

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 01991R2568-20161204
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
Consolidated text: Verordnung (EWG) Nr. 2568/91 der Kommission vom 11. Juli 1991 über die Merkmale von Olivenölen und Oliventresterölen sowie die Verfahren zu ihrer Bestimmung
Verordnung (EWG) Nr. 2568/91 der Kommission vom 11. Juli 1991 über die Merkmale von Olivenölen und Oliventresterölen sowie die Verfahren zu ihrer Bestimmung

01991R2568 — DE — 04.12.2016 — 031.005
Dieser Text dient lediglich zu Informationszwecken und hat keine Rechtswirkung. Die EU-Organe übernehmen keine Haftung für seinen Inhalt. Verbindliche Fassungen der betreffenden Rechtsakte einschließlich ihrer Präambeln sind nur die im Amtsblatt der Europäischen Union veröffentlichten und auf EUR-Lex verfügbaren Texte. Diese amtlichen Texte sind über die Links in diesem Dokument unmittelbar zugänglich
|
VERORDNUNG (EWG) Nr. 2568/91 DER KOMMISSION vom 11. Juli 1991 über die Merkmale von Olivenölen und Oliventresterölen sowie die Verfahren zu ihrer Bestimmung (ABl. L 248 vom 5.9.1991, S. 1) |
Geändert durch:
Berichtigt durch:
VERORDNUNG (EWG) Nr. 2568/91 DER KOMMISSION
vom 11. Juli 1991
über die Merkmale von Olivenölen und Oliventresterölen sowie die Verfahren zu ihrer Bestimmung
Artikel 1
(1) Native Olivenöle im Sinne von Nummer 1 Buchstaben a) und b) des Anhangs der Verordnung Nr. 136/66/EWG sind Öle, deren Merkmale mit den in Anhang I Nummern 1 und 2 dieser Verordnung genannten Merkmalen übereinstimmen.
(2) Lampantöl im Sinne von Nummer 1 Buchstabe c) des Anhangs der Verordnung Nr. 136/66/EWG ist Öl, dessen Merkmale mit den in Anhang I Nummer 3 dieser Verordnung genannten Merkmalen übereinstimmen.
(3) Raffiniertes Olivenöl im Sinne von Nummer 2 des Anhangs der Verordnung Nr. 136/66/EWG ist Olivenöl, dessen Merkmale mit den in Anhang I Nummer 4 dieser Verordnung genannten Merkmalen übereinstimmen.
(4) Olivenöl, bestehend aus raffiniertem und nativem Olivenöl, im Sinne von Nummer 3 des Anhangs der Verordnung Nr. 136/66/EWG ist Öl, dessen Merkmale mit den in Anhang I Nummer 5 dieser Verordnung genannten Merkmalen übereinstimmen.
(5) Rohes Oliventresteröl im Sinne von Nummer 4 des Anhangs der Verordnung Nr. 136/66/EWG ist Olivenöl, dessen Merkmale mit den in Anhang I Nummer 6 dieser Verordnung genannten Merkmalen übereinstimmen.
(6) Raffiniertes Oliventresteröl im Sinne der Nummer 5 des Anhangs der Verordnung Nr. 136/66/EWG ist Olivenöl, dessen Merkmale mit den in Anhang I Nummer 7 dieser Verordnung genannten Merkmalen übereinstimmen.
(7) Oliventresteröl im Sinne von Nummer 6 des Anhangs der Verordnung Nr. 136/66/EWG ist Olivenöl, dessen Merkmale mit den in Anhang I Nummer 8 dieser Verordnung genannten Merkmalen übereinstimmen.
Artikel 2
(1) Die Merkmale der Öle gemäß Anhang I dieser Verordnung werden nach folgenden Analysenverfahren bestimmt:
a) freie Fettsäuren, berechnet in Prozent Ölsäure, nach dem Verfahren des Anhangs II;
b) Peroxidzahl nach dem Verfahren des Anhangs III;
c) Wachsgehalt nach dem Verfahren des Anhangs IV;
d) Zusammensetzung und Gehalt an Sterinen und Triterpen-Dialkoholen durch Kapillar-Gaschromatographie nach dem Verfahren des Anhangs V;
e) prozentualer Anteil an 2-Glycerinmonopalmitat nach dem Verfahren des Anhangs VII;
f) spektrophotometrische Analyse nach dem Verfahren des Anhangs IX;
g) Fettsäurezusammensetzung nach dem Verfahren des Anhangs X;
h) flüchtige halogenierte Lösungsmittel nach dem Verfahren des Anhangs XI;
i) Bestimmung der organoleptischen Merkmale nativer Olivenöle nach dem Verfahren des Anhangs XII;
j) Stigmasterinnachweis nach dem Verfahren des Anhangs XVII;
k) Bestimmung der Zusammensetzung der Triglyceride mit ECN42 nach dem Verfahren des Anhangs XVIII;
l) Gehalt an aliphatischen und Triterpenalkoholen nach dem Verfahren des Anhangs XIX;
m) Gehalt an Wachsen, Fettsäuremethylestern und Fettsäureethylestern nach dem Verfahren des Anhangs XX.
▼M28 —————
(2) Die Prüfung der organoleptischen Merkmale nativer Olivenöle durch die einzelstaatlichen Behörden oder ihre Vertreter wird von durch die Mitgliedstaaten zugelassenen Prüfergruppen vorgenommen.
Die organoleptischen Merkmale eines in Unterabsatz 1 genannten Olivenöls werden als mit der deklarierten Olivenölkategorie übereinstimmend angesehen, wenn eine von dem betreffenden Mitgliedstaat zugelassene Prüfergruppe die diesbezügliche Einstufung bestätigt.
Bestätigt die zugelassene Prüfergruppe die organoleptischen Merkmale der deklarierten Olivenölkategorie nicht, so fordern die einzelstaatlichen Behörden oder ihre Vertreter auf Antrag des Betroffenen zwei Gegenanalysen anderer zugelassener Prüfergruppen an, von denen mindestens eine von einer Prüfergruppe vorgenommen wird, die von dem betreffenden Erzeugermitgliedstaat zugelassen wurde. Die fraglichen Merkmale gelten als mit den deklarierten Merkmalen konform, wenn zwei Gegenanalysen die deklarierte Einstufung bestätigen. Im gegenteiligen Fall sind – unbeschadet der fälligen Sanktionen – die Kosten für die Gegenanalysen vom Betroffenen zu tragen.
(3) Bei der Überprüfung der Merkmale der in Absatz 1 genannten Öle durch die nationalen Behörden oder ihre Vertreter erfolgen die Probenahmen gemäß den internationalen Normen EN ISO 661 betreffend die Vorbereitung der Untersuchungsproben und EN ISO 5555 betreffend die Entnahme der Proben. Jedoch werden die Proben bei Partien, die aus Olivenöl in unmittelbaren Umschließungen bestehen, abweichend von Nummer 6.8 der Norm EN ISO 5555 gemäß den Bestimmungen von Anhang Ia dieser Verordnung entnommen. Bei unverpackten Ölen, bei denen die Probenahme nicht gemäß der Norm EN ISO 5555 durchgeführt werden kann, wird die Probe entsprechend den Anweisungen der zuständigen Behörde des Mitgliedstaats entnommen.
Unbeschadet der Bestimmungen der Norm EN ISO 5555 und des Kapitels 6 der Norm EN ISO 661 werden die Proben unverzüglich vor Licht und starker Hitze geschützt sowie spätestens am fünften Arbeitstag nach der Probenahme zur Analyse in das Laboratorium geschickt; ansonsten werden die Proben so aufbewahrt, dass sie während des Transports oder während der Lagerung vor dem Versand an das Laboratorium nicht verderben oder beschädigt werden.
(4) Bei der Überprüfung gemäß Absatz 3 erfolgen die Analysen bei verpackten Erzeugnissen gemäß den Anhängen II, III, IX, XII und XX sowie gegebenenfalls die in den einzelstaatlichen Rechtsvorschriften vorgesehenen Gegenanalysen vor Erreichen des Mindesthaltbarkeitsdatums. Bei unverpackten Ölen müssen die Proben spätestens im sechsten Monat nach der Probenahme analysiert werden.
Für die übrigen in dieser Verordnung vorgesehenen Analysen gelten keine Fristen.
Entsprechen die Analyseergebnisse nicht den Merkmalen der angemeldeten Kategorie Olivenöl bzw. Oliventresteröl, so wird der Beteiligte davon spätestens einen Monat vor Ablauf der in Unterabsatz 1 genannten Frist in Kenntnis gesetzt, es sei denn, die Probenahme ist weniger als zwei Monate vor dem Mindesthaltbarkeitsdatum erfolgt.
(5) Bei der nach einem der Verfahren in Absatz 1 Unterabsatz 1 vorgenommenen Bestimmung der Merkmale der Öle werden die Analyseergebnisse direkt mit den in dieser Verordnung vorgesehenen Grenzwerten verglichen.
Artikel 2a
(1) Für die Zwecke dieses Artikels bedeutet „vermarktetes Olivenöl“ die Gesamtmenge an Olivenöl und Oliventresteröl eines Mitgliedstaats, die in dem Mitgliedstaat verbraucht oder aus diesem Mitgliedstaat ausgeführt wird.
(2) Die Mitgliedstaaten sorgen dafür, dass selektiv, auf der Grundlage einer Risikoanalyse und mit angemessener Häufigkeit Konformitätsprüfungen durchgeführt werden, um sicherzustellen, dass das vermarktete Olivenöl der angegebenen Kategorie entspricht.
(3) Die Kriterien zur Beurteilung des Risikos können Folgendes umfassen:
a) die Ölkategorie, den Erzeugungszeitraum, den Preis der Öle im Vergleich zu anderen Pflanzenölen, die Mischungs- und Verpackungsvorgänge, die Lagereinrichtungen und -bedingungen, das Ursprungsland, das Bestimmungsland, das Transportmittel oder den Umfang der Partie;
b) den Platz der Marktteilnehmer in der Vermarktungskette, Menge bzw. Wert der von ihnen vermarkteten Erzeugnisse, die Palette der von ihnen vermarkteten Ölkategorien, die Art des Unternehmens (Mühle, Lagerung, Raffination, Mischen, Verpacken oder Einzelhandelsverkauf);
c) Feststellungen bei vorangegangenen Kontrollen, einschließlich der Anzahl und Art der aufgedeckten Mängel, der marktüblichen Qualität der vermarkteten Öle, des Niveaus der verwendeten technischen Ausrüstung;
d) die Verlässlichkeit der Qualitätssicherungssysteme oder Eigenkontrollsysteme der Marktteilnehmer im Zusammenhang mit der Einhaltung der Vermarktungsnormen;
e) den Ort, an dem die Kontrolle durchgeführt wird, insbesondere, ob es sich um die Eingangszollstelle für das Gebiet der EU, die Ausgangszollstelle der EU oder den Ort handelt, an dem die Öle erzeugt, verpackt, verladen oder an den Endverbraucher verkauft werden;
f) jede andere Information, die auf ein Risiko der Nichtkonformität hinweisen könnte.
(4) Die Mitgliedstaaten legen Folgendes im Voraus fest:
a) die Kriterien für die Beurteilung des Risikos der Nichtkonformität der Partien;
b) auf der Grundlage einer Risikoanalyse für jede Risikokategorie die Mindestzahl der Marktteilnehmer oder Partien und/oder Mengen, die einer Konformitätsprüfung unterzogen werden.
Pro Jahr wird in den Mitgliedstaaten mindestens eine Konformitätsprüfung je Tausend Tonnen vermarktetes Olivenöl durchgeführt.
(5) Die Mitgliedstaaten prüfen die Konformität
a) entweder durch die Durchführung der in Anhang I aufgeführten Analysen in beliebiger Reihenfolge oder
b) nach der Reihenfolge des Anhangs Ib über den schematisierten Entscheidungsablauf, bis eine der in diesem Ablauf vorgesehenen Entscheidungen erreicht ist.
▼M19 —————
Artikel 3
Wird festgestellt, dass ein Olivenöl nicht der beschriebenen Kategorie entspricht, so verhängt der betreffende Mitgliedstaat unbeschadet etwaiger anderer Sanktionen wirksame, verhältnismäßige und abschreckende Maßnahmen, deren Umfang sich nach der Schwere der festgestellten Unregelmässigkeit richtet.
Lassen die Prüfungen bedeutende Unregelmäßigkeiten erkennen, so erhöhen die Mitgliedstaaten die Häufigkeit der Kontrollen in Bezug auf Vermarktungsschritt, Ölkategorie, Ursprung oder andere Kriterien.
Artikel 4
(1) Zur Beurteilung und Kontrolle der organoleptischen Merkmale durch die einzelstaatlichen Behörden oder ihre Vertreter können die Mitgliedstaaten Prüfergruppen zulassen.
Die Zulassungsbedingungen werden von den Mitgliedstaaten so festgelegt, dass insbesondere gewährleistet ist, dass
— die Bedingungen gemäß Anhang XII Nummer 4 erfüllt sind;
— der Leiter der Prüfergruppe in einem Institut und unter Bedingungen geschult wird, die vom Mitgliedstaat zu diesem Zweck anerkannt sind;
— die Zulassung von den Ergebnissen abhängig gemacht wird, die die Gruppe im Rahmen eines vom Mitgliedstaat jährlich festgelegten Kontrollprogramms erzielt.
Die Mitgliedstaaten teilen der Kommission die Liste der zugelassenen Prüfergruppen und die nach Maßgabe dieses Absatzes getroffenen Maßnahmen mit.
(2) Ergeben sich in einem Mitgliedstaat Schwierigkeiten bei der Zulassung von Prüfergruppen, kann eine Prüfergruppe eingeschaltet werden, die in einem anderen Mitgliedstaat zugelassen ist.
(3) Jeder Mitgliedstaat erstellt ein Verzeichnis der von Organisationen des Berufsstands oder mehrerer Berufsstände unter den Bedingungen des Absatzes 1 eingeführten Prüfergruppen und achtet auf die Einhaltung dieser Bedingungen.
▼M19 —————
Artikel 6
(1) Der Ölgehalt von Trester und anderen Rückständen der Olivenölextraktion (KN-Codes 2306 90 11 und 2306 90 19 ) wird nach dem Verfahren des Anhangs XV bestimmt.
(2) Der Ölgehalt gemäß Absatz 1 wird in Massenprozenten, bezogen auf die Trockenmasse, berechnet.
Artikel 7
Es gelten die Gemeinschaftsvorschriften über die Anwesenheit von Kontaminanten.
Hinsichtlich des Gehalts an halogenierten Lösungsmitteln gelten für alle Kategorien von Olivenölen folgende Grenzwerte:
— Höchstgehalt an jedem festgestellten halogenierten Lösungsmittel 0,1 mg/kg
— Höchstgehalt der Summe der festgestellten halogenierten Lösungsmittel 0,2 mg/kg.
Artikel 7a
Die natürlichen und juristischen Personen sowie die Vereinigungen von Personen, in deren Besitz sich zur Ausübung ihres Berufes oder zu gewerblichen Zwecken Olivenöl und Oliventresteröl von der Extraktion in der Mühle bis einschließlich zur Abfüllung befindet, sind verpflichtet, Ein- und Ausgangsbücher für jede Ölkategorie zu führen.
Die Mitgliedstaaten stellen sicher, dass die in Absatz 1 genannte Verpflichtung in vollem Umfang eingehalten wird.
Artikel 8
(1) Die Mitgliedstaaten teilen der Kommission die Maßnahmen mit, die sie zur Durchführung dieser Verordnung getroffen haben. Sie unterrichten die Kommission auch über alle späteren diesbezüglichen Änderungen.
(2) Spätestens bis 31. Mai jedes Jahres übermitteln die Mitgliedstaaten der Kommission einen Bericht über die Durchführung dieser Verordnung im vorangegangenen Kalenderjahr. Der Bericht enthält mindestens die Ergebnisse der an Olivenölen durchgeführten Konformitätsprüfungen gemäß der Tabelle in Anhang XXI.
(3) Die in dieser Verordnung genannten Mitteilungen erfolgen nach der Verordnung (EG) Nr. 792/2009 der Kommission ( 1 ).
Artikel 9
Die Verordnung (EWG) Nr. 1058/77 wird aufgehoben.
Artikel 10
(1) Diese Verordnung tritt am dritten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Gemeinschaften in Kraft.
Das Verfahren des Anhangs XII ist jedoch, außer wenn es sich um Interventionsmaßnahmen handelt, ab ►M1 1. November 1992 ◄ anwendbar.
Diese Methode findet keine Anwendung auf native Olivenöle, die vor dem 1. November 1992 abgefüllt wurden.
(2) Diese Verordnung gilt nicht für Olivenöl und Oliventresteröl, das vor dem Tag des Inkrafttretens dieser Verordnung abgefüllt und bis zum 31. Oktober 1992 vermarktet wurde.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
ANHÄNGE
Inhalt
|
Anhang I: |
Merkmale von Olivenölen |
|
Anhang Ia: |
Probenahme bei olivenöl oder oliventresteröl, das in unmittelbaren verpackungseinheiten geliefert wird |
|
Anhang Ib: |
Schematisierter entscheidungsablauf für die prüfung der konformität einer olivenölprobe mit der deklarierten kategorie |
|
Anhang II: |
Bestimmung der freien Fettsäuren, Kaltverfahren |
|
Anhang III: |
Bestimmung der Peroxidzahl |
|
Anhang IV: |
Kapillargaschromatografische bestimmung des wachsgehalts |
|
Anhang V: |
Bestimmung der zusammensetzung und des gehalts an sterinen und triterpen-dialkoholen mit der kapillar - gaschromatographie |
|
Anhang VII: |
►M21 Bestimmung des prozentualen Gehalts an 2-Glycerinmonopalmitat ◄ |
|
▼M20 ————— |
|
|
Anhang IX: |
UV-spektrophotometrische Analyse |
|
Anhang X: |
Bestimmung des Gehalts an Fettsäuremethylestern durch Gaschromatografie |
|
Anhang XI: |
Bestimmung des Gehalts an flüchtigen halogenierten Lösungsmitteln in Olivenöl |
|
Anhang XII: |
Verfahren des internationalen olivenölrates für die organoleptische prüfung von nativen olivenölen |
|
▼M20 ————— |
|
|
▼M19 ————— |
|
|
Anhang XV: |
Ölgehalt der Oliventrester |
|
Anhang XVI: |
Bestimmung der Iodzahl |
|
Anhang XVII: |
Methode zur Bestimmung von Stigmastadienen in pflanzlichen Ölen |
|
Anhang XVIII: |
Bestimmung der differenz zwischen dem tatsächlichen und dem theoretischen gehalt an triglyceriden mit ECN 42 |
|
Anhang XIX: |
►M28 Bestimmung des Gehalts an aliphatischen und Triterpenalkoholen durch Kapillar-Gaschromatografie ◄ |
|
Anhang XX: |
Verfahren für die Bestimmung des Gehalts an Wachsen, Fettsäuremethylestern und Fettsäureethylestern durch Kapillargaschromatographie |
|
▼M28 ————— |
|
|
Anhang XXI: |
Ergebnisse der durchgeführten Konformitätskontrollen von Olivenölen gemäß Artikel 8 Absatz 2 |
ANHANG I
MERKMALE VON OLIVENÖLEN
Qualitätsmerkmale
|
Kategorie |
Säuregehalt (%) (*) |
Peroxidzahl meq O2/kg (*) |
K232 (*) |
K268 oder K270 (*) |
Delta-K (*) |
Sensorische Prüfung |
Fettsäureethylester mg/kg (*) |
|
|
Fehlermedian (Md) (*) |
Fruchtigkeitsmedian (Mf) (*) |
|||||||
|
1. Natives Olivenöl extra |
≤ 0,8 |
≤ 20 |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0 |
Mf > 0 |
≤ 35 |
|
2. Natives Olivenöl |
≤ 2,0 |
≤ 20 |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0 |
— |
|
3. Lampantöl |
> 2,0 |
— |
— |
— |
— |
Md > 3,5 (1) |
— |
— |
|
4. Raffiniertes Olivenöl |
≤ 0,3 |
≤ 5 |
— |
≤ 1,25 |
≤ 0,16 |
— |
— |
— |
|
5. Olivenöl — bestehend aus raffinierten und nativen Olivenölen |
≤ 1,0 |
≤ 15 |
— |
≤ 1,15 |
≤ 0,15 |
— |
— |
— |
|
6. Rohes Oliventresteröl |
— |
— |
— |
— |
— |
— |
— |
— |
|
7. Raffiniertes Oliventresteröl |
≤ 0,3 |
≤ 5 |
— |
≤ 2,00 |
≤ 0,20 |
— |
— |
— |
|
8. Oliventresteröl |
≤ 1,0 |
≤ 15 |
— |
≤ 1,70 |
≤ 0,18 |
— |
— |
— |
|
(1) Der Fehlermedian darf bis zu 3,5 betragen, wenn der Fruchtigkeitsmedian gleich 0 ist. |
||||||||
Reinheitsmerkmale
|
Kategorie |
Fettsäuregehalt (1) |
Summe transisomere Ölsäure (%) |
Summe transisomere Linol- und Linolensäure (%) |
Stigmastadienen mg/kg (2) |
►C4 ECN42-Differenz zwischen HPLC-Messwert und theoretischer Berechnung ◄ |
2- Glycerinmonopalmitat (%) |
|||||
|
Myristinsäure (%) |
Linolensäure (%) |
Arachninsäure (%) |
Eicosensäure (%) |
Behensäure (%) |
Lignocerinsäure (%) |
||||||
|
1. Natives Olivenöl extra |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,05 |
►C4 ≤ |0,2| ◄ |
≤ 0,9 wenn Gesamtgehalt an Palmitinsäure ≤ 14 % |
|
≤ 1,0 wenn Gesamtgehalt an Palmitinsäure > 14 % |
|||||||||||
|
2. Natives Olivenöl |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,05 |
►C4 ≤ |0,2| ◄ |
≤ 0,9 wenn Gesamtgehalt an Palmitinsäure ≤ 14 % |
|
≤ 1,0 wenn Gesamtgehalt an Palmitinsäure > 14 % |
|||||||||||
|
3. Lampantöl |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,10 |
≤ 0,50 |
►C4 ≤ |0,3| ◄ |
≤ 0,9 wenn Gesamtgehalt an Palmitinsäure ≤ 14 % |
|
≤ 1,1 wenn Gesamtgehalt an Palmitinsäure > 14 % |
|||||||||||
|
4. Raffiniertes Olivenöl |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
— |
►C4 ≤ |0,3| ◄ |
≤ 0,9 wenn Gesamtgehalt an Palmitinsäure ≤ 14 % |
|
≤ 1,1 wenn Gesamtgehalt an Palmitinsäure > 14 % |
|||||||||||
|
5. Olivenöl — bestehend aus raffinierten und nativen Olivenölen |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
— |
►C4 ≤ |0,3| ◄ |
≤ 0,9 wenn Gesamtgehalt an Palmitinsäure ≤ 14 % |
|
≤ 1,0 wenn Gesamtgehalt an Palmitinsäure > 14 % |
|||||||||||
|
6. Rohes Oliventresteröl |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
— |
►C4 ≤ |0,6| ◄ |
≤ 1,4 |
|
7. Raffiniertes Oliventresteröl |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
— |
►C4 ≤ |0,5| ◄ |
≤ 1,4 |
|
8. Oliventresteröl |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
— |
►C4 ≤ |0,5| ◄ |
≤ 1,2 |
|
Kategorie |
Zusammensetzung der Sterine |
Sterine insgesamt (mg/kg) |
Erythrodiol und Uvaol (%) (**) |
Wachse mg/kg (**) |
|||||
|
Cholesterin (%) |
Brassicasterin (%) |
Campesterin (3) (%) |
Stigmasterin (%) |
App. β-Sitosterin (%) (4) |
Delta-7-Stigmastenol (3) (%) |
||||
|
1. Natives Olivenöl extra |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C42+C44+C46 ≤ 150 |
|
2. Natives Olivenöl |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C42+C44+C46 ≤ 150 |
|
3. Lampantöl |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (5) |
C40+C42+C44+C46 ≤ 300 (5) |
|
4. Raffiniertes Olivenöl |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C40+C42+C44+C46 ≤ 350 |
|
5. Olivenöl — bestehend aus raffinierten und nativen Olivenölen |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C40+C42+C44+C46 ≤ 350 |
|
6. Rohes Oliventresteröl |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (6) |
C40+C42+C44+C46 > 350 (6) |
|
7. Raffiniertes Oliventresteröl |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
C40+C42+C44+C46 > 350 |
|
8. Oliventresteröl |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
C40+C42+C44+C46 > 350 |
(1) Gehalt an anderen Fettsäuren (%): Palmitinsäure: 7,50-20,00; Palmitoleinsäure: 0,30-3,50; Heptadecansäure: ≤ 0,40; Heptadecensäure: ≤ 0,60; Stearinsäure: 0,50-5,00; Ölsäure: 55,00-83,00; Linolsäure: 2,50-21,00.
(2) Summe der mittels Kapillarsäule (nicht) abtrennbaren Isomere.
(3) Siehe die Anlage zu diesem Anhang.
(4) App. β-Sitosterin: Delta-5,23-Stigmastadienol + Clerosterin + Beta-Sitosterin + Sitostanol + Delta-5-Avenasterin + Delta-5,24-Stigmastadienol.
(5) Öl mit einem Wachsgehalt zwischen 300 mg/kg und 350 mg/kg wird als Lampantöl eingestuft, wenn der Gesamtgehalt an aliphatischen Alkoholen höchstens 350 mg/kg oder der Gehalt an Erytrodiol und Uvaol höchstens 3,5 % beträgt.
(6) Öl mit einem Wachsgehalt zwischen 300 mg/kg und 350 mg/kg wird als rohes Oliventresteröl eingestuft, wenn der Gesamtgehalt an aliphatischen Alkoholen über 350 mg/kg oder der Gehalt an Erytrodiol und Uvaol über 3,5 % beträgt.
Anmerkungen:
a) Die Analyseergebnisse müssen bis auf die gleiche Anzahl Dezimalstellen angegeben werden wie die für jedes Merkmal vorgesehenen Werte. Ist die nächstfolgende Ziffer größer als 4, so ist die angegebene letzte Stelle aufzurunden.
b) Auch wenn nur ein einziges Merkmal nicht mit dem vorgesehenen Grenzwert übereinstimmt, muss das Öl einer anderen Kategorie zugeordnet werden oder als nicht seinen Reinheitskriterien entsprechend erklärt werden.
c) Die mit einem Sternchen (*) gekennzeichneten Ölqualitätsmerkmale bedeuten: im Fall von Lampantöl, dass von den beiden betreffenden Grenzwerte gleichzeitig abgewichen werden kann; im Fall nativer Olivenöle, dass die Nichterfüllung des Grenzwerts auch nur eines einzigen Merkmals eine Umstufung innerhalb der Kategorie der nativen Olivenöle zur Folge hat.
d) Bei den mit zwei Sternchen (**) gekennzeichneten Merkmalen kann im Fall aller Arten von Oliventresterölen von den beiden betreffenden Grenzwerte gleichzeitig abgewichen werden.
Anlage
SCHEMATISIERTER ENTSCHEIDUNGSABLAUF
Entscheidungsablauf Campesterin für native Olivenöle und native Olivenöle extra:

Die übrigen Parameter müssen die in dieser Verordnung festgelegten Grenzwerte einhalten.
Entscheidungsablauf Delta-7-Stigmastenol für:
— Native Olivenöle extra und native Olivenöle
—

Die übrigen Parameter müssen die in dieser Verordnung festgelegten Grenzwerte einhalten.
— Oliventresteröle (roh und raffiniert)
—

ANHANG Ia
PROBENAHME BEI OLIVENÖL ODER OLIVENTRESTERÖL, DAS IN UNMITTELBAREN VERPACKUNGSEINHEITEN GELIEFERT WIRD
Diese Methode der Probenahme wird für Partien von Olivenöl oder Oliventresteröl angewendet, das in unmittelbare Verpackungseinheiten abgefüllt wurde. Je nachdem, ob der Inhalt der unmittelbaren Verpackung größer ist als 5 l oder nicht, kommen unterschiedliche Probenahmeverfahren zur Anwendung.
Eine „Partie“ besteht aus mehreren Verkaufseinheiten, die unter solchen Umständen hergestellt und aufgemacht wurden, dass das in jeder dieser Einheiten enthaltene Öl in Bezug auf alle analytischen Eigenschaften als homogen gilt. Die Vereinzelung einer Partie muss gemäß der Richtlinie 2011/91/EU des Europäischen Parlaments und des Rates ( 8 ) erfolgen.
„Teilprobe“ ist die Ölmenge, die in einer unmittelbaren Verpackungseinheit enthalten ist und von einer beliebigen Stelle der Partie entnommen wird.
1. ZUSAMMENSETZUNG EINER EINZELPROBE
1.1. Unmittelbare Verpackungseinheiten mit einem Inhalt von höchstens 5 l
Eine „Einzelprobe“ für unmittelbare Verpackungseinheiten mit einem Inhalt von höchstens 5 l ist die Anzahl der einer Partie entnommenen Teilproben gemäß Tabelle 1.
Tabelle 1
Die Mindestgröße einer Einzelprobe muss sich wie folgt zusammensetzen
|
Verpackungseinheiten mit einem Inhalt von |
Die Einzelprobe besteht aus dem Öl von |
|
a) mindestens 1 l |
a) 1 unmittelbaren Verpackungseinheit; |
|
b) weniger als 1 l |
b) der Mindestanzahl Verpackungseinheiten, deren Gesamtinhalt mindestens 1,0 l beträgt |
Die Anzahl der in Tabelle 1 genannten Verpackungseinheiten, die eine Einzelprobe darstellen, kann von jedem Mitgliedstaat nach seinen eigenen Erfordernissen erhöht werden (beispielsweise Durchführung der organoleptischen Prüfung durch ein anderes Labor als das Labor, das die chemischen Analysen, Gegenanalysen usw. durchgeführt hat).
1.2. Unmittelbare Verpackungseinheiten mit einem Inhalt von mehr als 5 l
Eine „Einzelprobe“ für unmittelbare Verpackungseinheiten mit einem Inhalt von mehr als 5 l ist ein repräsentativer Teil der Gesamtteilproben, der durch ein Reduktionsverfahren gemäß Tabelle 2 gewonnen wird. Die Einzelprobe muss sich aus verschiedenen Mustern zusammensetzen.
Ein „Muster“ einer Einzelprobe ist jede der Verpackungseinheiten, aus denen die Einzelprobe besteht.
Tabelle 2
Mindestanzahl der auszuwählenden Teilproben
|
Anzahl der Verpackungseinheiten im Los |
Mindestanzahl der auszuwählenden Teilproben |
|
bis 10 |
1 |
|
von … 11 bis 150 |
2 |
|
von … 151 bis 500 |
3 |
|
von … 501 bis 1 500 |
4 |
|
von … 1 501 bis 2 500 |
5 |
|
> 2 500 je 1 000 Verpackungseinheiten |
1 weitere Teilprobe |
Um den Umfang der unmittelbaren Verpackungseinheiten für die Probenahme zu reduzieren, wird der Inhalt der Probenahmeteilproben zur Herstellung der Einzelprobe homogenisiert. Die Portionen der verschiedenen Teilproben werden unter Rühren so in einen gemeinsamen Behälter zur Homogenisierung gegeben, dass sie am besten vor Luft geschützt sind.
Der Inhalt der Einzelprobe ist in mehrere Verpackungen der Mindestkapazität von 1,0 l zu geben, von denen jede ein Muster der Einzelprobe darstellt.
Die Anzahl der Einzelproben kann von jedem Mitgliedstaat nach seinen eigenen Erfordernissen erhöht werden (beispielsweise Durchführung der organoleptischen Prüfung durch ein anderes Labor als das Labor, das die chemischen Analysen, Gegenanalysen usw. durchgeführt hat).
Jede Verpackung ist so zu befüllen, dass oben eine möglichst geringe Luftschicht vorhanden ist, und danach auf geeignete Weise zu verschließen und zu versiegeln, so dass das Produkt manipulationssicher ist.
Diese Muster sind zu kennzeichnen, so dass eine korrekte Identifizierung ermöglicht wird.
2. ANALYSEN UND ERGEBNISSE
2.1. Jede Einzelprobe wird gemäß Nummer 2.5 der Norm EN ISO 5555 in Laborproben unterteilt, die entsprechend der im schematisierten Entscheidungsablauf des Anhangs Ib ausgewiesenen Reihenfolge oder in einer anderen beliebigen Reihenfolge analysiert werden.
2.2. Stimmen alle Analyseergebnisse mit den Merkmalen der gemeldeten Olivenölkategorie überein, so wird die gesamte Partie als konform eingestuft.
Stimmt ein einziges Analyseergebnis nicht mit den Merkmalen der gemeldeten Olivenölkategorie überein, so wird die gesamte Partie als nicht konform eingestuft.
3. ÜBERPRÜFUNG DER KATEGORIE DER PARTIE
3.1. Zur Überprüfung der Kategorie der Partie kann die zuständige Behörde die Anzahl der an verschiedenen Stellen der Partie entnommenen Einzelproben nach folgender Tabelle erhöhen:
Tabelle 3
Anzahl der Einzelproben in Abhängigkeit von der Partiegröße
|
Größe der Partie (l) |
Anzahl der Einzelproben |
|
weniger als 7 500 |
2 |
|
von 7 500 bis weniger als 25 000 |
3 |
|
von 25 000 bis weniger als 75 000 |
4 |
|
von 75 000 bis weniger als 125 000 |
5 |
|
ab 125 000 |
6 + 1 je weitere 50 000 l |
Jede Teilprobe, die eine Einzelprobe darstellt, ist einer fortlaufenden Stelle in der Partie zu entnehmen; dabei ist der Ort jeder Einzelprobe zu notieren und eindeutig auszuweisen.
Jede Einzelprobe ist nach den Verfahren unter den Nummern 1.1 und 1.2 herzustellen.
Jede Einzelprobe wird anschließend den Analysen gemäß Artikel 2 Absatz 1 unterzogen.
3.2. Stimmen für mindestens eine Einzelprobe nicht alle Analyseergebnisse gemäß Artikel 2 Absatz 1 mit den Merkmalen der gemeldeten Olivenölkategorie überein, so wird die gesamte Partie als nicht konform eingestuft.
ANHANG Ib
SCHEMATISIERTER ENTSCHEIDUNGSABLAUF FÜR DIE PRÜFUNG DER KONFORMITÄT EINER OLIVENÖLPROBE MIT DER DEKLARIERTEN KATEGORIE
Schema 1

Schema 2
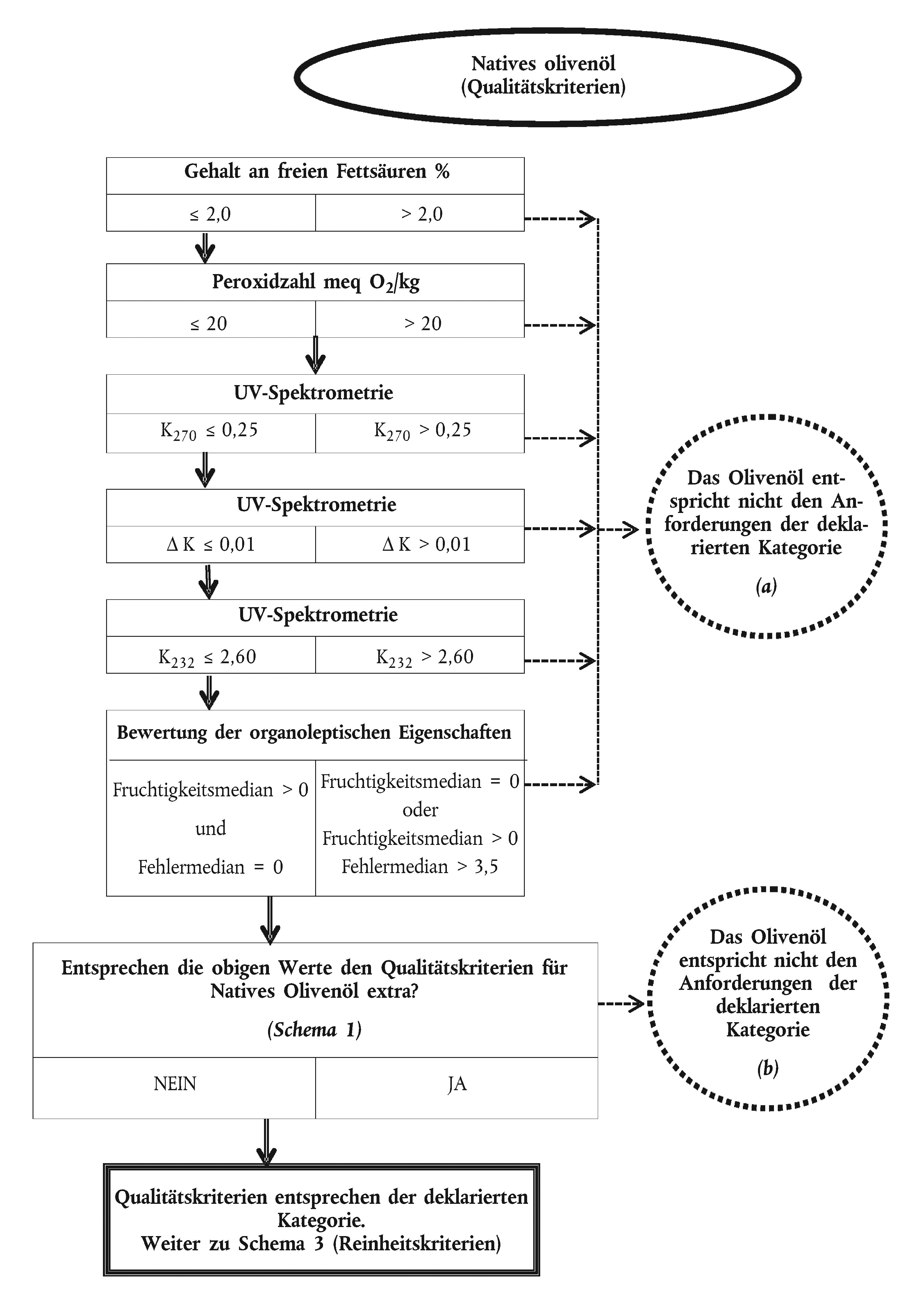
Schema 3
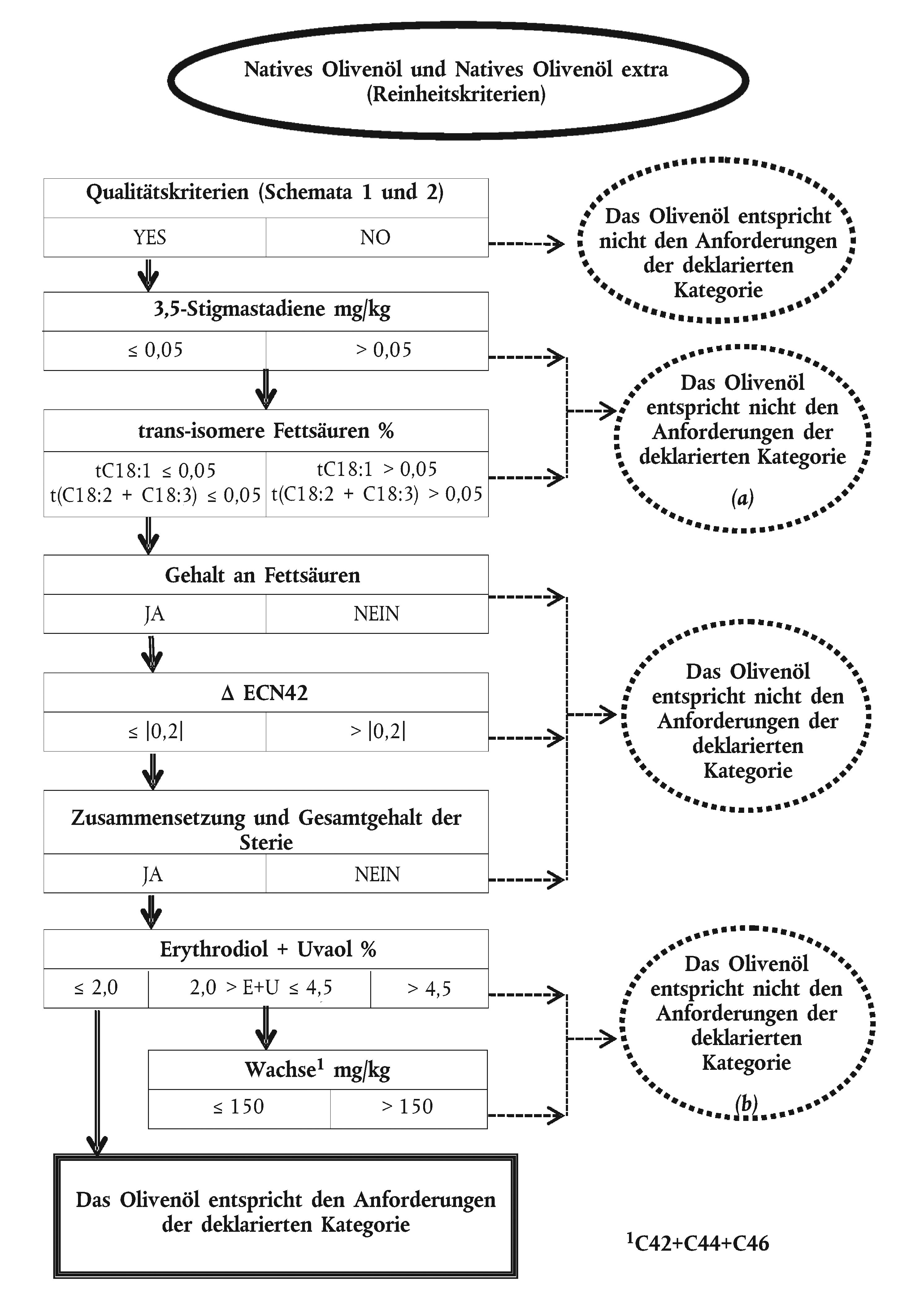
Anlage 1
Entsprechungen zwischen den Anhängen der vorliegenden Verordnung und den Analysen des Entscheidungsablaufs
|
— Säuregehalt |
Anhang II |
Bestimmung der freien fettsäuren, kaltverfahren |
|
— Peroxidzahl |
Anhang III |
Bestimmung der Peroxidzahl |
|
— UV-Spektrometrie |
Anhang IX |
Spektrophotometrische Analyse |
|
— Bewertung der organoleptischen Eigenschaften |
Anhang XII |
Bewertung der organoleptischen Eigenschaften von nativem Olivenöl |
|
— Ethylester |
Anhang XX |
Verfahren für die Bestimmung des Gehalts an Wachsen, Fettsäuremethylestern und Fettsäureethylestern durch Kapillargaschromatographie |
|
— Stigmasta-3,5-dien |
Anhang XVII |
Methode zur Bestimmung von Stigmastadienen in pflanzlichen Ölen |
|
— trans-isomere Fettsäuren |
Anhang X |
Bestimmung von Fettsäuremethylestern durch Gaschromatografie |
|
— Fettsäurenzusammensetzung |
Anhang X |
Bestimmung von Fettsäuremethylestern durch Gaschromatografie |
|
— ΔECN42 |
Anhang XVIII |
Bestimmung der Triglycerid-Zusammensetzung mit ECN42 (Differenz zwischen dem tatsächlichen und dem theoretischen Triglycerid-Gehalt) |
|
— Zusammensetzung und Gesamtgehalt der Sterine — Erythrodiol und Uvaol |
Anhang V |
Bestimmung der Zusammensetzung und des Gehalts an Sterinen und Triterpen-Dialkoholen mit Kapillar- Gaschromatografie |
|
— Wachse |
Anhang IV |
Kapillargaschromatografische bestimmung des wachsgehalts |
|
— Aliphatische und Triterpenalkohole |
Anhang XIX |
Bestimmung des Gehalts an aliphatischen und Triterpenalkoholen durch Kapillar-Gaschromatografie |
|
— Gesättigte Fettsäuren in 2- Stellung |
Anhang VII |
Bestimmung des prozentualen Gehalts an 2-Glycerinmonopalmitat |
ANHANG II
BESTIMMUNG DER FREIEN FETTSÄUREN, KALTVERFAHREN
1. GEGENSTAND UND ANWENDUNGSBEREICH
Dieses Verfahren beschreibt, wie die freien Fettsäuren in Olivenölen und Oliventresterölen bestimmt werden. Der Gehalt an freien Fettsäuren wird über den Säuregehalt in Prozent Ölsäure berechnet.
2. PRINZIP
Die Probe wird in einem geeigneten Lösungsmittelgemisch gelöst, und die vorhandenen freien Säuren werden mit Kaliumhydroxid- oder Natriumhydroxidlösung titriert.
3. REAGENZIEN
Alle Reagenzien müssen anerkannte Analysequalität aufweisen; als Wasser ist destilliertes Wasser oder Wasser gleicher Reinheit zu verwenden.
|
3.1. |
Diethylether; Ethanol 95 %ig (V/V), Mischung 1:1. Das Diethylether/Ethanol-Gemisch muss unmittelbar bei Gebrauch mit einer Kaliumhydroxidlösung (3.2) unter Zusatz von 0,3 ml Phenolphthaleinlösung (3.3) pro 100 ml Lösungsmittelgemisch neutralisiert werden. Anmerkung 1: Diethylether ist leicht brennbar und kann explosive Peroxide bilden. Es muss daher unter besonderen Vorsichtsmaßnahmen verwendet werden. Anmerkung 2: Kann kein Diethylether benutzt werden, so kann auch ein Lösungsmittelgemisch aus Ethanol und Toluol verwendet werden. Eventuell kann Ethanol auch durch 2-Propanol ersetzt werden. |
|
3.2. |
Titrierte ethanolische oder wässrige Kaliumhydroxid- bzw. Natriumhydroxidlösung, c(KOH) [bzw. c(NaOH)] etwa 0,1 mol/l oder, falls erforderlich, c(KOH) [bzw. c(NaOH)] etwa 0,5 mol/l. Entsprechende Lösungen sind im Handel erhältlich. Die genaue Konzentration der Kaliumhydroxidlösung (bzw. der Natriumhydroxidlösung) muss bekannt sein und vor der Verwendung überprüft werden. Die Lösung sollte mindestens fünf Tage vorher hergestellt und in eine braune Flasche, die mit einem Gummistopfen verschlossen wird, dekantiert werden. Die Lösung muss farblos oder strohgelb gefärbt sein. Kommt es bei der Verwendung einer wässrigen Kaliumhydroxidlösung (bzw. Natriumhydroxidlösung) zu einer Phasentrennung, muss die wässrige Lösung durch eine ethanolische Lösung ersetzt werden. Anmerkung 3: Eine stabile farblose Kaliumhydroxidlösung (bzw. Natriumhydroxidlösung) kann wie folgt hergestellt werden: 1 000 ml Ethanol oder Wasser mit 8 g Kaliumhydroxid (bzw. Natriumhydroxid) und 0,5 g Aluminiumspänen zum Kochen bringen und eine Stunde unter Rückfluss kochen. Sofort destillieren. Die erforderliche Menge Kaliumhydroxid (bzw. Natriumhydroxid) in dem Destillat lösen. Mehrere Tage stehen lassen und die klare überstehende Flüssigkeit von dem ausgefallenen Kaliumcarbonat (bzw. Natriumcarbonat) abdekantieren. Die Lösung kann auch ohne Destillation wie folgt hergestellt werden: 1 000 ml Ethanol (oder Wasser) mit 4 ml Aluminiumbutylat mehrere Tage stehen lassen. Die überstehende Flüssigkeit abdekantieren und die erforderliche Menge Kaliumhydroxid (bzw. Natriumhydroxid) darin lösen. Die Lösung ist gebrauchsfertig. |
|
3.3. |
Phenolphthalein, Lösung von 10 g/l in 95-96 %igem Ethanol (v/v) oder Alkaliblau 6B oder Thymolphthalein, Lösung von 20 g/l in 95-96 %igem Ethanol (v/v). Bei sehr stark gefärbten Ölen ist Alkaliblau oder Thymolphthalein zu verwenden. |
4. GERÄTE
Übliche Laborgeräte u. a.:
4.1. Analysenwaage;
4.2. 250-ml-Erlenmeyerkolben;
4.3. 10-ml-Bürette, Klasse A, mit 0,05-ml-Graduierung, oder gleichwertige automatische Bürette.
5. VERFAHREN
5.1. Vorbereitung der Probe zur Analyse
Wenn die Probe trüb ist, sollte sie gefiltert werden.
5.2. Probeneinwaage
Die Größe der Einwaage richtet sich nach dem zu erwartenden Säuregehalt gemäß nachstehender Tabelle:
|
Erwarteter Säuregehalt (Ölsäuregehalt g/100g) |
Masse der Probe (g) |
Wiegegenauigkeit (g) |
|
0 bis 2 |
10 |
0,02 |
|
> 2 bis 7,5 |
2,5 |
0,01 |
|
> 7,5 |
0,5 |
0,001 |
Die Probe in den Erlenmeyerkolben (4.2) einwiegen.
5.3. Bestimmung des Säuregehalts
Die Probe (5.2) in 50 bis 100 ml der zuvor neutralisierten Diethylether-Ethanol-Mischung (3.1) lösen.
Mit der Kaliumhydroxidlösung (bzw. der Natriumhydroxidlösung) 0,1 mol/l (3.2) (siehe Anmerkung 4) unter Schütteln bis zum Umschlag des Indikators titrieren (die Färbung des Farbindikators muss mindestens 10 Sekunden anhalten).
Anmerkung 4: Sind mehr als 10 ml der Kaliumhydroxidlösung (bzw. der Natriumhydroxidlösung) 0,1 mol/l erforderlich, ist die 0,5-mol/l-Lösung zu verwenden oder die Masse der Probe entsprechend dem erwarteten Gehalt an freien Säuren und der vorgeschlagenen Tabelle anzupassen.
Anmerkung 5: Wird die Lösung während der Titration trüb, so ist eine ausreichende Menge Lösungsmittel (3.1) zuzufügen, bis die Lösung wieder klar ist.
Eine zweite Bestimmung des Säuregehalts wird nur vorgenommen, wenn das erste Ergebnis über dem festgelegten Grenzwert für die entsprechende Ölkategorie liegt.
6. DARSTELLUNG DER ERGEBNISSE
Die Säure als Gehalt an Ölsäure in Gewichtsprozent berechnet sich wie folgt:

Dabei ist:
|
V |
= |
Volumen der verwendeten titrierten Kaliumhydroxidlösung (bzw. Natriumhydroxidlösung) in ml, |
|
c |
= |
genaue Konzentration der verwendeten titrierten Kaliumhydroxidlösung (bzw. Natriumhydroxidlösung) in mol/l, |
|
M |
= |
282 g/mol, die Molmasse in Gramm je mol Ölsäure, |
|
m |
= |
Masse der Probe in g. |
Der Ölsäuregehalt wird wie folgt angegeben:
a) auf zwei Dezimalstellen genau bei Werten von 0 bis einschließlich 1;
b) auf eine Dezimalstelle genau bei Werten von 1 bis einschließlich 100.
ANHANG III
BESTIMMUNG DER PEROXIDZAHL
1. Zweck
In diesem Anhang wird ein Verfahren zur Bestimmung der Peroxidzahl von tierischen und pflanzlichen Ölen und Fetten beschrieben.
2. Begriffsbestimmung
Die Peroxidzahl ist die Gesamtmenge solcher Substanzen in der Probe, ausgedrückt in Milliäquivalenten aktiven Sauerstoffs pro kg, die unter den beschriebenen Arbeitsbedingungen Kaliumiodid oxidieren.
3. Prinzip
Behandlung der in Essigsäure und Chloroform gelösten Probe mit einer Kaliumiodidlösung. Titration des freigesetzten Iods durch eine eingestellte Natriumthiosulfatlösung.
4. Geräte
Die Geräte müssen frei von reduzierenden oder oxidierenden Substanzen sein.
Anmerkung 1: Schliffflächen nicht einfetten.
4.1. Mikrobechergläschen, 3 ml.
4.2. Schliffkolben mit Stopfen, mit einem Fassungsvermögen von ca. 250 ml, die zuvor getrocknet und mit einem reinen, trockenen Inertgas (Stickstoff oder vorzugsweise Kohlendioxid) gefüllt wurden.
4.3. Bürette mit einem Fassungsvermögen von 5 ml, 10 ml oder 25 ml mit einer Graduierung von mindestens 0,05 ml, vorzugsweise mit automatischer Nullpunkteinstellung, oder gleichwertige automatische Bürette.
4.4. Analysenwaage.
5. Reagenzien
5.1. Chloroform, analytisch rein, durch Einleiten von reinem, trockenem Inertgas von Sauerstoff befreit.
5.2. Eisessig, analytisch rein, durch Einleiten von reinem, trockenem Inertgas von Sauerstoff befreit.
5.3. Kaliumiodid, gesättigte wässrige Lösung, frisch zubereitet, frei von Iod und Iodaten. Etwa 14 g Kaliumiodid in etwa 10 ml Wasser bei Raumtemperatur lösen.
5.4. Natriumthiosulfat, 0,01 mol/l (entspricht 0,01 N) wässrige Lösung, unmittelbar vor der Verwendung präzise eingestellt.
Die Natriumthiosulfatlösung 0,01 mol/l täglich vor Verwendung frisch aus einer Natriumthiosulfat-Standardlösung 0,1 mol/l zubereiten oder die genaue Normalität bestimmen. Die Erfahrung zeigt, dass die Stabilität begrenzt ist und vom pH-Wert sowie dem Gehalt an freiem Kohlendioxid abhängt. Nur frisch abgekochtes, eventuell mit Stickstoff gereinigtes Wasser zum Verdünnen verwenden.
Zur Bestimmung der genauen Normalität der Natriumthiosulfatlösung wird folgendes Verfahren empfohlen:
0,27 g bis 0,33 g Kaliumiodid (mKIO3) auf 0,001 g genau abwiegen, in einen Messkolben (250 ml oder 500 ml) geben und mit frisch abgekochtem und auf Raumtemperatur abgekühltem Wasser (V2) bis zur Markierung auffüllen. Mit Hilfe einer Pipette 5 ml oder 10 ml dieser Kaliumiodatlösung (V1) in einen 250-ml-Erlenmeyerkolben geben. 60 ml frisch abgekochtes Wasser, 5 ml Salzsäure 4 mol/l und 25 mg bis 50 mg Kaliumiodid oder 0,5 ml der gesättigten Kaliumiodidlösung hinzufügen. Diese Lösung mit der Natriumthiosulfatlösung (V3) titrieren, um die genaue Normalität der Natriumthiosulfatlösung zu bestimmen.
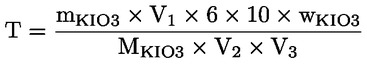
Dabei gilt:
|
mKIO3 |
= |
die Masse an Kaliumiodat in Gramm, |
|
V1 |
= |
das Volumen der Kaliumiodatlösung in Milliliter (5 ml oder 10 ml), |
|
V2 |
= |
das Gesamtvolumen der Kaliumiodatlösung in Milliliter (250 ml oder 500 ml), |
|
V3 |
= |
das Volumen der Natriumthiosulfatlösung in Milliliter, |
|
wKIO3 |
= |
die Reinheit des Kaliumiodats in g/100 g, |
|
MKIO3 |
= |
die Molekularmasse des Kaliumiodats (214 g/mol), |
|
T |
= |
die genaue Normalität der Natriumthiosulfatlösung (mol/l). |
5.5. Stärkelösung, wässrige Dispersion von 10 g/l, aus natürlicher löslicher Stärke frisch hergestellt. Es können auch gleichwertige Reagenzien verwendet werden.
6. Probe
Die Probe ist unter Lichtabschluss zu entnehmen und zu lagern und kühl in vollständig gefüllten Glasgefäßen, die mit Glas- oder Korkstopfen verschlossen sind, aufzubewahren.
7. Verfahren
Die Bestimmung ist bei diffusem Tageslicht oder künstlichem Licht durchzuführen. In ein Mikrobechergläschen (4.1) oder aber in einen Kolben (4.2) die Probemenge auf 0,001 g genau einwiegen, unter Berücksichtigung der erwarteten Peroxidzahl gemäß nachstehender Tabelle:
|
Erwartete Peroxidzahl (meq) |
Probemenge (g) |
|
0 bis 12 |
5,0 bis 2,0 |
|
12 bis 20 |
2,0 bis 1,2 |
|
20 bis 30 |
1,2 bis 0,8 |
|
30 bis 50 |
0,8 bis 0,5 |
|
50 bis 90 |
0,5 bis 0,3 |
Den Stopfen von dem Kolben (4.2) abnehmen und den Mikrobecher mit der Probemenge einführen. 10 ml Chloroform (5.1) hinzufügen. Die Probe schnell unter Rühren auflösen. 15 ml Essigsäure (5.2), dann 1 ml Kaliumiodidlösung (5.3) hinzufügen. Den Stopfen rasch einsetzen, eine Minute schütteln und genau fünf Minuten unter Lichtabschluss bei einer Temperatur von 15 bis 25 °C stehenlassen.
Etwa 75 ml destilliertes Wasser zugeben. Das freigesetzte Iod mit Natriumthiosulfatlösung (5.4) unter kräftigem Schütteln titrieren, dabei Stärkelösung (5.5) als Indikator verwenden.
Zwei Bestimmungen mit derselben Probe durchführen.
Gleichzeitig einen Blindversuch durchführen. Übersteigt das Ergebnis des Blindversuchs 0,05 ml einer 0,01-N-Natriumthiosulfatlösung (5.4), so sind die unreinen Reagenzien zu ersetzen.
8. Abfassung der Ergebnisse
Die Peroxidzahl (PV), ausgedrückt in Milliäquivalenten aktiven Sauerstoffs pro kg, wird nach folgender Formel errechnet:

Dabei gilt:
|
V |
= |
die Anzahl ml der eingestellten Natriumthiosulfatlösung (5.4), die für die Bestimmung verbraucht werden, korrigiert durch den entsprechenden Wert des Blindversuchs, |
|
T |
= |
die genaue Normalität der verwendeten Natriumthiosulfatlösung (5.4) in mol/l, |
|
m |
= |
das Gewicht der Probe in g. |
Als Ergebnis gilt das arithmetische Mittel der beiden so durchgeführten Bestimmungen.
Das Ergebnis der Bestimmung ist auf eine Dezimalstelle genau anzugeben.
ANHANG IV
KAPILLARGASCHROMATOGRAFISCHE BESTIMMUNG DES WACHSGEHALTS
1. GEGENSTAND
Diese Arbeitsvorschrift beschreibt ein Verfahren zur Bestimmung des Wachsgehalts von Olivenölen. Die Wachse werden nach der Zahl der Kohlenstoffatome getrennt. Das Verfahren kann insbesondere zur Unterscheidung zwischen abgepresstem und extrahiertem Olivenöl (Oliventresteröl) verwendet werden.
2. PRINZIP
Das Fett wird mit einem geeigneten internen Standard versetzt und chromatografisch über eine Kieselgelsäule fraktioniert; die unter Versuchsbedingungen zuerst eluierte Fraktion (schwächerer Polarität als Triglyceride) wird gesammelt und sofort kapillargaschromatografisch analysiert.
3. GERÄTE
|
3.1. |
Erlenmeyerkolben, 25 ml |
|
3.2. |
Glassäule für Gaschromatografie, Innendurchmesser 15,0 mm, Länge 30—40 cm, mit Hahn |
|
3.3. |
Gaschromatograf, geeignet für die Verwendung von Kapillarsäulen, mit Direkteinspritzung, bestehend aus:
|
|
3.4. |
Mikroliterspritze, 10 μl, zur Direkteinspritzung in die Säule, mit gehärteter Nadel |
|
3.5. |
Elektrorührwerk |
|
3.6. |
Rotationsverdampfer |
|
3.7. |
Muffelofen |
|
3.8. |
Analysenwaage mit einer Messgenauigkeit von + 0,1 mg |
|
3.9. |
Übliche Laborgeräte. |
4. REAGENZIEN
|
4.1. |
Kieselgel mit einer Korngröße zwischen 60 und 200 μm Das Kieselgel wird im Muffelofen vier Stunden auf eine Temperatur von 500 °C erhitzt und nach dem Abkühlen mit 2 % Wasser, bezogen auf die betreffende Menge Kieselgel, versetzt. Durch gründliches Schütteln homogenisieren. Vor Gebrauch mindestens 12 Stunden im Dunkeln aufbewahren. |
|
4.2. |
n-Hexan, zur Chromatografie |
|
4.3. |
Ethylether, zur Chromatografie |
|
4.4. |
n-Heptan, zur Chromatografie |
|
4.5. |
Standardlösung aus Laurylarachidat 0,1 % (m/V) in Hexan (interner Standard) (Es kann auch Palmitylpalmitat oder Myristylstearat verwendet werden.)
|
|
4.6. |
Trägergas: Wasserstoff oder Helium, rein, zur Gaschromatografie |
|
4.7. |
Hilfsgase — Wasserstoff, rein, zur Gaschromatografie — Luft, rein, zur Gaschromatografie |
5. VERFAHREN
5.1. Vorbereiten der Chromatografiesäule
15 g Kieselgel (4.1) werden in n-Hexan (4.2) suspendiert und in die Säule (3.2) aufgegeben. Nach der Spontansedimentation wird die Phase mit einem Elektrorührwerk (3.5) nachbehandelt, um eine möglichst homogene Chromatografieschicht zu erzielen, und zur Entfernung etwa enthaltener Verunreinigungen wird mit 30 ml n-Hexan gespült. Genau 500 mg der Probe werden mithilfe der Waage (3.8) in den 25-ml-Erlenmeyerkolben (3.1) eingewogen und entsprechend dem vermuteten Wachsgehalt mit der geeigneten Menge interner Standardlösung (4.5) versetzt. So werden z. B. bei Olivenöl 0,1 mg Laurylarachidat und bei Oliventresteröl 0,25 bis 0,5 mg zugesetzt. Die derart gewonnene Probe wird unter Verwendung von je zwei Teilen 2 ml n-Hexan (4.2) in die Chromatografiesäule überführt.
Das Lösungsmittel bis zu einem Stand von 1 mm über der oberen Absorbensgrenzfläche ablaufen lassen und dann zur Entfernung der natürlich enthaltenen n-Alkane noch mit 70 ml n-Hexan spülen. Mit der chromatografischen Elution beginnen und 180 ml des n-Hexan/Ethylether-Gemischs, Verhältnis 99:1, bei einem Durchsatz von etwa 15 Tropfen/10 Sekunden auffangen. Die Elution der Probe ist bei einer Umgebungstemperatur von 22 ± 4 °C durchzuführen.
Anmerkungen:
— Das n-Hexan/Ethylethergemisch (99:1) muss jeden Tag frisch zubereitet werden.
— Für eine visuelle Kontrolle der Elution der Wachse können der gelösten Probe 100 μl Sudan (1 % im Elutionsmittel) zugesetzt werden. Die Retentionszeit des Farbstoffs liegt zwischen derjenigen der Wachse und derjenigen der Triglyceride. Wenn die Färbung das Ende der Säule erreicht, sind daher alle Wachse eluiert und die Elution kann beendet werden.
Die derart gewonnene Fraktion wird im Rotationsverdampfer (3.6) so lange getrocknet, bis das Lösungsmittel nahezu restlos verdampft ist, wobei die letzten 2 ml im schwachen Stickstoffstrom abgeblasen werden; der Trocknungsrückstand wird mit 2—4 ml n-Heptan versetzt.
5.2. Gaschromatografische Analyse
5.2.1. Vorarbeiten
Die Säule in den Gaschromatografen (3.3) einsetzen, wobei der Säulenanfang an das On-column-System und das Säulenende an den Detektor angeschlossen wird. Sodann ist der Gaschromatograf auf Dichtigkeit der Gasleitungen, Betriebsbereitschaft des Detektors und des Schreibers usw. zu überprüfen.
Wird die Säule zum ersten Mal verwendet, wird empfohlen, sie einzufahren. Einen schwachen Gasstrom durch die Säule geben, den Gaschromatografen einschalten und allmählich über einen Zeitraum von etwa 4 Stunden auf eine Temperatur von 350 °C aufheizen. Die Temperatur ist mindestens 2 Stunden konstant zu halten; sodann sind die Analysebedingungen einzustellen (Gasstrom, Zünden der Flamme, Anschluss an den elektronischen Schreiber (3.3.4), Temperatur des Säulenofens, des Detektors usw.). Anschließend das Signal mit einer Empfindlichkeit aufzeichnen, die mindestens doppelt so groß ist wie bei Durchführung der Analyse. Die Grundlinie muss linear verlaufen, ohne Peaks oder Drift.
Eine negative Drift ist ein Indiz für einen undichten Anschluss der Säule, eine positive deutet auf ein mangelhaftes Einfahren der Säule hin.
5.2.2. Wahl der Arbeitsbedingungen
Anhaltspunkte für die Arbeitsbedingungen:
— Säulentemperatur:
—
|
|
20 °C/Min. |
|
5 °C/Min. |
|
20 °C/Min. |
|
|
Ausgangstemperatur 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— Detektortemperatur: 350 °C
— Einspritzvolumen: 1 μl der n-Heptanlösung (2—4 ml)
— Trägergas: Helium oder Wasserstoff mit der für das gewählte Gas optimalen linearen Strömungsgeschwindigkeit (vgl. Anlage)
— Geräteempfindlichkeit, die den nachstehenden Bedingungen genügt:
Diese Bedingungen können je nach Beschaffenheit der Säule und des Gaschromatografen abgewandelt werden, damit eine Trennung aller Wachse und eine ausreichende Auflösung der Peaks (siehe Abbildung) erzielt werden. Die Retentionszeit des internen Standards C32 muss 18 ± 3 Minuten betragen, und der größte Wachspeak muss mindestens 60 % des Vollausschlags erreichen.
Die Parameter für die Peakintegration sind so zu wählen, dass für die in Betracht kommenden Peaks korrekte Werte erzielt werden.
Anmerkung:In Anbetracht der hohen Endtemperatur ist eine positive Drift von höchstens 10 % der Skala zulässig.
5.3. Durchführung der Analyse
Mit der 10-μl-Spritze 1 μl Probelösung aufziehen und dabei den Kolben der Spritze so weit einziehen, dass die Nadel leer ist. Die Nadel in das Einspritzsystem einführen, nach 1—2 Sekunden schnell einspritzen, dann nach etwa 5 Sekunden die Nadel langsam herausziehen.
Das Chromatogramm aufzeichnen, bis alle vorhandenen Wachse eluiert sind.
Die Grundlinie muss stets den vorgeschriebenen Anforderungen genügen.
5.4. Identifizierung der Peaks
Die Peaks werden anhand der Retentionszeiten durch Vergleich mit Wachsgemischen identifiziert, deren Retentionszeiten bekannt sind und die unter denselben Bedingungen analysiert wurden.
Die Abbildung zeigt ein Chromatogramm der Wachsfraktion eines nativen Olivenöls.
5.5. Quantitative Bestimmung
Die Peakflächen des internen Standards und der aliphatischen C40- bis C46-Ester werden mithilfe eines Integrators ermittelt.
Der Wachsgehalt jedes einzelnen Esters in mg/kg Fett wird nach folgender Formel berechnet:
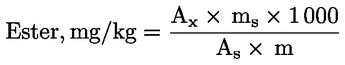
Dabei ist:
|
Ax |
= |
jeweilige Ester-Peakfläche, in mm2 |
|
As |
= |
die Peakfläche des internen Standards, in mm2 |
|
ms |
= |
das Gewicht des zugegebenen internen Standards, in mg |
|
m |
= |
Masse der zur Bestimmung entnommenen Probe in g. |
6. DARSTELLUNG DER ERGEBNISSE
Die Summe der Gehalte an den einzelnen Wachsen von C40 bis C46 wird in mg/kg Fett (ppm) angegeben.
AnmerkungDie mengenmäßig zu bestimmenden Bestandteile sind an den Peaks der Ester mit gerader Kohlenstoffzahl von C40 bis C46 abzulesen, wie als Beispiel im nachstehenden Chromatogramm der Wachse von Olivenöl dargestellt. Wenn der C46-Ester doppelt erscheint, ist zur Identifizierung die Wachsfraktion eines Oliventresteröls zu analysieren, bei dem der C46-Peak deutlich überwiegt und daher leicht zu erkennen ist.
Die Ergebnisse werden mit einer Dezimalstelle angegeben.
Abbildung
Chromatogramm der Wachsfraktion eines Olivenöls ( 9 )

Erläuterung:
|
I.S. |
= |
Laurylarachidat |
|
1. |
= |
Diterpenester |
|
2 + 2′ |
= |
C40-Ester |
|
3 + 3′ |
= |
C42-Ester |
|
4 + 4′ |
= |
C44-Ester |
|
5. |
= |
C46-Ester |
|
6. |
= |
Sterolester und Triterpenalkohole. |
Anlage
Bestimmung der linearen Strömungsgeschwindigkeit
In den auf Normalbedingungen eingestellten Gaschromatografen werden 1—3 μl Methan (oder Propan) eingespritzt, und die Säulendurchlaufzeit des Gases wird vom Zeitpunkt des Einspritzens bis zum Peak-Austritt (tM) gemessen.
Die lineare Strömungsgeschwindigkeit in cm/s ist durch die Beziehung L/tM definiert; dabei ist L die Länge der Säule in cm und tM die gemessene Zeit in Sekunden.
ANHANG V
BESTIMMUNG DER ZUSAMMENSETZUNG UND DES GEHALTS AN STERINEN UND TRITERPEN-DIALKOHOLEN MIT DER KAPILLAR - GASCHROMATOGRAPHIE
1. UMFANG UND ANWENDUNGSBEREICH
Diese Methode beschreibt ein Verfahren zur Bestimmung der Sterinzusammensetzung und des Steringehalts sowie des Gehalts an Triterpen-Dialkoholen von Olivenölen und Oliventresterölen.
2. KURZBESCHREIBUNG
Das Öl wird mit α-Cholestanol als innerem Standard versetzt, mit ethanolischer Kaliumhydroxidlösung verseift und das Unverseifbare mit Ethylether extrahiert.
Die Sterin- und Triterpen-Dialkohol-Fraktion wird dünnschichtchromatographisch über basische Kieselgelplatten vom Unverseifbaren abgetrennt. Die aus dem Kieselgel isolierten Fraktionen werden in Trimethylsilylether überführt und anschließend mit Hilfe der Kapillar-Gaschromatographie untersucht.
3. GERÄTE UND HILFSMITTEL
Übliche Laboreinrichtung und insbesondere nachstehende Geräte:
3.1. Kolben, 250 ml, mit Rückflusskühler und Schliffstopfen,
3.2. Scheidetrichter, 500 ml,
3.3. Kolben, 250 ml,
3.4. komplette Apparatur für die Dünnschichtchromatographie mit Glasplatten 20 × 20 cm,
3.5. UV-Lampe, Wellenlänge 254 oder 366 nm,
3.6. Mikroliterspritzen, 100 und 500 μl,
3.7. Glasfiltertiegel mit Porenfilter G 3 (Porosität 15-40 μm), etwa 2 cm Durchmesser, 5 cm Höhe, geeignet für die Vakuumfiltration mit Schliffmuffe,
3.8. Vakuumflasche, 50 ml, mit Schliffmuffe, für Glasfiltertiegel (Nummer 3.7),
3.9. 10-ml-Röhrchen mit konischem Boden und dicht schließendem Glasstopfen,
3.10. Gaschromatograph, geeignet für die Verwendung von Kapillarsäulen, mit Splitsystem, bestehend aus:
3.10.1. einem thermostatisierbaren Säulenofen, einstellbar auf die gewünschte Temperatur mit einer Genauigkeit von ± 1 °C,
3.10.2. einem temperaturregelbaren Injektor mit Verdampfer aus persilanisiertem Glas und Splitsystem,
3.10.3. einem Flammenionisations-Detektor (FID),
3.10.4. einem Datenerfassungssystem, geeignet zur Verwendung mit dem FID (Nummer 3.10.3), manuell integrierbar,
3.11. Glaskapillarsäule oder Fused-silica-Säule, 20 bis 30 m Länge, 0,25 bis 0,32 mm Innendurchmesser, Innenwand belegt mit 5 % Diphenyl- / 95 % Dimethylpolysiloxan (SE-52 oder SE-54 stationäre Phase oder gleichwertig), gleichmäßige Schichtdicke zwischen 0,10 und 0,30 μm,
3.12. Mikroliterspritze für die Gaschromatographie, 10 μl, mit gehärteter Nadel, geeignet für Split-Injektion,
3.13. Calciumdichlorid-Exsikkator.
4. REAGENZIEN
4.1. Kaliumhydroxid (Titer mindestens 85 %),
4.2. Kaliumhydroxid, ethanolische Lösung etwa 2 N:
130 g Kaliumhydroxid (Nummer 4.1) in 200 ml destilliertem Wasser unter Kühlen lösen und mit Ethanol auf 1 Liter auffüllen (Nummer 4.10). Die Lösung ist in gut verschlossenen Braunglasflaschen maximal zwei Tage haltbar,
4.3. Ethylether, analysenrein,
4.4. Kaliumhydroxid, ethanolische Lösung etwa 0,2 N:
13 g Kaliumhydroxid (Nummer 4.1) werden in 20 ml destilliertem Wasser gelöst und mit Ethanol auf 1 Liter aufgefüllt (Nummer 4.10),
4.5. wasserfreies Natriumsulfat, analysenrein,
4.6. kieselgelbeschichtete Glasplatten (20 x 20 cm) ohne Fluoreszenzindikator, Schichtdicke 0,25 mm (gebrauchsfertig im Handel erhältlich),
4.7. Toluol für die Chromatographie,
4.8. Aceton für die Chromatographie,
4.9. n-Hexan für die Chromatographie,
4.10. Ethylether für die Chromatographie,
4.11. Ethanol, analyserein,
4.12. Ethylacetat, analyserein,
4.13. Referenzlösung für die Dünnschichtchromatographie: Cholesterin oder Phytosterole, und Erythrodiol-Lösung 5 % in Ethylacetat (Nummer 4.11),
4.14. 2′, 7′-Dichlorfluorescein, 0,2%ige ethanolische Lösung, leicht alkalisch durch Zusatz einiger Tropfen alkoholischer 2-N-Kaliumhydroxid- Lösung (Nummer 4.2),
4.15. wasserfreies Pyridin für die Chromatographie (Anmerkung 5).
4.16. Hexamethyldisilazan, analyserein,
4.17. Trimethylchlorosilan, analyserein,
4.18. Probelösung von Sterin-Trimethylsilylethern,
unmittelbar vor Gebrauch ansetzen mit Sterinen und Erythrodiol aus Ölen, in denen sie enthalten sind,
4.19. α-Cholestanol, Reinheit mehr als 99 % (Reinheit mit GC-Analyse überprüfen),
4.20. α-Cholestanol-Lösung, interner Standard, 0,2 %ige Lösung (m/V) in Ethylacetat (Nummer 4.11).
4.21. Phenolphthalein-Lösung, 10 g/l in Ethanol (Nummer 4.10).
4.22. Trägergas: Wasserstoff oder Helium, rein, für die Gaschromatographie,
4.23. Hilfsgase: Wasserstoff, Helium, Stickstoff und Luft, rein, für die Gaschromatographie,
4.24. n-Hexan (Nummer 4.9)/Ethylether (Nummer 4.10)-Gemisch 65:35 (V/V),
4.25. Silanisierungsreagens, bestehend aus einem Gemisch aus Pyridin, Hexamethyldisilazan und Trimethylchlorsilan im Verhältnis 9:3:1 (V/V/V).
5. VERFAHREN
|
5.1. |
Herstellung des Unverseifbaren
|
|
5.2. |
Trennung der Sterin- und Triterpen-Dialkohol-Fraktion (Erythrodiol + Uvaol)
|
|
5.3. |
Herstellung der Trimethylsilylether (TMSE)
|
|
5.4. |
Gaschromatographische Analyse
|

6. ERGEBNISSE
6.1. Der Gehalt an den einzelnen Sterinen wird in mg/kg Öl angegeben, ihre Summe als „Gesamtsterine“.
Die Zusammensetzung der einzelnen Sterine sowie des Erythrodiols und des Uvaols wird mit einer Dezimalstelle angegeben.
Die Sterinzusammensetzung ist ohne Dezimalstelle anzugeben.
6.2. Der prozentuale Anteil jedes einzelnen Sterins errechnet sich aus dem Quotienten der Peakfläche des entsprechenden Peaks und der Summe der Peakflächen aller Sterine nach der Formel

Dabei ist
|
Ax |
= |
Peakfläche des Sterins x, |
|
ΣA |
= |
Summe der Peakfläche aller Sterine. |
6.3. Apparentes β-Sitosterin: Δ5-23-Stigmastadienol + Clerosterin + β-Sitosterin + Sitostanol + Δ5-Avenasterin + Δ5-24-Stigmastadienol.
6.4. Der prozentuale Anteil der Erythrodiols und Uvaols errechnet sich nach der Formel:
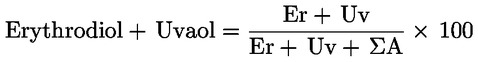
Darin bedeuten:
|
ΣA |
= |
Gesamtfläche von Sterin in Computer-Counts; |
|
Er |
= |
Fläche des Erythrodiols in Computer-Counts; |
|
Uv |
= |
Fläche des Uvaols in Computer-Counts; |
Anlage
Bestimmung der linearen Strömungsgeschwindigkeit
In den auf Normalbedingungen eingestellten Gaschromatographen werden 1 bis 3 μl Methan (oder Propan) eingespritzt und bis zum Peak-Austritt (tM) gemessen.
Die lineare Strömungsgeschwindigkeit in cm/s ist durch die Beziehung L/tM definiert; dabei ist L die Länge der Säule in cm und tM die gemessene Zeit in Sekunden.
Table 1
Relative Retentionszeit der Sterine
|
Peak |
Identifizierung |
Relative Retentionszeit |
||
|
SE 54 |
SE 52 |
|||
|
1 |
Cholesterin |
Δ-5-Cholesten-3ß-ol |
0,67 |
0,63 |
|
2 |
Cholestanol |
5α-Cholestan-3ß-ol |
0,68 |
0,64 |
|
3 |
Brassicasterin |
[24S]-24-Methyl-Δ-5,22-Cholestadien-3ß-ol |
0,73 |
0,71 |
|
* |
Ergosterin |
[24S] 24 Methy Δ5-7-22 Cholestatrien 3ß-ol |
0,78 |
0,76 |
|
4 |
24-Methylen-Cholesterin |
24-Methylen-Δ-5,24-Cholestadien-3ß-o1 |
0,82 |
0,80 |
|
5 |
Campesterin |
(24R)-24-Methyl-Δ-5-Cholesten-3ß-ol |
0,83 |
0,81 |
|
6 |
Campestanol |
(24R)-24-Methyl-Cholestan-3ß-ol |
0,85 |
0,82 |
|
7 |
Stigmasterin |
(24S)-24-Ethyl-Δ-5,22-Cholestadien-3ß-ol |
0,88 |
0,87 |
|
8 |
Δ-7-Campesterin |
(24R)-24-Methyl-Δ-7-Cholesten-3ß-ol |
0,93 |
0,92 |
|
9 |
Δ-5,23-Stigmastadienol |
(24R,S)-24-Ethyl-Δ-5,23-Cholestadien-3ß-ol |
0,95 |
0,95 |
|
10 |
Clerosterin |
(24S)-24-Ethyl-Δ-5,25-Cholestadien-3ß-ol |
0,96 |
0,96 |
|
11 |
ß-Sitosterin |
(24R)-24-Ethyl-Δ-5-Cholesten-3ß-ol |
1,00 |
1,00 |
|
12 |
Sitostanol |
24-Ethyl-Cholestan-3ß-ol |
1,02 |
1,02 |
|
13 |
Δ-5-Avenasterin |
(24Z)-24-ethylidene-Δ-Cholesten-3ß-ol |
1,03 |
1,03 |
|
14 |
Δ-5-24-Stigmastadienol |
(24R,S)-24-Ethyl-Δ-5,24-Cholestadien-3ß-ol |
1,08 |
1,08 |
|
15 |
Δ-7-Stigmastenol |
(24R,S)-24-Ethyl-Δ-7-Cholesten-3ß-ol |
1,12 |
1,12 |
|
16 |
Δ-7-Avenasterin |
(24Z)-24-Ethyliden-Δ-7-Cholesten-3ß-ol |
1,16 |
1,16 |
|
17 |
Erythrodiol |
5α Olean-12en-3ß28 diol |
1,41 |
1,41 |
|
18 |
Uvaol |
Δ12-Ursen-3ß28 diol |
1,52 |
1,52 |
Abbildung 1
Gaschromatogramm der Sterin- und Triterpen-Dialkohol-Fraktion von Lampantöl (mit dem internen Standard versetzt)

Abbildung 2
Gaschromatogramm der Sterin- und Triterpen-Dialkohol-Fraktion von raffiniertem Olivenöl (mit dem internen Standard versetzt)

Abbildung 3
Dünnschichtplatte Oliventresteröl mit dem Bereich, der zur Bestimmung der Sterine und Triterpen-Dialkohole abgekratzt werden muss

1 – Squalen
2 – Triterpen- und aliphatische Alkohole
3 – Sterine und triterpenische Dialkohole
4 – Beginn und freie Fettsäuren
▼M26 —————
ANHANG VII
BESTIMMUNG DES PROZENTUALEN GEHALTS AN 2-GLYCERINMONOPALMITAT
1. GEGENSTAND UND ANWENDUNGSBEREICH
Diese Arbeitsvorschrift beschreibt das Analyseverfahren für die Bestimmung des prozentualen Gehalts an Palmitinsäure in 2-Stellung der Triglyceride mittels Bestimmung von 2-Glycerinmonopalmitat.
Die Methode eignet sich für bei Raumtemperatur (20 °C) flüssige pflanzliche Öle.
2. PRINZIP
Die vorbereitete Olivenölprobe wird der Wirkung der Pankreaslipase ausgesetzt. Die hierdurch bewirkte partielle und spezifische Hydrolyse an den Positionen 1 und 3 des Triglyceridmoleküls ergibt ein 2-Monoglycerid. Der prozentuale Gehalt an 2-Glycerinmonopalmitat in der Monoglyceridfraktion wird nach Silylierung durch Kapillargaschromatografie bestimmt.
3. GERÄTE UND LABORAUSSTATTUNG
|
3.1. |
Erlenmeyerkolben, 25 ml |
|
3.2. |
Bechergläser, 100 ml, 250 ml und 300 ml |
|
3.3. |
Glassäule für Chromatografie, 21—23 mm Innendurchmesser, 400 mm Länge, mit gläsernem Frittenboden und Hahn |
|
3.4. |
Messkolben, 10, 50, 100 und 200 ml |
|
3.5. |
Rundkolben, 100 ml und 250 ml |
|
3.6. |
Rotationsverdampfer |
|
3.7. |
Zentrifugengläser mit konischem Boden, 10 ml, mit Schliffstopfen |
|
3.8. |
Zentrifuge für 10- und 100-ml-Gläser |
|
3.9. |
Thermostat, einstellbar auf 40 °C mit einer Genauigkeit von ± 0,5 °C |
|
3.10. |
Messpipetten, 1 ml und 2 ml |
|
3.11. |
Injektionsspritze, 1 ml |
|
3.12. |
Mikroliterspritze, 100 μl |
|
3.13. |
Trichter, 1 000 ml |
|
3.14. |
Kapillargaschromatograf mit „Cold-on-column“-Injektor zur Direkteinspritzung der Probe in die Säule und mit auf 1 °C genau einstellbarem Ofen |
|
3.15. |
„Cold-on-column“-Injektor zur Direkteinspritzung der Probe in die Säule |
|
3.16. |
Flammenionisationsdetektor und Elektrometer |
|
3.17. |
Für das Elektrometer geeigneter Integrator mit Schreiber, Ansprechzeit max. 1 Sekunde, variabler Papiervorschub |
|
3.18. |
Glaskapillarsäule oder Fused-silica-Säule, Länge 8—12 m, Innendurchmesser 0,25—0,32 mm, beschichtet mit Methylpolysiloxan oder Phenylmethylpolysiloxan 5 %, Schichtdicke 0,10—030 μm, verwendbar bis 370 °C |
|
3.19. |
Mikroliterspritze, 10 μl, mit gehärteter Nadel, Länge mindestens 7,5 cm, für Direkteinspritzung. |
4. REAGENZIEN
|
4.1. |
Kieselgel mit einer Korngröße zwischen 0,063 und 0,200 mm (70/280 mesh), hergestellt wie folgt: Kieselgel in eine Porzellanschale geben, im Trockenschrank 4 Stunden bei 160 °C trocknen, anschließend im Exsikkator auf Raumtemperatur abkühlen lassen. Ein 5 % der Kieselgelmasse entsprechendes Volumen Wasser wie folgt zugeben: 152 g Kieselgel in einen 500-ml-Erlenmeyerkolben einwiegen, 8 g destilliertes Wasser zugeben, den Kolben verschließen und leicht schütteln, bis eine gleichmäßige Verteilung des Wassers erreicht ist. Vor der Verwendung mindestens 12 Stunden ruhen lassen. |
|
4.2. |
n-Hexan, zur Chromatografie |
|
4.3. |
Isopropanol |
|
4.4. |
Isopropanol, wässrige Lösung 1/1 (V/V) |
|
4.5. |
Pankreaslipase: Die verwendete Lipase muss eine Aktivität zwischen 2,0 und 10 Lipaseeinheiten je mg haben (Pankreaslipasen mit einer Aktivität zwischen 2 und 10 Einheiten je mg Enzym sind im Handel erhältlich). |
|
4.6. |
tris-Hydroxymethylaminomethan-Pufferlösung: 1 M wässrige Lösung durch Zusatz von konzentrierter HCl (1/1 V/V) auf pH 8 gebracht (durch Potenziometer überprüfen) |
|
4.7. |
Natriumcholat, Enzymqualität, 0,1 %ige wässrige Lösung (diese Lösung ist innerhalb von 15 Tagen nach ihrer Herstellung zu verbrauchen) |
|
4.8. |
Calciumchlorid, 22 %ige wässrige Lösung |
|
4.9. |
Diethylether, zur Chromatografie |
|
4.10. |
Elutionsmittel: Gemisch von n-Hexan/Diethylether (87/13) (V/V) |
|
4.11. |
Natriumhydroxid, 12-Gew. %-Lösung |
|
4.12. |
Phenolphthalein, 1 %ige Lösung in Ethanol |
|
4.13. |
Trägergas: Wasserstoff oder Helium, zur Gaschromatografie |
|
4.14. |
Hilfsgase: Wasserstoff, Reinheit mindestens 99 %, frei von Feuchtigkeit und organischen Stoffen, und Luft, gleicher Reinheitsgrad, zur Gaschromatografie |
|
4.15. |
Silylierungsreagenz: Gemisch von Pyridin, Hexamethyldisilazan und Trimethylchlorsilan im Verhältnis 9/3/1 (V/V/V). (Gebrauchsfertige Lösungen sind im Handel erhältlich. Daneben gibt es auch andere Silylierungsreagenzien, z. B. bis-Trimethylsilyltrifluoracetamid + 1 % Trimethylchlorsilan, das mit gleichen Teilen wasserfreiem Pyridin gemischt werden muss.) |
|
4.16. |
Referenzproben: reine Monoglyceride oder Gemische von Monoglyceriden mit bekannter Zusammensetzung, die möglichst der Zusammensetzung der Probe ähnlich ist. |
5. VERFAHREN
5.1. Vorbereiten der Probe
|
5.1.1. |
Öle mit einem Gehalt an freien Säuren unter 3 % brauchen vor der Säulenchromatografie mit Kieselgel nicht neutralisiert zu werden. Öle mit einem Gehalt an freien Säuren über 3 % sind gemäß Ziffer 5.1.1.1 zu neutralisieren.
|
|
5.1.2. |
1,0 g des wie beschrieben vorbereiteten Öls in einen 25-ml-Erlenmeyerkolben (3.1) geben und in 10 ml Elutionsmittel (4.10) lösen. Die Lösung vor der Kieselgelsäulenchromatografie mindestens 15 Minuten ruhen lassen. Wenn die Lösung trüb ist, sollte sie zentrifugiert werden, um optimale Chromatografiebedingungen zu schaffen. (Es können gebrauchsfertige SPE-Kartuschen (500 mg) verwendet werden). |
|
5.1.3. |
Vorbereiten der Chromatografiesäule Etwa 30 ml Elutionsmittel (4.10) in die Säule (3.3) geben, einen Wattebausch mit Hilfe des Glasstabs bis auf den Grund der Säule schieben; dabei die Luft herausdrücken. In einem 100-ml-Becherglas 25 g Kieselgel (4.1) in etwa 80 ml Elutionsmittel suspendieren, dann mithilfe eines Trichters in die Säule geben. Zur restlosen Überführung des Kieselgels in die Säule ist das Becherglas mit dem Elutionsmittel (4.10) zu spülen. Hahn öffnen und soviel Elutionsmittel ablaufen lassen, bis der Elutionsmittelspiegel etwa 2 mm über dem Kieselgel liegt. |
|
5.1.4. |
Säulenchromatografie In einen 25-ml-Erlenmeyerkolben (3.1) wird genau 1,0 g der gemäß Ziffer 5.1 vorbereiteten Probe eingewogen. Die Probe in 10 ml Elutionsmittel (4.10) auflösen und die Lösung in die gemäß Ziffer 5.1.3 vorbereitete Chromatografiesäule geben. Nicht umrühren. Hahn öffnen und die Probenlösung bis zur Kieselgelschicht ablaufen lassen. Mit 150 ml Elutionsmittel eluieren. Die Durchlaufgeschwindigkeit auf 2 ml/Min. einstellen (so dass 150 ml die Säule in 60 bis 70 Minuten durchströmen). Eluat in einem zuvor austarierten 250-ml-Rundkolben auffangen. Das Lösungsmittel unter Vakuum eindampfen und letzte Lösungsmittelspuren im Stickstoffstrom entfernen. Den Rundkolben wiegen und den Extrakt berechnen. (Bei Verwendung von gebrauchsfertigen Kieselgel-SPE-Kartuschen ist wie folgt vorzugehen: 1 ml der Lösung (5.1.2) in die mit 3 ml n-Hexan vorbereiteten Kartuschen geben. Nach der Perkolation der Lösung mit 4 ml h-Hexan/Diethylether 9:1 (V/V) eluieren. Das Eluat in einem 10-ml-Glas auffangen und im Stickstoffstrom bis zur Trockne eindampfen. Die Pankreaslipase (5.2) auf den Rückstand einwirken lassen. Es ist wichtig, die Fettsäurezusammensetzung vor und nach dem Durchlaufen der SPE-Kartusche zu überprüfen). |
5.2. Hydrolyse durch Pankreaslipase
|
5.2.1. |
0,1 g des gemäß Ziffer 5.1 zubereiteten Öls in ein Zentrifugenglas einwiegen. 2 ml Pufferlösung (4.6), 0,5 ml Natriumcholatlösung (4.7) und 0,2 ml Calciumchloridlösung zugeben und nach jeder Zugabe gut schütteln. Das Glas mit dem Schliffstopfen verschließen und bei 40 + 0,5 °C in den Thermostaten stellen. |
|
5.2.2. |
20 mg Lipase zugeben, vorsichtig schütteln (Befeuchten des Stopfens vermeiden) und das Glas genau 2 Minuten in den Thermostaten stellen. Herausnehmen, genau 1 Minute kräftig schütteln und abkühlen lassen. |
|
5.2.3. |
1 ml Diethylether zugeben, das Glas verschließen und kräftig schütteln, dann zentrifugieren und die Etherlösung mithilfe einer Mikroliterspritze in ein sauberes und trockenes Glas überführen. |
5.3. Zubereitung der silylierten Derivate und Vorbereitung der Gaschromatografie
|
5.3.1. |
Mithilfe einer Mikroliterspritze 100 μl der Lösung (5.2.3.) in ein 10-ml-Zentrifugenglas mit konischem Boden geben. |
|
5.3.2. |
Das Lösungsmittel im schwachen Stickstoffstrom austreiben, 200 μl Silylierungsreagenz (4.15) zugeben, das Glas verschließen und 20 Minuten ruhen lassen. |
|
5.3.3. |
Nach 20 Minuten 1 bis 5 ml n-Hexan (je nach Chromatografiebedingungen) zugeben; die erhaltene Lösung ist bereit für die Gaschromatografie. |
5.4. Gaschromatografie
Betriebsbedingungen:
— Injektortemperatur („On-column-Injektor“) unter dem Siedepunkt des Lösungsmittels (68 °C),
— Detektortemperatur: 350 °C,
— Säulentemperatur: Temperaturprogramm des Ofens: 1 Minute 60 °C, dann Erhöhung um 15 °C pro Minute bis auf 180 °C, dann um 5 °C pro Minute bis auf 340 °C, dann 13 Minuten 340 °C,
— Trägergas: Wasserstoff oder Helium, mit für die Auflösung gemäß Abbildung 1 geeigneter linearer Strömungsgeschwindigkeit. Die Retentionszeit des Triglycerids C54 muss 40 ± 5 Minuten betragen (siehe Abbildung 2). (Die hier vorgeschlagenen Arbeitsbedingungen sind Anhaltspunkte. Bei der Durchführung der Analyse sind sie so zu optimieren, dass die gewünschte Auflösung erzielt wird. Die Höhe des 2-Glycerinmonopalmitat-Peaks muss mindestens 10 % der Skala des Schreibers entsprechen.)
— Einspritzmenge: 0,5—1 μl der (5 ml) n-Hexanlösung (5.3.3).
5.4.1. Identifizierung der Peaks
Die einzelnen Monoglyceride werden auf der Grundlage der erhaltenen Retentionszeiten bestimmt, die mit den Retentionszeiten von unter den gleichen Bedingungen analysierten Standardmischungen von Monoglyceriden verglichen werden.
5.4.2. Quantitative Bestimmung
Die einzelnen Peakflächen werden mithilfe eines elektronischen Integrators berechnet.
6. DARSTELLUNG DER ERGEBNISSE
Der prozentuale Gehalt an Glycerinmonopalmitat errechnet sich aus dem Quotienten der Peakfläche des entsprechenden Peaks und der Summe der Peakflächen aller Monoglyceride (vgl. Abbildung 2) nach folgender Formel:
Glycéryl monopalmitate (%):

(Glycéril monopalmitate = Glycerinmonopalmitat)
Dabei ist:
|
Ax |
= |
Peakfläche von Glycerinmonopalmitat |
|
ΣA |
= |
Summe der Peakflächen aller Monoglyceride |
Das Ergebnis wird mit einer Dezimalstelle angegeben.
7. ANALYSEBERICHT
Im Analysebericht ist Folgendes anzugeben:
— Verweis auf diese Methode,
— alle für die vollständige Identifizierung der Probe erforderlichen Angaben,
— Analyseergebnis,
— jede Abweichung von dieser Methode aufgrund einer Entscheidung der Beteiligten oder aus anderen Gründen,
— Angaben zum Labor, Analysedatum und Unterschrift der für die Analyse verantwortlichen Personen.
Abbildung 1
Chromatogramm der Produkte der Silylierungsreaktion, die durch Einwirkung der Lipase auf ein raffiniertes Olivenöl mit 20 %igem Zusatz an verestertem Öl (100 %) gewonnen wurden

Erläuterung: Acides gras libres = freie Fettsäuren; Huile d’olive raffinée + 20 % huile estérifiée = raffiniertes Olivenöl + 20 % verestertes Öl; 1-2 monopalmitoléine = 1-2 Monopalmitolein; 1-2 monopalmitine = 1-2 Monopalmitin; 1-2- monoC18 insat. = 1-2 MonoC18 unges.; Squalene = Squalen.
Abbildung 2
Chromatogramm von:
|
A) |
Olivenöl, nicht verestert; nach Lipase; nach Silylierung; unter diesen Bedingungen (Kapillarsäule 8—12 m) wird die Wachsfraktion gleichzeitig mit der Diglyceridfraktion oder kurze Zeit später eluiert. Nach der Lipase darf der Triglyceridgehalt 15 % nicht übersteigen. 
Erläuterung:
|
Chromatogramm von:
|
B) |
verestertem Öl nach Lipase; nach Silylierung; unter diesen Bedingungen (Kapillarsäule 8—12 m) wird die Wachsfraktion gleichzeitig mit der Diglyceridfraktion oder kurze Zeit später eluiert. Nach der Lipase darf der Triglyceridgehalt 15 % nicht übersteigen. 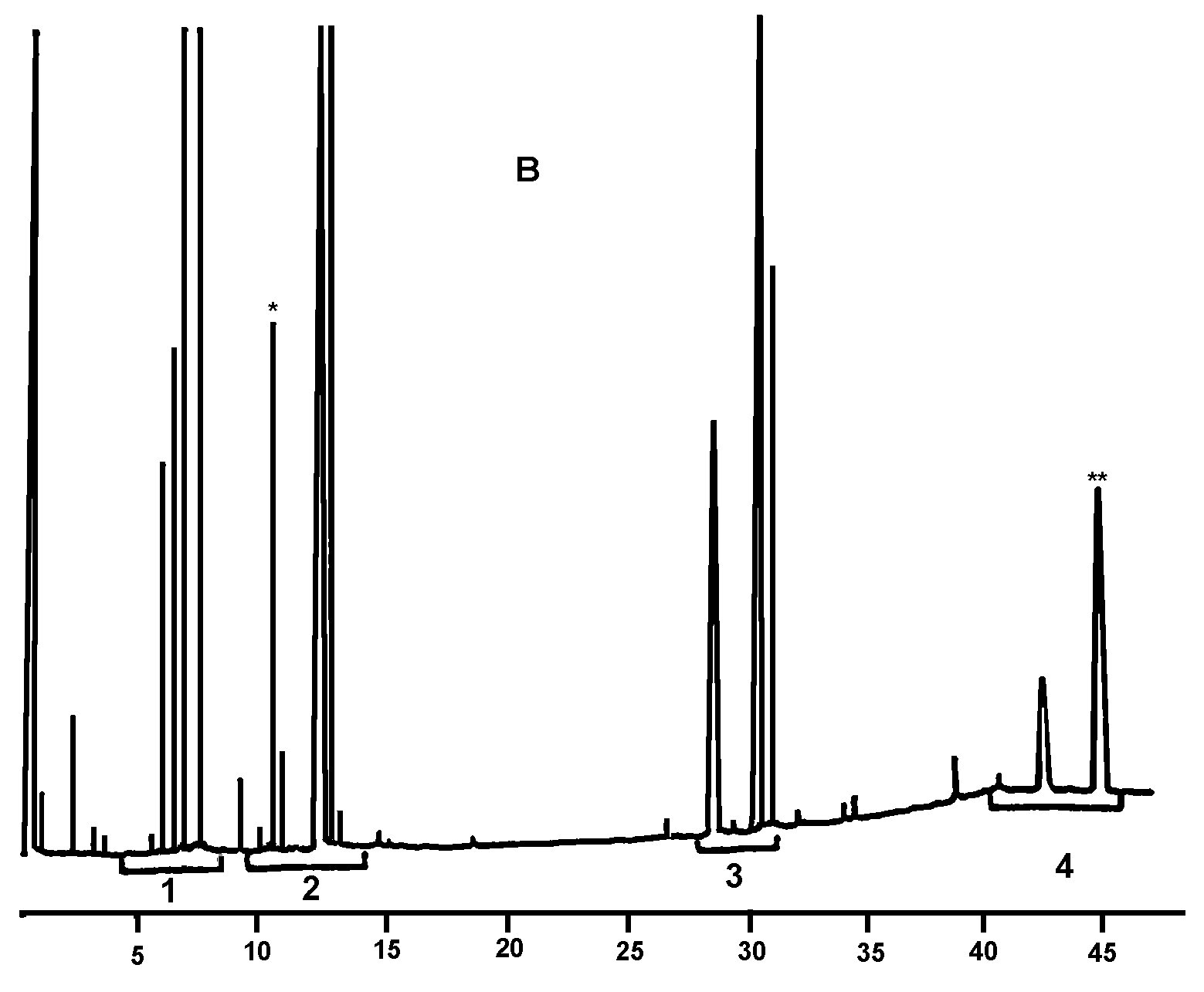
Erläuterung:
|
8. ANMERKUNGEN
HERSTELLUNG DER LIPASE
Lipasen mit ausreichender Aktivität sind im Handel erhältlich. Sie können aber auch im Labor wie folgt hergestellt werden:
5 kg frisches Schweinepankreas auf 0 °C abkühlen. Das umhüllende feste Fett und die anhängenden Gewebe entfernen und Pankreas im Mixer zu einem flüssigen Brei zerkleinern. Diesen Brei 4 bis 6 Stunden mit 2,5 l wasserfreiem Aceton rühren, anschließend zentrifugieren. Den Rückstand noch dreimal mit dem gleichen Volumen Aceton, dann zweimal mit einer Mischung von Aceton und Diethylether 1:1 (V/V) und zweimal mit Diethylether extrahieren.
Den Rückstand 48 Stunden im Vakuum trocknen, bis ein stabiles Pulver entsteht, das vor Feuchtigkeit geschützt im Kühlschrank aufbewahrt werden sollte.
PRÜFUNG DER LIPASEAKTIVITÄT
Durch Rühren (10 Min.) eines Gemischs von 165 ml Gummiarabikumlösung (100 g/l), 15 g gemahlenem Eis und 20 ml neutralisiertem Olivenöl mit einem geeigneten Rührgerät eine Ölemulsion herstellen.
10 ml dieser Emulsion in ein 50-ml-Becherglas geben, anschließend 0,3 ml Natriumcholatlösung (0,2 g/ml) und 20 ml destilliertes Wasser hinzufügen.
Das Becherglas bei 37 °C in einen Thermostaten stellen; die Elektroden des pH-Meters und den Spiralrührer einsetzen.
Mit Hilfe einer Bürette tropfenweise 0,1 N Natriumhydroxidlösung hinzufügen bis zu einem pH-Wert von 8,3.
Eine ausreichende Menge der wässrigen Lipasesuspension (0,1 g/ml Lipase) hinzufügen. Sobald das pH-Meter 8,3 anzeigt, die Stoppuhr anstellen und Natriumhydroxidlösung so zutropfen lassen, dass der pH-Wert bei 8,3 gehalten wird. Das Volumen der pro Minute verbrauchten Lösung notieren.
Die Werte in ein Koordinatensystem eintragen, und zwar die Zeitablesungen als Abszisse und die Anzahl ml 0,1 N Alkalilösung zur Aufrechterhaltung eines konstanten pH-Werts als Ordinate. Dabei muss sich eine Gerade ergeben.
Die Lipaseaktivität, ausgedrückt als Lipaseeinheiten je mg, wird mit folgender Formel angegeben:
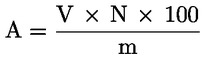
Dabei ist:
|
A |
die Aktivität in Lipaseeinheiten/mg |
|
V |
die Anzahl der pro Minute verbrauchten Milliliter 0,1 N Natriumhydroxidlösung (berechnet aus der grafischen Darstellung) |
|
N |
die Normalität der Natriumhydroxidlösung |
|
m |
das Gewicht der Lipaseprobe in mg. |
Die Lipaseeinheit wird als die Menge Enzym definiert, die 10 Mikroäquivalente Säure pro Minute freisetzt.
▼M20 —————
ANHANG IX
UV-SPEKTROFOTOMETRISCHE ANALYSE
VORWORT
Die spektrofotometrische Analyse im Ultraviolettlicht kann Angaben über die Qualität eines Fettes, seine Haltbarkeit und die infolge technologischer Verfahren eingetretenen Veränderungen erbringen. Die Absorption bei den in dem Verfahren vorgesehenen Wellenlängen ist bedingt durch das Vorhandensein konjugierter Dien- und Triensysteme infolge von Oxidationsprozessen und/oder Raffinantionsverfahren. Die Absorption wird als spezifische Extinktion
![]()
angegeben (Extinktion einer 1 %igen (m/v) Lösung des Fettes in dem vorgeschriebenen Lösungsmittel in einer 10-mm-Küvette). Sie wird üblicherweise als K bezeichnet (auch Extinktionskoeffizient genannt).
1. ANWENDUNGSBEREICH
In diesem Anhang wird die Durchführung der spektrofotometrischen Untersuchung von Olivenöl im Ultraviolettbereich beschrieben.
2. PRINZIP DER METHODE
Eine Stichprobe wird in dem vorgeschriebenen Lösungsmittel gelöst, anschließend wird die Absorption der Lösung bei den vorgeschriebenen Wellenlängen im Vergleich zum reinen Lösungsmittel bestimmt.
Die spezifischen Extinktionen bei 232 nm und 268 nm in Isooctan oder bei 232 nm und 270 nm in Cyclohexan werden bei einer Konzentration von 1 % (m/v) in einer 10-mm-Küvette errechnet.
3. GERÄTE
|
3.1. |
Spektrofotometer zur Messung bei Wellenlängen im UV-Bereich (220 nm bis 360 nm) mit der Möglichkeit, einzelne nanometrische Einheiten abzulesen. Es wird eine regelmäßige Kontrolle der Genauigkeit und Wiederholbarkeit der Absorptions- und Wellenlängenskalen sowie eine Überprüfung auf Streulicht empfohlen.
|
|
3.2. |
Rechteckige Quarzküvetten mit Deckel und einer optischen Weglänge von 10 mm, die für die Messung bei Wellenlängen im UV-Bereich (220 bis 360 nm) geeignet sind. Die Extinktionen der mit Wasser oder einem anderen geeigneten Lösungsmittel gefüllten Küvetten dürfen nicht mehr als 0,01 Einheiten voneinander abweichen. |
|
3.3. |
25-ml-Messkolben mit Füllmarkierung, Klasse A |
|
3.4. |
Analysenwaage, Ablesegenauigkeit 0,0001 g. |
4. REAGENZIEN
Soweit nicht anders angegeben, sind bei der Analyse ausschließlich Reagenzien von anerkannter Analysereinheit sowie destilliertes oder vollentsalztes Wasser oder Wasser von entsprechender Reinheit zu verwenden.
Lösungsmittel: Isooctan (2,2,4-Trimethylpentan) für die Messung bei 232 nm und 268 nm oder Cyclohexan für die Messung bei 232 nm und 270 nm, mit einer Absorption von weniger als 0,12 bei 232 nm und weniger als 0,05 bei 270 nm gegen destilliertes Wasser, gemessen in einer 10-mm-Küvette.
5. VERFAHREN
|
5.1. |
Die Probe muss völlig homogen und frei von suspendierten Verunreinigungen sein, anderenfalls muss sie bei einer Temperatur von etwa 30 °C durch Papier filtriert werden. |
|
5.2. |
Etwa 0,25 g (auf 1 mg genau) der so vorbereiteten Probe in einen 25-ml-Messkolben einwiegen, mit dem vorgeschriebenen Lösungsmittel auffüllen und homogenisieren. Die so hergestellte Lösung muss völlig klar sein. Wenn eine Opaleszenz oder Trübung vorliegt, ist die Lösung schnell durch Papier zu filtrieren. ANMERKUNG: Eine Menge von 0,25-0,30 g ist in der Regel ausreichend, um bei nativem Olivenöl und nativem Olivenöl extra die Absorption bei 268 nm und 270 nm zu messen. Für Messungen bei 232 nm ist in der Regel eine Probe von 0,05 g erforderlich, weshalb üblicherweise zwei unterschiedliche Lösungen vorbereitet werden. Für Absorptionsmessungen bei Oliventresteröl, raffiniertem Olivenöl und verfälschtem Olivenöl reicht wegen ihrer höheren Absorption in der Regel eine kleinere Probe, z. B. 0,1 g. |
|
5.3. |
Erforderlichenfalls ist die Basislinie (220-290 nm) mit Lösungsmittel in beiden Quarzküvetten (Probe und Referenzprobe) zu korrigieren; anschließend wird die Quarzküvette mit der Probe mit der Prüflösung gefüllt und die Extinktion bei 232, 268 oder 270 nm gegen das als Referenz verwendete Lösungsmittel gemessen. Die abgelesenen Extinktionswerte müssen im Bereich von 0,1 bis 0,8 oder im Bereich der Linearität des Spektrofotometers liegen, die überprüft werden sollte. Ist dies nicht der Fall, müssen die Messungen unter Verwendung von entsprechend stärker konzentrierten oder verdünnten Lösungen wiederholt werden. |
|
5.4. |
Nach Messung der Absorption bei 268 nm oder 270 nm ist die Absorption bei λmax, λmax + 4 und λmax – 4 zu messen. Anhand dieser Absorptionswerte wird die Schwankung der spezifischen Extinktion (ΔΚ) ermittelt. ANMERKUNG: Bei Verwendung von Isooctan als Lösungsmittel gilt 268 nm als λmax, bei Cyclohexan 270 nm. |
6. ABFASSUNG DER ERGEBNISSE
|
6.1. |
Angegeben werden die bei den verschiedenen Wellenlängen bestimmten spezifischen Extinktionen (Extinktionskoeffizienten), die wie folgt zu berechnen sind:
Dabei ist
Die Ergebnisse werden mit zwei Dezimalstellen angegeben. |

|
6.2. |
Schwankung der spezifischen Extinktion (ΔΚ) Die Schwankung des Absolutwerts der Extinktion (ΔΚ) wird nach folgender Gleichung berechnet:
Dabei ist Km die spezifische Extinktion bei der Wellenlänge für die maximale Absorption, die je nach verwendetem Lösungsmittel bei 270 nm oder 268nm liegt. Die Ergebnisse werden mit zwei Dezimalstellen angegeben. |

ANHANG X
BESTIMMUNG VON FETTSÄUREMETHYLESTERN DURCH GASCHROMATOGRAFIE
1. ANWENDUNGSBEREICH
Dieser Anhang enthält Anweisungen für die gaschromatografische Bestimmung der freien und der gebundenen Fettsäuren in pflanzlichen Fetten und Ölen nach ihrer Umwandlung in Fettsäuremethylester (FAME).
Die gebundenen Fettsäuren der Triacylglyceride (TAG) und, in Abhängigkeit vom Verfahren der Veresterung, die freien Fettsäuren (FFA) werden zu Fettsäuremethylestern (FAME) umgewandelt, die mit der Kapillar-Gaschromatografie bestimmt werden.
Mit dem in diesem Anhang beschriebenen Verfahren können FAME von C12 bis C24, einschließlich gesättigter Fettsäuremethylester, einfach ungesättigter cis- und trans-Fettsäuremethylester und mehrfach ungesättigter cis- und trans-Fettsäuremethylester bestimmt werden.
2. PRINZIP
Die Gaschromatografie wird für die quantitative Analyse von FAME angewendet. Die FAME werden nach Maßgabe von Teil A hergestellt und dann in den Injektor injiziert und in diesem eingedampft. Die Trennung der FAME wird an Analysesäulen mit spezifischer Polarität und Länge erreicht. Für den Nachweis der FAME wird ein Flammenionisationsdetektor (FID) angewendet. Die Analysebedingungen sind in Teil B beschrieben.
Bei der Gaschromatografie von FAME mit einem Flammenionisationdsdetektor kann Wasserstoff oder Helium als Trägergas (mobile Phase) verwendet werden. Wasserstoff beschleunigt die Trennung und führt zu schärferen Peaks. Die stationäre Phase ist eine mikroskopische Schicht eines dünnen Flüssigfilms an einer inerten festen Oberfläche aus Quarzglas
Die verdampften zu analysierenden Verbindungen interagieren beim Passieren durch die Kapillarsäule mit der stationären Phase, die die Innenseite der Säule auskleidet. Aufgrund dieser unterschiedlichen Interaktion verschiedener Verbindungen eluieren diese zu unterschiedlichen Zeitpunkten; dies wird als die Retentionszeit der Verbindung für einen gegebenen Satz von Analyseparametern bezeichnet. Die einzelnen Verbindungen werden durch den Vergleich der Retentionszeiten ermittelt.
TEIL A.
HERSTELLUNG DER FETTSÄUREMETHYLESTER VON OLIVENÖL UND OLIVENTRESTERÖL
1. GEGENSTAND
In diesem Teil ist die Herstellung der Fettsäuremethylester beschrieben. Er enthält Verfahren für die Herstellung der Fettsäuremethylester von Olivenöl und Oliventresteröl.
2. ANWENDUNGSBEREICH
Die Herstellung der Fettsäuremethylester von Olivenöl und Oliventresteröl erfolgt durch Umesterung mit methanolischer Kaliumhydroxidlösung bei Raumtemperatur. Ob die Probe vor der Umesterung gereinigt werden muss, hängt von ihrem Gehalt an freien Fettsäuren und dem zu bestimmenden Analyseparameter ab; dieser kann nach Maßgabe der folgenden Tabelle gewählt werden:
|
Ölkategorie |
Methode |
|
Natives Olivenöl mit einer Säure von ≤ 2,0 % |
1. Fettsäuren 2. trans-Fettsäuren 3. ΔECN42 (nach Reinigung mit Kieselgel-SPE) |
|
Raffiniertes Olivenöl |
|
|
Olivenöl — Gemisch aus raffiniertem und nativem Olivenöl |
|
|
Raffiniertes Oliventresteröl |
|
|
Oliventresteröl |
|
|
Natives Olivenöl mit einer Säure von > 2,0 % Rohes Oliventresteröl |
1. Fettsäuren (nach Reinigung mit Kieselgel-SPE) 2. trans-Fettsäuren (nach Reinigung mit Kieselgel-SPE) 3. ΔECN42 (nach der Reinigung mit Kieselgel-SPE) |
3. METHODIK
3.1. Umesterung mit methanolischer Kaliumhydroxidlösung bei Raumtemperatur
3.1.1. Prinzip
Methylester werden durch Umesterung mit methanolischer Kaliumhydroxidlösung als Zwischenprodukte vor der Verseifung gebildet.
3.1.2. Reagenzien
|
3.1.2.1. |
Methanol mit einem Massenanteil Wasser von nicht mehr als 0,5 % (m/m) |
|
3.1.2.2. |
Hexan, chromatografische Qualität |
|
3.1.2.3. |
Heptan, chromatografische Qualität |
|
3.1.2.4. |
Diethylether, stabilisiert zur Analyse |
|
3.1.2.5. |
Aceton, chromatografische Qualität |
|
3.1.2.6. |
Elutionsmittel zur Reinigung des Öls mittels Säulen-/SPE-Chromatografie: Gemisch aus Hexan und Diethylether im Volumenverhältnis 87/13 |
|
3.1.2.7. |
Kaliumhydroxid, etwa 2 N methanolische Lösung: 11,2 g Kaliumhydroxid in 100 ml Methanol lösen |
|
3.1.2.8. |
Kieselgelkartuschen, 1 g (6 ml), für die Festphasenextraktion |
3.1.3. Geräte
|
3.1.3.1. |
Probenröhrchen mit Schraubverschluss, 5 ml, Verschluss mit PTFE-Dichtung |
|
3.1.3.2. |
Messpipetten oder automatische Pipetten, 2 ml und 0,2 ml |
3.1.4. Reinigung der Ölproben
Die Ölproben werden bei Bedarf durch Festphasenextraktion an Kieselgelkartuschen gereinigt. In einen Elutionsapparat unter Vakuum eine Kieselgelkartusche (3.1.2.8) geben und mit 6 ml Hexan (3.1.2.2) waschen. Zur Wäsche wird das Vakuum unterbrochen. Dann eine Lösung von etwa 0,12 g Öl in 0,5 ml Hexan (3.1.2.2) auf die Säule laden. Nach Eindringen der Lösung mit 10 ml Hexan/Diethylether (Volumenverhältnis 87:13) (3.1.2.6) eluieren. Das gesamte Eluat homogenisieren und in zwei gleiche Teile teilen. Einen Teil des Eluats an einem Rotationsverdampfer unter reduziertem Druck bei Raumtemperatur bis zur Trockne abrotieren. Den Rückstand in 1 ml Heptan lösen. Die Lösung ist nun zur gaschromatografischen Analyse der Fettsäuren bereit. Zur Analyse der Triglyceride mit HPLC bei Bedarf den übrigen Teil des Eluats einrotieren und den Rückstand in 1 ml Aceton lösen.
3.1.5. Verfahren
In ein 5-ml-Probenröhrchen mit Schraubverschluss (3.1.3.1) etwa 0,1 g Ölprobe einwiegen. 2 ml Heptan (3.1.2.2) zufügen und schütteln. 0,2 ml der methanolischen Kaliumhydroxidlösung (3.1.2.7) zugeben, fest verschließen und 30 Sekunden kräftig schütteln. Absetzen lassen, bis sich der obere Teil der Lösung geklärt hat. Die obere Phase (mit den Methylestern) abdekantieren. Die Heptan-Lösung ist bereit zur Injektion in den Gaschromatografen. Es wird empfohlen, die Lösung bis zur gaschromatografischen Analyse im Kühlschrank und nicht länger als 12 Stunden aufbewahren.
TEIL B.
GASCHROMATOGRAFISCHE ANALYSE DER FETTSÄUREMETHYLESTER
1. GEGENSTAND
Dieser Teil enthält allgemeine Anweisungen zur Anwendung der Kapillar-Gaschromatografie zur Bestimmung der qualitativen und quantitativen Zusammensetzung eines Gemischs von Fettsäuremethylestern, die nach dem in Teil A beschriebenen Verfahren hergestellt wurden.
Er ist nicht auf polymerisierte Fettsäuren anwendbar.
2. REAGENZIEN
2.1. Trägergas
Inertgas (Helium oder Wasserstoff), vollständig getrocknet und mit einem Sauerstoffgehalt von weniger als 10 mg/kg.
Anmerkung 1: Wasserstoff kann die Analysegeschwindigkeit verdoppeln, das ist jedoch mit Gefahren verbunden. Sicherheitsvorrichtungen sind verfügbar.
2.2. Hilfsgase
|
2.2.1. |
Wasserstoff (Reinheit ≥ 99,9 %), frei von organischen Verunreinigungen |
|
2.2.2. |
Luft oder Sauerstoff, frei von organischen Verunreinigungen |
|
2.2.3. |
Stickstoff (Reinheit > 99 %) |
2.3. Referenzstandard
Ein Gemisch aus reinen Fettsäuremethylestern oder die Methylester eines Fetts mit bekannter Zusammensetzung, die möglichst der Zusammensetzung des zu analysierenden Fetts ähnlich sind. Cis- und trans-Isomere von Ölsäure-, Linolsäure- und Linolensäuremethylesthern sind bei der Bestimmung von trans-Isomeren ungesättigter Säuren hilfreich.
Eine Oxidation der mehrfach ungesättigten Fettsäuren ist zu vermeiden.
3. GERÄTE
Die vorliegenden Instruktionen gelten für die übliche Ausrüstung für die Gaschromatografie mit Kapillarsäule und Flammenionisationsdetektor.
3.1. Gaschromatograf
Der Gaschromatograf muss über folgende Bestandteile verfügen:
3.1.1. Injektionssystem
Bei Kapillarsäulen ist ein Injektor zu verwenden, der speziell für solche Säulen konzipiert ist. Möglich ist ein Split-Injektor oder ein Splitlos-Injektor für die On-Column-Injektion.
3.1.2. Ofen
Der Ofen sollte so ausgelegt sein, dass die Kapillarsäule auf mindestens 260 °C aufgeheizt und die Temperatur auf 0,1 °C genau gehalten werden kann. Letztere Bedingung ist vor allem dann wichtig, wenn eine Quarzsäule verwendet wird.
In jedem Fall, besonders aber bei Fettsäuren mit weniger als 16 Kohlenstoffatomen, wird eine Heizung mit Temperaturprogramm empfohlen.
3.1.3. Kapillarsäule
|
3.1.3.1. |
Kapillare aus einem Material, das nicht mit den zu analysierenden Stoffen reagiert (üblicherweise Glas oder Quarzglas („Fused Silica“)). Der Innendurchmesser sollte zwischen 0,20 und 0,32 mm liegen. Die Innenflächen müssen vor dem Auftragen der stationären Phase einer geeigneten Behandlung unterzogen werden (beispielsweise Oberflächenvorbereitung, Desaktivierung). Eine Länge von 60 m ist ausreichend für Fettsäuren und cis- und trans-Isomere von Fettsäuren. |
|
3.1.3.2. |
Als stationäre Phase sind vernetzte (cross linked) Säulen aus polarem Polysiloxan (Cyanopropylsilicon) geeignet. Anmerkung 2: Bei Verwendung von polaren Polysiloxanen können bei der Identifizierung und Trennung von Linolensäure und C20-Säuren Schwierigkeiten auftreten. Die Beschichtung sollte dünn, d. h. nur 0,1 bis 0,2 μm sein. |
|
3.1.3.3. |
Einbau und Vorbereitung der Säule Beim Einbau der Kapillarsäule müssen die üblichen Vorsichtsmaßnahmen, wie Anordnung der Säule im Ofen (Träger), Auswahl und Zusammenbau der Verbindungsstücke (Abdichtung), Ausrichten der Säulenenden im Injektor und im Detektor (Verringerung von Totvolumen), getroffen werden. Die Säule wird mit dem Trägergasstrom gespült (z. B. bei einer Säulenlänge von 25 m und einem Innendurchmesser von 0,3 mm mit 0,3 bar (30 kPa)). Zur Vorbereitung der Säule wird das Temperaturprogramm des Ofens auf 3 °C/min eingestellt und die Säule, ausgehend von der Raumtemperatur, auf eine Temperatur von 10 °C unter der Zersetzungsgrenze der stationären Phase erhitzt. Diese Ofentemperatur wird eine Stunde beibehalten, bis die Basislinie stabilisiert ist. Dann auf 180 °C zurückschalten und unter isothermen Bedingungen weiterarbeiten. Anmerkung 3: Entsprechend vorbehandelte Fertigsäulen können im Handel bezogen werden. |
3.1.4. Flammenionisations-Detektor mit Verstärker
3.2. Spritze
Die Spritze sollte eine maximale Kapazität von 10 μl und eine Graduierung in 0,1 μl haben.
3.3. Datenerfassungssystem
Online mit den Detektoren verbundenes Datenerfassungssystem, das unter einer Software für die Peak-Integration und -Normalisierung läuft.
4. VERFAHREN
Die in 4.1 bis 4.3 beschriebenen Verfahren beziehen sich auf den Gebrauch eines Flammenionisationsdetektors.
4.1. Prüfbedingungen
4.1.1. Ermittlung der optimalen Betriebsbedingungen für Kapillarsäulen
Aufgrund der Leistungsfähigkeit und Durchlässigkeit von Kapillarsäulen hängen die Trennung der Bestandteile und die Analysendauer weitgehend von der Durchflussrate des Trägergases in der Säule ab. Daher ist eine Optimierung der Betriebsbedingungen durch Anpassung dieses Parameters (oder einfach durch Kopfdruckminderung) notwendig, je nachdem, ob eine bessere Trennung oder eine schnellere Analyse gewünscht wird.
Die folgenden Bedingungen haben sich als geeignet für die Trennung von FAME (C4 bis C26) erwiesen. Beispiele für Chromatogramme sind in Anlage B enthalten:
|
Injektortemperatur: |
250 °C |
|
Detektortemperatur: |
250 °C |
|
Ofentemperatur: |
von 165 °C (8 Min.) auf 210 °C bei 2 °C/Min. |
|
Wasserstoff-Trägergas: |
Säulenkopfdruck: 179 kPa |
|
Gesamtdurchflussrate: |
154,0 ml/Min. |
|
Splitverhältnis: |
1:100 |
|
Injektionsvolumen: |
1 μl |
4.1.2. Bestimmung der Auflösung (siehe Anlage A)
Die Auflösung R zweier benachbarter Peaks I und II wird nach folgender Formel berechnet:
R = 2 × ((dr(II) – d r(I))/(ω(I) + ω(II))) oder R = 2 × ((tr(II) – t r(I))/(ω(I) + ω(II))) (USP) (United States Pharmacopeia)
oder
R = 1,18 × ((tr(II) – tr(I))/(ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB), (JP (Japanese Pharmacopeia), EP (Pharmacopée Européenne), BP (British Pharmacopeia).
Dabei ist
|
d r(I) |
die Retentionsstrecke von Peak I; |
|
d r(II) |
die Retentionsstrecke von Peak II; |
|
t r(I) |
die Retentionszeit von Peak I; |
|
t r(II) |
die Retentionszeit von Peak II; |
|
ω(I) |
die Breite von Peak I an der Basis; |
|
ω(II) |
die Breite von Peak II an der Basis; |
|
ω0,5 |
die Peakbreite der spezifizierten Verbindung auf halber Peakhöhe. |
Ist ω(I) ≈ ω(II), wird R nach folgender Gleichung berechnet:
R = (dr(II) – d r(I))/ω = (dr(II) – d r(I))/4σ
Dabei ist
|
σ |
die Standardabweichung (vgl. Anlage A, Abbildung 1). |
Ist der Abstand dr zwischen zwei Peaks d r(II) - d r(I) gleich 4σ, so ist der Auflösungsfaktor R = 1.
Bei zwei nicht vollständig getrennten Peaks schneiden sich die Tangenten zu den Wendepunkten der beiden Peaks am Punkt C. Damit die beiden Peaks vollständig getrennt sind, muss der Abstand zwischen den beiden Peaks Folgendes erfüllen:
d r(II) - d r(I) = 6 σ daraus ergibt sich R = 1,5 (siehe Anlage A, Abbildung 3).
5. DARSTELLUNG DER ERGEBNISSE
5.1. Qualitative Analyse
Die Methylester-Peaks der Probe werden aus dem Chromatogramm in Anlage B Abbildung 1 — wenn nötig durch Interpolation — oder durch Vergleich mit denen der Methylester-Referenzgemische (wie unter 2.3 beschrieben) identifiziert.
5.2. Quantitative Analyse
5.2.1. Bestimmung der Zusammensetzung
Der Massenanteil wi der einzelnen Fettsäuremethylester (ausgedrückt als der prozentuale Anteil der Masse der Methylester) wird wie folgt berechnet:
5.2.2. Berechnungsweise
5.2.2.1. Allgemeiner Fall
Der Gehalt eines Bestandteils i (ausgedrückt als prozentualer Anteil der Masse der Methylester) wird durch Bestimmung des prozentualen Anteils der jeweiligen Peakfläche im Verhältnis zur Summe aller Peakflächen nach folgender Gleichung berechnet:
wi = (Ai/ΣA) × 100
Dabei ist
|
Ai |
die Fläche des einzelnen Fettsäuremethylesters i; |
|
ΣA |
die Summe der Flächen aller Peaks von allen Fettsäuremethylestern. |
Die Ergebnisse werden mit zwei Dezimalstellen angegeben.
Anmerkung 4: Bei Fetten und Ölen ist der Massenanteil der Fettsäuremethylester gleich dem Massenanteil der Triacylglycerine in Gramm je 100 g. In Fällen, in denen diese Annahme nicht zulässig ist, siehe 5.2.2.2.
5.2.2.2. Anwendung von Korrekturfaktoren
In bestimmten Fällen, z. B. bei Vorhandensein von Fettsäuren mit weniger als acht Kohlenstoffatomen oder von Fettsäuren mit sekundären Gruppen, sind die Flächen mit spezifischen Korrekturfaktoren (Fci) zu korrigieren. Diese Faktoren sind für jedes einzelne Gerät zu bestimmen. Für diesen Zweck werden geeignete Referenzmaterialien mit zertifizierter Zusammensetzung der Fettsäure in dem betreffenden Bereich verwendet.
Anmerkung 5: Diese Korrekturfaktoren sind mit den in Anlage A angegebenen theoretischen FID-Korrekturfaktoren nicht identisch, weil sie auch die Leistung des Injektionssystems usw. umfassen. Jedoch sollte im Fall von größeren Abweichungen das ganze System hinsichtlich der Leistung überprüft werden.
Für dieses Referenzgemisch ist für den FAME i der Massenanteil nach folgender Gleichung zu berechnen:
w i = (mi /Σm) × 100
Dabei ist
|
mi |
die Masse des FAME i im Referenzgemisch, |
|
Σm |
die Gesamtheit der Massen der verschiedenen Komponenten als FAME des Referenzgemischs. |
Aus dem Chromatogramm des Referenzgemischs ist der prozentuale Flächenanteil für den FAME i nach folgender Gleichung zu berechnen:
wi = (Ai/ΣA) × 100
Dabei ist
|
Ai |
die Fläche des FAME i im Referenzgemisch, |
|
ΣA |
die Summe der Flächen sämtlicher FAME des Referenzgemischs. |
Der Korrekturfaktor Fc ist dann nach folgender Gleichung zu berechnen:
Fc = (mi × ΣA)/(Ai/Σm)
Für die Probe ist für jeden FAME i der Massenamteil nach folgender Gleichung zu berechnen:
wi = (Fi × Ai)/Σ (Fi × Ai)
Die Ergebnisse werden mit zwei Dezimalstellen angegeben.
Anmerkung 6: Der berechnete Wert entspricht dem Massenanteil der einzelnen Fettsäuren, berechnet als Triacylglycerin je 100 g Fett.
5.2.2.3. Verwendung eines inneren Standards
Für bestimmte Untersuchungen (z. B. wenn nicht alle Fettsäuren quantifiziert werden, wie beispielsweise dann, wenn Säuren mit vier und sechs Kohlenstoffatomen neben Säuren mit 16 und 18 Kohlenstoffatomen vorhanden sind, oder wenn es notwendig ist, die absolute Menge einer Fettsäure in einer Probe zu bestimmen) ist die Verwendung eines internen Standards erforderlich. Fettsäuren mit 5, 15 oder 17 Kohlenstoffatomen werden häufig verwendet. Der Korrekturfaktor (wenn überhaupt) sollte für den internen Standard bestimmt werden.
Der Massenanteil der Komponente i, angegeben als Methylester, ist dann nach folgender Gleichung zu berechnen:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
Dabei ist:
|
A i |
die Fläche des FAME i, |
|
A IS |
die Fläche des internen Standards; |
|
F i |
der Korrekturfaktor der Fettsäure i, angegeben als FAME; |
|
F IS |
der Korrekturfaktor des internen Standards; |
|
m |
die Masse der Einwaage in Milligramm; |
|
m IS |
die Masse des internen Standards in Milligramm. |
Die Ergebnisse werden mit zwei Dezimalstellen angegeben.
6. UNTERSUCHUNGSBERICHT
Im Untersuchungsbericht sind das angewandte Verfahren zur Herstellung der Methylester und das angewandte gaschromatografische Verfahren genau anzugeben. Darüber hinaus sind alle Arbeitsschritte, die nicht in diesem Standardverfahren aufgeführt wurden oder als fakultativ gelten, sowie alle Vorfälle, die das Untersuchungsergebnis beeinflusst haben können, zu nennen.
Der Untersuchungsbericht hat alle erforderlichen Informationen zur vollständigen Identifizierung der Probe zu enthalten.
7. PRÄSZISION DES VERFAHRENS
7.1. Ergebnisse des Ringversuchs
Einzelheiten eines Ringversuchs zur Präzision des Verfahrens sind in Anhang C der Norm IOC/T.20/Doc. Nr. 33 zusammengefasst. Die in diesem Ringversuch ermittelten Werte könnten auf andere Konzentrationsbereiche und Matrizes als die angegebenen nicht anwendbar sein.
7.2. Wiederholpräzision
Die absolute Differenz zwischen zwei einzelnen, voneinander unabhängigen Prüfergebnissen, die derselbe Bearbeiter mit dem gleichen Verfahren an identischem Untersuchungsmaterial in demselben Laboratorium mit derselben Geräteausstattung innerhalb der kürzest möglichen Zeitspanne erhält, wird nicht häufiger als in 5 % der Fälle den in den Tabellen 1 bis 14 in Anhang C der Norm IOC/T.20/Doc. Nr. 33 angegebenen Wert von r überschreiten.
7.3. Vergleichspräzision
Die absolute Differenz zwischen zwei einzelnen Prüfergebnissen, die verschiedene Bearbeiter mit dem gleichen Verfahren an identischem Untersuchungsmaterial in verschiedenen Laboratorien mit unterschiedlichen Geräteausstattungen erhalten, wird in nicht mehr als 5 % der Fälle den in den Tabellen 1 bis 14 in Anhang C der Norm IOC/T.20/Doc. Nr. 33 angegebenen Wert von R überschreiten.
Anlage A
Abbildung 1

Mit Breite ω0,5 auf halber Höhe des Dreiecks (ABC) und Breite b auf halber Höhe des Dreiecks (NPM).
|
Abbildung 2 |
Abbildung 3 |
|
|
|


Anlage B
Abbildung 1
Gaschromatogramm eines Oliventresteröls nach dem Kaltmethylierungsverfahren
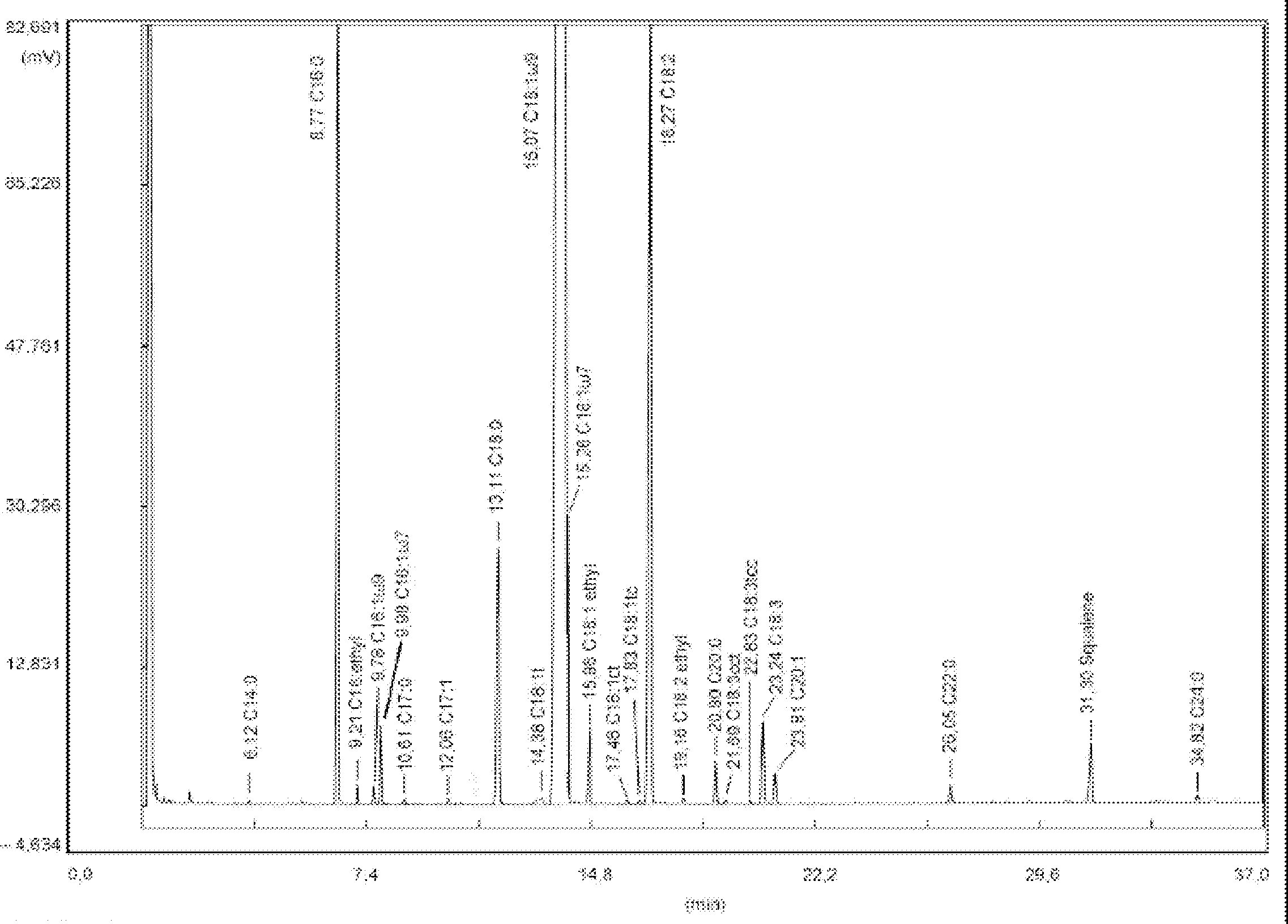
Die Peaks entsprechen den Methyl- und Ethylestern, soweit nichts anderes angegeben ist.
ANHANG XI
BESTIMMUNG DES GEHALTS AN FLÜCHTIGEN HALOGENIERTEN LÖSUNGSMITTELN IN OLIVENÖL
1. PRINZIP
Gaschromatographische Headspace-Analyse.
2. GERÄTE
|
2.1. |
Gaschromatograph mit Elektroneneinfangdetektor (ECD). |
|
2.2. |
Headspace-Vorrichtung. |
|
2.3. |
Gaschromatographische Glassäule von 2 m Länge und 2 mm Durchmesser. Stationäre Phase: 10 % OV 101 oder eine gleichwertige Phase, aufgezogen auf mit Säure gewaschenem und silanisiertem Kieselgur mit einer Korngröße von 80—100 Mesh. |
|
2.4. |
Träger- und Hilfsgas: Stickstoff für die Gaschromatographie, geeignet für Elektroneneinfangdetektoren. |
|
2.5. |
Glasfläschchen 10—15 ml, versehen mit einem Verschluß aus Aluminium und Teflon, der eine Entnahme mit Hilfe einer Spritze ermöglicht. |
|
2.6. |
Klemmen für absolut dichten Verschluß. |
|
2.7. |
Spritzen 0,5 bis 2 ml, zum Einspritzen von Gas. |
3. REAGENZIEN
Standard: Flüchtige halogenierte Lösungsmittel mit einem für die Gaschromatographie geeigneten Reinheitsgrad,
Pentan mit einem für die Gaschromatographie geeigneten Reinheitsgrad.
4. ANALYSENVERFAHREN
|
4.1. |
Etwa 3 g Öl in ein Glasfläschchen (das nicht wieder verwendet werden darf) genau einwiegen und das Fläschchen mit dem Verschluß absolut dicht verschließen. Das Fläschchen bei 70 °C eine Stunde in einen Thermostaten stellen. Mit Hilfe einer Spritze genau ein Volumen von 0,2 bis 0,5 ml aus dem Dampfraum entnehmen und auf die Säule des Gaschromatographen spritzen. Der Gaschromatograph ist wie folgt einzustellen: — Verdampfungstemperatur: 150 °C, — Säulentemperatur: 70 bis 80 °C, — Detektortemperatur: 200 bis 250 °C. Es können auch andere Temperaturen eingestellt werden, sofern dabei gleichwertige Ergebnisse erziehlt werden. |
|
4.2. |
Referenzlösungen: Standardlösungen mit unterschiedlichen Konzentrationen von flüchtigen halogenierten Lösungsmitteln zwischen 0,05 und 1 mg/kg herstellen unter Verwendung von Olivenöl, das frei von Lösungsmittelspuren ist. Falls erforderlich, werden die halogenierten Lösungsmittel mit Pentan verdünnt. |
|
4.3. |
Quantitative Auswertung. Das Verhältnis bilden zwischen den Peakflächen oder den Peakhöhen der Probe und der Standardlösung, deren Konzentration der erwarteten Konzentration am nächsten liegt. Liegt der relative Unterschied über 10 %, so muß die Analyse mit einer neuen Standardlösung wiederholt werden, bis ihre Konzentration innerhalb des obengenannten relativen Unterschieds liegt. Der Gehalt wird auf der Basis des Mittelwerts einzelner Einspritzungen ermittelt. |
|
4.4. |
Abfassung der Ergebnisse. Die Ergebnisse werden in mg/kg (ppm) angegeben. Die Nachweisgrenze des Verfahrens liegt bei 0,01 mg/kg. |
ANHANG XII
VERFAHREN DES INTERNATIONALEN OLIVENÖLRATES FÜR DIE ORGANOLEPTISCHE PRÜFUNG VON NATIVEN OLIVENÖLEN
1. ZWECK UND ANWENDUNGSBEREICH
Die in diesem Anhang beschriebene internationale Verfahrensvorschrift dient der Festlegung des Verfahrens für die Bewertung der organoleptischen Merkmale von nativen Olivenölen im Sinne von Anhang VII Teil VIII Nummer 1 der Verordnung (EU) Nr. 1308/2013 des Europäischen Parlaments und des Rates ( 10 ) und beschreibt die Methode für die Einstufung der Öle anhand dieser Merkmale. Das Verfahren umfasst zudem Hinweise für eine fakultative Kennzeichnung.
Die Verfahrensvorschrift gilt nur für native Olivenöle und deren Einstufung bzw. Kennzeichnung entsprechend dem Umfang der wahrgenommenen Mängel und der Fruchtigkeit, wie sie von einer Gruppe ausgewählter, geschulter und geprüfter Prüfer bestimmt werden.
Die in diesem Anhang angeführten IOR-Standards entsprechen der aktuellsten verfügbaren Fassung.
2. ALLGEMEINE GRUNDBEGRIFFE FÜR DIE SENSORISCHE PRÜFUNG
Vgl. dazu Standard IOC/T.20/Dok. Nr. 4 „Sensory Analysis: General Basic Vocabulary“
3. SPEZIFISCHE BEGRIFFE
3.1. Negative Attribute
Stichig/schlammig Typisches Flavour bei Ölen aus Oliven, die unter solchen Bedingungen geschichtet oder gelagert sind, dass sie eine fortgeschrittene anaerobe Gärung durchlaufen haben, oder bei Öl, das in Becken und Fässern mit Dekantier-„Schlämmen“ in Kontakt war, die ebenfalls eine anaerobe Gärung durchlaufen haben.
Modrig-feucht-erdig Typisches Flavour bei Ölen aus Früchten mit Schimmel- und Hefepilzbefall wegen mehrtägiger Lagerung unter feuchten Bedingungen, bzw. typisches Flavour bei Ölen, das von anhaftender Erde oder Schlamm ungewaschener Oliven herrührt.
Wein- oder essigartig/sauer-säuerlich Typisches Flavour bei bestimmten Ölen, an Wein oder Essig erinnernd und in erster Linie bedingt durch einen aeroben Gärungsprozess der Oliven oder Reste von Olivenpaste in nicht sachgemäß gewaschenen Pressmatten, bei dem Essigsäure, Ethylacetat und Ethanol entstehen.
Ranzig Flavour bei stark oxidierten Ölen.
Frostgeschädigte Oliven (feuchtes Holz) Typisches Flavour bei Ölen, die aus Oliven gewonnen wurden, die am Baum Frostschäden erlitten haben.
3.1.1. Sonstige negative Attribute
|
Brandig oder erhitzt |
Typisches Flavour bei Ölen aufgrund einer übermäßigen und/oder zu langen Erwärmung bei der Verarbeitung und insbesondere durch unsachgemäße Wärmebehandlung beim Rühren der Olivenpaste |
|
Heuartig-holzig |
Typisches Flavour bei bestimmten Ölen, die aus vertrockneten Oliven gewonnen wurden |
|
Roh |
Bezeichnung für bestimmte alte Öle, die im Mund einen dickflüssigen, pastösen Sinneseindruck hinterlassen |
|
Schmierölartig |
Flavour bei Ölen, das an Dieseltreibstoff, Fett oder Mineralöl erinnert |
|
Fruchtwasserartig |
Flavour bei Ölen, das von längerem Kontakt mit Fruchtwasser herrührt, das einen Gärungsprozess durchlaufen hat |
|
Lakig |
Flavour bei Ölen aus Oliven, die in Salzlake aufbewahrt wurden |
|
Metallisch |
An Metall erinnerndes Flavour, typisch für Öl, das beim Vermahlen, Schlagen, Pressen oder Lagern lange mit Metallflächen in Kontakt stand |
|
Espartograsartig |
Typisches Flavour bei Ölen aus Oliven, die mit Hilfe neuer Espartograsmatten gepresst wurden. Dieses Aroma kann in verschiedenen Nuancen auftreten, je nachdem, ob Matten aus grünem oder trockenem Espartogras verwendet wurden |
|
Wurmstichig |
Flavour bei Ölen aus stark von Larven der Olivenfliege (Bactrocera oleae) befallenen Oliven |
|
Gurkenartig |
Flavour bei Ölen, das von zu langem Lagern in luftdichten Behältnissen, insbesondere Weißblechdosen, und dem dadurch entstehenden 2,6-Nonadienal herrührt. |
3.2. Positive Attribute
|
Fruchtig |
Gesamtheit der von der Olivensorte abhängigen, unmittelbar und/oder retronasal wahrgenommenen charakteristischen Geruchsmerkmale eines Öls aus gesunden, frischen, reifen oder unreifen Früchten. |
|
Bitter |
Elementarer Geschmack, der typisch für Öle aus grünen oder in Reifung befindlichen Oliven ist und mit den auf der Zunge V-förmig angeordneten Wallpapillen wahrgenommen wird. |
|
Scharf |
Taktil empfundenes Prickeln, das typisch für Öle ist, die zu Beginn des Wirtschaftsjahres hauptsächlich aus noch unreifen Oliven gewonnen werden, und in der gesamten Mundhöhle und insbesondere in der Kehle wahrgenommen werden kann. |
3.3. Fakultative Terminologie bei der Etikettierung
Auf Antrag kann der Prüfungsleiter bescheinigen, dass die bewerteten Öle nach Intensität und Wahrnehmung der Attribute den Definitionen und Intervallen ausschließlich für die nachstehenden Bezeichnungen entsprechen.
Positive Attribute (fruchtig, bitter und scharf): Je nach Intensität der Wahrnehmung:
— intensiv, wenn der Median des betreffenden Attributs größer als 6 ist;
— mittel, wenn der Median des betreffenden Attributs zwischen 3 und 6 liegt;
— leicht, wenn der Median des betreffenden Attributs kleiner als 3 ist.
|
Fruchtigkeit |
Gesamtheit der von der Olivensorte abhängigen, unmittelbar und/oder retronasal wahrgenommenen charakteristischen Geruchsmerkmale eines Öls aus gesunden, frischen Oliven, bei dem weder grüne noch reife Fruchtigkeit vorherrscht. |
|
Grüne Fruchtigkeit |
Gesamtheit der von der Olivensorte abhängigen, unmittelbar und/oder retronasal wahrgenommenen charakteristischen Geruchsmerkmale eines Öls aus grünen, gesunden, frischen Oliven, das an grüne Früchte erinnert. |
|
Reife Fruchtigkeit |
Gesamtheit der von der Olivensorte abhängigen, unmittelbar und/oder retronasal wahrgenommenen charakteristischen Geruchsmerkmale eines Öls aus gesunden, frischen Oliven, das an reife Früchte erinnert. |
|
Ausgewogen |
Ein Öl, das nicht unausgewogen ist. Ausgewogenheit bezeichnet den olfaktorisch-gustatorischen und taktilen Sinneseindruck bei einem Öl, in dem der Median des Attributs „bitter“ und der des Attributs „scharf“ nicht mehr als zwei Punkte größer ist als der Median des Attributs „fruchtig“. |
|
Mildes Öl |
Ein Öl, in dem der Median des Attributs „bitter“ und der des Attributs „scharf“ kleiner oder gleich 2 sind. |
Bezeichnungen je nach Intensität der Wahrnehmung:
|
Bezeichnungen, für die eine Bescheinigung über eine organoleptische Prüfung vorzulegen ist |
Median des Attributs |
|
Fruchtigkeit |
— |
|
Reife Fruchtigkeit |
— |
|
Grüne Fruchtigkeit |
— |
|
Leichte Fruchtigkeit |
Kleiner als 3 |
|
Mittlere Fruchtigkeit |
Zwischen 3 und 6 |
|
Intensive Fruchtigkeit |
Größer als 6 |
|
Leichte reife Fruchtigkeit |
Kleiner als 3 |
|
Mittlere reife Fruchtigkeit |
Zwischen 3 und 6 |
|
Intensive reife Fruchtigkeit |
Größer als 6 |
|
Leichte grüne Fruchtigkeit |
Kleiner als 3 |
|
Mittlere grüne Fruchtigkeit |
Zwischen 3 und 6 |
|
Intensive grüne Fruchtigkeit |
Größer als 6 |
|
Leichte Bitterkeit |
Kleiner als 3 |
|
Mittlere Bitterkeit |
Zwischen 3 und 6 |
|
Intensive Bitterkeit |
Größer als 6 |
|
Leichte Schärfe |
Kleiner als 3 |
|
Mittlere Schärfe |
Zwischen 3 und 6 |
|
Intensive Schärfe |
Größer als 6 |
|
Ausgewogenes Öl |
Der Median des Attributs „bitter“ und der Median des Attributs „scharf“ sind nicht mehr als 2 Punkte größer als der Median des Attributs „fruchtig“. |
|
Mildes Öl |
Der Median des Attributs „bitter“ und der Median des Attributs „scharf“ sind nicht größer als 2. |
4. PRÜFGLAS FÜR ÖLE
Vgl. hierzu den Standard IOC/T.20/Dok. Nr. 5, „Glass for Oil Tasting“.
5. PRÜFRAUM
Vgl. hierzu den Standard IOC/T.20/Dok. Nr. 6, „Guide for the Installation of a Test Room“.
6. ZUBEHÖR
Jedem Prüfer ist in seiner Kabine das folgende zur Erfüllung seiner Aufgabe notwendige Zubehör zur Verfügung zu stellen:
— Gläser (genormt) mit den Proben, mit Code-Nummer versehen, mit einem Uhrglas abgedeckt und bei 28 °C ± 2 °C gehalten;
— Profilbeschreibung („Profile Sheet“, siehe Abbildung 1) auf Papier oder in elektronischer Form, sofern die Vorgaben für die Profilbeschreibung erfüllt sind, sowie gegebenenfalls Ausfüllanweisungen;
— Kugelschreiber oder dokumentenechte Tinte;
— Schälchen mit Apfelstücken und/oder Wasser, kohlensäurehaltiges Wasser und/oder Zwieback;
— Glas Wasser von Raumtemperatur;
— Hinweisbogen zu den allgemeinen Regeln in Abschnitt 8.4 und 9.1.1;
— Spucknäpfe.
7. PRÜFUNGSLEITER UND PRÜFER
7.1. Prüfungsleiter
Der Prüfungsleiter muss ausreichend vorgebildet sein und über das nötige Fachwissen für die zu Beurteilung anstehenden Olivenöle verfügen. Er ist die Schlüsselfigur der Prüfergruppe und trägt die Verantwortung für Organisation und Ablauf der Prüfungsarbeit.
Die Arbeit des Prüfungsleiters erfordert eine Grundausbildung im Umgang mit den Hilfsmitteln der sensorischen Analyse, sensorisches Feingefühl, Sorgfalt bei der Vorbereitung, Organisation und Durchführung der Prüfungen sowie das nötige Können und die Geduld für deren wissenschaftliche Planung und Abwicklung.
Der Prüfungsleiter ist alleinverantwortlich für die Auswahl, die Schulung und die Überwachung der Prüfer auf ihre Eignung. Somit obliegt ihm die Beurteilung der Prüfer, die stets objektiv sein muss und für die der Prüfleiter konkrete Verfahren entwickeln muss, die auf Tests und auf soliden Zulassungs- und Ausschlusskriterien beruhen. Siehe dazu den Standard IOC/T.20/Dok. Nr. 14, „Guide for the selection, training and monitoring of skilled virgin olive oil tasters“.
Der Prüfungsleiter ist verantwortlich für die Leistung der Gruppe und somit auch für ihre Bewertung, für die er zuverlässige objektive Nachweise erbringen muss. In jedem Falle muss er jederzeit nachweisen können, dass die Methodik und die Prüfer unter Aufsicht stehen. Es wird empfohlen, in regelmäßigen Abständen eine Neukalibrierung der Gruppe vorzunehmen (IOC/T.20/Dok. Nr. 14, § 5).
Er ist in letzter Instanz für das Führen der Aufzeichnungen der Gruppe verantwortlich. Diese Aufzeichnungen müssen stets nachverfolgbar sein. Sie müssen den Leistungs- und Qualitätsanforderungen internationaler Standards für die sensorische Prüfung entsprechen und jederzeit die Anonymität der Proben gewährleisten.
Der Prüfungsleiter ist verantwortlich für das Vorhandensein sowie die ordnungsgemäße Reinigung und Instandhaltung der Hilfsmittel und Ausrüstungsgegenstände, die für die vorschriftsmäßige Anwendung dieser Methode erforderlich sind, und führt darüber sowie über die Einhaltung der Prüfbedingungen Buch.
Ihm obliegen die Entgegennahme und Lagerung der im Labor eintreffenden Proben sowie deren Lagerung nach erfolgter Prüfung. Dabei hat er jederzeit sicherzustellen, dass die Proben anonym bleiben und ordnungsgemäß gelagert werden; zu diesem Zweck muss er ein schriftliches Verfahren entwickeln, das die Rückverfolgbarkeit des gesamten Prozesses gewährleistet und Sicherheiten bietet.
Darüber hinaus ist er verantwortlich für die Vorbereitung, die Kodierung und die Übergabe der Proben an die Prüfer unter Einhaltung eines geeigneten Versuchsplans und vorgegebener Protokolle sowie für das Einsammeln und die statistische Auswertung der von den Prüfern ausgehändigten Prüfdaten.
Er ist zuständig für die Entwicklung und Darlegung etwaiger sonstiger Verfahren, die in Ergänzung zu diesem Standard erforderlich sind, um eine ordnungsgemäße Arbeitsweise der Gruppe sicherzustellen.
Er bemüht sich um Vergleiche der Ergebnisse der Gruppe mit den Ergebnissen anderer Gruppen, die natives Olivenöl prüfen, um festzustellen, ob die Gruppe ordnungsgemäß arbeitet.
Aufgabe des Prüfungsleiters ist es, die Mitglieder zu motivieren und bei ihnen Interesse, Neugier und Wettbewerbsgeist zu wecken. Daher wird ihm nachdrücklich empfohlen, einen reibungslosen beiderseitigen Informationsfluss mit den Mitgliedern der Gruppe sicherzustellen, indem er sie ständig über seine Arbeitsaufgaben und über die gewonnenen Ergebnisse auf dem Laufenden hält. Zudem hat er sich einer Meinungsäußerung zu enthalten und muss sicherstellen, dass möglicherweise tonangebende Prüfer die anderen nicht beeinflussen.
Er lädt die Prüfer rechtzeitig ein und beantwortet etwaige Fragen hinsichtlich der Durchführung der Prüfungen, enthält sich aber jedweder Meinungsäußerung über die Proben.
7.1.1. Stellvertretender Prüfungsleiter
Der Prüfungsleiter kann aus berechtigten Gründen durch einen Stellvertreter ersetzt werden, der an seiner Stelle Aufgaben hinsichtlich der Durchführung der Prüfungen wahrnimmt. Dieser Stellvertreter muss über alle Fertigkeiten verfügen, die von einem Prüfungsleiter verlangt werden.
7.2. Prüfer
Die Personen, die an der organoleptischen Prüfung von Olivenöl als Prüfer teilnehmen, müssen dies freiwillig tun. Daher sind schriftliche Bewerbungen anzuraten. Die Bewerber werden von Prüfungsleiter entsprechend ihrer Eignung zur Unterscheidung ähnlicher Proben ausgewählt, geschult und begleitet, wobei zu berücksichtigen ist, dass ihre Urteilsfähigkeit im Zuge der Schulung zunimmt.
Die Prüfer müssen in sensorischer Hinsicht eine rein beobachtende Funktion ausüben, ihre persönlichen geschmacklichen Vorlieben außer Acht lassen und lediglich ihre Sinneseindrücke wiedergeben. Dabei kommt es darauf an, dass sie ihre Arbeit stets schweigend sowie entspannt und ohne Eile verrichten, um der zu prüfenden Probe maximale sensorische Aufmerksamkeit widmen zu können.
Für jede Prüfung werden 8 bis 12 Prüfer benötigt, nebst einigen Reserveprüfern zur Deckung von etwaigen Ausfällen.
8. PRÜFBEDINGUNGEN
8.1. Präsentation der Probe
Die zu prüfenden Olivenölproben werden in standardisierten Prüfgläsern dargereicht, die dem Standard IOC/T.20/Dok. Nr. 5 „Glass for oil tasting“ entsprechen.
Ein Glas enthält 14–16 ml Öl - bzw. 12,8 bis 14,6 g Öl, falls die Proben gewogen werden sollen – und ist mit einem Uhrglas bedeckt.
Jedes Glas wird mit einem willkürlich ausgewählten Code aus Ziffern oder aus einer Kombination von Buchstaben und Ziffern versehen. Der Code wird mit Hilfe eines geruchsfreien Systems angebracht.
8.2. Prüf- und Probentemperatur
Die zur Prüfung anstehenden Proben sind während der gesamten Prüfung im Glas auf einer Temperatur von 28 °C ± 2 °C zu halten. Diese Temperatur wurde gewählt, weil sich die organoleptischen Unterschiede auf diese Weise leichter erfassen lassen als bei Umgebungstemperatur und weil die Aromastoffe dieser Öle bei niedrigeren Temperaturen kaum zur Entfaltung kommen, während sich bei höheren Temperaturen die für erhitzte Öle typischen Aromastoffe bilden. Siehe dazu den Standard IOC/T.20/Dok. Nr. 5 „Glass for Oil Tasting“, in dem die Methode zur Erwärmung der Proben im Glas beschrieben wird.
Im Prüfraum muss eine Temperatur zwischen 20 °C und 25 °C herrschen (siehe IOC/T.20/Dok. Nr. 6).
8.3. Prüfungszeiten
Die für das Verkosten von Olivenöl geeignetste Tageszeit ist der Vormittag. Geruchs- und Geschmackssinn sind zu bestimmten Tageszeiten nachweislich besonders empfindlich. So ist die Empfindlichkeit von Geruchs- und Geschmackssinn vor den Mahlzeiten besonders groß und geht danach zurück.
Übertreibung ist aber auch hier zu vermeiden, da Hungergefühle die Prüfer ablenken und so ihr Unterscheidungsvermögen beeinträchtigen; es wird daher empfohlen, die Prüfgänge zwischen 10.00 und 12.00 Uhr durchzuführen.
8.4. Prüfer: allgemeine Verhaltensmaßregeln
Die folgenden Empfehlungen gelten für das Verhalten der Prüfer bei ihrer Arbeit.
Wer vom Prüfungsleiter zur Teilnahme an einer organoleptischen Prüfung aufgefordert wurde, muss zu der ihm genannten Uhrzeit zur Teilnahme in der Lage sein und hat dazu folgende Vorschriften zu beachten:
— Er stellt mindestens 30 Minuten vor der festgesetzten Uhrzeit das Rauchen und Kaffeetrinken ein.
— Parfüm, Kosmetika oder Seife, deren Geruch bei der Prüfung noch wahrnehmbar ist, darf nicht verwendet werden. Zum Händewaschen ist unparfümierte Seife zu verwenden; dabei sind die Hände so lange mit Wasser nachzuspülen und abzutrocknen, bis jedweder Geruch beseitigt ist.
— Der Prüfer muss bei Prüfungsantritt seit mindestens einer Stunde nüchtern sein.
— Wer an einer Unpässlichkeit und insbesondere einer Beeinträchtigung des Geruchs- oder Geschmacksempfindens oder an einer die Konzentrationsfähigkeit beeinflussenden psychischen Belastung leidet, sieht von der Verkostung ab und informiert den Prüfungsleiter entsprechend.
— Der die vorstehenden Vorschriften erfüllende Prüfer sucht die ihm zugewiesene Kabine auf; dies hat so leise und diskret wie möglich zu geschehen.
— Er liest die Hinweise auf dem Prüfbogen sorgfältig durch und beginnt erst dann mit der Prüfung der Proben, wenn er sich über seine Aufgabe vollends im Klaren ist (entspannt und ohne Eile). Unklarheiten sind mit dem Prüfungsleiter unter vier Augen zu klären.
— Er verrichtet seine Arbeit schweigend.
— Er hat sein Mobiltelefon für die gesamte Dauer der Prüfung abgeschaltet, damit die Kollegen nicht in ihrer Konzentration beeinträchtigt und bei ihrer Arbeit gestört werden.
9. VERFAHREN FÜR DIE ORGANOLEPTISCHE BEWERTUNG UND EINSTUFUNG VON NATIVEM OLIVENÖL
9.1. Verkostungsverfahren
9.1.1. Der Prüfer nimmt das Glas zur Hand, hält es dabei mit dem Uhrglas bedeckt schräg und schwenkt es dabei einmal ganz um, damit die Innenseite möglichst ganz benetzt wird. Danach lüftet er das Uhrglas und inhaliert das Bukett der Probe in ruhigen und langen Zügen durch die Nase, um das Öl zu bewerten. Das eigentliche Riechen sollte nicht länger als 30 Sekunden dauern. Gelangt der Prüfer innerhalb dieser Zeit nicht zu einem Urteil, legt er eine kleine Pause ein und macht einen weiteren Versuch.
Nach dem Riechen prüft er das Flavour (Gesamtsinneseindruck aus Geruchs-, Geschmacks- und Tastempfindung). Dazu nippt er einen kleinen Schluck Öl von etwa 3 ml. Sehr wichtig ist, dass alle geschmacksempfindlichen Teile des Mundes mit dem Öl benetzt werden, vom vorderen Teil des Mundes und der Zungenspitze über die Ränder des Zungenrückens bis zur Zungenwurzel und zur Kehle, da die Geschmackswahrnehmung und die taktilen Wahrnehmungen an verschiedenen Stellen der Zunge, des Gaumens und der Kehle unterschiedlich stark sind.
Es ist unbedingt darauf zu achten, dass genügend Olivenöl von der Zungenspitze bis zum Gaumen und zur Zungenwurzel langsam verteilt wird, wobei auf die Reihenfolge des Auftretens der Bitterkeit und der Schärfe zu achten ist. Anderenfalls kann es bei manchen Olivenölen vorkommen, dass beide Sinneseindrücke nicht wahrgenommen werden oder die Bitterkeit von der Schärfe verdeckt wird.
Durch kurzes, wiederholtes Einsaugen von Luft durch den Mund wird die Probe in der gesamten Mundhöhle verteilt, sodass die flüchtigen Aromastoffe zwangsläufig über den Gaumen in die Nase gelangen.
Anmerkung: Nimmt der Prüfer bei einer Probe keine Fruchtigkeit wahr und liegt die Intensität des die Einstufung bestimmenden negativen Attributs bei 3,5 oder weniger, kann der Prüfungsleiter beschließen, dass die Tester die Probe bei Raumtemperatur erneut analysieren (COI/T.20/Doc. Nr. 6/Rev. 1, September 2007, Abschnitt 3 — Allgemeine Vorschriften für die Einrichtung eines Testraums); dabei muss er die Umgebungsbedingungen und den Begriff der Raumtemperatur genau festlegen. Hat die Probe Raumtemperatur erreicht, sollte der Prüfer diese erneut bewerten, um ausschließlich zu prüfen, ob Fruchtigkeit wahrgenommen wird. Falls dies der Fall ist, sollte die entsprechende Intensität angegeben werden.
Auch die taktile Wahrnehmung von Schärfe sollte berücksichtigt werden. Dafür ist es ratsam, das Öl hinunterzuschlucken.
9.1.2. Es wird empfohlen, bei der organoleptischen Prüfung von nativem Olivenöl maximal VIER PROBEN in jedem Prüfgang zu bewerten und maximal drei Prüfgänge pro Tag durchzuführen, damit keine Kontrastwirkungen durch das sofortige Verkosten anderer Proben auftreten.
Da es bei aufeinanderfolgenden Prüfgängen zu Ermüdungserscheinungen oder zur Dämpfung der Sinneswahrnehmung durch die vorherigen Proben kommt, sind die Ölreste des vorherigen Prüfgangs mit einem geeigneten Mittel aus dem Mund zu entfernen.
Dazu empfiehlt sich ein kleines Stück Apfel, das nach dem Kauen ausgespuckt werden kann; anschließend ist der Mund mit Wasser von Zimmertemperatur zu spülen. Zwischen dem Ende eines Prüfgangs und dem Beginn des nächsten müssen mindestens 15 Minuten vergehen.
9.2. Verwendung der Profilbeschreibung durch den Prüfer
Die zu verwendende Profilbeschreibung ist in Abbildung 1 dieses Anhangs dargestellt.
Jeder zur Prüfergruppe gehörende Prüfer muss das zu untersuchende Öl zunächst riechen und dann verkosten ( 11 ). Anschließend trägt er auf der 10-cm-Skala in der vorliegenden Profilbeschreibung die Intensität der Wahrnehmung jedes negativen und positiven Attributs ein.
Werden negative Attribute wahrgenommen, die nicht in Abschnitt 4 aufgeführt sind, so sind diese unter Verwendung derjenigen Begriffe, mit denen sie am zutreffendsten beschrieben werden, im Feld „Sonstige“ anzugeben.
9.3. Verwendung der Angaben durch den Prüfungsleiter
Der Prüfungsleiter sammelt die ausgefüllten Profilbeschreibungen der einzelnen Prüfer ein, um die den einzelnen Attributen zugeteilten Intensitäten zu überprüfen; bei Feststellung einer Anomalie fordert er den Prüfer auf, seine Profilbeschreibung zu überarbeiten und den Prüfversuch erforderlichenfalls zu wiederholen.
Der Prüfungsleiter gibt die Bewertungsdaten der einzelnen Mitglieder der Prüfergruppe in ein Computerprogramm entsprechend dem Standard IOC/T.20/Doc. Nr. 15 ein, um eine statistische Berechnung der Analyseergebnisse auf Grundlage der Berechnung des Medians vorzunehmen; siehe dazu Abschnitt 9.4 und die Anlage zu diesem Anhang. Die Daten für eine Probe werden eingegeben anhand einer Matrix aus 9 Spalten, die jeweils den 9 sensorischen Attributen entsprechen, und n Zeilen, die den n Prüfern der Prüfergruppe entsprechen.
Wird ein von mindestens 50 % der Mitglieder der Prüfergruppe wahrgenommener Mangel unter „Sonstige“ eingetragen, so berechnet der Prüfungsleiter den Median dieses Mangels und nimmt die entsprechende Einstufung vor.
Der Wert des robusten Variationskoeffizienten, der die Einstufung bestimmt (Mangel mit der größten Intensität und Attribut „fruchtig“), muss kleiner oder gleich 20 % sein.
Ist dies nicht der Fall, muss der Prüfungsleiter für eine erneute Bewertung der spezifischen Probe in einem weiteren Prüfgang sorgen.
Sollte dies häufig vorkommen, wird dem Prüfungsleiter empfohlen, eine spezifische Fortbildung für die Prüfer durchzuführen (IOC/T.20/Doc. Nr. 14, § 5) und die Prüfungsleistung anhand des Wiederholbarkeitsindex und des Abweichungsindex zu beurteilen (IOC/T.20/Doc. Nr. 14, § 6).
9.4. Einstufung der Öle
Das Öl wird entsprechend dem Median der festgestellten Mängel und dem Median des Attributs „fruchtig“ in die nachstehenden Kategorien eingestuft. Der Median der Mängel ist definiert als der Median des mit der stärksten Intensität wahrgenommenen Mangels. Der Median der Mängel und der Median der Fruchtigkeit werden mit einer Dezimalstelle ausgedrückt.
Für die Einstufung des Öls wird der Wert des Medians der Mängel und des Medians der Fruchtigkeit mit den nachstehend aufgeführten Referenzintervallen verglichen. Die Grenzen dieser Intervalle wurden unter Berücksichtigung des Fehlers der Methode festgesetzt und gelten daher als absolut. Eine entsprechende Software gestattet eine visuelle Darstellung der Einstufung in tabellarischer oder grafischer Form.
a) Natives Olivenöl extra: Der Median der Mängel ist 0, und der Median des Attributs „fruchtig“ ist größer als 0.
b) Natives Olivenöl: Der Median der Mängel ist größer als 0, aber nicht größer als 3,5, und der Median des Attributs „fruchtig“ ist größer als 0.
c) Lampantöl: Der Median der Mängel ist größer als 3,5 oder der Median der Mängel ist nicht größer als 3,5, und der Median des Attributs „fruchtig“ ist gleich 0.
Anmerkung 1: Ist der Median des Attributs „bitter“ und/oder der des Attributs „scharf“ größer als 5,0, so vermerkt der Prüfungsleiter dies auf der Prüfbescheinigung.
Im Falle von Bewertungen im Rahmen von Konformitätskontrollen wird ein Test vorgenommen. Im Fall von Gegenbewertungen muss die Analyse zweimal in verschiedenen Prüfgängen stattfinden. Die Ergebnisse dieser Doppelanalyse müssen statistisch homogen sein (siehe Abschnitt 9.5). Ist dies nicht der Fall, muss die Probe erneut zweimal analysiert werden. Der endgültige Wert des Medians der Einstufungsattribute wird anhand des Durchschnitts der beiden Mediane berechnet.
9.5. Kriterien für die Annahme bzw. das Verwerfen von Doppelanalysen
Der nachstehend definierte standardisierte Fehler wird herangezogen, um zu ermitteln, ob die beiden Ergebnisse einer Doppelanalyse homogen bzw. statistisch verwertbar sind:

Dabei sind Me1 und Me2 die Mediane der Doppelanalyse (d. h. der ersten und der zweiten Prüfung) und U1 und U2 die erweiterten Unsicherheiten für die beiden Werte, die wie folgt gemäß der Anlage berechnet werden:
U
1 = c × s* und

Für die erweiterte Unsicherheit ist c gleich 1,96; folglich:
U1 = 0,0196 × CVr × Me1.
Dabei ist CVr der robuste Variationskoeffizient.
Damit die beiden ermittelten Werte als nicht statistisch unterschiedlich gelten, muss En gleich oder weniger als 1,0 sein.
Anlage
Methode zur berechnung des medians und der vertrauensintervalle
Median
![]()
Der Median ist definiert als die reelle Zahl Xm, gekennzeichnet durch die Tatsache, dass die Wahrscheinlichkeit (p), dass die Werte der Verteilung (X) unter dieser Zahl (Xm) liegen, geringer oder gleich 0,5 ist, und dass gleichzeitig die Wahrscheinlichkeit (p), dass die Werte der Verteilung (X) unter dieser Zahl Xm liegen oder ihr entsprechen, höher oder gleich 0,5 ist. Nach einer praktischeren Definition ist der Median das 50. Perzentil einer Zahlenverteilung in steigender Reihenfolge. Einfacher ausgedrückt: Der Median ist der Wert in der Mitte einer geordneten Datenreihe, wenn es sich um eine ungerade Anzahl von Datenwerten handelt, bzw. entspricht dem Durchschnitt der beiden mittleren Werte, wenn es sich um eine gerade Anzahl von Datenwerten handelt.
Robuste Standardabweichung
Um eine zuverlässige Schätzung für die Variabilität um den Mittelwert zu erhalten, ist der Schätzwert der robusten Standardabweichung nach Stuart und Kendall heranzuziehen (4). Die Formel ergibt die asymptotische Standardabweichung, d. h. den robusten Schätzwert der Variabilität der betreffenden Angaben, wobei N die Zahl der Beobachtungen und IQR der Quartilabstand ist, der genau 50 % der Fälle einer gegebenen Wahrscheinlichkeitsverteilung umfasst:

Zur Berechnung des Quartilabstands wird die Größe der Abweichung zwischen dem 75. und 25. Perzentil berechnet.
![]()
Dabei ist das Perzentil der Wert Xpc, gekennzeichnet durch die Tatsache, dass die Wahrscheinlichkeit (p), dass die Werte der Verteilung unter Xpc liegen, niedriger als ein bestimmtes Hundertstel ist oder ihm entspricht und dass gleichzeitig die Wahrscheinlichkeit (p), dass die Werte der Verteilung niedriger als Xpc sind oder ihm entsprechen, höher als das bestimmte Hundertstel ist oder ihm entspricht. Das Hundertstel gibt das gewählte Fraktil der Verteilung an. In Fall des Medians entspricht es 50/100.

In der Praxis ist das Perzentil der Verteilungswert, der einem bestimmten Bereich unterhalb der Verteilungs- oder Dichtekurve entspricht. Beispiel: das 25. Perzentil ist der Verteilungswert, der einem Bereich von 0,25 oder 25/100 entspricht.
Bei dieser Methode werden Perzentile auf der Grundlage der realen Werte berechnet, die in der Datenmatrix erscheinen (Perzentilberechnungsverfahren).
Robuster Variationskoeffizient (%)
Der CVr% stellt eine reine Zahl dar, die den Prozentsatz der Variabilität des analysierten Zahlensatzes angibt. Daher ist dieser Koeffizient zur Überprüfung der Zuverlässigkeit der Mitglieder der Prüfergruppe besonders geeignet.

Vertrauensintervalle des Medians bei 95 %
Das Vertrauensintervall von 95 % (der Wert des Fehlers erster Art entspricht 0,05 oder 5 %) ist das Intervall, in dem der Median, ausgehend von der Hypothese, dass sich der Versuch unendliche Male wiederholen ließe, schwanken könnte. In der Praxis gibt es das Variabilitätsintervall des Versuchs unter den vorgegebenen operationellen Bedingungen an, in der Annahme, dass der Versuch viele Male wiederholt werden könnte. Das Intervall ist, wie der CVr%, nützlich zur Beurteilung der Zuverlässigkeit des Versuchs.
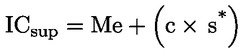

wobei C = 1,96 bei einem Vertrauensintervall auf 95-%-Niveau.
Ein Beispiel für den Berechnungsbogen ist in Anhang I des Standards IOC/T20/Dok. Nr. 15 enthalten.
Literaturhinweise
(1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.
(2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
(3) Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.
(4) Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
(5) McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
(6) IOC/T.28/Dok. Nr. 1 September 2007, Guidelines for the accreditation of sensory testing laboratories with particular reference to virgin olive oil according to standard ISO/IEC 17025:2005.
(7) IOC/T.20/Dok. Nr. 14.
(8) IOC/T.20/Dok. Nr. 15.
(9) ISO/IEC 17025:05.
▼M20 —————
▼M19 —————
ANHANG XV
1. ÖLGEHALT DER OLIVENTRESTER
1.1. Geräte
— Geeigneter Extraktionsapparat mit 200- bis 250-ml-Kolben,
— elektrisch beheiztes Bad (Sandbad, Wasserbad usw.) oder Heizplatte,
— Analysenwaage,
— Trockenschrank, eingestellt auf höchstens 80 °C,
— elektrischer Trockenschrank mit Thermostat, eingestellt auf 103 °C ± 2 °C, der unter Einblasen von Luft oder unter vermindertem Druck betrieben werden kann,
— mechanische Mühle, leicht zu reinigen, mit der die Trester ohne Erwärmung und ohne merkliche Verringerung ihres Gehalts an Wasser und Öl zerkleinert werden können,
— Extraktionshülse und Watte oder Filterpapier, frei von mit Hexan extrahierbaren Stoffen,
— Exsikkator,
— Sieb, Maschendurchmesser 1 mm,
— Siedesteinchen, zuvor getrocknet.
1.2. Reagenzien
— technisches n-Hexan; Rückstand bei vollständiger Verdampfung unter 0,002 g/100 ml.
2. ARBEITSVORSCHRIFT
2.1. Vorbereitung der Probe
Die Probe wird, wenn nötig, in der zuvor gut gereinigten mechanischen Mühle so weit gemahlen, daß die Teilchen vollständig das Sieb passieren können.
Etwa ein Zwanzigstel der Probe ist zur Reinigung der Mühle zu benutzen; dieses Mahlgut ist zu verwerfen. Der Rest ist fein zu mahlen, das Mahlgut aufzufangen, sorgfältig zu mischen und unverzüglich zu analysieren.
2.2. Untersuchungsprobe
Etwa 10 g der Probe werden nach dem Mahlen auf 0,01 g genau für die Untersuchung abgewogen.
2.3. Vorbereitung der Extraktionshülse
Die Probe wird in die Hülse gegeben und diese mit einem Wattebausch verschlossen. Bei Verwendung von Filterpapier wird das Mahlgut darin eingeschlagen.
2.4. Vertrocknung
Wenn die Trester sehr feucht sind (Gehalt an Wasser und flüchtigen Stoffen größer als 10 %), ist vorzutrocknen, wobei die gefüllte Hülse (bzw. das Filterpapier) so lange wie nötig in den auf höchstens 80 °C geheizten Trockenschrank gestellt wird, um den Gehalt an Wasser und flüchtigen Stoffen auf unter 10 % zu senken.
2.5. Vorbereitung des Kolbens
Der Kolben, der 1 bis 2 Siedesteinchen enthält und zuvor im Trockenschrank bei 103 °C ± 2 °C getrocknet und danach mindestens 1 Stunde lang im Exsikkator abgekühlt wurde, wird auf 1 mg genau gewogen.
2.6. Erste Extraktion
Die Hülse (bzw. das Filterpapier) mit der Probe wird in den Extraktionsapparat gestellt, die benötigte Menge Hexan in den Kolben gegeben, der Kolben an den Extraktionsapparat angeschlossen und das Ganze auf das elektrische Heizbad gestellt. Die Heizung ist so einzustellen, daß der Rückfluß mindestens 3 Tropfen in der Sekunde beträgt (mäßiges, nicht heftiges Sieden).
Nach 4stündiger Extraktion läßt man abkühlen. Die Hülse wird aus dem Extraktionsapparat genommen und in einen Luftstrom gestellt, um den größten Teil des Lösungsmittels, mit dem sie durchtränkt ist, zu entfernen.
2.7. Zweite Extraktion
Die Hülse wird in die Mikrokugelmühle entleert, und es wird so fein wie möglich gemahlen. Das Mahlgut wird quantitativ in die Hülse zurückgegeben und diese wieder in den Extraktionsapparat gestellt.
Es wird nochmals 2 Stunden extrahiert, wobei der die erste Extraktion enthaltene Kolben verwendet wird.
Die im Extraktionskolben enthaltene Lösung muß klar sein. Wenn nicht, ist sie über Filterpapier zu filtrieren, wobei der erste Kolben und das Filterpapier mehrmals mit Hexan gewaschen werden. Das Filtrat und die Waschflüssigkeit werden in einem zweiten, zuvor getrockneten und auf 1 mg abgewogenen Kolben aufgefangen.
2.8. Entfernung des Lösungsmittels und Wiegen des Extrakts
Durch Destillieren auf dem elektrischen Heizbad wird der größte Teil des Lösungsmittels entfernt. Die letzten Lösungsmittelspuren werden durch 20minütiges Erhitzen des Kolbens im Trockenschrank bei 103 °C ± 2 °C beseitigt. Die Beseitigung der Lösungsmittelreste wird durch zeitweiliges Einführen eines Luftstroms oder besser eines Inertgasstroms oder durch Arbeiten unter vermindertem Druck erleichtert.
Den Kolben läßt man wenigstens 1 Stunde im Exsikkator abkühlen und wiegt ihn dann auf 1 mg genau.
Danach wird der Kolben erneut 10 Minuten unter den gleichen Bedingungen erhitzt, im Exsikkator abgekühlt und gewogen.
Der Unterschied zwischen den Ergebnissen der zwei Wägungen darf höchstens 10 mg betragen. Wenn nicht, ist erneut 10 Minuten zu erhitzen, dann wieder abkühlen zu lassen und zu wiegen, bis der Gewichtsunterschied höchstens 10 mg beträgt. Das letzte Gewicht des Kolbens wird notiert.
Für jede Untersuchung werden mit der gleichen Probe zwei Bestimmungen durchgeführt.
3. ERGEBNISSE
3.1. Berechnung und Formel
a) Der Extrakt des Rohprodukts läßt sich in Gewichtsprozenten durch nachstehende Formel berechnen:

Dabei sind:
S = Gewichtsprozente des Extrakts des Rohprodukts,
m0 = Gewicht der Untersuchungsprobe in g,
m1 = Gewicht des Extrakts nach der Trocknung in g.
Als Ergebnis ist das arithmetische Mittel aus den beiden Bestimmungen zu nehmen, falls die Bedingungen der Wiederholbarkeit erfüllt sind.
Das Ergebnis wird auf eine Dezimalstelle angegeben.
b) Der Extrakt, bezogen auf den Trockenstoff, läßt sich berechnen durch die Formel:
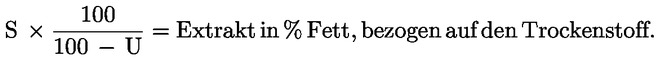
Dabei sind:
S = Gewichtsprozente des Extrakts des Rohprodukts (vgl. a)),
U = sein Gehalt an Wasser unf flüchtigen Stoffen.
3.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen von zwei gleichzeitig oder schnell nacheinander von ein und demselben Analytiker vorgenommenen Bestimmungen darf nicht mehr als 0,2 g Hexan-Extrakt je 100 g Probe betragen.
Andernfalls ist die Analyse mit zwei weiteren Untersuchungsproben zu wiederholen. Liegt die Differenz wieder über 0,2 g, so ist als Ergebnis das arithmetische Mittel aus allen vier Bestimmungen zu nehmen.
ANHANG XVI
BESTIMMUNG DER IODZAHL
1. ANWENDUNGSBEREICH
Diese Internationale Norm beschreibt ein Verfahren zur Bestimmung der Iodzahl von tierischen und pflanzlichen Fetten und Ölen, nachfolgend als „Fette“ bezeichnet.
2. DEFINITION
In dieser Internationalen Norm gilt folgende Definition:
|
2.1. |
Iodzahl: Iodmenge, die unter den in dieser Internationalen Norm beschriebenen Versuchsbedingungen von der Probe aufgenommen wird. Die Iodzahl wird in Gramm Iod pro 100 g Probe angegeben. |
3. PRINZIP
Die zu untersuchende Probe wird in dem Lösungsmittel gelöst und mit dem Wijs-Reagens versetzt. Nach einer bestimmten Zeit werden Kaliumiodidlösung und Wasser zugegeben und das freigesetzte Iod mit einer Natriumthiosulfatlösung titriert.
4. REAGENZIEN
Alle Reagenzien müssen von anerkannter analysenreiner Qualität sein.
|
4.1. |
Kaliumiodidlösung, 100 g/l, die kein Iodat oder freies Iod enthält. |
|
4.2. |
Stärkelösung 5 g lösliche Stärke mit 30 ml Wasser mischen und in 1 000 ml kochendes Wasser geben; 3 Minuten kochen und abkühlen lassen. |
|
4.3. |
Natriumthiosulfat, eingestellte Standardlösung c (Na2S2O3 · 5H2O) = 0,1 mol/l, höchstens 7 Tage vor der Verwendung eingestellt. |
|
4.4. |
Lösungsmittel, hergestellt durch Mischen gleicher Volumenteile Cyclohexan und Essigsäure. |
|
4.5. |
Wijs-Reagens, das Iodmonochlorid in Essigsäure enthält. Es soll das im Handel erhältliche Wijs-Reagens verwendet werden. Anmerkung: Das Reagens enthält 9 g ICl3 + 9 g I in Essigsäure. |
5. GERÄTE
Übliche Laboreinrichtung und insbesondere nachstehende Geräte:
|
5.1. |
Mikrobechergläschen, die sich für die zu untersuchende Probe und zum Einführen in die Kolben (5.2) eignen. |
|
5.2. |
500-ml-Erlenmeyerkolben mit Schliffstopfen, vollständig trocken. |
6. VORBEREITUNG DER ANALYSEPROBE
Die homogenisierte Probe wird über Natriumsulfat getrocknet und filtriert.
7. VERFAHREN
7.1. Probemenge
Die Menge der zu untersuchenden Probe hängt von der erwarteten Iodzahl ab (Tabelle 1).
Tabelle 1
|
Erwartete Iodzahl |
Masse der zu untersuchenden Probe |
|
weniger als 5 |
3,00 g |
|
5— 20 |
1,00 g |
|
21— 50 |
0,40 g |
|
51—100 |
0,20 g |
|
101—150 |
0,13 g |
|
151—200 |
0,10 g |
Die zu untersuchende Probe wird in einem Mikrobechergläschen (5.1) auf 0,1 mg genau eingewogen.
7.2. Bestimmung
Die Probe wird in einen 500-ml-Kolben (5.2) gegeben und mit 20 ml Lösungsmittel (4.5) versetzt, um das Fett zu lösen. Genau 25 ml Wijs-Reagens (4.6) zugeben, den Kolben mit dem Stopfen verschließen, schwenken und dunkel stellen. Für das Wijs-Reagens sollen keine Pipetten verwendet werden, die mit dem Mund aufgezogen werden müssen.
Dann wird in ähnlicher Weise eine Blindprobe vorbereitet; die das Lösemittel und das Reagens, nicht aber die Probe enthält.
Bei Proben mit Iodzahlen unter 150 muß der Kolben 1 Stunde dunkel gestellt werden; bei Iodzahlen über 150 sowie bei polymerisierten oder stark oxidierten Erzeugnissen sind es 2 Stunden.
Nach Ablauf dieser Zeit wird in jeden Kolben 20 ml Kaliumiodidlösung (4.1) und 150 ml Wasser gegeben.
Nun mit der eingestellten Natriumthiosulfat-Standardlösung (4.3) titrieren, bis die durch Iod bedingte Gelbfärbung kaum noch sichtbar ist. Einige Tropfen Stärkelösung (4.2) zugeben und weiter titrieren, bis die Blaufärbung unter starkem Schütteln verschwindet.
Anmerkung:
Eine potentiometrische Bestimmung des Endpunkts ist zulässig.
7.3. Zahl der Bestimmungen
Es werden zwei Bestimmungen mit der gleichen Probe durchgeführt.
8. DARSTELLUNG DER ERGEBNISSE
Die Iodzahl wird nach folgender Gleichung berechnet:
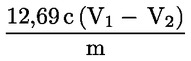
Hierin bedeuten:
c = die genaue zahlenmäßige Angabe der Konzentration der verwendeten Natriumthiosulfat-Standardlösung (4.3) in mol pro Liter;
V1 = Volumen der für den Blindversuch verwendeten Natriumthiosulfat-Standardlösung (4.3) in Millilitern;
V2 = Volumen der zur Bestimmung verwendeten Natriumthiosulfat-Standardlösung (4.3) in Millilitern;
m = Masse der untersuchten Probe (7.1) in Gramm.
Als Ergebnis ist das arithmetische Mittel der beiden Bestimmungen anzugeben.
ANHANG XVII
METHODE ZUR BESTIMMUNG VON STIGMASTADIENEN IN PFLANZLICHEN ÖLEN
1. ZWECK
Bestimmung von Stigmastadienen in pflanzlichen Ölen, die diese Kohlenwasserstoffe nur in geringer Konzentration enthalten, insbesondere in nativen Olivenölen und rohem Oliventresteröl.
2. ANWENDUNGSBEREICH
Das Verfahren läßt sich auf alle pflanzlichen Öle anwenden. Die Bestimmungen sind allerdings nur zuverlässig bei einer Konzentration dieses Kohlenwasserstoffs von 0,01 bis 4,0 mg/kg. Die Methode eignet sich besonders zum Nachweis von raffinierten pflanzlichen Ölen (Olivenöl, Oliventresteröl, Sonnenblumenöl, Palmöl usw.) in nativem Olivenöl, da raffinierte Öle im Gegensatz zu nativen Ölen Stigmastadien enthalten.
3. PRINZIP
Isolierung der unverseifbaren Bestandteile. Abtrennung der Steroid-Fraktion durch Säulenchromatographie an Kieselgel und Analyse durch Kapillargaschromatographie.
4. GERÄTE
|
4.1. |
250-ml-Stehkolben, zur Verwendung mit Rückflußkühler geeignet. |
|
4.2. |
200-ml-Scheidetrichter. |
|
4.3. |
100-ml-Rundkolben. |
|
4.4. |
Rotationsverdampfer. |
|
4.5. |
Chromatographiesäule aus Glas (innerer Durchmesser 1,5 bis 2,0 cm, Länge 50 cm) mit Teflonhahn und Glaswattebausch oder Sinterglasscheibe am unteren Ende. Zur Vorbereitung der Kieselgelsäule mit Chromatographiesäule bis zu einer Höhe von etwa 5 cm mit Hexan füllen und dann eine Suspension von 15 g Kieselgel in 40 ml Hexan mit Hilfe von mehreren Anteilen Hexan einschlämmen und unter leichtem Klopfen vollständig absetzen lassen. Anschließend wasserfreies Natriumsulfat bis zu einer Höhe von rund 0,5 cm zugeben und das überschüssige Hexan ablaufen lassen. |
|
4.6. |
Gaschromatograph mit Flammenionisationsdetektor, Split- oder cold On-column-Injektor und Ofen mit einer Einstellgenauigkeit von ± 1 °C. |
|
4.7. |
Fused-Silica-Kapillarsäule für Gaschromatographie (0,25 oder 0,32 mm Innendurchmesser und 25 m Länge), belegt mit 5 % Phenylmethylsilicon, Filmdicke 0,25 μm. Anmerkung 1: Andere Säulen mit vergleichbarer oder geringerer Polarität können ebenfalls verwendet werden. |
|
4.8. |
Aufzeichnungsgerät mit Integrator mit Möglichkeit der Integration zwischen zwei Minima. |
|
4.9. |
5-10-μl-Mikrospritze für Gaschromatographie mit fester Nadel. |
|
4.10. |
Pilzheizhaube oder Heizplatte. |
5. REAGENZIEN
Alle Reagenzien müssen, sofern nicht anders angegeben, von Analysequalität sein. Es ist destilliertes Wasser oder Wasser von mindestens gleichem Reinheitsgrad zu verwenden.
|
5.1. |
Hexan oder Alkanmischung mit einem Siedebereich von 65-70 °C, destilliert in einer Rektifiziersäule. Anmerkung 2: Das Lösungsmittel muß zur Entfernung von Verunreinigungen destilliert werden. |
|
5.2. |
Ethanol, 6 %ig (v/v). |
|
5.3. |
Wasserfreies Natriumsulfat. |
|
5.4. |
Ethanolische Kalilauge, 10 %ige Lösung. 10 ml Wasser zu 50 g Kaliumhydroxid geben, umrühren und die Mischung durch Zugabe von Ethanol lösen und mit Ethanol auf 500 ml auffüllen. Anmerkung 3: Ethanolische Kalilauge wird beim Stehen braun. Sie sollte täglich frisch zubereitet und in dunklen Glasflaschen gut verschlossen aufbewahrt werden. |
|
5.5. |
Kieselgel 60 für Säulenchromatographie, 70-230 mesh (Merck, Art. Nr. 7734 o. ä.). Anmerkung 4: Gewöhnlich kann Kieselgel direkt aus dem Behälter ohne Vorbehandlung verwendet werden. Manche Kieselgelpartien weisen jedoch eine geringe Aktivität auf, was zu unbefriedigenden chromatographischen Trennungen führt. In solchen Fällen ist wie folgt vorzugehen: das Kieselgel durch mindestens vierstündiges Erhitzen auf 550 °C aktivieren, nach dem Erhitzen in einen Exsikkator stellen und nach dem Abkühlen in einen verschließbaren Kolben überführen; 2 % Wasser zugeben und schütteln, bis keine Klumpen mehr sichtbar sind und das Pulver frei rieselt. Kieselgelpartien, die Chromatogramme mit sich überlappenden Peaks ergeben, sind wie oben beschrieben zu behandeln. Eine Alternative ist die Verwendung von extrareinem Kieselgel 60 (Merck, Art. Nr. 7754). |
|
5.6. |
Stammlösung (200 ppm) von Cholesta-3,5-dien (Sigma, 99 % rein) in Hexan (10 mg in 50 ml). |
|
5.7. |
Standardlösung von Cholesta-3,5-dien in Hexan in einer Konzentration von 20 ppm. Dazu die obengenannte Lösung verdünnen. Anmerkung 5: Bei Temperaturen unter 4 °C halten sich die Lösungen 5.6 und 5.7 mindestens vier Monate lang. |
|
5.8. |
Lösung aus n-Nonacosan in Hexan mit einer Konzentration von rund 100 ppm. |
|
5.9. |
Trägergas für die Gaschromatographie: Helium oder Wasserstoff (99,9990 % rein). |
|
5.10. |
Brenngase für den Flammenionisationsdetektor: Wasserstoff (99,9990 % rein) und gereinigte Luft. |
6. VERFAHREN
6.1. Herstellung des Unverseifbaren:
|
6.1.1. |
20 ± 0,1 g Öl in einen 250-ml-Kolben (4.1) einwiegen, 1 ml der Standardlösung von Cholesta-3,5-dien (20 μg) und 75 ml 10 %ige ethanolische Kalilauge zugeben, Rückflußkühler anschließen und 30 Minuten köcheln lassen. Den Kolben mit der Probe von der Hitzequelle nehmen und leicht abkühlen lassen (nicht vollständig abkühlen lassen, da die Probe sonst fest wird). 100 ml Wasser zugeben und die Lösung mit Hilfe von 100 ml Hexan in einen Scheidetrichter (4.2) geben. Die Mischung 30 Sekunden kräftig schütteln und zur Phasentrennung stehen lassen. Anmerkung 6: Bei Bildung einer Emulsion, die nicht gleich wieder verschwindet, kleine Mengen Ethanol zugeben. |
|
6.1.2. |
Die untere, wäßrige Phase in einen zweiten Scheidetrichter überführen und wiederum mit 100 ml Hexan extrahieren. Die untere Phase erneut ablaufen lassen und die Hexanextrakte zusammen in einem weiteren Scheidetrichter dreimal mit je 100 ml einer Ethanol-Wassermischung (1: 1) waschen, bis diese pH-neutral reagiert. |
|
6.1.3. |
Die Hexanlösung durch wasserfreies Natriumsulfat (50 g) laufen lassen, mit 20 ml Hexan waschen und in einem Rotationsverdampfer bei 30 °C im leichten Vakuum vollständig eindampfen. |
6.2. Abtrennung der Steroid-Fraktion:
|
6.2.1. |
Den Rückstand mit Hilfe von zwei Volumenanteilen Hexan (jeweils 1 ml) auf die Trennsäule aufgeben und soweit einziehen lassen, bis sich die Flüssigkeitsoberfläche bis über die Natriumsulfatschicht abgesenkt hat. Dann die Elution mit Hexan mit einer Flußrate von 1 ml/Minute beginnen. Die ersten 25 bis 30 ml des Eluats verwerfen, die folgenden 40 ml auffangen. Diese Fraktion in einen 100-ml-Rundkolben (4.3) überführen. Anmerkung 7: Die erste Fraktion enthält gesättigte Kohlenwasserstoffe (Abbildung 1a), die zweite Steroide. Danach werden Squalen und verwandte Verbindungen eluiert. Für eine gute Trennung zwischen den gesättigten Kohlenwasserstoffen und den Steroiden müssen die Fraktionsvolumina optimiert werden. Hierzu ist das Volumen der ersten Fraktion so zu regulieren, daß bei der Analyse der zweiten Fraktion nur kleine Peaks für gesättigte Kohlenwasserstoffe auftreten (siehe Abbildung 1c). Treten keine solchen Peaks auf und hat der Standardpeak gleichzeitig nur eine geringe Größe, so ist das Volumen der ersten Fraktion zu reduzieren. Im übrigen ist eine vollständige Trennung zwischen den Komponenten der ersten und zweiten Fraktion nicht erforderlich, da während der GC-Analyse unter den in Punkt 6.3.1 beschriebenen Arbeitsbedingungen eine Überlappung der Peaks nicht vorkommt. Eine Optimierung des Volumens der zweiten Fraktion ist in der Regel nicht notwendig, da sich die weiteren Komponenten gut abtrennen lassen. Ein großer Peak mit einer Retentionszeit von rund 1,5 Minuten unter dem des Standards ist jedoch auf das Vorhandensein von Squalen zurückzuführen und zeigt eine schlechte Trennung an. |
|
6.2.2. |
Die zweite Fraktion im Rotationsverdampfer bei 30 °C im leichten Vakuum vollständig eindampfen und den Rückstand sofort in 0,2 ml Hexan auflösen. Die Lösung bis zur Analyse im Kühlschrank aufbewahren. Anmerkung 8: Die Rückstände 6.1.3 und 6.2.2 sollten nicht trocken und nicht bei Zimmertemperatur aufbewahrt werden. Nach Gewinnung des Rückstands sofort das Lösungsmittel zugeben und die Lösung im Kühlschrank aufbewahren. |
6.3. Gaschromatographie
|
6.3.1. |
Arbeitsbedingungen für Split-Injektion: — Injektortemperatur: 300 °C, — Detektortemperatur: 320 °C, — Integrator: (Die Parameter für die Integration sind so zu wählen, daß die Peakflächen richtig beurteilt werden können. Empfohlen wird die Integration zwischen zwei Minima), — Empfindlichkeit: rund das 16-fache der Mindestdämpfung, — Einspritzvolumen: 1 μm Lösung, — Temperaturprogramm: 6 Minuten isotherm bei 235 °C, dann mit 2 °C/min auf 285 °C, — Injektor mit Stromteilventil (Splitverhältnis 1: 15), — Trägergasvordruck: ca. 120 kPa Helium oder Wasserstoff. Diese Bedingungen können entsprechend den Kenndaten des Chromatographen und der Säule derart geändert werden, daß die damit aufgezeichneten Chromatogramme folgende Bedingungen erfüllen: Der Peak des internen Standards muß innerhalb von 5 Minuten vor oder nach der unter 6.3.2 genannten Zeit erscheinen und mindestens 80 % Vollausschlag erreichen. Das GC-System ist durch Einspritzen einer Mischung aus der Cholestadien-Stammlösung (5.6) und der n-Nonacosan-Lösung (5.8) zu überprüfen. Der Cholesta-3,5-dien-Peak muß vor dem n-Nonacosan-Peak erscheinen (Abbildung 1c). Ist dies nicht der Fall, so kann die ursprüngliche Ofentemperatur verringert und/oder die GC-Säule durch eine weniger polare Säule ersetzt werden. |
|
6.3.2. |
Identifizierung der Peaks Der Peak der internen Standards erscheint nach rund 19 Minuten, derjenige von Stigmastadienen nach einer relativen Retentionszeit von rund 1,29 (Abbildung 1b). Das Stigmasta-3,5-dien tritt zusammen mit geringen Mengen eines Isomers auf; beide ergeben normalerweise aber nur einen Peak. Ist die Säule jedoch zu polar, oder hat sie eine große Trennleistung, kann das Isomer als kleiner Peak dicht vor dem des Stigmasta-3,5-dien erscheinen (Abbildung 2). Um sicherzugehen, daß die Stigmastadiene als ein Peak eluiert werden, empfiehlt es sich, die Säule durch eine weniger polare Säule oder eine Säule mit größerem Innendruchmesser zu ersetzen. Anmerkung 9: Ein Referenzchromatogramm für Stigmastadien erhält man durch die Analyse eines raffinierten pflanzlichen Öls mit einer kleineren Probe. Stigmastadiene rufen einen signifikanten, leicht identifizierbaren Peak hervor. |
|
6.3.3. |
Quantitative Analyse Der Gehalt an Stigmastadienen wird nach folgender Formel berechnet: mg/kg Stigmastadienen =
Nachweisgrenze: rund 0,01 mg/kg. |



Abb. 1:
Gaschromatogramme von Olivenölproben, analysiert auf einer Fused-Silica-Kapillarsäule (0,25 mm Innendurchmesser und 25 m Länge), belegt mit 5 % Phenylmethylsilicon (Filmdicke 0,25 μm).
a) Erste Fraktion (30 ml) eines nativen Olivenöls, mit dem Standard versetzt.
b) Zweite Fraktion (40 ml) eines Olivenöls mit 0,10 mg/kg Stigmastadienen.
c) Zweite Fraktion (40 ml) mit einem kleinen Anteil der ersten Fraktion.

Abb. 2:
Gaschromatogramm einer Probe raffinierten Olivenöls, analysiert auf einer DB-5-Säule, auf dem das Isomer von Stigmasta-3,5-dien zu erkennen ist.
ANHANG XVIII
BESTIMMUNG DER DIFFERENZ ZWISCHEN DEM TATSÄCHLICHEN UND DEM THEORETISCHEN GEHALT AN TRIGLYCERIDEN MIT ECN 42
1. ZWECK
Bestimmung der absoluten Differenz zwischen dem mittels Hochleistungsflüssigkeitschromatographie (HPLC) ermittelten Gehalt des Olivenöls an Triglyceriden (TAG) mit der äquivalenten Kohlenstoffzahl 42 (ECN42HPLC) und dem auf der Grundlage der Fettsäurezusammensetzung berechneten theoretischen Gehalt an Triglyceriden mit der äquivalenten Kohlenstoffzahl 42 (ECN42theoretisch).
2. ANWENDUNGSBEREICH
Diese Norm ist anwendbar auf Olivenöle. Das Verfahren dient dem Nachweis kleiner Mengen von (linolsäurereichen) Saatölen in Olivenölen aller Kategorien..
3. PRINZIP
Der mit Hilfe der HPLC-Analyse bestimmte Gehalt an Triglyceriden mit ECN 42 und der (auf der Grundlage der gaschromatographisch bestimmten Fettsäurezusammensetzung berechnete) theoretische Gehalt an Triglyceriden mit ECN 42 stimmen bei reinen Ölen bis zu einem gewissen Grad überein. Unterschiede, die über den für jeden Typ von Öl festgelegten Werten liegen, deuten darauf hin, dass das Öl Saatöle enthält.
4. VERFAHREN
Die Methode für die Berechnung des theoretischen Gehalts an Triglyceriden mit ECN 42 und der Differenz zwischen diesem und den HPLC-Daten besteht im Wesentlichen aus dem Vergleich von Analysedaten, die mit Hilfe anderer Methoden gewonnen wurden. Drei Verfahrensstufen sind zu unterscheiden: Bestimmung der Fettsäurezusammensetzung durch Kapillarsäulengaschromatographie, Berechnung des theoretischen Zusammensetzung der Triglyceride mit ECN 42 und HPLC-Bestimmung der Triglyceride mit ECN 42.
4.1. Geräte
|
4.1.1. |
250- und 500-ml-Rundkolben. |
|
4.1.2. |
100-ml-Bechergläser. |
|
4.1.3. |
Chromatographiesäule aus Glas, 21 mm innerer Durchmesser, 450 mm Länge, mit Hahn und Normschliffhülle am oberen Ende. |
|
4.1.4. |
Scheidetrichter 250 ml Inhalt, mit Normschliffkern am unteren Ende, geeignet zum Verbinden mit dem oberen Ende der Säule. |
|
4.1.5. |
Glasstab, 600 mm Länge. |
|
4.1.6. |
Glastrichter, 80 mm Durchmesser. |
|
4.1.7. |
50-ml-Messkolben. |
|
4.1.8. |
20-ml-Messkolben. |
|
4.1.9. |
Rotationsverdampfer. |
|
4.1.10. |
Hochleistungsflüssigchromatograph mit Säulenofen zur Regelung der Säulentemperatur. |
|
4.1.11. |
Einspritzsystem mit 10-μl-Probenschleife. |
|
4.1.12. |
Detektor: Differentialrefraktometer. Im Bereich der höchsten Empfindlichkeit müssen wenigstens 10–4 Einheiten des Brechungsindex erreicht werden. |
|
4.1.13. |
Säule: Säule aus rostfreiem Stahl, 250 mm Länge und 4,5 mm Innendurchmesser, gefüllt mit Kieselgel (Partikelgröße 5 μm) mit einer durchschnittlichen Kohlenstoffbelegung von 22—23 % in Form von Octodecylsilan. |
|
4.1.14. |
Datenverarbeitungssoftware. |
|
4.1.15. |
Phiolen mit etwa 2 ml Inhalt mit teflonbeschichtetem Septum und Schraubverschluss. |
4.2. Reagenzien
Die Reagenzien müssen analysenrein sein. Die Fließmittel müssen entgast sein. Sie können mehrere Male wiederverwendet werden, ohne dass dadurch die Trennleistungen beeinflusst werden.
|
4.2.1. |
Petrolether (40–60 °C) für die Chromatografie oder Hexan. |
|
4.2.2. |
Diethylether, peroxidfrei, frisch destilliert. |
|
4.2.3. |
Elutionsmittel zur Reinigung des Öls mittels Säulenchromatografie: Mischung aus Petrolether und Diethylether im Verhältnis 87/13 (v/v). |
|
4.2.4. |
Kieselgel, 70—230 mesh, Typ Merck 7734, standardisiert auf einem Wassergehalt von 5 % (w/w). |
|
4.2.5. |
Glaswatte. |
|
4.2.6. |
Aceton für die HPLC. |
|
4.2.7. |
Acetonitril oder Propionitril für die HPLC. |
|
4.2.8. |
HPLC-Fließmittel: Acetonitril + Aceton (das Mischungsverhältnis ist so einzustellen, dass damit die gewünschte Trennung erzielt wird; man beginnt mit einem Mischungsverhältnis von 1:1) oder Propionitril. |
|
4.2.9. |
Lösungsmittel: Aceton. |
|
4.2.10. |
Referenztriglyceride: Es können handelsübliche Triglyceride (Tripalmitin, Triolein usw.) verwendet werden, wobei ein Diagramm aus den Retentionszeiten und den äquivalenten Kohlenstoffzahlen aufgezeichnet wird, oder es werden Referenzchromatogramme von Sojaöl, einer Mischung von Sojaöl/Olivenöl (30:70) und reinem Olivenöl erstellt (siehe Anmerkungen 1 und 2 und Abbildungen 1 bis 4). |
|
4.2.11. |
Festphasenextraktionssäule mit Kieselgelphase 1 g, 6 ml. |
4.3. Vorbereitung der Proben
Da einige störende Substanzen zu falschen positiven Ergebnissen führen können, muss die Probe immer nach der IUPAC-Methode 2.507, die für die Bestimmung polarer Verbindungen in Bratfetten verwendet wird, gereinigt werden.
4.3.1. Vorbereitung der Chromatografiesäule
Etwa 30 ml Fließmittel (4.2.3) in die Säule (4.1.3) geben, Glaswattebausch (4.2.5) einführen und mit Hilfe des Glasstabs (4.1.5) bis auf den Grund der Säule schieben.
In einem 100-ml-Becherglas 25 g Kieselgel (4.2.4) in 80 ml Elutionsmittel (4.2.3) suspendieren, dann mit Hilfe eines Glastrichters (4.1.6) in die Säule geben.
Zur restlosen Überführung des Kieselgels in die Säule ist das Becherglas mit dem Elutionsmittel zu spülen und die Spülflüssigkeit ebenfalls in die Säule zu geben.
Hahn öffnen und soviel Fließmittel ablaufen lassen, bis der Fließmittelspiegel etwa 1 cm über dem Kieselgel liegt.
4.3.2. Säulenchromatografie
In einem 50-ml-Meßkolben (4.1.7) werden auf 0,001 g genau 2,5 ± 0,1 g filtriertes, homogenisiertes und erforderlichenfalls entwässertes Öl eingewogen.
Die Einwaage wird in etwa 20 ml Elutionsmittel (4.2.3) gelöst; erforderlichenfalls leicht erwärmen, damit sich das Öl besser löst. Auf Raumtemperatur erkalten lassen und mit Elutionsmittel auffüllen.
Mit Hilfe einer Meßpipette werden 20 ml der Lösung in die gemäß 4.3.1 vorbereitete Säule gegeben; Hahn öffnen und das Elutionsmittel bis zur Kieselgelschicht ablaufen lassen.
Mit Hilfe von 150 ml Elutionsmittel (4.2.3) bei einer Durchlaufgeschwindigkeit von etwa 2 ml/Minute eluieren, so dass 150 ml die Säule in 60 bis 70 Minuten durchströmen.
Eluat in einem zuvor im Ofen austarierten und genau gewogenen 250-ml-Rundkolben (4.1.1) auffangen. Lösungsmittel unter Unterdruck in einem Rotationsverdampfer (4.1.9) entfernen und Rückstand wägen, der zur Herstellung der Lösung für die HPLC-Analyse und für die Bereitung der Methylesterzubereitung verwendet wird.
Nach Durchlaufen der Säule muss im Fall von nativem Olivenöl extra, nativem Olivenöl, gewöhnlichem nativem Olivenöl, raffiniertem Olivenöl und Olivenöl die Probe zu 90 %, im Fall von Lampantöl und Tresteröl mindestens zu 80 % zurückgewonnen werden.
4.3.3. Reinigung mittels Festphasenextraktion
Die Kieselgel-Festphasenextraktionssäule wird durch Durchlauf von 6 ml Hexan (4.2.3) unter Unterdruck aktiviert, wobei ein Trockenfallen zu vermeiden ist.
0,12 g g ± 0,001 g werden in einer 2-ml-Phiole (4.1.15) eingewogen und in 0,5 ml Hexan (4.2.3) gelöst.
Die Festphasenextraktionssäule wird mit der Lösung befüllt und mit 10 ml Hexan-Diethylether-Mischung im Verhältnis 87: 13 (v/v) (4.2.3) unter Unterdruck eluiert.
Die aufgefangene Fraktion wird in einem Rotationsverdampfer (4.1.9) unter Unterdruck bei Raumtemperatur zur Trockene eingedampft. Der Rückstand wird in 2 ml Aceton (4.2.6) für die Triglyceridanalyse gelöst.
4.4. HPLC-Analyse
4.4.1. Vorbereitung der Proben für die Chromatografieanalyse
Von der zu analysierenden Probe wird eine 5%ige Lösung hergestellt, indem 0,5 g ± 0,001 g der Probe in einen 10-ml-Messkolben eingewogen und mit dem Lösungsmittel (4.2.9) auf 10 ml aufgefüllt werden.
4.4.2. Verfahren
Das HPLC-Gerät in Betrieb setzen. Das Fließmittel (4.2.8) mit einer Strömungsgeschwindigkeit von 1,5 ml/Minute durch die Säule pumpen, um das gesamte System zu spülen.
Sobald eine stabile Basislinie erreicht ist, werden 10 μl der gemäß 4.3 hergestellten Proben eingespritzt.
4.4.3. Berechnung und Abfassung der Ergebnisse
Es wird vorausgesetzt, dass die Summe der Peakflächen aller Triglyceride mit ECN 42 bis ECN 52 100 % entspricht (Flächenprozentmethode).
Der relative Anteil eines jeden Triglycerids berechnet sich nach folgender Formel:
![]()
.
Die Ergebnisse werden mit mindestens zwei Dezimalstellen angegeben.
Siehe Anmerkungen 1 bis 4.
4.5. Berechnung der Triglyceridzusammensetzung (Mol %) anhand der Daten zur Fettsäurezusammensetzung (Flächen %)
4.5.1. Bestimmung der Fettsäurezusammensetzung
Die Fettsäurezusammensetzung wird nach ISO 5508 unter Verwendung einer Kapillarsäule bestimmt. Die Methylester werden gemäß COI/T.20/Doc. No 24 vorbereitet.
4.5.2. Berechnungsrelevante Fettsäuren
Die Triglyceride werden nach ihrer äquivalenten Kohlenstoffzahl (ECN) unter Berücksichtigung folgender ECN/Fettsäureäquivalenzen zusammengefasst. Nur Fettsäuren mit 16 und 18 Kohlenstoffatomen werden berücksichtigt, da nur sie bei Olivenöl von Bedeutung sind. Die Fettsäuren sind auf 100 % zu normalisieren.
|
Fettsäure (FS) |
Abkürzung |
Molekulargewicht (MG) |
ECN |
|
Palmitinsäure |
P |
256,4 |
16 |
|
Palmitoleinsäure |
Po |
254,4 |
14 |
|
Stearinsäure |
S |
284,5 |
18 |
|
Ölsäure |
O |
282,5 |
16 |
|
Linolsäure |
L |
280,4 |
14 |
|
Linolensäure |
Ln |
278,4 |
12 |
4.5.3. Umrechnung der prozentualen Fläche in Mol für alle Fettsäuren (1)
|
|
|
|
|
|
|
|



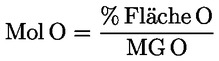


4.5.4. Normalisierung der Fettsäuren auf 100 % (2)
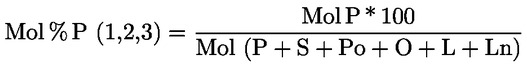
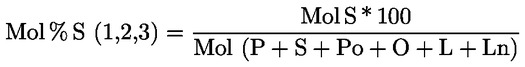
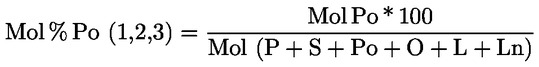



Als Ergebnis erhält man den prozentualen Anteil jeder Fettsäure in Molprozent in (1,2,3-) Positionen der TAG.
Anschließend wird die Summe der gesättigten Fettsäuren P und S (SFA) sowie der ungesättigten Fettsäuren Po, O, L und Ln (UFA) berechnet (3):
![]()
![]()
4.5.5. Berechnung der Fettsäurezusammensetzung in 2- und 1,3-Stellung der TAG
Die Fettsäuren werden wie folgt in drei Gruppen eingeteilt: eine Gruppe für 2-Stellung und zwei gleiche Gruppen für 1- und 3-Stellung, wobei unterschiedliche Koeffizienten für gesättigte (P und S) und ungesättigte Fettsäuren (Po, O, L und Ln) verwendet werden.
4.5.5.1. Gesättigte Fettsäuren in 2-Stellung [P (2) und S (2)] (4)
![]()
![]()
4.5.5.2. Ungesättigte Fettsäuren in 2-Stellung [PO(2), O(2), L(2) und Ln(2)] (5):
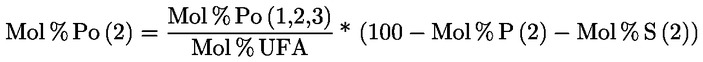


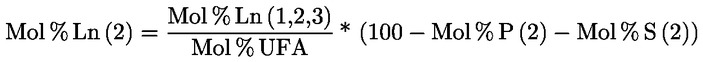
4.5.5.3. Fettsäuren in 1,3-Stellung [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) und Ln(1,3)] (6):



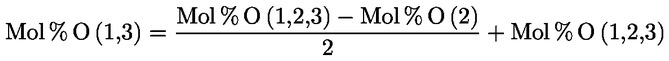
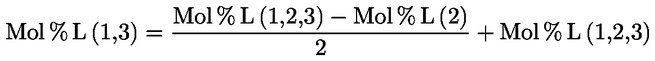

4.5.6. Berechnung der Triglyceride (TAG)
4.5.6.1. TAG mit einer Fettsäure (AAA, hier LLL, PoPoPo) (7)

4.5.6.2. TAG mit zwei Fettsäuren (AAB, hier PoPoL, PoLL) (8)
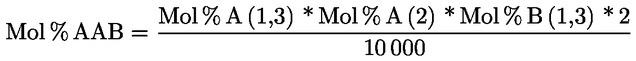

4.5.6.3. TAG mit drei unterschiedlichen Fettsäuren (ABC, hier OLLn, PLLn, PoOLn, PPoLn) (9)
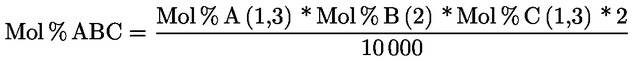


4.5.6.4. ECN42-haltige Triglyceride
Die folgenden Triglyceride mit ECN42 werden nach den Formeln 7, 8 und 9 in der Reihenfolge der bei der HPLC erwarteten Elution (normalerweise nur drei Peaks) berechnet.
LLL
PoLL und das Positionsisomer LPoL
OLLn und die Positionsisomere OLnL und LnOL
PoPoL und das Positionsisomer PoLPo
PoOLn und die Positionsisomere OPoLn und OLnPo
PLLn und die Positionsisomere LLnP und LnPL
PoPoPo
SLnLn und das Positionsisomer LnSLn
PPoLn und die Positionsisomere PLnPo und PoPLn
Die Triglyceride mit ECN42 ergeben sich aus der Summe der neun Triacylglyceride einschließlich ihrer Positionsisomere. Die Ergebnisse werden mit mindestens zwei Dezimalstellen angegeben.
5. AUSWERTUNG DER ERGEBNISSE
Der rechnerisch bestimmte theoretische Gehalt und der durch die HPLC-Analyse bestimmte Gehalt werden verglichen. Liegt die Differenz zwischen den Absolutwerten der HPLC-Daten und den theoretischen Daten über den in der Norm für die jeweilige Ölkategorie angegebenen Werten, so enthält die Probe Saatöl.
Die Ergebnisse werden mit zwei Dezimalstellen angegeben.
6. BEISPIEL (DIE NUMMERN BEZIEHEN SICH AUF DIE BETREFFENDEN TEXTABSCHNITTE DES VERFAHRENS)
— 4.5.1. Berechnung der Molprozente der Fettsäuren aus den GLC-Daten (normalisierte Flächenprozente)
Die Gaschromatographie ergibt folgende Fettsäurezusammensetzung:
|
FS |
P |
S |
Po |
O |
L |
Ln |
|
MG |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
% Fläche |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3 Umrechnung der prozentualen Fläche in Mol für alle Fettsäuren (siehe Formel (1))
|
Mol P |
= |
|

|
Mol S |
= |
|
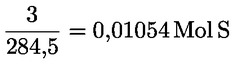
|
Mol Po |
= |
|

|
Mol O |
= |
|

|
Mol L |
= |
|

|
Mol Ln |
= |
|

|
Insgesamt |
= |
0,35821 Mol TAG |
— 4.5.4 Normalisierung der Fettsäuren auf 100 % (siehe Formel (2))
|
Mol % P(1,2,3) |
= |
|

|
Mol % S(1,2,3) |
= |
|

|
Mol % Po(1,2,3) |
= |
|
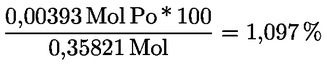
|
Mol % O(1,2,3) |
= |
|
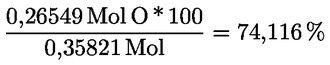
|
Mol % L(1,2,3) |
= |
|
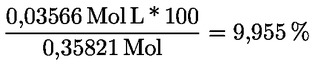
|
Mol % Ln(1,2,3) |
= |
|

|
Mol % insgesamt |
= |
100 % |
Summe der gesättigten und ungesättigten Fettsäuren in 1,2,3-Stellung der TAG (siehe Formel (3)):
![]()
![]()
— 4.5.5 Berechnung der Zusammensetzung der Fettsäuren in 2- und 1,3-Stellung der TAG
— 4.5.5.1 Gesättigte Fettsäuren in 2-Stellung [P(2) und S(2)] (siehe Formel (4))
![]()
![]()
— 4.5.5.2 Ungesättigte Fettsäuren in 2-Stellung [Po(1,3), O(1,3), L(1,3) und Ln(1,3)] (siehe Formel (5))



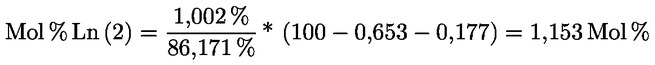
— 4.5.5.3 Fettsäuren in 1,3-Stellung [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) und Ln(1,3)] (siehe Formel (6))





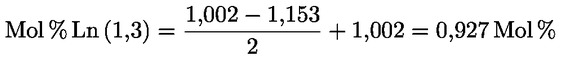
— 4.5.6. Berechnung der Triglyceride (TAG)
Anhand der berechneten Zusammensetzung der Fettsäuren in sn-2- und sn-1,3-Stellung
|
FS in |
1,3-Stellung |
2-Stellung |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Insgesamt |
100,0 % |
100,0 % |
werden folgende Triglyceride rechnerisch bestimmt:
LLL
PoPoPo
PoLL mit 1 Positionsisomer
SLnLn mit 1 Positionsisomer
PoPoL mit 1 Positionsisomer
PPoLn mit 2 Positionsisomeren
OLLn mit 2 Positionsisomeren
PLLn mit 2 Positionsisomeren
PoOLn mit 2 Positionsisomeren
— 4.5.6.1. TAG mit einer Fettsäure (LLL, PoPoPo) (siehe Fomel (7))
 = 0,09706 Mol LLL
= 0,09706 Mol LLL

— 4.5.6.2 TAG mit zwei Fettsäuren (PoLL, SLnLn, PoPoL) (siehe Formel (8))
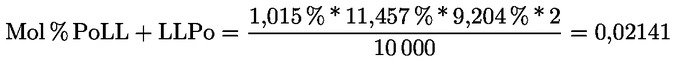

0,03210 Mol PoLL
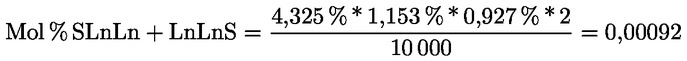

0,00094 Mol SLnLn
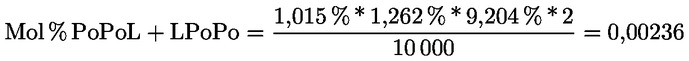

0,00354 Mol PoPoL
— 4.5.6.3 TAG mit drei unterschiedlichen Fettsäuren (PoPLn, OLLn, PLLn, PoOLn) siehe Formel (9)



0,00761 Mol PPoLn


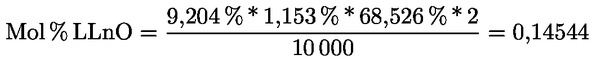
0,43655 Mol OLLn


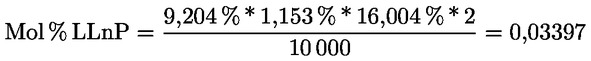
0,06907 Mol PLLn
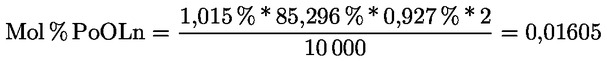
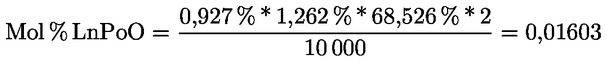
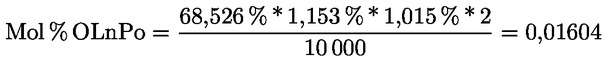
0,04812 Mol PoOLn
ECN42 = 0,69512 Mol TAGs
Anmerkung 1: Die Reihenfolge der Elution kann durch Berechnung der äquivalenten Kohlenstoffzahlen (ECN) bestimmt werden, die häufig durch die Beziehung
![]() definiert sind. Hierbei ist CN die Zahl der Kohlenstoffatome und n die Zahl der Doppelbindungen. Durch Berücksichtigung des Ursprungs der Doppelbindungen kann sie noch genauer bestimmt werden Wenn no, nl und nln die Zahlen der Doppelbindungen sind, die der Öl-, Linol- bzw. Linolensäure zugeordnet sind, so kann die äquivalente Kohlenstoffzahl nach folgender Formel berechnet werden:
definiert sind. Hierbei ist CN die Zahl der Kohlenstoffatome und n die Zahl der Doppelbindungen. Durch Berücksichtigung des Ursprungs der Doppelbindungen kann sie noch genauer bestimmt werden Wenn no, nl und nln die Zahlen der Doppelbindungen sind, die der Öl-, Linol- bzw. Linolensäure zugeordnet sind, so kann die äquivalente Kohlenstoffzahl nach folgender Formel berechnet werden:
![]()
Die Koeffizienten do, dl und dln können mit Hilfe der Referenztriglyceride berechnet werden. Unter den in dieser Methode beschriebenen Bedingungen gilt näherungsweise folgende Beziehung:
![]()
Anmerkung 2: Mit Hilfe verschiedener Triglyceride kann die Auflösung für Triolein unter Verwendung der reduzierten Retentionszeit
![]() Lösungsmittel wie folgt berechnet werden:
Lösungsmittel wie folgt berechnet werden:
![]()
Triolein
Der Graph von log α gegen f (Zahl der Doppelbindungen) ermöglicht es, die Retentionswerte aller Triglyceride zu bestimmen, die Fettsäuren enthalten, die in den Referenztriglyceriden vorkommen (siehe Abbildung 1).
Anmerkung 3: Die Trennfähigkeit der Säule muss eine klare Trennung des Trilinolein-Peaks von den Peaks der Triglyceride mit wenig unterschiedlicher Retentionszeit ermöglichen. Die Elution wird bis zum ECN52-Peak durchgeführt.
Anmerkung 4: Eine korrekte Bestimmung der Flächen aller für die Bestimmung interessierenden Peaks wird dadurch sichergestellt, dass der zweite Peak mit ECN50 50 % des Vollausschlags des Rekorders erreicht.
Abbildung 1
Graphische Darstellung von log α als Funktion von f (Zahl der Doppelbindungen)

Anzahl der Doppelbindungen
La: Laurinsäure; My: Myristinsäure; P: Palmitinsäure; S: Stearinsäure; O: Ölsäure; L: Linolsäure; Ln: Linolensäure
Abbildung 2
Olivenöl mit niedrigem Linolsäuregehalt
a)
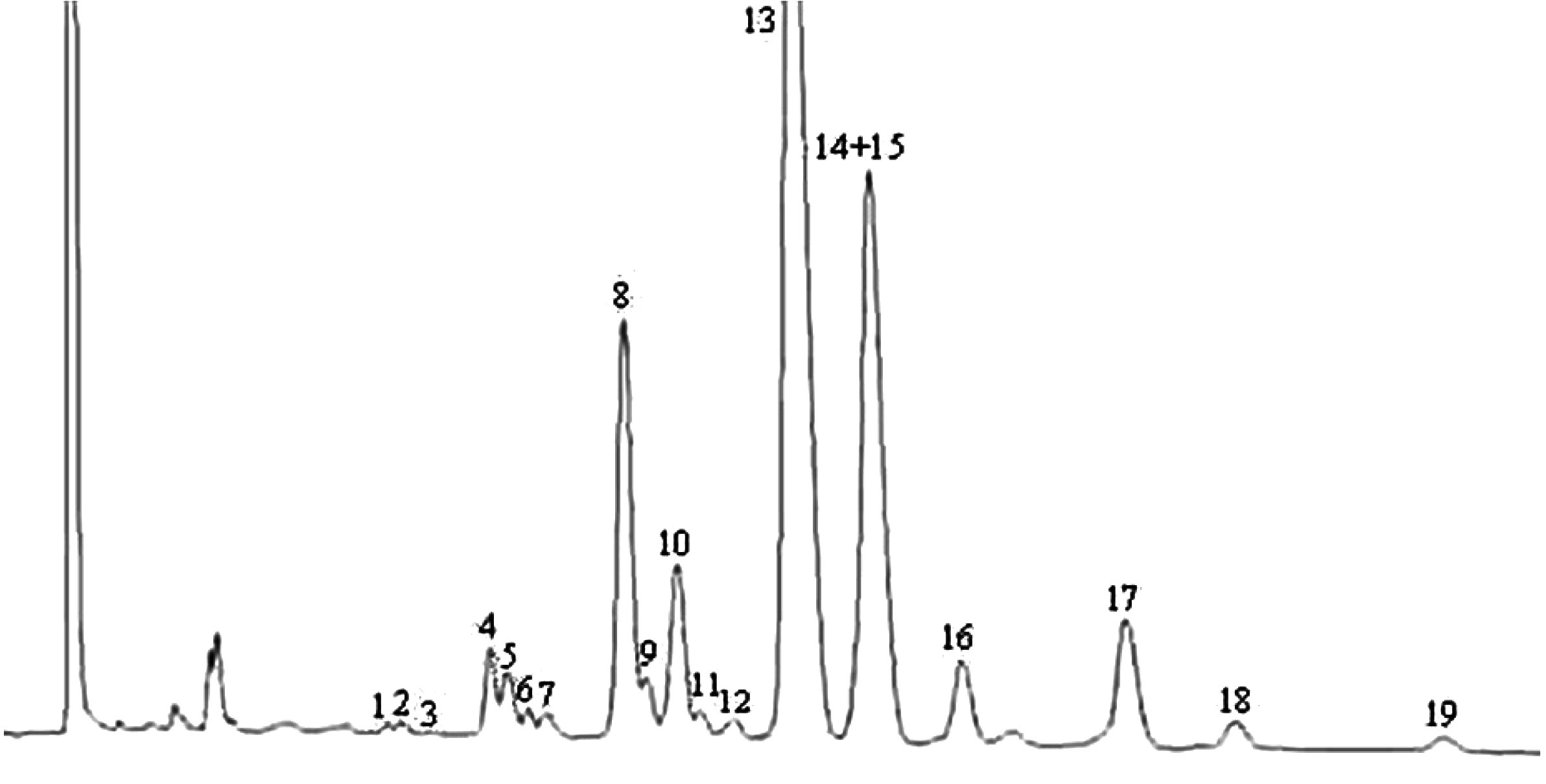
Mit Lösungsmittel: Aceton/Acetonitril.
PROFIL a: Hauptkomponenten der chromatografischen Peaks: ECN42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
b)

Mit Lösungsmittel: Propionitril
PROFIL b: Hauptkomponenten der chromatografischen Peaks: ECN42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS
Abbildung 3
Olivenöl mit hohem Linolsäuregehalt
a)

Mit Lösungsmittel: Aceton/Acetonitril (50:50).
Profil a: Hauptkomponenten der chromatografischen Peaks: ECN42: (1‘) LLL + PoLL; (2‘) OLLn + PoOLn; (3‘) PLLn; ECN44: (4‘) OLL + PoOL; (5‘) OOLn + PLL; (6‘) POLn + PPoPo; ECN46: (7‘) OOL + PoOO; (8‘) PLO + SLL + PoOP; (9‘) PLP + PoPP; ECN48: (10‘) OOO; (11‘) POO + SLL + PPoO; (12‘) POP + PLS; ECN50: (13‘) SOO; (14‘) POS + SLS
b)

Mit Lösungsmittel: Propionitril.
Profil b: Hauptkomponenten der chromatografischen Peaks: ECN42: (1) LLL; (2 + 2‘) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS; ECN52: (19) AOO.
ANHANG XIX
BESTIMMUNG DES GEHALTS AN ALIPHATISCHEN UND TRITERPENALKOHOLEN DURCH KAPILLAR-GASCHROMATOGRAFIE
1. GEGENSTAND
In diesem Anhang ist ein Verfahren zur Bestimmung des Gehalts an aliphatischen und Triterpenalkoholen in Ölen und Fetten beschrieben.
2. PRINZIP DER METHODE
Das Fett wird mit 1-Eicosanol als internem Standard versetzt, mit ethanolischer Kaliumhydroxidlösung verseift und das Unverseifbare mit Ethylether extrahiert. Die Alkoholfraktion wird dünnschichtchromatographisch über basische Kieselgelplatten vom Unverseifbaren abgetrennt. Die aus dem Kieselgel isolierten Alkohole werden in Trimethylsilylether überführt und mit Hilfe der Kapillar-Gaschromatographie untersucht.
3. GERÄTE
3.1. Kolben, 250 ml, mit Rückflusskühler und Schliffstopfen,
3.2. 500-ml-Scheidetrichter,
3.3. Kolben, 250 ml,
3.4. komplette Apparatur für die Dünnschichtchromatographie mit Glasplatten 20 × 20 cm,
3.5. UV-Lampe, Wellenlänge 366 oder 254 nm,
3.6. Mikroliterspritzen, 100 und 500 μl,
3.7. Glasfiltertiegel mit Porenfilter G 3 (Porosität 15-40 μm), etwa 2 cm Durchmesser, 5 cm Höhe, mit geeignetem Anschlussstück für die Vakuumfiltration und Schliffmuffe 12/21,
3.8. Vakuumflasche, 50 ml, mit Schliffmuffe 12/21, für Glasfiltertiegel (3.7),
3.9. 10 ml-Röhrchen mit konischem Boden und dicht schließendem Stopfen,
3.10. Gaschromatograph, geeignet für die Verwendung von Kapillarsäulen, mit Splitsystem, bestehend aus:
3.10.1. thermostatisierbarem Säulenofen, einstellbar auf die gewünschte Temperatur mit einer Genauigkeit von ± 1 °C,
3.10.2. temperaturregelbarer Injektionseinrichtung mit Verdampfer aus persilanisiertem Glas,
3.10.3. Flammenionisations-Detektor mit Verstärker,
3.10.4. Integrator mit Schreiber, an Verstärker zu koppeln, Reaktionszeit maximal 1 Sekunde, variabler Papiervorschub,
3.11. Kapillarsäule aus Glas oder fused silica, 20-30 m Länge, 0,25-0,32 mm Innendurchmesser, Innenwand belegt mit SE-52 oder SE-54 oder einer gleichwertigen stationären Phase, Schichtdicke 0,10-0,30 μm,
3.12. Mikroliterspritze für die Gaschromatographie, 10 μl, mit gehärteter Nadel,
3.13. Präzisionswaage mit einer Empfindlichkeit von 1 mg (Anzeigegenauigkeit 0,1 mg).
4. REAGENZIEN
4.1. Kaliumhydroxid, ethanolische Lösung 2 N: 130 g Kaliumhydroxid (Titer mindestens 85 %) in 200 ml destilliertem Wasser unter Kühlen lösen und mit Ethanol auf 1 Liter auffüllen. Die Lösung ist in gut verschlossenen braunen Glasflaschen aufzubewahren.
4.2. Ethylether, analysenrein,
4.3. wasserfreies Natriumsulfat, analysenrein,
4.4. kieselgelbeschichtete Glasplatten ohne Fluoreszenzindikator, Schichtdicke 0,25 mm (gebrauchsfertig im Handel erhältlich),
4.5. Kaliumhydroxid, ethanolische Lösung 0,2 N: 13 g Kaliumhydroxid in 20 ml destilliertem Wasser lösen und mit Ethanol auf 1 l auffüllen,
4.6. Benzol für die Chromatographie (siehe 5.2.2),
4.7. Aceton für die Chromatographie (siehe 5.2.2),
4.8. Hexan für die Chromatographie (siehe 5.2.2),
4.9. Ethylether für die Chromatographie (siehe 5.2.2),
4.10. Chloroform, analysenrein,
4.11. Referenzlösung für die Dünnschichtchromatografie: 0,5 %ige Lösung von C20-C28-Alkoholen in Chloroform oder einer Alkoholfraktion, die wie in Abschnitt 5.2 beschrieben aus den unverseifbaren Bestandteilen eines Oliventresteröls abgetrennt wurde.
4.12. 2',7'-Dichlorfluorescein, 0,2 %ige ethanolische Lösung, leicht alkalisch durch Zusatz einiger Tropfen alkoholischer 2-N-Kaliumhydroxid-Lösung,
4.13. wasserfreies Pyridin für die Chromatographie,
4.14. Hexamethyldisilazan,
4.15. Trimethylchlorsilan,
4.16. Standardlösung der Trimethylsilylether der aliphatischen Alkohole von C20 bis C28, unmittelbar vor Gebrauch ansetzen aus einer Mischung der reinen Alkohole,
4.17. 1-Eicosanol, 0,1 %ige Lösung (m/V) in Chloroform (interner Standard),
4.18. Trägergas: Wasserstoff oder Helium, rein, für die Gaschromatographie,
4.19. Hilfsgase: Stickstoff, rein, für die Gaschromatographie.
5. VERFAHREN
5.1. Herstellung des Unverseifbaren
|
5.1.1. |
Mit einer 500-Mikroliterspritze so viel von der 0,1 %igen 1-Eicosanollösung in Chloroform (4.17) in den 250-ml-Kolben geben, dass die Menge an 1-Eicosanol etwa 10 % des Gehalts an aliphatischen Alkoholen des zur Analyse verwandten aliquoten Teils der Probe entspricht. So werden z. B. für 5 g Olivenöl 250 μl und für Oliventresteröl 1 500 μl der 0,1 %igen 1-Eicosanollösung benötigt. Die Probe im Stickstoffstrom bis zur Trockne eindampfen. In den gleichen Kolben genau 5 g der trockenen und filtrierten Probe einwiegen. |
|
5.1.2. |
Die Probe bei aufgesetztem Rückflusskühler mit 50 ml 2-N-Ethanol-Kaliumhydroxidlösung versetzen, auf dem Wasserbad erhitzen und unter kräftigem Schütteln und schwachem Sieden verseifen (wobei die Lösung klar wird). Die Probe weitere 20 Minuten am Sieden halten und dann durch den Rückflusskühler mit 50 ml destilliertem Wasser versetzen; Rückflusskühler entfernen und den Kolben auf etwa 30 °C abkühlen. |
|
5.1.3. |
Den Kolbeninhalt quantitativ in einen 500-ml-Scheidetrichter überführen, wobei mehrfach mit insgesamt etwa 50 ml destilliertem Wasser nachgespült wird. Die Probe mit etwa 80 ml Ethylether versetzen, 30 Sekunden kräftig schütteln und absetzen lassen (Anmerkung 1). Die wässrige Phase in einen anderen Scheidetrichter ablassen und dann noch zweimal nach dem gleichen Verfahren mit jeweils 60-70 ml Ethylether extrahieren. Anmerkung 1: Etwaige Emulsionen können mit Hilfe einer Waschflasche durch Zusatz kleiner Mengen Ethylalkohol oder Methylalkohol zerstört werden. |
|
5.1.4. |
Die Etherauszüge in einem Scheidetrichter vereinigen und mehrmals mit jeweils 50 ml destilliertem Wasser waschen, bis das Waschwasser neutral reagiert. Das Waschwasser ablassen, die Etherphase mit wasserfreiem Natriumsulfat trocknen und über wasserfreiem Natriumsulfat in einen zuvor gewogenen 250-ml-Kolben filtrieren. Scheidetrichter und Filter mit kleinen Mengen Ethylether nachspülen. |
|
5.1.5. |
Den Ether bis auf wenige Milliliter abdestillieren, den Rückstand unter leichtem Vakuum oder im Stickstoffstrom trocknen, etwa 1/4 Stunde im Trockenschrank bei 100 °C nachtrocknen, im Exsikkator abkühlen lassen und wiegen. |
5.2. Abtrennen der Alkoholfraktion
|
5.2.1. |
Herstellung der basischen Platten: Die Kieselgelplatten (4.4) vollständig ca. 10 Sekunden lang in eine ethanolische 0,2-N-Kaliumhydroxidlösung (4.5) eintauchen, dann unter einem Abzug 2 Stunden trocknen lassen, anschließend noch für 1 Stunde bei 100 °C in den Trockenschrank gestellt. Die Platten aus den Trockenschrank nehmen und in einem Exsikkator über Calciumchlorid bis zum Gebrauch aufbewahren. (Derart behandelte Platten müssen innerhalb von 15 Tagen gebraucht werden.) Anmerkung 2: Bei Verwendung von basischen Kieselgelplatten zur Abtrennung der Alkoholfraktion erübrigt sich die Behandlung des Unverseifbaren mit Aluminiumoxid. Auf diese Weise bleiben sämtliche sauren Verbindungen (Fettsäuren und andere) an der Startlinie. So erhält man eine saubere Trennung der Zone der aliphatischen Alkohole und Terpenalkohole von den Sterinen. |
|
5.2.2. |
Die Entwicklerkammer bis zu einer Höhe von etwa 1 cm mit einem Hexan/Ethylether-Gemisch 65:35 (V/V) beschicken ( 12 ). Die Kammer mit einem geeigneten Deckel verschließen und mindestens eine halbe Stunde stehen lassen, damit sich ein Gleichgewicht zwischen Flüssigkeit und Dampfphase einstellt. An der Innenwand der Kammer können Filterpapierstreifen befestigt werden, die in das Fließmittel eintauchen. Dadurch kann die Laufzeit um ein Drittel verkürzt und eine gleichmäßige Elution der Komponenten erzielt werden. Anmerkung 3: Um gut reproduzierbare Trennungen zu erzielen, ist das Gemisch jedesmal frisch anzusetzen. |
|
5.2.3. |
Von einer 5 %igen Lösung des Unverseifbaren (5.1.5) in Chloroform mit einer 100-μl-Spritze 0,3 ml als durchgehenden, gleichmäßigen Strich 2 cm vom unteren Plattenrand entfernt auf die Dünnschichtplatte (5.2.1) auftragen. In der Verlängerung der Startlinie werden am Rande 2-3 μl der Standardlösung der aliphatischen Alkohole (4.11) aufgetragen, um die Alkoholzone nach der Entwicklung identifizieren zu können. |
|
5.2.4. |
Die Platte in die nach 5.2.2 vorbereitete Entwicklerkammer stellen. Die Temperatur der Umgebung sollte 15-20 °C betragen. Die Kammer mit dem Deckel verschließen und die Platte so lange entwickeln, bis die Fließmittelfront bis 1 cm unter den oberen Plattenrand gestiegen ist. Dann die Platte aus der Entwicklerkammer nehmen und das Lösungsmittel in einem warmen Luftstrom abdampfen oder die Platte eine geraume Zeit unter dem Abzug trocknen lassen. |
|
5.2.5. |
Die Platte vorsichtig und gleichmäßig mit 2′,7′-Dichlorfluoresceinlösung besprühen, wenn sie unter UV-Licht betrachtet wird. Die Lage der Zone der aliphatischen Alkohole kann mit Hilfe des Flecks der Referenzlösung bestimmt werden. Die Grenzen dieser Zone mit schwarzem Stift markieren. Die Zone der aliphatischen Alkohole und die unmittelbar darüber liegende Zone der Terpenalkohole zusammen umranden (Anmerkung 4). Anmerkung 4: Da aliphatische Alkohole in die Zone der Triterpenalkohole migrieren können, müssen die Zonen der aliphatischen und der Terpenalkohole zusammen erfasst werden. Ein Beispiel für die Dünnschichtchromatografie-Trennung ist in der Abbildung 1 der Anlage enthalten. |
|
5.2.6. |
Das in der markierten Zone liegende Kieselgel mit einem Metallspatel abkratzen, fein mahlen und in einen Glasfiltertiegel (3.7) überführen. Mit 10 ml warmem Chloroform versetzen, mit Hilfe des Metallspatels gründlich vermischen und unter Vakuum filtrieren. Das Filtrat in der an den Glasfiltertiegel angeschlossenen Vakuumflasche (3.8) auffangen. Das Kieselgel im Glasfiltertiegel dreimal mit je 10 ml Ethylether waschen und das Filtrat wiederum in der Vakuumflasche auffangen. Das Filtrat bis auf ein Volumen von etwa 4-5 ml eindampfen und den Rest der Lösung in das zuvor gewogene 10-ml-Röhrchen (3.9) überführen. Durch vorsichtiges Erhitzen unter einem schwachen Stickstoffstrom bis zur Trockne eindampfen. Mit einigen Tropfen Aceton versetzen, wieder bis zur Trockne eindampfen, etwa 10 Minuten im Trockenschrank auf 105 °C erhitzen, anschließend im Exsikkator abkühlen lassen und wiegen. Der Rückstand in dem Röhrchen enthält die Alkoholfraktion. |
5.3. Herstellung der Trimethylsilylether (TMSE)
|
5.3.1. |
Dem Probengläschen mit der Fraktion der aliphatischen Alkohole je Milligramm aliphatischem Alkohol 50 μl Silanisierungsreagenz, bestehend aus einem Gemisch aus Pyridin, Hexamethyldisilazan und Trimethylchlorsilan im Verhältnis 9:3:1 (V/V/V) (Anmerkung 5) zusetzen. Dabei darf es nicht zur Aufnahme von Wasser kommen (Anmerkung 6). Anmerkung 5: Gebrauchsfertige Lösungen sind im Handel erhältlich; daneben gibt es auch andere Silanisierungsreagenzien, wie z. B. N,O-bis-Trimethylsilyltrifluoracetamid + 1 % Trimethylchlorsilan, das mit gleichen Teilen wasserfreiem Pyridin gemischt werden muss. Anmerkung 6: Die Beobachtung einer leichten Opaleszenz ist normal und bedeutet keine Störung. Die Bildung eines weißen Niederschlags oder der Eindruck einer Rosafärbung sind Anzeichen der Anwesenheit von Feuchtigkeit oder der Zersetzung des Reagens. In diesem Fall ist der Test zu wiederholen. |
|
5.3.2. |
Das Probengläschen verschließen und gründlich, nicht zu stark schütteln, bis sich die aliphatischen Alkohole gelöst haben. Die Probe mindestens 15 Minuten bei Raumtemperatur stehen lassen und dann einige Minuten zentrifugieren. Die klare Lösung kann gaschromatographisch untersucht werden. |
5.4. Gaschromatographische Analyse
5.4.1. Vorbereiten, Einfahren der Säule
|
5.4.1.1. |
Die Säule in den Gaschromatographen einsetzen, wobei das Einlassteil an den mit dem Trennsystem verbundenen Injektor und das Auslassteil an den Detektor angeschlossen wird. Überprüfung des Gaschromatographen auf Dichtigkeit der Gasleitungen, Betriebsbereitschaft des Detektors, des Trennsystems, des Schreibers usw. |
|
5.4.1.2. |
Wird die Säule zum erstenmal verwendet, ist es ratsam, sie einzufahren. Einen schwachen Gasstrom durch die Säule geben, den Gaschromatographen einschalten und allmählich auf eine Temperatur von mindestens 20 °C über der Arbeitstemperatur (Anmerkung 7) aufheizen. Diese Temperatur mindestens 2 Stunden konstant halten und dann die Analysenbedingungen einstellen (Gasstrom, Split, Zünden der Flamme, Schreiberanschluss, Ofentemperatur, Injektor, Detektor). Eine Empfindlichkeit wählen, die mindestens doppelt so groß ist wie bei der Durchführung der Analyse. Die Grundlinie muss linear verlaufen, ohne Peaks oder Drift. Eine negative Drift ist ein Indiz für einen undichten Anschluss der Säule, eine positive deutet auf mangelhaftes Einfahren der Säule hin. Anmerkung 7: Die Einfahrtemperatur muss in jedem Fall 20 °C unter der für die stationäre Phase angegebenen Maximaltemperatur liegen. |
5.4.2. Wahl der Arbeitsbedingungen
5.4.2.1. Anhaltspunkte für die Arbeitsbedingungen:
— Säulentemperatur: anfangs isotherm 8 Minuten bei 180 °C, dann stufenweise um 5 °C pro Minute bis auf 260 °C ansteigend, anschließend 15 Minuten bei 260 °C,
— Verdampfertemperatur: 280 °C,
— Detektortemperatur: 290 °C
— Strömungsgeschwindigkeit des Trägergases: Helium 20-35 cm/s; Wasserstoff 30-50 cm/s,
— Splitverhältnis: 1:50 — 1:100,
— Geräteempfindlichkeit: das 4- bis 16fache der Mindestdämpfung,
— Detektorempfindlichkeit: 1-2 mV f. s.,
— Papiervorschub: 30-60 cm/Std.
— Einspritzvolumen: 0,5-1 μl TMSE-Lösung.
Diese Bedingungen können entsprechend den Kenndaten der Säule und des Gaschromatographen derart geändert werden, dass die damit aufgezeichneten Chromatogramme folgende Bedingungen erfüllen:
— Die Retentionszeit des C26-Alkohols muss 18 ± 5 Minuten betragen;
— der Peak des C22-Alkohols muss bei Olivenöl 80 ± 20 % und bei Samenölen 40 ± 20 % der gesamten Skala erreichen.
5.4.2.2. Zur Überprüfung der vorstehenden Bedingungen werden wiederholt Proben von TMSE-Alkohol-Gemischen eingespritzt und die Arbeitsbedingungen optimiert.
5.4.2.3. Die Parameter für die Peak-Integration sind so zu wählen, dass für die in Betracht kommenden Peaks korrekte Werte erzielt werden.
5.4.3. Durchführung der Analyse
5.4.3.1. Mit der 10-μl-Mikroliterspritze 1 μl Hexan entnehmen, 0,5 μl Luft und anschließend 0,5-1 μl Probenlösung aufziehen; dabei den Kolben der Spritze so weit einziehen, dass die Nadel leer ist. Die Nadel in die Membran des Injektors einführen, nach 1-2 Sekunden schnell einspritzen, dann nach etwa 5 Sekunden die Nadel langsam herausziehen.
5.4.3.2. Das Chromatogramm aufzeichnen, bis alle vorhandenen aliphatischen Alkohole eluiert sind. Die Grundlinie muss stets den vorgeschriebenen Anforderungen (5.4.1.2) genügen.
5.4.4. Identifizierung der Peaks
Die einzelnen Peaks werden anhand der Retentionszeiten und durch Vergleich mit dem unter denselben Bedingungen analysierten TSME-Gemisch der aliphatischen Alkohole identifiziert.
Beispiele des Chromatogramms der Alkoholfraktion eines raffinierten Olivenöls sind in den Abbildungen 2 und 3 der Anlage enthalten.
5.4.5. Quantitative Bestimmung
5.4.5.1. Die Peakflächen von 1-Eicosanol und der aliphatischen Alkohole C22, C24, C26 und C28 werden mit Hilfe eines Integrators ermittelt.
5.4.5.2. Der Gehalt an den einzelnen aliphatischen Alkoholen in mg/
1 000
g Fett wird nach folgender Formel berechnet:
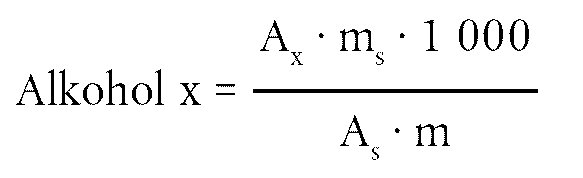
Hierin bedeuten:
Ax = Peakfläche des Alkohols x,
As = Peakfläche von 1-Eicosanol,
ms = zugesetzte Menge 1-Eicosanol in mg,
m = Menge der für die Bestimmung entnommenen Probe in g.
6. DARSTELLUNG DER ERGEBNISSE
Der Gehalt an den einzelnen aliphatischen Alkoholen wird in mg/1 000 g Fett angegeben und zum „Gesamtgehalt an aliphatischen Alkoholen“ addiert.
Anlage
Beispiel für DC-Trennung und Beispiele für Chromatogramme
Abbildung 1
Dünnschichtchromatografie-Platte der unverseifbaren Fraktion von mit Hexan/Ethylether (65/35) eluiertem Olivenöl

|
1 |
C26-Alkohol |
A |
Sterine |
|
2 |
C30-Alkohol |
B |
aliphatische Alkohole |
|
3 |
C20-Alkohol |
C |
Triterpenalkohole |
|
4 |
Gemisch aus C20-22-26-30-Alkoholen |
D |
Squalen |
|
5 |
Unverseifbares von nativem Olivenöl extra |
|
|
Abbildung 2
Chromatogramm der Alkoholfraktion eines raffinierten Olivenöls

|
1 = Phytol |
5 = C24-Alkohol |
9 = 24-Methylen-cycloartenol |
|
2 = Geranylgeraniol |
6 = C26-Alkohol |
10 = Citrostadienol |
|
3 = C20-Alkohol (IS) |
7 = C28-Alkohol |
11 = Cyclobranol |
|
4 = C22-Alkohol |
8 = Cycloartenol |
|
Abbildung 3
Aliphatische und Triterpenalkohole eines raffinierten Olivenöls und eines Olivenöls aus der zweiten Zentrifugation

|
1 = Phytol |
5 = C24-Alkohol |
9 = 24-Methylen-cycloartenol (24MeCA) |
|
2 = Geranylgeraniol |
6 = C26-Alkohol |
10 = Citrostadienol |
|
3 = C20-Alkohol |
7 = C28-Alkohol |
11 = Cyclobranol |
|
4 = C22-Alkohol |
8 = Cycloartenol (CA) |
|
ANHANG XX
Verfahren für die Bestimmung des Gehalts an Wachsen, Fettsäuremethylestern und Fettsäureethylestern durch Kapillargaschromatographie
1. ZWECK
Dieses Verfahren dient der Bestimmung des Gehalts an Wachsen, Fettsäuremethylestern und Fettsäureethylestern in Olivenölen. Die einzelnen Wachse und Alkylester werden nach der Zahl der Kohlenstoffatome getrennt. Das Verfahren wird empfohlen als Instrument für die Unterscheidung zwischen Olivenöl und Oliventresteröl sowie als Qualitätsparameter für native Olivenöle extra, indem es die Feststellung von betrügerischen Mischungen aus nativen Olivenölen extra und Ölen minderwertiger Qualität ermöglicht, unabhängig davon, ob es sich dabei um native Öle, Lampantöle oder desodorierte Öle handelt.
2. PRINZIP
Das Öl wird mit einem geeigneten internen Standard versetzt und chromatographisch über eine Kieselgelsäule fraktioniert. Die unter Versuchsbedingungen eluierte Fraktion (mit schwächerer Polarität als Triacylglycerine) wird rückgeführt und sofort kapillargaschromatographisch analysiert.
3. GERÄTE
|
3.1. |
Erlenmeyerkolben, 25 ml |
|
3.2. |
Glassäule für Flüssigchromatographie, Innendurchmesser 15 mm, Länge 30-40 cm, mit geeignetem Absperrhahn. |
|
3.3. |
Gaschromatograph, geeignet für die Verwendung von Kapillarsäulen, mit Direkteinspritzung, bestehend aus:
|
|
3.4. |
Mikroliterspritze, 10 μl, zur Direkteinspritzung in die Säule, mit gehärteter Nadel |
|
3.5. |
Elektrorührwerk |
|
3.6. |
Rotationsverdampfer |
|
3.7. |
Muffelofen |
|
3.8. |
Analysenwaage mit einer Messgenauigkeit von ± 0,1 mg |
|
3.9. |
Übliche Laborgeräte |
4. REAGENZIEN
|
4.1. |
Kieselgel, 60-200 μm mesh. Das Kieselgel im Muffelofen mindestens vier Stunden lang auf eine Temperatur von 500 °C erhitzen und nach dem Abkühlen mit 2 % Wasser, bezogen auf die verwendete Menge Kieselgel, versetzen. Durch gründliches Schütteln homogenisieren. Vor Gebrauch mindestens 12 Stunden im Exsikkator aufbewahren. |
|
4.2. |
n-Hexan, für die Chromatographie oder Rückstandsanalyse (die Reinheit muss überprüft werden). ACHTUNG - Die Dämpfe können sich entzünden. Von Wärmequellen, Funken oder offenem Feuer fernhalten. Die Flaschen müssen immer fest verschlossen sein. Bei der Verwendung ist für ausreichende Belüftung zu sorgen. Entstehung von Dämpfen verhindern und alle möglichen Geräte, von denen ein Brandrisiko ausgehen kann, z. B. nicht aus nicht entflammbaren Werkstoffen hergestellte Heizgeräte oder elektrische Geräte, entfernen. Schädlich beim Einatmen, da Nervenzellen geschädigt werden können. Dämpfe nicht einatmen. Erforderlichenfalls ein geeignetes Atemschutzgerät benutzen. Berührung mit den Augen und der Haut vermeiden. |
|
4.3. |
Ethylether, für die Chromatographie
ACHTUNG - Leichtentzündlich und mäßig toxisch. Reizt die Haut. Schädlich beim Einatmen. Kann die Augen schädigen. Die Wirkungen können mit Verzögerung eintreten. Der Stoff kann explosionsfähige Peroxide bilden. Die Dämpfe können sich entzünden. Von Wärmequellen, Funken oder offenem Feuer fernhalten. Die Flaschen müssen immer fest verschlossen sein. Bei der Verwendung ist für ausreichende Belüftung zu sorgen. Entstehung von Dämpfen verhindern und alle möglichen Geräte, von denen ein Brandrisiko ausgehen kann, z. B. nicht aus nicht entflammbaren Werkstoffen hergestellte Heizgeräte oder elektrische Geräte, entfernen. Nicht bis zur Trockne oder fast bis zur Trockne eindampfen. Die Peroxidbildung kann durch Zugabe von Wasser oder eines geeigneten Reduktionsmittels verringert werden. Nicht trinken. Dämpfe nicht einatmen. Längere oder wiederholte Berührung mit der Haut vermeiden. |
|
4.4. |
n-Heptan, für die Chromatographie, oder Iso-Octan ACHTUNG - Entzündlich. Schädlich beim Einatmen. Von Wärmequellen, Funken oder offenem Feuer fernhalten. Die Flaschen müssen immer fest verschlossen sein. Bei der Verwendung ist für ausreichende Belüftung zu sorgen. Dämpfe nicht einatmen. Längere oder wiederholte Berührung mit der Haut vermeiden. |
|
4.5. |
Standardlösung aus Laurylarachidat (Anmerkung 3) 0,05 % (m/v) in Heptan (interner Standard für Wachse) Anmerkung 3: Es kann auch Palmitylpalmitat, Myristylstearat oder Arachidyllaureat verwendet werden. |
|
4.6. |
Standardlösung aus Methylheptadecanoat, 0,02 % (m/v) in Heptan (interner Standard für Methyl- und Ethylester) |
|
4.7. |
Sudan 1 (1-Phenylazo-2-naphthol) |
|
4.8. |
Trägergas: Wasserstoff oder Helium, rein, für die Gaschromatographie ACHTUNG Wasserstoff: Leichtentzündlich, unter Druck. Von Wärmequellen, Funken, offenem Feuer oder elektrischen Geräten, die nicht aus nicht entflammbaren Werkstoffen hergestellt sind, fernhalten. Flaschenventil immer geschlossen halten, wenn das Gas nicht verwendet wird. Immer mit einem Druckregler verwenden. Vor dem Öffnen des Flaschenventils die Einstellfeder lockern. Beim Öffnen des Ventils nicht vor der Austrittsöffnung der Flasche stehen. Bei der Verwendung ist für ausreichende Belüftung zu sorgen. Wasserstoff nicht von einer Flasche in eine andere umfüllen. Gas nicht in der Flasche mischen. Die Flaschen gegen Umfallen sichern. Von Sonneneinstrahlung und Wärmequellen fernhalten. In korrosionsfreier Umgebung lagern. Keine beschädigten oder unetikettierten Flaschen verwenden. Helium: Verdichtetes Gas unter hohem Druck. Es verringert die zum Atmen zur Verfügung stehende Menge Sauerstoff. Flasche geschlossen halten. Bei der Verwendung ist für ausreichende Belüftung zu sorgen. Lagerbereiche nur betreten, wenn sie ordnungsgemäß belüftet werden. Immer mit einem Druckregler verwenden. Vor dem Öffnen des Flaschenventils die Einstellfeder lockern. Gas nicht von einer Flasche in eine andere umfüllen. Die Flaschen gegen Umfallen sichern. Beim Öffnen des Ventils nicht vor der Austrittsöffnung der Flasche stehen. Von Sonneneinstrahlung und Wärmequellen fernhalten. In korrosionsfreier Umgebung lagern. Keine beschädigten oder unetikettierten Flaschen verwenden. Nicht einatmen. Nur für technische Zwecke verwenden. |
|
4.9. |
Hilfsgase: — Wasserstoff, rein, für die Gaschromatographie — Luft, rein, für die Gaschromatographie ACHTUNG Luft: Verdichtetes Gas unter hohem Druck. In Gegenwart von brennbaren Stoffen vorsichtig verwenden, da bei den meisten organischen Verbindungen die Selbstentzündungstemperatur an der Luft unter hohem Druck erheblich niedriger ist. Flaschenventil immer geschlossen halten, wenn das Gas nicht verwendet wird. Immer einen Druckregler verwenden. Vor dem Öffnen des Flaschenventils die Einstellfeder lockern. Beim Öffnen des Ventils nicht vor der Austrittsöffnung der Flasche stehen. Gas nicht von einer Flasche in eine andere umfüllen. Gas nicht in der Flasche mischen. Die Flaschen gegen Umfallen sichern. Von Sonneneinstrahlung und Wärmequellen fernhalten. In korrosionsfreier Umgebung lagern. Keine beschädigten oder unetikettierten Flaschen verwenden. Für technische Zwecke bestimmte Luft darf nicht eingeatmet oder für Atemschutzgeräte verwendet werden. |
5. VERFAHREN
5.1. Vorbereiten der Chromatographiesäule
15 g Kieselgel (4.1) in n-Hexan (4.2) suspendieren und in die Säule (3.2) aufgeben. Nach der Spontansedimentation mit einem Elektrorührwerk nachbehandeln, um eine möglichst homogene Chromatographieschicht zu erzielen, und zur Entfernung etwa enthaltener Verunreinigungen mit 30 ml n-Hexan spülen. Mit Hilfe der Analysenwaage (3.8) genau 500 g der Probe in den 25-ml-Erlenmeyerkolben (3.1) einwiegen und entsprechend dem vermuteten Wachsgehalt mit der geeigneten Menge interner Standardlösung (4.5) versetzen. So werden bei Olivenöl 0,1 mg Laurylarachidat, bei Oliventresteröl 0,25 bis 0,50 mg und für Olivenöle 0,05 mg Methylheptadecanoat (4.6) zugesetzt.
Die derart gewonnene Probe unter Verwendung von je zwei Teilen 2 ml n-Hexan (4.2) in die gemäß Chromatographiesäule überführen.
Das Lösungsmittel bis zu einem Stand von 1 mm über der oberen Absorbensgrenzfläche ablaufen lassen. Weiter mit n-Hexan/Ethylether (99:1) spülen und 220 ml bei einem Durchsatz von etwa 15 Tropfen/10 Sekunden auffangen. (Diese Fraktion enthält die Methyl- und Ethylester sowie die Wachse). (Anmerkung 4) (Anmerkung 5).
Anmerkung 4: Das n-Hexan/Ethylethergemisch (99:1) sollte jeden Tag frisch zubereitet werden.
Anmerkung 5: Für eine visuelle Kontrolle der Elution der Wachse können der gelösten Probe 100 μl Sudan I (1 % im Elutionsmittel) zugesetzt werden.
Die Retentionszeit des Farbstoffs liegt zwischen der der Wachse und der der Triacylglycerine. Wenn der Farbstoff das Ende der Säule erreicht, sind daher alle Wachse eluiert und die Elution kann beendet werden.
Die derart gewonnenen Fraktionen im Rotationsverdampfer so lange trocknen, bis das Lösungsmittel nahezu restlos verdampft ist, wobei die letzten 2 ml im schwachen Stickstoffstrom abgeblasen werden. Die Fraktion, die die Methyl- und Ethylester enthält, wird mithilfe von 2-4 ml n-Heptan oder Iso-Octan als Verdünnungslösungsmittel aufgefangen.
5.2. Gaschromatographische Analyse
5.2.1. Vorarbeiten
Die Säule in den Gaschromatographen (3.3) einsetzen, wobei der Säulenanfang an das On-column-System und das Säulenende an den Detektor angeschlossen wird. Den Gaschromatographen auf Dichtigkeit der Gasleitungen, Betriebsbereitschaft des Detektors und des Schreibers usw. überprüfen.
Wird die Säule zum ersten Mal verwendet, wird empfohlen, sie einzufahren. Einen schwachen Gasstrom durch die Säule leiten, den Gaschromatographen einschalten und allmählich über einen Zeitraum von etwa 4 Stunden auf eine Temperatur von 350 °C aufheizen.
Die Temperatur ist mindestens 2 Stunden konstant zu halten; dann die Analysebedingungen einstellen (Gasstrom, Zünden der Flamme, Anschluss an den elektronischen Schreiber (3.3.4), Säulenofentemperatur, Detektor usw.). Das Signal mit einer Empfindlichkeit aufzeichnen, die mindestens doppelt so groß ist wie bei Durchführung der Analyse. Die Grundlinie muss linear verlaufen, ohne Peaks oder Drift.
Eine negative geradlinige Drift ist ein Indiz für einen fehlerhaften Anschluss der Säule, eine positive Drift deutet auf ein mangelhaftes Einfahren der Säule hin.
5.2.2. Wahl der Arbeitsbedingungen für Wachse sowie Methyl- und Ethylester (Anmerkung 6)
Anhaltspunkte für die Arbeitsbedingungen:
|
— |
Säulentemperatur: 20 °C/min 5 °C/min 80 °C Ausgangstemperatur (1′) |
|
— |
Detektortemperatur:350 °C. |
|
— |
Einspritzvolumen:1 μl der n-Heptanlösung (2-4 ml) |
|
— |
Trägergas:Helium oder Wasserstoff mit der für das gewählte Gas optimalen linearen Strömungsgeschwindigkeit (vgl. Anlage A) |
|
— |
Geräteempfindlichkeit,die den genannten Bedingungen genügt. |
Anmerkung 6: Wegen der hohen Endtemperatur ist eine positive Drift von höchstens 10 % der Skala zulässig.
Diese Bedingungen können je nach Beschaffenheit der Säule und des Gaschromatographen abgewandelt werden, damit eine Trennung aller Wachse und Fettsäuremethylester und -ethylester sowie eine ausreichende Auflösung der Peaks (siehe Abbildungen 2, 3 und 4) erzielt werden. Die Retentionszeit des internen Standards Laurylarachidat muss 18 ± 3 Minuten betragen, der größte Wachspeak muss mindestens 60 % des Vollausschlags erreichen und der interne Standard Methylheptadecanoat für die Methyl- und Ethylester muss den Vollausschlag erreichen.
Die Parameter für die Peakintegration sind so zu wählen, dass die in Betracht kommenden Peakflächen korrekt bewertet werden.
5.3. Durchführung der Analyse
Mit der 10-μl-Mikroliterspritze 10 μl Probelösung aufziehen und dabei den Kolben der Spritze so weit einziehen, bis die Nadel leer ist. Die Nadel in das Einspritzsystem einführen, nach 1-2 Sekunden schnell einspritzen, dann nach etwa 5 Sekunden die Nadel langsam herausziehen.
Das Chromatogramm aufzeichnen, bis je nach analysierter Fraktion alle Wachse oder Stigmastadiene eluiert sind.
Die Grundlinie muss stets den vorgeschriebenen Anforderungen genügen.
5.4. Identifizierung der Peaks
Die Peakflächen anhand der Retentionszeiten durch Vergleich mit Wachsgemischen identifizieren, deren Retentionszeiten bekannt sind und die unter denselben Bedingungen analysiert wurden. Die Alkylester werden anhand von Mischungen von Methyl- und Ethylestern der wichtigsten Fettsäuren in Olivenölen (Palmitinsäure und Ölsäure) identifiziert.
Abbildung 1 zeigt ein Chromatogramm der Wachse in einem nativen Olivenöl. Die Abbildungen 2 und 3 zeigen Chromatogramme von zwei nativen Olivenölen extra aus dem Einzelhandel, eines mit Methyl- und Ethylestern, das andere ohne. Abbildung 4 zeigt die Chromatogramme eines nativen Olivenöls extra höchster Qualität und des gleichen Öls, das mit 20 % desodoriertem Öl versetzt ist.
5.5. Quantitative Analyse der Wachse
Die Peakflächen des internen Standards Laurylarachidat und der aliphatischen C40- bis C46-Ester werden mit Hilfe eines Integrators ermittelt.
Der Gesamtwachsgehalt wird durch Addition jedes einzelnen Wachses, in mg/kg Fett, wie folgt bestimmt:
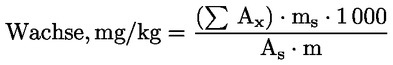
Dabei ist:
|
Ax |
= |
Peakfläche jedes einzelnen Esters in Computer Counts |
|
As |
= |
Peakfläche des internen Standards Laurylarachidat in Computer Counts |
|
ms |
= |
Masse des zugegebenen internen Standards Laurylarachidat in mg; |
|
m |
= |
Masse der für die Bestimmung entnommenen Probe in g. |
5.5.1. Quantitative Analyse der Methyl- und Ethylester
Die Peakflächen des internen Standards Methylheptadecanoat, der Methylester der C16- und C18-Fettsäuren und der Ethylester der C16- und C18-Fettsäuren werden mithilfe eines Integrators bestimmt.
Der Gehalt jedes Alkylesters in mg/kg Fett wird wie folgt bestimmt:

Dabei ist:
|
Ax |
= |
Peakfläche der einzelnen C16- und C18-Ester in Computer Counts |
|
As |
= |
Peakfläche des internen Standards Methylheptadecanoat in Computer Counts |
|
ms |
= |
Masse des zugegebenen internen Standards Methylheptadecanoat in mg; |
|
m |
= |
Masse der für die Bestimmung entnommenen Probe in g. |
6. DARSTELLUNG DER ERGEBNISSE
Die Summe der Gehalte an den einzelnen Wachsen von C40 bis C46 (Anmerkung 7) wird in mg/kg Fett angegeben.
Die Summe der Gehalte der Methyl- und der Ethylester von C16 bis C18 sowie die Summe der beiden angeben.
Die Ergebnisse sind auf das nächste mg/kg zu runden.
Anmerkung 7: Die mengenmäßig zu bestimmenden Bestandteile sind an den Peaks der Ester mit gerader Kohlenstoffzahl von C40 bis C46 abzulesen, wie als Beispiel im nachstehenden Chromatogramm der Wachse von Olivenöl dargestellt. Wenn der C46-Ester doppelt erscheint, ist zur Identifizierung die Wachsfraktion eines Oliventresteröls zu analysieren, bei dem der C46-Peak deutlich überwiegt und daher leicht zu erkennen ist.
Das Verhältnis zwischen Ethylestern und Methylestern angeben.
Abbildung 1
Beispiel eines Gaschromatogramms der Wachsfraktion eines Olivenöls ( 13 )

Peaks mit einer Retentionszeit der Fettsäuremethyl- und -ethylester von 5-8 Minuten.
Erläuterung:
|
I.S. |
= |
Laurylarachidat |
|
1 |
= |
Diterpenester |
|
2+2′ |
= |
C40-Ester |
|
3+3′ |
= |
C42-Ester |
|
4+4′ |
= |
C44-Ester |
|
5 |
= |
C46-Ester |
|
6 |
= |
Sterolester und Triterpenalkohole |
Abbildung 2
Methylester, Ethylester und Wachse in einem nativen Olivenöl
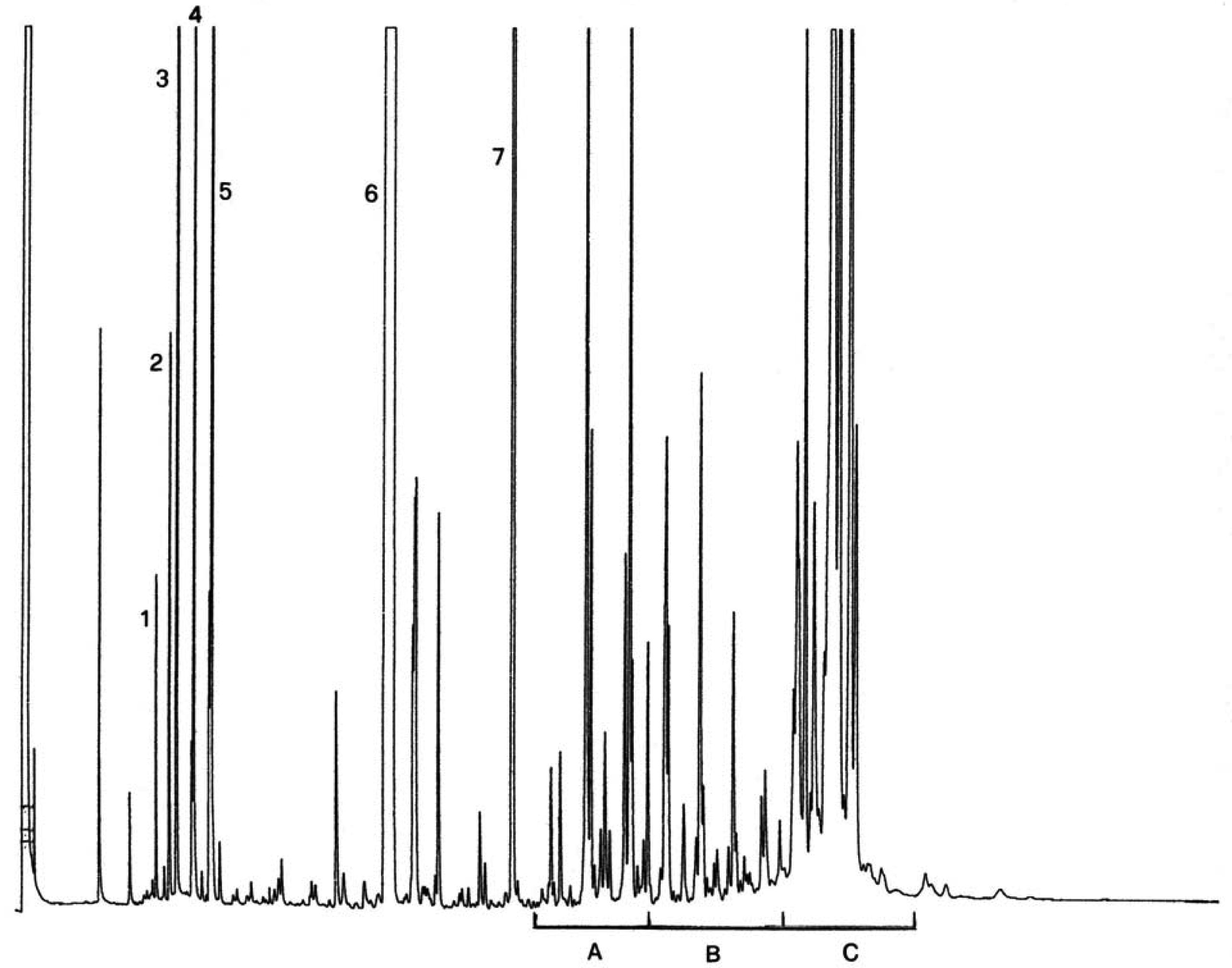
Erläuterung:
|
1 |
– |
Methyl C16 |
|
2 |
– |
Ethyl C16 |
|
3 |
– |
Methylheptadecanoat I.S. |
|
4 |
– |
Methyl C18 |
|
5 |
– |
Ethyl C18 |
|
6 |
– |
Squalen |
|
7 |
– |
Laurylarachidat I.S. |
|
A |
– |
Diterpenester |
|
B |
– |
Wachse |
|
C |
– |
Sterolester und Triterpenester |
Abbildung 3
Methylester, Ethylester und Wachse in einem nativen Olivenöl extra
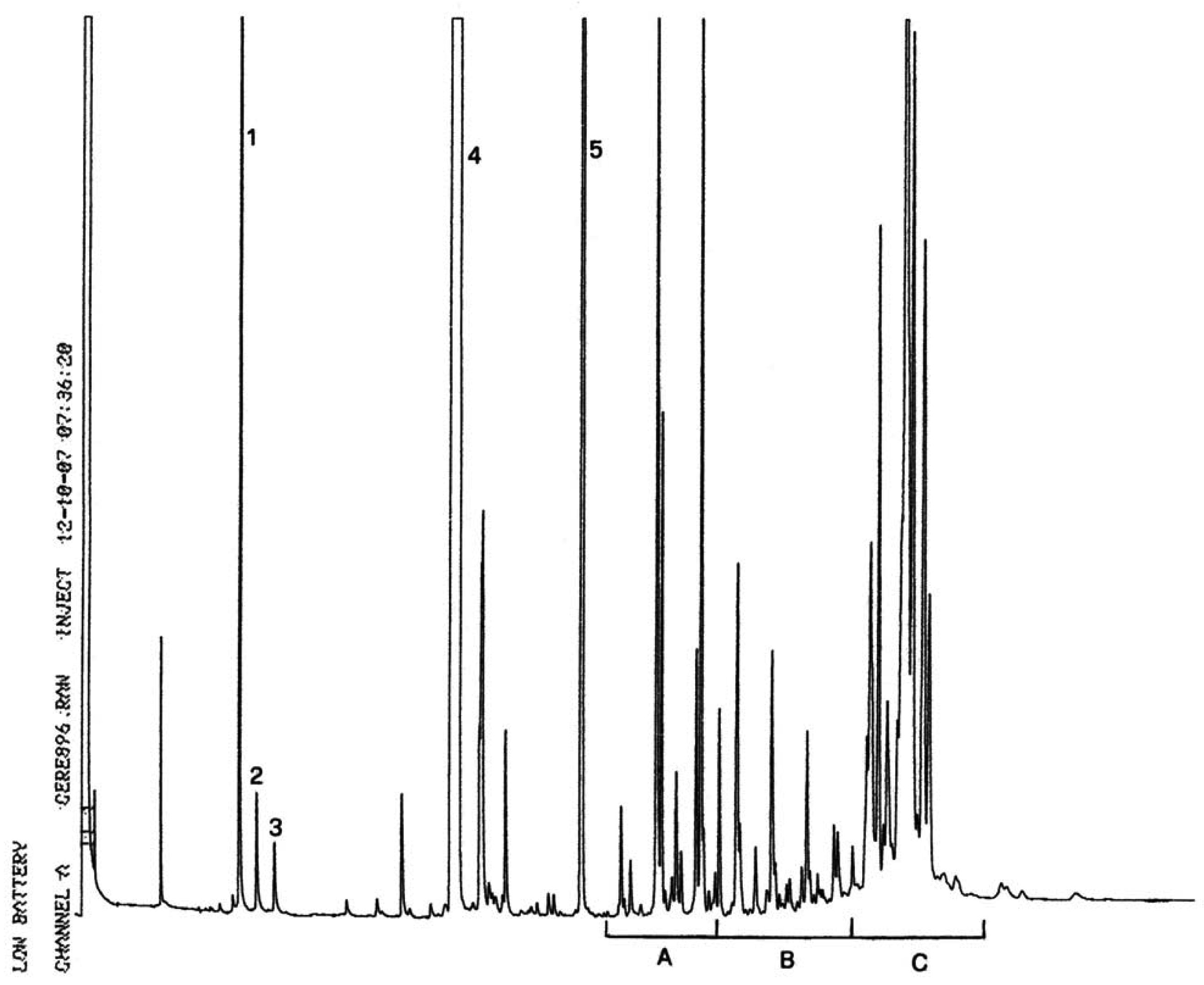
Erläuterung:
|
1 |
– |
Methylheptadecanoat I.S. |
|
2 |
– |
Methyl C18 |
|
3 |
– |
Ethyl C18 |
|
4 |
– |
Squalen |
|
5 |
– |
Laurylarachidat I.S. |
|
A |
– |
Diterpenester |
|
B |
– |
Wachse |
|
C |
– |
Sterolester und Triterpenester |
Abbildung 4
Teil eines Chromatogramms eines nativen Olivenöls extra und des gleichen Öls, das mit desodoriertem Öl versetzt ist

Erläuterung:
|
1 |
– |
Methylmyristat I.S. |
|
2 |
– |
Methylpalmitat |
|
3 |
– |
Ethylpalmitat |
|
4 |
– |
Methylheptadecanoat I.S. |
|
5 |
– |
Methyllinoleat |
|
6 |
– |
Methyloleat |
|
7 |
– |
Methylstearat |
|
8 |
– |
Ethyllinoleat |
|
9 |
– |
Ethyloleat |
|
10 |
– |
Ethylstearat |
Anlage A
Bestimmung der linearen Strömungsgeschwindigkeit des Gases
In den auf Normalbedingungen eingestellten Gaschromatographen 1-3 μl Methan (oder Propan) einspritzen und die Säulendurchlaufzeit des Gases vom Zeitpunkt des Einspritzens bis zum Peak-Austritt (tM) messen.
Die lineare Strömungsgeschwindigkeit in cm/s ist durch die Beziehung L/tM definiert; dabei ist L die Länge der Säule in cm und tM die gemessene Zeit in Sekunden.
▼M28 —————
ANHANG XXI
Ergebnisse der durchgeführten Konformitätskontrollen von Olivenölen gemäß Artikel 8 Absatz 2
|
|
Kennzeichnung |
Chemische Parameter |
Sensorische Merkmale (4) |
Endergebnis |
|||||||||||||
|
Stichprobe |
Kategorie |
Ursprungsland |
Ort der Inspektion (1) |
Offizielle Bezeichnung |
Ursprungsbezeichnung |
Lagerungsbedingungen |
Falsche Informationen |
Lesbarkeit |
K/NK (3) |
Parameter außerhalb der Grenzwerte J/N |
Wenn ja, Angabe der Parameter (2) |
K/NK (3) |
Fehlermedian |
Fruchtigkeitsmedian |
K/NK (3) |
Erforderliche Maßnahme |
Sanktion |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Binnenmarkt (Mühle, Abfüller, Einzelhandel), Ausfuhr, Einfuhr. (2) Jedes Merkmal des Olivenöls gemäß Anhang I erhält einen Code. (3) Konform/Nicht konform. (4) Nicht erforderlich für Olivenöl und Tresteröl. |
|||||||||||||||||
( 1 ) ABl. L 228 vom 1.9.2009, S. 3.
( 2 ) Gehalt an anderen Fettsäuren (%): Palmitinsäure: 7,50-20,00; Palmitoleinsäure: 0,30-3,50; Heptadecansäure: ≤ 0,40; Heptadecensäure: ≤ 0,60; Stearinsäure: 0,50-5,00; Ölsäure: 55,00-83,00; Linolsäure: 2,50-21,00.
( 3 ) Summe der mittels Kapillarsäule (nicht) abtrennbaren Isomere.
( 4 ) Siehe die Anlage zu diesem Anhang.
( 5 ) App. β-Sitosterin: Delta-5,23-Stigmastadienol + Clerosterin + Beta-Sitosterin + Sitostanol + Delta-5-Avenasterin + Delta-5,24-Stigmastadienol.
( 6 ) Öl mit einem Wachsgehalt zwischen 300 mg/kg und 350 mg/kg wird als Lampantöl eingestuft, wenn der Gesamtgehalt an aliphatischen Alkoholen höchstens 350 mg/kg oder der Gehalt an Erytrodiol und Uvaol höchstens 3,5 % beträgt.
( 7 ) Öl mit einem Wachsgehalt zwischen 300 mg/kg und 350 mg/kg wird als rohes Oliventresteröl eingestuft, wenn der Gesamtgehalt an aliphatischen Alkoholen über 350 mg/kg oder der Gehalt an Erytrodiol und Uvaol über 3,5 % beträgt.
( 8 ) Richtlinie 2011/91/EU des Europäischen Parlaments und des Rates vom 13. Dezember 2011 über Angaben oder Marken, mit denen sich das Los, zu dem ein Lebensmittel gehört, feststellen lässt (ABl. L 334 vom 16.12.2011, S. 1).
( 9 ) Nach der Elution der Sterolester darf das Chromatogramm keine signifikanten Peaks aufweisen (Triglyceride).
( 10 ) Verordnung (EU) Nr. 1308/2013 des Europäischen Parlaments und des Rates vom 17. Dezember 2013 über eine gemeinsame Marktorganisation für landwirtschaftliche Erzeugnisse und zur Aufhebung der Verordnungen (EWG) Nr. 922/72, (EWG) Nr. 234/79, (EG) Nr. 1037/2001 und (EG) Nr. 1234/2007 (ABl. L 347 vom 20.12.2013, S. 671).
( 11 ) Der Prüfer kann vom Verkosten eines Öls absehen, wenn er direkt über den Geruch ein extrem stark ausgeprägtes negatives Attribut feststellt. Er vermerkt diesen außergewöhnlichen Umstand in der Profilbeschreibung.
( 12 ) Insbesondere in diesem Fall ist zur sauberen Trennung der Zonen ein Benzol/Aceton-Gemisch 95:5 (V/V) als Elutionsmittel zu verwenden.
( 13 ) Nach der Elution der Sterolester darf das Chromatogramm keine signifikanten Peaks aufweisen (Triacylglycerine).