|
29.3.2008
|
DE
|
Amtsblatt der Europäischen Union
|
L 88/1
|
VERORDNUNG (EG) Nr. 273/2008 DER KOMMISSION
vom 5. März 2008
mit Durchführungsbestimmungen zu der Verordnung (EG) Nr. 1255/1999 des Rates hinsichtlich der Methoden für die Analyse und Qualitätsbewertung von Milch und Milcherzeugnissen
DIE KOMMISSION DER EUROPÄISCHEN GEMEINSCHAFTEN —
gestützt auf den Vertrag zur Gründung der Europäischen Gemeinschaft,
gestützt auf die Verordnung (EG) Nr. 1255/1999 des Rates vom 17. Mai 1999 über die gemeinsame Marktorganisation für Milch und Milcherzeugnisse (1), insbesondere auf die Artikel 10 und 15, Artikel 26 Absatz 3, Artikel 29 Absatz 1 und Artikel 31 Absatz 4,
in Erwägung nachstehender Gründe:
|
(1)
|
In der Verordnung (EG) Nr. 213/2001 der Kommission (2) wird die Umsetzung der Verordnung (EG) Nr. 1255/1999 des Rates hinsichtlich der Methoden zur Analyse und zur Qualitätsbewertung von Milch und Milcherzeugnissen ausführlich erläutert. In Anbetracht der technischen Entwicklungen im Bereich der Analysemethoden ist festzustellen, dass weitere erhebliche Änderungen vorgenommen werden müssen. Im Interesse der Klarheit und der Wirksamkeit sowie angesichts der Anzahl und der technischen Merkmale der Änderungen ist die Verordnung (EG) Nr. 213/2001 aufzuheben und durch eine neue Verordnung zu ersetzen.
|
|
(2)
|
Die Zusammensetzung und die Qualitätsmerkmale von Milch und Milcherzeugnissen sind gemäß der Verordnung (EG) Nr. 1255/1999 zu prüfen, um die genaue Übereinstimmung mit den vorgesehenen Anforderungen sicherzustellen.
|
|
(3)
|
Referenzmethoden für diese Prüfungen sind häufig von internationalen Organisationen wie etwa dem Europäischen Normenausschuss (CEN), der IDF (International Dairy Federation), der ISO (International Standards Organisation) und der AOAC International (Association of Official Analytical Chemists) veröffentlichte Methoden, die regelmäßig von diesen Organisationen aktualisiert werden. In bestimmten Fällen wird eine Referenzmethode der Gemeinschaft erlassen; in anderen Fällen enthalten die Gemeinschaftsvorschriften keine Referenzmethode. Um sicherzustellen, dass Referenzmethoden einheitlich angewendet werden, ist eine Liste mit Referenzmethoden zu erstellen und vorzusehen, dass die Kommission diese Liste gegebenenfalls anpasst.
|
|
(4)
|
Die Verwendung von Routinemethoden sollte nicht ausgeschlossen werden; entsprechend sind auch die maßgeblichen Mindestanforderungen zu spezifizieren.
|
|
(5)
|
Ferner sind gemeinsame Verfahren festzulegen, um eine einheitliche Praxis für die Bewertung der Analyseergebnisse herzustellen. Dies gilt auch für die sensorische Prüfung der betreffenden Erzeugnisse und die Überprüfung strittiger Ergebnisse.
|
|
(6)
|
Für die Durchführung bestimmter Analysen existieren zurzeit keine international anerkannten und validierten Referenzmethoden; daher sind keine Informationen über die Unterschiede zwischen den Analyseergebnissen der einzelnen Laboratorien verfügbar. Somit sind Methoden auf Gemeinschaftsebene festzulegen, die nach international festgelegten Regeln validiert wurden und als Referenzmethoden angewandt werden sollten.
|
|
(7)
|
Die Verordnung (EG) Nr. 1898/2005 der Kommission (3) enthält ausführliche Bestimmungen zur Anwendung der Verordnung (EG) Nr. 1255/1999 betreffend Maßnahmen zum Absatz von Rahm, Butter und Butterfett auf dem Gemeinschaftsmarkt und sieht in bestimmten Fällen die Kennzeichnung von Rahm, Butter und Butterfett vor, um eine bestimmungsgemäße Endverwendung dieser Erzeugnisse sicherzustellen. Die Kennzeichnung ist für ein reibungsloses Funktionieren des Systems wichtig. Um sicherzustellen, dass die beteiligten Marktteilnehmer gleich behandelt werden, sollten gemeinsame Methoden für die Bestimmung einiger dieser Kennzeichnungsmittel festgelegt werden.
|
|
(8)
|
Für die private Lagerhaltung von aus Schaf- oder Ziegenmilch hergestellten Käsesorten kann gemäß Artikel 9 der Verordnung (EG) Nr. 1255/1999 eine Beihilfe gewährt werden. Für diese Erzeugnisse kann gemäß Artikel 31 der genannten Verordnung außerdem eine besondere Erstattung gewährt werden. Für die Einfuhr von Käse aus Schaf-, Ziegen- oder Büffelmilch oder aus Gemischen von Schaf-, Ziegen- oder Büffelmilch aus bestimmten Drittländern in die Gemeinschaft gelten Präferenzregelungen. Wegen der genannten Vorschriften muss im Wege geeigneter Kontrollen nachgewiesen werden, dass dem betreffenden Erzeugnis keine Kuhmilch zugesetzt wurde. Es empfiehlt sich daher, eine gemeinschaftliche Referenzmethode für den Kuhmilchnachweis festzulegen, die auch mit Routinemethoden durchgeführt werden kann, wenn sie bestimmte Kriterien erfüllen.
|
|
(9)
|
Gemäß der Verordnung (EWG) Nr. 2921/90 der Kommission vom 10. Oktober 1990 über die Gewährung von Beihilfen für zur Herstellung von Kasein und Kaseinaten bestimmte Magermilch (4) muss nachgewiesen werden, dass keine coliformen Keime vorhanden sind. ISO 4831 ist international als Referenzmethode für den Nachweis coliformer Keime in Milch und Milcherzeugnissen anerkannt. Ausgehend von dieser Norm wird daher eine Referenzmethode der Gemeinschaft für die Untersuchung auf coliforme Keime beschrieben.
|
|
(10)
|
Die Verordnung (EWG) Nr. 2658/87 des Rates vom 23. Juli 1987 über die zolltarifliche und statistische Nomenklatur sowie den Gemeinsamen Zolltarif (5) sieht abhängig vom Anteil an Milcherzeugnissen unterschiedliche Zollsätze für Mischfuttermittel vor, die der Tarifposition Nr. 2309 zuzurechnen sind. Um eine einheitliche Anwendung der betreffenden Bestimmungen zu gewährleisten, ist eine allgemein anerkannte und für alle Mitgliedstaaten verbindliche Methode zur Bestimmung des Laktosegehalts festzulegen.
|
|
(11)
|
Gemäß der Verordnung (EG) Nr. 1255/1999 müssen bei Butter und Magermilchpulver, die zur Intervention bestimmt sind, bzw. bei Magermilchpulver zur Tierfütterung bestimmte Qualitätsbedingungen eingehalten werden. Daher sind Referenzmethoden für die Überprüfung der Einhaltung dieser Bedingungen festzulegen.
|
|
(12)
|
Einige Methoden werden in dieser Verordnung erstmals eingeführt. Nach Inkrafttreten dieser Verordnung sollte ausreichend Zeit eingeräumt werden, um diese Methoden ordnungsgemäß einführen und anwenden zu können. Wenn eine in Anhang I genannte Methode von der Normungsbehörde geändert und anschließend veröffentlicht wird, sollte den Laboratorien eine Frist von sechs Monaten zur Anpassung ihrer Analyseverfahren an die neue Norm gewährt werden.
|
|
(13)
|
Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Verwaltungsausschusses für Milch und Milcherzeugnisse —
|
HAT FOLGENDE VERORDNUNG ERLASSEN:
KAPITEL I
ALLGEMEINE BESTIMMUNGEN
Artikel 1
Gegenstand und Anwendungsbereich
(1) Mit dieser Verordnung werden bestimmte Referenzmethoden für die chemische, physikalische und mikrobiologische Analyse und für die sensorische Prüfung von Milch und Milcherzeugnissen gemäß den Regelungen im Rahmen der mit der Verordnung (EG) Nr. 1255/1999 festgelegten gemeinsamen Marktorganisation für Milch und Milcherzeugnisse erlassen und Regelungen für die Anwendung dieser Methoden beschrieben.
(2) Anhang I dieser Verordnung enthält eine Liste der Referenzmethoden, die bei den in Absatz 1 genannten Analysen anzuwenden sind.
(3) Die Kommission bringt die genannte Liste mindestens einmal jährlich nach dem Verfahren des Artikels 42 der Verordnung (EG) Nr. 1255/1999 auf den neusten Stand.
Artikel 2
Routinemethoden
Für die gemäß den Gemeinschaftsvorschriften erforderlichen Analysen dürfen Routinemethoden verwendet werden, wenn an die Routinemethoden die gleichen Maßstäbe wie an die Referenzmethode angelegt werden und die Routinemethoden regelmäßig überprüft werden. Die Ergebnisse werden unter Berücksichtigung des jeweiligen systematischen Fehlers sowie der Wiederholbarkeit und der Vergleichbarkeit verglichen.
Im Streitfalle sind die Ergebnisse der Referenzmethode maßgeblich.
Die Mitgliedstaaten benachrichtigen die Kommission über die Anwendung von Routinemethoden bei den in Artikel 1 genannten Analysen.
KAPITEL II
ANALYSEMETHODEN
Artikel 3
Bewertung der Konformität einer Partie mit den gesetzlichen Grenzwerten
Außer bei der Analyse von Kennzeichnungsmitteln ist die Konformität mit den gesetzlichen Anforderungen an die Zusammensetzung der Erzeugnisse nach Maßgabe von Anhang II dieser Verordnung nachzuweisen.
Artikel 4
Sensorische Prüfung
(1) Bei Milch und anderen Milcherzeugnissen als Butter, die zur öffentlichen Lagerhaltung vorgesehen sind, wenden die Mitgliedstaaten als Referenzmethode für die sensorische Prüfung entweder die Norm IDF 99C:1997 oder andere vergleichbare Methoden an, die sie der Kommission mitteilen.
Die Methoden gemäß Anhang III können für die Überprüfung der Leistung der Prüfpersonen und der Zuverlässigkeit der Ergebnisse verwendet werden.
(2) Bei Butter zur öffentlichen Lagerhaltung werden die Methoden gemäß Anhang III für die Überprüfung der Leistung der Prüfpersonen und der Zuverlässigkeit der Ergebnisse verwendet.
Die in Anhang IV beschriebene Methode wird als Referenzmethode für die sensorische Prüfung angewendet.
Artikel 5
Kennzeichnungsmittel
(1) Die in Anhang V beschriebene Analysemethode wird als Referenzmethode zur Bestimmung des Anteils an Önanthsäure-Triglycerid in Butter, Butterschmalz und Rahm verwendet.
(2) Die in Anhang VI beschriebene Analysemethode wird als Referenzmethode zur Bestimmung von Vanillin in Butterfett, Butter oder Rahm angewendet.
(3) Die in Anhang VII beschriebene Analysemethode wird als Referenzmethode zur Bestimmung des Gehalts an Beta-Apo-8'-Karotinsäure-Ethylester in Butterfett oder Butter angewendet.
(4) Die in Anhang VIII beschriebene Analysemethode wird als Referenzmethode zur Bestimmung des β-Sitosterin- oder Stigmasteringehalts von Butter und Butterfett angewendet.
(5) Butterfett und Rahm werden als gemäß der geltenden Gemeinschaftsregelung kennzeichnungspflichtig betrachtet, wenn die ermittelten Ergebnisse im Einklang mit den Spezifikationen der Nummern 10 und 11 in Anhang V und von Nummer 8 der Anhänge VI, VII und VIII stehen.
Artikel 6
Nachweis von Kuhmilchkasein
(1) Damit gewährleistet ist, dass ausschließlich aus Schaf-, Ziegen- oder Büffelmilch oder aus Gemischen von Schaf-, Ziegen- oder Büffelmilch herzustellender Käse kein Kuhmilchkasein enthält, ist für die Untersuchung die in Anhang IX beschriebene Referenzmethode anzuwenden.
Kuhmilchkasein gilt als nachgewiesen, wenn der festgestellte Kuhmilchkaseingehalt der Analyseprobe gleich dem Gehalt der in Anhang IX beschriebenen 1 % Kuhmilch enthaltenden Referenzprobe oder größer ist.
(2) Zum Nachweis von Kuhmilchkasein in Käse der in Absatz 1 genannten Kategorien dürfen Routinemethoden verwendet werden, wenn sie folgende Bedingungen erfüllen:
|
a)
|
Die Nachweisgrenze beträgt maximal 0,5 %,
|
|
b)
|
es wurden keine falsch-positiven Ergebnisse ermittelt,
|
|
c)
|
auch nach im Handel üblichen längeren Reifezeiten ist eine ausreichende Empfindlichkeit zum Nachweis von Kuhmilchkasein gegeben.
|
Wenn eine dieser Bedingungen nicht erfüllt ist, kommt die in Anhang IX beschriebene Referenzmethode zur Anwendung.
Artikel 7
Untersuchung auf coliforme Keime
Coliforme Keime in Butter, Magermilchpulver, Kasein und Kaseinaten werden gemäß der in Anhang X beschriebenen Referenzmethode nachgewiesen.
Artikel 8
Nachweis des Laktosegehalts
Der Laktosegehalt von Produkten des KN-Codes 2309 wird gemäß der in Anhang XI beschriebenen Referenzmethode bestimmt.
Artikel 9
Nachweis von Labmolke
(1) Labmolke in zur öffentlichen Lagerhaltung vorgesehenem Magermilchpulver ist gemäß der in Anhang XII beschriebenen Referenzmethode nachzuweisen.
(2) Labmolke in Magermilchpulver und in Gemischen zur Tierfütterung ist gemäß der in Anhang XII beschriebenen Referenzmethode nachzuweisen. Wenn Labmolke nachgewiesen wurde, ist gemäß Anhang XIII zu verfahren.
Artikel 10
Nachweis von Buttermilch
Buttermilch in Magermilchpulver ist gemäß der in Anhang XIV beschriebenen Referenzmethode nachzuweisen.
Artikel 11
Nachweis von Antimikrobiotika-Rückständen
Rückstände von Antimikrobiotika in Magermilchpulver sind mit der in Anhang XV beschriebenen Referenzmethode nachzuweisen.
Artikel 12
Ermittlung des Gehalts an Magermilchpulver
Der Gehalt an Magermilchpulver ist gemäß der in Anhang XVI beschriebenen Referenzmethode nachzuweisen.
Artikel 13
Nachweis von Stärke
Stärke in Magermilchpulver, denaturierten Milchpulver und Mischfutter ist gemäß der in Anhang XVII beschriebenen Referenzmethode nachzuweisen.
Artikel 14
Bestimmung der Feuchtigkeit von Trockenrahm
Die Feuchtigkeit von Trockenrahm ist gemäß der in Anhang XVIII beschriebenen Referenzmethode zu bestimmen.
Artikel 15
Bestimmung der Feuchtigkeit von saurem Buttermilchpulver
Die Feuchtigkeit von saurem Buttermilchpulver zur Tierfütterung ist gemäß der in Anhang XIX beschriebenen Referenzmethode zu bestimmen.
Artikel 16
Bestimmung der Reinheit von Milchfett
Die Reinheit von Milchfett ist gemäß der in Anhang XX beschriebenen Referenzmethode zu bestimmen.
KAPITEL III
SCHLUSSBESTIMMUNGEN
Artikel 17
Qualitätssicherung
Analysen werden in Laboratorien mit bestehendem System zur Sicherung der Analysequalität und mit internen Qualitätskontrollverfahren durchgeführt. Nicht zugelassene Laboratorien unterziehen sich mindestens einmal jährlich Leistungstests, und ihre Ergebnisse dürfen vom gemeinsamen Wert höchstens um 2σR (in der Referenzmethode vorgesehene Wiederholstandardabweichung) abweichen. Eine genaue Beschreibung der angewendeten Methode muss im Laboratorium verfügbar sein.
Gemäß den in Artikel 12 der Verordnung (EG) Nr. 882/2004 des Europäischen Parlaments und des Rates vom 29. April 2004 über amtliche Kontrollen zur Überprüfung der Einhaltung des Lebensmittel- und Futtermittelrechts sowie der Bestimmungen über Tiergesundheit und Tierschutz (6) genannten Anforderungen zugelassene Laboratorien sind von der Verpflichtung zur Teilnahme an Leistungsprüfungen befreit.
Artikel 18
Probenahme und Beanstandung der Analyseergebnisse
(1) Probenahmen erfolgen gemäß der geltenden Verordnung für das jeweils zu prüfende Erzeugnis. Wenn keine sonstigen Bestimmungen zur Probenahme genannt werden, sind die Bestimmungen in ISO 707|IDF 50, Milk and milk products — Guidance of sampling zu berücksichtigen.
(2) Laborberichte über die Ergebnisse der Analyse müssen genügend Informationen für eine Bewertung der Ergebnisse gemäß den Anhängen II und XXI beinhalten.
(3) Für die in der Gemeinschaftsregelung vorgesehenen Analysen sind Doppelproben zu entnehmen.
(4) Das in Anhang XXI dargestellte Verfahren wird in Fällen verwendet, in denen die Analyseergebnisse vom Marktteilnehmer nicht akzeptiert werden.
(5) Erbringt der Erzeuger innerhalb von fünf Tagen nach der Probenahme den Nachweis, dass diese nicht ordnungsgemäß durchgeführt wurde, muss die Probenahme möglichst wiederholt werden. Ist eine erneute Probenahme nicht möglich, wird die Partie angenommen.
Artikel 19
Übergangsfrist
Eine Bewertung der Konformität gemäß Anhang II dieser Verordnung erfolgt binnen 12 Monaten nach Inkrafttreten der Verordnung. Wenn in diesem Zeitraum gravierendere Probleme mit dem statistischen Kontrollverfahren auftreten, übermitteln die Mitgliedstaaten der Kommission gegebenenfalls unverzüglich einen entsprechenden Bericht.
Artikel 20
Aufgehobene Verordnungen
Die Verordnung (EG) Nr. 213/2001 wird aufgehoben.
Verweise auf die aufgehobene Verordnung sind als Verweise auf diese Verordnung zu verstehen und gemäß der Korrelationstabelle in Anhang XXII auszulegen.
Artikel 21
Inkrafttreten
Diese Verordnung tritt am dritten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Sie gilt ab dem 31. März 2008.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 5. März 2008
Für die Kommission
Mariann FISCHER BOEL
Mitglied der Kommission
(1) ABl. L 160 vom 26.6.1999, S. 48. Verordnung zuletzt geändert durch die Verordnung (EG) Nr. 1152/2007 (ABl. L 258 vom 4.10.2007, S. 3). Verordnung (EG) Nr. 1255/1999 wird ab 1. Juli 2008 durch die Verordnung (EG) Nr. 1234/2007 (ABl. L 299 vom 16.11.2007, S. 1) ersetzt.
(2) ABl. L 37 vom 7.2.2001, S. 1.
(3) ABl. L 308, 25.11.2005, S. 1. Verordnung zuletzt geändert durch die Verordnung (EG) Nr. 1546/2007 (ABl. L 337 vom 21.12.2007, S. 68).
(4) ABl. L 279, 11.10.1990, S. 22. Verordnung zuletzt geändert durch die Verordnung (EG) Nr. 1487/2006 (ABl. L 278 vom 10.10.2006, S. 8).
(5) ABl. L 256 vom 7.9.1987, S. 1. Verordnung zuletzt geändert durch die Verordnung (EG) Nr. 1352/2007 der Kommission (ABl. L 303 vom 21.11.2007, S. 3).
(6) ABl. L 165 vom 30.4.2004, S. 1.
ANHANG I
(Artikel 1)
LISTE DER REFERENZMETHODEN
Index Mind. = Mindestwert, Max. = Höchstwert, Anhang = Anhang der zitierten Verordnung, FFTM = Fettfreie Trockenmasse, PZ = Peroxidzahl, A = Aussehen, G = Geschmack, K = Konsistenz, TBC = Gesamtkolonienzahl, Therm = thermophile Keimzahl, MS = Mitgliedstaat, IDF = International Dairy Federation, ISO = International Standards Organisation, IUPAC = International Union of Pure and Applied Chemistry, ADPI = American Dairy Products Institute, LCS = gezuckerte Kondensmilch, LCC = eingedampfte Milch oder Sahne).
TEIL A
|
Verordnung der Kommission
|
Erzeugnis
|
Parameter
|
Grenzwert (1)
|
Referenzmethode
|
Anmerkung
|
|
Verordnung (EG) Nr. 2771/1999 — Öffentliche Lagerhaltung
|
Ungesalzene Butter
|
Fett
|
Mind. 82 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Wasser
|
Bis zu 16 % m/m
|
ISO 3727-1:2001|IDF 80-1:2001
|
|
|
|
|
FFTM
|
Bis zu 2 % m/m
|
ISO 3727-2:2001|IDF 80-2:2001
|
|
|
|
|
Säuregrad
|
1,2 mmol/100 g Fett
|
ISO 1740:2004|IDF 6:2004
|
|
|
|
|
PZ (max.)
|
0,3 mEq. Sauerstoff/1 000 g Fett
|
ISO 3976:2006|IDF 74:2006
|
Anmerkung 1
|
|
|
|
Coliforme Keime
|
Nicht nachweisbar in 1 g
|
Anhang X
|
Anmerkung 3
|
|
|
|
Fremdfett
|
Nicht nachweisbar durch Triglyceridanalyse
|
Anhang XX
|
|
|
|
|
Sterol-Kennzeichnungsmittel
|
Nicht nachweisbar, β-Sitosterin ≤40 mg/kg
|
Anhang VIII
|
|
|
|
|
Sonstige Kennzeichnungsmittel
|
|
|
|
|
|
|
|
Nicht nachweisbar
|
Anhang VI
|
|
|
|
|
|
—
|
Carotinsäure-Ethylester
|
|
≤6 mg/kg
|
Anhang VII
|
|
|
|
|
|
—
|
Önanthsäure-Triglyceride
|
|
Nicht nachweisbar
|
Anhang V
|
|
|
|
|
Sensorische Merkmale
|
Mindestens 4 von 5 Punkten für Aussehen, Geschmack und Konsistenz
|
Anhang IV
|
|
|
|
|
Wasserdispersion
|
Mindestens 4 Punkte
|
ISO 7586:1985|IDF 112A:1989
|
|
|
Verordnung (EG) Nr. 2771/1999 — Private Lagerhaltung
|
Ungesalzene Butter
|
Fett
|
Mind. 82 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Wasser
|
Bis zu 16 % m/m
|
ISO 3727-1:2001|IDF 80-1:2001
|
|
|
|
|
FFTM
|
Bis zu 2 % m/m
|
ISO 3727-2:2001|IDF 80-2:2001
|
|
|
Verordnung (EG) Nr. 2771/1999 — Private Lagerhaltung
|
Gesalzene Butter
|
Fett
|
Mind. 80 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Wasser
|
Bis zu 16 % m/m
|
ISO 3727-1:2001|IDF 80-1:2001
|
|
|
|
|
FFTM (ohne Salz)
|
Bis zu 2 % m/m
|
ISO 3727-2:2001|IDF 80-2:2001
|
|
|
|
|
Salz
|
Bis zu 2 % m/m
|
ISO 15648:2004|IDF 179:2004
|
|
|
Verordnung (EG) Nr. 1898/2005 Kapitel II
|
Ungesalzene Butter
|
Fett
|
Mind. 82 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Fremdfett
|
|
Anhang XX
|
|
|
|
|
Wasser
|
Bis zu 16 % m/m
|
ISO 3727-1 2001|IDF 80-1:2001
|
|
|
|
|
FFTM
|
Bis zu 2 % m/m
|
ISO 3727-2:2001|IDF 80-2:2001
|
|
|
|
|
Kennzeichnungsmittel
|
|
|
|
|
|
|
|
Siehe Anhang VIII
|
Anhang VIII
|
|
|
|
|
|
Siehe Anhang VI
|
Anhang VI
|
|
|
|
|
|
—
|
Carotinsäure-Ethylester
|
|
Siehe Anhang VII
|
Anhang VII
|
|
|
|
|
|
—
|
Önanthsäure-Triglyceride
|
|
Siehe Anhang V
|
Anhang V
|
|
|
Verordnung (EG) Nr. 1898/2005 Kapitel II
|
Gesalzene Butter
|
Fett
|
Mind. 80 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Fremdfett
|
|
Anhang XX
|
|
|
|
|
Wasser
|
Bis zu 16 % m/m
|
ISO 3727-1:2001|IDF 80-1:2001
|
|
|
|
|
FFTM (ohne Salz)
|
Bis zu 2 % m/m
|
ISO 3727-2:2001|IDF 80-2:2001
|
|
|
|
|
Salz
|
Bis zu 2 % m/m
|
ISO 15648:2004|IDF 179:2004
|
|
|
|
|
Kennzeichnungsmittel
|
|
|
|
|
|
|
|
Siehe Anhang VIII
|
Anhang VIII
|
|
|
|
|
|
Siehe Anhang VI
|
Anhang VI
|
|
|
|
|
|
—
|
Carotinsäure-Ethylester
|
|
Siehe Anhang VII
|
Anhang VII
|
|
|
|
|
|
—
|
Önanthsäure-Triglyceride
|
|
Siehe Anhang V
|
Anhang V
|
|
|
Verordnung (EG) Nr. 1898/2005 Kapitel II
|
Butterfett
|
Fett
|
Mind. 99,8 % m/m
|
|IDF 24:1964
|
|
|
|
|
Wasser und FFTM
|
Bis zu 0,2 % m/m
|
ISO 5536:2002|IDF 23:2002 (Feuchtigkeit)
IDF 24:1964 (FFTM)
|
|
|
|
|
Säuregrad
|
1,2 mmol/100 g Fett
|
ISO 1740:2004|IDF 6:2004
|
|
|
|
|
PZ (max.)
|
0,5 mEq Sauerstoff/1 000 g Fett
|
ISO 3976:2006|IDF 74:2006
|
Anmerkung 1
|
|
|
|
Fremdfett
|
Ohne
|
Anhang XX
|
|
|
Geschmack
|
Unverfälscht
|
|
Geruch
|
Kein Fremdgeruch
|
|
Sonst.
|
Keine Neutralisierungsmittel, Antioxidantien und Konservierungsmittel
|
|
|
|
Kennzeichnungsmittel
|
|
|
|
|
|
Siehe Anhang VIII
|
Anhang VIII
|
|
|
Siehe Anhang VI
|
Anhang VI
|
|
—
|
Carotinsäure-Ethylester
|
|
Siehe Anhang VII
|
Anhang VII
|
|
—
|
Önanthsäure-Triglyceride
|
|
Siehe Anhang V
|
Anhang V
|
|
Verordnung (EG) Nr. 1898/2005 Kapitel II
|
Rahm
|
Fett
|
Mind. 35 % m/m
|
ISO 2450:1999|IDF 16 C:1987
|
|
|
|
|
Fremdfett
|
|
Anhang XX
|
|
|
|
|
Kennzeichnungsmittel
|
|
|
|
|
|
Siehe Anhang VIII
|
|
Anmerkung 2
|
|
|
|
|
Siehe Anhang VI
|
Anhang VI
|
|
|
|
|
|
—
|
Carotinsäure-Ethylester
|
|
Siehe Anhang VII
|
|
Anmerkung 2
|
|
|
|
|
—
|
Önanthsäure-Triglyceride
|
|
Siehe Anhang V
|
Anhang V
|
|
|
Verordnung (EG) Nr. 1898/2005 Kapitel III
|
Butterfett
|
Fett
|
Mind. 96 % m/m
|
|
Anmerkung 2
|
|
|
|
Fremdfett
|
|
Anhang XX
|
|
|
|
|
FFTM
|
Bis zu 2 % m/m
|
|
Anmerkung 2
|
|
|
|
Kennzeichnungsmittel
|
|
|
|
|
—
|
Stigmasterin (95 % m/m)
|
|
15 g/100 kg Butterfett
|
Anhang VIII
|
|
|
|
|
—
|
Stigmasterin (85 % m/m)
|
|
17 g/100 kg Butterfett
|
Anhang VIII
|
|
|
|
|
|
—
|
Önanthsäure-Triglyceride
|
|
10,34 kg/t Butterfett
|
Anhang V
|
|
|
|
|
|
—
|
Ethylester der Buttersäure und Stigmasterin
|
|
|
|
—
|
Ethylester der Buttersäure
|
|
—
|
Stigmasterin: Anhang VIII
|
|
Anmerkung 2
|
|
|
|
|
—
|
Ethylester der Buttersäure und Önanthsäure-Triglyceride
|
|
|
|
—
|
Ethylester der Buttersäure
|
|
—
|
Önanthsäure-Triglyceride: Anhang V
|
|
Anmerkung 2
|
|
|
|
Lecithin (E 322)
|
Bis zu 0,5 % m/m
|
|
Anmerkung 2
|
|
|
|
NaCl
|
Bis zu 0,75 % m/m
|
ISO 15648:2004|IDF 179:2004
|
|
|
|
|
Säuregrad
|
1,2 mmol/100 g Fett
|
ISO 1740:2004|IDF 6:2004
|
|
|
|
|
PZ (max.)
|
Bis zu 0,5 mEq. Sauerstoff/1 000 g Fett
|
ISO 3976:2006|IDF 74:2006
|
Anmerkung 1
|
|
|
|
Geschmack
|
Unverfälscht
|
|
|
|
|
|
Geruch
|
Ohne Fremdgeruch
|
|
|
|
|
|
Sonstige
|
Ohne Neutralisierungsmittel, Antioxidantien und Konservierungsmittel
|
|
|
|
Verordnung (EG) Nr. 1898/2005 Kapitel IV
|
Ungesalzene Butter
|
Fett
|
Mind. 82 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Wasser
|
Bis zu 16 % m/m
|
ISO 3727-1:2001|IDF 80-1:2001
|
|
|
|
|
FFTM
|
Bis zu 2 % m/m
|
ISO 3727-2:2001|IDF 80-2:2001
|
|
|
Verordnung (EG) Nr. 1898/2005 Kapitel IV
|
Gesalzene Butter
|
Fett
|
Mind. 80 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Wasser
|
Bis zu 16 % m/m
|
ISO 3727-1:2001|IDF 80-1:2001
|
|
|
|
|
FFTM (ohne Salz)
|
Bis zu 2 % m/m
|
ISO 3727-2:2001|IDF 80-2:2001
|
|
|
|
|
Salz
|
Bis zu 2 % m/m
|
ISO 15648:2004|IDF 179:2004
|
|
|
Artikel 9 und Titel II der Verordnung (EG) Nr. 1255/1999
|
Käse aus Schafsmilch und/oder Ziegenmilch
|
Kuhmilch
|
<1 % m/m
|
Anhang IX
|
|
|
Verordnung (EWG) Nr. 2921/90
|
Anhang I — Säurekasein
|
Wasser
|
Bis zu 12,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Fett
|
Bis zu 1,75 % m/m
|
ISO 5543:2004|IDF127:2004
|
|
|
|
|
Freie Säure
|
Bis zu 0,30 ml 0,1 n NaOH-Lösung
|
ISO 5547:1978|IDF 91:1979
|
|
|
Verordnung (EWG) Nr. 2921/90
|
Anhang I — Labmolke
|
Wasser
|
Bis zu 12,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Fett
|
Bis zu 1,00 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Asche
|
Mind. 7,50 % m/m
|
ISO 5545:1978|IDF 90:1979
|
|
|
Verordnung (EWG) Nr. 2921/90
|
Anhang I — Kaseinate
|
Wasser
|
Bis zu 6,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Milcheiweiß
|
Mind. 88,00 % m/m
|
ISO 5549:1978|IDF 92:1979
|
|
|
|
|
Fett und Asche
|
Bis zu 6,00 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Gebundene Asche
|
|
ISO 5544:1978|IDF 89:1979
|
|
|
|
|
Asche
|
|
ISO 5545:1978|IDF 90:1979
|
|
|
Verordnung (EWG) Nr. 2921/90
|
Anhang II — Säurekasein
|
Wasser
|
Bis zu 10,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Fett
|
Bis zu 1,50 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Freie Säure
|
Bis zu 0,20 ml 0,1 n NaOH-Lösung/g
|
ISO 5547:1978|IDF 91:1979
|
|
|
|
|
TBC (max.)
|
30 000/g
|
ISO 4833:2003
|
Anmerkung 3
|
|
|
|
Coliforme Keime
|
Keine in 0,1 g
|
Anhang X
|
Anmerkung 3
|
|
|
|
Therm. (max.)
|
5 000/g
|
ISO 4833:2003
|
Anmerkungen 3 und 4
|
|
Verordnung (EWG) Nr. 2921/90
|
Anhang II — Labkasein
|
Wasser
|
Bis zu 8,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Fett
|
Bis zu 1,00 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Asche
|
Mind. 7,50 % m/m
|
ISO 5545:1978|IDF 90:1979
|
|
|
|
|
TBC (max.)
|
30 000/g
|
ISO 4833:2003
|
Anmerkung 3
|
|
|
|
Coliforme Keime
|
Keine in 0,1 g
|
Anhang X
|
Anmerkung 3
|
|
|
|
Therm. (max.)
|
5 000/g
|
ISO 4833:2003
|
Anmerkungen 3 und 4
|
|
Verordnung (EWG) Nr. 2921/90
|
Anhang II — Kaseinate
|
Wasser
|
Bis zu 6,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Milcheiweiß
|
Mind. 88,00 % m/m
|
ISO 5549:1978|IDF 92:1979
|
|
|
|
|
Fett und Asche
|
Bis zu 6,00 % m/m
|
ISO 5543:2004|IDF 127:2004
ISO 5544:1978|IDF 89:1979 oder
ISO 5545:1978|IDF 90:1979
|
|
|
|
|
TBC (max.)
|
30 000/g
|
ISO 4833:2003
|
Anmerkung 3
|
|
|
|
Coliforme Keime
|
Keine in 0,1 g
|
Anhang X
|
Anmerkung 3
|
|
|
|
Therm. (max.)
|
5 000/g
|
ISO 4833:2003
|
Anmerkungen 3 und 4
|
|
Verordnung (EWG) Nr. 2921/90
|
Anhang III — Kaseinate
|
Wasser
|
Bis zu 6,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Milcheiweiß
|
Mind. 85,00 % m/m
|
ISO 5549:1978|IDF 92:1979
|
|
|
|
|
Fett
|
Bis zu 1,50 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Laktose
|
Bis zu 1,00 % m/m
|
ISO 5548:2004|IDF 106:2004
|
|
|
|
|
Asche
|
Bis zu 6,50 % m/m
|
ISO 5544:1978|IDF 89:1979 oder ISO 5545:1978|IDF 90:1979
|
|
|
|
|
TBC (max.)
|
30 000/g
|
ISO 4833:2003
|
Anmerkung 3
|
|
|
|
Coliforme Keime
|
Keine in 0,1 g
|
Anhang X
|
Anmerkung 3
|
|
|
|
Therm. (max.)
|
5 000/g
|
ISO 4833:2003
|
Anmerkungen 3 und 4
|
|
Änderung der Verordnung (EG) Nr. 2799/1999
|
Mischfutter und Magermilchpulver (MMP) (Fütterung)
|
Wasser (saures Buttermilchpulver)
|
Bis zu 5 % m/m
|
Anhang XIX
|
|
|
|
|
Eiweiß
|
31,4 % m/m (mind.) der fettfreien Trockenmasse
|
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001
|
|
|
|
|
Wasser (MMP)
|
Bis zu 5 % m/m
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
Fette (MMP)
|
Bis zu 11 % m/m
|
ISO 1736:2000|IDF 9C:1987
|
|
|
|
|
Labmolke (MMP)
|
Keine
|
Anhang XIII
|
Anmerkung 6
|
|
|
|
Stärke (MMP)
|
Keine
|
Anhang XVII
|
|
|
|
|
Wasser (Mischungen)
|
Bis zu 5 % m/m der fettfreien Trockenmasse
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
Fett (Mischungen)
|
|
Kommissionsrichtlinie 84/4/EWG (ABl. L 15 vom 18.1.1984, S. 29)
|
|
|
|
|
Labmolke (Mischungen)
|
Keine
|
Anhang XIII
|
|
|
|
|
MMP-Gehalt (im Endprodukt)
|
Mind. 50 % m/m
|
Anhang XVI
|
|
|
|
|
Fett (im Endprodukt)
|
Mind. 2,5 % m/m oder 5 % m/m
|
Kommissionsrichtlinie 84/4/EWG (ABl. L 15 vom 18.1.1984, S. 29)
|
Anmerkung 7
|
|
|
|
Stärke (im Endprodukt)
|
Mind. 2 % m/m
|
Anhang XVII
|
Anmerkung 8
|
|
|
|
Kupfer (im Endprodukt)
|
25 ppm
|
Kommissionsrichtlinie 78/633/EWG (ABl. L 206 vom 26.7.1987, S. 43)
|
|
|
Verordnung (EG) Nr. 214/2001
|
MMP (sprühgetrocknet)
|
Fett
|
Bis zu 1,0 % m/m
|
ISO 1736:2000|IDF 9C:1987
|
|
|
|
|
Eiweiß
|
31,4 % (2)m/m (mind.) der fettfreien Trockenmasse
|
ISO 8968-1/2:2001|IDF 20-1/2:2001
|
|
|
|
|
Wasser
|
Bis zu 3,5 % m/m
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
Säuregrad
|
Bis zu 19,5 ml, 0,1 n NaOH, 10 g fettfreie Trockenmasse
|
ISO 6091:1980|IDF 86:1981
|
|
|
|
|
Laktate
|
Bis zu 150 mg/100 g fettfreie Trockenmasse
|
ISO 8069:2005|IDF 69:2005
|
|
|
|
|
Phosphatase
|
Negativ
|
ISO 11816-1:2006|IDF 155-1:2006
|
|
|
|
|
Löslichkeit
|
Bis zu 0,5 ml bei 24 °C
|
ISO 8156:2005|IDF 129:2005
|
|
|
|
|
Verbrannte Teilchen
|
Filterscheiben A oder B (15,0 mg)
|
ADPI (1990)
|
|
|
|
|
TBC
|
40 000/g
|
ISO 4833:2003
|
Anmerkung 3
|
|
|
|
Coliforme Keime
|
Negativ/0,1 g
|
Anhang X
|
Anmerkung 3
|
|
|
|
Buttermilch
|
Negativ
|
Anhang XIV
|
|
|
|
|
Molke-Lab
|
Negativ
|
Anhang XII
|
|
|
|
|
Molke-Säure
|
Negativ
|
|
Anmerkung 2
|
|
|
|
Antibakterielle Stoffe
|
|
Anhang XV
|
|
TEIL B
Die in Teil B genannten Referenzmethoden können bei der Analyse sämtlicher Produkte verwendet werden, die mindestens einer der in Spalte 1 genannten Verordnungen unterliegen.
|
Verordnung der Kommission
|
Produkt
|
KN-Code
|
Parameter
|
Grenzwert
|
Referenzmethode
|
Anmerkung
|
|
Verordnung (EWG) Nr. 2658/87
Verordnung (EG) Nr. 2535/2001
Verordnung (EG) Nr. 1282/2006
|
Milch und Rahm, weder eingedickt noch mit Zusatz von Zucker oder anderen Süßmitteln
|
0401
|
Fett (≤6 % m/m)
|
Die Grenzwerte wurden der Beschreibung des KN-Codes des jeweiligen Produkts bzw. Teil 9 der in der Verordnung (EWG) Nr. 3846/87 der Kommission (ABl. L 366 vom 24.12.1987, S. 1) enthaltenen Nomenklatur für Ausfuhrerstattungen oder der Verordnung (EG) Nr. 2535/2001 (ABl. L 341 vom 22.12.2001, S. 29) entnommen.
|
ISO 1211:2001|IDF 1D:1996
|
|
|
|
|
|
Fett (> 6 % m/m)
|
|
ISO 2450:1999|IDF 16C:1987
|
|
|
|
Milch und Rahm, eingedickt oder mit Zusatz von Zucker oder anderen Süßmitteln
|
0402
|
Fett (flüssig)
|
|
ISO 1737:1999|IDF 13C:1987
|
|
|
|
|
|
Fett (fest)
|
|
ISO 1736:2000|IDF 9C:1987
|
|
|
|
|
|
Eiweiß
|
|
ISO 8968-1|2|3:2001|IDF20-1|2|3:2001
|
|
|
|
|
|
Saccharose (normaler Gehalt)
|
|
ISO 2911:2004 |IDF 35:2004
|
|
|
|
|
|
Saccharose (niedriger Gehalt)
|
|
|
Anmerkung 2
|
|
|
|
|
Gesamtfeststoffe (LCS)
|
|
ISO 6734:1989|IDF 15B:1991
|
|
|
|
|
|
Gesamtfeststoffe (LCC)
|
|
ISO 6731:1989|IDF 21B:1987
|
|
|
|
|
|
Wasser (Milchpulver)
|
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
|
Wasser (Rahmpulver)
|
|
Anhang XVIII
|
|
|
|
Buttermilch, fermentierte oder gesäuerte Milch (einschließlich Rahm) eingedickt oder nicht eingedickt unter Zusatz von Zucker oder anderen Süßmitteln
|
0403
|
Fett
|
|
ISO 1211:2001|IDF 1D:1996
ISO 1736:2000|IDF 9C:1987
ISO 2450:1999|IDF 16 C:1987
ISO 7208:1999|IDF 22B:1987
ISO 8262-3:2005|IDF 124-3:2005
|
|
|
|
|
|
Eiweiß
|
|
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001
|
|
|
|
|
|
Saccharose (normaler Gehalt)
|
|
ISO 2911:2004|IDF 35:2004
|
|
|
|
|
|
Saccharose (niedriger Gehalt)
|
|
|
Anmerkung 2
|
|
|
|
|
Wasser (saures Buttermilchpulver)
|
|
Anhang XIX
|
|
|
|
|
|
Wasser (süßes Buttermilchpulver)
|
|
ISO 5537:2004|IDF26:2004
|
|
|
|
|
|
Gesamtfeststoffe (sonst. Produkte)
|
|
Von der zuständigen Behörde zugelassene Methoden
|
|
|
|
Molke, auch eingedickt oder mit Zusatz von Zucker oder sonstigen Süßmitteln; Erzeugnisse, die aus natürlichen Milchbestandteilen bestehen
|
0404
|
Fett
|
|
ISO 1736:2000|IDF 9C:1987
ISO 2450:1999|IDF 16C:1987
ISO 7208:1999|IDF 22B:1987
|
|
|
|
|
|
Eiweiß
|
|
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001
|
|
|
|
|
|
Saccharose (normaler Gehalt)
|
|
ISO 2911:2004|IDF 35:2004
|
|
|
|
|
|
Saccharose (niedriger Gehalt)
|
|
|
Anmerkung 2
|
|
|
|
0404 90
|
Eiweiß
|
|
ISO 8968 1/2 2001|IDF 20-1/2:2001
|
|
|
|
|
|
Wasser
|
|
IDF 21B:1987
|
|
|
|
|
|
Gesamtfeststoffe
|
|
ISO 6734:1989|IDF 15B:1991
|
|
|
|
|
|
(Eingedickte Produkte)
|
|
ISO 6731:1989|IDF 21B:1987
|
|
|
|
Butter und sonstige Fettstoffe aus Milch; Milchstreichfette
|
0405
|
Fett (wenn ≤ 85 % m/m)
|
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Butter
|
Wasser
|
|
ISO 3727-1:2001|IDF 80-1:2001
|
|
|
|
|
|
FFTM
|
|
ISO 3727-2:2001|IDF 80-2:2001
|
|
|
|
|
|
NaCl
|
|
ISO 15648:2004|IDF 179:2004
|
|
|
|
|
|
Fett (wenn > 99 % m/m)
|
|
IDF 24:1964
|
|
|
|
Butterschmalz
|
|
Wasser (wenn Fett < 99 % m/m)
|
|
ISO 5536:2002|IDF 23:2002
|
|
|
|
Käse und Quark/Topfen
|
0406
|
Fett
|
|
ISO 1735:2004|IDF 5:2004
|
|
|
|
|
|
Trockenmasse
|
|
ISO 5534:2004|IDF 4:2004
|
|
|
|
|
|
Trockenmasse (Ricotta)
|
|
ISO 2920:2004|IDF 58:2004
|
|
|
|
|
|
NaCl
|
|
ISO 5943:2006|IDF 88:2006
|
|
|
|
|
|
Laktose
|
|
ISO 5765-1/2:2002|IDF 79-1/2:2002
|
|
|
Verordnung (EWG) Nr. 2658/87
|
Mischfutter
|
2309
|
Laktose
|
|
Anhang XI
|
|
Anmerkungen zur Liste der EU-Referenzmethoden
Anmerkung 1: Milchfettisolierung siehe ISO 1740:1991 (Lichtschutz).
Anmerkung 2: Es wurde keine Referenzmethode festgelegt. Die Methoden wurden von der zuständigen Behörde zugelassen.
Anmerkung 3: Vorbereitung der Probe gemäß ISO 8261:2001|IDF 122:2001.
Anmerkung 4: Bebrütung 48 Stunden bei einer Temperatur von 55 °C; das Austrocknen des Nährmediums ist zu verhindern.
Anmerkung 5: % m/m FFTM = % m/m Trockenmasse — % m/m Fett.
Anmerkung 6: Richtlinie 84/4/EWG der Kommission.
Anmerkung 7: Verordnung (EG) Nr. 2799/1999 der Kommission (ABl. L 340 vom 31.12.1999, S. 3–27).
Anmerkung 8: Richtlinie 78/633/EWG der Kommission.
(1) Unbeschadet der Anforderungen der jeweiligen Verordnung.
(2) Der Mindesteiweißgehalt müsste zum 1. September 2009 34 % betragen.
ANHANG II
(Artikel 3)
BEWERTUNG DER KONFORMITÄT EINER PARTIE MIT DEN GESETZLICHEN GRENZWERTEN
1. KURZBESCHREIBUNG
Wenn die maßgeblichen Rechtsvorschriften ausführliche Verfahren für die Probenahme vorsehen, sind diese Verfahren einzuhalten. Ansonsten ist aus einer zur Kontrolle vorgelegten Partie grundsätzlich eine aus mindestens drei Probeneinheiten bestehende Stichprobe zu nehmen. Es kann eine Sammelprobe hergestellt werden. Das ermittelte Ergebnis wird mit den gesetzlichen Grenzwerten verglichen, indem ein Konfidenzintervall von 95 % (zweifache Standardabweichung) berechnet wird; die jeweilige Standardabweichung hängt davon ab, 1. ob die betreffende Methode im Wege einer internationalen Zusammenarbeit durch Werte für σr und σR validiert wurde bzw. 2. — bei interner Validierung — ob ein Wert für die interne Wiederholbarkeit berechnet wurde. Dieses Konfidenzintervall gibt dann Aufschluss über die Messunsicherheit des Ergebnisses.
2. DIE METHODE WIRD IM WEGE INTERNATIONALER ZUSAMMENARBEIT VALIDIERT
In diesem Fall wurden die Wiederholstandardabweichung σr und die Vergleichsstandardabweichung σR ermittelt, und das Laboratorium kann die Konformität mit den Leistungsmerkmalen der validierten Methode nachweisen.
Das arithmetische Mittel  der n wiederholten Messungen wird verglichen.
der n wiederholten Messungen wird verglichen.
Die erweiterte Messunsicherheit (k = 2) von 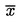 wird mit folgender Formel berechnet:
wird mit folgender Formel berechnet:
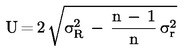
Wenn das Endergebnis x der Messung mit einer Gleichung der Form x = y
1 + y
2, x = y
1 – y
2, x = y
1 · y
2 oder x = y
1 / y
2 berechnet wird, sind die üblichen Verfahren zur Kombination von Standardabweichungen in diesen Fällen anzuwenden.
Die Partie wird als nicht konform mit dem oberen gesetzlichen Grenzwert UL eingestuft, wenn gilt:
 ;
;
Ansonsten ist die Konformität der Partie mit UL festzustellen.
Die Partie wird als nicht mit dem unteren gesetzlichen Grenzwert LL eingestuft, wenn
 ist.
ist.
Ansonsten wird die Konformität mit LL festgestellt.
3. INTERNE VALIDIERUNG UNTER BERECHNUNG DER INTERNEN VERGLEICHSSTANDARDABWEICHUNG
Wenn in dieser Verordnung nicht spezifizierte Methoden verwendet werden und keine Präzisionsmessungen durchgeführt wurden, ist eine interne Validierung vorzunehmen. Die interne Wiederholstandardabweichung sir und die interne Vergleichsstandardabweichung siR sind anstelle von σr bzw. σ
R
in der Formel zur Berechnung der erweiterten Messunsicherheit U zu verwenden.
Es gelten die in Absatz 1 genannten Entscheidungskriterien. Wenn die Partie allerdings als nicht mit den gesetzlichen Grenzwerten konform bewertet wird, müssen die Messungen mit der in dieser Verordnung genannten Methode wiederholt werden, und es ist eine Entscheidung gemäß Absatz 1 zu treffen.
ANHANG III
(Artikel 4)
BEWERTUNG DER PRÜFPERSONEN UND DER ZUVERLÄSSIGKEIT DER ERGEBNISSE BEI SENSORISCHEN PRÜFUNGEN
Von den Verfahren mit Skala (IDF-Norm 99C:1997) sind folgende Verfahren anwendbar:
A. BESTIMMUNG DES „WIEDERHOLINDEXES“
Während eines Zeitraums von 12 Monaten werden mindestens zehn Proben als Blind-Doppelanalysen durch eine Prüfperson analysiert. Diese Analyse erfolgt gewöhnlich zu verschiedenen Zeitpunkten. Die Ergebnisse für individuelle Produktmerkmale werden anhand folgender Formel bewertet:
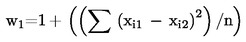
Dabei sind:
|
w1
|
:
|
Wiederholindex
|
|
xi1
|
:
|
Punktzahl für die erste Bewertung der Probe xi
|
|
xi2
|
:
|
Punktzahl für die zweite Bewertung der Probe xi
|
|
n
|
:
|
Anzahl Proben
|
Die zu beurteilenden Proben müssen einen breiten Qualitätsbereich umfassen. w1 darf den Wert 1,5 (5-Punkte-Skala) nicht überschreiten.
B. BESTIMMUNG DES „ABWEICHUNGSINDEXES“
Dieser Index ist zu verwenden um nachzuprüfen, ob eine Prüfperson die gleiche Skala für Qualitätsbewertung wie eine erfahrene Prüfpersonengruppe verwendet. Die von der Prüfperson erzielten Ergebnisse werden mit dem Durchschnitt der von der Prüfpersonengruppe erzielten Ergebnisse verglichen.
Folgende Formel wird für die Bewertung der Ergebnisse verwendet:

Dabei sind:
|
xi1; xi2
|
:
|
Siehe Abschnitt A)
|
|
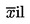
;

|
:
|
Durchschnittspunktzahl der Prüfpersonengruppe für die erste bzw. zweite Bewertung von Probe xi
|
|
n
|
:
|
Anzahl Proben (mindestens zehn innerhalb von 12 Monaten)
|
Die zu bewertenden Proben müssen einen weiten Qualitätsbereich erfassen. D1 darf den Wert 1,5 (5-Punkte-Skala) nicht überschreiten.
Die Mitgliedstaaten teilen alle Schwierigkeiten bei der Anwendung dieses Verfahrens mit.
Wenn festgestellt wird, dass bei einzelnen Prüfpersonen der Grenzwert 1,5 für den Abweichungs- oder den Wiederholungsindex überschritten wurde, müssen Experten der zuständigen Behörden binnen der jeweils nächsten Wochen mindestens eine Stichprobe unter den von diesen Prüfpersonen bewerteten Proben einer „erneuten Prüfung“ unterziehen oder mindestens eine weitere „begleitete“ Prüfung gemeinsam mit diesen Prüfpersonen durchführen. Eine sorgfältige Überwachung ist die Voraussetzung für die Entscheidung, ob diese Prüfpersonen weiter in Anspruch genommen werden sollen. Die Ergebnisse sollten dokumentiert und für etwaige Folgemaßnahmen als Nachweis aufbewahrt werden.
C. VERGLEICH DER IN VERSCHIEDENEN REGIONEN EINES MITGLIEDSTAATS UND IN VERSCHIEDENEN MITGLIEDSTAATEN ERZIELTEN ERGEBNISSE
Wenn durchführbar, wird mindestens einmal pro Jahr ein Versuch organisiert, um den Vergleich der Ergebnisse der Prüfpersonen verschiedener Regionen zu ermöglichen. Werden signifikante Unterschiede beobachtet, müssen die erforderlichen Maßnahmen getroffen werden, um die Gründe zu ermitteln und zu vergleichbaren Ergebnissen zu gelangen.
Die Mitgliedstaaten können Versuche organisieren, die den Vergleich der Ergebnisse ihrer eigenen Prüfpersonen mit den Ergebnissen von Prüfpersonen der benachbarten Mitgliedstaaten ermöglichen. Bei signifikanten Unterschieden muss eine gründliche Untersuchung erfolgen, um zu vergleichbaren Ergebnissen zu gelangen.
Die Mitgliedstaaten teilen der Kommission die Ergebnisse dieser Vergleiche mit.
ANHANG IV
(Artikel 4)
SENSORISCHE PRÜFUNG VON BUTTER
1. ANWENDUNGSBEREICH
Diese Arbeitsvorschrift beschreibt ein Verfahren für die sensorische Prüfung von Butter, das einheitlich in allen Mitgliedstaaten angewandt werden soll.
Nähere Informationen sind der geltenden Norm für Milch und Milcherzeugnisse IDF 99 — Teile 1, 2 und 3 über sensorische Prüfungen zu entnehmen.
2. BEGRIFFSBESTIMMUNGEN
Sensorische Prüfung (Sinnenprüfung): Beurteilung der Merkmale eines Erzeugnisses durch menschliche Sinne.
Prüfergruppe: Gruppe von ausgewählten Prüfern, bei deren Arbeit es nicht zu einem Gedankenaustausch oder einer gegenseitigen Beeinflussung kommen darf.
Prüfperson: eine Person, die wegen ihrer Fähigkeit zur Durchführung einer sensorischen Prüfung ausgewählt wurde; dieser Typ von Prüfpersonen kann über eingeschränkte Erfahrung verfügen.
Fachkundige Prüfperson: eine Person mit hoch entwickelter sensorischer Empfindlichkeit und ausgeprägter Erfahrung mit Methoden zur sensorischen Prüfung, die konsistente und zuverlässige sensorische Bewertungen unterschiedlicher Produkte vornehmen kann; dieser Typ von Prüfpersonen verfügt über ein gutes sensorisches Langzeitgedächtnis.
Bewertende Prüfung: sensorische Prüfung durch eine Prüfergruppe anhand einer Punkteskala; eine Terminologie zur Bezeichnung von Fehlern ist zu verwenden.
Benotende Prüfung: Qualitätseinstufung aufgrund der bewertenden Prüfung.
Prüfformulare: Formulare zur Aufzeichnung der Einzelmesswerte für jedes Merkmal und der Gesamtnote des Erzeugnisses. (Dieses Formular kann auch zur Aufzeichnung der chemischen Zusammensetzung verwendet werden.)
3. PRÜFRAUM
Siehe ISO 8589 und ISO/DIS 22935-2|IDF 99-2 Absatz 7.
Es ist dafür Sorge zu tragen, dass die Prüfer im Prüfraum nicht durch äußere Einflüsse gestört werden.
Der Prüfraum muss frei von Fremdgerüchen und einfach zu reinigen sein. Die Wände müssen hell sein und dürfen nicht reflektieren.
Der Prüfraum und seine Beleuchtung müssen so beschaffen sein, dass die zu prüfenden Produktmerkmale nicht beeinflusst werden.
Der Raum muss mit einer geeigneten Thermostatregelung ausgestattet sein, um eine gleich bleibende Temperatur der Butter sicherstellen zu können. Die Butter sollte bei der benotenden Prüfung eine Temperatur von 12 °C (±2 °C) haben.
4. AUSWAHL DER PRÜFPERSONEN
Die Prüfperson muss mit Buttererzeugnissen vertraut und zur sensorischen Prüfung geeignet sein. Ihre Eignung ist von der zuständigen Behörde regelmäßig zu überprüfen (mindestens einmal jährlich).
4.1. ISO/DIS 22935-1|IDF 99-1 Absatz 4 (Einstellung) und Absatz 5.1 sind bezüglich der allgemeinen Anforderungen sowie der Prüfungen zu beachten, die möglicherweise vor dem offiziellen Einsatz einer neuen Prüfperson durchzuführen sind.
Wesentlich ist, dass ständig Schulungsmaßnahmen durchgeführt werden, und regelmäßig sollten allgemeine Zusammenkünfte stattfinden. Bezüglich der Qualifizierung der Prüfergruppe ist ISO 8586-1 zu beachten.
4.2. Eine erste Qualifizierungsmaßnahme sollte folgende Inhalte zum Gegenstand haben:
|
—
|
allgemeine Theorie und praktische Bedeutung der sensorischen Prüfung;
|
|
—
|
Methoden, Punkte-Skalen und Beschreibung sensorischer Eindrücke;
|
|
—
|
Erkennung und Nachweis sensorischer Merkmale und spezifische sensorische Begriffe;
|
|
—
|
allgemeine Unterweisung zur Herstellung von Butter;
|
|
—
|
validierte Referenzen und Proben, die der Prüfperson helfen können, spezifische Qualitäten und Intensitäten des Produktgeschmacks zu bestimmen.
|
5. ANFORDERUNGEN AN DIE PRÜFERGRUPPE
Die Gruppe soll sich aus einer ungeraden Zahl von Prüfern zusammensetzen und mindestens drei Prüfer umfassen. Dabei muss es sich mehrheitlich um Bedienstete der zuständigen Behörde oder um eigens dazu befugte Personen handeln, die nicht für die Milchwirtschaft tätig sind.
Ein Prüfungsleiter ist für das gesamte Verfahren verantwortlich; er kann der Prüfergruppe angehören.
Vor der Prüfung müssen mehrere Faktoren berücksichtigt werden, damit die Prüfer bestmögliche Leistungen erbringen können:
|
—
|
Die Prüfer dürfen an keiner Krankheit leiden, die ihre Leistungen beeinträchtigen könnte. Ansonsten muss ein anderer Prüfer in die Gruppe aufgenommen werden;
|
|
—
|
die Prüfer müssen sich pünktlich zur Prüfung einfinden und dafür sorgen, dass sie genügend Zeit für ihre Prüfung haben;
|
|
—
|
die Prüfer sollten keine stark riechenden Erzeugnisse wie Parfüm, Rasierwasser, Deodorants usw. benutzen und keine stark gewürzten Speisen gegessen haben;
|
|
—
|
die Prüfer dürfen während der letzten halben Stunde vor der Prüfung nicht rauchen und nicht essen und ausschließlich Wasser trinken.
|
6. LEISTUNGSFÄHIGKEIT
Alle Prüfpersonen sollten regelmäßig in Gruppen zur Durchführung sensorischer Prüfungen eingesetzt werden, damit sie ihre Kompetenz nicht verlieren. Die Häufigkeit wird von der Menge und dem Durchsatz der zu prüfenden Butter abhängen; nach Möglichkeit sollte pro Monat mindestens eine Prüfergruppe zusammenkommen.
Auch erfahrenere Prüfpersonen sollten jährlich an mehreren Prüfergruppen beteiligt werden (möglichst mindestens einmal pro Vierteljahr).
7. PROBENAHME UND VORBEREITUNG DER PROBE
Entscheidend ist, dass keine störenden Rückschlüsse auf die Identität der Proben gezogen werden können; die Proben sollten codiert werden.
Die entsprechenden Vorkehrungen sollten vor der Prüfung getroffen werden. Die Butter muss während der Beförderung zum Prüfraum entsprechend temperiert sein (6 °C ± 2 °C).
Erfolgt die sensorische Bewertung in einem Kühlhaus, wird die Probe mit Hilfe eines Butterbohrers entnommen. Findet die sensorische Bewertung an einem anderen Ort statt, sollte mindestens eine 500-g-Probe entnommen werden. Während der Prüfung sollte die Butter eine Temperatur von 12 °C (±2 °C) haben; (in ISO/DIS 22935-2|IDF 99-2 wird die Prüftemperatur für Butter mit 14 °C ±2 °C angegeben.) Größere Abweichungen sind unter allen Umständen zu vermeiden.
8. BEWERTUNG DER PRÜFMERKMALE
8.1. Die sensorische Prüfung bezieht sich auf drei Merkmale: Aussehen, Konsistenz und Geschmack.
Das „Aussehen“ wird durch folgende Kriterien bestimmt: Farbe, augenscheinliche Reinheit, Fehlen physikalischer Verunreinigungen, Fehlen von Schimmelbildung und Einheitlichkeit der Wasserdispersion. Die Wasserdispersion wird gemäß der IDF-Norm 112A/1989 geprüft.
Die „Konsistenz“ ergibt sich aus folgenden Kriterien: Struktur, Textur undFestigkeit. Die Streichfähigkeit kann physisch geprüft werden, wenn ein einzelner Mitgliedstaat dies wünscht, um den Anforderungen der Kunden nachzukommen. Die Kommission kann beschließen, die Methodik künftig zu harmonisieren.
Der Begriff „Struktur“ bezieht sich auf die Stabilität des Produktes in der vom Verbraucher verwendeten Form. Im Allgemeinen besteht ein Zusammenhang zwischen Struktur, Festigkeit und Streichfähigkeit, und die Struktur sollte im Allgemeinen im gesamten Produkt einheitlich sein. Dieses Merkmal steht in engem Zusammenhang auch mit der Textur und beschreibt die Formstabilität des Produktes unter dem Eigengewicht. Die Struktur zeigt sich in der Schnittfestigkeit und kann mechanisch sowie mit dem Mund und den Fingern erfühlt werden.
Als „Geschmack“ wird die im Mund, vornehmlich durch die Geschmacksknospen der Zunge, wahrnehmbare Beschaffenheit bezeichnet.
Als „Aroma“ wird die mit der Nase und mit dem Geruchssinn wahrnehmbare Beschaffenheit bezeichnet.
Bei einer deutlichen Abweichung von der empfohlenen Temperatur ist eine zuverlässige Prüfung der Konsistenz und des Geschmacks nicht möglich. Die Temperatur ist von größter Bedeutung.
Die benotende Prüfung von Butter muss auf einen späteren Zeitpunkt verschoben werden, wenn die Temperatur außerhalb der empfohlenen Bandbreite liegt.
8.2. Jedes Merkmal ist separat zu beurteilen. Die Bewertung der Merkmale erfolgt gemäß Tabelle 1.
8.3. Vor dem Beginn der Prüfung sollten die Prüfer eine gemeinsame Beurteilung einer oder zweier Kontrollproben auf Aussehen, Konsistenz und Geschmack bzw. Aroma durchführen, um Übereinstimmung hinsichtlich der Kriterien zu erzielen.
8.4. Für die Akzeptanz erforderliche Bewertung
Siehe Teil 7 — Terminologie und Beschreibung der jeweils maßgeblichen Bewertungskriterien.
|
|
Höchstwert
|
Mindestanforderung
|
|
Aussehen
|
5
|
4
|
|
Konsistenz
|
5
|
4
|
|
Geschmack/Aroma
|
5
|
4
|
|
—
|
Wird die erforderliche Mindestpunktzahl nicht erreicht, ist der jeweilige Mangel zu beschreiben.
|
|
—
|
Die von den einzelnen Prüfern vergebenen Punktzahlen sind auf dem Prüfformular zu vermerken.
|
|
—
|
Das Erzeugnis wird aufgrund der Mehrheitsentscheidung akzeptiert oder verworfen.
|
|
—
|
Fälle, in denen zwischen den einzelnen bewertenden Prüfungen auf die jeweiligen Merkmale größere Unterschiede auftreten als zwischen benachbarten Punkten, sollten Ausnahmen darstellen (höchstens eine von 20 Proben). Anderenfalls ist die Eignung der Prüfergruppe vom Prüfungsleiter zu überprüfen.
|
9. ÜBERWACHUNG
Der Prüfungsleiter, bei dem es sich um einen offiziellen Bediensteten der zuständigen Behörde handeln muss und der auch Mitglied der Prüfergruppe sein kann, muss generell für das gesamte Verfahren verantwortlich sein. Er muss alle für jedes Merkmal vergebenen und auf dem Prüfformular vermerkten Bewertungen erfassen und bescheinigen, dass das Erzeugnis akzeptiert oder zurückgewiesen wurde.
10. TERMINOLOGIE
Siehe Anhang, Tabelle 2.
11. LITERATUR
FIL-|IDF 99C:1997 Sensory evaluation of dairy products by scoring — Reference method.
ISO/DIS 22935|IDF 99 Milch und Milcherzeugnisse — Sensorische Analyse — Teile 1-3.
ISO 8586-1 Sensorische Analyse; Allgemeine Richtlinien für die Auswahl, Schulung und Überprüfung von Prüfpersonen, Teil 1.
ISO 8589 Sensorische Analyse; Allgemeine Richtlinien für die Gestaltung von Prüfräumen.
FIL-|IDF 112A:1989 Butter — Determination of Wasser dispersion value.
Tabelle 1
Bewertende Prüfung von Butter
|
Aussehen
|
Konsistenz
|
Geschmack + Aroma
|
|
Punktzahlen
|
Nr. (1)
|
Bewertung
|
Punktzahlen (Qualitätsklasse)
|
Nr. (1)
|
Bewertung
|
Punktzahl (Qualitätsklasse)
|
Nr. (1)
|
Bewertung
|
|
5
|
|
Sehr gut
Idealtyp
Höchste Qualität
(uniform, seco)
|
5
|
|
Sehr gut
Idealtyp
Höchste Qualität
(gut streichfähig)
|
5
|
|
Sehr gut
Idealtyp
Höchste Qualität
(absolut rein, feinstes Aroma)
|
|
4
|
|
Gut
(2)
Keine offensichtlichen Mängel
|
4
|
17
18
|
Gut
(2)
hart
weich
|
4
|
|
Gut
(2)
keine offensichtlichen Mängel
|
|
3
|
1
2
3
4
5
6
7
8
|
Genügend (geringe Mängel)
wasserlässig
zweifarbig (bunt)
streifig
geflammt, marmoriert
fleckig
ausgeölt
überfärbt
locker, nicht kompakt
|
3
|
14
15
16
17
18
|
Genügend (geringe Mängel)
kurz, spröde, krümelig
salbig, teigig, schmierig
klebrig
hart
weich
|
3
|
21
22
25
27
33
34
35
|
Genügend (geringe Mängel)
unrein
Fremdgeschmack
sauer
Kochgeschmack
Futtergeschmack herb
bitter
versalzen
|
|
2
|
1
3
4
5
6
10
11
12
|
Mangelhaft (offensichtliche Mängel)
wasserlässig
streifig
geflammt, marmoriert
fleckig
ausgeölt
Fremdbestandteile
schimmelig
ungelöstes Salz
|
2
|
14
15
16
17
18
|
Mangelhaft (offensichtliche Mängel)
kurz, spröde, krümelig
salbig, schmierig, teigig
klebrig
hart
weich
|
2
|
21
22
23
25
32
33
34
35
36
38
|
Ungenügend (wahrnehmbare Mängel)
unrein
Fremdgeschmack
schal
sauer
Oxydationsgeschmack, Metallgeschmack
Futtergeschmack
herb, bitter
versalzen
dumpf, muffig, faulig
Chemikaliengeschmack
|
|
1
|
1
3
4
5
6
7
9
10
11
12
|
Schlecht (ausgeprägte Mängel)
wasserlässig
streifig
geflammt, marmoriert
fleckig
ausgeölt
überfärbt
körnig
Fremdbestandteile
schimmelig
ungelöstes Salz
|
1
|
14
15
16
17
18
|
Sehr mangelhaft (ausgeprägte Mängel)
kurz, bröckelig, krümelig
salbig, schmierig, teigig
klebrig
hart
weich
|
1
|
22
24
25
26
28
29
30
31
32
34
35
36
37
38
|
Sehr mangelhaft (ausgeprägte Mängel)
Fremdgeschmack
käsig, käsigsauer
sauer
hefeartig
Schimmelgeschmack
ranzig
ölig, fischig
talgig
Oxidationsgeschmack, Metallgeschmack
grob, bitter
versalzen
dumpf, muffig, faulig
Malzig
Chemikaliengeschmack
|
Tabelle 2
Mängelbezeichnungen bei Butter
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8.
|
Locker (nicht kompakt) (mit Löchern und Spalten)
|
|
|
|
|
|
|
|
|
|
|
|
|
14.
|
Kurz, spröde, krümelig, bröckelig
|
|
|
15.
|
Salbig, teigig, schmierig
|
|
|
|
|
|
|
|
|
III.
|
Geruch und Geschmack
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b)
|
Anbrenngeschmack (brandig)
|
|
|
|
|
|
|
|
|
|
|
32. a)
|
Oxydationsgeschmack
|
|
|
|
|
33.
|
Futtermittel-Geschmack
|
|
|
|
|
|
|
36.
|
Dumpf, muffig, faulig
|
|
|
|
|
38.
|
Chemikalien-Geschmack
|
|
(1) Tabelle 2.
(2) Die unter der Bewertung „gut“ genannten Mängel sind nur als sehr geringfügige Abweichungen vom Idealtyp zu betrachten.
(3) Diese Mängelbezeichnung soll möglichst wenig verwendet und auf die Fälle beschränkt werden, in denen der Geschmacksmangel nicht genauer umschrieben werden kann.
ANHANG V
(Artikel 5)
BESTIMMUNG DES GEHALTS AN ÖNANTHSÄURE-TRIGLYCERID IN BUTTER, BUTTERSCHMALZ UND RAHM DURCH GASCHROMATOGRAFISCHE ANALYSE DER TRIGLYCERIDE
1. ANWENDUNGSBEREICH
Im Folgenden wird eine Methode zur Bestimmung des Gehalts an Önanthsäure-Triglycerid in Butterschmalz, Butter und Rahm beschrieben.
2. BEGRIFFSBESTIMMUNGEN
Önanthsäure-Gehalt: Der mit dem in dieser Methode beschriebenen Verfahren ermittelte Gehalt an Önanthsäure-Triglycerid.
Anmerkung: Der Önanthsäure-Gehalt wird bei Butterschmalz und Butter ausgedrückt in kg/t Produkt und bei Rahm in kg/t Milchfett.
3. KURZBESCHREIBUNG
Milchfett wird aus den verschiedenen Produkten gemäß ISO 14156|IDF 172:2001 extrahiert. Die quantitative Bestimmung des Gehalts an Önanthsäure-Triglycerid im gewonnenen Fett erfolgt durch Kapillarsäulen-Gaschromatografie. Das für die Probe ermittelte Ergebnis wird unter Annahme des Capronsäure-Triglycerids als interne Standardprobe bewertet.
Anmerkung: Auch Tributyrin hat sich als ausreichende interne Standardprobe erwiesen.
4. REAGENZIEN
Es sind ausschließlich anerkannt analysenreine Reagenzien zu verwenden.
4.1. n-Hexan
4.2. Standard-Capronsäure-Triglycerid, Reinheit mindestens 99 %
4.3. Standard-Önanthsäure-Triglycerid, Reinheit mindestens 99 %
4.4. Wasserfreies Natriumsulfat (Na2SO4)
5. APPARATUR
Normale Laborausrüstung sowie insbesondere:
5.1. eine Analysewaage, Genauigkeit 1 mg
5.2. Messkolben, Füllmengen 10 ml und 20 ml
5.3. Zentrifugenröhrchen, Füllmenge 30 ml
5.4. ein Rotationsverdampfer
5.5. ein Ofen, in dem eine gleich bleibende Temperatur von 50 °C ±5 °C aufrechterhalten werden kann
5.6. Filterpapier, mittlere Porengröße, Durchmesser ca. 15 cm
Gaschromatografie-Ausrüstung:
5.7.1. ein Gaschromatograf mit Injektor (split/splitless oder on-column) und Flammenionisations-Detektor (FID);
eine GC-Säule, mit einer stationären Phase, in der erfolgreich eine Triglycerid-Abtrennung durchgeführt wurde (100 % Dimethylpolysiloxan oder 5 % Phenyl — 95 % Methylpolysiloxan). Die stationäre Phase, die Säulenhöhe (4 bis 15 m), der Innendurchmesser (0,22 bis 0,50 mm) und die Filmstärke (mindestens 0,12 µm) sind aufgrund von Erfahrungen im Laboratorium sowie abhängig vom jeweiligen Injektionssystem zu wählen. In jedem Fall müssen mit der gewählten Säule der Lösungsmittel-Peak und der Capronsäure-Triglycerid-Peak klar getrennt dargestellt und eine Basislinien-Auflösung zwischen dem Capronsäure-Triglycerid- und dem Önanthsäure-Triglycerid-Peak ermöglicht wird. Im Folgenden werden exemplarisch einige mögliche Aufbauten beschrieben.
5.7.2.1. Beispiele möglicher Aufbauten mit Split-Injektor:
|
—
|
Kopfdruck Säule: 100 kPa;
|
|
—
|
Säule: 12 m Höhe, 0,5 mm Innendurchmesser, 0,1 µm Filmstärke Quarzglassäule;
|
|
—
|
stationäre Phase: 100 % Dimethylpolysiloxan oder 5 % Phenyl/95 % Dimethylpolysiloxan (z. B. HT5);
|
|
—
|
Säulentemperatur: Ausgangstemperatur 130 °C, Dauer 1 Minute, steigend um 20 °C/min bis auf 260 °C; anschließend weiterer Anstieg um 30 °C/min bis auf 360 °C; 10 min bei 360 °C;
|
|
—
|
Detektortemperatur: 370 °C;
|
|
—
|
Injektortemperatur: 350 °C;
|
|
—
|
injiziertes Probenvolumen: 1 µl;
|
5.7.2.2. Beispiel eines möglichen Aufbaus mit On-Column-Injektor:
|
—
|
Trägergas: Wasserstoff (Constant-Flow-System);
|
|
—
|
Kopfdruck Säule: 89 kPa;
|
|
—
|
Säule: 4 m Höhe, 0,32 mm Innendurchmesser, 0,25 µm Filmstärke, Quarzglassäule;
|
|
—
|
stationäre Phase: 5 % Phenyl/95 % Dimethylpolysiloxan;
|
|
—
|
Säulentemperatur: Ausgangstemperatur 60 °C, Dauer 2 min, ansteigend um 35 °C/min auf 340 °C, Beibehaltung dieser Temperatur 5 min;
|
|
—
|
Detektortemperatur: 350 °C;
|
|
—
|
injiziertes Probenvolumen: 1 µl;
|
5.8. eine Injektionsspritze, Fassungsvermögen 5 µl.
6. PROBENAHME
Wichtig ist, dass das Laboratorium eine Probe erhält, die tatsächlich repräsentativ ist und bei Transport oder Lagerung nicht beschädigt oder verändert wurde.
Die Probenahme ist nicht Bestandteil der in dieser internationalen Norm spezifizierten Methode. Eine empfohlene Methode zur Probenahme ist IDF-Norm 50C: 1995 bzw. ISO 707-1997 — „Milch und Milchprodukte — Leitfaden zur Probenahme“ zu entnehmen.
7. VERFAHREN
7.1. Herstellung der Testprobe und der Probeneinwaage
Verfahren gemäß ISO 14156|IDF 172:2001
7.1.1. Butterschmalz, Butter
7.1.1.1. 50 bis 100 g der Testprobe werden im Ofen (5.5) geschmolzen.
7.1.1.2. 0,5 bis 1,0 g wasserfreies Natriumsulfat (5.4) werden auf ein Faltenfilterpapier gegeben.
7.1.1.3. Das Fett wird durch das Filterpapier mit dem wasserfreien Natriumsulfat gefiltert, und das entstehende Filtrat in einem im Ofen (5.5) vorgehaltenen Becherglas aufgefangen. Beim Dekantieren der geschmolzenen Butter auf das Filterpapier ist darauf zu achten, dass kein Serum übertragen wird.
7.1.2. Rahm
7.1.2.1. Die Testprobe wird auf 20 °C ±2 °C erwärmt.
7.1.2.2. Die Probe wird gründlich gemischt oder gerührt.
7.1.2.3. Von der Testprobe wird eine geeignete Menge aufgelöst, so dass sich eine Probeneinwaage von 100 ml mit einem Fettmasseanteil von etwa 4 % ergibt.
7.1.2.4. Anschließend ist das Fett wie bei Rohmilch und bei homogenisierter Milch (siehe ISO 14156|IDF 172:2001, Absatz 8.3) aus dem Rahm abzutrennen.
7.1.2.5. In einen 10-ml-Messkolben (5.2) wird 1 g des gewonnenen Fetts gegeben (Toleranz 1 mg). 1 ml der in Absatz 7.2.2 genannten Lösung wird hinzugegeben. Anschließend ist der Messkolben unter Zugabe von n-Hexan (4.1) auf eine Füllmenge von 10 ml aufzufüllen und zu homogenisieren.
7.1.2.6. 1 ml der in Absatz 7.1.1.2 genannten Lösung wird in einen 10-ml-Messkolben (5.2) gegeben; anschließend wird mit n-Hexan (4.1) auf 10 ml aufgefüllt.
7.2. Vorbereitung der Kalibrierungsstandards
7.2.1. 100 mg Önanthsäure-Triglycerid (4.3) werden in 10 ml n-Hexan (4.1) aufgelöst.
7.2.2. 100 mg Capronsäure-Triglycerid (4.2) werden in 10 ml n-Hexan (4.1) aufgelöst.
7.2.3. 1 ml der in Absatz 7.2.2 genannten Lösung wird in einen 10-ml-Messkolben (5.2) gegeben; anschließend wird mit n-Hexan (4.1) auf 10 ml aufgefüllt.
7.2.4. Jeweils 1 ml der in Absatz 7.2.1 und der in Absatz 7.2.2 genannten Lösung wird in einen 10-ml-Messkolben (5.2) gegeben; anschließend wird mit n-Hexan (4.1) auf 10 ml aufgefüllt.
7.2.5. 1 ml der in Absatz 7.2.4 genannten Lösung wird in einen 10-ml-Messkolben (5.2) gegeben; anschließend wird mit n-Hexan (4.1) auf 10 ml aufgefüllt.
7.3. Chromatografische Bestimmung
7.3.1. Von der in Absatz 7.2.5 genannten Standardlösung wird zweimal 1 µl injiziert.
7.3.2. Von jeder Probenlösung ist jeweils 1 µl zu injizieren.
Anmerkung: Wenn der On-Column-Injektor eingesetzt wird, sollten sowohl die Standardlösung als auch die Probenlösungen in einer erhöhten Verdünnung verwendet werden.
7.3.3. Das in Absatz 7.3.1 genannte Verfahren ist bei jeder dritten Probe zu wiederholen, damit jeweils am Anfang und am Ende einer Probenreihe eine Standardinjektion erfolgen kann. Die Ergebnisse beruhen auf den mittleren durchschnittlichen Kalibrierfaktoren der Standardchromatogramme.
8. BERECHNUNG DER ERGEBNISSE
Bei jedem Chromatogramm sind die Peak-Flächen des Önanthsäure- und des Capronsäure-Triglycerids zu integrieren.
Diese Anweisungen sind für alle Probenfolgen zu wiederholen, bei denen jeweils vorher und nachher die Standardlösung injiziert wird. (Unmittelbar vor jeder Probenreihe wird zweimal die Standardlösung STD1 injiziert, und unmittelbar nach jeder Probenreihe wird zweimal die Standardlösung STD2 eingespritzt.)
8.1. Kalibrierung
8.1.1. Jeweils für die zweite Probe von STD1, Rf1(a) und Rf1(b) ist der Reaktionsfaktor zu berechnen.
Rf1 (a) oder (b) = (Peak-Fläche Capronsäure-Triglycerid/Peak-Fläche Önanthsäure-Triglycerid) × 100
Der mittlere durchschnittliche Reaktionsfaktor Rf1 wird berechnet:
Rf1 = (Rf1(a) + Rf1(b)) / 2
8.1.2. Ähnlich wird der mittlere durchschnittliche Reaktionsfaktor für STD2 und Rf2 berechnet.
8.1.3. Der mittlere durchschnittliche Reaktionsfaktor Rf wird berechnet:
Rf = (Rf1 + Rf2) /2
8.2. Testproben
Für jedes ermittelte Proben-Chromatogramm zwischen STD1 und STD2 wird der Önanthsäuregehalt (C (in kg/t)) berechnet:
C = (Peak-Fläche für Önanthsäure-Triglycerid × Rf × 100)/(Peak-Fläche Capronsäure-Triglycerid × Wt × 1 000)
Dabei sind:
|
—
|
Wt = Gewicht des gewonnenen Fetts (g),
|
|
—
|
100 = Verdünnungsvolumen der Probe,
|
|
—
|
1 000 = Umrechnungsfaktor (μg/g in kg/t).
|
Bei Butterproben wird der Fettgehalt der Butter berücksichtigt und ein bereinigter Konzentrationswert CButter (kg/t Butter) berechnet
CButter = CFett × F
Dabei ist: F = Fettgehalt der Butter.
9. GENAUIGKEIT
Nähere Informationen zu einem Leistungstest mit Butter gemäß ISO 5725-1 und ISO 5725-2 (Genauigkeit der Methode) sind Ziffer 12 zu entnehmen.
Die Grenzwerte für die Wiederholbarkeit und die Vergleichbarkeit werden für das Konfidenzintervall 95 % ausgedrückt und sind auf andere als die genannten Konzentrationsbereiche und Matrizen möglicherweise nicht übertragbar.
9.1. Wiederholbarkeit
Die absoluten Unterschiede zwischen zwei einzelnen Testergebnissen, die nach derselben Methode bei identischem Testmaterial im selben Laboratorium von demselben Analytiker und mit derselben Ausrüstung binnen eines kurzen Zeitraums ermittelt wurden, liegen in höchstens 5 % aller Fälle über 0,35 kg/t.
9.2. Vergleichbarkeit
Die absoluten Unterschiede zwischen zwei einzelnen Testergebnissen, die nach derselben Methode bei identischem Testmaterial in verschiedenen Laboratorien von verschiedenen Analytikern und mit unterschiedlicher Ausrüstung ermittelt wurden, liegen in höchstens 5 % aller Fälle über 0,66 kg/t.
10. TOLERANZGRENZEN: UNTERGRENZE (BEI UNZUREICHENDEN MENGEN)
10.1. Zur Prüfung der richtigen Kennzeichnung des Erzeugnisses sind dem gekennzeichneten Erzeugnis drei Proben zu entnehmen.
10.2. Butter und Butterfett
10.2.1. Die Beimischungsquote beträgt 11 kg Önanthsäure-Triglycerid mit einer Reinheit von 95 % pro Tonne Butter (d. h. 10,45 kg/t).
10.2.2. Die Ergebnisse von drei Proben, die aus der Analyse des Produkts ermittelt wurden, werden zur Prüfung der Quote und der Homogenität der Beimischung des Kennzeichnungsmittels verwendet; das niedrigste dieser Ergebnisse wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
9,51 kg/t (95 % der Mindestquote der Beimischung von Önanthsäure-Triglycerid mit einer Reinheit von 95 %, einfache Bestimmung);
|
|
—
|
6,89 kg/t (70 % der Mindestquote der Beimischung von Önanthsäure-Triglycerid mit einer Reinheit von 95 %, einfache Bestimmung);
|
|
—
|
es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das man durch Interpolierung zwischen 9,51 kg und 6,89 kg erhält.
|
10.3. Rahm
10.3.1. Die Beimischung beträgt 10 kg Önanthsäure-Triglycerid mit einer Reinheit von mindestens 95 % pro Tonne Milchfett (d. h. 9,50 kg/t gekennzeichnetes Milchfett).
10.3.2. Aufgrund der Ergebnisse der drei in der Produktanalyse untersuchten Proben wird die Homogenität der Beimischung des Kennzeichnungsmittels geprüft; das niedrigste Ergebnis wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
8,60 kg/t (95 % der Mindestquote der Beimischung von Önanthsäure-Triglycerid mit einer Reinheit von 95 %, einfache Bestimmung);
|
|
—
|
6,23 kg/t (70 % der Mindestquote der Beimischung von Önanthsäure-Triglycerid mit einer Reinheit von 95 %, einfache Bestimmung);
|
|
—
|
es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das man durch Interpolierung zwischen 8,60 kg und 6,23 kg erhält.
|
11. TOLERANZGRENZEN: OBERGRENZEN (BEI ÜBERSCHREITUNG DER MENGE UM MEHR ALS 20 %)
11.1. Zur Prüfung der richtigen Kennzeichnung des Erzeugnisses sind dem gekennzeichneten Erzeugnis drei Proben zu entnehmen.
11.2. Butter und Butterfett
11.2.1. Die Ergebnisse von drei Proben, die aus der Analyse des Produkts ermittelt wurden, werden zur Prüfung der Quote und der Homogenität der Beimischung des Kennzeichnungsmittels verwendet; das niedrigste dieser Ergebnisse wird bezogen auf den folgenden Grenzwert beurteilt:
|
—
|
oberer Grenzwert 12,96 kg/t.
|
11.3. Rahm
11.3.1. Die Ergebnisse von drei Proben, die aus der Analyse des Produkts ermittelt wurden, werden zur Prüfung der Quote und der Homogenität der Beimischung des Kennzeichnungsmittels verwendet; das niedrigste dieser Ergebnisse wird bezogen auf den folgenden Grenzwert beurteilt:
|
—
|
oberer Grenzwert 11,82 kg/t.
|
12. WEITERE INFORMATIONEN: STATISTISCHE ANALYSE VON ERGEBNISSEN BEI DER BESTIMMUNG VON ÖNANTHSÄURETRIGLYCERID IN BUTTERFETT DURCH TRIGLYCERIDANALYSE
Zur Bestimmung des Önanthsäuretriglycerid-Gehalts in gekennzeichneter Butter wurden vier gemeinsame Versuche durchgeführt.
Am ersten Ringtest haben sich neun Laboratorien beteiligt; es wurden keine Spezifikationen bezüglich der anzuwendenden Analysemethoden vorgegeben.
Am zweiten Ringtest nahmen zehn Laboratorien teil; es wurden vier unterschiedliche Methoden angewendet:
|
—
|
quantitative Methylheptanoat-Bestimmung mit n-Nonan oder Methylnonanoat als interner Standardprobe;
|
|
—
|
quantitative Önanthsäuretriglycerid-Bestimmung mit Tricaproat als interner Standardprobe;
|
|
—
|
quantitative Methylheptanoat-Bestimmung mit einer Kalibrierungsprobe/Gemisch;
|
|
—
|
quantitative Methylheptanoat-Bestimmung mit einem Kalibrierungsgemisch.
|
Wenn FAME analysiert würde, müssten zudem zwei verschiedene Verfahren zum Methylierungsnachweis (De Francesco und Christopherson & Glass) eingesetzt werden.
Wegen der ermittelten Ergebnisse wurden zwei Methoden zur Durchführung des dritten Ringtests gewählt:
|
—
|
quantitative Methylheptanoat-Bestimmung mit n-Nonan oder Methylnonanoat als interner Standardprobe;
|
|
—
|
quantitative Önanthsäuretriglycerid-Bestimmung mit Tricaproat als interner Standardprobe.
|
Die Ergebnisse von sieben Laboratorien zeigten, dass sich mit der FAME-Methode eine höhere Variabilität ergab, und entsprechend wurde entschieden, ausschließlich die Bestimmung mit Önanthsäuretriglycerid als Triglycerid gemäß dem Verfahren zum quantitativen Önanthsäuretriglycerid-Nachweis mit Tricaproat als interner Standardprobe zu verwenden. Zudem muss die Triglyceridanalyse mit einer Kapillarsäule durchgeführt werden.
Im vierten Ringtest wurden vier Proben (A, B, C und D) verteilt, und neun Laboratorien übermittelten Ergebnisse (Tabellen 1-2).
Zwei Laboratorien (DE und EU) analysierten die Proben nach der FAME-Methode.
Wegen der geringeren Anzahl der Laboratorien wurde die statistische Berechnung sowohl für die gesamte Datengruppe (Abbildungen 1-2) einschließlich der FAME-Ergebnisse als auch für die aus der TG-Analyse ermittelten Daten durchgeführt.
Tests auf Ausreißer
|
—
|
Probe A: In Dixon-, Cochran- und Grubbs-Tests mit 1 und 5 % wurde ein Ausreißer in einem Laboratorium festgestellt.
|
|
—
|
Probe B: Im Grubbs-Test mit 5 % wurde ein Ausreißer in einem Laboratorium festgestellt.
|
|
—
|
Probe C: In Dixon- und Grubbs-Tests mit 1 und 5 % wurde in einem Labor ein Ausreißer festgestellt.
|
|
—
|
Probe D: In Dixon- und Grubbs-Tests mit 1 und 5 % wurde in einem Laboratorium ein Ausreißer festgestellt.
|
Der Ausreißer wurde bei der Berechnung nicht berücksichtigt.
Die mit der FAME-Methode ermittelten Ergebnisse wurden mit den durchgeführten Tests nie als Ausreißer gewertet.
Parameter für die Genauigkeit
In den Tabellen 1 und 2 sind die Ergebnisse aller Laboratorien und die Parameter für die Genauigkeit genannt, die für eine annehmbare Anzahl (8) an Laboratorien berechnet wurden; die Ergebnisse beruhen leider aber nicht alle auf derselben Analysemethode.
In den Tabellen 3 und 4 sind ausschließlich mit der TG-Methode ermittelte Ergebnisse sowie die entsprechenden Genauigkeitsparameter zusammengestellt. Die Annahme dieser Parameter unterliegt dem Vorbehalt der Annahme der geringen Anzahl an Laboratorien (6).
Aus den Abbildungen 2 und 3 gehen die Trends der berechneten Werte für Sr und SR der vier Proben der beiden beschriebenen Datenreihen hervor.
In Tabelle 5 sind die Werte für Sr und SR mit den entsprechenden Pool-Werten und den Gesamtparametern r und R zusammengestellt.
Außerdem wurde die kritische Differenz bei einem Konfidenzintervall von 95 % berechnet.
Tabelle 1
Statistische Ergebnisse TG- und FAME*-Methode
|
Probe A
|
|
R1
|
R2
|
Mittelwert
|
Verbleibende Laboratorien nach Ausscheiden von Ausreißern
|
8
|
|
RENNES
|
FR1
|
11,0
|
11,1
|
11,1
|
Anz. Ausreißer
|
1
|
|
RIKILT
|
NL
|
11,2
|
11,2
|
11,2
|
Ausreißer
|
DК
|
|
ZPLA
|
DE*
|
11,6
|
11,8
|
11,7
|
Mittelwert
|
11,3
|
|
ADAS
|
GB
|
11,4
|
11,2
|
11,3
|
Tatsächlicher Wert
|
11,0
|
|
CNEVA
|
FR2
|
11,4
|
11,4
|
11,4
|
Wiederholstandardabweichung (Sr)
|
0,09
|
|
LODI
|
IT
|
11,1
|
11,3
|
11,2
|
Relative Wiederholbarkeit sd (RSDr %)
|
0,80
|
|
ËÈLA
|
FI
|
11,3
|
11,2
|
11,3
|
Wiederholbarkeit < r (95 %)
|
0,26
|
|
ISPRA
|
EU*
|
11,0
|
11,0
|
11,0
|
Relative Wiederholbarkeit r %
|
2,24
|
|
D.V.F.A.
|
DK
|
13,3
|
11,8
|
12,6
|
Vergleichsstandardabweichung (SR)
|
0,23
|
|
|
|
|
|
|
Relative Vergleichbarkeit sd (RSDR %)
|
2,04
|
|
|
|
|
|
|
Vergleichbarkeit R (95 %)
|
0,84
|
|
|
|
|
|
|
Relative Vergleichbarkeit R %
|
5,71
|
|
Probe B
|
|
R1
|
R2
|
Mittelwert
|
Verbleibende Laboratorien nach Ausschluss von Ausreißern
|
8
|
|
RENNES
|
FR1
|
12,7
|
12,8
|
12,8
|
Anz. Ausreißer
|
1
|
|
RIKILT
|
NL
|
13,5
|
13,3
|
13,4
|
Ausreißer
|
DK
|
|
ZPLA
|
DE*
|
14,0
|
13,8
|
13,9
|
Mittelwert
|
13,4
|
|
ADAS
|
GB
|
13,4
|
13,5
|
13,5
|
Tatsächlicher Wert
|
13,5
|
|
CNEVA
|
FR2
|
13,3
|
13,4
|
13,4
|
Wiederholstandardabweichung (Sr)
|
0,14
|
|
LODI
|
IT
|
13,9
|
13,5
|
13,7
|
Relative Wiederholbarkeit sd (RSDr %)
|
1,04
|
|
EELA
|
FI
|
13,4
|
13,2
|
13,3
|
Wiederholbarkeit r (95 %)
|
0,40
|
|
ISPRA
|
EU*
|
13,2
|
13,3
|
13,3
|
Relative Wiederholbarkeit r %
|
2,91
|
|
D.V.F.A.
|
DK
|
14,1
|
14,8
|
14,5
|
Vergleichsstandardabweichung (SR)
|
0,35
|
|
|
|
|
|
|
Relative Vergleichbarkeit sd (RSDR %)
|
2,61
|
|
|
|
|
|
|
Vergleichbarkeit R (95 %)
|
0,99
|
|
|
|
|
|
|
Relative Vergleichbarkeit R %
|
7,31
|
Tabelle 2
Statistische Ergebnisse TG- und FAME*-Methode
|
Probe C
|
|
R1
|
R2
|
Mittelwert
|
Verbleibende Laboratorien nach Ausschluss von Ausreißern
|
8
|
|
RENNES
|
FR1
|
8,9
|
9,2
|
9,1
|
Anz. Ausreißer
|
1
|
|
RIKILT
|
NL
|
9,2
|
9,3
|
9,3
|
Ausreißer
|
DK
|
|
ZPLA
|
DE*
|
9,2
|
9,4
|
9,3
|
Mittelwert
|
9,3
|
|
ADAS
|
GB
|
9,5
|
9,3
|
9,4
|
Tatsächlicher Wert
|
9,3
|
|
CNEVA
|
FR2
|
9,4
|
9,4
|
9,4
|
Wiederholstandardabweichung (Sr)
|
0,14
|
|
LODI
|
IT
|
9,2
|
9,5
|
9,4
|
Relative Wiederholbarkeit sd (RSDr %)
|
1,50
|
|
EELA
|
FI
|
9,4
|
9,6
|
9,5
|
Wiederholbarkeit r (95 %)
|
0,40
|
|
ISPRA
|
EU*
|
9,4
|
9,3
|
9,4
|
Relative Wiederholbarkeit r %
|
4,20
|
|
D.V.F.A.
|
DK
|
10,7
|
10,9
|
10,8
|
Vergleichsstandardabweichung (SR)
|
0,17
|
|
|
|
|
|
|
Relative Vergleichbarkeit sd (RSDR %)
|
1,82
|
|
|
|
|
|
|
Vergleichbarkeit R (95 %)
|
0,47
|
|
|
|
|
|
|
Relative Vergleichbarkeit R %
|
5,10
|
|
Probe D
|
|
R1
|
R2
|
Mittelwert
|
Verbleibende Laboratorien nach Ausschluss von Ausreißern
|
8
|
|
RENNES
|
R1
|
1,6
|
1,6
|
1,6
|
Anz. Ausreißer
|
1
|
|
RIKILT
|
NL
|
2,1
|
2,1
|
2,1
|
Ausreißer
|
DK
|
|
ZPLA
|
DE*
|
2,3
|
2,3
|
2,3
|
Mittelwert
|
2,1
|
|
ADAS
|
GB
|
2,1
|
2,2
|
2,2
|
Tatsächlicher Wert
|
2,1
|
|
CNEVA
|
FR2
|
2,1
|
2,1
|
2,1
|
Wiederholstandardabweichung (Sr)
|
0,08
|
|
LODI
|
IT
|
2,2
|
1,9
|
2,1
|
Relative Wiederholbarkeit sd (RSDr %)
|
3,81
|
|
EELA
|
FI
|
2,3
|
2,3
|
2,3
|
Wiederholbarkeit r (95 %)
|
0,22
|
|
ISPRA
|
EU*
|
2,3
|
2,3
|
2,3
|
Relative Wiederholbarkeit r %
|
10,67
|
|
D.V.F.A.
|
DK
|
3,4
|
2,9
|
3,2
|
Vergleichsstandardabweichung (SR)
|
0,24
|
|
|
|
|
|
|
Relative Vergleichbarkeit sd (RSDR %)
|
11,43
|
|
|
|
|
|
|
Vergleichbarkeit R (95 %)
|
0,67
|
|
|
|
|
|
|
Relative Vergleichbarkeit R %
|
32,00
|
Tabelle 3
Statistische Ergebnisse TG- und FAME*-Methode
|
Probe A
|
|
R1
|
R2
|
Mittelwert
|
Verbleibende Laboratorien nach Ausschluss von Ausreißern
|
6
|
|
RENNES
|
FR1
|
11,0
|
11,1
|
11,1
|
Anz. Ausreißer
|
1
|
|
RIKILT
|
NL
|
11,2
|
11,2
|
11,2
|
Ausreißer
|
DК
|
|
ADAS
|
GB
|
11,4
|
11,2
|
11,3
|
Mittelwert
|
11,2
|
|
CNEVA
|
FR2
|
11,4
|
11,4
|
11,4
|
Tatsächlicher Wert
|
11,0
|
|
LODI
|
IT
|
11,1
|
11,3
|
11,2
|
Wiederholstandardabweichung (Sr)
|
0,09
|
|
ËÈLA
|
FI
|
11,3
|
11,2
|
11,3
|
Relative Wiederholbarkeit sd (RSDr %)
|
0,80
|
|
D.V.F.A.
|
DK
|
13,3
|
11,8
|
12,6
|
Wiederholbarkeit r (95 %)
|
0,25
|
|
|
|
|
|
|
Relative Wiederholbarkeit r %
|
2,24
|
|
|
|
|
|
|
Vergleichsstandardabweichung (SR)
|
0,13
|
|
|
|
|
|
|
Relative Vergleichbarkeit sd (RSDR %)
|
1,16
|
|
|
|
|
|
|
Vergleichbarkeit R (95 %)
|
0,36
|
|
|
|
|
|
|
Relative Vergleichbarkeit R %
|
3,25
|
|
Probe B
|
|
R1
|
R2
|
Mittelwert
|
Verbleibende Laboratorien nach Ausschluss von Ausreißern
|
6
|
|
RENNES
|
FR1
|
12,7
|
12,8
|
12,8
|
Anz. Ausreißer
|
1
|
|
RIKILT
|
NL
|
13,5
|
13,3
|
13,4
|
Ausreißer
|
DК
|
|
ADAS
|
GB
|
13,4
|
13,5
|
13,5
|
Mittelwert
|
13,3
|
|
CNEVA
|
FR2
|
13,3
|
13,4
|
13,4
|
Tatsächlicher Wert
|
13,5
|
|
LODI
|
IT
|
13,9
|
13,5
|
13,7
|
Wiederholstandardabweichung (Sr)
|
0,15
|
|
EELA
|
FI
|
13,4
|
13,2
|
13,3
|
Relative Wiederholbarkeit sd (RSDr %)
|
1,13
|
|
D.V.F.A.
|
DK
|
14,1
|
14,8
|
14,5
|
Wiederholbarkeit r (95 %)
|
0,42
|
|
|
|
|
|
|
Relative Wiederholbarkeit r %
|
3,16
|
|
|
|
|
|
|
Vergleichsstandardabweichung (SR)
|
0,33
|
|
|
|
|
|
|
Relative Vergleichbarkeit sd (RSDR %)
|
2,48
|
|
|
|
|
|
|
Vergleichbarkeit R (95 %)
|
0,93
|
|
|
|
|
|
|
Relative Vergleichbarkeit R %
|
6,94
|
Tabelle 4
Statistische Ergebnisse TG-Methode
|
Probe C
|
|
R1
|
R2
|
Mittelwert
|
Verbleibende Anzahl Laboratorien nach Ausschluss von Ausreißern
|
6
|
|
RENNES
|
FR1
|
8,9
|
9,2
|
9,1
|
Anz. Ausreißer
|
1
|
|
RIKILT
|
NL
|
9,2
|
9,3
|
9,3
|
Ausreißer
|
DК
|
|
ADAS
|
GB
|
9,5
|
9,3
|
9,4
|
Mittelwert
|
9,3
|
|
CNEVA
|
FR2
|
9,4
|
9,4
|
9,4
|
Tatsächlicher Wert
|
9,3
|
|
LODI
|
IT
|
9,2
|
9,5
|
9,4
|
Wiederholstandardabweichung (Sr)
|
0,15
|
|
ËÈLA
|
FI
|
9,4
|
9,6
|
9,5
|
Relative Wiederholbarkeit sd (RSDr %)
|
1,61
|
|
D.V.F.A.
|
DK
|
10,7
|
10,9
|
10,8
|
Wiederholbarkeit r (95 %)
|
0,42
|
|
|
|
|
|
|
Relative Wiederholbarkeit r %
|
4,51
|
|
|
|
|
|
|
Vergleichsstandardabweichung (SR)
|
0,19
|
|
|
|
|
|
|
Relative Vergleichbarkeit sd (RSDR %)
|
2,04
|
|
|
|
|
|
|
Vergleichbarkeit R (95 %)
|
0,53
|
|
|
|
|
|
|
Relative Vergleichbarkeit R %
|
5,71
|
|
Probe D
|
|
R1
|
R2
|
Mittelwert
|
Verbleibende Anzahl Laboratorien nach Ausschluss von Ausreißern
|
6
|
|
RENNES
|
FR1
|
1,6
|
1,6
|
1,6
|
Anz. Ausreißer
|
1
|
|
RIKILT
|
NL
|
2,1
|
2,1
|
2,1
|
Ausreißer
Mittelwert
|
DK
|
|
|
|
|
|
|
Mittelwert
|
2,1
|
|
ADAS
|
GB
|
2,1
|
2,2
|
2,2
|
Tatsächlicher Wert
|
2,1
|
|
CNEVA
|
FR2
|
2,1
|
2,1
|
2,1
|
Wiederholstandardabweichung (Sr)
|
0,09
|
|
LODI
|
IT
|
2,2
|
1,9
|
2,1
|
Relative Wiederholbarkeit sd (RSDr %)
|
4,29
|
|
EELA
|
FI
|
2,3
|
2,3
|
2,3
|
Wiederholbarkeit r (95 %)
|
0,26
|
|
D.V.F.A.
|
DK
|
3,4
|
2,9
|
3,2
|
Relative Wiederholbarkeit r%
|
12,01
|
|
|
|
|
|
|
Vergleichsstandardabweichung (SR)
|
0,25
|
|
|
|
|
|
|
Relative Vergleichbarkeit sd (RSDR %)
|
11,90
|
|
|
|
|
|
|
Vergleichbarkeit R (95 %)
|
0,69
|
|
|
|
|
|
|
Relative Vergleichbarkeit R %
|
33,32
|
Tabelle 5
Wiederholbarkeit und Vergleichbarkeit (mit FAME)
|
|
Anz. Laboratorien
|
Ausreißer
|
Wiederholbarkeit
Sr (95 %)
|
Vergleichbarkeit
SR (95 %)
|
|
Probe A
|
8
|
1
|
0,09
|
0,23
|
|
Probe Β
|
8
|
1
|
0,14
|
0,35
|
|
Probe C
|
8
|
1
|
0,14
|
0,17
|
|
Probe D
|
8
|
1
|
0,08
|
0,24
|
|
Pool-Wert
|
|
|
0,116
|
0,256
|
|
|
|
|
r
|
R
|
|
Pool-Wert * 2,8
|
|
|
0,324
|
0,716
|
|
CrD95 = 0,40
Erklärte Mindestreinheit für Önanthsäuretriglycerid = 95 %
Erklärter unterer Grenzwert für Önanthsäuretriglycerid in Butterfett = 11 kg/t
In Anbetracht der kritischen Differenz bei einem Konfidenzintervall von 95 % darf der Mittelwert dieser beiden Ergebnisse folgende Werte nicht unterschreiten:
|
|
bei Beimischung von Önanthsäuretriglycerid mit einer Reinheit von 95 % 10,05 kg/t.
|
|
Wiederholbarkeit und Vergleichbarkeit (ohne FAME)
|
|
Anz. Laboratorien
|
Ausreißer
|
Wiederholbarkeit
Sr (95 %)
|
Vergleichbarkeit
SR (95 %)
|
|
Probe A
|
6
|
1
|
0,09
|
0,13
|
|
Probe B
|
6
|
1
|
0,15
|
0,33
|
|
Probe C
|
6
|
1
|
0,15
|
0,19
|
|
Probe D
|
6
|
1
|
0,09
|
0,25
|
|
Pool-Wert
|
|
|
0,124
|
0,237
|
|
|
|
|
r
|
R
|
|
Pool-Wert * 2,8
|
|
|
0,347
|
0,663
|
|
CrD95 = 0,36
Erklärte Mindestreinheit für Önanthsäuretriglycerid = 95 %
Erklärter unterer Grenzwert für Önanthsäuretriglycerid in Butterfett = 11 kg/t
In Anbetracht der kritischen Differenz bei einem Konfidenzintervall von 95 % darf der Mittelwert dieser beiden Ergebnisse folgende Werte nicht unterschreiten:
|
|
bei Beimischung von Önanthsäuretriglycerid mit einer Reinheit von 95 % 10,09 kg/t.
|
|
Abbildung 1 (1)
Versuchsergebnisse Probe A

Versuchsergebnisse Probe B

Versuchsergebnisse Probe C

Versuchsergebnisse Probe D
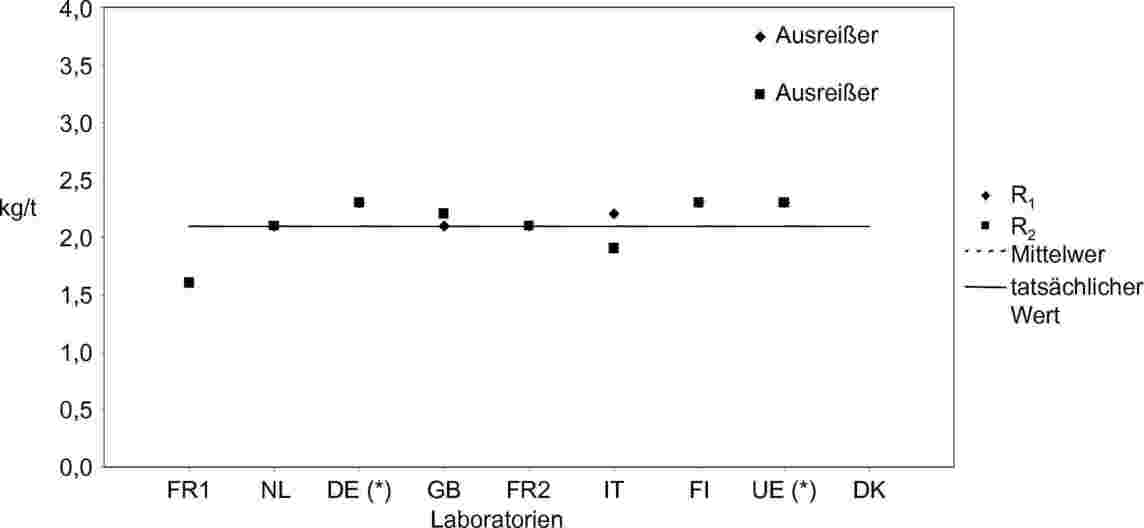
Abbildung 2
Wiederholstandardabweichung und Vergleichsstandardabweichung bei verschiedenen Konzentrationen (TG und FAME)
Abbildung 3
Wiederholstandardabweichung und Vergleichsstandardabweichung bei verschiedenen Konzentrationen (TG)

Abbildung 4
Beispiel unter Verwendung eines On-Column-Injektors

(1) = FAME-Methode.
ANHANG VI
(Artikel 5)
BESTIMMUNG DES VANILLIN-GEHALTS IN BUTTERFETT, BUTTER ODER RAHM DURCH HPLC
1. GEGENSTAND UND ANWENDUNGSBEREICH
Beschrieben wird ein Verfahren zur quantitativen Bestimmung von Vanillin in Butter, Butterfett oder Rahm.
2. KURZBESCHREIBUNG
Extraktion einer bekannten Probemenge mit Hilfe eines Isopropanol-Ethanol-Acetonitril-Gemisches (1:1:2); Abscheidung des Hauptteils des Fetts durch Kühlung bei –15 °C bis –20 °C und anschließendes Zentrifugieren.
Nach Verdünnung mit Wasser Bestimmung des Vanillingehalts durch Hochleistungs-Flüssigkeitschromatografie (HPLC).
3. APPARATUR
Normale Laborausstattung und insbesondere:
3.1. Gefrierschrank zur Kühlung auf einen Temperaturbereich von –15 bis –20 °C;
3.2. Einwegspritzen mit einem Fassungsvermögen von 2 ml;
3.3. Membranmikrofilter (Porengröße 0,45 µm), beständig gegen eine Lösung mit 5 % Extraktionslösung (4.4);
3.4. Flüssigkeitschromatografie-System, bestehend aus einer Pumpe (Durchfluss von 1,0 ml/min), einem Injektor (20 µl Injektionsvolumen, automatisch oder manuell), einem UV-Detektor (306 nm, 0,01 Å gesamter Bereich), einem Aufzeichnungsgerät oder Integrator und einem Säulenthermostat für eine Betriebstemperatur von 25 °C;
3.5. Analysesäule (250 mm × 4,6 mm ID), gepackt mit LiChrospher RP 18 (Merck, 5 µm) oder gleichwertigem Material;
3.6. Vorsäule (ca. 20 mm × 3 mm ID), trocken gepackt mit LiChrospher RP 18 (5 bis 10 µm) oder gleichwertigem Material;
3.7. Zentrifuge (2 000 Umin-1).
4. REAGENZIEN
Alle verwendeten Reagenzien müssen analysenrein sein.
4.1. Isopropanol
4.2. Ethanol 96 % (v/v)
4.3. Acetonitril
4.4. Extraktionslösung
Gemisch aus Isopropanol (4.1), Ethanol (4.2) und Acetonitril (4.3) im Verhältnis 1:1:2 (v/v)
Vanillin (4-Hydroxy-3-methoxy-benzaldehyd) ≥98 %
4.5.1. Vanillin-Stammlösung (= 500 µg/ml)
Etwa 50 mg Vanillin (4.5) auf 0,1 mg (CM mg) werden genau in einen 100-ml-Messkolben eingewogen; anschließend werden 25 ml Extraktionslösung (4.4) hinzugefügt und mit Wasser aufgefüllt.
4.5.2. Vanillin-Standardlösung (= 10 µg/ml)
5 ml der Vanillin-Stammlösung (4.5.1) werden in einen 250-ml-Messkolben pipettiert und mit Wasser aufgefüllt.
4.5.3. Methanol, HPLC-Qualität
4.5.4. Eisessigsäure
4.5.5. Wasser, HPLC-Qualität
4.5.6. HPLC, mobile Phase
300 ml Methanol (4.5.3) werden mit etwa 500 ml Wasser (4.5.5) und 20 ml Essigsäure (4.5.4) in einem 1 000-ml-Messkolben gemischt und mit Wasser (4.5.5) aufgefüllt. Dieses Gemisch wird durch einen Filter mit einer Porengröße von 0,45 µm (3.3) gefiltert.
5. VERFAHREN
5.1. Vorbereitung der Probe
5.1.1. Butter
Die Probe wird erhitzt, bis sie zu schmelzen beginnt. Eine Überhitzung auf etwa 30 °C ist zu vermeiden. Möglicherweise trennt sich die Butter nicht in zwei Phasen. Wenn die Probe ausreichend pastös ist, ist sie durch Schütteln zu homogenisieren. Nach 15-minütigem Rühren der Butter wird eine Probe genommen. Etwa 5 g (SM g) Butter werden auf 1 mg genau in einen 100-ml-Messkolben eingewogen.
5.1.2. Butterfett
Unmittelbar vor der Probenahme wird der Behälter mit dem Butterfett in einen Ofen mit einer Temperatur von 40 bis 50 °C gegeben und dort belassen, bis das Butterfett vollständig geschmolzen ist. Die Probe wird durch Schütteln oder Rühren vermischt; dabei ist die Bildung von Blasen durch zu starkes Rühren zu vermeiden. Etwa 4 g (SM g) Butterfett werden auf 1 mg genau in einen 100-ml-Messkolben eingewogen.
5.1.3. Rahm
Die Probe wird in einem Wasserbad oder einem Inkubator auf 35 bis 40 °C erwärmt. Anschließend wird das Fett durch Schütteln bzw. wenn nötig durch Rühren homogenisiert. Die Probe wird rasch auf 20 ±2 °C abgekühlt. Nun müsste die Probe homogen aussehen; ansonsten ist das Verfahren zu wiederholen. Etwa 10 g (SM g) Rahm werden auf 1 mg genau in einen 100-ml-Messkolben eingewogen.
5.2. Vorbereitung der Versuchslösung
Zur Probe (5.1.1, 5.1.2 oder 5.1.3) werden etwa 75 ml der Extraktionslösung (4.4) hinzugefügt; anschließend wird ca. 15 Minuten umgerührt oder stark geschüttelt und mit Extraktionslösung (4.4) aufgefüllt. Etwa 10 ml dieses Extrakts werden in ein Reagenzglas mit Stopfen gegeben. Das Reagenzglas wird in den Gefrierschrank (3.1) gesetzt und ca. 30 Minuten stehen gelassen. Der kalte Extrakt wird 5 Minuten bei etwa 2 000 min-1 zentrifugiert und sofort abgegossen. Die abgegossene Lösung muss auf Zimmertemperatur abkühlen. 5 ml der abgegossenen Lösung werden in einen 100-ml-Messkolben pipettiert; danach wird mit Wasser aufgefüllt. Ein Aliquot wird mit einer Spritze (3.2) durch einen Membran-Mikrofilter (3.3) gefiltert. Das Filtrat ist nun für die HPLC bereit.
5.3. Kalibrierung
5 ml der Vanillin-Standardlösung (4.5.2) werden in einen 100-ml-Messkolben pipettiert. 5 ml Extraktionslösung (4.4) werden hinzugefügt; anschließend wird bis zur Markierung mit Wasser aufgefüllt. Diese Lösung enthält 0,5 µg/ml Vanillin.
5.4. Bestimmung durch HPLC
Das Chromatografiesystem wird zur Stabilisierung etwa 30 Minuten stehen gelassen. Die Standardlösung (5.3) wird eingespritzt. Dieser Vorgang wird wiederholt, bis die Differenz der Peak-Flächen oder Peak-Höhen zwischen zwei aufeinanderfolgenden Einspritzungen weniger als 2 % beträgt. Unter den beschriebenen Bedingungen beträgt die Retentionszeit von Vanillin etwa 9 Minuten. Die Kalibrierlösung (5.3) wird doppelt analysiert, indem 20 µl eingespritzt werden. 20 µl der Versuchslösungen (5.2) werden eingespritzt. Die Peak-Flächen oder -Höhen für Vanillin werden ermittelt. Nach 10 Einspritzungen der Testproben (5.2) wird erneut zweimal die Kalibrierlösung (5.3) eingespritzt.
6. BERECHNUNG DER ERGEBNISSE
Für jede Partie Versuchslösungen wird die durchschnittliche Peak-Fläche (oder -Höhe) (AC) der Vanillin-Peaks in Zusammenhang mit den Doppeleinspritzungen der Kalibrierlösung jeweils vor und nach einer Probenreihe berechnet (insgesamt 4 Flächen oder Höhen).
Der Kalibrierfaktor (R) wird wie folgt berechnet:

Dabei ist CM die Masse des Vanillins in mg (4.5.1).
Der Vanillin-Anteil (C) der Testprobe (in mg/kg) wird wie folgt berechnet:

Dabei sind:
|
AS
|
=
|
Peak-Fläche des Vanillin-Peaks der Testprobe
|
|
SM
|
=
|
Masse der Testprobe in g (5.1.1, 5.1.2 oder 5.1.3)
|
Anmerkung: Bei der Untersuchung von Rahm auf Vanillin wird die Kennzeichnungsmittelkonzentration als mg Kennzeichnungsmittel/kg Milchfett ausgedrückt. Dazu wird C mit 100/f multipliziert. Mit f wird der Fettgehalt von Rahm in Prozent (m/m) bezeichnet.
|
20
|
=
|
Faktor zur Berücksichtigung der Verdünnung der Standard- und der Testprobe
|
|
0,96
|
=
|
Korrekturfaktor für den Fettgehalt in der ersten Verdünnung der Testprobe
|
Anmerkung: Anstelle von Peak-Flächen können auch Peak-Höhen verwendet werden (siehe 8.3).
7. GENAUIGKEIT DER METHODE
7.1. Wiederholbarkeit (r)
Die Ergebnisse zweier Bestimmungen, die von demselben Analytiker mit denselben Geräten am gleichen Testmaterial so rasch wie möglich nacheinander ausgeführt worden sind, dürfen um nicht mehr als 16 mg/kg voneinander abweichen.
7.2. Vergleichbarkeit (R)
Die Ergebnisse zweier Bestimmungen, die von Analytikern in verschiedenen Laboratorien mit verschiedenen Geräten am gleichen Testmaterial durchgeführt worden sind, dürfen um nicht mehr als 27 mg/kg voneinander abweichen.
8. TOLERANZGRENZEN
8.1. Dem gekennzeichneten Erzeugnis sind drei Proben zu entnehmen, um die Homogenität zu prüfen.
Das Kennzeichnungsmittel wird entweder aus Vanille oder aus synthetischem Vanillin gewonnen.
8.2.1. Die Beimischungsquote für 4-Hydroxy-3-methoxy-benzaldehyd beträgt 250 g je Tonne Butterfett oder Butter. Bei der Kennzeichnung von Rahm beträgt die Beimischungsquote 250 g je Tonne Milchfett.
8.2.2. Die Ergebnisse der drei in der Produktanalyse gewonnenen Proben werden verwendet, um die Quote und die Homogenität der Beimischung des Kennzeichnungsmittels zu bestimmen; das niedrigste ermittelte Ergebnis wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
220,8 mg/kg (95 % der Beimischungsmindestquote),
|
|
—
|
158,3 mg/kg (70 % der Beimischungsmindestquote).
|
Es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das man durch Interpolierung zwischen 220,8 mg/kg und 158,3 mg/kg erhält.
Das Kennzeichnungsmittel wird ausschließlich aus Vanilleschoten oder deren vollständigen Auszügen gewonnen.
8.3.1. Die Beimischungsquote für 4-Hydroxy-3-methoxy-benzaldehyd beträgt 100 g je Tonne Butterfett oder Butter. Bei der Kennzeichnung von Rahm beträgt die Beimischungsquote 100 g je Tonne Milchfett.
8.3.2. Aufgrund der Ergebnisse der drei aus der Produktanalyse gewonnenen Proben werden die Quote und die Homogenität der Beimischung des Kennzeichnungsmittels geprüft; das niedrigste dieser Ergebnisse wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
78,3 mg/kg (95 % der Beimischungsmindestquote),
|
|
—
|
53,3 mg/kg (70 % der Beimischungsmindestquote).
|
Es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das man durch Interpolierung zwischen 78,3 mg/kg und 53,3 mg/kg erhält.
9. ANMERKUNGEN
9.1. Die Wiederfindung von Vanillinzusätzen liegt bei einer Menge von 250 mg/kg Butterschmalz zwischen 97 und 103,8 %. Durchschnittlich wurde ein Gehalt von 99,9 % mit einer Standardabweichung von 2,7 % ermittelt.
9.2. Die Standardlösung enthält 5 % Extraktionslösung, um die Peak-Erweiterung durch die in den Proben vorhandenen 5 % Extraktionslösung zu kompensieren. Dadurch wird eine Quantifizierung über die Peak-Höhen möglich.
9.3. Die Analyse basiert auf einer linearen Kalibrierungslinie mit Nullachsenabschnitt.
9.4. Die Linearität sollte bei der ersten Analyse und später in regelmäßigen Abständen sowie nach Änderungen oder Reparaturen der HPLC-Geräte durch Verwendung geeigneter Verdünnungen der Standardlösung (4.5.2) geprüft werden. Vanillin kann durch in unpasteurisiertem Rahm oder in Erzeugnissen aus unpasteurisiertem Rahm enthaltene Enzyme zu Vanillinsäure, Divanillin und sonstigen Verbindungen abgebaut werden.
ANHANG VII
(Artikel 5)
BESTIMMUNG VON BETA-APO-8'-CAROTINSÄURE-ETHYLESTER IN BUTTERFETT UND IN BUTTER DURCH SPEKTROMETRIE
1. GEGENSTAND UND ANWENDUNGSBEREICH
Die Methode beschreibt ein Verfahren für die quantitative Bestimmung von Beta-Apo-8'-Carotinsäure-Ethylester (Apo-Carotin-Ester) in Butterfett und Butter. Apo-Carotin-Ester stellt die Summe aller Stoffe in einem Extrakt von Proben dar, die unter den in der Methode dargestellten Bedingungen gewonnen werden und Licht bei 440 nm absorbieren.
2. KURZBESCHREIBUNG
Das Butterfett wird in Petrolether gelöst; anschließend wird die Absorption bei 440 nm gemessen. Der Apo-Carotinester-Gehalt wird bezogen auf eine externe Standardprobe bestimmt.
3. APPARATUR
3.1. Messpipetten mit Fassungsvermögen von 0,25, 0,50, 0,75 und 1,0 ml
3.2. Spektrophotometer, geeignet für die Verwendung bei 440 nm (und 447-449 nm), ausgestattet mit Küvetten mit optischer Weglänge von 1 cm
3.3. Messkolben, 20 ml und 100 ml
3.4. Analysewaage, Empfindlichkeit 0,1 mg, Messgenauigkeit 1 mg, Skalenteilung 0,1 mg
3.5. Ofen, 45 °C ±1 °C
3.6. Aschefreie Schnellfilter
4. REAGENZIEN
Alle Reagenzien müssen analysenrein sein.
Apo-Carotinester-Suspension (ungefähr 20 %)
4.1.1. Der Gehalt der Suspension ist wie folgt zu bestimmen:
Die Suspension wird auf 45 bis 50 °C erwärmt und in der ungeöffneten Originalverpackung homogenisiert. Etwa 400 mg werden genau in einen 100-ml-Messkolben eingewogen und in 20 ml Chloroform (4.4) aufgelöst; anschließend wird bis zur Marke mit Cyclohexan (4.5) aufgefüllt. 5 ml dieser Lösung werden mit Cyclohexan auf 100 ml aufgefüllt (Lösung A). 5 ml der Lösung A werden mit Cyclohexan auf 100 ml verdünnt. Die Absorption wird bei 447-449 nm gemessen. (Das Maximum im Vergleich zu Cyclohexan unter Verwendung von 1-cm-Küvetten wird als Blindprobe gemessen.)
P Apo-Carotin-Ester-Gehalt (%) = (Absmax × 40 000) / (Msusp × 2 550) bzw. (Absmax / 2 550) × (100 / 5) × (100 / 5) × (100 / Msusp)
|
Absmax
|
=
|
Höchstabsorption der Kalibrierlösung
|
|
Msusp
|
=
|
Masse der Suspension (g)
|
|
2 550
|
=
|
Referenzlaboratorien (1 %, 1 cm)
|
|
P
|
=
|
Reinheit (Gehalt) der Suspension (%)
|
Anmerkung: Apo-Carotin-Ester-Suspension ist empfindlich gegenüber Luft, Hitze und Licht. In der ungeöffneten Originalverpackung (unter Stickstoffsiegel) und an einem kühlen Platz kann sie etwa zwölf Monate aufbewahrt werden. Nach Öffnung ist der Inhalt innerhalb kurzer Zeit zu verwenden.
4.1.2. Apo-Carotin-Ester-Standardlösung, ungefähr 0,2 mg/ml
0,100 g Apo-Carotin-Ester-Suspension (4.1.1) (W) werden auf 1 mg genau abgewogen, in Petroleumbenzin (4.2) aufgelöst und quantitativ in einen 100-ml-Messkolben gegeben; anschließend wird bis zur Marke mit Petroleumbenzin aufgefüllt.
Diese Lösung enthält (W × P) 10 mg/ml Apo-Carotin-Ester.
Anmerkung: Die Lösung muss kühl und dunkel aufbewahrt werden. Nicht verwendete Lösung ist nach einem Monat zu beseitigen.
4.2. Petroleumbenzin (40-60 °C)
4.3. Natriumsulfat, wasserfrei, granular, zuvor 2 Stunden lang bei 102 °C getrocknet.
4.4. Chloroform
4.5. Cyclohexan
5. VERFAHREN
5.1. Vorbereitung der Probe
5.1.1. Butterfett
Die Probe wird in einem Ofen bei etwa 45 °C geschmolzen.
5.1.2. Butter
Die Probe wird in einem Ofen bei etwa 45 °C geschmolzen und ein repräsentativer Teil durch einen Filter mit etwa 10 g wasserfreiem Natriumsulfat (4.3) gefiltert; diese Schritte werden unter Abschirmung von starkem natürlichem oder künstlichem Licht und bei einer gleich bleibenden Temperatur von 45 °C durchgeführt. Es wird eine geeignete Menge Butterfett entnommen.
5.2. Bestimmung
Etwa 1 g Butterfett oder Milchfett (5.1.2) (M) werden auf 1 mg genau abgewogen. Das Fett wird unter Verwendung von Petroleumbenzin (4.2) quantitativ in einen 20-ml-(V)-Messkolben gegeben; anschließend wird bis zur Marke aufgefüllt und gründlich gemischt.
Ein Aliquot dieses Gemisches wird in eine 1-cm-Küvette gebracht; dann wird die Absorption bei 440 nm im Vergleich zur Blindprobe mit Petroleumbenzin gemessen. Die Konzentration des Apo-Carotin-Esters in der Lösung wird durch Vergleich mit der Kalibrierkurve C (μg/ml) ermittelt.
5.3. Kalibrierung
0, 0,25, 0,5, 0,75 und 1,0 ml der Apo-Carotin-Ester-Standardlösung (4.1.2) werden in fünf 100-ml-Messkolben pipettiert. Anschließend wird mit Petroleumbenzin (4.2) aufgefüllt und gemischt.
Die ungefähren Konzentrationen der Lösungen liegen im Bereich 0 bis 2 µg/ml und werden im Vergleich mit der Konzentration der Standardlösung (4.1.2) (W × P) / 10 mg/ml genau berechnet. Die Absorptionswerte bei 440 nm werden bezogen auf die Petroleumbenzin-Blindprobe (4.2) gemessen.
Die Absorptionswerte auf der y-Achse und die jeweiligen Apo-Carotin-Ester-Konzentrationen werden auf der x-Achse eintragen. Die Formel der Kalibrierkurve wird berechnet.
6. BERECHNUNG DER ERGEBNISSE
6.1. Der Apo-Carotin-Ester-Gehalt, ausgedrückt in mg/kg des Erzeugnisses, wird mit den folgenden Formeln berechnet:
Butterfett: Butterfett (C × V)/M
Butter: 0,82 (C × V)M
Dabei sind:
|
C
|
=
|
Apo-Carotin-Ester-Gehalt in µg/ml, von der Kalibrierkurve (5.3) abzulesen
|
|
V
|
=
|
Volumen (ml) der Versuchslösung (5.2)
|
|
M
|
=
|
Masse (g) der Testprobe (5.2)
|
|
0,82
|
=
|
Berichtigungsfaktor für den Milchfettgehalt der Butter
|
7. GENAUIGKEIT DER METHODE
7.1. Wiederholbarkeit
7.1.1. Butteranalyse
Die Ergebnisse zweier Bestimmungen, die von demselben Analytiker mit denselben Geräten am gleichen Testmaterial so rasch wie möglich nacheinander ausgeführt worden sind, dürfen um nicht mehr als 1,4 mg/kg voneinander abweichen.
7.1.2. Butterfettanalyse
Die Ergebnisse zweier Bestimmungen, die von demselben Analytiker mit denselben Geräten am gleichen Testmaterial so rasch wie möglich nacheinander ausgeführt worden sind, dürfen um nicht mehr als 1,6 mg/kg voneinander abweichen.
7.2. Vergleichbarkeit
7.2.1. Butteranalyse
Die Ergebnisse zweier Bestimmungen, die von Analytikern in verschiedenen Laboratorien mit verschiedenen Geräten am gleichen Testmaterial durchgeführt worden sind, dürfen um nicht mehr als 4,7 mg/kg voneinander abweichen.
7.3. Butterfettanalyse
Die Ergebnisse zweier Bestimmungen, die von Analytikern in verschiedenen Laboratorien mit verschiedenen Geräten am gleichen Testmaterial durchgeführt worden sind, dürfen um nicht mehr als 5,3 mg/kg voneinander abweichen.
7.4. Herkunft der Präzisionsdaten
Die Präzisionsdaten stammen aus einem 1995 unter Beteiligung von elf Laboratorien und zwölf gekennzeichneten Proben (sechs Blindduplikate) für Butter und zwölf gekennzeichneten Proben (sechs Blindduplikate) für Butterfett durchgeführten Versuch.
8. TOLERANZGRENZEN
8.1. Zur Prüfung der richtigen Kennzeichnung des Erzeugnisses sind dem gekennzeichneten Erzeugnis drei Proben zu entnehmen.
8.2. Butter
8.2.1. Der Beimischungsanteil für Butter beträgt unter Berücksichtigung der Hintergrundabsorption 22 mg/kg.
8.2.2. Die Ergebnisse der drei aus der Produktanalyse erhaltenen Proben werden verwendet, um die Quote und die Homogenität der Beimischung des Kennzeichnungsmittels zu prüfen; das niedrigste dieser Ergebnisse wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
17,7 mg/kg (95 % der Beimischungsmindestquote),
|
|
—
|
12,2 mg/kg (70 % der Beimischungsmindestquote).
|
Es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das durch Interpolierung zwischen 17,7 mg/kg und 12,2 mg/kg ermittelt wurde.
8.3. Butterfett
8.3.1. Der Beimischungsanteil für Butterfett beträgt unter Berücksichtigung der Hintergrundabsorption 24 mg/kg.
Die Ergebnisse der drei aus der Produktanalyse erhaltenen Proben werden verwendet, um die Quote und die Homogenität der Beimischung des Kennzeichnungsmittels zu prüfen; das niedrigste dieser Ergebnisse wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
19,2 mg/kg (95 % der Beimischungsmindestquote),
|
|
—
|
13,2 mg/kg (70 % der Beimischungsmindestquote).
|
Es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das durch Interpolierung zwischen 13,2 mg/kg und 19,2 mg/kg ermittelt wurde.
ANHANG VIII
(Artikel 5)
BESTIMMUNG DES SITOSTERIN- ODER DES STIGMASTERIN-GEHALTS IN BUTTER ODER BUTTERFETT DURCH KAPILLARSÄULEN-GASCHROMATOGRAFIE
1. GEGENSTAND UND ANWENDUNGSBEREICH
Die Methode beschreibt ein Verfahren zur quantitativen Bestimmung von Sitosterin bzw. Stigmasterin in Butterfett und Butter. Sitosterin ist dabei die Summe von β-Sitosterin und 22-Dihydro-β-Sitosterin; die anderen Sitosterine werden als unbedeutend betrachtet.
2. KURZBESCHREIBUNG
Das Butterfett bzw. die Butter wird mit ethanolischer Kaliumhydroxidlösung verseift. Die unverseifbaren Bestandteile werden mit Ether extrahiert.
Die Sterine werden zu Trimethylsilylethern derivatisiert und durch Kapillarsäulen-Gaschromatografie mit Betulin als interner Standardprobe analysiert.
3. APPARATUR
3.1. 150-ml-Verseifungskolben mit Rückflusskühler und Schliffverbindungen
3.2. 500-ml- Scheidetrichter
3.3. 250-ml-Kolben
3.4. Druckausgleichtrichter (etwa 250 ml) zum Auffangen von überschüssigem Ether
3.5. Glassäule, 350 mm × 20 mm, mit Sinterglasfritte
3.6. Wasserbad oder Heizhaube
3.7. Reaktionsglas, 2 ml
Zur Verwendung mit einer Kapillarsäule geeigneter Gaschromatograf, ausgerüstet mit einem „Split-System“ und folgenden Bestandteilen:
3.8.1. Säulenofen, der die gewünschte Temperatur mit einer Genauigkeit von ±1 °C halten kann;
3.8.2. Verdampfer mit einstellbarer Temperatur;
3.8.3. Flammenionisationsdetektor und einem Signal-Verstärker;
3.8.4. Aufzeichnungsgerät mit Integrator für den Signal-Verstärker (3.8.3).
3.9. Quarzglas-Kapillarsäule, vollständig mit BP1 oder einem gleichwertigen Material beschichtet (oder eine sonstige Säule mit mindestens gleicher Auflösung) mit einheitlicher Stärke von 0,25 µm; die Säule muss die Trimethylsilylderivate von Lanosterin und Sitosterin trennen können. Geeignet ist eine mit BP 1 beschichtete Säule von 12 m Länge und 0,2 mm Innendurchmesser.
3.10. 1-μl-Gaschromatografie-Mikrospritze mit gehärteter Nadel.
4. REAGENZIEN
Alle Reagenzien müssen analysenrein sein. Das verwendete Wasser muss destilliert sein oder einen mindestens gleichwertigen Reinheitsgrad aufweisen.
4.1. Ethanol, Reinheitsgrad mindestens 95 %
4.2. 60 %ige Kaliumhydroxid-Lösung (600 g Kaliumhydroxid (mindestens 85 %) werden in Wasser gelöst; die Lösung wird anschließend mit Wasser bis auf einen Liter aufgefüllt
Betulin, Reinheitsgrad mindestens 99 %
Betulinlösungen in Diethylether (4.4)
4.3.1.1. Die zur Bestimmung von Sitosterin verwendete Betulinlösung muss eine Konzentration von 1,0 mg/ml aufweisen.
4.3.1.2. Die zur Bestimmung von Stigmasterin verwendete Betulinlösung muss eine Konzentration von 0,4 mg/ml aufweisen.
4.4. Diethylether, analysenrein (frei von Peroxiden oder Rückständen)
4.5. Natriumsulfat, wasserfrei, granular, zuvor 2 Stunden bei 102 °C getrocknet
4.6. Silylierungsreagens, z. B. TRI-SIL (erhältlich von Pierce Chemical Co., Kat.-Nr. 49001) oder vergleichbares Reagenz. (Achtung: TRI-SIL ist entflammbar, giftig, ätzend und potenziell kanzerogen. Das Laborpersonal muss mit den Sicherheitsdaten zu TRI-SIL vertraut sein und die erforderlichen Vorsichtsmaßnahmen treffen.)
4.7. Lanosterol
Sitosterin, bekannter Reinheitsgrad mindestens 90 % (P)
Anmerkung 1: Die Reinheit der Standardstoffe für die Kalibrierung ist durch Normalisierung zu bestimmen. Dazu wird angenommen, dass das Chromatogramm alle in der Probe vorhandenen Sterine wiedergibt, dass die Gesamtfläche der Peaks 100 % der Sterinbestandteile darstellt und dass alle Sterine ein gleich starkes Detektorsignal hervorrufen. Es ist sicherzustellen, dass das System in den maßgeblichen Konzentrationsbereichen linear verläuft.
4.8.1. Sitosterin-Standardlösung: Sitosterin (4.8) wird in Diethylether (4.4) mit einer Konzentration von etwa 0,5 mg/ml (Genauigkeit 0,001 mg/ml) gelöst;
Stigmasterin, bekannter Reinheitsgrad mindestens 90 % (P);
4.9.1. Stigmasterin-Standardlösung: Stigmasterin (4.9) wird in Diethylether (4.4) mit einer Konzentration von etwa 0,2 mg/ml (Genauigkeit 0,001 mg/ml) gelöst;
4.10. Testlösung für die Prüfung der Auflösung: Lanosterin (4.7) (0,05 mg/ml) und Sitosterin (4.8) (0,5 mg/ml) werden in Diethylether (4.4) gelöst.
5. METHODE
5.1. Herstellung der Standardlösungen für die Chromatografie:
Die interne Standardlösung (4.3.1) muss der entsprechenden Sterin-Standardlösung und der verseiften Probe gleichzeitig zugesetzt werden (siehe 5.2.2).
5.1.1. Sitosterin-Standardlösung für die Chromatografie: je 1 ml der Sitosterin-Standardlösung (4.8.1) werden in 2 Reaktionsgläser (3.7) gegeben; der Ether ist durch einen Stickstoffstrom zu entfernen. 1 ml der internen Standardlösung (4.3.1.1) werden hinzugegeben und der Ether durch einen Stickstoffstrom entfernt.
5.1.2. Stigmasterin-Standardlösung für die Chromatografie: je 1 ml der Stigmasterin-Standardlösung (4.9.1) wird in 2 Reaktionsgläser (3.7) gegeben und der Ether durch einen Stickstoffstrom entfernt. 1 ml der internen Standardlösung (4.3.1.2) wird hinzugegeben und der Ether durch einen Stickstoffstrom entfernt.
5.2. Vorbereitung der unverseifbaren Bestandteile
5.2.1. Die Butterprobe wird bei höchstens 35 °C geschmolzen und unter Rühren gründlich durchgemischt.
Ungefähr 1 g Butter (W2) oder Butterfett (W2) werden auf 1 mg genau in einen 150-ml-Kolben (3.1) eingewogen. 50 ml Ethanol (4.1) und 10 ml Kaliumhydroxid-Lösung (4.2) werden hinzugegeben. Der Rückflusskühler wird angebracht und 30 Minuten auf etwa 75 °C erhitzt. Der Kühler wird abgenommen; danach wird gewartet, bis sich der Kolben auf Raumtemperatur abgekühlt hat.
5.2.2. 1,0 ml der internen Standardlösung (4.3.1.1 bei der Bestimmung von Sitosterin bzw. 4.3.1.2 bei der Bestimmung von Stigmasterin) werden in den Kolben gegeben. Die Lösung wird gründlich gemischt. Der Inhalt des Kolbens wird quantitativ in einen 500-ml-Scheidetrichter (3.2) gegeben, in dem der Kolben nacheinander mit 50 ml Wasser und 250 ml Diethylether (4.4) ausgespült wird. Der Scheidetrichter wird zwei Minuten kräftig geschüttelt; anschließend wird die Phasentrennung abgewartet. Die untere Wasserschicht wird abgelassen und die Etherschicht viermal mit je 100 ml Wasser ausgeschüttelt.
Anmerkung 2: Damit keine Emulsion entsteht, ist es wichtig, die ersten beiden Waschvorgänge mit Wasser vorsichtig auszuführen (10 Drehungen). Beim dritten Waschvorgang kann 30 Sekunden lang kräftig geschüttelt werden. Sollte sich eine Emulsion bilden, kann diese durch Zugabe von 5-10 ml Ethanol aufgelöst werden. Bei Zugabe von Ethanol muss unbedingt noch zweimal kräftig mit Wasser ausgeschüttelt werden.
5.2.3. Die klare seifenfreie Etherschicht wird durch eine Glassäule (3.5) mit 30 g wasserfreiem Natriumsulfat (4.5) eluiert. Der Ether wird in einem 250-ml-Kolben (3.3) aufgefangen. Ein Siedesteinchen wird hinzugegeben; anschließend wird die Flüssigkeit auf einem Wasserbad oder einer Heizhaube fast bis zur völligen Trockenheit eingedampft; dabei wird das überschüssige Lösungsmittel aufgefangen.
Anmerkung 3: Werden die Probenextrakte bei zu hoher Temperatur vollständig getrocknet, können Sterinverluste auftreten.
5.3. Vorbereitung der Trimethylsilylether
5.3.1. Die im Kolben verbliebene Etherlösung mit 2 ml Ether wird in ein 2-ml-Reaktionsglas (3.7) gegeben und der Ether durch einen Stickstoffstrom entfernt. Der Kolben wird mit 2 weiteren 2-ml-Aliquoten Ether ausgespült; dabei wird jeweils der Inhalt in das Reaktionsglas umgefüllt und der Ether durch einen Stickstoffstrom entfernt.
5.3.2. Die Probe wird durch Zugabe von 1 ml TRI-SIL (4.6) silyliert. Das Glas wird verschlossen und kräftig geschüttelt, damit sich die Probe auflöst. Wenn sich die Probe nicht vollständig auflöst, wird die Probe auf 65-70 °C erwärmt. Danach wird die Probe mindestens fünf Minuten stehen gelassen, bevor sie in den Gaschromatografen injiziert wird. Die Standardlösungen werden in gleicher Weise wie die Proben silyliert. Die Mischung zur Prüfung der Auflösung (4.10) wird ebenfalls in gleicher Weise wie die Proben silyliert.
Anmerkung 4: Die Silylierung ist in wasserfreier Umgebung vorzunehmen. Die unvollständige Silylierung von Betulin wird durch einen zweiten Peak dicht neben dem von Betulin angezeigt.
Ethanol-Rückstände in der Silylierungsphase können die Silylierung beeinträchtigen. Möglicherweise wurde in der Extraktionsphase nicht hinreichend gespült. In diesem Fall muss bei der Extraktion ein fünftes Mal gespült werden (30 Sekunden kräftiges Schütteln).
5.4. Analyse durch Gaschromatografie
5.4.1. Herstellung der Arbeitsbedingungen
Der Gaschromatograf wird gemäß der Herstelleranweisung vorbereitet.
Als Leitlinie gelten die folgenden Arbeitsbedingungen:
|
—
|
Säulentemperatur: 265 °C
|
|
—
|
Injektortemperatur: 265 °C
|
|
—
|
Detektortemperatur: 300 °C
|
|
—
|
Durchflussgeschwindigkeit des Trägergases: 0,6 ml/min
|
|
—
|
Wasserstoffdruck: 84 kPa
|
|
—
|
Splitverhältnis: 10:1 bis 50:1; das Splitverhältnis ist nach den Herstelleranweisungen zu optimieren; anschließend wird die Linearität des Detektorsignals im interessierenden Konzentrationsbereich überprüft.
Anmerkung 5: Es ist unbedingt darauf zu achten, dass die Injektionsröhre regelmäßig gereinigt wird.
|
|
—
|
Menge der eingespritzten Substanz: 1 µl TMSE-Lösung.
|
Vor Beginn jeglicher Analysen muss gewartet werden, bis das System äquilibriert ist und ein hinreichend stabiles Response-Signal liefert.
Diese Bedingungen können je nach Beschaffenheit der Säule und des Gaschromatografen abgewandelt werden, damit Chromatogramme entstehen, die den folgenden Anforderungen genügen:
|
—
|
Der Sitosterin-Peak muss vom Lanosterin-Peak genügend abgesetzt sein. Abbildung 1 zeigt ein typisches Chromatogramm, das sich aus der silylierten Testlösung zur Prüfung der Auflösung ergeben sollte (4.10).
|
|
—
|
Die relativen Retentionszeiten der folgenden Sterine sollten ungefähr betragen:
|
|
—
|
Die Retentionszeit für Betulin sollte etwa 24 Minuten betragen.
|
5.4.2. Analyseverfahren
1 µl der silylierten Standardlösung (Stigmasterin bzw. Sitosterin) wird eingespritzt; danach sind die Parameter für die Kalibrierung des Integrators einzustellen.
Darauf wird nochmals 1 µl der silylierten Standardlösung eingespritzt, um die Response-Faktoren in Bezug auf Betulin zu bestimmen.
1 µl der silylierten Probelösung wird eingespritzt; danach werden die Peak-Flächen gemessen. Am Anfang und am Ende einer jeden gaschromatografischen Analysenfolge muss die Standardlösung eingespritzt werden.
Als Leitlinie sollte nach je sechs Probelösungen die Standardlösung eingespritzt werden.
Anmerkung 6: Bei der Integration des Stigmasterin-Peaks ist auch das durch die Punkte 1, 2 und 3 in Abbildung 2b markierte Tailing mit zu erfassen.
Bei der Integration des Sitosterin-Peaks ist die Fläche des Peaks von 22-Dihydro-β-Sitosterin (Stigmastanin), das unmittelbar nach Sitosterin eluiert wird, mit zu erfassen, wenn das gesamte Sitosterin berechnet werden soll.
6. BERECHNUNG DER ERGEBNISSE
6.1. Die Fläche der Sterin-Peaks und der Betulin-Peaks für die zu Beginn und am Ende eines Durchlaufs eingespritzten Standardlösungen bestimmen und R1 berechnen:
R1 = (durchschnittliche Fläche des Sterin-Peaks der Standardlösung)/(durchschnittliche Fläche des Betulin-Peaks der Standardlösung)
Die Fläche des Sterin-Peaks (Stigmasterin und Sitosterin) und des Betulin-Peaks der Probe werden bestimmt, und R2 wird berechnet:
R2 = (Fläche des Sterin-Peaks der Probe)/(Fläche des Betulin-Peaks der Probe)
|
W1
|
=
|
Steringehalt der Standardprobe (mg) in 1 ml Standardlösung (4.8.1 oder 4.9.1)
|
|
W2
|
=
|
Gewicht der Probe (g) (5.2.1)
|
|
P
|
=
|
Reinheitsgrad des Standardsterins (4.8 oder 4.9)
|
Steringehalt der Probe (mg/kg) = ((R
2) / (R
1)) × ((W
1) / (W
2)) × P × 10
7. GENAUIGKEIT DER METHODE
7.1. Butter
7.1.1. Wiederholbarkeit
7.1.1.1. Stigmasterin
Die Ergebnisse zweier Bestimmungen, die von demselben Analytiker mit denselben Geräten am gleichen Testmaterial so rasch wie möglich nacheinander ausgeführt worden sind, dürfen um nicht mehr als 19,3 mg/kg voneinander abweichen.
7.1.1.2. Sitosterin
Die Ergebnisse zweier Bestimmungen, die von demselben Analytiker mit denselben Geräten am gleichen Testmaterial so rasch wie möglich nacheinander ausgeführt worden sind, dürfen um nicht mehr als 23,0 mg/kg voneinander abweichen.
7.1.2. Vergleichbarkeit
7.1.2.1. Stigmasterin
Die Ergebnisse zweier Bestimmungen, die von Analytikern in verschiedenen Laboratorien mit verschiedenen Geräten am gleichen Testmaterial durchgeführt worden sind, dürfen um nicht mehr als 31,9 mg/kg voneinander abweichen.
7.1.2.2. Sitosterin
Die Ergebnisse zweier Bestimmungen, die von Analytikern in verschiedenen Laboratorien mit verschiedenen Geräten am gleichen Testmaterial durchgeführt worden sind, dürfen um nicht mehr als 8,7 % des Durchschnitts der Bestimmungen voneinander abweichen.
7.1.3. Herkunft der Präzisionsdaten
Die Präzisionsdaten stammen aus einem 1992 durchgeführten Versuch, an dem 8 Laboratorien beteiligt waren und bei dem 6 Proben (3 Blindproben) auf Stigmasterin und 6 Proben (3 Blindproben) auf Sitosterin untersucht wurden.
7.2. Butterfett
7.2.1. Wiederholbarkeit
7.2.1.1. Stigmasterin
Die Ergebnisse zweier Bestimmungen, die von demselben Analytiker mit denselben Geräten am gleichen Testmaterial so rasch wie möglich nacheinander ausgeführt worden sind, dürfen um nicht mehr als 10,2 mg/kg voneinander abweichen.
7.2.1.2. Sitosterin
Die Ergebnisse zweier Bestimmungen, die von demselben Analytiker mit denselben Geräten am gleichen Testmaterial so rasch wie möglich nacheinander ausgeführt worden sind, dürfen um nicht mehr als 3,6 % des Durchschnitts der Bestimmungen voneinander abweichen.
7.2.2. Vergleichbarkeit
7.2.2.1. Stigmasterin
Die Ergebnisse zweier Bestimmungen, die von Analytikern in verschiedenen Laboratorien mit verschiedenen Geräten am gleichen Testmaterial durchgeführt worden sind, dürfen um nicht mehr als 25,3 mg/kg voneinander abweichen.
7.2.2.2. Sitosterin
Die Ergebnisse zweier Bestimmungen, die von Analytikern in verschiedenen Laboratorien mit verschiedenen Geräten am gleichen Testmaterial durchgeführt worden sind, dürfen um nicht mehr als 8,9 % des Durchschnitts der Bestimmungen voneinander abweichen.
7.2.3. Herkunft der Präzisionsdaten
Die Präzisionsdaten stammen aus einem 1991 durchgeführten Versuch, an dem neun Laboratorien beteiligt waren und bei dem 6 Proben (3 Blindproben) auf Stigmasterin und 6 Proben (3 Blindproben) auf Sitosterin untersucht wurden.
8. TOLERANZBEREICH
8.1. Zur Prüfung der richtigen Kennzeichnung des Erzeugnisses sind dem gekennzeichneten Erzeugnis drei Proben zu entnehmen.
8.2. Butter
8.2.1. Stigmasterin
8.2.1.1. Der Beimischungsanteil für Stigmasterin beträgt 150 g bei mindestens 95 % reinem Stigmasterin je Tonne Butter, d. h. 142,5 mg/kg, bzw. 170 g bei mindestens 85 % reinem Stigmasterin je Tonne Butter, d. h. 144,5 mg/kg.
8.2.1.2. Die in der Produktanalyse für die drei Proben ermittelten Ergebnisse werden verwendet, um die Quote und die Homogenität der Beimischung des Kennzeichnungsmittels zu prüfen; das niedrigste dieser Ergebnisse wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
115,8 mg/kg (95 % der Beimischungsmindestquote bei 95 % reinem Sitosterin),
|
|
—
|
117,7 mg/kg (95 % der Beimischungsmindestquote bei 85 % reinem Stigmasterin).
|
|
—
|
80,1 mg/kg (70 % der Beimischungsmindestquote bei 95 % reinem Stigmasterin),
|
|
—
|
81,5 mg/kg (70 % der Beimischungsmindestquote bei 85 % reinem Stigmasterin).
|
Es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das durch Interpolierung zwischen 115,8 mg/kg und 80,1 mg/kg bzw. 117,7 mg/kg und 81,5 mg/kg ermittelt wurde.
8.2.2. Sitosterin
8.2.2.1. Der Beimischungsanteil für Sitosterin beträgt 600 g bei mindestens 90 % reinem Sitosterin je Tonne Butter, d. h. 540 mg/kg.
8.2.2.2. Die in der Produktanalyse für die drei Proben ermittelten Ergebnisse werden verwendet, um die Quote und die Homogenität der Beimischung des Kennzeichnungsmittels zu prüfen; das niedrigste dieser Ergebnisse wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
482,6 mg/kg (95 % der Beimischungsmindestquote bei 90 % reinem Sitosterin),
|
|
—
|
347,6 mg/kg (70 % der Beimischungsmindestquote bei 90 % reinem Sitosterin).
|
Es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das man durch Interpolierung zwischen 482,6 mg/kg und 347,6 mg/kg erhält.
8.3. Butterfett
8.3.1. Stigmasterin
8.3.1.1. Der Beimischungsanteil für Sitosterin beträgt 150 g bei mindestens 95 % reinem Sitosterin je Tonne Butterfett, d. h. 142,5 mg/kg oder 170 g bei mindestens 85 % reinem Stigmasterin je Tonne Butterfett, d. h. 144,5 mg/kg
8.3.1.2. Aufgrund der Ergebnisse der drei in der Produktanalyse untersuchten Proben wird die Homogenität der Beimischung des Kennzeichnungsmittels geprüft; das niedrigste Ergebnis wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
118,5 mg/kg (95 % der Beimischungsmindestquote bei 95 % reinem Sitosterin),
|
|
—
|
120,4 mg/kg (95 % der Beimischungsmindestquote bei 85 % reinem Stigmasterin),
|
|
—
|
82,9 mg/kg (70 % der Beimischungsmindestquote bei 95 % reinem Stigmasterin),
|
|
—
|
84,3 mg/kg (70 % der Beimischungsmindestquote bei 85 % reinem Stigmasterin).
|
Es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das man durch Interpolierung zwischen 118,5 mg/kg und 82,9 mg/kg bzw. 120,4 mg/kg und 84,3 mg/kg erhält.
8.3.2. Sitosterin
8.3.2.1. Der Beimischungsanteil für Sitosterin beträgt 600 g bei mindestens 90 % reinem Sitosterin je Tonne Butterfett, d. h. 540 mg/kg.
8.3.2.2. Aufgrund der Ergebnisse der drei in der Produktanalyse untersuchten Proben wird die Homogenität der Beimischung des Kennzeichnungsmittels geprüft; das niedrigste Ergebnis wird bezogen auf die folgenden Grenzwerte beurteilt:
|
—
|
480,9 mg/kg (95 % der Beimischungsmindestquote bei 90 % reinem Sitosterin),
|
|
—
|
345,9 mg/kg (70 % der Beimischungsmindestquote bei 90 % reinem Sitosterin).
|
Es wird die Kennzeichnungsmittelkonzentration der Probe mit dem niedrigsten Ergebnis verwendet, das man durch Interpolierung zwischen 480,9 mg/kg und 345,9 mg/kg erhält.
Abbildung 1
Chromatogramm Auflösung Testgemisch
Wünschenswert ist eine vollständige Auflösung, d. h. der Peak-Verlauf muss zur Basislinie zurückführen, bevor der nächste Sitosterin-Peak beginnt; allerdings ist auch eine unvollständige Auflösung annehmbar.

|
Abbildung 2a
|
Abbildung 2b
|
|
Stigmasterol standard
|
Butter sample denatured with stigmasterol
|
|
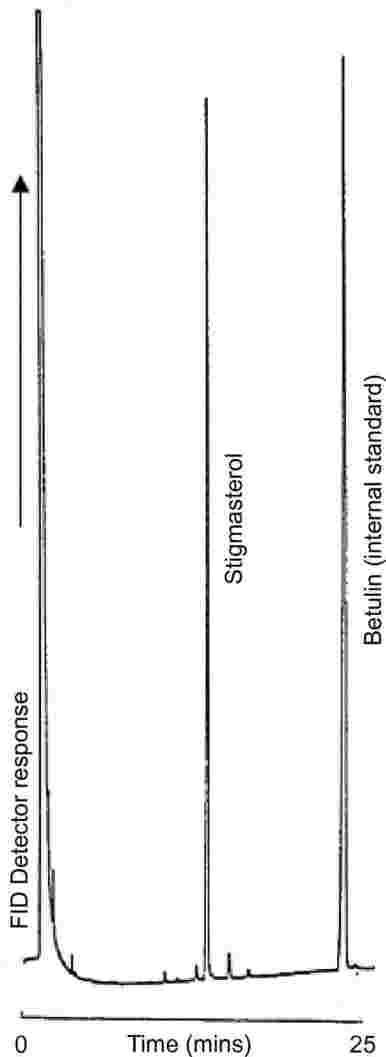
|

|
|
Hinweis: Die Integration des Stigmasterin-Peaks muss etwaige Tailings gemäß den Ziffern 1, 2 und 3 beinhalten.
|
|
Abbildung 3a
|
Abbildung 3b
|
|
Sistosterol standard
|
Butter sample denatured with β-Sitosterol
|
|

|

|
|
Hinweis: β-Sitosterin enthält häufig eine Verunreinigung (als Stigmastanol), die unmittelbar nach dem β-Sitosterin eluiert. Die Flächen dieser beiden Peaks sind bei der Bewertung des Gesamtgehalts an β-Sitosterin zusammenzufassen.
|
ANHANG IX
(Artikel 6)
REFERENZMETHODE ZUM NACHWEIS VON KUHMILCH UND KASEIN IN KÄSE AUS SCHAF-, ZIEGEN- ODER BÜFFELMILCH ODER AUS GEMISCHEN VON SCHAF-, ZIEGEN- ODER BÜFFELMILCH
1. ANWENDUNGSBEREICH
Diese Arbeitsvorschrift beschreibt ein Verfahren zum Nachweis von Kuhmilch und Kasein in Käse aus Schaf-, Ziegen- oder Büffelmilch oder aus Gemischen von Schaf-, Ziegen- oder Büffelmilch mit Hilfe der isoelektrischen Fokussierung der γ-Kaseine nach Plasminolyse.
2. ANWENDBARKEIT
Dieses Verfahren dient dem empfindlichen und spezifischen Nachweis von Kuhmilch und Kasein (jeweils mit und ohne Wärmebehandlung) in frischem und gereiftem Käse aus Schaf-, Ziegen- oder Büffelmilch oder aus Gemischen von Schaf-, Ziegen- oder Büffelmilch. Es ist ungeeignet für den Nachweis von Milch- und Käsefälschung durch hitzebehandeltes Rindermolkekonzentrat.
3. KURZBESCHREIBUNG
3.1. Das Kasein wird aus Käse und Referenzproben isoliert.
3.2. Das isolierte Kasein wird gelöst; anschließend wird eine Plasminspaltung vorgenommen (EC.3.4.21.7).
3.3. Die plasminbehandelten Kaseine werden in Anwesenheit von Harnstoff und unter Anfärben der Proteine isoelektrisch fokussiert.
3.4. Die Auswertung erfolgt durch Vergleich der gefärbten γ3- und γ2-Kaseinbanden (Kuhmilchnachweis) zwischen der zu bestimmenden Probe und der im selben Gel laufenden, 0 % und 1 % Kuhmilch enthaltenden Referenzproben.
4. REAGENZIEN
Soweit nicht anders angegeben, sind analysenreine Reagenzien zu verwenden. Das Wasser muss zweimal destilliert oder von entsprechender Reinheit sein.
Anmerkung: Die folgenden Angaben gelten für im Labor hergestellte harnstoffhaltige Polyacrylamidgele im Format 265 × 125 × 0,25 mm. Bei Verwendung anderer Gelrezepturen und -formate müssen die Elektrophoresebedingungen gegebenenfalls angepasst werden.
Isoelektrische Fokussierung
4.1. Reagenzien zur Herstellung der harnstoffhaltigen Polyacrylamidgele
4.1.1. Gelstammlösung
4,85 g Acrylamid,
0,15 g N, N-Methylen-bis-acrylamid (BIS),
48,05 g Harnstoff und
15,00 g Glycerin (87 % w/w)
werden in Wasser aufgelöst; anschließend wird die Lösung auf 100 ml aufgefüllt, in Braunglasflaschen gegeben und im Kühlschrank aufbewahrt.
Anmerkung: Statt der angegebenen Festeinwaagen der neurotoxischen Acrylamide kann vorzugsweise eine der handelsüblichen Acrylamid-Fertiglösungen verwendet werden. Bei einer solchen Lösung mit 30 % (w/w) Acrylamid und 0,8 % (w/v) BIS müssen anstelle der Festeinwaagen 16,2 ml für die genannte Rezeptur eingesetzt werden. Die Haltbarkeit der Gelstammlösung beträgt maximal 10 Tage. Beträgt die Leitfähigkeit mehr als 5 µS, wird die Lösung mit 2 g Amberlite MB-3 versetzt, unter Rühren 30 Minuten lang entionisiert und dann membranfiltriert (0,45 µm).
4.1.2. Gellösung
Aus der Gelstammlösung (siehe 4.1.1) wird durch Versetzen mit verschiedenen Ampholyten und Additiven eine Gellösung hergestellt:
9,0 ml Gelstammlösung,
24 mg β-Alanin,
500 µl Ampholyt pH 3,5-9,5 (1),
250 µl Ampholyt pH 5-7 (1),
250 µl Ampholyt pH 6-8 (1).
Die Gellösung wird durchmischt und 2 bis 3 Minuten im Ultraschallbad oder im Vakuum entgast.
Anmerkung: Die Gellösung darf erst unmittelbar vor dem Gießen (siehe 6.2) angesetzt werden.
4.1.3. Katalysatorlösungen
4.1.3.1. TEMED: N, N, N', N'-Tetramethylethylendiamin
4.1.3.2. 40 % (w/w) PER: Ammoniumpersulfat
800 mg PER werden mit Wasser versetzt und auf 2 ml aufgefüllt.
Anmerkung: Der PER-Lösung muss immer frisch angesetzt werden.
4.2. Kontaktflüssigkeit
Kerosin oder n-Decan.
4.3. Anodenlösung
5,77 g Phosphorsäure (85 % w/w) werden mit Wasser auf 100 ml aufgefüllt.
4.4. Kathodenlösung
2,00 g Natriumhydroxid werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
Probenvorbereitung
4.5. Reagenzien zur Proteinisolierung
4.5.1. Verdünnte Essigsäure (25,0 ml Eisessigsäure werden mit Wasser auf 100 aufgefüllt.)
4.5.2. Dichlormethan
4.5.3. Aceton
4.6. Protein-Lösungspuffer
5,75 g Glycerin (87 % w/w),
24,03 g Harnstoff und
250 mg Dithiothreitol
werden in destilliertem Wasser aufgelöst und auf 50 ml aufgefüllt.
Anmerkung: Die Lösung ist im Kühlschrank aufzubewahren und höchstens 1 Woche haltbar.
4.7. Reagenzien für die Plasminspaltung des Kaseins
4.7.1. Ammoniumcarbonat-Pufferlösung
0,2 mol/l NH4HCO3-Lösung (1,58 g/100 ml Wasser), die mit 0,05 mol/l Ethylendiamintetraessigsäure (EDTA 1,46 g/100 ml) versetzt wurde, wird mit 0,2 mol/l (NH4)2CO3-Lösung (1,92 g/100 ml Wasser), die mit 0,05 mol/l EDTA versetzt wurde, auf pH 8 titriert.
4.7.2. Bovines Plasmin (EC. 3.4.21.7), Aktivität mindestens 5 U/ml
4.7.3. ε-Aminocapronsäure zur Enzyminaktivierung
2,624 g ε-Aminocapronsäure (6-Amino-n-Hexansäure) werden in 100 ml 40 %igem Ethanol (v/v) gelöst.
4.8. Standardproben
4.8.1. Zertifizierte Standardproben eines Gemisches von mit Lab versetztem Schaf- und Ziegen-Magermilchpulver, das jeweils 0 % und 1 % Kuhmilch enthält, sind erhältlich beim Kommissionsinstitut für Referenzmaterialien und Messwesen, B-2440 Geel, Belgien.
4.8.2. Ansetzen der Interim-Standardproben von labversetzter Büffelmilch mit 0 % bzw. 1 % Kuhmilch
Zur Gewinnung von Magermilch wird eine rohe Büffel- bzw. Kuhsammelmilch bei einer Temperatur von 37 °C mit 2 500 g 20 Minuten lang zentrifugiert. Nach raschem Abkühlen des Zentrifugenbechers nebst Inhalt auf 6 bis 8 °C wird die Fettschwimmschicht restlos entfernt. Zum Ansetzen der 1 %igen Standardprobe werden 495 ml Büffelmagermilch in einem 1-l-Becherglas mit 5,00 ml Kuhmagermilch versetzt und durch Zusetzen verdünnter Milchsäure (10 % w/w) auf einen pH-Wert von 6,4 eingestellt. Die Temperatur wird auf 35 °C eingestellt; anschließend werden 100 µl Kälberlab (Labstärke 1: 10 000, ca. 3 000 U/ml) hinzugegeben. 1 Minute wird gerührt; danach wird das Becherglas mit einer Aluminiumfolie abgedeckt eine Stunde bei einer Temperatur von 35 °C stehen gelassen, damit sich der Quark/Topfen bilden kann. Anschließend wird die gesamte dickgelegte Milchprobe ohne vorheriges Homogenisieren oder Abscheiden der Molke gefriergetrocknet. Nach dem Gefriertrocknen wird das Produkt gleichmäßig und homogen vermahlen. Zum Ansetzen der 0 %igen Standardprobe wird das gleiche Verfahren mit reiner Büffelmagermilch durchgeführt. Die Standardproben sind bei –20 °C aufzubewahren.
Anmerkung: Es empfiehlt sich, die Reinheit der Büffelmilch durch isoelektrische Fokussierung der plasminbehandelten Kaseine vor dem Ansetzen der Standardproben zu prüfen.
Reagenzien zur Proteinfärbung
4.9. Fixierlösung
150 g Trichloressigsäure werden in Wasser gelöst und auf 1 000 ml aufgefüllt.
4.10. Entfärbelösung
500 ml Methanol und 200 ml Eisessig werden mit destilliertem Wasser auf 2 000 ml aufgefüllt.
Anmerkung: Die Entfärbelösung ist jeden Tag frisch anzusetzen; sie kann zweckmäßigerweise aus Vorratslösungen 50 % (v/v) Methanol und 20 % (v/v) Eisessig durch Mischen gleicher Volumina zubereitet werden.
4.11. Färbelösungen
4.11.1. Färbelösungen (Vorratslösung 1)
3,0 g Coomassie Brilliant Blau G-250 (C.I. 42655) werden in 1 000 ml 90 % (v/v) Methanol mit einem Magnetrührer (etwa 45 Minuten) verrührt und anschließend durch zwei mittelschnelle Faltenfilter gefiltert.
4.11.2. Färbelösung (Vorratslösung 2)
5,0 g Kupfersulfatpentahydrat werden in 1 000 ml 20 %iger Essigsäure (v/v) gelöst.
4.11.3. Färbelösung (gebrauchsfertig)
Unmittelbar vor dem Färben werden jeweils 125 ml der Vorratslösungen (4.11.1 und 4.11.2) miteinander vermischt.
Anmerkung: Die Färbelösung ist am Tag des Ansetzens aufzubrauchen.
5. AUSRÜSTUNG
5.1. Glasplatten (265 × 125 × 4 mm); Gummiroller mit Walzenbreite von 15 cm; Nivelliertisch
5.2. Gelträgerfolie (265 × 125 mm)
5.3. Gegenfolie (280 × 125 mm); beide Längsseiten werden mit je einem Klebestreifen (280 × 6 × 0,25 mm) abgeklebt (Abbildung 1).
5.4. Elektrofokussierkammer mit Kühlplatte (z. B. 265 × 125 mm) mit geeignetem Spannungsgeber (≥ 2,5 kV) oder Elektrophoreseautomat
5.5. Umlaufkryostat, thermostatregelbar auf 12 ± 0,5 °C
5.6. Zentrifuge, einstellbar auf 3 000 g
5.7. Elektrodenstreifen (≥265 mm lang)
5.8. Tropfflaschen aus Kunststoff, für Anoden- und Kathodenlösung
5.9. Probenapplikatoren 10 × 5 mm (Viskose oder Filterpapier mit geringer Proteinadsorption)
5.10. Scheren, Skalpelle und Pinzetten aus Edelstahl
5.11. Färbe- und Entfärbeschalen aus Edelstahl oder Glas (z. B. Instrumentenschalen 280 × 150 mm)
5.12. Regelbarer Homogenisierstab (Schaftdurchmesser 10 mm), Drehzahlbereich 8 000-20 000 min-1
5.13. Magnetrührer
5.14. Ultraschallbad
5.15. Folienschweißgerät
5.16. 25-μl-Mikropipetten
5.17. Vakuumzentrifuge oder Gefriertrockner
5.18. Thermostatregelbares Wasserbad, einstellbar auf 35 und 40 ± 1 °C, mit Schüttelvorrichtung
5.19. Densitometerausrüstung, Registrierung bei λ = 634 nm
6. VERFAHREN
6.1. Probenvorbereitung
6.1.1. Kaseinisolierung
Eine 5 g Trockenmasse entsprechende Menge Käse oder die Referenzstandardproben werden in ein 100-ml-Zentrifugenröhrchen gegeben; 60 ml destilliertes Wasser werden hinzugegeben; anschließend wird die Probe mit einem Homogenisierstab (8 000 bis 10 000 Umin-1 homogenisiert. Mit verdünnter Essigsäure (4.5.1) wird der pH-Wert 4,6 eingestellt; dann wird die Probe zentrifugiert (5 Minuten, 3 000 g). Fette und Molke werden dekantiert und der Rückstand in 40 ml destilliertem Wasser, das mit verdünnter Essigsäure (4.5.1) auf einen pH-Wert von 4-5 eingestellt wurde, homogenisiert (20 000 Umin-1), mit 20 ml Dichlormethan (4.5.2) versetzt, nochmals homogenisiert und dann zentrifugiert (5 Minuten bei 3 000 g). Die zwischen wässriger und organischer Phase schwimmende Kaseinschicht (siehe Abbildung 2) wird mit einem Spatel angehoben, damit beide Phasen dekantiert werden können. Das Kasein wird nochmals in 40 ml destilliertem Wasser (siehe oben) und 20 ml Dichlormethan homogenisiert und zentrifugiert. Dieser Vorgang ist zu wiederholen, bis beide Extraktionsphasen farblos sind (zwei- bis dreimal). Der Proteinrückstand wird mit 50 ml Aceton (4.5.3) homogenisiert und über ein mittelschnelles Faltenpapierfilter abfiltriert. Das Filterretentat wird zweimal mit je 25 ml Aceton gewaschen, an der Luft oder im Stickstoffstrom getrocknet und in einer Reibschale fein zerrieben.
Anmerkung: Trockene Kaseinisolate sind bei –20 °C aufzubewahren.
6.1.2. Plasminspaltung der β-Kaseine zur Anreicherung der γ-Kasein-Konzentration
25 mg Kaseinisolat (6.1.1) werden in 0,5 ml Ammoniumcarbonatpufferlösung (4.7.1) suspendiert und 20 Minuten z. B. im Ultraschallbad homogenisiert. Das Homogenisat wird auf 40 °C erwärmt, mit 10 µl Plasmin (4.7.2) versetzt, durchmischt und eine Stunde bei 40 °C unter ständigem Schütteln bebrütet. Zur Enzyminhibierung werden 20 µl ε-Aminocapronsäurelösung (4.7.3), danach 200 mg fester Harnstoff und schließlich 2 mg Dithiothreitol zugesetzt.
Anmerkung: Zur Verbesserung der Symmetrie der fokussierten Kaseinbanden empfiehlt es sich, die Lösung nach Zusatz von ε-Aminocapronsäure gefrierzutrocknen und den Probenrückstand in 0,5 ml Harnstoffpufferlösung (4.6) zu lösen.
6.2. Herstellen der harnstoffhaltigen Polyacrylamidgele
Auf eine Glasplatte (5.1) wird die Gelträgerfolie (5.2) mit Hilfe einiger Tropfen Wasser aufgewalzt; ausgetretenes Wasser ist mit einem Papiertuch aufzunehmen. In gleicher Weise wird die mit Abstandshaltern (0,25 mm) versehene Gegenfolie (5.3) auf eine weitere Glasplatte aufgewalzt. Diese wird dann horizontal auf einem Nivelliertisch ausgerichtet.
Die frisch angesetzte, entgaste Gellösung (4.1.2) wird mit je 10 µl der Katalysatorlösungen TEMED und PER (4.1.3.1) versetzt, kurz durchmischt und gleichmäßig auf die Mitte der Gegenfolie ausgegossen. Die Gelträgerplatte wird (mit der Folie nach unten) mit einer Kante auf die nivellierte Gegenfolienplatte aufgesetzt und so langsam abgesenkt, dass sich zwischen den Folien ein Gelfilm bilden und sich gleichmäßig und blasenfrei ausbreiten kann (Abbildung 3). Die Gelträgerfolienplatte wird mit Hilfe eines dünnen Spatels vollständig aufgesetzt; drei weitere Glasplatten werden als Andruckgewichte aufgelegt. Nach vollständiger Polymerisierung (ca. 60 Minuten) wird das auf der Gelträgerfolie anpolymerisierte Gel mitsamt Gegenfolie durch Verkanten der Glasplatten herausgelöst. Die Rückseite der Trägerfolie wird sorgfältig von Gelresten und Harnstoff gesäubert. Das „Gel-Sandwich“ wird nun in einen Folienschlauch eingeschweißt und im Kühlschrank (maximal 6 Wochen) aufbewahrt.
Anmerkung: Die Gegenfolie mit den Abstandshaltern kann wiederverwendet werden. Das Polyacrylamidgel kann auf kleinere Formate zugeschnitten werden; dies ist bei kleinem Probenumfang oder bei Verwendung eines Elektrophoreseautomaten zu empfehlen (2 Gele im Format 4,5 × 5 cm).
6.3. Isoelektrische Fokussierung
Der Kühlthermostat wird auf 12 °C eingestellt. Nach Abwischen der Gelträgerfolienrückseite mit Kerosin (4.2) werden einige Tropfen Kerosin auf die Mitte des Kühlblocks aufgebracht. Das „Gel-Sandwich“ wird mit der Trägerseite nach unten blasenfrei aufgewalzt. Ausgetretenes Kerosin wird abgewischt und die Gegenfolie abgezogen. Die Elektrodenstreifen werden mit den entsprechenden Elektrodenlösungen (4.3, 4.4) getränkt, auf die Gellänge zugeschnitten und auf die dafür vorgesehenen Stellen gelegt (Elektrodenabstand 9,5 cm).
Die Fokussierung ist unter folgenden Bedingungen durchzuführen:
6.3.1. Gelformat 265 × 125 × 0,25 mm
|
Schritt
|
Dauer
(min)
|
Spannung
(V)
|
Stromstärke
(mA)
|
Leistung
(W)
|
Voltstunden
(Vh)
|
|
|
30
|
max. 2 500
|
max. 15
|
konstant 4
|
ca. 300
|
|
2.
|
Probenfokussierung (2)
|
|
60
|
max. 2 500
|
max. 15
|
konstant 4
|
ca. 1 000
|
|
|
60
|
max. 2 500
|
max. 5
|
max. 20
|
ca. 3 000
|
|
40
|
max. 2 500
|
max. 6
|
max. 20
|
ca. 3 000
|
|
30
|
max. 2 500
|
max. 7
|
max. 25
|
ca. 3 000
|
Anmerkung: Bei Änderung der Stärke oder Breite der Gele sind Strom und Leistung entsprechend anzupassen (z. B. Verdopplung von Stromstärke und Leistung bei Verwendung eines 0,5 mm starken Gels im Format 265 × 125 × 0,5 mm).
6.3.2. Beispiel eines Spannungsprogramms für einen Elektrophoreseautomaten (2 Gele je 5,0 × 4,5 cm); direkte Aufbringung der Elektroden auf das Gel (ohne Elektrodenstreifen)
|
Schritt
|
Spannung
|
Stromstärke
|
Leistung
|
Temp.
|
Voltstunden
|
|
|
1 000 V
|
10,0 mA
|
3,5 W
|
8 °C
|
85 Vh
|
|
|
250 V
|
5,0 mA
|
2,5 W
|
8 °C
|
30 Vh
|
|
|
1 200 V
|
10,0 mA
|
3,5 W
|
8 °C
|
80 Vh
|
|
|
1 500 V
|
5,0 mA
|
7,0 W
|
8 °C
|
570 Vh
|
In Schritt 2 wird der Probenapplikator bei 0 Vh aufgesetzt.
In Schritt 2 wird der Probenapplikator bei 30 Vh abgenommen.
6.4. Proteinanfärbung
6.4.1. Fixieren der Proteine
Unmittelbar nach Abschalten des Stroms werden die Elektrodenstreifen entfernt. Das Gel wird sofort in eine mit 200 ml Fixierlösung (4.9) gefüllte Färbe- und Entfärbeschale gegeben und 15 Minuten geschüttelt.
6.4.2. Waschen und Färben der Gelplatte
Die Fixierlösung wird restlos abgegossen und die Gelplatte zweimal je 30 Sekunden mit 100 ml Entfärbelösung (4.10) gespült. Die Entfärbelösung wird abgegossen und die Schale mit 250 ml Färbelösung (4.11.3) gefüllt; die Färbelösung wird 45 Minuten leicht geschwenkt, um die Lösung einwirken zu lassen.
6.4.3. Entfärben der Gelplatte
Die Färbelösung wird abgegossen, die Gelplatte zweimal mit je 100 ml Entfärbelösung (4.10) gespült und anschließend mindestens 2 x 15 Minuten in 200 ml Entfärbelösung geschwenkt, bis der Hintergrund klar und farblos ist. Die Gelplatte wird dann mit destilliertem Wasser (zweimal 2 Minuten) gespült und an der Luft (2 bis 3 Stunden) oder mit dem Fön (ca. 10 bis 15 Minuten) getrocknet.
Anmerkung 1: Fixierung, Färben und Entfärben erfolgen bei einer Temperatur von 20 °C. Bei höheren Temperaturen dürfen diese Schritte nicht ausgeführt werden.
Anmerkung 2: Wird einer empfindlicheren Silberfärbelösung (z. B. Silberfärbelösungs-Kit, Protein Pharmacia Biotech, Code Nr. 17-1150-01) der Vorzug gegeben, sind die plasminbehandelten Kaseinproben auf 5 mg/ml zu verdünnen.
7. BEWERTUNG
Die Bewertung erfolgt durch Vergleich der Proteinbandenmuster der unbekannten Probe mit den Referenzstandardproben auf demselben Gel. Der Nachweis von Kuhmilch in Käsen aus Schaf, Ziegen- oder Büffelmilch und in Gemischen aus Schaf-, Ziegen- oder Büffelmilch erfolgt aufgrund der γ3- und der γ2-Kaseine, bei denen die isoelektischen Punkte im pH-Wert-Bereich 6,5 bis 7,5 liegen (Abbildungen 4a und b und Abbildung 5). Die Nachweisgrenze des Verfahrens liegt unter 0,5 %.
7.1. Visuelle Bestimmung
Zur visuellen Bestimmung des Kuhmilchanteils empfiehlt es sich, die Konzentration der Proben und der Standardproben auf die Stärke der Schaf-, Ziegen- und/oder Büffel-γ2 und -γ3-Kaseine einzustellen (siehe „γ2 E,G,B“ und „γ3 E,G,B“ in den Abbildungen 4a und 4b und Abbildung 5). Nur dann kann der Kuhmilchgehalt (kleiner, gleich oder größer als 1 %) in der zu beurteilenden Probe durch direkten Vergleich der Stärken der Rinder-γ2- und -γ3-Kaseinzonen (siehe „γ3 C“ und „γ2 C“ in den Abbildungen 4a, 4b und 5) mit denen der 0 %- und 1 %-Standardproben (Schaf, Ziege) oder laboreigenen Interims-Standardproben (Büffelmilch) bestimmt werden.
7.2. Densitometrische Bestimmung
Nach Möglichkeit ist mit Hilfe eines Densitometers (5.19) das Peak-Flächen-Verhältnis der γ2- und γ3-Kaseine (siehe Abbildung 5) zwischen Rind und Schaf, Ziege und/oder Büffel zu ermitteln. Dieser Wert ist mit dem γ2- und γ3-Kasein-Peak-Flächen-Verhältnis der 1 %igen Standardprobe (Schaf, Ziege) oder der im selben Gel laufenden laboreigenen Interims-Standardprobe (Büffel) zu vergleichen.
Anmerkung: Reagiert die 1 % Kuhmilch enthaltende Standardprobe eindeutig positiv sowohl auf γ2- als auch auf γ3-Kaseine, die 0 %ige Standardprobe dagegen nicht, liefert die Methode zuverlässige Ergebnisse. Ansonsten ist das Verfahren unter genauer Beachtung der Verfahrensvorschrift zu optimieren.
Eine Stichprobe gilt als positiv, wenn die Kuhmilchkaseine γ2 und γ3 oder die entsprechenden Peak-Flächen-Verhältnisse zumindest den Zahlen für die 1 %ige Standardprobe entsprechen.
8. LITERATUR
|
1.
|
Addeo F., Moio L., Chianese L., Stingo C., Resmini P., Berner I, Krause I., Di Luccia A., Bocca A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft 45, 708-711 (1990).
|
|
2.
|
Addeo F., Nicolai M.A., Chianese L., Moio L., Spagna Musso S., Bocca A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft 50, 83-85 (1995).
|
|
3.
|
Krause I., Berner I, Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte — and carrier ampholyte/immobilized pH gradient — isoelectric focusing of γ-caseins using plasmin as signal amplifier; in: Electrophoresis-Forum 89 (B. J. Radola, Hrsg.) S. 389-393, Bode-Verlag, München (1989).
|
|
4.
|
Krause Ι., Belitz H.-D., Kaiser K.-P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. -käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen. Z. Lebensm. Unters. Forsch. 174, 195-199 (1982).
|
|
5.
|
Radola B.J.: Ultrathin-layer isoelectric focusing in 50-100 µm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis 1, 43-56 (1980).
|
Abbildung 1
Schematische Darstellung der Gegenfolie
Abbildung 2
Kaseinschwimmschicht zwischen wässriger und organischer Phase nach dem Zentrifugieren

Abbildung 3
Klapptechnik zur Herstellung ultradünner Polyacrylamidgele
a = Abstandshalter (0,25 mm); b = Gegenplatte (5.3); c, e = Glasplatten (5.1); d = Gellösung (4.1.2); f = Gelträgerplatte (5.2)
Abbildung 4a
Isoelektrische Fokussierung plasminbehandelter Kaseine von Schaf- und Ziegenmilchkäse mit unterschiedlichem Kuhmilchgehalt

% CM = Kuhmilchgehalt C = Kuh, E = Schaf, G = Ziege.
Abgebildet ist die obere Hälfte des IEF-Gels.
Abbildung 4b
Isoelektrische Fokussierung plasminbehandelter Kaseine von Käse, der aus Gemischen von Schaf-, Ziegen- und Büffelmilch mit unterschiedlichem Kuhmilchgehalt hergestellt wurde

% CM = Prozentanteil Kuhmilch; 1 + = Probe mit 1 % Kuhmilch, versetzt mit reinem Kuhmilchkasein in der Mitte des Durchlaufs C = Kuh, E = Schaf, G = Ziege, B = Büffel.
Abgebildet ist der gesamte Trennungsabstand des IEF-Gels.
Abbildung 5
Densitogrammreihe der Standardproben (STD) und Käseproben aus Gemischen von Schaf- und Ziegenmilch nach isoelektrischer Fokussierung

a, b = Standardproben mit 0 und 1 % Kuhmilch; c-g = Käseproben mit 0, 1, 2, 3 und 7 % Kuhmilch; C = Kuh, E = Schaf, G = Ziege
Die obere Hälfte des IEF-Gels wurde bei λ = 634 nm analysiert.
(1) Die Produkte Ampholine® pH 3,5-9,5 (Pharmacia) und Resolyte® pH 5-7 und pH 6-8 (BDH, Merck) haben sich als besonders geeignet für die erforderliche Trennung der γ-Kaseine erwiesen.
(2) Probenauftrag: Nach der Vorfokussierung (Schritt 1) werden 18 µl der Probe und der Standardlösungen auf die Probenapplikatoren (10 × 5 mm) pipettiert und in Abständen von 1 mm voneinander sowie von 5 mm in Längsrichtung von der Anode auf das Gel gebracht und leicht angedrückt. Die Fokussierung erfolgt unter den beschriebenen Bedingungen; nach der 60-minütigen Probenfokussierung sind die Probenapplikatoren vorsichtig abzunehmen.
ANHANG X
(Artikel 7)
REFERENZMETHODE ZUM NACHWEIS COLIFORMER KEIME IN BUTTER, MAGERMILCHPULVER, KASEIN UND KASEINATEN
1. VORBEREITUNG DER PROBEN
ISO-Norm 8261
2. VERFAHREN
ISO-Norm 4831
Proben entsprechend 1 g Butter oder 0,1 g Magermilchpulver oder Kasein/Kaseinate werden in das Kulturmedium geimpft.
Je Probe sind drei Röhrchen zu impfen.
3. ERGEBNISSE
Wenn für die 3 Röhrchen 3 negative Befunde zu verzeichnen ist, lautet das Ergebnis „konform“.
Führen die 3 Röhrchen zu 2 oder 3 positiven Befunden, lautet das Ergebnis „nicht konform“.
Werden für die 3 Röhrchen 2 negative Befunde festgestellt, wird die Analyse zweimal (mit zwei Röhrchen) wiederholt.
|
—
|
Sind auch diese beiden Ergebnisse negativ, lautet das Ergebnis „konform“.
|
|
—
|
Ist mindestens ein Ergebnis positiv, lautet das Gesamtergebnis „nicht konform“.
|
ANHANG XI
(Artikel 8)
LAKTOSENACHWEIS IN MISCHFUTTER
1. GEGENSTAND UND ANWENDUNGSBEREICH
Bestimmung des Laktosegehalts in Mischfutter
2. HINWEIS
Unter dem Laktosegehalt wird der nach diesem Verfahren bestimmte Massenanteil in Prozent verstanden.
3. BEGRIFFSBESTIMMUNG
Der Gehalt an wasserfreier Laktose wird in g/100 g ausgedrückt.
4. KURZBESCHREIBUNG
Das Mischfutter wird in Wasser rekonstituiert. Eine „Biggs“-Lösung wird zu einem verdünnten abgewogenen Aliquot hinzugegeben, um das Fett und die Proteinfraktionen des Mischfutters auszufällen. Die Probe wird gefiltert (oder zentrifugiert), und das Filtrat (oder der Überstand) wird auf eine HPLC-Säule mit Kationenaustauscher (Blei) mit Wasser in HPLC-Qualität als mobiler Phase injiziert. Die eluierte Laktose wird mit einem Differenzrefraktometer bestimmt (1).
5. REAGENZIEN
5.1. Kurzbeschreibung
Wenn nicht ausdrücklich anderweitig angegeben, sind ausschließlich Reagenzien von anerkannter Analysequalität und entgastes Wasser in HPLC-Qualität zu verwenden.
5.2. Laktose
D-Laktose-Monohydrat ((C12H22)O11H2O) kann die erhöhte Feuchtigkeit aufnehmen. Vor Gebrauch ist der tatsächliche Wasseranteil nach der Karl-Fisher-Methode zu messen bzw. die erhöhte Feuchtigkeit zu verdampfen, indem die Laktose 8 Stunden in einen Ofen mit einer Temperatur von 105 °C gegeben wird. (Der Kristallwasseranteil der Laktose bleibt bei dieser Behandlung erhalten.)
5.3. Konzentrierte Biggs/Szijarto-Lösung (2)
9,10 g Zinkacetatdihydrat (Zn(CH3COO)2.2H2O) und 5,46 g Phosphorwolframsäure-Monohydrat (H3[P(W3O10)4xH2O]) werden in etwa 70 ml Wasser in HPLC-Qualität (6.8) in einem 100-ml-Messkolben aufgelöst.
5,81 ml Eisessigsäure (CH3COOH) werden hinzugegeben. Anschließend wird die Lösung bis zur 100-ml-Marke mit Wasser in HPLC-Qualität (6.8) aufgefüllt und gemischt. Die Lösung kann bei Raumtemperatur 1 Jahr gelagert werden.
5.4. Verdünnte Biggs/Szijarto-Lösung
25 ml konzentrierte Biggs/Szijarto-Lösung (5.3) werden mit Wasser in einem Messkolben bis auf 500 ml aufgefüllt. Die Lösung kann bei Raumtemperatur einen Monat gelagert werden.
5.5. Vorbereitung des Wassers in HPLC-Qualität
Das ultrareine Wasser (6.8) wird einer Vakuumfiltration (6.9) unterzogen. Um die Pumpleistung zu verbessern und eine stabile Basislinie zu erhalten, ist die mobile Phase täglich durch eines der verfügbaren Verfahren (z. B. Heliumeinleitung, Ultraschallbehandlung, Vakuum- oder In-line-Entgasung) zu entgasen.
Anmerkung: Um die Lebensdauer der Säule zu verlängern, muss sichergestellt werden, dass der Kohlendioxidanteil des Elutionsmittels möglichst gering ist und dass eine erneute Aufnahme ausgeschlossen werden kann.
6. APPARATUR
Normale Laborausstattung und insbesondere:
6.1. HPLC-Ionenaustauscher-Harzsäule
Säulenpackung: 8 % quervernetztes Polystyrol-divinylbenzol-Copolymer funktionalisiert mit Kationenaustauschergruppen in der Bleiform;
Säulenabmessungen: Länge 300 mm, Innendurchmesser ca. 8 mm.
Andere Durchmesser können verwendet werden, wenn der Durchfluss entsprechend angepasst wird.
6.2. Vorsäule
Die Vorsäule besteht aus einem getrennten Kationenaustauscher (H+) kombiniert mit einem Anionen-Austauscher (CO3-), jeweils in Säulen von ca. 30 mm × 4,6 mm (H × ID) gepackt (z. B. Mikrovorsäulen in einem Mikrovorsäulenhalter); die Säulen sind in Reihen hintereinander oder in Form eines gemischten Betts mit AG 50W-X4, -400 mesh (H+) und AG3-X4A, 200-400 mesh (OH-) im Verhältnis 35:65 (m/m) angeordnet und werden von Hand in eine ca. 20 × 9 mm (L × ID) große Säule gepackt.
6.3. Säulenofen
Mit dem Ofen muss eine gleich bleibende Temperatur von 85 °C ±1 °C hergestellt werden können.
6.4. HPLC-Pumpe
Die Pumpe muss einen gleich bleibenden Durchfluss von 0,2-1,0 m/min (bei Schwankungen < 0,5 %) erzielen können.
6.5. HPLC-Injektionssystem
Der Autosampler muss ein Volumen von 25 µl injizieren können; die Wiederholbarkeit muss < 0,5 % sein.
Alternativ kann ein manuelles System eingesetzt werden, das die auch an den Autosampler gestellten Anforderungen erfüllt.
6.6. HPLC-Detektor
Benötigt wird ein hoch empfindlicher Refraktionsindex-Detektor mit einem Rauschpegel von < 5,10-9 RI.
6.7. Integrator
Für die Datenerfassung sowie zum Erzeugen von Peak-Flächen und Peak-Höhen, die in Laktose-Konzentrationen umgerechnet werden können, wird eine Software oder ein eigener Integrator verwendet.
6.8. Wasserreinigungssystem
Das System muss ultrareines Wasser (Typ 1) mit einem spezifischen Widerstand > 14 MΩ.cm herstellen können.
6.9. Lösungsmittelfiltersystem
Das System muss das Wasser über einen Membranfilter mit einer Porengröße von 0,45 µm filtern können.
Anmerkung: Viele Wasserreinigungssysteme (6.8) besitzen einen integrierten 0,45- oder 0,2-µm-Filter. Eine weitere Filtration kann unterbleiben, wenn das Wasser sofort verwendet wird.
6.10. Analysewaage
Waage mit einer Empfindlichkeit von 0,1 mg
6.11. Wasserbad
Im Wasserbad muss eine Temperatur von 40 °C (±0,5 °C) aufrechterhalten werden können.
6.12. Zentrifuge
Die Zentrifuge muss Eppendorf-Röhrchen oder gleichwertige bzw. größere Röhrchentypen mit mindestens 3 000 g beschleunigen können.
6.13. Messkolben 50 ml
Füllmenge 50 ml, Klasse A
Anmerkung: Bei Berücksichtigung des jeweiligen Volumenfaktors können Kolben mit sonstigen Füllmengen verwendet werden.
6.14. Messkolben 100 ml
Fassungsvermögen 100 ml, Klasse A
6.15. Messpipette
Messpipette, 10 ml
Anmerkung: Alternativ kann eine manuelle Pipettierhilfe mit einem Fassungsvermögen von 5 ml verwendet werden; in diesem Fall müssen zweimal 5 ml des Reagens verwendet werden (5.3).
7. PROBENAHME
Wichtig ist, dass das Labor eine Probe erhält, die gemäß ISO 707|IDF 50 (3) entnommen wurde, die tatsächlich repräsentativ ist und die bei Transport oder Lagerung nicht beschädigt wurde.
8. VORBEREITUNG DER LAKTOSE-STANDARDLÖSUNG
8.1. Standardprobe 1
Eine auf 0,1 mg genau abgewogene Menge von ca. 50 mg Laktose-Monohydrat (5.2) wird in einem 100-ml-Messkolben (6.14) aufgelöst; anschließend wird bis zur Füllmarke mit Wasser aufgefüllt.
8.2. Standardprobe 2
Eine auf 0,1 mg genau abgewogene Menge von ca. 100 mg Laktose-Monohydrat (5.2) wird in einem 100-ml-Messkolben (6.14) aufgelöst; anschließend wird mit Wasser aufgefüllt.
Anmerkung: Die Standardlösungen können bei einer Temperatur von ca. 5 °C höchstens 1 Woche gelagert werden.
9. VORBEREITUNG DER PROBE
9.1. Rekonstituierung der Probe
Ca. 5 g des Pulvers werden in einen 50-ml-Kolben (6.13) eingewogen; das Gewicht wird auf 1 mg genau bestimmt (W1 (11)). Nach Zugabe von 50 ml Wasser wird die Zunahme des Gewichts (W2 (11)) auf 0,01 g genau ermittelt. Der verschlossene Kolben wird für 30 Minuten in das Wasserbad (6.11) gebracht und während dieser Zeit einige Male gestürzt. Anschließend muss der Kolben auf Raumtemperatur abkühlen.
9.2. Probenbehandlung
Ca. 1 g dieser Lösung wird entnommen und in einen 50-ml-Messkolben (6.13) gegeben; anschließend wird das Gewicht (W3 (11)) auf 1 mg genau bestimmt; danach werden zunächst 20 ml Wasser und dann 10 ml verdünnte Biggs/Szijarto-Lösung (5.4) hinzugefügt. Die Lösung wird mit Wasser bis zur Marke aufgefüllt. Der Kolben wird in den ersten 30 Minuten fünfmal vorsichtig gestürzt.
Nach 1 Stunde wird eine Aliquote entnommen und 10 Minuten mit 3 000 g zentrifugiert (6.12). (Bei entsprechend kürzerer Zeit sind auch höhere Beschleunigungen möglich.) Eine Aliquote des Überstands wird für die HPLC-Analyse verwendet.
10. HPLC-ANALYSE
10.1. Vorläufige Vorbereitung für die HPLC
10.1.1. Aufbau der Hauptsäule und der Vorsäule
Die Vorsäule (6.2) wird außerhalb des Säulenofens (6.3) angeordnet und die Hauptsäule (6.1) in den Ofen gebracht.
Anmerkung: Wenn der Ofen nicht mit Röhren zur Vorheizung des Elutionsmittels ausgestattet ist, muss das Elutionsmittel durch ein etwa 15 cm langes Edelstahlrohr in den Ofen geleitet werden, bevor es in die Säule gelangt. (Das Elutionsmittel muss vor dem Eintritt in die Säule erwärmt worden sein; ansonsten werden die Peaks unangemessen breit.)
10.1.2. Detektor und Ausgangsdurchfluss
Um eine stabile Basislinie zu erhalten, wird der Detektor (6.6) mindestens 24 Stunden vor Beginn der Analyse eingeschaltet. Die Innentemperatur des Detektors wird auf 35 °C geregelt. Mindestens 20 Minuten wird ein Durchfluss von 0,2 ml/min (6.4) eingestellt; dabei wird der Säulenofen (6.3) auf Raumtemperatur geregelt.
10.1.3. Säulenofen und endgültiger Durchfluss
Der Säulenofen (6.3) wird auf 85 °C eingestellt. Wenn diese Temperatur erreicht ist, wird nach 30 Minuten allmählich der Durchfluss von 0,2 ml/min auf 0,6 ml/min erhöht (6.4). Es wird gewartet, bis das Gerät sich bei diesem Durchfluss und bei einer Temperatur von 85 °C äquilibriert hat (2 Stunden bzw. bis zum Erreichen einer stabilen Basislinie).
10.1.4. Integration
Die Parameter für Erfassung und Integration der Daten (6.7) (z. B. Datenrate, Empfindlichkeit, Zeitkonstante, Peak-Breite und Schwellenwert) sind sorgfältig auszuwählen.
Die Retentionszeit beträgt bei Laktose etwa 11 Minuten.
Anmerkung: Viele Software-Programme zur Datenerfassung (6.7) ermöglichen die bequeme Messung der theoretischen Plattenzahl. Die theoretische Plattenzahl von Standardlösung 1 (8.1) ist regelmäßig zu messen; wenn die Plattenzahl den Ausgangswert einer neuen Säule um mindestens 25 % unterschreitet, ist die Säule (6.1) zu ersetzen.
10.1.5. Vorsäulentest
Regelmäßig (d. h. in jeder Analysereihe mindestens einmal) muss geprüft werden, ob die Vorsäule (6.2) Salze aus der Probe abtrennen kann; dazu werden 25 µl einer 0,05 %igen Natriumchloridlösung injiziert. Wenn Peaks auftreten, sollte die Vorsäule ersetzt werden.
10.2. Messen der Standardproben
Jeweils zu Beginn einer Analysereihe werden 25 µl (6.5) von Standardprobe 1 (8.1) und anschließend von Standardprobe 2 (8.2) injiziert. Diese Injektionen werden jeweils nach 10 bis 20 Proben sowie am Ende einer Analysereihe wiederholt.
10.3. Messen der Proben
25 µl des Überstands (9.2) der Probe werden injiziert.
11. BERECHNUNG UND DARSTELLUNG DER ERGEBNISSE
11.1. Kalibrierung
Normalerweise werden die Ergebnisse aufgrund von Peak-Höhen berechnet; bei zu starkem Rauschpegel können auch die Peak-Flächen verwendet werden. (Bei der quantitativen Bestimmung anhand der Peak-Höhe bestehen geringere Beeinträchtigungen durch Peaks von in niedriger Konzentration vorliegenden Bestandteilen, die nur teilweise und nicht hinreichend von Laktose-Peak abgegrenzt werden können.)
Die Software (6.7) muss eine lineare Kalibrierkurve berechnen, die durch das Ausgangsniveau führt. Die Kurve ist auf eine mögliche Linearitätsabweichung zu prüfen; (scheinbare Linearitätsabweichungen sind meist auf einen Fehler bei der Vorbereitung der Standardprobe 1 (8.1) oder 2 (8.2), auf eine schlechte Integration oder — seltener — auf einen defekten Injektor zurückzuführen.)
Als Ausgangsdaten werden die berechneten Laktosekonzentrationen in mg/ml der Standardprobe 1 (8.1) und 2 (8.2) als wasserfreie Laktose angenommen.
Die Steigung (RF) der Kalibrierungslinie hängt vom Verhältnis zwischen der Fläche und der Konzentration in mg/ml ab.
11.2. Proben
Das Analyseergebnis wird in g/100 g ausgedrückt und mit der Software (6.7) bzw. mit der folgenden Formel berechnet:

Dabei bedeuten:
|
C
|
:
|
Laktosekonzentration in g/100 g Pulver
|
|
H
|
:
|
Höhe der Laktose-Peaks der Proben
|
|
RF
|
:
|
Kalibrierfaktor (oder Steigung) der Kalibrierkurve in mV/mg/ml
|
|
W1
|
:
|
Gewicht der Pulverprobe in g (9.1)
|
|
W2
|
:
|
Gewicht des zur Pulverprobe hinzugefügten Wassers in g (9.1)
|
|
W3
|
:
|
Gewicht der rekonstituierten Lösung des Probenpulvers in g (9.2)
|
|
50
|
:
|
Volumen des verwendeten Messkolbens (9.2)
|
|
0,1
|
:
|
Umrechnung des Ergebnisses in g/100 g
|
12. PRÄZISION
Die in diesem Leistungstest ermittelten Werte sind möglicherweise nur auf die hier genannten Konzentrationsbereiche und Matrizen anwendbar. Die Werte für die Wiederholbarkeit und die Vergleichbarkeit stützen sich auf die Ergebnisse eines gemäß ISO 5725 (4) durchgeführten Leistungstests.
12.1. Wiederholbarkeit
Der absolute Unterschied zwischen zwei einzelnen Testergebnissen, die nach derselben Methode bei identischem Testmaterial in verschiedenen Laboratorien von verschiedenen Analytikern und mit unterschiedlicher Ausrüstung ermittelt wurden, liegt in höchstens 5 % aller Fälle über xxx (durch gemeinsame Versuche zu überprüfen) (5).
12.2. Vergleichbarkeit
Der absolute Unterschied zwischen zwei einzelnen Testergebnissen, die nach derselben Methode bei identischem Testmaterial in verschiedenen Laboratorien von verschiedenen Analytikern und mit unterschiedlicher Ausrüstung ermittelt wurden, liegt in höchstens 5 % aller Fälle über 0,5 g/100 g (durch gemeinsame Versuche zu überprüfen).
13. LITERATUR
(1) J. Koops en C. Olieman, Netherlands Milk and Dairy Journal, 39 (1985) 89-106.
(2) D.A. Biggs en L. Szijarto, Journal of Dairy Science, 46 (1963) 1196.
(3) ISO 707 (IDF 50), „Milchprodukte — Leitfaden. zur Probenahme“.
(4) ISO 5725-1, Genauigkeit (Richtigkeit und Präzision) von Messverfahren und Messergebnissen — Teil 1: Allgemeine Grundlagen und Begriffe.
(5) ISO 5725-2, Genauigkeit (Richtigkeit und Präzision) von Messverfahren und Messergebnissen. Teil 2: Ein grundlegendes Verfahren für die Ermittlung der Wiederhol- und Vergleichpräzision von festgelegten Messverfahren.
ANHANG XII
(Artikel 9)
BESTIMMUNG VON LABMOLKE IN MAGERMILCHPULVER ZUR ÖFFENTLICHEN LAGERHALTUNG UNTER NACHWEIS VON KASEINMAKROPEPTIDEN DURCH HOCHLEISTUNGS-FLÜSSIGKEITSCHROMATOGRAFIE (HPLC)
1. GEGENSTAND UND ANWENDUNGSBEREICH
Durch die Bestimmung der Kaseinmakropeptide ermöglicht diese Methode den Nachweis von Labmolke in zur öffentlichen Lagerung bestimmtem Magermilchpulver.
2. LITERATUR
Internationale Norm ISO 707 — Milch und Milcherzeugnisse — Probenahmetechniken, gemäß den Angaben in Anhang I Ziffer 2 Buchstabe c letzter Unterabsatz.
3. BEGRIFFSBESTIMMUNG
Unter dem Gehalt an Labmolkepulver wird der unter Nachweis von Kaseinmakropeptiden nach diesem Verfahren bestimmte Massenanteil in Prozent verstanden.
4. KURZBESCHREIBUNG
|
—
|
Rekonstitution des Magermilchpulvers in warmem Wasser, Entfernen des Fettanteils und der Proteine mit Trichloressigsäure und anschließende Zentrifugierung oder Filtrierung.
|
|
—
|
Bestimmung der im Überstand vorhandenen Kaseinmakropeptidmenge (CMP) durch Hochleistungsflüssigkeitschromatografie (HPLC).
|
|
—
|
Beurteilung des Ergebnisses durch den Vergleich mit Standardproben aus Magermilchpulver, ohne oder mit Zusatz eines bekannten Anteils an Labmolkepulver.
|
5. REAGENZIEN
Alle Reagenzien müssen analysenrein sein. Das verwendete Wasser muss destilliert sein oder einen mindestens gleichwertigen Reinheitsgrad aufweisen.
5.1. Trichloressigsäure
240 g Trichloressigsäure (CCl3COOH) werden in Wasser aufgelöst; anschließend wird die Lösung auf 1 000 ml aufgefüllt. Die Lösung sollte klar und farblos sein.
5.2. Elutionsmittel, pH 6,0
1,74 g Di-Kaliumphosphat (K2HPO4), 12,37 g Mono-Kaliumphosphat (KH2PO4) und 21,41 g Natriumsulfat (Na2SO4) werden in etwa 700 ml Wasser gelöst. Wenn erforderlich, ist der pH-Wert mit Phosphorsäure- oder Kaliumhydroxidlösung auf 6,0 zu korrigieren.
Die Lösung wird mit Wasser auf 1 000 ml aufgefüllt und gut durchmischt.
Anmerkung: Die Zusammensetzung des Elutionsmittels kann so geändert werden, dass die im Zertifikat der Standardlösungen vorgegebenen Anforderungen oder die Herstelleranweisungen bzw. die Anforderungen an das Packmaterial der Säule erfüllt werden.
Vor der Verwendung wird die Elutionslösung durch einen Membranfilter mit einer Porengröße von 0,45 µm filtriert.
5.3. Waschlösung
Ein Teil Acetonitril (CH3CN) wird mit 9 Teilen Wasser gemischt, und die Lösung vor Gebrauch durch einen Membranfilter mit 0,45 µm Porengröße filtriert.
Anmerkung: Es kann auch eine beliebige sonstige Waschlösung mit bakterizider Wirkung verwendet werden, die die Trennleistung der Säulen nicht beeinträchtigt.
5.4. Standardproben
5.4.1. Magermilchpulver gemäß den Anforderungen dieser Verordnung d. h. [0].
5.4.2. Das gleiche Magermilchpulver, verfälscht durch Zugabe von 5 % (m/m) Labmolkepulver von durchschnittlicher Zusammensetzung, d. h. [5].
6. APPARATUR
6.1. Analysewaage
6.2. (Fakultativ:) Zentrifuge, mit der eine Zentrifugalkraft von 2 200 g erreicht werden kann, ausgerüstet mit Zentrifugenröhrchen mit Verschlussstopfen oder -kappe und einem Fassungsvermögen von 50 ml
6.3. Mechanische Schüttelvorrichtung
6.4. Magnetrührer
6.5. Glastrichter, ca. 7 cm Durchmesser
6.6. Filterpapier, mittlere Filtriergeschwindigkeit, ca. 12,5 cm Durchmesser
6.7. Filtriervorrichtung aus Glas mit Membranfilter, 0,45 µm Porendurchmesser
6.8. Messpipetten, Nennvolumen 10 ml (ISO 648, Klasse A oder ISO/R 835), bzw. ein Pipettiersystem, mit dem in 2 Minuten ein Volumen von 10,0 ml übertragen werden kann
6.9. Aufgabesystem zur Abgabe von 20,0 ml Wasser mit einer Temperatur von ca. 50 °C
6.10. Thermostatisierbares Wasserbad, eingestellt auf 25 ±0,5 °C
HPLC-Ausrüstung, bestehend aus:
6.11.1. einer Pumpe;
6.11.2. einem manuellem Injektor oder einem Autosampler mit 15 bis 30 µl Fassungsvermögen;
6.11.3. 2 TSK 2 000-SW-Säulen in Reihe (Länge 30 cm, Innendurchmesser 0,75 cm) oder gleichwertige Säulen (z. B. einmal TSK 2 000-SWXL, einmal Agilent Technologies Zorbax GF 250) und einer Vorsäule (3 cm × 0,3 cm) gepackt mit I 125 oder Material mit gleichwertiger Effizienz;
6.11.4. einem thermostatisierbarem Säulenofen, einstellbar auf 35 ±1 °C;
6.11.5. einem UV-Detektor mit variabler Wellenlänge für Messungen bei 205 nm mit einer Empfindlichkeit von 0,008 A und
6.11.6. einem Integrator zur Messung der Peak-Höhe.
Anmerkung: Die Säulen können auch bei Raumtemperatur eingesetzt werden. Allerdings ist die Trennleistung dann etwas geringer. In diesem Fall müssen die Temperaturschwankungen während einer Analysenreihe unter ±5 °C betragen.
7. PROBENAHME
7.1. Die Probenahme erfolgt nach dem von der internationalen Norm ISO 707 vorgesehenen Verfahren. Die Mitgliedstaaten können jedoch ein anderes Probenahmeverfahren anwenden, wenn dieses die Anforderungen der genannten Norm erfüllt.
7.2. Die Probe ist so aufzubewahren, dass sie unversehrt bleibt und ihre Zusammensetzung sich nicht ändert.
8. VERFAHREN
8.1. Vorbereitung der Probe
Das Milchpulver wird in einen ungefähr das doppelte Volumen fassenden Behälter mit luftdichtem Verschluss gegeben und der Behälter sofort verschlossen. Das Milchpulver wird durch mehrmaliges Stürzen des Behälters vollständig durchmischt.
8.2. Probeneinwaage
2,000 g ±0,001 g der Probe werden in ein Zentrifugenröhrchen (6.2) oder ein geeignetes verschließbares Gefäß (50 ml) eingewogen.
8.3. Abtrennen von Fett und Eiweiß
8.3.1. Die Probeneinwaage wird mit 20,0 ml warmem Wasser (50 °C) versetzt. Das Pulver wird durch fünfminutiges Bewegen auf dem Schüttelgerät (6.3) rekonstituiert. Das Röhrchen wird in ein Wasserbad (6.10) gegeben und bei 25 °C stehen gelassen.
8.3.2. Innerhalb von 2 Minuten werden 10 ml Trichloressigsäure (5.1) unter ständigem Rühren mit dem Magnetrührer (6.4) hinzugegeben. Das Röhrchen wird in ein Wasserbad (6.10) gestellt und 60 Minuten stehen gelassen.
8.3.3. Danach wird die Probe bei 2 200 g 10 Minuten zentrifugiert (6.2) oder durch Filterpapier (6.6) gefiltert. Die ersten 5 ml des Filtrats sind zu verwerfen.
8.4. Chromatografische Bestimmung
8.4.1. Ein genau abgemessenes Volumen zwischen 15 und 30 µl vom Überstand oder Filtrat (8.3.3) wird in das HPLC-Gerät (6.10) injiziert; währenddessen wird das Elutionsmittel (5.2) mit einer Durchflussgeschwindigkeit von 1,0 ml pro Minute zugeführt.
Anmerkung 1: Abhängig vom Innendurchmesser der verwendeten Säulen bzw. den Anweisungen des Säulenherstellers kommen möglicherweise auch sonstige Durchflussgeschwindigkeiten in Betracht.
Anmerkung 2: Das Elutionsmittel (5.2) sollte während der gesamten chromatografischen Analyse eine Temperatur von 85 °C haben, um das Elutionsmittel gasfrei zu halten und unerwünschtes Bakterienwachstum zu verhindern. Ergänzend können auch sonstige Maßnahmen mit vergleichbarer Wirkung durchgeführt werden.
Anmerkung 3: Bei jeder Unterbrechung sind die Säulen mit Wasser zu spülen. Das Elutionsmittel darf niemals in den Säulen bleiben (5.2).
Vor jeder Unterbrechung von mehr als 24 Stunden sind die Säulen mit Wasser zu spülen und mindestens 3 Stunden lang bei einem Durchfluss von 0,2 ml pro Minute mit der in Ziffer 5.3 genannten Lösung zu waschen.
8.4.2. Die Ergebnisse der chromatogaphischen Analyse der Proben [E] werden in Form eines Chromatogramms gewonnen, in dem jeder Peak durch seine Retentionszeit RT identifiziert wird, d. h.:
|
Peak II:
|
Zweiter Peak des Chromatogramms mit RT etwa 12,5 Minuten.
|
|
Peak III:
|
Dritter Peak des Chromatogramms (entsprechend CMP) mit RT 15,5 Minuten.
|
Die Qualität der Säulen kann die Retentionszeit der verschiedenen Peaks beeinflussen.
Der Integrator (6.11.6) berechnet automatisch die Fläche A der einzelnen Peaks, d. h.:
|
AII:
|
Fläche von Peak II
|
|
AIII:
|
Fläche von Peak III
|
Vor der quantitativen Interpretation muss das Aussehen des Chromatogramms untersucht werden, um gegebenenfalls Anomalien infolge einer Störung des Gerätes oder der Säulen oder infolge der Art und Herkunft der analysierten Probe feststellen zu können.
Im Zweifelsfall ist die Analyse zu wiederholen.
8.5. Kalibrierung
8.5.1. Für die Standardproben (5.4) ist das unter den Punkten 8.2 bis 8.4.2 beschriebene Verfahren genau einzuhalten.
Es sind frisch zubereitete Lösungen zu verwenden, da die CMP im 8 %igen Trichloressigsäure-Milieu abgebaut werden. Der Verlust wird bei 30 °C auf 0,2 % pro Stunde geschätzt.
8.5.2. Vor jeder chromatografischen Bestimmung der Proben sind die Säulen durch wiederholte Injektionen der Lösung (8.5.1) der Standardproben (5.4.1) vorzubehandeln, bis die Fläche und die Retentionszeit des dem CMP entsprechenden Peaks konstant bleiben.
8.5.3. Die Kalibrierfaktoren R werden durch Injektion des auch bei den Proben verwendeten Filtratvolumens (8.5.1) bestimmt.
9. DARSTELLUNG DER ERGEBNISSE
9.1. Berechnungsformel
9.1.1. Berechnung der Kalibrierfaktoren R:
|
Peak II:
|
RII = 100/(AII[0])
|
Dabei sind:
|
RII
|
=
|
Kalibrierfaktoren der Peaks II
|
|
AII [0]
|
=
|
Flächen der Peaks II der gemäß Absatz 8.5.3 hergestellten Standardprobe [0]
|
|
Peak III:
|
RIII = W/(AIII[5] – AIII[0])
|
Dabei sind:
|
RIII
|
=
|
Kalibrierfaktor für Peak III
|
|
AIII [0] und AIII [5]
|
=
|
Flächen von Peak III in den gemäß Absatz 8.5.3 hergestellten Standardproben [0] und [5]
|
|
W
|
=
|
Menge der Labmolke in der Standardprobe [5], d. h. 5
|
9.1.2. Berechnung der relativen Flächen der Peaks der Probe [E]:
SII[E] = RII × AII[E]
SIII[E] = RIII × AIII[E]
SIV[E] = RIV × AIV[E]
Dabei sind:
|
SII [E], SIII [E], SIV [E]
|
=
|
relative Flächen der Peaks II, III und IV der Probe [E]
|
|
AII [E], AIII [E]
|
=
|
Flächen der Peaks II und III der gemäß Absatz 8.4.2 hergestellten Probe [E]
|
|
RII, RIII
|
=
|
gemäß Absatz 9.1.1 berechnete Kalibrierfaktoren
|
9.1.3. Berechnung der relativen Retentionszeit für PeakIII der Probe [E] RRTIII[E] = (RTIII[E])/(RTIII[5])
Dabei sind:
|
RRTIII [E]
|
=
|
relative Retentionszeit von Peak III in Probe [E]
|
|
RTIII [E]
|
=
|
Retentionszeit von Peak III in der gemäß Absatz 8.4.2 hergestellten Probe [E]
|
|
RTIII [5]
|
=
|
Retentionszeit von Peak III der gemäß Absatz 8.5.3 hergestellten Kontrollprobe [5]
|
9.1.4. In Versuchen ist nachgewiesen worden, dass zwischen der relativen Retentionszeit von Peak III, d. h. RRTIII [E], und der zugesetzten Menge an Labmolkepulver bis zu einer Konzentration von 10 % eine lineare Abhängigkeit besteht:
|
—
|
Die RRTIII [E] ist < 1,000, wenn der Gehalt an Labmolke > 5 % ist;
|
|
—
|
die RRTIII [E] ist ≥ 1,000, wenn der Gehalt an Labmolke ≤ 5 % ist.
|
Die zulässige Toleranz für die Werte von RRTIII liegt bei ±0,002.
In der Regel weicht der Wert für RRTIII [0] kaum von 1,034 ab. Je nach dem Zustand der Säulen kann sich dieser Wert 1,000 annähern, muss jedoch immer größer sein.
9.2. Berechnung des Gehalts an Labmolkepulver in der Probe
W = SIII[E] – [1, 3 + (SIII[0] – 0, 9)]
Dabei sind:
|
W
|
=
|
Prozentanteil m/m der Labmolke in der Probe [E]
|
|
SIII [E]
|
=
|
relative Fläche von Peak III der gemäß Absatz 9.1.2 hergestellten Testprobe
|
|
1,3
|
=
|
relative durchschnittliche Fläche von Peak III in Gramm Labmolke pro 100 g ermittelt in unverfälschtem Magermilchpulver unterschiedlicher Herkunft. Der Wert wurde experimentell bestimmt
|
|
SIII [0]
|
=
|
relative Fläche von Peak III; diese ist gleich RIII × AIII [0]; diese Werte wurden mit den in den Absätzen 9.1.1 und 8.5.3 beschriebenen Verfahren bestimmt
|
|
(SIII [0] – 0,9)
|
=
|
an der relativen durchschnittlichen Fläche vorzunehmende Korrektur (1,3), wenn SIII [0] nicht gleich 0,9 ist; im Versuch wurde für die relative durchschnittliche Fläche von Peak III der Kontrollprobe [0] der Wert 0,9 ermittelt
|
9.3. Genauigkeit des Verfahrens
9.3.1. Wiederholbarkeit
Die Differenz der Ergebnisse zweier Untersuchungen, die mit identischem Untersuchungsmaterial unter denselben Versuchsbedingungen von demselben Untersucher gleichzeitig oder unmittelbar nacheinander durchgeführt werden, darf Massenanteil von 0,2 % nicht überschreiten.
9.3.2. Vergleichbarkeit
Die Differenz zwischen zwei einzelnen unabhängigen Ergebnissen, die zwei Bearbeiter, welche in verschiedenen Labors arbeiten, an identischem Probenmaterial erhalten, darf 0,4 % (m/m) nicht überschreiten.
9.4. Interpretation
9.4.1. Es muss darauf geschlossen werden, dass keine Labmolke enthalten ist, wenn die relative Fläche von Peak III, SIII [E], ausgedrückt in Gramm Labmolke pro 100 g des Erzeugnisses, ≤ 2,0 + (SIII[0] – 0,9) ist.
|
2,0
|
=
|
in Anbetracht der relativen Fläche von Peak III (d. h. 1,3), der durch Schwankungen in der Zusammensetzung des Magermilchpulvers bedingten Unsicherheit und der Vergleichbarkeit der Methode (9.3.2) der höchstens zulässige Wert für die relative Fläche von Peak III
|
|
(SIII [0] – 0,9)
|
=
|
vorzunehmende Korrektur, wenn die Fläche SIII [0] nicht gleich 0,9 ist (siehe Absatz 9.2)
|
9.4.2. Wenn die relative Fläche von Peak III SIII [E] > 2,0 + (SIII[0] – 0,9) und die relative Fläche von Peak II SII [E] ≤ 160 ist, muss der Labmolkeanteil wie in Absatz 9.2 beschrieben bestimmt werden.
Ist die relative Fläche von Peak III SIII [E] > 2,0 + (SIII[0] – 0,9) und die relative Fläche von Peak II SII [E] ≤ 160, wird der Gesamteiweißgehalt (P %) bestimmt; anschließend ist eine Bewertung aufgrund der Abbildungen 1 und 2 vorzunehmen.
9.4.3.1. Die nach Analyse von Proben von unverfälschtem Magermilchpulver gewonnenen Daten mit hohem Gesamteiweißgehalt sind in den Abbildungen 1 und 2 zusammengefasst.
Die durchgezogene Gerade ist die Gerade der linearen Regression, deren Koeffizienten nach dem Verfahren der kleinsten Quadrate errechnet wurden.
Die gestrichelte Gerade gibt die Obergrenze der relativen Oberfläche von Peak III mit einer Wahrscheinlichkeit an, die in 90 % der Fälle nicht überschritten wird.
Die Formeln der gestrichelten Geraden der Abbildungen 1 und 2 lauten jeweils:
|
SIII = 0,376 P % –10,7
|
(Abbildung 1)
|
|
SIII = 0,0123 SII [E] + 0,93
|
(Abbildung 2)
|
dabei ist jeweils
|
SIII
|
=
|
die relative Fläche von Peak III berechnet entweder aufgrund des Gesamteiweißgehalts oder aufgrund der relativen Fläche von Peak SII [E]
|
|
P %
|
=
|
der Gesamteiweißgehalt in Gewichtsprozent
|
|
SII [E]
|
=
|
die relative Fläche der gemäß Absatz 9.1.2 berechneten Probe
|
Die Gleichungen stimmen bei dem Wert 1,3 gemäß Punkt 9.2 überein.
Der Abstand (T1 und T2) zwischen der gefundenen relativen Fläche SIII [E] und der relativen Oberfläche SIII ergibt sich aus folgenden Formeln: T1 = SIII [E] – [(0,376 P% –10,7) + (SIII [0] – 0,9)]T2 = SIII [E] – [(0,0123 SII [E] + 0,93) + (SIII [0] – 0,9)]
|
Wenn T1 und/oder T2
|
Null oder kleiner sind, kann ein Labmolke-Nachweis nicht geführt werden.
|
|
Sind T1 und T2
|
größer als Null, enthält die Probe Labmolke.
|
Der Gehalt an Labmolke wird mit der Formel W = T2+0,91 berechnet.
Dabei ist:
0,91 der Abstand zwischen der durchgezogenen Geraden und der gestrichelten Geraden auf der vertikalen Achse.
Magermilchpulver
Magermilchpulver

ANHANG XIII
(Artikel 9)
BESTIMMUNG VON LABMOLKEPULVER IN MAGERMILCHPULVER UND IN DEN IN DER VERORDNUNG (EG) NR. 2799/1999 GENANNTEN GEMISCHEN
1. GEGENSTAND: NACHWEIS DES ZUSATZES VON LABMOLKEPULVER ZU:
|
a)
|
Magermilchpulver im Sinne von Artikel 2 der Verordnung (EG) Nr. 2799/1999 und
|
|
b)
|
Mischungen im Sinne von Artikel 4 der Verordnung (EG) Nr. 2799/1999.
|
2. LITERATUR: INTERNATIONALE NORM ISO 707
3. BEGRIFFSBESTIMMUNG
Unter dem Gehalt an Labmolkepulver wird der nach diesem Verfahren bestimmte Massenanteil an Kaseinmakropeptiden in Prozent verstanden.
4. KURZBESCHREIBUNG
Der Gehalt an Kaseinmakropeptid wird gemäß Anhang XII bestimmt. Proben mit positivem Ergebnis werden mit dem HPLC-Verfahren (Umkehrphasen-Hochleistungsflüssigkeitschromatografie) auf Kaseinmakropeptid A (CKPA) untersucht. Alternativ können die Proben auch sofort durch Umkehrphasen-HPLC analysiert werden. Das Ergebnis wird im Vergleich zu Standardproben aus molkefreiem und molkehaltigem Magermilchpulver mit bekanntem Gehalt ausgewertet. Labmolkepulver gilt als nachgewiesen, wenn der Massenanteil größer als 1 % ist.
5. REAGENZIEN
Alle Reagenzien müssen analysenrein sein. Das verwendete Wasser muss destilliert sein oder einen mindestens gleichwertigen Reinheitsgrad aufweisen. Die Reinheit von Acetonitril muss den Anforderungen der Spektroskopie bzw. der HPLC genügen.
Die für das Verfahren erforderlichen Reagenzien sind in Anhang XII dieser Verordnung genannt.
Reagenzien für die Umkehrphasen-HPLC:
5.1. Trichloressigsäure-Lösung
240 g Trichloressigsäure (CCl3COOH) werden in Wasser aufgelöst; anschließend wird die Lösung auf 1 000 ml aufgefüllt. Die Lösung muss klar und farblos sein.
5.2. Elutionsmittel A und B
Elutionsmittel A: 150 ml Acetonitril (CH3CN), 20 ml Isopropanol (CH3CHOHCH3) und 1,00 ml Trifluoressigsäure (TFA, CF3COOH) werden in einen 1 000-ml-Messkolben gegeben und mit Wasser auf 1 000 ml aufgefüllt.
Elutionsmittel B: 550 ml Acetonitril, 20 ml Isopropanol und 1,00 ml TFA werden in einen 1 000-ml-Messkolben gegeben und mit Wasser auf 1 000 ml aufgefüllt. Vor der Verwendung wird die Elutionslösung durch einen Membranfilter mit 0,45 µm Porendurchmesser filtriert.
5.3. Aufbewahrung der Säule
Nach den Analysen wird die Säule mit Elutionsmittel B (über einen Gradienten) und danach mit Acetonitril (30 Minuten, ebenfalls über einen Gradienten) gespült. Die Säule wird in Acetonitril aufbewahrt.
5.4. Standardproben
5.4.1. Magermilchpulver entsprechend den Anforderungen für die öffentliche Lagerhaltung (d. h. [0]).
5.4.2. Das gleiche Magermilchpulver, verfälscht durch Zusatz von 5 % (m/m) Labmolkepulver von durchschnittlicher Zusammensetzung (d. h. [5]).
5.4.3. Das gleiche Magermilchpulver, verfälscht durch Zusatz von 50 % (m/m) Labmolkepulver von durchschnittlicher Zusammensetzung (d. h [50].) (1).
6. APPARATUR
Die für das beschriebene Verfahren erforderliche Apparatur wird in Anhang XII dieser Verordnung spezifiziert.
6.1. Analysewaage
6.2. (Fakultativ:) Zentrifuge zur Herstellung einer Zentrifugalkraft von 2 200 g, Zentrifugenröhrchen mit Stopfen oder Kappe und einem Fassungsvermögen von etwa 50 ml
6.3. Mechanische Schüttelvorrichtung
6.4. Magnetrührer
6.5. Glastrichter, ca. 7 cm Durchmesser
6.6. Filterpapier, mittlere Filtriergeschwindigkeit, ca. 12,5 cm Durchmesser
6.7. Filtriervorrichtung aus Glas mit Membranfilter, 0,45 µm Porendurchmesser
6.8. Messpipetten, Nennvolumen 10 ml (ISO 648, Klasse A oder ISO/R 835) bzw. ein Pipettiersystem zur Aufgabe von 10,0 ml in 2 Minuten
6.9. Aufgabesystem zur Übertragung von 20,0 ml Wasser bei einer Temperatur von ca. 50 °C
6.10. Thermostatisierbares Wasserbad, eingestellt auf 25 ± 0,5 °C
HPLC-Ausrüstung, bestehend aus:
6.11.1. einem Pumpensystem für binäre Gradienten;
6.11.2. einem manuellen Injektor oder einem Autosampler mit einem Nennvolumen 100 µl;
6.11.3. einer Säule (Agilent Technologies Zorbax 300 SB-C3, 25 cm × 0,46 cm (Höhe x Innendurchmesser)) oder einer gleichwertigen großporigen Umkehrphasen-Säule auf Silicagelbasis;
6.11.4. einem thermostatisierbaren Säulenofen, einstellbar auf 35 ± 1 °C;
6.11.5. einem UV-Detektor mit variabler Wellenlängeneinstellung zur Messung bei 210 nm mit einer Empfindlichkeit von 0,02 Å (ggf. auch Wellenlängen bis zu 220 nm) und
6.11.6. einem Integrator, mit dem eine Integration auf die allgemeine Basislinie oder von Tal zu Tal vorgenommen werden kann.
Anmerkung: Die Säule kann auch bei Raumtemperatur betrieben werden, sofern diese um höchstens 1 °C schwankt. (Ansonsten ändert sich die Retentionszeit für CMPA zu stark.)
7. PROBENAHME
7.1. Die Probenahme erfolgt nach dem von der internationalen Norm ISO 707 vorgesehenen Verfahren. Die Mitgliedstaaten können jedoch ein anderes Probenahmeverfahren anwenden, sofern es den Prinzipien der genannten Norm entspricht.
7.2. Die Probe ist so aufzubewahren, dass sie unversehrt bleibt und ihre Zusammensetzung sich nicht ändert.
8. VERFAHREN
8.1. Vorbereitung der Probe
Das Milchpulver wird in einen ungefähr das doppelte Volumen fassenden Behälter mit luftdichtem Verschluss gegeben und der Behälter sofort verschlossen. Das Milchpulver wird durch mehrmaliges Stürzen des Behälters vollständig durchmischt.
8.2. Probeneinwaage
2,00 ± 0,001 g der Probe werden in ein Zentrifugenröhrchen (6.2) oder ein geeignetes verschließbares Gefäß (50 ml) eingewogen.
Anmerkung: Bei Gemischen ist die Testprobe in einer Menge einzuwiegen, bei der die entfettete Probeneinwaage einem Gewicht von 2,00 g entspricht.
8.3. Abtrennen von Fett und Eiweiß
8.3.1. Die Probeneinwaage wird mit 20,0 ml warmem Wasser (50 °C) versetzt. Zum Auflösen wird das Pulver mit Hilfe eines mechanischen Schüttelgerätes (6.3) 5 Minuten bzw. bei saurer Buttermilch 30 Minuten gerüttelt. Das Röhrchen wird in ein Wasserbad (6.10) gegeben und bei 25 °C stehen gelassen.
8.3.2. 10,0 ml Trichloressigsäurelösung mit einer Temperatur von 25 °C (5.1) werden innerhalb von 2 Minuten unter kräftigem Rühren mit Hilfe des Magnetrührers (6.4) gleichmäßig zugesetzt. Das Röhrchen wird in ein Wasserbad (6,10) gestellt und 60 Minuten stehen gelassen.
8.3.3. Bei 2 200 g wird 10 Minuten lang zentrifugiert (6.2) oder durch Filterpapier (6.6) filtriert, wobei die ersten 5 ml des Filtrats zu verwerfen sind.
8.4. Chromatografische Bestimmung
8.4.1. Eine HPLC-Analyse wird gemäß Anhang XII durchgeführt. Bei negativem Befund enthält die analysierte Probe keine nachweisbaren Mengen an Labmolkepulver. Bei positivem Befund ist die im Folgenden beschriebene Umkehrphasen-HPLC durchzuführen. Alternativ kann auch sofort eine Umkehrphasen-HPLC vorgenommen werden. Anteile an saurem Buttermilchpulver können bei Anwendung der in Anhang XII beschriebenen Methode falsch-positive Ergebnisse zur Folge haben. Dies ist bei der Umkehrphasen-HPLC ausgeschlossen.
8.4.2. Bevor die Umkehrphasen-HPLC durchgeführt wird, sind die Gradientenbedingungen zu optimieren. Eine CMPA-Retentionszeit von 26 Minuten ±2 Minuten ist optimal für Gradientensysteme mit einem Totvolumen von ca. 6 ml (Volumen ab der Stelle, an der die Lösungsmittel zusammentreffen, bis einschließlich Volumen der Probenschleife). Bei Gradientensystemen mit geringerem Totvolumen (z. B. 2 ml) ist eine Retentionszeit von 22 Minuten optimal.
Zu analysieren sind Lösungen labmolkefreier Standardproben (5.4) und von Standardproben mit 50 % Labmolkeanteil.
100 µl des Überstandes bzw. des Filtrates (8.3.3) werden in die HPLC-Apparatur injiziert, die unter den in Tabelle 1 genannten Bedingungen (Testgradient) betrieben wird.
Tabelle 1
Testgradientenbedingungen für eine optimale chromatografische Bestimmung
|
Zeit
(min)
|
Durchfluss
(ml/min)
|
% A
|
% B
|
Kurve
|
|
Anfänglich
|
1,0
|
90
|
10
|
*
|
|
27
|
1,0
|
60
|
40
|
linear
|
|
32
|
1,0
|
10
|
90
|
linear
|
|
37
|
1,0
|
10
|
90
|
linear
|
|
42
|
1,0
|
90
|
10
|
linear
|
Beim Vergleich beider Chromatogramme müsste die Lage des CMPA-Peaks festzustellen sein.
Mit der folgenden Formel kann die anfängliche Zusammensetzung des für den Normalgradienten zu verwendenden Lösungsmittels (siehe Absatz 8.4.3) berechnet werden: % B = 10-2,5 + (13,5 + (RTcmpA – 26) / 6) × 30 / 27 % B = 7,5 + (13,5 + (RTcmpA – 26) / 6) × 1,11
Dabei bedeuten:
|
RTcmpA
|
:
|
CMP-Retentionszeit im Testgradienten
|
|
10
|
:
|
Ausgangs-% B des Testgradienten
|
|
2,5
|
:
|
2,5 % B zur halben Laufzeit abzüglich % B beim Start im Normalgradienten
|
|
13,5
|
:
|
halbe Laufzeit des Testgradienten
|
|
26
|
:
|
erforderliche Retentionszeit für CMPA
|
|
6
|
:
|
Verhältnis der Steigungen von Testgradient und Normalgradient
|
|
30
|
:
|
% B zu Beginn abzüglich % B nach 27 Minuten im Testgradienten
|
|
27
|
:
|
Laufzeit des Testgradienten
|
8.4.3. Analyse von Lösungen der Testproben
Genau 100 µl des Überstandes bzw. Filtrates (8.3.3) werden in die HPLC-Apparatur injiziert, in die das Elutionsmittel (5.2) mit 1,0 ml/Minute geleitet wird.
Die Zusammensetzung des Elutionsmittels beim Beginn der Analyse wurde in Absatz 8.4.2 beschrieben. Normalerweise entspricht sie einem Verhältnis von A:B = 76:24 (5.2). Sofort nach der Injektion kommt es zur Ausbildung eines linearen Gradienten, der nach 27 Minuten einen um 5 % höheren prozentualen Anteil von B ergibt. Danach beginnt ein Gradient, bei dem sich die Zusammensetzung des Elutionsmittels in 5 Minuten auf 90 % B einstellt. Diese Zusammensetzung bleibt 5 Minuten konstant, um dann innerhalb von 5 Minuten mit einem linearen Gradienten wieder auf die Zusammensetzung beim Start abzufallen. Je nach Fassungsvermögen des Pumpensystems kann die nächste Injektion 15 Minuten nach Erreichen der Ausgangsbedingungen durchgeführt werden.
Anmerkung 1: Die Retentionszeit des CMPA muss 26 ± 2 Minuten betragen. Dies kann durch Veränderung der Ausgangs- und Endbedingungen des ersten Gradienten erreicht werden. Die prozentuale Differenz von B am Anfang und Ende des ersten Gradienten muss jedoch in jedem Fall 5 % B betragen.
Anmerkung 2: Die Elutionsmittel müssen ausreichend entgast werden und in diesem Zustand gehalten werden. Dies ist für den einwandfreien Betrieb des Gradientenpumpsystems von wesentlicher Bedeutung. Die Standardabweichung der Retentionszeit für den CMPA-Peak sollte <0,1 Minute (n = 10) sein.
Anmerkung 3: Bei jeder fünften Probe ist die Standardprobe [5] zu injizieren und erneut der Kalibrierfaktor R (9.1.1) zu bestimmen.
8.4.4. Im Chromatogramm der Testprobe [E] hat der CMPA-Peak eine Retentionszeit von ca. 26 Minuten.
Der Integrator (6.11.6) errechnet automatisch die Höhe H des CMPA-Peaks. Die Lage der Basislinie ist bei jedem Chromatogramm zu prüfen. Bei unrichtiger Lage der Basislinie ist die Analyse bzw. die Integration erneut durchzuführen.
Anmerkung: Wenn der CMPA-Peak hinreichend von anderen Peaks abgegrenzt ist, sollte eine Zuordnung gemäß der Basislinie von Tal zu Tal erfolgen; ansonsten sind Senkrechten auf einer gemeinsamen Basislinie einzuzeichnen, die in der Nähe des CMPA-Peak (und somit nicht bei t = 0 min!) beginnen sollte. Für die Standardlösung und die Probenlösungen ist derselbe Integrationstyp zu verwenden; bei gemeinsamer Basislinie muss die Konsistenz für die einzelnen Proben und für die Standardlösung geprüft werden.
Vor der quantitativen Bestimmung sind die Chromatogramme unbedingt auf Unregelmäßigkeiten zu überprüfen, die sich durch Betriebsstörungen der Apparatur bzw. der Säule oder aufgrund von Herkunft oder Art der analysierten Probe ergeben können. Im Zweifelsfall ist die Analyse zu wiederholen.
8.5. Kalibrierung
8.5.1. Die Standardproben (5.4.1 und 5.4.2) werden dem in den Absätzen 8.2 bis 8.4.4 beschriebenen Verfahren unterzogen. Dabei sind frisch angesetzte Lösungen zu verwenden, da CMP in 8 %iger Trichloressigsäure bei Raumtemperatur abgebaut wird. Die Lösung ist bei 4 °C 24 Stunden haltbar. Bei langen Versuchsreihen ist die Verwendung einer gekühlten Probenschale im Autosampler zu empfehlen.
Anmerkung: 8.4.2 kann entfallen, sofern der prozentuale Anteil von B beim Beginn der Analyse aus vorangehenden Analysen bekannt ist.
Das Chromatogramm der Referenzprobe [5] sollte wie in Abbildung 1 aussehen. Diese Abbildung zeigt zwei kleine Peaks vor dem CMPA-Peak. Es ist unbedingt für eine analoge Trennung zu sorgen.
8.5.2. Vor der chromatografischen Bestimmung der Proben sind 100 µl der labmolkefreien Standardprobe [0] (5.4.1) zu injizieren.
Das entsprechende Chromatogramm darf bei der Retentionszeit des CMPA-Peaks keinen Peak aufweisen.
8.5.3. Der Kalibrierfaktor R ist nach Injektion des gleichen Filtratvolumens wie bei den Proben (8.5.1) zu bestimmen.
9. DARSTELLUNG DER ERGEBNISSE
9.1. Methode und Berechnungsformel
9.1.1. Berechnung des Kalibrierfaktors R
CMPA-Peak: R = W/H
Dabei bedeuten:
|
R
|
=
|
Kalibrierfaktor des CMPA-Peaks
|
|
H
|
=
|
Höhe des CMPA-Peaks
|
|
W
|
=
|
Labmolkegehalt der Standardprobe [5]
|
9.2. Berechnung des Labmolkepulvergehalts der Probe in Prozent
W[E] = R × H[E]
Dabei bedeuten:
|
W[E]
|
=
|
prozentualer Massenanteil an Labmolkepulver in der Probe [E]
|
|
R
|
=
|
Kalibrierfaktor des CMPA-Peaks (9.1.1)
|
|
H[E]
|
=
|
Höhe des CMPA-Peaks der Probe (E)
|
Ist W[E] größer als 1 % und ist die Differenz zwischen der entsprechenden Retentionszeit und der der Standardprobe [5] kleiner als 0,2 Minuten, enthält die Probe Labmolkepulver.
9.3. Genauigkeit des Verfahrens
9.3.1. Wiederholbarkeit
Die Differenz der Ergebnisse zweier Untersuchungen, die mit identischem Untersuchungsmaterial unter denselben Versuchsbedingungen von demselben Analytiker gleichzeitig oder unmittelbar nacheinander durchgeführt werden, darf einen Massenanteil von 0,2 % nicht überschreiten.
9.3.2. Vergleichbarkeit
Nicht bestimmt.
9.3.3. Linearität
Im Bereich von 0 bis 16 % Labmolkepulveranteil muss Linearität bei einem Korrelationskoeffizienten von > 0,99 gegeben sein.
9.4. Interpretation
Die 1-%-Grenze wurde gemäß den Vorschriften des Anhangs XIX Absätze 9.2 und 9.4.1 der Verordnung (EG) Nr. 214/2001 festgesetzt und beinhaltet die durch die Vergleichbarkeit bedingte Unsicherheit.
Table 1
Ni — 4,6 standard
(1) Labmolkepulver der Standardzusammensetzung sowie das verfälschte Magermilchpulver sind zu beziehen von NIZO, Kernhemseweg 2, PO Box 20 — NL-6710 BA Ede. Es können aber auch sonstige Pulver verwendet werden, mit denen entsprechende Ergebnisse wie mit den NIZO-Pulvern erzielt werden.
ANHANG XIV
(Artikel 10)
GEHALT AN MAGERMILCHPULVER: QUANTITATIVE BESTIMMUNG VON PHOSPHATIDYLSERIN UND PHOSPHATIDYLETHANOLAMIN
Methode: Umkehrphasen-HPLC
1. GEGENSTAND UND ANWENDUNGSBEREICH
Diese Methode beschreibt ein Verfahren zur quantitativen Bestimmung von Phosphatidylserin (PS) und Phosphatidylethanolamin (PE) in Magermilchpulver (MMP) und ist auch für den Nachweis von Buttermilch-Feststoffen in Magermilchpulver geeignet.
2. BEGRIFFSBESTIMMUNG
Gehalt an PS + PE: Massenfraktion der mit dieser Methode festgestellten Substanz; das Ergebnis wird in mg Phosphatidylethanolamin-Dipalmitoyl (PEDP) je 100 g Pulver ausgedrückt.
3. KURZBESCHREIBUNG
Extraktion von Aminophosholipiden aus dem rekonstituierten Milchpulver mit Hilfe von Methanol; Bestimmung von PS und PE als o-Phthaldialdehyd (OPA)-Derivat durch Umkehrphasen(RP)-HPLC und Fluoreszenzdetektion; Quantifizierung des PS- und PE-Gehalts der Testprobe anhand einer Referenzstandardprobe mit einem bekannten PEDP-Gehalt.
4. REAGENZIEN
Alle Reagenzien müssen analysenrein sein. Wasser muss destilliert oder von mindestens gleicher Reinheit sein, wenn nicht anderweitig angegeben.
4.1. Standardmaterial: PEDP, Reinheit mindestens 99 %.
Anmerkung: Das Standardmaterial ist bei –18 °C zu lagern.
4.2. Reagenzien zur Vorbereitung der Standard- und der Testproben
4.2.1. Methanol in HPLC-Qualität
4.2.2. Chloroform in HPLC-Qualität
4.2.3. Tryptaminmonohydrochlorid
4.3. Reagenzien für die Derivatisierung des o-Phthaldialdehyds
4.3.1. Natriumhydroxid, 12 M wässrige Lösung
4.3.2. Borsäure, 0,4 M wässrige Lösung, mit Natriumhydroxyd (4.3.1) auf pH 10,0 eingestellt
4.3.3. 2-Mercaptoethanol
4.3.4. o-Phthaldialdehyd (OPA)
4.4. HPLC-Elutionsmittel
4.4.1. Die Elutionsmittel müssen mit Reagenzien in HPLC-Qualität zubereitet werden.
4.4.2. Wasser in HPLC-Qualität
4.4.3. Methanol für die fluorimetrische Reinheitsmessung
4.4.4. Tetrahydrofuran
4.4.5. Natriumdihydrogenphosphat
4.4.6. Natriumacetat
4.4.7. Essigsäure
5. APPARATUR
5.1. Analysewaage, Messgenauigkeit 1 mg, Teilung 0,1 mg
5.2. Bechergläser, 25 und 100 ml
5.3. Pipetten, Aufgabemengen 1 und 10 ml
5.4. Magnetrührer
5.5. Messpipetten, Aufgabemengen 0,2, 0,5 und 5 ml
5.6. Messkolben, 10, 50 und 100 ml
5.7. Spritzen, 20 und 100 µl
5.8. Ultraschallbad
5.9. Zentrifuge, Zentrifugalkraft 27 000 × g
5.10. Glasfläschchen, etwa 5 ml
5.11. Messzylinder, 25 ml
5.12. pH-Messgerät, Genauigkeit 0,1 pH
HPLC-Ausrüstung
5.13.1. Gradientenpumpe, Pumpleistung 1,0 ml/min bei 200 bar
5.13.2. Autosampler mit Derivatisierungsfunktion
5.13.3. Säulenheizung zur Regelung einer Säulentemperatur von 30 °C ±1 °C
5.13.4. Fluoreszenz-Messgerät, Messbereich 330 nm, Emissionsbereich 440 nm
5.13.5. Integrator oder Datenverarbeitungs-Software zur Messung von Peak-Flächen
5.13.6. Lichrosphere-100-Säule (250 × 4,6 mm) oder gleichwertige mit Octadecylsilan (C 18) gepackte Säule, Partikelgröße 5 µm
6. PROBENAHME
Die Probenahme ist nach der ISO-Norm 707 durchzuführen.
7. VERFAHREN
7.1. Zubereitung der internen Standardlösung
7.1.1. 30,0 mg (±0,1 mg) Tryptamin-monohydrochlorid (4.2.3) werden in einen 100-ml-Messkolben (5.6) eingewogen und bis zur Marke mit Methanol (4.2.1) aufgefüllt.
7.1.2. Von dieser Lösung wird 1 ml in einen 10-ml-Messkolben (5.6) einpipettiert (5.3) und bis zur Marke mit Methanol (4.2.1) aufgefüllt, um eine Tryptaminkonzentration von 0,15 mM zu erhalten.
7.2. Zubereitung der Testprobenlösung
7.2.1. 1,000 ±0,001 g der Magermilchpulver-Probe werden in ein 25-ml-Becherglas (5.2) eingewogen. 10 ml destilliertes Wasser mit einer Temperatur von 40 °C ±1 °C werden mit einer Pipette (5.3) hinzugegeben und mit einem Magnetrührer (5.4) 30 Minuten gerührt, um ggf. vorhandene Klumpen aufzulösen.
7.2.2. Danach werden 0,2 ml der rekonstituierten Milch (5.5) in einen 10-ml-Messkolben (5.6) einpipettiert und 100 µl der 0,15 mM-Tryptaminlösung (7.1) mit einer Spritze (5.7) zugegeben und mit Methanol (4.2.1) bis zur Marke aufgefüllt. Die Lösung wird durch Stürzen sorgfältig gemischt und 15 Minuten mit Ultraschall (5.8) behandelt.
7.2.3. Danach ist die Lösung 10 Minuten mit einer Zentrifuge (5.9) bei 27 000 g zu beschleunigen und der Überstand in einem Glasfläschchen (5.10) aufzunehmen.
Anmerkung: Die Testprobenlösung wird bis zur HPLC-Analyse bei 4 °C aufbewahrt.
7.3. Zubereitung der externen Standardlösung
7.3.1. 55,4 mg PEDP (4.1) werden in einen 50-ml-Messkolben (5.6) eingewogen und etwa 25 ml Chloroform (4.2.2) mit einem Messzylinder (5.11) zugegeben. Der verschlossene Kolben wird auf 50 °C erhitzt und der Inhalt sorgfältig gemischt, bis sich das PEDP aufgelöst hat. Danach wird der Kolben auf 20 °C gekühlt, mit Methanol (4.2.1) bis zur Marke aufgefüllt und durch Stürzen des Kolbens gemischt.
7.3.2. 1 ml dieser Lösung wird in einen 100-ml-Messkolben (5.6) einpipettiert (5.3) und bis zur Marke mit Methanol (4.2.1) aufgefüllt. Von dieser Lösung wird 1 ml in einen 10-ml-Messkolben (5.6) einpipettiert (5.3), mit 100 µl (5.7) der 0,15 mM Tryptaminlösung (7.1) versetzt und bis zur Marke mit Methanol (4.2.1) aufgefüllt. Die Lösung wird durch Stürzen des Kolbens gemischt.
Anmerkung: Die Standardprobenlösung ist bis zur HPLC-Analyse bei 4 °C aufzubewahren.
7.4. Vorbereitung des Derivatisierungsreagens
25,0 mg (±0,1 mg) OPA (4.3.4) werden in einen 10-ml-Messkolben (5.6) gegeben, mit 0,5 ml (5.5) Methanol (4.2.1) versetzt und sorgfältig gemischt, bis sich das OPA gelöst hat. Danach wird bis zur Marke mit der Borsäurelösung (4.3.2) aufgefüllt; mit einer Spritze (5.7) werden 20 µl 2-Mercaptoethanol (4.3.3) zugegeben.
Anmerkung: Das Derivatisierungsreagens kann ohne Stabilitätsverlust eine Woche bei 4 °C in einer braunen Glasflasche aufbewahrt werden.
7.5. Bestimmung durch HPLC
7.5.1. Elutionslösungen (4.4)
Lösungsmittel A: 0,3 mM Natriumdihydrogenphosphat und 3 mM Natriumacetatlösung (mit Essigsäure auf pH 6,5 ±0,1 eingestellt): Methanol: Tetrahydrofuran = 558:440:2 (v/v/v)
Lösungsmittel B: Methanol
7.5.2. Empfohlener Elutionsgradient:
|
Zeit
(min)
|
Lösungsmittel A
(%)
|
Lösungsmittel B
(%)
|
Durchfluss
(ml/min)
|
|
Anfänglich
|
40
|
60
|
0
|
|
0,1
|
40
|
60
|
0,1
|
|
5,0
|
40
|
60
|
0,1
|
|
6,0
|
40
|
60
|
1,0
|
|
6,5
|
40
|
60
|
1,0
|
|
9,0
|
36
|
64
|
1,0
|
|
10,0
|
20
|
80
|
1,0
|
|
11,5
|
16
|
84
|
1,0
|
|
12,0
|
16
|
84
|
1,0
|
|
16,0
|
10
|
90
|
1,0
|
|
19,0
|
0
|
100
|
1,0
|
|
20,0
|
0
|
100
|
1,0
|
|
21,0
|
40
|
60
|
1,0
|
|
29,0
|
40
|
60
|
1,0
|
|
30,0
|
40
|
60
|
0
|
Anmerkung: Der Elutionsgradient muss unter Umständen geringfügig verändert werden, damit die in Abbildung 1 gezeigte Auflösung erreicht wird.
Säulentemperatur: 30 °C
7.5.3. Einspritzvolumen: 50 µl Derivatisierungsmittel und 50 µl Probelösung.
7.5.4. Säulenäquilibrierung
Wird die Säule täglich neu gestartet, muss sie 15 Minuten lang mit Lösungsmittel B (100 %) gespült, dann bei einem Verhältnis A:B von 40:60 eingestellt und mit einem Durchfluss von 1 ml/min 15 Minuten lang äquilibriert werden. Danach wird ein Blindlauf unter Einspritzung von Methanol (4.2.1) durchgeführt.
Anmerkung: Bevor die Säule für längere Zeit gelagert wird, sollte sie 30 Minuten mit einer Mischung von Methanol und Chloroform im Verhältnis 80:20 (v/v) gespült werden.
7.5.5. Bestimmung des PS- und des PE-Gehalts des Testprobe
7.5.6. Die chromatografischen Analysen werden in der entsprechenden Reihenfolge durchgeführt, wobei die Intervalle zwischen den Durchläufen konstant bleiben müssen, um konstante Retentionszeiten zu erhalten. Die externe Standardlösung (7.3) wird in alle 5-10 Testproben eingespritzt, damit der Kalibrierfaktor bewertet werden kann.
Anmerkung: Die Säule ist nach ungefähr 20-25 Durchläufen 30 Minuten mit 100 %iger Lösung B (7.5.1) zu spülen.
7.6. Integrationsbetrieb
7.6.1. PEDP-Peak
Das PEDP wird als einzelner Peak eluiert. Die Peak-Fläche wird durch Integration von Tal zu Tal ermittelt.
7.6.2. Tryptamin-Peak
Das Tryptamin wird als einzelner Peak (siehe Abbildung 1) eluiert. Die Peak-Fläche wird durch Integration von Tal zu Tal ermittelt.
7.6.3. PS- und PE-Peak-Gruppen
Unter den beschriebenen Bedingungen (Abbildung 1) ergibt PS zwei teilweise getrennte Haupt-Peaks, denen ein kleinerer Peak vorausgeht. PE ergibt drei teilweise getrennte Haupt-Peaks. Die Gesamtfläche des jeweiligen Peak-Bündels wird ermittelt, indem die Basislinie wie in Abbildung 1 zu sehen gezogen wird.
8. BERECHNUNG UND DARSTELLUNG DER ERGEBNISSE
Der PS- und PE-Gehalt der Testprobe wird wie folgt berechnet: C = 55,36 × ((A2)/(A1)) × ((T1)/(T2))
Dabei sind:
|
C
|
=
|
PS- oder PE-Gehalt (mg/100 g Pulver in der Testprobe)
|
|
A1
|
=
|
Peak-Fläche PEDP der Standard-Probenlösung (7.3)
|
|
A2
|
=
|
Peak-Fläche PS oder PE der Testprobe (7.2)
|
|
T1
|
=
|
Peak-Fläche Tryptamin der Standard-Probenlösung (7.3)
|
|
T2
|
=
|
Peak-Fläche Tryptamin der Testprobe (7.2)
|
9. GENAUIGKEIT DER METHODE
Anmerkung: Die Werte für die Wiederholbarkeit wurden nach der Internationalen IDF-Norm berechnet (1). Der vorläufige Vergleichbarkeitsgrenzwert wurde gemäß Anhang III Buchstabe b berechnet.
9.1. Wiederholbarkeit
Die relative Wiederholstandardabweichung beschreibt die Variabilität der Ergebnisse, die von dem gleichen Analytiker mit den gleichen Geräten unter den gleichen Bedingungen mit der gleichen Probe in kurzen Zeitabständen unabhängig voneinander erzielt wurden; ein Relativwert von 2 % sollte nicht überschritten werden. Werden zwei Bestimmungen unter diesen Bedingungen durchgeführt, sollte der relative Unterschied zwischen den beiden Ergebnissen nicht mehr als 6 % des arithmetischen Mittels der Ergebnisse betragen.
9.2. Vergleichbarkeit
Werden zwei Bestimmungen von Analytikern in unterschiedlichen Laboratorien mit unterschiedlichen Geräten unter unterschiedlichen Bedingungen mit der gleichen Testprobe durchgeführt, sollte der relative Unterschied zwischen den beiden Ergebnissen nicht mehr als 11 % des arithmetischen Mittels der Ergebnisse betragen.
10. LITERATUR
10.1. Resmini P., Pellegrino L., Hogenboom J.A., Sadini V., Rampilli M., „Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids.“ Sci. Tecn. Latt.-Cas., 39,395 (1988).
Abbildung 1
HPLC-Ergebnis für die OPA-Derivate Phosphatidylserin (PS) und Phosphatidylethanolamin (PE) in einem Methanol-Extrakt aus rekonstituiertem Magermilchpulver; die Integrationsform für die Peaks von PS, PE und Tryptamin (interner Standard) ist eingezeichnet

(1) Internationale |IDF-Norm 135B/1991: Milk and milk products. Precision characteristics of analytical methods. Outline of collaborative study procedure.
ANHANG XV
(Artikel 11)
NACHWEIS VON ANTIMIKROBIOTIKA-RÜCKSTÄNDEN IN MAGERMILCHPULVER
Es muss ein Hemmstoff-Screening-Test für Antimikrobiotika mit Geobacillus stearothermophilus var. calidolactis (entsprechend Stamm C953) als Testorganismus durchgeführt werden, der empfindlich genug ist, um 4 µg Benzylpenicillin pro kg Milch und 100 µg Sulfonamide pro kg Milch nachzuweisen. Handelsübliche Tests können verwendet werden, wenn sie für Benzylpenicillin und Sulfonamid ausreichend empfindlich sind.
Für den Test wird rekonstituiertes Magermilchpulver verwendet (1 g Pulver +9 ml destilliertes Wasser). Der Test wird wie in ISO/TS 26844:2006 Milk and milk products — Determination of antimicrobial residues — Tube diffusion test IDF — Bulletin Nr. 258/1991, Abschnitt 1, Kapitel 2, beschrieben oder gemäß den Anweisungen des Test-Kit-Herstellers (1) durchgeführt.
Positive Ergebnisse sind wie folgt zu interpretieren:
|
1.
|
β-Laktame können durch Wiederholung des Tests unter Zugabe von Penicillinase zum Testsystem nachgewiesen werden (2).
Negatives Ergebnis: Der Hemmstoff ist ein Beta-Lactam-Antibiotikum.
Positives Ergebnis: Der Hemmstoff kann mit diesem Verfahren nicht nachgewiesen werden; entsprechend ist mit Schritt 2 fortzufahren.
|
|
2.
|
Sulfonamide können durch Wiederholung des Tests unter Zugabe von p-Aminobenzoesäure zum Testsystem nachgewiesen werden.
Negatives Ergebnis: Der Hemmstoff ist ein Sulfonamid.
Erneut positives Ergebnis: Der Hemmstoff kann mit diesem Verfahren nicht nachgewiesen werden; weiter mit Schritt 3.
|
|
3.
|
β-Laktame und Sulfonamid können nachgewiesen werden, indem der Test unter Zugabe von Pencillinase + p-Aminobenzoesäure zum Testsystem wiederholt wird.
Negatives Ergebnis: Der Hemmstoff ist ein Beta-Lactam-Antibiotikum und ein Sulfonamid.
Positives Ergebnis: Der Hemmstoff kann mit diesem Verfahren nicht nachgewiesen werden.
|
(1) Achtung: Bei der Analyse von Magermilchpulver können falsch-positive Ergebnisse auftreten. Es muss daher unbedingt sichergestellt werden, dass der Test keine falsch-positiven Ergebnisse erbringen kann.
(2) Einige β-Laktame sind weniger empfindlich gegenüber β-Laktamase. In diesen Fällen ist eine zusätzliche vorherige Erwärmung der Probe (1 ml Testprobe mit 0,3 ml Penase-Konzentrat bei 37 °C über 2 Std.) zu empfehlen.
ANHANG XVI
(Artikel 12)
BESTIMMUNG DES GEHALTS AN MAGERMILCHPULVER IN EINEM MISCHFUTTERMITTEL ÜBER PARAKASEIN NACH ENZYMATISCHER GERINNUNG
1. GEGENSTAND
Bestimmung des Gehalts an Magermilchpulver in einem Mischfuttermittel durch enzymatische Parakasein-Gerinnung
2. ANWENDUNGSBEREICH
Diese Methode gilt für Mischfuttermittel mit einem Anteil an Magermilchpulver von mindestens 10 %; das Vorhandensein größerer Mengen von Buttermilch und/oder bestimmter Nichtmilchproteine kann Interferenzen zur Folge haben.
3. KURZBESCHREIBUNG
3.1. Lösung des im Mischfuttermittel enthaltenen Kaseins durch Extraktion mit einer Natriumcitrat-Lösung
3.2. Anpassung der zur Ausfällung des Kaseins notwendigen Kalziumionenkonzentration unter Zugabe von Lab
3.3. Der Stickstoffgehalt des ausgefällten Parakaseins wird nach der Kjeldahl-Methode bestimmt, wie in der ISO-Norm 8968-2:2001|IDF 20-2:2001 beschrieben; der Gehalt an Magermilchpulver wird aufgrund eines Kaseingehalts von mindestens 27,5 % (siehe Absatz 8.1) berechnet.
4. REAGENZIEN
Die Reagenzien müssen den zu Analysezwecken erforderlichen Reinheitsgrad aufweisen. Das verwendete Wasser muss destilliert sein oder einen mindestens gleichwertigen Reinheitsgrad aufweisen. Mit Ausnahme des Labs (4.5) müssen alle verwendeten Reagenzien und Lösungen frei von stickstoffhaltigen Stoffen sein.
4.1. Trinatriumcitrat als Dihydrat (Lösung zu 1 % w/v)
4.2. Calciumchlorid (Lösung etwa 5 M)
75 g CaCl2 . 2H2O werden in 100 ml destilliertem Wasser unter Schütteln aufgelöst (Vorsicht: exotherme Reaktion). Die Lösung bleibt über Nacht stehen und wird dann gefiltert. Die Lösung muss im Kühlschrank gelagert werden.
4.3. 0,1 n Natriumhydroxid
4.4. 0,1 n Chlorwasserstoffsäure
4.5. Flüssiges Kälberlab (Stärke etwa 100 IMCU/ml gemäß ISO-Norm 11815|IDF 157); die Lösung ist im Kühlschrank bei 4 bis 6 °C zu lagern.
4.6. Reagenzien für die quantitative Stickstoffbestimmung nach der Kjeldahl-Methode, wie in ISO 8968-2:2001|IDF 20-2:2001 erläutert.
5. APPARATUR
Übliche Laborgeräte, insbesondere:
5.1. Reibschale oder Homogenisiergerät
5.2. Analysewaage mit einer Genauigkeit von 1 mg mit 0,1-mg-Teilung
5.3. Tischzentrifuge (500 g bzw. 2 000 bis 3 000 Umin–1) mit 50-m-Röhrchen, Zentrifugalkraft 2 000 g
5.4. Magnetrührer mit Rührstäben von 10 bis 15 mm
5.5. Bechergläser 150 bis 200 ml
5.6. 250- und 500-ml-Kolben
5.7. Glastrichter, Durchmesser 60 bis 80 mm
5.8. Aschefreie Schnellfilter mit einem Durchmesser von 150 mm (Whatman Nr. 41 oder gleichwertig)
5.9. Pipetten verschiedener Größe
5.10. Thermostatisierbares Wasserbad, einstellbar auf 37 °C ±1 °C
5.11. pH-Messgerät, Genauigkeit 0,1 pH
5.12. Thermometer, Genauigkeit 1 °C
6. VERFAHREN
6.1. Vorbereitung der Probe
10 bis 20 g Probe werden in einer Reibschale zerkleinert oder im Homogenisiergerät behandelt, um eine homogene Mischung herzustellen.
6.2. Das Milchpulver wird aufgelöst und der nichtlösliche Rückstand abgetrennt.
6.2.1. 1,000 ± 0,002 g gut homogenisiertes Mischfutter (6.1) werden direkt in ein Zentrifugenröhrchen mit einem Fassungsvermögen von 50 ml eingewogen. 30 ml Trinatriumcitrat-Lösung (4.1) werden auf 45 °C ±2 °C erwärmt und hinzugegeben. Die Lösung wird mit dem Magnetrührer mindestens fünf Minuten gemischt oder kräftig von Hand geschüttelt.
6.2.2. Die Probe wird 10 Minuten bei 500 g (2 000 bis 3 000 Umin–1) zentrifugiert und der Überstand in ein Becherglas mit einem Fassungsvermögen von 150 bis 200 ml abgegossen. Beim Abgießen ist darauf zu achten, dass kein Material verloren geht.
6.2.3. Nach dem gleichen Verfahren werden zwei weitere Extraktionen aus dem Rückstand durchgeführt; die beiden neuen Extrakte werden zum ersten Extrakt hinzugegeben.
6.2.4. Scheidet sich Fett ab, ist die Probe bis zur Verfestigung der Fettphase abzukühlen und das verfestigte Fett mit einem Spatel zu entfernen.
6.3. Gerinnung des Kaseins durch Labenzyme
6.3.1. Unter ständigem Rühren werden tropfenweise 2 ml Calciumchlorid (4.2) zum gesamten wässerigen Extrakt (etwa 100 ml) hinzugegeben. Der pH-Wert wird mit NaOH- (4.3) oder HCl-Lösung (4.4) auf 6,4-6,5 eingestellt. Die Lösung wird 15 bis 20 Minuten in einem auf 37 °C ±1 °C eingestellten thermostatisierbaren Wasserbad gelassen, bis sich das Salzgleichgewicht eingestellt hat. Wenn die Lösung ein milchiges Aussehen angenommen hat, ist das Gleichgewicht erreicht.
6.3.2. Die Flüssigkeit wird in ein (oder zwei) Zentrifugenröhrchen umgefüllt und 10 Minuten bei 2 000 g zentrifugiert, um die Ausfällungen zu entfernen. Der Überstand wird in ein (oder zwei) Zentrifugenröhrchen abgegossen, ohne den Niederschlag auszuwaschen.
6.3.3. Der Überstand wird wieder auf 37 °C ±1 °C erwärmt. Unter Rühren werden tropfenweise 0,5 ml des flüssigen Labs (4.5) zum Extrakt gegeben. Die Koagulierung erfolgt binnen zwei Minuten.
6.3.4. Die Probe wird ins Wasserbad zurückgestellt und 15 Minuten bei einer Temperatur von 37 °C ±1 °C stehen gelassen. Danach wird die Probe aus dem Wasserbad genommen und das Koagulum durch Umrühren aufgebrochen. 10 Minuten wird die Probe bei 2 000 g zentrifugiert. Der Überstand wird durch ein geeignetes Filterpapier (5.8) gefiltert und das Filterpapier aufbewahrt. Der Niederschlag wird im Zentrifugenröhrchen mit 50 ml Wasser bei einer Temperatur von etwa 35 °C durch Umrühren gewaschen.
Anschließend erfolgt nochmals eine Zentrifugierung über 10 Minuten mit 2 000 g. Der Überstand wird durch das aufbewahrte Filterpapier gefiltert.
6.4. Bestimmung des Kaseinstickstoffs
6.4.1. Nach dem Waschen wird der Niederschlag unter Verwendung von destilliertem Wasser quantitativ auf das gemäß Absatz 6.3.4 aufbewahrte Filterpapier gebracht. Das Filterpapier wird in den Kjeldahl-Kolben gegeben. Der Stickstoffgehalt wird nach der Kjeldahl-Methode bestimmt, wie in ISO-Norm 8968-2:2001|IDF 20-2:2001 beschrieben.
7. BLINDVERSUCH
7.1. Regelmäßig wird ein Blindversuch durchgeführt, indem die Probe nach der Kjeldahl-Methode wie in ISO-Norm 8968-2:2001|IDF 20-2:2001 beschrieben mineralisiert wird. Ein aschefreies Filterpapier (5.8) wird mit einem Gemisch aus 90 ml Natriumcitrat (4.1), 2 ml Calciumchlorid-Lösung (4.2) und 0,5 ml flüssigem Lab (4.5) befeuchtet und dreimal mit 15 ml destilliertem Wasser gewaschen.
7.2. Das für den Blindversuch erforderliche Säurevolumen (4.4) ist von dem zur Titration der Probe verbrauchten Volumen abzuziehen.
8. DARSTELLUNG DER ERGEBNISSE
8.1. Der Gehalt an Magermilchpulver in einem Mischfuttermittel wird nach folgender Formel berechnet:

wobei gilt:
N = Prozentsatz des Parakasein-Stickstoffes;
27,5 = Faktor zur Umrechnung des ermittelten Kaseins in Prozent Magermilchpulver;
2,81 und 0,908 = aus der Regressionsanalyse erhaltene Berichtigungsfaktoren.
9. GENAUIGKEIT DER METHODE
9.1. Wiederholbarkeit
In mindestens 95 % der untersuchten Fälle darf der Unterschied zwischen zwei einzelnen mit der gleichen Probe im gleichen Labor vom gleichen Analytiker erhaltenen Ergebnissen 2,3 g Magermilchpulver auf 100 g geprüftes Mischfutter nicht übersteigen.
9.2. Vergleichbarkeit
In mindestens 95 % der untersuchten Fälle darf der Unterschied zwischen den von zwei Laboratorien mit einer gleichen Probe erhaltenen Ergebnissen 6,5 g Magermilchpulver auf 100 g untersuchtes Mischfutter nicht übersteigen.
10. BEOBACHTUNGEN
10.1. Die Zugabe größerer Mengen bestimmter Fremdproteine, insbesondere Sojaproteine, kann — wenn sie mit der Milch erwärmt worden sind — erhöhte Werte zur Folge haben, da diese Stoffe gleichzeitig mit Milchparakasein ausgefällt werden.
10.2. Die Zugabe von Buttermilch kann zu niedrigen Werten führen, da bei der Bestimmung nur der fettfreie Anteil erfasst wird. Die Zugabe bestimmter aus Sauerrahm gewonnener Buttermilcharten kann zu deutlich niedrigeren Werten führen, da sie in der Citrat-Lösung nur unvollständig gelöst werden.
10.3. Die Zugabe von mindestens 0,5 % Lecithin kann ebenfalls zu niedrige Ergebnisse zur Folge haben.
10.4. Die Einbeziehung von auf hohe Temperaturen erwärmtem Magermilchpulver (high-heat) kann überhöhte Werte zur Folge haben, da bestimmte Molkeproteine mit dem Parakasein der Milch ausfällen.
ANHANG XVII
(Artikel 13)
BESTIMMUNG VON STÄRKE IN MAGERMILCHPULVER, DENATURIERTEM MILCHPULVER UND MISCHFUTTER
1. ANWENDUNGSBEREICH
Diese Methode beschreibt ein Verfahren zum Nachweis von Stärke als Kennzeichnungsmittel in denaturierten Milchpulvern.
Die Nachweisgrenze dieser Methode liegt bei etwa 0,05 g Stärke je 100 g Probe.
2. KURZBESCHREIBUNG
Dieses Verfahren beruht auf einer iodometrischen Reaktion:
|
—
|
Fixierung des freien Iods durch die Kolloide in wässriger Lösung,
|
|
—
|
Absorption durch die Stärke-Micellen und Farbentwicklung.
|
3. REAGENZIEN
3.1. Iodlösung
|
—
|
destilliertes Wasser: 100 ml.
|
|
—
|
1,0 g Iod und 2,0 g Kaliumiod werden in einem 100-ml-Messkolben mit Füllmarkierung in Wasser aufgelöst. Die Lösung wird bis zur 100-ml-Marke mit Wasser gefüllt und gemischt.
|
4. APPARATUR
4.1. Analysewaage
4.2. Siedendes Wasserbad
4.3. Teströhrchen, 25 mm × 200 mm
5. VERFAHREN
1,0 g der Probe wird mit einer Genauigkeit von 0,1 g in das Reagenzglas (4.3) eingewogen.
Die Einwaage wird mit 20 ml destilliertem Wasser versetzt und durch Schütteln aufgelöst.
Das Reagenzglas wird ins siedende Wasserbad (4.2) gestellt und dort fünf Minuten belassen.
Die Probe wird aus dem Wasserbad genommen, um sie bei Raumtemperatur abkühlen zu lassen.
0,5 ml Iodlösung (3.1) werden hinzugegeben; anschließend wird die Probe geschüttelt und die Farbe beurteilt.
6. DARSTELLUNG DER ERGEBNISSE
Eine Blaufärbung ist als Anzeichen für native Stärke in der Probe zu werten.
Enthält die Probe modifizierte Stärke, tritt nicht unbedingt eine Blaufärbung ein.
7. ANMERKUNGEN
Je nach Ursprung der nativen Stärke (z. B. Mais oder Kartoffel) und der Art der modifizierten Stärke in der Probe fallen Farbe, Farbintensität und mikroskopisches Erscheinungsbild der Stärke unterschiedlich aus.
Wenn modifizierte Stärke enthalten ist, erfolgt ein Farbumschlag nach Violett, Rot oder Braun, je nach Grad der Modifizierung der Kristallstruktur der nativen Stärke.
ANHANG XVIII
(Artikel 14)
BESTIMMUNG DES FEUCHTIGKEITSGEHALTS VON TROCKENRAHM
1. ANWENDUNGSBEREICH
Dieser Anhang beschreibt ein Verfahren zur Bestimmung des Feuchtigkeitsgehalts von Trockenrahm.
2. BEGRIFFSBESTIMMUNGEN
In diesem Anhang werden die folgenden Begriffe verwendet:
Feuchtigkeitsgehalt: Mit dem in dieser internationalen Norm beschriebenen Verfahren ermittelter Masseverlust;
der Feuchtigkeitsgehalt wird als Massegehalt ausgedrückt.
3. KURZBESCHREIBUNG
Eine Probeneinwaage wird bei 102 ± 2 °C auf eine konstante Masse getrocknet und gewogen, um den Masseverlust zu bestimmen.
4. APPARATUR
Normale Laborausstattung und insbesondere:
4.1. Analysewaage, Messgenauigkeit 1 mg, Teilung 0,1 mg
4.2. Ofen, gut belüftet, mit einem Thermostaten regelbar auf konstant 102 ± 2 °C im gesamten Arbeitsbereich
4.3. Exsikkator mit frisch getrocknetem Silicagel mit Feuchtigkeitsanzeige oder ein sonstiges wirksames Trockenmittel
4.4. Schalen mit flachem Boden, Tiefe etwa 25 mm, Durchmesser etwa 50 mm, aus geeignetem Material (z. B. Glas, Edelstahl, Nickel oder Aluminium), mit gut sitzenden und leicht abnehmbaren Deckeln
4.5. Flaschen mit passgenau sitzenden Verschlüssen zum Mischen der Laborproben
5. PROBENAHME
Wichtig ist, dass das Laboratorium eine Testprobe erhält, die tatsächlich repräsentativ ist und nicht bei Transport oder Lagerung beschädigt oder verändert wurde.
Die Probenahme ist nicht Bestandteil der in dieser internationalen Norm beschriebenen Methode. Eine empfohlene Methode zur Probenahme wird in ISO 707|IDF 50 erläutert.
Die Probe wird so aufbewahrt, dass eine Beeinträchtigung oder Änderung der Zusammensetzung verhindert wird.
6. VORBEREITUNG DER TESTPROBE
Die Testprobe wird gründlich gemischt, indem sie mehrfach geschüttelt und der Probenbehälter gestürzt wird (ggf. nach Übertragung sämtlicher Testproben in einen luftdichten Behälter mit hinreichendem Fassungsvermögen für die Durchführung dieses Schritts).
Wenn auf diese Weise eine vollständige Homogenität nicht erzielt werden kann, sind (für zwei getrennte Bestimmungen) zwei Einwaagen der vorbereiteten Testprobe zu verwenden, die an zwei möglichst weit voneinander entfernten Punkten genommen wurden.
7. VERFAHREN
7.1. Vorbereitung der Schale
7.1.1. Eine unbedeckte Schale und der dazugehörige Deckel (4.4) werden im auf 102 ± 2 °C eingestellten Ofen (4.2) mindestens 1 Stunde erwärmt.
7.1.2. Der Deckel wird auf die Schale gesetzt und die verschlossene Schale in den Exsikkator (4.3) gegeben; anschließend muss die Schale auf die Temperatur des Raums abkühlen, in dem sich die Waage befindet. Die Schale wird auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert.
7.2. Probeneinwaage
1 bis 3 g der vorbereiteten Testrobe (6) werden in die Schale gegeben; der Deckel wird geschlossen. Die Schale wird auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert.
7.3. Bestimmung
7.3.1. Der Deckel wird abgenommen und die Schale mit dem dazugehörigen Deckel 2 Stunden in den auf 102 ± 2 °C gestellten Ofen gesetzt.
7.3.2. Der Deckel wird auf die Schale gesetzt und die verschlossene Schale in den Exsikkator gegeben; anschließend muss die Schale auf die Temperatur des Raums abkühlen, in dem sich die Waage befindet. Die Schale wird auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert.
7.3.3. Der Deckel der Schale wird geöffnet und die Schale mit dem dazugehörigen Deckel nochmals 1 Stunde im Ofen erwärmt. Anschließend wird Schritt 7.3.2 wiederholt.
7.3.4. Die Schale wird so oft erwärmt und neu gewogen, bis die Masse nach dem erneuten Wiegen nur noch um höchstens 1 mg ab- oder zunimmt.
Für die Berechnung ist die niedrigste protokollierte Masse anzunehmen.
8. BERECHNUNG UND DARSTELLUNG DER ERGEBNISSE
8.1. Berechnung
Der Feuchtigkeitsgehalt, ausgedrückt in g/100 g, wird wie folgt berechnet:

Dabei sind:
m0
= Masse der Schale und des Deckels (7.1.2) in Gramm;
m1
= Masse der Schale, des Deckels und der Probeneinwaage vor dem Trocknen (7.2) in Gramm;
m2
= Masse der Schale, des Deckels und der Probeneinwaage nach dem Trocknen (7.3.4) in Gramm.
Das Ergebnis wird auf zwei Dezimalstellen genau protokolliert.
9. GENAUIGKEIT
Anmerkung: Die Werte für die Wiederholbarkeit und die Vergleichbarkeit wurden aus den Ergebnissen eines Leistungstests (siehe Steiger, G. Bulletin of IDF Nr. 285/1993, S. 21-28) abgeleitet, der gemäß IDF-Norm 135B:1991 Milk and milk products — Precision characteristics of analytical methods — Outline of collaborative study procedure durchgeführt wurde.
9.1. Wiederholbarkeit
Der absolute Unterschied zwischen zwei einzelnen und voneinander unabhängigen Testergebnissen, die nach derselben Methode bei identischem Testmaterial im selben Laboratorium von demselben Analytiker und mit derselben Ausrüstung binnen eines kurzen Zeitraums ermittelt wurden, liegt in höchstens 5 % aller Fälle über 0,20 g Feuchtigkeit pro 100 g Produkt.
9.2. Vergleichbarkeit
Der absolute Unterschied zwischen zwei einzelnen und voneinander unabhängigen Testergebnissen, die nach derselben Methode bei identischem Testmaterial in verschiedenen Laboratorien von verschiedenen Analytikern und mit unterschiedlicher Ausrüstung ermittelt wurden, liegt in höchstens 5 % aller Fälle über 0,40 g Feuchtigkeit pro 100 g Produkt.
10. PRÜFPROTOKOLL
Das Prüfprotokoll enthält folgende Informationen:
|
—
|
sämtliche Informationen, die zur Bestimmung der Probe benötigt werden;
|
|
—
|
die verwendete Probenahmemethode (wenn bekannt);
|
|
—
|
die verwendete Prüfmethode unter Bezug auf diese internationale Norm;
|
|
—
|
sämtliche Arbeitsbedingungen, die in dieser internationalen Norm nicht genannt sind oder als fakultativ bewertet werden, sowie nähere Informationen zu sämtlichen Vorfällen, die sich auf die Testergebnisse ausgewirkt haben könnten;
|
die ermittelten Testergebnisse sowie — wenn die Wiederholbarkeit geprüft wurde — das ermittelte Endergebnis.
ANHANG XIX
(Artikel 15)
BESTIMMUNG DES FEUCHTIGKEITSGEHALTS VON SAUREM BUTTERMILCHPULVER
1. ANWENDUNGSBEREICH
Ermittlung des Wassergehaltes von saurem Buttermilchpulver für die Tierfütterung
2. KURZBESCHREIBUNG
Die Probe wird unter Vakuum getrocknet und der Gewichtsverlust durch Wiegen ermittelt.
3. APPARATUR
3.1. Analysewaage, Messgenauigkeit 1 mg, Teilung 0,1 mg;
3.2. Schalen aus korrosionsbeständigem Metall oder Glas, mit luftdicht schließenden Deckeln; auf der nutzbaren Fläche muss die Probe so verteilt werden können, dass etwa 0,3 g auf 1 cm2 kommen;
3.3. Einstellbarer, elektrisch beheizter Vakuumofen mit Ölpumpe und einem Mechanismus zur Zuführung heißer Luft, die in einem Turm getrocknet wird, der z. B. mit Calciumoxid oder mit Calciumsulfat gefüllt ist (mit Feuchtigkeitsanzeige);
3.4. Exsikkator mit einem wirksamen Trocknungsmittel;
3.5. Thermostatisierbarer Ofen mit Lüftung, 102 ± 2 °C.
4. VERFAHREN
Eine Schale (3.2) wird zusammen mit dem Deckel im Ofen (3.5) mindestens 1 Stunde lang erhitzt. Nach Aufsetzen des Deckels wird die Schale unverzüglich in einen Exsikkator (3.4) gestellt; die Schale wird herausgenommen und kühlt auf Raumtemperatur ab; anschließend wird die Schale auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert.
Die Schale wird geöffnet, und etwa 5 g der Probe werden auf die Schale gegeben; die Schale wird mit einer Toleranz von 1 mg auf 0,1 mg genau gewogen und das Ergebnis protokolliert. Die Schale wird mit Deckel in den Vakuumofen (3.3) gesetzt und auf 83 °C erwärmt. Um ein übermäßiges Absinken der Ofentemperatur zu verhindern, ist die Schale möglichst rasch in den Ofen zu stellen.
Der Druck wird auf 100 Torr (13,3 kPa) erhöht; die Probe wird bei diesem Druck in einem heißen trockenen Luftstrom zum Trocknen im Ofen belassen, bis sich ein konstantes Gewicht einstellt (etwa nach 4 Stunden).
Die Trocknungszeit wird ab dem Moment gerechnet, in dem der Ofen wieder eine Temperatur von 83 °C erreicht hat. Der Druck im Ofen wird vorsichtig wieder auf Atmosphärendruck reduziert. Nach dem Öffnen des Ofens wird die Schale sofort wieder verschlossen und aus dem Ofen genommen; die Schale wird 30 bis 45 Minuten in einen Exsikkator (3.4) gestellt. Anschließend wird sie auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert. Die Probe wird nochmals 30 Minuten bei 83 °C im Vakuumofen (3.3) getrocknet und erneut gewogen. Die Schale wird so oft erwärmt und neu gewogen, bis die Masse nach dem erneuten Wiegen nur noch um höchstens 1 mg ab- oder zunimmt. Für die Berechnung ist die niedrigste protokollierte Masse anzunehmen.
5. BERECHNUNG
% Feuchtigkeit = (m1 – m2) / (m1 – m0) × 100 %
Dabei ist
|
m0
|
die Masse der Schale mit Deckel;
|
|
m1
|
die Masse von Schale, Deckel und Probeneinwaage vor dem Trocknen;
|
|
m2
|
die Masse von Schale, Deckel und Probeneinwaage nach dem Trocknen.
|
Das Ergebnis ist mit einer Genauigkeit von 0,1 g/100 g zu protokollieren.
6. GENAUIGKEIT
6.1. Wiederholbarkeit
Der absolute Unterschied zwischen zwei einzelnen und voneinander unabhängigen Testergebnissen, die nach derselben Methode bei identischem Testmaterial im selben Laboratorium von demselben Analytiker und mit derselben Ausrüstung binnen eines kurzen Zeitraums ermittelt wurden, liegt in höchstens 5 % aller Fälle über 0,4 g Wasser/100 g Buttermilchpulver.
6.2. Vergleichbarkeit
Der absolute Unterschied zwischen zwei einzelnen und voneinander unabhängigen Testergebnissen, die nach derselben Methode bei identischem Testmaterial in verschiedenen Laboratorien von verschiedenen Analytikern und mit unterschiedlicher Ausrüstung ermittelt wurden, liegt in höchstens 5 % aller Fälle über 0,6 g Wasser/100 g saures Buttermilchpulver.
6.3. Herkunft der Präzisionsdaten
Die Präzisionsdaten stammen aus einem 1995 durchgeführten Versuch, an dem 8 Labors beteiligt waren und in dem 12 Proben (6 Blindproben) untersucht wurden.
ANHANG XX
(Artikel 16)
REFERENZMETHODE ZUR BESTIMMUNG DER REINHEIT VON MILCHFETT DURCH GASCHROMATOGRAFIE — FASSUNG 2
1. GEGENSTAND UND ANWENDUNGSBEREICH
Diese Norm beschreibt eine Referenzmethode zur Bestimmung der Reinheit von Milchfett mittels einer Triglyceridbestimmung durch Gaschromatografie. Nachgewiesen werden können sowohl pflanzliche als auch tierische Fette (z. B. Rindertalg und Schmalz).
Anhand definierter Triglycerid-Formeln wird die Zusammensetzung des Milchfetts geprüft. Grundsätzlich ist diese Methode auf rohe Kuhmilch und auf Erzeugnisse anzuwenden, die aus roher Kuhmilch hergestellt wurden; dabei sind die Fütterungs- und Aufzuchtbedingungen ebenso unerheblich wie eine etwaige Laktation. Nur eine Fütterung mit reinen pflanzlichen Ölen in äußerst großem Umfang (z. B. mit Rapsöl) kann zu falsch-positiven Befunden führen. Milcherzeugnisse einzelner Kühle können ebenfalls falsch-positive Ergebnisse zur Folge haben.
Insbesondere ist die Methode bei der Untersuchung von Fett anzuwenden, das aus Milcherzeugnissen extrahiert wurde, die reines Milchfett in unveränderter Zusammensetzung enthalten sollten (z. B. Butter, Rahm, Milch und Milchpulver). Die technische Behandlung von Milchfett z. B. durch Abtrennung von Cholesterol oder durch Fraktionierung kann falsch-positive Ergebnisse zur Folge haben. Dies gilt auch für Milchfett, das aus Magermilch oder aus Buttermilch gewonnen wurde. Bei aus Käse extrahierten Fetten ist die Methode nicht immer anwendbar, weil der Reifungsprozess die Fettzusammensetzung derart verändern kann, dass falsch-positive Befunde ermittelt werden können.
Anmerkung 1: Buttersäure (n-Buttersäure, C4) kommt ausschließlich in Milchfett vor und ermöglicht quantitative Schätzungen niedriger bis mäßiger Anteile an Milchfett in pflanzlichem und tierischem Fett. Wegen der sehr unterschiedlichen C4-Anteile (Massegehalt ungefähr 3,1 % bis 3,8 %) sind qualitative und quantitative Angaben zu Fremdfetten in reinen Milchfett-Massenfraktionen von bis zu 20 % jedoch problematisch [1].
Anmerkung 2: Quantitative Ergebnisse können in der Praxis nicht aus dem Sterolgehalt von pflanzlichen Fetten abgeleitet werden; diese sind nämlich von den Produktions- und Verarbeitungsbedingungen abhängig. Außerdem ist die qualitative Bestimmung von Fremdfetten mit Sterolen nicht eindeutig.
2. BEGRIFFSBESTIMMUNG
Milchfettreinheit: Freiheit von pflanzlichen und tierischen Fetten, nachgewiesen durch das in dieser Norm genannte Verfahren.
Anmerkung: Die Reinheit wird mit S-Werten ermittelt, die aufgrund der Triglycerid-Zusammensetzung berechnet werden. Die Triglycerid-Massenfraktionen werden in Prozentanteilen ausgedrückt.
3. KURZBESCHREIBUNG
Das aus Milch oder Milcherzeugnissen extrahierte Fett wird durch Gaschromatografie in einer gepackten Säule oder einer kurzen Kapillarsäule analysiert, um die Triglyceride (TG) zu bestimmen; eine Unterscheidung erfolgt nach der Gesamtzahl der Kohlenstoffatome. Durch Einsetzen der Massenfraktion (als Prozentanteil) von Fettmolekülen unterschiedlicher Größen (C24 bis C54, nur Geradzahlige) in geeignete TG-Formeln werden S-Werte berechnet. Eine Überschreitung der mit reinem Milchfett bestimmten Grenzwerte durch die S-Werte gilt als Fremdfett-Nachweis.
Anmerkung 1: Die Eignung und die Gleichwertigkeit von gepackten Säulen und Kapillarsäulen wurde bereits nachgewiesen [2-4].
Anmerkung 2: Der S-Wert ist die Summe von TG-Massenfraktionen multipliziert mit bestimmten definierten Faktoren.
4. REAGENZIEN
Alle Reagenzien müssen analysenrein sein.
4.1. Trägergas: Stickstoff oder Helium oder Wasserstoff; jeweils mit einer Reinheit von mindestens 99,995 %
Fett-Standardproben zur Herstellung einer Milchfett-Standardprobe gemäß Absatz 7.3.3
4.2.1. Triglycerid-Standards, gesättigt; geeignete Erzeugnisse sind im Handel erhältlich.
4.2.2. Cholesterol-Standardlösung
4.3. Methanol (CH3OH), wasserfrei
4.4. n-Hexan (CH3(CH2)4CH3)
4.5. n-Heptan (CH3(CH2)5CH3)
4.6. Sonstige Gase: Wasserstoff, Reinheit mindestens 99,995 %, frei von organischen Verunreinigungen (CnHm <1 µl/l); synthetische Luft, frei von organischen Verunreinigungen (CnHm <1 µl/l)
4.7. Wasserfreies Natriumsulfat (Na2SO4)
5. APPARATUR
Normale Laborausstattung und insbesondere:
5.1. Hochtemperatur-Gaschromatograf
Der Hochtemperatur-Gaschromatograf muss für Temperaturen von mindestens 400 °C ausgelegt und mit einem Flammenionisations-Detektor (FID) ausgerüstet sein. Die im Injektor verwendeten Septa sind gegenüber hohen Temperaturen beständig und zeichnen sich durch sehr geringe Verluste („bleeding“) aus. Wenn ein Gaschromatograf mit Kapillarrohr eingesetzt wird, ist ein On-Column-Injektor zu verwenden. Säule, Injektor und/oder Detektoreinsätze (soweit vorhanden) werden immer unter Verwendung von Graphitdichtungen verbunden.
5.2. Chromatografiesäule
5.2.1. Gepackte Säule
Für die Analyse wird eine Glassäule mit einem Innendurchmesser von 2 mm und einer Länge von 500 mm, gepackt mit einer stationären Phase (3 % OV-1 on 125 bis 150 µm, 100-120 mesh, Gas ChromQ (1) verwendet. Die Vorbereitung, Silanisierung, Packung und Vorbehandlung der gepackten Säule wird in Anhang A beschrieben.
Alternativ kann eine Kapillarsäule verwendet werden (5.2.2).
5.2.2. Kapillarsäule
Für die Analysen wird eine kurze Kapillarsäule (z. B. 5 m) mit nicht-polarer stationärer Phase verwendet, die für Temperaturen bis zu mindestens 400 °C ausgelegt ist (2). Die Säule ist durch 20 Analysen einer Milchfettlösung (7.2) im Laufe von 2 bis 3 Tagen unter Verwendung der in Absatz 7.3.4.2. genannten Einstellungen vorzubehandeln. Anschließend müssen die Kalibrierfaktoren (7.3.3) etwa 1 betragen und in jedem Fall unter 1,20 liegen.
Anmerkung: Säulen mit anderen Abmessungen und eine sonstige nicht-polare, gegenüber hohen Temperaturen beständige Phase können verwendet werden, wenn sie die Anforderungen dieser Norm erfüllen (siehe 7.3.4.2).
5.3. Extrelut-Säule, Fassungsvermögen 1-3 ml, gefüllt mit Silicagel; benötigt zur Extraktion von Milchfett ausschließlich gemäß Absatz 7.1.3
5.4. Graphitdichtungen, beständig gegenüber Temperaturen von mindestens 400 °C; zur Verbindung der GC-Säule sowie des Injektors und/oder der Detektoreinsätze
5.5. Wasserbad; regelbar auf eine konstante Temperatur von 50 °C ± 2 °C
5.6. Ofen, regelbar auf konstante Temperaturen von 50 °C ± 2 °C und 100 °C ± 2 °C
5.7. µl-Pipette
5.8. Messpipette mit einem Fassungsvermögen von 5 ml
5.9. Rundbodenkolben, Fassungsvermögen 50 ml
5.10. Erlenmeyer-Kolben, Nennvolumen 250 ml
5.11. Trichter
5.12. Feinporiges Filterpapier
5.13. Rotationsverdampfer
5.14. Ampullen, Nennvolumen 1 ml, mit Crimp-Verschluss aus Aluminium mit Polytetrafluorethylen-Beschichtung oder mit Schraubverschluss
5.15. Injektionsspritze, bei der der Kolben nicht zur Spitze reicht (Gaschromatograf mit gepackter Säule)
Anmerkung: Mit diesen Spritzen wird bei den Ergebnissen eine bessere Wiederholbarkeit erreicht.
5.16. Analysewaage, Messgenauigkeit 1 mg, Teilung 0,1 mg
6. PROBENAHME
Eine repräsentative Probe wird an das Laboratorium geschickt. Die Probe darf beim Transport und während der Lagerung nicht beschädigt oder verändert worden sein.
Die Probenahme ist nicht Bestandteil der in dieser internationalen Norm beschriebenen Methode. Eine empfohlene Probenahmemethode wird in ISO 707|IDF50 [5] erläutert.
7. VERFAHREN
7.1. Vorbereitung der Testprobe
Die Testprobe ist nach einer der drei folgenden Methoden zur Extraktion von Milchfett vorzubereiten.
7.1.1. Trennung von Butter und Butterschmalz
50 bis 100 g der Testprobe werden in einem Wasserbad (5.5) oder einem Ofen (5.6) bei einer Temperatur von 50 °C geschmolzen. 0,5 bis 1,0 g Natriumsulfat (4.7) werden auf ein Faltenpapierfilter (5.12) gegeben. Ein 250-ml-Erlenmeyer-Kolben (5.10) und ein Trichter (5.11) mit dem eingelegten Filterpapier werden im Ofen (5.6) erwärmt; der Ofen wurde auf 50 °C eingestellt. Nach erfolgter Erwärmung werden Kolben, Trichter und eingelegtes Filterpapier im Ofen belassen; dort wird die Fettschicht der geschmolzenen Probe abfiltriert. Dabei ist darauf zu achten, dass keine Serumübertragung erfolgt.
Wenn die Testprobe nur in einer begrenzten Menge verfügbar ist, kann eine kleinere Testprobe verwendet werden, und das Verfahren sollte entsprechend angepasst werden. Bei Verwendung einer kleineren Probeeinwaage besteht jedoch das erhöhte Risiko, dass die Probe nicht mehr repräsentativ ist.
Anmerkung 1: Aus Rahm kann durch Schlagen und durch Spülen der entstehenden Klümpchen Butter hergestellt werden.
Anmerkung 2: Das mit dem in Absatz 7.1.1 beschriebenen Verfahren erhaltene Milchfett ist nahezu frei von Phospholipiden.
7.1.2. Extraktion nach der gravimetrischen Röse-Gottlieb-Methode
Die Fettfraktion der Testprobe wird nach der in den Normen ISO 1211|IDF001D, ISO 2450|IDF016C oder ISO 7328|IDF116A beschriebenen gravimetrischen Methode extrahiert.
Anmerkung: Wenn das erhaltene Milchfett Phospholipide enthält, erscheint ein um etwa 0,1 % erhöhter Cholesterol-Peak. Die auf 100 % (einschließlich Cholesterol) standardisierte TG-Zusammensetzung wird dadurch nur in vernachlässigbarem Umfang beeinträchtigt.
7.1.3. Extraktion von Milch mit Silicagelsäulen
Mit einer µl-Pipette (5.7) werden 0,7 ml der auf 20 °C temperierten Testprobe in eine Extrelut-Säule mit einem Fassungsvermögen von 1 bis 3 ml (5.3) gegeben. In etwa 5 Minuten verteilt sich die Probe gleichmäßig auf dem Silicagel.
Um die Proteinlipid-Komplexe zu denaturieren, werden mit der Messpipette (5.8) 1,5 ml Methanol (4.3) in die Extrelut-Säule gegeben. Anschließend wird mit 20 ml n-Hexan (4.4) die Fettfraktion aus der Testprobe extrahiert. Das n-Hexan wird langsam in geringen Anteilen hinzugegeben. Das ablaufende Lösungsmittel wird in einem 50-ml-Kolben mit Rundboden (5.9) aufgefangen, der vorher auf eine konstante, bekannte Masse getrocknet wurde; die Masse wurde auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert.
Nach der Extraktion wird gewartet, bis die Flüssigkeit vollständig aus der Säule abgelaufen ist. Die Lösungsmittel werden in einem Rotationsverdampfer (5.13) aus dem Eluat destilliert; das Wasserbad des Verdampfers wurde auf eine Temperatur von 40 bis 50 °C eingestellt. Nachdem die Lösungsmittel ausdestilliert wurden, wird der Rundbodenkolben getrocknet; anschließend wird der Kolben mit Inhalt auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert. Die erhaltene Fettmasse wird bestimmt, indem die Masse des getrockneten leeren Rundbodenkolbens von der ermittelten Masse abgezogen wird.
Anmerkung: Fettextraktionen nach den Methoden von Gerber, Weibull-Berntrop oder Schmid-Bondzynski-Ratzlaff sowie die Abscheidung des Milchfetts durch Fettdetergentien (BDI-Methode) sind für TG-Analysen nicht geeignet, da es bei diesen Methoden zum Übertritt mehr oder weniger großer Mengen an Partialglyceriden oder an Phospholipiden in die Fettphase kommt. Entsprechend ist diese internationale Norm bei bestimmten Produkten, insbesondere bei Käse, nur eingeschränkt anwendbar.
7.2. Vorbereitung der Probenlösung
Zur Gaschromatografie mit einer gepackten Säule wird eine 5 %ige Lösung des Fettes (Volumenfraktion) (hergestellt gemäß Absatz 7.1) in n-Hexan (4.4) oder n-Heptan (4.5) aufgelöst. Abhängig von den Abmessungen der Säule ist bei der On-Column-Injektion in Verbindung mit einer Kapillarsäule eine höchstens 1 %ige Konzentration (0,53 mm, weiter ID) zu verwenden.
Je nach verwendeter Säule und nach der Masse des gemäß der Beschreibung in Absatz 7.1.3 erhaltenen Fetts wird ermittelt, in welcher Menge das Lösungsmittel (4.4 oder 4.5) zur Testprobe in den Kolben zu geben ist; dazu wird das Material auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert. Das übrige Material ist vollständig aufzulösen.
Etwa 1 ml der Probenlösung wird in eine Ampulle (5.14) gegeben.
7.3. Chromatografische Triglycerid-Bestimmung
7.3.1. Basislinien-Verschiebung
Um den Anstieg der Basislinie zu minimieren, wird die Säule vorbereitet, wie in Absatz 5.2.2 (Kapillarsäule) bzw. in Anhang A.4 (gepackte Säule) beschrieben.
Anmerkung: Wegen der hohen Säulentemperatur ergibt sich bei der TG-Analyse besonders leicht ein Anstieg der Basislinie bei hohen Kohlenstoff-Isotopwerten.
7.3.2. Einspritztechnik
7.3.2.1. Gepackte Säule
Zur Vermeidung von Diskriminierungseffekten und Erzielung besserer quantitativer Ergebnisse mit den hochsiedenden Triglyceridkomponenten wird die „Heißeinspritztechnik“ verwendet. Die Nadel wird mit Luft gefüllt, indem die Fettlösung auf die Spritze gezogen wird. Die Nadel wird in das Aufgabesystem eingesetzt. Vor der Injektion wird die Nadel etwa 3 s erhitzt. Danach wird der Inhalt der Spritze umgehend injiziert.
7.3.2.2. Kapillarsäule
Wenn die Aufgabe mit einem kalten On-Column-System (7.3.4.2) erfolgt, wird die Spritzennadel eingeführt und der Inhalt sofort injiziert. Die Verweildauer der Nadel in der Einspritzöffnung sollte so gehalten sein, dass beim Lösungsmittel-Peak kein breites Tailing auftritt.
Anmerkung: Die optimale Verweildauer liegt in der Regel bei etwa 3 s.
7.3.3. Kalibrierung
7.3.3.1. Kurzbeschreibung
Zur Kalibrierung der Testproben wird jeweils am Beginn eines Tages standardisiertes Milchfett zwei- bis dreimal analysiert. Aufgrund der letzten Analyse des standardisierten Milchfetts werden die Kalibrierfaktoren (RFsi
) (Massenfraktion/Flächenfraktion) der TG und des Cholesterols bestimmt und für die folgenden Testproben (siehe Absatz 9.1) angenommen:
 (1)
(1)
Dabei bedeuten:
|
w
si
|
Massenfraktion in Prozent des TG oder des Cholesterols des standardisierten Milchfetts und
|
|
A
si
|
Wert (numerisch) der Peak-Fläche der einzelnen TG oder des Cholesterols im standardisierten Milchfett.
|
Standardisiertes Milchfett mit bekannter TG-Zusammensetzung wird nach den Beschreibungen in Absatz 7.3.3.2 bzw. in Absatz 7.3.3.3 hergestellt.
7.3.3.2. Handelsübliche Milchfett-Standardprobe
Am besten wird der Kalibrierfaktor der einzelnen Bestandteile der Testprobe unter Verwendung eines standardisierten Milchfetts mit zertifizierter TG-Zusammensetzung bestimmt.
Anmerkung: Eine geeignete Norm ist etwa CRM 519 (wasserfreies Milchfett) (zu beziehen über das Institute for Reference Materials and Measurements (IRMM), in Geel, Belgien) (3).
7.3.3.3. Laborstandardprobe Milchfett
Es wird etwa 1 g eines Fettstandard-Gemischs hergestellt (siehe Absatz 4.2); um eine TG-Zusammensetzung ähnlich wie bei Milchfett zu erhalten, werden die gesättigte TG, C24, C30, C36, C42, C48 und C54 und Cholesterol sowie vorzugsweise C50 und C52 verwendet. Das Gewicht wird auf 1 mg gewogen und das Ergebnis auf 0,1 mg genau protokolliert.
Mehrfach wird eine Lösung des Fettstandard-Gemischs in n-Hexan (4.4) oder n-Heptan (4.5) analysiert, wie in Absatz 7.3.4 beschrieben. In der betreffenden Analysereihe ist auch mehrfach Milchfett mit durchschnittlicher Zusammensetzung zu analysieren.
Die TG-Kalibrierfaktoren des Fettstandard-Gemischs werden bestimmt. Vorläufige Kalibrierfaktoren von TG, die in dem Gemisch nicht enthalten sind, können mathematisch interpoliert werden. Die ermittelten Kalibrierfaktoren werden auf das Milchfett übertragen, um eine standardisierte Zusammensetzung zu erhalten. Das so hergestellte standardisierte Milchfett ist bei einer Temperatur von höchstens –18 °C in Stickstoff mehrere Jahre lagerfähig.
7.3.4. Bedingungen für die chromatografische Analyse
Anmerkung: Die Verwendung von gepackten Säulen oder von Kapillarsäulen ergibt gewöhnlich eine Auflösung ähnlich wie in Abbildung 1 dargestellt. Eine Aufteilung der TG mit geraden Isotopzahlen erfolgt normalerweise nicht und ist zu vermeiden.
7.3.4.1. Gepackte Säule
|
a)
|
Temperaturprogramm: Die Ausgangstemperatur des Ofens wird auf 210 °C eingestellt. Diese Temperatur wird 1 Minute aufrechterhalten. Danach wird die Temperatur um 6 °C/min auf 350 °C erhöht. Diese (End-)Temperatur wird 5 Minuten beibehalten.
|
|
b)
|
Temperaturen von Detektor und Injektor jeweils 370 °C
|
|
c)
|
Trägergas: Stickstoff wird mit einer gleich bleibenden Durchflussgeschwindigkeit von etwa 40 ml/min zugeführt. Der Trägergasstrom wird so eingestellt, dass C54 bei 341 °C eluiert wird.
|
|
d)
|
Dauer der Analyse: 29,3 min
|
|
e)
|
Einspritzvolumen: 0,5 µl einer 5 %igen Probenlösung (Volumenfraktion) werden injiziert.
|
Wenn keine TG-Analysen durchgeführt werden, ist die unter Buchstabe a genannte anfängliche Ofentemperatur aufrechtzuerhalten; im Detektor und im Injektor werden die unter Buchstabe b genannten Temperaturen beibehalten, und der Trägergas-Durchfluss wird in der unter Buchstabe c genannten Höhe aufrechterhalten (auch über Nacht und an Wochenenden und arbeitsfreien Tagen). Auf diese Weise ist gewährleistet, dass die Säule bestmöglich funktioniert.
7.3.4.2. Kapillarsäule
|
a)
|
Temperaturprogramm: Die Ausgangstemperatur des Ofens wird auf 80 °C eingestellt. Diese Temperatur wird 0,5 Minuten aufrechterhalten. Danach wird die Temperatur zunächst um 50 °C/min auf 190 °C und dann um 6 °C/min auf 350 °C erhöht. Diese (End-)Temperatur wird 5 Minuten aufrechterhalten.
|
|
b)
|
Detektortemperatur: 370 °C
|
|
c)
|
Trägergas: Stickstoff mit einem Durchfluss von konstant etwa 3 ml/min
|
|
d)
|
Dauer der Analyse: 34,4 min
|
|
e)
|
Einspritzvolumen: 0,5 µl einer 1 %igen Probenlösung (Volumenfraktion) werden injiziert.
|
Diese Einstellungen werden auch im Standby-Betrieb beibehalten, um ein bestmögliches Funktionieren des Systems zu gewährleisten (siehe Absatz 7.3.4.1).
Die in Absatz 7.3.4.2 genannten Analyseeinstellungen sind für eine Säule mit großem Innendurchmesser (0,53 mm) (siehe 5.2.2) vorgesehen. Bei anderen Säulenabmessungen oder Phasen müssen die Einstellungen möglicherweise angepasst werden.
8. INTEGRATION, BEWERTUNG UND KONTROLLE DES ANALYSEVERHALTENS
Die Chromatogramm-Peaks werden mit einem Integrationssystem bewertet, das eine Basislinie erstellen und eine Rückintegrierung ausführen kann. Abbildung 1 zeigt ein ordnungsgemäß integriertes Chromatogramm; das Chromatogramm in Abbildung 2 hingegen weist einen sporadischen Fehler in der Basislinie nach C54 auf, der sich auf die Prozentanteile aller TG auswirkt. Nach C54 auftretende Peaks werden allerdings in der Analyse nicht berücksichtigt.
TG mit ungerader Acyl-C-Zahl (2n + 1) sind mit dem jeweils vorangehenden geradzahligen TG (2n) zu kombinieren. Der niedrige C56-Gehalt wird nicht berücksichtigt. Die Flächen-Prozentanteile der übrigen TG einschließlich Cholesterol werden mit den entsprechenden Kalibrierfaktoren des standardisierten Milchfetts (letze Kalibrierung) multipliziert und insgesamt gemäß Absatz 9.1 auf 100 % normalisiert:
Abbildung 1
Beispiel eines Milchfett-TG-Chromatogramms mit ordnungsgemäßer Basislinie
Abbildung 2
Beispiel eines Milchfett-TG-Chromatogramms mit verschobener Basislinie incorrect baseline

Um die Messbedingungen zu kontrollieren, werden die Werte mit den in Tabelle 1 genannten Variationskoeffizienten (VK, in Prozent) der verschiedenen TG verglichen; die Koeffizienten beruhen auf 19 nacheinander ausgeführten Analysen derselben Milchfettprobe.
Wenn die VK erheblich höher als die in Tabelle 1 genannten Werte sind, müssen die Chromatografiebedingungen als ungeeignet betrachtet werden.
Anmerkung: Die Werte in Tabelle 1 sind nicht bindend und geben nur einen Anhalt für die Qualitätskontrolle.
Auch wenn höhere VK-Werte berücksichtigt werden, müssen die in Absatz 10 genannten Werte für die Wiederholbarkeit und die Reproduzerbarkeit eingehalten werden.
Tabelle 1
Variationskoeffizienten von Triglycerid-Bestandteilen (19 nacheinander durchgeführte Analysen)
|
Triglycerid
|
CV %
|
|
C24
|
10,00
|
|
C 26
|
2,69
|
|
C 28
|
3,03
|
|
C 30
|
1,76
|
|
C 32
|
1,03
|
|
C 34
|
0,79
|
|
C 36
|
0,25
|
|
C 38
|
0,42
|
|
C 40
|
0,20
|
|
C 42
|
0,26
|
|
C 44
|
0,34
|
|
C 46
|
0,37
|
|
C 48
|
0,53
|
|
C 50
|
0,38
|
|
C 52
|
0,54
|
|
C 54
|
0,60
|
9. BERECHNUNG UND DARSTELLUNG DER ERGEBNISSE
9.1. Triglycerid-Zusammensetzung
9.1.1. Berechnung
Die Massenfraktion der einzelnen TG (i = C24, C26, C28, C30, C32, C34, C36, C38, C40, C42, C44, C46, C48, C50, C52 und C54) sowie von Cholesterol wi
werden mit der folgenden Formel berechnet und in Prozent des TG-Gesamtgehalts der Testprobe ausgedrückt:
 (2)
(2)
Dabei bedeutet:
|
Ai
|
Zahlenwert der Peak-Fläche der einzelnen TG der Testprobe und
|
|
RF
si
|
durch Kalibrierung gemäß 7.3.3 ermittelter Kalibrierfaktor der einzelnen TG.
|
9.1.2. Darstellung der Testergebnisse
Die Testergebnisse sind auf zwei Dezimalstellen genau anzugeben.
9.2. S-Werte
9.2.1. Berechnung
9.2.1.1. Der S-Wert in Prozent wird berechnet, indem der berechnete Wert für wi
(9.1.1) der betreffenden TG-Prozentanteile in die Formeln 3 bis 7 eingesetzt wird. Alle Formeln sind unabhängig von der Art der mutmaßlichen Fremdfette zu verwenden.
9.2.1.2. Formeln für Sojaöl, Sonnenblumenöl, Olivenöl, Rapsöl, Leinöl, Weizenkeimöl, Maiskeimöl, Baumwollöl und Fischöl
S = 2,098 3 · w
C30 + 0,728 8 · w
C34 + 0,692 7 · w
C36 + 0,635 3 · w
C38 + 3,745 2 · w
C40 – 1,292 9 · w
C42 + 1,354 4 · w
C44 + 1,701 3 · w
C46 + 2,528 3 · w
C50 (3)
9.2.1.3. Kokos- und Palmkernfett
S = 3,745 3 · w
C32 + 1,113 4 · w
C36 + 1,364 8 · w
C38 + 2,154 4 · w
C42 + 0,427 3 · w
C44 + 0,580 9 · w
C46 + 1,292 6 · w
C48 + 1,030 6 · w
C50 + 0,995 3 · w
C52 + 1,239 6 · w
C54 (4)
9.2.1.4. Palmöl und Rindertalg
S = 3,664 4 · w
C28 + 5,229 7 · w
C30 – 12,507 3 · w
C32 + 4,428 5 · w
C34 – 0,201 0 · w
C36 + 1,279 1 · w
C38 + 6,743 3 · w
C40 – 4,271 4 · w
C42 +6,373 9 · w
C46 (5)
9.2.1.5. Schmalz
S = 6,512 5 · w
C26 + 1,205 2 · w
C32 + 1,733 6 · w
C34 + 1,755 7 · w
C36 + 2,232 5 · w
C42 + 2,800 6 · w
C46 + 2,543 2 · w
C52 + 0,989 2 · w
C54 (6)
9.2.1.6. Gesamt
S = – 2,757 5 · w
C26 + 6,407 7 · w
C28 + 5,543 7 · w
C30 – 15,324 7 · w
C32 + 6,260 0 · w
C34 + 8,010 8 · w
C40 – 5,033 6 · w
C42 + 0,635 6 · w
C44 + 6,017 1 · w
C46 (7)
9.2.2. Darstellung der Testergebnisse
Die Testergebnisse sind auf zwei Dezimalstellen genau anzugeben.
9.3. Nachweis von Fremdfett
Die fünf gemäß Absatz 9.2.1 ermittelten S-Werte werden mit den entsprechenden S-Grenzwerten in Tabelle 2 verglichen.
Die Testprobe ist als reines Milchfett zu betrachten, wenn alle fünf S-Werte innerhalb der in Tabelle 2 genannten Grenzwerte liegen. Überschreitet ein S-Wert die betreffenden Grenzwerte, ist davon auszugehen, dass die Probe Fremdfett enthält.
Mit einigen der unter 3 bis 6 genannten Formeln können bestimmte Fremdfette zwar leichter nachgewiesen werden als mit der Formel für den Gesamtfettanteil (7) (siehe Tabelle B.1); ein positiver Befund mit nur einer der Formeln 3 bis 6 lässt jedoch keine Rückschlüsse auf die Art des Fremdfetts zu.
In Anhang B wird ein Verfahren zur Berechnung des Gehalts an pflanzlichem und tierischem Fett in verfälschtem Milchfett beschrieben. Dieses Verfahren wurde nicht validiert und wird nur zur Information erläutert.
Tabelle 2
S-Grenzwerte für reine Milchfette
|
Fremdfett
|
Formel
|
S-Grenzwerte (4)
|
|
Sojaöl, Sonnenblumenöl, Olivenöl, Rapsöl, Leinöl, Weizenkeimöl, Maiskeimöl, Baumwollöl und Fischöl
|
(3)
|
98,05 bis 101,95
|
|
Kokos- und Palmkernfett
|
(4)
|
99,42 bis 100,58
|
|
Palmöl und Rindertalg
|
(5)
|
95,90 bis 104,10
|
|
Schmalz
|
(6)
|
97,96 bis 102,04
|
|
Gesamt
|
(7)
|
95,68 bis 104,32
|
10. GENAUIGKEIT
10.1. Leistungstest
Die Werte für die Wiederholbarkeit und die Vergleichbarkeit wurden mit den Formeln 3 bis 7 für reines Milchfett bestimmt und sind auf andere als die hier genannten Matrizen unter Umständen nicht anwendbar.
10.2. Wiederholbarkeit
Der absolute Unterschied zwischen zwei einzelnen Testergebnissen, die nach derselben Methode bei identischem Testmaterial im selben Laboratorium von demselben Analytiker und mit derselben Ausrüstung binnen eines kurzen Zeitraums ermittelt wurden, liegt in höchstens 5 % aller Fälle über den in Tabelle 3 genannten Grenzwerten.
Tabelle 3
Wiederholbarkeit, r, für die Formeln 3 bis 7
|
Fremdfett
|
Formel
|
r:%
|
|
Sojaöl, Sonnenblumenöl, Olivenöl, Rapsöl, Leinöl, Weizenkeimöl, Maiskeimöl, Baumwollöl und Fischöl
|
(3)
|
0,67
|
|
Kokos- und Palmkernfett
|
(4)
|
0,12
|
|
Palmöl und Rindertalg
|
(5)
|
1,20
|
|
Schmalz
|
(6)
|
0,58
|
|
Gesamt
|
(7)
|
1,49
|
10.3. Vergleichbarkeit
Der absolute Unterschied zwischen zwei einzelnen Testergebnissen, die nach derselben Methode bei identischem Testmaterial in verschiedenen Laboratorien von verschiedenen Analytikern und mit unterschiedlicher Ausrüstung ermittelt wurden, liegt in höchstens 5 % aller Fälle über den in Tabelle 4 genannten Grenzwerten.
Tabelle 4
Vergleichbarkeit, R, für die Formeln 3 bis 7
|
Fremdfett
|
Formel
|
R%
|
|
Sojaöl, Sonnenblumenöl, Olivenöl, Rapsöl, Leinöl, Weizenkeimöl, Maiskeimöl, Baumwollöl und Fischöl
|
(3)
|
1,08
|
|
Kokos- und Palmkernfett
|
(4)
|
0,40
|
|
Palmöl und Rindertalg
|
(5)
|
1,81
|
|
Schmalz
|
(6)
|
0,60
|
|
Gesamt
|
(7)
|
2,07
|
11. UNSICHERHEIT DER MESSUNGEN
Aus der Wiederholbarkeit, r, und der Vergleichbarkeit, R, kann die erweiterte Messunsicherheit eines S-Werts berechnet werden.
Die Berücksichtigung der erweiterten Messunsicherheit (aufgrund von Doppelanalysen) bei den S-Grenzwerten in Tabelle 2 führt zu erweiterten S-Grenzwerten; diese erweiterten Werte sind in Tabelle 5 zusammengestellt:
Tabelle 5
Erweiterte S-Grenzwerte für reine Milchfette unter Berücksichtigung der erweiterten Messunsicherheit
|
Fremdfett
|
Formel
|
Erweiterte S-Grenzwerte
|
|
Sojaöl, Sonnenblumenöl, Olivenöl, Rapsöl, Leinöl, Weizenkeimöl, Maiskeimöl, Baumwollöl und Fischöl
|
(3)
|
97,36 bis 102,64
|
|
Kokos- und Palmkernfett
|
(4)
|
99,14 bis 100,86
|
|
Palmöl und Rindertalg
|
(5)
|
94,77 bis 105,23
|
|
Schmalz
|
(6)
|
97,65 bis 102,35
|
|
Gesamt
|
(7)
|
94,42 bis 105,58
|
12. PRÜFPROTOKOLL
Das Prüfprotokoll enthält folgende Informationen:
|
—
|
sämtliche Informationen, die zur Bestimmung der Probe benötigt werden;
|
|
—
|
die verwendete Probenahmemethode (wenn bekannt);
|
|
—
|
die verwendete Prüfmethode unter Bezug auf diese internationale Norm;
|
|
—
|
sämtliche Arbeitsbedingungen, die in dieser internationalen Norm nicht genannt sind oder als fakultativ bewertet werden, sowie nähere Informationen zu sämtlichen Vorfällen, die sich auf die Testergebnisse ausgewirkt haben könnten;
|
|
—
|
die ermittelten Testergebnisse sowie — wenn die Wiederholbarkeit geprüft wurde — das ermittelte Endergebnis.
|
(1) Beispiel eines geeigneten handelsüblichen Produkts; diese Angabe soll Benutzern dieser internationalen Norm zur Orientierung dienen und ist nicht als Befürwortung der Verwendung dieses Produktes zu verstehen.
(2) CP-Ultimetal SimDist (5 m x 0,53 mm × 0,17 µm) z. B. ist ein handelsübliches geeignetes Produkt. Diese Angabe soll Benutzern dieser internationalen Norm zur Orientierung dienen und ist nicht als Befürwortung der Verwendung dieses Produktes zu verstehen.
(3) Beispiel eines geeigneten handelsüblichen Produkts; diese Angabe soll Benutzern dieser internationalen Norm zur Orientierung dienen und ist nicht als Befürwortung der Verwendung dieses Produktes zu verstehen.
(4) Berechnet mit einem Konfidenzintervall von 99 %; die Zugabe von Fremdfetten ist nur dann anzunehmen, wenn die Nachweisgrenzen der betreffenden Formel überschritten werden (siehe Tabelle B.1).
ANHANG A
(normativ)
VORBEREITUNG DER GEPACKTEN SÄULE
A.1. REAGENZIEN UND APPARATUR
A.1.1. Toluol (C6H5CH3)
A.1.2. Dimethyldichlorsilan-Lösung [Si(CH3)2Cl2]
50 ml Dimethyldichlorsilan werden in 283 ml Toluol (A.1.1) aufgelöst.
A.1.3. Kakaobutter-Lösung mit einer Massenfraktion von 5 % Kakaobutter in n-Hexan (4.4) oder n-Heptan (4.5)
A.1.4. Stationäre Phase, 3 % OV-1 auf 125 bis 150 µm (100 bis 120 mesh) Gas ChromQ (1)
Anmerkung: Die Korngrößenangabe wurde gemäß BS 410 (alle Teile) in Mikrometer umgerechnet [6].
A.1.5. Glassäule, Innendurchmesser 2 mm, Länge 500 mm, U-förmig
Apparatur, zum Befüllen der gepackten Säule
A.1.6.1. Aufgabesäule mit Schraubkappen, versehen mit einer Markierung, bis zu der die Säule mit der stationären Phase zu befüllen ist
A.1.6.2. Feines Sieb, Maschenweite etwa 100 µm, mit Schraubkappe zum Verschließen der Glassäule wie in Abbildung A.3 dargestellt
A.1.6.3. Silanisierte Glaswolle, deaktiviert
A.1.6.4. Vibrator zur gleichmäßigen Verteilung der stationären Phase beim Befüllen
A.1.6.5. Silanisierungssysteme, zum Silanisieren der Glasoberfläche der Säule
A.1.6.6. Woulff-Flasche
A.1.6.7. Wasser-Absaugpumpe
A.2. SILANISIERUNG (DEAKTIVIERUNG DER GLASOBERFLÄCHE)
Nach dem Anschließen der Woulff-Flasche (A.1.6.6) an die Wasserstrahlpumpe (A.1.6.7) wird Schlauch 2 (siehe Abbildung A.1) in die Dimethyldichlorsilan-Lösung (A.1.2) getaucht. Die Säule (A.1.5) wird mit dieser Lösung befüllt, indem der Absperrhahn geschlossen wird. Nach dem erneuten Öffnen des Absperrhahns werden die beiden Schläuche wieder herausgenommen. Die Säule wird auf einem Ständer befestigt. Anschließend wird die Säule mit einer Pipette vollständig mit Dimethyldichlorsilan-Lösung (A.1.2) gefüllt.
Abbildung A.1
Aufbau für die Silanisierung
Legende
|
6
|
Dimethyldichlorsilan und Toluol
|
Die Säule wird 20 bis 30 Minuten stehen gelassen. Danach wird die Woulff-Flasche durch einen Saugflasche ersetzt. Die Säule wird entleert, indem sie an die Wasserstrahlpumpe (A.1.6.7) angeschlossen wird (siehe Abbildung A.2). Die entleerte Säule wird anschließend mit 75 ml Toluol (A.1.1) und mit 50 ml Methanol (4.3) gespült, indem Schlauch 2 in das Lösungsmittel getaucht wird. Die gespülte Säule wird bei einer Temperatur von 100 °C etwa 30 Minuten im Ofen (5.6) getrocknet.
Abbildung A.2
Spül-Apparatur
Legende
A.3. BEFÜLLEN
Die Säule wird mit der in Abbildung A.3 dargestellten Apparatur befüllt. Die stationäre Phase (A.1.4) wird bis zur Marke in die Aufgabesäule (A.1.6.1) gegeben. Der Boden der zu befüllenden Glassäule wird mit einem etwa 1 cm langen Stopfen aus silanisierter und komprimierter Glaswolle (A.1.6.3) verschlossen. Die Säule wird mit einem feinen Sieb (A.1.6.2) verschlossen.
Abbildung A.3
Befüllen der Glassäule

Legende
|
2
|
Aufgabesäule, bis zur Marke mit OV-1 zu füllen
|
|
4
|
Schraubkappe mit Filter; hier liegt der durch die Glasfaser und die stationäre Phase ausgeübte Druck an.
|
Die Säule wird unter Druck (300 kPa unter Stickstoffzufuhr) mit der stationären Phase befüllt. Zur Erzielung einer homogenen, gleichmäßigen und festen Packung ist während des Befüllens ein Vibrator in der Säule auf- und abzuführen. Nach dem Befüllen wird ein fester Stopfen aus silanisierter Glaswolle (A.1.6.3) auf das andere Ende der gepackten Säule gesetzt. Das überstehende Material wird abgeschnitten. Der Stopfen wird mit einem Spatel einige Millimeter in die Säule gedrückt.
A.4. VORBEHANDLUNG
Um Verunreinigungen auszuschließen, darf das hintere Ende der Säule während der Durchführung der Schritte a bis c nicht mit dem Detektor verbunden werden. Die befüllte Säule (A.3) ist wie folgt vorzubehandeln:
|
a)
|
Die Säule wird 15 Minuten mit Stickstoff gespült (Durchfluss 40 ml/min); der GC-Ofen wird auf 50 °C eingestellt;
|
|
b)
|
die Säule wird mit 1 °C/min auf 355 °C erwärmt; der Stickstoffdurchfluss beträgt 10 ml/min;
|
|
c)
|
die Säulentemperatur von 355 °C wird 12 bis 15 Stunden aufrechterhalten;
|
|
d)
|
in die Säule wird zweimal 1 µl Kakaobutter-Lösung (A.1.3) injiziert; dabei ist das in Absatz 7.3.4.1 genannte Temperaturprogramm für die gepackte Säule zu berücksichtigen;
Anmerkung: Kakaobutter besteht nahezu ausschließlich aus den hoch siedenden C50- bis C56-Triglyceriden und reduziert daher den erforderlichen Aufwand für die Vorbehandlung der Säule entsprechend den jeweiligen Kalibrierfaktoren.
|
|
e)
|
im Laufe von 2 bis 3 Tagen werden zwanzigmal jeweils 0,5 µl einer gemäß 7.2 hergestellten Milchfettlösung injiziert; dabei sind die in Absatz 7.3.4.1 genannten Einstellungen für die gepackte Säule vorzunehmen.
|
—
|
Zur Analyse von Testproben dürfen ausschließlich Säulen mit Kalibrierfaktoren von etwa 1 verwendet werden. Die Kalibrierfaktoren sollten nicht größer als 1,20 sein.
|
|
(1) Beispiel eines geeigneten handelsüblichen Produkts; diese Angabe soll Benutzern dieser internationalen Norm zur Orientierung dienen und ist nicht dahin gehend auszulegen, dass die ISO oder der |IDF dieses Produkt unterstützen würde.
ANHANG B
(zur Information)
QUANTIFIZIERUNG DES FREMDFETTGEHALTS
B.1. KURZBESCHREIBUNG
In Tabelle B.1 sind die mit einem Konfidenzintervall von 99 % berechneten Nachweisgrenzen für verschiedene Fremdfette zusammengestellt. Der mittleren Spalte sind die Nachweisgrenzen der jeweils besten Formel unter den Einzelformeln 3 bis 6 zu entnehmen.
Die Nachweisgrenzen der Gesamtformel 7 (in der rechten Spalte) sind etwas höher. Prinzipiell wird Formel 7 nur zur Quantifizierung von Fremdfetten benötigt.
Mit allen Formeln können auch Kombinationen der verschiedenen Fremdfette nachgewiesen werden. Die Schwankungsbreiten der Triglyceride der verschiedenen Fremdfette eines Typs haben keinen größeren Einfluss auf die Nachweisgrenzen.
Sowohl bei Verwendung der Einzelformeln als auch bei der Gesamtformel sind die Nachweisgrenzen der Einzelformeln zu beachten. In bestimmten Fällen wird jedoch der S-Wert der Gesamtformel zur Quantifizierung benötigt (B.2).
Tabelle B.1
Grenzwerte für den Nachweis der Zugabe von Fremdfetten zu Milch in Prozent (Konfidenzintervall 99 %)
|
Fremdfett
|
Einzelformel
%
|
Gesamtformel
%
|
|
Sojaöl
|
2,1
|
4,4
|
|
Sonnenblumenöl
|
2,3
|
4,8
|
|
Olivenöl
|
2,4
|
4,7
|
|
Kokosfett
|
3,5
|
4,3
|
|
Palmöl
|
4,4
|
4,7
|
|
Palmkernfett
|
4,6
|
5,9
|
|
Rapsöl
|
2,0
|
4,4
|
|
Leinöl
|
2,0
|
4,0
|
|
Weizenkeimöl
|
2,7
|
6,4
|
|
Maiskeimöl
|
2,2
|
4,5
|
|
Baumwollöl
|
3,3
|
4,4
|
|
Schmalz
|
2,7
|
4,7
|
|
Rindertalg
|
5,2
|
5,4
|
|
Hydrogenisiertes Fischöl
|
5,4
|
6,1
|
B.2. BERECHNUNG
Eine quantitative Fremdfettbestimmung ist nur dann vorzunehmen, wenn mindestens einer der S-Grenzwerte (Tabellen 2 oder 5) überschritten wird. Um quantitative Informationen zu erhalten, werden die in der verwendeten Testprobe enthaltene Massenfraktion des Fremdfetts oder der Fremdfettmischung in Prozent w
f mit der folgenden Formel berechnet:

Dabei bedeuten:
|
S
|
unter Verwendung von TG-Daten von Milchfett, zu dem ein Fremdfrett oder eine Fremdfettmischung hinzugegeben wurde, mit einer der Formeln 3 bis 7 ermitteltes Ergebnis;
|
|
S
f
|
eine vom Typ des zugesetzten Fremdfetts abhängige Konstante.
|
Wenn der Typ des zum Milchfett zugesetzten Fremdfetts nicht bekannt ist, wird ein allgemeiner S
f-Wert von 7,46 (Tabelle B.2) verwendet. Der mit Formel 7 ermittelte S-Wert ist selbst dann anzunehmen, wenn nicht die S-Werte dieser Formel, sondern die Grenzwerte einer anderen Formel überschritten werden.
Wenn die Fremdfetttypen bekannt sind, werden die jeweiligen S
f-Werte (Tabelle B.2) in Formel B.1 eingesetzt. Zur Berechnung von S wird die betreffende Fremdfettformel unter den Formeln 3 bis 6 ausgewählt.
Tabelle B.2
Sf-Werte verschiedener Fremdfette
|
Fremdfett
|
Sf
|
|
Unbekannt
|
7,46
|
|
Sojaöl
|
8,18
|
|
Sonnenblumenöl
|
9,43
|
|
Olivenöl
|
12,75
|
|
Kokosöl
|
118,13
|
|
Palmöl
|
7,55
|
|
Palmkernfett
|
112,32
|
|
Rapsöl
|
3,30
|
|
Leinöl
|
4,44
|
|
Weizenkeimöl
|
27,45
|
|
Maiskeimöl
|
9,29
|
|
Baumwollöl
|
41,18
|
|
Schmalz
|
177,55
|
|
Rindertalg
|
17,56
|
|
Fischöl
|
64,12
|
B.3. DARSTELLUNG DER TESTERGEBNISSE
Die Testergebnisse sind auf zwei Dezimalstellen genau anzugeben.
Literatur
|
(1)
|
Molkentin, J., Precht, D. Representative determination of the butyric acid content in European milk fats. Milchwissenschaft, 52, 1987, S. 82-85.
|
|
(2)
|
Precht, D., Molkentin, J. Quantitative triglyceride analysis using short capillary columns. Chrompack News, 4, 1993, S. 16-17.
|
|
(3)
|
Molkentin, J., Precht, D. Comparison of packed and capillary columns for quantitative gas chromatography of triglycerides in milk fat. Chromatographia, 39, 1994, S. 265-270.
|
|
(4)
|
Molkentin, J., Precht, D. Equivalence of packed and capillary GC columns with respect to suitability for foreign fat detection in butter using the triglyceride formula method. Chromatographia, 52, 2000, S. 791-797.
|
|
(5)
|
ISO 707|IDF50, Milch und Milchprodukte — Leitfaden zur Probenahme.
|
|
(6)
|
BS 410:1988, Test sieves — Technical requirements and testing.
|
|
(7)
|
Precht, D. Control of milk fat purity by gas chromatographic triglyceride analysis. Kieler Milchwirtsch. Forschungsber., 43, 1991, S. 219-242.
|
|
(8)
|
Precht, D. Detection of adulterated milk fat by fatty acid and triglyceride analyses. Fat Sci. Technol., 93, 1991, S. 538-544.
|
|
(9)
|
DIN 10336:1994, Nachweis und Bestimmung von Fremdfetten in Milchfett anhand einer gaschromatografischen Triglyceridanalyse.
|
|
(10)
|
Kommission der Europäischen Gemeinschaften: Auswertung der Ergebnisse des ersten, zweiten, dritten, vierten, fünften und sechsten EG-Ringversuchs: Nachweis von Triglyceriden in Milchfett; Dok. Nr. VI/2644/91, VI/8.11.91, VI/1919/92, VI 3842/92. VI/5317/92, VI/4604/93.
|
|
(11)
|
Molkentin, J. Detection of foreign fat in milk fat from different continents by triacylglycerol analysis. Eur. J. Lipid Sci. Technol., 109, 2007, S. 505-510.
|
ANHANG XXI
(Artikel 18)
VERFAHREN BEI STRITTIGEN ANALYSEERGEBNISSEN (CHEMISCHE ANALYSE)
1. Auf Antrag eines Erzeugers wird in einem anderen von der zuständigen Stelle zugelassenen Laboratorium eine weitere Analyse nach der jeweiligen Methode durchgeführt; wenn versiegelte Duplikatproben des Erzeugnisses vorliegen und bei den zuständigen Stellen unter angemessenen Bedingungen gelagert worden sind. Der Antrag ist binnen sieben Werktagen nach Notifizierung der Befunde der ersten Analyse zu stellen. Die Analyse wird binnen 21 Werktagen nach Eingang des Antrags durchgeführt. Die zuständige Stelle übersendet diese Proben auf Antrag und Kosten des Marktteilnehmers an ein zweites Laboratorium. Dieses muss zur Durchführung amtlicher Analysen ermächtigt sein und nachgewiesene Befähigung für solche Analysen besitzen.
2. Für die erweiterten Messunsicherheiten (k = 2) des Mittelwerts 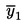 der
der  wiederholten Messungen in Laboratorium 1 und des Mittelwerts
wiederholten Messungen in Laboratorium 1 und des Mittelwerts  der
der 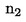 wiederholten Messungen in Laboratorium 2 gilt:
wiederholten Messungen in Laboratorium 2 gilt:
3.  und
und  ; dabei ist
; dabei ist 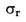 die Wiederholstandardabweichung und
die Wiederholstandardabweichung und  die Vergleichsstandardabweichung der jeweiligen Methode. Wenn das Endergebnis y der Messung in den Laboratorien mit einer Formel der Form
die Vergleichsstandardabweichung der jeweiligen Methode. Wenn das Endergebnis y der Messung in den Laboratorien mit einer Formel der Form  ,
,  ,
,  oder
oder  berechnet wird, sind die üblichen Verfahren zur Kombination von Standardabweichungen in derartigen Fällen einzuhalten, um die Unsicherheit zu ermitteln.
berechnet wird, sind die üblichen Verfahren zur Kombination von Standardabweichungen in derartigen Fällen einzuhalten, um die Unsicherheit zu ermitteln.
4. Um zu prüfen, ob die Ergebnisse der beiden Laboratorien für die Methode im Bereich der Vergleichsstandardabweichung  liegen, wird die erweiterte Messunsicherheit der Differenz
liegen, wird die erweiterte Messunsicherheit der Differenz  berechnet:
berechnet:
5.  Wenn der absolute Wert der Differenz der Mittelwerte der Laboratorien,
Wenn der absolute Wert der Differenz der Mittelwerte der Laboratorien, 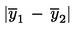 , nicht größer ist als die Unsicherheit
, nicht größer ist als die Unsicherheit  ,
,
 ,
,
erfüllen die Ergebnisse der beiden Laboratorien die Anforderungen an die Vergleichsstandardabweichung,  und das arithmetische Mittel der Mittelwerte der beiden Laboratorien,
und das arithmetische Mittel der Mittelwerte der beiden Laboratorien,
 ,
,
wird als Endergebnis angenommen. Die erweiterte Messunsicherheit beträgt
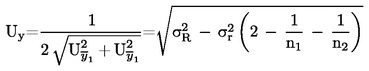 .
.
Die betreffende Partie wird als nicht konform mit einem oberen rechtlich zulässigen Grenzwert UL zurückgewiesen, wenn
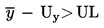 ;
;
ansonsten wird die Partie als konform mit UL betrachtet.
Die Partie wird als nicht konform mit einem unteren rechtlich zulässigen Grenzwert LL zurückgewiesen, wenn
 ;
;
ansonsten wird die Partie als konform mit LL betrachtet.
Wenn der absolute Wert der Differenz der Mittelwerte der beiden Laboratorien, 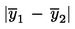 , größer als die betreffende Unsicherheit
, größer als die betreffende Unsicherheit 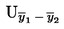 ist, gilt:
ist, gilt:
 ,
,
Die Ergebnisse der beiden Laboratorien erfüllen die Anforderungen an die Vergleichsstandardabweichung nicht.
In diesem Fall wird die Partie als nicht konform zurückgewiesen, wenn die zweite Analyse die erste Analyse bestätigt. Ansonsten wird die Partie als konform angenommen.
Das Endergebnis teilt die zuständige Stelle dem Erzeuger möglichst umgehend mit. Die Kosten der zweiten Analyse trägt der Erzeuger nur dann, wenn die Partie zurückgewiesen wurde.
ANHANG XXII
KORRELATIONSTABELLE
|
Verordnung (EG) Nr. 213/2001
|
Diese Verordnung
|
|
Artikel 1
|
Artikel 1
|
|
Artikel 2
|
Artikel 1
|
|
Artikel 3
|
Artikel 2
|
|
—
|
Artikel 3
|
|
Artikel 4
|
—
|
|
Artikel 5
|
—
|
|
Artikel 6
|
Artikel 4
|
|
Artikel 7
|
Artikel 18
|
|
Artikel 8
|
—
|
|
Artikel 9
|
Artikel 5
|
|
Artikel 10
|
Artikel 6
|
|
Artikel 11
|
Artikel 7
|
|
Artikel 12
|
Artikel 8
|
|
Artikel 13
|
Artikel 9
|
|
Artikel 14
|
Artikel 10
|
|
Artikel 15
|
Artikel 11
|
|
Artikel 16
|
Artikel 12
|
|
Artikel 17
|
Artikel 13
|
|
—
|
Artikel 14
|
|
Artikel 18
|
Artikel 15
|
|
Artikel 19
|
Artikel 16
|
|
|
Artikel 17
|
|
|
Artikel 19
|
|
Artikel 20
|
—
|
|
Artikel 21
|
—
|
|
Artikel 22
|
Artikel 20
|
|
Artikel 23
|
Artikel 21
|