|
19.
|
Se analiza el nivel de expresión de CD86 y la viabilidad celular utilizando la citometría de flujo. Las células se representan dentro de una gráfica de puntos de tamaño (FSC) y granularidad (SSC) a escala logartítmica, a fin de identificar claramente a la población en una primera ventana R1 y eliminar los desechos. El objetivo es medir un total de 10 000 células en la ventana R1 para cada pocillo. Las células de la misma ventana R1 se representan dentro de una gráfica de puntos FL3 o FL4 / SSC. Las células viables se definen mediante la separación de una segunda ventana R2 que seleccione la población de células negativas para el yoduro de propidio (canal FL3 o FL4). La viabilidad celular puede calcularse con la siguiente ecuación por el programa de análisis del citómetro. Cuando la viabilidad celular es baja, podrán medirse hasta 20 000 células, incluidas las células muertas. Como alternativa, también es posible medir los datos durante un minuto tras el inicio del análisis.

A continuación se mide el porcentaje de células FL1-positivas entre estas células viables seleccionadas en R2 (dentro de R1). La expresión en la superficie celular del CD86 se analiza en una gráfica de puntos de FL1/SSC seleccionada en células viables (R2).
En el caso de los pocillos de medio completo / IgG1, el marcador de análisis se sitúa cerca de la población principal, de manera que los controles del medio completo tengan IgG1 dentro de la zona diana del 0,6 al 0,9 %.
La interferencia del color se define como un desplazamiento de la gráfica de puntos de IgG1 marcada con FITC (media geométrica del IE de IgG1 FL1 ≥ 150 %).
Los índices de estimulación (IE) del CD86 en las células del control (sin tratar o en un 0,4 % de DMSO) y en las células tratadas con producto se calculan de acuerdo con la ecuación siguiente:.

% de células del control sin tratar IgG1+: indicado como porcentaje de las células IgG1 FL1-positivas definidas con el marcador de análisis (intervalo aceptado de ≥ 0,6 % y < 1,5 %, véase el punto 22) entre las células viables sin tratar.
% de células control/tratadas IgG1+/CD86+: indicado como porcentaje de células IgG1/CD86 FL1-positivas medidas sin desplazar el marcador de análisis entre las células control/tratadas viables.
|
DATOS E INFORME
Evaluación de los datos
|
20.
|
En el ensayo U-SENS™ se calculan los parámetros siguientes: valor de CV70, es decir, concentración que muestra el 70 % de la supervivencia celular de U937 (30 % de citotoxicidad) y valor de EC150, es decir, la concentración a la que los productos problema han inducido un índice de estimulación (IE) de CD86 del 150 %.
CV70 se calcula por interpolación semilogarítmica mediante la ecuación siguiente:
CV70 = C1 + [(V1 - 70) / (V1 – V2) * (C2 – C1)]
donde:
V1 es el valor mínimo de viabilidad celular superior al 70 %;
V2 es el valor máximo de viabilidad celular inferior al 70 %;
C1 y C2 son las concentraciones que muestran los valores de viabilidad celular V1 y V2, respectivamente.

Pueden utilizarse otros enfoques para obtener el valor de CV70 siempre que se demuestre que esto no afecta a los resultados (por ejemplo, sometiendo a ensayo las sustancias para demostrar la competencia).
EC150 se calcula por interpolación semilogarítmica mediante la ecuación siguiente:
EC150 = C1 + [(150 – IE1) / (IE2 – IE1) * (C2 – C1)]
donde:
C1 es la concentración más alta en μg/ml con un IE de CD86 < 150 % (IE 1)
C2 es la concentración más baja en μg/ml con un IE de CD86 ≥ 150 % (IE 2).

Se calculan los valores de CE150 y CV70 según lo siguiente:
|
—
|
con cada tanda: los valores individuales de CE150 y CV70 se utilizan como herramientas para investigar el efecto de respuesta a la concentración como aumento del CD86 (véase el punto 14),
|
|
—
|
sobre la base de las viabilidades medias, se determina el valor global de CV70 (12),
|
|
—
|
sobre la base de los valores medios del IE de CD86, se determina la CE150 global del producto problema clasificado como POSITIVO con el U-SENS™ (véase el punto 21) (12).
|
|
Modelo de asignación
|
21.
|
Para la medición de la expresión de CD86, cada producto problema se somete en al menos dos concentraciones al menos a dos tandas independientes (realizadas en días diferentes) a fin de obtener una única asignación (POSITIVA o NEGATIVA).
|
—
|
La conclusión individual de una tanda de U-SENS™ se considera negativa (en lo sucesivo, "N") si el IE de CD86 es inferior al 150 % en todas las concentraciones no citotóxicas (viabilidad celular ≥ 70 %) y si no se observa interferencia [citotoxicidad, solubilidad (véase el punto 18) o color (véase el punto 19), con independencia de las concentraciones no citotóxicas a las que se detecte la interferencia]. En todos los demás casos (con el IE de CD86 superior o igual al 150 % y/o con interferencias observadas), la conclusión individual de una tanda U-SENS™ se considera positiva (en lo sucesivo, "P").
|
|
—
|
Una asignación U-SENS™ se considera NEGATIVA si al menos dos tandas independientes son negativas (N) (figura 1). Si las dos primeras tandas son negativas (N), la asignación de U-SENS™ se considera NEGATIVA y no es necesario realizar la tercera tanda.
|
|
—
|
Una asignación U-SENS™ se considera POSITIVA si al menos dos tandas independientes son positivas (P) (figura 1). Si las dos primeras tandas son positivas (P), la asignación de U-SENS™ se considera POSITIVA y no es necesario realizar la tercera tanda.
|
|
—
|
Dado que no se ha realizado un ensayo de determinación de la dosis, hay una excepción si, en la primera tanda, el IE de CD86 es superior o igual al 150 % únicamente a la concentración no citotóxica más alta. La tanda se considera entonces NO CONCLUYENTE (NC), y deben someterse a ensayo, en tandas adicionales, otras concentraciones (entre la concentración no citotóxica más alta y la concentración citotóxica más baja, véase el punto 20). En caso de que una tanda se identifique como NC, deberán realizarse al menos dos tandas adicionales, y una cuarta tanda en caso de que las tandas 2 y 3 no sean concordantes (N y/o P independientemente) (figura 1). Las tandas de seguimiento se considerarán positivas aunque solo una concentración no citotóxica dé un CD86 igual o superior al 150 %, ya que la concentración se ha ajustado para el producto problema específico. La asignación final se basará en el resultado mayoritario de las tres o cuatro tandas individuales (es decir, 2 de 3 o 2 de 4) (figura 1).
|

Figura 1 Modelo de asignación utilizado en el ensayo U-SENS™. Las asignaciones obtenidas con el U-SENS™ deben interpretarse en el marco de un enfoque IATA y de conformidad con las disposiciones del punto 4 y de los puntos 7,8 y 9 de la introducción general.
N: Tanda sin observación de resultado positivo del CD86 o de interferencia;
P: Tanda con observación de resultado positivo del CD86 y/o de interferencia o interferencias;
NC: No concluyente. Primera tanda no concluyente, cuando CD86 es positivo solo a la concentración no citotóxica más alta;
#: Un resultado individual no concluyente (NC) atribuido únicamente a la primera tanda implica automáticamente la necesidad de efectuar una tercera tanda para alcanzar una mayoría de resultados positivos (P) o negativos (N) en al menos 2 de 3 tandas independientes.
$: Los recuadros muestran las combinaciones pertinentes de resultados de las tres tandas sobre la base de los resultados obtenidos en las dos primeras, indicadas en el recuadro superior.
°: Los recuadros muestran las combinaciones pertinentes de resultados de las cuatro tandas sobre la base de los resultados obtenidos en las tres primeras, indicadas en el recuadro superior.
|
Criterios de aceptación
|
22.
|
Cuando se use el ensayo U-SENS™ deben cumplirse los siguientes criterios de aceptación (12).
|
—
|
Al final del período de exposición de 45 ± 3 horas, la viabilidad media de las células U937 sin tratar en triplicado ha de ser superior al 90 % y no debe observarse ninguna deriva en la expresión de CD86. La expresión basal de CD86 de las células U937 sin tratar debe estar comprendida entre ≥ 2 % y ≤ 25 %.
|
|
—
|
Cuando se utiliza el DMSO como disolvente, la validez del control del vehículo DMSO se evalúa calculando un valor del IE con DMSO comparado con el de las células no tratadas, y la viabilidad media de las células en triplicado debe ser superior al 90 %. El control del vehículo DMSO es válido si la media de los tres valores del IE de CD86 con él es inferior al 250 % de la media de los tres valores del IE de CD86 de las células U937 sin tratar.
|
|
—
|
Las tandas se consideran válidas si al menos dos de los tres valores de IgG1 de células de U937 sin tratar caen dentro del intervalo de ≥ 0,6 % y < 1,5 %.
|
|
—
|
El testigo negativo (ácido láctico) en paralelo se considera válido si al menos dos de las tres réplicas son negativas (IE de CD86 < 150 %) y no citotóxicas (viabilidad celular ≥ 70 %).
|
|
—
|
El testigo positivo (TNBS) se considera válido si al menos dos de las tres réplicas son positivas (IE de CD86 ≥ 150 %) y no citotóxicas (viabilidad celular ≥ 70 %).
|
|
Informe del ensayo
|
23.
|
El informe del ensayo debe incluir la información siguiente:
|
Sustancia problema
Sustancias de un solo componente
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
Aspecto físico, solubilidad en el medio completo, solubilidad en DMSO, peso molecular, y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Justificación de la elección del disolvente o vehículo para cada producto problema.
|
Sustancias de componentes múltiples, sustancias UVCB y mezclas:
|
Caracterización, en la medida de lo posible, mediante, por ejemplo, la identidad química (véase más arriba), pureza, presencia cuantitativa y propiedades fisicoquímicas pertinentes de los componentes (véase más arriba), en la medida de lo posible;
|
|
Aspecto físico, solubilidad en el medio completo, solubilidad en DMSO y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible;
|
|
Peso molecular o peso molecular aparente en el caso de mezclas/polímeros de composición conocida u otra información pertinente para la realización del estudio;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Justificación de la elección del disolvente o vehículo para cada producto problema.
|
|
|
Testigos y controles
Testigo positivo
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
Aspecto físico, solubilidad en DMSO, peso molecular y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible y en los casos aplicables;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Referencia a los resultados de los testigos positivos históricos que demuestren unos criterios adecuados de aceptación de las tandas, si procede.
|
Testigo negativo y control del disolvente o vehículo
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Aspecto físico, peso molecular, y propiedades fisicoquímicas pertinentes adicionales en el caso de que se utilicen unos controles de disolvente o vehículo distintos de los especificados en las directrices del ensayo y en la medida de lo posible;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Justificación de la elección del disolvente o vehículo para cada producto problema.
|
|
|
Condiciones de ensayo
|
Nombre y dirección del promotor, laboratorio y director del estudio;
|
|
Descripción del ensayo utilizado;
|
|
Línea celular utilizada, sus condiciones de conservación y su origen (por ejemplo, instalación en la que se ha obtenido);
|
|
Citometría de flujo utilizada (por ejemplo, modelo), incluidos los ajustes instrumentales, los anticuerpos y el marcador de citotoxicidad utilizados;
|
|
Procedimiento utilizado para demostrar la competencia del laboratorio en la realización del ensayo mediante el ensayo de sustancias destinadas a tal fin, y el procedimiento utilizado para demostrar que el ensayo tiene un comportamiento reproducible a lo largo del tiempo, por ejemplo datos de testigos históricos o datos históricos de controles de la reactividad.
|
|
|
Criterios de aceptación del ensayo
|
Valores de la viabilidad celular y del IE de CD86 obtenidos con el control del disolvente o vehículo en comparación con los intervalos de aceptación;
|
|
Valores de la viabilidad celular y del IE obtenidos con el testigo positivo en comparación con los intervalos de aceptación;
|
|
Viabilidad celular de todas las concentraciones sometidas a ensayo del producto problema.
|
|
|
Procedimiento de ensayo
|
Número de tandas utilizadas;
|
|
Concentraciones del producto problema, tiempo de aplicación y de exposición utilizado (si es diferente del recomendado);
|
|
Duración de la exposición;
|
|
Descripción de los criterios de evaluación y decisión seguidos;
|
|
Descripción de las eventuales modificaciones del procedimiento del ensayo.
|
|
|
Resultados
|
Tabulación de los datos, incluidos la CV70 (si procede), el IE, los valores de viabilidad celular, los valores de CE150 (si procede) obtenidos con el producto problema y con el testigo positivo en cada tanda, y una indicación de la calificación del producto problema según el modelo de asignación;
|
|
Descripción de cualesquiera otras observaciones pertinentes, si procede.
|
|
|
Discusión de los resultados
|
Discusión de los resultados obtenidos con el ensayo U-SENS™;
|
|
Consideración de los resultados del ensayo obtenidos en el contexto de un enfoque IATA, si se dispone de otra información pertinente.
|
|
Conclusiones
|
BIBLIOGRAFÍA
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Puede consultarse en: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2787/815737. Disponible en: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2760/588955. Disponible en: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OCDE (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1683-1696.
|
|
(11)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33 pp. Puede consultarse en: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OCDE (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
Naciones Unidas (2015). Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York & Geneva: Publicaciones de las Naciones Unidas. Disponible en: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OCDE (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. Disponible en: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Apéndice 2.1
DEFINICIONES
Exactitud
: Grado de coincidencia entre los resultados del ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del ensayo y es un aspecto de su pertinencia. Este término y el de concordancia se suelen usar indistintamente para indicar la proporción de resultados correctos de un ensayo (14).
Ruta de resultados adversos (AOP, Adverse Outcome Pathway)
: Secuencia de fenómenos desde la estructura química de un producto diana o grupo de productos similares, pasando por el fenómeno molecular desencadenante, hasta un resultado in vivo de interés (15).
Respuesta del CD86 a la concentración
: Se produce dependencia de la concentración (o respuesta a la concentración) cuando una concentración positiva (con IE de CD86 ≥ 150) va seguida de una concentración con un aumento del IE de CD86.
Producto
: Sustancia o mezcla.
CV70
: Concentración estimada que muestra una viabilidad celular del 70 %.
Deriva
: La deriva se define por lo siguiente i) el valor corregido del % CD86+ de la réplica 3 del testigo no tratado es inferior al 50 % de la media del valor corregido del % CD86+ de las réplicas 1 y 2 de los testigos no tratados; y ii) el valor corregido del % CD86+ de la réplica 3 del testigo negativo es inferior al 50 % de la media del valor corregido del % CD86+ de las réplicas 1 y 2 de los testigos negativos.
CE150
: Concentraciones estimadas que muestran un IE de expresión del CD86 del 150 %.
Citometría de flujo
: Técnica de citometría en la que las células suspendidas en un fluido fluyen una a una a través de un foco de luz excitadora, que se dispersa en patrones característicos según las células y sus componentes; las células están marcadas frecuentemente con marcadores fluorescentes, de modo que la luz se absorbe primero y, a continuación, se emite con una frecuencia modificada.
Peligro
: Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente.
IATA (Enfoque integrado de pruebas y evaluación, Integrated Approach to Testing and Assessment)
: Enfoque estructurado para la identificación del peligro (potencial), caracterización del peligro (potencia) o evaluación de la seguridad (potencial/potencia y exposición) de un producto o grupo de productos, que integra y pondera de forma estratégica todos los datos pertinentes para fundamentar una decisión normativa relativa a posibles peligros o riesgos o a la necesidad de realizar ensayos más específicos y, por tanto, mínimos.
Mezcla
: Mezcla o solución compuesta de dos o más sustancias.
Sustancias de un solo componente
: Sustancia, definida por su composición cuantitativa, en la que un solo componente principal representa al menos el 80 % (p/p).
Sustancia de componentes múltiples
: Sustancia, definida por su composición cuantitativa, en la que hay varios componentes principales presentes a una concentración ≥ 10 % (p/p) y < 80 % (p/p). Una sustancia de componentes múltiples es el resultado de un proceso de fabricación. La diferencia entre mezcla y sustancia de componentes múltiples es que una mezcla se obtiene mezclando dos o más sustancias sin reacción química. Una sustancia de componentes múltiples es el resultado de una reacción química.
Testigo positivo
: Réplica que contiene todos los componentes de un sistema de ensayo y que se trata con una sustancia de la que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta positiva.
Pre-haptenos
: Productos que pasan a ser sensibilizantes por una transformación abiótica, p. ej. mediante oxidación.
Pro-haptenos
: Productos que requieren activación enzimática para ejercer su potencial de sensibilización cutánea.
Pertinencia
: Descripción de la relación del ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un ensayo (14).
Fiabilidad
: Medida del grado en que un ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios (14).
Tanda
: Una tanda consta del ensayo de uno o más productos de forma simultánea con un control del disolvente o vehículo y con un testigo positivo.
Sensibilidad
: Proporción de todos los productos activos/positivos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un ensayo (14).
IE
: Índice de estimulación. Valores relativos de la media geométrica de la intensidad de la fluorescencia de las células tratadas con el producto en comparación con las células tratadas con disolvente.
Control del disolvente o vehículo
: Muestra no tratada que contiene todos los componentes de un sistema de ensayo, excepto el producto problema, pero incluido el disolvente o vehículo utilizado. Se utiliza para establecer la respuesta de referencia para las muestras tratadas con el producto problema disuelto o disperso de forma estable en el mismo disolvente o vehículo. Cuando se somete a ensayo con un control del medio en paralelo, esta muestra pone de manifiesto también si el disolvente o vehículo interactúa con el sistema de ensayo.
Especificidad
: Proporción de todos los productos inactivos/negativos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un ensayo (14).
Solución amortiguadora de tinción
: Solución salina amortiguadora de fosfato con un 5 % de suero de ternera fetal.
Sustancia
: Elemento químico y sus compuestos naturales u obtenidos mediante algún proceso de producción, incluidos los eventuales aditivos necesarios para mantener su estabilidad y las eventuales impurezas que se produzcan en el proceso, con exclusión de los eventuales disolventes que puedan separarse sin afectar a la estabilidad de la sustancia ni modificar su composición;
Producto problema
: Sustancia o mezcla estudiada con este ensayo.
Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de las Naciones Unidas)
: Sistema que propone la clasificación de los productos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (16).
UVCB
: Sustancias de composición desconocida o variable, productos de reacción compleja o materiales biológicos.
Ensayo válido
: Ensayo del que se considera que tiene suficiente pertinencia y fiabilidad con un fin específico y que se basa en principios sólidos desde el punto de vista científico. Un ensayo nunca es válido en un sentido absoluto, sino únicamente en relación con un fin determinado (14).
Apéndice 2.2
SUSTANCIAS UTILIZADAS PARA DEMOSTRAR LA COMPETENCIA
Antes de proceder al uso sistemático del ensayo descrito en el presente apéndice del método B.71, los laboratorios deben demostrar su competencia técnica mediante la correcta obtención con el U-SENS™ de la clasificación prevista de las diez sustancias recomendadas en el cuadro 1, y la obtención de valores de CV70 y CE150 que estén dentro del intervalo de referencia respectivo con al menos ocho de las diez sustancias utilizadas para demostrar la competencia. Estas sustancias para demostrar la competencia se han seleccionado a fin de representar la gama de respuestas correspondientes a los peligros de sensibilización cutánea. Otros criterios de selección fueron que las sustancias estén disponibles en el mercado y que se disponga de datos de referencia in vivo de alta calidad, así como de datos in vitro de alta calidad obtenidos con el ensayo U-SENS™. Asimismo, ha de disponerse de datos de referencia publicados sobre el ensayo U-SENS™ (1) (8).
Cuadro 1
Sustancias recomendadas para demostrar la competencia técnica con el ensayo U-SENS™
|
Sustancias para la competencia
|
N.o CAS
|
Estado físico
|
Asignación in vivo
(106)
|
Disolvente/Vehículo de U-SENS™
|
Intervalo de referencia de CV70 en μg/ml (107) de U-SENS™
|
Intervalo de referencia de CE150 en μg/ml (107) de U-SENS™
|
|
4-Fenilendiamina
|
106-50-3
|
Sólido
|
Sensibilizante (fuerte)
|
Medio completo (108)
|
< 30
|
Positivo (≤ 10)
|
|
Ácido picrilsulfónico
|
2508-19-2
|
Líquido
|
Sensibilizante (fuerte)
|
Medio completo
|
> 50
|
Positivo (≤ 50)
|
|
Maleato de dietilo
|
141-05-9
|
Líquido
|
Sensibilizante (moderado)
|
DMSO
|
10-100
|
Positivo (≤ 20)
|
|
Resorcinol
|
108-46-3
|
Sólido
|
Sensibilizante (moderado)
|
Medio completo
|
> 100
|
Positivo (≤ 50)
|
|
Alcohol cinámico
|
104-54-1
|
Sólido
|
Sensibilizante (débil)
|
DMSO
|
> 100
|
Positivo (10-100)
|
|
4-Alilanisol
|
140-67-0
|
Líquido
|
Sensibilizante (débil)
|
DMSO
|
> 100
|
Positivo (< 200)
|
|
Sacarina
|
81-07-2
|
Sólido
|
No sensibilizante
|
DMSO
|
> 200
|
Negativo (> 200)
|
|
Glicerol
|
56-81-5
|
Líquido
|
No sensibilizante
|
Medio completo
|
> 200
|
Negativo (> 200)
|
|
Ácido láctico
|
50-21-5
|
Líquido
|
No sensibilizante
|
Medio completo
|
> 200
|
Negativo (> 200)
|
|
Ácido salicílico
|
69-72-7
|
Sólido
|
No sensibilizante
|
DMSO
|
> 200
|
Negativo (> 200)
|
|
Abreviaturas: No CAS = número de registro del Chemical Abstracts Service.
|
Apéndice 3
SENSIBILIZACIÓN CUTÁNEA IN VITRO: ENSAYO IL-8 LUC
CONSIDERACIONES INICIALES Y LIMITACIONES
|
1.
|
A diferencia de los ensayos que analizan la expresión de los marcadores de superficie celular, el ensayo IL-8 Luc cuantifica los cambios en la expresión de la IL-8, que es una citocina asociada a la activación de células dendríticas (DC). En la línea celular con el marcador IL-8 derivada de la THP-1 (THP-G8, establecida a partir de la línea celular de la leucemia monocítica aguda humana, THP-1), se mide la expresión de la IL-8 tras la exposición a los sensibilizantes (1). La expresión de la luciferasa se utiliza entonces para ayudar a discriminar entre los sensibilizantes cutáneos y los no sensibilizantes.
|
|
2.
|
Se ha evaluado el ensayo IL-8 Luc en un estudio de validación (2) realizado por el Centro Japonés para la Validación de Métodos Alternativos (JaCVAM, Japanese Centre for the Validation of Alternatives Methods), el Ministerio de Economía, Comercio e Industria (METI, Ministry of Economy, Trade and Industry) y la Sociedad Japonesa de Alternativas a Experimentos con Animales (JSAAE, Japanese Society for Alternatives to Animal Experiments) y posteriormente se ha sometido a una revisión independiente por pares (3) bajo los auspicios del JaCVAM y el Ministerio de Salud, Trabajo y Bienestar (MHLW), con el apoyo de la Cooperación Internacional sobre Métodos Alternativos de Ensayo (ICATM, International Cooperation on Alternative Test Methods). Teniendo en cuenta todas las pruebas disponibles y las aportaciones de los reguladores y de las partes interesadas, se considera que el ensayo IL-8 Luc es útil como parte de un enfoque IATA para justificar la discriminación entre sensibilizantes y no sensibilizantes a efectos de la clasificación de los peligros y el etiquetado. En la bibliografía se recogen ejemplos del uso de datos del ensayo IL-8 Luc en combinación con otra información (4) (5) (6).
|
|
3.
|
El ensayo con IL-8 IL- ha demostrado ser transferible a los laboratorios con experiencia en el cultivo celular y la medición de la luciferasa. Las reproducibilidades dentro de un laboratorio y entre laboratorios eran del 87,7 % y del 87,5 %, respectivamente (2). Los resultados obtenidos en el estudio de validación (2) y otras obras publicadas (1) (6) indican que, frente al LLNA, el ensayo IL-8 Luc consideró que 118 de 143 productos eran positivos o negativos y que 25 productos no eran concluyentes, y que la exactitud del ensayo IL-8 Luc en cuanto a la distinción entre sensibilizantes cutáneos (categoría 1 del SGA de las Naciones Unidas y del CLP) y no sensibilizantes (sin categoría del SGA de las Naciones Unidas y del CLP) es del 86 % (101/118) con una sensibilidad del 96 % (92/96) y una especificidad del 41 % (9/22). Excluidas las sustancias que no están en el ámbito de aplicabilidad que se describe más abajo (punto 5), el ensayo IL-8 Luc consideró positivos o negativos 113 de 136 productos y consideró no concluyentes 23 productos, y la exactitud del ensayo IL-8 Luc es del 89 % (101/113), con una sensibilidad del 96 % (92/96) y una especificidad del 53 % (9/17). Utilizando los datos humanos citados en Urbisch et al. (7), el ensayo IL-8 Luc consideró positivos o negativos 76 de 90 productos, y consideró no concluyentes 14 productos, y la exactitud es del 80 % (61/76), la sensibilidad es del 93 % (54/58) y la especificidad es del 39 % (7/18). Excluidas las sustancias que no están en el ámbito de aplicabilidad, el ensayo IL-8 Luc consideró positivos o negativos 71 de 84 productos y consideró no concluyentes 13 productos, y la exactitud es del 86 % (61/71), con una sensibilidad del 93 % (54/58) y una especificidad del 54 % (7/13). Es más probable que se den asignaciones negativas falsas con el ensayo IL-8 Luc en relación con los productos que muestran una potencia de sensibilización cutánea baja o moderada (es decir, subcategoría 1B del SGA de las Naciones Unidas y del CLP) que con los productos que presentan una potencia elevada (es decir, subcategoría 1A del SGA de las Naciones Unidas y del CLP) (6). En conjunto, la información avala la función del ensayo IL-8 Luc para identificar los peligros de sensibilización cutánea. Los valores de la exactitud aquí indicados para el ensayo IL-8 Luc utilizado como ensayo único solo son indicativos, ya que el ensayo debe considerarse en combinación con otras fuentes de información en el contexto de un enfoque IATA y de conformidad con las disposiciones de los puntos 7 y 8 de la introducción general. Además, a la hora de evaluar los ensayos de la sensibilización cutánea sin animales, debe tenerse en cuenta que el ensayo LLNA y otros ensayos con animales pueden no reflejar plenamente la situación en los seres humanos.
|
|
4.
|
Sobre la base de los datos actualmente disponibles, se ha demostrado que el ensayo IL-8 Luc es aplicable a productos problema que abarcan diversos grupos funcionales orgánicos, mecanismos de reacción, potencias de sensibilización cutánea (determinada en estudios in vivo) y propiedades fisicoquímicas (2) (6).
|
|
5.
|
Aunque el ensayo IL-8 Luc utiliza X-VIVO™ 15 como disolvente, con él se han evaluado correctamente productos con un log Kow > 3,5 y los que tienen una hidrosolubilidad de alrededor de 100 μg/ml, según lo calculado por el EPI SuiteTM, y su comportamiento para detectar sensibilizantes con baja hidrosolubilidad es superior al del ensayo IL-8 Luc con dimetilsulfóxido (DMSO) como disolvente (2). Sin embargo, los resultados negativos de los productos problema que no se disuelven a 20 mg/ml pueden ser resultados negativos falsos debido a su incapacidad de disolverse en X-VIVOTM 15. Por tanto, no deben tenerse en cuenta los resultados negativos de estos productos. En el estudio de validación se observó una elevada tasa de falsos resultados negativos. Por otra parte, debido a la limitada capacidad metabólica de la línea celular (8) y a las condiciones experimentales, pueden dar resultados negativos en el ensayo los pro-haptenos (sustancias que requieren activación metabólica) y los pre-haptenos (sustancias activadas por oxidación con el aire). Sin embargo, aunque deben interpretarse con cautela los resultados negativos de posibles pre/pro-haptenos, el ensayo IL-8 Luc ha considerado correctamente 11 de 11 pre-haptenos, 6/6 pro-haptenos y 6/8 pre/pro-haptenos en el conjunto de datos del ensayo con IL-8 Luc (2). Sobre la base de la reciente revisión global de tres ensayos sin animales (el DPRA, el KeratinoSens™ y el h-CLAT) para detectar pre y pro-haptenos (9), y sobre la base del hecho de que las células THP-G8 utilizadas en el ensayo IL-8 Luc son una línea celular derivada de la THP-1 que se utiliza en el ensayo h-CLAT, el ensayo IL-8 Luc puede también contribuir a aumentar la sensibilidad de los ensayos sin animales a fin de detectar pre y pro-haptenos en la combinación de otros ensayos. Los tensioactivos sometidos a ensayo hasta ahora han dado resultados positivos (falsos) independientemente de su tipo (por ejemplo, catiónicos, aniónicos o no iónicos). Por último, los productos que interfieren con la luciferasa pueden confundir su actividad/medición, provocando una mayor luminiscencia o una inhibición aparente (10). Por ejemplo, se ha informado de que unas concentraciones de fitoestrógenos superiores a 1 μM interfieren con las señales de luminiscencia en otros ensayos de luciferasa con gen marcador debido a la sobreactivación del gen marcador de la luciferasa. En consecuencia, debe examinarse detenidamente la expresión de la luciferasa obtenida a altas concentraciones de fitoestrógenos o de compuestos sospechosos de provocar la activación fitoestrogenoide del gen marcador de la luciferasa (11). Sobre la base de lo anterior, los tensioactivos, los anhídridos y los productos que interfieren con la luciferasa quedan fuera del ámbito de aplicación de este ensayo. En los casos en que haya pruebas que demuestren la inaplicabilidad del ensayo IL-8 Luc a otras categorías específicas de productos problema, no deberá utilizarse este ensayo con tales categorías específicas.
|
|
6.
|
Tal como se ha descrito anteriormente, el ensayo IL-8 Luc sirve para justificar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes. Es necesario seguir trabajando, preferiblemente sobre la base de datos humanos, para determinar si los resultados del ensayo IL-8 Luc pueden contribuir a la evaluación de la potencia cuando se considere en combinación con otras fuentes de información.
|
|
7.
|
En el apéndice 3.1 se dan las definiciones pertinentes.
|
PRINCIPIO DEL ENSAYO
|
8.
|
El ensayo IL-8 Luc hace uso de la línea celular THP-1 de la leucemia monocítica humana, que se obtuvo de la American Type Culture Collection (Manassas, VA, EE. UU.). Utilizando esta línea celular, el Departamento de Dermatología de la Facultad de Medicina de la Universidad de Tohoku, estableció una línea celular con el marcador IL-8 derivada de la THP-1, la THP-G8, que alberga los genes del naranja estable de luciferasa (SLO, Stable Luciferase Orange) y del rojo estable de luciferasa (SLR, Stable Luciferase Red), bajo el control del promotor de la IL-8 y del de la gliceraldehído-3-fosfato-deshidrogenasa (GAPDH), respectivamente (1). Esto permite la medición cuantitativa de la inducción del gen de la luciferasa mediante la detección de la luminiscencia a partir de sustratos de luciferasa bien establecidos que emiten luz como indicador de la actividad de la IL-8 y del GAPDH en células tras la exposición a productos sensibilizantes.
|
|
9.
|
El sistema de ensayo de doble color incluye una luciferasa que emite luz naranja (SLO; λmáx = 580 nm) (12) correspondiente a la expresión génica del promotor de la IL-8, así como una luciferasa que emite luz roja (SLR; λmáx = 630 nm) (13) correspondiente a la expresión génica del promotor del testigo interno, GAPDH. Las dos luciferasas emiten distintos colores al reaccionar con la D-luciferina de la luciérnaga, y su luminiscencia se mide simultáneamente en una reacción de una sola fase, dividiendo la emisión de la mezcla de ensayo mediante un filtro óptico (14) (apéndice 3.2).
|
|
10.
|
Las células THP-G8 se tratan durante 16 horas con el producto problema, tras lo cual se mide la actividad de la luciferasa SLO (SLO-LA), que refleja la actividad del promotor de la IL-8, y la actividad de la luciferasa SLR (SLR-LA), que refleja la actividad del promotor de la GAPDH. Para que las abreviaturas sean fáciles de comprender, SLO-LA y SLR-LA se designan como IL8LA y GAPLA, respectivamente. El cuadro 1 presenta una descripción de los términos asociados a la actividad de la luciferasa en el ensayo IL-8 Luc. Los valores medidos se utilizan para calcular el valor normalizado de IL8LA (nIL8LA), que es la relación entre IL8LA y GAPLA; el factor de inducción de nIL8LA (Ind-IL8LA), que es la relación entre las medias aritméticas de los valores cuádruples medidos de la nIL8LA en las células THP-G8 tratadas con un producto problema y los valores de la nIL8LA de células THP-G8 sin tratar; y la inhibición de la GAPLA (Inh-GAPLA), que es la relación entre las medias aritméticas de los valores cuádruples medidos de la GAPLA en las células THP-G8 tratadas con un producto problema y los valores de la GAPLA de células THP-G8 sin tratar, y que se utiliza como indicador de la citotoxicidad.
Cuadro 1
Descripción de los términos asociados a la actividad de la luciferasa en el ensayo IL-8 Luc
|
Abreviaturas
|
Definición
|
|
GAPLA
|
Actividad de la luciferasa SLR que refleja la actividad del promotor de la GAPDH
|
|
IL8LA
|
Actividad de la luciferasa SLO que refleja la actividad del promotor de la IL-8
|
|
nIL8LA
|
IL8LA/GAPLA
|
|
Ind-IL8LA
|
nIL8LA de células THP-G8 tratadas con productos / nIL8LA de células sin tratar
|
|
Inh-GAPLA
|
GAPLA de células THP-G8 tratadas con productos / GAPLA de células sin tratar
|
|
CV05
|
Concentración más baja del producto a la que el valor de Inh-GAPLA se hace < 0,05.
|
|
|
11.
|
Se dispone de normas de comportamiento (NC) (15) para facilitar la validación de ensayos de la luciferasa IL-8 in vitro modificados similares al ensayo IL-8 Luc y que tienen en cuenta la modificación oportuna de las directrices de ensayo 442E de la OCDE para su inclusión. Solo se garantizará la aceptación mutua de datos de la OCDE (MAD, Mutual Acceptance of Data) respecto a los ensayos validados con arreglo a las NC, si estos ensayos han sido revisados e incluidos en las directrices de ensayo 442E por la OCDE (16).
|
DEMOSTRACIÓN DE LA COMPETENCIA
|
12.
|
Antes de proceder al uso sistemático del ensayo descrito en el presente apéndice del método de ensayo B.71, los laboratorios deben demostrar su competencia técnica, utilizando las diez sustancias para la prueba de la competencia que figuran en el apéndice 3.3, de conformidad con las buenas prácticas de los métodos in vitro (17). Por otra parte, los usuarios del ensayo deben mantener una base de datos históricos que contenga los generados en los controles de reactividad (véase el punto 15) y con los testigos positivos y los controles del disolvente o vehículo (véanse los puntos 21-24), y utilizar estos datos para confirmar que la reproducibilidad del ensayo en su laboratorio se mantiene a lo largo del tiempo.
|
PROCEDIMIENTO
|
13.
|
Está disponible el procedimiento normalizado de trabajo (PNT) para el ensayo IL-8 Luc y debe utilizarse al realizar el ensayo (18). Los laboratorios dispuestos a realizar el ensayo pueden obtener la línea celular THP-G8 recombinante de la empresa CPG Lab. Co. Ltd., Tottori, Japón, previa firma de un acuerdo de transferencia de material (MTA) segúnlas condiciones del modelo de la OCDE. En los puntos siguientes se recoge una descripción de los principales componentes y procedimientos del ensayo.
|
Preparación de las células
|
14.
|
La línea celular THP-G8 de GPC Lab. Co. Ltd., Tottori, Japón, es la que debe utilizarse para realizar el ensayo IL-8 Luc (véanse los puntos 8 y 13). Una vez recibidas, las células se propagan (2-4 pases) y se conservan congeladas como una población homogénea. Las células de esta población pueden propagarse hasta un máximo de 12 pases o un máximo de 6 semanas. El medio utilizado para la propagación es el medio de cultivo RPMI-1640 que contiene un 10 % de suero bovino fetal (FBS), solución antibiótica/antimicótica (100 U/ml de penicilina G, 100 μg/ml de estreptomicina y 0,25 μg/ml de anfotericina B en una solución salina del 0,85 %) (p. ej., GIBCO Cat # 15240-062), 0,15 μg/ml de puromicina (p. ej., n.o CAS 58-58-2) y 300 μg/ml de G418 (p. ej., n.o CAS 108321-42-2).
|
|
15.
|
Antes de utilizarse para el ensayo, las células deben ser objeto de un control de reactividad. Este control debe realizarse 1-2 semanas o 2-4 pases después de la descongelación, utilizando el testigo positivo, el bromuro de 4-nitrobencilo (4-NBB) (n.o CAS 100-11-8, ≥ 99 % de pureza) y el testigo negativo, el ácido láctico (LA) (n.o CAS 50-21-5, ≥ 85 % de pureza). El 4-NBB debe producir una respuesta positiva de Ind-IL8LA (≥ 1,4), mientras que el LA debe dar una respuesta negativa de Ind-IL8LA (< 1,4). Solo se pueden utilizar para el ensayo las células que hayan superado el control de reactividad. Este control debe realizarse con arreglo a los procedimientos descritos en los puntos 22 a 24.
|
|
16.
|
Para el ensayo, se siembran células THP-G8 a una densidad de 2-5 × 105 células/ml, y se precultivan en matraces de cultivo durante 48-96 horas. El día del ensayo, se lavan las células recolectadas del matraz de cultivo utilizando RPMI-1640 con un 10 % de FBS sin antibióticos y, a continuación, se vuelven a suspender con RPMI-1640 con un 10 % de FBS sin antibióticos a la densidad de 1 × 106 células/ml. A continuación, se distribuyen las células en una placa negra de fondo plano de 96 pocillos (p. ej., Costar Cat#3603) a razón de 50 μl (5 × 104 células) por pocillo.
|
Preparación del producto problema y de las sustancias testigo
|
17.
|
El producto problema y las sustancias testigo se preparan el día del ensayo. Para el ensayo IL-8 Luc, los productos problema se disuelven en X-VIVOTM 15, un medio exento de suero disponible en el mercado (Lonza, 04-418Q), hasta la concentración final de 20 mg/ml. Se añade X-VIVOTM 15 a 20 mg de producto problema (con independencia de la solubilidad del producto) en un tubo de microcentrifugadora, y se lleva al volumen de 1 ml; a continuación se mezcla enérgicamente en vórtex y se agita en un rotor a una velocidad máxima de 8 rpm durante 30 minutos a una temperatura ambiente de unos 20oC. Además, si los productos sólidos siguen sin disolverse, el tubo se somete a ultrasonidos hasta que el producto se disuelve totalmente o se dispersa de forma estable. En el caso de productos problema solubles en X-VIVOTM 15, la solución se diluye por un factor de 5 con X-VIVOTM 15 y se utiliza como solución madre del producto problema en X-VIVOTM 15 (4 mg/ml). En el caso de productos problema insolubles en X-VIVOTM 15, la mezcla se pone en rotación de nuevo durante al menos 30 minutos, y se centrifuga después a 15000 rpm (≈ 20000 g) durante 5 min. El sobrenadante resultante se utiliza como solución madre del producto problema en X-VIVOTM 15. Debe aportarse una justificación científica del uso de otros disolventes, como el DMSO, el agua o el medio de cultivo. El procedimiento detallado para la disolución de productos figura en el apéndice 3.5. Las soluciones en X-VIVOTM 15 descritas en los puntos 18-23 se mezclan 1: 1 (v/v) con las suspensiones celulares preparadas en la placa negra de fondo plano de 96 pocillos (véase el punto 16).
|
|
18.
|
La primera tanda del ensayo tiene por objeto determinar la concentración citotóxica y examinar el potencial de sensibilización cutánea de los productos. Utilizando el X-VIVOTM 15, se hacen diluciones en serie de las soluciones madre de los productos problema en X- VIVOTM 15, con un factor de dilución de dos (véase el apéndice 3.5) utilizando un bloque de ensayo de 96 pocillos (p. ej., Costar Cat # EW-01729-03). A continuación, se añaden 50 μl/pocillo de la solución diluida a 50 μl de la suspensión celular en una placa negra de fondo plano de 96 pocillos. Por lo tanto, en el caso de los productos problema solubles en X-VIVOTM 15, las concentraciones finales de los productos problema van de 0,002 a 2 mg/ml (apéndice 3.5). En el caso de los productos problema que no son solubles en X-VIVOTM 15 a la concentración de 20 mg/ml, solo se determinan los factores de dilución que van de 2 a 210, si bien las concentraciones finales reales de los productos problema siguen siendo inciertas y dependen de la concentración saturada de los productos problema en la solución madre en X-VIVOTM 15.
|
|
19.
|
En tandas de ensayo posteriores (es decir, las réplicas segunda, tercera y cuarta), la solución madre de X-VIVOTM 15 se hará a una concentración 4 veces superior a la concentración de viabilidad celular 05 (CV05, concentración más baja a la que el valor de Inh-GAPLA se hace < 0,05) del primer experimento. Si el valor de Inh-GAPLA no se reduce por debajo de 0,05 con la concentración más elevada en la primera tanda, la solución madre en X-VIVOTM 15 se hace a la concentración más alta de la primera tanda. La concentración CV05 se calcula dividiendo la concentración de la solución madre en la primera tanda por el primer factor de dilución correspondiente a CV05 (X) [factor de dilución CV05 (X), que es el factor de dilución necesario para diluir la solución madre hasta CV05] (véase el apéndice 3.5). En el caso de sustancias problema insolubles en X-VIVO a 20 mg/ml, el valor de CV05 se determina mediante la concentración de la solución madre × 1/X. En las tandas 2 a 4, se prepara una segunda solución madre a 4 × CV05 (apéndice 3.5).
|
|
20.
|
De las segundas soluciones madre en X-VIVOTM 15 se realizan diluciones en serie con un factor de dilución de 1,5 utilizando un bloque de ensayo de 96 pocillos. A continuación, se añaden 50 μl/pocillo de la solución diluida a 50 μl de la suspensión celular en los pocillos de una placa negra de fondo plano de 96 pocillos. Cada concentración de cada producto problema debe ensayarse en 4 pocillos. A continuación, las muestras se mezclan con un agitador de placas y se incuban durante 16 horas a 37oC y con 5 % de CO2, tras lo cual se mide la actividad de la luciferasa como se describe a continuación.
|
|
21.
|
El control del disolvente es la mezcla de 50 μl/pocillo de X-VIVO™ 15 y de 50 μl/pocillo de suspensión celular en RPMI-1640 con un 10 % de FBS.
|
|
22.
|
El testigo positivo recomendado es el 4-NBB. Se preparan 20 mg de 4-NBB en un tubo de microcentrifugación de 1,5 ml, al que se añade X-VIVOTM 15 hasta completar 1 ml. Se mezcla enérgicamente en vórtex el contenido del tubo y se agita en un rotor a la velocidad máxima de 8 rpm durante al menos 30 minutos. Tras la centrifugación a 20000 g durante 5 min, el sobrenadante se diluye por un factor de 4 con X-VIVOTM 15, y se transfieren 500 μl del sobrenadante diluido a un pocillo de un bloque de ensayo de 96 pocillos. El sobrenadante diluido se diluye más con X-VIVOTM 15 por factores de 2 y 4, y se añaden 50 μl de la solución a 50 μl de la suspensión de células THP-G8 en los pocillos de una placa negra de fondo plano de 96 pocillos (apéndice 3.6). Cada concentración del testigo positivo debe ensayarse en 4 pocillos. La placa se agita en un agitador de placas y se incuba en una incubadora de CO2 durante 16 horas (37oC, 5 % de CO2), tras lo cual se mide la actividad de la luciferasa como se describe en el punto 29.
|
|
23.
|
El testigo negativo recomendado es el LA. Se preparan 20 mg de este en un tubo de microcentrifugación de 1,5 ml, al que se añade X-VIVOTM 15 hasta completar 1 ml (20 mg/ ml). Una solución de 20 mg/ml de solución de LA se diluye por un factor de 5 con X-VIVOTM 15 (4 mg/ml); se transfieren 500 μl de esta solución de LA de 4 mg/ml a un pocillo de un bloque de ensayo de 96 pocillos. Esta solución se diluye por un factor de 2 con X-VIVOTM 15 y después se diluye de nuevo por un factor de 2 para producir soluciones de 2 mg/ml y 1 mg/ml. Se añaden 50 μl de estas 3 soluciones y el control del vehículo (X-VIVOTM 15) a 50 μl de suspensión de células THP-G8 en los pocillos de una placa negra de fondo plano de 96 pocillos. Cada concentración del testigo negativo debe ensayarse en 4 pocillos. La placa se agita en un agitador de placas y se incuba en una incubadora de CO2 durante 16 horas (37oC, 5 % de CO2), tras lo cual se mide la actividad de la luciferasa como se describe en el punto 29.
|
|
24.
|
Podrán utilizarse otros testigos positivos o negativos adecuados si se dispone de datos históricos para obtener criterios comparables de aceptación de tandas.
|
|
25.
|
Deben tomarse precauciones para evitar la evaporación de los productos problema volátiles y la contaminación cruzada entre pocillos por productos problema, por ejemplo tapando la placa antes de la incubación con los productos problema.
|
|
26.
|
Los productos problema y el control de disolvente requieren de 2 a 4 tandas para obtener una asignación positiva o negativa (véase el cuadro 2). Cada tanda se realiza un día diferente con nueva solución madre de los productos problema en X-VIVOTM 15 y células recolectadas de forma independiente. Las células pueden proceder del mismo pase.
|
Mediciones de la actividad de la luciferasa
|
27.
|
La luminiscencia se mide mediante un luminómetro de microplacas de 96 pocillos, equipado con filtros ópticos, como, p. ej., Phios (ATTO, Tokio, Japón), Tristan 941 (Berthold, Bad Wildbad, Alemania) y la serie ARVO (PerkinElmer, Waltham, MA, EE. UU.). El luminómetro deberá calibrarse para cada ensayo, a fin de garantizar la reproducibilidad (19). Para esta calibración se dispone de luciferasas recombinantes que emiten luz de color naranja y rojo.
|
|
28.
|
A cada pocillo de la placa que contenga la suspensión celular tratada con o sin producto se transfieren 100 μl del reactivo precalentado de ensayo de la luciferasa Tripluc® (Tripluc). Se agita la placa durante 10 minutos a una temperatura ambiente de 20oC aproximadamente. La placa se coloca en el luminómetro para medir la actividad de la luciferasa. Se mide la bioluminiscencia durante 3 segundos tanto en ausencia (F0) como en presencia (F1) del filtro óptico. Debe justificarse el uso de otras condiciones, por ejemplo en función del modelo de luminómetro utilizado.
|
|
29.
|
Los parámetros de cada concentración se calculan a partir de los valores medidos, por ejemplo IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, la media ± DT de IL8LA, la media ± DT de GAPLA, la media ± DT de nIL8LA, la media ± DT de Ind-IL8LA, la media ± DT de Inh-GAPLA, y el intervalo de confianza del 95 % de Ind-IL8LA. Las definiciones de los parámetros utilizados en este punto figuran en los apéndices 3.1 y 3.4.
|
|
30.
|
Antes de proceder a la medición, la discriminación del color en los ensayos con marcadores multicolor se consigue generalmente utilizando detectores (luminómetro y lector de placas) equipados con filtros ópticos, tales como filtros de corte agudo (de paso largo o de paso corto) o filtros de paso de banda. Los coeficientes de transmisión de los filtros para cada color de la señal de luminiscencia deben calibrarse antes del ensayo, según el apéndice 3.2.
|
DATOS E INFORME
Evaluación de los datos
|
31.
|
Los criterios para una decisión positiva o negativa exigen que, en cada una de las fases:
|
—
|
se considere positiva una asignación del ensayo IL-8 Luc si un producto problema tiene un Ind-IL8LA ≥ 1,4 y el límite inferior del intervalo de confianza del 95 % del Ind-IL8LA ≥ 1,0;
|
|
—
|
se considere negativa una asignación del ensayo IL-8 Luc si un producto problema tiene un Ind-IL8LA < 1,4 y/o el límite inferior del intervalo de confianza del 95 % de Ind-IL8LA < 1,0.
|
|
Modelo de asignación
|
32.
|
Los productos problema que proporcionan dos resultados positivos de entre las tandas primera, segunda, tercera o cuarta se identifican como positivos, mientras que los que dan tres resultados negativos de entre las tandas primera, segunda, tercera o cuarta se identifican como supuestos negativos (cuadro 2). Entre los productos supuestos negativos, los productos que se disuelven a la concentración de 20 mg/ml de X-VIVOTM 15 se consideran negativos, mientras que los productos que no se disuelven a la concentración de 20 mg/ml de X-VIVOTM 15 no deben tenerse en cuenta (figura 1).
Cuadro 2
Criterios para identificar positivos y supuestos negativos
|
1.a tanda
|
2.a tanda
|
3.a tanda
|
4.a tanda
|
Clasificación final
|
|
Positivo
|
Positivo
|
–
|
–
|
Positivo
|
|
Negativo
|
Positivo
|
–
|
Positivo
|
|
Negativo
|
Positivo
|
Positivo
|
|
Negativo
|
Supuesto negativo
|
|
Negativo
|
Positivo
|
Positivo
|
–
|
Positivo
|
|
Negativo
|
Positivo
|
Positivo
|
|
Negativo
|
Supuesto negativo
|
|
Negativo
|
Positivo
|
Positivo
|
Positivo
|
|
Negativo
|
Supuesto negativo
|
|
Negativo
|
–
|
Supuesto negativo
|
|
Figura 1
Modelo de asignación para la decisión final

Criterios de aceptación
|
33.
|
Deben cumplirse los siguientes criterios de aceptación cuando se use el ensayo IL-8 Luc.
|
—
|
El valor de Ind-IL8LA debe ser superior a 5,0 a al menos una concentración del testigo positivo, 4-NBB, en cada tanda.
|
|
—
|
El valor de Ind-IL8LA debe ser inferior a 1,4 a cualquier concentración del testigo negativo, ácido láctico, en cada tanda.
|
|
—
|
Deben rechazarse los datos de las placas en las que la GAPLA de los pocillos testigo con células y Tripluc pero sin productos sea inferior a 5 veces la de los pocillos con solo medio de ensayo (50 μl/pocillo de RPMI-1640 con un 10 % de FBS y 50 μl/pocillo de X-VIVOTM 15).
|
|
—
|
Deben rechazarse los datos de las placas en las que la Inh-GAPLA de todas las concentraciones de los productos problema o testigo sea inferior a 0,05. En este caso, debe repetirse el primer ensayo de manera que la concentración final más alta del ensayo repetido sea la concentración final más baja del ensayo anterior.
|
|
Informe del ensayo
|
34.
|
El informe del ensayo debe incluir la información siguiente:
|
Productos problema
Sustancias de un solo componente:
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
Aspecto físico, hidrosolubilidad, peso molecular, y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Solubilidad en X-VIVOTM 15. En el caso de los productos que sean insolubles en X-VIVOTM 15, si se observa precipitación o flotación después de la centrifugación;
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Justificación de la elección del disolvente o vehículo para cada producto problema si no se ha utilizado X-VIVOTM 15.
|
Sustancias de componentes múltiples, sustancias UVCB y mezclas:
|
Caracterización, en la medida de lo posible, mediante, por ejemplo, la identidad química (véase más arriba), pureza, presencia cuantitativa y propiedades fisicoquímicas pertinentes de los componentes (véase más arriba), en la medida de lo posible;
|
|
Aspecto físico, hidrosolubilidad, y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible;
|
|
Peso molecular o peso molecular aparente en el caso de mezclas/polímeros de composición conocida u otra información pertinente para la realización del estudio;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Solubilidad en X-VIVOTM 15. En el caso de los productos que sean insolubles en X-VIVOTM 15, si se observa precipitación o flotación después de la centrifugación;
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible.
|
|
Justificación de la elección del disolvente o vehículo para cada producto problema si no se ha utilizado X-VIVOTM 15.
|
|
|
Testigos
Testigo positivo:
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, código SMILES o InChI, fórmula estructural, y/u otros identificadores;
|
|
Aspecto físico, hidrosolubilidad, peso molecular, y otras propiedades fisicoquímicas pertinentes, en la medida de lo posible y en los casos aplicables;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Tratamiento antes del ensayo, en su caso (p. ej., calentamiento, trituración);
|
|
Concentración o concentraciones estudiadas;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Referencia a los resultados de los testigos positivos históricos que demuestren unos criterios adecuados de aceptación, si procede.
|
Testigo negativo:
|
Identificación química, como nombre o nombres IUPAC o CAS, número o números CAS, y/u otros identificadores;
|
|
Pureza, identidad química de las impurezas según convenga y sea factible en la práctica, etc.;
|
|
Aspecto físico, peso molecular, y propiedades fisicoquímicas pertinentes adicionales en el caso de que se utilicen unos testigos negativos distintos de los especificados en las directrices del ensayo y en la medida de lo posible;
|
|
Condiciones de conservación y estabilidad en la medida de lo posible;
|
|
Justificación de la elección del disolvente para cada producto problema.
|
|
|
Condiciones de ensayo
|
Nombre y dirección del promotor, laboratorio y director del estudio;
|
|
Descripción del ensayo utilizado;
|
|
Línea celular utilizada, sus condiciones de conservación y su origen (por ejemplo, laboratorio donde se ha obtenido);
|
|
Número de lote y origen del FBS, nombre del proveedor, número de lote de la placa negra de fondo plano de 96 pocillos y número de lote del reactivo Tripluc;
|
|
Número de pases y densidad celular utilizados para el ensayo;
|
|
Método de recuento de células utilizado para la siembra antes del ensayo, y medidas adoptadas para garantizar la homogeneidad de la distribución del número de células;
|
|
Luminómetro utilizado (por ejemplo, modelo), incluidos los ajustes instrumentales, sustrato de luciferasa utilizado, y demostración de las mediciones adecuadas de luminiscencia sobre la base del ensayo de control descrito en el apéndice 3.2;
|
|
Procedimiento utilizado para demostrar la competencia del laboratorio en cuanto a la realización del ensayo (por ejemplo, mediante el ensayo de sustancias de la prueba de la competencia) o para demostrar que el ensayo tiene un comportamiento reproducible a lo largo del tiempo.
|
|
|
Procedimiento de ensayo
|
Número de réplicas y de tandas efectuadas;
|
|
Concentraciones del producto problema, procedimiento de aplicación y tiempo de exposición (si son diferentes de los recomendados);
|
|
Descripción de los criterios de evaluación y decisión seguidos;
|
|
Descripción de los criterios de aceptación del estudio seguidos;
|
|
Descripción de las eventuales modificaciones del procedimiento del ensayo.
|
|
|
Resultados
|
Mediciones de IL8LA y GAPLA;
|
|
Cálculos de nIL8LA, Ind-IL8LA e Inh-GAPLA;
|
|
Intervalo de confianza del 95 % de Ind-IL8LA;
|
|
Gráfico que muestre las curvas dosis-respuesta en cuanto a la inducción de la actividad de la luciferasa y la viabilidad;
|
|
Descripción de cualesquiera otras observaciones pertinentes, si procede.
|
|
|
Discusión de los resultados
|
Discusión de los resultados obtenidos con el ensayo IL-8 Luc;
|
|
Consideración de los resultados del ensayo en el contexto de un enfoque IATA, si se dispone de otra información pertinente.
|
|
Conclusión
|
BIBLIOGRAFÍA
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OCDE (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OCDE (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OCDE (2016). Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OCDE (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, Publicaciones de la OCDE, París. Disponible en: http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OCDE (2017). To be published - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OCDE, París, Francia.
|
|
(16)
|
OCDE (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OCDE, París, Francia.
|
|
(17)
|
OCDE (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organización para la Cooperación y el Desarrollo Económico, París. Disponible en: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, Disponible en: http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OCDE (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OCDE, París, Francia.
|
|
(21)
|
Naciones Unidas (2015). Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA). Sexta edición revisada. Nueva York & Ginebra: Publicaciones de las Naciones Unidas. ISBN: 978-92-1-117006-1. Disponible en: http://www.unece.org/es/trans/danger/publi/ghs/ghs_rev05/05files_s.html
|
Apéndice 3.1
DEFINICIONES
Exactitud
: Grado de coincidencia entre los resultados del ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del ensayo y es un aspecto de su pertinencia. Este término y el de concordancia se suelen usar indistintamente para indicar la proporción de resultados correctos de un ensayo (16).
Ruta de resultados adversos (AOP, Adverse Outcome Pathway)
: Secuencia de fenómenos desde la estructura química de un producto diana o grupo de productos similares, pasando por el fenómeno molecular desencadenante, hasta un resultado in vivo de interés (20).
Producto
: Sustancia o mezcla.
CV05
: Viabilidad celular 05, es decir, concentración mínima a la que los productos muestran un valor de Inh-GAPLA inferior a 0,05.
FInSLO-LA
: Abreviatura utilizada en el informe de validación y en publicaciones anteriores en relación con el ensayo IL-8 Luc para referirse a la Ind-IL8LA. Véase la definición de Ind-IL8LA.
GAPLA
: Actividad de luciferasa del rojo estable de luciferasa (SLR) (λmáx = 630 nm), regulada por el promotor de la GAPDH y que demuestra la viabilidad celular y el número de células viables.
Peligro
: Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente.
IATA (Enfoque integrado de pruebas y evaluación, Integrated Approach to Testing and Assessment)
: Enfoque estructurado para la identificación del peligro (potencial), caracterización del peligro (potencia) o evaluación de la seguridad (potencial/potencia y exposición) de un producto o grupo de productos, que integra y pondera de forma estratégica todos los datos pertinentes para fundamentar una decisión normativa relativa a posibles peligros o riesgos o a la necesidad de realizar ensayos más específicos y, por tanto, mínimos.
II-SLR-LA
: Abreviatura utilizada en el informe de validación y en publicaciones anteriores en relación con el ensayo IL-8 Luc para referirse a la Inh-GAPLA. Véase la definición de Inh-GAPLA.
IL-8 (interleucina-8)
: Citocina derivada de células endoteliales, fibroblastos, queratinocitos, macrófagos y monocitos que provoca quimiotaxia de neutrófilos y linfocitos T.
IL8LA
: Actividad de la luciferasa del naranja estable de luciferasa (SLO) (λmáx = 580 nm), regulada por el promotor de la IL-8.
Ind-IL8LA
: Factor multiplicador de la nIL8LA. Se obtiene dividiendo el valor de la nIL8LA de las células THP-G8 tratadas con los productos por el valor de la de las células THP-G8 no estimuladas y representa la inducción de la actividad del promotor de la IL-8 por los productos.
Inh-GAPLA
: Inhibición de la GAPLA. Se obtiene dividiendo el valor de la GAPLA de las células THP-G8 tratadas con productos por el de la GAPLA de THP-G8 no tratadas y representa la citotoxicidad de los productos.
Umbral mínimo de inducción (MIT)
: Concentración más baja a la que un producto cumple los criterios positivos.
Mezcla
: Mezcla o solución compuesta de dos o más sustancias.
Sustancia de un solo componente
: Sustancia, definida por su composición cuantitativa, en la que un solo componente principal representa al menos el 80 % (p/p).
Sustancia de componentes múltiples
: Sustancia, definida por su composición cuantitativa, en la que más de uno de los componentes principales están presentes a una concentración ≥ 10 % (p/p) y < 80 % (p/p). Una sustancia de componentes múltiples es el resultado de un proceso de fabricación. La diferencia entre mezcla y sustancia de componentes múltiples es que una mezcla se obtiene mezclando dos o más sustancias sin reacción química. Una sustancia de componentes múltiples es el resultado de una reacción química.
nIL8LA
: Actividad de la luciferasa SLO que refleja la actividad del promotor de la IL-8 (IL8LA), normalizada por la actividad de la luciferasa SLR, que refleja la actividad del promotor de la GAPDH (GAPLA). Representa la actividad del promotor de la IL-8 tras considerar la viabilidad celular o el número de células.
nSLO-LA
: Abreviatura utilizada en el informe de validación y en publicaciones anteriores en relación con el ensayo IL-8 Luc para referirse a la nIL8LA. Véase la definición de nIL8LA.
Testigo positivo
: Réplica que contiene todos los componentes de un sistema de ensayo y que se trata con una sustancia de la que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta positiva.
Pre-haptenos
: Productos que pasan a ser sensibilizantes por una transformación abiótica.
Pro-haptenos
: Productos que requieren activación enzimática para ejercer su potencial de sensibilización cutánea.
Pertinencia
: Descripción de la relación del ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un ensayo (16).
Fiabilidad
: Medida del grado en que un ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios (16).
Tanda
: Una tanda consta del ensayo de uno o más productos de forma simultánea con un control del disolvente o vehículo y con un testigo positivo.
Sensibilidad
: Proporción de todos los productos activos/positivos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un ensayo (16).
SLO-LA
: Abreviatura utilizada en el informe de validación y en publicaciones anteriores en relación con el ensayo IL-8 Luc para referirse a la IL8LA. Véase la definición de IL8LA.
SLR-LA
: Abreviatura utilizada en el informe de validación y en publicaciones anteriores en relación con el ensayo IL-8 Luc para referirse a la GAPLA. Véase la definición de GAPLA.
Control del disolvente o vehículo
: Muestra no tratada que contiene todos los componentes de un sistema de ensayo, excepto el producto problema, pero incluido el disolvente o vehículo utilizado. Se utiliza para establecer la respuesta de referencia para las muestras tratadas con el producto problema disuelto o disperso de forma estable en el mismo disolvente o vehículo. Cuando se somete a ensayo con un control de medio en paralelo, esta muestra pone de manifiesto también si el disolvente o vehículo interactúa con el sistema de ensayo.
Especificidad
: Proporción de todos los productos inactivos/negativos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un ensayo (16).
Sustancia
: Elementos químicos y sus compuestos en estado natural u obtenidos mediante cualquier proceso de producción, incluidos los aditivos necesarios para mantener la estabilidad del producto y las eventuales impurezas derivadas del proceso empleado, pero excluidos los disolventes que se puedan separar sin influir en la estabilidad de la sustancia ni cambiar su composición.
Tensioactivo
: También denominado agente tensioactivo, es una sustancia, como un detergente, que puede reducir la tensión superficial de un líquido y, de este modo, permitir que forme espuma o penetre en los sólidos. También se conoce como agente humectante (TG437).
Producto problema
: Sustancia o mezcla estudiada con este método.
THP-G8
: Línea celular con el marcador IL-8 utilizada en el ensayo IL-8 Luc. La línea celular humana THP-1, similar a los macrófagos, fue transinfectada con los genes de la luciferasa SLO y SLR bajo el control de los promotores de la IL-8 y de la GAPDH, respectivamente.
Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de las Naciones Unidas)
: Sistema que propone la clasificación de los productos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (21).
UVCB
: Sustancias de composición desconocida o variable, productos de reacción compleja o materiales biológicos.
Método de ensayo válido
: Ensayo del que se considera que tiene suficiente pertinencia y fiabilidad con un fin específico y que se basa en principios sólidos desde el punto de vista científico. Un ensayo nunca es válido en un sentido absoluto, sino únicamente en relación con un fin determinado.
Apéndice 3.2
PRINCIPIO DE LA MEDICIÓN DE LA ACTIVIDAD DE LA LUCIFERASA Y DETERMINACIÓN DE LOS COEFICIENTES DE TRANSMISIÓN DEL FILTRO ÓPTICO RESPECTO A SLO Y SLR
El sistema de ensayo multimarcador (Tripluc) puede utilizarse con un luminómetro de microplacas con un sistema de detección multicolor que puede combinarse con un filtro óptico [por ejemplo, Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)]. El filtro óptico utilizado en la medición es un filtro de paso largo o corto de 600-620 nm o un filtro de paso de banda de 600-700 nm.
Medición de luciferasas de dos colores con un filtro óptico.
En el ejemplo aquí presentado se utiliza Phelios AB-2350 (ATTO). Este luminómetro está equipado con un filtro de paso largo (LP, long pass) de 600 nm (R60 HOYA Co., 600 nm LP, Filtro 1) para separar la luminiscencia SLO (λmáx = 580 nm) de la SLR (λmáx = 630 nm).
Para determinar los coeficientes de transmisión del LP de 600 nm, primero, utilizando enzimas luciferasa SLO y SLR purificadas, ha de medirse: i) la intensidad de la bioluminiscencia SLO y SLR sin filtro (F0), ii) la intensidad de la bioluminiscencia SLO y SLR que ha pasado por el LP de 600 nm (filtro 1), y iii) calcular los coeficientes de transmisión del LP de 600 nm respecto a SLO y SLR que se recogen más abajo.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κOR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
Si las intensidades de SLO y SLR en la muestra problema se expresan como O y R, respectivamente, i) la intensidad de la luz sin filtro (totalmente óptica) F0 y ii) la intensidad de la luz que se transmite por el LP de 600 nm (filtro 1) F1 se describen como se indica a continuación:
F0 = O + R
F1=κOR60 x O + κRR60 x R
Estas fórmulas pueden expresarse también de la siguiente manera:

A continuación, utilizando los factores de transmitancia calculados (κOR60 y κRR60) y los valores medidos de F0 y F1, podrán calcularse los valores de O y R como sigue:

Materiales y métodos para determinar el factor de transmitancia
|
1)
|
Reactivos
Enzimas luciferasa aisladas y purificadas:
|
Enzima SLO purificada y liofilizada
|
|
Enzima SLR purificada y liofilizada
|
|
(que para el trabajo de validación se obtuvieron de GPC Lab. GPC Lab. Co. Ltd., Tottori, Japón, con la línea celular THP-G8).
|
Reactivo de ensayo:
|
Reactivo de ensayo de la luciferasa Tripluc® (por ejemplo, de TOYOBO Cat#MRA-301)
|
Medio: para el ensayo de la luciferasa (30 ml, conservado a 2-8oC)
|
|
Reactivo
|
Conc.
|
Conc. final en medio
|
Cantidad necesaria
|
|
RPMI-1640
|
-
|
-
|
27 ml
|
|
FBS
|
-
|
10 %
|
3 ml
|
|
2)
|
Preparación de la solución de enzima:
Disolver la enzima luciferasa purificada y liofilizada en un tubo mediante la adición de 200 μl de Tris/HCl o Hepes/HCl 10 ~ 100 mM (pH 7,5 ~ 8,0), complementados con glicerol al 10 % (p/v); dividir la solución de enzima en alícuotas de 10 μl puestas en tubos desechables de 1,5 ml y conservarlas en congelador a -80oC; la solución de enzima congelada podrá utilizarse durante un máximo de 6 meses. Cuando se utiliza, se añade 1 ml de medio para el ensayo de la luciferasa (RPMI-1640 con un 10 % de FBS) a cada tubo que contiene las soluciones de enzima (solución diluida de enzima) y se mantiene en hielo para evitar la desactivación.
|
|
3)
|
Medición de la bioluminiscencia
Descongelar el reactivo de ensayo de la luciferasa Tripluc® (Tripluc) y mantenerlo a temperatura ambiente en un baño de agua o al aire. Encender el luminómetro 30 min antes del inicio de la medición para permitir que se estabilice el fotomultiplicador. Pasar 100 μl de la solución diluida de enzima a una placa negra de 96 pocillos (fondo plano) (la muestra de referencia de SLO a #B1, #B2, #B3, la muestra de referencia de SLR a #D1, #D2, #D3). A continuación, pasar 100 μl de Tripluc precalentado a cada pocillo de la placa que contiene la solución diluida de enzima, utilizando una micropipeta. Agitar la placa durante 10 minutos a temperatura ambiente (alrededor de 25oC) utilizando un agitador de placas. Eliminar las eventuales burbujas que aparezcan en las soluciones de los pocillos. Colocar la placa en el luminómetro para medir la actividad de la luciferasa. Se mide la bioluminiscencia durante 3 segundos tanto en ausencia (F0) como en presencia (F1) del filtro óptico.
Se calcula el coeficiente de transmisión del filtro óptico de la manera siguiente:
Coeficiente de transmisión [SLO (κOR60)] = (#B1 de F1 + #B2 de F1 + #B3 de F1) / (#B1 de F0 + #B2 de F0 + #B3 de F0)
Coeficiente de transmisión [SLR (κRR60)] = (#D1 de F1 + #D2 de F1 + #D3 de F1) / (#D1 de F0 + #D2 de F0 + #D3 de F0)
Los factores de transmitancia calculados se utilizan para todas las mediciones realizadas con el mismo luminómetro.
|
Control de calidad del equipo
Se utilizarán los procedimientos descritos en el protocolo IL-8 (18).
Apéndice 3.3
SUSTANCIAS UTILIZADAS PARA DEMOSTRAR LA COMPETENCIA
Antes de proceder al uso sistemático del ensayo descrito en el presente apéndice del método B.71, los laboratorios deben demostrar su competencia técnica mediante la obtención con el ensayo IL-8 Luc de la clasificación prevista de las nueve sustancias recomendadas en el cuadro 1, y la obtención de valores que estén dentro del intervalo de referencia respectivo con al menos ocho de las nueve sustancias utilizadas para demostrar la competencia (seleccionadas para representar la gama de respuestas correspondientes a los peligros de sensibilización cutánea). Otros criterios de selección fueron que las sustancias estén disponibles en el mercado y que se disponga de datos de referencia in vivo de alta calidad, así como de datos in vitro de alta calidad obtenidos con el ensayo IL-8 Luc. Asimismo, ha de disponerse de datos de referencia publicados sobre el ensayo IL-8 Luc (6) (1).
Cuadro 1
Sustancias recomendadas para demostrar la competencia técnica con el ensayo IL-8 Luc
|
Sustancias para la competencia
|
No CAS
|
Estado físico
|
Solubilidad en X-VIVO15 a 20 mg/ml
|
Asignación in vivo
(109)
|
Asignación con IL-8 Luc (110)
|
Intervalo de referencia (μg/ml) (111)
|
|
|
CV05
(112)
|
MIT con IL-8 Luc (113)
|
|
2,4-Dinitroclorobenceno
|
97-00-7
|
Sólido
|
Insoluble
|
Sensibilizante (extremo)
|
Positivo
|
2,3-3,9
|
0,5-2,3
|
|
Formaldehído
|
50-00-0
|
Líquido
|
Soluble
|
Sensibilizante (fuerte)
|
Positivo
|
9-30
|
4-9
|
|
2-Mercaptobenzotiazol
|
149-30-4
|
Sólido
|
Insoluble
|
Sensibilizante (moderado)
|
Positivo
|
250-290
|
60-250
|
|
Etilendiamina
|
107-15-3
|
Líquido
|
Soluble
|
Sensibilizante (moderado)
|
Positivo
|
500-700
|
0,1-0,4
|
|
Dimetacrilato de etilenglicol
|
97-90-5
|
Líquido
|
Insoluble
|
Sensibilizante (débil)
|
Positivo
|
> 2000
|
0,04-0,1
|
|
4-Alilanisol (estragol)
|
140-67-0
|
Líquido
|
Insoluble
|
Sensibilizante (débil)
|
Positivo
|
> 2000
|
0,01-0,07
|
|
Sulfato de estreptomicina
|
3810-74-0
|
Sólido
|
Soluble
|
No sensibilizante
|
Negativo
|
> 2000
|
> 2000
|
|
Glicerol
|
56-81-5
|
Líquido
|
Soluble
|
No sensibilizante
|
Negativo
|
> 2000
|
> 2000
|
|
Isopropanol
|
67-63-0
|
Líquido
|
Soluble
|
No sensibilizante
|
Negativo
|
> 2000
|
> 2000
|
|
Abreviaturas: No CAS = número de registro del Chemical Abstracts Service.
|
Apéndice 3.4
ÍNDICES Y CRITERIOS DE DECISIÓN
nIL8LA (nSLO-LA)
Se mide la repetición j (j = 1-4) de la concentración i (i = 0-1) en el caso de la IL8LA (SLO-LA) y la GAPLA (SLR-LA) respectivamente. La IL8LA normalizada, denominada nIL8LA (nSLO-LA), se define como:
nIL8LAij = IL8LAij/GAPLAij
Esta es la unidad básica de medición de este ensayo.
Ind-IL8LA (FInSLO-LA)
El número de veces que aumenta el promedio de nIL8LA (nSLO-LA) en la repetición a la concentración i con respecto a su valor a la concentración 0, Ind-IL8LA, es la medida primaria de este ensayo. Esta relación se expresa con la siguiente fórmula:

El laboratorio principal ha propuesto que un valor de 1,4 corresponde a un resultado positivo para el producto problema. Este valor se basa en la investigación de los datos históricos del laboratorio principal. El equipo de gestión de datos utilizó después este valor a través de todas las fases del estudio de validación. El resultado primario, Ind-IL8LA, es el cociente de dos medias aritméticas, tal como se indica en la ecuación.
Intervalo de confianza del 95 % (IC del 95 %)
Puede estimarse que el intervalo de confianza del 95 % (IC del 95 %) basado en el cociente muestra la precisión de la medida de este resultado primario. Un límite inferior del IC del 95 % ≥ 1 indica que la nIL8LA a la concentración i es significativamente mayor que la del control del disolvente. Hay varias formas de construir el IC del 95 %. Hemos utilizado en este estudio el método conocido como teorema de Fieller. Este teorema sobre el intervalo de intervalo de confianza del 95 % se obtiene de la siguiente fórmula:

donde: .

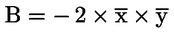






es el porcentil 97,5 de la distribución t central con la ν del grado de libertad, donde
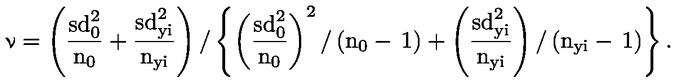
Inh-GAPLA (II-SLR-LA)
La Inh-GAPLA es una relación entre el promedio de la GAPLA (SLR-LA) correspondiente a la repetición de la concentración i en comparación con la del control del disolvente, y su valor viene dado por:
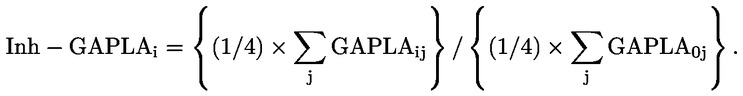
Como la GAPLA es el denominador de la nIL8LA, un valor sumamente pequeño causa una gran variación en el nIL8LA. Por tanto, los valores de Ind-IL8LA con un valor extremadamente bajo de Inh-GAPLA (menos de 0,05) pueden considerarse de escasa precisión.
Apéndice 3.5
ESQUEMA DE LOS MÉTODOS DE DISOLUCIÓN DE LOS PRODUCTOS PARA EL ENSAYO IL-8 LUC
|
a)
|
En el caso de los productos disueltos en X-VIVOTM 15 a 20 mg/ml

|
|
b)
|
En el caso de los productos insolubles en X-VIVOTM 15 a 20 mg/ml
|
Apéndice 3.6
ESQUEMA DEL MÉTODO DE DISOLUCIÓN DEL 4-NBB PARA EL TESTIGO POSITIVO DEL ENSAYO IL-8 LUC

» |