ISSN 1977-0642
Amtsblatt
der Europäischen Union
L 272

Ausgabe in deutscher Sprache
Rechtsvorschriften
64. Jahrgang
30. Juli 2021
|
ISSN 1977-0642 |
||
|
Amtsblatt der Europäischen Union |
L 272 |
|
|
|
||
|
Ausgabe in deutscher Sprache |
Rechtsvorschriften |
64. Jahrgang |
|
|
|
|
|
(1) Text von Bedeutung für den EWR. |
|
DE |
Bei Rechtsakten, deren Titel in magerer Schrift gedruckt sind, handelt es sich um Rechtsakte der laufenden Verwaltung im Bereich der Agrarpolitik, die normalerweise nur eine begrenzte Geltungsdauer haben. Rechtsakte, deren Titel in fetter Schrift gedruckt sind und denen ein Sternchen vorangestellt ist, sind sonstige Rechtsakte. |
II Rechtsakte ohne Gesetzescharakter
VERORDNUNGEN
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/1 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/1241 DES RATES
vom 29. Juli 2021
zur Durchführung des Artikels 21 Absatz 2 der Verordnung (EU) 2016/44 über restriktive Maßnahmen angesichts der Lage in Libyen und zur Aufhebung der Verordnung (EU) Nr. 204/2011
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) 2016/44 des Rates vom 18. Januar 2016 über restriktive Maßnahmen angesichts der Lage in Libyen und zur Aufhebung der Verordnung (EU) Nr. 204/2011 (1), insbesondere auf Artikel 21 Absatz 2,
auf Vorschlag des Hohen Vertreters der Union für Außen- und Sicherheitspolitik,
in Erwägung nachstehender Gründe:
|
(1) |
Am 18. Januar 2016 hat der Rat die Verordnung (EU) 2016/44 angenommen. |
|
(2) |
Der Rat die in Anhang III jener Verordnung enthaltene Liste der benannten Personen und Organisationen gemäß Artikel 21 Absatz 6 der Verordnung (EU) 2016/44 überprüft. |
|
(3) |
Der Rat ist zu dem Schluss gelangt, dass der Eintrag für eine Person gestrichen werden sollte, da sie verstorben ist, und dass die restriktiven Maßnahmen gegen alle Personen und Organisationen in der Liste in Anhang III der Verordnung (EU) 2016/44 aufrechterhalten werden sollten. Darüber hinaus sollten die Angaben zur Identität einer Person aktualisiert werden. |
|
(4) |
Die Verordnung (EU) 2016/44 sollte daher entsprechend geändert werden — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang III der Verordnung (EU) 2016/44 wird gemäß dem Anhang der vorliegenden Verordnung geändert.
Artikel 2
Diese Verordnung tritt am Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Geschehen zu Brüssel am 29. Juli 2021.
Im Namen des Rates
Der Präsident
G. DOVŽAN
ANHANG
In Anhang III (Liste der natürlichen und juristischen Personen, Organisationen und Einrichtungen nach Artikel 6 Absatz 2) der Verordnung (EU) 2016/44 wird Abschnitt A (Personen) wie folgt geändert:
|
1. |
Eintrag 3 (betreffend General TOHAMI, Khaled) wird gestrichen. |
|
2. |
Eintrag 6 (betreffend AL-MAHMOUDI, Baghdadi) erhält folgende Fassung:
|
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/4 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/1242 DES RATES
vom 29. Juli 2021
zur Durchführung der Verordnung (EU) Nr. 267/2012 über restriktive Maßnahmen gegen Iran
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) Nr. 267/2012 des Rates vom 23. März 2012 über restriktive Maßnahmen gegen Iran und zur Aufhebung der Verordnung (EU) Nr. 961/2010 (1), insbesondere auf Artikel 46 Absatz 2,
auf Vorschlag des Hohen Vertreters der Union für Außen- und Sicherheitspolitik,
in Erwägung nachstehender Gründe:
|
(1) |
Der Rat hat am 23. März 2012 die Verordnung (EU) Nr. 267/2012 angenommen. |
|
(2) |
Der Rat hat am 18. Juni 2020 die Verordnung (EU) 2020/847 (2) zur Durchführung der Verordnung (EU) Nr. 267/2012 angenommen. |
|
(3) |
Nach dem Urteil des Gerichts in der Rechtssache T-580/19 (3) sollte Sayed Shamsuddin Borborudi von der in Anhang IX der Verordnung (EU) Nr. 267/2012 enthaltenen Liste der Personen und Einrichtungen, gegen die restriktive Maßnahmen verhängt wurden, gestrichen werden. |
|
(4) |
Darüber hinaus sollten aufgrund der Überprüfung des Anhangs II des Beschlusses 2010/413/GASP des Rates (4) die restriktiven Maßnahmen gegen alle Personen und Einrichtungen, die in der darin enthaltenen Liste aufgeführt sind, beibehalten werden, soweit ihre Namen nicht in Anhang VI jenes Beschlusses aufgeführt sind, und 21 Einträge in Anhang IX der Verordnung (EU) Nr. 267/2012 sollten aktualisiert werden. |
|
(5) |
Die Verordnung (EU) Nr. 267/2012 sollte daher entsprechend geändert werden — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang IX der Verordnung (EU) Nr. 267/2012 wird gemäß dem Anhang der vorliegenden Verordnung geändert.
Artikel 2
Diese Verordnung tritt am Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Geschehen zu Brüssel am 29. Juli 2021.
Im Namen des Rates
Der Präsident
G. DOVŽAN
(1) ABl. L 88 vom 24.3.2012, S. 1.
(2) Durchführungsverordnung (EU) 2020/847 des Rates vom 18. Juni 2020 zur Durchführung der Verordnung (EU) Nr. 267/2012 über restriktive Maßnahmen gegen Iran (ABl. L 196 vom 19.6.2020, S. 1).
(3) Urteil des Gerichts vom 9. Juni 2021, Sayed Shamsuddin Borborudi gegen Rat der Europäischen Union, T-580/19, ECLI:EU:T:2021:330.
(4) Beschluss 2010/413/GASP des Rates vom 26. Juli 2010 über restriktive Maßnahmen gegen Iran und zur Aufhebung des Gemeinsamen Standpunkts 2007/140/GASP (ABl. L 195 vom 27.7.2010, S. 39).
ANHANG
Anhang IX der Verordnung (EU) Nr. 267/2012 wird wie folgt geändert:
|
1. |
Unter der Überschrift „I. Personen und Einrichtungen, die an nuklearen Tätigkeiten oder Tätigkeiten im Zusammenhang mit ballistischen Flugkörpern beteiligt sind, sowie Personen und Einrichtungen, die die iranische Regierung unterstützen“ wird der folgende Eintrag gestrichen: „25. Sayed Shamsuddin Borborudi“. |
|
2. |
Unter der Überschrift „I. Personen und Einrichtungen, die an nuklearen Tätigkeiten oder Tätigkeiten im Zusammenhang mit ballistischen Flugkörpern beteiligt sind, sowie Personen und Einrichtungen, die die iranische Regierung unterstützen“ ersetzen die folgenden Einträge die entsprechenden Einträge in der Liste unter der Unterüberschrift „A. Personen“:
|
|
3. |
Unter der Überschrift „I. Personen und Einrichtungen, die an nuklearen Tätigkeiten oder Tätigkeiten im Zusammenhang mit ballistischen Flugkörpern beteiligt sind, sowie Personen und Einrichtungen, die die iranische Regierung unterstützen“ ersetzen die folgenden Einträge die entsprechenden Einträge in der Liste unter der Unterüberschrift „B. Einrichtungen“:
|
|
4. |
Unter der Überschrift „II. Korps der Iranischen Revolutionsgarden (IRGC)“ ersetzen die folgenden Einträge die entsprechenden Einträge in der Liste unter der Unterüberschrift „A. Personen“:
|
|
5. |
Unter der Überschrift „II. Korps der Iranischen Revolutionsgarden (IRGC)“ ersetzt der folgende Eintrag den entsprechenden Eintrag in der Liste unter der Unterüberschrift „B. Einrichtungen“:
|
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/11 |
DELEGIERTE VERORDNUNG (EU) 2021/1243 DER KOMMISSION
vom 19. April 2021
zur Ergänzung der Verordnung (EU) 2019/2144 des Europäischen Parlaments und des Rates durch Festlegung detaillierter Vorschriften für die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre in Kraftfahrzeugen und zur Änderung des Anhangs II der genannten Verordnung
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) 2019/2144 des Europäischen Parlaments und des Rates vom 27. November 2019 über die Typgenehmigung von Kraftfahrzeugen und Kraftfahrzeuganhängern sowie von Systemen, Bauteilen und selbstständigen technischen Einheiten für diese Fahrzeuge im Hinblick auf ihre allgemeine Sicherheit und den Schutz der Fahrzeuginsassen und von ungeschützten Verkehrsteilnehmern, zur Änderung der Verordnung (EU) 2018/858 des Europäischen Parlaments und des Rates und zur Aufhebung der Verordnungen (EG) Nr. 78/2009, (EG) Nr. 79/2009 und (EG) Nr. 661/2009 des Europäischen Parlaments und des Rates sowie der Verordnungen (EG) Nr. 631/2009, (EU) Nr. 406/2010, (EU) Nr. 672/2010, (EU) Nr. 1003/2010, (EU) Nr. 1005/2010, (EU) Nr. 1008/2010, (EU) Nr. 1009/2010, (EU) Nr. 19/2011, (EU) Nr. 109/2011, (EU) Nr. 458/2011, (EU) Nr. 65/2012, (EU) Nr. 130/2012, (EU) Nr. 347/2012, (EU) Nr. 351/2012, (EU) Nr. 1230/2012 und (EU) 2015/166 der Kommission (1), insbesondere auf Artikel 4 Absatz 6 und Artikel 6 Absatz 6,
in Erwägung nachstehender Gründe:
|
(1) |
Artikel 6 der Verordnung (EU) 2019/2144 sieht vor, dass Kraftfahrzeuge der Klassen M und N mit bestimmten hochentwickelten Fahrerassistenzsystemen, u. a. einer Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre, ausgerüstet sein müssen. In Anhang II der genannten Verordnung sind grundlegende Anforderungen für die Typgenehmigung von Kraftfahrzeugen in Bezug auf die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre festgelegt. |
|
(2) |
Die alkoholempfindliche Wegfahrsperre erhöht die Verkehrssicherheit, indem verhindert wird, dass Personen mit einer Alkoholkonzentration im Blut, die einen festgelegten Grenzwert überschreitet, ein Kraftfahrzeug führen können. |
|
(3) |
Es ist erforderlich, spezifische technische Anforderungen für die Genehmigung von Kraftfahrzeugen im Hinblick auf die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre festzulegen. |
|
(4) |
Die Europäische Normenreihe EN 50436 legt Prüfmethoden und grundlegende Leistungsanforderungen für alkoholempfindliche Wegfahrsperren fest und bietet Leitlinien für Behörden, Entscheidungsträger, Käufer und Nutzer. Die Normen dieser Reihe enthalten auch besondere Bestimmungen für Kraftfahrzeuge, um den Einbau von alkoholempfindlichen Wegfahrsperren zu erleichtern. |
|
(5) |
Alkoholempfindliche Wegfahrsperren sind hauptsächlich für den Einbau im Anschlussmarkt bestimmt. Zu diesem Zweck werden sie an die Strom- und Steuerstromkreise des Fahrzeugs angeschlossen. Eine entsprechende Vorrichtung sollte die ordnungsgemäße Leistung oder Wartung sowie die Sicherheit des Fahrzeugs nicht beeinträchtigen und sollte so einfach wie möglich durch spezialisierte und geschulte Installateure zu bedienen sein. |
|
(6) |
Daher müssen die Fahrzeughersteller dazu verpflichtet werden, auf ihren Websites ein Dokument mit klaren Anweisungen für den Einbau der alkoholempfindlichen Wegfahrsperren (im Folgenden „Einbauanleitung“) zur Verfügung zu stellen, damit die Techniker in einem bestimmten Fahrzeugmodell eine alkoholempfindliche Wegfahrsperre ordnungsgemäß installieren können. |
|
(7) |
Da sich einige der in der Einbauanleitung enthaltenen Informationen möglicherweise auf sicherheitsbezogene Fahrzeugreparatur- und -wartungsinformationsdienste beziehen, sollten sie nur denjenigen unabhängigen Marktteilnehmern zur Verfügung stehen, die von akkreditierten Stellen gemäß Anhang X Anlage 3 der Verordnung (EU) 2018/858 des Europäischen Parlaments und des Rates (2) zugelassen wurden. |
|
(8) |
Die Tabelle mit der Liste der Anforderungen in Anhang II der Verordnung (EU) 2019/2144 enthält keinen Verweis auf Rechtsakte in Bezug auf die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre. Daher ist es erforderlich, einen Verweis auf die vorliegende Verordnung in diesen Anhang aufzunehmen. |
|
(9) |
Die Verordnung (EU) 2019/2144 sollte daher entsprechend geändert werden. |
|
(10) |
Da die Verordnung (EU) 2019/2144 ab dem 6. Juli 2022 gelten soll, sollte die vorliegende Verordnung ab demselben Zeitpunkt gelten. |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anforderungen an die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre
Die Typgenehmigung von Kraftfahrzeugen hinsichtlich der Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre unterliegt den Anforderungen des Anhangs I.
Artikel 2
Änderung der Verordnung (EU) 2019/2144
Anhang II der Verordnung (EU) 2019/2144 wird gemäß Anhang II dieser Verordnung geändert.
Artikel 3
Inkrafttreten und Anwendung
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Sie gilt ab dem 6. Juli 2022.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 19. April 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 325 vom 16.12.2019, S. 1.
(2) Verordnung (EU) 2018/858 des Europäischen Parlaments und des Rates vom 30. Mai 2018 über die Genehmigung und die Marktüberwachung von Kraftfahrzeugen und Kraftfahrzeuganhängern sowie von Systemen, Bauteilen und selbstständigen technischen Einheiten für diese Fahrzeuge, zur Änderung der Verordnungen (EG) Nr. 715/2007 und (EG) Nr. 595/2009 und zur Aufhebung der Richtlinie 2007/46/EG (ABl. L 151 vom 14.6.2018, S. 1).
ANHANG I
Technische Anforderungen
1.
Die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre muss den Einbau oder die Nachrüstung einer alkoholempfindlichen Wegfahrsperre ermöglichen, die den europäischen Normen EN 50436-1:2014 oder EN 50436-2:2014+A1:2015 entspricht.
2.
Das Fahrzeugsystem muss hinsichtlich der Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre in jedem Kraftfahrzeug der Klassen M und N dem entsprechenden Fahrzeugtyp entsprechen, das in dem Einbaudokument für die alkoholempfindliche Wegfahrsperre (im Folgenden „Einbaudokument“) gemäß der Europäischen Norm EN 50436-7:2016 festgelegt ist. Zu diesem Zweck muss das Einbaudokument mindestens eine der Optionen in EN 50436-7:2016 Anhang C (3a, 3b oder 3c) abdecken. Der Fahrzeughersteller kann im Einvernehmen mit der Genehmigungsbehörde und dem technischen Dienst das Einbaudokument entsprechend den späteren Überarbeitungen der Europäischen Norm vorlegen.
3.
Einbaudokument
3.1.
Das Einbaudokument muss eine ausführliche Beschreibung, Diagramme und Bilder zur Erläuterung des Einbaus einer alkoholempfindlichen Wegfahrsperre enthalten, die einen der folgenden Informationssätze umfasst:|
a) |
Angaben zu den Elementen: Batterieladezustand, Erdung, Betriebsbereitschaft des Fahrzeugs, Anlasseraktivierung; |
|
b) |
Angaben zu den Elementen: Batterieladezustand, Erdung, Betriebsbereitschaft des Fahrzeugs, Eingangs- und Ausgangs-Leitung für die Anlasseraktivierung oder -deaktivierung sowie eine optionale Erkennung der Antriebsfähigkeit (laufender Motor) bzw. Signal für Fahrzeugbewegung; oder |
|
c) |
Angaben zu den Elementen Batterieladezustand, Erdung und Verbindung zum Datenbus. |
3.2.
Alle zusätzlichen Software- und Hardware-Komponenten oder Verfahren, die für den Einbau einer alkoholempfindlichen Wegfahrsperre in ein Standardfahrzeug erforderlich sind, sind zu kennzeichnen und im Einbaudokument anzugeben.
3.3.
Die alkoholempfindliche Wegfahrsperre befindet sich standardmäßig im Sperrzustand. Der Sperrzustand der alkoholempfindlichen Wegfahrsperre wird entweder durch ein offenes Ausgangsrelais, ein entsprechendes Ausgangs-Signal oder durch eine entsprechende Digitalbusmeldung aktiviert. Das Schließen dieses Relais oder die Änderung des Sperrungs-Ausgangssignals in das Entsperrungs-Ausgangssignal oder die Übertragung der entsprechenden Entsperrungs-Nachricht an den Digitalbus erfolgt nach Abgabe einer akzeptierten Atemprobe mit einer Alkoholkonzentration unterhalb eines vordefinierten Schwellenwerts.
3.4.
Eine eingebaute alkoholempfindliche Wegfahrsperre darf nur in den Prozess des Motoranlassens bzw. die Freigabe einer selbsttätigen Bewegung des Fahrzeugs beim Einschalten des Hauptkontrollschalters des Fahrzeugs eingreifen, nicht hingegen bei laufendem Motor bzw. bei einem Fahrzeug in Bewegung.
4.
Zugang zu Informationen über die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre
4.1.
Die Fahrzeughersteller treffen die erforderlichen Vorkehrungen und richten die erforderlichen Verfahren ein, um sicherzustellen, dass Informationen über die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre in Form der einschlägigen Angaben des standardisierten Einbaudokuments gemäß Anhang X der Verordnung (EU) 2018/858 zugänglich sind. Da sich einige der in dem Einbaudokument enthaltenen Informationen möglicherweise auf sicherheitsbezogene Fahrzeugreparatur- und -wartungsinformationsdienste beziehen, sollten sie nur denjenigen unabhängigen Marktteilnehmern zur Verfügung stehen, die das in der Anlage 3 dieses Anhangs festgelegte Verfahren einhalten.
5.
Der Fahrzeughersteller fügt dem Einbaudokument eine Bescheinigung gemäß dem Muster der Anlage zu diesem Anhang bei.
Anlage
Bescheinigung des Herstellers
(Hersteller):
…
(Anschrift des Herstellers):
…
bescheinigt Folgendes:
Im Einklang mit Artikel 1 der Delegierten Verordnung (EU) 2021/1243 der Kommission (1) wird das Einbaudokument für die alkoholempfindliche Wegfahrsperre für die (den) nachfolgend aufgeführte(n) Fahrzeugmarken und Fahrzeugtyp(en) zur Verfügung gestellt: ...
Die Haupt-Website(s), auf denen das Einbaudokument verfügbar ist, ist (sind) in Anhang A dieser Bescheinigung angegeben. Die Kontaktdaten des Bevollmächtigten des Herstellers, der diese Bescheinigung unterzeichnet hat, sind in Anhang B dieser Bescheinigung aufgeführt.
Ort: …
Datum: …
[Unterschrift] [Funktion]
Anhang A: Internetadresse(n);
Anhang B: Kontaktdaten
(1) Delegierte Verordnung (EU) 2021/1243 der Kommission vom 19. April 2021 zur Ergänzung der Verordnung (EU) 2019/2144 des Europäischen Parlaments und des Rates durch Festlegung detaillierter Vorschriften für die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre in Kraftfahrzeugen und zur Änderung des Anhangs II der genannten Verordnung (ABl. L 272 vom …, S. 11).
ANHANG II
Änderung der Verordnung (EU) 2019/2144
In Anhang II der Verordnung (EU) 2019/2144 erhält die Zeile für die Anforderung E1 folgende Fassung:
|
„E1 Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre |
Delegierte Verordnung (EU) 2021/1243 der Kommission (*1) |
|
B |
B |
B |
B |
B |
B |
|
|
|
|
|
|
(*1) Delegierte Verordnung (EU) 2021/1243 der Kommission vom 19. April 2021 zur Ergänzung der Verordnung (EU) 2019/2144 des Europäischen Parlaments und des Rates durch Festlegung detaillierter Vorschriften für die Vorrichtung zum Einbau einer alkoholempfindlichen Wegfahrsperre in Kraftfahrzeugen und zur Änderung des Anhangs II der genannten Verordnung (ABl. L 272 vom …, S. 11).“
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/16 |
DELEGIERTE VERORDNUNG (EU) 2021/1244 DER KOMMISSION
vom 20. Mai 2021
zur Änderung des Anhangs X der Verordnung (EU) 2018/858 des Europäischen Parlaments und des Rates hinsichtlich des standardisierten Zugangs zu Fahrzeug-OBD-Informationen und zu Reparatur- und Wartungsinformationen sowie der Anforderungen und Verfahren für den Zugang Sicherheitsinformationen des Fahrzeugs
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) 2018/858 des Europäischen Parlaments und des Rates vom 30. Mai 2018 über die Genehmigung und die Marktüberwachung von Kraftfahrzeugen und Kraftfahrzeuganhängern sowie von Systemen, Bauteilen und selbstständigen technischen Einheiten für diese Fahrzeuge, zur Änderung der Verordnungen (EG) Nr. 715/2007 und (EG) Nr. 595/2009 und zur Aufhebung der Richtlinie 2007/46/EG (1), insbesondere auf Artikel 61 Absatz 11,
in Erwägung nachstehender Gründe:
|
(1) |
Nach Artikel 61 Absatz 2 der Verordnung (EG) 2018/858 müssen die Fahrzeughersteller die Fahrzeug-OBD-Informationen sowie die Reparatur- und Wartungsinformationen („vehicle repair and maintenance information“ — RMI) auf ihren Webseiten veröffentlichen. Es gibt jedoch keine harmonisierten Kriterien für die Art und Weise, in der diese Informationen zur Verfügung zu stellen sind; dies zwingt die unabhängigen Wirtschaftsakteure zur Anpassung an zahlreiche und unterschiedliche Webdienste und uneinheitliche Terminologie. |
|
(2) |
Im Bericht der Kommission an das Europäische Parlament und den Rat vom 9. Dezember 2016 über das Funktionieren der Regelung für den Zugang zu Reparatur- und Wartungsinformationen (2) wurde der Schluss gezogen, dass der Aufwand für die unabhängigen Wirtschaftsakteure durch Standardisierung der Webseiten und der entsprechenden Terminologie verringert werden könnte. |
|
(3) |
Da der Zugang zu Fahrzeug-OBD-Informationen und zu Reparatur- und Wartungsinformationen unabhängig vom Typ des Antriebsstrangs eines Fahrzeugs möglich sein sollte, ist es erforderlich, klarzustellen, dass dieser Zugang nicht nur für emissionsrelevante Anforderungen obligatorisch ist. |
|
(4) |
Am 15. September 2014 veröffentlichte das Europäische Komitee für Normung (CEN) Teile 1 bis 5 der Norm EN ISO 18541 „Straßenfahrzeuge — Standardisierter Zugang zu Reparatur- und Wartungsinformationen (RMI)“. Mit diesen Teilen soll der Austausch von Fahrzeug-OBD-Informationen und von Reparatur- und Wartungsinformationen zwischen Herstellern und unabhängigen Wirtschaftsakteuren durch Festlegung der technischen Anforderungen und der Verfahren zur Erleichterung des Zugangs zu diesen Informationen erleichtert werden. Daher sollte in Anhang X der Verordnung (EU) 2018/858 auf die Anforderungen der Teile 1 bis 5 der Norm EN ISO 18541-2014 verwiesen werden. |
|
(5) |
Da die Fahrzeug-OBD-Informationen und die Reparatur- und Wartungsinformationen Angaben enthalten, die wichtig für die Gewährleistung der Sicherheit des Fahrzeugs sind, sollte der Zugang zu bestimmten Sicherheitsmerkmalen der Fahrzeuge nur denjenigen unabhängigen Wirtschaftsakteuren gewährt werden, die die Anforderungen dieses Anhangs erfüllen. |
|
(6) |
Gemäß den Empfehlungen des in Artikel 66 Absatz 1 der Verordnung (EU) 2018/858 genannten Forums für den Zugang zu Fahrzeuginformationen sollten diese Anforderungen die Zulassung der betreffenden unabhängigen Wirtschaftsakteure und die Autorisierung ihrer Mitarbeiter, die die einschlägigen Tätigkeiten ausüben, durch akkreditierte Stellen umfassen. Daher ist es notwendig, das Verfahren für die Zulassung und Autorisierung des Zugangs unabhängiger Wirtschaftsakteure zu Sicherheitsmerkmalen der Fahrzeuge festzulegen, das auf dem „System für die Akkreditierung, Genehmigung und Autorisierung des Zugangs zu sicherheitsrelevanten Reparatur- und Wartungsinformationen (RMI)“ beruhen sollte, das am 19. Mai 2016 von der Europäischen Kooperation für Akkreditierung validiert wurde. Außerdem ist zu sicherzustellen, dass diese Akteure nicht an unrechtmäßigen Geschäftstätigkeiten beteiligt sind. |
|
(7) |
Darüber hinaus müssen die Rolle und die Zuständigkeiten der Stellen festgelegt werden, die an der Zulassung und Autorisierung des Zugangs zu sicherheitsrelevanten Reparatur- und Wartungsinformationen für unabhängige Wirtschaftsakteure und ihre Mitarbeiter beteiligt sind. |
|
(8) |
Damit die Mitgliedstaaten und nationalen Behörden sowie die Wirtschaftsakteure sich auf die Anwendung der durch diese Verordnung eingeführten neuen Vorschriften einstellen können, sollte der Geltungsbeginn aufgeschoben werden. |
|
(9) |
Anhang X der Verordnung (EU) 2018/858 sollte daher entsprechend geändert werden — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang X der Verordnung (EU) 2018/858 wird gemäß dem Anhang der vorliegenden Verordnung geändert.
Artikel 2
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Sie gilt ab dem 30. Juli 2023.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 20. Mai 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 151 vom 14.6.2018, S. 1.
(2) Bericht der Kommission an das Europäische Parlament und den Rat über das Funktionieren der Regelung für den Zugang zu Reparatur- und Wartungsinformationen für Fahrzeuge, eingerichtet durch die Verordnung (EG) Nr. 715/2007 über die Typgenehmigung von Kraftfahrzeugen hinsichtlich der Emissionen von leichten Personenkraftwagen und Nutzfahrzeugen (Euro 5 und Euro 6) und über den Zugang zu Reparatur- und Wartungsinformationen für Fahrzeuge, COM(2016) 782 final.
ANHANG
Anhang X der Verordnung (EU) 2018/858 wird wie folgt geändert:
|
1. |
Nummer 2.1 erhält folgende Fassung:
|
|
2. |
Nummer 2.5.2 erhält folgende Fassung:
|
|
3. |
In Nummer 2.9 erhält der erste Absatz folgende Fassung: „Für die Zwecke von Fahrzeug-OBD-Diagnose-, Reparatur-, Wartungs-, Überwachungs- und Inspektionszwecken ist der unmittelbare Datenstrom des Fahrzeugs einschließlich Fehlercodes und Diagnosefunktionen über die serielle Schnittstelle auf dem Standard-Datenübertragungsanschluss gemäß Absatz 6.5.1.4 sowie den Spezifikationen in Anhang 11 Anlage 1 Abschnitt 6.5.3 der Regelung Nr. 83 der Wirtschaftskommission der Vereinten Nationen für Europa (UNECE) (*1) und gemäß Anhang 9B Absatz 4.7.3 sowie den in Anhang 9B Anlage 6 genannten Bezugsnormentexten der Regelung Nr. 49 der Wirtschaftskommission der Vereinten Nationen für Europa (UNECE) (*2) zur Verfügung zu stellen. (*1) Regelung Nr. 83 der Wirtschaftskommission der Vereinten Nationen für Europa (UNECE) — Einheitliche Bedingungen für die Genehmigung der Fahrzeuge hinsichtlich der Emission von Schadstoffen aus dem Motor entsprechend den Kraftstofferfordernissen des Motors (ABl. L 42 vom 15.2.2012, S. 1)." (*2) Regelung Nr. 49 der Wirtschaftskommission der Vereinten Nationen für Europa (UNECE) — Einheitliche Bedingungen für die Genehmigung der Motoren mit Selbstzündung, der mit Erdgas betriebenen und der mit Flüssiggas betriebenen Motoren mit Fremdzündung sowie der mit einem Motor mit Selbstzündung, einem mit Erdgas betriebenen oder einem mit Flüssiggas betriebenen Motor mit Fremdzündung ausgestatteten Fahrzeuge hinsichtlich der Emissionen von Schadstoffen aus dem Motor (ABl. L 180 vom 8.7.2011, S. 53).“" |
|
4. |
In Nummer 6.1 erhält der erste Absatz folgende Fassung: „Die Einhaltung der Verpflichtung des Herstellers, Fahrzeug-OBD-Informationen sowie Reparatur- und Wartungsinformationen auf seiner Webseite in einem standardisierten Format zur Verfügung zu stellen, wird durch Einhaltung der in Nummer 2.1 genannten Teile der Norm EN ISO 18541 vermutet.“ |
|
5. |
Nummer 6.2 erhält folgende Fassung: „Der Zugang zu Sicherheitsmerkmalen der Fahrzeuge muss auch unabhängigen Wirtschaftsakteuren offenstehen, wobei für den Schutz durch Sicherheitstechnik nach folgenden Anforderungen zu sorgen ist:“ |
|
6. |
Nummer 6.3. wird wie folgt geändert:
|
|
7. |
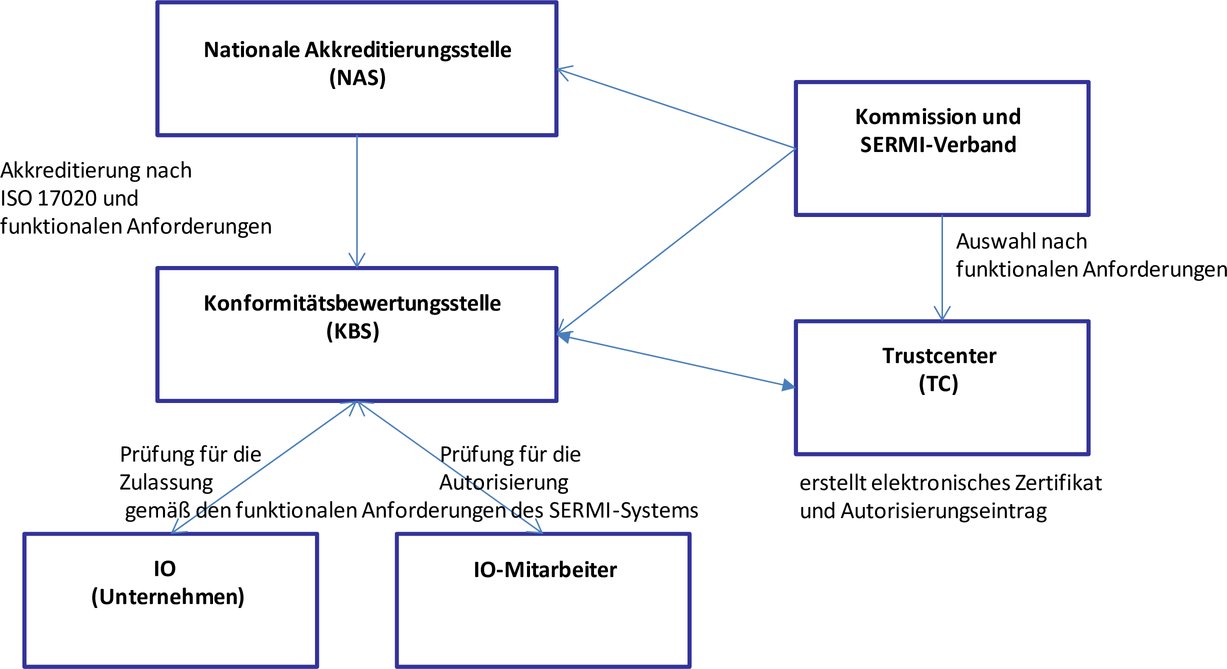
Die folgende Anlage 3 wird eingefügt: „Anlage 3 Verfahren für die Zulassung und Autorisierung des Zugangs unabhängiger Wirtschaftsakteure zu den Sicherheitsmerkmalen der Fahrzeuge (*5) 1. Geltungsbereich Diese Anlage enthält die Anforderungen für die Zulassung und Autorisierung unabhängiger Wirtschaftsakteure, die Zugang zu sicherheitsrelevanten Reparatur- und Wartungsinformationen (RMI) benötigen. In ihr werden die zuständigen Stellen bzw. erforderlichen Verfahren für die Zulassung und Autorisierung unabhängiger Wirtschaftsakteure festgelegt, damit diesen Zugang zu sicherheitsrelevanten Reparatur- und Wartungsinformationen für leichte Personenkraftwagen und Nutzfahrzeuge sowie schwere Nutzfahrzeuge gewährt werden kann. 2. Begriffsbestimmungen und Abkürzungen 2.1. Begriffsbestimmungen Für die Zwecke dieser Anlage gelten die folgenden Begriffsbestimmungen: 2.1.1. „Akkreditierung“ „Akkreditierung“ bezeichnet die Akkreditierung im Sinne von Artikel 2 Nummer 10 der Verordnung (EG) Nr. 765/2008. 2.1.2. „IO-Mitarbeiter“ „IO-Mitarbeiter“ bezeichnet den Mitarbeiter eines zugelassenen unabhängigen Wirtschaftsakteurs („independent operator“ — IO), der mit Autorisierung der zuständigen Konformitätsbewertungsstelle (KBS) Zugang zu sicherheitsrelevanten RMI hat. 2.1.3. „Sicherheitsrelevante Reparatur- und Wartungsinformationen“ oder „sicherheitsrelevante RMI“ „Sicherheitsrelevante Reparatur- und Wartungsinformationen“ oder „sicherheitsrelevante RMI“ bezeichnet die Informationen, Software, Funktionen und Dienstleistungen, die für die Reparatur und Wartung der vom Hersteller in einem Fahrzeug eingebauten Funktionen erforderlich sind, um zu verhindern, dass das Fahrzeug gestohlen oder weggefahren wird bzw. um zu ermöglichen, dass das Fahrzeug zurückverfolgt und wieder in Besitz genommen werden kann. 2.1.4. Prüfbescheinigung über die Zulassung „Prüfbescheinigung über die Zulassung“ bezeichnet die Bescheinigung, die die Konformitätsbewertungsstelle den unabhängigen Wirtschaftsakteuren ausstellt, die die Kriterien für die Zulassung gemäß dieser Anlage erfüllen; durch sie wird bestätigt, dass diese unabhängigen Wirtschaftsakteure zugelassen sind und dass IO-Mitarbeiter die Autorisierung für den Zugang zu sicherheitsrelevanten RMI beantragen können. 2.1.5. Prüfbescheinigung über die Autorisierung „Prüfbescheinigung über die Autorisierung“ bezeichnet die Bescheinigung, die die Konformitätsbewertungsstelle den IO-Mitarbeitern ausstellt, die die Kriterien für die Autorisierung gemäß dieser Anlage erfüllen; durch sie wird bestätigt, dass diese IO-Mitarbeiter autorisiert sind, den Zugang zu sicherheitsrelevanten RMI auf der Webseite eines Fahrzeugherstellers zu beantragen. 2.1.6. „Trustcenter“ oder „TC“ „Trustcenter“ oder „TC“ bezeichnet die vom SERMI benannte und von der Kommission zugelassene Stelle, die für Folgendes zuständig ist:
2.1.7. „Sicherheitstoken“ „Sicherheitstoken“ bezeichnet ein Gerät, das eine sichere Authentifizierung eines unabhängigen Wirtschaftsakteurs ermöglicht. 2.1.8. „Elektronisches Zertifikat“ „Elektronisches Zertifikat“ bezeichnet ein elektronisches Zertifikat, das eine digitale Signatur des ausstellenden Trustcenters erfordert, um einen öffentlichen Schlüssel gemäß der Norm ISO 9594 mit der Identität des IO-Mitarbeiters zu verbinden. 2.1.9. „Datenbank für die Autorisierungen“ „Datenbank für die Autorisierungen“ bezeichnet eine vom Trustcenter geführte Datenbank, in der die Autorisierungsdaten der anonymisierten autorisierten IO-Mitarbeiter und die Registrierung der zugelassenen unabhängigen Wirtschaftsakteure gespeichert sind. 2.1.10. „Datenbank mit den Zertifizierungen“ „Datenbank mit den Zertifizierungen“ bezeichnet eine vom Trustcenter geführte Datenbank zur Verwaltung der Gültigkeit der elektronischen Zertifikate und der Kennungen der autorisierten IO-Mitarbeiter. 2.1.11. „Europäische Kooperation für Akkreditierung“ oder „EA“ „Europäische Kooperation für Akkreditierung“ oder „EA“ bezeichnet die von der Kommission gemäß Artikel 14 der Verordnung (EG) Nr. 765/2008 anerkannte Stelle, die für die Entwicklung, Aufrechterhaltung und Umsetzung der Akkreditierung in der Union zuständig ist. 2.1.12. „Forum für den Zugang zu sicherheitsrelevanten Reparatur- und Wartungsinformationen“ oder „SERMI“ Das „Forum für den Zugang zu sicherheitsrelevanten Reparatur- und Wartungsinformationen“ oder „SERMI“ bezeichnet die Stelle, die für die Koordinierung und die Beratung der Kommission in Bezug auf die Umsetzung der Akkreditierungs-, Zulassungs- und Autorisierungsverfahren für den Zugang zu sicherheitsrelevanten RMI zuständig ist. 2.1.13. „Zuständige Behörden“ „Zuständige Behörden“ bezeichnet die Behörden, die rechtlich befugt sind, im Bereich des Schutzes vor sowie der Ermittlung und Verfolgung von Straftaten im Bereich der Fahrzeugsicherheit tätig zu werden. 3. Akkreditierung von Konformitätsbewertungsstellen, Zulassung von unabhängigen Wirtschaftsakteuren und Autorisierung von IO-Mitarbeitern Nur Konformitätsbewertungsstellen, die von der nationalen Akkreditierungsstelle (NAS) gemäß Artikel 2 Nummer 11 der Verordnung (EG) Nr. 765/2008 des Mitgliedstaats, in dem sie niedergelassen sind, akkreditiert sind, stellen Prüfbescheinigungen über die Zulassung aus, in denen bescheinigt wird, dass ein unabhängiger Wirtschaftsakteur zugelassen ist, und Prüfbescheinigungen über die Autorisierung, in denen bescheinigt wird, dass ein IO-Mitarbeiter Zugang zu sicherheitsrelevanten RMI hat. Die Zulassung der unabhängigen Wirtschaftsakteure und die Autorisierung der IO-Mitarbeiter werden für einen Zeitraum von 60 Monaten ab dem Datum der Ausstellung der entsprechenden Prüfbescheinigungen erteilt. Unabhängige Wirtschaftsakteure, die Zugang zu sicherheitsrelevanten RMI erhalten möchten, beantragen bei einer Konformitätsbewertungsstelle, die von der nationalen Akkreditierungsstelle des Mitgliedstaats, in dem der unabhängige Wirtschaftsakteur niedergelassen ist, akkreditiert ist, eine Prüfbescheinigung über die Zulassung. IO-Mitarbeiter, die Zugriff auf sicherheitsrelevante RMI erhalten sollen, beantragen bei einer Konformitätsbewertungsstelle, die durch die nationale Akkreditierungsstelle des Mitgliedstaats, in dem der unabhängige Wirtschaftsakteur niedergelassen ist, akkreditiert ist, eine Prüfbescheinigung über die Autorisierung. Die Konformitätsbewertungsstellen unterrichten die Trustcenter über alle ausgestellten Prüfbescheinigungen über die Zulassung bzw. über die Autorisierung, woraufhin die Trustcenter einen Autorisierungseintrag erstellen und einen Sicherheitstoken sowie ein elektronisches Zertifikat ausgeben, anhand derer die IO-Mitarbeiter auf der RMI-Webseite des Fahrzeugherstellers eindeutig identifiziert werden können. Die Konformitätsbewertungsstellen stellen den jeweiligen IO-Mitarbeitern einen Sicherheitstoken und das elektronische Zertifikat zur Verfügung. Fahrzeughersteller können eine Gebühr für die Registrierung von IO-Mitarbeitern auf der RMI-Webseite des jeweiligen Fahrzeugherstellers und für den Zugang zu sicherheitsrelevanten RMI verlangen. Diese Gebühr muss in einem angemessenen Verhältnis zu den Kosten für die Registrierung und die Bereitstellung des Zugangs stehen. Die zu entrichtenden Gebühren sind auf den RMI-Webseiten des Fahrzeugherstellers anzugeben. Jegliche Übermittlung von digitalen Daten zwischen unabhängigen Wirtschaftsakteuren, Trustcentern und Konformitätsbewertungsstellen erfolgt zeitnah als Business-to-Business-Transaktion (B2B) unter Verwendung sicherer Protokolle.
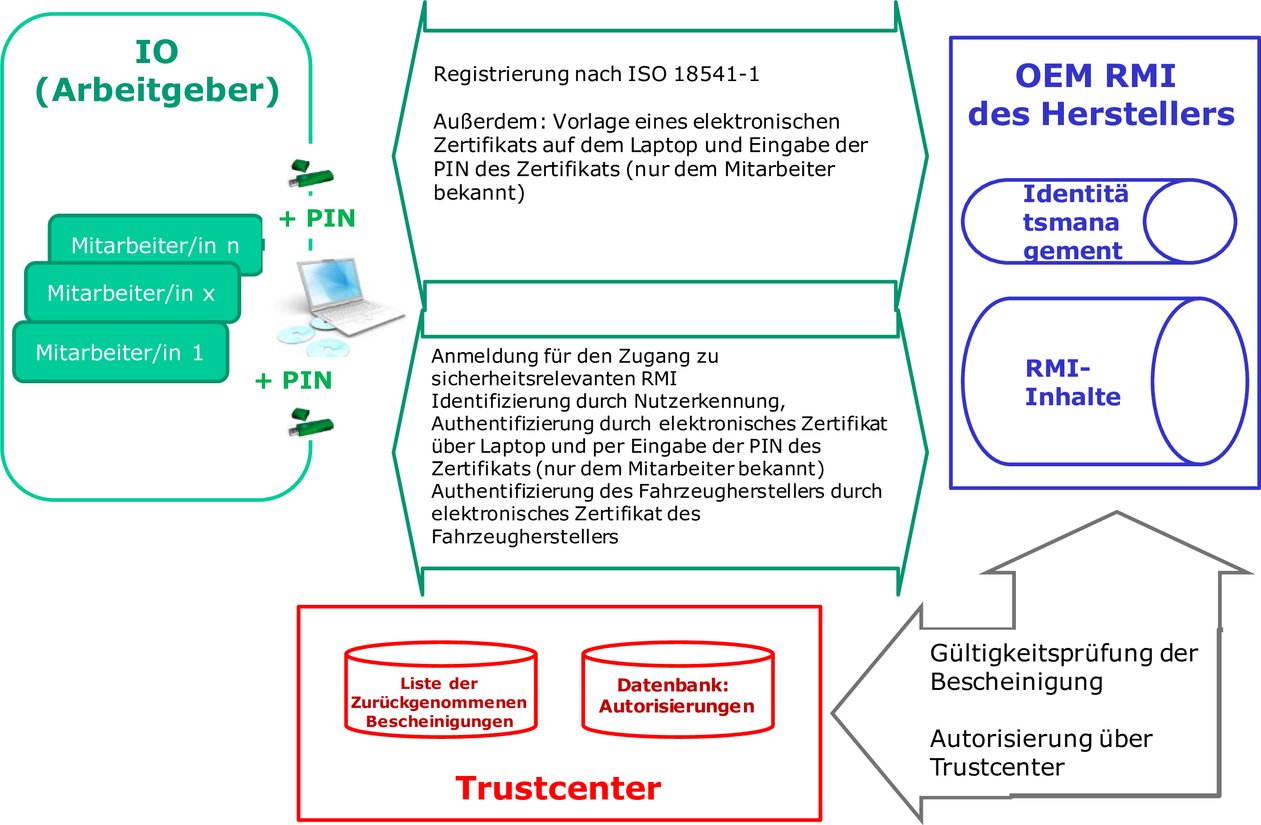
Eine Erklärung zur Bescheinigung, dass der unabhängige Wirtschaftsakteur einer legitimen Geschäftstätigkeit im Sinne von Nummer 6.3 dieses Anhangs nachgeht, ist von dem unabhängigen Wirtschaftsakteur zu unterzeichnen, der die Autorisierung durch die Konformitätsbewertungsstelle beantragt. Ein unabhängiger Wirtschaftsakteur wird erst nach Prüfung durch die Konformitätsbewertungsstelle zugelassen; dabei wird überprüft, ob die Erklärung unterzeichnet wurde und ob der unabhängige Wirtschaftsakteur sowie die jeweiligen IO-Mitarbeiter die Anforderungen dieser Anlage erfüllen. Die jeweiligen IO-Mitarbeiter dürfen erst nach Prüfung durch eine Konformitätsbewertungsstelle autorisiert werden. Die Konformitätsbewertungsstellen prüfen die vorgelegten Unterlagen und überprüfen, ob der betreffende IO-Mitarbeiter einen früheren Antrag auf Autorisierung gestellt hat, der von der betreffenden oder einer anderen Konformitätsbewertungsstelle auf Unionsebene abgelehnt wurde. Die Konformitätsbewertungsstellen übermitteln dem Trustcenter alle Daten, die das Trustcenter benötigt, um das elektronische Zertifikat und den Sicherheitstoken zu erstellen; beides übermittelt die Konformitätsbewertungsstelle an die IO-Mitarbeiter. IO-Mitarbeiter, die autorisiert wurden, erhalten von den jeweiligen Konformitätsbewertungsstellen die mit dem elektronischen Zertifikat verknüpfte PIN.
3.1. Überblick über den Zugang zu sicherheitsrelevanten RMI Die Fahrzeughersteller gewähren über ihre RMI-Webseite Zugang zu sicherheitsrelevanten RMI, sofern die IO-Mitarbeiter autorisiert sind und ihre Prüfbescheinigung über die Autorisierung vorlegen können und sofern der unabhängige Wirtschaftsakteur, für den die IO-Mitarbeiter tätig sind, über eine Prüfbescheinigung über die Zulassung verfügt. Die Hersteller können autorisierten IO-Mitarbeitern, die für zugelassene unabhängige Wirtschaftsakteure tätig sind, mittels einer speziellen Anwendung, die mit der RMI-Webseite verknüpft ist, den Zugang zu einer Online-Bestellmöglichkeit für sicherheitsrelevante Teile anbieten. Geht ein Antrag auf Zugang zu einer RMI-Webseite ein, erfordern die Webseiten des Fahrzeugherstellers die Identifizierung anhand der eindeutigen Kennung des IO-Mitarbeiters und fordern die Authentifizierungsdaten an. Die Authentifizierung von IO-Mitarbeitern erfolgt ausschließlich unter Verwendung elektronischer Zertifikate. Nach Erhalt eines elektronischen Zertifikats überprüft die RMI-Webseite des Fahrzeugherstellers durch Kommunikation mit dem im elektronischen Zertifikat angegebenen Trustcenter die eindeutige Kennung des IO-Mitarbeiters sowie den aktuellen Status des elektronischen Zertifikats und der Autorisierung. Jegliche Übermittlung von digitalen Daten zwischen unabhängigen Wirtschaftsakteuren, Fahrzeugherstellern, Trustcentern und Konformitätsbewertungsstellen erfolgt zeitnah als Business-to-Business-Transaktion (B2B) unter Verwendung sicherer Protokolle. Sobald die eindeutige Kennung und der Autorisierungsstatus des IO-Mitarbeiters überprüft worden sind, stellt der Fahrzeughersteller auf seiner Webseite den Zugang zu den erforderlichen sicherheitsrelevanten RMI bereit.
4. Detaillierte Vorschriften für den Zugang zu sicherheitsrelevanten RMI 4.1. Die Rolle des SERMI 4.1.1. Zuständigkeiten und Verpflichtungen Das SERMI überwacht die Umsetzung des Akkreditierungsverfahrens in allen Mitgliedstaaten und unterrichtet die Kommission entsprechend. Das SERMI berät die Kommission über Anträge auf Änderung des Akkreditierungsverfahrens.
4.1.2. Auswahl der Trustcenter Das Trustcenter wird vom SERMI ausgewählt und der Kommission zur Genehmigung mitgeteilt. Die ausgewählten Trustcenter müssen der Norm ETSI TS 319 411-3 entsprechen und die Anforderungen an elektronische Signaturen gemäß der Verordnung (EU) Nr. 910/2014 des Europäischen Parlaments und des Rates (*6) sowie die Anforderungen unter Nummer 4.6 dieser Anlage erfüllen. Darüber hinaus müssen die Trustcenter:
4.2. Die Rolle der nationalen Akkreditierungsstellen Die nationale Akkreditierungsstelle ist für die Akkreditierung von Konformitätsbewertungsstellen zum Zwecke der Zulassung von unabhängigen Wirtschaftsakteuren und der Autorisierung von IO-Mitarbeitern für den Zugang zu sicherheitsrelevanten RMI zuständig. 4.2.1. Zuständigkeiten und Anforderungen Die Zuständigkeiten und Anforderungen der nationalen Akkreditierungsstellen sind in den Artikeln 8 bis 12 der Verordnung (EG) Nr. 765/2008 festgelegt. 4.2.2. Kriterien für die Akkreditierung der Konformitätsbewertungsstelle Konformitätsbewertungsstellen müssen als Prüfstellen des Typs A gemäß ISO/IEC 17020:2012 akkreditiert sein. Konformitätsbewertungsstellen müssen die Anforderungen in Bezug auf das höchste Maß an Unabhängigkeit erfüllen. Darüber hinaus bewerten die nationalen Akkreditierungsstellen die Fähigkeiten der Konformitätsbewertungsstellen zur Einhaltung der Anforderungen der Nummern 4.3.1 bis 4.3.4. Das für die Prüfung unabhängiger Wirtschaftsakteure zuständige Personal muss über für die von ihm wahrgenommenen Aufgaben angemessene Kenntnisse im Bereich des Reparatur- und Wartungsgeschäfts für Kraftfahrzeuge und der Besonderheiten des Kfz-Anschlussmarktes (Aftermarket) verfügen. 4.3. Die Rolle der Konformitätsbewertungsstellen Die Konformitätsbewertungsstelle ist für die Prüfung der unabhängigen Wirtschaftsakteure und ihrer jeweiligen IO-Mitarbeiter sowie für die Ausstellung von Prüfbescheinigungen über die Zulassung bzw. über die Autorisierung gemäß dieser Anlage und für die Rücknahme dieser Bescheinigungen zuständig. 4.3.1. Zuständigkeiten und Anforderungen
4.3.2. Erneuerung der Zulassung Die Konformitätsbewertungsstellen führen auf Antrag eines unabhängigen Wirtschaftsakteurs oder sechs Monate vor Ablauf der Gültigkeit der Zulassung eine Vor-Ort-Prüfung durch und erneuern im Falle eines positiven Prüfergebnisses die Zulassung. Die Konformitätsbewertungsstellen stellen einem unabhängigen Wirtschaftsakteur, der die Zulassungskriterien erfüllt, eine neue Prüfbescheinigung über die Zulassung aus. Die Konformitätsbewertungsstellen bewerten die Anträge auf Erneuerung der Autorisierungen und stellen den IO-Mitarbeitern, die die Autorisierungskriterien erfüllen, eine Prüfbescheinigung über die Autorisierung aus. 4.3.3. Kriterien über die Zulassung der unabhängigen Wirtschaftsakteure durch die Konformitätsbewertungsstelle Vor der Zulassung eines unabhängigen Wirtschaftsakteurs und bei einer Vor-Ort-Prüfung während der Gültigkeitsdauer der Zulassung prüfen die Konformitätsbewertungsstellen Folgendes:
4.3.4. Kriterien für die Autorisierung der IO-Mitarbeiter durch die Konformitätsbewertungsstelle Vor der Autorisierung eines Mitarbeiters als IO-Mitarbeiter und bei einer Vor-Ort-Prüfung während der Gültigkeitsdauer der Zulassung stellen die Konformitätsbewertungsstellen sicher, dass:
4.4. Rolle der unabhängigen Wirtschaftsakteure 4.4.1. Zuständigkeiten und Anforderungen
4.5. Rolle der IO-Mitarbeiter 4.5.1. Zuständigkeiten und Anforderungen
4.6. Rolle des Trustcenters Die Trustcenter erstellen und versenden die elektronischen Zertifikate über die jeweiligen Konformitätsbewertungsstellen an die unabhängigen Wirtschaftsakteure und die IO-Mitarbeiter. Die Trustcenter führen eine Datenbank über die Prüfbescheinigungen über die Autorisierung, die ausgestellt worden sind. Die Trustcenter stellen den Fahrzeugherstellern eine Schnittstelle zur Verfügung, durch die der Status der elektronischen Zertifikate und der Prüfbescheinigungen über die Autorisierung überprüft werden kann. Die Trustcenter müssen die Informationen über IO-Mitarbeiter für einen zusätzlichen Zeitraum von höchstens 60 Monaten in der Datenbank für die Autorisierungen aufbewahren. Dieser Zeitraum darf nicht länger sein als die verbleibende Gültigkeitsdauer der Zulassung des unabhängigen Wirtschaftsakteurs, für den der IO-Mitarbeiter tätig ist. 4.6.1. Zuständigkeiten und Anforderungen
4.7. Rolle der Fahrzeughersteller Die Fahrzeughersteller gewähren allen zugelassenen unabhängigen Wirtschaftsakteuren und allen autorisierten IO-Mitarbeitern Zugang zu sicherheitsrelevanten Reparatur- und Wartungsinformationen. Die Fahrzeughersteller kommunizieren mit den Trustcentern, um den Autorisierungs- und Authentifizierungsstatus der IO-Mitarbeiter, die den Zugang zu diesen Informationen anfordern, zu überprüfen. 4.7.1. Zuständigkeiten und Anforderungen
4.7.2. Verfahrensvorschriften für Fahrzeughersteller Fahrzeughersteller gewähren nur dann Zugang zu sicherheitsrelevanten RMI, wenn alle folgenden Verfahrensvorschriften erfüllt sind:
(*5) Die Anforderungen dieser Anlage beruhen auf dem durch die Europäische Kooperation für Akkreditierung am 19. Mai 2016 festgelegten System für die Akkreditierung, Zulassung und Autorisierung für den Zugang zu sicherheitsrelevanten Reparatur- und Wartungsinformationen (‚Scheme for accreditation, approval and authorization to Access Security-related Repair and Maintenance Information‘ — RMI) (https://www.vehiclesermi.eu/)." (*6) Verordnung (EU) Nr. 910/2014 des Europäischen Parlaments und des Rates vom 23. Juli 2014 über elektronische Identifizierung und Vertrauensdienste für elektronische Transaktionen im Binnenmarkt und zur Aufhebung der Richtlinie 1999/93/EG (ABl. L 257 vom 28.8.2014, S. 73)." |
(*1) Regelung Nr. 83 der Wirtschaftskommission der Vereinten Nationen für Europa (UNECE) — Einheitliche Bedingungen für die Genehmigung der Fahrzeuge hinsichtlich der Emission von Schadstoffen aus dem Motor entsprechend den Kraftstofferfordernissen des Motors (ABl. L 42 vom 15.2.2012, S. 1).
(*2) Regelung Nr. 49 der Wirtschaftskommission der Vereinten Nationen für Europa (UNECE) — Einheitliche Bedingungen für die Genehmigung der Motoren mit Selbstzündung, der mit Erdgas betriebenen und der mit Flüssiggas betriebenen Motoren mit Fremdzündung sowie der mit einem Motor mit Selbstzündung, einem mit Erdgas betriebenen oder einem mit Flüssiggas betriebenen Motor mit Fremdzündung ausgestatteten Fahrzeuge hinsichtlich der Emissionen von Schadstoffen aus dem Motor (ABl. L 180 vom 8.7.2011, S. 53).“
(*3) Im Sinne des Artikels 3 Absatz 10 der Verordnung (EG) Nr. 715/2007.
(*4) Im Sinne des Artikels 3 Absatz 8 der Verordnung (EG) Nr. 595/2009.“
(*5) Die Anforderungen dieser Anlage beruhen auf dem durch die Europäische Kooperation für Akkreditierung am 19. Mai 2016 festgelegten System für die Akkreditierung, Zulassung und Autorisierung für den Zugang zu sicherheitsrelevanten Reparatur- und Wartungsinformationen (‚Scheme for accreditation, approval and authorization to Access Security-related Repair and Maintenance Information‘ — RMI) (https://www.vehiclesermi.eu/).
(*6) Verordnung (EU) Nr. 910/2014 des Europäischen Parlaments und des Rates vom 23. Juli 2014 über elektronische Identifizierung und Vertrauensdienste für elektronische Transaktionen im Binnenmarkt und zur Aufhebung der Richtlinie 1999/93/EG (ABl. L 257 vom 28.8.2014, S. 73).“
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/29 |
VERORDNUNG (EU) 2021/1245 DER KOMMISSION
vom 23. Juli 2021
zur Genehmigung einer Änderung der Spezifikation einer geschützten Ursprungsbezeichnung oder einer geschützten geografischen Angabe („Coteaux du Pont du Gard“ (g. g. A.))
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) Nr. 1308/2013 des Europäischen Parlaments und des Rates vom 17. Dezember 2013 über eine gemeinsame Marktorganisation für landwirtschaftliche Erzeugnisse und zur Aufhebung der Verordnungen (EWG) Nr. 922/72, (EWG) Nr. 234/79, (EG) Nr. 1037/2001 und (EG) Nr. 1234/2007 des Rates (1), insbesondere auf Artikel 99,
in Erwägung nachstehender Gründe:
|
(1) |
Die Kommission hat den Antrag auf Genehmigung einer Änderung der Spezifikation der geschützten geografischen Angabe „Coteaux du Pont du Gard“ geprüft, den Frankreich gemäß Artikel 105 der Verordnung (EU) Nr. 1308/2013 gestellt hat. |
|
(2) |
Die Kommission hat den Antrag auf Genehmigung einer Änderung der Spezifikation gemäß Artikel 97 Absatz 3 der Verordnung (EU) Nr. 1308/2013 im Amtsblatt der Europäischen Union (2) veröffentlicht. |
|
(3) |
Bei der Kommission ist kein Einspruch gemäß Artikel 98 der Verordnung (EU) Nr. 1308/2013 eingegangen. |
|
(4) |
Die Änderung der Spezifikation sollte daher gemäß Artikel 99 der Verordnung (EU) Nr. 1308/2013 genehmigt werden. |
|
(5) |
Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ausschusses für die gemeinsame Organisation der Agrarmärkte — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Die im Amtsblatt der Europäischen Union veröffentlichte Änderung der Spezifikation für den Namen „Coteaux du Pont du Gard“ (g. g. A.) wird genehmigt.
Artikel 2
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 23. Juli 2021
Für die Kommission
Im Namen der Präsidentin
Janusz WOJCIECHOWSKI
Mitglied der Kommission
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/30 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/1246 DER KOMMISSION
vom 28. Juli 2021
zur Änderung der Verordnung (EG) Nr. 1484/95 in Bezug auf die Festsetzung der repräsentativen Preise in den Sektoren Geflügelfleisch und Eier sowie für Eieralbumin
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) Nr. 1308/2013 des Europäischen Parlaments und des Rates vom 17. Dezember 2013 über eine gemeinsame Marktorganisation für landwirtschaftliche Erzeugnisse und zur Aufhebung der Verordnungen (EWG) Nr. 922/72, (EWG) Nr. 234/79, (EG) Nr. 1037/2001 und (EG) Nr. 1234/2007 des Rates (1), insbesondere auf Artikel 183 Buchstabe b,
gestützt auf die Verordnung (EU) Nr. 510/2014 des Europäischen Parlaments und des Rates vom 16. April 2014 über die Handelsregelung für bestimmte aus landwirtschaftlichen Erzeugnissen hergestellte Waren und zur Aufhebung der Verordnungen (EG) Nr. 1216/2009 und (EG) Nr. 614/2009 des Rates (2), insbesondere auf Artikel 5 Absatz 6 Buchstabe a,
in Erwägung nachstehender Gründe:
|
(1) |
Mit der Verordnung (EG) Nr. 1484/95 der Kommission (3) wurden Durchführungsbestimmungen zur Regelung der zusätzlichen Einfuhrzölle in den Sektoren Geflügelfleisch und Eier sowie für Eieralbumin festgelegt und die diesbezüglichen repräsentativen Preise festgesetzt. |
|
(2) |
Aus der regelmäßig durchgeführten Kontrolle der Angaben, auf die sich die Festsetzung der repräsentativen Preise für Erzeugnisse der Sektoren Geflügelfleisch und Eier sowie für Eieralbumin stützt, geht hervor, dass die repräsentativen Preise für die Einfuhren bestimmter Erzeugnisse unter Berücksichtigung der von ihrem Ursprung abhängigen Preisschwankungen zu ändern sind. |
|
(3) |
Die Verordnung (EG) Nr. 1484/95 sollte daher entsprechend geändert werden. |
|
(4) |
Da sicherzustellen ist, dass diese Maßnahme so bald wie möglich, nachdem die aktualisierten Angaben vorliegen, Anwendung findet, sollte diese Verordnung am Tag ihrer Veröffentlichung in Kraft treten — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang I der Verordnung (EG) Nr. 1484/95 erhält die Fassung des Anhangs der vorliegenden Verordnung.
Artikel 2
Diese Verordnung tritt am Tag ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 28. Juli 2021
Für die Kommission,
im Namen der Präsidentin,
Wolfgang BURTSCHER
Generaldirektor
Generaldirektion Landwirtschaft und ländliche Entwicklung
(1) ABl. L 347 vom 20.12.2013, S. 671.
(2) ABl. L 150 vom 20.5.2014, S. 1.
(3) Verordnung (EG) Nr. 1484/95 der Kommission vom 28. Juni 1995 mit Durchführungsbestimmungen zur Regelung der zusätzlichen Einfuhrzölle und zur Festsetzung der repräsentativen Preise in den Sektoren Geflügelfleisch und Eier sowie für Eieralbumin und zur Aufhebung der Verordnung Nr. 163/67/EWG (ABl. L 145 vom 29.6.1995, S. 47).
ANHANG
„ANHANG I
|
KN-Code |
Warenbezeichnung |
Repräsentativer Preis (EUR/100 kg) |
Sicherheit gemäß Artikel 3 (EUR/100 kg) |
Ursprung (1) |
|
0207 14 10 |
Geflügelteilstücke ohne Knochen der Art Gallus domesticus, gefroren |
140,3 176,2 |
60 42 |
BR TH |
(1) Nomenklatur der Länder gemäß der Verordnung (EU) Nr. 1106/2012 der Kommission vom 27. November 2012 zur Durchführung der Verordnung (EG) Nr. 471/2009 des Europäischen Parlaments und des Rates über Gemeinschaftsstatistiken des Außenhandels mit Drittländern hinsichtlich der Aktualisierung des Verzeichnisses der Länder und Gebiete (ABl. L 328 vom 28.11.2012, S. 7).“
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/33 |
VERORDNUNG (EU) 2021/1247 DER KOMMISSION
vom 29. Juli 2021
zur Änderung des Anhangs II der Verordnung (EG) Nr. 396/2005 des Europäischen Parlaments und des Rates hinsichtlich der Höchstgehalte an Rückständen von Mandestrobin in Trauben und Erdbeeren
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EG) Nr. 396/2005 des Europäischen Parlaments und des Rates vom 23. Februar 2005 über Höchstgehalte an Pestizidrückständen in oder auf Lebens- und Futtermitteln pflanzlichen und tierischen Ursprungs und zur Änderung der Richtlinie 91/414/EWG des Rates (1), insbesondere auf Artikel 14 Absatz 1 Buchstabe a,
in Erwägung nachstehender Gründe:
|
(1) |
Für Mandestrobin wurden in Anhang II der Verordnung (EG) Nr. 396/2005 Rückstandshöchstgehalte (im Folgenden „RHG“) festgelegt. |
|
(2) |
Gemäß Artikel 6 Absätze 2 und 4 der Verordnung (EG) Nr. 396/2005 wurde ein Antrag auf eine Einfuhrtoleranz für Mandestrobin bezüglich der Anwendung bei Erdbeeren und Trauben in Kanada gestellt. Der Antragsteller macht geltend, dass die zulässigen Anwendungen dieses Stoffs bei solchen Kulturen in Kanada zu Rückständen führen, die die RHG gemäß der Verordnung (EG) Nr. 396/2005 übersteigen, und dass die RHG erhöht werden sollten, um Handelshemmnisse bei der Einfuhr von Trauben und Erdbeeren zu vermeiden. |
|
(3) |
Dieser Antrag wurde gemäß Artikel 8 der Verordnung (EG) Nr. 396/2005 von dem betreffenden Mitgliedstaat bewertet, und der Bewertungsbericht wurde an die Kommission weitergeleitet. |
|
(4) |
Die Europäische Behörde für Lebensmittelsicherheit (im Folgenden die „Behörde“) hat den Antrag und den Bewertungsbericht, insbesondere im Hinblick auf die Risiken für Verbraucher und gegebenenfalls für Tiere, geprüft und eine mit Gründen versehene Stellungnahme zu den vorgeschlagenen RHG (2) abgegeben. Diese Stellungnahme wurde dem Antragsteller, der Kommission und den Mitgliedstaaten übermittelt und der Öffentlichkeit zugänglich gemacht. |
|
(5) |
Die Behörde gelangte zu dem Schluss, dass sämtliche Anforderungen in Bezug auf Daten erfüllt sind und die vom Antragsteller gewünschten RHG-Änderungen im Hinblick auf die Verbrauchersicherheit, basierend auf einer Bewertung der Verbraucherexposition für 27 spezifische europäische Verbrauchergruppen, akzeptiert werden können. Dabei wurden die neuesten Erkenntnisse über die toxikologischen Eigenschaften des Stoffs berücksichtigt. Im Hinblick auf die lebenslange Exposition gegenüber diesem Stoff durch den Verzehr aller Lebensmittelerzeugnisse, die ihn enthalten können, wurde nachgewiesen, dass kein Risiko einer Überschreitung der zulässigen täglichen Aufnahme besteht. Des Weiteren kam die Behörde zu dem Schluss, dass wegen der geringen akuten Toxizität des Stoffs keine akute Referenzdosis festgelegt werden muss. |
|
(6) |
Die mit Gründen versehene Stellungnahme der Behörde und die Prüfung der relevanten Faktoren haben ergeben, dass die vorgeschlagenen Änderungen der RHG die Anforderungen von Artikel 14 Absatz 2 der Verordnung (EG) Nr. 396/2005 erfüllen. |
|
(7) |
Die Verordnung (EG) Nr. 396/2005 sollte daher entsprechend geändert werden. |
|
(8) |
Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Ausschusses für Pflanzen, Tiere, Lebensmittel und Futtermittel — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang II der Verordnung (EG) Nr. 396/2005 wird gemäß dem Anhang der vorliegenden Verordnung geändert.
Artikel 2
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 29. Juli 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 70 vom 16.3.2005, S. 1.
(2) Wissenschaftliche Berichte der EFSA online abrufbar unter http://www.efsa.europa.eu/de/ Reasoned opinion on the setting of import tolerances for mandestrobin in strawberries and table and wine grapes. EFSA Journal 2018;16(8):5395.
ANHANG
In Anhang II der Verordnung (EG) Nr. 396/2005 erhält die Spalte für Mandestrobin folgende Fassung:
„Rückstände von Schädlingsbekämpfungsmitteln und Rückstandshöchstgehalte (mg/kg)
|
Code-Nummer |
Gruppen und Beispiele von Einzelerzeugnissen, für die die Rückstandshöchstgehalte gelten (1) |
Mandestrobin |
||
|
(1) |
(2) |
(3) |
||
|
0100000 |
FRÜCHTE, FRISCH ODER GEFROREN; SCHALENFRÜCHTE |
|
||
|
0110000 |
Zitrusfrüchte |
0,01 (*1) |
||
|
0110010 |
Grapefruits |
|
||
|
0110020 |
Orangen |
|
||
|
0110030 |
Zitronen |
|
||
|
0110040 |
Limetten |
|
||
|
0110050 |
Mandarinen |
|
||
|
0110990 |
Sonstige (2) |
|
||
|
0120000 |
Schalenfrüchte |
0,01 (*1) |
||
|
0120010 |
Mandeln |
|
||
|
0120020 |
Paranüsse |
|
||
|
0120030 |
Kaschunüsse |
|
||
|
0120040 |
Esskastanien |
|
||
|
0120050 |
Kokosnüsse |
|
||
|
0120060 |
Haselnüsse |
|
||
|
0120070 |
Macadamia-Nüsse |
|
||
|
0120080 |
Pekannüsse |
|
||
|
0120090 |
Pinienkerne |
|
||
|
0120100 |
Pistazien |
|
||
|
0120110 |
Walnüsse |
|
||
|
0120990 |
Sonstige (2) |
|
||
|
0130000 |
Kernobst |
0,01 (*1) |
||
|
0130010 |
Äpfel |
|
||
|
0130020 |
Birnen |
|
||
|
0130030 |
Quitten |
|
||
|
0130040 |
Mispeln |
|
||
|
0130050 |
Japanische Wollmispeln |
|
||
|
0130990 |
Sonstige (2) |
|
||
|
0140000 |
Steinobst |
|
||
|
0140010 |
Aprikosen |
2 |
||
|
0140020 |
Kirschen (süß) |
3 |
||
|
0140030 |
Pfirsiche |
2 |
||
|
0140040 |
Pflaumen |
0,5 |
||
|
0140990 |
Sonstige (2) |
0,01 (*1) |
||
|
0150000 |
Beeren und Kleinobst |
|
||
|
0151000 |
|
5 |
||
|
0151010 |
Tafeltrauben |
|
||
|
0151020 |
Keltertrauben |
|
||
|
0152000 |
|
3 |
||
|
0153000 |
|
0,01 (*1) |
||
|
0153010 |
Brombeeren |
|
||
|
0153020 |
Kratzbeeren |
|
||
|
0153030 |
Himbeeren (rot und gelb) |
|
||
|
0153990 |
Sonstige (2) |
|
||
|
0154000 |
|
0,01 (*1) |
||
|
0154010 |
Heidelbeeren |
|
||
|
0154020 |
Cranbeeren/Großfrüchtige Moosbeeren |
|
||
|
0154030 |
Johannisbeeren (schwarz, rot und weiß) |
|
||
|
0154040 |
Stachelbeeren (grün, rot und gelb) |
|
||
|
0154050 |
Hagebutten |
|
||
|
0154060 |
Maulbeeren (schwarz und weiß) |
|
||
|
0154070 |
Azarole/Mittelmeermispel |
|
||
|
0154080 |
Holunderbeeren |
|
||
|
0154990 |
Sonstige (2) |
|
||
|
0160000 |
Sonstige Früchte mit |
0,01 (*1) |
||
|
0161000 |
|
|
||
|
0161010 |
Datteln |
|
||
|
0161020 |
Feigen |
|
||
|
0161030 |
Tafeloliven |
|
||
|
0161040 |
Kumquats |
|
||
|
0161050 |
Karambolen |
|
||
|
0161060 |
Kakis/Japanische Persimonen |
|
||
|
0161070 |
Jambolans |
|
||
|
0161990 |
Sonstige (2) |
|
||
|
0162000 |
|
|
||
|
0162010 |
Kiwis (grün, rot, gelb) |
|
||
|
0162020 |
Lychees (Litschis) |
|
||
|
0162030 |
Passionsfrüchte/Maracujas |
|
||
|
0162040 |
Stachelfeigen/Kaktusfeigen |
|
||
|
0162050 |
Sternäpfel |
|
||
|
0162060 |
Amerikanische Persimonen/Virginia-Kakis |
|
||
|
0162990 |
Sonstige (2) |
|
||
|
0163000 |
|
|
||
|
0163010 |
Avocadofrüchte |
|
||
|
0163020 |
Bananen |
|
||
|
0163030 |
Mangos |
|
||
|
0163040 |
Papayas |
|
||
|
0163050 |
Granatäpfel |
|
||
|
0163060 |
Cherimoyas |
|
||
|
0163070 |
Guaven |
|
||
|
0163080 |
Ananas |
|
||
|
0163090 |
Brotfrüchte |
|
||
|
0163100 |
Durianfrüchte |
|
||
|
0163110 |
Saure Annonen/Guanabanas |
|
||
|
0163990 |
Sonstige (2) |
|
||
|
0200000 |
GEMÜSE, FRISCH ODER GEFROREN |
0,01 (*1) |
||
|
0210000 |
Wurzel- und Knollengemüse |
|
||
|
0211000 |
|
|
||
|
0212000 |
|
|
||
|
0212010 |
Kassawas/Kassaven/Manioks |
|
||
|
0212020 |
Süßkartoffeln |
|
||
|
0212030 |
Yamswurzeln |
|
||
|
0212040 |
Pfeilwurz |
|
||
|
0212990 |
Sonstige (2) |
|
||
|
0213000 |
|
|
||
|
0213010 |
Rote Rüben |
|
||
|
0213020 |
Karotten |
|
||
|
0213030 |
Knollensellerie |
|
||
|
0213040 |
Meerrettiche/Kren |
|
||
|
0213050 |
Erdartischocken |
|
||
|
0213060 |
Pastinaken |
|
||
|
0213070 |
Petersilienwurzeln |
|
||
|
0213080 |
Rettiche |
|
||
|
0213090 |
Haferwurz/Purpur-Bocksbart |
|
||
|
0213100 |
Kohlrüben |
|
||
|
0213110 |
Weiße Rüben |
|
||
|
0213990 |
Sonstige (2) |
|
||
|
0220000 |
Zwiebelgemüse |
|
||
|
0220010 |
Knoblauch |
|
||
|
0220020 |
Zwiebeln |
|
||
|
0220030 |
Schalotten |
|
||
|
0220040 |
Frühlingszwiebeln/grüne Zwiebeln und Winterzwiebeln |
|
||
|
0220990 |
Sonstige (2) |
|
||
|
0230000 |
Fruchtgemüse |
|
||
|
0231000 |
|
|
||
|
0231010 |
Tomaten |
|
||
|
0231020 |
Paprikas |
|
||
|
0231030 |
Auberginen/Eierfrüchte |
|
||
|
0231040 |
Okras/Griechische Hörnchen |
|
||
|
0231990 |
Sonstige (2) |
|
||
|
0232000 |
|
|
||
|
0232010 |
Schlangengurken |
|
||
|
0232020 |
Gewürzgurken |
|
||
|
0232030 |
Zucchinis |
|
||
|
0232990 |
Sonstige (2) |
|
||
|
0233000 |
|
|
||
|
0233010 |
Melonen |
|
||
|
0233020 |
Kürbisse |
|
||
|
0233030 |
Wassermelonen |
|
||
|
0233990 |
Sonstige (2) |
|
||
|
0234000 |
|
|
||
|
0239000 |
|
|
||
|
0240000 |
Kohlgemüse (außer Kohlwurzeln und Baby-Leaf-Salaten aus Kohlgemüse) |
|
||
|
0241000 |
|
|
||
|
0241010 |
Broccoli |
|
||
|
0241020 |
Blumenkohle |
|
||
|
0241990 |
Sonstige (2) |
|
||
|
0242000 |
|
|
||
|
0242010 |
Rosenkohle/Kohlsprossen |
|
||
|
0242020 |
Kopfkohle |
|
||
|
0242990 |
Sonstige (2) |
|
||
|
0243000 |
|
|
||
|
0243010 |
Chinakohle |
|
||
|
0243020 |
Grünkohle |
|
||
|
0243990 |
Sonstige (2) |
|
||
|
0244000 |
|
|
||
|
0250000 |
Blattgemüse, Kräuter und essbare Blüten |
|
||
|
0251000 |
|
|
||
|
0251010 |
Feldsalate |
|
||
|
0251020 |
Grüne Salate |
|
||
|
0251030 |
Kraussalate/Breitblättrige Endivien |
|
||
|
0251040 |
Kressen und andere Sprossen und Keime |
|
||
|
0251050 |
Barbarakraut |
|
||
|
0251060 |
Salatrauken/Rucola |
|
||
|
0251070 |
Roter Senf |
|
||
|
0251080 |
Baby-Leaf-Salate (einschließlich der Brassica-Arten) |
|
||
|
0251990 |
Sonstige (2) |
|
||
|
0252000 |
|
|
||
|
0252010 |
Spinat |
|
||
|
0252020 |
Portulak |
|
||
|
0252030 |
Mangold |
|
||
|
0252990 |
Sonstige (2) |
|
||
|
0253000 |
|
|
||
|
0254000 |
|
|
||
|
0255000 |
|
|
||
|
0256000 |
|
|
||
|
0256010 |
Kerbel |
|
||
|
0256020 |
Schnittlauch |
|
||
|
0256030 |
Sellerieblätter |
|
||
|
0256040 |
Petersilie |
|
||
|
0256050 |
Salbei |
|
||
|
0256060 |
Rosmarin |
|
||
|
0256070 |
Thymian |
|
||
|
0256080 |
Basilikum und essbare Blüten |
|
||
|
0256090 |
Lorbeerblätter |
|
||
|
0256100 |
Estragon |
|
||
|
0256990 |
Sonstige (2) |
|
||
|
0260000 |
Hülsengemüse |
|
||
|
0260010 |
Bohnen (mit Hülsen) |
|
||
|
0260020 |
Bohnen (ohne Hülsen) |
|
||
|
0260030 |
Erbsen (mit Hülsen) |
|
||
|
0260040 |
Erbsen (ohne Hülsen) |
|
||
|
0260050 |
Linsen |
|
||
|
0260990 |
Sonstige (2) |
|
||
|
0270000 |
Stängelgemüse |
|
||
|
0270010 |
Spargel |
|
||
|
0270020 |
Kardonen |
|
||
|
0270030 |
Stangensellerie |
|
||
|
0270040 |
Fenchel |
|
||
|
0270050 |
Artischocken |
|
||
|
0270060 |
Porree |
|
||
|
0270070 |
Rhabarber |
|
||
|
0270080 |
Bambussprossen |
|
||
|
0270090 |
Palmherzen |
|
||
|
0270990 |
Sonstige (2) |
|
||
|
0280000 |
Pilze, Moose und Flechten |
|
||
|
0280010 |
Kulturpilze |
|
||
|
0280020 |
Wilde Pilze |
|
||
|
0280990 |
Moose und Flechten |
|
||
|
0290000 |
Algen und Prokaryonten |
|
||
|
0300000 |
HÜLSENFRÜCHTE |
0,01 (*1) |
||
|
0300010 |
Bohnen |
|
||
|
0300020 |
Linsen |
|
||
|
0300030 |
Erbsen |
|
||
|
0300040 |
Lupinen |
|
||
|
0300990 |
Sonstige (2) |
|
||
|
0400000 |
ÖLSAATEN UND ÖLFRÜCHTE |
0,01 (*1) |
||
|
0401000 |
Ölsaaten |
|
||
|
0401010 |
Leinsamen |
|
||
|
0401020 |
Erdnüsse |
|
||
|
0401030 |
Mohnsamen |
|
||
|
0401040 |
Sesamsamen |
|
||
|
0401050 |
Sonnenblumenkerne |
|
||
|
0401060 |
Rapssamen |
|
||
|
0401070 |
Sojabohnen |
|
||
|
0401080 |
Senfkörner |
|
||
|
0401090 |
Baumwollsamen |
|
||
|
0401100 |
Kürbiskerne |
|
||
|
0401110 |
Saflorsamen |
|
||
|
0401120 |
Borretschsamen |
|
||
|
0401130 |
Leindottersamen |
|
||
|
0401140 |
Hanfsamen |
|
||
|
0401150 |
Rizinusbohnen |
|
||
|
0401990 |
Sonstige (2) |
|
||
|
0402000 |
Ölfrüchte |
|
||
|
0402010 |
Oliven für die Gewinnung von Öl |
|
||
|
0402020 |
Ölpalmenkerne |
|
||
|
0402030 |
Ölpalmenfrüchte |
|
||
|
0402040 |
Kapok |
|
||
|
0402990 |
Sonstige (2) |
|
||
|
0500000 |
GETREIDE |
0,01 (*1) |
||
|
0500010 |
Gerste |
|
||
|
0500020 |
Buchweizen und anderes Pseudogetreide |
|
||
|
0500030 |
Mais |
|
||
|
0500040 |
Hirse |
|
||
|
0500050 |
Hafer |
|
||
|
0500060 |
Reis |
|
||
|
0500070 |
Roggen |
|
||
|
0500080 |
Sorghum |
|
||
|
0500090 |
Weizen |
|
||
|
0500990 |
Sonstige (2) |
|
||
|
0600000 |
TEES, KAFFEE, KRÄUTERTEES, KAKAO UND JOHANNISBROT |
0,05 (*1) |
||
|
0610000 |
Tees |
|
||
|
0620000 |
Kaffeebohnen |
|
||
|
0630000 |
Kräutertees aus |
|
||
|
0631000 |
|
|
||
|
0631010 |
Kamille |
|
||
|
0631020 |
Hibiskus |
|
||
|
0631030 |
Rose |
|
||
|
0631040 |
Jasmin |
|
||
|
0631050 |
Linde |
|
||
|
0631990 |
Sonstige (2) |
|
||
|
0632000 |
|
|
||
|
0632010 |
Erdbeere |
|
||
|
0632020 |
Rooibos |
|
||
|
0632030 |
Mate |
|
||
|
0632990 |
Sonstige (2) |
|
||
|
0633000 |
|
|
||
|
0633010 |
Baldrian |
|
||
|
0633020 |
Ginseng |
|
||
|
0633990 |
Sonstige (2) |
|
||
|
0639000 |
|
|
||
|
0640000 |
Kakaobohnen |
|
||
|
0650000 |
Johannisbrote/Karuben |
|
||
|
0700000 |
HOPFEN |
0,05 (*1) |
||
|
0800000 |
GEWÜRZE |
|
||
|
0810000 |
Samengewürze |
0,05 (*1) |
||
|
0810010 |
Anis/Anissamen |
|
||
|
0810020 |
Schwarzkümmel |
|
||
|
0810030 |
Sellerie |
|
||
|
0810040 |
Koriander |
|
||
|
0810050 |
Kreuzkümmel |
|
||
|
0810060 |
Dill |
|
||
|
0810070 |
Fenchel |
|
||
|
0810080 |
Bockshornklee |
|
||
|
0810090 |
Muskatnuss |
|
||
|
0810990 |
Sonstige (2) |
|
||
|
0820000 |
Fruchtgewürze |
0,05 (*1) |
||
|
0820010 |
Nelkenpfeffer |
|
||
|
0820020 |
Szechuanpfeffer |
|
||
|
0820030 |
Kümmel |
|
||
|
0820040 |
Kardamom |
|
||
|
0820050 |
Wacholderbeere |
|
||
|
0820060 |
Pfeffer (schwarz, grün und weiß) |
|
||
|
0820070 |
Vanille |
|
||
|
0820080 |
Tamarinde |
|
||
|
0820990 |
Sonstige (2) |
|
||
|
0830000 |
Rindengewürze |
0,05 (*1) |
||
|
0830010 |
Zimt |
|
||
|
0830990 |
Sonstige (2) |
|
||
|
0840000 |
Wurzel- und Rhizomgewürze |
|
||
|
0840010 |
Süßholzwurzeln |
0,05 (*1) |
||
|
0840020 |
Ingwer (10) |
|
||
|
0840030 |
Kurkuma |
0,05 (*1) |
||
|
0840040 |
Meerrettich/Kren (11) |
|
||
|
0840990 |
Sonstige (2) |
0,05 (*1) |
||
|
0850000 |
Knospengewürze |
0,05 (*1) |
||
|
0850010 |
Nelken |
|
||
|
0850020 |
Kapern |
|
||
|
0850990 |
Sonstige (2) |
|
||
|
0860000 |
Blütenstempelgewürze |
0,05 (*1) |
||
|
0860010 |
Safran |
|
||
|
0860990 |
Sonstige (2) |
|
||
|
0870000 |
Samenmantelgewürze |
0,05 (*1) |
||
|
0870010 |
Muskatblüte |
|
||
|
0870990 |
Sonstige (2) |
|
||
|
0900000 |
ZUCKERPFLANZEN |
0,01 (*1) |
||
|
0900010 |
Zuckerrübenwurzeln |
|
||
|
0900020 |
Zuckerrohre |
|
||
|
0900030 |
Wurzeln der gewöhnlichen Wegwarte |
|
||
|
0900990 |
Sonstige (2) |
|
||
|
1000000 |
ERZEUGNISSE TIERISCHEN URSPRUNGS - LANDTIERE |
|
||
|
1010000 |
Waren von |
0,01 (*1) |
||
|
1011000 |
|
|
||
|
1011010 |
Muskel |
|
||
|
1011020 |
Fett |
|
||
|
1011030 |
Leber |
|
||
|
1011040 |
Nieren |
|
||
|
1011050 |
Genießbare Schlachtnebenerzeugnisse (außer Leber und Nieren) |
|
||
|
1011990 |
Sonstige (2) |
|
||
|
1012000 |
|
|
||
|
1012010 |
Muskel |
|
||
|
1012020 |
Fett |
|
||
|
1012030 |
Leber |
|
||
|
1012040 |
Nieren |
|
||
|
1012050 |
Genießbare Schlachtnebenerzeugnisse (außer Leber und Nieren) |
|
||
|
1012990 |
Sonstige (2) |
|
||
|
1013000 |
|
|
||
|
1013010 |
Muskel |
|
||
|
1013020 |
Fett |
|
||
|
1013030 |
Leber |
|
||
|
1013040 |
Nieren |
|
||
|
1013050 |
Genießbare Schlachtnebenerzeugnisse (außer Leber und Nieren) |
|
||
|
1013990 |
Sonstige (2) |
|
||
|
1014000 |
|
|
||
|
1014010 |
Muskel |
|
||
|
1014020 |
Fett |
|
||
|
1014030 |
Leber |
|
||
|
1014040 |
Nieren |
|
||
|
1014050 |
Genießbare Schlachtnebenerzeugnisse (außer Leber und Nieren) |
|
||
|
1014990 |
Sonstige (2) |
|
||
|
1015000 |
|
|
||
|
1015010 |
Muskel |
|
||
|
1015020 |
Fett |
|
||
|
1015030 |
Leber |
|
||
|
1015040 |
Nieren |
|
||
|
1015050 |
Genießbare Schlachtnebenerzeugnisse (außer Leber und Nieren) |
|
||
|
1015990 |
Sonstige (2) |
|
||
|
1016000 |
|
|
||
|
1016010 |
Muskel |
|
||
|
1016020 |
Fett |
|
||
|
1016030 |
Leber |
|
||
|
1016040 |
Nieren |
|
||
|
1016050 |
Genießbare Schlachtnebenerzeugnisse (außer Leber und Nieren) |
|
||
|
1016990 |
Sonstige (2) |
|
||
|
1017000 |
|
|
||
|
1017010 |
Muskel |
|
||
|
1017020 |
Fett |
|
||
|
1017030 |
Leber |
|
||
|
1017040 |
Nieren |
|
||
|
1017050 |
Genießbare Schlachtnebenerzeugnisse (außer Leber und Nieren) |
|
||
|
1017990 |
Sonstige (2) |
|
||
|
1020000 |
Milch |
0,01 (*1) |
||
|
1020010 |
Rinder |
|
||
|
1020020 |
Schafe |
|
||
|
1020030 |
Ziegen |
|
||
|
1020040 |
Pferde |
|
||
|
1020990 |
Sonstige (2) |
|
||
|
1030000 |
Vogeleier |
0,01 (*1) |
||
|
1030010 |
Huhn |
|
||
|
1030020 |
Ente |
|
||
|
1030030 |
Gans |
|
||
|
1030040 |
Wachtel |
|
||
|
1030990 |
Sonstige (2) |
|
||
|
1040000 |
Honig und sonstige Imkereierzeugnisse (7) |
0,05 (*1) |
||
|
1050000 |
Amphibien und Reptilien |
0,01 (*1) |
||
|
1060000 |
Wirbellose Landtiere |
0,01 (*1) |
||
|
1070000 |
Wildlebende Landwirbeltiere |
0,01 (*1) |
||
|
1100000 |
ERZEUGNISSE TIERISCHEN URSPRUNGS - FISCH, FISCHEREIERZEUGNISSE UND SONSTIGE VON MEERES- ODER SÜSSWASSERTIEREN GEWONNENE LEBENSMITTEL (8) |
|
||
|
1200000 |
AUSSCHLIESSLICH ZUR FUTTERMITTELHERSTELLUNG VERWENDETE ERZEUGNISSE ODER TEILE VON ERZEUGNISSEN (8) |
|
||
|
1300000 |
VERARBEITETE LEBENSMITTEL (9) |
|
(*1) Untere analytische Bestimmungsgrenze
(1) Für die vollständige Liste der Erzeugnisse pflanzlichen und tierischen Ursprungs, für die Rückstandshöchstgehalte gelten, sollte auf Anhang I verwiesen werden.“
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/46 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/1248 DER KOMMISSION
vom 29. Juli 2021
über Maßnahmen zur guten Vertriebspraxis für Tierarzneimittel gemäß der Verordnung (EU) 2019/6 des Europäischen Parlaments und des Rates
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) 2019/6 des Europäischen Parlaments und des Rates vom 11. Dezember 2018 über Tierarzneimittel und zur Aufhebung der Richtlinie 2001/82/EG (1), insbesondere auf Artikel 99 Absatz 6,
in Erwägung nachstehender Gründe:
|
(1) |
Gemäß Artikel 101 Absatz 5 der Verordnung (EU) 2019/6 müssen sich Großhändler an die gute Vertriebspraxis für Tierarzneimittel, wie von der Kommission angenommen, halten. |
|
(2) |
Die Maßnahmen zur guten Vertriebspraxis sollten so gestaltet sein, dass die Identität, Unversehrtheit, Rückverfolgbarkeit und Qualität von Tierarzneimitteln in der gesamten Lieferkette gewährleistet sind. Darüber hinaus sollte mit diesen Maßnahmen sichergestellt werden, dass Tierarzneimittel angemessen gelagert, transportiert und gehandhabt werden und dass sie während der Lagerung und des Transports innerhalb der legalen Lieferkette verbleiben. |
|
(3) |
Für Humanarzneimittel existieren mehrere internationale Standards und Leitlinien für die gute Vertriebspraxis (2) (3) (4) (5). Auf Unionsebene wurden Leitlinien für die gute Vertriebspraxis lediglich für Humanarzneimittel angenommen. (6) Bei entsprechenden Maßnahmen in Bezug auf Tierarzneimittel sollten die Erfahrungen mit der Anwendung des derzeitigen Systems im Rahmen der Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates (7) im Lichte der Ähnlichkeiten und potenziellen Unterschiede zwischen den Anforderungen für die gute Vertriebspraxis für Human- und Tierarzneimittel berücksichtigt werden. |
|
(4) |
Großhändler handeln häufig sowohl mit Human- als auch mit Tierarzneimitteln. Zudem werden Inspektionen im Hinblick auf die gute Vertriebspraxis für beide Arten von Arzneimitteln häufig von denselben Sachverständigen der zuständigen Behörde durchgeführt. Um unnötigen Verwaltungsaufwand für die Industrie und die zuständigen Behörden zu vermeiden, ist es daher sinnvoll, in Bezug auf Tierarzneimittel ähnliche Maßnahmen wie in Bezug auf Humanarzneimittel anzuwenden, sofern nicht besondere Erfordernisse etwas anderes vorschreiben. |
|
(5) |
Um die Verfügbarkeit von Tierarzneimitteln in der Union nicht zu beeinträchtigen, sollten die Anforderungen für die gute Vertriebspraxis für Tierarzneimittel nicht strenger sein als die entsprechenden Anforderungen für Humanarzneimittel. |
|
(6) |
Die in der vorliegenden Verordnung festgelegten Maßnahmen zur guten Vertriebspraxis für Tierarzneimittel sollten die Kohärenz mit den Durchführungsmaßnahmen zur guten Herstellungspraxis für Tierarzneimittel und für als Ausgangsstoffe verwendete Wirkstoffe gemäß Artikel 93 Absatz 2 der Verordnung (EU) 2019/6 sowie zur guten Vertriebspraxis für als Ausgangsstoffe in Tierarzneimitteln verwendete Wirkstoffe gemäß Artikel 95 Absatz 8 der genannten Verordnung gewährleisten und diese ergänzen. |
|
(7) |
Jede Person, die als Großhändler für Tierarzneimittel tätig ist, muss gemäß Artikel 99 Absatz 1 der Verordnung (EU) 2019/6 über eine Großhandelsvertriebserlaubnis verfügen und gemäß Artikel 101 Absatz 5 der genannten Verordnung die gute Vertriebspraxis für Tierarzneimittel einhalten. Gemäß Artikel 99 Absatz 5 der genannten Verordnung ermöglicht eine Herstellungserlaubnis den Großhandelsvertrieb mit Tierarzneimitteln, für die diese Herstellungserlaubnis gilt. Daher müssen Hersteller, die solche Vertriebstätigkeiten mit ihren eigenen Tierarzneimitteln durchführen, auch die gute Vertriebspraxis für Tierarzneimittel einhalten. |
|
(8) |
Von der Definition des Begriffs „Großhandelsvertrieb“ in Artikel 4 Absatz 36 der Verordnung (EU) 2019/6 sind Großhändler, die im Rahmen bestimmter Zollregelungen, z. B. Freizonen oder Zolllager, niedergelassen oder tätig sind, nicht ausgeschlossen. Daher gelten alle Verpflichtungen im Zusammenhang mit Großhandelsvertriebstätigkeiten (wie Ausfuhr, Lagerung oder Abgabe) auch für diese Großhändler in Bezug auf die gute Vertriebspraxis für Tierarzneimittel. |
|
(9) |
Die einschlägigen Abschnitte der guten Vertriebspraxis für Tierarzneimittel sollten auch von dritten Akteuren, die am Großhandelsvertrieb von Tierarzneimitteln beteiligt sind, eingehalten werden und Teil ihrer vertraglichen Verpflichtungen sein. Um Tierarzneimittelfälschungen wirksam bekämpfen zu können, müssen alle Teilnehmer der Lieferkette einem einheitlichen Ansatz folgen. |
|
(10) |
Um sicherzustellen, dass die Ziele der guten Vertriebspraxis erreicht werden, bedarf es eines Qualitätssicherungssystems, in dem die Zuständigkeiten, Abläufe und die Grundsätze des Risikomanagements in Bezug auf die Großhandelsvertriebstätigkeiten klar dargelegt sind. Zuständig für dieses Qualitätssicherungssystem sollten die Geschäftsführer der Organisation sein, welche die Leitung übernehmen und sich aktiv beteiligen sollten; außerdem sollte das Personal das System unterstützen. |
|
(11) |
Der ordnungsgemäße Vertrieb von Tierarzneimitteln hängt in erheblichem Maße davon ab, dass ausreichend fachkundiges Personal zur Verfügung steht, um die Tätigkeiten, für die der Großhändler verantwortlich ist, tatsächlich auszuführen. Die Mitarbeiter sollten sich über die einzelnen Zuständigkeiten — die auch aufgezeichnet werden sollten — im Klaren sein. |
|
(12) |
Die Personen, die Tierarzneimittel vertreiben, sollten über geeignete und ausreichende Betriebsräume, Anlagen und Ausrüstungen verfügen, um eine ordnungsgemäße Lagerung und einen ordnungsgemäßen Vertrieb der Tierarzneimittel zu gewährleisten. |
|
(13) |
Gute Dokumentation sollte ein wesentlicher Bestandteil eines jeden Qualitätssicherungssystems sein. Es sollte eine schriftliche Dokumentation verlangt werden, um Irrtümer, die sich in die mündliche Kommunikation einschleichen können, zu vermeiden und um zu ermöglichen, die relevanten Arbeitsgänge beim Großhandelsvertrieb von Tierarzneimitteln nachzuvollziehen. Alle Arten von Unterlagen sollten definiert und beachtet werden. |
|
(14) |
In den Verfahren sollten alle Vertriebstätigkeiten beschrieben werden, die sich auf die Identität, Rückverfolgbarkeit und Qualität der Tierarzneimittel auswirken. |
|
(15) |
Es sollten Aufzeichnungen über alle wichtigen Tätigkeiten oder Vorkommnisse erstellt und aufbewahrt werden, um die Rückverfolgbarkeit der Herkunft und des Bestimmungsorts der Tierarzneimittel sowie die Ermittlung aller Lieferanten der Tierarzneimittel bzw. aller mit den Tierarzneimitteln Belieferten zu gewährleisten. Diese Aufzeichnungen sollten erforderlichenfalls den Rückruf einer Charge eines Tierarzneimittels sowie die Untersuchung von gefälschten Tierarzneimitteln bzw. von Verdachtsfällen gefälschter Tierarzneimittel erleichtern. |
|
(16) |
Bei der Verarbeitung personenbezogener Daten von Angestellten, Beschwerdeführern oder anderen natürlichen Personen sollte die Verordnung (EU) 2016/679 des Europäischen Parlaments und des Rates (8) zum Schutz natürlicher Personen bei der Verarbeitung personenbezogener Daten und zum freien Datenverkehr gelten. |
|
(17) |
Im Qualitätssicherungssystem sollten alle Schlüsselvorgänge anhand geeigneter Aufzeichnungen genau beschrieben sein. |
|
(18) |
Beschwerden, Rückgaben, Verdachtsfälle gefälschter Tierarzneimittel und Rückrufe sollten dokumentiert und gemäß den etablierten Verfahren bearbeitet werden. Die entsprechenden Aufzeichnungen sollten den zuständigen Behörden zugänglich gemacht werden. Bevor zurückgegebene Tierarzneimittel wieder zum Verkauf freigegeben werden, sollte eine Bewertung der Produkte erfolgen. |
|
(19) |
Alle Tätigkeiten, die unter die gute Vertriebspraxis für Tierarzneimittel fallen und ausgelagert werden, sollten definiert, abgestimmt und kontrolliert werden, damit Missverständnisse, die die Unversehrtheit des Tierarzneimittels beeinträchtigen könnten, vermieden werden. In einem schriftlichen Vertrag zwischen dem Auftraggeber und dem Auftragnehmer sollten die Pflichten jeder Partei klar festgelegt sein. |
|
(20) |
Unabhängig von der Transportweise sollte es möglich sein nachzuweisen, dass die Tierarzneimittel keinerlei Bedingungen ausgesetzt wurden, die ihre Qualität oder Unversehrtheit beeinträchtigen könnten. Der Planung des Transports und dem eigentlichen Transport von Tierarzneimitteln sollte ein risikobasierter Ansatz zugrunde liegen. |
|
(21) |
Zur Überwachung der Umsetzung und Einhaltung der guten Vertriebspraxis für Tierarzneimittel und um erforderliche Korrektur- und Präventivmaßnahmen vorschlagen zu können, sind regelmäßige Selbstinspektionen erforderlich. |
|
(22) |
Die in dieser Verordnung vorgesehenen Vorschriften entsprechen der Stellungnahme des in Artikel 145 der Verordnung (EU) 2019/6 genannten Ständigen Ausschusses für Tierarzneimittel — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
KAPITEL I
ALLGEMEINE BESTIMMUNGEN
Artikel 1
Gegenstand und Geltungsbereich
(1) In der vorliegenden Verordnung sind die Maßnahmen zur guten Vertriebspraxis für Tierarzneimittel festgelegt.
(2) Diese Verordnung gilt für Inhaber einer Herstellungserlaubnis, die den Großhandelsvertrieb mit den unter diese Herstellungserlaubnis fallenden Tierarzneimitteln durchführen, sowie für Inhaber einer Großhandelsvertriebserlaubnis, einschließlich derjenigen, die im Rahmen bestimmter Zollregelungen, z. B. Freizonen oder Zolllager, niedergelassen oder tätig sind.
Artikel 2
Begriffsbestimmungen
Für die Zwecke dieser Verordnung bezeichnet der Ausdruck
|
a) |
„gute Vertriebspraxis für Tierarzneimittel“ den Teil der Qualitätssicherung entlang der Lieferkette, mit dessen Hilfe gewährleistet wird, dass die Qualität von Tierarzneimitteln während sämtlicher Etappen der Lieferkette — vom Herstellungsort bis zu den in Artikel 101 Absatz 2 der Verordnung (EU) 2019/6 genannten Personen — erhalten bleibt; |
|
b) |
„Freizone“ jede von den Mitgliedstaaten gemäß Artikel 243 der Verordnung (EU) Nr. 952/2013 des Europäischen Parlaments und des Rates (9) bestimmte Freizone; |
|
c) |
„Zolllager“ jedes Lager gemäß Artikel 240 Absatz 1 der Verordnung (EU) Nr. 952/2013; |
|
d) |
„Qualitätssicherungssystem“ die Summe aller Teile eines Systems zur Umsetzung der Qualitätspolitik eines Unternehmens und Gewährleistung des Erreichens der Qualitätsziele; |
|
e) |
„Qualitätsrisikomanagement“ ein systematisches, sowohl proaktiv als auch retrospektiv angewandtes Verfahren für die Bewertung, Kontrolle, Überprüfung und Mitteilung von Risiken in Bezug auf die Qualität des Tierarzneimittels über den gesamten Lebenszyklus des Produkts hinweg; |
|
f) |
„Validierung“ ein dokumentiertes Programm, das ein hohes Maß an Gewissheit bietet, dass ein bestimmter Prozess, eine bestimmte Methode oder ein bestimmtes System beständig Ergebnisse hervorbringt, die im Voraus festgelegte Akzeptanzkriterien erfüllen; |
|
g) |
„Verfahren“ eine dokumentierte Beschreibung der durchzuführenden Operationen, der zu treffenden Vorsichtsmaßnahmen und der zu ergreifenden Maßnahmen, die direkt oder indirekt im Zusammenhang mit dem Vertrieb eines Tierarzneimittels stehen; |
|
h) |
„Dokumentation“ schriftlich niedergelegte Verfahren, Anweisungen, Verträge, Berichte und Daten auf Papier oder in elektronischer Form; |
|
i) |
„Beschaffung“ das Erlangen, den Erwerb oder Kauf von Tierarzneimitteln von Herstellern, Importeuren oder anderen Großhändlern; |
|
j) |
„Lagerung“ die Aufbewahrung von Tierarzneimitteln; |
|
k) |
„Lieferung“ alle Tätigkeiten der Bereitstellung, des Verkaufs oder der Schenkung von Tierarzneimitteln an die in Artikel 101 Absatz 2 der Verordnung (EU) 2019/6 genannten Personen; |
|
l) |
„Transport“ das Verbringen von Tierarzneimitteln von einem Ort an einen anderen, ohne dass sie unterwegs für ungerechtfertigte Zeiträume zwischengelagert werden; |
|
m) |
„Abweichung“ eine Abweichung von genehmigten Unterlagen oder von einem festgelegten Standard; |
|
n) |
„gefälschtes Tierarzneimittel“ jedes Tierarzneimittel, bei dem Folgendes gefälscht wurde:
|
|
o) |
„Kontamination“ das unerwünschte Einbringen von Verunreinigungen chemischer oder mikrobiologischer Natur oder von Fremdstoffen in oder auf Tierarzneimittel während der Produktion, Probenahme, Verpackung oder Umverpackung, Lagerung oder des Transports; |
|
p) |
„Kalibrierung“ eine Reihe von Arbeitsschritten zum Abgleich der Messergebnisse eines Messinstruments oder Messsystems oder der Werte eines Prüfnormals mit den entsprechenden bekannten Werten eines Referenzstandards unter vorgegebenen Bedingungen; |
|
q) |
„Qualifizierung“ den Nachweis, dass Ausrüstungen korrekt funktionieren und mit ihnen tatsächlich die erwarteten Ergebnisse erreicht werden können; |
|
r) |
„abgezeichnet“ die Abzeichnung der Person, die eine bestimmte Maßnahme oder eine bestimmte Überprüfung vorgenommen hat. Dabei kann es sich um Initialen, eine vollständige handschriftliche Unterschrift, ein persönliches Siegel oder eine fortgeschrittene elektronische Signatur im Sinne von Artikel 3 Absatz 11 der Verordnung (EU) Nr. 910/2014 des Europäischen Parlaments und des Rates (10) handeln; |
|
s) |
„Charge“ eine bestimmte Menge an Ausgangsstoff, Verpackungsmaterial oder Produkt, die in einem einzigen Prozess oder einer Reihe von Prozessen so verarbeitet wird, dass davon auszugehen ist, dass sie homogen ist; |
|
t) |
„Verfalldatum“ das auf der Verpackung eines Tierarzneimittels angegebene Datum, das die Zeit angibt, während der das Tierarzneimittel innerhalb der festgelegten Laufzeitspezifikationen bleiben sollte, wenn es unter definierten Bedingungen gelagert wird, und nach dem es nicht mehr verwendet werden sollte. |
|
u) |
„Chargennummer“ eine unverwechselbare Kombination von Zahlen oder Buchstaben zur eindeutigen Identifizierung einer Charge. |
KAPITEL II
QUALITÄTSMANAGEMENT
Artikel 3
Entwicklung und Unterhalt eines Qualitätssicherungssystems
(1) Die in Artikel 1 Absatz 2 genannten Personen entwickeln und unterhalten ein Qualitätssicherungssystem.
(2) Das Qualitätssicherungssystem trägt dem Umfang, der Struktur und der Komplexität der Tätigkeiten dieser Personen und den für diese Tätigkeiten vorgesehenen Änderungen Rechnung.
(3) Die in Artikel 1 Absatz 2 genannten Personen sorgen dafür, dass für alle Teilbereiche des Qualitätssicherungssystems ausreichend kompetentes Personal sowie geeignete und ausreichende Räumlichkeiten, Ausrüstungen und Einrichtungen zur Verfügung stehen.
Artikel 4
Anforderungen an das Qualitätssicherungssystem
(1) Im Qualitätssicherungssystem sind die Zuständigkeiten, Abläufe und die Grundsätze des Risikomanagements im Zusammenhang mit den Tätigkeiten der in Artikel 1 Absatz 2 genannten Personen festgelegt. Alle Großhandelsvertriebstätigkeiten werden klar definiert und systematisch überprüft. Alle kritischen Abschnitte der Großhandelsvertriebstätigkeiten und wesentlichen Änderungen werden begründet und unterliegen, soweit erforderlich, der Validierung.
(2) Das Qualitätssicherungssystem umfasst die Organisationsstruktur, Verfahren, Prozesse und Ressourcen sowie Tätigkeiten, mit denen sichergestellt werden kann, dass Qualität und Unversehrtheit der gelieferten Tierarzneimittel beibehalten werden und sie während Transport oder Lagerung in der legalen Lieferkette verbleiben.
(3) Das Qualitätssicherungssystem wird vollständig dokumentiert. Seine Wirksamkeit wird überwacht. Alle Tätigkeiten im Zusammenhang mit dem Qualitätssicherungssystem werden definiert und dokumentiert.
(4) Es wird ein Qualitäts-Handbuch oder eine ähnliche Dokumentation erstellt, mit einer Beschreibung etwaiger Unterschiede im Qualitätssicherungssystem in Bezug auf die Handhabung von Tierarzneimitteln unterschiedlicher Art.
(5) Es wird ein Änderungskontrollsystem eingerichtet, das die Grundsätze des Qualitätsrisikomanagements berücksichtigt und verhältnismäßig sowie wirksam ist.
(6) Mit dem Qualitätssicherungssystem wird gewährleistet, dass die folgenden Verpflichtungen erfüllt werden:
|
a) |
Die Beschaffung, Lagerung, Lieferung, der Transport und die Ausfuhr von Tierarzneimitteln entsprechen den Anforderungen der guten Vertriebspraxis für Tierarzneimittel, wie sie in der vorliegenden Verordnung festgelegt sind; |
|
b) |
die Zuständigkeiten der Geschäftsführung sind klar definiert; |
|
c) |
die Tierarzneimittel werden innerhalb einer angemessen Frist an die richtigen Empfänger geliefert; |
|
d) |
Aufzeichnungen werden zeitnah erstellt; |
|
e) |
Abweichungen werden dokumentiert und untersucht; |
|
f) |
geeignete Korrektur- bzw. Vorbeugemaßnahmen (corrective and preventive measures, „CAPA“) werden gemäß den Grundsätzen des Qualitätsrisikomanagements ergriffen; |
|
g) |
Änderungen, die die Lagerung und den Vertrieb von Tierarzneimitteln beeinträchtigen können, werden bewertet. |
Artikel 5
Verwaltung ausgelagerter Tätigkeiten
Das Qualitätssicherungssystem erstreckt sich auf die Kontrolle und Überprüfung jeglicher ausgelagerten Tätigkeiten im Zusammenhang mit dem Großhandelsvertrieb von Tierarzneimitteln. Diese Kontrolle und Überprüfung umfassen das Qualitätsrisikomanagement und beinhalten Folgendes:
|
a) |
Bewertung der Eignung und Kompetenz des Auftragnehmers für die Durchführung der Tätigkeit sowie erforderlichenfalls Überprüfung seiner Berechtigung; |
|
b) |
Festlegung der Zuständigkeiten und der Kommunikationsprozesse für die qualitätsbezogenen Tätigkeiten der beteiligten Parteien; |
|
c) |
regelmäßige Überwachung und Überprüfung der Leistung des Auftragnehmers und regelmäßige Bestimmung und Implementierung jeglicher erforderlicher Verbesserungen. |
Artikel 6
Überprüfung und Überwachung durch die Geschäftsführung
(1) Die Geschäftsführung der in Artikel 1 Absatz 2 genannten Personen richtet formelle Prozesse für eine regelmäßige Überprüfung des Qualitätssicherungssystems ein und setzt diese um.
(2) Die Überprüfung umfasst Folgendes:
|
a) |
Messung des Erreichens der Ziele des Qualitätssicherungssystems; |
|
b) |
Bewertung von:
|
|
c) |
neu auftretende Regelungs-, Anleitungs- oder Qualitätsprobleme, die Auswirkungen auf das Qualitätssicherungssystem haben können; |
|
d) |
Innovationen, mit denen das Qualitätssicherungssystem verbessert werden könnte; |
|
e) |
Änderungen des Unternehmensumfeldes oder der Unternehmensziele. |
(3) Die Ergebnisse jeder Überprüfung des Qualitätssicherungssystems durch das Management werden zeitnah dokumentiert und wirksam intern kommuniziert.
Artikel 7
Qualitätsrisikomanagement
(1) Die in Artikel 1 Absatz 2 genannten Personen wenden das Qualitätsrisikomanagement an.
(2) Mithilfe des Qualitätsrisikomanagements wird sichergestellt, dass die Bewertung der Risiken für die Qualität auf wissenschaftlichen Erkenntnissen und Erfahrung mit den entsprechenden Prozessen beruht und letztlich den Schutz des behandelten Tieres bzw. der behandelten Gruppe von Tieren, der für das Tier und die Behandlung zuständigen Personen, des Verbrauchers eines der Lebensmittelgewinnung dienenden Tieres und der Umwelt fördert.
(3) Der Detaillierungsgrad und die Dokumentation des Qualitätsrisikomanagementprozesses entsprechen dem Qualitätsrisikoniveau.
KAPITEL III
ANFORDERUNGEN AN DAS PERSONAL
Artikel 8
Pflichten der für den Großhandelsvertrieb verantwortlichen Personen
(1) Die für den Großhandelsvertrieb verantwortlichen Personen gemäß Artikel 101 Absatz 3 der Verordnung (EU) 2019/6 (im Folgenden „verantwortliche Personen“) gewährleisten die Einhaltung der guten Vertriebspraxis für Tierarzneimittel. Zusätzlich zu der Anforderung gemäß Artikel 100 Absatz 2 Buchstabe a der genannten Verordnung verfügen die verantwortlichen Personen in Bezug auf die Einhaltung der guten Vertriebspraxis für Tierarzneimittel über angemessene Kompetenz und Erfahrung sowie über Kenntnisse und eine Ausbildung.
(2) Die verantwortlichen Personen sind persönlich für die Wahrnehmung ihrer Pflichten verantwortlich und jederzeit erreichbar.
(3) Die verantwortlichen Personen können ihre Aufgaben delegieren, nicht aber ihre Verantwortung.
(4) Stehen die verantwortlichen Personen nicht zur Verfügung, so benennen die in Artikel 1 Absatz 2 genannten Personen für den erforderlichen Zeitraum einen Vertreter, damit die Kontinuität des Geschäftsbetriebs gewährleistet ist.
(5) In der schriftlichen Arbeitsplatzbeschreibung für die verantwortlichen Personen sollte klar festgelegt sein, dass sie die Befugnis haben, die für die Wahrnehmung ihrer Zuständigkeiten erforderlichen Entscheidungen zu treffen. Die in Artikel 1 Absatz 2 genannten Personen übertragen den verantwortlichen Personen die zur Erfüllung ihrer Aufgaben erforderliche Weisungsbefugnis und die erforderlichen Ressourcen und Zuständigkeiten.
(6) Die verantwortlichen Personen führen ihre Aufgaben so aus, dass die in Artikel 1 Absatz 2 genannten einschlägigen Personen die Einhaltung der guten Vertriebspraxis für Tierarzneimittel nachweisen können und dass die Pflichten gemäß Artikel 101 Absatz 4 der Verordnung (EU) 2019/6 erfüllt werden.
(7) Der Verantwortungsbereich der verantwortlichen Personen umfasst Folgendes:
|
a) |
Sicherstellung der Implementierung und Unterhaltung eines Qualitätsmanagementsystems; |
|
b) |
Konzentration auf die Durchführung genehmigter Tätigkeiten, sowie auf Genauigkeit und Qualität der Aufzeichnungen; |
|
c) |
Sicherstellung, dass regelmäßig ein- und weiterführende Schulungsprogramme durchgeführt werden; |
|
d) |
Koordinierung und unmittelbare Durchführung jeglicher Tierarzneimittel-Rückrufaktion; |
|
e) |
Sicherstellung, dass einschlägige Kundenbeschwerden angemessen bearbeitet werden; |
|
f) |
Sicherstellung, dass Lieferanten und Kunden zugelassen sind; |
|
g) |
Genehmigung aller ausgelagerten Tätigkeiten, die Auswirkungen auf die gute Vertriebspraxis für Tierarzneimittel haben können; |
|
h) |
Sicherstellung, dass in angemessen regelmäßigen Abständen Selbstinspektionen nach einem vorab festgelegten Programm durchgeführt und die erforderlichen CAPA ergriffen werden; |
|
i) |
Führen angemessener Aufzeichnungen über sämtliche delegierten Aufgaben; |
|
j) |
Entscheidung über den endgültigen Verbleib zurückgegebener, zurückgewiesener, zurückgerufener oder gefälschter Tierarzneimittel; |
|
k) |
Genehmigung sämtlicher Wiederaufnahmen in den verkaufsfähigen Bestand; |
|
l) |
Gewährleistung, dass alle in den nationalen Rechtsvorschriften festgelegten zusätzlichen Auflagen für bestimmte Tierarzneimittel eingehalten werden; |
|
m) |
Dokumentation von Abweichungen und Ergreifen von CAPA zu ihrer Korrektur und zur Vermeidung eines erneuten Auftretens sowie Überwachung der Wirksamkeit dieser CAPA. |
Artikel 9
Sonstiges Personal
(1) Auf allen Ebenen des Großhandelsvertriebs von Tierarzneimitteln steht eine angemessene Zahl kompetenter Mitarbeiter zur Verfügung. Die erforderliche Mitarbeiterzahl entspricht dem Umfang der Tätigkeiten und dem Tätigkeitsbereich.
(2) Die Organisationsstruktur der in Artikel 1 Absatz 2 genannten Personen ist in einem Organigramm aufgezeichnet. Die einzelnen Rollen, Zuständigkeiten und Beziehungen aller Mitarbeiter zueinander sind in diesem Organigramm klar angegeben. Jeder Mitarbeiter versteht seine jeweilige Rolle und seine jeweiligen Zuständigkeiten.
(3) Rolle und Zuständigkeiten der Mitarbeiter in Schlüsselpositionen werden — ebenso wie die entsprechenden Vertretungsregeln — in schriftlichen Arbeitsplatzbeschreibungen festgehalten.
Artikel 10
Schulung der Mitarbeiter
(1) Alle Mitarbeiter, die an den Großhandelsvertriebstätigkeiten beteiligt sind, sind hinsichtlich der Anforderungen der guten Vertriebspraxis für Tierarzneimittel geschult. Ferner verfügen sie vor Aufnahme ihrer Tätigkeit über die erforderliche Kompetenz und Erfahrung.
(2) Jeder Mitarbeiter wird vor Übernahme seiner Rolle speziell für diese geschult und danach ständig weitergebildet; dies sollte auf der Grundlage von Verfahren und eines schriftlich festgelegten Schulungsprogramms geschehen. Die verantwortlichen Personen halten ihre Kompetenz im Bereich der guten Vertriebspraxis für Tierarzneimittel durch regelmäßige Fortbildung immer auf dem neuesten Stand.
(3) In den Schulungen wird auf die Identifizierung von gefälschten Tierarzneimitteln eingegangen sowie darauf, wie verhindert werden kann, dass diese in die Lieferkette gelangen.
(4) Personen, die mit Tierarzneimitteln zu tun haben, für die strengere Handhabungsbedingungen gelten, darunter gefährliche Produkte, Produkte mit besonders hohem Missbrauchsrisiko, einschließlich Betäubungsmittel und psychotrope Substanzen, und temperaturempfindliche Produkte, werden speziell dafür geschult.
(5) Die in Artikel 1 Absatz 2 genannten Personen führen über alle Schulungen Aufzeichnungen und überprüfen und dokumentieren regelmäßig ihre Wirksamkeit.
Artikel 11
Hygiene
Die in Artikel 1 Absatz 2 genannten Personen führen den Tätigkeiten angepasste Verfahren für die Personalhygiene, einschließlich Personalgesundheit und angemessener Kleidung, ein. Diese Verfahren werden von den Mitarbeitern eingehalten.
KAPITEL IV
BETRIEBSRÄUME UND AUSRÜSTUNGEN
Artikel 12
Betriebsräume
(1) Die Betriebsräume sind so konzipiert bzw. eingerichtet, dass die erforderlichen Lagerbedingungen eingehalten werden. Sie sind ausreichend sicher und baulich einwandfrei und sie verfügen über ausreichende Kapazität für die sichere Lagerung und Handhabung der Tierarzneimittel. Die Lagerbereiche sind ausreichend beleuchtet, damit alle Tätigkeiten fehlerfrei und sicher durchgeführt werden können. Tierarzneimittel werden in einem angemessenen Abstand gelagert, um eine Reinigung und Inspektion zu ermöglichen. Paletten werden in sauberem und einwandfreiem Zustand gehalten.
(2) Unterstehen die Betriebsräume nicht unmittelbar den in Artikel 1 Absatz 2 genannten Personen, besteht ein entsprechender Vertrag. Die in Artikel 1 Absatz 2 genannten Personen dürfen die von einem Vertrag umfassten Betriebsräume nur dann nutzen, wenn diese Betriebsräume von einer separaten Großhandelsvertriebserlaubnis abgedeckt sind.
(3) Tierarzneimittel werden in gesonderten, deutlich gekennzeichneten Bereichen gelagert, zu denen nur befugtes Personal Zugang hat.
(4) Jedes System, das eine physische Trennung ersetzt, beispielsweise ein computergestütztes System zur Trennung, sollte eine vergleichbare Sicherheit bieten und einer angemessenen Validierung unterliegen.
(5) Tierarzneimittel, über deren Entsorgung entschieden werden soll, oder Tierarzneimittel, die aus dem verkaufsfähigen Bestand entfernt wurden, werden gesondert gelagert, und zwar entweder physisch abgesondert oder mit einem vergleichbaren elektronischen System abgegrenzt, sofern ein solches System vorhanden ist; dies betrifft auch zurückgegebene Tierarzneimittel.
(6) Tierarzneimittel aus einem Drittland, die nicht für den Unionsmarkt bestimmt sind, werden physisch und, sofern ein elektronisches System vorhanden ist, elektronisch gesondert gelagert.
(7) Alle abgelaufenen, zurückgerufenen und zurückgewiesenen Tierarzneimittel werden unverzüglich physisch und, sofern ein elektronisches System vorhanden ist, elektronisch abgesondert und in einem speziell dafür eingerichteten Bereich abseits von den übrigen Tierarzneimittel gelagert. In diesen Bereichen finden geeignete Sicherheitsmaßnahmen Anwendung, damit solche Artikel vom verkaufsfähigen Bestand getrennt bleiben. Diese Bereiche werden deutlich markiert.
(8) Die Betriebsräume sind so konzipiert bzw. eingerichtet, dass Tierarzneimittel, für die besondere Lager- und Handhabungsmaßnahmen gelten, wie Betäubungsmittel oder psychotrope Substanzen, nach schriftlichen Anweisungen gelagert werden und geeigneten Sicherheitsmaßnahmen unterliegen.
(9) Für die Lagerung gefährlicher sowie leicht brennbarer oder explosiver Tierarzneimittel, z. B. medizinische Gase, Brennstoffe, brennbare Flüssigkeiten oder Feststoffe, sind ein oder mehrere spezielle Bereiche vorzusehen und geeignete Sicherheitsmaßnahmen zu treffen.
(10) Warenannahme- und Versandstelle sind so gestaltet, dass die Tierarzneimittel vor Wettereinflüssen geschützt sind. Annahme- und Versandstelle sind angemessen vom Lagerbereich abgegrenzt. Es sind Verfahren zur Kontrolle der ein- und ausgehenden Waren vorhanden. Die Bereiche, in denen eingehende Waren nach der Annahme überprüft werden, sind gekennzeichnet und angemessen ausgestattet.
(11) Der Zugang Unbefugter zu den von der Genehmigung erfassten Betriebsräumen wird anhand geeigneter Einrichtungen wie einer personell überwachten Einbruchmeldeanlage und einer angemessenen Zugangskontrolle verhindert. Besucher werden stets begleitet.
(12) Betriebs- und Lagerräume sind sauber und frei von Müll und Staub. Es existieren Reinigungsprogramme, -anweisungen und -berichte. Es werden geeignete Reinigungsutensilien und Reinigungsmittel gewählt, damit sich daraus keine Kontamination ergeben kann.
(13) Die Betriebsräume sind trocken und angemessen temperiert.
(14) Es sind geeignete Verfahren für die Reinigung von ausgetretenem Material vorhanden, um sicherzustellen, dass jegliches Kontaminationsrisiko vollständig beseitigt wird.
(15) Fahrzeuge werden regelmäßig gereinigt. Aus den für die Reinigung von Fahrzeugen gewählten Utensilien ergibt sich keine Kontamination.
(16) Die Betriebsräume sind so gestaltet und eingerichtet, dass keine Insekten, Nagetiere und anderen Tiere eindringen können. Es gibt ein vorbeugendes Schädlingsbekämpfungsprogramm.
(17) Die Ruhe-, Wasch- und Erfrischungsräume für das Personal sind von den Lagerbereichen angemessen abgetrennt. Lebensmittel, Getränke, Tabakwaren oder Arzneimittel zum persönlichen Gebrauch des Personals sind in den Lagerbereichen untersagt.
Artikel 13
Temperatur- und Umgebungskontrolle
(1) Es werden geeignete Ausrüstungen und Verfahren für die Kontrolle der Umgebung, in der Tierarzneimittel gelagert werden, eingesetzt. Zu beachtende Umgebungsfaktoren sind dabei z. B. Temperatur, Licht, Feuchtigkeit und Sauberkeit der Betriebsräume.
(2) Vor Inbetriebnahme der Räumlichkeiten wird eine Temperaturverteilungsstudie unter repräsentativen Bedingungen im Lagerbereich durchgeführt. Die Temperaturüberwachungsanlage wird gemäß den Ergebnissen der Temperaturverteilungsstudie so eingerichtet, dass Überwachungsgeräte vor allem in den Bereichen mit den größten Temperaturschwankungen aufgestellt werden. Die Temperaturverteilungsstudie wird auf der Basis einer entsprechenden Risikobewertung bzw. immer dann wiederholt, wenn erhebliche Veränderungen an den Räumlichkeiten oder der Temperaturüberwachungsanlage vorgenommen werden. In kleineren Betriebsräumen, die nur wenige Quadratmeter groß sind und in denen Raumtemperatur herrscht, wird eine Bewertung des potenziellen Risikos, z. B. durch Heizkörper, durchgeführt, und es werden entsprechende Temperaturüberwachungsgeräte aufgestellt.
Artikel 14
Ausrüstungen
(1) Alle Ausrüstungen, die einen Einfluss auf die Lagerung und den Vertrieb der Tierarzneimittel haben, werden nach einem ihrem Zweck entsprechenden Standard konzipiert, platziert und gewartet. Für Ausrüstungen, die für die Funktionsfähigkeit des Betriebs unbedingt erforderlich sind, wird ein Wartungsplan aufgestellt.
(2) Ausrüstungen, die zur Kontrolle oder zur Überwachung der Lagerumgebung der Tierarzneimittel benutzt werden, unterliegen in bestimmten Abständen einer Kalibrierung auf der Grundlage einer Risiko- und Zuverlässigkeitsbewertung.
(3) Die Kalibrierung der Ausrüstungen beruht auf einer nationalen oder internationalen Messnorm. Es bestehen geeignete Alarmsysteme, die Abweichungen von den prädefinierten Lagerbedingungen melden. Die Alarmstufen werden angemessen festgelegt und die Alarmsysteme werden regelmäßig auf ordnungsgemäße Funktionsweise überprüft.
(4) Die Reparatur, Wartung und Kalibrierung von Ausrüstungen werden so durchgeführt, dass die Unversehrtheit der Tierarzneimittel nicht beeinträchtigt wird.
(5) Defekte Fahrzeuge und Ausrüstungen dürfen nicht verwendet werden und werden entweder als defekt gekennzeichnet oder außer Betrieb genommen.
(6) Ausrüstungen, die für die Großhandelsvertriebstätigkeiten nicht relevant sind, dürfen nicht im selben Bereich wie die Tierarzneimittel gelagert werden.
(7) Über die Reparatur, Wartung und Kalibrierung der wichtigsten Ausrüstungen, darunter Kühlräume, personell überwachte Einbruchmeldeanlagen, Zugangskontrollsysteme, Kühlschränke, Thermo-Hygrometer und andere Temperatur- und Feuchtigkeitsaufzeichnungssysteme, Klimaanlagen und alle Gerätschaften im Zusammenhang mit der weiterführenden Lieferkette, werden angemessene Aufzeichnungen geführt und aufbewahrt.
Artikel 15
Computergestützte Systeme
(1) Bevor ein computergestütztes System in Betrieb genommen wird, wird durch eine geeignete Validierung oder durch Verifikationsstudien nachgewiesen, dass das System in der Lage ist, die gewünschten Ergebnisse genau, kontinuierlich und reproduzierbar zu erreichen.
(2) Eine schriftliche, detaillierte Beschreibung des computergestützten Systems, einschließlich gegebenenfalls Diagramme, liegt vor. Diese Beschreibung wird stets auf dem neuesten Stand gehalten. In dem Dokument werden die Grundlagen, Ziele, Sicherheitsmaßnahmen, der Erfassungsbereich des Systems und seine wichtigsten Funktionalitäten beschrieben, sowie wie es verwendet wird und wie es sich in andere Systeme einbinden lässt.
(3) Daten werden nur von hierzu befugten Personen in das computergestützte System eingegeben oder geändert.
(4) Die Daten sind physisch oder elektronisch gesichert und gegen versehentliche oder unbefugte Änderung geschützt. Es wird regelmäßig überprüft, dass Zugang zu den gespeicherten Daten besteht. Es werden regelmäßige Sicherheitskopien der Daten erstellt. Sicherheitskopien werden wenigstens fünf Jahre lang oder für den im anwendbaren nationalen Recht vorgesehenen Zeitraum, wenn dieser Zeitraum mehr als fünf Jahre beträgt, an einem separaten und gesicherten Ort aufbewahrt.
(5) Es werden Verfahren für den Fall eines Systemausfalls oder -zusammenbruchs festgelegt. Dies schließt Systeme für die Wiederherstellung der Daten ein.
Artikel 16
Qualifizierung und Validierung
(1) Die in Artikel 1 Absatz 2 genannten Personen bestimmen, welche wesentlichen Ausrüstungsqualifizierungen und wesentlichen Verfahrensvalidierungen für die korrekte Einrichtung und den korrekten Betrieb erforderlich sind. Art und Umfang solcher Qualifizierungs- und Validierungstätigkeiten wie Lagerung und Verfahren für Warenkommissionierung und Verpackung werden auf der Grundlage eines dokumentierten Risikobewertungsansatzes festgelegt.
(2) Ausrüstungen und Verfahren unterliegen vor der Benutzung bzw. Anwendung sowie nach jeder wesentlichen Änderung, z. B. Reparatur oder Wartung, der Qualifizierung bzw. Validierung.
(3) Es werden Qualifizierungs- und Validierungsberichte erstellt, in denen die Ergebnisse zusammengefasst und etwaige Abweichungen kommentiert werden. Erforderlichenfalls werden die CAPA-Grundsätze angewendet. Nachweise der zufriedenstellenden Validierung und Abnahme eines Verfahrens oder eines Ausrüstungsgegenstandes werden von hierfür qualifizierten Mitarbeitern erstellt und genehmigt.
KAPITEL V
DOKUMENTATION, VERFAHREN UND AUFZEICHNUNGEN
Artikel 17
Dokumentationsanforderungen
(1) Die Dokumentation entspricht folgenden Anforderungen:
|
a) |
Sie ist leicht zugänglich bzw. abrufbar; |
|
b) |
sie ist in Bezug auf den Tätigkeitsbereich der in Artikel 1 Absatz 2 genannten Personen umfassend genug; |
|
c) |
sie ist in einer für das Personal verständlichen Sprache verfasst; |
|
d) |
sie ist klar und eindeutig formuliert. |
(2) Die Dokumentation wird von den entsprechend befugten Personen je nach Bedarf genehmigt, abgezeichnet und datiert. Sie wird nicht handschriftlich verfasst, es sei denn, handschriftliche Einträge sind aus praktischen Gründen gerechtfertigt. In diesem Fall wird ausreichend Platz für entsprechende handschriftliche Einträge gelassen.
(3) Werden Fehler in den Unterlagen festgestellt, so werden diese unverzüglich berichtigt, wobei klar nachvollziehbar ist, wer sie berichtigt hat und wann.
(4) Jede Änderung an der Dokumentation wird mit Unterschrift und Datum versehen. Die Originalfassung bleibt trotz der Änderung weiterhin erkennbar. Erforderlichenfalls wird der Grund der Änderung festgehalten.
(5) Unterlagen werden wenigstens fünf Jahre lang oder für den im anwendbaren nationalen Recht vorgesehenen Zeitraum, wenn dieser Zeitraum mehr als fünf Jahre beträgt, aufbewahrt. Personenbezogene Daten werden gelöscht, sobald ihre Speicherung für die Zwecke der Vertriebstätigkeiten nicht mehr erforderlich ist.
(6) Jeder Angestellte hat direkten Zugang zu allen für seine Aufgaben erforderlichen Unterlagen.
(7) Bei allen papiergestützten, elektronischen und hybriden Systemen werden die Beziehungen und Kontrollmaßnahmen für Originaldokumente und amtliche Kopien, Datenverarbeitung und Aufzeichnungen angegeben.
Artikel 18
Verfahren
(1) Die Verfahren beschreiben die Großhandelsvertriebstätigkeiten, die sich auf die Qualität von Tierarzneimitteln auswirken. Diese Tätigkeiten umfassen:
|
a) |
Annahme und Überprüfung der Lieferungen; Kontrolle der Lieferanten und Kunden; |
|
b) |
Lagerung; |
|
c) |
Reinigung und Instandhaltung der Betriebsräume und Ausrüstungen, einschließlich Schädlingsbekämpfung; |
|
d) |
Kontrolle und Aufzeichnung der Lagerbedingungen; |
|
e) |
Schutz der Tierarzneimittel während des Transports; |
|
f) |
Sicherung der Bestände vor Ort und der Durchfuhrsendungen; |
|
g) |
Entfernung aus dem verkaufsfähigen Bestand; |
|
h) |
Handhabung zurückgegebener Tierarzneimittel; |
|
i) |
Planung von Rückrufaktionen; |
|
j) |
Qualifizierung und Validierung; |
|
k) |
Verfahren und Maßnahmen für die Entsorgung unbrauchbarer Tierarzneimittel; |
|
l) |
Verfahren zur Untersuchung und Lösung von Beschwerden; |
|
m) |
Verfahren zur Identifizierung von Tierarzneimitteln, bei denen der Verdacht auf Fälschung besteht. |
(2) Verfahren werden von der verantwortlichen Person genehmigt, abgezeichnet und datiert.
(3) Es werden gültige und genehmigte Verfahren angewendet. Die Unterlagen sind klar und angemessen detailliert. Titel, Art und Zweck der Unterlagen werden angegeben. Die Unterlagen werden regelmäßig überprüft und stets auf dem neuesten Stand gehalten. Bei den Verfahren wird eine Versionskontrolle angewendet. Es existiert ein System, um nach Überarbeitung eines Dokuments sicherzustellen, dass nicht aus Versehen nicht mehr gültige Versionen verwendet werden. Nicht mehr gültige oder überholte Verfahren werden von den Arbeitsplätzen entfernt und archiviert.
Artikel 19
Aufzeichnungen
(1) Alle Transaktionen mit Tierarzneimitteln — Eingang oder Lieferung — werden entweder in der Form von Einkaufs- bzw. Verkaufsrechnungen und Lieferscheinen oder in elektronischer Form aufgezeichnet.
(2) Zusätzlich zu den detaillierten Aufzeichnungen gemäß Artikel 101 Absatz 7 der Verordnung (EU) 2019/6 enthalten Aufzeichnungen gegebenenfalls alle zusätzlichen Anforderungen, die im nationalen Recht festgelegt sind.
(3) Die Aufzeichnungen erfolgen zeitgleich mit der Durchführung der Tätigkeiten. Handschriftliche Aufzeichnungen sind in klarer, leserlicher und unauslöschlicher Handschrift verfasst.
KAPITEL VI
BETRIEB
Artikel 20
Anforderungen an den Betrieb
(1) Die in Artikel 1 Absatz 2 genannten Personen sorgen dafür, dass die Identität des Tierarzneimittels während des Großhandelsvertriebs nicht verloren geht, und bedienen sich aller verfügbaren Mittel, um das Risiko zu minimieren, dass gefälschte Tierarzneimittel in die legale Lieferkette gelangen.
(2) Die in Artikel 1 Absatz 2 genannten Personen stellen sicher, dass der Großhandelsvertrieb der Tierarzneimittel gemäß den Angaben auf der äußeren Umhüllung erfolgt.
(3) Die in Artikel 1 Absatz 2 genannten Personen sorgen dafür, dass alle Tierarzneimittel, die sie in der Union vertreiben,
|
a) |
über eine von der zuständigen Behörde bzw. der Kommission erteilte Zulassung verfügen; |
|
b) |
durch eine von einer zuständigen Behörde erteilte Registrierung abgedeckt sind; |
|
c) |
unter eine von der zuständigen Behörde gewährte Ausnahme von den Anforderungen für die Zulassung fallen; |
|
d) |
durch eine von der zuständigen Behörde des Bestimmungsmitgliedstaats erteilte Genehmigung für den Parallelhandel abgedeckt sind; |
|
e) |
durch eine Anwendungserlaubnis gemäß Artikel 110 Absätze 2 und 3 der Verordnung (EU) 2019/6 abgedeckt sind, oder |
|
f) |
im Falle von Produkten, die gemäß Artikel 112 Absatz 2, Artikel 113 Absatz 2 oder Artikel 114 Absatz 4 der Verordnung (EU) 2019/6 verwendet werden sollen, von Inhabern einer Herstellungserlaubnis eingeführt wurden, die gemäß Artikel 90 jener Verordnung oder gegebenenfalls nach den in Artikel 106 Absatz 3 jener Verordnung genannten Verfahren erteilt wurde. |
(4) Alle wesentlichen Vorgänge durch die in Artikel 1 Absatz 2 genannten Personen werden im Qualitätssicherungssystem anhand geeigneter Aufzeichnungen genau beschrieben.
Artikel 21
Überprüfung der Eignung und Zulassung von Lieferanten
(1) Stammen die Tierarzneimittel von einer in Artikel 1 Absatz 2 genannten Person, überprüft der die Produkte empfangende Großhändler, ob der Lieferant die gute Vertriebspraxis für Tierarzneimittel im Sinne der vorliegenden Verordnung einhält und über eine Genehmigung verfügt. Diese Informationen werden von den zuständigen nationalen Behörden eingeholt oder aus der in Artikel 91 Absatz 1 der Verordnung (EU) 2019/6 genannten Datenbank der Union für Herstellung, Einfuhr und Großhandelsvertrieb beschafft. Vor jeglicher Beschaffung von Tierarzneimitteln erfolgt eine geeignete Überprüfung der Eignung und Zulassung der Lieferanten. Dieser Prozess wird mittels eines Verfahrens kontrolliert und die Ergebnisse werden aufgezeichnet und gemäß den Grundsätzen des Qualitätsrisikomanagements regelmäßig überprüft.
(2) Bevor ein Großhändler ein Vertragsverhältnis mit einem neuen Lieferanten eingeht, führen die in Artikel 1 Absatz 2 genannten Personen sogenannte Due-Diligence-Prüfungen durch, um Eignung, Kompetenz und Zuverlässigkeit der anderen Partei zu bewerten. Folgendes ist bei den Due-Diligence-Prüfungen zu berücksichtigen:
|
a) |
Ansehen oder Zuverlässigkeit des Lieferanten; |
|
b) |
Angebote von Tierarzneimitteln, bei denen die Wahrscheinlichkeit einer Fälschung höher ist; |
|
c) |
Angebote über große Mengen an Tierarzneimitteln, die im Allgemeinen nur in begrenzter Menge zur Verfügung stehen; |
|
d) |
ungewöhnlich hohe Vielfalt an Tierarzneimitteln, die vom Lieferanten gehandhabt werden; |
|
e) |
ungewöhnlich niedrige Preise. |
Artikel 22
Überprüfung der Eignung und Zulassung von Kunden
(1) Die in Artikel 1 Absatz 2 genannten Personen nehmen erste und gegebenenfalls regelmäßige Überprüfungen vor, um festzustellen, ob ihre Kunden die Anforderungen gemäß Artikel 101 Absatz 2 der Verordnung (EU) 2019/6 erfüllen. Dies kann das Ersuchen um Kopien der Genehmigungen gemäß nationalem Recht, über die der Kunde verfügt, die Überprüfung des Status auf der Website der zuständigen Behörde sowie das Ersuchen um Nachweise der Qualifikationen oder Befugnisse nach nationalem Recht umfassen.
(2) Die in Artikel 1 Absatz 2 genannten Personen überwachen ihre Transaktionen und gehen jeder Unregelmäßigkeit in den üblichen Verkaufsmustern für Betäubungsmittel, psychotrope Substanzen oder andere Gefahrstoffe nach. Ungewöhnliche Verkaufsmuster, die auf eine Abzweigung oder einen Missbrauch von Tierarzneimitteln hindeuten können, werden untersucht und erforderlichenfalls den zuständigen Behörden gemeldet.
Artikel 23
Annahme von Tierarzneimitteln
(1) Die für die Annahme von Tierarzneimitteln verantwortlichen Personen stellen sicher, dass die eingehende Sendung korrekt ist, die Tierarzneimittel von zugelassenen Lieferanten stammen und dass sie während des Transports nicht sichtbar beschädigt wurden.
(2) Tierarzneimittel, die eine spezielle Lagerung oder besondere Sicherheitsmaßnahmen erfordern, werden prioritär behandelt und nach Abschluss der angemessenen Kontrollen sofort in geeignete Lagerräume gebracht.
(3) Chargen von Tierarzneimitteln, die für den Unionsmarkt bestimmt sind, werden nicht in den verkaufsfähigen Bestand aufgenommen, bevor nicht verfahrensgemäß sichergestellt ist, dass sie zum Verkauf zugelassen sind. Bei Chargen aus einem anderen Mitgliedstaat werden vor ihrer Aufnahme in den verkaufsfähigen Bestand der in Artikel 97 Absätze 6 und 9 der Verordnung (EU) 2019/6 genannte Prüfbericht und gegebenenfalls die Ergebnisse der erforderlichen Prüfungen gemäß Artikel 97 Absatz 7 jener Verordnung oder ein anderer, auf einem gleichwertigen System beruhender Nachweis der Marktzulassung von angemessen ausgebildetem Personal sorgfältig geprüft.
Artikel 24
Lagerung
(1) Tierarzneimittel werden getrennt von anderen Produkten gelagert, welche diese möglicherweise verändern könnten, und vor den negativen Auswirkungen von Licht, Temperatur, Feuchtigkeit und anderen externen Faktoren geschützt. Besondere Aufmerksamkeit wird Produkten gewidmet, die besonderer Lagerbedingungen bedürfen.
(2) Angelieferte Tierarzneimittelbehälter werden, falls erforderlich, vor der Lagerung gereinigt. Alle Tätigkeiten, die an den eingehenden Waren durchgeführt werden, dürfen sich nicht auf die Qualität der Tierarzneimittel auswirken.
(3) Im Rahmen des Lagerverfahrens wird gewährleistet, dass geeignete Lagerbedingungen eingehalten werden und die Bestände angemessen gesichert werden können.
(4) Die Lagerbestände werden nach dem Prinzip „first expired — first out“ (die Waren, deren Verfalldatum zuerst überschritten wird, verlassen das Lager zuerst) verwaltet. Ausnahmen werden dokumentiert.
(5) Tierarzneimittel werden so gehandhabt und gelagert, dass ein Austreten, Zu-Bruch-Gehen, eine Kontaminierung oder Verwechselung weitestgehend vermieden werden. Tierarzneimittel werden nicht direkt auf dem Boden gelagert, es sei denn, die Verpackung ist für eine solche Lagerung gedacht, beispielsweise bestimmte Flaschen für medizinische Gase.
(6) Tierarzneimittel, die kurz vor ihrem Verfalldatum liegen, werden sofort physisch oder, sofern ein vergleichbares elektronisches System verfügbar ist, mittels elektronischer Trennung aus dem verkaufsfähigen Bestand entfernt.
(7) Es werden regelmäßige Inventuren durchgeführt, wobei jeweils die nationalen rechtlichen Anforderungen zu berücksichtigen sind. Unregelmäßigkeiten im Bestand werden untersucht und dokumentiert.
Artikel 25
Vernichtung veralteter Tierarzneimittel
(1) Tierarzneimittel, die vernichtet werden sollen, werden angemessen gekennzeichnet, gesondert gelagert und verfahrensgemäß gehandhabt.
(2) Die Vernichtung von Tierarzneimitteln geschieht im Einklang mit den geltenden Anforderungen an Handhabung, Transport und Entsorgung solcher Produkte.
(3) Über alle vernichteten Tierarzneimittel werden für einen bestimmten Zeitraum, der in dem in Artikel 3 genannten Qualitätssicherungssystem bestimmt ist, Aufzeichnungen aufbewahrt.
Artikel 26
Warenkommissionierung
Es gibt Kontrollen, mit denen gewährleistet werden kann, dass das richtige Tierarzneimittel entnommen wird. Das entnommene Tierarzneimittel verfügt noch über eine angemessen lange Haltbarkeit und wurde während der Lagerung nicht beschädigt.
Artikel 27
Lieferung
(1) Jeder Lieferung wird ein elektronisches oder physisches Dokument beigefügt, das zusätzlich zu den Angaben gemäß Artikel 101 Absatz 7 der Verordnung (EU) 2019/6 eine eindeutige Nummer zur Identifizierung des Lieferauftrags, die geltenden Transport- und Lagerbedingungen sowie zusätzliche Anforderungen nach nationalem Recht enthält.
(2) Elektronische oder physische Aufzeichnungen werden so geführt, dass der tatsächliche Lagerort des Tierarzneimittels jederzeit erkennbar ist.
Artikel 28
Ausfuhr
(1) Bei der Ausfuhr von Tierarzneimitteln, für die weder die zuständige nationale Behörde noch gegebenenfalls die Kommission eine Zulassung gemäß Kapitel III der Verordnung (EU) 2019/6 erteilt hat, treffen die Großhändler die notwendigen Vorkehrungen, damit diese Tierarzneimittel nicht auf den Unionsmarkt gelangen.
(2) Liefern die in Artikel 1 Absatz 2 genannten Personen Tierarzneimittel an Personen in Drittländern, sorgen sie dafür, dass eine solche Lieferung nur an Personen erfolgt, die ermächtigt oder befugt sind, Tierarzneimittel zum Großhandelsvertrieb oder zur Abgabe an die Öffentlichkeit gemäß den anwendbaren Rechts- und Verwaltungsvorschriften des betreffenden Drittlandes zu erhalten.
KAPITEL VII
BESCHWERDEN, RÜCKGABEN, VERDACHTSFÄLLE GEFÄLSCHTER TIERARZNEIMITTEL UND RÜCKRUFE
Artikel 29
Beschwerden
(1) Beschwerden werden in allen ursprünglich angegebenen Einzelheiten aufgezeichnet. Dabei wird nach Beschwerden über die Qualität des Tierarzneimittels und Beschwerden über den Großhandelsvertrieb unterschieden.
Von einer Beschwerde über die Qualität eines Tierarzneimittels und einen möglichen Produktmangel wird der Hersteller oder der Zulassungsinhaber umgehend in Kenntnis gesetzt.
Jeder Beschwerde über den Vertrieb eines Tierarzneimittels wird gründlich nachgegangen, um die Ursache oder den Grund der Beschwerde festzustellen.
(2) Für die Bearbeitung von Beschwerden wird eine Person benannt und es wird genügend Personal zu deren Unterstützung zur Verfügung gestellt.
(3) Wenn nötig, werden nach Untersuchung und Bewertung der Beschwerde geeignete Folgemaßnahmen ergriffen (einschließlich CAPA), zu denen erforderlichenfalls auch eine Meldung an die zuständigen nationalen Behörden gehören kann.
Artikel 30
Rückgaben
(1) Zurückgegebene Tierarzneimittel werden gemäß einem schriftlich niedergelegten, risikobasierten Prozess gehandhabt, bei dem die Art des betroffenen Tierarzneimittels, eventuell erforderliche besondere Lagerbedingungen und die seit der Lieferung verstrichene Zeit berücksichtigt werden. Rückgaben erfolgen gemäß nationalem Recht und im Einklang mit den zwischen den Parteien bestehenden Vertragsbestimmungen.
(2) Tierarzneimittel, die die Verfügungsgewalt der in Artikel 1 Absatz 2 genannten Personen verlassen haben, werden nur dann wieder in den verkaufsfähigen Bestand aufgenommen, wenn folgende Voraussetzungen erfüllt sind:
|
a) |
Die Tierarzneimittel befinden sich noch in ihrer ungeöffneten und unbeschädigten Sekundärverpackung und sind in gutem Zustand; |
|
b) |
das Verfalldatum der Tierarzneimittel ist nicht überschritten und sie sind nicht Gegenstand eines Rückrufs; |
|
c) |
die Tierarzneimittel, die von einem Kunden, der nicht über eine Großhandelsvertriebserlaubnis verfügt, oder von Apotheken oder Personen, die gemäß dem nationalen Recht des betreffenden Mitgliedstaats zur Abgabe von Tierarzneimitteln an die Öffentlichkeit befugt sind, zurückgegeben wurden, sind innerhalb einer bestimmten, nach den Grundsätzen des Qualitätsrisikomanagements festgelegten akzeptablen Frist zurückgegeben worden; |
|
d) |
die Tierarzneimittel wurden nicht vom Tierhalter an die Apotheke oder an andere Personen zurückgegeben, die gemäß dem nationalen Recht des betreffenden Mitgliedstaats zur Abgabe von Tierarzneimitteln an die Öffentlichkeit befugt sind, es sei denn, eine solche Rückgabe ist nach dem nationalen Recht des betreffenden Mitgliedstaats zulässig; |
|
e) |
der Kunde hat nachgewiesen, dass die Tierarzneimittel gemäß den für sie geltenden besonderen Lagerbedingungen transportiert, gelagert und gehandhabt wurden; |
|
f) |
die Tierarzneimittel wurden von einer ausreichend geschulten, sachkundigen und entsprechend befugten Person untersucht und bewertet; |
|
g) |
die in Artikel 1 Absatz 2 genannten Personen haben nach dem nationalen Recht erforderliche angemessene Nachweise (Kopien des Lieferscheins oder Verweise auf Rechnungsnummern, Chargennummern, Verfalldatum usw.) über die tatsächliche Auslieferung des Tierarzneimittels an den Kunden, der das Tierarzneimittel zurückgibt, und es gibt keinen Grund zu der Annahme, dass das Tierarzneimittel gefälscht ist. |
(3) Tierarzneimittel, die eine Lagerung bei besonderen (beispielsweise niedrigen) Temperaturen erfordern, werden darüber hinaus nur dann in den verkaufsfähigen Bestand zurückgeführt, wenn dokumentierte Nachweise vorliegen, dass das Produkt während der in den Buchstaben a bis f genannten Zeiträume unter den genehmigten Lagerbedingungen gelagert wurde. Gab es irgendwelche Abweichungen, wird eine Risikobewertung durchgeführt, mit der die Unversehrtheit des Tierarzneimittels nachgewiesen wird. Es sind Nachweise über alle folgenden Schritte erforderlich:
|
a) |
Lieferung an den Kunden; |
|
b) |
Untersuchung des Tierarzneimittels; |
|
c) |
Öffnen der Transportverpackung; |
|
d) |
Zurücklegen des Tierarzneimittels in die Verpackung; |
|
e) |
Abholung und Rückgabe an die in Artikel 1 Absatz 2 genannten Personen; |
|
f) |
Rückführung in die Kühleinrichtung des Großhändlers. |
(4) Produkte, die wieder in den verkaufsfähigen Bestand aufgenommen wurden, werden nach dem System „first expired — first out“ gehandhabt.
(5) Gestohlene Tierarzneimittel, die wieder aufgefunden werden, werden weder wieder in den verkaufsfähigen Bestand aufgenommen noch an Kunden verkauft.
Artikel 31
Gefälschte Tierarzneimittel
(1) Zusätzlich zu der Unterrichtung gemäß Artikel 101 Absatz 6 der Verordnung (EU) 2019/6 stellen Großhändler den Vertrieb von Tierarzneimitteln, die sie als gefälscht erkennen oder von denen sie vermuten, dass sie gefälscht sind, unverzüglich ein und handeln entsprechend den Anweisungen der zuständigen Behörden. Ein entsprechendes Verfahren ist vorhanden. Der Vorfall wird in allen Einzelheiten aufgezeichnet und untersucht.
(2) Alle Verdachtsfälle gefälschter Tierarzneimittel, die sich in der Lieferkette finden, werden unverzüglich physisch oder, sofern ein vergleichbares elektronisches System verfügbar ist, elektronisch abgesondert. Alle gefälschten Arzneimittel, die sich in der Lieferkette finden, werden unverzüglich physisch abgesondert, in einem speziell dafür eingerichteten Bereich, abseits von den übrigen Tierarzneimitteln, gelagert und entsprechend gekennzeichnet. Alle einschlägigen Maßnahmen, die in Bezug auf solche Produkte ergriffen werden, werden dokumentiert und die Aufzeichnungen aufbewahrt.
Artikel 32
Rückrufe
(1) Es sind eine Dokumentation und Verfahren vorhanden, um sicherzustellen, dass die eingegangenen und vertriebenen Tierarzneimittel für die Zwecke eines Produktrückrufs rückverfolgbar sind.
(2) Im Falle eines Tierarzneimittelrückrufs unterrichten die in Artikel 1 Absatz 2 genannten Personen mit der gebotenen Dringlichkeit und mit klaren Handlungsanweisungen alle betroffenen Kunden, an die das Produkt vertrieben wurde.
(3) Die in Artikel 1 Absatz 2 genannten Personen unterrichten die zuständige nationale Behörde über alle Tierarzneimittelrückrufe. Im Falle einer Ausfuhr des Tierarzneimittels unterrichten die in Artikel 1 Absatz 2 genannten Personen die Kunden bzw. zuständigen Behörden in dem betreffenden Drittland gemäß den nationalen Rechtsvorschriften über den Rückruf.
(4) Die in Artikel 1 Absatz 2 genannten Personen bewerten auf der Grundlage von Grundsätzen des Qualitätsrisikomanagements regelmäßig die Wirksamkeit der Vorkehrungen für Tierarzneimittelrückrufe.
(5) Die in Artikel 1 Absatz 2 genannten Personen gewährleisten, dass Rückrufaktionen jederzeit und zügig eingeleitet werden können.
(6) Die in Artikel 1 Absatz 2 genannten Personen befolgen die Anweisungen einer Rückrufnachricht, die, falls erforderlich, von den zuständigen Behörden genehmigt wird.
(7) Jede Rückrufaktion wird zeitgleich mit der Durchführung dokumentiert. Die entsprechenden Aufzeichnungen werden den zuständigen Behörden leicht zugänglich gemacht.
(8) Die Aufzeichnungen über den Vertrieb, einschließlich der Aufzeichnungen über ausgeführte Tierarzneimittel und Tierarzneimittelproben, sind den für den Rückruf zuständigen Personen leicht zugänglich und enthalten ausreichende Angaben zu den Händlern und direkt belieferten Kunden (Anschriften, Telefonnummern und elektronische Kommunikationsmittel während und außerhalb der Geschäftszeiten, rechtlich vorgeschriebene Chargennummer und gelieferte Mengen).
(9) Der Fortschritt der Rückrufaktion, einschließlich des Abgleichs der ausgelieferten mit der zurückerhaltenen Menge des zurückgerufenen Tierarzneimittels, wird in einem Abschlussbericht festgehalten.
KAPITEL VIII
AUSGELAGERTE TÄTIGKEITEN
Artikel 33
Pflichten des Auftraggebers
(1) Der Auftraggeber ist für alle ausgelagerten Tätigkeiten verantwortlich.
(2) Der Auftraggeber ist dafür verantwortlich, die Fähigkeit des Auftragnehmers zur erfolgreichen Durchführung der Arbeiten zu bewerten und mittels Vertragsgestaltung und Audits sicherzustellen, dass die Grundsätze der guten Vertriebspraxis für Tierarzneimittel eingehalten werden. Der Auftragnehmer führt vor Aufnahme der ausgelagerten Tätigkeiten ein Audit des Auftragnehmers durch und überwacht und überprüft ihre Ausführung durch den Auftragnehmer. Die Häufigkeit der Audits wird auf Risikobasis je nach Art der ausgelagerten Tätigkeiten festgelegt. Bei einer Änderung der ausgelagerten Tätigkeiten wendet der Auftraggeber im Rahmen der Änderungskontrolle eine Risikobewertung an, um festzustellen, ob ein erneutes Audit erforderlich ist. Der Auftragnehmer gestattet dem Auftragnehmer die Durchführung von Audits der ausgelagerten Tätigkeiten.
(3) Der Auftraggeber stellt dem Auftragnehmer alle Informationen zu Verfügung, die zur Durchführung der ausgelagerten Tätigkeiten gemäß den spezifischen Anforderungen für das Tierarzneimittel und anderen einschlägigen Anforderungen erforderlich sind.
Artikel 34
Pflichten des Auftragnehmers
(1) Der Auftragnehmer verfügt über angemessene Ausrüstungen, Verfahren, Fachkenntnis und Erfahrung sowie über fachkundiges Personal zur Durchführung der in Auftrag gegebenen Tätigkeiten und, falls für die Tätigkeit erforderlich, über angemessene Betriebsräume.
(2) Der Auftragnehmer vergibt die ihm im Rahmen des Vertrags übertragenen Aufgaben nur dann an eine dritte Partei weiter, wenn der Auftraggeber diese Vereinbarung zuvor bewertet und gutgeheißen hat und die Drittpartei entweder vom Auftraggeber oder vom Auftragnehmer auditiert worden ist. In der zwischen dem Auftragnehmer und einer dritten Partei getroffenen Vereinbarung ist festgelegt, dass die Informationen über den Großhandelsvertrieb ebenso zur Verfügung gestellt werden wie in dem ursprünglichen Vertrag zwischen Auftraggeber und Auftragnehmer vorgesehen.
(3) Der Auftragnehmer führt keinerlei Tätigkeiten aus, die die Qualität der im Namen des Auftraggebers gehandhabten Tierarzneimittel beeinträchtigen könnten.
(4) Der Auftragnehmer leitet dem Auftraggeber gemäß den Anforderungen des Vertrags jede Information weiter, die einen Einfluss auf die Qualität des Tierarzneimittels haben könnte.
KAPITEL IX
SELBSTINSPEKTIONEN
Artikel 35
Selbstinspektionsprogramm
Es wird ein Selbstinspektionsprogramm durchgeführt, in dessen Rahmen alle Aspekte der guten Vertriebspraxis für Tierarzneimittel und die Einhaltung der vorliegenden Verordnung und Verfahren innerhalb eines bestimmten Zeitraums überprüft werden.
Artikel 36
Durchführung und Aufzeichnung der Selbstinspektionen
(1) Selbstinspektionen können in mehrere einzelne Selbstinspektionen geringeren Umfangs unterteilt werden.
(2) Selbstinspektionen werden von hierfür benannten, kompetenten Mitarbeitern genau und unvoreingenommen durchgeführt. Von externen Experten durchgeführte Audits können die Selbstinspektion nicht ersetzen.
(3) Über alle Selbstinspektionen werden Aufzeichnungen geführt. Die Berichte enthalten alle im Rahmen der Inspektion gemachten Beobachtungen. Der Geschäftsführung und anderen zuständigen Personen wird eine Kopie des Berichts weitergeleitet.
(4) Werden Unregelmäßigkeiten oder Mängel festgestellt, werden die Gründe hierfür untersucht und die CAPA werden dokumentiert und weiterverfolgt. Die Wirksamkeit der CAPA wird überprüft.
KAPITEL X
TRANSPORT
Artikel 37
Transportanforderungen
(1) Die in Artikel 1 Absatz 2 genannten Personen, die Tierarzneimittel liefern, sind für den Schutz der Tierarzneimittel vor Bruch, Beeinträchtigung und Diebstahl verantwortlich; zudem stellen sie sicher, dass die Temperaturbedingungen sich während des Transports in einem akzeptablen Bereich bewegen, und überwachen diese Bedingungen nach Möglichkeit.
(2) Während des Transports werden die erforderlichen Lager- bzw. Transportbedingungen für Tierarzneimittel innerhalb der vom Hersteller und Zulassungsinhaber oder auf der äußeren Umhüllung angegebenen Grenzen gehalten.
(3) Kommt es während des Transports zu Abweichungen, wie einer Temperaturabweichung, oder zu einer Beschädigung des Tierarzneimittels, werden die in Artikel 1 Absatz 2 genannten Personen und der Empfänger der betroffenen Tierarzneimittel davon unterrichtet, damit diese die potenziellen Auswirkungen auf die Qualität der betreffenden Tierarzneimittel bewerten können. Es gibt ein Verfahren für die Untersuchung und die Handhabung von Abweichungen von der Referenztemperatur.
(4) Die in Artikel 1 Absatz 2 genannten Personen stellen sicher, dass die für den Vertrieb, die Lagerung oder die Handhabung von Tierarzneimitteln verwendeten Fahrzeuge und Ausrüstungen für die betreffende Verwendung geeignet und so ausgestattet sind, dass die Tierarzneimittel keinen Bedingungen ausgesetzt werden, durch die ihre Qualität beeinträchtigt oder ihre Verpackung beschädigt werden könnte.
(5) Für Betrieb und Wartung aller Fahrzeuge und Ausrüstungen, die im Vertriebsprozess verwendet werden, gibt es Verfahren, die auch die Reinigung und Sicherheitsvorkehrungen erfassen.
(6) Aus den für die Reinigung von Fahrzeugen gewählten Utensilien ergibt sich keine Kontamination.
(7) Um festzustellen, wo Temperaturkontrollen erforderlich sind, wird eine Risikobewertung der Transportwege durchgeführt. Ausrüstungen zur Temperaturüberwachung beim Transport in Fahrzeugen oder Behältern werden in regelmäßigen Abständen, die auf der Grundlage der Grundsätze des Qualitätsrisikomanagements festgelegt werden, gewartet und kalibriert.
(8) Für die Handhabung sowohl von Tierarzneimitteln als auch von Humanarzneimitteln werden, soweit möglich, diesem Zweck vorbehaltene Fahrzeuge und Ausrüstungen verwendet. Werden Fahrzeuge und Ausrüstungen benutzt, die nicht diesem Zweck vorbehalten sind, bestehen Verfahren, mit denen gewährleistet werden kann, dass die Qualität der Tierarzneimittel nicht beeinträchtigt wird.
(9) Die Lieferung erfolgt an die auf dem Lieferschein angegebene Adresse und zu Händen oder an die Betriebsräume des Empfängers. Tierarzneimittel dürfen zu keinem Zeitpunkt in anderen Betriebsräumen abgegeben werden.
(10) Für dringende Lieferungen außerhalb der üblichen Bürozeiten werden Personen benannt und es bestehen Verfahren.
(11) Übernimmt eine dritte Partei den Transport, beinhaltet der mit ihr abgeschlossene Vertrag die Anforderungen der Artikel 33 und 34; in diesem Vertrag sind zudem eindeutig die Pflichten der dritten Partei zur Gewährleistung der Einhaltung der guten Vertriebspraxis für Tierarzneimittel festgelegt. Die in Artikel 1 Absatz 2 genannten Personen weisen die Transportdienstleister auf die für die Sendung geltenden Transportbedingungen hin.
(12) Umfasst der Transportweg das Abladen, Umladen oder die Zwischenlagerung an einem Verkehrsumschlagplatz, ist jedes Zwischenlager sauber und sicher und ermöglicht gegebenenfalls die Überwachung der Temperatur.
(13) Es werden Vorkehrungen getroffen, um die Dauer von Zwischenlagerungen bis zur nächsten Transportetappe zu minimieren.
Artikel 38
Behälter, Verpackung und Etikettierung
(1) Tierarzneimittel werden in Behältern transportiert, die keine negativen Auswirkungen auf die Qualität der Tierarzneimittel haben und einen angemessenen Schutz vor äußeren Einflüssen und Kontamination bieten.
(2) Die Wahl der Behälter und der Verpackung erfolgt nach
|
a) |
den Lagerungs- und Transportanforderungen für die Tierarzneimittel; |
|
b) |
dem Platz, der für die Tierarzneimittelmenge benötigt wird; |
|
c) |
pharmazeutischer Form, einschließlich Arzneimittel-Vormischungen; |
|
d) |
den zu erwartenden äußeren Temperaturextremen; |
|
e) |
der zu erwartenden maximalen Transportdauer einschließlich Zwischenlagerung beim Zoll; |
|
f) |
dem Qualifikationsstatus der Verpackung; |
|
g) |
dem Validierungsstatus der Versandbehälter. |
(3) Behälter sind mit Etiketten versehen, die ausreichende Informationen über die Handhabungs- und Lagerbedingungen sowie Vorsichtsmaßnahmen enthalten, damit sichergestellt ist, dass die Tierarzneimittel jederzeit korrekt gehandhabt werden und gesichert sind. Anhand der Behälter lassen sich ihr Inhalt und ihre Herkunft bestimmen.
Artikel 39
Produkte, für die besondere Bedingungen gelten
(1) Bei Lieferungen, die Tierarzneimittel enthalten, die besonderen Bedingungen unterliegen, wie Betäubungsmittel oder psychotrope Substanzen, sorgen die in Artikel 1 Absatz 2 genannten Personen für eine sichere und geschützte Lieferkette für diese Produkte gemäß den Rechtsvorschriften des betreffenden Mitgliedstaats. Für die Lieferung solcher Produkte gibt es zusätzliche Kontrollsysteme. Für den Fall eines Diebstahls gibt es ein spezifisches Protokoll.
(2) Tierarzneimittel, die hochaktives Material enthalten, werden in sicheren, geschützten und hierfür bestimmten Behältern und Fahrzeugen im Einklang mit den geltenden Sicherheitsmaßnahmen transportiert.
(3) Für temperaturempfindliche Tierarzneimittel werden einer Qualifizierung unterliegende Ausrüstungen, z. B. Thermalverpackungen, temperierte Behälter oder Fahrzeuge mit Temperaturkontrolle, verwendet, damit auf dem Weg vom Hersteller zum Großhändler und zum Kunden ordnungsgemäße Transportbedingungen gewährleistet sind, es sei denn, die Stabilität des Produkts wurde bei anderen Transportbedingungen nachgewiesen.
(4) Werden Fahrzeuge mit Temperaturkontrolle verwendet, werden die während des Transports zur Überwachung der Temperatur verwendeten Ausrüstungen in regelmäßigen Abständen gewartet und kalibriert. Es wird eine Temperaturverteilungsstudie unter repräsentativen Bedingungen durchgeführt, bei der auch die jahreszeitlich bedingten Schwankungen berücksichtigt werden.
(5) Auf hinreichend begründetes Verlangen eines Kunden und in jedem Fall bei Zwischenfällen legen die in Artikel 1 Absatz 2 genannten Personen ihm Angaben vor, aus denen hervorgeht, dass für die Tierarzneimittel die Lager- bzw. Transportbedingungen in Bezug auf die Temperatur eingehalten wurden.
(6) Werden Kühlakkus in Isolierbehältern verwendet, werden sie so platziert, dass die Tierarzneimittel nicht in direkten Kontakt mit den Kühlakkus kommen.
(7) Das Personal ist in den Verfahren für die (je nach Jahreszeit unterschiedliche) Montage von Isolierbehältern und der Wiederverwendung von Kühlakkus geschult.
(8) Die in Artikel 1 Absatz 2 genannten Personen verfügen über ein System zur Kontrolle der Wiederverwendung der Kühlakkus, mit dem ausgeschlossen werden kann, dass versehentlich unzureichend gekühlte Akkus verwendet werden. Die in Artikel 1 Absatz 2 genannten Personen stellen sicher, dass eine angemessene physische Trennung von tiefgekühlten und gekühlten Kühlelementen besteht.
(9) Die in Artikel 1 Absatz 2 genannten Personen beschreiben den Lieferungsprozess empfindlicher Tierarzneimittel und die Kontrolle jahreszeitlich bedingter Temperaturschwankungen in einem Verfahren.
KAPITEL XI
SCHLUSSBESTIMMUNGEN
Artikel 40
Inkrafttreten und Anwendung
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 29. Juli 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 4 vom 7.1.2019, S. 43.
(2) Good storage and distribution practices for medical products (Gute Lager- und Vertriebspraxis für Arzneimittel): Sachverständigenausschuss der Weltgesundheitsorganisation für Spezifikationen für pharmazeutische Zubereitungen: 54. Bericht. Genf: Weltgesundheitsorganisation; 2020: Anhang 7 (WHO Technical Report Series, No. 1025 (Technischer Bericht Nr. 1025 der Weltgesundheitsorganisation)).
(3) Guide to good storage practices for pharmaceuticals (Leitfaden für die gute Lagerpraxis für Arzneimittel): Sachverständigenausschuss der Weltgesundheitsorganisation für Spezifikationen für pharmazeutische Zubereitungen: 37. Bericht. Genf: Weltgesundheitsorganisation; 2003: Anhang 9 (WHO Technical Report Series, No. 908 (Technischer Bericht Nr. 908 der Weltgesundheitsorganisation)).
(4) Model guidance for the storage and transport of time- and temperature-sensitive pharmaceutical products (Musterleitlinien für die Lagerung und den Transport von zeit- und temperaturempfindlichen Arzneimitteln): Sachverständigenausschuss der Weltgesundheitsorganisation für Spezifikationen für pharmazeutische Zubereitungen: 45. Bericht. Genf: Weltgesundheitsorganisation; 2011: Anhang 9 (WHO Technical Report Series, No. 961 (Technischer Bericht Nr. 961 der Weltgesundheitsorganisation)).
(5) Pharmaceutical Inspection Convention (Übereinkommen zur gegenseitigen Anerkennung von Inspektionen betreffend die Herstellung pharmazeutischer Produkte, PIC) — Pharmaceutical Inspection Co-operation Scheme (Kooperationsprogramm zu Inspektionen im pharmazeutischen Bereich, PIC/S): PIC/S Guide to good distribution practice for medicinal products (Leitlinien für die gute Vertriebspraxis für Arzneimittel), PIC/S, PE 011-1, 1. Juni 2014.
(6) Leitlinien vom 5. November 2013 für die gute Vertriebspraxis von Humanarzneimitteln (2013/C 343/01) (ABl. C 343 vom 23.11.2013, S. 1).
(7) Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel (ABl. L 311 vom 28.11.2001, S. 67).
(8) Verordnung (EU) 2016/679 des Europäischen Parlaments und des Rates vom 27. April 2016 zum Schutz natürlicher Personen bei der Verarbeitung personenbezogener Daten, zum freien Datenverkehr und zur Aufhebung der Richtlinie 95/46/EG (Datenschutz-Grundverordnung) (ABl. L 119 vom 4.5.2016, S. 1).
(9) Verordnung (EU) Nr. 952/2013 des Europäischen Parlaments und des Rates vom 9. Oktober 2013 zur Festlegung des Zollkodex der Union (ABl. L 269 vom 10.10.2013, S. 1).
(10) Verordnung (EU) Nr. 910/2014 des Europäischen Parlaments und des Rates vom 23. Juli 2014 über elektronische Identifizierung und Vertrauensdienste für elektronische Transaktionen im Binnenmarkt und zur Aufhebung der Richtlinie 1999/93/EG (ABl. L 257 vom 28.8.2014, S. 73).
BESCHLÜSSE
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/67 |
BESCHLUSS (EU) 2021/1249 DES RATES
vom 26. Juli 2021
über den Standpunkt, der im Namen der Europäischen Union im Gemeinsamen EWR-Ausschuss zur Änderung des Protokolls 31 über die Zusammenarbeit in bestimmten Bereichen außerhalb der vier Freiheiten zum EWR-Abkommen zu vertreten ist (Haushaltslinie 07 20 03 01 — Soziale Sicherheit)
(Text von Bedeutung für den EWR)
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union, insbesondere auf die Artikel 46 und 48 in Verbindung mit Artikel 218 Absatz 9,
gestützt auf die Verordnung (EG) Nr. 2894/94 des Rates vom 28. November 1994 mit Durchführungsvorschriften zum Abkommen über den Europäischen Wirtschaftsraum (1), insbesondere auf Artikel 1 Absatz 3,
auf Vorschlag der Europäischen Kommission,
in Erwägung nachstehender Gründe:
|
(1) |
Das Abkommen über den Europäischen Wirtschaftsraum (2) (im Folgenden „EWR-Abkommen“) trat am 1. Januar 1994 in Kraft. |
|
(2) |
Gemäß Artikel 98 des EWR-Abkommens kann der Gemeinsame EWR-Ausschuss auch eine Änderung von Protokoll 31 über die Zusammenarbeit in bestimmten Bereichen außerhalb der vier Freiheiten (im Folgenden „Protokoll 31“) im Anhang des EWR-Abkommens beschließen. |
|
(3) |
Es empfiehlt sich, die Zusammenarbeit der Vertragsparteien des EWR-Abkommens bei aus dem Gesamthaushalt der Europäischen Union finanzierten Unionsmaßnahmen in den Bereichen Freizügigkeit der Arbeitnehmer, Koordinierung der Systeme der sozialen Sicherheit und Maßnahmen für Migranten, einschließlich Migranten aus Drittländern, fortzusetzen. |
|
(4) |
Protokoll 31 zum EWR-Abkommen sollte daher geändert werden, damit eine solche erweiterte Zusammenarbeit ab dem 1. Januar 2021 fortgesetzt werden kann. |
|
(5) |
Der Standpunkt der Union im Gemeinsamen EWR-Ausschuss sollte auf dem Beschlussentwurf des Gemeinsamen EWR-Ausschusses beruhen — |
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Der Standpunkt, der im Namen der Union im Gemeinsamen EWR-Ausschuss zur vorgeschlagenen Änderung des Protokolls 31 über die Zusammenarbeit in bestimmten Bereichen außerhalb der vier Freiheiten im Anhang des EWR-Abkommens zu vertreten ist, beruht auf dem Entwurf eines Beschlusses des Gemeinsamen EWR-Ausschusses (3).
Artikel 2
Dieser Beschluss tritt am Tag seiner Annahme in Kraft.
Geschehen zu Brüssel am 26. Juli 2021.
Im Namen des Rates
Der Präsident
G. DOVŽAN
(1) ABl. L 305 vom 30.11.1994, S. 6.
(2) ABl. L 1 vom 3.1.1994, S. 3.
(3) Siehe Dokument ST 10507/21 unter http://register.consilium.europa.eu.
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/69 |
BESCHLUSS (EU) 2021/1250 DES RATES
vom 26. Juli 2021
über den im Namen der Europäischen Union im Gemeinsamen EWR-Ausschuss zur Änderung des dem EWR-Abkommen als Anhang beigefügte Protokolls 31 über die Zusammenarbeit in bestimmten Bereichen außerhalb der vier Freiheiten zu vertretenden Standpunkt (Europäischer Verteidigungsfonds)
(Text von Bedeutung für den EWR)
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union, insbesondere auf Artikel 173 Absatz 3, Artikel 182 Absatz 4, Artikel 183 und Artikel 188 Absatz 2 in Verbindung mit Artikel 218 Absatz 9,
gestützt auf die Verordnung (EG) Nr. 2894/94 des Rates vom 28. November 1994 mit Durchführungsvorschriften zum Abkommen über den Europäischen Wirtschaftsraum (1), insbesondere auf Artikel 1 Absatz 3,
auf Vorschlag der Europäischen Kommission,
in Erwägung nachstehender Gründe:
|
(1) |
Das Abkommen über den Europäischen Wirtschaftsraum (2) (im Folgenden „EWR-Abkommen“) trat am 1. Januar 1994 in Kraft. |
|
(2) |
Gemäß Artikel 98 des EWR-Abkommens kann der Gemeinsame EWR-Ausschuss unter anderem Änderungen des dem EWR-Abkommen als Anhang beigefügten Protokolls 31 über die Zusammenarbeit in bestimmten Bereichen außerhalb der vier Freiheiten (im Folgenden „Protokoll 31“) beschließen. |
|
(3) |
Die Verordnung (EU) 2021/697 des Europäischen Parlaments und des Rates (3) ist in das EWR-Abkommen aufzunehmen. |
|
(4) |
Protokoll 31 zum EWR-Abkommen sollte daher entsprechend geändert werden. |
|
(5) |
Der von der Union im Gemeinsamen Ausschuss zu vertretende Standpunkt sollte daher auf dem Entwurf eines Beschlusses des Gemeinsamen EWR-Ausschusses beruhen — |
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Der Standpunkt, der im Namen der Europäischen Union im Gemeinsamen EWR-Ausschuss zur vorgeschlagenen Änderung des dem EWR-Abkommen als Anhang beigefügten Protokolls 31 über die Zusammenarbeit in bestimmten Bereichen außerhalb der vier Freiheiten zu vertreten ist, beruht auf dem Entwurf eines Beschlusses des Gemeinsamen EWR-Ausschusses (4).
Artikel 2
Dieser Beschluss tritt am Tag seiner Annahme in Kraft.
Geschehen zu Brüssel am 26. Juli 2021.
Im Namen des Rates
Der Präsident
G. DOVŽAN
(1) ABl. L 305 vom 30.11.1994, S. 6.
(2) ABl. L 1 vom 3.1.1994, S. 3.
(3) Verordnung (EU) 2021/697 des Europäischen Parlaments und des Rates vom 29. April 2021 zur Einrichtung des Europäischen Verteidigungsfonds und zur Aufhebung der Verordnung (EU) 2018/1092 (ABl. L 170 vom 12.5.2021, S. 149).
(4) Siehe Dokument ST 10693/21 unter http://register.consilium.europa.eu.
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/71 |
BESCHLUSS (GASP) 2021/1251 DES RATES
vom 29. Juli 2021
zur Änderung des Beschlusses (GASP) 2015/1333 über restriktive Maßnahmen angesichts der Lage in Libyen
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Europäische Union, insbesondere auf Artikel 29,
auf Vorschlag des Hohen Vertreters der Union für Außen- und Sicherheitspolitik,
in Erwägung nachstehender Gründe:
|
(1) |
Am 31. Juli 2015 hat der Rat den Beschluss (GASP) 2015/1333 (1) angenommen. |
|
(2) |
Der Rat hat die in den Anhängen II und IV jenes Beschlusses enthaltene Liste der benannten Personen und Organisationen gemäß Artikel 17 Absatz 2 des Beschlusses (GASP) 2015/1333 überprüft. |
|
(3) |
Der Rat ist zu dem Schluss gelangt, dass die Einträge zu einer Person, die verstorben ist, und einer weiteren Person, für die bis zum 2. April 2021 restriktive Maßnahmen galten, gestrichen werden sollten, und dass die restriktiven Maßnahmen gegen alle anderen Personen und Organisationen in den Listen in den Anhängen II und IV des Beschlusses (GASP) 2015/1333 aufrechterhalten werden sollten. Darüber hinaus sollten die Angaben zur Identität einer Person aktualisiert werden. |
|
(4) |
Der Beschluss (GASP) 2015/1333 sollte daher entsprechend geändert werden — |
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Der Beschluss (GASP) 2015/1333 wird wie folgt geändert:
|
1. |
In Artikel 17 werden die Absätze 3 und 4 gestrichen. |
|
2. |
Die Anhänge II und IV werden gemäß dem Anhang des vorliegenden Beschlusses geändert. |
Artikel 2
Dieser Beschluss tritt am Tag nach seiner Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Geschehen zu Brüssel am 29. Juli 2021.
Im Namen des Rates
Der Präsident
G. DOVŽAN
(1) Beschluss (GASP) 2015/1333 des Rates vom 31. Juli 2015 über restriktive Maßnahmen angesichts der Lage in Libyen und zur Aufhebung des Beschlusses 2011/137/GASP (ABl. L 206 vom 1.8.2015, S. 34).
ANHANG
Der Beschluss (GASP) 2015/1333 wird wie folgt geändert:
|
1. |
Anhang II (Liste der Personen und Organisationen nach Artikel 8 Absatz 2) Abschnitt A (Personen) wird wie folgt geändert:
|
|
2. |
Anhang IV (Liste der Personen und Organisationen nach Artikel 9 Absatz 2) Abschnitt A (Personen) wird wie folgt geändert:
|
|
30.7.2021 |
DE |
Amtsblatt der Europäischen Union |
L 272/73 |
BESCHLUSS (GASP) 2021/1252 DES RATES
vom 29. Juli 2021
zur Änderung des Beschlusses 2010/413/GASP über restriktive Maßnahmen gegen Iran
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Europäische Union, insbesondere auf Artikel 29,
auf Vorschlag des Hohen Vertreters der Union für Außen- und Sicherheitspolitik,
in Erwägung nachstehender Gründe:
|
(1) |
Der Rat hat am 26. Juli 2010 den Beschluss 2010/413/GASP (1) über restriktive Maßnahmen gegen Iran angenommen. |
|
(2) |
Der Rat hat am 18. Juni 2020 den Beschluss (GASP) 2020/849 (2) zur Änderung des Beschlusses 2010/413/GASP angenommen. |
|
(3) |
Nach dem Urteil des Gerichts in der Rechtssache T-580/19 (3) sollte Sayed Shamsuddin Borborudi von der in Anhang II des Beschlusses 2010/413/GASP enthaltenen Liste der Personen und Einrichtungen, gegen die restriktive Maßnahmen verhängt wurden, gestrichen werden. |
|
(4) |
Gemäß Artikel 26 Absatz 3 des Beschlusses 2010/413/GASP hat der Rat die in Anhang II des genannten Beschlusses enthaltene Liste der benannten Personen und Einrichtungen überprüft. |
|
(5) |
Aufgrund dieser Überprüfung sollten die restriktiven Maßnahmen gegen alle Personen und Einrichtungen in der Liste in Anhang II des Beschlusses 2010/413/GASP beibehalten werden, soweit ihre Namen nicht in Anhang VI jenes Beschlusses aufgeführt sind, und 21 Einträge in Anhang II sollten aktualisiert werden. |
|
(6) |
Der Beschluss 2010/413/GASP sollte daher entsprechend geändert werden — |
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Anhang II des Beschlusses 2010/413/GASP wird gemäß dem Anhang des vorliegenden Beschlusses geändert.
Artikel 2
Dieser Beschluss tritt am Tag nach seiner Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Geschehen zu Brüssel am 29. Juli 2021.
Im Namen des Rates
Der Präsident
G. DOVŽAN
(1) Beschluss 2010/413/GASP des Rates vom 26. Juli 2010 über restriktive Maßnahmen gegen Iran und zur Aufhebung des Gemeinsamen Standpunkts 2007/140/GASP (ABl. L 195 vom 27.7.2010, S. 39).
(2) Beschluss (GASP) 2020/849 des Rates vom 18. Juni 2020 zur Änderung des Beschlusses 2010/413/GASP über restriktive Maßnahmen gegen Iran (ABl. L 196 vom 19.6.2020, S. 8).
(3) Urteil des Gerichts vom 9. Juni 2021, Sayed Shamsuddin Borborudi gegen Rat der Europäischen Union, T-580/19, ECLI:EU:T:2021:330.
ANHANG
Anhang II des Beschlusses 2010/413/GASP wird wie folgt geändert:
|
1. |
Unter der Überschrift „I. Personen und Einrichtungen, die an nuklearen Tätigkeiten oder Tätigkeiten im Zusammenhang mit ballistischen Raketen beteiligt sind, und Personen und Einrichtungen, die die Regierung Irans unterstützen“ wird der folgende Eintrag gestrichen: „25. Sayed Shamsuddin Borborudi“. |
|
2. |
Unter der Überschrift „I. Personen und Einrichtungen, die an nuklearen Tätigkeiten oder Tätigkeiten im Zusammenhang mit ballistischen Raketen beteiligt sind, und Personen und Einrichtungen, die die Regierung Irans unterstützen“ ersetzen die folgenden Einträge die entsprechenden Einträge in der Liste unter der Unterüberschrift „A. Personen“:
|
|
3. |
Unter der Überschrift „I. Personen und Einrichtungen, die an nuklearen Tätigkeiten oder Tätigkeiten im Zusammenhang mit ballistischen Raketen beteiligt sind, und Personen und Einrichtungen, die die Regierung Irans unterstützen“ ersetzen die folgenden Einträge die entsprechenden Einträge in der Liste unter der Unterüberschrift „B. Einrichtungen“:
|
|
4. |
Unter der Überschrift „II. Korps der Islamischen Revolutionsgarden (IRGC)“ ersetzen die folgenden Einträge die entsprechenden Einträge in der Liste unter der Unterüberschrift „A. Personen“:
|
|
5. |
Unter der Überschrift „II. Korps der Islamischen Revolutionsgarden (IRGC)“ ersetzt der folgende Eintrag den entsprechenden Eintrag in der Liste unter der Unterüberschrift „B. Einrichtungen“:
|