|
(8)
|
In Teil B werden die folgenden Kapitel angefügt:
„B.63 SCREENING-PRÜFUNG ZUR BEWERTUNG DER REPRODUKTIONS-/ENTWICKLUNGSTOXIZITÄT
EINLEITUNG
|
1.
|
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 421 (2016). Die OECD-Richtlinien für die Prüfung von Chemikalien werden regelmäßig unter Berücksichtigung des wissenschaftlichen Fortschritts überarbeitet. Die ursprüngliche Screening-Prüfrichtlinie 421 wurde im Jahr 1995 angenommen. Sie beruhte auf einem Protokoll für einen Vorversuch zur Bewertung der Reproduktionstoxizität, der in zwei Expertensitzungen im Jahr 1990 in London (1) und im Jahr 1992 in Tokio (2) erörtert wurde.
|
|
2.
|
Diese Prüfmethode wurde als Folgemaßnahme der 1998 bei der OECD eingeleiteten prioritären Maßnahme zur Änderung bestehender Prüfrichtlinien und zur Entwicklung neuer Prüfrichtlinien für das Screening und die Prüfung potenzieller endokriner Disruptoren unter Festlegung von für einen endokrinen Disruptor relevanten Endpunkten aktualisiert (3). OECD TG 407 (28-Tage-Toxizitätsstudie mit wiederholter oraler Verabreichung an Nagern, Kapitel B.7 dieses Anhangs) beispielsweise wurde 2008 um geeignete Parameter zum Nachweis einer endokrinen Wirkung von Prüfchemikalien aktualisiert. Ziel der Aktualisierung von TG 421 war die Einbeziehung einiger für endokrine Disruptoren relevanter Endpunkte in Screening-TGs, bei denen die Expositionszeiträume einige der empfindlichsten Entwicklungszeiträume (Zeiträume vor oder kurz nach der Geburt) beinhalten.
|
|
3.
|
Die ausgewählten für endokrine Disruptoren relevanten zusätzlichen Endpunkte, die Bestandteil auch von TG 443 (Erweiterte Eingenerationen-Prüfung auf Reproduktionstoxizität, Kapitel B.56 dieses Anhangs) sind, wurden auf der Grundlage einer Machbarkeitsstudie zur Untersuchung wissenschaftlicher und technischer Fragen im Zusammenhang mit deren Berücksichtigung sowie mit möglichen Anpassungen des für deren Einbeziehung erforderlichen Prüfprotokolls in TG 421 aufgenommen (4).
|
|
4.
|
Mit dieser Prüfmethode sollen begrenzte Informationen über die Wirkungen einer Prüfchemikalie auf die Reproduktionsleistung männlicher und weiblicher Tiere (Funktion der Keimdrüsen, Paarungsverhalten, Empfängnis, Entwicklung des Conceptus, Geburt usw.) ermittelt werden. Sie ist nicht als alternative Prüfmethode und auch nicht als Ersatz für die bereits existierenden Prüfmethoden B.31, B.34, B.35 und B.56 zu betrachten.
|
AUSGANGSÜBERLEGUNGEN
|
5.
|
Diese Screening-Prüfmethode kann verwendet werden, um erste Informationen über mögliche reproduktions- und/oder entwicklungsrelevante Auswirkungen entweder bereits früh bei der Bewertung toxikologischer Eigenschaften von Chemikalien oder bei Besorgnis erregenden Chemikalien zu erhalten. Außerdem kann sie als Teil einer Reihe von Vorversuchen für existierende Chemikalien verwendet werden, für die nur wenig oder keinerlei toxikologische Informationen verfügbar sind, oder beispielsweise als Dosisfindungsstudie im Zusammenhang mit umfassenderen Reproduktions-/Entwicklungsstudien dienen. Bei der Prüfung sollten die im ‚OECD Guidance Document no 19 on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluation‘ (5) genannten Grundsätze und Erwägungen beachtet werden.
|
|
6.
|
Diese Prüfmethode liefert keine umfassenden Informationen über sämtliche Reproduktions- und Entwicklungsaspekte. Insbesondere bietet sie nur beschränkte Möglichkeiten zur Erkennung postnataler Ausprägungen einer pränatalen Exposition oder zur Feststellung von Wirkungen, die bei postnataler Exposition induziert werden können. Aufgrund (unter anderem) der verhältnismäßig geringen Anzahl an Versuchstieren in den Dosisgruppen sowie der Selektivität der Endpunkte und der kurzen Dauer der Studie werden mit dieser Methode keine Nachweise dafür erlangt, dass keine Wirkungen eintreten. Wenn keine Daten aus anderen Prüfungen zur Bewertung der Reproduktions-/Entwicklungstoxizität vorliegen, sind positive Ergebnisse zudem hilfreich für eine erste Gefährdungsabschätzung und können in Entscheidungen über die Notwendigkeit und die zeitliche Gestaltung weiterer Prüfungen beitragen.
|
|
7.
|
Die Ergebnisse in Bezug auf die endokrinen Parameter sind im Kontext des ‚OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals‘ (6) auszuwerten. Dieser Conceptual Framework enthält die verbesserte OECD TG 421 in Niveau 4 als In-vivo-Prüfung zur Erlangung von Daten über negative Wirkungen auf endokrin relevante Endpunkte. Ein endokrin relevantes Signal kann jedoch nicht an sich als hinreichender Beleg dafür betrachtet werden, dass die betreffende Prüfchemikalie als endokriner Disruptor wirkt.
|
|
8.
|
Bei dieser Prüfmethode wird angenommen, dass die Prüfchemikalie oral verabreicht wird. Bei anderen Expositionswegen können Änderungen erforderlich sein.
|
|
9.
|
Bevor die Prüfmethode an einem Gemisch für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs rechtlich vorgeschrieben ist.
|
|
10.
|
Die Definitionen der verwendeten Begriffe sind Anlage 1 zu entnehmen.
|
PRINZIP DER PRÜFMETHODE
|
11.
|
Die Prüfchemikalie wird verschiedenen Gruppen von männlichen und weiblichen Tieren in abgestuften Dosen verabreicht. Männlichen Tieren sollte die Prüfchemikalie mindestens vier Wochen bis zum Tag vor der vorgesehenen Tötung (einschließlich) verabreicht werden. (Dieser Zeitraum umfasst mindestens zwei Wochen vor der Paarung, die Paarungszeit und etwa zwei Wochen nach der Paarung). Wegen des begrenzten Verabreichungszeitraums vor der Paarung ist die Fruchtbarkeit bei männlichen Tieren möglicherweise kein besonders empfindlicher Indikator für eine testikuläre Toxizität. Daher ist eine eingehende histologische Untersuchung der Hoden von wesentlicher Bedeutung. Die Kombination eines Verabreichungszeitraums von zwei Wochen mit anschließenden Beobachtungen des Paarungsverhaltens und der Fertilität mit einem Verabreichungszeitraum von insgesamt mindestens vier Wochen und einer anschließenden detaillierten Histopathologie der männlichen Keimdrüsen wird als ausreichend für den Nachweis der meisten Wirkungen auf die männliche Fruchtbarkeit und die Spermatogenese betrachtet.
|
|
12.
|
Weiblichen Tieren sollte die Prüfchemikalie während der gesamten Dauer der Studie verabreicht werden. Dazu zählen zwei Wochen vor der Paarung (damit mindestens zwei vollständige Östruszyklen erfasst werden), die variable Zeit bis zur Empfängnis und die Dauer der Gravidität sowie mindestens 13 Tage nach der Geburt bis zum Tag vor der vorgesehenen Tötung (einschließlich).
|
|
13.
|
Die Dauer der Studie nach der Eingewöhnung und einer Bewertung des Östruszyklus vor der Verabreichung der Prüfchemikalie hängt von der Leistungsfähigkeit des weiblichen Tiers ab; im Allgemeinen beträgt sie etwa 63 Tage [mindestens 14 Tage vor der Paarung, (bis zu) 14 Tage Paarungszeit, 22 Tage Gravidität, 13 Tage Laktation].
|
|
14.
|
Während des Verabreichungszeitraums werden die Tiere täglich sorgfältig auf Toxizitätszeichen beobachtet. Tiere, die während der Dauer der Prüfung sterben, und vorzeitig getötete Tiere werden seziert; die nach Abschluss der Prüfung überlebenden Tiere werden getötet und ebenfalls seziert.
|
BESCHREIBUNG DER METHODE
Auswahl von Versuchstierarten
|
15.
|
Diese Prüfmethode ist für die Verwendung von Ratten ausgelegt. Wenn die in dieser Prüfmethode genannten Parameter an einer anderen Nagetierart untersucht werden, ist dies ausführlich zu begründen. Im internationalen Validierungsprogramm für den Nachweis von endokrinen Disruptoren in OECD TG 407 (entsprechend Kapitel B.7 in diesem Anhang) wurden ausschließlich Ratten als Versuchstiere verwendet. Stämme mit geringer Fruchtbarkeit oder bekannter hoher Häufigkeit von Entwicklungsdefekten sind nicht zu verwenden. Es sind gesunde jungfräuliche Tiere zu verwenden, die zuvor nicht in anderen Versuchen eingesetzt wurden. Die Versuchstiere sind nach Tierart, Tierstamm, Geschlecht, Gewicht und Alter auszuwählen. Bei Beginn der Studie sollten die Gewichtsunterschiede der Tiere möglichst gering sein und 20 % des geschlechtsspezifischen Durchschnittsgewichts nicht überschreiten. Wenn die Studie als Vorstudie zu einer Langzeitstudie oder einer Eingenerationsstudie durchgeführt wird, sollten in beiden Studien vorzugsweise Tiere desselben Stamms und derselben Herkunft verwendet werden.
|
Haltung und Fütterung
|
16.
|
Bei allen Verfahren sind die örtlichen Standards der Versuchstierpflege einzuhalten. Die Temperatur im Tierversuchsraum muss 22 °C (± 3 °) betragen. Die relative Luftfeuchtigkeit sollte mindestens 30 % betragen und – außer beim Reinigen des Raums - 70 % nicht überschreiten; anzustreben ist ein Wert von 50-60 %. Die Beleuchtung sollte künstlich sein, und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, wobei eine unbegrenzte Trinkwasserversorgung zu gewährleisten ist. Die Auswahl des Futters wird eventuell dadurch beeinflusst, dass eine geeignete Beimischung der Prüfchemikalie sichergestellt werden muss, wenn die Prüfchemikalie auf diese Art verabreicht werden soll.
|
|
17.
|
Die Tiere sollten in kleinen gleichgeschlechtlichen Gruppen untergebracht werden; sie können auch einzeln gehalten werden, wenn dies wissenschaftlich gerechtfertigt ist. Bei Gruppenhaltung sollten maximal fünf Tiere in einem Käfig untergebracht sein. Die Verpaarung erfolgt in für diesen Zweck geeigneten Käfigen. Trächtigen weiblichen Tieren sollte in Einzelhaltung Material für den Nestbau bereitgestellt werden. Säugende weibliche Tiere werden in Käfigen einzeln mit ihren Nachkommen gehalten.
|
|
18.
|
Das Futter ist regelmäßig auf Schadstoffe zu analysieren. Eine Probe des Futters ist bis zur Fertigstellung des Abschlussberichts aufzubewahren.
|
Vorbereitung der Versuchstiere
|
19.
|
Gesunde und geschlechtsreife junge Tiere werden randomisiert und den einzelnen Kontroll- bzw. Behandlungsgruppen zugeteilt. Die Käfige sind so aufzustellen, dass etwaige standortbedingte Auswirkungen möglichst gering sind. Die Tiere werden eindeutig gekennzeichnet und vor Beginn der Studie in ihren Käfigen über einen Zeitraum von mindestens fünf Tagen unter Laborbedingungen eingewöhnt.
|
Vorbereitung der Dosierung
|
20.
|
Die Prüfchemikalie sollte oral verabreicht werden, wenn nicht andere Verabreichungswege als besser geeignet erscheinen. Auf oralem Weg wird die Prüfchemikalie gewöhnlich mit einer Sonde verabreicht. Alternativ können Prüfchemikalien aber auch über die Nahrung oder das Trinkwasser zugeführt werden.
|
|
21.
|
Bei Bedarf wird die Prüfchemikalie in einem geeigneten Vehikel gelöst oder suspendiert. Es empfiehlt sich, nach Möglichkeit zunächst die Verwendung einer wässrigen Lösung/Suspension, dann eine Lösung/Emulsion in Öl (z. B. Maisöl) und erst dann eine Lösung in einem anderen Vehikel in Betracht zu ziehen. Bei anderen Vehikeln als Wasser müssen deren toxische Merkmale bekannt sein. Die Stabilität und die Homogenität der Prüfchemikalie in dem Vehikel sind zu bestimmen.
|
VERFAHREN
Anzahl und Geschlecht der Versuchstiere
|
22.
|
Jede Gruppe sollte mit mindestens 10 männlichen und 12-13 weiblichen Tieren begonnen werden. Bei den weiblichen Tieren wird vor der Exposition der Östruszyklus untersucht, und Tiere, bei denen nicht der typische Zyklus von 4-5 Tagen festzustellen ist, werden nicht in die Studie einbezogen. Daher wird die Vorhaltung weiterer weiblicher Tiere empfohlen, damit pro Gruppe tatsächlich 10 weibliche Tiere verfügbar sind. Außer bei ausgeprägten toxischen Wirkungen dürften dann pro Gruppe mindestens 8 Graviditäten entstehen; dies ist gewöhnlich die erforderliche Mindestzahl an trächtigen Tieren pro Gruppe. So soll gewährleistet werden, dass genügend Graviditäten und Nachkommen erzeugt werden, um eine aussagekräftige Bewertung des Schädigungspotenzials der Prüfchemikalie in Bezug auf die Fertilität, die Gravidität, das Verhalten des Muttertiers, das Säugen, das Wachstum und die Entwicklung der F1-Generation von der Empfängnis bis zu Tag 13 nach der Geburt vornehmen zu können.
|
Dosierung
|
23.
|
Im Allgemeinen sollten mindestens drei Versuchsgruppen und eine Kontrollgruppe gewählt werden. Die Dosierungen können auf Informationen aus akuten Toxizitätsprüfungen oder auf Ergebnissen von Studien mit Wiederholungsdosen beruhen. Abgesehen von der Behandlung mit der Prüfchemikalie sollen die Tiere in der Kontrollgruppe unter identischen Bedingungen behandelt werden wie die Versuchstiere in der Prüfgruppe. Wird die Prüfchemikalie mit einem Vehikel verabreicht, muss die Kontrollgruppe das Vehikel mit dem höchsten verwendeten Volumen erhalten.
|
|
24.
|
Bei der Wahl der Dosisstufen sollten sämtliche vorliegenden Daten zur Toxizität und (Toxiko)kinetik berücksichtigt werden. Außerdem sollte berücksichtigt werden, dass trächtige und nicht trächtige Tiere unterschiedlich empfindlich sein können. Die höchste Dosis sollte so gewählt werden, dass zwar toxische Wirkungen, aber keine Todesfälle oder schweres Leiden hervorgerufen werden. Anschließend soll eine absteigende Folge von Dosierungsstufen ausgewählt werden, um dosisabhängige Wirkungen und die niedrigste Dosis ohne zu beobachtende schädliche Wirkungen (NOAEL) zu bestimmen. Zwei- bis vierfache Abstände erweisen sich häufig als optimale Dosisabstufungen, gegenüber der Verwendung sehr großer Intervalle (z. B. mehr als Faktor 10) ist die Hinzunahme einer vierten Prüfgruppe häufig vorzuziehen.
|
|
25.
|
Bei allgemeiner Toxizität (z. B. vermindertes Körpergewicht, Wirkungen auf Leber, Herz, Lungen oder Nieren usw.) oder anderen Veränderungen, die möglicherweise keine toxischen Reaktionen darstellen (z. B. verminderte Futteraufnahme, Lebervergrößerung), sind die beobachteten Wirkungen auf endokrine Endpunkte mit Vorsicht zu bewerten.
|
Limit-Prüfung
|
26.
|
Ergibt eine orale Prüfung mit einer einzigen Dosierung von mindestens 1 000 mg/kg Körpergewicht/Tag oder bei Verabreichung in Futter oder Trinkwasser ein gleichwertiger prozentualer Anteil im Futter oder Trinkwasser nach den für diese Studie beschriebenen Verfahren keine wahrnehmbare Toxizität und ist aufgrund von Daten strukturverwandter Stoffe keine Toxizität zu erwarten, kann auf eine vollständige Studie mit verschiedenen Dosisabstufungen gegebenenfalls verzichtet werden. Die Limit-Prüfung kann angebracht sein, es sei denn, die Exposition beim Menschen lässt die Prüfung bei einer höheren oralen Dosis angezeigt erscheinen. Bei anderen Verabreichungsformen, z. B. Inhalation oder dermale Applikation, wird die maximal erzielbare Konzentration in vielen Fällen durch die physikalisch-chemischen Eigenschaften der Prüfchemikalien bestimmt.
|
Verabreichung der Dosen
|
27.
|
Den Tieren wird die Prüfchemikalie für die Dauer einer Woche (7 Tage) täglich verabreicht. Wird die Prüfchemikalie über eine Sonde verabreicht, so sollte dies in einer einmaligen Dosis unter Verwendung einer Magensonde oder einer geeigneten Intubationskanüle erfolgen. Das maximale Flüssigkeitsvolumen, das einem Versuchstier jeweils verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte 1 ml/100 g Körpergewicht nicht überschreiten, bei wässrigen Lösungen kommen aber auch 2 ml/100 g Körpergewicht in Betracht. Außer für Reizungen auslösende oder ätzende Prüfchemikalien, die in der Regel bei höheren Konzentrationen eine Verschlimmerung bewirken, sollte die Variabilität des Prüfvolumens durch Anpassung der Konzentration möglichst gering gehalten werden, um ein konstantes Volumen bei allen Dosen zu gewährleisten.
|
|
28.
|
Bei mit dem Futter oder dem Trinkwasser verabreichten Prüfchemikalien ist unbedingt sicherzustellen, dass die Mengen der jeweiligen Prüfchemikalie die normale Nahrungsaufnahme oder den Wasserhaushalt nicht beeinträchtigen. Wenn die Prüfchemikalie im Futter verabreicht wird, kann entweder eine konstante Konzentration im Futter (ppm) oder eine konstante Dosis, bezogen auf das Körpergewicht des Tieres, verwendet werden; die jeweils gewählte Verfahrensweise sollte angegeben werden. Eine mit einer Magensonde verabreichte Dosis soll jeweils zu denselben Tageszeiten gegeben und mindestens einmal pro Woche so angepasst werden, dass eine konstante Dosis im Verhältnis zum Körpergewicht aufrechterhalten wird.
|
Versuchsplan
|
29.
|
Die Verabreichung sollte für beide Geschlechter mindestens 2 Wochen vor der Paarung beginnen, nachdem sich die Tiere mindestens 5 Tage lang eingewöhnt haben und nachdem die weiblichen Tiere auf einen normalen Östruszykus (in einem 2-wöchigen Zeitraum vor der Behandlung) untersucht wurden. Die Studie sollte so geplant werden, dass mit der Bewertung des Östruszykus bald nach Erreichen der vollständigen Geschlechtsreife begonnen wird. Der Beginn dieser Zeiträume kann je nach Rattenstämmen in den einzelnen Labors leicht variieren und beispielsweise bei Sprague-Dawley-Ratten bei einem Alter von 10 Wochen und bei Wistar-Ratten etwa bei einem Alter von 12 Wochen liegen. Muttertiere sollten an Tag 13 nach der Geburt oder kurz darauf getötet werden. Der Tag der Geburt (d. h. der Tag, an dem der Geburtsvorgang abgeschlossen wurde) wird als Tag 0 nach der Geburt bezeichnet. Weibliche Tiere, bei denen keine Besamung festzustellen ist, werden 24-26 Tage nach dem letzten Tag der Paarungszeit getötet. Die Verabreichung wird bei beiden Geschlechtern während der Paarungszeit fortgesetzt. Männliche Tiere werden auch nach der Paarungszeit noch mindestens über den vollständigen Verabreichungszeitraum von 28 Tagen behandelt. Anschließend werden sie getötet oder alternativ weiter gehalten und behandelt, damit eine zweite Paarung erfolgen kann, wenn dies als angemessen betrachtet wird.
|
|
30.
|
Die tägliche Behandlung der weiblichen Muttertiere sollte während der Gravidität sowie mindestens bis zu Tag 13 nach der Geburt bzw. zum Tag vor der Tötung (einschließlich) fortgesetzt werden. Bei Studien, bei denen die Prüfchemikalie durch Inhalation oder dermal verabreicht wird, sollte die Verabreichung mindestens bis zu Tag 19 der Trächtigkeit (einschließlich) fortgesetzt und so bald wie möglich (spätestens PND 4) wieder aufgenommen werden.
|
|
31.
|
Der Versuchsplan ist in Anlage 2 in einem Diagramm dargestellt.
|
Verpaarung
|
32.
|
In der Regel sollten bei dieser Studie männliche und weibliche Tiere im Verhältnis 1:1 (ein männliches auf ein weibliches Tier) eingesetzt werden. Ausnahmen können sich gelegentlich ergeben, wenn männliche Tiere sterben. Das weibliche Tier sollte mit demselben männlichen Tier gehalten werden, bis Anzeichen für eine Paarung festgestellt werden bzw. bis zwei Wochen vergangen sind. Jeden Morgen werden die weiblichen Tiere auf Sperma oder Vaginalpfröpfe untersucht. Tag 0 der Trächtigkeit wird als der Tag festgelegt, an dem die Besamung (durch Vaginalpfropf oder Spermaspuren) nachgewiesen werden kann. Bei einer erfolglosen Verpaarung kann gegebenenfalls die erneute Verpaarung weiblicher Tiere mit bewährten männlichen Tieren der gleichen Gruppe erwogen werden.
|
Wurfgröße
|
33.
|
Am PND 4 kann die Größe eines jeden Wurfs angepasst werden, indem überschüssige Jungtiere nach dem Zufallsprinzip aussortiert werden, um je nach normalem Umfang eines Wurfs beim verwendeten Rattenstamm pro Wurf nach Möglichkeit vier oder fünf Jungtiere pro Geschlecht und Wurf zu erhalten. Von zwei der überschüssigen Jungtiere sollten Blutproben entnommen, in Pools zusammengefasst und zur Bestimmung von Serumspiegeln (T4) verwendet werden. Die selektive Eliminierung von Jungtieren, z. B. auf der Grundlage des Körpergewichts oder des anogenitalen Abstands (AGD), wird nicht empfohlen. Wenn es wegen der Anzahl männlicher bzw. weiblicher Jungtiere nicht möglich ist, pro Wurf jeweils vier oder fünf Jungtiere eines jeden Geschlechts zu erhalten, ist auch eine grobe Anpassung (beispielsweise sechs männliche und vier weibliche Tiere) akzeptabel. Wenn die Anzahl der Tiere eines Wurfs die Eliminierungsgrenze (8 oder 10 Jungtiere pro Wurf) unterschreitet, werden keine Jungtiere aussortiert. Wenn die Anzahl der Jungtiere um nur ein Tier über der Eliminierungsgrenze liegt, wird nur ein Jungtier aussortiert und zur Blutentnahme für mögliche Bewertungen der Serumspiegel (T4) verwendet.
|
|
34.
|
Wenn die Größe des Wurfs nicht geändert wird, werden an Tag 4 nach der Geburt zwei Jungtiere pro Wurf human getötet und Blutproben zur Bestimmung der Konzentration der Schilddrüsenhormone im Serum entnommen. Nach Möglichkeit sollten diese zwei Jungtiere je Wurf weibliche Jungtiere sein, damit männliche Jungtiere zur Prüfung der Brustwarzenretention für den Fall aufgespart werden können, dass nach dem Aussortieren dieser Jungtiere keine weiblichen Tiere zur abschließenden Bewertung mehr verbleiben würden. Wenn ein Wurf weniger als 8 bzw. 10 Jungtiere hat (je nach Größe eines normalen Wurfs beim betreffenden Rattenstamm), werden keine Jungtiere aussortiert. Wenn die Anzahl der Jungtiere um nur ein Tier über der normalen Größe eines Wurfs liegt, wird nur ein Jungtier aussortiert und zur Blutentnahme für mögliche Bewertungen der Serumspiegel (T4) verwendet.
|
Beobachtungen am lebenden Tier
Klinische Beobachtungen
|
35.
|
Während der gesamten Versuchsdauer sollten allgemeine klinische Beobachtungen mindestens einmal täglich durchgeführt werden, bei Anzeichen für Toxizität häufiger. Die Beobachtungen sollen vorzugsweise jeden Tag zur gleichen Uhrzeit erfolgen, wobei die Zeit der nach der Verabreichung erwarteten maximalen Wirkungen zu berücksichtigen ist. Auffallende Verhaltensstörungen und Anzeichen einer schweren oder verzögerten Geburt sowie alle Vergiftungserscheinungen einschließlich Mortalität sind aufzuzeichnen. In den Aufzeichnungen sind auch der Zeitpunkt des erstmaligen Auftretens, des Umfangs und der Dauer der Anzeichen für eine Toxizität zu vermerken.
|
Körpergewicht und Futter-/Trinkwasserverbrauch
|
36.
|
Männliche und weibliche Tiere der P-Generation müssen am ersten Tag der Behandlung und anschließend mindestens in wöchentlichen Abständen sowie bei Versuchsende gewogen werden. Während der Gravidität sollten weibliche Tiere an den Tagen 0, 7, 14 und 20 sowie innerhalb von 24 Stunden nach der Geburt (Tag 0 oder Tag 1 nach der Geburt) und mindestens an den Tagen 4 und 13 nach der Geburt gewogen werden. Diese Beobachtungen sind für jedes ausgewachsene Tier einzeln zu protokollieren.
|
|
37.
|
Vor der Paarung, vor der Gravidität und vor der Laktation sollte die Futteraufnahme mindestens einmal wöchentlich gemessen werden. Die Messung der Futteraufnahme während der Paarungszeit ist fakultativ. Wenn die Prüfchemikalie mit dem Trinkwasser verabreicht wird, sollte in diesen Zeiträumen auch die Wasseraufnahme gemessen werden.
|
Östruszyklen
|
38.
|
Östruszyklen sollten vor Beginn der Behandlung überwacht werden, damit für die Studie weibliche Tiere mit regelmäßigem Zyklus ausgewählt werden können (Nummer 22). Außerdem sollten ab Beginn des Behandlungszeitraums bis zu Anzeichen für eine Besamung täglich Vaginalabstriche untersucht werden. Wenn Bedenken hinsichtlich akuter Auswirkungen von Stress bestehen, die bei Beginn der Verabreichung die Östruszyklen verändern könnten, können die Versuchstiere in den Labors zwei Wochen behandelt werden, um anschließend die Östruszyklen vor der Paarungszeit über mindestens zwei Wochen und anschließend bis zu Anzeichen für eine Besamung in der Paarungszeit anhand täglicher Vaginalabstriche zu überwachen. Bei der Entnahme vaginaler/zervikaler Zellen ist sorgfältig darauf zu achten, dass die Schleimhaut nicht gereizt und eine Pseudogravidität eingeleitet werden könnte (7) (8).
|
Parameter für die Nachkommen
|
39.
|
Die Trächtigkeitsdauer wird vom Tag 0 der Gravidität an berechnet und sollte vermerkt werden. Jeder Wurf ist so bald wie möglich nach der Geburt zu untersuchen, um die Anzahl und das Geschlecht der Nachkommen, Totgeburten, Lebendgeburten und Kümmerlinge (Jungtiere, die erheblich kleiner sind als Jungtiere einer vergleichbaren Kontrollgruppe) sowie auffallende Anomalien feststellen zu können.
|
|
40.
|
Innerhalb von 24 Stunden nach der Geburt (Tag 0 oder Tag 1 nach der Geburt) und mindestens an den Tagen 4 und 13 nach der Geburt sollten lebende Jungtiere gezählt und nach Geschlechtern unterschieden und die Würfe jeweils gewogen werden. Zusätzlich zu den in Nummer 35 genannten Beobachtungen sollte ungewöhnliches Verhalten der Nachkommen vermerkt werden.
|
|
41.
|
Der anogenitale Abstand (AGD) sollte bei jedem Jungtier zwischen PND 0 und PND 4 am selben Tag gemessen werden. Das Körpergewicht des Jungtiers wird am Tag der Messung des AGD erfasst, der auf Jungtiergröße – vorzugsweise die Quadratwurzel des Körpergewichts – genormt sein sollte (9). Die Anzahl Brustwarzen/Warzenhöfe bei männlichen Jungtieren ist wie in OECD GD 151 empfohlen am PND 12 oder 13 zu kontrollieren (10).
|
Klinisch-biochemische Untersuchungen
|
42.
|
Nach dem folgenden Plan werden Blutproben an einer vorgegebenen Stelle entnommen:
|
—
|
von mindestens zwei Jungtieren pro Wurf an Tag 4 nach der Geburt, wenn die Anzahl der Jungtiere dies zulässt (Nummern 33 und 34),
|
|
—
|
von allen Muttertieren und von mindestens zwei Jungtieren pro Wurf bei Beendigung des Versuchs an Tag 13 und
|
|
—
|
von allen adulten männlichen Tieren bei Beendigung des Versuchs.
|
|
Alle Blutproben werden unter geeigneten Bedingungen gelagert. Bei Blutproben der Jungtiere vom Tag 13 sowie bei Blutproben der adulten männlichen Tiere werden die Serumspiegel auf Schilddrüsenhormone (T4) untersucht. Wenn relevant, wird außerdem eine Bewertung von T4 bei Blutproben der Muttertiere sowie bei Jungtieren an Tag 4 vorgenommen. Ebenfalls wenn relevant können auch weitere Hormone gemessen werden. Das Blut der Jungtiere kann zur Analyse der Schilddrüsenhormone nach Würfen zu Pools zusammengefasst werden. Die Schilddrüsenhormone (T4 und TSH) sollten vorzugsweise „insgesamt“ gemessen werden.
|
43.
|
Die folgenden Faktoren können die Variabilität und die absoluten Konzentrationen der Hormonbestimmungen beeinflussen:
|
—
|
der Zeitpunkt der Tötung wegen Schwankungen der Hormonkonzentrationen im Tagesverlauf,
|
|
—
|
Methode der Tötung, die gewählt wird, um übermäßigen Stress für die Tiere zu vermeiden, der die Hormonkonzentrationen beeinflussen könnte,
|
|
—
|
Prüfkits für Hormonbestimmungen mit unterschiedlichen Standardkurven.
|
|
|
44.
|
Plasmaproben, die speziell zur Hormonbestimmung vorgesehen sind, sollten immer zur gleichen Tageszeit gewonnen werden. Die verschiedenen im Handel erhältlichen Assay-Kits können bei der Analyse der Hormonkonzentration unterschiedliche numerische Werte ergeben.
|
Pathologie
Autopsie
|
45.
|
Zum Zeitpunkt der Tötung oder bei vorzeitigem Tod im Verlauf der Studie sind die adulten Tiere makroskopisch auf etwaige Anomalien oder pathologische Veränderungen zu untersuchen. Dabei ist besonders auf die Organe des Fortpflanzungssystems zu achten. Die Anzahl der Implantationsstellen sollte vermerkt werden. Am Morgen des Tags der Sektion ist ein Vaginalabstrich zu untersuchen, um das Stadium des Östruszyklus zu bestimmen und eine Korrelation zur histopathologischen Untersuchung der Ovarien zu ermöglichen.
|
|
46.
|
Die Hoden und die Nebenhoden sowie die Prostata und die Samenbläschen zusammen mit den Koagulationsdrüsen aller adulter männlicher Tiere sollten ggf. von anhaftendem Gewebe befreit und möglichst umgehend nach der Sektion gewogen werden, bevor das Material eintrocknet. Als Organgewichte können ferner die Gewichte des Muskelkomplex Levator ani/bulbospongiosus, der Cowperschen Drüsen und der Glans penis bei männlichen Tieren sowie der Ovarien (Nassgewicht) und des Uterus mit Zervix bei weiblichen Tieren gemessen werden; wenn diese Gewichte berücksichtigt werden sollen, sollten die Messungen möglichst umgehend nach der Sektion vorgenommen werden.
|
|
47.
|
Tote Jungtiere und Jungtiere, die an Tag 13 nach der Geburt oder kurz danach getötet wurden, sollten zumindest äußerlich sorgfältig auf auffallende Anomalien untersucht werden. Besondere Beachtung erfordern die externen Genitalien, die auf Anzeichen für eine veränderte Entwicklung zu untersuchen sind. An Tag 13 sollte die Schilddrüse eines männlichen und eines weiblichen Jungtieres pro Wurf konserviert werden.
|
|
48.
|
Bei allen adulten Tieren sollten die Ovarien, Hoden, akzessorischen Geschlechtsorgane (Uterus und Zervix, Nebenhoden, Prostata, Samenbläschen und Koagulationsdrüsen), Schilddrüse und alle Organe mit makroskopischen Veränderungen konserviert werden. Eine Fixation in Formalin ist für die regelmäßige Prüfung von Hoden und Nebenhoden nicht zu empfehlen. Eine annehmbare Methode besteht in der Verwendung von Bouinscher Lösung oder modifizierter Davidson-Lösung für diese Gewebe (11). Um ein rasches Eindringen des Fixierungsmittels zu ermöglichen, kann die Tunica albuginea an beiden Enden des Organs vorsichtig und flach mit einer Nadel punktiert werden.
|
Histopathologie
|
49.
|
Bei den Tieren der höchsten Dosisgruppe und der Kontrollgruppe sollten die Ovarien, Hoden und Nebenhoden (unter besonderer Berücksichtigung der Stadien der Spermatogenese und der Histopathologie der interstitiellen testikuären Zellstruktur) einer detaillierten histologischen Untersuchung unterzogen werden. Die übrigen konservierten Organe einschließlich der Schilddrüse von Jungtieren und von adulten Tieren können erforderlichenfalls untersucht werden. Das Gewicht der Schilddrüse kann nach der Fixierung bestimmt werden. Anhaftendes Gewebe ist sehr vorsichtig und erst nach der Fixierung zu entfernen, um Gewebeschäden zu vermeiden. Eine Gewebeschädigung könnte die histopathologische Analyse beeinträchtigen. Die Untersuchungen sind auch auf die Tiere anderer Dosisgruppen auszudehnen, wenn in der Gruppe mit der höchsten Dosis Veränderungen festgestellt werden. Der Leitfaden zur Histopathologie (11) enthält zusätzliche Informationen zur Sektion, Fixierung, Schnittherstellung und Histopathologie endokriner Gewebe.
|
DATEN UND BERICHTERSTATTUNG
Daten
|
50.
|
Es sollen Daten zu den einzelnen Tieren bereitgestellt werden. Außerdem sollten sämtliche Daten in tabellarischer Form zusammengefasst werden; dabei werden für jede Prüfgruppe die Anzahl der Tiere zu Beginn der Prüfung, die Anzahl der während der Prüfung tot aufgefundenen Tiere beziehungsweise der aus humanen Gründen getöteten Tiere, der jeweilige Zeitpunkt des Todes beziehungsweise der Tötung, die Anzahl der fruchtbaren Tiere, die Anzahl der trächtigen weiblichen Tiere, die Anzahl der Tiere mit Anzeichen von Toxizität, eine Beschreibung der beobachteten Anzeichen von Toxizität, einschließlich des Zeitpunkts, zu dem die toxischen Wirkungen eingetreten sind, deren Dauer und Schweregrad, die Arten von histopathologischen Veränderungen und alle relevanten Daten zu den Würfen angegeben. Anlage 3 enthält ein tabellarisches Berichtsformat, das sich als sehr hilfreich für die Evaluierung der reproduktions- bzw. entwicklungstoxischen Auswirkungen erwiesen hat.
|
|
51.
|
Wegen des begrenzten Umfangs der Studie sind statistische Analysen in Form von „Signifikanztests“ für viele Endpunkte, insbesondere für reproduktionsbezogene Endpunkte, nur von begrenzter Bedeutung. Wenn statistische Analysen durchgeführt werden, sollte die gewählte Methode der Verteilung der zu untersuchenden Variable angemessen sein und vor Beginn der Studie festgelegt werden. Statistische Analysen des AGD und der Brustwarzenretention sollten auf den Daten einzelner Jungtiere unter Berücksichtigung wurfbedingter Auswirkungen beruhen. Gegebenenfalls sind die jeweiligen Würfe als Analyseeinheit anzunehmen. Statistische Analysen des Körpergewichts der Jungtiere sollten auf den Daten einzelner Jungtiere unter Berücksichtigung der Wurfgröße beruhen. In Anbetracht des geringen Umfangs der Gruppe kann ggf. auch die Einbeziehung historischer Kontrolldaten (z. B. für die Wurfgröße) bei der Auswertung der Studie hilfreich sein.
|
Auswertung der Ergebnisse
|
52.
|
Die Befunde dieser Toxizitätsstudie sind im Hinblick auf die beobachteten Wirkungen sowie auf Nekropsie- und Mikroskopiebefunde zu bewerten. Die Bewertung beinhaltet den vorhandenen beziehungsweise nicht vorhandenen Zusammenhang zwischen der Dosierung der Prüfchemikalie und vorhandenen Anomalien sowie deren Häufigkeit und Schwere, einschließlich makroskopischer Läsionen, identifizierter Zielorgane, Unfruchtbarkeit, klinischer Anomalien, beeinträchtigter Reproduktions- und Wurfleistungen, Körpergewichtsveränderungen, Auswirkungen auf die Mortalität und etwaiger sonstiger toxischer Auswirkungen.
|
|
53.
|
Wegen der kurzen Dauer der Behandlung der männlichen Tiere sollte bei der Bewertung der reproduktionstoxischen Auswirkungen die Histopathologie der Hoden und der Nebenhoden zusammen mit den Fertilitätsdaten berücksichtigt werden. Die Einbeziehung verfügbarer historischer Kontrolldaten zur Reproduktions-/Entwicklungstoxizität (z. B. für Wurfgröße, AGD, Brustwarzenretention und Serumspiegel (T4)) kann bei der Auswertung der Studie ebenfalls hilfreich sein.
|
|
54.
|
Für die Qualitätskontrolle wird vorgeschlagen, historische Kontrolldaten zu sammeln und Variationskoeffizienten für numerische Daten zu berechnen, insbesondere für die Parameter, die mit dem Nachweis der Störung des endokrinen Systems zusammenhängen. Diese Daten können für Vergleichszwecke verwendet werden, wenn tatsächliche Studien bewertet werden.
|
Prüfbericht
|
55.
|
Der Prüfbericht sollte folgende Angaben enthalten:
|
|
Prüfchemikalie:
|
—
|
Herkunft, Chargennummer, ggf. begrenztes Verwendungsdatum;
|
|
—
|
Stabilität der Prüfchemikalie, falls bekannt.
|
|
|
|
Einkomponentiger Stoff:
|
—
|
physikalisches Erscheinungsbild, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
|
|
—
|
chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer, SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.
|
|
|
|
Mehrkomponentiger Stoff, UVCB-Stoffe und Gemische:
|
—
|
so weit wie möglich charakterisiert durch die chemische Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Komponenten.
|
|
|
|
Vehikel (wenn verwendet):
|
—
|
Begründung der Auswahl des Vehikels, falls kein Wasser verwendet wurde;
|
|
|
|
Versuchstiere:
|
—
|
Anzahl, Alter und Geschlecht der Tiere;
|
|
—
|
Herkunft der Tiere, Haltungsbedingungen, Futter usw.;
|
|
—
|
Gewicht der einzelnen Tiere bei Versuchsbeginn;
|
|
—
|
Begründung für die Wahl einer anderen Art als der Ratte.
|
|
|
|
Prüfbedingungen:
|
—
|
Begründung der gewählten Dosisstufen;
|
|
—
|
Angaben zur Zubereitungsform der Prüfchemikalie/des Futters, der erreichten Konzentrationen, Stabilität und Homogenität der Zubereitung,
|
|
—
|
Angaben zur Verabreichung der Prüfchemikalie;
|
|
—
|
gegebenenfalls Angaben zur Umrechnung der Konzentration der Prüfchemikalie im Futter/Wasser (ppm) in die entsprechende Dosis (mg/kg Körpergewicht/Tag),
|
|
—
|
Angaben über Futter- und Wasserqualität;
|
|
—
|
genaue Beschreibung des Verfahrens für die Zufallsauswahl von Jungtieren zwecks Tötung (wenn Jungtiere getötet werden).
|
|
|
|
Ergebnisse:
|
—
|
Körpergewicht/Änderungen des Körpergewichts,
|
|
—
|
Angaben zur Futter- und Wasseraufnahme (wenn verfügbar);
|
|
—
|
Daten über toxische Reaktionen nach Geschlecht und Dosierung, einschließlich Fruchtbarkeit, Trächtigkeit und sonstige Anzeichen von Toxizität;
|
|
—
|
toxische oder sonstige Wirkungen auf Reproduktion, Nachkommen, postnatales Wachstum usw.;
|
|
—
|
Art, Schweregrad und Dauer der klinischen Beobachtungen (mit Angaben zur Reversibilität);
|
|
—
|
Zahl adulter weiblicher Tiere mit normalem oder abnormalem Östruszyklus sowie Zyklusdauer;
|
|
—
|
Anzahl der Lebendgeburten und der Postimplantationsverluste;
|
|
—
|
Angaben zum Körpergewicht der Jungtiere;
|
|
—
|
AGD aller Jungtiere (und Körpergewicht am Tag der AGD-Messung);
|
|
—
|
Brustwarzenretention bei männlichen Jungtieren;
|
|
—
|
Schilddrüsenhormonspiegel, Jungtiere an Tag 13 und adulte männliche Tiere (sowie Muttertiere und Jungtiere an Tag 4, wenn gemessen)
|
|
—
|
Anzahl der Jungtiere mit sichtbaren erheblichen Anomalien, allgemeine Bewertung externer Genitalien, Anzahl der Kümmerlinge;
|
|
—
|
Zeitpunkt des Todes im Verlauf der Studie oder Angabe, ob Tiere bis zum Schluss überlebt haben;
|
|
—
|
Anzahl der Implantate, Wurfgröße und Wurfgewichte zum Zeitpunkt der Aufzeichnung;
|
|
—
|
Körpergewicht bei Tötung sowie Organgewichtsdaten für die Elterntiere;
|
|
—
|
ausführliche Beschreibung aller histopathologischen Befunde;
|
|
—
|
Absorptionsdaten, soweit vorhanden;
|
|
—
|
statistische Auswertung der Ergebnisse, wenn möglich.
|
|
Erörterung der Ergebnisse.
Schlussfolgerungen
|
Auswertung der Ergebnisse
|
56.
|
Mit der Studie wird die Reproduktions-/Entwicklungstoxizität bei wiederholter Verabreichung (Nummern 5 und 6) bewertet. Die Studie könnte Aufschluss über die Notwendigkeit der Durchführung weiterer Untersuchungen geben und zu Empfehlungen für die Gestaltung von Folgestudien führen. Als Hilfe bei der Auswertung der reproduktions- und entwicklungstoxischen Ergebnisse ist das OECD Guidance Document Nr. 43 zu Rate zu ziehen (12). OECD Guidance Document Nr. 106 über die histologische Auswertung von Prüfungen zur Feststellung endokriner und reproduktionstoxischer Wirkungen bei Nagern (11) enthält Informationen zur Vorbereitung und Auswertung von (endokrinen) Organen und von Vaginalabstrichen, die für diese TG hilfreich sein könnten.
|
LITERATUR
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Auf Anfrage erhältlich bei der Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokio, 27.-29. Oktober 1992. Auf Anfrage erhältlich bei der Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10. und 11. März 1998. Auf Anfrage erhältlich bei der Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(7)
|
Goldman, J.M., Murr, A.S., Buckalew, A.R., Ferrell, J.M. und Cooper, R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), S. 84-97.
|
|
(8)
|
Sadleir, R.M.F.S (1979). Cycles and Seasons, in Auston, C.R. und Short, R.V. (Hrsg.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan, R.H. Jr, Holson, J.F., Stump, D.G., Knapp, J.F. und Reynolds, V.L. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No106.), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
Anlage 1
BEGRIFFSBESTIMMUNGEN (SIEHE AUCH OECD GD 150 (6))
Androgene Wirkung: Die Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches androgenes Hormon zu wirken (z. B. Testosteron).
Antiandrogene Wirkung: Die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen androgenen Hormons (z. B. Testosteron) in einem Säugetierorganismus zu unterdrücken.
Antiöstrogene Wirkung: Die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen östrogenen Hormons (z. B. Östradiol 17ß) in einem Säugetierorganismus zu unterdrücken.
Antithyroide Wirkung. Die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen Schilddrüsenhormons (z. B. T3) in einem Säugetierorganismus zu unterdrücken.
Chemikalie: Ein Stoff oder Gemisch.
Dosierung: Ein allgemeiner Begriff, der die Dosis, ihre Häufigkeit und die Dauer der Verabreichung umfasst.
Dosis: Die Menge der verabreichten Prüfchemikalie. Die Dosis wird ausgedrückt als Masse der Prüfchemikalie je Einheit Körpergewicht des Versuchstiers pro Tag (z. B. mg/kg Körpergewicht/Tag) oder als konstante Konzentration im Futter.
Entwicklungstoxizität: Das Auftreten einer reproduktionstoxischen Wirkung mit prä-, peri- und postnatalen, strukturellen oder funktionellen Störungen bei der Nachkommenschaft.
Fertilitätsströungen: Störungen der männlichen oder weiblichen Reproduktionsfunktionen oder -fähigkeit.
Maternale Toxizität: Schädliche Wirkungen auf trächtige weibliche Tiere, die entweder spezifisch (direkte Wirkungen) oder nicht spezifisch (indirekte Wirkungen) auftreten.
NOAEL: Abkürzung für No Observed Adverse Effect Level. Dies ist die höchste Dosis, bei der keine schädigenden behandlungsbedingten Wirkungen festgestellt werden.
Offensichtliche Toxizität: Ein allgemeiner Begriff zur Beschreibung deutlicher Toxizitätszeichen nach Verabreichung einer Prüfchemikalie. Diese Zeichen sollten für eine Bewertung der Gefährdung ausreichen und so schwerwiegend sein, dass bei einer Steigerung der verabreichten Dosis die Entwicklung schwerer Toxizitätszeichen und der wahrscheinliche Tod zu erwarten wären.
Östrogene Wirkung: Die Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches östrogenes Hormon zu wirken (z. B. Östradiol 17ß).
Prüfchemikalie: Ein beliebiger Stoff oder eine beliebige Mischung, der/die nach dieser Methode geprüft wird.
Reproduktionstoxizität: Schädliche Wirkungen auf die Nachkommenschaft und/oder Beeinträchtigung der männlichen und weiblichen Reproduktionsfunktionen oder -kapazität.
Thyroide Wirkung: Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches Schilddrüsenhormon (z. B. T3) zu wirken.
Validierung: Ein wissenschaftlicher Prozess zur Beschreibung der operationellen Anforderungen und Grenzen einer Prüfmethode und zum Nachweis ihrer Zuverlässigkeit und Eignung für einen bestimmten Zweck.
Anlage 2
DIAGRAMM ZUM VERSUCHSPLAN MIT ANGABEN ZUR MAXIMALEN DAUER DER STUDIE AUSGEHEND VON DER VOLLSTÄNDIGEN 14-TÄGIGEN PAARUNGSZEIT

Anlage 3
TABELLARISCHE ZUSAMMENFASSUNG DER AUSWIRKUNGEN AUF DIE REPRODUKTION/ENTWICKLUNG
|
BEOBACHTUNGEN
|
WERTE
|
|
|
|
Dosierung (Einheiten)
|
0 (Kontrolle)
|
…
|
…
|
…
|
…
|
|
Ausgangspaarungen (N)
|
|
|
|
|
|
|
Östruszyklus (mindestens mittlere Dauer und Häufigkeit unregelmäßiger Zyklen)
|
|
|
|
|
|
|
Weibliche Tiere mit Anzeichen für eine Besamung (N)
|
|
|
|
|
|
|
Trächtige weibliche Tiere (N)
|
|
|
|
|
|
|
Empfängnistage 1-5 (N)
|
|
|
|
|
|
|
Empfängnistage 6- (N)..(
(21)
) (N)
|
|
|
|
|
|
|
Gravidität ≤ 21 Tage (N)
|
|
|
|
|
|
|
Gravidität = 22 Tage (N)
|
|
|
|
|
|
|
Gravidität ≥ 23 Tage (N)
|
|
|
|
|
|
|
Muttertiere mit lebenden Jungtieren (N)
|
|
|
|
|
|
|
Muttertiere mit lebenden Jungtieren an Tag 4 pp (N)
|
|
|
|
|
|
|
Implantate/Muttertier (Mittelwert)
|
|
|
|
|
|
|
Lebende Jungtiere/Muttertiere bei Geburt (Mittelwert)
|
|
|
|
|
|
|
Lebende Jungtiere/Muttertiere an Tag 4 (Mittelwert)
|
|
|
|
|
|
|
Geschlechterverhältnis (m/w) bei Geburt (Mittelwert)
|
|
|
|
|
|
|
Geschlechterverhältnis (m/w) an Tag 4 (Mittelwert)
|
|
|
|
|
|
|
Wurfgewicht bei Geburt (Mittelwert)
|
|
|
|
|
|
|
Wurfgewicht an Tag 4 (Mittelwert)
|
|
|
|
|
|
|
Gewicht der Jungtiere bei Geburt (Mittelwert)
|
|
|
|
|
|
|
Gewicht der Jungtiere zum Zeitpunkt der AGD-Messung (Mittelwert männliche Jungtiere, Mittelwert weibliche Jungtiere)
|
|
|
|
|
|
|
AGD der Jungtiere am selben Tag nach der Geburt, Geburt – Tag 4 (Mittelwert männliche Jungtiere, Mittelwert weibliche Jungtiere, PND vermerken)
|
|
|
|
|
|
|
Gewicht der Jungtiere an Tag 4 (Mittelwert)
|
|
|
|
|
|
|
Brustwarzenretention bei männlichen Jungtieren an Tag 13 (Mittelwert)
|
|
|
|
|
|
|
Gewicht der Jungtiere an Tag 13 (Mittelwert)
|
|
|
|
|
|
|
|
|
JUNGTIERE MIT ANOMALIEN
|
|
Muttertiere mit 0
|
|
|
|
|
|
|
Muttertiere mit 1
|
|
|
|
|
|
|
Muttertiere mit ≥2
|
|
|
|
|
|
|
|
|
VERLUST AN NACHKOMMEN
|
|
|
|
Pränatal / nach der Implantation (Implantationen abzüglich Lebendgeburten)
|
|
Weibliche Tiere mit 0
|
|
|
|
|
|
|
Weibliche Tiere mit 1
|
|
|
|
|
|
|
Weibliche Tiere mit 2
|
|
|
|
|
|
|
Weibliche Tiere mit ≥ 3
|
|
|
|
|
|
|
|
|
Postnatal (Lebendgeburten abzüglich lebender Tiere an Tag 13 nach der Geburt)
|
|
Weibliche Tiere mit 0
|
|
|
|
|
|
|
Weibliche Tiere mit 1
|
|
|
|
|
|
|
Weibliche Tiere mit 2
|
|
|
|
|
|
|
Weibliche Tiere mit ≥ 3
|
|
|
|
|
|
B.64 KOMBINIERTE SCREENING-PRÜFUNG MIT WIEDERHOLTER VERABREICHUNG ZUR BEWERTUNG DER REPRODUKTIONS-/ENTWICKLUNGSTOXIZITÄT
EINLEITUNG
|
1.
|
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 422 (2016). Die OECD-Richtlinien für die Prüfung von Chemikalien werden regelmäßig unter Berücksichtigung des wissenschaftlichen Fortschritts überarbeitet. Die ursprüngliche Screening-Prüfrichtlinie 422 wurde im Jahr 1996 angenommen. Sie beruhte auf einem Protokoll für eine kombinierte Screening-Prüfung mit wiederholter Verabreichung zur Bewertung der Reproduktions-/Entwicklungstoxizität, die in zwei Expertensitzungen im Jahr 1990 in London (1) und im Jahr 1992 in Tokio (2) erörtert wurde.
|
|
2.
|
Bei dieser Prüfmethode wird eine Screening-Prüfung zur Bewertung der Reproduktions-/Entwicklungstoxizität, die auf Erfahrungen in Mitgliedstaaten aufgrund der Verwendung der ursprünglichen Methode bei in großen Mengen hergestellten Chemikalien und bei Untersuchungen mit Positivkontrollstoffen (3) (4) basiert, mit einer Toxizitätsprüfung mit wiederholter Verabreichung nach der OECD-Prüfrichtlinie 407 (28-Tage-Toxizitätsstudie mit wiederholter oraler Verabreichung an Nagern, Kapitel B.7 dieses Anhangs) kombiniert.
|
|
3.
|
Diese Prüfmethode wurde als Folgemaßnahme der 1998 bei der OECD eingeleiteten prioritären Maßnahme zur Änderung bestehender Prüfrichtlinien und zur Entwicklung neuer Prüfrichtlinien für das Screening und die Prüfung potenzieller endokriner Disruptoren unter Festlegung von für einen endokrinen Disruptor relevanten Endpunkten aktualisiert (5). In diesem Zusammenhang wurde TG 407 (entsprechend Kapitel B.7 in diesem Anhang) im Jahr 2008 durch Parameter verbessert, mit denen eine endokrine Wirkung von Prüfchemikalien nachgewiesen werden kann. Ziel der Aktualisierung von TG 422 war die Einbeziehung einiger für endokrine Disruptoren relevanter Endpunkte in Screening-TGs, bei denen die Expositionszeiträume einige der empfindlichsten Entwicklungszeiträume (Zeiträume vor oder kurz nach der Geburt) beinhalten.
|
|
4.
|
Die ausgewählten für endokrine Disruptoren relevanten zusätzlichen Endpunkte, die Bestandteil auch von TG 443 (Erweiterte Eingenerationen-Prüfung auf Reproduktionstoxizität, entsprechend Kapitel B.56 dieses Anhangs) sind, wurden auf der Grundlage einer Machbarkeitsstudie zur Untersuchung wissenschaftlicher und technischer Fragen im Zusammenhang mit deren Berücksichtigung sowie mit möglichen Anpassungen des für deren Einbeziehung erforderlichen Prüfprotokolls in TG 422 aufgenommen (6).
|
|
5.
|
Mit dieser Prüfmethode sollen begrenzte Informationen über die Wirkungen einer Prüfchemikalie auf die Reproduktionsleistung männlicher und weiblicher Tiere (Funktion der Keimdrüsen, Paarungsverhalten, Empfängnis, Entwicklung des Conceptus, Geburt usw.) ermittelt werden. Sie ist nicht als alternative Prüfmethode und auch nicht als Ersatz für die bereits existierenden Prüfmethoden B.31, B.34, B.35 und B.56 zu betrachten.
|
AUSGANGSÜBERLEGUNGEN
|
6.
|
Bei der Beurteilung und Bewertung der toxischen Merkmale einer Prüfchemikalie kann die orale Toxizität nach wiederholter Verabreichung des Stoffs bestimmt werden, nachdem zunächst durch Prüfungen auf akute Wirkungen die ersten Toxizitätsdaten gewonnen wurden. Diese Prüfung gibt Aufschluss über mögliche Gesundheitsgefahren infolge wiederholter Exposition über einen begrenzten Zeitraum. Die Methode umfasst die Basisstudie zur Prüfung auf Toxizität bei wiederholter Verabreichung, die für chemische Stoffe, bei denen eine 90-Tage-Studie nicht gerechtfertigt ist (z. B. wenn das Produktionsvolumen bestimmte Grenzen nicht überschreitet), oder als Vorstudie zu einer Langzeitstudie verwendet werden kann. Bei der Prüfung sollten die im „OECD Guidance No. 19 on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluation“ (7) genannten Grundsätze und Erwägungen beachtet werden.
|
|
7.
|
Außerdem beinhaltet die Prüfung eine Screening-Prüfung auf Reproduktions-/Entwicklungstoxizität und kann daher auch durchgeführt werden, um ersten Aufschluss über mögliche Wirkungen auf die Reproduktionsleistung männlicher und weiblicher Tiere (Funktion der Keimdrüsen, Paarungsverhalten, Empfängnis, Entwicklung des Conceptus, Geburt usw.) entweder bereits früh bei der Bewertung toxikologischer Eigenschaften von Prüfchemikalien oder bei Besorgnis erregenden Prüfchemikalien zu erhalten. Diese Prüfmethode liefert keine umfassenden Informationen über sämtliche Reproduktions- und Entwicklungsaspekte. Insbesondere bietet sie nur beschränkte Möglichkeiten zur Erkennung postnataler Ausprägungen einer pränatalen Exposition oder zur Feststellung von Wirkungen, die bei postnataler Exposition induziert werden können. Aufgrund (unter anderem) der Selektivität der Endpunkte und der kurzen Dauer der Studie werden mit dieser Methode keine Nachweise dafür erlangt, dass keine reproduktions-/entwicklungstoxische Wirkungen eintreten. Wenn keine Daten aus anderen Prüfungen zur Bewertung der Reproduktions-/Entwicklungstoxizität vorliegen, sind positive Ergebnisse zudem hilfreich für eine erste Gefährdungsabschätzung und können bei Entscheidungen über die Notwendigkeit und die zeitliche Gestaltung weiterer Prüfungen berücksichtigt werden.
|
|
8.
|
Die Ergebnisse in Bezug auf die endokrinen Parameter sind im Kontext des „OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals“ (8) zu interpretieren. Dieser Conceptual Framework enthält die verbesserte OECD TG 422 in Niveau 4 als In-vivo-Prüfung zur Erlangung von Daten über negative Wirkungen auf endokrin relevante Endpunkte. Ein endokrin relevantes Signal kann jedoch nicht an sich als hinreichender Beleg dafür betrachtet werden, dass die betreffende Prüfchemikalie als endokriner Disruptor wirkt.
|
|
9.
|
Auch bei dieser Prüfmethode wird das Hauptaugenmerk auf neurologische Wirkungen als spezifischer Endpunkt gelegt; außerdem ist die Notwendigkeit einer sorgfältigen klinischen Beobachtung der Tiere zu betonen, um möglichst umfangreiche Daten zu erfassen. Die Methode sollte chemische Stoffe mit neurotoxischem Potenzial aufspüren, die dann gegebenenfalls eine eingehendere Untersuchung dieses Aspektes erfordern. Außerdem kann die Methode grundlegenden Aufschluss über immunologische Wirkungen ergeben.
|
|
10.
|
Wenn keine Daten aus anderen Prüfungen zur Bewertung der systemischen Toxizität, der Reproduktions-/Entwicklungstoxizität, der Neurotoxizität und/oder der Immunotoxizität vorliegen, sind positive Ergebnisse hilfreich für eine erste Gefährdungsabschätzung und können bei Entscheidungen über die Notwendigkeit und die zeitliche Gestaltung weiterer Prüfungen berücksichtigt werden. Die Prüfung kann insbesondere als Bestandteil des SIDS-Dossiers (Screening Information Data Set) der OECD für die Bewertung existierender Chemikalien hilfreich sein, für die nur wenig oder keinerlei toxikologische Informationen verfügbar sind; außerdem kann sie anstelle zweier getrennter Prüfungen zur Bewertung der Toxizität bei wiederholter Verabreichung (OECD TG 407, entsprechend Kapitel B.7 dieses Anhangs) bzw. der Reproduktions-/Entwicklungstoxizität (OECD TG 421, entsprechend Kapitel B.63 dieses Anhangs) durchgeführt werden. Ferner kann sie als Dosisfindungsstudie für umfassendere Untersuchungen der Reproduktions- und Entwicklungstoxizität dienen sowie dann durchgeführt werden, wenn sie aus sonstigen Gründen als relevant betrachtet wird.
|
|
11.
|
Im Allgemeinen wird davon ausgegangen, dass trächtige und nicht trächtige Tiere unterschiedlich empfindlich sind. Dosierungen, die sowohl zur Bewertung der allgemeinen systemischen Toxizität als auch der spezifischen Reproduktions-/Entwicklungstoxizität geeignet sind, sind daher mit dieser kombinierten Prüfung u. U. schwieriger zu ermitteln als mit einzelnen Prüfungen, die getrennt durchgeführt werden. Außerdem kann die Auswertung der Prüfergebnisse hinsichtlich der allgemeinen systemischen Toxizität schwieriger sein als bei Durchführung einer getrennten Prüfung mit wiederholter Verabreichung, insbesondere wenn in der Prüfung nicht gleichzeitig serumbezogene und histopathologische Parameter bewertet werden. In Anbetracht dieser technischen Schwierigkeiten erfordert die Durchführung dieser kombinierten Screening-Prüfung beträchtliche Erfahrung mit Toxizitätsprüfungen. Abgesehen von der geringeren Anzahl benötigter Prüftiere kann mit der kombinierten Prüfung allerdings möglicherweise besser zwischen direkten reproduktions-/entwicklungsbezogenen Wirkungen und Wirkungen unterschieden werden, die als sekundäre Wirkungen anderer (systemischer) Wirkungen zu betrachten sind.
|
|
12.
|
Bei dieser Prüfung ist der Verabreichungszeitraum länger als bei einer herkömmlichen 28-Tage-Prüfung mit wiederholter Verabreichung, Allerdings werden von jedem Geschlecht pro Gruppe weniger Tiere verwendet als dann, wenn eine herkömmliche 28-Tage-Prüfung mit wiederholter Verabreichung zusätzlich zu einer Screening-Prüfung auf Reproduktions-/Entwicklungstoxizität durchgeführt wird.
|
|
13.
|
Bei dieser Prüfmethode wird angenommen, dass die Prüfchemikalie oral verabreicht wird. Bei anderen Expositionswegen können Änderungen erforderlich sein.
|
|
14.
|
Bevor die Prüfmethode an einem Gemisch für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs rechtlich vorgeschrieben ist.
|
|
15.
|
Die verwendeten Begriffe sind in Anlage 1 definiert.
|
PRINZIP DER PRÜFMETHODE
|
16.
|
Die Prüfchemikalie wird verschiedenen Gruppen von männlichen und weiblichen Tieren in abgestuften Dosen verabreicht. Männlichen Tieren sollte die Prüfchemikalie mindestens vier Wochen bis zum Tag vor der vorgesehenen Tötung (einschließlich) verabreicht werden. (Dieser Zeitraum umfasst mindestens zwei Wochen vor der Paarung, die Paarungszeit und etwa zwei Wochen nach der Paarung). Wegen des begrenzten Verabreichungszeitraums vor der Paarung ist die Fruchtbarkeit bei männlichen Tieren möglicherweise kein besonders empfindlicher Indikator für eine testikuläre Toxizität. Daher ist eine eingehende histologische Untersuchung der Hoden von wesentlicher Bedeutung. Die Kombination eines Verabreichungszeitraums von zwei Wochen vor der Paarung mit anschließenden Beobachtungen des Paarungsverhaltens und der Fertilität mit einem Verabreichungszeitraum von insgesamt mindestens vier Wochen und einer anschließenden detaillierten Histopathologie der männlichen Keimdrüsen wird als ausreichend für den Nachweis der meisten Wirkungen auf die männliche Fruchtbarkeit und die Spermatogenese betrachtet.
|
|
17.
|
Weiblichen Tieren sollte die Prüfchemikalie während der gesamten Dauer der Studie verabreicht werden. Dazu zählen zwei Wochen vor der Paarung (damit mindestens zwei vollständige Östruszyklen erfasst werden), die variable Zeit bis zur Empfängnis und die Dauer der Gravidität sowie mindestens 13 Tage nach der Geburt bis zum Tag vor der vorgesehenen Tötung (einschließlich).
|
|
18.
|
Die Dauer der Studie nach der Eingewöhnung und einer Bewertung des Östruszyklus vor der Verabreichung der Prüfchemikalie hängt von der Leistungsfähigkeit des weiblichen Tiers ab; im Allgemeinen beträgt sie etwa 63 Tage [mindestens 14 Tage vor der Paarung, (bis zu) 14 Tage Paarungszeit, 22 Tage Gravidität, 13 Tage Laktation].
|
|
19.
|
Während des Verabreichungszeitraums werden die Tiere täglich sorgfältig auf Toxizitätszeichen beobachtet. Tiere, die im Verlauf der Prüfung sterben, und vorzeitig getötete Tiere werden seziert; die nach Abschluss der Prüfung überlebenden Tiere werden getötet und ebenfalls seziert.
|
BESCHREIBUNG DER METHODE
Auswahl von Versuchstierarten
|
20.
|
Diese Prüfmethode ist für die Verwendung von Ratten vorgesehen. Wenn die in dieser TG 422 genannten Parameter an einer anderen Nagetierart untersucht werden, ist dies ausführlich zu begründen. Im internationalen Validierungsprogramm für den Nachweis von endokrinen Disruptoren mit TG 407 wurde nur die Ratte als Versuchstier verwendet. Stämme mit geringer Fruchtbarkeit oder bekannter hoher Häufigkeit von Entwicklungsdefekten sind nicht zu verwenden. Es sind gesunde jungfräuliche Tiere zu verwenden, die zuvor nicht in anderen Versuchen eingesetzt wurden. Die Versuchstiere sind nach Tierart, Tierstamm, Geschlecht, Gewicht und Alter zu beschreiben. Bei Beginn der Studie sollten die Gewichtsunterschiede der Tiere möglichst gering sein und ± 20 % des geschlechtsspezifischen Durchschnittsgewichts nicht überschreiten. Wenn die Studie als Vorstudie zu einer Langzeitstudie oder einer Eingenerationsstudie durchgeführt wird, sollten in beiden Studien vorzugsweise Tiere desselben Stamms und derselben Herkunft verwendet werden.
|
Haltung und Fütterung
|
21.
|
Bei allen Verfahren sind die örtlichen Standards der Versuchstierpflege einzuhalten. Die Temperatur im Versuchstierraum sollte 22 °C (± 3 °C) betragen. Die relative Luftfeuchtigkeit sollte mindestens 30 % betragen und – außer beim Reinigen des Raums – 70 % nicht überschreiten. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, wobei eine unbegrenzte Trinkwasserversorgung zu gewährleisten ist. Die Auswahl des Futters wird eventuell dadurch beeinflusst, dass eine geeignete Beimischung der Prüfchemikalie sichergestellt werden muss, wenn die Prüfchemikalie auf diesem Weg verabreicht werden soll.
|
|
22.
|
Die Tiere sollten in kleinen gleichgeschlechtlichen Gruppen untergebracht werden; sie können auch einzeln gehalten werden, wenn dies wissenschaftlich gerechtfertigt ist. Bei Gruppenhaltung sollten maximal fünf Tiere in einem Käfig untergebracht sein. Die Verpaarung erfolgt in für diesen Zweck geeigneten Käfigen. Trächtigen weiblichen Tieren sollte in Einzelhaltung Material für den Nestbau bereitgestellt werden. Säugende weibliche Tiere werden in Käfigen einzeln mit ihren Nachkommen gehalten.
|
|
23.
|
Das Futter ist regelmäßig auf Schadstoffe zu analysieren. Eine Probe des Futters ist bis zur Fertigstellung des Abschlussberichts aufzubewahren.
|
Vorbereitung der Versuchstiere
|
24.
|
Gesunde junge adulte Tiere werden randomisiert und in Behandlungsgruppen und auf Käfige verteilt. Die Käfige sind so aufzustellen, dass etwaige standortbedingte Auswirkungen möglichst gering sind. Die Tiere werden eindeutig gekennzeichnet und vor Beginn der Studie in ihren Käfigen über einen Zeitraum von mindestens fünf Tagen unter Laborbedingungen eingewöhnt.
|
Vorbereitung der Dosierung
|
25.
|
Die Prüfchemikalie sollte oral verabreicht werden, wenn nicht andere Verabreichungswege als besser geeignet erscheinen. Auf oralem Weg wird die Prüfchemikalie gewöhnlich mit einer Sonde verabreicht. Alternativ können Prüfchemikalien aber auch über die Nahrung oder das Trinkwasser zugeführt werden.
|
|
26.
|
Bei Bedarf wird der Prüfstoff in einem geeigneten Vehikel gelöst oder suspendiert. Es empfiehlt sich, nach Möglichkeit zunächst die Verwendung einer wässrigen Lösung/Suspension, dann eine Lösung/Suspension in Öl (z. B. Maisöl) und erst dann eine Lösung in einem anderen Vehikel in Betracht zu ziehen. Bei nicht wässrigen Vehikeln müssen deren toxische Merkmale bekannt sein. Die Stabilität und die Homogenität der Prüfchemikalie in dem Vehikel sind zu bestimmen.
|
VERFAHREN
Anzahl und Geschlecht der Versuchstiere
|
27.
|
Jede Gruppe sollte mit mindestens 10 männlichen und 12-13 weiblichen Tieren begonnen werden. Bei den weiblichen Tieren wird vor der Exposition der Östruszyklus untersucht, und Tiere, bei denen nicht der typische Zyklus von 4-5 Tagen festzustellen ist, werden nicht in die Studie einbezogen. Daher wird die Vorhaltung weiterer weiblicher Tiere empfohlen, damit pro Gruppe tatsächlich 10 weibliche Tiere verfügbar sind. Außer bei ausgeprägten toxischen Wirkungen dürften dann pro Gruppe mindestens 8 Graviditäten entstehen; dies ist gewöhnlich die erforderliche Mindestzahl an trächtigen Tieren pro Gruppe. So soll gewährleistet werden, dass genügend Graviditäten und Nachkommen erzeugt werden, um eine aussagekräftige Bewertung des Schädigungspotenzials der Prüfchemikalie in Bezug auf die Fertilität, die Gravidität, das Verhalten des Muttertiers, das Säugen, das Wachstum und die Entwicklung der F1-Generation von der Empfängnis bis zu Tag 13 nach der Geburt vornehmen zu können. Sollen Tiere bereits im Verlauf der Prüfung getötet werden, ist die Anzahl der Tiere um die Zahl der Tiere zu erhöhen, die noch vor Abschluss der Studie getötet werden sollen. Zur Beobachtung der Reversibilität, der Persistenz oder des verzögerten Auftretens systemischer toxischer Wirkungen für mindestens 14 Tage nach der Behandlung ist die Einbeziehung einer zusätzlichen Satellitengruppe von fünf Tieren je Geschlecht in der Kontrollgruppe und in der Gruppe mit der höchsten Dosis in Betracht zu ziehen. Tiere der Satellitengruppen werden nicht verpaart und daher auch nicht für die Bewertung der Reproduktions-/Entwicklungstoxizität berücksichtigt.
|
Dosierung
|
28.
|
Im Allgemeinen sollten mindestens drei Versuchsgruppen und eine Kontrollgruppe gewählt werden. Liegen keine geeigneten allgemeinen Toxizitätsdaten vor, kann eine Dosisfindungsstudie (Tiere desselben Stamms und derselben Herkunft) durchgeführt werden, um die zu verwendenden Dosen zu bestimmen. Abgesehen von der Behandlung mit der Prüfchemikalie sollten die Tiere in der Kontrollgruppe unter identischen Bedingungen behandelt werden wie die Versuchstiere in der Prüfgruppe. Wird der Prüfstoff mit einem Vehikel verabreicht, muss die Kontrollgruppe das Vehikel im höchsten verwendeten Volumen erhalten.
|
|
29.
|
Bei der Wahl der Dosisstufen sollten sämtliche vorliegenden Daten zur Toxizität und (Toxiko)kinetik berücksichtigt werden. Außerdem sollte berücksichtigt werden, dass trächtige und nicht trächtige Tiere unterschiedlich empfindlich sein können. Die höchste Dosis sollte so gewählt werden, dass zwar toxische Wirkungen, aber keine Todesfälle oder offensichtliches Leiden hervorgerufen werden. Anschließend sollte eine absteigende Folge von Dosisstufen gewählt werden, um dosisabhängige Wirkungen und die niedrigste Dosis ohne zu beobachtende schädliche Wirkungen nachzuweisen. Häufig sind zwei bis vier Intervalle optimal, und die Ergänzung durch eine vierte Versuchsgruppe ist häufig der Anwendung sehr langer Intervalle (z. B. größer als Faktor 10) zwischen den Verabreichungen vorzuziehen.
|
|
30.
|
Bei allgemeiner Toxizität (z. B. vermindertes Körpergewicht, Wirkungen auf Leber, Herz, Lungen oder Nieren usw.) oder anderen Veränderungen, die möglicherweise keine toxischen Reaktionen darstellen (z. B. verminderte Futteraufnahme, Lebervergrößerung), sind die beobachteten Wirkungen auf endokrine Endpunkte mit Vorsicht zu bewerten.
|
Limit-Prüfung
|
31.
|
Verursacht die Prüfung unter oraler Verabreichung einer Dosis von mindestens 1 000 mg/kg Körpergewicht pro Tag bzw. eine entsprechende Konzentration im Futter (je nach Körpergewicht) unter Verwendung der für diese Studie beschriebenen Verfahren keine feststellbaren toxischen Wirkungen, und ist aufgrund der Daten strukturverwandter Stoffe keine Toxizität zu erwarten, kann auf eine vollständige Studie mit mehreren Dosisstufen gegebenenfalls verzichtet werden. Die Limit-Prüfung kann angebracht sein, es sei denn, die Exposition beim Menschen lässt die Prüfung bei einer höheren oralen Dosis angezeigt erscheinen. Bei anderen Verabreichungsformen, z. B. Inhalation oder dermale Applikation, wird die maximal erzielbare Exposition in vielen Fällen durch die physikalisch-chemischen Eigenschaften der Prüfchemikalien bestimmt.
|
Verabreichung der Dosen
|
32.
|
Den Tieren wird die Prüfchemikalie für die Dauer einer Woche (7 Tage) täglich verabreicht. Wird der Prüfstoff über eine Sonde verabreicht, so sollte dies in einer einmaligen Dosis unter Verwendung einer Schlundsonde oder einer geeigneten Intubationskanüle erfolgen. Das maximale Flüssigkeitsvolumen, das einem Versuchstier jeweils verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte 1 ml/100 g Körpergewicht nicht überschreiten, bei wässrigen Lösungen können aber auch 2 ml/100 g Körpergewicht in Betracht gezogen werden. Außer bei reizenden oder ätzenden Prüfchemikalien, die in der Regel bei höheren Konzentrationen eine Verschlimmerung bewirken, sollte die Variabilität des Prüfvolumens durch Anpassung der Konzentration möglichst gering gehalten werden, um ein konstantes Volumen bei allen Dosen zu gewährleisten.
|
|
33.
|
Bei mit dem Futter oder dem Trinkwasser verabreichten Prüfchemikalien ist unbedingt sicherzustellen, dass die Mengen der jeweiligen Prüfchemikalie die normale Nahrungsaufnahme oder den Wasserhaushalt nicht beeinträchtigen. Wenn die Prüfchemikalie im Futter verabreicht wird, kann entweder eine konstante Konzentration im Futter (ppm) oder eine konstante Dosis, bezogen auf das Körpergewicht des Tieres, verwendet werden; die jeweils gewählte Verfahrensweise sollte angegeben werden. Eine mit einer Magensonde verabreichte Dosis sollte jeweils zu denselben Tageszeiten gegeben und mindestens einmal pro Woche so angepasst werden, dass eine konstante Dosis im Verhältnis zum Körpergewicht aufrechterhalten wird. Wird die kombinierte Prüfung als Vorstudie für eine Langzeitstudie über chronische Toxizität verwendet, sollte in beiden Prüfungen die gleiche Nahrung verabreicht werden.
|
Versuchsplan
|
34.
|
Die Verabreichung sollte für beide Geschlechter mindestens 2 Wochen vor der Paarung beginnen, nachdem sich die Tiere mindestens 5 Tage lang eingewöhnt haben und nachdem die weiblichen Tiere auf einen normalen Östruszykus (in einem 2-wöchigen Zeitraum vor der Behandlung) untersucht wurden. Die Studie sollte so geplant werden, dass mit der Bewertung des Östruszykus bald nach Erreichen der vollständigen Geschlechtsreife begonnen wird. Der Beginn dieser Zeiträume kann je nach Rattenstämmen in den einzelnen Labors leicht variieren und beispielsweise bei Sprague-Dawley-Ratten bei einem Alter von 10 Wochen und bei Wistar-Ratten etwa bei einem Alter von 12 Wochen liegen. Muttertiere sollten an Tag 13 nach der Geburt oder kurz darauf getötet werden. Um eine nächtliche Futterkarenz der Muttertiere vor der Blutentnahme (wenn diese Option bevorzugt wird) zu ermöglichen, müssen Muttertiere und ihre Nachkommen nicht unbedingt am selben Tag getötet werden. Der Tag der Geburt (d. h. der Tag, an dem der Geburtsvorgang abgeschlossen wurde) wird als Tag 0 nach der Geburt bezeichnet. Weibliche Tiere, bei denen keine Besamung festzustellen ist, werden 24-26 Tage nach dem letzten Tag der Paarungszeit getötet. Die Verabreichung wird bei beiden Geschlechtern während der Paarungszeit fortgesetzt. Männliche Tiere werden auch nach der Paarungszeit noch mindestens über den vollständigen Verabreichungszeitraum von 28 Tagen behandelt. Anschließend werden sie getötet oder alternativ weiter gehalten und behandelt, damit eine zweite Paarung erfolgen kann, wenn dies als angemessen betrachtet wird.
|
|
35.
|
Die tägliche Behandlung der weiblichen Muttertiere sollte während der Gravidität sowie mindestens bis zu Tag 13 nach der Geburt bzw. zum Tag vor der Tötung (einschließlich) fortgesetzt werden. Bei Studien, bei denen die Prüfchemikalie durch Inhalation oder dermal verabreicht wird, sollte die Verabreichung mindestens bis zu Tag 19 der Trächtigkeit (einschließlich) fortgesetzt und so bald wie möglich (spätestens an Tag 4 nach der Geburt (PND 4)) wieder aufgenommen werden.
|
|
36.
|
Tiere in einer Satellitengruppe, bei denen Nachfolgebeobachtungen vorgesehen sind, (wenn überhaupt berücksichtigt) werden nicht verpaart. Sie sollten für mindestens weitere 14 Tage nach der ersten vorgesehenen Tötung von Muttertieren ohne Behandlung gehalten werden, um ein verzögertes Auftreten, die Persistenz oder die Reversibilität toxischer Wirkungen festzustellen.
|
|
37.
|
Der Versuchsplan ist in Anlage 2 in einem Diagramm dargestellt.
|
Östruszyklen
|
38.
|
Östruszyklen sollten vor Beginn der Behandlung überwacht werden, damit für die Studie weibliche Tiere mit regelmäßigem Zyklus ausgewählt werden können (Nummer 27). Außerdem sollten ab Beginn des Behandlungszeitraums bis zu Anzeichen für eine Besamung täglich Vaginalabstriche untersucht werden. Wenn Bedenken hinsichtlich akuter Auswirkungen von Stress bestehen, die bei Beginn der Verabreichung die Östruszyklen verändern könnten, können die Versuchstiere in den Labors zwei Wochen behandelt werden, um anschließend die Östruszyklen vor der Paarungszeit über mindestens zwei Wochen und anschließend bis zu Anzeichen für eine Besamung in der Paarungszeit anhand täglicher Vaginalabstriche zu überwachen. Bei der Entnahme vaginaler/zervikaler Zellen ist sorgfältig darauf zu achten, dass die Schleimhaut nicht gereizt wird, damit es nicht zu Pseudograviditäten kommt (8) (9).
|
Verpaarung
|
39.
|
In der Regel sollten bei dieser Studie männliche und weibliche Tiere im Verhältnis 1:1 (ein männliches auf ein weibliches Tier) eingesetzt werden. Ausnahmen können sich gelegentlich ergeben, wenn männliche Tiere sterben. Das weibliche Tier sollte mit demselben männlichen Tier gehalten werden, bis Anzeichen für eine Paarung festgestellt werden bzw. bis zwei Wochen vergangen sind. Jeden Morgen werden die weiblichen Tiere auf Sperma oder Vaginalpfröpfe untersucht. Tag 0 der Trächtigkeit ist der Tag, an dem die Besamung (durch Vaginalpfropf oder Spermaspuren) nachgewiesen werden kann. Bei einer erfolglosen Verpaarung kann gegebenenfalls die erneute Verpaarung weiblicher Tiere mit bewährten männlichen Tieren der gleichen Gruppe erwogen werden.
|
Wurfgröße
|
40.
|
Am PND 4 kann die Größe eines jeden Wurfs angepasst werden, indem überschüssige Jungtiere nach dem Zufallsprinzip aussortiert werden, um je nach normalem Umfang eines Wurfs beim verwendeten Rattenstamm pro Wurf nach Möglichkeit vier oder fünf Jungtiere pro Geschlecht und Wurf zu erhalten. Von zwei der überschüssigen Jungtiere sollten Blutproben entnommen, in Pools zusammengefasst und zur Bestimmung von Serumspiegeln (T4) verwendet werden. Die selektive Eliminierung von Jungtieren, z. B. auf der Grundlage des Körpergewichts oder des anogenitalen Abstands (AGD), wird nicht empfohlen. Wenn es wegen der Anzahl männlicher bzw. weiblicher Jungtiere nicht möglich ist, pro Wurf jeweils vier oder fünf Jungtiere eines jeden Geschlechts zu erhalten, ist auch eine grobe Anpassung (beispielsweise sechs männliche und vier weibliche Tiere) akzeptabel. Wenn die Anzahl der Tiere eines Wurfs die Eliminierungsgrenze (8 oder 10 Jungtiere pro Wurf) unterschreitet, werden keine Jungtiere aussortiert. Wenn die Anzahl der Jungtiere um nur ein Tier über der Eliminierungsgrenze liegt, wird nur ein Jungtier aussortiert und zur Blutentnahme für mögliche Bewertungen der Serumspiegel (T4) verwendet.
|
|
41.
|
Wenn die Größe des Wurfs nicht geändert wird, werden an Tag 4 nach der Geburt zwei Jungtiere pro Wurf human getötet und Blutproben zur Bestimmung der Konzentration der Schilddrüsenhormone im Serum entnommen. Nach Möglichkeit sollten diese zwei Jungtiere je Wurf zwei weibliche Jungtiere sein, damit männliche Jungtiere zur Prüfung der Brustwarzenretention aufgespart werden können, es sei denn, dass nach dem Aussortieren dieser Jungtiere keine weiblichen Tiere zur abschließenden Bewertung mehr verbleiben würden. Es werden keine Jungtiere aussortiert, wenn die Größe des Wurfs dadurch auf weniger als 8 bzw. 10 Jungtiere sinkt (je nach Größe eines normalen Wurfs beim betreffenden Rattenstamm). Wenn die Anzahl der Jungtiere um nur ein Tier über der normalen Größe eines Wurfs liegt, wird nur ein Jungtier aussortiert und zur Blutentnahme für mögliche Bewertungen der Serumspiegel (T4) verwendet.
|
Beobachtungen
|
42.
|
Allgemeine klinische Beobachtungen sollten mindestens einmal täglich, vorzugsweise jeweils zur gleichen Zeit, und unter Berücksichtigung des voraussichtlichen Zeitraums nach der Verabreichung vorgenommen werden, in welchem der Wirkungsgipfel zu erwarten ist. Der Gesundheitszustand der Tiere ist zu dokumentieren. Mindestens zweimal täglich erfolgt eine Beobachtung aller Tiere auf Erkrankungen oder Todesfälle.
|
|
43.
|
Bei allen Muttertieren sollte einmal vor der Exposition (um intraindividuelle Vergleiche zu ermöglichen) und danach mindestens einmal wöchentlich eine eingehende klinische Untersuchung erfolgen. Die Beobachtungen sind außerhalb des Käfigs, in dem die Tiere gehalten werden, in stets gleicher Umgebung und vorzugsweise stets zur gleichen Zeit täglich vorzunehmen. Sie sind sorgfältig zu dokumentieren, am besten nach einer speziell vom Prüflabor entwickelten Bewertungsskala. Durch geeignete Maßnahmen ist sicherzustellen, dass die Prüfbedingungen möglichst konstant bleiben und dass die Beobachtungen vorzugsweise durch Untersucher erfolgen, denen die Behandlung der Tiere nicht bekannt ist. Zu achten ist insbesondere auf Veränderungen an Haut, Fell, Augen, Schleimhäuten, auf Sekrete und Exkrete und auf autonome Aktivitäten (z. B. Tränensekretion, Piloerektion, Pupillengröße, ungewöhnliche Atemmuster). Gang- und Haltungsstörungen, ferner Reaktionen auf die Behandlung der Tiere sowie etwaige klonische oder tonische Bewegungen, Stereotypien (z. B. übermäßiges Putzen, wiederholte Kreisbewegungen), schwere oder verzögerte Geburt oder abnormes Verhalten (z. B. Selbstverstümmelung, Rückwärtsgehen) sollten ebenfalls dokumentiert werden (10).
|
|
44.
|
Einmal während der Untersuchung sollte bei fünf männlichen und fünf weiblichen Tieren, die zufällig aus den Gruppen ausgewählt wurden, die sensorische Reaktivität auf Reize verschiedener Art (z. B. akustische, visuelle und propriozeptive Reize) (8) (9) (11), die Greifkraft (12) und die motorische Aktivität (13) bewertet werden. Weitere Einzelheiten zu den möglichen Untersuchungen finden sich in der Literatur. Allerdings können auch andere als dort genannte Verfahren angewendet werden. Bei männlichen Tieren sollten diese funktionellen Beobachtungen gegen Ende des Verabreichungszeitraums kurz vor der vorgesehenen Tötung und in jedem Fall vor der Blutentnahme zur hämatologischen und klinisch-chemischen Untersuchung erfolgen (Nummern 53-56 einschließlich Fußnote 1). Weibliche Tiere sollten sich bei diesen Funktionsprüfungen in einem physiologisch ähnlichen Zustand befinden und vorzugsweise einmal in der letzten Woche der Laktation (z. B. LD 6-13) kurz vor der vorgesehenen Tötung untersucht werden. Die Zeiträume der Trennung von Muttertieren und Jungtieren sollten möglichst kurz sein.
|
|
45.
|
Die einmalig gegen Ende der Untersuchung vorgenommenen funktionellen Beobachtungen können bei Vorstudien für eine nachfolgende Prüfung auf subchronische Toxizität (90 Tage) und bei Langzeitstudien entfallen. In diesem Fall sollten die funktionellen Beobachtungen im Rahmen dieser Folgestudie vorgenommen werden. Andererseits könnten die Daten über funktionelle Beobachtungen aus der Untersuchung mit wiederholter Verabreichung aber die Wahl der Dosisstufen für eine nachfolgende Prüfung auf subchronische oder langfristige Toxizität erleichtern.
|
|
46.
|
In Ausnahmefällen können funktionelle Beobachtungen auch bei Gruppen entfallen, die so starke sonstige Toxizitätsanzeichen aufweisen, dass die Leistungen in Funktionsprüfungen dadurch signifikant beeinträchtigt würden.
|
|
47.
|
Die Trächtigkeitsdauer wird von Tag 0 der Gravidität an berechnet und sollte vermerkt werden. Jeder Wurf ist so bald wie möglich nach der Geburt zu untersuchen, um die Anzahl und das Geschlecht der Nachkommen, Totgeburten, Lebendgeburten und Kümmerlinge (Jungtiere, die erheblich kleiner sind als Jungtiere einer vergleichbaren Kontrollgruppe) sowie auffallende Anomalien feststellen zu können.
|
|
48.
|
Innerhalb von 24 Stunden nach der Geburt (Tag 0 oder Tag 1 nach der Geburt) und mindestens an den Tagen 4 und 13 nach der Geburt sollten lebende Jungtiere gezählt und nach Geschlechtern unterschieden und die Würfe jeweils gewogen werden. Zusätzlich zu den Beobachtungen bei Elterntieren (Nummern 43 und 44) sollte ungewöhnliches Verhalten der Nachkommen dokumentiert werden.
|
|
49.
|
Der anogenitale Abstand (AGD) sollte bei jedem Jungtier zwischen PND 0 und PND 4 am selben Tag gemessen werden. Das Körpergewicht des Jungtiers wird am Tag der Messung des AGD erfasst, der auf Jungtiergröße — vorzugsweise die Quadratwurzel des Körpergewichts — genormt sein sollte (14). Die Anzahl der Brustwarzen/Warzenhöfe bei männlichen Jungtieren ist wie in OECD GD 151 empfohlen an PND 12 oder 13 zu kontrollieren (15).
|
Körpergewicht und Futter-/Trinkwasserverbrauch
|
50.
|
Männliche und weibliche Tiere der P-Generation müssen am ersten Tag der Behandlung und anschließend mindestens in wöchentlichen Abständen sowie bei Versuchsende gewogen werden. Während der Gravidität sollten weibliche Tiere an den Tagen 0, 7, 14 und 20 sowie innerhalb von 24 Stunden nach der Geburt (Tag 0 oder Tag 1 nach der Geburt) und mindestens an den Tagen 4 und 13 nach der Geburt gewogen werden. Diese Beobachtungen sind für jedes ausgewachsene Tier einzeln zu protokollieren.
|
|
51.
|
Vor der Paarung, vor der Gravidität und vor der Laktation sollte die Futteraufnahme mindestens einmal wöchentlich gemessen werden. Die Messung der Futteraufnahme während der Paarungszeit ist fakultativ. Wenn die Prüfchemikalie mit dem Trinkwasser verabreicht wird, sollte in diesen Zeiträumen auch die Wasseraufnahme gemessen werden.
|
Hämatologie
|
52.
|
Bei fünf männlichen und fünf weiblichen Tieren, die aus jeder Gruppe zufällig ausgewählt wurden, sollten während der Untersuchung einmal die folgenden hämatologischen Parameter bestimmt werden: Hämatokrit, Hämoglobinkonzentration, Erythrozytenzahl, Retikulozyten, Gesamt- und Differential-Leukozytenzahl, Thrombozytenzahl und Blutgerinnungszeit/-fähigkeit. Wenn die Prüfchemikalie oder ihre möglichen Metaboliten oxidierende Eigenschaften haben oder diese vermutet werden, sollten zusätzlich die Methämoglobinkonzentration und die Heinz-Körper bestimmt werden.
|
|
53.
|
Blutproben sollten an einer benannten Stelle entnommen werden. Die weiblichen Tiere sollten sich bei der Entnahme der Proben in einem physiologisch ähnlichen Zustand befinden. Um bei der praktischen Handhabung Schwierigkeiten aufgrund des unterschiedlichen Beginns der Trächtigkeit zu vermeiden, kann die Blutentnahme bei weiblichen Tieren alternativ zur Probenahme unmittelbar vor dem Verfahren zur humanen Tötung der Tiere bzw. im Rahmen des Verfahrens ausschließlich am Ende des Zeitraums vor der Paarung erfolgen. Bei männlichen Tieren sollten die Blutproben vorzugsweise unmittelbar vor dem Verfahren zur humanen Tötung der Tiere bzw. im Rahmen des Verfahrens entnommen werden. Alternativ kann die Blutentnahme bei männlichen Tieren auch am Ende des Zeitraums vor der Paarung erfolgen, wenn dieser Zeitpunkt auch für die weiblichen Tiere angenommen wurde.
|
|
54.
|
Die Blutproben sind unter geeigneten Bedingungen zu lagern.
|
Klinisch-biochemische Untersuchungen
|
55.
|
Klinisch-biochemische Bestimmungen zur Untersuchung der wichtigsten toxischen Wirkungen in Geweben, insbesondere der Wirkungen auf Nieren und Leber, sind an Blutproben durchzuführen, die von den ausgewählten fünf männlichen und fünf weiblichen Tieren je Gruppe entnommen werden. Es empfiehlt sich eine Futterkarenz der Tiere über Nacht, bevor die Blutproben entnommen werden. (22) Die Plasma- oder Serumuntersuchungen sollten die Parameter Natrium, Kalium, Glucose, Gesamtcholesterin, Harnstoff, Kreatinin, Gesamtprotein und Albumin, mindestens zwei Enzyme, die auf hepatozelluläre Wirkungen schließen lassen, (wie Alanin-Aminotransferase, Aspartat-Aminotransferase und Sorbitolhydrogenase) sowie Gallensäuren umfassen. Die Bestimmung weiterer Enzyme (aus der Leber oder anderen Organen) sowie von Bilirubin kann unter bestimmten Umständen ebenfalls wertvolle Hinweise liefern.
|
|
56.
|
Nach dem folgenden Plan werden Blutproben an einer vorgegebenen Stelle entnommen:
|
—
|
von mindestens zwei Jungtieren pro Wurf an Tag 4 nach der Geburt, wenn die Anzahl der Jungtiere dies zulässt (Nummern 40 und 41),
|
|
—
|
von allen Muttertieren und von mindestens zwei Jungtieren pro Wurf bei Beendigung des Versuchs an Tag 13 und
|
|
—
|
von allen adulten männlichen Tieren bei Beendigung des Versuchs.
|
Alle Blutproben werden unter geeigneten Bedingungen gelagert. Bei Blutproben der Jungtiere von Tag 13 sowie bei Blutproben der adulten männlichen Tiere werden die Serumspiegel auf Schilddrüsenhormone (T4) untersucht. Wenn relevant, wird außerdem eine Bewertung von T4 bei Blutproben der Muttertiere sowie bei Jungtieren an Tag 4 vorgenommen. Ebenfalls wenn relevant können auch weitere Hormone gemessen werden. Das Blut der Jungtiere kann zur Analyse der Schilddrüsenhormone nach Würfen zu Pools zusammengefasst werden. Die Schilddrüsenhormone (T4 und TSH) sollten vorzugsweise „insgesamt“ gemessen werden.
|
|
57.
|
Optional können in der letzten Woche der Studie am Urin, der zu festgelegten Zeiten gesammelt wird, bei fünf zufällig ausgewählten männlichen Tieren je Gruppe folgende Parameter untersucht werden: Aussehen, Volumen, Osmolalität oder spezifisches Gewicht, pH-Wert, Eiweiß, Glucose und Blut/Blutzellen.
|
|
58.
|
Darüber hinaus sollten Untersuchungen zur Bestimmung von Serummarkern für eine allgemeine Gewebeschädigung erwogen werden. Des Weiteren sollten, wenn die bekannten Eigenschaften der Prüfchemikalie im Verdacht stehen, die entsprechenden Stoffwechselprofile zu beeinflussen, die Parameter Calcium, Phosphat, Nüchtern-Triglyzeride und -Glucose, spezifische Hormone, Methämoglobin und Cholinesterase bestimmt werden. Die Bestimmung muss auf Einzelfallbasis erfolgen.
|
|
59.
|
Die folgenden Faktoren können die Variabilität und die absoluten Konzentrationen der Hormonbestimmungen beeinflussen:
|
—
|
der Zeitpunkt der Tötung wegen Schwankungen der Hormonkonzentrationen im Tagesverlauf,
|
|
—
|
Methode der Tötung, die gewählt wird, um übermäßigen Stress für die Tiere zu vermeiden, der die Hormonkonzentrationen beeinflussen könnte,
|
|
—
|
Testkits für Hormonbestimmungen mit unterschiedlichen Standardkurven.
|
|
|
60.
|
Plasmaproben, die speziell zur Hormonbestimmung vorgesehen sind, sollten immer zur gleichen Tageszeit gewonnen werden. Die verschiedenen im Handel erhältlichen Assay-Kits können bei der Analyse der Hormonkonzentration unterschiedliche numerische Werte ergeben.
|
|
61.
|
Erweisen sich die vorliegenden Ausgangsdaten als ungeeignet, sollte eine Bestimmung hämatologischer und klinisch-biochemischer Parameter vor Versuchsbeginn oder vorzugsweise an einer nicht in die Versuchsgruppen einbezogenen Gruppe von Tieren in Betracht gezogen werden. Für weibliche Tiere müssen die Daten während der Laktation ermittelt werden.
|
PATHOLOGIE
Autopsie
|
62.
|
Alle Versuchstiere sollten einer vollständigen, eingehenden Autopsie unterzogen werden. Sie umfasst eine sorgfältige Untersuchung der äußeren Körperoberfläche, aller Körperöffnungen sowie der Hirn-, Brust- und Bauchhöhle und ihres Inhalts. Dabei ist besonders auf die Organe des Fortpflanzungssystems zu achten. Die Anzahl der Implantationsstellen sollte vermerkt werden. Am Tag der Sektion ist ein Vaginalabstrich zu untersuchen, um das Stadium des Östruszyklus zu bestimmen und eine Korrelation zur histopathologischen Untersuchung der weiblichen Fortpflanzungsorgane zu ermöglichen.
|
|
63.
|
Die Hoden und die Nebenhoden sowie die Prostata und die Samenbläschen zusammen mit den Koagulationsdrüsen aller adulten männlichen Tiere sollten ggf. von anhaftendem Gewebe befreit und möglichst umgehend nach der Sektion gewogen werden, bevor das Material eintrocknet. Als Organgewichte können ferner die Gewichte des Muskelkomplex Levator ani/bulbospongiosus, der Cowperschen Drüsen und der Glans penis bei männlichen Tieren sowie der Ovarien (Nassgewicht) und des Uterus mit Zervix bei weiblichen Tieren gemessen werden; wenn diese Gewichte berücksichtigt werden sollen, sollten die Messungen möglichst umgehend nach der Sektion vorgenommen werden. Von allen adulten Tieren sollten die Ovarien, Hoden, Nebenhoden, akzessorischen Geschlechtsorgane und alle Organe aufbewahrt werden, die makroskopische Veränderungen aufweisen.
|
|
64.
|
Von allen adulten männlichen und weiblichen Tieren sowie von einem männlichen und einem weiblichen Jungtier an Tag 13 je Wurf sollten die Schilddrüsen in dem am besten geeigneten Fixierungsmittel für die vorgesehene histopathologische Untersuchung aufbewahrt werden. Das Gewicht der Schilddrüse kann nach der Fixierung bestimmt werden. Anhaftendes Gewebe ist sehr vorsichtig und erst nach der Fixierung zu entfernen, um Gewebeschäden zu vermeiden. Eine Gewebeschädigung könnte die histopathologische Analyse beeinträchtigen. Die Blutproben müssen an einer benannten Stelle unmittelbar vor oder bei der Tötung der Tiere entnommen und fachgerecht gelagert werden (Nummer 56).
|
|
65.
|
Außerdem sollten bei mindestens fünf zufällig ausgewählten adulten männlichen und weiblichen Tieren je Gruppe (außer bei moribunden Tieren und/oder bei vor Ende der Untersuchung human getöteten Tieren) Leber, Nieren, Nebennieren, Thymus, Milz, Hirn und Herz von anhaftendem Gewebe befreit und möglichst umgehend nach der Sektion gewogen werden, bevor das Material eintrocknet. Die folgenden Gewebe sind in dem für die betreffende Gewebeart und die vorgesehene anschließende histopathologische Untersuchung am besten geeigneten Fixierungsmittel aufzubewahren: alle Gewebe mit makroskopischen Veränderungen, Hirn (typische Regionen einschließlich Cerebrum, Cerebellum und Pons), Rückenmark, Auge, Magen, Dünn- und Dickdarm (mit Peyer’schen-Platten), Leber, Nieren, Nebennieren, Milz, Herz, Thymus, Trachea und Lungen (konserviert durch Inflation mit Fixierungsmittel und Immersion), Gonaden (Hoden und Ovarien), akzessorische Geschlechtsorgane (Uterus und Zervix, Nebenhoden, Prostata und Samenbläschen mit Koagulationsdrüsen), Vagina, Harnblase, Lymphknoten (je nach Erfahrung des Labors sollte neben dem proximalsten drainierenden Lymphknoten ein weiterer Lymphknoten entnommen werden (16)), periphere Nerven (N. ischiadicus oder N. tibialis) vorzugsweise in der Nähe des Muskels, Skelettmuskel und Knochen mit Knochenmark (Schnitt oder wahlweise ein frisch fixiertes Knochenmarkaspirat). Es wird empfohlen, die Hoden durch Immersion in Bouin’scher Lösung oder modifizierter Davidson-Lösung zu fixieren (16) (17) (18). Eine Fixation in Formalin ist für diese Gewebe nicht zu empfehlen. Um ein rasches Eindringen des Fixierungsmittels zu ermöglichen, kann die Tunica albuginea an beiden Enden des Organs vorsichtig und flach mit einer Nadel punktiert werden. Die klinischen und sonstigen Befunde können weitere Gewebeuntersuchungen erforderlich machen. Auch Organe, die aufgrund der bekannten Eigenschaften des Prüfstoffs als mögliche Zielorgane in Frage kommen, sollten aufbewahrt werden.
|
|
66.
|
Die folgenden Gewebe können wichtige Hinweise auf endokrine Wirkungen geben: Gonaden (Ovarien und Hoden), akzessorische Geschlechtsorgane (Uterus mit Zervix, Nebenhoden, Samenbläschen mit Koagulationsdrüsen, Prostata dorsolateral und ventral), Vagina, Hypophose, männliche Brustdrüse und Nebennieren. Veränderungen der männlichen Brustdrüsen sind nicht ausreichend dokumentiert, bei diesem Parameter können aber sehr empfindliche Reaktionen auf Stoffe mit östrogener Wirkung vorkommen. Die Beobachtung von nicht unter Nummer 65 genannten Organen/Geweben ist fakultativ.
|
|
67.
|
Tote Jungtiere und Jungtiere, die an Tag 13 nach der Geburt oder kurz danach getötet wurden, sollten zumindest äußerlich sorgfältig auf auffallende Anomalien untersucht werden. Besondere Beachtung erfordern die externen Genitalien, die auf Anzeichen für eine veränderte Entwicklung zu untersuchen sind.
|
Histopathologie
|
68.
|
Die konservierten Organe und Gewebe der ausgewählten Tiere der Kontrollgruppe und der Hochdosisgruppe (unter besonderer Berücksichtigung der Spermatogenese in den männlichen Keimdrüsen und der Histopathologie der interstitiellen testikulären Zellstruktur) sollten einer vollständigen histopathologischen Untersuchung unterzogen werden. Die Schilddrüsen von Jungtieren und von den übrigen adulten Tieren können erforderlichenfalls untersucht werden. Diese Untersuchungen sind auch auf die Tiere anderer Dosisgruppen auszudehnen, wenn behandlungsbedingte Veränderungen in der Gruppe mit der höchsten Dosis festgestellt werden. Der Leitfaden zur Histopathologie (10) enthält zusätzliche Informationen zur Sektion, Fixierung, Schnittherstellung und Histopathologie endokriner Gewebe.
|
|
69.
|
Alle makroskopischen Läsionen sind zu untersuchen. Um die Ermittlung der Konzentration ohne beobachtete schädliche Wirkungen (NOAEL) zu erleichtern, sollten Zielorgane in anderen Dosisgruppen untersucht werden, insbesondere in Gruppen, für die niedrige NOAEL-Werte angegeben wurden.
|
|
70.
|
Umfasst eine Prüfung auch eine Satellitengruppe, sind bei den Tieren dieser Gruppe die Gewebe und Organe histopathologisch zu untersuchen, bei denen in den Behandlungsgruppen Wirkungen aufgetreten sind.
|
DATEN UND BERICHTERSTATTUNG
Daten
|
71.
|
Es sollten Daten zu den einzelnen Tieren bereitgestellt werden. Außerdem sollten sämtliche Daten in tabellarischer Form zusammengefasst werden; dabei werden für jede Prüfgruppe die Anzahl der Tiere zu Beginn der Prüfung, die Anzahl der während der Prüfung tot aufgefundenen Tiere beziehungsweise der aus humanen Gründen getöteten Tiere, der jeweilige Zeitpunkt des Todes beziehungsweise der Tötung, die Anzahl der fruchtbaren Tiere, die Anzahl der trächtigen weiblichen Tiere, die Anzahl der Tiere mit Anzeichen von Toxizität, eine Beschreibung der beobachteten Anzeichen von Toxizität, einschließlich des Zeitpunkts, zu dem die toxischen Wirkungen eingetreten sind, deren Dauer und Schweregrad, die Arten von histopathologischen Veränderungen und alle relevanten Daten zu den Würfen angegeben. Anlage 3 enthält ein tabellarisches Berichtsformat, das sich als sehr hilfreich für die Evaluierung der reproduktions- bzw. entwicklungstoxischen Wirkungen erwiesen hat.
|
|
72.
|
Wenn möglich, sollten die numerischen Ergebnisse mittels einer geeigneten und allgemein anerkannten statistischen Methode ausgewertet werden. Durch Vergleiche der Wirkungen in einem Dosisbereich sollte die Anwendung multipler t-Tests vermieden werden. Die statistischen Methoden sollten bei der Planung der Studie festgelegt werden. Statistische Analysen des AGD und der Brustwarzenretention sollten auf den Daten einzelner Jungtiere unter Berücksichtigung der Wurfgröße beruhen. Gegebenenfalls sind die jeweiligen Würfe als Analyseeinheit anzunehmen. Statistische Analysen des Körpergewichts der Jungtiere sollten auf den Daten einzelner Jungtiere unter Berücksichtigung der Wurfgröße beruhen. Wegen des begrenzten Umfangs der Studie sind statistische Analysen in Form von „Signifikanzprüfungen“ für viele Endpunkte, insbesondere für reproduktionsbezogene Endpunkte, nur von begrenzter Bedeutung. Einige der am weitesten verbreiteten Methoden, besonders parametrische Untersuchungen anhand von Maßen der zentralen Tendenz, sind nicht geeignet. Wenn statistische Analysen durchgeführt werden, sollte die gewählte Methode der Verteilung der zu untersuchenden Variable angemessen sein und vor Beginn der Studie festgelegt werden.
|
Auswertung der Ergebnisse
|
73.
|
Die Befunde dieser Toxizitätsstudie sind im Hinblick auf die beobachteten Wirkungen sowie auf Nekropsie- und Mikroskopiebefunde zu bewerten. Die Bewertung beinhaltet den vorhandenen beziehungsweise nicht vorhandenen Zusammenhang zwischen der Dosierung der Prüfchemikalie und vorhandenen Anomalien sowie deren Häufigkeit und Schwere, einschließlich makroskopischer Läsionen, identifizierter Zielorgane, Unfruchtbarkeit, klinischer Anomalien, beeinträchtigter Reproduktions- und Wurfleistungen, Körpergewichtsveränderungen, Auswirkungen auf die Mortalität und etwaiger sonstiger toxischer Auswirkungen.
|
|
74.
|
Wegen der kurzen Dauer der Behandlung der männlichen Tiere sollte bei der Bewertung der reproduktionstoxischen Wirkungen bei männlichen Tieren die Histopathologie der Hoden und der Nebenhoden zusammen mit den Fertilitätsdaten berücksichtigt werden. Die Einbeziehung verfügbarer historischer Kontrolldaten zur Reproduktions-/Entwicklungstoxizität (z. B. für Wurfgröße, AGD, Brustwarzenretention und Serumspiegel (T4)) kann bei der Auswertung der Studie ebenfalls hilfreich sein.
|
|
75.
|
Für die Qualitätskontrolle wird vorgeschlagen, historische Kontrolldaten zu sammeln und Variationskoeffizienten für numerische Daten zu berechnen, insbesondere für die Parameter, die mit dem Nachweis der Störung des endokrinen Systems zusammenhängen. Diese Daten können für Vergleichszwecke verwendet werden, wenn tatsächliche Studien bewertet werden.
|
Prüfbericht
|
76.
|
Der Prüfbericht sollte folgende Angaben enthalten:
|
|
Prüfchemikalie:
|
—
|
Herkunft, Partienummer, ggf. begrenztes Verwendungsdatum;
|
|
—
|
Stabilität der Prüfchemikalie, falls bekannt.
|
|
|
|
Einkomponentiger Stoff:
|
—
|
physikalisches Erscheinungsbild, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
|
|
—
|
chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer, SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.
|
|
|
|
Mehrkomponentiger Stoff, UVCB-Stoffe und Gemische:
|
—
|
möglichst weitgehende Charakterisierung anhand der chemischen Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Komponenten.
|
|
|
|
Vehikel (wenn verwendet):
|
—
|
Begründung der Auswahl des Vehikels, falls kein Wasser verwendet wurde.
|
|
|
|
Versuchstiere:
|
—
|
Anzahl, Alter und Geschlecht der Tiere;
|
|
—
|
Herkunft der Tiere, Haltungsbedingungen, Futter usw.;
|
|
—
|
Gewicht der einzelnen Tiere bei Versuchsbeginn;
|
|
—
|
Begründung für die Wahl einer anderen Art als der Ratte.
|
|
|
|
Prüfbedingungen:
|
—
|
Begründung der gewählten Dosisstufen;
|
|
—
|
Angaben zur Zubereitungsform des Prüfstoffs/des Futters, der erreichten Konzentration, Stabilität und Homogenität der Zubereitung;
|
|
—
|
Angaben zur Verabreichung der Prüfchemikalie;
|
|
—
|
gegebenenfalls Angaben zur Umrechnung der Konzentration des Prüfstoffs im Futter/Wasser (ppm) in die entsprechende Dosis (mg/kg Körpergewicht/Tag);
|
|
—
|
Angaben über Futter- und Wasserqualität;
|
|
—
|
genaue Beschreibung des Verfahrens für die Zufallsauswahl von Jungtieren zwecks Tötung (wenn Jungtiere getötet werden).
|
|
|
|
Ergebnisse
|
—
|
Körpergewicht/Änderungen des Körpergewichts;
|
|
—
|
gegebenenfalls Angaben zur Futter- und Wasseraufnahme;
|
|
—
|
Daten über toxische Reaktionen nach Geschlecht und Dosierung, einschließlich Fruchtbarkeit, Trächtigkeit und sonstige Anzeichen von Toxizität;
|
|
—
|
toxische oder sonstige Wirkungen auf Reproduktion, Nachkommen, postnatales Wachstum usw.;
|
|
—
|
Art, Schweregrad und Dauer der klinischen Beobachtungen (mit Angaben zur Reversibilität);
|
|
—
|
Bewertung von sensorischer Aktivität, Greifkraft und motorischer Aktivität;
|
|
—
|
hämatologische Parameter mit entsprechenden Ausgangswerten;
|
|
—
|
klinisch-biochemische Parameter mit entsprechenden Ausgangswerten;
|
|
—
|
Zahl adulter weiblicher Tiere mit normalem oder abnormalem Östruszyklus sowie Zyklusdauer;
|
|
—
|
Anzahl der Lebendgeburten und der Postimplantationsverluste;
|
|
—
|
Anzahl der Jungtiere mit sichtbaren erheblichen Anomalien, allgemeine Bewertung externer Genitalien, Anzahl der Kümmerlinge;
|
|
—
|
Zeitpunkt des Todes im Verlauf der Studie oder Angabe, ob Tiere bis zum Schluss überlebt haben;
|
|
—
|
Anzahl der Implantate, Wurfgröße und Wurfgewichte zum Zeitpunkt der Aufzeichnung;
|
|
—
|
Angaben zum Körpergewicht der Jungtiere;
|
|
—
|
AGD aller Jungtiere (und Körpergewicht am Tag der AGD-Messung);
|
|
—
|
Brustwarzenretention bei männlichen Jungtieren;
|
|
—
|
Schilddrüsenhormonspiegel, Jungtiere an Tag 13 und adulte männliche Tiere (sowie Muttertiere und Jungtiere an Tag 4, wenn gemessen);
|
|
—
|
Körpergewicht bei Tötung sowie Organgewichtsdaten für die Elterntiere;
|
|
—
|
ausführliche Beschreibung aller histopathologischen Befunde;
|
|
—
|
Absorptionsdaten, soweit vorhanden;
|
|
—
|
statistische Auswertung der Ergebnisse, wenn möglich.
|
|
|
|
Erörterung der Ergebnisse.
|
|
Auswertung der Ergebnisse
|
77.
|
Mit der Studie wird die Reproduktions-/Entwicklungstoxizität bei wiederholter Verabreichung bewertet. Insbesondere da der Schwerpunkt sowohl auf die allgemeine Toxizität als auch auf reproduktions-/entwicklungstoxische Endpunkte gelegt wird, ermöglichen die Prüfungsergebnisse eine Unterscheidung zwischen den Wirkungen auf die reproduktions-/entwicklungstoxische Wirkung, die ohne allgemeine Toxizität auftreten, und Veränderungen, die nur bei Dosen vorkommen, die auch bei Elterntieren toxisch wirken (Nummern 7-11). Die Untersuchung könnte Aufschluss über die Notwendigkeit der Durchführung weiterer Untersuchungen geben und zu Empfehlungen für die Gestaltung von Folgestudien führen. Als Hilfe bei der Auswertung der reproduktions- und entwicklungstoxischen Ergebnisse ist OECD Guidance Document Nr. 43 zu Rate zu ziehen (19). OECD Guidance Document Nr. 106 über die histologische Auswertung von Prüfungen zur Feststellung endokriner und reproduktionstoxischer Wirkungen bei Nagern (16) enthält Informationen zur Vorbereitung und Auswertung von (endokrinen) Organen und von Vaginalabstrichen, die für diese Prüfmethode hilfreich sein könnten.
|
LITERATUR
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Auf Anfrage erhältlich bei der Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokio, 27.-29. Oktober 1992. Auf Anfrage erhältlich bei der Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(3)
|
Mitsumori, K., Kodama, Y., Uchida, O., Takada, K., Saito, M. Naito, K., Tanaka, S., Kurokawa, Y., Usami, M., Kawashima, K., Yasuhara, K., Toyoda, K., Onodera, H., Furukawa, F., Takahashi, M. und Hayashi, Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol. Sci., 19, 141-149.
|
|
(4)
|
Tanaka, S., Kawashima, K., Naito, K., Usami, M., Nakadate, M., Imaida, K., Takahashi, M., Hayashi, Y., Kurokawa, Y. und Tobe, M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10. und 11. März 1998. Auf Anfrage erhältlich bei der Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(8)
|
Goldman, J.M., Murr, A.S., Buckalew, A.R., Ferrell, J.M. und Cooper, R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), S. 84-97.
|
|
(9)
|
Sadleir, R.M.F.S. (1979). Cycles and Seasons, in Auston, C.R. und Short, R.V. (Hrsg.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No. 60).
|
|
(11)
|
Moser, V.C., McDaniel, K.M. und Phillips, P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Toxic. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer, O.A., Tilson, H.A., Byrd, W.C. und Riley, M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rates and Mice. A Method for the Routine Assessment of Fore- and Hind- limb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton, K.M., Haward, J.L., Moser, V.C., M.W., Reiter, L.W., Tilson, H.A. und MacPhail, R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan, R.H. Jr., J.F. Holson, D.G. Stump, J.F. Knapp und V.L. Reynolds. (1999). „Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights“, Reproductive Toxicology, 13: S. 383-390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(16)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment, (No. 106), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(17)
|
Hess, R.A. und Moore, B.J. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin, R.E. und Heindel, J.J. (Hrsg.). Academic Press: San Diego, CA, S. 52-85.
|
|
(18)
|
Latendresse, J.R., Warbrittion, A.R., Jonassen, H., Creasy, D.M. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
Anlage 1
BEGRIFFSBESTIMMUNGEN (Siehe Auch (20) Oecd Gd 150)
Androgene Wirkung Die Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches androgenes Hormon zu wirken (z. B. Testosteron).
Antiandrogene Wirkung Die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen androgenen Hormons (z. B. Testosteron) in einem Säugetierorganismus zu unterdrücken.
Antiöstrogene Wirkung Die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen östrogenen Hormons (z. B. 17ß-Estradiol) in einem Säugetierorganismus zu unterdrücken.
Antithyroide Wirkung Die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen Schilddrüsenhormons (z. B. T3) in einem Säugetierorganismus zu unterdrücken.
Chemikalie Ein Stoff oder Gemisch.
Entwicklungstoxizität Das Auftreten einer reproduktionstoxischen Wirkung mit prä-, peri- und postnatalen, strukturellen oder funktionellen Störungen bei der Nachkommenschaft.
Dosis Die Menge der verabreichten Prüfchemikalie. Die Dosis wird ausgedrückt als Masse der Prüfchemikalie je Einheit Körpergewicht des Versuchstiers pro Tag (z. B. mg/kg Körpergewicht/Tag) oder als konstante Konzentration im Futter.
Dosierung Ein allgemeiner Begriff, der die Dosis, ihre Häufigkeit und die Dauer der Verabreichung umfasst.
Offensichtliche Toxizität Ein allgemeiner Begriff zur Beschreibung deutlicher Toxizitätszeichen nach Verabreichung einer Prüfchemikalie. Diese Zeichen sollten für eine Bewertung der Gefährdung ausreichen und so schwerwiegend sein, dass bei einer Steigerung der verabreichten Dosis die Entwicklung schwerer Toxizitätszeichen und der wahrscheinliche Tod zu erwarten wären.
Fertilitätsströungen Störungen der männlichen oder weiblichen Reproduktionsfunktionen oder -fähigkeit.
Maternale Toxizität Schädliche Wirkungen auf trächtige weibliche Tiere, die entweder spezifisch (direkte Wirkungen) oder nicht spezifisch (indirekte Wirkungen) auftreten und mit dem Zustand der Gravidität in Zusammenhang stehen.
NOAEL Abkürzung für No Observed Adverse Effect Level. Dies ist die höchste Dosis, bei der keine schädigenden behandlungsbedingten Wirkungen festgestellt werden.
Östrogene Wirkung Die Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches östrogenes Hormon zu wirken (z. B. 17ß-Estradiol).
Reproduktionstoxizität Schädliche Wirkungen auf die Nachkommenschaft und/oder Beeinträchtigung der männlichen und weiblichen Reproduktionsfunktionen oder -kapazität.
Prüfchemikalie Ein beliebiger Stoff oder ein beliebiges Gemisch, der/das nach dieser Methode geprüft wird.
Thyroide Wirkung Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches Schilddrüsenhormon (z. B. T3) zu wirken.
Validierung Ein wissenschaftlicher Prozess zur Beschreibung der operationellen Anforderungen und Grenzen einer Prüfmethode und zum Nachweis ihrer Zuverlässigkeit und Eignung für einen bestimmten Zweck.
Anlage 2
DIAGRAMM ZUM VERSUCHSPLAN MIT ANGABEN ZUR MAXIMALEN DAUER DER STUDIE AUSGEHEND VON DER VOLLSTÄNDIGEN 14-TÄGIGEN PAARUNGSZEIT

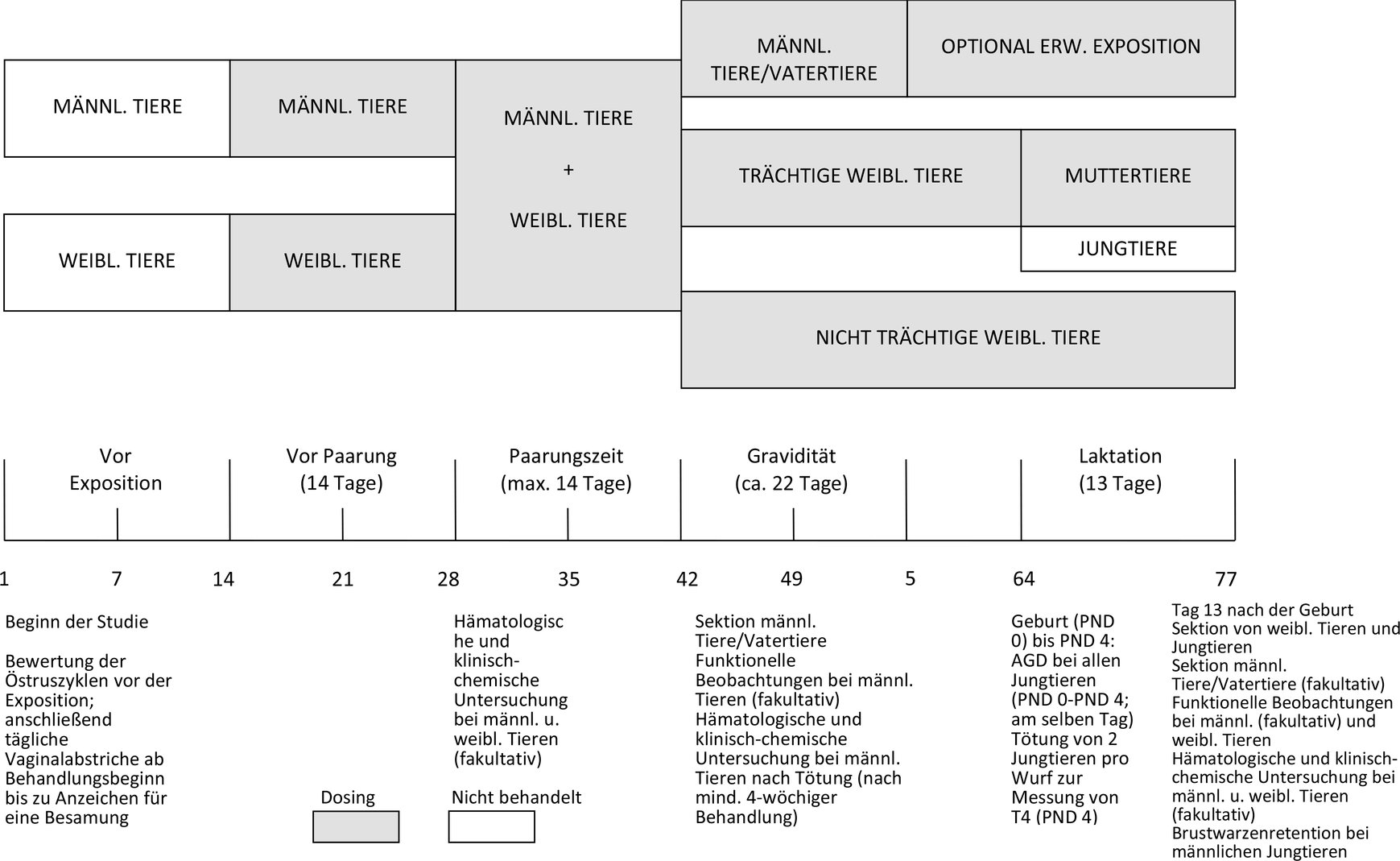
Anlage 3
TABELLARISCHE ZUSAMMENFASSUNG DER AUSWIRKUNGEN AUF DIE REPRODUKTION/ENTWICKLUNG
|
BEOBACHTUNGEN
|
WERTE
|
|
Dosierung (Einheiten).
|
0 (Kontrolle)
|
…
|
…
|
…
|
…
|
|
Ausgangspaarungen (N)
|
|
|
|
|
|
|
Östruszyklus (mindestens mittlere Dauer und Häufigkeit unregelmäßiger Zyklen)
|
|
|
|
|
|
|
Weibliche Tiere mit Anzeichen für eine Besamung (N)
|
|
|
|
|
|
|
Trächtige weibliche Tiere (N)
|
|
|
|
|
|
|
Empfängnistage 1-5 (N)
|
|
|
|
|
|
|
Empfängnistage 6- (N)..([1]
(23)
) (N)
|
|
|
|
|
|
|
Gravidität ≤ 21 Tage (N)
|
|
|
|
|
|
|
Gravidität = 22 Tage (N)
|
|
|
|
|
|
|
Gravidität ≥ 23 Tage (N)
|
|
|
|
|
|
|
Muttertiere mit lebenden Jungtieren (N)
|
|
|
|
|
|
|
Muttertiere mit lebenden Jungtieren an Tag 4 pp (N)
|
|
|
|
|
|
|
Implantate/Muttertier (Mittelwert)
|
|
|
|
|
|
|
Lebende Jungtiere/Muttertiere bei Geburt (Mittelwert)
|
|
|
|
|
|
|
Lebende Jungtiere/Muttertiere an Tag 4 (Mittelwert)
|
|
|
|
|
|
|
Geschlechterverhältnis (m/w) bei Geburt (Mittelwert)
|
|
|
|
|
|
|
Geschlechterverhältnis (m/w) an Tag 4 (Mittelwert)
|
|
|
|
|
|
|
Wurfgewicht bei Geburt (Mittelwert)
|
|
|
|
|
|
|
Wurfgewicht an Tag 4 (Mittelwert)
|
|
|
|
|
|
|
Gewicht der Jungtiere bei Geburt (Mittelwert)
|
|
|
|
|
|
|
Gewicht der Jungtiere zum Zeitpunkt der AGD-Messung (Mittelwert männliche Jungtiere, Mittelwert weibliche Jungtiere)
|
|
|
|
|
|
|
AGD der Jungtiere am selben Tag nach der Geburt, Geburt – Tag 4 (Mittelwert männliche Jungtiere, Mittelwert weibliche Jungtiere, PND vermerken)
|
|
|
|
|
|
|
Gewicht der Jungtiere an Tag 4 (Mittelwert)
|
|
|
|
|
|
|
Gewicht der Jungtiere an Tag 13 (Mittelwert)
|
|
|
|
|
|
|
Brustwarzenretention bei männlichen Jungtieren an Tag 13 (Mittelwert)
|
|
|
|
|
|
|
JUNGTIERE MIT ANOMALIEN
|
|
Muttertiere mit 0
|
|
|
|
|
|
|
Muttertiere mit 1
|
|
|
|
|
|
|
Muttertiere mit ≥ 2
|
|
|
|
|
|
|
VERLUST AN NACHKOMMEN
|
|
Pränatal (Implantationen abzüglich Lebendgeburten)
|
|
Weibliche Tiere mit 0
|
|
|
|
|
|
|
Weibliche Tiere mit 1
|
|
|
|
|
|
|
Weibliche Tiere mit 2
|
|
|
|
|
|
|
Weibliche Tiere mit ≥ 3
|
|
|
|
|
|
|
Postnatal (Lebendgeburten abzüglich lebender Tiere an Tag 13 nach der Geburt)
|
|
Weibliche Tiere mit 0
|
|
|
|
|
|
|
Weibliche Tiere mit 1
|
|
|
|
|
|
|
Weibliche Tiere mit 2
|
|
|
|
|
|
|
Weibliche Tiere mit ≥ 3
|
|
|
|
|
|
B.65
IN-ITRO-PRÜFMETHODE ZUR UNTERSUCHUNG DER MEMBRAN-BARRIERE AUF HAUTVERÄTZUNGEN
EINLEITUNG
|
1.
|
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 435 (2015). Als Hautverätzung wird das Auslösen einer irreversiblen Hautschädigung, d. h. einer sichtbaren, bis in das Corium reichenden Nekrose der Epidermis nach Applikation einer Prüfchemikalie im Sinne der Definition des Global harmonisierten Systems zur Einstufung und Kennzeichnung von Gefahrstoffen der UN (GHS) (1) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP-Verordnung) (24)] bezeichnet. Diese Prüfmethode entspricht der aktualisierten OECD Prüfrichtlinie 435 und besteht in einer In-vitro-Prüfmethode zur Untersuchung der Membranbarriere, mit der ätzende Chemikalien ermittelt werden können. Bei dieser Prüfmethode wird eine künstliche Membran verwendet, die in situ auf ätzende Chemikalien ähnlich reagiert wie Tierhaut.
|
|
2.
|
Hautverätzungen wurden gewöhnlich durch Auftragen der Prüfchemikalie auf die Haut lebender Tiere und durch anschließende Bewertung des Umfangs von Gewebeschäden nach einem bestimmten Zeitraum bewertet (2). Außer dieser Prüfmethode wurden alternativ (3) (4) zum In-vivo-Standardverfahren zur Untersuchung von Kaninchenhaut (Kapitel B.4, entsprechend OECD TG 404) noch mehrere weitere In-vitro-Prüfmethoden zur Ermittlung ätzender Chemikalien (2) angenommen. In der abgestuften Prüf- und Bewertungsstrategie nach dem UN GHS zur Bewertung und Beurteilung von Hautverätzungen und im OECD-Leitliniendokument über integrierte Prüfungs- und Bewertungsansätze (IATA = Integrated Approaches to Testing and Assessment) zur Untersuchung von Hautverätzungen und -reizungen wird die Verwendung validierter und akzeptierter In-vitro-Prüfmethoden in den Modulen 3 und 4 beschrieben (1) (5). Die IATA beschreiben mehrere Module, in denen Informationsquellen und Analyseinstrumente zu Gruppen zusammengefasst werden. Sie enthalten i) Leitlinien dazu, wie vorhandene Daten aus Versuchen und aus anderen Quellen integriert und verwendet werden können, um das Hautreizungs- und das Hautverätzungspotenzial von Chemikalien zu bewerten, und ii) einen Vorschlag zur Durchführung ggf. erforderlicher weiterer Versuche (u. a. bei negativen Ergebnissen) (5). Bei diesem modularen Ansatz können Chemikalien aufgrund von positiven Ergebnissen, die in In-vitro-Prüfmethoden ermittelt wurden, als ätzend eingestuft werden, ohne Tierversuche durchführen zu müssen; dadurch wird die Verwendung von Tieren reduziert und optimiert, um Schmerzen und Qualen zu vermeiden, denen für diesen Zweck zu verwendende Tiere ausgesetzt würden.
|
|
3.
|
Das im Handel unter der Bezeichnung Corrositex® erhältliche In-vitro-Membran-Barrieremodell (6) (7) (8) wurde in Validierungsstudien untersucht; dabei wurden für eine Datenbank mit 163 Stoffen und Gemischen eine Gesamtgenauigkeit zur Vorhersage von Hautverätzungen von 79 % (128/163), eine Empfindlichkeit von 85 % (76/89) und eine Spezifizität von 70 % (52/74) ermittelt (7). Aufgrund ihrer anerkannten Validität wurde diese validierte Referenzmethode (VRM) zur Verwendung im Rahmen einer abgestuften Prüfstrategie zur Bewertung des hautätzenden Gefährdungspotenzials von Chemikalien empfohlen (5) (7). Bevor ein In-vitro-Membran-Barrieremodell zur Untersuchung auf Hautverätzungen für regulatorische Zwecke verwendet werden kann, sollten nach den Anforderungen der Leistungsstandards (10) Verlässlichkeit, Relevanz (Genauigkeit) und die Grenzen des Verfahrens für den vorgeschlagenen Verwendungszweck bestimmt werden, um sicherzustellen, dass es nach den vordefinierten Leistungsstandards (PS) mit der VRM vergleichbar ist (9). Die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen wird erst dann garantiert, wenn vorgeschlagene neue oder aktualisierte Methoden auf der Grundlage der Leistungsstandards überprüft und in die gleichwertige OECD-Prüfrichtlinie aufgenommen wurden. Gegenwärtig ist das im Handel unter der Bezeichnung Corrositex® angebotene Modell die einzige In-vitro-Methode, die der OECD-Prüfrichtlinie 435 entspricht.
|
|
4.
|
Andere Prüfmethoden zur Untersuchung auf Hautverätzungen beruhen auf der Verwendung rekonstituierter menschlicher Haut (OECD TG 431) (3) und isolierter Rattenhaut (OECD TG 430) (4). Diese Prüfrichtlinie sieht die Möglichkeit der Einstufung ätzender Chemikalien nach ihrer Verätzungswirkung in drei UN-GHS-Unterkategorien und nach der Verätzungsgefahr in drei UN-Verpackungsgruppen vor. Die OECD-Prüfrichtlinie wurde ursprünglich im Jahr 2006 angenommen und 2015 unter Berücksichtigung des IATA-Leitliniendokuments sowie zur Anpassung der Liste der Leistungsstoffe aktualisiert.
|
BEGRIFFSBESTIMMUNGEN
|
5.
|
Es gelten die Begriffsbestimmungen der Anlage.
|
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
|
6.
|
Die in dieser Prüfmethode beschriebene Untersuchung ermöglicht die Ermittlung ätzender Prüfchemikalien und die Unterkategorisierung ätzender Prüfchemikalien nach dem UN GHS bzw. der CLP-Verordnung (Tabelle 1). Außerdem kann eine solche Prüfmethode als Entscheidungsgrundlage für die Beurteilung einer ätzenden bzw. nicht ätzenden Wirkung spezifischer Chemikalienklassen (z. B. organischer und anorganischer Säuren, Säurederivate (25) und Basen für die Zwecke der Untersuchung bestimmter Transportwirkungen) dienen (7) (11) (12). Diese Prüfmethode beschreibt ein allgemeines Verfahren ähnlich der validierten Referenzprüfmethode (7). Sie liefert keine geeigneten Informationen über Hautreizungen; mit PM B.46 (gleichwertig OECD TG 439) werden jedoch gezielt die gesundheitlichen Auswirkungen von Hautreizungen in vitro untersucht (13). Eine umfassende Evaluierung lokaler Auswirkungen auf die Haut nach einer einmaligen dermalen Exposition ist dem OECD-Leitliniendokument über integrierte Prüfungs- und Bewertungsansätze (IATA) zu entnehmen (5).
Tabelle 1
Die Hautverätzungskategorien und -unterkategorien des UN GHS (1) (26)
|
Verätzungskategorie (Kategorie 1) (für Behörden, die keine Unterkategorien verwenden)
|
Potenzielle Unterkategorien von Verätzungswirkungen (26) (für Behörden, die Unterkategorien (u. a. die der CLP-Verordnung) verwenden)
|
Hautätzend bei ≥ 1 von 3 Tieren
|
|
Exposition
|
Beobachtung
|
|
Hautätzend
|
Hautätzend Unterkategorie 1A
|
≤ 3 Minuten
|
≤ 1 Stunde
|
|
Hautätzend Unterkategorie 1B
|
> 3 Minuten / ≤ 1 Stunde
|
≤ 14 Tage
|
|
Hautätzend Unterkategorie 1C
|
> 1 Stunde / ≤ 4 Stunden
|
≤ 14 Tage
|
|
|
7.
|
Eine Begrenzung der validierten Referenzmethode (7) besteht darin, dass viele nicht ätzende Chemikalien und einige ätzende Chemikalien aufgrund der Ergebnisse der ursprünglichen Kompatibilitätsprüfung für eine Untersuchung möglicherweise nicht in Betracht kommen (Nummer 13). Wässerige Chemikalien mit pH-Werten im Bereich von 4,5 bis 8,5 können häufig nicht untersucht werden; allerdings erwiesen sich 85 % der in diesem pH-Bereich untersuchten Chemikalien in Tierversuchen als nicht ätzend (7). Die Prüfmethode mit der In-vitro-Membranbarriere kann zur Untersuchung von (in Wasser löslichen oder nicht löslichen) Feststoffen, (wässerigen oder nicht wässerigen) Flüssigkeiten und Emulsionen verwendet werden. Prüfchemikalien, die bei der Kompatibilitätsprüfung jedoch keine nachweisbare Änderung bewirken (d. h. keine farbliche Veränderung im CDS (Chemical Detection System) der validierten Referenzprüfmethode) können mit der Prüfmethode unter Verwendung der Membranbarriere nicht untersucht werden. Bei diesen Chemikalien sollten andere Prüfmethoden zur Anwendung kommen.
|
PRINZIP DER PRÜFMETHODE
|
8.
|
Das Prüfsystem umfasst zwei Bestandteile: eine synthetische makromolekulare Biobarriere und ein CDS (Chemical Detection System); bei dieser Prüfmethode werden mit dem CDS Schäden der Membranbarriere erkannt, die durch ätzende Prüfchemikalien nach der Aufbringung der Prüfchemikalien auf die synthetische makromolekulare Membranarriere hervorgerufen wurden (7) und die vermutlich auf denselben Verätzungsmechanismus bzw. dieselben Verätzungsmechanismen zurückzuführen sind, die auch auf lebende Haut einwirken.
|
|
9.
|
Die Durchdringung (oder der Durchbruch) der Membranbarriere kann mit verschiedenen Verfahren oder mit einem CDS gemessen werden, u. a. anhand von Verfärbungen eines pH-Indikatorfarbstoffs oder anderer Merkmale der Indikatorlösung unter der Barriere.
|
|
10.
|
Die Validität der Membranbarriere, d. h. die Eignung und die Zuverlässigkeit für die beabsichtigte Verwendung, sollte nachgewiesen werden. In diesem Zusammenhang ist auch sicherzustellen, dass unterschiedliche Zubereitungen hinsichtlich der Barriereeigenschaften einheitlich sind (z. B. dass sie eine Barriere gegenüber nicht ätzenden Chemikalien aufrechterhalten können, oder dass sie die Verätzungsmerkmale von Chemikalien in den verschiedenen Unterkategorien von Verätzungswirkungen nach dem UN GHS unterscheiden können (1). Die vorgenommene Einstufung richtet sich nach dem Zeitraum, den eine Chemikalie benötigt, um durch die Membranbarriere in die Indikatorlösung zu gelangen.
|
NACHWEIS DER LEISTUNGFÄHIGKEIT
|
11.
|
Vor der routinemäßigen Anwendung der Prüfmethode mit der In-vitro-Membranbarriere nach dieser Prüfmethode sollten Labors die erforderliche technische Befähigung durch eine ordnungsgemäße Einstufung der zwölf in Tabelle 2 empfohlenen Leistungsstoffe nachweisen. Wenn ein dort genannter Stoff nicht verfügbar ist bzw. wenn dies gerechtfertigt ist, kann ein anderer Stoff verwendet werden, für den geeignete In-vivo- und In-vitro-Referenzdaten verfügbar sind (z. B. aus der Liste der Referenzchemikalien (10)), sofern die in Tabelle 1 beschriebenen Auswahlkriterien berücksichtigt werden.
Tabelle 2
Leistungsstoffe
(27)
|
Stoff (28)
|
CAS-Nr.
|
Chemikalienklasse
|
In-vivo-UN-GHS-Unterkategorie (29)
|
In-vitro-UN-GHS-Unterkategorie (29)
|
|
Bortrifluoriddihydrat
|
13319-75-0
|
Anorganische Säuren
|
1A
|
1A
|
|
Salpetersäure
|
7697-37-2
|
Anorganische Säuren
|
1A
|
1A
|
|
Phosphorpentachlorid
|
10026-13-8
|
Ausgangsstoffe anorganischer Säuren
|
1A
|
1A
|
|
Valerylchlorid
|
638-29-9
|
Säurechloride
|
1B
|
1B
|
|
Natriumhydroxid
|
1310-73-2
|
Anorganische Basen
|
1B
|
1B
|
|
1-(2-Aminoethyl)piperazin
|
140-31-8
|
Aliphatische Amine
|
1B
|
1B
|
|
Benzolsulfonylchlorid
|
98-09-9
|
Säurechloride
|
1C
|
1C
|
|
N,N-Dimethylbenzylamin
|
103-83-3
|
Aniline
|
1C
|
1C
|
|
Tetraethylenepentamin
|
112-57-2
|
Aliphatische Amine
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Phenole
|
NC
|
NC
|
|
Nonylacrylat
|
2664-55-3
|
Acrylate/Methacrylate
|
NC
|
NC
|
|
Natriumbicarbonat
|
144-55-8
|
Anorganische Salze
|
NC
|
NC
|
|
VERFAHREN
|
12.
|
In den folgenden Absätzen werden die Elemente und Verfahren einer Prüfmethode mit einer künstlichen Membranbarriere zur Beurteilung der Verätzungswirkung (7) (15) auf der Grundlage der aktuellen VRM (d. h. mit der im Handel erhältlichen Membran Corrositex®) beschrieben. Die Membranbarriere und die Kompatibilitäts-/Indikator- und Kategorisierungslösungen können hergestellt, zubereitet und im Handel bezogen werden. Dies gilt beispielsweise im Falle der VRM für Corrositex®. Für die validierte Referenzprüfmethode wurde ein Prüfmethodenprotokoll als Beispiel beschrieben (7). Die Prüfung sollte bei Umgebungstemperatur (17-25 °C) vorgenommen werden, und die verwendeten Elemente sollten die folgenden Anforderungen erfüllen.
|
Kompatibilitätsprüfung der Prüfchemikalie
|
13.
|
Vor der Durchführung der Prüfung mit der Membranbarriere wird eine Kompatibilitätsprüfung vorgenommen, um festzustellen, ob die Prüfchemikalie vom CDS erkannt wird. Wenn das CDS die Prüfchemikalie nicht erkennt, ist die Prüfmethode mit der Membranbarriere zur Beurteilung der potenziellen Verätzungswirkung dieser Prüfchemikalie nicht geeignet. In diesem Fall sollte eine andere Prüfmethode gewählt werden. Das CDS und die Expositionsbedingungen bei der Kompatibilitätsprüfung sollten der Exposition in der anschließenden Prüfung mit der Membranbarriere entsprechen.
|
Prüfung zur Ermittlung der Zeitkategorie für Prüfchemikalien
|
14.
|
Wenn für die Prüfmethode relevant, sollten in der Kompatibilitätsprüfung als geeignet befundene Prüfchemikalien einer Prüfung zur Ermittlung der Zeitkategorie, d. h. einer Screening-Prüfung zur Unterscheidung zwischen schwachen und starken Säuren bzw. Basen, unterzogen werden. Bei der validierten Referenzprüfmethode beispielsweise wird durch eine Zeitkategorieprüfung anhand des Nachweises einer starken sauren bzw. alkalischen Reserve festgestellt, welche der beiden Zeitkategorien anzunehmen ist. Die Verätzungswirkung und die UN-GHS-Unterkategorie der hautverätzenden Wirkung sollten je nach der sauren oder alkalischen Reserve der Prüfchemikalie mit zwei unterschiedlichen Durchbruchzeiten bestimmt werden.
|
ELEMENTE DER PRÜFMETHODE UNTER VERWENDUNG EINER MEMBRANBARRIERE
Membranbarriere
|
15.
|
Die Membranbarriere besteht aus zwei Elementen: einem makromolekularen wässerigen Protein-Gel und einer permeablen Trägermembran. Das Protein-Gel sollte für Flüssigkeiten und Feststoffe undurchlässig sein. Eine Durchlässigkeit kann aber infolge einer Verätzung entstehen. Die vollständige Membranbarriere sollte unter festgelegten Bedingungen aufbewahrt werden, bei der sich die Beschaffenheit des Gels nachweislich nicht verändert (etwa durch Austrocknung, Wachstum von Mikroorganismen, Verrutschen oder Risse, die das Verhalten der Barriere beeinträchtigen würden). Wie lange die Membranbarriere aufbewahrt werden kann, sollte ermittelt werden. Nach Ablauf der betreffenden Frist sollten die zubereiteten Membranbarrieren nicht mehr verwendet werden.
|
|
16.
|
Die permeable Trägermembran dient beim Geliervorgang und bei der Exposition gegenüber der Prüfchemikalie als mechanische Unterlage für das Proteingel. Die Trägermembran sollte dafür sorgen, dass das Gel nicht sackt oder verrutscht und für alle Prüfchemikalien leicht durchlässig sein.
|
|
17.
|
Das Protein-Gel (bestehend z. B. aus Keratin, Kollagen oder Gemischen von Proteinen, die eine Gelmatrix bilden) fungiert als Zielmaterial für die Prüfchemikalie. Das Proteinmaterial wird auf die Trägermembran aufgebracht, wo es gelieren kann, bevor die Indikatorlösung mit der Membranbarriere verschlossen wird. Das Protein-Gel sollte eine einheitliche Stärke und Dichte haben und keine Lufteinschlüsse oder Schäden aufweisen, die seine Funktion beeinträchtigen könnten.
|
CDS (Chemical Detection System)
|
18.
|
Die Indikatorlösung (die gleiche Lösung, die auch für die Kompatibilitätsprüfung verwendet wurde), sollte das Vorhandensein von Prüfchemikalien anzeigen. Für die Prüfung sollten ein pH-Indikatorfarbstoff oder eine Kombination von Farbstoffen (z. B. Kresolrot und Methylorange) verwendet werden, deren Farbe sich verändert, wenn eine Prüfchemikalie vorhanden ist. Das Messsystem kann visuell oder elektronisch sein.
|
|
19.
|
Detektionssysteme, die zur Detektion des Durchgangs der Prüfchemikalie durch die Membranbarriere entwickelt wurden, sollten in Bezug auf ihre Relevanz und Zuverlässigkeit geprüft werden, um das Spektrum der erkennbaren Chemikalien und die quantitativen Detektionsgrenzen nachzuweisen.
|
DURCHFÜHRUNG DER PRÜFUNG
Anordnung der Elemente der Prüfmethode
|
20.
|
Die Membranbarriere wird so in ein Fläschchen (oder Röhrchen) mit der Indikatorlösung gegeben, dass die Trägermembran vollflächig mit der Indikatorlösung in Berührung kommt und keine Luftblasen vorhanden sind. Dabei ist sorgfältig darauf zu achten, dass die Barriere nicht beschädigt wird.
|
Applikation der Prüfchemikalie
|
21.
|
Eine geeignete Menge der Prüfchemikalie (z. B. 500 μl einer Flüssigkeit oder 500 mg eines fein vermahlenen Feststoffs (7) wird vorsichtig auf die Oberfläche der Membranbarriere aufgetragen und gleichmäßig verteilt. Für jede Prüfchemikalie und für die entsprechenden Kontrollen wird eine angemessene Anzahl an Replikaten (z. B. vier) (7) zubereitet (Nummern 23 bis 25). Der Zeitpunkt, zu dem die Prüfchemikalie auf die Membranbarriere aufgetragen wird, ist zu protokollieren. Um sicherzustellen, dass kurze Verätzungszeiten präzise protokolliert werden, wird die Prüfchemikalie zeitlich abgestuft auf die Replikatfläschchen gegeben.
|
Messung der Durchdringung der Membranbarriere
|
22.
|
Die Fläschchen werden jeweils angemessen überwacht, und der Zeitpunkt der ersten Änderung der Indikatorlösung (d. h. der Zeitpunkt der Durchdringung der Barriere) wird protokolliert, und die Zeit vom Auftragen der Chemikalie bis zur Durchdringung der Membranbarriere wird ermittelt.
|
Kontrollen
|
23.
|
Bei Prüfungen, bei denen in Verbindung mit der Prüfchemikalie ein Vehikel oder ein Lösungsmittel eingesetzt wird, sollte das Vehikel bzw. das Lösungsmittel kompatibel mit dem Membranbarrieresystem sein, d. h. es sollte die Integrität des Membranbarrieresystems nicht verändern und keine Auswirkungen auf die Verätzungswirkung der Prüfchemikalie haben. Die Lösungsmittelkontrolle (bzw. die Vehikelkontrolle) sollte ggf. gleichzeitig mit der Prüfchemikalie geprüft werden, um die Kompatibilität des Lösungsmittels mit dem Membranbarrieresystem nachzuweisen.
|
|
24.
|
Eine positive (hautätzende) Kontrolle mit mittlerer Verätzungswirkung (z. B. 110 ± 15 mg Natriumhydroxid (Verätzung der UN-GHS-Unterkategorie 1B)) (7) sollte gleichzeitig mit der Prüfchemikalie geprüft werden, um festzustellen, ob die Leistung des Prüfsystems annehmbar ist. Eine zweite Positivkontrolle derselben Chemikalienklasse wie die Prüfchemikalie kann zur Beurteilung des relativen Verätzungspotenzials einer ätzenden Prüfchemikalie hilfreich sein. Die ausgewählte/n) Positivkontrolle(n) sollte(n) eine mittlere Verätzungswirkung haben (z. B. UN-GHS-Unterkategorie 1B), damit Änderungen der Durchdringungszeit erkannt werden können, die den ermittelten Referenzwert in unannehmbarer Weise über- oder unterschreiten und damit darauf hindeuten, dass das Prüfsystem nicht ordnungsgemäß funktioniert. Für diesen Zweck sind sehr stark ätzende Chemikalien (UN-GHS-Unterkategorie 1A) oder nicht ätzende Chemikalien nur begrenzt verwendbar. Eine ätzende Chemikalie der UN-GHS-Unterkategorie 1B würde die Erkennung einer zu kurzen oder zu langen Durchbruchzeit ermöglichen. Eine schwach ätzende Chemikalie der UN-GHS-Unterkategorie 1C könnte als Positivkontrolle eingesetzt werden, um zu prüfen, ob mit der Prüfmethode konsistent zwischen schwach ätzenden und nicht ätzenden Chemikalien unterschieden werden kann. Unabhängig vom verwendeten Ansatz sollte ein annehmbarer Reaktionsbereich der Positivkontrolle auf der Grundlage des historischen Bereichs der Durchbruchzeiten bei der (den) eingesetzten Positivkontrolle(n) entwickelt werden (beispielsweise der Mittelwert ± 2-3 Standardabweichungen). Bei jeder Prüfung sollte die genaue Durchbruchzeit der Positivkontrolle ermittelt werden, damit Abweichungen außerhalb des annehmbaren Bereichs erkannt werden können.
|
|
25.
|
Gleichzeitig mit der Prüfchemikalie sollte als weitere Maßnahme zur Qualitätskontrolle auch eine Negativkontrolle (nicht ätzend) (z. B. 10 %ige Zitronensäure oder 6 %ige Proprionsäure) (7) geprüft werden, um die uneingeschränkte Funktion der Membranbarriere nachzuweisen.
|
Akzeptanzkriterien der Untersuchung
|
26.
|
Auf der Grundlage der ermittelten Zeitparameter der UN-GHS-Unterkategorien der Verätzungswirkung wird anhand des Zeitraums (in Minuten) vom Auftragen einer Prüfchemikalie auf die Membranbarriere bis zur Durchdringung der Barriere die Verätzungswirkung der Prüfchemikalie vorhergesagt. Damit eine Prüfung als annehmbar betrachtet werden kann, sollte sich bei der gleichzeitigen Positivkontrolle die erwartete Reaktionszeit für die Durchdringung ergeben (z. B. für Natriumhydroxid als Positivkontrolle 8-16 Minuten); außerdem sollte die gleichzeitige Negativkontrolle nicht ätzend sein, und die gleichzeitige Lösungsmittelkontrolle sollte – wenn berücksichtigt – weder ätzend sein noch sich auf das Verätzungspotenzial der Prüfchemikalie auswirken. Vor der routinemäßigen Durchführung einer Prüfung nach dieser Prüfmethode sollten Labors die erforderliche technische Befähigung anhand der zwölf in Tabelle 2 empfohlenen Leistungsstoffe nachweisen. Die Zuverlässigkeit und die Genauigkeit neuer vergleichbarer Methoden („Me-too-Methoden“), die ausgehend von dieser Prüfmethode entwickelt wurden und strukturell und funktionell mit der validierten Referenzmethode vergleichbar sind, (14) sollten vor ihrer Verwendung für vorgeschriebene Prüfungen anhand der vordefinierten Leistungsstandards nachgewiesen werden (10).
|
Auswertung von Ergebnissen und Einstufung der Verätzungswirkung von Prüfchemikalien
|
27.
|
Anhand der Zeit (in Minuten) von der Auftragung der Prüfchemikalie auf die Membranbarriere bis zur Durchdringung der Barriere werden die Prüfchemikalien nach ihrer Verätzungswirkung nach UN-GHS-Unterkategorien (1) sowie ggf. nach UN-Verpackungsgruppen eingestuft (16). Bei allen vorgeschlagenen Prüfmethoden sind für jede der drei Unterkategorien ätzender Chemikalien die Schwellenwerte der Zeiträume zu ermitteln. Bei der endgültigen Entscheidung über diese Schwellenwerte ist zu beachten, dass zu niedrige Einstufungen einer Verätzungsgefahr (d. h. falsch negative Ergebnisse) möglichst minimiert werden müssen. Bei dieser Prüfmethode sollten die Schwellenwerte für Corrositex® aus Tabelle 3 angenommen werden, da die Prüfung mit Corrositex® gegenwärtig die einzige Prüfung ist, die unter die Prüfrichtlinie fällt (7).
Tabelle 3
Vorhersagemodell Corrositex®
|
Mittlere Durchbruchzeit (min.)
|
UN-GHS-Vorhersage (32)
|
|
Prüfchemikalien der Kategorie 1 (30) (mit dem Einstufungstest der Methode ermittelt)
|
Prüfchemikalien der Kategorie 2 (31) (mit dem Einstufungstest der Methode ermittelt)
|
|
0-3 min.
|
0-3 min.
|
Hautätzend opt. Unterkategorie 1A
|
|
> 3 bis 60 min.
|
> 3 bis 30 min.
|
Hautätzend opt. Unterkategorie 1B
|
|
> 60 bis 240 min.
|
> 30 bis 60 min.
|
Hautätzend opt. Unterkategorie 1C
|
|
> 240 min.
|
> 60 min.
|
Nicht hautätzend
|
|
DATEN UND BERICHTERSTATTUNG
Daten
|
28.
|
Die Zeiten (in Minuten) von der Auftragung bis zur Durchdringung der Barriere bei der Prüfchemikalie und der (den) Positivkontrolle(n) sollten in einer Tabelle zum einen für die einzelnen Replikate und zum anderen als Mittelwerte ± der einzelnen Versuche angegeben werden.
|
Prüfbericht
|
29.
|
Der Prüfbericht sollte folgende Angaben enthalten:
|
|
Prüfchemikalien und Kontrollchemikalien:
|
—
|
Einkomponentiger Stoff: chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer, SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
—
|
mehrkomponentiger Stoff, UVCB und Gemische: möglichst weit gehende Charakterisierung durch die chemische Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Komponenten;
|
|
—
|
physikalisches Erscheinungsbild, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
|
|
—
|
Herkunft, Chargennummer, sofern vorhanden;
|
|
—
|
Behandlung der Prüfchemikalien/Kontrollstoffe vor der Prüfung, sofern relevant (z. B. Erwärmen, Mahlen);
|
|
—
|
Stabilität der Prüfchemikalie, letztes Verwendungsdatum oder Datum für erneute Analyse, soweit bekannt;
|
|
|
|
Vehikel:
|
—
|
Angaben zur Identität, (gegebenenfalls) Konzentration; Einsatzvolumen;
|
|
—
|
Begründung der Auswahl des Vehikels.
|
|
|
|
In-vitro-Membran-Barrieremodell und verwendetes Protokoll einschließlich nachgewiesener Genauigkeit und Zuverlässigkeit
|
|
|
Prüfbedingungen:
|
—
|
Beschreibung der verwendeten Geräte und der Versuchsvorbereitung;
|
|
—
|
Herkunft und Zusammensetzung der verwendeten In-vitro-Membranbarriere;
|
|
—
|
Zusammensetzung und Merkmale der Indikatorlösung;
|
|
—
|
Mengen der Prüfchemikalie und der Kontrollchemikalien;
|
|
—
|
Beschreibung und Begründung der Prüfung zur Einstufung der Zeiten;
|
|
—
|
Beschreibung der angewandten Bewertungs- und Einstufungskriterien;
|
|
—
|
Nachweis der Befähigung zur Durchführung der Prüfmethode vor der regelmäßigen Verwendung durch die Prüfung von Leistungschemikalien.
|
|
|
|
Ergebnisse:
|
—
|
Tabellarische Darstellung einzelner Rohdaten aus einzelnen Prüfungen und Kontrollen für die einzelnen Replikate;
|
|
—
|
Beschreibung anderer beobachteter Wirkungen;
|
|
—
|
abgeleitete Einstufung mit Bezug auf das Vorhersagemodell und/oder angewandte Entscheidungskriterien.
|
|
Erörterung der Ergebnisse.
Schlussfolgerungen.
|
LITERATUR
|
(1)
|
Vereinte Nationen (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), fünfte überarbeitete Ausgabe, UN New York und Genf, 2013. Abrufbar unter:http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Kapitel B.4 dieses Anhangs, Akute Hautreizung/-verätzung.
|
|
(3)
|
Kapitel B.40bis dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: Prüfmethode mit rekonstruierter humaner Epidermis (RHE).
|
|
(4)
|
Kapitel B.40 dieses Anhangs, In-vitro-Hautverätzung: TER-Prüfmethode.
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhütter, H.-G. und Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. In Vitro Test Methods for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No. 99-4495).
|
|
(8)
|
Gordon, V.C., Harvell, J.D. und Maibach, H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (No 34).
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. Abrufbar unter:http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italien. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). 20. September 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Kapitel B.46 dieses Anhangs, In-vitro-Hautreizung: Prüfmethode mit rekonstruierter humaner Epidermis. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Abrufbar unter:http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Abrufbar unter:http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
Vereinte Nationen (UN) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Abrufbar unter:http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Anlage
BEGRIFFSBESTIMMUNGEN
CDS (Chemical Detection System)
: Ein visuelles oder elektronisches Messsystem mit einer Indikatorlösung, die bei Vorhandensein einer Prüfchemikalie reagiert (beispielsweise durch eine Verfärbung eines pH-Indikatorfarbstoffs oder einer Kombination von Farbstoffen aufgrund des Vorliegens der Prüfchemikalie oder durch andere chemische oder elektrochemische Reaktionen).
Chemikalie
: Ein Stoff oder ein Gemisch.
Einkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorliegt.
Gemisch
: Ein Gemisch oder eine Lösung, die aus zwei oder mehr Stoffen besteht.
Genauigkeit
: Der Grad an Übereinstimmung zwischen Testergebnissen und akzeptierten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von „Übereinstimmung“ verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (9).
GHS (Globally Harmonized System of Classification and Labelling of Chemicals = Global harmonisiertes System zur Einstufung und Kennzeichnung)
: Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenbögen, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1).
Hautverätzung, in vivo
: Das Auslösen einer irreversiblen Hautschädigung, d. h. einer sichtbaren, bis in das Corium reichenden Nekrose der Epidermis nach Applikation einer Prüfchemikalie für die Dauer von bis zu 4 Stunden. Verätzungsreaktionen sind gekennzeichnet durch Geschwüre, Blutungen, blutige Verschorfungen und am Ende des Beobachtungszeitraums von 14 Tagen durch eine auf ein Ausbleichen der Haut zurückzuführende Verfärbung, komplett haarlose Bereiche und Narben. Bei der Beurteilung fragwürdiger Schädigungen ist die Histopathologie mit zu berücksichtigen.
IATA
: Integrated Approach to Testing and Assessment = integrierter Prüfungs- und Bewertungsansatz.
Leistungsstandards
: Auf einer validierten Prüfmethode beruhende Normen, auf deren Grundlage die Vergleichbarkeit einer vorgeschlagenen, mechanistisch und funktionell ähnlichen Prüfmethode bewertet werden kann. Sie umfassen (i) wesentliche Elemente der Prüfmethode, (ii) ein Mindestverzeichnis von Referenzchemikalien, ausgewählt aus den Chemikalien, die zum Nachweis der akzeptablen Leistung der validierten Referenzmethode verwendet werden, und (iii) je nach den für die validierte Referenzmethode erzielten Ergebnissen die vergleichbaren Zuverlässigkeits- und Genauigkeitswerte, die die vorgeschlagene Prüfmethode bei der Bewertung anhand des Mindestverzeichnisses von Referenzchemikalien demonstrieren sollte (9).
Mehrkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens ≥ 10 % w/w und < 80 % w/w vorhanden sind. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet.
NC
: Nicht hautätzend
Prüfchemikalie
: Stoff oder Gemisch, der/das mit dieser Prüfmethode untersucht wird.
Relevanz
: Dieser Begriff bezieht sich auf das Verhältnis zwischen der Prüfmethode und der betreffenden Wirkung und auf die Frage, ob er aussagekräftig und nützlich für einen bestimmten Zweck ist. Er beschreibt das Ausmaß, in dem die Prüfmethode die untersuchte biologische Wirkung korrekt misst oder vorhersagt. Die Relevanz schließt eine Beurteilung der Genauigkeit (Übereinstimmung) einer Prüfmethode ein (9).
Sensitivität
: Der Anteil aller positiven/wirkenden Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Sensitivität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (9).
Spezifizität
: Der Anteil aller negativen/wirkungslosen Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (9).
Stoff
: Ein chemisches Element und seine Verbindungen in natürlicher Form oder gewonnen durch ein Herstellungsverfahren, einschließlich der zur Wahrung seiner Stabilität notwendigen Zusatzstoffe und der durch das angewandte Verfahren bedingten Verunreinigungen, aber mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können.
Übereinstimmung
: Die Übereinstimmung ist ein Maß der Leistung einer Prüfmethode für Prüfverfahren mit kategorialen Ergebnissen und ein Aspekt der Relevanz. Der Begriff wird gelegentlich gleichbedeutend mit Genauigkeit verwendet und als Anteil aller geprüften Chemikalien definiert, die korrekt als positiv oder negativ eingestuft werden. Die Übereinstimmung hängt in hohem Maße von der Prävalenz bei den Typen der untersuchten Prüfchemikalien ab (9).
UVCB
: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Zuverlässigkeit
: Beschreibung der Beziehung zwischen der Prüfung und der untersuchten Wirkung und ob die Prüfung aussagekräftig und nützlich für einen bestimmten Zweck ist. Sie wird durch Berechnung der Intra- und Inter-Labor-Reproduzierbarkeit bewertet (9).
B.66 STTA-IN-VITRO-ASSAYS ZUM NACHWEIS VON ÖSTROGENREZEPTOR-AGONISTEN UND -ANTAGONISTEN
ALLGEMEINE EINLEITUNG
Leistungsbezogene OECD-Prüfrichtlinie
|
1.
|
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 455 (2016). TG 455 ist eine PBTG (Performance-Based Test Guideline = leistungsbezogene Prüfrichtlinie), die die Methode der STTA-in-vitro-Assays (Stably Transfected Transactivation = Transaktivierung durch stabile Transfektion) zum Nachweis von Östrogenrezeptor-Agonisten und -Antagonisten (Estrogen Receptor (ER) Agonists and Antagonists) (ER-TA-Assays) beschreibt. Sie umfasst mehrere mechanisch und funktionell ähnliche Prüfmethoden zum Nachweis von Östrogenrezeptor-(oder ERα- und/oder ERβ-)Agonisten und -Antagonisten und sollte die Entwicklung neuer ähnlicher oder modifizierter Prüfmethoden nach den Validierungsgrundsätzen im OECD Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment erleichtern (1). Die folgenden vollständig validierten Referenzprüfmethoden (Anlagen 2 und 3) sind Grundlage dieser PBTG:
|
—
|
Der STTA-Assay (2) unter Verwendung der (h) ERα-HeLa-9903-Zelllinie und
|
|
—
|
der VM7Luc-ER-TA-Assay (3) unter Verwendung der VM7Luc4E2-Zelllinie, (33) mit dem in erster Linie hERα sowie in beschränktem Umfang hERββ(4) (5) exprimiert wird.
|
Zur Entwicklung und Validierung ähnlicher Assays für denselben Gefahren-Endpunkt sollten die verfügbaren Leistungsstandards (PS) (6) (7) verwendet werden. Sie ermöglichen eine frühzeitige Änderung der PBTG 455 zur Aufnahme neuer ähnlicher Assays in eine aktualisierte Fassung der PBTG; ähnliche Assays werden jedoch erst dann aufgenommen, wenn sie von der OECD geprüft wurden und von der OECD bestätigt wurde, dass die Leistungsstandards erfüllt werden. Die in TG 455 aufgenommenen Assays erfüllen die Anforderungen der OECD-Mitgliedstaaten an Prüfergebnisse bei Östrogenrezeptor-Transaktivierungen alle gleichermaßen und ermöglichen die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen.
|
Hintergrund und Grundsätze der in diese Prüfmethode aufgenommenen Assays
|
2.
|
Die OECD leitete 1998 mit hoher Priorität die Überarbeitung bestehender Prüfrichtlinien für Screening-Prüfungen und Untersuchungen potenziell endokriner Disruptoren und die Entwicklung neuer Prüfrichtlinien ein. Der „OECD Conceptual Framework (CF) for the Testing and Assessment of Endocrine Disrupting Chemicals“ wurde 2012 überarbeitet. Die ursprünglichen und überarbeiteten CFs wurden dem OECD Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (8) als Anhänge beigefügt. Der CF umfasst fünf Ebenen mit jeweils eigener biologischer Komplexität. Die in dieser Prüfmethode beschriebenen ER-TA-Assays (ER-TA = Estrogen Receptor Agonists and Antagonists Transactivation) sind Ebene 2 (In-vitro-Assays zur Ermittlung von Daten über ausgewählte endokrine Mechanismen/Wirkungspfade) zuzuordnen. Diese Prüfmethode wurde für In-vitro-Transaktivierungs-Assays (TA-Assays) zur Ermittlung von Östrogenrezeptor-Agonisten und -Antagognisten (ER-Agonisten und -Antagonisten) entwickelt.
|
|
3.
|
Das Zusammenwirken von Östrogenen mit ER kann die Transkription von Genen beeinträchtigen, die durch Östrogene gesteuert werden. Dadurch können Zellprozesse ausgelöst oder gehemmt werden (u. a. für die Zellproliferation, für die normale fetale Entwicklung und für die Reproduktion erforderliche Prozesse) (9) (10) (11). Störungen normaler östrogener Systeme können Beeinträchtigungen der normalen Entwicklung (Ontogenese), der reproduktiven Gesundheit und des Reproduktionssystems auslösen.
|
|
4.
|
In-vitro-TA-Assays beruhen auf einer mittelbaren oder unmittelbaren Wechselwirkung der Stoffe mit einem spezifischen Rezeptor, der die Transkription eines Reporter-Genpodukts steuert. Diese Assays wurden in großem Umfang zur Beurteilung der durch spezifische nukleäre Rezeptoren (z. B. ER) gesteuerten Gen-Expression verwendet (12) (13) (14) (15) (16). Sie wurden zur Erkennung einer durch die ER gesteuerten östrogenen Transaktivierung vorgeschlagen (17) (18) (19). Bei nukleären ER sind zumindest die zwei großen Untertypen α und β zu unterscheiden, die durch verschiedene Gene kodiert werden. Die betreffenden Proteine haben unterschiedliche biologische Funktionen, Gewebeverteilungen und Ligandenbindungsaffinitäten (20) (21) (22) (23) (24) (25) (26). Nukleäre ERα vermitteln die klassische Östrogenreaktion (27) (28) (29) (30). Daher beziehen sich die meisten gegenwärtig zur Messung der Aktivierung oder Hemmung von ER entwickelten Modelle speziell auf ERα. Die Assays werden zur Ermittlung von Chemikalien verwendet, die nach erfolgter Ligandenbindung die ER aktivieren, was dazu führt, dass der Rezeptor-Ligandenkomplex eine Bindung mit spezifischen DNA-Response-Elementen eingeht und ein Reporter-Gen transaktiviert; dies wiederum bewirkt eine verstärkte zelluläre Expression eines Markerproteins. Für diese Assays können unterschiedliche Reporter-Reaktionen berücksichtigt werden. Bei auf Luciferase basierenden Systemen wandelt das Enzym Luziferase das Luciferinsubstrat in ein biolumineszierendes Produkt um, das mit einem Luminometer einer quantitativen Messung unterzogen werden kann. Weitere Beispiele verbreiteter Reporter-Gene sind fluoreszierende Proteine und das LacZ-Gen, das β-Galactosidase kodiert. Dieses Enzym kann das farblose Substrat X-gal (5-Brom-4-chlor-indolyl-galactopyranosid) in ein blaues Produkt umwandeln, das mit einem Spektralfotometer quantitativ bestimmt werden kann. Diese Reporter-Gene können rasch und kostengünstig mit im Handel erhältlichen Test-Kits bewertet werden.
|
|
5.
|
In Studien zur Validierung des STTA- und des VM7Luc-TA-Assays wurden die Relevanz und die Zuverlässigkeit im Hinblick auf die vorgesehene Verwendung nachgewiesen (3) (4) (5) (30). Im ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (VM7Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals (3) werden Leistungsstandards für luminiszenzbasierte ER-TA-Assays mit Brust-Zelllinien berücksichtigt. Diese Leistungsstandards wurden so modifiziert, dass sie sowohl beim STTA- als auch beim VM7Luc-TA-Assay angewendet werden können (2).
|
|
6.
|
Die in der vorliegenden Prüfmethode verwendeten Begriffe und Abkürzungen werden in Anlage 1 erläutert.
|
Anwendungsbereich und Begrenzungen bei TA-Assays
|
7.
|
Diese Assays werden für Screening- und Priorisierungszwecke vorgeschlagen, können aber auch zu mechanistischen Erkenntnissen führen, die im Rahmen von Beweiskraftermittlungen verwendet werden können. Gegenstand der Assays sind durch TA ausgelöste chemische Bindungen an die ER in einem In-vitro-System. Daher sollten Ergebnisse nicht unmittelbar für die komplexen Signal- und Steuerungswirkungen des intakten endokrinen In-vivo-Systems extrapoliert werden.
|
|
8.
|
Durch ER vermittelte TA gelten als einer der zentralen Mechanismen endokriner Störungen (ED), wenngleich ED auch durch andere Mechanismen hervorgerufen werden können, darunter (i) Wechselwirkungen mit anderen Rezeptoren und Enzymsystemen des endokrinen Systems, (ii) Hormonsynthesen, (iii) Stoffwechselaktivierungen und/oder die Deaktivierung von Hormonen, (iv) die Verteilung von Hormonen an Zielgewebe und (v) die Beseitigung von Hormonen im Körper. Keiner der Assays im Rahmen dieser Prüfmethode hat diese Wirkungsweisen zum Gegenstand.
|
|
9.
|
Gegenstand dieser Prüfmethode ist die Fähigkeit von Chemikalien, die ER-gesteuerte Transkription zu aktivieren (d. h. als Agonisten zu wirken) und zu unterdrücken (d. h. als Antagonisten zu fungieren). Einige Chemikalien können je nach Zelltyp sowohl als Agonisten als auch als Antagonisten wirken und sind als selektive Östrogenrezeptor-Modulatoren (SERM) bekannt. Chemikalien, für die in diesen Assays negative Ergebnisse ermittelt wurden, könnten einem Assay zur Prüfung der ER-Bindung unterzogen werden, bevor festgestellt wird, dass sie keine Bindung mit dem Rezeptor eingehen. Außerdem geben die Assays wegen der begrenzten Metabolisierung von In-vitro-Zellsystemen Aufschluss vermutlich nur über die Aktivität der jeweiligen Ausgangsverbindung. Da bei der Validierung nur einzelne Stoffe verwendet wurden, wurde die Eignung für Prüfgemische nicht untersucht. Dennoch ist die Prüfmethode theoretisch auch für die Prüfung von mehrkomponentigen Stoffen, Stoffen mit unbekannter oder schwankender Zusammensetzung, komplexen Reaktionsprodukten oder biologischen Materialien (UVCB) und Gemischen geeignet. Bevor die Prüfmethode bei mehrkomponentigen Stoffen, UVCB oder Gemischen für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob, und falls ja, warum, sie für den beabsichtigten Zweck geeignete Ergebnisse liefert. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs von Rechts wegen vorgeschrieben ist.
|
|
10.
|
Tabelle 1 enthält eine Übersicht über die Agonisten-Ergebnisse der 34 Stoffe, die mit den beiden in dieser Prüfmethode beschriebenen vollständig validierten Referenzprüfmethoden untersucht wurden. Von diesen Stoffen wurden nach veröffentlichten Berichten (u. a. aufgrund von In-vitro-Assays zur Prüfung auf ER-Bindungen und TA und/oder aufgrund des Uterotrophen Bioassays) (2) (3) (18) (31) (32) (33) (34) 26 Stoffe sicher als ER-Agonisten und 8 als negativ eingestuft. Tabelle 2 enthält eine Übersicht über die Antagonisten-Ergebnisse der 15 Stoffe, die mit den beiden in dieser Prüfmethode beschriebenen vollständig validierten Referenzprüfmethoden untersucht wurden. Von diesen Stoffen wurden nach veröffentlichten Berichten (u. a. aufgrund von In-vitro-Assays zur Prüfung auf ER-Bindungen und TA) (2) (3) (18) (31) 4 Stoffe sicher/vermutlich als ER-Agonisten und 10 als negativ eingestuft. Nach den in den Tabellen 1 und 2 zusammengefassten Daten ergab sich hinsichtlich der Einstufungen aller Stoffe mit Ausnahme eines Stoffes (Mifepristone) beim Antagonisten-Assay eine 100 %ige Übereinstimmung zwischen den beiden Referenzprüfmethoden, und alle Stoffe wurden zutreffend als ER-Agonisten/-Antagonisten bzw. als negativ eingestuft. Ergänzende Informationen über diese Gruppe von Chemikalien sowie über weitere Chemikalien, die im Rahmen der Validierungsstudien mit den STTA- und den VM7Luc-ER-TA-Assays untersucht wurden, sind den Leistungsstandards für die ER-TA (6) (7), Anlage 2 (Tabellen 1, 2 und 3) zu entnehmen.
|
Tabelle 1
Übersicht über die Ergebnisse von STTA- und VM7Luc-ER-TA-Assays bei Stoffen, die in beiden Agonisten-Prüfungen untersucht und als ER-Agonisten (POS) oder als negativ (NEG) eingestuft wurden
|
|
Stoff
|
CAS-Nr.
|
STTA-Assay (35)
|
VM7Luc-ER-TA-Assay (36)
|
Datenquelle für die Einstufung (38)
|
|
ER-TA Aktivität
|
PC10-Wert (M)
|
PC50-Wert (2) (M)
|
ER-TA-Aktivität
|
EC50-Wert (2), (37) (M)
|
Sonstige ER-TAs (34)
|
ER Bindung
|
Uterotroph
|
|
1
|
17ß-Estradiol (1)
|
50-28-2
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
5,63 × 10-12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-Estradiol (1)
|
57-91-0
|
POS
|
7,24 × 10-11
|
6,44 × 10-10
|
POS
|
1,40 × 10-9
|
POS (11/11)
|
POS
|
POS
|
|
3
|
17α-Ethinylestradiol (1)
|
57-63-6
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
7,31 × 10-12
|
POS (22/22)
|
POS
|
POS
|
|
4
|
17β-Trenbolon
|
10161-33-8
|
POS
|
1,78 × 10-8
|
2,73 × 10-7
|
POS
|
4,20 × 10-8
|
POS (2/2)
|
NT
|
NT
|
|
5
|
19-Nortestosteron (1)
|
434-22-0
|
POS
|
9,64 × 10-9
|
2,71 × 10-7
|
POS
|
1,80 × 10-6
|
POS (4/4)
|
POS
|
POS
|
|
6
|
4-Cumylphenol (1)
|
599-64-4
|
POS
|
1,49 × 10-7
|
1,60 × 10-6
|
POS
|
3,20 × 10-7
|
POS (5/5)
|
POS
|
NT
|
|
7
|
4-tert-Octylphenol (1)
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
POS
|
3,19 × 10-8
|
POS (21/24)
|
POS
|
POS
|
|
8
|
Apigenin (1)
|
520-36-5
|
POS
|
1,31 × 10-7
|
5,71 × 10-7
|
POS
|
1,60 × 10-6
|
POS (26/26)
|
POS
|
NT
|
|
9
|
Atrazin (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Bisphenol A (1)
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
POS
|
5,33 × 10-7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisphenol B (1)
|
77-40-7
|
POS
|
2,36 × 10-8
|
2,11 × 10-7
|
POS
|
1,95 × 10-7
|
POS (6/6)
|
POS
|
POS
|
|
12
|
Butylbenzylphthalat (1)
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
POS
|
1,98 × 10-6
|
POS (12/14)
|
POS
|
NEG
|
|
13
|
Corticosteron (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (6/6)
|
NEG
|
NT
|
|
14
|
Cumestrol (1)
|
479-13-0
|
POS
|
1,23 × 10-9
|
2,00 × 10-8
|
POS
|
1,32 × 10-7
|
POS (30/30)
|
POS
|
NT
|
|
15
|
Daidzein (1)
|
486-66-8
|
POS
|
1,76 × 10-8
|
1,51 × 10-7
|
POS
|
7,95 × 10-7
|
POS (39/39)
|
POS
|
POS
|
|
16
|
Diethylstilbestrol (1)
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POS
|
3,34 × 10-11
|
POS (42/42)
|
POS
|
NT
|
|
17
|
Di-n-butylphthalat
|
84-74-2
|
POS
|
4,09 × 10-6
|
|
POS
|
4,09 × 10-6
|
POS (6/11)
|
POS
|
NEG
|
|
18
|
Ethylparaben
|
120-47-8
|
POS
|
5,00 × 10-6
|
(ohne PC50)
|
POS
|
2,48 × 10-5
|
POS
|
|
NT
|
|
19
|
Estron (1)
|
53-16-7
|
POS
|
3,02 × 10-11
|
5,88 × 10-10
|
POS
|
2,34 × 10-10
|
POS (26/28)
|
POS
|
POS
|
|
20
|
Genistein (1)
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
POS
|
2,71 × 10-7
|
POS (100/102)
|
POS
|
POS
|
|
21
|
Haloperidol
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kaempferol (1)
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
POS
|
3,99 × 10-6
|
POS (23/23)
|
POS
|
NT
|
|
23
|
Kepon (1)
|
143-50-0
|
POS
|
7,11 × 10-7
|
7,68 × 10-6
|
POS
|
4,91 × 10-7
|
POS (14/18)
|
POS
|
NT
|
|
24
|
Ketoconazol
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Linuron (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
meso-Hexestrol (1)
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POS
|
1,65 × 10-11
|
POS (4/4)
|
POS
|
NT
|
|
27
|
Methyltestosteron (1)
|
58-18-4
|
POS
|
1,73 × 10-7
|
4,11 × 10-6
|
POS
|
2,68 × 10-6
|
POS (5/6)
|
POS
|
NT
|
|
28
|
Morin
|
480-16-0
|
POS
|
5,43 × 10-7
|
4,16 × 10-6
|
POS
|
2,37 × 10-6
|
POS (2/2)
|
POS
|
NT
|
|
29
|
Norethynodrel (1)
|
68-23-5
|
POS
|
1,11 × 10-11
|
1,50 × 10-9
|
POS
|
9,39 × 10-10
|
POS (5/5)
|
POS
|
NT
|
|
30
|
p,p’-Methoxychlor (1)
|
72-43-5
|
POS
|
1,23 × 10-6
|
(ohne PC50) (2)
|
POS
|
1,92 × 10-6
|
POS (24/27)
|
POS
|
POS
|
|
31
|
Phenobarbital (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
32
|
Reserpin
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
33
|
Spironolacton (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
34
|
Testosteron
|
58-22-0
|
POS
|
2,82 × 10-8
|
9,78 × 10-6
|
POS
|
1,75 × 10-5
|
POS (5/10)
|
POS
|
NT
|
|
Abkürzungen: CAS-NR. = CAS-Registrierungsnummer); M = molar; EC50 = die Hälfte der maximalen Wirkungskonzentration eines Prüfstoffs; NEG = negativ; POS = positiv; NT = nicht geprüft; PC10 (und PC50) = Konzentration eines Prüfstoffs, bei der die Reaktion 10 % (bzw. 50 % bei PC50) der Reaktion entspricht, die auf allen Platten mit der Positivkontrolle (E2, 1 nm) ausgelöst wird.
|
Tabelle 2
Vergleich der Ergebnisse von STTA- und VM7Luc-ER-TA-Assays bei Stoffen, die in beiden Antagonisten-Prüfungen untersucht und als ER-Antagonisten (POS) oder als negativ (NEG) eingestuft wurden
|
|
Stoff (3)
|
CAS-Nr.
|
ER-STTA-Assay (40)
|
VM7Luc-ER-STTA-Assay (41)
|
Wirkungen Bei ER-STTA-Kandidaten (43)
|
ICCVAM (44) -Konsensklassifikation
|
MeSH (45) -Chemikalienklasse
|
Produktklasse (46)
|
|
ER-TA-Aktivität
|
IC50-Wert (39) (M)
|
ER-TA-Aktivität
|
IC50-Wert (39), (42) (M)
|
|
1
|
4-Hydroxytamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
POS
|
2,08 × 10-7
|
mäßig POS
|
POS
|
Kohlenwasserstoff (cyclisch)
|
Pharmazeutisches Erzeugnis
|
|
2
|
Dibenzo(a,h)anthracen
|
53-70-3
|
POS
|
Ohne IC50
|
POS
|
Ohne IC50
|
POS
|
PP
|
Polycyclische Verbindung
|
Laborchemikalie, Naturprodukt
|
|
3
|
Mifepriston
|
84371-65-3
|
POS
|
5,61 × 10-6
|
NEG
|
—
|
schwach POS
|
NEG
|
Steroid
|
Pharmazeutisches Erzeugnis
|
|
4
|
Raloxifen-HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
POS
|
1,19 × 10-9
|
mäßig POS
|
POS
|
Kohlenwasserstoff (cyclisch)
|
Pharmazeutisches Erzeugnis
|
|
5
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
POS
|
8,17 × 10-7
|
POS
|
POS
|
Kohlenwasserstoff (cyclisch)
|
Pharmazeutisches Erzeugnis
|
|
6
|
17β-Estradiol
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
7
|
Apigenin
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Heterocyclische Verbindung
|
Farbstoff, Naturprodukt, pharmazeutisches Zwischenprodukt
|
|
8
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Heterocyclische Verbindung
|
Herbizid
|
|
9
|
Di-n-butylphthalat
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Ester, Phthalsäure
|
Kosmetischer Inhaltsstoff, Industriechemikalie, Weichmacher
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
nicht geprüft
|
PN
|
Heterocyclische Verbindung, Pyrimidin
|
Fungizid
|
|
11
|
Flavon
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Flavonoid, heterocyclische Verbindung
|
Naturprodukt, pharmazeutisches Erzeugnis
|
|
12
|
Flutamid
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Amid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
13
|
Genistein
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Flavonoid, heterocyclische Verbindung
|
Naturprodukt, pharmazeutisches Erzeugnis
|
|
14
|
p-n-Nonylphenol
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
nicht geprüft
|
NEG
|
Phenol
|
Chemisches Zwischenprodukt
|
|
15
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Kohlenwasserstoff (cyclisch)
|
Naturprodukt
|
|
Abkürzungen: CASRN = CAS-Registrierungsnummer; M = molar; IC50 = die Hälfte der maximalen Hemmkonzentration eines Prüfstoffs; NEG = negativ; PN = vermutlich negativ; POS = positiv; PP = vermutlich positiv.
|
ELEMENTE DES ER-TA-ASSAYS
Wesentliche Elemente des Assays
|
11.
|
Diese Prüfmethode gilt für Assays unter Verwendung eines stabil transfizierten oder endogenen ERα-Rezeptors und eines stabil transfizierten Reporter-Genprodukts unter Steuerung durch eines oder mehrere Östrogen-Response-Elemente; es können aber auch andere Rezeptoren (z. B. ERβ) vorhanden sein. Der Assay beinhaltet die folgenden wesentlichen Elemente:
|
Kontrollen
|
12.
|
Die Grundlage der vorgeschlagenen gleichzeitigen Referenzstandards für die einzelnen Agonisten- und Antagonisten-Assays sollte erläutert werden. Gegebenenfalls dienen gleichzeitige Kontrollen (Negativ-, Lösungsmittel- und Positivkontrolle) als Anzeichen dafür, dass der Assay unter den Prüfbedingungen funktioniert; außerdem bilden sie eine Grundlage für versuchsübergreifende Vergleiche. In der Regel sind sie Bestandteil der Akzeptanzkriterien für die Versuche (1).
|
Standard-Qualitätskontrollverfahren
|
13.
|
Die einzelnen Assays sollten Standard-Qualitätskontrollverfahren unterzogen werden, wie jeweils beschrieben, um sicherzustellen, dass die Zelllinie auch nach mehreren Durchgängen stabil bleibt, kein Mycoplasma aufweist (d. h. frei von Bakterienkontaminationen ist) und unverändert in der Lage ist, die erwarteten durch ER vermittelten Reaktionen zu entwickeln. Außerdem sollten die Identität der Zelllinien geprüft und Untersuchungen auf andere Verunreinigungen (z. B. Pilze, Hefen und Viren) vorgenommen werden.
|
Nachweis der Eignung des Labors
|
14.
|
Vor der Prüfung unbekannter Chemikalien mit einem der Assays dieser Prüfmethode sollte jedes Labor nachweisen, dass es in der Lage ist, den betreffenden Assay durchzuführen. Zum Nachweis seiner Leistungsfähigkeit sollte jedes Labor die 14 Leistungsstoffe in Tabelle 3 für den Agonisten-Assay und die 10 Leistungsstoffe in Tabelle 4 für den Antagonisten-Assay prüfen. Mit dieser Eignungsprüfung wird auch die Empfindlichkeit des Prüfsystems nachgewiesen. Die Liste der Leistungsstoffe ist eine Untergruppe der Referenzstoffe der Leistungsstandards für die ER-TA-Assays (6). Diese Stoffe sind im Handel erhältlich und entsprechen den Chemikalienklassen, bei denen gewöhnlich eine Aktivität von ER-Agonisten bzw. -Antagonisten zu verzeichnen ist. Außerdem zeigen sie ein geeignetes und für ER-Agonisten bzw. -Antagonisten zu erwartendes Wirkungsspektrum (d. h. stark bis schwach) und umfassen auch Negativstoffe. Die Untersuchung der Leistungsstoffe sollte an unterschiedlichen Tagen mindestens zweimal wiederholt werden. Die Leistungsfähigkeit wird durch die ordnungsgemäße Einstufung (positiv/negativ) der einzelnen Leistungsstoffe nachgewiesen. Beim Erlernen der Assays sollte die Eignungsprüfung von jedem einzelnen Labortechniker wiederholt werden. Je nach Zelltyp können sich einige dieser Leistungsstoffe wie selektive Östrogenrezeptor-Modulatoren (SERM) verhalten und sowohl als Agonisten als auch als Antagonisten wirken. Die Leistungsstoffe werden in den Tabellen 3 und 4 jedoch nach ihrer bekannten vorwiegenden Aktivität eingestuft, die auch für die Bewertung der Leistungsfähigkeit angenommen werden sollte.
|
|
15.
|
Zum Nachweis der Leistungsfähigkeit und zu Qualitätskontrollzwecken sollte jedes Labor Datenbanken mit historischen Daten zu Agonisten und Antagonisten mit Daten zu Referenzstandards (z. B. 17β-Estradiol und Tamoxifen) sowie zu Positiv- und Negativkontrollchemikalien und Lösungsmittelkontrollen (z. B. DMSO) erstellen. Die Datenbank sollte beginnend mit Prüfläufen mit mindestens 10 unabhängigen Agonisten (z. B. 17β-Estradiol) und 10 unabhängigen Antagonisten (z. B. Tamoxifen) erstellt werden. Anschließend sollten die Ergebnisse künftiger Analysen dieser Referenzstandards und Lösungsmittelkontrollen in die Datenbank aufgenommen werden, um die Konsistenz und die Leistungsfähigkeit des im jeweiligen Labor im Laufe der Zeit durchgeführten Bioassays sicherzustellen.
|
Tabelle 3
Liste mit (14) Leistungsstoffen für Agonisten-Assays
(54)
|
Nr. (53)
|
Stoff
|
CAS-Nr.
|
Erwartete Reaktion (47)
|
STTA-Assay
|
VM7Luc-ER-TA-Assay
|
MeSH-Chemikalienklasse (51)
|
Produktklasse (52)
|
|
PC10-Wert (M) (48)
|
PC50-Wert (M) (48)
|
Prüfkonzentrations- bereich (M)
|
VM7Luc-EC50-Wert (M) (49)
|
Höchstkonzentration für Vorversuch (M) (50)
|
|
14
|
Diethylstilbestrol
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Kohlenwasserstoff (cyclisch)
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
12
|
17α-Estradiol
|
57-91-0
|
POS
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
15
|
meso-Hexestrol
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Kohlenwasserstoff (cyclisch), Phenol
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
11
|
4-tert-Octylphenol
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Phenol
|
Chemisches Zwischenprodukt
|
|
9
|
Genistein
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoid, heterocyclische Verbindung
|
Naturprodukt, pharmazeutisches Erzeugnis
|
|
6
|
Bisphenol A
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Phenol
|
Chemisches Zwischenprodukt
|
|
2
|
Kaempferol
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoid, heterocyclische Verbindung
|
Naturprodukt
|
|
3
|
Butylbenzylphthalat
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Carboxylsäure, Ester, Phthalsäure
|
Weichmacher, Industriechemikalie
|
|
4
|
p,p’- Methoxychlor
|
72-43-5
|
POS
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Kohlenwasserstoff (halogeniert)
|
Pestizid, veterinärmediznisches Agens
|
|
1
|
Ethylparaben
|
120-47-8
|
POS
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Carboxylsäure, Phenol
|
Pharmazeutisches Erzeugnis, Konservierungsmittel
|
|
17
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Heterocyclische Verbindung
|
Herbizid
|
|
20
|
Spironolacton
|
52-01-7
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Lacton, Steroid
|
Pharmazeutisches Erzeugnis
|
|
21
|
Ketoconazol
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Heterocyclische Verbindung
|
Pharmazeutisches Erzeugnis
|
|
22
|
Reserpin
|
50-55-5
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Heterocyclische Verbindung, Indol
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
Abkürzungen: CAS-NR. = CAS-Registrierungsnummer); EC50 = die Hälfte der maximalen Wirkungskonzentration eines Prüfstoffs; NEG = negativ; POS = positiv; PC10 (und PC50) = Konzentration eines Prüfstoffs, bei der die Reaktion 10 % (bzw. 50 % bei PC50) der Reaktion entspricht, die auf allen Platten mit der Positivkontrolle (E2, 1 nm) ausgelöst wird.
|
Tabelle 4
Liste mit (10) Leistungsstoffen für Antagonisten-Assays
|
|
Stoff (4)
|
CAS-Nr.
|
ER-STTA-Assay (55)
|
VM7Luc-ER-STTA-Assay (56)
|
Wirkungen Bei ER-STTA (55) -Kandidaten
|
ICCVAM (59) -Konsensklassifikation
|
MeSH (60) -Chemikalienklasse
|
Produktklasse (61)
|
|
ER-TA-Aktivität
|
IC50 (M)
|
Prüfkonzentrationsbereich (M)
|
ER-TA-Aktivität
|
IC50
(57) (M)
|
Höchstkonzentration für Vorversuch (M) (58)
|
|
1
|
4-Hydroxytamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
10-12 – 10-7
|
POS
|
2,08 × 10-7
|
2,58 × 10-4
|
mäßig POS
|
POS
|
Kohlenwasserstoff (cyclisch)
|
Pharmazeutisches Erzeugnis
|
|
2
|
Raloxifen-HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
10-12 – 10-7
|
POS
|
1,19 × 10-9
|
1,96 × 10-4
|
mäßig POS
|
POS
|
Kohlenwasserstoff (cyclisch)
|
Pharmazeutisches Erzeugnis
|
|
3
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
10-10 – 10-5
|
POS
|
8,17 × 10-7
|
2,69 × 10-4
|
POS
|
POS
|
Kohlenwasserstoff (cyclisch)
|
Pharmazeutisches Erzeugnis
|
|
4
|
17β-Estradiol
|
50-28-2
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,67 × 10-3
|
vermutlich negativ (*4)
|
PN
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
5
|
Apigenin
|
520-36-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Heterocyclische Verbindung
|
Farbstoff, Naturprodukt, pharmazeutisches Zwischenprodukt
|
|
6
|
Di-n-butylphthalat
|
84-74-2
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Ester, Phthalsäure
|
Kosmetischer Inhaltsstoff, Industriechemikalie, Weichmacher
|
|
7
|
Flavon
|
525-82-6
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,50 × 10-4
|
vermutlich negativ (*4)
|
PN
|
Flavonoid, heterocyclische Verbindung
|
Naturprodukt, pharmazeutisches Erzeugnis
|
|
8
|
Genistein
|
446-72-0
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
vermutlich negativ (*4)
|
NEG
|
Flavonoid, heterocyclische Verbindung
|
Naturprodukt, pharmazeutisches Erzeugnis
|
|
9
|
p-n-Nonylphenol
|
104-40-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
4,54 × 10-4
|
nicht geprüft
|
NEG
|
Phenol
|
Chemisches Zwischenprodukt
|
|
10
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,38 × 10-4
|
vermutlich negativ (*4)
|
NEG
|
Kohlenwasserstoff (cyclisch)
|
Naturprodukt
|
|
Abkürzungen: CASRN = CAS-Registrierungsnummer; M = molar; IC50 = die Hälfte der maximalen Hemmkonzentration eines Prüfstoffs; NEG = negativ; PN = vermutlich negativ; POS = positiv.
|
Akzeptanzkriterien für Prüfläufe
|
16.
|
Ob ein Prüflauf akzeptiert wird, hängt von der Bewertung der Ergebnisse mit den jeweils verwendeten Referenzstandards und Kontrollen ab. Die PC50- (EC50-) bzw. die IC50-Werte bei den Referenzstandards sollten die Akzeptanzkriterien des betreffenden Assays erfüllen (zum STTA-Assay siehe Anlage 2 und zum VM7Luc-ER-TA-Assay siehe Anlage 3), und alle positiven/negativen Kontrollen sollten für jeden akzeptierten Versuch ordnungsgemäß eingestuft werden. Die Fähigkeit zur konsistenten Durchführung der Assays sollte durch den Aufbau und die Pflege einer historischen Datenbank für die Referenzstandards und Kontrollen nachgewiesen werden (Nummer 15). Standardabweichungen (SD) oder Variationskoeffizienten (VK) der Mittelwerte von Parametern zur Anpassung der Referenzstandardkurven aus mehreren Versuchen können als Maßstab für die Reproduzierbarkeit innerhalb eines Labors herangezogen werden. Darüber hinaus sollten für Akzeptanzkriterien die folgenden Grundsätze gelten:
|
—
|
Die Daten sollten hinreichend für eine quantitative Bewertung der ER-Aktivierung beim Agonisten-Assay) bzw. zur ER-Unterdrückung (beim Antagonisten-Assay) (d. h. zur Bewertung der Wirksamkeit und Leistungsfähigkeit) sein.
|
|
—
|
Die mittlere Reporter-Aktivität bei der Referenzkonzentration des Referenzöstrogens sollte wenigstens der bei den betreffenden Assays für die Vehikelkontrolle (Lösungsmittelkontrolle) spezifizierten Mindestaktivität entsprechen, damit eine angemessene Empfindlichkeit gewährleistet ist. Beim STTA- und beim VM7Luc-ER-TA-Assay beträgt diese Aktivität das Vierfache der Aktivität der mittleren Vehikelkontrolle der jeweiligen Platte.
|
|
—
|
Die geprüften Konzentrationen sollten im Löslichkeitsbereich der Prüfchemikalien bleiben und nicht zytotoxisch wirken.
|
|
Analyse der Daten
|
17.
|
Positive bzw. negative Reaktionen sollten nach dem für den jeweiligen Assay festgelegten Verfahren zur Auswertung der Daten analysiert werden.
|
|
18.
|
Die Erfüllung der Akzeptanzkriterien (Nummer 16) deutet darauf hin, dass ein Assay ordnungsgemäß funktioniert. Dies gewährleistet jedoch nicht, dass ein bestimmter Prüflauf tatsächlich exakte Daten ergibt. Die Wiederholung der Ergebnisse des ersten Prüflaufs ist die beste Bestätigung dafür, dass exakte Daten ermittelt wurden. Wenn zwei Prüfläufe zu reproduzierbaren Ergebnissen führen (d. h. wenn für eine Prüfchemikalie in beiden Prüfläufen ein positives Ergebnis ermittelt wurde), ist ein dritter Prüflauf nicht erforderlich.
|
|
19.
|
Führen zwei Prüfläufe zu unterschiedlichen Ergebnissen (z. B. bei einer einzigen Prüfchemikalie einmal ein positives und einmal ein negatives Ergebnis) oder wenn eine höhere Gewissheit hinsichtlich des Ergebnisses benötigt wird, sollten mindestens drei unabhängige Prüfläufe durchgeführt werden. In diesem Fall wird die Einstufung anhand der beiden übereinstimmenden Ergebnisse von drei Prüfläufen vorgenommen.
|
Allgemeine Kriterien für die Auswertung von Daten
|
20.
|
Zurzeit besteht keine allgemein anerkannte Methode zur Auswertung der Daten aus ER-TA-Assays. Allerdings sollten qualitative (z. B. positiv/negativ) und/oder quantitative (z. B. EC50, PC50 und IC50) Bewertungen einer durch ER vermittelten Aktivität auf empirischen Daten und fundierten wissenschaftlichen Beurteilungen beruhen. Nach Möglichkeit sollten positive Ergebnisse sowohl unter Angabe der Größenordnung der Wirkung im Vergleich zur Vehikelkontrolle (Lösungsmittelkontrolle) oder zum Referenzöstrogen als auch der Konzentration beschrieben werden, bei der die Wirkung eintritt (z. B. EC50, PC50, RPCMax oder IC50).
|
Prüfbericht
|
21.
|
Der Prüfbericht sollte folgende Angaben enthalten:
|
|
Gehalt:
|
—
|
Kontrolle/Referenzstandard/Prüfchemikalie;
|
|
—
|
Herkunft, Partienummer, ggf. begrenztes Verwendungsdatum;
|
|
—
|
Stabilität der Prüfchemikalie, falls bekannt;
|
|
—
|
Löslichkeit und Stabilität der Prüfchemikalie in Lösungsmittel, falls bekannt;
|
|
—
|
Messung des pH-Werts, Osmolalität und ggf. Niederschlag im Kulturmedium, dem die Prüfchemikalie zugegeben wurde.
|
|
|
|
Einkomponentiger Stoff:
|
—
|
physikalisches Erscheinungsbild, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
|
|
—
|
chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer, SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.
|
|
|
|
Mehrkomponentiger Stoff, UVCB-Stoffe und Gemische:
|
—
|
so weit wie möglich charakterisiert durch die chemische Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Komponenten.
|
|
|
|
Lösungsmittel/Vehikel:
|
—
|
Beschreibung (Art, Lieferant und Charge);
|
|
—
|
Begründung der Auswahl des Lösungsmittels/Vehikels;
|
|
—
|
Löslichkeit und Stabilität der Prüfchemikalie in Lösungsmittel/in einem Vehikel, falls bekannt.
|
|
|
|
Zellen:
|
—
|
Art und Herkunft der Zellen:
|
—
|
Wurde ER endogen exprimiert? Wenn nicht, welche Rezeptor(en) wurden transfiziert?
|
|
—
|
verwendete(s) Reporter-Genprodukt(e) (einschließlich Spezies);
|
|
—
|
(ggf.) Auswahlmethode zur Aufrechterhaltung einer stabilen Transfektion);
|
|
—
|
ist die Transfektionsmethode für stabile Linien relevant?
|
|
|
—
|
ggf. Passagenanzahl (nach dem Auftauen);
|
|
—
|
Passagenanzahl beim Auftauen;
|
|
—
|
zum Erhalt der Zellkultur verwendete Verfahren.
|
|
|
|
Prüfbedingungen:
|
—
|
Beschreibung der eingesetzten Methoden zur Bewertung der Viabilität;
|
|
—
|
Medienzusammensetzung, CO2-Konzentration;
|
|
—
|
Konzentrationen der Prüfchemikalie;
|
|
—
|
Volumen des Vehikels und der beigegebenen Prüfchemikalie;
|
|
—
|
Inkubationstemperatur und Feuchte;
|
|
—
|
Zelldichte zu Beginn und während der Behandlung;
|
|
—
|
positive und negative Referenzstandards;
|
|
—
|
Reporter-Reagenzien (Produktbezeichnung, Hersteller und Charge);
|
|
—
|
Kriterien zur Einstufung der Prüfläufe als positiv, negativ oder nicht eindeutig.
|
|
|
|
Akzeptanzprüfung:
|
—
|
n-fache Induktionen der einzelnen Assay-Platten und Angabe, ob damit die Mindestanforderungen des betreffenden Assays nach historischen Kontrollen erfüllt werden;
|
|
—
|
tatsächliche Werte der Akzeptanzkriterien, z. B. log10-, EC50, log10-, PC50-, logIC50- und Hillslope-Werte für gleichzeitige Positivkontrollen/Referenzstandards.
|
|
|
|
Ergebnisse:
|
—
|
Rohdaten und normalisierte Daten;
|
|
—
|
höchste n-fache Induktion;
|
|
—
|
wenn vorhanden, niedrigste Wirkungskonzentration (LEC);
|
|
—
|
ggf. RPCMax, PCMax, PC50, IC50 und/oder EC50;
|
|
—
|
nach Möglichkeit Konzentrations-Wirkungs-Verhältnis;
|
|
—
|
wenn vorhanden, statistische Analysen sowie Messabweichung und Konfidenzwerte (z. B. SEM, SD, VK oder 95 % CI) sowie Angaben dazu, wie diese Werte ermittelt wurden.
|
|
|
|
Erörterung der Ergebnisse.
|
|
LITERATUR
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 34), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol, P., u. a. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): S. 5367-5373.
|
|
(5)
|
Rogers, J.M. und Denison, M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): S. 67-82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(8)
|
OECD (2012). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 150), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: S. 20-26.
|
|
(10)
|
Welboren, W.J., u. a. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): S. 1073-1089.
|
|
(11)
|
Younes, M. und Honma, N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): S. 63-66.
|
|
(12)
|
Jefferson, W.N., u. a. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): S. 179-189.
|
|
(13)
|
Sonneveld, E., u. a. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): S. 173-187.
|
|
(14)
|
Takeyoshi, M., u. a. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): S. 91- 98.
|
|
(15)
|
Combes, R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals, 28(1): S. 81-118.
|
|
(16)
|
Escande, A., u. a. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol, 71(10): S. 1459-1469.
|
|
(17)
|
Gray, L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson, J.Ö. (1999). Estrogen Receptor ß – A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): S. 379-383.
|
|
(21)
|
Ogawa, S., u. a. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERαIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): S. 122-126.
|
|
(22)
|
Enmark, E., u. a. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism, 82(12): S. 4258-4265.
|
|
(23)
|
Ball, L.J., u. a. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): S. 204-211.
|
|
(24)
|
Barkhem, T., u. a. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): S. 105-112.
|
|
(25)
|
Deroo, B.J. und Buensuceso, A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): S. 1703-1714.
|
|
(26)
|
Harris, D.M., u. a. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): S. 558-568.
|
|
(27)
|
Anderson, J.N., Clark, J.H. und Peck, E.J. Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): S. 1460-1468.
|
|
(28)
|
Toft, D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): S. 515-522.
|
|
(29)
|
Gorski, J., u. a. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: S. 45-80.
|
|
(30)
|
Jensen, E.V., u. a. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3): S. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno, J., u. a. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno, J., u. a. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose-Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno, J., u. a. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger u. a. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin u. a. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. u. a. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. und Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Anlage 1
BEGRIFFSBESTIMMUNGEN UND ABKÜRZUNGEN
Agonist
: Ein Stoff, der eine Wirkung (z. B. eine Transkription) hervorruft, wenn er eine Bindung mit einem bestimmten Rezeptor eingeht.
Aktivkohle-/Dextranbehandlung
: Behandlung von bei Zellkulturen verwendeten Seren. Bei der Behandlung mit Aktivkohle/Dextran (häufig auch als „Stripping“ bezeichnet) werden endogene Hormone und hormonbindende Proteine abgetrennt.
Akzeptanzkriterien
: Mindeststandards für Kontrollen und Referenzstandards. Damit ein Versuch als gültig betrachtet werden kann, sollten alle Akzeptanzkriterien erfüllt sein.
Antagonist
: Eine Art Rezeptor-Ligand oder eine Chemikalie, die nach der Bindung an einen Rezeptor an sich keine biologische Reaktion auslöst, aber durch Agonisten vermittelte Reaktionen blockiert oder dämpft.
Antiöstrogene Aktivität
: Fähigkeit einer Chemikalie zur Unterdrückung der durch Östrogenrezeptoren vermittelten Wirkung von 17β-Estradiol.
Assay
: Im Zusammenhang mit dieser Prüfmethode wird als Assay eine Methode bezeichnet, die die beschriebenen Leistungskriterien erfüllt und daher als gültige Methode anerkannt wird. Elemente eines Assays sind beispielsweise die spezifische Zelllinie mit den entsprechenden Wachstumsbedingungen, spezifische Medien, in denen die Untersuchung erfolgt, die Konfiguration der Platten, Anordnung und Verdünnungen von Prüfchemikalien sowie alle sonstigen erforderlichen Qualitätskontrollmaßnahmen und mit der Untersuchung verbundenen Schritte zur Auswertung der Daten.
CF
: OECD Conceptual Framework for the Testing and Evaluation of Endocrine Disrupters.
Chemikalie
: Ein Stoff oder ein Gemisch.
DCC-FBS
: Mit dextranbeschichteter Aktivkohle behandeltes fetales Rinderserum.
DMEM
: Dulbecco's modifiziertes Eagle-Medium.
DMSO
: Dimethylsulfoxid.
E2
: 17β-Estradiol.
EC50
: Die Hälfte der maximalen Wirkungskonzentration einer Prüfchemikalie.
ED
: Endokrine Störung.
EFM
: Östrogenfreies Medium. Dulbecco's modifiziertes Eagle-Medium (DMEM), ergänzt um 4,5 % mit Aktivkohle/Dextran behandeltes fetales Rinderserum (FBS), 1,9 % L-Glutamin und 0,9 % Pen/Strep.
Eignung
: Die nachgewiesene Fähigkeit zur ordnungsgemäßen Durchführung eines Assays vor der Prüfung unbekannter Stoffe.
ER
: Östrogenrezeptor.
ERE
: Östrogen-Response-Element.
ER-TA
: Östrogenrezeptor-Transaktivierung
FBS
: Fetales Rinderserum.
Genauigkeit (Übereinstimmung)
: Der Grad an Übereinstimmung zwischen den Ergebnissen eines Assays und akzeptierten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung eines Assays und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von „Übereinstimmung“ verwendet und bezeichnet den Anteil der korrekten Ergebnisse eines Assays (1).
HeLa
: Eine unsterbliche humane Zervikalkarzinom-Zelllinie.
HeLa9903
: Ein Subklon der HeLa-Zelllinie, in den hERαα und ein Luciferase-Reporter-Gen stabil transfiziert wurden.
hERß
: Humaner Östrogenrezeptor ß.
hERα
: Humaner Östrogenrezeptor α.
IC50
: Die Hälfte der maximalen Wirkungskonzentration einer Prüfchemikalie mit hemmender Wirkung.
ICCVAM
: Interagency Coordinating Committee on the Validation of Alternative Methods (das US-amerikanische Validierungszentrum).
Inter-Labor-Reproduzierbarkeit
: Das Ausmaß, in dem unterschiedliche qualifizierte Laboratorien, die dasselbe Protokoll verwenden und dieselben Prüfstoffe untersuchen, qualitativ und quantitativ vergleichbare Ergebnisse erzielen können. Die Inter-Labor-Reproduzierbarkeit wird während der Prävalidierungs- und Validierungsverfahren ermittelt und zeigt das Maß an, in dem ein Assay erfolgreich zwischen Laboratorien übertragen werden kann (1).
Intra-Labor-Reproduzierbarkeit
: Das Ausmaß, in dem qualifizierte Personen innerhalb desselben Labors, die dasselbe spezifische Protokoll zu unterschiedlichen Zeiten verwenden, erfolgreich dieselben Ergebnisse replizieren können. Auch als „laborinterne Reproduzierbarkeit“ bezeichnet (1).
LEC
: Als niedrigste Wirkungskonzentration (Lowest Effective Concentration) wird die niedrigste Konzentration einer Prüfchemikalie bezeichnet, die eine Wirkung hervorruft (d. h. die niedrigste Konzentration einer Prüfchemikalie, bei der die n-fache Induktion sich statistisch von der gleichzeitigen Vehikelkontrolle unterscheidet).
Leistungsstandards
: Auf einem validierten Assay beruhende Normen, auf deren Grundlage die Vergleichbarkeit einem vorgeschlagenen, mechanistisch und funktionell ähnlichen Assay bewertet werden kann. Sie umfassen (1) wesentliche Elemente des Assays; (2) ein Mindestverzeichnis von Referenzchemikalien, ausgewählt aus den Chemikalien, die zum Nachweis der akzeptablen Leistung der validierten Referenzmethode verwendet werden, und (3) je nach den für die validierte Referenzmethode erzielten Ergebnissen die vergleichbaren Genauigkeits- und Zuverlässigkeitswerte, die der vorgeschlagene Assay bei der Bewertung anhand des Mindestverzeichnisses von Referenzchemikalien ergeben sollte (1).
Leistungsstoffe
: Eine Untergruppe der in die Leistungsstandards einbezogenen Referenzstoffe, die von Labors bei standardisierten Prüfmethoden zum Nachweis der fachlichen Kompetenz eingesetzt werden können. Auswahlkriterien für diese Stoffe sind u. a., dass sie den Reaktionsbereich abdecken und im Handel erhältlich sein müssen und dass für diese Stoffe hochwertige Referenzdaten verfügbar sein müssen.
Me-Too-Prüfung
: [Im englischen Sprachraum gemeinsprachliche] Bezeichnung einer Prüfmethode, die strukturell und funktionell mit einer validierten und akzeptierten Referenzprüfmethode vergleichbar ist. Gleichbedeutend mit „vergleichbare Prüfmethode“ verwendet.
MMTV
: Maus-Mammatumorvirus.
MT
: Metallothionein.
OHT
: 4-Hydroxytamoxifen.
Östrogenaktivität
: Fähigkeit einer Chemikalie, wie 17β-Estradiol Bindungen Östrogenrezeptoren einzugehen und Östrogenrezeptoren zu aktivieren. Mit dieser Prüfmethode kann durch hERα vermittelte Östrogenaktivität nachgewiesen werden.
PBTG
: Leistungsbezogene Prüfrichtlinie.
PC10
: Die Konzentration einer Prüfchemikalie, bei der die gemessene Aktivität in einem Agonisten-Assay auf jeder einzelnen Platte 10 % der durch die PK (1 nM E2 beim STTA-Assay) induzierten maximalen Aktivität beträgt.
PC50
: Die Konzentration einer Prüfchemikalie, bei der die gemessene Aktivität in einem Agonisten-Assay auf jeder einzelnen Platte 50 % der durch die PK (E2 in der für die jeweilige Prüfmethode spezifizierten Referenzkonzentration) induzierten maximalen Aktivität beträgt.
PCMax
: Die Konzentration einer Prüfchemikalie, die RPCMax induziert.
PK (Positivkontrolle)
: Ein starker Wirkstoff, vorzugsweise 17ß-Estradiol, der in alle Prüfungen einbezogen wird, um das ordnungsgemäße Funktionieren der Assays sicherzustellen.
Prüfchemikalie
: Stoff oder Gemisch, der/das mit dieser Prüfmethode untersucht wird.
Prüflauf
: Ein einzelner Versuch zur Bewertung der Wirkung einer Chemikalie auf das biologische Ergebnis eines Assays. Jeder Prüflauf besteht aus einem vollständigen Versuch an Replikat-Wells mit Zellen, die gleichzeitig aus einem gemeinsamen Zellpool plattiert wurden.
Referenz-Östrogen (Positivkontrolle, PK)
: 17β-Estradiol (E2, CAS 50-28-2).
Referenzprüfmethode
: Die Assays, auf denen die PBTG 455 beruht.
Referenzstandard
: Ein Referenzstoff zum Nachweis der Eignung eines Assays. 17β-Estradiol ist der Referenzstandard für den STTA- und den VM7Luc-ER-TA-Assay.
Relevanz
: Dieser Begriff bezieht sich auf das Verhältnis zwischen dem Assay und der betreffenden Wirkung und auf die Frage, ob er aussagekräftig und nützlich für einen bestimmten Zweck ist. Er beschreibt das Ausmaß, in dem der Assay die untersuchte biologische Wirkung korrekt misst oder vorhersagt. Die Relevanz schließt eine Beurteilung der Genauigkeit (Übereinstimmung) eines Assays ein (1).
RLU
: Relative Lichteinheiten.
RNA
: Ribonukleinsäure.
RPCMax
: Durch eine Prüfchemikalie induzierte maximale Reaktion, ausgedrückt als Prozentanteil der durch 1 nM E2 auf derselben Platte induzierten Reaktion.
RPMI
: RPMI-1640-Medium, ergänzt um 0,9 % Pen/Strep und 8,0 % fetales Rinderserum (FBS).
Schwache Positivkontrolle
: Ein schwacher Wirkstoff aus der Liste der Referenzchemikalien, der in alle Prüfungen einbezogen wird, um das ordnungsgemäße Funktionieren der Assays sicherzustellen.
SD
: Standardabweichung,
Sensitivität
: Der Anteil aller positiven/wirkenden Prüfstoffe, die durch den Assay korrekt eingestuft werden. Die Sensitivität ist ein Maß der Genauigkeit eines Assays mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung seiner Relevanz (1).
Spezifität
: Der Anteil aller negativen/wirkungslosen Prüfstoffe, die durch die Prüfung korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit eines Assays mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung seiner Relevanz (1).
Stabile Transfektion
: Wenn DNA so in Zellkulturen transfiziert wird, dass sie stabil in das Zellgenom eingefügt wird, erfolgt eine stabile Expression der betreffenden Gene. Klone stabil transfizierter Zellen werden unter Verwendung stabiler Marker (z. B. Resistenz gegenüber G418) selektioniert.
Stoff
: In der REACH-Verordnung (62) werden Stoffe definiert als ein chemisches Element und seine Verbindungen in natürlicher Form oder gewonnen durch ein Herstellungsverfahren, einschließlich der zur Wahrung seiner Stabilität notwendigen Zusatzstoffe und der durch das angewandte Verfahren bedingten Verunreinigungen, aber mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können. Eine sehr ähnliche Begriffsbestimmung wird im Zusammenhang mit dem UN GHS verwendet (1).
STTA-Assay
: Assay mit transkriptioneller ERα-Aktivierung unter Verwendung der Zelllinie HeLa 9903.
Studie/Untersuchung
: Eine umfassende experimentelle Untersuchung zur Bewertung eines einzelnen, spezifischen Stoffs mit einem spezifischen Assay. Eine Studie umfasst sämtliche Schritte einschließlich Untersuchungen von Verdünnungen eines Prüfstoffs im Prüfmedium, Vorversuchen zur Dosisfindung, allen erforderlichen Hauptversuchen, Datenanalysen, Qualitätssicherung, Zytotoxizitätsbewertungen usw. Aufgrund einer Studie kann die mit dem jeweiligen Assay zu untersuchende Wirkung der Prüfchemikalie auf das Zielmaterial einer Toxizitätsprüfung bewertet werden (als aktiv, nicht aktiv oder nicht eindeutig) und die Stärke der Wirkung bezogen auf die positive Referenzchemikalie abgeschätzt werden.
TA (Transaktivierung): Die Auslösung einer mRNA-Synthese als Reaktion auf ein bestimmtes chemisches Signal wie beispielsweise die Bindung eines Östrogens an einen Östrogenrezeptor.
Transkription
: mRNA-Synthese.
Unabhängiger Prüflauf
: Ein getrennter unabhängiger Versuch, mit dem die Wirkung einer Chemikalie auf das biologische Ergebnis eines Assays mit Zellen aus einem anderen Pool sowie mit frisch verdünnten Chemikalien an unterschiedlichen Tagen oder am selben Tag durch unterschiedliche Labortechniker bewertet wird.
UVCB
: Chemische Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Validierte Prüfmethode
: Ein Assay, für den zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien durchgeführt wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht so genau und zuverlässig ist, dass sie für den vorgeschlagenen Zweck akzeptiert werden kann (1).
Validierung
: Prozess, mit dem die Zuverlässigkeit und die Relevanz eines bestimmten Ansatzes, einer Methode, eines Assays, eines Prozesses oder einer Bewertung für einen bestimmten Zweck festgestellt wird (1).
VK (Vehikelkontrolle)
: Das zur Lösung der Prüf- und der Kontrollchemikalien verwendete Lösungsmittel wird ohne gelöste Chemikalie nur als Vehikel geprüft.
VK
: Variationskoeffizient.
VM7
: Eine immortalisierte Adenokarzinom-Zelllinie, die einen Östrogenrezeptor endogen exprimiert.
VM7Luc4E2
: Die VM7Luc4E2-Zelllinie wurde aus immortalisierten VM7-Zellen (humanen Adenokarzinom-Zellen) abgeleitet, die beide Formen von Endogenrezeptoren (ERα und ERβ) endogen exprimieren und mit dem Plasmid pGudLuc7.ERE stabil transfiziert wurden. Dieses Plasmid enthält vier Kopien eines synthetischen Oligonucleotids mit dem Östrogen-Response-Element vor dem MMTV-Promoter (MMTV = Maus-Mammatumorvirus) und dem Leuchtkäfer-Luciferase-Gen.
Zellmorphologie
: Form und Aussehen von Zellen, die in einer Monolayer-Kultur in einem einzelnen Well einer Gewebekultur-Platte gewachsen sind. Absterbende Zellen weisen häufig eine abnormale Zellmorphologie auf.
Zuverlässigkeit
: Maß der Verlässlichkeit der Reproduzierbarkeit eines Assays innerhalb von und zwischen Laboratorien (Intra- und Inter-Labor-Reproduzierbarkeit) in einem bestimmten Zeitintervall bei einheitlichem Protokoll. Sie wird durch Berechnung der Intra- und Inter-Labor-Reproduzierbarkeit bewertet.
Zytotoxizität
: Gefährliche Wirkungen auf Zellstrukturen oder Funktionen, die letztlich zum Absterben von Zellen führen und sich in einer Reduzierung der Anzahl der Zellen auf dem Well am Ende des Expositionszeitraums oder in einer reduzierten Messbarkeit von Zellfunktionen im Vergleich zur gleichzeitigen Vehikelkontrolle äußern können.
Anlage 2
ASSAY ZUR TRANSAKTIVIERUNG DES STABIL TRANSFIZIERTEN HUMANEN ÖSTROGENREZEPTORS Α ZUM NACHWEIS DER WIRKUNG VON CHEMIKALIEN AUF ÖSTROGEN-AGONISTEN UND -ANTAGONISTEN MIT DER HERΑ-HELA-9903-ZELLLINIE
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
1.
|
Bei diesem Transaktivierungs-Assay (TA-Assay) wird die hERα-HeLa-9903-Zelllinie zum Nachweis einer durch den humanen Östrogenrezeptor α (hERα) vermittelten Östrogen-Agonisten-Aktivität verwendet. Mit der Validierungsstudie des japanischen Chemikalienevaluierungs- und -forschungsinstituts CERI zum STTA-Assay (STTA = Stably Transfected Transactivation) unter Verwendung der hERα-HeLa-9903-Zelllinie zum Nachweis der durch den humanen Östrogenrezeptor α (hERα) vermittelten Östrogen-Agonisten- und -Antagonisten-Aktivität wurden die Relevanz des Assays für den vorgesehenen Zweck und seine Zuverlässigkeit nachgewiesen (1).
|
|
2.
|
Dieser Assay wurde speziell zum Nachweis von hERα und einer durch hERα vermittelten TA anhand einer Messung der Chemiluminiszenz als Endpunkt entwickelt. Bei Phytoöstrogenkonzentrationen von mehr als 1 μM wurden infolge der Überaktivierung des Luciferase-Reporter-Gens Luminiszenzsignale festgestellt, die nicht durch Rezeptoren vermittelt wurden (2) (3). Die Dosis-Reaktions-Kurve zeigt, dass die tatsächliche Aktivierung des ER-Systems bei niedrigeren Konzentrationen erfolgt; bei stabil transfizierten ER-TA-Assay-Systemen muss die Luciferase-Expression bei hohen Konzentrationen von Phytoöstrogenen oder ähnlichen Vereinbarungen, bei denen vermutet wird, dass sie eine phytoöstrogenartige Überaktivierung des Luciferase-Reportergens verursachen, jedoch sorgfältig untersucht werden (Anlage 1).
|
|
3.
|
Vor der Verwendung dieses Assays für rechtliche Zwecke sollten die Abschnitte „ALLGEMEINE EINLEITUNG“ und „ELEMENTE DES ER-TA-ASSAYS“ gelesen werden. Die in dieser Prüfrichtlinie verwendeten Begriffe und Abkürzungen werden in Anlage 2.1 erläutert.
|
PRINZIP DES ASSAYS (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
4.
|
Der Assay wird zur Feststellung von Bindungen des Östrogenrezeptors mit einem Liganden verwendet. Nach der Bindung an den Liganden transloziert der Rezeptor-Liganden-Komplex in den Zellkern, wo er spezifische DNA-Response-Elemente bindet und ein Leuchtkäfer-Luciferase-Reporter-Gen transaktiviert. Dies führt zu einer verstärkten zellulären Expression des Luciferase-Enzyms. Luciferin ist ein Substrat, das durch das Luciferase-Enzym in ein Biolumineszenz-Produkt umgewandelt wird, das mit einem Luminometer quantitativ gemessen werden kann. Die Luciferase-Aktivität kann rasch und kostengünstig mit verschiedenen im Handel erhältlichen Test-Kits bewertet werden.
|
|
5.
|
Bei diesem Prüfsystem wird die aus einem humanen Zervikaltumor abgeleitete hERα-HeLa-9903-Zelllinie mit zwei stabil eingefügten Produkten verwendet: (i) der hERαα-Expression (mit in voller Länge kodiertem humanem Rezeptor) und (ii) einem Leuchtkäfer-Luciferase-Reporterprodukt mit fünf Tandem-Repeats eines auf ein Maus-Metallothionein(MT)-Promoter-TA-Element reagierenden Vitellogenin-Östrogen-Response-Elements (ERE). Da mit dem Maus-MT-TATA-Genprodukt nachweislich die besten Ergebnisse erzielt werden, wird gewöhnlich dieses Produkt verwendet. Mit dieser hERα-HeLa-9903-Zelllinie kann die Fähigkeit einer Prüfchemikalie zur Induzierung einer durch hERα vermittelten Transaktivierung einer Luciferasegen-Expression gemessen werden.
|
|
6.
|
Beim ER-Agonisten-Assay hängt die Auswertung der Daten davon ab, ob die durch eine Prüfchemikalie induzierte maximale Reaktion größer oder gleich einer Agonisten-Reaktion im Umfang von 10 % der Reaktion ist, die durch eine Konzentration der Positivkontrolle (PK) 17β-Estradiol (E2) mit maximaler Induktion (1 nM) (d. h. PC10) induziert wird. Beim ER-Antagonisten-Assay wird die Auswertung der Daten davon bestimmt, ob die Reaktion mindestens in einer 30 %igen Reduzierung der Aktivität infolge der Spike-in-Kontrolle (25 pM E2) ohne Zytotoxizität besteht. Analysen und Auswertungen der Daten siehe Nummern 34-48.
|
VERFAHREN
Zelllinien
|
7.
|
Für den Assay sollte die stabil transfizierte hERα-HeLa-9903-Zelllinie verwendet werden. Diese Zelllinie kann von der JCRB-Zellbank (JRCB = Japanese Collection of Research Bioresources) (63) nach Unterzeichnung einer Materialübertragungsvereinbarung (Material Transfer Agreement, MTA) bezogen werden.
|
|
8.
|
Für die Prüfungen sollten ausschließlich als mycoplasmafrei charakterisierte Zellen verwendet werden. Die RT-PCR (Echtzeit-Polymerase-Kettenreaktion) ist die Methode der Wahl für den empfindlichen Nachweis einer Mycoplasma-Infektion (4) (5) (6).
|
Stabilität der Zelllinie
|
9.
|
Zur Überwachung der Stabilität der Zelllinie sollten E2, 17α-Estradiol, 17α-Methyltestosteron und Corticosteron als Referenzstandards für den Agonisten-Assay verwendet werden, und bei jeder Durchführung des Assays sollte mindestens eine vollständige Konzentrations-Reaktions-Kurve in dem in Tabelle 1 genannten Prüfkonzentrationsbereich gemessen werden. Dabei sollten die in Tabelle 1 genannten Ergebnisse ermittelt werden.
|
|
10.
|
Beim Antagonisten-Assays sollten für jeden Prüflauf gleichzeitig vollständige Konzentrationskurven für die beiden Referenzstandards Tamoxifen und Flutamid gemessen werden. Die ordnungsgemäße Einstufung der beiden Chemikalien als positiv oder negativ sollte überwacht werden.
|
Bedingungen für die Haltung der Zellkulturen und für die Plattierung
|
11.
|
Die Zellen werden in Eagle’s Minimum Essential Medium (EMEM) ohne Phenolrot ergänzt mit 60 mg/l des Antibiotikums Kanamycin und 10 % mit dextranbeschichteter Aktivkohle behandeltem fetalem Rinderserumdextran (DCC-FBS) in einem CO2-Inkubator (5 % CO2) bei 37 ± 1 °C gehalten. Wenn eine Konfluenz von 75-90 % erreicht ist, können Zellen mit 10 ml 0,4 × 105 – 1 × 105 Zellen/ml auf einer 100-mm-Zellkulturschale subkultiviert werden. Dann werden die Zellen mit 10 % FBS-EMEM (identisch mit EMEM mit DCC-FBS) suspendiert und anschließend mit einer Dichte von 1 × 104 Zellen/(100 μl × Well) in Wells einer Mikrotiterplatte plattiert. Danach werden die Zellen in einem CO2-Inkubator (5 %) bei 37 ± 1 °C drei Stunden vorinkubiert, bevor sie der zu untersuchenden Chemikalie ausgesetzt werden. Die Verbrauchsmaterialien aus Kunststoff sollten keine Östrogenaktivität aufweisen.
|
|
12.
|
Um die Integrität der Reaktion aufrechtzuerhalten, sollten die Zellen im konditionierten Medium über mehr als eine Passage höchstens aber bis zu 40 Passagen des tiefgefrorenen Materials vermehrt werden. Bei der hERα-HeLa-9903-Zelllinie dauert dies weniger als drei Monate. Das Zellwachstum kann jedoch verringert sein, wenn die Zellen unter ungeeigneten Kulturbedingungen vermehrt werden.
|
|
13.
|
Das DCC-FBS kann wie in Anlage 2.2 beschrieben zubereitet oder im Handel bezogen werden.
|
Akzeptanzkriterien
Positive und negative Referenzstandards für den ER-Agonisten-Assay
|
14.
|
Vor und während der Untersuchung sollte die Reaktion des Prüfsystems mit den jeweils geeigneten Konzentrationen eines starken Östrogens verifiziert werden: E2, ein schwaches Östrogen (17α-Estradiol), ein sehr schwacher Agonist (17α-Methyltestosteron) und ein Negativstoff (Corticosteron). Tabelle 1 sind annehmbare Bereichswerte angegeben, die aus der Validierungsstudie (1) entnommen wurden. Diese 4 gleichzeitigen Referenzstandards sollten in jeden Versuch einbezogen werden, und die Ergebnisse sollten jeweils im annehmbaren Bereich liegen. Ansonsten sollten die Ursachen dafür ermittelt werden, dass die Akzeptanzkriterien nicht erfüllt werden (z. B. die Handhabung der und der Assay sollte wiederholt werden. Wenn die Akzeptanzkriterien erfüllt werden, ist die konsistente Verwendung von Materialien für die Kultivierung der Zellen von entscheidender Bedeutung, um eine möglichst geringe Variabilität der EC50-, PC50- und PC10-Werte sicherzustellen. Die vier gleichzeitigen Referenzstandards, die in alle (unter identischen Bedingungen u. a. in Bezug auf die Materialien, die Passagenanzahl und die Labortechniker durchzuführenden) Versuche einbezogen werden sollten, können die Empfindlichkeit des Assays gewährleisten, da die PC10-Werte der drei positiven Referenzstandards ebenso wie die PC50- und die EC50-Werte (wenn diese berechnet werden können) im annehmbaren Bereich liegen sollten (siehe Tabelle 1).
|
Tabelle 1
Werte im annehmbaren Bereich für die vier Referenzstandards beim ER-Agonisten-Assay
|
Name
|
logPC50
|
logPC10
|
logPC50
|
Hillslope
|
Prüfbereich
|
|
17β-Estradiol (E2) CAS-Nr.: 50-28-2
|
-11,4~-10,1
|
< -11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-Estradiol CAS-Nr.: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Corticosteron CAS-Nr.: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-Methyltestosteron CAS-Nr.: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5M
|
Positive und negative Referenzstandards für den ER-Antagonisten-Assay
|
15.
|
Vor und während der Untersuchung sollte die Reaktion des Prüfsystems mit den jeweils geeigneten Konzentrationen eines Positivstoffs (Tamoxifen) und eines Negativstoffs (Flutamid) verifiziert werden: Tabelle 2 sind annehmbare Bereichswerte angegeben, die aus der Validierungsstudie (1) entnommen wurden. Diese beiden gleichzeitigen Referenzstandards sollten in jeden Versuch einbezogen werden, und die Ergebnisse sollten anhand der Kriterien als akzeptabel bewertet werden. Andernfalls sollten die Ursachen dafür ermittelt werden, dass die Kriterien nicht erfüllt werden (z. B. die Handhabung der Zellen sowie die Qualität und die Konzentration des Serums und der Antibiotika), und der Assay sollte wiederholt werden. Außerdem sollten die IC50-Werte eines Positivstoffs (Tamoxifen) berechnet werden. Die Ergebnisse sollten im annehmbaren Bereich liegen. Wenn die Akzeptanzkriterien erfüllt werden, ist die konsistente Verwendung von Materialien für die Kultivierung der Zellen von entscheidender Bedeutung, um eine möglichst geringe Variabilität der IC50-Werte sicherzustellen. Die beiden gleichzeitigen Referenzstandards, die in jeden (unter identischen Bedingungen u. a. in Bezug auf die Materialien, die Passagenanzahl und die Labortechniker durchzuführenden) Versuch einbezogen werden sollten, können die Empfindlichkeit des Assays sicherstellen (siehe Tabelle 2).
|
Tabelle 2
Kriterien und Werte im annehmbaren Bereich für die beiden Referenzstandards beim ER-Antagonisten-Assay
|
Name
|
Kriterium
|
LogIC50
|
Prüfbereich
|
|
Tamoxifen CAS-Nr.: 10540-29-1
|
Positiv: IC50 sollte berechnet werden.
|
-5,942~ -7,596
|
10-10~10-5 M
|
|
Flutamid CAS-Nr.: 13311-84-7
|
Negativ: IC30 sollte nicht berechnet werden.
|
—
|
10-10 ~10-5 M
|
Positivkontrolle und Vehikelkontrolle
|
16.
|
Die Positivkontrolle (PK) des ER-Agonisten-Assays (1 nM E2) und des ER-Antagonisten-Assays (10μM TAM) sollte pro Platte mindestens dreimal geprüft werden. Auch das zur Auflösung einer Prüfchemikalie verwendete Vehikel ist als Vehikelkontrolle (VK) pro Platte mindestens dreimal zu prüfen. Wenn bei der PK nicht die Prüfchemikalie als Vehikel verwendet wird, sollte zusätzlich zu dieser VK auf der Platte mit der PK eine weitere VK ebenfalls mindestens dreimal geprüft werden.
|
Qualitätskriterien für den ER-Agonisten-Assay
|
17.
|
Die mittlere Luciferase-Aktivität der Positivkontrolle (1 nM E2) sollte mindestens das Vierfache der mittleren VK auf jeder Platte betragen. Dieses Kriterium wird anhand der Zuverlässigkeit der Endpunktwerte der Validierungsstudie geprüft. (In der Regel werden Werte zwischen dem 4- und dem 30-Fachen ermittelt.)
|
|
18.
|
Bei der Qualitätskontrolle für diesen Assay sollte die n-fache Induktion entsprechend dem PC10-Wert der gleichzeitigen PK (1 nM E2) größer als 1+2 SD des Wertes der n-fachen Induktion (= 1) der gleichzeitigen VK sein. Bei der Priorisierung kann der PC10-Wert die erforderliche Datenanalyse im Vergleich zu einer statistischen Analyse erleichtern. Eine statistische Analyse liefert wichtige Informationen, ist aber nicht als quantitativer Parameter hinsichtlich eines konzentrationsabhängigen Potenzials zu betrachten und für Priorisierungszwecke daher weniger hilfreich.
|
Qualitätskriterien für den ER-Antagonisten-Assay
|
19.
|
Die mittlere Luciferase-Aktivität der Spike-in-Kontrolle (25 nM E2) sollte mindestens das 4-Fache der mittleren VK auf jeder Platte betragen. Dieses Kriterium wird anhand der Zuverlässigkeit der Endpunktwerte der Validierungsstudie geprüft.
|
|
20.
|
Bei der Qualitätskontrolle des Assays sollte die relative transkriptionelle Aktivierung (RTA) von 1 nM E2 mehr als 100 % betragen; die RTA von 1 μM 4-Hydroxytamoxifen (OHT) sollte bei weniger als 40,6 % und die RTA von 100 μM Digitonin (Dig) bei weniger als 0 % liegen.
Zum Nachweis der Eignung des Labors siehe Nummer 14 und Tabellen 3 und 4 im Abschnitt „ELEMENTE DES ER-TA-ASSAYS“ bei dieser Prüfmethode.
|
Vehikel
|
21.
|
Als gleichzeitige VK sollte Dimethylsulfoxid (DMSO) bzw. ein anderes geeignetes Lösungsmittel in der Konzentration verwendet werden, die auch bei den verschiedenen Positiv- und Negativkontrollen sowie bei den Prüfchemikalien zum Einsatz kommt. Die Prüfchemikalien sollten in einem Lösungsmittel gelöst werden, das die betreffende Prüfchemikalie löst und sich mit dem Zellmedium mischt. Geeignete Vehikel sind Wasser, Ethanol (Reinheit 95-100 %) und DMSO. Wenn DMSO verwendet wird, sollte die Konzentration höchstens 0,1 % (v/v) betragen. Bei jedem Vehikel sollte nachgewiesen werden, dass die verwendete Höchstmenge nicht zytotoxisch ist und keine Auswirkungen auf den Assay hat.
|
Zubereitung der Prüfchemikalien
|
22.
|
Im Allgemeinen sollten die Prüfchemikalien in DMSO oder in einem anderen geeigneten Lösungsmittel gelöst und mit diesem Lösungsmittel seriell im üblichen Verhältnis von 1:10 gelöst werden, um Lösungen zur Verdünnung mit Medien zuzubereiten.
|
Löslichkeit und Zytotoxizität: Hinweise zur Dosisfindung
|
23.
|
Mit einem Vorversuch sollte der geeignete Konzentrationsbereich der zu prüfenden Chemikalie ermittelt und festgestellt werden, ob bei der Prüfchemikalie Probleme hinsichtlich der Löslichkeit und der Zytotoxizität bestehen. Zunächst werden die Chemikalien bis zur maximalen Konzentration von 1 μl/ml, 1 mg/ml bzw. 1 mM geprüft. (Maßgeblich ist der niedrigste Wert.) Je nach dem im Vorversuch festgestellten Grad der Zytotoxizität bzw. nach der festgestellten mangelnden Löslichkeit sollte die Prüfchemikalie beim ersten gültigen Prüflauf in logarithmischen, seriellen Verdünnungen beginnend mit der maximal annehmbaren Konzentration (1 mM, 100 μM, 10 μM usw.) untersucht werden. Die Bildung von Trübungen oder Niederschlag sowie eine festgestellte Zytotoxizität sollten protokolliert werden. Die Konzentrationen im zweiten und ggf. im dritten Prüflauf sollten ggf. angepasst werden, um die Konzentrations-Reaktions-Kurve besser zu beschreiben und um Konzentrationen zu vermeiden, bei denen keine Lösung mehr erfolgt oder eine übermäßige Zytotoxizität induziert wird.
|
|
24.
|
Bei ER-Agonisten und -Antagonisten kann eine zunehmende Zytotoxizität die typische sigmoidale Reaktion erheblich verändern oder vollständig beseitigen und sollte daher bei der Auswertung der Daten berücksichtigt werden. Für die Prüfungen sollten Methoden zur Untersuchung der Zytotoxizität verwendet werden, aus denen Informationen für eine Zellviabilität von 80 % zu entnehmen sind. Die Untersuchung sollte mit einem nach Laborerfahrungen geeigneten Assay durchgeführt werden.
|
|
25.
|
Wenn in der Zytotoxizitätsprüfung festgestellt wird, dass die Konzentration der Prüfchemikalie die Anzahl der Zellen um 20 % oder mehr reduziert hat, ist die betreffende Konzentration als zytotoxisch zu betrachten, und Verdünnungen ab der zytotoxischen Konzentration aufwärts sollten bei der Auswertung nicht berücksichtigt werden.
|
Chemische Exposition und Anordnung auf der Assay-Platte
|
26.
|
Das Verfahren für chemische Verdünnungen (Schritte 1 und 2) sowie die Exposition der Zellen (Schritt 3) können wie folgt durchgeführt werden:
|
Schritt 1: Jede Prüfchemikalie sollte seriell in DMSO oder einem anderen geeigneten Lösungsmittel verdünnt und in die Wells einer Mikrotiterplatte gegeben werden, um die endgültigen seriellen Verdünnungen herzustellen, die in der Dosisfindungsstudie als maßgeblich ermittelt wurde (in der Regel z. B. 1 mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM und 10 pM (10-3-10-11 M)) bei dreifacher Prüfung.
Schritt 2: Chemische Verdünnung: Zunächst sind 1,5 μl der Prüfchemikalie im Lösungsmittel auf ein Medienvolumen von 500 μl zu verdünnen.
Schritt 3: Exposition der Zellen gegenüber der Chemikalie: 50 μl der (in Schritt 2 zubereiteten) Verdünnung mit dem Medium werden in einen Assay-Well mit 104 Zellen/100 μl/Well gegeben.
Für jedes Well wird ein endgültiges Medienvolumen von 150 μl als erforderlich empfohlen. Die zu untersuchenden Proben und Referenzstandards können angeordnet werden wie in den Tabellen 3 und 4 angegeben.
Tabelle 3
Beispiel für die Anordnung von Konzentrationen der Referenzstandards auf der Assay-Platte des ER-Agonisten-Assays
|
Reihe
|
17α-Methyltestosteron
|
Chemikalie
|
17α-Estradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
Konz. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
Konz. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
Konz. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
Konz. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
Konz. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
Konz. 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
Konz. 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VK
|
→
|
→
|
→
|
→
|
→
|
PK
|
→
|
→
|
→
|
→
|
→
|
|
VK: Vehikelkontrolle (0,1 % DMSO); PK: Positivkontrolle (1 nM E2)
|
|
27.
|
Die Referenzstandards (E2, 17α-Estradiol, 17- Methyltestosteron und Corticosteron) sollten bei jedem Prüflauf untersucht werden (Tabelle 3). PK-Wells mit 1 nM E2, die in ausschließlich mit DMSO (oder mit einem anderen geeigneten Lösungsmittel) behandelten E2- und VK-Wells eine maximale Induktion verursachen können, sollten in alle Assay-Platten einbezogen werden (Tabelle 4). Wenn in einem Versuch Zellen unterschiedlicher Herkunft (unterschiedliche Passagenzahl, unterschiedliche Chargen usw.) verwendet werden, sollten die Referenzstandards für die jeweilige Herkunft der Zellen geprüft werden.
|
Tabelle 4
Beispiel für die Anordnung von Konzentrationen der Prüf- und der Kontrollchemikalien auf der Assay-Platte des ER-Agonisten-Assays
|
Reihe
|
Prüfchemikalie 1
|
Prüfchemikalie 2
|
Prüfchemikalie 3
|
Prüfchemikalie 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
Konz. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
Konz. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
Konz. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
Konz. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
Konz. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
Konz. 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
Konz. 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VK
|
→
|
→
|
→
|
→
|
→
|
PK
|
→
|
→
|
→
|
→
|
→
|
|
VK: Vehikelkontrolle (0,1 % DMSO); PK: Positivkontrolle (1 nM E2)
|
Tabelle 5
Beispiel für die Anordnung von Konzentrationen der Referenzstandards auf der Assay-Platte des ER-Antagonisten-Assays

|
28.
|
Um die Antagonisten-Aktivität von Chemikalien zu ermitteln, sollten in Reihen von A bis G angeordnete Assay-Wells mit 25 pM E2 gespikt werden. Bei jedem Prüflauf sollten auch die Referenzstandards (Tamoxifen und Flutamid) geprüft werden. Jede Assay-Platte sollte mit 1 nM E2 behandelte PK-Wells, die als Qualitätskontrolle für die hERα-HeLa-9903-Zelllinie dienen kann, mit DMSO (oder einem anderen geeigneten Lösungsmittel) behandelte VK-Wells, Wells mit 0,1 % DMSO, die zusätzlich zum Spiken von E2 durch Zugabe von DMSO entsprechend der Spike-in-Kontrolle behandelt wurden, mit der Endkonzentration von 1 μM OHT behandelte Wells und mit 100 μM Dig behandelte Wells enthalten (Tabelle 5). Die nächste Assay-Platte sollte die gleiche Anordnung aufweisen, allerdings ohne die Wells mit den Referenzstandards (Tabelle 6). Wenn in einem Versuch Zellen unterschiedlicher Herkunft (unterschiedliche Passagenzahl, unterschiedliche Chargen usw.) verwendet werden, sollten die Referenzstandards für die jeweilige Herkunft der Zellen geprüft werden.
|
Tabelle 6
Beispiel für die Anordnung von Konzentrationen der Prüf- und der Kontrollchemikalien auf der Assay-Platte des ER-Antagonisten-Assays

|
29.
|
Dass keine Randeffekte auftreten, sollte ggf. bestätigt werden, und wenn das Auftreten von Randeffekten als möglich betrachtet wird, sollte die Anordnung der Wells auf den Platten geändert werden, um diese Effekte zu vermeiden. Beispielsweise könnte eine Anordnung gewählt werden, bei der die Rand-Wells nicht berücksichtigt werden.
|
|
30.
|
Nach Zugabe der Chemikalien sollten die Assay-Platten 20-24 Stunden in einem Inkubator mit 5 % CO2 bei 37 ± 1 oC inkubiert werden, um die Bildung von Reporter-Genprodukten zu induzieren.
|
|
31.
|
Bei stark flüchtigen Verbindungen ist besondere Vorsicht geboten. Bei diesen Verbindungen kann es bei unmittelbar benachbarten Kontroll-Wells zu falsch positiven Ergebnissen kommen. Dies sollte im Hinblick auf erwartete und historische Kontrollwerte beachtet werden. In den seltenen Fällen, in denen die Flüchtigkeit problematisch sein könnte, können Plattenversiegelungen helfen, einzelne Wells während der Prüfung zu isolieren. Dies ist in den betreffenden Fällen zu empfehlen.
|
|
32.
|
Um unabhängige Ergebnisse sicherzustellen, sollten die Hauptprüfungen einer Chemikalie an unterschiedlichen Tagen wiederholt werden.
|
Luciferase-Assay
|
33.
|
Für diesen Assay kann ein im Handel erhältliches Reagens für Luciferase-Assays [z. B. Steady-Glo® Luciferase Assay System (Promega, E2510 oder gleichwertig)] oder ein Luciferase-Assay-Standardsystem (z. B. Promega, E1500 oder gleichwertig) verwendet werden, wenn das Reagens die Akzeptanzkriterien erfüllt. Die Reagenzien für den Assay sollten nach der Empfindlichkeit des zu verwendenden Luminometers ausgewählt werden. Wenn das Luciferase-Assay-Standardsystem verwendet wird, sollte vor der Zugabe des Substrats der Luciferase Zellkultur-Lysispuffer (Cell Culture Lysis Reagent) (z. B. Promega, E1531 oder gleichwertig) hinzugegeben werden. Die Zugabe des Luciferase-Reagens sollte nach den Herstelleranweisungen erfolgen.
|
ANALYSE DER DATEN
ER-Agonisten-Assay
|
34.
|
Zur Erzielung einer relativen transkriptionellen Aktivität bei der PK (1 nM E2) können bei einem ER-Agonisten-Assay die Luminiszenssignale derselben Platte in den folgenden Schritten analysiert werden (wobei auch gleichwertige mathematische Verfahren annehmbar sind):
|
Schritt 1: Für die VK wird der Mittelwert berechnet.
Schritt 2: Der Mittelwert der VK wird vom Wert jedes einzelnen Wells abgezogen, um die Daten zu normalisieren.
Schritt 3: Für die normalisierte PK wird der Mittelwert berechnet.
Schritt 4: Der normalisierte Wert jedes einzelnen Wells auf der Platte wird durch den Mittelwert der normalisierten PK (PK = 100 %) geteilt.
Der endgültige Wert der einzelnen Wells ist die relative transkriptionelle Aktivität dieses Wells im Vergleich zur Reaktion der PK.
Schritt 5: Der Mittelwert der relativen transkriptionellen Aktivität der einzelnen Konzentrationsgruppen der Prüfchemikalie wird berechnet. Bei der Reaktion sind zwei Dimensionen zu beachten: die gemittelte transkriptionelle Aktivität (Reaktion) und die Konzentration, bei der die Reaktion auftritt (siehe folgender Abschnitt).
Hinweise zur Induktion bei EC50, PC50 und PC10
|
35.
|
Zur Berechnung des EC50-Werts wird die vollständige Konzentrations-Reaktions-Kurve benötigt. Die Erstellung dieser Kurve ist aufgrund von Begrenzungen des Prüfkonzentrationsbereichs (beispielsweise infolge von Zytotoxizität oder von Problemen hinsichtlich der Löslichkeit) allerdings u. U. nicht immer möglich oder praktikabel. Da der EC50-Wert und die maximale Induktion (entsprechend dem Höchstwert der Hill-Gleichung) relevante Parameter sind, sollten diese Parameter nach Möglichkeit angegeben werden. Zur Berechnung des EC50-Wertes und der maximalen Induktion sollte eine geeignete Statistik-Software verwendet werden (z. B. Graphpad Prism). Wenn die Konzentrations-Reaktions-Daten für die Hill-Logistik-Gleichung verwendet werden können, sollte der EC50-Wert mit der folgenden Gleichung (7) berechnet werden:
Y = Bottom + (Top-Bottom) / (1+10 exp ((log EC50 -X) × Hillslope)), wobei gilt:
X = Logarithmus der Konzentration und
Y = Reaktion; Y beginnt beim unteren Niveau (Bottom) und steigt in einer Sigmoidkurve bis zum oberen Niveau (Top). Das untere Niveau ist bei der Hill-Logistik-Gleichung gleich Null.
|
|
36.
|
Bei den Prüfchemikalien ist jeweils Folgendes anzugeben:
RPCMax (die durch eine Prüfchemikalie induzierte maximale Reaktion, ausgedrückt als Prozentanteil der durch 1 nM E2 auf derselben Platte induzierten Reaktion) sowie PCMax (die Konzentration bei RPCMax) und
(bei positiven Chemikalien) die Konzentrationen, bei denen PC10 und ggf. PC50 induziert wird.
|
|
37.
|
Der PCx-Wert kann durch Interpolation zwischen zwei Punkten auf der XY-Koordinate, einem unmittelbar über und einem unmittelbar unter einem PCx-Wert, berechnet werden. Wenn die Datenpunkte unmittelbar über und unter dem PCx-Wert die Koordinaten (a,b) und (c,d) haben, kann der PCx-Wert mit folgender Gleichung berechnet werden:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
Erläuterungen zu den PK-Werten sind der folgenden Abbildung 1 zu entnehmen.
Abbildung 1
Beispiel zur Ableitung von PK-Werten – Die PK (1 nM E2) wird auf jeder Assay-Platte berücksichtigt.
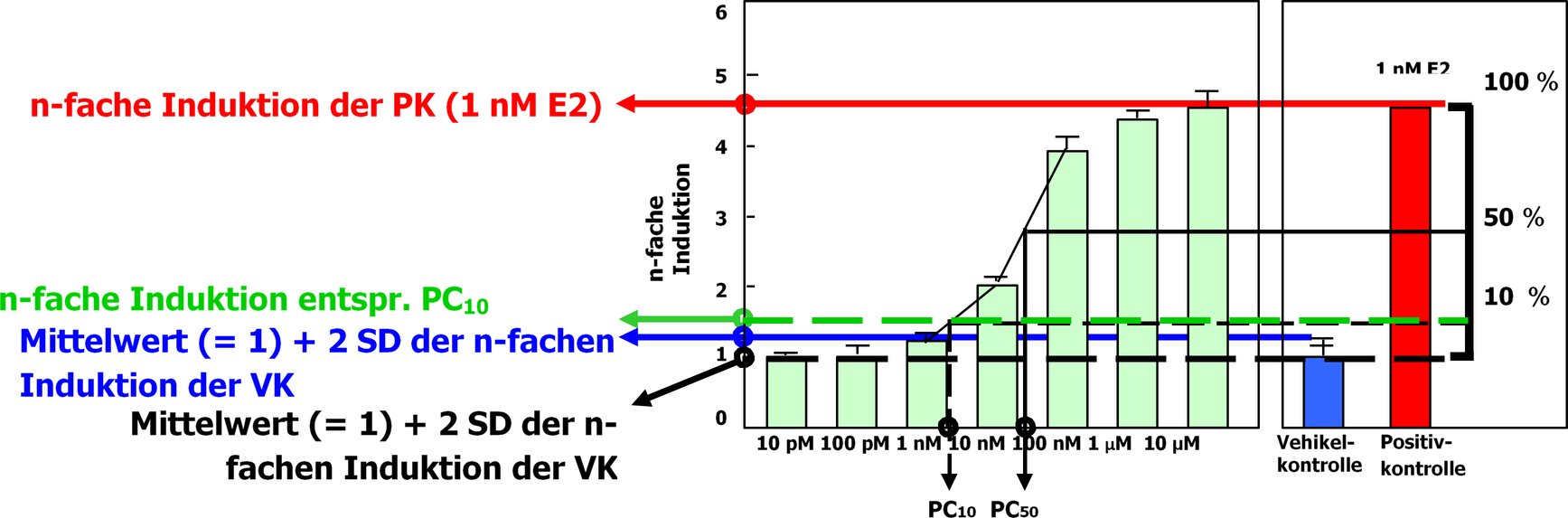
|
ER- Antagonisten-Assay
|
39.
|
Zur Erzielung einer relativen transkriptionellen Aktivität (RTA) der Spike-in-Kontrolle (25 pM E2) können bei einem ER-Antagonisten-Assay die Luminiszenssignale derselben Platte in den folgenden Schritten analysiert werden (wobei auch gleichwertige mathematische Verfahren annehmbar sind):
Schritt 1: Für die VK wird der Mittelwert berechnet.
Schritt 2: Der Mittelwert der VK wird vom Wert jedes einzelnen Wells abgezogen, um die Daten zu normalisieren. Schritt 3: Für die normalisierte Spike-in-Kontrolle wird der Mittelwert berechnet.
Schritt 4: Der normalisierte Wert jedes einzelnen Wells auf der Platte wird durch den Mittelwert der normalisierten Spike-in-Kontrolle (Spike-in-Kontrolle = 100 %) geteilt.
Der endgültige Wert der einzelnen Wells ist die relative transkriptionelle Aktivität dieses Wells im Vergleich zur Reaktion der Spike-in-Kontrolle.
Schritt 5: Der Mittelwert der relativen transkriptionellen Aktivität der einzelnen Behandlungen wird berechnet.
|
Hinweise zur Induktion bei IC30 und IC50
|
40.
|
Bei positiven Chemikalien sollten die Konzentrationen angegeben werden, bei denen IC30 und ggf. IC50 induziert wird.
|
|
41.
|
Der ICx-Wert kann durch Interpolation zwischen zwei Punkten auf der XY-Koordinate, einem unmittelbar über und einem unmittelbar unter einem ICx-Wert, berechnet werden. Wenn die Datenpunkte unmittelbar über und unter dem ICx-Wert die Koordinaten (c,d) und (a,b) haben, kann der ICx-Wert mit folgender Gleichung berechnet werden:
lin ICx = a-(b-(100-x)) (a-c) /(b-d)
Abbildung 2
Beispiel zur Ableitung von IC-Werten – Der Spike (25 pM E2) wird auf jeder Assay-Platte berücksichtigt.

RTA: relative transkriptionelle Aktivität
|
|
42.
|
Die Ergebnisse sollten auf zwei (oder drei) unabhängigen Prüfläufen beruhen. Wenn bei zwei Prüfläufen vergleichbare und daher auch reproduzierbare Ergebnisse erzielt werden, ist ein dritter Prüflauf nicht erforderlich. Ergebnisse können dann akzeptiert werden, wenn die folgenden Anforderungen erfüllt sind:
|
—
|
Die Ergebnisse erfüllen die Akzeptanzkriterien (Nummern 14-20)
|
|
—
|
und sind reproduzierbar.
|
|
Kriterien für die Auswertung von Daten
Tabelle 7
Kriterien für ein positives oder ein negatives Ergebnis beim ER-Agonisten-Assay
|
Positiv
|
Wenn RPCMax bei mindestens zwei von zwei oder zwei von drei Prüfläufen größer oder gleich 10 % der Reaktion der Positivkontrolle ist.
|
|
Negativ
|
Wenn RPCMax bei zwei von zwei oder zwei von drei Prüfläufen nicht mindestens gleich 10 % der Reaktion der Positivkontrolle ist.
|
Tabelle 8
Kriterien für ein positives oder ein negatives Ergebnis beim ER-Antgonisten-Assay
|
Positiv
|
Wenn bei mindestens zwei von zwei oder zwei von drei Prüfläufen IC30 berechnet wird.
|
|
Negativ
|
Wenn IC30 bei zwei von zwei oder zwei von drei Prüfläufen nicht berechnet wird.
|
|
43.
|
Kriterien zur Auswertung der Daten sind den Tabellen 7 und 8 zu entnehmen. Positive Ergebnisse werden sowohl durch die Größenordnung der Wirkung als auch durch die Konzentration beschrieben, bei der die Wirkung eintritt. Die Beschreibung von Ergebnissen in Konzentrationen bei denen 50 % (PC50) bzw. 10 % (PC10) der PK-Werte beim Agonisten-Assay erreicht und 50 % (IC50) bzw. 30 % (IC30) des Wertes der Spike-in-Kontrolle beim Antagonisten-Assay gehemmt werden, erfüllt beide Anforderungen. Eine Prüfchemikalie wird jedoch als positiv eingestuft, wenn die durch die Prüfchemikalie induzierte maximale Reaktion (RPCMax) bei mindestens zwei von zwei oder zwei von drei Prüfläufen größer oder gleich 10 % der Reaktion der PK ist. Als negativ werden Prüfchemikalien eingestuft, wenn RPCMax in zwei von zwei oder zwei von drei Prüfläufen nicht mindestens 10 % der Reaktion der Positivkontrolle beträgt.
|
|
44.
|
Die Berechnungen von PC10, PC50 und PCMax beim ER-Agonisten-Assay und von IC30 und IC50 beim ER-Antagonisten-Assay kann anhand einer Tabelle vorgenommen werden, die auf der öffentlichen Website der OECD (64) mit der Prüfrichtlinie bereitgestellt wird.
|
|
45.
|
Es sollte hinreichend sein, die PC10- oder PC50- bzw. IC30- oder IC50-Werte mindestens zweimal zu bestimmen. Wenn die ermittelten Normalwerte von Daten im selben Konzentrationsbereich jedoch Unterschiede mit einem unannehmbar hohen Variationskoeffizienten (VK, %) aufweisen, können die Daten nicht als zuverlässig betrachtet werden. In diesem Fall sollte untersucht werden, worauf die hohe Variabilität zurückzuführen ist. Der VK der dreifachen Rohdaten (d. h. der Daten zur Leuchtdichte) der zur Berechnung von PC10 verwendeten Datenpunkte sollte weniger als 20 % betragen.
|
|
46.
|
Die Erfüllung der Akzeptanzkriterien deutet darauf hin, dass das Assay-System ordnungsgemäß funktioniert. Dies gewährleistet jedoch nicht, dass ein bestimmter Prüflauf tatsächlich exakte Daten ergibt. Die Wiederholung der Ergebnisse des ersten Prüflaufs ist die beste Bestätigung dafür, dass exakte Daten ermittelt wurden.
|
|
47.
|
Wenn beim ER-Agonisten-Assay über die für die Screening- und Priorisierungszwecke dieser Prüfrichtlinie benötigten Daten zu positiven Prüfchemikalien hinaus weitere Informationen benötigt werden (insbesondere für PC10- bis PC49-Chemikalien sowie bei Chemikalien, bei denen vermutet wird, dass es zu einer Überstimulation der Luciferase kommt), kann mit einem ERα-Antagonisten (siehe Anlage 2.1) bestätigt werden, dass die beobachtete Luciferase-Aktivität ausschließlich eine ERα-spezifische Reaktion ist.
|
PRÜFBERICHT
|
48.
|
Siehe Nummer 20 im Abschnitt „ELEMENTE DES ER-TA-ASSAYS“.
|
LITERATUR
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(2)
|
Escande, A., u. a. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1459-1469.
|
|
(3)
|
Kuiper, G.G., u. a. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4252-4263.
|
|
(4)
|
Spaepen, M., u. a. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi, H., u. a. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769-771.
|
|
(6)
|
Dussurget, O. und Roulland-Dussoix, D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-959.
|
|
(7)
|
De Lean, A., Munson, P.J. und Rodbard, D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Anlage 2.1
FALSCH POSITIVE: BEWERTUNG VON NICHT DURCH REZEPTOREN VERMITTELTEN LUMINISZENZSIGNALEN
|
1.
|
Falsch positive Ergebnisse beim ER-Agonisten-Assay können auf eine nicht durch ER vermittelte Aktivierung des Luciferase-Gens, auf eine direkte Aktivierung des Genprodukts oder eine sonstige Fluoreszenz zurückzuführen sein. Diese Wirkungen äußern sich in einer unvollständigen oder ungewöhnlichen Dosis-Reaktions-Kurve. Wenn solche Effekte vermutet werden, sollte der Effekt eines ER-Antagonisten (z. B. 4-Hydroxytamoxifen (OHT) in einer nicht toxischen Konzentration) auf die Reaktion untersucht werden. Der reine Antagonist ICI 1827 80 ist für diesen Zweck möglicherweise nicht geeignet, da eine hinreichende Konzentration von ICI 1827 80 eine Reduzierung des VK-Werts bewirken kann und da dies die Datenanalyse beeinträchtigt.
|
|
2.
|
Um die Validität dieses Ansatzes sicherzustellen, sollten auf derselben Platte die folgenden Parameter geprüft werden:
|
—
|
Agonisten-Aktivität der unbekannten Chemikalie mit/ohne 10 μM OHT,
|
|
—
|
1 nM E2 (dreifach) als Agonisten-PK und
|
|
—
|
1 nM E2 + OHT (dreifach).
|
|
Kriterien für die Auswertung von Daten
Hinweis: Alle Wells sollten mit dem Vehikel in der gleichen Konzentration behandelt werden.
|
—
|
Wenn die Agonisten-Aktivität der unbekannten Chemikalie durch die Behandlung mit dem ER-Antagonisten NICHT beeinträchtigt wird, ist sie als „negativ“ einzustufen.
|
|
—
|
Wird die Agonisten-Aktivität der unbekannten Chemikalie vollständig gehemmt, sind die Entscheidungskriterien zu berücksichtigen.
|
|
—
|
Ist die Agonisten-Aktivität bei der niedrigsten Konzentration größer oder gleich der Reaktion bei PC10 ist, wird die unbekannte Chemikalie mindestens in der Größenordnung der Reaktion bei PC10 gehemmt. Die Differenz der Reaktionen zwischen den nicht behandelten und den mit dem ER-Antagonisten behandelten Wells wird berechnet. Diese Differenz ist als tatsächliche Reaktion zu betrachten und sollte zur Berechnung der betreffenden Parameter verwendet werden, damit über die Einstufung entschieden werden kann.
|
Analyse der Messdaten
Der Leistungsstandard wird geprüft.
Der VK zwischen unter gleichen Bedingungen behandelten Wells wird geprüft.
|
1.
|
Der Mittelwert der VK wird berechnet.
|
|
2.
|
Vom Wert der einzelnen nicht mit OHT behandelten Wells wird der Mittelwert der VK abgezogen.
|
|
3.
|
Der OHT-Mittelwert wird berechnet.
|
|
4.
|
Vom Wert der einzelnen mit OHT behandelten Wells wird der Mittelwert der VK abgezogen.
|
|
5.
|
Der Mittelwert der PK wird berechnet.
|
|
6.
|
Die relative transkriptionelle Aktivität aller übrigen Wells wird bezogen auf die PK berechnet.
|
Anlage 2.2
ZUBEREITUNG VON MIT DEXTRANBESCHICHTETER AKTIVKOHLE (DCC) BEHANDELTEM SERUM
|
1.
|
Die Behandlung von Serum mit dextranbeschichteter Aktivkohle (DDC) ist ein allgemeines Verfahren zur Entfernung von Östrogenverbindungen aus zu einem Zellmedium hinzugegebenem Serum, um verfälschte Reaktionen aufgrund von im Serum verbliebenem Östrogen auszuschließen. Mit diesem Verfahren können 500 ml fetales Rinderserum (FBS) behandelt werden.
|
Elemente
|
2.
|
Für das Verfahren werden die folgende Ausrüstung und die folgenden Materialien benötigt:
|
Materialien
|
Magnesiumchlorid-Hexahydrat (MgCl2·6H2O)
|
|
1 M HEPES-Pufferlösung) (pH 7,4)
|
|
Mit einem Filtersystem hergestelltes ultrareines Wasser
|
Ausrüstung
Autoklaviertes Glasgefäß mit geeigneter Größe, normale Laborzentrifuge (die auf eine Temperatur von 4 oC eingestellt werden kann)
Verfahren
|
3.
|
Das folgende Verfahren wird für die Verwendung von 50-ml-Zentrifugenröhrchen angepasst:
[Tag 1] Zur Zubereitung einer Suspension mit dextranbeschichteter Aktivkohle wird 1 l ultrareines Wasser mit 1,5 mM MgCl2, 0,25 M Saccharose, 2,5 g Aktivkohle, 0,25 g Dextran und 5 mM HEPES gemischt, bei 4 oC umgerührt und über Nacht stehen gelassen.
[Tag 2] Die Suspension wird in 50-ml-Zentrifugenröhrchen gegeben und bei 4 oC 10 Minuten mit 10 000 rpm zentrifugiert. Der Überstand wird entfernt, und die Hälfte des Aktivkohlesediments zur Verwendung an Tag 3 bei 4 oC aufbewahrt. Die andere Hälfte der Aktivkohle wird mit FBS suspendiert, das zur Vermeidung von Ausfällungen vorsichtig aufgetaut, 30 Minuten bei 56 oC durch Wärme inaktiviert und dann in ein autoklaviertes Glasgefäß (z. B. einen Erlenmeyer-Kolben) gegeben wurde. Diese Suspension wird über Nacht bei 4 oC vorsichtig umgerührt.
[Tag 3] Die Suspension wird in Zentrifugenröhrchen gegeben und bei 4 oC 10 Minuten mit 10 000 rpm zentrifugiert. Das FBS wird entnommen und in das an Tag 2 zubereitete und aufbewahrte frische Aktivkohlesediment gegeben. Das Aktivkohlesediment wird suspendiert und die erhaltene Suspension vorsichtig in einem autoklavierten Glasgefäß über Nacht bei 4 oC umgerührt.
[Tag 4] Die Suspension wird 10 Minuten bei einer Temperatur von 4 oC mit 10 000 rpm zentrifugiert. Anschließend wird der Überstand durch einen sterilen 0,2-μm-Filter filtriert. Dieses mit DCC behandelte FBS ist bei -20 oC zu lagern und kann dann innerhalb eines Zeitraums von bis zu einem Jahr verwendet werden.
|
Anlage 3
VM7LUC-ER-TA-ASSAY ZUR BESTIMMUNG VON ÖSTROGENREZEPTOR-AGONISTEN UND -ANTAGONISTEN
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
1.
|
Für diesen Assay wird die VM7Luc4E2-Zelllinie verwendet. (65) Diese Zelllinie wurde vom National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) und vom Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) validiert (1). Die VM7Luc-Zelllinien exprimieren hauptsächlich den endogenen ERα sowie in geringerer Menge den endogenen ERβ (2) (3) (4).
|
|
2.
|
Dieser Assay ist bei zahlreichen Stoffen anwendbar, die in Dimethylsulfoxid (DMSO; CASRN 67-68-5) gelöst werden können, nicht mit DMSO oder mit dem Zellkulturmedium reagieren und bei der zu untersuchenden Konzentration nicht zytotoxisch wirken. Wenn DMSO nicht verwendet werden kann, kommen auch andere Vehikel wie beispielsweise Ethanol oder Wasser in Betracht (Nummer 12). Angesichts der nachgewiesenen Leistungsfähigkeit des VM7Luc-ER-TA-(Ant)agonisten-Assays ist davon auszugehen, dass mit diesem Assay generierte Daten Aufschluss über durch ER vermittelte Wirkungsmechanismen geben und bei der Priorisierung von Stoffen für weitere Untersuchungen berücksichtigt werden können.
|
|
3.
|
Dieser Assay wurde speziell zum Nachweis von hERα und einer durch hERß vermittelten TA anhand einer Messung der Chemiluminiszenz als Endpunkt entwickelt. Die Berücksichtigung der Chemiluminiszenz in Bioassays ist allgemein üblich, da die Luminiszenz ein ausgeprägtes Signal-Rausch-Verhältnis ergibt. Die Aktivität der Leuchtkäfer-Luciferase bei zellbasierten Assays kann jedoch mit der Aktivität von Stoffen verwechselt werden, die das Luciferase-Enzym hemmen und infolge der Proteinstabilisierung sowohl eine erkennbare Hemmung als auch eine erhöhte Luminiszenz bewirken. Außerdem wurden bei einige luciferasebasierten EN-Reporter-Gen-Assays bei Phytoöstrogenkonzentrationen von mehr als 1 μM infolge der Überaktivierung des Luciferase-Reporter-Gens Luminiszenzsignale festgestellt, die nicht durch Rezeptoren vermittelt wurden (9) (11). Die Dosis-Reaktions-Kurve zeigt, dass die tatsächliche Aktivierung des ER-Systems bei niedrigeren Konzentrationen erfolgt; bei stabil transfizierten ER-TA-Assay-Systemen muss die Luciferase-Expression bei hohen Konzentrationen von Phytoöstrogenen oder ähnlichen Verbindungen, bei denen vermutet wird, dass sie eine phytoöstrogenartige Überaktivierung des Luciferase-Reportergens verursachen, jedoch sorgfältig untersucht werden.
|
|
4.
|
Vor der Verwendung dieses Assays für rechtliche Zwecke sollten die Abschnitte „ALLGEMEINE EINLEITUNG“ und „ELEMENTE DES ER-TA-ASSAYS“ gelesen werden. Die in der vorliegenden Prüfmethode verwendeten Begriffe und Abkürzungen werden in Anlage 1 erläutert.
|
PRINZIP DES ASSAYS (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
5.
|
Mit dem Assay werden ER-Liganden-Bindungen nach einer Translokation des Rezeptor-Liganden-Komplexes an den Zellkern nachgewiesen. Im Zellkern geht der Rezeptor-Liganden-Komplex eine Bindung mit spezifischen DNA-Response-Elements ein und transaktiviert das Reporter-Gen (luc). Dadurch wird die Bildung von Luciferase angeregt und anschließend die Emission von Licht bewirkt, das mit einem Luminometer gemessen werden kann. Die Luciferase-Aktivität kann rasch und kostengünstig mit verschiedenen im Handel erhältlichen Test-Kits bewertet werden. Beim VM7Luc-ER-TA-Assay wird eine auf ER reagierende humane Brustadenokarzinom-Zelllinie (VM7) verwendet, die mit einem Leuchtkäfer-luc-Reporter-Genprodukt stabil transfiziert wurde, das kontrolliert durch vier Östrogen-Response-Elemente vor dem Maus-Mammatumorvirus-Promoter (MMTV) angeordnet wurde, um anhand der In-vitro-Aktivität eines ER-Agonisten oder -Antagonisten Stoffe nachzuweisen. Dieser MMTV-Promotor bewirkt nur eine geringe Kreuzreaktivität mit anderen Steroiden und nicht steroiden Hormonen (8). Kriterien für die Auswertung der Daten werden in Nummer 41 eingehend beschrieben. Eine positive Reaktion ist kurz gesagt an einer Konzentrations-Reaktions-Kurve mit mindestens drei Punkten mit sich nicht überschneidenden Fehlerbalken (Mittelwert ± SD) sowie an einer Änderung der Amplitude (normalisierte relative Lichteinheiten [RLU]) von mindestens 20 % des Höchstwerts für den betreffenden Referenzstandard (17β-Estradiol [E2; CASRN 50-28-2] beim Agonsten-Assay und Raloxifen HCl [Ral; CASRN 84449-90-1]/E2 beim Antagonisten-Assay) zu erkennen.
|
VERFAHREN
Zelllinie
|
6.
|
Für den Assay sollte die stabil transfizierte VM7Luc4E2-Zelllinie verwendet werden. Diese Zelllinie kann gegenwärtig nur nach Maßgabe einer technischen Lizenzvereinbarung von der University of California, Davis, Kalifornien, USA (66) und von Xenobiotic Detection Systems Inc., Durham, North Carolina, USA, (67) bezogen werden.
|
Stabilität der Zelllinie
|
7.
|
Um die Stabilität und die Integrität der Zelllinie aufrechtzuerhalten, sollten die Zellen im Erhaltungsmedium über mehr als eine Passage des tiefgefrorenen Materials vermehrt werden (Nummer 9). Die Kultivierung sollte sich auf höchstens 30 Passagen beschränken. Bei der VM7Luc4E2-Zelllinie werden für 30 Passagen etwa drei Monate benötigt.
|
Bedingungen für die Haltung der Zellkulturen und für die Plattierung
|
8.
|
Die in der Guidance on Good Cell Culture Practice spezifizierten Verfahren (5) (6) sollten eingehalten werden, damit die Qualität aller Materialien und Methoden gewährleistet ist und die Integrität, die Validität und die Reproduzierbarkeit aller durchgeführten Arbeiten gewahrt werden.
|
|
9.
|
VM7Luc4E2-Zellen werden im RPMI-1640-Medium ergänzt um 0,9 % Pen/Strep und 8,0 % fetales Rinderserum (FBS) in einem eigenen Inkubator für Gewebekulturen bei einer Temperatur von 37 ± 1 oC, 90 ± 5 % Luftfeuchtigkeit und 5,0 ± 1 % CO2/Luft gehalten.
|
|
10.
|
Nach Erreichen einer Konfluenz von ~80 % werden die VM7Luc4E2-Zellen subkultiviert und 48 Stunden in einer östrogenfreien Umgebung konditioniert, bevor sie in 96-Well-Platten plattiert werden, wo sie den Prüfchemikalien ausgesetzt und auf eine öströgenbedingte Induzierung einer Luciferase-Aktivität untersucht werden. Das östrogenfreie Medium (EFM) enthält Dulbecco's modifiziertes Eagle-Medium (DMEM) ohne Phenolrot, ergänzt um 4,5 % mit Aktivkohle/Dextran behandeltes fetales Rinderserum (FBS), 1,9 % L-Glutamin und 0,9 % Pen/Strep. Die Kunststoffmaterialien sollten keine Östrogenaktivität entwickeln [siehe detailliertes Protokoll (7)].
|
Akzeptanzkriterien
|
11.
|
Ob eine Prüfung akzeptiert oder verworfen wird, hängt von der Auswertung der Ergebnisse mit den Referenzstandards und mit den Kontrollen aller auf einer 96-Well-Platte durchgeführten Prüfungen ab. Jeder Referenzstandard wird in mehreren Konzentrationen geprüft, und von allen Konzentrationen der Referenzstandards und der Kontrollen sind mehrere Proben verfügbar. Die Ergebnisse werden mit Qualitätskontrollen (QC) der Parameter verglichen, die aus Datenbanken mit historischen Daten zu Agonisten und Antagonisten abgeleitet wurden, die zum Nachweis ihrer Leistungsfähigkeit von den einzelnen Labors ermittelt wurden. Die Datenbanken mit historischen Datenbanken werden unter Eingabe der Werte der Referenzstandards und der Kontrollen kontinuierlich aktualisiert. Nach Änderungen der Ausrüstung oder der Laborbedingungen müssen die Datenbanken mit historischen Daten unter Umständen aktualisiert werden.
|
Agonisten-Prüfung
Dosisfindungsstudie
|
•
|
Induktion: Zur Messung der Platteninduktion wird der höchste mittlere RLU-Wert (RLU = relative Lichteinheit) des E2-Referenzstandards durch den mittleren RLU-Wert der DMSO-Kontrolle geteilt. In der Regel wird eine 5-fache Induktion erreicht; als Akzeptanzkriterium gilt jedoch, dass die Induktion größer oder gleich der 4-fachen Induktion sein sollte.
|
|
•
|
Ergebnisse der DMSO-Kontrolle: Die RLU-Werte der Lösungsmittelkontrolle sollten das 2,5-Fache der Standardabweichung des historischen mittleren RLU-Werts der Lösungsmittelkontrolle betragen.
|
|
•
|
Eine Prüfung, die keines der beiden Akzeptanzkriterien erfüllt, sollte verworfen und wiederholt werden.
|
Hauptversuch
Der Hauptversuch beinhaltet Akzeptanzkriterien aus der Agonisten-Dosisfindungsstudie sowie Folgendes:
|
•
|
Ergebnisse der Referenzstandards: Die Konzentrations-Reaktions-Kurve des E2-Referenzstandards sollte sigmoidal verlaufen und mindestens drei Werte im linearen Bereich enthalten.
|
|
•
|
Ergebnisse der Positivkontrolle: Die RLU-Werte der Methoxychlor-Kontrolle sollten höher sein als der Mittelwert für DMSO zuzüglich der 3-fachen Standardabweichung vom DMSO-Mittelwerts.
|
|
•
|
Eine Prüfung, die keines der Akzeptanzkriterien erfüllt, sollte verworfen und wiederholt werden.
|
Antagonisten-Prüfung
Dosisfindungsstudie
|
•
|
Reduktion: Zur Messung der Plattenreduktion wird der höchste mittlere RLU-Wert des Ral/E2-Referenzstandards durch den mittleren RLU-Wert der DMSO-Kontrolle geteilt. In der Regel wird eine 5-fache Reduktion erreicht; als Akzeptanzkriterium gilt jedoch, dass die Reduktion größer oder gleich der 3-fachen Reduktion sein sollte.
|
|
•
|
Ergebnisse der E2-Kontrolle: Die RLU-Werte der E2-Kontrolle sollten das 2,5-Fache der Standardabweichung des historischen mittleren RLU-Werts der E2-Kontrolle betragen.
|
|
•
|
Ergebnisse der DMSO-Kontrolle: Die RLU-Werte der DMSO-Kontrolle sollten das 2,5-Fache der Standardabweichung des historischen mittleren RLU-Werts der Lösungsmittelkontrolle betragen.
|
|
•
|
Eine Prüfung, die keines der Akzeptanzkriterien erfüllt, wird verworfen und wiederholt.
|
Hauptversuch
Der Hauptversuch beinhaltet Akzeptanzkriterien aus der Antagonisten-Dosisfindungsstudie sowie Folgendes:
|
•
|
Ergebnisse der Referenzstandards: Die Konzentrations-Reaktions-Kurve des Ral/E2-Referenzstandards sollte sigmoidal verlaufen und mindestens drei Werte im linearen Bereich enthalten.
|
|
•
|
Ergebnisse der Positivkontrolle: Die RLU-Werte der Tamoxifen/E2-Kontrolle sollten höher sein als der Mittelwert der E2-Kontrolle zuzüglich der 3-fachen Standardabweichung vom Mittelwert der E2-Kontrolle.
|
|
•
|
Eine Prüfung, die keines der Akzeptanzkriterien erfüllt, wird verworfen und wiederholt.
|
Referenzstandards, Positivkontrolle und Vehikelkontrolle
Vehikelkontrolle (Agonisten- und Antagonisten-Assay)
|
12.
|
Das zur Auflösung einer Prüfchemikalie verwendete Vehikel sollte als Vehikelkontrolle geprüft werden. Bei der Validierung des VM7Luc-ER-TA-Assays wurde 1 % (v/v) Dimethylsulfoxid (DMSO, CASRN 67-68-5) verwendet (Nummer 24). Wenn ein anderes Vehikel als DMSO verwendet wird, sollten alle Referenzstandards, Kontrollen und Prüfchemikalien mit diesem Vehikel geprüft werden.
|
Referenzstandard (Dosisfindung beim Agonisten-Assay)
|
13.
|
Als Referenzstandard wird E2 (CASRN 50-28-2) verwendet. Der Referenzstandard für die Dosisfindungsstudie besteht aus einer seriellen Verdünnung mit vier Konzentrationen E2 (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 und 2,87 x 10-12 M), wobei jede Konzentration in zwei Wells geprüft wird.
|
Referenzstandard (Dosisfindung beim Agonisten-Assay)
|
14.
|
E2 für den Hauptversuch besteht aus einer seriellen Verdünnung 1:2 mit 11 E2-Konzentrationen (von 3,67 x 10-10 bis 3,59 x 10-13 M) jeweils in zwei Wells.
|
Referenzstandard (Dosisfindung beim Antagonisten-Assay)
|
15.
|
Der Referenzstandard besteht aus einer Kombination von Ral (CASRN 84449-90-1) und E2 (CASRN 50-28-2). Ral/E2 für die Dosisfindungsstudie umfasst eine serielle Verdünnung mit Ral in drei Konzentrationen (3,06 x 10-9, 7,67 x 10-10 und 1,92 x 10-10 M) sowie einer festen Konzentration (9,18 × 10-11 M) E2 in zwei Wells.
|
Referenzstandard (Agonisten-Hauptversuch)
|
16.
|
Ral/E2 für den Hauptversuch besteht aus einer seriellen Verdünnung von Ral (1:2) mit neun Ral/E2-Konzentrationen von 2,45x 10-8 bis 9,57 x 10-11 M sowie einer festen Konzentration (9,18 × 10-11 M) E2 jeweils in zwei Wells.
|
Schwache Positivkontrolle (Agonist)
|
17.
|
Die schwache Positivkontrolle besteht aus 9,06 x 10-6 M p,p’-Methoxychlor (Methoxychlor; CASRN 72-43-5) in einem EFM.
|
Schwache Positivkontrolle (Antagonist)
|
18.
|
Die schwache Positivkontrolle besteht aus 3,36 x 10-6 M Tamoxifen (CASRN 10540-29-1) mit 9,18 × 10-11 M E2 in einem EFM.
|
E2-Kontrolle (nur Antagonisten-Assay)
|
19.
|
Die E2-Kontrolle besteht aus 9,18 × 10-11 M E2 in einem EFM und dient als Normalwert-Negativkontrolle.
|
n-fache Induktion (Agonist)
|
20.
|
Die Induktion der Luciferase-Aktivität des Referenzstandards (E2) wird gemessen, indem der höchste mittlere RLU-Wert des E2-Referenzstandards durch den mittleren RLU-Wert der DMSO-Kontrolle geteilt wird; dabei sollte das Ergebnis mehr als das 4-Fache betragen.
|
n-fache Reduktion (Antagonist)
|
21.
|
Die mittlere Luciferase-Aktivität des Referenzstandards (Ral/E2) wird gemessen, indem der höchste mittlere RLU-Wert des Ral/E2-Referenzstandards durch den mittleren RLU-Wert der DMSO-Kontrolle geteilt wird; dabei sollte das Ergebnis mehr als das 3-Fache betragen.
Zum Nachweis der Eignung des Labors siehe Nummer 14 und Tabellen 3 und 4 im Abschnitt „ELEMENTE DES ER-TA-ASSAYS“ bei dieser Prüfmethode.
|
Vehikel
|
22.
|
Die Prüfchemikalien sollten in einem Lösungsmittel gelöst werden, das die betreffende Prüfchemikalie löst und sich mit dem Zellmedium mischt. Geeignete Vehikel sind Wasser, Ethanol (Reinheit 95-100 %) und DMSO. Wenn DMSO verwendet wird, sollte die Konzentration höchstens 1 % (v/v) betragen. Bei jedem Vehikel sollte nachgewiesen werden, dass die verwendete Höchstmenge nicht zytotoxisch ist und keine Auswirkungen auf den Assay hat. Die Referenzstandards und die Kontrollen werden in 100 % Lösungsmittel gelöst und dann in einem EFM auf geeignete Konzentrationen verdünnt.
|
Zubereitung der Prüfchemikalien
|
23.
|
Die Referenzstandards und die Kontrollen werden in 100 % DMSO (oder einem anderen geeigneten Lösungsmittel) gelöst und dann in einem EFM auf geeignete Konzentrationen verdünnt. Alle Prüfchemikalien sollten sich vor der Lösung und Verdünnung an die Raumtemperatur anpassen können. Die Lösungen der Prüfchemikalien sollten für jeden Versuch neu zubereitet werden. In den Lösungen sollten kein Niederschlag und keine Trübung zu erkennen sein. Der Referenzstandard und die Kontrollen können in größeren Mengen zubereitet und vorgehalten werden. Der endgültige Referenzstandard, die Verdünnungen der Kontrollen und die Prüfchemikalien sollten jedoch für jeden Versuch frisch zubereitet und innerhalb von 24 Stunden nach der Zubereitung verwendet werden.
|
Löslichkeit und Zytotoxizität: Hinweise zur Dosisfindung
|
24.
|
Die Dosisfindungsstudie umfasst serielle Verdünnungen (1:10) mit sieben Verdünnungspunkten, die jeweils zweimal geprüft werden. Zunächst werden die Prüfchemikalien bis zur Höchstkonzentration von 1 mg/ml (~1 mM) beim Agonisten-Assay bzw. von 20 μg/ml (~10 μM) beim Antagonisten-Assay geprüft. Mit den Dosisfindungsstudien werden
|
|
—
|
die im Hauptversuch zu verwendenden Ausgangskonzentrationen der Prüfchemikalien und
|
|
—
|
die im Hauptversuch zu verwendenden Verdünnungen der Prüfchemikalien (1:2 oder 1:5) ermittelt.
|
|
25.
|
Die Prüfprotokolle des Agonisten- und der Antagonisten-Assays umfassen eine Bewertung der Zellviabilität/Zytotoxizität (7) im Rahmen der Dosisfindung und des Hauptversuchs. Die Methode zur Ermittlung der Zytotoxizität zur Bewertung der Zellviabilität bei der Validierung des VM7Luc-ER-TA-Assays (1) bestand in einer skalierten qualitativen visuellen Beobachtung; allerdings kann die Zytotoxizität auch mit einer quantitativen Methode bestimmt werden (siehe Protokoll (7)). Daten aufgrund von Konzentrationen der Prüfchemikalien, bei denen die Viabilität um mehr als 20 % beeinträchtigt wird, können nicht berücksichtigt werden.
|
Prüfung der chemischen Exposition und Anordnung auf der Assay-Platte
|
26.
|
Die Zellen werden gezählt und in 96-Well-Gewebekulturplatten (2 x 105 Zellen pro Well) in ein EFM gegeben und 24 Stunden inkubiert, damit die Zellen an der Platte anhaften können. Das EFM wird entfernt und durch die Prüf- und Referenzchemikalien im EFM ersetzt und 19-24 Stunden inkubiert. Besondere Aufmerksamkeit ist bei stark flüchtigen Stoffen geboten, da durch benachbarte Kontroll-Wells falsch positive Ergebnisse entstehen können. In diesen Fällen können „Plattenversiegelungen“ helfen, die einzelnen Wells während der Prüfung zu isolieren. Dies ist in den betreffenden Fällen zu empfehlen.
|
Dosisfindungsstudien
|
27.
|
Bei Dosisfindungsstudien werden alle Wells der 96-Well-Platte verwendet, um bis zu sechs Prüfchemikalien in seriellen Verdünnungen (1:10) mit sieben Verdünnungspunkten jeweils zweimal zu prüfen (siehe Abbildungen 1 und 2).
|
|
—
|
Bei der Dosisfindungsstudie für Agonisten werden als Referenzstandard vier E2-Konzentrationen (jeweils zwei) und vier Replikat-Wells für die DMSO-Kontrolle verwendet.
|
|
—
|
Bei der Dosisfindungsstudie für Antagonisten werden als Referenzstandard drei Ral/E2-Konzentrationen mit 9,18 × 10-11 M E2 (jeweils zwei) und drei Replikat-Wells für die E2- und die DMSO-Kontrolle verwendet.
|
Abbildung 1
Anordnung bei Prüfungen zur Dosisfindung bei Agonisten mit einer 96-Well-Platte
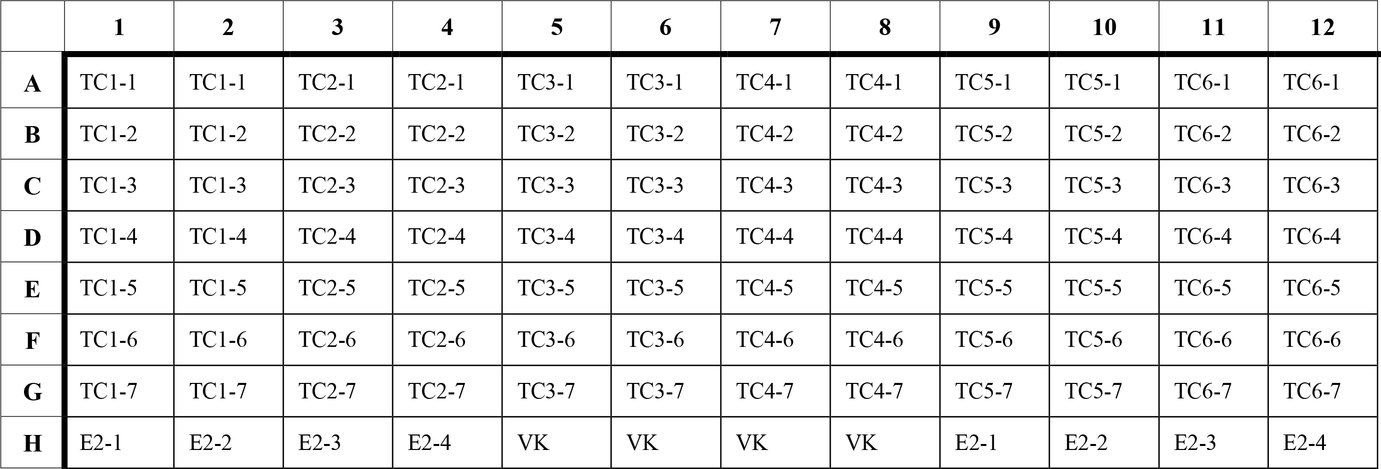
Abkürzungen: E2-1 bis E2-4 = Konzentrationen des E2-Referenzstandards (hoch bis niedrig); TC1-1 bis TC1-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 1 (TC1); TC2-1 bis TC2-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 2 (TC2); TC3-1 bis TC3-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 3 (TC3); TC4-1 bis TC4-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 4 (TC4); TC5-1 bis TC5-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 5 (TC5); TC6-1 bis TC6-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 6 (TC6); VK = Vehikelkontrolle (DMSO [1 % v/v EFM.]).
Abbildung 2
Anordnung bei Prüfungen zur Dosisfindung bei Antagonisten mit einer 96-Well-Platte
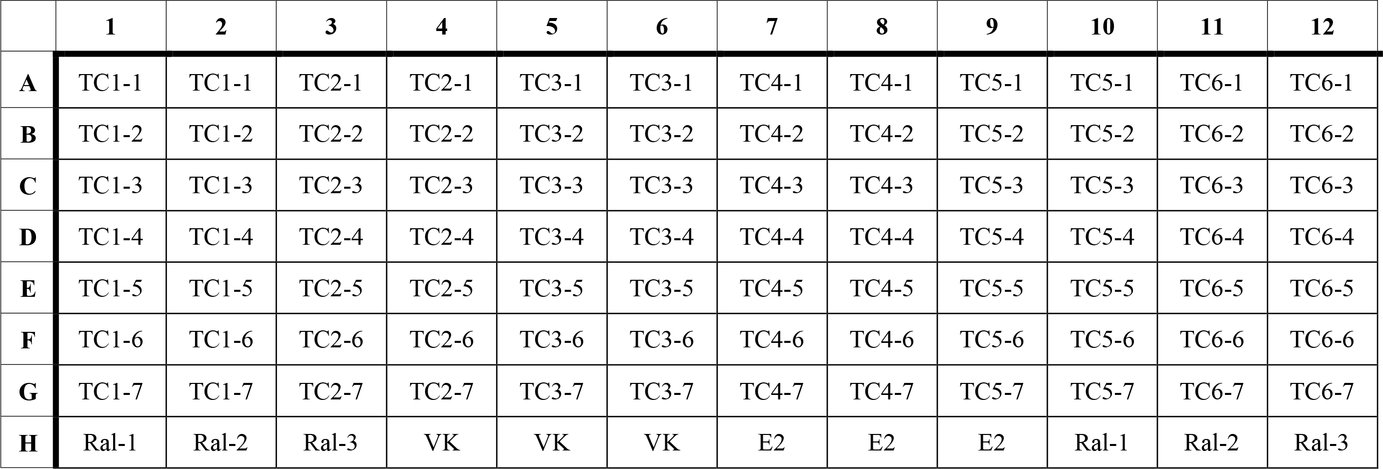
Abkürzungen: E2 = E2-Kontrolle; Ral-1 bis Ral-3 = Konzentrationen des Raloxifen/E2-Referenzstandards (hoch bis niedrig); TC1-1 bis TC1-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 1 (TC1); TC2-1 bis TC2-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 2 (TC2); TC3-1 bis TC3-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 3 (TC3); TC4-1 bis TC4-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 4 (TC4); TC5-1 bis TC5-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 5 (TC5); TC6-1 bis TC6-7 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 6 (TC6); VK = Vehikelkontrolle (DMSO [1 % v/v EFM.]).
Hinweis: Alle Prüfchemikalien werden mit 9,18 × 10-11 M E2 geprüft.
|
28.
|
Für jedes Well wird ein endgültiges Medienvolumen von 200 μl als erforderlich empfohlen. Für die Prüfung sind ausschließlich Prüfplatten zu verwenden, bei denen die Zellviabilität in allen Wells mindestens 80 % beträgt.
|
|
29.
|
Die Bestimmung von Ausgangskonzentrationen für
Agonisten
-Hauptversuche wird im Protokoll für Agonisten-Assays eingehend beschrieben (7). Dabei sind die folgenden Kriterien zu berücksichtigen:
|
|
—
|
Wenn die Konzentrationskurve der Prüfchemikalie keine Punkte enthält, die über dem Mittelwert zuzüglich der 3-fachen Standardabweichung der DMSO-Kontrolle liegen, wird der Hauptversuch an einer seriellen Verdünnung (1:2) mit 11 Verdünnungspunkten beginnend mit der maximalen löslichen Konzentration vorgenommen.
|
|
—
|
Wenn die Konzentrationskurve der Prüfchemikalie keine Punkte enthält, die über dem Mittelwert zuzüglich der 3-fachen Standardabweichung der DMSO-Kontrolle liegen, sollte die beim Hauptversuch zu verwendende Verdünnungsreihe mit 11 Verdünnungspunkten um eine logarithmische Einheit höher als die Konzentration sein, bei der beim Vorversuch der höchste bereinigte RLU-Wert ermittelt wurde. Die Verdünnungsreihe mit 11 Verdünnungspunkten beruht auf Verdünnungen im Verhältnis 1:2 oder 1:5; das Verdünnungsverhältnis richtet sich nach den folgenden Kriterien:
|
Eine serielle Verdünnung im Verhältnis 1:2 mit 11 Verdünnungspunkten sollte verwendet werden, wenn der sich ergebende Konzentrationsbereich das gesamte Spektrum der Reaktionen aufgrund der in der Dosisfindungsstudie ermittelten Konzentrations-Reaktions-Kurve abdeckt. Ansonsten ist eine Verdünnung im Verhältnis 1:5 zu verwenden.
|
|
|
—
|
Wenn eine Prüfchemikalie in der Dosisfindungsstudie eine biphasige Konzentrations-Reaktions-Kurve ergibt, sollten die beiden Phasen auch im Hauptversuch berücksichtigt werden.
|
|
30.
|
Die Bestimmung von Ausgangskonzentrationen für
Antagonisten
-Hauptversuche wird im Protokoll für Antagonisten-Assays eingehend beschrieben (7). Dabei sind die folgenden Kriterien zu berücksichtigen:
|
|
—
|
Wenn die Konzentrationskurve der Prüfchemikalie keine Punkte enthält, die unter dem Mittelwert abzüglich der 3-fachen Standardabweichung der E2-Kontrolle liegen, wird der Hauptversuch mit einer seriellen Verdünnung (1:2) mit 11 Verdünnungspunkten beginnend mit der maximalen löslichen Konzentration vorgenommen.
|
|
—
|
Wenn die Konzentrationskurve der Prüfchemikalie keine Punkte enthält, die unter dem Mittelwert abzüglich der 3-fachen Standardabweichung der E2-Kontrolle liegen, sollte beim Hauptversuch für die Verdünnungsreihe mit 11 Verdünnungspunkten eine der folgenden Ausgangskonzentrationen verwendet werden:
|
—
|
die Konzentration mit dem niedrigsten angepassten RLU-Wert bei der Dosisfindung,
|
|
—
|
die maximal lösliche Konzentration (siehe Protokoll zum Antagonisten-Assay (7), Abbildung 14-2),
|
|
—
|
die niedrigste zytotoxische Konzentration (Beispiel siehe Antagonisten-Protokoll (7), Abbildung 14-3).
|
|
|
—
|
Die Verdünnungsreihe mit 11 Verdünnungspunkten beruht auf Verdünnungen im Verhältnis 1:2 oder 1:5; das Verdünnungsverhältnis richtet sich nach den folgenden Kriterien:
|
Eine serielle Verdünnung im Verhältnis 1:2 mit 11 Verdünnungspunkten sollte verwendet werden, wenn der sich ergebende Konzentrationsbereich das gesamte Spektrum der Reaktionen aufgrund der in der Dosisfindungsstudie ermittelten Konzentrations-Reaktions-Kurve abdeckt. Ansonsten sollte eine Verdünnung im Verhältnis 1:5 verwendet werden.
|
|
Hauptversuche
|
31.
|
Ein Hauptversuch besteht aus seriellen Verdünnungen mit 11 Verdünnungspunkten (je nach Ausgangskonzentration für die Kriterien des Hauptversuchs entweder 1:2 oder 1:5), wobei jede Konzentration an drei Wells der 96-Well-Platte zu prüfen ist (siehe Abbildungen 3 und 4).
|
|
—
|
Bei den Agonisten-Hauptversuchen wird E2 in 11 Konzentrationen jeweils zweifach als Referenzstandard geprüft. Jede Platte enthält 4 Replikat-Wells mit der DMSO-Kontrolle und 4 Replikat-Wells mit der Methoxychlor-Kontrolle (9,06 x 10-6 M).
|
|
—
|
Bei den Antagonisten-Hauptversuchen werden Ral/E2 in 9 Konzentrationen mit 9,18 × 10-11 M E2 jeweils zweifach als Referenzstandard, 4 Replikat-Wells mit der 9,18 x 10-11 M E2 als Kontrolle, 4 Replikat-Wells als DMSO-Kontrolle und 4 Replikat-Wells mit 3,36 x 10-6 M Tamoxifen geprüft.
|
Abbildung 3
Anordnung bei Agonisten-Hauptversuchen mit 96-Well-Platten

Abkürzungen: TC1-1 bis TC1-11 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 1; TC2-1 bis TC2-11 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 2; E2-1 bis E2-11 = Konzentrationen des E2-Referenzstandards (hoch bis niedrig); Meth = p,p’-Methoxychlor, schwach positive Kontrolle; VK = DMSO (1 % v/v) EFM Vehikelkontrolle.
Abbildung 4
Anordnung bei Antagonisten-Hauptversuchen mit 96-Well-Platten
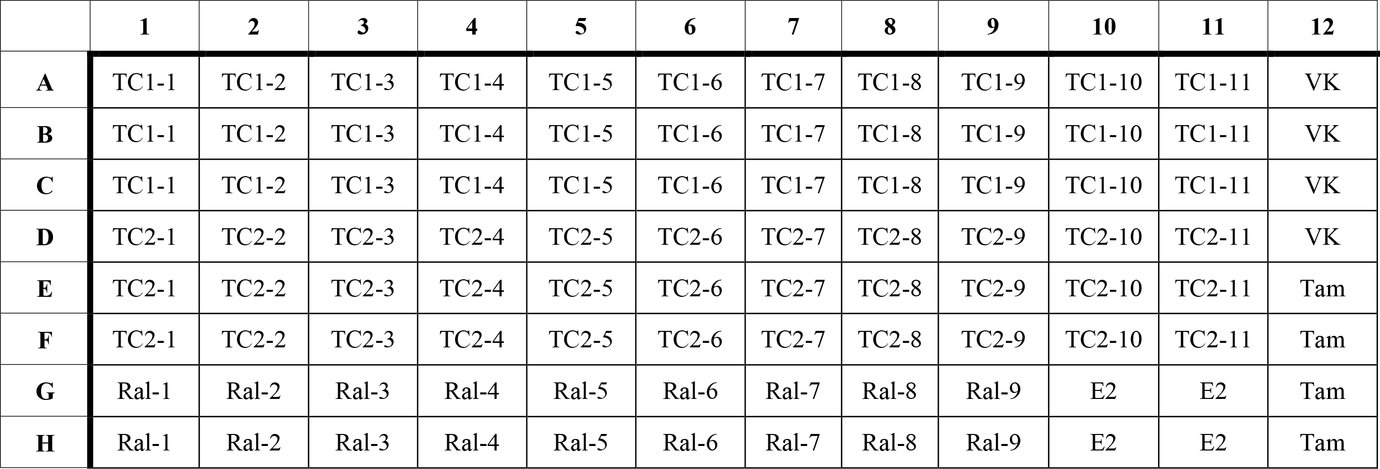
Abkürzungen: E2 = E2-Kontrolle; Ral-1 bis Ral-9 = Konzentrationen des Raloxifen-/E2-Referenzstandards (hoch bis niedrig); Tam = Tamoxifen/E2, schwach positive Kontrolle; TC1-1 bis TC1-11 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 1 (TC1); TC2-1 bis TC2-11 = Konzentrationen (hoch bis niedrig) der Prüfchemikalie 2 (TC2); VK = Vehikelkontrolle (DMSO [1 % v/v EFM.]).
Hinweis: Alle Referenz- und alle Prüf-Wells enthalten jeweils E2 in einer bestimmten Konzentration (9,18 x 10-11 M).
|
32.
|
Um unabhängige Ergebnisse sicherzustellen, sollten die Hauptversuche zur Prüfung der Chemikalien an unterschiedlichen Tagen wiederholt werden. In jedem Fall sollten mindestens zwei Hauptversuche durchgeführt werden. Wenn die Prüfergebnisse einander widersprechen (z. B. eine positives und ein negatives Ergebnis) oder wenn eine der durchgeführten Prüfungen nicht berücksichtigt werden kann, sollte eine dritte Prüfung durchgeführt werden.
|
Messung der Luminiszenz
|
33.
|
Die Luminiszenz wird im Bereich von 300-650 nm mit einem Injektionsluminometer und einer Software zur Steuerung des Injektionsvolumens und des Messintervalls gemessen (7). Die Lichtemission der einzelnen Wells wird in RLU pro Well angegeben.
|
ANALYSE DER DATEN
Bestimmung von EC50 /IC50
|
34.
|
Aus den Konzentrations-Reaktions-Daten werden der EC50-Wert (die Hälfte der maximalen Wirkungskonzentration einer Prüfchemikalie [Agonisten]) und der IC50-Wert (die Hälfte der maximalen Hemmkonzentration einer Prüfchemikalie [Antagonisten]) bestimmt. Bei Prüfchemikalien, die in einer oder mehreren Konzentrationen positiv bewertet wurden, wird die Konzentration, die die Hälfte der maximalen Wirkung auslöst, (IC50 bzw. EC50) mit einer Hill-Funktion oder mit einer geeigneten alternativen Funktion berechnet. Die Hill-Funktion ist ein logistisches mathematisches Modell mit vier Parametern, bei dem die Konzentration einer Prüfchemikalie mit der folgenden Formel in Bezug zu einer Wirkung (die gewöhnlich einer Sigmoidkurve folgt) gesetzt wird:
|

Dabei gilt:
Y= Wirkung (d. h. RLU-Werte),
X= Logarithmus der Konzentration,
Bottom= Mindestwirkung,
Top= maximale Wirkung,
lg EC50 (bzw. lg IC50)= Logarithmus von X als Mittelwert der Mindestwirkung und der maximalen Wirkung und
Hillslope= Steilheit der Kurve.
Mit dem Modell wird die beste Anpassung (best fit) für die Parameter Top, Bottom, Hillslope, und IC50 und EC50 ermittelt. Für die Berechnung der EC50- und der IC50-Werte sollte eine geeignete Statistik-Software verwendet werden (z. B. Graphpad PrismR).
Bestimmung von Ausreißern
|
35.
|
Eine fundierte statistische Bewertung könnte (u. a.) durch die Einbeziehung des Q-Tests (siehe Protokolle zum Agonisten- und zum Antagonisten-Assay (7)) zur Ermittlung nicht verwendbarer und aus der Datenanalyse auszuschließender Wells erleichtert werden.
|
|
36.
|
Beim E2-Referenzstandard (zwei Proben) werden alle angepassten RLU-Werte eines Replikat-Wells bei einer bestimmten E2-Konzentration als Ausreißer betrachtet, bei denen der Wert mehr als 20 % über oder unter dem angepassten RLU-Wert dieser Konzentration in der Datenbank der historischen Daten liegt.
|
Erfassung und Anpassung von Luminometer-Daten für die Dosisfindungsstudie
|
37.
|
Die Rohdaten des Luminometers werden in ein Tabellenformular für den Assay übertragen. Dabei sollte geprüft werden, ob die Daten auszuschließende Ausreißer beinhalten (siehe Akzeptanzkriterien der Prüfung in Bezug auf die in den Analysen ermittelten Parameter). Folgende Berechnungen sind durchzuführen:
|
Agonisten
|
Schritt 1
|
Der Mittelwert der DMSO-Vehikelkontrolle (VK) wird berechnet.
|
|
Schritt 2
|
Der Mittelwert der DMSO-VK wird vom Wert jedes einzelnen Wells abgezogen, um die Daten zu normalisieren.
|
|
Schritt 3
|
Für den Referenzstandard (E2) wird die mittlere n-fache Induktion ermittelt.
|
|
Schritt 4
|
Für die Prüfchemikalien wird der mittlere EC50-Wert ermittelt.
|
Antagonisten
|
Schritt 1
|
Der Mittelwert der DMSO-VK wird berechnet.
|
|
Schritt 2
|
Der Mittelwert der DMSO-VK wird vom Wert jedes einzelnen Wells abgezogen, um die Daten zu normalisieren.
|
|
Schritt 3
|
Für den Referenzstandard (E2) wird die mittlere n-fache Reduktion ermittelt.
|
|
Schritt 4
|
Der Mittelwert des E2-Referenzstandards wird berechnet.
|
|
Schritt 5
|
Für die Prüfchemikalien wird der mittlere IC50-Wert ermittelt.
|
Erfassung und Anpassung von Luminometer-Daten für Hauptversuche
|
38.
|
Die Rohdaten des Luminometers werden in ein Tabellenformular für den Assay übertragen. Dabei sollte geprüft werden, ob die Daten auszuschließende Ausreißer beinhalten (siehe Akzeptanzkriterien der Prüfung in Bezug auf die in den Analysen ermittelten Parameter). Folgende Berechnungen sind durchzuführen:
|
Agonisten
|
Schritt 1
|
Der Mittelwert der DMSO-VK wird berechnet.
|
|
Schritt 2
|
Der Mittelwert der DMSO-VK wird vom Wert jedes einzelnen Wells abgezogen, um die Daten zu normalisieren.
|
|
Schritt 3
|
Für den Referenzstandard (E2) wird die mittlere n-fache Induktion ermittelt.
|
|
Schritt 4
|
Für die Prüfchemikalien wird der mittlere EC50-Wert ermittelt.
|
|
Schritt 5
|
Für Methoxychlor wird der mittlere angepasste RLU-Wert berechnet.
|
Antagonisten
|
Schritt 1
|
Der Mittelwert der DMSO-VK wird berechnet.
|
|
Schritt 2
|
Der Mittelwert der DMSO-VK wird vom Wert jedes einzelnen Wells abgezogen, um die Daten zu normalisieren.
|
|
Schritt 3
|
Für den Referenzstandard (Ral/E2) wird die mittlere n-fache Induktion ermittelt.
|
|
Schritt 4
|
Für Ral/E2 und für die Prüfchemikalien wird der mittlere IC50-Wert ermittelt.
|
|
Schritt 5
|
Für Tamoxifen wird der mittlere angepasste RLU-Wert berechnet.
|
|
Schritt 6
|
Der Mittelwert des E2-Referenzstandards wird berechnet.
|
Kriterien für die Auswertung von Daten
|
39.
|
Der VM7Luc-ER-TA-Assay soll im Rahmen einer Beweiskraftermittlung die Priorisierung von Stoffen für die In-vivo-Prüfung von ED unterstützen. In diesem Verfahren werden die Prüfchemikalien hinsichtlich der Aktivität von ER-Agonisten oder -Antagonisten als positiv oder negativ eingestuft. Die Kriterien für die Einstufung als positiv oder negativ in der Studie zur Validierung des VM7Luc-ER-TA-Assays werden in Tabelle 1 beschrieben:
|
Tabelle 1
Kriterien für die Einstufung als positiv oder als negativ
|
|
|
AGONISTEN-AKTIVITÄT
|
|
Positiv
|
|
—
|
Bei allen hinsichtlich der ER-Agonisten-Aktivität als positiv eingestuften Prüfchemikalien sollte die Konzentrations-Reaktions-Kurve eine Basislinie gefolgt von einer positiven Steigung aufweisen und in einem Plateau oder einem Peak schließen. Manchmal sind vielleicht nur zwei dieser Merkmale (Basislinie – positive Steigung oder positive Steigung – Peak) ausgeprägt.
|
|
—
|
Der Kurvenverlauf im Bereich der positiven Steigung sollte mindestens drei Punkte mit einander nicht überschneidenden Fehlerbalken enthalten (Mittelwert ± SD). Die Punkte auf der Basislinie werden nicht berücksichtigt. Der lineare Verlauf der Kurve kann jedoch den Peak oder den ersten Punkt des Plateaus beinhalten.
|
|
—
|
Eine Einstufung als positiv setzt eine bestimmte Reaktionsamplitude voraus, d. h. eine Differenz zwischen Basislinie und Peak von mindestens 20 % des maximalen Wertes beim Referenzstandard E2 (mindestens 2 000 RLU, wenn der maximale Reaktionswert der Referenzstandards [E2] auf 10 000 RLU angepasst wurde).
|
|
—
|
Nach Möglichkeit sollte für jede positiv eingestufte Prüfchemikalie ein EC50-Wert berechnet werden.
|
|
|
Negativ
|
Der mittlere angepasste RLU-Wert einer bestimmten Konzentration entspricht höchstens dem mittleren RLU-Wert der DMSO-Kontrolle zuzüglich der 3-fachen Standardabweichung des DMSO-RLU-Werts.
|
|
Ungeeignet
|
Daten, die aufgrund erheblicher qualitativer oder quantitativer Einschränkungen nicht als gültig zum Nachweis des Vorliegens oder des Fehlens einer Aktivität akzeptiert werden können, sind als ungeeignet zu betrachten und können für die Entscheidung, ob eine Prüfchemikalie als positiv oder als negativ einzustufen ist, nicht herangezogen werden. In diesen Fällen sollten die betreffenden Chemikalien nochmals geprüft werden.
|
|
|
|
ANTAGONISTEN-AKTIVITÄT
|
|
Positiv
|
|
—
|
Die Prüfchemikalie ergibt eine Konzentrations-Reaktions-Kurve bestehend aus einer Basislinie mit anschließender negativer Steigung.
|
|
—
|
Der Kurvenverlauf im Bereich der negativen Steigung sollte mindestens drei Punkte mit einander nicht überschneidenden Fehlerbalken enthalten. Die Punkte auf der Basislinie werden nicht berücksichtigt. Der lineare Bereich der Kurve kann jedoch den ersten Punkt des Plateaus enthalten.
|
|
—
|
Die Aktivität des Werts der maximalen Reaktion beim Referenzstandard Ral/E2 sollte um mindestens 20 % reduziert werden (d. h. auf höchstens 8 000 RLU, wenn der Wert der maximalen Reaktion des Referenzstandards [Ral/E2] auf 10 000 RLU angepasst wurde).
|
|
—
|
Die höchsten nicht zytotoxischen Konzentrationen der Prüfchemikalie sollten höchstens 1x10-5 M betragen.
|
|
—
|
Nach Möglichkeit sollte für jede positiv eingestufte Prüfchemikalie ein IC50-Wert berechnet werden.
|
|
|
Negativ
|
Alle Datenpunkte liegen bei Konzentrationen unter 1,0 x 10-5 M über dem EC80-Wert (80 % der E2-Rektion oder 8 000 RLU).
|
|
Ungeeignet
|
Daten, die aufgrund erheblicher qualitativer oder quantitativer Einschränkungen nicht als gültig zum Nachweis des Vorliegens oder des Fehlens einer Aktivität akzeptiert werden können, sind als ungeeignet zu betrachten und können für die Entscheidung, ob eine Prüfchemikalie als positiv oder als negativ einzustufen ist, nicht herangezogen werden. In diesen Fällen sollten die betreffenden Chemikalien nochmals geprüft werden.
|
|
40.
|
Positive Ergebnisse werden möglichst sowohl durch die Größenordnung der Wirkung als auch durch die Konzentration beschrieben, bei der die Wirkung eintritt. Den Abbildungen 5 und 6 sind Beispiele für als positiv, negativ und nicht geeignet zu bewertende Daten zu entnehmen.
|
Abbildung 5
Beispiele Agonisten: Als positiv, negativ und nicht geeignet zu bewertende Daten

Die gestrichelte Linie bezeichnet 20 % der E2-Reaktion, 2 000 angepasste und normalisierte RLU-Werte.
Abbildung 6
Beispiele Antagonisten: Als positiv, negativ und nicht geeignet zu bewertende Daten

Die gestrichelte Linie bezeichnet 80 % der Ral/E2-Reaktion, 8 000 angepasste und normalisierte RLU-Werte.
Die durchgehende Linie bezeichnet die Konzentration 1,00 x 10-5 M. Eine als positiv einzustufende Reaktion sollte bei Konzentrationen unter 1,00 x 10-5 M unter der 8 000-RLU-Linie liegen.
Bei mit einem Sternchen gekennzeichneten Konzentrationen im meso-Hexestrol-Diagramm liegt die Viabilität bei mindestens 2.
Die Prüfergebnisse für meso-Hexestrol werden als nicht geeignete Daten betrachtet, da die einzige Reaktion unter 8 000 RLU bei einer Konzentration von 1,00 x 10-5 M erfolgt.
|
41.
|
Die Berechnung der EC50- und der IC50-Werte kann mit einer Hill-Funktion mit vier Parametern vorgenommen werden (siehe Protokolle zu Agonisten- und Antagonisten-Assays (7)). Die Erfüllung der Akzeptanzkriterien deutet darauf hin, dass das System ordnungsgemäß funktioniert. Dies gewährleistet jedoch nicht, dass ein bestimmter Prüflauf tatsächlich exakte Daten ergibt. Die Wiederholung der Ergebnisse des ersten Prüflaufs ist die beste Bestätigung dafür, dass exakte Daten ermittelt wurden (siehe Nummer 19 im Abschnitt „ELEMENTE DES ER-TA-ASSAYS“).
|
PRÜFBERICHT
|
42.
|
Siehe Nummer 20 im Abschnitt „ELEMENTE DES ER-TA-ASSAYS“.
|
LITERATUR
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5367-5373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): S 270-273.
|
|
(6)
|
Coecke, S., u. a. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: S. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7850.
|
|
(8)
|
Rogers, J.M., Denison, M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol., 13(1):67-82.
|
|
(9)
|
Escande, A., u. a. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1459-1469.
|
|
(10)
|
Thorne, N., Inglese, J., Auld, D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-657.
|
|
(11)
|
Kuiper, G.G, u. a. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10):4252-4263.
|
|
(12)
|
Geisinger u. a. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin u. a. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., u. a. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, , J.M. und Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
ANLAGE 4
ASSAY ZUR TRANSAKTIVIERUNG DES STABIL TRANSFIZIERTEN HUMANEN ÖSTROGENREZEPTORS Α ZUM NACHWEIS DER WIRKUNG VON CHEMIKALIEN AUF ÖSTROGEN-AGONISTEN UND -ANTAGONISTEN MIT DER ERΑ-CALUX-ZELLLINIE
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
1.
|
Beim ERα-CALUX-Transaktivierungs-Assay wird die humane U2OS-Zelllinie zum Nachweis einer durch den humanen Östrogenrezeptor α (hERα) vermittelten Östrogen-Agonisten- und -Antagonisten-Aktivität verwendet. In der Studie zur Validierung des ERα-CALUX-Bioassays mit stabil transfiziertem ERα durch BioDetection Systems BV (Amsterdam, Niederlande) wurden die Relevanz und die Zuverlässigkeit des Assays für den vorgesehenen Zweck nachgewiesen (1). Die ERα-CALUX-Zelllinie exprimiert ausschließlich den humanen ERα (2) (3),
|
|
2.
|
Dieser Assay wurde speziell zum Nachweis von hERα und einer durch hERα vermittelten Transaktivierung anhand einer Messung der Bioluminiszenz als Endpunkt entwickelt. In Anbetracht des hohen Signal-Rausch-Verhältnisses ist bei Bioassays die Berücksichtigung der Bioluminiszenz allgemein üblich (4).
|
|
3.
|
Bei Phytoöstrogen-Konzentrationen von mehr als 1 μM wurde eine Überaktivierung des Luciferase-Reportergens mit der Folge einer nicht durch den Rezeptor vermittelten Lumineszenz festgestellt (5) (6) (7). Daher müssen höhere Konzentrationen von Phytoöstrogenen oder ähnlichen Verbindungen sorgfältig untersucht werden, die bei Transaktivierungs-Assays mit stabil transfiziertem ER zu einer Überaktivierung der Luciferase-Expression führen können (siehe Anlage 2).
|
|
4.
|
Vor der Verwendung dieses Assays für rechtliche Zwecke sollten die Abschnitte „ALLGEMEINE EINLEITUNG“ und „ELEMENTE DES ER-TA-ASSAYS“ gelesen werden. Die in der vorliegenden Prüfmethode verwendeten Begriffe und Abkürzungen werden in Anlage 1 erläutert.
|
PRINZIP DES ASSAYS (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
5.
|
Mit dem Bioassay werden ER-Liganden-Bindungen nach einer Translokation des Rezeptor-Liganden-Komplexes an den Zellkern bewertet. Im Zellkern bindet der Rezeptor-Liganden-Komplex spezifische DNA-Response-Elemente und transaktiviert ein Leuchtkäfer-Luciferase-Reporter-Gen. Dies führt zu einer verstärkten zellulären Expression des Luciferase-Enzyms. Nach der Zugabe des Luciferase-Substrats Luciferin wird das Luciferin in ein biolumineszierendes Produkt umgewandelt. Das emittierte Licht kann mit einem Luminometer leicht erkannt und gemessen werden.
|
|
6.
|
Für das Prüfsystem werden stabil transfizierte ERα-CALUX-Zellen verwendet. Die ERα-CALUX-Zellen stammen aus der humanen Osteoblasten-Osteosarkom-Zelllinie U2OS. Die humanen U2OS-Zellen wurden durch Copräzipitation mit Calciumphosphat mit 3xHRE-TATA-Luc und pSG5-neo-hERα stabil transfiziert. Die U2OS-Zelllinie wurde als am besten geeignete Responsive-Reporter-Zelllinie für Östrogen (und andere Steroidhormone) ausgewählt, da diese Zelllinie nur eine geringe bzw. keinerlei endogene Rezeptor-Aktivität entwickelte. Das Fehlen endogener Rezeptoren wurde nur mit Luciferase-Reporter-Plasmiden geprüft, bei denen nach Zugabe von Rezeptor-Liganden keine Aktivität festgestellt werden konnte. Außerdem stimulierte diese Zelllinie starke hormonvermittelte Reaktionen bei vorübergehender Zugabe verwandter Rezeptoren (2) (3) (8).
|
|
7.
|
Die Prüfung von Chemikalien auf eine östrogene bzw. antiöstrogene Aktivität mit der ERα-CALUX-Zelllinie besteht aus einem Vorversuch mit anschließenden Hauptversuchen. Im Vorversuch werden die Löslichkeit, die Zytotoxizität und ein engerer Konzentrationsbereich von Prüfchemikalien für den Hauptversuch ermittelt. Bei den Hauptversuchen werden die engeren Konzentrationsbereiche der Prüfchemikalien zunächst mit den ERα-CALUX-Bioassays untersucht. Anschließend erfolgt die Einstufung der Prüfchemikalien aufgrund ihrer Wirkung als Agonisten oder Antagonisten.
|
|
8.
|
Kriterien für die Auswertung der Daten werden in Nummer 59 eingehend beschrieben. Eine Prüfchemikalie wird kurz gesagt dann als Agonist eingestuft, wenn zwei aufeinander folgende Konzentrationen der Prüfchemikalie eine Reaktion induzieren, die größer oder gleich 10 % der maximalen Reaktion des Referenzstandards 17β-Estradiol (PC10) ist. Entsprechend wird eine Prüfchemikalie als Antagonist eingestuft, wenn zwei aufeinander folgende Konzentrationen der Prüfchemikalie eine Reaktion induzieren, die kleiner oder gleich 80 % der maximalen Reaktion des Referenzstandards Tamoxifen (PC80) ist.
|
VERFAHREN
Zelllinien
|
9.
|
Für den Assay sollte die stabil transfizierte U2OS-ERα-CALUX-Zelllinie verwendet werden. Die Zelllinie kann nach Maßgabe einer technischen Lizenzvereinbarung von BioDetection Systems BV, Amsterdam, Niederlande, bezogen werden.
|
|
10.
|
Für die Prüfung sollten ausschließlich mycoplasmafreie Zellkulturen verwendet werden. Die zu verwendenden Zellchargen sollten entweder in Bezug auf Mycoplasma-Kontaminationen als negativ zertifiziert sein oder vor der Verwendung auf Mycoplasma untersucht werden. Die RT-PCR (Echtzeit-Polymerase-Kettenreaktion) sollte für den empfindlichen Nachweis einer Mycoplasma-Infektion verwendet werden (9).
|
Stabilität der Zelllinie
|
11.
|
Um die Stabilität und die Integrität der CALUX-Zellen zu erhalten, sollten die Zellen in flüssigem Stickstoff (bei -80 0C) aufbewahrt werden. Nach dem Auftauen der Zellen zum Ansetzen einer neuen Kultur sollten die Zellen mindestens zweimal subkultiviert werden, bevor sie zur Bewertung der Agonisten- bzw. Antagonisten-Aktivität von Chemikalien verwendet werden. Die Subkultivierung sollte sich auf höchstens 30 Passagen beschränken.
|
|
12.
|
Zur Überwachung der Stabilität der Zelllinie sollte die Empfindlichkeit des Systems zur Prüfung der Agonisten- bzw. Antagonisten-Wirkung anhand der EC50- bzw. der IC50-Werte des Referenzstandards untersucht werden. Außerdem sollten die relative Induktion der Positivkontrollprobe (PK) und der Negativkontrollprobe (NK) überwacht werden. Die Ergebnisse sollten mit den Akzeptanzkriterien des ERα-CALUX-Bioassays zur Untersuchung der Agonisten- (Tabelle 3C) bzw. der Antagonisten-Wirkung (Tabelle 4C) übereinstimmen. Die Referenzstandards sowie die Positiv- und die Negativkontrollen für Agonisten und Antagonisten sind in den Tabellen 1 und 2 angegeben.
|
Bedingungen für die Haltung der Zellkulturen und für die Plattierung
|
13.
|
Die U2OS-Zellen sollten im Nährmedium (DMEM/F12 (1:1) mit Phenolrot als pH-Indikator ergänzt um fetales Rinderserum (7,5 %), nicht essentielle Aminosäuren (1 %), 10 Einheiten/ml Penicillin, Streptomycin und Geneticin (G-418) als Selektionsmarker) kultiviert werden. Anschließend werden die Zellen bei 37 °C und 100 % Luftfeuchtigkeit in einem CO2-Inkubator (5 % CO2) inkubiert. Wenn die Zellen eine Konfluenz von 85-95 % erreicht haben, werden sie entweder subkultiviert oder zum Überimpfen in 96-Well-Mikrotiterplatten vorbereitet. Im letztgenannten Fall sind 1x105 Zellen/ml im östrogenfreien Assay-Medium (DMEM/F12 (1:1) (ohne Phenolrot und ergänzt um mit dextranbeschichteter Aktivkohle behandeltes fetales Rinderserum (5 % v/v), nicht essentielle Aminosäuren (1 % v/v) und 10 Einheiten/ml Penicillin und Streptomycin) neu zu suspendieren und in die Wells der 96-Well-Mikrotiterplatten (100 μl homogenisierte Zellsuspension) zu plattieren. Vor der Exposition werden die Zellen 24 Stunden in einem CO2-Inkubator (5 % CO2, 37 0C, 100 % Luftfeuchtigkeit) vorinkubiert. Verwendetes Kunststoffmaterial muss östrogenfrei sein.
|
Akzeptanzkriterien
|
14.
|
Die Agonisten- und die Antagonisten-Aktivität der Prüfchemikalie(n) werden in Prüfreihen untersucht. Eine Prüfreihe umfasst höchstens 6 Mikrotiterplatten. Jede Prüfreihe beinhaltet mindestens eine vollständige Verdünnungsreihe eines Referenzstandards, eine Positivkontrollprobe, eine Negativkontrollprobe und Lösungsmittelkontrollen. In den Abbildungen 1 und 2 ist die Anordnung auf den Platten für Prüfreihen zur Ermittlung der Agonisten- bzw. Antagonisten-Aktivität dargestellt.
|
|
15.
|
Jede Verdünnung der Referenzstandards, der Prüfchemikalien und aller Lösungsmittelkontrollen sowie alle Positiv- und alle Negativkontrollen sollten jeweils dreimal untersucht werden. Jede der drei Untersuchungen sollte die in den Tabellen 3A und 4A genannten Anforderungen erfüllen.
|
|
16.
|
Bei allen Prüfreihen wird jeweils auf der ersten Platte eine vollständige Verdünnungsreihe des Referenzstandards (17β-Estradiol für Agonisten und Tamoxifen für Antagonisten) gemessen. Um die Ergebnisse der Untersuchungen der übrigen 5 Mikrotiterplatten mit den Ergebnissen der ersten Mikrotiterplatte mit der vollständigen Konzentrations-Reaktions-Kurve des Referenzstandards vergleichen zu können, sollten alle Platten 3 Kontrollproben enthalten: die Lösungsmittelkontrolle, die höchste geprüfte Konzentration des Referenzstandards und die ungefähre EC50- (Agonisten) bzw. IC50-Konzentration (Antagonisten) des Referenzstandards. Das Verhältnis der durchschnittlichen Kontrollproben auf der ersten Platte und den übrigen 5 Platten sollte die Anforderungen in Tabelle 3C (Agonisten) bzw. in Tabelle 4C (Antagonisten) erfüllen.
|
|
17.
|
Für jede der Mikrotiterplatten einer Prüfreihe wird der z-Faktor berechnet (10). Die Berechnung des z-Faktors ist anhand der Reaktionen bei der höchsten und bei der niedrigsten Konzentration des Referenzstandards vorzunehmen. Eine Mikrotiterplatte ist dann gültig, wenn sie die Anforderungen in Tabelle 3C (Agonisten) bzw. in Tabelle 4C (Antagonisten) erfüllt.
|
|
18.
|
Die Dosis-Reaktions-Kurve des Referenzstandards muss sigmoidal verlaufen. Die aus der Reaktion der Verdünnungsreihe des Referenzstandards abgeleiteten EC50- bzw. IC50-Werte sollten die Anforderungen in Tabelle 3C (Agonisten) bzw. Tabelle 4C (Antagonisten) erfüllen.
|
|
19.
|
Jede Prüfreihe sollte mindestens eine Positivkontrollprobe und eine Negativkontrollprobe enthalten. Die berechnete relative Induktion der Positiv- und der Negativkontrollprobe sollte die Anforderungen in Tabelle 3C (Agonisten) bzw. in Tabelle 4C (Antagonisten) erfüllen.
|
|
20.
|
Bei allen Messungen wird der Induktionsfaktor der höchsten Konzentration des Referenzstandards gemessen, indem der höchste mittlere RLU-Wert des 17β-Estradiol-Referenzstandards durch den mittleren RLU-Wert der Referenz-Lösungsmittelkontrolle geteilt wird. Dieser Induktionsfaktor sollte die Mindestanforderungen für die n-fache Induktion in Tabelle 3C (Agonisten) bzw. in Tabelle 4C (Antagonisten) erfüllen.
|
|
21.
|
Nur Mikrotiterplatten, die alle oben genannten Akzeptanzkriterien erfüllen, werden als gültig betrachtet und können zur Bewertung der Reaktion der Prüfchemikalien berücksichtigt werden.
|
|
22.
|
Die Akzeptanzkriterien gelten sowohl für Vorversuche als auch für die Hauptversuche.
|
Tabelle 1
Konzentrationen des Referenzstandards, der Positivkontrolle (PK) und der Negativkontrolle (NK) beim CALUX-Antagonisten-Bioassay
|
|
Stoff
|
CAS-Nr.
|
Prüfbereich (M)
|
|
Referenzstandard
|
17β-Estradiol
|
50-28-2
|
1*10-13 - 1*10-10
|
|
Positivkontrolle (PK)
|
17α-Methyltestosteron
|
58-18-4
|
3*10-6
|
|
Negativkontrolle (NK)
|
Corticosteron
|
50-22-6
|
1*10-8
|
Tabelle 2
Konzentrationen des Referenzstandards, der Positivkontrolle (PK) und der Negativkontrolle (NK) beim CALUX-Agonisten-Bioassay
|
|
Stoff
|
CAS-Nr.
|
Prüfbereich (M)
|
|
Referenzstandard
|
Tamoxifen
|
10540-29-1
|
3*10-9 - 1*10-5
|
|
Positivkontrolle (PK)
|
4-Hydroxytamoxifen
|
68047-06-3
|
1*10-9
|
|
Negativkontrolle (NK)
|
Resveratrol
|
501-36-0
|
1*10-5
|
Tabelle 3
Akzeptanzkriterien des ERα-CALUX-Agonisten-Bioassays
|
A – einzelne Proben auf einer Platte
|
Kriterium
|
|
1
|
Maximale %SD bei 3 Wells (NK und PK sowie alle Verdünnungen der Prüfchemikalie und des Referenzstandards außer C0)
|
< 15 %
|
|
2
|
Maximale %SD bei 3 Wells (Referenzstandard und Lösungsmittelkontrollen der Prüfchemikalie (C0, SC))
|
< 30 %
|
|
3
|
Maximale LDH-Freisetzung als Maß der Zytotoxizität
|
< 120 %
|
|
B – innerhalb einer einzelnen Mikrotiterplatte
|
|
|
4
|
Verhältnis der Lösungsmittelkontrolle des Referenzstandards (C0, Platte 1) und der Lösungsmittelkontrolle der Prüfchemikalie (SC, Platten 2 bis x)
|
0,5 bis 2,0
|
|
5
|
Verhältnis zwischen der ungefähren EC50 und den höchsten Konzentrationen des Referenzstandards auf Platte 1 und der ungefähren EC50 und den höchsten Konzentrationen des Referenzstandards auf den Platten 2 bis x (C4, C8)
|
0,70 bis 1,30
|
|
6
|
z-Faktor der einzelnen Platten
|
> 0,6
|
|
C – innerhalb einer Untersuchungsreihe (alle Platten einer Reihe)
|
|
|
7
|
Sigmoidkurve des Referenzstandards
|
Ja (17ß-Estradiol)
|
|
8
|
EC50-Bereich des Referenzstandards 17ß-Estradiol
|
4*10-12 – 4*10-11 M
|
|
9
|
Geringste n-fache Induktion der höchsten 17ß-Estradiol-Konzentration bezogen auf die Lösungsmittelkontrolle des Referenzstandards
|
5
|
|
10
|
Relative Induktion (%) PK
|
> 30 %
|
|
11
|
Relative Induktion (%) NK
|
< 10 %
|
PK: Positivkontrolle; NK: Negativkontrolle; SC: Lösungsmittelkontrolle der Prüfchemikalie; C0: Lösungsmittelkontrolle des Referenzstandards; SD: Standardabweichung; LDH: Laktatdehydrogenase
Tabelle 4
Akzeptanzkriterien des ERα-CALUX-Antagonisten-Bioassays
|
A – einzelne Proben auf einer Platte
|
Kriterium
|
|
1
|
Maximale %SD bei 3 Wells (NK und PK sowie alle Verdünnungen der Prüfchemikalie und des Referenzstandards, Lösungsmittelkontrolle (C0))
|
< 15 %
|
|
2
|
Maximale %SD bei 3 Wells (Vehikelkontrolle (VK) und höchste Konzentration des Referenzstandards (C8))
|
< 30 %
|
|
3
|
Maximale LDH-Freisetzung als Maß der Zytotoxizität
|
< 120 %
|
|
B – innerhalb einer einzelnen Mikrotiterplatte
|
|
|
4
|
Verhältnis der Lösungsmittelkontrolle des Referenzstandards (C0, Platte 1) und der Lösungsmittelkontrolle der Prüfchemikalie (SC, Platten 2 bis x)
|
0,70 bis 1,30
|
|
5
|
Verhältnis zwischen der ungefähren IC50-Konzentrationen des Referenzstandards auf Platte 1 und den ungefähren IC50-Konzentrationen des Referenzstandards auf den Platten 2 bis x (C8)
|
0,70 bis 1,30
|
|
6
|
Verhältnis der höchsten Konzentrationen des Referenzstandards auf Platte 1 und den höchsten Konzentrationen des Referenzstandards auf den Platten 2 bis x (C8)
|
0,50 bis 2,0
|
|
7
|
z-Faktor der einzelnen Platten
|
> 0,6
|
|
C – innerhalb einer Untersuchungsreihe (alle Platten einer Reihe)
|
|
|
8
|
Sigmoidkurve des Referenzstandards
|
Ja (Tamoxifen)
|
|
9
|
IC50-Bereich des Referenzstandards (Tamoxifen)
|
1*10-8 - 1*10-7 M
|
|
10
|
Niedrigste n-fache Induktion der Lösungsmittelkontrolle des Referenzstandards in Bezug auf die höchste Tamoxifen-Konzentration
|
2,5
|
|
11
|
Relative Induktion (%) PK
|
< 70 %
|
|
12
|
Relative Induktion (%) NK
|
< 85 %
|
PK: Positivkontrolle; NK: Negativkontrolle; VK: Vehikelkontrolle (Lösungsmittelkontrolle ohne feste Konzentration des Agonisten-Referenzstandards); SC: Lösungsmittelkontrolle der Prüfchemikalie; C0: Lösungsmittelkontrolle des Referenzstandards; SD: Standardabweichung; LDH: Laktatdehydrogenase
Lösungsmittel-/Vehikelkontrolle, Referenzstandards, Positivkontrollen, Negativkontrollen
|
23.
|
Sowohl für den Vorversuch als auch für die Hauptversuche sollten die gleichen Lösungsmittel-/Vehikelkontrollen, Referenzstandards, Positivkontrollen und Negativkontrollen verwendet werden. Außerdem sollte die Konzentration von Referenzstandards, Positivkontrollen und Negativkontrollen identisch sein.
|
Lösungsmittelkontrolle
|
24.
|
Das zur Auflösung einer Prüfchemikalie verwendete Lösungsmittel sollte als Lösungsmittelkontrolle geprüft werden. Bei der Validierung des ERα-CALUX-Bioassays wurde Dimethylsulfoxid (DMSO, 1 % (v/v); CASRN 67-68-5) als Vehikel verwendet. Wenn ein anderes Vehikel als DMSO verwendet wird, sollten alle Referenzstandards, Kontrollen und Prüfchemikalien mit diesem Vehikel geprüft werden. Dabei ist zu beachten, dass die Lösungsmittelkontrolle bei den Agonisten-Untersuchungen den Agonisten-Referenzstandard 17β-Estradiol in einer bestimmten Konzentration (etwa EC50) enthält. Zur Untersuchung des bei Antagonisten-Assays zu verwendenden Lösungsmittels sollte eine Vehikelkontrolle zubereitet und geprüft werden.
|
Vehikelkontrolle (Antagonisten)
|
25.
|
Dabei ist zu beachten, dass zum Assay-Medium der Agonisten-Referenzstandard 17β-Estradiol in einer bestimmten Konzentration (etwa EC50) hinzugegeben wurde. Um das zur Lösung der Prüfchemikalien für Antagonisten zu verwendende Lösungsmittel zu prüfen, ist ein Assay-Medium ohne bestimmte Konzentration des Agonisten-Referenzstandards 17β-Estradiol zuzubereiten. Diese Kontrollprobe wird als Vehikelkontrolle bezeichnet. Bei der Validierung des ERα-CALUX-Bioassays wurde Dimethylsulfoxid (DMSO, 1 % (v/v); CASRN 67-68-5) als Vehikel verwendet. Wenn ein anderes Vehikel als DMSO verwendet wird, sollten alle Referenzstandards, Kontrollen und Prόfchemikalien mit diesem Vehikel geprόft werden.
|
Referenzstandards
|
26.
|
Der Referenzstandard für Agonisten ist 17β-Estradiol (Tabelle 1). Die Referenzstandards umfassen eine Verdünnungsreihe mit 17β-Estradiol in acht Konzentrationen (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11 und 1*10-10 M).
|
|
27.
|
Der Referenzstandard für Antagonisten ist Tamoxifen (Tabelle 2). Die Referenzstandards umfassen eine Verdünnungsreihe mit Tamoxifen in acht Konzentrationen (3*10-9, 1*10-8, 3*10-8, 1*10-7, 3*10-7, 1*10-6, 3*10-6 und 1*10-5 M). Jede der Konzentrationen des Antagonisten-Referenzstandards wird parallel mit einer bestimmten Konzentration des Agonisten-Referenzstandards 17β-Estradiol (3*10-12 M) inkubiert.
|
Positivkontrolle
|
28.
|
Als Positivkontrolle für Agonisten-Assays wird 17α-Methyltestosteron verwendet (Tabelle 1).
|
|
29.
|
Die Positivkontrolle für Antagonisten-Assays besteht aus 4-Hydroxytamoxifen (Tabelle 2). Die Antagonisten-Positivkontrolle wird parallel mit einer bestimmten Konzentration des Agonisten-Referenzstandards 17β-Estradiol (3*10-12 M) inkubiert.
|
Negativkontrolle
|
30.
|
Als Negativkontrolle für Agonisten-Assays wird Corticosteron verwendet (Tabelle 1).
|
|
31.
|
Die Negativkontrolle für Antagonisten-Assays besteht aus Resveratrol (Tabelle 2). Die Antagonisten-Negativkontrolle wird parallel mit einer bestimmten Konzentration des Agonisten-Referenzstandards 17β-Estradiol (3*10-12 M) inkubiert.
|
Zum Nachweis der Eignung des Labors siehe Nummer 14 und Tabellen 3 und 4 im Abschnitt „ELEMENTE DES ER-TA-ASSAYS“ bei dieser Prüfmethode.
Vehikel
|
32.
|
Das Vehikel zur Lösung von Prüfchemikalien sollte die Prüfchemikalien vollständig auflösen und sich mit dem Zellmedium mischen. Geeignete Lösungsmittel sind DMSO, Wasser und Ethanol (mit einer Reinheit 95-100 %). Wenn DMSO als Lösungsmittel verwendet wird, sollte die maximale DMSO-Konzentration nicht mehr als 1 % (v/v) betragen. Vor der Verwendung sollte in einer Prüfung sichergestellt werden, dass das Lösungsmittel nicht zytotoxisch wirkt und die Durchführung des Assays nicht beeinflusst.
|
Zubereitung der Referenzstandards, Positivkontrollen, Negativkontrollen und Prüfchemikalien
|
33.
|
Referenzstandards, Positivkontrollen, Negativkontrollen und Prüfchemikalien werden in 100 % DMSO (oder einem sonstigen geeigneten Lösungsmittel) gelöst. Anschließend werden von diesem Lösungsmittel geeignete (serielle) Verdünnungen zubereitet. Vor der Lösung sollten sich alle Stoffe an die Raumtemperatur anpassen können. Frisch zubereitete Vorratslösungen der Referenzstandards, der Positivkontrollen, der Negativkontrollen und der Prüfchemikalien sollten keine erkennbaren Niederschläge oder Trübungen aufweisen. Vorräte des Referenzstandards und der Kontrollen können in größeren Mengen zubereitet werden. Vorratslösungen der Prüfchemikalien sollten für jeden Versuch frisch zubereitet werden. Endverdünnungen von Referenzstandards, Positivkontrollen, Negativkontrollen und Prüfchemikalien sollten für jeden Versuch frisch zubereitet und innerhalb von 24 Stunden nach der Zubereitung verwendet werden.
|
Löslichkeit, Zytotoxizität und Dosisfindung
|
34.
|
Beim Vorversuch wird die Löslichkeit der Prüfchemikalien im gewählten Lösungsmittel bestimmt. Für den Versuch wird eine Vorratslösung mit einer Konzentration von maximal 0,1 M zubereitet. Bei Lösungsproblemen bei dieser Konzentration sollten Vorratslösungen mit niedrigeren Konzentrationen zubereitet werden, bis die Prüfchemikalien vollständig gelöst sind. Beim Vorversuch werden serielle Verdünnungen der Prüfchemikalie im Verhältnis 1:10 untersucht. Bei der Prüfung von Agonisten und Antagonisten liegt die maximale Assay-Konzentration bei 1 mM. Nach dem Vorversuch wird ein geeigneter angepasster Konzentrationsbereich für die Prüfchemikalien ermittelt, der dann bei den Hauptversuchen zu untersuchen ist. Für Hauptversuche sollten folgende Verdünnungen verwendet werden: 1x, 3x, 10x, 30x, 100x, 300x, 1000x und 3000x.
|
|
35.
|
Im Protokoll des Agonisten- und des Antagonisten-Assays ist eine Zytotoxizitätsprüfung vorgesehen (11). Die Zytotoxizitätsprüfung ist Bestandteil sowohl des Vorversuchs als auch der Hauptversuche. Bei der Validierung des ERα-CALUX-Bioassays wurde die Zytotoxizität nach der Exposition gegenüber den Prüfchemikalien anhand einer Untersuchung auf freigesetzte Laktatdehydrogenase (LDH) in Verbindung mit einer qualitativen visuellen Prüfung der Zellen (siehe Anlage 4.1) bewertet. Andere quantitative Methoden zur Bestimmung der Zytotoxizität (z. B. eine auf Tetrazolium beruhende kolorimetrische (MTT-)Prüfung oder ein CALUX-Bioassay zur Zytotoxizitätsprüfung) können aber ebenfalls zur Anwendung kommen. Im Allgemeinen werden Konzentrationen von Prüfchemikalien, die die Zellviabilität um mehr als 20 % beeinträchtigen, als zytotoxisch betrachtet und können bei der Auswertung der Daten daher nicht berücksichtigt werden. Bei der Untersuchung auf freigesetztes LDH ist die jeweilige Konzentration der Prüfchemikalie dann als zytotoxisch zu betrachten, wenn der Anteil des freigesetzten LDH mehr als 120 % beträgt.
|
Prüfung der chemischen Exposition und Anordnung auf der Assay-Platte
|
36.
|
Nach der Trypsinierung eines Fläschchens konfluenter kultivierter Zellen werden die Zellen mit einer Konzentration von 1x105 Zellen/ml in einem östrogenfreien Assay-Medium resuspendiert. 100 μl resuspendierte Zellen werden in die inneren Wells einer 96-Well-Mikrotiterplatte resuspendiert. In die äußeren Wells werden 200 μl Phosphatpuffer (PBS) gegeben (siehe Abbildungen 1 und 2). Die plattierten Zellen werden 24 Stunden in einem CO2-Inkubator (5 % CO2, 37 °C, 100 % Luftfeuchtigkeit) vorinkubiert.
|
|
37.
|
Nach der Vorinkubierung werden die Platten auf sichtbare Anzeichen von Zytotoxizität (siehe Anlage 4.1) sowie auf Kontaminationen und auf Konfluenz untersucht. Nur Platten ohne sichtbare Anzeichen für eine Zytotoxizität und für Kontaminationen sowie Platten mit einer Konfluenz von mindestens 85 % werden für die Versuche verwendet. Das Medium aus den inneren Wells wird vorsichtig entnommen und durch 200 μl östrogenfreies Assay-Medium mit geeigneten seriellen Verdünnungen von Referenzstandards, Prüfchemikalien, Positivkontrollen, Negativkontrollen und Lösungsmittelkontrollen ersetzt (Tabelle 5: Agonisten-Assays; Tabelle 6: Antagonisten-Assays). Alle Referenzstandards, Prüfchemikalien, Positivkontrollen, Negativkontrollen und Lösungsmittelkontrollen werden dreimal geprüft. In Abbildung 1 ist die Anordnung auf der Platte bei Agonisten-Assays dargestellt. Abbildung 2 ist die Anordnung auf der Platte bei Antagonisten-Assays zu entnehmen. Die Anordnung auf den Platten beim Vorversuch und beim Hauptversuch ist identisch. Bei der Untersuchung von Agonisten enthalten alle inneren Wells mit Ausnahme der Wells mit der Vehikelkontrolle (VK) auch eine bestimmte Konzentration des Agonisten-Referenzstandards 17β-Estradiol (3*10-12 M). Auf alle Platten mit der Prüfchemikalie (TC) sollten auch die Referenzstandards C8 und C4 gegeben werden.
|
|
38.
|
Nachdem die Zellen allen Chemikalien ausgesetzt wurden, werden die 96-Well-Mikrotiterplatten weitere 24 Stunden in einem CO2-Inkubator (5 % CO2, 37 °C, 100 % Luftfeuchtigkeit) inkubiert.
|
Abbildung 1
Anordnung auf der 96-Well-Mikrotiterplatte für Vorversuche und für die endgültige Bewertung der Agonisten-Wirkung
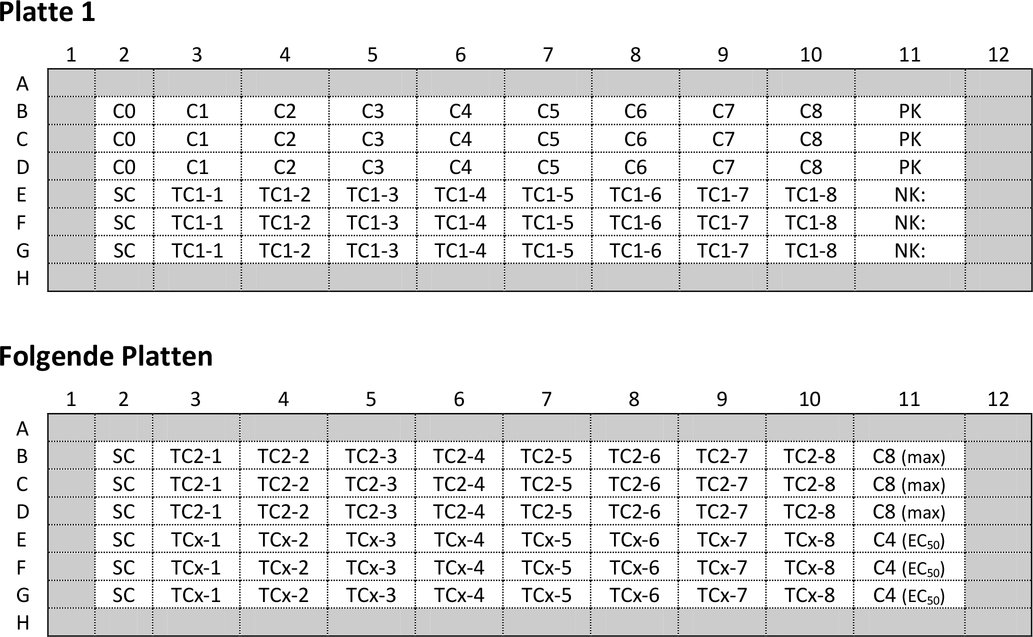
C0 = Lösungsmittel Referenzstandard
C(1-8) = Verdünnungsreihe (1-8, niedrige bis hohe Konzentrationen) des Referenzstandards
PK = Positivkontrolle
NK = Negativkontrolle
TCx-(1-8) = Verdünnungen (1-8, niedrige bis hohe Konzentrationen) der Prüfchemikalie für den Vorversuch und für die Bewertung der Agonisten-Wirkung der Prüfchemikalie x.
SC = Lösungsmittelkontrolle der Prüfchemikalie (möglichst desselben Lösungsmittels wie bei C0, wobei dies aber aus einer anderen Charge stammen kann)
Graue Zellen: = Äußere Wells, aufgefüllt mit 200 μl PBS
Abbildung 2
Anordnung auf der 96-Well-Mikrotiterplatte für Vor- und Hauptversuche zur Bewertung der Antagonisten-Wirkung

C0 = Lösungsmittel Referenzstandard
C(1-8) = Verdünnungsreihe (1-8, niedrige bis hohe Konzentrationen) des Referenzstandards
NK = Negativkontrolle
PK = Positivkontrolle
TCx-(1-8) = Verdünnungen (1-8, niedrige bis hohe Konzentrationen) der Prüfchemikalie für den Vorversuch und für die Bewertung der Agonisten-Wirkung der Prüfchemikalie x.
SC = Lösungsmittelkontrolle der Prüfchemikalie (möglichst desselben Lösungsmittels wie bei C0, wobei dies aber aus einer anderen Charge stammen kann)
VK = Vehikelkontrolle (Lösungsmittelkontrolle ohne feste Konzentration des Agonisten-Referenzstandards 17β-Estradiol).
Graue Zellen: = Äußere Wells, aufgefüllt mit 200 μl PBS
Hinweis: Die inneren Wells mit Ausnahme der Wells mit der Vehikelkontrolle (VK) enthalten alle auch eine bestimmte Konzentration des Agonisten-Referenzstandards 17β-Estradiol (3,0*10-12 M).
Messung der Luminiszenz
|
39.
|
Die Messung der Luminiszenz wird im Protokoll zum Agonisten- und zum Atagonisten-Assay (10) eingehend beschrieben. Das Medium wird aus den Wells entnommen. Nach 24-stündiger Inkuation werden die Zellen lysiert, um die Zellmembran aufzuschließen und die Luciferase-Aktivität messen zu können.
|
|
40.
|
Zur Messung der Luminiszenz wird für dieses Verfahren ein Luminometer mit zwei Injektoren benötigt. Die Luciferase-Reaktion wird durch Injektion des Luciferinsubstrats induziert. Durch Zugabe von 0,2 M NaOH wird die Reaktion gestoppt. Dadurch werden Luminiszenzübertragungen zwischen den Wells verhindert.
|
|
41.
|
Das von den einzelnen Wells emittierte Licht wird in RLU (relativen Lichteinheiten) pro Well ausgedrückt.
|
Vorversuch
|
42.
|
Anhand der Vorversuche wird ein optimierter Konzentrationsbereich der Prüfchemikalien für den Hauptversuch ermittelt. Die Auswertung der Ergebnisse des Vorversuchs und die Ermittlung des optimierten Konzentrationsbereichs der Prüfchemikalien für den Hauptversuch werden eingehend im Protokoll zum Agonisten- und zum Antagonisten-Assay (10) beschrieben. Im Folgenden werden die Verfahren zur Ermittlung des Konzentrationsbereichs der Prüfchemikalien für die Agonisten- und die Antagonisten-Prüfung beschrieben. Informationen zur Gestaltung der Verdünnungsreihen sind den Tabellen 5 und 6 zu entnehmen.
|
Auswahl von Konzentrationen zur Bewertung agonistischer Wirkungen
|
43.
|
Im Vorversuch sollten die Prüfchemikalien mit den in den Tabellen 5 (Agonisten) und 6 (Antagonisten) genannten Verdünnungen geprüft werden. Alle Konzentrationen sind in drei Wells zu prüfen, die auf den Platten angeordnet werden, wie in Abbildung 1 (Agonisten) bzw. Abbildung 2 (Antagonisten) beschrieben.
|
|
44.
|
Nur Untersuchungsergebnisse, die die in Tabelle 3 genannten Akzeptanzkriterien erfüllen, werden als gültig betrachtet und können zur Bewertung der Reaktion der Prüfchemikalien berücksichtigt werden. Wenn bei einer Untersuchung eine oder mehrere Mikrotiterplatten die Akzeptanzkriterien nicht erfüllen, sollten die betreffenden Mikrotiterplatten nochmals geprüft werden. Erfüllt die erste Platte mit der vollständigen Verdünnungsreihe des Referenzstandards die Akzeptanzkriterien nicht, muss die gesamte Prüfreihe (6 Platten) neu geprüft werden.
|
|
45.
|
In folgenden Fällen sollten die Ausgangskonzentrationsbereiche der Prüfchemikalien angepasst und der Vorversuch wiederholt werden:
|
—
|
In der Prüfung wurde eine zytotoxische Wirkung festgestellt. Der Vorversuch sollte mit niedrigeren nicht zytotoxischen Konzentrationen der Prüfchemikalie wiederholt werden.
|
|
—
|
Im Vorversuch mit der Prüfchemikalie wurde keine vollständige Dosis-Reaktions-Kurve ermittelt, weil bei den geprüften Konzentrationen bereits die maximale Induktion erreicht wird. Der Vorversuch sollte mit niedrigeren Konzentrationen der Prüfchemikalie wiederholt werden.
|
|
|
46.
|
Wenn eine gültige dosisabhängige Reaktion festgestellt wird, sollte die (niedrigste) Konzentration mit maximaler Induktion, aber ohne Anzeichen einer Zytotoxizität, gewählt werden. Die in den Hauptversuchen zu prüfende höchste Konzentration der Prüfchemikalie wird dreimal mit dieser gewählten Konzentration geprüft.
|
|
47.
|
Beginnend mit der oben ermittelten maximalen Konzentration wird eine vollständige optimierte Verdünnungsreihe der Prüfchemikalie mit den in Tabelle 5 angegebenen Verdünnungsschritten zubereitet.
|
|
48.
|
Prüfchemikalien, bei denen keine agonistische Wirkung festgestellt wurde, werden in den Hauptversuchen beginnend mit der im Vorversuch ermittelten maximalen nicht zytotoxischen Konzentration geprüft.
|
Auswahl von Konzentrationen zur Bewertung antagonistischer Wirkungen
|
49.
|
Nur Untersuchungsergebnisse, die die in Tabelle 4 genannten Akzeptanzkriterien erfüllen, werden als gültig betrachtet und können zur Bewertung der Reaktion der Prüfchemikalien berücksichtigt werden. Wenn bei einer Untersuchung eine oder mehrere Mikrotiterplatten die Akzeptanzkriterien nicht erfüllen, sollten die betreffenden Mikrotiterplatten nochmals geprüft werden. Erfüllt die erste Platte mit der vollständigen Verdünnungsreihe des Referenzstandards die Akzeptanzkriterien nicht, muss die gesamte Prüfreihe (6 Platten) neu geprüft werden.
|
|
50.
|
In folgenden Fällen sollten die Ausgangskonzentrationsbereiche der Prüfchemikalien angepasst und der Vorversuch wiederholt werden:
|
—
|
In der Prüfung wurde eine zytotoxische Wirkung festgestellt. Der Vorversuch sollte mit niedrigeren nicht zytotoxischen Konzentrationen der Prüfchemikalie wiederholt werden.
|
|
—
|
Im Vorversuch mit der Prüfchemikalie wurde keine vollständige Dosis-Reaktions-Kurve ermittelt, weil bei den geprüften Konzentrationen bereits die maximale Hemmung erreicht wird. Der Vorversuch sollte mit niedrigeren Konzentrationen der Prüfchemikalie wiederholt werden.
|
|
|
51.
|
Wenn eine gültige dosisabhängige Reaktion festgestellt wird, sollte die (niedrigste) Konzentration mit maximaler Hemmung, aber ohne Anzeichen einer Zytotoxizität, gewählt werden. Die in den Hauptversuchen zu prüfende höchste Konzentration der Prüfchemikalie wird dreimal mit dieser gewählten Konzentration geprüft.
|
|
52.
|
Beginnend mit der oben ermittelten maximalen Konzentration wird eine vollständige optimierte Verdünnungsreihe der Prüfchemikalie mit den in Tabelle 6 angegebenen Verdünnungsschritten zubereitet.
|
|
53.
|
Prüfchemikalien, bei denen keine antagonistische Wirkung festgestellt wurde, werden in den Hauptversuchen beginnend mit der im Vorversuch geprüften maximalen nicht zytotoxischen Konzentration geprüft.
|
Hauptversuche
|
54.
|
Nach der Bestimmung der optimalen Konzentrationsbereiche sollten die Prüfchemikalien mit den in den Tabellen 5 (Agonisten) und 6 (Antagonisten) genannten Verdünnungen geprüft werden. Alle Konzentrationen sind in drei Wells zu prüfen, die auf den Platten angeordnet werden, wie in Abbildung 1 (Agonisten) bzw. Abbildung 2 (Antagonisten) beschrieben.
|
|
55.
|
Nur Untersuchungsergebnisse, die die in den Tabellen 3 und 4 genannten Akzeptanzkriterien erfüllen, werden als gültig betrachtet und können zur Bewertung der Reaktion der Prüfchemikalien berücksichtigt werden. Wenn bei einer Untersuchung eine oder mehrere Mikrotiterplatten die Akzeptanzkriterien nicht erfüllen, sollten die betreffenden Mikrotiterplatten nochmals geprüft werden. Erfüllt die erste Platte mit der vollständigen Verdünnungsreihe des Referenzstandards die Akzeptanzkriterien nicht, muss die gesamte Prüfreihe (6 Platten) neu geprüft werden.
|
Tabelle 5
Konzentrationen und Verdünnungen von Referenzstandards, Kontrollen und Prüfchemikalien für die Agonisten-Prüfung
|
Referenzstandard 17β-Estradiol
|
TCx – Vorversuch
|
TCx – Vorversuch
|
Kontrollen
|
|
Konz. (M)
|
Verdünnung
|
Verdünnung
|
Konz. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PK
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx –Prüfchemikalie x
PK –Positivkontrolle (17α-Methyltestosteron)
NK –Negativkontrolle (Corticosteron)
C0 –Lösungsmittelkontrolle Referenzstandard
SC –Lösungsmittelkontrolle Prüfchemikalie
|
Tabelle 6
Konzentrationen und Verdünnungen von Referenzstandards, Kontrollen und Prüfchemikalien für die Antagonisten-Prüfung
|
Referenzstandard Tamoxifen
|
TCx – Vorversuch
|
TCx – Vorversuch
|
Kontrollen
|
|
Konz. (M)
|
Verdünnung
|
Verdünnung
|
Konz. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PK
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NK
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-7
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Hinzugegebener Agonist
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
Konz. (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-Estradiol
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx –Prüfchemikalie x
PK –Positivkontrolle (4-Hydroxytamoxifen)
NK –Negativkontrolle (Resveratrol)
C0 –Lösungsmittelkontrolle Referenzstandard
SC –Lösungsmittelkontrolle Prüfchemikalie
VK –Vehikelkontrolle (enthält keine bestimmte Konzentration des Agonisten-Referenzstandards 17β-Estradiol (3,0*10-12 M))
|
Erfassung und Auswertung der Daten
|
56.
|
Nach dem Vorversuch und dem Hauptversuch sollten EC10, EC50, PC10, PC50 und die maximale Induktion (TCxmax) der Prüfchemikalien für Agonisten-Prüfungen ermittelt werden. Bei Antagonisten-Prüfungen sollten IC20, IC50, PC80, PC50 und die niedrigste Induktion (TCxmin) berechnet werden. In den Abbildungen 3 (Agonisten) und 4 (Antagonisten) werden diese Parameter grafisch dargestellt. Die benötigten Parameter werden ausgehend von der relativen Induktion der einzelnen Prüfchemikalien (bezogen auf die maximale Induktion des Referenzstandards (= 100 %) berechnet. Zur Auswertung der Daten wird eine nicht lineare Regression (variable Steigung, 4 Parameter) mit der folgenden Formel vorgenommen.

Dabei gilt:
X = Logarithmus der Dosis bzw. Konzentration
Y = Reaktion (relative Induktion (%))
Top = Maximale Induktion (%)
Bottom = Niedrigste Induktion (%)
LogEC50 = Logarithmus der Konzentration, bei der 50 % der maximalen Reaktion beobachtet werden
HillSlope = Steigungsfaktor oder Hillslope
|
|
57.
|
Die Rohdaten des Luminometers in RLU (relative Lichteinheiten) werden in die Tabelle zur Analyse der Daten der Vorversuche und der Hauptversuche übertragen. Die Rohdaten sollten die in den Tabellen 3A und 3B (Agonisten) und 4A und 4B (Antagonisten) genannten Akzeptanzkriterien erfüllen. Wenn die Rohdaten die Akzeptanzkriterien erfüllen, werden mit den folgenden Schritten die benötigten Parameter berechnet:
|
Agonisten
|
—
|
Die durchschnittlichen RLU der Lösungsmittelkontrolle des Referenzstandards werden von den Rohdaten der Untersuchung der Referenzstandards abgezogen.
|
|
—
|
Die durchschnittlichen RLU der Lösungsmittelkontrolle der Prüfchemikalie werden von den Rohdaten der Untersuchung der Prüfchemikalien abgezogen.
|
|
—
|
Die relative Induktion der einzelnen Konzentrationen des Referenzstandards wird berechnet. Die Induktion der höchsten Konzentration des Referenzstandards wird als 100 % angenommen.
|
|
—
|
Die relative Induktion der einzelnen Konzentrationen der Prüfchemikalie wird in Bezug zur höchsten Konzentration des als 100 % angenommenen Referenzstandards gesetzt.
|
|
—
|
Die Untersuchungsergebnisse werden durch nicht lineare Regression (variable Steigung, 4 Parameter) berechnet.
|
|
—
|
EC50 und EC10 des Referenzstandards werden berechnet.
|
|
—
|
EC50 und EC10 der Prüfchemikalien werden berechnet.
|
|
—
|
Die maximale relative Induktion der Prüfchemikalie (TCmax) wird berechnet.
|
|
—
|
PC10 und PC50 der Prüfchemikalien werden berechnet.
|
Bei Prüfchemikalien kann infolge einer bestehenden Zytotoxizität oder aufgrund von Löslichkeitsproblemen möglicherweise nicht immer eine vollständige Dosis-Reaktions-Kurve dargestellt werden. In diesen Fällen können EC50, EC10 und PC50 nicht bestimmt werden, und die Darstellung beschränkt sich auf PC10 und TCmax.
Antagonisten
|
—
|
Die durchschnittlichen RLU der höchsten Konzentration des Referenzstandards werden von den Rohdaten der Untersuchung der Referenzstandards abgezogen.
|
|
—
|
Die durchschnittlichen RLU der höchsten Konzentration des Referenzstandards werden von den Rohdaten der Untersuchung der Prüfchemikalien abgezogen.
|
|
—
|
Die relative Induktion der einzelnen Konzentrationen des Referenzstandards wird berechnet. Die Induktion der niedrigsten Konzentration des Referenzstandards wird als 100 % angenommen.
|
|
—
|
Die relative Induktion der einzelnen Konzentrationen der Prüfchemikalie wird in Bezug zur niedrigsten Konzentration des als 100 % angenommenen Referenzstandards gesetzt.
|
|
—
|
Die Untersuchungsergebnisse werden durch nicht lineare Regression (variable Steigung, 4 Parameter) berechnet.
|
|
—
|
IC50 und IC20 des Referenzstandards werden berechnet.
|
|
—
|
IC50 und IC20 der Prüfchemikalien werden berechnet.
|
|
—
|
Die niedrigste relative Induktion der Prüfchemikalie (TCmax) wird berechnet.
|
|
—
|
PC80 und PC50 der Prüfchemikalien werden berechnet.
|
Abbildung 3
Im Agonisten-Assay bestimmte Parameter

EC10 = Konzentration eines Stoffs, bei der 10 % seiner maximalen Reaktion beobachtet werden
EC50 = Konzentration eines Stoffs, bei der 50 % seiner maximalen Reaktion beobachtet werden
PC10 = Konzentration einer Prüfchemikalie, bei der deren Reaktion EC10 des Referenzstandards entspricht
PC50 = Konzentration einer Prüfchemikalie, bei der deren Reaktion EC50 des Referenzstandards entspricht
TCxmax = maximale relative Induktion der Prüfchemikalie
Abbildung 4
Im Antagonisten-Assay bestimmte Parameter
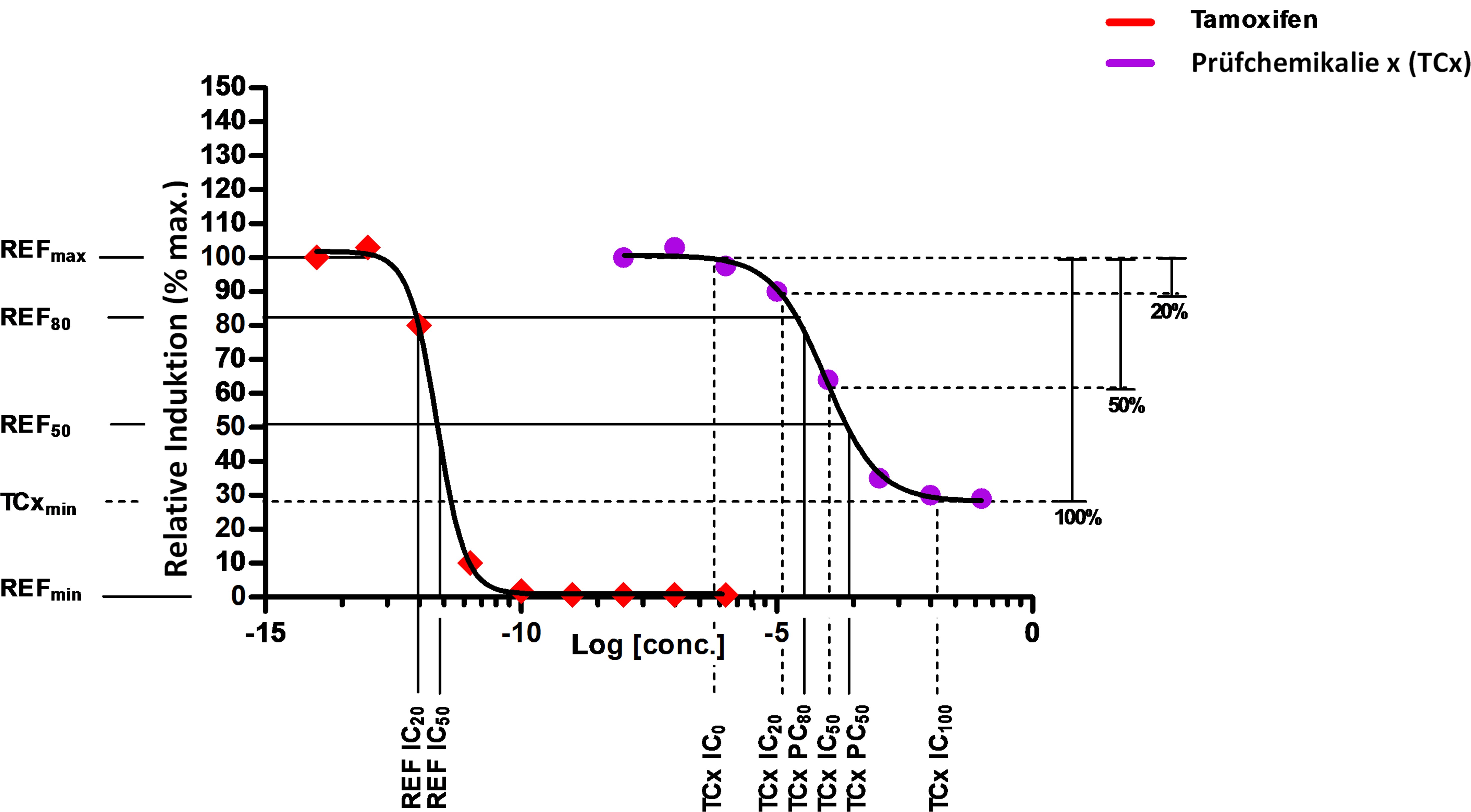
IC20 = Konzentration eines Stoffs, bei der 80 % seiner maximalen Reaktion (Hemmung 20 %) beobachtet werden
IC50 = Konzentration eines Stoffs, bei der 50 % seiner maximalen Reaktion (Hemmung 50 %) beobachtet werden
PC80 = Konzentration einer Prüfchemikalie, bei der deren Reaktion IC20 des Referenzstandards entspricht
PC50 = Konzentration einer Prüfchemikalie, bei der deren Reaktion IC50 des Referenzstandards entspricht
TCxmin = niedrigste relative Induktion der Prüfchemikalie
Bei Prüfchemikalien kann infolge einer bestehenden Zytotoxizität oder aufgrund von Löslichkeitsproblemen möglicherweise nicht immer eine vollständige Dosis-Reaktions-Kurve dargestellt werden. In diesen Fällen können IC50, IC20 und PC50 nicht bestimmt werden, und die Darstellung beschränkt sich auf PC20 und TCmin.
|
58.
|
Die Ergebnisse sollten auf zwei (oder drei) unabhängigen Prüfläufen beruhen. Wenn bei zwei Prüfläufen vergleichbare und daher auch reproduzierbare Ergebnisse erzielt werden, ist ein dritter Prüflauf nicht erforderlich. Ergebnisse können dann akzeptiert werden, wenn die folgenden Anforderungen erfüllt sind:
|
—
|
Die Ergebnisse erfüllen die Akzeptanzkriterien (Nummern 14-22)
|
|
—
|
und sind reproduzierbar.
|
|
Kriterien für die Auswertung von Daten
|
59.
|
Bei der Auswertung der Daten und der Einstufung einer Prüfchemikalie als positiv oder negativ sind die folgenden Kriterien zu berücksichtigen:
|
Bei jedem Hauptversuch wird eine Prüfchemikalie als positiv betrachtet, wenn die folgenden Bedingungen erfüllt sind:
|
1
|
TCmax ist größer oder gleich 10 % der maximalen Reaktion des Referenzstandards (REF10).
|
|
2
|
Mindestens 2 aufeinander folgende Konzentrationen der Prüfchemikalie sind größer oder gleich REF10.
|
|
|
Bei jedem Hauptversuch wird eine Prüfchemikalie als negativ betrachtet, wenn die folgenden Bedingungen erfüllt sind:
|
1
|
TCmax beträgt nicht mehr als 10 % der maximalen Reaktion des Referenzstandards (REF10).
|
|
2
|
Weniger als 2 aufeinander folgende Konzentrationen der Prüfchemikalie sind größer oder gleich REF10.
|
|
|
Bei jedem Hauptversuch wird eine Prüfchemikalie als positiv betrachtet, wenn die folgenden Bedingungen erfüllt sind:
|
1
|
TCmin ist kleiner oder gleich 80 % der maximalen Reaktion des Referenzstandards (REF80 = Hemmung 20 %).
|
|
2
|
Mindestens 2 aufeinander folgende Konzentrationen der Prüfchemikalie sind kleiner oder gleich REF80.
|
|
|
Bei jedem Hauptversuch wird eine Prüfchemikalie als negativ betrachtet, wenn die folgenden Bedingungen erfüllt sind:
|
1
|
TCmin ist größer als 80 % der maximalen Reaktion des Referenzstandards (REF80 = Hemmung 20 %).
|
|
2
|
Weniger als 2 aufeinander folgende Konzentrationen der Prüfchemikalie sind kleiner oder gleich REF80.
|
|
|
|
60.
|
Zur Beschreibung der Wirkung der positiven Reaktion einer Prüfchemikalie sollten die Wirkstärke (Agonisten: TCmax; Antagonisten: TCmin) und die Konzentration angegeben werden, bei der die Wirkung eintritt (Agonisten: EC10, EC50, PC10, PC50; Antagonisten: IC20, IC50, PC80, PC50).
|
PRÜFBERICHT
|
61.
|
Siehe Nummer 20 im Abschnitt „ELEMENTE DES ER-TA-ASSAYS“.
|
LITERATUR
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay – transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(2)
|
Sonneveld, E., Jansen, H.J., Riteco, J.A., Brouwer, A., van der Burg, B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers, M.E., van den Brink, C.E., Wissink, S., Schreurs, R.H.M.M., Gustafsson, J.A., van der Saag, P.T. und van der Burg, B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne, N, Inglese, J. und Auld, D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-657.
|
|
(5)
|
Escande, A., Pillon, A., Servant, N., Cravedi, J.P., Larrea, F., Muhn, P., Nicolas, J.C., Cavaillès, V. und Balaguer, P.. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper, G.G., Lemmen, J.G., Carlsson, B., Corton, J.C., Safe, S.H., van der Saag, P.T., van der Burg, B. und Gustafsson, J.A. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca, A.M., Bovee, T.F.H., Brand, W., Velikova, N., Boeren, S., Murk, A.J., Vervoort, J., Rietjens, I.M.C.M. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204-211.
|
|
(8)
|
Sonneveld, E., Riteco, J.A.C., Jansen, H.J., Pieterse, B., Brouwer, A., Schoonen, W.G. und van der Burg, B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173-187.
|
|
(9)
|
Kobayashi, H., Yamamoto, K., Eguchi, M., Kubo, M., Nakagami, S., Wakisaka, S., Kaizuka, M. und Ishii, H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang, J.-H., Chung,T.D.Y. und Oldenburg, K.R. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Sci., 4, 67-73.
|
|
(11)
|
Besselink, H., Middelhof, I. und Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, Niederlande.
|
Anlage 4.1
VISUELLE PRÜFUNG DER ZELLVIABILITÄT
B.67
IN-VITRO-GENMUTATIONSPRÜFUNG AN SÄUGETIERZELLEN ANHAND DES THYMIDINKINASE-GENS
EINLEITUNG
|
1.
|
Diese Prüfmethode (PM) entspricht der OECD-Prüfrichtlinie (TG) 490 (2016). Die Prüfmethoden werden regelmäßig geprüft und überarbeitet, um dem wissenschaftlichen Fortschritt, den rechtlichen Anforderungen und den Belangen des Tierschutzes gerecht zu werden. Der Maus-Lymphom-Assay (MLA) und die TK6-Prüfung mit dem Thymidinkinase-Lokus (TK-Lokus) waren ursprünglich Bestandteil der Prüfmethode B.17. Die MLA Expert Workgroup des Internationalen Workshops für Genotoxizitätsprüfungen (International Workshop on Genotoxicity Testing, IWGT) hat später jedoch international harmonisierte Empfehlungen zu Akzeptanzkriterien für den Assay und zur Auswertung der Daten des MLA entwickelt (1) (2) (3) (4) (5). Diese Empfehlungen wurden in dieser neuen Prüfmethode B.67 berücksichtigt. Diese Prüfmethode wurde für den MLA sowie (da dabei ebenfalls der TK-Lokus verwendet wird) für die TK6-Prüfung entwickelt. Der MLA ist für rechtliche Zwecke allgemein üblich. Die TK6-Prüfung hingegen ist erheblich weniger verbreitet. Ungeachtet der Ähnlichkeit der Endpunkte sind die beiden Zelllinien allerdings nicht austauschbar, und in rechtlichen Vorgaben kann im Zusammenhang mit einem bestimmten rechtlichen Zweck durchaus eine Präferenz für die eine oder andere Zelllinie zum Ausdruck gebracht werden. Beispielsweise wurde mit der Validierung des MLA nachgewiesen, dass mit dem Assay nicht nur Genmutationen erkannt werden können, sondern dass auch die Fähigkeit einer Prüfchemikalie zur Induzierung struktureller Chromosomenschäden festgestellt werden kann. Diese Prüfmethode ist Bestandteil einer Reihe von Prüfmethoden zur genetischen Toxikologie. Die OECD hat ein Dokument erstellt, das kurz gefasste und hilfreiche Informationen zu Untersuchungen zur genetischen Toxikologie sowie eine Übersicht über die jüngsten Änderungen der OECD-Prüfrichtlinien zur genetischen Toxikologie enthält (6).
|
|
2.
|
Die In-vitro-Genmutationsprüfungen an Säugetierzellen werden zum Nachweis chemisch induzierter Genmutationen verwendet. Mit den in diesen Prüfungen verwendeten Zelllinien können Vorwärtsmutationen an Reporter-Genen, insbesondere am endogenen Thymidinkinase-Gen (TK bei menschlichen Zellen und Tk bei Nagerzellen, bei dieser Prüfmethode gemeinsam als TK bezeichnet) gemessen werden. Diese Prüfmethode ist zur Verwendung mit zwei Zelllinien vorgesehen: der Maus-Lymphom-Zelllinie L5178Y TK+/--3.7.2C (im Allgemeinen als L5178Y-Zelllinie bezeichnet) und der humanen Lymphoblastoid-Zelllinie TK6 (allgemein als TK6-Zelllinie bezeichnet). Die beiden Zelllinien unterscheiden sich zwar u. a. in Bezug auf ihre Herkunft, das Zellwachstum und den p53-Status. Die TK-Genmutationsprüfungen können jedoch bei beiden Zelltypen ähnlich durchgeführt werden, wie in dieser Prüfmethode beschrieben.
|
|
3.
|
Die autosomale und heterozygote Beschaffenheit des Thymidin-Kinase-Gens ermöglicht die Erkennung lebensfähiger Kolonien von Zellen, denen infolge einer Mutation von TK+/-
nach TK-/-
das Enzym Thymidin-Kinase fehlt. Das Fehlen des Enzyms kann auf genetische Prozesse zurückzuführen sein, die das TK-Gen beeinträchtigen (u. a. auf Genmutationen (Punktmutationen, Frame-Shift-Mutationen, kleine Deletionen usw.) und auf Chromosomenveränderungen (große Deletionen, Chromosomen-Rearragements und mitotische Rekombinationen)). Letztere äußern sich in einem Verlust der Heterozygotie, der bei der humanen Tumorgenese häufig mit einer genetischen Veränderung von Tumorsuppressorgenen einhergeht. Theoretisch kann der Verlust des gesamten Chromosoms mit dem TK-Gen infolge der Hemmung der Spindelfaserbildung und/oder durch mitotische Non-disjunction mit dem MLA nachgewiesen werden. Eine kombinierte zytogenetische und molekulare Untersuchung zeigt tatsächlich eindeutig, dass einige MLA-TK-Mutanten auf eine Non-disjunction zurückzuführen sind. Mit TK-Genmutationsprüfungen werden Aneugene durch die Berücksichtigung von Standardkriterien zur Feststellung einer Zytotoxizität (wie bei dieser Prüfmethode beschrieben) jedoch nachweislich nicht zuverlässig erkannt. Daher sind diese Prüfungen zum Nachweis von Aneugenen nicht geeignet (7) (8) (9).
|
|
4.
|
Bei den TK-Genmutationsprüfungen werden zwei verschiedene Phänotypklassen von TK-Mutanten erzeugt: die normal wachsenden Mutanten, die so schnell wachsen wie die heterozygoten TK-Zellen, und die langsam wachsenden Mutanten mit längeren Verdopplungszeiten. Die normal und die langsam wachsenden Mutanten werden beim MLA als Mutanten mit starker bzw. geringer Koloniebildung und bei der TK6-Prüfung als Mutanten mit früh bzw. spät auftretender Koloniebildung festgestellt. Die molekulare und zellgenetische Beschaffenheit von MLA-Mutanten mit starker bzw. geringer Koloniebildung wurde eingehend untersucht (8) (10) (11) (12) (13). Die molekulare und zellgenetische Beschaffenheit der früh und der spät auftretenden TK6-Mutanten wurde ebenfalls eingehend untersucht (14) (15) (16) (17). Langsam wachsende Mutanten bei beiden Zelltypen entwickelten genetische Schäden (einschließlich möglicherweise wachstumsregulierender Gene in der Nähe des TK-Locus), die zu längeren Verdopplungszeiten und zur späteren Bildung kleinerer Kolonien führen (18). Eine Induzierung langsam wachsender Mutanten erfolgt vor allem bei Chemikalien, die erhebliche strukturelle Änderungen auf der Chromosomenebene hervorrufen. Zellen, bei denen die Schäden die möglicherweise wachstumsregulierenden Gene in der Nähe des TK-Locus nicht betreffen, teilen sich ähnlich schnell wie die Elternzellen und entwickeln sich zu normal wachsenden Mutanten. Eine Induzierung normal wachsender Mutanten erfolgt bei Chemikalien, die hauptsächlich als Punktmutagene wirken. Daher ist entscheidend, dass sowohl langsam als auch normal wachsende Mutanten gezählt werden, um alle Mutanten zu erfassen und Aufschluss über den Typ (die Typen) der Schäden (Mutagene vs. Klastogene) zu erhalten, die durch die Prüfchemikalie induziert werden (10) (12) (18) (19).
|
|
5.
|
Die Prüfmethode wurde so gestaltet, dass sie allgemeine Informationen sowohl beim MLA als auch bei der TK6-Prüfung liefert. Außerdem enthält sie spezifische Hinweise zur Durchführung der einzelnen Prüfungen.
|
|
6.
|
Es gelten die Begriffsbestimmungen in Anlage 1.
|
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
|
7.
|
In vitro durchgeführte Prüfungen erfordern in der Regel die Verwendung eines exogenen Fremdstoff-Metabolisierungssystems. Mit diesem exogenen Stoffwechselaktivierungssystem lassen sich die In-vivo-Bedingungen jedoch nicht gänzlich nachvollziehen.
|
|
8.
|
Bedingungen, die zu künstlich verursachten Positivergebnissen führen könnten (d. h. mögliche Wechselwirkungen mit dem Prüfsystem), die nicht von einer Interaktion zwischen der Prüfchemikalie und dem genetischen Material der Zelle herrühren, sind unbedingt zu vermeiden; zu solchen Bedingungen gehören Veränderungen des pH-Wertes bzw. der Osmolalität, eine Interaktion mit einzelnen Komponenten des Mediums (20) (21) oder eine hochgradige Zytotoxizität (22) (23) (24). Zytotoxizitätswerte, die die empfohlenen Höchstwerte nach Nummer 28 überschreiten, werden für den MLA und TK6-Prüfung als zu hoch betrachtet. Außerdem ist zu beachten, dass Prüfchemikalien, bei denen es sich um Thymidin-Nachbildungen handelt oder die sich wie Thymidin-Nachbildungen verhalten, bei der Behandlung der Zellen infolge der selektiven Vermehrung der spontanen Hintergrundmutationen die Mutantenhäufigkeit erhöhen und Untersuchungen mit weiteren Prüfmethoden erforderlich machen können, um eine angemessene Auswertung zu ermöglichen (25).
|
|
9.
|
Für hergestellte Nanomaterialien sind möglicherweise spezielle Anpassungen dieser Prüfmethode erforderlich, die an dieser Stelle jedoch nicht beschrieben werden.
|
|
10.
|
Bevor die Prüfmethode zur Untersuchung eines Gemischs für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs rechtlich vorgeschrieben ist.
|
|
11.
|
Mutantenzellen, die infolge der Mutation von TK
+/- zu TK
-/- keine Aktivität des Enzyms Thymidinkinase (TK) entwickeln, sind gegenüber der zytotoxischen Wirkung des Pyrimidinanalogons Trifluorthymidin (TFT) resistent. Bei Anwesenheit von TK sind Zellen hingegen empfindlich gegenüber TFT, das die Hemmung des Zellstoffwechsels verursacht und eine weitere Zellteilung verhindert. Somit können Mutantenzellen sich in Anwesenheit von TFT vermehren und erkennbare Kolonien bilden. Zellen, die das Enzym TK enthalten, sind dazu nicht in der Lage.
|
PRINZIP DER PRÜFMETHODE
|
12.
|
Zellen in Suspensionskultur werden über einen angemessenen Zeitraum (Nummer 19) mit und ohne exogenes Fremdstoff-Metabolisierungssystem (Nummer 33) mit der Prüfchemikalie behandelt und anschließend subkultiviert, um die Zytotoxizität zu bestimmen und vor der Mutantenselektion die Expression des Phänotyps zu ermöglichen. Die Zytotoxizität wird anhand des relativen Gesamtwachstums (RTG) (Nummer 25) beim MLA bzw. anhand der relativen Überlebensrate (RS) (Nummer 26) bei der TK6-Prüfung ermittelt. Die behandelten Kulturen werden für einen ausreichenden Zeitraum, der für den jeweils gewählten Zelltyp charakteristisch ist (Nummer 37), in einem Wachstumsmedium gehalten, um eine annähernd optimale Expression des Phänotyps der induzierten Mutationen zu ermöglichen. Nach der Expression des Phänotyps wird die Mutantenhäufigkeit bestimmt, indem eine bekannte Anzahl von Zellen auf ein Medium mit dem selektierenden Agens zur Bestimmung der Mutantenkolonien und auf ein Medium ohne selektierendes Agens zur Bestimmung der Klonierungseffizienz (Lebensfähigkeit) geimpft wird. Nach einer geeigneten Inkubationszeit werden die Kolonien gezählt. Die Mutantenhäufigkeit wird anhand der entsprechend der Klonierungseffizienz bei der Mutantenselektion bereinigten Anzahl der Mutantenkolonien berechnet.
|
BESCHREIBUNG DER METHODE
Vorbereitungen
Zellen
|
13.
|
MLA: Da der MLA mit der TK
+/--3.7.2C-Zelllinie als Unterzelllinie der L5178Y-Zelllinie entwickelt und beschrieben wurde, ist für den MLA diese spezielle Unterzelllinie zu verwenden. Die L5178Y-Zelllinie wurde aus einem durch Methylcholanthren induzierten Thymuslymphom einer DBA-2-Maus abgeleitet (26). Clive u. a. behandelten L5178Y-Zellen (von Clive als TK+/+-3 bezeichnet) mit Ethylmethansulfonat und isolierten einen TK-/--Klon (bezeichnet als TK-/--3.7-Klon) mit Bromodeoxyuridin als selektierendes Agens. Aus dem TK-/--Klon wurden ein spontaner TK+/--Klon (als TK+/--3.7.2. bezeichnet) und ein Subklon (bezeichnet als TK+/--3.7.2C) zur Verwendung im MLA isoliert und charakterisiert (27). Der Karyotyp der Zelllinie wurde veröffentlicht (28) (29) (30) (31). Der Modalwert der Chromosomen beträgt 40. Das metazentrische Chromosom (t12;13) ist als ein Chromosom zu zählen. Der TK-Locus bei Mäusen befindet sich am distalen Ende des Chromosoms 11. Die Zelllinie L5178Y TK
+/--3.7.2C hat Mutationen auf den p53-Allelen und erzeugt mutantes p53-Protein (32) (33). Der p53-Status der TK+/--3.7.2C-Zelllinie ist wahrscheinlich die Ursache dafür, dass mit der Prüfung größere Schäden erkannt werden können (17).
|
|
14.
|
TK6: TK6 ist eine humane lymphoblastoide Zelllinie. Die Elternzelllinie ist eine mit dem Epstein-Barr-Virus transformierte Zelllinie (WI-L2), die ursprünglich von einem 5-jährigen Jungen mit hereditärer Sphärozytose abgeleiet wurde. Der erste isolierte Klon (HH4) wurde mit ICR191 mutagenisiert; dabei wurde eine heterozygote TK-Zelllinie (TK6) erzeugt (34). TK6-Zellen sind annähernd diploid; der entsprechende Karyotyp ist 47, XY, 13+, t(14; 20), t(3; 21) (35). Der TK-Locus beim Menschen befindet sich am langen Arm des Chromosoms 17. TK6 ist eine p53-kompetente Zelllinie, da sie auf beiden Allelen eine Wildtyp-p53-Sequenz aufweist und ausschließlich Wildtyp-p53-Protein exprimiert (36).
|
|
15.
|
Sowohl beim MLA als auch bei der TK6-Prüfung empfiehlt sich, dass das Prüflabor beim erstmaligen Herstellen oder beim Auffüllen der Master-Zellenbestände sicherstellt, dass keine Mycoplasma-Kontamination vorliegt, sowie dass es den Karyotyp der Zellen bestimmt oder die Chromosomen markiert, auf denen sich der TK-Locus befindet, und dass es die Zeiten für die Verdopplung der Populationen ermittelt. Die normale Dauer des Zellzyklus sollte bei den im Prüflabor verwendeten Zellen bekannt sein und mit veröffentlichten Zellcharakteristiken übereinstimmen (16) (19) (37). Dieser Master-Zellenbestand sollte bei -150 °C (oder noch niedrigeren Temperaturen) aufbewahrt und zur Herstellung aller Arbeitsstämme verwendet werden.
|
|
16.
|
Entweder vor der Herstellung einer großen Anzahl an kryokonservierten Arbeitsstämmen oder unmittelbar vor der Verwendung in einem Versuch müssen die Zellkulturen möglicherweise von noch vorhandenen Mutanten gereinigt werden [wenn die Mutantenhäufigkeit (MF) der Lösungsmittelkontrolle nicht bereits im akzeptablen Bereich liegt – für MLA siehe Tabelle 2]. Dazu wird Methotrexat (Aminopterin) zur Selektion von Zellen ohne TK verwendet und Thymidin, Hypoxanthin und Glycin (L5178Y) bzw. 2’-Deoxycytidin (TK6) zur Kultur hinzugegeben, um ein optimales Wachstum der TK-kompetenten Zellen sicherzustellen (19) (38) (39) bzw. (40) für TK6. Allgemeine Hinweise zu bewährten Verfahren bei der Haltung von Zellkulturen sowie spezifische Hinweise zu L5178Y- und TK6-Zellen siehe (19) (31) (37) (39) und (41). Für Labors, die Master-Zellenbestände als Ausgangszellen für den MLA oder die TK6-Prüfung benötigen, sowie allgemein, wenn neue Master-Zellenbestände benötigt werden, ist ein Zellendepot mit gut charakterisierten Zellen verfügbar (37).
|
Medien und Kulturbedingungen
|
17.
|
Bei beiden Prüfungen erfordert die Kultivierung geeignete Kulturmedien und Inkubationsbedingungen (z. B. Kulturgefäße, eine befeuchtete Atmosphäre mit einer CO2-Konzentration von 5 % und eine Inkubationstemperatur von 37 °C). Die Zellkulturen sollten immer unter Bedingungen gehalten werden, bei denen das Wachstum in der Log-Phase sichergestellt ist. Vor allem ist für Medien- und Kulturbedingungen zu sorgen, die ein optimales Zellwachstum während der Expressionszeit und der Klonierung der mutierenden und nichtmutierenden Zellen gewährleisten. Für den MLA und die TK6-Prüfung ist ferner wichtig, dass die Kulturlösungen ein optimales Wachstum sowohl der großen Kolonien bzw. frühzeitig auftretender TK-Mutanten als auch der kleinen Kolonien bzw. spät auftretender TK-Mutanten gewährleisten. Nähere Informationen zur Haltung der Kulturen einschließlich der Notwendigkeit der Inaktivierung von Pferdeserum durch angemessene Erwärmung (wenn bei der Mutantenselektion RPMI-Medium verwendet wird) siehe (19) (31) (38) (39) (40) und (42).
|
Vorbereitung der Kulturen
|
18.
|
Die Zellen werden aus Stammkulturen gewonnen und im Kulturmedium in einer solchen Dichte überimpft, dass die Suspensionskulturen während der Behandlung und der Expressionszeit weiterhin exponentiell wachsen.
|
Stoffwechselaktivierung
|
19.
|
Bei der Verwendung von L5178Y- und TK6-Zellen sollten exogene metabolisierende Systeme verwendet werden, da diese Zellen eine unzulängliche endogene Stoffwechselkapazität entwickeln. Das gängigste und – sofern nicht anders begründet – standardmäßig empfohlene System, ist eine durch Ko-Faktoren ergänzte post-mitochondriale Fraktion (S9) aus der Leber von Nagetieren (in der Regel Ratten), die mit enzyminduzierenden Agenzien wie Aroclor 1254 (43) (44) (45) oder einer Kombination aus Phenobarbiton und β-Naphtoflavon (46) (47) (48) (49) (50) (51) vorbehandelt wurde. Die letztgenannte Kombination verstößt nicht gegen das Stockholmer Übereinkommen über persistente organische Schadstoffe (52) und hat sich für die Induktion von Multifunktionsoxidasen als ebenso wirksam wie Aroclor 1254 erwiesen (45) (46) (47) (48) (49). Die S9-Fraktion wird im Endmedium in der Regel in Konzentrationen von 1-2 % verwendet, kann jedoch auf 10 % v/v erhöht werden. Die Wahl der Art und Konzentration des exogenen Metabolisierungssystems oder metabolischen Agens ist möglicherweise von der Klasse der Prüfchemikalien abhängig.
|
Vorbereitung der Prüfchemikalie
|
20.
|
Feste Prüfchemikalien sollten vor der Behandlung der Zellen in geeigneten Lösungsmitteln gelöst und ggf. verdünnt werden (Nummer 21). Flüssige Prüfchemikalien können vor der Behandlung direkt zum Versuchssystem gegeben und/oder verdünnt werden. Gasförmige oder flüchtige Prüfchemikalien sind durch entsprechende Modifikationen der Standardprotokolle zu prüfen, z. B. durch Behandlung in hermetisch verschlossenen Kulturgefäßen (53) (54) (55). Zubereitungen der Prüfchemikalie sollten kurz vor der Behandlung hergestellt werden, es sei denn, die Stabilität der Chemikalie bei der Lagerung wird nachgewiesen.
|
PRÜFBEDINGUNGEN
Lösungsmittel
|
21.
|
Das Lösungsmittel sollte so gewählt werden, dass zum einen eine optimale Löslichkeit der Prüfchemikalie gewährleistet ist, zum anderen aber die Durchführung der Prüfung nicht beeinträchtigt wird, z. B. durch Veränderung des Zellwachstums, Beeinträchtigung der Integrität der Prüfchemikalie, Reaktion mit Kulturgefäßen oder Behinderung des Metabolisierungssystems. Als erste Wahl ist möglichst die Verwendung eines wässrigen Lösungsmittels (oder Kulturmediums) zu empfehlen. Gründlich erprobte Lösungsmittel sind Wasser oder Dimethylsulfoxid. Organische Lösungsmittel sollten 1 % (v/v) und wässrige Lösungsmittel (Kochsalzlösung oder Wasser) 10 % (v/v) im Endmedium möglichst nicht überschreiten. Kommen weniger gründlich erprobte Lösungsmittel zum Einsatz (z. B Ethanol oder Aceton), ist deren Verwendung anhand von Daten zu begründen, die ihre Verträglichkeit mit den Prüfchemikalien und dem Versuchssystem und ihre nicht gentoxische Wirkung in der verwendeten Konzentration belegen. Sind keine entsprechenden Belege verfügbar, sollten unbedingt unbehandelte Kontrollen (siehe Anlage 1, Begriffsbestimmungen) einbezogen werden, um nachzuweisen, dass durch die gewählten Lösungsmittel keine schädlichen oder mutagenen Wirkungen ausgelöst werden.
|
MESSUNG DER ZYTOTOXIZITÄT UND AUSWAHL DER BEHANDLUNGSKONZENTRATIONEN
|
22.
|
Bei der Bestimmung der höchsten Konzentration der Prüfchemikalie sind Konzentrationen zu vermeiden, die zu künstlich positiven Reaktionen führen können, z. B. zu übermäßiger Zytotoxizität (Nummer 28), Niederschlag (Nummer 29) oder ausgeprägten Veränderungen des pH-Werts oder der Osmolalität (Nummer 8). Wenn die Prüfchemikalie zum Zeitpunkt der Zugabe den pH-Wert des Mediums erheblich verändert, lässt sich der pH-Wert auch durch Anwendung eines Puffers im Endmedium einstellen, damit künstlich positive Reaktionen vermieden und geeignete Kulturbedingungen aufrechterhalten werden.
|
|
23.
|
Die Konzentrationen werden u. a. nach der Zytotoxizität ausgewählt (Nummern 27-30). Wenngleich die Bewertung der Zytotoxizität im Rahmen eines Vorversuchs nützlich sein kann, um die im Hauptversuch verwendeten Konzentrationen besser bestimmen zu können, ist ein Vorversuch nicht erforderlich. Auch wenn die anfängliche Zytotoxizität bewertet wird, ist im Hauptversuch die Zytotoxizität jeder einzelnen Kultur zu messen. Wenn ein Dosisfindungsversuch durchgeführt wird, sollte dieser ein breites Spektrum an Konzentrationen abdecken. Der Dosisfindungsversuch kann entweder an Tag 1 nach der Behandlung abgebrochen werden oder über die 2-tägige Expression bis zur Mutantenselektion fortgesetzt werden (wenn sich die verwendeten Konzentrationen als geeignet erweisen sollten).
|
|
24.
|
Die Zytotoxizität sollte für jede Prüfkultur und für jede Kontrollkultur separat ermittelt werden. Hinsichtlich der Methoden für den MLA (2) und die TK6-Prüfung (15) ist die international anerkannte Praxis maßgeblich.
|
|
25.
|
Für die Durchführung des MLA mit der Agar- und der Microwell-Methode gilt: Die Zytotoxizität sollte anhand des relativen Gesamtwachstums (RTG) ermittelt werden (erstmals im Jahr 1975 von Clive und Spector beschrieben (2)). Dieser Parameter beinhaltet das relative Suspensionswachstum (RSG: Prüfkultur vs. Lösungsmittelkontrolle) während der Behandlung der Zellen, die Expressionszeit und die relative Klonierungseffizienz (RCE: Prüfkultur vs. Lösungsmittelkontrolle) zum Zeitpunkt der Mutantenselektion (2). Das RSG beinhaltet alle Zellverluste in der Prüfkultur während der Behandlung (Formel siehe Anlage 2).
|
|
26.
|
Für die TK6-Prüfung gilt: Die Zytotoxizität sollte anhand der relativen Überlebensrate (RS) (d. h. der Klonierungseffizienz von unmittelbar nach der Behandlung ausplattierten Zellen) ermittelt werden; dabei ist eine Bereinigung um Zellverluste während der Behandlung auf der Grundlage der Zellzahl im Vergleich zur Negativkontrolle (mit einer Überlebensrate von 100 %) vorzunehmen (Formel siehe Anlage 2).
|
|
27.
|
Es sollten mindestens vier Prüfkonzentrationen (ausgenommen das Lösungsmittel und Positivkontrollen) ausgewertet werden, die die Akzeptanzkriterien erfüllen (geeignete Zytotoxizität, Anzahl der Zellen usw.). Die Verwendung von Zweifachkulturen ist zu empfehlen, doch können für jede überprüfte Konzentration auch Replikat- oder Einfachkulturen herangezogen werden. Die Ergebnisse aus Replikatkulturen bei einer gegebenen Konzentration sollten getrennt angegeben werden, können zu Datenanalysezwecken aber auch gepoolt werden (55). Bei Prüfchemikalien mit geringer Zytotoxizität oder ohne zytotoxische Wirkung sind in der Regel Konzentrationsintervalle mit zwei- bis dreifacher Konzentration geeignet. Wenn Zytotoxizität auftritt, sollten die Konzentrationen einen Zytotoxizitätsbereich ausgehend vom Wert, bei dem Zytotoxizität auftritt (siehe Beschreibung unter Nummer 28), bis zu Konzentrationen mit mäßiger oder geringer oder nicht vorhandener Toxizität umfassen. Viele Prüfchemikalien zeigen steile Konzentrations-Reaktions-Kurven. Um Daten zu mäßiger oder geringer Toxizität zu erhalten oder um die Dosis-Reaktions-Beziehungen im Einzelnen auszuwerten, kann es daher erforderlich sein, Konzentrationen mit kleineren Abständen und/oder mehr als vier Konzentrationen zu verwenden, insbesondere in Fällen, in denen ein Wiederholungsversuch erforderlich ist (Nummer 70). Die Verwendung von mehr als 4 Konzentrationen kann besonders bei Einfachkulturen wichtig sein.
|
|
28.
|
Wenn die Höchstkonzentration auf der Zytotoxizität beruht, sollte bei der Höchstkonzentration beim MLA ein relatives Gesamtwachstum von 10-20 % und bei der TK6-Prüfung eine relative Überlebensrate von 10 % erreicht werden (Nummer 67).
|
|
29.
|
Im Falle schwer löslicher Chemikalien, die bei Konzentrationen unterhalb der niedrigsten unlöslichen Konzentration nicht zytotoxisch sind, sollte die höchste analysierte Konzentration am Ende der Behandlung mit der Prüfchemikalie eine Trübung oder eine mit bloßem Auge oder mithilfe eines Inversmikroskops erkennbare Ausfällung bewirken. Auch wenn Zytotoxizität oberhalb der niedrigsten unlöslichen Konzentration auftritt, ist es ratsam, nur eine Konzentration zu testen, bei der es zu einer Trübung oder sichtbaren Ausfällung kommt, da infolge dieser Ausfällungen künstliche Wirkungen auftreten können. Da beim MLA und bei der TK6-Prüfung Suspensionskulturen verwendet werden, ist bei der Konzentration, bei der es zu einer Ausfällung kommt, unbedingt sicherzustellen, dass die Ausfällung nicht die Durchführung des Versuchs beeinträchtigt. Unter Umständen empfiehlt es sich, die Löslichkeit im Kulturmedium vor dem Versuch zu bestimmen.
|
|
30.
|
Wird keine Ausfällung bzw. keine grenzwertige Zytotoxizität beobachtet, sollte die höchste Versuchskonzentration 10 mM, 2 mg/ml oder 2 μl/ml entsprechen, je nachdem, welcher Wert der niedrigere ist (57) (58). Wenn die Zusammensetzung der Prüfchemikalie nicht genau definiert ist (bei Stoffen mit unbekannter oder schwankender Zusammensetzung, komplexen Reaktionsprodukten oder biologischen Materialien (UVCB), Umweltextrakten usw.), muss die höchste Konzentration möglicherweise höher angesetzt werden (z. B. 5 mg/ml), sofern keine ausreichende Zytotoxizität vorhanden ist, um die Konzentration der einzelnen Bestandteile zu erhöhen. Es sei jedoch darauf hingewiesen, dass diese Anforderungen sich von denen für Humanpharmazeutika unterscheiden können (59).
|
Kontrollen
|
31.
|
Bei allen Versuchsbedingungen sind jeweils gleichzeitige Negativkontrollen (Nummer 21) anzulegen, bei denen das Behandlungsmedium lediglich Lösungsmittel enthält, die ansonsten aber auf die gleiche Weise behandelt wurden wie die Behandlungskulturen.
|
|
32.
|
Die Eignung des Labors zum Nachweis von Mutagenen unter den Bedingungen des verwendeten Prüfprotokolls und (ggf.) die Wirksamkeit des exogenen Stoffwechselaktivierungssystems sowie eine angemessene Erkennung sowohl kleiner/spät auftretender als auch großer/früh auftretender TK-Mutanten sind anhand gleichzeitiger Positivkontrollen nachzuweisen. Beispiele für Positivkontrollen sind Tabelle 1 zu entnehmen. In begründeten Fällen können andere geeignete Positivkontrollstoffe verwendet werden. Da In-vitro-Tests auf Genotoxizität in Säugetierzellen für die gleichzeitigen Kurzzeitbehandlungen (3-4 Stunden) mit und ohne Zusatz eines Stoffwechselaktivierungssystems über die gleiche Behandlungsdauer ausreichend standardisiert sind, kann sich die Hinzuziehung von Positivkontrollen auf ein Mutagen beschränken, das eine Stoffwechselaktivierung erfordert. In diesem Fall wird durch die Reaktion einer einzelnen Positivkontrolle sowohl die Aktivität des Stoffwechselaktivierungssystems als auch die Empfindlichkeit des Prüfsystems nachgewiesen. Im Falle einer Langzeitbehandlung (d. h. 24 Stunden ohne S9) sollte jedoch eine gesonderte Positivkontrolle erfolgen, da der Versuch mit Stoffwechselaktivierung eine andere Behandlungsdauer hat. Jede Positivkontrolle sollte bei einer oder mehreren Konzentrationen durchgeführt werden, die voraussichtlich eine reproduzierbare und erkennbare Zunahme gegenüber dem Hintergrund ergeben (womit sich die Empfindlichkeit des Versuchssystems nachweisen lässt), und die Wirkung sollte nicht durch einen Zytotoxizitätswert beeinträchtigt werden, der die in dieser Prüfmethode vorgegebenen Grenzen überschreitet (Nummer 28).
|
Tabelle 1
Zur Beurteilung der Eignung des Labors und zur Wahl der Positivkontrollen empfohlene Referenzstoffe
|
Kategorie
|
Stoff
|
CAS-Nr.
|
|
1. Mutagene, die ohne Stoffwechselaktivierung wirken
|
|
Methylmethansulfonat
|
66-27-3
|
|
Mitomycin C
|
50-07-7
|
|
4-Nitroquinolin-N-oxid
|
56-57-5
|
|
2. Mutagene, die eine Stoffwechselaktivierung erfordern
|
|
Benzo(a)pyren
|
50-32-8
|
|
Cyclophosphamid(monohydrat)
|
50-18-0 (6055-19-2)
|
|
7,12-Dimethylbenzanthracen
|
57-97-6
|
|
3-Methylcholanthren
|
56-49-5
|
VERFAHREN
Behandlung mit der Prüfchemikalie
|
33.
|
Proliferierende Zellen werden mit und ohne Stoffwechselaktivierungssystem mit der Prüfchemikalie behandelt. Die Exposition sollte über einen geeigneten Zeitraum (gewöhnlich 3-4 Stunden) erfolgen. Es sei jedoch darauf hingewiesen, dass diese Anforderungen sich von denen für Humanpharmazeutika unterscheiden können (59). Wenn die Kurzzeit-Behandlung zu negativen Ergebnissen führt und verfügbare Informationen eine längere Behandlung als erforderlich erscheinen lassen [z. B. bei Nucleosidanalogen oder schlecht löslichen Chemikalien (5) (59)], ist beim MLA die Durchführung der Prüfung mit längeren Behandlungszeiten (d. h. 24 Stunden ohne S9) in Erwägung zu ziehen.
|
|
34.
|
Wie viele Zellen für jede Prüfung und für jedes Stadium der Prüfung (Kontrolle und behandelte Kultur) mindestens zu verwenden sind, hängt von der Spontanmutationshäufigkeit ab. Als Faustregel gilt, dass in jeder Versuchskultur ausreichend Zellen behandelt werden sollten und eine hinreichende Passagenanzahl erreicht werden sollte, um in allen Stadien der Prüfung (Behandlung, Expression des Phänotyps und Mutantenselektion) mindestens 10 bzw. im Idealfall 100 Spontanmutationen zu erhalten (56).
|
|
35.
|
Beim MLA liegt eine annehmbare Spontanmutationshäufigkeit bei 35-140 × 10-6 (Agar-Methode) bzw. bei 50-170 × 10-6 (Microwell-Methode) (siehe Tabelle 2). Um pro Prüfkultur mindestens 10 und im Idealfall 100 Spontanmutationen zu erhalten, die die Behandlung überleben, müssen mindestens 6 × 106 Zellen behandelt werden. Die Behandlung dieser Anzahl an Zellen und der Erhalt einer hinreichenden Anzahl an Zellen während der Expression und beim Klonen für die Mutantenselektion führt in allen Phasen des Versuchs zu einer hinreichenden Anzahl an Spontanmutationen (mindestens 10) – selbst bei Kulturen, die mit Konzentrationen behandelt wurden, bei denen sich eine Zytotoxizität von 90 % ergibt (nachgewiesen anhand eines RTG von 10 %) (19) (38) (39).
|
|
36.
|
Die Spontanmutationshäufigkeit liegt bei Tk6 in der Regel zwischen 2 und 10 × 10-6. Um pro Prüfkultur mindestens 10 Spontanmutationen zu erhalten, die die Behandlung überleben, müssen in jeder Kultur mindestens 20 × 106 Zellen behandelt werden. Die Behandlung dieser Anzahl an Zellen führt selbst bei Kulturen, die mit Konzentrationen behandelt wurden, bei denen sich eine Zytotoxizität von 90 % (RS 10 %) ergibt, zu einer hinreichenden Anzahl an Spontanmutationen (mindestens 10). Außerdem muss während der Expressionszeit eine hinreichende Anzahl an Zellen kultiviert und zur Mutantenselektion plattiert werden (60).
|
Expressionszeit des Phänotyps und Messung der Zytotoxizität und der Mutantenhäufigkeit
|
37.
|
Im Anschluss an die Behandlungsphase werden die Zellen über einen bestimmten Zeitraum kultiviert, um für jede Zelllinie eine annähernd optimale spezifische Expression des Phänotyps neu induzierter Mutanten zu ermöglichen. Beim MLA beträgt die Expressionszeit des Phänotyps 2 Tage. Bei der TK6-Prüfung dauert die Expression des Phänotyps 3-4 Tage. Bei einer 24-stündigen Behandlung beginnt die Expressionszeit am Ende der Behandlung.
|
|
38.
|
Während der Expressionszeit des Phänotyps werden die Zellen täglich gezählt. Beim MLA wird anhand der täglichen Zellzählungen das tägliche Suspensionswachstum (SG) berechnet. Nach der 2-tägigen Expressionszeit werden die Zellen mit und ohne selektierendes Agens zur Bestimmung der Mutantenzahl (Selektionsplatten) bzw. der Klonierungseffizienz (Viabilitätsplatten) im Medium suspendiert. Beim MLA sind zwei Methoden zum Klonen für die Mutantenselektion annehmbar: die Soft-Agar-Methode und die Methode mit dem flüssigen Medium in 96-Well-Platten (19) (38) (39). Die Klonierung bei der TK6-Prüfung erfolgt mit flüssigen Medien und einer 96-Well-Platte (16).
|
|
39.
|
Als selektierendes Agens für TK-Mutanten wird ausschließlich Triflurothymidin (TFT) empfohlen (61).
|
|
40.
|
Beim MLA werden die Agar- und die Microwell-Platten nach 10- bis 12-tägiger Inkubation gezählt. Bei der TK6-Prüfung werden die Kolonien der früh auftretenden Mutanten auf den Microwell-Platten nach 10-14 Tagen gezählt. Zur Rückgewinnung der langsam wachsenden (spät auftretenden) TK6-Mutanten müssen die Zellen nach dem Zählen der früh auftretenden Mutanten nochmals mit dem Wachstumsmedium versorgt und mit TFT behandelt werden. Anschließend werden die Platten weitere 7-10 Tage inkubiert (62). Hinweise zur Zählung langsam und normal wachsender TK-Mutanten siehe Nummern 42 und 44.
|
|
41.
|
Die Berechnungen bei den beiden Untersuchungen mit den beiden Methoden (Agar- und Microwell-Methode) beim MLA werden in Anlage 2 erläutert. Beim MLA mit der Agar-Methode werden die Kolonien gezählt, und die Anzahl der Mutantenkolonien wird entsprechend der Klonierungseffizienz bereinigt, um die Mutantenhäufigkeit (MF) zu berechnen. Beim MLA mit der Microwell-Methode und bei der TK6-Prüfung wird die Klonierungseffizienz sowohl der Platten zur Selektion als auch der Platten zur Bestimmung der Klonierungseffizienz nach der Poisson-Verteilung bestimmt (63). Aufgrund der jeweils ermittelten Klonierungseffizienz wird die MF berechnet.
|
Charakterisierung der Mutantenkolonien
|
42.
|
Wenn die Prüfchemikalie beim MLA einen positiven Befund ergibt (Nummern 62 und 63), sollte die Charakterisierung nach Größe und Wachstumsentwicklung der Kolonien zumindest bei einer der Prüfkulturen (im Allgemeinen der mit der höchsten positiven Konzentration) sowie bei den Negativ- und den Positivkontrollen erfolgen. Ergibt sich bei der Prüfchemikalie ein negativer Befund (Nummer 64), so ist eine Charakterisierung der Mutantenkolonien bei den Negativ- und Positivkontrollen vorzunehmen. Beim MLA mit der Microwell-Methode werden Mutantenkolonien als klein betrachtet, die weniger als 25 % des Durchmessers eines Wells bedecken. Als groß gelten Mutantenkolonien, die mehr als 25 % des Well-Durchmessers bedecken. Bei der Agar-Methode werden die Mutantenkolonien und die Koloniegrößen mit einem automatischen Kolonienzähler bestimmt. Die Ansätze zur Bestimmung von Koloniegrößen werden in der Fachliteratur erläutert (19) (38) (40). Die Charakterisierung der Kolonien auf der Negativ- und der Positivkontrolle wird als Nachweis dafür benötigt, dass die Untersuchungen ordnungsgemäß durchgeführt wurden.
|
|
43.
|
Für eine Prüfchemikalie kann kein negativer Befund festgestellt werden, wenn in der Positivkontrolle weder große noch kleine Mutantenkolonien angemessen nachgewiesen werden können. Die Charakterisierung der Kolonien kann allgemein Aufschluss darüber geben, ob eine Prüfchemikalie Punktmutationen und/oder Chromosomenveränderungen auslösen kann (Nummer 4).
|
|
44.
|
TK6: Normal und langsam wachsende Mutanten unterscheiden sich durch die Inkubationszeit (Nummer 40). Bei der TK6-Prüfung werden sowohl früh als auch spät auftretende Mutanten bei allen Kulturen einschließlich der Negativ- und der Positivkontrollen bewertet. Die Charakterisierung der Kolonien der Negativ- und der Positivkontrolle wird als Nachweis dafür benötigt, dass die Untersuchungen ordnungsgemäß durchgeführt wurden. Für eine Prüfchemikalie kann kein negativer Befund festgestellt werden, wenn in der Positivkontrolle weder früh noch spät auftretende Mutantenkolonien angemessen nachgewiesen werden können. Die Charakterisierung der Kolonien kann allgemein Aufschluss darüber geben, ob eine Prüfchemikalie Punktmutationen und/oder Chromosomenveränderungen auslösen kann (Nummer 4).
|
Kompetenz des Labors
|
45.
|
Um ausreichende Erfahrungen mit der Prüfung nachzuweisen, bevor routinemäßige Prüfungen erfolgen, sollte das Labor eine Reihe von Versuchen mit positiven Referenzstoffen durchgeführt haben, die sich unterschiedlicher Mechanismen (mindestens ein aktiver Mechanismus mit und ein aktiver Mechanismus ohne Stoffwechselaktivierung, ausgewählt aus den in Tabelle 1 aufgeführten Stoffen) und verschiedener Negativkontrollen (unter Verwendung verschiedener Lösungsmittel/Vehikel) bedienen. Die Reaktionen dieser Positiv- und Negativkontrollen sollten mit der Literatur im Einklang stehen. Dies gilt nicht für erfahrene Laboratorien, d. h. für Laboratorien, die über eine historische Datenbank im Sinne der Nummern 47-50 verfügen. Bei der MLA sollten die für die Positiv- und die Negativkontrollen ermittelten Werte im Einklang mit den IWGT-Empfehlungen stehen (siehe Tabelle 2).
|
|
46.
|
Bei kurzer Behandlungsdauer sowie dann, wenn keine Stoffwechselaktivierung erfolgt ist, sollte eine Auswahl von Positivkontrollstoffen (siehe Tabelle 1) mit kurzer und (bei längeren Behandlungen) mit langer Behandlungsdauer untersucht werden, um die Fähigkeit zur Erkennung mutagener Chemikalien nachzuweisen und die Wirksamkeit des Stoffwechselaktivierungssystems zu ermitteln und zum einen die Eignung der Bedingungen für das Zellwachstum während der Behandlung sowie für die phänotypische Expression und für die Mutantenselektion und zum anderen die Eignung der Auswertungsverfahren zu belegen. Zum Nachweis der Empfindlichkeit und dynamischen Bandbreite des Versuchssystems sollte eine Spanne der Konzentrationen der ausgewählten Stoffe festgelegt werden, um reproduzierbare und konzentrationsabhängige Zunahmen gegenüber dem Hintergrund zu erhalten.
|
Historische Kontrolldaten
|
47.
|
Das Labor sollte Folgendes bestimmen:
|
—
|
den Bereich und die Verteilung historischer Positivkontrollen und
|
|
—
|
den Bereich und die Verteilung historischer Negativkontrollen (unbehandelt, Lösungsmittel).
|
|
|
48.
|
Bei der erstmaligen Erfassung von Daten zur Verteilung einer historischen Negativkontrolle sollten gleichzeitige Negativkontrollen veröffentlichten Negativkontrolldaten entsprechen. Werden weitere Versuchsdaten zur Verteilung der Kontrollen hinzugefügt, sollten gleichzeitige Negativkontrollen idealerweise innerhalb der Kontrollgrenzen von 95 % der gewählten Verteilung liegen (64) (65).
|
|
49.
|
Die Datenbank des Labors zu historischen Negativkontrollen sollte zunächst mit mindestens 10 Versuchen angelegt werden. Vorzugsweise sollte sie jedoch aus mindestens 20 Versuchen bestehen, die unter vergleichbaren Versuchsbedingungen durchgeführt wurden. Labors sollten Qualitätskontrollverfahren anwenden, wie z. B. Qualitätsregelkarten (z. B. C-Karten oder X-Bar-Karten) (65), um zu ermitteln, wie variabel ihre Positiv- und Negativkontrolldaten sind, und um nachzuweisen, dass die Methodik in ihrem Labor kontrolliert wird (66). Nähere Informationen und Empfehlungen zum Sammeln und zur Verwendung historischer Daten sind der Fachliteratur zu entnehmen (64).
|
|
50.
|
Negativkontrolldaten sollten die Mutantenhäufigkeit aus einer Einzelkultur oder vorzugsweise aus Replikatkulturen erfassen, wie in Nummer 27 beschrieben. Gleichzeitige Negativkontrollen sollten idealerweise innerhalb der Kontrollgrenzen von 95 % der Verteilung in der Datenbank des Labors zu historischen Negativkontrollen liegen. Sofern Negativkontrolldaten außerhalb der Kontrollgrenze von 95 % liegen, ist es zulässig, sie in die historische Kontrollverteilung aufzunehmen, solange es sich bei den Daten nicht um „extreme Ausreißer“ handelt und nachgewiesen werden kann, dass das Prüfsystem kontrolliert wird (Nummer 49) und dass kein technisches oder menschliches Versagen vorliegt.
|
|
51.
|
Sämtliche Änderungen am Versuchsprotokoll sind auf Übereinstimmung mit den bereits vorhandenen historischen Kontrolldaten zu prüfen. Bei größeren Inkonsistenzen sollte eine neue Datenbank historischer Kontrolldaten erstellt werden.
|
DATEN UND BERICHTERSTATTUNG
Darstellung der Prüfergebnisse
|
52.
|
Die Darstellung der Daten des MLA und der TK6-Prüfung sollte für behandelte Kulturen und für Kontrollkulturen die erforderlichen Daten für die Berechnung der Zytotoxizität (RTG bzw. RS) und die Mutantenhäufigkeit beinhalten wie im Folgenden beschrieben.
|
|
53.
|
Beim MLA sollten für die jeweiligen Kulturen das RSG und das RTG, die Kolonierungseffizienz zum Zeitpunkt der Mutantenselektion und die Anzahl der Mutantenkolonien (Agar-Methode) bzw. die Anzahl der leeren Wells (Microwell-Methode) angegeben werden. Unter der Mutantenhäufigkeit ist der zahlenmäßige Anteil der Mutantenzellen pro Million überlebender Zellen zu verstehen. Bei einer positiven Reaktion sollten die Mutantenhäufigkeiten kleiner und großer Kolonien (und/oder die Prozentanteile der Gesamtmutationshäufigkeit) für mindestens eine Konzentration der Prüfchemikalie (im Allgemeinen die höchste Konzentration mit positivem Befund) sowie für die Negativ- und die Positivkontrolle angegeben werden. Bei einer negativen Reaktion sollte die Mutantenhäufigkeit für die Negativ- und für die Positivkontrolle angegeben werden.
|
|
54.
|
Bei der TK6-Prüfung sollten für die einzelnen Kulturen jeweils die relative Überlebensrate (RS), die Klonierungseffizienz zum Zeitpunkt der Mutantenselektion und die Anzahl der leeren Wells bei früh und bei spät auftretenden Mutanten angegeben werden. Die MF sollte als Anzahl der Mutantenzellen bezogen auf die Anzahl der überlebenden Zellen angegeben werden und die Gesamtmutationshäufigkeit sowie die Mutantenhäufigkeit (und/oder den Prozentanteil der Gesamtmutationshäufigkeit) der früh und der spät auftretenden Mutanten beinhalten.
|
Akzeptanzkriterien
|
55.
|
Beim MLA und bei der TK6-Prüfung sollten die folgenden Anforderungen erfüllt sein, bevor die Gesamtergebnisse einer Prüfchemikalie ermittelt werden.
|
—
|
Beide Versuchsanordnungen (kurzzeitige Behandlung mit und ohne Stoffwechselaktivierung – siehe Nummer 33) wurden geprüft, sofern nicht bereits mit einer Versuchsanordnung ein positiver Befund ermittelt wurde.
|
|
—
|
Für die Untersuchung ist eine angemessene Anzahl an Zellen und Konzentrationen verfügbar (Nummern 27 und 34-36).
|
|
—
|
Die Kriterien für die Auswahl der höchsten Konzentration entsprechen den Kriterien unter den Nummern 28-30.
|
|
Akzeptanzkriterien für Positiv- und Negativkontrollen
|
56.
|
Die von der IWGT Expert MLA Workgroup durchgeführte Untersuchung umfangreicher MLA-Daten führte zu einem internationalen Konsens in Bezug auf bestimmte Akzeptanzkriterien für den MLA (1) (2) (3) (4) (5). Daher enthält die Prüfmethode spezifische Empfehlungen für die Akzeptanz negativer und positiver Kontrollen und für die Bewertung der Ergebnisse einzelner Stoffe beim MLA. Bei der TK6-Prüfung ist die Datenbasis erheblich kleiner, und die verfügbaren Daten wurden nicht von der Arbeitsgruppe evaluiert.
|
|
57.
|
Beim MLA sollte bei jedem Versuch geprüft werden, ob die nicht behandelte Kultur/Lösungsmittelkontrolle die Akzeptanzkriterien der IWGT MLA Workgroup ((4) und folgende Tabelle 2) in Bezug auf die folgenden Punkte erfüllt: (1) MF (wobei zu beachten ist, dass die IWGT beim MLA je nach Verwendung der Agar- oder der Microwell-Methode unterschiedliche MFs ansetzt), (2) Klonierungseffizienz (CE) zum Zeitpunkt der Mutantenselektion und (3) Suspensionswachstum (SG) der Lösungsmittelkontrolle (Formel siehe Anlage 2).
Tabelle 2
Akzeptanzkritieren des MLA
|
Parameter
|
Soft-Agar-Methode
|
Microwell-Methode
|
|
Mutantenhäufigkeit
|
35-140 × 10-6
|
50-170 × 10-6
|
|
Klonierungseffizienz
|
65-120 %
|
65-120 %
|
|
Suspensionswachstum
|
8- bis 32-fach (3- bis 4-stündige Behandlung) 32- bis 180-fach (24-stündige Behandlung, wenn durchgeführt)
|
8- bis 32-fach (3- bis 4-stündige Behandlung) 32- bis 180-fach (24-stündige Behandlung, wenn durchgeführt)
|
|
|
58.
|
Beim MLA sollte bei jeder Prüfung auch bewertet werden, ob die Positivkontrolle/n mindestens eines der beiden folgenden von der IWGT-Arbeitsgruppe entwickelten Akzeptanzkriterien erfüllt/erfüllen:
|
—
|
Bei der Positivkontrolle ist eine absolute Zunahme der Gesamtmutationshäufigkeit (d. h. eine Zunahme über die Häufigkeit der spontanen Hintergrundmutationen hinaus [eine induzierte MF (IMF)] von mindestens 300 × 10-6) festzustellen. Mindestens 40 % der IMF sollten sich in der Häufigkeit kleiner Mutantenkolonien widerspiegeln.
|
|
—
|
Die Positivkontrolle führt zu einer Zunahme der Häufigkeit kleiner Mutantenkolonien um mindestens 150 × 10-6 gegenüber der Häufigkeit bei der gleichzeitigen unbehandelten Kontrolle/Lösungsmittelkontrolle (eine induzierte Häufigkeit kleiner Mutantenkolonien von 150 × 10-6).
|
|
|
59.
|
Bei der TK-Prüfung gilt eine Untersuchung als annehmbar, wenn die gleichzeitige Negativkontrolle als zulässig für die Aufnahme in die Datenbank des Labors mit historischen Negativkontrolldaten betrachtet wird (Nummern 48 und 49). Außerdem sollten die gleichzeitigen Positivkontrollen (Nummer 32) Reaktionen hervorrufen, die mit den Reaktionen kompatibel sind, die in der Datenbank für historische Positivkontrollen erzeugt werden, und gegenüber den gleichzeitigen Negativkontrollen eine statistisch signifikante Zunahme aufweisen.
|
|
60.
|
Bei beiden Prüfungen sollte der obere Grenzwert der bei der Positivkontrolle festgestellten Zytotoxizität identisch mit dem Wert der Versuchskulturen sein. Das RTG bzw. die RS darf nicht weniger als 10 % betragen. Die Verwendung einer einzelnen Konzentration (bzw. einer der Konzentrationen der Positivkontrollkulturen, wenn mehrere Konzentrationen verwendet werden) ist hinreichend für den Nachweis, dass die Akzeptanzkriterien der Positivkontrolle erfüllt wurden. Außerdem muss die MF der Positivkontrolle im für das Labor festgestellten annehmbaren Bereich liegen.
|
Beurteilung und Auswertung der Ergebnisse
|
61.
|
Im Zusammenhang mit der MLA hat die Mauslymphom-Sachverständigengruppe (Mouse Lymphoma Expert Workgroup) der IWGT umfangreiche Arbeiten zur biologischen Relevanz und zu den Kriterien für eine positive Reaktion durchgeführt (4). Daher enthält diese Prüfmethode spezifische Empfehlungen für die Auswertung der mit dem MLA ermittelten Ergebnisse der Prüfchemikalien (Nummern 62-64). Bei der TK6-Prüfung ist die Datenbasis erheblich kleiner, und die verfügbaren Daten wurden nicht von der Arbeitsgruppe evaluiert. Daher sind die Empfehlungen zur Auswertung der Daten bei der TK6-Prüfung allgemeiner gehalten (Nummern 65 und 66). Weitere Empfehlungen gelten für beide Prüfungen (Nummern 67-71).
|
MLA
|
62.
|
Ein Ansatz zur Festlegung positiver und negativer Reaktionen wird empfohlen, um sicherzustellen, dass die erhöhte MF biologisch relevant ist. Anstelle der bei anderen Prüfungen üblichen statistischen Analysen wird bei dieser Prüfung von der Verwendung einer festgelegten induzierten Mutantenhäufigkeit (d. h. einer Zunahme der MF gegenüber der gleichzeitigen Kontrolle) ausgegangen; dieser globale Bewertungsfaktor (GEF) beruht auf der Analyse der Verteilung der MF-Daten der Negativkontrolle der beteiligten Labors (4). Beim MLA unter Verwendung der Agar-Methode beträgt der GEF 90 × 10-6, und bei der Microwell-Methode wird ein GEF von 126 × 10-6 angenommen.
|
|
63.
|
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn bei einer der getesteten Versuchsbedingungen (Nummer 33) die über den gleichzeitigen Hintergrund hinausgehende MF-Zunahme den GEF überschreitet und die Zunahme konzentrationsabhängig ist. (Dies wird beispielsweise mit einem Trendtest festgestellt.) Es wird dann davon ausgegangen, dass die Prüfchemikalie Mutationen in diesem Versuchssystem auslösen kann.
|
|
64.
|
Sofern alle Akzeptanzkriterien erfüllt sind, wird eine Prüfchemikalie als eindeutig negativ bewertet, wenn bei allen geprüften Versuchsbedingungen (Nummer 33) keine konzentrationsabhängige Reaktion festgestellt wurde bzw. – wenn eine Zunahme der MF zu verzeichnen ist – wenn die Zunahme der MF den GEF nicht überschreitet. Es wird dann davon ausgegangen, dass die Prüfchemikalie keine Mutationen in diesem Versuchssystem auslösen kann.
|
TK6
|
65.
|
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn bei einer der getesteten Versuchsbedingungen (siehe Nummer 33):
|
—
|
mindestens eine der Versuchskonzentrationen weist gegenüber der gleichzeitigen Negativkontrolle eine statistisch signifikante Zunahme auf,
|
|
—
|
ein geeigneter Trendtest zeigt, dass die Zunahme konzentrationsabhängig ist (Nummer 33),
|
|
—
|
Ergebnisse liegen außerhalb der Verteilung der historischen Negativkontrolldaten (z. B. auf einer Poisson-Verteilung beruhende Kontrollgrenze von 95 %, siehe Nummer 48).
|
Sind all diese Kriterien erfüllt, wird davon ausgegangen, dass die Prüfchemikalie in diesem Versuchssystem Mutationen auslösen kann. Für Empfehlungen zu den am besten geeigneten statistischen Methoden siehe Literaturhinweise (66) (67).
|
|
66.
|
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig negativ, wenn in allen getesteten Versuchsbedingungen (siehe Punkt 33):
|
—
|
keine der Versuchskonzentrationen eine statistisch signifikante Zunahme gegenüber der gleichzeitigen Negativkontrolle aufweist,
|
|
—
|
die Bewertung anhand einer geeigneten Trendprüfung keine konzentrationsabhängige Zunahme ergibt,
|
|
—
|
alle Ergebnisse innerhalb der Verteilung der historischen Negativkontrolldaten liegen (z. B. auf einer Poisson-Verteilung beruhende Kontrollgrenze von 95 %, siehe Nummer 48).
|
Es wird dann davon ausgegangen, dass die Prüfchemikalie keine Mutationen in diesem Versuchssystem auslösen kann.
|
MLA und TK6:
|
67.
|
Wenn die Höchstkonzentration auf der Zytotoxizität beruht, sollte bei der Höchstkonzentration ein relatives Gesamtwachstum bzw. eine relative Überlebensrate von 10-20 % erreicht werden. Allgemeiner Konsens ist, dass bei der Auswertung positiver Ergebnisse ausschließlich mit RTG- bzw. RS-Werten im Bereich zwischen 10 und 20 % Vorsicht geboten ist und dass Ergebnisse dann nicht als positiv zu bewerten sind, wenn die Zunahme der MF ausschließlich im Umfang von höchstens eines RTG- bzw. RS-Werts von 10 % (wenn ermittelt) erfolgt (2) (59).
|
|
68.
|
In einigen Fällen können zusätzliche Informationen bei der Feststellung hilfreich sein, dass eine Prüfchemikalie nicht mutagen wirkt, wenn keine Kultur vorliegt, bei der ein RTG-Wert im Bereich von 10-20 % der RTG-/RS-Werte ermittelt wird. Dies gilt für folgende Fälle: (1) Es gibt bei einer Reihe von Datenpunkten im Bereich von 20-100 % der RTG-/RS-Werte keine Anzeichen für eine Mutagenität (z. B. keine Dosis-Reaktion, keine Mutantenhäufigkeiten über die in der gleichzeitigen Negativkontrolle oder die historisch ermittelten Häufigkeiten hinaus), und mindestens ein Datenpunkt liegt im Bereich von 20-25 % der RTG-/RS-Werte. (2) Es gibt bei einer Reihe von Datenpunkten im Bereich von 25-100 % der RTG-/RS-Werte keine Anzeichen für eine Mutagenität (z. B. keine Dosis-Reaktion, keine Mutantenhäufigkeiten über die in der gleichzeitigen Negativkontrolle festgestellten Häufigkeiten oder über die historisch ermittelten Häufigkeiten hinaus) und einen negativen Datenpunkt geringfügig unter 10 % der RTG-/RS-Werte. In beiden Fällen kann für die Prüfchemikalie ein negativer Befund festgestellt werden.
|
|
69.
|
Bei einer eindeutig positiven oder negativen Reaktion ist eine Verifizierung nicht erforderlich.
|
|
70.
|
In den Fällen, in denen die Reaktion, wie oben beschrieben, weder eindeutig negativ noch eindeutig positiv ist, und/oder um die biologische Relevanz eines Ergebnisses zu untermauern, sollten die Daten durch eine fachkundige Beurteilung und/oder anhand weiterer Untersuchungen bewertet werden. Die Durchführung eines Wiederholungsversuchs, möglicherweise unter veränderten Versuchsbedingungen [z. B. Abstände der Konzentrationen, um die Wahrscheinlichkeit der Ermittlung von Datenpunkten im RTG-/RS-Bereich von 10-20 % zu erhöhen, sowie die Verwendung anderer Stoffwechselaktivierungsbedingungen (d. h. Konzentration oder Herkunft von S9) und eine Änderung der Behandlungsdauer], könnte hilfreich sein.
|
|
71.
|
In seltenen Fällen erlaubt der Datensatz selbst nach weiteren Untersuchungen keine definitive Aussage zu positiven oder negativen Ergebnissen. Daher sollte die Reaktion der Prüfchemikalie als nicht eindeutig eingestuft werden (d. h. für ein positives und für ein negatives Ergebnis besteht die gleiche Wahrscheinlichkeit).
|
PRÜFBERICHT
|
72.
|
Der Prüfbericht sollte folgende Angaben enthalten:
|
|
Prüfchemikalie:
|
—
|
Herkunft, Partienummer, ggf. begrenztes Verwendungsdatum;
|
|
—
|
Stabilität der Prüfchemikalie, falls bekannt;
|
|
—
|
Löslichkeit und Stabilität der Prüfchemikalie im Lösungsmittel, falls bekannt;
|
|
—
|
Messung des pH-Werts, der Osmolalität und ggf. des Niederschlags im Kulturmedium, dem die Prüfchemikalie zugegeben wurde.
|
|
|
Einkomponentiger Stoff:
|
—
|
physikalisches Erscheinungsbild, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
|
|
—
|
chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer, SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.
|
|
|
|
Mehrkomponentiger Stoff, UVCB-Stoffe und Gemische:
|
—
|
So weit wie möglich Charakterisierung durch die chemische Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Komponenten.
|
|
|
|
|
Lösungsmittel:
|
—
|
Begründung der Auswahl des Lösungsmittels;
|
|
—
|
Anteil des Lösungsmittels im endgültigen Kulturmedium.
|
|
|
|
Zellen:
|
|
Bei Labor-Masterkulturen:
|
—
|
Art und Herkunft der Zellen und Historie des Prüflabors;
|
|
—
|
Karyotypmerkmale und/oder Modalzahl der Chromosomen;
|
|
—
|
zum Erhalt der Zellkultur verwendete Verfahren;
|
|
—
|
Nichtvorhandensein von Mycoplasma;
|
|
—
|
Verdopplungszeit der Zellen.
|
|
|
|
|
Prüfbedingungen:
|
—
|
Begründung der Wahl der Konzentrationen und der Anzahl der Zellkulturen darunter z. B. Angaben zur Zytotoxizität und Löslichkeitsgrenze;
|
|
—
|
Medienzusammensetzung, CO2-Konzentration, Feuchtigkeit;
|
|
—
|
Konzentration der Prüfchemikalie, ausgedrückt als Endkonzentration im Kulturmedium (z. B. μg oder mg/ml oder mM des Kulturmediums);
|
|
—
|
Konzentration (und/oder Volumen) des Lösungsmittels und der beigegebenen Prüfchemikalie;
|
|
—
|
Zelldichte während der Behandlung;
|
|
—
|
Art und Zusammensetzung des Stoffwechselaktivierungssystems (Herkunft von S9, Zubereitungsmethode des S9-Gemisches, Konzentration oder Volumen des S9-Gemisches und S9 im Endmedium, Qualitätskontrollen von S9);
|
|
—
|
Positiv- und Negativkontrollstoffe, Endkonzentrationen für die jeweiligen Behandlungsbedingungen;
|
|
—
|
Dauer der Expressionszeit (ggf. einschließlich Anzahl der überimpften Zellen, Subkulturen und Medienwechsel);
|
|
—
|
Bezeichnung und Konzentration des selektierenden Agens;
|
|
—
|
beim MLA verwendete Methode (Agar oder Microwell);
|
|
—
|
Akzeptanzkriterien für die Prüfungen;
|
|
—
|
Methoden zur Zählung der lebensfähigen und mutierten Zellen;
|
|
—
|
Methoden zur Bestimmung der Zytotoxizität;
|
|
—
|
evtl. zusätzliche Angaben zur Zytotoxizität und zum verwendeten Verfahren;
|
|
—
|
Inkubationszeiten nach dem Plattieren;
|
|
—
|
Angaben zur Berücksichtigung der Kolonien nach Größe und Typ (ggf. einschließlich Kriterien für „kleine“ und „große“ Kolonien);
|
|
—
|
Kriterien zur Einstufung der Studien als positiv, negativ oder nicht eindeutig;
|
|
—
|
Methoden zur Bestimmung des pH-Werts, der Osmolalität (wenn durchgeführt) und des Niederschlags (wenn relevant).
|
|
|
|
Ergebnisse:
|
—
|
Anzahl der behandelten Zellen und Anzahl der subkultivierten Zellen der einzelnen Kulturen;
|
|
—
|
Toxizitätsparameter (RTG beim MLA und RS bei der TK6-Prüfung);
|
|
—
|
Anzeichen für Ausfällungen und Bestimmungszeit;
|
|
—
|
Anzahl der plattierten Zellen im selektierenden und im nicht selektierenden Medium
|
|
—
|
Anzahl der Kolonien im nicht selektierenden Medium und Anzahl der resistenten Kolonien im selektierenden Medium sowie entsprechende Mutantenhäufigkeiten;
|
|
—
|
Koloniegröße bei Negativ- und Positivkontrollen und die Angabe, ob die Prüfchemikalie zumindest in einer Konzentration positiv ist, sowie die entsprechenden Mutantenhäufigkeiten;
|
|
—
|
nach Möglichkeit Dosis-Reaktions-Verhältnis;
|
|
—
|
Daten zu gleichzeitigen Negativkontrollen (Lösungsmittelkontrollen) und Positivkontrollen (Konzentrationen und Lösungsmittel);
|
|
—
|
historische Daten zu Negativkontrollen (Lösungsmittelkoontrollen) und Positivkontrollen (Konzentrationen und Lösungsmittel) mit Bereichen, Mittelwerten und Standardabweichungen; Anzahl der Prüfungen, auf denen die historischen Kontrollen beruhen;
|
|
—
|
statistische Analysen (für einzelne Kulturen und (ggf.) für gepoolte Replikatkulturen) sowie ggf. p-Werte; beim MLa zusätzlich die GEF-Bewertung.
|
|
|
|
Erörterung der Ergebnisse.
|
|
LITERATUR
|
(1)
|
Moore, M.M., Honma, M., Clements, J. (Berichterstatter), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. und Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35( 3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. und Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. und Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. und Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur, A.K. und Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment No. 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. und O'Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. und Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. und Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A. und Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. und Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. und Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. und Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber, H.L., Call, K.M. und Little, J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li, C.Y., Yandell, D.W. und Little, J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma, M., Hayashi, M. und Sofuni, T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. und Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-214.
|
|
(18)
|
Amundson, S.A. und Liber, H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler, M.R., Moore, M.M. und Gollapudi, B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. und Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. und Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick, D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. und Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. und Myhr, B.C. (1991). Genotoxicity Under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang, J., Heflich, R.H. und Moore, M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. und Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. und Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer, J.R., Moore, M.M. und Hozier, J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. und Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. und Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. und DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark, L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. und Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek, T.R., Liber, H.L., Penman, B.W. und Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411–416.
|
|
(35)
|
Honma, M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. und Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuskript in Vorbereitung).
|
|
(38)
|
Lloyd, M. und Kidd, D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, Hrsg. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei, N., Guo, X. und Moore, M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan, Z, und Caldwell (Hrsg.) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber, H.L. und Thilly, W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. und Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore, M.M. und Howard, B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames, B.N., McCann, J. und Yamasaki, E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron, D.M. und Ames, B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, , A.T., Tates, , A.D, Van Buul, , P.P.W., Meijers, M. und de Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka, A., Hayashi, M. und Ishidate, M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong, T.M., u. a. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. und Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. und Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., u. a., Hrsg., Elsevier, North-Holland, S. 85-88.
|
|
(50)
|
Galloway, S.M., u. a. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson, T.E., Umbenhauer, D.R. und Galloway, S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholmer Übereinkommen über persistente organische Schadstoffe, Umweltprogramm der Vereinten Nationen (UNEP).
|
|
(53)
|
Krahn, D.F., Barsky, F.C. und McCooey, K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Hrsg.). New York, Plenum, S. 91-103.
|
|
(54)
|
Zamora, , P.O., Benson, , J.M., Li, , A.P. und Brooks, , A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura, M., Sasaki, T., Sugiyama, T., Arito, H., Fukushima, S. und Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett, C.F., u. a. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Hrsg.), Cambridge University Press, S. 66-101.
|
|
(57)
|
Morita, T., Honma, M. und Morikawa, K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire, L., Chen, J.J. und Levy, D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Abrufbar unter:[https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma, M. und Hayashi, M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. und Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber, H.L., Yandell, D.W. und Little, J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth, E.E., Thilly, W.G., Penman, B.W., Liber, H.L. und Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. und Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan, T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental Health and Safety Publications, Series on Testing and Assessment, (No 199.), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(67)
|
Fleiss, J.L., Levin, B. und Paik, M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Anlage 1
BEGRIFFSBESTIMMUNGEN
Aneugen
: Eine Chemikalie oder ein Prozess, die/der durch Wechselwirkung mit den Komponenten des mitotischen oder meiotischen Zellteilungszyklus zu Aneuploidie in Zellen oder Organismen führt.
Aneuploidie
: Eine Abweichung von der normalen diploiden (oder haploiden) Chromosomenzahl durch ein einziges Chromosom oder mehr, nicht aber durch einen ganzen (oder mehrere) Chromosomensatz/-sätze (Polyploidie).
Basenpaaraustauschmutagene
: Chemikalien, die den Austausch von Basenpaaren in der DNA verursachen.
Chemikalie
: Ein Stoff oder ein Gemisch.
Expressionszeit des Phänotyps
: Die Zeit nach der Behandlung, in der sich die genetische Alteration im Genom ausprägt und bereits vorhandene Genprodukte so weit abgebaut werden, dass die phänotypischen Eigenschaften verändert werden.
Genotoxisch
: Ein allgemeiner Begriff, der alle Typen von DNA- oder Chromosomenschädigungen umfasst, einschließlich DNA-Brüchen, Addukt-Neubildungen, Mutationen, Chromosomenaberrationen sowie Aneuploidie. Nicht alle genotoxischen Effekte führen zu Mutationen oder stabilen Chromosomenschäden.
Klastogen
: Chemikalie oder Prozess, die/der strukturelle Chromosomenaberrationen in Zell- oder Organismenpopulationen verursacht.
Klonierungseffizienz
: Der Prozentanteil der mit einer niedrigen Dichte plattierten Zellen, die sich zu einer zählbaren Kolonie entwickeln können.
Lösungsmittelkontrolle
: Veränderung der Chromosomenstruktur, nachweisbar durch mikroskopische Untersuchung des Metaphase-Stadiums der Zellteilung, äußert sich in Form von Deletionen und Fragmenten, intrachromosomalen oder reziproken Translokationen.
Mitotische Rekombination
: Während der Mitose Rekombination homologer Chromatiden, die zur Induktion von DNA-Doppelstrangbrüchen oder einem Verlust der Heterozygotie führen kann.
Mutagen
: Auslöser einer Erbgutveränderung der DNA-Basenpaarsequenz(en) in Genen oder in der Chromosomenstruktur (Chromosomenaberrationen).
Mutantenhäufigkeit (MF)
: Verhältnis der Anzahl der mutierten Zellen zur Anzahl der lebensfähigen Zellen.
Prüfchemikalie
: Stoff oder Gemisch, der/das mit dieser Prüfmethode untersucht wird.
Rasterschubmutagene
: Chemikalien, die die Addition oder Deletion eines oder mehrerer Basenpaare im DNA-Molekül verursachen.
Relative Überlebensrate (RS)
: Die relative Überlebensrate wird als Maßstab für die behandlungsbedingte Zytotoxizität bei der TK6 angenommen. Dabei handelt es sich um die relative Klonierungseffizienz (CE) von unmittelbar nach der Behandlung ausplattierten Zellen bereinigt um Zellverluste während der Behandlung bezogen auf die Klonierungseffizienz der Negativkontrolle.
Relatives Gesamtwachstum (RTG)
: Das relative Gesamtwachstum ist ein Maß der behandlungsbedingten Zytotoxizität beim MLA. Das RTG ist ein Maß für das relative (auf die Vehikelkontrolle bezogene) Wachstum der Prüfkulturen während der Behandlung, der zweitägigen Expression und der Klonierung der selektionierten Mutanten im Laufe der Prüfung. Das RSG der einzelnen Prüfkulturen wird mit der relativen Klonierungseffizienz der Prüfkultur zum Zeitpunkt der Mutantenselektion multipliziert und bezogen auf die Klonierungseffizienz der Negativkontrolle/Lösungsmittelkontrolle ausgedrückt (Clive und Spector, 1975).
Relatives Suspensionswachstum (RSG)
: Beim MLA das innerhalb von zwei Tagen zu verzeichnende relative Gesamtwachstum der Suspension der Prüfkultur bezogen auf das Gesamtwachstum der Suspension der Negativkontrolle/Lösungsmittelkontrolle in zwei Tagen (Clive und Spector, 1975). Das RSG sollte das relative Wachstum der Prüfkultur bezogen auf die Negativkontrolle/Lösungsmittelkontrolle während der Behandlungszeit beinhalten.
S9-Gemisch
: Gemisch aus der S9-Leberfraktion und für die metabolische Enzymaktivität notwendigen Ko-Faktoren.
S9-Leberfraktion
: Überstand des Leberhomogenats nach Zentrifugieren mit 9 000 g, d. h. Rohleberextrakt.
Suspensionswachstum (SG)
: Die n-fache Zunahme der Anzahl der Zellen im Laufe der Behandlung und der Expression während des MLA. Das SG wird durch Multiplikation der n-fachen Zunahme an Tag 1 mit der n-fachen Zunahme an Tag 2 bei der kurzzeitigen (3- bis 4-stündigen) Behandlung berechnet. Bei einer 24-stündigen Behandlung ist das SG die n-fache Zunahme während der 24-stündigen Behandlung multipliziert mit den n-fachen Zunahmen an den Expressionstagen 1 und 2.
Unbehandelte Kontrollen
: Kulturen, die nicht behandelt werden (d. h. weder mit der Prüfchemikalie noch mit Lösungsmittel), jedoch in gleicher Weise aufgearbeitet werden wie die Kulturen, die mit der Prüfchemikalie behandelt werden.
Vorwärtsmutation
: Genmutation vom Elterntyp zur mutierten Form, die eine Veränderung oder den Ausfall der Enzymaktivität oder der Funktion des kodierten Proteins bewirkt.
Zytotoxizität
: Bei den Assays im Rahmen dieser Prüfmethode wird die Zytotoxizität als Reduzierung des relativen Gesamtwachstums (RTG) beim MLA bzw. der relativen Überlebensrate (RS) bei der TK6-Prüfung bezeichnet.
Anlage 2
FORMELN
Relative Populationsverdopplung (RPD)
Für die Durchführung des MLA mit der Agar- und der Microwell-Methode gilt:
Zytotoxizität ist das relative Gesamtwachstum (RTG); dieses beinhaltet das relative Suspensionswachstum (RSG) während der zweitägigen Expressionszeit und die zum Zeitpunkt der Mutantenselektion ermittelte relative Klonierungseffizienz (RCE). RTG, RSG und RCE werden in Prozent ausgedrückt.
Berechnung des RSG: Als Suspensionswachstum 1 (SG1) wird das Wachstum zwischen Tag 0 und Tag 1 bezeichnet (Zellkonzentration an Tag 1 / Zellkonzentration an Tag 0). Suspensionswachstum 2 (SG2) ist das Wachstum zwischen Tag 1 und Tag 2 (Zellkonzentration an Tag 2 / Zellkonzentration an Tag 1). Als RSG wird das Gesamtwachstum der Suspension (SG1 × SG2) der behandelten Kultur bezogen auf die unbehandelte Kontrolle / Lösungsmittelkontrolle bezeichnet. Dabei gilt: RSG = [SG1(test) × SG2(test)] / [SG1(control) × SG2(control)] SG1 sollte anhand der Ausgangszellkonzentration bei Beginn der Behandlung berechnet werden. Damit werden unterschiedliche Zytotoxizitätswirkungen in der Prüfkultur / den Prüfkulturen während der Behandlung berücksichtigt.
Als RCE wird die relative Klonierungseffizienz der Prüfkultur bezogen auf die relative Klonierungseffizenz der unbehandelten Kontrolle / Lösungsmittelkontrolle zum Zeitpunkt der Mutantenselektion bezeichnet.
Relatives Gesamtwachstum (RTG):
RTG = RSG × RCE
TK6
Relative Überlebensrate (RS):
Die Zytotoxizität wird anhand der relativen Überlebensrate ermittelt, d. h. anhand der Klonierungseffizienz (CE) von Zellen, die nach der Behandlung umgehend plattiert werden; dabei ist eine Bereinigung um Zellverluste während der Behandlung im Vergleich zur Klonierungseffizienz bei Negativkontrollen (mit einer Überlebensrate von 100 %) vorzunehmen. Die Bereinigung um Zellverluste bei der Behandlung kann wie folgt vorgenommen werden:

Die RS einer mit einer Prüfchemikalie behandelten RS wird wie folgt berechnet:

Mutantenhäufigkeit bei MLA und TK6
Als Mutantenhäufigkeit (MF) wird die Klonierungseffizienz von Mutantenkolonien im selektierenden Medium (CEM) bereinigt um die Klonierungseffizienz im nicht selektierenden Medium zum Zeitpunkt der Mutantenselektion (CEV) bezeichnet (d. h. MF = CEM/CEV). Im Folgenden wird die Berechnung der Klonierungseffizienz-Werte beim Klonieren mit der Agar-Methode und der Microwell-Methode beschrieben.
MLA mit der Agar-Methode: Beim MLA mit der Soft-Agar-Methode werden die Anzahl der Kolonien auf der Platte zur Mutantenselektion (CM) und die Anzahl der Kolonien auf der Platte, die nicht zur Selektion von Mutanten, sondern zur Bestimmung der Klonierungseffizienz (Anzahl der lebensfähigen Klone) (CV) vorgesehen ist, unmittelbar durch Zählen der Klone ermittelt. Wenn 600 Zellen zur Bestimmung der Klonierungseffizienz (CE) auf der Platte zur Mutantenselektion (CEM) und auf der Platte, die nicht zur Selektion von Mutanten, sondern zur Bestimmung der Klonierungseffizienz (Anzahl der lebensfähigen Klone) (CEV) vorgesehen ist, ausplattiert wurden, werden 3 × 106 Zellen zur Mutantenselektion verwendet.
CEM = CM / (3 × 106) = (CM / 3) × 10-6
CEV = CV / 600
MLA und TK6 – Microwell-Methode:
Wenn der MLA mit der Microwell-Methode durchgeführt wird, werden CM und CV als Produkt der Gesamtzahl der Microwells (TW) und der wahrscheinlichen Anzahl der Kolonien pro Well (P) auf den Microwell-Platten ermittelt.
CM = PM × TWM
CV = PV × TWV
Beim Null-Term der Poisson-Verteilung (Furth u. a., 1981) wird P wie folgt berechnet:
P = - ln (EW / TW)
Wobei gilt: EW = leere Wells und TW = Gesamtzahl der Wells. Daher ist
CEM = CM / TM = (PM × TWM) / TM
CEV = CV / TV = (PV × TWV) / TV
Bei Durchführung des MLA mit der Microwell-Methode wird die Mutantenhäufigkeit (kleine und große Kolonien) für kleine und große Kolonien auf die gleiche Weise jeweils anhand der Anzahl leerer Wells berechnet.
Bei der TK6-Prüfung wird die Mutantenhäufigkeit (kleine und große Kolonien) danach bestimmt, ob die Mutanten früh oder später auftreten.
B.68 IN-VITRO-PRÜFMETHODE MIT KURZZEITEXPOSITION ZUR ERMITTLUNG VON i) CHEMIKALIEN, DIE SCHWERE AUGENSCHÄDEN VERURSACHEN, UND ii) CHEMIKALIEN, DIE KEINE EINSTUFUNG ALS AUGENREIZEND ODER SCHWER AUGENSCHÄDIGEND ERFORDERN
EINLEITUNG
|
1.
|
Diese Prüfmethode (PM) entspricht der OECD-Prüfrichtlinie 491 (2017). Die Prüfmethode zur Untersuchung unter Kurzzeitexposition ist eine In-Vitro-Methode, die unter bestimmten Umständen und mit bestimmten Einschränkungen zur Einstufung und Kennzeichnung von Chemikalien (Stoffen und Gemischen) verwendet werden kann, die schwere Augenschädigungen bzw. Augenreizungen im Sinne des GHS (Globally Harmonized System of Classification and Labelling of Chemicals = Global harmonisiertes System zur Einstufung und Kennzeichnung) (UN) (1) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP-Verordnung) (68) induzieren.
|
|
2.
|
Über viele Jahre wurde das augengefährdende Potenzial von Chemikalien in erster Linie mit einem In-vivo-Kaninchenaugentest (PM B.5 (8), entsprechend OECD TG 405) geprüft. Es herrscht allgemeiner Konsens darüber, dass der In-vivo-Kaninchenaugentest in absehbarer Zukunft nicht in vollem Umfang durch eine einzelne alternative In-vitro-Prüfung ersetzt werden kann, die geeignet wäre, das gesamte Spektrum augenreizender bzw. schwer augenschädigender Reaktionen auf unterschiedliche Chemikalienklassen vorherzusagen. Unter Umständen könnte der Kaninchenaugentest jedoch durch strategische Kombinationen mehrerer alternativer Prüfmethoden im Rahmen einer (gestuften) Prüfstrategie vollständig ersetzt werden (2). Der Top-down-Ansatz ist zur Prüfung von Chemikalien ausgelegt, bei denen nach verfügbaren Informationen davon ausgegangen werden kann, dass sie stark reizend wirken oder schwere Augenschäden hervorrufen können. Mit dem Bottom-up-Ansatz werden Chemikalien geprüft, bei denen ausgehend von verfügbaren Informationen angenommen werden kann, dass sie keine Augenreizungen in einem Umfang induzieren, der eine Einstufung erfordern würde. Die Kurzzeit-Prüfmethode wird nicht als geeignet betrachtet, den In-vivo-Kaninchenaugentest vollständig zu ersetzen. Sie kann jedoch im Rahmen einer gestuften Prüfstrategie zu gesetzgeberischen Einstufungs- und Kennzeichnungszwecken verwendet werden (beispielsweise mit dem Top-down-/Bottom-up-Ansatz), um (i) Chemikalien, die schwere Augenschädigungen (UN-GHS-/CLP-Kategorie 1) induzieren, und (ii) Chemikalien (außer stark flüchtigen Stoffen und allen festen Chemikalien mit Ausnahme von Tensiden), die keine Einstufung als augenreizend oder schwer augenschädigend (UN-GHS-/CLP-Kategorie Keine Einstufung) erfordern, ohne weitere Prüfungen zu erkennen (1) (2). Bei Chemikalien, für die aufgrund der Kurzzeit-Prüfmethode weder eine schwer augenschädigende Wirkung (UN-GHS-/CLP-Kategorie 1) vorhergesagt noch erwartet wird, dass sie Augenreizungen oder schwere Augenschädigungen induzieren (UN-GHS-/CLP-Kategorie Keine Einstufung), müssen weitere Prüfungen durchgeführt werden, um eine endgültige Einstufung vornehmen zu können. Außerdem sollten die zuständigen Regulierungsbehörden konsultiert werden, bevor die Kurzzeitprüfung mit einem Bottom-Up-Ansatz unter Berücksichtigung einer anderen Klassifizierungsregelung als der des UN GHS bzw. der CLP-Verordnung durchgeführt wird. Die Auswahl der am besten geeigneten Prüfmethode und die Anwendung dieser Prüfmethode sollten unter Berücksichtigung des OECD-Leitliniendokuments über integrierte Prüfungs- und Bewertungsansätze (IATA = Integrated Approaches to Testing and Assessment) zur Untersuchung von schweren Augenschädigungen und von Augenreizungen (14) erfolgen.
|
|
3.
|
Diese Prüfmethode beschreibt die Verfahrensschritte zur Beurteilung des augengefährdenden Potenzials einer Prüfchemikalie anhand ihrer Fähigkeit, in der Kurzzeitprüfung eine zytotoxische Wirkung hervorzurufen. Die zytotoxische Wirkung von Chemikalien auf Hornhaut-Epithelzellen ist eine erhebliche Wirkungsweise, die zu Schädigungen des Hornhautepithels und zu Augenreizungen führt. Die Zellviabilität wird bei der Kurzzeit-Prüfmethode durch Enzymkonversion des Vitalfarbstoffs MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid, auch als Thiazolyl-Blau-Tetrazoliumbromid bezeichnet) zu einem blauen Formazan-Salz gemessen, das nach seiner Extraktion aus Geweben quantifiziert wird (3). Die ermittelte Zellviabilität wird mit der Lösungsmittelkontrolle verglichen (relative Viabilität) und zur Abschätzung der potenziellen Augengefährdung durch die Prüfchemikalie verwendet. Prüfchemikalien sind dann als UN-GHS-/CLP-Kategorie 1 einzustufen, wenn sowohl die 5- als auch die 0,05 %ige Konzentration zu einer Zellviabilität von kleiner oder gleich (≤) 70 % führt. Die UN-GHS-/CLP-Kategorie Keine Einstufung ist für Prüfchemikalien anzunehmen, bei denen sowohl die 5- als auch die 0,05 %ige Konzentration eine Zellviabilität von mehr als (>) 70 % ergibt.
|
|
4.
|
Der Begriff „Prüfchemikalie“ bezeichnet bei dieser Prüfmethode das, was untersucht wird, und bezieht sich nicht auf die Anwendbarkeit der Kurzzeit-Prüfmethode zur Prüfung von Stoffen und/oder Gemischen. Es gelten die Begriffsbestimmungen der Anlage.
|
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
|
5.
|
Diese Prüfmethode beruht auf einem von der Kao Corporation entwickelten Protokoll (4), das zwei getrennten Validierungsstudien unterzogen wurde: einer durch den Validierungsausschuss der Japanischen Gesellschaft für Alternativen zu Tierversuchen (JSAAE = Japanese Society for Alternative to Animal Experiments) (5) und einer durch das Japanische Zentrum zur Validierung alternativer Methoden (JaCVAM = Japanese Center for the Validation of Alternative Methods) (6). Das NICEATM und der ICCVAM haben ausgehend von den Berichten über die Validierungsstudien und von Background Review Documents zur Prüfmethode einen Peer-Review durchgeführt (7).
|
|
6.
|
Bei der Ermittlung von Chemikalien (Stoffen und Gemischen), die schwere Augenschädigungen hervorrufen (UN-GHS-/CLP-Kategorie 1) (1), ergaben sich für die mit der Kurzzeit-Prüfmethode an 125 Chemikalien (sowohl Stoffe als auch Gemische) ermittelten Daten eine Gesamtgenauigkeit von 83 % (104/125), eine Falsch-positiv-Rate von 1 % (1/86) und eine Falsch-negativ-Rate von 51 % (20/39) im Vergleich zum In-vivo-Kaninchenaugentest (7). Die ermittelte Falsch-negativ-Rate ist in diesem Zusammenhang nicht maßgeblich, da alle Prüfchemikalien, bei denen eine Zellviabilität von ≤ 70 % bei einer Konzentration von 5 % und von > 70 % bei einer Konzentration von 0,05 % gemessen wird, anschließend je nach rechtlichen Anforderungen und gemäß der sequenziellen Prüfstrategie und den gegenwärtig empfohlenen evidenzbasierten Bewertungsansätzen (weight-of-evidence approach) ohnehin mit anderen angemessen validierten In-vitro-Prüfmethoden bzw. – als letzte Möglichkeit – mit dem In-vivo-Kaninchenaugentest geprüft werden (1) (8). Hauptsächlich wurden einkomponentige Stoffe geprüft; begrenzte Daten liegen jedoch auch über die Prüfung von Gemischen vor. Die Prüfmethode ist dennoch für die Prüfung von mehrkomponentigen Stoffen und Gemischen technisch geeignet. Bevor diese Prüfmethode jedoch für die Generierung von Daten für einen bestimmten Regelungszweck verwendet wird, sollte geprüft werden, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefert, und wenn dem so ist, warum. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs gesetzlich vorgeschrieben ist. Im Hinblick auf die Eignung der Kurzzeit-Prüfmethode zur Entscheidung über die Einstufung von Prüfchemikalien in die UN-GHS-/CLP-Kategorie 1 wurden keine sonstigen spezifischen Defizite festgestellt. Prüfer können diese Prüfmethode für Prüfchemikalien einsetzen, und eine Zellviabilität von ≤ 70 % bei Konzentrationen von 5 % und von 0,05 % sollte als Indikator für eine schwer augenschädigende Wirkung betrachtet werden, nach der ohne weitere Prüfungen eine Einstufung in die UN-GHS-/CLP-Kategorie 1 vorzunehmen ist.
|
|
7.
|
Bei der Ermittlung von Chemikalien (Stoffen und Gemischen), bei denen eine Einstufung aufgrund von Augenreizungen und schweren Augenschädigungen nicht erforderlich ist (UN-GHS-/CLP-Kategorie Keine Einstufung), ergaben sich für die mit der Kurzzeit-Prüfmethode an 130 Chemikalien (sowohl Stoffe als auch Gemische) ermittelten Daten eine Gesamtgenauigkeit von 85 % (110/130), eine Falsch-negativ-Rate von 12 % (9/73) und eine Falsch-positiv-Rate von 19 % (11/57) im Vergleich zum In-vivo-Kaninchenaugentest (7). Wenn stark flüchtige Stoffe und feste Stoffe (außer Tensiden) aus dem Datensatz ausgeschlossen werden, erhöht sich die Gesamtgenauigkeit auf 90 % (92/102), die Falsch-negativ-Rate sinkt auf 2 % (1/54), und die Falsch-positiv-Rate bleibt bei 19 % (9/48) (7). Insoweit bestehen die potenziellen Defizite der Kurzzeit-Prüfmethode bei der Ermittlung von Prüfchemikalien, bei denen im Hinblick auf die Augenreizungen und schwere Augenschädigungen keine Einstufung vorzunehmen ist (UN-GHS-/CLP-Kategorie Keine Einstufung) in einer hohen Falsch-negativ-Rate bei i) stark flüchtigen Stoffen mit Dampfdrücken über 6 kPa und ii) festen Chemikalien (Stoffen und Gemischen) mit Ausnahme von Tensiden und Gemischen, die ausschließlich Tenside beinhalten. Bei diesen Chemikalien ist die Kurzzeit-Prüfmethode nicht anzuwenden (7).
|
|
8.
|
Zusätzlich zu den in den Nummern 6 und 7 genannten Chemikalien enthält der mit der Kurzzeit-Prüfmethode generierte Datensatz auch interne Daten zu 40 Gemischen, mit denen sich im Vergleich zum In-vivo-Draize-Augentest eine Genauigkeit von 88 % (35/40), eine Falsch-positiv-Rate von 50 % (5/10) und eine Falsch-negativ-Rate von 0 % (0/30) bei den Vorhersagen für Gemische ergaben und bei denen eine Einstufung nach dem UN GHS bzw. der CLP-Verordnung nicht erforderlich ist (9). Daher kann die Kurzzeit-Prüfmethode nicht nur bei festen Stoffen, sondern auch bei Gemischen (außer bei festen Gemischen mit Ausnahme von Gemischen, die ausschließlich Tenside enthalten) verwendet werden, um zu ermitteln, ob eine Einstufung nicht erforderlich ist (UN-GHS-/CLP-Kategorie Keine Einstufung). Auch Gemische mit Stoffen mit Dampfdrücken von über 6 kPa sollten mit besonderer Sorgfalt untersucht werden, damit Gefahren nicht unterschätzt werden. Ob die Verwendung der Kurzzeit-Prüfmethode in Betracht kommt, sollte daher auf Einzelfallbasis entschieden werden.
|
|
9.
|
Da die Wirkung einer erheblichen Anzahl von in die UN-GHS-/CLP-Kategorie 1 einzustufenden Chemikalien unterschätzt wurde und die Chemikalien fälschlich in die Kategorien 2, 2A oder 2B eingestuft wurden bzw. da die Wirkung einer erheblichen Anzahl von Chemikalien, für die eine Einstufung nicht erforderlich ist (UN-GHS-/CLP-Kategorie Keine Einstufung), überschätzt wurde und die Chemikalien fälschlich in die Kategorien 2, 2A oder 2B eingestuft wurden, ist die Kurzzeit-Prüfmethode nicht zur Ermittlung von Prüfchemikalien geeignet, die in die UN-GHS-/CLP-Kategorie 2 oder in die UN-GHS-Kategorien 2A (augenreizend) oder 2B (leicht augenreizend) einzustufen sind (7). Bei diesen Chemikalien können weitere Prüfungen mit einer anderen geeigneten Methode erforderlich sein.
|
|
10.
|
Die Kurzzeit-Prüfmethode ist für Prüfchemikalien geeignet, die gelöst oder mindestens 5 Minuten in physiologischer Kochsalzlösung, in 5 % Dimethylsulfoxid (DMSO) in Kochsalz oder in Mineralöl gleichmäßig suspendiert wurden. Für nicht lösliche Prüfchemikalien und für Prüfchemikalien, die nicht mindestens 5 Minuten gleichmäßig in physiologischer Kochsalzlösung, 5 % DMSO in Kochsalz oder in Mineralöl suspendiert werden können, ist die Kurzzeit-Prüfmethode nicht geeignet. In Anbetracht der kurzzeitigen Exposition kann bei der Kurzzeit-Prüfung Mineralöl verwendet werden. Daher ist die Kurzzeit-Prüfmethode für die Vorhersage des augengefährdenden Potenzials wasserunlöslicher Prüfchemikalien (z. B. langkettiger Fettalkohole oder Ketone) geeignet, sofern die Chemikalien in mindestens einem der drei oben genannten Lösungsmittel gemischt werden können (4).
|
|
11.
|
Der Begriff „Prüfchemikalie“ bezeichnet bei dieser Prüfmethode das, was untersucht wird, (69) und bezieht sich nicht auf die Anwendbarkeit der Kurzzeit-Prüfmethode zur Prüfung von Stoffen und/oder Gemischen.
|
PRINZIP DER PRÜFMETHODE
|
12.
|
Die Kurzzeit-Prüfmethode ist ein zytotoxizitätsbasierter In-vitro-Assay, der an einem konfluenten Monolayer von SRIC-Zellen (Kaninchen-Corneazellen des Statens Serum Institut) durchgeführt wird, die auf einer Polycarbonat-Mikrotiterplatte mit 96 Wells kultiviert wurden (4). Nach einer fünfminütigen Exposition gegenüber einer Prüfchemikalie wird die relative Viabilität von SIRC-Zellen mit der MTT-Prüfung gemessen (4). Eine reduzierte Zellviabilität ist ein Indikator für potenzielle schädliche Wirkungen, die zu Augenschäden führen können.
|
|
13.
|
Es wurde berichtet, dass 80 % einer auf ein Kaninchenauge applizierten Lösung innerhalb von drei bis vier Minuten bzw. beim Menschen sogar mehr als 80 % innerhalb von einer bis zwei Minuten über den Bindehautsack ausgeschieden werden (10). Bei der Kurzzeit-Prüfmethode wird versucht, diese Expositionszeiten in etwa einzuhalten; dabei wird die Zytotoxizität als Endpunkt für die Beurteilung der Schädigung der SIRC-Zellen nach einer fünfminütigen Exposition gegenüber der Prüfchemikalie angenommen.
|
NACHWEIS DER LEISTUNGSFÄHIGKEIT
|
14.
|
Vor der routinemäßigen Anwendung der Kurzzeitprüfung nach dieser Prüfmethode sollten Labors die erforderliche technische Befähigung durch eine ordnungsgemäße Einstufung der elf in Tabelle 1 empfohlenen Stoffe nachweisen. Diese Stoffe wurden als repräsentativ für das gesamte Spektrum an Reaktionen mit schweren Augenschädigungen oder Augenreizungen nach den Ergebnissen von In-vivo-Kaninchenaugentests (TG 405) und nach der UN-GHS-/CLP-Klassifizierung ausgewählt (1). Weitere Auswahlkriterien waren die Verfügbarkeit der Stoffe im Handel sowie das Vorliegen hochwertiger In-vivo-Referenzdaten und hochwertiger In-vitro-Daten, die mit der Kurzzeit-Prüfmethode ermittelt wurden (3). Wenn ein dort genannter Stoff nicht verfügbar ist bzw. wenn dies gerechtfertigt ist, kann ein anderer Stoff verwendet werden, für den geeignete In-vivo- und In-vitro-Referenzdaten verfügbar sind, sofern die hier beschriebenen Auswahlkriterien berücksichtigt werden.
Tabelle 1
Liste der Leistungsstoffe
|
Stoff
|
CAS-Nr.
|
Chemikalienklasse (70)
|
Aggregatzustand
|
In-vivo-UN-GHS-/CLP-Kat. (71)
|
Lösungsmittel in der Kurzzeitprüfung
|
UN-GHS-/CLP-Kat. nach Kurzzeitexposition
|
|
Benzalkoniumchlorid (10 %, wässrig)
|
8001-54-5
|
Oniumverbindung
|
Flüssigkeit
|
Kategorie 1
|
Kochsalzlösung
|
Kategorie 1
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Ether
|
Flüssigkeit
|
Kategorie 1
|
Kochsalzlösung
|
Kategorie 1
|
|
Acid Red 92
|
18472-87-2
|
Heterocyclische Verbindung, Bromverbindung, Chlorverbindung
|
Feststoff
|
Kategorie 1
|
Kochsalzlösung
|
Kategorie 1
|
|
Natriumhydroxid
|
1310-73-2
|
Lauge, anorganische Chemikalie
|
Feststoff
|
Kategorie 1 (72)
|
Kochsalzlösung
|
Kategorie 1
|
|
Butyrolacton
|
96-48-0
|
Lacton, heterocyclische Verbindung
|
Flüssigkeit
|
Kategorie 2A (nach CLP Kategorie 2)
|
Kochsalzlösung
|
Keine Vorhersage möglich
|
|
1-Octanol
|
111-87-5
|
Alkohol
|
Flüssigkeit
|
Kategorie 2A/B (73) (nach CLP Kategorie 2)
|
Mineralöl
|
Keine Vorhersage möglich
|
|
Cyclopentanol
|
96-41-3
|
Alkohol, Kohlenwasserstoff, cyclisch
|
Flüssigkeit
|
Kategorie 2A/B (74) (nach CLP Kategorie 2)
|
Kochsalzlösung
|
Keine Vorhersage möglich
|
|
2-Ethoxyethylacetat
|
111-15-9
|
Alkohol, Ether
|
Flüssigkeit
|
Keine Einstufung
|
Kochsalzlösung
|
Keine Einstufung
|
|
Dodecan
|
112-40-3
|
Kohlenwasserstoff (acyclisch)
|
Flüssigkeit
|
Keine Einstufung
|
Mineralöl
|
Keine Einstufung
|
|
Methylisobutylketon
|
108-10-1
|
Keton
|
Flüssigkeit
|
Keine Einstufung
|
Mineralöl
|
Keine Einstufung
|
|
1,1-Dimethylguanidinsulfat
|
598-65-2
|
Amidin, Schwefelverbindung
|
Feststoff
|
Keine Einstufung
|
Kochsalzlösung
|
Keine Einstufung
|
|
Abkürzungen: CAS-Nr. = Registernummer des Chemical Abstracts Service
|
|
PRÜFVERFAHREN
Vorbereitung des Zellmonolayers
|
15.
|
Die Kurzzeitprüfung sollte mit SRIC-Zellen (Kaninchen-Corneazellen des Statens Serum Institut) durchgeführt werden. Die SIRC-Zellen sollten aus einer gut qualifizierten Zellbank bezogen werden (z. B. American Type Culture Collection (ATCC), CCL 60).
|
|
16.
|
SIRC-Zellen werden bei 37 °C in 5 % CO2 in befeuchteter Atmosphäre in einem Kulturkolben mit einem Kulturmedium (d. h. Eagle's MEM) kultiviert, das mit 10 % fetalem Rinderserum (FBS), 2 mM L-Glutamin, 50-100 Einheiten/ml Penicillin und 50-100 μg/ml Streptomycin versetzt wurde. Zellen, die sich im Kulturkolben konfluent entwickelt haben, sollten mit Trypsin-Ethylendiamintetraessigsäure-Lösung (ggf. unter Verwendung eines Zellschabers) abgenommen werden. Bevor sie für Routineprüfungen verwendet werden, sind die Zellen in einem Kulturkolben zu propagieren (z. B. mit 2 bis 3 Passagen); nach dem Auftauen sollten nicht mehr als 25 Passagen erfolgen.
|
|
17.
|
Die für die Kurzzeitprüfung zu verwendende Zellen werden dann mit geeigneter Dichte präpariert und in 96-Well-Platten ausgesät. Zum Aussäen in einem Kulturvolumen von 200 μl wird eine Zelldichte von 6,0 × 103 Zellen pro Well empfohlen, wenn die Zellen vier Tage nach dem Aussäen verwendet werden, bzw. 3,0 × 103 Zellen bei Verwendung der Zellen fünf Tage nach dem Aussäen. Zellen, die zur Kurzzeitprüfung mit angemessener Dichte in ein Kulturmedium ausgesät wurden, erreichen zum Zeitpunkt der Prüfung (d. h. vier oder fünf Tage nach dem Aussäen) eine Konfluenz von mehr als 80 %.
|
Applikation der Prüfchemikalien und Kontrollstoffe
|
18.
|
Erste Wahl als Lösungsmittel zum Auflösen oder Suspendieren von Prüfchemikalien ist physiologische Kochsalzlösung. Wenn eine Prüfchemikalie nur schwach oder überhaupt nicht löslich ist oder in Kochsalzlösung nicht mindestens fünf Minuten gleichmäßig suspendiert wird, ist als zweite Wahl 5 % DMSO (CAS-Nr. 67-68-5) in Kochsalzlösung als Lösungsmittel zu verwenden. Bei Prüfchemikalien, die in Kochsalzlösung oder in 5 % DMSO in Kochsalzlösung nicht gelöst oder mindestens fünf Minuten gleichmäßig suspendiert werden, wird als dritte Wahl Mineralöl (CAS-Nr. 8042-47-5) als Lösungsmittel verwendet.
|
|
19.
|
Die Prüfchemikalien werden im jeweils ausgewählten Lösungsmittel in einer Konzentration von 5 % (w/w) gelöst oder gleichmäßig suspendiert und anschließend in Verdünnungsreihen mit jeweils 10 Verdünnungen auf Konzentrationen von 0,5 % und 0,05 % verdünnt. Jede einzelne Prüfchemikalie ist in beiden Konzentrationen (5 % und 0,05 %) zu untersuchen. In der 96-Well-Platte kultivierte Zellen werden bei Raumtemperatur fünf Minuten pro Well mit 200 μl der Prüfchemikalienlösung (bzw. der Suspension) in Konzentrationen von 5 bzw. 0,05 % behandelt. Prüfchemikalien (einkomponentige oder mehrkomponentige Stoffe oder Gemische) werden je nach Methode unabhängig von ihrer tatsächlichen Reinheit als reine Stoffe betrachtet und verdünnt bzw. suspendiert.
|
|
20.
|
Das in Nummer 16 beschriebene Kulturmedium wird bei jeder einzelnen Replikatplatte als Mediumkontrolle verwendet. Außerdem werden die Zellen auf jeder einzelnen Replikatplatte mit Proben der Lösungsmittelkontrolle behandelt. Für die in Nummer 18 genannten Lösungsmittel wurde bestätigt, dass die Viabilität von SRIC-Zellen nicht beeinträchtigt wird.
|
|
21.
|
Bei der Kurzzeit-Prüfmethode ist bei allen Replikatplatten 0,01 % Natriumlaurylsulfat (SLS) in Kochsalzlösung als Positivkontrolle zu verwenden. Zur Berechnung der Zellviabilität der Positivkontrolle muss jede Replikatplatte auch eine Kochsalzlösung als Kontrolle beinhalten.
|
|
22.
|
Zur Bestimmung des Ausgleichs der optischen Dichte wird eine Blindkontrolle benötigt. Die Blindkontrolle ist in Wells zu prüfen, die ausschließlich calcium- und magnesiumfreien Phosphatpuffer oder Zellen enthalten.
|
|
23.
|
Jede Probe (die Prüfchemikalie in Konzentrationen von > 5 % und 0,05 %, die Mediumkontrolle, die Lösungsmittelkontrolle und die Positivkontrolle) sollte bei jeder Wiederholung jeweils dreimal geprüft werden, indem die Zellen fünf Minuten bei Raumtemperatur mit 200 μl der betreffenden Prüf- bzw. Kontrollchemikalie behandelt werden.
|
|
24.
|
Referenzstoffe erleichtern die Evaluierung des augenreizenden Potenzials unbekannter Chemikalien einer bestimmten Chemikalien- oder Produktklasse und die Evaluierung des relativen Reizpotenzials eines Augenreizstoffes innerhalb einer spezifischen Spanne von Reizwirkungen.
|
Messung der Zellviabilität
|
25.
|
Nach der Behandlung werden die Zellen zweimal mit 200 μl PBS gewaschen; anschließend werden 200 μl MTT-Lösung (0,5 mg MTT/ml Kulturmedium) zugesetzt. Nach einer zweistündigen Reaktionszeit in einem Inkubator (37 °C, 5 % CO2) wird die MTT-Lösung dekantiert. Danach wird das MTT-Formazan unter Zugabe von 200 μl 0,04 n Salzsäure/Isopropanol 60 Minuten im Dunkeln bei Raumtemperatur extrahiert. Darauf wird mit einem Platten-Lesegerät die Absorption der MTT-Formazan-Lösung bei 570 nm gemessen. Bei der MTT-Prüfung treten Interferenzen durch die Prüfchemikalien (aufgrund von Farbstoffen oder durch direkte MTT-Reduktionsmittel) nur dann auf, wenn nach dem Waschen im Anschluss an die Behandlung eine Prüfchemikalie noch in erheblichem Umfang im Prüfsystem enthalten ist. Dies ist etwa bei rekonstruiertem humanem 3D-Hornhaut- oder Epidermisgewebe der Fall. Für die bei der Kurzzeit-Prüfmethode verwendeten 2D-Zellkulturen sind diese Interferenzen jedoch nicht von Bedeutung.
|
Auswertung der Ergebnisse und Prädiktionsmodell
|
26.
|
Die für jede Prüfchemikalie ermittelten OD-Werte (OD = optische Dichte) werden anschließend zur Berechnung der prozentualen Viabilität im Vergleich zur Lösungsmittelkontrolle herangezogen, die gleich 100 % gesetzt wird. Die relative Zellviabilität wird als Prozentanteil ausgedrückt. Dieser Prozentanteil wird durch Division der OD der Prüfchemikalie durch die OD der Lösungsmittelkontrolle ermittelt, nachdem von beiden Werten jeweils die OD der Blindkontrolle abgezogen wurde.
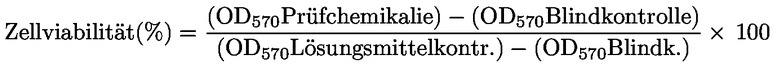
Ähnlich wird die relative Zellviabilität der einzelnen Lösungsmittelkontrollen als Prozentanteil ausgedrückt. Dieser Prozentanteil wird durch Division der OD der einzelnen Lösungsmittelkontrollen durch die OD der einzelnen Mediumkontrollen ermittelt, nachdem von beiden Werten jeweils die OD der Blindkontrolle abgezogen wurde.
|
|
27.
|
Bei der Prüfung sollten drei unabhängige Wiederholungen jeweils mit drei Replikat-Wells (d. h. n = 9) durchgeführt werden. Das arithmetische Mittel der relativen Zellviabilität wird anhand des arithmetischen Mittels der drei Wells jeder einzelnen Prüfchemikalie und jeder einzelnen Lösungsmittelkontrolle der einzelnen unabhängigen Wiederholungen ermittelt. Das endgültige arithmetische Mittel der Zellviabilität wird anhand der drei unabhängigen Wiederholungen berechnet.
|
|
28.
|
Die Schwellenwerte der Zellviabilität zur Identifizierung von schwer augenschädigenden Chemikalien (UN-GHS-/CLP-Kategorie 1) und von Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern (keine Einstufung nach UN GHS/CLP), sind nachfolgend aufgeführt:
|
Tabelle 2
Vorhersagemodell der Kurzzeit-Prüfmethode
|
Zellviabilität
|
UN-GHS-/CLP-Kategorie
|
Anwendbarkeit
|
|
5 %
|
0,05 %
|
|
> 70 %
|
> 70 %
|
Keine Einstufung
|
Stoffe und Gemische außer i) stark flüchtigen Stoffen mit Dampfdrücken über 6 kPa (75) und ii) festen Chemikalien (Stoffen und Gemischen) mit Ausnahme von Tensiden und Gemischen, die ausschließlich Tenside enthalten
|
|
> 70 %
|
> 70 %
|
Keine Vorhersage möglich
|
Nicht anwendbar
|
|
> 70 %
|
> 70 %
|
Kategorie 1
|
Stoffe und Gemische (76)
|
Akzeptanzkriterien
|
29.
|
Die Prüfergebnisse sind annehmbar, wenn die folgenden Kriterien vollständig erfüllt sind:
|
a)
|
Die optische Dichte der (mit dem Kulturmedium behandelten) Mediumkontrolle nach Abzug der optischen Dichte der Blindkontrolle beträgt mindestens 0,3.
|
|
b)
|
Die Viabilität der Lösungsmittelkontrolle liegt bei mindestens 80 % der Viabilität der Mediumkontrolle. Wenn bei den Wiederholungen jeweils mehrere Lösungsmittelkontrollen verwendet werden, sind die Prüfchemikalien zur Untersuchung mit den betreffenden Lösungsmitteln nur dann geeignet, wenn die Zellviabilität der Lösungsmittelkontrollen jeweils mehr als 80 % beträgt.
|
|
c)
|
Die Zellviabilität der Positivkontrolle (0,01 % SLS) liegt im Bereich von zwei Standardabweichungen vom historischen Mittelwert. Die obere und die untere Akzeptanzgrenze der Positivkontrolle werden häufig aktualisiert (d. h. alle drei Monate bzw. in Labors, in denen die Prüfungen seltener durchgeführt werden (weniger als einmal pro Monat), immer dann, wenn eine akzeptable Prüfung durchgeführt wurde). Wenn Versuche in einem Labor nicht in hinreichender Anzahl durchgeführt werden, um eine statistisch robuste Verteilung der Positivkontrollen zu ermitteln, kann unter Berücksichtigung der vom Entwickler der Prüfmethode festgelegten oberen und unteren Grenzwerte (d. h. 21,1 % und 62,3 %, je nach den historischen Daten des betreffenden Labors) nach den ersten routinemäßigen Prüfungen eine interne Verteilung ermittelt werden.
|
|
d)
|
Die Standardabweichung der aus drei unabhängigen Wiederholungen ermittelten endgültigen Zellviabilität beträgt bei beiden Konzentrationen der Prüfchemikalie (5 % und 0,05 %) weniger als 15 %.
Wenn mindestens eines dieser Kriterien nicht erfüllt ist, sollten die Ergebnisse nicht berücksichtigt werden. In diesem Fall sind drei weitere unabhängige Wiederholungen durchzuführen.
|
|
DATEN UND BERICHTERSTATTUNG
Daten
|
30.
|
Die Daten der einzelnen Wells (z. B. die Werte der Zellviabilität) der jeweiligen Wiederholungen sowie der Gesamtmittelwert, die Standardabweichung und die Einstufung sind anzugeben.
|
Prüfbericht
|
31.
|
Der Prüfbericht sollte folgende Angaben enthalten:
|
|
Prüfchemikalien und Kontrollstoffe
|
—
|
Einkomponentige Stoffe: Chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
—
|
mehrkomponentige Stoffe, UVCB-Stoffe und Gemische: Charakterisierung, so weit wie möglich, z. B. durch die chemische Zusammensetzung (siehe oben), Reinheit, das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften (siehe oben) der einzelnen Elemente, soweit verfügbar;
|
|
—
|
Aggregatzustand, Flüchtigkeit, pH-Wert, log P, Molekulargewicht, Chemikalienklasse und weitere für die Untersuchung relevante physikalische und chemische Eigenschaften, soweit verfügbar;
|
|
—
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
—
|
Behandlung vor der Untersuchung, sofern relevant (z. B. Erwärmen, Mahlen);
|
|
—
|
Lagerbedingungen und Stabilität, soweit verfügbar.
|
|
|
|
Prüfbedingungen und -verfahren
|
—
|
Name und Anschrift des Auftraggebers, der Prüfanstalt und des Studienleiters;
|
|
—
|
Identifikation der verwendeten Prüfmethode;
|
|
—
|
verwendete Zelllinie, Herkunft, Passagenzahl und Konfluenz der für die Untersuchung verwendeten Zellen;
|
|
—
|
Details des angewandten Prüfverfahrens;
|
|
—
|
Anzahl der Wiederholungen und verwendeten Replikate
|
|
—
|
Konzentrationen der verwendeten Prüfchemikalie (falls von den Empfehlungen abweichend);
|
|
—
|
Begründung der Auswahl des Lösungsmittels für jede Prüfchemikalie;
|
|
—
|
Dauer der Exposition gegenüber der Prüfchemikalie (falls von den Empfehlungen abweichend);
|
|
—
|
Beschreibung etwaiger Änderungen am Prüfverfahren;
|
|
—
|
Beschreibung der angewandten Bewertungs- und Entscheidungskriterien;
|
|
—
|
Angabe des historischen Mittelwerts der Kontrollgruppe und der Standardabweichung (SD);
|
|
—
|
Nachweis der Leistungsfähigkeit des Labors hinsichtlich der Durchführung der Prüfmethode (z. B. durch Prüfen von Leistungsstoffen) oder zum Nachweis der reproduzierbaren Durchführung der Prüfmethode im Zeitverlauf.
|
|
|
|
Ergebnisse
|
—
|
Für alle Prüfchemikalien und Kontrollstoffe sowie für jede geprüfte Konzentration sind die einzelnen OD-Werte pro Replikat-Well, das arithmetische Mittel der OD-Werte der einzelnen unabhängigen Wiederholungen, die Zellviabilität der einzelnen unabhängigen Wiederholungen in % und das endgültige arithmetische Mittel der Zellviabilität in % sowie die Standardabweichung bei drei Wiederholung in einer Tabelle anzugeben;
|
|
—
|
Ergebnisse der Medium-, der Lösungsmiottel- und der Positivkontrolle als Nachweis für die Erfüllung geeigneter Akzeptanzkriterien;
|
|
—
|
Beschreibung anderer beobachteter Wirkungen;
|
|
—
|
insgesamt abgeleitete Einstufung mit Bezug auf das Vorhersagemodell und/oder angewandte Entscheidungskriterien.
|
|
|
|
Erörterung der Ergebnisse.
|
|
LITERATUR
|
(1)
|
Vereinte Nationen (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals = Global harmonisiertes System zur Einstufung und Kennzeichnung) (GHS): fünfte überarbeitete Auflage, New York und Genf: United Nations Publications. ISBN: 978-92-1-117006-1. Abrufbar unter:http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott, L, u. a. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi, Y., u. a. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi, H., u. a. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima, H., u. a. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, S. 1 855-1 869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Abrufbar unter:[http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Kapitel B.5 dieses Anhangs, Akute Augenreizung/-verätzung.
|
|
(9)
|
Saito, K, u. a. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson, T.J., Chrai, S.S., und Robinson. J.R. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1 648-1 653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brüssel, Belgien.
|
|
(12)
|
Gautheron, P., u. a. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam. Appl. Toxicol. 18, 442–449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
Anlage
BEGRIFFSBESTIMMUNGEN
Augenreizung
: Erzeugen von Veränderungen am Auge nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation vollständig reversibel sind. Synonym zu den Einstufungen „reversible Wirkungen am Auge“ und „UN-GHS-/CLP-Kategorie 2“.
Bottom-up-Ansatz
: Schrittweiser Ansatz für eine Prüfchemikalie, von der vermutet wird, dass sie keine Einstufung als augenreizend oder schwer augenschädigend erfordert. Dabei werden zunächst Chemikalien, die keine Einstufung erfordern (negatives Ergebnis), von anderen Chemikalien (positives Ergebnis) unterschieden.
Chemikalie
: Ein Stoff oder Gemisch.
Einkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorliegt.
Falsch-Negativ-Rate
: Der Anteil aller positiven Chemikalien, die mit einer Prüfmethode fälschlicherweise als negativ identifiziert werden. Die Falsch-Negativ-Rate ist ein Indikator für die Leistungsfähigkeit einer Prüfmethode.
Falsch-Positiv-Rate
: Der Anteil aller negativen Chemikalien, die mit einer Prüfmethode fälschlicherweise als positiv identifiziert werden. Die Falsch-Positiv-Rate ist ein Indikator für die Leistungsfähigkeit einer Prüfmethode.
Gefahr
: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen.
Gemisch
: Ein Gemisch oder eine Lösung, die aus zwei oder mehr Stoffen besteht.
Genauigkeit
: Der Grad an Übereinstimmung zwischen Testergebnissen und akzeptierten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von „Übereinstimmung“ verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (13).
Gestufte Prüfstrategie
: Eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Bewertungsansatz (weight-of-evidence) vorgegangen wird, um feststellen zu können, ob genügend Informationen für eine Gefahreneinstufung vorliegen, bevor zur nächsten Stufe übergegangen wird. Wenn das Reizpotenzial einer Prüfchemikalie auf Basis der vorliegenden Informationen ermittelt werden kann, sind keine weiteren Untersuchungen erforderlich. Ist dies nicht der Fall, müssen schrittweise sequenzielle Tierversuche durchgeführt werden, bis eine eindeutige Einstufung vorgenommen werden kann.
Lösungsmittel-/Vehikelkontrolle
: Eine unbehandelte Probe, die alle Elemente eines Prüfsystems enthält, einschließlich des mit der Prüfchemikalie behandelten Lösungsmittels oder Vehikels und anderer Kontrollproben, und die verwendet wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben zu bestimmen, die im selben Lösungsmittel oder Vehikel aufgelöst wurden. Bei der Prüfung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Prüfsystem interagiert.
Mediumkontrolle
: Ein unbehandeltes Replikat, das alle Elemente eines Prüfsystems enthält. Diese Probe wird mit einer Prüfchemikalie behandelten Proben und anderen Kontrollproben mitgeführt, um festzustellen, ob das Lösungsmittel mit dem Prüfsystem interagiert.
Mehrkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens ≥ 10 % w/w und < 80 % w/w vorhanden ist. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet.
MTT
: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid; Thiazolyl-Blau-Tetrazoliumbromid.
OD
: Optische Dichte.
Positivkontrolle
: Ein Replikat, das alle Elemente eines Prüfsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prüfchemikalie
: Stoff oder Gemisch, der/das mit dieser Prüfmethode untersucht wird.
Referenzstoff
: Ein zum Vergleich mit einer Prüfchemikalie verwendeter Stoff. Referenzstoffe sollten die folgenden Eigenschaften haben: (i) einheitliche und zuverlässige Herkunft; (ii) strukturelle und funktionelle Ähnlichkeit mit der Klasse der zu prüfenden Stoffe; (iii) bekannte physikalisch-chemische Eigenschaften; (iv) unterstützende Daten zu bekannten Wirkungen und (v) bekannte Wirksamkeit im Bereich der erwünschten Reaktion.
Relevanz
: Dieser Begriff bezieht sich auf das Verhältnis zwischen der Prüfmethode und der betreffenden Wirkung und auf die Frage, ob er aussagekräftig und nützlich für einen bestimmten Zweck ist. Er beschreibt das Ausmaß, in dem mit einer Prüfung die untersuchte biologische Wirkung korrekt gemessen oder vorhersagt wird. Die Relevanz schließt eine Beurteilung der Genauigkeit (Übereinstimmung) einer Prüfmethode ein (10).
Schwere Augenschädigung
: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Synonym zu den Einstufungen „irreversible Wirkungen am Auge“ und „UN-GHS-/CLP-Kategorie 1“.
Sensitivität
: Der Anteil aller positiven/wirkenden Chemikalien, die durch die Prüfung korrekt eingestuft werden. Die Sensitivität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (10).
Spezifität
: Der Anteil aller negativen/wirkungslosen Chemikalien, die durch die Prüfung korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (13).
Stoff
: Ein chemisches Element und seine Verbindungen in natürlicher Form oder gewonnen durch ein Herstellungsverfahren, einschließlich der zur Wahrung seiner Stabilität notwendigen Zusatzstoffe und der durch das angewandte Verfahren bedingten Verunreinigungen, aber mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können.
Tensid
: Auch als oberflächenaktiver Stoff bezeichnet; Stoffe, wie waschaktive Stoffe (Detergenzien), die die Oberflächenspannung einer Flüssigkeit herabsetzen und so die Bildung von Schaum oder das Eindringen in feste Stoffe ermöglichen; auch bekannt als Netzmittel.
Top-down-Ansatz
: Schrittweiser Ansatz bei einer Chemikalie, von der vermutet wird, dass sie schwere Augenschäden verursacht. Dabei werden zunächst Chemikalien, die schwere Augenschäden verursachen (positives Ergebnis), von anderen Chemikalien (negatives Ergebnis) unterschieden.
UN GHS (United Nations Globally Harmonized System of Classification and Labelling of Chemicals = Global harmonisiertes System zur Einstufung und Kennzeichnung der Vereinten Nationen)
: Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenbögen, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1).
UN-GHS/CLP-Kategorie 1
: Siehe „Schwere Augenschädigung“.
UN-GHS/CLP-Kategorie 2
: Siehe „Augenreizung“.
UN-GHS-/CLP-Kategorie Keine Einstufung
: Chemikalien, die nicht in die UN-GHS-/CLP-Kategorien 1 oder 2 (oder die UN-GHS-Kategorien 2A oder 2B) eingestuft wurden.
UVCB
: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Zuverlässigkeit
: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit bewertet (13).
B.69 PRÜFMETHODE UNTER VERWENDUNG VON REKONSTRUIERTEM MENSCHLICHEM HORNHAUTARTIGEM EPITHEL (RhCE) ZUR ERMITTLUNG VON CHEMIKALIEN, BEI DENEN EINE EINSTUFUNG UND KENNZEICHNUNG ALS AUGENREIZEND ODER SCHWER AUGENSCHÄDIGEND NICHT ERFORDERLICH IST
EINLEITUNG
|
1.
|
Diese Prüfmethode (PM) entspricht der OECD-Prüfrichtlinie (TG) 492 (2017). Als schwere Augenschädigung werden nach dem Global harmonisierten System (UN GHS) zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (1) und nach Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP-Verordnung) (77) das Erzeugen von Gewebeschäden im Auge oder schwerwiegende Verschlechterungen des Sehvermögens nach Applikation eines Prüfstoffes auf die Oberfläche des Auges bezeichnet, die innerhalb von 21 Tagen nach Applikation nicht vollständig reversibel sind. Ebenfalls nach dem UN GHS und der CLP-Verordnung bezeichnet Augenreizung das Erzeugen von Veränderungen am Auge nach Applikation eines Prüfstoffes auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation vollständig reversibel sind. Prüfchemikalien, die schwere Augenschädigungen auslösen, werden als UN-GHS-/CLP-Kategorie 1 eingestuft. Chemikalien die Augenreizungen hervorrufen, werden als UN-GHS-/CLP-Kategorie 2 eingestuft. Prüfchemikalien, die nicht als augenreizende oder schwer augenschädigende Stoffe eingestuft werden, sind als Stoffe definiert, bei denen die Kriterien für eine Einstufung in die UN-GHS-/CLP-Kategorien 1 oder 2 (2A oder 2B) nicht erfüllt sind, d. h. sie werden als Stoffe der UN-GHS-/CLP-Kategorie Keine Einstufung bezeichnet.
|
|
2.
|
Zur Beurteilung von schweren Augenschädigungen bzw. Augenreizungen wurden bislang in der Regel Labortiere verwendet (PM B.5 (2)). Die Auswahl der am besten geeigneten Prüfmethode und die Anwendung dieser Prüfmethode sollten unter Berücksichtigung des OECD-Leitliniendokuments über integrierte Prüfungs- und Bewertungsansätze (IATA = Integrated Approaches to Testing and Assessment) zur Untersuchung von schweren Augenschädigungen und von Augenreizungen (39) erfolgen.
|
|
3.
|
Die Prüfmethode beschreibt ein In-vitro-Verfahren zur Ermittlung von Chemikalien (Stoffen und Gemischen), die keine Einstufung und Kennzeichnung als augenreizend oder schwer augenschädigend nach dem UN GHS und der CLP-Verordnung erfordern. Für diese Prüfmethode wird rekonstruiertes menschliches hornhautartiges Epithel (RhCE) verwendet, das die histologischen, morphologischen, biochemischen und physiologischen Eigenschaften des menschlichen Hornhautepithels weitgehend widerspiegelt. Vier weitere In-vitro-Prüfmethoden wurden validiert, als wissenschaftlich fundiert bewertet und als PM B.47 (3), B.48 (4), B.61 (5) und B.68 (6) zur Bewertung der Endpunkte für schwere Augenschädigungen bzw. für Augenreizungen beim Menschen angenommen.
|
|
4.
|
Bei dieser Prüfmethode kommen zwei validierte im Handel erhältliche RhCE-Modelle zum Einsatz. Zur Beurteilung von Augenreizungen / schweren Augenschädigungen anhand des EpiOcular™ Eye Irritation Test (EIT) und des SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT) wurden Validierungsstudien durchgeführt (7) (8) (9) (10) (11) (12) (13). Bei diesen Prüfungen wurden jeweils im Handel erhältliche RhCE-Gewebemodelle als Prüfsystem verwendet. Diese Prüfungen werden im Folgenden die validierten Referenzmethoden (VRM 1 bzw. VRM 2) genannt. Aufgrund dieser Validierungsstudien und nach dem unabhängigen Peer-Review dieser Studien (9) (12) wurde festgestellt, dass Chemikalien (Stoffe und Gemische), die keine Einstufung und Kennzeichnung als augenreizend bzw. schwer augenschädigend nach dem UN GHS erfordern, mit dem EpiOcular™ EIT und dem SkinEthic™ HCE EIT korrekt ermittelt werden. Daher wurden diese Prüfungen für diesen Zweck als wissenschaftlich fundiert empfohlen (13).
|
|
5.
|
Gegenwärtig herrscht allgemeiner Konsens darüber, dass der In-vivo-Draize-Augentest in absehbarer Zukunft nicht in vollem Umfang durch eine einzelne In-vitro-Prüfmethode ersetzt werden kann (2) (14), die geeignet wäre, das gesamte Spektrum augenreizender bzw. schwer augenschädigender Reaktionen auf unterschiedliche Chemikalienklassen vorherzusagen. Unter Umständen ist es jedoch möglich, den Draize-Augentest durch strategische Kombinationen mehrerer alternativer Prüfmethoden im Rahmen (gestufter) Prüfstrategien (z. B. des Bottom-up- oder des Top-down-Ansatzes) vollständig zu ersetzen (15). Der Bottom-up-Ansatz (15) wird verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie Augenreizungen nicht in einem Umfang auslöst, der eine Einstufung erfordern würde. Umgekehrt wird der Top-down-Ansatz (15) verwendet, wenn nach den vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie schwere Augenschädigungen hervorruft. Der EpiOcular™ EIT und der SkinEthic™ HCE EIT werden empfohlen, um im Rahmen einer Prüfstrategie (z. B. des von Scott u. a. empfohlenen Bottom-up- oder Top-down-Ansatzes) etwa in einem ersten Schritt bei einem Bottom-up-Ansatz oder in einem der letzten Schritte bei einem Top-down-Ansatz ohne weitere Prüfung Chemikalien zu ermitteln, bei denen eine Einstufung als augenreizend oder schwer augenschädigend nach dem UN GHS bzw. der CLP-Verordnung nicht erforderlich ist (Keine Einstufung) (15). Zur Unterscheidung zwischen der UN-GHS-/CLP-Kategorie 1 (schwere Augenschädigung) und der UN-GHS-/CLP-Kategorie 2 (Augenreizung) sind der EpiOcular™ EIT und der SkinEthic™ HCE EIT jedoch nicht vorgesehen. Diese Unterscheidung muss in einem weiteren Schritt einer Prüfstrategie vorgenommen werden (15). Eine Prüfchemikalie, bei der die Durchführung des EpiOcular™ EIT oder SkinEthic™ HCE EIT zur Einstufung als augenreizend oder schwer augenschädigend als erforderlich betrachtet wird, muss daher einer weiteren Untersuchung (in vitro und/oder in vivo) unterzogen werden (z. B. durch die PM B.47, B.48, B.61 oder B.68), um zu einem endgültigen Ergebnis (UN-GHS-/CLP-Kategorie Keine Einstufung, UN-GHS-/CLP-Kategorie 2 oder UN-GHS-/CLP-Kategorie 1) zu gelangen.
|
|
6.
|
Diese Prüfmethode beschreibt das Verfahren zur Beurteilung des augengefährdenden Potenzials einer Prüfchemikalie anhand ihrer mit der MTT-Prüfung (16) (Nummer 21) gemessenen Fähigkeit, bei einem RhCE-Gewebemodell eine zytotoxische Wirkung hervorzurufen. Die Viabilität des RhCE-Gewebes nach Behandlung mit einer Prüfchemikalie wird im Vergleich zu Geweben ermittelt, die mit dem Negativkontrollstoff behandelt wurde (Viabilität in %). Nach dieser Viabilität wird das Augengefährdungspotenzial der Prüfchemikalie angegeben.
|
|
7.
|
Verfügbare Leistungsstandards (17) erleichtern die Validierung neuer oder modifizierter In-vitro-Prüfungen unter Verwendung von RhCE ähnlich dem EpiOcular™ EIT und dem SkinEthic™ HCE EIT nach den Grundsätzen von OECD Guidance Document Nr. 34 (18) und ermöglichen eine frühzeitige Änderung der OECD TG 492 zur entsprechenden Berücksichtigung. Die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen wird nur für Prüfungen garantiert, die gemäß diesen Leistungsnormen validiert wurden, sofern diese Prüfungen von der OECD überprüft und in die entsprechende Prüfrichtlinie aufgenommen wurden.
|
BEGRIFFSBESTIMMUNGEN
|
8.
|
Es gelten die Begriffsbestimmungen in Anlage 1.
|
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
|
9.
|
Diese Prüfmethode beruht auf im Handel erhältlichen dreidimensionalen RhCE-Gewebemodellen, die entweder mit primären humanen epidermalen Keratinozyten (d. h. EpiOcular™ OCL-200) oder mit humanen immortalisierten Hornhaut-Epithelzellen (d. h. SkinEthic™ HCE/S) hergestellt wurden. Die RhCE-Gewebemodelle EpiOcular™ OCL-200 und SkinEthic™ HCE/S spiegeln das dreidimensionale In-vivo-Hornhaut-Epithelgewebe wider und werden aus Zellen der zu untersuchenden Spezies hergestellt (19) (20). Mit den Prüfungen werden die Zytotoxizität infolge der Durchdringung der Hornhaut durch die Chemikalien und die Induzierung von Zell- und Gewebeschäden direkt gemessen. Die gemessene zytotoxische Reaktion gibt dann Aufschluss über den Gesamtumfang der in vivo auftretenden Augenreizungen bzw. schweren Augenschädigungen. Zellschädigungen können aufgrund unterschiedlicher Wirkungsweisen auftreten (Nummer 20). Der Zytotoxizität kommt jedoch eine wichtige, wenn nicht gar die entscheidende, grundlegende Bedeutung bei der Ermittlung des Gesamtumfangs der schweren Augenschädigungen bzw. der Augenreizungen aufgrund der Wirkung einer Chemikalie zu, die sich unabhängig von den den Gewebeschädigungen zugrunde liegenden physikalisch-chemischen Prozessen in vivo in einer Hornhauttrübung, einer Iritis, einer Bindehautrötung und/oder einem Bindehautödem (Chemose) äußert.
|
|
10.
|
In der dieser Prüfmethode zugrunde liegenden Validierungsstudie wurden vielfältige Chemikalien sowie zahlreiche unterschiedliche Chemikalientypen, Chemikalienklassen, Molekulargewichte, log-P-Werte, chemische Strukturen usw. berücksichtigt. Die Validierungsdatenbank für den EpiOcular™ EIT enthielt insgesamt 113 Chemikalien, darunter 95 verschiedene organische Funktionsgruppen entsprechend einer Analyse mit der QSAR-Toolbox der OECD (8). Bei diesen Chemikalien handelte es sich meist um einkomponentige Stoffe; in der Studie wurden aber auch mehrere mehrkomponentige Stoffe berücksichtigt (darunter 3 Homopolymere, 5 Copolymere und 10 Quasipolymere). Hinsichtlich des Aggregatzustands und der UN-GHS-/CLP-Kategorien verteilten sich die 113 Prüfchemikalien wie folgt: 13 Flüssigkeiten der Kategorie 1, 15 Feststoffe der Kategorie 1, 6 Flüssigkeiten der Kategorie 2A, 10 Feststoffe der Kategorie 2A, 7 Flüssigkeiten der Kategorie 2B, 7 Feststoffe der Kategorie 2B, 27 Flüssigkeiten der Kategorie Keine Einstufung und 28 Feststoffe der Kategorie Keine Einstufung (8). Die Validierungsdatenbank für den SkinEthic™ HCE EIT enthielt insgesamt 200 Chemikalien, darunter 165 verschiedene organische Funktionsgruppen (8) (10) (11). Bei diesen Chemikalien handelte es sich meist um einkomponentige Stoffe; in der Studie wurden aber auch mehrere mehrkomponentige Stoffe berücksichtigt (darunter 10 Polymere). Hinsichtlich des Aggregatzustands und der UN-GHS-/CLP-Kategorien verteilten sich die 200 Prüfchemikalien wie folgt: 27 Flüssigkeiten der Kategorie 1, 24 Feststoffe der Kategorie 1, 19 Flüssigkeiten der Kategorie 2A, 10 Feststoffe der Kategorie 2A, 9 Flüssigkeiten der 2B, 8 Feststoffe der Kategorie 2B, 50 Flüssigkeiten der Kategorie Keine Einstufung und 53 Feststoffe der Kategorie Keine Einstufung (10) (11).
|
|
11.
|
Diese Prüfmethode ist bei Stoffen und Gemischen sowie bei Feststoffen, Flüssigkeiten, halbfesten Stoffen und Wachsen anwendbar. Flüssigkeiten können wässrig oder nicht wässrig sein, Feststoffe in Wasser löslich oder unlöslich. Sofern möglich, sollten Feststoffe vor der Applikation zu Feinpulver gemahlen werden; eine weitere Behandlung der Probe ist nicht nötig. In den Validierungsstudien wurden keine Gase und Aerosole bewertet. Obwohl deren Prüfung mit der RhCE-Technologie prinzipiell vorstellbar ist, erlaubt die vorliegende Prüfmethode das Untersuchen von Gasen und Aerosolen nicht.
|
|
12.
|
Prüfchemikalien, die Licht im selben Spektrum absorbieren können wie MTT-Formazan (natürlich oder nach einer Behandlung), und Prüfchemikalien, die in der Lage sind, den lebenswichtigen Farbstoff MTT zu reduzieren (zu MTT-Formazan), können die Messungen der Gewebeviabilität stören und erfordern die Verwendung geeigneter Kontrollen zur Korrektur. Die Art der möglicherweise erforderlichen geeigneten Kontrollen hängt von der Art der durch die Prüfchemikalie ausgelösten Interferenzen und vom Verfahren zur Quantifizierung von MTT-Formazan ab (Nummern 36-42).
|
|
13.
|
Die Ergebnisse von Vorvalidierungsstudien (21) (22) und von umfassenden Validierungsstudien (8) (10) (11) haben gezeigt, dass sowohl der EpiOcular™ EIT als auch der SkinEthic™ HCE EIT auch in Labors durchgeführt werden können, die noch nicht über Erfahrungen mit der Durchführung dieser Assays verfügen, und dass die Ergebnisse sowohl innerhalb der Labors als auch zwischen den Labors reproduzierbar sind. Diesen Studien zufolge liegt die zu erwartende Reproduzierbarkeit hinsichtlich der Übereinstimmung der Vorhersagen beim EpiOcular™ EIT mit Daten zu 113 Chemikalien bei 95 % (innerhalb von Labors) bzw. 93 % (zwischen Labors). Die zu erwartende Reproduzierbarkeit hinsichtlich der Übereinstimmung der Vorhersagen beim SkinEthic™ HCE EIT mit Daten zu 120 Chemikalien beträgt 92 % (innerhalb von Labors) bzw. 95 % (zwischen Labors).
|
|
14.
|
Der EpiOcular™ EIT kann auch zur Identifizierung von Chemikalien angewendet werden, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß dem UN GHS und der CLP-Verordnung erfordern. Aufgrund der in der Validierungsstudie ermittelten Daten (8) ergeben sich für den EpiOcular™ EIT eine Gesamtgenauigkeit von 80 % (bei 112 Chemikalien), eine Sensitivität von 96 % (bei 57 Chemikalien), eine Falsch-negativ-Rate von 4 % (bei 57 Chemikalien), eine Spezifität von 63 % (bei 55 Chemikalien) und eine Falsch-negativ-Rate von 37 % (bei 55 Chemikalien) im Vergleich zu den Daten des In-vivo-Kaninchenaugentests (PM B.5) (2) (14) bei Einstufung nach dem UN GHS und der CLP-Verordnung. In einer Studie, in der 97 flüssige agrochemische Zubereitungen mit dem EpiOcular™ EIT geprüft wurden, wurde für diese Art von Gemischen eine ähnliche Leistungsfähigkeit der Prüfmethode festgestellt wie in der Validierungsstudie (23). Die 97 Zubereitungen verteilten sich wie folgt: 21 Kategorie 1, 19 Kategorie 2A, 14 Kategorie 2B und 43 Keine Einstufung bei Einstufung nach dem UN GHS bezogen auf Referenzdaten des In-vivo-Kaninchenaugentests (PM B.5) (2) (14). Folgende Werte wurden ermittelt: Gesamtgenauigkeit 82 % (97 Zubereitungen), Sensitivität 91 % (54 Zubereitungen), Falsch-negativ-Rate 9 % (54 Zubereitungen), Spezifität 72 % (43 Zubereitungen) und Falsch-positiv-Rate 28 % (43 Zubereitungen) (23).
|
|
15.
|
Der SkinEthic™ HCE EIT kann auch zur Identifizierung von Chemikalien angewendet werden, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß dem UN GHS und der CLP-Verordnung erfordern. Aufgrund der in der Validierungsstudie ermittelten Daten (10) (11) ergeben sich für den SkinEthic™ HCE EIT eine Gesamtgenauigkeit von 84 % (bei 200 Chemikalien), eine Sensitivität von 95 % (bei 97 Chemikalien), eine Falsch-negativ-Rate von 5 % (bei 97 Chemikalien), eine Spezifität von 72 % (bei 103 Chemikalien) und eine Falsch-negativ-Rate von 28 % (bei 103 Chemikalien) im Vergleich zu den Daten des In-vivo-Kaninchenaugentests (PM B.5) (2) (14) bei Einstufung nach dem UN GHS und der CLP-Verordnung.
|
|
16.
|
Die mit beiden RhCE-Prüfungen mit Stoffen oder Gemischen ermittelten Falsch-negativ-Raten liegen bei 12 % der Gesamtwahrscheinlichkeit, dass Chemikalien im In-vivo-Draize-Augentest bei wiederholten Prüfungen entweder als UN-GHS-/CLP-Kategorie 2 oder als UN-GHS-/CLP-Kategorie Keine Einstufung ermittelt werden; dies ist auf die prüfungsimmanente Variabilität der Methode zurückzuführen (24). Die mit beiden RhCE-Prüfmethoden sowohl bei Stoffen als auch bei Gemischen ermittelten Falsch-positiv-Raten sind bei dieser Prüfmethode nicht kritisch, da alle Prüfchemikalien, für die sich eine Gewebeviabilität von kleiner oder gleich den festgelegten Grenzwerten ergibt (Nummer 44), je nach den rechtlichen Anforderungen im Rahmen einer sequenziellen Prüfstrategie nach einem evicdenzbasierten Ansatz weiteren In-vitro-Prüfmethoden bzw. als letzte Möglichkeit Kaninchentests unterzogen werden müssen. Diese Prüfmethoden können für alle Arten von Chemikalien eingesetzt werden, wobei ein Negativergebnis als Indikator dafür akzeptiert werden sollte, dass die betreffenden Chemikalien nicht als augenreizend oder schwer augenschädigend einzustufen sind (UN-GHS-/CLP-Kategorie Keine Einstufung). Außerdem sollten die zuständigen Regulierungsbehörden konsultiert werden, bevor die Kurzzeitprüfung mit dem EpiOcular™ EIT und dem SkinEthic™ HCE EIT unter Berücksichtigung einer anderen Klassifizierungsregelung als der des UN GHS bzw. der CLP-Verordnung durchgeführt wird.
|
|
17.
|
Eine Einschränkung besteht bei dieser Prüfmethode darin, dass nicht zwischen Augenreizungen / reversiblen Wirkungen am Auge (Kategorie 2) und schweren Augenschädigungen / irreversiblen Wirkungen am Auge (Kategorie 1) im Sinne des UN GHS und der CLP-Verordnung und nicht zwischen einer augenreizenden Wirkung (optionale Kategorie 2A) und einer schwach augenreizenden Wirkung (optionale Kategorie 2B) im Sinne des UN GHS unterschieden werden kann (1). Um diese Unterscheidungen vornehmen zu können, müssen weitere Untersuchungen mit anderen In-vitro-Prüfmethoden vorgenommen werden.
|
|
18.
|
Der Begriff „Prüfchemikalie“ bezeichnet bei dieser Prüfmethode das, was geprüft wird, (78) und bezieht sich nicht auf die Anwendbarkeit der RhCE-Prüfmethode zur Prüfung von Stoffen und/oder Gemischen.
|
PRINZIP DER PRÜFMETHODE
|
19.
|
Die Prüfchemikalie wird oberflächlich auf zwei dreidimensionale RhE-Modelle appliziert. Nach der Exposition und der Inkubation im Anschluss an die Behandlung wird die Gewebeviabilität gemessen. Die RhCE-Gewebe werden aus primären humanen epidermalen Keratinozyten oder humanen immortalisierten Hornhaut-Epithelzellen rekonstruiert, die mehrere Tage bis zur Bildung eines geschichteten Plattenepithels mit ausgeprägter Differenzierung kultiviert wurden, das das menschliche Hornhaut-Epithel widerspiegelt. Das RhCE-Gewebemodell des EpiOcular™ EIT besteht aus mindestens drei lebensfähigen Zellschichten mit nicht verhornter Oberfläche mit hornhautartiger Struktur entsprechend der In-vivo-Struktur. Das RhCE-Gewebemodell des SkinEthic™ HCE EIT umfasst mindestens vier lebensfähige Zellschichten mit säulenartigen Basalzellen, Flügelzellen (Übergangszellen) und oberflächlichen Plattenepithelzellen ähnlich dem normalen menschlichen Hornhaut-Epithel (20) (26).
|
|
20.
|
Chemisch induzierte Augenreizungen bzw. schwere Augenschädigungen, die sich in vivo vorwiegend in einer Hornhauttrübung, Irititis, Bindehautrötung und/oder einem Bindehautödem (Chemose) äußern, sind auf eine Kaskade von Ereignissen zurückzuführen, an deren Beginn das Eindringen der Chemikalien in die Hornhaut und/oder die Bindehaut und die Schädigung von Zellen stehen. Zellschädigungen können aufgrund unterschiedlicher Wirkungsweisen entstehen, darunter eine Lyse der Zellmembran (z. B. durch Tenside oder organische Lösungsmittel), die Koagulation von Makromolekülen (insbesondere Proteinen) (z. B. durch Tenside, organische Lösungsmittel, Laugen und Säuren), die Verseifung von Lipiden (z. B. durch Laugen) und Alkylierungen oder sonstige kovalente Wechselwirkungen mit Makromolekülen (z. B. durch Bleichmittel, Peroxide und Alkylierungsmittel) (15) (27) (28). Allerdings wurde nicht nachgewiesen, dass die Zytotoxizität unabhängig von den der Gewebeschädigung zugrunde liegenden physikalisch-chemischen Prozessen eine wichtige, wenn nicht die primäre, ursächliche Rolle bei der Ermittlung der Gesamtreaktion im Hinblick auf Augenreizungen bzw. schwere Augenschädigungen durch eine Chemikalie spielt (29) (30). Das Potenzial einer Chemikalie, eine Augenreizung oder eine schwere Augenschädigung zu verursachen, hängt vom Umfang der Ausgangsverletzung (31) ab, der mit dem Umfang, in dem Zellen absterben, (29) und mit dem Umfang der anschließenden Reaktionen und der endgültigen Ergebnisse (32) korreliert. Leichte Reizungen betreffen daher gewöhnlich nur das oberflächliche Hornhaut-Epithel, geringe und mäßige Reizungen schädigen hauptsächlich das Epithel und das oberflächliche Stroma, und schwere Reizungen führen zu Schädigungen des Epithels, des tiefen Stromas und gelegentlich des Hornhaut-Endothels (30) (33). Die Messung der Viabilität des RhCE-Gewebemodells nach der oberflächlichen Applikation einer Prüfchemikalie zur Ermittlung von Chemikalien, bei denen eine Einstufung als augenreizend oder schwer augenschädigend nicht erforderlich ist (UN-GHS-/CLP-Kategorie Keine Einstufung), beruht auf der Annahme, dass alle Chemikalien, die Augenreizungen oder schwere Augenschädigungen verursachen, im Hornhaut-Epithel und/oder in der Bindehaut eine zytotixische Wirkung induzieren.
|
|
21.
|
Die Viabilität des RhCE-Gewebes wird gewöhnlich anhand der durch die lebensfähigen Zellen des Gewebes bewirkten Enzymkonversion des Vitalfarbstoffs MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid, Thiazolyl-Blau-Tetrazoliumbromid; CAS-Nummer 298-93-1] zu einem blauen MTT-Formazan-Salz gemessen, das nach seiner Extraktion aus Geweben quantifiziert wird (16). Chemikalien, bei denen eine Einstufung und Kennzeichnung nach dem UN GHS bzw. der CLP-Verordnung nicht erforderlich ist (Kategorie Keine Einstufung), sind die Chemikalien, bei denen die Viabilität des Gewebes eine festgelegte Grenze nicht unterschreitet (d. h. Gewebeviabilität > 60 % beim EpiOcular™ EIT und beim SkinEthic™ HCE EITL (79) und > 50 % beim SkinEthic™ HCE EITS (80)) (Nummer 44).
|
NACHWEIS DER LEISTUNGSFÄHIGKEIT
|
22.
|
Vor der routinemäßigen Durchführung von RhCE-Prüfungen für rechtliche Zwecke sollten Labors die erforderliche technische Befähigung durch eine ordnungsgemäße Einstufung der 15 in Tabelle 1 genannten Chemikalien nachweisen. Diese Chemikalien wurden unter den in den Validierungsstudien der VRM verwendeten Chemikalien ausgewählt (8) (10) (11). Die Auswahl beinhaltet nach Möglichkeit Chemikalien, die (i) verschiedene Aggregatzustände abdecken, (ii) das gesamte Spektrum an in vivo auftretenden Augenreizungen bzw. schweren Augenschädigungen nach hochwertigen Ergebnissen des In-vivo-Kaninchenaugen-Referenztests (PM B.5) (2) (14) sowie nach dem UN GHS (d. h. Kategorien 1, 2A, 2B und Keine Einstufung) (1) und nach der CLP-Verordnung (d. h. Kategorien 1 oder 2 bzw. Keine Einstufung) abdecken, (iii) die verschiedenen In-vivo-Einstufungsparameter abdecken (24) (25), (iv) repräsentativ für die in der Validierungsstudie verwendeten Chemikalienklassen sind (8) (10) (11), (v) organische Funktionsgruppen gut und umfassend repräsentieren (8) (10) (11), (vi) gut definierte chemische Strukturen aufweisen (8) (10) (11), (vii) farbig und/oder direkte MTT-Reduktionsmittel sind, (viii) bei der Validierung zu reproduzierbaren Ergebnissen geführt haben, (ix) mit RhCE-Prüfmethoden in den jeweiligen Validierungsstudien korrekt vorhergesagt wurden, (x) das gesamte Spektrum an In-vitro-Reaktionen abdecken, das mit hochwertigen RhCE-Prüfmethoden ermittelt wurde (Viabilität 0-100 %), (xi) im Handel erhältlich sind und (xii) nicht mit untragbar hohen Beschaffungs- und/oder Entsorgungskosten verbunden sind. Wenn eine genannte Chemikalie nicht erhältlich ist oder aus sonstigen berechtigten Gründen nicht verwendet werden kann, kann eine andere Chemikalie eingesetzt werden, die die oben beschriebenen Anforderungen erfüllt (z. B. eine bei der Validierung der VRM verwendete Chemikalie). Solche Abweichungen sollten nur in gerechtfertigten Fällen vorkommen.
|
Tabelle 1
Liste der Leistungschemikalien
|
Chemische Bezeichnung
|
CAS-Nr.
|
Organische Funktionsgruppe (81)
|
Aggregatzustand
|
Viabilität VRM 1 (%) (82)
|
Viabilität VRM 2 (%) (83)
|
Vorhersage VRM
|
MTT-Reduktionsmittel
|
Farbinterf.
|
|
In-vivo-Kategorie 1 (84)
|
|
|
Methylmercaptoacetat (Methylthioglykolat)
|
2365-48-2
|
Carbonsäureester, Thioalkohol
|
L
|
10,9 ± 6,4
|
5,5 ± 7,4
|
Keine Vorhersage möglich
|
J (stark)
|
N
|
|
Hydroxyethylacrylat
|
818-61-1
|
Acrylat,Alkohol
|
L
|
7,5 ± 4,7 (85)
|
1,6 ± 1,0
|
Keine Vorhersage möglich
|
N
|
N
|
|
2,5-Dimethyl-2,5-hexanediol
|
110-03-2
|
Alkohol
|
S
|
2,3 ± 0,2
|
0,2 ± 0,1
|
Keine Vorhersage möglich
|
N
|
N
|
|
Natriumoxalat
|
62-76-0
|
Oxocarbonsäure
|
S
|
29,0 ± 1,2
|
5,3 ± 4,1
|
Keine Vorhersage möglich
|
N
|
N
|
|
In-vivo-Kategorie 2A (84)
|
|
|
2,4,11,13-Tetraazatetradecan-diimidamid, N,N″-Bis(4-Chlorphenyl)-3,12-diimino-, di-D-gluconat (20 %, wässrig) (86)
|
18472-51-0
|
Aromatisches heterozyklisches Halogenid, Aryl-Halogenid, Dihydroxyl-Gruppe, Guanidin
|
L
|
4,0 ± 1,1
|
1,3 ± 0,6
|
Keine Vorhersage möglich
|
N
|
J (schwach)
|
|
Natriumbenzoat
|
532-32-1
|
Aryl, Carboxylsäure
|
S
|
3,5 ± 2,6
|
0,6 ± 0,1
|
Keine Vorhersage möglich
|
N
|
N
|
|
In-vivo-Kategorie 2B (84)
|
|
|
Diethyltoluamid
|
134-62-3
|
Benzamid
|
L
|
15,6 ± 6,3
|
2,8 ± 0,9
|
Keine Vorhersage möglich
|
N
|
N
|
|
2,2-Dimethyl-3-methylenbicyclo[2.2.1]-heptan;
|
79-92-5
|
Alkan, verzweigt mit tertiärem Kohlenstoff; Alken; Bicycloheptan; Kohlenstoffzyklen mit Brückenbindung; Cycloalkan
|
S
|
4,7 ± 1,5
|
15,8 ± 1,1
|
Keine Vorhersage möglich
|
N
|
N
|
|
In Vivo Keine Einstufung (84)
|
|
|
1-Ethyl-3-methylimidazoliumethylsulfat
|
342573-75-5
|
Alkoxy, Ammoniumsalz, Aryl; Imidazol, Sulfat
|
L
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Keine Einst.
|
N
|
N
|
|
Dicaprylylether
|
629-82-3
|
Alkoxy, Ether
|
L
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Keine Einst.
|
N
|
N
|
|
Piperonylbutoxid
|
51-03-6
|
Alkoxy, Benzodioxol, Benzyl, Ether
|
L
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Keine Einst.
|
N
|
N
|
|
Polyethylenglycol (PEG-40) hydriertes Rizinusöl
|
61788-85-0
|
Acylal, Alkohol, Allyl, Ether
|
Viskose Flüssigkeit
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Keine Einst.
|
N
|
N
|
|
1-(4-Chlorphenyl)-3-(3,4-dichlorphenyl)-harnstoff
|
101-20-2
|
Aromatisches heterozyklisches Halogenid, Aryl-Halogenid, Harnstoffderivate
|
S
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Keine Einst.
|
N
|
N
|
|
2,2′-Methylen-bis-(6-(2H-benzotriazol-2-yl)-4-(1,1,3,3-tetramethylbutyl)phenol)
|
103597-45-1
|
Alkan verzweigt mit quartärem Kohlenstoff, kondensierte carbocyclische aromatische Verbindungen, kondensierte gesättigte Heterocyclen, Vorläuferchemikalien quinoider Verbindungen, tert-Butyl
|
S
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Keine Einst.
|
N
|
N
|
|
Kaliumtetrafluoroborat
|
14075-53-7
|
Anorganische Salze
|
S
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Keine Einst.
|
N
|
N
|
|
Abkürzungen:
CASRN = Registernummern des Chemical Abstracts Service.; UN GHS = United Nations Globally Harmonized System of Classification and Labelling of Chemicals (1); VRM 1 = validierte Referenzmethode, EpiOcular™ EIT; VRM 2 = validierte Referenzmethode, SkinEthic™ HCE EIT; Farbinterf. = Farbinterferenz mit der Standardabsorption (optische Dichte (OD)) Messung von MTT-Formazan.
|
PRÜFVERFAHREN
|
23.
|
Im Rahmen des Nachweises der Leistungsfähigkeit sollten die Benutzer die vom Hersteller des RhCE-Gewebemodells spezifizierten Barriereeigenschaften der Gewebe nach Erhalt überprüfen (Nummern 25, 27 und 30). Dies ist besonders dann wichtig, wenn die Gewebe über große Entfernungen/Zeiträume transportiert werden. Sobald eine Prüfung erfolgreich etabliert und ihre Leistungsfähigkeit festgestellt und nachgewiesen wurde, ist diese Überprüfung nicht mehr routinemäßig erforderlich. Allerdings empfiehlt es sich auch bei routinemäßig durchgeführten Prüfungen, die Barriereeigenschaften in regelmäßigen Abständen zu kontrollieren.
|
|
24.
|
Gegenwärtig entsprechen der EpiOcular™ EIT und der SkinEthic™ HCE EIT als wissenschaftlich fundierte Prüfungen dieser Prüfmethode (9) (12) (13); diese Prüfungen werden auch als validierte Referenzmethoden (VRM 1 bzw. VRM 2) bezeichnet. Für die RhCE-Prüfmethoden liegen Standardarbeitsanweisungen vor, die bei der Umsetzung und Verwendung dieser Prüfmethoden im Labor angewendet werden sollten (34) (35). In den folgenden Abschnitten sowie in Anlage 2 werden die wesentlichen Elemente und Verfahren der RhCE-Prüfungen beschrieben.
|
ELEMENTE DER RHCE-PRÜFMETHODE
Allgemeine Bedingungen
|
25.
|
Zur Rekonstruktion des aus progressiv geschichteten, aber nicht verhornten Zellen bestehenden dreidimensionalen hornhautartigen Epithelgewebes sind die betreffenden menschlichen Zellen zu verwenden. Das RhCE-Gewebemodell wird in Einsätzen mit einer porösen synthetischen Membran konstruiert, durch die Nährstoffe in die Zellen gelangen können. Das rekonstruierte hornhautartige Epithel sollte mehrere Schichten lebensfähiger nicht verhornter Epithelzellen enthalten. Beim RhCE-Gewebemodell sollte die Epitheloberfläche unmittelbar mit der Luft in Berührung kommen, damit eine oberflächliche Exposition gegenüber den Prüfchemikalien ähnlich wie in vivo beim natürlichen Hornhaut-Epithel erfolgen kann. Das RhCE-Gewebemodell sollte eine funktionsfähige Barriere bilden, die so robust ist, dass sie eine rasche Durchdringung zytotoxischer Referenzstoffe (z. B. Triton X-100 oder Natriumdodecylsulfat (SDS)) verhindert. Die Barrierefunktion sollte nachgewiesen werden. Zur Beurteilung der Barrierewirkung kann entweder die Expositionszeit, bei der nach Applikation eines Referenzstoffs in einer bestimmten festgelegten Konzentration (z. B. 100 μl 0,3 % (v/v) Triton X-100) die Viabilität eines Gewebes um 50 % reduziert wird (ET50), oder die Konzentration bestimmt werden, bei der ein Referenzstoff nach einer bestimmten Expositionszeit (z. B. nach 30-minütiger Behandlung mit 50 μl SDS) die Viabilität der Gewebe um 50 % reduziert (IC50) (Nummer 30). Die Rückhalteeigenschaften des RhCE-Gewebemodells müssen ausschließen, dass die Prüfchemikalie rund um die Hornschicht in lebensfähiges Gewebe eindringt und die Modellierung der Hornhautexposition beeinträchtigt. Die zur Herstellung des RhCE-Gewebemodells verwendeten menschlichen Zellen müssen frei von Verunreinigungen durch Bakterien, Viren, Mycoplasma und Pilze sein. Der Hersteller prüft die Sterilität des Gewebemodells und stellt sicher, dass das Modell nicht durch Pilze oder Bakterien verunreinigt ist.
|
Funktionale Bedingungen
Viabilität
|
26.
|
Die Größenordnung der Viabilität der Gewebe wird mit der MTT-Prüfung bestimmt (16). Die lebensfähigen Zellen des RhCE-Gewebemodells reduzieren den lebenswichtigen Farbstoff MTT zu einem blauen MTT-Formazan-Niederschlag, der dann mit Isopropanol (oder einem ähnlichen Lösungsmittel) aus dem Gewebe extrahiert wird. Das extrahierte MTT-Formazan kann durch eine Standard-(OD)-Absorptionsmessung oder mit einem HPLC/UPLC-Spektrometrieverfahren (OD = optische Dichte) (36) quantifiziert werden. Die optische Dichte (OD) des reinen Extraktionslösungsmittels sollte ausreichend gering sein, d. h. OD < 0,1. Das RhCE-Gewebemodell sollte sicherstellen, dass jede Charge des verwendeten RhCE-Gewebemodells die vorgegebenen Kriterien für die Negativkontrolle erfüllt. Die Akzeptanzspannen der OD-Werte der Negativkontrolle für die VRM sind Tabelle 2 zu entnehmen. Bei Durchführung einer HPLC/UPLC-Spektrophotometrie sollten die in Tabelle 2 genannten OD-Spannen der Negativkontrolle als Akzeptanzkriterium für die Negativkontrolle angenommen werden. Im Prüfbericht sollte dokumentiert werden, dass die mit dem Negativkontrollstoff behandelten Gewebe über den gesamten Expositionszeitraum stabil bleiben (bzw. dass eine vergleichbare Gewebeviabilität gemessen wird). Entsprechend verfährt der Hersteller des Gewebes im Rahmen der Qualitätskontrolle zur Freigabe der Gewebecharge. Dabei können allerdings andere als die in Tabelle 2 genannten Akzeptanzkriterien maßgeblich sein. Der Entwickler/Hersteller des RhCE-Gewebemodells sollte eine Akzeptanzspanne (oberer und unterer Grenzwert) für die OD-Werte der Negativkontrolle (unter den Bedingungen bei der Qualitätskontrolle der Prüfmethode) festlegen.
|
Tabelle 2
Akzeptanzspannen für OD-Werte der Negativkontrolle zur Kontrolle der Chargenqualität (für die Anwender der Prüfung)
|
Prüfung
|
Untere Akzeptanzgrenze
|
Obere Akzeptanzgrenze
|
|
EpiOcular™ EIT (OCL-200) – VRM 1 (Protokolle für Flüssigkeiten und für Feststoffe)
|
> 0,8 (87)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM 2 (Protokolle für Flüssigkeiten und für Feststoffe)
|
> 1,0
|
≤ 2,5
|
Barrierefunktion
|
27.
|
Das RhCE-Gewebemodell sollte so dick und robust sein, dass es ein rasches Eindringen der zytotoxischen Referenzstoffe verhindert; als Maßstab sind die Werte z. B. für ET50 (Triton X-100) oder IC50 (SDS) (Tabelle 3) anzunehmen. Die Barrierefunktion der einzelnen Chargen des verwendeten RhCE-Gewebemodells sollte nach der Lieferung der Gewebe an den Endverwender vom Entwickler/Hersteller des RhCE-Gewebemodells nachgewiesen werden (Nummer 30).
|
Morphologie
|
28.
|
Anhand einer histologischen Untersuchung des RhCE-Gewebemodells sollte die Ähnlichkeit des Epithelgewebes mit der menschlichen Hornhaut (bei mindestens 3 Schichten lebensfähiger Epithelzellen und nicht verhornter Oberfläche) nachgewiesen werden. Für die VRM hat der Entwickler/Hersteller eine geeignete Morphologie nachgewiesen. Daher brauchen die Anwender der Prüfmethode diesen Nachweis nicht für jede einzelne verwendete Gewebecharge neu zu führen.
|
Reproduzierbarkeit
|
29.
|
Die Ergebnisse der Positivkontrolle und der Negativkontrollen der Prüfmethode sollen die Reproduzierbarkeit über längere Zeit belegen.
|
Qualitätskontrolle (QK)
|
30.
|
Das RhCE-Gewebemodell sollte nur dann verwendet werden, wenn der Entwickler/Hersteller nachweist, dass jede Charge des verwendeten RhCE-Gewebemodells bestimmten Freigabekriterien genügt, von denen die Kriterien der Viabilität (Nummer 26) und der Barrierefunktion (Nummer 27) die wichtigsten sind. Der Entwickler/Hersteller des RhCE-Gewebemodells legt eine Akzeptanzspanne (oberer und unterer Grenzwert) für die Barrierefunktionen anhand der Werte für ET50 oder IC50 (Nummern 25 und 26) fest. Die vom Entwickler/Hersteller der RhCE-Gewebemodelle bei der Qualitätskontrolle für die Freigabe einer Charge angenommenen (und für die VRM verwendeten) Akzeptanzspannen für ET50 und IC50 sind Tabelle 3 zu entnehmen. Der Entwickler/Hersteller des RhCE-Gewebemodells stellt den Anwendern der Prüfmethode Daten zur Verfügung, die die Erfüllung sämtlicher Freigabekriterien belegen, damit die Anwender die betreffenden Informationen im Prüfbericht berücksichtigen können. Für die zuverlässige Vorhersage, dass Chemikalien eine Einstufung und Kennzeichnung im Hinblick auf eine augenreizende oder schwer augenschädigende Wirkung nach dem UN GHS bzw. der CLP-Verordnung nicht erfordern, können ausschließlich Ergebnisse akzeptiert werden, die mit Geweben ermittelt wurden, die diese Freigabekriterien vollständig erfüllen.
|
Tabelle 3
Chargenfreigabekriterien im Rahmen der Qualitätskontrolle
|
Prüfung
|
Untere Akzeptanzgrenze
|
Obere Akzeptanzgrenze
|
|
EpiOcular™ EIT (OCL-200) – VRM 1 (100 μl 0,3 % (v/v) Triton X-100)
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM 2 (30-minütige Behandlung mit 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Applikation der Prüfchemikalie und der Kontrollstoffe
|
31.
|
Für jede Prüfchemikalie und für jeden Prüfstoff sollten bei jedem Prüflauf mindestens zwei Gewebereplikate verwendet werden. Zwei unterschiedliche Behandlungsprotokolle werden verwendet (jeweils eines für flüssige und eines für feste Prüfchemikalien) (34) (35). Bei beiden Methoden und Protokollen ist die Oberfläche des Gewebemodells vor der Applikation von Prüfchemikalien mit calcium- und magnesiumfreiem Dulbecco-Phosphatpuffer (Ca2+/Mg2+-freiem DPBS) zu befeuchten, um eine Feuchtigkeit ähnlich wie am menschlichen Auge herzustellen. Die Behandlung der Gewebe beginnt mit der Exposition gegenüber den Prüfchemikalien und den Kontrollstoffen. Nach den beiden Behandlungsprotokollen der beiden VRM sind die Prüfchemikalien und Kontrollstoffe so ausreichend aufzutragen, dass die gesamte Epitheloberfläche gleichmäßig bedeckt, eine unklare Dosierung jedoch vermieden wird (Nummern 32 und 33) (Anlage 2).
|
|
32.
|
Prüfchemikalien, die bei höchstens 37 °C pipettiert werden können (erforderlichenfalls mit einer Direktverdränger-Pipette), werden in den VRM als Flüssigkeiten behandelt; ansonsten sind die Prüfchemikalien als Feststoffe zu betrachten (Nummer 33). Bei den VRM wird die flüssige Prüfchemikalie gleichmäßig auf die Gewebeoberfläche aufgetragen (mindestens 60 μl/cm2) (siehe Anlage 2, (33) (34)). Kapillareffekte (Wirkungen der Oberflächenspannung), die aufgrund der geringen Volumina der in den Einsatz (auf die Gewebeoberfläche) aufgetragenen Chemikalien auftreten könnten, sollten möglichst vermieden werden, um eine korrekte Dosierung bei der Behandlung des Gewebes sicherzustellen. Mit flüssigen Prüfchemikalien behandelte Gewebe werden 30 Minuten bei Standard-Kulturbedingungen (37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relative Luftfeuchtigkeit) inkubiert. Am Ende des Expositionszeitraums werden die flüssige Prüfchemikalie und die Kontrollstoffe unter gründlichem Waschen mit Ca2+/Mg2+-freiem DPBS bei Raumtemperatur vorsichtig von der Gewebeoberfläche entfernt. Nach der Behandlung und nach dem Waschen erfolgt eine Immersion des Gewebes bei Raumtemperatur für einen bestimmten, je nach VRM unterschiedlichen Zeitraum in ein frisches Medium, um im Gewebe absorbierte Rückstände der Prüfchemikalien zu entfernen. Nur bei VRM 1 werden die Zellen nach der Exposition und vor der Durchführung der MTT-Prüfung in einem frischen Medium bei Standard-Kulturbedingungen inkubiert (siehe Anlage 2, (34) (35)).
|
|
33.
|
Prüfchemikalien, die bei Temperaturen bis zu 37 °C nicht pipettiert werden können, werden in den VRM als Feststoffe behandelt. Die Prüfchemikalie sollte in ausreichender Menge aufgetragen werden, um die gesamte Oberfläche des Gewebes zu bedecken (d. h. mindestens 60 mg/cm2 (Anlage 2)). Feststoffe sollten nach Möglichkeit als Feinpulver geprüft werden. Mit festen Prüfchemikalien behandelte Gewebe werden über einen festgelegten Zeitraum (je nach VRM) bei Standard-Kulturbedingungen inkubiert (siehe Anlage 2, (34) (35)). Am Ende des Expositionszeitraums werden die feste Prüfchemikalie und die Kontrollstoffe unter gründlichem Waschen mit Ca2+/Mg2+-freiem DPBS bei Raumtemperatur vorsichtig von der Gewebeoberfläche entfernt. Nach der Behandlung und nach dem Waschen erfolgt eine Immersion des Gewebes bei Raumtemperatur für einen bestimmten, je nach VRM unterschiedlichen, Zeitraum in ein frisches Medium, um im Gewebe absorbierte Rückstände der Prüfchemikalien zu entfernen. Nach der Exposition und vor der Durchführung der MTT-Prüfung werden die Zellen in frischem Medium bei Standard-Kulturbedingungen inkubiert (siehe Anlage 2, (34) (35)).
|
|
34.
|
Bei jedem Prüflauf sind gleichzeitige Negativ- und Positivkontrollen zu berücksichtigen, um nachzuweisen, dass die Werte für die Viabilität (mit der Negativkontrolle ermittelt) und die Sensitivität (mit der Positivkontrolle ermittelt) der Gewebe innerhalb der anhand historischer Daten festgelegten Akzeptanzspannen liegen. Die gleichzeitige Negativkontrolle dient auch als Referenzwert (Gewebeviabilität 100 %) zur Berechnung der relativen Viabilität der mit der Prüfchemikalie behandelten Gewebe (% ViabilitätPrüfung). Der für die VRM empfohlene Positivkontrollstoff ist reines Methylacetat (CAS-Nr. 79-20-9, im Handel erhältlich beispielsweise von Sigma-Aldrich, Bestellnr. 45997, flüssig). Als Negativkontrollstoffe für VRM 1 und VRM 2 werden ultrareines H2O bzw. Ca2+/Mg2+-freier DPBS empfohlen. Diese Kontrollstoffe wurden in den Studien zur Validierung der VRM verwendet und für diese Kontrollstoffe liegen die umfangreichsten historischen Daten vor. Die Verwendung geeigneter alternativer Positiv- oder Negativkontrollstoffe sollte wissenschaftlich begründet und angemessen gerechtfertigt sein. Negativ- und Positivkontrollen sollten mit denselben Protokollen wie die im jeweiligen Prüflauf (d. h. für Flüssigkeiten und/oder Feststoffe) verwendeten Prüfchemikalien geprüft werden. Vor der Durchführung der MTT-Prüfung (Nummer 35) ist hinsichtlich der Exposition und ggf. der Immersion nach der Behandlung das Protokoll so einzuhalten, wie für die gleichzeitig durchgeführten Kontrollen bei flüssigen Prüfchemikalien (Nummer 32) bzw. bei festen Prüfchemikalien (Nummer 33) beschrieben (34) (35). Eine einzelne Gruppe von Negativ- und Positivkontrollen ist für alle in einem Prüflauf berücksichtigten Prüfchemikalien jeweils eines Aggregatzustands (flüssig oder fest) ausreichend.
|
Messungen der Gewebeviabilität
|
35.
|
Die MTT-Prüfung ist eine standardisierte quantitative Methode (16), die zur Messung der Gewebeviabilität nach dieser Prüfmethode angewandt werden sollte. Sie ist zur Anwendung in einem dreidimensionalen Gewebemodell geeignet. Die MTT-Prüfung wird unmittelbar im Anschluss an die Inkubation nach der Behandlung durchgeführt. Bei den VRM wird die Probe des RhCE-Gewebemodells 180 ± 15 Minuten bei Standard-Kulturbedingungen in 0,3 ml MTT-Lösung (1 mg/ml) gegeben. Der Vitalfarbstoff MTT wird durch die lebensfähigen Zellen des RhCE-Gewebemodells zu einem blauen MTT-Formazan-Niederschlag reduziert. Der blaue MTT-Formazan-Niederschlag wird dann mit einer geeigneten Menge Isopropanol (oder einem ähnlichen Lösungsmittel) aus dem Gewebe extrahiert (34) (35). Mit flüssigen Prüfchemikalien geprüfte Gewebe sind sowohl oben als auch unten aus dem Gewebe zu extrahieren. Mit festen Prüfchemikalien und mit gefärbten Flüssigkeiten geprüfte Gewebe dagegen sind ausschließlich aus dem unteren Bereich zu extrahieren (um die Gefahr einer Verunreinigung der Isoropanol-Extraktionslösung durch möglicherweise im Gewebe verbliebene Prüfchemikalien zu minimieren). Bei Geweben, die mit flüssigen Prüfchemikalien geprüft wurden, die nicht ohne Weiteres auswaschbar sind, muss die Extraktion unter Umständen ebenfalls ausschließlich aus dem unteren Bereich des Gewebes erfolgen. Die gleichzeitig geprüften Negativ- und Positivkontrollstoffe sind wie die geprüfte Chemikalie zu behandeln. Das extrahierte MTT-Formazan kann entweder durch eine Standardabsorptions-(OD-)Messung bei 570 nm innerhalb einer Filter-Bandbreite von maximal ± 30 nm oder mit einem HPLC/UPLC-Spektrophotometrieverfahren gemessen werden (Nummer 42) (11) (36).
|
|
36.
|
Die optischen Eigenschaften der Prüfchemikalie bzw. ihre chemische Wirkung auf das MTT können die MTT-Formazan-Messung beeinträchtigen und dazu führen, dass die Gewebeviabilität falsch eingeschätzt wird. Prüfchemikalien können die MTT-Formazan-Messung durch direkte Reduzierung des MTT zu blauem MTT-Formazan und/oder durch Farbinterferenzen stören, wenn die Prüfchemikalie an sich oder infolge von Behandlungsverfahren in demselben OD-Bereich wie das MTT-Formazan (570 nm) absorbiert. Vor der eigentlichen Prüfung sind Vorprüfungen durchzuführen, um direkt wirkende potenzielle MTT-Reduziermittel und/oder Chemikalien zu ermitteln, die Farbinterferenzen verursachen können. Außerdem sollten weitere Kontrollen verwendet werden, um potenzielle Interferenzen aufgrund dieser Prüfchemikalien erkennen und korrigieren zu können (Nummern 37-41). Dies ist besonders dann wichtig, wenn eine bestimmte Prüfchemikalie nicht vollständig aus dem RhCE-Gewebemodell ausgewaschen wurde oder das hornhautartige Epithel durchdringt und daher bei Durchführung der MTT-Prüfung auch in den RhCE-Gewebemodellen enthalten ist. Bei Prüfchemikalien, die (an sich oder nach einer Behandlung) Licht im selben Bereich wie MTT-Formazan absorbieren und wegen übermäßiger Interferenzen (d. h. einer Absorption bei 570 ± 30 nm) nicht mit der Standardabsorptions-(OD-)Messung bei MTT-Formazan im Einklang stehen, kann eine MTT-Formazan-Messung mit einem HPLC/UPLC-Spektrophotometrieverfahren vorgenommen werden (Nummern 41 und 42) (11) (36). Eine genaue Beschreibung der Vorgehensweise zur Erkennung und Korrektur einer direkten MTT-Reduktion sowie von Interferenzen durch Färbemittel ist den Standardarbeitsanweisungen für die VRM zu entnehmen (34) (35). Die Flussdiagramme in den Anhängen III und IV veranschaulichen die Vorgehensweise zur Erkennung und Behandlung von direkt wirkenden MTT-Reduktionsmitteln und von Farbinterferenzen verursachenden Chemikalien für die VRM 1 und 2.
|
|
37.
|
Um potenzielle Interferenzen durch Prüfchemikalien zu ermitteln, die (an sich oder nach einer Behandlung) Licht im selben Spektrum wie MTT-Formazan absorbieren, und um entscheiden zu können, ob weitere Kontrollen erforderlich sind, wird die Prüfchemikalie zu Wasser und/oder Isopropanol hinzugegeben und über einen angemessenen Zeitraum bei Raumtemperatur inkubiert (siehe Anlage 2, (34) (35)). Wenn die Prüfchemikalie bei VRM 1 in Wasser und/oder Isopropanol ausreichend Licht im Bereich von 570 ± 20 nm absorbiert (siehe Anlage 3), oder wenn bei VRM 2 eine farbige Lösung entsteht, wenn die Prüfchemikalie mit Wasser gemischt wird (siehe Anlage 4), ist anzunehmen, dass die Prüfchemikalie bei der Standardabsorptions-(OD-)Messung von MTT-Formazan Interferenzen verursacht; in diesem Fall sind weitere Farbkontrollen oder eine HPLC/UPLC-Spektrophotometrie vorzunehmen. Die betreffenden Kontrollen werden dann nicht benötigt (Nummern 41 und 42 sowie Anhänge III und IV) (34) (35). Bei Durchführung der Standardabsorptions-(OD-)Messung wird jede interferierende Prüfchemikalie auf mindestens zwei lebensfähige Gewebereplikate aufgetragen; diese Replikate werden dem vollständigen Prüfverfahren unterzogen, aber während der MTT-Inkubation nicht mit der MTT-Lösung, sondern mit einem Medium inkubiert, um in lebendem Gewebe eine nicht spezifische Farbe (NSClebend) zu erzeugen (34) (35). Die NSClebend-Kontrolle muss gleichzeitig mit der Untersuchung des Prüffarbstoffs durchgeführt werden, und bei mehreren Prüfungen ist wegen der inhärenten biologischen Variabilität lebender Gewebe für jede durchgeführte Prüfung (bei jedem Prüflauf) eine unabhängige NSClebend-Kontrolle zu prüfen. Die tatsächliche Viabilität des Gewebes wird als Prozentanteil der ermittelten Viabilität bei lebendem Gewebe nach der Exposition gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit der MTT-Lösung (% ViabilitätPrüfung) abzüglich des Prozentanteils der nicht spezifischen Farbe berechnet, die bei gleichzeitig mit der zu korrigierenden Prüfung untersuchten lebenden Geweben nach Exposition gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit einem Medium ohne MTT entstanden ist (% NSClebend), d. h. tatsächliche Gewebeviabilität = [% ViabilitätPrüfung] - [% NSClebend].
|
|
38.
|
Um direkte MTT-Reduktionsmittel zu identifizieren, sollten die Prüfchemikalien jeweils zu frisch hergestellter MTT-Lösung hinzugegeben werden. Eine geeignete Menge einer Prüfchemikalie wird zu einer MTT-Lösung hinzugegeben und etwa 3 Stunden unter Standard-Kulturbedingungen inkubiert (siehe Anhänge III und IV) (34) (35). Wenn sich das MTT-Gemisch mit der Prüfchemikalie (bzw. die Suspension bei nicht lösliche Prüfchemikalien) blau/violett färbt, ist davon auszugehen, dass die Prüfchemikalie das MTT direkt reduziert; anschließend sollte unabhängig von der Durchführung der Standardabsorptions-(OD-)Messung oder einer HPLC/UPLC-Spektrophotometrie eine weitere Funktionsprüfung der nicht lebensfähigen RhCE-Gewebemodelle vorgenommen werden. Bei dieser zusätzlichen Funktionsprüfung werden abgetötete Gewebe mit nur noch residualer metabolischer Aktivität eingesetzt, die die Prüfchemikalie in ähnlicher Weise wie lebensfähige Gewebe absorbieren und binden. Bei der VRM 1 werden Gewebe abgetötet, indem sie einer niedrigen Temperatur ausgesetzt werden („durch Gefrieren abgetötet“). Bei der VRM 2 werden Gewebe abgetötet, indem sie über einen längeren Zeitraum (z. B. mindestens 24 ± 1 Std.) in Wasser inkubiert und anschließend bei einer niedrigen Temperatur aufbewahrt werden („in Wasser abgetötet“). Jede MTT reduzierende Prüfchemikalie wird auf mindestens zwei abgetötete Gewebereplikate aufgetragen, die dem gesamten Prüfverfahren unterzogen werden, um eine nicht spezifische MTT-Reduktionskontrolle (NSMTT-Reduktion) herzustellen (34) (35). Pro Prüfchemikalie ist unabhängig von der Anzahl der durchgeführten unabhängigen Prüfungen/Prüfläufe eine einzelne NSMTT-Kontrolle ausreichend. Anschließend wird die tatsächliche Viabilität des Gewebes als Prozentanteil der bei lebenden Geweben nach Exposition gegenüber dem MTT-Reduktionsmittel gemessenen Viabilität (% ViabilitätPrüfung) abzüglich des Prozentanteils der nicht spezifischen MTT-Reduktion durch dasselbe MTT-Reduktionsmittel bei abgetöteten Geweben bezogen auf die gleichzeitig mit der zu korrigierenden Prüfung untersuchte Negativkontrolle (% NSMTT), d. h. tatsächliche Gewebeviabilität = [% ViabilitätPrüfung] - [% NSMTT], berechnet.
|
|
39.
|
Bei Prüfchemikalien, die sowohl Farbinterferenzen (Nummer 37) als auch eine direkte MTT-Reduktion (Nummer 38) verursachen, wird für die Standardabsorptions-(OD-)Messung außer der NSMTT-Kontrolle und der NSClebend-Kontrolle (siehe vorstehende Abschnitte) eine dritte Gruppe von Kontrollen benötigt. Dies ist gewöhnlich bei dunklen Prüfchemikalien der Fall, die beim MTT-Versuch Licht im Bereich von 570 ± 30 nm (z. B. blau, violett oder schwarz) absorbieren, weil deren Fähigkeit zur direkten MTT-Reduktion durch die inhärente Farbqualität dieser Chemikalien beeinträchtigt wird (Nummer 38). In diesen Fällen sind zusammen mit den NSClebend-Kontrollen regelmäßig NSMTT-Kontrollen zu verwenden. Prüfchemikalien, bei denen sowohl NSMTT-Kontrollen als auch NSClebend-Kontrollen untersucht wurden, können von lebenden und von abgetöteten Geweben absorbiert und gebunden werden. Daher kann in diesen Fällen bei der NSMTT-Kontrolle eine Korrektur nicht nur der potentiellen direkten MTT-Reduktion durch die Prüfchemikalie, sondern auch der Farbinterferenz infolge der Absorption und der Bindung der Prüfchemikalie an abgetötete Gewebe erfolgen. Dies kann eine doppelte Korrektur der Farbinterferenz zur Folge haben, da mit der NSClebend-Kontrolle bereits eine Korrektur der Farbinterferenz aufgrund der Absorption und der Bindung der Prüfchemikalie durch lebende Gewebe erfolgt. Um eine mögliche doppelte Korrektur von Farbinterferenzen zu vermeiden, muss eine dritte Kontrolle mit nicht spezifischen Farben mit abgetöteten Geweben (NSCabgetötet) untersucht werden (siehe Anhänge III und IV) (34) (35). Bei dieser zusätzlichen Kontrolle wird die Prüfchemikalie auf mindestens zwei abgetötete Gewebereplikate aufgetragen, die dem vollständigen Prüfverfahren zu unterziehen sind, wobei die Gewebe jedoch während der MTT-Inkubation nicht mit der MTT-Lösung, sondern mit einem Medium inkubiert werden. Unabhängig von der Anzahl der durchgeführten unabhängigen Prüfungen/Prüfläufe ist pro Prüfchemikalie eine einzige NSCabgetötet-Kontrolle hinreichend. Diese Kontrolle sollte jedoch gleichzeitig mit der NSMTT-Kontrolle sowie mit derselben Gewebecharge untersucht werden. Die tatsächliche Viabilität des Gewebes wird dann als Prozentanteil der ermittelten Viabilität bei lebendem Gewebe nach der Exposition gegenüber der Prüfchemikalie (% ViabilitätPrüfung) abzüglich % NSMTT und abzüglich % NSClebend zuzüglich des Prozentanteils der nicht spezifischen Farbe berechnet, die nach Exposition der abgetöteten Gewebe gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit einem Medium ohne MTT entstanden ist; dieser Prozentanteil wird bezogen auf die gleichzeitig mit der zu korrigierenden Prüfung untersuchte Negativkontrolle (% NSCabgetötet), d. h. tatsächliche Gewebeviabilität = [% ViabilitätPrüfung] - [% NSMTT] - [% NSClebend] + [% NSCabgetötet], ermittelt.
|
|
40.
|
Wichtig ist, dass eine nicht spezifische MTT-Reduktion und nicht spezifische Farbinterferenzen die optische Dichte (bei Standard-Absorptionsmessungen) und die Peak-Flächen für MTT-Formazan (bei HPLC/UPLC-Spektrophotometriemessungen) der Gewebeextrate über die Linearitätsspanne des Spektrophotometers hinaus verstärken bzw. vergrößern können. Daher muss jedes Labor vor der Untersuchung von Prüfchemikalien für rechtliche Zwecke die Linearitätsspanne der OD/Peak-Fläche seines Spektrophotometers mit im Handel (z. B. bei Sigma-Aldrich (Bestellnr. M2003) erhältlichem MTT-Formazan (CAS-Nr. 57360-69-7) ermitteln.
|
|
41.
|
Die Standardabsorptions-(OD-)Messung mit einem Spektrophotometer ist zur Beurteilung von Prüfchemikalien geeignet, die als direkte MTT-Reduktionsmittel wirken bzw. Farbinterferenzen verursachen, sofern die bei der MTT-Formazan-Messung beobachtete Interferenz nicht zu stark ausgeprägt ist (d. h. wenn die OD-Werte der Gewebeextrakte mit der Prüfchemikalie ohne Korrektur für eine direkte MTT-Reduktion und/oder Farbinterferenzen innerhalb der Linearitätsspanne des Spektrophotometers liegen). Ergebnisse bei Prüfchemikalien mit % NSMTT und/oder % NSClebend ≥ 60 % (VRM 1 sowie VRM 2 beim Protokoll für Flüssigkeiten) bzw. 50 % (VRM 2 beim Protokoll für Feststoffe) der Negativkontrolle sind jedoch mit Sorgfalt zu bewerten, da dies die festgelegten und in den VRM verwendeten Schwellenwerte sind, nach denen darüber entschieden wird, ob bei Chemikalien eine Einstufung erforderlich ist (Nummer 44). Die Standardabsorption (OD) kann bei übermäßiger Interferenz mit der MTT-Formazan-Messung (d. h. wenn die Interferenz dazu führt, dass nicht korrigierte OD-Werte der Prüfgewebeextrakte außerhalb der Linearitätsspanne des Spektrophotometers liegen) jedoch nicht gemessen werden. Prüffarbstoffe, die sich bei Kontakt mit Wasser oder Isopropanol färben und die Standardabsorptions-(OD-)Messung von MTT-Formazan übermäßig beeinträchtigen, können durch HPLC/UPLC-Spektrophotometrie gemessen werden (siehe Anhänge III und IV). Eine Messung durch HPLC/UPLC-Spektrophotometrie ist deshalb möglich, weil bei der HPLC/UPLC-Spektrophotometrie das MTT-Formazan vor der Quantifizierung von der Prüfchemikalie getrennt werden kann (36). Daher werden unabhängig von der zu prüfenden Chemikalie keine NSClebend- oder NSCabgetötet-Kontrollen benötigt, wenn eine HPLC/UPLC-Spektrophotometrie vorgenommen wird. Allerdings sollten NSMTT-Kontrollen verwendet werden, wenn vermutet wird, dass eine Prüfchemikalie MTT direkt reduziert (Verfahren wie in Nummer 38 beschrieben). NSMTT-Kontrollen sollten jedoch auch bei Prüfchemikalien mit Färbungen (an sich oder in Wasser auftretend) verwendet werden, wenn vermutet wird, dass eine Prüfchemikalie MTT direkt reduziert oder wenn die Farbe einer Prüfchemikalie die Beurteilung ihrer Fähigkeit zur direkten MTT-Reduktion beeinträchtigt (Nummer 38). Wenn MTT-Formazan durch HPLC/UPLC-Spektrophotometrie gemessen wird, ist der Prozentanteil der Viabilität des Gewebes als Prozentanteil der MTT-Formazan-Peak-Fläche, die mit lebenden Geweben nach Exposition gegenüber der Prüfchemikalie ermittelt wurde, bezogen auf die MTT-Formazan-Peak-Fläche der gleichzeitigen Negativkontrolle zu berechnen. Bei Prüfchemikalien, die MTT direkt reduzieren können, wird die tatsächliche Gewebeviabilität wie folgt berechnet: % ViabilitätPrüfung
abzüglich % NSMTT, wie in Nummer 38 im letzten Satz beschrieben. Und schließlich ist festzustellen, dass direkte MTT-Reduktionsmittel sowie auch Farbinterferenzen verursachende direkte MTT-Reduktionsmittel, die nach der Behandlung in den Geweben gebunden sein und MTT so stark reduzieren können, dass es (bei Standard-OD-Messungen) zu OD-Werten oder (bei der Untersuchung der Gewebeextrakte durch UPLC-/HPLC-Spektrophotometrie) zu Peak-Flächen außerhalb der Linearitätsspanne des Spektrophotometers und zu Farbinterferenzen kommen kann, mit Prüfmethoden unter Verwendung von RhCE nicht bewertet werden können. Solche Fälle sind allerdings sehr selten.
|
|
42.
|
MTT-Formazan-Messungen können bei allen Arten von Prüfchemikalien (gefärbt, nicht gefärbt, MTT-Reduktionsmittel und Mittel, die keine MTT-Reduktion bewirken) durch HPLC/UPLC-Spektrophotometrie vorgenommen werden (11) (36). Aufgrund der Vielfalt der HPLC/UPLC-Spektrophotometriesysteme können nicht für alle Systeme exakt identische Bedingungen festgelegt werden. Daher sollte die Eignung des jeweiligen HPLC/UPLC-Spektrophotometriesystems vor der Verwendung zur Quantifizierung von MTT-Formazan in Gewebextrakten durch Erfüllung der Akzeptanzkriterien für eine Reihe von Standard-Qualifikationsparametern auf der Grundlage der Branchenleitlinien der US-amerikanischen Food and Drug Administration (FDA) für die Validierung bioanalytischer Methoden nachgewiesen werden (36) (38). Diese Schlüsselparameter sowie die jeweiligen Akzeptanzkriterien sind Anlage 5 zu entnehmen. Wenn die in Anlage 5 beschriebenen Akzeptanzkriterien erfüllt sind, wird das betreffende HPLC/UPLC-Spektrophotometriesystem als geeignet für die Messung von MTT-Formazan unter den in dieser Prüfmethode beschriebenen Versuchsbedingungen betrachtet.
|
Akzeptanzkriterien
|
43.
|
Bei allen Prüfläufen mit RhCE-Gewebechargen, die die Anforderungen der Qualitätskontrolle erfüllen, (Nummer 30) müssen alle mit dem Negativkontrollstoff behandelten Gewebe OD-Werte aufweisen, die der Qualität der Gewebe nach der Lieferung sowie nach Durchführung aller Schritte und aller im Protokoll vorgesehenen Prozesse entsprechen und nicht außerhalb der in Tabelle 2 genannten historisch ermittelten Grenzen liegen (Nummer 26). Die mittlere Gewebeviabilität von mit dem Positivkontrollstoff (d. h. mit Methylacetat) behandelten Geweben muss bei Anwendung der Protokolle für Flüssigkeiten oder für Feststoffe < 50 % der Negativkontrolle bei der VRM 1 und ≤ 30 % (beim Protokoll für Flüssigkeiten) bzw. ≤ 20 % (beim Protokoll für Feststoffe) der Negativkontrolle bei der VRM 2 sein und damit belegen, dass die Gewebe unter den Bedingungen der Prüfmethode auf eine reizende Prüfchemikalie reagieren können (34) (35). Die Variabilität zwischen Gewebereplikaten bei Prüfchemikalien und Kontrollstoffen muss innerhalb der akzeptierten Grenzen liegen (d. h. die Viabilität von zwei Gewebereplikaten muss unter 20 % liegen, bzw. bei drei Gewebereplikaten darf die Standardabweichung (SD) höchstens 18 % betragen). Wenn die Negativ- oder die Positivkontrolle bei einem Prüflauf Werte außerhalb der akzeptierten Spannen ergibt, ist der Prüflauf als nicht geeignet zu betrachten und sollte wiederholt werden. Liegt die Variabilität zwischen Gewebereplikaten bei einer Prüfchemikalie außerhalb der akzeptierten Spanne, ist die Prüfung als nicht geeignet zu betrachten und die Prüfchemikalie erneut zu prüfen.
|
Auswertung der Ergebnisse und Prädiktionsmodell
|
44.
|
Die OD-Werte/Peak-Flächen der Replikatgewebe-Extrakte für die einzelnen Prüfchemikalien werden zur Berechnung der mittleren Gewebeviabilität in Prozent (Mittelwert der Gewebereplikate) bezogen auf die mit 100 % angesetzte Negativkontrolle verwendet. Der prozentuale Grenzwert der Gewebeviabilität bei der Ermittlung von Prüfchemikalien, bei denen eine Einstufung als augenreizend oder schwer augenschädigend (UN-GHS-/CLP-Kategorie Keine Einstufung) nicht erforderlich ist, sind Tabelle 4 zu entnehmen. Die Ergebnisse sind wie folgt zu bewerten:
|
—
|
Bei einer Prüfchemikalie sind eine Einstufung und Kennzeichnung nach dem UN GHS bzw. der CLP-Verordnung nicht erforderlich (Keine Einstufung), wenn die mittlere prozentuale Gewebeviabilität nach der Exposition und nach der Inkubation im Anschluss an die Exposition den in Tabelle 4 genannten Grenzwert der Gewebeviabilität in Prozent überschreitet (>). In diesem Fall sind weitere Untersuchungen mit anderen Prüfmethoden nicht erforderlich.
|
|
—
|
Ist die mittlere prozentuale Gewebeviabilität nach der Exposition und nach der Inkubation im Anschluss an die Exposition kleiner oder gleich (≤) dem in Tabelle 4 genannten Grenzwert der Gewebeviabilität in Prozent, kann keine Vorhersage getroffen werden. In diesem Fall sind weitere Untersuchungen mit anderen Prüfmethoden vorzunehmen, weil bei den Prüfmethoden unter Verwendung von RhCE ein bestimmter Anteil falsch positiver Ergebnisse ermittelt wird (Nummern 14 und 15) und nicht zwischen den UN-GHS-/CLP-Kategorien 1 und 2 unterschieden werden kann (Nummer 17).
|
Tabelle 4
Vorhersagemodelle nach dem UN-GHS und der CLP-Verordnung
|
VRM
|
Keine Einstufung
|
Keine Vorhersage möglich
|
|
VRM 1 – EpiOcular™ EIT (beide Protokolle)
|
Mittlere Gewebeviabilität > 60 %
|
Mittlere Gewebeviabilität ≤ 60 %
|
|
VRM 2 – SkinEthic™ HCE EIT (Protokoll für Flüssigkeiten)
|
Mittlere Gewebeviabilität > 60 %
|
Mittlere Gewebeviabilität ≤ 60 %
|
|
VRM 2 – SkinEthic™ HCE EIT (Protokoll für Feststoffe)
|
Mittlere Gewebeviabilität > 50 %
|
Mittlere Gewebeviabilität ≤ 50 %
|
|
|
45.
|
Kann eine eindeutige Klassifizierung vorgenommen werden, so ist eine einzige, Prüfung mit mindestens zwei Geweben ausreichend. Bei Grenzergebnissen, wie z. B. nicht übereinstimmenden Replikatmessungen und/oder einer mittleren prozentualen Gewebeviabilität von 60 ± 5 % (VRM 1 sowie VRM 2 beim Protokoll für Flüssigkeiten) bzw. 50 ± 5 % (VRM 2 beim Protokoll für Feststoffe), sollte eine zweite bzw. bei abweichenden Ergebnissen der ersten beiden Prüfungen eine dritte Prüfung in Betracht gezogen werden.
|
|
46.
|
Wenn angemessen und begründet, können bei bestimmten Arten von Gemischen für Prüfchemikalien, bei denen eine Einstufung vorgenommen wurde, und für Prüfchemikalien, bei denen eine Einstufung nicht erforderlich ist, unterschiedliche prozentuale Grenzwerte für die Gewebeviabilität angenommen werden, um die Leistungsfähigkeit der Prüfmethode für die betreffende Art von Gemischen insgesamt zu erhöhen (Nummer 14). Referenzchemikalien können die Beurteilung des augenreizenden bzw. schwer augenschädigenden Potenzials unbekannter Prüfchemikalien oder Produktklassen und die Bewertung des relativen augentoxischen Potenzials einer klassifizierten Chemikalie innerhalb einer bestimmten Spanne positiver Reaktionen erleichtern.
|
DATEN UND BERICHTERSTATTUNG
Daten
|
47.
|
Die Daten einzelner Replikatgewebe eines Prüflaufs (z. B. OD-Werte/MTT-Formazan-Peak-Flächen und berechnete prozentuale Daten der Gewebeviabilität für die Prüfchemikalie und für Kontrollen sowie die endgültige Vorhersage mit der RhCE-Prüfmethode) sind für jede Prüfchemikalie (ggf. einschließlich der Daten von Wiederholungsprüfungen) tabellarisch darzustellen. Außerdem sind für jede einzelne Prüfchemikalie und für alle Kontrollen die mittlere prozentuale Gewebeviabilität und der Unterschied der Viabilität zwischen zwei Gewebereplikaten (bei n = 2 Replikatgewebe) bzw. die Standardabweichung (SD) (bei n ≥ 3 Replikatgeweben) anzugeben. Wenn bei einer Prüfchemikalie bei der Messung von MTT-Formazan anhand der direkten MTT-Reduktion eine Interferenz festgestellt wird und/oder eine farbliche Interferenz auftritt, ist dies für jede einzelne geprüfte Chemikalie anzugeben.
|
Prüfbericht
|
48.
|
Der Prüfbericht enthält die folgenden Angaben:
Prüfstoff
|
|
Einkomponentiger Stoff:
|
—
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung(en), CAS-Registriernummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
—
|
Aggregatzustand, Flüchtigkeit, pH-Wert, log P, Molekulargewicht, Chemikalienklasse und weitere für die Untersuchung relevante physikalische und chemische Eigenschaften, soweit verfügbar;
|
|
—
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
—
|
Behandlung vor der Untersuchung, sofern relevant (z. B. Erwärmen, Mahlen);
|
|
—
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
|
|
mehrkomponentige Stoffe, UVCB-Stoffe und Gemische:
|
—
|
Charakterisierung, so weit wie möglich, z. B. durch die chemische Zusammensetzung (siehe oben), Reinheit, das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften (siehe oben) der einzelnen Elemente, soweit verfügbar;
|
|
—
|
Aggregatzustand sowie weitere für die Untersuchung relevante physikalische und chemische Eigenschaften, soweit verfügbar;
|
|
—
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
—
|
Behandlung vor der Untersuchung, sofern relevant (z. B. Erwärmen, Mahlen);
|
|
—
|
Lagerbedingungen und Stabilität, soweit verfügbar.
|
|
Positiv- und Negativkontrollstoffe
|
—
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung(en), CAS-Registriernummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
—
|
Aggregatzustand, Flüchtigkeit, Molekulargewicht, Chemikalienklasse und weitere für die Untersuchung relevante physikalische und chemische Eigenschaften, soweit verfügbar;
|
|
—
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
—
|
Behandlung vor der Prüfung, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
—
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
—
|
ggf. Begründung für die Verwendung einer anderen Negativkontrolle als ultrareines H2O oder Ca2+/Mg2+-freier DPBS;
|
|
—
|
ggf. Begründung der Verwendung einer anderen Positivkontrolle als reines Methylacetat;
|
|
—
|
Verweis auf historische Ergebnisse von Positiv- und Negativkontrollen, die geeignete Akzeptanzkriterien für einen Prüfdurchlauf dokumentieren.
|
Informationen zu Auftraggeber und Prüfanstalt
|
—
|
Name und Anschrift des Auftraggebers, der Prüfanstalt und des Studienleiters.
|
|
—
|
RhCE-Gewebemodell und verwendetes Protokoll (ggf. mit Begründung für die getroffene Auswahl)
|
Bedingungen der Prüfmethode
|
—
|
Verwendetes RhCE-Gewebemodell einschließlich Chargennummer;
|
|
—
|
Wellenlänge und Bandbreite (wenn relevant) für die Quantifizierung von MTT-Formazan und die Linearitätsspanne des Messgeräts (z. B. eines Spektrophotometers);
|
|
—
|
Beschreibung der verwendeten Methode zur Quantifizierung von MTT-Formazan;
|
|
—
|
Beschreibung des HPLC/UPLC-Spektrophotometriesystems (wenn relevant);
|
|
—
|
umfassende Begleitdokumentation für das betreffende RhCE-Gewebemodell, einschließlich seiner Leistung. Diese sollte unter anderem die folgenden Elemente und Angaben beinhalten:
|
i)
|
Qualitätskontrolle der Viabilität (Hersteller)
|
|
ii)
|
Viabilität unter den Bedingungen der Prüfmethode (Anwender);
|
|
iii)
|
Qualitätskontrolle der Barrierefunktion;
|
|
v)
|
Reproduzierbarkeit und Vorhersagefähigkeit;
|
|
vi)
|
ggf. sonstige Qualitätskontrollen des RhCE-Gewebemodells;
|
|
|
—
|
Verweis auf historische Daten zum RhCE-Gewebemodell. Unter anderem sollte die Akzeptierbarkeit der QK-Daten auf der Grundlage historischer Chargendaten spezifiziert werden;
|
|
—
|
Erklärung, dass die Prüfeinrichtung ihre Befähigung zur Durchführung der Prüfmethode vor der regelmäßigen Untersuchung der Leistungschemikalien nachgewiesen hat.
|
Akzeptanzkriterien für Prüfläufe und Prüfungen
|
—
|
Mittelwerte und Akzeptanzspannen der Positiv- und der Negativkontrollen auf der Grundlage historischer Daten;
|
|
—
|
akzeptable Variabilität zwischen Gewebereplikaten für Positiv- und Negativkontrollen;
|
|
—
|
akzeptable Variabilität zwischen Gewebereplikaten für die Prüfchemikalie.
|
Prüfverfahren
|
—
|
Einzelheiten der angewandten Methode;
|
|
—
|
Dosierungen der verwendeten Prüfchemikalien und Kontrollstoffe;
|
|
—
|
Expositionszeitraum und -temperatur, Dauer der Immersion nach der Exposition und Inkubationszeit nach der Exposition (wenn relevant);
|
|
—
|
Beschreibung etwaiger Änderungen am Prüfverfahren;
|
|
—
|
Angabe verwendeter Kontrollen für direkte MTT-Reduktionsmittel und/oder Prüffarbstoffe (wenn relevant);
|
|
—
|
Anzahl der pro Prüfchemikalie und pro Kontrolle (PK, Negativkontrolle und NSMTT, NSClebend und NSCabgetötet, wenn relevant) verwendeten Gewebereplikate.
|
Ergebnisse
|
—
|
Tabellarische Darstellung der Daten für die einzelnen Prüfchemikalien und Kontrollstoffe für die einzelnen Prüfläufe (ggf. einschließlich Wiederholungsprüfungen) und Replikatmessungen einschließlich der OD-Werte bzw. der MTT-Formazan-Peak-Flächen, der prozentualen Gewebeviabilität, der mittleren prozentualen Gewebeviabilität, Unterschieden zwischen Gewebereplikaten bzw. der Standardabweichung und der endgültigen Vorhersagen;
|
|
—
|
gegebenenfalls Ergebnisse verwendeter Kontrollen für direkte MTT-Reduktionsmittel und/oder Prüffarbstoffe einschließlich OD-Werten oder MTT-Formazan-Peak-Flächen, % NSMTT, % NSClebend, % NSCabgetötet, Unterschieden zwischen Gewebereplikaten bzw. der Standardabweichung, der endgültigen korrekten prozentualen Gewebeviabilität und der endgültigen Vorhersagen;
|
|
—
|
mit der Prüfchemikalie (den Prüfchemikalien) und den Kontrollstoffen ermittelte Ergebnisse bezogen auf die definierten Akzeptanzkriterien für Prüfläufe und Prüfungen;
|
|
—
|
Beschreibung weiterer festgestellter Wirkungen (z. B. Verfärbung der Gewebe durch Prüffarbstoffe).
|
Erörterung der Ergebnisse.
Schlussfolgerung.
|
LITERATUR
|
(1)
|
UN (2015). GHS (Globally Harmonized System of Classification and Labelling of Chemicals = Global harmonisiertes System zur Einstufung und Kennzeichnung) der Vereinten Nationen, ST/SG/AC.10/30/Rev.6, Sechste überarbeitete Auflage, New York und Genf: Vereinte Nationen. Abrufbar unter:http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Kapitel B.5 dieses Anhangs, Akute Augenreizung/-verätzung.
|
|
(3)
|
Kapitel B.47 dieses Anhangs, Trübungs- und Durchlässigkeitstest an der Rinderhornhaut zwecks Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern.
|
|
(4)
|
Kapitel B.48 dieses Anhangs, Test am isolierten Hühnerauge zur Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung erfordern.
|
|
(5)
|
Kapitel B.61 dieses Anhangs Fluorescein-Leckage-Prüfmethode zur Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung
|
|
(6)
|
Kapitel B.68 dieses Anhangs, In-vitro-Prüfmethode mit Kurzzeitexposition zur Ermittlung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern.
|
|
(7)
|
Freeman, S.J., Alépée, N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2 010,261-266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM – Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28 125 EN; doi:10.2787/41680. Abrufbar unter:http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03, 17. November 2014; EUR 28 173 EN; doi: 10.2787/043697. Abrufbar unter:http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02, 24. Juni 2016; EUR 28 175 EN; doi: 10.2787/390390. Abrufbar unter:http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuskript in Vorbereitung).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OECD (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(18)
|
OECD (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Hrsg.), Alternative Toxicological Methods, CRC Press, S. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1 476-1 488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli, F.N., und Maibach, H.I. (Hrsg.), 4. Auflage, S. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D. (Ed.), 7. Auflage, S. 665–697. Withby, O.N., Kanada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922–936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Toxicol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115-130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2 610–2 625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41-54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (5. März 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Abrufbar unter:[https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (20. Juli 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Abrufbar unter:https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. Mai 2001. Abrufbar unter:http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications. Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
Anlage 1
BEGRIFFSBESTIMMUNGEN
Augenreizung
: Erzeugen von Veränderungen am Auge nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation vollständig reversibel sind. Synonym zu den Einstufungen „reversible Wirkungen am Auge“ und „UN-GHS-/CLP-Kategorie 2“.
Bottom-up-Ansatz
: Schrittweiser Ansatz für eine Prüfchemikalie, von der vermutet wird, dass sie keine Einstufung und Kennzeichnung als augenreizend oder schwer augenschädigend erfordert. Dabei werden zunächst Chemikalien, die keine Einstufung und Kennzeichnung erfordern (negatives Ergebnis), von anderen Chemikalien (positives Ergebnis) unterschieden.
Chemikalie
: Ein Stoff oder Gemisch.
Dev
: Abweichung
Einkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorliegt.
EIT
: Eye Irritation Test (Prüfung auf Augenreizung).
Ersatzprüfung
: Eine Prüfung, die eine routinemäßig angewandte Prüfung zur Identifikation von Gefahren und/oder zur Risikobewertung ersetzen soll, und die im Vergleich zur akzeptierten Prüfung nachweislich in allen möglichen Prüfsituationen und mit allen Stoffen einen gleichwertigen oder besseren Schutz der Gesundheit von Mensch oder Tier bzw. der Umwelt gewährleistet (18).
ET50
: Expositionszeit, die erforderlich ist, um die Gewebeviabilität bei Anwendung einer Referenzchemikalie in einer vorgegebenen festen Konzentration um 50 % zu reduzieren.
EURL ECVAM
: Referenzlabor der Europäischen Union für alternative Methoden zu Tierversuchen.
Evidenzbasierte Bewertung
: Prüfung der Stärken und Schwächen verschiedener Informationen, um über das Gefahrenpotenzial einer Prüfchemikalie entscheiden zu können und diese Entscheidung zu untermauern.
Falsch-Negativ-Rate
: Der Anteil aller Positivstoffe, die mit einer Prüfmethode fälschlicherweise als Negativstoffe identifiziert werden. Die Falsch-Negativ-Rate ist ein Indikator für die Leistungsfähigkeit einer Prüfmethode.
Falsch-Positiv-Rate
: Der Anteil aller Negativstoffe, die mit einer Prüfmethode fälschlicherweise als Positivstoffe identifiziert werden. Die Falsch-Positiv-Rate ist ein Indikator für die Leistungsfähigkeit einer Prüfmethode.
Gefahr
: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen.
Gemisch
: Ein Gemisch oder eine Lösung, die aus zwei oder mehr Stoffen besteht.
Genauigkeit
: Der Grad an Übereinstimmung zwischen Testergebnissen und akzeptierten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von „Übereinstimmung“ verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (18).
Gestufte Prüfstrategie
: Eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Bewertungsansatz (weight-of-evidence) vorgegangen wird, um feststellen zu können, ob genügend Informationen für eine Gefahreneinstufung vorliegen, bevor zur nächsten Stufe der Strategie übergegangen wird. Wenn das Gefahrenpotenzial/die gefahrenrelevante Wirksamkeit einer Prüfchemikalie auf Basis der vorliegenden Informationen ermittelt werden kann, sind keine weiteren Untersuchungen erforderlich (18).
Gewebeviabilität
: Parameter zur Messung der Gesamtaktivität einer Zellenpopulation (bei einem rekonstruierten Gewebe die Fähigkeit, den Vitalfarbstoff zu reduzieren), der je nach gemessenem Endpunkt und verwendetem Testaufbau mit der Gesamtzahl und/oder Vitalität lebender Zellen korreliert.
Gültige Prüfmethode
: Eine Prüfmethode, die eine ausreichende Relevanz und Zuverlässigkeit für einen bestimmten Zweck aufweist und auf wissenschaftlich fundierten Grundsätzen beruht. Eine Prüfmethode ist nie im absoluten Sinn, sondern nur in Bezug auf einen definierten Zweck gültig (18).
HCE
: Menschliches Hornhautepithel SkinEthic™.
Hornhaut
: Der Iris und Pupille überdeckende transparente vordere Teil des Augapfels, über den Licht ins Augeninnere übertragen wird.
HPLC
: Hochleistungs-Flüssigkeitschromatografie.
IC50
: Konzentration, bei der eine Referenzchemikalie die Viabilität der Gewebe nach einer vorgegebenen Expositionsdauer um 50 % verringert (z. B. nach einer 30-minütigen Behandlung mit SDS).
Irreversible Wirkungen am Auge
: Siehe „Schwere Augenschädigung“.
Keine Einstufung
: Chemikalien, die nicht als augenreizend (UN-GHS-/CLP-Kategorie 2, UN-GHS-/Kategorien 2A oder 2B) oder schwer augenschädigend (UN-GHS-/CLP-Kategorie 1) eingestuft werden. Synonym zu „UN-GHS-/CLP-Kategorie Keine Einstufung“.
Leistungsstandards
: Auf einer validierten und als wissenschaftlich fundiert betrachteten Prüfmethode beruhende Normen, auf deren Grundlage die Vergleichbarkeit einer vorgeschlagenen, mechanistisch und funktionell ähnlichen Prüfmethode bewertet werden kann. Sie umfassen (i) wesentliche Elemente der Prüfmethode; (ii) ein Mindestverzeichnis von Referenzchemikalien, ausgewählt aus den Chemikalien, die zum Nachweis der akzeptablen Leistung der validierten Referenzmethode verwendet werden; und (iii) je nach den für die validierte Referenzmethode erzielten Ergebnissen die vergleichbaren Genauigkeits- und Zuverlässigkeitswerte, die die vorgeschlagene Prüfmethode bei der Bewertung anhand des Mindestverzeichnisses von Referenzchemikalien erreichen sollte (18).
LLOQ
: Untere Quantifizierungsgrenze.
log P
: Logarithmus des Octanol-Wasser-Verteilungskoeffizienten.
Mehrkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens ≥ 10 % w/w und < 80 % w/w vorhanden ist. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet.
MTT
: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid; Thiazolyl-Blau-Tetrazoliumbromid.
Negativkontrolle
: Eine Probe, die alle Elemente eines Prüfsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Diese Probe wird mit Proben, die mit einer Prüfchemikalie behandelt wurden, und mit anderen Kontrollproben mitgeführt, um eine Gewebeviabilität von 100 % zu ermitteln.
NSCabgetötet
: Nicht spezifische Farbe bei abgetöteten Geweben.
NSClebend
: Nicht spezifische Farbe bei lebenden Geweben.
NSMTT
: Nicht spezifische MTT-Reduktion.
OD
: Optische Dichte.
Positivkontrolle
: Eine Probe, die alle Elemente eines Prüfsystems enthält und mit einem Stoff behandelt wird, der im Prüfsystem bekanntermaßen eine positive Reaktion hervorruft. Diese Probe wird mit Proben, die mit einer Prüfchemikalie behandelt wurden, und mit anderen Kontrollproben mitgeführt. Um sicherzustellen, dass Abweichungen der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prüfchemikalie
: Stoff oder Gemisch, der/das mit dieser Prüfmethode untersucht wird.
Prüflauf
: Ein Prüflauf besteht aus einer oder mehreren Prüfchemikalien, die gleichzeitig mit einer Negativ- und einer Positivkontrolle geprüft werden.
Prüfung
: Eine einzelne Prüfchemikalie, die gleichzeitig an mindestens zwei Gewebereplikaten im Sinne der jeweiligen Standardarbeitsanweisungen geprüft wird.
Referenzchemikalie
: Eine zum Vergleich mit einer Prüfchemikalie verwendete Chemikalie. Eine Referenzchemikalie sollte die folgenden Eigenschaften aufweisen: (i) einheitliche und zuverlässige Herkunft; (ii) strukturelle und funktionelle Ähnlichkeit mit der Klasse der zu prüfenden Stoffe; (iii) bekannte physikalisch-chemische Eigenschaften; (iv) unterstützende Daten zu bekannten Wirkungen und (v) bekannte Wirksamkeit im Bereich der erwünschten Reaktion.
Relevanz
: Dieser Begriff bezieht sich auf das Verhältnis zwischen der Prüfmethode und der betreffenden Wirkung und auf die Frage, ob dieses Verhältnis aussagekräftig und nützlich für einen bestimmten Zweck ist. Er beschreibt das Ausmaß, in dem mit einer Prüfung die untersuchte biologische Wirkung korrekt gemessen oder vorhersagt wird. Die Relevanz schließt eine Beurteilung der Genauigkeit (Übereinstimmung) einer Prüfmethode ein (18).
Reproduzierbarkeit
: Die Übereinstimmung von Ergebnissen, die mit wiederholten Prüfungen derselben Prüfchemikalie nach demselben Prüfprotokoll ermittelt wurde (siehe „Zuverlässigkeit“) (18).
Reversible Wirkungen am Auge
: Siehe „Augenreizung“.
RhCE
: Rekonstruiertes menschliches hornhautartiges Epithel.
Schwere Augenschädigung
: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation eines Prüfstoffs auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Synonym zu den Einstufungen „irreversible Wirkungen am Auge“ und „UN-GHS-/CLP-Kategorie 1“.
SD
: Standardabweichung
Sensitivität
: Der Anteil aller positiven/wirkenden Prüfchemikalien, die durch die Prüfung korrekt eingestuft werden. Die Sensitivität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (18).
Spezifität
: Der Anteil aller negativen/wirkungslosen Prüfchemikalien, die durch die Prüfung korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (18).
Standardarbeitsanweisungen
: Förmliche, schriftliche Verfahrensbeschreibungen, in denen detailliert erläutert wird, wie bestimmte regelmäßige und prüfungsspezifische Tätigkeiten im Labor durchgeführt werden sollten. Standardarbeitsanweisungen sind nach guter Laborpraxis (GLP) vorgeschrieben.
Stoff
: Ein chemisches Element und seine Verbindungen in natürlicher Form oder gewonnen durch ein Herstellungsverfahren, einschließlich der zur Wahrung seiner Stabilität notwendigen Zusatzstoffe und der durch das angewandte Verfahren bedingten Verunreinigungen, aber mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können.
Top-down-Ansatz
: Schrittweiser Ansatz bei einer Chemikalie, von der vermutet wird, dass sie schwere Augenschäden verursacht. Dabei werden zunächst Chemikalien, die schwere Augenschäden verursachen (positives Ergebnis), von anderen Chemikalien (negatives Ergebnis) unterschieden.
Übereinstimmung
: Siehe „Genauigkeit“.
Überschüssige Dosis
: Die Menge der auf das RhCE-Gewebemodell aufgetragenen Prüfchemikalie, die über die zur vollständigen und gleichmäßigen Bedeckung der Hautoberfläche erforderliche Menge hinausgeht.
ULOQ
: Obere Quantifizierungsgrenze.
UN GHS (United Nations Globally Harmonized System of Classification and Labelling of Chemicals = Global harmonisiertes System zur Einstufung und Kennzeichnung der Vereinten Nationen)
: Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenbögen, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1).
UN-GHS/CLP-Kategorie 1
: Siehe „Schwere Augenschädigung“.
UN-GHS/CLP-Kategorie 2
: Siehe „Augenreizung“.
UN-GHS-/CLP-Kategorie Keine Einstufung
: Chemikalien, die die Anforderungen für eine Einstufung in die UN-GHS-/CLP-Kategorien 1 oder 2 (oder die UN-GHS-Kategorien 2A oder 2B) nicht erfüllen. Entspricht der Kategorie „Keine Einstufung“.
UPLC
: Ultra-Hochleistungs-Flüssigkeitschromatografie.
UVCB
: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Validierte Prüfmethode
: Eine Prüfmethode, für die zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien abgeschlossen wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht genau und zuverlässig genug ist, um für den vorgeschlagenen Zweck akzeptiert zu werden (18).
VK
: Variationskoeffizient.
VRM 1
: Der EpiOcular™ EIT wird als validierte Referenzmethode 1 bezeichnet.
VRM 2
: Der SkinEthic™ HCE EIT wird als validierte Referenzmethode 2 bezeichnet.
VRM
: Validierte Referenzmethode.
Zuverlässigkeit
: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit bewertet (18).
Anlage 2
WESENTLICHE ELEMENTE DER PRÜFMETHODE UNTER VERWENDUNG VON REKONSTRUIERTEM MENSCHLICHEM HORNHAUTARTIGEM EPITHEL (RHCE) ZUR ERMITTLUNG VON CHEMIKALIEN, BEI DENEN EINE EINSTUFUNG UND KENNZEICHNUNG ALS AUGENREIZEND ODER SCHWER AUGENSCHÄDIGEND NICHT ERFORDERLICH IST
|
Elemente des Prüfmodells
|
EpiOcular™ EIT
(VRM 1)
|
SkinEthic™ HCE EIT
(VRM 2)
|
|
Protokolle
|
Flüssigkeiten
(15 Minuten bei Temperaturen von höchstens 37 ± 1 oC pipettierbar)
|
Feststoffe
(nicht pipettierbar)
|
Flüssigkeiten und viskose Flüssigkeiten:
(pipettierbar)
|
Feststoffe
(nicht pipettierbar)
|
|
Modelloberfläche
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Anzahl der Gewebereplikate
|
Mindestens 2
|
Mindestens 2
|
Mindestens 2
|
Mindestens 2
|
|
Vorabprüfung auf Farbinterferenzen
|
50 μl + 1 ml H2O 60 min bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit (nicht farbige Prüfchemikalien) bzw. 50 μl + 2 ml Isopropanol 2-3 h bei Raumtemperatur gemischt (Prüffarbstoffe)
|
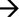 Wenn die OD der Prüfchemikalie bei 570 ± 20 nM nach Subtraktion der OD von Isopropanol oder Wasser > 0,08 ist (was etwa 5 % der mittleren OD der Negativkontrolle entspricht), sollten Prüfungen an geeigneten lebenden Kontrollen durchgeführt werden.
Wenn die OD der Prüfchemikalie bei 570 ± 20 nM nach Subtraktion der OD von Isopropanol oder Wasser > 0,08 ist (was etwa 5 % der mittleren OD der Negativkontrolle entspricht), sollten Prüfungen an geeigneten lebenden Kontrollen durchgeführt werden.
|
|
50 mg + 1 ml H2O 60 min bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit (nicht farbige Prüfchemikalien)
und/oder
50 mg + 2 ml Isopropanol 2-3 h bei Raumtemperatur gemischt (Prüffarbstoffe und nicht farbige Prüfchemikalien)
|
 Wenn die OD der Prüfchemikalie bei 570 ± 20 nM nach Subtraktion der OD von Isopropanol oder Wasser > 0,08 ist (was etwa 5 % der mittleren OD der Negativkontrolle entspricht), sollten Prüfungen an geeigneten lebenden Kontrollen durchgeführt werden.
Wenn die OD der Prüfchemikalie bei 570 ± 20 nM nach Subtraktion der OD von Isopropanol oder Wasser > 0,08 ist (was etwa 5 % der mittleren OD der Negativkontrolle entspricht), sollten Prüfungen an geeigneten lebenden Kontrollen durchgeführt werden.
|
|
10 μl + 90 μl H2O 30 ± 2 min bei Raumtemperatur (RT) gemischt (RT = 18-28 oC)
|
 Wenn sich die Prüfchemikalie färbt, sollten Prüfungen an geeigneten lebenden Kontrollen durchgeführt werden.
Wenn sich die Prüfchemikalie färbt, sollten Prüfungen an geeigneten lebenden Kontrollen durchgeführt werden.
|
|
10 mg + 90 μl H2O 30 ± 2 min gemischt bei Raumtemperatur
|
 Wenn sich die Prüfchemikalie färbt, sollten Prüfungen an geeigneten lebenden Kontrollen durchgeführt werden.
Wenn sich die Prüfchemikalie färbt, sollten Prüfungen an geeigneten lebenden Kontrollen durchgeführt werden.
|
|
|
Vorabprüfung auf direkte MTT-Reduktion
|
50 μl + 1 ml MTT 1 mg/ml Lösung, 180 ± 15 min bei 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % relative Luftfeuchtigkeit
|
 wenn sich die Lösung blau/violett färbt, sollten geeignete durch Gefrieren abgetötete Kontrollen geprüft werden
wenn sich die Lösung blau/violett färbt, sollten geeignete durch Gefrieren abgetötete Kontrollen geprüft werden
50 μl steriles entionisiertes Wasser in MTT-Lösung).
|
|
50 mg + 1 ml MTT 1 mg/ml Lösung, 180 ± 15 min bei 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % relative Luftfeuchtigkeit
|
 Wenn die Lösung sich blau/violett färbt, sollten durch Gefrieren abgetötete Kontrollen untersucht werden
Wenn die Lösung sich blau/violett färbt, sollten durch Gefrieren abgetötete Kontrollen untersucht werden
(Negativkontrolle = 50 μl steriles entionisiertes Wasser in MTT-Lösung).
|
|
30 μl + 300 μl MTT 1 mg/ml Lösung, 180 ± 15 min bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
 Wenn die Lösung sich blau/violett färbt, sollten geeignete in Wasser abgetötete Kontrollen untersucht werden
Wenn die Lösung sich blau/violett färbt, sollten geeignete in Wasser abgetötete Kontrollen untersucht werden
(Negativkontrolle = 30 μl steriles entionisiertes Wasser in MTT-Lösung).
|
|
30 mg + 300 μl MTT 1 mg/ml Lösung, 180 ± 15 min bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
 Wenn die Lösung sich blau/violett färbt, sollten geeignete in Wasser abgetötete Kontrollen untersucht werden
Wenn die Lösung sich blau/violett färbt, sollten geeignete in Wasser abgetötete Kontrollen untersucht werden
(Negativkontrolle = 30 μl steriles entionisiertes Wasser in MTT-Lösung).
|
|
|
Vorbehandlung
|
20 μl Ca2+/Mg2+-freier DPBS
30 ± 2 min bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit, lichtgeschützt.
|
20 μl Ca2+/Mg2+ -freier DPBS
30 ± 2 min bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit, lichtgeschützt.
|
–
|
–
|
|
Behandlungsdosierungen und Auftragung
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) mit einem geeichten Werkzeug (z. B. einem gestrichenen Löffel, der 50 mg Natriumchlorid fasst).
|
10 μl Ca2+/Mg2+-freier DPBS
Bei viskosen Flüssigkeiten ist ein Nylon-Maschenfilter zu verwenden.
|
30 μl Ca2+/Mg2+-freier DPBS
|
|
Expositionszeitraum und temperatur
|
30 min (± 2 min)
im Kulturmedium
bei 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % relative Luftfeuchtigkeit
|
6 h (± 0,25 h)
im Kulturmedium
bei 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % relative Luftfeuchtigkeit
|
30 min (± 2 min)
im Kulturmedium
bei 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % relative Luftfeuchtigkeit
|
4 h (± 0,1 h)
im Kulturmedium
bei 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % relative Luftfeuchtigkeit
|
|
Waschen bei Raumtemperatur
|
Dreimal in 100 ml Ca2+/Mg2+-freiem DPBS
|
Dreimal in 100 ml Ca2+/Mg2+-freiem DPBS
|
20 μl Ca2+/Mg2+-freiem DPBS
|
25 μl Ca2+/Mg2+-freiem DPBS
|
|
Immersion nach der Exposition
|
12 min (± 2 min) bei Raumtemperatur im Kulturmedium
|
25 min (± 2 min) bei Raumtemperatur im Kulturmedium
|
30 min (± 2 min) bei 37 oC, 5 % CO2, 95 % relative Luftfeuchtigkeit im Kulturmedium
|
30 min (± 2 min) bei Raumtemperatur im Kulturmedium
|
|
Inkubation nach der Exposition
|
120 min (± 15 min) im Kulturmedium bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
18 h (± 0,25 h) im Kulturmedium bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
–
|
18 h (± 0,5 h) im Kulturmedium bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
|
Negativkontrolle
|
50 μl H2O
Gleichzeitig geprüft
|
50 μl H2O
Gleichzeitig geprüft
|
30 ± 2 μl Ca2+/Mg2+-freier DPBS
Gleichzeitig geprüft
|
30 ± 2 μl Ca2+/Mg2+-freier DPBS
Gleichzeitig geprüft
|
|
Positivkontrolle
|
50 μl Methylacetat
Gleichzeitig geprüft
|
50 μl Methylacetat
Gleichzeitig geprüft
|
30 ± 2 μl Methylacetat
Gleichzeitig geprüft
|
30 ± 2 μl Methylacetat
Gleichzeitig geprüft
|
|
MTT-Lösung
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
MTT Inkubationszeit and temperature
|
180 min (± 15 min) im Kulturmedium bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
180 min (± 15 min) im Kulturmedium bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
180 min (± 15 min) im Kulturmedium bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
180 min (± 15 min) im Kulturmedium bei 37 ± 2 oC, 5 ± 1 % CO2, ≥95 % relative Luftfeuchtigkeit
|
|
Extraktionslösung
|
2 ml Isopropanol
(Extraktion oben und unten aus dem Einsatz unter Durchstechen des Gewebes)
|
2 ml Isopropanol
(Extraktion unten aus dem Einsatz unter Durchstechen des Gewebes)
|
1,5 ml Isopropanol
(Extraktion oben und unten aus dem Einsatz)
|
1,5 ml Isopropanol
(Extraktion unten aus dem Einsatz)
|
|
Expositionszeitraum and temperature
|
2-3 h unter Schütteln (~120 rpm) bei Raumtemperatur oder über Nacht bei 4-10 oC
|
2-3 h unter Schütteln (~120 rpm) bei Raumtemperatur oder über Nacht bei 4-10 oC
|
4 h unter Schütteln (~120 rpm) bei Raumtemperatur oder über mindestens Nacht ohne Schütteln bei 4-10 oC
|
Mindestens 2 h unter Schütteln (~120 rpm) bei Raumtemperatur
|
|
OD-Anzeige
|
570 nM (550 - 590 nM)
ohne Referenzfilter
|
570 nM (550-590 nM)
ohne Referenzfilter
|
570 nM (540 - 600 nM)
ohne Referenzfilter
|
570 nM (540 - 600 nM)
ohne Referenzfilter
|
|
Kontrolle der Gewebequalität
|
Behandlung mit 100 μl 0,3 % (v/v) Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
Behandlung mit 100 μl 0,3 % (v/v) Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
30-minütige Behandlung mit SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
30-minütige Behandlung mit SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Akzeptanzkriterien
|
|
1.
|
Die mittlere OD der mit der Negativkontrolle behandelten Gewebereplikate sollte ≥ 0,8 und ≤ 2,5 sein.
|
|
2.
|
Die mittlere Viabilität der Gewebereplikate nach 30-minütiger Exposition gegenüber der Positivkontrolle, ausgedrückt in % der Negativkontrolle, sollte < 50 % sein.
|
|
3.
|
Die Viabilität zweier Gewebereplikate sollte sich um weniger als 20 % unterscheiden.
|
|
|
1.
|
Die mittlere OD der mit der Negativkontrolle behandelten Gewebereplikate sollte ≥ 0,8 und ≤ 2,5 sein.
|
|
2.
|
Die mittlere Viabilität der Gewebereplikate nach 6-stündiger Exposition gegenüber der Positivkontrolle, ausgedrückt in % der Negativkontrolle, sollte < 50 % sein.
|
|
3.
|
Die Viabilität zweier Gewebereplikate sollte sich um weniger als 20 % unterscheiden.
|
|
|
1.
|
Die mittlere OD der mit der Negativkontrolle behandelten Gewebereplikate sollte > 1,0 und ≤ 2,5 sein.
|
|
2.
|
Die mittlere Viabilität der Gewebereplikate nach 30-minütiger Exposition gegenüber der Positivkontrolle, ausgedrückt in % der Negativkontrolle, sollte < 30 % sein.
|
|
3.
|
Die Viabilität zweier Gewebereplikate sollte sich um weniger als 20 % unterscheiden.
|
|
|
1.
|
Die mittlere OD der mit der Negativkontrolle behandelten Gewebereplikate sollte > 1,0 und ≤ 2,5 sein.
|
|
2.
|
Die mittlere Viabilität der Gewebereplikate nach 4-stündiger Exposition gegenüber der Positivkontrolle, ausgedrückt in % der Negativkontrolle, sollte ≤ 20 % sein.
|
|
3.
|
Die Viabilität zweier Gewebereplikate sollte sich um weniger als 20 % unterscheiden.
|
|
Anlage 3
FLUSSDIAGRAMM ZUR VERANSCHAULICHUNG DER ERMITTLUNG UND BEHANDLUNG VON DIREKTEN MTT-REDUKTIONSMITTELN UND/ODER VON CHEMIKALIEN, DIE FARBINTERFERENZEN VERURSACHEN, NACH DEN STANDARDARBEITSANWEISUNGEN DER VRM 1
Anlage 4
FLUSSDIAGRAMM ZUR VERANSCHAULICHUNG DER ERMITTLUNG UND BEHANDLUNG VON DIREKTEN MTT-REDUKTIONSMITTELN UND/ODER VON CHEMIKALIEN, DIE FARBINTERFERENZEN VERURSACHEN, NACH DEN STANDARDARBEITSANWEISUNGEN DER VRM 2

Anlage 5
SCHLÜSSELPARAMETER UND AKZEPTANZKRITERIEN FÜR DIE QUALIFIZIERUNG EINES HPLC/UPLC-SPEKTROPHOTOMETRIESYSTEMS ZUR MESSUNG VON AUS RhCE-GEWEBEMODELLEN EXTRAHIERTEM MTT-FORMAZAN
|
Parameter
|
Protokoll abgeleitet aus FDA-Leitlinie (36) (38)
|
Akzeptanzkriterien
|
|
Selektivität
|
Analyse von Isopropanol, Blindkontrolle lebend (Isopropanol-Extrakt aus lebenden RhCE-Gewebemodellen ohne Behandlung), Blindkontrolle abgetötet (Isopropanol-Extrakt aus abgetöteten RhCE-Gewebemodellen ohne Behandlung) und einem Farbstoff (z. B. Methylenblau)
|
FlächeInterferenz ≤ 20 % FlächeLLOQ
(88)
|
|
Präzision
|
Qualitätskontrollen (d. h. MTT-Formazan bei 1,6 μg/ml, 16 μg/ml und 160 μg/ml) in Isopropanol (n=5)
|
VK ≤ 15 % bzw. ≤ 20 % für LLOQ
|
|
Genauigkeit
|
Qualitätskontrollen mit Isopropanol (n=5)
|
% Abw. ≤ 15 % bzw. ≤ 20 % für LLOQ
|
|
Matrixeffekt
|
Qualitätskontrollen mit Blindkontrolle lebend (n=5)
|
85 % ≤ Matrixeffekt % ≤ 115 %
|
|
Übertragung
|
Analyse Isopropanol nach einem ULOQ (89) -Standard
|
FlächeInterferenz ≤ 20 % FlächeLLOQ
|
|
Reproduzierbarkeit (innerhalb eines Tages)
|
3 unabhängige Eichkurven (aufgrund von 6 aufeinander folgenden Verdünnungen von MTT-Formazan in Isopropanol im Verhältnis 1:3 beginnend mit der ULOQ, d. h. 200 μg/ml);
Qualitätskontrollen mit Isopropanol (n=5)
|
Eichkurven: % Abw. ≤ 15 % bzw. ≤ 20 % für LLOQ
Qualitätskontrollen: % Abw. ≤ 15 % und VK ≤ 15 %
|
|
Reproduzierbarkeit (innerhalb eines Tages)
|
Tag 1: 1 Eichkurve und Qualitätskontrollen in Isopropanol (n=3)
Tag 2: 1 Eichkurve und Qualitätskontrollen in Isopropanol (n=3)
Tag 3: 1 Eichkurve und Qualitätskontrollen in Isopropanol (n=3)
|
|
Kurzzeitstabilität von MTT-Formazan in RhCE-Gewebeextrakt
|
Qualitätskontrollen in Blindkontrollen lebend (n=3), die am Tag der Vorbereitung sowie nach 24-stündiger Lagerung bei Raumtemperatur analysiert wurden.
|
% Abw. ≤ 15 %
|
|
Langzeitstabilität von MTT-Formazan in RhCE-Gewebeextrakt (erforderlichenfalls)
|
Qualitätskontrollen in Blindkontrollen lebend (n=3), die am Tag der Vorbereitung sowie nach mehrtägiger Lagerung bei -20 °C analysiert wurden.
|
% Abw. ≤ 15 %
|
B.70
IN-VITRO-ASSAYS MIT EINEM HUMANEN REKOMBINANTEN ÖSTROGENREZEPTOR (hrER) ZUR ERMITTLUNG VON CHEMIKALIEN MIT ER-BINDUNGSAFFINITÄT
ALLGEMEINE EINLEITUNG
Leistungsbezogene OECD-Prüfrichtlinie
|
1.
|
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 493 (2015). TG 493 ist eine leistungsbezogene Prüfrichtlinie (PBTG), die die Methode für In-vitro-Assays unter Verwendung eines humanen rekombinanten Östrogenrezeptors zur Ermittlung von Stoffen mit Bindungsaffinität für Östrogenrezeptoren (hrER-Bindungsassays) beschreibt. Sie umfasst zwei mechanistisch und funktionell ähnliche Assays zum Nachweis von Östrogenrezeptor-Bindern (d. h. ERα-Bindern) und sollte die Entwicklung neuer ähnlicher oder modifizierter Prüfmethoden nach den Validierungsgrundsätzen im OECD Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment erleichtern (1). Die folgenden vollständig validierten Referenzprüfmethoden (Anlagen 2 und 3) sind Grundlage dieser PBTG:
|
—
|
der Freyberger-Wilson-(FW-)In-vitro-Östrogenrezeptor-(ER-)Bindungsassay mit dem in voller Länge rekombinanten humanen ERα (2) und
|
|
—
|
der vom Chemical Evaluation and Research Institute (CERI) beschriebene In-vitro-Östrogenrezeptor-Bindungsassay mit einem Ligandenbindungs-Domänen-Protein des humanen rekombinanten ERα (2).
|
Leistungsstandards (PS) (3) sollen die Entwicklung und die Validierung ähnlicher Prüfmethoden für denselben Gefahren-Endpunkt erleichtern und eine rechtzeitige Änderung der PBTG 493 ermöglichen, damit ähnliche Assays in einer aktualisierten PBTG berücksichtigt werden können. Ähnliche Assays werden jedoch erst dann berücksichtigt, wenn die OECD die Assays geprüft und die Erfüllung der Leistungsstandards bestätigt hat. Die in TG 493 aufgenommenen Assays erfüllen die Anforderungen der OECD-Mitgliedstaaten an Prüfergebnisse bei Östrogenrezeptor-Bindungen alle gleichermaßen und ermöglichen die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen.
|
Hintergrund und Grundsätze der in diese Prüfmethode aufgenommenen Assays
|
2.
|
Die OECD leitete 1998 mit hoher Priorität die Überarbeitung bestehender Prüfrichtlinien für Screening-Prüfungen und Untersuchungen potenziell endokriner Disruptoren und die Entwicklung neuer Prüfrichtlinien ein. Der „OECD Conceptual Framework (CF) for the Testing and Assessment of Endocrine Disrupting Chemicals“ wurde 2012 überarbeitet. Die ursprünglichen und überarbeiteten CFs wurden dem Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (4) als Anhänge beigefügt. Der CF umfasst fünf Ebenen mit jeweils eigener biologischer Komplexität. Die in dieser Prüfmethode beschriebenen ER-Bindungsassays sind Ebene 2 (In-vitro-Assays zur Ermittlung von Daten über ausgewählte endokrine Mechanismen/Wirkungspfade) zuzuordnen. Diese Prüfmethode betrifft In-vitro-Rezeptor-Bindungsassays zur Ermittlung von Liganden für den humanen Östrogenrezeptor alpha (ERα).
|
|
3.
|
Die Relevanz des In-vitro-ER-Bindungsassays für biologische Funktionen wurde eindeutig nachgewiesen. ER-Bindungsassays sind zur Ermittlung von Chemikalien vorgesehen, die den Östrogen-Hormonweg unterbrechen können. Sie wurden in den letzten beiden Jahrzehnten in großem Umfang zur Charakterisierung der Verteilung von ER in Geweben und zur Identifizierung von ER-Agonisten/-Antagonisten durchgeführt. Diese Assays spiegeln das Zusammenwirken zwischen Liganden und Rezeptoren als ersten Schritt des Östrogen-Signalwegs wider und sind bei allen Wirbeltieren wesentlich für die Reproduktionsfunktion.
|
|
4.
|
Das Zusammenwirken von Östrogenen mit ER kann die Transkription von Genen beeinträchtigen, die durch Östrogene gesteuert werden, und nicht genomische Wirkungen induzieren. Dadurch können Zellprozesse ausgelöst oder gehemmt werden (u. a. für die Zellproliferation, für die normale fetale Entwicklung und für die Reproduktion erforderliche Prozesse) (5) (6) (7). Störungen normaler östrogener Systeme können Beeinträchtigungen der normalen Entwicklung (Ontogenese), der reproduktiven Gesundheit und des Reproduktionssystems auslösen. Eine unangemessene Übertragung von ER-Signalen kann beispielsweise das Risiko einer hormonbedingten Krebserkrankung erhöhen, die Fruchtbarkeit beeinträchtigen und das Wachstum und die Entwicklung von Föten beeinflussen (8).
|
|
5.
|
In-vitro-Bindungsassays beruhen auf einer unmittelbaren Wechselwirkung eines Stoffs mit einer spezifischen Rezeptorliganden-Bindungsstelle, die die Transkription eines Reporter-Genprodukts steuert. Wesentliches Element des hrERα-Bindungsassays (hrERα = humaner rekombinanter Östrogenrezeptor alpha) ist die Fähigkeit eines radioaktiv markierten Liganden ([3H]-17β-Estradiol), bei zunehmenden Konzentrationen einer Prüfchemikalie (z. B. eines Bindungskonkurrenten) eine Bindung mit dem ER einzugehen. Prüfchemikalien mit einer starken ER-Affinität konkurrieren mit dem radioaktiv markierten Liganden bei einer niedrigeren Konzentration als Chemikalien mit einer geringeren Affinität für diesen Rezeptor. Dieser Assay beinhaltet zwei wesentliche Elemente: einen Versuch zur Ermittlung von Sättigungsbindungen zur Charakterisierung der Parameter der Wechselwirkung zwischen Rezeptoren und Liganden und zur Dokumentation der ER-Spezifität und einen anschließenden Versuch zur Ermittlung konkurrierender Bindungen, mit dem die Konkurrenz zwischen einer Prüfchemikalie und einem radioaktiv markierten Liganden im Hinblick auf die Bindung an den ER beschrieben wird.
|
|
6.
|
In Studien zur Validierung der CERI- und der FW-Bindungsassays wurden die Relevanz und die Zuverlässigkeit im Hinblick auf die vorgesehene Verwendung nachgewiesen (2).
|
|
7.
|
Die in der vorliegenden Prüfmethode verwendeten Begriffe und Abkürzungen werden in Anlage 1 erläutert.
|
Anwendungsbereich und Einschränkungen der Rezeptor-Bindungsassays
|
8.
|
Diese Assays werden für Screening- und Priorisierungszwecke vorgeschlagen, können aber auch Aufschluss über molekulare Auslösungsereignisse (MIE = Molecular Initiation Events) geben. Die betreffenden Informationen können im Rahmen von Beweiskraftermittlungen verwendet werden. Die Assays beruhen auf der chemischen Bindung an die Ligandenbindungs-Domäne des ERα in einem In-vitro-System. Daher sollten Ergebnisse nicht unmittelbar für die komplexen Signal- und Steuerungswirkungen des intakten endokrinen In-vivo-Systems extrapoliert werden.
|
|
9.
|
Die Bindung des natürlichen Liganden 17β-Estradiol ist das erste einer Reihe molekularer Ereignisse, die die Transkription von Zielgenen aktivieren und schließlich zu einer physiologischen Veränderung führen (9). Die Bindung an die Ligandenbindungs-Domäne des ERα ist also als einer der Schlüsselmechanismen einer durch ER vermittelten endokrinen Störung (ED = Endocrine Disruption) zu betrachten; allerdings können ED auch infolge anderer Mechanismen auftreten, darunter (i) Wechselwirkungen mit anderen ER-Bindungsstellenα als die Liganden-Bindungstasche, (ii) Wechselwirkungen mit anderen für die Übertragung von Östrogensignalen relevanten Rezeptoren, ER-β und G-Protein-gekoppelten Östrogenrezeptoren, sonstigen Rezeptoren und Enzymsystem innerhalb des Hormonsystems, (iii) die Hormonsynthese, (iv) die metabolische Aktivierung/Deaktivierung von Hormonen, (v) die Verteilung von Hormonen an Zielgewebe und (vi) die Beseitigung von Hormonen im Körper. Keiner der Assays im Rahmen dieser Prüfmethode hat diese Wirkungsweisen zum Gegenstand.
|
|
10.
|
Gegenstand dieser Prüfmethode ist die Fähigkeit von Stoffen, Bindungen mit dem humanen ERα einzugehen. Dabei wird nicht zwischen ERα-Agonisten und ERα-Antagonisten unterschieden. Nachgelagerte Ereignisse wie Gentranskriptionen oder physiologische Veränderungen werden bei diesen Assays nicht berücksichtigt. Da bei der Validierung nur einzelne einkomponentige Stoffe verwendet wurden, wurde die Eignung für Prüfgemische nicht untersucht. Dennoch sind die Assays theoretisch auch für die Prüfung von mehrkomponentigen Stoffen und Gemischen geeignet. Bevor die Prüfmethode für die Generierung von Daten für einen bestimmten rechtlichen Zweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs von Rechts wegen vorgeschrieben ist.
|
|
11.
|
Die zellfreien Rezeptorsysteme können an sich keine Stoffwechselaktivität entwickeln und wurden in Verbindung mit metabolischen Enzymsystemen nicht validiert. Eine Stoffwechselaktivität könnte bei der Auslegung einer Studie berücksichtigt werden. Dazu wären jedoch weitere Validierungsarbeiten erforderlich.
|
|
12.
|
Chemikalien, die in der Lage sind, das Protein (d. h. das Rezeptor-Protein) zu denaturieren (beispielsweise ein Tensid oder Chemikalien, die den pH-Wert des Assay-Puffers verändern können), können nicht bzw. nur in Konzentrationen geprüft werden, bei denen diese Wechselwirkungen nicht auftreten. Ansonsten beschränkt sich der Konzentrationsbereich, der mit diesen Assays geprüft werden kann, auf die Löslichkeit der betreffenden Prüfchemikalie im Assay-Puffer.
|
|
13.
|
Tabelle 1 enthält eine Übersicht über die Ergebnisse der 24 Stoffe, die mit den beiden in dieser Prüfmethode beschriebenen vollständig validierten Assays untersucht wurden. Von diesen Stoffen wurden nach veröffentlichten Berichten (u. a. aufgrund von In-vitro-Assays zur Prüfung auf transkriptionelle ER-Aktivierung und/oder aufgrund des Uterotrophen Bioassays) (9) (10) (11) (12) (13) (14) (15) 17 Stoffe als ER-Binder eingestuft; 6 Stoffe wurden als Nicht-Binder eingestuft. Nach den in Tabelle 1 zusammengefassten Daten ergab sich hinsichtlich der Einstufungen aller Stoffe bis zu 10-4 M beim Antagonisten-Assay eine nahezu 100 %ige Übereinstimmung zwischen den beiden Assays, und alle Stoffe wurden zutreffend als ER-Binder bzw. als Nicht-Binder eingestuft. Ergänzende Informationen über diese Gruppe von Stoffen sowie über weitere Stoffe, die im Rahmen der Validierungsstudien mit den ER-Bindungsassays untersucht wurden, sind den Leistungsstandards für den hrER-Bindungsassay (3), Anlage 2 (Tabellen 1, 2 und 3), zu entnehmen.
Tabelle 1
Einstufung von Stoffen als ER-Binder oder -Nicht-Binder nach der erwarteten Reaktion bei Prüfung mit dem FW- und dem CERI-hrER-Bindungsassay
|
|
Bezeichnung des Stoffs
|
CAS-Nr.
|
Erwartete Reaktion
|
FW-Assay
|
CERI-Assay
|
MeSH-Chemikalie Klasse
|
Produktklasse
|
|
|
Konzentrations- bereich (M)
|
Einstufung
|
Konzentrations- bereich (M)
|
Einstufung
|
|
1
|
17β-Estradiol
|
50-28-2
|
Binder
|
1×10-11 – 1×10-6
|
Binder
|
1×10-11 – 1×10-6
|
Binder
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
2
|
Norethynodrel
|
68-23-5
|
Binder
|
3×10-9 – 30×10-4
|
Binder
|
3×10-9 – 30×10-4
|
Binder
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
3
|
Norethindron
|
68-22-4
|
Binder
|
3×10-9 – 30×10-4
|
Binder
|
3×10-9 – 30×10-4
|
Binder
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
4
|
Di-n-butylphthalat
|
84-74-2
|
Nicht-Binder (*5)
|
1×10-10 – 1×10-4
|
Nicht-Binder (*6)
(1)
|
1×10-10 – 1×10-4
|
Nicht-Binder (*6)
(1)
|
Kohlenwasserstoff (cyclisch), Ester
|
Weichmacher, chemisches Zwischenprodukt
|
|
5
|
DES
|
56-53-1
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kohlenwasserstoff (cyclisch), Phenol
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
6
|
17α-Ethinylestradiol
|
57-63-6
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
7
|
meso-Hexestrol
|
84-16-2
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kohlenwasserstoff (cyclisch), Phenol
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
8
|
Genistein
|
446-72-0
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kohlenwasserstoff (heterocyclisch), Flavonoid
|
Naturprodukt
|
|
9
|
Equol
|
531-95-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Phytoestrogen-Metabolit
|
Naturprodukt
|
|
10
|
Butylparaben (n-Butyl-4-hydroxybenzoat)
|
94-26-8
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Paraben
|
Konservierungsmittel
|
|
11
|
Nonylphenol (Gemisch)
|
84852-15-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Alkylphenol
|
Zwischenproduktverbindung
|
|
12
|
o,p’-DDT
|
789-02-6
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
chlororganisches
|
Insektizid
|
|
13
|
Corticosteron
|
50-22-6
|
Nicht-Binder (*5)
|
1×10-10 – 1×10-4
|
Nicht-Binder
|
1×10-10 – 1×10-4
|
Nicht-Binder
|
Steroid
|
Naturprodukt
|
|
14
|
Zearalenon
|
17924-92-4
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kohlenwasserstoff (heterocyclisch), Lacton
|
Naturprodukt
|
|
15
|
Tamoxifen
|
10540-29-1
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kohlenwasserstoff (cyclisch)
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
16
|
5α-Dihydrotestosteron
|
521-18-6
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Steroid, nicht phenolisch
|
Naturprodukt
|
|
17
|
Bisphenol A
|
80-05-7
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Phenol
|
Chemisches Zwischenprodukt
|
|
18
|
4-n-Heptylphenol
|
1987-50-4
|
Binder
|
1×10-10 – 1×10-3
|
Nicht eindeutig (5)
|
1×10-10 – 1×10-3
|
Binder
|
Alkylphenol
|
Zwischendrehzahl
|
|
19
|
Kepon (Chlordecon)
|
143-50-0
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kohlenwasserstoff (halogeniert)
|
Pestizid
|
|
20
|
Benz(a)anthracen
|
56-55-3
|
Nicht-Binder
|
1×10-10 – 1×10-3
|
Nicht-Binder (6)
|
1×10-10 – 1×10-3
|
Nicht-Binder (6)
|
Aromatischer Kohlenwasserstoff
|
Zwischenprodukt
|
|
21
|
Enterolacton
|
78473-71-9
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Phytoestrogen
|
Naturprodukt
|
|
22
|
Progesteron
|
57-83-0
|
Nicht-Binder (*5)
|
1×10-10 – 1×10-4
|
Nicht-Binder
|
1×10-10 – 1×10-4
|
Nicht-Binder
|
Steroid
|
Naturprodukt
|
|
23
|
Octyltriethoxysilan
|
2943-75-1
|
Nicht-Binder
|
1×10-10 – 1×10-3
|
Nicht-Binder
|
1×10-10 – 1×10-3
|
Nicht-Binder
|
Silan
|
Oberflächen-Modifiziermittel
|
|
24
|
Atrazin
|
1912-24-9
|
Nicht-Binder (*5)
|
1×10-10 – 1×10-4
|
Nicht-Binder
|
1×10-10 – 1×10-4
|
Nicht-Binder
|
Heterocyclische Verbindung
|
Herbizid
|
|
ELEMENTE DES hrER-BINDUNGSASSAYS
Wesentliche Elemente des Assays
|
14.
|
Diese Prüfmethode gilt für Assays mit einem ER-Rezeptor und einem ausreichend starken Liganden für diesen Rezeptor, der als Marker/Tracer für den Assay verwendet und durch zunehmende Konzentrationen einer Prüfchemikalie verdrängt werden kann. Die Bindungsassays beinhalten die beiden folgenden wesentlichen Elemente: 1) einen Versuch zur Ermittlung von Sättigungsbindungen und 2) einen Versuch zur Ermittlung konkurrierender Bindungen. Mit dem Assay zur Ermittlung von Sättigungsbindungen werden die Spezifität und die Aktivität der Rezeptor-Zubereitungen bestätigt. Mit der Untersuchung zur Ermittlung konkurrierender Bindungen wird die Fähigkeit einer Prüfchemikalie zur Bindung an den hrER beurteilt.
|
Kontrollen
|
15.
|
Die Grundlage der vorgeschlagenen gleichzeitig geprüften Referenzöstrogens und der Kontrollen sollte erläutert werden. Gegebenenfalls dienen gleichzeitige Kontrollen (Lösungsmittel- (Vehikel-), Positiv- (ER-Binder; starke und geringe Affinität) und Negativkontrollen (Nicht-Binder)) als Nachweis dafür, dass der Assay unter den Prüfbedingungen funktioniert; außerdem bilden sie eine Grundlage für versuchsübergreifende Vergleiche. In der Regel sind sie Bestandteil der Akzeptanzkriterien für die Versuche (1). Bei jedem Prüflauf sollten auf jeweils einer Platte vollständige Konzentrationskurven des Referenzöstrogens und der Kontrollen (d. h. schwache Binder und Nicht-Binder) verwendet werden. Alle anderen Platten sollten Folgendes enthalten: 1) eine hohe (etwa vollständige Verdrängung des radioaktiv markierten Liganden) und eine mittlere Konzentration (etwa IC50) sowohl von E2 als auch des schwachen Binders, jeweils dreifach, und 2) eine Lösungsmittelkontrolle und einen nicht spezifischen Binder, ebenfalls jeweils dreifach.
|
Standard-Qualitätskontrollverfahren
|
16.
|
Standard-Qualitätskontrollverfahren sollten wie für den jeweiligen Assay beschrieben durchgeführt werden, um gewährleisten zu können, dass aktive Rezeptoren, die korrekten Chemikalienkonzentrationen und auch über mehrere Wiederholungen stabile Toleranzgrenzen verwendet werden und sichergestellt ist, dass die erwarteten ER-Bindungsreaktionen im Zeitverlauf festgestellt werden können.
|
Nachweis der Eignung des Labors
|
17.
|
Vor der Prüfung unbekannter Chemikalien mit einem der Assays dieser Prüfmethode sollte jedes Labor nachweisen, dass es in der Lage ist, den betreffenden Assay durchzuführen. Dazu sind Sättigungsprüfungen zur Bestätigung der Spezifität und der Aktivität der ER-Zubereitung sowie Assays zur Ermittlung konkurrierender Bindungen mit dem Referenzöstrogen und den Kontrollen (schwacher Binder und Nicht-Binder) durchzuführen. Das Labor sollte eine historische Datenbank mit Ergebnissen des Referenzöstrogens und der Kontrollen aufgrund von 3-5 unabhängigen Versuchen an unterschiedlichen Tagen erstellen. Die betreffenden Versuche bilden die Grundlage für das Referenzöstrogen und die historischen Kontrollen des Labors und werden zur partiellen Beurteilung der Akzeptierbarkeit des Assays bei künftigen Prüfläufen berücksichtigt.
|
|
18.
|
Die Reaktionsfähigkeit des Prüfsystems wird auch durch eine Prüfung der in Tabelle 2 genannten Leistungsstoffe bestätigt. Die Liste der Leistungsstoffe ist eine Untergruppe der Referenzstoffe der Leistungsstandards für die ER-Bindungsassays (3). Diese Stoffe sind im Handel erhältlich und entsprechen den Chemikalienklassen, bei denen gewöhnlich eine ER-Bindungsaktivität zu verzeichnen ist. Außerdem zeigen sie ein geeignetes Wirkungsspektrum (d. h. stark bis schwach) für ER-Binder und für Nicht-Binder (d. h. Negativstoffe). Bei allen Leistungsstoffen sollten die zu geprüften Konzentrationen den gesamten in Tabelle 2 genannten Bereich abdecken. Für jeden Stoff sollten mindestens drei Versuche durchgeführt werden, und die Ergebnisse sollten mit der erwarteten chemischen Aktivität in Einklang stehen. Jeder Versuch sollte unabhängig (d. h. mit frischen Verdünnungen des Rezeptors, der Chemikalien und des Reagens) und mit drei Wiederholungen pro Konzentration durchgeführt werden. Die Leistungsfähigkeit wird durch die ordnungsgemäße Einstufung (positiv/negativ) der einzelnen Leistungsstoffe nachgewiesen. Beim Erlernen der Assays sollte die Eignungsprüfung von jedem einzelnen Labortechniker durchgeführt werden.
Tabelle 2
Liste der Kontroll- und der Leistungsstoffe für die Assays zur Prüfung auf konkurrierende hrER-Bindungen
(90)
|
Nr.
|
Bezeichnung des Stoffs
|
CAS-Nr. (91)
|
Erwartete Reaktion (92)
(93)
|
Prüfkonzentrationsbereich (M)
|
MeSH-Chemikalienklasse (94)
|
Produktklasse (95)
|
|
Kontrollen (Referenzöstrogen, schwacher Binder, Nicht-Binder)
|
|
1
|
17-Εστραδιολ
|
50-28-2
|
Binder
|
1×10-11 – 1×10-6
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
2
|
Norethynodrel (oder) Norethindron
|
68-23-5 (oder) 68-22-4
|
Binder
|
3×10-9 – 30×10-6
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
3
|
Octyltriethoxysilan
|
2943-75-1
|
Nicht-Binder
|
1×10-10 – 1×10-3
|
Silan
|
Oberflächen-Modifiziermittel
|
|
Leistungsstoffe (95)
|
|
4
|
Diethylstilbestrol
|
56-53-1
|
Binder
|
1×10-11 – 1×10-6
|
Kohlenwasserstoff (cyclisch), Phenol
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
5
|
17α-Ethinylestradiol
|
57-63-6
|
Binder
|
1×10-11 – 1×10-6
|
Steroid
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
6
|
meso-Hexestrol
|
84-16-2
|
Binder
|
1×10-11 – 1×10-6
|
Kohlenwasserstoff (cyclisch), Phenol
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
7
|
Tamoxifen
|
10540-29-1
|
Binder
|
1×10-11 – 1×10-6
|
Kohlenwasser-stoff (cyclisch)
|
Pharmazeutisches Erzeugnis, veterinärmediznisches Agens
|
|
8
|
Genistein
|
446-72-0
|
Binder
|
1×10-10 – 1×10-3
|
Heterocyclische Verbindung, Flavonoid
|
Naturprodukt
|
|
9
|
Bisphenol A
|
80-05-7
|
Binder
|
1×10-10 – 1×10-3
|
Phenol
|
Chemisches Zwischenprodukt
|
|
10
|
Zearalenon
|
17924-92-4
|
Binder
|
1×10-11 – 1×10-3
|
Heterocyclische Verbindung, Lacton
|
Naturprodukt
|
|
11
|
Butylparaben
|
94-26-8
|
Binder
|
1×10-11 – 1×10-3
|
Carboxylsäure, Phenol
|
Konservierungsmittel
|
|
12
|
Atrazin
|
1912-24-9
|
Nicht-Binder
|
1×10-11 – 1×10-6
|
Heterocyclische Verbindung
|
Herbizid
|
|
13
|
Di-n-butylphthalat (DBP) (96)
|
84-74-2
|
Nicht-Binder (97)
|
1×10-10 – 1×10-4
|
Kohlenwasserstoff (cyclisch), Ester
|
Weichmacher, chemisches Zwischenprodukt
|
|
14
|
Corticosteron
|
50-22-6
|
Nicht-Binder
|
1×10-11 – 1×10-4
|
Steroid
|
Naturprodukt
|
|
Löslichkeitsprüfungen und Dosisfindungsversuche für die Prüfchemikalien
|
19.
|
In einem Vorversuch sollten die Löslichkeitsgrenze der einzelnen Prüfchemikalien ermittelt und der jeweils geeignete Konzentrationsbereich für die Prüfung bestimmt werden. Die Löslichkeitsgrenze der einzelnen Prüfchemikalien wird zunächst im Lösungsmittel bestimmt und anschließend unter den Bedingungen des Assays bestätigt. Die im Assay geprüfte Endkonzentration sollte höchstens bei 1 mM liegen. Für die Dosisfindungsprüfung werden eine Lösungsmittelkontrolle sowie acht logarithmische serielle Verdünnungen ausgehend von der maximal akzeptierbaren Konzentration (z. B. 1 mM oder weniger, je nach Löslichkeitsgrenze) verwendet. Das Auftreten einer Trübung oder die Bildung eines Niederschlags werden protokolliert. Die Konzentrationen beim zweiten und beim dritten Versuch sollten ggf. angepasst werden, um die Konzentrations-Reaktionskurve besser zu beschreiben.
|
Akzeptanzkriterien für Prüfläufe
|
20.
|
Ob ein Prüflauf akzeptiert wird, hängt von der Bewertung der Ergebnisse mit dem jeweils verwendeten Referenzöstrogen und den Kontrollen ab. Zunächst sollten für Platte 1 die vollständigen Konzentrationskurven der Referenzkontrollen jedes einzelnen Versuchs mit den Werten der Leistungsmessungen bei den Parametern für die Kurvenanpassung (z. B. IC50 und Hillslope) übereinstimmen; der Vergleich erfolgt anhand der für die Protokolle des CERI- und des FW-Assays angegebenen Werte (Anlagen 2 und 3) und der historischen Kontrolldaten des Labors, das die Prüfungen durchführt. Bei allen Versuchen sollten alle Kontrollen (Referenzöstrogen, schwacher Binder und Nicht-Binder) korrekt eingestuft werden. Anschließend sind die Kontrollen auf allen folgenden Platten auf Übereinstimmung mit Platte 1 zu prüfen. Um den Höchstwert der Kurve der konkurrierenden Bindungen eindeutig zu ermitteln, sollte ein ausreichendes Spektrum an Konzentrationen der Prüfchemikalie berücksichtigt werden. Die Variabilität zwischen Wiederholungen bei den einzelnen Konzentrationen der Prüfchemikalie sowie zwischen den drei unabhängigen Prüfläufen sollte angemessen und wissenschaftlich vertretbar sein. Die Fähigkeit zur konsistenten Durchführung der Assays sollte durch den Aufbau und die Pflege einer historischen Datenbank für das Referenzöstrogen und die Kontrollen nachgewiesen werden. Standardabweichungen (SD) oder Variationskoeffizienten (VK) der Mittelwerte von Parametern zur Anpassung der Kurven für das Referenzöstrogen und die Kontrolle mit dem schwachen Binder aus mehreren Versuchen können als Maßstab für die Reproduzierbarkeit innerhalb eines Labors herangezogen werden. Die Ergebnisse der Kontrollen auf den Platten der einzelnen Prüfläufe sowie die Ergebnisse für die einzelnen Prüfchemikalien sind einer qualifizierten fachlichen Beurteilung zu unterziehen.
Darüber hinaus sollten für Akzeptanzkriterien die folgenden Grundsätze gelten:
|
—
|
Die Daten sollten für eine quantitative Bewertung der ER-Bindung ausreichen.
|
|
—
|
Die geprüften Konzentrationen sollten im Löslichkeitsbereich der Prüfchemikalien bleiben.
|
|
Analyse der Daten
|
21.
|
Das beschriebene Verfahren zur Analyse der Daten zur Sättigung und zu konkurrierenden Bindungen sollte unter Berücksichtigung der wesentlichen Grundsätze der Charakterisierung von Wechselwirkungen zwischen Rezeptoren und Liganden durchgeführt werden. In der Regel werden Daten zur Sättigungsbindung mit einem nicht linearen Regressionsmodell analysiert, das die Bindungswirkung insgesamt ebenso wie nicht spezifische Bindungen berücksichtigt. Bei der Bestimmung von Bmax und Kd ist möglicherweise eine Korrektur für den Abbau von Liganden (z. B. Swillens, 1995 (19)) vorzunehmen. Daten aus Untersuchungen zur Ermittlung konkurrierender Bindungen werden gewöhnlich umgewandelt (z. B. spezifische Bindung in Prozent und Konzentration der Prüfchemikalie (log M)). Mit einer geeigneten Software zur nicht linearen Kurvenanpassung sollten die Schätzwerte für log (IC50) für die einzelnen Prüfchemikalien zur Verwendung in einer Hill-Gleichung mit vier Parametern ermittelt werden. Nach einer ersten Analyse sollten die Parameter zur Kurvenanpassung geprüft werden; außerdem sollte geprüft werden, in welchem Umfang die Bindungsdaten mit der erzeugten Kurve der konkurrierenden Bindungen übereinstimmen. Manchmal müssen weitere Analysen vorgenommen werden, um die optimale Kurvenanpassung zu ermitteln (z. B. indem Höchst- und/oder Mindestwerte für die Kurve festgelegt werden oder die 10-%-Regel angewendet wird (siehe Anlage 4 und (2) (Abschnitt III.A.2).
|
|
22.
|
Die Erfüllung der Akzeptanzkriterien (Nummer 20) deutet darauf hin, dass ein Assaysystem ordnungsgemäß funktioniert. Dies gewährleistet jedoch nicht, dass eine bestimmte Prüfung tatsächlich exakte Daten ergibt. Die Wiederholung der korrekten Ergebnisse der ersten Prüfung ist die beste Bestätigung dafür, dass exakte Daten ermittelt wurden.
|
Allgemeine Kriterien für die Auswertung von Daten
|
23.
|
Zurzeit besteht keine allgemein anerkannte Methode zur Auswertung der Daten aus ER-Bindungsassays. Allerdings sollten qualitative (z. B. Binder/Nicht-Binder) und/oder quantitative (IC50, relative Bindungsaffinität (RBA) usw.) Bewertungen einer durch hrER vermittelten Aktivität auf empirischen Daten und fundierten wissenschaftlichen Beurteilungen beruhen.
|
Prüfbericht
|
24.
|
Der Prüfbericht sollte folgende Angaben enthalten:
|
|
Kontrolle/Referenz/Prüfchemikalie
|
—
|
Herkunft, Partienummer, ggf. begrenztes Verwendungsdatum;
|
|
—
|
Stabilität der Prüfchemikalie, falls bekannt;
|
|
—
|
Löslichkeit und Stabilität der Prüfchemikalie in Lösungsmittel, falls bekannt;
|
|
—
|
Messung des pH-Werts, der Osmolalität und ggf. des Niederschlags im Kulturmedium, dem die Prüfchemikalie zugegeben wurde;
|
|
|
|
einkomponentiger Stoff
|
—
|
physikalisches Erscheinungsbild, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
|
|
—
|
chemische Bezeichnung, wie z. B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer, SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
|
|
mehrkomponentiger Stoff, UVCB-Stoffe und Gemische:
|
—
|
So weit wie möglich Charakterisierung durch die chemische Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Elemente.
|
|
|
|
Lösungsmittel/Vehikel
|
—
|
Beschreibung (Art, Lieferant und Charge);
|
|
—
|
Begründung der Auswahl des Lösungsmittels/Vehikels;
|
|
—
|
Löslichkeit und Stabilität der Prüfchemikalie in Lösungsmittel/in einem Vehikel, falls bekannt.
|
|
|
|
Rezeptoren
|
—
|
Herkunft der Rezeptoren (Hersteller, Bestellnummer, Charge, Art des Rezeptors, vom Hersteller gelieferte aktive Rezeptorkonzentration, Zertifizierung des Herstellers);
|
|
—
|
Beschreibung der Rezeptoren (einschl. ermittelte Sättigungsbindungen): Kd, Bmax;
|
|
—
|
Lagerung der Rezeptoren;
|
|
—
|
radioaktiv markierter Ligand;
|
|
—
|
Hersteller, Bestellnummer, Charge, spezifische Aktivität.
|
|
|
|
Prüfbedingungen
|
—
|
Einschränkungen hinsichtlich der Löslichkeit unter Assay-Bedingungen;
|
|
—
|
Zusammensetzung des Bindungspuffers;
|
|
—
|
Konzentration des Rezeptors;
|
|
—
|
Konzentration des Tracers (d. h. des radioaktiv markierten Liganden);
|
|
—
|
Konzentrationen der Prüfchemikalie;
|
|
—
|
Prozentanteil des Vehikels im endgültigen Assay;
|
|
—
|
Inkubationstemperatur und -dauer;
|
|
—
|
Methode zur Trennung von gebundenen/nicht gebundenen Chemikalien;
|
|
—
|
positive und negative Kontrollen/Referenzstandards;
|
|
—
|
Kriterien zur Einstufung der Prüfungen als positiv, negativ oder nicht eindeutig.
|
|
|
|
Akzeptanzprüfung
|
—
|
tatsächliche IC50- und Hillslope-Werte für gleichzeitige Positivkontrollen/Referenzstoffe.
|
|
|
|
Ergebnisse
|
—
|
Rohdaten und Daten zu gebundenen/nicht gebundenen Chemikalien;
|
|
—
|
ggf. Bestätigung durch Prüfung der Denaturierung;
|
|
—
|
wenn vorhanden, niedrigste Wirkungskonzentration (LEC);
|
|
—
|
RBA- und/oder IC50-Werte;
|
|
—
|
nach Möglichkeit Dosis-Reaktions-Verhältnis;
|
|
—
|
wenn vorhanden, statistische Analysen sowie Messabweichung und Konfidenzwerte (z. B. SEM, SD, VK oder 95 % CI) sowie Angaben dazu, wie diese Werte ermittelt wurden.
|
|
|
|
Erörterung der Ergebnisse
|
—
|
Anwendung der 10-%-Regel.
|
|
Schlussfolgerung.
|
LITERATUR
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 34), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisation für Wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(4)
|
OECD (2012). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 150), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(5)
|
Cavailles, V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: S. 20-26.
|
|
(6)
|
Welboren, W.J., u. a. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer, 16(4): S. 1073-1089.
|
|
(7)
|
Younes, M., und Honma, N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): S. 63-66.
|
|
(8)
|
Diamanti-Kandarakis u. a. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document: Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori, Y., u. a. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity – The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. S. 1-188.
|
|
(15)
|
Yamasaki, K.; Noda, S.; Imatanaka, N.; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer, V.; Maskova, J; Zraly, Z.; Neca, J.; Simeckova, P.; Vondracek, J.; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, J.M.; Nestor, K.M.; Fasco, M.J.; Pentecost, B.T.; Arcaro, K.F. Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Anlage 1
BEGRIFFSBESTIMMUNGEN UND ABKÜRZUNGEN
10-%-Regel
: Bei den Analysen Möglichkeit des Ausschlusses von Datenpunkten, wenn bei Wiederholungen der Mittelwert der prozentualen spezifischen [3H]-17β-Estradiol-Bindung mindestens 10 % über dem beobachteten Mittelwert bei einer geringeren Konzentration liegt (siehe Anlage 4).
Akzeptanzkriterien
: Mindeststandards für Kontrollen und Referenzstandards. Damit ein Versuch als gültig betrachtet werden kann, sollten alle Akzeptanzkriterien erfüllt sein.
CF
: OECD Conceptual Framework for the Testing and Evaluation of Endocrine Disrupters.
Chemikalie
: Ein Stoff oder ein Gemisch.
E2
: 17β-Estradiol
ED
: Endokrine Störung.
Eignung
: Die nachgewiesene Fähigkeit zur ordnungsgemäßen Durchführung eines Assays vor der Prüfung unbekannter Stoffe.
ER
: Östrogenrezeptor.
Genauigkeit (Übereinstimmung)
: Der Grad an Übereinstimmung zwischen den Ergebnissen eines Assays und akzeptierten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung eines Assays und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von „Übereinstimmung“ verwendet und bezeichnet den Anteil der korrekten Ergebnisse eines Assays (1).
hERα
: Humaner Östrogenrezeptor α.
IC
50
: Die Hälfte der maximalen Wirkungskonzentration einer Prüfchemikalie mit hemmender Wirkung.
ICCVAM
: Interagency Coordinating Committee on the Validation of Alternative Methods (das US-amerikanische Validierungszentrum).
Inter-Labor-Reproduzierbarkeit
: Das Ausmaß, in dem unterschiedliche qualifizierte Labors, die dasselbe Protokoll verwenden und dieselben Prüfstoffe untersuchen, qualitativ und quantitativ vergleichbare Ergebnisse erzielen können. Die Inter-Labor-Reproduzierbarkeit wird während der Prävalidierungs- und Validierungsverfahren ermittelt und zeigt das Maß an, in dem ein Assay erfolgreich zwischen Labors übertragen werden kann (1).
Intra-Labor-Reproduzierbarkeit
: Das Ausmaß, in dem qualifizierte Personen innerhalb desselben Labors, die dasselbe spezifische Protokoll zu unterschiedlichen Zeiten verwenden, erfolgreich dieselben Ergebnisse replizieren können. Auch als „laborinterne Reproduzierbarkeit“ bezeichnet (1).
LEC
: Als niedrigste Wirkungskonzentration (Lowest Effective Concentration) wird die niedrigste Konzentration einer Prüfchemikalie bezeichnet, die eine Wirkung hervorruft (d. h. die niedrigste Konzentration einer Prüfchemikalie, bei der die n-fache Induktion sich statistisch von der gleichzeitigen Vehikelkontrolle unterscheidet).
Leistungsstandards
: Auf einer validierten Prüfmethode beruhende Normen, auf deren Grundlage die Vergleichbarkeit einem vorgeschlagenen, mechanistisch und funktionell ähnlichen Assay bewertet werden kann. Sie umfassen (1) wesentliche Elemente des Assays; (2) ein Mindestverzeichnis von Referenzchemikalien, ausgewählt aus den Chemikalien, die zum Nachweis der akzeptablen Leistung der validierten Referenzmethode verwendet werden, und (3) je nach den für die validierte Referenzmethode erzielten Ergebnissen die vergleichbaren Genauigkeits- und Zuverlässigkeitswerte, die der vorgeschlagene Assay bei der Bewertung anhand des Mindestverzeichnisses von Referenzchemikalien ergeben sollte (1).
Leistungsstoffe
: Eine Untergruppe der in die Leistungsstandards einbezogenen Referenzstoffe, die von Labors bei standardisierten Assays zum Nachweis der fachlichen Kompetenz eingesetzt werden können. Auswahlkriterien für diese Stoffe sind u. a., dass sie den Reaktionsbereich abdecken und im Handel erhältlich sein müssen und dass für diese Stoffe hochwertige Referenzdaten verfügbar sein müssen.
Me-Too-Prüfung
: [Im englischen Sprachraum gemeinsprachliche] Bezeichnung einer Prüfmethode, die strukturell und funktionell mit einer validierten und akzeptierten Referenzprüfmethode vergleichbar ist. Gleichbedeutend mit „vergleichbare Prüfmethode“ verwendet.
Östrogenaktivität
: Die Fähigkeit einer Chemikalie, die Bindung von Östrogenrezeptoren durch 17β-Estradiol nachzubilden. Mit dieser Prüfmethode kann die Bindung an den hERα festgestellt werden.
PBTG
: Leistungsbezogene Prüfrichtlinie.
Prüfchemikalie
: Stoff oder Gemisch, der/das mit dieser Prüfmethode untersucht wird.
RBA
: Relative Bindungsaffinität. Die RBA eines Stoffs wird als Prozentanteil von log (IC50) des Stoffs im Vergleich zu log (IC50) bei 17β-Estradiol berechnet.
Referenzöstrogen
: 17ß-Estradiol (E2, CAS 50-28-2).
Referenzprüfmethode
: Die Assays, auf denen die PBTG 493 beruht.
Relevanz
: Dieser Begriff bezieht sich auf das Verhältnis zwischen dem Assay und der betreffenden Wirkung und auf die Frage, ob er aussagekräftig und nützlich für einen bestimmten Zweck ist. Er beschreibt das Ausmaß, in dem der Assay die untersuchte biologische Wirkung korrekt misst oder vorhersagt. Die Relevanz schließt eine Beurteilung der Genauigkeit (Übereinstimmung) eines Assays ein (1).
SD
: Standardabweichung,
Validierte Prüfmethode
: Eine Prüfmethode, für die zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien abgeschlossen wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht genau und zuverlässig genug ist, um für den vorgeschlagenen Zweck akzeptiert zu werden (1).
Validierung
: Prozess, mit dem die Zuverlässigkeit und die Relevanz eines bestimmten Ansatzes, einer Methode, eines Assays, eines Prozesses oder einer Bewertung für einen bestimmten Zweck festgestellt wird (1).
VK
: Variationskoeffizient.
Zuverlässigkeit
: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Sie wird durch Berechnung der Intra- und Inter-Labor-Reproduzierbarkeit bewertet (13).
Anlage 2
DER FREYBERGER-WILSON-IN-VITRO-ÖSTROGENREZEPTOR-(ERΑ-)BINDUNGSASSAY MIT IN VOLLER LÄNGE REKOMBINANTEM ERΑ ZUR ERMITTLUNG VON SÄTTIGUNGSBINDUNGEN UND KONKURRIERENDEN BINDUNGEN
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
1.
|
Bei diesem In-vitro-Östrogenrezeptor-(ERα)-Bindungsassay zur Ermittlung von Sättigungsbindungen und konkurrierenden Bindungen wird der in voller Länge rekombinante humane ERαα(hrERα) verwendet. Dieser Östrogenrezeptor wird in mit einem Baculovirus infizierten Insektenzellen erzeugt und aus diesen Zellen isoliert. Das von Freyberger und Wilson entwickelte Protokoll wurde einer internationalen Validierungsstudie durch mehrere Labors unterzogen (2). Dabei wurden die Relevanz und die Zuverlässigkeit des Protokolls für den vorgesehenen Zweck des Assays nachgewiesen.
|
|
2.
|
Der Assay besteht in einem Prüfverfahren zur Ermittlung von Stoffen, die eine Bindung mit dem hrERα über die volle Länge eingehen können. Mit dem Assay kann untersucht werden, ob eine Prüfchemikalie mit 17β-Estradiol um Bindungen an den hrERα konkurrieren kann. Als quantitative Ergebnisse können mit dem Assay u. a. die Werte für IC50 (als Maßstab für die zur Verdrängung der Hälfte des [3H]-17β-Estradiols vom hrERα durch eine Prüfchemikalie erforderliche Konzentration) und die relative Affinität von Prüfchemikalien für Bindungen an den hrERα im Vergleich zu 17β-Estradiol ermittelt werden. Mit Blick auf die Verwendung zur Prüfung von Chemikalien kann ein akzeptables qualitatives Ergebnis des Assays auch darin bestehen, dass Prüfchemikalien nach ihrer Bindung an den hrERα und nach den genannten Kriterien für die Bindungskurven als Binder, Nicht-Binder oder nicht eindeutig eingestuft werden.
|
|
3.
|
Da bei diesem Assay ein radioaktiver Ligand verwendet wird, benötigen die Labors eine Genehmigung für die Verwendung von radioaktivem Material. Bei allen Verfahren unter Verwendung von Radioisotopen und von gefährlichen Chemikalien sind die nach den nationalen Rechtsvorschriften geltenden Bestimmungen und die dort vorgesehenen Verfahren einzuhalten.
|
|
4.
|
Vor der Verwendung dieses Assays für rechtliche Zwecke sollten die Abschnitte „ALLGEMEINE EINLEITUNG“ und „ELEMENTE DES hrER-BINDUNGSASSAYS“ gelesen werden. Die in dieser Prüfrichtlinie verwendeten Begriffe und Abkürzungen werden in Anlage 1 erläutert.
|
PRINZIPIEN DES ASSAYS (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
5.
|
Mit dem hrERα-Bindungsassay wird die Fähigkeit eines radioaktiv markierten Liganden ([3H]-17β-Estradiol) gemessen, bei zunehmenden Konzentrationen einer Prüfchemikalie (z. B. eines Bindungskonkurrenten) eine Bindung mit dem ER einzugehen. Prüfchemikalien mit einer starken ER-Affinität konkurrieren mit dem radioaktiv markierten Liganden bei einer niedrigen Konzentration als Chemikalien mit einer geringeren Affinität für diesen Rezeptor.
|
|
6.
|
Dieser Assay beinhaltet zwei wesentliche Elemente: einen Versuch zur Ermittlung von Sättigungsbindungen zur Charakterisierung der Parameter der Wechselwirkung zwischen Rezeptor und Ligand und einen anschließenden Versuch zur Ermittlung konkurrierender Bindungen, mit dem die Konkurrenz zwischen einer Prüfchemikalie und einem radioaktiv markierten Liganden im Hinblick auf die Bindung an den ER beschrieben wird.
|
|
7.
|
Zur Vorbereitung der Prüfung konkurrierender Bindungen soll anhand einer Ermittlung der Sättigungsbindung eine bestimmte Rezeptorcharge hinsichtlich ihrer Bindungsaffinität geprüft und quantitativ bewertet werden. Mit der Prüfung konkurrierender Bindungen werden unter Gleichgewichtsbedingungen die Affinität einer bestimmten Konzentration des Östrogenrezeptors für seinen natürlichen Liganden (ausgedrückt durch die Dissoziationskonstante Kd) und die Konzentration der aktiven Rezeptor-Bindungsstellen (Bmax) gemessen.
|
|
8.
|
Mit der Prüfung konkurrierender Bindungen wird die Affinität eines Stoffs für Bindungen an den ER im Hinblick auf die Konkurrenz mit [3H]-17β-Estradiol bewertet. Die Affinität wird anhand der Konzentration der Prüfchemikalie quantifiziert, die unter Gleichgewichtsbedingungen 50 % der spezifischen Bindung von [3H]-17β-Estradiol hemmt („Hemmkonzentration 50 %“ oder IC50). Diese Affinität kann auch anhand der relativen Bindungsaffinität (RBA, bezogen auf IC50 Estradiol, im selben Prüflauf getrennt gemessen) beurteilt werden. Bei der Prüfung konkurrierender Bindungen wird die Bindung von [3H]-17β-Estradiol in einer bestimmten Konzentration an zahlreiche (acht Größenordnungen) Konzentrationen einer Prüfchemikalie gemessen. Die ermittelten Daten werden dann möglichst entsprechend der Hill-Gleichung (Hill, 1910) angepasst, die die Verdrängung des Radioliganden durch einen für eine bestimmte Bindungsstelle spezifischen konkurrierenden Binder beschreibt. Anhand des Umfangs der Verdrängung des radioaktiv markierten Estradiols bei Gleichgewichtsbedingungen wird die Prüfchemikalie als Binder, Nicht-Binder oder nicht eindeutig eingestuft.
|
PRÜFVERFAHREN
Nachweis einer akzeptablen Leistungsfähigkeit bei Untersuchungen des hrERα-Proteins
|
9.
|
Vor der regelmäßigen Durchführung der Assays zur Ermittlung von Sättigungsbindungen und konkurrierenden Bindungen ist jeweils mit einer neuen Charge des hrERα nachzuweisen, dass in dem Labor, in dem der Rezeptor verwendet wird, korrekte Ergebnisse ermittelt werden. Die Leistungsfähigkeit sollte in zwei Schritten nachgewiesen werden:
|
—
|
Durchführung des [3H]-17β-Estradiol-Bindungsassays zum Nachweis der Spezifität und der Sättigung für den hrERα; anhand einer nicht linearen Regressionsanalyse der ermittelten Daten (z. B. BioSoft; McPherson, 1985; Motulsky, 1995) und des anschließenden Scatchard-Diagramms sollten die Bindungsaffinität des [3H]-17β-Estradiols (Kd) für den hrERα und die Anzahl der Rezeptoren (Bmax) der einzelnen hrERα-Chargen dokumentiert werden;
|
|
—
|
Durchführung eines Assays zur Prüfung konkurrierender Bindungen mit Kontrollstoffen (Referenzöstrogen (17β-Estradiol)), einem schwachen Binder (z. B. Norethynodrel oder Norethindron) und einem Nicht-Binder (Octyltriethoxysilan, OTES). Jedes Labor sollte eine historische Datenbank zum Nachweis der Konsistenz der IC50-Werte und sonstiger relevanter Werte zwischen den einzelnen Versuchen sowie zwischen den einzelnen hrERα-Chargen bei Versuchen mit dem Referenzöstrogen und einem schwachen Binder aufbauen. Die Parameter der Kurven der konkurrierenden Bindungen bei den Kontrollstoffen sollten innerhalb des 95-%-Konfidenzintervalls liegen (siehe Tabelle 1), das anhand von Daten von an der Validierungsstudie zu diesem Assay beteiligten Labors festgelegt wurde (2).
|
|
Tabelle 1
Leistungskriterien für das Referenzöstrogen und den schwachen Binder, FW-hrER-Bindungsassay
|
Stoff
|
Parameter
|
Mittelwert (7)
|
Standardabweichung (n)
|
95-%-Konfidenzintervalle (8)
|
|
Untere Grenze
|
Obere Grenze
|
|
17β-Estradiol
|
Höchstwert (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Mindestwert (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Hillslope
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
Log IC50 (M)
|
-8,92 (9)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Norethynodrel
|
Höchstwert (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Mindestwert (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Hillslope
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
IC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Norethindron (9)
|
Höchstwert (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Mindestwert (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Hillslope
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
Log IC50(M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Nachweis der Eignung des Labors
|
10.
|
Siehe Nummern 17 und 18 sowie Tabelle 2 im Abschnitt „ELEMENTE DES hrER-BINDUNGSASSAYS“ für diese Prüfmethode. Die Assays (zur Ermittlung von Sättigungsbindungen und konkurrierenden Bindungen) sollten jeweils drei unabhängige Prüfläufe (d. h. mit frischen Rezeptor-, Chemikalien- und Reagenzienverdünnungen) an unterschiedlichen Tagen jeweils mit drei Wiederholungen umfassen.
|
Ermittlung der Rezeptorkonzentration (hrERα)
|
11.
|
Die Konzentration des aktiven Rezeptors ist je nach Charge und Lagerungsbedingungen leicht unterschiedlich. Daher sollte die Konzentration des vom Hersteller gelieferten aktiven Rezeptors bestimmt werden. Daraus ergibt sich die geeignete Konzentration des aktiven Rezeptors beim Prüflauf.
|
|
12.
|
Unter den Bedingungen bei der konkurrierenden Bindung (d. h. 1 nM [3H]-Estradiol) werden Nennkonzentrationen von 0,25, 0,5, 0,75 und 1 nM Rezeptor einmal ohne (Gesamtbindung) und einmal mit (nicht spezifische Bindung) 1 μM nicht markiertem Estradiol inkubiert. Die spezifische Bindung wird berechnet als Differenz zwischen der Gesamtbindung und der nicht spezifischen Bindung bezogen auf die Rezeptor-Nennkonzentration in einem Diagramm dargestellt. Die Rezeptorkonzentration, bei der sich spezifische Bindungswerte entsprechend einem Anteil von 20 % des zugesetzten radioaktiven Liganden ergeben, wird auf die entsprechende Rezeptor-Nennkonzentration bezogen; diese Rezeptorkonzentration ist bei den Versuchen zur Ermittlung von Sättigungsbindungen und konkurrierenden Bindungen zu verwenden. Diese Anforderung wird häufig mit einer endgültigen hrER-Konzentration von 0,5 nM erfüllt.
|
|
13.
|
Wenn das 20-%-Kriterium mehrfach nicht erfüllt werden kann, sollte der Versuchsaufbau auf mögliche Fehlerquellen geprüft werden. Dass das 20-%-Kriterium nicht erfüllt werden kann, deutet möglicherweise auch darauf hin, dass die rekombinante Charge den aktiven Rezeptor nur in einem sehr geringen Anteil enthält. In diesem Fall sollte die Verwendung einer anderen Rezeptor-Charge in Betracht gezogen werden.
|
Assay zur Ermittlung der Sättigungsbindung
|
14.
|
Acht zunehmende [3H]-17β-Estradiol-Konzentrationen sollten jeweils dreifach unter den drei folgenden Bedingungen geprüft werden (siehe Tabelle 2):
|
—
|
ohne nicht markiertes 17β-Estradiol und mit dem ER, um anhand einer Messung der Radioaktivität der Wells, die ausschließlich [3H]-17β-Estradiol enthalten, die Gesamtbindung zu ermitteln;
|
|
—
|
bei einer 1 000-mal höheren Konzentration des nicht markierten 17β-Estradiols im Vergleich zum markierten 17β-Estradiol und zu Prüfungen mit dem ER; dadurch sollen die aktiven Bindungsstellen mit nicht markiertem 17β-Estradiol gesättigt werden, um anhand einer Messung der Radioaktivität in den Wells die nicht spezifische Bindung zu bestimmen. Verbleibendes warmes Estradiol, das Bindungen mit dem Rezeptor eingehen kann, wird als Bindung an eine nicht spezifischen Bindungsstelle betrachtet, da das kalte Estradiol in einer derart hohen Konzentration vorliegen sollte, dass es an allen verfügbaren spezifischen Bindungsstellen des Rezeptors gebunden ist;
|
|
—
|
ohne nicht markiertes 17β-Estradiol und ohne den ER (Ermittlung der Gesamt-Radioaktivität).
|
|
Zubereitung der Lösungen mit [3H]-17β-Estradiol und mit nicht markiertem 17β-Estradiol
|
15.
|
Unter Zugabe eines Assay-Puffers zu einer 12-nM-Vorratslösung [3H]-17β-Estradiol wird [3H]-17β-Estradiol auf Konzentrationen von anfänglich 0,12 nM bis 12 nM verdünnt. Durch Zugabe von 40 μl dieser Lösungen in die jeweiligen Wells einer 96-Well-Mikrotiterplatte (mit einem Endvolumen von 160 μl) werden die endgültigen Assay-Konzentrationen von 0,03-3,0 nM hergestellt. Die Zubereitung des Assay-Puffers, der [3H]-17β-Estradiol-Vorratslösung und der Verdünnungen sowie die Bestimmung der Konzentrationen werden im FW-Protokoll eingehend beschrieben (2).
|
|
16.
|
Die Ethanol-17β-Estradiol-Lösungen sind unter Zugabe des Assay-Puffers so zu verdünnen, dass sich acht zunehmende Ausgangskonzentrationen von 0,06-6 μM ergeben. Durch Zugabe von 80 μl dieser Lösungen in die jeweiligen Wells einer 96-Well-Mikrotiterplatte (mit einem Endvolumen von 160 μl) werden die endgültigen Assay-Konzentrationen von 0,03-3,0 μM hergestellt. Die Endkonzentrationen des nicht markierten 17β-Estradiols in den einzelnen Assays zur Ermittlung der nicht spezifischen Bindung sollten das 1 000-Fache der markierten [3H]-17β-Estradiol-Konzentration betragen. Die Zubereitung der nicht markierten 17β-Estradiol-Lösungen wird eingehend im FW-Protokoll beschrieben (2).
|
|
17.
|
Für die Versuche ist die Nennkonzentration des Rezeptors zu verwenden, bei der sich eine spezifische Bindung von 20 ± 5 % ergibt (Nummern 12 und 13). Die hrERα-Lösung ist unmittelbar vor Gebrauch zuzubereiten.
|
|
18.
|
Die 96-Well-Mikrotiterplatten werden mit 3 Wiederholungen pro Konzentration vorbereitet, wie in Tabelle 2 dargestellt. Anlage 2.2 enthält ein Beispiel für die Anordnung der Konzentrationen und der Volumina von [3H]-17β-Estradiol, nicht markiertem 17β-Estradiol, des Puffers und des Rezeptors.
|
Tabelle 2
Anordnung auf der Mikrotiterplatte beim Assay zur Ermittlung der Sättigungsbindung
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Gesamtbindung (Lösungsmittel)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER
+ 0,10 μM E2
|
Nicht spezifische Bindung
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
[3H] E2: [3H]-17β-Estradiol
ER: = Östrogenrezeptor.
E2: nicht markiertes 17β-Estradiol
|
|
19.
|
Die für den Assay verwendeten Mikrotiterplatten werden 16-20 Stunden bei 2-8 °C auf einem Karussell inkubiert.
|
Messung des an den hrERα gebundenen [3H]-17β-Estradiols
|
20.
|
Das an den hrERα gebundene [3H]-17β-Estradiol ist von dem nicht gebundenen [3H]-17β-Estradiol zu trennen. Dazu werden in jedes Well 80 μl kalte DCC-Suspension gegeben; anschließend werden die Mikrotiterplatten 10 Minuten geschüttelt und danach 10 Minuten mit etwa 2 500 rpm zentrifugiert. Um bei diesem Prozess eine Dissoziation des gebundenen [3H]-17β-Estradiols vom hrERα zu minimieren, ist von entscheidender Bedeutung, dass bei den Puffer- und den Assay-Wells eine Temperatur von 2-8 °C aufrechterhalten wird und dass alle Schritte rasch durchgeführt werden. Für eine effiziente und rasche Behandlung der Platten wird eine Schüttelvorrichtung für Mikrotiterplatten benötigt.
|
|
21.
|
50 μl des Überstands des an den hrERα gebundenen [3H]-17β-Estradiols sind anschließend mit äußerster Sorgfalt zu entnehmen, damit Verunreinigungen der Wells durch Berührung mit der DCC-Lösung verhindert werden. Der Überstand wird auf eine zweite Mikrotiterplatte gegeben.
|
|
22.
|
Danach werden in jedes Well (A1-B12 und D1-E12) 200 μl einer Szintillationsflüssigkeit gegeben, die die kinetische Energie radioaktiver Emissionen in Lichtenergie umwandeln kann. Die Wells G1-H12 (= DPM gesamt) enthalten serielle Verdünnungen des [3H]-17β-Estradiols (40 μl), die unmittelbar in die Szintillationsflüssigkeit in den Wells der Messplatte zu geben sind, wie in Tabelle 3 angegeben (d. h. die Wells enthalten jeweils nur 200 μl Szintillationsflüssigkeit und [3H]-17β-Estradiol in der jeweiligen Verdünnung). Anhand der Zerfallsereignisse pro Minute (DPM) wird so nachgewiesen, wie viel [3H]-17β-Estradiol jeweils in die Wells zur Ermittlung der Gesamtbindung und der nicht spezifischen Bindung gegeben wurde.
|
Tabelle 3
Anordnung auf der Mikrotiterplatte beim Assay zur Ermittlung der Sättigungsbindung, Messung der Radioaktivität
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Gesamtbindung (Lösungsmittel)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + + ER
+ 0,10 μM E2
|
Nicht spezifische Bindung
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3H] E2 + ER + + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [ 3H] E2 (DPM gesamt)
|
0,06 nM [ 3H] E2
|
0,08 nM [ 3H] E2
|
0,10 nM [ 3H] E2
|
DPM gesamt (*7)
|
|
H
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2: [3H]-17β-Estradiol
ER: Östrogenrezeptor.
E2: nicht markiertes 17β-Estradiol
DPM: Zerfallsereignisse pro Minute
|
|
23.
|
Die Messungen werden mit einer Verzögerung von mindestens zwei Stunden durchgeführt; die Zählung sollte 40 Minuten pro Well andauern. Zur Bestimmung der DPM-Werte pro Well ist ein Mikrotiterplatten-Szintillationszähler mit Querempfindlichkeitskorrektur zu verwenden. Wenn kein Szintillationszähler für Mikrotiterplatten verfügbar ist, können die Proben auch mit einem sonstigen Zähler gemessen werden. In diesem Fall kann die Dauer der Zählung reduziert werden.
|
Assay zur Ermittlung konkurrierender Bindungen
|
24.
|
Mit dem Assay zur Ermittlung konkurrierender Bindungen wird die Bindung von [3H]-17β- Estradiol einer einzelnen Konzentration bei zunehmenden Konzentrationen einer Prüfchemikalie gemessen. Bei jedem Prüflauf sind für jede Konzentration drei gleichzeitige Replikate zu verwenden. Außerdem werden für jede zu prüfende Chemikalie drei nicht gleichzeitige Prüfläufe durchgeführt. Der Assay ist auf zwei oder mehr 96-Well-Mikrotiterplatten durchzuführen.
|
Kontrollen
|
25.
|
Bei der Durchführung des Assays sind die Lösungsmittelkontrolle und die sonstigen Kontrollen (d. h. die Kontrollen mit dem Referenzöstrogen, dem schwachen Binder und dem Nicht-Binder) gleichzeitig zu prüfen. Bei jedem Prüflauf sollten auf jeweils einer Platte vollständige Konzentrationskurven des Referenzöstrogens und der Kontrollen (d. h. schwache Binder und Nicht-Binder) verwendet werden. Alle anderen Platten sollten Folgendes enthalten: (i) eine hohe Konzentration (maximale Verdrängung) und eine mittlere Konzentration (etwa IC50) sowohl von E2 als auch des schwachen Binders jeweils dreifach, (ii) eine Lösungsmittelkontrolle und einen nicht spezifischen Binder, ebenfalls jeweils mindestens dreifach. Die Verfahren für die Zubereitung des Assay-Puffers, der Kontrollen, des [3H]-17β-Estradiols, des hrERα und der Prüfchemikalienlösungen werden in (2) (Anlage K, siehe Protokoll zum FW-Assay) beschrieben.
|
Lösungsmittelkontrolle
|
26.
|
Die Lösungsmittelkontrolle bestätigt, dass das Lösungsmittel nicht mit dem Prüfsystem reagiert und die Gesamtbindung (TB) misst. Bevorzugtes Lösungsmittel ist Ethanol. Wenn die höchste Konzentration der Prüfchemikalie in Ethanol nicht löslich ist, kann alternativ DMSO verwendet werden. Die Ethanol- bzw. ggf. die DMSO-Konzentration in den endgültigen Assay-Wells sollte bei 1,5 % liegen und darf höchstens 2 % betragen.
|
Pufferkontrolle
|
27.
|
Die Pufferkontrolle (PK) darf weder Lösungsmittel noch die Prüfchemikalie, jedoch alle sonstigen im Assay verwendeten Elemente enthalten. Die Ergebnisse der Pufferkontrolle werden mit der Lösungsmittelkontrolle verglichen, um sicherzustellen, dass das verwendete Lösungsmittel das Assay-System nicht beeinträchtigt.
|
Starker Binder (Referenzöstrogen)
|
28.
|
17β-Estradiol (CAS 50-28-2) bindet sich als endogener Ligand mit hoher Affinität an den ER Untertyp alpha. Für jeden hrERα-Assay zur Ermittlung konkurrierender Bindungen wird eine Standardkurve mit nicht markiertem 17β-Estradiol erstellt, damit die Variabilität bei Durchführung des Assays in einem Labor im Zeitverlauf beurteilt werden kann. Aus nicht markiertem 17β-Estradiol werden acht Lösungen in Ethanol hergestellt. Das Spektrum der Konzentrationen in den Assay-Wells reicht von 100 nM – 10 pM (-7[log M] bis -11[log M]) mit den folgenden Einzelkonzentrationen: (-7[log M], -8[log M], -8,5[log M], -9[log M], -9,5[log M], -10[log M], -11[log M]). Die höchste Konzentration des nicht markierten 17β-Estradiols (1 μM) dient auch als nicht spezifischer Bindungsindikator. In Tabelle 4 wird diese Konzentration als „NSB“ bezeichnet, obwohl sie Teil der Standardkurve ist.
|
Schwacher Binder
|
29.
|
Um die Empfindlichkeit der einzelnen Versuche nachzuweisen und um die Variabilität bei der Durchführung des Assays im Laufe der Zeit beurteilen zu können, sollte ein schwacher Binder (Norethynodrel (CAS68-23-5) oder Norethindron (CAS 68-22-4)) einbezogen werden. Aus dem schwachen Binder werden acht Lösungen in Ethanol hergestellt. Das Spektrum der Konzentrationen in den Assay-Wells reicht von 3 nM – 30 pM (-8,5[log M] bis -4,5[log M]) mit den folgenden Einzelkonzentrationen: -4,5[log M], -5[log M], -5,5[log M], -6[log M], -6,5[log M], -7[log M],-7,5[log M], -8,5[log M].
|
Nicht-Binder
|
30.
|
Als Negativkontrolle (Nicht-Binder) wird Octyltriethoxysilan (OTES, CAS 2 943-75-1) verwendet. Die Negativkontrolle gewährleistet, dass mit dem Assay unter den gegebenen Bedingungen erkannt wird, wenn Prüfchemikalien sich nicht an den hrERα binden. Aus dem Nicht-Binder werden acht Lösungen in Ethanol hergestellt. Das Spektrum der Konzentrationen in den Assay-Wells reicht von 0,1 nM – 1 000 pM (-10[log M] bis -3[log M]) in logarithmischen Schritten. Als Nicht-Binder kann zur Kontrolle alternativ auch Di-n-butylphtalat (DBP) verwendet werden. Die maximale Löslichkeit wurde für -4[log M] ermittelt.
|
hrERα-Konzentration
|
31.
|
Für die Versuche ist die Menge des Rezeptors zu verwenden, bei der sich eine spezifische Bindung von 20 ± 5 % bezogen auf 1 nM des Radioliganden ergibt (Nummern 12 und 13 in Anlage 2). Die hrERα-Lösung ist unmittelbar vor Gebrauch zuzubereiten.
|
[3H]-17β-Estradiol
|
32.
|
In den Assay-Wells ist [3H]-17β-Estradiol in einer Konzentration von 1,0 nM zu verwenden.
|
Prüfchemikalien
|
33.
|
Zunächst muss anhand einer Löslichkeitsprüfung die Löslichkeitsgrenze der einzelnen Prüfchemikalien ermittelt und ein geeigneter Konzentrationsbereich für die Durchführung des Prüfprotokolls bestimmt werden. Die Löslichkeitsgrenze der einzelnen Prüfchemikalien wird zunächst im Lösungsmittel bestimmt und anschließend unter den Bedingungen des Assays bestätigt. Mit dem Assay sind Endkonzentrationen höchstens bis zu 1 mM zu prüfen. Der Vorversuch zur Dosisfindung wird mit einer Lösungsmittelkontrolle und 8 logarithmischen seriellen Verdünnungen beginnend mit der maximal akzeptierbaren Konzentration (z. B. 1 mM oder weniger, je nach Löslichkeitsgrenze) durchgeführt; dabei ist das Auftreten einer Trübung oder eines Niederschlags zu protokollieren (Nummer 35). Die Prüfchemikalie wird anhand der Kurven für 8 im Vorversuch ermittelte logarithmische Konzentrationskurven untersucht. Die Konzentrationen im zweiten und im dritten Versuch sind ggf. so anzupassen, dass die Konzentrations-Reaktionskurve besser charakterisiert wird.
|
|
34.
|
Verdünnungen der Prüfchemikalien werden mit dem jeweils geeigneten Lösungsmittel hergestellt (Nummer 26 in Anlage 2). Wenn die höchste Konzentration der Prüfchemikalie weder in Ethanol noch in DMSO löslich ist und die Zugabe von weiterem Lösungsmittel dazu führen würde, dass die Lösungsmittelkonzentration im letzten Röhrchen die Akzeptanzgrenze überschreiten würde, kann die nächstgeringere Konzentration als höchste Konzentration angenommen werden. In diesem Fall kann am unteren Ende der Konzentrationsreihe eine weitere Konzentration eingefügt werden. Die übrigen Konzentrationen der Reihe bleiben unverändert.
|
|
35.
|
Die Lösungen mit der Prüfchemikalie werden beim Einbringen in die Assay-Wells sorgfältig beobachtet, da die Prüfchemikalien nach dem Einbringen in die Assay-Wells einen Niederschlag bilden können. Die Daten aller Wells mit einem Niederschlag werden bei der Kurvenanpassung nicht berücksichtigt. Der Grund für den Ausschluss der betreffenden Wells ist zu vermerken.
|
|
36.
|
Wenn bereits Informationen aus anderen Quellen mit einem log (IC50) für eine Prüfchemikalie verfügbar sind, können geometrische Verdünnungsstufen (in 0,5-log-Einheiten um den erwarteten log (IC50) angemessen sein. Am Ende sollte ein ausreichender Abstand der Konzentrationen auf beiden Seiten von log (IC50) einschließlich der höchsten und der niedrigsten Konzentration erreicht sein, damit die Bindungskurve angemessen beschrieben werden kann.
|
Vorbereitung der Assay-Platte
|
37.
|
Mit Etiketten versehene Mikrotiterplatten werden für sechs Inkubationen mit Codes für die Lösungsmittelkontrolle, die höchste Konzentration des Referenzöstrogens (die auch als Indikator für die nicht spezifische Bindung (NSB) dient) und die Pufferkontrolle vorbereitet; außerdem werden für drei Inkubationen Etiketten mit Codes für jede einzelne der acht Konzentrationen der Nicht-Binder-Kontrolle (Octyltriethyxysilan) und für die 7 geringeren Konzentrationen des Referenzöstrogens, die 8 Konzentrationen des schwachen Binders und die 8 Konzentrationen der einzelnen Prüfchemikalien (TC) vorbereitet. Ein Beispiel für die Gestaltung des Plattendiagramms für die vollständigen Konzentrationskurven des Referenzöstrogens und der Kontrolle ist Tabelle 4 zu entnehmen. Für die Prüfchemikalien werden weitere Mikrotiterplatten verwendet; diese Platten sollten Plattenkontrollen, d. h. 1) eine hohe (maximale Verdrängung) und eine mittlere Konzentration (etwa die IC50-Konzentration) von E2 sowie des schwachen Binders (dreifach) und 2) eine Lösungsmittelkontrolle und eine nicht spezifische Bindung (jeweils sechsfach) umfassen (Tabelle 5). Ein Beispiel für die Anordnung auf einer Mikrotiterplatte für einen Assay zur Untersuchung konkurrierender Bindungen mit drei unbekannten Prüfchemikalien ist Anlage 2.3 zu entnehmen. Die in den Tabellen 4 und 5 genannten Konzentrationen sind die Endkonzentrationen des Assays. Die maximale E2-Konzentration beträgt 1×10-7 M. Für den schwachen Binder ist die höchste Konzentration zu verwenden, auch für den schwachen Binder auf Platte 1 verwendet wurde. Die IC50-Konzentration wird vom Labor anhand der Datenbank mit historischen Kontrollen des Labors bestimmt. Dieser Wert sollte sich in etwa mit dem in der Validierungsstudie ermittelten Wert decken (siehe Tabelle 1).
|
Tabelle 4
Anordnung der Mikrotiterplatte für einen Assay zur Untersuchung konkurrierender Bindungen, vollständige Konzentrationskurven für das Referenzöstrogen und die Kontrollen (Platte 1).
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (nur Lösungsmittel)
|
TB (nur Lösungsmittel)
|
NSB
|
NSB
|
|
B
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
C
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Blindkontrolle (*8)
|
|
D
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
E
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
F
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
H
|
Blindkontrolle (für warme Wells) (*9)
|
Blindkontrolle (für warme Wells) (*9)
|
Pufferkontrolle
|
Pufferkontrolle
|
|
Bei diesem Beispiel wird Norethinodrel (NE) als schwacher Binder verwendet.
|
Tabelle 5
Anordnung der Mikrotiterplatte für einen Assay zur Untersuchung konkurrierender Bindungen, vollständige Konzentrationskurven für die Prüfchemikalien und die Plattenkontrollen.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (nur Lösungsmittel)
|
TB (nur Lösungsmittel)
|
NSB
|
NSB
|
|
B
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
C
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
D
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
E
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
F
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
H
|
NE (IC50)
|
NE (1×10-4,5)
|
E2 (IC50)
|
E2 (1×10-7)
|
|
Bei diesem Beispiel wird Norethinodrel (NE) als schwacher Binder verwendet.
|
Abschluss des Assays zur Ermittlung konkurrierender Bindungen
|
38.
|
Wie in Tabelle 6 dargestellt, werden 80 μl der Lösungsmittelkontrolle, der Pufferkontrolle, des Referenzöstrogens, des schwachen Binders, des Nicht-Binders und der im Assay-Puffer zubereiteten Prüfchemikalien in die Wells gegeben. Anschließend werden in jeden Well 40 μl einer Lösung mit 4 nM [3H]-17β-Estradiol gegeben. Nach sanftem Drehen über einen Zeitraum von 10-15 Minuten bei Temperaturen zwischen 2 und 8 °C werden 40 μl der hrERα-Lösung hinzugegeben. Die für den Assay verwendeten Mikrotiterplatten werden 16-20 Stunden bei 2-8 °C auf einem Karussell inkubiert.
|
Tabelle 6
Volumina der Elemente des hrER-Assays zur Ermittlung konkurrierender Bindungen auf den Mikrotiterplatten
|
Volumen (μl)
|
Element
|
|
80
|
Nicht markiertes 17β-Estradiol, Norethynodrel, OTES, Prüfchemikalien, Lösungsmittel oder Puffer
|
|
40
|
4 nM [3H]-17β-Estradiol-Lösung
|
|
40
|
hrERα-Lösung, Konzentration wie ermittelt
|
|
160
|
Gesamtvolumen in den einzelnen Assay-Wells
|
|
39.
|
Nach der Trennung des an den hrERα gebundenen [3H]-17β-Estradiols von dem freien [3H]-17β-Estradiol durch Zugabe von 80 μl kalter DCC-Suspension zu jedem einzelnen Well (wie in den Nummern 20-23 für den Assay zur Ermittlung der Sättigungsbindung beschrieben) wird die Bindung von [3H]-17β-Estradiol an den hrERα quantifiziert.
|
|
40.
|
Die Wells H1-6 (in Tabelle 4 als Blindkontrollen (für warme Wells) gekennzeichnet) geben Aufschluss über die Zerfallsereignisse pro Minute (DPM) des markierten [3H]-Estradiols in einer Aliquote von 40 μl. Die Aliquote von 40 μl wird unmittelbar in die Szintillationsflüssigkeit in den Wells H1-H6 gegeben.
|
Akzeptanzkriterien
Assay zur Ermittlung der Sättigungsbindung
|
41.
|
Die spezifische Bindungskurve sollte mit zunehmenden Konzentrationen des [3H]-17β-Estradiols (soweit verwendet) ein Plateau erreichen. Dieses Plateau zeigt, dass der hrERα mit dem Liganden gesättigt wurde.
|
|
42.
|
Die spezifische Bindung bei 1 nM [3H]-17β-Estradiol sollte im akzeptablen Bereich von 15-25 % des Durchschnitts der insgesamt gemessenen Radioaktivität liegen, um die sich die Radioaktivität in allen Prüfläufen erhöht hat. Gelegentliche geringfügige Über- oder Unterschreitungen dieses Bereichs sind akzeptabel. Wenn Prüfläufe aber durchgängig außerhalb dieses Bereichs liegen bzw. ein bestimmter Prüflauf erheblich außerhalb dieses Bereichs liegt, sollte die Proteinkonzentration angepasst und der Assay zur Ermittlung der Sättigungsbindung wiederholt werden.
|
|
43.
|
Aus den Daten sollte sich ein lineares Scatchard-Diagramm ergeben.
|
|
44.
|
Die nicht spezifische Bindung sollte nicht überhöht sein. In der Regel sollte der Wert der nicht spezifischen Bindung weniger als 35 % der Gesamtbindung betragen. Diese Grenze kann jedoch in Einzelfällen überschritten werden, wenn bei der niedrigsten Konzentration des geprüften 17β-Estradiols sehr wenige Zerfallsereignisse pro Minute gemessen werden.
|
Assay zur Ermittlung konkurrierender Bindungen
|
45.
|
Zunehmende Konzentrationen nicht markierten 17β-Estradiols sollten [3H]-17β-Estradiol aus dem Rezeptor verdrängen; die Verdrängung sollte im Einklang mit einer konkurrierenden Bindung an einer einzigen Bindungsstelle stehen.
|
|
46.
|
Der IC50-Wert des Referenzöstrogens (17β-Estradiol) sollte in etwa mit der molaren Konzentration von [3H]-17β-Estradiol zuzüglich der im Assay zur Ermittlung der Sättigungsbindung berechneten Dissoziationskonstante (Kd) übereinstimmen.
|
|
47.
|
Die spezifische Gesamtbindung sollte einheitlich im akzeptablen Bereich von 20 ± 5 % liegen, wenn die gemessene durchschnittliche Konzentration, um die sich die Radioaktivität in jedem einzelnen Well erhöht hat, im Verlauf der Prüfläufe bei 1 nM lag. Gelegentliche geringfügige Über- oder Unterschreitungen dieses Bereichs sind akzeptabel. Wenn Prüfläufe aber durchgängig außerhalb dieses Bereichs liegen bzw. ein bestimmter Prüflauf erheblich außerhalb dieses Bereichs liegt, sollte die Proteinkonzentration angepasst werden.
|
|
48.
|
Das Lösungsmittel darf keinen Einfluss auf die Empfindlichkeit oder die Reproduzierbarkeit des Assays haben. Die Ergebnisse der Lösungsmittelkontrolle werden mit der Pufferkontrolle verglichen, um sicherzustellen, dass das verwendete Lösungsmittel das Assay-System nicht beeinträchtigt. Die Ergebnisse für die Gesamtbindung (TB) und die Pufferkontrolle sollten vergleichbar sein, wenn das Lösungsmittel den Assay nicht beeinflusst.
|
|
49.
|
Bei Prüfungen mit bis zu 10-3 M (OTES) bzw. 10-4 M (DBP) darf der Nicht-Binder nicht mehr als 25 % des [3H]-17β-Estradiols vom hrERα verdrängen.
|
|
50.
|
Aufgrund von Daten der Studie zur Validierung des FW-Bindungsassays mit hrER wurden Leistungskriterien für das Referenzöstrogen und zwei schwache Binder (z. B. Norethynodrel und Norethindron) entwickelt (Anhang N in (2)). Für den Mittelwert (n) +/- SD aller Kontrolldurchläufe in den an der Validierungsstudie beteiligten Labors werden 95-%-Konfidenzintervalle angegeben. 95-%-Konfidenzintervalle wurden für die Parameter zur Kurvenanpassung (Höchstwert, Mindestwert, Hillslope und log IC50) für das Referenzöstrogen und für schwache Binder sowie für log10 RBA der schwachen Binder bezogen auf das Referenzöstrogen berechnet. Die Konfidenzintervalle werden als Leistungskriterien für die Positivkontrollen angegeben. Tabelle 1 enthält die erwarteten Spannen der Kurvenanpassungsparameter, die als Leistungskriterien verwendet werden können. In der Praxis kann die IC50-Spanne je nach Kd der Rezeptorzubereitung und der Ligandenkonzentration leicht variieren.
|
|
51.
|
In Anbetracht der großen Anzahl potenzieller Prüfchemikalien und der Unterschiede hinsichtlich der potenziellen Affinitäten und Ergebnisse (vollständige Kurve, Teilkurve, keine Kurvenanpassung usw.) wurden keine Leistungskriterien für die Kurvenanpassungsparameter der Prüfchemikalien entwickelt. Die Ergebnisse der einzelnen Prüfläufe sowie die Ergebnisse für die einzelnen Prüfchemikalien sind jedoch einer fachlich qualifizierten Beurteilung zu unterziehen. Um den Höchstwert der Kurve der konkurrierenden Bindungen (z. B. eine Bindungsquote von 90-100 %) eindeutig zu ermitteln, sollte ein ausreichendes Spektrum an Konzentrationen der Prüfchemikalie berücksichtigt werden. Die Variabilität zwischen Wiederholungen bei den einzelnen Konzentrationen der Prüfchemikalie sowie zwischen den drei nicht gleichzeitigen Prüfläufen sollte angemessen und wissenschaftlich vertretbar sein. Die Kontrollen der einzelnen Prüfläufe einer Prüfchemikalie sollten sich im Bereich der Messungen der für diesen FW-Assay angegebenen Leistungen bewegen und mit historischen Kontrolldaten der jeweiligen Labors übereinstimmen.
|
ANALYSE DER DATEN
Assay zur Ermittlung der Sättigungsbindung
|
52.
|
Gemessen werden sowohl die Gesamtbindung als auch die nicht spezifische Bindung. Aufgrund dieser Werte wird die spezifische Bindung bei zunehmenden Konzentrationen von [3H]-17β-Estradiol unter Gleichgewichtsbedingungen ermittelt, indem die nicht spezifische Bindung von der Gesamtbindung abgezogen wird. In der grafischen Darstellung der spezifischen Bindung im Vergleich zur Konzentration von [3H]-17β-Estradiol sollte sich bei der maximalen spezifischen Konzentration ein Plateau als Bestätigung für die erfolgte Sättigung des hrERα mit [3H]-17β-Estradiol ergeben. Außerdem sollte in der Analyse der Daten die Bindung des [3H]-17β-Estradiols an eine einzelne Bindungsstelle mit hoher Affinität dokumentiert werden. Die nicht spezifische Bindung, die Gesamtbindung und die spezifische Bindung sind auf einer Sättigungskurve darzustellen. Darüber hinaus sollte eine nicht lineare Regressionsanalyse (z. B. mit BioSoft; McPherson, 1985; Motulsky, 1995) vorgenommen werden. Die ermittelten Daten sind in einem endgültigen Scatchard-Diagramm darzustellen.
|
|
53.
|
Bei der Analyse sind Bmax und Kd ausschließlich anhand der Daten zur Gesamtbindung zu ermitteln. Dabei ist von einem linearen Verlauf der nicht spezifischen Bindung auszugehen. Eine anderweitige Vorgehensweise ist zu begründen. Ferner sollte eine robuste Regression zur Ermittlung der Best-Fit-Werte vorgenommen werden, sofern nicht eine Begründung für ein anderweitiges Vorgehen angegeben wird. Die gewählte Methode für die Durchführung der robusten Regression ist anzugeben. Bei der Ermittlung von Bmax und Kd aufgrund von Daten zur Sättigungsbindung ist immer eine Korrektur aufgrund des Ligandenabbaus vorzunehmen (z. B. nach der Methode von Swillens 1995).
|
Assay zur Ermittlung konkurrierender Bindungen
|
54.
|
Die Kurve zur Darstellung konkurrierender Bindungen wird als spezifische Bindung von [3H]-17β-Estradiol im Vergleich zur Konzentration (log10-Stufen) des Bindungskonkurrenten dargestellt. Die Konzentration der Prüfchemikalie, die die maximale spezifische Bindung von [3H]-17β-Estradiol um 50 % hemmt, wird als IC50-Wert bezeichnet.
|
|
55.
|
Schätzungen der log-(IC50)-Werte der positiven Kontrollen (z. B. Referenzöstrogen und ein schwacher Binder) sollten mit einer geeigneten Software zur nicht linearen Kurvenanpassung für eine Hill-Gleichung mit 4 Parametern (z. B. BioSoft; McPherson, 1985; Motulsky, 1995) vorgenommen werden. Die Höchstwerte, die Mindestwerte, die Hillslope-Werte und die log-(IC50)-Werte sollten bei der Anpassung dieser Kurven allgemein nicht beschränkt werden. Zur Ermittlung der Best-Fit-Werte sollte eine robuste Regression vorgenommen werden, sofern nicht eine Begründung für ein anderweitiges Vorgehen angegeben wird. Eine Korrektur aufgrund des Ligandenabbaus sollte nicht vorgenommen werden. Nach der Ausgangsanalyse sollte jede einzelne Bindungskurve nochmals geprüft werden, um eine angemessene Anpassung an das Modell sicherzustellen. Die relative Bindungsaffinität (RBA) des schwachen Binders sollte als Prozentanteil von log (IC50) für den schwachen Binder bezogen auf log (IC50) für 17β-Estradiol berechnet werden. Die Ergebnisse der Positivkontrollen und der Nicht-Binder-Kontrolle sind anhand der Kriterien für die Leistungsfähigkeit des Assays (Nummern 45-50 in Anlage 2) zu bewerten.
|
|
56.
|
Die Daten aller Prüfchemikalien werden schrittweise analysiert, um eine angemessene Vorgehensweise und eine korrekte Einstufung der einzelnen Kurven zum konkurrierenden Bindungsverhalten sicherzustellen. Für jeden Prüflauf einer Prüfchemikalie sollte zunächst eine standardisierte Datenanalyse nach dem Verfahren vorgenommen werden, das auch zur Analyse der Kontrollen mit dem Referenzöstrogen und dem schwachen Binder durchgeführt wird (Nummer 55). Nach dieser Analyse sollten die Parameter zur Kurvenanpassung fachlich geprüft werden; außerdem sollte geprüft werden, in welchem Umfang die Daten mit der erzeugten Kurve der konkurrierenden Bindungen übereinstimmen. Bei dieser fachlichen Prüfung sind die Feststellung eines konzentrationsabhängigen Rückgangs der prozentualen spezifischen Bindung von [3H]-17β-Estradiol, eine geringe Variabilität der technischen Wiederholungen bei den einzelnen Chemikalienkonzentrationen und die Konsistenz der Anpassungsparameter bei den drei Prüfläufen gute Indikatoren für die ordnungsgemäße Durchführung des Assays und der Datenanalysen.
|
Datenauswertung
|
57.
|
Sofern alle Akzeptanzkriterien erfüllt sind, wird eine Prüfchemikalie als Binder für den hrERα betrachtet, wenn eine Bindungskurve angepasst werden kann und der niedrigste Punkt auf der Reaktionskurve im Datenbereich weniger als 50 % beträgt (Abbildung 1).
|
|
58.
|
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als Nicht-Binder für den hrERα, wenn die folgenden Bedingungen gegeben sind:
|
—
|
Eine Bindungskurve kann angepasst werden, und der niedrigste Punkt auf der angepassten Reaktionskurve innerhalb des Datenbereichs liegt über 75 %, oder
|
|
—
|
es kann keine Anpassung der Bindungskurve vorgenommen werden, und der niedrigste nicht korrigierte Durchschnittswert der prozentualen Bindung der Konzentrationsgruppen im Datenbereich liegt über 75 %.
|
|
|
59.
|
Prüfchemikalien werden als nicht eindeutig betrachtet, wenn keine der genannten Bedingungen erfüllt ist (z. B. wenn der niedrigste Punkt auf der angepassten Reaktionskurve zwischen 76 und 51 % liegt).
|
Tabelle 7
Kriterien für eine Einstufung nach der Kurve des konkurrierenden Bindungsverhaltens bei einer Prüfchemikalie
|
Einstufung
|
Kriterien
|
|
Bindera
|
Eine Bindungskurve kann angepasst werden. Der niedrigste Punkt auf der Reaktionskurve im Datenbereich liegt bei weniger als 50 %.
|
|
Nicht-Binder b
|
Wenn eine Bindungskurve angepasst werden kann: Der niedrigste Punkt auf der angepassten Reaktionskurve im Datenbereich liegt über 75 %. Wenn keine Bindungskurve angepasst werden kann: Der niedrigste nicht korrigierte Durchschnittswert der prozentualen Bindung der Konzentrationsgruppen im Datenbereich liegt über 75 %.
|
|
Nicht eindeutigc
|
Alle analysierbaren Prüfläufe, bei denen die Prüfchemikalie weder als Binder noch als Nicht-Binder eingestuft wird (beispielsweise wenn der niedrigste Punkt auf der angepassten Reaktionskurve zwischen 76 und 51 % liegt).
|
Abbildung 1
Beispiele für die Einstufung von Prüfchemikalien anhand der Kurve der konkurrierenden Bindungen

|
60.
|
Mehrere in einem Labor mit einer Prüfchemikalie durchgeführte Prüfläufe werden unter Zuweisung von nummerischen Werten zu den einzelnen Prüfläufen und unter Ermittlung des Durchschnitts der Prüfläufe durchgeführt (siehe Tabelle 8). Die Ergebnisse der kombinierten Prüfläufe in den einzelnen Labors werden mit der erwarteten Einstufung der einzelnen Prüfchemikalien verglichen.
|
Tabelle 8
Methode zur Einstufung von Prüfchemikalien aufgrund mehrerer Prüfläufe in einem Labor
|
Zuweisung eines Wertes zu den einzelnen Prüfläufen:
|
|
|
Einstufung
|
Numerischer Wert
|
|
Binder
|
2
|
|
Nicht eindeutig
|
1
|
|
Nicht-Binder
|
0
|
|
Einstufung des durchschnittlichen numerischen Werts für mehrere Prüfläufe:
|
|
|
Einstufung
|
Numerischer Wert
|
|
Binder
|
Durchschnitt ≥ 1,5
|
|
Nicht eindeutig
|
0,5 ≤ Durchschnitt < 1,5
|
|
Nicht-Binder
|
Durchschnitt ≥ 0,5
|
PRÜFBERICHT
|
61.
|
Siehe Nummer 24 im Abschnitt „ELEMENTE DES hrER-BINDUNGSASSAYS“ für diese Prüfmethode.
|
Anlage 2.1
BEGRIFFSBESTIMMUNGEN
[3H]E2
: 17β-Estradiol mit Tritium radioaktiv markiert.
Assay-Puffer
: 10 mM Tris, 10 mg Rinderserumalbumin/ml, 2 mM DTT, 10 % Glycerin, 0,2 mM Leupeptin, pH 7,5.
DCC
: Dextranbeschichtete Aktivkohle.
E2
: Nicht markiertes 17β-Estradiol (inert).
hrERα
: Humaner rekombinanter Östrogenrezeptor alpha.
Prüflauf
: Eine vollständige Reihe gleichzeitig zu prüfender Mikrotiterplatten-Wells, denen alle Informationen zu entnehmen sind, die zur Charakterisierung der Bindung einer Prüfchemikalie an den hrERα benötigt werden (d. h. insgesamt zum Well hinzugegebenes [3H]-17β-Estradiol, maximale Bindung von [3H]-17β-Estradiol an den hrERα, nicht spezifische Bindung und Gesamtbindung bei verschiedenen Konzentrationen der Prüfchemikalie). Ein Prüflauf kann bereits aus einem einzigen Well (d. h. einer einzigen Wiederholung) pro Konzentration bestehen. Da nach diesem Protokoll jedoch eine dreifache Untersuchung vorzunehmen ist, umfasst ein Prüflauf drei Wells pro Konzentration. Außerdem sind nach diesem Protokoll drei unabhängige (d. h. nicht gleichzeitige) Prüfläufe pro Chemikalie vorzunehmen.
Wiederholung (Replikat)
: Eines von mehreren Wells mit denselben Inhalten in denselben Konzentrationen, die in einem einzelnen Prüflauf gleichzeitig untersucht werden. Bei diesem Protokoll wird jede Konzentration der Prüfchemikalie dreimal geprüft, d. h. je Konzentration der Prüfchemikalie werden drei Wiederholungen gleichzeitig untersucht.
Anlage 2.2
TYPISCHER ASSAY ZUR ERMITTLUNG DER SÄTTIGUNGSBINDUNG MIT [3H]-17Β-ESTRADIOL MIT DREI REPLIKAT-WELLS
|
Typischer Assay zur Ermittlung der Sättigungsbindung mit [3H]-17β-Estradiol mit drei Replikat-Wells
|
|
Position
|
Replikat
|
Well-Typ-Code
|
Ausgangskonzentration warmes E2 (nM)
|
Volumen warmes E2 (μl)
|
Endkonzentration warmes E2 (nM)
|
Ausgangskonzentration kaltes E2 (μM)
|
Volumen kaltes E2 (μl)
|
Ausgangskonzentration kaltes E2 (μM)
|
Puffer-Volumen (μl)
|
Rezeptor-Volumen (μl)
|
Ausgangsvolumen in Wells
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Warm
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Warm
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Warm
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Warm
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Warm
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Warm
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Warm
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Warm
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Warm
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Warm
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Warm
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Warm
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Warm
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Warm
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Warm
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Warm
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Warm
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Warm
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Warm
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Warm
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Warm
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Warm
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Warm
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Warm
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Die „warmen“ Wells sind bei der Inkubation leer. Die 40 μl werden nur für die Szintillationszählung hinzugegeben.
|
Anlage 2.3
ANORDNUNG DER WELLS BEIM ASSAY ZUR ERMITTLUNG KONKURRIERENDER BINDUNGEN
|
Platte
|
Position
|
Replikat
|
Well-Typ
|
Well-Code
|
Konzentrationscode
|
Ausgangskonzentration des Bindungskonkurrenten (M)
|
hrER-Vorratslösung (μl)
|
Puffer-Volumen (μl)
|
Tracer-Volumen (warmes E2) (μl)
|
Volumen der Verdünnungsplatte (μl)
|
Endvolumen (μl)
|
Endkonzentration des Bindungskonkurrenten (M)
|
|
S
|
A1
|
1
|
Gesamtbindung
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
Gesamtbindung
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
Gesamtbindung
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
Gesamtbindung
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
Gesamtbindung
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
Gesamtbindung
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
A8
|
2
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
A9
|
3
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
A10
|
1
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
A11
|
2
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
A12
|
3
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
B1
|
1
|
kaltes E2
|
S
|
S1
|
2,0E-7
|
40
|
—
|
40
|
80
|
160
|
1,0E-7
|
|
S
|
B2
|
2
|
kaltes E2
|
S
|
S1
|
2,0E-7
|
40
|
—
|
40
|
80
|
160
|
1,0E-7
|
|
S
|
B3
|
3
|
kaltes E2
|
S
|
S1
|
2,0E-7
|
40
|
—
|
40
|
80
|
160
|
1,0E-7
|
|
S
|
B4
|
1
|
kaltes E2
|
S
|
S2
|
2,0E-8
|
40
|
—
|
40
|
80
|
160
|
1,0E-8
|
|
S
|
B5
|
2
|
kaltes E2
|
S
|
S2
|
2,0E-8
|
40
|
—
|
40
|
80
|
160
|
1,0E-8
|
|
S
|
B6
|
3
|
kaltes E2
|
S
|
S2
|
2,0E-8
|
40
|
—
|
40
|
80
|
160
|
1,0E-8
|
|
S
|
B7
|
1
|
kaltes E2
|
S
|
S3
|
6,0E-9
|
40
|
—
|
40
|
80
|
160
|
3,0E-9
|
|
S
|
B8
|
2
|
kaltes E2
|
S
|
S3
|
6,0E-9
|
40
|
—
|
40
|
80
|
160
|
3,0E-9
|
|
S
|
B9
|
3
|
kaltes E2
|
S
|
S3
|
6,0E-9
|
40
|
—
|
40
|
80
|
160
|
3,0E-9
|
|
S
|
B10
|
1
|
kaltes E2
|
S
|
S4
|
2,0E-9
|
40
|
—
|
40
|
80
|
160
|
1,0E-9
|
|
S
|
B11
|
2
|
kaltes E2
|
S
|
S4
|
2,0E-9
|
40
|
—
|
40
|
80
|
160
|
1,0E-9
|
|
S
|
B12
|
3
|
kaltes E2
|
S
|
S4
|
2,0E-9
|
40
|
—
|
40
|
80
|
160
|
1,0E-9
|
|
S
|
C1
|
1
|
kaltes E2
|
S
|
S5
|
6,0E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
kaltes E2
|
S
|
S5
|
6,0E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
kaltes E2
|
S
|
S5
|
6,0E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E
-10
|
|
S
|
C4
|
1
|
kaltes E2
|
S
|
S6
|
2,0E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
kaltes E2
|
S
|
S6
|
2,0E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
kaltes E2
|
S
|
S6
|
2,0E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
kaltes E2
|
S
|
S7
|
2,0E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
kaltes E2
|
S
|
S7
|
2,0E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
kaltes E2
|
S
|
S7
|
2,0E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
Blindkontrolle
|
Blindkontrolle
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
Blindkontrolle
|
Blindkontrolle
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
Blindkontrolle
|
Blindkontrolle
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
Norethynodrel
|
NE
|
WP1
|
6,0E-5
|
40
|
—
|
40
|
80
|
160
|
3,0E-5
|
|
S
|
D2
|
1
|
Norethynodrel
|
NE
|
WP1
|
6,0E-5
|
40
|
—
|
40
|
80
|
160
|
3,0E-5
|
|
S
|
D3
|
1
|
Norethynodrel
|
NE
|
WP1
|
6,0E-5
|
40
|
—
|
40
|
80
|
160
|
3,0E-5
|
|
S
|
D4
|
1
|
Norethynodrel
|
NE
|
WP2
|
2,0E-5
|
40
|
—
|
40
|
80
|
160
|
1,0E-5
|
|
S
|
D5
|
1
|
Norethynodrel
|
NE
|
WP2
|
2,0E-5
|
40
|
—
|
40
|
80
|
160
|
1,0E-5
|
|
S
|
D6
|
1
|
Norethynodrel
|
NE
|
WP2
|
2,0E-5
|
40
|
—
|
40
|
80
|
160
|
1,0E-5
|
|
S
|
D7
|
1
|
Norethynodrel
|
NE
|
WP3
|
6,0E-6
|
40
|
—
|
40
|
80
|
160
|
3,0E-6
|
|
S
|
D8
|
1
|
Norethynodrel
|
NE
|
WP3
|
6,0E-6
|
40
|
—
|
40
|
80
|
160
|
3,0E-6
|
|
S
|
D9
|
1
|
Norethynodrel
|
NE
|
WP3
|
6,0E-6
|
40
|
—
|
40
|
80
|
160
|
3,0E-6
|
|
S
|
D10
|
1
|
Norethynodrel
|
NE
|
WP4
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
D11
|
1
|
Norethynodrel
|
NE
|
WP4
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
D12
|
1
|
Norethynodrel
|
NE
|
WP4
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
E1
|
1
|
Norethynodrel
|
NE
|
WP
|
6,0E-7
|
40
|
|
40
|
80
|
160
|
3,0E-7
|
|
S
|
E2
|
2
|
Norethynodrel
|
NE
|
WP
|
6,0E-7
|
40
|
|
40
|
80
|
160
|
3,0E-7
|
|
S
|
E3
|
3
|
Norethynodrel
|
NE
|
WP
|
6,0E-7
|
40
|
|
40
|
80
|
160
|
3,0E-7
|
|
S
|
E4
|
1
|
Norethynodrel
|
NE
|
WP
|
2,0E-7
|
40
|
|
40
|
80
|
160
|
1,0E-7
|
|
S
|
E5
|
2
|
Norethynodrel
|
NE
|
WP
|
2,0E-7
|
40
|
|
40
|
80
|
160
|
1,0E-7
|
|
S
|
E6
|
3
|
Norethynodrel
|
NE
|
WP
|
2,0E-7
|
40
|
|
40
|
80
|
160
|
1,0E-7
|
|
S
|
E7
|
1
|
Norethynodrel
|
NE
|
WP
|
6,0E-8
|
40
|
—
|
40
|
80
|
160
|
3,0E-8
|
|
S
|
E8
|
2
|
Norethynodrel
|
NE
|
WP
|
6,0E-8
|
40
|
—
|
40
|
80
|
160
|
3,0E-8
|
|
S
|
E9
|
3
|
Norethynodrel
|
NE
|
WP
|
6,0E-8
|
40
|
—
|
40
|
80
|
160
|
3,0E-8
|
|
S
|
E10
|
1
|
Norethynodrel
|
NE
|
WP
|
6,0E-9
|
40
|
—
|
40
|
80
|
160
|
3,0E-9
|
|
S
|
E11
|
2
|
Norethynodrel
|
NE
|
WP
|
6,0E-9
|
40
|
—
|
40
|
80
|
160
|
3,0E-9
|
|
S
|
E12
|
3
|
Norethynodrel
|
NE
|
WP
|
6,0E-9
|
40
|
—
|
40
|
80
|
160
|
3,0E-9
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,0E-3
|
40
|
—
|
40
|
80
|
160
|
1,0E-3
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,0E-3
|
40
|
—
|
40
|
80
|
160
|
1,0E-3
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,0E-3
|
40
|
—
|
40
|
80
|
160
|
1,0E-3
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,0E-4
|
40
|
—
|
40
|
80
|
160
|
1,0E-4
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,0E-4
|
40
|
—
|
40
|
80
|
160
|
1,0E-4
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,0E-4
|
40
|
—
|
40
|
80
|
160
|
1,0E-4
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,0E-5
|
40
|
—
|
40
|
80
|
160
|
3,0E-5
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,0E-5
|
40
|
—
|
40
|
80
|
160
|
3,0E-5
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,0E-5
|
40
|
—
|
40
|
80
|
160
|
3,0E-5
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,0E-7
|
40
|
—
|
40
|
80
|
160
|
3,0E-7
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,0E-7
|
40
|
—
|
40
|
80
|
160
|
3,0E-7
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,0E-7
|
40
|
—
|
40
|
80
|
160
|
3,0E-7
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,0E-8
|
40
|
—
|
40
|
80
|
160
|
1,0E-8
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,0E-8
|
40
|
—
|
40
|
80
|
160
|
1,0E-8
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,0E-8
|
40
|
—
|
40
|
80
|
160
|
1,0E-8
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,0E-9
|
40
|
—
|
40
|
80
|
160
|
1,0E-9
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,0E-9
|
40
|
—
|
40
|
80
|
160
|
1,0E-9
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,0E-9
|
40
|
—
|
40
|
80
|
160
|
1,0E-9
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,0E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,0E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,0E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
Warm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
Warm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
Warm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
Warm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
Warm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
Warm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
Pufferkontrolle
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
Pufferkontrolle
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
Pufferkontrolle
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
Pufferkontrolle
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
Pufferkontrolle
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
Pufferkontrolle
|
PK
|
PK
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Die „warmen“ Wells sind bei der Inkubation leer. Die 40 μl werden nur für die Szintillationszählung hinzugegeben.
|
Anordnung der Wells beim Assay zur Ermittlung konkurrierender Bindungen
|
|
Platte
|
Position
|
Replikat
|
Well-Typ
|
Well-Code
|
Konzentrationscode
|
Ausgangskonzentration des Bindungskonkurrenten (M)
|
hrER-Vorratslösung (μl)
|
Puffer-Vol. (μl)
|
Tracer-Volumen (warmes E2) (μl)
|
Volumen der Verdünnungsplatte (μl)
|
Endvolumen (μl)
|
Endkonzentration des Bindungskonkurrenten (M)
|
|
P1
|
A1
|
1
|
Gesamtbindung
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
Gesamtbindung
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
Gesamtbindung
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
Gesamtbindung
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
Gesamtbindung
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
Gesamtbindung
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
A8
|
2
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
A9
|
3
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
A10
|
1
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
A11
|
2
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
A12
|
3
|
kaltes E2 (hoch)
|
NSB
|
S0
|
2,0E-6
|
40
|
—
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
B1
|
1
|
Prüfchemikalie 1
|
TC1
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
B2
|
2
|
Prüfchemikalie 1
|
TC1
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
B3
|
3
|
Prüfchemikalie 1
|
TC1
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
B4
|
1
|
Prüfchemikalie 1
|
TC1
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
B5
|
2
|
Prüfchemikalie 1
|
TC1
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
B6
|
3
|
Prüfchemikalie 1
|
TC1
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
B7
|
1
|
Prüfchemikalie 1
|
TC1
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
B8
|
2
|
Prüfchemikalie 1
|
TC1
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
B9
|
3
|
Prüfchemikalie 1
|
TC1
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
B10
|
1
|
Prüfchemikalie 1
|
TC1
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
B11
|
2
|
Prüfchemikalie 1
|
TC1
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
B12
|
3
|
Prüfchemikalie 1
|
TC1
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
C1
|
1
|
Prüfchemikalie 1
|
TC1
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
C2
|
2
|
Prüfchemikalie 1
|
TC1
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
C3
|
3
|
Prüfchemikalie 1
|
TC1
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
C4
|
1
|
Prüfchemikalie 1
|
TC1
|
6
|
2,0E-8
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
C5
|
2
|
Prüfchemikalie 1
|
TC1
|
6
|
2,0E-8
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
C6
|
3
|
Prüfchemikalie 1
|
TC1
|
6
|
2,0E-8
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
C7
|
1
|
Prüfchemikalie 1
|
TC1
|
7
|
2,0E-9
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
C8
|
2
|
Prüfchemikalie 1
|
TC1
|
7
|
2,0E-9
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
C9
|
3
|
Prüfchemikalie 1
|
TC1
|
7
|
2,0E-9
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
C10
|
1
|
Prüfchemikalie 1
|
TC1
|
8
|
2,0E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
Prüfchemikalie 1
|
TC1
|
8
|
2,0E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
Prüfchemikalie 1
|
TC1
|
8
|
2,0E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
Prüfchemikalie 2
|
TC2
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
D2
|
2
|
Prüfchemikalie 2
|
TC2
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
D3
|
3
|
Prüfchemikalie 2
|
TC2
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
D4
|
1
|
Prüfchemikalie 2
|
TC2
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
D5
|
2
|
Prüfchemikalie 2
|
TC2
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
D6
|
3
|
Prüfchemikalie 2
|
TC2
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
D7
|
1
|
Prüfchemikalie 2
|
TC2
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
D8
|
2
|
Prüfchemikalie 2
|
TC2
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
D9
|
3
|
Prüfchemikalie 2
|
TC2
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
D10
|
1
|
Prüfchemikalie 2
|
TC2
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
D11
|
2
|
Prüfchemikalie 2
|
TC2
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
D12
|
3
|
Prüfchemikalie 2
|
TC2
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
E1
|
1
|
Prüfchemikalie 2
|
TC2
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
E2
|
2
|
Prüfchemikalie 2
|
TC2
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
E3
|
3
|
Prüfchemikalie 2
|
TC2
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
E4
|
1
|
Prüfchemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
E5
|
2
|
Prüfchemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
E6
|
3
|
Prüfchemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
E7
|
1
|
Prüfchemikalie 2
|
TC2
|
7
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
E8
|
2
|
Prüfchemikalie 2
|
TC2
|
7
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
E9
|
3
|
Prüfchemikalie 2
|
TC2
|
7
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
E10
|
1
|
Prüfchemikalie 2
|
TC2
|
8
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
Prüfchemikalie 2
|
TC2
|
8
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
Prüfchemikalie 2
|
TC2
|
8
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
Prüfchemikalie 3
|
TC3
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
F2
|
2
|
Prüfchemikalie 3
|
TC3
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
F3
|
3
|
Prüfchemikalie 3
|
TC3
|
1
|
2,0E-3
|
40
|
0
|
40
|
80
|
160
|
1,0E-3
|
|
P1
|
F4
|
1
|
Prüfchemikalie 3
|
TC3
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
F5
|
2
|
Prüfchemikalie 3
|
TC3
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
F6
|
3
|
Prüfchemikalie 3
|
TC3
|
2
|
2,0E-4
|
40
|
0
|
40
|
80
|
160
|
1,0E-4
|
|
P1
|
F7
|
1
|
Prüfchemikalie 3
|
TC3
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
F8
|
2
|
Prüfchemikalie 3
|
TC3
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
F9
|
3
|
Prüfchemikalie 3
|
TC3
|
3
|
2,0E-5
|
40
|
0
|
40
|
80
|
160
|
1,0E-5
|
|
P1
|
F10
|
1
|
Prüfchemikalie 3
|
TC3
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
F11
|
2
|
Prüfchemikalie 3
|
TC3
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
F12
|
3
|
Prüfchemikalie 3
|
TC3
|
4
|
2,0E-6
|
40
|
0
|
40
|
80
|
160
|
1,0E-6
|
|
P1
|
G1
|
1
|
Prüfchemikalie 3
|
TC3
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
G2
|
2
|
Prüfchemikalie 3
|
TC3
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
G3
|
3
|
Prüfchemikalie 3
|
TC3
|
5
|
2,0E-7
|
40
|
0
|
40
|
80
|
160
|
1,0E-7
|
|
P1
|
G4
|
1
|
Prüfchemikalie 3
|
TC3
|
6
|
2,0E-8
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
G5
|
2
|
Prüfchemikalie 3
|
TC3
|
6
|
2,0E-8
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
G6
|
3
|
Prüfchemikalie 3
|
TC3
|
6
|
2,0E-8
|
40
|
0
|
40
|
80
|
160
|
1,0E-8
|
|
P1
|
G7
|
1
|
Prüfchemikalie 3
|
TC3
|
7
|
2,0E-9
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
G8
|
2
|
Prüfchemikalie 3
|
TC3
|
7
|
2,0E-9
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
G9
|
3
|
Prüfchemikalie 3
|
TC3
|
7
|
2,0E-9
|
40
|
0
|
40
|
80
|
160
|
1,0E-9
|
|
P1
|
G10
|
1
|
Prüfchemikalie 3
|
TC3
|
8
|
2,0E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
Prüfchemikalie 3
|
TC3
|
8
|
2,0E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
Prüfchemikalie 3
|
TC3
|
8
|
2,0E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
Norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
Norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
Norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
Norethynodrel
|
NE
|
|
1,0E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
Norethynodrel
|
NE
|
|
1,0E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
Norethynodrel
|
NE
|
|
1,0E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
kaltes E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
kaltes E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
kaltes E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
kaltes E2 S
|
|
|
1,0E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
kaltes E2 S
|
|
|
1,0E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
kaltes E2 S
|
|
|
1,0E-7
|
40
|
0
|
40
|
80
|
160
|
|
Anlage 3
DER IN-VITRO-ÖSTROGENREZEPTOR-BINDUNGSASSAY DES CHEMICAL EVALUATION AND RESEARCH INSTITUTE (CERI) MIT EINEM LIGANDENBINDUNGS-DOMÄNEN-PROTEIN DES HUMANEN REKOMBINANTEN ERΑ
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
1.
|
Bei diesem In-vitro-Assay zur Ermittlung der Sättigung mit dem Östrogenrezeptor ERα und zur Ermittlung konkurrierender Bindungen wird eine Ligandenbindungs-Domäne (LBD) des humanen ERα (hrERα) verwendet. Dieses Proteinmodell wurde vom Chemicals Evaluation Research Institute (CERI), Japan, hergestellt und wird als GST-Fusionsprotein (GST = Glutathion-S-transferase) in E. coli exprimiert. Das CERI-Protokoll wurde einer internationalen Validierungsstudie durch mehrere Labors unterzogen (2). Dabei wurden die Relevanz und die Zuverlässigkeit des Protokolls für den vorgesehenen Zweck des Assays nachgewiesen.
|
|
2.
|
Der Assay besteht in einem Prüfverfahren zur Ermittlung von Stoffen, die eine Bindung mit dem hrERα eingehen können. Mit dem Assay kann untersucht werden, ob eine Prüfchemikalie mit 17β-Estradiol um Bindungen an die hrERα-LBD konkurrieren kann. Als quantitative Ergebnisse können mit dem Assay u. a. die Werte für IC50 (als Maßstab für die zur Verdrängung der Hälfte des [3H]-17β-Estradiols vom hrERα durch eine Prüfchemikalie) und die relative Affinität von Prüfchemikalien für Bindungen an den hrERα im Vergleich zu 17β-Estradiol ermittelt werden. Mit Blick auf die Verwendung zur Prüfung von Chemikalien kann ein akzeptables qualitatives Ergebnis des Assays auch darin bestehen, dass Prüfchemikalien nach ihrer Bindung an den hrERα und nach den genannten Kriterien für die Bindungskurven als Binder, Nicht-Binder oder nicht eindeutig eingestuft werden.
|
|
3.
|
Da bei diesem Assay ein radioaktiver Ligand verwendet wird, benötigen die Labors eine Genehmigung für die Verwendung von radioaktivem Material. Bei allen Verfahren unter Verwendung von Radioisotopen und von gefährlichen Chemikalien sind die nach den nationalen Rechtsvorschriften geltenden Bestimmungen und die dort vorgesehenen Verfahren einzuhalten.
|
|
4.
|
Vor der Verwendung dieses Assays für rechtliche Zwecke sollten die Abschnitte „ALLGEMEINE EINLEITUNG“ und „ELEMENTE DES hrER-BINDUNGSASSAYS“ gelesen werden. Die in dieser Prüfrichtlinie verwendeten Begriffe und Abkürzungen werden in Anlage 1 erläutert.
|
PRINZIPIEN DES ASSAYS (SIEHE AUCH ALLGEMEINE EINLEITUNG)
|
5.
|
Mit dem hrERα-Bindungsassay wird die Fähigkeit eines radioaktiv markierten Liganden ([3H]-17β-Estradiol) gemessen, bei zunehmenden Konzentrationen einer Prüfchemikalie (z. B. eines Bindungskonkurrenten) eine Bindung mit dem ER einzugehen. Prüfchemikalien mit einer starken ER-Affinität konkurrieren mit dem radioaktiv markierten Liganden bei einer niedrigeren Konzentration als Chemikalien mit einer geringeren Affinität für diesen Rezeptor.
|
|
6.
|
Dieser Assay beinhaltet zwei wesentliche Elemente: einen Versuch zur Ermittlung von Sättigungsbindungen zur Charakterisierung der Parameter der Wechselwirkung zwischen Rezeptor und Ligand und einen anschließenden Versuch zur Ermittlung konkurrierender Bindungen, mit dem die Konkurrenz zwischen einer Prüfchemikalie und einem radioaktiv markierten Liganden im Hinblick auf die Bindung an den ER beschrieben wird.
|
|
7.
|
Zur Vorbereitung der Prüfung konkurrierender Bindungen soll anhand einer Ermittlung der Sättigungsbindung eine bestimmte Rezeptorcharge hinsichtlich ihrer Bindungsaffinität geprüft und quantitativ bewertet werden. Mit der Prüfung konkurrierender Bindungen werden unter Gleichgewichtsbedingungen die Affinität einer bestimmten Konzentration des Östrogenrezeptors für seinen natürlichen Liganden (ausgedrückt durch die Dissoziationskonstante Kd) und die Konzentration der aktiven Rezeptor-Bindungsstellen (Bmax) gemessen.
|
|
8.
|
Ferner wird mit der Prüfung konkurrierender Bindungen die Affinität eines Stoffs für Bindungen an den ER im Hinblick auf die Konkurrenz mit [3]17β-Estradiol bewertet. Die Affinität wird anhand der Konzentration der Prüfchemikalie quantifiziert, die unter Gleichgewichtsbedingungen 50 % der spezifischen Bindung von [3]17β-Estradiol („Hemmkonzentration 50 %“ oder IC50) hemmt. Diese Affinität kann auch anhand der relativen Bindungsaffinität (RBA, bezogen auf IC50 Estradiol, im selben Prüflauf getrennt gemessen) beurteilt werden. Bei der Prüfung konkurrierender Bindungen wird die Bindung von [3H]-17β-Estradiol in einer bestimmten Konzentration an zahlreiche (acht Größenordnungen) Konzentrationen einer Prüfchemikalie gemessen. Die ermittelten Daten werden dann möglichst entsprechend der Hill-Gleichung (Hill, 1910) angepasst, die die Verdrängung des Radioliganden durch einen für eine bestimmte Bindungsstelle spezifischen konkurrierenden Binder beschreibt. Anhand des Umfangs der Verdrängung des radioaktiv markierten Estradiols bei Gleichgewichtsbedingungen wird die Prüfchemikalie als Binder, Nicht-Binder oder nicht eindeutig eingestuft.
|
PRÜFVERFAHREN
Nachweis einer akzeptablen Leistungsfähigkeit bei Untersuchungen des hrERα-Proteins
|
9.
|
Vor der regelmäßigen Durchführung der Assays zur Ermittlung von Sättigungsbindungen und konkurrierenden Bindungen ist jeweils mit einer neuen Charge des hrERα nachzuweisen, dass in dem Labor, in dem der Rezeptor verwendet wird, korrekte Ergebnisse ermittelt werden. Die Leistungsfähigkeit sollte in zwei Schritten nachgewiesen werden:
|
—
|
Durchführung des [3H]-17β-Estradiol-Bindungsassays zum Nachweis der Spezifität und der Sättigung für den hrERα; anhand einer nicht linearen Regressionsanalyse der ermittelten Daten (z. B. BioSoft; McPherson, 1985; Motulsky, 1995) und des anschließenden Scatchard-Diagramms sollten die Bindungsaffinität des [3H]-17β-Estradiols (Kd) für den hrERα und die Anzahl der Rezeptoren (Bmax) einer bestimmten hrERα-Charge dokumentiert werden;
|
|
—
|
Durchführung eines Assays zur Prüfung konkurrierender Bindungen mit Kontrollstoffen (Referenzöstrogen (17β-Estradiol), einem schwachen Binder (z. B. Norethynodrel oder Norethindron) und einem Nicht-Binder (Octyltriethoxysilan, OTES). Jedes Labor sollte eine historische Datenbank zum Nachweis der versuchs- und hrERα-Chargen-übergreifenden Konsistenz der IC50-Werte und der relevanten Werte aus den Versuchen mit dem Referenzöstrogen und einem schwachen Binder aufbauen. Außerdem sollten die Parameter der Kurven der konkurrierenden Bindungen bei den Kontrollstoffen innerhalb des 95-%-Konfidenzintervalls liegen (siehe Tabelle 1), das anhand von Daten von an der Validierungsstudie zu diesem Assay beteiligten Labors festgelegt wurde (2).
|
Tabelle 1
Leistungskriterien für das Referenzöstrogen und den schwachen Binder beim hrER-Bindungsassay des CERI
|
Stoff
|
Parameter
|
Mittelwert (10)
|
Standardabweichung(en)
|
95-%-Konfidenzintervalle (11)
|
|
Obere Grenze
|
Untere Grenze
|
|
17β-Estradiol
|
Höchstwert
|
104,74
|
13.12 (70)
|
101,6
|
107,9
|
|
Mindestwert
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Hillslope
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
log IC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Norethynodrel
|
Höchstwert
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Mindestwert
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Hillslope
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
log IC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Norethindron (12)
|
Höchstwert
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Mindestwert
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Hillslope
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
log IC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Nachweis der Eignung des Labors
|
10.
|
Siehe Nummern 17 und 18 sowie Tabelle 2 im Abschnitt „ELEMENTE DES hrER-BINDUNGSASSAYS“ für diese Prüfmethode. Die Assays (zur Ermittlung von Sättigungsbindungen und konkurrierenden Bindungen) sollten jeweils drei unabhängige Prüfläufe (d. h. mit frischen Rezeptor-, Chemikalien- und Reagenzienverdünnungen) an unterschiedlichen Tagen jeweils mit drei Wiederholungen umfassen.
|
Ermittlung der Rezeptorkonzentration (hrERα)
|
11.
|
Die Konzentration des aktiven Rezeptors ist je nach Charge und Lagerungsbedingungen leicht unterschiedlich. Daher sollte die Konzentration des vom Hersteller gelieferten aktiven Rezeptors bestimmt werden. Daraus ergibt sich die geeignete Konzentration des aktiven Rezeptors beim Prüflauf.
|
|
12.
|
Unter den Bedingungen bei der konkurrierenden Bindung (d. h. 0,5 nM [3H]-Estradiol) werden Nennkonzentrationen von 0,1, 0,2, 0,4 und 0,6 nM Rezeptor einmal ohne (Gesamtbindung) und einmal mit (nicht spezifische Bindung) 1 μM nicht markiertem Estradiol inkubiert. Die spezifische Bindung wird berechnet als Differenz zwischen der Gesamtbindung und der nicht spezifischen Bindung bezogen auf die Rezeptor-Nennkonzentration in einem Diagramm dargestellt. Die Konzentration des Rezeptors, bei der sich spezifische Bindungswerte entsprechend einem Anteil von 40 % des zugesetzten radioaktiven Liganden ergeben, wird auf die entsprechende Rezeptor-Konzentration bezogen; diese Rezeptorkonzentration ist bei den Versuchen zur Ermittlung von Sättigungsbindungen und konkurrierenden Bindungen zu verwenden. Diese Anforderung wird häufig mit einer hrER-Endkonzentration von 0,2 nM erfüllt.
|
|
13.
|
Wenn das 40-%-Kriterium mehrfach nicht erfüllt werden kann, sollte der Versuchsaufbau auf mögliche Fehlerquellen geprüft werden. Dass das 40-%-Kriterium nicht erfüllt werden kann, deutet möglicherweise auch darauf hin, dass die rekombinante Charge den aktiven Rezeptor nur in einem sehr geringen Anteil enthält. In diesem Fall sollte die Verwendung einer anderen Rezeptor-Charge in Betracht gezogen werden.
|
Assay zur Ermittlung der Sättigungsbindung
|
14.
|
Acht zunehmende [3H]-17β-Estradiol-Konzentrationen sollten jeweils dreifach unter den drei folgenden Bedingungen geprüft werden (siehe Tabelle 2):
|
a.
|
ohne nicht markiertes 17β-Estradiol und mit dem ER, um anhand einer Messung der Radioaktivität der Wells, die ausschließlich [3H]-17β-Estradiol enthalten, die Gesamtbindung zu ermitteln;
|
|
b.
|
bei einer 2000-mal höheren Konzentration des nicht markierten 17β-Estradiols im Vergleich zum markierten 17β-Estradiol und zu Prüfungen mit dem ER; dadurch sollen die aktiven Bindungsstellen mit nicht markiertem 17β-Estradiol gesättigt werden, um anhand einer Messung der Radioaktivität in den Wells die nicht spezifische Bindung zu bestimmen. Verbleibendes warmes Estradiol, das Bindungen mit dem Rezeptor eingehen kann, wird als Bindung an eine nicht spezifischen Bindungsstelle betrachtet, da das kalte Estradiol in einer derart hohen Konzentration vorliegen sollte, dass es an allen verfügbaren spezifischen Bindungsstellen des Rezeptors gebunden ist;
|
|
c.
|
ohne nicht markiertes 17β-Estradiol und ohne den ER (Ermittlung der Gesamt-Radioaktivität).
|
Zubereitung der Lösungen mit [3H]-17β-Estradiol und mit nicht markiertem 17β-Estradiol und dem hrERα
|
15.
|
Aus einer Vorratslösung mit 1 μM [3H]-17β-Estradiol in DMSO wird unter Zugabe von DMSO (zur Herstellung von 200 nM) und des Assay-Puffers (zur Herstellung von 40 nM) bei Raumtemperatur eine Lösung mit einer molaren Masse von 40 nM [3H]-17β-Estradiol hergestellt. Mit dieser Lösung mit einer Konzentration von 40 nM werden mit dem Assay-Puffer bei Raumtemperatur serielle Verdünnungen von [3H]-17β-Estradiol im Bereich von 0,313 nM bis 40 nM hergestellt (siehe Zeile 12 in Tabelle 2). Um die endgültigen Assay-Konzentrationen von 0,0313-4,0 nM herzustellen, werden 10 μl dieser Lösungen in die Wells einer 96-Well-Mikrotiterplatte gegeben (siehe Tabellen 2 und 3). Die Zubereitung des Assay-Puffers, die Berechnung der Ausgangskonzentration der [3H]-17β-Estradiol-Vorratslösung und der Verdünnungen sowie die Bestimmung der Konzentrationen werden im CERI-Protokoll eingehend beschrieben (2).
|
|
16.
|
Die nicht markierten 17β-Estradiol-Lösungen werden aus einer 1-nM-17β-Estradiol-Vorratslösung unter Zugabe des Assay-.Puffers so verdünnt, dass sich acht zunehmende Ausgangskonzentrationen von 0,625-80 μM ergeben. Um die endgültigen Assay-Konzentrationen von 0,0625-8 μM herzustellen, werden 10 μl dieser Lösungen in die Wells einer 96-Well-Mikrotiterplatte zur Messung nicht spezifischer Bindungen gegeben (siehe Tabellen 2 und 3). Die Zubereitung der nicht markierten 17â-Estradiol-Lösungen wird eingehend im CERI-Protokoll beschrieben (2).
|
|
17.
|
Für die Versuche ist die Rezeptorkonzentration zu verwenden, bei der sich eine spezifische Bindung von 40 ± 10 % ergibt (Nummern 12 und 13). Die hrERα-Lösung wird mit eiskaltem Assay-Puffer unmittelbar vor der Verwendung hergestellt, d. h. nachdem alle Wells zur Ermittlung der Gesamtbindung, der nicht spezifischen Bindung und der warmen Liganden vorbereitet wurden.
|
|
18.
|
Die 96-Well-Mikrotiterplatten werden mit 2-3 Wiederholungen pro Konzentration des [3H]-17β-Estradiols vorbereitet, wie in Tabelle 2 dargestellt. Die Anordnung der Volumina von [3H]-17β-Estradiol, nicht markiertem 17β-Estradiol, des Puffers und des Rezeptors ist Tabelle 3 zu entnehmen.
Tabelle 2
Anordnung auf der Mikrotiterplatte beim Assay zur Ermittlung der Sättigungsbindung
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
Zur Messung der TB
|
Zur Messung der NSB
|
Zur Ermittlung nur des warmen Liganden
|
|
Nicht markierte E2-Verdünnungen für die Plattenreihen 4-6.
|
[3H]E2-Verdünnungen für die Plattenreihen 1-9
|
|
A
|
0,0313 nM [3H] E2+ ER
|
0,0313 nM [3H] E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H] E2+ ER
|
0,0625 nM [3H] E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H] E2+ ER
|
0,125 nM [3H] E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H] E2+ ER
|
0,250 nM [3H] E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [3H] E2+ ER
|
0,50 nM [3H] E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H] E2+ ER
|
1,00 nM [3H] E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H] E2+ ER
|
2,00 nM [3H] E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H] E2+ ER
|
4,00 nM [3H] E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
-
|
Gesamtbindung
|
|
NSB
|
-
|
Nicht spezifische Bindung
|
|
[3H] E2
|
-
|
[3H]-17β-Estradiol
|
|
E2
|
-
|
nicht markiertes 17β-Estradiol
|
|
Tabelle 3
Volumina der Reagenzien auf der Mikrotiterplatte zur Ermittlung der Sättigungsbindung
|
Plattenreihe
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Vorbereitungsschritte
|
TB-Wells
|
NSB-Wells
|
Nur warme Liganden
|
|
Volumen der Bestandteile für die genannten Reaktions-Wells und Reihenfolge der Zugabe des …
|
… Puffers
|
60 μl
|
50 μl
|
90 μl
|
|
… nicht markierten E2 aus Reihe 11 in Tabelle 2
|
–
|
10 μl
|
–
|
|
…[3H]E2 aus Reihe 12 in Tabelle 2
|
10 μl
|
10 μl
|
10 μl
|
|
… hrERα
|
30 μl
|
30 μl
|
–
|
|
Gesamtreaktionsvolumen
|
100 μl
|
100 μl
|
100 μl
|
|
Inkubation
|
REAKTION NACH 2-STÜNDIGER INKUBATION
|
Quantifizierung der Radioaktivität unmittelbar nach der Vorbereitung Keine Inkubation
|
|
Behandlung mit 0,4 % DCC
|
Ja
|
Ja
|
Nein
|
|
Volumen 0,4 % DCC
|
100 μl
|
100 μl
|
–
|
|
Filtration
|
Ja
|
Ja
|
Nein
|
|
MESSUNG DER DPM
|
|
Quantifizierung des dem Szintillationscocktail zuzusetzenden Volumens
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Assay-Mikrotiterplatten zur Ermittlung der Gesamtbindung und der nicht spezifischen Bindung werden zwei Stunden bei Raumtemperatur (22-28 °C) inkubiert.
|
|
Messung des an den hrERα gebundenen [3H]-17β-Estradiols
|
20.
|
Nach der 2-stündigen Inkubationszeit wird das an den hrERα gebundene [3H]-17β-Estradiol unter Zugabe von 100 μl einer eiskalten Suspension mit 0,4 % DCC zu den Wells vom freien [3H]-17β-Estradiol getrennt. Danach werden die Platten 10 Minuten auf Eis gelegt. Um das DCC zu entfernen, werden das Reaktionsgemisch und die DCC-Suspension durch Übertragung auf einen Mikrotiterplatten-Filter gefiltert. 100 μl des Filtrats werden zur Szintillationsflüssigkeit in Flüssigszintillationsfläschchen hinzugegeben, um anhand von Flüssigkeitsszintillationszählungen den Abbau pro Minute (DPM) pro Fläschchen zu ermitteln.
|
|
21.
|
Wenn kein Mikrotiterplatten-Filter verfügbar ist, kann das DCC auch durch Zentrifugieren entfernt werden. 50 μl des Überstands des an den hrERα gebundenen [3H]-17β-Estradiols sind anschließend mit äußerster Sorgfalt zu entnehmen, damit Verunreinigungen der Wells durch Berührung mit der DCC-Lösung vermieden werden. Der Überstand wird zur Szintillationszählung verwendet.
|
|
22.
|
Anhand der Wells, die ausschließlich den warmen Liganden enthalten, wird der Zerfall des zu den Assay-Wells hinzugegebenen [3H]-17β-Estradiols (DPM = Zerfallsereignisse pro Minute) bestimmt. Die Radioaktivität wird erst nach der Zubereitung ermittelt. Die betreffenden Wells werden nicht inkubiert und nicht mit der DCC-Suspension behandelt; ihr Inhalt wird direkt in die Szintillationsflüssigkeit gegeben. Anhand der Zerfallsereignisse pro Minute (DPM) wird so nachgewiesen, wieviel [3H]-17β-Estradiol jeweils in die Wells zur Ermittlung der Gesamtbindung und der nicht spezifischen Bindung gegeben wurde.
|
Assay zur Ermittlung konkurrierender Bindungen
|
23.
|
Mit dem Assay zur Ermittlung konkurrierender Bindungen wird die Bindung von [3H]-17β- Estradiol einer einzelnen Konzentration bei zunehmenden Konzentrationen einer Prüfchemikalie gemessen. Bei jedem Prüflauf sind für jede Konzentration drei gleichzeitige Replikate zu verwenden. Außerdem werden für jede zu prüfende Chemikalie drei nicht gleichzeitige Prüfläufe durchgeführt. Der Assay ist auf zwei oder mehr 96-Well-Mikrotiterplatten durchzuführen.
|
Kontrollen
|
24.
|
Bei der Durchführung des Assays sind die Lösungsmittelkontrolle und die sonstigen Kontrollen (d. h. die Kontrollen mit dem Referenzöstrogen, dem schwachen Binder und dem Nicht-Binder) gleichzeitig zu prüfen. Bei jedem Prüflauf sollten auf jeweils einer Platte vollständige Konzentrationskurven des Referenzöstrogens und der Kontrollen (d. h. schwache Binder und Nicht-Binder) verwendet werden. Alle anderen Platten sollten Folgendes enthalten: (i) eine hohe Konzentration (maximale Verdrängung, d. h. etwa die Konzentration, bei der der radioaktiv markierte Ligand vollständig verdrängt wird) und eine mittlere Konzentration (etwa IC50) sowohl von E2 als auch des schwachen Binders jeweils dreifach, (ii) eine Lösungsmittelkontrolle und einen nicht spezifischen Binder, ebenfalls jeweils dreifach. Die Verfahren für die Zubereitung des Assay-Puffers, des [3H]-17β-Estradiols, des hrERα und der Prüfchemikalienlösungen werden im CERI-Protokoll eingehend beschrieben (2).
|
Lösungsmittelkontrolle
|
25.
|
Die Lösungsmittelkontrolle bestätigt, dass das Lösungsmittel nicht mit dem Prüfsystem reagiert und die Gesamtbindung (TB) misst. Bevorzugtes Lösungsmittel ist DMSO. Wenn die höchste Konzentration der Prüfchemikalie in DMSO nicht löslich ist, kann alternativ Ethanol verwendet werden. Die DMSO-Konzentration in den endgültigen Assay-Wells sollte 2,05 % betragen und kann auf bis zu 2,5 % erhöht werden, wenn sich die Prüfchemikalie nicht löst. DMSO-Konzentrationen über 2,5 % sollten nicht verwendet werden, da der Assay durch höhere Lösungsmittelkonzentrationen beeinträchtigt werden kann. Bei Prüfchemikalien, die in DMSO nicht löslich sind, sich aber in Ethanol lösen, kann der Assay ohne Beeinträchtigungen mit Höchstkonzentrationen von bis zu 2 % Ethanol durchgeführt werden.
|
Pufferkontrolle
|
26.
|
Die Pufferkontrolle (PK) darf weder Lösungsmittel noch die Prüfchemikalie, jedoch alle sonstigen im Assay verwendeten Elemente enthalten. Die Ergebnisse der Pufferkontrolle werden mit der Lösungsmittelkontrolle verglichen, um sicherzustellen, dass das verwendete Lösungsmittel das Assay-System nicht beeinträchtigt.
|
Starker Binder (Referenzöstrogen)
|
27.
|
17β-Estradiol (CAS 50-28-2) bindet sich als endogener Ligand mit hoher Affinität an den ER Untertyp alpha. Für jeden hrERα-Assay zur Ermittlung konkurrierender Bindungen wird eine Standardkurve mit nicht markiertem 17β-Estradiol erstellt, damit die Variabilität bei Durchführung des Assays in einem Labor im Laufe der Zeit beurteilt werden kann. In DMSO und im Assay-Puffer werden acht Lösungen mit nicht markiertem 17β-Estradiol mit den folgenden Endkonzentrationen in den Assay-Wells für die Erstellung der Standardkurve hergestellt: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10 und 10–11 M. Die höchste Konzentration mit nicht markiertem 17β-Estradiol (1 μM) sollte als Indikator für nicht spezifische Bindungen verwendet werden. In Tabelle 4 wird diese Konzentration als „NSB“ bezeichnet, obwohl sie Teil der Standardkurve ist.
|
Schwacher Binder
|
28.
|
Um die Empfindlichkeit der einzelnen Versuche nachzuweisen und um die Variabilität bei der Durchführung des Assays im Laufe der Zeit beurteilen zu können, sollte ein schwacher Binder (Norethynodrel (CAS68-23-5) oder Norethindron (CAS 68-22-4)) einbezogen werden. In DMSO und im Assay-Puffer werden acht Lösungen des schwachen Binders mit den folgenden Endkonzentrationen hergestellt: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 und 10–9 M.
|
Nicht-Binder
|
29.
|
Als Negativkontrolle (Nicht-Binder) wird Octyltriethoxysilan (OTES, CAS 2943-75-1) verwendet. Die Negativkontrolle gewährleistet, dass mit dem Assay unter den gegebenen Bedingungen erkannt wird, wenn Prüfchemikalien sich nicht an den hrERα binden. In DMSO und im Assay-Puffer werden acht Lösungen des Nicht-Binders mit den folgenden Endkonzentrationen hergestellt: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9 und 10–10 M. Als alternativer Nicht-Binder kann Di-n-butylphthalat (DBP, CAS 84-72-2) verwendet werden, das allerdings nur bei Konzentrationen bis zu höchstens 10–4 M zu prüfen ist. Die maximale Löslichkeit von DBP wurde im Assay bei einer Konzentration von 10–4 M festgestellt.
|
hrERα-Konzentration
|
30.
|
Für die Versuche ist die Menge des Rezeptors zu verwenden, bei der sich eine spezifische Bindung von 40 ± 10 % ergibt (Nummern 12 und 13 in Anlage 3). Die hrERα-Lösung ist unmittelbar vor der Verwendung durch Verdünnung des funktionalen hrERα in eiskaltem Assay-Puffer herzustellen.
|
[3H]-17β-Estradiol
|
31.
|
In den Assay-Wells ist [3H]-17β-Estradiol in einer Endkonzentration von 0,5 nM zu verwenden.
|
Prüfchemikalien
|
32.
|
Zunächst muss anhand einer Löslichkeitsprüfung die Löslichkeitsgrenze der einzelnen Prüfchemikalien ermittelt und ein geeigneter Konzentrationsbereich für die Durchführung des Prüfprotokolls bestimmt werden. Die Löslichkeitsgrenze der einzelnen Prüfchemikalien wird zunächst im Lösungsmittel bestimmt und anschließend unter den Bedingungen des Assays bestätigt. Mit dem Assay sind Endkonzentrationen höchstens bis zu 1 mM zu prüfen. Der Vorversuch zur Dosisfindung wird mit einer Lösungsmittelkontrolle und mindestens 8 logarithmischen seriellen Verdünnungen beginnend mit der maximal akzeptierbaren Konzentration (z. B. 1 mM oder weniger, je nach Löslichkeitsgrenze) durchgeführt; dabei ist das Auftreten einer Trübung oder eines Niederschlags zu protokollieren (Nummer 35 in Anlage 3). Nachdem der Konzentrationsbereich für die Prüfungen ermittelt wurde, sollte eine Prüfchemikalie mit 8 im Vorversuch ermittelten logarithmischen Konzentrationen geprüft werden. Die im zweiten und im dritten Versuch geprüften Konzentrationen sind ggf. so anzupassen, dass die Konzentrations-Reaktionskurve ggf. besser charakterisiert wird.
|
|
33.
|
Verdünnungen der Prüfchemikalien werden mit dem jeweils geeigneten Lösungsmittel hergestellt (Nummer 25 in Anlage 3). Wenn die höchste Konzentration der Prüfchemikalie weder in DMSO noch in Ethanol löslich ist und die Zugabe von weiterem Lösungsmittel dazu führen würde, dass die Lösungsmittelkonzentration im letzten Röhrchen die Akzeptanzgrenze überschreiten würde, kann die nächstgeringere Konzentration als höchste Konzentration angenommen werden. In diesem Fall kann am unteren Ende der Konzentrationsreihe eine weitere Konzentration eingefügt werden. Die übrigen Konzentrationen der Reihe bleiben unverändert.
|
|
34.
|
Die Lösungen mit der Prüfchemikalie werden beim Einbringen in die Assay-Wells sorgfältig beobachtet, da die Prüfchemikalien nach dem Einbringen in die Assay-Wells einen Niederschlag bilden können. Die Daten aller Wells mit einem Niederschlag werden bei der Kurvenanpassung nicht berücksichtigt. Der Grund für den Ausschluss der betreffenden Wells ist zu vermerken.
|
|
35.
|
Wenn bereits Informationen aus anderen Quellen mit einem log (IC50) für eine Prüfchemikalie verfügbar sind, können geometrische Verdünnungsstufen enger um den erwarteten log (IC50) (in 0,5 log-Einheiten) angemessen sein. Am Ende sollte ein ausreichender Abstand der Konzentrationen auf beiden Seiten von log (IC50) einschließlich der höchsten und der niedrigsten Konzentration erreicht sein, damit die Bindungskurve angemessen beschrieben werden kann.
|
Vorbereitung der Assay-Platte
|
36.
|
Mit Etiketten versehene Mikrotiterplatten werden für sechs Inkubationen für die Lösungsmittelkontrolle, die höchste Konzentration des Referenzöstrogens (E2) (die auch als Indikator für die nicht spezifische Bindung (NSB) dient), die Pufferkontrolle, drei Inkubationen für jede der acht Konzentrationen der Nicht-Binder-Kontrolle (Octyltriethoxysilan), die 7 geringeren Konzentrationen des Referenzöstrogens (E2), die 8 Konzentrationen des schwachen Binders und die 8 Konzentrationen der einzelnen Prüfchemikalien (TC) vorbereitet. Ein Beispiel für die Gestaltung des Plattendiagramms für die vollständigen Konzentrationskurven des Referenzöstrogens und die Kontrollen ist Tabelle 4 zu entnehmen. Für die Prüfchemikalie werden weitere Mikrotiterplatten verwendet; diese Platten sollten Plattenkontrollen, d. h. (i) eine hohe (maximale Verdrängung) und eine mittlere Konzentration (etwa die IC50-Konzentration) von E2 sowie des schwachen Binders (dreifach) und (ii) eine Lösungsmittelkontrolle (als Gesamtbindung) und eine nicht spezifische Bindung (jeweils sechsfach), umfassen (Tabelle 5). Ein Beispiel für die Anordnung auf einer Mikrotiterplatte für einen Assay zur Untersuchung konkurrierender Bindungen mit drei unbekannten Prüfchemikalien ist Anlage 3.3 zu entnehmen. Die auf dem Arbeitsblatt und in den Tabellen 4 und 5 genannten Konzentrationen sind die Endkonzentrationen in den jeweiligen Assay-Wells. Die maximale E2-Konzentration beträgt 1×10–7 M. Für den schwachen Binder ist die höchste Konzentration zu verwenden, die auch für den schwachen Binder auf Platte 1 verwendet wurde. Die IC50-Konzentration wird vom Labor anhand der Datenbank mit historischen Kontrollen des Labors bestimmt. Dieser Wert sollte sich in etwa mit dem in der Validierungsstudie ermittelten Wert decken (siehe Tabelle 1).
|
Tabelle 4
Anordnung der Mikrotiterplatte für einen Assay zur Untersuchung konkurrierender Bindungen
(98)
(99)
, vollständige Konzentrationskurven für das Referenzöstrogen und die Kontrollen (Platte 1).
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Pufferkontrolle und Positivkontrolle (E2)
|
Schwach positiv (Norethynodrel)
|
Negativkontrolle (OTES)
|
TB und NSB
|
|
A
|
Blindkontrolle (*14)
|
1×10–9 M
|
1×10–10 M
|
TB (Lösungsmittelkontrolle) (2,05 % DMSO)
|
|
B
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
C
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
E
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Pufferkontrolle
|
|
F
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
G
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Blindkontrolle (für warme Wells) (*15)
|
|
H
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
Tabelle 5
Anordnung der Mikrotiterplatte für einen Assay zur Untersuchung konkurrierender Bindungen, weitre Platten für Prüfchemikalien (TC) und Plattenkontrollen.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Prüfchemikalie 1 (TC-1)
|
Prüfchemikalie 2 (TC-2)
|
Prüfchemikalie 3 (TC-3)
|
Kontrollen
|
|
A
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10
-7
M)
|
|
B
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10–4,5)
|
|
D
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (10-6 M E2)
|
|
F
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
G
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
TB (Lösungsmittelkontrolle)
|
|
H
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
Bei diesem Beispiel wird Norethinodrel (NE) als schwacher Binder verwendet.
|
Abschluss des Assays zur Ermittlung konkurrierender Bindungen
|
37.
|
Mit Ausnahme der Wells zur Ermittlung der Gesamtbindung und zur Prüfung der Blindkontrollen (für warme Wells) (siehe Tabelle 6) sollten in jedes Well 50 μl des Assay-Puffer:s gegeben und mit 10 μl der Lösungsmittelkontrolle, des Referenzöstrogens (E2), des schwachen Binders, des Nicht-Binders und der Prüfchemikalien bzw. mit 10 μl einer 5-nM-[3H]-17β-Estradiollösung gemischt werden. Anschließend werden 30 μl der eiskalten Rezeptorlösung zu den Platten gegeben und vorsichtig gemischt. Als letztes Reagens ist die hrERα-Lösung hinzuzugeben. Die Assay-Mikrotiterplatten sollten 2 Stunden bei Raumtemperatur (22-28 °C) inkubiert werden.
Tabelle 6
Volumina der Elemente des hrER-Assays zur Ermittlung konkurrierender Bindungen auf den Mikrotiterplatten
|
Vorbereitungsschritte
|
Wells mit Ausnahme der TB-Wells
|
TB-Wells
|
Blindkontrolle (für warme Wells)
|
|
Volumen der Bestandteile für die genannten Reaktions-Wells und Reihenfolge der Zugabe des …
|
Assay-Puffers bei Raumtemperatur
|
50 μl
|
60 μl
|
90 μl
|
|
nicht markierten E2, des schwachen Binders, des Nicht-Binders, des Lösungsmittels und der Prüfchemikalien (*16)
|
10 μl
|
–
|
–
|
|
[3H]-17β-Estradiols zur Herstellung einer Endkonzentration von 0,5 nM (d. h. 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
der ermittelten hrERα-Konzentration (Nummern 12 und 13)
|
30 μl
|
30 μl
|
–
|
|
Gesamtvolumen in den einzelnen Assay-Wells
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Nach der Trennung des an den hrERα gebundenen [3H]-17β-Estradiols von dem freien [3H]-17β-Estradiol durch Zugabe von 100 μl kalter DCC-Suspension zu jedem einzelnen Well wie in den Nummern 21-23 in Anlage 3 für den Assay zur Ermittlung der Sättigungsbindung beschrieben, wird anschließend die Bindung von [3H]-17β-Estradiol an den hrERα quantifiziert.
|
|
39.
|
Die Wells G10-12 und H10-12 (in Tabelle 4 als Blindkontrollen (für warme Wells) gekennzeichnet) geben Aufschluss über die Zerfallsereignisse pro Minute (DPM) des markierten [3H]-Estradiols in einer Aliquote von 10 μl. Die Aliquote von 10 μl wird unmittelbar in die Szintillationsflüssigkeit gegeben.
|
Akzeptanzkriterien
Assay zur Ermittlung der Sättigungsbindung
|
40.
|
Die spezifische Bindungskurve sollte mit zunehmenden Konzentrationen des [3H]-17β-Estradiols (soweit verwendet) ein Plateau erreichen. Dieses Plateau zeigt, dass der hrERα mit dem Liganden gesättigt wurde.
|
|
41.
|
Die spezifische Bindung bei 0,5 nM [H]-17β-Estradiol sollte im akzeptablen Bereich von 30-50 % des Durchschnitts der insgesamt gemessenen Radioaktivität liegen, um die sich die Radioaktivität in allen Prüfläufen erhöht hat. Gelegentliche geringfügige Über- oder Unterschreitungen dieses Bereichs sind akzeptabel. Wenn Prüfläufe aber durchgängig außerhalb dieses Bereichs liegen bzw. wenn ein bestimmter Prüflauf erheblich außerhalb dieses Bereichs liegt, sollte die Proteinkonzentration angepasst und der Assay zur Ermittlung der Sättigungsbindung wiederholt werden.
|
|
42.
|
Aus den Daten sollte sich ein lineares Scatchard-Diagramm ergeben.
|
|
43.
|
Die nicht spezifische Bindung sollte nicht überhöht sein. In der Regel sollte der Wert der nicht spezifischen Bindung weniger als 35 % der Gesamtbindung betragen. Diese Grenze kann jedoch in Einzelfällen überschritten werden, wenn bei der niedrigsten Konzentration des geprüften 17β-Estradiols sehr wenige Zerfallsereignisse pro Minute gemessen werden.
|
Assay zur Ermittlung konkurrierender Bindungen
|
44.
|
Zunehmende Konzentrationen nicht markierten 17β-Estradiols sollten [3H]-17β-Estradiol aus dem Rezeptor verdrängen; die Verdrängung sollte im Einklang mit einer konkurrierenden Bindung an einer einzigen Bindungsstelle stehen.
|
|
45.
|
Der IC50-Wert des Referenzöstrogens (17β-Estradiol) sollte in etwa mit der molaren Konzentration von [3H]-17β-Estradiol zuzüglich der im Assay zur Ermittlung der Sättigungsbindung berechneten Dissoziationskonstante (Kd) übereinstimmen.
|
|
46.
|
Die spezifische Gesamtbindung sollte einheitlich im akzeptablen Bereich von 40 ± 10 % liegen, wenn die gemessene durchschnittliche Konzentration, um die sich die Radioaktivität in jedem einzelnen Well erhöht hat, im Verlauf der Prüfläufe bei 0,5 nM lag. Gelegentliche geringfügige Über- oder Unterschreitungen dieses Bereichs sind akzeptabel. Wenn Prüfläufe aber durchgängig außerhalb dieses Bereichs liegen bzw. wenn ein bestimmter Prüflauf erheblich außerhalb dieses Bereichs liegt, sollte die Proteinkonzentration angepasst werden.
|
|
47.
|
Das Lösungsmittel darf keinen Einfluss auf die Empfindlichkeit oder die Reproduzierbarkeit des Assays haben. Die Ergebnisse der Lösungsmittelkontrolle werden mit der Pufferkontrolle verglichen, um sicherzustellen, dass das verwendete Lösungsmittel das Assay-System nicht beeinträchtigt. Die Ergebnisse für die Gesamtbindung (TB) und die Pufferkontrolle sollten vergleichbar sein, wenn das Lösungsmittel den Assay nicht beeinflusst.
|
|
48.
|
Bei Prüfungen mit bis zu 10–3 M (OTES) bzw. 10–4 M (DBP) darf der Nicht-Binder nicht mehr als 25 % des [3H]-17β-Estradiols vom hrERα verdrängen.
|
|
49.
|
Aufgrund von Daten der Studie zur Validierung des CERI-Bindungsassays mit hrER wurden Leistungskriterien für das Referenzöstrogen und zwei schwache Binder (z. B. Norethynodrel und Norethindron) entwickelt (Anhang N in (2)). Für den Mittelwert ± SD (n) aller Kontrolldurchläufe in den vier an der Validierungsstudie beteiligten Labors werden 95-%-Konfidenzintervalle angegeben. 95-%-Konfidenzintervalle wurden für die Parameter zur Kurvenanpassung (Höchstwert, Mindestwert, Hillslope und log IC50) für das Referenzöstrogen und für schwache Binder sowie für log10 RBA der schwachen Binder bezogen auf das Referenzöstrogen berechnet. Tabelle 1 enthält die erwarteten Spannen der Kurvenanpassungsparameter, die als Leistungskriterien verwendet werden können. In der Praxis kann die IC50-Spanne je nach der im Versuch ermittelten Kd der Rezeptorzubereitung und der Ligandenkonzentration für den Assay leicht variieren.
|
|
50.
|
In Anbetracht der großen Anzahl potenzieller Prüfchemikalien und der Unterschiede hinsichtlich der potenziellen Affinitäten und Ergebnisse (vollständige Kurve, Teilkurve, keine Kurvenanpassung usw.) wurden keine Leistungskriterien für die Kurvenanpassungsparameter der Prüfchemikalien entwickelt. Die Ergebnisse der einzelnen Prüfläufe sowie die Ergebnisse für die einzelnen Prüfchemikalien sind jedoch einer fachlich qualifizierten Beurteilung zu unterziehen. Um den Höchstwert der Kurve der konkurrierenden Bindungen (z. B. eine Bindungsquote von 90-100 %) eindeutig zu ermitteln, sollte ein ausreichendes Spektrum an Konzentrationen der Prüfchemikalie berücksichtigt werden. Die Variabilität zwischen Wiederholungen bei den einzelnen Konzentrationen der Prüfchemikalie sowie zwischen den drei nicht gleichzeitigen Prüfläufen sollte angemessen und wissenschaftlich vertretbar sein. Die Kontrollen der einzelnen Prüfläufe einer Prüfchemikalie sollten sich im Bereich der Messungen der für diesen CERI-Assay angegebenen Leistungen bewegen und mit historischen Kontrolldaten der jeweiligen Labors übereinstimmen.
|
ANALYSE DER DATEN
Assay zur Ermittlung der Sättigungsbindung
|
51.
|
Gemessen werden sowohl die Gesamtbindung als auch die nicht spezifische Bindung. Aufgrund dieser Werte wird die spezifische Bindung bei zunehmenden Konzentrationen von [3H]-17β-Estradiol unter Gleichgewichtsbedingungen ermittelt, indem die nicht spezifische Bindung von der Gesamtbindung abgezogen wird. In der grafischen Darstellung der spezifischen Bindung im Vergleich zur Konzentration von [3H]-17β-Estradiol sollte sich bei der maximalen spezifischen Konzentration ein Plateau als Bestätigung für die erfolgte Sättigung des hrERα mit [3H]-17β-Estradiol ergeben. Außerdem sollte in der Analyse der Daten die Bindung des [3H]-17β-Estradiols an eine einzelne Bindungsstelle mit hoher Affinität dokumentiert werden. Die nicht spezifische Bindung, die Gesamtbindung und die spezifische Bindung sind auf einer Sättigungskurve darzustellen. Darüber hinaus sollte eine nicht lineare Regressionsanalyse (z. B. mit BioSoft; McPherson, 1985; Motulsky, 1995) vorgenommen werden. Die ermittelten Daten sind in einem endgültigen Scatchard-Diagramm darzustellen.
|
|
52.
|
Bei der Analyse sind Bmax und Kd ausschließlich anhand der Daten zur Gesamtbindung zu ermitteln. Dabei ist von einem linearen Verlauf der nicht spezifischen Bindung auszugehen. Ansonsten ist eine Begründung für eine anderweitige Vorgehensweise anzugeben. Ferner sollte eine robuste Regression zur Ermittlung der Best-Fit-Werte vorgenommen werden, sofern nicht eine Begründung für ein anderweitiges Vorgehen angegeben wird. Die gewählte Methode für die Durchführung der robusten Regression ist anzugeben. Bei der Ermittlung von Bmax und Kd aufgrund von Daten zur Sättigungsbindung ist immer eine Korrektur aufgrund des Ligandenabbaus vorzunehmen (z. B. nach der Methode von Swillens 1995).
|
Assay zur Ermittlung konkurrierender Bindungen
|
53.
|
Die Kurve zur Darstellung konkurrierender Bindungen wird als spezifische Bindung von [3H]-17β-Estradiol im Vergleich zur Konzentration (log10-Stufen) des Bindungskonkurrenten dargestellt. Die Konzentration der Prüfchemikalie, die die maximale spezifische Bindung von [3H]-17β-Estradiol um 50 % hemmt, wird als IC50-Wert bezeichnet.
|
|
54.
|
Schätzungen der log(IC50)-Werte der positiven Kontrollen (z. B. Referenzöstrogen und ein schwacher Binder) sollten mit einer geeigneten Software zur nicht linearen Kurvenanpassung für eine Hill-Gleichung mit 4 Parametern (z. B. BioSoft; McPherson, 1985; Motulsky, 1995) vorgenommen werden. Die Höchstwerte, die Mindestwerte, die Hillslope-Werte und die log(IC50)-Werte sollten bei der Anpassung dieser Kurven allgemein nicht beschränkt werden. Zur Ermittlung der Best-Fit-Werte sollte eine robuste Regression vorgenommen werden, sofern nicht eine Begründung für ein anderweitiges Vorgehen angegeben wird. Eine Korrektur aufgrund des Ligandenabbaus sollte nicht vorgenommen werden. Nach der Ausgangsanalyse sollte jede einzelne Bindungskurve nochmals geprüft werden, um eine angemessene Anpassung an das Modell sicherzustellen. Die relative Bindungsaffinität (RBA) des schwachen Binders sollte als Prozentanteil von log (IC50) für den schwachen Binder bezogen auf log (IC50) für 17β-Estradiol berechnet werden. Die Ergebnisse der Positivkontrollen und der Nicht-Binder-Kontrolle sind anhand der Kriterien für die Leistungsfähigkeit des Assays (Nummern 44-49 in Anlage 3) zu bewerten.
|
|
55.
|
Die Daten aller Prüfchemikalien werden schrittweise analysiert, um eine angemessene Vorgehensweise und eine korrekte Einstufung der einzelnen Kurven zum konkurrierenden Bindungsverhalten sicherzustellen. Für jeden Prüflauf einer Prüfchemikalie sollte zunächst eine standardisierte Datenanalyse nach dem Verfahren vorgenommen werden, das auch zur Analyse der Kontrollen mit dem Referenzöstrogen und dem schwachen Binder durchgeführt wird (Nummer 54 in Anlage 3). Nach dieser Analyse sollten die Parameter zur Kurvenanpassung fachlich geprüft werden; außerdem sollte geprüft werden, in welchem Umfang die Daten mit der erzeugten Kurve der konkurrierenden Bindungen übereinstimmen. Bei dieser fachlichen Prüfung sind die Feststellung eines konzentrationsabhängigen Rückgangs der prozentualen spezifischen Bindung von [3H]-17β-Estradiol, eine geringe Variabilität der technischen Wiederholungen bei den einzelnen Prüfchemikalienkonzentrationen und die Konsistenz der Anpassungsparameter bei den drei Prüfläufen gute Indikatoren für die ordnungsgemäße Durchführung des Assays und der Datenanalysen.
|
Datenauswertung
|
56.
|
Sofern alle Akzeptanzkriterien erfüllt sind, wird eine Prüfchemikalie als Binder für den hrERα betrachtet, wenn eine Bindungskurve angepasst werden kann und der niedrigste Punkt auf der Reaktionskurve im Datenbereich bei weniger als 50 % liegt (Abbildung 1).
|
|
57.
|
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als Nicht-Binder für den hrERα, wenn die folgenden Bedingungen gegeben sind:
|
—
|
Eine Bindungskurve kann angepasst werden, und der niedrigste Punkt auf der angepassten Reaktionskurve innerhalb des Datenbereichs liegt über 75 %, oder
|
|
—
|
es kann keine Anpassung der Bindungskurve vorgenommen werden, und der niedrigste nicht korrigierte Durchschnittswert der prozentualen Bindung der Konzentrationsgruppen im Datenbereich liegt über 75 %.
|
|
|
58.
|
Prüfchemikalien werden als nicht eindeutig betrachtet, wenn keine der genannten Bedingungen erfüllt ist (z. B. wenn der niedrigste Punkt auf der angepassten Reaktionskurve zwischen 76 und 51 % liegt).
|
Tabelle 7
Kriterien für eine Einstufung nach der Kurve des konkurrierenden Bindungsverhaltens bei einer Prüfchemikalie
|
Einstufung
|
Kriterien
|
|
Bindera
|
Eine Bindungskurve kann angepasst werden.
Der niedrigste Punkt auf der Reaktionskurve im Datenbereich liegt bei weniger als 50 %.
|
|
Nicht-Binder b
|
Wenn eine Bindungskurve angepasst werden kann:
Der niedrigste Punkt auf der angepassten Reaktionskurve im Datenbereich liegt über 75 %.
Wenn keine Bindungskurve angepasst werden kann:
Der niedrigste nicht korrigierte Durchschnittswert der prozentualen Bindung der Konzentrationsgruppen im Datenbereich liegt über 75 %.
|
|
Nicht eindeutigc
|
Alle analysierbaren Prüfläufe, bei denen die Prüfchemikalie weder als Binder noch als Nicht-Binder eingestuft wird (beispielsweise wenn der niedrigste Punkt auf der angepassten Reaktionskurve zwischen 76 und 51 % liegt).
|
Abbildung 1
Beispiele für die Einstufung von Prüfchemikalien anhand der Kurve der konkurrierenden Bindungen

|
59.
|
Mehrere in einem Labor mit einer Prüfchemikalie durchgeführte Prüfläufe werden unter Zuweisung von nummerischen Werten zu den einzelnen Prüfläufen und unter Ermittlung des Durchschnitts der Prüfläufe durchgeführt (siehe Tabelle 8). Die Ergebnisse der kombinierten Prüfläufe in den einzelnen Labors werden mit der erwarteten Einstufung der einzelnen Prüfchemikalien verglichen.
|
Tabelle 8
Methode zur Einstufung von Prüfchemikalien aufgrund mehrerer Prüfläufe in einem Labor
|
Zuweisung eines Wertes zu einem Prüflauf:
|
|
Einstufung
|
Numerischer Wert
|
|
Binder
|
2
|
|
Nicht eindeutig
|
1
|
|
Nicht-Binder
|
0
|
|
Einstufung des durchschnittlichen numerischen Werts für mehrere Prüfläufe:
|
|
Einstufung
|
Numerischer Wert
|
|
Binder
|
Durchschnitt ≥ 1,5
|
|
Nicht eindeutig
|
0,5 ≤ Durchschnitt < 1,5
|
|
Nicht-Binder
|
Durchschnitt ≥ 0,5
|
PRÜFBERICHT
|
60.
|
Siehe Nummer 24 im Abschnitt „ELEMENTE DES hrER-BINDUNGSASSAYS“ für diese Prüfmethode.
|
Anlage 3.1
BEGRIFFSBESTIMMUNGEN
[3H]E2
: 17β-Estradiol mit Tritium radioaktiv markiert.
Assay-Puffer
: 10 mM Tris-HCl, pH 7,4, mit 1 mM EDTA, 1mM EGTA, 1 mM NaVO3, 10 % Glycerin, 0,2 mM Leupeptin, 1 mM Dithiothreitol und 10 mg/ml Rinderseriumalbumin.
DCC
: Dextranbeschichtete Aktivkohle.
E2
: Nicht markiertes 17β-Estradiol (inert).
hrERα
: Humaner rekombinanter Östrogenrezeptor alpha (Lingandenbindungs-Domäne).
Prüflauf
: Eine vollständige Reihe gleichzeitig zu prüfender Mikrotiterplatten-Wells, denen alle Informationen zu entnehmen sind, die zur Charakterisierung der Bindung einer Prüfchemikalie an den hrERα benötigt werden (d. h. insgesamt zum Well hinzugegebenes [3H]-17β-Estradiol, maximale Bindung von [3H]-17β-Estradiol an den hrERα, nicht spezifische Bindung und Gesamtbindung bei verschiedenen Konzentrationen der Prüfchemikalie). Ein Prüflauf kann bereits aus einem einzigen Well (d. h. einer einzigen Wiederholung) pro Konzentration bestehen. Da nach diesem Protokoll jedoch eine dreifache Untersuchung vorzunehmen ist, umfasst ein Prüflauf drei Wells pro Konzentration. Außerdem sind nach diesem Protokoll drei unabhängige (d. h. nicht gleichzeitige) Prüfläufe pro Chemikalie vorzunehmen.
Wiederholung (Replikat)
: Eines von mehreren Wells mit denselben Inhalten in denselben Konzentrationen, die in einem einzelnen Prüflauf gleichzeitig untersucht werden. Bei diesem Protokoll wird jede Konzentration der Prüfchemikalie dreimal geprüft, d. h. je Konzentration der Prüfchemikalie werden drei Wiederholungen gleichzeitig untersucht.
Anlage 3.2
ANORDNUNG DER WELLS BEIM ASSAY ZUR ERMITTLUNG KONKURRIERENDER BINDUNGEN
|
Platte
|
Position
|
Replikat
|
Well-Typ
|
Well-Code
|
Konzentrationscode
|
Ausgangskonzentration des Bindungskonkurrenten (M)
|
hrER-Vorratslösung (μl)
|
Puffer-Vol. (μl)
|
Tracer-Volumen (warmes E2) (μl)
|
Volumen der Verdünnungsplatte (μl)
|
Endvolumen (μl)
|
Endkonzentration des Bindungskonkurrenten (M)
|
|
S
|
A1
|
1
|
Blindkontrolle
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Blindkontrolle
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Blindkontrolle
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
kaltes E2
|
S
|
S1
|
1,0E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B2
|
2
|
kaltes E2
|
S
|
S1
|
1,0E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B3
|
3
|
kaltes E2
|
S
|
S1
|
1,0E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
C1
|
1
|
kaltes E2
|
S
|
S2
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C2
|
2
|
kaltes E2
|
S
|
S2
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C3
|
3
|
kaltes E2
|
S
|
S2
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
D1
|
1
|
kaltes E2
|
S
|
S3
|
3,16E-9
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D2
|
2
|
kaltes E2
|
S
|
S3
|
3,16E-9
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D3
|
3
|
kaltes E2
|
S
|
S3
|
3,16E-9
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
E1
|
1
|
kaltes E2
|
S
|
S4
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
E2
|
2
|
kaltes E2
|
S
|
S4
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
E3
|
3
|
kaltes E2
|
S
|
S4
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
F1
|
1
|
kaltes E2
|
S
|
S5
|
3,16E-8
|
30
|
50
|
10
|
10
|
100
|
3,2E-9
|
|
S
|
F2
|
2
|
kaltes E2
|
S
|
S5
|
3,16E-8
|
30
|
50
|
10
|
10
|
100
|
3,2E-9
|
|
S
|
F3
|
3
|
kaltes E2
|
S
|
S5
|
3,16E-8
|
30
|
50
|
10
|
10
|
100
|
3,2E-9
|
|
S
|
G1
|
1
|
kaltes E2
|
S
|
S6
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
G2
|
2
|
kaltes E2
|
S
|
S6
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
G3
|
3
|
kaltes E2
|
S
|
S6
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
H1
|
1
|
kaltes E2
|
S
|
S7
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
H2
|
2
|
kaltes E2
|
S
|
S7
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
H3
|
3
|
kaltes E2
|
S
|
S7
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
A4
|
1
|
Norethynodrel
|
NE
|
WP1
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
A5
|
2
|
Norethynodrel
|
NE
|
WP1
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
A6
|
3
|
Norethynodrel
|
NE
|
WP1
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
B4
|
1
|
Norethynodrel
|
NE
|
WP2
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
B5
|
2
|
Norethynodrel
|
NE
|
WP2
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
B6
|
3
|
Norethynodrel
|
NE
|
WP2
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
C4
|
1
|
Norethynodrel
|
NE
|
WP3
|
3,16E-7
|
30
|
50
|
10
|
10
|
100
|
3,2E-8
|
|
S
|
C5
|
2
|
Norethynodrel
|
NE
|
WP3
|
3,16E-7
|
30
|
50
|
10
|
10
|
100
|
3,2E-8
|
|
S
|
C6
|
3
|
Norethynodrel
|
NE
|
WP3
|
3,16E-7
|
30
|
50
|
10
|
10
|
100
|
3,2E-8
|
|
S
|
D4
|
1
|
Norethynodrel
|
NE
|
WP4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
D5
|
2
|
Norethynodrel
|
NE
|
WP4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
D6
|
3
|
Norethynodrel
|
NE
|
WP4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
E4
|
1
|
Norethynodrel
|
NE
|
WP5
|
3,16E-6
|
30
|
50
|
10
|
10
|
100
|
3,2E-7
|
|
S
|
E5
|
2
|
Norethynodrel
|
NE
|
WP5
|
3,16E-6
|
30
|
50
|
10
|
10
|
100
|
3,2E-7
|
|
S
|
E6
|
3
|
Norethynodrel
|
NE
|
WP5
|
3,16E-6
|
30
|
50
|
10
|
10
|
100
|
3,2E-7
|
|
S
|
F4
|
1
|
Norethynodrel
|
NE
|
WP6
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
F5
|
2
|
Norethynodrel
|
NE
|
WP6
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
F6
|
3
|
Norethynodrel
|
NE
|
WP6
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
G4
|
1
|
Norethynodrel
|
NE
|
WP7
|
3,16E-5
|
30
|
50
|
10
|
10
|
100
|
3,2E--6
|
|
S
|
G5
|
2
|
Norethynodrel
|
NE
|
WP7
|
3,16E-5
|
30
|
50
|
10
|
10
|
100
|
3,2E--6
|
|
S
|
G6
|
3
|
Norethynodrel
|
NE
|
WP7
|
3,16E-5
|
30
|
50
|
10
|
10
|
100
|
3,2E--6
|
|
S
|
H4
|
1
|
Norethynodrel
|
NE
|
WP8
|
3,16E-4
|
30
|
50
|
10
|
10
|
100
|
3,2E--5
|
|
S
|
H5
|
2
|
Norethynodrel
|
NE
|
WP8
|
3,16E-4
|
30
|
50
|
10
|
10
|
100
|
3,2E--5
|
|
S
|
H6
|
3
|
Norethynodrel
|
NE
|
WP8
|
3,16E-4
|
30
|
50
|
10
|
10
|
100
|
3,2E--5
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
S
|
A10
|
1
|
Gesamtbindung
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
Gesamtbindung
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
Gesamtbindung
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
Gesamtbindung
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B11
|
5
|
Gesamtbindung
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
Gesamtbindung
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
kaltes E2 (hoch)
|
NSB
|
S1
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
C11
|
2
|
kaltes E2 (hoch)
|
NSB
|
S2
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
C12
|
3
|
kaltes E2 (hoch)
|
NSB
|
S3
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
D10
|
4
|
kaltes E2 (hoch)
|
NSB
|
S4
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
D11
|
5
|
kaltes E2 (hoch)
|
NSB
|
S5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
D12
|
6
|
kaltes E2 (hoch)
|
NSB
|
S6
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
S
|
E10
|
1
|
Pufferkontrolle
|
PK
|
PK1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
Pufferkontrolle
|
PK
|
PK2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
Pufferkontrolle
|
PK
|
PK3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
Pufferkontrolle
|
PK
|
PK4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
Pufferkontrolle
|
PK
|
PK5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F12
|
6
|
Pufferkontrolle
|
PK
|
PK6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
Blindkontrolle (für warme Wells)
|
Warm
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G11 (*17)
|
2
|
Blindkontrolle (für warme Wells)
|
Warm
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
Blindkontrolle (für warme Wells)
|
Warm
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
Blindkontrolle (für warme Wells)
|
Warm
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
Blindkontrolle (für warme Wells)
|
Warm
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12
|
6
|
Blindkontrolle (für warme Wells)
|
Warm
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Unbekannt 1
|
U1
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
Unbekannt 1
|
U1
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
Unbekannt 1
|
U1
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
Unbekannt 1
|
U1
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
B2
|
2
|
Unbekannt 1
|
U1
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
B3
|
3
|
Unbekannt 1
|
U1
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
C1
|
1
|
Unbekannt 1
|
U1
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
C2
|
2
|
Unbekannt 1
|
U1
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
C3
|
3
|
Unbekannt 1
|
U1
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
D1
|
1
|
Unbekannt 1
|
U1
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
D2
|
2
|
Unbekannt 1
|
U1
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
D3
|
3
|
Unbekannt 1
|
U1
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
E1
|
1
|
Unbekannt 1
|
U1
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
E2
|
2
|
Unbekannt 1
|
U1
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
E3
|
3
|
Unbekannt 1
|
U1
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
F1
|
1
|
Unbekannt 1
|
U1
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
F2
|
2
|
Unbekannt 1
|
U1
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
F3
|
3
|
Unbekannt 1
|
U1
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
G1
|
1
|
Unbekannt 1
|
U1
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
G2
|
2
|
Unbekannt 1
|
U1
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
G3
|
3
|
Unbekannt 1
|
U1
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
H1
|
1
|
Unbekannt 1
|
U1
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
H2
|
2
|
Unbekannt 1
|
U1
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
H3
|
3
|
Unbekannt 1
|
U1
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
A4
|
1
|
Unbekannt 2
|
U2
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
Unbekannt 2
|
U2
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
Unbekannt 2
|
U2
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
Unbekannt 2
|
U2
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
B5
|
2
|
Unbekannt 2
|
U2
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
B6
|
3
|
Unbekannt 2
|
U2
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
C4
|
1
|
Unbekannt 2
|
U2
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
C5
|
2
|
Unbekannt 2
|
U2
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
C6
|
3
|
Unbekannt 2
|
U2
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
D4
|
1
|
Unbekannt 2
|
U2
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
D5
|
2
|
Unbekannt 2
|
U2
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
D6
|
3
|
Unbekannt 2
|
U2
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
E4
|
1
|
Unbekannt 2
|
U2
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
E5
|
2
|
Unbekannt 2
|
U2
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
E6
|
3
|
Unbekannt 2
|
U2
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
F4
|
1
|
Unbekannt 2
|
U2
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
F5
|
2
|
Unbekannt 2
|
U2
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
F6
|
3
|
Unbekannt 2
|
U2
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
G4
|
1
|
Unbekannt 2
|
U2
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
G5
|
2
|
Unbekannt 2
|
U2
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
G6
|
3
|
Unbekannt 2
|
U2
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
H4
|
1
|
Unbekannt 2
|
U2
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
H5
|
2
|
Unbekannt 2
|
U2
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
H6
|
3
|
Unbekannt 2
|
U2
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
A7
|
1
|
Unbekannt 3
|
U3
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
Unbekannt 3
|
U3
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
Unbekannt 3
|
U3
|
1
|
1,0E-9
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
Unbekannt 3
|
U3
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
B8
|
2
|
Unbekannt 3
|
U3
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
B9
|
3
|
Unbekannt 3
|
U3
|
2
|
1,0E-8
|
30
|
50
|
10
|
10
|
100
|
1,0E-9
|
|
P1
|
C7
|
1
|
Unbekannt 3
|
U3
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
C8
|
2
|
Unbekannt 3
|
U3
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
C9
|
3
|
Unbekannt 3
|
U3
|
3
|
1,0E-7
|
30
|
50
|
10
|
10
|
100
|
1,0E-8
|
|
P1
|
D7
|
1
|
Unbekannt 3
|
U3
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
D8
|
2
|
Unbekannt 3
|
U3
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
D9
|
3
|
Unbekannt 3
|
U3
|
4
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
E7
|
1
|
Unbekannt 3
|
U3
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
E8
|
2
|
Unbekannt 3
|
U3
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
E9
|
3
|
Unbekannt 3
|
U3
|
5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
F7
|
1
|
Unbekannt 3
|
U3
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
F8
|
2
|
Unbekannt 3
|
U3
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
F9
|
3
|
Unbekannt 3
|
U3
|
6
|
1,0E-4
|
30
|
50
|
10
|
10
|
100
|
1,0E-5
|
|
P1
|
G7
|
1
|
Unbekannt 3
|
U3
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
G8
|
2
|
Unbekannt 3
|
U3
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
G9
|
3
|
Unbekannt 3
|
U3
|
7
|
1,0E-3
|
30
|
50
|
10
|
10
|
100
|
1,0E-4
|
|
P1
|
H7
|
1
|
Unbekannt 3
|
U3
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
H8
|
2
|
Unbekannt 3
|
U3
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
H9
|
3
|
Unbekannt 3
|
U3
|
8
|
1,0E-2
|
30
|
50
|
10
|
10
|
100
|
1,0E-3
|
|
P1
|
A10
|
1
|
Kontrolle E2 (max)
|
S
|
E2max1
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
A11
|
2
|
Kontrolle E2 (max)
|
S
|
E2max2
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
A12
|
3
|
Kontrolle E2 (max)
|
S
|
E2max3
|
1,0E-6
|
30
|
50
|
10
|
10
|
100
|
1,0E-7
|
|
P1
|
B10
|
1
|
Kontrolle E2 (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Kontrolle E2 (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Kontrolle E2 (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Kontrolle NE (max)
|
S
|
Nemax1
|
1,0E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,0E-4,5
|
|
P1
|
C11
|
2
|
Kontrolle NE (max)
|
S
|
Nemax2
|
1,0E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,0E-4,5
|
|
P1
|
C12
|
3
|
Kontrolle NE (max)
|
S
|
Nemax3
|
1,0E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,0E-4,5
|
|
P1
|
D10
|
1
|
Kontrolle NE (IC50)
|
S
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Kontrolle NE (IC50)
|
S
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Kontrolle NE (IC50)
|
S
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
kaltes E2 (hoch)
|
NSB
|
S1
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
E11
|
2
|
kaltes E2 (hoch)
|
NSB
|
S2
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
E12
|
3
|
kaltes E2 (hoch)
|
NSB
|
S3
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
F10
|
4
|
kaltes E2 (hoch)
|
NSB
|
S4
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
F11
|
5
|
kaltes E2 (hoch)
|
NSB
|
S5
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
F12
|
6
|
kaltes E2 (hoch)
|
NSB
|
S6
|
1,0E-5
|
30
|
50
|
10
|
10
|
100
|
1,0E-6
|
|
P1
|
G10
|
1
|
Gesamtbindung
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
Gesamtbindung
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
Gesamtbindung
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
Gesamtbindung
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
Gesamtbindung
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
Gesamtbindung
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
–
|
Anlage 4
HINWEISE ZUR ANALYSE DER DATEN AUS DEM hrER-ASSSAY ZUR ERMITTLUNG KONKURRIERENDER BINDUNGEN
|
1.
|
Mit dem hrERα-Assay zur Ermittlung konkurrierender Bindungen wird die Bindung von [3H]-17β-Estradiol einer einzelnen Konzentration bei zunehmenden Konzentrationen einer Prüfchemikalie gemessen. Die Kurve zur Darstellung konkurrierender Bindungen wird als spezifische Bindung von [3H]-17β-Estradiol im Vergleich zur Konzentration (log10-Stufen) des Bindungskonkurrenten dargestellt. Die Konzentration der Prüfchemikalie, die die maximale spezifische Bindung von [3H]-17β-Estradiol um 50 % hemmt, wird als IC50 bezeichnet.
|
Analyse der Daten zum Referenzöstrogen und zum schwachen Binder (1)
|
2.
|
Daten aus den Prüfläufen mit den Kontrollen werden für weitere Analysen umgewandelt (prozentuale [3H]-17β-Estradiol-spezifische Bindung und Log-Konzentration der Kontrollchemikalie). Schätzungen der log-(IC50)-Werte der positiven Kontrollen (z. B. Referenzöstrogen und ein schwacher Binder) sollten mit einer geeigneten Software zur nicht linearen Kurvenanpassung für eine Hill-Gleichung mit 4 Parametern (z. B. BioSoft; GraphPad Prism) vorgenommen werden (2). Die Höchstwerte, die Mindestwerte, die Hillslope-Werte und die log-(IC50)-Werte brauchen bei der Anpassung dieser Kurven allgemein nicht beschränkt zu werden. Zur Ermittlung der Best-Fit-Werte sollte eine robuste Regression vorgenommen werden, sofern nicht eine Begründung für ein anderweitiges Vorgehen angegeben wird. Die gewählte Methode für die Durchführung der robusten Regression ist anzugeben. Beim FW- und beim CERI-hrER-Assay waren Korrekturen für den Ligandenabbau nicht erforderlich; solche Korrekturen können aber ggf. in Betracht gezogen werden. Nach der Ausgangsanalyse sollte jede einzelne Bindungskurve nochmals geprüft werden, um eine angemessene Anpassung an das Modell sicherzustellen. Die relative Bindungsaffinität (RBA) des schwachen Binders kann als Prozentanteil von log (IC50) für den schwachen Binder bezogen auf log (IC50) für 17β-Estradiol berechnet werden. Die Ergebnisse der Positivkontrollen und der Nicht-Binder-Kontrolle sollten anhand von Parametern für die Leistungsfähigkeit des Assays sowie aufgrund von Akzeptanzkriterien bewertet werden. Im Zusammenhang mit dieser Prüfmethode (Nummer 20), in Anlage 2 (FW-Assay, Nummern 41-51) und in Anlage 3 (CERI-Assay, Nummern 41-51) wurden Beispiele für solche Akzeptanzkriterien genannt. Abbildung 1 enthält Beispiele aus 3 Prüfläufen mit dem Referenzöstrogen und dem schwachen Binder.
|
Abbildung 1
Beispiele für Kurven konkurrierender Bindungen für das Referenzöstrogen un die Kontrolle mit dem schwachen Binder

Analyse der Daten zu den Prüfchemikalien
|
3.
|
Die Daten aller Prüfchemikalien werden schrittweise analysiert, um eine angemessene Vorgehensweise und eine korrekte Einstufung der einzelnen Kurven zum konkurrierenden Bindungsverhalten sicherzustellen. Für jeden Prüflauf einer Prüfchemikalie sollte zunächst eine standardisierte Datenanalyse nach dem Verfahren vorgenommen werden, das auch zur Analyse der Kontrollen mit dem Referenzöstrogen und dem schwachen Binder durchgeführt wird. Nach dieser Analyse sollten die Parameter zur Kurvenanpassung fachlich geprüft werden; außerdem sollte geprüft werden, in welchem Umfang die Daten mit der erzeugten Kurve der konkurrierenden Bindungen übereinstimmen. Bei dieser fachlichen Prüfung sind die Feststellung eines konzentrationsabhängigen Rückgangs der prozentualen spezifischen Bindung von [3H]-17β-Estradiol, eine geringe Variabilität der technischen Wiederholungen bei den einzelnen Chemikalienkonzentrationen und die Konsistenz der Anpassungsparameter bei den drei Prüfläufen gute Indikatoren für die ordnungsgemäße Durchführung des Assays und der Datenanalysen. Die Ergebnisse der einzelnen Prüfläufe mit einer Prüfchemikalie sollten einer fachlichen Bewertung unterzogen werden, und die zur Einstufung der einzelnen Prüfchemikalien als Binder oder als Nicht-Binder verwendeten Daten sollten wissenschaftlich fundiert sein.
|
|
4.
|
Gelegentlich liegen möglicherweise Daten vor, die besondere Aufmerksamkeit erfordern, wenn angemessene Analysen und Auswertungen der Daten zu hrER-Bindungen sichergestellt werden sollen. In früheren Studien wurden Fälle festgestellt, in denen die Analyse und die Auswertung von Daten zur Bindung an konkurrierende Rezeptoren durch eine Zunahme der prozentualen spezifischen Bindung bei den höchsten Konzentrationen der betreffenden Prüfchemikalien erschwert werden (Abbildung 2). Dieses Problem ist hinlänglich bekannt und wurde bei der Verwendung von Protokollen für mehrere Assays zur Untersuchung konkurrierender Rezeptorbindungen deutlich (3). In diesen Fällen wird eine konzentrationsabhängige Reaktion bei niedrigeren Konzentrationen festgestellt; wenn sich die Konzentration der Prüfchemikalie jedoch der Löslichkeitsgrenze nähert, nimmt die Verdrängung von [3H]-17β-Estradiol nicht mehr weiter ab. In diesen Fällen deuten die Daten bei den höheren Konzentrationen darauf hin, dass die biologische Grenze des Assays erreicht ist. Häufig hängt dieses Phänomen damit zusammen, dass Chemikalien bei hohen Konzentrationen nicht mehr löslich sind und sich niederschlagen. Außerdem kann dies darauf zurückzuführen sein, dass die Kapazität der dextranbeschichteten Aktivkohle zur Bindung des ungebundenen radioaktiv markierten Liganden während des Trennverfahrens bei den höchsten Chemikalienkonzentrationen überschritten wird. Wenn die betreffenden Datenpunkte bei der Anpassung der Daten zu konkurrierenden Bindungen an eine Sigmoidkurve beibehalten werden, kann es zu Fehleinstufungen des Potenzials der jeweiligen Chemikalien für ER-Bindungen kommen (Abbildung 2). Um dies zu vermeiden, sieht das Protokoll des FW- und des CERI-Bindungsassays mit dem hrER die Möglichkeit vor, Datenpunkte aus den Analysen auszuschließen, bei denen der Mittelwert der Wiederholungen für die prozentuale [3H]-17β-Estradiol-spezifische Bindung um mindestens 10 % über dem Mittelwert bei einer niedrigeren Konzentration liegt. (Diese Regel wird allgemein als 10-%-Regel bezeichnet.) Bei jeder Kurve kann diese Regel nur einmal angewendet werden. Außerdem ist eine Anwendung nur dann möglich, wenn Daten für mindestens 6 weitere Konzentrationen verbleiben, damit die Kurve korrekt eingestuft werden kann.
|
Abbildung 2
Beispiele für die Kurven konkurrierender Binder mit und ohne Anwendung der 10-%-Regel
|
5.
|
Ob es angemessen ist, die 10-%-Regel zur Korrektur dieser Kurven anzuwenden, sollte sorgfältig geprüft werden. Die Anwendung sollte auf die Fälle beschränkt werden, in denen starke Anhaltspunkte für das Vorliegen eines hrER-Binders vorliegen. Bei der Durchführung von Versuchen für die Studie zur Validierung des FW-hrER-Bindungsassays wurde festgestellt, dass die 10-%-Regel manchmal nicht beabsichtigte und unvorhergesehene Folgen hatte. Chemikalien, bei denen keine Wechselwirkung mit dem Rezeptor auftrat (d. h. echte Nicht-Binder), zeigten häufig eine Variabilität von mehr als 10 % über das Spektrum der geprüften Konzentrationen, wenn der Wert der Radioliganden-Bindung bei nahe 100 % lag. Wenn der niedrigste Wert bei einer niedrigen Konzentration auftrat, konnten nach der 10-%-Regel die Daten aller höheren Konzentrationen aus der Analyse möglicherweise gelöscht werden. Allerdings konnten die Daten auch dieser Konzentrationen hilfreich für die Einstufung der betreffenden Chemikalie als Nicht-Binder sein. Abbildung 3 enthält Beispiele, bei denen die Anwendung der 10-%-Regel nicht angemessen wäre.
|
Abbildung 3
Beispiele für Daten konkurrierender Binder, bei denen eine Anwendung der 10-%-Regel nicht angemessen wäre


LITERATUR
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation für Wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
|
|
(2)
|
Motulsky, H., und Christopoulos, A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, S. 187-191. www.graphpad.com/manuals/Prism4/RegressionBook.pdf.
|
|
(3)
|
Laws, S.C., Yavanhxay, S., Cooper, R.L., Eldridge, J.C. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71
IN-VITRO-TESTS ZUR SENSIBILISIERUNG DER HAUT IN BEZUG AUF DEN SCHLÜSSELVORGANG DER AKTIVIERUNG VON DENDRITISCHEN ZELLEN AUF DEM ADVERSE OUTCOME PATHWAY (AOP) FÜR DIE SENSIBILISIERUNG DER HAUT
ALLGEMEINE EINLEITUNG
Prüfmethode auf der Grundlage des Schlüsselvorgangs der Aktivierung von dendritischen Zellen
|
1.
|
Ein Hautallergen ist gemäß Definition des Globalen Harmonisierten Systems zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN-GHS) (1) und der Verordnung (EU) Nr. 1272/2008 der Europäischen Union über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP) (100) ein Stoff, der bei Hautkontakt eine allergische Reaktion auslöst. Über die biologischen Schlüsselvorgänge, die der Sensibilisierung der Haut zugrunde liegen, besteht allgemeines Einvernehmen. Das derzeitige Wissen über die chemischen und biologischen Mechanismen im Zusammenhang mit der Hautsensibilisierung wurde in Form eines Adverse Outcome Pathway (AOP) unter dem OECD-AOP-Programm (2), ausgehend von dem auslösenden molekularen Ereignis über die Zwischenvorgänge bis hin zur schädlichen Auswirkung auf die Gesundheit, nämlich die allergische Kontaktdermatitis, zusammengefasst. In diesem Fall ist das auslösende molekulare Ereignis (d. h. der erste Schlüsselvorgang) die kovalente Bindung von elektrophilen Stoffen an nukleophile Zentren in Hautproteinen. Der zweite Schlüsselvorgang in diesem AOP findet in den Keratinozyten statt und umfasst entzündliche Reaktionen sowie Änderungen der Genexpression in Verbindung mit spezifischen Zellsignalketten wie beispielsweise ARE (Antioxidant/Electrophile Response Element)-abhängigen Ketten. Der dritte Schlüsselvorgang ist die Aktivierung von dendritischen Zellen (DC), die normalerweise durch Expression von spezifischen Zelloberflächenmarkern, Chemokinen und Cytokinen bewertet werden. Der vierte Schlüsselvorgang ist die T–Zellaktivierung und -proliferation, die indirekt im Lokalen Lymphknotentest (LLNA) an der Maus (3) bewertet wird.
|
|
2.
|
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 442E (2017). Sie beschreibt die In-vitro-Tests in Bezug auf Mechanismen im Rahmen des Schlüsselvorgangs der Aktivierung von dendritischen Zellen des AOP für die Sensibilisierung der Haut (2). Die Testmethode umfasst Tests, die zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren gemäß UN-GHS und CLP durchgeführt werden.
|
|
Die in dieser Prüfmethode beschriebenen Tests umfassen:
|
—
|
Aktivierungstest mit humanen Zelllinien (h-CLAT)
|
|
—
|
U937 Aktivierungstest mit Zelllinien (U-SENS™)
|
|
—
|
Interleukin-8 Reportergen-Test (IL-8 Luc-Test)
|
|
|
|
3.
|
Die Tests in dieser Prüfmethode und die entsprechenden OECD-TG können sich in Bezug auf das Verfahren zur Erzeugung der Daten und der gemessenen Werte unterscheiden, können aber gleichermaßen verwendet werden, um die länderspezifischen Anforderungen an die Prüfergebnisse des Schlüsselvorgangs zur Aktivierung dendritischer Zellen des AOP für die Hautsensibilisierung zu erfüllen; zugleich findet die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen auf sie Anwendung.
|
Hintergrund und Prinzipien der Tests, die in der auf den Schlüsselvorgang gestützten Prüfmethode enthalten sind
|
4.
|
Die Bewertung der Hautsensibilisierung erfolgte normalerweise an Labortieren. Bei den klassischen Methoden an Meerschweinchen, dem Maximierungstest an Meerschweinchen (Guinea Pig Maximisation Test, GPMT) nach Magnusson/Kligman und dem Bühler-Test (Prüfmethode B.6) (4), werden die Induktions- und die Auslösephase der Hautsensibilisierung bewertet. Ein Test an der Maus, der Lokale Lymphknotentest (LLNA, Prüfmethode B.42) (3) und die beiden nicht radioaktiven Abwandlungen dieser Tests, LLNA: DelR (Prüfmethode B.50) (5) und LLNA: BrdU-ELISA (Prüfmethode B.51) (6), bei denen jeweils nur die Induktionsreaktion bewertet wird, haben ebenfalls an Akzeptanz gewonnen, da sie sowohl in Bezug auf den Tierschutz als auch die objektive Messung der Induktionsphase der Hautsensibilisierung Vorteile gegenüber Tests am Meerschweinchen bieten.
|
|
5.
|
Kürzlich wurden mechanistisch basierte In-chemico- und In-vitro-Prüfmethoden in Bezug auf den ersten Schlüsselvorgang (Prüfmethode B.59; Direkt-Peptidreaktivitätstest (7)) und den zweiten Schlüsselvorgang (Prüfmethode B.60; ARE-Nrf2 Luciferase-Prüfmethode (8)) des Hautsensibilisierungs-AOP als Beitrag zur Bewertung des Gefahrenpotenzials für die Hautsensibilisierung von Chemikalien eingeführt.
|
|
6.
|
Die in dieser Prüfmethode beschriebenen Tests quantifizieren entweder die Änderung der Expression von Zelloberflächenmarkern im Zusammenhang mit dem Aktivierungsprozess von Monozyten und dendritischen Zellen (DC) nach Exposition gegenüber Allergenen (z. B. CD54, CD86) oder die Änderungen der IL-8-Expression, einem Zytokin in Verbindung mit der Aktivierung von DC. Es wurde berichtet, dass Hautallergene die Expression von Zellmembranmarkern wie CD40, CD54, CD80, CD83 und CD86 zusätzlich zur Induktion von proinflammatorischen Zytokinen wie IL-1β und TNF-α und mehreren Chemokine wie IL-8 (CXCL8) und CCL3 (9) (10) (11) (12) im Zusammenhang mit der DC-Aktivierung induzieren (2).
|
|
7.
|
Da die DC-Aktivierung jedoch nur einen einzigen Schlüsselvorgang des Hautsensibilisierungs-AOP darstellt (2) (13), reichen Informationen, die mit Tests erzeugt werden, die nur Marker der DC-Aktivierung messen, möglicherweise nicht aus, um auf das Vorhandensein oder Fehlen eines Hautsensibilisierungspotenzials von Chemikalien zu schließen. Daher sollten die mit dieser Prüfmethode ermittelten Daten zur Unterstützung der Unterscheidung zwischen Hautallergenen (d. h. UN-GHS/CLP Kategorie 1) und Nichtsensibilisatoren im Rahmen integrierter Ansätze (IATA) eingesetzt und mit anderen ergänzenden Informationen, die beispielsweise aus In-vitro-Tests in Bezug auf andere Schlüsselvorgänge des Hautsensibilisierungs-AOP abgeleitet werden, sowie mit anderen Nicht-Prüfmethoden, einschließlich der Übertragung von Informationen (read across) zu chemischen Analoga (13), kombiniert werden. Beispiele zur Verwendung von Daten, die mit diesen Tests im Rahmen definierter Ansätze erzeugt wurden, d. h. standardisierter Ansätze sowohl in Bezug auf den Satz der verwendeten Informationsquellen als auch in Bezug auf das Verfahren zur Ableitung von Vorhersagen, wurden veröffentlicht (13) und können innerhalb des IATA als nützliche Elemente verwendet werden.
|
|
8.
|
Die in dieser Prüfmethode beschriebenen Tests können alleine weder zur Einstufung von Hautallergenen in die Unterkategorien 1A und 1B gemäß Definition in UN-GHS/CLP (für Behörden, die diese zwei optionalen Unterkategorien einführen), noch zur Vorhersage des Potenzials im Rahmen von Sicherheitsbewertungsentscheidungen verwendet werden. Jedoch können mithilfe dieser Methoden erzeugte positive Ergebnisse je nach Rechtsrahmen alleine zur Einstufung einer Chemikalie in die UN-GHS/CLP-Kategorie 1 herangezogen werden.
|
|
9.
|
Der Begriff „Prüfchemikalie“ bezeichnet bei dieser Prüfmethode das, was getestet (101) wird, und bezieht sich nicht auf die Anwendbarkeit der Tests zur Prüfung von einkomponentigen Stoffen, mehrkomponentigen Stoffen und/oder Gemischen. Gegenwärtig stehen nur begrenzte Informationen über die Anwendbarkeit der Tests für mehrkomponentige Stoffe/Gemische zur Verfügung (14) (15). Die Tests sind dennoch für die Prüfung von mehrkomponentigen Stoffen und Gemischen technisch geeignet. Bevor diese Prüfmethode jedoch an einem Gemisch für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, sollte geprüft werden, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefert, und wenn dem so ist, warum. (102) Solche Erwägungen entfallen, wenn die Prüfung des Gemischs gesetzlich vorgeschrieben ist. Zudem sollte bei mehrkomponentigen Stoffen oder Gemischen die mögliche Interferenz der zytotoxischen Komponenten mit den beobachteten Reaktionen beachtet werden.
|
LITERATURHINWEISE
|
(1)
|
Vereinte Nationen (UN) (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sechste überarbeitete Auflage. New York und Genf: United Nations Publications. ISBN: 978-92-1-117006-1. Verfügbar unter:https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Verfügbar unter:http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Kapitel B.42 in diesem Anhang: Lokaler Lymphknotentest.
|
|
(4)
|
Kapitel B.6 in diesem Anhang: Sensibilisierung der Haut.
|
|
(5)
|
Kapitel B.50 in diesem Anhang: Sensibilisierung der Haut: Lokaler Lymphknotentest: DA.
|
|
(6)
|
Kapitel B.51 in diesem Anhang: Sensibilisierung der Haut: Lokaler Lymphknotentest: BrdU-ELISA.
|
|
(7)
|
Kapitel B.59 in diesem Anhang: In-chemico-Hautsensibilisierung: Direkt-Peptidreaktivitätstest (DPRA).
|
|
(8)
|
Kapitel B.60 in diesem Anhang: In-vitro-Hautsensibilisierung: ARE-Nrf2 Luciferase-Prüfmethode.
|
|
(9)
|
Steinman, RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271–96.
|
|
(10)
|
Caux, C., Vanbervliet B., Massacrier, C., Azuma, M., Okumura, K., Lanier, LL. und Banchereau, J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841–7.
|
|
(11)
|
Aiba, S., Terunuma, A., Manome, H. und Tagami, H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031–8.
|
|
(12)
|
Aiba, S., Manome, H., Nakagawa, S., Mollah, ZU., Mizuashi, M., Ohtani, T., Yoshino, Y. und Tagami, H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390–8.
|
|
(13)
|
OECD (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter:https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga, T., Sakaguchi, H., Sono, S., Kosaka, N., Ishikawa, M., Nukada, Y., Miyazawa, M., Ito, Y., Nishiyama, N., Itagaki, H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275–284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901–916.
|
Anlage 1
IN-VITRO-HAUTSENSIBILISIERUNG: AKTIVIERUNGSTEST MIT HUMANEN ZELLLINIEN (H-CLAT)
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
|
1.
|
Der h-CLAT quantifiziert die Änderungen der Expression von Zelloberflächenmarkern in Zusammenhang mit dem Aktivierungsprozess von Monozyten und dendritischen Zellen (DC) (d. h. CD86 und CD54) in der humanen monozytischen Leukämie-Zelllinie THP-1 nach Exposition gegenüber Allergenen (1)(2). Die gemessenen Expressionswerte der Zelloberflächenmarker CD86 und CD54 werden dann zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren verwendet.
|
|
2.
|
Der h-CLAT wurde in einer Validierungsstudie unter der Leitung eines Europäischen Referenzlabors für Alternativen zu Tierversuchen (EURL ECVAM) und in einer anschließenden unabhängigen Peer-Review des Wissenschaftlich Beratenden Ausschusses (ESAC) des EURL ECVAM bewertet. Unter Berücksichtigung sämtlicher verfügbarer Nachweise und Informationen von Regulierungsbehörden und Interessengruppen wurde der h-CLAT von EURL ECVAM (3) empfohlen, um im Rahmen eines IATA die Unterscheidung zwischen Hautallergenen und Nichtsensibilatoren im Hinblick auf die Gefahreneinstufung und -kennzeichnung zu unterstützen. Beispiele für die Verwendung von h-CLAT-Daten in Kombination mit anderen Informationen werden in der Literatur beschrieben (4)(5)(6)(7)(8)(9)(10)(11).
|
|
3.
|
Der h-CLAT erwies sich als übertragbar auf Labore mit Erfahrung auf dem Gebiet der Zellkulturtechniken und der Flusszytometrieanalyse. Der Grad der Reproduzierbarkeit, der bei den Tests erwartet werden kann, liegt in der Größenordnung von 80 % innerhalb von und zwischen Labors (3)(12). Die in der Validierungsstudie (13) und anderen veröffentlichten Studien (14) erzeugten Ergebnisse deuten insgesamt darauf hin, dass die Genauigkeit bei der Unterscheidung zwischen Hautallergenen (d. h. UN-GHS/CLP Kat.1) und Nichtsensibilisatoren im Vergleich zu den LLNA-Ergebnissen bei 85 % (N = 142) mit einer Empfindlichkeit von 93 % (94/101) und einer Spezifität von 66 % (27/41) liegt (gestützt auf eine erneute Analyse durch EURL ECVAM (12), wobei alle bestehenden Daten berücksichtigt und alle negativen Ergebnisse für Chemikalien mit log KOW von mehr als 3,5 wie in Abschnitt 4 beschrieben nicht berücksichtigt wurden). Bei falsch-negativen Vorhersagen mit dem h-CLAT ist die Wahrscheinlichkeit höher, dass Chemikalien mit geringer bis mittlerer Hautsensibilisierungspotenz betroffen sind (d. h. UN-GHS/CLP-Unterkategorie 1B), als Chemikalien mit hoher Hautsensibilisierungspotenz (d. h. UN-GHS/CLP-Unterkategorie 1A) (4)(13)(15). Insgesamt deuten diese Informationen auf die Zweckmäßigkeit der h-CLAT-Methode für die Erkennung der Gefahr einer Hautsensibilisierung hin. Jedoch sind die Genauigkeitswerte, die hier für den h-CLAT als eigenständiger Tests angegeben werden, lediglich als Anhaltspunkte zu betrachten, da der Test in Kombination mit anderen Informationsquellen im Rahmen eines IATA sowie gemäß den Bestimmungen unter Nummer 7 und 8 der Allgemeinen Einleitung betrachtet werden sollte. Darüber hinaus sollte bei der Bewertung von Prüfmethoden zur Hautsensibilisierung ohne Tierversuche beachtet werden, dass der LLNA-Test sowie andere Tierversuche die Situation bei Menschen nicht vollständig widerspiegeln.
|
|
4.
|
Auf der Grundlage der gegenwärtig verfügbaren Daten über die h-CLAT-Methode wurde nachgewiesen, dass der Test bei Prüfchemikalien, die eine Vielzahl an organischen Funktionsgruppen, Reaktionsmechanismen, Hautsensibilisierungspotenzialen (wie in In-vivo-Studien festgestellt) und physikalisch-chemischen Eigenschaften abdecken, anwendbar ist (3)(14)(15). Die h-CLAT-Methode ist anwendbar bei Prüfchemikalien, die löslich sind oder eine stabile Dispersion (d. h. ein Kolloid oder eine Suspension, in denen die Prüfchemikalie sich nicht absetzt oder sich vom Lösungsmittel/Vehikel in verschiedene Phasen trennt) in einem geeigneten Lösungsmittel/Vehikel bilden (siehe Abschnitt 14). Prüfchemikalien mit einem log Kow von mehr als 3,5 tendieren dazu, falsch negative Ergebnisse hervorzurufen (14). Deshalb sollten negative Ergebnisse bei Prüfchemikalien mit einem log Kow von mehr als 3,5 nicht berücksichtigt werden. Positive Ergebnisse für Prüfchemikalien mit einem log Kow von mehr als 3,5 könnten jedoch noch verwendet werden, um die Identifizierung der Prüfchemikalie als Hautallergen zu unterstützen. Außerdem können Prohaptene (d. h. Stoffe, die beispielsweise über P450-Enzyme aktiviert werden müssen) und Prähaptene (d. h. Stoffe, die durch Oxidation aktiviert werden) aufgrund der begrenzten Stoffwechselfähigkeit der verwendeten Zelllinie (16) sowie der Versuchsbedingungen, insbesondere bei geringer Oxidationsgeschwindigkeit, zu negativen Ergebnissen des h-CLAT (15) führen. Fluoreszierende Prüfchemikalien können mit h-CLAT (17) bewertet werden; dennoch beeinträchtigen stark fluoreszierende Prüfchemikalien, die mit derselben Wellenlänge ausstrahlen wie Fluorescein-Isothiocyanat (FITC) oder Propidiumjodid (PI), den Nachweis mittels Durchflusszytometrie und können daher nicht korrekt mithilfe von mit FITC-konjugierten Antikörpern oder PI bewertet werden. In einem solchen Fall können andere mit Fluorochrom markierte Antikörper bzw. andere Zytotoxizitätsmarker verwendet werden, solange aufgezeigt werden kann, dass sie ähnliche Ergebnisse liefern wie die mit FITC markierten Antikörper (siehe Abschnitt 24) oder PI (siehe Abschnitt 18), z. B. durch Testen der Leistungsstoffe in Anlage 1–2. Angesichts der vorstehenden Ausführungen sollten negative Ergebnisse im Kontext der angegebenen Grenzen und in Verbindung mit anderen Informationsquellen im Rahmen von IATA interpretiert werden. In Fällen, in denen die Nichtanwendbarkeit der h-CLAT-Methode bei anderen spezifischen Kategorien von Prüfchemikalien nachgewiesen wird, sollte sie bei diesen spezifischen Kategorien nicht verwendet werden.
|
|
5.
|
Die h-CLAT-Methode unterstützt, wie bereits oben erwähnt, die Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren. Er kann jedoch bei Verwendung im Rahmen von integrierten Ansätzen wie IATA möglicherweise auch zur Bewertung der Sensibilisierungspotenz beitragen (4)(5)(9). Es sind dennoch weitere Untersuchungen erforderlich, vorzugsweise auf der Grundlage von Humandaten, um herauszufinden, inwieweit die Ergebnisse des h-CLAT möglicherweise zur Potenzbewertung herangezogen werden können.
|
|
6.
|
Definitionen sind Anlage 1.1 zu entnehmen.
|
PRINZIP DER PRÜFMETHODE
|
7.
|
Die h-CLAT-Methode ist ein In-vitro-Test, der Änderungen der Expression von Zelloberflächenmarkern (d. h. CD86 und CD54) auf einer humanen monozytischen Leukämiezelllinie, THP-1-Zellen, nach einer 24-stündigen Exposition gegenüber der Prüfchemikalie quantifiziert. Diese Oberflächenmoleküle sind typische Marker der monotypischen THP-1-Aktivierung und können die DC-Aktivierung eventuell nachahmen, die eine kritische Rolle bei der T-Zellenvorbehandlung spielt. Die Änderungen der Expression von Oberflächenmarkern werden mittels Durchflusszytometrie nach Zellfärbung mit Antikörpern, die mit Fluorochrom markiert wurden, gemessen. Die Zytotoxizitätsmessung wird ebenfalls zur gleichen Zeit durchgeführt, um zu beurteilen, ob die Hochregulierung der Expression von Oberflächenmarkern bei Konzentrationen unterhalb des zytotoxischen Werts auftritt. Die relative Fluoreszenzintensität der Oberflächenmarker im Vergleich zur Lösungsmittel-/Vehikelkontrolle wird berechnet und im Vorhersagemodell verwendet (siehe 26), um die Unterscheidung zwischen Allergenen und Nichtsensibilisatoren zu unterstützen.
|
NACHWEIS DER KOMPETENZ
|
8.
|
Vor der routinemäßigen Verwendung des unter dieser Anlage zur Prüfmethode B.71 beschriebenen Tests sollten Laboratorien ihre technische Kompetenz anhand der zehn in Anlage 1.2 aufgeführten Leistungsstoffe nachweisen. Darüber hinaus sollten Prüfungsnutzer eine historische Datenbank der mit den Reaktivitätsprüfungen (siehe Abschnitt 11) und mit den positiven sowie Lösungsmittel-/Vehikelkontrollen (siehe Abschnitte 20–22) erzeugten Daten pflegen und diese Daten nutzen, um zu bestätigen, dass die Reproduzierbarkeit der Prüfung in ihrem Labor im Lauf der Zeit aufrechterhalten wird.
|
VERFAHREN
|
9.
|
Diese Prüfmethode basiert auf dem h-CLAT-DataBase-Dienst für alternative Methoden zur Durchführung von Tierversuchen (DB-ALM) Protokoll Nr. 158 (18), das für die vom EURL ECVAM koordinierte Validierungsstudie verwendet wurde. Es wird empfohlen, dieses Protokoll bei der Durchführung und Anwendung der h-CLAT-Methode im Labor zugrunde zu legen. Nachfolgend werden die wichtigsten Komponenten und Verfahren der h-CLAT-Methode beschrieben, die zwei Schritte umfasst: Test zur Dosisfindung und CD86/CD54-Expressionsmessung.
|
Vorbereitung der Zellen
|
10.
|
Bei Ausführung der h-CLAT-Methode sollte die humane monozytische Leukämie-Zelllinie THP-1 verwendet werden. Es wird empfohlen, dass Zellen (TIB-202™) aus einer gut qualifizierten Zellbank wie der American Type Culture Collection entnommen werden.
|
|
11.
|
THP-1-Zellen werden bei 37°C unter 5 % CO2 und einer befeuchteten Atmosphären in einem mit 10 % fetalem Kälberserum (FBS), 0,05 mM 2-Mercaptoethanol, 100 Einheiten/ml Penicillin und 100 μg/ml Streptomycin angereicherten RPMI-1640-Medium kultiviert. Die Verwendung von Penicillin und Streptomycin im Kulturmedium kann vermieden werden. In einem solchen Fall sollten Benutzer jedoch verifizieren, dass das Nichtvorhandensein von Antibiotika im Kulturmedium keinen Einfluss auf die Ergebnisse hat, beispielsweise durch Testen der in Anlage 1.2 aufgeführten Leistungsstoffe. Um das Kontaminationsrisiko zu minimieren, sollten bewährte Zellkulturpraktiken auf jeden Fall unabhängig vom Vorhandensein oder Nichtvorhandensein von Antibiotika im Zellkulturmedium befolgt werden. THP-1-Zellen werden regelmäßig alle 2–3 Tage bei einer Dichte von 0,1 bis 0,2 × 106 Zellen/ml ausgesät. Es sollten Zelldichten von 0,1 bis 1,0 × 106 Zellen/ml gewahrt werden. Vor ihrer Verwendung zu Testzwecken sollten die Zellen qualifiziert werden, indem ein Reaktivitätstest durchgeführt wird. Der Reaktivitätstest der Zellen sollte mithilfe der positiven Kontrollen, 2,4-Dinitrochlorbenzol (DNCB) (CAS Nr. 97-00-7, ≥99 % Reinheit) und Nickelsulfat (NiSO4) (CAS Nr. 10101-97-0, ≥99 % Reinheit) und der negativen Kontrolle, Milchsäure (LA) (CAS Nr. 50-21-5, ≥85 % Reinheit) zwei Wochen nach dem Auftauen durchgeführt werden. DNCB und NiSO4 sollten eine positive Reaktion auf CD86- und CD54-Zelloberflächenmarker ergeben und LA sollte eine negative Reaktion auf CD86- und CD54-Zelloberflächenmarker ergeben. Nur die Zellen, welche den Reaktivitätstest bestanden haben, dürfen für den Test verwendet werden. Zellen können bis zu zwei Monate nach dem Auftauen gewonnen werden. Die Passagenanzahl sollte 30 nicht übersteigen. Der Reaktivitätstest sollte gemäß den in den Abschnitten 20–24 beschriebenen Verfahren durchgeführt werden.
|
|
12.
|
Zur Prüfung werden die THP-1-Zellen bei einer Dichte von entweder 0,1 × 106 Zellen/ml oder 0,2 × 106 Zellen/ml ausgesät und in Kulturkolben 72 bzw. 48 Stunden lang vorgezüchtet. Es ist wichtig, dass die Zelldichte im Kulturkolben direkt nach dem Vorkulturzeitraum bei jedem Versuch so konstant wie möglich ist (indem eine der zwei oben beschriebenen Vorkulturbedingungen angewandt wird), da die Zelldichte im Kulturkolben direkt nach der Vorkultur die von Allergenen induzierte CD86/CD54-Expression beeinträchtigen könnte (19). Am Tag des Tests werden die Zellen, die aus dem Kulturkolben geerntet wurden, mit einem frischen Kulturmedium mit 2 × 106 Zellen/ml neu angesetzt. Dann werden die Zellen in eine flache Platte mit 24 Mulden (500 μl, 1 × 106 Zellen/Mulde) oder eine flache Platte mit 96 Mulden (80 μl, 1,6 × 105 Zellen/Mulde) verteilt.
|
Test zur Dosisfindung
|
13.
|
Ein Test zur Dosisfindung wird durchgeführt, um CV75 zu bestimmen. Dies ist die Prüfchemikalienkonzentration, die im Vergleich zur Lösungsmittel-/Vehikelkontrolle zu einer Zellviabilität (CV) von 75 % führt. Der CV75-Wert wird verwendet, um die Konzentration der Prüfchemikalien für die CD86/CD54-Expressionsmessung zu bestimmen (siehe Abschnitte 20–24).
|
Vorbereitung der Prüfchemikalien und Kontrollstoffe
|
14.
|
Die Prüfchemikalien sowie die Kontrollstoffe werden am Tag des Tests vorbereitet. Bei der h-CLAT-Method werden die Prüfchemikalien in Kochsalzlösung oder einem Medium, als erste Lösungsmittel-/Vehikeloptionen, oder Dimethylsulfoxid (DMSO,≥ 99 % Reinheit), als zweite Lösungsmittel-/Vehikeloption, aufgelöst oder stabil verteilt (siehe auch Abschnitt 4), falls die Prüfchemikalie in den zwei zuvor genannten Lösungsmitteln/Vehikeln mit endgültigen Konzentrationen von 100 mg/ml (in Kochsalzlösung oder Medium) oder 500 mg/ml (in DMSO) nicht löslich ist oder keine stabile Verteilung ausbildet. Andere als die oben beschriebenen Lösungsmittel/Vehikel können verwendet werden, wenn eine ausreichende wissenschaftliche Begründung gegeben wird. Die Stabilität der Prüfchemikalie im endgültigen Lösungsmittel/Vehikel sollte berücksichtigt werden.
|
|
15.
|
Ausgehend von den 100 mg/ml- (in Kochsalzlösung oder Medium) oder 500 mg/ml- (in DMSO) Stammlösungen der Prüfchemikalien sollten die folgenden Verdünnungsschritte durchgeführt werden:
|
—
|
Für Kochsalzlösung oder Medium als Lösungsmittel/Vehikel: Acht Stammlösungen (acht Konzentrationen) werden nach zweifachen seriellen Verdünnungen mithilfe des entsprechenden Lösungsmittels/Vehikels vorbereitet. Diese Stammlösungen werden dann im Kulturmedium 50-fach weiter verdünnt (Arbeitslösungen). Wenn die obere endgültige Konzentration in der Platte von 1 000 μg/ml nicht toxisch ist, sollte die maximale Konzentration erneut bestimmt werden, indem eine neue Zytotoxizitätsprüfung durchgeführt wird. Die endgültige Konzentration in der Platte sollte 5 000 μg/ml für Prüfchemikalien, die in Kochsalzlösung oder einem Medium gelöst oder stabil verteilt wurden, nicht überschreiten.
|
|
—
|
Für DMSO als Lösungsmittel/Vehikel: Acht Stammlösungen (acht Konzentrationen) werden nach zweifachen seriellen Verdünnungen mithilfe des entsprechenden Lösungsmittels/Vehikels vorbereitet. Diese Stammlösungen werden dann im Kulturmedium 250-fach weiter verdünnt (Arbeitslösungen). Die endgültige Konzentration in der Platte sollte 1 000 μg/ml nicht überschreiten, auch wenn die Konzentration nicht toxisch ist.
|
Die Arbeitslösungen werden schließlich zur Exposition verwendet, indem eine gleiche Menge an Arbeitslösung zum Volumen der THP-1-Zellsuspension in der Platte hinzugefügt wird (siehe auch Abschnitt 17), um eine weitere zweifache Verdünnung zu erhalten (normalerweise beträgt der endgültige Bereich an Konzentrationen in der Platte 7,81–1 000 μg/ml).
|
|
16.
|
Die bei der h-CLAT-Methode verwendete Lösungsmittel-/Vehikelkontrolle ist das Kulturmedium (für mit Medium oder Kochsalzlösung löslich gemachte oder stabil verteilte (siehe Abschnitt 4) Prüfchemikalien) oder DMSO (für in DMSO löslich gemachte oder stabil verteilte Prüfchemikalien), die bei einer einzigen endgültigen Konzentration in der Platte von 0,2 % geprüft wurden. Hier wird dasselbe Verdünnungsverfahren verwendet, das für Arbeitslösungen in Abschnitt 15 beschrieben wird.
|
Applikation der Prüfchemikalien und Kontrollstoffe
|
17.
|
Das Kulturmedium oder die Arbeitslösungen, die in den Abschnitten 15 und 16 beschrieben werden, werden 1:1 (v/v) mit den in der flachen Platte mit 24 oder 96 Mulden vorbereiteten Zellsuspensionen gemischt (siehe Abschnitt 12). Die behandelten Platten werden dann 24 ± 0,5 Stunden bei 37oC unter 5 % CO2 inkubiert. Es ist darauf zu achten, dass eine Verdunstung der flüchtigen Prüfchemikalien sowie eine Kreuzkontamination zwischen Mulden durch die Prüfchemikalien vermieden werden, indem die Platte beispielsweise vor der Inkubation mit den Prüfchemikalien versiegelt wird (20).
|
Färben mit Propidiumjodid (PI)
|
18.
|
Nach einer Expositionszeit von 24 ± 0,5 Stunden werden die Zellen in Probenröhrchen übertragen und durch Zentrifugierung gesammelt. Die Überstände werden entsorgt und die verbleibenden Zellen werden mit 200 μl (im Falle von 96 Mulden) oder 600 μl (im Falle von 24 Mulden) einer mit Phosphat gepufferten Kochsalzlösung mit 0,1 % Rinderserum-Albumin (Färbepuffer) resuspensiert. 200 μl der Zellsuspension werden in eine Platte mit gewölbtem Boden und 96 Mulden (im Falle von 96 Mulden) oder ein Mikroröhrchen (im Falle von 24 Mulden) gegeben und zweimal mit 200 μl (im Falle von 96 Mulden) oder 600 μl (im Falle von 24 Mulden) Färbepuffer gewaschen. Schließlich werden die Zellen in Färbepuffer (z. B. 400 μl) resuspensiert und es wird PI-Lösung (z. B. 20 μl) hinzugefügt (zum Beispiel ist die endgültige Konzentration von PI 0,625 μg/ml). Andere Zytotoxizitätsmarker wie 7-Aminoactinomycin D (7-AAD), Trypan blau oder andere können verwendet werden, wenn gezeigt werden kann, dass die alternativen Farbstoffe ähnliche Ergebnisse liefern wie PI, zum Beispiel durch Testen der Leistungsstoffe in Anlage 1.2.
|
Zytotoxizitätsmessung anhand von Durchflusszytometrie und Schätzung von CV75-Wert
|
19.
|
Die PI-Aufnahme wird anhand der Durchflusszytometrie mit dem Aufnahmekanal FL-3 analysiert. Es werden insgesamt 10 000 lebende Zellen (PI negativ) aufgenommen. Die Zellviabilität kann anhand der folgenden Gleichung über das Zytometeranalyseprogramm berechnet werden. Wenn die Zellviabilität gering ist, sollten bis zu 30 000 Zellen einschließlich toter Zellen aufgenommen werden. Alternativ können die Daten eine Minute nach der Einleitung der Analyse aufgenommen werden.

Der CV75-Wert (siehe Abschnitt 13), d. h. eine Konzentration, die 75 % des THP-1-Zellüberlebens (25 % Zytotoxizität) zeigt, wird anhand der log-linearen Interpolation mit der folgenden Gleichung berechnet:

Dabei gilt:
a ist der Mindestwert der Zellviabilität über 75 %
c ist der Höchstwert der Zellviabilität unter 75 %
b und d sind die Konzentrationen, die den Wert der Zellviabilität a bzw. c zeigen

Weitere Ansätze zur Ableitung von CV75 können angewandt werden, solange demonstriert wird, dass dies keinen Einfluss auf die Ergebnisse hat (z. B. durch Testen der Leistungsstoffe).
|
CD86/CD54-Expressionsmessung
Vorbereitung der Prüfchemikalien und Kontrollstoffe
|
20.
|
Das geeignete Lösungsmittel/Vehikel (Kochsalzlösung, Medium oder DMSO; siehe Abschnitt 14) wird verwendet, um die Prüfchemikalien zu lösen oder stabil zu verteilen. Die Prüfchemikalien werden zuerst auf die Konzentration verdünnt, die dem 100-fachen (für Kochsalzlösung oder Medium) oder 500-fachen (für DMSO) von 1,2 × CV75 entspricht, der im Test zur Dosisfindung bestimmt wurde (siehe Abschnitt 19). Wenn der CV75 nicht bestimmt werden kann (d. h. wenn keine ausreichende Zytotoxizität im Test zur Dosisfindung beobachtet werden kann), sollte die höchste lösliche oder stabil verteilte Konzentration der Prüfchemikalie, die mit jedem Lösungsmittel/Vehikel vorbereitet wird, als Ausgangskonzentration verwendet werden. Bitte beachten Sie, dass die endgültige Konzentration in der Platte 5 000 μg/ml (im Falle von Kochsalzlösung oder Medium) oder 1 000 μg/ml (im Falle von DMSO) nicht überschreiten sollte. Dann werden 1,2-fache serielle Verdünnungen mit dem entsprechenden Lösungsmittel/Vehikel hergestellt, um die Stammlösungen zu erhalten (acht Konzentrationen von 100 × 1,2 × CV75 bis 100 × 0,335 × CV75 (für Kochsalzlösung oder Medium) oder von 500 × 1,2 × CV75 bis 500 × 0,335 × CV75 (für DMSO)), die in der h-CLAT-Methode geprüft werden sollen (siehe DB-ALM-Protokoll Nr. 158 für ein Beispiel eines Dosierungsschemas). Die Stammlösungen werden dann im Kulturmedium 50-fach (für Kochsalzlösung oder Medium) oder 250-fach (für DMSO) weiter verdünnt (Arbeitslösungen). Diese Arbeitslösungen werden schließlich für die Exposition mit einem weiteren endgültigen zweifachen Verdünnungsfaktor in der Platte verwendet. Wenn die Ergebnisse die in den Abschnitten 29 und 30 beschriebenen Akzeptanzkriterien zur Zellviabilität nicht erfüllen, kann der Test zur Dosisfindung wiederholt werden, um einen präziseren CV75 zu bestimmen. Bitte beachten Sie, dass nur Platten mit 24 Mulden für die CD86/CD54-Expressionsmessung verwendet werden können.
|
|
21.
|
Die Lösungsmittel-/Vehikelkontrolle wird wie in Abschnitt 16 beschrieben vorbereitet. Die bei der h-CLAT-Methode verwendete Positivkontrolle ist DNCB (siehe Abschnitt 11), für die Stammlösungen in DMSO vorbereitet und wie für die Stammlösungen in Abschnitt 20 beschrieben verdünnt werden. DNCB sollte als Positivkontrolle für die CD86/CD54-Expressionsmessung mit einer endgültigen Konzentration in der Platte verwendet werden (normalerweise 4,0 μg/ml). Um eine Konzentration von 4,0 μg/ml DNCB in der Platte zu erhalten, wird eine Stammlösung von 2 mg/ml DNCB in DMSO vorbereitet und weiter 250-fach mit dem Kulturmedium in eine Arbeitslösung von 8 μg/ml verdünnt. Alternativ könnte auch der CV75 von DNCB, der in jeder Prüfeinrichtung bestimmt wird, als Positivkontrollkonzentration verwendet werden. Andere geeignete Positivkontrollen können verwendet werden, sofern historische Daten für die Ableitung vergleichbarer Akzeptanzkriterien für einen Testdurchlauf zur Verfügung stehen. Bei Positivkontrollen sollte die endgültige Einzelkonzentration in der Platte 5 000 μg/ml (im Falle von Kochsalzlösung oder Medium) oder 1 000 μg/ml (im Falle von DMSO) nicht überschreiten. Die Durchlaufakzeptanzkriterien entsprechen denen, die für die Prüfchemikalien beschrieben werden (siehe Abschnitt 29), ausgenommen für das letzte Akzeptanzkriterium, da die Positivkontrolle mit einer einzelnen Konzentration geprüft wird.
|
Applikation der Prüfchemikalien und Kontrollstoffe
|
22.
|
Für jede Prüfchemikalie und jeden Kontrollstoff ist ein Experiment erforderlich, um eine Vorhersage zu ermitteln. Jedes Experiment besteht aus mindestens zwei unabhängigen Durchläufen für die CD86/CD54-Expressionsmessung (siehe Abschnitte 26–28). Jeder unabhängige Durchlauf wird an einem anderen Tag bzw. am selben Tag durchgeführt, vorausgesetzt, dass für jeden Durchlauf: a) unabhängige frische Stammlösungen und Arbeitslösungen der Prüfchemikalie und Antikörperlösungen vorbereitet werden und b) unabhängig geerntete Zellen verwendet werden (d. h. Zellen, die aus verschiedenen Kulturkolben entnommen werden); Zellen können jedoch aus derselben Passage stammen. Prüfchemikalien und Kontrollstoffe, die als Arbeitslösungen (500 μl) vorbereitet wurden, werden mit 500 μl der suspensierten Zellen (1 × 106 Zellen) im Verhältnis von 1:1 gemischt und die Zellen werden 24 ± 0,5 Stunden wie in den Abschnitten 20 und 21 beschrieben inkubiert. In jedem Durchlauf reicht ein einziges Replikat für jede Konzentration der Prüfchemikalie und des Kontrollstoffs aus, da eine Vorhersage in mindestens zwei unabhängigen Durchläufen ermittelt wird.
|
Zellfärbung und Analyse
|
23.
|
Nach einer Expositionsdauer von mindestens 24 ± 0,5 Stunden werden die Zellen von einer Platte mit 24 Mulden in Probenröhrchen gegeben, mithilfe von Zentrifugierung gesammelt und dann zweimal mit 1 ml Färbepuffer gewaschen (wenn erforderlich können zusätzliche Waschschritte durchgeführt werden). Nach dem Waschen werden die Zellen mit 600 μl Blockierungslösung blockiert (Färbepuffer mit 0,01 % (w/v) Globulin (Cohn-Fraktion II, III, human; SIGMA, #G2388-10G oder ähnliches)) und bei 4oC 15 Minuten lang inkubiert. Nach der Blockierung werden die Zellen in drei Aliquote von je 180 μl in eine Platte mit gewölbtem Boden und 96 Mulden oder ein Mikroröhrchen aufgeteilt.
|
|
24.
|
Nach der Zentrifugierung werden die Zellen mit 50 μl der mit FITC markierten anti-CD86-, anti-CD54- oder Maus-IgG1-Antikörper (Isotyp) bei 4oC 30 Minuten lang gefärbt. Die im h-CLAT DB-ALM Protokoll Nr. 158 (18) beschriebenen Antikörper sollten verwendet werden, indem sie 3:25 v/v (für CD86 (BD-PharMingen, #555657; Klon: Fun-1)) oder 3:50 v/v (für CD54 (DAKO, #F7143; Klon: 6.5B5) und IgG1 (DAKO, #X0927)) mit Färbepuffer verdünnt werden.Diese Antikörperverdünnungsfaktoren wurden von den Entwicklern des Tests als diejenigen definiert, die den besten Rauschabstand bieten. Nach Erfahrung der Testentwickler ist die Fluoreszenzintensität der Antikörper normalerweise zwischen verschiedenen Chargen konsistent. Benutzer können jedoch erwägen, die Antikörper unter den Bedingungen im eigenen Labor zu titrieren, um die besten Gebrauchskonzentrationen zu definieren. Andere mit Fluorochrom markierte anti-CD86- und/oder anti-CD54-Antikörper können verwendet werden, wenn gezeigt werden kann, dass sie ähnliche Ergebnisse wie mit FITC konjugierte Antikörper liefern, beispielsweise durch Testen der Leistungsstoffe in Anlage 1.2.Anzumerken ist, dass die Änderung des Klons oder des Lieferanten der Antikörper, wie dies im h-CLAT DB-ALM Protokoll Nr. 158 (18) beschrieben wird, die Ergebnisse beeinträchtigen kann. Nach dem zwei- oder mehrfachen Waschen mit 150 μl Färbepuffer werden die Zellen in Färbepuffer resuspensiert (z. B. 400 μl) und die PI-Lösung (z. B. 20 μl, um eine endgültige Konzentration von 0,625 μg/ml zu erhalten) oder eine andere Zytotoxizitätsmarkerlösung (siehe Abschnitt 18) wird hinzugefügt. Die Expressionswerte von CD86 und CD54 sowie die Zellviabilität werden mithilfe der Durchflusszytometrie analysiert.
|
DATEN UND BERICHTERSTATTUNG
Datenauswertung
|
25.
|
Die Expression von CD86 und CD54 wird mithilfe der Durchflusszytometrie mit dem Aufnahmekanal FL-1 analysiert. Auf der Grundlage der geometrischen mittleren Fluoreszenzintensität (MFI) werden die relative Fluoreszenzintensität (RFI) von CD86 und CD54 für Positivkontrollzellen (ctrl) und chemisch behandelte Zellen nach der folgenden Gleichung berechnet:
Die Zellviabilität aus den Isotypkontrollzellen (ctrl) (die mit Maus-IgG1-Antikörpern (Isotyp) gefärbt werden) wird ebenfalls nach der in Abschnitt 19 beschriebenen Gleichung berechnet.
|
Vorhersagemodell
|
26.
|
Bei der CD86/CD54-Expressionsmessung wird jede Prüfchemikalie in mindestens zwei unabhängigen Durchläufen geprüft, um eine einzige Vorhersage abzuleiten (POSITIV oder NEGATIV). Eine h-CLAT-Vorhersage gilt als POSITIV, wenn mindestens eine der folgenden Bedingungen in 2 von 2 oder in mindestens 2 von 3 unabhängigen Durchläufen erfüllt ist, andernfalls wird die h-CLAT-Vorhersage als NEGATIV angesehen (Abbildung 1):
|
—
|
Die RFI von CD86 ist bei jeder geprüften Konzentration gleich oder größer als 150 % (mit einer Zellviabilität ≥50 %);
|
|
—
|
die RFI von CD54 ist bei jeder geprüften Konzentration gleich oder größer als 200 % (mit einer Zellviabilität ≥50 %).
|
|
|
27.
|
Auf dieser Grundlage ist die h-CLAT-Vorhersage als POSITIV anzusehen, wenn die ersten zwei Durchläufe positiv für CD86 und/oder positiv für CD54 waren; ein dritter Durchlauf ist dann nicht mehr erforderlich. Analog ist die h-CLAT-Vorhersage als NEGATIV anzusehen (unter angemessener Berücksichtigung der Bestimmungen in Abschnitt 30), wenn die ersten zwei Durchläufe für beide Marker negativ sind; ein dritter Durchlauf ist dann nicht mehr erforderlich. Wenn die ersten zwei Durchläufe jedoch für mindestens einen der Marker nicht konkordant ist (CD54 oder CD86), dann ist ein dritter Durchlauf erforderlich und die endgültige Vorhersage basiert auf dem Mehrheitsergebnis der drei individuellen Durchläufe (d. h. 2 von 3). In dieser Hinsicht ist zu beachten, dass ein dritter Durchlauf erforderlich ist, wenn zwei unabhängige Durchläufe durchgeführt werden und einer nur für CD86 (nachfolgend „P1“ genannt) und der andere nur für CD54 (nachfolgend „P2“ genannt) positiv ist. Wenn dieser dritte Durchlauf für beide Marker negativ ist (nachfolgend „N“ genannt), dann wird die h-CLAT-Vorhersage als NEGATIV angesehen. Wenn der dritte Durchlauf andererseits für einen der Marker (P1 oder P2) oder für beide Marker (nachfolgend „P12“ genannt) positiv ist, dann ist die h-CLAT-Vorhersage als POSITIV anzusehen.

Abb. 1: Bei der h-CLAT-Methode verwendetes Vorhersagemodell. Eine h-CLAT-Vorhersage sollte im Rahmen eines IATA sowie gemäß den Nummern 7 und 8 der Allgemeinen Einleitung in Betracht gezogen werden.
P1: Durchlauf mit nur CD86 positiv; P2; Durchlauf mit nur CD54 positiv; P12: Durchlauf mit CD86 und CD54 positiv; N: Durchlauf mit weder CD86 noch CD54 positiv.
*Die Kästchen zeigen die relevanten Kombinationen der Ergebnisse aus den ersten beiden Durchläufen unabhängig von der Reihenfolge, in der sie ermittelt wurden.
#Die Kästchen zeigen die relevanten Kombinationen der Ergebnisse aus den drei Durchläufen auf der Grundlage der Ergebnisse aus den ersten zwei Durchläufen, wie im obigen Kästchen dargestellt, aber spiegeln nicht die Reihenfolge wider, in der sie ermittelt wurden.
|
|
28.
|
Für die Prüfchemikalien, die mit h-CLAT als POSITIV vorhergesehen wurden, können optional zwei effektive Konzentrationswerte (EC) bestimmt werden, EC150 für CD86 und EC200 für CD54, d. h. die Konzentration, bei der die Prüfchemikalien eine RFI von 150 bzw. 200 induzierten. Diese EC-Werte können jedoch bei Verwendung im Rahmen von integrierten Ansätzen wie IATA (4) (5) (6) (7) (8) möglicherweise auch zur Bewertung der Sensibilisierungspotenz beitragen (9). Sie können mithilfe der folgenden Gleichungen berechnet werden:
EC 150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
Dabei gilt:
Aconc ist die niedrigste Konzentration in μg/ml mit RFI > 150 (CD86) oder 200 (CD54)
Bconc ist die höchste Konzentration in μg/ml mit RFI < 150 (CD86) oder 200 (CD54)
ARFI ist die RFI bei der niedrigsten Konzentration mit RFI > 150 (CD86) oder 200 (CD54)
BRFI ist die RFI bei der höchsten Konzentration mit RFI > 150 (CD86) oder 200 (CD54)
Zum Zwecke der präziseren Ableitung der EC150- und EC200-Werte können drei unabhängige Durchläufe für die CD86/CD54-Expressionsmessung erforderlich sein. Die endgültigen EC150- und EC200-Werte werden dann als Mittelwert der aus den drei unabhängigen Durchläufen berechneten ECs bestimmt. Wenn nur zwei der drei unabhängigen Durchläufe die Kriterien für Positivität (siehe Abschnitte 26–27) erfüllen, wird der höhere EC150 oder EC200 der zwei berechneten Werte übernommen.
|
Akzeptanzkriterien
|
29.
|
Bei der Verwendung der h-CLAT-Methode (22) (27) sollten die folgenden Akzeptanzkriterien erfüllt werden.
|
—
|
Die Zellviabilitäten der Medium- und Lösungsmittel-/Vehikelkontrollen sollten höher als 90 % sein.
|
|
—
|
Bei der Lösungsmittel-/Vehikelkontrolle sollten die RFI-Werte von CD86 und CD54 die positiven Kriterien nicht überschreiten (CD86 RFI ≥ 150 % und CD54 RFI ≥ 200 %). Die RFI-Werte der Lösungsmittel-/Vehikelkontrolle werden berechnet, indem die in Abschnitt 25 beschriebene Formel verwendet wird („MFI der Chemikalie“ sollte durch „MFI des Lösungsmittels/Vehikels“ ersetzt werden und „MFI des Lösungsmittels/Vehikels“ sollte durch „MFI der (Medium-)Kontrolle“ ersetzt werden).
|
|
—
|
Bei den Medium- und Lösungsmittel-/Vehikelkontrollen sollte das MFI-Verhältnis von CD86 und CD54 zur Isotypkontrolle >105 % betragen.
|
|
—
|
Bei der Positivkontrolle (DNCB) sollten die RFI-Werte von CD86 und CD54 die Positivkriterien (CD86 RFI ≥ 150 und CD54 RFI ≥ 200) erfüllen und die Zellviabilität sollte mehr als 50 % betragen.
|
|
—
|
Für die Prüfchemikalie sollte die Zellviabilität in mindestens vier geprüften Konzentrationen in jedem Durchlauf mehr als 50 % betragen.
|
|
|
30.
|
Negative Ergebnisse sind nur für Prüfchemikalien mit einer Zellviabilität von weniger als 90 % bei der höchsten geprüften Konzentration zulässig (d. h. 1,2 × CV75 gemäß dem in Abschnitt 20 beschriebenen seriellen Verdünnungsschema). Wenn die Zellviabilität bei 1,2 × CV75 90 % oder mehr beträgt, sollte das negative Ergebnis verworfen werden. In einem solchen Fall wird empfohlen, die Dosiswahl durch Wiederholung der CV75-Bestimmung zu präzisieren. Es sollte beachtet werden, dass bei der Verwendung von 5 000 μg/ml in Kochsalzlösung (oder Medium oder anderen Lösungsmitteln/Vehikeln), 1 000 μg/ml in DMSO oder der höchsten löslichen Konzentration als maximale Prüfkonzentration einer Prüfchemikalie ein negatives Ergebnis auch dann akzeptabel ist, wenn die Zellviabilität über 90 % liegt.
|
Prüfbericht
|
31.
|
Der Prüfbericht sollte folgende Angaben enthalten.
|
Prüfchemikalie
|
Einkomponentiger Stoff:
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
physikalisches Erscheinungsbild, log Kow, Löslichkeit in Wasser, Löslichkeit in DMSO, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern verfügbar;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
Begründung der Auswahl des Lösungsmittels/Vehikels für jede Prüfchemikalie.
|
|
|
Mehrkomponentiger Stoff, UVCB-Stoff und Gemisch
|
Charakterisierung, so weit wie möglich, z. B. durch die chemische Zusammensetzung (siehe oben), Reinheit, das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften (siehe oben) der einzelnen Komponenten, soweit verfügbar;
|
|
physikalisches Erscheinungsbild, Löslichkeit in Wasser, Löslichkeit in DMSO und weitere relevante physikalisch-chemische Eigenschaften, sofern verfügbar;
|
|
Molekulargewicht oder scheinbares Molekulargewicht im Fall von Gemischen/Polymeren mit bekannter Zusammensetzung oder andere für die Durchführung der Studie relevante Informationen;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
Begründung der Auswahl des Lösungsmittels/Vehikels für jede Prüfchemikalie.
|
|
|
|
Kontrollen
|
Positivkontrolle
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
physikalisches Erscheinungsbild, log Kow, Löslichkeit in Wasser, Löslichkeit in DMSO, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern verfügbar und falls zutreffend;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
ggf. Verweis auf historische Ergebnisse von Positivkontrollen, die geeignete Akzeptanzkriterien für einen Testdurchlauf dokumentieren.
|
|
|
|
Negativ- und Lösungsmittel-/Vehikelkontrolle:
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
Aussehen, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern andere Kontroll-Lösungsmittel/Vehikel als die in der Prüfrichtlinie genannten verwendet werden und soweit verfügbar;
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
Begründung der Auswahl des Lösungsmittels/Vehikels für jede Prüfchemikalie.
|
|
|
Prüfbedingungen:
|
Name und Anschrift des Auftraggebers, der Prüfanstalt und des Studienleiters;
|
|
Beschreibung des verwendeten Tests;
|
|
verwendete Zelllinie, zugehörige Lagerbedingungen und Quelle (z. B. Einrichtung, von der sie gewonnen wurden);
|
|
die verwendete Durchflusszytometrie (z. B. Modell), einschließlich der Geräteeinstellungen, Globulin, Antikörper und verwendeten Zytotoxizitätsmarker;
|
|
das angewandte Verfahren zum Nachweis der Kompetenz des Labors bei der Durchführung des Tests durch Prüfen der Leistungsstoffe) und das angewandte Verfahren zum Nachweis der reproduzierbaren Leistung des Tests im Zeitverlauf, z. B. historische Kontrolldaten und/oder Daten zu historischen Reaktivitätstests.
|
|
|
Validitätskriterien
|
Zellviabilität, MFI- und RFI-Werte aus der Lösungsmittel-/Vehikelkontrolle im Vergleich zu den Validitätsbereichen;
|
|
Zellviabilität und RFI-Werte aus der Positivkontrolle im Vergleich zu den Validitätsbereichen;
|
|
Zellviabilität aller geprüften Konzentrationen der geprüften Chemikalie.
|
|
|
Prüfverfahren
|
Anzahl der vorgenommenen Durchläufe;
|
|
Konzentrationen der Prüfchemikalie, Applikationen und angewandte Expositionsdauer (falls von den Empfehlungen abweichend)
|
|
Beschreibung der angewandten Bewertungs- und Entscheidungskriterien;
|
|
Beschreibung etwaiger Änderungen am Prüfverfahren.
|
|
|
Ergebnisse
|
Tabellierung der Daten, einschließlich CV75 (falls anwendbar), individueller geometrischer MFI, RFI, Zellviabilitätswerte, EC150/EC200-Werte (falls anwendbar) für die Prüfchemikalie und für die Positivkontrolle in jedem Durchlauf und eine Anzeige der Einstufung der Prüfchemikalie gemäß dem Vorhersagemodell;
|
|
Beschreibung etwaiger sonstiger relevanter Beobachtungen, falls zutreffend.
|
|
|
Erörterung der Ergebnisse
|
Erörterung der anhand der h-CLAT-Methode erhaltenen Ergebnisse;
|
|
Analyse der Prüfergebnisse im Rahmen eines IATA, sofern sonstige relevante Informationen vorliegen.
|
|
|
LITERATURHINWEISE
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767–773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428–437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Verfügbar unter:https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318–1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333–1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489–504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371–379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337–351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355–2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150–60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Verfügbar unter:https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report. Verfügbar unter:https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599–609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275–284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683–1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81–88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23ff. Verfügbar unter:http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70–82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, , Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89–96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organization for Economic Cooperation and Development, Paris, Frankreich, 2005, 96 ff.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Verfügbar unter: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fünfte überarbeitete Fassung. New York und Genf: United Nations Publications. ISBN: 978-92-1-117006-1. Verfügbar unter:http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27–35.
|
Anlage 1.1
DEFINITIONEN
Genauigkeit
: Grad der Übereinstimmung zwischen Prüfergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der „Relevanz“. Der Begriff wird oft im Sinne von „Übereinstimmung“ verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (21).
AOP (Adverse Outcome Pathway)
: Abfolge von Vorgängen, ausgehend von der chemischen Struktur einer Zielchemikalie oder Zielgruppe ähnlicher Chemikalien, über den molekularen auslösenden Vorgang bis hin zu einem In-vivo-Ergebnis von Interesse (22).
Chemikalie
: Stoff oder Gemisch.
CV75
: Die geschätzte Konzentration, die 75 % Zellviabilität zeigt.
EC150
: die Konzentrationen, die die RFI-Werte von 150 in der CD86-Expression zeigen
EC200
: die Konzentrationen, die die RFI-Werte von 200 in der CD54-Expression zeigen
Durchflusszytometrie
: eine zytometrische Technik, bei der Zellen, die in einem Fluid suspendiert sind, einzeln durch eine Lichtstrahl fließen, das in für die Zellen und ihre Komponenten typischen Muster gestreut ist; Zellen werden häufig mit fluoreszierenden Markern gekennzeichnet, damit das Licht zuerst absorbiert und dann in geänderten Frequenzen ausgestrahlt wird.
Gefahr
: Inhärente Eigenschaft eines Stoffes oder einer Situation, potenziell schädigende Auswirkungen zu haben, wenn ein Organismus, ein System oder eine (Teil-)Population diesem Stoff ausgesetzt ist.
IATA (Integrated Approach to Testing and Assessment)
: Ein strukturierter Ansatz zur Gefahrenidentifizierung (Potenzial), Gefahrencharakterisierung (Potenz) und/oder Sicherheitsbeurteilung (Potenzial/Potenz und Exposition) einer Chemikalie oder Gruppe von Chemikalien, der strategisch sämtliche relevanten Daten für eine informierte regulatorische Entscheidung hinsichtlich potenziellen Gefahren und/oder Risiken und/oder des Bedürfnisses weiterer gezielter und somit minimaler Tests integriert und abwägt.
Mediumkontrolle
: Ein unbehandeltes Replikat, das alle Komponenten eines Prüfsystems enthält. Diese Probe wird mit prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt, um festzustellen, ob das Lösungsmittel/Vehikel mit dem Prüfsystem interagiert.
Gemisch
: Ein Gemisch oder eine Lösung, die aus zwei oder mehreren Stoffen besteht.
Einkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorhanden ist.
Mehrkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens ≥ 10 % w/w und < 80 % w/w vorhanden sind. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet.
Positivkontrolle
: Ein Replikat, das alle Komponenten eines Prüfsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prähaptene
: Chemikalien, die erst nach einer abiotischen Umwandlung zu Sensibilisatoren werden
Prohaptene
: Chemikalien, die ihr Hautsensibilisierungspotenzial erst durch eine enzymatische Bioaktivierung erlangen
Relative Fluoreszenzintensität (RFI)
: Relative Werte der geometrischen mittleren Fluoreszenzintensität (MFI) in chemisch behandelten Zellen im Vergleich zu MFI in mit Lösungsmittel/Vehikel behandelten Zellen.
Relevanz
: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Sie berücksichtigt auch die Genauigkeit (Übereinstimmung) einer Prüfmethode (21).
Zuverlässigkeit
: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet (21).
Durchlauf
: Ein Durchlauf besteht aus einer oder mehreren hintereinander getesteten Prüfchemikalien mit einer Lösungsmittel-/Vehikelkontrolle und mit einer Positivkontrolle.
Sensitivität
: Der Anteil aller positiven/wirkungsvollen Chemikalien, die durch den Test korrekt eingestuft werden. Sie ist ein Maß der Genauigkeit eines Tests mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (21).
Färbepuffer
: Eine mit Phosphat gepufferte Kochsalzlösung mit 0,1 % Rinderserumalbumin.
Lösungsmittel-/Vehikelkontrolle
: Eine unbehandelte Probe, die sämtliche Komponenten eines Prüfsystems enthält, außer der Prüfchemikalie, aber einschließlich des verwendeten Lösungsmittels/Vehikels. Sie wird verwendet, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst oder stabil verteilt wurden, zu bestimmen. Wenn mit einer simultanen Medienkontrolle geprüft wird, zeigt diese Probe außerdem, ob das Lösungsmittel oder Vehikel mit dem Prüfsystem interagiert.
Spezifität
: Der Anteil aller negativen/wirkungslosen Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode (21).
Stoff
: Ein chemisches Element und seine Verbindungen im natürlichen Zustand oder durch ein Produktionsverfahren, die einen Zusatzstoff induzieren, der zur Erhaltung seiner Stabilität erforderlich ist, sowie Verunreinigungen, die aus dem verwendeten Prozess stammen, aber mit Ausnahme von Lösungsmitteln, die getrennt sein können, ohne die Stabilität des Stoffes zu beeinträchtigen oder seine Zusammensetzung zu ändern.
Prüfchemikalie
: Stoffe oder Gemische, die mit dieser Methode geprüft wurden.
Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS)
: Ein System, das die Einstufung von Chemikalien (Stoffen und Gemischen) gemäß normierten Typen und Stufen physikalischer, gesundheitlicher und Umweltgefahren vorschlägt und entsprechende Kommunikationselemente wie Piktogramme, Signalwörter, Gefahrenberichte, Berichte zu Vorsichtsmaßnahmen und Sicherheitsdatenblätter anspricht, damit Informationen zu Nebenwirkungen mit Blick auf den Schutz von Menschen (einschließlich Mitarbeitern, Arbeitern, Logistikern, Verbrauchern und Ersthelfern) und der Umwelt zu übermitteln (23).
UVCB
: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Gültiger Test
: Ein Test, von der angenommen wird, dass sie über ein ausreichendes Maß an Relevanz und Zuverlässigkeit für einen bestimmten Zweck verfügt und die auf wissenschaftlich bewährten Prinzipien basieren. Ein Test ist niemals in einem absoluten Sinn gültig, sondern nur im Verhältnis zu einem definierten Zweck (21).
Anlage 1.2
LEISTUNGSSTOFFE
Vor der routinemäßigen Anwendung des in dieser Anlage zur Prüfmethode B.71 beschriebenen Tests sollten Labors ihre technische Leistungsfähigkeit demonstrieren, indem sie die erwartete h-CLAT-Vorhersage für die in Tabelle 1 empfohlenen 10 Leistungsstoffe korrekt ermitteln und die CV75-, EC150- und EC200-Werte, die in den betreffenden Referenzbereich fallen, bei mindestens 8 der 10 Leistungsstoffe bestimmen. Diese Leistungsstoffe wurden so ausgewählt, dass sie die Bandbreite von Reaktionen im Hinblick auf die Gefahr einer Hautsensibilisierung repräsentieren. Weitere Auswahlkriterien betrafen die Erhältlichkeit der Chemikalien im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und die Verfügbarkeit hochwertiger In-vitro-Daten aus der h-CLAT-Prüfmethode. Außerdem sind veröffentlichte Referenzdaten für die h-CLAT-Methode verfügbar (3) (14).
Tabelle 1
Empfohlene Stoffe zum Nachweis der technischen Leistungsfähigkeit mit der h-CLAT-Methode
|
Leistungsstoffe
|
CAS-Nr.
|
Aggregat-zustand
|
In-vivo-Vorhersage
(103)
|
CV75 Referenz Bereich in μg/ml
(104)
|
h-CLAT-Ergebnisse für CD86 (EC150-Referenzbereich in μg/ml)
(104)
|
h-CLAT-Ergebnisse für CD54 (EC200-Referenzbereich in μg/ml)
(104)
|
|
2,4-Dinitrochlorbenzol
|
97-00-7
|
fest
|
Sensibilisator (extrem)
|
2–12
|
positiv (0,5–10)
|
positiv (0,5–15)
|
|
4-Phenylenediamin
|
106-50-3
|
fest
|
Allergen (stark)
|
5–95
|
positiv (< 40)
|
negativ (> 1,5) (105)
|
|
Nickelsulfat
|
10101-97-0
|
fest
|
Allergen (mäßig)
|
30–500
|
positiv (< 100)
|
positiv (10–100)
|
|
2-Mercaptbenzothiazol
|
149-30-4
|
fest
|
Allergen (mäßig)
|
30–400
|
negativ (> 10) (105)
|
positiv (10–140)
|
|
R(+)-Limonen
|
5989-27-5
|
flüssig
|
Allergen (schwach)
|
> 20
|
negativ (> 5) (105)
|
positiv (< 250)
|
|
Imidazolidinylharnstoff
|
39236-46-9
|
fest
|
Sensibilisator (schwach)
|
25–100
|
positiv (20–90)
|
positiv (20–75)
|
|
Isopropanol
|
67-63-0
|
flüssig
|
Nichsensibilisa-tor
|
> 5 000
|
negativ (> 5 000 )
|
negativ (> 5 000 )
|
|
Glyzerin
|
56-81-5
|
flüssig
|
Nichtsensibilisa-tor
|
> 5 000
|
negativ (> 5 000 )
|
negativ (> 5 000 )
|
|
Milchsäure
|
50-21-5
|
flüssig
|
Nichsensibilisa-tor
|
1500–5000
|
negativ (> 5000)
|
negativ (> 5000)
|
|
4-Aminobenzoesäure
|
150-13-0
|
fest
|
Nichsensibilisa-tor
|
> 1 000
|
negativ (> 1 000 )
|
negativ (> 1000)
|
|
Abkürzungen: CAS-Nr. = Registernummer des Chemical Abstracts Service
|
Anlage 2
IN-VITRO-HAUTSENSIBILISIERUNG: U937-AKTIVIERUNGSTEST MIT ZELLLINIEN (U-SENS™)
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
|
1.
|
Der U-SENS™-Test quantifiziert die Änderungen der Expression von Zelloberflächenmarkern in Zusammenhang mit dem Aktivierungsprozess von Monozyten und dendritischen Zellen (DC) (d. h. CD86) in der humanen histiozytären Lymphom-Zelllinie U937 nach einer Exposition gegenüber Allergenen (1). Die gemessenen Expressionswerte des Zelloberflächenmarkers CD86 in der Zelllinie U937 werden dann zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren verwendet.
|
|
2.
|
Der U-SENS™-Test wurde in einer von L‘Oreal koordinierten Validierungsstudie (2) und anschließend in einer unabhängigen Begutachtung durch den EURL ECVAM wissenschaftlichen beratenden Ausschuss (ESAC) (3) des Referenzlabors der europäischen Union für Alternativen zu Tierversuchen (EURL ECVAM) bewertet. Unter Berücksichtigung sämtlicher verfügbarer Nachweise und Informationen von Behörden und Interessenträgern wurde U-SENS™ von EURL ECVAM (4) empfohlen, um als Teil eines IATA verwendet zu werden, um die Unterscheidung zwischen Allergenen und Nichtsensibilisatoren zum Zweck der Gefahreneinstufung und Etikettierung zu unterstützen. In ihrem Leitfaden zur Berichterstattung strukturierter Ansätze zur Datenintegration und individuellen Informationsquellen, die innerhalb von IATA für die Hautsensibilisierung verwendet werden, diskutiert die OECD derzeit eine Anzahl an Fallstudien, die verschiedene Teststrategien und Vorhersagemodelle beschreiben. Einer der anders definierten Ansätze basiert auf dem U-SENS-Test (5). Beispiele für die Verwendung der U-SENS™-Daten in Kombination mit anderen Informationen, einschließlich historischer Daten und vorhandener gültiger menschlicher Daten (6), werden auch an anderer Stelle in der Literatur aufgeführt (4) (5) (7).
|
|
3.
|
Der U-SENS™-Test erwies sich als übertragbar auf Labore mit Erfahrung auf dem Gebiet der Zellkulturtechniken und der Durchflusszytometrieanalyse haben. Der Grad der Reproduzierbarkeit, der bei der Prüfmethode erwartet werden kann, liegt in der Größenordnung von 90 % und 84 % innerhalb und zwischen Labors (8). Die Ergebnisse der Validierungsstudie (8) und veröffentlichter Studien (1) weisen insgesamt darauf hin, dass die Genauigkeit bei der Unterscheidung von Hautsensibilatoren (d. h. UN-GHS-/CLP-Kategorie 1) gegenüber Nichtsensibilisatoren 86 % (N = 166) bei einer Empfindlichkeit von 91 % (118/129) und einer Spezifität von 65 % (24/37) im Vergleich zu den Ergebnissen des LLNA beträgt. Verglichen mit menschlichen Ergebnissen beträgt die Genauigkeit bei der Unterscheidung von Hautallergenen (d. h. UN-GHS/CLP Kat.1) von Nichtsensibilisatoren 77 % (N = 101) mit einer Sensitivität von 100 % (58/58) und einer Spezifität von 47 % (20/43). Im Vergleich zu LLNA ist bei falsch negativen Vorhersagen mit dem U-SENS™ die Chance größer, dass Chemikalien mit geringer bis mittlerer Hautsensibilisierungspotenz betroffen sind (d. h. UN-GHS/CLP-Unterkategorie 1B), als Chemikalien mit hoher Hautsensibilisierungspotenz (d. h. UN-GHS/CLP-Unterkategorie 1A) (1) (8) (9). Zusammenfassend geben diese Informationen an, wie nützlich sich der U-SENS™-Test bei der Beteiligung an der Identifikation von Hautsensibilisierungsgefahren erweist. Jedoch sind die Genauigkeitswerte, die hier für U-SENS™ als eigenständiger Test angegeben werden, lediglich als Anhaltspunkte zu betrachten, da die Prüfmethode in Kombination mit anderen Informationsquellen im Rahmen eines IATA sowie gemäß den Bestimmungen unter Nummer 7 und 8 der allgemeinen Einleitung betrachtet werden sollte. Darüber hinaus sollte bei der Bewertung von Prüfmethoden zur Hautsensibilisierung ohne Tierversuche beachtet werden, dass der LLNA-Test sowie andere Tierversuche die Situation bei Menschen nicht vollständig widerspiegeln.
|
|
4.
|
Auf der Basis der aktuell verfügbaren Daten wurde gezeigt, dass der U-SENS™-Test auf Prüfchemikalien angewandt werden kann (einschließlich Zutaten von Kosmetika, z. B. Konservierungsmittel, Tenside, Aktivstoffe, Farbstoffe), die eine Vielfalt an organischen Funktionsgruppen, physikalisch-chemischen Eigenschaften, Hautsensibilisierungspotenzial (wie in In-vivo-Studien festgelegt) und das Spektrum der Wirkmechanismen, das für seine Assoziation mit der Hautsensibilisierung bekannt ist, abdecken (d. h. Michael-Akzeptor, Schiff-Basisbildung, Acylübertragungsmittel, Substitution nukleophil bimolekular [SN2] oder nukleophile aromatische Substitution [SNAr]) (1) (8) (9) (10). Der U-SENS™-Test gilt für Prüfchemikalien, die löslich sind oder eine stabile Dispersion (d. h. ein Kolloid oder eine Suspension, in denen die Prüfchemikalie sich nicht absetzt oder vom Lösungsmittel/Vehikel in verschiedene Phasen trennt) in einem geeigneten Lösungsmittel/Vehikel bilden (siehe Abschnitt 13). Im Datensatz eingetragene Chemikalien, die Prähaptene (d. h. durch Oxidierung aktivierte Stoffe) oder Prohaptene (d. h. Stoffe, die eine enzymatische Aktivierung erfordern, wie beispielsweise über P450-Enzyme) sind, wurden von U-SENS™ korrekt vorhergesagt (1) (10).Membranschädigende Stoffe können aufgrund einer nichtspezifischen Erhöhung der CD86-Expression zu falsch positiven Ergebnissen führen, da 3 von 7 falsch positiven Ergebnissen in Bezug auf die In-vivo-Referenzeinstufung Tenside waren (1). Solche positiven Ergebnisse mit Tensiden sollen mit Vorsicht betrachtet werden, während negative Ergebnisse mit Tensiden weiterhin genutzt werden können, um die Identifizierung der Prüfchemikalie als Nichtsensibilisator zu unterstützen. Fluoreszierende Prüfchemikalien können mit U-SENS™ (1) bewertet werden. Dennoch beeinträchtigen stark fluoreszierende Prüfchemikalien, die mit derselben Wellenlänge ausstrahlen wie Fluorescein-Isothiocyanat (FITC) oder Propidiumjodid (PI) die durchflusszytometrische Erkennung und können daher nicht korrekt mithilfe von mit FITC-konjugierten Antikörpern (potenziell falsch negativ) oder PI (Viabilität nicht messbar) bewertet werden. In einem solchen Fall können andere mit Fluorochrom markierte Antikörper bzw. andere Zytotoxizitätsmarker verwendet werden, solange aufgezeigt werden kann, dass sie ähnliche Ergebnisse liefern wie die mit FITC markierten Antikörper oder PI (siehe Abschnitt 18), z. B. durch Testen der Leistungsstoffe in Anlage 2.2. Angesichts der vorstehenden Ausführungen sollten positive Ergebnisse mit Tensiden und negative Ergebnisse mit stark fluoreszierenden Prüfchemikalien im Kontext der angegebenen Grenzen und in Verbindung mit anderen Informationsquellen im Rahmen von IATA interpretiert werden. In Fällen, in denen die Nichtanwendbarkeit des U-SENS™-Tests bei anderen spezifischen Kategorien von Prüfchemikalien nachgewiesen wird, sollte sie bei diesen spezifischen Kategorien nicht verwendet werden.
|
|
5.
|
Der U-SENS™-Test unterstützt, wie bereits oben erwähnt, die Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren. Er kann jedoch bei Verwendung im Rahmen von integrierten Ansätzen wie IATA möglicherweise auch zur Bewertung der Sensibilisierungspotenz beitragen. Es sind allerdings weitere Untersuchungen erforderlich, vorzugsweise auf der Grundlage von Humandaten, um herauszufinden, inwieweit die Ergebnisse von U-SENS™ möglicherweise zur Potenzbewertung herangezogen werden können.
|
|
6.
|
Definitionen sind Anlage 2.1 zu entnehmen.
|
Prinzip Der Prüfmethode
|
7.
|
Der U-SENS™-Test ist ein In-vitro-Test, der Änderungen der Expression von CD86-Zelloberflächenmarkern auf einer humanen histiozytären Lymphom-Zelllinie, U937-Zellen, nach einer Exposition von 45 ± 3 Stunden gegenüber der Prüfchemikalie quantifiziert. Der Oberflächenmarker CD86 ist ein typischer Marker der U937-Aktivierung. CD86 ist dafür bekannt, ein ko-stimulierendes Molekül zu sein, das eine monozytische Aktivierung nachahmen kann, die eine wichtige Rolle beim T-Zellen-Priming spielt. Die Änderungen der Expression von Oberflächenmarkern von CD86 werden anhand der Durchflusszytometrien nach einer Zellfärbung, die typischerweise mit Antikörpern erfolgt, die mit Fluorescein-Isothiocyanat (FITC) markiert wurden, gemessen. Die Zytotoxizitätsmessung wird ebenfalls zur gleichen Zeit durchgeführt (z. B. durch die Verwendung von PI), um zu beurteilen, ob die Hochregulierung der Expression von CD86-Zelloberflächenmarkern bei Konzentrationen unterhalb des zytotoxischen Werts auftritt. Der Stimulationsindex (S.I.) des CD86-Zelloberflächenmarkers im Vergleich zur Lösungsmittel-/Vehikelkontrolle wird berechnet und im Vorhersagemodell verwendet (siehe Abschnitt 19), um die Unterscheidung zwischen Allergenen und Nichsensibilisatoren zu unterstützen.
|
Nachweis Der Kompetenz
|
8.
|
Vor der routinemäßigen Verwendung des unter dieser Anlage zur Prüfmethode B.71 beschriebenen Tests sollten Laboratorien ihre technische Kompetenz anhand der zehn in Anlage 2.2 aufgeführten Leistungsstoffe in Übereinstimmung mit bewährten In-vitro-Verfahren nachweisen (11). Darüber hinaus sollten Testbenutzer eine historische Datenbank der mit den Reaktivitätstests (siehe Abschnitt 11) und mit den positiven and Lösungsmittel-/Vehikelkontrollen (siehe Abschnitte 15-16) erzeugten Daten pflegen und diese Daten nutzen, um zu bestätigen, dass die Reproduzierbarkeit des Tests in ihrem Labor mit der Zeit aufrechterhalten wird.
|
Verfahren
|
9.
|
Dieser Test basiert auf dem U-SENS™ DataBase-Dienst zu Alternativmethoden für Tierversuche (DB-ALM) Protokoll Nr. 183 (12). Die Standardarbeitsanweisungen (SOP) sollten bei der Umsetzung und Verwendung des U-SENS™-Tests im Labor angewandt werden. Es kann ein automatisiertes System zur Durchführung von U-SENS™ verwendet werden, wenn aufgezeigt werden kann, dass es ähnliche Ergebnisse liefert, beispielsweise durch Testen der Leistungsstoffe in Anlage 2.2. Nachfolgend werden die wichtigsten Komponenten und Verfahren für den U-SENS™-Test beschrieben.
|
Vorbereitung der Zellen
|
10.
|
Die humane histiozytäre Lymphom-Zelllinie U937 (13) sollte zur Durchführung des U-SENS™-Tests verwendet werden. Zellen (Klon CRL1593.2) sollten aus einer gut qualifizierten Zellbank wie der American Type Culture Collection entnommen werden.
|
|
11.
|
U937-Zellen werden bei 37 °C unter 5 % CO2 und einer befeuchteten Atmosphären in einem mit 10 % fetales Kälberserum (FCS), 2 mM L-Glutamin, 100 Einheiten/ml Penicillin und 100 μg/ml Streptomycin angereicherten RPMI-1 640-Medium (vollständiges Medium) kultiviert. U937-Zellen werden regelmäßig alle 2–3 Tage bei einer Dichte von 1,5 bzw. 3 × 105 Zellen/ml passagiert. Die Zelldichte sollte 2 × 106 Zellen/ml nicht überschreiten und die Zellviabilität, die anhand des Trypanblau-Ausschlusses gemessen wird, sollte ≥ 90 % betragen (nach dem Auftauen nicht an der ersten Passage anwenden). Vor ihrer Verwendung zu Testzwecken sollte jede Charge von Zellen, FCS oder Antikörpern qualifiziert werden, indem ein Reaktivitätstest durchgeführt wird. Der Reaktivitätstest der Zellen sollte mithilfe der Positivkontrolle, Picrylsulfonsäure (2,4,6-Trinitrobenzolsulfonsäure: TNBS) (CASRN 2 508-19-2, ≥ 99 % Reinheit) und der Negativkontrolle Milchsäure (LA) (CASRN 50-21-5, ≥ 85 % Reinheit) mindestens eine Woche nach dem Auftauen durchgeführt werden. Beim Reaktivitätstest sollten sechs endgültige Konzentrationen für jede der 2 Kontrollen geprüft werden (TNBS: 1, 12,5, 25, 50, 75, 100 μg/ml und LA: 1, 10, 20, 50, 100, 200 μg/ml). In vollständigem Medium gelöste TNBS sollte eine positive und von der Konzentration abhängige Wirkung auf CD86 zeigen (z. B. wenn auf eine positive Konzentration, CD86 S.I. ≥ 150, eine Konzentration mit steigendem CD86 S.I folgt), und in vollständigem Medium gelöste LA sollte eine negative Wirkung auf CD86 zeigen (siehe Abschnitt 21). Nur die Chargen von Zellen, welche den Reaktivitätstest zwei Mal bestanden haben, dürfen für den Test verwendet werden. Zellen können bis zu sieben Wochen nach dem Auftauen gewonnen werden. Die Anzahl an Passagen sollte 21 nicht übersteigen. Der Reaktivitätstest sollte gemäß den in den Abschnitten 18–22 beschriebenen Verfahren durchgeführt werden.
|
|
12.
|
Zur Prüfung werden die U937-Zellen bei einer Dichte von entweder 3 × 105 Zellen/ml oder 6 × 105 Zellen/ml ausgesät und in Kulturkolben 2 bzw. 1 Tag lang vorgezüchtet. Andere vorgezüchtete Bedingungen als diejenigen, die oben beschrieben werden, können verwendet werden, wenn eine ausreichende wissenschaftliche Begründung vorliegt und wenn gezeigt werden kann, dass ähnliche Ergebnisse geliefert werden, beispielsweise durch Testen der Leistungsstoffe in Anlage 2.2. Am Testtag werden die Zellen, die aus dem Kulturkolben geerntet wurden, mit einem frischen Kulturmedium mit 5 × 105 Zellen/ml neu angesetzt. Dann werden die Zellen in einer flachen Platte mit 96 Mulden mit 100 μl verteilt (endgültige Zelldichte von 0,5 × 105 Zellen/Mulde).
|
Vorbereitung der Prüfchemikalien und Kontrollstoffe
|
13.
|
Die Beurteilung der Löslichkeit wird vor dem Test vorgenommen. Zu diesem Zweck werden die Prüfchemikalien bei einer Konzentration von 50 mg/ml in vollständigem Medium als erste Lösungsmitteloption oder Dimethylsulfoxid (DMSO,≥ 99 % Reinheit) als zweite Lösungsmittel-/Vehikeloption gelöst oder stabil verteilt, wenn die Prüfchemikalie nicht im Lösungsmittel/Vehikel des vollständigen Mediums löslich ist. Beim Test wird die Prüfchemikalie auf eine endgültige Konzentration von 0,4 mg/ml im vollständigen Medium gelöst, wenn die Chemikalie in diesem Lösungsmittel/Vehikel löslich ist. Wenn die Chemikalie nur in DMSO löslich ist, wird die Chemikalie bei einer Konzentration von 50 mg/ml gelöst. Andere als die oben beschriebenen Lösungsmittel/Vehikel können verwendet werden, wenn eine ausreichende wissenschaftliche Begründung gegeben ist. Die Stabilität der Prüfchemikalie im endgültigen Lösungsmittel/Vehikel sollte berücksichtigt werden.
|
|
14.
|
Die Prüfchemikalien sowie die Kontrollstoffe werden am Tag des Tests vorbereitet. Da kein Test zur Dosisfindung durchgeführt wird, sollten für den ersten Durchlauf 6 endgültige Konzentrationen (1, 10, 20, 50, 100 und 200 μg/ml) im entsprechenden Lösungsmittel/Vehikel entweder in vollständigem Medium oder in 0,4 % DMSO im Medium geprüft werden. Bei den nachfolgenden Durchläufen sollten, ausgehend von den 0,4 mg/ml im vollständigen Medium oder 50 mg/ml in DMSO-Lösungen der Prüfchemikalie, mindestens 4 Arbeitslösungen mithilfe des entsprechenden Lösungsmittels/Vehikels vorbereitet werden (d. h. mindestens 4 Konzentrationen). Die Arbeitslösungen werden schließlich für die Behandlung verwendet, indem eine gleiche Menge der U937-Zellsuspension (siehe Abschnitt 11 oben) zum Volumen der Arbeitslösung in der Platte hinzugefügt wird, um eine weitere zweifache Verdünnung zu erhalten (12). Die Konzentrationen (mindestens 4 Konzentrationen) für weitere Durchläufe werden auf der Grundlage der individuellen Ergebnisse sämtlicher vorheriger Durchläufe gewählt (8). Die nutzbaren endgültigen Konzentrationen sind 1, 2, 3, 4, 5, 7.5, 10, 12.5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 und 200 μg/ml. Die höchste endgültige Konzentration beträgt 200 μg/ml.Wenn ein CD86 positiver Wert bei 1 μg/ml beobachtet wird, dann werden 0,1 μg/ml bewertet, um die Konzentration der Prüfchemikalie zu finden, die CD86 nicht über dem positiven Schwellenwert induziert. Bei jedem Durchlauf wird der EC150 (Konzentration, bei der eine Chemikalie den positiven Schwellenwert für CD86 von 150 % erreicht, siehe Abschnitt 19) berechnet, falls eine positive Konzentrationswirkung für CD86 beobachtet wird. Wo die Prüfchemikalie eine positive CD86-Wirkung induziert, die nicht von der Konzentration abhängig ist, ist eine Berechnung von EC150 eventuell nicht relevant, wie dies im U-SENS™ DB-ALM Protokoll Nr. 183 (12) beschrieben wird. Bei jedem Durchlauf wird CV70 (Konzentration, bei der eine Chemikalie die Zytotoxizitätsschwelle von 70 % erreicht, siehe Abschnitt 19) wo immer möglich berechnet (12). Um den Konzentrationswirkungseffekt der CD86-Steigung zu untersuchen sollten Konzentrationen aus den verwendbaren Konzentrationen gleichmäßig zwischen EC150 (oder der höchsten CD86 negativen, nicht zytotoxischen Konzentration) und CV70 (oder der höchsten zulässigen Konzentration, d. h. 200 μg/ml) verteilt gewählt werden. Es sollten pro Durchlauf mindestens 4 Konzentrationen geprüft werden, wobei zu Vergleichszwecken mindestens 2 Konzentrationen mit den vorherigen Durchläufen übereinstimmen sollten.
|
|
15.
|
Die im U-SENS™-Test verwendete Lösungsmittel-/Vehikelkontrolle ist ein vollständiges Medium (für gelöste oder stabil im vollständigen Medium verteilte Prüfchemikalien) (siehe Abschnitt 4) oder 0,4 % DMSO in vollständigem Medium (für in DMSO gelöste oder stabil verteilte Prüfchemikalien).
|
|
16.
|
Die im U-SENS™-Test verwendete Positivkontrolle ist TNBS (siehe Abschnitt 11), die in vollständigem Medium vorbereitet wird. TNBS sollte als Positivkontrolle für die CD86-Expressionsmessung mit einer endgültigen einzelnen Konzentration in der Platte (50 μg/ml) verwendet werden, die > 70 % der Zellviabilität erbringt. Um eine Konzentration von 50 μg/ml TNBS in der Platte zu erhalten, wird eine Stammlösung von 1M (d. h. 293 mg/ml) TNBS im vollständigen Medium vorbereitet und 2 930-fach mit dem vollständigen Medium in einer Arbeitslösung von 100 μg/ml verdünnt. Milchsäure (LA, CAS 50-21-5) sollte bei 200 μg/ml in vollständigem Medium gelöst als Negativkontrolle verwendet werden (von einer Stammlösung von 0,4 mg/ml). In jeder Platte jedes Durchlaufs werden drei Replikate der unbehandelten Kontrolle, Lösungsmittel-/Vehikelkontrolle, Positiv- und Negativkontrollen im vollständigen Medium vorbereitet (12). Andere geeignete Positivkontrollen können verwendet werden, sofern historische Daten für die Ableitung vergleichbarer Akzeptanzkriterien für einen Testdurchlauf zur Verfügung stehen. Die Akzeptanzkriterien des Durchlaufs entsprechen denen, die für die Prüfchemikalie beschrieben wurden (siehe Abschnitt 12).
|
Applikation der Prüfchemikalien und Kontrollstoffe
|
17.
|
Die Lösungsmittel-/Vehikelkontrolle oder die Arbeitslösungen, die in den Abschnitten 14–16 beschrieben werden, werden 1:1 (v/v) mit den in der flachen Platte mit 96 Mulden vorbereiteten Zellsuspensionen gemischt (siehe Abschnitt 12). Die behandelten Platten werden dann 45 ± 3 Stunden bei 37 °C unter 5 % CO2 inkubiert. Vor der Inkubation werden die Platten mit einer semipermeablen Membran versiegelt, um eine Verdampfung von flüchtigen Prüfchemikalien und eine Kreuzkontaminierung zwischen den mit den Prüfchemikalien behandelten Zellen zu vermeiden (12).
|
Zellfärbung
|
18.
|
Nach einer Expositionszeit von 45 ± 3 Stunden werden die Zellen in eine V-förmige Mikrotiterplatte übertragen und durch Zentrifugierung gesammelt. Die Löslichkeitsinterferenz ist definiert als Kristalle oder Tropfen, die 45 ± 3 Stunden nach der Behandlung (vor der Zellfärbung) unter dem Mikroskop beobachtet werden. Die Tenside werden entsorgt und die verbleibenden Zellen werden einmal mit 100 μl eiskalter mit Phosphat gepufferter Kochsalzlösung (PBS) mit 5 % fetalem Kälberserum (Färbepuffer) gewaschen. Nach der Zentrifugierung werden die Zellen mit 100 μl Färbepuffer resuspensiert und mit 5 μl (z. B. 0,25 μg) mit FITC markierten anti-CD86- oder Maus-IgG1-Antikörpern (Isotyp) bei 4 °C 30 Minuten lang vor Licht geschützt. Die im U-SENS™ DB-ALM Protokoll Nr. 183 (12) beschriebenen Antikörper sollten verwendet werden (für CD86: BD-PharMingen #5556 57 Klon: Fun-1, oder Caltag/Invitrogen # MHCD8601 Klon: BU63; und für IgG1: BD-PharMingen #5557 48, oder Caltag/Invitrogen # GM4992). Nach Erfahrung der Testentwickler ist die Fluoreszenzintensität der Antikörper normalerweise zwischen verschiedenen Chargen konsistent. Andere Klone oder Lieferanten der Antikörper, welche den Reaktivitätstest bestanden haben, können für den Test verwendet werden (siehe Abschnitt 11). Benutzer können jedoch erwägen, die Antikörper unter den Bedingungen im eigenen Labor zu titrieren, um die beste Gebrauchskonzentration zu definieren. Andere Erkennungssystem, z. B.mit Fluorochrom markierte anti-CD86-Antikörper, können verwendet werden, wenn gezeigt werden kann, dass sie ähnliche Ergebnisse wie mit FITC konjugierte Antikörper liefern, beispielsweise durch Testen der Leistungsstoffe in Anlage 2.2. Die Zellen werden zweimal mit 100 μl Färbepuffer und einmal mit 100 μl einer eiskalten PBS gewaschen und anschließend in eiskalter PBS resuspensiert (z. B. 125 μl für Proben, die manuell Röhrchen für Röhrchen analysiert werden, oder 50 μl mithilfe einer Autosampler-Platte) und PI-Lösung wird hinzugefügt (endgültige Konzentration von 3 μg/ml). Andere Zytotoxizitätsmarker wie 7-Aminoactinomycin D (7-AAD) oder Trypanblau können verwendet werden, wenn gezeigt werden kann, dass die alternativen Farbstoffe ähnliche Ergebnisse liefern wie PI, zum Beispiel durch Testen der Leistungsstoffe in Anlage 2.2.
|
Durchflusszytometrieanalyse
|
19.
|
Die Expressionswerte von CD86 und die Zellviabilität werden mithilfe der Durchflusszytometrie analysiert. Zellen werden in einem Größen- (FSC) und Granularitäts- (SSC) Dot-Plot angezeigt, der auf eine log-Skala eingestellt ist, um die Population eindeutig in einem ersten Gate R1 zu identifizieren und die Verschmutzung zu beseitigen. Für jede Mulde wird eine Sollgesamtmenge von 10 000 Zellen in Gate R1 aufgenommen. Zellen vom selben R1-Gate werden in einem FL3- oder FL4 / SSC-Dot-Plot angezeigt. Lebensfähige Zellen werden dargestellt, indem ein zweites Gate R2 platziert wird, das die Population von Propidiumjodid-negativen Zellen auswählt (FL3- oder FL4-Kanal). Die Zellviabilität kann anhand der folgenden Gleichung über das Zytometeranalyseprogramm berechnet werden. Wenn die Zellviabilität gering ist, könnten bis zu 20 000 Zellen einschließlich toter Zellen aufgenommen werden. Alternativ können die Daten eine Minute nach der Einleitung der Analyse aufgenommen werden.

Die Prozentzahl der FL1-positiven Zellen wird dann unter diesen lebensfähigen Zellen gemessen, die in R2 eingeschlossen sind (innerhalb von R1). Die Zelloberflächenexpression von CD86 wird in einem FL1- / SSC-Dot-Plot analysiert, das auf lebensfähigen Zellen eingeschlossen ist (R2).
Bei den vollständiges Medium/IgG1-Mulden befindet sich der Analysemarker in der Nähe der Hauptpopulation, sodass die Kontrollen des vollständigen Mediums einen IgG1 im Sollbereich von 0,6 bis 0,9 % aufweisen.
Die Farbinterferenz wird definiert als eine Verlagerung des mit FITC markierten IgG1-Dot-Plots (IgG1 FL1 geo. Mittelw. S.I. ≥ 150 %).
Der Stimulationsindex (S.I.) von CD86 für Kontrollzellen (unbehandelt oder in 0,4 % DMSO) und chemisch behandelte Zellen wird gemäß der folgenden Gleichung berechnet:

% von IgG1+ unbehandelte Kontrollzellen: bezeichnet als Prozentzahl der FL1-positiven IgG1-Zellen, die mit dem Analysemarker (zulässiger Bereich von ≥ 0,6 % und < 1,5 %, siehe Abschnitt 22) unter den lebensfähigen unbehandelten Zellen definiert wurden.
% von IgG1+/CD86+ Kontrolle/behandelte Zellen: bezeichnet als Prozentzahl von FL1-positiven IgG1/CD86-Zellen, die ohne Verlagerung des Analysemarkers unter den lebensfähigen Kontroll-/behandelten Zellen gemessen werden.
|
DATEN UND BERICHTERSTATTUNG
Datenauswertung
|
20.
|
Die folgenden Parameter werden im U-SENS™-Test berechnet: CV70-Wert, d. h. eine Konzentration, die 70 % des U937-Zellüberlebens (30 % Zytotoxizität) zeigt, und der EC150-Wert, d. h. die Konzentration, bei der die Prüfchemikalien einen CD86-Stimulationsindex (S.I.) von 150 % induzierten.
CV70 wird anhand der log-linearen Interpolation mithilfe der folgenden Gleichung berechnet:
CV70 = C1 + [(V1 - 70) / (V1 – V2) * (C2 – C1)]
Dabei gilt:
V1 ist der Mindestwert der Zellviabilität über 70 %
V2 ist der Höchstwert der Zellviabilität unter 70 %
C1 und C2 sind die Konzentrationen, die den Wert der Zellviabilität V1 bzw. V2 zeigen.

Weitere Ansätze zur Ableitung von CV70 können angewandt werden, solange demonstriert wird, dass dies keinen Einfluss auf die Ergebnisse hat (z. B. durch Testen der Leistungsstoffe).
EC150 wird anhand der log-linearen Interpolation mithilfe der folgenden Gleichung berechnet:
EC150 = C1 + [(150 – S.I.1) / (S.I.2 – S.I.1) * (C2 – C1)]
Dabei gilt:
C1 ist die höchste Konzentration in μg/ml mit einem CD86 S.I. < 150 % (S.I. 1)
C2 ist die niedrigste Konzentration in μg/ml mit einem CD86 S.I. ≥ 150 % (S.I. 2)
Die EC150- und CV70-Werte wurden für jeden
|
—
|
Durchlauf berechnet: die individuellen EC150- und CV70-Werte wurden als Werkzeuge zur Untersuchung des Konzentrationswirkungseffekts der CD86-Steigung verwendet (siehe Abschnitt 14),
|
|
—
|
gestützt auf die durchschnittlichen Lebensfähigkeiten wird der CV70-Gesamtwert bestimmt (12),
|
|
—
|
gestützt auf die durchschnittlichen S.I. der CD86-Werte wird der EC150-Gesamtwert für die mit U-SENS™ als POSITIV vorhergesagte Prüfchemikalie bestimmt (siehe Abschnitt 21) (12).
|
|
Vorhersagemodell
|
21.
|
Bei der CD86-Expressionsmessung wird jede Prüfchemikalie in mindestens vier Konzentrationen und in mindestens zwei unabhängigen Durchläufen geprüft (an unterschiedlichen Tagen durchgeführt), um eine einzelne Vorhersage abzuleiten (NEGATIV oder POSITIV).
|
—
|
Die individuelle Schlussfolgerung eines U-SENS™-Durchlaufs wird als Negativ (nachfolgend als „N“ bezeichnet) angesehen, wenn der S.I. von CD86 bei allen nicht-zytotoxischen Konzentrationen weniger als 150 % beträgt (Zellviabilität ≥ 70 %) und wenn keine Beeinträchtigung beobachtet wird (Zytotoxizität, Löslichkeit: siehe Abschnitt 18 oder Farbe: siehe Abschnitt 19 unabhängig der nicht-zytotoxischen Konzentrationen, bei denen die Beeinträchtigung erkannt wird). In allen anderen Fällen: S.I. von CD86 höher als oder gleich 150 % und/oder Beeinträchtigungen beobachtet, dann wird die individuelle Schlussfolgerung eines U-SENS™-Durchlaufs als Positiv (nachfolgend als „P“ bezeichnet) angesehen.
|
|
—
|
Eine U-SENS™-Vorhersage wird als NEGATIV angesehen, wenn mindestens zwei unabhängige Durchläufe negativ (N) sind (Abbildung 1). Wenn die ersten zwei Durchläufe beide negativ (N) sind, dann wird die U-SENS™-Vorhersage als NEGATIV angesehen und ein dritter Durchlauf ist nicht erforderlich.
|
|
—
|
Eine U-SENS™-Vorhersage wird als POSITIV angesehen, wenn mindestens zwei unabhängige Durchläufe positiv (P) sind (Abbildung 1). Wenn die ersten zwei Durchläufe beide positiv (P) sind, dann wird die U-SENS™-Vorhersage als POSITIV angesehen und ein dritter Durchlauf ist nicht erforderlich.
|
|
—
|
Da kein Test zur Dosisfindung durchgeführt wird, gibt es eine Ausnahme, wenn der S.I. von CD86 im ersten Durchlauf höher als oder gleich 150 % bei ausschließlich der höchsten, nicht-zytotoxischen Konzentration ist. Der Durchlauf wird dann als NICHT SCHLÜSSIG angesehen und zusätzliche Konzentrationen (zwischen der höchsten nicht-zytotoxischen Konzentration und der niedrigsten nicht-zytotoxischen Konzentration – siehe Abschnitt 20) sollten in zusätzlichen Durchläufen geprüft werden. Wenn ein Durchlauf als nicht schlüssig identifiziert wird, sollten mindestens 2 zusätzliche Durchläufe vorgenommen werden; ein vierter Durchlauf ist erforderlich, falls die Durchläufe 2 und 3 nicht konkordant sind (N und/oder P unabhängig voneinander) (Abbildung 1). Folgedurchläufe werden auch dann als positiv angesehen, wenn nur eine nicht-zytotoxische Konzentration ein CD86 von 150 % oder mehr ergibt, da die Konzentrationseinstellung für die spezifische Prüfchemikalie angepasst wurde. Die endgültige Vorhersage basiert auf dem Mehrheitsergebnis der drei oder vier individuellen Durchläufe (d. h. 2 von 3 oder 2 von 4) (Abbildung 1).
|

Abb. 1: Beim U-SENS™-Test verwendetes Vorhersagemodell. Eine U-SENS™-Vorhersage sollte im Rahmen eines IATA sowie gemäß den Bestimmungen der Nummer 4 sowie der Nummern 7, 8 und 9 der allgemeinen Einleitung in Betracht gezogen werden
N: Durchlauf ohne Feststellung von CD86 als positiv oder mit Interferenz;
P: Durchlauf mit Feststellung von CD86 als positiv und/oder mit Interferenz(en);
NC: Nicht schlüssig. Erster Durchlauf als nicht schlüssig, wenn CD86 nur bei der höchsten nicht-zytotoxischen Konzentration positiv ist;
#: Eine nicht schlüssige individuelle Schlussfolgerung, die ausschließlich dem ersten Durchlauf zugeschrieben wird, führt automatisch zum Erfordernis eines dritten Durchlaufs, um eine Mehrheit von entweder Positiven (P) oder Negativen (N) Schlussfolgerungen in mindestens 2 von 3 unabhängigen Durchläufen zu erzielen.
$: Die Kästchen zeigen die relevanten Kombinationen der Ergebnisse aus den drei Durchläufen auf der Grundlage der Ergebnisse aus den ersten zwei Durchläufen, die im obigen Kästchen dargestellt sind.
°: Die Kästchen zeigen die relevanten Kombinationen der Ergebnisse aus den vier Durchläufen auf der Grundlage der Ergebnisse aus den ersten drei Durchläufen, die im obigen Kästchen dargestellt sind.
|
Akzeptanzkriterien
|
22.
|
Bei der Verwendung des U-SENS™-Tests sollten die folgenden Akzeptanzkriterien erfüllt werden (12).
|
—
|
Am Ende des Expositionszeitraums von 45 ± 3 Stunden musste die Hauptviabilität der dreifach ausgefertigten unbehandelten U937-Zellen > 90 % sein und es wurde keine Verschiebung der CD86-Expression beobachtet. Die basale CD86-Expression unbehandelter U937-Zellen musste im Bereich von ≥ 2 % und ≤ 25 % liegen.
|
|
—
|
Wenn DMSO als ein Lösungsmittel verwendet wird, dann wird die Gültigkeit der DMSO-Vehikelkontrolle durch die Berechnung eines DMSO S.I. im Vergleich zu unbehandelten Zellen bewertet und die Viabilität der dreifach ausgeführten Zellen musste > 90 % liegen. Die DMSO-Vehikelkontrolle ist gültig, wenn der Mittelwert des dreifach ausgeführten CD86 S.I. kleiner als 250 % des Mittelwerts des dreifach ausgeführten CD86 S.I. von unbehandelten U937-Zellen war.
|
|
—
|
Die Durchläufe werden als gültig angesehen, wenn mindestens zwei von drei IgG1-Werte von unbehandelten U937-Zellen in den Bereich von ≥ 0,6 % und < 1,5 % gefallen sind.
|
|
—
|
Die gleichzeitig geprüfte Negativkontrolle (Milchsäure) wird als gültig angesehen, wenn mindestens zwei der drei Replikate negativ (CD86 S.I. < 150 %) und nicht-zytotoxisch waren (Zellviabilität ≥ 70 %).
|
|
—
|
Die Positivkontrolle (TNBS) wurde als gültig angesehen, wenn mindestens zwei der drei Replikate positiv (CD86 S.I. ≥ 150 %) und nicht-zytotoxisch waren (Zellviabilität ≥ 70 %).
|
|
Prüfbericht
|
23.
|
Der Prüfbericht sollte folgende Angaben enthalten.
|
Prüfchemikalie
Einkomponentiger Stoff:
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
physikalisches Erscheinungsbild, Löslichkeit in Wasser, Löslichkeit in DMSO, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, im verfügbaren Umfang;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
Begründung der Auswahl des Lösungsmittels/Vehikels für jede Prüfchemikalie.
|
Mehrkomponentiger Stoff, UVCB-Stoff und Gemisch:
|
Charakterisierung, so weit wie möglich, z. B. durch die chemische Zusammensetzung (siehe oben), Reinheit, das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften (siehe oben) der einzelnen Komponenten, soweit verfügbar;
|
|
physikalisches Erscheinungsbild, Löslichkeit in Wasser, Löslichkeit in DMSO und weitere relevante physikalisch-chemische Eigenschaften, im verfügbaren Umfang;
|
|
Molekulargewicht oder scheinbares Molekulargewicht im Fall von Gemischen/Polymeren mit bekannter Zusammensetzung oder andere für die Durchführung der Studie relevante Informationen;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
Begründung der Auswahl des Lösungsmittels/Vehikels für jede Prüfchemikalie.
|
|
|
Kontrollen
Positivkontrolle
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
physikalisches Erscheinungsbild, Löslichkeit in DMSO, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern verfügbar und falls zutreffend;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
ggf. Verweis auf historische Ergebnisse von Positivkontrollen, die geeignete Akzeptanzkriterien für einen Testdurchlauf dokumentieren.
|
Negativ- und Lösungsmittel-/Vehikelkontrolle:
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
Aussehen, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern andere Kontroll-Lösungsmittel/Vehikel als die in der Prüfrichtlinie genannten verwendet werden und soweit verfügbar;
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
Begründung der Auswahl des Lösungsmittels/Vehikels für jede Prüfchemikalie.
|
|
|
Prüfbedingungen
|
Name und Anschrift des Auftraggebers, der Prüfanstalt und des Studienleiters;
|
|
Beschreibung des verwendeten Prüfprotokolls;
|
|
verwendete Zelllinie, zugehörige Lagerbedingungen und Quelle (z. B. Einrichtung, von der sie gewonnen wurden);
|
|
die verwendete Durchflusszytometrie (z. B. Modell), einschließlich der Geräteeinstellungen, Antikörper und verwendeten Zytotoxizitätsmarker;
|
|
das angewandte Verfahren zum Nachweis der Kompetenz des Labors bei der Durchführung des Tests durch Prüfen der Leistungsstoffe) und das angewandte Verfahren zum Nachweis der reproduzierbaren Leistung des Tests im Zeitverlauf, z. B. historische Kontrolldaten und/oder Daten zu historischen Reaktivitätstests.
|
|
|
Validitätskriterien
|
Zellviabilität und CD86 S.I.-Werte aus der Lösungsmittel-/Vehikelkontrolle im Vergleich zu den Validitätsbereichen;
|
|
Zellviabilität und S.I.-Werte aus der Positivkontrolle im Vergleich zu den Validitätsbereichen;
|
|
Zellviabilität aller geprüften Konzentrationen der geprüften Chemikalie.
|
|
|
Prüfverfahren
|
Anzahl der vorgenommenen Durchläufe;
|
|
Konzentrationen der Prüfchemikalie, Applikationen und angewandte Expositionsdauer (falls von den Empfehlungen abweichend)
|
|
Beschreibung der angewandten Bewertungs- und Entscheidungskriterien;
|
|
Beschreibung etwaiger Änderungen am Prüfverfahren.
|
|
|
Ergebnisse
|
Tabellierung der Daten, einschließlich CV70 (falls anwendbar), S.I., Zellviabilitätswerte, EC150-Werte (falls anwendbar) für die Prüfchemikalie und für die Positivkontrolle in jedem Durchlauf und eine Anzeige der Einstufung der Prüfchemikalie gemäß dem Vorhersagemodell;
|
|
Beschreibung etwaiger sonstiger relevanter Beobachtungen, falls zutreffend.
|
|
|
Erörterung der Ergebnisse
|
Erörterung der anhand des U-SENS™-Tests erhaltenen Ergebnisse;
|
|
Analyse der Prüfergebnisse im Rahmen eines IATA, sofern sonstige relevante Informationen vorliegen.
|
|
Schlussfolgerungen
|
LITERATURHINWEISE
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901–916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Verfügbar unter:http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. Verfügbar unter: [ http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Verfügbar unter:https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter: [http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337–351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259–270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683–1 696.
|
|
(11)
|
OECD. (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter:http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33ff. Verfügbar unter: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565–577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter:http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
Vereinte Nationen (UN) (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sechste überarbeitete Fassung, New York und Genf: United Nations Publications. Verfügbar unter:http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter:http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. Verfügbar unter:https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Anlage 2.1
DEFINITIONEN
Genauigkeit
: Grad der Übereinstimmung zwischen Prüfergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der „Relevanz“. Der Begriff wird oft im Sinne von „Übereinstimmung“ verwendet und bezeichnet den Anteil der korrekten Ergebnisse eines Tests (14).
AOP (Adverse Outcome Pathway)
: Abfolge von Vorgängen, ausgehend von der chemischen Struktur einer Zielchemikalie oder Zielgruppe ähnlicher Chemikalien, über den molekularen auslösenden Vorgang bis hin zu einem In-vivo-Ergebnis von Interesse (15).
CD86-Konzentrationswirkung
: Es liegt eine Konzentrationsabhängigkeit (oder Konzentrationswirkung) vor, wenn auf eine positive Konzentration (CD86 S.I. ≥ 150) eine Konzentration mit einem steigenden CD86 S.I. folgt.
Chemikalie
: Stoff oder Gemisch.
CV70
: Die geschätzte Konzentration, die 70 % Zellviabilität zeigt.
Verschiebung
: Eine Verschiebung wird definiert durch i) den korrigierten %CD86+-Wert des unbehandelten Kontrollreplikats 3 beträgt weniger als 50 % des Mittelwerts des korrigierten %CD86+ Werts der unbehandelten Kontrollreplikate 1 und 2; und ii) der korrigierte %CD86+-Wert des Negativkontrollreplikats 3 beträgt weniger als 50 % des Mittelwerts des korrigierten %CD86+ Werts der Negativkontrollreplikate 1 und 2.
EC150
: die geschätzten Konzentrationen, die 150 % S.I. der CD86-Expression aufzeigen.
Durchflusszytometrie
: eine zytometrische Technik, bei der Zellen, die in einem Fluid suspendiert sind, einzeln durch eine Lichtstrahl fließen, das in für die Zellen und ihre Komponenten typischen Muster gestreut ist; Zellen werden häufig mit fluoreszierenden Markern gekennzeichnet, damit das Licht zuerst absorbiert und dann in geänderten Frequenzen ausgestrahlt wird.
Gefahr
: Inhärente Eigenschaft eines Stoffes oder einer Situation, potenziell schädigende Auswirkungen zu haben, wenn ein Organismus, ein System oder eine (Teil-)Population diesem Stoff ausgesetzt ist.
IATA (Integrated Approach to Testing and Assessment)
: Ein strukturierter Ansatz zur Gefahrenidentifizierung (Potenzial), Gefahrencharakterisierung (Potenz) und/oder Sicherheitsbeurteilung (Potenzial/Potenz und Exposition) einer Chemikalie oder Gruppe von Chemikalien, der strategisch sämtliche relevanten Daten für eine informierte regulatorische Entscheidung hinsichtlich potenziellen Gefahren und/oder Risiken und/oder des Bedürfnisses weiterer gezielter und somit minimaler Tests integriert und abwägt.
Gemisch
: Ein Gemisch oder eine Lösung, die aus zwei oder mehreren Stoffen besteht.
Einkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorhanden ist.
Mehrkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens ≥ 10 % w/w und < 80 % w/w vorhanden sind. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet.
Positivkontrolle
: Ein Replikat, das alle Komponenten eines Prüfsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prähaptene
: Chemikalien, die durch abiotische Transformation zu Allergenen werden, z. B. durch Oxidierung.
Prohaptene
: Chemikalien, die eine enzymatische Aktivierung erfordern, um ihr Hautsensibilisierungspotenzial auszuschöpfen.
Relevanz
: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Die Relevanz umfasst die Einbeziehung der Genauigkeit (Übereinstimmung) eines Tests (14).
Zuverlässigkeit
: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Labors über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet (14).
Durchlauf
: Ein Durchlauf besteht aus einer oder mehreren hintereinander getesteten Prüfchemikalien mit einer Lösungsmittel-/Vehikelkontrolle und mit einer Positivkontrolle.
Sensitivität
: Der Anteil aller positiven/wirkungsvollen Chemikalien, die durch den Test korrekt eingestuft werden. Die Sensitivität ist ein Maß der Genauigkeit eines Tests mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (14).
S.I.
: Stimulationsindex. Relative Werte der geometrischen mittleren Fluoreszenzintensität in chemisch behandelten Zellen im Vergleich zu mit Lösungsmittel behandelten Zellen.
Lösungsmittel-/Vehikelkontrolle
: Eine unbehandelte Probe, die sämtliche Komponenten eines Prüfsystems enthält, außer der Prüfchemikalie, aber einschließlich des verwendeten Lösungsmittels/Vehikels. Sie wird verwendet, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst oder stabil verteilt wurden, zu bestimmen. Wenn mit einer simultanen Medienkontrolle geprüft wird, zeigt diese Probe außerdem, ob das Lösungsmittel oder Vehikel mit dem Prüfsystem interagiert.
Spezifität
: Der Anteil aller negativen/wirkungslosen Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit eines Tests mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (14).
Färbepuffer
: Eine mit Phosphat gepufferte Kochsalzlösung mit 5 % fetalem Kälberserum.
Stoff
: Ein chemisches Element und seine Verbindungen im natürlichen Zustand oder durch ein Produktionsverfahren, die einen Zusatzstoff induzieren, der zur Erhaltung seiner Stabilität erforderlich ist, sowie Verunreinigungen, die aus dem verwendeten Prozess stammen, aber mit Ausnahme von Lösungsmitteln, die getrennt sein können, ohne die Stabilität des Stoffes zu beeinträchtigen oder seine Zusammensetzung zu ändern.
Prüfchemikalie
: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS)
: Ein System, das die Einstufung von Chemikalien (Stoffen und Gemischen) gemäß normierten Typen und Stufen physikalischer, gesundheitlicher und Umweltgefahren vorschlägt und entsprechende Kommunikationselemente wie Piktogramme, Signalwörter, Gefahrenberichte, Berichte zu Vorsichtsmaßnahmen und Sicherheitsdatenblätter anspricht, damit Informationen zu Nebenwirkungen mit Blick auf den Schutz von Menschen (einschließlich Mitarbeitern, Arbeitern, Logistikern, Verbrauchern und Ersthelfern) und der Umwelt zu übermitteln (16).
UVCB
: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Gültiger Test
: Ein Test, von der angenommen wird, dass sie über ein ausreichendes Maß an Relevanz und Zuverlässigkeit für einen bestimmten Zweck verfügt und die auf wissenschaftlich bewährten Prinzipien basieren. Ein Test ist niemals in einem absoluten Sinn gültig, sondern im Verhältnis zu einem definierten Zweck (14).
Anlage 2.2
LEISTUNGSSTOFFE
Vor der routinemäßigen Anwendung des in dieser Anlage zu Prüfmethode B.71 beschriebenen Tests sollten Labors ihre technische Leistungsfähigkeit demonstrieren, indem sie die erwartete U-SENS™-Vorhersage für die in Tabelle 1 empfohlenen 10 Stoffe korrekt ermitteln und die CV70- und EC150-Werte, die in den betreffenden Referenzbereich fallen, bei mindestens 8 der 10 Leistungsstoffe bestimmen. Diese Leistungsstoffe wurden so ausgewählt, dass sie die Bandbreite von Reaktionen im Hinblick auf die Gefahr einer Hautsensibilisierung repräsentieren. Weitere Auswahlkriterien betrafen die Erhältlichkeit der Stoffe im Handel, und dass qualitativ hochwertige In-vivo-Referenzdaten sowie qualitativ hochwertige In-vitro-Daten aus dem U-SENS™-Test verfügbar sind. Außerdem sind veröffentlichte Referenzdaten für den U-SENS™-Test verfügbar (1) (8).
Tabelle 1
Empfohlene Stoffe zum Nachweis der technischen Leistungsfähigkeit beim U-SENS™-Test
|
Leistungsstoffe
|
CAS-Nr.
|
Aggregatzustand
|
In-vivo-Vorhersage (106)
|
U-SENS Lösungs-mittel/Vehikel
|
U-SENS CV70 Referenz Bereich in μg/ml (107)
|
U-SENS EC150 Referenz Bereich in μg/ml (107)
|
|
4-Phenylenediamin
|
106-50-3
|
fest
|
Allergen (stark)
|
Vollständiges Medium (108)
|
< 30
|
positiv (≤ 10)
|
|
Picrylschwefelsäure
|
2508-19-2
|
flüssig
|
Allergen (stark)
|
Vollständiges Medium
|
> 50
|
positiv (≤ 50)
|
|
Diethylmaleat
|
141-05-9
|
flüssig
|
Allergen (mäßig)
|
DMSO
|
10–100
|
positiv (≤ 20)
|
|
Resorcin
|
108-46-3
|
fest
|
Allergen (mäßig)
|
Vollständiges Medium
|
> 100
|
positiv (≤ 50)
|
|
Zimtalkohol
|
104-54-1
|
fest
|
Allergen (schwach)
|
DMSO
|
> 100
|
positiv (10–100)
|
|
4-Allylanisol
|
140-67-0
|
flüssig
|
Sensibilisator (schwach)
|
DMSO
|
> 100
|
positiv (< 200)
|
|
Saccharin
|
81-07-2
|
fest
|
Nichtsensibili-sator
|
DMSO
|
> 200
|
negativ (> 200)
|
|
Glyzerin
|
56-81-5
|
flüssig
|
Nichtaensibili-sator
|
Vollständiges Medium
|
> 200
|
negativ (> 200)
|
|
Milchsäure
|
50-21-5
|
flüssig
|
Nichtsensibili-sator
|
Vollständiges Medium
|
> 200
|
negativ (> 200)
|
|
Salicylsäure
|
69-72-7
|
fest
|
Nichtsensibili-sator
|
DMSO
|
> 200
|
negativ (> 200)
|
|
Abkürzungen: CAS-Nr. = Registernummer des Chemical Abstracts Service
|
Anlage 3
IN-VITRO-HAUTSENSIBILISIERUNG: IL-8 LUC-TEST
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
|
1.
|
Im Gegensatz zu Tests, die die Expression von Zelloberflächenmarkern analysieren, quantifiziert der IL8-Luc-Test die Änderungen des IL-8-Expressions, ein Zytokin im Zusammenhang mit der Aktivierung dendritischer Zellen (DC). In der von THP-1 abgeleiteten IL-8-Reporter-Zelllinie (THP-G8, etabliert aus der humanen akuten monozytischen Leukämiezelllinie THP-1) wird die IL-8-Expression nach der Exposition gegenüber Allergenen gemessen (1). Die Expression der Luciferase wird dann verwendet, um bei der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren zu helfen.
|
|
2.
|
Der IL-8-Luc-Test wurde in einer Validierungsstudie (2), das vom Japanischen Zentrum für die Validierung von Alternativmethoden (JaCVAM), dem Ministerium für Wirtschaft, Handel und Industrie (METI) und der Japanischen Gesellschaft für Alternativen zu Tierversuchen (JSAAE) durchgeführt wurde, bewertet und anschließend einer unabhängigen Begutachtung (3) unter der Aufsicht des JaCVAM und des Ministeriums für Gesundheit, Arbeit und Wohlstand (MHLW) mit Unterstützung der Internationalen Kooperation für alternative Prüfmethoden (ICATM) unterzogen. Unter Berücksichtigung sämtlicher verfügbarer Nachweise und Informationen von Behörden und Interessenträgern wurde der IL-8-Luc-Test als Teil eines IATA als nützlich angesehen, zwischen Allergenen und Nichtsensibilisatoren zum Zweck der Gefahreneinstufung und Etikettierung zu unterscheiden. Beispiele für die Verwendung von IL-8-Luc-Test-Daten in Kombination mit anderen Informationen werden in der Literatur beschrieben (4) (5) (6).
|
|
3.
|
Der IL-8-Luc-Test stellte sich als übertragbar auf Labore heraus, die mit Zellkulturen und Luciferasemessungen Erfahrung haben. Laborreproduzierbarkeiten lagen zwischen 87,7 % bzw. 87,5 % (2). Daten, die in der Validierungsstudie (2) und anderen veröffentlichten Arbeiten (1) (6) erzeugt wurden, zeigen, dass der IL-8-Luc-Test im Vergleich zu LLNA 118 von 143 Chemikalien als positiv oder negativ und 25 Chemikalien als nicht schlüssig eingestuft hat und die Genauigkeit des IL-8-Luc-Tests bei der Unterscheidung von Hautallergenen (UN-GHS/CLP Kat. 1) von Nichtsensibilisatoren (UN-GHS/CLP Nr. Kat.) beträgt 86 % (101/118) mit einer Sensitivität von 96 % (92/96) und einer Spezifität von 41 % (9/22). Außer den unten beschriebenen Stoffen außerhalb des Anwendungsbereichs (Abschnitt 5) stufte der IL-8-Luc-Test 113 von 136 Chemikalien als positiv oder negativ und 23 Chemikalien als nicht schlüssig ein und die Genauigkeit des IL-8-Luc-Tests liegt bei 89 % (101/113) mit einer Sensitivität von 96 % (92/96) und einer Spezifität von 53 % (9/17). Unter Anwendung der in Urbisch et al. zitierten Humandaten (7) stufte der IL-8-Luc-Test 76 von 90 Chemikalien als positiv oder negativ und 14 Chemikalien als nicht schlüssig ein und die Genauigkeit liegt bei 80 % (61/76), die Sensitivität bei 93 % (54/58) und die Spezifität bei 39 % (7/18). Außer den Stoffen außerhalb des Anwendungsbereichs stufte der IL-8-Luc-Test 71 von 84 Chemikalien als positiv oder negativ und 13 Chemikalien als nicht schlüssig ein und die Genauigkeit liegt bei 86 % (61/71)mit einer Sensitivität von 93 % (54/58) und einer Spezifität von 54 % (7/13). Falsch negative Vorhersagen mit dem IL-8-Luc-Test treten wahrscheinlicher bei Chemikalien auf, die eine niedrige/mittlere Hautsensibilisierungspotenz (UN-GHS/CLP Unterkategorie 1B) aufweisen als bei denjenigen mit einer hohen Potenz (UN-GHS/CLP Unterkategorie 1A) (6). Zusammen unterstützen die Informationen die Zweckdienlichkeit des IL-8-Luc-Tests bei der Identifizierung von Hautsensibilisierungsgefahren. Jedoch sollte die Genauigkeit, die hier für den IL-8-Luc-Test als eigenständiger Test angegeben wird, in Kombination mit anderen Informationsquellen im Rahmen eines IATA sowie gemäß den Bestimmungen unter Nummer 7 und 8 oben in der Allgemeinen Einleitung betrachtet werden. Darüber hinaus sollte bei der Bewertung von Tests zur Hautsensibilisierung ohne Tierversuche beachtet werden, dass der LLNA sowie andere Tierversuche die Situation bei Menschen nicht vollständig widerspiegeln.
|
|
4.
|
Auf der Grundlage der gegenwärtig verfügbaren Daten über den IL-8-Luc-Test wurde nachgewiesen, dass er bei Prüfchemikalien, die eine Vielzahl an organischen Funktionsgruppen, Reaktionsmechanismen, Hautsensibilisierungspotenzial (wie in In-vivo-Studien festgestellt) und physikalisch-chemischen Eigenschaften abdecken, anwendbar ist (2) (6).
|
|
5.
|
Obwohl der IL-8-Luc-Test X-VIVOTM 15 als Lösungsmittel verwendet, bewertete es Chemikalien mit einem Log Kow > 3,5 und diejenigen mit einer Wasserlöslichkeit von ungefähr 100 μg/ ml wie von der EPI SuiteTM berechnet korrekt und seine Leistung bei der Erkennung von Allergenen mit schlechter Wasserlöslichkeit ist besser als die des IL-8-Luc-Tests, das Dimethylsulfoxid (DMSO) als Lösungsmittel verwendet (2). Negative Ergebnisse für Prüfchemikalien, die nicht bei 20 mg/ml gelöst werden, können falsch negative Ergebnisse hervorrufen, da sie nicht in X-VIVOTM 15 löslich sind. Deshalb sollten negative Ergebnisse für diese Chemikalien nicht berücksichtigt werden. Bei der Validierungsstudie gab es eine hohe Falsch-Negativ-Rate für Anhydride. Darüber hinaus können Prohaptene (Stoffe, die eine Stoffwechselaktivierung erfordern) und Prähaptene (Stoffe, die durch Oxidation aktiviert werden) aufgrund der eingeschränkten Stoffwechselfähigkeit der Zelllinie (8) und der Versuchsbedingungen falsch negative Ergebnisse hervorrufen. Obwohl negative Ergebnisse für mutmaßliche Prä-/Prohaptene jedoch mit Vorsicht interpretiert werden sollten, hat der IL-8-Luc-Test 11 von 11 Prähaptene, 6/6 Prohaptene und 6/8 Prä-/Prohaptene im IL-8-Luc-Test-Datensatz korrekt eingestuft (2). Auf der Grundlage der kürzlichen umfassenden Begutachtung von drei Tests ohne Tierversuche (DPRA, KeratinoSens™ und h-CLAT) zur Erkennung von Prä- und Prohaptenen (9) und basierend auf der Tatsache, dass THP-G8-Zellen im IL-8-Luc-Test verwendet werden, wird eine Zelllinie von THP-1 abgeleitet, die in h-CLAT verwendet wird; der IL-8-Luc-Test kann auch dazu beitragen, die Sensitivität von Tests ohne Tierversuche zur Erkennung von Prä- und Prohaptenen in der Kombination anderer Tests zu steigern. Bisher geprüfte Tenside führten unabhängig von ihrem Typ (z. B. kationisch, anionisch oder nichtionisch) zu (falsch) positiven Ergebnissen. Schließlich können Chemikalien, die mit Luciferase interferieren, die Aktivität/Messung durcheinander bringen, was zu einer offensichtlichen Hemmung oder einer erhöhten Lumineszenz führt (10). Beispielweise wurde eingetragen, dass Phytoöstrogenkonzentrationen > 1 μM die Lumineszenzsignale in anderen auf Luciferase basierenden Reporter-Gen-Tests aufgrund der Überaktivierung des Luciferase-Reporter-Gens stören. Infolgedessen muss die Luciferase-Expression, die bei hohen Konzentrationen von Phytoöstrogenen oder Verbindungen, die vermutlich eine mit Phytoöstrogen vergleichbare Überaktivierung des Luciferase-Reporter-Gens bewirken, sorgfältig untersucht werden (11). Basierend auf den obigen Aussagen liegen Tenside, Anhydride und Chemikalien, die Luciferase beeinträchtigen, außerhalb des Anwendungsbereichs dieser Tests. In Fällen, in denen die Nichtanwendbarkeit des IL-8-Luc-Tests bei anderen spezifischen Kategorien von Prüfchemikalien nachgewiesen wird, sollte der Test bei diesen spezifischen Kategorien nicht verwendet werden.
|
|
6.
|
Der IL-8-Luc-Test unterstützt, wie bereits oben erwähnt, die Unterscheidung von Hautallergenen und Nichtsensibilisatoren. Weitere Arbeiten, vorzugsweise basierend auf menschlichen Daten, sind erforderlich, um zu bestimmen, ob die Ergebnisse von IL-8-Luc zur Potenzbewertung beitragen können, wenn sie in Kombination mit anderen Informationsquellen berücksichtigt werden.
|
|
7.
|
Definitionen sind Anlage 3.1 zu entnehmen.
|
PRINZIP DER PRÜFMETHODE
|
8.
|
Der IL-8-Luc-Test nutzt eine humane monozytische Leukämie-Zelllinie THP-1, die von der American Type Culture Collection (Manassas, VA, USA) erhalten wurde. Mit dieser Zelllinie etablierte die Abteilung für Dermatologie der Tohoku University School of Medicine eine von THP-1 abgeleitete IL-8-Reporter-Zelllinie, THP-G8, die die Luciferase-Gene „Stable Luciferase Orange (SLO)“ und „Stable Luciferase Red (SLR)“ unter der Kontrolle von IL-8 bzw. Glycerinaldehyd-3-phosphat-Dehydrogenase (GAPDH)-Trägern beherbergt (1). Dies ermöglicht die quantitative Messung der Luciferase-Geninduktion durch Lumineszenzerkennung unter Verwendung von bekannten lichterzeugenden Luciferase-Substraten als Indikator für die Aktivität des IL-8 und GAPDH in Zellen nach Exposition gegenüber sensibilisierenden Chemikalien.
|
|
9.
|
Das zweifarbige Prüfsystem besteht aus einer orange emittierenden Luciferase (SLO; λmax = 580 nm) (12) für die Genexpression des IL-8-Trägers sowie einer rot emittierenden Luciferase (SLR; λmax = 630 nm) (13) für die Genexpression des internen Kontrollträgers GAPDH. Die zwei Luciferasen strahlen bei der Reaktion mit Leuchtkäfer-d-Luciferin verschiedene Farben aus und ihre Lumineszenz wird gleichzeitig in einer einstufigen Reaktion gemessen, indem die Emission von der Testmischung mithilfe eines optischen Filters geteilt wird (14) (Anlage 3.2).
|
|
10.
|
THP-G8-Zellen werden 16 Stunden lang mit der Prüfchemikalie behandelt und danach wird die SLO-Luciferase-Aktivität (SLO-LA) gemessen, die die IL-8-Trägeraktivität widerspiegelt, sowie die SLR-Luciferase-Aktivität (SLR-LA), die die GAPDH-Trägeraktivität widerspiegelt. Um die Abkürzungen leicht verständlich zu machen, wenden SLO-LA und SLR-LA als IL8LA bzw. GAPLA bezeichnet. Tabelle 1 bietet eine Beschreibung der Ausdrücke im Zusammenhang mit der Luciferase-Aktivität im IL-8-Luc-Test. Die gemessenen Werte werden verwendet, um das normalisierte IL8LA (nIL8LA) zu berechnen, das das Verhältnis von IL8LA zu GAPLA ist; die Induktion von nIL8LA (Ind-IL8LA), das das Verhältnis des arithmetischen Mittels der in vierfacher Ausfertigung gemessenen Wert des nIL8LA von mit einer Prüfchemikalie behandelten THP-G8-Zellen zu den Werten des nIL8LA der unbehandelten THP-G8-Zellen ist; und die Hemmung von GAPLA (Inh-GAPLA), das das Verhältnis des arithmetischen Mittels der in vierfacher Ausfertigung gemessenen Werte des GAPLA von mit einer Prüfchemikalie behandelten THP-G8-Zellen zu den Werten des GAPLA der unbehandelten THP-G8-Zellen ist und als Indikator für die Zytotoxizität verwendet wird.
Tabelle 1
Beschreibung der Ausdrücke im Zusammenhang mit der Luciferase-Aktivität im IL-8-Luc-Test
|
Abkürzungen
|
Definition
|
|
GAPLA
|
SLR-Luciferase-Aktivität, die die GAPDH-Trägeraktivität widerspiegelt
|
|
IL8LA
|
SLO-Luciferase-Aktivität, die die IL-8-Trägeraktivität widerspiegelt
|
|
nIL8LA
|
IL8LA / GAPLA
|
|
Ind-IL8LA
|
nIL8LA von mit Chemikalien behandelten THP-G8-Zellen / nIL8LA von unbehandelten Zellen
|
|
Inh-GAPLA
|
GAPLA von mit Chemikalien behandeltem THP-G8 / GAPLA von unbehandelten Zellen
|
|
CV05
|
Die niedrigste Konzentration der Chemikalie, bei der Inh-GAPLA < 0,05 wird.
|
|
|
11.
|
Es stehen Leistungsnormen (15) zur Verfügung, die die Validierung geänderter In-vitro-IL-8-Luciferase-Tests ähnlich dem IL-8-Luc-Test sowie die rechtzeitige Anpassung der OECD-Prüfrichtlinie 442E für deren Einbeziehung ermöglichen. Die gegenseitige Anerkennung der Daten gemäß OECD wird nur für Tests garantiert, die gemäß diesen Leistungsnormen validiert wurden, sofern diese Tests von der OECD überprüft und in die Prüfrichtlinie 442E aufgenommen wurden (16).
|
NACHWEIS DER KOMPETENZ
|
12.
|
Vor der routinemäßigen Verwendung des unter dieser Anlage zur Prüfmethode B.71 beschriebenen Tests sollten Laboratorien ihre technische Kompetenz anhand der zehn in Anlage 3.3 aufgeführten Leistungsstoffe in Übereinstimmung mit bewährten In-vitro-Verfahren nachweisen (17). Darüber hinaus sollten Testbenutzer eine historische Datenbank der mit den Reaktivitätstests (siehe Abschnitt 15) und mit den positiven and Lösungsmittel-/Vehikelkontrollen (siehe Abschnitte 21–24) erzeugten Daten pflegen und diese Daten nutzen, um zu bestätigen, dass die Reproduzierbarkeit des Tests in ihrem Labor mit der Zeit aufrechterhalten wird.
|
VERFAHREN
|
13.
|
Die Standardarbeitsanweisung (SOP) für den IL-8-Luc-Test ist verfügbar und sollte bei der Durchführung des Tests verwendet werden (18). Labore, die der Durchführung des Tests zugestimmt haben, können die rekombinante THP-G8-Zelllinie von GPC Lab. Co. Ltd., Tottori, Japan, nach Unterzeichnung einer Materialübertragungsvereinbarung (MTA) in Übereinstimmung mit den Bedingungen der OECD-Vorlage erhalten. Nachfolgend werden die Hauptkomponenten und Verfahren des Tests beschrieben.
|
Vorbereitung der Zellen
|
14.
|
Die THP-G8-Zelllinie vom GPC Lab. Co. Ltd., Tottori, Japan, sollte für die Durchführung des IL-8-Luc-Tests verwendet werden (siehe Abschnitte 8 und 13). Bei Erhalt werden die Zellen gewonnen (2 bis 4 Passagen) und als homogener Stamm eingefroren aufbewahrt. Zellen von diesem Stamm können bis zu einem Höchstwert von 12 Passagen oder maximal 6 Wochen gewonnen werden. Das für die Vermehrung verwendete Medium ist das RPMI-1640-Kulturmedium mit 10 % fetalem Kälberserum (FBS), antibiotischer/antimyzotischer Lösung (100 U/ml Penicillin G, 100 μg/ml Streptomycin und 0,25 μg/ml Amphotericin B in 0,85 % Kochsalzlösung) (z. B. GIBCO Kat. Nr. 15240-062), 0,15 μg/ml Puromycin (z. B. CAS:58-58-2) und 300 μg/ml G418 (z. B. CAS:108321-42-2).
|
|
15.
|
Vor ihrer Verwendung zu Testzwecken sollten die Zelle qualifiziert werden, indem ein Reaktivitätstest durchgeführt wird. Dieser Test sollte 1–2 Wochen oder 2–4 Passagen nach dem Auftauen mithilfe einer Positivkontrolle, 4-Nitrobenzylbromid (4-NBB) (CAS:100-11-8, ≥ 99 % Reinheit) und der Negativkontrolle Milchsäure (LA) (CAS:50-21-5, ≥ 85 % Reinheit) durchgeführt werden. 4-NBB sollte eine positive Wirkung auf Ind-IL8LA (≥ 1,4) hervorrufen, während LA eine negative Wirkung auf Ind-IL8LA (< 1,4) hervorrufen sollte. Nur Zellen, die den Reaktivitätstest bestehen, werden für den Test verwendet. Der Test sollte gemäß den in den Abschnitten 22–24 beschriebenen Verfahren durchgeführt werden.
|
|
16.
|
Zu Testzwecken werden THP-G8-Zellen bei einer Dichte von 2 bis 5 × 105 Zellen/ml ausgesät und 48 bis 96 Stunden in Kulturkolben vorgezüchtet. Am Tag des Tests werden die vom Kulturkolben geernteten Zellen mit RPMI-1640 mit 10 % FBS ohne Antibiotika gewaschen und anschließend mit RPMI-1640 mit 10 % FBS ohne Antibiotika bei 1 × 106 Zellen/ml resuspensiert. Die Zellen werden dann in einer flachen schwarzen Platte mit 96 Mulden (z. B. Costar Kat. Nr. 3603) mit 50 μl (5 × 104 Zellen/well) verteilt.
|
Vorbereitung der Prüfchemikalie und Kontrollstoffe
|
17.
|
Die Prüfchemikalie sowie die Kontrollstoffe werden am Tag des Tests vorbereitet. Bei dem IL-8-Luc-Test werden Prüfchemikalien in X-VIVOTM 15 gelöst, einem im Handel erhältlichen, serumfreien Medium (Lonza, 04-418Q), bis eine endgültige Konzentration von 20 mg/ml erreicht wurde. X-VIVOTM 15 wird in einem Mikrozentrifugenröhrchen zu 20 mg/ml der Prüfchemikalie hinzugefügt (unabhängig von der Löslichkeit der Chemikalie) und auf ein Volumen von 1 ml gebracht und dann kräftig geschüttelt und auf einem Rotor bei einer Höchstgeschwindigkeit von 8 U/min 30 Minuten lang bei einer Umgebungstemperatur von ungefähr 20 oC geschüttelt. Wenn feste Chemikalien noch immer nicht löslich sind, wird das Röhrchen darüber hinaus im Ultraschallbad behandelt, bis die Chemikalie sich komplett gelöst hat oder stabil verteilt wurde. Bei Prüfchemikalien, die in X-VIVOTM 15 löslich sind, wird die Lösung mit einem Faktor von 5 mit X-VIVOTM 15 verdünnt und dann als X-VIVOTM 15-Stammlösung der Prüfchemikalie verwendet (4 mg/ml). Bei Prüfchemikalien, die nicht in X-VIVOTM 15 löslich sind, wird das Gemisch erneut mindestens 30 Minuten lang rotiert und dann bei 15000 U/min (≈ 20000 g) 5 Minuten lang zentrifugiert; das sich daraus ergebende Tensid wird als X-VIVOTM 15-Stammlösung der Prüfchemikalie verwendet. Für den Einsatz anderer Lösungsmittel wie DMSO, Wasser oder Kulturmedium sollten ausreichende wissenschaftliche Gründe vorgelegt werden. Das detaillierte Verfahren zur Lösung von Chemikalien wird in Anlage 3.5 dargestellt. Die X-VIVOTM 15-Lösungen, die in den Abschnitten 18–23 beschrieben werden, werden 1:1 (v/v) mit den in der flachen Platte mit 96 Mulden vorbereiteten Zellsuspensionen gemischt (siehe Abschnitt 16).
|
|
18.
|
Der erste Testdurchlauf zielt darauf ab, die zytotoxische Konzentration zu bestimmen und das Hautsensibilisierungspotenzial der Chemikalien zu untersuchen. Mithilfe von X-VIVOTM 15 werden serielle Verdünnungen der X-VIVOTM 15-Stammlösungen der Prüfchemikalien mit einem Verdünnungsfaktor von zwei (siehe Anlage 3.5) in einem Testblock mit 96 Mulden (z. B. Costar Kat. Nr. EW-01729-03) hergestellt. Dann werden 50 μl/Mulde verdünnte Lösung zu 50 μl der Zellsuspension in einer flachen schwarzen Platte mit 96 Mulden hinzugefügt. Damit reicht die endgültige Konzentration für Prüfchemikalien, die in X-VIVOTM 15 löslich sind, von 0,002 bis 2 mg/ml (Anlage 3.5). Bei Prüfchemikalien, die bei 20 mg/ml nicht in X-VIVOTM 15 löslich sind, werden nur die Verdünnungsfaktoren bestimmt, die von 2 bis 210 reichen, obwohl die tatsächlichen endgültigen Konzentrationen der Prüfchemikalien unbestimmt bleiben und von der gesättigten Konzentration der Prüfchemikalien in der X-VIVOTM 15-Stammlösung abhängen.
|
|
19.
|
In nachfolgenden Testdurchläufen (d. h. das zweite, dritte und vierte Replikat) wird die X-VIVOTM 15-Stammlösung im ersten Versuch bei einer Konzentration hergestellt, die 4 Mal höher ist als die Konzentration der Zellviabilität 05 (CV05; die niedrigste Konzentration, bei der Inh-GAPLA < 0,05 wird). Wenn Inh-GAPLA bei der höchsten Konzentration im ersten Durchlauf nicht unter 0,05 fällt, wird die X-VIVOTM 15-Stammlösung bei der höchsten Konzentration im ersten Durchlauf hergestellt. Die Konzentration von CV05 wird berechnet, indem die Konzentration der Stammlösung im ersten Durchlauf durch den Verdünnungsfaktor für CV05 (X) (Verdünnungsfaktor CV05 (X) geteilt wird; der Verdünnungsfaktor, der erforderlich ist, um die Stammlösung auf CV05 zu verdünnen) (siehe Anlage 3.5). Bei Prüfstoffen, die bei 20 mg/ml nicht in X-VIVO löslich sind, wird CV05 anhand der Konzentration der Stammlösung × 1/X bestimmt. Für die Durchläufe 2 bis 4 wird eine zweite Stammlösung wie 4 × CV05 vorbereitet (Anlage 3.5).
|
|
20.
|
Serielle Verdünnungen der zweiten X-VIVOTM 15-Stammlösungen werden mit einem Verdünnungsfaktor von 1,5 mit einem Testblock mit 96 Mulden hergestellt. Dann werden 50 μl/Mulde verdünnte Lösung zu 50 μl der Zellsuspension in den Mulden einer flachen schwarzen Platte mit 96 Mulden hinzugefügt. Jede Konzentration jeder Prüfchemikalie sollte in 4 Mulden geprüft werden. Die Proben werden dann auf einem Platten-Schüttler gemischt und 16 Stunden bei 37 oC und 5 % CO2 gemischt und anschließend wird die Luciferase-Aktivität wie unten beschrieben gemessen.
|
|
21.
|
Die Lösungsmittelkontrolle ist das Gemisch von 50 μl/Mulde von X-VIVOTM 15 und 50 μl/Mulde der Zellsuspension in RPMI-1640 mit 10 % FBS.
|
|
22.
|
Die empfohlene Positivkontrolle ist 4-NBB. 20 mg von 4-NBB werden in einem 1,5-ml-Mikrofugenröhrchen vorbereitet, zu dem X-VIVOTM 15 bis auf 1 ml hinzugefügt wird. Das Röhrchen wird kräftig geschüttelt und auf einem Rotor bei einer Höchstgeschwindigkeit von 8 U/min mindestens 30 Minuten lang geschüttelt. Nach der Zentrifugierung bei 20000 g für 5 Minuten wird das Tensid mit einem Faktor von 4 mit X-VIVOTM 15 verdünnt und 500 μl des verdünnten Tensids werden in eine Mulde in einem Testblock mit 96 Mulden übertragen. Das verdünnte Tensid wird mit X-VIVOTM 15 mit Faktoren von 2 und 4 weiter verdünnt und 50 μl der Lösung wird zu 50 μl der THP-G8-Zellsuspension in den Mulden der flachen schwarzen Platte mit 96 Mulden hinzugefügt (Anlage 3.6). Jede Konzentration der Positivkontrolle sollte in 4 Mulden geprüft werden. Die Platte wird auf einem Platten-Schüttler geschüttelt und in einem CO2-Inkubator 16 Stunden lang inkubiert (37 oC, 5 % CO2), anschließend wird die Luciferase-Aktivität wie in Abschnitt 29 beschrieben gemessen.
|
|
23.
|
Die empfohlene Negativkontrolle ist LA. 20 mg von LA werden in einem 1,5-ml-Mikrofugenröhrchen vorbereitet, zu dem X-VIVOTM 15 bis auf 1 ml hinzugefügt wird (20 mg/ml). 20 mg/ml der LA-Lösung werden um einen Faktor von 5 mit X-VIVOTM 15 verdünnt (4 mg/ml); 500 μl dieser 4 mg/ml-LA-Lösung werden in eine Mulde eines Testblocks mit 96 Mulden übertragen. Diese Lösung wird um einem Faktor von 2 mit X-VIVOTM 15 verdünnt und dann erneut um einen Faktor von 2 verdünnt, um 2 mg/ml- und 1 mg/ml-Lösungen herzustellen. 50 μl dieser 3 Lösungen und Vehikelkontrollen (X-VIVOTM 15) werden zu 50 μl der THP-G8-Zellsuspension in den Mulden der flachen schwarzen Platte mit 96 Mulden hinzugefügt. Jede Konzentration der Negativkontrolle wird in 4 Mulden geprüft. Die Platte wird auf einem Platten-Schüttler geschüttelt und in einem CO2-Inkubator 16 Stunden lang inkubiert (37 oC, 5 % CO2), anschließend wird die Luciferase-Aktivität wie in Abschnitt 29 beschrieben gemessen.
|
|
24.
|
Andere geeignete Positiv- oder Negativkontrollen können verwendet werden, sofern historische Daten für die Ableitung vergleichbarer Akzeptanzkriterien für einen Testdurchlauf zur Verfügung stehen.
|
|
25.
|
Es ist darauf zu achten, dass eine Verdunstung der flüchtigen Prüfchemikalien sowie eine Kreuzkontamination zwischen Mulden durch die Prüfchemikalien vermieden werden, indem die Platte beispielsweise vor der Inkubation mit den Prüfchemikalien versiegelt wird.
|
|
26.
|
Bei den Prüfchemikalien und der Lösungsmittelkontrolle sind 2 bis 4 Durchläufe erforderlich, um eine positive oder negative Vorhersage ableiten zu können (siehe Tabelle 2). Jeder Durchlauf wird an einem anderen Tag mit einer frischen X-VIVOTM 15-Stammlösung der Prüfchemikalie und unabhängig voneinander gewonnenen Zellen vorgenommen. Die Zellen können aus derselben Passage stammen.
|
Messungen der Luciferase-Aktivität
|
27.
|
Die Lumineszenz wird mithilfe eines Mikroplattenluminometers mit 96 Mulden gemessen, das mit optischen Filtern ausgestattet ist, z. B. Phelios (ATTO, Tokyo, Japan), Tristan 941 (Berthold, Bad Wildbad, Deutschland) und die ARVO-Serie (PerkinElmer, Waltham, MA, USA). Das Luminometer muss für jeden Test kalibriert werden, um Reproduzierbarkeit zu gewähren (19). Rekombinante orange und rot emittierende Luciferasen sind für diese Kalibrierung verfügbar.
|
|
28.
|
100 μl des vorgewärmten Tripluc® Luciferase-Test-Reagenz (Tripluc) wird in jede Mulde der Platte übertragen, die die mit oder ohne Chemikalien behandelte Zellsuspension enthält. Die Platte wird 10 Minuten lang bei einer Umgebungstemperatur von ungefähr 20 oC geschüttelt. Die Platte wird auf dem Luminometer platziert, um die Luciferase-Aktivität zu messen. Die Biolumineszenz wird jeweils 3 Sekunden lang in der Abwesenheit (F0) und Anwesenheit (F1) des optischen Filters gemessen. Bei Verwendung anderer Einstellungen, z. B. je nach verwendetem Luminometermodell, sollten diese begründet werden.
|
|
29.
|
Parameter für jede Konzentration werden aus den gemessenen Werten berechnet, z. B. IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, die mittlere ±SD von IL8LA, die mittlere ±SD von GAPLA, die mittlere ±SD von nIL8LA, die mittlere ±SD von Ind-IL8LA, die mittlere ±SD von Inh-GAPLA und das 95 % Konfidenzintervall von Ind-IL8LA. Definitionen der in diesem Abschnitt verwendeten Parameter werden in den Anlagen 3.1 bzw. 3.4 bereitgestellt.
|
|
30.
|
Vor der Messung wird die Farbunterscheidung in mehrfarbigen Reporter-Tests allgemein durch die Verwendung von Detektoren erzielt (Luminometer und Plattenleser), die mit optischen Filtern ausgestattet sind, wie beispielsweise Short-Cut-Filtern (Langpass oder Kurzpass) oder Bandpassfiltern. Die Übertragungskoeffizienten der Filter für jede Biolumineszenzsignalfarbe sollte vor dem Test gemäß Anlage 3.2 kalibriert werden.
|
DATEN UND BERICHTERSTATTUNG
Datenauswertung
|
31.
|
Kriterien für eine positive/negative Entscheidung erfordern, dass in jedem Durchlauf:
|
—
|
eine IL-8-Luc-Test-Vorhersage als positiv eingestuft wird, wenn eine Prüfchemikalie über einen Ind-IL8LA ≥ von 1,4 verfügt und die untere Grenze des 95 %-Konfidenzintervalls von Ind-IL8LA ≥ 1,0 beträgt
|
|
—
|
eine IL-8-Luc-Test-Vorhersage als negativ eingestuft wird, wenn eine Prüfchemikalie über einen Ind-IL8LA < 1,4 verfügt und/oder die untere Grenze des 95 %-Konfidenzintervalls von Ind-IL8LA < 1,0 beträgt
|
|
Vorhersagemodell
|
32.
|
Prüfchemikalien, die zwei positive Ergebnisse in den 1., 2., 3. oder 4. Durchläufen liefern, werden als positiv identifiziert, während diejenigen, die drei negative Ergebnisse in den 1., 2., 3. oder 4. Durchläufen liefern, als vermeintlich negativ identifiziert werden (Tabelle 2). Unter den vermeintlich negativen Chemikalien werden Chemikalien, die bei 20 mg/ml von X-VIVOTM 15 gelöst werden, als negativ eingestuft, während Chemikalien, die nicht bei 20 mg/ml von X-VIVOTM 15 gelöst werden, nicht berücksichtigt werden sollten (Abbildung 1).
Tabelle 2
Kriterien zur Identifizierung von positiv und vermeintlich negativ
|
1. Durchlauf
|
2. Durchlauf
|
3. Durchlauf
|
4. Durchlauf
|
Endgültige Vorhersage
|
|
positiv
|
positiv
|
–
|
–
|
positiv
|
|
negativ
|
positiv
|
–
|
positiv
|
|
negativ
|
positiv
|
positiv
|
|
negativ
|
Vermeintlich negativ
|
|
negativ
|
positiv
|
positiv
|
–
|
positiv
|
|
negativ
|
positiv
|
positiv
|
|
negativ
|
Vermeintlich negativ
|
|
negativ
|
positiv
|
positiv
|
positiv
|
|
negativ
|
Vermeintlich negativ
|
|
negativ
|
–
|
Vermeintlich negativ
|
|
Abb. 1
Vorhersagemodell für endgültiges Urteil
Akzeptanzkriterien
|
33.
|
Bei der Verwendung des IL-8-Luc-Tests sollten die folgenden Akzeptanzkriterien erfüllt werden:
|
—
|
Ind-IL8LA sollte in mindestens einer Konzentration der Positivkontrolle, 4-NBB, in jedem Durchlauf mehr als 5,0 betragen.
|
|
—
|
Ind-IL8LA sollte in jeder Konzentration der Negativkontrolle, Milchsäure, in jedem Durchlauf weniger als 1,4 betragen.
|
|
—
|
Daten aus Platten, für die die GAPLA der Kontrollmulden mit Zellen und Tripluc aber ohne Chemikalien weniger als 5 Mal des Wertes der Mulde, die nur das Prüfmedium enthält (50 μl/Mulde von RPMI-1640 mit 10 % FBS und 50 μl/Mulde von X-VIVOTM 15), beträgt, sollten abgelehnt werden.
|
|
—
|
Daten aus Platten, für die die Inh-GAPLA aller Konzentrationen des Tests oder Kontrollchemikalien weniger als 0,05 beträgt, sollten abgelehnt werden. In diesem Fall sollte der erste Test wiederholt werden, damit die höchste endgültige Konzentration des wiederholten Tests die niedrigste endgültige Konzentration des vorherigen Tests ist.
|
|
Prüfbericht
|
34.
|
Der Prüfbericht sollte folgende Angaben enthalten:
|
Prüfchemikalien
Einkomponentiger Stoff:
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
Aussehen, Wasserlöslichkeit, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, soweit verfügbar;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
Löslichkeit in X-VIVOTM 15. Bei Chemikalien, die in X-VIVOTM 15 nicht löslich sind, ob ein Niederschlag oder ein Aufschwemmen nach der Zentrifugierung beobachtet wird;
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
Begründung der Auswahl des Lösungsmittels/Vehikels für jede Prüfchemikalie, wenn X-VIVOTM 15 nicht verwendet wurde.
|
Mehrkomponentiger Stoff, UVCB-Stoff und Gemisch:
|
Charakterisierung, so weit wie möglich, z. B. durch die chemische Zusammensetzung (siehe oben), Reinheit, das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften (siehe oben) der einzelnen Komponenten, soweit verfügbar;
|
|
Physikalisches Erscheinungsbild, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften, soweit verfügbar;
|
|
Molekulargewicht oder scheinbares Molekulargewicht im Fall von Gemischen/Polymeren mit bekannter Zusammensetzung oder andere für die Durchführung der Studie relevante Informationen;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
Löslichkeit in X-VIVOTM 15. Bei Chemikalien, die in X-VIVOTM 15 nicht löslich sind, ob ein Niederschlag oder ein Aufschwemmen nach der Zentrifugierung beobachtet wird;
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar.
|
|
Begründung der Auswahl des Lösungsmittels/Vehikels für jede Prüfchemikalie, wenn X-VIVOTM 15 nicht verwendet wurde.
|
|
|
Kontrollen
Positivkontrolle:
|
Chemische Bezeichnung, wie z. B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer(n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
|
|
physikalisches Erscheinungsbild, Löslichkeit in Wasser, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern verfügbar und falls zutreffend;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
Behandlung vor dem Test, soweit zutreffend (z. B. Erwärmung, Zerkleinerung);
|
|
geprüfte Konzentration(en);
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
ggf. Verweis auf historische Ergebnisse von Positivkontrollen, die geeignete Akzeptanzkriterien dokumentieren.
|
Negativkontrolle:
|
Chemische Kennung, wie beispielsweise IUPAC- oder CAS-Bezeichnung(en), CAS-Nummer(n) und/oder sonstige Kennungen;
|
|
Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
|
|
physikalisches Erscheinungsbild, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, falls andere als die in der Prüfrichtlinie genannten Negativkontrollen verwendet werden, und sofern verfügbar;
|
|
Lagerbedingungen und Stabilität, soweit verfügbar;
|
|
Begründung der Auswahl des Lösungsmittels für jede Prüfchemikalie.
|
|
|
Prüfbedingungen
|
Name und Anschrift des Auftraggebers, der Prüfanstalt und des Studienleiters;
|
|
Beschreibung des verwendeten Prüfprotokolls;
|
|
verwendete Zelllinie, zugehörige Lagerbedingungen und Quelle (z. B. Einrichtung, von der sie gewonnen wurde);
|
|
Chargennummer und Herkunft von FBS, Lieferantenname, Chargennummer der flachen schwarzen Platte mit 96 Mulden und Chargennummer des Tripluc-Reagenz;
|
|
Passagenanzahl und Zelldichte für den Test;
|
|
angewandte Zellzählungsmethode bei der Beimpfung vor dem Test und ergriffene Maßnahmen zur Gewährleistung einer homogenen quantitativen Verteilung der Zellen;
|
|
verwendetes Luminometer (z. B. Modell), einschließlich Geräteeinstellungen, verwendetes Luciferase-Substrat und Nachweis geeigneter Lumineszenzmessungen basierend auf der in Anlage 3.2 beschriebenen Kontrollprüfung;
|
|
angewandtes Verfahren zum Nachweis der Leistungsfähigkeit des Labors hinsichtlich der Durchführung des Tests (z. B. durch Prüfen von Leistungsstoffen) oder zum Nachweis der reproduzierbaren Durchführung des Tests im Zeitverlauf.
|
|
|
Prüfverfahren
|
Anzahl an Replikaten und ausgeführten Durchläufen;
|
|
Konzentrationen der Prüfchemikalie, Applikationsverfahren und Expositionsdauer (falls von den Empfehlungen abweichend);
|
|
Beschreibung der angewandten Bewertungs- und Entscheidungskriterien;
|
|
Beschreibung der angewandten Studienakzeptanzkriterien;
|
|
Beschreibung etwaiger Änderungen am Prüfverfahren.
|
|
|
Ergebnisse
|
Messungen von IL8LA und GAPLA;
|
|
Berechnungen für nIL8LA, Ind-IL8LA und Inh-GAPLA;
|
|
das 95 %ige Konfidenzintervall von Ind-IL8LA;
|
|
Diagramm zur Darstellung der Dosis-Wirkungs-Kurven für die Induktion der Luciferase-Aktivität und Viabilität;
|
|
Beschreibung etwaiger sonstiger relevanter Beobachtungen, falls zutreffend.
|
|
|
Erörterung der Ergebnisse
|
Erörterung der anhand des IL-8-Luc-Tests erhaltenen Ergebnisse;
|
|
Erörterung der Test-Ergebnisse im Rahmen eines IATA, sofern andere sachdienliche Informationen verfügbar sind.
|
|
Schlussfolgerung
|
LITERATURHINWEISE
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359–69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Aasay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371–9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816–30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337–51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275–84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147–155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646–57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286–91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271–9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891–4.
|
|
(15)
|
OECD (2017). To be published - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OECD, Paris, Frankreich
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OECD, Paris, Frankreich.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Verfügbar unter: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, Verfügbar unter. http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046–9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OECD, Paris, Frankreich.
|
|
(21)
|
Vereinte Nationen (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sechste überarbeitete Fassung. New York und Genf: United Nations Publications. ISBN: 978-92-1-117006-1. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Anlage 3.1
DEFINITIONEN
Genauigkeit
: Grad der Übereinstimmung zwischen Prüfergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der „Relevanz“. Der Begriff wird oft im Sinne von „Übereinstimmung“ verwendet und bezeichnet den Anteil der korrekten Ergebnisse eines Tests (16).
AOP (Adverse Outcome Pathway)
: Abfolge von Vorgängen, ausgehend von der chemischen Struktur einer Zielchemikalie oder Zielgruppe ähnlicher Chemikalien, über den molekularen auslösenden Vorgang bis hin zu einem In-vivo-Ergebnis von Interesse (20).
Chemikalie
: Stoff oder Gemisch.
CV05
: Zellviabilität 05, d. h. die Mindestkonzentration, bei der die Chemikalien weniger als 0,05 von Inh-GAPLA aufweisen.
FInSLO-LA
: Abkürzung, die im Validierungsbericht und in vorherigen Veröffentlichungen bezüglich des IL-8-Luc-Tests verwendet wurde, um sich auf Ind-IL8LA zu beziehen. Siehe Ind-IL8LA für eine Definition.
GAPLA
: Luciferase-Aktivität der „Stable Luciferase Red (SLR)“ (λmax = 630 nm), reguliert durch den GAPDH-Träger und zeigt die Zellviabilität und die Anzahl der lebensfähigen Zellen auf.
Gefahr
: Inhärente Eigenschaft eines Stoffes oder einer Situation, potenziell schädigende Auswirkungen zu haben, wenn ein Organismus, ein System oder eine (Teil-)Population diesem Stoff ausgesetzt ist.
IATA (Integrated Approach to Testing and Assessment)
: Ein strukturierter Ansatz zur Gefahrenidentifizierung (Potenzial), Gefahrencharakterisierung (Potenz) und/oder Sicherheitsbeurteilung (Potenzial/Potenz und Exposition) einer Chemikalie oder Gruppe von Chemikalien, der strategisch sämtliche relevanten Daten für eine informierte regulatorische Entscheidung hinsichtlich potenziellen Gefahren und/oder Risiken und/oder des Bedürfnisses weiterer gezielter und somit minimaler Tests integriert und abwägt.
II-SLR-LA
: Abkürzung, die im Validierungsbericht und in vorherigen Veröffentlichungen bezüglich des IL-8-Luc-Tests verwendet wurde, um sich auf Inh-GAPLA zu beziehen. Siehe Inh-GAPLA für eine Definition
IL-8 (Interleukin-8)
: Ein aus Endothelzellen, Fibroblasten, Keratinozyten, Makrophagen und Monozyten abgeleitetes Zytokin, das Chemotaxis von Neutrophilen und T-Zell-Lymphozyten verursacht.
IL8LA
: Luciferase-Aktivität von „Stable Luciferase Orange (SLO)“ (λmax = 580 nm), reguliert durch IL-8-Träger.
Ind-IL8LA
: n-fache Induktion von nIL8LA. Der Wert wird erhalten, indem der nIL8LA der THP-G8-Zellen, die mit Chemikalien behandelt wurden, durch den der nicht stimulierten THP-G8-Zellen geteilt wird und die Induktion der IL-8-Trägeraktivität durch Chemikalien darstellt.
Inh-GAPLA
: Hemmung von GAPLA. Der Wert wird erhalten, indem GAPLA des mit Chemikalien behandelten THP-G8 durch GAPLA des nicht behandelten THP-G8 geteilt wird und die Zytotoxizität der Chemikalien darstellt.
Minimaler Induktionsschwellenwert (MIT)
: die niedrigste Konzentration, bei der eine Chemikalie die positiven Kriterien erfüllt
Gemisch
: Ein Gemisch oder eine Lösung, die aus zwei oder mehreren Stoffen besteht.
Einkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorhanden ist.
Mehrkomponentiger Stoff
: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von ≥ 10 % w/w und < 80 % w/w vorhanden sind. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet.
nIL8LA
: Die SLO-Luciferase-Aktivität, die die IL-8-Trägeraktivität (IL8LA) widerspiegelt, die durch die SLR-Luciferase-Aktivität normalisiert wird, die die GAPDH-Trägeraktivität (GAPLA) widerspiegelt. Der Wert stellt die IL-8-Trägeraktivität nach Berücksichtigung der Zellviabilität oder der Zellanzahl dar.
nSLO-LA
: Abkürzung, die im Validierungsbericht und in vorherigen Veröffentlichungen bezüglich des IL-8-Luc-Tests verwendet wurde, um sich auf nIL8LA zu beziehen. Siehe nIL8LA für eine Definition
Positivkontrolle
: Ein Replikat, das alle Komponenten eines Prüfsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prähaptene
: Chemikalien, die durch abiotische Transformation zu Allergenen werden.
Prohaptene
: Chemikalien, die eine enzymatische Aktivierung erfordern, um ihr Hautsensibilisierungspotenzial auszuschöpfen.
Relevanz
: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Die Relevanz umfasst die Einbeziehung der Genauigkeit (Übereinstimmung) eines Tests (16).
Zuverlässigkeit
: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Labors über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet (16).
Durchlauf
: Ein Durchlauf besteht aus einer oder mehreren hintereinander getesteten Prüfchemikalien mit einer Lösungsmittel-/Vehikelkontrolle und mit einer Positivkontrolle.
Sensitivität
: Der Anteil aller positiven/wirkungsvollen Chemikalien, die durch den Test korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit eines Tests mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (16).
SLO-LA
: Abkürzung, die im Validierungsbericht und in vorherigen Veröffentlichungen bezüglich des IL-8-Luc-Tests verwendet wurde, um sich auf IL8LA zu beziehen. Siehe IL8LA für eine Definition.
SLR-LA
: Abkürzung, die im Validierungsbericht und in vorherigen Veröffentlichungen bezüglich des IL-8-Luc-Tests verwendet wurde, um sich auf GAPLA zu beziehen. Siehe GAPLA für eine Definition.
Lösungsmittel-/Vehikelkontrolle
: Eine unbehandelte Probe, die sämtliche Komponenten eines Prüfsystems enthält, außer der Prüfchemikalie, aber einschließlich des verwendeten Lösungsmittels/Vehikels. Sie wird verwendet, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst oder stabil verteilt wurden, zu bestimmen. Wenn mit einer simultanen Medienkontrolle geprüft wird, zeigt diese Probe außerdem, ob das Lösungsmittel oder Vehikel mit dem Prüfsystem interagiert.
Spezifität
: Der Anteil aller negativen/wirkungslosen Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit eines Tests mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (16).
Stoff
: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität erforderlichen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können.
Tensid
: Dies eine Substanz, auch oberflächenaktives Mittel genannt, wie z. B. ein Waschmittel, die die Oberflächenspannung einer Flüssigkeit reduzieren und ihr somit ermöglichen kann, zu schäumen oder Feststoffe zu durchdringen; ein Tensid wird auch als Netzmittel bezeichnet. (TG437)
Prüfchemikalie
: Stoffe oder Gemische, die mit dieser Methode geprüft wurden.
THP-G8
: Eine IL-8-Reporter-Zelllinie, die im IL-8-Luc-Test verwendet wird. Die humane makrophagenähnliche Zelllinie THP-1 wurde unter der Kontrolle der IL-8- bzw. GAPDH-Träger mit den SLO- und SLR-Luciferasegenen transfiziert.
Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS)
: Ein System, das die Einstufung von Chemikalien (Stoffen und Gemischen) gemäß normierten Typen und Stufen physikalischer, gesundheitlicher und Umweltgefahren vorschlägt und entsprechende Kommunikationselemente wie Piktogramme, Signalwörter, Gefahrenberichte, Berichte zu Vorsichtsmaßnahmen und Sicherheitsdatenblätter anspricht, damit Informationen zu Nebenwirkungen mit Blick auf den Schutz von Menschen (einschließlich Mitarbeitern, Arbeitern, Logistikern, Verbrauchern und Ersthelfern) und der Umwelt zu übermitteln (21).
UVCB
: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Gültige Prüfmethode
: Ein Test, von der angenommen wird, dass sie über ein ausreichendes Maß an Relevanz und Zuverlässigkeit für einen bestimmten Zweck verfügt und die auf wissenschaftlich bewährten Prinzipien basieren. Ein Test ist niemals in einem absoluten Sinn gültig, sondern im Verhältnis zu einem definierten Zweck.
Anlage 3.2
GRUNDSATZ DER MESSUNG DER LUCIFERASE-AKTIVITÄT UND BESTIMMUNG DER ÜBERTRAGUNGSKOEFFIZIENTEN DES OPTISCHEN FILTERS FÜR SLO UND SLR
Das MultiReporter-Prüfsystem -Tripluc- kann mit einem Luminometer des Typs Mikroplatte mit einem mehrfarbigen Erkennungssystem verwendet werden, das einen optischen Filter ausstatten kann (z. B. Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). Der bei Messungen verwendete optische Filter ist ein 600–620 nm Lang- oder Kurzpassfilter oder ein 600–700 nm Bandpassfilter.
Messung der zweifarbigen Luciferasen mit einem optischen Filter.
Dies ist ein Beispiel, das Phelios AB-2350 (ATTO) verwendet. Dieses Luminometer ist mit einem 600 nm Langpassfilter (R60 HOYA Co., 600 nm LP, Filter 1) zum Splitten der SLO- (λmax = 580 nm) und SLR-Lumineszenz (λmax = 630 nm) ausgestattet.
Um die Übertragungskoeffizienten des 600 nm LP zu bestimmen muss zuerst mit aufbereiteten SLO- und SLR-Luciferase-Enzymen i) die SLO- und SLR-Biolumineszenzintensität ohne Filter (F0), ii) die SLO- und SLR-Biolumineszenzintensität, die durch den 600 nm LP (Filter 1) passiert ist, gemessen werden und iii) die Übertragungskoeffizienten des 600 nm LP für die unten aufgeführten SLO und SLR berechnet werden.
|
Übertragungskoeffizienten
|
Abkürzung
|
Definition
|
|
SLO
|
Filter 1 Übertragungskoeffizient
|
κOR60
|
Definition Übertragungskoeffiziet des Filters für die SLO
|
|
SLR
|
Filter 1 Übertragungskoeffizient
|
κRR60
|
Definition Übertragungskoeffiziet des Filters für die SLR
|
Wenn die Intensität von SLO und SLR in Prüfproben als O bzw. R definiert ist, werden i) die Intensität des Lichts ohne Filter (alle optisch) F0 und ii) die Intensität des Lichts, das durch den 600 nm LP (Filter 1) F1 geschickt wird, wie unten beschrieben.
F0 = O + R
F1 = κOR60 x O + κRR60 x R
Diese Formeln können wie folgt umformuliert werden:

Dann können anhand der berechneten Transmissionsfaktoren (κOR60 und κRR60) der gemessenen F0 and F1 die O- und R-Werte wie folgt berechnet werden:

Materialien und Methoden zur Bestimmung des Transmissionsfaktors
|
(1)
|
Reagenzien
Einzelne aufbereitete Luciferase-Enzyme:
|
Lyophilisiertes aufbereitetes SLO-Enzym
|
|
Lyophilisiertes aufbereitetes SLR-Enzym
|
|
(die für die Validierungsarbeiten von GPC Lab. Co. Ltd., Tottori, Japan mit THP-G8-Zelllinie erhalten wurden)
|
Test-Reagenz:
|
Tripluc® Luciferase-Test-Reagenz (zum Beispiel aus TOYOBO Kat. Nr. MRA-301)
|
Medium: für Luciferase-Test (30 ml, gelagert bei 2–8°C)
|
|
Reagenz
|
Konz.
|
Finale Konz. in Medium
|
Erforder-liche Menge
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
(2)
|
Herstellung der Enzymlösung
Das lyophilisierte, aufbereitete Luciferase-Enzym im Röhrchen lösen, indem 200 μl 10 ~ 100 mM Tris/HCl oder Hepes/HCl (pH 7,5 ~ 8,0) mit 10 % (w/v) Glycerol hinzugefügt werden, die Enzymlösung in 10 μl Aliquoten in Einwegröhrchen mit einem Volumen von 1,5 ml aufgeteilt wird und diese bei -80 °C in einem Gefrierschrank gelagert werden. Die gefrorene Enzymlösung kann bis zu 6 Monate verwendet werden. Wenn es verwendet wird, dann 1 ml des Mediums für den Luciferase-Test (RPMI-1640 mit 10 % FBS) zu jedem Röhrchen mit den Enzymlösungen (verdünnte Enzymlösung) hinzufügen und diese auf Eis halten, um eine Deaktivierung zu verhindern.
|
|
(3)
|
Messung der Biolumineszenz
Tripluc®-Luciferase-Test-Reagenz (Tripluc) auftauen und bei Raumtemperatur entweder in einem Wasserbad oder bei Umgebungslufttemperatur aufbewahren. Das Luminometer 30 Minuten vor Beginn der Messungen einschalten, damit sich der Fotovervielfacher stabilisieren kann. 100 μl der verdünnten Enzymlösung in eine schwarze Platte mit 96 Mulden übertragen (flache Unterseite) (die SLO-Referenzprobe zu #B1, #B2, #B3, die SLR-Referenzprobe zu #D1, #D2, #D3). Anschließend 100 μl vorgewärmtes Tripluc mithilfe eines Pipetman in jede Mulde der Platte geben, die die verdünnte Enzymlösung enthält. Die Platte 10 Minuten lang bei Raumtemperatur (ca. 25 °C) mit einem Platten-Schüttler schütteln. Die Luftblasen aus den Lösungen in den Mulden entfernen, wenn diese erscheinen. Die Platte im Luminometer platzieren, um die Luciferase-Aktivität zu messen. Die Biolumineszenz wird jeweils 3 Sekunden lang in der Abwesenheit (F0) und Anwesenheit (F1) des optischen Filters gemessen.
Der Übertragungskoeffizient des optischen Filters wurde wie folgt berechnet:
Übertragungskoeffizient (SLO (κOR60))= (Anz. B1 von F1+ Anz. B2 von F1+ Anz. B3 von F1) / (Anz. B1 von F0+ Anz. B2 von F0+ Anz. B3 von F0)
Übertragungskoeffizient (SLR (κRR60))= (Anz. D1 von F1+ Anz. D2 von F1+ Anz. D3 von F1) / (Anz. D1 von F0+ Anz. D2 von F0+ Anz. D3 von F0)
Die berechneten Transmissionsfaktoren werden für alle Messungen verwendet, die mit demselben Luminometer durchgeführt werden.
|
Qualitätskontrolle der Ausrüstung
Es sollten die im IL-8-Luc-Protokoll beschriebenen Verfahren verwendet werden (18).
Anlage 3.3
LEISTUNGSSTOFFE
Vor der routinemäßigen Anwendung des in dieser Anlage zu Prüfmethode B.71 beschriebenen Tests sollten Labors ihre technische Leistungsfähigkeit demonstrieren, indem sie die erwartete IL-8-Luc-Vorhersage für die in Tabelle 1 empfohlenen 9 Stoffe ermitteln und die Werte, die in den betreffenden Referenzbereich fallen, bei mindestens 8 der 9 Leistungsstoffe bestimmen (ausgewählt, um den Reaktionsbereich für Hautsensibilisierungsgefahren zu repräsentieren). Weitere Auswahlkriterien betrafen die Erhältlichkeit der Stoffe im Handel, und dass qualitativ hochwertige In-vivo-Referenzdaten sowie qualitativ hochwertige In-vitro-Daten aus dem IL-8-Luc-Test verfügbar sind. Außerdem sind veröffentlichte Referenzdaten für den IL-8-Luc-Test verfügbar (6) (1).
Tabelle 1
Empfohlene Stoffe zum Nachweis der technischen Leistungsfähigkeit beim IL-8-Luc-Test
|
Leistungsstoffe
|
CAS Nr.
|
Zustand
|
Löslichkeit in X-VIVO15 bei 20 mg/ml
|
In-vivo-Vorhersage (109)
|
IL-8 Luc-Vorhersage (110)
|
Referenzbereich (μg/ml) (111)
|
|
|
CV05
(112)
|
IL-8 Luc MIT (113)
|
|
2,4-Dinitrochlorbenzol
|
97-00-7
|
fest
|
unlöslich
|
Sensibilisator (extrem)
|
positiv
|
2,3–3,9
|
0,5–2,3
|
|
Formaldehyd
|
50-00-0
|
flüssig
|
löslich
|
Sensibilisator (stark)
|
positiv
|
9–30
|
4–9
|
|
2-Mercaptobenzothiazol
|
149-30-4
|
fest
|
unlöslich
|
Sensibilisator (mäßig)
|
positiv
|
250–290
|
60–250
|
|
Ethylendiamin
|
107-15-3
|
flüssig
|
löslich
|
Sensibilisator (mäßig)
|
positiv
|
500–700
|
0,1–0,4
|
|
Ethylenglykoldimethacrylat
|
97-90-5
|
flüssig
|
unlöslich
|
Sensibilisator (schwach)
|
positiv
|
> 2 000
|
0,04–0,1
|
|
4-Allylanisol (Estragol)
|
140-67-0
|
flüssig
|
unlöslich
|
Sensibilisator (schwach)
|
positiv
|
> 2 000
|
0,01–0,07
|
|
Streptomycinsulfat
|
3810-74-0
|
fest
|
löslich
|
Nichtsensibi-lisator
|
negativ
|
> 2 000
|
> 2 000
|
|
Glyzerin
|
56-81-5
|
flüssig
|
löslich
|
Nichtssensibi-lisator
|
negativ
|
> 2 000
|
> 2 000
|
|
Isopropanol
|
67-63-0
|
flüssig
|
löslich
|
Nichtsensibi-lisator
|
negativ
|
> 2 000
|
> 2 000
|
|
Abkürzungen: CAS-Nr. = Registernummer des Chemical Abstracts Service
|
Anlage 3.4
INDIZES UND URTEILSKRITERIEN
nIL8LA (nSLO-LA)
Die j. Wiederholung (j = 1–4) der i. Konzentration (i = 0–11) wird für IL8LA (SLO-LA) bzw. GAPLA (SLR-LA) gemessen. Die normalisierte IL8LA wird bezeichnet als nIL8LA (nSLO-LA) und definiert als:
nIL8LAij = IL8LAij/GAPLAij
Dies ist die Grundmaßeinheit in diesem Test.
Ind-IL8LA (FInSLO-LA)
Die vielfache Steigerung der gemittelten nIL8LA (nSLO-LA) für die Wiederholung auf der i. Konzentration im Vergleich zu ihr bei der 0-Konzentration, Ind-IL8LA, ist die primäre Messung dieser Tests. Das Verhältnis wird durch die folgende Formel bestimmt:

Das Hauptlabor hat vorgeschlagen, dass ein Wert von 1,4 einem positiven Ergebnis für die geprüfte Chemikalie entspricht. Dieser Wert basiert auf der Untersuchung der historischen Daten des Hauptlabors. Das Datenmanagement-Team nutzte diesen Wert dann in allen Phasen der Validierungsstudie. Das primäre Ergebnis, Ind-IL8LA, ist das Verhältnis von 2 arithmetischen Mitteln, wie sie in der Gleichung dargestellt werden.
95 % Konfidenzintervall (95 % CI)
Das auf dem Verhältnis basierende 95 % Konfidenzintervall (95 % CI) kann so eingeschätzt werden, dass es die Präzision dieser primären Ergebnismessung zeigt. Die untere Grenze des 95 % CI ≥ 1 zeigt an, dass die nIL8LA mit der i. Konzentration deutlich größer ist als die mit Lösungsmittelkontrolle. Es gibt verschiedene Arten, auf die das 95 % CI erstellt wird. Wir nutzten die Methode, die in dieser Studie als Fiellers Theorem bekannt ist. Das Theorem des 95 % Konfidenzintervalls wird aus der folgenden Formel erhalten:

Dabei gilt:
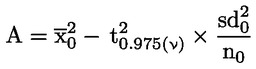
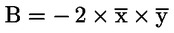


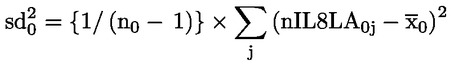


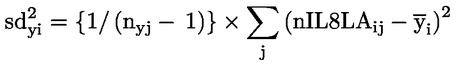
t0,975(ν) ist das 97,5te Perzentil der zentralen t-Verteilung mit dem ν des Freiheitsgrades, wobei
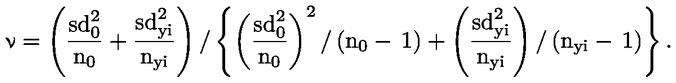
Inh-GAPLA (II-SLR-LA)
Die Inh-GAPLA ist ein Verhältnis der gemittelten GAPLA (SLR-LA) für die Wiederholung der i. Konzentration im Vergleich zu der mit Lösungsmittelkontrolle und dies wird folgendermaßen bestimmt:
Da die GAPLA der Nenner der nIL8LA ist, verursacht ein extrem kleiner Wert eine große Schwankung der nIL8LA. Deshalb können Ind-IL8LA-Werte mit einem extrem kleinen Wert von Inh-GAPLA (weniger als 0,05) als eine schlechte Präzision angesehen werden.
Anlage 3.5
DAS SCHEMA DER METHODEN ZUM LÖSEN VON CHEMIKALIEN FÜR DEN IL-8-LUC-TEST.
|
(a)
|
Bei Chemikalien, die bei 20 mg/ml in X-VIVOTM 15 gelöst sind

|
|
(b)
|
Bei Chemikalien, die bei 20 mg/ml in X-VIVOTM 15 unlöslich sind

|
Anlage 3.6
DAS SCHEMA DER METHODE ZUM LÖSEN VON 4-NBB FÜR DIE POSITIVKONTROLLE DES IL-8-LUC-TESTS.

“ |