ISSN 1977-0642
Amtsblatt
der Europäischen Union
L 180

Ausgabe in deutscher Sprache
Rechtsvorschriften
64. Jahrgang
21. Mai 2021
|
ISSN 1977-0642 |
||
|
Amtsblatt der Europäischen Union |
L 180 |
|
|
|
||
|
Ausgabe in deutscher Sprache |
Rechtsvorschriften |
64. Jahrgang |
|
|
|
|
|
(1) Text von Bedeutung für den EWR. |
|
DE |
Bei Rechtsakten, deren Titel in magerer Schrift gedruckt sind, handelt es sich um Rechtsakte der laufenden Verwaltung im Bereich der Agrarpolitik, die normalerweise nur eine begrenzte Geltungsdauer haben. Rechtsakte, deren Titel in fetter Schrift gedruckt sind und denen ein Sternchen vorangestellt ist, sind sonstige Rechtsakte. |
II Rechtsakte ohne Gesetzescharakter
VERORDNUNGEN
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/1 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/804 DES RATES
vom 20. Mai 2021
zur Durchführung des Artikels 15 Absatz 3 der Verordnung (EU) Nr. 747/2014 über restriktive Maßnahmen angesichts der Lage in Sudan
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) Nr. 747/2014 des Rates vom 10. Juli 2014 über restriktive Maßnahmen angesichts der Lage in Sudan und zur Aufhebung der Verordnungen (EG) Nr. 131/2004 und (EG) Nr. 1184/2005 (1), insbesondere auf Artikel 15 Absatz 3,
auf Vorschlag des Hohen Vertreters der Union für Außen- und Sicherheitspolitik,
in Erwägung nachstehender Gründe:
|
(1) |
Am 10. Juli 2014 hat der Rat die Verordnung (EU) Nr. 747/2014 angenommen. |
|
(2) |
Am 5. März 2021 hat der Ausschuss des Sicherheitsrats der Vereinten Nationen (VN), der gemäß der Resolution 1591 (2005) des Sicherheitsrats der VN eingesetzt wurde, die Streichung einer Person von der Liste der Personen und Einrichtungen, die restriktiven Maßnahmen unterliegen, genehmigt. |
|
(3) |
Anhang I der Verordnung (EU) Nr. 747/2014 sollte daher entsprechend geändert werden — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang I der Verordnung (EU) Nr. 747/2014 wird gemäß dem Anhang dieser Verordnung geändert.
Artikel 2
Diese Verordnung tritt am Tag ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Geschehen zu Brüssel am 20. Mai 2021.
Im Namen des Rates
Der Präsident
A. SANTOS SILVA
ANHANG
In der Liste in Anhang I der Verordnung (EU) Nr. 747/2014 wird der Eintrag zu folgender Person gestrichen:
|
3. |
SHAREIF, Adam. |
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/3 |
DELEGIERTE VERORDNUNG (EU) 2021/805 DER KOMMISSION
vom 8. März 2021
zur Änderung von Anhang II der Verordnung (EU) 2019/6 des Europäischen Parlaments und des Rates
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) 2019/6 des Europäischen Parlaments und des Rates vom 11. Dezember 2018 über Tierarzneimittel und zur Aufhebung der Richtlinie 2001/82/EG (1), insbesondere auf Artikel 146 Absatz 2,
in Erwägung nachstehender Gründe:
|
(1) |
Die Anforderungen in Anhang II der Verordnung (EU) 2019/6, in dem die Dossieranforderungen von Anhang I der Richtlinie 2001/82/EG des Europäischen Parlaments und des Rates (2) übernommen wurden, sollten grundlegend aktualisiert werden, da in der genannten Verordnung die Dossieranforderungen zum Zeitpunkt der Aufhebung dieser Richtlinie nicht aktualisiert wurden. Die in Anhang I der Richtlinie 2001/82/EG aufgeführten Dossieranforderungen wurden zuletzt 2009 aktualisiert. Daher sollte Anhang II geändert werden, um dem wissenschaftlichen Fortschritt und den Entwicklungen seit 2009 Rechnung zu tragen, einschließlich der Leitlinien der Internationalen Konferenz zur Harmonisierung der technischen Anforderungen an die Zulassung von Tierarzneimitteln (VICH) und der Weltgesundheitsorganisation (WHO) sowie der Standards der Organisation für wirtschaftliche Zusammenarbeit und Entwicklung (OECD). |
|
(2) |
Außerdem sollten Anforderungen an biologische Tierarzneimittel und Tierarzneimittel für neuartige Therapien festgelegt werden, die mit der Verordnung (EU) 2019/6 als neue Kategorien von Tierarzneimitteln eingeführt wurden. Für diese Produkte sollten spezifische technische Anforderungen festgelegt werden, die bei der Beantragung einer Zulassung vorzulegen sind. |
|
(3) |
In Anerkennung der Tatsache, dass antimikrobielle Resistenzen gegen Arzneimittel in der Union und weltweit ein wachsendes Gesundheitsproblem darstellen, wurden mit der Verordnung (EU) 2019/6 spezifische rechtliche Bestimmungen eingeführt, die darauf abzielen, das Risiko der Entwicklung antimikrobieller Resistenzen gegen Arzneimittel zu begrenzen. Daher ist es angebracht, spezifische technische Anforderungen für antimikrobielle Tierarzneimittel einzuführen. |
|
(4) |
Gemäß Artikel 153 Absatz 3 der Verordnung (EU) 2019/6 sollte diese Verordnung ab dem 28. Januar 2022 gelten. |
|
(5) |
Die Verordnung (EU) 2019/6 sollte daher entsprechend geändert werden — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang II der Verordnung (EU) 2019/6 erhält die Fassung des Anhangs der vorliegenden Verordnung.
Artikel 2
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Sie gilt ab dem 28. Januar 2022.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 8. März 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 4 vom 7.1.2019, S. 43.
(2) Richtlinie 2001/82/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Tierarzneimittel (ABl. L 311 vom 28.11.2001, S. 1).
ANHANG
„ANHANG II
ANFORDERUNGEN GEMÄß ARTIKEL 8 ABSATZ 1 BUCHSTABE B
Inhalt
|
ABSCHNITT I |
ALLGEMEINE GRUNDSÄTZE UND ANFORDERUNGEN | 11 |
|
I.1. |
Allgemeine Grundsätze | 11 |
|
I.2. |
Anforderungen an die Zusammensetzung des Dossiers | 11 |
|
I.2.1. |
Teil 1: Zusammenfassung der Unterlagen | 11 |
|
I.2.2. |
Teil 2: Qualitätsbezogene Unterlagen (physikalisch-chemische, biologische oder mikrobiologische Informationen) | 12 |
|
I.2.3. |
Teil 3: Unterlagen zur Unbedenklichkeit (Unbedenklichkeits- und Rückstandsversuche) | 13 |
|
I.2.4. |
Teil 4: Unterlagen zur Wirksamkeit (vorklinische Studien und klinische Prüfungen) | 13 |
|
I.2.5. |
Detaillierte Anforderungen an unterschiedliche Arten von Tierarzneimitteln oder Dossiers von Zulassungsanträgen | 14 |
|
ABSCHNITT II |
ANFORDERUNGEN FÜR NICHT BIOLOGISCHE TIERARZNEIMITTEL | 14 |
|
II.1. |
TEIL 1: Zusammenfassung der Unterlagen | 14 |
|
II.2. |
TEIL 2: Qualitätsbezogene Unterlagen (physikalisch-chemische, biologische oder mikrobiologische Informationen) | 14 |
|
II.2A. |
Produktbeschreibung | 14 |
|
II.2A1. |
Qualitative und quantitative Zusammensetzung | 14 |
|
II.2A2. |
Produktentwicklung | 16 |
|
II.2B. |
Angaben über die Herstellungsweise | 16 |
|
II.2C. |
Herstellung und Kontrolle der Ausgangsstoffe | 16 |
|
II.2C1. |
Wirkstoff(e) | 17 |
|
II.2C1.1. |
In Arzneibüchern aufgeführte Wirkstoffe | 18 |
|
II.2C1.2. |
In Arzneibüchern nicht aufgeführte Wirkstoffe | 18 |
|
II.2C1.3. |
Physikalisch-chemische Eigenschaften, die die Bioverfügbarkeit beeinflussen können | 18 |
|
II.2C2. |
Arzneiträgerstoffe | 19 |
|
II.2C3. |
Verpackung (Behältnisse und Verschlusssysteme) | 19 |
|
II.2C3.1. |
Wirkstoff | 19 |
|
II.2C3.2. |
Fertigprodukt | 19 |
|
II.2C4. |
Stoffe biologischer Herkunft | 20 |
|
II.2D. |
Kontrollen, die während des Herstellungsprozesses an isolierten Zwischenprodukten durchgeführt werden | 20 |
|
II.2E. |
Kontrollen am Fertigprodukt | 20 |
|
II.2E1. |
Allgemeine Merkmale des Fertigprodukts | 21 |
|
II.2E2. |
Identitätsnachweis und Gehaltsbestimmung der Wirkstoffe | 21 |
|
II.2E3. |
Identitätsnachweis und Gehaltsbestimmung der Bestandteile des Arzneiträgerstoffs | 21 |
|
II.2E4. |
Mikrobiologische Kontrollen | 21 |
|
II.2E5. |
Gleichbleibende Qualität der Chargen | 21 |
|
II.2E6. |
Sonstige Kontrollen | 22 |
|
II.2F. |
Haltbarkeitsversuche | 22 |
|
II.2F1. |
Wirkstoff(e) | 22 |
|
II.2F2. |
Fertigprodukt | 22 |
|
II.2G. |
Sonstige Informationen | 23 |
|
II.3 |
Teil 3: Unterlagen zur Unbedenklichkeit (Unbedenklichkeits- und Rückstandsversuche) | 23 |
|
II.3A. |
Unbedenklichkeitsversuche | 23 |
|
II.3A1. |
Genaue Identifizierung des Arzneimittels und seiner Wirkstoffe | 24 |
|
II.3A2. |
Pharmakologie | 24 |
|
II.3A2.1. |
Pharmakodynamik | 24 |
|
II.3A2.2. |
Pharmakokinetik | 25 |
|
II.3A3. |
Toxikologie | 25 |
|
II.3A4. |
Sonstige Anforderungen | 26 |
|
II.3A.4.1. |
Spezielle Untersuchungen | 26 |
|
II.3A.4.2. |
Beobachtungen beim Menschen | 26 |
|
II.3A.4.3. |
Resistenzentwicklung und damit verbundenes Risiko für den Menschen | 27 |
|
II.3A5. |
Anwendersicherheit | 27 |
|
II.3A6. |
Umweltverträglichkeitsprüfung | 27 |
|
II.3B. |
Rückstandsversuche | 28 |
|
II.3B1. |
Identifizierung des Arzneimittels | 28 |
|
II.3B2. |
Abbau von Rückständen (Stoffwechsel und Rückstandskinetik) | 28 |
|
II.3B3. |
Methoden zur Rückstandsanalyse | 29 |
|
II.4. |
TEIL 4: Unterlagen zur Wirksamkeit (vorklinische Studien und klinische Prüfungen) | 29 |
|
II.4A. |
Vorklinische Studien | 29 |
|
II.4A1. |
Pharmakologie | 29 |
|
II.4A.1.1. |
Pharmacodynamics | 29 |
|
II.4A.1.2. |
Pharmakokinetik | 29 |
|
II.4A2. |
Resistenzentwicklung und damit verbundenes Risiko bei Tieren | 30 |
|
II.4A3. |
Dosisermittlung und -bestätigung | 30 |
|
II.4A4. |
Verträglichkeit bei den Zieltierarten | 30 |
|
II.4B. |
Klinische Prüfung(en) | 31 |
|
II.4B1. |
Allgemeine Grundsätze | 31 |
|
II.4B2. |
Dokumentation | 31 |
|
II.4B2.1. |
Ergebnisse der vorklinischen Studien | 31 |
|
II.4B2.2. |
Ergebnisse der klinischen Prüfungen | 32 |
|
ABSCHNITT III |
ANFORDERUNGEN FÜR BIOLOGISCHE TIERARZNEIMITTEL | 32 |
|
ABSCHNITT IIIa |
ANFORDERUNGEN FÜR NICHT IMMUNOLOGISCHE BIOLOGISCHE TIERARZNEIMITTEL | 33 |
|
IIIa.1. |
TEIL 1: Zusammenfassung der Unterlagen | 33 |
|
IIIa.2. |
TEIL 2: Qualitätsbezogene Unterlagen (physikalisch-chemische, biologische oder mikrobiologische Informationen) | 33 |
|
IIIa.2A. |
Produktbeschreibung | 33 |
|
IIIa.2A1. |
Qualitative und quantitative Zusammensetzung | 33 |
|
IIIa.2A2. |
Produktentwicklung | 34 |
|
IIIa.2A3. |
Charakterisierung | 34 |
|
IIIa.2A3.1. |
Erläuterung der Struktur und anderer Merkmale | 34 |
|
IIIa.2A3.2. |
Verunreinigungen | 35 |
|
IIIa.2B. |
Angaben über die Herstellungsweise | 35 |
|
IIIa.2C. |
Herstellung und Kontrolle der Ausgangsstoffe | 35 |
|
IIIa.2C1. |
In Arzneibüchern aufgeführte Ausgangsstoffe | 36 |
|
IIIa.2C2. |
Nicht in einem Arzneibuch aufgeführte Ausgangsstoffe | 36 |
|
IIIa.2C2.1. |
Ausgangsstoffe biologischer Herkunft | 36 |
|
IIIa.2C2.2. |
Ausgangsstoffe nicht biologischer Herkunft | 37 |
|
IIIa.2D. |
Kontrollen während des Herstellungsprozesses | 37 |
|
IIIa.2E. |
Kontrollen am Fertigprodukt | 38 |
|
IIIa.2E1 |
Spezifikation des Fertigprodukts | 38 |
|
IIIa.2E2 |
Methodenbeschreibungen und Validierung von Freigabeprüfungen | 38 |
|
IIIa.2E3. |
Referenzstandards oder -materialien | 39 |
|
IIIa.2F. |
Gleichbleibende Qualität der Chargen | 39 |
|
IIIa.2F1. |
Wirkstoff | 39 |
|
IIIa.2F2. |
Fertigprodukt | 39 |
|
IIIa.2G. |
Haltbarkeitsversuche | 39 |
|
IIIa.2H. |
Sonstige Informationen | 40 |
|
IIIa.3. |
TEIL 3: Unterlagen zur Unbedenklichkeit (Unbedenklichkeits- und Rückstandsversuche) | 40 |
|
IIIa.3A. |
Unbedenklichkeitsversuche | 41 |
|
IIIa.3A1. |
Genaue Identifizierung des Arzneimittels und seiner Wirkstoffe: | 41 |
|
IIIa.3A2. |
Pharmakologie | 41 |
|
IIIa.3A2.1. |
Pharmakodynamik | 42 |
|
IIIa.3A2.2. |
Pharmakokinetik | 42 |
|
IIIa.3A3. |
Toxikologie | 42 |
|
IIIa.3A3.1. |
Toxizität bei einmaliger Verabreichung | 42 |
|
IIIa.3A3.2. |
Toxizität bei wiederholter Verabreichung | 42 |
|
IIIa.3A3.3. |
Verträglichkeit bei der Zieltierart | 43 |
|
IIIa.3A3.4. |
Reproduktions- und Entwicklungstoxizität | 43 |
|
IIIa.3A3.5. |
Genotoxizität | 43 |
|
IIIa.3A3.6. |
Kanzerogenität | 43 |
|
IIIa.3A3.7. |
Ausnahmen | 43 |
|
IIIa.3A4. |
Sonstige Anforderungen | 44 |
|
IIIa.3A4.1. |
Spezielle Untersuchungen | 44 |
|
IIIa.3A4.2. |
Beobachtungen beim Menschen | 44 |
|
IIIa.3A4.3. |
Resistenzentwicklung und damit verbundenes Risiko beim Menschen | 44 |
|
IIIa.3A5. |
Anwendersicherheit | 45 |
|
IIIa.3A6. |
Umweltverträglichkeitsprüfung | 45 |
|
IIIa.3A6.1. |
Umweltverträglichkeitsprüfung für Tierarzneimittel, die genetisch veränderte Organismen weder enthalten noch aus solchen bestehen | 45 |
|
IIIa.3A6.2. |
Umweltverträglichkeitsprüfung für Tierarzneimittel, die genetisch veränderte Organismen enthalten oder aus solchen bestehen | 45 |
|
IIIa.3B. |
Rückstandsversuche | 46 |
|
IIIa.3B1. |
Identifizierung des Arzneimittels | 46 |
|
IIIa.3B2. |
Abbau von Rückständen | 46 |
|
IIIa.3B3. |
Methoden zur Rückstandsanalyse | 46 |
|
IIIa.4. |
TEIL 4: Unterlagen zur Wirksamkeit (vorklinische Studien und klinische Prüfungen) | 47 |
|
IIIa.4A. |
Vorklinische Studien | 47 |
|
IIIa.4A1. |
Pharmakologie | 47 |
|
IIIa.4A1.1. |
Pharmakodynamik | 47 |
|
IIIa.4A1.2. |
Pharmakokinetik | 47 |
|
IIIa.4A2. |
Resistenzentwicklung und damit verbundenes Risiko bei Tieren | 48 |
|
IIIa.4A3. |
Dosisermittlung und -bestätigung | 48 |
|
IIIa.4A4. |
Verträglichkeit bei den Zieltierarten | 48 |
|
IIIa.4B. |
Klinische Prüfungen | 48 |
|
IIIa.4B1. |
Allgemeine Grundsätze | 48 |
|
IIIa.4B2. |
Allgemeine Grundsätze | 49 |
|
IIIa.4B2.1. |
Ergebnisse der vorklinischen Studien | 49 |
|
IIIa.4B2.2. |
Ergebnisse der klinischen Prüfungen | 49 |
|
ABSCHNITT IIIb |
ANFORDERUNGEN FÜR IMMUNOLOGISCHE TIERARZNEIMITTEL | 50 |
|
IIIb.1. |
TEIL 1: Zusammenfassung der Unterlagen | 50 |
|
IIIb.2. |
TEIL 2: Qualitätsbezogene Unterlagen (physikalisch-chemische, biologische und mikrobiologische Informationen) | 50 |
|
IIIb.2.A. |
Produktbeschreibung | 50 |
|
IIIb.2A1. |
Qualitative und quantitative Zusammensetzung | 50 |
|
IIIb.2A2. |
Produktentwicklung | 51 |
|
IIIb.2B. |
Angaben über die Herstellungsweise | 52 |
|
IIIb.2C. |
Herstellung und Kontrolle der Ausgangsstoffe | 52 |
|
IIIb.2C1. |
In Arzneibüchern aufgeführte Ausgangsstoffe | 53 |
|
IIIb.2C2. |
Nicht in einem Arzneibuch aufgeführte Ausgangsstoffe | 53 |
|
IIIb.2C2.1. |
Ausgangsstoffe biologischer Herkunft | 53 |
|
IIIb.2C2.2. |
Ausgangsstoffe nicht biologischer Herkunft | 54 |
|
IIIb.2D. |
Kontrollen während des Herstellungsprozesses | 54 |
|
IIIb.2E. |
Kontrollen am Fertigprodukt | 55 |
|
IIIb.2F. |
Gleichbleibende Qualität der Chargen | 56 |
|
IIIb.2G. |
Haltbarkeitsversuche | 56 |
|
IIIb.2H. |
Sonstige Informationen | 57 |
|
IIIb.3. |
TEIL 3: Unterlagen zur Unbedenklichkeit (Unbedenklichkeits- und Rückstandsversuche) | 57 |
|
IIIb.3A. |
Allgemeine Vorschriften | 57 |
|
IIIb.3B. |
Vorklinische Studien | 58 |
|
IIIb.3C. |
Klinische Prüfungen | 60 |
|
IIIb.3D. |
Umweltverträglichkeitsprüfung | 60 |
|
IIIb.3E. |
Beurteilungspflicht für Tierarzneimittel, die genetisch modifizierte Organismen enthalten oder aus ihnen bestehen | 61 |
|
IIIb.3F. |
Rückstandsversuche, die in die vorklinischen Studien einbezogen werden müssen | 61 |
|
IIIb.4. |
TEIL 4: Unterlagen zur Wirksamkeit (vorklinische Studien und klinische Prüfungen) | 61 |
|
IIIb.4A. |
Allgemeine Vorschriften | 61 |
|
IIIb.4B. |
Vorklinische Studien | 62 |
|
IIIb.4C. |
Klinische Prüfungen | 63 |
|
ABSCHNITT IV |
ANFORDERUNGEN FÜR BESTIMMTE ZULASSUNGSANTRÄGE | 64 |
|
IV.1. |
Anträge für Tierarzneimittel-Generika | 64 |
|
IV.2. |
Anträge für hybride Tierarzneimittel | 65 |
|
IV.3. |
Anträge für Tierarzneimittel aus kombinierten Wirkstoffen | 66 |
|
IV.4. |
Anträge aufgrund einer in Kenntnis der Sachlage erteilten Einwilligung | 66 |
|
IV.5. |
Anträge auf bibliografischer Grundlage | 66 |
|
IV.6. |
Anträge für begrenzte Märkte | 68 |
|
IV.7. |
Anträge unter außergewöhnlichen Umständen | 68 |
|
ABSCHNITT V |
ANFORDERUNGEN FÜR ZULASSUNGSANTRÄGE FÜR BESONDERE TIERARZNEIMITTEL | 68 |
|
V.1. |
Tierarzneimittel für neuartige Therapien | 68 |
|
V.1.1 |
Allgemeine Vorschriften | 68 |
|
V.1.2. |
Qualitätsanforderungen | 69 |
|
V.1.3. |
Sicherheitsanforderungen | 70 |
|
V.1.4. |
Wirksamkeitsanforderungen | 70 |
|
V.1.5. |
Spezifische Datenanforderungen für bestimmte Arten von Arzneimitteln für neuartige Therapien | 70 |
|
V.1.5.1. |
Grundlagen | 70 |
|
V.1.5.2. |
Tierarzneimittel für die Gentherapie | 70 |
|
V.1.5.3. |
Für die regenerative Medizin, die Gewebezüchtung oder die Zelltherapie entwickelte Tierarzneimittel | 71 |
|
V.1.5.4. |
Speziell für die Phagentherapie entwickeltes Tierarzneimittel | 72 |
|
V.1.5.5. |
Tierarzneimittel aus der Nanotechnologie | 72 |
|
V.1.5.6. |
Antisense-RNA-Therapie und RNA-Interferenz-Therapieprodukte | 73 |
|
V.2. |
Impfantigen-Stammdokumentation | 74 |
|
V.3. |
Multi-Strain-Dossier | 75 |
|
V.4. |
Impfstoff-Plattformtechnologie | 75 |
|
V.5. |
Zugelassene homöopathische Tierarzneimittel | 76 |
ABSCHNITT I
ALLGEMEINE GRUNDSÄTZE UND ANFORDERUNGEN
I.1. Allgemeine Grundsätze
|
I.1.1. |
Die Unterlagen, die einem Zulassungsantrag gemäß Artikel 8 und Artikel 18 bis 25 beiliegen müssen, sind entsprechend den Anforderungen dieses Anhangs vorzulegen und müssen sich an den von der Kommission veröffentlichen Leitlinien und den von der Agentur veröffentlichten Anforderungen an das elektronische Format orientieren. |
|
I.1.2. |
Bei der Zusammenstellung des Dossiers für einen Antrag auf Zulassung müssen die Antragsteller auch den aktuellsten Wissensstand der Veterinärmedizin und die von der Agentur veröffentlichten wissenschaftlichen Leitlinien für die Qualität, Unbedenklichkeit und Wirksamkeit von Tierarzneimitteln berücksichtigen. |
|
I.1.3. |
Für Tierarzneimittel sind für die entsprechenden Teile des Dossiers alle einschlägigen Monografien des Europäischen Arzneibuchs, einschließlich der allgemeinen Monografien und Kapitel, maßgeblich. |
|
I.1.4. |
Die Herstellungsverfahren für den/die Wirkstoff(e) und das Fertigprodukt müssen der guten Herstellungspraxis entsprechen. |
|
I.1.5. |
Dem Antrag sind alle für die Bewertung des betreffenden Tierarzneimittels zweckdienlichen Angaben beizufügen, ob diese nun günstig oder ungünstig für das Arzneimittel sind. Insbesondere sind alle zweckdienlichen Einzelheiten über etwaige unvollständige oder abgebrochene Studien bzw. Prüfungen im Zusammenhang mit dem Tierarzneimittel vorzulegen. |
|
I.1.6. |
Pharmakologische, toxikologische und Rückstandsuntersuchungen sowie vorklinische Studien sind nach den Bestimmungen der Guten Laborpraxis (GLP) durchzuführen, die in den Richtlinien 2004/10/EG (1) und 2004/9/EG (2) des Europäischen Parlaments und des Rates festgelegt sind. |
|
I.1.7. |
Alle Tierversuche müssen unter Berücksichtigung der in der Richtlinie 2010/63/EU festgelegten Grundsätze durchgeführt werden, ungeachtet des Ortes, an dem die Versuche durchgeführt werden. |
|
I.1.8. |
Das Dossier muss als separates Dokument die Umweltverträglichkeitsprüfung in Verbindung mit der Freisetzung von Tierarzneimitteln umfassen, die genetisch veränderte Organismen (GVO) im Sinne von Artikel 2 der Richtlinie 2001/18/EG enthalten oder aus ihnen bestehen. Diese Angaben sind entsprechend der Richtlinie 2001/18/EG zu machen und es sind dabei sämtliche von der Kommission veröffentlichte Leitfäden zu berücksichtigen. |
|
I.1.9. |
Der Antragsteller muss in Teil 1 des Dossiers eines Zulassungsantrags bestätigen, dass alle vorgelegten Daten, die für die Qualität, Unbedenklichkeit und Wirksamkeit des Tierarzneimittels von Bedeutung sind, einschließlich öffentlich zugänglicher Daten, nicht dem Schutz technischer Unterlagen unterliegen. |
I.2. Anforderungen an die Zusammensetzung des Dossiers
Jedes Dossier für einen Zulassungsantrag eines Tierarzneimittels muss aus den folgenden Teilen bestehen:
I.2.1. Teil 1: Zusammenfassung der Unterlagen
Teil 1 enthält folgende in Anhang I aufgeführten administrativen Angaben:
|
a) |
Teil 1A: Punkte 1 bis 4 und 6.1 bis 6.4; |
|
b) |
Teil 1B: Punkt 5; |
|
c) |
Teil 1C: Punkt 6.5. |
In Bezug auf Teil 1B Punkt 5.1 in Verbindung mit Artikel 35 Absatz 1 Ziffer i) muss ein Antrag, in dem die Einstufung eines Tierarzneimittels als ‚nicht verschreibungspflichtig‘ vorgeschlagen wird, eine kritische Prüfung der Merkmale des Tierarzneimittels enthalten, um die Eignung einer solchen Einstufung unter Berücksichtigung der Sicherheit von Ziel- und Nichtzieltierarten, der öffentlichen Gesundheit und des Umweltschutzes gemäß den in Artikel 34 Absatz 3 Buchstaben a bis g genannten Kriterien zu begründen.
Jeder kritische Expertenbericht ist auf der Grundlage des wissenschaftlichen Kenntnisstands zum Zeitpunkt der Antragstellung auszuarbeiten. Er muss eine Bewertung der verschiedenen Prüfungen und Versuche enthalten, die Bestandteile des Antragsdossiers sind, und alle für die Beurteilung von Qualität, Sicherheit und Wirksamkeit des Tierarzneimittels relevanten Aspekte behandeln. Die Ergebnisse der vorgelegten Prüfungen und Versuche sind detailliert aufzuführen und es sind genaue Quellenangaben zu machen. Kopien der zitierten Quellenangaben sind beizufügen.
Die kritischen Expertenberichte sind von den Autoren der Berichte zu unterzeichnen und zu datieren; außerdem sind Angaben zum Ausbildungsprofil und zur Berufserfahrung des Verfassers zu machen. Die beruflichen Beziehungen des Verfassers zum Antragsteller sind darzulegen.
Die kritischen Expertenberichte und die Anlagen dazu müssen präzise und eindeutige Querverweise zu den Angaben in den technischen Unterlagen aufweisen.
Wenn Teil 2 im CTD-Format (Common Technical Document) vorgelegt wird, ist für den kritischen Expertenbericht die Zusammenfassung der pharmazeutischen Qualität zu verwenden.
Für die Teile 3 und 4 muss der kritische Expertenbericht auch eine tabellarische Zusammenfassung aller eingereichten technischen Unterlagen und sachdienlichen Daten enthalten.
I.2.2. Teil 2: Qualitätsbezogene Unterlagen (physikalisch-chemische, biologische oder mikrobiologische Informationen)
|
(1) |
Die pharmazeutischen (physikalisch-chemischen, biologischen oder mikrobiologischen) qualitätsbezogenen Daten müssen sowohl für die Wirkstoffe als auch für das fertige Tierarzneimittel alle einschlägigen Angaben zu Herstellungsprozess, kennzeichnenden Merkmalen und Eigenschaften, Verfahren für und Anforderungen an die Qualitätskontrolle sowie Angaben zur Haltbarkeit enthalten und eine Beschreibung von Zusammensetzung, Entwicklung und Darreichung des Fertigarzneimittels umfassen. |
|
(2) |
Es gelten alle Monografien, einschließlich der spezifischen und allgemeinen Monografien und allgemeinen Kapitel des Europäischen Arzneibuchs. Für immunologische Tierarzneimittel gelten alle Monografien, einschließlich der spezifischen und allgemeinen Monografien und allgemeinen Kapitel des Europäischen Arzneibuchs; anderenfalls ist dies zu begründen. Gibt es keine entsprechende Monografie des Europäischen Arzneibuchs, kann die Monografie eines Arzneibuchs eines Mitgliedstaats verwendet werden. Sofern ein Stoff weder im Europäischen Arzneibuch noch im Arzneibuch eines Mitgliedstaats beschrieben ist, kann die Übereinstimmung mit der Monografie des Arzneibuchs eines Drittlandes akzeptiert werden, wenn sich deren Eignung nachweisen lässt; in solchen Fällen hat der Antragsteller eine Kopie der Monografie, falls erforderlich zusammen mit einer Übersetzung, einzureichen. Es ist der Nachweis zu erbringen, dass sich die Monografie zur angemessenen Qualitätskontrolle des Stoffes eignet. |
|
(3) |
Werden andere als die in einem Arzneibuch angegebenen Prüfungen durchgeführt, so ist dies zu begründen und der Nachweis zu erbringen, dass die Ausgangsstoffe, wenn sie in Übereinstimmung mit dem Arzneibuch getestet worden wären, den Qualitätsanforderungen der einschlägigen Monografie des Arzneibuchs entsprechen würden. |
|
(4) |
Bei allen Prüfverfahren für die Analyse und Kontrolle der Qualität sind bestehende Leitlinien und Anforderungen zu berücksichtigen. Die Ergebnisse der Validierungsstudien sind vorzulegen. Alle Prüfverfahren sind so detailliert zu beschreiben, dass sie bei Kontrollversuchen, die auf Verlangen der zuständigen Behörde durchgeführt werden, reproduzierbar sind, um von der zuständigen Behörde ordnungsgemäß bewertet werden zu können. Von allen gegebenenfalls verwendeten besonderen Geräten und Anlagen sind angemessene Beschreibungen und, sofern sachdienlich, eine grafische Darstellung beizufügen. Die Formeln der Laborreagenzien sind gegebenenfalls durch die Herstellungsmethode zu ergänzen. Bei Prüfverfahren, die im Europäischen Arzneibuch oder im Arzneibuch eines Mitgliedstaats enthalten sind, kann diese Beschreibung durch einen detaillierten Verweis auf das betreffende Arzneibuch ersetzt werden. |
|
(5) |
Sofern verfügbar, sind die im Europäischen Arzneibuch verzeichneten chemischen und biologischen Referenzmaterialien zu verwenden. Falls andere Referenzzubereitungen und -standards verwendet werden, sind diese anzugeben und ausführlich zu beschreiben. |
|
(6) |
Die pharmazeutischen (physikalisch-chemischen, biologischen oder mikrobiologischen) Daten zur Qualität des Wirkstoffs und/oder des Fertigprodukts können im CTD-Format in das Dossier aufgenommen werden. |
|
(7) |
Für biologische Tierarzneimittel, einschließlich immunologischen Tierarzneimitteln, muss das Dossier auch Informationen über die Lösungsmittel enthalten, die zur Zubereitung des Fertigprodukts erforderlich sind. Ein biologisches Tierarzneimittel gilt als ein einziges Arzneimittel, selbst wenn mehrere Lösungsmittel benötigt werden, sodass sich verschiedene Zubereitungen des Fertigarzneimittels herstellen lassen, die für verschiedene Verabreichungswege oder -arten gedacht sind. Lösungsmittel, die zusammen mit biologischen Tierarzneimitteln geliefert werden, können zusammen mit den Wirkstoffampullen oder getrennt verpackt sein. |
|
(8) |
Gemäß der Richtlinie 2010/63/EU und dem Europäischen Übereinkommen zum Schutz der für Versuche und andere wissenschaftliche Zwecke verwendeten Wirbeltiere sind die Versuche so durchzuführen, dass möglichst wenige Tiere verwendet und möglichst wenig Schmerzen, Leiden, Ängste oder dauerhafte Schäden verursacht werden. Falls verfügbar, ist ein alternativer In-vitro-Test zu verwenden, wenn dies zum Ersatz oder zur Verringerung des Einsatzes von Tieren oder zur Verringerung des Leidens führt. |
I.2.3. Teil 3: Unterlagen zur Unbedenklichkeit (Unbedenklichkeits- und Rückstandsversuche)
|
(1) |
Das Dossier zu den Unbedenklichkeitsversuchen muss Folgendes beinhalten:
|
|
(2) |
Das Dossier muss Folgendes enthalten:
|
I.2.4. Teil 4: Unterlagen zur Wirksamkeit (vorklinische Studien und klinische Prüfungen)
|
(1) |
Das Dossier über die Wirksamkeit muss alle vorklinischen und klinischen Unterlagen enthalten, unabhängig davon, ob sie günstig oder ungünstig für die Tierarzneimittel ausgefallen sind, damit eine objektive Gesamtbeurteilung des Nutzen-Risiko-Verhältnisses des Arzneimittels ermöglicht wird. |
|
(2) |
Das Dossier zu den Wirksamkeitsstudien muss Folgendes beinhalten:
|
|
(3) |
Das Dossier muss Folgendes enthalten:
|
|
(4) |
Zweck der in diesem Teil beschriebenen Versuche ist der Nachweis der Wirksamkeit des Tierarzneimittels. Alle vom Antragsteller in Bezug auf die Eigenschaften, die Wirkungen und die Anwendung des Arzneimittels gemachten Behauptungen sind durch die im Zulassungsantrag enthaltenen Ergebnisse aus spezifischen Versuchen umfassend zu erhärten. |
|
(5) |
Alle Versuche zur Wirksamkeit sind im Einklang mit einem vollständig überprüften detaillierten und vor Versuchsbeginn schriftlich niedergelegten Protokoll durchzuführen. Das Wohlergehen der Versuchstiere muss der tierärztlichen Aufsicht unterliegen und ist bei Ausarbeitung jedes Versuchsprotokolls sowie während des gesamten Versuchs in vollem Umfang zu berücksichtigen. |
|
(6) |
Klinische Prüfungen (Feldversuche) sind gemäß den festgelegten Grundsätzen der guten klinischen Praxis durchzuführen, sofern nicht anders begründet. |
|
(7) |
Vor Beginn jedes Feldversuchs ist die in voller Kenntnis der Sachlage erteilte Zustimmung des Eigentümers der Versuchstiere einzuholen und zu dokumentieren. Insbesondere muss der Eigentümer der Tiere schriftlich über die Folgen einer Teilnahme an den Versuchen im Hinblick auf die anschließende Beseitigung behandelter Tiere oder die Herstellung von Lebensmitteln aus behandelten Tieren informiert werden. |
I.2.5. Detaillierte Anforderungen an unterschiedliche Arten von Tierarzneimitteln oder Dossiers von Zulassungsanträgen
|
(1) |
Detaillierte Anforderungen an unterschiedliche Arten von Tierarzneimitteln oder spezifische Arten von Dossiers von Zulassungsanträgen sind in den folgenden Abschnitten dieses Anhangs enthalten:
|
ABSCHNITT II
ANFORDERUNGEN FÜR NICHT BIOLOGISCHE TIERARZNEIMITTEL
Die nachstehenden detaillierten Anforderungen gelten für Tierarzneimittel, bei denen es sich nicht um biologische Tierarzneimittel handelt, sofern in Abschnitt IV nicht anders vorgesehen.
II.1. Teil 1: Zusammenfassung der Unterlagen
Es wird auf Abschnitt I verwiesen.
II.2. Teil 2: Qualitätsbezogene Unterlagen (physikalisch-chemische, biologische oder mikrobiologische Informationen)
II.2A. Produktbeschreibung
II.2A1. Qualitative und quantitative Zusammensetzung
|
(1) |
Unter ‚qualitativer Zusammensetzung‘ aller Bestandteile des Arzneimittels versteht man die Bezeichnung oder Beschreibung:
|
|
(2) |
Für die gebräuchlichen Bezeichnungen zur Beschreibung der Bestandteile eines Tierarzneimittels ist unbeschadet der in Artikel 8 vorgesehenen Angaben Folgendes maßgeblich:
|
|
(3) |
Was die quantitative Zusammensetzung aller Wirkstoffe und Arzneiträgerstoffe der Tierarzneimittel betrifft, so ist je nach der Darreichungsform für jeden Wirkstoff oder Arzneiträgerstoff die Masse oder die Zahl der Einheiten biologischer Aktivität je Einnahme-, Masse- oder Volumeneinheit anzugeben. |
|
(4) |
Einheiten der biologischen Aktivität sind für chemisch nicht zu definierende Stoffe anzuwenden. Sofern eine internationale Einheit biologischer Aktivität festgelegt wurde, ist diese anzuwenden. Falls keine internationale Einheit festgelegt wurde, sind die Einheiten der biologischen Aktivität unter Verwendung der Einheiten des Europäischen Arzneibuchs so auszudrücken, dass sie eindeutig Aufschluss über die Wirksamkeit der Stoffe geben. |
|
(5) |
Die Zusammensetzung nach Menge ist zu ergänzen:
|
|
(6) |
Wirkstoffe in Form von Verbindungen oder Derivaten sind quantitativ durch ihre Gesamtmasse und — sofern erforderlich oder sachdienlich — durch die Masse der aktiven Moleküleinheit(en) angegeben. |
|
(7) |
Für Tierarzneimittel, die einen Wirkstoff enthalten, für den erstmals in der Europäischen Union ein Zulassungsantrag gestellt wird, ist die Zusammensetzung nach Menge eines Wirkstoffs als Salz oder Hydrat systematisch als Masse des aktiven Bestandteils des Moleküls anzugeben. Bei allen später in den Mitgliedstaaten genehmigten Tierarzneimitteln ist die mengenmäßige Zusammensetzung für denselben Wirkstoff in derselben Weise anzugeben. |
II.2A2. Produktentwicklung
|
(1) |
Es müssen erläuternde Angaben über die Wahl der Zusammensetzung, der Bestandteile, der Verpackung, die beabsichtigte Funktion der Arzneiträgerstoffe im Fertigprodukt und das Herstellungsverfahren gemacht werden, einschließlich einer Begründung für die Wahl des Verfahrens und Einzelheiten zu den Sterilisierungsverfahren und/oder aseptischen Verfahren, die beim Fertigprodukt angewandt werden. Diese Aussagen sind durch wissenschaftliche Daten über die pharmazeutische Entwicklung zu erhärten. Etwaige Wirkstoffzuschläge sind anzugeben und zu begründen. Die mikrobiologischen Merkmale (mikrobiologische Reinheit und antimikrobielle Eigenschaften) und die Anwendungshinweise müssen nachweislich für den Verwendungszweck des Tierarzneimittels geeignet sein, der im Antragsdossier angegeben wurde. |
|
(2) |
Eine Beschreibung der Wechselwirkung von Fertigprodukt und Primärverpackung ist in allen Fällen vorzulegen, in denen ein solches Risiko denkbar ist, insbesondere, wenn es sich um injizierbare Präparate handelt. |
|
(3) |
Die vorgeschlagenen Packungsgrößen sind in Bezug auf den vorgeschlagenen Verabreichungsweg, die Posologie und die Zieltierarten, insbesondere für antimikrobielle (Wirk-) Stoffe zu begründen. |
|
(4) |
Wenn dem Fertigprodukt eine Dosiervorrichtung beigefügt ist, muss die Genauigkeit der Dosierung(en) nachgewiesen werden. |
|
(5) |
Wenn ein Begleittest für das Fertigprodukt empfohlen wird (z. B. ein Diagnoseversuch), müssen einschlägige Informationen über den Test angegeben werden. |
|
(6) |
Für zur Herstellung von Fütterungsarzneimitteln bestimmte Tierarzneimittel sind Angaben zu Einmischraten, Herstellung und Kompatibilität/Eignung der Mischfuttermittel zu machen. |
II.2B. Angaben über die Herstellungsweise
|
(1) |
Die Angaben über die Herstellungsweise, die dem Zulassungsantrag gemäß Artikel 8 beizufügen sind, müssen einen geeigneten Überblick über die Art der Herstellungsabläufe geben. |
|
(2) |
Zu diesem Zweck umfassen sie mindestens:
|
II.2C. Herstellung und Kontrolle der Ausgangsstoffe
|
(1) |
‚Ausgangsstoffe‘ im Sinne dieses Punktes sind Wirkstoffe, Arzneiträgerstoffe und Verpackungen (Primärverpackung mit ihrem Verschlusssystem und gegebenenfalls die äußere Umhüllung sowie jede mit dem Tierarzneimittel gelieferte Dosiervorrichtung). |
|
(2) |
Das Dossier muss die Spezifikationen und Informationen für die Prüfungen umfassen, die zur Qualitätskontrolle aller Ausgangsstoffchargen durchzuführen sind. |
|
(3) |
Die Routineprüfungen der Ausgangsstoffe sind in der gleichen Weise durchzuführen, wie im Dossier angegeben. |
|
(4) |
Hat die Europäische Direktion für Arzneimittelqualität ein Eignungszertifikat für einen Ausgangsstoff, Wirkstoff oder Arzneiträgerstoff ausgestellt, gilt dieses als Bezugnahme auf die einschlägige Monografie des Europäischen Arzneibuchs. |
|
(5) |
Wird auf ein Eignungszertifikat Bezug genommen, versichert der Hersteller dem Antragsteller schriftlich, dass das Herstellungsverfahren seit der Erteilung des Eignungszertifikats durch die Europäische Direktion für Arzneimittelqualität nicht geändert wurde. Ist das Feld ‚Zugangsfeld‘ des Zertifikats ausgefüllt und unterzeichnet, gilt diese Anforderung als erfüllt, ohne dass eine zusätzliche Zusicherung erforderlich ist. |
|
(6) |
Für die Ausgangsstoffe sind Analysezertifikate zum Nachweis der Einhaltung der festgelegten Spezifikation vorzulegen. |
II.2C1. Wirkstoff(e)
|
(1) |
Die erforderlichen Daten sind auf eine der drei unter den Punkten 2 bis 4 genannten Arten zu übermitteln. |
|
(2) |
Die folgenden Angaben sind zu übermitteln:
|
|
(3) |
Wirkstoff-Stammdokumentation
Bei einem nicht biologischen Wirkstoff trägt der Antragsteller dafür Sorge, dass Informationen zum Wirkstoff unter Punkt 2 in Form einer Wirkstoff-Stammdokumentation vom Hersteller des Wirkstoffs direkt an die zuständigen Behörden übermittelt werden. In diesem Fall muss der Hersteller des Wirkstoffs dem Antragsteller alle Angaben vorlegen (in dem dem Antragsteller vorbehaltenen Teil der Wirkstoff-Stammdokumentation), die dieser benötigt, um die Verantwortung für das Tierarzneimittel zu übernehmen. Eine Kopie der Daten, die der Hersteller des Wirkstoffs dem Antragsteller zur Verfügung stellt, ist dem Arzneimitteldossier beizufügen. Der Hersteller des Wirkstoffs muss dem Antragsteller schriftlich bestätigen, dass er die Einheitlichkeit der einzelnen Chargen gewährleistet und den Herstellungsprozess oder die Spezifikationen nicht verändert, ohne den Antragsteller darüber in Kenntnis zu setzen. |
|
(4) |
Eignungszertifikat, ausgestellt von der Europäischen Direktion für die Arzneimittelqualität
Das Eignungszertifikat und alle zusätzlichen, für die Darreichungsform relevanten Daten, die nicht im Eignungszertifikat enthalten sind, sind vorzulegen. |
II.2C1.1. In Arzneibüchern aufgeführte Wirkstoffe
|
(1) |
Die Bestimmungen des Artikels 8 gelten als hinreichend erfüllt, wenn die Wirkstoffe den Vorschriften des Europäischen Arzneibuchs oder in Ermangelung einer Monografie des Europäischen Arzneibuchs des Arzneibuchs eines der Mitgliedstaaten entsprechen. In diesem Fall kann die Beschreibung der Analysemethoden und -verfahren im jeweiligen Abschnitt durch eine geeignete Bezugnahme auf das betreffende Arzneibuch ersetzt werden. |
|
(2) |
Die zuständigen Behörden können von dem Antragsteller geeignete Spezifikationen, darunter auch Akzeptanzkriterien für bestimmte Verunreinigungen samt validierten Prüfverfahren, verlangen, wenn eine Spezifikation einer Monografie des Europäischen Arzneibuchs oder des Arzneibuchs eines Mitgliedstaats unter Umständen nicht genügt, um die Qualität des Ausgangsstoffes zu gewährleisten. |
|
(3) |
Die zuständigen Behörden setzen die für das betreffende Arzneibuch zuständigen Behörden davon in Kenntnis. Der Zulassungsinhaber muss den für das betreffende Arzneibuch zuständigen Behörden alle Einzelheiten bezüglich der angeblichen Unzulänglichkeit und der zusätzlichen angewandten Spezifikationen vorlegen. |
II.2C1.2. In Arzneibüchern nicht aufgeführte Wirkstoffe
|
(1) |
Für die in keinem Arzneibuch aufgeführten Wirkstoffe ist eine Monografie anzufertigen, die Folgendes umfasst:
|
|
(2) |
Diese Daten müssen belegen, dass die vorgeschlagenen Prüfverfahren die Kontrolle der Qualität des Wirkstoffs einer bestimmten Herkunft gewährleisten. |
II.2C1.3. Physikalisch-chemische Eigenschaften, die die Bioverfügbarkeit beeinflussen können
Die nachfolgenden Daten über Wirkstoffe sind als Teil der allgemeinen Beschreibung der Wirkstoffe anzugeben, wenn sie sich auf die Bioverfügbarkeit des Tierarzneimittels auswirken:
|
a) |
Kristallform und Löslichkeit, |
|
b) |
Partikelgröße, |
|
c) |
Hydrationsgrad, |
|
d) |
Öl-Wasser-Verteilungskoeffizient, |
|
e) |
pK- und pH-Wert. |
Die Buchstaben a bis c gelten nicht für nur als Lösung verwendete Stoffe.
II.2C2. Arzneiträgerstoffe
|
(1) |
Die Bestimmungen des Artikels 8 gelten als erfüllt, wenn die Arzneiträgerstoffe den Vorschriften des Europäischen Arzneibuchs oder in Ermangelung einer Monografie des Europäischen Arzneibuchs des Arzneibuchs eines der Mitgliedstaaten entsprechen. In diesem Fall kann die Beschreibung der Analysemethoden und -verfahren im jeweiligen Abschnitt durch eine geeignete Bezugnahme auf das betreffende Arzneibuch ersetzt werden. Gegebenenfalls müssen die Vorschriften der Monografie durch zusätzliche Prüfungen zur Kontrolle von Parametern wie Partikelgröße, Sterilität und/oder Lösungsmittelrückstände ergänzt werden. |
|
(2) |
Gibt es keine Arzneibuchmonografie, muss eine Spezifikation vorgeschlagen und begründet werden. Für den Wirkstoff gelten die Anforderungen für Spezifikationen gemäß Teil II.2C1.2 Punkt 1 Buchstaben a bis e. Die vorgeschlagenen Methoden und die entsprechenden Validierungsdaten sind darzustellen. |
|
(3) |
Es ist eine Erklärung vorzulegen, in der bestätigt wird, dass die Farbstoffe, die Tierarzneimitteln hinzugefügt werden sollen, den Vorschriften der Richtlinie 2009/35/EG des Europäischen Parlaments und des Rates (3) genügen; dies gilt nicht für bestimmte Tierarzneimittel zur topischen Anwendung, wie wirkstoffhaltige Halsbänder und Ohrmarken. |
|
(4) |
Es ist eine Erklärung vorzulegen, in der bestätigt wird, dass die verwendeten Farbstoffe die Reinheitskriterien gemäß Verordnung (EU) Nr. 231/2012 der Kommission (4) erfüllen. |
|
(5) |
Bei neuartigen Arzneiträgerstoffen, die in der Union erstmalig in einem Tierarzneimittel eingesetzt werden, oder bei Arzneiträgerstoffen, bei denen dies durch eine neue Art der Anwendung geschieht, sind umfassende Angaben zur Herstellung, zur Charakterisierung und zu den Kontrollen zu machen, wobei Querverweise sowohl auf die klinischen als auch auf die nichtklinischen Daten zur Unbedenklichkeit zu machen sind. Für Farbstoffe werden die Konformitätserklärungen in den Punkten 3 und 4 als ausreichend angesehen. |
II.2C3. Verpackung (Behältnisse und Verschlusssysteme)
II.2C3.1. Wirkstoff
|
(1) |
Es sind Informationen über das für den Wirkstoff verwendete Behältnis und sein Verschlusssystem, einschließlich der Identität jeder Primärverpackung und ihrer Spezifikationen bereitzustellen. Welche Informationen bereitzustellen sind, hängt vom Aggregatzustand (flüssig oder fest) des Wirkstoffes ab. |
|
(2) |
Liegt für den Wirkstoff aus der vorgeschlagenen Bezugsquelle ein Eignungszertifikat vor und werden darin ein Behältnis und sein Verschlusssystem genannt, so können die ausführlichen Informationen über letztere für den Wirkstoff aus dieser Quelle durch einen Verweis auf das gültige Eignungszertifikat ersetzt werden. |
|
(3) |
Liegt eine Wirkstoff-Stammdokumentation aus der vorgeschlagenen Bezugsquelle vor und werden darin ein Behältnis und sein Verschlusssystem genannt, so können die ausführlichen Informationen über letztere für den Wirkstoff aus dieser Quelle durch einen Verweis auf die Wirkstoff-Stammdokumentation ersetzt werden. |
II.2C3.2. Fertigprodukt
|
(1) |
Eine Beschreibung des Behältnisses und seines Verschlusssystems sowie jeder etwaigen Vorrichtung für das Fertigprodukt, einschließlich der Angabe aller Bestandteile des Primärverpackungsmaterials und ihrer Spezifikationen, ist vorzulegen. Welche Informationen bereitzustellen sind, hängt vom Verabreichungsweg des Tierarzneimittels und vom Aggregatzustand (flüssig oder fest) der Darreichungsform ab. |
|
(2) |
Gibt es keine Arzneibuchmonografie, muss eine Spezifikation für das Verpackungsmaterial vorgeschlagen und begründet werden. |
|
(3) |
Für Verpackungsmaterial, das erstmalig in der Europäischen Union verwendet wird und mit dem Arzneimittel in Kontakt kommt, sind Informationen über Zusammensetzung, Herstellung und Unbedenklichkeit vorzulegen. |
II.2C4. Stoffe biologischer Herkunft
|
(1) |
Es sind Informationen über die Herkunft, Verarbeitung, Charakterisierung und Kontrolle aller Materialien biologischen Ursprungs (menschlich, tierisch, pflanzlich oder aus Mikroorganismen), die bei der Herstellung der Tierarzneimittel verwendet werden, vorzulegen, einschließlich Daten zur viralen Unbedenklichkeit, gemäß den einschlägigen Leitlinien. |
|
(2) |
Durch Vorlage entsprechender Unterlagen ist nachzuweisen, dass das Material, das von Tierarten stammt, die transmissible spongiforme Enzephalopathien (TSE) übertragen können, mit den Vorschriften der Leitlinien für die Minimierung des Risikos der Übertragung von Erregern der spongiformen Enzephalopathie tierischen Ursprungs durch Human- und Tierarzneimittel sowie mit der entsprechenden Monografie des Europäischen Arzneibuchs in Einklang stehen. Dieser Nachweis kann durch Eignungszertifikate erbracht werden, die von der Europäischen Direktion für die Arzneimittelqualität für die einschlägige Monografie des Europäischen Arzneibuchs ausgestellt werden. |
II.2D. Kontrollen, die während des Herstellungsprozesses an isolierten Zwischenprodukten durchgeführt werden
|
(1) |
Im Sinne dieses Abschnitts gilt als ‚isoliertes Zwischenprodukt‘ teilweise verarbeitetes Material, das für eine bestimmte Zeit gelagert werden kann und weitere Verarbeitungsschritte durchläuft, bevor es zum Fertigprodukt wird. |
|
(2) |
Für jedes Zwischenprodukt ist eine Spezifikation festzulegen, und die Analysemethoden sind zu beschreiben und gegebenenfalls zu validieren. |
|
(3) |
Es sind Angaben zur Primärverpackung des Zwischenproduktes zu machen, wenn sich diese von derjenigen des Fertigprodukts unterscheidet. |
|
(4) |
Die Haltbarkeitsdauer und die Lagerungsbedingungen des Zwischenprodukts werden auf der Grundlage der Daten errechnet, die sich aus Haltbarkeitsstudien ergeben haben. |
II.2E. Kontrollen am Fertigprodukt
|
(1) |
Im Zusammenhang mit der Kontrolle des Fertigprodukts bedeutet eine Charge eines Fertigprodukts die Gesamtheit der Einheiten einer Darreichungsform, die aus derselben Menge an Ausgangsmaterial stammen und derselben Serie von Herstellungs- und/oder Sterilisierungsprozessen unterworfen wurden. Bei kontinuierlicher Herstellung kann die Chargengröße durch einen Zeitraum oder eine Produktmenge angegeben und in Bereichen ausgedrückt werden. |
|
(2) |
Die Prüfungen, die am Fertigprodukt durchgeführt werden, sind aufzuführen. Die vorgeschlagene Spezifikation ist zu begründen. Die Häufigkeit der nicht routinemäßig durchgeführten Prüfungen ist anzugeben und zu rechtfertigen. Akzeptanzkriterien für die Freigabe müssen angegeben werden. |
|
(3) |
Das Dossier muss Angaben zu Kontrollen enthalten, die am Fertigprodukt bei der Freigabe und Validierung vorgenommen wurden. Die Angaben und Unterlagen müssen folgenden Anforderungen entsprechen. |
|
(4) |
Wenn andere als in den einschlägigen Monografien und den allgemeinen Kapiteln des Europäischen Arzneibuchs oder, falls nicht vorhanden, des Arzneibuchs eines Mitgliedstaats aufgeführte Prüfverfahren und Akzeptanzkriterien angewandt werden, sind diese Verfahren und Kriterien anhand des Nachweises zu begründen, dass das Fertigprodukt bei einer Prüfung im Einklang mit jenen Monografien den Qualitätsanforderungen des entsprechenden Arzneibuchs an die betreffende Darreichungsform genügen würde. |
II.2E1. Allgemeine Merkmale des Fertigprodukts
|
(1) |
Bestimmte Kontrollen allgemeiner Merkmale eines Arzneimittels müssen immer am Fertigprodukt durchgeführt werden. Diese Kontrollen erstrecken sich gegebenenfalls auf die Bestimmung der Durchschnittsmassen/-volumina und der zulässigen Abweichungen, auf mechanische oder physikalische Versuche, auf das Erscheinungsbild, die physikalischen Eigenschaften wie Dichte, pH-Wert oder die Partikelgröße. Für jede dieser Eigenschaften müssen die Standards und Akzeptanzkriterien vom Antragsteller spezifiziert werden. |
|
(2) |
Die Prüfbedingungen und gegebenenfalls die verwendeten Geräte/Einrichtungen sowie die Standards sind, sofern sie nicht im Europäischen Arzneibuch oder im Arzneibuch eines Mitgliedstaats angegeben sind, detailliert zu beschreiben; das Gleiche gilt für den Fall, dass die in diesen Arzneibüchern vorgesehenen Verfahren nicht anwendbar sind. |
II.2E2. Identitätsnachweis und Gehaltsbestimmung der Wirkstoffe
|
(1) |
Der Identitätsnachweis und die Gehaltsbestimmung des oder der Wirkstoffe sind bei einer Durchschnittsprobe, die für die Charge repräsentativ ist, oder bei einer bestimmten Anzahl gesondert betrachteter Gebrauchseinheiten durchzuführen. |
|
(2) |
Ohne angemessene Begründung dürfen die zulässigen Fehlerbreiten der Wirkstoffe im Fertigprodukt bei der Herstellung ± 5 % nicht überschreiten. |
|
(3) |
In bestimmten Fällen besonders komplexer Mischungen, bei denen die Bestimmung zahlreicher oder in äußerst geringen Mengen vorhandener Wirkstoffe schwierige Prüfungen, die sich kaum bei jeder einzelnen Herstellungscharge durchführen lassen, erforderlich machen würde, ist es zulässig, dass die Gehaltsbestimmung eines oder mehrerer Wirkstoffe im Fertigprodukt unterbleibt, dies jedoch unter der ausdrücklichen Bedingung, dass diese Gehaltsbestimmungen am Zwischenprodukt durchgeführt werden. Diese vereinfachte Methode darf nicht auf die Charakterisierung der betreffenden Stoffe ausgedehnt werden. Sie wird durch eine Methode der quantitativen Bestimmung vervollständigt, die es den zuständigen Behörden ermöglicht nachzuprüfen, ob das Arzneimittel nach der Zulassung mit seinen Spezifikationen im Einklang ist. |
|
(4) |
Eine biologische Bestimmung in vivo oder in vitro ist erforderlich, sofern die physikalisch-chemischen Methoden nicht ausreichen, um Auskunft über die Qualität des Arzneimittels zu erhalten. Solch eine Bestimmung sollte möglichst Referenzmaterialien und statistische Analysen mit Berechnung der Sicherheitskoeffizienten umfassen. Sofern diese Versuche nicht am Fertigprodukt durchgeführt werden können, ist es möglich, dass sie in einem Zwischenstadium, möglichst gegen Ende des Herstellungsverfahrens, erfolgen. |
|
(5) |
Der annehmbare Höchstwert unmittelbar nach der Herstellung ist für Einzel- und Gesamtabbauprodukte anzugeben. Die Gründe für die Aufnahme oder den Ausschluss von Abbauprodukten in die Spezifikation sind darzulegen. |
II.2E3. Identitätsnachweis und Gehaltsbestimmung der Bestandteile des Arzneiträgerstoffs
Eine Identitätsprüfung und die Bestimmung des oberen und des unteren Grenzwerts sind für jeden einzelnen antimikrobiellen Konservierungsstoff und für alle Arzneiträgerstoffe vorgeschrieben, die die Bioverfügbarkeit des Wirkstoffs beeinträchtigen könnten, sofern die Bioverfügbarkeit nicht durch andere geeignete Prüfungen gewährleistet ist. Die Identitätsprüfung und die Bestimmung eines oberen Grenzwerts sind für alle Antioxidantien und alle Arzneiträgerstoffe erforderlich, die die physiologischen Funktionen beeinträchtigen könnten; außerdem ist für Antioxidantien zum Zeitpunkt der Freigabe auch ein unterer Grenzwert zu bestimmen.
II.2E4. Mikrobiologische Kontrollen
Angaben zu mikrobiologischen Prüfungen in Bezug auf Sterilität und bakterielle Endotoxine sind in den analytischen Unterlagen zu leisten, soweit die Prüfungen routinemäßig zur Kontrolle der Qualität des Arzneimittels durchgeführt werden müssen.
II.2E5. Gleichbleibende Qualität der Chargen
Um sicherzustellen, dass die Qualität des Produkts von Charge zu Charge gleichbleibend ist, und um die Konformität mit der Spezifikation nachzuweisen, sind Chargendaten vorzulegen, die die Ergebnisse aller Prüfungen enthalten, die im Allgemeinen an [3] Chargen, die in der/den vorgeschlagenen Produktionsstätte(n) nach dem beschriebenen Herstellungsverfahren hergestellt wurden, durchgeführt worden sind.
II.2E6. Sonstige Kontrollen
Jede andere Prüfung, die zur Bestätigung der Qualität des Arzneimittels für erforderlich gehalten wird, ist zu kontrollieren.
II.2F. Haltbarkeitsversuche
II.2F1. Wirkstoff(e)
|
(1) |
Es sind ein Überprüfungszeitraum und Lagerungsbedingungen für den Wirkstoff festzulegen, es sei denn, der Hersteller des Fertigprodukts unterzieht den Wirkstoff unmittelbar vor seiner Verwendung im Rahmen der Herstellung des Fertigprodukts einer vollständigen Überprüfung. |
|
(2) |
Es sind Haltbarkeitsdaten vorzulegen, um nachzuweisen, wie sich die Qualität eines Wirkstoffs im Laufe der Zeit unter dem Einfluss verschiedener Umweltfaktoren verändert, und um gegebenenfalls den festgelegten Überprüfungszeitraum und die Lagerungsbedingungen zu erhärten. Die Art der durchgeführten Haltbarkeitsstudien, die verwendeten Versuchsprotokolle und Analyseverfahren sowie ihre Validierung sind zusammen mit den ausführlichen Ergebnissen vorzulegen. |
|
(3) |
Liegt für den Wirkstoff aus der vorgeschlagenen Bezugsquelle ein Eignungszertifikat vor und werden darin ein Überprüfungszeitraum und Lagerungsbedingungen genannt, so sind für den Wirkstoff aus dieser Bezugsquelle keine Haltbarkeitsdaten erforderlich. |
|
(4) |
Liegt eine Wirkstoff-Stammdokumentation aus der vorgeschlagenen Bezugsquelle vor und werden darin Haltbarkeitsdaten genannt, können die ausführlichen Informationen über die Haltbarkeitsdaten für den Wirkstoff aus dieser Quelle durch einen Verweis auf die Wirkstoff-Stammdokumentation ersetzt werden. |
II.2F2. Fertigprodukt
|
(1) |
Zu beschreiben sind die Untersuchungen, die es ermöglicht haben, die Haltbarkeitsdauer, die empfohlenen Lagerungsbedingungen und die Spezifikationen bei Ablauf der Haltbarkeitsdauer, wie vom Antragsteller vorgeschlagen, zu erhärten. |
|
(2) |
Die Art der durchgeführten Haltbarkeitsstudien, die verwendeten Versuchsprotokolle und Analyseverfahren sowie ihre Validierung sind zusammen mit den ausführlichen Ergebnissen vorzulegen. |
|
(3) |
Sofern bei einem Fertigprodukt vor der Verabreichung eine Rekonstituierung oder Verdünnung erforderlich ist, sind detaillierte Angaben zu der vorgeschlagenen Haltbarkeitsdauer und zu den Spezifikationen für das rekonstituierte/verdünnte Arzneimittel zu machen und durch sachdienliche Haltbarkeitsdaten zu erhärten. |
|
(4) |
Bei Multidosisbehältnissen sind gegebenenfalls Haltbarkeitsdaten vorzulegen, um die Haltbarkeitsdauer nach erstmaliger Entnahme oder Öffnung des Arzneimittels zu begründen, und es ist eine Spezifikation für das im Gebrauch befindliche Behältnis festzulegen. |
|
(5) |
Ist damit zu rechnen, dass sich bei einem Fertigprodukt Abbauprodukte bilden, so muss der Antragsteller diese Produkte mitteilen und angeben, welche Methoden für ihre Identifizierung und welche Prüfverfahren angewandt werden. |
|
(6) |
Zeigen die Haltbarkeitsdaten, dass der Wirkstoffgehalt sich bei Lagerung verringert, muss die Beschreibung der Methoden zur Kontrolle des Fertigprodukts gegebenenfalls die chemische Prüfung und erforderlichenfalls die toxikologisch-pharmakologische Prüfung der bei diesem Stoff eingetretenen Veränderungen umfassen; hier sind gegebenenfalls die Abbauprodukte zu charakterisieren und/oder zu bestimmen. |
|
(7) |
Der annehmbare Höchstwert für Einzel- und Gesamtabbauprodukte bei Ablauf der Haltbarkeitsdauer ist anzugeben und zu begründen. |
|
(8) |
Auf der Grundlage der Ergebnisse der Haltbarkeitsprüfung sind die Prüfungen und ihre Akzeptanzkriterien, die am Fertigprodukt im Verlauf der Haltbarkeitsdauer durchgeführt werden, aufzulisten und zu begründen. |
|
(9) |
Die Schlussfolgerungen müssen die Analyseergebnisse enthalten, die die vorgeschlagene Haltbarkeitsdauer und gegebenenfalls die Haltbarkeitsdauer nach der ersten Entnahme unter den empfohlenen Lagerungsbedingungen rechtfertigen. |
|
(10) |
Außerdem sind für zur Herstellung von Fütterungsarzneimitteln bestimmte Tierarzneimittel Angaben zur Haltbarkeit und zur vorgeschlagenen Haltbarkeitsdauer nach der Einmischung zu machen. Es ist auch eine Spezifikation für das Fütterungsarzneimittel vorzulegen, das unter Verwendung dieser Tierarzneimittel gemäß der empfohlenen Gebrauchsanweisung hergestellt wird. |
II.2G. Sonstige Informationen
Informationen über die Qualität des Tierarzneimittels, die nicht an anderer Stelle in diesem Teil behandelt werden, können unter diesem Punkt in das Dossier aufgenommen werden.
II.3 Teil 3: Unterlagen zur Unbedenklichkeit (Unbedenklichkeits- und Rückstandsversuche)
|
(1) |
Der Studienbericht enthält Folgendes:
|
|
(2) |
Veröffentlichte Studien können akzeptiert werden, wenn sie eine ausreichende Menge an Daten und genügend Details enthalten, um eine unabhängige Bewertung zu ermöglichen. Die Versuchsmethoden sind so genau zu beschreiben, dass sie reproduziert werden können, und der Prüfer muss ihre Validität nachweisen. Zusammenfassungen von Studien, zu denen keine detaillierten Berichte vorliegen, stellen keine vollständigen Unterlagen dar. Wenn der Stoff zuvor für die Festsetzung von Rückstandshöchstmengen (maximum residues limit, MRL) bewertet wurde, um bestimmte Sicherheitsanforderungen zu erfüllen, kann auf die EPMAR (European Public MRL Assessment Reports) Bezug genommen werden. Wenn auf die EPMAR verwiesen wird, müssen keine Studien vorgelegt werden, die bereits im Rahmen der Rückstandshöchstmengen-Bewertung bewertet wurden; es sind nur neue Studien vorzulegen, die für die Bewertung der Rückstandshöchstmengen nicht verfügbar waren. Wenn der Verabreichungsweg (z. B. für den Anwender) nicht identisch ist mit dem gemäß der Verordnung (EU) 2018/782 der Kommission (5) verwendeten Weg, können neue Studien erforderlich sein. |
II.3A. Unbedenklichkeitsversuche
|
(1) |
Die Unbedenklichkeitsunterlagen müssen zur Bewertung von Folgendem angemessen sein:
|
|
(2) |
In bestimmten Fällen ist es erforderlich, die Metaboliten der Ausgangsverbindung zu prüfen, wenn es sich bei diesen um die bedenklichen Rückstände handelt. |
|
(3) |
Ein Arzneiträgerstoff, der erstmalig in einem Tierarzneimittel eingesetzt wird, oder ein Arzneiträgerstoff, bei dem dies durch eine neue Art der Anwendung geschieht, ist wie ein Wirkstoff zu behandeln. |
II.3A1. Genaue Identifizierung des Arzneimittels und seiner Wirkstoffe
|
a) |
Internationaler Freiname (INN), |
|
b) |
IUPAC-Bezeichnung, |
|
c) |
Nummer des Chemical Abstracts Service (CAS-Nummer), |
|
d) |
therapeutische, pharmakologische und chemische Klassifizierung, |
|
e) |
Synonyme und Abkürzungen, |
|
f) |
Strukturformel, |
|
g) |
Molekularformel, |
|
h) |
Molekulargewicht, |
|
i) |
Reinheitsgrad, |
|
j) |
qualitative und quantitative Zusammensetzung der Verunreinigungen, |
|
k) |
Beschreibung physikalischer Eigenschaften,
|
|
l) |
Formulierung des Arzneimittels. |
II.3A2. Pharmakologie
|
(1) |
Pharmakologische Untersuchungen sind zur Klärung der Mechanismen, die die therapeutischen Wirkungen des Tierarzneimittels herbeiführen, von fundamentaler Bedeutung, weshalb an Versuchs- und Zieltierarten durchgeführte pharmakologische Untersuchungen aufgenommen werden sollten. Gegebenenfalls kann ein Querverweis auf die in Teil 4 des Dossiers eingereichten Studien erfolgen. |
|
(2) |
Wenn ein Tierarzneimittel bei Ausbleiben einer toxischen Reaktion oder bei Dosen, die so niedrig sind, dass kein toxischer Effekt auftritt, pharmakologische Wirkungen aufweist, sind Letztere bei der Bewertung der Unbedenklichkeit des Tierarzneimittels in Betracht zu ziehen. |
|
(3) |
Den Unterlagen zur Unbedenklichkeit sind in jedem Fall ausführliche Angaben über an Versuchstieren durchgeführte pharmakologische Untersuchungen sowie sämtliche einschlägigen Angaben zu den klinischen Prüfungen am Zieltier voranzuschicken. |
II.3A2.1 Pharmakodynamik
Zum besseren Verständnis etwaiger Nebenwirkungen in den Tierstudien sind Informationen über den Wirkungsmechanismus der Wirkstoffe sowie über die primären und sekundären pharmakodynamischen Wirkungen vorzulegen. Detaillierte Angaben zu den pharmakodynamischen Eigenschaften, die sich auf die therapeutische Wirksamkeit beziehen, sind in Teil 4A des Dossiers zu melden.
II.3A2.2 Pharmakokinetik
Es sind Daten zum Verhalten des Wirkstoffs und seiner Metaboliten bei den Versuchstieren vorzulegen, die Angaben zu Resorption, Verteilung, Metabolisierung und Ausscheidung (ADME) enthalten. Zur Ermittlung der entsprechenden Exposition sind die Daten mit den Dosis-Wirkungs-Ergebnissen der pharmakologischen und toxikologischen Studien in Beziehung zu setzen.
II.3A3. Toxikologie
|
(1) |
Die Toxikologieunterlagen müssen den Leitlinien der Agentur über den allgemeinen Ansatz für Prüfungen sowie den Leitlinien für spezielle Studien entsprechen. Generell sind die Toxizitätsstudien nicht mit dem formulierten Produkt, sondern mit den Wirkstoffen durchzuführen, sofern nicht spezifisch etwas anderes vorgeschrieben ist. |
|
(2) |
Tierexperimentelle Studien sind an etablierten Stämmen von Versuchstieren durchzuführen, für die (vorzugsweise) historische Daten vorliegen. |
|
(3) |
Toxizität bei einmaliger Verabreichung
Die Prüfungen der Toxizität bei einmaliger Verabreichung können eingesetzt werden zur Prognose:
Prüfungen der Toxizität bei einmaliger Verabreichung haben die akuten toxischen Wirkungen des Stoffs und den zeitlichen Ablauf ihres Einsetzens und Abklingens aufzuzeigen. Die durchzuführenden Prüfungen werden im Hinblick auf Informationen zur Anwendersicherheit ausgewählt; ist z. B. mit einer erheblichen Exposition des Benutzers gegenüber dem Tierarzneimittel durch Inhalation oder Hautkontakt zu rechnen, sollten diese Verabreichungswege untersucht werden. |
|
(4) |
Toxizität bei wiederholter Verabreichung
Die Prüfungen der Toxizität bei wiederholter Verabreichung haben zum Ziel, physiologische und/oder pathologische Veränderungen infolge wiederholter Verabreichung eines wirksamen Bestandteils bzw. einer Wirkstoffkombination festzustellen und die Dosierungen zu ermitteln, die für das Auftreten dieser Veränderungen verantwortlich sind. Normalerweise ist eine Prüfung der Toxizität bei wiederholter Verabreichung an einer Versuchstierart ausreichend. Diese Untersuchung kann durch eine Untersuchung am Zieltier ersetzt werden. Häufigkeit und Weg der Verabreichung sowie die Dauer des Versuchs sind unter Berücksichtigung der vorgeschlagenen klinischen Anwendungsbedingungen und/oder Exposition der Anwender anzusetzen. Der Antragsteller muss Umfang und Dauer der Studien sowie die gewählten Dosierungen begründen. |
|
(5) |
Verträglichkeit bei der Zieltierart
Alle Anzeichen von Unverträglichkeit, die bei der Zieltierart im Verlauf der üblicherweise mit der endgültigen Formulierung durchgeführten Versuche aufgetreten sind, müssen gemäß den Anforderungen von Teil II.4A4 (Verträglichkeit bei den Zieltierarten) zusammenfassend angegeben werden. Die betreffenden Untersuchungen, die Dosierungen, die die Unverträglichkeit hervorgerufen haben, sowie die betreffenden Arten und Rassen sind zu beschreiben. Ferner sind alle unerwarteten physiologischen Veränderungen im Detail anzugeben. Teil 4 des Dossiers muss die vollständigen Berichte dieser Studien enthalten. |
|
(6) |
Reproduktions- und Entwicklungstoxizität
Studie zu den Wirkungen auf die Fortpflanzung Für Produkte, die zur Verwendung bei Zuchttieren bestimmt sind, sind Reproduktionsstudien in Übereinstimmung mit VICH GL43 vorzulegen. Studien zur Reproduktionstoxizität an Versuchstieren werden für die Bewertung der Auswirkungen auf den Anwender nicht erwartet. |
|
(7) |
Studie zur Entwicklungstoxizität
Für die Bewertung der Wirkungen bei der Zieltierart sind Studien zur Entwicklungstoxizität nicht erforderlich für Produkte, die nur zur Verwendung bei nicht zu Zuchtzwecken gehaltenen Tieren bestimmt sind. Für andere Produkte ist eine Studie zur Entwicklungstoxizität an mindestens einer Tierart durchzuführen, die die Zieltierart sein kann. Wird die Studie an der Zieltierart durchgeführt, so ist hier eine Zusammenfassung zu geben, und der vollständige Bericht zur Studie ist in Teil 4 des Dossiers aufzunehmen. Zur Bewertung der Anwendersicherheit sind in allen Fällen, in denen eine signifikante Exposition des Anwenders zu erwarten ist, Standardtests zur Entwicklungstoxizität in Übereinstimmung mit Standardtests auf der Grundlage etablierter Leitlinien (einschließlich VICH GL32 und OECD-Prüfungen) durchzuführen. |
|
(8) |
Genotoxizität
Das genotoxische Potenzial ist zu untersuchen, um etwaige Veränderungen zu ermitteln, die ein Stoff am genetischen Material von Zellen verursachen kann. Alle Stoffe, die zum ersten Mal in einem Tierarzneimittel verwendet werden, sind auf ihre genotoxischen Eigenschaften hin zu beurteilen. An dem/den Wirkstoff(en) ist eine Standardbatterie von Genotoxizitätstests in Übereinstimmung mit Standardtests auf der Grundlage festgelegter Leitlinien (einschließlich VICH GL23 und OECD-Tests) durchzuführen. |
|
(9) |
Kanzerogenität
Bei der Entscheidung über die Notwendigkeit einer Prüfung auf Kanzerogenität sind die Ergebnisse von Genotoxizitätsprüfungen, die Struktur-Wirkungs-Beziehungen und die Ergebnisse von Untersuchungen auf Toxizität bei wiederholter Verabreichung zu berücksichtigen, die das Potenzial für hyper-/neoplastische Veränderungen haben können. Es sind alle bekannten artspezifischen Toxizitätsmechanismen sowie die Unterschiede im Metabolismus zwischen der in den Prüfungen verwendeten Tierart, der Zieltierart und dem Menschen zu berücksichtigen. Die Prüfung auf Kanzerogenität ist nach Standardprüfungen auf der Grundlage festgelegter Leitlinien (einschließlich VICH GL28 und OECD-Prüfungen) durchzuführen. |
|
(10) |
Ausnahmen
Sofern ein Tierarzneimittel zur topischen Anwendung bestimmt ist, muss die systemische Absorption an der Zieltierart geprüft werden. Wenn nachgewiesen ist, dass die systemische Absorption unerheblich ist, kann auf die Prüfungen der Toxizität bei wiederholter Verabreichung, die Versuche zur Reproduktions- und Entwicklungstoxizität und die Prüfungen auf Kanzerogenität verzichtet werden, es sei denn,
|
II.3A4. Sonstige Anforderungen
II.3A.4.1 Spezielle Untersuchungen
Bei bestimmten Stoffgruppen oder wenn die im Verlauf der Prüfungen bei wiederholter Verabreichung an Tiere beobachteten Wirkungen Veränderungen umfassen, die beispielsweise auf Immunotoxizität, Neurotoxizität oder endokrine Dysfunktion hindeuten, sind weitere Prüfungen erforderlich, z. B. Sensibilisierungsstudien oder Prüfungen auf verzögerte Neurotoxizität. Je nach Art des Arzneimittels kann es erforderlich sein, zusätzliche Studien zur Beurteilung der Mechanismen durchzuführen, die der toxischen Wirkung oder dem Reizpotenzial zugrunde liegen.
Für Produkte, bei denen es zu einer Exposition von Haut und Augen kommen kann, sind Studien zur Reizung und Sensibilisierung vorzulegen. Diese Studien werden mit der endgültigen Formulierung durchgeführt.
Bei der Konzeption solcher Untersuchungen und der Bewertung ihrer Ergebnisse sind der Stand der neuesten wissenschaftlichen Kenntnisse sowie die festgelegten Leitlinien zu berücksichtigen.
II.3A.4.2. Beobachtungen beim Menschen
Es sind Informationen vorzulegen, aus denen hervorgeht, ob die pharmakologisch wirksamen Stoffe des Tierarzneimittels als Arzneimittel in der Humantherapie verwendet werden. Trifft dies zu, sind alle am Menschen festgestellten Wirkungen (einschließlich Nebenwirkungen) und deren Ursachen zusammenzustellen, sofern sie für die Beurteilung der Unbedenklichkeit des Tierarzneimittels von Bedeutung sein können; dabei sind gegebenenfalls die Ergebnisse veröffentlichter Studien zu berücksichtigen. Sofern Bestandteile der Tierarzneimittel als solche nicht bzw. nicht mehr als Arzneimittel in der Humantherapie angewandt werden, ist dies zu begründen, sofern diese Informationen öffentlich verfügbar sind.
II.3A.4.3. Resistenzentwicklung und damit verbundenes Risiko für den Menschen
Die in diesem Punkt beschriebenen Datenanforderungen beziehen sich auf antibakterielle Stoffe und sind möglicherweise nicht in vollem Umfang auf andere Arten von antimikrobiellen Wirkstoffen (nämlich Virostatika, Antimykotika und Antiprotozoika) anwendbar, obwohl die Anforderungen im Prinzip befolgt werden können, sofern sie zutreffen.
Für diese Produkte sind Daten über das mögliche Auftreten resistenter Bakterien oder Resistenzdeterminanten erforderlich, die für die menschliche Gesundheit von Bedeutung sind und mit der Verwendung von Tierarzneimitteln in Zusammenhang stehen. Dabei ist besonders wichtig, durch welchen Mechanismus sich derartige Resistenzen entwickeln und selektieren. Der Antragsteller muss erforderlichenfalls Maßnahmen vorschlagen, durch die sich die Entwicklung einer Resistenz infolge der vorgesehenen Verwendung des Tierarzneimittels begrenzen lässt.
Sind solche Resistenzdaten für die klinische Anwendung des Produkts bei Zieltierarten relevant, ist dies gemäß Teil II.4A2 zu behandeln. Gegebenenfalls sind Querverweise zu den in Teil II.4A2 aufgeführten Angaben zu machen.
|
(1) |
Für zur Lebensmittelerzeugung genutzte Tiere muss die Risikobewertung Folgendes umfassen:
|
|
(2) |
Bei Heimtieren ist das Risiko für die menschliche oder öffentliche Gesundheit wie folgt zu prüfen:
|
|
(3) |
Resistenzen in der Umwelt müssen angegangen werden. |
II.3A5. Anwendersicherheit
In diesem Abschnitt ist eine Bewertung der in Teil II.3A bis II.3A4 festgestellten Wirkungen aufzunehmen, und diese Wirkungen sind mit Art und Umfang der menschlichen Exposition gegenüber dem Arzneimittel in Beziehung zu setzen, damit zweckdienliche Warnhinweise für die Anwender formuliert und andere Risikomanagement-Maßnahmen ergriffen werden können.
Die Anwendersicherheit ist in Übereinstimmung mit den Leitlinien des Ausschusses für Tierarzneimittel (CVMP) zu behandeln.
II.3A6. Umweltverträglichkeitsprüfung
|
(1) |
Es ist eine Umweltverträglichkeitsprüfung zur Beurteilung potenziell schädlicher Wirkungen, die sich durch die Anwendung des Tierarzneimittels für die Umwelt ergeben können, sowie zur Ermittlung der Risiken solcher Wirkungen durchzuführen. Im Rahmen der Beurteilung sind außerdem die zur Herabsetzung solcher Risiken gegebenenfalls erforderlichen Vorsichtsmaßnahmen festzustellen. |
|
(2) |
Diese Beurteilung besteht aus zwei Phasen. Die erste Beurteilungsphase ist in jedem Fall durchzuführen. Die Einzelheiten zur Beurteilung sind gemäß den von der Agentur veröffentlichten Leitlinien anzugeben. Darin ist auszuführen, wie eine potenzielle Exposition der Umwelt gegenüber diesem Arzneimittel aussehen kann und wie hoch das mit dieser Exposition verbundene Risiko ist, wobei insbesondere auf folgende Punkte einzugehen ist:
|
|
(3) |
In der zweiten Phase sind weitere Untersuchungen über das Verhalten und die Wirkungen des Arzneimittels auf besondere Ökosysteme gemäß den von der Agentur veröffentlichten Leitlinien erforderlich. Das Ausmaß der Exposition der Umwelt gegenüber dem Arzneimittel und die verfügbaren Informationen über die physikalisch/chemischen, pharmakologischen und/oder toxikologischen Eigenschaften der betreffenden Wirkstoffe einschließlich der Metaboliten bei einem erkannten Risiko, die im Verlauf der sonstigen gemäß dieser Verordnung erforderlichen Versuche und Prüfungen gewonnen wurden, sind zu berücksichtigen. |
|
(4) |
Bei Produkten, die für zur Lebensmittelerzeugung genutzte Tierarten bestimmt sind, werden persistente, bioakkumulierbare und toxische (PBT) oder sehr persistente und sehr bioakkumulierbare (vPvB) Stoffe gemäß den Kriterien in Anhang XIII der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates (6) (REACH-Verordnung) eingestuft und gemäß den von der Agentur veröffentlichten Leitlinien für die PBT- und vPvB-Bewertung von Stoffen in Tierarzneimitteln bewertet. |
II.3B. Rückstandsversuche
|
(1) |
Für diesen Punkt gelten die Begriffsbestimmungen der Verordnung (EG) Nr. 470/2009 des Rates. |
|
(2) |
Mit der Untersuchung des Abbaus von Rückständen in essbaren Geweben oder von Eiern, Milch und Honig (gegebenenfalls Wachs), die von behandelten Tieren gewonnen wurden, soll festgestellt werden, unter welchen Bedingungen und in welchem Umfang Rückstände in von behandelten Tieren stammenden Lebensmitteln bestehen bleiben. Außerdem dienen diese Untersuchungen dazu, Wartezeiten festzulegen. |
|
(3) |
Bei Tierarzneimitteln, die für zur Lebensmittelerzeugung genutzte Tiere bestimmt sind, muss aus den Rückstandsunterlagen Folgendes hervorgehen:
|
II.3B1. Identifizierung des Arzneimittels
Die bei den Versuchen verwendeten Tierarzneimittel sind anhand folgender Merkmale zu identifizieren:
|
a) |
Zusammensetzung; |
|
b) |
physikalische und chemische Versuchsergebnisse (Stärke und Reinheit) für die betreffenden Chargen; |
|
c) |
Identifizierung der Charge. |
II.3B2. Abbau von Rückständen (Stoffwechsel und Rückstandskinetik)
|
(1) |
Zweck dieser Versuche, durch die die Geschwindigkeit gemessen wird, mit der Rückstände im Zieltier nach der letzten Verabreichung des Tierarzneimittels abgebaut werden, ist die Bestimmung von Wartezeiten, die erforderlich sind, um sicherzustellen, dass in Lebensmitteln, die von behandelten Tieren stammen, keine Rückstände vorhanden sind, die eine Gefahr für den Verbraucher darstellen können. |
|
(2) |
Der aktuelle Stand der Rückstandshöchstmengen für die Bestandteile des Tierarzneimittels bei den betreffenden Zieltierarten ist zu melden. |
|
(3) |
Die Menge der vorhandenen Rückstände ist zu einer ausreichenden Anzahl von Zeitpunkten zu bestimmen, nachdem die Versuchstiere die letzte Dosis des Tierarzneimittels erhalten haben. Die Studien an Säugetieren und Vögeln sind gemäß VICH GL48 und anderen einschlägigen Leitlinien durchzuführen. Rückstandsuntersuchungen in Honig sind gemäß VICH GL56 und Untersuchungen über den Rückstandsabbau von im Wasser lebenden Tierarten gemäß VICH GL57 durchzuführen. |
|
(4) |
Auf der Grundlage der Bewertung ist die Begründung für die vorgeschlagene Wartezeit zu behandeln. |
II.3B3. Methoden zur Rückstandsanalyse
Die Studie(n) über den Abbau von Rückständen, das/die Analyseverfahren und dessen/deren Validierung sind gemäß VICH GL49 durchzuführen.
Die Analysemethode trägt dem wissenschaftlichen und technischen Kenntnisstand zum Zeitpunkt der Antragstellung Rechnung.
II.4. Teil 4: Unterlagen zur Wirksamkeit (vorklinische Studien und klinische Prüfungen)
II.4A. Vorklinische Studien
Vorklinische Studien dienen der Untersuchung der Unbedenklichkeit und Wirksamkeit des Produkts bei der Zieltierart und sind erforderlich, um die pharmakologische Aktivität, die pharmakokinetischen Eigenschaften, die Dosis und das Dosierungsintervall, die (etwaigen) Resistenzen und die Verträglichkeit des Produkts bei der Zieltierart zu ermitteln.
II.4A1. Pharmakologie
II.4A.1.1. Pharmacodynamics
|
(1) |
Die pharmakodynamischen Wirkungen der im Tierarzneimittel enthaltenen Wirkstoffe sind zu charakterisieren. |
|
(2) |
Die Wirkungsweise und die pharmakologischen Auswirkungen, auf denen die empfohlene praktische Anwendung basiert, wird angemessen beschrieben, was auch für die (etwaigen) Nebenwirkungen gilt. Generell sind die Wirkungen auf die wichtigsten Körperfunktionen zu untersuchen. Die Ergebnisse sind quantitativ darzustellen (beispielsweise durch Dosis-Wirkungs-Kurven und/oder Zeit-Wirkungs-Kurven usw.) und, soweit möglich, im Vergleich zu einem Stoff, dessen Wirkung gut bekannt ist (wird für einen Stoff eine stärkere Wirksamkeit als beim bekannten Stoff behauptet, ist der Unterschied nachzuweisen und aufzuzeigen, dass er statistisch signifikant ist). |
|
(3) |
Jeder Einfluss der anderen Eigenschaften des Arzneimittels (wie etwa der Verabreichungsweg oder die Formulierung) auf die pharmakologische Aktivität des Wirkstoffes muss untersucht werden. |
|
(4) |
Sofern es sich bei den Versuchsmethoden nicht um Standardverfahren handelt, sind sie so genau zu beschreiben, dass sie reproduzierbar sind, und ihre Validierung nachgewiesen wird. Die Versuchsergebnisse sind klar darzulegen und das Ergebnis etwaiger statistischer Vergleiche ist zu unterbreiten. |
|
(5) |
Sofern keine stichhaltigen Gegenargumente vorgelegt werden können, ist jede quantitative Änderung von Reaktionen aufgrund wiederholter Verabreichung des Stoffes ebenfalls zu untersuchen. |
II.4A.1.2. Pharmakokinetik
|
(1) |
Im Rahmen einer Beurteilung der klinischen Unbedenklichkeit und Wirksamkeit des Tierarzneimittels bei der Zieltierart sind Angaben über die grundlegenden pharmakokinetischen Abläufe bei einem Wirkstoff erforderlich, insbesondere wenn es sich um einen neuen Stoff oder eine neue Formulierung handelt. |
|
(2) |
Die Zielsetzung pharmakokinetischer Studien bei der Zieltierart kann in folgende vier Kernbereiche unterteilt werden:
|
|
(3) |
Bei der Zieltierart sind pharmakokinetische Studien in der Regel als Ergänzung der pharmakodynamischen Studien für eine fundierte Festlegung von sicheren und wirksamen Dosierungsschemata (Verabreichungsweg und -ort, Dosis, Dosierungsintervalle, Anzahl der Verabreichungen usw.) erforderlich. Zusätzliche pharmakokinetische Studien können erforderlich werden, um Dosierungsschemata im Einklang mit bestimmten Populationsvariablen festlegen zu können. |
|
(4) |
Wurden pharmakokinetische Studien gemäß Teil 3 des Dossiers eingereicht, können Querverweise auf diese Studien aufgenommen werden. Für fixe Kombinationen wird auf Abschnitt IV verwiesen. |
II.4A2. Resistenzentwicklung und damit verbundenes Risiko bei Tieren
|
(1) |
Für betroffene Tierarzneimittel (z. B. antimikrobielle Wirkstoffe, Antiparasitika) sind Angaben über derzeitige Resistenzen (falls zutreffend) und über das potenzielle Auftreten von Resistenzen von klinischer Relevanz für die beanspruchte Indikation bei der Zieltierart vorzulegen. Soweit möglich, sind Informationen über die Resistenzmechanismen, die molekulargenetische Grundlage der Resistenzen und die Rate der Übertragung von Resistenzdeterminanten vorzulegen. Soweit relevant, müssen Informationen zu Ko- und Kreuzresistenzen vorgelegt werden. Der Antragsteller muss Maßnahmen vorschlagen, durch die sich die Resistenzentwicklung bei Organismen von klinischer Relevanz für den Verwendungszweck des Tierarzneimittels begrenzen lässt. |
|
(2) |
Resistenzen, die ein Risiko für den Menschen darstellen können, sind gemäß Teil II.3A4 Punkt 3 zu behandeln. Gegebenenfalls sind Querverweise zu den in Teil II.3A4 Punkt 3 aufgeführten Angaben zu machen. |
II.4A3. Dosisermittlung und -bestätigung
Es sind geeignete Daten vorzulegen, um die vorgeschlagene Dosis, das Dosierungsintervall, die Dauer der Behandlung und ein etwaiges Wiederbehandlungsintervall zu begründen.
Für Studien, die unter Feldbedingungen durchgeführt werden, sind sachdienliche Informationen gemäß Teil II.4B vorzulegen, außer bei Vorliegen hinreichender Gründe.
II.4A4. Verträglichkeit bei den Zieltierarten
Die lokale und systemische Verträglichkeit des Tierarzneimittels ist bei den Zieltierarten zu untersuchen. Zweck der Unbedenklichkeitsversuche für Zieltierarten ist es, Anzeichen von Unverträglichkeit zu beschreiben und einen angemessenen Sicherheitsspielraum bei Anwendung der empfohlenen Verabreichungswege aufzuzeigen. Dies lässt sich durch Erhöhung der Dosis und/oder durch Verlängerung der Behandlungsdauer erreichen. Der Studienbericht muss detaillierte Angaben zu allen erwarteten pharmakologischen Wirkungen und allen Nebenwirkungen enthalten. Die Durchführung von Unbedenklichkeitsversuchen für Zieltierarten erfolgt in Übereinstimmung mit den internationalen Leitlinien der Internationalen Konferenz zur Harmonisierung der technischen Anforderungen an die Zulassung von Tierarzneimitteln (VICH) und den einschlägigen von der Agentur veröffentlichten Leitlinien. Andere vorklinische Studien, einschließlich Studien gemäß Teil 3, und klinische Prüfungen sowie einschlägige Informationen aus der veröffentlichten Literatur können ebenfalls Informationen zur Sicherheit bei der Zieltierart liefern. An der Zieltierart durchgeführte Studien zur Entwicklungstoxizität sind hier aufzunehmen, und eine Zusammenfassung ist in Teil 3 des Dossiers zu geben.
II.4B. Klinische Prüfung(en)
II.4B1. Allgemeine Grundsätze
|
(1) |
Klinische Prüfungen sind unter angemessener Berücksichtigung der internationalen Leitlinien der VICH zur guten klinischen Praxis und der von der Agentur veröffentlichten einschlägigen Leitlinien zu planen, durchzuführen und zu melden. Daten aus klinischen Prüfungen, die außerhalb der Union durchgeführt wurden, dürfen nur dann zur Bewertung eines Zulassungsantrags herangezogen werden, wenn diese Daten ausreichend repräsentativ für die Situation in der EU sind. |
|
(2) |
Experimentelle Daten, wie z. B. Sondierungs-/Pilotversuche, oder Ergebnisse aus nichtexperimentellen Ansätzen müssen durch klinische Prüfungen bestätigt werden; anderenfalls ist dies zu begründen. |
|
(3) |
Zweck der klinischen Prüfung ist es, unter Feldbedingungen die Sicherheit für die Zieltierart und die Wirksamkeit eines Tierarzneimittels unter normalen Bedingungen der Tierhaltung und/oder im Rahmen einer normalen tierärztlichen Praxis zu untersuchen. Sie müssen die Wirkung des Tierarzneimittels nach Verabreichung an die vorgesehene Zieltierart unter Verwendung des vorgeschlagenen Dosierungsschemas und des/der vorgeschlagenen Verabreichungsweges/-wege nachweisen. Der Aufbau der Prüfung zielt darauf ab, Indikationen zu erhärten und Gegenanzeigen je nach Tierart, Alter, Rasse und Geschlecht sowie die Anweisungen zum Gebrauch des Tierarzneimittels und mögliche Nebenwirkungen zu spezifizieren. |
|
(4) |
Alle veterinärklinischen Prüfungen sind entsprechend einem detaillierten Prüfplan durchzuführen. |
|
(5) |
Bei Formulierungen, die zur Anwendung in veterinärklinischen Prüfungen in der Union bestimmt sind, ist der Hinweis ‚Nur für veterinärklinische Prüfungen‘ deutlich sichtbar und unlöschbar auf der Etikettierung anzubringen. |
|
(6) |
Von anderweitig begründeten Ausnahmen abgesehen, sind klinische Prüfungen mit Kontrolltieren (kontrollierte klinische Prüfungen) durchzuführen. Die Ergebnisse, die zur Wirksamkeit des neuen Produkts erzielt wurden, sollten mit jenen bei Zieltierarten verglichen werden, die ein in der Union zugelassenes Tierarzneimittel, das einen annehmbaren Grad an Wirksamkeit gezeigt hat und für die vorgeschlagenen Indikationen bei derselben Zieltierart zugelassen ist, oder ein Placebo oder gar keine Behandlung erhalten haben. Alle erzielten Ergebnisse, ob positiv oder negativ, sind zu melden. |
|
(7) |
Bei Entwurf des Prüfplans, Analyse und Bewertung der klinischen Prüfungen sind feststehende statistische Grundsätze in Übereinstimmung mit den von der Agentur veröffentlichten einschlägigen Leitlinien zu befolgen, sofern nicht anders begründet. |
II.4B2. Dokumentation
II.4B2.1. Ergebnisse der vorklinischen Studien
Sofern möglich, sind Angaben zu den Ergebnissen folgender Versuche zu machen:
|
a) |
Versuche zum Nachweis pharmakologischer Aktivität, einschließlich Versuchen zum Nachweis der pharmakodynamischen Mechanismen, die der therapeutischen Wirkung zugrunde liegen, und Versuchen zum Nachweis des pharmakokinetischen Hauptprofils; |
|
b) |
Versuche und Untersuchungen zu Resistenzen, falls zutreffend; |
|
c) |
Versuche zum Nachweis der Sicherheit für die Zieltiere; |
|
d) |
Versuche zur Bestimmung und Bestätigung der Dosis (einschließlich Dosisintervall, Behandlungsdauer und etwaiges Wiederbehandlungsintervall). |
Sofern während der Versuche unerwartete Ergebnisse auftreten, sind diese Ergebnisse im Detail anzugeben. Werden diese Daten gar nicht oder nur teilweise angegeben, ist dies zu begründen. In allen vorklinischen Studienberichten sind folgende Angaben vorzulegen:
|
a) |
eine Zusammenfassung; |
|
b) |
ein Studienprotokoll; |
|
c) |
eine detaillierte Beschreibung der Ziele, der Konzeption und Durchführung, einschließlich angewandter Methoden, Geräte und Materialien, mit Angaben wie Tierart, Alter, Gewicht, Geschlecht, Anzahl, Rasse oder Stamm der Tiere, Identifizierung der Tiere, Dosis sowie Verabreichungsweg und -schema; |
|
d) |
eine statistische Analyse der Ergebnisse, falls zutreffend; |
|
e) |
eine objektive Erörterung der erzielten Ergebnisse mit Schlussfolgerungen zu Unbedenklichkeit und Wirksamkeit des Tierarzneimittels für die Zieltierart. |
II.4B2.2. Ergebnisse der klinischen Prüfungen
Alle Angaben sind von jedem Versuchsleiter bei individueller Behandlung auf einzelnen Datenblättern und bei Behandlung von Tiergruppen auf kollektiven Datenblättern festzuhalten.
Der Zulassungsinhaber des Tierarzneimittels muss alle notwendigen Vorkehrungen treffen, um sicherzustellen, dass die Originalunterlagen, die die Grundlage der vorgelegten Daten darstellen, mindestens fünf Jahre nach Ablauf der Zulassung aufbewahrt werden.
Für jede klinische Prüfung müssen die klinischen Beobachtungen in einer Gesamtschau der Versuche und ihrer Ergebnisse unter besonderer Berücksichtigung folgender Angaben zusammengefasst werden:
|
a) |
Anzahl der Kontroll- und Versuchstiere, die entweder individuell oder kollektiv behandelt wurden, mit einer Aufschlüsselung in Bezug auf Art, Rasse oder Stamm, Alter und Geschlecht; |
|
b) |
Anzahl der vorzeitig aus den Prüfungen herausgenommenen Tiere mit entsprechender Begründung; |
|
c) |
bei Kontrolltieren die Angabe, ob sie:
|
|
d) |
Häufigkeit des Auftretens der beobachteten Nebenwirkungen; |
|
e) |
gegebenenfalls Beobachtungen zur Auswirkung auf die Nutzleistung der Tiere; |
|
f) |
ausführliche Angaben zu Versuchstieren, die wegen ihres Alters sowie der Zucht- oder Fütterungsmethoden oder angesichts ihres Verwendungszwecks größeren Risiken ausgesetzt sein können, oder zu Tieren, deren physiologischer oder pathologischer Zustand besondere Aufmerksamkeit erfordert; |
|
g) |
eine statistische Auswertung der Ergebnisse. |
Der Hauptversuchsleiter muss allgemeine Schlussfolgerungen über die Wirksamkeit und Unbedenklichkeit des Tierarzneimittels für die Zieltierart unter den vorgeschlagenen Anwendungsbedingungen und insbesondere unter Berücksichtigung aller Informationen zu Indikationen und Gegenanzeigen, Dosierung und durchschnittlicher Behandlungsdauer sowie gegebenenfalls aller beobachteten Wechselwirkungen mit anderen Arzneimitteln oder Zusatzstoffen in Futtermitteln sowie aller während der Behandlung zu treffenden besonderen Vorkehrungen und der möglicherweise festgestellten klinischen Anzeichen einer Überdosis ziehen.
ABSCHNITT III
ANFORDERUNGEN FÜR BIOLOGISCHE TIERARZNEIMITTEL
Unbeschadet der besonderen Anforderungen des Unionsrechts über die Eindämmung und Bekämpfung bestimmter infektiöser Tierkrankheiten gelten für biologische Tierarzneimittel die nachstehenden Anforderungen, es sei denn die Arzneimittel sind, wie in Abschnitt IV und V und in den einschlägigen Leitlinien festgelegt, für die Anwendung bei gewissen Arten und bestimmten Indikationen bestimmt.
ABSCHNITT IIIa
ANFORDERUNGEN FÜR NICHT IMMUNOLOGISCHE BIOLOGISCHE TIERARZNEIMITTEL
Die nachstehenden Anforderungen gelten für biologische Tierarzneimittel gemäß der in Artikel 4 Absatz 6 enthaltenen Definition, bei denen es sich nicht um Produkte handelt, die in Artikel 4 Absatz 5 definiert sind, bzw. sofern in Abschnitt IV nicht anders vorgesehen.
Hinsichtlich der Einhaltung der in diesem Abschnitt festgelegten Anforderungen ist Flexibilität zulässig, doch müssen Abweichungen von den Anforderungen dieses Anhangs wissenschaftlich begründet sein und auf spezifischen Eigenschaften des biologischen Produkts beruhen. Für bestimmte Stoffe können je nach Art des Produkts zusätzlich zu den in diesem Abschnitt aufgeführten Anforderungen weitere Sicherheitsdaten erforderlich sein.
IIIa.1. Teil 1: Zusammenfassung der Unterlagen
Es wird auf Abschnitt I verwiesen.
IIIa.2. Teil 2: Qualitätsbezogene Unterlagen (physikalisch-chemische, biologische oder mikrobiologische Informationen)
IIIa.2A. Produktbeschreibung
IIIa.2A1. Qualitative und quantitative Zusammensetzung
|
(1) |
Die qualitative und quantitative Zusammensetzung des Tierarzneimittels muss angegeben sein. Dieser Abschnitt enthält Informationen über:
|
|
(2) |
Was die quantitative Zusammensetzung aller Wirkstoffe und Arzneiträgerstoffe der Tierarzneimittel betrifft, so ist je nach der Darreichungsform für jeden Wirkstoff oder Arzneiträgerstoff die Masse oder die Zahl der Einheiten biologischer Aktivität je Einnahme-, Masse- oder Volumeneinheit anzugeben. |
|
(3) |
Wenn möglich, ist die biologische Aktivität je Masse- oder Volumeneinheit anzugeben. Sofern eine internationale Einheit biologischer Aktivität festgelegt wurde, ist diese anzuwenden; anderenfalls ist dies zu begründen. Falls keine internationale Einheit festgelegt wurde, sind die Einheiten der biologischen Aktivität unter Verwendung der Einheiten des Europäischen Arzneibuchs so auszudrücken, dass sie eindeutig Aufschluss über die Wirksamkeit der Stoffe geben. |
|
(4) |
Für die gebräuchlichen Bezeichnungen zur Beschreibung der Bestandteile eines biologischen Tierarzneimittels ist unbeschadet der in Artikel 8 vorgesehenen Angaben Folgendes maßgeblich:
|
IIIa.2A2. Produktentwicklung
Es ist eine Erklärung abzugeben, die unter anderem Folgendes umfasst:
|
a) |
die Auswahl der Zusammensetzung und die Auswahl der Bestandteile, insbesondere hinsichtlich ihrer jeweiligen Funktion und Konzentration; |
|
b) |
die Aufnahme eines Konservierungsmittels in die Zusammensetzung ist zu begründen; |
|
c) |
die Primärverpackung und die Eignung des Behältnisses und seines Verschlusssystems für Lagerung und Verwendung des Fertigprodukts. Eine Beschreibung der Wechselwirkung von Fertigprodukt und Primärverpackung ist in allen Fällen vorzulegen, in denen ein solches Risiko denkbar ist, insbesondere, wenn es sich um injizierbare Präparate handelt; |
|
d) |
die mikrobiologischen Merkmale (mikrobiologische Reinheit und antimikrobielle Eigenschaften) und die Anwendungshinweise; |
|
e) |
die mögliche weitere Verpackung, gegebenenfalls eine äußere Umhüllung; |
|
f) |
die vorgeschlagenen Packungsgrößen in Bezug auf den vorgeschlagenen Verabreichungsweg, die Posologie und die Zieltierarten; |
|
g) |
etwaige Zuschläge in der Formulierung, um die Mindestwirksamkeit bei Ablauf der Haltbarkeit zu gewährleisten, mit Begründung; |
|
h) |
die Auswahl des Herstellungsprozesses des Wirkstoffs und des Fertigprodukts; |
|
i) |
Unterschiede zwischen den Herstellungsprozessen, die zur Herstellung der in den klinischen Prüfungen verwendeten Chargen verwendet werden, und dem im Zulassungsantrag beschriebenen Prozess sind zu erörtern; |
|
j) |
wenn dem Fertigprodukt eine Dosiervorrichtung beigefügt ist, muss die Genauigkeit der Dosierung(en) nachgewiesen werden; |
|
k) |
wenn ein Begleittest für das Fertigprodukt empfohlen wird (z. B. ein Diagnoseversuch), müssen einschlägige Informationen über den Test angegeben werden. |
|
l) |
Diese Erläuterungen sind durch wissenschaftliche Daten über die Produktentwicklung zu erhärten. |
IIIa.2A3. Charakterisierung
IIIa.2A3.1. Erläuterung der Struktur und anderer Merkmale
|
(1) |
Die Charakterisierung eines biotechnologischen oder biologischen Stoffes (die die Bestimmung der physikalisch-chemischen Eigenschaften, der biologischen Wirkung, der immunochemischen Eigenschaften, der Reinheit und der Verunreinigungen einschließt) mit geeigneten Techniken ist notwendig, um eine geeignete Spezifikation erstellen zu können. Eine ausschließliche Bezugnahme auf Literaturdaten ist nicht akzeptabel, es sei denn, dies ist durch Vorwissen von ähnlichen Molekülen für Änderungen, bei denen keine Sicherheitsbedenken bestehen, gerechtfertigt. Eine angemessene Charakterisierung muss in der Entwicklungsphase und, falls erforderlich, nach wesentlichen Prozessänderungen durchgeführt werden. |
|
(2) |
Es sind alle verfügbaren einschlägigen Informationen über die Primär-, Sekundär- und übergeordnete Struktur, einschließlich posttranslationaler (z. B. Glykoformen) und anderer Änderungen des Wirkstoffs, anzugeben. |
|
(3) |
Es sind Angaben zur biologischen Wirkung (d. h. zur spezifischen Fähigkeit oder Kapazität eines Produkts, eine definierte biologische Wirkung zu erzielen) zu machen. In der Regel muss die biologische Wirkung mit einer geeigneten, zuverlässigen und qualifizierten Methode bestimmt oder bewertet werden. Das Fehlen einer solchen Bestimmung ist zu begründen. Es ist bekannt, dass der Umfang der Charakterisierungsdaten während der Entwicklung zunehmen wird. |
|
(4) |
Die Gründe für die Auswahl der zur Charakterisierung verwendeten Methoden sind anzugeben und ihre Eignung ist zu begründen. |
IIIa.2A3.2. Verunreinigungen
|
(1) |
Es sind prozessbedingte Verunreinigungen (z. B. Proteine und DNA der Wirtszelle, Rückstände von Mitteln, auslaugbare Säulenbestandteile) und produktbedingte Verunreinigungen (z. B. Vorstufen, gespaltene Formen, Abbauprodukte, Aggregate) zu berücksichtigen. Es sind quantitative Angaben zu Verunreinigungen, einschließlich der Höchstmenge für die höchste Dosis, zu machen. Bei bestimmten prozessbedingten Verunreinigungen (z. B. Antischaummittel) kann eine Schätzung der Clearance gerechtfertigt sein. |
|
(2) |
Falls für bestimmte Verunreinigungen nur qualitative Daten vorgelegt werden, ist dies zu begründen. |
IIIa.2B. Angaben über die Herstellungsweise
|
(1) |
Die Angaben über die Herstellungsweise, die dem Zulassungsantrag gemäß Artikel 8 beizufügen sind, sind so abzufassen, dass sie eine angemessene Beschreibung der Art der verwendeten Abläufe gestattet. |
|
(2) |
Es sind der Name, die Anschrift und die Zuständigkeiten jedes Herstellers, einschließlich der Auftragnehmer, sowie jeder vorgeschlagene Produktionsstandort bzw. jede Anlage anzugeben, die an der Herstellung, Prüfung und Chargenfreigabe beteiligt sind. |
|
(3) |
Die Beschreibung des Herstellungsprozesses umfasst mindestens:
|
|
(4) |
Beschreibung, Dokumentation und Ergebnisse der Validierungs- und/oder Bewertungsstudien sind für wichtige Verfahrensschritte oder Gehaltsbestimmungen, die im Herstellungsprozess verwendet werden, vorzulegen (z. B. Validierung des Sterilisierungsverfahren oder der aseptischen Verarbeitung oder Abfüllung), und die Validierung des gesamten Produktionsprozesses ist durch Vorlage der Ergebnisse von drei aufeinanderfolgenden Chargen, die nach der beschriebenen Methode hergestellt wurden, nachzuweisen. |
IIIa.2C. Herstellung und Kontrolle der Ausgangsstoffe
|
(1) |
‚Ausgangsstoffe‘ im Sinne dieses Teils sind alle Bestandteile, einschließlich Wirkstoffen, die zur Herstellung des biologischen Tierarzneimittels verwendet werden. Zur Herstellung der Wirkstoffe verwendete Kulturmedien gelten als ein einziger Ausgangsstoff. |
|
(2) |
Die qualitative und quantitative Zusammensetzung muss angegeben werden, falls die Behörden der Auffassung sind, dass diese Informationen wichtig für die Qualität des Fertigprodukts und etwaige dadurch bedingte Risiken sind. |
|
(3) |
Wird für die Zubereitung dieser Kulturmedien Material tierischen Ursprungs verwendet, müssen auch die Tierart und das verwendete Gewebe daraus hervorgehen und die Übereinstimmung mit den einschlägigen Monografien, einschließlich der allgemeinen Monografien und allgemeinen Kapitel des Europäischen Arzneibuchs, ist nachzuweisen. |
|
(4) |
Der Antragsteller hat Unterlagen vorzulegen, aus denen hervorgeht, dass die Ausgangsstoffe, einschließlich Saatmaterial, Zellkulturen, Serumchargen und sonstigen Materials von Tierarten, die TSE übertragen könnten, sowie die Herstellung des Tierarzneimittels den Anforderungen der Leitlinien für die Minimierung des Risikos der Übertragung von Erregern der spongiformen Enzephalopathie tierischen Ursprungs durch Human- und Tierarzneimittel sowie den Anforderungen der entsprechenden Monografie des Europäischen Arzneibuchs entsprechen. |
|
(5) |
Dieser Nachweis kann durch Eignungszertifikate erbracht werden, die von der Europäischen Direktion für die Arzneimittelqualität für die einschlägige Monografie des Europäischen Arzneibuchs ausgestellt werden. |
|
(6) |
Das Dossier muss die Spezifikationen sowie Informationen über die Qualitätskontrollprüfungen, die für alle Chargen von Ausgangsstoffen durchzuführen sind, und die Ergebnisse für eine Charge aller verwendeten Bestandteile umfassen und ist gemäß den folgenden Bestimmungen vorzulegen. |
|
(7) |
Für die Ausgangsstoffe sind Analysezertifikate zum Nachweis der Einhaltung der festgelegten Spezifikation vorzulegen. |
|
(8) |
Die Farbstoffe müssen in allen Fällen den Anforderungen der Richtlinie 2009/35/EG genügen. |
|
(9) |
Die Verwendung von Antibiotika bei der Herstellung und von Konservierungsmitteln muss dem Europäischen Arzneibuch entsprechen. |
|
(10) |
Bei neuartigen Arzneiträgerstoffen, die erstmalig in der Union in einem Tierarzneimittel eingesetzt werden, oder bei Arzneiträgerstoffen, bei denen dies durch eine neue Art der Anwendung geschieht, sind umfassende Angaben zur Herstellung, zur Charakterisierung und zu den Kontrollen zu machen, wobei Querverweise sowohl auf die klinischen als auch auf die nichtklinischen Daten zur Unbedenklichkeit zu machen sind. Für Farbstoffe werden die Konformitätserklärungen in Teil II.2C2 Punkte 3 und 4 als ausreichend angesehen. |
IIIa.2C1. In Arzneibüchern aufgeführte Ausgangsstoffe
|
(1) |
Die Monografien des Europäischen Arzneibuchs gelten für alle darin aufgeführten Ausgangsstoffe; anderenfalls ist dies angemessen zu begründen. |
|
(2) |
Bei allen anderen Stoffen kann jeder Mitgliedstaat verlangen, dass bei der in seinem Hoheitsgebiet erfolgenden Herstellung die Vorschriften seines Arzneibuchs eingehalten werden. |
|
(3) |
Die Beschreibung der Analysemethoden kann durch eine detaillierte Bezugnahme auf das betreffende Arzneibuch ersetzt werden. |
|
(4) |
Die bei jeder einzelnen Charge der Ausgangsstoffe durchzuführenden Routineprüfungen entsprechen den Angaben im Zulassungsantrag. Werden andere als die im Arzneibuch angegebenen Prüfungen durchgeführt, so ist der Nachweis zu erbringen, dass die Ausgangsstoffe den Qualitätsanforderungen dieses Arzneibuchs entsprechen. |
|
(5) |
Die zuständigen Behörden können vom Antragsteller geeignetere Spezifikationen verlangen, wenn eine Spezifikation oder sonstige Bestimmungen einer Monografie des Europäischen Arzneibuchs oder des Arzneibuchs eines Mitgliedstaats unter Umständen nicht genügen, um die Qualität der Stoffe zu gewährleisten. Die für das betreffende Arzneibuch zuständigen Behörden werden auf den vermuteten Mangel hingewiesen. |
IIIa.2C2. Nicht in einem Arzneibuch aufgeführte Ausgangsstoffe
IIIa.2C2.1. Ausgangsstoffe biologischer Herkunft
|
(1) |
Sofern Ausgangsstoffe wie Mikroorganismen, Gewebe pflanzlichen oder tierischen Ursprungs, Zellen oder Flüssigkeiten (einschließlich Blut) menschlichen oder tierischen Ursprungs oder biotechnologische Zellstrukturen für die Herstellung von Tierarzneimitteln verwendet werden, sind Herkunft (auch die geografische Herkunft) und Entstehung der Ausgangsstoffe zu beschreiben und durch Unterlagen zu belegen. Es sind die Herkunft, der allgemeine Gesundheitszustand und der immunologische Status der für die Herstellung verwendeten Tiere anzugeben und das Ausgangsmaterial muss aus genau definierten Bezugsquellen stammen. |
|
(2) |
Die Abwesenheit von Fremderregern (Bakterien, Mykoplasmen, Pilzen und Viren) ist gemäß dem Europäischen Arzneibuch für Saatmaterial, einschließlich Zellkulturen und Serumpools, und, soweit möglich, für das Ausgangsmaterial, aus dem sie gewonnen wurden, nachzuweisen. |
|
(3) |
Angaben sind zu allen Stoffen biologischer Herkunft zu machen, die in einer der Herstellungsstufen verwendet werden. Die Angaben umfassen den Herstellungsvorgang, die Reinigungs-/Inaktivierungsverfahren sowie deren Validierung und alle prozessbegleitenden Kontrollverfahren, durch die die Qualität, Unbedenklichkeit und gleichbleibende Qualität des Fertigprodukts sichergestellt werden soll, sowie Einzelheiten zu allen Kontaminationsprüfungen, die an jeder Charge des Stoffes durchgeführt wurden. Etwaige besondere Vorsichtsmaßnahmen, die bei der Lagerung des Ausgangsstoffs erforderlich sind, und, falls erforderlich, dessen Haltbarkeit sind anzugeben. |
|
(4) |
Sofern Ausgangsstoffe tierischen oder menschlichen Ursprungs verwendet werden, sind die Maßnahmen zu beschreiben, die getroffen werden, um sicherzustellen, dass sie frei von Fremderregern sind. Wird das Vorhandensein von Fremderregern nachgewiesen oder vermutet, ist das entsprechende Material zu beseitigen oder so zu verarbeiten, dass das Risiko des Vorhandenseins mit einer validierten Behandlung verringert wird. Wird nach der Verarbeitung das Vorhandensein von Fremderregern nachgewiesen oder vermutet, ist das entsprechende Material nur unter der Voraussetzung zu verwenden, dass durch die weitere Verarbeitung des Produkts ihre Beseitigung und/oder Inaktivierung sichergestellt ist; die Beseitigung und/oder Inaktivierung solcher Fremderreger ist nachzuweisen. |
|
(5) |
Beim Einsatz von Zellkulturen ist nachzuweisen, dass die Zelleigenschaften bis zu der für die Produktion verwendeten Passagenzahl unverändert geblieben sind. |
|
(6) |
Für gentechnisch veränderte Ausgangsstoffe müssen diese Angaben Einzelheiten enthalten, wie die Beschreibung der Ausgangszellen oder -stämme, den Aufbau des Expressionsvektors (Name, Herkunft, Funktion des Replikons, Promotor, Enhancer und andere Regulatoren), Kontrolle der effektiv eingefügten DNA- oder RNA-Sequenz, oligonukleotide Plasmidvektorsequenzen von Zellen, zur Kotransfektion verwendetes Plasmid, hinzugefügte oder entfernte Gene, biologische Eigenschaften des fertigen Konstrukts und der exprimierten Gene, Vervielfältigungszahl und genetische Stabilität. |
|
(7) |
Bei Tierarzneimitteln, die genetisch veränderte Organismen (GVO) enthalten oder aus solchen bestehen, sind dem qualitativen Teil des Zulassungsantrags die gemäß Richtlinie 2001/18/EG vorgeschriebenen Unterlagen beizufügen. |
|
(8) |
Sofern erforderlich, sind Proben der biologischen Ausgangsstoffe oder der in den Prüfverfahren verwendeten Reagenzien vorzulegen, sodass die zuständige Behörde Kontrollprüfungen durchführen lassen kann. |
IIIa.2C2.2. Ausgangsstoffe nicht biologischer Herkunft
|
(1) |
Die Beschreibung muss in Form einer Monografie unter folgenden Titeln erfolgen:
|
IIIa.2D. Kontrollen während des Herstellungsprozesses
|
(1) |
Das Dossier muss Angaben zu den Kontrollen während der Herstellung enthalten, die an Zwischenherstellungsphasen durchgeführt werden, um die Konstanz des Herstellungsverfahrens und die gleichbleibende Qualität des Fertigprodukts festzustellen. Für jede Kontrolle sind Spezifikationen festzulegen und die Analysemethoden zu beschreiben. Eine Validierung der Kontrollen ist vorzusehen, sofern nicht anders begründet. |
|
(2) |
In der Spezifikation für die Wirkstoffcharge(n) sind Akzeptanzkriterien zusammen mit den Kontrollen festzulegen, die zur ausreichenden Kontrolle der Wirkstoffqualität verwendet werden. Eine Kontrolle auf biologische Aktivität ist vorzusehen, sofern nicht anders begründet. Für die Verunreinigungen sind unter Berücksichtigung von Sicherheitsaspekten Obergrenzen festzulegen. Die mikrobiologische Qualität des Wirkstoffs muss angegeben werden. Die Abwesenheit von Fremderregern (Bakterien, Mykoplasmen, Pilzen und Viren) ist gemäß dem Europäischen Arzneibuch nachzuweisen. |
|
(3) |
Gemäß der Richtlinie 2010/63/EU und dem Europäischen Übereinkommen zum Schutz der für Versuche und andere wissenschaftliche Zwecke verwendeten Wirbeltiere sind die Versuche so durchzuführen, dass möglichst wenige Tiere verwendet und möglichst wenig Schmerzen, Leiden, Ängste oder dauerhafte Schäden verursacht werden. Falls verfügbar, ist ein alternativer In-vitro-Test zu verwenden, wenn dies zum Ersatz oder zur Verringerung des Einsatzes von Tieren oder zur Verringerung des Leidens führt. |
IIIa.2E. Kontrollen am Fertigprodukt
IIIa.2E1 Spezifikation des Fertigprodukts
Bei allen Kontrollen muss die Beschreibung der Verfahren zur Analyse des Fertigprodukts hinreichend detailliert für eine Qualitätsbeurteilung sein.
Sofern Testverfahren und Grenzwerte angewandt werden, die nicht in den Monografien des Europäischen Arzneibuchs oder andernfalls in dem Arzneibuch eines Mitgliedstaats aufgeführt sind, muss, wenn geeignete Monografien vorliegen, der Nachweis erbracht werden, dass das Fertigprodukt den Qualitätsanforderungen jenes Arzneibuchs für die betreffende Darreichungsform entsprechen würde, wenn es im Einklang mit diesen Monografien geprüft würde. In dem Zulassungsantrag sind die Prüfungen anzugeben, die an repräsentativen Proben jeder Charge des Fertigprodukts durchgeführt werden. Die Häufigkeit der Prüfungen, die am endgültigen unabgefüllten Produkt anstelle der daraus hergestellten Charge oder Chargen durchgeführt werden, ist gegebenenfalls anzugeben. Die Häufigkeit der nicht routinemäßig durchgeführten Prüfungen ist zu rechtfertigen. Akzeptanzkriterien für die Freigabe müssen angegeben und begründet werden. Die Validierung der am Fertigprodukt durchgeführten Kontrollen ist vorzulegen.
Für die Verunreinigungen sind unter Berücksichtigung von Sicherheitsaspekten Obergrenzen festzulegen.
IIIa.2E2 Methodenbeschreibungen und Validierung von Freigabeprüfungen
|
(1) |
Allgemeine Merkmale
Die Prüfung von allgemeinen Merkmalen erstreckt sich gegebenenfalls auf die Erscheinung des Fertigprodukts und physikalische, oder chemische Prüfungen, physikalische Merkmale wie pH-Wert, Osmolalität usw. Für jedes dieser Merkmale müssen vom Antragsteller in jedem einzelnen Fall Spezifikationen mit geeigneten Sicherheitskoeffizienten erstellt werden. |
|
(2) |
Identifizierung und Potenzprüfung
Erforderlichenfalls ist auch eine spezifische Prüfung zur Identifizierung des Wirkstoffs durchzuführen. Gegebenenfalls kann die Identitätsprüfung mit der Potenzprüfung kombiniert werden. Es ist eine Prüfung der Wirkung oder eine Prüfung zur Quantifizierung des Wirkstoffs oder eine Prüfung zur quantitativen Messung der Funktion (biologische/funktionelle Wirkung), die mit relevanten biologischen Eigenschaften verbunden ist, durchzuführen, um nachzuweisen, dass jede Charge die entsprechende Potenz enthält, um ihre Sicherheit und Wirksamkeit zu gewährleisten. Eine biologische Bestimmung ist erforderlich, sofern die physikalisch-chemischen Methoden nicht ausreichen, um Auskunft über die Qualität des Arzneimittels zu erhalten. Solch eine Bestimmung sollte möglichst Referenzmaterialien und statistische Analysen mit Berechnung der Sicherheitskoeffizienten umfassen. Sofern diese Versuche nicht am Fertigprodukt durchgeführt werden können, ist es möglich, dass sie in einem Zwischenstadium, möglichst gegen Ende des Herstellungsverfahrens, erfolgen. Kommt es bei der Herstellung des Fertigprodukts zu Abbauprozessen, ist der annehmbare Höchstwert unmittelbar nach der Herstellung für Einzel- und Gesamtabbauprodukte anzugeben. |
|
(3) |
Identitätsnachweis und Gehaltsbestimmung der Bestandteile des Arzneiträgerstoffs
Soweit erforderlich, sind an den Arzneiträgerstoffen zumindest Prüfungen zur Identifizierung durchzuführen. Die Bestimmung des oberen und des unteren Grenzwerts ist für Konservierungsmittel zwingend vorgeschrieben. Die Bestimmung des oberen Grenzwerts ist für alle anderen Bestandteile von Arzneiträgerstoffen, die im Verdacht stehen, zu Nebenwirkungen zu führen, zwingend vorgeschrieben. Falls zutreffend, sind Menge und Art des Adjuvans und seiner Bestandteile am Fertigprodukt zu überprüfen; anderenfalls ist dies zu begründen. |
|
(4) |
Prüfungen auf Sterilität und Reinheit
Die Abwesenheit von Fremderregern (Bakterien, Mykoplasmen, Pilzen und bakterielle Endotoxine, soweit relevant) ist gemäß dem Europäischen Arzneibuch nachzuweisen. Es sind geeignete Prüfungen zum Nachweis der Freiheit von Kontamination durch andere Stoffe entsprechend der Art des biologischen Tierarzneimittels, der Herstellungsmethode und -bedingungen durchzuführen. Werden routinemäßig weniger Prüfungen je Charge durchgeführt, als in dem einschlägigen Europäischen Arzneibuch vorgeschrieben sind, müssen diese für die Einhaltung der Monografie maßgeblich sein. Es sind Nachweise vorzulegen, dass das biologische Tierarzneimittel die Anforderungen erfüllen würde, würde es in vollem Umfang gemäß der Monografie geprüft. |
|
(5) |
Feuchtigkeitsrückstände
Jede Charge eines lyophilisierten Produkts oder einer Tablette ist auf Feuchtigkeitsrückstände zu prüfen. |
|
(6) |
Füllvolumen
Geeignete Prüfungen zum Nachweis des korrekten Füllvolumens müssen durchgeführt werden. |
IIIa.2E3. Referenzstandards oder -materialien
Es sind Informationen über den Herstellungsprozess des Referenzmaterials anzugeben. Wenn während der Produktentwicklung mehr als ein Referenzstandard für eine bestimmte Prüfung verwendet wurde, muss eine Qualifizierungshistorie vorgelegt werden, die beschreibt, wie die Beziehung zwischen den verschiedenen Normen aufrechterhalten wurde.
Falls andere Referenzzubereitungen und -standards als im Europäischen Arzneimittelbuch vorgesehen verwendet werden, sind diese anzugeben und ausführlich zu beschreiben.
IIIa.2F. Gleichbleibende Qualität der Chargen
IIIa.2F1. Wirkstoff
Damit eine gleichbleibende Qualität des Wirkstoffs von Charge zu Charge gewährleistet ist und die Übereinstimmung mit den Spezifikationen nachgewiesen wird, sind Daten von repräsentativen Chargen vorzulegen.
IIIa.2F2. Fertigprodukt
Damit eine gleichbleibende Qualität von Charge zu Charge gewährleistet ist und die Übereinstimmung mit den Spezifikationen nachgewiesen wird, ist ein vollständiges Protokoll für drei aufeinanderfolgende und für die Routineproduktion repräsentative Chargen vorzulegen.
IIIa.2G. Haltbarkeitsversuche
|
(1) |
Die Haltbarkeitsversuche umfassen die Haltbarkeit des Wirkstoffs und des Fertigprodukts, einschließlich gegebenenfalls des/der Lösungsmittel(s). Wenn der/die Wirkstoff(e) gelagert wird/werden, müssen die vorgesehenen Bedingungen und die Dauer der Lagerung auf der Grundlage von Haltbarkeitsdaten festgelegt werden; sie können entweder durch Prüfung der Wirkstoffe selbst oder durch entsprechende Prüfung des Fertigprodukts gewonnen werden. |
|
(2) |
Zu beschreiben sind die Prüfungen, die durchgeführt wurden, um die Haltbarkeitsdauer, die empfohlenen Lagerungsbedingungen und die Spezifikationen bei Ablauf der Haltbarkeitsdauer, wie vom Antragsteller vorgeschlagen, zu erhärten. Diese Versuche müssen immer Echtzeitstudien sein; sie sind an mindestens drei repräsentativen Chargen durchzuführen, die im Einklang mit dem beschriebenen Produktionsverfahren hergestellt wurden, sowie an Produkten, die im endgültigen Behältnis gelagert sind; diese Versuche umfassen biologische und physikalisch-chemische Haltbarkeitsprüfungen, die in regelmäßigen Abständen durchgeführt werden, für das Fertigprodukt bis zum Ablauf der angegebenen Haltbarkeitsdauer. |
|
(3) |
Die Schlussfolgerungen müssen die Analyseergebnisse enthalten, die die vorgeschlagene Haltbarkeitsdauer unter allen vorgeschlagenen Lagerungsbedingungen rechtfertigen. Die Ergebnisse der Haltbarkeitsstudie sind bei der Festlegung geeigneter Formulierungs- und Freigabespezifikationen zu berücksichtigen, um die Konformität des Produkts mit der angegebenen Haltbarkeitsdauer sicherzustellen. |
|
(4) |
Bei Arzneimitteln, die über das Futter verabreicht werden, müssen die Angaben bezüglich der Haltbarkeit, falls erforderlich, auch für die verschiedenen Stadien des Mischvorgangs bei Einhaltung der empfohlenen Anweisungen gemacht werden. |
|
(5) |
Muss das Fertigprodukt vor der Verabreichung rekonstituiert oder muss es in Trinkwasser verabreicht werden, sind genaue Angaben zur vorgeschlagenen Haltbarkeitsdauer für das entsprechend den Anweisungen rekonstituierte Produkt erforderlich. Zur Untermauerung der vorgeschlagenen Haltbarkeitsdauer für das rekonstituierte Produkt sind Daten vorzulegen. |
|
(6) |
Bei Multidosisbehältnissen sind gegebenenfalls Haltbarkeitsdaten vorzulegen, um die Haltbarkeitsdauer nach erstmaliger Entnahme oder Öffnung des Arzneimittels zu begründen, und es ist eine Spezifikation für das im Gebrauch befindliche Behältnis festzulegen. |
|
(7) |
Ist damit zu rechnen, dass sich bei einem Fertigprodukt Abbauprodukte bilden, so muss der Antragsteller diese Produkte mitteilen und angeben, welche Methoden für ihre Identifizierung und welche Prüfverfahren angewandt werden. |
|
(8) |
Bei kombinierten Arzneimitteln gewonnene Haltbarkeitsdaten können bei hinreichender Begründung für Derivate dienen, die eines oder mehrere der gleichen Bestandteile enthalten. |
|
(9) |
Die Wirksamkeit jeder Konservierungsart ist nachzuweisen. Angaben zur Wirksamkeit der Konservierungsmittel bei anderen, ähnlichen biologischen Tierarzneimitteln desselben Herstellers sind unter Umständen ausreichend. |
IIIa.2H. Sonstige Informationen
In das Dossier können auch Informationen über die Qualität des biologischen Tierarzneimittels aufgenommen werden, die in Teil IIIa.2 bis IIIa2G nicht erfasst sind.
IIIa.3. Teil 3: Unterlagen zur Unbedenklichkeit (Unbedenklichkeits- und Rückstandsversuche)
|
(1) |
Der Studienbericht enthält Folgendes:
|
|
(2) |
Veröffentlichte Studien können akzeptiert werden, wenn sie eine ausreichende Menge an Daten und genügend Details enthalten, um eine unabhängige Bewertung zu ermöglichen. Die Versuchsmethoden sind so genau zu beschreiben, dass sie reproduziert werden können, und der Prüfer muss ihre Validität nachweisen. Zusammenfassungen von Studien, zu denen keine detaillierten Berichte vorliegen, stellen keine vollständigen Unterlagen dar. Zur Erfüllung bestimmter Sicherheitsanforderungen kann auf die EPMAR verwiesen werden, wenn der Stoff zuvor für die Festlegung von Rückstandshöchstmengen bewertet wurde. Wenn auf die EPMAR verwiesen wird, müssen keine Studien vorgelegt werden, die bereits im Rahmen der Rückstandshöchstmengen-Bewertung bewertet wurden; es sind nur neue Studien vorzulegen, die für die Bewertung der Rückstandshöchstmengen nicht verfügbar waren. Wenn der Verabreichungsweg (z. B. für den Anwender) nicht identisch ist mit dem gemäß der Verordnung (EU) 2018/78 verwendeten Weg, können neue Studien erforderlich sein. |
IIIa.3A. Unbedenklichkeitsversuche
|
(1) |
Die Unbedenklichkeitsunterlagen müssen zur Bewertung von Folgendem angemessen sein:
|
|
(2) |
In bestimmten Fällen ist es erforderlich, die Metaboliten der Ausgangsverbindung zu prüfen, wenn es sich bei diesen um die bedenklichen Rückstände handelt. |
|
(3) |
Ein Arzneiträgerstoff, der erstmalig in einem Tierarzneimittel eingesetzt wird, oder ein Arzneiträgerstoff, bei dem dies durch ein neue Darreichungsform geschieht, ist wie ein Wirkstoff zu behandeln. |
|
(4) |
Alle in Teil IIIa.3A aufgeführten Abschnitte müssen behandelt werden. Je nach Art des Produkts sind bestimmte Abschnitte möglicherweise nicht relevant und Studien können, sofern gerechtfertigt, weggelassen werden. |
IIIa.3A1. Genaue Identifizierung des Arzneimittels und seiner Wirkstoffe:
|
a) |
internationaler Freiname (INN), |
|
b) |
IUPAC-Bezeichnung, |
|
c) |
Nummer des Chemical Abstracts Service (CAS-Nummer), |
|
d) |
therapeutische, pharmakologische und chemische Klassifizierung, |
|
e) |
Synonyme und Abkürzungen, |
|
f) |
Strukturformel, |
|
g) |
Molekularformel, |
|
h) |
Molekulargewicht, |
|
i) |
Verunreinigungsgrad, |
|
j) |
qualitative und quantitative Zusammensetzung der Verunreinigungen, |
|
k) |
Beschreibung physikalischer Eigenschaften, |
|
l) |
Löslichkeit in Wasser und organischen Lösungsmitteln, ausgedrückt in g/l mit Temperaturangabe, |
|
m) |
Lichtbrechung, optische Rotation usw., |
|
n) |
Formulierung des Arzneimittels. |
IIIa.3A2. Pharmakologie
|
(1) |
Pharmakologische Untersuchungen sind zur Klärung der Mechanismen, die die therapeutischen Wirkungen des Tierarzneimittels herbeiführen, von fundamentaler Bedeutung, weshalb an Zieltierarten und gegebenenfalls Nichtzieltierarten durchgeführte pharmakologische Untersuchungen aufgenommen werden sollten. Gegebenenfalls kann ein Querverweis auf die in Teil 4 des Dossiers eingereichten Studien erfolgen. |
|
(2) |
Pharmakologische Untersuchungen können auch zum Verständnis toxikologischer Phänomene beitragen. Wenn ein Tierarzneimittel bei Ausbleiben einer toxischen Reaktion oder bei Dosen, die so niedrig sind, dass kein toxischer Effekt auftritt, pharmakologische Wirkungen aufweist, sind Letztere bei der Bewertung der Unbedenklichkeit des Tierarzneimittels in Betracht zu ziehen. |
|
(3) |
Den Unterlagen zur Unbedenklichkeit sind in jedem Fall ausführliche Angaben über an Versuchstieren durchgeführte pharmakologische Untersuchungen sowie sämtliche einschlägigen Angaben zu den klinischen Prüfungen am Zieltier voranzuschicken. |
IIIa.3A2.1. Pharmakodynamik
Zum besseren Verständnis etwaiger Nebenwirkungen in den Tierstudien sind Informationen über den Wirkungsmechanismus der Wirkstoffe sowie über die primären und sekundären pharmakodynamischen Wirkungen vorzulegen. Detaillierte Angaben zu den pharmakodynamischen Eigenschaften, die sich auf die therapeutische Wirksamkeit beziehen, sind in Teil 4A des Dossiers zu melden.
IIIa.3A2.2. Pharmakokinetik
Es sind Daten zum Verhalten des Wirkstoffs und seiner Metaboliten bei den Versuchstieren vorzulegen, die Angaben zu Resorption, Verteilung, Metabolisierung und Ausscheidung (ADME) enthalten. Zur Ermittlung der entsprechenden Exposition sind die Daten mit den Dosis-Wirkungs-Ergebnissen der pharmakologischen und toxikologischen Studien in Beziehung zu setzen.
IIIa.3A3. Toxikologie
|
(1) |
Die Toxikologieunterlagen müssen den Leitlinien der Agentur über den allgemeinen Ansatz für Prüfungen sowie den Leitlinien für spezielle Studien entsprechen. Dieser Leitfaden enthält toxikologische Daten, die für die Feststellung der Anwendersicherheit und die Bewertung der schädlichen Auswirkungen bei Zieltieren und auf die Umwelt erforderlich sind. |
|
(2) |
Die Toxizitätsstudien sind nicht mit dem formulierten Produkt, sondern mit den Wirkstoffen durchzuführen, sofern nicht spezifisch etwas anderes vorgeschrieben ist. |
|
(3) |
Tierexperimentelle Studien sind an etablierten Stämmen von Versuchstieren durchzuführen, für die (vorzugsweise) historische Daten vorliegen. |
IIIa.3A3.1. Toxizität bei einmaliger Verabreichung
Die Prüfungen der Toxizität bei einmaliger Verabreichung können eingesetzt werden zur Prognose:
|
a) |
der möglichen Wirkungen einer akuten Überdosis bei der Zieltierart; |
|
b) |
der möglichen Wirkungen einer unbeabsichtigten Verabreichung an Menschen; |
|
c) |
der Dosen, die bei Prüfungen der Toxizität bei wiederholter Verabreichung sinnvoll verwendet werden können. |
Prüfungen der Toxizität bei einmaliger Verabreichung haben die akuten toxischen Wirkungen des Stoffs und den zeitlichen Ablauf ihres Einsetzens und Abklingens aufzuzeigen.
Die durchzuführenden Prüfungen werden im Hinblick auf Informationen zur Anwendersicherheit ausgewählt; ist z. B. mit einer erheblichen Exposition des Benutzers gegenüber dem Tierarzneimittel durch Inhalation oder Hautkontakt zu rechnen, sollten diese Verabreichungswege untersucht werden.
IIIa.3A3.2. Toxizität bei wiederholter Verabreichung
Die Prüfungen der Toxizität bei wiederholter Verabreichung haben zum Ziel, physiologische und/oder pathologische Veränderungen infolge wiederholter Verabreichung eines wirksamen Bestandteils bzw. einer Wirkstoffkombination festzustellen und die Dosierungen zu ermitteln, die für das Auftreten dieser Veränderungen verantwortlich sind.
Normalerweise ist eine Prüfung der Toxizität bei wiederholter Verabreichung an einer Versuchstierart ausreichend. Diese Untersuchung kann durch eine Untersuchung am Zieltier ersetzt werden. Häufigkeit und Weg der Verabreichung sowie die Dauer des Versuchs sind unter Berücksichtigung der vorgeschlagenen klinischen Anwendungsbedingungen und/oder Exposition der Anwender anzusetzen. Der Antragsteller muss Umfang und Dauer der Studien sowie die gewählten Dosierungen begründen.
IIIa.3A3.3. Verträglichkeit bei der Zieltierart
Alle Anzeichen von Unverträglichkeit, die bei der Zieltierart im Verlauf der üblicherweise mit der endgültigen Formulierung durchgeführten Versuche aufgetreten sind, müssen gemäß den Anforderungen in Teil IIIa.4A4 (Verträglichkeit bei den Zieltierarten) zusammenfassend angegeben werden. Die betreffenden Untersuchungen, die Dosierungen, die die Unverträglichkeit hervorgerufen haben, sowie die betreffenden Arten und Rassen sind zu beschreiben. Ferner sind alle unerwarteten physiologischen Veränderungen im Detail anzugeben. Teil 4 des Dossiers muss die vollständigen Berichte dieser Studien enthalten.
IIIa.3A3.4. Reproduktions- und Entwicklungstoxizität
|
(1) |
Studie zu den Wirkungen auf die Fortpflanzung
Für Produkte, die zur Verwendung bei Zuchttieren bestimmt sind, sind Reproduktionsstudien in Übereinstimmung mit VICH GL43 vorzulegen. Studien zur Reproduktionstoxizität an Versuchstieren werden für die Bewertung der Auswirkungen auf den Anwender nicht erwartet. |
|
(2) |
Studie zur Entwicklungstoxizität
Für die Bewertung der Wirkungen bei der Zieltierart sind Studien zur Entwicklungstoxizität nicht erforderlich für Produkte, die nur zur Verwendung bei nicht zu Zuchtzwecken gehaltenen Tieren bestimmt sind. Für andere Produkte ist eine Studie zur Entwicklungstoxizität an mindestens einer Tierart durchzuführen, die die Zieltierart sein kann. Zur Bewertung der Anwendersicherheit sind in allen Fällen, in denen eine signifikante Exposition des Anwenders zu erwarten ist, Standardtests zur Entwicklungstoxizität in Übereinstimmung mit Standardtests auf der Grundlage etablierter Leitlinien (einschließlich VICH GL32 und OECD-Prüfungen) durchzuführen. |
IIIa.3A3.5. Genotoxizität
Das genotoxische Potenzial ist zu untersuchen, um etwaige Veränderungen zu ermitteln, die ein Stoff am genetischen Material von Zellen verursachen kann; anderenfalls ist dies zu begründen. Alle Stoffe, die zum ersten Mal in einem Tierarzneimittel verwendet werden, sind auf ihre genotoxischen Eigenschaften hin zu beurteilen.
An dem/den Wirkstoff(en) ist in der Regel eine Standardbatterie von Genotoxizitätstests in Übereinstimmung mit Standardtests auf der Grundlage festgelegter Leitlinien (einschließlich VICH GL23 und OECD-Tests) durchzuführen.
IIIa.3A3.6. Kanzerogenität
Bei der Entscheidung über die Notwendigkeit einer Prüfung auf Kanzerogenität sind die Ergebnisse von Genotoxizitätsprüfungen, die Struktur-Wirkungs-Beziehungen und die Ergebnisse von Untersuchungen auf Toxizität bei wiederholter Verabreichung zu berücksichtigen, die das Potenzial für hyper-/neoplastische Veränderungen haben können.
Es sind alle bekannten artspezifischen Toxizitätsmechanismen sowie die Unterschiede im Metabolismus zwischen der in den Prüfungen verwendeten Tierart, der Zieltierart und dem Menschen zu berücksichtigen.
Die Prüfung der Kanzerogenität ist nach Standardprüfungen auf der Grundlage festgelegter Leitlinien (einschließlich VICH GL28 und OECD-Prüfungen) durchzuführen.
IIIa.3A3.7. Ausnahmen
Sofern ein Tierarzneimittel zur topischen Anwendung bestimmt ist, muss die systemische Absorption an der Zieltierart geprüft werden. Wenn nachgewiesen ist, dass die systemische Absorption unerheblich ist, kann auf die Prüfungen der Toxizität bei wiederholter Verabreichung, die Versuche zur Entwicklungstoxizität und die Kanzerogenitätsprüfungen verzichtet werden, es sei denn, dass
|
a) |
unter den festgelegten Anwendungsbedingungen ist eine orale Aufnahme des Tierarzneimittels durch das Tier zu erwarten oder |
|
b) |
unter den festgelegten Anwendungsbedingungen ist eine orale Exposition des Anwenders des Tierarzneimittels zu erwarten. |
IIIa.3A4. Sonstige Anforderungen
IIIa.3A4.1. Spezielle Untersuchungen
Bei bestimmten Stoffgruppen oder wenn die im Verlauf der Prüfungen bei wiederholter Verabreichung an Tiere beobachteten Wirkungen Veränderungen umfassen, die beispielsweise auf Immunogenität, Immunotoxizität, Neurotoxizität oder endokrine Dysfunktion hindeuten, sind weitere Prüfungen erforderlich, z. B. Sensibilisierungsstudien oder Prüfungen auf verzögerte Neurotoxizität. Je nach Art des Arzneimittels kann es erforderlich sein, zusätzliche Studien zur Beurteilung der Mechanismen durchzuführen, die der toxischen Wirkung oder dem Reizpotenzial zugrunde liegen.
Für Produkte, bei denen es zu einer Exposition von Haut und Augen kommen kann, sind Studien zur Reizung und Sensibilisierung vorzulegen. Diese Studien werden normalerweise mit der endgültigen Formulierung durchgeführt.
Bei der Konzeption solcher Untersuchungen und der Bewertung ihrer Ergebnisse sind der Stand der wissenschaftlichen Kenntnisse sowie die festgelegten Leitlinien zu berücksichtigen.
IIIa.3A4.2. Beobachtungen beim Menschen
Es sind Informationen dazu vorzulegen, ob die pharmakologisch wirksamen Stoffe des Tierarzneimittels als Arzneimittel in der Humantherapie verwendet werden. Trifft dies zu, sind alle am Menschen in veröffentlichen Studien festgestellten Wirkungen (einschließlich Nebenwirkungen) und deren Ursachen zusammenzustellen, sofern sie für die Beurteilung der Unbedenklichkeit des Tierarzneimittels von Bedeutung sein können; wenn Bestandteile der Tierarzneimittel als solche bzw. aus Sicherheitsgründen nicht mehr als Arzneimittel in der Humantherapie angewandt werden, ist dies zu begründen, sofern diese Daten öffentlich zugänglich sind.
IIIa.3A4.3. Resistenzentwicklung und damit verbundenes Risiko beim Menschen
Die in diesem Punkt genannten Datenanforderungen beziehen sich auf antibakterielle Stoffe und sind möglicherweise nicht in vollem Umfang auf andere Arten von antimikrobiellen Wirkstoffen (nämlich Virostatika, Antimykotika und Antiprotozoika); für andere als antibakterielle Stoffe, für die das Vorhandensein einer antimikrobiellen Resistenz belegt ist, können gegebenenfalls dieselben Anforderungen befolgt werden.
Es sind Daten über das mögliche Auftreten resistenter Bakterien oder Resistenzdeterminanten erforderlich, die für die menschliche Gesundheit von Bedeutung sind und mit der Verwendung von Tierarzneimitteln in Zusammenhang stehen. Dabei ist besonders wichtig, durch welchen Mechanismus sich derartige Resistenzen entwickeln und selektieren. Falls erforderlich, müssen Maßnahmen vorgeschlagen werden, durch die sich die Entwicklung einer Resistenz gegen den Verwendungszweck des Tierarzneimittels begrenzen lässt.
Sind solche Resistenzdaten für die klinische Anwendung des Produkts bei Zieltierarten relevant, ist dies gemäß Teil III.4A2 zu behandeln. Gegebenenfalls sind Querverweise zu den in Teil IIIa.4A2 aufgeführten Angaben zu machen.
|
(1) |
Für zur Lebensmittelerzeugung genutzte Tiere muss die Risikobewertung Folgendes umfassen:
|
|
(2) |
Bei Heimtieren ist das Risiko für die menschliche oder öffentliche Gesundheit wie folgt zu prüfen:
|
|
(3) |
Resistenzen in der Umwelt müssen angegangen werden. |
IIIa.3A5. Anwendersicherheit
In diesem Abschnitt zur Anwendersicherheit ist eine Bewertung der in Teil IIIa.3A bis IIIa.3A4 festgestellten Wirkungen aufzunehmen, und diese Wirkungen sind mit Art und Umfang der menschlichen Exposition gegenüber dem Arzneimittel in Beziehung zu setzen, damit zweckdienliche Warnhinweise für die Anwender formuliert und andere Risikomanagement-Maßnahmen ergriffen werden können.
Die Anwendersicherheit ist in Übereinstimmung mit den Richtlinien des CVMP zu behandeln.
IIIa.3A6. Umweltverträglichkeitsprüfung
IIIa.3A6.1. Umweltverträglichkeitsprüfung für Tierarzneimittel, die genetisch veränderte Organismen weder enthalten noch aus solchen bestehen
|
(1) |
Es ist eine Umweltverträglichkeitsprüfung zur Beurteilung potenziell schädlicher Wirkungen, die sich durch die Anwendung des Tierarzneimittels für die Umwelt ergeben können, sowie zur Ermittlung der Risiken solcher Wirkungen durchzuführen. Im Rahmen der Beurteilung sind außerdem die zur Herabsetzung solcher Risiken gegebenenfalls erforderlichen Vorsichtsmaßnahmen festzustellen. |
|
(2) |
Diese Beurteilung besteht aus zwei Phasen. Die erste Beurteilungsphase ist in jedem Fall durchzuführen. Die Einzelheiten zur Beurteilung sind gemäß den von der Agentur veröffentlichten Leitlinien anzugeben. Die potenzielle Exposition der Umwelt gegenüber dem Arzneimittel sowie das Ausmaß des mit einer solchen Exposition verbundenen Risikos sind unter Berücksichtigung folgender Elemente anzugeben:
|
|
(3) |
In der zweiten Phase sind weitere Untersuchungen über das Verhalten und die Wirkungen des Arzneimittels auf besondere Ökosysteme gemäß den von der Agentur veröffentlichten Leitlinien erforderlich. Das Ausmaß der Exposition der Umwelt gegenüber dem Arzneimittel und die verfügbaren Informationen über die physikalisch/chemischen, pharmakologischen und/oder toxikologischen Eigenschaften der betreffenden Wirkstoffe, einschließlich der Metaboliten bei einem erkannten Risiko, die im Verlauf der sonstigen gemäß dieser Verordnung erforderlichen Versuche und Prüfungen gewonnen wurden, sind zu berücksichtigen.
Bei Produkten, die für zur Lebensmittelerzeugung genutzte Tierarten bestimmt sind, werden persistente, bioakkumulierbare und toxische (PBT) oder sehr persistente und sehr bioakkumulierbare (vPvB) Stoffe gemäß den Kriterien in Anhang XIII der REACH-Verordnung eingestuft und gemäß den von der Agentur veröffentlichten Leitlinien für die PBT- und vPvB-Bewertung von Stoffen in Tierarzneimitteln bewertet. |
IIIa.3A6.2. Umweltverträglichkeitsprüfung für Tierarzneimittel, die genetisch veränderte Organismen enthalten oder aus solchen bestehen
|
(1) |
Bei einem Tierarzneimittel, das genetisch veränderte Organismen enthält oder aus solchen besteht, muss der Antrag die Unterlagen gemäß Artikel 2 und Teil C der Richtlinie 2001/18/EG umfassen. |
|
(2) |
Potenzielle nachteilige Auswirkungen auf die menschliche Gesundheit und die Umwelt, die durch den Gentransfer von GVO auf andere Organismen auftreten können oder sich aus genetischen Veränderungen ergeben, sind von Fall zu Fall genau zu bewerten. Ziel einer solchen Umweltverträglichkeitsprüfung ist es, mögliche direkte und indirekte, sofortige oder spätere schädliche Auswirkungen des GVO auf die menschliche Gesundheit und die Umwelt (einschließlich Pflanzen und Tiere) zu ermitteln und zu bewerten; sie ist nach den Grundsätzen von Anhang II der Richtlinie 2001/18/EG durchzuführen. |
IIIa.3B. Rückstandsversuche
|
(1) |
Für diesen Punkt gelten die Begriffsbestimmungen der Verordnung (EG) Nr. 470/2009 des Rates. |
|
(2) |
Mit der Untersuchung des Abbaus von Rückständen in essbaren Geweben oder von Eiern, Milch und Honig (gegebenenfalls Wachs), die von behandelten Tieren gewonnen wurden, soll festgestellt werden, unter welchen Bedingungen und in welchem Umfang Rückstände in von behandelten Tieren stammenden Lebensmitteln bestehen bleiben. Außerdem dienen diese Untersuchungen dazu, Wartezeiten festzulegen. |
|
(3) |
Bei Tierarzneimitteln, die für zur Lebensmittelerzeugung genutzte Tiere bestimmt sind, muss aus den Rückstandsunterlagen Folgendes hervorgehen:
|
IIIa.3B1. Identifizierung des Arzneimittels
Die bei den Versuchen verwendeten Tierarzneimittel sind anhand folgender Merkmale zu identifizieren:
|
a) |
Zusammensetzung; |
|
b) |
physikalische und chemische Versuchsergebnisse (Stärke und Reinheit) für die betreffenden Chargen; |
|
c) |
Identifizierung der Charge. |
IIIa.3B2. Abbau von Rückständen
|
(1) |
Zweck dieser Versuche, durch die die Geschwindigkeit gemessen wird, mit der Rückstände im Zieltier nach der letzten Verabreichung des Tierarzneimittels abgebaut werden, ist die Bestimmung von Wartezeiten, die erforderlich sind, um sicherzustellen, dass in Lebensmitteln, die von behandelten Tieren stammen, keine Rückstände vorhanden sind, die eine Gefahr für den Verbraucher darstellen könnten. |
|
(2) |
Der aktuelle Stand der Rückstandshöchstmengen für die Bestandteile des Tierarzneimittels bei den betreffenden Zieltierarten ist zu melden. |
|
(3) |
Die Menge der vorhandenen Rückstände ist zu einer ausreichenden Anzahl von Zeitpunkten zu bestimmen, nachdem die Versuchstiere die letzte Dosis des Tierarzneimittels erhalten haben. Die Studien an Säugetieren und Vögeln sind gemäß VICH GL48 und anderen einschlägigen Leitlinien durchzuführen. Rückstandsuntersuchungen bei Honig sind gemäß VICH GL56 und Untersuchungen über den Rückstandsabbau bei im Wasser lebenden Tierarten gemäß VICH GL57 durchzuführen. |
|
(4) |
Die Begründung für die vorgeschlagene Wartezeit ist auf die Bewertung zu stützen. |
IIIa.3B3. Methoden zur Rückstandsanalyse
|
(1) |
Die Studie(n) über den Abbau von Rückständen, das/die Analyseverfahren und dessen/deren Validierung sind gemäß VICH GL49 durchzuführen. |
|
(2) |
Die Eignung der vorgeschlagenen Analysemethoden ist in Bezug auf den wissenschaftlichen und technischen Kenntnisstand zum Zeitpunkt der Antragstellung zu bewerten. |
IIIa.4. Teil 4: Unterlagen zur Wirksamkeit (vorklinische Studien und klinische Prüfungen)
IIIa.4A. Vorklinische Studien
Vorklinische Studien dienen der Untersuchung der Unbedenklichkeit und Wirksamkeit des Produkts bei der Zieltierart und sind erforderlich, um die pharmakologische Aktivität, die pharmakokinetischen Eigenschaften, die Dosis und das Dosierungsintervall, die (etwaigen) Resistenzen und die Verträglichkeit des Produkts bei der Zieltierart zu ermitteln.
IIIa.4A1. Pharmakologie
IIIa.4A1.1. Pharmakodynamik
|
(1) |
Die pharmakodynamischen Wirkungen der im Tierarzneimittel enthaltenen Wirkstoffe sind zu charakterisieren. |
|
(2) |
Die Wirkungsweise und die pharmakologischen Auswirkungen, auf denen die empfohlene praktische Anwendung basiert, wird angemessen beschrieben, was auch für die (etwaigen) Nebenwirkungen gilt. Generell sind die Wirkungen auf die wichtigsten Körperfunktionen zu untersuchen. Die Ergebnisse sind quantitativ darzustellen (beispielsweise durch Dosis-Wirkungs-Kurven, Zeit-Wirkungs-Kurven usw.) und sofern möglich durch Vergleich mit einem Stoff, dessen Wirkung gut bekannt ist. Wird für einen Stoff eine stärkere Wirkung behauptet, ist der Unterschied nachzuweisen und aufzuzeigen, dass er statistisch signifikant ist. |
|
(3) |
Jeder Einfluss der anderen Eigenschaften des Arzneimittels (wie etwa der Verabreichungsweg oder die Formulierung) auf die pharmakologische Aktivität des Wirkstoffes muss untersucht werden. |
|
(4) |
Sofern es sich bei den Versuchsmethoden nicht um Standardverfahren handelt, sind sie so genau zu beschreiben, dass sie reproduzierbar sind und ihre Validierung nachgewiesen wird. Die Versuchsergebnisse sind klar darzulegen und das Ergebnis etwaiger statistischer Vergleiche ist zu unterbreiten. |
|
(5) |
Sofern keine angemessenen Gegenargumente vorgelegt werden können, ist jede quantitative Änderung von Reaktionen aufgrund wiederholter Verabreichung des Stoffes ebenfalls zu untersuchen. |
IIIa.4A1.2. Pharmakokinetik
|
(1) |
Im Rahmen einer Beurteilung der klinischen Unbedenklichkeit und Wirksamkeit des Tierarzneimittels bei der Zieltierart sind Angaben über die grundlegenden pharmakokinetischen Abläufe bei einem Wirkstoff erforderlich, insbesondere wenn es sich um einen neuen Stoff oder eine neue Formulierung handelt. |
|
(2) |
Die Zielsetzung pharmakokinetischer Studien bei der Zieltierart kann in folgende vier Kernbereiche unterteilt werden:
|
|
(3) |
Bei der Zieltierart sind pharmakokinetische Studien in der Regel als Ergänzung der pharmakodynamischen Studien für eine fundierte Festlegung von sicheren und wirksamen Dosierungsschemata (Verabreichungsweg und -ort, Dosis, Dosierungsintervalle, Anzahl der Verabreichungen usw.) erforderlich. Zusätzliche pharmakokinetische Studien können erforderlich werden, um Dosierungsschemata im Einklang mit bestimmten Populationsvariablen festlegen zu können. |
|
(4) |
Wurden pharmakokinetische Studien gemäß Teil 3 des Dossiers eingereicht, können Querverweise auf diese Studien aufgenommen werden. |
|
(5) |
Für fixe Kombinationen wird auf Abschnitt IV verwiesen. |
IIIa.4A2. Resistenzentwicklung und damit verbundenes Risiko bei Tieren
|
(1) |
Für betroffene biologische Tierarzneimittel (z. B. antimikrobielle Wirkstoffe und Antiparasitika) sind Angaben über derzeitige Resistenzen (falls zutreffend) und über das potenzielle Auftreten von Resistenzen von klinischer Relevanz für die vorgesehene Indikation bei der Zieltierart vorzulegen. Soweit möglich, sind Informationen über die Resistenzmechanismen, die molekulargenetische Grundlage der Resistenzen und die Rate der Übertragung von Resistenzdeterminanten vorzulegen. Soweit relevant, müssen Informationen zu Ko- und Kreuzresistenzen vorgelegt werden. Der Antragsteller muss Maßnahmen vorschlagen, durch die sich die Resistenzentwicklung bei Organismen von klinischer Relevanz infolge der vorgesehenen Verwendung des Tierarzneimittels begrenzen lässt. |
|
(2) |
Resistenzen, die ein Risiko für den Menschen darstellen können, sind in Teil 3 des Antragsdossiers zu behandeln. Gegebenenfalls sind Querverweise zu den in Teil 3 des Dossiers aufgeführten Angaben zu machen. |
IIIa.4A3. Dosisermittlung und -bestätigung
|
(1) |
Es sind geeignete Daten vorzulegen, um die vorgeschlagene Dosis, das Dosierungsintervall, die Dauer der Behandlung und ein etwaiges Wiederbehandlungsintervall zu begründen. |
|
(2) |
Für Studien, die unter Feldbedingungen durchgeführt werden, sind sachdienliche Informationen vorzulegen, wie bei den klinischen Studien dargestellt. |
IIIa.4A4. Verträglichkeit bei den Zieltierarten
|
(1) |
Die lokale und systemische Verträglichkeit des Tierarzneimittels ist bei den Zieltierarten zu untersuchen. Zweck der Unbedenklichkeitsversuche bei Zieltierarten ist es, Anzeichen von Unverträglichkeit zu beschreiben und einen angemessenen Sicherheitsspielraum bei Anwendung der empfohlenen Verabreichungswege aufzuzeigen. Dies lässt sich durch Erhöhung der Dosis und/oder durch Verlängerung der Behandlungsdauer erreichen. |
|
(2) |
Der Studienbericht muss detaillierte Angaben zu allen erwarteten pharmakologischen Wirkungen und allen Nebenwirkungen enthalten. Die Durchführung von Unbedenklichkeitsversuchen bei Zieltierarten erfolgt in Übereinstimmung mit den VICH-Leitlinien und den einschlägigen von der Agentur veröffentlichten Leitlinien. Andere vorklinische und klinische Studien sowie einschlägige Informationen aus der veröffentlichten Literatur können ebenfalls Informationen zur Sicherheit bei der Zieltierart liefern. |
IIIa.4B. Klinische Prüfungen
IIIa.4B1. Allgemeine Grundsätze
|
(1) |
Klinische Prüfungen sind unter Berücksichtigung der VICH-Leitlinien und der von der Agentur veröffentlichten sachdienlichen Leitlinien zu planen und durchzuführen und es ist darüber zu berichten. Daten aus klinischen Prüfungen, die außerhalb der Union durchgeführt wurden, dürfen nur dann zur Bewertung eines Zulassungsantrags herangezogen werden, wenn diese Daten ausreichend repräsentativ für die Situation in der EU sind. |
|
(2) |
Experimentelle Daten, wie z. B. Sondierungs-/Pilotversuche, oder Ergebnisse aus nichtexperimentellen Ansätzen müssen durch Daten, die unter normalen Feldbedingungen erhalten wurden, bestätigt werden; anderenfalls ist dies zu begründen. |
|
(3) |
Zweck der klinischen Prüfung ist es, unter Feldbedingungen die Sicherheit für die Zieltierart und die Wirksamkeit eines Tierarzneimittels unter normalen Bedingungen der Tierhaltung und/oder im Rahmen einer normalen tierärztlichen Praxis zu untersuchen. Sie müssen die Wirkung des Tierarzneimittels nach Verabreichung an die vorgesehene Zieltierart unter Verwendung des vorgeschlagenen Dosierungsschemas und des/der vorgeschlagenen Verabreichungsweges/-wege nachweisen. Der Aufbau der Prüfung zielt darauf ab, Indikationen zu erhärten und Gegenanzeigen je nach Tierart, Alter, Rasse und Geschlecht sowie die Anweisungen zum Gebrauch des Tierarzneimittels und mögliche Nebenwirkungen zu spezifizieren. |
|
(4) |
Alle veterinärklinischen Prüfungen sind entsprechend einem detaillierten Prüfplan durchzuführen. Bei Formulierungen, die zur Anwendung in veterinärklinischen Prüfungen in der Union bestimmt sind, ist der Hinweis ‚Nur für veterinärklinische Prüfungen‘ deutlich sichtbar und unlöschbar auf der Etikettierung anzubringen. |
|
(5) |
Von anderweitig begründeten Ausnahmen abgesehen, sind klinische Prüfungen mit Kontrolltieren (kontrollierte klinische Prüfungen) durchzuführen. Die Ergebnisse, die zur Wirksamkeit des neuen Produkts erzielt wurden, sollten mit jenen bei Zieltierarten verglichen werden, die ein in der Union zugelassenes Tierarzneimittel, das einen annehmbaren Grad an Wirksamkeit gezeigt hat und für die vorgeschlagenen Indikationen bei derselben Zieltierart zugelassen ist, oder ein Placebo oder gar keine Behandlung erhalten haben. Alle erzielten Ergebnisse, ob positiv oder negativ, sind zu melden. |
|
(6) |
Bei Entwurf des Prüfplans, Analyse und Bewertung der klinischen Prüfungen sind feststehende statistische Grundsätze in Übereinstimmung mit den von der Agentur veröffentlichten einschlägigen Leitlinien zu befolgen, sofern nicht anders begründet. |
IIIa.4B2. Dokumentation
Das Dossier über die Wirksamkeit muss alle vorklinischen und klinischen Unterlagen enthalten, unabhängig davon, ob sie günstig oder ungünstig für die Tierarzneimittel ausgefallen sind, damit eine objektive Gesamtbeurteilung des Nutzen-Risiko-Verhältnisses des Arzneimittels ermöglicht wird.
IIIa.4B2.1. Ergebnisse der vorklinischen Studien
Sofern möglich, sind Angaben zu den Ergebnissen folgender Versuche zu machen:
|
a) |
Versuche zum Nachweis pharmakologischer Aktivität; |
|
b) |
Versuche zum Nachweis der pharmakodynamischen Mechanismen, die der therapeutischen Wirkung zugrunde liegen; |
|
c) |
Versuche zum Nachweis des pharmakokinetischen Hauptprofils; |
|
d) |
Versuche zum Nachweis der Sicherheit für die Zieltiere; |
|
e) |
Versuche zur Bestimmung und Bestätigung der Dosis (einschließlich Dosisintervall, Behandlungsdauer und etwaiges Wiederbehandlungsintervall); |
|
f) |
Versuche und Untersuchungen zu Resistenzen, falls zutreffend. |
Sofern während der Versuche unerwartete Ergebnisse auftreten, sind diese Ergebnisse ausreichend anzugeben. Außerdem sind in allen vorklinischen Studienberichten folgende Angaben vorzulegen:
|
a) |
eine Zusammenfassung; |
|
b) |
ein Studienprotokoll; |
|
c) |
eine detaillierte Beschreibung der Ziele, der Konzeption und Durchführung, einschließlich angewandter Methoden, Geräte und Materialien, mit Angaben wie Tierart, Alter, Gewicht, Geschlecht, Anzahl, Rasse oder Stamm der Tiere, Identifizierung der Tiere, Dosis sowie Verabreichungsweg und -schema; |
|
d) |
eine statistische Analyse der Ergebnisse; |
|
e) |
eine objektive Erörterung der erzielten Ergebnisse mit Schlussfolgerungen zu Unbedenklichkeit und Wirksamkeit des Tierarzneimittels für die Zieltierart. |
Werden diese Daten gar nicht oder nur teilweise angegeben, ist dies zu begründen.
IIIa.4B2.2. Ergebnisse der klinischen Prüfungen
Alle Angaben sind von jedem Versuchsleiter bei individueller Behandlung auf einzelnen Datenblättern und bei Behandlung von Tiergruppen auf kollektiven Datenblättern festzuhalten.
Der Zulassungsinhaber des Tierarzneimittels muss alle notwendigen Vorkehrungen treffen, um sicherzustellen, dass die Originalunterlagen, die die Grundlage der vorgelegten Daten darstellen, mindestens fünf Jahre nach Ablauf der Zulassung aufbewahrt werden.
Für jede klinische Prüfung müssen die klinischen Beobachtungen in einer Gesamtschau der Versuche und ihrer Ergebnisse unter besonderer Berücksichtigung folgender Angaben zusammengefasst werden:
|
a) |
Anzahl der Kontroll- und Versuchstiere, die entweder individuell oder kollektiv behandelt wurden, mit einer Aufschlüsselung in Bezug auf Art, Rasse oder Stamm, Alter und Geschlecht; |
|
b) |
Anzahl der vorzeitig aus den Prüfungen herausgenommenen Tiere mit entsprechender Begründung; |
|
c) |
bei Kontrolltieren die Angabe, ob sie:
|
|
d) |
Häufigkeit des Auftretens der beobachteten Nebenwirkungen; |
|
e) |
gegebenenfalls Beobachtungen zur Auswirkung auf die Nutzleistung der Tiere; |
|
f) |
ausführliche Angaben zu Versuchstieren, die wegen ihres Alters sowie der Zucht- oder Fütterungsmethoden oder angesichts ihres Verwendungszwecks größeren Risiken ausgesetzt sein können, oder zu Tieren, deren physiologischer oder pathologischer Zustand besondere Aufmerksamkeit erfordert; |
|
g) |
eine statistische Auswertung der Ergebnisse. |
Der Hauptversuchsleiter muss allgemeine Schlussfolgerungen über die Wirksamkeit und Unbedenklichkeit des Tierarzneimittels für die Zieltierart unter den vorgeschlagenen Anwendungsbedingungen und insbesondere alle Informationen zu Indikationen und Gegenanzeigen, Dosierung und durchschnittlicher Behandlungsdauer sowie gegebenenfalls allen beobachteten Wechselwirkungen mit anderen Arzneimitteln oder Zusatzstoffen in Futtermitteln sowie allen während der Behandlung zu treffenden besonderen Vorkehrungen und die möglicherweise beobachteten klinischen Anzeichen einer Überdosierung ziehen.
ABSCHNITT IIIb
ANFORDERUNGEN FÜR IMMUNOLOGISCHE TIERARZNEIMITTEL
Die nachstehenden Anforderungen gelten für immunologische Tierarzneimittel gemäß der in Artikel 4 Absatz 5 enthaltenen Definition, sofern in Abschnitt IV nicht anders vorgesehen.
IIIb.1. Teil 1: Zusammenfassung der Unterlagen
Es wird auf Abschnitt I verwiesen.
IIIb.2. Teil 2: Qualitätsbezogene Unterlagen (physikalisch-chemische, biologische und mikrobiologische Informationen)
IIIb.2.A. Produktbeschreibung
IIIb.2A1. Qualitative und quantitative Zusammensetzung
|
(1) |
Unter ‚qualitativer Zusammensetzung‘ aller Bestandteile des immunologischen Tierarzneimittels versteht man die Bezeichnung oder Beschreibung:
|
|
(2) |
Diese unter Punkt 1 genannten Daten sind durch alle anderen sachdienlichen Daten über die Art der Primärverpackung und gegebenenfalls der äußeren Umhüllung sowie erforderlichenfalls über die Art ihres Verschlusses zu ergänzen; dazu gehören auch ausführliche Angaben zu den Vorrichtungen, mit denen das immunologische Tierarzneimittel angewandt oder verabreicht wird und die zusammen mit dem Arzneimittel abgegeben werden. Wird die Vorrichtung nicht mit dem immunologischen Tierarzneimittel geliefert, sind zweckdienliche Angaben zur Vorrichtung zu machen, sofern dies für die Beurteilung des Arzneimittels erforderlich ist. |
|
(3) |
Für die gebräuchlichen Bezeichnungen zur Beschreibung der Bestandteile eines immunologischen Tierarzneimittels ist unbeschadet der in Artikel 8 vorgesehenen Angaben Folgendes maßgeblich:
|
|
(4) |
Was die quantitative Zusammensetzung der Wirkstoffe eines immunologischen Tierarzneimittels betrifft, so ist, sofern möglich, die Anzahl der Organismen, der spezifische Proteingehalt, die Masse, die Zahl der internationalen Einheiten oder Einheiten biologischer Aktivität entweder je Dosierungseinheit oder Volumen und, in Bezug auf das Adjuvans und die Bestandteile der Arzneiträgerstoffe, die Masse oder das Volumen von jedem einzelnen von ihnen unter angemessener Berücksichtigung der im Teil IIb.2B. genannten Einzelheiten genau anzugeben. |
|
(5) |
Sofern eine internationale Einheit biologischer Aktivität festgelegt wurde, ist diese anzuwenden. |
|
(6) |
Die Einheiten biologischer Aktivität, für die keine veröffentlichten Daten bestehen, sind so auszudrücken, dass eine eindeutige Aussage über die Wirksamkeit der Bestandteile getroffen wird, z. B. der Ergebnisse der Titer- und Potenztests des Fertigprodukts. |
|
(7) |
Die Zusammensetzung ist in Form von Mindestmengen und gegebenenfalls mit Höchstmengen anzugeben. |
IIIb.2A2. Produktentwicklung
|
(1) |
Es sind Erläuterungen zu geben, die sich unter anderem auf folgende Punkte beschränken:
|
|
(2) |
Diese Erläuterungen sind durch wissenschaftliche Daten über die Produktentwicklung zu erhärten. |
IIIb.2B. Angaben über die Herstellungsweise
|
(1) |
Die Angaben über die Herstellungsweise, die dem Zulassungsantrag gemäß Artikel 8 beizufügen sind, sind so abzufassen, dass sie eine geeignete Beschreibung der Art der verwendeten Abläufe gestattet, darunter die Bestimmung der Schlüsselphasen im Produktionsprozess. |
|
(2) |
Die Beschreibung des Herstellungsprozesses umfasst mindestens:
|
|
(3) |
Die Validierung aller im Herstellungsprozess verwendeten Kontrollmethoden ist zu beschreiben und zu dokumentieren, und die Ergebnisse sind vorzulegen, sofern nicht anders begründet. Die Validierung der Hauptproduktionsschritte und die Validierung des gesamten Produktionsprozesses sind nachzuweisen, wobei die Ergebnisse von drei nach dem beschriebenen Verfahren in Folge produzierten Chargen vorzulegen sind. |
IIIb.2C. Herstellung und Kontrolle der Ausgangsstoffe
|
(1) |
‚Ausgangsstoffe‘ im Sinne dieses Teils sind alle Bestandteile, die zur Herstellung des immunologischen Tierarzneimittels verwendet werden. |
|
(2) |
Handelsübliche, gebrauchsfertige Hilfsstoffsysteme, die mit einem Markennamen gekennzeichnet sind, sowie zur Herstellung des Wirkstoffes verwendete Kulturmedien, die sich aus mehreren Bestandteilen zusammensetzen, gelten als ein einziger Ausgangsstoff. Trotzdem muss die qualitative und quantitative Zusammensetzung jedes Kulturmediums angegeben werden, falls die Behörden der Auffassung sind, dass diese Informationen wichtig für die Qualität des Fertigprodukts und etwaige dadurch bedingte Risiken sind. |
|
(3) |
Wird für die Zubereitung dieser Kulturmedien oder Hilfsstoffsysteme Material tierischen Ursprungs verwendet, müssen auch die Tierart und das verwendete Gewebe daraus hervorgehen und die Übereinstimmung mit den einschlägigen Monografien, einschließlich der allgemeinen Monografien und allgemeinen Kapitel des Europäischen Arzneibuchs, ist nachzuweisen. |
|
(4) |
Der Antragsteller hat Unterlagen vorzulegen, aus denen hervorgeht, dass die Ausgangsstoffe, einschließlich Saatmaterial, Zellkulturen, Serumchargen und sonstigen Materials von Tierarten, die TSE übertragen könnten, sowie die Herstellung des Tierarzneimittels den Anforderungen der Leitlinien für die Minimierung des Risikos der Übertragung von Erregern der spongiformen Enzephalopathie tierischen Ursprungs durch Human- und Tierarzneimittel sowie den Anforderungen der entsprechenden Monografie des Europäischen Arzneibuchs entsprechen. Dieser Nachweis kann durch Eignungszertifikate erbracht werden, die von der Europäischen Direktion für die Arzneimittelqualität für die einschlägige Monografie des Europäischen Arzneibuchs ausgestellt werden. |
|
(5) |
Das Dossier muss die Spezifikationen sowie Informationen über die Qualitätskontrollprüfungen, die für alle Chargen von Ausgangsstoffen durchzuführen sind, und die Ergebnisse für eine Charge aller verwendeten Bestandteile umfassen und ist gemäß den Anforderungen dieses Teils vorzulegen. |
|
(6) |
Für die Ausgangsstoffe sind Analysezertifikate zum Nachweis der Einhaltung der festgelegten Spezifikation vorzulegen. |
|
(7) |
Die Farbstoffe müssen in allen Fällen den Anforderungen der Richtlinie 2009/35/EG genügen. |
|
(8) |
Die Verwendung von Antibiotika bei der Herstellung und die Aufnahme von Konservierungsmitteln in die Zusammensetzung des Fertigprodukts müssen begründet sein und dem Europäischen Arzneibuch entsprechen. |
|
(9) |
Bei neuartigen Arzneiträgerstoffen, die erstmalig in der Union in einem Tierarzneimittel eingesetzt werden, oder bei Arzneiträgerstoffen, bei denen dies durch eine neue Art der Anwendung geschieht, sind umfassende Angaben zur Herstellung, zur Charakterisierung und zu den Kontrollen zu machen, wobei Querverweise sowohl auf die klinischen als auch auf die nichtklinischen Daten zur Unbedenklichkeit zu machen sind. Für Farbstoffe werden die Konformitätserklärungen in Teil II.2C2 Punkte 3 und 4 als ausreichend angesehen. |
IIIb.2C1. In Arzneibüchern aufgeführte Ausgangsstoffe
|
(1) |
Die Monografien des Europäischen Arzneibuchs gelten für alle darin aufgeführten Ausgangsstoffe; anderenfalls ist dies angemessen zu begründen. |
|
(2) |
Bei allen anderen Stoffen kann jeder Mitgliedstaat verlangen, dass bei der in seinem Hoheitsgebiet erfolgenden Herstellung die Vorschriften seines Arzneibuchs eingehalten werden. |
|
(3) |
Die Beschreibung der Analysemethoden kann durch eine detaillierte Bezugnahme auf das betreffende Arzneibuch ersetzt werden. |
|
(4) |
Die bei jeder einzelnen Charge der Ausgangsstoffe durchzuführenden Routineprüfungen entsprechen den Angaben im Zulassungsantrag. Werden andere als die im Arzneibuch angegebenen Prüfungen durchgeführt, so ist der Nachweis zu erbringen, dass die Ausgangsstoffe den Qualitätsanforderungen dieses Arzneibuchs entsprechen. |
|
(5) |
Die zuständigen Behörden können vom Antragsteller geeignetere Spezifikationen verlangen, wenn eine Spezifikation oder sonstige Bestimmungen einer Monografie des Europäischen Arzneibuchs oder des Arzneibuchs eines Mitgliedstaats unter Umständen nicht genügen, um die Qualität der Stoffe zu gewährleisten. Die für das betreffende Arzneibuch zuständigen Behörden werden auf den vermuteten Mangel hingewiesen. |
IIIb.2C2. Nicht in einem Arzneibuch aufgeführte Ausgangsstoffe
IIIb.2C2.1. Ausgangsstoffe biologischer Herkunft
|
(1) |
Die Beschreibung muss in Form einer Monografie erfolgen. |
|
(2) |
Die Herstellung von Impfstoffen erfolgt möglichst auf der Grundlage eines Saatsystems und angelegter Zellkulturen. Für die Herstellung immunologischer Tierarzneimittel, die aus einem Serum bestehen, sind die Herkunft, der allgemeine Gesundheitszustand und der immunologische Status der für die Herstellung verwendeten Tiere anzugeben und das Ausgangsmaterial muss aus genau definierten Bezugsquellen stammen. |
|
(3) |
Die Herkunft, auch die geografische Herkunft, und die Entstehung der Ausgangsstoffe sind zu beschreiben und zu dokumentieren. |
|
(4) |
Für gentechnisch veränderte Ausgangsstoffe müssen diese Angaben Einzelheiten enthalten, wie die Beschreibung der Ausgangszellen oder -stämme, den Aufbau des Expressionsvektors (Name, Herkunft, Funktion des Replikons, Promotor, Enhancer und andere Regulatoren), Kontrolle der effektiv eingefügten DNA- oder RNA-Sequenz, oligonukleotide Plasmidvektorsequenzen von Zellen, zur Kotransfektion verwendetes Plasmid, hinzugefügte oder entfernte Gene, biologische Eigenschaften des fertigen Konstrukts und der exprimierten Gene, Vervielfältigungszahl und genetische Stabilität. |
|
(5) |
Bei Tierarzneimitteln, die genetisch veränderte Organismen (GVO) enthalten oder aus solchen bestehen, sind dem qualitativen Teil des Zulassungsantrags die gemäß Richtlinie 2001/18/EG vorgeschriebenen Unterlagen beizufügen. |
|
(6) |
Saatmaterial, einschließlich Zellkulturen und Rohserum für die Antiserumherstellung, ist zum Nachweis seiner Identität zu prüfen, und die Freiheit von Fremderregern ist gemäß Europäischem Arzneibuch nachzuweisen. |
|
(7) |
Angaben sind zu allen Stoffen biologischer Herkunft zu machen, die in einer der Herstellungsstufen verwendet werden. Die Informationen müssen Folgendes umfassen:
|
|
(8) |
Wird das Vorhandensein von Fremderregern nachgewiesen oder vermutet, ist das entsprechende Material zu beseitigen oder so zu verarbeiten, dass das Risiko des Vorhandenseins mit einer validierten Behandlung verringert wird. Wird nach der Verarbeitung das Vorhandensein von Fremderregern nachgewiesen oder vermutet, ist das entsprechende Material nur unter der Voraussetzung zu verwenden, dass durch die weitere Verarbeitung des Produkts ihre Beseitigung und/oder Inaktivierung sichergestellt ist; die Beseitigung und/oder Inaktivierung solcher Fremderreger ist nachzuweisen. |
|
(9) |
Beim Einsatz von Zellkulturen ist nachzuweisen, dass die Zelleigenschaften bis zu der für die Produktion verwendeten Passagenzahl unverändert geblieben sind. |
|
(10) |
Für lebende attenuierte Impfstoffe ist der Nachweis der Stabilität der Attenuierungseigenschaften der Zellen zu erbringen. Sofern keine spezifische Eigenschaft mit der Attenuierung in Zusammenhang steht (z. B. genetischer Marker, thermische Stabilität), wird diese typischerweise durch die fehlende Reversion der Virulenz bei der Zieltierart erreicht. |
|
(11) |
Sofern erforderlich, sind Proben der biologischen Ausgangsstoffe oder der in den Prüfverfahren verwendeten Reagenzien vorzulegen, sodass die zuständige Behörde Kontrollprüfungen durchführen lassen kann. |
IIIb.2C2.2. Ausgangsstoffe nicht biologischer Herkunft
Die Beschreibung muss in Form einer Monografie unter folgenden Titeln erfolgen:
|
a) |
der Name des Ausgangsstoffs, der den Vorschriften von Teil IIIb.2A1 Punkt 3 entspricht, ist durch handelsübliche oder wissenschaftliche Synonyme zu ergänzen; |
|
b) |
die Beschreibung des Ausgangsstoffs, in ähnlicher Form abgefasst wie eine Beschreibung im Europäischen Arzneibuch; |
|
c) |
die Funktion des Ausgangsstoffs; |
|
d) |
Identifizierungsmethoden; |
|
e) |
etwaige besondere Vorsichtsmaßnahmen, die bei der Lagerung des Ausgangsmaterials erforderlich sind, und, falls erforderlich, dessen Haltbarkeit sind anzugeben. |
IIIb.2D. Kontrollen während des Herstellungsprozesses
|
(1) |
Das Dossier muss Angaben zu den Kontrollen enthalten, die in Zwischenherstellungsphasen durchgeführt werden, um die Konstanz des Herstellungsverfahrens und die gleichbleibende Qualität des Fertigprodukts festzustellen. Für jede Kontrolle sind Spezifikationen festzulegen und die Analysemethoden zu beschreiben. Eine Validierung der Kontrollen der Parameter, die als entscheidend für das Herstellungsverfahren erachtet werden, ist vorzusehen, sofern nicht anders begründet. |
|
(2) |
Bei inaktivierten oder entgifteten Impfstoffen sind Inaktivierung oder Entgiftung während jedes Produktionslaufs möglichst rasch nach dem Inaktivierungs- oder Entgiftungsvorgang und gegebenenfalls nach der Neutralisierung, aber noch vor dem nächsten Produktionsschritt zu prüfen. |
|
(3) |
Gemäß den Bestimmungen der Richtlinie 2010/63/EU und dem Europäischen Übereinkommen zum Schutz der für Versuche und andere wissenschaftliche Zwecke verwendeten Wirbeltiere sind die Versuche so durchzuführen, dass möglichst wenige Tiere verwendet und möglichst wenig Schmerzen, Leiden, Ängste oder dauerhafte Schäden verursacht werden. Falls verfügbar, ist ein alternativer In-vitro-Test zu verwenden, wenn dies zum Ersatz oder zur Verringerung des Einsatzes von Tieren oder zur Verringerung ihres Leidens führt. |
IIIb.2E. Kontrollen am Fertigprodukt
|
(1) |
Bei allen Kontrollen muss die Beschreibung der Verfahren zur Analyse des Fertigprodukts hinreichend detailliert für eine Qualitätsbeurteilung sein. |
|
(2) |
Sofern Testverfahren und Grenzwerte angewandt werden, die nicht in den Monografien des Europäischen Arzneibuchs oder andernfalls in dem Arzneibuch eines Mitgliedstaats aufgeführt sind, muss, wenn geeignete Monografien vorliegen, der Nachweis erbracht werden, dass das Fertigprodukt den Qualitätsanforderungen jenes Arzneibuchs für die betreffende Darreichungsform entsprechen würde, wenn es im Einklang mit diesen Monografien geprüft würde. In dem Zulassungsantrag sind die Prüfungen anzugeben, die an repräsentativen Proben jeder Charge des Fertigprodukts durchgeführt werden. Die Häufigkeit der Prüfungen, die am endgültigen unabgefüllten Impfstoff anstelle der daraus hergestellten Charge oder Chargen durchgeführt werden, ist anzugeben. Außerdem sind Freigabegrenzwerte anzugeben und zu begründen. Die Validierung der am Fertigprodukt durchgeführten Kontrollen ist vorzulegen. |
|
(3) |
Es sind Informationen über die Erstellung und den Ersatz von Referenzmaterial bereitzustellen. Wenn während der Produktentwicklung mehr als ein Referenzstandard verwendet wurde, muss eine Qualifizierungshistorie vorgelegt werden, die beschreibt, wie die Beziehung zwischen den verschiedenen Normen aufrechterhalten wurde. |
|
(4) |
Sofern verfügbar, sind die im Europäischen Arzneibuch verzeichneten chemischen und biologischen Referenzmaterialien zu verwenden. Falls andere Referenzzubereitungen und -standards verwendet werden, sind diese anzugeben und ausführlich zu beschreiben. |
|
(5) |
Gemäß den Bestimmungen der Richtlinie 2010/63/EU und dem Europäischen Übereinkommen zum Schutz der für Versuche und andere wissenschaftliche Zwecke verwendeten Wirbeltiere sind die Versuche so durchzuführen, dass möglichst wenige Tiere verwendet und möglichst wenig Schmerzen, Leiden, Ängste oder dauerhafte Schäden verursacht werden. Falls verfügbar, ist ein alternativer In-vitro-Test zu verwenden, wenn dies zum Ersatz oder zur Verringerung des Einsatzes von Tieren oder zur Verringerung des Leidens führt. |
|
(6) |
Allgemeine Merkmale des Fertigprodukts
Die Prüfung von allgemeinen Merkmalen erstreckt sich gegebenenfalls auf die Erscheinung und physikalische oder chemische Prüfungen, physikalische Merkmale wie Leitfähigkeit, pH-Wert, Viskosität usw. Für jedes dieser Merkmale müssen vom Antragsteller Spezifikationen mit geeigneten Akzeptanzgrenzen erstellt werden. |
|
(7) |
Identitätsnachweis der Wirkstoffe
Erforderlichenfalls ist auch eine spezifische Prüfung zu Identifizierungszwecken durchzuführen. Gegebenenfalls kann die Identitätsprüfung mit der Prüfung des Titers oder der Potenz der Charge kombiniert werden. |
|
(8) |
Titer oder Potenz der Charge
An jeder Charge ist eine Quantifizierung des Wirkstoffs vorzunehmen, um nachzuweisen, dass der Titer oder die Potenz in jeder Charge geeignet ist, ihre Unbedenklichkeit und Wirksamkeit sicherzustellen. |
|
(9) |
Identitätsnachweis und Gehaltsbestimmung von Adjuvanzien
Menge und Art des Adjuvans und seiner Bestandteile am Fertigprodukt sind zu überprüfen; anderenfalls ist dies zu begründen. |
|
(10) |
Identitätsnachweis und Gehaltsbestimmung der Bestandteile des Arzneiträgerstoffs
Soweit erforderlich, sind an den Arzneiträgerstoffen zumindest Prüfungen zur Identifizierung durchzuführen. Die Bestimmung des oberen und des unteren Grenzwerts ist für Konservierungsmittel zwingend vorgeschrieben. Die Bestimmung des oberen Grenzwerts ist für alle anderen Bestandteile von Arzneiträgerstoffen, die im Verdacht stehen, zu Nebenwirkungen zu führen, zwingend vorgeschrieben. |
|
(11) |
Prüfungen auf Sterilität und Reinheit
Die Abwesenheit von Fremderregern (Bakterien, Mykoplasmen, Pilzen und bakterielle Endotoxine, soweit relevant) ist für parenteral verabreichte Produkte gemäß dem Europäischen Arzneibuch nachzuweisen. Für nicht flüssige, nicht parenteral verabreichte Produkte kann bei entsprechender Begründung die Einhaltung eines maximalen Biobelastungsgrenzwertes anstelle einer Sterilitätsprüfung akzeptabel sein. Es sind geeignete Prüfungen zum Nachweis der Freiheit von Kontamination durch Fremderreger oder andere Stoffe entsprechend der Art des immunologischen Tierarzneimittels, der Herstellungsmethode und -bedingungen durchzuführen. Es ist ein risikobasierter Ansatz zum Nachweis der Freiheit von Fremderregern, wie im Europäischen Arzneibuch beschrieben, zu verwenden. |
|
(12) |
Feuchtigkeitsrückstände
Jede Charge eines lyophilisierten Produkts ist auf Feuchtigkeitsrückstände zu prüfen. |
|
(13) |
Füllvolumen
Geeignete Prüfungen zum Nachweis des korrekten Füllvolumens müssen durchgeführt werden. |
IIIb.2F. Gleichbleibende Qualität der Chargen
Damit eine gleichbleibende Qualität von Charge zu Charge gewährleistet ist und die Übereinstimmung mit den Spezifikationen nachgewiesen wird, ist für drei aufeinanderfolgende und für die Routineproduktion repräsentative Chargen ein vollständiges Protokoll mit den Ergebnissen aller Prüfungen vorzulegen, die im Verlauf der Produktion und am Fertigprodukt durchgeführt worden sind. Bei kombinierten Arzneimitteln gewonnene Daten zur gleichbleibenden Qualität können bei hinreichender Begründung für Derivate dienen, die einen oder mehrere der gleichen Bestandteile enthalten.
IIIb.2G. Haltbarkeitsversuche
|
(1) |
Die Haltbarkeitsversuche umfassen die Haltbarkeit des Wirkstoffs und des Fertigprodukts, einschließlich gegebenenfalls des/der Lösungsmittel(s). |
|
(2) |
Zu beschreiben sind die Prüfungen, die durchgeführt wurden, um die Haltbarkeitsdauer, die empfohlenen Lagerungsbedingungen und die Spezifikationen bei Ablauf der Haltbarkeitsdauer, die für den Wirkstoff und das Fertigprodukt vorgeschlagen werden, zu erhärten. Diese Versuche müssen immer Echtzeitstudien sein.
Werden Zwischenprodukte, die auf verschiedenen Stufen des Herstellungsprozesses anfallen, gelagert, so sind die vorgesehenen Lagerungsbedingungen und die Lagerungsdauer auf der Grundlage der verfügbaren Haltbarkeitsdaten angemessen zu begründen. |
|
(3) |
Haltbarkeitsprüfungen des Fertigprodukts sind an mindestens drei repräsentativen Chargen durchzuführen, die im Einklang mit dem beschriebenen Produktionsverfahren hergestellt wurden, sowie an Produkten, die im endgültigen Behältnis gelagert sind; diese Versuche umfassen biologische und physikalisch-chemische Haltbarkeitsprüfungen, die in regelmäßigen Abständen durchgeführt werden, für das Fertigprodukt bis drei Monate nach Ablauf der angegebenen Haltbarkeitsdauer. |
|
(4) |
Die Schlussfolgerungen müssen die Analyseergebnisse enthalten, die die vorgeschlagene Haltbarkeitsdauer unter allen vorgeschlagenen Lagerungsbedingungen rechtfertigen. Die Ergebnisse der Haltbarkeitsstudie sind bei der Festlegung geeigneter Formulierungs- und Freigabespezifikationen zu berücksichtigen, um die Konformität des Produkts mit der angegebenen Haltbarkeitsdauer sicherzustellen |
|
(5) |
Bei Arzneimitteln, die über das Futter verabreicht werden, müssen die Angaben bezüglich der Haltbarkeit, falls erforderlich, auch für die verschiedenen Stadien des Mischvorgangs bei Einhaltung der empfohlenen Anweisungen gemacht werden. |
|
(6) |
Muss das Fertigprodukt vor der Verabreichung rekonstituiert oder muss es in Trinkwasser verabreicht werden, sind genaue Angaben zur vorgeschlagenen Haltbarkeitsdauer für das entsprechend den Anweisungen rekonstituierte Produkt erforderlich. Zur Untermauerung der vorgeschlagenen Haltbarkeitsdauer für das rekonstituierte Produkt sind Daten vorzulegen. |
|
(7) |
Bei kombinierten Arzneimitteln gewonnene Haltbarkeitsdaten können bei hinreichender Begründung für Derivate dienen, die einen oder mehrere der gleichen Bestandteile enthalten. |
|
(8) |
Bei Multidosisbehältnissen sind gegebenenfalls Haltbarkeitsdaten vorzulegen, um die Haltbarkeitsdauer nach erstmaliger Entnahme oder Öffnung des Arzneimittels zu begründen, und es ist eine Spezifikation zur Haltbarkeit für das im Gebrauch befindliche Behältnis festzulegen. |
|
(9) |
Die Wirksamkeit jeder Konservierungsart ist nachzuweisen. |
|
(10) |
Angaben zur Wirksamkeit der Konservierungsmittel bei anderen, ähnlichen immunologischen Tierarzneimitteln desselben Herstellers sind unter Umständen ausreichend. |
|
(11) |
Wenn die Wirkstoffe gelagert werden, müssen die vorgesehenen Bedingungen und die Dauer der Lagerung auf der Grundlage von Haltbarkeitsdaten festgelegt werden. Diese Daten können entweder durch Prüfung der Wirkstoffe selbst oder durch entsprechende Prüfung des Fertigprodukts gewonnen werden. |
IIIb.2H. Sonstige Informationen
In das Dossier können auch Informationen über die Qualität des immunologischen Tierarzneimittels aufgenommen werden, die in diesem Abschnitt nicht erfasst sind.
IIIb.3. Teil 3: Unterlagen zur Unbedenklichkeit (Unbedenklichkeits- und Rückstandsversuche)
IIIb.3A. Allgemeine Vorschriften
|
(1) |
Die Unbedenklichkeitsunterlagen sind angemessen zur Bewertung folgender Aspekte:
|
|
(2) |
Vorklinische Studien müssen in Übereinstimmung mit den Anforderungen der Guten Laborpraxis (GLP) durchgeführt werden.
Studien, die nicht der GLP entsprechen, können bei Studien an Nichtzieltierarten sowie bei Studien zur Bewertung immunologischer, biologischer oder genetischer Eigenschaften der Impfstoffstämme unter angemessen kontrollierten Bedingungen akzeptiert werden. Andere Abweichungen sind zu begründen. |
|
(3) |
Alle Versuche zur Unbedenklichkeit sind im Einklang mit einem vollständig überprüften detaillierten und vor Versuchsbeginn schriftlich niedergelegten Protokoll durchzuführen. Das Wohlergehen der Versuchstiere muss der tierärztlichen Aufsicht unterliegen und ist bei Ausarbeitung jedes Versuchsprotokolls sowie während des gesamten Versuchs in vollem Umfang zu berücksichtigen. |
|
(4) |
Im Vorfeld schriftlich festgelegte systematische Verfahren für Organisation, Durchführung, Datenerfassung, Dokumentation und Kontrolle der Versuche zur Unbedenklichkeit sind vorgeschrieben. |
|
(5) |
Klinische Prüfungen (Feldversuche) sind gemäß den festgelegten Grundsätzen der guten klinischen Praxis durchzuführen. Abweichungen sind zu begründen. |
|
(6) |
Die Unbedenklichkeitsversuche müssen mit den einschlägigen Vorschriften des Europäischen Arzneibuchs übereinstimmen. Abweichungen sind zu begründen. |
|
(7) |
Die Unbedenklichkeitsversuche sind an den Zielarten durchzuführen. Die anzuwendende Dosis muss der Menge des Arzneimittels entsprechen, die für die Verwendung zu empfehlen ist, und die für die Unbedenklichkeitsversuche verwendete Charge muss von einer Charge bzw. Chargen stammen, die im Einklang mit dem in Teil 2 des Zulassungsantrags beschriebenen Verfahren hergestellt sind. |
|
(8) |
Bei den in den Abschnitten B.1, B.2 und B.3 beschriebenen Laboruntersuchungen muss die Dosis des Tierarzneimittels den maximalen Titer, Antigengehalt oder die maximale Potenz enthalten. Falls erforderlich, kann die Konzentration des Antigens so angepasst werden, dass die vorgeschriebene Dosis erreicht wird. |
|
(9) |
Die Sicherheit eines immunologischen Tierarzneimittels ist für jede Kategorie der für die Impfung empfohlenen Zieltierarten nachzuweisen, und zwar auf jedem empfohlenen Verabreichungsweg und jeder empfohlenen Verabreichungsmethode und unter Anwendung des vorgeschlagenen Verabreichungsschemas. Ein Worst-Case-Szenario für Verabreichungsweg und -methode kann verwendet werden, wenn dies wissenschaftlich begründet ist. |
|
(10) |
Bei immunologischen Tierarzneimitteln, die aus lebenden Organismen bestehen, sind besondere Anforderungen unter B.6 aufgeführt. |
|
(11) |
Die Angaben und Unterlagen, die dem Zulassungsantrag beizufügen sind, müssen den Vorschriften für vorklinische Studien und klinische Prüfungen entsprechen, die in den Teilen IIIb.4B Punkt 4 und IIIb.4C Punkt 3 beschrieben sind. |
IIIb.3B. Vorklinische Studien
|
(1) |
Unbedenklichkeit der Verabreichung einer einzigen Dosis
Das immunologische Tierarzneimittel muss in der empfohlenen Dosis und auf jedem der empfohlenen Verabreichungswege und -methoden an Tiere jeder Art und jede betroffene Kategorie (z. B. gegebenenfalls Mindestalter, trächtige Tiere) verabreicht werden, für die es bestimmt ist. Die Tiere sind täglich auf Anzeichen systemischer und lokaler Reaktionen hin zu beobachten und zu untersuchen, bis solche Reaktionen nicht mehr zu erwarten sind, mindestens aber über einen Zeitraum von 14 Tagen nach der letzten Verabreichung. Gegebenenfalls müssen diese Studien detaillierte makroskopische und mikroskopische Untersuchungen post mortem an der Injektionsstelle umfassen. Sonstige objektive Kriterien, wie Rektaltemperatur und Leistungsmessungen, sind anzugeben. Diese Untersuchung kann Teil der gemäß Punkt 3 vorgeschriebenen Studie zur wiederholten Verabreichung sein, es kann aber auch auf sie verzichtet werden, wenn die Ergebnisse der gemäß Punkt 2 vorgeschriebenen Überdosis-Studie keine wesentlichen Anzeichen für systemische oder lokale Reaktionen ergeben haben. Wenn dies nicht der Fall ist, werden die in der Überdosis-Studie beobachteten systemischen oder lokalen Reaktionen als Grundlage für die Beschreibung der Sicherheit des Produkts in der Zusammenfassung der Merkmale des Arzneimittels herangezogen. |
|
(2) |
Unbedenklichkeit der Verabreichung einer einzigen Überdosis
Nur bei immunologischen Tierarzneimitteln mit lebenden Organismen sind Versuche zur Überdosis erforderlich. Eine Überdosis des immunologischen Tierarzneimittels, die normalerweise aus zehn Dosen besteht, ist auf jedem empfohlenen Verabreichungsweg und mit jeder empfohlenen Verabreichungsmethode an Tiere der empfindlichsten Kategorien der Zielarten zu verabreichen, es sei denn, es wird begründet, warum nur der heikelste von mehreren Verabreichungswegen ausgewählt wurde. Bei immunologischen Tierarzneimitteln, die injiziert werden, muss bei der Auswahl von Dosis und Verabreichungswegen und -methoden das maximale Volumen berücksichtigt werden, das an einer einzigen Injektionsstelle verabreicht werden kann. Die Tiere sind über einen Zeitraum von mindestens 14 Tagen täglich nach der letzten Verabreichung auf Anzeichen systemischer und lokaler Reaktionen hin zu beobachten und zu untersuchen. Sonstige Kriterien, wie Rektaltemperatur und Leistungsmessungen, sind anzugeben. Gegebenenfalls müssen diese Versuche detaillierte makroskopische und mikroskopische Untersuchungen post mortem an der Injektionsstelle umfassen, sofern dies nicht bereits gemäß Punkt 1 erfolgt ist. |
|
(3) |
Unbedenklichkeit der wiederholten Verabreichung einer Einzeldosis
Bei immunologischen Tierarzneimitteln, die im Rahmen eines Basisverabreichungsschemas mehrmals verabreicht werden sollen, ist eine Studie zur wiederholten Verabreichung einer Einzeldosis vorgeschrieben, um dabei auftretende Nebenwirkungen festzustellen. Dieser Versuch ist an den empfindlichsten Kategorien der Zieltierart (etwa bei bestimmten Rassen oder Altersgruppen) unter Anwendung jedes empfohlenen Verabreichungsweges und jeder empfohlenen Verabreichungsmethode durchzuführen. Die Anzahl der Verabreichungen darf nicht geringer sein als die empfohlene Höchstzahl; bei Impfstoffen ist dabei die Anzahl der Verabreichungen für die Erstimpfung und die erste Auffrischungsimpfung zu berücksichtigen. Der Abstand zwischen den Verabreichungen kann kürzer sein als in der Zusammenfassung der Merkmale des Arzneimittels angegeben. Der gewählte Abstand muss im Hinblick auf die vorgeschlagenen Einsatzbedingungen begründet werden. Die Tiere sind über einen Zeitraum von mindestens 14 Tagen täglich nach der letzten Verabreichung auf Anzeichen systemischer und lokaler Reaktionen hin zu beobachten und zu untersuchen. Sonstige objektive Kriterien, wie Rektaltemperatur und Leistungsmessungen, sind anzugeben. |
|
(4) |
Untersuchung der Fortpflanzungsfähigkeit
Die Untersuchung der Fortpflanzungsfähigkeit ist zu erwägen, wenn das immunologische Tierarzneimittel zur Verwendung bei trächtigen Tieren oder Legehennen bestimmt ist oder verwendet werden kann und wenn Daten darauf hinweisen, dass das Ausgangsmaterial des Arzneimittels einen potenziellen Risikofaktor darstellen kann. Die Fortpflanzungsfähigkeit von männlichen sowie nicht trächtigen und trächtigen weiblichen Tieren ist mit der empfohlenen Dosierung und auf dem heikelsten Verabreichungsweg und der heikelsten Verarbeitungsmethode zu untersuchen. Bei immunologischen Tierarzneimitteln, die für die Anwendung bei trächtigen Tieren empfohlen werden, muss sich die Prüfung der Fortpflanzungsfähigkeit auf die Unbedenklichkeit der Verabreichung während der gesamten Trächtigkeitsdauer oder während eines bestimmten Zeitraums der Trächtigkeit beziehen, unter Berücksichtigung des vorgesehenen Verwendungszwecks des Arzneimittels. Der Beobachtungszeitraum ist bis zur Geburt zu verlängern, um schädliche Wirkungen auf die Nachkommenschaft sowie teratogene und Aborte induzierende Wirkungen zu untersuchen. Diese Versuche können Teil der unter den Punkten 1, 2 und 3 beschriebenen Unbedenklichkeitsversuche oder der gemäß Abschnitt IIIb.3C vorgeschriebenen Feldversuche sein. |
|
(5) |
Untersuchung immunologischer Funktionen
Sofern das immunologische Tierarzneimittel die Immunreaktion des geimpften Tieres oder seiner Nachkommenschaft beeinträchtigen könnte, sind geeignete Versuche über die immunologische Funktion durchzuführen. |
|
(6) |
Besondere Vorschriften für Lebendimpfstoffe |
(1) Übertragung des Impfstammes
Die Übertragung des Impfstammes von geimpften auf ungeimpfte Zieltiere ist zu untersuchen, und zwar bei Anwendung des empfohlenen Verabreichungsweges, der am wahrscheinlichsten eine solche Übertragung mit sich bringt. Außerdem kann es erforderlich sein, die Übertragung auf Nichtzieltierarten zu untersuchen, die für lebende Impfstämme möglicherweise hochgradig empfänglich sind. Es ist eine Bewertung der Anzahl der Passagen von Tier zu Tier, die unter normalen Verwendungsbedingungen auftreten können, und der möglichen Folgen vorzulegen.
(2) Verbreitung im geimpften Tier
Fäkalien, Urin, Milch, Eier, orale, nasale oder sonstige Sekretionen sind auf Anwesenheit des Organismus zu prüfen, sofern es zweckdienlich ist. Weitere Untersuchungen der Verbreitung des Impfstammes im Tierkörper können erforderlich sein, wobei besonderes Augenmerk auf jenen Stellen liegen muss, an denen sich der Organismus hauptsächlich repliziert. Bei Lebendimpfstoffen gegen Zoonosen im Sinne der Richtlinie 2003/99/EG des Europäischen Parlaments und des Rates, die an zur Lebensmittelherstellung genutzten Tieren angewandt werden sollen, müssen diese Versuche insbesondere die Persistenz der Organismen an der Injektionsstelle berücksichtigen.
(3) Steigerung der Virulenz
Die Steigerung und Reversion der Virulenz ist an den Master-Zellen zu untersuchen. Sind Master-Zellen nicht in ausreichender Menge vorhanden, so sind die in der Produktion eingesetzten Zellen mit der niedrigsten Passagenzahl zu untersuchen. Wird in Bezug auf die Passagen eine andere Option gewählt, ist dies zu begründen. Die erste Impfung ist über den Verabreichungsweg und mit der Verabreichungsmethode vorzunehmen, die am wahrscheinlichsten zu einer Steigerung der Virulenz führen, der auf eine Reversion der Virulenz hinweist. An den Zieltieren sind Passagereihen in fünf Tiergruppen durchzuführen, es sei denn, es besteht Veranlassung dazu, mehr Passagen durchzuführen, oder der Organismus ist bereits vorher nicht mehr in den Versuchstieren vorhanden. Falls sich der Organismus nur unzureichend repliziert, sind so viele Passagen wie möglich an der Zieltierart durchzuführen.
(4) Biologische Eigenschaften des Impfstammes
Weitere Versuche können erforderlich sein, um kongenitale biologische Eigenschaften des Impfstammes (z. B. Neurotropismus) zu bestimmen.
Bei Impfstoffen, die lebende genetisch veränderte Organismen enthalten und bei denen das Produkt eines fremden Gens als Strukturprotein in den Stamm eingebaut wird, ist das Risiko einer Veränderung des Tropismus oder der Virulenz des Stammes zu berücksichtigen und erforderlichenfalls sind spezifische Tests durchzuführen.
(5) Rekombination oder Genom-Reassortment von Stämmen
Die Wahrscheinlichkeit einer Rekombination oder eines Genom-Reassortments mit Feld- oder anderen Stämmen ist zu bewerten und die Folgen solcher Ereignisse sind zu erörtern.
|
(7) |
Anwendersicherheit
In diesem Abschnitt ist eine Erörterung der in Teil IIIb.3A bis IIIb.3B festgestellten Wirkungen aufzunehmen, die diese Wirkungen mit Art und Umfang der menschlichen Exposition gegenüber dem Arzneimittel in Beziehung setzen soll, damit zweckdienliche Warnhinweise für die Anwender formuliert und andere Risikomanagement-Maßnahmen ergriffen werden können. Die Anwendersicherheit ist in Übereinstimmung mit den von der Agentur veröffentlichten einschlägigen Leitlinien zu behandeln. |
|
(8) |
Wechselwirkungen
Enthält die Zusammenfassung der Merkmale des Arzneimittels eine Aussage über die Kompatibilität mit anderen Tierarzneimitteln, ist die Unbedenklichkeit der Kombination zu untersuchen. Alle weiteren bekannten Wechselwirkungen mit Tierarzneimitteln sind zu beschreiben. |
IIIb.3C. Klinische Prüfungen
Sofern nicht anders begründet, müssen die Ergebnisse der vorklinischen Studien durch Daten aus klinischen Prüfungen ergänzt werden, bei denen Chargen verwendet werden, die mit dem im Zulassungsantrag beschriebenen Herstellungsverfahren übereinstimmen. Sowohl die Sicherheit als auch die Wirksamkeit können in denselben klinischen Prüfungen untersucht werden.
IIIb.3D. Umweltverträglichkeitsprüfung
|
(1) |
Es ist eine Umweltverträglichkeitsprüfung zur Beurteilung potenziell schädlicher Wirkungen, die sich durch die Anwendung des Tierarzneimittels für die Umwelt ergeben können, sowie zur Ermittlung der Risiken solcher Wirkungen durchzuführen. Im Rahmen der Beurteilung sind außerdem die zur Herabsetzung solcher Risiken gegebenenfalls erforderlichen Vorsichtsmaßnahmen festzustellen. |
|
(2) |
Diese Beurteilung besteht aus zwei Phasen. Die erste Beurteilungsphase ist in jedem Fall durchzuführen. Die Einzelheiten zur Beurteilung sind gemäß den von der Agentur veröffentlichten Leitlinien anzugeben. Darin ist auszuführen, wie eine potenzielle Exposition der Umwelt gegenüber diesem Arzneimittel aussehen kann und wie hoch das mit dieser Exposition verbundene Risiko ist, wobei insbesondere auf folgende Punkte einzugehen ist:
|
|
(3) |
Bei Stämmen für Lebendimpfstoffe, die zoonotisch wirken können, muss die Gefahr für den Menschen bewertet werden. |
|
(4) |
Deuten die Schlussfolgerungen der ersten Phase auf eine betreffende potenzielle Umweltexposition durch das Arzneimittel hin, muss der Antragsteller in einer zweiten Phase bewerten, welche Gefahren das Tierarzneimittel möglicherweise für die Umwelt birgt. Erforderlichenfalls sind weitere Untersuchungen über die Auswirkungen des Arzneimittels (Boden, Wasser, Luft, aquatische Systeme, Nicht-Zielorganismen) durchzuführen. |
|
(5) |
Bei DNA-Impfstoffen besteht ein besonderes Sicherheitsrisiko in der möglichen Migration der DNA in das Gonadengewebe und dem möglichen DNA-Transfer in die Keimbahnzellen geimpfter männlicher und weiblicher Tiere und damit der möglichen Übertragung auf Nachkommen. Der Antragsteller muss potenzielle Risiken, die solche immunologischen Tierarzneimittel für die menschliche Gesundheit und die Umwelt (einschließlich Pflanzen und Tieren) darstellen könnten, bewerten und erörtern. Werden potenzielle Risiken festgestellt, so sind Untersuchungen über die Auswirkungen des Impfstoffs in Abhängigkeit von seiner Verwendung bei Heimtieren oder bei zur Lebensmittelerzeugung genutzten Tieren durchzuführen, um Informationen zu diesem Punkt zu erhalten. |
IIIb.3E. Beurteilungspflicht für Tierarzneimittel, die genetisch modifizierte Organismen enthalten oder aus ihnen bestehen
|
(1) |
Bei Tierarzneimitteln, die genetisch veränderte Organismen (GVO) enthalten oder aus solchen bestehen, sind dem Zulassungsantrag die gemäß Artikel 2 und Teil C der Richtlinie 2001/18/EG sowie den spezifischen Leitlinien für GVO vorgeschriebenen Unterlagen beizufügen. |
|
(2) |
Potenzielle nachteilige Auswirkungen auf die menschliche Gesundheit und die Umwelt, die durch den Gentransfer von GVO auf andere Organismen auftreten können oder sich aus genetischen Veränderungen ergeben, sind von Fall zu Fall genau zu bewerten. Ziel einer solchen Umweltverträglichkeitsprüfung ist es, mögliche direkte und indirekte, sofortige oder spätere schädliche Auswirkungen des GVO auf die menschliche Gesundheit und die Umwelt (einschließlich Pflanzen und Tieren) zu ermitteln und zu bewerten; sie ist nach den Grundsätzen von Anhang II der Richtlinie 2001/18/EG durchzuführen. |
IIIb.3F. Rückstandsversuche, die in die vorklinischen Studien einbezogen werden müssen
|
(1) |
Für immunologische Tierarzneimittel ist es normalerweise nicht erforderlich, Rückstandsversuche durchzuführen. |
|
(2) |
Werden bei der Herstellung immunologischer Tierarzneimittel, die für zur Lebensmittelerzeugung genutzte Tiere bestimmt sind, Antibiotika, Adjuvantien, Konservierungsmittel oder andere Arzneiträgerstoffe verwendet und/oder sind diese in der endgültigen Formulierung enthalten, ist die Möglichkeit einer Exposition der Verbraucher gegenüber Rückständen in Lebensmitteln, die von behandelten Tieren stammen, sowie die Einhaltung der Rechtsvorschriften über Rückstandshöchstmengen zu berücksichtigen. Die Auswirkungen auf die Verbrauchersicherheit, die sich aus ihrem möglichen Vorhandensein im Fertigprodukt ergeben, sind zu berücksichtigen. |
|
(3) |
Bei Lebendimpfstoffen gegen gut etablierte Zoonosen kann neben den Untersuchungen zur Verbreitung auch die Bestimmung von Restimpfstofforganismen an der Injektionsstelle erforderlich sein. Falls erforderlich, sind die Wirkungen solcher Rückstände zu untersuchen. |
|
(4) |
Es ist eine Wartezeit vorzuschlagen, und deren Angemessenheit ist im Zusammenhang mit allen durchgeführten Rückstandsversuchen zu erörtern. |
IIIb.4. Teil 4: Unterlagen zur Wirksamkeit (vorklinische Studien und klinische Prüfungen)
IIIb.4A. Allgemeine Vorschriften
|
(1) |
Die folgenden allgemeinen Vorschriften müssen eingehalten werden:
|
|
(2) |
Generell müssen vorklinische Studien durch unter Feldbedingungen durchgeführte Versuche bestätigt werden.
Wenn vorklinische Studien die Angaben in der Zusammenfassung der Merkmale des Arzneimittels vollständig unterstützen, sind Versuche unter Feldbedingungen nicht erforderlich. Von begründeten Ausnahmen abgesehen, müssen die Ergebnisse der vorklinischen Studien durch Daten aus klinischen Prüfungen ergänzt werden, bei denen Chargen verwendet werden, die mit dem im Zulassungsantrag beschriebenen Herstellungsverfahren übereinstimmen. Sowohl die Sicherheit als auch die Wirksamkeit können in denselben klinischen Prüfungen untersucht werden. |
|
(3) |
Alle Prüfungen sind so ausführlich zu beschreiben, dass sie von den zuständigen Behörden ordnungsgemäß bewertet werden können. Die Gültigkeit aller in der Prüfung verwendeten Techniken muss nachgewiesen werden. |
|
(4) |
Alle erzielten Ergebnisse, ob günstig oder ungünstig, sind vorzulegen.
|
IIIb.4B. Vorklinische Studien
|
(1) |
Grundsätzlich muss der Nachweis der Wirksamkeit unter kontrollierten Laborbedingungen durch eine Provokationsprobe nach Verabreichung des immunologischen Tierarzneimittels an das Zieltier unter den empfohlenen Anwendungsbedingungen erfolgen. Soweit möglich, müssen die Bedingungen, unter denen die Belastungsinfektion erfolgt, die natürlichen Infektionsbedingungen simulieren. Es sind genaue Angaben zum Infektionsstamm und zu seiner Relevanz zu machen. |
|
(2) |
Bei Lebendimpfstoffen ist das für die Wirksamkeitsprüfung verwendete Produkt einer oder mehreren Chargen mit dem geringsten Titer bzw. der geringsten Potenz zu entnehmen. Bei anderen Arzneimitteln sind die Chargen zu verwenden, die den am Ende der Gültigkeitsdauer zu erwartenden Mindestwirkstoffgehalt oder die zu erwartende Potenz enthalten, sofern nicht anders begründet. |
|
(3) |
Der Immunmechanismus (zellständige/humorale, lokale/allgemeine Immunglobulinklassen), der nach der Verabreichung des immunologischen Tierarzneimittels an Zieltiere auf dem empfohlenen Verabreichungsweg einsetzt, ist, wenn möglich, zu spezifizieren und zu dokumentieren. |
|
(4) |
Für alle vorklinischen Studien ist Folgendes vorzulegen:
|
IIIb.4C. Klinische Prüfungen
|
(1) |
Sofern nicht anders begründet, müssen die Ergebnisse der vorklinischen Studien durch Daten aus Feldversuchen ergänzt werden, bei denen Chargen verwendet werden, die mit dem im Zulassungsantrag beschriebenen Herstellungsverfahren übereinstimmen. Sowohl die Unbedenklichkeit als auch die Wirksamkeit können in demselben Feldversuch untersucht werden. |
|
(2) |
Soweit die Wirksamkeit durch vorklinische Studien nicht bestätigt werden kann, ist die alleinige Durchführung von Feldversuchen annehmbar. |
|
(3) |
Die Angaben zu den Feldversuchen müssen so ausführlich sein, dass eine objektive Beurteilung möglich ist. Sie müssen Folgendes enthalten:
|
ABSCHNITT IV
ANFORDERUNGEN FÜR BESTIMMTE ZULASSUNGSANTRÄGE
IV.1. Anträge für Tierarzneimittel-Generika
|
IV.1.1. |
Anträge aufgrund von Artikel 18 (generische Tierarzneimittel) müssen die in Abschnitt II Teile 1 und 2 dieses Anhangs genannten Daten enthalten. Falls erforderlich, ist gemäß Artikel 18 Absatz 7 eine Umweltverträglichkeitsprüfung beizufügen. Außerdem enthält das Dossier Daten, die belegen, dass das Arzneimittel dieselbe qualitative und quantitative Wirkstoffzusammensetzung und dieselbe Darreichungsform wie das Referenzarzneimittel aufweist; und Daten zur Bioäquivalenz mit dem Referenztierarzneimittel bzw. eine Begründung, warum keine Studien unter Beachtung festgelegter Leitfäden durchgeführt wurden. Alle oralen Darreichungsformen mit sofortiger Wirkstofffreigabe gelten als dieselbe Darreichungsform.
Bei biologischen (einschließlich immunologischen) Tierarzneimitteln wird der standardmäßige generische Ansatz grundsätzlich nicht als angemessen erachtet, und es ist ein hybrider Ansatz zu verfolgen (siehe Teil IV.2.). |
|
IV.1.2. |
Bei generischen Tierarzneimitteln müssen die ausführlichen kritischen Zusammenfassungen über die Unbedenklichkeit und Wirksamkeit insbesondere auf folgende Elemente abstellen:
|
|
IV.1.3. |
Für eine generische Tierarzneimittelanwendung, die einen antimikrobiellen Stoff enthält, sind Informationen über den Grad der Resistenz, wie er aus bibliografischen Daten bekannt ist, vorzulegen. |
|
IV.1.4. |
Für eine generische Tierarzneimittelanwendung, die Antiparasitika enthält, sind Informationen über den Grad der Resistenz, wie er aus bibliografischen Daten bekannt ist, vorzulegen. |
|
IV.1.5. |
Bei generischen Tierarzneimitteln, die intramuskulär, subkutan oder transdermal verabreicht werden sollen, sind folgende zusätzliche Daten vorzulegen:
|
IV.2. Anträge für hybride Tierarzneimittel
|
IV.2.1. |
Anträge aufgrund von Artikel 19 (hybride Tierarzneimittel) betreffen Tierarzneimittel, die einem Referenztierarzneimittel ähnlich sind, aber die Bedingungen der Definition eines generischen Tierarzneimittels nicht erfüllen. |
|
IV.2.2. |
Für solche Anwendungen sind die folgenden Angaben zu machen:
|
|
IV.2.3. |
Bei biologischen (auch immunologischen) Tierarzneimitteln ist eine umfassende Vergleichbarkeitsprüfung durchzuführen, welche die Aspekte Qualität, Sicherheit und Wirksamkeit abdeckt. |
|
IV.2.4. |
Wird auf Daten verwiesen, die von einem anderen zugelassenen Tierarzneimittel stammen, ist eine Begründung für die Verwendung und die Relevanz dieser Daten für das neue Arzneimittel vorzulegen. |
|
IV.2.5. |
Der Umfang der neuen Daten, die zum Nachweis der Sicherheit und Wirksamkeit erforderlich sind, hängt von den spezifischen Merkmalen des einzelnen neuen Produkts und seinen Unterschieden zum Referenztierarzneimittel ab und wird von Fall zu Fall festgelegt. Es sind neue vorklinische und klinische Daten für das neue Produkt zu allen Aspekten vorzulegen, für die das Referenztierarzneimittel keine einschlägigen Daten bietet. |
|
IV.2.6. |
Werden neue Studien mit Chargen eines in einem Drittland genehmigten Referenztierarzneimittels durchgeführt, weist der Antragsteller nach, dass Referenztierarzneimittel gemäß Anforderungen zugelassen wurden, die den Anforderungen in der Union entsprechen, und dass sie sich so stark ähneln, dass sie einander in den vorklinischen Studien und klinischen Prüfungen substituieren können. |
IV.3. Anträge für Tierarzneimittel aus kombinierten Wirkstoffen
|
IV.3.1. |
Ein Antrag für eine fixe Arzneimittelkombination mit einzelnen Wirkstoffen, die bereits Gegenstand einer Zulassung eines Tierarzneimittels im EWR waren, ist gemäß Artikel 20 vorzulegen.
Ein Antrag für eine fixe Arzneimittelkombination, die mindestens einen neuen Wirkstoff enthält, der noch nicht für ein Tierarzneimittel im EWR zugelassen ist, ist gemäß Artikel 8 vorzulegen. |
|
IV.3.2. |
Bei Anträgen, die gemäß Artikel 20 gestellt werden, ist ein vollständiges Dossier mit den Teilen 1, 2, 3 und 4 vorzulegen. |
|
IV.3.3. |
Es ist eine fundierte wissenschaftliche Begründung auf der Grundlage gültiger therapeutischer Grundsätze für die Kombination von Wirkstoffen, einschließlich klinischer Daten, vorzulegen, aus der die Notwendigkeit und der Beitrag aller Wirkstoffe zum Zeitpunkt der Behandlung hervorgehen. |
|
IV.3.4. |
Im Allgemeinen sind alle Daten zur Sicherheit und Wirksamkeit für die fixe Arzneimittelkombination vorzulegen; Daten zur Sicherheit und Wirksamkeit für die einzelnen Wirkstoffe allein sind nicht erforderlich, außer zur Klärung ihrer individuellen pharmakologischen Eigenschaften. |
|
IV.3.5. |
Wenn dem Antragsteller ausreichend detaillierte Daten über die Sicherheit und Wirksamkeit eines einzelnen bekannten Wirkstoffs zur Verfügung stehen, könnten diese Daten vorgelegt werden, um einige Studien mit der fixen Arzneimittelkombination überflüssig zu machen, oder einschlägige Informationen beizusteuern. In diesem Fall müssen auch mögliche Wechselwirkungen zwischen den Wirkstoffen untersucht werden. |
|
IV.3.6. |
Mit der fixen Arzneimittelkombination sind eine Sicherheitsbewertung für den Anwender, eine Umweltverträglichkeitsprüfung, Studien zum Abbau von Rückständen und klinische Studien durchzuführen. |
|
IV.3.7. |
Sofern die Auslassung nicht begründet ist, muss ein Unbedenklichkeitsversuch für Zieltiere mit der endgültigen Formulierung vorgelegt werden. |
IV.4. Anträge aufgrund einer in Kenntnis der Sachlage erteilten Einwilligung
|
IV.4.1. |
Anträge aufgrund von Artikel 21 betreffen Produkte, deren Zusammensetzung, Darreichungsform und Herstellungsverfahren (einschließlich Roh- und Ausgangsstoffen, Prozessparametern und Produktionsstätten) mit denen der bereits zugelassenen Tierarzneimittel identisch sind. |
|
IV.4.2. |
Das Dossier für solche Anträge enthält nur Daten für Teil 1A und 1B gemäß Anhang I (Nummern 1 bis 6.4), sofern der Inhaber der Zulassung des bereits genehmigten Tierarzneimittels dem Antragsteller seine schriftliche Zustimmung erteilt hat, auf den Inhalt der Teile 1C, 2, 3 und 4 des Dossiers dieses Arzneimittels Bezug zu nehmen. In diesem Fall brauchen keine kritischen Expertenberichte über die Qualität, Unbedenklichkeit und Wirksamkeit vorgelegt zu werden. Der Antragsteller hat den Nachweis der schriftlichen Zustimmung mit seinem Antrag zu erbringen. |
IV.5. Anträge auf bibliografischer Grundlage
|
IV.5.1. |
Bei Tierarzneimitteln mit Wirkstoffen, deren tiermedizinische Anwendung gemäß Artikel 22 etabliert ist und die eine anerkannte Wirksamkeit sowie einen annehmbaren Grad an Unbedenklichkeit aufweisen, gelten die folgenden Sonderregelungen. |
|
IV.5.2. |
Es ist ein vollständiges Dossier (mit den Teilen 1, 2, 3 und 4) vorzulegen. Der Antragsteller muss die Teile 1 und 2, wie in diesem Anhang beschrieben, vorlegen. Für die Teile 3 und 4 ist eine detaillierte wissenschaftliche Bibliografie zusammen mit Informationen vorzulegen, die die Verbindung zwischen bibliografischen Referenzen und dem Tierarzneimittel angemessen belegen, um die Sicherheit und Wirksamkeit zu erhärten. Die bibliografischen Daten müssen möglicherweise durch einige produktspezifische Unterlagen ergänzt werden, z. B. Bewertungen der Anwendersicherheit und Umweltverträglichkeitsprüfungen oder Daten aus Rückstandsversuchen, um die vorgeschlagene(n) Wartezeit(en) zu rechtfertigen. |
|
IV.5.3. |
Für den Nachweis der etablierten tiermedizinischen Anwendung gelten die spezifischen Regeln in Teil IV.5.3.1 bis IV.5.3.12. |
|
IV.5.3.1. |
Für den Nachweis, dass Bestandteile von Tierarzneimitteln allgemein tiermedizinisch verwendet werden, sind folgende Faktoren maßgeblich:
|
|
IV.5.3.2. |
Bei verschiedenen Wirkstoffen können zum Nachweis ihrer allgemeinen tiermedizinischen Verwendung auch unterschiedliche Zeiträume erforderlich sein. Der Nachweis der etablierten tiermedizinischen Anwendung eines Arzneimittelbestandteils darf frühestens zehn Jahre, nachdem der betreffende Stoff erstmals systematisch und dokumentiert in der EU als Tierarzneimittel verwendet wurde, erbracht werden. |
|
IV.5.3.3. |
‚Tiermedizinische Verwendung‘ bedeutet nicht ausschließlich die Verwendung als zugelassenes Tierarzneimittel. Die etablierte tiermedizinische Verwendung bezieht sich auf die Verwendung für einen spezifischen therapeutischen Zweck bei der Zieltierart. |
|
IV.5.3.4. |
Wenn ein Stoff mit etablierter Anwendung für völlig neue therapeutische Indikationen vorgeschlagen wird, ist es nicht möglich, ausschließlich auf eine etablierte tiermedizinische Verwendung zu verweisen. Es sind zusätzliche Daten über das neue therapeutische Anwendungsgebiet zusammen mit geeigneten Unbedenklichkeits- und Rückstandsversuchen sowie vorklinischen und klinischen Daten vorzulegen; in einem solchen Fall ist eine Verwendung auf der Grundlage von Artikel 21 nicht möglich. |
|
IV.5.3.5. |
Die vom Antragsteller eingereichten veröffentlichten Unterlagen müssen der Öffentlichkeit frei zugänglich sein und von einer angesehenen Quelle, vorzugsweise nach Peer-Review, veröffentlicht werden. |
|
IV.5.3.6. |
Die Dokumentation muss genügend Details enthalten, um eine unabhängige Beurteilung zu ermöglichen. |
|
IV.5.3.7. |
In den Unterlagen ist auf alle Aspekte der Beurteilung der Unbedenklichkeit und/oder Wirksamkeit des Arzneimittels für die vorgeschlagene Indikation bei der Zieltierart unter Verwendung des vorgeschlagenen Verabreichungswegs und Dosierungsschemas einzugehen. Sie umfassen einen Überblick über die einschlägigen Veröffentlichungen bzw. verweisen auf einen solchen; dabei sind vor und nach der Zulassung durchgeführte Studien und wissenschaftliche Veröffentlichungen über die Erfahrungen aus epidemiologischen Studien, insbesondere vergleichenden epidemiologischen Studien, zu berücksichtigen. |
|
IV.5.3.8. |
Alle Unterlagen, sowohl günstige als auch ungünstige, sind vorzulegen. In Bezug auf die Vorschriften über die allgemeine tiermedizinische Verwendung ist insbesondere zu präzisieren, dass auch ein bibliografischer Verweis auf andere Informationsquellen (beispielsweise Untersuchungen nach der Zulassung, epidemiologische Studien usw.) und nicht nur Daten von Versuchen und Prüfungen als gültiger Nachweis für die Unbedenklichkeit und Wirksamkeit eines Arzneimittels dienen können, wenn der Antragsteller erläutert und begründet, warum diese Nachweise angeführt werden. |
|
IV.5.3.9. |
Öffentliche Beurteilungsberichte oder Zusammenfassungen können nicht als ausreichende Informationsquelle angesehen werden, abgesehen von dem Beurteilungsbericht, der von der Agentur nach der Beurteilung eines Antrags auf Festsetzung von Rückstandshöchstmengen veröffentlicht wird, der in angemessener Weise als wissenschaftliche Dokumentation, insbesondere für Unbedenklichkeitsversuche, verwendet werden kann. |
|
IV.5.3.10. |
Besondere Aufmerksamkeit ist auf etwaige fehlende Informationen zu richten und es ist zu begründen, warum der Nachweis eines annehmbaren Grades an Unbedenklichkeit und/oder Wirksamkeit erbracht werden kann, obwohl bestimmte Informationen fehlen. |
|
IV.5.3.11. |
Aus den kritischen Expertenberichten über die Unbedenklichkeit und Wirksamkeit hat hervorzugehen, inwiefern vorgelegte Daten, die ein anderes als das in Verkehr zu bringende Arzneimittel betreffen, relevant sind. Es ist zu beurteilen, ob das in der Bibliografie geprüfte Arzneimittel trotz der bestehenden Unterschiede als demjenigen Arzneimittel befriedigend oder wissenschaftlich nachweisbar gleich betrachtet werden kann, für das der Zulassungsantrag gestellt wurde. |
|
IV.5.3.12. |
Nach dem Inverkehrbringen gemachte Erfahrungen mit anderen Arzneimitteln, die die gleichen Bestandteile enthalten, sind von besonderer Bedeutung, und die Antragsteller müssen diesen Aspekt besonders berücksichtigen. |
IV.6. Anträge für begrenzte Märkte
|
IV.6.1. |
Eine Zulassung kann bei Fehlen umfassender Daten zur Sicherheit und/oder Wirksamkeit für einen begrenzten Markt erteilt werden, wenn der Antragsteller gemäß Artikel 23 nachweist, dass das Produkt für die Verwendung in einem begrenzten Markt bestimmt ist und dass der Nutzen der Verfügbarkeit des neuen Produkts das Risiko überwiegt, das mit dem Fehlen einiger der in diesem Anhang vorgeschriebenen Daten zur Sicherheit oder Wirksamkeit verbunden ist. |
|
IV.6.2. |
Für diese Anträge muss der Antragsteller die Teile 1 und 2, wie in diesem Anhang beschrieben, vorlegen. |
|
IV.6.3. |
Für die Teile 3 und 4 können einige der in diesem Anhang geforderten Daten zur Sicherheit oder Wirksamkeit weggelassen werden. Hinsichtlich des Umfangs der Sicherheits- und Wirksamkeitsdaten, die weggelassen werden können, sind die von der Agentur veröffentlichten einschlägigen Leitlinien zu berücksichtigen. |
IV.7. Anträge unter außergewöhnlichen Umständen
|
IV.7.1. |
Unter außergewöhnlichen Umständen im Zusammenhang mit der Gesundheit von Mensch und Tier kann gemäß Artikel 25 eine Zulassung eines Tierarzneimittels vorbehaltlich bestimmter spezifischer Verpflichtungen, Bedingungen und/oder Einschränkungen erteilt werden. |
|
IV.7.2. |
Bei solchen Anträgen muss der Antragsteller den in diesem Anhang beschriebenen Teil 1 vorlegen, zusammen mit einer Begründung, warum der Nutzen der sofortigen Verfügbarkeit des Tierarzneimittels auf dem Markt das Risiko überwiegt, das dadurch bedingt ist, dass bestimmte Unterlagen zur Qualität, Sicherheit und Wirksamkeit nicht vorgelegt wurden. |
|
IV.7.3. |
Für die Teile 2 und 4 können bestimmte der in diesem Anhang geforderten Daten zu Qualität, Sicherheit und Wirksamkeit weggelassen werden, wenn der Antragsteller begründet, dass diese Daten zum Zeitpunkt der Einreichung nicht vorgelegt werden können. Bei der Festlegung der grundlegenden Anforderungen für alle derartigen Anwendungen sind die von der Agentur veröffentlichten einschlägigen Leitlinien zu berücksichtigen. |
|
IV.7.4. |
Studien nach der Zulassung können als Teil der Bedingungen für die Zulassung verlangt werden und müssen gemäß den in diesem Anhang dargelegten allgemeinen Grundsätzen für Qualitäts-, Sicherheits- und Wirksamkeitsprüfungen sowie gemäß den einschlägigen Leitliniendokumenten konzipiert, durchgeführt, analysiert und vorgelegt werden, je nachdem, welche Fragestellung in der Studie behandelt werden soll. |
ABSCHNITT V
ANFORDERUNGEN FÜR ZULASSUNGSANTRÄGE FÜR BESONDERE TIERARZNEIMITTEL
In diesem Abschnitt werden für bestimmte Tierarzneimittel je nach Eigenart der darin enthaltenen Wirkstoffe spezifische Anforderungen festgelegt.
V.1. Tierarzneimittel für neuartige Therapien
V.1.1. Allgemeine Vorschriften
|
V.1.1.1. |
Je nach Wirkstoff und Wirkungsweise könnte ein Tierarzneimittel für neuartige Therapien unter eine der drei Produktkategorien fallen:
|
|
V.1.1.2. |
Im Allgemeinen müssen Anträge auf Zulassung von Tierarzneimitteln für neuartige Therapien gemäß der Definition in Artikel 4 Absatz 43 dem Format und den Datenanforderungen entsprechen, die in Abschnitt II oder III dieses Anhangs beschrieben sind, je nachdem, wie die neuartige Therapie eingestuft wird. Ein vollständiges Dossier, das die Teile 1, 2, 3 und 4 enthält, ist in der Regel in Übereinstimmung mit den in Abschnitt II oder III beschriebenen Anforderungen und allen von der Agentur veröffentlichten einschlägigen Leitlinien einzureichen. Abweichungen von den Anforderungen dieses Anhangs sind in begründeten Fällen möglich. Gegebenenfalls und unter Berücksichtigung der Besonderheiten von Arzneimitteln für neuartige Therapien können zusätzliche Anforderungen für bestimmte Arten von Arzneimitteln relevant sein. |
|
V.1.1.3. |
Die Herstellungsverfahren für Tierarzneimittel für neuartige Therapien müssen den Grundsätzen der Guten Herstellungspraxis entsprechen, die erforderlichenfalls angepasst werden, um die Besonderheiten dieser Produkte zu berücksichtigen. Es sind spezifische Leitlinien für Tierarzneimittel für neuartige Therapien zu erstellen, die die Besonderheiten ihres Herstellungsverfahrens angemessen berücksichtigen. |
|
V.1.1.4. |
Je nach der spezifischen Beschaffenheit eines Arzneimittels für neuartige Therapien kann die Verwendung des Produkts möglicherweise mit spezifischen Risiken verbunden sein. Diese Risiken werden unter Anwendung einer Risikoprofilierungsmethode ermittelt, um die mit dem spezifischen Produkt verbundenen Risiken und die Risikofaktoren, die zu diesen Risiken beitragen, zu identifizieren. In diesem Zusammenhang wären Risiken alle potenziellen ungünstigen Auswirkungen, die mit der Verwendung des neuartigen Therapieprodukts verbunden sein können und die für die Zielbevölkerung und/oder den Anwender, den Verbraucher und/oder die Umwelt von Bedeutung sind. Die Risikoanalyse kann sich auf die gesamte Entwicklung beziehen. Zu den Risikofaktoren, die berücksichtigt werden können, gehören die Herkunft des Ausgangsmaterials (Zellen usw.), die Wirkungsweise im Tier (Proliferation, Auslösung einer Immunantwort, Verbleib im Körper usw.), der Grad der Zellmanipulation (z. B. das Herstellungsverfahren), die Kombination des Wirkstoffs mit bioaktiven Molekülen oder Strukturmaterialien, das Ausmaß der Replikationskompetenz der in vivo verwendeten Viren oder Mikroorganismen, der Grad der Integration von Nukleinsäuresequenzen oder Genen in das Genom, die Langzeitfunktionsfähigkeit, das Onkogenitätsrisiko, die Off-Target-Effekte und die Art der Verabreichung oder Verwendung. |
|
V.1.1.5. |
Auf der Grundlage der Auswertung der Informationen über die ermittelten Risiken und Risikofaktoren wird ein spezifisches Profil jedes einzelnen Risikos erstellt, das mit einem bestimmten Produkt verbunden ist und dazu verwendet werden kann, zu bestimmen und zu begründen, inwiefern der vorgelegte Datensatz die erforderlichen Garantien für Qualität, Sicherheit und Wirksamkeit bietet und geeignet ist, einen Zulassungsantrag zu unterstützen, insbesondere für diejenigen Aspekte der Arzneimittel für neuartige Therapien, die über den derzeitigen Kenntnisstand hinausgehen. |
|
V.1.1.6. |
Um Datenlücken oder Unsicherheiten zum Zeitpunkt der Produktzulassung zu überwinden, kann die Durchführung von Maßnahmen oder Studien nach der Zulassung von Fall zu Fall in Betracht gezogen werden. Um frühe oder verzögerte Signale von Nebenwirkungen zu erkennen, klinische Folgen solcher Reaktionen zu vermeiden und eine rechtzeitige Behandlung zu gewährleisten sowie Informationen über die langfristige Sicherheit und Wirksamkeit von Arzneimitteln für neuartige Therapien zu gewinnen, sind in einem Risikomanagementplan die vorgesehenen Maßnahmen zur Sicherstellung einer solchen Weiterverfolgung im Einzelnen darzulegen. |
|
V.1.1.7. |
Für jedes Arzneimittel für neuartige Therapien, insbesondere für solche, die in der Veterinärmedizin als im Entstehen begriffen gelten, wird empfohlen, rechtzeitig vor der Einreichung des Zulassungsdossiers den Rat der Agentur einzuholen, um das Produkt zu klassifizieren, die anzuwendende Dossierstruktur zu bestimmen und sachdienliche Informationen über den zusätzlichen Datensatz zu erhalten, der zur Erhärtung von Qualität, Sicherheit und Wirksamkeit erforderlich sein kann. |
V.1.2. Qualitätsanforderungen
|
V.1.2.1. |
Im Allgemeinen sind eine Beschreibung der Zusammensetzung, des Herstellungsverfahrens, der Konsistenz der Produktion, der Kontrollen der Ausgangsstoffe, der während des Herstellungsverfahrens durchgeführten Kontrollen, der Prüfung des Fertigprodukts einschließlich der Durchführung einer Prüfung der Wirkung oder einer Quantifizierung des Wirkstoffs sowie Haltbarkeitsdaten vorzulegen. |
|
V.1.2.2. |
Die Datenanforderungen für die Herstellung und Prüfung von Tierarzneimitteln für neuartige Therapien biologischen Ursprungs, die als biologisches oder immunologisches Produkt eingestuft sind, entsprechen im Allgemeinen denen für biologische oder immunologische Arzneimittel (wie in Abschnitt III dieses Anhangs beschrieben), einschließlich der Notwendigkeit einer entsprechenden Potenzprüfung. Es kann Fälle geben, in denen zusätzliche Anforderungen gelten, z. B. für Zellen und Vektorgenkonstrukte. |
|
V.1.2.3. |
Für Tierarzneimittel für neuartige Therapien, die durch chemische Synthese hergestellt werden, gelten im Allgemeinen die gleichen Datenanforderungen wie für Tierarzneimittel, die keine biologischen Arzneimittel sind (wie in Abschnitt II dieses Anhangs beschrieben). Es kann Fälle geben, in denen zusätzliche Anforderungen gelten, z. B. die Anforderung einer einschlägigen Potenzprüfung. |
V.1.3. Sicherheitsanforderungen
|
V.1.3.1. |
Je nach Art des Produkts und seines Verwendungszwecks können weitere Daten zur Bewertung der Sicherheit für das Zieltier, den Anwender, den Verbraucher oder die Umwelt sachdienlich sein, wie in einer Risikoanalyse im Einzelfall zu ermitteln ist. |
|
V.1.3.2. |
Die Vorschriften gemäß Richtlinie 2001/18/EG sind zu berücksichtigen, wenn das behandelte Tier selbst ein genetisch veränderter Organismus werden könnte. Die Richtlinie 2001/18/EG gilt zwar für Fertigprodukte, die genetisch veränderte Organismen enthalten, sie ist aber nach wie vor der beste derzeit verfügbare technische Leitfaden für die Auflistung der erforderlichen Daten. Insbesondere geht es um die Integrationsrate der DNA in Keimzellen (und damit die Übertragbarkeit auf die Nachkommen) bzw. die mögliche Übertragung der gentechnisch veränderten Zellen auf die Nachkommen. Es sei auch darauf hingewiesen, dass diese Problematik bei der Betrachtung von Heimtieren und für zur Lebensmittelgewinnung genutzten Tieren (menschlicher Verzehr von Produkten, die gentechnisch veränderte Organismen enthalten) nicht völlig gleich ist. |
|
V.1.3.3. |
Für Stoffe, die zur Integration in das Genom oder zur Genom-Editierung bestimmt sind, müssen geeignete Tests durchgeführt werden, um das Risiko von Off-Target-Veränderungen und/oder der Insertionsmutagenese zu bewerten. |
V.1.4. Wirksamkeitsanforderungen
|
V.1.4.1. |
Die Anforderungen an die Wirksamkeitsdaten unterscheiden sich vor allem in Abhängigkeit von den beabsichtigten Indikationen für die Anwendung bei den Zieltierarten. Je nach Einstufung des Arzneimittels für neuartige Therapien und Verwendungszweck bei der Zieltierart können die in den Abschnitten II oder III genannten Anforderungen an die Wirksamkeit für ein Arzneimittel für neuartige Therapien gelten. |
|
V.1.4.2. |
Die beanspruchten Indikationen müssen durch entsprechende Daten bei der Zieltierart belegt werden. |
V.1.5. Spezifische Datenanforderungen für bestimmte Arten von Arzneimitteln für neuartige Therapien
V.1.5.1. Grundlagen
|
V.1.5.1.1. |
Unter Berücksichtigung der Besonderheiten von Arzneimitteln für neuartige Therapien können zusätzlich zu den Standardanforderungen an die Bewertung von Qualität, Sicherheit und Wirksamkeit spezifische Anforderungen angebracht sein. |
|
V.1.5.1.2. |
In den folgenden Abschnitten werden spezifische Anforderungen hervorgehoben, die für bestimmte Arten von Arzneimitteln für neuartige Therapien zu berücksichtigen sind. Diese spezifischen Anforderungen, die für einen bestimmten Typ eines Arzneimittels für neuartige Therapien festgelegt wurden, stellen eine nicht erschöpfende Liste von Anforderungen dar, die möglicherweise von Fall zu Fall und auf der Grundlage einer Risikoanalyse an das betreffende Produkt angepasst werden müssen. |
|
V.1.5.1.3. |
In allen Fällen und insbesondere bei neuartigen Therapien, die im Bereich der Tiermedizin als im Entstehen begriffen gelten, müssen die Antragsteller den aktuellen Stand der veterinärmedizinischen Kenntnisse und die von der Agentur und der Kommission veröffentlichten wissenschaftlichen Leitlinien im Einklang mit Abschnitt I dieses Anhangs berücksichtigen. |
V.1.5.2. Tierarzneimittel für die Gentherapie
|
V.1.5.2.1. |
Gentherapeutika sind biologische Tierarzneimittel, die einen Wirkstoff enthalten, der eine rekombinante Nukleinsäure enthält oder daraus besteht, der im Tier verwendet oder ihm verabreicht wird, um eine Nukleinsäuresequenz zu regulieren, zu reparieren, zu ersetzen, hinzuzufügen oder zu entfernen. Ihre therapeutische, prophylaktische oder diagnostische Wirkung steht in unmittelbarem Zusammenhang mit der rekombinanten Nukleinsäuresequenz, die sie enthalten, oder mit dem Produkt, das aus der Expression dieser Sequenz resultiert. |
|
V.1.5.2.2. |
Zusätzlich zu den Anforderungen in Bezug auf die Daten der Abschnitte II und III gelten folgende Anforderungen:
|
V.1.5.3. Für die regenerative Medizin, die Gewebezüchtung oder die Zelltherapie entwickelte Tierarzneimittel
|
V.1.5.3.1. |
Unter regenerativer Medizin versteht man einen weiten Bereich von Produkten und Therapien mit dem allgemeinen Ziel der Wiederherstellung von Funktionen. Zu diesen Arzneimitteln gehören zellbasierte Therapien, zu denen auch Produkte aus Gewebezüchtung zählen. |
|
V.1.5.3.2. |
Tierarzneimittel für Zelltherapien sind biologische Tierarzneimittel, die aus Zellen oder Geweben bestehen, die in Bezug auf ihre Natur oder Funktion substanziell bearbeitet wurden, sodass biologische Merkmale, physiologische Funktionen oder strukturelle Eigenschaften, die für die beabsichtigte klinische Verwendung relevant sind, verändert wurden, oder aus Zellen oder Geweben, die im Empfänger im Wesentlichen nicht der-/denselben Funktion(en) dienen sollen wie im Spender, oder es enthält derartige Zellen oder Gewebe. Ihnen werden Eigenschaften zur Behandlung, Vorbeugung oder Diagnose von Krankheiten durch pharmakologische, immunologische oder metabolische Wirkungen der enthaltenen Zellen oder Gewebe zugeschrieben und es wird zu diesem Zweck im Tier verwendet oder ihm verabreicht oder um ein Gewebe zu regenerieren, zu reparieren oder zu ersetzen. |
|
V.1.5.3.3. |
Zusätzlich zu den Anforderungen in Bezug auf die Daten der Abschnitte II und III gelten folgende Anforderungen:
|
V.1.5.4. Speziell für die Phagentherapie entwickeltes Tierarzneimittel
|
V.1.5.4.1. |
Bakteriophagen sind Viren, die zur Vermehrung auf bakterielle Wirte angewiesen sind und sehr spezifisch auf bestimmte Bakterienstämme wirken. Die Phagentherapie kann z. B. als Alternative zu Antibiotika eingesetzt werden. Im Allgemeinen bestehen Bakteriophagen aus einem Genom, das aus ein- oder doppelsträngiger DNA oder RNA besteht und von einem Proteinkapsid eingekapselt ist. Aufgrund der Vielfalt der vorgesehenen Behandlungsziele und der Spezifität der Bakteriophagen wird es notwendig sein, den geeigneten Bakteriophagenstamm gegen den krankheitsverursachenden Bakterienstamm von Fall zu Fall für den individuellen Krankheitsausbruch auszuwählen. |
|
V.1.5.4.2 |
Die Qualität und Quantität der Bakteriophagen, die im Fertigprodukt verwendet werden sollen, sind normalerweise variabel. Eine feste qualitative und quantitative Zusammensetzung von Bakteriophagen wird daher nicht der Normalfall sein, da die Phagen laufend angepasst werden müssen. Darauf aufbauend muss ein Zellenbestand an Bakteriophagenstämmen etabliert und gepflegt werden (vergleichbar mit einem Multi-Strain-Ansatz). |
|
V.1.5.4.3. |
Bakteriophagen sowie Wirtsbakterien/Master-Zellbänke für die Herstellung sollen vorzugsweise auf Basis eines Master-Zellsystems hergestellt werden. Es ist zu bestätigen, dass der verwendete Bakteriophage lytisch ist. |
|
V.1.5.4.4. |
Das Nichtvorhandensein von Resistenzgenen und das Nichtvorhandensein von Genen, die für Virulenzfaktoren kodieren, muss auf allen Stammsamen nachgewiesen werden. |
|
V.1.5.4.5. |
Die Indikation dient der prophylaktischen, metaphylaktischen und/oder therapeutischen Behandlung einer oder mehrerer spezifischer Infektion(en) oder Infektionskrankheit(en). Die Wirksamkeit der Behandlung ist mit der lytischen Wirkung der Phagen verbunden, die denjenigen Bakteriophagen bakterizide Wirkung verleiht, die für den betreffenden Bakterienstamm spezifisch sind. |
|
V.1.5.4.6. |
Bei genetisch veränderten Phagen ist die genetische Veränderung zu beschreiben. |
V.1.5.5. Tierarzneimittel aus der Nanotechnologie
|
V.1.5.5.1. |
Nanotechnologien werden in erster Linie als Technologie zur Erzeugung von Trägern für chemisch synthetisierte Stoffe gesehen, können aber auch Träger für biologische Stoffe sein. Die Verwendung von Nanopartikeln kann eine Möglichkeit sein, die Abgabe von schwer löslichen oder toxischen Stoffen zu steuern. |
|
V.1.5.5.2. |
Die ‚Nanotechnologie‘ entspricht dem Design, der Charakterisierung und der Produktion von Nanomaterialien durch die Kontrolle von Form und Größe im Nanobereich (bis zu etwa 100 nm). |
|
V.1.5.5.3. |
Als ‚Nanopartikel‘ werden solche bezeichnet, die zwei oder mehr Dimensionen auf der Nanoskala haben. |
|
V.1.5.5.4. |
Im Bereich der Veterinärmedizin sind Nanopartikel für das Arzneimittelabgabesystem als ‚Produkte aus der Nanotechnologie‘ von Belang: Nanopartikel werden mit Stoffen konjugiert, um die pharmakokinetischen und/oder pharmakodynamischen Eigenschaften zu verändern. mRNA-Medikamente werden eher in Nanopartikel-Anwendungen verkapselt. |
|
V.1.5.5.5. |
Zusätzlich zu den Anforderungen in Bezug auf die Daten zur Qualität der Abschnitte II und III gelten folgende Anforderungen:
|
|
V.1.5.5.6. |
Im Hinblick auf die Sicherheit kann die Art der Gefahren, die durch die Verwendung von Nanopartikeln für die Arzneimittelverabreichung eingeführt werden, über die konventionellen Gefahren hinausgehen, die durch Chemikalien in klassischen Verabreichungsmatrizen entstehen. Daher sind die folgenden Aspekte hinsichtlich der Sicherheit zu berücksichtigen:
|
V.1.5.6. Antisense-RNA-Therapie und RNA-Interferenz-Therapieprodukte
|
V.1.5.6.1. |
Antisense-Therapie- und Interferenztherapie-Produkte können durch Synthese oder durch rekombinante Techniken erzeugt werden. |
|
V.1.5.6.2. |
Antisense-RNA ist eine einzelsträngige RNA, die komplementär zu einer proteincodierenden Boten-RNA ist, mit der sie hybridisiert und dadurch deren Translation in Protein blockiert. |
|
V.1.5.6.3. |
RNA-Interferenz ist ein biologischer Prozess, bei dem RNA-Moleküle die Genexpression oder -translation hemmen, indem sie gezielt mRNA-Moleküle neutralisieren. |
|
V.1.5.6.4. |
Zusätzlich zu den Anforderungen in Bezug auf die Daten der Abschnitte II und III gelten folgende Anforderungen:
|
V.2. Impfantigen-Stammdokumentation
Für besondere immunologische Tierarzneimittel und abweichend von Abschnitt IIIb Teil 2 wird das Konzept einer Impfantigen-Stammdokumentation eingeführt.
|
V.2.1. |
Grundlagen |
|
V.2.1.1. |
Im Sinne dieses Anhangs ist unter einer Impfantigen-Stammdokumentation ein eigenständiger Teil des Antragsdossiers für einen Impfstoff zu verstehen, in dem alle sachdienlichen Angaben zur Qualität jedes einzelnen Wirkstoffs, der Bestandteil des Tierarzneimittels ist, enthalten sind. Dieser eigenständige Teil kann für einen oder für mehrere monovalente und/oder kombinierte Impfstoffe gemeinsam gelten, die vom gleichen Antragsteller oder Zulassungsinhaber eingereicht werden. |
|
V.2.1.2. |
Die Verwendung der Impfantigen-Stammdokumentation ist optional. Bei kombinierten Impfstoffen sind die Impfantigene anzugeben, die in die Impfantigen-Stammdokumentation aufgenommen werden sollen, und für jedes dieser Antigene ist eine eigene Impfantigen-Stammdokumentation erforderlich. |
|
V.2.1.3. |
Antrag und Genehmigung einer Impfantigen-Stammdokumentation müssen mit den von der Agentur veröffentlichten einschlägigen Leitlinien übereinstimmen. |
|
V.2.2. |
Inhalt
Das Dossier der Impfantigen-Stammdokumentation muss die Angaben in den Teilen V.2.2.1 bis V.2.3.3 enthalten, die den entsprechenden Abschnitten von Teil 1 (Zusammenfassung der Unterlagen) und Teil 2 (Qualitätsbezogene Unterlagen) gemäß Abschnitt IIIb dieses Anhangs entnommen sind: |
|
V.2.2.1. |
Zusammenfassung der Unterlagen (Teil 1)
Es sind der Name und die Anschrift des Herstellers und der Unternehmensstandorte, die an den verschiedenen Stufen der Herstellung und Kontrolle des Wirkstoffs beteiligt sind, zusammen mit Kopien der entsprechenden Herstellungserlaubnisse anzugeben. |
|
V.2.2.2. |
Qualitative und quantitative Zusammensetzung der Bestandteile (Teil 2.A)
Die vollständige und genaue Bezeichnung des Wirkstoffs (z. B. Virus- oder Bakterienstamm, Antigen) ist anzugeben, so wie sie auch bei jedem Fertigprodukt genannt wird. Es sind Informationen über die für den Wirkstoff relevante Produktentwicklung vorzulegen. |
|
V.2.2.3. |
Angaben über die Herstellungsweise (Teil 2.B)
Es ist Angaben über die Herstellungsweise für den Wirkstoff vorzulegen, einschließlich der Validierung der wichtigsten Produktionsstufen und gegebenenfalls einer Begründung für die vorgeschlagene Zwischenlagerung. Für inaktivierte Impfstoffe sind die für die Inaktivierung des Wirkstoffs relevanten Daten, einschließlich der Validierung des Inaktivierungsprozesses, vorzulegen. |
|
V.2.2.4. |
Herstellung und Kontrolle der Ausgangsstoffe (Teil 2.C) |
|
V.2.2.4.1. |
Es gelten die in Abschnitt IIIb.2C beschriebenen und für den Wirkstoff relevanten Standardanforderungen. |
|
V.2.2.4.2. |
Es sind Informationen über den Wirkstoff (z. B. Virus-/Bakterienstamm), Substrat (Zellen, Kulturmedium) und alle bei der Herstellung des Wirkstoffs verwendeten Rohstoffe (die in einem Arzneibuch aufgeführt sind oder nicht, biologischer oder nicht biologischer Natur) anzugeben. |
|
V.2.2.4.3. |
Das Dossier muss die Spezifikationen sowie Informationen über die durchgeführten Prozesse und Qualitätskontrollprüfungen, die für alle Chargen von Ausgangsstoffen durchzuführen sind, und die Ergebnisse für eine Charge aller verwendeten Bestandteile umfassen. |
|
V.2.2.4.4. |
Gegebenenfalls ist eine Risikobewertung für TSE und Fremderreger vorzulegen. Es ist zu beachten, dass für die TSE- und Fremderreger-Risikobewertung die Zieltierart zu berücksichtigen ist, die für die Fertigprodukte unter Bezugnahme auf die Impfantigen-Stammdokumentation beibehalten wird. Warnhinweise oder Verwendungsbeschränkungen können auf der Ebene der Impfantigen-Stammdokumentation eingebracht werden, je nach den vorliegenden Informationen, die während der Risikoanalyse auf der Ebene des Fertigprodukts abgeschwächt werden können. |
|
V.2.2.4.5. |
Wird der Wirkstoff durch rekombinante Techniken gewonnen, sind alle entsprechenden relevanten Daten über das gentechnisch veränderte Virus/Bakterium vorzulegen. |
|
V.2.2.5. |
Kontrollen während des Herstellungsprozesses (Teil 2.D)
Die in Abschnitt IIIb.2D beschriebenen Standardanforderungen gelten für die prozessbegleitenden Kontrollen, die während der Herstellung des Wirkstoffs durchgeführt werden, einschließlich der Validierungen von Schlüsselkontrollprüfungen und, falls relevant, einer vorgeschlagenen Zwischenlagerung (vor der Mischung). |
|
V.2.2.6. |
Gleichbleibende Qualität der Chargen (Teil 2.F)
Für den Nachweis der gleichbleibenden Qualität bei der Herstellung des Antigens gelten die in Abschnitt IIIb.2F beschriebenen Standardanforderungen. |
|
V.2.2.7. |
Haltbarkeit (Teil 2.G)
Es gelten die in Abschnitt IIIb.2G beschriebenen Standardanforderungen zum Nachweis der Haltbarkeit des Antigens und gegebenenfalls der Zwischenlagerung. |
|
V.2.3. |
Beurteilung und Zertifizierung |
|
V.2.3.1. |
Für Impfstoffe, die neue Impfantigene enthalten und für die es noch keine Impfantigen-Stammdokumentation gibt, legt der Antragsteller der Agentur ein vollständiges Dossier für den Zulassungsantrag vor, das alle Impfantigen-Stammdokumentationen für jedes einzelne Impfantigen enthält, für das eine Impfantigen-Stammdokumentation verwendet werden soll. Die Agentur nimmt eine wissenschaftliche und technische Beurteilung jeder Impfantigen-Stammdokumentation vor. Fällt die Beurteilung positiv aus, so wird für jede Impfantigen-Stammdokumentation eine Bescheinigung über die Einhaltung der Unionsrechtsvorschriften ausgestellt, der der Beurteilungsbericht beigefügt wird. Diese Bescheinigung ist in der gesamten Europäischen Union gültig. |
|
V.2.3.2. |
Teil V.2.3.1 gilt ebenso für jeden Impfstoff, der aus einer neuartigen Kombination von Impfantigenen besteht, und zwar ungeachtet dessen, ob eines oder mehrere dieser Impfantigene Bestandteil von in der EU bereits zugelassenen Impfstoffen ist/sind. |
|
V.2.3.3. |
Änderungen am Inhalt einer Impfantigen-Stammdokumentation für einen in der Europäischen Union zugelassenen Impfstoff werden von der Agentur einer wissenschaftlichen und technischen Beurteilung unterzogen. Fällt die Beurteilung positiv aus, erteilt die Agentur für die Impfantigen-Stammdokumentation eine Bescheinigung über deren Einhaltung der Unionsvorschriften. Diese Bescheinigung ist in der gesamten Europäischen Union anwendbar. |
V.3. Multi-Strain-Dossier
|
V.3.1. |
Für bestimmte immunologische Tierarzneimittel wird abweichend von den Bestimmungen für Wirkstoffe in Teil 2 Abschnitt IIIb das Konzept eines Multi-Strain-Dossiers eingeführt. |
|
V.3.2. |
Unter einem Multi-Strain-Dossier ist ein Einzeldossier zu verstehen, das die sachdienlichen Daten für eine einmalige gründliche wissenschaftliche Beurteilung der verschiedenen möglichen Stämme/Kombinationen von Stämmen enthält und die Genehmigung von Impfstoffen gegen antigenvariable inaktivierte Viren oder Bakterien ermöglicht, für die eine rasche oder häufige Änderung der Zusammensetzung von Impfstoffformulierungen erforderlich ist, um die Wirksamkeit im Hinblick auf die epidemiologische Situation vor Ort zu gewährleisten. Je nach der epidemiologischen Situation, in der der Impfstoff verwendet werden soll, könnte eine Reihe von Stämmen aus den im Dossier enthaltenen ausgewählt werden, um ein Fertigprodukt zu formulieren. |
|
V.3.3. |
Jedes Multi-Strain-Dossier gilt nur für eine Virusart, Bakteriengattung oder einen Vektor für eine bestimmte Krankheit; Mischungen verschiedener Viren, die zu unterschiedlichen Familien, Gattungen oder Arten gehören, oder Bakterien, die zu unterschiedlichen Familien oder Gattungen gehören, können im Rahmen eines Multi-Strain-Dossiers nicht zugelassen werden. |
|
V.3.4. |
Bei neuen Anträgen auf Zulassung mit Multi-Strain-Dossier, bei denen es für ein bestimmtes Virus/Bakterium/eine bestimmte Krankheit noch keinen zugelassenen Multi-Strain-Impfstoff gibt, ist die Eignung für das Multi-Strain-Dossier-Konzept von der Agentur vor Einreichung des Antrags zu bestätigen. |
|
V.3.5. |
Die Einreichung von Multi-Strain-Dossiers muss den von der Agentur veröffentlichten einschlägigen Leitlinien entsprechen. |
V.4. Impfstoff-Plattformtechnologie
|
V.4.1. |
Grundlagen |
|
V.4.1.1. |
Die Impfstoff-Plattformtechnologie ist eine Sammlung von Technologien, denen die Verwendung eines ‚Gerüst‘-Trägers oder Vektors gemeinsam ist, der für jeden von der Plattform abgeleiteten Impfstoff mit einem anderen Antigen oder einer anderen Gruppe von Antigenen modifiziert wird. Dazu gehören unter anderem proteinbasierte Plattformen (virusähnliche Partikel), DNA-Impfstoffplattformen, mRNA-basierte Plattformen, Replikons (selbstreplizierende RNA) sowie virale und bakterielle Vektorimpfstoffe. |
|
V.4.1.2. |
Bei Zulassungsanträgen für immunologische Tierarzneimittel, die auf der Grundlage von Impfstoff-Plattformtechnologien hergestellt werden, wird davon ausgegangen, dass die Datenanforderungen reduziert werden können. Für das erste Produkt eines Herstellers, das auf einer bestimmten Plattformtechnologie für eine bestimmte Zieltierart basiert, ist ein vollständiges Dossier erforderlich. Zum Zeitpunkt der Einreichung des ersten (vollständigen) Dossiers auf der Grundlage der Plattformtechnologie kann der Antragsteller parallel dazu eine ‚Plattformtechnologie-Stammdokumentation‘ einreichen, die alle Daten in Bezug auf die Plattform enthält, für die mit hinreichender wissenschaftlicher Sicherheit davon ausgegangen werden kann, dass sie unabhängig von den betroffenen Antigenen/Genen, die der Plattform hinzugefügt werden, unverändert bleiben. Die Art der Daten, die in die Plattformtechnologie-Stammdokumentation aufzunehmen sind, hängt von der Art der Plattform ab. |
|
V.4.1.3. |
Sobald eine Plattformtechnologie-Stammdokumentation zertifiziert ist, kann das Zertifikat verwendet werden, um die entsprechenden Datenanforderungen in nachfolgenden Zulassungsanträgen zu erfüllen, die auf derselben Plattform basieren und für dieselbe Zieltierart bestimmt sind. |
V.4.2. Beurteilung und Zertifizierung
|
V.4.2.1. |
Die Einreichung der Plattformtechnologie-Stammdokumentationen muss den von der Agentur veröffentlichten einschlägigen Leitlinien entsprechen. Die Agentur nimmt eine wissenschaftliche und technische Beurteilung jeder Plattformtechnologie-Stammdokumentation vor. Fällt die Beurteilung positiv aus, so wird eine Bescheinigung über die Einhaltung der geltenden Unionsvorschriften für die Stammdokumentation der Plattformtechnologie ausgestellt, der der Beurteilungsbericht beizufügen ist. Diese Bescheinigung ist in der gesamten Europäischen Union gültig. |
|
V.4.2.2. |
Änderungen am Inhalt einer Plattformtechnologie-Stammdokumentation für einen in der Europäischen Union zugelassenen Impfstoff werden von der Agentur einer wissenschaftlichen und technischen Beurteilung unterzogen. |
|
V.4.2.3. |
Fällt die Bewertung positiv aus, erteilt die Agentur für die Stammdokumentation der Plattformtechnologie eine Bescheinigung über deren Einhaltung der Unionsvorschriften. |
V.5. Zugelassene homöopathische Tierarzneimittel
|
V.5.1. |
Qualität (Teil 2)
Die Bestimmungen von Abschnitt II.2. Teil 2 gilt für die in Artikel 85 Absatz 2 genannten Unterlagen zur Zulassung homöopathischer Tierarzneimittel mit den folgenden Änderungen. |
|
V.5.2. |
Terminologie
Die lateinische Bezeichnung der im Antragsdossier beschriebenen homöopathischen Ursubstanz muss mit der lateinischen Bezeichnung des Europäischen Arzneibuchs oder — sollte diese darin fehlen — eines amtlichen Arzneibuchs eines Mitgliedstaats übereinstimmen. Gegebenenfalls ist anzugeben, welche traditionellen Benennungen in den einzelnen Mitgliedstaaten verwendet werden. |
|
V.5.3. |
Kontrolle der Ausgangsstoffe
Die dem Antrag beigefügten Angaben und Unterlagen zu den Ausgangsstoffen, d. h. zu allen verwendeten Stoffen, einschließlich der Rohstoffe und Zwischenprodukte bis hin zur endgültigen Verdünnung, die in dem zugelassenen homöopathischen Fertigtierarzneimittel verarbeitet werden sollen, sind durch zusätzliche Daten zur homöopathischen Ursubstanz zu ergänzen. Die allgemeinen Qualitätsanforderungen gelten für sämtliche Ausgangs- und Rohstoffe sowie für alle Zwischenschritte des Herstellungsprozesses bis hin zur endgültigen Verdünnung, die in dem homöopathischen Fertigprodukt verarbeitet werden sollen. Falls ein toxischer Bestandteil enthalten ist, ist dieser möglichst in der endgültigen Verdünnung zu kontrollieren. Ist dies aufgrund des hohen Verdünnungsgrades nicht möglich, ist dieser toxische Bestandteil üblicherweise in einem früheren Stadium zu kontrollieren. Jeder Herstellungsschritt von den Ausgangsstoffen bis hin zur endgültigen Verdünnung, die in dem Fertigprodukt verarbeitet werden sollen, ist vollständig zu beschreiben. Im Fall von Verdünnungen sollten die Verdünnungsschritte in Übereinstimmung mit den homöopathischen Herstellungsverfahren erfolgen, die in der einschlägigen Monografie des Europäischen Arzneibuchs oder, falls dort nicht vorhanden, in einem offiziellen Arzneibuch eines Mitgliedstaats festgelegt sind. |
|
V.5.4. |
Kontrollprüfungen des Fertigarzneimittels
Die allgemeinen Qualitätsanforderungen gelten für das fertige homöopathische Tierarzneimittel. Jede Ausnahme ist vom Antragsteller hinreichend zu begründen Die Identität und der Gehalt aller toxikologisch relevanten Bestandteile sind zu bestimmen. Lässt sich begründen, dass eine Identitäts-/Gehaltsbestimmung aller toxikologisch relevanten Bestandteile z. B. wegen ihrer Verdünnung im Fertigarzneimittel nicht möglich ist, so ist die Qualität durch eine vollständige Validierung des Herstellungs- und Verdünnungsprozesses nachzuweisen. |
|
V.5.5. |
Haltbarkeitsversuche
Die Haltbarkeit des Fertigprodukts ist nachzuweisen. Haltbarkeitsdaten der homöopathischen Ursubstanzen sind in der Regel auch auf daraus gewonnene Verdünnungen/Potenzierungen übertragbar. Ist aufgrund des Verdünnungsgrades keine Identitäts-/Gehaltsbestimmung des Wirkstoffs möglich, können die Haltbarkeitsdaten der Darreichungsform berücksichtigt werden. |
|
V.5.6. |
Sicherheitsdokumentation (Teil 3)
Teil 3 gilt für homöopathische Tierarzneimittel gemäß Artikel 4 Absatz 10 der vorliegenden Verordnung mit der folgenden Spezifikation, unbeschadet der Bestimmungen der Verordnung (EU) Nr. 37/2010 der Kommission (7) über pharmakologisch wirksame Stoffe und ihre Einstufung hinsichtlich der Rückstandshöchstmengen in Lebensmitteln tierischen Ursprungs. Fehlende Informationen sind zu begründen, z. B. ist zu begründen, warum der Nachweis eines annehmbaren Grades an Unbedenklichkeit und/oder Wirksamkeit angenommen werden kann, obwohl bestimmte Studien fehlen. |
(1) Richtlinie 2004/10/EG des Europäischen Parlaments und des Rates vom 11. Februar 2004 zur Angleichung der Rechts- und Verwaltungsvorschriften für die Anwendung der Grundsätze der Guten Laborpraxis und zur Kontrolle ihrer Anwendung bei Versuchen mit chemischen Stoffen (ABl. L 50 vom 20.2.2004, S. 44).
(2) Richtlinie 2004/9/EG des Europäischen Parlaments und des Rates vom 11. Februar 2004 über die Inspektion und Überprüfung der Guten Laborpraxis (GLP) (ABl. L 50 vom 20.2.2004, S. 28).
(3) Richtlinie 2009/35/EG des Europäischen Parlaments und des Rates vom 23. April 2009 über die Stoffe, die Arzneimitteln zum Zwecke der Färbung hinzugefügt werden dürfen (ABl. L 109 vom 30.4.2009, S. 10).
(4) Verordnung (EU) Nr. 231/2012 vom 9. März 2012 mit Spezifikationen für die in den Anhängen II und III der Verordnung (EG) Nr. 1333/2008 des Europäischen Parlaments und des Rates aufgeführten Lebensmittelzusatzstoffe (ABl. L 83 vom 22.3.2012, S. 1).
(5) Verordnung (EU) 2018/782 vom 29. Mai 2018 zur Festlegung der Grundsätze zur Methodik der Risikobewertung und der Empfehlungen für das Risikomanagement gemäß der Verordnung (EG) Nr. 470/2009 (ABl. L 132, 30.5.2018, S. 5).
(6) Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates vom 18. Dezember 2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH), zur Schaffung einer Europäischen für chemische Stoffe, zur Änderung der Richtlinie 1999/45/EG und zur Aufhebung der Verordnung (EWG) Nr. 793/93 des Rates, der Verordnung (EG) Nr. 1488/94 der Kommission, der Richtlinie 76/769/EWG des Rates sowie der Richtlinien 91/155/EWG, 93/67/EWG, 93/105/EG und 2000/21/EG der Kommission (ABl. L 396 vom 30.12.2006, S. 1).
(7) Verordnung (EU) Nr. 37/2010 der Kommission vom 22. Dezember 2009 über pharmakologisch wirksame Stoffe und ihre Einstufung hinsichtlich der Rückstandshöchstmengen in Lebensmitteln tierischen Ursprungs (ABl. L 15 vom 20.1.2010, S. 1).
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/78 |
DELEGIERTE VERORDNUNG (EU) 2021/806 DER KOMMISSION
vom 10. März 2021
zur Änderung der Verordnung (EU) Nr. 528/2012 des Europäischen Parlaments und des Rates zwecks Aufnahme des Wirkstoffs Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, in Anhang I
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) Nr. 528/2012 des Europäischen Parlaments und des Rates vom 22. Mai 2012 über die Bereitstellung auf dem Markt und die Verwendung von Biozidprodukten (1), insbesondere auf Artikel 28 Absatz 1,
in Erwägung nachstehender Gründe:
|
(1) |
Mit der Delegierten Verordnung (EU) Nr. 1062/2014 der Kommission (2) wurde eine Liste der alten Wirkstoffe festgelegt, die im Hinblick auf ihre mögliche Genehmigung zur Verwendung in Biozidprodukten bewertet werden sollen. Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, ist in dieser Liste enthalten. |
|
(2) |
Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, wurde in Bezug auf seine Verwendung in Biozidprodukten der in Anhang V der Verordnung (EU) Nr. 528/2012 beschriebenen Produktart 19 (Repellentien und Lockmittel) bewertet. |
|
(3) |
Frankreich wurde zum Bericht erstattenden Mitgliedstaat bestimmt, und seine zuständige bewertende Behörde übermittelte der Europäischen Chemikalienagentur (im Folgenden die „Agentur“) am 18. September 2019 den Bewertungsbericht und seine Schlussfolgerungen. |
|
(4) |
Gemäß Artikel 7 Absatz 2 der Delegierten Verordnung (EU) Nr. 1062/2014 nahm der Ausschuss für Biozidprodukte am 16. Juni 2020 unter Berücksichtigung der Schlussfolgerungen der zuständigen bewertenden Behörde die Stellungnahme der Agentur (3) an. |
|
(5) |
Nach dieser Stellungnahme kann davon ausgegangen werden, dass Biozidprodukte der Produktart 19, in denen Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, verwendet wird, die Anforderungen gemäß Artikel 19 Absatz 1 Buchstabe b der Verordnung (EU) Nr. 528/2012 erfüllen. In der Stellungnahme der Agentur wurde auch der Schluss gezogen, dass dieser Wirkstoff keinen Anlass zur Besorgnis gibt und für die Aufnahme in Anhang I der Verordnung (EU) Nr. 528/2012 in Betracht kommt. |
|
(6) |
Unter Berücksichtigung der Stellungnahme der Agentur ist es daher angezeigt, Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, in Anhang I der Verordnung (EU) Nr. 528/2012 aufzunehmen. Da Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, auf der Grundlage eines Wirkstoffdossiers geprüft wurde, welches den Anforderungen von Artikel 11 Absatz 1 der Richtlinie 98/8/EG des Europäischen Parlaments und des Rates (4) entspricht, sollte Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, in Kategorie 6 des Anhangs I der Verordnung (EU) Nr. 528/2012, „Stoffe, für die ein Mitgliedstaat ein Wirkstoffdossier gemäß Artikel 7 Absatz 3 der vorliegenden Verordnung validiert oder ein solches Dossier gemäß Artikel 11 Absatz 1 der Richtlinie 98/8/EG akzeptiert hat“ aufgenommen werden. |
|
(7) |
Artikel 89 Absatz 3 der Verordnung (EU) Nr. 528/2012 enthält Übergangsmaßnahmen für den Fall, dass ein alter Wirkstoff, der im Arbeitsprogramm zur systematischen Prüfung der alten Wirkstoffe enthalten ist, gemäß der genannten Verordnung genehmigt wird. Im Hinblick auf Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, für die Produktart 19 sollte das Datum der Genehmigung für die Zwecke des Artikels 89 Absatz 3 der genannten Verordnung auf den 1. Juli 2022 festgesetzt werden, um ausreichend Zeit für Anträge auf Zulassung zu gewähren, die gemäß Artikel 89 Absatz 3 Unterabsatz 2 der genannten Verordnung zu stellen sind — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang I der Verordnung (EU) Nr. 528/2012 wird gemäß dem Anhang der vorliegenden Verordnung geändert.
Artikel 2
Für die Zwecke des Artikels 89 Absatz 3 der Verordnung (EU) Nr. 528/2012 gilt als Datum der Genehmigung von Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, für die Produktart 19 der 1. Juli 2022.
Artikel 3
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 10. März 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 167 vom 27.6.2012, S. 1.
(2) Delegierte Verordnung (EU) Nr. 1062/2014 der Kommission vom 4. August 2014 über das Arbeitsprogramm zur systematischen Prüfung aller in Biozidprodukten enthaltenen alten Wirkstoffe gemäß der Verordnung (EU) Nr. 528/2012 des Europäischen Parlaments und des Rates (ABl. L 294 vom 10.10.2014, S. 1).
(3) Stellungnahme des Ausschusses für Biozidprodukte (BPC) zum Antrag auf Genehmigung des Wirkstoffs: Carbon dioxide generated from propane, butane or a mixture of both by combustion, Product type: 19, ECHA/BPC/249/2020, angenommen am 16. Juni 2020.
(4) Richtlinie 98/8/EG des Europäischen Parlaments und des Rates vom 16. Februar 1998 über das Inverkehrbringen von Biozid-Produkten (ABl. L 123 vom 24.4.1998, S. 1).
ANHANG
In Anhang I der Verordnung (EU) Nr. 528/2012 wird unter Kategorie 6 der Liste der unter Artikel 25 Buchstabe a fallenden Wirkstoffe folgender Eintrag angefügt:
|
EG-Nummer |
Name/Gruppe |
Einschränkung |
Bemerkung |
|
„204-696-9 |
Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung (*1) |
|
CAS Nr. 124-38-9 |
(*1) Für die Zwecke des Artikels 89 Absatz 3 gilt als Datum der Genehmigung von Kohlendioxid, hergestellt aus Propan, Butan oder einer Mischung beider Stoffe mittels Verbrennung, für die Produktart 19 der 1. Juli 2022.“
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/81 |
DELEGIERTE VERORDNUNG (EU) 2021/807 DER KOMMISSION
vom 10. März 2021
zur Änderung der Verordnung (EU) Nr. 528/2012 des Europäischen Parlaments und des Rates zwecks Aufnahme des Wirkstoffs Kaliumsorbat in Anhang I
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) Nr. 528/2012 des Europäischen Parlaments und des Rates vom 22. Mai 2012 über die Bereitstellung auf dem Markt und die Verwendung von Biozidprodukten (1), insbesondere auf Artikel 28 Absatz 1,
in Erwägung nachstehender Gründe:
|
(1) |
Kalium-(E,E)-hexa-2,4-dienoat (Kaliumsorbat) wurde im Rahmen des in Artikel 89 Absatz 1 der Verordnung (EU) Nr. 528/2012 genannten und gemäß der Delegierten Verordnung (EU) Nr. 1062/2014 der Kommission (2) durchgeführten Arbeitsprogramms für die systematische Prüfung aller in Biozidprodukten enthaltenen alten Wirkstoffe als alter Wirkstoff bewertet. |
|
(2) |
Gemäß Artikel 7 Absatz 2 der Delegierten Verordnung (EU) Nr. 1062/2014 wurde die Stellungnahme der Europäischen Chemikalienagentur (im Folgenden die „Agentur“) unter Berücksichtigung der Schlussfolgerungen der bewertenden zuständigen Behörde am 4. Dezember 2014 vom Ausschuss für Biozidprodukte (3) angenommen. Da die Bewertung durch die zuständige Behörde am 20. Oktober 2010 abgeschlossen wurde, wurde der Antrag auf Genehmigung von Kaliumsorbat gemäß Artikel 90 Absatz 2 der Verordnung (EU) Nr. 528/2012 gemäß der Richtlinie 98/8/EG des Europäischen Parlaments und des Rates (4) geprüft, und die Agentur kam in ihrer Stellungnahme zu dem Schluss, dass davon ausgegangen werden kann, dass Biozidprodukte der Produktart 8, die Kaliumsorbat enthalten, die Anforderungen des Artikels 5 der Richtlinie 98/8/EG erfüllen. |
|
(3) |
Kaliumsorbat wurde daher mit der Durchführungsverordnung (EU) 2015/1729 der Kommission (5) als Wirkstoff zur Verwendung in Biozidprodukten der Produktart 8 genehmigt. |
|
(4) |
Kaliumsorbat ist noch im Arbeitsprogramm zur systematischen Prüfung aller in Biozidprodukten enthaltenen alten Wirkstoffe aufgrund seiner Verwendung in Biozidprodukten der Produktart 6 enthalten. |
|
(5) |
In der Stellungnahme vom 4. Dezember 2014 zog die Agentur auch den Schluss, dass Kaliumsorbat die Kriterien für die Aufnahme in Anhang I der Verordnung (EU) Nr. 528/2012 erfüllt. |
|
(6) |
Unter Berücksichtigung der Stellungnahme der Agentur ist es angezeigt, Kaliumsorbat in Anhang I der Verordnung (EU) Nr. 528/2012 aufzunehmen. Da Kaliumsorbat auf der Grundlage eines Wirkstoffdossiers geprüft wurde, das den Anforderungen gemäß Artikel 11 Absatz 1 der Richtlinie 98/8/EG genügt, sollte Kaliumsorbat in Kategorie 6 des Anhangs I der Verordnung (EU) Nr. 528/2012 aufgenommen werden. |
|
(7) |
Artikel 89 Absatz 3 der Verordnung (EU) Nr. 528/2012 enthält Übergangsmaßnahmen für den Fall, dass ein alter Wirkstoff, der im Arbeitsprogramm zur systematischen Prüfung der alten Wirkstoffe enthalten ist, gemäß der genannten Verordnung genehmigt wird. Im Hinblick auf Kaliumsorbat für die Produktart 6 sollte das Datum der Genehmigung für die Zwecke des Artikels 89 Absatz 3 der genannten Verordnung auf den 1. Februar 2023 festgesetzt werden, um ausreichend Zeit für Anträge auf Zulassung zu gewähren, die gemäß Artikel 89 Absatz 3 Unterabsatz 2 der genannten Verordnung zu stellen sind — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang I der Verordnung (EU) Nr. 528/2012 wird gemäß dem Anhang der vorliegenden Verordnung geändert.
Artikel 2
Für die Zwecke des Artikels 89 Absatz 3 der Verordnung (EU) Nr. 528/2012 gilt als Datum der Genehmigung von Kaliumsorbat für die Produktart 6 der 1. Februar 2023.
Artikel 3
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 10. März 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 167 vom 27.6.2012, S. 1.
(2) Delegierte Verordnung (EU) Nr. 1062/2014 der Kommission vom 4. August 2014 über das Arbeitsprogramm zur systematischen Prüfung aller in Biozidprodukten enthaltenen alten Wirkstoffe gemäß der Verordnung (EU) Nr. 528/2012 des Europäischen Parlaments und des Rates (ABl. L 294 vom 10.10.2014, S. 1).
(3) Stellungnahme des Ausschusses für Biozidprodukte zum Antrag auf Genehmigung des Wirkstoffs: Kaliumsorbat, Produktart 8, ECHA/BPC/37/2014, angenommen am 4. Dezember 2014.
(4) Richtlinie 98/8/EG des Europäischen Parlaments und des Rates vom 16. Februar 1998 über das Inverkehrbringen von Biozid-Produkten (ABl. L 123 vom 24.4.1998, S. 1).
(5) Durchführungsverordnung (EU) 2015/1729 der Kommission vom 28. September 2015 zur Genehmigung von Kaliumsorbat als alten Wirkstoff zur Verwendung in Biozidprodukten der Produktart 8 (ABl. L 252 vom 29.9.2015, S. 24).
ANHANG
In Anhang I der Verordnung (EU) Nr. 528/2012 wird unter Kategorie 6 der Liste der unter Artikel 25 Buchstabe a fallenden Wirkstoffe folgender Eintrag angefügt:
|
EG-Nummer |
Name/Gruppe |
Beschränkung |
Anmerkung |
|
„246-376-1 |
Kalium-(E,E)-hexa-2,4-dienoat (Kaliumsorbat) (*1) |
Mindestreinheit des Wirkstoffs (*2): 990 g/kg |
CAS-Nr. 24634-61-5 |
(*1) Das Datum der Genehmigung von Kaliumsorbat für die Produktart 6 für die Zwecke des Artikels 89 Absatz 3 ist der 1. Februar 2023.
(*2) Die in dieser Spalte angegebene Reinheit war die Mindestreinheit des bewerteten Wirkstoffs. Der Wirkstoff in dem in Verkehr gebrachten Produkt kann dieselbe oder eine andere Reinheit aufweisen, sofern er nachgewiesenermaßen technisch äquivalent zu dem bewerteten Wirkstoff ist.“
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/84 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/808 DER KOMMISSION
vom 22. März 2021
über Leistungskriterien für Analysemethoden für Rückstände pharmakologisch wirksamer Stoffe in zur Lebensmittelerzeugung genutzten Tieren und über die Auswertung von Ergebnissen sowie über die für Probenahmen anzuwendenden Methoden und zur Aufhebung der Entscheidungen 2002/657/EG und 98/179/EG
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) 2017/625 des Europäischen Parlaments und des Rates vom 15. März 2017 über amtliche Kontrollen und andere amtliche Tätigkeiten zur Gewährleistung der Anwendung des Lebens- und Futtermittelrechts und der Vorschriften über Tiergesundheit und Tierschutz, Pflanzengesundheit und Pflanzenschutzmittel, zur Änderung der Verordnungen (EG) Nr. 999/2001, (EG) Nr. 396/2005, (EG) Nr. 1069/2009, (EG) Nr. 1107/2009, (EU) Nr. 1151/2012, (EU) Nr. 652/2014, (EU) 2016/429 und (EU) 2016/2031 des Europäischen Parlaments und des Rates, der Verordnungen (EG) Nr. 1/2005 und (EG) Nr. 1099/2009 des Rates sowie der Richtlinien 98/58/EG, 1999/74/EG, 2007/43/EG, 2008/119/EG und 2008/120/EG des Rates und zur Aufhebung der Verordnungen (EG) Nr. 854/2004 und (EG) Nr. 882/2004 des Europäischen Parlaments und des Rates, der Richtlinien 89/608/EWG, 89/662/EWG, 90/425/EWG, 91/496/EWG, 96/23/EG, 96/93/EG und 97/78/EG des Rates und des Beschlusses 92/438/EWG des Rates (Verordnung über amtliche Kontrollen) (1), insbesondere auf Artikel 34 Absatz 6,
in Erwägung nachstehender Gründe:
|
(1) |
Die Verordnung (EU) 2017/625 regelt die amtlichen Kontrollen und die anderen amtlichen Tätigkeiten, die von den zuständigen Behörden der Mitgliedstaaten durchgeführt werden, um zu überprüfen, ob das Unionsrecht unter anderem im Bereich der Lebensmittelsicherheit auf allen Produktions-, Verarbeitungs- und Vertriebsstufen eingehalten wird. Sie enthält spezifische Vorschriften für die amtlichen Kontrollen in Bezug auf Stoffe, deren Verwendung zu Rückständen in Lebens- und Futtermitteln führen kann, und legt allgemeine Anforderungen an die Methoden fest, die für die Probenahmen, Laboranalysen und Tests im Rahmen der amtlichen Kontrollen und anderen amtlichen Tätigkeiten anzuwenden sind. |
|
(2) |
Die Entscheidung 2002/657/EG der Kommission (2) enthält Anforderungen an die Durchführung von Analysemethoden und die Auswertung von Ergebnissen der Analysen bestimmter Stoffe und deren Rückstände in lebenden Tieren und tierischen Erzeugnissen, und in der Entscheidung 98/179/EG der Kommission (3) sind Durchführungsvorschriften für die amtlichen Probenahmen zur Kontrolle von lebenden Tieren und tierischen Erzeugnissen auf bestimmte Stoffe und deren Rückstände festgelegt. Beide Entscheidungen wurden auf der Grundlage der Richtlinie 96/23/EG des Rates (4) erlassen, die durch die Verordnung (EU) 2017/625 aufgehoben wurde. In Anbetracht neuer wissenschaftlicher Entwicklungen sollten diese Vorschriften aktualisiert und in den mit der Verordnung (EU) 2017/625 festgelegten Rahmen für amtliche Kontrollen integriert werden. |
|
(3) |
Gemäß Artikel 1 Absatz 2 der Entscheidung 2002/657/EG gilt die genannte Entscheidung nicht für Stoffe, für die in anderen Rechtsvorschriften der Union speziellere Regelungen festgelegt worden sind. Bei diesen Stoffen handelt es sich um Mykotoxine in Lebensmitteln, Dioxine und dioxinähnliche polychlorierte Biphenyle (PCB) in Lebensmitteln sowie Blei, Cadmium, Quecksilber und Benzo(a)pyren in Lebensmitteln. Mykotoxine in Lebensmitteln müssen die in der Verordnung (EG) Nr. 401/2006 der Kommission (5) zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Kontrolle des Mykotoxingehalts von Lebensmitteln festgelegten Anforderungen erfüllen. Die Verordnung (EU) 2017/644 der Kommission (6) zur Festlegung der Probenahmeverfahren und Analysemethoden für die Kontrolle der Gehalte an Dioxinen, dioxinähnlichen PCB und nicht dioxinähnlichen PCB in bestimmten Lebensmitteln findet im Fall von Dioxinen und dioxinähnlichen PCB Anwendung. Vorschriften für die Probenahme und Analyse im Rahmen der amtliche Kontrolle in Bezug auf Blei, Cadmium, Quecksilber und Benzo(a)pyren in Lebensmitteln sind in der Verordnung (EG) Nr. 333/2007 der Kommission (7) festgelegt. |
|
(4) |
Im Interesse der Klarheit und der Rechtssicherheit sollten die für die Probenahme und Analyse in Bezug auf pharmakologisch wirksame Stoffe geltenden Vorschriften in einem einzigen Rechtsakt zusammengeführt werden, wie im Fall von Mykotoxinen, Dioxinen, dioxinähnlichen PCB, Blei, Cadmium, Quecksilber und Benzo(a)pyren in Lebensmitteln bereits geschehen. |
|
(5) |
Die Entscheidungen 98/179/EG und 2002/657/EG sollten daher aufgehoben und durch die vorliegende Verordnung ersetzt werden. |
|
(6) |
Gemäß der Verordnung (EG) Nr. 1831/2003 des Europäischen Parlaments und des Rates (8) dürfen Kokzidiostatika und Histomonostatika als Futtermittelzusatzstoffe verwendet werden; somit gilt für Analysen ihres Gehalts in Futtermitteln die Verordnung (EG) Nr. 152/2009 der Kommission (9) zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln. Hingegen sollte die vorliegende Verordnung gelten für Analysen von Futtermitteln im Rahmen von Folgemaßnahmen bei Nachforschungen bezüglich der Quelle nicht konformer Proben in Fällen mutmaßlicher oder festgestellter Verstöße gegen Unionsvorschriften über die Verwendung oder über Rückstände pharmakologisch wirksamer Stoffe, die in Tierarzneimitteln oder als Futtermittelzusatzstoffe zugelassen sind, bzw. gegen Unionsvorschriften über die Verwendung oder über Rückstände verbotener oder nicht zugelassener pharmakologisch wirksamer Stoffe. |
|
(7) |
Um die Kontinuität der Durchführung amtlicher Kontrollen und anderer amtlicher Tätigkeiten in Bezug auf Rückstände pharmakologisch wirksamer Stoffe zu gewährleisten und um zu vermeiden, dass sämtliche Verfahren und Methoden gleichzeitig revalidiert werden müssen, dürfen Verfahren und Methoden, die vor Inkrafttreten der vorliegenden Verordnung validiert wurden, vorbehaltlich der Anforderungen in Anhang I Nummern 2 und 3 der Entscheidung 2002/657/EG während eines begrenzten Zeitraums weiter angewandt werden. Daher sollte den Mitgliedstaaten ausreichend Zeit eingeräumt werden, damit die in der vorliegenden Verordnung festgelegten Anforderungen bei allen Analysemethoden erfüllt werden können. |
|
(8) |
Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Ausschusses für Pflanzen, Tiere, Lebensmittel und Futtermittel — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Gegenstand und Geltungsbereich
Diese Verordnung enthält Vorschriften zu den Analysemethoden für Probenahmen und Laboranalysen in Bezug auf Rückstände pharmakologisch wirksamer Stoffe in zur Lebensmittelerzeugung genutzten Tieren, deren Körperteilen und -flüssigkeiten, Exkrementen und Geweben, in von ihnen gewonnenen Erzeugnissen tierischen Ursprungs und tierischen Nebenprodukten sowie in Futtermitteln und Tränkwässer für diese Tiere. Des Weiteren sind dort Vorschriften für die Auswertung der Ergebnisse dieser Laboranalysen festgelegt.
Diese Verordnung gilt für amtliche Kontrollen zur Überprüfung der Einhaltung der Anforderungen bezüglich Rückständen pharmakologisch wirksamer Stoffe.
Artikel 2
Begriffsbestimmungen
Für die Zwecke dieser Verordnung gelten die Begriffsbestimmungen in Artikel 2 der Delegierten Verordnung (EU) 2019/2090 der Kommission (10), in der Verordnung (EU) 2019/1871 der Kommission (11), in Artikel 2 der Verordnung (EG) Nr. 470/2009 des Europäischen Parlaments und des Rates (12) sowie in der Verordnung (EWG) Nr. 315/93 des Rates (13).
Ferner gelten folgende Begriffsbestimmungen:
|
(1) |
„absolute Wiederfindungsrate“ bezeichnet die in der letzten Phase eines analytischen Prozesses gemessene Menge eines Analyten, dividiert durch die Menge des Analyten in der ursprünglichen Probe, ausgedrückt in Prozent; |
|
(2) |
„Genauigkeit“ bezeichnet den Grad der Übereinstimmung zwischen einem Testergebnis und dem anerkannten richtigen Referenzwert, bestimmt durch Richtigkeit und Präzision (14); |
|
(3) |
„Alpha-Fehler (α-Fehler)“ bezeichnet die Wahrscheinlichkeit, dass die untersuchte Probe konform ist, obwohl ein nicht konformes Messergebnis erzielt wurde; |
|
(4) |
„Analyt“ bezeichnet die zu analysierende Komponente eines Systems; |
|
(5) |
„zugelassener Stoff“ bezeichnet einen pharmakologisch wirksamen Stoff, der zur Verwendung bei zur Lebensmittelerzeugung genutzten Tieren zugelassen ist, im Sinne der Richtlinie 2001/82/EG des Europäischen Parlaments und des Rates (15); |
|
(6) |
„Beta-Fehler (β-Fehler)“ bezeichnet die Wahrscheinlichkeit, dass die untersuchte Probe tatsächlich nicht konform ist, obwohl ein konformes Messergebnis erzielt wurde; |
|
(7) |
„Bias“ bezeichnet den Unterschied zwischen dem erwarteten Wert des Testergebnisses und einem anerkannten Referenzwert; |
|
(8) |
„Kalibrierstandard“ bezeichnet eine rückführbare Referenz für Messungen, welche die Menge des fraglichen Stoffs so darstellt, dass ihr Wert mit einer Referenzbasis in Beziehung gesetzt wird; |
|
(9) |
„zertifiziertes Referenzmaterial“ bezeichnet ein Referenzmaterial mit Unterlagen, die von einer autorisierten Stelle herausgegeben wurden, und das einen oder mehrere spezifizierte Merkmalswerte mit beigeordneten Unsicherheiten und Rückführbarkeiten liefert, unter Anwendung gültiger Verfahren (16); |
|
(10) |
„Co-Chromatografie“ bezeichnet eine Technik, bei der ein unbekannter Stoff zusammen mit einer bekannten Verbindung oder mehreren bekannten Verbindungen einer chromatografischen Trennung unterzogen wird, mit der Erwartung, dass das relative Verhalten des unbekannten Stoffs und der bekannten Stoffe zur Identifizierung des unbekannten Stoffs beiträgt; |
|
(11) |
„Methodenvergleichsstudie“ bezeichnet die Analyse derselben Probe(n) mittels derselben Methode, um Leistungseigenschaften der Methode in verschiedenen Laboren zu bestimmen, wobei die Studie die Berechnung der zufälligen Messabweichung und der systematischen Abweichung für die angewandte Methode ermöglicht; |
|
(12) |
„Bestätigungsmethode“ bezeichnet eine Methode, die vollständige oder ergänzende Daten dafür liefert, dass der Stoff eindeutig identifiziert und, falls erforderlich, auf einem der folgenden Niveaus quantifiziert werden kann:
|
|
(13) |
„Erweiterungsfaktor (k)“ bezeichnet eine Zahl, die das erwünschte Konfidenzniveau ausdrückt und zur Ermittlung der erweiterten Messunsicherheit dient; |
|
(14) |
„Entscheidungsgrenze für die Bestätigung (CCα)“ bezeichnet den Grenzwert, an und über dem mit einer Fehlerwahrscheinlichkeit von α geschlussfolgert werden kann, dass eine Probe nicht konform ist. Der Wert 1–α drückt die statistische Sicherheit in Prozent aus, dass der zulässige Grenzwert überschritten ist; |
|
(15) |
„Nachweisvermögen von Screening (CCβ)“ bezeichnet den niedrigsten Analytengehalt, der mit einer Fehlerwahrscheinlichkeit von β in einer Probe nachgewiesen oder quantifiziert werden kann:
|
|
(16) |
„dotiertes Probenmaterial“ bezeichnet eine Probe, die mit einer bekannten Menge des nachzuweisenden oder zu quantifizierenden Analyten angereichert ist; |
|
(17) |
„Laborvergleichsstudie“ bezeichnet die Organisation, Durchführung und Bewertung von Tests mit derselben Probe bzw. denselben Proben durch zwei oder mehr Labore unter zuvor festgelegten Bedingungen, um die Leistungsfähigkeit der Tests zu bewerten; dabei kann es sich um eine Methodenvergleichsstudie oder eine Eignungsprüfung handeln; |
|
(18) |
„interner Standard (IS)“ bezeichnet einen nicht in der Probe enthaltenen Stoff mit physikalisch-chemischen Eigenschaften, die denen des zu identifizierenden oder quantifizierenden Analyten möglichst ähnlich sind; |
|
(19) |
„relevantes Konzentrationsniveau“ bezeichnet die Konzentration eines Stoffs oder Analyten in einer Probe, die wesentlich ist, um die Einhaltung der Rechtsvorschriften in Bezug auf Folgendes zu bestimmen:
|
|
(20) |
„niedrigstes Dotierniveau“ („LCL“, lowest calibrated level) bezeichnet die geringste Konzentration, für die das Messsystem kalibriert wurde; |
|
(21) |
„Matrix“ bezeichnet das Material, aus dem eine Probe entnommen wird; |
|
(22) |
„Matrixeffekt“ bezeichnet die Differenz des Analyseergebnisses zwischen einem im Lösungsmittel gelösten Standard und einem matrix-angepassten Standard, entweder ohne Korrektur mittels eines internen Standards oder einschließlich einer Korrektur mittels eines internen Standards; |
|
(23) |
„matrix-angepasster Standard“ bezeichnet eine (analytfreie) Leerwertmatrix, der nach der Probenaufarbeitung der Analyt in verschiedenen Konzentrationen zugesetzt wird; |
|
(24) |
„Matrixstandard“ bezeichnet eine (analytfreie) Leerwertmatrix, die vor der Lösungsmittelextraktion und der Probenaufarbeitung mit dem Analyten in verschiedenen Konzentrationen dotiert wird; |
|
(25) |
„Messgröße“ bezeichnet die spezielle Größe, die Gegenstand der Messung ist; |
|
(26) |
„Messunsicherheit“ bezeichnet einen nicht-negativen Parameter, der die Streuung der Werte kennzeichnet, die der Messgröße auf der Grundlage der genutzten Information beigeordnet ist; |
|
(27) |
„Leistungskriterien“ bezeichnet die Anforderungen an ein Leistungsmerkmal, nach denen beurteilt werden kann, ob die Analysemethode für den vorgesehenen Zweck geeignet ist und zuverlässige Ergebnisse liefert; |
|
(28) |
„Präzision“ bezeichnet den Grad der Übereinstimmung zwischen unabhängigen, unter festgelegten Bedingungen erzielten Testergebnissen und wird als Standardabweichung oder Variationskoeffizient der Testergebnisse ausgedrückt; |
|
(29) |
„qualitative Methode“ bezeichnet eine Analysemethode, die einen Stoff oder eine Stoffgruppe aufgrund seiner bzw. ihrer chemischen, biologischen oder physikalischen Eigenschaften bestimmt oder identifiziert; |
|
(30) |
„quantitative Methode“ bezeichnet eine Analysemethode, welche die Menge oder den Massenanteil eines Stoffs so bestimmt, dass sie bzw. er als numerischer Wert geeigneter Einheiten ausgedrückt werden kann; |
|
(31) |
„Wiederfindungsrate“ bezeichnet die wiederfindungskorrigierte Menge eines Analyten, dividiert durch die dotierte Menge des Analyten in der Matrixprobe, ausgedrückt in Prozent; |
|
(32) |
„Wiederfindungskorrektur“ bezeichnet die Anwendung interner Standards, die Verwendung einer Matrixkalibrierkurve und die Anwendung eines Wiederfindungskorrekturfaktors sowie eine Kombination dieser Ansätze; |
|
(33) |
„Referenzmaterial“ bezeichnet ein hinsichtlich einer festgelegten Eigenschaft oder mehrerer festgelegter Eigenschaften ausreichend homogenes und stabiles Material, das für die vorgesehene Verwendung in einem Messverfahren oder bei einer Untersuchung nominaler Eigenschaften als geeignet erachtet wurde (19); |
|
(34) |
„relativer Matrixeffekt“ bezeichnet die Differenz des Analyseergebnisses zwischen einem im Lösungsmittel gelösten Standard und einem matrixbezogenen Standard einschließlich einer Korrektur mittels eines internen Standards; |
|
(35) |
„Wiederholpräzision“ bezeichnet Präzision unter Bedingungen, bei denen unabhängige Testergebnisse mittels derselben Methode an identischem Testmaterial im selben Labor durch denselben Untersuchenden mit derselben Ausrüstung kurz nacheinander erzielt werden; |
|
(36) |
„Reproduzierbarkeit“ bezeichnet Präzision unter Bedingungen, bei denen Testergebnisse mittels derselben Methode an identischem Testmaterial in verschiedenen Laboren durch verschiedene Untersuchende mit unterschiedlicher Ausrüstung erzielt werden (20); |
|
(37) |
„Robustheit“ bezeichnet die Anfälligkeit einer Analysemethode gegenüber Änderungen in den Versuchsbedingungen, unter denen die Methode wie beschrieben oder mit festgelegten geringfügigen Änderungen angewandt werden kann; |
|
(38) |
„Screeningmethode“ bezeichnet eine Methode, die für das Screening eines Stoffs oder einer Klasse von Stoffen am jeweils relevanten Konzentrationsniveau verwendet wird; |
|
(39) |
„Screening-Zielkonzentration“ („STC“, screening target concentration) bezeichnet die Konzentration unterhalb oder gleich dem CCβ, bei der ein Screeningergebnis die Probe als potenziell nicht konform („Screening-positiv“) einstuft und das einen Bestätigungstest veranlasst; |
|
(40) |
„Selektivität“ bezeichnet die Fähigkeit einer Methode, zwischen dem gemessenen Analyten und anderen Stoffen zu unterscheiden; |
|
(41) |
„laborinterne Validierungsstudie“ bezeichnet eine analytische Studie in einem einzelnen Labor unter Anwendung einer einzigen Methode zur Analyse desselben Testmaterials oder verschiedener Testmaterialien unter unterschiedlichen Bedingungen und über angemessen lange Zeiträume; |
|
(42) |
„Standardaddition“ bezeichnet ein Verfahren, bei dem ein Teil der Probe als solcher analysiert wird und den anderen Analysenproben vor der Analyse bekannte Mengen der Standardsubstanz zugesetzt werden; |
|
(43) |
„Standardsubstanz“ bezeichnet einen Analyten mit bekanntem und zertifiziertem Gehalt bzw. bekannter und zertifizierter Reinheit, der bei der Analyse als Referenz zu verwenden ist; |
|
(44) |
„Substanz“ bezeichnet einen Stoff mit konstanter Zusammensetzung, der durch die Einheiten, aus denen er sich zusammensetzt, sowie durch bestimmte physikalische Eigenschaften gekennzeichnet ist; |
|
(45) |
„Analysenprobe“ bezeichnet die Menge des entnommenen Materials einer Probe, an der die Analyse oder Beobachtung durchgeführt wird; |
|
(46) |
„Richtigkeit“ bezeichnet den Grad der Übereinstimmung zwischen dem aus einer langen Serie von Testergebnissen ermittelten Mittelwert und einem akzeptierten Referenzwert; |
|
(47) |
„Einheiten“ bezeichnet die in ISO 80000 (21) und in der Richtlinie 80/181/EWG des Rates (22) beschriebenen Einheiten; |
|
(48) |
„Validierung“ bezeichnet die Demonstration durch Untersuchung und die Erbringung eines effektiven Nachweises, dass die besonderen Anforderungen eines bestimmten vorgesehenen Verwendungszwecks durch eine laborinterne Validierungsstudie oder eine Methodenvergleichsstudie erfüllt sind (23); |
|
(49) |
„laborinterne Reproduzierbarkeit“ oder „In-Haus-Vergleichspräzision“ bezeichnet Messpräzision unter einer Reihe laborinterner Bedingungen in einem bestimmten Labor. |
Artikel 3
Analysemethoden
Die Mitgliedstaaten gewährleisten, dass die gemäß Artikel 34 der Verordnung (EU) 2017/625 entnommenen Proben anhand von Methoden analysiert werden, die folgende Anforderungen erfüllen:
|
(1) |
Sie sind in Prüfvorschriften dokumentiert, vorzugsweise gemäß den Anhängen von ISO 78-2:1999 (Chemie — Gestaltung von Normen — Teil 2: Verfahren der chemischen Analyse) (24); |
|
(2) |
sie erfüllen die Leistungskriterien und die anderen Anforderungen an Analysemethoden, wie in Anhang I Kapitel 1 dieser Verordnung festgelegt; |
|
(3) |
sie wurden gemäß den Anforderungen des Anhangs I Kapitel 2 und 4 dieser Verordnung validiert; |
|
(4) |
sie ermöglichen die Durchsetzung der in der Verordnung (EU) 2019/1871 festgelegten Referenzwerte für Maßnahmen (RPA), den eindeutigen Nachweis verbotener und nicht zugelassener Stoffe sowie die Durchsetzung von Höchstgehalten (ML), die auf Grundlage der Verordnungen (EWG) Nr. 315/93 und (EG) Nr. 124/2009 festgelegt wurden, und von Rückstandshöchstmengen (MRL), die auf Grundlage der Verordnungen (EG) Nr. 1831/2003 und (EG) Nr. 470/2009 festgelegt wurden. |
Artikel 4
Qualitätskontrolle
Die Mitgliedstaaten gewährleisten die Qualität der Ergebnisse der gemäß der Verordnung (EU) 2017/625 durchgeführten Analysen, insbesondere durch die Überwachung der Validität der Test- oder der Kalibrierergebnisse nach ISO/IEC 17025:2017 (Allgemeine Anforderungen an die Kompetenz von Prüf- und Kalibrierlaboratorien) und Anwendung der Anforderungen an die Qualitätskontrolle bei Routineanalysen, wie in Anhang I Kapitel 3 der vorliegenden Verordnung festgelegt.
Artikel 5
Auswertung der Ergebnisse
(1) Das Ergebnis einer Analyse gilt als nicht konform, wenn es größer als die oder gleich der Entscheidungsgrenze für die Bestätigung (CCα) ist.
(2) Bei zugelassenen Stoffen, für die ein MRL oder ein ML festgelegt wurde, gilt als Entscheidungsgrenze für die Bestätigung (CCα) die Konzentration, bei oder über der mit einer statistischen Sicherheit des numerischen Wertes 1–α gesagt werden kann, dass der zulässige Grenzwert überschritten ist.
(3) Bei nicht zugelassenen oder verbotenen Stoffen oder bei zugelassenen Stoffen, für die in Bezug auf eine bestimmte Tierart oder ein bestimmtes Erzeugnis kein RHG oder HG festgelegt wurde, gilt als Entscheidungsgrenze für die Bestätigung (CCα) die niedrigste Konzentration, bei der mit einer statistischen Sicherheit des numerischen Wertes 1–α gesagt werden kann, dass der betreffende Analyt vorhanden ist.
(4) Bei nicht zugelassenen oder verbotenen pharmakologisch wirksamen Stoffen muss der α-Fehler kleiner oder gleich 1 % sein. Bei allen anderen Stoffen muss der α-Fehler kleiner oder gleich 5 % sein.
Artikel 6
Methoden für die Probenahme
Die Mitgliedstaaten stellen sicher, dass Proben nach den detaillierten Methoden für die Probenahme, wie in Anhang II dieser Verordnung festgelegt, entnommen, gehandhabt und gekennzeichnet werden.
Artikel 7
Aufhebungen und Übergangsmaßnahmen
Die Entscheidungen 2002/657/EG und 98/179/EG werden mit Inkrafttreten dieser Verordnung aufgehoben.
Die Anforderungen gemäß Anhang I Abschnitte 2 und 3 der Entscheidung 2002/657/EG gelten jedoch bis zum 10. Juni 2026 weiterhin für Methoden, die vor Inkrafttreten dieser Verordnung validiert wurden.
Artikel 8
Inkrafttreten
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 22. März 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 95 vom 7.4.2017, S. 1.
(2) Entscheidung 2002/657/EG der Kommission vom 14. August 2002 zur Umsetzung der Richtlinie 96/23/EG des Rates betreffend die Durchführung von Analysemethoden und die Auswertung von Ergebnissen (ABl. L 221 vom 17.8.2002, S. 8).
(3) Entscheidung 98/179/EG der Kommission vom 23. Februar 1998 mit Durchführungsvorschriften für die amtlichen Probenahmen zur Kontrolle von lebenden Tieren und tierischen Erzeugnissen auf bestimmte Stoffe und ihre Rückstände (ABl. L 65 vom 5.3.1998, S. 31).
(4) Richtlinie 96/23/EG des Rates vom 29. April 1996 über Kontrollmaßnahmen hinsichtlich bestimmter Stoffe und ihrer Rückstände in lebenden Tieren und tierischen Erzeugnissen und zur Aufhebung der Richtlinien 85/358/EWG und 86/469/EWG und der Entscheidungen 89/187/EWG und 91/664/EWG (ABl. L 125 vom 23.5.1996, S. 10).
(5) Verordnung (EG) Nr. 401/2006 der Kommission vom 23. Februar 2006 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Kontrolle des Mykotoxingehalts von Lebensmitteln (ABl. L 70 vom 9.3.2006, S. 12).
(6) Verordnung (EU) 2017/644 der Kommission vom 5. April 2017 zur Festlegung der Probenahmeverfahren und Analysemethoden für die Kontrolle der Gehalte an Dioxinen, dioxinähnlichen PCB und nicht dioxinähnlichen PCB in bestimmten Lebensmitteln sowie zur Aufhebung der Verordnung (EU) Nr. 589/2014 (ABl. L 92 vom 6.4.2017, S. 9).
(7) Verordnung (EG) Nr. 333/2007 der Kommission vom 28. März 2007 zur Festlegung der Probenahme- und Analysemethoden für die Kontrolle des Gehalts an Spurenelementen und Prozesskontaminanten in Lebensmitteln (ABl. L 88 vom 29.3.2007, S. 29).
(8) Verordnung (EG) Nr. 1831/2003 des Europäischen Parlaments und des Rates vom 22. September 2003 über Zusatzstoffe zur Verwendung in der Tierernährung (ABl. L 268 vom 18.10.2003, S. 29).
(9) Verordnung (EG) Nr. 152/2009 der Kommission vom 27. Januar 2009 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln (ABl. L 54 vom 26.2.2009, S. 1).
(10) Delegierte Verordnung (EU) 2019/2090 der Kommission vom 19. Juni 2019 zur Ergänzung der Verordnung (EU) 2017/625 des Europäischen Parlaments und des Rates in Bezug auf mutmaßliche oder festgestellte Verstöße gegen Unionsvorschriften über die Verwendung oder über Rückstände pharmakologisch wirksamer Stoffe, die in Tierarzneimitteln oder als Futtermittelzusatzstoffe zugelassen sind, bzw. gegen Unionsvorschriften über die Verwendung oder über Rückstände verbotener oder nicht zugelassener pharmakologisch wirksamer Stoffe (ABl. L 317 vom 9.12.2019, S. 28).
(11) Verordnung (EU) 2019/1871 der Kommission vom 7. November 2019 betreffend die Referenzwerte für Maßnahmen für nicht zulässige pharmakologisch wirksame Stoffe, die in Lebensmitteln tierischen Ursprungs enthalten sind, und zur Aufhebung der Entscheidung 2005/34/EG (ABl. L 289 vom 8.11.2019, S. 41).
(12) Verordnung (EG) Nr. 470/2009 des Europäischen Parlaments und des Rates vom 6. Mai 2009 über die Schaffung eines Gemeinschaftsverfahrens für die Festsetzung von Höchstmengen für Rückstände pharmakologisch wirksamer Stoffe in Lebensmitteln tierischen Ursprungs, zur Aufhebung der Verordnung (EWG) Nr. 2377/90 des Rates und zur Änderung der Richtlinie 2001/82/EG des Europäischen Parlaments und des Rates und der Verordnung (EG) Nr. 726/2004 des Europäischen Parlaments und des Rates (ABl. L 152 vom 16.6.2009, S. 11).
(13) Verordnung (EWG) Nr. 315/93 des Rates vom 8. Februar 1993 zur Festlegung von gemeinschaftlichen Verfahren zur Kontrolle von Kontaminanten in Lebensmitteln (ABl. L 37 vom 13.2.1993, S. 1).
(14) ISO 3534-1: 2006 Statistik — Begriffe und Formelzeichen — Teil 1: Wahrscheinlichkeit und allgemeine statistische Begriffe (Kapitel 1).
(15) Richtlinie 2001/82/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Tierarzneimittel (ABl. L 311 vom 28.11.2001, S. 1).
(16) JCGM 200:2008, International vocabulary of metrology — Basic and general concepts and associated terms (VIM), Third Edition 2008: https://www.iso.org/sites/JCGM/VIM-JCGM200.htmhttps://www.iso.org/sites/JCGM/VIM-JCGM200.htm
(17) Verordnung (EG) Nr. 124/2009 der Kommission vom 10. Februar 2009 zur Festlegung von Höchstgehalten an Kokzidiostatika und Histomonostatika, die in Lebensmitteln aufgrund unvermeidbarer Verschleppung in Futtermittel für Nichtzieltierarten vorhanden sind (ABl. L 40 vom 11.2.2009, S. 7).
(18) Verordnung (EU) Nr. 37/2010 der Kommission vom 22. Dezember 2009 über pharmakologisch wirksame Stoffe und ihre Einstufung hinsichtlich der Rückstandshöchstmengen in Lebensmitteln tierischen Ursprungs (ABl. L 15 vom 20.1.2010, S. 1).
(19) Codex-Alimentarius-Kommission, Ernährungs- und Landwirtschaftsorganisation der Vereinten Nationen/Weltgesundheitsorganisation, Guidelines on analytical terminology (CAC/GL 72-2009).
(20) ISO 5725-1:1994 Genauigkeit (Richtigkeit und Präzision) von Messverfahren und Messergebnissen — Teil 1: Allgemeine Grundlagen und Begriffe (Kapitel 3).
(21) ISO 80000-1:2009 Größen und Einheiten — Teil 1: Allgemeines (Einleitung).
(22) Richtlinie 80/181/EWG des Rates vom 20. Dezember 1979 zur Angleichung der Rechtsvorschriften der Mitgliedstaaten über die Einheiten im Messwesen und zur Aufhebung der Richtlinie 71/354/EWG (ABl. L 39 vom 15.2.1980, S. 40).
(23) ISO/IEC 17025:2017 Allgemeine Anforderungen an die Kompetenz von Prüf- und Kalibrierlaboratorien (Kapitel 3).
(24) ISO 78-2: 1999 Chemie — Gestaltung von Normen — Teil 2: Verfahren der chemischen Analyse (Anhänge).
ANHANG I
KAPITEL 1
LEISTUNGSKRITERIEN UND SONSTIGE ANFORDERUNGEN FÜR ANALYSEMETHODEN
1.1. Anforderungen an Screeningmethoden
1.1.1. Kategorien geeigneter Screeningmethoden
Qualitative, semi-quantitative oder quantitative Methoden sind als Screeningmethoden geeignet.
1.1.2. Anforderungen an biologische, biochemische oder physikalisch-chemische Screeningmethoden
Bei verbotenen oder nicht zugelassenen Stoffen muss das CCβ so niedrig wie möglich (ALARA-Prinzip) sein, in jedem Fall aber unter dem RPA liegen, sofern für den betreffenden Stoff mit der Verordnung (EU) 2019/1871 ein solcher Wert festgelegt wurde.
Bei zugelassenen pharmakologisch wirksamen Stoffen muss das CCβ unter dem MRL oder dem ML liegen.
Nur solche Analysemethoden, für die nachvollziehbar dokumentiert werden kann, dass sie validiert sind und eine nicht konforme („falsch negative“) Rate von 5 % oder darunter (β-Fehler) aufweisen, dürfen für Screeningzwecke eingesetzt werden. Bei Verdacht auf ein nicht konformes Ergebnis muss dieses Ergebnis durch eine Bestätigungsmethode abgesichert werden.
Quantitative Screeningmethoden, die sowohl für Screening- als auch für Bestätigungszwecke eingesetzt werden, müssen denselben Anforderungen an Genauigkeit, Bereich und Präzision genügen, wie unter 1.2.2.1 und 1.2.2.2 beschrieben.
1.2. Anforderungen an Bestätigungsmethoden
1.2.1. Allgemeine Anforderungen an Bestätigungsmethoden
Bei verbotenen oder nicht zugelassenen Stoffen muss die CCα so niedrig wie nach vernünftigem Ermessen erreichbar sein. Bei verbotenen oder nicht zugelassenen Stoffen, für die mit der Verordnung (EU) 2019/1871 ein RPA festgelegt wurde, muss die CCα kleiner oder gleich diesem Referenzwert für Maßnahmen sein.
Bei zugelassenen Stoffen muss die CCα über dem MRL oder dem ML liegen, diesem jedoch so nahe wie möglich sein.
Für Bestätigungszwecke dürfen nur solche Analysemethoden eingesetzt werden, für die nachvollziehbar dokumentiert werden kann, dass sie validiert sind und eine falsch nicht konforme („falsch positive“) Rate von 1 % oder darunter (α-Fehler) bei verbotenen oder nicht zugelassenen Stoffen bzw. von 5 % oder darunter bei zugelassenen Stoffen aufweisen.
Bestätigungsmethoden müssen Informationen über die chemische Struktur des Analyten liefern. Folglich sind Bestätigungsmethoden, die sich auf chromatografische Techniken ohne massenspektrometrische Detektion stützen, für sich allein genommen als Bestätigungsmethoden für verbotene oder nicht zugelassene pharmakologisch wirksame Stoffe nicht geeignet. Sollte für zugelassene Stoffe die Massenspektrometrie nicht geeignet sein, so können auch andere Methoden wie zum Beispiel HPLC-DAD und HPLC-FLD oder eine Kombination aus beiden angewandt werden.
Wenn es die Bestätigungsmethode vorschreibt, muss zu Beginn des Extraktionsverfahrens der Analysenprobe ein geeigneter interner Standard zugesetzt werden. Je nach Verfügbarkeit werden entweder stabile isotopenmarkierte Formen des Analyten, die besonders für die massenspektrometrische Detektion geeignet sind, oder analoge Verbindungen, die mit dem Analyten strukturell eng verwandt sind, verwendet. Wenn kein geeigneter interner Standard verwendet werden kann, muss die Identifizierung des Analyten vorzugsweise durch Co-Chromatografie (1) bestätigt werden. In diesem Fall darf nur ein Peak erhalten werden, wobei dann die Zunahme der Peakhöhe (oder -fläche) der Menge des zugesetzten Analyten entspricht. Ist dies nicht praktikabel, so sind matrix-angepasste Standards oder Matrixstandards zu verwenden.
1.2.2. Allgemeine Leistungskriterien für Bestätigungsmethoden
1.2.2.1. Richtigkeit durch Wiederfindung
Bei wiederholten Analysen eines zertifizierten Referenzmaterials muss die Abweichung des experimentell bestimmten wiederfindungskorrigierten mittleren Masseanteils vom zertifizierten Wert den in Tabelle 1 aufgeführten Mindestwerten der Richtigkeit entsprechen.
Tabelle 1
Mindestwerte der Richtigkeit quantitativer Methoden
|
Massenanteil |
Bereich |
|
≤ 1 μg/kg |
–50 % bis +20 % |
|
> 1 μg/kg bis 10 μg/kg |
–30 % bis +20 % |
|
≥ 10 μg/kg |
–20 % bis +20 % |
Wenn keine zertifizierten Referenzmaterialien zur Verfügung stehen, ist die Bestimmung der Richtigkeit der Messungen auf anderem Wege akzeptabel, zum Beispiel durch die Verwendung von Materialien mit zugewiesenen Werten (Referenzwerten) aus Laborvergleichsstudien oder durch den Zusatz bekannter Mengen des/der Analyten zu einer Leerwertmatrix.
1.2.2.2. Präzision
Bei wiederholter Analyse eines Referenzmaterials oder dotierten Materials darf der Variationskoeffizient (CV) unter laborinternen Reproduzierbarkeitsbedingungen den anhand der Horwitz-Gleichung berechneten Wert nicht überschreiten. Diese Gleichung lautet:
CV = 2(1-0.5 log C)
Dabei ist C der Massenanteil, ausgedrückt als Zehnerpotenz (Exponent) (z. B. 1 mg/g = 10-3). Für Massenanteile unter 120 μg/kg liefert die Horwitz-Gleichung unangemessen hohe Werte. Deshalb darf der zulässige maximale Variationskoeffizient nicht über den in Tabelle 2 aufgeführten Werten liegen.
Tabelle 2
Akzeptabler Variationskoeffizient
|
Massenanteil |
Reproduzierbarkeits-CV (%) |
|
> 1 000 μg/kg |
16 (gemäß der Horwitz-Gleichung) |
|
> 120 μg/kg — 1 000 μg/kg |
22 (gemäß der Horwitz-Gleichung) |
|
10-120 μg/kg |
25 (*) |
|
< 10 μg/kg |
30 (*) |
Bei Analysen unter Wiederholbedingungen soll der Variationskoeffizient unter Wiederholbedingungen zwei Drittel der in Tabelle 2 aufgeführten Werte oder weniger betragen.
1.2.3. Anforderungen an die chromatografische Trennung
Bei der Flüssigchromatografie (LC) oder der Gaschromatografie (GC) muss die Mindestretentionszeit für den/die untersuchten Analyten das Doppelte der Retentionszeit für das Totvolumen der Säule betragen. Die Retentionszeit des Analyten im Extrakt muss mit einer Toleranz von ± 0,1 Minute derjenigen des Kalibrierstandards, eines matrix-angepassten Standards oder eines Matrixstandards entsprechen. Bei schneller Chromatografie, bei der die Retentionszeit unter 2 Minuten beträgt, ist eine Abweichung von weniger als 5 % der Retentionszeit akzeptabel. Für alle Methoden, die ab Inkrafttreten dieser Verordnung validiert werden, muss bei Verwendung eines internen Standards das Verhältnis der chromatografischen Retentionszeit des Analyten zu derjenigen des internen Standards, d. h. die relative Retentionszeit des Analyten, derjenigen des Kalibrierstandards, des matrix-angepassten Standards oder des Matrixstandards mit einer maximalen Abweichung von 0,5 % im Fall von Gaschromatografie und 1 % im Fall von Flüssigchromatografie entsprechen.
1.2.4. Spezifische Leistungskriterien für die Massenspektrometrie
1.2.4.1. Massenspektrometrische Detektion
Zur Durchführung massenspektrometrischer Detektionen stehen unter anderem folgende Datenaufnahmearten zur Verfügung:
|
1. |
Aufzeichnung vollständiger Massenspektren (Full Scan); |
|
2. |
Selected Ion Monitoring (SIM); |
|
3. |
sequentielle massenspektrometrische Verfahren (MSn), zum Beispiel Selected Reaction Monitoring (SRM); |
|
4. |
Kombination aus massenspektrometrischen Verfahren (MS) oder sequentiellen massenspektrometrischen Verfahren (MSn) mit entsprechenden Ionisierungsarten. |
Sowohl die niedrig auflösende Massenspektrometrie (LRMS, mit Nominalmassenauflösung) als auch die hochauflösende Massenspektrometrie (HRMS), inklusive zum Beispiel doppelfokussierende Sektorfeld-, Flugzeit- und Orbitrap-Instrumente, sind hierfür geeignete Techniken.
Zur Bestätigung der Identität eines Analyten bei der hochauflösenden Massenspektrometrie (HRMS) muss die Massenabweichung aller diagnostischen Ionen weniger als 5 ppm (oder im Fall von m/z < 200 weniger als 1 mDa) betragen. Basierend hierauf sollte die effektive Massenauflösung zweckbezogen gewählt werden. Sie muss für den gesamten Massenbereich typischerweise höher als 10 000 (bei der 10 % Tal Definition) oder höher 20 000 (bei voller Halbwertsbreite, FWHM) sein.
Wenn die massenspektrometrische Bestimmung im Full Scan (Aufzeichnung vollständiger Spektren, sowohl LRMS als auch HRMS) durchgeführt wird, sind als diagnostische Ionen nur Ionen mit einer relativen Intensität von mehr als 10 % im Referenzspektrum des Kalibrierstandards, des matrix-angepassten Standards oder des Matrixstandards geeignet. Zu den diagnostischen Ionen gehören das Molekül-Ion (falls es mit einer Intensität von ≥ 10 % des Basispeaks detektiert wird) und die weiteren charakteristischen Fragment- oder Produkt-Ionen.
Auswahl der Vorläufer-Ionen: Wenn die massenspektrometrische Bestimmung durch Fragmentierung von Vorläufer-Ionen erfolgt, wird die Vorläufer-Ionenauswahl mit mindestens Nominalmassenauflösung vorgenommen. Bei dem ausgewählten Vorläufer-Ion muss es sich um das Molekül-Ion, charakteristische Addukte des Molekül-Ions, charakteristische Produkt-Ionen oder eines ihrer Isotopen-Ionen handeln. Ist der Massenauswahlbereich bei der Vorläufer-Ionenauswahl größer als ein Dalton (z. B. im Fall der „Data Independent Acquisition“-Methode), so gilt die Methode als Full-Scan-Bestätigungsanalyse.
Auswahl der Fragment- und Produkt-Ionen: Als Fragment- oder Produkt-Ionen sind für den gemessenen Analyten/das gemessene Produkt diagnostische Fragmente auszuwählen. Nichtselektive Übergänge (z. B. das Tropylium-Kation oder Wasserverlust) sind dabei weitestmöglich zu vermeiden. Die Signalintensität der einzelnen diagnostischen Ionen ist anhand der Peakfläche oder -höhe integrierter extrahierter Ionen-Chromatogramme zu bestimmen. Dies gilt auch, wenn Full-Scan-Messungen zur Identifizierung vorgenommen werden. Das Signal-Rausch-Verhältnis (S/N) aller diagnostischen Ionen muss größer oder gleich drei zu eins (3:1) sein.
Relative Intensitäten: Die relativen Intensitäten der diagnostischen Ionen (Ionenverhältnis) werden in Prozent der Intensität des häufigsten Ions oder Übergangs ausgedrückt. Das Ionenverhältnis ist durch Vergleich von Spektren oder durch Integration der Signale der extrahierten Einzelmassen zu ermitteln. Das Ionenverhältnis des zu bestätigenden Analyten muss den Ionenverhältnissen der matrix-angepassten Standards, der Matrixstandards oder von Standardlösungen mit vergleichbaren Konzentrationen, die unter denselben Bedingungen gemessen wurden, mit einer relativen Abweichung von maximal ± 40 % entsprechen.
Für alle massenspektrometrischen Analysen ist mindestens ein Ionenverhältnis zu bestimmen. Dabei handelt es sich vorzugsweise um mittels eines einzelnen Scans gewonnene Ionen, doch sie können auch aus verschiedenen Scans derselben Injektion stammen (d. h. Full Scan und Fragmentierungsscan).
1.2.4.2. Identifizierung
Für die Auswahl geeigneter Datenaufnahmearten und Auswertungskriterien ist ein System von Identifizierungspunkten zu verwenden. Zur Bestätigung der Identität von Stoffen in einer Matrix, für die ein MRL festgelegt ist (zulässige Verwendung), sind mindestens 4 Identifizierungspunkte erforderlich. Für nicht zugelassene oder verbotene Stoffe sind 5 Identifizierungspunkte nötig. Einer dieser Punkte kann aus der chromatografischen Trennung stammen. Tabelle 3 zeigt die Anzahl der Identifizierungspunkte, die die einzelnen Verfahren liefern. Die zur Bestätigung benötigten Identifizierungspunkte können durch Einsatz verschiedener Techniken erhalten worden sein.
|
1. |
Alle massenspektrometrischen Analysen sind mit einem Trennverfahren zu kombinieren, das ein ausreichendes Trennvermögen und eine ausreichende Selektivität für die spezielle Anwendung aufweist. Geeignete Trennverfahren sind unter anderem die Flüssig- und Gaschromatografie, die Kapillarelektrophorese (CE) und die überkritische Flüssigchromatographie (SFC). Falls zu dem Analyten isobare oder isomere Verbindungen existieren, müssen die Akzeptanzkriterien für Retentionszeitabweichungen (d. h. ± 0,5 % bei GC und ± 1 % bei LC und SFC) für die Bestätigung der Identität des Analyten erfüllt sein. |
|
2. |
Maximal drei separate Verfahren können kombiniert werden, um die Mindestanzahl an Identifizierungspunkten zu erreichen. |
|
3. |
Verschiedene Ionisierungsarten (z. B. Elektronenstoßionisation und chemische Ionisierung) gelten als verschiedene Verfahren. Tabelle 3 Identifizierungspunkte pro Verfahren
Tabelle 4 Beispiele für die Anzahl an Identifizierungspunkten für spezifische Verfahren und Kombinationen aus Verfahren (n = ganze Zahl)
|
1.2.5. Spezifische Leistungskriterien für die Bestimmung eines Analyten mittels Flüssigchromatografie mit Detektion, ausgenommen Massenspektrometrie
Folgende Verfahren dürfen ausschließlich für zugelassene Stoffe alternativ zu massenspektrometrischen Methoden angewandt werden, sofern die für diese Verfahren relevanten Kriterien erfüllt sind:
|
1. |
Full-Scan-Diodenarray-Spektralfotometrie (DAD) gekoppelt mit HPLC; |
|
2. |
Fluoreszenzdetektion (FLD) gekoppelt mit HPLC. |
Flüssigchromatografie mit UV/VIS-Detektion (eine Wellenlänge) ist für sich allein nicht als Bestätigungsmethode geeignet.
1.2.5.1. Leistungskriterien für die Full-Scan-Diodenarray-Spektralfotometrie
Die Leistungskriterien für die chromatografische Trennung gemäß Kapitel 1.2.3 müssen erfüllt sein.
Die Absorptionsmaxima im UV-Spektrum des Analyten müssen mit einer maximalen Toleranz, die durch das Auflösungsvermögen des Detektionssystems bestimmt wird, bei denselben Wellenlängen wie denjenigen des Kalibrierstandards in der Matrix liegen. Bei der Dioden-Array-Detektion beträgt diese maximale Toleranz typischerweise ± 2 nm. Das Analytspektrum oberhalb von 220 nm darf sich für diese Teile der beiden Spektren mit einem relativen Absorptionsvermögen größer oder gleich 10 % optisch nicht vom Spektrum des Kalibrierstandards unterscheiden. Dieses Kriterium ist erfüllt, wenn zum einen die gleichen Maxima vorliegen und zum anderen der Unterschied zwischen den beiden Spektren an keinem Punkt mehr als 10 % des Absorptionsvermögens des Kalibrierstandards beträgt. Wenn mittels computergestützter Bibliothekssuche ein Spektrenvergleich durchgeführt wird, muss das Ergebnis des Spektrenvergleichs zwischen amtlichen Proben und Kalibrierlösung einen kritischen Übereinstimmungsgrad überschreiten. Dieser Übereinstimmungsgrad wird bei der Validierung für jeden Analyten auf der Basis von Spektren bestimmt, für welche die oben beschriebenen Kriterien erfüllt sind. Schwankungen in den Spektren, die durch die Probenmatrix und die Leistungsfähigkeit des Detektors verursacht werden, müssen überprüft werden.
1.2.5.2. Leistungskriterien für die Fluoreszenzdetektion
Die Leistungskriterien für die chromatografische Trennung gemäß Kapitel 1.2.3 müssen erfüllt sein.
Die Anregungs- und Emissionswellenlängen müssen in Verbindung mit den chromatografischen Bedingungen so gewählt werden, dass die Wirkungen störender Bestandteile in Leerwertprobenextrakten auf ein Mindestmaß reduziert werden. Zwischen den Anregungs- und Emissionswellenlängen sollten mindestens 50 Nanometer liegen.
Der jeweilige Analytpeak muss von dem nächstgelegenen Peakmaximum im Chromatogramm durch mindestens eine volle Peakbreite bei 10 % der maximalen Höhe des Analytpeaks getrennt sein.
Dies betrifft Moleküle, die eine natürliche Fluoreszenz aufweisen, und Moleküle, die nach Transformation oder Derivatisierung eine Fluoreszenz zeigen.
KAPITEL 2
VALIDIERUNG
2.1. Für die Analysemethoden zu bestimmende Leistungsmerkmale
Durch die Validierung der Methode ist nachzuweisen, dass die relevanten Leistungsmerkmale der Analysemethode die jeweils dafür gültigen Kriterien erfüllen. Unterschiedliche Kontrollzwecke erfordern unterschiedliche Arten von Methoden. Aus Tabelle 5 geht hervor, welche Leistungsmerkmale für welchen Methodentyp bestimmt werden müssen; nähere Erläuterungen zu jedem Parameter finden sich in diesem Kapitel.
Tabelle 5
Klassifikation von Analysenmethoden nach den zu bestimmenden Leistungsmerkmalen
|
Methode |
Bestätigung |
Screening |
|||
|
qualitativ |
quantitativ |
qualitativ |
semi-quantitativ |
quantitativ |
|
|
Stoffe |
A |
A, B |
A, B |
A, B |
A, B |
|
Identifizierung gemäß 1.2 |
x |
x |
|
|
|
|
CCα |
x |
x |
|
|
|
|
CCβ |
- |
|
x |
x |
x |
|
Richtigkeit |
x |
|
|
x |
|
|
Präzision |
x |
|
(x) |
x |
|
|
Relativer Matrixeffekt/absolute Wiederfindungsrate (*) |
x |
|
|
x |
|
|
Selektivität/Spezifität |
x |
x |
x |
x |
|
|
Stabilität (#) |
x |
x |
x |
x |
|
|
Robustheit |
x |
x |
x |
x |
|
|
x: Durch die Validierung muss nachgewiesen werden, dass die Anforderungen bezüglich der Leistungsmerkmale erfüllt sind. (x) Die Anforderungen bezüglich der Präzision gemäß Kapitel 1.2.2.2 brauchen bei semi-quantitativen Screeningmethoden nicht erfüllt zu sein. Die Präzision ist jedoch zu bestimmen, um nachzuweisen, dass sich die Methode zur Vermeidung nicht konformer Analyseergebnisse eignet. A: verbotene oder nicht zugelassene Stoffe B: zugelassene Stoffe |
|||||
2.2. Richtigkeit, Wiederholpräzision und laborinterne Reproduzierbarkeit
Dieses Kapitel enthält Beispiele für bzw. Verweise auf Validierungsverfahren. Andere Ansätze zum Nachweis, dass die Methode die Leistungskriterien erfüllt, sind zulässig, sofern sie Informationen im selben Umfang und in derselben Qualität liefern.
2.2.1. Klassische Validierung
Zur Berechnung der Parameter nach den klassischen Validierungsmethoden müssen mehrere Einzeluntersuchungen durchgeführt werden. Jedes Leistungsmerkmal muss für jede größere Änderung bestimmt werden (siehe Abschnitt 2.4). Bei Multianalytmethoden können mehrere Analyten gleichzeitig analysiert werden, sofern möglicherweise relevante Störungen ausgeschlossen wurden. Mehrere Leistungsmerkmale können auf ähnliche Weise bestimmt werden. Daher empfiehlt es sich zur Reduzierung des Aufwands, die Untersuchungen soweit wie möglich zu kombinieren (z. B. Wiederholpräzision und laborinterne Reproduzierbarkeit mit der Spezifität, die Analyse von Leerwertproben zur Bestimmung der Entscheidungsgrenze zur Bestätigung und Prüfung auf Spezifität).
2.2.1.1. Richtigkeit auf der Grundlage eines zertifizierten Referenzmaterials
Die Richtigkeit einer Analysemethode ist vorzugsweise anhand eines zertifizierten Referenzmaterials zu bestimmen. Das Verfahren hierfür ist in ISO 5725-4:1994 (2) beschrieben.
Ein Beispiel findet sich nachstehend:
|
1. |
Analyse von sechs Replikaten des zertifizierten Referenzmaterials gemäß der Prüfvorschrift für die Methode; |
|
2. |
Ermittlung der Konzentration des Analyten in jedem Replikat; |
|
3. |
Berechnung des Mittelwerts, der Standardabweichung und des Variationskoeffizienten (%) für diese sechs Replikate; |
|
4. |
Berechnung der Richtigkeit, indem die gemessene mittlere Konzentration durch den zertifizierten Wert (gemessen als Konzentration) dividiert, mit 100 multipliziert und das Ergebnis als Prozentwert angegeben wird. |
Richtigkeit (%) = (mittlere wiederfindungskorrigierte gemessene Konzentration) x 100/zertifizierter Wert
2.2.1.2. Richtigkeit auf der Grundlage dotierter Proben
Ist kein zertifiziertes Referenzmaterial verfügbar, so muss die Richtigkeit der Methode durch Untersuchungen mit dotierter Leerwertmatrix bestimmt werden, und zwar mindestens wie folgt:
|
1. |
Bei Methoden, die ab Inkrafttreten dieser Verordnung validiert werden, ist Leerwertmatrix auszuwählen und auf folgenden Konzentrationsniveaus zu dotieren:
|
|
2. |
Für jedes Konzentrationsniveau ist die Analyse mit mindestens sechs Replikaten durchzuführen. |
|
3. |
Analyse der Proben. |
|
4. |
Berechnung der in jeder Probe festgestellten Konzentration. |
|
5. |
Berechnung der Richtigkeit für jede Probe anhand der nachstehenden Gleichung und anschließend Berechnung der mittleren Richtigkeit und des Variationskoeffizienten für die sechs Ergebnisse bei jeder Konzentration. |
Richtigkeit (%) = (mittlere wiederfindungskorrigierte gemessene Konzentration) x 100/Dotierungsniveau
Bei Methoden für zugelassene Stoffe, die vor dem Geltungsbeginn dieser Verordnung validiert wurden, ist eine Bestimmung der Richtigkeit der Methode anhand von 6 dotierten Aliquoten beim 0,5fachen, 1,0fachen und 1,5fachen des MRL oder des ML ausreichend.
2.2.1.3. Wiederholpräzision
|
1. |
Bei Methoden, die ab Inkrafttreten dieser Verordnung validiert werden, ist ein Satz identischer Proben (identische Leerwertmatrix, identische Tierart) vorzubereiten. Diese sind mit dem Analyten zu folgenden Konzentrationsniveaus zu dotieren:
|
|
2. |
Für jedes Konzentrationsniveau ist die Analyse mit mindestens sechs Replikaten durchzuführen. |
|
3. |
Analyse der Proben. |
|
4. |
Berechnung der in jeder Probe festgestellten Konzentration. |
|
5. |
Berechnung der mittleren Konzentration, der Standardabweichung und des Variationskoeffizienten (%) der dotierten Proben. |
|
6. |
Mindestens zweimalige Wiederholung dieser Schritte. |
|
7. |
Berechnung der mittleren Konzentrationen, der Standardabweichungen (durch Berechnung des Durchschnitts der quadrierten Standardabweichung und Ziehung der Quadratwurzel hieraus) und der Variationskoeffizienten der dotierten Proben insgesamt.
Bei Methoden für zugelassene Stoffe, die vor Inkrafttreten dieser Verordnung validiert wurden, ist eine Bestimmung der Wiederholpräzision auf Konzentrationsniveaus des 0,5fachen, 1,0fachen und 1,5fachen des MRL oder des ML ausreichend (dotierte Leerwertmatrices). Alternativ kann die Berechnung der Wiederholpräzision gemäß ISO 5725-2:2019 (7) erfolgen. |
2.2.1.4. Laborinterne Reproduzierbarkeit
|
1. |
Bei Validierungen, die nach Inkrafttreten dieser Verordnung durchgeführt werden, ist ein Satz Proben mit definiertem Prüfmaterial (identische oder verschiedene Matrices) vorzubereiten, die mit dem/den Analyten zu folgenden Konzentrationsniveaus dotiert werden:
|
|
2. |
In jeder Konzentration ist die Analyse mit mindestens sechs Replikaten von Leerwertmaterial durchzuführen. |
|
3. |
Analyse der Proben. |
|
4. |
Berechnung der in jeder Probe festgestellten Konzentration. |
|
5. |
Mindestens zweimalige Wiederholung dieser Schritte mit verschiedenen Chargen Leerwertmaterials, anderen Bearbeitenden und möglichst vielen unterschiedlichen Umgebungsbedingungen, beispielsweise anderen Chargen von Reagenzien, anderen Lösungsmitteln, anderen Raumtemperaturen, anderen Geräten oder anderen Parametern. |
|
6. |
Bestimmung der mittleren Konzentration, der Standardabweichung und des Variationskoeffizienten (%) der dotierten Proben.
Bei Methoden für zugelassene Stoffe, die vor Inkrafttreten dieser Verordnung validiert wurden, ist eine Bestimmung der laborinternen Reproduzierbarkeit anhand dotierter Matrices in Konzentrationen des 0,5fachen, 1,0fachen und 1,5fachen des MRL oder des ML ausreichend. Alternativ kann die Berechnung der laborinternen Reproduzierbarkeit/der Laborpräzision auch gemäß ISO 5725-2:2019, ISO 11843-1:1997 (8), Codex CAC/GL 59-2006 (9) erfolgen. |
2.2.2. Validierung nach alternativen Modellen
Zur Berechnung der Parameter nach alternativen Modellen muss ein Versuchsplan erstellt werden. Dieser Versuchsplan muss in Abhängigkeit von der Anzahl der verschiedenen Tierarten und der verschiedenen zu untersuchenden Faktoren angelegt sein. Deshalb muss als erster Schritt des Validierungsvorhabens ermittelt werden, welche Probenarten künftig im Labor analysiert werden sollen, um die wichtigsten Tierarten und Faktoren auszuwählen, welche die Messergebnisse beeinflussen können. Der faktorielle Ansatz erlaubt die Einschätzung der Messunsicherheit der Testergebnisse, die unter verschiedenen Bedingungen, wie zum Beispiel verschiedene Bearbeitende, verschiedene Geräte, verschiedene Chargen von Reagenzien, verschiedene Matrices, verschiedene Bearbeitungsdauern und Umgebungsbedingungen in einem bestimmten Labor erzielt wurden. Anschließend muss der Konzentrationsbereich anwendungsbezogen entsprechend dem MRL bzw. dem ML bei zugelassenen Stoffen oder dem RPA bzw. dem niedrigsten Dotierniveau bei verbotenen oder nicht zugelassenen Stoffen gewählt werden.
Ziel des faktoriellen Ansatzes ist die Ermittlung verlässlicher Präzisionsdaten und Messergebnisse durch die gleichzeitig stattfindende kontrollierte Variation der ausgewählten Faktoren. Er ermöglicht die Auswertung der kombinierten Wirkung faktorieller und zufälliger Effekte. Der Versuchsplan erlaubt außerdem die Untersuchung der Robustheit (10) der Analysemethode und die Bestimmung der laborinternen Reproduzierbarkeit unter Einbeziehung verschiedener Matrices.
Nachstehend folgt ein Beispiel für einen alternativen Validierungsansatz unter Verwendung eines orthogonalen Versuchsplans.
Es können bis zu sieben Faktoren („Rauschfaktoren“) untersucht werden. Die Untersuchung ist so konzipiert, dass Präzision, Richtigkeit (auf Basis dotierter Proben), Sensitivität, Messunsicherheit und kritische Konzentrationen durch die Verwendung des Versuchsplans gleichzeitig bestimmt werden können.
Tabelle 6
Beispiel für einen orthogonalen Versuchsplan mit 7 Faktoren (I — VII), jeweils auf zwei Faktorstufen (A/B) variiert, in einer Validierungsstudie mit acht Läufen (Faktor-Stufen-Kombinationen)
|
Faktor |
I |
II |
III |
IV |
V |
VI |
VII |
|
Lauf 01 |
A |
A |
A |
A |
A |
A |
A |
|
Lauf 02 |
A |
A |
B |
A |
B |
B |
B |
|
Lauf 03 |
A |
B |
A |
B |
A |
B |
B |
|
Lauf 04 |
A |
B |
B |
B |
B |
A |
A |
|
Lauf 05 |
B |
A |
A |
B |
B |
A |
B |
|
Lauf 06 |
B |
A |
B |
B |
A |
B |
A |
|
Lauf 07 |
B |
B |
A |
A |
B |
B |
A |
|
Lauf 08 |
B |
B |
B |
A |
A |
A |
B |
Die Berechnung der Methodeneigenschaften ist gemäß der Beschreibung von Jülicher et al. (11) vorzunehmen.
2.2.3. Andere Validierungsansätze
Andere Ansätze zum Nachweis, dass die Methode die Leistungskriterien für die Leistungsmerkmale erfüllt, sind zulässig, sofern sie Informationen im selben Umfang und in derselben Qualität liefern. Die Validierung kann auch erfolgen, indem eine Laborvergleichsstudie zum Beispiel gemäß Codex Alimentarius, ISO oder IUPAC (12) durchgeführt wird, oder mittels alternativer Methoden, zum Beispiel laborinterner Validierungsstudien (13). Wenn andere Validierungsverfahren angewandt werden, sind das zugrunde liegende Modell und die Strategie sowie die jeweiligen Voraussetzungen, Annahmen und Formeln im Validierungsprotokoll darzulegen oder es ist zumindest auf die entsprechenden Quellen zu verweisen.
2.3. Selektivität/Spezifität
Das Unterscheidungsvermögen zwischen dem Analyten und eng verwandten Stoffen ist im bestmöglichen Umfang zu bestimmen. Störungen durch Homologe, Isomere, Abbauprodukte, endogene Stoffe, Analoga, Metabolite des interessierenden Rückstands, Matrixbestandteile oder sonstige möglicherweise störende Stoffe sind zu ermitteln. Falls nötig ist die Methode dahingehend zu ändern, dass die ermittelten Störungen vermieden werden. Die Spezifität der Methode muss anhand des folgenden Ansatzes bestimmt werden:
|
1. |
Es ist eine Reihe chemisch verwandter Verbindungen oder anderer Stoffe auszuwählen, die wahrscheinlich zusammen mit der interessierenden Verbindung, die in den Proben vorhanden sein kann, vorkommen, und zu prüfen, ob sie die Analyse des/der Zielanalyten beeinträchtigen könnten. |
|
2. |
Eine geeignete Anzahl an repräsentativen Leerwertproben, z. B. verschiedene Chargen einer Tierart oder verschiedene Chargen verschiedener Tierarten (n ≥ 20), ist zu analysieren und in dem interessierenden Bereich, in dem die Elution des Zielanalyten zu erwarten ist, auf Störungen (Signale, Peaks oder Ionenspuren) zu untersuchen. |
|
3. |
Repräsentative Leerwertproben sind in einer relevanten Konzentration mit Stoffen zu dotieren, welche die Identifizierung und/oder Quantifizierung des Analyten beeinträchtigen könnten, und zu untersuchen, ob der zugesetzte Stoff
|
2.4. Robustheit
Die Gültigkeit der Methodenkenngrößen ist unter verschiedenen Versuchsbedingungen zu prüfen, wozu beispielsweise unterschiedliche Probenahmebedingungen und geringfügige Änderungen gehören, die sich bei Routineanalysen ergeben können. Bei dieser Prüfung der Robustheit der Methode sollten die Versuchsbedingungen nur geringfügig geändert werden. Der Einfluss dieser Änderungen auf die Leistungsfähigkeit der Methode ist zu bewerten. Für alle geringfügigen Änderungen, die nachweislich einen wesentlichen Einfluss auf die Leistungsfähigkeit des Tests haben, muss jedes Leistungsmerkmal bestimmt werden.
2.5. Stabilität
Die Stabilität des Kalibrierstandards, des matrix-angepassten Standards und/oder des Matrixstandards sowie des Analyten oder der Matrixbestandteile in der Probe während der Lagerung oder der Analyse ist zu bestimmen, da Instabilitäten die Testergebnisse beeinflussen könnten.
Normalerweise ist die Analytstabilität unter verschiedenen Lagerungsbedingungen gut charakterisiert. Die zur Überwachung der Lagerungsbedingungen von Standards und Proben durchgeführten Untersuchungen, die im Rahmen der normalen Laborakkreditierung und des Qualitätskontrollsystems erfolgen, können die erforderlichen Informationen liefern. Stehen für die Analyten in der Matrix Stabilitätsdaten zur Verfügung (z. B. auf Grundlage von Informationen aus den EU-Referenzlaboratorien oder von veröffentlichten Daten), so brauchen diese Daten nicht von jedem Labor ermittelt zu werden. Ein Verweis auf verfügbare Stabilitätsdaten für Analyten in Lösung und in Matrix ist jedoch nur akzeptabel, wenn identische Bedingungen angewandt werden.
Falls die erforderlichen Stabilitätsdaten nicht verfügbar sind, sollten die nachstehenden Ansätze verfolgt werden.
2.5.1. Bestimmung der Stabilität des Analyten in Lösung
|
1. |
Herstellung frischer Stammlösungen des/der Analyten und Verdünnung gemäß den Angaben in der Prüfvorschrift, um ausreichend Aliquote (z. B. 40) jeder gewählten Konzentration zu erhalten. Die Proben sind herzustellen aus
|
|
2. |
Messung des Analytgehalts in der frisch hergestellten Lösung gemäß der Prüfvorschrift. |
|
3. |
Abfüllung entsprechender Volumina in geeignete Behältnisse, Etikettierung und Lagerung gemäß den Licht- und Temperaturbedingungen in Tabelle 7. Die Lagerungszeit ist unter Berücksichtigung der angewandten analytischen Praxis zu wählen, idealerweise bis die ersten Abbauerscheinungen bei der Identifizierung und/oder Quantifizierung festzustellen sind. Ist während der Stabilitätsprüfung kein Abbau festzustellen, so gilt die Lagerungsdauer der Stabilitätsprüfung als identisch mit der maximalen Lagerungsdauer der Lösung. |
|
4. |
Berechnung der Konzentration des/der Analyten in jedem Aliquot im Vergleich zur Konzentration des Analyten in der frisch hergestellten Lösung anhand folgender Formel:
Restanalyt (%) = Ci × 100/Cfrisch Ci = Konzentration zum Zeitpunkt i Cfrisch = Konzentration der frischen Lösung Der Mittelwert von fünf gelagerten Replikaten darf maximal um 15 % vom Mittelwert von fünf frisch hergestellten Replikaten abweichen. Der Mittelwert der fünf frisch hergestellten Lösungen ist als Grundlage für die Berechnung der prozentualen Differenz heranzuziehen. Tabelle 7 Schema für die Bestimmung der Analytstabilität in Lösung
|
2.5.2. Bestimmung der Stabilität des/der Analyten in Matrix
|
1. |
Nach Möglichkeit ist gewachsenes Probenmaterial zu verwenden. Wenn kein solches Material zur Verfügung steht, ist eine mit dem Analyten dotierte Leerwertmatrix zu verwenden. |
|
2. |
Wenn gewachsenes Probenmaterial zur Verfügung steht, ist die Konzentration in der Matrix zu bestimmen, solange diese noch frisch ist. Weitere Aliquote des homogenisierten gewachsenen Probenmaterials sind bei -20 °C oder erforderlichenfalls darunter zu lagern, und die Konzentrationen des Analyten sind zu bestimmen, solange die Probe im Labor gelagert wird. |
|
3. |
Wenn kein gewachsenes Probenmaterial zur Verfügung steht, ist eine ausreichende Menge Leerwertmatrix zu homogenisieren und in 5 Aliquote aufzuteilen. Jedes Aliquot ist mit dem Analyten zu dotieren, der vorzugsweise in einer geringen Menge wässriger Lösung gelöst werden sollte. Ein Aliquot der so dotierten Leerwertmatrix ist sofort zu analysieren. Die verbleibenden Aliquote sind bei mindestens -20 °C oder erforderlichenfalls darunter zu lagern und nach kurzfristiger, mittelfristiger und langfristiger Lagerung mit geeigneten Analysenmethoden zu analysieren. |
|
4. |
Die maximal akzeptable Lagerungszeit und die optimalen Lagerungsbedingungen sind zu dokumentieren.
Der Mittelwert von fünf gelagerten Replikaten darf maximal um die laborinterne Reproduzierbarkeit der Methode vom Mittelwert von fünf frisch hergestellten Replikaten abweichen. Der Mittelwert der fünf frisch hergestellten Replikate ist als Grundlage für die Berechnung der prozentualen Differenz heranzuziehen. |
2.6. Entscheidungsgrenze für die Bestätigung (CCα)
Die CCα ist für Bestätigungsmethoden zu bestimmen. Die CCα muss unter Bedingungen bestimmt werden, die den Anforderungen für die Identifizierung oder die Identifizierung plus Quantifizierung entsprechen, wie in Kapitel 1 („Leistungskriterien und sonstige Anforderungen für Analysemethoden“) dargelegt.
Zur Kontrolle der Konformität der Proben wird die kombinierte Standardmessunsicherheit beim CCα-Wert (Entscheidungsgrenze für die Bestätigung) bereits berücksichtigt.
|
1. |
Für nicht zugelassene oder verbotene pharmakologisch wirksame Stoffe ist die CCα wie folgt zu berechnen:
Im Fall von Methoden, die vor Inkrafttreten dieser Verordnung validiert wurden, darf Methode 2 zur Berechnung der CCα nur bis zum 1. Januar 2026 angewandt werden. Im Fall von Methoden, die nach Inkrafttreten dieser Verordnung validiert werden, dürfen nur die Methoden 1 und 3 angewandt werden. |
|
2. |
Bei zugelassenen Stoffen ist die CCα wie folgt zu berechnen:
|
2.7. Nachweisvermögen von Screeningverfahren (CCβ)
Das CCβ ist für Screeningmethoden zu bestimmen. Das CCβ ist wie in Kapitel 1 („Leistungskriterien und sonstige Anforderungen für Analysemethoden“) dieses Anhangs beschrieben und gemäß den Anforderungen in Tabelle 5 zu bestimmen. Bei Screeningmethoden brauchen jedoch die Anforderungen bezüglich der Identifizierung (siehe 1.2.3, 1.2.4 und 1.2.5) nicht vollständig erfüllt zu werden.
|
1. |
Bei nicht zugelassenen oder verbotenen pharmakologisch wirksamen Stoffen ist ein maximaler β-Fehler von 5 % sicherzustellen. Das CCβ ist wie folgt zu berechnen:
|
|
2. |
Bei zugelassenen Stoffen ist ein maximaler β-Fehler von 5 % sicherzustellen. Das CCβ ist wie folgt zu berechnen:
Bei pharmakologisch wirksamen Stoffen, bei denen der MRL für die Summe verschiedener Stoffe festgelegt ist, muss das CCβ des Stoffs mit der höchsten Konzentration in der Probe als CCβ für die Bewertung der Summe der Stoffe in der gemessenen Probe verwendet werden. |
2.8. Kalibrierkurven
Wenn Kalibrierkurven zur Quantifizierung verwendet werden, gilt Folgendes:
|
(1) |
Mindestens fünf — vorzugsweise in gleichmäßigen Schritten festgelegte — Konzentrationsstufen (einschließlich null) sollten zur Erstellung der Kurve verwendet werden; |
|
(2) |
der Arbeitsbereich der Kurve ist zu beschreiben; |
|
(3) |
die mathematische Formel der Kurve und die Anpassungsgüte der Daten (Bestimmtheitsmaß R2) an die Kurve sind zu beschreiben; |
|
(4) |
Akzeptanzbereiche für die Parameter der Kurve sind zu beschreiben. |
Für Kalibrierkurven auf der Grundlage von Standardlösungen, matrix-angepasster Standards oder Matrixstandards sind Akzeptanzbereiche für die Parameter der Kalibrierkurve anzugeben, die von Serie zu Serie variieren können.
2.9. Absolute Wiederfindungsrate
Die absolute Wiederfindungsrate der Methode ist zu bestimmen, wenn kein interner Standard oder kein Matrixstandard verwendet wird.
Wenn die Anforderungen bezüglich der Richtigkeit, wie in Tabelle 1 angegeben, erfüllt sind, kann ein fester Korrekturfaktor verwendet werden. Ansonsten ist der für die betreffende Probenserie ermittelte Wiederfindungsfaktor zu verwenden. Alternativ muss anstatt eines Wiederfindungskorrekturfaktors das Standardadditionsverfahren (16) oder ein interner Standard verwendet werden.
Die absolute Wiederfindungsrate muss für mindestens sechs repräsentative Matrixchargen berechnet werden.
Ein Aliquot eines Leerwertmaterials ist vor der Extraktion mit dem Analyten zu dotieren, ein zweites Aliquot eines Leerwertmaterials ist nach der Probenvorbereitung in einer relevanten Konzentration zu dotieren, und die Konzentration des Analyten ist zu bestimmen.
Die Wiederfindungsrate ist wie folgt zu berechnen:
Rec (Analyt) = (Signalfäche Matrixstandard)/(Signalfläche matrix-angepasster Standard) × 100
2.10. Relative Matrixeffekte
Der relative Matrixeffekt muss in allen Fällen bestimmt werden. Dies kann entweder im Rahmen der Validierung oder in getrennten Untersuchungen erfolgen. Die Berechnung des relativen Matrixeffekts muss für mindestens 20 verschiedene Leerwertproben (Matrix/Tierart-Kombinationen) durchgeführt werden, entsprechend des Anwendungsbereichs der Methode (z. B. Erfassung verschiedener Tierarten).
Die Leerwertmatrix sollte nach der Extraktion mit dem Analyten in der Konzentration des RPA, des MRL oder des ML dotiert und zusammen mit einer reinen Lösung des Analyten analysiert werden.
Der relative Matrixeffekt oder Matrixfaktor (MF) wird wie folgt berechnet:

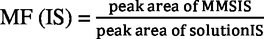

IS: interner Standard
MMS: matrix-angepasster Standard
Der Variationskoeffizient für den MF (für den IS normalisierter Standard) darf nicht mehr als 20 % betragen.
KAPITEL 3
QUALITÄTSKONTROLLE IN DER ROUTINEANALYTIK — LAUFENDE ÜBERPRÜFUNG DER LEISTUNGSFÄHIGKEIT DER METHODE
Die Anforderungen bezüglich der Qualitätssicherung von Analyseergebnissen gemäß Kapitel 7.7 von ISO/IEC 17025:2017 (17) müssen erfüllt sein.
Bei der Routineanalyse ist die Analyse zertifizierter Referenzmaterialien die bevorzugte Option zum Nachweis der Leistungsfähigkeit der Methode. Da zertifizierte Referenzmaterialien, welche die relevanten Analyten in den erforderlichen Konzentrationen enthalten, nur selten verfügbar sind, können alternativ auch Referenzmaterialien verwendet werden, die von den EU-Referenzlaboratorien oder von Laboratorien mit einer Akkreditierung gemäß ISO/IEC 17043:2010 (18) bereitgestellt und charakterisiert wurden. Eine weitere Alternative ist die Verwendung interner Referenzmaterialien, die aber regelmäßig überprüft werden müssen.
Die laufende Überwachung der Leistungsfähigkeit der Methode in der Routineanalytik sollte während des Probenscreenings und der Probenbestätigung erfolgen.
|
1. |
In der Screeningphase: Für jede Serie durchgeführter Analysen ist ein Satz folgender Qualitätskontrollproben gleichzeitig zu analysieren:
|
|
2. |
In der Bestätigungsphase: Für jede Serie durchgeführter Analysen ist ein Satz folgender Qualitätskontrollproben gleichzeitig zu analysieren:
|
Für die Qualitätskontrollproben wird folgende Reihenfolge empfohlen: Systemkontrollprobe, konforme Kontrollprobe, zu bestätigende Probe(n), konforme Kontrollprobe und dotierte Qualitätskontrollprobe (nicht konforme Kontrollproben).
Bei quantitativen Methoden ist für jede Serie amtlicher Proben vor oder nach den oben aufgeführten Proben eine Kalibrierkurve zu analysieren und zu messen.
Soweit praktikabel, ist die Richtigkeit (auf der Grundlage dotierter Proben) aller Zielanalyten in den nicht konformen Kontrollproben anhand von Qualitätskontrollkarten gemäß Kapitel 7.7 von ISO/IEC 17025:2017 zu bewerten. Falls dies eine unverhältnismäßig hohe Anzahl an Bestimmungen der Richtigkeit erfordert, kann die Zahl der Analyten auf eine Anzahl repräsentativer Analyten reduziert werden.
KAPITEL 4
ERWEITERUNG DES VALIDIERTEN ANWENDUNGSBEREICHS EINER BEREITS ZUVOR VALIDIERTEN METHODE
Bisweilen ist es erforderlich, den Anwendungsbereich einer bereits zuvor umfassend validierten Methode zu erweitern. In diesen Fällen sollte eine Erweiterung des Anwendungsbereichs auf effiziente und analytisch fundierte Weise erfolgen. Erreicht werden kann dies durch die Validierung einer im Vergleich zu einer vollständigen Validierung reduzierten Anzahl von Proben (z. B. der halben Probenzahl).
Allerdings müssen Art und Anzahl der zu validierenden Änderungen in einem einzelnen reduzierten Validierungsplan stets auf Expertenwissen und Erfahrung basieren; so würde beispielsweise eine Änderung im Detektionsverfahren in jedem Fall eine vollständige Validierung erfordern.
Generell muss zur Gewährleistung der Validität der Methode deren Leistungsfähigkeit laufend überwacht und mit den ursprünglich erzielten Validierungsparametern verglichen werden. Idealerweise ist diese laufende Kontrolle der Leistungsfähigkeit der Methode so angelegt, dass die für eine vollständige Validierung fehlenden Daten im Lauf der Zeit erhoben werden können (z. B. mit einigen Datenpunkten aus Qualitätskontrollproben in jeder Analysenserie).
4.1. Erweiterungen von Methoden in Bezug auf den Konzentrationsbereich
Aufgrund von Änderungen des MRL, des ML oder des RPA kann sich die Notwendigkeit ergeben, den Konzentrationsbereich, für den eine Methode validiert ist, anzupassen. In solchen Fällen ist die Anwendung eines reduzierten Validierungsplans akzeptabel.
Kalibrierkurven für den geänderten Bereich sollten gemäß dem validierten Verfahren erstellt werden. Es sollten verschiedene Chargen Leerwertmatrix, die in unterschiedlichen Konzentrationen (siehe 2.2.1 und 2.2.2) dotiert sind, analysiert werden. Die Richtigkeit, die Wiederholpräzision und die laborinterne Reproduzierbarkeit/die Laborpräzision sollten verglichen mit den entsprechenden Werten der ursprünglich validierten Methode innerhalb eines akzeptablen Bereichs liegen. Falls relevant, sollte eine Neuberechnung des CCβ (Screeningmethoden) und der CCα (Bestätigungsmethoden) vorgenommen werden.
4.2. Erweiterungen von Methoden in Bezug auf zusätzliche Stoffe
Generell ist die Erweiterung einer Methode auf zusätzliche Verbindungen nur bei Analyten möglich, die eine ähnliche Struktur und ähnliche Eigenschaften wie die bereits in der Analysemethode erfassten Analyten aufweisen. In solchen Fällen ist die Anwendung eines reduzierten Validierungsplans akzeptabel. Eine Abweichung von der Methodenbeschreibung ist nicht erlaubt.
Kalibrierkurven für die zusätzlichen Stoffe sollten gemäß dem validierten Verfahren erstellt werden. Es sollten verschiedene Chargen mit Leerwertmatrix, die in unterschiedlichen Konzentrationen (siehe 2.2.1 und 2.2.2) dotiert sind, analysiert werden. Die Richtigkeit, die Wiederholpräzision und die laborinterne Reproduzierbarkeit/die Laborpräzision sollten innerhalb eines Bereichs liegen, der mit den entsprechenden Werten der in der ursprünglich validierten Methode erfassten anderen Analyten vergleichbar ist und den Anforderungen gemäß 1.2.2 entspricht. Für die neuen Analyten muss eine Berechnung des CCβ (Screeningmethoden) und der CCα (Bestätigungsmethoden) vorgenommen werden.
4.3. Erweiterungen von Methoden in Bezug auf Matrices/Tierarten
Über die Aufnahme neuer Matrices oder Tierarten in eine bereits validierte Analysemethode ist stets im Einzelfall zu entscheiden, basierend auf dem Wissen über die Methode, den bisherigen Erfahrungen sowie Vorversuchen zur Bewertung potenzieller Matrixeffekte und Interferenzen. Generell wird dies nur bei Matrices mit ähnlichen Eigenschaften und bei nicht-kritischen Analyten (Stabilität, Nachweisbarkeit) möglich sein.
Kalibrierkurven (Standard oder Matrix) sollten gemäß dem validierten Verfahren erstellt werden. Es sollten verschiedene Chargen mit Leerwertmatrix, die in unterschiedlichen Konzentrationen (siehe 2.2.1 und 2.2.2) dotiert sind, analysiert werden. Die Richtigkeit, die Wiederholpräzision und die laborinterne Reproduzierbarkeit/die Laborpräzision sollten innerhalb eines Bereichs liegen, der verglichen mit den entsprechenden Werten der ursprünglich validierten Methode akzeptabel ist und den Anforderungen gemäß 1.2.2 entspricht. Abhängig vom Validierungsansatz könnte eine Neuberechnung des CCβ (Screeningmethoden) oder der CCα (Bestätigungsmethoden) erforderlich sein.
Wenn die Ergebnisse verglichen mit den Werten für die ursprüngliche Matrix nicht innerhalb eines akzeptablen Bereichs liegen, ist eine zusätzliche vollständige Validierung erforderlich, um die für die Matrix/Tierart spezifischen Leistungsparameter zu bestimmen.
Wenn MRL für einen spezifischen Stoff bei bestimmten Matrices abweichen, wird es wahrscheinlich schwierig sein, den Anwendungsbereich der Methode an die zusätzliche Matrix/Tierart und Konzentration anzupassen, da in diesem Fall zwei Änderungen berücksichtigt werden müssen. In solchen Fällen wird eine vollständige Validierung empfohlen.
(1) Co-Chromatografie ist eine Technik, bei der der Probenextrakt vor dem (den) chromatografischen Schritt(en) in zwei Teile aufgeteilt wird. Der eine Teil wird normal chromatografiert. Der zweite Teil wird mit der zu messenden Standardsubstanz gemischt. Dieses Gemisch wird dann ebenfalls chromatografiert. Die Menge an zugesetzter Standardsubstanz muss etwa gleich groß wie die geschätzte Menge des Analyten im Extrakt sein. Co-Chromatografie soll die Identifizierung eines Analyten verbessern, wenn chromatografische Methoden eingesetzt werden, besonders wenn kein geeigneter interner Standard verwendet werden kann.
(*) Der angegebene Reproduzierbarkeits-CV (in %) stellt einen Richtwert dar und sollte so niedrig wie möglich sein (ALARA-Prinzip).
(a) Für die Vorläufer-Ionenauswahl gibt es keinen zusätzlichen Identifizierungspunkt, wenn dieses Vorläufer-Ion dasselbe Ion (oder ein Addukt oder Isotop) ist wie das bereits beim Full Scan berücksichtigte HRMS-Ion.
(#) Stehen für die Analyten in einer Matrix aus der wissenschaftlichen Literatur oder von einem anderen Labor Stabilitätsdaten zur Verfügung, so brauchen diese Daten von dem betreffenden Labor nicht nochmals ermittelt zu werden. Dagegen ist ein Verweis auf verfügbare Stabilitätsdaten für Analyten in Lösung nur akzeptabel, wenn identische Bedingungen vorliegen.
(*) Relevant für MS-Methoden, um durch die Validierung nachzuweisen, dass die Anforderungen bezüglich der Leistungsmerkmale erfüllt sind. Der relative Matrixeffekt der Methode ist zu bestimmen, wenn dieser nicht im Rahmen des Validierungsverfahrens bestimmt wurde. Die absolute Wiederfindungsrate der Methode ist zu bestimmen, wenn kein interner Standard oder kein Matrixstandard verwendet wird.
(2) ISO 5725-4:2020 Genauigkeit (Richtigkeit und Präzision) von Messverfahren und Messergebnissen — Teil 4: Grundlegende Methoden für die Ermittlung der Richtigkeit eines vereinheitlichten Messverfahrens (Satz 3).
(3) Ist für die Validierung eines nicht zugelassenen pharmakologisch wirksamen Stoffes eine Konzentration des 0,5fachen des RPA nicht mit angemessenem Aufwand erreichbar, so kann die Konzentration des 0,5fachen RPA ersetzt werden durch die niedrigste Konzentration zwischen dem 0,5fachen und dem 1,0fachen des RPA, die sinnvoll erreichbar ist.
(4) Ist für die Validierung eines spezifischen pharmakologisch wirksamen Stoffes eine Konzentration des 0,1fachen des MRL nicht mit angemessenem Aufwand erreichbar, so kann die Konzentration des 0,1fachen des MRL ersetzt werden durch die niedrigste Konzentration zwischen dem 0,1fachen und dem 0,5fachen des RHG, die sinnvoll erreichbar ist.
(5) Ist für die Validierung eines nicht zugelassenen pharmakologisch wirksamen Stoffes eine Konzentration des 0,5fachen des RPA nicht mit angemessenem Aufwand erreichbar, so kann die Konzentration des 0,5fachen des RPA ersetzt werden durch die niedrigste Konzentration zwischen dem 0,5fachen und dem 1,0fachen des Referenzwerts für Maßnahmen, die sinnvoll erreichbar ist.
(6) Ist für die Validierung eines spezifischen pharmakologisch wirksamen Stoffes eine Konzentration des 0,1fachen des MRL nicht mit angemessenem Aufwand erreichbar, so kann die Konzentration des 0,1fachen des MRL ersetzt werden durch die niedrigste Konzentration zwischen dem 0,1fachen und dem 0,5fachen des MRL, die sinnvoll erreichbar ist.
(7) ISO 5725-2:2019 Genauigkeit (Richtigkeit und Präzision) von Messverfahren und Messergebnissen — Teil 2: Grundlegende Methode für die Ermittlung der Wiederhol- und Vergleichpräzision eines vereinheitlichten Messverfahrens (Satz 3).
(8) ISO 11843-1:1997 Erkennungsfähigkeit — Teil 1: Begriffe.
(9) Codex-Alimentarius-Kommission, Ernährungs- und Landwirtschaftsorganisation der Vereinten Nationen/Weltgesundheitsorganisation, Guidelines on estimation of uncertainty of results (CAC/GL 59-2006).
(10) Die einschlägigen Änderungen in den Versuchsbedingungen können das Probenmaterial, die Analyten, die Lagerbedingungen sowie Umgebung- und/oder Probenvorbereitungsbedingungen betreffen. Für alle Versuchsbedingungen, die in der Praxis möglicherweise schwanken können (beispielsweise Stabilität der Reagenzien, Zusammensetzung der Probe, pH-Werte, Temperatur), ist anzugeben, welche Schwankungen das Analysenergebnis beeinflussen könnten.
(11) Jülicher, B., Gowik, P. and Uhlig, S. (1998) Assessment of detection methods in trace analysis by means of a statistically based in-house validation concept. Analyst, 120, 173.
(12) IUPAC (1995), Protocol for the design, conduct and interpretation of method-performance studies, Pure & Applied Chem, 67, 331.
(13) Gowik, P., Jülicher, B. and Uhlig, S. (1998) Multi-residue method for non-steroidal anti-inflammatory drugs in plasma using high performance liquid chromatography-photodiode-array detection. Method description and comprehensive in-house validation. J. Chromatogr., 716, 221.
(14) ISO 11843-1:1997 Erkennungsfähigkeit — Teil 1: Begriffe.
(15) Durchführungsverordnung (EU) 2018/470 der Kommission vom 21. März 2018 mit ausführlichen Vorschriften zu den Rückstandshöchstmengen, die bei Kontrollen von Lebensmitteln zu berücksichtigen sind, die von in der EU gemäß Artikel 11 der Richtlinie 2001/82/EG behandelten Tieren stammen (ABl. L 79 vom 22.3.2018, S. 16).
(16) Die Menge des zugesetzten Standardanalyten kann beispielsweise zwischen dem Zweifachen und dem Fünffachen der geschätzten Menge des Analyten in der Probe betragen. Dieses Verfahren dient dazu, den Gehalt eines Analyten in einer Probe unter Berücksichtigung der Wiederfindung des Analyseverfahrens zu bestimmen.
(17) ISO/IEC 17025:2017 Allgemeine Anforderungen an die Kompetenz von Prüf- und Kalibrierlaboratorien (Kapitel 7.7).
(18) ISO/IEC 17043:2010 Konformitätsbewertung — Allgemeine Anforderungen an Eignungsprüfungen.
ANHANG II
Probenahmeverfahren und Behandlung amtlicher Proben
1. Probenmenge
Die Mindestprobenmengen sind im nationalen Rückstandsüberwachungsprogramm festzulegen. Die Mindestprobenmengen müssen ausreichend sein, um den zugelassenen Laboratorien die Durchführung der erforderlichen Screening- und Bestätigungsanalysen zu ermöglichen. Speziell bei Geflügel, Tieren in Aquakultur, Kaninchen, Zuchtwild, Reptilien und Insekten umfasst eine Probe je nach den Anforderungen der Analysemethoden ein Tier oder mehrere Tiere. Bei Eiern umfasst eine Probe je nach der angewandten Analysemethode mindestens 12 Eier. Falls mehrere Stoffkategorien in einer einzigen Probe mittels verschiedener Analysemethoden analysiert werden müssen, ist der Probenumfang entsprechend zu erweitern.
2. Aufteilung in Teilproben
Jede Probe muss in mindestens zwei gleiche Teilproben aufgeteilt werden, die jeweils die vollständige Durchführung des Analyseverfahrens ermöglichen, es sei denn, eine solche Aufteilung ist technisch nicht möglich oder nach nationalem Recht nicht erforderlich. Die Unterteilung kann am Ort der Probenahme oder im Labor vorgenommen werden.
3. Rückverfolgbarkeit
Jede Probe ist so zu nehmen, dass deren Rückverfolgung zum Herkunftsbetrieb und gegebenenfalls zur Charge mit den betreffenden Tieren oder zum einzelnen Tier jederzeit möglich ist. Bei Milch können die Proben nach Wahl des Mitgliedstaats an einem der folgenden Orte genommen werden:
|
1. |
im Betrieb aus dem Sammelbehälter; |
|
2. |
im Molkereibetrieb vor Entleerung des Behälters. |
4. Probenbehältnisse
Die Proben müssen in geeigneten Behältnissen verpackt werden, um ihre Integrität zu gewährleisten und ihre Rückverfolgbarkeit zu ermöglichen. Insbesondere müssen die Behältnisse einen Austausch, eine Kreuzkontamination oder eine Zersetzung der Proben ausschließen. Die Behältnisse sind amtlich zu versiegeln.
5. Probenahmebericht
Nach jeder Probenahme ist ein Bericht anzufertigen.
Der Probenahmebericht enthält mindestens folgende Informationen:
|
1. |
Anschrift der zuständigen Behörden; |
|
2. |
Name oder Identifikationscode des Inspektors; |
|
3. |
amtliche Code-Nummer der Probe; |
|
4. |
Datum der Probenahme; |
|
5. |
Name und Anschrift des Eigentümers oder Besitzers der Tiere oder der tierischen Erzeugnisse; |
|
6. |
Name und Anschrift des Herkunftsbetriebs des Tieres (bei Probenahme im Betrieb); |
|
7. |
Registriernummer des Betriebs/Schlachthofs; |
|
8. |
Identifikation des Tieres oder Erzeugnisses; |
|
9. |
Tierart(en); |
|
10. |
Probenmatrix; |
|
11. |
gegebenenfalls in den letzten vier Wochen vor der Probenahme verabreichte Arzneimittel (bei Probenahme im Betrieb); |
|
12. |
zu untersuchender Stoff bzw. zu untersuchende Stoffgruppen; |
|
13. |
besondere Anmerkungen. |
Je nach Probenahmeverfahren sind Papierfassungen oder elektronische Fassungen des Berichts bereitzustellen. Der Probenahmebericht und seine Fassungen sind so zu erstellen, dass ihre Echtheit und Rechtsgültigkeit sichergestellt sind; zu diesem Zweck kann die Unterzeichnung der Dokumente durch den Inspektor erforderlich sein. Bei Probenahmen im Betrieb kann der Landwirt oder sein Stellvertreter aufgefordert werden, den Original-Probenahmebericht zu unterzeichnen.
Das Original des Probenahmeberichts verbleibt bei der zuständigen Behörde, die gewährleisten muss, dass keine unbefugten Personen Zugang zu diesem Originalbericht haben.
Falls nötig, kann der Landwirt oder der Eigentümer des Betriebs über die Probenahmen unterrichtet werden.
6. Probenahmebericht für das Labor
Der von den zuständigen Behörden erstellte Probenahmebericht für das Labor muss den Anforderungen des Kapitels 7 von ISO/IEC 17025:2017 (1) genügen und mindestens folgende Informationen enthalten:
|
1. |
Anschrift der zuständigen Behörden oder benannten Stellen; |
|
2. |
Name oder Identifikationscode des Inspektors; |
|
3. |
amtliche Code-Nummer der Probe; |
|
4. |
Datum der Probenahme; |
|
5. |
Tierart(en); |
|
6. |
Probenmatrix; |
|
7. |
zu untersuchende Stoffe bzw. Stoffgruppen; |
|
8. |
besondere Anmerkungen. |
Der Probenahmebericht für das Labor muss mit der Probe zusammen an das Labor geschickt werden.
7. Transport und Lagerung
Die Rückstandsüberwachungsprogramme müssen geeignete Lagerungs- und Transportbedingungen für jede Analyten-Matrix-Kombination festlegen, damit Analytenstabilität und Probenintegrität gewährleistet sind. Die Transportzeit muss so kurz wie möglich gehalten werden und die Temperatur beim Transport geeignet sein, die Analytenstabilität zu gewährleisten.
Besondere Aufmerksamkeit ist den Transportbehältnissen, der Transporttemperatur und den Lieferzeiten zum zuständigen Labor zu widmen.
Sind Anforderungen des Überwachungsprogramms nicht erfüllt, so unterrichtet das Labor unverzüglich die zuständige Behörde.
(1) ISO/IEC 17025:2017 Allgemeine Anforderungen an die Kompetenz von Prüf- und Kalibrierlaboratorien (Kapitel 7.7).
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/110 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/809 DER KOMMISSION
vom 20. Mai 2021
über die Nichtgenehmigung von fermentiertem Extrakt aus den Blättern von Symphytum officinale L. (Beinwell) als Grundstoff gemäß der Verordnung (EG) Nr. 1107/2009 des Europäischen Parlaments und des Rates über das Inverkehrbringen von Pflanzenschutzmitteln
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EG) Nr. 1107/2009 des Europäischen Parlaments und des Rates vom 21. Oktober 2009 über das Inverkehrbringen von Pflanzenschutzmitteln und zur Aufhebung der Richtlinien 79/117/EWG und 91/414/EWG des Rates (1), insbesondere auf Artikel 13 Absatz 2 in Verbindung mit Artikel 23 Absatz 5,
in Erwägung nachstehender Gründe:
|
(1) |
Am 22. Januar 2015 ging bei der Kommission ein Antrag des Unternehmens Greenprotech auf Genehmigung von „Beinwell-Jauche“ („comfrey steeping“) als Grundstoff zur Verwendung als Insektenabwehrmittel und als Auslöser der eigenen Abwehrmechanismen der Pflanze in Obstbäumen, Gras und Gemüse ein. Dem Antrag waren die gemäß Artikel 23 Absatz 3 Unterabsatz 2 der Verordnung (EG) Nr. 1107/2009 vorgeschriebenen Informationen beigefügt. |
|
(2) |
Die Kommission ersuchte die Europäische Behörde für Lebensmittelsicherheit (im Folgenden die „Behörde“) um wissenschaftliche Unterstützung. Die Behörde legte der Kommission am 28. November 2019 einen technischen Bericht zu „Beinwell-Jauche“ („comfrey steeping“) vor. (2) Auf der Grundlage dieses technischen Berichts und der vom Antragsteller vorgelegten Unterlagen sollte der Geltungsbereich des Antrags so definiert werden, dass er sich auf den Wirkstoff „fermentierter Extrakt aus Blättern von Symphytum officinale L. (Beinwell)“ bezieht. |
|
(3) |
Die Angaben des Antragstellers zu dem fermentierten Extrakt aus Blättern von Symphytum officinale L. (Beinwell) reichten nicht aus, um nachzuweisen, dass er die Kriterien für ein Lebensmittel im Sinne des Artikels 2 der Verordnung (EG) Nr. 178/2002 des Europäischen Parlaments und des Rates (3) erfüllt. |
|
(4) |
Die Behörde kam zu dem Schluss, dass die Spezifikation des fermentierten Extrakts aus Blättern von Symphytum officinale L. (Beinwell) nicht klar definiert ist. Die Behörde wies ferner darauf hin, dass Beinwell bekanntermaßen genotoxische und karzinogene Bestandteile enthält und keine Informationen über die Konzentration von genotoxischen und karzinogenen Verbindungen im fermentierten Extrakt aus Blättern von Symphytum officinale L. vorgelegt wurden. Daher konnte nicht der Schluss gezogen werden, dass der fermentierte Extrakt aus Blättern von Symphytum officinale L. nicht als bedenklicher Stoff im Sinne des Artikels 23 Absatz 1 Buchstabe a der Verordnung (EG) Nr. 1107/2009 anzusehen ist. |
|
(5) |
Darüber hinaus war die Behörde auf der Grundlage der verfügbaren Informationen über den fermentierten Extrakt aus Blättern von Symphytum officinale L. nicht in der Lage, in Bezug auf das Risiko einer nicht ernährungsbedingten Exposition und das Risiko für die Verbraucher zu einer abschließenden Bewertung zu gelangen. Außerdem waren keine ausreichenden Informationen über die Umweltexposition und die Risiken für Nichtzielorganismen verfügbar. |
|
(6) |
Es lagen keine einschlägigen, gemäß anderen Unionsvorschriften durchgeführten Bewertungen nach Artikel 23 Absatz 2 der Verordnung (EG) Nr. 1107/2009 vor. |
|
(7) |
Am 19. Mai 2020 bzw. am 26. Januar 2021 unterbreitete die Kommission dem Ständigen Ausschuss für Pflanzen, Tiere, Lebensmittel und Futtermittel den Überprüfungsbericht (4) und den Entwurf einer Verordnung; die endgültigen Fassungen legte sie diesem Ausschuss anlässlich seiner Sitzung am 24. März 2021 vor. |
|
(8) |
Die Kommission forderte den Antragsteller auf, zum technischen Bericht der Behörde und zum Entwurf des Überprüfungsberichts der Kommission Stellung zu nehmen. Die daraufhin vom Antragsteller vorgelegte Stellungnahme wurde eingehend geprüft. |
|
(9) |
Die Bedenken in Bezug auf den Stoff konnten jedoch trotz der vom Antragsteller vorgebrachten Argumente nicht ausgeräumt werden. |
|
(10) |
Es ist folglich nicht erwiesen, dass die Bedingungen des Artikels 23 der Verordnung (EG) Nr. 1107/2009 erfüllt sind. Daher ist es angezeigt, fermentierten Extrakt aus Blättern von Symphytum officinale L. (Beinwell) nicht als Grundstoff zu genehmigen. |
|
(11) |
Die vorliegende Verordnung steht der Einreichung eines weiteren Antrags auf Genehmigung von fermentiertem Extrakt aus Blättern von Symphytum officinale L. (Beinwell) als Grundstoff gemäß Artikel 23 Absatz 3 der Verordnung (EG) Nr. 1107/2009 oder einem Antrag auf Genehmigung als Wirkstoff gemäß Artikel 7 der genannten Verordnung nicht entgegen. |
|
(12) |
Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Ausschusses für Pflanzen, Tiere, Lebensmittel und Futtermittel — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Der Stoff fermentierter Extrakt aus Blättern von Symphytum officinale L. (Beinwell) wird nicht als Grundstoff genehmigt.
Artikel 2
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 20. Mai 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 309 vom 24.11.2009, S. 1.
(2) EFSA (Europäische Behörde für Lebensmittelsicherheit), 2019. Technischer Bericht über das Ergebnis der Konsultation der Mitgliedstaaten und der EFSA zum Antrag auf Zulassung von Beinwell-Jauche („comfrey steeping“) zur Verwendung im Pflanzenschutz als Insektenabwehrmittel und als Auslöser der eigenen Abwehrmechanismen der Pflanze in Obstbäumen, Gras und Gemüse. EFSA supporting publication 2019:EN-1753. 64 S. doi:10.2903/sp.efsa.2019.EN-1753.
(3) Verordnung (EG) Nr. 178/2002 des Europäischen Parlaments und des Rates vom 28. Januar 2002 zur Festlegung der allgemeinen Grundsätze und Anforderungen des Lebensmittelrechts, zur Errichtung der Europäischen Behörde für Lebensmittelsicherheit und zur Festlegung von Verfahren zur Lebensmittelsicherheit (ABl. L 31 vom 1.2.2002, S. 1).
(4) https://ec.europa.eu/food/plant/pesticides/eu-pesticides-db_en
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/112 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/810 DER KOMMISSION
vom 20. Mai 2021
zur Änderung der Durchführungsverordnung (EU) 2021/2021/808 hinsichtlich der Übergangsbestimmungen für bestimmte in Anhang II der Entscheidung 2002/657/EG aufgeführte Stoffe
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) 2017/625 des Europäischen Parlaments und des Rates vom 15. März 2017 über amtliche Kontrollen und andere amtliche Tätigkeiten zur Gewährleistung der Anwendung des Lebens- und Futtermittelrechts und der Vorschriften über Tiergesundheit und Tierschutz, Pflanzengesundheit und Pflanzenschutzmittel, zur Änderung der Verordnungen (EG) Nr. 999/2001, (EG) Nr. 396/2005, (EG) Nr. 1069/2009, (EG) Nr. 1107/2009, (EU) Nr. 1151/2012, (EU) Nr. 652/2014, (EU) 2016/429 und (EU) 2016/2031 des Europäischen Parlaments und des Rates, der Verordnungen (EG) Nr. 1/2005 und (EG) Nr. 1099/2009 des Rates sowie der Richtlinien 98/58/EG, 1999/74/EG, 2007/43/EG, 2008/119/EG und 2008/120/EG des Rates und zur Aufhebung der Verordnungen (EG) Nr. 854/2004 und (EG) Nr. 882/2004 des Europäischen Parlaments und des Rates, der Richtlinien 89/608/EWG, 89/662/EWG, 90/425/EWG, 91/496/EWG, 96/23/EG, 96/93/EG und 97/78/EG des Rates und des Beschlusses 92/438/EWG des Rates (Verordnung über amtliche Kontrollen) (1), insbesondere auf Artikel 34 Absatz 6,
in Erwägung nachstehender Gründe:
|
(1) |
Mit der Durchführungsverordnung (EU) 2021/2021/808 der Kommission (2) wird unter anderem die Entscheidung 2002/657/EG der Kommission (3) aufgehoben. In Artikel 4 der genannten Entscheidung in Verbindung mit deren Anhang II sind die geforderten Mindestleistungsgrenzen für die pharmakologisch wirksamen Stoffe Chloramphenicol, Nitrofuranmetaboliten, Medroxy-Progesteron-Acetat und Malachitgrün in bestimmten Matrices festgelegt. |
|
(2) |
Artikel 8 der Verordnung (EU) 2019/1871 der Kommission (4) enthält Übergangsbestimmungen für Referenzwerte für Maßnahmen für verbotene pharmakologisch wirksame Stoffe. Bis zum 27. November 2022 sollten die in Anhang II der Entscheidung 2002/657/EG aufgeführten Mindestleistungsgrenzen für Chloramphenicol, Nitrofuranmetaboliten und die Summe von Malachitgrün und Leukomalachitgrün als Referenzwerte für Maßnahmen für Lebensmittel tierischen Ursprungs, die aus Drittländern eingeführt werden, und für Lebensmittel tierischen Ursprungs, die in der Union hergestellt werden, gelten. |
|
(3) |
Für die in Artikel 8 der Verordnung (EU) 2019/1871 genannten Zwecke sollte Anhang II der Entscheidung 2002/657/EG daher bis zum 27. November 2022 gelten. |
|
(4) |
Um Kontinuität zu wahren, sollte diese Verordnung ab demselben Datum gelten wie die Durchführungsverordnung (EU) 2021/2021/808. |
|
(5) |
Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Ausschusses für Pflanzen, Tiere, Lebensmittel und Futtermittel — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Artikel 7 der Durchführungsverordnung (EU) 2021/2021/808 erhält folgende Fassung:
„Artikel 7
Aufhebungen und Übergangsmaßnahmen
Die Entscheidungen 2002/657/EG und 98/179/EG werden mit Inkrafttreten dieser Verordnung aufgehoben.
Die Anforderungen gemäß Anhang I Abschnitte 2 und 3 der Entscheidung 2002/657/EG gelten jedoch bis zum 10. Juni 2026 weiterhin für Methoden, die vor Inkrafttreten dieser Verordnung validiert wurden.
Für die in Artikel 8 Absatz 2 der Verordnung (EU) 2019/1871 genannten Zwecke sollte Anhang II der Entscheidung 2002/657/EG bis zum 27. November 2022 gelten.“
Artikel 2
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 20. Mai 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 95 vom 7.4.2017, S. 1.
(2) Durchführungsverordnung (EU) 2021/2021/808 der Kommission vom 22. März 2021 über Leistungskriterien für Analysemethoden für Rückstände pharmakologisch wirksamer Stoffe in zur Lebensmittelerzeugung genutzten Tieren und über die Auswertung von Ergebnissen sowie über die für Probenahmen anzuwendenden Methoden und zur Aufhebung der Entscheidungen 2002/657/EG und 98/179/EG (siehe Seite 84 dieses Amtsblatts).
(3) Entscheidung 2002/657/EG der Kommission vom 14. August 2002 zur Umsetzung der Richtlinie 96/23/EG des Rates betreffend die Durchführung von Analysemethoden und die Auswertung von Ergebnissen (ABl. L 221 vom 17.8.2002, S. 8).
(4) Verordnung (EU) 2019/1871 der Kommission vom 7. November 2019 betreffend die Referenzwerte für Maßnahmen für nicht zulässige pharmakologisch wirksame Stoffe, die in Lebensmitteln tierischen Ursprungs enthalten sind, und zur Aufhebung der Entscheidung 2005/34/EG (ABl. L 289 vom 8.11.2019, S. 41).
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/114 |
DURCHFÜHRUNGSVERORDNUNG (EU) 2021/811 DER KOMMISSION
vom 20. Mai 2021
zur Änderung des Anhangs I der Durchführungsverordnung (EU) 2021/605 mit besonderen Maßnahmen zur Bekämpfung der Afrikanischen Schweinepest
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EU) 2016/429 des Europäischen Parlaments und des Rates vom 9. März 2016 zu Tierseuchen und zur Änderung und Aufhebung einiger Rechtsakte im Bereich der Tiergesundheit („Tiergesundheitsrecht“ (1)), insbesondere auf Artikel 71 Absatz 3,
in Erwägung nachstehender Gründe:
|
(1) |
Die Afrikanische Schweinepest ist eine ansteckende Viruserkrankung, die gehaltene und wilde Schweine befällt und schwerwiegende Auswirkungen auf die betroffene Tierpopulation sowie die Rentabilität der Landwirtschaft haben kann, was zu Störungen von Verbringungen von Sendungen dieser Tiere und ihrer Erzeugnisse innerhalb der Union sowie von Ausfuhren in Drittländer führen kann. |
|
(2) |
Die Durchführungsverordnung (EU) 2021/605 der Kommission (2) wurde im Rahmen der Verordnung (EU) 2016/429 erlassen und enthält besondere Seuchenbekämpfungsmaßnahmen in Bezug auf die Afrikanische Schweinepest, die von den in Anhang I der genannten Verordnung aufgeführten Mitgliedstaaten in den in demselben Anhang aufgeführten Sperrzonen für einen begrenzten Zeitraum anzuwenden sind. Die in Anhang I der Durchführungsverordnung (EU) 2021/605 als Sperrzonen I, II und III aufgeführten Gebiete beruhen auf der Seuchenlage in Bezug auf die Afrikanische Schweinepest in der Union. Anhang I der Durchführungsverordnung (EU) 2021/605 wurde durch die Durchführungsverordnung (EU) 2021/687 der Kommission (3) geändert, um die Kontinuität und Kohärenz der besonderen Seuchenbekämpfungsmaßnahmen in Bezug auf die Afrikanische Schweinepest in der Union nach dem Auslaufen des Durchführungsbeschlusses 2014/709/EU der Kommission (4) und dem Geltungsbeginn der Durchführungsverordnung (EU) 2021/605 am 21. April 2021 zu gewährleisten. |
|
(3) |
Jegliche Änderungen der Sperrzonen I, II und III in Anhang I der Durchführungsverordnung (EU) 2021/605 sollten sich auf die Seuchenlage in Bezug auf die Afrikanische Schweinepest in den von dieser Seuche betroffenen Gebieten und die allgemeine Seuchenlage in Bezug auf die Afrikanische Schweinepest in dem betroffenen Mitgliedstaat, das Risikoniveau hinsichtlich der weiteren Ausbreitung dieser Seuche, wissenschaftlich fundierte Grundsätze und Kriterien für die geografische Abgrenzung von Zonen aufgrund der Afrikanischen Schweinepest und die Leitlinien der Union stützen, die mit den Mitgliedstaaten im Rahmen des Ständigen Ausschusses für Pflanzen, Tiere, Lebensmittel und Futtermittel vereinbart wurden und auf der Website der Kommission (5) öffentlich zugänglich sind. Diese Änderungen sollten auch internationalen Standards wie dem Gesundheitskodex für Landtiere (6) der Weltorganisation für Tiergesundheit (OIE-Kodex) und den von den zuständigen Behörden der betroffenen Mitgliedstaaten vorgelegten Begründungen für die Abgrenzung der Zonen Rechnung tragen. |
|
(4) |
Seit dem Erlass der Durchführungsverordnung (EU) 2021/687 ist es zu neuen Ausbrüchen der Afrikanischen Schweinepest bei Wildschweinen in der Slowakei und in Polen gekommen. |
|
(5) |
Im April 2021 wurde ein Fall der Afrikanischen Schweinepest bei einem Wildschwein im Bezirk Rimavska Sobota in der Slowakei in einem derzeit in Anhang I der Durchführungsverordnung (EU) 2021/605 als Sperrzone I aufgeführten Gebiet festgestellt. Durch diesen neuen Ausbruch der Afrikanischen Schweinepest bei einem Wildschwein erhöht sich das Risiko, was sich in dem genannten Anhang widerspiegeln sollte. Dementsprechend sollte dieses von diesem jüngsten Ausbruch der Afrikanischen Schweinepest betroffene Gebiet in der Slowakei, das in dem genannten Anhang derzeit als Sperrzone I aufgeführt ist, in diesem Anhang nun stattdessen als Sperrzone II aufgeführt werden. |
|
(6) |
Im Mai 2021 wurde ein Fall der Afrikanischen Schweinepest bei einem Wildschwein im Powiat Szamotulski in Polen in einem derzeit in Anhang I der Durchführungsverordnung (EU) 2021/605 als Sperrzone I aufgeführten Gebiet festgestellt. Durch diesen neuen Ausbruch der Afrikanischen Schweinepest bei einem Wildschwein erhöht sich das Risiko, was sich in dem genannten Anhang widerspiegeln sollte. Dementsprechend sollte dieses von diesem jüngsten Ausbruch der Afrikanischen Schweinepest betroffene Gebiet in Polen, das in dem genannten Anhang derzeit als Sperrzone I aufgeführt ist, in diesem Anhang nun stattdessen als Sperrzone II aufgeführt werden. |
|
(7) |
Nach diesen jüngsten Ausbrüchen der Afrikanischen Schweinepest bei Wildschweinen in der Slowakei und in Polen und unter Berücksichtigung der derzeitigen Seuchenlage in Bezug auf die Afrikanische Schweinepest in der Union wurde die Abgrenzung der Zonen in diesen Mitgliedstaaten neu bewertet und aktualisiert. Darüber hinaus wurden auch die bestehenden Risikomanagementmaßnahmen neu bewertet und aktualisiert. Diese Änderungen sollten sich in Anhang I der Durchführungsverordnung (EU) 2021/605 widerspiegeln. |
|
(8) |
Um den jüngsten epidemiologischen Entwicklungen in Bezug auf die Afrikanische Schweinepest in der Union Rechnung zu tragen und die mit der Ausbreitung dieser Seuche verbundenen Risiken proaktiv anzugehen, sollten in der Slowakei und in Polen neue, ausreichend große Sperrzonen abgegrenzt und ordnungsgemäß als Sperrzonen II in Anhang I der Durchführungsverordnung (EU) 2021/605 aufgenommen werden. Da sich die Lage in Bezug auf die Afrikanische Schweinepest in der Union laufend ändert, wurde bei der Abgrenzung dieser neuen Sperrzonen der Lage in den umliegenden Gebieten Rechnung getragen. |
|
(9) |
Angesichts der Dringlichkeit der Seuchenlage in der Union in Bezug auf die Ausbreitung der Afrikanischen Schweinepest ist es wichtig, dass die mit der vorliegenden Durchführungsverordnung an Anhang I der Durchführungsverordnung (EU) 2021/605 vorzunehmenden Änderungen so bald wie möglich wirksam werden. |
|
(10) |
Die in der vorliegenden Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Ausschusses für Pflanzen, Tiere, Lebensmittel und Futtermittel — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Anhang I der Durchführungsverordnung (EU) 2021/605 erhält die Fassung des Anhangs der vorliegenden Verordnung.
Artikel 2
Diese Verordnung tritt am Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 20. Mai 2021
Für die Kommission
Die Präsidentin
Ursula VON DER LEYEN
(1) ABl. L 84 vom 31.3.2016, S. 1.
(2) Durchführungsverordnung (EU) 2021/605 der Kommission vom 7. April 2021 mit besonderen Maßnahmen zur Bekämpfung der Afrikanischen Schweinepest (ABl. L 129 vom 15.4.2021, S. 1).
(3) Durchführungsverordnung (EU) 2021/687 der Kommission vom 26. April 2021 zur Änderung des Anhangs I der Durchführungsverordnung (EU) 2021/605 mit besonderen Maßnahmen zur Bekämpfung der Afrikanischen Schweinepest (ABl. L 143 vom 27.4.2021, S. 11).
(4) Durchführungsbeschluss 2014/709/EU der Kommission vom 9. Oktober 2014 mit tierseuchenrechtlichen Maßnahmen zur Bekämpfung der Afrikanischen Schweinepest in bestimmten Mitgliedstaaten und zur Aufhebung des Durchführungsbeschlusses 2014/178/EU (ABl. L 295 vom 11.10.2014, S. 63).
(5) Arbeitsunterlage SANTE/7112/2015/Rev. 3 „Grundsätze und Kriterien für die geografische Definition der ASP-Regionalisierung”.https://ec.europa.eu/food/animals/animal-diseases/control-measures/asf_enhttps://ec.europa.eu/food/animals/animal-diseases/control-measures/asf_en
(6) OIE-Gesundheitskodex für Landtiere, 28. Ausgabe, 2019. ISBN von Band I: 978-92-95108-85-1; ISBN von Band II: 978-92-95108-86-8.https://www.oie.int/standard-setting/terrestrial-code/access-online/https://www.oie.int/standard-setting/terrestrial-code/access-online/
ANHANG
Anhang I der Durchführungsverordnung (EU) 2021/605 erhält folgende Fassung:
„ANHANG I
SPERRZONEN
TEIL I
1. Deutschland
Die folgenden Sperrzonen I in Deutschland:
Bundesland Brandenburg:
|
— |
Landkreis Dahme-Spreewald:
|
|
— |
Landkreis Märkisch-Oderland:
|
|
— |
Landkreis Oder-Spree:
|
|
— |
Landkreis Spree-Neiße:
|
Bundesland Sachsen:
|
— |
Landkreis Bautzen
|
|
— |
Landkreis Görlitz:
|
2. Estland
Die folgenden Sperrzonen I in Estland:
|
— |
Hiiu maakond. |
3. Griechenland
Die folgenden Sperrzonen I in Griechenland:
|
— |
in the regional unit of Drama:
|
|
— |
in the regional unit of Xanthi:
|
|
— |
in the regional unit of Rodopi:
|
|
— |
in the regional unit of Evros:
|
|
— |
in the regional unit of Serres:
|
4. Lettland
Die folgenden Sperrzonen I in Lettland:
|
— |
Pāvilostas novada Vērgales pagasts, |
|
— |
Stopiņu novada daļa, kas atrodas uz rietumiem no autoceļa V36, P4 un P5, Acones ielas, Dauguļupes ielas un Dauguļupītes, |
|
— |
Grobiņas novada Medzes, Grobiņas un Gaviezes pagasts. Grobiņas pilsēta, |
|
— |
Rucavas novada Rucavas pagasts, |
|
— |
Nīcas novads. |
5. Litauen
Die folgenden Sperrzonen I in Litauen:
|
— |
Klaipėdos rajono savivaldybė: Agluonėnų, Dovilų, Gargždų, Priekulės, Vėžaičių, Kretingalės ir Dauparų-Kvietinių seniūnijos, |
|
— |
Palangos miesto savivaldybė. |
6. Ungarn
Die folgenden Sperrzonen I in Ungarn:
|
— |
Békés megye 950950, 950960, 950970, 951950, 952050, 952750, 952850, 952950, 953050, 953150, 953650, 953660, 953750, 953850, 953960, 954250, 954260, 954350, 954450, 954550, 954650, 954750, 954850, 954860, 954950, 955050, 955150, 955250, 955260, 955270, 955350, 955450, 955510, 955650, 955750, 955760, 955850, 955950, 956050, 956060, 956150 és 956160 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Bács-Kiskun megye 600150, 600850, 601550, 601650, 601660, 601750, 601850, 601950, 602050, 603250, 603750 és 603850 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Budapest 1 kódszámú, vadgazdálkodási tevékenységre nem alkalmas területe, |
|
— |
Csongrád-Csanád megye 800150, 800160, 800250, 802220, 802260, 802310 és 802450 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Fejér megye 400150, 400250, 400351, 400352, 400450, 400550, 401150, 401250, 401350, 402050, 402350, 402360, 402850, 402950, 403050, 403250, 403350, 403450, 403550, 403650, 403750, 403950, 403960, 403970, 404570, 404650, 404750, 404850, 404950, 404960, 405050, 405750, 405850, 405950, |
|
— |
406050, 406150, 406550, 406650 és 406750 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Jász-Nagykun-Szolnok megye 750150, 750160, 750260, 750350, 750450, 750460, 754450, 754550, 754560, 754570, 754650, 754750, 754950, 755050, 755150, 755250, 755350 és 755450 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Komárom-Esztergom megye 250150, 250250, 250350, 250450, 250460, 250550, 250650, 250750, 250850, 250950, 251050, 251150, 251250, 251350, 251360, 251450, 251550, 251650, 251750, 251850, 252150 és 252250, kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Pest megye 571550, 572150, 572250, 572350, 572550, 572650, 572750, 572850, 572950, 573150, 573250, 573260, 573350, 573360, 573450, 573850, 573950, 573960, 574050, 574150, 574350, 574360, 574550, 574650, 574750, 574850, 574860, 574950, 575050, 575150, 575250, 575350, 575550, 575650, 575750, 575850, 575950, 576050, 576150, 576250, 576350, 576450, 576650, 576750, 576850, 576950, 577050, 577150, 577350, 577450, 577650, 577850, 577950, 578050, 578150, 578250, 578350, 578360, 578450, 578550, 578560, 578650, 578850, 578950, 579050, 579150, 579250, 579350, 579450, 579460, 579550, 579650, 579750, 580250 és 580450 kódszámú vadgazdálkodási egységeinek teljes területe. |
7. Polen
Die folgenden Sperrzonen I in Polen:
w województwie warmińsko-mazurskim:
|
— |
gminy Wielbark i Rozogi w powiecie szczycieńskim, |
|
— |
gminy Janowiec Kościelny, Janowo i część gminy Kozłowo położona na południe od linii wyznaczonej przez linię kolejową w powiecie nidzickim, |
|
— |
gminy Iłowo – Osada, Lidzbark, Płośnica, miasto Działdowo, część gminy Rybno położona na południe od linii wyznaczonej przez drogę kolejową, część gminy wiejskiej Działdowo położona na południe od linii wyznaczonej przez linie kolejowe biegnące od wschodniej do zachodniej granicy gminy w powiecie działdowskim, |
|
— |
gminy Kisielice, Susz i część gminy wiejskiej Iława położona na zachód od linii wyznaczonej przez drogę nr 521 biegnącą od zachodniej granicy gminy do skrzyżowania z drogą łączącą miejscowości Szymbark - Ząbrowo - Segnowy – Laseczno – Gulb, a następnie na zachód od linii wyznaczonej przez drogę łączącą miejscowości Szymbark - Ząbrowo - Segnowy – Laseczno - Gulb biegnącą do południowej granicy gminy w powiecie iławskim, |
|
— |
gminy Biskupiec, Kurzętnik, część gminy wiejskiej Nowe Miasto Lubawskie położona na południe od linii wyznaczonej przez drogę biegnącą od zachodniej granicy gminy do miejscowości Lekarty, a następnie na południowy - zachód od linii wyznaczonej przez drogę łączącą miejscowości Lekarty – Nowy Dwór Bratiański biegnącą do północnej granicy gminy miejskiej Nowe Miasto Lubawskie oraz na południe od linii wyznaczonej przez drogę nr 538, część gminy Grodziczno położona na południe od linii wyznaczonej przez drogę nr 538 w powiecie nowomiejskim. |
w województwie podlaskim:
|
— |
gminy Wysokie Mazowieckie z miastem Wysokie Mazowieckie, Czyżew i część gminy Kulesze Kościelne położona na południe od linii wyznaczonej przez linię koleją w powiecie wysokomazowieckim, |
|
— |
gminy Miastkowo, Nowogród, Śniadowo i Zbójna w powiecie łomżyńskim, |
|
— |
gminy Szumowo, Zambrów z miastem Zambrów i część gminy Kołaki Kościelne położona na południe od linii wyznaczonej przez linię kolejową w powiecie zambrowskim, |
|
— |
gminy Grabowo, Kolno i miasto Kolno, Turośl w powiecie kolneńskim, |
w województwie mazowieckim:
|
— |
powiat ostrołęcki, |
|
— |
powiat miejski Ostrołęka, |
|
— |
gminy Bielsk, Brudzeń Duży, Bulkowo, Drobin, Gąbin, Łąck, Nowy Duninów, Radzanowo, Słupno, Staroźreby i Stara Biała w powiecie płockim, |
|
— |
powiat miejski Płock, |
|
— |
powiat ciechanowski, |
|
— |
gminy Baboszewo, Dzierzążnia, Joniec, Nowe Miasto, Płońsk i miasto Płońsk, Raciąż i miasto Raciąż, Sochocin w powiecie płońskim, |
|
— |
powiat sierpecki, |
|
— |
powiat żuromiński, |
|
— |
gminy Andrzejewo, Brok, Stary Lubotyń, Szulborze Wielkie, Wąsewo, Ostrów Mazowiecka z miastem Ostrów Mazowiecka, część gminy Małkinia Górna położona na północ od rzeki Brok w powiecie ostrowskim, |
|
— |
powiat mławski, |
|
— |
powiat przasnyski, |
|
— |
powiat makowski, |
|
— |
powiat pułtuski, |
|
— |
powiat wyszkowski, |
|
— |
powiat węgrowski, |
|
— |
gminy Dąbrówka, Jadów, Klembów, Poświętne, Radzymin, Strachówka Wołomin i Tłuszcz w powiecie wołomińskim, |
|
— |
gminy Mokobody i Suchożebry w powiecie siedleckim, |
|
— |
gminy Dobre, Jakubów, Kałuszyn, Stanisławów w powiecie mińskim, |
|
— |
gminy Bielany i gmina wiejska Sokołów Podlaski w powiecie sokołowskim, |
|
— |
gminy Kowala, Wierzbica, część gminy Wolanów położona na południe od linii wyznaczonej przez drogę nr 12 w powiecie radomskim, |
|
— |
powiat miejski Radom, |
|
— |
gminy Jastrząb, Mirów, Orońsko w powiecie szydłowieckim, |
|
— |
powiat gostyniński, |
w województwie podkarpackim:
|
— |
gminy Pruchnik, Rokietnica, Roźwienica, w powiecie jarosławskim, |
|
— |
gminy Fredropol, Krasiczyn, Krzywcza, Medyka, Orły, Żurawica, Przemyśl w powiecie przemyskim, |
|
— |
powiat miejski Przemyśl, |
|
— |
gminy Gać, Jawornik Polski, Kańczuga, część gminy Zarzecze położona na południe od linii wyznaczonej przez rzekę Mleczka w powiecie przeworskim, |
|
— |
powiat łańcucki, |
|
— |
gminy Trzebownisko, Głogów Małopolski i część gminy Sokołów Małopolski położona na południe od linii wyznaczonej przez drogę nr 875 w powiecie rzeszowskim, |
|
— |
gminy Dzikowiec, Kolbuszowa, Niwiska i Raniżów w powiecie kolbuszowskim, |
|
— |
gminy Borowa, Czermin, Gawłuszowice, Mielec z miastem Mielec, Padew Narodowa, Przecław, Tuszów Narodowy w powiecie mieleckim, |
w województwie świętokrzyskim:
|
— |
powiat opatowski, |
|
— |
powiat sandomierski, |
|
— |
gminy Bogoria, Łubnice, Oleśnica, Osiek, Połaniec, Rytwiany i Staszów w powiecie staszowskim, |
|
— |
gminy Bliżyn, Skarżysko – Kamienna, Suchedniów i Skarżysko Kościelne w powiecie skarżyskim, |
|
— |
gmina Wąchock, część gminy Brody położona na zachód od linii wyznaczonej przez drogę nr 9 oraz na południowy - zachód od linii wyznaczonej przez drogi: nr 0618T biegnącą od północnej granicy gminy do skrzyżowania w miejscowości Lipie, drogę biegnącą od miejscowości Lipie do wschodniej granicy gminy oraz na północ od drogi nr 42 i część gminy Mirzec położona na zachód od linii wyznaczonej przez drogę nr 744 biegnącą od południowej granicy gminy do miejscowości Tychów Stary a następnie przez drogę nr 0566T biegnącą od miejscowości Tychów Stary w kierunku północno - wschodnim do granicy gminy w powiecie starachowickim, |
|
— |
powiat ostrowiecki, |
|
— |
gminy Fałków, Ruda Maleniecka, Radoszyce, Smyków, część gminy Końskie położona na zachód od linii kolejowej, część gminy Stąporków położona na południe od linii kolejowej w powiecie koneckim, |
|
— |
gminy Mniów i Zagnańsk w powiecie kieleckim, |
w województwie łódzkim:
|
— |
gminy Łyszkowice, Kocierzew Południowy, Kiernozia, Chąśno, Nieborów, część gminy wiejskiej Łowicz położona na północ od linii wyznaczonej przez drogę nr 92 biegnącej od granicy miasta Łowicz do zachodniej granicy gminy oraz część gminy wiejskiej Łowicz położona na wschód od granicy miasta Łowicz i na północ od granicy gminy Nieborów w powiecie łowickim, |
|
— |
gminy Cielądz, Rawa Mazowiecka z miastem Rawa Mazowiecka w powiecie rawskim, |
|
— |
gminy Bolimów, Głuchów, Godzianów, Lipce Reymontowskie, Maków, Nowy Kawęczyn, Skierniewice, Słupia w powiecie skierniewickim, |
|
— |
powiat miejski Skierniewice, |
|
— |
gminy Mniszków, Paradyż, Sławno i Żarnów w powiecie opoczyńskim, |
|
— |
gminy Czerniewice, Inowłódz, Lubochnia, Rzeczyca, Tomaszów Mazowiecki z miastem Tomaszów Mazowiecki i Żelechlinek w powiecie tomaszowskim, |
|
— |
gmina Aleksandrów w powiecie piotrkowskim, |
w województwie pomorskim:
|
— |
gminy Ostaszewo, miasto Krynica Morska oraz część gminy Nowy Dwór Gdański położona na południowy - zachód od linii wyznaczonej przez drogę nr 55 biegnącą od południowej granicy gminy do skrzyżowania z drogą nr 7, następnie przez drogę nr 7 i S7 biegnącą do zachodniej granicy gminy w powiecie nowodworskim, |
|
— |
gminy Lichnowy, Miłoradz, Nowy Staw, Malbork z miastem Malbork w powiecie malborskim, |
|
— |
gminy Mikołajki Pomorskie, Stary Targ i Sztum w powiecie sztumskim, |
|
— |
powiat gdański, |
|
— |
Miasto Gdańsk, |
|
— |
powiat tczewski, |
|
— |
powiat kwidzyński, |
w województwie lubuskim:
|
— |
gminy Przytoczna, Pszczew, Skwierzyna i część gminy Trzciel położona na północ od linii wyznaczonej przez drogę nr 92 w powiecie międzyrzeckim, |
|
— |
gminy Lubniewice i Krzeszyce w powiecie sulęcińskim, |
|
— |
gminy Bogdaniec, Deszczno, Lubiszyn i część gminy Witnica położona na północny - wschód od drogi biegnącej od zachodniej granicy gminy od miejscowości Krześnica, przez miejscowości Kamień Wielki - Mościce -Witnica - Kłopotowo do południowej granicy gminy w powiecie gorzowskim, |
w województwie dolnośląskim:
|
— |
gminy Bolesławiec z miastem Bolesławiec, Gromadka i Osiecznica w powiecie bolesławieckim, |
|
— |
gmina Węgliniec w powiecie zgorzeleckim, |
|
— |
gmina Chocianów i część gminy Przemków położona na południe od linii wyznaczonej przez drogę nr 12 w powiecie polkowickim, |
|
— |
gmina Góra , Wąsosz, część gminy Niechlów położona na północny – wschód od linii wyznaczonej przez rzekę Barycz i część gminy Jemielno położona na wschód od linii wyznaczonej przez drogę nr 323 w powiecie górowskim, |
|
— |
gmina Wińsko w powiecie wołowskim, |
|
— |
gminy Ścinawa i Lubin z miastem Lubin w powiecie lubińskim, |
w województwie wielkopolskim:
|
— |
gminy Krzemieniewo, Osieczna, Rydzyna, część gminy Lipno położona na wschód od linii wyznaczonej przez drogę nr S5, część gminy Święciechowa położona na południe od linii wyznaczonej przez drogę nr 12 oraz na wschód od linii wyznaczonej przez drogę nr S5 w powiecie leszczyńskim, |
|
— |
powiat miejski Leszno, |
|
— |
gminy Chrzypsko Wielkie, Międzychód, część gminy Sieraków położona na zachód od linii wyznaczonej przez drogę nr 186 biegnącą od południowej granicy gminy do miejscowości Lutomek, a następnie na zachód od linii wyznaczonej przez drogę biegnącą od skrzyżowania z drogą nr 186 w miejscowości Lutomek biegnącą do skrzyżowania z ul. Leśną w miejscowości Lutom i dalej na zachód od ul. Leśnej biegnącej do wschodniej granicy gminy, część gminy Kwilcz położona na zachód linii wyznaczonej przez drogę nr 186 biegnącą od północnej granicy gminy do skrzyżowania z drogą nr 24, następnie na południe od linii wyznaczonej przez drogę nr 24 biegnącą od skrzyżowania z drogą nr 186 do skrzyżowania z drogą w miejscowości Pólko, i dalej na zachód od linii wyznaczonej przez drogę biegnącą od miejscowości Pólko przez miejscowość Wituchowo do południowej granicy gminy, w powiecie międzychodzkim, |
|
— |
gminy Lwówek, Kuślin, Opalenica, część gminy Miedzichowo położona na północ od linii wyznaczonej przez drogę nr 92, część gminy Nowy Tomyśl położona na wschód od linii wyznaczonej przez drogę nr 305 w powiecie nowotomyskim, |
|
— |
gminy Granowo, Grodzisk Wielkopolski i część gminy Kamieniec położona na wschód od linii wyznaczonej przez drogę nr 308 w powiecie grodziskim, |
|
— |
gminy Czempiń, Kościan i miasto Kościan, Krzywiń, część gminy Śmigiel położona na wschód od linii wyznaczonej przez drogę nr S5 w powiecie kościańskim, |
|
— |
powiat miejski Poznań, |
|
— |
gminy Buk, Dopiewo, Komorniki, Tarnowo Podgórne, Stęszew, Swarzędz, Pobiedziska, Czerwonak, Mosina, miasto Luboń, miasto Puszczykowo i część gminy Kórnik położona na zachód od linii wyznaczonych przez drogi: nr S11 biegnącą od północnej granicy gminy do skrzyżowania z drogą nr 434 i drogę nr 434 biegnącą od tego skrzyżowania do południowej granicy gminy, część gminy Rokietnica położona na południowy zachód od linii kolejowej biegnącej od północnej granicy gminy w miejscowości Krzyszkowo do południowej granicy gminy w miejscowości Kiekrz oraz część gminy wiejskiej Murowana Goślina położona na południe od linii kolejowej biegnącej od północnej granicy miasta Murowana Goślina do północno-wschodniej granicy gminy w powiecie poznańskim, |
|
— |
gmina Kiszkowo i część gminy Kłecko położona na zachód od rzeki Mała Wełna w powiecie gnieźnieńskim, |
|
— |
gminy Lubasz, Czarnków z miastem Czarnków, część gminy Połajewo na położona na północ od drogi łączącej miejscowości Chraplewo, Tarnówko-Boruszyn, Krosin, Jakubowo, Połajewo - ul. Ryczywolska do północno-wschodniej granicy gminy oraz część gminy Wieleń położona na południe od linii kolejowej biegnącej od wschodniej granicy gminy przez miasto Wieleń i miejscowość Herburtowo do zachodniej granicy gminy w powiecie czarnkowsko-trzcianeckim, |
|
— |
gmina Kaźmierz część gminy Duszniki położona na południowy – wschód od linii wyznaczonej przez drogę nr 306 biegnącą od północnej granicy gminy do miejscowości Duszniki, a następnie na południe od linii wyznaczonej przez ul. Niewierską oraz drogę biegnącą przez miejscowość Niewierz do zachodniej granicy gminy, część gminy Ostroróg, położona na wschód od linii wyznaczonej przez drogę nr 186 i 184 biegnące od granicy gminy do miejscowości Ostroróg, a następnie od miejscowości Ostroróg przez miejscowości Piaskowo – Rudki do południowej granicy gminy, część gminy Wronki położona na północ od linii wyznaczonej przez drogi nr 182 i 186, miasto Szamotuły i część gminy Szamotuły położona na wschód od linii wyznaczonej przez drogę nr 306 do linii wyznaczonej przez wschodnią granicę miasta Szamotuły i na południe od linii kolejowej biegnącej od południowej granicy miasta Szamotuły, do południowo-wschodniej granicy gminy oraz część gminy Obrzycko położona na zachód od drogi nr 185 łączącej miejscowości Gaj Mały, Słopanowo i Obrzycko do północnej granicy miasta Obrzycko, a następnie na zachód od drogi przebiegającej przez miejscowość Chraplewo w powiecie szamotulskim, |
|
— |
gmina Budzyń w powiecie chodzieskim, |
|
— |
gminy Mieścisko, Skoki i Wągrowiec z miastem Wągrowiec w powiecie wągrowieckim, |
|
— |
powiat pleszewski, |
|
— |
gmina Zagórów w powiecie słupeckim, |
|
— |
gmina Pyzdry w powiecie wrzesińskim, |
|
— |
gminy Kotlin, Żerków i część gminy Jarocin położona na wschód od linii wyznaczonej przez drogi nr S11 i 15 w powiecie jarocińskim, |
|
— |
gmina Rozdrażew, część gminy Koźmin Wielkopolski położona na wschód od linii wyznaczonej przez drogę nr 15, część gminy Krotoszyn położona na wschód od linii wyznaczonej przez drogę nr 15 oraz na wschód od granic miasta Krotoszyn w powiecie krotoszyńskim, |
|
— |
gminy Nowe Skalmierzyce, Raszków, Ostrów Wielkopolski z miastem Ostrów Wielkopolski w powiecie ostrowskim, |
|
— |
powiat miejski Kalisz, |
|
— |
gminy Blizanów, Stawiszyn, Żelazków, Ceków – Kolonia, Godziesze Wielkie, Koźminek, Lisków, Mycielin, Opatówek, Szczytniki w powiecie kaliskim, |
|
— |
gmina Malanów i część gminy Tuliszków położona na zachód od linii wyznaczonej przez drogę nr 72 w powiecie tureckim, |
|
— |
gminy Rychwał, Rzgów, Grodziec, część gminy Stare Miasto położona na południe od linii wyznaczonej przez autostradę nr A2 w powiecie konińskim, |
w województwie zachodniopomorskim:
|
— |
część gminy Dębno położona na wschód od linii wyznaczonej przez drogę nr 126 biegnącą od zachodniej granicy gminy do skrzyżowania z drogą nr 23 w miejscowości Dębno, następnie na wschód od linii wyznaczonej przez drogę nr 23 do skrzyżowania z ul. Jana Pawła II w miejscowości Cychry, następnie na północ od ul. Jana Pawła II do skrzyżowania z ul. Ogrodową i dalej na północ od linii wyznaczonej przez ul. Ogrodową, której przedłużenie biegnie do wschodniej granicy gminy w powiecie myśliborskim, |
|
— |
gminy Chojna, Trzcińsko - Zdrój oraz część gminy Cedynia położona na północ od linii wyznaczonej przez drogę nr 124 biegnącą od zachodniej granicy gminy do miasta Cedynia, a następnie na północ od linii wyznaczonej przez drogę nr 125 biegnącą od miasta Cedynia do wschodniej granicy gminy w powiecie gryfińskim. |
8. Slowakei
Die folgenden Sperrzonen I in der Slowakei:
|
— |
the whole district of Vranov nad Topľou, except municipalities included in part II, |
|
— |
the whole district of Humenné, except municipalities included in part II, |
|
— |
the whole district of Snina, |
|
— |
the whole district of Medzilaborce |
|
— |
the whole district of Stropkov |
|
— |
the whole district of Svidník, except municipalities included in part II, |
|
— |
the whole district of Stará Ľubovňa, except municipalities included in part II, |
|
— |
the whole district of whole Kežmarok, |
|
— |
the whole district of Poprad, |
|
— |
the whole district of Veľký Krtíš, except municipalities included in part II, |
|
— |
in the whole district of Zvolen, except municipalities included in part II, |
|
— |
the whole district of Detva, except municipalities included in part II, |
|
— |
the whole district of Krupina, except municipalities included in part II, |
|
— |
the whole district of Brezno. |
TEIL II
1. Bulgarien
Die folgenden Sperrzonen II in Bulgarien:
|
— |
the whole region of Haskovo, |
|
— |
the whole region of Yambol, |
|
— |
the whole region of Stara Zagora, |
|
— |
the whole region of Pernik, |
|
— |
the whole region of Kyustendil, |
|
— |
the whole region of Plovdiv, |
|
— |
the whole region of Pazardzhik, |
|
— |
the whole region of Smolyan, |
|
— |
the whole region of Dobrich, |
|
— |
the whole region of Sofia city, |
|
— |
the whole region of Sofia Province, |
|
— |
the whole region of Blagoevgrad, |
|
— |
the whole region of Razgrad, |
|
— |
the whole region of Kardzhali, |
|
— |
the whole region of Burgas excluding the areas in Part III, |
|
— |
the whole region of Varna excluding the areas in Part III, |
|
— |
the whole region of Silistra, excluding the areas in Part III, |
|
— |
the whole region of Ruse, excluding the areas in Part III, |
|
— |
the whole region of Veliko Tarnovo, excluding the areas in Part III, |
|
— |
the whole region of Pleven, excluding the areas in Part III, |
|
— |
the whole region of Targovishte, excluding the areas in Part III, |
|
— |
the whole region of Shumen, excluding the areas in Part III, |
|
— |
the whole region of Sliven, excluding the areas in Part III, |
|
— |
the whole region of Vidin, excluding the areas in Part III. |
2. Deutschland
Die folgenden Sperrzonen II in Deutschland:
Bundesland Brandenburg:
|
— |
Landkreis Oder-Spree:
|
|
— |
Landkreis Dahme-Spreewald:
|
|
— |
Landkreis Spree-Neiße:
|
|
— |
Landkreis Märkisch-Oderland:
|
|
— |
kreisfreie Stadt Frankfurt (Oder), |
Bundesland Sachsen:
|
— |
Landkreis Görlitz:
|
3. Estland
Die folgenden Sperrzonen II in Estland:
|
— |
Eesti Vabariik (välja arvatud Hiiu maakond). |
4. Lettland
Die folgenden Sperrzonen II in Lettland:
|
— |
Ādažu novads, |
|
— |
Aizputes novada Aizputes, Cīravas un Lažas pagasts, Kalvenes pagasta daļa uz rietumiem no ceļa pie Vārtājas upes līdz autoceļam A9, uz dienvidiem no autoceļa A9, uz rietumiem no autoceļa V1200, Kazdangas pagasta daļa uz rietumiem no ceļa V1200, P115, P117, V1296, Aizputes pilsēta, |
|
— |
Aglonas novads, |
|
— |
Aizkraukles novads, |
|
— |
Aknīstes novads, |
|
— |
Alojas novads, |
|
— |
Alsungas novads, |
|
— |
Alūksnes novads, |
|
— |
Amatas novads, |
|
— |
Apes novads, |
|
— |
Auces novads, |
|
— |
Babītes novads, |
|
— |
Baldones novads, |
|
— |
Baltinavas novads, |
|
— |
Balvu novads, |
|
— |
Bauskas novads, |
|
— |
Beverīnas novads, |
|
— |
Brocēnu novads, |
|
— |
Burtnieku novads, |
|
— |
Carnikavas novads, |
|
— |
Cēsu novads |
|
— |
Cesvaines novads, |
|
— |
Ciblas novads, |
|
— |
Dagdas novads, |
|
— |
Daugavpils novads, |
|
— |
Dobeles novads, |
|
— |
Dundagas novads, |
|
— |
Durbes novads, |
|
— |
Engures novads, |
|
— |
Ērgļu novads, |
|
— |
Garkalnes novads, |
|
— |
Grobiņas novada Bārtas pagasts, |
|
— |
Gulbenes novads, |
|
— |
Iecavas novads, |
|
— |
Ikšķiles novads, |
|
— |
Ilūkstes novads, |
|
— |
Inčukalna novads, |
|
— |
Jaunjelgavas novads, |
|
— |
Jaunpiebalgas novads, |
|
— |
Jaunpils novads, |
|
— |
Jēkabpils novads, |
|
— |
Jelgavas novads, |
|
— |
Kandavas novads, |
|
— |
Kārsavas novads, |
|
— |
Ķeguma novads, |
|
— |
Ķekavas novads, |
|
— |
Kocēnu novads, |
|
— |
Kokneses novads, |
|
— |
Krāslavas novads, |
|
— |
Krimuldas novads, |
|
— |
Krustpils novads, |
|
— |
Kuldīgas novada, Laidu pagasta daļa uz ziemeļiem no autoceļa V1296, Padures, Rumbas, Rendas, Kabiles, Vārmes, Pelču, Ēdoles, Īvandes, Kurmāles, Turlavas, Gudenieku un Snēpeles pagasts, Kuldīgas pilsēta, |
|
— |
Lielvārdes novads, |
|
— |
Līgatnes novads, |
|
— |
Limbažu novads, |
|
— |
Līvānu novads, |
|
— |
Lubānas novads, |
|
— |
Ludzas novads, |
|
— |
Madonas novads, |
|
— |
Mālpils novads, |
|
— |
Mārupes novads, |
|
— |
Mazsalacas novads, |
|
— |
Mērsraga novads, |
|
— |
Naukšēnu novads, |
|
— |
Neretas novads, |
|
— |
Ogres novads, |
|
— |
Olaines novads, |
|
— |
Ozolnieku novads, |
|
— |
Pārgaujas novads, |
|
— |
Pāvilostas novada Sakas pagasts, Pāvilostas pilsēta, |
|
— |
Pļaviņu novads, |
|
— |
Preiļu novads, |
|
— |
Priekules novads, |
|
— |
Priekuļu novads, |
|
— |
Raunas novads, |
|
— |
republikas pilsēta Daugavpils, |
|
— |
republikas pilsēta Jelgava, |
|
— |
republikas pilsēta Jēkabpils, |
|
— |
republikas pilsēta Jūrmala, |
|
— |
republikas pilsēta Rēzekne, |
|
— |
republikas pilsēta Valmiera, |
|
— |
Rēzeknes novads, |
|
— |
Riebiņu novads, |
|
— |
Rojas novads, |
|
— |
Ropažu novads, |
|
— |
Rucavas novada Dunikas pagasts, |
|
— |
Rugāju novads, |
|
— |
Rundāles novads, |
|
— |
Rūjienas novads, |
|
— |
Salacgrīvas novads, |
|
— |
Salas novads, |
|
— |
Salaspils novads, |
|
— |
Saldus novads, |
|
— |
Saulkrastu novads, |
|
— |
Sējas novads, |
|
— |
Siguldas novads, |
|
— |
Skrīveru novads, |
|
— |
Skrundas novada Raņķu pagasta daļa uz ziemeļiem no autoceļa V1272 līdz robežai ar Ventas upi, Skrundas pagasta daļa no Skrundas uz ziemeļiem no autoceļa A9 un austrumiem no Ventas upes, |
|
— |
Smiltenes novads, |
|
— |
Stopiņu novada daļa, kas atrodas uz austrumiem no autoceļa V36, P4 un P5, Acones ielas, Dauguļupes ielas un Dauguļupītes, |
|
— |
Strenču novads, |
|
— |
Talsu novads, |
|
— |
Tērvetes novads, |
|
— |
Tukuma novads, |
|
— |
Vaiņodes novada Vaiņodes pagasts un Embūtes pagasta daļa uz dienvidiem autoceļa P116, P106, |
|
— |
Valkas novads, |
|
— |
Varakļānu novads, |
|
— |
Vārkavas novads, |
|
— |
Vecpiebalgas novads, |
|
— |
Vecumnieku novads, |
|
— |
Ventspils novads, |
|
— |
Viesītes novads, |
|
— |
Viļakas novads, |
|
— |
Viļānu novads, |
|
— |
Zilupes novads. |
5. Litauen
Die folgenden Sperrzonen II in Litauen:
|
— |
Alytaus miesto savivaldybė, |
|
— |
Alytaus rajono savivaldybė, |
|
— |
Anykščių rajono savivaldybė, |
|
— |
Akmenės rajono savivaldybė, |
|
— |
Birštono savivaldybė, |
|
— |
Biržų miesto savivaldybė, |
|
— |
Biržų rajono savivaldybė, |
|
— |
Druskininkų savivaldybė, |
|
— |
Elektrėnų savivaldybė, |
|
— |
Ignalinos rajono savivaldybė, |
|
— |
Jonavos rajono savivaldybė, |
|
— |
Joniškio rajono savivaldybė, |
|
— |
Jurbarko rajono savivaldybė: Eržvilko, Girdžių, Jurbarko miesto, Jurbarkų, Raudonės, Šimkaičių, Skirsnemunės, Smalininkų, Veliuonos ir Viešvilės seniūnijos, |
|
— |
Kaišiadorių rajono savivaldybė, |
|
— |
Kalvarijos savivaldybė, |
|
— |
Kauno miesto savivaldybė, |
|
— |
Kauno rajono savivaldybė: Akademijos, Alšėnų, Batniavos, Ežerėlio, Domeikavos, Garliavos, Garliavos apylinkių, Karmėlavos, Kulautuvos, Lapių, Linksmakalnio, Neveronių, Raudondvario, Ringaudų, Rokų, Samylų, Taurakiemio, Vandžiogalos, Užliedžių, Vilkijos, ir Zapyškio seniūnijos, Babtų seniūnijos dalis į rytus nuo kelio A1, ir Vilkijos apylinkių seniūnijos dalis į vakarus nuo kelio Nr. 1907, |
|
— |
Kazlų rūdos savivaldybė, |
|
— |
Kelmės rajono savivaldybė, |
|
— |
Kėdainių rajono savivaldybė: Dotnuvos, Gudžiūnų, Kėdainių miesto, Krakių, Pelėdnagių, Surviliškio, Šėtos, Truskavos, Vilainių ir Josvainių seniūnijos dalis į šiaurę ir rytus nuo kelio Nr. 229 ir Nr. 2032, |
|
— |
Klaipėdos rajono savivaldybė: Judrėnų, Endriejavo ir Veiviržėnų seniūnijos, |
|
— |
Kupiškio rajono savivaldybė, |
|
— |
Kretingos rajono savivaldybė, |
|
— |
Lazdijų rajono savivaldybė, |
|
— |
Marijampolės savivaldybė, |
|
— |
Mažeikių rajono savivaldybė, |
|
— |
Molėtų rajono savivaldybė, |
|
— |
Pagėgių savivaldybė, |
|
— |
Pakruojo rajono savivaldybė, |
|
— |
Panevėžio rajono savivaldybė, |
|
— |
Panevėžio miesto savivaldybė, |
|
— |
Pasvalio rajono savivaldybė, |
|
— |
Radviliškio rajono savivaldybė, |
|
— |
Rietavo savivaldybė, |
|
— |
Prienų rajono savivaldybė, |
|
— |
Plungės rajono savivaldybė: Žlibinų, Stalgėnų, Nausodžio, Plungės miesto, Šateikių ir Kulių seniūnijos, |
|
— |
Raseinių rajono savivaldybė: Betygalos, Girkalnio, Kalnujų, Nemakščių, Pagojukų, Paliepių, Raseinių miesto, Raseinių, Šiluvos, Viduklės seniūnijos, |
|
— |
Rokiškio rajono savivaldybė, |
|
— |
Skuodo rajono savivaldybės: Aleksandrijos, Ylakių, Lenkimų, Mosėdžio, Skuodo ir Skuodo miesto seniūnijos, |
|
— |
Šakių rajono savivaldybė, |
|
— |
Šalčininkų rajono savivaldybė, |
|
— |
Šiaulių miesto savivaldybė, |
|
— |
Šiaulių rajono savivaldybė, |
|
— |
Šilutės rajono savivaldybė, |
|
— |
Širvintų rajono savivaldybė, |
|
— |
Šilalės rajono savivaldybė, |
|
— |
Švenčionių rajono savivaldybė, |
|
— |
Tauragės rajono savivaldybė, |
|
— |
Telšių rajono savivaldybė, |
|
— |
Trakų rajono savivaldybė, |
|
— |
Ukmergės rajono savivaldybė, |
|
— |
Utenos rajono savivaldybė, |
|
— |
Varėnos rajono savivaldybė, |
|
— |
Vilniaus miesto savivaldybė, |
|
— |
Vilniaus rajono savivaldybė, |
|
— |
Vilkaviškio rajono savivaldybė, |
|
— |
Visagino savivaldybė, |
|
— |
Zarasų rajono savivaldybė. |
6. Ungarn
Die folgenden Sperrzonen II in Ungarn:
|
— |
Békés megye 950150, 950250, 950350, 950450, 950550, 950650, 950660, 950750, 950850, 950860, 951050, 951150, 951250, 951260, 951350, 951450, 951460, 951550, 951650, 951750, 952150, 952250, 952350, 952450, 952550, 952650, 953250, 953260, 953270, 953350, 953450, 953550, 953560, 953950, 954050, 954060, 954150, 956250, 956350, 956450, 956550, 956650 és 956750 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Borsod-Abaúj-Zemplén megye valamennyi vadgazdálkodási egységének teljes területe, |
|
— |
Fejér megye 403150, 403160, 403260, 404250, 404550, 404560, 405450, 405550, 405650, 406450 és 407050 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Hajdú-Bihar megye valamennyi vadgazdálkodási egységének teljes területe, |
|
— |
Heves megye valamennyi vadgazdálkodási egységének teljes területe, |
|
— |
Jász-Nagykun-Szolnok megye 750250, 750550, 750650, 750750, 750850, 750970, 750980, 751050, 751150, 751160, 751250, 751260, 751350, 751360, 751450, 751460, 751470, 751550, 751650, 751750, 751850, 751950, 752150, 752250, 752350, 752450, 752460, 752550, 752560, 752650, 752750, 752850, 752950, 753060, 753070, 753150, 753250, 753310, 753450, 753550, 753650, 753660, 753750, 753850, 753950, 753960, 754050, 754150, 754250, 754360, 754370, 754850, 755550, 755650 és 755750 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Komárom-Esztergom megye: 251950, 252050, 252350, 252450, 252460, 252550, 252650, 252750, 252850, 252860, 252950, 252960, 253050, 253150, 253250, 253350, 253450 és 253550 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Nógrád megye valamennyi vadgazdálkodási egységeinek teljes területe, |
|
— |
Pest megye 570150, 570250, 570350, 570450, 570550, 570650, 570750, 570850, 570950, 571050, 571150, 571250, 571350, 571650, 571750, 571760, 571850, 571950, 572050, 573550, 573650, 574250, 577250, 580050 és 580150 kódszámú vadgazdálkodási egységeinek teljes területe, |
|
— |
Szabolcs-Szatmár-Bereg megye valamennyi vadgazdálkodási egységének teljes területe. |
7. Polen
Die folgenden Sperrzonen II in Polen:
w województwie warmińsko-mazurskim:
|
— |
gminy Kalinowo, Stare Juchy, Prostki oraz gmina wiejska Ełk w powiecie ełckim, |
|
— |
powiat elbląski, |
|
— |
powiat miejski Elbląg, |
|
— |
powiat gołdapski, |
|
— |
powiat piski, |
|
— |
powiat bartoszycki, |
|
— |
gminy Biskupiec, Jeziorany, Kolno, część gminy Olsztynek położona na południe od linii wyznaczonej przez drogę nr S51 biegnącą od wschodniej granicy gminy do miejscowości Ameryka oraz na zachód od linii wyznaczonej przez drogę biegnącą od skrzyżowania z drogą S51 do północnej granicy gminy, łączącej miejscowości Mańki – Mycyny – Ameryka w powiecie olsztyńskim, |
|
— |
powiat ostródzki, |
|
— |
powiat olecki, |
|
— |
powiat giżycki, |
|
— |
powiat braniewski, |
|
— |
powiat kętrzyński, |
|
— |
gminy Lubomino i Orneta w powiecie lidzbarskim, |
|
— |
gmina Nidzica i część gminy Kozłowo położona na północ od linii wyznaczonej przez linię kolejową w powiecie nidzickim, |
|
— |
gminy Dźwierzuty, Jedwabno, Pasym, Szczytno i miasto Szczytno i Świętajno w powiecie szczycieńskim, |
|
— |
powiat mrągowski, |
|
— |
gminy Lubawa, miasto Lubawa, Zalewo, miasto Iława i część gminy wiejskiej Iława położona na wschód od linii wyznaczonej przez drogę nr 521 biegnącą od zachodniej granicy gminy do skrzyżowania z drogą łączącą miejscowości Szymbark - Ząbrowo - Segnowy – Laseczno – Gulb, a następnie na wschód od linii wyznaczonej przez drogę łączącą miejscowości Szymbark - Ząbrowo - Segnowy – Laseczno - Gulb biegnącą do południowej granicy gminy w powiecie iławskim, |
|
— |
część gminy wiejskiej Nowe Miasto Lubawskie położona na północ od linii wyznaczonej przez drogę biegnącą od zachodniej granicy gminy do miejscowości Lekarty, a następnie na północny -wschód od linii wyznaczonej przez drogę łączącą miejscowości Lekarty – Nowy Dwór Bratiański biegnącą do północnej granicy gminy miejskiej Nowe Miasto Lubawskie oraz na północ od linii wyznaczonej przez drogę nr 538, część gminy Grodziczno położona na północ od linii wyznaczonej przez drogę nr 538 w powiecie nowomiejskim, |
|
— |
powiat węgorzewski, |
|
— |
część gminy Rybno położona na północ od linii kolejowej, część gminy wiejskiej Działdowo położona na północ od linii wyznaczonej przez linie kolejowe biegnące od wschodniej do zachodniej granicy gminy w powiecie działdowskim, |
w województwie podlaskim:
|
— |
powiat bielski, |
|
— |
powiat grajewski, |
|
— |
powiat moniecki, |
|
— |
powiat sejneński, |
|
— |
gminy Łomża, Piątnica, Jedwabne, Przytuły i Wizna w powiecie łomżyńskim, |
|
— |
powiat miejski Łomża, |
|
— |
powiat siemiatycki, |
|
— |
powiat hajnowski, |
|
— |
gminy Ciechanowiec, Klukowo, Szepietowo, Kobylin-Borzymy, Nowe Piekuty, Sokoły i część gminy Kulesze Kościelne położona na północ od linii wyznaczonej przez linię kolejową w powiecie wysokomazowieckim, |
|
— |
gmina Rutki i część gminy Kołaki Kościelne położona na północ od linii wyznaczonej przez linię kolejową w powiecie zambrowskim, |
|
— |
gminy Mały Potok i Stawiski w powiecie kolneńskim, |
|
— |
powiat białostocki, |
|
— |
powiat suwalski, |
|
— |
powiat miejski Suwałki, |
|
— |
powiat augustowski, |
|
— |
powiat sokólski, |
|
— |
powiat miejski Białystok, |
w województwie mazowieckim:
|
— |
gminy Domanice, Korczew, Kotuń, Mordy, Paprotnia, Przesmyki, Siedlce, Skórzec, Wiśniew, Wodynie, Zbuczyn w powiecie siedleckim, |
|
— |
powiat miejski Siedlce, |
|
— |
gminy Ceranów, Jabłonna Lacka, Kosów Lacki, Repki, Sabnie, Sterdyń w powiecie sokołowskim, |
|
— |
powiat łosicki, |
|
— |
powiat sochaczewski, |
|
— |
gminy Policzna, Przyłęk, Tczów i Zwoleń w powiecie zwoleńskim, |
|
— |
powiat kozienicki, |
|
— |
gminy Chotcza i Solec nad Wisłą w powiecie lipskim, |
|
— |
gminy Gózd, Jastrzębia, Jedlnia Letnisko, Pionki z miastem Pionki, Skaryszew, Jedlińsk, Przytyk, Zakrzew, część gminy Iłża położona na zachód od linii wyznaczonej przez drogę nr 9, część gminy Wolanów położona na północ od drogi nr 12 w powiecie radomskim, |
|
— |
gminy Bodzanów, Słubice, Wyszogród i Mała Wieś w powiecie płockim, |
|
— |
powiat nowodworski, |
|
— |
gminy Czerwińsk nad Wisłą, Naruszewo, Załuski w powiecie płońskim, |
|
— |
gminy: miasto Kobyłka, miasto Marki, miasto Ząbki, miasto Zielonka w powiecie wołomińskim, |
|
— |
gminy Borowie, Garwolin z miastem Garwolin, Miastków Kościelny, Parysów, Pilawa, część gminy Wilga położona na północ od linii wyznaczonej przez rzekę Wilga biegnącą od wschodniej granicy gminy do ujścia do rzeki Wisły, część gminy Górzno położona na północ od linii wyznaczonej przez drogę łączącą miejscowości Łąki i Górzno biegnącą od wschodniej granicy gminy, następnie od miejscowości Górzno na północ od drogi nr 1328W biegnącej do drogi nr 17, a następnie na północ od linii wyznaczonej przez drogę biegnącą |
|
— |
od drogi nr 17 do zachodniej granicy gminy przez miejscowości Józefów i Kobyla Wola w powiecie garwolińskim, |
|
— |
gminy Boguty – Pianki, Zaręby Kościelne, Nur i część gminy Małkinia Górna położona na południe od rzeki Brok w powiecie ostrowskim, |
|
— |
gminy Chlewiska i Szydłowiec w powiecie szydłowieckim, |
|
— |
gminy Cegłów, Dębe Wielkie, Halinów, Latowicz, Mińsk Mazowiecki i miasto Mińsk Mazowiecki, Mrozy, Siennica, miasto Sulejówek w powiecie mińskim, |
|
— |
powiat otwocki, |
|
— |
powiat warszawski zachodni, |
|
— |
powiat legionowski, |
|
— |
powiat piaseczyński, |
|
— |
powiat pruszkowski, |
|
— |
powiat grójecki, |
|
— |
powiat grodziski, |
|
— |
powiat żyrardowski, |
|
— |
powiat białobrzeski, |
|
— |
powiat przysuski, |
|
— |
powiat miejski Warszawa, |
w województwie lubelskim:
|
— |
powiat bialski, |
|
— |
powiat miejski Biała Podlaska, |
|
— |
gminy Batorz, Godziszów, Janów Lubelski, Modliborzyce i Potok Wielki w powiecie janowskim, |
|
— |
gminy Janowiec, Kazimierz Dolny, Końskowola, Kurów, Markuszów, Nałęczów, Puławy z miastem Puławy, Wąwolnica i Żyrzyn w powiecie puławskim, |
|
— |
gminy Nowodwór, miasto Dęblin i część gminy Ryki położona na południe od linii wyznaczonej przez linię kolejową powiecie ryckim, |
|
— |
gminy Adamów, Krzywda, Stoczek Łukowski z miastem Stoczek Łukowski, Wola Mysłowska, Trzebieszów, Stanin, Wojcieszków, gmina wiejska Łuków i miasto Łuków w powiecie łukowskim, |
|
— |
powiat lubelski, |
|
— |
powiat miejski Lublin, |
|
— |
gminy Niedźwiada, Ostrówek, Ostrów Lubelski, Serniki, Uścimów i Lubartów z miastem Lubartów w powiecie lubartowskim, |
|
— |
powiat łęczyński, |
|
— |
powiat świdnicki, |
|
— |
gminy Fajsławice, Gorzków, Izbica, Krasnystaw z miastem Krasnystaw, Kraśniczyn, Łopiennik Górny, Siennica Różana i część gminy Żółkiewka położona na północ od linii wyznaczonej przez drogę nr 842 w powiecie krasnostawskim, |
|
— |
gminy Chełm, Ruda – Huta, Sawin, Rejowiec, Rejowiec Fabryczny z miastem Rejowiec Fabryczny, Siedliszcze, Wierzbica, Żmudź, Dorohusk, Dubienka, Kamień, Leśniowice, Wojsławice w powiecie chełmskim, |
|
— |
powiat miejski Chełm, |
|
— |
powiat kraśnicki, |
|
— |
powiat opolski, |
|
— |
powiat parczewski, |
|
— |
powiat włodawski, |
|
— |
powiat radzyński, |
|
— |
powiat miejski Zamość, |
|
— |
gminy Sitno, Skierbieszów, Stary Zamość, Zamość w powiecie zamojskim |
w województwie podkarpackim:
|
— |
powiat stalowowolski, |
|
— |
gminy Oleszyce, Lubaczów z miastem Lubaczów, Wielkie Oczy w powiecie lubaczowskim, |
|
— |
część gminy Kamień położona na zachód od linii wyznaczonej przez drogę nr 19, część gminy Sokołów Małopolski położona na północ od linii wyznaczonej przez drogę nr 875 w powiecie rzeszowskim, |
|
— |
gminy Cmolas i Majdan Królewski w powiecie kolbuszowskim, |
|
— |
gminy Grodzisko Dolne, część gminy wiejskiej Leżajsk położona na południe od miasta Leżajsk oraz na zachód od linii wyznaczonej przez rzekę San, w powiecie leżajskim, |
|
— |
gmina Jarocin, część gminy Harasiuki położona na północ od linii wyznaczona przez drogę nr 1048 R, część gminy Ulanów położona na północ od linii wyznaczonej przez rzekę Tanew, część gminy Nisko położona na zachód od linii wyznaczonej przez drogę nr 19 oraz na północ od linii wyznaczonej przez linię kolejową biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 19, część gminy Jeżowe położona na zachód od linii wyznaczonej przez drogę nr 19 w powiecie niżańskim, |
|
— |
powiat tarnobrzeski, |
|
— |
część gminy wiejskiej Przeworsk położona na zachód od miasta Przeworsk i na zachód od linii wyznaczonej przez autostradę A4 biegnącą od granicy z gminą Tryńcza do granicy miasta Przeworsk, część gminy Zarzecze położona na zachód od linii wyznaczonej przez drogę nr 1594R biegnącą od północnej granicy gminy do miejscowości Zarzecze oraz na południe od linii wyznaczonej przez drogi nr 1617R oraz 1619R biegnącą do południowej granicy gminy oraz na północ od linii wyznaczonej przez rzekę Mleczka w powiecie przeworskim, |
w województwie pomorskim:
|
— |
gminy Dzierzgoń i Stary Dzierzgoń w powiecie sztumskim, |
|
— |
gmina Stare Pole w powiecie malborskim, |
|
— |
gminy Stegny, Sztutowo i część gminy Nowy Dwór Gdański położona na północny - wschód od linii wyznaczonej przez drogę nr 55 biegnącą od |
|
— |
południowej granicy gminy do skrzyżowania z drogą nr 7, następnie przez drogę nr 7 i S7 biegnącą do zachodniej granicy gminy w powiecie nowodworskim, |
w województwie świętokrzyskim:
|
— |
gmina Tarłów i część gminy Ożarów położona na północ od linii wyznaczonej przez drogę nr 74 w powiecie opatowskim, |
|
— |
część gminy Brody położona na zachód od linii kolejowej biegnącej od miejscowości Marcule i od północnej granicy gminy przez miejscowości Klepacze i Karczma Kunowska do południowej granicy gminy oraz na wschód od linii wyznaczonej przez drogę nr 9 i na północny - wschód od linii wyznaczonej przez drogę nr 0618T biegnącą od północnej granicy gminy do skrzyżowania w miejscowości Lipie oraz przez drogę biegnącą od miejscowości Lipie do wschodniej granicy gminy i część gminy Mirzec położona na wschód od linii wyznaczonej przez drogę nr 744 biegnącą od południowej granicy gminy do miejscowości Tychów Stary a następnie przez drogę nr 0566T biegnącą od miejscowości Tychów Stary w kierunku północno – wschodnim do granicy gminy w powiecie starachowickim, |
|
— |
gmina Gowarczów, część gminy Końskie położona na wschód od linii kolejowej, część gminy Stąporków położona na północ od linii kolejowej w powiecie koneckim, |
w województwie lubuskim:
|
— |
powiat wschowski, |
|
— |
gmina Kostrzyn nad Odrą i część gminy Witnica położona na południowy zachód od drogi biegnącej od zachodniej granicy gminy od miejscowości Krześnica, przez miejscowości Kamień Wielki - Mościce - Witnica - Kłopotowo do południowej granicy gminy w powiecie gorzowskim, |
|
— |
gminy Gubin z miastem Gubin, Maszewo i część gminy Bytnica położona na zachód od linii wyznaczonej przez drogę nr 1157F w powiecie krośnieńskim, |
|
— |
powiat słubicki, |
|
— |
gminy Słońsk, Sulęcin i Torzym w powiecie sulęcińskim, |
|
— |
gminy Bledzew i Międzyrzecz w powiecie międzyrzeckim, |
|
— |
gminy Kolsko, część gminy Kożuchów położona na południe od linii wyznaczonej przez drogę 283 biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 290 i na południe od linii wyznaczonej przez drogę nr 290 biegnącej od miasta Mirocin Dolny do zachodniej granicy gminy, część gminy Bytom Odrzański położona na północny zachód od linii wyznaczonej przez drogi nr 293 i 326, część gminy Nowe Miasteczko położona na zachód od linii wyznaczonych przez drogi 293 i 328, część gminy Siedlisko położona na północny zachód od linii wyznaczonej przez drogę biegnącą od rzeki Odry przy południowe granicy gminy do drogi nr 326 łączącej się z drogą nr 325 biegnącą w kierunku miejscowości Różanówka do skrzyżowania z drogą nr 321 biegnącą od tego skrzyżowania w kierunku miejscowości Bielawy, a następnie przedłużoną przez drogę przeciwpożarową biegnącą od drogi nr 321 w miejscowości Bielawy do granicy gminy w powiecie nowosolskim, |
|
— |
gminy Nowogród Bobrzański, Trzebiechów, część gminy Bojadła położona na północ od linii wyznaczonej przez drogę nr 278 biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 282 i na północ od linii |
|
— |
wyznaczonej przez drogę nr 282 biegnącej od miasta Bojadła do zachodniej granicy gminy, część gminy Sulechów położona na wschód od linii wyznaczonej przez drogę nr S3 oraz na południe od linii wyznaczonej przez drogę łączącą miejscowości Kępsko - Buków biegnącą od zachodniej granicy gminy do miejscowości Buków, a następnie na wschód od linii wyznaczonej przez drogę łączącą miejscowości Buków – Miłkowo biegnącą od miejscowości Buków do północnej granicy gminy w powiecie zielonogórskim, |
|
— |
powiat żarski, |
|
— |
gminy Brzeźnica, Iłowa, Małomice, Szprotawa, Wymiarki, Żagań, miasto Żagań, miasto Gozdnica, część gminy Niegosławice położona na zachód od linii wyznaczonej przez drogę nr 328 w powiecie żagańskim, |
|
— |
gmina Łagów, część gminy Lubrza położona na północ od linii wyznaczonej przez autostradę A2 i część gminy Świebodzin położona na północ od linii wyznaczonej przez autostradę A2w powiecie świebodzińskim, |
w województwie dolnośląskim:
|
— |
gmina Pęcław, część gminy Kotla położona na północ od linii wyznaczonej przez rzekę Krzycki Rów, część gminy wiejskiej Głogów położona na wschód od linii wyznaczonej przez drogi nr 12, 319 oraz 329, część miasta Głogów położona na wschód od linii wyznaczonej przez drogę nr 12 w powiecie głogowskim, |
|
— |
gminy Grębocice i Polkowice w powiecie polkowickim, |
|
— |
gmina Rudna w powiecie lubińskim, |
|
— |
część gminy Niechlów położona na południowy – zachód od linii wyznaczonej przez rzekę Barycz, część gminy Jemielno położona na zachód od linii wyznaczonej przez drogę nr 323 w powiecie górowskim, |
w województwie wielkopolskim:
|
— |
gminy Przemęt i Wolsztyn w powiecie wolsztyńskim, |
|
— |
gmina Wielichowo część gminy Kamieniec położona na zachód od linii wyznaczonej przez drogę nr 308 i część gminy Rakoniewice położona na zachód od linii wyznaczonej przez drogę nr 305 w powiecie grodziskim, |
|
— |
gminy Wijewo, Włoszakowice, część gminy Lipno położona na zachód od linii wyznaczonej przez drogę nr S5 i część gminy Święciechowa położona na północ od linii wyznaczonej przez drogę nr 12 oraz na zachód od linii wyznaczonej przez drogę nr S5 w powiecie leszczyńskim, |
|
— |
część gminy Śmigiel położona na zachód od linii wyznaczonej przez drogę nr S5, w powiecie kościańskim, |
|
— |
powiat obornicki, |
|
— |
część gminy Połajewo na położona na południe od drogi łączącej miejscowości Chraplewo, Tarnówko-Boruszyn, Krosin, Jakubowo, Połajewo - ul. Ryczywolska do północno-wschodniej granicy gminy w powiecie czarnkowsko-trzcianeckim, |
|
— |
gmina Suchy Las, część gminy wiejskiej Murowana Goślina położona na północ od linii kolejowej biegnącej od północnej granicy miasta Murowana Goślina do północno-wschodniej granicy gminy oraz część gminy Rokietnica położona na północ i na wschód od linii kolejowej biegnącej od północnej |
|
— |
granicy gminy w miejscowości Krzyszkowo do południowej granicy gminy w miejscowości Kiekrz w powiecie poznańskim, |
|
— |
gmina Pniewy, część gminy Duszniki położona na północny – zachód od linii wyznaczonej przez drogę nr 306 biegnącą od północnej granicy gminy do miejscowości Duszniki, a następnie na północ od linii wyznaczonej przez ul. Niewierską oraz drogę biegnącą przez miejscowość Niewierz do zachodniej granicy gminy, część gminy Ostroróg położona na zachód od linii wyznaczonej przez drogę nr 186 i 184 biegnące od granicy gminy do miejscowości Ostroróg, a następnie od miejscowości Ostroróg przez miejscowości Piaskowo – Rudki do południowej granicy gminy, część gminy Wronki położona na południe od linii wyznaczonej przez drogi nr 182 i 186, część gminy Szamotuły położona na zachód od linii wyznaczonej przez drogę nr 306 oraz na wschód od wschodniej granicy miasta Szamotuły i na północ od linii kolejowej biegnącej od południowej granicy miasta Szamotuły do południowo-wschodniej granicy gminy oraz część gminy Obrzycko położona na wschód od drogi nr 185 łączącej miejscowości Gaj Mały, Słopanowo i Obrzycko do północnej granicy miasta Obrzycko, a następnie na wschód od drogi przebiegającej przez miejscowość Chraplewo w powiecie szamotulskim, |
|
— |
część gminy Sieraków położona na wschód od linii wyznaczonej przez drogę nr 186 biegnącą od południowej granicy gminy do miejscowości Lutomek, a następnie na wschód od linii wyznaczonej przez drogę biegnącą od skrzyżowania z drogą nr 186 w miejscowości Lutomek biegnącą do skrzyżowania z ul. Leśną w miejscowości Lutom i dalej na wschód od ul. Leśnej biegnącej do wschodniej granicy gminy, część gminy Kwilcz położona na wschód od linii wyznaczonej przez drogę nr 186 biegnącą od północnej granicy gminy do skrzyżowania z drogą nr 24, następnie na północ od linii wyznaczonej przez drogę nr 24 biegnącą od skrzyżowania z drogą nr 186 do skrzyżowania z drogą w miejscowości Pólko, i dalej na wschód od linii wyznaczonej przez drogę biegnącą od miejscowości Pólko przez miejscowość Wituchowo do południowej granicy gminy w powiecie międzychodzkim |
w województwie łódzkim:
|
— |
gminy Białaczów, Drzewica, Opoczno i Poświętne w powiecie opoczyńskim, |
|
— |
gminy Biała Rawska, Regnów i Sadkowice w powiecie rawskim, |
|
— |
gmina Kowiesy w powiecie skierniewickim, |
w województwie zachodniopomorskim:
|
— |
gmina Boleszkowice i część gminy Dębno położona na zachód od linii wyznaczonej przez drogę nr 126 biegnącą od zachodniej granicy gminy do skrzyżowania z drogą nr 23 w miejscowości Dębno, następnie na zachód od linii wyznaczonej przez drogę nr 23 do skrzyżowania z ul. Jana Pawła II w miejscowości Cychry, następnie na południe od ul. Jana Pawła II do skrzyżowania z ul. Ogrodową i dalej na południe od linii wyznaczonej przez ul. Ogrodową, której przedłużenie biegnie do wschodniej granicy gminy w powiecie myśliborskim, |
|
— |
gminy Mieszkowice, Moryń, część gminy Cedynia położona na południe od linii wyznaczonej przez drogę nr 124 biegnącą od zachodniej granicy gminy do miasta Cedynia, a następnie na południe od linii wyznaczonej przez drogę nr 125 biegnącą od miasta Cedynia do wschodniej granicy gminy w powiecie gryfińskim. |
8. Slowakei
Die folgenden Sperrzonen II in der Slowakei:
|
— |
the whole district of Gelnica, |
|
— |
the whole district of Spišská Nová Ves, |
|
— |
the whole district of Levoča, |
|
— |
in the whole district of Michalovce, |
|
— |
the whole district of Košice-okolie, |
|
— |
the whole district of Rožnava, |
|
— |
the whole city of Košice, |
|
— |
the whole district of Sobrance, |
|
— |
in the district of Vranov nad Topľou, the whole municipalities of Zámutov, Rudlov, Jusková Voľa, Banské, Cabov, Davidov, Kamenná Poruba, Vechec, Čaklov, Soľ, Komárany, Čičava, Nižný Kručov, Vranov nad Topľou, Sačurov, Sečovská Polianka, Dlhé Klčovo, Nižný Hrušov, Poša, Nižný Hrabovec, Hencovce, Kučín, Majerovce, Sedliská, Kladzany and Tovarnianska Polianka, Herrmanovce nad Topľou, Petrovce, Pavlovce, Hanušovce nad Topľou, Medzianky, Radvanovce, Babie, Vlača, Ďurďoš, Prosačov, Remeniny, |
|
— |
Skrabské, Bystré, Petkovce, Michalok, Vyšný Žipov, Čierne nad Topľou, Zlatník, Hlinné, Jastrabie nad Topľou, Merník, Ondavské Maťašovce, Tovarné, |
|
— |
in the district of Humenné the whole municipalities of Hudcovce, Brekov, Jasenov, Ptičie, Chlmec, Porúbka, |
|
— |
the whole district of Prešov, |
|
— |
in the whole district of Sabinov, |
|
— |
in the district of Svidník, the whole municipalities of Dukovce, Želmanovce, Kuková, Kalnište, Lužany pri Ondave, Lúčka, Giraltovce, Kračúnovce, Železník, Kobylince, Mičakovce, |
|
— |
the whole district of Bardejov, |
|
— |
in the district of Stará Ľubovňa, the whole municipalities of Kyjov, Pusté Pole, Šarišské Jastrabie, Čirč, Ruská Voľa nad Popradom, Obručné, Vislanka, Ďurková, Plaveč, Ľubotín, Orlov, |
|
— |
the whole district of Revúca, |
|
— |
the whole district of Rimavská Sobota, |
|
— |
in the district of Veľký Krtíš, the whole municipalities of Ľuboriečka, Muľa, Dolná Strehová, Závada, Pravica, Chrťany, Senné, Brusník, Horná Strehová, Slovenské Kľačany, Vieska, Veľký Lom, Suché Brezovo, Horné Strháre, Dolné Strháre, Modrý Kameň,Veľký Krtíš, Veľké Zlievce, Malé Zlievce, Veľké Stračiny, Malé Stračiny, Bušince, Čeláre, Gabušovce, Zombor, Olováry, Malý Krtíš, Nová Ves, Šuľa, Červeňany, Sucháň, Dačov Lom, |
|
— |
the whole district of Lučenec, |
|
— |
the whole district of Poltár |
|
— |
in the district of Zvolen, the whole municipalities Lešť, Pliešovce |
|
— |
in the district of Detva, the whole municipalities of Stará Huta, Vígľašská Huta,- Kalinka, Slatinské Lazy, Stožok, Klokoč, Vígľaš, Detva, |
|
— |
in the district of Krupina the whole municipalities of Senohrad, Horné Mladonice, Dolné Mladonice, Čekovce, Lackov. |
TEIL III
1. Bulgarien
Die folgenden Sperrzonen III in Bulgarien:
|
— |
the whole region of Gabrovo, |
|
— |
the whole region of Lovech, |
|
— |
the whole region of Montana, |
|
— |
the Pleven region:
|
|
— |
the Ruse region:
|
|
— |
the Shumen region:
|
|
— |
the Silistra region:
|
|
— |
the Sliven region:
|
|
— |
the Targovishte region:
|
|
— |
the Vidin region,
|
|
— |
the Veliko Tarnovo region:
|
|
— |
the whole region of Vratza, |
|
— |
in Varna region:
|
|
— |
in Burgas region:
|
2. Italien
Die folgenden Sperrzonen III in Italien:
|
— |
tutto il territorio della Sardegna. |
3. Lettland
Die folgenden Sperrzonen III in Lettland:
|
— |
Aizputes novada Kalvenes pagasta daļa uz austrumiem no ceļa pie Vārtājas upes līdz autoceļam A9, uz ziemeļiem no autoceļa A9, uz austrumiem no autoceļa V1200, Kazdangas pagasta daļa uz austrumiem no ceļa V1200, P115, P117, V1296, |
|
— |
Kuldīgas novada, Laidu pagasta daļa uz dienvidiem no autoceļa V1296, |
|
— |
Skrundas novada Rudbāržu, Nīkrāces pagasts, Raņķu pagasta daļa uz dienvidiem no autoceļa V1272 līdz robežai ar Ventas upi, Skrundas pagasts (izņemot pagasta daļa no Skrundas uz ziemeļiem no autoceļa A9 un austrumiem no Ventas upes), Skrundas pilsēta, |
|
— |
Vaiņodes novada Embūtes pagasta daļa uz ziemeļiem autoceļa P116, P106. |
4. Litauen
Die folgenden Sperrzonen III in Litauen:
|
— |
Jurbarko rajono savivaldybė: Seredžiaus ir Juodaičių seniūnijos, |
|
— |
Kauno rajono savivaldybė: Čekiškės seniūnija, Babtų seniūnijos dalis į vakarus nuo kelio A1ir Vilkijos apylinkių seniūnijos dalis į rytus nuo kelio Nr. 1907, |
|
— |
Kėdainių rajono savivaldybė: Pernaravos seniūnija ir Josvainių seniūnijos pietvakarinė dalis tarp kelio Nr. 229 ir Nr. 2032, |
|
— |
Plungės rajono savivaldybė: Alsėdžių, Babrungo, Paukštakių, Platelių ir Žemaičių Kalvarijos seniūnijos, |
|
— |
Raseinių rajono savivaldybė: Ariogalos ir Ariogalos miesto seniūnijos, |
|
— |
Skuodo rajono savivaldybės: Barstyčių, Notėnų ir Šačių seniūnijos. |
5. Polen
Die folgenden Sperrzonen III in Polen:
w województwie warmińsko-mazurskim:
|
— |
gminy Kiwity i Lidzbark Warmiński z miastem Lidzbark Warmiński w powiecie lidzbarskim, |
|
— |
gminy Barczewo, Gietrzwałd, Jonkowo, Dywity, Dobre Miasto, Purda, Stawiguda, Świątki, część gminy Olsztynek położona na północ od linii wyznaczonej przez drogę nr S51 biegnącą od wschodniej granicy gminy do miejscowości Ameryka oraz na wschód od linii wyznaczonej przez drogę biegnącą od skrzyżowania z drogą S51 do północnej granicy gminy, łączącej miejscowości Mańki – Mycyny – Ameryka w powiecie olsztyńskim, |
|
— |
powiat miejski Olsztyn, |
w województwie mazowieckim:
|
— |
gminy Łaskarzew z miastem Łaskarzew, Maciejowice, Sobolew, Trojanów, Żelechów, część gminy Wilga położona na południe od linii wyznaczonej przez rzekę Wilga biegnącą od wschodniej granicy gminy do ujścia do rzeki Wisły, część gminy Górzno położona na południe od linii wyznaczonej przez drogę łączącą miejscowości Łąki i Górzno biegnącą od wschodniej granicy gminy, następnie od miejscowości Górzno na południe od drogi nr 1328W biegnącej do drogi nr 17, a następnie na południe od linii wyznaczonej przez drogę biegnącą od drogi nr 17 do zachodniej granicy gminy przez miejscowości Józefów i Kobyla Wola w powiecie garwolińskim, |
|
— |
część gminy Iłża położona na wschód od linii wyznaczonej przez drogę nr 9 w powiecie radomskim, |
|
— |
gmina Kazanów w powiecie zwoleńskim, |
|
— |
gminy Ciepielów, Lipsko, Rzeczniów i Sienno w powiecie lipskim, |
w województwie lubelskim:
|
— |
powiat tomaszowski, |
|
— |
gmina Białopole w powiecie chełmskim, |
|
— |
gmina Rudnik i część gminy Żółkiewka położona na południe od linii wyznaczonej przez drogę nr 842 w powiecie krasnostawskim, |
|
— |
gminy Adamów, Grabowiec, Komarów – Osada, Krasnobród, Łabunie, Miączyn, Nielisz, Radecznica, Sułów, Szczebrzeszyn, Zwierzyniec w powiecie zamojskim, |
|
— |
powiat biłgorajski, |
|
— |
powiat hrubieszowski, |
|
— |
gminy Dzwola i Chrzanów w powiecie janowskim, |
|
— |
gmina Serokomla w powiecie łukowskim, |
|
— |
gminy Abramów, Kamionka, Michów, Firlej, Jeziorzany, Kock w powiecie lubartowskim, |
|
— |
gminy Kłoczew, Stężyca, Ułęż i część gminy Ryki położona na północ od linii wyznaczonej przez linię kolejową w powiecie ryckim, |
|
— |
gmina Baranów w powiecie puławskim, |
w województwie podkarpackim:
|
— |
gminy Cieszanów, Horyniec – Zdrój, Narol i Stary Dzików w powiecie lubaczowskim, |
|
— |
gminy Kuryłówka, Nowa Sarzyna, miasto Leżajsk, część gminy wiejskiej Leżajsk położona na północ od miasta Leżajsk oraz część gminy wiejskiej Leżajsk położona na wschód od linii wyznaczonej przez rzekę San, w powiecie leżajskim, |
|
— |
gminy Krzeszów, Rudnik nad Sanem, część gminy Harasiuki położona na południe od linii wyznaczona przez drogę nr 1048 R, część gminy Ulanów położona na południe od linii wyznaczonej przez rzekę Tanew, część gminy Nisko położona na wschód od linii wyznaczonej przez drogę nr 19 oraz na południe od linii wyznaczonej przez linię kolejową biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 19, część gminy Jeżowe położona na wschód od linii wyznaczonej przez drogę nr 19 w powiecie niżańskim, |
|
— |
gminy Chłopice, Jarosław z miastem Jarosław, Laszki, Wiązownica, Pawłosiów, Radymno z miastem Radymno, w powiecie jarosławskim, |
|
— |
gmina Stubno w powiecie przemyskim, |
|
— |
część gminy Kamień położona na wschód od linii wyznaczonej przez drogę nr 19 w powiecie rzeszowskim, |
|
— |
gminy Adamówka, Sieniawa, Tryńcza, miasto Przeworsk, część gminy wiejskiej Przeworsk położona na wschód od miasta Przeworsk i na wschód od linii wyznaczonej przez autostradę A4 biegnącą od granicy z gminą Tryńcza do granicy miasta Przeworsk, część gminy Zarzecze położona na wschód od linii wyznaczonej przez drogę nr 1594R biegnącą od północnej granicy gminy do miejscowości Zarzecze oraz na północ od linii wyznaczonej przez drogi nr 1617R oraz 1619R biegnącą do południowej granicy gminy w powiecie przeworskim, |
w województwie lubuskim:
|
— |
gminy Nowa Sól i miasto Nowa Sól, Otyń oraz część gminy Kożuchów położona na północ od linii wyznaczonej przez drogę nr 283 biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 290 i na północ od linii wyznaczonej przez drogę nr 290 biegnącej od miasta Mirocin Dolny do zachodniej granicy gminy, część gminy Bytom Odrzański położona na południowy wschód od linii wyznaczonej przez drogi nr 293 i 326, część gminy Nowe Miasteczko położona na wschód od linii wyznaczonych przez drogi 293 i 328, część gminy Siedlisko położona na południowy wschód od linii wyznaczonej przez drogę biegnącą od rzeki Odry przy południowe granicy gminy do drogi nr 326 łączącej się z drogą nr 325 biegnącą w kierunku miejscowości Różanówka do skrzyżowania z drogą nr 321 biegnącą od tego skrzyżowania w kierunku miejscowości Bielawy, a następnie przedłużoną przez drogę przeciwpożarową biegnącą od drogi nr 321 w miejscowości Bielawy do granicy gminy w powiecie nowosolskim, |
|
— |
gminy Babimost, Czerwieńsk, Kargowa, Świdnica, Zabór, część gminy Bojadła położona na południe od linii wyznaczonej przez drogę nr 278 biegnącą od wschodniej granicy gminy do skrzyżowania z drogą nr 282 i na południe od linii wyznaczonej przez drogę nr 282 biegnącej od miasta Bojadła do zachodniej granicy gminy i część gminy Sulechów położona na zachód od linii wyznaczonej przez drogę nr S3 oraz na północ od linii wyznaczonej przez |
|
— |
drogę łączącą miejscowości Kępsko - Buków biegnącą od zachodniej granicy gminy do miejscowości Buków, a następnie na zachód od linii wyznaczonej przez drogę łączącą miejscowości Buków – Miłkowo biegnącą od miejscowości Buków do północnej granicy gminy w powiecie zielonogórskim, |
|
— |
część gminy Niegosławice położona na wschód od linii wyznaczonej przez drogę nr 328 w powiecie żagańskim, |
|
— |
powiat miejski Zielona Góra, |
|
— |
gminy Skąpe, Szczaniec, Zbąszynek , część gminy Lubrza położona na południe od linii wyznaczonej przez autostradę A2 i część gminy Świebodzin położona na południe od linii wyznaczonej przez autostradę A2 w powiecie świebodzińskim, |
|
— |
gminy Bobrowice, Dąbie, Krosno Odrzańskie i część gminy Bytnica położona na wschód od linii wyznaczonej przez drogę nr 1157F w powiecie krośnieńskim, |
|
— |
część gminy Trzciel położona na południe od linii wyznaczonej przez drogę nr 92 w powiecie międzyrzeckim, |
w województwie wielkopolskim:
|
— |
gmina Zbąszyń, część gminy Miedzichowo położona na południe od linii wyznaczonej przez drogę nr 92, część gminy Nowy Tomyśl położona na zachód od linii wyznaczonej przez drogę nr 305 w powiecie nowotomyskim, |
|
— |
gmina Siedlec w powiecie wolsztyńskim, |
|
— |
część gminy Rakoniewice położona na wschód od linii wyznaczonej przez drogę nr 305 w powiecie grodziskim, |
w województwie dolnośląskim:
|
— |
gminy Jerzmanowa, Żukowice, część gminy Kotla położona na południe od linii wyznaczonej przez rzekę Krzycki Rów, część gminy wiejskiej Głogów położona na zachód od linii wyznaczonej przez drogi nr 12, 319 oraz 329, część miasta Głogów położona na zachód od linii wyznaczonej przez drogę nr 12 w powiecie głogowskim, |
|
— |
gminy Gaworzyce, Radwanice i część gminy Przemków położona na północ od linii wyznaczonej prze drogę nr 12 w powiecie polkowickim, |
w województwie świętokrzyskim:
|
— |
część gminy Brody położona na wschód od linii kolejowej biegnącej od miejscowości Marcule i od północnej granicy gminy przez miejscowości Klepacze i Karczma Kunowska do południowej granicy gminy w powiecie starachowickim. |
6. Rumänien
Die folgenden Sperrzonen III in Rumänien:
|
— |
Zona orașului București, |
|
— |
Județul Constanța, |
|
— |
Județul Satu Mare, |
|
— |
Județul Tulcea, |
|
— |
Județul Bacău, |
|
— |
Județul Bihor, |
|
— |
Județul Bistrița Năsăud, |
|
— |
Județul Brăila, |
|
— |
Județul Buzău, |
|
— |
Județul Călărași, |
|
— |
Județul Dâmbovița, |
|
— |
Județul Galați, |
|
— |
Județul Giurgiu, |
|
— |
Județul Ialomița, |
|
— |
Județul Ilfov, |
|
— |
Județul Prahova, |
|
— |
Județul Sălaj, |
|
— |
Județul Suceava |
|
— |
Județul Vaslui, |
|
— |
Județul Vrancea, |
|
— |
Județul Teleorman, |
|
— |
Judeţul Mehedinţi, |
|
— |
Județul Gorj, |
|
— |
Județul Argeș, |
|
— |
Judeţul Olt, |
|
— |
Judeţul Dolj, |
|
— |
Județul Arad, |
|
— |
Județul Timiș, |
|
— |
Județul Covasna, |
|
— |
Județul Brașov, |
|
— |
Județul Botoșani, |
|
— |
Județul Vâlcea, |
|
— |
Județul Iași, |
|
— |
Județul Hunedoara, |
|
— |
Județul Alba, |
|
— |
Județul Sibiu, |
|
— |
Județul Caraș-Severin, |
|
— |
Județul Neamț, |
|
— |
Județul Harghita, |
|
— |
Județul Mureș, |
|
— |
Județul Cluj, |
|
— |
Județul Maramureş. |
7. Slowakei
Die folgenden Sperrzonen III in der Slowakei:
|
— |
the whole district of Trebišov. |
BESCHLÜSSE
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/147 |
BESCHLUSS (EU) 2021/812 DES RATES
vom 10. Mai 2021
über den Standpunkt, der im Namen der Europäischen Union im Assoziationsausschuss in der Zusammensetzung „Handel“ sowie im Assoziationsrat, eingerichtet durch das Assoziierungsabkommen zwischen der Europäischen Union und der Europäischen Atomgemeinschaft und ihren Mitgliedstaaten einerseits und Georgien andererseits, in Bezug auf eine befürwortende Stellungnahme zu dem von der Regierung Georgiens gebilligten umfassenden Fahrplan für die Umsetzung der Rechtsvorschriften für das öffentliche Beschaffungswesen und in Bezug auf die Anerkennung des Abschlusses der in Anhang XVI-B dieses Assoziierungsabkommens genannten Phase 1 zu vertreten ist
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union, insbesondere auf Artikel 207 Absatz 4 Unterabsatz 1 in Verbindung mit Artikel 218 Absatz 9,
auf Vorschlag der Europäischen Kommission,
in Erwägung nachstehender Gründe:
|
(1) |
Das Assoziierungsabkommen zwischen der Europäischen Union und der Europäischen Atomgemeinschaft und ihren Mitgliedstaaten einerseits und Georgien andererseits (1) (im Folgenden „Abkommen“) wurde von der Union mit dem Beschluss (EU) 2016/838 des Rates (2) geschlossen und ist am 1. Juli 2016 in Kraft getreten. |
|
(2) |
Artikel 145 Absatz 1 des Abkommens bestimmt, dass Georgien dem Assoziationsausschuss in der Zusammensetzung „Handel“ einen umfassenden Fahrplan für die Umsetzung der Rechtsvorschriften für das öffentliche Beschaffungswesen in Georgien mit zeitlichen Vorgaben und Etappenzielen übermittelt, der sämtliche Reformen im Zusammenhang mit der Annäherung der Rechtsvorschriften an den Besitzstand der Union beinhaltet. |
|
(3) |
Nach Artikel 145 Absatz 2 des Abkommens ist eine befürwortende Stellungnahme des Assoziationsausschusses in der Zusammensetzung „Handel“ erforderlich, damit der umfassende Fahrplan als das Referenzdokument für die Umsetzung, d. h. für die Annäherung der Rechtsvorschriften von Georgien im Bereich des öffentlichen Beschaffungswesens an den Besitzstand der Union, dienen kann. |
|
(4) |
Nach Artikel 146 Absatz 2 des Abkommens erfolgt die Annäherung an den Besitzstand der Union in mehreren Phasen entsprechend dem Zeitplan in Anhang XVI-B des Abkommens. Die Umsetzung jeder Phase wird vom Assoziationsausschuss in der in Artikel 408 Absatz 4 genannten Zusammensetzung „Handel“ bewertet und nach dessen positiver Einschätzung mit der gegenseitigen Gewährung des Marktzugangs verbunden, wie in Anhang XVI-B festgelegt. |
|
(5) |
Der Assoziationsausschuss in der Zusammensetzung „Handel“ nimmt einen Beschluss nach Artikel 11 Absatz 2 seiner in Anhang II des Beschlusses Nr. 1/2014 des Assoziationsrates EU-Georgien (3) festgelegten Geschäftsordnung an, in dem er eine Stellungnahme zu dem von der Regierung Georgiens gebilligten Fahrplan sowie eine Einschätzung der bisherigen Annäherung des Rechts Georgiens an den Besitzstand der Union bei Abschluss der in Anhang XVI-B des Abkommens genannten Phase 1 abgibt. Die Billigung des Fahrplans erfolgte durch den von der georgischen Regierung am 31. März 2016 verabschiedeten Regierungserlass Nr. 536 „betreffend die geplanten Änderungen im Bereich des öffentlichen Beschaffungswesens, die im Einklang mit den Verpflichtungen zwischen Georgien und der EU im Rahmen des vertieften und umfassenden Freihandelsabkommens vorgesehen sind“, geändert durch die am 22. Januar 2018 bzw. am 12. Juni 2020 verabschiedeten Regierungserlasse Nr. 154 und Nr. 974. |
|
(6) |
Nach Anerkennung des Abschlusses der Phase 1 gemäß Anhang XVI-B des Abkommens fasst der Assoziationsrat nach Artikel 11 Absatz 2 seiner Geschäftsordnung gemäß Anhang I des Beschlusses Nr. 1/2014 des Assoziationsrates EU-Georgien einen Beschluss über die gegenseitige Gewährung des Marktzugangs für Beschaffungen für zentrale Regierungsbehörden gemäß Anhang XVI-B des Abkommens. |
|
(7) |
Es ist angebracht, den im Namen der Union im Assoziationsausschuss in der Zusammensetzung „Handel“ und im Assoziationsrat zu vertretenden Standpunkt festzulegen, da die vorgesehenen Beschlüsse für die Union verbindlich sein werden — |
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Der Standpunkt, der im Namen der Union im Assoziationsausschuss in der Zusammensetzung „Handel“ in Bezug auf den von der Regierung Georgiens gebilligten umfassenden Fahrplan und den Abschluss der Phase 1 gemäß Anhang XVI-B des Abkommens zu vertreten ist, beruht auf dem Entwurf eines Beschlusses des Assoziationsausschusses in der Zusammensetzung „Handel“ (4).
Artikel 2
Der Standpunkt, der im Namen der Union im Assoziationsrat in Bezug auf die gegenseitige Gewährung des Marktzugangs gemäß Anhang XVI-B des Abkommens zu vertreten ist, beruht auf dem Entwurf eines Beschlusses des Assoziationsrates (4).
Artikel 3
Dieser Beschluss tritt am Tag seiner Annahme in Kraft.
Geschehen zu Brüssel am 10. Mai 2021.
Im Namen des Rates
Der Präsident
J. BORRELL FONTELLES
(1) ABl. L 261 vom 30.8.2014, S. 4.
(2) Beschluss (EU) 2016/838 des Rates vom 23. Mai 2016 über den Abschluss des Assoziierungsabkommens zwischen der Europäischen Union und der Europäischen Atomgemeinschaft und ihren Mitgliedstaaten einerseits und Georgien andererseits im Namen der Europäischen Union (ABl. L 141 vom 28.5.2016, S. 26).
(3) Beschluss Nr. 1/2014 des Assoziationsrates EU-Georgien vom 17. November 2014 zur Annahme seiner Geschäftsordnung sowie der Geschäftsordnung des Assoziationsausschusses und der Unterausschüsse [2015/2261] (ABl. L 321 vom 5.12.2015, S. 60).
(4) Siehe Dokument ST 7791/21 unter http://register.consilium.europa.euhttp://register.consilium.europa.eu
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/149 |
BESCHLUSS (GASP) 2021/813 DES RATES
vom 20. Mai 2021
zur Änderung des Beschlusses 2014/486/GASP über die Beratende Mission der Europäischen Union für eine Reform des zivilen Sicherheitssektors in der Ukraine (EUAM Ukraine)
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Europäische Union, insbesondere auf die Artikel 42 Absatz 4 und 43 Absatz 2,
auf Vorschlag des Hohen Vertreters der Union für Außen- und Sicherheitspolitik,
in Erwägung nachstehender Gründe:
|
(1) |
Am 22. Juli 2014 hat der Rat den Beschluss 2014/486/GASP über die Beratende Mission der Europäischen Union für eine Reform des zivilen Sicherheitssektors in der Ukraine (EUAM Ukraine) (1) angenommen. |
|
(2) |
Am 13. Mai 2019 hat der Rat den Beschluss (GASP) 2019/761 (2) angenommen, mit dem das Mandat der EUAM Ukraine bis zum 31. Mai 2021 verlängert wurde. |
|
(3) |
Im Rahmen einer strategischen Überprüfung der EUAM Ukraine ist das Politische und Sicherheitspolitische Komitee übereingekommen, dass die EUAM Ukraine bis zum 31. Mai 2024 verlängert werden und nach zwei Jahren eine strategische Bewertung mit Schwerpunkt auf der Entwicklung der politischen Dimension durchgeführt werden sollte. |
|
(4) |
Der Beschluss 2014/486/GASP sollte daher bis zum 31. Mai 2024 verlängert werden. |
|
(5) |
Die EUAM Ukraine wird in einer Lage durchgeführt, die sich verschlechtern kann und die Erreichung der Ziele des auswärtigen Handelns der Union nach Artikel 21 des Vertrags behindern könnte — |
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Der Beschluss 2014/486/GASP wird wie folgt geändert:
|
1. |
In Artikel 14 Absatz 1 wird folgender Unterabsatz angefügt: „Der als finanzieller Bezugsrahmen dienende Betrag zur Deckung der Kosten der EUAM Ukraine für den Zeitraum vom 1. Juni 2021 bis zum 31. Mai 2024 beläuft sich auf 88 500 000 EUR.“ |
|
2. |
Artikel 18 erhält folgende Fassung: „Artikel 18 Strategische Überprüfung Nach dem 31. Mai 2023 wird eine strategische Bewertung der EUAM Ukraine durchgeführt, die sich in erster Linie mit der Entwicklung der politischen Dimension befasst.“ |
|
3. |
Artikel 19 Absatz 2 erhält folgende Fassung: „Er gilt bis zum 31. Mai 2024.“ |
Artikel 2
Dieser Beschluss tritt am Tag seiner Annahme in Kraft.
Geschehen zu Brüssel am 20. Mai 2021.
Im Namen des Rates
Der Präsident
A. SANTOS SILVA
(1) ABl. L 217 vom 23.7.2014, S. 42.
(2) Beschluss (GASP) 2019/761 des Rates vom 13. Mai 2019 zur Änderung des Beschlusses 2014/486/GASP über die Beratende Mission der Europäischen Union für eine Reform des zivilen Sicherheitssektors in der Ukraine (EUAM Ukraine) (ABl. L 125 vom 14.5.2019, S. 16).
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/151 |
BESCHLUSS (GASP) 2021/814 DES RATES
vom 20. Mai 2021
zur Änderung des Beschlusses (GASP) 2017/915 über Outreach-Maßnahmen der Union zur Unterstützung der Durchführung des Vertrags über den Waffenhandel
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Europäische Union, insbesondere auf Artikel 28 Absatz 1 und Artikel 31 Absatz 1,
auf Vorschlag des Hohen Vertreters der Union für Außen- und Sicherheitspolitik,
in Erwägung nachstehender Gründe:
|
(1) |
Am 29. Mai 2017 hat der Rat den Beschluss (GASP) 2017/915 (1) erlassen. |
|
(2) |
Am 30. Juli 2020 hat der Rat den Beschluss (GASP) 2020/1134 (2) zur Änderung des Beschlusses (GASP) 2017/915 und zur Verlängerung des Durchführungszeitraums der in Artikel 1 des Beschlusses (GASP) 2017/915 genannten Maßnahmen bis zum 30. Juni 2021 erlassen. |
|
(3) |
Am 31. März 2021 hat das Bundesamt für Wirtschaft und Ausfuhrkontrolle und am 6. April 2021 hat Expertise France in ihrer jeweiligen Eigenschaft als Durchführungsstelle die Ermächtigung durch die Union beantragt, aufgrund der anhaltenden Herausforderungen infolge der COVID-19-Pandemie die Durchführung des Beschlusses (GASP) 2017/915 zum zweiten Mal, bis zum 31. Januar 2022, zu verlängern. |
|
(4) |
Die Fortsetzung der in Artikel 1 des Beschlusses (GASP) 2017/915 genannten Maßnahmen bis zum 31. Januar 2022 erfordert keine zusätzlichen Finanzmittel — |
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Artikel 5 des Beschlusses (GASP) 2017/915 erhält folgende Fassung:
„Artikel 5
Dieser Beschluss tritt am Tag seiner Annahme in Kraft.
Seine Geltungsdauer endet am 31. Januar 2022.“
Artikel 2
Dieser Beschluss tritt am Tag seiner Annahme in Kraft.
Geschehen zu Brüssel am 20. Mai 2021.
Im Namen des Rates
Der Präsident
A. SANTOS SILVA
(1) Beschluss (GASP) 2017/915 des Rates vom 29. Mai 2017 über Outreach-Maßnahmen der Union zur Unterstützung der Durchführung des Vertrags über den Waffenhandel (ABl. L 139 vom 30.5.2017, S. 38).
(2) Beschluss (GASP) 2020/1134 des Rates vom 30. Juli 2020 zur Änderung des Beschlusses (GASP) 2017/915 des Rates über Outreach-Maßnahmen der Union zur Unterstützung der Durchführung des Vertrags über den Waffenhandel (ABl. L 247 vom 31.7.2020, S. 24).
|
21.5.2021 |
DE |
Amtsblatt der Europäischen Union |
L 180/152 |
DURCHFÜHRUNGSBESCHLUSS (GASP) 2021/815 DES RATES
vom 20. Mai 2021
zur Durchführung des Beschlusses 2014/450/GASP über restriktive Maßnahmen angesichts der Lage in Sudan
DER RAT DER EUROPÄISCHEN UNION —
gestützt auf den Vertrag über die Europäische Union, insbesondere auf Artikel 31 Absatz 2,
gestützt auf den Beschluss 2014/450/GASP des Rates vom 10. Juli 2014 über restriktive Maßnahmen angesichts der Lage in Sudan und zur Aufhebung des Beschlusses 2011/423/GASP (1), insbesondere auf Artikel 6,
auf Vorschlag des Hohen Vertreters der Union für Außen- und Sicherheitspolitik,
in Erwägung nachstehender Gründe:
|
(1) |
Am 10. Juli 2014 hat der Rat den Beschluss 2014/450/GASP angenommen. |
|
(2) |
Am 5. März 2021 hat der Ausschuss des Sicherheitsrats der Vereinten Nationen (VN), der gemäß der Resolution 1591 (2005) des Sicherheitsrats der VN eingesetzt wurde, die Streichung einer Person von der Liste der Personen und Einrichtungen, die restriktiven Maßnahmen unterliegen, genehmigt. |
|
(3) |
Der Anhang des Beschlusses 2014/450/GASP sollte daher entsprechend geändert werden — |
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Der Anhang des Beschlusses 2014/450/GASP wird gemäß dem Anhang dieses Beschlusses geändert.
Artikel 2
Dieser Beschluss tritt am Tag seiner Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Geschehen zu Brüssel am 20. Mai 2021.
Im Namen des Rates
Der Präsident
A. SANTOS SILVA
ANHANG
In der Liste in Anhang des Beschlusses 2014/450/GASP wird der Eintrag zu folgender Person gestrichen:
|
3. |
SHAREIF, Adam. |