

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32009R0152
Commission Regulation (EC) No 152/2009 of 27 January 2009 laying down the methods of sampling and analysis for the official control of feed (Text with EEA relevance)
Verordnung (EG) Nr. 152/2009 der Kommission vom 27. Januar 2009 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln (Text von Bedeutung für den EWR)
Verordnung (EG) Nr. 152/2009 der Kommission vom 27. Januar 2009 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln (Text von Bedeutung für den EWR)
OJ L 54, 26.2.2009, p. 1–130
(BG, ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
Special edition in Croatian: Chapter 03 Volume 006 P. 3 - 132
 In force: This act has been changed. Current consolidated version: 04/04/2024
In force: This act has been changed. Current consolidated version: 04/04/2024
- Date of document:
- 27/01/2009
- Date of effect:
- 18/03/2009; Inkrafttreten Datum der Veröffentlichung + 20 Siehe Art. 7
- Date of effect:
- 26/08/2009; Anwendung Siehe Art. 7
- Date of end of validity:
- No end date
- Author:
- Europäische Kommission
- Responsible body:
- Directorate-General for Health and Food Safety
- Form:
- Verordnung
- Additional information:
- Bedeutung für den EWR
- Treaty:
- Vertrag zur Gründung der Europäischen Gemeinschaft
- Legal basis:
-
- 32004R0882 - A11P4PTA) 32004R0882 - A11P4PTB) 32004R0882 - A11P4PTC)
- Link
- Link
- Link
- Select all documents mentioning this document No data available in the table
- Modifies:
-
Relation Act Comment Subdivision concerned From To Repeal 31971L0250 Repeal 31971L0393 Repeal 31972L0199 Repeal 31973L0046 Repeal 31976L0371 Repeal 31976L0372 Repeal 31978L0633 Implicit repeal 31981L0680 26/08/2009 Repeal 31981L0715 Implicit repeal 31984L0004 26/08/2009 Repeal 31984L0425 Repeal 31986L0174 Implicit repeal 31992L0089 26/08/2009 Implicit repeal 31992L0095 26/08/2009 Implicit repeal 31993L0028 26/08/2009 Repeal 31993L0070 Repeal 31993L0117 Implicit repeal 31994L0014 26/08/2009 Implicit repeal 31998L0054 26/08/2009 Repeal 31998L0064 Repeal 31999L0027 Repeal 31999L0076 Implicit repeal 31999L0079 26/08/2009 Repeal 32000L0045 Repeal 32002L0070 Repeal 32003L0126 Implicit repeal 32005L0006 26/08/2009 Implicit repeal 32005L0007 26/08/2009
No data available in the table
- Modified by:
-
Relation Act Comment Subdivision concerned From To Corrected by 32009R0152R(01) (HU) Corrected by 32009R0152R(02) (HU) Modified by 32012R0278 Ersetzung Anhang 5 P.B 18/04/2012 Modified by 32013R0051 Ersetzung Anhang VI 12/02/2013 Modified by 32013R0691 Ersetzung Artikel 1 01/01/2014 Modified by 32013R0691 Ersetzung Anhang I 01/01/2014 Modified by 32013R0691 Ersetzung Anhang II 01/01/2014 Modified by 32014R0709 Ersetzung Anhang V B 17/07/2014 Modified by 32017R0645 Ersetzung (LV) Anhang I Nummer 5.1.3 TABL FOOTNOTE Text 26/04/2017 Modified by 32017R0645 Ersetzung (LV) Anhang I Nummer 5.1.1 TABL FOOTNOTE Text 26/04/2017 Modified by 32017R0645 Ersetzung (LV) Anhang I Nummer 5.1.5 TABL FOOTNOTE Text 26/04/2017 Modified by 32017R0771 Ersetzung Anhang V S. B 24/05/2017 Modified by 32020R1560 Zusatz Anhang VI Nummer 2.1.2.2 Nummer 2.1.2.2.9 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.3.3.1 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.4.3 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.4.1 Satz 1 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.2.1.3.2 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.3.3.2 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.5 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.1 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.3.1 16/11/2020 Modified by 32020R1560 Zusatz Anhang VI Nummer 2.1.2.2 Nummer 2.1.2.2.10 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.2.2.2 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.2.2.3 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.4.2 16/11/2020 Modified by 32020R1560 Zusatz Anhang VI Nummer 2.1.2.2 Nummer 2.1.2.2.11 16/11/2020 Modified by 32020R1560 Ersetzung Anhang VI Nummer 2.1.3.3.4 nicht nummerierter Absatz 16/11/2020 Modified by 32022R0893 Ersetzung Anhang VI Nummer 2.1 28/06/2022 Modified by 32022R0893 Ersetzung Anhang VI Nummer 1 28/06/2022 Modified by 32024R0771 Ersetzung Anhang VII 04/04/2024 Modified by 32024R0771 Ersetzung Anhang I 04/04/2024 Modified by 32024R0771 Ersetzung Anhang II 04/04/2024 Modified by 32024R0771 Ersetzung Artikel 1 nicht nummerierter Absatz 1 04/04/2024 Modified by 32024R0771 Ersetzung Anhang IV 04/04/2024 Modified by 32024R0771 Ersetzung Anhang V 04/04/2024 Modified by 32024R0771 Streichung Anhang VIII 04/04/2024 Modified by 32024R0771 Ersetzung Anhang III 04/04/2024 - Link
- EUROVOC descriptor:
- Subject matter:
- Directory code:
-
- 03.50.10.00 Landwirtschaft / Rechtsangleichung und Viehseuchenrecht / Futtermittel
|
26.2.2009 |
DE |
Amtsblatt der Europäischen Union |
L 54/1 |
VERORDNUNG (EG) Nr. 152/2009 DER KOMMISSION
vom 27. Januar 2009
zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln
(Text von Bedeutung für den EWR)
DIE KOMMISSION DER EUROPÄISCHEN GEMEINSCHAFTEN —
gestützt auf den Vertrag zur Gründung der Europäischen Gemeinschaft,
gestützt auf die Verordnung (EG) Nr. 882/2004 des Europäischen Parlaments und des Rates vom 29. April 2004 über amtliche Kontrollen zur Überprüfung der Einhaltung des Lebensmittel- und Futtermittelrechts sowie der Bestimmungen über Tiergesundheit und Tierschutz (1), insbesondere auf Artikel 11 Absatz 4 Buchstaben a, b und c,
in Erwägung nachstehender Gründe:
|
(1) |
Die nachstehenden Rechtsakte wurden zur Durchführung der Richtlinie 70/373/EWG erlassen und bleiben gemäß Artikel 61 Absatz 2 der Verordnung (EG) Nr. 882/2004 in Kraft:
|
|
(2) |
Da die Richtlinie 70/373/EWG durch die Verordnung (EG) Nr. 882/2004 ersetzt wurde, ist es angezeigt, die Durchführungsrechtsakte zu dieser Richtlinie aufzuheben und in einer einzigen Verordnung zusammenzufassen. Gleichzeitig sollten die Methoden und Verfahren entsprechend den neuesten wissenschaftlichen und technologischen Erkenntnissen angepasst werden. Methoden, die zu ihrem Zweck nicht mehr verwendet werden, sollten gestrichen werden. Es ist geplant, die Probenahmebestimmungen zu gegebener Zeit zu aktualisieren, um den jüngsten Entwicklungen bei der Erzeugung, Lagerung, Beförderung und Vermarktung von Futtermitteln Rechnung zu tragen. Dennoch ist es angezeigt, die bestehenden Probenahmebestimmungen vorläufig beizubehalten. |
|
(3) |
Die Richtlinien 71/250/EWG, 71/393/EWG, 72/199/EWG, 73/46/EWG, 76/371/EWG, 76/372/EWG, 78/633/EWG, 81/715/EWG, 84/425/EWG, 86/174/EWG, 93/70/EWG, 93/117/EG, 98/64/EG, 1999/27/EG, 1999/76/EG, 2000/45/EG, 2002/70/EG und 2003/126/EG sollten daher aufgehoben werden. |
|
(4) |
Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Ausschusses für die Lebensmittelkette und Tiergesundheit — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Die Probenahmen für die amtliche Untersuchung von Futtermitteln auf ihre Bestandteile, Zusatzstoffe und unerwünschte Stoffe, ausgenommen Rückstände von Schädlingsbekämpfungsmitteln und Mikroorganismen, erfolgen nach den in Anhang I aufgeführten Verfahren.
Artikel 2
Die Vorbereitung der Analyseproben und die Formulierung der Ergebnisse erfolgen nach den in Anhang II aufgeführten Methoden.
Artikel 3
Die Analyse für die amtliche Untersuchung von Futtermitteln erfolgt nach den Methoden, die in Anhang III (Analysemethoden zur Untersuchung der Zusammensetzung von Futtermittel-Ausgangserzeugnissen und Mischfuttermitteln), Anhang IV (Analysemethoden zur Untersuchung von Futtermitteln auf ihren Gehalt an zugelassenen Zusatzstoffen), Anhang V (Analysemethoden zur Untersuchung von Futtermitteln auf unerwünschte Stoffe) und Anhang VI (Analysemethoden zur Bestimmung der Bestandteile tierischen Ursprungs bei der amtlichen Untersuchung von Futtermitteln) aufgeführt sind.
Artikel 4
Der Energiegehalt von Mischfuttermitteln für Geflügel wird in Übereinstimmung mit Anhang VII berechnet.
Artikel 5
Die in Anhang VIII aufgeführten Analysemethoden für die Untersuchung auf rechtswidriges Vorhandensein von nicht mehr zugelassenen Zusatzstoffen in Futtermitteln werden zu Bestätigungszwecken verwendet.
Artikel 6
Die Richtlinien 71/250/EWG, 71/393/EWG, 72/199/EWG, 73/46/EWG, 76/371/EWG, 76/372/EWG, 78/633/EWG, 81/715/EWG, 84/425/EWG, 86/174/EWG, 93/70/EWG, 93/117/EG, 98/64/EG, 1999/27/EG, 1999/76/EG, 2000/45/EG, 2002/70/EG und 2003/126/EG werden aufgehoben.
Verweise auf die aufgehobenen Richtlinien gelten als Verweise auf die vorliegende Verordnung nach den Entsprechungstabellen in Anhang IX.
Artikel 7
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Sie gilt ab dem 26. August 2009.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 27. Januar 2009
Für die Kommission
Androulla VASSILIOU
Mitglied der Kommission
(1) ABl. L 165 vom 30.4.2004, S. 1. Berichtigte Fassung im ABl. L 191 vom 28.5.2004, S. 1.
(2) ABl. L 155 vom 12.7.1971, S. 13.
(3) ABl. L 279 vom 20.12.1971, S. 7.
(4) ABl. L 123 vom 29.5.1972, S. 6.
(5) ABl. L 83 vom 30.3.1973, S. 21.
(6) ABl. L 102 vom 15.4.1976, S. 1.
(7) ABl. L 102 vom 15.4.1976, S. 8.
(8) ABl. L 206 vom 29.7.1978, S. 43.
(9) ABl. L 257 vom 10.9.1981, S. 38.
(10) ABl. L 238 vom 6.9.1984, S. 34.
(11) ABl. L 130 vom 16.5.1986, S. 53.
(12) ABl. L 234 vom 17.9.1993, S. 17.
(13) ABl. L 329 vom 30.12.1993, S. 54.
(14) ABl. L 257 vom 19.9.1998, S. 14.
(15) ABl. L 118 vom 6.5.1999, S. 36.
(16) ABl. L 207 vom 6.8.1999, S. 13.
(17) ABl. L 174 vom 13.7.2000, S. 32.
(18) ABl. L 209 vom 6.8.2002, S. 15.
(19) ABl. L 339 vom 24.12.2003, S. 78.
ANHANG I
PROBENAHMEVERFAHREN
1. ZWECK UND ANWENDUNGSBEREICH
Die zur amtlichen Untersuchung bestimmten Futtermittelproben werden gemäß den nachstehenden Verfahren entnommen. Die dabei erhaltenen Proben gelten als repräsentativ für die betreffende Partie.
2. PROBENAHMEPERSONAL
Die Probenahme erfolgt durch die von den Mitgliedstaaten zu diesem Zweck bevollmächtigten Personen.
3. BEGRIFFSBESTIMMUNGEN
Partie: Futtermittelmenge, die eine Einheit bildet und von der angenommen wird, dass sie einheitliche Merkmale besitzt.
Einzelprobe: Menge, die an einer Stelle der Partie entnommen wird.
Sammelprobe: Gesamtmenge von aus einer Partie entnommenen Einzelproben.
Reduzierte Sammelprobe: Repräsentative Teilmenge der Sammelprobe, die nach mengenmäßiger Verringerung erhalten wird.
Endprobe: Teilmenge der reduzierten Sammelprobe oder der homogenisierten Sammelprobe.
4. GERÄTE
|
4.1. |
Die Geräte zur Probenahme müssen aus einem Material bestehen, das die zu beprobenden Erzeugnisse nicht beeinflusst. Diese Geräte können von den Mitgliedstaaten zugelassen werden. |
4.2. Empfohlene Geräte für die Probenahme fester Futtermittel
4.2.1. Manuelle Probenahme
|
4.2.1.1. |
Schaufel mit ebenem Boden und rechteckig hochgebogenem Rand. |
|
4.2.1.2. |
Probestecher mit langem Schlitz oder Kammerstecher. Die Größe des Probestechers ist den Merkmalen der Partie (Tiefe des Behälters, Größe des Sacks usw.) und der Größe der Futtermittelteilchen anzupassen. |
4.2.2. Mechanische Probenahme
Zugelassene mechanische Geräte dürfen zur Probenahme aus in Bewegung befindlichen Futtermitteln verwendet werden.
4.2.3. Probenteiler
Zur Zerlegung der Probe in ungefähr gleiche Teile bestimmte Geräte dürfen zur Herstellung der Einzelproben sowie zur Vorbereitung von reduzierten Sammelproben und Endproben verwendet werden.
5. MENGENMÄSSIGE ANFORDERUNGEN
|
5.A. |
Bei der Untersuchung auf Stoffe oder Erzeugnisse, die gleichmäßig im Futtermittel verteilt sind |
|
|
5.A.1. |
Partie Die Partie darf nur so groß sein, dass von allen Teilen, aus denen die Partie besteht, Proben entnommen werden können. |
|
|
5.A.2. |
Einzelproben |
|
|
5.A.2.1. |
Lose Futtermittel: |
Mindestanzahl der Einzelproben: |
|
5.A.2.1.1. |
Partien bis zu 2,5 t |
7 |
|
5.A.2.1.2. |
Partien von mehr als 2,5 t |
√ 20 Mal die Zahl der Tonnen, aus denen die Partie besteht (1), begrenzt auf höchstens 40 Einzelproben |
|
5.A.2.2. |
Verpackte Futtermittel: |
Mindestanzahl der zu beprobenden Packungen (2): |
|
5.A.2.2.1. |
Packungen von mehr als 1 kg Inhalt |
|
|
5.A.2.2.1.1. |
Partien aus 1-4 Packungen |
Alle Packungen |
|
5.A.2.2.1.2. |
Partien aus 5-16 Packungen |
4 |
|
5.A.2.2.1.3. |
Partien aus mehr als 16 Packungen |
√ Anzahl der Packungen, aus denen die Partie besteht (1), begrenzt auf höchstens 20 Packungen |
|
5.A.2.2.2. |
Packungen bis 1 kg Inhalt |
4 |
|
5.A.2.3. |
Flüssige oder halbflüssige Futtermittel: |
Mindestanzahl der zu beprobenden Behälter (2): |
|
5.A.2.3.1. |
Behälter von mehr als 1 l Inhalt |
|
|
5.A.2.3.1.1. |
Partien aus 1-4 Behältern |
Alle Behälter |
|
5.A.2.3.1.2. |
Partien aus 5-16 Behältern |
4 |
|
5.A.2.3.1.3. |
Partien aus mehr als 16 Behältern |
√ Anzahl der Behälter, aus denen die Partie besteht (1), begrenzt auf höchstens 20 Behälter |
|
5.A.2.3.2. |
Behälter bis zu 1 l Inhalt |
4 |
|
5.A.2.4. |
Futterblöcke und Lecksteine |
Mindestanzahl der zu beprobenden Futterblöcke oder Lecksteine (2): 1 Futterblock oder Leckstein je Partie von 25 Einheiten, begrenzt auf höchstens 4 Futterblöcke oder Lecksteine |
|
5.A.3. |
Sammelproben Je Partie ist eine einzige Sammelprobe erforderlich. Die Gesamtmenge der Einzelproben, aus denen sich die Sammelprobe zusammensetzt, darf nicht unter den nachstehend festgesetzten Mindestmengen liegen: |
|
|
5.A.3.1. |
Lose Futtermittel: |
4 kg |
|
5.A.3.2. |
Verpackte Futtermittel: |
|
|
5.A.3.2.1. |
Packungen von mehr als 1 kg |
4 kg |
|
5.A.3.2.2. |
Packungen bis zu 1 kg |
Gewicht des Inhalts von 4 Originalpackungen |
|
5.A.3.3. |
Flüssige oder halbflüssige Futtermittel: |
|
|
5.A.3.3.1. |
Behälter von mehr als 1 l Inhalt |
4 l |
|
5.A.3.3.2. |
Behälter bis zu 1 l Inhalt |
Volumen des Inhalts von 4 Originalbehältern |
|
5.A.3.4. |
Futterblöcke oder Lecksteine: |
|
|
5.A.3.4.1. |
mit einem Einzelgewicht von mehr als 1 kg |
4 kg |
|
5.A.3.4.2. |
mit einem Einzelgewicht bis zu 1 kg |
Gewicht von 4 Originalblöcken oder -steinen |
|
5.A.4. |
Endprobe Die Sammelprobe dient, sofern erforderlich nach Reduzierung, der Herstellung der Endproben. Mindestens eine Endprobe ist zu untersuchen. Die Menge jeder zur Untersuchung bestimmten Endprobe darf nicht unter den nachstehend festgesetzten Mindestmengen liegen: |
|
|
|
Feste Futtermittel |
500 g |
|
|
Flüssige oder halbflüssige Futtermittel |
500 ml |
|
5.B. |
Bei der Untersuchung auf unerwünschte Stoffe oder Erzeugnisse, die ungleichmäßig im Futtermittel verteilt sein können, wie z. B. Aflatoxine, Mutterkorn, Ricinus und Crotalaria in Einzelfuttermitteln (3) |
|
|
5.B.1. |
Partie: siehe 5.A.1 |
|
|
5.B.2. |
Einzelproben |
|
|
5.B.2.1. |
Lose Futtermittel: siehe 5.A.2.1 |
|
|
5.B.2.2. |
Verpackte Futtermittel: |
Mindestanzahl der zu beprobenden Packungen: |
|
5.B.2.2.1. |
Partien aus 1-4 Packungen |
Alle Packungen |
|
5.B.2.2.2. |
Partien aus 5-16 Packungen: |
4 |
|
5.B.2.2.3. |
Partien aus mehr als 16 Packungen |
√ Anzahl der Packungen, aus der die Partie besteht (1), begrenzt auf höchstens 40 Packungen |
|
5.B.3. |
Sammelproben Die Anzahl der Sammelproben hängt von der Größe der Partie ab. Die Mindestanzahl der Sammelproben je Partie ist nachstehend angegeben. Die Gesamtmenge der Einzelproben, aus denen sich die Sammelprobe zusammensetzt, darf nicht unter 4 kg liegen. |
|
|
5.B.3.1. |
Lose Futtermittel: |
|
|
|
Gewicht der Partie (t) |
Mindestanzahl der Sammelproben je Partie: |
|
|
bis zu 1 t |
1 |
|
|
mehr als 1 bis 10 t |
2 |
|
|
mehr als 10 bis 40 t |
3 |
|
|
mehr als 40 t |
4 |
|
5.B.3.2. |
Verpackte Futtermittel: |
|
|
|
Anzahl der Packungen, die die Partie bilden: |
Mindestanzahl der Sammelproben je Partie: |
|
|
von 1 bis 16 |
1 |
|
|
17 bis 200 |
2 |
|
|
201 bis 800 |
3 |
|
|
mehr als 800 |
4 |
|
5.B.4. |
Endprobe Jede Sammelprobe ergibt nach Reduzierung die Endproben. Mindestens eine Endprobe je Sammelprobe ist zu untersuchen. Das Gewicht der zur Untersuchung bestimmten Endproben darf nicht unter 500 g liegen. |
|
6. VORSCHRIFTEN FÜR DIE ENTNAHME, VORBEREITUNG UND VERPACKUNG DER PROBEN
6.1. Allgemeines
Die Proben sind so schnell wie möglich zu entnehmen und vorzubereiten, wobei mit der angemessenen Sorgfalt vorzugehen ist, damit das Erzeugnis weder verändert noch verunreinigt wird. Die für die Probenahme bestimmten Geräte, Flächen und Behälter müssen sauber und trocken sein.
6.2. Einzelproben
6.2.A. Bei der Untersuchung auf Stoffe oder Erzeugnisse, die gleichmäßig im Futtermittel verteilt sind
Die Einzelproben sind nach dem Zufallsprinzip aus der gesamten Partie zu entnehmen. Ihre Größe muss ungefähr gleich sein.
6.2.A.1.
Die Partie ist künstlich in ungefähr gleiche Teile aufzuteilen. Nach dem Zufallsprinzip ist entsprechend der Anzahl der unter 5.A.2 vorgesehenen Einzelproben eine Anzahl Teile zu wählen und jedem dieser Teile mindestens eine Probe zu entnehmen.
Die Probenahme kann auch bei einer Partie erfolgen, die sich in Bewegung (Aufladen bzw. Abladen) befindet.
6.2.A.2.
Die erforderliche Anzahl der zu beprobenden Packungen ist nach 5.A.2 festgelegt; aus jeder dieser Packungen ist ein Teil des Inhalts mit einem Probestecher oder einer Schaufel zu entnehmen. Gegebenenfalls sind die Proben zu entnehmen, nachdem die Packungen getrennt entleert worden sind. Wenn erforderlich, sind bei jeder einzelnen Sammelprobe Klumpen zu zerdrücken; dazu werden diese gegebenenfalls von dem übrigen Material abgetrennt und anschließend wieder untergemischt.
6.2.A.3.
Die erforderliche Anzahl der zu beprobenden Behälter ist nach 5.A.2 festgelegt; aus jedem dieser Behälter ist eine Probe zu entnehmen, nachdem sein Inhalt, falls nötig, homogenisiert worden ist.
Die Einzelproben können auch beim Ablassen des Erzeugnisses entnommen werden.
6.2.A.4.
Die erforderliche Anzahl der zu beprobenden Behälter ist nach 5.A.2 festgelegt; die Proben sind verschiedenen Höhen zu entnehmen.
Die Proben können ebenfalls während des Ablassens eines Erzeugnisses nach Beseitigung der ersten Bestandteile entnommen werden.
In beiden Fällen muss das Gesamtvolumen der entnommenen Proben mindestens 10 l betragen.
6.2.A.5.
Die erforderliche Anzahl der zu beprobenden Futterblöcke oder Lecksteine ist nach 5.A.2 festgelegt; jedem Block oder Stein ist ein Teil zu entnehmen.
6.2.B. Bei der Untersuchung auf unerwünschte Stoffe oder Erzeugnisse, die ungleichmäßig im Futtermittel verteilt sein können, wie z. B. Aflatoxine, Mutterkorn, Ricinus und Crotalaria in Futtermittel-Ausgangserzeugnissen
Die Partie ist entsprechend der Anzahl der unter 5.B.3 vorgesehenen Sammelproben künstlich in eine Anzahl ungefähr gleicher Teile aufzuteilen. Falls diese Anzahl größer als 1 ist, ist die Gesamtanzahl der unter 5.B.2 vorgesehenen Einzelproben ungefähr gleich auf die verschiedenen Teile zu verteilen. Anschließend sind Proben ungefähr gleicher Größe (4) dergestalt zu ziehen, dass das Gesamtgewicht der Proben jedes Teiles nicht unter 4 kg liegt, wie dies für jede Sammelprobe erforderlich ist. Die aus verschiedenen Teilen stammenden Einzelproben dürfen nicht miteinander vereinigt werden.
6.3. Vorbereitung der Sammelproben
6.3.A. Bei der Untersuchung auf Stoffe oder Erzeugnisse, die gleichmäßig im Futtermittel verteilt sind
Die Einzelproben sind zu einer einzigen Sammelprobe zusammenzufassen.
6.3.B. Bei der Untersuchung auf unerwünschte Stoffe oder Erzeugnisse, die ungleichmäßig im Futtermittel verteilt sein können, wie z. B. Aflatoxine, Mutterkorn, Ricinus und Crotalaria in Futtermittel-Ausgangserzeugnissen
Die Einzelproben aus jedem Teil der Partie sind zu mischen, und daraus ist die in 5.B.3 vorgesehene Anzahl Sammelproben herzustellen. Dabei ist darauf zu achten, dass die Herkunft jeder Sammelprobe angegeben wird.
6.4. Vorbereitung der Endproben
Jede Sammelprobe ist sorgfältig zu mischen, bis sie homogen ist (5). Wenn nötig, wird die Sammelprobe zuerst auf bis zu mindestens 2 kg bzw. 2 l entweder mittels eines mechanischen oder automatischen Probenteilers oder durch das Vierteilungsverfahren reduziert (reduzierte Sammelprobe).
Dann werden mindestens 3 ungefähr gleich große Endproben entsprechend den mengenmäßigen Anforderungen unter 5.A.4 oder 5.B.4 hergestellt. Jede Probe ist in einen geeigneten Behälter zu füllen. Es sind alle notwendigen Vorkehrungen zu treffen, damit jede Veränderung der Zusammensetzung bzw. jede Verunreinigung oder Verfälschung der Probe während des Transports oder der Lagerung vermieden wird.
6.5. Verschließung und Kennzeichnung der Endproben
Die Behälter oder Packungen sind so zu versiegeln/zu plombieren und zu kennzeichnen, dass sie nicht ohne Beschädigung des Siegels/der Plombe geöffnet werden können, wobei die gesamte Kennzeichnung von dem Siegel/der Plombe mit erfasst werden muss.
7. PROBENAHMEPROTOKOLL
Für jede Probenahme ist ein Probenahmeprotokoll zu erstellen, aus dem die Identität der beprobten Partie eindeutig hervorgeht.
8. VERWENDUNG DER PROBEN
Für jede Sammelprobe wird so schnell wie möglich mindestens eine Endprobe — zusammen mit den Angaben, die für die Untersuchung erforderlich sind — an das zugelassene Laboratorium gesandt.
(1) Wenn die Zahl einen Bruch ergibt, ist dieser auf die nächsthöhere ganze Zahl aufzurunden.
(2) Für Packungen oder Behälter bis zu 1 kg oder 1 l Inhalt sowie für Futterblöcke oder Lecksteine bis zu 1 kg bildet der Inhalt einer Originalverpackung oder eines Originalbehälters, ein Futterblock oder ein Leckstein eine Einzelprobe.
(3) Die unter 5.A vorgesehenen Methoden sind bestimmt für die Untersuchung auf Aflatoxine, Mutterkorn, Ricinus und Crotalaria in Allein- und Ergänzungsfuttermitteln.
(4) Bei den verpackten Futtermitteln ist ein Teil des zu beprobenden Inhalts mittels eines Probestechers oder einer Schaufel zu entnehmen, nachdem die Packungen gegebenenfalls getrennt geleert wurden.
(5) Wenn erforderlich, sind bei jeder einzelnen Sammelprobe Klumpen zu zerdrücken; dazu werden diese gegebenenfalls von dem übrigen Material abgetrennt und anschließend wieder untergemischt.
ANHANG II
ALLGEMEINE BESTIMMUNGEN HINSICHTLICH DER METHODEN ZUR ANALYSE VON FUTTERMITTELN
A. VORBEREITUNG DER PROBEN ZUR ANALYSE
1. Zweck
Die nachfolgend beschriebenen Verfahren beziehen sich auf die Vorbereitung der zur Analyse bestimmten Endproben, die nach der Probenahme gemäß den Bestimmungen von Anhang I an die Kontrolllaboratorien gesandt wurden.
Die Vorbereitung dieser Proben ist so vorzunehmen, dass die in den Analysemethoden vorgesehenen Einwaagen homogen und repräsentativ für die Endproben sind.
2. Vorsichtsmaßnahmen
Die Wahl des Verfahrens für die Probenvorbereitung hängt von der Art der angewandten Analysemethode ab. Daher ist es von größter Bedeutung, sicherzustellen, dass das angewandte Probenvorbereitungsverfahren für die jeweilige Analysemethode geeignet ist.
Alle notwendigen Schritte sind so durchzuführen, dass eine Verunreinigung der Probe und eine Veränderung ihrer Zusammensetzung so weit wie möglich vermieden werden.
Das Zerkleinern, Mischen und Sieben ist möglichst rasch durchzuführen, wobei die Probe möglichst wenig Luft und Licht ausgesetzt wird. Die Verwendung von Mühlen oder Zerkleinerungsgeräten, die zu einer merklichen Erwärmung der Probe führen könnten, ist zu vermeiden.
Für besonders hitzeempfindliche Futtermittel wird manuelles Zerkleinern empfohlen. Es ist außerdem darauf zu achten, dass die verwendeten Geräte keine Kontaminationsquelle für Spurenelemente bilden.
Kann die Probe nicht ohne eine signifikante Veränderung ihres Feuchtigkeitsgehalts vorbereitet werden, so ist dieser vor und nach der Vorbereitung gemäß dem in Anhang III Teil A festgelegten Verfahren zu bestimmen.
3. Verfahren
Die Probe ist für Analyse- bzw. Referenzzwecke mittels angemessener Teilungsmethoden wie alternierendes Schaufeln, Riffel- oder Rotationsteilung in geeignete Teilproben zu teilen. Das Kegeln und das Vierteilungsverfahren werden nicht empfohlen, da mit diesen eine hohe Teilungsfehlerquote bei den Teilproben einhergehen kann. Die Referenzproben sind in einem geeigneten sauberen und trockenen, luftdicht verschließbaren Behälter aufzubewahren. Die für die Analyse bestimmten Teilproben von mindestens 100 g sind wie nachfolgend angegeben vorzubereiten.
3.1. Futtermittel, die direkt gemahlen werden können
Sofern keine besonderen Angaben in der Analysemethode gemacht werden, wird die gesamte Probe nach dem Zerkleinern erforderlichenfalls durch ein Sieb mit einer Maschenweite von 1 mm Seitenlänge (entsprechend der Empfehlung ISO R 565) passiert. Zu starkes Zerkleinern ist zu vermeiden.
Die gesiebte Probe wird gemischt und in einen geeigneten sauberen, trockenen, luftdicht verschließbaren Behälter abgefüllt. Unmittelbar vor der Einwaage muss die Probe erneut gemischt werden.
3.2. Futtermittel, die nach Trocknung gemahlen werden können
Sofern keine besonderen Angaben in der Analysemethode gemacht werden, wird die Probe bis auf einen Feuchtigkeitsgehalt von 8 bis 12 % unter Anwendung des unter Nummer 4.3 der Methode zur Bestimmung des Feuchtigkeitsgehalts (siehe Anhang III Teil A) vorgesehenen Vortrocknungsverfahrens getrocknet. Die weitere Vorbereitung erfolgt gemäß Nummer 3.1.
3.3. Flüssige oder halbflüssige Futtermittel
Die Probe wird in einen geeigneten sauberen, trockenen, luftdicht verschließbaren Behälter abgefüllt und unmittelbar vor der Einwaage gründlich gemischt.
3.4. Andere Futtermittel
Proben, die nach keinem der oben genannten Verfahren vorbereitet werden können, sind durch ein anderes Verfahren zu behandeln, das eine homogene und repräsentative Einwaage der Endprobe gestattet.
4. Aufbewahrung der Proben
Die Proben müssen bei einer Temperatur gelagert werden, die ihre Zusammensetzung nicht beeinflusst. Für Proben, die zur Analyse von Vitaminen oder besonders lichtempfindlichen Stoffen bestimmt sind, werden braune Glasgefäße verwendet.
B. BESTIMMUNGEN ÜBER IN ANALYSEVERFAHREN VERWENDETE REAGENZIEN UND GERÄTE
|
1. |
Vorbehaltlich besonderer Angaben in den Analysemethoden müssen alle zur Analyse verwendeten Reagenzien von analysenreiner (p. a.) Qualität sein. Bei der Analyse von Spurenelementen muss die Reinheit der Reagenzien in einem Reagenzienblindversuch überprüft werden. Je nach dem erhaltenen Wert kann eine weitere Reinigung der Reagenzien erforderlich sein. |
|
2. |
Bei jedem in den Analysemethoden erwähnten Lösungs-, Verdünnungs-, Spül- oder Auswaschvorgang, bei dem keine Angabe über die Art des zu verwendenden Lösungs- oder Verdünnungsmittels gemacht wird, ist Wasser zu verwenden. Im Allgemeinen wird destilliertes oder demineralisiertes Wasser verwendet. In besonderen in den Analysemethoden angegebenen Fällen muss das Wasser speziellen Reinigungsverfahren unterzogen werden. |
|
3. |
Da die übliche Ausstattung von Kontrolllaboratorien vorausgesetzt wird, werden nur besondere Instrumente und Geräte oder solche, die speziellen Anforderungen entsprechen müssen, in den Analysemethoden aufgeführt. Sie müssen gut gereinigt sein, vor allem bei der Bestimmung von sehr geringen Stoffmengen. |
C. ANWENDUNG VON ANALYSEMETHODEN UND FORMULIERUNG DER ERGEBNISSE
1. Extraktionsverfahren
Einige Methoden geben ein spezifisches Extraktionsverfahren vor. Generell können andere Extraktionsverfahren als das in der Methode genannte angewandt werden unter der Bedingung, dass das verwendete Extraktionsverfahren für die analysierte Matrix eine vergleichbare Extraktionseffizienz aufweist wie das in der Methode genannte Verfahren.
2. Clean-up-Verfahren
Einige Methoden geben ein spezifisches Clean-up-Verfahren vor. Generell können andere Clean-up-Verfahren als das in der Methode genannte angewandt werden unter der Bedingung, dass das verwendete Clean-up-Verfahren für die analysierte Matrix zu vergleichbaren Analyseergebnissen führt wie das in der Methode genannte Verfahren.
3. Berichterstattung über die angewandte Analysemethode
Im Allgemeinen wird zur Bestimmung eines Stoffes in Futtermitteln eine einzige Analysemethode festgelegt. Werden mehrere Methoden genannt, muss die vom Kontrolllaboratorium angewandte Methode im Analysebericht angegeben werden.
4. Anzahl der Bestimmungen
Das im Analysebericht angegebene Ergebnis soll den Mittelwert aus mindestens 2 Bestimmungen von separaten Einwaagen der Probe mit ausreichender Wiederholbarkeit darstellen.
Allerdings sind bei der Analyse auf unerwünschte Stoffe — sofern das Ergebnis der ersten Bestimmung deutlich (> 50 %) unter dem zu kontrollierenden Sollwert liegt — keine weiteren Bestimmungen erforderlich unter der Bedingung, dass die geeigneten Qualitätsverfahren angewandt werden.
Bei der Kontrolle des angegebenen Gehalts an einem Stoff oder einer Zutat sind — sofern das Ergebnis der ersten Bestimmung den angegebenen Gehalt bestätigt, d. h., wenn das Analyseergebnis innerhalb des akzeptablen Toleranzbereichs für den angegebenen Gehalt liegt — keine weiteren Bestimmungen erforderlich unter der Bedingung, dass die geeigneten Qualitätsverfahren angewandt werden.
In einigen Fällen ist dieser Toleranzbereich in Rechtsvorschriften wie beispielsweise in der Richtlinie 79/373/EWG des Rates (1) definiert.
5. Bericht über die Analyseergebnisse
Das Ergebnis ist gemäß den Angaben in den Analysemethoden mit einer angemessenen Zahl an signifikanten Ziffern anzugeben und, falls nötig, auf den Feuchtigkeitsgehalt der Endprobe vor deren Vorbereitung umzurechnen.
6. Messunsicherheit und Wiederfindungsrate bei der Analyse auf unerwünschte Stoffe
Hinsichtlich unerwünschter Stoffe im Sinne der Richtlinie 2002/32/EG, einschließlich Dioxinen und dioxinähnlichen PCB, erfüllt ein zur Verfütterung bestimmtes Erzeugnis die Bestimmung bezüglich des festgelegten Höchstgehalts nicht, wenn das Analyseergebnis unter Berücksichtigung der erweiterten Messunsicherheit und der Berichtigung um die Wiederfindungsrate den Höchstgehalt überschreitet. Zur Beurteilung, ob die Höchstgehalte eingehalten werden, wird die um die Wiederfindungsrate berichtigte gemessene Konzentration sowie die vom Analyseergebnis subtrahierte erweiterte Messunsicherheit herangezogen. Dieses Verfahren wird nur dann angewandt, wenn die Analysemethode die Schätzung der Messunsicherheit und die Berichtigung um die Wiederfindungsrate ermöglicht (z. B. nicht möglich bei mikroskopischer Analyse).
Das Analyseergebnis ist wie folgt anzugeben (soweit die verwendete Analysemethode die Schätzung der Messunsicherheit und der Wiederfindungsrate ermöglicht):
|
a) |
Berichtigung um die Wiederfindungsrate, wobei diese anzugeben ist. Eine Berichtigung um die Wiederfindungsrate ist nicht erforderlich, wenn Letztere zwischen 90 und 110 % beträgt; |
|
b) |
als „x +/– U“, wobei x das Analyseergebnis und U die erweiterte Messunsicherheit bezeichnen und ein Faktor von 2 verwendet wird, der zu einem Konfidenzniveau von ca. 95 % führt. |
Liegt jedoch das Analyseergebnis deutlich (> 50 %) unter dem zu kontrollierenden Sollwert — und unter der Bedingung, dass die geeigneten Qualitätsverfahren angewandt werden und die Analyse lediglich dem Zweck der Überprüfung der Einhaltung der Rechtsvorschriften dient —, können die Analyseergebnisse ohne Berichtigung um die Wiederfindungsrate angegeben werden, und die Angabe der Wiederfindungsrate und der Messunsicherheit kann in diesen Fällen entfallen.
ANHANG III
ANALYSEMETHODEN ZUR UNTERSUCHUNG DER ZUSAMMENSETZUNG VON FUTTERMITTEL-AUSGANGSERZEUGNISSEN UND MISCHFUTTERMITTELN
A. BESTIMMUNG DES FEUCHTIGKEITSGEHALTS
1. Zweck und Anwendungsbereich
Diese Methode erlaubt die Bestimmung des Feuchtigkeitsgehalts von Futtermitteln. Bei Futtermitteln, die flüchtige Stoffe wie z. B. organische Säuren enthalten, ist zu beachten, dass zusammen mit dem Feuchtigkeitsgehalt auch bedeutende Mengen an flüchtigen Stoffen bestimmt werden.
Sie betrifft nicht die Untersuchung von Milcherzeugnissen als Futtermittel-Ausgangserzeugnisse, die Untersuchung von Mineralstoffen und Mischungen, die überwiegend aus Mineralstoffen bestehen, die Untersuchung von tierischen und pflanzlichen Fetten und Ölen oder die Untersuchung von Ölsaaten und Ölfrüchten.
2. Prinzip
Die Probe wird unter definierten Bedingungen getrocknet, die von der Art des Futtermittels abhängen. Der Gewichtsverlust wird durch Wiegen bestimmt. Bei festen Futtermitteln mit einem hohen Feuchtigkeitsgehalt ist eine zusätzliche Vortrocknung erforderlich.
3. Geräte
|
3.1. |
Zerkleinerungsgerät aus einem Material, das keine Feuchtigkeit absorbiert, leicht zu reinigen ist, eine schnelle und gleichmäßige Zerkleinerung ermöglicht, ohne eine merkliche Erwärmung hervorzurufen, das so weit wie möglich den Kontakt mit der Außenluft verhindert und den unter 4.1.1 und 4.1.2 gestellten Anforderungen entspricht (z. B. Mikroschlagkreuzmühlen, Mikrozerkleinerer mit Wasserkühlung, zerlegbare Kegelmühlen, langsam laufende Kegelmühlen oder Zahnscheibenmühlen). |
|
3.2. |
Analysenwaage, Genauigkeit 1 mg. |
|
3.3. |
Trockene Gefäße aus korrosionsbeständigem Metall oder Glas, mit luftdicht schließenden Deckeln; die Nutzfläche muss eine Verteilung der Probe von ca. 0,3 g/cm2 ermöglichen. |
|
3.4. |
Elektrisch beheizter, temperaturgeregelter Trockenschrank (± 2 oC), der eine schnelle Regelung der Temperatur gewährleistet und eine gute Lüftung besitzt (1). |
|
3.5. |
Elektrisch beheizter regulierbarer Vakuumtrockenschrank mit einer Ölpumpe, der entweder mit einer Vorrichtung für die Zufuhr warmer und getrockneter Luft oder mit einem Trocknungsmittel (z. B. Calciumoxid) ausgestattet ist. |
|
3.6. |
Exsikkator mit dicker, perforierter Platte aus Metall oder Porzellan, der ein wirksames Trocknungsmittel enthält. |
4. Verfahren
|
Anmerkung |
: |
Die Verfahren, die in diesem Abschnitt beschrieben werden, müssen unverzüglich nach dem Öffnen der Packungen, die die Proben enthalten, durchgeführt werden. Die Analysen sind mindestens doppelt auszuführen. |
4.1. Vorbereitung
4.1.1.
Mindestens 50 g der Probe werden entnommen und erforderlichenfalls unter Vermeidung von Feuchtigkeitsänderungen entsprechend zerkleinert oder geteilt (siehe 6).
4.1.2.
Mindestens 50 g der Probe werden entnommen. Diese Menge wird so gemahlen, dass zumindest 50 % der Teilchen durch ein Sieb mit einer Maschenweite von 0,5 mm passiert werden können und dass beim Passieren durch ein Rundlochsieb mit einem Lochdurchmesser von 1 mm ein Rückstand von höchstens 10 % verbleibt.
4.1.3.
Etwa 25 g der Probe, auf 10 mg genau gewogen, werden entnommen, mit einer entsprechenden, auf 10 mg genau gewogenen Menge wasserfreiem Sand vermischt, bis ein homogenes Produkt entsteht.
4.2. Trocknung
4.2.1.
Ein Gefäß (3.3) wird mit seinem Deckel auf 1 mg genau gewogen. Etwa 5 g der Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird ohne Deckel in den auf 103 oC vorgeheizten Trockenschrank gestellt. Damit die Temperatur des Trockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen. Es wird 4 h lang getrocknet, wobei die Trocknungszeit von dem Zeitpunkt an gerechnet wird, an dem die Temperatur im Trockenschrank erneut 103 oC erreicht hat. Nach Öffnen des Trockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, 30 bis 45 min lang zum Abkühlen in den Exsikkator (3.6) gestellt und anschließend auf 1 mg genau gewogen.
Bei Futtermitteln, die überwiegend aus Ölen und Fetten bestehen, wird eine zusätzliche Trocknung von 30 min im Trockenschrank bei 130 oC vorgenommen. Der Unterschied zwischen den beiden Wägeergebnissen darf höchstens 0,1 % Feuchtigkeit betragen.
4.2.2.
Ein Gefäß (3.3) wird mit seinem Deckel auf 0,5 mg genau gewogen. Etwa 5 g der zerkleinerten Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird ohne Deckel in den auf 130 oC vorgeheizten Trockenschrank gestellt. Damit die Temperatur des Trockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen. Es wird 2 h lang getrocknet, wobei die Trocknungszeit von dem Zeitpunkt an gerechnet wird, an dem die Temperatur im Trockenschrank erneut 130 oC erreicht hat. Nach Öffnen des Trockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, 30 bis 45 min lang zum Abkühlen in den Exsikkator (3.6) gestellt und anschließend auf 1 mg genau gewogen.
|
4.2.3. |
Mischfuttermittel mit einem Saccharose- oder Lactosegehalt von mehr als 4 %: Futtermittel-Ausgangserzeugnisse wie z. B. Johannisbrotschrot, hydrolysierte Getreideerzeugnisse, Malzkeime, Zuckerrübenschnitzel, Fisch- und Zuckerpresssäfte; Mischfuttermittel mit einem Gehalt an kristallwasserhaltigen Mineralstoffen von mehr als 25 %. Ein Gefäß (3.3) wird mit seinem Deckel auf 0,5 mg genau gewogen. Etwa 5 g der Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird ohne Deckel in den auf 80 bis 85 oC vorgeheizten Vakuumtrockenschrank (3.5) gestellt. Damit die Temperatur des Vakuumtrockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen. Der Druck wird auf 100 Torr eingestellt. Die Probe wird 4 h lang bei diesem Druck entweder unter Zufuhr von heißer trockener Luft oder mittels eines Trocknungsmittels (etwa 300 g für 20 Proben) getrocknet. Im letzteren Fall wird beim Erreichen des vorgeschriebenen Drucks die Verbindung zur Vakuumpumpe unterbrochen. Die Trocknungszeit wird von dem Zeitpunkt an gerechnet, an dem die Temperatur im Trockenschrank erneut 80 bis 85 oC erreicht hat. Nach Ablauf der Trocknungszeit wird der Vakuumtrockenschrank vorsichtig wieder auf atmosphärischen Druck gebracht. Nach Öffnen des Vakuumtrockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, zum Abkühlen 30 bis 45 min lang in den Exsikkator (3.6) gestellt und anschließend auf 1 mg genau gewogen. Es wird im Vakuumtrockenschrank weitere 30 min bei 80 bis 85 oC getrocknet und erneut gewogen. Der Unterschied zwischen den beiden Wägeergebnissen darf höchstens 0,1 % Feuchtigkeit betragen. |
4.3. Vortrocknung
4.3.1.
Bei festen Futtermitteln mit einem hohen Feuchtigkeitsgehalt, deren Zerkleinerung schwierig ist, ist eine Vortrocknung wie folgt vorzunehmen:
Von der nicht zerkleinerten Probe (sofern erforderlich können gepresste oder klumpenhaltige Futtermittel grob zerkleinert werden) werden 50 g auf 10 mg genau in ein geeignetes Gefäß (z. B. eine Aluminiumschale von 20 × 12 cm mit einem Rand von 0,5 cm) eingewogen und in einem Trockenschrank bei einer Temperatur von 60 bis 70 oC getrocknet, bis der Feuchtigkeitsgehalt einen Wert zwischen 8 und 12 % erreicht hat. Anschließend wird das Gefäß aus dem Trockenschrank genommen, im Labor 1 h lang offen abkühlen gelassen und dann auf 10 mg genau gewogen. Im Folgenden wird die Probe unverzüglich, wie unter 4.1.1 angegeben, zerkleinert und je nach Art des Futtermittels entsprechend den Angaben unter 4.2.1 oder 4.2.3 getrocknet.
4.3.2.
Körner, deren Feuchtigkeitsgehalt höher als 17 % ist, müssen wie folgt vorgetrocknet werden:
Von den nicht gemahlenen Körnern werden 50 g auf 10 mg genau in ein geeignetes Gefäß (z. B. in eine Aluminiumschale von 20 × 12 cm mit einem Rand von 0,5 cm) eingewogen und in einem Trockenschrank bei einer Temperatur von 130 oC 5 bis 7 min getrocknet. Anschließend wird das Gefäß aus dem Trockenschrank genommen, im Labor 2 h lang offen abkühlen gelassen und dann auf 10 mg genau gewogen. Im Folgenden wird die Probe unverzüglich, wie unter 4.1.2 angegeben, gemahlen und entsprechend den Angaben unter 4.2.2 getrocknet.
5. Berechnung der Ergebnisse
Der Feuchtigkeitsgehalt (X) der Probe als Prozentsatz wird nach folgenden Formeln berechnet:
5.1. Trocknung ohne Vortrocknung

wobei:
|
m |
= |
Anfangsgewicht der Probe in g, |
|
m0 |
= |
Gewicht der trockenen Probe in g. |
5.2. Trocknung mit Vortrocknung

wobei:
|
m |
= |
Anfangsgewicht der Probe in g, |
|
m1 |
= |
Gewicht der Probe nach der Vortrocknung in g, |
|
m2 |
= |
Gewicht der Probe nach der Zerkleinerung oder dem Zermahlen in g, |
|
m0 |
= |
Gewicht der trockenen Probe in g. |
5.3. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 0,2 % Feuchtigkeit (absolut) nicht überschreiten.
6. Bemerkung
Wenn eine Zerkleinerung sich als notwendig erweist und dabei mit einer Änderung des Feuchtigkeitsgehalts des Materials gerechnet werden muss, so sind die Analyseergebnisse, die die Bestandteile des Futtermittels betreffen, auf den Feuchtigkeitsgehalt der Originalprobe umzurechnen.
B. BESTIMMUNG DES FEUCHTIGKEITSGEHALTS IN TIERISCHEN UND PFLANZLICHEN FETTEN UND ÖLEN
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Wasser und flüchtigen Stoffen in tierischen und pflanzlichen Fetten und Ölen.
2. Prinzip
Die Probe wird bei 103 oC getrocknet, bis kein Gewichtsverlust mehr eintritt (der Gewichtsverlust zwischen 2 aufeinanderfolgenden Wägungen darf 1 mg nicht überschreiten). Der Gewichtsverlust wird durch Wiegen bestimmt.
3. Geräte
|
3.1. |
Schale mit flachem Boden, aus korrosionsbeständigem Material, Durchmesser: 8 bis 9 cm, Höhe ca. 3 cm. |
|
3.2. |
Thermometer mit verstärkter Kugel und Ausdehnungsraum am oberen Ende, von ca. 80 bis mindestens 110 oC graduiert, Länge ca. 10 cm. |
|
3.3. |
Sandbad oder elektrische Heizplatte. |
|
3.4. |
Exsikkator, der ein wirksames Trocknungsmittel enthält. |
|
3.5. |
Analysenwaage. |
4. Verfahren
Etwa 20 g der homogenisierten Probe werden auf 1 mg genau in die trockene, gewogene Schale (3.1.) eingewogen, die das Thermometer (3.2) enthält, und auf dem Sandbad oder der elektrischen Heizplatte (3.3) unter ständigem Rühren mit dem Thermometer so erhitzt, dass in ca. 7 min eine Temperatur von 90 oC erreicht wird.
Entsprechend der Häufigkeit, mit der Gasblasen vom Boden der Schale aufsteigen, wird die Heizintensität verringert. Die Temperatur darf 105 oC nicht überschreiten. Unter ständigem Abkratzen des Bodens der Schale wird weiter umgerührt, bis sich keine Blasen mehr bilden.
Um sicherzustellen, dass keine Feuchtigkeit mehr vorhanden ist, wird mehrmals auf 103 oC ± 2 oC erhitzt und zwischen aufeinanderfolgenden Erhitzungen jeweils auf 93 oC gekühlt. Anschließend wird bis zur Raumtemperatur im Exsikkator (3.4) abkühlen gelassen und gewogen. Dieses Verfahren ist so oft zu wiederholen, bis der Gewichtsverlust zwischen 2 aufeinanderfolgenden Wägungen 2 mg nicht mehr überschreitet.
|
Anmerkung |
: |
Eine Gewichtserhöhung der Probe nach wiederholter Erwärmung zeigt eine Oxidation des Fettes an. In diesem Fall wird der Berechnung das Ergebnis der Wägung zugrunde gelegt, die unmittelbar vor dem Auftreten der Gewichtszunahme ausgeführt wurde. |
5. Berechnung der Ergebnisse
Der Feuchtigkeitsgehalt (X) der Probe als Prozentsatz wird nach folgender Formel berechnet:
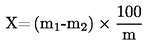
wobei:
|
m |
= |
Probeneinwaage in g, |
|
m1 |
= |
Gewicht der Schale mit Inhalt in g vor dem Erhitzen, |
|
m2 |
= |
Gewicht der Schale mit Inhalt in g nach dem Erhitzen. |
Ergebnisse unter 0,05 % sollten mit der Bezeichnung „weniger als 0,05 %“ angegeben werden.
Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 0,05 % Feuchtigkeit (absolut) nicht überschreiten.
C. BESTIMMUNG DES ROHPROTEINGEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Berechnung des Rohproteingehalts von Futtermitteln anhand des nach Kjeldahl bestimmten Stickstoffgehalts.
2. Prinzip
Die Probe wird durch Schwefelsäure in Anwesenheit eines Katalysators aufgeschlossen. Der saure Aufschluss wird durch eine Natriumhydroxidlösung alkalisiert. Das Ammoniak wird durch Destillation abgetrennt und in einer definierten Menge Schwefelsäure aufgefangen, deren Überschuss durch eine Standard-Natriumhydroxidlösung titriert wird.
Alternativ wird das freigesetzte Ammoniak in überschüssiger Borsäurelösung destilliert, gefolgt von einer Titration mit Salz- oder Schwefelsäurelösung.
3. Reagenzien
|
3.1. |
Kaliumsulfat. |
|
3.2. |
Katalysator: Kupfer(II)-oxid, CuO, oder Kupfer(II)-sulfatpentahydrat, CuSO4 5H2O. |
|
3.3. |
Zinkgranulat. |
|
3.4. |
Schwefelsäure, ρ20 = 1,84 g/ml. |
|
3.5. |
Schwefelsäure, volumetrische Standardlösung, c(H2SO4) = 0,25 mol/l. |
|
3.6. |
Schwefelsäure, volumetrische Standardlösung, c(H2SO4) = 0,10 mol/l. |
|
3.7. |
Schwefelsäure, volumetrische Standardlösung, c(H2SO4) = 0,05 mol/l. |
|
3.8. |
Methylrot-Indikator; 300 mg Methylrot werden in 100 ml Ethanol (σ = 95 bis 96 %) gelöst. |
|
3.9. |
Natriumhydroxidlösung (Verwendung technischer Qualität möglich), β = 40 g/100 ml (40 %). |
|
3.10. |
Natriumhydroxid, volumetrische Standardlösung, c(NaOH) = 0,25 mol/l. |
|
3.11. |
Natriumhydroxid, volumetrische Standardlösung, c(NaOH) = 0,10 mol/l. |
|
3.12. |
Bimssteinkörner, mit Salzsäure gewaschen und geglüht. |
|
3.13. |
Acetanilid (Schmelzpunkt = 114 oC, N-Gehalt = 10,36 %). |
|
3.14. |
Saccharose (stickstofffrei). |
|
3.15. |
Borsäure (H3BO3). |
|
3.16. |
Methylrot-Indikatorlösung: 100 mg Methylrot werden in 100 ml Ethanol oder Methanol gelöst. |
|
3.17. |
Bromkresolgrünlösung: 100 mg Bromkresolgrün werden in 100 ml Ethanol oder Methanol gelöst. |
|
3.18. |
Borsäurelösung (10 bis 40 g/l in Abhängigkeit vom eingesetzten Gerät) Bei einer kolorimetrischen Endpunktbestimmung sind der Borsäurelösung Methylrot- und Bromkresolindikatoren zuzufügen. Wird 1 l Borsäurelösung zubereitet, sind vor dem Auffüllen zur Marke 7 ml Methylrot-Indikatorlösung (3.16) und 10 ml Bromkresolgrünlösung (3.17) zuzufügen. In Abhängigkeit von dem verwendeten Wasser kann sich der pH-Wert von einer Borsäurelösung zur anderen unterscheiden. Häufig ist eine Einstellung des pH-Werts mithilfe einer geringen Menge Alkali erforderlich, um eine positive Blindprobe zu erhalten.
|
|
3.19. |
Salzsäure, volumetrische Standardlösung, c(HCl) = 0,10 mol/l.
|
4. Geräte
Für Aufschluss, Destillation und Titration nach dem Kjeldahl-Verfahren geeignete Geräte.
5. Verfahren
5.1. Aufschluss
Von der Probe wird 1 g auf 0,001 g genau gewogen und in den Kolben des Aufschlussgeräts gegeben. Anschließend werden 15 g Kaliumsulfat (3.1), eine geeignete Katalysatormenge (3.2) (0,3 bis 0,4 g Kupfer(II)-oxid oder 0,9 bis 1,2 g Kupfer(II)-sulfatpentahydrat), 25 ml Schwefelsäure (3.4) und ggf. einige Bimssteinkörnchen (3.12) zugesetzt und vermischt.
Der Kolben wird, wenn notwendig, unter regelmäßigem Schwenken zunächst mäßig erhitzt, bis die Substanz verkohlt ist und das Schäumen aufgehört hat. Dann wird die Flüssigkeit stärker erhitzt und gleichmäßig am Sieden gehalten. Bei korrekter Erhitzung kondensiert die siedende Säure auf der Kolbenwand. Ein Überhitzen der Kolbenwände und Ansetzen organischer Partikel ist zu vermeiden.
Sobald die Lösung klar ist und sich hellgrün färbt, wird sie noch weitere 2 h am Sieden gehalten und danach abkühlen gelassen.
5.2. Destillation
Es wird vorsichtig so viel Wasser zugegeben, dass die Sulfate vollständig gelöst werden. Nach dem Abkühlen werden ggf. einige Körner Zinkgranulat (3.3) zugesetzt. Es wird entweder gemäß 5.2.1 oder 5.2.2 weiterverfahren.
5.2.1.
In den Auffangkolben des Destillationsapparats werden je nach dem zu erwartenden Stickstoffgehalt genau 25 ml Schwefelsäure (3.5 oder 3.7) gebracht und einige Tropfen Methylrot-Indikator (3.8) hinzugefügt.
Der Aufschlusskolben wird mit dem Kühler des Destillationsapparats verbunden und das Ende des Kühlers mindestens 1 cm tief in die Flüssigkeit des Auffangkolbens gesenkt (siehe Bemerkung 8.3). Dann werden 100 ml Natriumhydroxidlösung (3.9) langsam in den Aufschlusskolben eingefüllt, wobei kein Ammoniak entweichen darf (siehe Bemerkung 8.1). Der Kolben wird so lange erhitzt, bis alles Ammoniak überdestilliert ist.
5.2.2.
Wird der Ammoniakgehalt des Destillats von Hand titriert, findet das im Folgenden beschriebene Verfahren Anwendung. Bei einem voll automatisierten Destilliergerät, das auch die Titration des Ammoniakgehalts des Destillats vornimmt, ist die Betriebsanleitung des Herstellers zu befolgen.
Ein Auffangkolben mit 25 bis 30 ml Borsäurelösung (3.18) wird so unter den Ablauf des Kühlers gestellt, dass sich das Ablaufrohr unter der Oberfläche der überschüssigen Borsäurelösung befindet. Das Destilliergerät wird so eingestellt, dass es 50 ml Natriumhydroxidlösung (3.9) abgibt. Die Bedienung des Destilliergeräts erfolgt entsprechend den Anleitungen des Herstellers. Anschließend wird das durch die Zugabe der Natriumhydroxidlösung freigesetzte Ammoniak abdestilliert. Das Destillat wird in der Borsäure (Auffangsäure) aufgefangen. Die Destillatmenge (Dauer der Dampfdestillation) hängt von der in der Probe enthaltenen Menge Stickstoff ab. Hierbei sind die Anleitungen des Herstellers zu befolgen.
|
Anmerkung |
: |
In einem halbautomatischen Destilliergerät erfolgen die Zugabe von überschüssigem Natriumhydroxid und die Dampfdestillation automatisch. |
5.3. Titration
Es wird entweder gemäß 5.3.1 oder 5.3.2 weiterverfahren.
5.3.1.
Die überschüssige Schwefelsäure im Auffangkolben wird mit Natriumhydroxidlösung (3.10 oder 3.11) in Abhängigkeit von der Konzentration der verwendeten Schwefelsäure) titriert, bis der Endpunkt erreicht ist.
5.3.2.
Der Inhalt des Auffangkolbens wird mit der volumetrischen Standardlösung der Salzsäure (3.19) oder mit der volumetrischen Standardlösung der Schwefelsäure (3.6) unter Verwendung einer Bürette titriert und die Menge des eingesetzten Titrationsmittels abgelesen.
Bei der kolorimetrischen Endpunktbestimmung ist der Endpunkt erreicht, wenn sich der Inhalt erstmals pink verfärbt. Die Bürette ist auf 0,05 ml genau abzulesen. Eine beleuchtete Magnetrührplatte oder ein fotometrischer Detektor können bei der Visualisierung des Endpunkts hilfreich sein.
Dies kann automatisch erfolgen bei Verwendung eines Wasserdampfdestillators mit automatischer Titration.
Hinsichtlich des Betriebs des jeweiligen Destillators bzw. des Destillier-/Titriergeräts sind die Anleitungen des Herstellers zu befolgen.
|
Anmerkung |
: |
Bei Verwendung eines automatischen Titrationssystems beginnt die Titration unmittelbar mit dem Start der Destillation, und es wird die 1 %ige Borsäurelösung (3.18) eingesetzt. Wird ein vollautomatisches Destilliergerät eingesetzt, kann die automatische Titration des Ammoniaks auch mittels Endpunktbestimmung unter Verwendung eines potenziometrischen pH-Systems durchgeführt werden. In diesem Fall wird eine automatische Titriervorrichtung mit einem pH-Meter verwendet. Das pH-Meter ist nach dem üblichen Labor-pH-Kalibrierverfahren ordnungsgemäß im Bereich pH 4 bis ph 7 zu kalibrieren. Der pH-Endpunkt der Titration ist bei einem pH-Wert von 4,6 erreicht, dem steilsten Punkt der Titrationskurve (Wendepunkt). |
5.4. Blindversuch
Zur Bestätigung, dass die Reagenzien stickstofffrei sind, wird ein Blindversuch (Aufschluss, Destillation und Titration) mit 1 g Saccharose (3.14) anstelle der Probe durchgeführt.
6. Berechnung der Ergebnisse
Die Berechnungen werden gemäß 6.1 oder 6.2 durchgeführt.
6.1. Berechnung für die Titration nach 5.3.1
Der Rohproteingehalt, ausgedrückt als Massenanteil, wird nach folgender Formel berechnet:
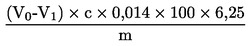
wobei:
|
V0 |
= |
Volumen NaOH (3.10 oder 3.11), das im Blindversuch eingesetzt wurde, in ml, |
|
V1 |
= |
Volumen NaOH (3.10 oder 3.11), das in der Titration der Probe eingesetzt wurde, in ml, |
|
c |
= |
Konzentration des Natriumhydroxids (3.10 oder 3.11), in mol/l, |
|
m |
= |
Probeneinwaage in g. |
6.2. Berechnung für die Titration nach 5.3.2
6.2.1.
Der Rohproteingehalt, ausgedrückt als Massenanteil, wird nach folgender Formel berechnet:

wobei:
|
m |
= |
Probeneinwaage in g, |
|
c |
= |
Konzentration der volumetrischen Standardlösung von Salzsäure (3.19), in mol/l, |
|
V0 |
= |
Volumen der Salzsäure, die im Blindversuch eingesetzt wurde, in ml, |
|
V1 |
= |
Volumen der Salzsäure, die für die Probenmenge eingesetzt wurde, in ml. |
6.2.2.
Der Rohproteingehalt, ausgedrückt als Massenanteil, wird nach folgender Formel berechnet:

wobei:
|
m |
= |
Probeneinwaage in g, |
|
c |
= |
Konzentration der volumetrischen Standardlösung von Schwefelsäure (3.6), in mol/l, |
|
V0 |
= |
Volumen der Schwefelsäure (3.6), die im Blindversuch eingesetzt wurde, in ml, |
|
V1 |
= |
Volumen der Schwefelsäure (3.6), die für die Probenmenge eingesetzt wurde, in ml. |
7. Beurteilung des Verfahrens
7.1. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
|
— |
0,2 % absolut bei Gehalten von weniger als 20 % Rohprotein, |
|
— |
1,0 % relativ zum höheren Wert bei Gehalten von 20 bis 40 % Rohprotein, |
|
— |
0,4 % absolut bei Gehalten von mehr als 40 % Rohprotein. |
7.2. Genauigkeit
Die Analyse (Aufschluss, Destillation und Titration) wird mit 1,5 bis 2,0 g Acetanilid (3.13) in Gegenwart von 1 g Saccharose (3.14) durchgeführt; 1 g Acetanilid verbraucht 14,80 ml Schwefelsäure (3.5). Die Wiederfindung muss mindestens 99 % betragen.
8. Bemerkungen
|
8.1. |
Es können manuelle, halbautomatische oder automatische Geräte verwendet werden. Bei Geräten, die ein Umfüllen zwischen Aufschluss und Destillation erfordern, ist darauf zu achten, dass dies verlustlos geschieht. Verfügt der Destillationsapparat nicht über einen Tropftrichter, so erfolgt die Zugabe der Natriumhydroxidlösung unmittelbar vor dem Anschließen des Kolbens an den Kühler; die Flüssigkeit ist in diesem Fall langsam an den Kolbenwänden entlang laufen zu lassen. |
|
8.2. |
Kommt es während des Aufschlusses zur Verfestigung der Mischung, ist mit der Bestimmung von vorn zu beginnen und eine größere als die oben genannte Menge an Schwefelsäure (3.4) zu verwenden. |
|
8.3. |
Bei stickstoffarmen Proben kann die in den Auffangkolben einzufüllende Menge Schwefelsäure (3.7) gegebenenfalls auf 10 oder 15 ml verringert und mit Wasser auf 25 ml aufgefüllt werden. |
|
8.4. |
Für Routineanalysen können auch andere Methoden zur Bestimmung des Rohproteins herangezogen werden; die in diesem Teil C beschriebene Kjeldahl-Methode ist jedoch die Referenzmethode. Die Gleichwertigkeit der Ergebnisse der alternativen Methode (z. B. DUMAS) im Vergleich zur Referenzmethode muss für jede Matrix einzeln nachgewiesen werden. Da die Ergebnisse einer alternativen Methode auch nach Feststellung ihrer Gleichwertigkeit leicht von den Ergebnissen der Referenzmethode abweichen können, muss im Analysebericht angegeben werden, welche Analyse zur Bestimmung des Rohproteins verwendet wurde. |
D. BESTIMMUNG DES HARNSTOFFGEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Harnstoffgehalts von Futtermitteln.
2. Prinzip
Die Probe wird unter Zusatz eines Klärungsmittels in Wasser suspendiert. Die Suspension wird filtriert. Nach Zugabe von 4-Dimethylaminobenzaldehyd (4-DMAB) wird der Gehalt an Harnstoff im Filtrat durch Messung der Extinktion bei einer Wellenlänge von 420 nm bestimmt.
3. Reagenzien
|
3.1. |
4-Dimethylaminobenzaldehydlösung: 1,6 g 4-DMAB werden in 100 ml 96 %igen Ethanols gelöst und 10 ml Salzsäure (ρ20 = 1,19 g/ml) hinzugefügt. Das Reagenz ist höchstens 2 Wochen haltbar. |
|
3.2. |
Carrez-Lösung I: 21,9 g Zinkazetat, Zn(CH3COO)2·2H2O und 3 g Eisessig werden in Wasser gelöst und auf 100 ml mit Wasser aufgefüllt. |
|
3.3. |
Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), K4Fe(CN)6·3H2O, werden in Wasser gelöst und auf 100 ml mit Wasser aufgefüllt. |
|
3.4. |
Aktivkohle, keinen Harnstoff adsorbierend (zu prüfen). |
|
3.5. |
Harnstofflösung (Massenkonzentration = 0,1 %). |
4. Geräte
|
4.1. |
Mechanisches Schüttelgerät, ca. 35 bis 40 min-1. |
|
4.2. |
Reagenzgläser: 160 × 16 mm, mit Schliffstopfen. |
|
4.3. |
Spektralfotometer. |
5. Verfahren
5.1. Analysengang der Probe
Von der Probe werden 2 g auf 1 mg genau eingewogen und mit 1 g Aktivkohle (3.4) in einen 500-ml-Messkolben gebracht. Hierzu werden 400 ml Wasser und 5 ml Carrez-Lösung I (3.2) gegeben, ca. 30 s gemischt und dann 5 ml Carrez-Lösung II (3.3) zugesetzt. Das Ganze wird 30 min lang im Schüttelgerät rotiert. Dann wird mit Wasser zur Marke aufgefüllt, geschüttelt und filtriert.
Von den klaren und farblosen Filtraten werden je 5 ml in je ein Reagenzglas mit Schliffstopfen pipettiert, 5 ml 4-DMAB-Lösung (3.1) zugesetzt und gemischt. Die Gläser werden in einem Wasserbad bei 20 oC (+/– 4 oC) temperiert. Nach 15 min wird die Extinktion der Probenlösung im Vergleich mit der Blindprobenlösung der Reagenzien im Spektralfotometer bei 420 nm gemessen.
5.2. Kalibrationskurve
Volumen von 1, 2, 4, 5 bzw. 10 ml Harnstofflösung (3.5) werden entnommen, in je 100-ml-Messkolben gebracht und mit Wasser zur Marke aufgefüllt. Von jeder Lösung werden 5 ml mit je 5 ml 4-DMAB-Lösung (3.1) gemischt. Die Extinktion wird, wie oben angegeben, im Vergleich mit einer Lösung, die 5 ml 4-DMAB und 5 ml harnstofffreies Wasser enthält, gemessen. Es wird eine Kalibrationskurve aufgestellt.
6. Berechnung der Ergebnisse
Mithilfe der Kalibrationskurve ist die Menge an Harnstoff in der Versuchsprobe zu bestimmen.
Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
7. Bemerkungen
|
7.1. |
Bei einem Harnstoffgehalt von mehr als 3 % ist die Einwaage auf 1 g zu reduzieren bzw. die Anfangslösung so weit zu verdünnen, dass in 500 ml höchstens 50 mg Harnstoff enthalten sind. |
|
7.2. |
Bei niedrigen Harnstoffgehalten wird die Einwaage erhöht, soweit das Filtrat klar und farblos bleibt. |
|
7.3. |
Enthält die Probe einfache Stickstoffverbindungen, insbesondere Aminosäuren, so ist die Extinktion bei 435 nm zu messen. |
E. BESTIMMUNG DES GEHALTS AN FLÜCHTIGEN STICKSTOFFHALTIGEN BASEN
I. DURCH MIKRODIFFUSION
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an flüchtigen stickstoffhaltigen Basen, ausgedrückt als Ammoniak, in Futtermitteln.
2. Prinzip
Die Probe wird mit Wasser extrahiert, die Lösung geklärt und filtriert. Die flüchtigen stickstoffhaltigen Basen werden nach Zusatz von Kaliumcarbonatlösung durch Mikrodiffusion abgetrennt, in einer Borsäurelösung aufgefangen und mit Schwefelsäure titriert.
3. Reagenzien
|
3.1. |
Trichloressigsäurelösung (Massenkonzentration = 20 %). |
|
3.2. |
Indikator: 33 mg Bromkresolgrün und 65 mg Methylrot werden in 100 ml Ethanol (Volumenkonzentration = 95 bis 96 %) gelöst. |
|
3.3. |
Borsäurelösung: 10 g Borsäure werden in einem 1-l-Messkolben in 200 ml Ethanol (Volumenkonzentration = 95 bis 96 %) und 700 ml Wasser gelöst. Vom Indikator (3.2) werden 10 ml hinzugefügt. Die Lösung wird gemischt und nötigenfalls unter Zusatz von Natriumhydroxidlösung auf eine hellrote Farbe gebracht. Von dieser Lösung kann 1 ml bis zu 300 μg NH3 binden. |
|
3.4. |
Gesättigte Kaliumkarbonatlösung: 100 g Kaliumcarbonat werden in 100 ml siedendem Wasser gelöst. Nach dem Abkühlen wird die Lösung filtriert. |
|
3.5. |
0,01 mol/l Schwefelsäure. |
4. Geräte
|
4.1. |
Mechanisches Schüttelgerät, ca. 35 bis 40 min-1. |
|
4.2. |
Conwayschalen (vgl. Skizze) aus Glas oder Plastik. |
|
4.3. |
Mikrobüretten mit 0,01-ml-Einteilung. |
5. Verfahren
Von der Probe werden 10 g auf 1 mg genau gewogen, mit 100 ml Wasser in einen 200-ml-Messkolben gegeben und 30 min lang im Schüttelgerät gemischt oder verrührt. Dann werden 50 ml Trichloressigsäurelösung (3.1) hinzugefügt, mit Wasser zur Marke aufgefüllt, kräftig geschüttelt und durch einen Faltenfilter filtriert.
In die Mitte der Conwayschale wird 1 ml Borsäurelösung (3.3) und in den Ring der Schale 1 ml des Probenfiltrats pipettiert. Die Schale wird mit dem angefetteten Deckel teilweise bedeckt; dann wird schnell 1 ml der gesättigten Kaliumcarbonatlösung (3.4) in den Ring gegeben und die Schale luftdicht verschlossen. Um mit Sicherheit eine Mischung der beiden Reagenzien zu erreichen, wird die Schale vorsichtig in waagerechter Stellung gedreht. Darauf lässt man das Ganze mindestens 4 h lang bei Raumtemperatur oder 1 h lang bei 40 oC stehen.
Die in der Borsäurelösung enthaltenen flüchtigen Basen werden anschließend mit Schwefelsäure (3.5) unter Verwendung einer Mikrobürette (4.3) titriert.
Nach demselben Verfahren ist ein Blindversuch ohne die zu analysierende Probe durchzuführen.
6. Berechnung der Ergebnisse
1 ml H2SO4 (0,01 mol/l) entspricht 0,34 mg Ammoniak.
Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
|
— |
10 % relativ bei Ammoniakgehalten von weniger als 1,0 %, |
|
— |
0,1 % absolut bei Ammoniakgehalten von 1,0 % oder mehr. |
7. Bemerkung
Enthält die Probe mehr als 0,6 % Ammoniak, ist das ursprüngliche Filtrat zu verdünnen.
CONWAY CELL
Scale 1/1
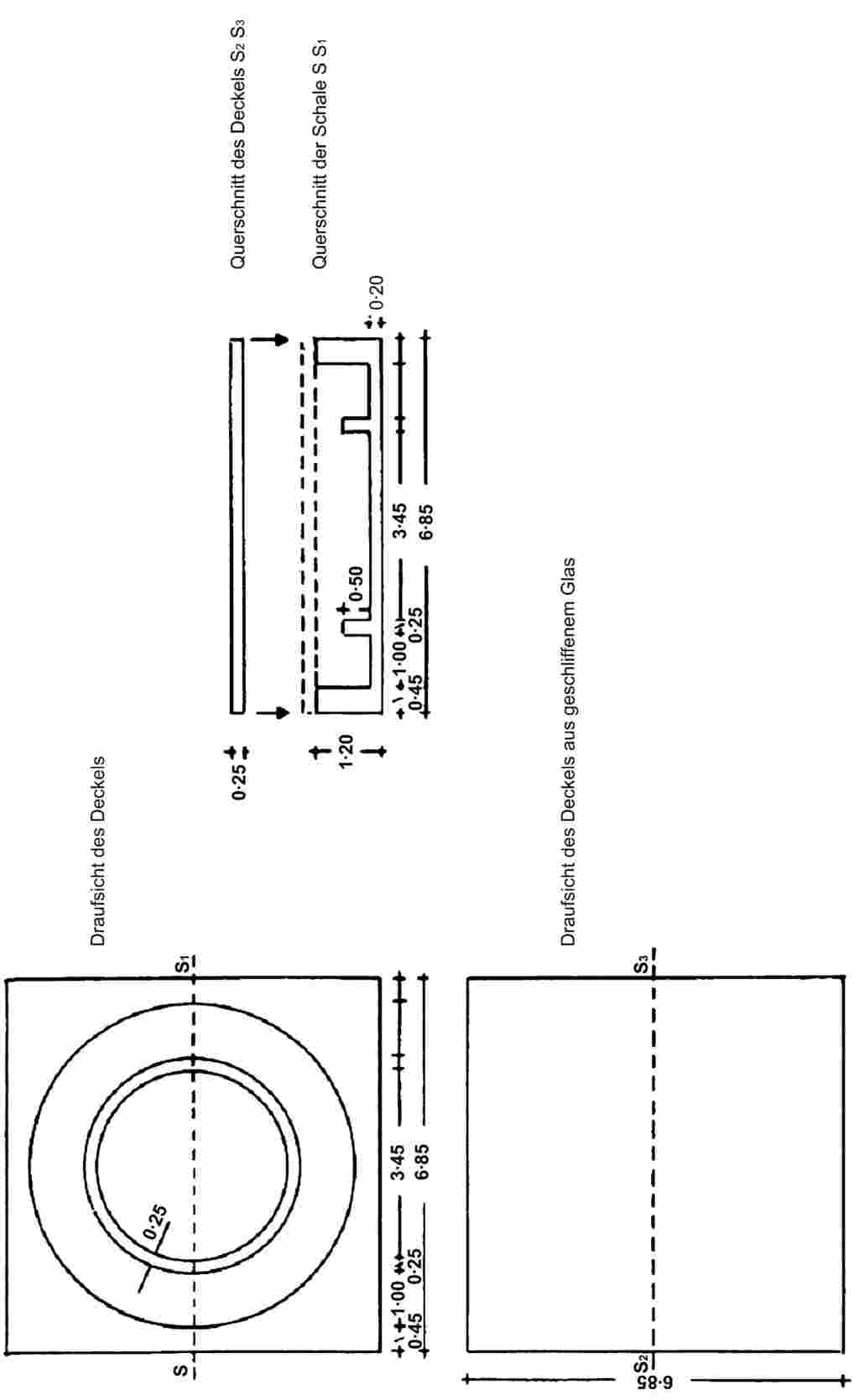
II. DURCH DESTILLATION
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an flüchtigen stickstoffhaltigen Basen, ausgedrückt als Ammoniak, in Fischmehl, das praktisch keinen Harnstoff enthält. Sie ist nur anzuwenden bei Gehalten < 0,25 % Ammoniak.
2. Prinzip
Die Probe wird mit Wasser extrahiert, die Lösung geklärt und filtriert. Die flüchtigen stickstoffhaltigen Basen werden nach Zusatz von Magnesiumoxid in der Siedehitze abgetrennt und in einer definierten Menge Schwefelsäure aufgefangen, deren Überschuss durch Natriumhydroxidlösung zurücktitriert wird.
3. Reagenzien
|
3.1. |
Trichloressigsäurelösung (Massenkonzentration = 20 %). |
|
3.2. |
Magnesiumoxid. |
|
3.3. |
Antischaumemulsion (z. B. Silikon). |
|
3.4. |
0,05 mol/l Schwefelsäure. |
|
3.5. |
0,1 mol/l Natriumhydroxidlösung. |
|
3.6. |
Methylrotlösung 0,3 %, in Ethanol (Volumenkonzentration = 95 bis 96 %). |
4. Geräte
|
4.1. |
Mechanisches Schüttelgerät, ca. 35 bis 40 min-1. |
|
4.2. |
Destillationsapparatur nach Kjeldahl. |
5. Verfahren
Von der Probe werden 10 g auf 1 mg genau gewogen, mit 100 ml Wasser in einen 200-ml-Messkolben gegeben und 30 min lang im Schüttelgerät gemischt oder gerührt. Dann werden 50 ml Trichloressigsäurelösung (3.1) hinzugefügt, es wird mit Wasser zur Marke aufgefüllt, kräftig geschüttelt und durch einen Faltenfilter filtriert.
Je nach dem vermuteten Gehalt an flüchtigen stickstoffhaltigen Basen wird eine entsprechende Menge (im Allgemeinen 100 ml) des klaren Filtrats auf 200 ml verdünnt und 2 g Magnesiumoxid (3.2) sowie einige Tropfen Antischaumemulsion (3.3) hinzugefügt. Die Lösung muss gegen Lackmuspapier alkalisch reagieren; anderenfalls ist noch Magnesiumoxid (3.2) zuzusetzen. Es ist gemäß 5.2 und 5.3 der Analysenmethode zur Bestimmung des Rohproteingehalts (Teil C dieses Anhangs) weiterzuverfahren.
Nach demselben Verfahren ist ein Blindversuch ohne die zu analysierende Probe durchzuführen.
6. Berechnung der Ergebnisse
1 ml H2SO4 (0,05 mol/l) entspricht 1,7 mg Ammoniak.
Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 10 % (relativ) Ammoniak nicht überschreiten.
F. BESTIMMUNG DES GEHALTS AN AMINOSÄUREN (AUSSER TRYPTOPHAN)
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an freien (synthetischen und natürlichen) sowie der gesamten (peptidgebundenen und freien) Aminosäuren in Futtermitteln unter Verwendung eines Aminosäureanalysators. Die Methode ist für folgende Aminosäuren anwendbar: Cyst(e)in, Methionin, Lysin, Threonin, Alanin, Arginin, Asparaginsäure, Glutaminsäure, Glycin, Histidin, Isoleucin, Leucin, Phenylalanin, Prolin, Serin, Tyrosin und Valin.
Die Methode unterscheidet nicht zwischen Salzen von Aminosäuren und dient auch nicht zur Unterscheidung der D- und L-Formen der Aminosäuren. Sie ist nicht zur Bestimmung des Gehalts an Tryptophan oder Hydroxyanaloga der Aminosäuren anwendbar.
2. Prinzip
2.1. Freie Aminosäuren
Die freien Aminosäuren werden mit verdünnter Salzsäure extrahiert. Mitextrahierte stickstoffhaltige Makromoleküle werden mit Sulfosalicylsäure ausgefällt und durch Filtrieren entfernt. Die filtrierte Lösung wird auf einen pH-Wert von 2,20 eingestellt. Die Aminosäuren werden durch Ionenaustauschchromatografie getrennt und nach Reaktion mit Ninhydrin durch fotometrischen Nachweis bei 570 nm bestimmt.
2.2. Gesamtaminosäuren
Die gewählte Methode hängt von den zu untersuchenden Aminosäuren ab. Cyst(e)in und Methionin müssen vor der Hydrolyse zu Cysteinsäure bzw. Methioninsulfon oxidiert werden. Tyrosin muss im Hydrolysat der nicht oxidierten Probe bestimmt werden. Alle übrigen unter 1 genannten Aminosäuren können aus dem Hydrolysat der oxidierten oder der nicht oxidierten Probe bestimmt werden.
Die Oxidation wird bei 0 oC mit einer Perameisensäure-Phenol-Mischung durchgeführt. Überschüssiges Oxidationsreagenz wird mit Natriumdisulfit zerstört. Die oxidierte bzw. die nicht oxidierte Probe wird mit Salzsäure (3.20) 23 h lang hydrolysiert. Das Hydrolysat wird auf einen pH-Wert von 2,20 eingestellt. Die Aminosäuren werden durch Ionenaustauschchromatografie getrennt und nach Reaktion mit Ninhydrin fotometrisch bei 570 nm (bei Prolin 440 nm) bestimmt.
3. Reagenzien
Es ist bidestilliertes Wasser oder Wasser gleichwertiger Qualität zu verwenden (Leitfähigkeit < 10 μS/cm).
|
3.1. |
Wasserstoffperoxid, w (Massenanteil) = 30 %. |
|
3.2. |
Ameisensäure, w (Massenanteil) = 98 bis 100 %. |
|
3.3. |
Phenol. |
|
3.4. |
Natriumdisulfit. |
|
3.5. |
Natriumhydroxid. |
|
3.6. |
5-Sulfosalicylsäure-Dihydrat. |
|
3.7. |
Salzsäure, Dichte ca. 1,18 g/ml. |
|
3.8. |
Tri-Natriumcitrat-Dihydrat. |
|
3.9. |
2,2′-Thiodiethanol (Thiodiglycol). |
|
3.10. |
Natriumchlorid. |
|
3.11. |
Ninhydrin. |
|
3.12. |
Petrolether, Siedeintervall 40 bis 60 oC. |
|
3.13. |
Norleucin oder sonstige für die Verwendung als interner Standard geeignete Verbindung. |
|
3.14. |
Stickstoffgas (< 10 ppm Sauerstoff). |
|
3.15. |
1-Octanol. |
|
3.16. |
Aminosäuren. |
|
3.16.1. |
Standardstoffe gemäß Auflistung unter 1. Es dürfen nur reine Verbindungen ohne Kristallwasser verwendet werden. Sie sind vor der Verwendung im Vakuum über P2O5 oder H2SO4 1 Woche lang zu trocknen. |
|
3.16.2. |
Cysteinsäure. |
|
3.16.3. |
Methioninsulfon. |
|
3.17. |
Natriumhydroxidlösung, c = 7,5 mol/l: 300 g NaOH (3.5) in Wasser lösen und auf 1 l auffüllen. |
|
3.18. |
Natriumhydroxidlösung, c = 1 mol/l: 40 g NaOH (3.5) in Wasser lösen und auf 1 l auffüllen. |
|
3.19. |
Phenolhaltige Ameisensäurelösung: 889 g Ameisensäure (3.2) werden mit 111 g Wasser gemischt; anschließend werden 4,73 g Phenol (3.3) hinzugefügt. |
|
3.20. |
Hydrolysemischung, c = 6 mol HCl/l unter Zusatz von 1 g Phenol/l: Zu 492 ml HCl (3.7) wird 1 g Phenol (3.3) hinzugefügt und mit Wasser auf 1 l aufgefüllt. |
|
3.21. |
Extraktionslösung, c = 0,1 mol HCl/l, 2 % Thiodiglycol enthaltend: 8,2 ml HCl (3.7) werden in ca. 900 ml Wasser gegeben, 20 ml Thiodiglycol (3.9) hinzugefügt, und es wird mit Wasser auf 1 l aufgefüllt (3.7 und 3.9 dürfen nicht unmittelbar gemischt werden). |
|
3.22. |
5-Sulfosalicylsäurelösung, β = 6 %: 60 g 5-Sulfosalicylsäure (3.6) in Wasser lösen und mit Wasser auf 1 l auffüllen. |
|
3.23. |
Oxidationsmischung (Perameisensäure-Phenol): 0,5 ml Wasserstoffperoxid (3.1) werden mit 4,5 ml phenolhaltiger Ameisensäurelösung (3.19) in einem kleinen Becherglas gemischt. Es wird bei 20 bis 30 oC 1 h stehen gelassen, um die Bildung von Perameisensäure zu bewirken. Anschließend wird die Lösung 15 min in ein Eisbad gestellt, bevor sie der Probe zugegeben wird. Vorsicht: Hautkontakt vermeiden und Schutzkleidung tragen. |
|
3.24. |
Citratpufferlösung, c = 0,2 mol Na+/l, pH 2,20: 19,61 g Natriumcitrat (3.8), 5 ml Thiodiglycol (3.9), 1 g Phenol (3.3) und 16,50 ml HCl (3.7) werden in ca. 800 ml Wasser gelöst. Der pH-Wert wird auf 2,20 eingestellt. Mit Wasser auf 1 l auffüllen. |
|
3.25. |
Elutionspuffer entsprechend den Anforderungen des verwendeten Gerätes (4.9). |
|
3.26. |
Ninhydrinreagenz entsprechend den Bedingungen des verwendeten Gerätes (4.9). |
|
3.27. |
Aminosäuren-Standardlösungen: Diese Lösungen sind bei < 5 oC aufzubewahren. |
|
3.27.1. |
Aminosäuren-Standard-Stammlösung (3.16.1): c = 2,5 μmol/ml je Aminosäure in Salzsäure Diese Lösung ist im Handel erhältlich. |
|
3.27.2. |
Standard-Stammlösung von Cysteinsäure und Methioninsulfon, c = 1,25 μmol/ml: 0,2115 g Cysteinsäure (3.16.2) und 0,2265 g Methioninsulfon (3.16.3) werden in Citratpufferlösung (3.24) in einem 1 000-ml-Messkolben gelöst, und es wird mit Citratpufferlösung zur Marke aufgefüllt. Die Lösung ist bei < 5 oC höchstens 12 Monate haltbar. Sie wird nicht benötigt, falls die Standard-Stammlösung (3.27.1) bereits Cysteinsäure und Methioninsulfon enthält. |
|
3.27.3. |
Stammlösung des internen Standards, z. B. Norleucin, c = 20 μmol/ml: 0,6560 g Norleucin (3.13) werden in Citratpufferlösung (3.24) in einem Messkolben gelöst, und es wird mit Citratpufferlösung auf 250 ml aufgefüllt. Diese Lösung ist bei < 5 oC höchstens 6 Monate haltbar. |
|
3.27.4. |
Aminosäurenkalibrierlösung zur Verwendung bei Hydrolysaten, c = 5 nmol/50 μl Cysteinsäure und Methioninsulfon und c = 10 nmol/50 μl der übrigen Aminosäuren: 2,2 g Natriumchlorid (3.10) werden in einem 100-ml-Becherglas in 30 ml Citratpufferlösung (3.24) gelöst. Es werden 4,00 ml Standard-Stammlösung der Aminosäuren (3.27.1), 4,00 ml Standard-Stammlösung von Cysteinsäure und Methinoninsulfon (3.27.2), und, falls verwendet, 0,50 ml Stammlösung des internen Standards (3.27.3) hinzugefügt. Der pH-Wert wird mit Natriumhydroxid (3.18) auf 2,20 eingestellt. Die Lösung wird quantitativ in einen 50-ml-Messkolben überführt, und es wird mit Citratpufferlösung (3.24) zur Marke aufgefüllt und gemischt. Die Lösung ist bei < 5 oC höchstens 3 Monate haltbar. Siehe auch Bemerkungen unter 9.1. |
|
3.27.5. |
Aminosäurenkalibrierlösung zur Verwendung bei Hydrolysaten gemäß 5.3.3.1 und bei Extrakten gemäß 5.2. Die Kalibrierlösung wird gemäß 3.27.4 hergestellt, jedoch ohne Zugabe von Natriumchlorid. Die Lösung ist bei < 5 oC höchstens 3 Monate haltbar. |
4. Geräte
|
4.1. |
100- oder 250-ml-Rundkolben mit Rückflusskühler. |
|
4.2. |
100-ml-Borosilikat-Glasflaschen mit Schraubverschluss mit Gummidichtung/teflonbeschichteter Dichtung (z. B. Duran, Schott). |
|
4.3. |
Trockenschrank mit forcierter Umluft, auf ± 2 oC regelbar. |
|
4.4. |
pH-Meter (auf drei Dezimalstellen genau). |
|
4.5. |
Membranfilter, 0,22 μm Porengröße. |
|
4.6. |
Zentrifuge. |
|
4.7. |
Vakuum-Rotationsverdampfer. |
|
4.8. |
Mechanischer Schüttler oder Magnetrührer. |
|
4.9. |
Aminosäureanalysator oder HPLC-Einrichtung mit Ionenaustauschersäule, Einrichtung für Ninhydrin-Nachsäulenderivatisierung und Fotometer. Die Säule wird mit sulfoniertem Polystyrolharz gefüllt, das die Trennung der einzelnen Aminosäuren und deren Abtrennung von sonstigen Ninhydrin-positiven Substanzen ermöglicht. Die Pumpen für die Ninhydrin- und für die Pufferlösungen müssen über den Zeitraum des Kalibrationslaufes und des Probenlaufes eine Flussstabilität von ±0,5 % gewährleisten. Bei einigen Aminosäureanalysatoren können auch Hydrolyseverfahren angewandt werden, wenn die Hydrolysate Natriumionenkonzentrationen von c = 0,8 mol/l aufweisen und die restliche Ameisensäure des Oxidationsschrittes enthalten. Andere Analysatoren bieten keine befriedigende Abtrennung bestimmter Aminosäuren, falls das Hydrolysat überschüssige Ameisensäure und/oder hohe Natriumionenkonzentrationen aufweist. In diesem Fall wird das Säurevolumen nach der Hydrolyse und vor der pH-Wert-Einstellung durch Einengen auf ca. 5 ml reduziert. Das Einengen ist unter Vakuum bei höchstens 40 oC vorzunehmen. |
5. Verfahren
5.1. Vorbereitung der Probe
Die Probe wird so fein vermahlen, dass sie ein Sieb mit 0,5 mm Maschenweite passieren kann. Proben mit hohem Feuchtigkeitsgehalt müssen vor dem Vermahlen entweder bei einer Temperatur von höchstens 50 oC luftgetrocknet oder aber gefriergetrocknet werden. Proben mit hohem Fettgehalt sind vor dem Vermahlen mit Petrolether (3.12) zu extrahieren.
5.2. Bestimmung des Gehalts an freien Aminosäuren in Futtermitteln und Vormischungen
Eine geeignete Menge (1 bis 5 g) der vorbereiteten Probe (5.1) auf 0,2 mg genau in einen Erlenmeyerkolben einwiegen und 100,0 ml Extraktionslösung (3.21) hinzufügen. Mit einem mechanischen Schüttler 60 min schütteln bzw. mit einem Magnetrührer (4.8) rühren. Nach dem Absetzen lassen werden mit einer Pipette 10,0 ml der überstehenden Lösung in ein 100-ml-Becherglas gegeben.
Es werden 5,0 ml Sulfosalicylsäurelösung (3.22) unter Rühren hinzugefügt, und mithilfe des Magnetrührers wird 5 min weitergerührt. Die überstehende Lösung wird filtriert oder zentrifugiert, um alle Ausfällungen zu entfernen. Von der klaren Lösung werden 10,0 ml in ein 100-ml-Becherglas gegeben, und unter Verwendung von Natriumhydroxidlösung (3.18) wird der pH-Wert auf 2,20 eingestellt. Die Lösung wird mit Citratpufferlösung (3.24) in einen Messkolben geeigneter Größe überführt und mit Citratpufferlösung (3.24) zur Marke aufgefüllt.
Bei Verwendung eines internen Standards werden vor dem Auffüllen 1,00 ml der Stammlösung des internen Standards (3.27.3) für jeweils 100 ml Endlösung hinzugefügt und mit Citratpufferlösung (3.24) zur Marke aufgefüllt.
Anschließend wird die Chromatografie gemäß 5.4 durchgeführt.
Werden die Extrakte nicht am selben Tag chromatografiert, sind sie bei < 5 oC aufzubewahren.
5.3. Bestimmung des Gehalts an Gesamtaminosäuren
5.3.1.
0,1 bis 1 g der vorbereiteten Probe (5.1) werden auf 0,2 mg genau eingewogen
|
— |
in einen 100-ml-Rundkolben (4.1) für die offene Hydrolyse (5.3.2.3) oder |
|
— |
in einen 250-ml-Rundkolben (4.1), falls eine niedrige Natriumionenkonzentration (5.3.3.1) erforderlich ist, oder |
|
— |
in eine 100-ml-Flasche mit Schraubverschluss (4.2) für die geschlossene Hydrolyse (5.3.2.4). |
Die Einwaage muss einen Stickstoffgehalt von etwa 10 mg und einen Feuchtigkeitsgehalt von höchstens 100 mg haben.
Der Kolben/die Flasche wird in ein Eisbad gestellt und auf 0 oC abgekühlt. Dann werden 5 ml der Oxidationsmischung (3.23) hinzugefügt, und es wird mithilfe eines Glasspatels mit gebogener Spitze gemischt. Der Behälter, der den Spatel enthält, wird mit einer luftdichten Folie verschlossen, das Eiswasserbad mit dem verschlossenen Behälter in einen Kühlschrank mit einer Temperatur von 0 oC gestellt und 16 h stehen gelassen. Nach 16 h wird der Behälter aus dem Kühlschrank genommen und das überschüssige Oxidationsreagenz durch Zusatz von 0,84 g Natriumdisulfit (3.4) zersetzt.
Es wird gemäß 5.3.2.1 weiterverfahren.
5.3.2.
5.3.2.1.
Zu der oxidierten Probe (die gemäß 5.3.1 vorbereitet wurde) werden 25 ml Hydrolysemischung (3.20) gegeben. Dabei ist darauf zu achten, dass alle Probenrückstände, die an den Seitenwänden des Gefäßes sowie am Spatel haften, abgewaschen werden.
Je nach dem verwendeten Hydrolyseverfahren wird gemäß 5.3.2.3 oder 5.3.2.4 weiterverfahren.
5.3.2.2.
In einen 100-ml- oder 250-ml-Rundkolben (4.1) bzw. in eine 100-ml-Flasche mit Schraubverschluss (4.2) werden 0,1 bis 1 g der vorbereiteten Probe (5.1) auf 0,2 mg genau eingewogen. Die Einwaage muss einen Stickstoffgehalt von ca. 10 mg aufweisen. Vorsichtig 25 ml Hydrolysemischung (3.20) hinzufügen und mit der Probe vermischen. Es wird entweder gemäß 5.3.2.3 oder gemäß 5.3.2.4 weiterverfahren.
5.3.2.3.
Zu der nach 5.3.2.1 oder 5.3.2.2 hergestellten Mischung werden 3 Glasperlen hinzugegeben. Sie wird zum Sieden erhitzt und unter Rückfluss genau 23 h am Sieden gehalten. Nach Beendigung der Hydrolyse wird der Rückflusskühler mit 5 ml Citratpufferlösung (3.24) gespült und der abgehängte Kolben im Eisbad gekühlt.
Es wird gemäß 5.3.3 weiterverfahren.
5.3.2.4.
Die Flasche mit der nach 5.3.2.1 oder 5.3.2.2 vorbereiteten Mischung wird bei 110 oC in den Trockenschrank (4.3) gestellt. Um einen durch Gasentwicklung hervorgerufenen Druckaufbau zu vermeiden und eine Explosion zu verhindern, wird der Schraubverschluss während der ersten Stunde nur lose auf die Flasche gelegt. Die Flasche darf nicht verschlossen werden. Nach 1 h wird die Flasche mit dem Deckel fest verschlossen und 23 h im Trockenschrank (4.3) belassen. Nach Beendigung der Hydrolyse wird die Flasche aus dem Trockenschrank genommen, der Verschluss vorsichtig geöffnet, die Flasche in ein Eisbad gestellt und abkühlen gelassen.
Je nach dem Verfahren für die pH-Wert-Einstellung (5.3.3) wird der Inhalt der Flasche quantitativ mit Citratpufferlösung (3.24) in ein 250-ml-Becherglas oder in einen 250-ml-Rundkolben überführt.
Es wird gemäß 5.3.3 weiterverfahren.
5.3.3.
Je nach der Natriumionentoleranz des Aminosäureanalysators (4.9) wird die pH-Wert-Einstellung gemäß 5.3.3.1 oder 5.3.3.2 vorgenommen.
5.3.3.1.
Werden Aminosäureanalysatoren verwendet, bei denen eine geringe Natriumionenkonzentration erforderlich ist (wenn also das Säurevolumen reduziert werden muss), wird die Verwendung einer Stammlösung eines internen Standards (3.27.3) empfohlen.
In diesem Fall sind dem Hydrolysat vor dem Verdampfen 2,00 ml der internen Standardlösung (3.27.3) hinzuzufügen.
Zu dem nach 5.3.2.3 oder 5.3.2.4 erhaltenen Hydrolysat werden 2 Tropfen 1-Octanol (3.15) gegeben.
Das Volumen wird am Rotationsverdampfer (4.7) unter Vakuum bei 40 oC auf 5 bis 10 ml reduziert. Wurde das Volumen beim Einengen versehentlich auf weniger als 5 ml reduziert, wird das Hydrolysat verworfen und die Analyse wiederholt.
Der pH-Wert wird mit Natriumhydroxidlösung (3.18) auf 2,20 eingestellt, und es wird gemäß 5.3.4 weiterverfahren.
5.3.3.2.
Die nach 5.3.2.3 oder 5.3.2.4 erhaltenen Hydrolysate werden durch vorsichtige Zugabe unter Rühren von 17 ml Natriumhydroxidlösung (3.17) teilweise neutralisiert, wobei eine Temperatur von unter 40 oC eingehalten werden muss.
Der pH-Wert wird mit Natriumhydroxidlösung (3.17 und anschließend 3.18) bei Raumtemperatur auf 2,20 eingestellt, und es wird gemäß 5.3.4 weiterverfahren.
5.3.4.
Das auf pH 2,20 eingestellte Hydrolysat (5.3.3.1 oder 5.3.3.2) wird mit Citratpufferlösung (3.24) quantitativ in einen 200-ml-Messkolben überführt und mit Citratpufferlösung (3.24) zur Marke aufgefüllt.
Falls bisher noch kein interner Standard hinzugefügt worden ist, werden vor dem Auffüllen 2,00 ml des internen Standards (3.27.3) hinzugegeben und dann mit Citratpufferlösung (3.24) zur Marke aufgefüllt. Es wird sorgfältig gemischt.
Anschließend wird die Chromatografie gemäß 5.4 durchgeführt.
Falls die Probenlösungen nicht am selben Tag chromatografiert werden, sind sie bei < 5 oC aufzubewahren.
5.4. Chromatografie
Falls erforderlich, wird der Extrakt (5.2) oder das Hydrolysat (5.3.4) vor der Chromatografie auf Raumtemperatur gebracht. Die Mischung wird geschüttelt und eine geeignete Menge durch einen 0,22-μm-Membranfilter (4.5) filtriert. Die so erhaltene klare Lösung wird der Ionenaustauschchromatografie unter Verwendung eines Aminosäureanalysators bzw. einer HPLC-Einrichtung (4.9) unterzogen.
Die Einspritzung kann von Hand oder automatisch erfolgen. Wesentlich ist, dass jeweils die gleiche Menge ±0,5 % der Standardlösung und der Probenlösung eingespritzt wird, es sei denn, es wird ein interner Standard verwendet. Das Verhältnis der Konzentrationen von Natriumionen und Aminosäuren sollte in der Standard- und der Probenlösung so ähnlich wie möglich sein.
Die Häufigkeit von Kalibrationsläufen hängt von der Stabilität des Ninhydrinreagenzes und des Analysatorsystems ab. Die Standardlösung oder die Probenlösung wird mit Citratpufferlösung (3.24) so verdünnt, dass die Peakfläche des Standards etwa 30 bis 200 % der Peakfläche der einzelnen Aminosäuren in der Probenlösung ausmacht.
Die Chromatografie der Aminosäuren unterscheidet sich leicht je nach Art des verwendeten Analysators und des eingesetzten Harzes. Das System muss in der Lage sein, die Aminosäuren vollständig voneinander und von den sonstigen Ninhydrin-positiven Substanzen zu trennen. Im Betriebsbereich muss das chromatografische System bei Änderungen der Aminosäuremengen, die der Säule hinzugefügt wird, eine lineare Reaktion aufweisen.
Wird eine equimolare Lösung der zu bestimmenden Aminosäuren analysiert, sollten bei der Chromatografie die unten angegebenen Verhältnisse von Tal zu Peakhöhe zwischen den überlappenden Peaks erreicht werden. Diese equimolare Lösung muss mindestens 30 % der maximalen Menge jeder Aminosäure enthalten, die mit dem jeweiligen Aminosäureanalysator (4.9) noch präzise bestimmt werden kann.
Für die Trennung von Threonin-Serin darf das Verhältnis von Tal zur Peakhöhe des niedrigeren der 2 sich überlappenden Peaks auf dem Chromatogramm nicht mehr als 2:10 betragen (wenn nur Cyst(e)in, Methionin, Threonin und Lysin bestimmt werden, beeinträchtigt eine unzureichende Trennung von angrenzenden Peaks die Bestimmung). Für alle übrigen Aminosäuren sollte die Trennung besser als 1:10 sein.
Das System muss gewährleisten, dass Lysin von „Lysinartefakten“ und Omithin abgetrennt wird.
6. Berechnung der Ergebnisse
Die Flächen der Proben- und der Kalibrationsstandardpeaks werden für jede einzelne Aminosäure bestimmt und der Gehalt (X) in g Aminosäure je kg Probe berechnet:
Bei Verwendung eines internen Standards ist zu multiplizieren mit 
|
A |
= |
Peakfläche, Hydrolysat oder Extrakt |
|
B |
= |
Peakfläche, Kalibrierstandardlösung |
|
C |
= |
Peakfläche, interner Standard im Hydrolysat oder Extrakt |
|
D |
= |
Peakfläche, interner Standard, Kalibrierstandardlösung |
|
M |
= |
Molmasse der zu bestimmenden Aminosäure |
|
c |
= |
Konzentration des Standards in μmol/ml |
|
m |
= |
Probeneinwaage (auf Originalgewicht berichtigt, falls getrocknet oder entfettet), in g |
|
V |
= |
Gesamtvolumen des Hydrolysats (5.3.4) in ml oder errechnetes Gesamtverdünnungsvolumen des Extrakts (6.1) in ml |
Sowohl Cystin als auch Cystein werden im Hydrolysat der oxidierten Probe als Cysteinsäure bestimmt, jedoch als Cystin (C6H12N2O4S2, M = 240,30 g/mol) unter Verwendung der Molmasse von 120,15 g/mol (0,5 × 240,30 g/mol) berechnet.
Methionin wird als Methioninsulfon in Hydrolysaten der oxidierten Probe bestimmt, aber unter Verwendung des M von Methionin (149,21 g/mol) als Methionin berechnet.
Zugesetztes freies Methionin wird nach Extraktion als Methionin bestimmt, wobei für die Berechnung das gleiche M verwendet wird.
|
6.1. |
Das Gesamtverdünnungsvolumen der Extrakte (F) für die Bestimmung des Gehalts an freien Aminosäuren (5.2) wird wie folgt berechnet: 
|
7. Beurteilung des Verfahrens
Das Verfahren ist in einem auf internationaler Ebene im Jahr 1990 durchgeführten Vergleichstest an 4 verschiedenen Futtermitteln (Mischfuttermittel für Schweine, Mischfuttermittel für Mastküken, Eiweißkonzentrat und einer Vormischung) erprobt worden. Die nach der Eliminierung von Ausreißern erhaltenen Ergebnisse (Mittelwerte und Standardabweichungen) sind in den Tabellen unter diesem Punkt dargestellt.
Mittelwerte in g/kg
|
Referenzmaterial |
Aminosäure |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Schweinemischfutter |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|||
|
Mastkükenmischfutter |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|||
|
Eiweißkonzentrat |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|||
|
Vormischung |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|||
|
|||||||
7.1. Wiederholbarkeit
Für die untersuchten Aminosäuren wurde die Wiederholbarkeit ermittelt. Sie ist, ausgedrückt als Standardabweichung der Wiederholbarkeit, für den oben genannten Vergleichstest in der folgenden Tabelle dargestellt.
Standardabweichung der Wiederholbarkeit (sr) in g/kg
|
Referenzmaterial |
Aminosäure |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Schweinemischfutter |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|||
|
Mastkükenmischfutter |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|||
|
Eiweißkonzentrat |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|||
|
Vormischung |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|||
|
|||||||
Variationskoeffizient (%) für die Standardabweichung der Wiederholbarkeit (sr)
|
Referenzmaterial |
Aminosäure |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Schweinemischfutter |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|||
|
Mastkükenmischfutter |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|||
|
Eiweißkonzentrat |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|||
|
Vormischung |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|||
|
|||||||
7.2. Vergleichbarkeit
Die Ergebnisse hinsichtlich der Standardabweichung der Vergleichbarkeit, die sich aus den oben genannten Untersuchungen ergeben, sind in nachstehender Tabelle dargestellt.
Standardabweichung der Vergleichbarkeit (sR) in g/kg
|
Referenzmaterial |
Aminosäure |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Schweinemischfutter |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|||
|
Mastkükenmischfutter |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|||
|
Eiweißkonzentrat |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|||
|
Vormischung |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|||
|
|||||||
Variationskoeffizient (%) für die Standardabweichung der Vergleichbarkeit (sR)
|
Referenzmaterial |
Aminosäure |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Schweinemischfutter |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|||
|
Mastkükenmischfutter |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|||
|
Eiweißkonzentrat |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|||
|
Vormischung |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|||
|
|||||||
8. Verwendung von Referenzmaterialien
Die korrekte Anwendung der Methode ist durch Mehrfachbestimmungen an zertifizierten Referenzmaterialien (soweit verfügbar) zu überprüfen. Für die Kalibration wird die Verwendung einer zertifizierten Aminosäurestandardlösung empfohlen.
9. Bemerkungen
|
9.1. |
Wegen der Unterschiede zwischen den Aminosäureanalysatoren sind die Endkonzentrationen der Aminosäurenkalibrierlösungen (3.27.4 und 3.27.5) und der Hydrolysate (5.3.4) als Richtwerte anzusehen. Der lineare Reaktionsbereich des Geräts muss für alle Aminosäuren geprüft werden. Die Standardlösung wird mit Citratpufferlösung verdünnt, um Peakflächen in der Mitte des Bereichs zu erhalten. |
|
9.2. |
Falls Geräte für die Hochleistungsflüssigkeitschromatografie zur Analyse der Hydrolysate eingesetzt werden, sind die Versuchsbedingungen gemäß den Empfehlungen des Herstellers zu optimieren. |
|
9.3. |
Wird diese Methode für Futtermittel angewandt, die mehr als 1 % Chlorid enthalten (Kraftfuttermittel, Mineralfuttermittel, Ergänzungsfuttermittel), so könnte der Methioningehalt unterschätzt werden, und es ist eine modifizierte Oxidation erforderlich. |
G. BESTIMMUNG DES TRYPTOPHANGEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gesamtgehalts an Tryptophan und des Gehalts an freiem Tryptophan in Futtermitteln. Sie dient nicht zur Unterscheidung zwischen D- und L-Formen.
2. Prinzip
Zur Bestimmung des Gesamtgehalts an Tryptophan wird die Probe unter alkalischen Bedingungen mit gesättigter Bariumhydroxid-Lösung hydrolysiert, auf 110 oC erhitzt und 20 h auf dieser Temperatur gehalten. Nach der Hydrolyse wird ein interner Standard zugegeben.
Zur Bestimmung des Gehalts an freiem Tryptophan wird die Probe unter leicht sauren Bedingungen in Gegenwart eines internen Standards extrahiert.
Tryptophan und der interne Standard im Hydrolysat bzw. im Extrakt werden durch HPLC mit Fluoreszenzdetektor bestimmt.
3. Reagenzien
|
3.1. |
Es ist bidestilliertes Wasser oder Wasser gleichwertiger Qualität zu verwenden (Leitfähigkeit < 10 μS/cm). |
|
3.2. |
Standardsubstanz: Tryptophan (Reinheit/Gehalt ≥ 99 %), im Vakuum über Phosphorpentoxid getrocknet. |
|
3.3. |
Interner Standard: α-Methyl-Tryptophan (Reinheit/Gehalt ≥ 99 %), im Vakuum über Phosphorpentoxid getrocknet. |
|
3.4. |
Bariumhydroxid-Octahydrat (das Ba(OH)2·8H2O darf nicht übermäßig der Luft ausgesetzt werden, um die Bildung von BaCO3 zu vermeiden, das die Bestimmung beeinträchtigen könnte) (siehe Bemerkung 9.3). |
|
3.5. |
Natriumhydroxid. |
|
3.6. |
Orthophosphorsäure, w (Massenanteil) = 85 %. |
|
3.7. |
Salzsäure, ρ20 = 1,19 g/ml. |
|
3.8. |
Methanol, HPLC-Qualität. |
|
3.9. |
Petrolether, Siedeintervall 40 bis 60 oC. |
|
3.10. |
Natriumhydroxidlösung, c = 1 mol/l: 40,0 g NaOH (3.5) in Wasser lösen und mit Wasser (3.1) auf 1 l auffüllen. |
|
3.11. |
Salzsäure, c = 6 mol/l: 492 ml HCl (3.7) mit Wasser auf 1 l auffüllen. |
|
3.12. |
Salzsäure, c = 1 mol/l: 82 ml HCl (3.7) mit Wasser auf 1 l auffüllen. |
|
3.13. |
Salzsäure, c = 0,1 mol/l: 8,2 ml HCl (3.7) mit Wasser auf 1 l auffüllen. |
|
3.14. |
Orthophosphorsäure, c = 0,5 mol/l: 34 ml Orthophosphorsäure (3.6) mit Wasser (3.1) auf 1 l auffüllen. |
|
3.15. |
Konzentrierte Tryptophanlösung (3.2), c = 2,50 μmol/ml: 0,2553 g Tryptophan (3.2) in einem 500-ml-Messkolben in Salzsäure (3.13) lösen und mit Salzsäure (3.13) zur Marke auffüllen. Bei — 18 oC höchstens 4 Wochen aufbewahren. |
|
3.16. |
Konzentrierte interne Standardlösung, c = 2,50 μmol/ml: 0,2728 g α-Methyl-Tryptophan (3.3) in einem 500-ml-Messkolben in Salzsäure (3.13) lösen und mit Salzsäure (3.13) zur Marke auffüllen. Bei — 18 oC höchstens 4 Wochen aufbewahren. |
|
3.17. |
Kalibrierstandardlösung von Tryptophan und internem Standard: 2,00 ml konzentrierte Tryptophanlösung (3.15) und 2,00 ml konzentrierte interne Standardlösung (α-Methyl-Tryptophan) (3.16) mit Wasser (3.1) und Methanol (3.8) auf etwa dasselbe Volumen und etwa dieselbe Methanolkonzentration (10 bis 30 %) wie das fertige Hydrolysat verdünnen. Diese Lösung muss vor Gebrauch frisch hergestellt werden. Während der Zubereitung vor direktem Sonnenlicht schützen. |
|
3.18. |
Essigsäure. |
|
3.19. |
1,1,1-Trichlor-2-methyl-2-propanol. |
|
3.20. |
Ethanolamin, w (Massenanteil) > 98 %. |
|
3.21. |
Lösung von 1 g 1,1,1-Trichlor-2-methyl-2-propanol (3.19) in 100 ml Methanol (3.8). |
|
3.22. |
Mobile Phase für die HPLC: 3,00 g Essigsäure (3.18) + 900 ml Wasser (3.1) +50,0 ml Lösung (3.21) von 1,1,1-Trichlor-2-methyl-2-propanol (3.19) in Methanol (3.8) (1g/100ml). Der pH-Wert wird mit Ethanolamin (3.20) auf 5,00 eingestellt. Mit Wasser (3.1) auf 1 l auffüllen. |
4. Geräte
|
4.1. |
HPLC-Einrichtung mit Fluoreszenzdetektor. |
|
4.2. |
HPLC-Trennsäule, 125 × 4 mm, C18, 3 μm Korngröße, oder vergleichbare Säule. |
|
4.3. |
pH-Meter. |
|
4.4. |
Polypropylen-Kolben, 125 ml, Weithals mit Schraubverschluss. |
|
4.5. |
Membranfilter, 0,45 μm Porengröße. |
|
4.6. |
Autoklav, 110 (± 2) oC, 1,4 (±0,1) bar. |
|
4.7. |
Mechanischer Schüttler oder Magnetrührer. |
|
4.8. |
Vortex-Schüttelgerät. |
5. Verfahren
5.1. Vorbereitung der Proben
Die Probe wird so fein vermahlen, dass sie ein Sieb mit 0,5 mm Maschenweite passieren kann. Proben mit hohem Feuchtigkeitsgehalt müssen vor dem Vermahlen entweder bei einer Temperatur von höchstens 50 oC luftgetrocknet oder aber gefriergetrocknet werden. Proben mit hohem Fettgehalt sind vor dem Vermahlen mit Petrolether (3.9) zu extrahieren.
5.2. Bestimmung des Gehalts an freiem Tryptophan (Extrakt)
Eine geeignete Menge (1 bis 5 g) der vorbereiteten Probe (5.1) auf 1 mg genau in einen Erlenmeyerkolben einwiegen. 100,0 ml Salzsäure (3.13) und 5,00 ml konzentrierte interne Standardlösung (3.16) zugeben. Mit einem mechanischen Schüttler oder einem Magnetrührer (4.7) 60 min schütteln bzw. mischen. Absetzen lassen und 10,0 ml der überstehenden Lösung in ein Becherglas pipettieren. 5 ml Orthophosphorsäure (3.14) zugeben. Der pH-Wert wird mit Natriumhydroxidlösung (3.10) auf 3 eingestellt. Ausreichend Methanol (3.8) für eine Methanolkonzentration von 10 bis 30 % im Endvolumen zugeben. In einen Messkolben mit ausreichendem Volumen überführen und mit Wasser auf das für die Chromatografie notwendige Volumen verdünnen (entspricht etwa dem Volumen der Kalibrierstandardlösung (3.17)).
Einige ml der Lösung werden vor der Einspritzung in die HPLC-Säule durch einen 0,45-μm-Membranfilter (4.5) filtriert. Die Chromatografie gemäß 5.4 durchführen.
Standardlösung und Extrakte sind vor direktem Sonnenlicht zu schützen. Falls es nicht möglich ist, die Extrakte am selben Tag zu analysieren, können sie bei 5 oC maximal 3 Tage aufbewahrt werden.
5.3. Bestimmung des Gehalts an Gesamttryptophan (Hydrolysat)
0,1 bis 1 g der vorbereiteten Probe (5.1) werden auf 0,2 mg genau in den Polypropylenkolben (4.4) eingewogen. Die Einwaage muss einen Stickstoffgehalt von ca. 10 mg aufweisen. 8,4 g Bariumhydroxid-Octahydrat (3.4) und 10 ml Wasser zugeben. Mithilfe eines Vortex-Schüttelgeräts (4.8) oder Magnetrührgeräts (4.7) mischen. Den teflonbeschichteten Magneten in der Mischung lassen. Die Gefäßwände mit 4 ml Wasser abspülen. Schraubkappe aufsetzen und Kolben locker verschließen. In einem Autoklaven (4.6) 30 bis 60 min mit kochendem Wasser erhitzen. Den Autoklaven schließen und 20 h bei 110 (+/– 2) oC autoklavieren.
Vor dem Öffnen des Autoklaven ist die Temperatur auf knapp unter 100 oC zu senken. Um Kristallisation des Ba(OH)2·8HO2 zu vermeiden, sind der warmen Mischung 30 ml Wasser mit Zimmertemperatur zuzugeben. Leicht schütteln oder rühren. 2,00 ml konzentrierte interne Standardlösung (α-Methyl-Tryptophan) (3.16) zugeben. Die Gefäße im Wasser-/Eisbad 15 min kühlen.
Anschließend 5 ml Orthophosphorsäure (3.14) zugeben. Das Gefäß im Kühlbad belassen und unter Rühren mit HCl (3.11) neutralisieren; der pH-Wert wird mit HCl (3.12) auf 3,0 eingestellt. Ausreichend Methanol für eine Methanolkonzentration von 10 bis 30 % im Endvolumen zugeben. In einen Messkolben mit ausreichendem Volumen überführen und mit Wasser auf das für die Chromatografie notwendige Volumen verdünnen (z. B. 100 ml). Die Zugabe von Methanol darf keine Präzipitation verursachen.
Einige ml der Lösung werden vor der Einspritzung in die HPLC-Säule durch einen 0,45-μm-Membranfilter (4.5) filtriert. Die Chromatografie gemäß 5.4 durchführen.
Standardlösung und Extrakte sind vor direktem Sonnenlicht zu schützen. Falls es nicht möglich ist, die Hydrolysate am selben Tag zu analysieren, können sie bei 5 oC höchstens 3 Tage aufbewahrt werden.
5.4. HPLC-Bestimmung
Die folgenden Angaben für eine isokratische Trennung sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen (siehe auch Bemerkungen 9.1 und 9.2):
|
HPLC-Trennsäule (4.2): |
125 × 4 mm, C18, 3 μm Korngröße, oder vergleichbare Säule |
|
Säulentemperatur: |
Raumtemperatur |
|
Mobile Phase (3.22): |
3,00 g Essigsäure (3.18) + 900 ml Wasser (3.1) +50,0 ml Lösung (3.21) von 1,1,1-Trichlor-2-methyl-2-propanol (3.19) in Methanol (3.8) (1 g/100 ml). Den pH-Wert mit Ethanolamin (3.20) auf 5,0 einstellen. Mit Wasser (3.1) auf 1 l auffüllen. |
|
Durchflussrate: |
1 ml/min |
|
Gesamtlaufzeit: |
ca. 34 min |
|
Detektionswellenlänge: |
Anregung: 280 nm, Emission: 356 nm |
|
Einspritzvolumen: |
20 μl |
6. Berechnung der Ergebnisse
Die Menge von Tryptophan (X) in g je 100 g Probe wird wie folgt berechnet:

|
A |
= |
Peakfläche des internen Standards; Kalibrierstandardlösung (3.17) |
|
B |
= |
Peakfläche von Tryptophan, Extrakt (5.2) oder Hydrolysat (5.3) |
|
V1 |
= |
Volumen der konzentrierten Tryptophanlösung (3.15), das der Kalibrierlösung (3.17) zugegeben wurde, in ml (2 ml) |
|
c |
= |
Konzentration der konzentrierten Tryptophanlösung (3.15), die der Kalibrierlösung (3.17) zugegeben wurde, in μmol/ml (= 2,50) |
|
V2 |
= |
Volumen der konzentrierten internen Standardlösung (3.16), das bei der Extraktion (5.2) (= 5,00 ml) oder dem Hydrolysat (5.3) (= 2,00 ml) zugegeben wurde, in ml |
|
C |
= |
Peakfläche des internen Standards, Extrakt (5.2) oder Hydrolysat (5.3) |
|
D |
= |
Peakfläche von Tryptophan, Kalibrierstandardlösung (3.17) |
|
V3 |
= |
Volumen der konzentrierten internen Standardlösung (3.16), das der Kalibrierstandardlösung (3.17) zugegeben wurde, in ml (2,00 ml) |
|
m |
= |
Probeneinwaage (korrigiert auf Originalgewicht, falls getrocknet und/oder entfettet), in g |
|
M |
= |
Molmasse von Tryptophan (= 204,23 g/mol) |
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 10 % des höheren Werts nicht überschreiten.
8. Ergebnisse eines Ringversuchs
Es wurde ein EG-Ringversuch (4. Vergleichstest) durchgeführt, bei dem 3 Proben von bis zu 12 Laboratorien analysiert wurden, um die Hydrolysemethode zu zertifizieren. Jede Probe wurde mehrfach (5-mal) analysiert. Die Ergebnisse sind in nachstehender Tabelle zusammengefasst:
|
|
Probe 1 Schweinefutter |
Probe 2 Schweinefutter mit Zusatz von L-Tryptophan |
Probe 3 Kraftfutter für Schweine |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Mittelwert [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
VKr [ %] |
1,9 |
1,6 |
1,9 |
|
sR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
VKR [ %] |
6,3 |
6,0 |
2,2 |
|
L |
= |
Zahl der Laboratorien, die Ergebnisse übermittelt haben |
|
n |
= |
Zahl der Einzelergebnisse ohne Ausreißer (Ausreißertest von Cochran, Dixon) |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
r |
= |
Wiederholbarkeit |
|
R |
= |
Vergleichbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit, in % |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit, in % |
Es wurde ein weiterer EG-Ringversuch (3. Vergleichstest) durchgeführt, bei dem 2 Proben von bis zu 13 Laboratorien analysiert wurden, um die Methode zur Extraktion von freiem Tryptophan zu zertifizieren. Jede Probe wurde mehrfach (5-mal) analysiert. Die Ergebnisse sind in nachstehender Tabelle zusammengefasst:
|
|
Probe 4 Mischung aus Weizen und Soja |
Probe 5 Mischung aus Weizen und Soja (= Probe 4) mit Tryptophan-Zusatz (0,457 g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Mittelwert [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
VKr [ %] |
1,34 |
1,34 |
|
sR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
VKR [ %] |
4,71 |
5,11 |
|
L |
= |
Zahl der Laboratorien, die Ergebnisse übermittelt haben |
|
n |
= |
Zahl der Einzelergebnisse ohne Ausreißer (Ausreißertest von Cochran, Dixon) |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
r |
= |
Wiederholbarkeit |
|
R |
= |
Vergleichbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
Es wurde ein weiterer EG-Vergleichstest durchgeführt, bei dem 4 Proben von bis zu 7 Laboratorien analysiert wurden, um die Tryptophan-Hydrolyse zu zertifizieren. Die Ergebnisse sind in nachstehender Tabelle zusammengefasst. Jede Probe wurde mehrfach (5-mal) analysiert.
|
|
Probe 1 Schweine mischfuttermittel (CRM 117) |
Probe 2 Fischmehl mit geringem Fettgehalt (CRM 118) |
Probe 3 Sojamehl (CRM 119) |
Probe 4 Magermilch pulver (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Mittelwert [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
VKr [ %] |
1,04 |
1,15 |
1,30 |
0,76 |
|
sR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
VKR [ %] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
Zahl der Laboratorien, die Ergebnisse übermittelt haben |
|
n |
= |
Zahl der Einzelergebnisse ohne Ausreißer (Ausreißertest von Cochran, Dixon) |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
r |
= |
Wiederholbarkeit |
|
R |
= |
Vergleichbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
9. Bemerkungen
|
9.1. |
Mit den folgenden besonderen Chromatografiebedingungen kann eine bessere Trennung von Tryptophan und α-Methyl-Tryptophan erreicht werden. Isokratische Trennung, gefolgt von der Reinigung der Gradientensäule:
|
||||||||||||||||||||||||||||||||||||||||||||
|
9.2. |
Die Durchführung der Chromatografie variiert je nach Art der HPLC und der verwendeten Säulenpackung. Das System muss so gewählt werden, dass eine Grundlinientrennung zwischen Tryptophan und dem internen Standard möglich ist. Die Abbauprodukte müssen deutlich von Tryptophan und dem internen Standard getrennt werden. Zur Untersuchung der Grundlinie unter dem internen Standard auf Verunreinigungen sind Hydrolysate ohne internen Standard zu analysieren. Die Laufzeit muss lang genug sein, dass alle Abbauprodukte eluiert werden, andernfalls können späte Elutionspeaks anschließende Chromatografiedurchgänge beeinträchtigen. Im Betriebsbereich muss das Chromatografiesystem eine lineare Reaktion ergeben. Die lineare Reaktion ist bei einer konstanten (der normalen) Konzentration des internen Standards und unterschiedlichen Tryptophankonzentrationen zu messen. Die Größe des Tryptophan- und des internen Standardpeaks müssen sich innerhalb des linearen Bereichs des HPLC-/Fluoreszenzsystems befinden. Sollten der Tryptophan- und/oder der interne Standardpeak/die internen Standardpeaks zu klein oder zu hoch sein, ist die Analyse mit einer anderen Probengröße und/oder einem anderen Endvolumen zu wiederholen. |
|
9.3. |
Bariumhydroxid
Älteres Bariumhydroxid ist schwerer löslich. Dies kann zur Folge haben, dass die Lösung für die HPLC-Bestimmung nicht klar ist und zu niedrigen Tryptophan-Werten führen kann. |
H. BESTIMMUNG DES GEHALTS AN ROHÖLEN UND -FETTEN
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Rohöl- und Rohfettgehalts von Futtermitteln. Sie erstreckt sich nicht auf die Analyse von Ölsaaten und Ölfrüchten.
Je nach Art und Zusammensetzung des Futtermittels sowie in Abhängigkeit vom Zweck der Untersuchung ist eines der beiden nachstehend beschriebenen Verfahren anzuwenden.
1.1. Verfahren A — Direkt extrahierbare Rohöle und Rohfette
Das Verfahren ist anzuwenden bei Futtermittel-Ausgangserzeugnissen pflanzlicher Herkunft mit Ausnahme derjenigen, die in den Anwendungsbereich von Verfahren B fallen.
1.2. Verfahren B — Gesamtgehalt an Rohölen und Rohfetten
Das Verfahren ist anzuwenden bei Futtermittel-Ausgangserzeugnissen tierischen Ursprungs und bei allen Mischfuttermitteln. Es ist außerdem bei allen Erzeugnissen anzuwenden, aus denen Öle und Fette ohne vorherige Hydrolyse nicht vollständig extrahiert werden können (z. B. Kleber, Hefen, Kartoffeleiweiß und Erzeugnisse, die Verfahren unterzogen wurden wie Extrudieren, Flockieren und Erhitzen).
1.3. Auswertung der Ergebnisse
In allen Fällen, in denen mit Verfahren B ein höheres Ergebnis erzielt wird als mit Verfahren A, gilt das mit Verfahren B erhaltene Ergebnis als der richtige Wert.
2. Prinzip
2.1. Verfahren A
Die Probe wird mit Petrolether extrahiert. Das Lösungsmittel wird abdestilliert und der Rückstand getrocknet und gewogen.
2.2. Verfahren B
Die Probe wird unter Erhitzen mit Salzsäure behandelt. Die Mischung wird abgekühlt und filtriert. Der gewaschene und getrocknete Rückstand wird nach Verfahren A weiterbehandelt.
3. Reagenzien
|
3.1. |
Petrolether, Siedeintervall 40 bis 60 oC. Die Bromzahl muss weniger als 1 und der Abdampfrückstand weniger als 2 mg/100 ml betragen. |
|
3.2. |
Natriumsulfat, wasserfrei. |
|
3.3. |
Salzsäure, c = 3 mol/l. |
|
3.4. |
Filterhilfsmittel, z. B. Kieselgur, Hyflo-Supercel. |
4. Geräte
|
4.1. |
Extraktionsapparat: Sofern das Gerät mit einem Siphon (Soxhletapparat) ausgestattet ist, muss die Rückflussmenge so bemessen sein, dass das Lösungsmittel mindestens 10-mal in der Stunde abläuft. Wenn es sich um ein Gerät ohne Siphon handelt, muss die Rückflussmenge etwa 10 ml/min betragen. |
|
4.2. |
Extraktionshülsen: Diese müssen frei sein von petroletherlöslichen Stoffen und eine Porosität besitzen, die den Anforderungen nach 4.1 Genüge leistet. |
|
4.3. |
Trockenschrank: Vakuumtrockenschrank, eingestellt auf 75 oC (± 3 oC,) oder Lufttrockenschrank, eingestellt auf 100 oC (± 3 oC). |
5. Verfahren
5.1. Verfahren A (siehe Bemerkung 8.1)
Von der Probe werden 5 g auf 1 mg genau abgewogen, dann in eine Extraktionshülse (4.2) überführt und mit einem fettfreien Wattebausch abgedeckt.
Die Hülse wird in einen Extraktionsapparat (4.1) eingesetzt und 6 h lang mit Petrolether (3.1) extrahiert. Der Petroletherextrakt wird in einem trockenen, mit einigen Körnern Bimsstein (2) versehenen, tarierten Kolben aufgefangen.
Nach beendeter Extraktion wird das Lösungsmittel abdestilliert. Anschließend wird der Rückstand mit Kolben 1,5 h im Trockenschrank (4.3) getrocknet und nach dem Abkühlen in einem Exsikkator gewogen. Durch eine zweite Trocknung von 30 min Dauer ist zu prüfen, ob das Gewicht der Öle und Fette konstant geblieben ist (der Gewichtsverlust zwischen 2 aufeinanderfolgenden Wägungen darf 1 mg nicht überschreiten).
5.2. Verfahren B
Von der Probe (siehe Bemerkung 8.2) werden 2,5 g auf 1 mg genau abgewogen und in ein 400-ml-Becherglas oder einen 300-ml-Erlenmeyerkolben gebracht. Anschließend werden 100 ml Salzsäure (3.3) und einige Körner Bimsstein hinzugefügt. Das Becherglas wird mit einem Uhrglas abgedeckt, bzw. dem Erlenmeyerkolben wird ein Rückflusskühler aufgesetzt. Das Gemisch wird auf kleiner Flamme oder auf einer Heizplatte bis zum Siedepunkt erhitzt und 1 h schwach sieden gelassen. Es ist zu verhindern, dass das Material sich an den Wänden des Gefäßes festsetzt.
Dann wird abgekühlt und so viel vom Filterhilfsmittel (3.4) zugesetzt, dass beim Filtrieren kein Öl- bzw. Fettverlust eintritt. Filtriert wird unter Verwendung eines feuchten, fettfreien doppelten Filterpapiers. Der Rückstand wird bis zur neutralen Reaktion mit kaltem Wasser gewaschen. Danach ist zu überprüfen, dass das Filtrat keine Öle und Fette mehr enthält. Bei Vorhandensein von Ölen oder Fetten muss vor der Hydrolyse eine Extraktion der Probe mit Petrolether nach Verfahren A vorgenommen werden.
Das doppelte Filterpapier mit dem Rückstand ist auf ein Uhrglas zu legen und ca. 1,5 h bei 100 oC (± 3 oC) im Lufttrockenschrank (4.3) zu trocknen.
Das doppelte Filterpapier mit dem trockenen Rückstand wird in eine Extraktionshülse (4.2) überführt und mit einem fettfreien Wattebausch abgedeckt. Die Hülse wird in einen Extraktionsapparat (4.1) eingesetzt. Dann wird, wie unter 5.1 Absätze 2 und 3 beschrieben, weiterverfahren.
6. Berechnung der Ergebnisse
Das Gewicht des Rückstands wird als Prozentsatz der Probe ausgedrückt.
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen eines Analytikers darf bei ein und derselben Probe die folgenden Werte nicht überschreiten:
|
— |
0,2 % absolut bei Gehalten an Rohölen und -fetten von unter 5 %, |
|
— |
4,0 % relativ zum höchsten Wert bei Gehalten von 5 bis 10 %, |
|
— |
0,4 % absolut bei Gehalten über 10 %. |
8. Bemerkungen
|
8.1. |
Bei Erzeugnissen mit hohem Öl- und Fettgehalt, die schwer zu zerkleinern sind oder sich zur Entnahme einer reduzierten homogenen Probe nicht eignen, ist folgendermaßen zu verfahren: Von der Probe werden 20 g auf 1 mg genau abgewogen und mit 10 g oder mehr wasserfreiem Natriumsulfat (3.2) gemischt. Dann wird mit Petrolether (3.1), wie unter 5.1 beschrieben, extrahiert. Der dabei aufgefangene Extrakt wird mit Petrolether (3.1) auf 500 ml aufgefüllt und gemischt. Von der Lösung werden 50 ml entnommen und in einen kleinen, trockenen, mit Bimssteinkörnchen versehenen und tarierten Kolben gebracht. Das Lösungsmittel wird abdestilliert und dann wird, wie unter 5.1 letzter Absatz beschrieben, getrocknet und weiterverfahren. Der Extraktionsrückstand in der Hülse wird vom Lösungsmittel befreit, auf 1 mm zerkleinert und wieder zurück in die Extraktionshülse gebracht (ohne Zugabe von Natriumsulfat). Dann wird, wie unter 5.1 Absätze 2 und 3 beschrieben, fortgefahren. Der Gehalt an Ölen und Fetten, ausgedrückt als Prozentsatz der Probe, wird nach folgender Formel berechnet: (10 m1 + m2) × 5 wobei:
|
|
8.2. |
Bei öl- und fettarmen Erzeugnissen kann mit einer Einwaage von 5 g gearbeitet werden. |
|
8.3. |
Heimtierfutter mit hohem Wassergehalt muss vor der Hydrolyse und Extraktion nach Verfahren B möglicherweise mit wasserfreiem Natriumsulfat gemischt werden. |
|
8.4. |
Bei dem Verfahren nach 5.2 kann die Verwendung von heißem statt kaltem Wasser zum Waschen des Rückstands nach der Filtration möglicherweise wirksamer sein. |
|
8.5. |
Die Trocknungszeit von 1,5 h muss bei einigen Futtermitteln möglicherweise verlängert werden. Übermäßiges Trocknen ist zu vermeiden, da dies zu niedrigen Ergebnissen führen kann. Es kann auch ein Mikrowellengerät verwendet werden. |
|
8.6. |
Bei einem Rohöl/-fettgehalt von mehr als 15 % wird vor der Hydrolyse eine Extraktion nach Verfahren A und danach eine erneute Extraktion nach Verfahren B empfohlen. Dies hängt zum Teil von der Art des Futtermittels und der Art des Rohfetts im Futtermittel ab. |
I. BESTIMMUNG DES ROHFASERGEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an säure- und alkaliunlöslichen, fettfreien organischen Bestandteilen in Futtermitteln, herkömmlicherweise als Rohfaser bezeichnet.
2. Prinzip
Die gegebenenfalls entfettete Probe wird nacheinander mit kochender Schwefelsäurelösung und kochender Kaliumhydroxidlösung definierter Konzentrationen behandelt. Der Rückstand wird durch Filtration auf einem gesinterten Glasfilter getrennt, gewaschen, getrocknet, gewogen und bei 475 bis 500 oC verascht. Der Gewichtsverlust bei dieser Veraschung entspricht der Rohfaser der Einwaage.
3. Reagenzien
|
3.1. |
Schwefelsäure, c = 0,13 mol/l. |
|
3.2. |
Antischaummittel (z. B. n-Oktanol). |
|
3.3. |
Filterhilfsmittel (Celite 545 oder gleichwertiges Mittel), 4 h auf 500 oC erhitzt (8.6). |
|
3.4. |
Aceton. |
|
3.5. |
Petrolether, Siedeintervall 40 bis 60 oC. |
|
3.6. |
Salzsäure, c = 0,5 mol/l. |
|
3.7. |
Kaliumhydroxidlösung, c = 0,23 mol/l. |
4. Geräte
|
4.1. |
Heizvorrichtung für den Aufschluss mit Schwefelsäure und Kaliumhydroxidlösung, ausgestattet mit einer Halterung für den Glasfiltertiegel (4.2) und einem Abflussrohr mit Hahn zum Flüssigkeitsablauf, für den Vakuumanschluss und, sofern möglich, auch mit einem Anschluss für die Zufuhr von Druckluft. Vor der täglichen Inbetriebnahme muss die gesamte Apparatur mit Wasser 5 min erhitzt werden. |
|
4.2. |
Glasfiltertiegel mit eingeschmolzenem gesintertem Glasfilter (Porengröße 40 bis 90 μm). Vor dem erstmaligen Gebrauch wird der Tiegel einige Minuten bei 500 oC geglüht und dann abkühlen gelassen (8.6). |
|
4.3. |
Siedezylinder (mindestens 270 ml) mit einem Rückflusskühler. |
|
4.4. |
Trockenschrank mit Thermostat. |
|
4.5. |
Muffelofen mit Thermostat. |
|
4.6. |
Extraktionsvorrichtung mit einer Halterung für den Glasfiltertiegel (4.2) und einem Abflussrohr mit Hahn für den Flüssigkeitsablauf und den Vakuumanschluss. |
|
4.7. |
Verbindungsringe zur Verbindung von Heizvorrichtung (4.1), Filtertiegel (4.2) und Siedezylinder (4.3), Verbindungsringe zur Verbindung von Kaltextraktionsvorrichtung (4.6) und Filtertiegel. |
5. Verfahren
Von der vorbereiteten Probe werden 1 g auf 1 mg genau in den Glasfiltertiegel (4.2) eingewogen (siehe Bemerkungen 8.1, 8.2 und 8.3), und es wird 1 g Filterhilfsmittel (3.3) hinzugefügt.
Der Glasfiltertiegel (4.2) wird in die Heizvorrichtung (4.1) eingesetzt und mit dem Siedezylinder (4.3) verbunden. 150 ml auf Siedetemperatur erhitzte Schwefelsäure (3.1) werden in den mit dem Tiegel verbundenen Siedezylinder überführt, und es werden erforderlichenfalls einige Tropfen Antischaummittel (3.2) hinzugefügt.
Die Flüssigkeit wird innerhalb von 5 ± 2 min zum Sieden erhitzt und genau 30 min im kräftigen Sieden gehalten.
Der Hahn im Abflussrohr (4.1) wird geöffnet und die Schwefelsäure mithilfe von Vakuum durch den Glasfiltertiegel abgesaugt. Der Filterrückstand wird 3-mal mit je 30 ml kochendem Wasser gewaschen. Nach jedem Waschvorgang ist der Rückstand trocken zu saugen.
Der Auslaufhahn wird geschlossen, und es werden 150 ml siedende Kaliumhydroxidlösung (3.7) und einige Tropfen Antischaummittel (3.2) in den mit dem Siedezylinder verbundenen Glasfiltertiegel gegeben. Die Flüssigkeit wird innerhalb von 5 ± 2 min zum Sieden erhitzt und genau 30 min im kräftigen Sieden gehalten. Die Filtration und das Waschen des Rückstands werden in der gleichen Weise durchgeführt wie nach der Behandlung mit Schwefelsäure.
Nach dem letzten Waschen wird der Rückstand trocken gesaugt und der Tiegel mit Inhalt vom Siedezylinder gelöst und an die Kaltextraktionsvorrichtung (4.6) angeschlossen. Unter Anlegen von Vakuum wird der Rückstand 3-mal mit je 25 ml Aceton (3.4) gewaschen, wobei der Rückstand nach jedem Waschvorgang trocken zu saugen ist.
Der Filtertiegel mit Inhalt wird im Trockenschrank bei 130 oC bis zur Massekonstanz getrocknet, in einem Exsikkator abgekühlt und anschließend rasch gewogen. Danach wird der Filtertiegel mit Inhalt in einen Muffelofen gebracht und mindestens 30 min bei einer Temperatur von 475 bis 500 oC bis zur Massekonstanz verascht (der Gewichtsverlust in 2 aufeinanderfolgenden Wägungen darf 2 mg nicht überschreiten).
Nach dem Erhitzen wird der Tiegel zunächst im Muffelofen und dann im Exsikkator abgekühlt und anschließend gewogen.
Es wird ein Blindversuch ohne Probe durchgeführt. Der beim Veraschen auftretende Gewichtsverlust darf 4 mg nicht überschreiten.
6. Berechnung der Ergebnisse
Der Gehalt an Rohfaser als Prozentsatz der Probe wird nach folgender Formel berechnet:

wobei:
|
m |
= |
Einwaage in g, |
|
m0 |
= |
Gewichtsverlust nach dem Veraschen während der Bestimmung, in g, |
|
m1 |
= |
Gewichtsverlust nach dem Veraschen während des Blindversuchs, in g. |
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
|
— |
0,6 % absolut bei einem Rohfasergehalt von weniger als 10 %, |
|
— |
6 % relativ zum höheren Wert bei einem Rohfasergehalt von 10 % und mehr. |
8. Bemerkungen
|
8.1. |
Futtermittel, welche mehr als 10 % Rohfett enthalten, müssen vor der Bestimmung mit Petrolether (3.5) entfettet werden. Dazu wird der Glasfiltertiegel (4.2) mit Inhalt an die Kaltextraktionsvorrichtung (4.6) angeschlossen und unter Anlegen von Vakuum 3-mal mit je 30 ml Petrolether gewaschen, wobei sichergestellt wird, dass der Rückstand trocken ist. Der Tiegel mit Inhalt wird an die Heizvorrichtung (4.1) angeschlossen. Danach ist gemäß 5 fortzufahren. |
|
8.2. |
Futtermittel, die Fette enthalten, welche nicht direkt mit Petrolether (3.5) extrahiert werden können, müssen, wie unter 8.1 angegeben, entfettet werden und nach dem Sieden mit Säure ein weiteres Mal entfettet werden. Nach dem Sieden mit Säure und dem anschließenden Waschvorgang wird der Tiegel mit Inhalt an die Kaltextraktionsvorrichtung (4.6) angeschlossen und 3-mal mit je 30 ml Aceton und danach weitere 3-mal mit je 30 ml Petrolether entfettet. Der Filtertiegel (4.2) wird trocken gesaugt und die Analyse, wie unter 5 beschrieben, fortgesetzt, beginnend mit der Behandlung mit Kaliumhydroxidlösung. |
|
8.3. |
Bei Futtermitteln, die mehr als 5 % Carbonate, ausgedrückt als Calciumcarbonat, enthalten, wird der Tiegel (4.2) mit der eingewogenen Probemenge an die Heizvorrichtung (4.1) angeschlossen. Die Probe wird 3-mal mit je 30 ml Salzsäure (3.6) gewaschen. Nach jeder Zugabe wird die Probe vor dem Absaugen etwa 1 min stehen gelassen. Anschließend wird 1-mal mit 30 ml Wasser gewaschen. Danach ist, wie unter 5 angegeben, fortzufahren. |
|
8.4. |
Wenn ein Gerät benutzt wird, bei dem mehrere Glasfiltertiegel an derselben Heizvorrichtung gleichzeitig befestigt sind, dürfen 2 Einzelbestimmungen derselben Probe nicht in derselben Untersuchungsreihe vorgenommen werden. |
|
8.5. |
Wenn nach dem Sieden mit Säure bzw. Lauge Schwierigkeiten bei der Filtration eintreten, wird Druckluft durch das Auslassrohr der Heizvorrichtung zugeführt und anschließend die Filtration fortgesetzt. |
|
8.6. |
Die Veraschungstemperatur darf 500 oC nicht überschreiten, um die Haltbarkeit der Glasfiltertiegel zu verlängern. Beim Erhitzen und Abkühlen sind vor allem übermäßige Temperatursprünge zu vermeiden. |
J. BESTIMMUNG DES ZUCKERGEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an reduzierenden Zuckern und des Gesamtzuckers nach Inversion, ausgedrückt als Glucose oder gegebenenfalls nach Multiplikation mit dem Faktor 0,95 als Saccharose. Sie wird bei Mischfuttermitteln angewendet. Für andere Futtermittel sind besondere Verfahren vorgesehen. Gegebenenfalls muss der Gehalt an Lactose getrennt bestimmt und dieses Ergebnis bei der Berechnung berücksichtigt werden.
2. Prinzip
Die Zucker werden in verdünntem Ethanol gelöst; die Lösung wird mit den Lösungen Carrez I und II geklärt. Nach dem Verdunsten des Ethanols werden vor und nach der Inversion die Bestimmungen nach der Luff-Schoorl-Methode durchgeführt.
3. Reagenzien
|
3.1. |
Ethanollösung (Volumenkonzentration = 40 %), Dichte: 0,948 g/ml bei 20 oC, auf den Umschlagspunkt von Phenolphthalein eingestellt. |
|
3.2. |
Carrez-Lösung I: 21,9 g Zinkacetat, Zn(CH3COO)2·2H2O, und 3 g Eisessig werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt. |
|
3.3. |
Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), K4Fe(CN)6·3H2O, werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt. |
|
3.4. |
Methylorangelösung (Massenkonzentration = 0,1 %). |
|
3.5. |
Salzsäure 4 mol/l. |
|
3.6. |
Salzsäure 0,1 mol/l. |
|
3.7. |
Natriumhydroxidlösung 0,1 mol/l. |
|
3.8. |
Reagenz nach Luff-Schoorl: Die Zitronensäurelösung (3.8.2) ist unter vorsichtigem Rühren in die Natriumcarbonatlösung (3.8.3) zu gießen. Nach Zugabe der Kupfersulfatlösung (3.8.1) ist mit Wasser auf 1 l aufzufüllen. Über Nacht absetzen lassen und dann filtrieren. Die Konzentration des so erhaltenen Reagenz (Cu 0,05 mol/l; Na2CO3 1 mol/l) muss geprüft werden, siehe 5.4 letzter Absatz. Der pH-Wert der Lösung muss bei etwa 9,4 liegen. |
|
3.8.1. |
Kupfersulfatlösung: 25 g Kupfersulfat, CuSO4·5H2O, eisenfrei, werden in 100 ml Wasser gelöst. |
|
3.8.2. |
Zitronensäurelösung: 50 g Zitronensäure, C6H8O7·H2O, werden in 50 ml Wasser gelöst. |
|
3.8.3. |
Natriumcarbonatlösung: 143,8 g Natriumcarbonat, wasserfrei, werden in ca. 300 ml warmem Wasser gelöst. Die Lösung wird abkühlen gelassen. |
|
3.9. |
Natriumthiosulfatlösung 0,1 mol/l. |
|
3.10. |
Stärkelösung: eine Aufschlämmung von 5 g löslicher Stärke in 30 ml Wasser wird zu 1 l siedendem Wasser hinzugefügt und 3 min lang im Sieden gehalten; dann wird abkühlen gelassen und gegebenenfalls 10 mg Quecksilberiodid als Konservierungsmittel hinzugefügt. |
|
3.11. |
Schwefelsäure 3 mol/l. |
|
3.12. |
Kaliumiodidlösung (Massenkonzentration = 30 %). |
|
3.13. |
Bimssteinkörner, mit Salzsäure ausgekocht, mit Wasser gewaschen und getrocknet. |
|
3.14. |
3-Methylbutan-1-ol. |
4. Geräte
Mechanisches Schüttelgerät, ca. 35 bis 40 min-1.
5. Verfahren
5.1. Extraktion der Probe
Von der Probe werden 2,5 g auf 1 mg genau in einen 250-ml-Messkolben eingewogen. Nach Zugabe von 200 ml Ethanol (3.1) wird der Kolben 1 h lang im Schüttelgerät gemischt. Anschließend werden 5 ml Carrez-Lösung I (3.2) hinzugefügt, und es wird ca. 30 s lang gerührt. Dann werden 5 ml Carrez-Lösung II (3.3) hinzugefügt und 1 min lang gerührt; nun wird mit Ethanol (3.1) zur Marke aufgefüllt, homogenisiert und filtriert. Vom Filtrat werden 200 ml abpipettiert und auf ungefähr die Hälfte des Volumens eingeengt, um den größten Teil des Ethanols zur Verdunstung zu bringen. Der Abdampfrückstand wird mit warmem Wasser in einen 200-ml-Messkolben übergespült, abgekühlt, mit Wasser zur Marke aufgefüllt, homogenisiert und, falls erforderlich, filtriert. Diese Lösung wird für die Bestimmung des Gehalts an reduzierenden Zuckern und nach Inversion zur Bestimmung des Gesamtzuckers verwendet.
5.2. Bestimmung des Gehalts an reduzierenden Zuckern
Eine Menge von höchstens 25 ml der Lösung, die weniger als 60 mg reduzierende Zucker, ausgedrückt als Glucose, enthält, wird abpipettiert, falls erforderlich mit destilliertem Wasser auf 25 ml aufgefüllt und der Gehalt an reduzierendem Zucker nach der Luff-Schoorl-Methode bestimmt. Das Ergebnis wird als Prozentsatz Glucose in der Probe ausgedrückt.
5.3. Bestimmung des Gehalts an Gesamtzucker nach Inversion
Von der Lösung werden 50 ml in einen 100-ml-Messkolben pipettiert, einige Tropfen Methylorangelösung (3.4) hinzugefügt und dann vorsichtig unter ständigem Rühren Salzsäure (3.5) bis zum eindeutigen Umschlag nach Rot hinzugegeben. Dann werden 15 ml Salzsäure (3.6) hinzugefügt, und der Kolben wird 30 min lang in ein Bad mit kräftig siedendem Wasser gestellt, dann schnell auf die Temperatur von etwa 20 oC abgekühlt und 15 ml Natriumhydroxidlösung (3.7) hinzugegeben. Der Kolben wird mit Wasser auf 100 ml aufgefüllt, geschüttelt und eine Menge von höchstens 25 ml entnommen, die weniger als 60 mg reduzierende Zucker, ausgedrückt als Glucose, enthält. Falls erforderlich, wird mit destilliertem Wasser auf 25 ml aufgefüllt und der Gehalt an reduzierenden Zuckern nach der Luff-Schoorl-Methode bestimmt. Das Ergebnis wird als Prozentsatz Glucose oder gegebenenfalls nach Multiplikation mit dem Faktor 0,95 als Saccharose ausgedrückt.
5.4. Titration nach Luff-Schoorl
In einen 300-ml-Erlenmeyerkolben werden 25 ml Reagenz nach Luff-Schoorl (3.8) pipettiert und anschließend genau 25 ml der geklärten Zuckerlösung hinzugefügt. Nach Zugabe von 2 Körnchen Bimsstein (3.13) wird unter Schütteln mit der Hand über freier Flamme von mittlerer Höhe erhitzt, so dass die Flüssigkeit in etwa 2 min zum Sieden kommt. Anschließend wird der Erlenmeyerkolben sofort auf ein Drahtnetz mit einer Asbestscheibe, in die ein Loch von etwa 6 cm Durchmesser gestanzt wurde, gestellt; vorher wird unter dem Drahtnetz eine Flamme entzündet und so eingestellt, dass der Kolben nur am Boden erhitzt wird. Dann wird der Kolben mit einem Rückflusskühler verbunden. Von diesem Augenblick an wird der Kolben genau 10 min sieden gelassen und anschließend sofort in kaltem Wasser abgekühlt. Nach etwa 5 min wird wie folgt titriert:
Der Flüssigkeit werden 10 ml Kaliumiodidlösung (3.12) und unmittelbar danach, aber vorsichtig (wegen der Gefahr des übermäßigen Schäumens), 25 ml Schwefelsäure (3.11) zugegeben. Dann wird mit Natriumthiosulfatlösung (3.9) zunächst bis zum Auftreten einer mattgelben Farbe titriert und nach Zugabe von Stärkelösung (3.10) als Indikator die Titration zu Ende geführt.
Die gleiche Titration wird in einer Mischung aus genau 25 ml Reagenz nach Luff-Schoorl (3.8) und 25 ml Wasser nach Zugabe von 10 ml Kaliumiodidlösung (3.12) und 25 ml Schwefelsäure (3.11) durchgeführt, jedoch ohne vorheriges Erhitzen.
6. Berechnung der Ergebnisse
Aus der unten angeführten Tabelle wird die Menge Glucose in mg abgelesen, die der Differenz der Ergebnisse beider Titrationen, ausgedrückt in ml Natriumthiosulfatlösung (0,1 mol/l), entspricht. Das Ergebnis wird als Prozentsatz der Probe angegeben.
7. Sonderverfahren
|
7.1. |
Bei sehr stark melassehaltigen Futtermitteln und anderen wenig homogenen Futtermitteln werden 20 g in einen 1-l-Messkolben eingewogen, 500 ml Wasser hinzugefügt und 1 h lang im Schüttelgerät gemischt. Danach wird — jedoch mit jeweils der 4-fachen Menge der Carrez-Lösungen I (3.2) und II (3.3) — wie unter 5.1 beschrieben geklärt. Anschließend wird mit Ethanol (Volumenkonzentration = 80 %) zur Marke aufgefüllt. Die Lösung wird homogenisiert und filtriert. Das Ethanol wird, wie unter 5.1 beschrieben, entfernt. Bei Abwesenheit verkleisterter Stärke wird mit Wasser zur Marke aufgefüllt. |
|
7.2. |
Bei Melasse und Futtermittel-Ausgangserzeugnissen, die reich an Zucker, aber praktisch stärkefrei sind (Johannisbrot, Zuckerrübentrockenschnitzel usw.), werden 5 g in einen 250-ml-Messkolben eingewogen und 200 ml destilliertes Wasser hinzugefügt. Dann wird 1 h lang oder, falls erforderlich, länger im Schüttelgerät gemischt. Danach wird mit den Carrez-Lösungen I (3.2) und II (3.3), wie unter 5.1 beschrieben, geklärt und anschließend mit Wasser zur Marke aufgefüllt, homogenisiert und filtriert. Zur Bestimmung des Gesamtzuckergehalts wird, wie unter 5.3 beschrieben, verfahren. |
8. Bemerkungen
|
8.1. |
Es wird empfohlen, vor dem Erhitzen mit dem Luff-Schoorl-Reagenz (unabhängig vom Volumen) etwa 1 ml 3-Methylbutan-1-ol (3.14) hinzuzufügen, um die Schaumbildung zu vermeiden. |
|
8.2. |
Die Differenz zwischen dem Gehalt an Gesamtzucker nach Inversion, ausgedrückt als Glucose, und dem Gehalt an reduzierenden Zuckern, ausgedrückt als Glucose, ergibt nach Multiplikation mit dem Faktor 0,95 das Ergebnis als Prozentsatz Saccharose. |
|
8.3. |
Zur Bestimmung des Gehalts an reduzierenden Zuckern, mit Ausnahme von Milchzucker (Lactose), gibt es 2 Möglichkeiten: |
|
8.3.1. |
Für eine annähernde Berechnung wird der aus einer getrennten Bestimmung ermittelte Lactosegehalt mit 0,675 multipliziert und das Ergebnis von dem Gehalt an reduzierenden Zuckern abgezogen. |
|
8.3.2. |
Für eine genaue Berechnung der reduzierenden Zucker (mit Ausnahme von Lactose) ist es notwendig, dass beiden endgültigen Bestimmungen die gleiche Menge der Einwaage zugrunde liegt. Die eine Bestimmung wird in einem Teil der nach 5.1. hergestellten Lösung durchgeführt, die zweite Bestimmung in einem Teil der Lösung, die bei der Bestimmung des Lactosegehalts mittels der für diesen Zweck vorgesehenen Methode (nach Vergärung der anderen Zuckerarten und Klärung) hergestellt wird. In beiden Fällen wird der anwesende Zucker nach der Luff-Schoorl-Methode bestimmt und in mg Glucose berechnet. Diese beiden Werte werden voneinander subtrahiert und die Differenz wird als Prozentsatz der Probe ausgedrückt. Beispiel: Die beiden abpipettierten Mengen entsprechen bei jeder Bestimmung einer Probeneinwaage von 250 mg. Im ersten Fall werden 17 ml Natriumthiosulfatlösung 0,1 mol/l, entsprechend 44,2 mg Glucose, verbraucht; im zweiten Fall werden 11 ml, entsprechend 27,6 mg Glucose, verbraucht. Die Differenz beträgt also 16,6 mg Glucose. Der Gehalt an reduzierenden Zuckern (mit Ausnahme von Lactose), berechnet als Glucose, ist demnach
|
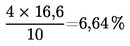
Tabelle für 25 ml Reagenz nach Luff-Schoorl
ml Na2 S2 O30,1 mol/l, 2 min erhitzen, 10 min sieden
|
Na2 S2 O3 0,1 mol/l |
Glucose, Fructose, Invertzucker C6 H12 O6 |
Lactose C12 H22 O11 |
Maltose C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
Differenz |
mg |
Differenz |
Mg |
Differenz |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. BESTIMMUNG DES LACTOSEGEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Lactosegehalts von Futtermitteln, die mehr als 0,5 % Lactose enthalten.
2. Prinzip
Die Zucker werden in Wasser gelöst. Die Lösung wird mit Hefe — Saccharomyces cerevisiae — vergoren, wobei die Lactose nicht angegriffen wird. Nach Klärung und Filtration wird der Gehalt an Lactose im Filtrat nach der Luff-Schoorl-Methode bestimmt.
3. Reagenzien
|
3.1. |
Saccharomyces-cerevisiae-Suspension: 25 g frische Hefe werden in 100 ml Wasser suspendiert. Im Kühlschrank ist die Suspension höchstens 1 Woche haltbar. |
|
3.2. |
Carrez-Lösung I: 21,9 g Zinkacetat, Zn (CH3COO)2·2H2O, und 3 g Eisessig werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt. |
|
3.3. |
Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), K4Fe(CN)6·3H2O, werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt. |
|
3.4. |
Reagenz nach Luff-Schoorl: Die Zitronensäurelösung (3.4.2) wird unter vorsichtigem Rühren in die Natriumcarbonatlösung (3.4.3) gegossen. Nach Zugabe der Kupfersulfatlösung (3.4.1) wird auf 1 l mit Wasser aufgefüllt. Über Nacht absetzen lassen und dann filtrieren. Die Konzentration des so erhaltenen Reagenz (Cu 0,05 mol/l; Na2CO3 1 mol/l) muss geprüft werden. Der pH-Wert muss bei etwa 9,4 liegen. |
|
3.4.1. |
Kupfersulfatlösung: 25 g Kupfersulfat, CuSO4·5H2O, eisenfrei, werden in 100 ml Wasser gelöst, |
|
3.4.2. |
Zitronensäurelösung: 50 g Zitronensäure, C6H8O7·H2O, werden in 50 ml Wasser gelöst, |
|
3.4.3. |
Natriumcarbonatlösung: 143,8 g Natriumcarbonat, wasserfrei, werden in ca. 300 ml warmem Wasser gelöst. Die Lösung wird abkühlen gelassen. |
|
3.5. |
Bimssteinkörner mit Salzsäure ausgekocht, mit Wasser gewaschen und getrocknet. |
|
3.6. |
Kaliumiodidlösung (Massenkonzentration = 30 %). |
|
3.7. |
Schwefelsäure 3 mol/l. |
|
3.8. |
Natriumthiosulfatlösung 0,1 mol/l. |
|
3.9. |
Stärkelösung: Eine Aufschlämmung von 5 g löslicher Stärke in 30 ml Wasser wird zu 1 l siedendem Wasser hinzugefügt und 3 min lang im Sieden gehalten; dann wird abgekühlt und gegebenenfalls 10 mg Quecksilberiodid als Konservierungsmittel hinzugefügt. |
4. Geräte
Wasserbad mit Thermostat, auf 38 bis 40 oC eingestellt.
5. Verfahren
Von der Probe wird 1 g auf 1 mg genau eingewogen und mit 25 bis 30 ml Wasser in einen 100-ml-Messkolben gebracht. Der Messkolben wird 30 min lang in ein Bad mit siedendem Wasser gestellt und auf eine Temperatur von etwa 35 oC abgekühlt. Anschließend werden 5 ml Hefesuspension (3.1) zugefügt. Dann wird der Kolben geschüttelt, 2 h lang in einem Wasserbad bei 38 bis 40 oC stehen gelassen und auf etwa 20 oC abgekühlt.
Danach werden 2,5 ml Carrez-Lösung I (3.2) hinzugefügt, und es wird 30 s lang gerührt. Dann werden 2,5 ml Carrez-Lösung II (3.3) hinzugefügt, und es wird nochmals 30 s lang gerührt. Nun wird mit Wasser auf 100 ml aufgefüllt, gemischt und filtriert. Vom Filtrat wird eine Menge von höchstens 25 ml, die möglichst 40 bis 80 mg Lactose enthält, in einen 300-ml-Erlenmeyerkolben abpipettiert. Gegebenenfalls wird mit Wasser auf 25 ml aufgefüllt.
Auf dieselbe Weise wird ein Blindversuch mit 5 ml Hefesuspension (3.1) ausgeführt. Dann wird der Gehalt an Lactose nach der Luff-Schoorl-Methode wie folgt bestimmt: Nach Zugabe von genau 25 ml des Reagenz nach Luff-Schoorl (3.4) und von 2 Körnchen Bimsstein (3.5) wird unter Schütteln mit der Hand über freier Flamme von mittlerer Höhe erhitzt, so dass die Flüssigkeit in etwa 2 min zum Sieden kommt. Anschließend wird der Erlenmeyerkolben sofort auf ein Drahtnetz mit einer Asbestscheibe, in die ein Loch von etwa 6 cm Durchmesser gestanzt wurde, gestellt; vorher wird unter dem Drahtnetz eine Flamme entzündet und so eingestellt, dass der Kolben nur am Boden erhitzt wird. Dann wird der Kolben mit einem Rückflusskühler verbunden. Von diesem Augenblick an wird der Kolben genau 10 min sieden gelassen und anschließend sofort in kaltem Wasser abgekühlt. Nach etwa 5 min wird wie folgt titriert:
Zu der Flüssigkeit werden 10 ml Kaliumiodidlösung (3.6) und unmittelbar danach, aber vorsichtig (wegen der Gefahr des übermäßigen Schäumens), 25 ml Schwefelsäure (3.7) hinzugegeben. Dann wird mit Natriumthiosulfatlösung (3.8) zunächst bis zum Auftreten einer mattgelben Farbe titriert und nach Zugabe von Stärkelösung (3.9) als Indikator die Titration zu Ende geführt.
Die gleiche Titration wird in einer Mischung aus genau 25 ml Reagenz nach Luff-Schoorl (3.4) und 25 ml Wasser nach Zugabe von 10 ml Kaliumiodidlösung (3.6) und 25 ml Schwefelsäure (3.7) durchgeführt, jedoch ohne vorheriges Erhitzen.
6. Berechnung der Ergebnisse
Aus der unten angeführten Tabelle wird die Menge Lactose in mg abgelesen, die der Differenz der Ergebnisse beider Titrationen, ausgedrückt in ml Natriumthiosulfatlösung (0,1 mol/l), entspricht.
Das Ergebnis entspricht dem Gehalt an wasserfreier Lactose und wird als Prozentsatz der Probe ausgedrückt.
7. Bemerkung
Wenn mehr als 40 % vergärbare Zucker vorhanden sind, werden mehr als 5 ml Hefesuspension (3.1) verwendet.
Tabelle für 25 ml Reagenz nach Luff-Schoorl
ml Na2 S2 O30,1 mol/l, 2 min erhitzen, 10 min sieden
|
Na2 S2 O3 0,1 mol/l |
Glucose, Fructose, Invertzucker C6 H12 O6 |
Lactose C12 H22 O11 |
Maltose C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
Differenz |
mg |
Differenz |
Mg |
Differenz |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. BESTIMMUNG DES STÄRKEGEHALTS
POLARIMETRISCHES VERFAHREN
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Stärke und ihrer Abbauprodukte mit hoher Molmasse in Futtermitteln zum Zweck der Prüfung ihrer Übereinstimmung mit dem angegebenen Energiegehalt (Anhang VII) und mit der Richtlinie 96/25/EG des Rates (3).
2. Prinzip
Die Methode basiert auf einer doppelten Bestimmung. Bei der ersten Bestimmung wird die Probe mit verdünnter Salzsäure behandelt. Nach Klärung und Filtration wird die optische Drehung der Lösung polarimetrisch gemessen.
Bei der zweiten Bestimmung wird die Probe mit 40 %igem Ethanol extrahiert. Nach Behandlung des Filtrats mit Salzsäure wird geklärt, filtriert und die optische Drehung unter den gleichen Bedingungen wie bei der ersten Bestimmung gemessen.
Der Unterschied zwischen den beiden Messungen, multipliziert mit einem bekannten Faktor, ergibt den Stärkegehalt der Probe.
3. Reagenzien
|
3.1. |
Salzsäure, Lösung (Massenanteil = 25 %) Dichte: 1,126 g/ml. |
|
3.2. |
Salzsäure, Lösung (Massenkonzentration = 1,13 %). Die Konzentration muss durch Titration mit Natriumhydroxidlösung (0,1 mol/l) in Gegenwart von Methylrot (Massenkonzentration = 0,1 %) in Ethanol (Volumenkonzentration = 94 %) geprüft werden. Für die Neutralisierung von 10 ml werden 30,94 ml NaOH (0,1 mol/l) benötigt. |
|
3.3. |
Carrez-Lösung I: 21,9 g Zinkacetat, Zn (CH3COO)2·2H2O, und 3 g Eisessig werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt. |
|
3.4. |
Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), K4Fe(CN)6·3H2O, werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt. |
|
3.5. |
Ethanol, Lösung (Volumenkonzentration = 40 %), Dichte: 0,948 g/ml bei 20 oC. |
4. Geräte
|
4.1. |
250-ml-Erlenmeyerkolben mit Schliffstopfen und Rückflusskühler. |
|
4.2. |
Polarimeter oder Saccharimeter. |
5. Verfahren
5.1. Vorbereitung der Probe
Die Probe muss so fein gemahlen werden, dass sie vollständig durch ein Rundlochsieb mit einem Lochdurchmesser von 0,5 mm passiert werden kann.
5.2. Bestimmung der gesamten optischen Drehung (P oder S) (siehe Bemerkung 7.1)
Von der Probe werden 2,5 g auf 1 mg genau in einen 100-ml-Messkolben eingewogen und 25 ml Salzsäure (3.2) hinzugefügt. Der Kolben wird geschüttelt, bis sich der Stoff gleichmäßig verteilt hat; dann werden weitere 25 ml Salzsäure (3.2) hinzugegeben. Der Kolben wird in ein Bad mit kochendem Wasser gestellt und während der ersten 3 min kräftig und regelmäßig geschüttelt, um die Bildung von Klumpen zu verhindern. Die in dem Wasserbad enthaltene Wassermenge muss ausreichen, um das Wasser auch dann noch über dem Siedepunkt zu halten, wenn der Kolben eingetaucht wird. Während des Schüttelns darf der Kolben nicht aus dem Wasser herausgenommen werden. Nach genau 15 min wird der Kolben aus dem Wasserbad entfernt, 30 ml kaltes Wasser hinzugefügt und unverzüglich auf 20 oC abgekühlt.
Danach werden 5 ml Carrez-Lösung I (3.3) hinzugefügt, und es wird ca. 30 s lang geschüttelt. Anschließend werden 5 ml Carrez-Lösung II (3.4) hinzugefügt, und es wird nochmals 30 s geschüttelt. Dann wird mit Wasser zur Marke aufgefüllt, gemischt und filtriert. Ist das Filtrat nicht vollständig klar (was selten vorkommt), muss die Bestimmung mit größeren Mengen Carrez-Lösungen I und II, z. B. 10 ml, wiederholt werden.
Anschließend wird die optische Drehung der Lösung in einem 200-mm-Rohr mit einem Polarimeter oder einem Saccharimeter gemessen.
5.3. Bestimmung der optischen Drehung (P' oder S') der in 40 %igem Ethanol löslichen Substanzen
Von der Probe werden 5 g auf 1 mg genau in einen 100-ml-Messkolben eingewogen und etwa 80 ml Ethanol (3.5) hinzugefügt (siehe Bemerkung 7.2). Anschließend wird der Kolben 1 h bei Raumtemperatur stehen gelassen und währenddessen 6-mal kräftig geschüttelt, damit sich die Testprobe gründlich mit dem Ethanol vermischt. Dann wird mit Ethanol (3.5) zur Marke aufgefüllt, geschüttelt und filtriert.
Von dem Filtrat werden 50 ml (= 2,5 g der Probe) in einen 250-ml-Erlenmeyerkolben abpipettiert und 2,1 ml Salzsäure (3.1) hinzugefügt; der Kolben wird kräftig geschüttelt, an einen Rückflusskühler angeschlossen und in ein Bad mit siedendem Wasser gestellt. Nach genau 15 min wird der Erlenmeyerkolben aus dem Wasserbad herausgenommen und der Inhalt in einen 100-ml-Messkolben mit einer kleinen Menge von kaltem Wasser überspült; anschließend wird auf eine Temperatur von 20 oC abgekühlt.
Danach wird mit den Carrez-Lösungen I (3.3) und II (3.4) geklärt, mit Wasser zur Marke aufgefüllt, geschüttelt und filtriert und die optische Drehung gemessen, wie unter 5.2 zweiter und dritter Absatz beschrieben.
6. Berechnung der Ergebnisse
Der Stärkegehalt (%) wird wie folgt berechnet:
6.1. Polarimetrische Messung
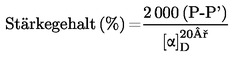
|
P |
= |
Optische Drehung insgesamt in Winkelgrad |
||||||||||||||
|
P' |
= |
Optische Drehung in Winkelgrad der in 40 %igem Ethanol löslichen Substanzen |
||||||||||||||
|
|
= |
Spezifische optische Drehung von reiner Stärke. Für diesen Faktor D werden die nachstehenden, allgemein anerkannten Werte angewendet:
|

6.2. Saccharimetrische Messung

|
S |
= |
Optische Drehung insgesamt in Saccharimeter-Grad |
|
S' |
= |
Optische Drehung in Saccharimeter-Grad der in 40 %igem Ethanol löslichen Substanzen |
|
N |
= |
Gewicht (g) von Saccharose in 100 ml Wasser, das bei Messung mit einem 200-mm-Rohr eine optische Drehung von 100 Saccharimeter-Grad bewirkt: 16,29 g für die französischen Saccharimeter, 26,00 g für die deutschen Saccharimeter, 20,00 g für gemischte Saccharimeter, |
|
|
= |
Spezifische optische Drehung von reiner Stärke (siehe 6.1). |
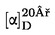
6.3. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 0,4 % (absolut) bei Stärkegehalten von weniger als 40 % und 1 % (relativ) bei Stärkegehalten von 40 % und mehr nicht überschreiten.
7. Bemerkungen
|
7.1. |
Enthält die Probe mehr als 6 % Carbonate, berechnet als Calciumcarbonat, so müssen diese vor der Bestimmung der gesamten optischen Drehung durch Behandlung mit der genau notwendigen Menge verdünnter Schwefelsäure zerstört werden. |
|
7.2. |
Bei Erzeugnissen mit hohem Lactosegehalt, z. B. bei Molkenpulver oder Magermilchpulver, wird nach Zusatz von 80 ml Ethanol (3.5) wie folgt verfahren: Der Messkolben wird an einen Rückflusskühler angeschlossen und 30 min lang in ein Wasserbad von 50 oC gestellt. Nach dem Abkühlen wird das Verfahren, wie unter 5.3 beschrieben, fortgeführt. |
|
7.3. |
Folgende Futtermittel-Ausgangserzeugnisse führen, sofern sie in größeren Mengen in Futtermitteln vorhanden sind, erwiesenermaßen zu Interferenzen bei der Bestimmung des Stärkegehalts durch das polarimetrische Verfahren und könnten so falsche Ergebnisse zur Folge haben:
|
M. BESTIMMUNG DES ROHASCHEGEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Rohaschegehalts von Futtermitteln.
2. Prinzip
Die Probe wird bei 550 oC verascht; der Rückstand wird gewogen.
3. Reagenzien
Ammoniumnitrat, Lösung (Massenkonzentration = 20 %).
4. Geräte
|
4.1. |
Heizplatte. |
|
4.2. |
Elektrischer Muffelofen mit Thermostat. |
|
4.3. |
Veraschungsschale aus Quarzglas, Porzellan oder Platin, entweder rechteckig (ca. 60 × 40 × 25 mm) oder rund (Durchmesser: 60 bis 75 mm, Höhe: 20 bis 40 mm). |
5. Verfahren
Etwa 5 g (2,5 g bei Stoffen, die zur Blasenbildung neigen) der Probe werden auf 1 mg genau in eine vorher bei 550 oC geglühte, abgekühlte und tarierte Veraschungsschale eingewogen. Die Schale wird auf der Heizplatte allmählich bis zum Verkohlen des Stoffes erhitzt. Die Veraschung erfolgt gemäß 5.1 oder 5.2.
|
5.1. |
Die Schale wird in den auf 550 oC eingestellten Muffelofen gebracht und darin bei dieser Temperatur so lange gehalten, bis sich eine weiße, hellgraue oder rötliche Asche bildet, die offensichtlich frei von Kohlepartikeln ist. Dann wird die Schale in einen Exsikkator gestellt und unmittelbar nach dem Abkühlen gewogen. |
|
5.2. |
Die Schale wird 3 h lang in den auf 550 oC eingestellten Muffelofen gebracht. Dann wird die Schale in einen Exsikkator gestellt und unmittelbar nach dem Abkühlen gewogen. Anhand einer zweiten Veraschung von 30 min ist zu prüfen, ob das Gewicht der Asche konstant geblieben ist (der Gewichtsverlust zwischen 2 aufeinanderfolgenden Wägungen darf 1 mg nicht überschreiten). |
6. Berechnung der Ergebnisse
Das Gewicht des Rückstands wird berechnet, indem das Leergewicht abgezogen wird.
Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
7. Bemerkungen
|
7.1. |
Bei schwer zerstörbaren Stoffen werden der abgekühlten Rohasche nach einer ersten Veraschung von mindestens 3 h einige Tropfen 20 %iger Ammoniumnitratlösung oder Wasser zugefügt (jedoch vorsichtig, um ein Zerstäuben oder Verkleben der Asche zu vermeiden). Nach Trocknung im Trockenschrank wird die Veraschung weitergeführt. Der Vorgang wird gegebenenfalls bis zur vollständigen Veraschung wiederholt. |
|
7.2. |
Bei Stoffen, die der unter 7.1 beschriebenen Behandlung widerstehen, ist wie folgt vorzugehen: Nach einer Veraschung von 3 h wird die Asche mit warmem Wasser aufgenommen und durch einen kleinen aschefreien Filter abfiltriert. In der ursprünglichen Schale werden Filter und dessen Inhalt verascht. Das Filtrat wird in die abgekühlte Schale gebracht, bis zur Trockne eingedampft, verascht und gewogen. |
|
7.3. |
Bei Ölen und Fetten wird eine Probe von 25 g in eine Schale entsprechender Abmessung genau eingewogen. Es wird dann durch Anzünden des Stoffes mit einem Streifen aus aschefreiem Filterpapier verkohlt. Nach der Verkohlung wird mit möglichst geringer Menge Wasser angefeuchtet. Anschließend wird getrocknet und die Veraschung, wie unter 5 beschrieben, zu Ende geführt. |
N. BESTIMMUNG DES GEHALTS AN IN SALZSÄURE UNLÖSLICHER ASCHE
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an in Salzsäure unlöslichen, mineralischen Bestandteilen von Futtermitteln. Abhängig von der Art der Probe sind 2 Verfahren vorgesehen.
|
1.1. |
Verfahren A: anzuwenden bei organischen Futtermittel-Ausgangserzeugnissen und bei den meisten Mischfuttermitteln. |
|
1.2. |
Verfahren B: anzuwenden bei Mineralstoffen und Mineralstoffmischungen sowie bei allen Mischfuttermitteln, deren Gehalt an in Salzsäure unlöslicher Asche bei der Bestimmung nach Verfahren A mehr als 1 % beträgt. |
2. Prinzip
|
2.1. |
Verfahren A: Die Probe wird verascht. Die Asche wird in Salzsäure zum Sieden gebracht und der unlösliche Rückstand abfiltriert und gewogen. |
|
2.2. |
Verfahren B: Die Probe wird mit Salzsäure behandelt. Die Lösung wird abfiltriert, der Rückstand wird verascht, und die so erhaltene Asche nach Verfahren A weiterbehandelt. |
3. Reagenzien
|
3.1. |
Salzsäure 3 mol/l. |
|
3.2. |
Trichloressigsäure, Lösung (Massenkonzentration = 20 %). |
|
3.3. |
Trichloressigsäure, Lösung (Massenkonzentration = 1 %). |
4. Geräte
|
4.1. |
Heizplatte. |
|
4.2. |
Elektrischer Muffelofen mit Thermostat. |
|
4.3. |
Veraschungsschalen aus Quarzglas, Porzellan oder Platin, entweder rechteckig (ca. 60 × 40 × 25 mm) oder rund (Durchmesser: 60 bis 75 mm, Höhe: 20 bis 40 mm). |
5. Verfahren
5.1. Verfahren A
Die Einwaage wird unter den Bedingungen, die für die Bestimmung des Rohaschegehalts beschrieben sind, verascht. Es kann auch die bei dieser Analyse erhaltene Asche verwendet werden.
Die Asche wird zusammen mit 75 ml Salzsäure (3.1) in ein Becherglas von 250 bis 400 ml Inhalt überführt. Es wird vorsichtig zum Sieden erhitzt und 15 min weiter in schwachem Sieden gehalten. Die warme Lösung wird durch einen aschefreien Papierfilter filtriert und der Rückstand mit warmem Wasser bis zum Ausbleiben der sauren Reaktion ausgewaschen. Der Filter mit dem Rückstand wird nach dem Trocknen bei einer Temperatur von mindestens 550 oC und höchstens 700 oC in einer tarierten Veraschungsschale verascht und anschließend im Exsikkator gekühlt und gewogen.
5.2. Verfahren B
Von der Analysenprobe werden 5 g auf 1 mg genau in ein Becherglas von 250 bis 400 ml Fassungsvermögen eingewogen. Dann werden nacheinander 25 ml Wasser und 25 ml Salzsäure (3.1) zugegeben, gemischt, und es wird bis zum Nachlassen des Aufbrausens gewartet. Zuletzt werden weitere 50 ml Salzsäure (3.1) zugegeben. Nach dem Nachlassen der eventuell erneut auftretenden Gasentwicklung wird das Becherglas 30 min lang oder, wenn notwendig, länger in ein siedendes Wasserbad gestellt, bis die vollständige Hydrolyse eventuell vorhandener Stärke eingetreten ist. Es wird warm durch einen aschefreien Filter filtriert. Der Filter wird mit etwa 50 ml warmem Wasser ausgewaschen (siehe Bemerkung 7). Anschließend wird der Filter mit Rückstand in eine Veraschungsschale gestellt, getrocknet und bei einer Temperatur von mindestens 550 oC und höchstens 700 oC verascht. Die Asche wird mit 75 ml Salzsäure (3.1) in ein Becherglas von 250 bis 400 ml Fassungsvermögen überführt und anschließend, wie unter 5.1 zweiter Absatz beschrieben, weiterbehandelt.
6. Berechnung der Ergebnisse
Das Gewicht des Rückstands wird berechnet, indem das Leergewicht abgezogen wird. Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
7. Bemerkung
Treten bei der Filtration Schwierigkeiten auf, so wird die Bestimmung wiederholt. Hierbei werden anstelle von 50 ml Salzsäure (3.1) 50 ml 20 %iger Trichloressigsäurelösung (3.2) zugegeben; der Filter wird mit einer warmen 1 %igen Trichloressigsäurelösung (3.3) ausgewaschen.
O. BESTIMMUNG DES GEHALTS AN CARBONATEN
1. Zweck und Anwendungsbereich
Die Methode erlaubt es, in den meisten Futtermitteln den Gehalt an Carbonaten, herkömmlicherweise als Calciumcarbonat bezeichnet, zu bestimmen.
In bestimmten Fällen (z. B. bei Eisencarbonat) ist jedoch eine besondere Methode anzuwenden.
2. Prinzip
Die Carbonate werden mit Salzsäure zersetzt; die frei gewordene Kohlensäure wird in einem graduierten Rohr aufgefangen. Das erhaltene Gasvolumen wird mit demjenigen verglichen, welches unter den gleichen Bedingungen von einer bekannten Menge Calciumcarbonat freigesetzt wird.
3. Reagenzien
|
3.1. |
Salzsäure, Dichte 1,10 g/ml. |
|
3.2. |
Calciumcarbonat. |
|
3.3. |
Schwefelsäure, etwa 0,05 mol/l, mit Methylrot angefärbt. |
4. Geräte
Apparatur nach Scheibler-Dietrich (siehe Skizze) oder gleichwertiges Gerät.
5. Verfahren
Je nach dem Carbonatgehalt der Probe wird eine Einwaage wie folgt eingewogen:
|
— |
0,5 g bei Erzeugnissen mit einem Anteil von 50 bis 100 % Carbonaten, ausgedrückt als Calciumcarbonat, |
|
— |
1 g bei Erzeugnissen mit einem Anteil von 40 bis 50 % Carbonaten, ausgedrückt als Calciumcarbonat, |
|
— |
2 bis 3 g bei anderen Erzeugnissen. |
Die Einwaage wird in die Spezialflasche (4) der Apparatur gebracht, die mit einem Röhrchen aus unzerbrechlichem Material versehen ist, das 10 ml Salzsäure (3.1) enthält. Anschließend wird die Flasche an die Apparatur angeschlossen. Dann wird der Drei-Weg-Hahn (5) so gedreht, dass das Rohr (1) mit der Außenluft in Verbindung steht. Mit dem beweglichen Rohr (2), das mit angefärbter Schwefelsäure (3.3) gefüllt und mit dem graduierten Rohr (1) verbunden ist, wird das Niveau der Flüssigkeit auf den Nullpunkt der Skala eingestellt. Der Hahn (5) wird so gedreht, dass das Rohr (1) mit dem Rohr (3) verbunden ist. Anschließend wird der Nullpunkt kontrolliert.
Die Salzsäure (3.1) wird durch Kippen der Flasche (4) langsam über die Einwaage laufen gelassen. Durch Absenken des beweglichen Rohrs (2) wird der Druck ausgeglichen. Die Flasche (4) wird so lange hin und her bewegt, bis die Kohlensäureentwicklung ganz aufgehört hat.
Der Druckausgleich erfolgt, indem die Flüssigkeit in den Rohren (1) und (2) wieder auf gleiches Niveau gebracht wird. Nach einigen Minuten — sobald das Volumen konstant ist — kann abgelesen werden.
Es ist ein Vergleichsversuch mit 0,5 g Calciumcarbonat (3.2) unter denselben Bedingungen durchzuführen.
6. Berechnung der Ergebnisse
Der Gehalt an Carbonaten, ausgedrückt als Calciumcarbonat, wird nach folgender Formel berechnet:

wobei!
|
X |
= |
Massenanteil (%) an Carbonaten in der Probe, ausgedrückt als Calziumkarbonat, |
|
V |
= |
ml CO2, von der Einwaage freigesetzt, |
|
V1 |
= |
ml CO2, aus 0,5 g CaCO3 freigesetzt, |
|
m |
= |
Probeneinwaage in g. |
7. Bemerkungen
|
7.1. |
Beträgt die Einwaage mehr als 2 g, werden vor Beginn des Versuchs 15 ml destilliertes Wasser in die Flasche (4) zugegeben und gemischt. Beim Vergleichsversuch ist dieselbe Wassermenge zu verwenden. |
|
7.2. |
Bei Verwendung eines Apparats, dessen Volumen von dem der Scheibler-Dietrich-Apparatur abweicht, sind die Einwaage, die Vergleichsmenge und die Berechnung der Ergebnisse entsprechend anzupassen.
|

P. BESTIMMUNG DES GESAMTPHOSPHORGEHALTS
FOTOMETRISCHE METHODE
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Gesamtphosphor in Futtermitteln. Sie ist besonders für die Untersuchung von Futtermitteln mit einem geringen Gehalt an Phosphor geeignet. In bestimmten Fällen (phosphorreiche Erzeugnisse) kann eine gravimetrische Methode angewandt werden.
2. Prinzip
Die Probe wird entweder auf trockenem Wege (bei organischen Futtermitteln) oder auf nassem Wege (bei Mineralstoffen und flüssigen Futtermitteln) aufgeschlossen und in Säure gelöst. Die Lösung wird mit dem Vanadat-Molybdat-Reagenz behandelt. Die Extinktion der gelb gefärbten Lösung wird bei 430 nm im Spektralfotometer gemessen.
3. Reagenzien
|
3.1. |
Calciumcarbonat. |
|
3.2. |
Salzsäure, ρ20 = 1,10 g/ml (ca. 6 mol/l). |
|
3.3. |
Salpetersäure, ρ20 = 1,045 g/ml. |
|
3.4. |
Salpetersäure, ρ20 = 1,38 bis 1,42 g/ml. |
|
3.5. |
Schwefelsäure, ρ20 = 1,84 g/ml. |
|
3.6. |
Vanadat-Molybdat-Reagenz: 200 ml Ammoniumheptamolybdatlösung (3.6.1) werden mit 200 ml Ammoniummonovanadatlösung (3.6.2) und 134 ml Salpetersäure (3.4) in einem l-l-Messkolben gemischt und mit Wasser zur Marke aufgefüllt. |
|
3.6.1. |
Ammoniumheptamolybdatlösung: 100 g Ammoniumheptamolybdat, (NH4) 6Mo7O24 4H2O, werden in heißem Wasser gelöst. 10 ml Ammoniakwasser (D: 0,91 g/ml) werden hinzugefügt, und das Volumen wird mit Wasser auf 1 l aufgefüllt. |
|
3.6.2. |
Ammoniummonovanadatlösung: 2,35 g Ammoniummonovanadat, NH4VO3, werden in 400 ml heißem Wasser gelöst. 20 ml verdünnte Salpetersäure (7 ml HNO3 (3.4) + 13 ml H2O) werden unter ständigem Rühren langsam hinzugefügt, und das Volumen wird mit Wasser auf 1 l aufgefüllt. |
|
3.7. |
Phosphor-Standardlösung 1 mg je ml: 4,387 g Kaliumdihydrogenphosphat, KH2PO4, werden in Wasser gelöst. Das Volumen wird mit Wasser auf 1 l aufgefüllt. |
4. Geräte
|
4.1. |
Veraschungsschalen aus Quarzglas, Platin oder Porzellan. |
|
4.2. |
Elektrischer Muffelofen mit Thermostat auf 550 oC eingestellt. |
|
4.3. |
Kjeldahl-Kolben, 250 ml. |
|
4.4. |
Messkolben und Präzisions-Pipetten. |
|
4.5. |
Spektralfotometer. |
|
4.6. |
Reagenzgläser, ca. 16 mm Durchmesser, mit Stopfen graduiert bis zu 14,5 mm Durchmesser; Fassungsvermögen: 25 bis 30 ml. |
5. Verfahren
5.1. Vorbereitung der Lösung
Je nach der Art der Probe wird die Lösung, wie unter 5.1.1 oder 5.1.2 angegeben, vorbereitet.
5.1.1.
Von der Probe werden 1 g oder mehr auf 1 mg genau in einen Kjeldahl-Kolben eingewogen und mit 20 ml Schwefelsäure (3.5) versetzt; dann wird geschüttelt, um das Material vollständig mit der Säure zu benetzen und um zu verhindern, dass es an den Wandungen des Kolbens anhaftet; anschließend wird der Kolben erhitzt und 10 min im Sieden gehalten. Nach geringem Abkühlen werden 2 ml Salpetersäure (3.4) hinzugefügt; es wird schwach erhitzt und wieder etwas abgekühlt. Dann wird noch ein wenig Salpetersäure (3.4) hinzugegeben und erneut zum Sieden erhitzt. Das Verfahren wird so oft wiederholt, bis die Lösung farblos geworden ist. Hierauf wird abgekühlt, etwas Wasser zugesetzt und die Flüssigkeit in einen 500-ml-Messkolben überführt, wobei der Kjeldahl-Kolben mit heißem Wasser ausgespült wird. Nach Abkühlen wird mit Wasser zur Marke aufgefüllt, homogenisiert und filtriert.
5.1.2.
Von der Probe werden 2,5 g auf 1 mg genau in eine Veraschungsschale eingewogen und mit 1 g Calciumcarbonat (3.1) vollständig vermischt. Im Muffelofen wird bei 550 oC verascht, bis die Asche weiß oder grau ist (kleine Mengen Kohle stören nicht). Die Asche wird in ein 250-ml-Becherglas gebracht, 20 ml Wasser und so viel Salzsäure (3.2) zugefügt, bis das Aufbrausen ausbleibt. Zuletzt werden weitere 10 ml Salzsäure (3.2) zugegeben. Dann wird das Becherglas auf ein Sandbad gestellt und bis zur Trockne eingedampft, um die Kieselsäure unlöslich zu machen. Der Rückstand wird in 10 ml Salpetersäure (3.3) aufgenommen und 5 min lang auf dem Sandbad oder der Heizplatte zum Sieden erhitzt, wobei der Rückstand nicht ganz trocken werden darf. Die Flüssigkeit wird in einen 500-ml-Messkolben übergespült, wobei das Becherglas mehrmals mit heißem Wasser ausgewaschen wird. Nach Abkühlen wird mit Wasser zur Marke aufgefüllt, homogenisiert und filtriert.
5.2. Entwicklung der Färbung und Messung der Extinktion
Ein aliquoter Teil des nach 5.1.1 oder 5.1.2 erhaltenen Filtrats wird verdünnt, um eine Konzentration an Phosphor von höchstens 40 μg/ml zu erhalten. Von dieser Lösung werden 10 ml in ein Reagenzglas (4.6) pipettiert und 10 ml Vanadat-Molybdat-Reagenz (3.6) hinzugefügt. Das Reagenzglas wird geschüttelt und dann mindestens 10 min bei einer Temperatur von 20 oC stehen gelassen. Anschließend wird die Extinktion im Spektralfotometer bei 430 nm gegen eine Lösung von 10 ml Wasser und 10 ml Vanadat-Molybdat-Reagenz (3.6) gemessen.
5.3. Kalibrationskurve
Aus der Standardlösung (3.7) werden Lösungen hergestellt, die 5, 10, 20, 30 bzw. 40 μg Phosphor je ml enthalten. Zu jeweils 10 ml dieser Lösungen werden 10 ml Vanadat-Molybdat-Reagenz (3.6) hinzugefügt. Das Reagenzglas wird geschüttelt und dann mindestens 10 min bei einer Temperatur von 20 oC stehen gelassen. Dann wird die Extinktion entsprechend den unter 5.2 genannten Bedingungen gemessen. Die Kalibrationskurve wird aufgestellt, indem die Extinktionswerte auf der Ordinate und die entsprechende Phosphormenge auf der Abszisse aufgetragen werden. Bei Phosphorkonzentrationen von 0 bis 40 μg/ml ist die Kurve linear.
6. Berechnung der Ergebnisse
Der Phosphorgehalt der Analysenprobe wird mithilfe der Kalibrationskurve bestimmt.
Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
|
— |
3 % relativ zum höheren Wert bei einem Phosphorgehalt von unter 5 %, |
|
— |
0,15 % absolut bei einem Phosphorgehalt von mehr als 5 %. |
Q. BESTIMMUNG DES CHLORGEHALTS AUS CHLORIDEN
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Chlorsgehalts aus wasserlöslichen Chloriden, herkömmlicherweise als Natriumchlorid bezeichnet. Sie ist bei allen Futtermitteln anwendbar.
2. Prinzip
Die Chloride werden in Wasser gelöst. Handelt es sich um Erzeugnisse, die organische Stoffe enthalten, wird eine Klärung vorgenommen. Die Lösung wird mittels Salpetersäure schwach angesäuert, und die Chloride werden mit einer Silbernitratlösung als Silberchlorid gefällt. Der Silbernitratüberschuss wird mit einer Ammoniumthiocyanatlösung nach der Volhard-Methode zurücktitriert.
3. Reagenzien
|
3.1. |
Ammoniumthiocyanatlösung 0,1 mol/l. |
|
3.2. |
Silbernitratlösung 0,1 mol/l. |
|
3.3. |
Gesättigte Ammonium-Eisen(III)-Sulfatlösung (NH4)Fe(SO4)2. |
|
3.4. |
Salpetersäure, Dichte: 1,38 g/ml. |
|
3.5. |
Diethylether. |
|
3.6. |
Aceton. |
|
3.7. |
Carrez-Lösung I: 21,9 g Zinkacetat, Zn (CH3COO)2·2H2O, und 3 g Eisessig werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt. |
|
3.8. |
Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat(II), K4Fe(CN)6·3H2O, werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt. |
|
3.9. |
Aktivkohle, chloridfrei und keine Chloride adsorbierend. |
4. Geräte
Mechanisches Schüttelgerät, ca. 35 bis 40 min-1.
5. Verfahren
5.1. Vorbereitung der Lösung
Je nach der Art der Probe wird, wie unter 5.1.1, 5.1.2 oder 5.1.3 beschrieben, eine Lösung hergestellt.
Gleichzeitig ist ein Blindversuch ohne die zu analysierende Probe durchzuführen.
5.1.1.
Eine Menge von höchstens 10 g der Analysenprobe mit einem Gehalt von höchstens 3 g Chlor in Form von Chloriden wird auf 1 mg genau abgewogen und in einen 500-ml-Messkolben mit 400 ml Wasser von etwa 20 oC gebracht. Dann wird 30 min lang in dem Schüttelgerät rotiert, zur Marke aufgefüllt, homogenisiert und filtriert.
5.1.2.
Etwa 5 g der Analysenprobe werden auf 1 mg genau eingewogen und mit 1 g Aktivkohle in einen 500-ml-Messkolben gebracht. Hierzu werden 400 ml Wasser von etwa 20 oC und 5 ml Carrez-Lösung I (3.7) gegeben, 30 s gerührt und anschließend 5 ml Carrez-Lösung II (3.8) zugesetzt. Dann wird 30 min lang in dem Schüttelgerät rotiert, zur Marke aufgefüllt, homogenisiert und filtriert.
5.1.3.
Die Lösung wird, wie unter 5.1.2 beschrieben, zubereitet, jedoch nicht filtriert. Es wird dekantiert (falls erforderlich, zentrifugiert); dann werden 100 ml der überstehenden Lösung in einen 200-ml-Messkolben pipettiert. Es wird mit Aceton (3.6) gemischt und mit demselben Lösungsmittel zur Marke aufgefüllt, homogenisiert und filtriert.
5.2. Titration
Von dem Filtrat, das nach 5.1.1, 5.1.2 oder 5.1.3 erhalten wurde, werden (je nach dem zu erwartenden Chlorgehalt) 25 bis 100 ml in einen Erlenmeyerkolben pipettiert. Die aliquote Menge darf höchstens 150 mg Chlor (Cl) enthalten. Falls erforderlich, wird mit Wasser auf mindestens 50 ml verdünnt. Dann werden 5 ml Salpetersäure (3.4), 20 ml der gesättigten Ammonium-Eisen(III)-Sulfatlösung (3.3) und 2 Tropfen Ammoniumthiocyanatlösung (3.1) aus einer bis zum Nullpunkt gefüllten Bürette hinzugesetzt. Anschließend wird aus einer Bürette Silbernitratlösung (3.2) zugegeben, bis ein Überschuss von 5 ml vorhanden ist. Dann werden 5 ml Diethylether (3.5) zugegeben, und es wird kräftig geschüttelt, um den Niederschlag zum Ausflocken zu bringen. Der Überschuss an Silbernitrat wird mit Ammoniumthiocyanatlösung (3.1) titriert, bis eine rotbraune Farbe auftritt, die 1 min beständig ist.
6. Berechnung der Ergebnisse
Die Menge Chlor (X), ausgedrückt als % Natriumchlorid, wird nach folgender Formel berechnet:

wobei:
|
V1 |
= |
zugegebene Silbernitratlösung 0,1 mol/l, in ml, |
|
V2 |
= |
Ammoniumthiocyanatlösung 0,1 mol/l, die für die Titration verbraucht werden, in ml, |
|
m |
= |
Probeneinwaage in g. |
Falls der Blindversuch einen Verbrauch an Silbernitratlösung 0,1 mol/l anzeigt, wird dieser Wert von dem Volumen (V1-V2) abgezogen.
7. Bemerkungen
|
7.1. |
Die Titration kann auch mit der potenziometrischen Methode erfolgen. |
|
7.2. |
Bei sehr fettreichen Erzeugnissen ist eine vorherige Entfettung mit Diethylether oder Petrolether durchzuführen. |
|
7.3. |
Bei Fischmehl kann die Titration nach der Mohr-Methode durchgeführt werden. |
(1) Für die Trocknung von Getreide, Mehl, Grütze und Grieß muss der Trockenschrank eine Wärmekapazität haben, mit der er nach Beladung mit der Höchstzahl gleichzeitig zu trocknender Proben die voreingestellte Temperatur von 131 oC in weniger als 45 min wieder erreicht. Die Lüftung muss so beschaffen sein, dass die Ergebnisse nach 2-stündiger Trocknung einer sein gesamtes Fassungsvermögen auslastenden Anzahl an Weichweizenproben von den Ergebnissen nach 4-stündiger Trocknung um weniger als 0,15 % abweichen.
(2) Soll das Öl oder Fett später einer Qualitätsprüfung unterzogen werden, sind die Bimssteinkörner durch Glasperlen zu ersetzen.
ANHANG IV
ANALYSEMETHODEN ZUR UNTERSUCHUNG VON FUTTERMITTELN AUF IHREN GEHALT AN ZUGELASSENEN ZUSATZSTOFFEN
A. BESTIMMUNG DES VITAMIN-A-GEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Vitamin-A-(Retinol-)Gehalts in Futtermitteln und Vormischungen. Unter Vitamin A wird der nach dieser Methode ermittelte Gehalt an all-trans-Vitamin-A-Alkohol und seinen cis-Isomeren verstanden. Der Vitamin-A-Gehalt wird in Internationalen Einheiten (IE) je kg angegeben. Eine IE entspricht der Aktivität von 0,300 μg all-trans-Vitamin-A-Alkohol oder 0,344 μg all-trans-Vitamin-A-Acetat oder 0,550 μg all-trans-Vitamin-A-Palmitat.
Die Bestimmungsgrenze beträgt 2 000 IE Vitamin A/kg.
2. Prinzip
Die Probe wird mit ethanolischer Kaliumhydroxidlösung hydrolysiert, und das Vitamin A wird mit Petrolether extrahiert. Das Lösungsmittel wird eingedampft; der Rückstand wird in Methanol gelöst und, falls notwendig, auf die erforderliche Konzentration verdünnt. Der Vitamin-A-Gehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (RP-HPLC) unter Verwendung eines UV- oder Fluoreszenzdetektors bestimmt. Die chromatografischen Bedingungen werden so gewählt, dass keine Ruftrennung zwischen all-trans-Vitamin-A-Alkohol und seinen cis-Isomeren erfolgt.
3. Reagenzien
|
3.1. |
Ethanol, σ = 96 %. |
|
3.2. |
Petrolether, Siedeintervall 40 bis 60 oC. |
|
3.3. |
Methanol. |
|
3.4. |
Kaliumhydroxid-Lösung, c = 50 g/100 ml. |
|
3.5. |
Natriumascorbat-Lösung, c = 10 g/100 ml (siehe Bemerkung 7.7). |
|
3.6. |
Natriumsulfid, Na2S · x H2O (x = 7 bis 9) |
|
3.6.1. |
Natriumsulfid-Lösung, c = 0,5 mol/l in Glycerin, β = 120 g/l (für x = 9) (siehe Bemerkung 7.8). |
|
3.7. |
Phenolphthalein-Lösung, c = 2 g/100 ml in Ethanol (3.1). |
|
3.8. |
2-Propanol. |
|
3.9. |
Mobile Phase für die HPLC: Mischung von Methanol (3.3) und Wasser, z. B. 980 + 20 (V+V). Das Mischungsverhältnis ist der jeweils verwendeten Säule anzupassen. |
|
3.10. |
Stickstoff, sauerstofffrei. |
|
3.11. |
All-trans-Vitamin-A-Acetat, reinst, mit zertifizierter Aktivität, z. B. 2,80 × 106 IE/g |
|
3.11.1. |
Stammlösung von all-trans-Vitamin-A-Acetat: 50 mg Vitamin-A-Acetat (3.11) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In 2-Propanol (3.8) lösen und zur Marke mit demselben Lösungsmittel auffüllen. Der Sollgehalt dieser Lösung beträgt 1 400 IE Vitamin A je ml. Der genaue Gehalt ist gemäß 5.6.3.1 zu bestimmen. |
|
3.12. |
All-trans-Vitamin-A-Palmitat, reinst, mit zertifizierter Aktivität, z. B. 1,80 × 106 IE/g |
|
3.12.1. |
Stammlösung von all-trans-Vitamin-A-Palmitat: 80 mg Vitamin-A-Palmitat (3.12) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In 2-Propanol (3.8) lösen und mit demselben Lösungsmittel zur Marke auffüllen. Der Sollgehalt dieser Lösung beträgt 1 400 IE Vitamin A je ml. Der genaue Gehalt ist gemäß 5.6.3.2 zu bestimmen. |
|
3.13. |
2,6-Di-tert-butyl-4-methylphenol (BHT) (siehe Bemerkung 7.5). |
4. Geräte
|
4.1. |
Vakuum-Rotationsverdampfer. |
|
4.2. |
Braunglasgeräte |
|
4.2.1. |
Stehkolben oder Erlenmeyerkolben, 500 ml, mit Schliffhülse. |
|
4.2.2. |
Messkolben mit Schliffstopfen, enghalsig, 10, 25, 100 und 500 ml. |
|
4.2.3. |
Scheidetrichter, konische Form, 1 000 ml, mit Schliffstopfen. |
|
4.2.4. |
Spitzkolben, 250 ml, mit Schliffhülsen. |
|
4.3. |
Allihn-Rückflusskühler, Mantellänge 300 mm, Kernschliff mit Adapter für Gaseinleitung. |
|
4.4. |
Phasentrennungsfaltenfilter, Durchmesser 185 mm (z. B. Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
HPLC-Einrichtung mit Injektionssystem |
|
4.5.1. |
HPLC-Trennsäule, 250 × 4 mm, C18, 5 oder 10 μm Korngröße, oder vergleichbare Säule (Leistungskriterium: nur ein Peak für alle Retinol-Isomeren unter diesen HPLC-Bedingungen). |
|
4.5.2. |
UV- oder Fluoreszenzdetektor mit variabler Wellenlängeneinstellung. |
|
4.6. |
Spektralfotometer mit Quarz-Küvetten von 10 mm Schichtdicke. |
|
4.7. |
Wasserbad mit Magnetrührer. |
|
4.8. |
Extraktionsapparat (Abbildung 1) bestehend aus: |
|
4.8.1. |
1-l-Standzylinder mit Schliff und Schliffstopfen, |
|
4.8.2. |
Schliffeinsatz mit Seitenarm und einem in der Höhe verschiebbaren Rohr in der Mitte. Das verschiebbare Rohr sollte ein U-förmiges unteres Ende und eine Düse am entgegengesetzten Ende haben, so dass die obere Flüssigkeitsphase aus dem Zylinder in den Scheidetrichter überführt werden kann. |
5. Verfahren
|
Anmerkung |
: |
Vitamin A ist empfindlich gegenüber Licht (UV-Strahlung) und Oxidation. Es muss deshalb unter Ausschluss von Licht (Verwendung von Braunglasgeräten oder von mit Aluminiumfolie umhüllten Glasgeräten) und Sauerstoff (Stickstoffspülung) gearbeitet werden. Während der Extraktion muss das Luftpolster über der Flüssigkeit durch Stickstoff ersetzt werden (zur Vermeidung von Überdruck Stopfen rechtzeitig lüften). |
5.1. Vorbereitung der Probe
Die Probe wird unter Vermeidung von Erwärmung so fein vermahlen, dass sie ein Sieb mit 1 mm Maschenweite passieren kann. Die Zerkleinerung darf erst unmittelbar vor dem Einwiegen und Verseifen erfolgen, da sonst Vitamin-A-Verluste auftreten können.
5.2. Verseifung
Je nach Vitamin-A-Gehalt 2 bis 25 g der Probe auf 1 mg genau in einen 500-ml-Steh- oder Erlenmeyerkolben (4.2.1) einwiegen. Nacheinander unter Schwenken 130 ml Ethanol (3.1), etwa 100 mg BHT (3.13), 2 ml Natriumascorbat-Lösung (3.5) und 2 ml Natriumsulfid-Lösung (3.6) zusetzen. Den Kolben mit einem Rückflusskühler (4.3) verbinden und in ein Wasserbad mit Magnetrührer (4.7) geben. Bis zum Sieden erhitzen und 5 min am Rückflusskühler kochen. Anschließend 25 ml Kaliumhydroxidlösung (3.4) durch den Rückflusskühler (4.3) zugeben und unter ständigem Rühren und schwachem Stickstoffstrom 25 min weiterkochen. Den Kühler mit etwa 20 ml Wasser ausspülen und den Kolbeninhalt auf Zimmertemperatur abkühlen.
5.3. Extraktion
Die Verseifungslösung wird mit insgesamt 250 ml Wasser quantitativ in einen 1 000-ml-Scheidetrichter (4.2.3) oder in den Extraktionsapparat (4.8) überspült. Der Verseifungskolben wird mit 25 ml Ethanol (3.1) und anschließend mit 100 ml Petrolether (3.2) nachgewaschen, und die Spülflüssigkeiten werden ebenfalls in den Scheidetrichter oder den Extraktionsapparat überführt. Das Wasser/Ethanol-Verhältnis in den vereinigten Lösungen muss etwa 2:1 betragen. 2 min lang kräftig schütteln und dann 2 min lang absetzen lassen.
5.3.1.
Nach Phasentrennung (siehe Bemerkung 7.3) wird die Petroletherphase in einen anderen Scheidetrichter (4.2.3) überführt. Diese Extraktion 2-mal mit je 100 ml Petrolether (3.2) und 2-mal mit je 50 ml Petrolether (3.2) wiederholen.
Die vereinigten Extrakte werden im Scheidetrichter zunächst unter leichtem Schwenken (Vermeidung von Emulsionsbildung) 2-mal mit jeweils 100 ml Wasser gespült und dann durch mehrmaliges Schütteln mit weiteren Portionen von 100 ml Wasser so oft gewaschen, bis das Waschwasser bei Zugabe von Phenolphthalein-Lösung (3.7) farblos bleibt (4-maliges Waschen ist normalerweise ausreichend). Zur Entfernung eventuell suspendierten Wassers wird der gewaschene Extrakt durch einen trockenen Phasentrennungsfaltenfilter (4.4) in einen 500-ml-Messkolben (4.2.2) filtriert. Scheidetrichter und Filter mit 50 ml Petrolether (3.2) nachwaschen, mit Petrolether (3.2) zur Marke auffüllen und gut schütteln.
5.3.2.
Nach Phasentrennung (siehe Bemerkung 7.3) wird der Schliffstopfen des Standzylinders (4.8.1) durch den Schliffeinsatz (4.8.2) ersetzt und das verschiebbare Rohr so eingestellt, dass sich das U-förmige untere Ende gerade über dem Niveau der Grenzfläche befindet. Durch Druckausübung von einer am Seitenarm angebrachten Stickstoffleitung wird die obere Petroletherphase in einen 1 000-ml-Scheidetrichter (4.2.3) überführt. 100 ml Petrolether (3.2) werden in den Standzylinder gegeben, der mit einem Stopfen verschlossen und gründlich geschüttelt wird. Nach der Phasentrennung wird die obere Phase wie zuvor in den Scheidetrichter überführt. Diese Extraktion wird noch 1-mal mit 100 ml Petrolether (3.2) und 2-mal mit je 50 ml Petrolether (3.2) wiederholt, und die Petroletherphasen werden in den Scheidetrichter überführt.
Die vereinigten Petroletherextrakte, wie unter 5.3.1 erläutert, waschen und wie dort beschrieben weiterverfahren.
5.4. Vorbereitung der Probenlösung für die HPLC
Ein aliquoter Teil der Petrolether-Lösung (gemäß 5.3.1 oder 5.3.2) wird in einen 250-ml-Spitzkolben (4.2.4) pipettiert. Das Lösungsmittel wird unter vermindertem Druck und bei einer Badtemperatur von höchstens 40 oC am Rotationsverdampfer (4.1) fast bis zur Trockne eingedampft. Danach wird unter Stickstoff (3.10) Druckausgleich geschaffen und der Kolben vom Rotationsverdampfer abgenommen. Das restliche Lösungsmittel wird im Stickstoffstrom (3.10) abgeblasen, und der Rückstand wird sofort in einer definierten Menge (10 bis 100 ml) Methanol (3.3) aufgenommen (die Konzentration von Vitamin A muss etwa 5 bis 30 IE/ml betragen).
5.5. Bestimmung durch HPLC
Die Abtrennung von Vitamin A erfolgt mithilfe einer Umkehrphasen-C18-Säule (4.5.1), und die Konzentration wird mithilfe eines UV-Detektors (325 nm) oder eines Fluoreszenzdetektors (Anregung: 325 nm, Emission: 475 nm) (4.5.2) gemessen.
Hierzu wird ein aliquoter Teil (z. B. 20 μl) der nach 5.4 erhaltenen methanolischen Lösung auf die Trennsäule gegeben und mit der mobilen Phase (3.9) eluiert. Die mittlere Peakhöhe (-fläche) von mehreren Einspritzungen derselben Probenlösung wird ermittelt. In gleicher Weise werden auch die mittleren Peakhöhen (-flächen) von mehreren Einspritzungen der Kalibrierlösungen (5.6.2) bestimmt.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
|
HPLC-Trennsäule (4.5.1): |
250 × 4 mm, C18, 5 oder 10 μm Korngröße, oder vergleichbare Säule |
|
Mobile Phase (3.9): |
Mischung von Methanol (3.3) und Wasser, z. B. 980 + 20 (V+V). |
|
Durchflussrate: |
1 bis 2 ml/min |
|
Detektor (4.5.2): |
UV-Detektor (325 nm) oder Fluoreszenzdetektor (Anregung: 325 nm, Emission: 475 nm) |
5.6. Kalibrierung
5.6.1.
Von der Vitamin-A-Acetat-Stammlösung (3.11.1) oder der Vitamin-A-Palmitat-Stammlösung (3.12.1) 20 ml in einen 500-ml-Steh- oder Erlenmeyerkolben (4.2.1) pipettieren und, wie unter 5.2 beschrieben, aber ohne Zugabe von BHT, hydrolysieren. Anschließend mit Petrolether (3.2) gemäß 5.3 extrahieren und mit Petrolether (3.2) auf 500 ml auffüllen. 100 ml dieses Extrakts werden am Rotationsverdampfer (siehe 5.4) fast bis zur Trockne eingedampft. Das verbleibende Lösungsmittel wird im Stickstoffstrom (3.10) abgeblasen, und der Rückstand wird in 10,0 ml Methanol (3.3) aufgenommen. Der Sollgehalt dieser Lösung beträgt 560 IE Vitamin A je ml. Der genaue Gehalt ist nach 5.6.3.3 zu bestimmen. Die Gebrauchsstandardlösung muss vor Gebrauch frisch hergestellt werden.
Von dieser Gebrauchsstandardlösung werden 2,0 ml in einen 20-ml-Messkolben pipettiert. Es wird mit Methanol (3.3) zur Marke aufgefüllt und gemischt. Der Sollgehalt dieser verdünnten Gebrauchsstandardlösung beträgt 56 IE Vitamin A je ml.
5.6.2.
Von der verdünnten Gebrauchsstandardlösung werden 1,0 ml, 2,0 ml, 5,0 ml bzw. 10,0 ml in jeweils einen 20-ml-Messkolben pipettiert. Es wird mit Methanol (3.3) zur Marke aufgefüllt und gemischt. Der Sollgehalt dieser Lösungen beträgt 2,8 IE, 5,6 IE, 14,0 IE bzw. 28,0 IE Vitamin A je ml.
Von jeder Kalibrierlösung werden mehrmals 20 μl eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Anhand der so ermittelten mittleren Peakhöhen (-flächen) und unter Berücksichtigung der Ergebnisse der UV-Kontrolle (5.6.3.3) wird eine Kalibrationskurve erstellt.
5.6.3.
5.6.3.1.
Von der Vitamin-A-Acetat-Stammlösung (3.11.1) werden 2,0 ml in einen 50-ml-Messkolben (4.2.2) pipettiert, und es wird mit 2-Propanol (3.8) zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 56 IE Vitamin A je ml. Von dieser verdünnten Vitamin-A-Acetat-Lösung werden 3,0 ml in einen 25-ml-Messkolben pipettiert; es wird mit 2-Propanol (3.8) zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin A je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol (3.8) im Spektralfotometer (4.6) zwischen 300 und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E326 × 19,0
(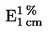 E für Vitamin-A-Acetat = 1 530 bei 326 nm in 2-Propanol)
E für Vitamin-A-Acetat = 1 530 bei 326 nm in 2-Propanol)
5.6.3.2.
Von der Vitamin-A-Palmitat-Stammlösung (3.12.1) werden 2,0 ml in einen 50-ml-Messkolben (4.2.2) pipettiert, und es wird mit 2-Propanol (3.8) zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 56 IE Vitamin A je ml. Von dieser verdünnten Vitamin-A-Palmitat-Lösung werden 3,0 ml in einen 25-ml-Messkolben pipettiert; es wird zur Marke mit 2-Propanol (3.8) aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin A je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol (3.8) im Spektralfotometer (4.6) zwischen 300 und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E326 × 19,0
(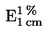 für Vitamin-A-Palmitat = 957 bei 326 nm in 2-Propanol)
für Vitamin-A-Palmitat = 957 bei 326 nm in 2-Propanol)
5.6.3.3.
Von der gemäß 5.6.1 hergestellten, unverdünnten Vitamin-A-Gebrauchsstandardlösung werden 3,0 ml in einen 50-ml-Messkolben (4.2.2) pipettiert, und es wird mit 2-Propanol (3.8) zur Marke aufgefüllt. 5,0 ml dieser Lösung werden in einen 25-ml-Messkolben pipettiert; es wird mit 2-Propanol (3.8) zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin A je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol (3.8) im Spektralfotometer (4.6) zwischen 300 und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E325 × 18,3
( E für Vitamin-A-Alkohol = 1 821 bei 325 nm in 2-Propanol)
E für Vitamin-A-Alkohol = 1 821 bei 325 nm in 2-Propanol)
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Vitamin-A-Peaks der Probenlösung wird anhand der Kalibrationskurve (5.6.2) die Konzentration der Probenlösung in IE/ml bestimmt.
Der Vitamin-A-Gehalt w (in IE/kg) der Probe wird nach folgender Formel berechnet:
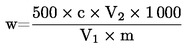 [IE/kg]
[IE/kg]
wobei:
|
c |
= |
Vitamin-A-Konzentration der Probenlösung (5.4) in IE/ml, |
|
V1 |
= |
Volumen der Probenlösung (5.4) in ml, |
|
V2 |
= |
Volumen des unter 5.4 entnommenen aliquoten Teils in ml, |
|
m |
= |
Probeneinwaage in g. |
7. Bemerkungen
|
7.1. |
Bei Proben mit niedriger Vitamin-A-Konzentration kann es zweckmäßig sein, die Petroletherextrakte von 2 Verseifungsansätzen (Einwaage 25 g) zu einer Probenlösung für die HPLC-Bestimmung zu vereinigen. |
|
7.2. |
Die Einwaage für die Analyse darf höchstens 2 g Fett enthalten. |
|
7.3. |
Bei schlechter Phasentrennung können etwa 10 ml Ethanol (3.1) zur Zerstörung der Emulsion zugegeben werden. |
|
7.4. |
Bei Lebertran oder anderen reinen Fetten ist die Verseifungsdauer auf 45 bis 60 min zu erhöhen. |
|
7.5. |
Statt BHT kann Hydrochinon verwendet werden. |
|
7.6. |
Bei Verwendung einer Normalphasensäule ist die Trennung der Retinolisomeren möglich. Allerdings müssen in diesem Fall für die Berechnungen die Peakhöhen (-flächen) aller cis- und trans-Isomeren addiert werden. |
|
7.7. |
Statt Natriumascorbat-Lösung können etwa 150 mg Ascorbinsäure verwendet werden. |
|
7.8. |
Statt Natriumsulfid-Lösung können etwa 50 mg EDTA verwendet werden. |
|
7.9. |
Bei der Bestimmung des Gehalts an Vitamin A in Milchaustausch-Futtermitteln sind folgende Aspekte zu berücksichtigen:
Um festzustellen, ob die angewandte Analysemethode zuverlässige Ergebnisse für diese spezifische Matrix (Milchaustausch-Futtermittel) liefert, ist ein Wiederfindungstest an einer weiteren Probeneinwaage durchzuführen. Ist die Wiederfindungsrate kleiner als 80 %, muss das Analyseergebnis um die Wiederfindung korrigiert werden. |
8. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 15 % des höheren Werts nicht überschreiten.
9. Ergebnisse eines Ringversuchs (1)
|
|
Vormischungen |
Fertigfuttervormischungen |
Mineralfutter |
Einweißkonzentrat |
Ferkelaufzuchtfutter |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
Mittelwert [IE/kg] |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
sr [IE/kg] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [IE/kg] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
VKr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
SR [IE/kg] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [IE/kg] |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
VKR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
Anzahl der Laboratorien |
|
n |
= |
Anzahl der Einzelwerte |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
r |
= |
Wiederholbarkeit |
|
R |
= |
Vergleichbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |

B. BESTIMMUNG DES VITAMIN-E-GEHALTS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Vitamin-E-Gehalts von Futtermitteln und Vormischungen. Der Vitamin-E-Gehalt wird in mg DL-α-Tocopherol-Acetat je kg angegeben. 1 mg DL-α-Tocopherol-Acetat entspricht 0,91 mg DL-α-Tocopherol (Vitamin E).
Die Bestimmungsgrenze beträgt 2 mg Vitamin E/kg. Diese Grenze wird nur mit dem Fluoreszenzdetektor erreicht. Bei einem UV-Detektor beträgt die Bestimmungsgrenze 10 mg/kg.
2. Prinzip
Die Probe wird mit ethanolischer Kaliumhydroxidlösung hydrolysiert, und das Vitamin E wird mit Petrolether extrahiert. Das Lösungsmittel wird eingedampft; der Rückstand wird in Methanol gelöst und, falls notwendig, auf die erforderliche Konzentration verdünnt. Der Vitamin-E-Gehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (RP-HPLC) unter Verwendung eines UV- oder Fluoreszenzdetektors bestimmt.
3. Reagenzien
|
3.1. |
Ethanol, σ = 96 %. |
|
3.2. |
Petrolether, Siedeintervall 40 bis 60 oC. |
|
3.3. |
Methanol. |
|
3.4. |
Kaliumhydroxid-Lösung, c = 50 g/100 ml. |
|
3.5. |
Natriumascorbat-Lösung, c = 10 g/100 ml (siehe Bemerkung 7.7). |
|
3.6. |
Natriumsulfid, Na2S · x H2O (x = 7 bis 9) |
|
3.6.1. |
Natriumsulfid-Lösung, c = 0,5 mol/l in Glycerin, β = 120 g/l (für x = 9) (siehe Bemerkung 7.8). |
|
3.7. |
Phenolphthalein-Lösung, c = 2 g/100 ml in Ethanol (3.1). |
|
3.8. |
Mobile Phase für die HPLC: Mischung von Methanol (3.3) und Wasser, z. B. 980 + 20 (V+V). Das Mischungsverhältnis ist der jeweils verwendeten Säule anzupassen. |
|
3.9. |
Stickstoff, sauerstofffrei. |
|
3.10. |
DL-α-Tocopherol-Acetat, reinst, mit zertifizierter Aktivität |
|
3.10.1. |
DL-α-Tocopherol-Acetat-Stammlösung: 100 mg DL-α-Tocopherol-Acetat (3.10) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In Ethanol (3.1) lösen und mit demselben Lösungsmittel zur Marke auffüllen. 1 ml dieser Lösung enthält 1 mg DL-α-Tocopherol-Acetat. (UV-Kontrolle siehe 5.6.1.3; Stabilisierung siehe Bemerkung 7.4). |
|
3.11. |
DL-α-Tocopherol, reinst, mit zertifizierter Aktivität |
|
3.11.1. |
DL-α-Tocopherol-Stammlösung: Von DL-α-Tocopherol (3.11) werden 100 mg auf 0,1 mg genau in einen 100-ml Messkolben eingewogen. In Ethanol (3.1) lösen und mit demselben Lösungsmittel zur Marke auffüllen. 1 ml dieser Lösung enthält 1 mg DL-α-Tocopherol. (UV-Kontrolle siehe 5.6.2.3; Stabilisierung siehe Bemerkung 7.4). |
|
3.12. |
2,6-Di-tert-butyl-4-methylphenol (BHT) (siehe Bemerkung 7.5). |
4. Geräte
|
4.1. |
Rotationsfilmverdampfer. |
|
4.2. |
Braunglasgeräte |
|
4.2.1. |
Stehkolben oder Erlenmeyerkolben, 500 ml, mit Schliffhülse. |
|
4.2.2. |
Messkolben mit Schliffstopfen, enghalsig, 10, 25, 100 und 500 ml. |
|
4.2.3. |
Scheidetrichter, konische Form, 1 000 ml, mit Schliffstopfen. |
|
4.2.4. |
Spitzkolben, 250 ml, mit Schliffhülsen. |
|
4.3. |
Allihn-Rückflusskühler, Mantellänge 300 mm, Kernschliff mit Adapter für Gaseinleitung. |
|
4.4. |
Phasentrennungsfaltenfilter, Durchmesser 185 mm (z. B. Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
HPLC-Einrichtung mit Injektionssystem. |
|
4.5.1. |
HPLC-Trennsäule, 250 × 4 mm, C18, 5 oder 10 μm Korngröße, oder vergleichbare Säule, |
|
4.5.2. |
UV- oder Fluoreszenzdetektor mit variabler Wellenlängeneinstellung. |
|
4.6. |
Spektralfotometer mit Quarz-Küvetten von 10 mm Schichtdicke. |
|
4.7. |
Wasserbad mit Magnetrührer. |
|
4.8. |
Extraktionsapparat (Abbildung 1) bestehend aus: |
|
4.8.1. |
1-l-Standzylinder mit Schliff und Schliffstopfen, |
|
4.8.2. |
Schliffeinsatz mit Seitenarm und einem in der Höhe verschiebbaren Rohr in der Mitte. Das verschiebbare Rohr muss ein U-förmiges unteres Ende und eine Düse am entgegengesetzten Ende haben, so dass die obere Flüssigkeitsphase aus dem Zylinder in den Scheidetrichter überführt werden kann. |
5. Verfahren
|
Anmerkung |
: |
Vitamin E ist empfindlich gegenüber Licht (UV-Strahlung) und Oxidation. Daher muss unter Ausschluss von Licht (Verwendung von Braunglasgeräten oder von mit Aluminiumfolie umhüllten Glasgeräten) und Sauerstoff (Stickstoffspülung) gearbeitet werden. Während der Extraktion muss das Luftpolster über der Flüssigkeit durch Stickstoff ersetzt werden (zur Vermeidung von Überdruck Stopfen immer wieder lüften). |
5.1. Vorbereitung der Probe
Die Probe wird unter Vermeidung von Erwärmung so fein vermahlen, dass sie ein Sieb mit 1 mm Maschenweite passieren kann. Die Zerkleinerung darf erst unmittelbar vor dem Einwiegen und Verseifen erfolgen, da sonst Vitamin-E-Verluste auftreten können.
5.2. Verseifung
Je nach Vitamin-E-Gehalt 2 bis 25 g der Probe auf 0,01 g genau in einen 500-ml-Steh- oder Erlenmeyerkolben (4.2.1) einwiegen. Nacheinander unter Schwenken 130 ml Ethanol (3.1), etwa 100 mg BHT (3.12), 2 ml Natriumascorbat-Lösung (3.5) und 2 ml Natriumsulfid-Lösung (3.6) zusetzen. Den Kolben mit dem Rückflusskühler (4.3) verbinden und in ein Wasserbad mit Magnetrührer (4.7) geben. Bis zum Sieden erhitzen und 5 min am Rückflusskühler kochen. Anschließend 25 ml Kaliumhydroxidlösung (3.4) durch den Rückflusskühler (4.3) zugeben, und unter ständigem Rühren und schwachem Stickstoffstrom 25 min weiterkochen. Den Kühler mit etwa 20 ml Wasser ausspülen und den Kolbeninhalt auf Zimmertemperatur abkühlen.
5.3. Extraktion
Die Verseifungslösung wird mit insgesamt 250 ml Wasser quantitativ in einen 1 000-ml-Scheidetrichter (4.2.3) oder in den Extraktionsapparat (4.8) überspült. Der Verseifungskolben wird mit 25 ml Ethanol (3.1) und anschließend mit 100 ml Petrolether (3.2) nachgewaschen, und die Spülflüssigkeiten werden ebenfalls in den Scheidetrichter oder den Extraktionsapparat überführt. Das Wasser/Ethanol-Verhältnis in den vereinigten Lösungen muss etwa 2:1 betragen. 2 min kräftig schütteln und dann 2 min absetzen lassen.
5.3.1.
Nach Phasentrennung (siehe Bemerkung 7.3) wird die Petroletherphase in einen anderen Scheidetrichter (4.2.3) überführt. Diese Extraktion 2-mal mit je 100 ml Petrolether (3.2) und 2-mal mit je 50 ml Petrolether (3.2) wiederholen.
Die vereinigten Extrakte werden im Scheidetrichter unter leichtem Schwenken (Vermeidung von Emulsionsbildung) 2-mal mit jeweils 100 ml Wasser gespült und dann durch mehrmaliges Schütteln mit weiteren Portionen von 100 ml Wasser so oft gewaschen, bis das Waschwasser bei Zugabe von Phenolphthalein-Lösung (3.7) farblos bleibt (4-maliges Waschen ist normalerweise ausreichend). Zur Entfernung eventuell suspendierten Wassers wird der gewaschene Extrakt durch einen trockenen Phasentrennungsfaltenfilter (4.4) in einen 500-ml-Messkolben (4.2.2) filtriert. Scheidetrichter und Filter mit 50 ml Petrolether (3.2) nachwaschen, mit Petrolether (3.2) zur Marke auffüllen und gut schütteln.
5.3.2.
Nach Phasentrennung (siehe Bemerkung 7.3) wird der Stopfen des Standzylinders (4.8.1) durch den Schliffeinsatz (4.8.2) ersetzt und das verschiebbare Rohr so eingestellt, dass sich das U-förmige untere Ende gerade über dem Niveau der Grenzfläche befindet. Durch Druckausübung von einer am Seitenarm angebrachten Stickstoffleitung wird die obere Petroletherphase in einen 1 000-ml-Scheidetrichter (4.2.3) überführt. 100 ml Petrolether (3.2) werden in den Standzylinder gegeben, der mit einem Stopfen verschlossen und gründlich geschüttelt wird. Nach der Phasentrennung wird die obere Phase wie zuvor in den Scheidetrichter überführt. Diese Extraktion wird noch 1-mal mit 100 ml Petrolether (3.2) und 2-mal mit je 50 ml Petrolether (3.2) wiederholt, und die Petroletherphasen werden in den Scheidetrichter überführt.
Die vereinigten Petroletherextrakte, wie unter 5.3.1 erläutert, waschen und wie dort beschrieben weiterverfahren.
5.4. Vorbereitung der Probenlösung für die HPLC
Ein aliquoter Teil der Petrolether-Lösung (gemäß 5.3.1 oder 5.3.2) wird in einen 250-ml-Spitzkolben (4.2.4) pipettiert. Das Lösungsmittel wird unter vermindertem Druck und bei einer Badtemperatur von höchstens 40 oC am Rotationsverdampfer (4.1) fast bis zur Trockne eingedampft. Danach wird unter Stickstoff (3.9) Druckausgleich geschaffen und der Kolben vom Rotationsverdampfer abgenommen. Das restliche Lösungsmittel wird im Stickstoffstrom (3.9) abgeblasen, und der Rückstand wird sofort in einer definierten Menge (10 bis 100 ml) Methanol (3.3) aufgenommen (die Konzentration von DL-α-Tocopherol muss etwa 5 bis 30 μg/ml betragen).
5.5. Bestimmung durch HPLC
Die Abtrennung von Vitamin E erfolgt mithilfe einer Umkehrphasen-C18-Säule (4.5.1), und die Konzentration wird mithilfe eines Fluoreszenzdetektors (Anregung: 295 nm, Emission: 330 nm) oder eines UV-Detektors (292 nm) (4.5.2) gemessen.
Hierzu wird ein aliquoter Teil (z. B. 20 μl) der nach 5.4 erhaltenen methanolischen Lösung auf die Trennsäule gegeben und mit der mobilen Phase (3.8) eluiert. Die mittlere Peakhöhe (-fläche) von mehreren Einspritzungen derselben Probenlösung wird ermittelt. In gleicher Weise werden auch die mittleren Peakhöhen (-flächen) von mehreren Einspritzungen der Kalibrierlösungen (5.6.2) bestimmt.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
|
HPLC-Trennsäule (4.5.1): |
250 × 4 mm, C18, 5 oder 10 μm Korngröße, oder vergleichbare Säule |
|
Mobile Phase (3.8): |
Mischung von Methanol (3.3) und Wasser z. B. 980 + 20 (V+V) |
|
Durchflussrate: |
1 bis 2 ml/min |
|
Detektor (4.5.2) |
Fluoreszenzdetektor (Anregung: 295 nm; Emission 330 nm) oder UV-Detektor (292 nm) |
5.6. Kalibrierung (DL-α-Tocopherol-Acetat oder DL-α-Tocopherol)
5.6.1.
5.6.1.1.
Von der DL-α-Tocopherol-Acetat-Stammlösung (3.10.1) werden 25 ml in einen 500-ml-Steh- oder Erlenmeyerkolben (4.2.1) pipettiert und, wie unter 5.2 beschrieben, hydrolysiert. Anschließend mit Petrolether (3.2) gemäß 5.3 extrahieren und mit Petrolether auf 500 ml auffüllen. Von diesem Extrakt werden 25 ml am Rotationsverdampfer (siehe 5.4) fast bis zur Trockne eingedampft. Das verbleibende Lösungsmittel wird im Stickstoffstrom (3.9) abgeblasen, und der Rückstand wird in 25,0 ml Methanol (3.3) aufgenommen. Der Sollgehalt dieser Lösung beträgt 45,5 μg DL-α-Tocopherol je ml; das entspricht 50 μg DL-α-Tocopherol-Acetat je ml. Die Gebrauchsstandardlösung muss vor Gebrauch frisch hergestellt werden.
5.6.1.2.
Von der Gebrauchsstandardlösung werden 1,0 ml, 2,0 ml, 4,0 ml bzw. 10,0 ml in jeweils einen 20-ml-Messkolben pipettiert; es wird mit Methanol (3.3) zur Marke aufgefüllt und gemischt. Der Sollgehalt dieser Lösungen beträgt 2,5 μg/ml, 5,0 μg/ml, 10,0 μg/ml bzw. 25,0 μg/ml DL-α-Tocopherol-Acetat, d. h. 2,28 μg/ml, 4,55 μg/ml, 9,10 μg/ml bzw. 22,8 μg/ml DL-α-Tocopherol.
Von jeder Kalibrierlösung werden mehrmals 20 μl eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Anhand der so ermittelten mittleren Peakhöhen (-flächen) wird eine Kalibrationskurve erstellt.
5.6.1.3.
Von der DL-α-Tocopherol-Acetat-Stammlösung (3.10.1) werden 5,0 ml mit Ethanol auf 25,0 ml aufgefüllt. Das UV-Spektrum dieser Lösung wird gegen Ethanol (3.1) im Spektralfotometer (4.6) zwischen 250 und 320 nm gemessen.
Das Extinktionsmaximum muss bei 284 nm liegen:
 = 43,6 bei 284 nm in Ethanol
= 43,6 bei 284 nm in Ethanol
Bei der vorliegenden Verdünnung muss eine Extinktion von 0,84 bis 0,88 erzielt werden.
5.6.2.
5.6.2.1.
Von der DL-α-Tocopherol-Stammlösung (3.11.1) werden 2 ml in einen 50-ml-Messkolben pipettiert und in Methanol (3.3) gelöst; es wird mit Methanol zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 40 μg DL-α-Tocopherol je ml; das entspricht 44,0 μg DL-α-Tocopherol-Acetat je ml. Die Gebrauchsstandardlösung muss vor Gebrauch frisch hergestellt werden.
5.6.2.2.
Von der Gebrauchsstandardlösung werden 1,0 ml, 2,0 ml, 4,0 ml bzw. 10,0 ml in jeweils einen 20-ml-Messkolben pipettiert; es wird mit Methanol (3.3) zur Marke aufgefüllt und gemischt. Der Sollgehalt dieser Lösungen beträgt 2,0 μg/ml, 4,0 μg/ml, 8,0 μg/ml bzw. 20,0 μg/ml DL-α-Tocopherol, d. h. 2,20 μg/ml, 4,40 μg/ml, 8,79 μg/ml bzw. 22,0 μg/ml DL-α-Tocopherol-Acetat.
Von jeder Kalibrierlösung werden mehrmals 20 μl eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Anhand der so ermittelten mittleren Peakhöhen (-flächen) wird eine Kalibrationskurve erstellt.
5.6.2.3.
Von der DL-α-Tocopherol-Stammlösung (3.11.1) werden 2,0 ml mit Ethanol auf 25,0 ml aufgefüllt; das UV-Spektrum dieser Lösung wird gegen Ethanol (3.1) im Spektralfotometer (4.6) zwischen 250 und 320 nm gemessen. Das Extinktionsmaximum muss bei 292 nm liegen:
 = 75,8 bei 292 nm in Ethanol
= 75,8 bei 292 nm in Ethanol
Bei der vorliegenden Verdünnung muss eine Extinktion von 0,6 erzielt werden.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Vitamin-E-Peaks der Probenlösung wird anhand der Kalibrationskurve (5.6.1.2 oder 5.6.2.2) die Konzentration der Probenlösung in μg/ml (berechnet als α-Tocopherol-Acetat) bestimmt.
Der Vitamin-E-Gehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
 [mg/kg]
[mg/kg]
wobei:
|
c |
= |
Vitamin-E-Konzentration (als α-Tocopherol-Acetat) der Probenlösung (5.4) in μg/ml, |
|
V1 |
= |
Volumen der Probenlösung (5.4) in ml, |
|
V2 |
= |
Volumen des unter 5.4 entnommenen aliquoten Teils in ml, |
|
m |
= |
Probeneinwaage in g. |
7. Bemerkungen
|
7.1. |
Bei Proben mit niedriger Vitamin-E-Konzentration kann es zweckmäßig sein, die Petroletherextrakte von 2 Verseifungsansätzen (Einwaage 25 g) zu einer Probenlösung für die HPLC-Bestimmung zu vereinigen. |
|
7.2. |
Die Einwaage für die Analyse darf höchstens 2 g Fett enthalten. |
|
7.3. |
Bei schlechter Phasentrennung können etwa 10 ml Ethanol (3.1) zur Zerstörung der Emulsion zugegeben werden. |
|
7.4. |
Nach der spektralfotometrischen Vermessung der DL-α-Tocopherol-Acetat- oder der DL-α-Tocopherol-Lösung nach 5.6.1.3 bzw. 5.6.2.3 empfiehlt es sich, der Lösung (3.10.1 bzw. 3.10.2) etwa 10 mg BHT (3.12) zuzusetzen und die Lösung im Kühlschrank aufzubewahren (Haltbarkeit höchstens 4 Wochen). |
|
7.5. |
Statt BHT kann Hydrochinon verwendet werden. |
|
7.6. |
Bei Verwendung einer Normalphasensäule ist die Trennung von α-, β-, γ- und δ-Tocopherol möglich. |
|
7.7. |
Statt Natriumascorbat-Lösung können etwa 150 mg Ascorbinsäure verwendet werden. |
|
7.8. |
Statt Natriumsulfid-Lösung können etwa 50 mg EDTA verwendet werden. |
|
7.9. |
Vitamin-E-Acetat hydrolysiert unter alkalischen Bedingungen sehr schnell und ist deshalb sehr oxidationsanfällig, insbesondere in Gegenwart von Spurenelementen wie Eisen oder Kupfer. Bei der Bestimmung des Vitamin-E-Gehalts in Vormischungen, bei denen ein Gehalt von 5 000 mg/kg überschritten wird, könnte es zu einem Abbau von Vitamin E kommen. Deshalb wird zur Bestätigung eine HPLC-Methode mit einem enzymatischen Aufschluss der Vitamin-E-Formel ohne einen alkalischen Verseifungsschritt empfohlen. |
8. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 15 % des höheren Werts nicht überschreiten.
9. Ergebnisse eines Ringversuchs (2)
|
|
Vormischungen |
Fertigfuttervormischungen |
Mineralfutter |
Eiweißkonzentrat |
Ferkelaufzuchtfutter |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
Mittelwert [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
VKr [ %] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
SR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
VKR [ %] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
Anzahl der Laboratorien |
|
n |
= |
Anzahl der Einzelwerte |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
r |
= |
Wiederholbarkeit |
|
R |
= |
Vergleichbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |

C. BESTIMMUNG DES GEHALTS AN DEN SPURENELEMENTEN EISEN, KUPFER, MANGAN UND ZINK
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an den Spurenelementen Eisen, Kupfer, Mangan und Zink in Futtermitteln. Die Bestimmungsgrenzen liegen bei:
|
— |
Eisen (Fe): 20 mg/kg, |
|
— |
Kupfer (Cu): 10 mg/kg, |
|
— |
Mangan (Mn): 20 mg/kg, |
|
— |
Zink (Zn): 20 mg/kg. |
2. Prinzip
Die Probe wird nach Zerstörung eventuell vorhandener organischer Substanz in Salzsäure gelöst. Die Elemente Eisen, Kupfer, Mangan und Zink werden nach entsprechender Verdünnung der Analysenlösung mittels Atomabsorptionsspektrometrie bestimmt.
3. Reagenzien
Vorbemerkungen
Das zur Herstellung der Reagenzien und Analysenlösungen verwendete Wasser muss „frei“ von den zu bestimmenden Kationen sein, d. h., das Wasser muss entweder in einer Borsilikatglas- oder Quarzapparatur 2-fach destilliert oder durch doppelten Ionenaustausch gereinigt worden sein.
Die zur Analyse verwendeten Reagenzien müssen mindestens von analysenreiner Qualität sein. Die Abwesenheit des zu bestimmenden Elements wird mittels eines Blindversuchs kontrolliert. Falls erforderlich, sind die Reagenzien einer zusätzlichen Reinigung zu unterziehen.
Anstelle der nachfolgend beschriebenen Standardlösungen können auch handelsübliche Standardlösungen verwendet werden, deren Reinheit garantiert und vor der Verwendung kontrolliert wurde.
|
3.1. |
Salzsäure (D: 1,19 g/ml). |
|
3.2. |
Salzsäure (6 mol/l). |
|
3.3. |
Salzsäure (0,5 mol/l). |
|
3.4. |
Fluorwasserstoffsäure (Volumenkonzentration = 38 bis 40 %); Eisengehalt: weniger als 1 mg/l; Glührückstand (als Sulfat): weniger als 10 mg/l. |
|
3.5. |
Schwefelsäure (D: 1,84 g/ml). |
|
3.6. |
Wasserstoffperoxid, ca. 100 Volumina Sauerstoff (Massenanteil = 30 %). |
|
3.7. |
Eisen-Standardlösung (1 000 μg Fe/ml), wie folgt zubereitet, oder eine gleichwertige handelsübliche Lösung: 1 g Eisendraht wird in 200 ml 6 mol/l Salzsäure (3.2) gelöst. Es werden 16 ml Wasserstoffperoxid (3.6) zugegeben; dann wird mit Wasser auf 1 l aufgefüllt: |
|
3.7.1. |
Eisen-Gebrauchsstandardlösung (100 μg Fe/ml): Die Standardlösung (3.7) wird im Verhältnis 1:9 mit Wasser verdünnt. |
|
3.8. |
Kupfer-Standardlösung (1 000 μg Cu/ml), wie folgt zubereitet, oder eine gleichwertige handelsübliche Lösung:
|
|
3.8.1. |
Kupfer-Gebrauchsstandardlösung (10 μg Cu/ml): Die Standardlösung (3.8) wird im Verhältnis 1:9 mit Wasser verdünnt; anschließend wird die so entstandene Lösung wiederum im Verhältnis 1:9 mit Wasser verdünnt. |
|
3.9. |
Mangan-Standardlösung (1 000 μg Mn/ml), wie folgt zubereitet, oder eine gleichwertige handelsübliche Lösung:
|
|
3.9.1. |
Mangan-Gebrauchsstandardlösung (10 μg Mn/ml): Die Standardlösung (3.9) wird im Verhältnis 1:9 mit Wasser verdünnt; anschließend wird die so entstandene Lösung wiederum im Verhältnis 1:9 mit Wasser verdünnt. |
|
3.10. |
Zink-Standardlösung (1 000 μg Zn/ml), wie folgt zubereitet, oder eine gleichwertige handelsübliche Lösung:
|
|
3.10.1. |
Zink-Gebrauchsstandardlösung (10 μg Zn/ml): Die Standardlösung (3.10) wird im Verhältnis 1:9 mit Wasser verdünnt; anschließend wird die so entstandene Lösung wiederum im Verhältnis 1:9 mit Wasser verdünnt. |
|
3.11. |
Lanthanchloridlösung: In 150 ml Wasser wird 12 g Lanthanoxid gelöst und 100 ml 6 mol/l Salzsäure (3.2) zugegeben. Dann wird mit Wasser auf 1 l aufgefüllt. |
4. Geräte
|
4.1. |
Muffelofen mit Regelvorrichtung und gegebenenfalls mit Temperaturanzeige. |
|
4.2. |
Glasgeräte müssen aus resistentem Borsilikatglas sein. Es wird empfohlen, Glasgeräte zu benutzen, die ausschließlich der Bestimmung des Gehalts an Spurenelementen vorbehalten bleiben. |
|
4.3. |
Atomabsorptions-Spektralfotometer; das Gerät muss innerhalb des vorgesehenen Messbereichs hinsichtlich seiner Empfindlichkeit und Genauigkeit den Anforderungen der jeweiligen Methode genügen. |
5. Verfahren (3)
5.1. Proben mit organischen Bestandteilen
5.1.1. (4)
|
5.1.1.1. |
Von der Probe werden 5 bis 10 g (auf 0,2 mg genau abgewogen) in eine Quarz- oder Platinschale gegeben (siehe Anmerkung b) und im Trockenschrank bei 105 oC getrocknet; dann wird die Schale in den kalten Muffelofen (4.1) verbracht. Der Ofen wird geschlossen (siehe Anmerkung c) und die Temperatur innerhalb von ca. 90 min langsam auf 450 bis 475 oC erhöht. Diese Temperatur wird 4 bis 16 h (z. B. über Nacht) aufrechterhalten, um Kohleteilchen zu entfernen; dann wird der Ofen geöffnet und abkühlen gelassen (siehe Anmerkung d). Die Asche wird mit Wasser angefeuchtet und in ein 250-ml-Becherglas gegeben. Die Schale wird mit insgesamt etwa 5 ml Salzsäure (3.1) ausgespült, die anschließend langsam und vorsichtig (da es aufgrund von CO2-Bildung eventuell zu einer heftigen Reaktion kommen kann) in das Becherglas gegeben wird. Unter Schütteln wird Salzsäure (3.1) tropfenweise zugegeben, bis die Schaumbildung aufhört. Die Salzsäure wird unter gelegentlichem Umrühren mit einem Glasstab bis zur Trockne abgedampft. Dem Rückstand werden 15 ml 6 mol/l Salzsäure (3.2) und anschließend etwa 120 ml Wasser zugefügt. Umgerührt wird mit dem Glasstab, der in dem Becherglas zu belassen ist. Letzteres wird mit einem Uhrglas abgedeckt. Die Flüssigkeit wird langsam zum Sieden gebracht und so lange im Sieden gehalten, bis die Asche vollständig gelöst ist. Dann wird durch ein aschefreies Filterpapier filtriert und das Filtrat in einem 250-ml-Messkolben aufgefangen. Das Becherglas und der Filter werden mit 5 ml heißer 6 mol/l Salzsäure (3.2) und 2-mal mit kochendem Wasser ausgewaschen. Anschließend wird der Messkolben mit Wasser zur Marke aufgefüllt (HCl-Konzentration etwa 0,5 mol/l). |
|
5.1.1.2. |
Sollte der Filterrückstand schwarz aussehen (Kohlenstoff), so wird er im Trockenschrank abermals bei 450 bis 475 oC verascht. Diese Veraschung, die nur wenige Stunden erfordert (etwa 3 bis 5 h) ist abgeschlossen, wenn die Asche weiß oder nahezu weiß aussieht. Der Rückstand wird mit etwa 2 ml Salzsäure (3.1) aufgenommen, zur Trockne eingedampft und mit 5 ml 6 mol/l Salzsäure (3.2) versetzt. Nach dem Erwärmen wird die Lösung in den Messkolben filtriert, und Letzterer mit Wasser zur Marke aufgefüllt (HCl-Konzentration etwa 0,5 mol/l). Anmerkungen:
|
5.1.2.
5.1.2.1.
Aus den Gebrauchsstandardlösungen 3.7.1, 3.8.1, 3.9.1 und 3.10.1 werden für jedes der zu bestimmenden Spurenelemente eine Reihe von Kalibrierlösungen hergestellt. Jede dieser Kalibrierlösungen hat eine HCl-Konzentration von etwa 0,5 mol/l und (im Falle von Eisen, Mangan und Zink) einen Lanthanchlorid-Gehalt, der einer Massenkonzentration von 0,1 % La entspricht.
Die gewählten Spurenelementkonzentrationen müssen im Empfindlichkeitsbereich des verwendeten Spektralfotometers liegen. Die nachfolgenden Tabellen zeigen Beispiele für die Zusammensetzung typischer Reihen von Kalibrierlösungen. Je nach Typ und Empfindlichkeit des verwendeten Spektralfotometers kann es jedoch erforderlich sein, für die Kalibrierlösungen eine andere Konzentration zu wählen.
Eisen
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml Gebrauchsstandardlösung (3.7.1) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
10 ml Lanthanchloridlösung (3.11) zugeben und mit Wasser auf 100 ml auffüllen. |
|||||||
Kupfer
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml Gebrauchsstandardlösung (3.8.1) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
Mangan
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml Gebrauchsstandardlösung (3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
10 ml Lanthanchloridlösung (3.11) zugeben und mit Wasser auf 100 ml auffüllen. |
|||||||
Zink
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml Gebrauchsstandardlösung (3.10.1) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
10 ml Lanthanchloridlösung (3.11) zugeben und mit Wasser auf 100 ml auffüllen. |
|||||||
5.1.2.2.
Für die Bestimmung des Kupfergehalts kann die nach 5.1.1 hergestellte Analysenlösung in der Regel direkt verwendet werden. Gegebenenfalls wird ein aliquoter Teil dieser Analysenlösung in einen 100-ml-Messkolben pipettiert und mit 0,5 mol/l Salzsäure (3.3) zur Marke aufgefüllt, um ihre Konzentration in den Bereich der Kalibrierlösungen zu bringen.
Für die Bestimmung des Gehalts an Eisen, Mangan und Zink wird ein aliquoter Teil der nach 5.1.1 hergestellten Lösung in einen 100-ml-Messkolben pipettiert. Es werden 10 ml Lanthanchloridlösung (3.11) zugegeben und mit 0,5 mol/l Salzsäure (3.3) zur Marke aufgefüllt (siehe Bemerkung 8).
5.1.2.3.
Es wird ein Blindversuch ausgeführt, der alle Verfahrensschritte umfassen muss, nur dass das Probematerial selbst weggelassen wird. Die Kalibrierlösung „0“ ersetzt nicht den Blindversuch.
5.1.2.4.
Die Atomabsorption der Kalibrierlösungen und der Analysenlösungen ist mit oxydierender Luft-Azetylen-Flamme bei folgenden Wellenlängen zu messen:
|
|
Fe: 248,3 nm, |
|
|
Cu: 324,8 nm, |
|
|
Mn: 279,5 nm, |
|
|
Zn: 213,8 nm. |
Jede Messung ist 4-mal auszuführen.
5.2. Mineralfutter
Enthält die Probe keine organischen Substanzen, so erübrigt sich eine vorherige Veraschung. In diesem Fall wird dann ab 5.1.1.1 zweiter Absatz weiterverfahren. Ein Abrauchen mit Fluorwasserstoffsäure kann entfallen.
6. Berechnung der Ergebnisse
Die Spurenelementkonzentration in der Analysenlösung wird mithilfe einer Kalibrationskurve berechnet und das Ergebnis in mg Spurenelement/kg der Probe (ppm) ausgedrückt.
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen eines Analytikers bei ein und derselben Probe darf die folgenden Werte nicht überschreiten:
|
— |
5 mg/kg absolut für Spurenelementgehalte bis 50 mg/kg, |
|
— |
10 % des höheren Werts für Spurenelementgehalte von 50 bis 100 mg/kg, |
|
— |
10 mg/kg absolut für Spurenelementgehalte von 100 bis 200 mg/kg, |
|
— |
5 % des höheren Werts für Spurenelementgehalte von mehr als 200 mg/kg. |
8. Bemerkung
Das Vorhandensein von größeren Phosphatmengen kann die Bestimmung des Gehalts an Eisen, Mangan und Zink beeinträchtigen. Diese Interferenzen sind durch die Zugabe von Lanthanchloridlösung (3.11) zu korrigieren. Weist die Probe jedoch das Gewichtsverhältnis Ca + Mg/P > 2 auf, kann auf die Zugabe von Lanthanchloridlösung (3.11) zu der Analysenlösung und den Kalibrierlösungen verzichtet werden.
D. BESTIMMUNG DES HALOFUGINONGEHALTS
DL-trans-7-Brom-6-chlor-3-(3-(3-hydroxy-2-piperidyl)acetonyl)-4(3H)-chinazolinon-hydrobromid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Halofuginongehalts von Futtermitteln. Die Bestimmungsgrenze beträgt 1 mg/kg.
2. Prinzip
Nach der Behandlung mit heißem Wasser wird Halofuginon als freie Base mit Ethylacetat extrahiert und anschließend durch Ausschütteln mit Salzsäure in das Hydrochlorid überführt. Der Extrakt wird durch Ionenaustauschchromatografie gereinigt. Der Halofuginongehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
|
3.1. |
Acetonitril, HPLC-Qualität. |
|
3.2. |
Amberlite XAD-2-Harz. |
|
3.3. |
Ammoniumacetat. |
|
3.4. |
Ethylacetat. |
|
3.5. |
Essigsäure (Eisessig). |
|
3.6. |
Halofuginon-Standardsubstanz: DL-trans-7-Brom-6-chlor-3-[3-(3-hydroxy-2-piperidyl)acetonyl]-4(3H)-chinazolinon-hydrobromid, E 764 |
|
3.6.1. |
Von Halofuginon (3.6) werden 50 mg auf 0,1 mg genau in einen 500-ml-Messkolben eingewogen und in Ammoniumacetat-Pufferlösung (3.18) gelöst. Es wird mit der Pufferlösung zur Marke aufgefüllt und gemischt. Diese Lösung ist 3 Wochen haltbar, wenn sie im Dunkeln bei 5 oC aufbewahrt wird. |
|
3.6.2. |
Von der Standard-Stammlösung (3.6.1) werden 1,0, 2,0, 3,0, 4,0 bzw. 6,0 ml jeweils in einen 100-ml-Messkolben überführt. Es wird mit der mobilen Phase (3.21) zur Marke aufgefüllt und durchmischt. Diese Lösungen enthalten Halofuginon in Konzentrationen von 1,0, 2,0, 3,0, 4,0 bzw. 6,0 μg/ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden; |
|
3.7. |
Salzsäure (ρ20 = ca. 1,16 g/ml). |
|
3.8. |
Methanol. |
|
3.9. |
Silbernitrat. |
|
3.10. |
Natriumascorbat. |
|
3.11. |
Natriumcarbonat (Soda). |
|
3.12. |
Natriumchlorid. |
|
3.13. |
EDTA (Ethylendiamintetraessigsäure, Dinatriumsalz). |
|
3.14. |
Wasser, HPLC-Qualität. |
|
3.15. |
Natriumcarbonatlösung, c = 10 g/100 ml. |
|
3.16. |
Mit Natriumchlorid gesättigte Natriumcarbonatlösung, c = 5 g/100 ml 50 g Natriumcarbonat (3.11) werden in Wasser gelöst und auf 1 l verdünnt; dann wird Natriumchlorid (3.12) zugegeben, bis die Lösung gesättigt ist. |
|
3.17. |
Salzsäure, ca. 0,1 mol/l 10 ml Salzsäure (3.7) werden mit Wasser auf 1 l verdünnt. |
|
3.18. |
Ammoniumacetat-Pufferlösung, ca. 0,25 mol/l 19,3 g Ammoniumacetat (3.3) und 30 ml Essigsäure (3.5) werden in Wasser (3.14) gelöst und auf 1 l verdünnt. |
|
3.19. |
Vorbereitung des Amberlite XAD-2-Harzes Eine ausreichende Menge Amberlite (3.2) wird mit Wasser chloridfrei gewaschen. Die Prüfung der Waschflüssigkeit erfolgt mit Silbernitratlösung (3.20). Danach wird das Harz mit 50 ml Methanol (3.8) gewaschen. Das Methanol wird verworfen und das Harz in frischem Methanol aufbewahrt. |
|
3.20. |
Silbernitratlösung, ca. 0,1 mol/l 0,17 g Silbernitrat (3.9) werden in 10 ml Wasser gelöst. |
|
3.21. |
Mobile Phase für die HPLC: 500 ml Acetonitril (3.1) werden mit 300 ml Ammoniumacetat-Pufferlösung (3.18) und 1 200 ml Wasser (3.14) gemischt. Der pH-Wert wird mit Essigsäure (3.5) auf 4,3 eingestellt. Die Lösung wird durch einen 0,22-μm-Filter (4.8) filtriert und entgast (z. B. durch 10-minütige Ultraschallbehandlung). Diese Lösung ist 1 Monat lang haltbar, wenn sie in einem verschlossenen Gefäß im Dunkeln aufbewahrt wird. |
4. Geräte
|
4.1. |
Ultraschallbad. |
|
4.2. |
Rotationsverdampfer. |
|
4.3. |
Zentrifuge. |
|
4.4. |
HPLC-Einrichtung mit UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor |
|
4.4.1. |
HPLC-Trennsäule, 300 × 4 mm, C18, 10 μm Korngröße, oder vergleichbare Säule. |
|
4.5. |
Glassäule (300 × 10 mm) mit gesintertem Glasfilter und Absperrhahn. |
|
4.6. |
Glasfaserfilter, 150 mm Durchmesser. |
|
4.7. |
Membranfilter, 0,45 μm Porengröße. |
|
4.8. |
Membranfilter, 0,22 μm Porengröße. |
5. Verfahren
|
Anmerkung |
: |
Halofuginon ist als freie Base in Alkali- und Ethylacetat-Lösungen instabil. Es darf höchstens 30 min in Ethylacetat bleiben. |
5.1. Allgemeines
|
5.1.1. |
Zur Prüfung, dass weder Halofuginon noch Störsubstanzen vorhanden sind, ist eine Blindprobe zu untersuchen. |
|
5.1.2. |
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Halofuginon angereichert wurde. Die zugesetzte Menge an Halofuginon sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 3 mg/kg werden 300 μl der Standard-Stammlösung (3.6.1) zu 10 g der Blindprobe gegeben. Es wird gemischt und 10 min gewartet, bevor mit der Extraktion (5.2) fortgefahren wird.
|
5.2. Extraktion
Von der vorbereiteten Probe werden 10 g auf 0,1 g genau in ein 200-ml-Zentrifugenglas eingewogen. Es werden 0,5 g Natriumascorbat (3.10), 0,5 g EDTA (3.13) und 20 ml Wasser hinzugefügt, und es wird gemischt. Das Zentrifugenglas wird 5 min in ein 80 oC heißes Wasserbad gestellt. Nach dem Abkühlen auf Raumtemperatur werden 20 ml Natriumcarbonatlösung (3.15) hinzugefügt, und es wird gemischt. Unmittelbar danach werden 100 ml Ethylacetat (3.4) hinzugefügt, und es wird 15 s kräftig von Hand geschüttelt. Danach wird das Zentrifugenglas mit gelockertem Stopfen 3 min in ein Ultraschallbad (4.1) gestellt. Es wird 2 min zentrifugiert und die Ethylacetatphase durch einen Glasfaserfilter (4.6) in einen 500-ml-Scheidetrichter dekantiert. Die Extraktion der Probe wird mit weiteren 100 ml Ethylacetat wiederholt. Die vereinigten Extrakte werden 1 min lang mit 50 ml der mit Natriumchlorid gesättigten Natriumcarbonatlösung (3.16) gewaschen. Die wässrige Phase wird verworfen.
Die organische Phase wird 1 min mit 50 ml Salzsäure (3.17) extrahiert. Die untere Säurephase wird in einen 250-ml-Scheidetrichter abgelassen. Die organische Phase wird erneut 1,5 min mit weiteren 50 ml Salzsäure extrahiert. Die beiden Säureextrakte werden vereinigt und durch ca. 10 s langes Schütteln mit 10 ml Ethylacetat (3.4) gewaschen.
Die wässrige Phase wird quantitativ in einen 250-ml-Rundkolben überführt und die organische Phase verworfen. Das restliche in der sauren Lösung enthaltene Ethylacetat wird mithilfe des Rotationsverdampfers (4.2) entfernt. Die Temperatur des Wasserbads darf 40 oC nicht überschreiten. Bei einem Unterdruck von ca. 25 mbar wird das restliche Ethylacetat bei 38 oC innerhalb von 5 min entfernt.
5.3. Clean-up
5.3.1.
Für jeden Probenextrakt wird eine XAD-2-Säule vorbereitet. Von dem vorbereiteten Amberlite (3.19) werden 10 g mit Methanol (3.8) in eine Glassäule (4.5) eingefüllt. Auf das obere Ende des Harzbettes wird ein kleiner Glaswattebausch gebracht. Das Methanol wird aus der Säule ablaufen gelassen und das Harz mit 100 ml Wasser gewaschen. Sobald die Flüssigkeit das obere Ende des Harzbettes erreicht hat, wird der Absperrhahn geschlossen. Vor Gebrauch ist die Säule 10 min zu äquilibrieren. Die Säule darf nie trockenlaufen.
5.3.2.
Der Extrakt (5.2) wird quantitativ auf die vorbereitete Amberlitesäule (5.3.1) aufgebracht und eluiert. Das Eluat wird verworfen. Die Elutionsgeschwindigkeit darf 20 ml/min nicht überschreiten. Der Rundkolben wird mit 20 ml Salzsäure (3.17) gespült und die Austauschersäule mit der Spülflüssigkeit gewaschen. Die eventuell auf der Säule verbliebene saure Lösung wird mit einem Luftstrom restlos ausgeblasen. Die Waschlösungen werden verworfen. Es werden 100 ml Methanol (3.8) auf die Säule gegeben und ein Eluat von 5 bis 10 ml in einem 250-ml-Rundkolben aufgefangen. Das restliche Methanol wird 10 min lang zur Äquilibrierung auf dem Harz belassen; anschließend wird die Elution mit einer Elutionsgeschwindigkeit von höchstens 20 ml/min fortgesetzt und das Eluat in demselben Rundkolben aufgefangen. Das Methanol wird am Rotationsverdampfer (4.2) abgedampft, wobei die Temperatur des Wasserbads 40 oC nicht übersteigen darf. Der Rückstand wird unter Verwendung der mobilen Phase (3.21) quantitativ in einen 10-ml-Messkolben überführt. Es wird mit der mobilen Phase zur Marke aufgefüllt und gemischt. Ein aliquoter Teil wird durch einen Membranfilter (4.7) filtriert. Diese Lösung wird für die HPLC-Bestimmung (5.4) aufbewahrt.
5.4. HPLC-Bestimmung
5.4.1.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
HPLC-Trennsäule (4.4.1),
Mobile Phase für die HPLC (3.21),
Durchflussrate: 1,5 bis 2 ml/min,
Detektionswellenlänge: 243 nm,
Einspritzvolumen: 40 bis 100 μl.
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.6.2), die 3,0 μg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.4.2.
Jede Kalibrierlösung (3.6.2) wird mehrmals eingespritzt, und die Peakhöhen (-flächen) für die einzelnen Konzentrationen werden gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen oder -flächen der Kalibrierlösungen auf der Ordinate und die dazugehörigen Konzentrationen in μg/ml auf der Abszisse aufgetragen werden.
5.4.3.
Der Probenextrakt (5.3.2) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird, und die mittlere Peakhöhe (-fläche) der Halofuginonpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Peakhöhe (-fläche) der Halofuginonpeaks der Probenlösung wird anhand der Kalibrationskurve (5.4.2) die Konzentration der Probenlösung in μg/ml bestimmt.
Der Halofuginongehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:

wobei:
|
c |
= |
Halofuginonkonzentration der Probenlösung in μg/ml, |
|
m |
= |
Probeneinwaage in g. |
7. Überprüfung der Ergebnisse
7.1. Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung und der Kalibrierlösung (3.6.2), die 6,0 μg/ml enthält, verglichen werden.
7.1.1.
Eine Probenlösung wird mit einer geeigneten Menge einer Kalibrierlösung (3.6.2) versetzt. Die Menge des zugesetzten Halofuginons muss dem erwarteten Halofuginongehalt der Probenlösung entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Halofuginonpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2.
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
|
a) |
Die Wellenlängen bei maximaler Absorption des Proben- und des Standardspektrums an der Peakspitze des Chromatogramms müssen innerhalb eines Bereichs übereinstimmen, der durch das Auflösungsvermögen des Detektionssystems bestimmt wird. Für die Diodenarray-Detektion beträgt dieser Bereich in der Regel ± 2 nm. |
|
b) |
Zwischen 225 und 300 nm dürfen sich das Proben- und das Standardspektrum an den Peakspitzen des Chromatogramms in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den beiden Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Standardanalyten beträgt. |
|
c) |
Zwischen 225 und 300 nm dürfen sich die Spektren des Probenextrakts im Anstieg, an der Spitze und im Abstieg des Probenpeaks in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung der Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Spektrums am Peakmaximum beträgt. |
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf bei einem Halofuginongehalt von bis zu 3 mg/kg 0,5 mg/kg nicht überschreiten.
7.3. Wiederfindungsrate
Für eine angereicherte Blindprobe muss die Wiederfindung mindestens 80 % betragen.
8. Ergebnisse eines Ringversuchs
Es wurde ein Ringversuch (5) durchgeführt, bei dem 3 Proben von 8 Laboratorien untersucht wurden. Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst:
Ergebnisse
|
|
Probe A (Blindprobe) nach Erhalt |
Probe B (Mehl) |
Probe C (Pellets) |
||
|
|
|
Nach Erhalt |
Nach 2 Monaten |
Nach Erhalt |
Nach 2 Monaten |
|
Mittelwert [mg/kg] |
NN |
2,80 |
2,42 |
2,89 |
2,45 |
|
sR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
VKR [ %] |
— |
16 |
18 |
14 |
17 |
|
Wiederfindung [ %] |
|
86 |
74 |
88 |
75 |
|
NN |
= |
nicht nachweisbar |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
E. BESTIMMUNG DES ROBENIDINGEHALTS
1,3-bis[(4-Chlorobenzyliden)amino]-guanidin-hydrochlorid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Robenidingehalts von Futtermitteln. Die Bestimmungsgrenze beträgt 5 mg/kg.
2. Prinzip
Die Probe wird mit angesäuertem Methanol extrahiert. Der Extrakt wird getrocknet und ein aliquoter Teil zum Clean-up auf eine Aluminiumoxidsäule gegeben. Robenidin wird mit Methanol von der Säule eluiert, eingeengt und mit der mobilen Phase auf ein geeignetes Volumen gebracht. Der Robenidingehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
|
3.1. |
Methanol |
|
3.2. |
Angesäuertes Methanol In einen 500-ml-Messkolben werden 4,0 ml Salzsäure (ρ20 = 1,18 g/ml) überführt. Es wird mit Methanol (3.1) zur Marke aufgefüllt und durchmischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden. |
|
3.3. |
Acetonitril, HPLC-Qualität |
|
3.4. |
Molekularsieb Typ 3A, 8 bis 12 Mesh (Korngröße 1,6 bis 2,5 mm, kristallines Aluminiumsilicat, Porendurchmesser 0,3 nm). |
|
3.5. |
Aluminiumoxid, sauer, Aktivitätsstufe I, für die Säulenchromatografie 100 g Aluminiumoxid werden in ein geeignetes Gefäß gegeben und mit 2,0 ml Wasser versetzt. Das Gefäß wird verschlossen und etwa 20 min geschüttelt. Das Aluminiumoxid ist in einem gut verschlossenen Behälter aufzubewahren. |
|
3.6. |
Kaliumdihydrogenphosphatlösung, c = 0,025 mol/l In einem 1 000-ml-Messkolben werden 3,40 g Kaliumdihydrogenphosphat in Wasser (HPLC-Qualität) gelöst. Es wird zur Marke aufgefüllt und durchmischt. |
|
3.7. |
Dinatriumhydrogenphosphatlösung, c = 0,025 mol/l In einem 1 l-Messkolben werden 3,55 g wasserfreies Dinatriumhydrogenphosphat (oder 4,45 g Dihydrat oder 8,95 g Dodecahydrat) in Wasser (HPLC-Qualität) gelöst. Es wird zur Marke aufgefüllt und durchmischt. |
|
3.8. |
Mobile Phase für die HPLC Folgende Reagenzien werden gemischt:
Die Lösung wird durch einen 0,22-μm-Filter (4.6) filtriert und entgast (z. B. durch 10-minütige Ultraschallbehandlung). |
|
3.9. |
Standardsubstanz Robenidin 1,3-bis[(4-Chlorobenzyliden)amino]-guanidin-hydrochlorid, rein |
|
3.9.1. |
Von der Robenidin-Standardsubstanz (3.9) werden 30 mg auf 0,1 mg genau eingewogen, in einem 100-ml-Messkolben in angesäuertem Methanol (3.2) gelöst und mit demselben Lösungsmittel zur Marke aufgefüllt und durchmischt. Der Messkolben wird mit Aluminiumfolie umhüllt und im Dunkeln aufbewahrt. |
|
3.9.2. |
Von der Standard-Stammlösung (3.9.1) werden 10,0 ml in einen 250-ml-Messkolben überführt. Es wird mit der mobilen Phase (3.8) zur Marke aufgefüllt und durchmischt. Der Messkolben wird mit Aluminiumfolie umhüllt und im Dunkeln aufbewahrt. |
|
3.9.3. |
Von der Robenidin-Standardlösung (3.9.2) werden 5,0, 10,0, 15,0, 20,0 bzw. 25,0 ml jeweils in einen 50-ml-Messkolben überführt. Es wird mit der mobilen Phase (3.8) zur Marke aufgefüllt und durchmischt. Diese Lösungen enthalten Robenidin in Konzentrationen von 1,2, 2,4, 3,6, 4,8 bzw. 6,0 μg/ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden. |
|
3.10. |
Wasser, HPLC-Qualität |
4. Geräte
|
4.1. |
Glassäule Braunglas mit Absperrhahn und Reservoir mit einem Fassungsvermögen von ca. 150 ml, Innendurchmesser 10 bis 15 mm, Länge 250 mm. |
|
4.2. |
Mechanischer Schüttler oder Magnetrührer. |
|
4.3. |
Rotationsfilmverdampfer. |
|
4.4. |
HPLC-Einrichtung mit UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor mit einem Messbereich von 250 bis 400 nm |
|
4.4.1. |
HPLC-Trennsäule: 300 × 4 mm, C18, 10 μm Korngröße, oder vergleichbare Säule. |
|
4.5. |
Glasfaser-Filterpapier, z. B. Whatman GF/A oder gleichwertig. |
|
4.6. |
Membranfilter, 0,22 μm Porengröße. |
|
4.7. |
Membranfilter, 0,45 μm Porengröße. |
5. Verfahren
|
Anmerkung |
: |
Robenidin ist lichtempfindlich. Bei allen Verfahrensschritten sind Braunglasgeräte zu verwenden. |
5.1. Allgemeines
|
5.1.1. |
Zur Prüfung, dass weder Robenidin noch Störsubstanzen vorhanden sind, ist eine Blindprobe zu untersuchen. |
|
5.1.2. |
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe (5.1.1) untersucht wird, die mit Robenidin angereichert wurde. Die zugesetzte Menge an Robenidin sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 60 mg/kg werden 3,0 ml der Standard-Stammlösung (3.9.1) in einen 250-ml-Erlenmeyerkolben gegeben. Die Lösung wird in einem Stickstoffstrom auf ca. 0,5 ml eingeengt. Dann werden 15 g der Blindprobe zugegeben. Es wird gemischt und 10 min stehen gelassen, bevor mit der Extraktion (5.2) fortgefahren wird.
|
5.2. Extraktion
Von der vorbereiteten Probe werden 15 g auf 0,01 g genau in einen 250-ml-Erlenmeyerkolben eingewogen, mit 100,0 ml angesäuertem Methanol (3.2) versetzt und nach Verschließen des Kolbens 1 h auf dem Schüttler (4.2) geschüttelt. Die Lösung wird über Glasfaserfilterpapier (4.5) filtriert und das gesamte Filtrat in einem 150-ml-Erlenmeyerkolben aufgefangen. Es werden 7,5 g Molekularsieb (3.4) zugegeben, und nach Verschließen des Kolbens wird 5 min geschüttelt. Anschließend wird sofort durch ein Glasfaserfilterpapier filtriert. Diese Lösung wird für die Reinigung (5.3) aufbewahrt.
5.3. Reinigung
5.3.1.
In das untere Ende der Glassäule (4.1) wird ein Glaswattebausch eingebracht, der mit einem Glasstab zusammengedrückt wird. Von dem vorbereiteten Aluminiumoxid (3.5) werden 11,0 g auf die Säule aufgebracht. Dabei ist darauf zu achten, dass der Kontakt mit der Luft so gering wie möglich gehalten wird. Durch leichtes Klopfen an das untere Ende der befüllten Säule wird das Aluminiumoxid verdichtet.
5.3.2.
Von dem Probenextrakt (5.2) werden 5,0 ml mit einer Pipette auf die Säule gegeben, wobei die Pipettenspitze die Säulenwand berühren soll. Nach Absorption der Lösung durch das Aluminiumoxid wird das Robenidin mit 100 ml Methanol (3.1) bei einer Flussrate von 2-3 ml/min von der Säule eluiert und das Eluat in einem 250-ml-Rundkolben aufgefangen. Die Methanollösung wird am Rotationsverdampfer (4.3) bei 40 oC und unter vermindertem Druck bis zur Trockne eingeengt. Der Rückstand wird mit 3 bis 4 ml der mobilen Phase (3.8) erneut gelöst und quantitativ in einen 10-ml-Messkolben überführt. Der Kolben wird mehrmals mit je 1 bis 2 ml der mobilen Phase gespült, und die Spüllösungen werden in den Messkolben gegeben. Es wird mit der mobilen Phase zur Marke aufgefüllt und gemischt. Ein aliquoter Teil wird durch einen 0,45-μm-Membranfilter (4.7) filtriert. Diese Lösung wird für die HPLC-Bestimmung (5.4) aufbewahrt.
5.4. HPLC-Bestimmung
5.4.1.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
HPLC-Trennsäule (4.4.1),
mobile Phase für die HPLC (3.8),
Durchflussrate: 1,5 bis 2 ml/min,
Detektionswellenlänge: 317 nm,
Einspritzvolumen: 20 bis 50 μl).
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.9.3), die 3,6 μg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.4.2.
Jede Kalibrierlösung (3.9.3) wird mehrmals eingespritzt, und die Peakhöhen (-flächen) für die einzelnen Konzentrationen werden gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) der Kalibrierlösungen auf der Ordinate und die dazugehörigen Konzentrationen in μg/ml auf der Abszisse aufgetragen werden.
5.4.3.
Der Probenextrakt (5.3.2) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird, und die mittlere Peakhöhe (-fläche) der Robenidinpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Robenidinpeaks der Probenlösung wird anhand der Kalibrationskurve (5.4.2) die Konzentration der Probenlösung in μg/ml bestimmt.
Der Robenidingehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:

wobei:
|
c |
= |
Robenidinkonzentration der Probenlösung in μg/ml, |
|
m |
= |
Probeneinwaage in g. |
7. Überprüfung der Ergebnisse
7.1. Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung und der Kalibrierlösung (3.9.3), die 6 μg/ml enthält, verglichen werden.
7.1.1.
Eine Probenlösung wird mit einer geeigneten Menge Kalibrierlösung (3.9.3) versetzt. Die Menge des zugesetzten Robenidins muss dem erwarteten Robenidingehalt der Probenlösung entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Robenidinpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2.
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
|
a) |
Die Wellenlängen bei maximaler Absorption des Proben- und des Standardspektrums an der Peakspitze des Chromatogramms müssen innerhalb eines Bereichs übereinstimmen, der durch das Auflösungsvermögen des Detektionssystems bestimmt wird. Für die Diodenarray-Detektion beträgt dieser Bereich in der Regel ± 2 nm. |
|
b) |
Zwischen 250 und 400 nm dürfen sich das Proben- und das Standardspektrum an den Peakspitzen des Chromatogramms in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den beiden Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Standardanalyten beträgt. |
|
c) |
Zwischen 250 und 400 nm dürfen sich die Spektren des Probenextrakts im Anstieg, an der Spitze und im Abstieg des Probenpeaks in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung der Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Spektrums am Peakmaximum beträgt. |
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Bei einem Robenidingehalt von mehr als 15 mg/kg darf die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe 10 % des höheren Werts nicht überschreiten.
7.3. Wiederfindungsrate
Für eine angereicherte Blindprobe muss die Wiederfindungsrate mindestens 85 % betragen.
8. Ergebnisse eines Ringversuchs
Bei einem EG-Ringversuch wurden 4 Proben von Geflügel- und Kaninchenfutter in Form von Mehl oder Pellets von 12 Laboratorien analysiert. Jede Probe wurde doppelt analysiert. Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst:
|
|
Geflügelfutter |
Kaninchenfutter |
||
|
|
Mehl |
Pellets |
Mehl |
Pellets |
|
Mittelwert [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
VKr [ %] |
5,4 |
4,5 |
3,3 |
4,1 |
|
sR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
VKR [ %] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Wiederfindung [ %] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
F. BESTIMMUNG DES DICLAZURILGEHALTS
2,6-Dichlor-alpha-(4-chlorophenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3H)-yl)benzacetonitril
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Diclazurilgehalts von Futtermitteln und Vormischungen. Die Nachweisgrenze beträgt 0,1 mg/kg, die Bestimmungsgrenze 0,5 mg/kg.
2. Prinzip
Nach Hinzufügen eines internen Standards wird die Probe mit angesäuertem Methanol extrahiert. Bei Futtermitteln wird ein aliquoter Teil des Extrakts mit einer C18-Festphasen-Extraktionskartusche gereinigt. Diclazuril wird aus der Kartusche mit einem Gemisch aus angesäuertem Methanol und Wasser eluiert. Nach dem Einengen wird der Rückstand in DMF/Wasser gelöst. Bei Vormischungen wird der Extrakt eingeengt und der Rückstand in DMF/Wasser gelöst. Der Diclazurilgehalt wird mittels ternärer Gradienten-Hochleistungsflüssigkeitschromatografie (HPLC) über eine Umkehrphase unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
|
3.1. |
Wasser, HPLC-Qualität. |
|
3.2. |
Ammoniumacetat. |
|
3.3. |
Tetrabutylammoniumhydrogensulfat (TBHS). |
|
3.4. |
Acetonitril, HPLC-Qualität. |
|
3.5. |
Methanol, HPLC-Qualität. |
|
3.6. |
N,N-Dimethylformamid (DMF). |
|
3.7. |
Salzsäure, ρ20 = 1,19 g/ml. |
|
3.8. |
Standardsubstanz: Diclazuril II-24: 2,6-Dichlor-alpha-(4-chlorophenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3H)-yl)benzacetonitril, rein, E 771. |
|
3.8.1. |
Von der Diclazuril-Standardsubstanz (3.8) werden 25 mg auf 0,1 mg genau in einen 50-ml-Messkolben eingewogen und in DMF (3.6) gelöst; es wird mit DMF (3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 oC ist diese Lösung 1 Monat haltbar. |
|
3.8.2. |
Von der Standard-Stammlösung (3.8.1) werden 5,00 ml in einen 50-ml-Messkolben überführt; es wird mit DMF (3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 oC ist diese Lösung 1 Monat haltbar. |
|
3.9. |
Interne Standardsubstanz: 2,6 Dichloro-α-(4-chlorophenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3H)-yl)-α-methylbenzacetonitril |
|
3.9.1. |
Von der internen Standardsubstanz (3.9) werden 25 mg auf 0,1 mg genau in einen 50-ml-Messkolben eingewogen und in DMF (3.6) gelöst; es wird mit DMF (3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 oC ist diese Lösung 1 Monat haltbar. |
|
3.9.2. |
Von der internen Standard-Stammlösung (3.9.1.) werden 5,00 ml in einen 50-ml-Messkolben überführt; es wird mit DMF (3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 oC ist diese Lösung 1 Monat haltbar. |
|
3.9.3. |
(p = Diclazuril-Sollgehalt in der Vormischung in mg/kg) Von der internen Standardsubstanz werden p/10 mg auf 0,1 mg genau in einen 100-ml-Messkolben eingewogen und mittels Ultraschallbad (4.6) in DMF (3.6) gelöst; es wird mit DMF (3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 oC ist diese Lösung 1 Monat haltbar. |
|
3.10. |
Kalibrierlösung, 2 μg/ml In einen 50-ml-Messkolben werden 2,00 ml Diclazuril-Standardlösung (3.8.2) und 2,00 ml interne Standardlösung (3.9.2) pipettiert. Es werden 16 ml DMF (3.6) hinzugefügt; anschließend wird mit Wasser zur Marke aufgefüllt und gemischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden. |
|
3.11. |
C18-Festphasen-Extraktionskartusche, z. B. Bond Elut, Größe: 1 cc, Sorptionsmenge: 100 mg. |
|
3.12. |
Extraktionslösung: angesäuertes Methanol 5,0 ml Salzsäure (3.7) werden in 1 000 ml Methanol (3.5) pipettiert und gemischt. |
|
3.13. |
Mobile Phase für die HPLC: |
|
3.13.1. |
Elutionsmittel A: Ammoniumacetat-Tetrabutylammoniumhydrogensulfat-Lösung, 5 g Ammoniumacetat (3.2) und 3,4 g TBHS (3.3) werden in 1 000 ml Wasser (3.1) gelöst und gemischt, |
|
3.13.2. |
Elutionsmittel B: Acetonitril (3.4), |
|
3.13.3. |
Elutionsmittel C: Methanol (3.5). |
4. Geräte
|
4.1. |
Mechanischer Schüttler. |
|
4.2. |
HPLC-Einrichtung für ternären Gradienten |
|
4.2.1. |
HPLC-Trennsäule, 3 μm Korngröße, 100 × 4,6 mm, z. B. Hypersil ODS, oder vergleichbare Säule. |
|
4.2.2. |
UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor. |
|
4.3. |
Rotationsfilmverdampfer. |
|
4.4. |
Membranfilter, 0,45 μm Porengröße. |
|
4.5. |
Vakuumeinrichtung. |
|
4.6. |
Ultraschallbad. |
5. Verfahren
5.1. Allgemeines
5.1.1.
Zur Prüfung, dass weder Diclazuril noch Störsubstanzen vorhanden sind, ist eine Blindprobe zu untersuchen. Die Blindprobe muss ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Diclazuril oder Störsubstanzen dürfen nicht nachweisbar sein.
5.1.2.
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Diclazuril angereichert wurde. Die zugesetzte Menge sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 1 mg/kg werden 0,1 ml der Standard-Stammlösung (3.8.1) zu 50 g der Blindprobe gegeben. Es wird gründlich gemischt, 10 min stehen gelassen und nochmals mehrfach gemischt, bevor mit der Extraktion (5.2) fortgefahren wird.
Ist eine der zu untersuchenden Probe ähnliche Blindprobe nicht verfügbar (siehe 5.1.1), so kann ein Wiederfindungstest mithilfe des Additionsverfahrens durchgeführt werden. In diesem Fall wird die zu untersuchende Probe mit einer Diclazurilmenge angereichert, die der bereits in der Probe vorhandenen Menge entspricht. Diese Probe wird zusammen mit der nicht angereicherten Probe untersucht, und die Wiederfindungsrate kann durch Subtraktion ermittelt werden.
5.2. Extraktion
5.2.1.
Von der Probe werden 50 g auf 0,01 g genau eingewogen und in einen 500-ml-Erlenmeyerkolben überführt. Es werden 1,00 ml der internen Standardlösung (3.9.2) und 200 ml Extraktionslösung (3.12) hinzugefügt. Anschließend wird der Kolben verschlossen. Die Mischung wird über Nacht auf dem Schüttler (4.1) geschüttelt und anschließend 10 min stehen gelassen. Ein aliquoter Teil von 20 ml der überstehenden Lösung wird in einen geeigneten Glasbehälter überführt und mit 20 ml Wasser verdünnt. Diese Lösung wird auf eine Extraktionskartusche (3.11) gegeben und mittels Vakuum (4.5) hindurch gesaugt. Die Kartusche wird mit 25 ml einer Mischung aus Extraktionslösung (3.12) und Wasser, 65 + 35 (V+V), ausgewaschen. Die gesammelten Fraktionen werden verworfen, und der Wirkstoff und der interne Standard werden mit 25 ml einer Mischung aus Extraktionslösung (3.12) und Wasser, 80 + 20 (V+V), eluiert. Diese Fraktion wird mithilfe eines Rotationsverdampfers (4.3) bei 60 oC zur Trockne eingeengt. Der Rückstand wird in 1,0 ml DMF (3.6) gelöst; es werden 1,5 ml Wasser (3.1) hinzugefügt, gemischt und durch einen Membranfilter (4.4) filtriert. Anschließend wird die HPLC-Bestimmung (5.3) durchgeführt.
5.2.2.
Von der Probe wird 1 g auf 0,001 g genau eingewogen und in einen 500-ml-Erlenmeyerkolben überführt. Es werden 1,00 ml interne Standardlösung (3.9.3) und 200 ml Extraktionslösung (3.12) hinzugefügt, und der Kolben wird verschlossen. Die Mischung wird über Nacht auf dem Schüttler (4.1) geschüttelt und anschließend 10 min stehen gelassen. Ein 10 000/p-ml-Aliquot (p = Diclazuril-Sollgehalt in der Vormischung in mg/kg) der überstehenden Lösung wird in einen Rundkolben geeigneter Größe überführt. Es wird mithilfe eines Rotationsverdampfers (4.3) bei vermindertem Druck und 60 oC zur Trockne eingeengt. Der Rückstand wird in 10,0 ml DMF (3.6) gelöst; es werden 15,0 ml Wasser (3.1) hinzugefügt und gemischt. Anschließend wird die HPLC-Bestimmung (5.3) durchgeführt.
5.3. HPLC-Bestimmung
5.3.1.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
|
HPLC-Trennsäule (4.2.1.): |
100 × 4,6 mm, Hypersil ODS, 3 μm Korngröße, oder vergleichbare Säule |
|
||||||
|
Mobile Phase: |
Elutionsmittel A (3.13.1): |
wässrige Lösung von Ammoniumacetat und Tetrabutylammonium-Hydrogensulfat |
||||||
|
|
Elutionsmittel B (3.13.2): |
Acetonitril |
||||||
|
|
Elutionsmittel C (3.13.3): |
Methanol |
||||||
|
Elutionsmodus: |
Mit Elutionsmittel B 10 min spülen. |
|||||||
|
Durchflussrate: |
1,5 bis 2 ml/min |
|
||||||
|
Einspritzvolumen: |
20 μl |
|
||||||
|
Detektionswellenlänge: |
280 nm |
|
||||||
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.10), die 2 μg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.3.2.
Von der Kalibrierlösung (3.10) werden 20 μl mehrmals eingespritzt, und für Diclazuril und den internen Standard wird die mittlere Peakhöhe (-fläche) bestimmt.
5.3.3.
Von der Probenlösung (5.2.1 oder 5.2.2) werden mehrmals 20 μl eingespritzt, und für Diclazuril und den internen Standard wird die mittlere Peakhöhe (-fläche) bestimmt.
6. Berechnung der Ergebnisse
6.1. Futtermittel
Der Diclazurilgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
 [mg/kg]
[mg/kg]
wobei:
|
hd,s |
= |
Peakhöhe (-fläche) von Diclazuril in der Probenlösung (5.2.1), |
|
hi,s |
= |
Peakhöhe (-fläche) des internen Standards in der Probenlösung (5.2.1), |
|
hd,c |
= |
Peakhöhe (-fläche) von Diclazuril in der Kalibrierlösung (3.10), |
|
hi,c |
= |
Peakhöhe (-fläche) des internen Standards in der Kalibrierlösung (3.10), |
|
cd,c |
= |
Diclazurilkonzentration in der Kalibrierlösung in μg/ml (3.10), |
|
m |
= |
Probeneinwaage in g, |
|
V |
= |
Volumen der Probenextrakts gemäß 5.2.1 (d. h. 2,5 ml). |
6.2. Vormischungen
Der Diclazurilgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
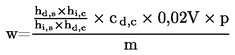 [mg/kg]
[mg/kg]
wobei:
|
hd,c |
= |
Peakhöhe (-fläche) von Diclazuril in der Kalibrierlösung (3.10), |
|
hi,c |
= |
Peakhöhe (-fläche) des internen Standards in der Kalibrierlösung (3.10), |
|
hd,s |
= |
Peakhöhe (-fläche) von Diclazuril in der Probenlösung (5.2.2), |
|
hi,s |
= |
Peakhöhe (-fläche) des internen Standards in der Probenlösung (5.2.2), |
|
cd,c |
= |
Diclazurilkonzentration in der Kalibrierlösung in μg/ml (3.10), |
|
m |
= |
Probeneinwaage in g, |
|
V |
= |
Volumen des Probenextrakts gemäß 5.2.2 (d. h. 25 ml), |
|
p |
= |
Diclazuril-Sollgehalt in der Vormischung in mg/kg. |
7. Überprüfung der Ergebnisse
7.1. Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung (5.2.1 oder 5.2.2) und der Kalibrierlösung (3.10) verglichen werden.
7.1.1.
Eine nach 5.2.1 oder 5.2.2 hergestellte Probenlösung wird mit einer geeigneten Menge Kalibrierlösung (3.10) angereichert. Die Menge des zugesetzten Diclazurils muss dem erwarteten Diclazurilgehalt der Probenlösung entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Diclazurilpeaks und des Peaks des internen Standards vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des ursprünglichen Diclazurilpeaks oder Standardpeaks des nicht angereicherten Probenextrakts abweichen.
7.1.2.
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
|
a) |
Die Wellenlängen bei maximaler Absorption des Proben- und des Standardspektrums an der Peakspitze des Chromatogramms müssen innerhalb eines Bereichs übereinstimmen, der durch das Auflösungsvermögen des Detektionssystems bestimmt wird. Für die Diodenarray-Detektion beträgt dieser Bereich in der Regel ± 2 nm. |
|
b) |
Zwischen 230 und 320 nm dürfen sich das Proben- und das Standardspektrum an den Peakspitzen des Chromatogramms in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den beiden Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Standardanalyten beträgt. |
|
c) |
Zwischen 230 und 320 nm dürfen sich die Spektren des Probenextrakts im Anstieg, an der Spitze und im Abstieg des Probenpeaks in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Spektrums am Peakmaximum beträgt. |
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf folgende Werte nicht überschreiten:
|
— |
30 % relativ zum höheren Wert bei Diclazurilgehalten zwischen 0,5 und 2,5 mg/kg, |
|
— |
0,75 mg/kg bei Diclazurilgehalten zwischen 2,5 und 5 mg/kg, |
|
— |
15 % relativ zum höheren Wert bei Diclazurilgehalten von mehr als 5 mg/kg. |
7.3. Wiederfindungsrate
Bei einer angereicherten (Blind-)Probe muss die Wiederfindungsrate mindestens 80 % betragen.
8. Ergebnisse eines Ringversuchs
Bei einem Ringversuch wurden 5 Proben von 11 Laboratorien untersucht. Diese Proben bestanden aus 2 Vormischungen, von denen eine mit einer organischen Matrix (O 100) und die andere mit einer anorganischen Matrix (A 100) gemischt war. Der theoretische Diclazurilgehalt liegt bei 100 mg/kg. Die 3 Geflügelmischfuttermittel stammten von 3 verschiedenen Herstellern (NL) (L1/Z1/K1). Der theoretische Diclazurilgehalt liegt bei 1 mg/kg. Die Laboratorien wurden beauftragt, jede der Proben 1- oder 2-mal zu untersuchen. (Nähere Informationen zu diesem Ringversuch sind dem Journal of AOAC International, Band 77, Nr. 6, 1994, S. 1359-1361, zu entnehmen.) Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst:
|
|
Probe 1 A 100 |
Probe 2 O 100 |
Probe 3 L1 |
Probe 4 Z1 |
Probe 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Mittelwert |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
VKr ( %) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
sR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
VKR ( %) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Sollgehalt (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
Anzahl der Laboratorien |
|
n |
= |
Anzahl der Einzelwerte |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
9. Bemerkungen
Es muss vorab erwiesen sein, dass das Diclazurilsignal im linearen Konzentrationsbereich liegt.
G. BESTIMMUNG DES GEHALTS AN LASALOCID-NATRIUM
Monocarboxylsäure-Polyether-Natriumsalz, gebildet durch Streptomyces lasaliensis
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Lasalocid-Natrium in Futtermitteln und Vormischungen. Die Nachweisgrenze beträgt 5 mg/kg, die Bestimmungsgrenze 10 mg/kg.
2. Prinzip
Lasalocid-Natrium wird aus der Probe mit angesäuertem Methanol extrahiert und mittels Umkehrphasen-Hochleistungsflüssigchromatografie (RP-HPLC) unter Verwendung eines spektrofluorometrischen Detektors bestimmt.
3. Reagenzien
|
3.1. |
Kaliumdihydrogenphosphat (KH2PO4). |
|
3.2. |
Orthophosphorsäure, w (Massenanteil) = 85 %. |
|
3.3. |
Orthophosphorsäurelösung, c = 20 % 23,5 ml Orthophosphorsäure (3.2) werden mit Wasser auf 100 ml aufgefüllt. |
|
3.4. |
6-Methyl-2-Heptylamin (1,5-Dimethylhexylamin), w (Massenanteil) = 99 %. |
|
3.5. |
Methanol, HPLC-Qualität. |
|
3.6. |
Salzsäure, Dichte = 1,19 g/ml. |
|
3.7. |
Phosphatpufferlösung, c = 0,01 mol/l In 500 ml Wasser (3.11) werden 1,36 g KH2PO4 (3.1) gelöst, und es werden 3,5 ml Orthophosphorsäure (3.2) sowie 10,0 ml 6-Methyl-2-Heptylamin (3.4) hinzugefügt. Den pH-Wert mit Orthophosphorsäurelösung (3.3) auf pH 4,0 einstellen und mit Wasser (3.11) auf 1 000 ml auffüllen. |
|
3.8. |
Angesäuertes Methanol In einen 1 000-ml-Messkolben werden 5,0 ml Salzsäure (3.6) gegeben, und es wird mit Methanol (3.5) zur Marke aufgefüllt und gemischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden. |
|
3.9. |
Mobile Phase für die HPLC: Phosphatpuffer-Methanollösung 5 + 95 (V+V) Von der Phosphatpufferlösung (3.7) werden 5 ml mit 95 ml Methanol (3.5) vermischt. |
|
3.10. |
Lasalocid-Natrium-Standardsubstanz, garantiert rein, C34H53O8Na (Monocarboxylsäure-Polyether-Natriumsalz, gebildet durch Streptomyces lasaliensis), E 763 |
|
3.10.1. |
Von Lasalocid-Natrium (3.10) werden 50 mg auf 0,1 mg genau in einen 100-ml-Messkolben eingewogen und in angesäuertem Methanol (3.8) gelöst. Es wird mit demselben Lösungsmittel zur Marke aufgefüllt und gemischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden. |
|
3.10.2. |
Von der Standard-Stammlösung (3.10.1) werden 10,0 ml in einen 100-ml-Messkolben pipettiert; es wird mit angesäuertem Methanol (3.8) zur Marke aufgefüllt und gemischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden. |
|
3.10.3. |
Von der Lasalocid-Natrium-Standardlösung (3.10.2) werden 1,0, 2,0, 4,0, 5,0 bzw. 10,0 ml in jeweils einen 50-ml-Messkolben überführt. Es wird mit angesäuertem Methanol (3.8) zur Marke aufgefüllt und durchmischt. Diese Kalibrierlösungen enthalten jeweils 1,0, 2,0, 4,0, 5,0 bzw. 10,0 μg Lasalocid-Natrium je ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden. |
|
3.11. |
Wasser, HPLC-Qualität. |
4. Geräte
|
4.1. |
Ultraschallbad (oder Schüttel-Wasserbad) mit Temperatursteuerung. |
|
4.2. |
Membranfilter, 0,45 μm Porengröße. |
|
4.3. |
HPLC-Einrichtung mit Injektionssystem für Einspritzvolumina von 20 μl |
|
4.3.1. |
HPLC-Trennsäule, 125 × 4 mm, mit Umkehrphase C18, 5 μm Korngröße, oder vergleichbare Säule. |
|
4.3.2. |
Spektrofluorometer mit variabler Einstellung für Anregungs- und Emissionswellenlängen. |
5. Verfahren
5.1. Allgemeines
5.1.1.
Für die Durchführung des Wiederfindungstests (5.1.2) ist zur Prüfung, dass weder Lasalocid-Natrium noch Störsubstanzen vorhanden sind, eine Blindprobe zu untersuchen. Die Blindprobe muss ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Lasalocid-Natrium oder Störsubstanzen dürfen nicht nachweisbar sein.
5.1.2.
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Lasalocid-Natrium angereichert wurde. Die zugesetzte Menge sollte in etwa der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 100 mg/kg werden 10,0 ml der Standard-Stammlösung (3.10.1) in einen 250-ml-Erlenmeyerkolben überführt. Die Lösung wird auf ca. 0,5 ml eingeengt. Dann werden 50 g der Blindprobe zugegeben. Es wird gründlich gemischt, 10 min stehen gelassen und erneut mehrmals gemischt, bevor mit der Extraktion (5.2) fortgefahren wird.
Ist eine der zu untersuchenden Probe ähnliche Blindprobe nicht verfügbar (siehe 5.1.1), so kann ein Wiederfindungstest mithilfe des Additionsverfahrens durchgeführt werden. In diesem Fall wird die zu untersuchende Probe mit einer Lasalocid-Natrium-Menge angereichert, die in etwa der bereits in der Probe vorhandenen Menge entspricht. Diese Probe wird zusammen mit der nicht angereicherten Probe untersucht und die Wiederfindungsrate kann durch Subtraktion ermittelt werden.
5.2. Extraktion
5.2.1.
Von der Probe werden 5 bis 10 g auf 0,01 g genau in einen 250-ml-Erlenmeyerkolben mit Stopfen eingewogen und 100,0 ml angesäuertes Methanol (3.8) mit einer Pipette hinzugefügt. Der Stopfen wird lose aufgesetzt und der Inhalt zum Dispergieren geschwenkt. Der Kolben wird 20 min in ein Ultraschallbad (4.1) mit einer Temperatur von ca. 40 oC gestellt, dann entnommen und auf Raumtemperatur abkühlen gelassen. Der Kolben wird etwa 1 h stehen gelassen, bis die Schwebstoffe sich abgesetzt haben. Dann wird ein aliquoter Teil über einen 0,45-μm-Membranfilter (4.2) in ein geeignetes Gefäß filtriert. Anschließend wird die HPLC-Bestimmung (5.3) durchgeführt.
5.2.2.
Rund 2 g der nicht gemahlenen Vormischung werden auf 0,001 g genau in einen 250-ml-Messkolben gegeben. Es werden 100,0 ml angesäuertes Methanol (3.8) hinzugefügt; der Inhalt wird zum Dispergieren geschwenkt. Der Kolben wird mit Inhalt 20 min lang in ein Ultraschallbad (4.1) mit einer Temperatur von ca. 40 oC gestellt, dann entnommen und auf Raumtemperatur abkühlen gelassen. Es wird mit angesäuertem Methanol (3.8) zur Marke aufgefüllt und sorgfältig gemischt. Der Kolben wird etwa 1 h stehen gelassen, bis die Schwebstoffe sich abgesetzt haben. Dann wird ein aliquoter Teil durch einen 0,45-μm-Membranfilter (4.2) filtriert. Ein aliquoter Teil des klaren Filtrats wird mit angesäuertem Methanol (3.8) verdünnt, um eine Lösung mit einer Konzentration von etwa 4 μg/ml Lasalocid-Natrium zu erhalten. Anschließend wird die HPLC-Bestimmung (5.3) durchgeführt.
5.3. HPLC-Bestimmung
5.3.1.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
|
HPLC-Trennsäule (4.3.1): |
125 × 4 mm, Umkehrphase C18, 5 μm Korngröße, oder vergleichbare Säule |
|
Mobile Phase (3.9) |
Mischung aus Phosphatpufferlösung (3.7) und Methanol (3.5), 5 + 95 (V+V) |
|
Durchflussrate: |
1,2 ml/min |
|
Detektionswellenlänge: |
|
|
Anregung: |
310 nm |
|
Emission |
419 nm |
|
Einspritzvolumen: |
20 μl |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.10.3), die 4,0 μg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.3.2.
Jede Kalibrierlösung (3.10.3) wird mehrmals eingespritzt, und es werden die mittleren Peakhöhen (-flächen) für die einzelnen Konzentrationen gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) auf der Ordinate und die dazugehörigen Konzentrationen in μg/ml auf der Abszisse aufgetragen werden.
5.3.3.
Die nach 5.2.1 bzw. 5.2.2 gewonnenen Probenextrakte werden mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird. Die mittlere Peakhöhe (-fläche) der Lasalocid-Natrium-Peaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Peakhöhe (-fläche) der Probenlösung (5.3.3) wird anhand der Kalibrationskurve die Konzentration an Lasalocid-Natrium (μg/ml) bestimmt.
6.1. Futtermittel
Der Gehalt an Lasalocid-Natrium w (in mg/kg) der Probe wird nach folgender Formel berechnet:
 [mg/kg]
[mg/kg]
wobei:
|
c |
= |
Lasalocid-Natrium-Konzentration der Probenlösung (5.2.1) in μg/ml, |
|
V1 |
= |
Volumen des Probenextrakts gemäß 5.2.1 (d. h. 100 ml), |
|
m |
= |
Probeneinwaage in g. |
6.2. Vormischungen
Der Gehalt an Lasalocid-Natrium w (in mg/kg) der Probe wird nach folgender Formel berechnet:
 [mg/kg]
[mg/kg]
wobei:
|
c |
= |
Lasalocid-Natrium-Konzentration der Probenlösung (5.2.2) in μg/ml, |
|
V2 |
= |
Volumen des Probenextrakts gemäß 5.2.2 in ml (d. h. 250 ml), |
|
f |
= |
Verdünnungsfaktor gemäß 5.2.2, |
|
m |
= |
Probeneinwaage in g. |
7. Überprüfung der Ergebnisse
7.1. Identität
Nachweis- und Bestimmungsverfahren, denen die Fluoreszenzdetektion zugrunde liegt, sind im Vergleich zur UV-Detektion weniger interferenzanfällig. Die Identität des Analyten kann durch Co-Chromatografie bestätigt werden.
7.1.1.
Ein Probenextrakt (5.2.1 oder 5.2.2) wird mit einer entsprechenden Menge Kalibrierlösung (3.10.3) angereichert. Die zugesetzte Lasalocid-Natrium-Menge muss in etwa dem Lasalocid-Natrium-Gehalt des Probenextrakts entsprechen. Unter Berücksichtigung der zugesetzten Lasalocid-Natrium-Menge und der Verdünnung des Extrakts darf nur die Höhe des Lasalocid-Natrium-Peaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der ursprünglichen Peakbreite abweichen, die sich bei dem nicht angereicherten Probenextrakt ergibt.
7.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
|
— |
15 % relativ zum höheren Wert bei Lasalocid-Natrium-Gehalten zwischen 30 und 100 mg/kg, |
|
— |
15 mg/kg bei Lasalocid-Natrium-Gehalten zwischen 100 und 200 mg/kg, |
|
— |
7,5 % des höheren Werts bei Lasalocid-Natrium-Gehalten von mehr als 200 mg/kg. |
7.3. Wiederfindungsrate
Bei einer angereicherten (Blind-)Probe muss die Wiederfindungsrate bei Futtermitteln mindestens 80 % betragen. Bei angereicherten Vormischungsproben muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
In einem Ringversuch (6) wurden 2 Vormischungen (Proben 1 und 2) und 5 Futtermittel (Proben 3 bis 7) in 12 Laboratorien untersucht. Jede Probe wurde doppelt analysiert. Die Ergebnisse sind in nachstehender Tabelle zusammengefasst:
|
|
Probe 1 Vormischung Hühnerfutter |
Probe 2 Vormischung Truthahnfutter |
Probe 3 Truthahnpellets |
Probe 4 Hühnerkrümelfutter |
Probe 5 Truthahnfutter |
Probe 6 Geflügelfutter A |
Probe 7 Geflügelfutter B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Mittelwert [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
VKr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
sR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
VKR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Sollgehalt [mg/kg] |
5 000 (7) |
16 000 (7) |
80 (7) |
105 (7) |
120 (7) |
50 (8) |
35 (8) |
|
L |
= |
Anzahl der Laboratorien |
|
n |
= |
Anzahl der Einzelwerte |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
(1) Durchgeführt von der Fachgruppe Futtermittel des Verbands deutscher landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(2) Durchgeführt von der Fachgruppe Futtermittel des Verbands deutscher landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(3) Andere Aufschlussmethoden können verwendet werden, sofern erwiesen ist, dass sie zu vergleichbaren Ergebnissen führen (z. B. Mikrowellen-Druckaufschluss).
(4) Grünfutter (frisch oder getrocknet) kann größere Mengen pflanzlicher Kieselsäure enthalten, welche Spurenelemente binden kann und daher entfernt werden muss. Bei Proben dieser Futtermittel muss also nach folgendem geänderten Verfahren vorgegangen werden: Das Verfahren 5.1.1.1 wird bis zur Filtration durchgeführt. Das den unlöslichen Rückstand enthaltende Filterpapier wird 2-mal mit kochendem Wasser ausgewaschen, in eine Quarz- oder Platinschale gegeben und im Muffelofen (4.1) bei weniger als 550 oC bis zur vollständigen Elimination aller Kohlepartikel verascht. Nach dem Abkühlen werden einige Tropfen Wasser und danach 10-15 ml Fluorwasserstoffsäure (3.4) zugegeben und bei ca. 150 oC zur Trockne eingedampft. Verbleibt im Rückstand noch Kieselsäure, so wird diese erneut in einigen ml Fluorwasserstoffsäure (3.4) gelöst und anschließend zur Trockne eingedampft. Dann werden 5 Tropfen Schwefelsäure (3.5) zugesetzt und so lange erhitzt, bis keine weißen Dämpfe mehr auftreten. Nach Zugabe von 5 ml 6 mol/l Salzsäure (3.2) und etwa 30 ml Wasser wird erhitzt und die Lösung in den 250-ml-Messkolben filtriert. Letzterer wird mit Wasser zur Marke aufgefüllt (HCl-Konzentration etwa 0,5 mol/l). Anschließend wird wie ab 5.1.2 beschrieben weiterverfahren.
(5) The Analyst 108, 1983, S. 1252-1256.
(6) Analyst, 1995, 120, S. 2175-2180.
(7) Gehalt nach Angabe des Herstellers.
(8) Im Laboratorium zubereitetes Futter.
ANHANG V
ANALYSEMETHODEN ZUR UNTERSUCHUNG VON FUTTERMITTELN AUF UNERWÜNSCHTE STOFFE
A. BESTIMMUNG DES GEHALTS AN FREIEM UND GESAMTGOSSYPOL
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an freiem Gossypol, Gesamtgossypol und chemisch verwandten Substanzen in Samen, Mehl und Kuchen von Baumwollsaat sowie in Mischfuttermitteln, die diese Futtermittel-Ausgangsstoffe enthalten, sofern mehr als 20 mg/kg an freiem Gossypol, Gesamtgossypol und chemisch verwandten Substanzen vorhanden sind.
2. Prinzip
Gossypol wird in Gegenwart von 3-Amino-1-Propanol entweder mit einer Isopropanol-Hexan-Mischung (zur Bestimmung des Gehalts an freiem Gossypol) oder mit Dimethylformamid (zur Bestimmung des Gesamtgossypolgehalts) extrahiert und mittels Anilin zu Gossypoldianilin umgesetzt, dessen Extinktion bei 440 nm gemessen wird.
3. Reagenzien
|
3.1. |
Isopropanol-Hexan-Mischung: Isopropanol (Volumenanteil = 60 %) wird mit n-Hexan (Volumenanteil = 40 % gemischt. |
|
3.2. |
Lösungsmittel A: In einen 1-l-Messkolben werden ca. 500 ml Isopropanol-Hexan-Mischung (3.1), 2 ml 3-Amino-1-Propanol, 8 ml Eisessig und 50 ml Wasser gegeben, und es wird mit Isopropanol-Hexan-Mischung (3.1) zur Marke aufgefüllt. Dieses Reagenz ist 1 Woche lang haltbar. |
|
3.3. |
Lösungsmittel B: In einen 100-ml-Messkolben werden 2 ml 3-Amino-1-Propanol und 10 ml Eisessig pipettiert, auf Raumtemperatur abgekühlt und mit N, N-Dimethylformamid zur Marke aufgefüllt. Dieses Reagenz ist 1 Woche lang haltbar. |
|
3.4. |
Anilin: Wenn die Extinktion im Blindversuch 0,022 überschreitet, ist das Anilin über Zinkstaub unter Verwerfung der ersten und der letzten 10 % des Destillats zu destillieren. In einem braunen, geschlossenen Gefäß ist dieses Reagenz im Kühlschrank einige Monate haltbar. |
|
3.5. |
Gossypol-Standardlösung A: In einen 250-ml-Messkolben werden 27,9 mg Gossypol-Acetat eingewogen und mit Lösungsmittel A (3.2) gelöst. Dann wird mit Lösungsmittel A (3.2) zur Marke aufgefüllt. Von dieser Lösung werden 50 ml in einen 250-ml-Messkolben pipettiert, und es wird mit Lösungsmittel A (3.2) zur Marke aufgefüllt. Die Gossypol-Konzentration dieser Lösung beträgt 0,02 mg/ml. Vor Gebrauch 1 h lang bei Raumtemperatur stehen lassen. |
|
3.6. |
Gossypol-Standardlösung B: In einen 50-ml-Messkolben werden 27,9 mg Gossypol-Acetat eingewogen und mit Lösungsmittel B (3.3) gelöst. Dann wird mit Lösungsmittel B (3.3) zur Marke aufgefüllt. Die Gossypol-Konzentration dieser Lösung beträgt 0,5 mg/ml. Die Standard-Gossypol-Lösungen A und B sind vor Lichteinwirkung geschützt 24 h lang haltbar. |
4. Geräte
|
4.1. |
Mechanisches Schüttelgerät, ca. 35 min-1. |
|
4.2. |
Spektralfotometer. |
5. Verfahren
5.1. Einwaage
Die Menge der Einwaage richtet sich nach dem vermuteten Gehalt an Gossypol in der Probe. Dabei sollte vorzugsweise mit einer geringen Einwaage und einem relativ großen aliquoten Teil des Filtrats gearbeitet werden, um genügend Gossypol für eine genaue fotometrische Messung zu erhalten. Für die Bestimmung des Gehalts an freiem Gossypol in Samen, Mehl und Kuchen von Baumwollsaat darf höchstens 1 g eingewogen werden; bei Mischfuttermitteln bis zu 5 gn. Ein aliquoter Teil des Filtrats von 10 ml ist in den meisten Fällen geeignet; er muss 50 bis 100 μg Gossypol enthalten. Für die Bestimmung des Gehalts an Gesamtgossypol muss 0,5 bis 5 g eingewogen werden, so dass in einem aliquoten Teil des Filtrats von 2 ml 40 bis 200 μg Gossypol enthalten sind.
Die Analyse ist bei einer Raumtemperatur von etwa 20 oC auszuführen.
5.2. Bestimmung des Gehalts an freiem Gossypol
Die Einwaage wird in einen 250-ml-Kolben mit Schliffhals, dessen Boden mit Glassplittern bedeckt ist, überführt. Mit Hilfe einer Pipette werden 50 ml des Lösungsmittels A (3.2) hinzugegeben. Anschließend wird der Kolben verschlossen und 1 h lang im Schüttelgerät geschüttelt. Dann wird durch einen trockenen Filter filtriert und das Filtrat in einem kleinen Kolben mit Schliffhals aufgefangen. Während der Filtration wird der Trichter mit einem Uhrglas bedeckt.
Es werden gleich große aliquote Teile des Filtrats, die 50 bis 100 μg Gossypol enthalten, in 2 25-ml-Messkolben (A und B) pipettiert und gegebenenfalls mit Lösungsmittel A (3.2) auf 10 ml aufgefüllt. Dann wird der Inhalt des Kolbens (A) mit der Isopropanol-Hexan-Mischung (3.1) zur Marke aufgefüllt. Diese Lösung wird als Vergleichslösung für die Messung der Probenlösung verwendet.
Danach werden je 10 ml des Lösungsmittels A (3.2) in 2 weitere 25-ml-Messkolben (C und D) pipettiert. Der Inhalt des Kolbens (C) wird mit der Isopropanol-Hexan-Mischung (3.1) zur Marke aufgefüllt. Diese Lösung wird als Vergleichslösung für die Messung der Blindprobenlösung verwendet.
Zu den Messkolben (D) und (B) werden je 2 ml Anilin (3.4) zugegeben. Die Kolben werden dann 30 min lang über einem Bad mit siedendem Wasser zur Entwicklung der Färbung erhitzt, auf Raumtemperatur abgekühlt, mit der Isopropanol-Hexan-Mischung (3.1) jeweils zur Marke aufgefüllt, geschüttelt und 1 h lang stehen gelassen.
Dann werden im Spektralfotometer bei 440 nm unter Verwendung von 1-cm-Glasküvetten die Extinktion der Blindprobe (D) im Vergleich mit der Vergleichslösung (C) und die Extinktion der Probenlösung (B) im Vergleich mit der Vergleichslösung (A) gemessen.
Die Extinktion der Blindprobenlösung wird von der Extinktion der Probenlösung subtrahiert (= korrigierte Extinktion). Anhand des so ermittelten Werts wird der Gehalt an freiem Gossypol, wie unter 6 angegeben, berechnet.
5.3. Bestimmung des Gehalts an Gesamtgossypol
Eine Einwaage, die 1 bis 5 mg Gossypol enthält, wird in einen 50-ml-Messkolben gegeben; dann werden 10 ml des Lösungsmittels B (3.3) hinzugefügt. Gleichzeitig wird ein Blindversuch mit 10 ml des Lösungsmittels B (3.3) in einem anderen 50-ml-Messkolben vorbereitet. Beide Messkolben werden 30 min lang über einem Bad mit siedendem Wasser erhitzt, auf Raumtemperatur abkühlen gelassen und mit der Isopropanol-Hexan-Mischung (3.1) jeweils zur Marke aufgefüllt. Es wird geschüttelt, 10 bis 15 min stehen gelassen, dann filtriert und das Filtrat in Kolben mit Schliffhals aufgefangen.
Es werden jeweils 2 ml des Filtrats der Probe in 2 25-ml-Messkolben und jeweils 2 ml des Filtrats des Blindversuchs in 2 andere 25-ml-Messkolben pipettiert. Je 1 Kolben jeder Versuchsreihe wird mit der Isopropanol-Hexan-Mischung (3.1) auf 25 ml aufgefüllt. Diese Lösungen werden als Vergleichslösungen verwendet.
Zu den beiden anderen Messkolben werden je 2 ml Anilin (3.4) hinzugefügt. Anschließend werden die Kolben 30 min lang über einem Bad mit siedendem Wasser zur Entwicklung der Färbung erhitzt, auf Raumtemperatur abkühlen gelassen, mit der Isopropanol-Hexan-Mischung (3.1) auf jeweils 25 ml aufgefüllt, geschüttelt und 1 h lang stehen gelassen.
Die Extinktionen werden, wie unter 5.2 für freies Gossypol angegeben, gemessen. Anhand des so ermittelten Werts wird der Gehalt an Gesamtgossypol, wie unter 6 angegeben, berechnet.
6. Berechnung der Ergebnisse
Die Berechnung der Ergebnisse kann anhand der spezifischen Extinktion (6.1) oder anhand einer Kalibrationskurve (6.2) erfolgen.
6.1. Anhand der spezifischen Extinktion
Die spezifischen Extinktionen berechnen sich unter den beschriebenen Bedingungen wie folgt:
|
Freies Gossypol |
|
|
Gesamtgossypol: |
|


Der Gehalt an freiem bzw. an Gesamtgossypol der Probe wird nach folgender Formel berechnet:

wobei:
|
E |
= |
korrigierte Extinktion, ermittelt gemäß 5.2, |
|
p |
= |
Einwaage in g, |
|
a |
= |
aliquoter Teil des Filtrats in ml. |
6.2. Anhand einer Kalibrationskurve
6.2.1.
Es werden 2 Reihen von je 5 25-ml-Messkolben vorbereitet. In beide Reihen werden jeweils 2,0, 4,0, 6,0, 8,0 bzw. 10,0 der Gossypol-Standardlösung A (3.5) pipettiert; das Volumen wird mit dem Lösungsmittel A (3.2) auf 10 ml aufgefüllt. Jeder Reihe wird ein weiterer 25-ml-Messkolben hinzugefügt, der nur 10 ml des Lösungsmittels A (3.2) enthält (Blindversuch).
Die Kolben der ersten Reihe einschließlich des Kolbens für den Blindversuch werden mit der Isopropanol-Hexan-Mischung (3.1) auf 25 ml aufgefüllt (Vergleichsreihe).
Den Kolben der zweiten Reihe, einschließlich des Blindversuchs, werden jeweils 2 ml Anilin (3.4) zugesetzt. Anschließend werden die Kolben 30 min lang über einem Bad mit siedendem Wasser zur Entwicklung der Färbung erhitzt, auf Raumtemperatur abgekühlt, mit der Isopropanol-Hexan-Mischung (3.1) zur Marke aufgefüllt, geschüttelt und 1 h lang stehen gelassen (Standardreihe).
Die Extinktion der Lösungen der Standardreihe wird gemäß 5.2 durch einen Vergleich mit den entsprechenden Lösungen der Vergleichsreihe ermittelt. Die Kalibrationskurve wird aufgestellt, indem die Extinktionswerte auf der Ordinate und die entsprechende Gossypolmenge (in μg) auf der Abszisse aufgetragen werden.
6.2.2.
Es werden 6 50-ml-Messkolben vorbereitet. In den ersten werden 10 ml des Lösungsmittels B (3.3) und in die übrigen je 2,0, 4,0, 6,0, 8,0 bzw. 10,0 ml der Gossypol-Standardlösung B (3.6) pipettiert. Der Inhalt eines jeden Kolbens wird mit dem Lösungsmittel B (3.3) auf 10 ml aufgefüllt. Anschließend werden die Kolben 30 min lang über einem Bad mit siedendem Wasser erhitzt, auf Raumtemperatur abgekühlt, mit der Isopropanol-Hexan-Mischung (3.1) zur Marke aufgefüllt und geschüttelt.
In 2 Reihen von je 6 25-ml Messkolben werden jeweils 2,0 ml dieser Lösung pipettiert. Die Kolben der ersten Reihe werden mit der Isopropanol-Hexan-Mischung (3.1) auf 25 ml aufgefüllt. (Vergleichsreihe).
Den Kolben der zweiten Reihe werden jeweils 2 ml Anilin (3.4) zugesetzt. Dann werden sie 30 min lang über einem Bad mit siedendem Wasser erhitzt, auf Raumtemperatur abgekühlt, mit der Isopropanol-Hexan-Mischung (3.1) zur Marke aufgefüllt, geschüttelt und 1 h lang stehen gelassen (Standardreihe).
Die Extinktion der Lösungen der Standardreihe wird gemäß 5.2 durch einen Vergleich mit den entsprechenden Lösungen der Vergleichsreihe ermittelt. Die Kalibrationskurve wird aufgestellt, indem die Extinktionswerte auf der Ordinate und die entsprechende Gossypolmenge (in μg) auf der Abszisse aufgetragen werden.
6.3. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
|
— |
15 % relativ zum höheren Wert bei Gossypol-Gehalten unter 500 ppm, |
|
— |
75 ppm absolut bei Gossypol-Gehalten zwischen 500 und 750 ppm, |
|
— |
10 % relativ zum höheren Wert bei Gossypol-Gehalten über 750 ppm. |
B. BESTIMMUNG DES GEHALTS AN DIOXINEN (PCDD/PCDF) UND DIOXINÄHNLICHEN PCB
I. PROBENAHMEVERFAHREN UND AUSWERTUNG VON ANALYSEERGEBNISSEN
1. Zweck und Anwendungsbereich
Die Proben für die amtliche Untersuchung des Gehalts an Dioxinen (polychlorierte Dibenzo-p-dioxine (PCDD) und polychlorierte Dibenzofurane (PCDF)) sowie dioxinähnlichen polychlorierten Biphenylen (PCB) (1) in Futtermitteln werden nach den in Anhang I beschriebenen Verfahren genommen. Hinsichtlich der Untersuchung auf Stoffe oder Erzeugnisse, die in Futtermitteln gleichmäßig verteilt sind, gelten die quantitativen Anforderungen gemäß Anhang I Punkt 5.A. Die mit diesem Verfahren gewonnenen Sammelproben sind als repräsentativ für die betreffenden Partien oder Teilpartien anzusehen. Anhand der in den Laborproben bestimmten Gehalte wird festgestellt, ob die in der Richtlinie 2002/32/EG des Europäischen Parlaments und des Rates (2) festgesetzten Höchstgehalte eingehalten werden.
2. Übereinstimmung der Partie bzw. Teilpartie mit den Höchstgehalten
Die Partie wird angenommen, wenn das Ergebnis einer Einzelanalyse den entsprechenden Höchstgehalt gemäß Richtlinie 2002/32/EG unter Berücksichtigung der Messunsicherheit nicht überschreitet.
Die Partie entspricht nicht dem in der Richtlinie 2002/32/EG festgelegten Höchstgehalt, wenn die Obergrenze (3) („Upperbound“) des Ergebnisses, das durch eine Zweitanalyse (4) bestätigt wird, unter Berücksichtigung der Messunsicherheit den Höchstgehalt zweifelsfrei überschreitet.
Die Messunsicherheit kann auf eine der beiden folgenden Arten berücksichtigt werden:
|
— |
durch Berechnung der erweiterten Messunsicherheit unter Verwendung eines Faktors von 2, was ein Konfidenzniveau von ca. 95 % ergibt. Eine Partie ist zu beanstanden, wenn der gemessene Wert minus U über dem zulässigen Höchstgehalt liegt. Bei einer getrennten Bestimmung des Gehalts an Dioxinen und dioxinähnlichen PCB ist die Summe der geschätzten erweiterten Messunsicherheit der getrennten Analyseergebnisse der Dioxine und dioxinähnlichen PCB für die Summe der Dioxine und dioxinähnlichen PCB anzulegen; |
|
— |
durch Bestimmung der Entscheidungsgrenze (CCα) gemäß den Bestimmungen der Entscheidung 2002/657/EG der Kommission (5) (Nummer 3.1.2.5 des Anhangs — Fall von Stoffen mit einem festgelegten zulässigen Grenzwert). Eine Partie ist zu beanstanden, wenn der gemessene Wert gleich CCα ist oder diesen Wert übersteigt. |
Diese Auslegungsvorschriften gelten für das Analyseergebnis der zur amtlichen Untersuchung entnommenen Probe. Das Recht der Mitgliedstaaten, für die Analyse zu Verteidigungs- oder Schiedszwecken nationale Vorschriften anzuwenden, bleibt davon unberührt.
II. PROBENVORBEREITUNG UND ANFORDERUNGEN AN ANALYSEMETHODEN ZUR AMTLICHEN UNTERSUCHUNG DES GEHALTS AN DIOXINEN (PCDD/PCDF) UND DIOXINÄHNLICHEN PCB
1. Zweck und Anwendungsbereich
Diese Anforderungen gelten, wenn Futtermittel-Ausgangserzeugnisse und Futtermittel zur Bestimmung ihres Gehalts an Dioxinen (polychlorierte Dibenzo-p-dioxine (PCDD) und polychlorierte Dibenzofurane (PCDF)) sowie dioxinähnlichen polychlorierten Biphenylen (PCB) untersucht werden.
Bei der Überwachung des Dioxingehalts in Futtermitteln kann ein Screening-Verfahren angewandt werden, mit dessen Hilfe diejenigen Proben mit einem Gehalt an Dioxinen und dioxinähnlichen PCB ausgewählt werden, die weniger als 25 % unter der interessierenden Konzentration oder darüber liegen. Die Konzentration der Dioxine in denjenigen Proben mit signifikanten Werten muss dann durch ein Bestätigungsverfahren ermittelt/bestätigt werden.
Screening-Verfahren sind Verfahren, die zum Nachweis des Vorhandenseins von Dioxinen und dioxinähnlichen PCB in der interessierenden Konzentration verwendet werden. Diese Verfahren ermöglichen einen hohen Probendurchsatz und werden eingesetzt, um eine große Anzahl von Proben auf mögliche positive Ergebnisse zu sichten. Sie sind speziell dafür ausgelegt, falsch negative Ergebnisse zu vermeiden.
Bestätigungsverfahren sind Verfahren, die vollständige oder ergänzende Daten liefern, damit Dioxine und dioxinähnliche PCB in der interessierenden Konzentration eindeutig identifiziert und quantifiziert werden können.
2. Hintergrund
Da Umweltproben und biologische Proben (einschließlich Proben von Futtermittel-Ausgangserzeugnissen/Futtermitteln) im Allgemeinen komplexe Mischungen verschiedener Dioxin-Kongenere enthalten, wurde zur Erleichterung der Risikobewertung das Konzept der Toxizitätsäquivalenzfaktoren (TEF) entwickelt. Durch diese TEF werden Konzentrationen aus Gemischen aus 2,3,7,8-substituierten PCDD und PCDF und einige nicht-ortho- und mono-orthochlorsubstituierte PCB mit dioxinähnlicher Aktivität in Toxizitätsäquivalenten (TEQ) von 2,3,7,8-TCDD ausgedrückt. Die Konzentrationen der einzelnen Substanzen in einer bestimmten Probe werden mit ihren jeweiligen TEF multipliziert und anschließend addiert, woraus sich die Gesamtkonzentration an dioxinähnlichen Verbindungen, ausgedrückt in TEQ, ergibt.
Ausschließlich für die Zwecke dieser Verordnung gilt als akzeptierte spezifische Bestimmungsgrenze eines einzelnen Kongeners die Konzentration eines Analyts in einem Probenextrakt, die ein Signal des Messgeräts bei 2 verschiedenen, charakteristischen Ionen (Fragmentionen) hervorruft, die mit einem Signal-Rausch-Verhältnis von 3:1 bei dem weniger empfindlichen Signal verbunden sind. Weiterhin müssen die Grundanforderungen erfüllt werden, wie z. B. Retentionszeit und Isotopenverhältnis gemäß dem Bestimmungsverfahren nach der Beschreibung in der EPA-Methode 1613 Revision B.
3. Anforderungen an die Qualitätssicherung bei der Probenvorbereitung
Es gelten die allgemeinen Bestimmungen hinsichtlich der Vorbereitung der Proben zur Analyse gemäß Anhang II.
Darüber hinaus sind folgende Anforderungen zu erfüllen:
|
— |
Die Proben sind in Glas-, Aluminium-, Polypropylen- oder Polyethylen-Behältern zu lagern und zu transportieren. Spuren von Papierstaub sind vom Probenbehälter zu entfernen. Gläser sind mit Lösungsmitteln auszuspülen, die zuvor auf das Vorhandensein von Dioxinen überprüft wurden. |
|
— |
Es ist eine Blindanalyse vorzunehmen, indem das gesamte Analyseverfahren durchgeführt und nur die Probe dabei weggelassen wird. |
|
— |
Das Gewicht der für die Extraktion verwendeten Probe muss ausreichend groß sein, um die Anforderungen an die Messempfindlichkeit zu erfüllen. |
4. Anforderungen an Laboratorien
|
— |
Die Laboratorien haben den Nachweis der Leistungsfähigkeit eines Verfahrens im Bereich der interessierenden Konzentration, z. B. 0,5 ×, 1 × und 2 × die interessierende Konzentration mit einem akzeptablen Abweichungskoeffizienten für wiederholte Untersuchung, zu führen. Näheres zu den Akzeptanzkriterien siehe 5. |
|
— |
Die Bestimmungsgrenze liegt beim Bestätigungsverfahren im Bereich von etwa einem Fünftel der interessierenden Konzentration, damit sichergestellt ist, dass im Bereich der interessierenden Konzentration akzeptable Abweichungskoeffizienten eingehalten werden. |
|
— |
Als interne Qualitätssicherungsmaßnahmen werden regelmäßige Blindkontrollen und Experimente mit gespikten Proben oder Analysen von Kontrollproben (sofern erhältlich, vorzugsweise zertifiziertes Referenzmaterial) durchgeführt. |
|
— |
Die erfolgreiche Teilnahme an Laborvergleichsuntersuchungen zur Bewertung der Leistung von Laboratorien ist der beste Weg, Kompetenz bei spezifischen Untersuchungen nachzuweisen. Allerdings belegt eine erfolgreiche Teilnahme an Laborvergleichsuntersuchungen beispielsweise in Bezug auf Boden- oder Abwasserproben nicht zwangsläufig auch eine Kompetenz im Bereich Lebensmittel- oder Futtermittelproben, die eine geringere Kontamination aufweisen. Daher ist die kontinuierliche Teilnahme an Laborvergleichsuntersuchungen zur Bestimmung des Gehalts an Dioxin und dioxinähnlichen PCB in den entsprechenden Futtermittel-/Lebensmittelmatrizen obligatorisch. |
|
— |
Laboratorien müssen von einer anerkannten Stelle akkreditiert sein, die nach ISO/IEC-Leitfaden 58 arbeitet, damit sichergestellt ist, dass die Laboratorien bei ihren Untersuchungen Qualitätssicherungsverfahren anwenden. Die Laboratorien müssen gemäß der Norm ISO/IEC/17025 akkreditiert sein. |
5. Anforderungen an Methoden zur Analyse auf Dioxine und dioxinähnliche PCB
Grundsätzliche Anforderungen an Untersuchungsverfahren:
|
— |
Große Messempfindlichkeit und niedrige Nachweisgrenze. Bei PCDD und PCDF müssen die nachweisbaren Mengen wegen der extrem hohen Toxizität einiger dieser Verbindungen im Bereich Pikogramm TEQ (10-12 g) liegen. Der Gehalt an PCB ist bekanntlich höher als derjenige an PCDD und PCDF. Bei den meisten PCB-Kongeneren ist eine Messempfindlichkeit im Bereich Nanogramm (10-9 g) bereits ausreichend. Zur Messung der toxischeren dioxinähnlichen PCB-Kongenere (insbesondere der nicht-ortho-substituierten Kongenere) muss jedoch die gleiche Messempfindlichkeit erreicht werden wie für die PCDD und PCDF. |
|
— |
Hohe Selektivität (Spezifizität). PCDD, PCDF und dioxinähnliche PCB müssen von einer Vielzahl anderer, gemeinsam extrahierter und möglicherweise interferierender Verbindungen unterschieden werden, die in Konzentrationen von bis zu mehreren Größenordnungen höher als diejenigen der zu prüfenden Analyten vorhanden sind. Bei Gaschromatografie/Massenspektrometrie-(GC/MS-)Verfahren ist eine Unterscheidung zwischen verschiedenen Kongeneren erforderlich, wie beispielsweise zwischen toxischen (z. B. die 17 2,3,7,8-substituierten PCDD und PCDF sowie dioxinähnliche PCB) und anderen Kongeneren. Bioassays müssen eine selektive Bestimmung der TEQ-Werte als Summe aus PCDD, PCDF und dioxinähnlichen PCB ermöglichen. |
|
— |
Hohe Genauigkeit (Richtigkeit und Präzision). Die Bestimmung muss eine valide und zuverlässige Schätzung der tatsächlichen Konzentration in einer Probe ermöglichen. Hohe Genauigkeit (Messgenauigkeit: der Grad der Übereinstimmung zwischen dem Ergebnis einer Messung und dem wahren oder ermittelten Wert der Messgröße) ist notwendig, damit die Zurückweisung des Ergebnisses einer Probenuntersuchung aufgrund der geringen Zuverlässigkeit der TEQ-Schätzung vermieden wird. Die Genauigkeit des Analyseverfahrens wird angegeben durch die Richtigkeit (Differenz zwischen dem gemessenen Mittelwert eines Analyten in einem zertifizierten Material und seinem zertifizierten Wert, ausgedrückt als Prozentsatz dieses Wertes) und der Präzision (RSDR; relative Standardabweichung, berechnet aus unter Wiederholbarkeitsbedingungen ermittelten Ergebnissen). |
Screening-Verfahren können Bioassays und GC/MS-Verfahren umfassen; Bestätigungsverfahren sind hochauflösende Gaschromatografie-/hochauflösende Massenspektrometrie-Verfahren (HRGC/HRMS).
Die folgenden Kriterien müssen vom Gesamt-TEQ-Wert erfüllt werden:
|
|
Screening-Verfahren |
Bestätigungsverfahren |
|
Falsch negativer Anteil |
< 1 % |
|
|
Richtigkeit |
|
- 20 bis + 20 % |
|
Präzision RSDR |
< 30 % |
< 15 % |
6. Spezielle Anforderungen an GC/MS-Verfahren, wenn sie zu Screening- oder Bestätigungszwecken eingesetzt werden
|
— |
Die Addition von 13C-markierten 2,3,7,8-chlorsubstituierten internen PCDD/F-Standards und 13C-markierten internen dioxinähnlichen PCB-Standards ist gleich zu Beginn des Analyseverfahrens, z. B. vor der Extraktion, durchzuführen, damit das Analyseverfahren validiert werden kann. Bei jeder der tetra- bis octa-chlorierten homologen Gruppen von PCDD/F (und bei jeder der homologen Gruppen von dioxinähnlichen PCB, sofern dioxinähnliche PCB zu bestimmen sind) muss mindestens ein Kongener zugegeben werden (alternativ dazu mindestens ein Kongener je massenspektrometrisch ausgewählter Ionenaufzeichnungsfunktion zur Überwachung von PCDD/F und dioxinähnlichen PCB). Im Fall der Bestätigungsverfahren ist die Verwendung aller 17 13C-markierten 2,3,7,8-substituierten internen PCDD/F-Standards und aller 12 13C-markierten internen dioxinähnlichen PCB-Standards eindeutig vorzuziehen. |
|
— |
Die relativen Responsefaktoren sind mittels geeigneter Kalibrierlösungen auch für diejenigen Kongenere zu bestimmen, bei denen kein 13C-markiertes Analogon zugegeben ist. |
|
— |
Bei Futtermitteln pflanzlichen Ursprungs und Futtermitteln tierischen Ursprungs, die weniger als 10 % Fett enthalten, ist die Addition der internen Standards vor der Extraktion obligatorisch. Bei Futtermitteln tierischen Ursprungs, die mehr als 10 % Fett enthalten, können die internen Standards entweder vor der Extraktion oder nach der Fettextraktion zugegeben werden. Die Extraktionseffizienz ist auf geeignete Weise zu validieren, je nachdem, auf welcher Stufe interne Standards zugegeben und ob die Ergebnisse auf Produkt- oder Fettbasis angegeben werden. |
|
— |
Vor der GC/MS-Analyse sind 1 oder 2 Wiederfindungs-(Surrogat-) Standard(s) zu addieren. |
|
— |
Es ist eine Kontrolle der Wiederfindungsrate erforderlich. Bei Bestätigungsverfahren müssen die Wiederfindungsraten der einzelnen internen Standards im Bereich von 60 bis 120 % liegen. Geringere oder höhere Wiederfindungsraten für einzelne Kongenere, insbesondere für einige hepta- und octa-chlorierte Dibenzodioxine und Dibenzofurane, können unter der Bedingung akzeptiert werden, dass ihr Beitrag zum TEQ-Wert 10 % des gesamten TEQ-Werts (basierend auf der Summe von PCDD/F und dioxinähnlichen PCB) nicht übersteigt. Bei Screening-Verfahren müssen die Wiederfindungsraten im Bereich von 30 bis 140 % liegen. |
|
— |
Die Dioxine sind von interferierenden chlorierten Verbindungen, wie z. B. nicht dioxinähnlichen PCB und chlorierten Diphenylethern, mittels geeigneter chromatografischer Verfahren abzutrennen (vorzugsweise mit Florisil-, Aluminiumoxid- und/oder Aktivkohlensäule). |
|
— |
Die gaschromatografische Auftrennung der Isomere ist ausreichend (< 25 % von Peak zu Peak zwischen 1,2,3,4,7,8-HxCDF und 1,2,3,6,7,8-HxCDF). |
|
— |
Die Bestimmung ist nach der EPA-Methode 1613 Revision B („Tetra- through octa-chlorinated dioxins and furans by isotope dilution HRGC/HRMS“) oder nach einer anderen Methode mit gleichwertigen Leistungskriterien durchzuführen. |
|
— |
Bei Futtermitteln, deren Dioxinkontamination im Bereich des Höchstwerts oder darüber liegt, darf die Differenz zwischen Ober- und Untergrenze höchstens 20 % betragen. Bei Futtermitteln mit einer Kontamination, die deutlich unter der Höchstgrenze liegt, kann die Differenz im Bereich von 25 bis 40 % liegen. |
7. Screening-Verfahren
7.1. Einführung
Ein Screening-Verfahren kann zu unterschiedlichen analytischen Zwecken eingesetzt werden: zum reinen Screening und zur quantitativen Untersuchung.
Das Messsignal der Proben wird mit demjenigen einer Referenzprobe bei der interessierenden Konzentration verglichen. Proben mit einem niedrigeren Wert als demjenigen der Referenzprobe werden als negativ erklärt, diejenigen mit einem höheren Signal als positiv vermutet. Weitere Anforderungen sind:
|
— |
Bei jeder Testreihe sind eine Blind- und eine Referenzprobe/mehrere Referenzproben einzubeziehen, die zur gleichen Zeit und unter den gleichen Bedingungen extrahiert und untersucht werden. Die Referenzprobe(n) muss/müssen im Vergleich zur Blindprobe ein deutlich erhöhtes Messsignal aufweisen. |
|
— |
Zusätzliche Referenzproben, deren Konzentration das 0,5- und 2-fache der interessierenden Konzentration beträgt, sind einzubeziehen, damit die ordnungsgemäße Durchführung des Tests in dem für die Kontrolle der interessierenden Konzentration relevanten Bereich nachgewiesen werden kann. |
|
— |
Bei der Untersuchung anderer Matrizen ist die Eignung der Referenzprobe(n) nachzuweisen, vorzugsweise durch die Aufnahme von Proben, bei denen sich durch HRGC/HRMS ein TEQ-Gehalt vergleichbar mit dem der Referenzprobe ergeben hat, oder andernfalls durch die Aufnahme einer Blindprobe, die bis zu dieser Höhe gespikt wurde. |
|
— |
Da in Bioassays keine internen Standards verwendet werden können, sind Wiederholbarkeitstests zur Information über die Standardabweichung innerhalb einer Testreihe sehr wichtig. Der Abweichungskoeffizient muss unter 30 % liegen. |
|
— |
Bei Bioassays sind die zu bestimmenden Analyten, mögliche auftretende Störungen und der maximal akzeptable Blindwert zu definieren. |
Zur quantitativen Untersuchung sind Standardverdünnungsreihen, 2- oder 3-fache Clean-ups und Messungen der Proben sowie Blind- und Wiederfindungskontrollen erforderlich. Das Ergebnis kann in TEQ ausgedrückt werden, wobei davon ausgegangen wird, dass die für das Signal verantwortlichen Verbindungen dem TEQ-Prinzip entsprechen. Eine Kalibrierungskurve ergibt sich durch die Verwendung von TCDD (oder einer Standardmischung aus Dioxinen/Furanen/dioxinähnlichen PCB). Damit kann der TEQ-Wert im Extrakt und somit in der Probe errechnet werden. Dieser TEQ-Wert wird anschließend um den für eine Blindprobe (zur Berücksichtigung von Verunreinigungen durch Lösungsmittel und Chemikalien) errechneten TEQ-Wert und um eine Wiederfindung (errechnet aus dem TEQ-Wert in einer Qualitätskontrollprobe mit etwa der interessierenden Konzentration) korrigiert. Es sei hier darauf hingewiesen, dass der offensichtliche Wiederfindungsverlust teilweise auf Matrixeffekte und/oder auf die Unterschiede zwischen den TEF-Werten in den Bioassays und den amtlichen TEF-Werten der WHO zurückzuführen sein kann.
7.2. Anforderungen an zum Screening verwendete Untersuchungsverfahren
|
— |
Zum Screening können GC/MS-Verfahren und Bioassays verwendet werden. Bei GC/MS-Verfahren sind die unter 6 festgelegten Anforderungen heranzuziehen. Spezielle Anforderungen sind für zellbasierte Bioassays und Kit-basierte Bioassays unter 7.3 bzw. 7.4 festgelegt. |
|
— |
Es sind Informationen über die Anzahl falsch positiver und falsch negativer Ergebnisse eines großen Probensatzes unterhalb und oberhalb der Höchstgehalte oder der Auslösewerte, im Vergleich zum TEQ-Gehalt erforderlich, der durch ein Bestätigungsverfahren bestimmt wurde. Der tatsächliche Anteil der falsch negativen Ergebnisse muss unter 1 % liegen. Der Anteil der falsch positiven Proben muss so gering sein, dass ein Screening von Vorteil ist. |
|
— |
Positive Ergebnisse sind immer durch ein Bestätigungsverfahren abzusichern (HRGC/HRMS). Außerdem sind die Proben aus einem großen TEQ-Bereich durch HRGC/HRMS (ca. 2 bis 10 % der negativen Proben) zu bestätigen. Informationen über Übereinstimmungen von Bioassay- und HRGC/HRMS-Ergebnissen sind zur Verfügung zu stellen. |
7.3. Spezielle Anforderungen an zellbasierte Bioassays
|
— |
Für jeden Testlauf in einem Bioassay ist eine Referenzkonzentrationsreihe von TCDD oder einem Dioxin-Furan-Gemisch (vollständige Dosis-Response-Kurve mit R2 > 0,95) erforderlich. Zu Screening-Zwecken kann eine erweiterte Kurve im Niedrigkonzentrationsbereich zur Untersuchung von Proben im Niedriggehaltbereich verwendet werden. |
|
— |
Zum Beleg der Richtigkeit der Ergebnisse des Bioassays über einen konstanten Zeitraum hinweg sollte eine TCDD-Referenzkonzentration (etwa 3 × die Bestimmungsgrenze) auf einem Qualitätskontrollblatt verwendet werden. Eine Alternative dazu wäre die relative Response einer Referenzprobe im Vergleich zur TCDD-Kalibrierungslinie, da die Response der Zellen von vielen Faktoren abhängen kann. |
|
— |
Für jeden Typ Referenzmaterial sind Qualitätskontroll-Charts aufzuzeichnen und zu prüfen, damit sichergestellt ist, dass das Ergebnis mit den Leitlinien übereinstimmt. |
|
— |
Insbesondere bei quantitativen Berechnungen muss die Induktion der Probenverdünnung innerhalb des linearen Teils der Response-Kurve liegen. Über dem linearen Teil der Response-Kurve liegende Proben sind zu verdünnen und neu zu testen. Daher wird empfohlen, immer mindestens 3 oder mehr Verdünnungen zur gleichen Zeit zu testen. |
|
— |
Die Standardabweichung darf bei einer 3-fachen Bestimmung einer Probenlösung höchstens 15 % betragen und zwischen 3 unabhängigen Versuchen höchstens 30 %. |
|
— |
Die Nachweisgrenze kann auf 3 × die Standardabweichung der Blindlösung oder des Hintergrundsignals festgelegt werden. Eine andere Möglichkeit wäre, einen über dem Hintergrund liegenden Messwert anzuwenden (Induktionsfaktor 5 × der Blindwert des Lösungsmittels), der anhand der Kalibrationskurve des Tages berechnet wird. Die Bestimmungsgrenze kann auf 5- bis 6 × die Standardabweichung der Blindlösung oder des Hintergrundsignals festgelegt werden, oder es kann ein Signal über dem Hintergrund (Induktionsfaktor 10 × der Blindwert des Lösungsmittels) angewandt werden, der anhand der Kalibrationskurve des Tages berechnet wird. |
7.4. Spezielle Anforderungen an Kit-basierte Bioassays
|
— |
Es muss sichergestellt werden, dass die Kit-basierten Bioassays ausreichend empfindlich und zuverlässig für Futtermittel sind. |
|
— |
Bei der Vorbereitung und Untersuchung der Proben sind die Anweisungen des Herstellers zu beachten. |
|
— |
Testkits, deren Haltbarkeitsdatum abgelaufen ist, dürfen nicht mehr verwendet werden. |
|
— |
Materialien oder Bestandteile, die zur Verwendung mit anderen Kits bestimmt sind, dürfen nicht verwendet werden. |
|
— |
Die Testkits sind bei den angegebenen Lagertemperaturen aufzubewahren und bei der angegebenen Betriebstemperatur zu verwenden. |
|
— |
Die Nachweisgrenze für Immunoassays wird bestimmt als Summe aus dem Mittelwert und der 3-fachen Standardabweichung von 10 Untersuchungen des Blindwerts, dividiert durch den Steigungsbeitrag der linearen Regressionsgleichung. |
|
— |
Für die Labortests sind Referenzstandards zu verwenden, damit sichergestellt ist, dass das Messsignal des Standards im Test innerhalb eines akzeptablen Bereichs liegt. |
8. Bericht über die Ergebnisse
Sofern das Untersuchungsverfahren dies zulässt, müssen die Untersuchungsergebnisse die Werte der einzelnen PCDD/F- und PCB-Kongenere enthalten und als Obergrenze („Upperbound“), Untergrenze („Lowerbound“) und Mittelwert („Mediumbound“) vorgelegt werden, damit möglichst viele Informationen in den Untersuchungsberichten enthalten sind und die Ergebnisse somit entsprechend den speziellen Anforderungen interpretiert werden können.
In dem Bericht muss auch der Lipidgehalt der Probe sowie das zur Lipidextraktion verwendete Verfahren genannt werden.
Die Wiederfindungsraten der einzelnen internen Standards sind zur Verfügung zu stellen, sofern die Wiederfindungen außerhalb des unter 6 genannten Bereichs liegen oder sofern die Gehalte in den Proben den Höchstgehalt überschreiten bzw. gegebenenfalls auch auf Nachfrage.
Da die Messunsicherheit bei der Entscheidung über die Konformität einer Probe zu berücksichtigen ist, muss dieser Parameter ebenfalls vorgelegt werden. Das Analyseergebnis ist als x +/– U anzugeben, wobei x das Analyseergebnis und U die erweiterte Messunsicherheit unter Verwendung eines Faktors von 2, der zu einem Konfidenzwert von ca. 95 % führt, darstellen. Bei einer getrennten Bestimmung des Gehalts an Dioxinen und dioxinähnlichen PCB ist die Summe der geschätzten erweiterten Messunsicherheit der getrennten Analyseergebnisse der Dioxine und dioxinähnlichen PCB für die Summe der Dioxine und dioxinähnlichen PCB zu berechnen.
Wird die Messunsicherheit durch Anwendung des CCα (vgl. Abschnitt I. 2 dieses Teils B) berücksichtigt, so ist dieser Parameter anzugeben.
(1) Tabelle der TEF (= Toxizitätsäquivalenzfaktoren) für Dioxine, Furane und dioxinähnliche PCB:
|
Kongener |
TEF-Wert |
Kongener |
TEF-Wert |
|
Dibenzo-p-dioxine („PCDD“) |
|
„Dioxinähnliche“ PCB: |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
Nicht-ortho PCB |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0001 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,01 |
|
OCDD |
0,0001 |
Mono-ortho PCB |
|
|
|
|
PCB 105 |
0,0001 |
|
Dibenzofurane („PCDF“) |
|
PCB 114 |
0,0005 |
|
2,3,7,8-TCDF |
0,1 |
PCB 118 |
0,0001 |
|
1,2,3,7,8-PeCDF |
0,05 |
PCB 123 |
0,0001 |
|
2,3,4,7,8-PeCDF |
0,5 |
PCB 156 |
0,0005 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 157 |
0,0005 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00001 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 189 |
0,0001 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
|
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
|
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0001 |
|
|
|
Abkürzungen: „T“ = tetra; „Pe“ = penta; „Hx“ = hexa; „Hp“ = hepta; „O“ = octa; „CDD“ = Chlordibenzo-p-dioxin, „CDF“ = Chlorodibenzofuran; „CB“ = Chlorbiphenyl. |
|||
(2) ABl. L 140 vom 30.5.2002, S. 10.
(3) Zur Berechnung der Obergrenze („Upperbound“) wird der Beitrag jedes nicht quantifizierten Kongeners zum TEQ der Bestimmungsgrenze gleichgesetzt.
Das Konzept der Untergrenze („Lowerbound“) setzt voraus, dass der Beitrag jedes nicht quantifizierten Kongeners zum TEQ mit 0 veranschlagt wird.
Das Konzept des Mittelwerts („Mediumbound“) setzt voraus, dass der Beitrag jedes nicht quantifizierten Kongeners zum TEQ mit der Hälfte der Bestimmungsgrenze gleichgesetzt wird.
(4) Die Zweitanalyse ist erforderlich, um eine interne Kreuzkontamination oder eine versehentliche Vermischung der Proben auszuschließen. Mit der Erstanalyse, welche die Messunsicherheit berücksichtigt, wird die Einhaltung der Höchstgehalte überprüft.
Bei einer Untersuchung wegen einer Dioxinkontamination kann auf die Bestätigung durch Zweitanalyse verzichtet werden, wenn sich die untersuchten Proben auf das Kontaminationsereignis zurückverfolgen lassen.
ANHANG VI
ANALYSEMETHODEN ZUR BESTIMMUNG DER BESTANDTEILE TIERISCHEN URSPRUNGS BEI DER AMTLICHEN UNTERSUCHUNG VON FUTTERMITTELN
Bedingungen für den mikroskopischen Nachweis, die Identifizierung oder die Schätzung von Bestandteilen tierischen Ursprungs in Futtermitteln
1. Zweck und Anwendungsbereich
Diese Bedingungen gelten für den Nachweis von Bestandteilen tierischen Ursprungs (definiert als Erzeugnisse aus der Verarbeitung von Tierkörpern oder Teilen von Tierkörpern von Säugetieren, Geflügel und Fischen) in Futtermitteln durch mikroskopische Untersuchung im Rahmen des koordinierten Kontrollprogramms im Bereich der Futtermittel gemäß der Verordnung (EG) Nr. 882/2004 des Europäischen Parlaments und des Rates (1). Unter der Voraussetzung, dass die in diesem Anhang aufgeführten Methoden bei allen amtlichen Untersuchungen angewandt werden, kann auch eine zweite Untersuchung mittels abweichender oder alternativer Methoden durchgeführt werden, um den Nachweis bestimmter Arten tierischer Bestandteile zu verbessern oder um den Ursprung der tierischen Bestandteile weiter zu spezifizieren. Außerdem kann bei der Untersuchung bestimmter spezifischer Bestandteile tierischen Ursprungs, wie z. B. Plasma oder Knochen in Talg (siehe auch 9), ein anderes Protokoll verwendet werden, sofern diese Analysen zusätzlich zu den im koordinierten Kontrollprogramm vorgesehenen durchgeführt werden.
2. Empfindlichkeit
Je nach Art der Bestandteile tierischen Ursprungs können in Futtermitteln sehr geringe Mengen (< 0,1 %) festgestellt werden.
3. Prinzip
Eine gemäß den Bestimmungen in Anhang I entnommene Probe wird nach geeigneter Aufbereitung zur Identifizierung verwendet. Das nachfolgende Protokoll eignet sich für die Handhabung von Futtermitteln mit geringem Feuchtigkeitsgehalt. Futtermittel mit einem Feuchtigkeitsgehalt von über 14 % sind vor der Handhabung zu trocknen (zu kondensieren). Spezielle Futtermittel oder Einzelfuttermittel (z. B. Fette, Öle) müssen gezielt behandelt werden (siehe 9). Identifiziert werden die tierischen Bestandteile anhand charakteristischer, mikroskopisch erkennbarer Merkmale (d. h. Muskelfasern und andere Fleischpartikel, Knorpel, Knochen, Horn, Haare, Borsten, Blut, Federn, Eierschalen, Gräten, Schuppen). Die Identifizierung wird sowohl in der Siebfraktion (6.1) als auch im konzentrierten Sediment (6.2) der Probe durchgeführt.
4. Reagenzien
4.1. Einbettungsmittel
|
4.1.1. |
Chloralhydrat (wässrig, Massenkonzentration = 60 %). |
|
4.1.2. |
Lauge (NaOH, Massenkonzentration = 2,5 %, oder KOH, Massenkonzentration = 2,5 %) für die Siebfraktionen. |
|
4.1.3. |
Paraffinöl oder Glyzerin (Viskosität: 68 bis 81) für mikroskopische Beobachtungen im Sediment. |
4.2. Spülmittel
|
4.2.1. |
96 %iger Alkohol. |
|
4.2.2. |
Aceton. |
4.3. Konzentrationsmittel
|
4.3.1. |
Tetrachlorethylen (Dichte 1,62). |
4.4. Nachweisreagenzien
|
4.4.1. |
Iod-/Iodkalium-Lösung (2 g Iodkalium in 100 ml Wasser lösen und unter häufigem Schütteln 1 g Iod zufügen). |
|
4.4.2. |
Alizarinrot (2,5 ml 1 M Salzsäure in 100 ml Wasser lösen und dieser Lösung 200 mg Alizarinrot zufügen). |
|
4.4.3. |
Cystin-Reagenz (2 g Bleiacetat, 10 g NaOH/100 ml H2O). |
|
4.4.4. |
Iod-/Iodkalium-Lösung (gelöst in 70 %igem Ethanol). |
4.5. Bleichmittel
|
4.5.1. |
Handelsübliche Natriumhypochlorit-Lösung (9,6 % aktives Chlor). |
5. Geräte und Hilfsmittel
|
5.1. |
Analysenwaage (Genauigkeit von 0,01 g, außer für das konzentrierte Sediment: 0,001 g). |
|
5.2. |
Zerkleinerungsgeräte (Mühle oder Mörser, insbesondere für Futtermittel > 15 % Fett bei Analyse). |
|
5.3. |
Sieb mit einer Maschenweite von höchstens 0,50 mm. |
|
5.4. |
Scheidetrichter oder Absetzglas mit konischem Boden. |
|
5.5. |
Stereomikroskop (mindestens 40-fache Vergrößerung). |
|
5.6. |
Zusammengesetztes Mikroskop (mindestens 400-fache Vergrößerung), mit Durchlicht oder Polarisierungseinrichtung. |
|
5.7. |
Standardglaswaren für Laboratorien Alle Geräte sind gründlich zu reinigen. Scheidetrichter und Glaswaren sind in einer Spülmaschine zu waschen. Siebe sind mit einer steifborstigen Bürste zu reinigen. |
6. Verfahren
Pelletierte Futtermittel können vorgesiebt werden, sofern beide Fraktionen als getrennte Probe untersucht werden.
Mindestens 50 g der Untersuchungsprobe werden behandelt (mittels geeigneter Zerkleinerungsgeräte (5.2) vorsichtig zerkleinert, sofern dies zur Erreichung einer geeigneten Struktur erforderlich ist). Das zerkleinerte Material wird in 2 repräsentative Teile geteilt, einen für die Siebfraktion (mindestens 5 g) (6.1) und einen für das konzentrierte Sediment (mindestens 5 g) (6.2). Färbungen mit Nachweisreagenzien (6.3) können zur Identifizierung zusätzlich verwendet werden.
Zur Angabe der Art des tierischen Proteins und des Ursprungs der Partikel kann ein System zur Unterstützung der Entscheidungsfindung, wie z. B. ARIES, verwendet werden, und Referenzproben können dokumentiert werden.
6.1. Identifizierung von Bestandteilen tierischen Ursprungs in den Siebfraktionen
Mindestens 5 g der Probe werden durch Sieben (5.3) in 2 Fraktionen getrennt.
Die Siebfraktion(en) mit den großen Partikeln (oder ein repräsentativer Teil der Fraktion) wird in einer dünnen Schicht auf einer geeigneten Unterlage aufgebracht und unter dem Stereomikroskop (5.5) bei verschiedenen Vergrößerungen systematisch auf Bestandteile tierischen Ursprungs untersucht.
Präparate mit der/den Feinpartikel-Siebfraktion(en) werden systematisch unter dem zusammengesetzten Mikroskop (5.6) bei verschiedenen Vergrößerungen auf Bestandteile tierischen Ursprungs untersucht.
6.2. Identifizierung von Bestandteilen tierischen Ursprungs im konzentrierten Sediment
Mindestens 5 g der Probe werden (auf 0,01 g genau) in einen Scheidetrichter oder einen Absetzbecher mit konischem Boden eingewogen und mit mindestens 50 ml Tetrachlorethylen (4.3.1) versetzt. Die Mischung wird wiederholt geschüttelt oder umgerührt.
|
— |
Wird ein geschlossener Scheidetrichter verwendet, so wird das Sediment nach einer ausreichenden Standzeit (mindestens 3 min) abgetrennt. Es wird noch einmal geschüttelt und nach weiteren mindestens 3 min Standzeit noch einmal abgetrennt. |
|
— |
Wird ein offener Absetzbecher verwendet, so wird das Sediment nach einer Standzeit von mindestens 5 min abgetrennt. |
Das gesamte Sediment wird getrocknet und anschließend ausgewogen (auf 0,001 g genau). Das Wiegen ist nur notwendig, wenn eine Schätzung erforderlich ist. Besteht das Sediment aus vielen großen Partikeln, kann es durch ein Sieb (5.3) in 2 Fraktionen gesiebt werden. Das trockene Sediment wird unter dem Stereomikroskop (5.5) und unter dem zusammengesetzten Mikroskop (5.6) auf Knochenbestandteile untersucht.
6.3. Verwendung von Einbettungsmitteln und Nachweisreagenzien
Die mikroskopische Identifizierung der Bestandteile tierischen Ursprungs kann durch spezielle Einbettungsmittel und Nachweisreagenzien unterstützt werden.
Chloralhydrat (4.1.1): Durch vorsichtiges Erhitzen können Zellstrukturen eindeutiger erkannt werden, weil Stärketeilchen gelatinieren und unerwünschter Zelleninhalt entfernt wird.
Lauge (4.1.2): Natriumhydroxid und Kaliumhydroxid klären das Material und tragen so zum Nachweis von Muskelfasern, Haaren und anderen Keratinstrukturen bei.
Paraffinöl und Glyzerin (4.1.3.): Knochenbestandteile sind in diesem Einbettungsmittel gut nachzuweisen, weil die meisten Lakunen mit Luft gefüllt bleiben und als schwarze etwa 5 bis 15 μm große Löcher zu erkennen sind.
Iod-/Iodkalium-Lösung (4.4.1): Wird für den Nachweis von Stärke (Blau-Violettfärbung) und Eiweiß (Gelb-Orangefärbung) verwendet. Die Lösung kann bei Bedarf verdünnt werden.
Alizarinrotlösung (4.4.2): Rot-/Pinkfärbung von Knochen, Gräten und Schuppen. Das gesamte Sediment wird vor der Trocknung (siehe 6.2) in ein Reagenzglas gefüllt und 2-mal mit etwa 5 ml Alkohol (4.2.1) gespült (jedes Mal wird verwirbelt, nach etwa 1 min Absetzzeit wird das Lösungsmittel abgegossen). Vor der Verwendung dieses Nachweisreagenzes wird das Sediment durch Zusatz von mindestens 1 ml Natriumhydroxidlösung (4.5.1) gebleicht. Die Reaktionszeit beträgt etwa 10 min. Danach wird das Reagenzglas mit Wasser gefüllt, nach einer Absetzzeit des Sediments von 2 bis 3 min werden das Wasser und die suspendierten Partikel abgeschüttet. Das Sediment wird noch 2-mal mit etwa 10 ml Wasser gespült (dabei wird verwirbelt, absetzen gelassen, und das Wasser wird jedes Mal abgegossen). Von der Alizarinrot-Lösung werden dann 2 bis 10 oder mehr Tropfen (je nach Rückstandsmenge) zugefügt. Die Mischung wird geschüttelt, und die Reaktion kann ein paar Sekunden lang stattfinden. Das eingefärbte Sediment wird 2-mal mit etwa 5 ml Alkohol (4.2.1) gespült und anschließend 1-mal mit Aceton (4.2.2) (jedes Mal wird verwirbelt, das Lösungsmittel wird nach einer Absetzzeit von 1 min abgegossen. Danach ist das Sediment fertig zur Trocknung.
Cystin-Reagenz (4.4.3.): Durch vorsichtiges Erhitzen werden cystinhaltige Bestandteile (Haare, Federn usw.) schwarzbraun gefärbt.
6.4. Untersuchung von Futtermitteln, die möglicherweise Fischmehl enthalten
Mindestens 1 Objektträger mit der feinen Siebfraktion und der feinen Fraktion des Sediments wird unter dem zusammengesetzten Mikroskop untersucht (siehe 6.1 und 6.2).
Ist auf der Etikettierung Fischmehl als Zutat angegeben oder wird das Vorhandensein von Fischmehl vermutet oder in der ersten Untersuchung nachgewiesen, werden mindestens 2 weitere Objektträger mit der feinen Siebfraktion der ursprünglichen Probe sowie die gesamte Sedimentfraktion untersucht.
7. Berechnung und Auswertung
Die Mitgliedstaaten stellen sicher, dass die unter dieser Nummer beschriebenen Verfahren angewandt werden, wenn eine amtliche Analyse zur Schätzung des Anteils (und nicht nur des Vorhandenseins) von Bestandteilen tierischen Ursprungs durchgeführt wird.
Die Berechnung kann nur erfolgen, wenn die tierischen Bestandteile Knochenfragmente enthalten.
Knochenfragmente warmblütiger Landtiere (d. h. Säugetiere und Vögel) sind von den verschiedenen Fischknochen im mikroskopischen Präparat durch die charakteristischen Lakunen (Knochenzellenhöhlen) zu unterscheiden. Die Schätzung des Gehalts an tierischen Bestandteilen im Probenmaterial erfolgt unter Berücksichtigung
|
— |
des geschätzten Gehalts (Massenanteil) an Knochenfragmenten im konzentrierten Sediment und |
|
— |
des Knochengehalts (Massenanteil) in den Bestandteilen tierischen Ursprungs. |
Die Schätzung ist auf der Grundlage von (wenn möglich) mindestens 3 Präparaten und mindestens 5 Feldern pro Präparat durchzuführen. Bei Mischfuttermitteln enthält das konzentrierte Sediment in der Regel neben Knochenfragmenten von Landtieren und Fischknochenfragmenten auch andere Partikel mit hohem spezifischen Gewicht, z. B. Mineralien, Sand, verholzte Pflanzenfragmente usw.
7.1. Geschätzter Anteil der Knochenfragmente
Knochenfragmente von Landtieren ( %) = (S × c)/W
Fischknochen- und Schuppenfragmente ( %) = (S × d)/W
|
(S = |
Sediment (mg), c = Korrekturfaktor ( %) für den geschätzten Anteil von Landtierknochen im Sediment, d = Korrekturfaktor ( %) für den geschätzten Anteil von Fischknochen- und Schuppenfragmenten im Sediment, W = Einwaage des Probenmaterials für die Sedimentation (mg)). |
7.2. Geschätzter Anteil an Bestandteilen tierischen Ursprungs
Der Knochenanteil in tierischen Erzeugnissen kann sehr stark variieren. (Der Prozentsatz liegt im Fall von Knochenmehlen in der Größenordnung von 50 bis 60 % und im Fall von Fleischmehlen bei 20 bis 30 %; bei Fischmehlen variiert der Knochen- und Schuppenanteil je nach Kategorie und Ursprung des Fischmehls und liegt normalerweise bei 10 bis 20 %.)
Ist die Art des Tiermehls in der Probe bekannt, so ist es möglich, den Anteil zu schätzen:
Geschätzter Anteil an Bestandteilen von Landtiererzeugnissen ( %) = (S × c)/(W × f) × 100,
geschätzter Anteil an Bestandteilen von Fischerzeugnissen ( %) = (S × d)/(W × f) × 100,
|
(S = |
Sediment (mg), c = Korrekturfaktor ( %) für den geschätzten Anteil von Landtierknochenbestandteilen im Sediment, d = Korrekturfaktor ( %) für den geschätzten Anteil von Fischknochen- und Schuppenfragmenten im Sediment, f = Korrekturfaktor für den Knochenanteil der Bestandteile tierischen Ursprungs der Untersuchungsprobe, W = Einwaage des Probenmaterials für die Sedimentation (mg)). |
8. Darstellung der Untersuchungsergebnisse
Der Bericht enthält zumindest Informationen über das Vorhandensein von Bestandteilen, die von Landtieren oder von Fischmehl stammen. Die verschiedenen Befunde können wie folgt dargestellt werden:
|
8.1. |
Hinsichtlich des Vorhandenseins von Bestandteilen von Landtieren:
|
|
8.2. |
Und hinsichtlich des Vorhandenseins von Fischmehl:
Sofern von Fischen oder Landtieren stammende Bestandteile festgestellt werden, enthält der Bericht über die Untersuchungsergebnisse erforderlichenfalls auch eine Schätzung der Menge an nachgewiesenen Bestandteilen (x %, < 0,1 %, 0,1 bis 0,5 %, 0,5 bis 5 % oder > 5 %), nach Möglichkeit eine nähere Bestimmung der Art der Landtiere und der identifizierten tierischen Bestandteile (Muskelfasern, Knorpel, Knochen, Horn, Haare, Borsten, Federn, Blut, Eierschalen, Fischknochen, Schuppen). Wird die Menge an tierischen Bestandteilen geschätzt, ist der verwendete Korrekturfaktor f anzugeben. Werden Knochenbestandteile von Landtieren identifiziert, muss der Bericht folgenden Zusatz enthalten: „Es kann nicht ausgeschlossen werden, dass es sich um Bestandteile von Säugetieren handelt.“ Dieser Zusatz ist nicht notwendig in den Fällen, in denen die Knochenfragmente von Landtieren spezifiziert sind in Knochenfragmente von Geflügel oder von Säugetieren. |
9. Fakultatives Protokoll zur Analyse von Fett oder Öl
Folgendes Protokoll kann zur Analyse von Fett oder Öl verwendet werden:
|
— |
Handelt es sich um festes Fett, wird es beispielsweise in einem Mikrowellenofen erhitzt, bis es flüssig ist. |
|
— |
Der Probe werden mittels einer Pipette am Boden 40 ml Fett entnommen und in ein Zentrifugenröhrchen gegeben. |
|
— |
10 min bei 4 000 min-1 zentrifugieren. |
|
— |
Ist das Fett nach der Zentrifugierung fest, wird es noch einmal in einem Ofen erwärmt, bis es flüssig ist. Danach ist die Zentrifugierung 5 min lang bei 4 000 min-1 zu wiederholen. |
|
— |
Mithilfe eines kleinen Löffels oder eines Spatels wird eine Hälfte der abgegossenen Verunreinigungen auf eine kleine Petrischale oder einen Objektträger zur mikroskopischen Identifizierung eines möglichen Gehalts an tierischen Bestandteilen aufgebracht (Fleischfasern, Federn, Knochenfragmente usw.). Als Einbettungsmittel für die mikroskopische Untersuchung wird Paraffinöl oder Glyzerin empfohlen. |
|
— |
Die verbleibenden Verunreinigungen werden zur Sedimentierung gemäß 6.2 verwendet. |
(1) ABl. L 165 vom 30.4.2004, S. 1. Berichtigte Fassung im ABl. L 191 vom 28.5.2004, S. 1.
ANHANG VII
METHODE ZUR BERECHNUNG DES ENERGIEGEHALTS VON FUTTERMITTELN FÜR GEFLÜGEL
1. Berechnungsmethode und Formel des Energiegehalts
Der Energiegehalt von Mischfuttermitteln für Geflügel wird anhand der prozentualen Anteile bestimmter analytischer Bestandteile der Futtermittel nach nachstehender Formel berechnet. Dieser Wert wird in Megajoules (MJ) umsetzbarer, N-korrigierter Energie (ME) je kg Mischfuttermittel ausgedrückt und nach folgender Formel berechnet:
MJ ME/kg = 0,1551 × % Rohprotein +0,3431 × % Rohfett +0,1669 × % Stärke +0,1301 × % Gesamtzucker (ausgedrückt als Saccharose).
2. Toleranzen auf die angegebenen Gehalte
Ergibt sich bei den amtlichen Untersuchungen eine energieerhöhende oder energievermindernde Abweichung zwischen dem Kontrollergebnis und dem angegebenen Gehalt, so gilt eine Mindesttoleranz von 0,4 MJ ME/kg.
3. Ausdruck der Ergebnisse
Das nach vorstehender Formel errechnete Ergebnis ist mit nur einer Dezimalstelle anzugeben.
4. Probenahmeverfahren und Analysemethoden
Die Probenahme beim Mischfuttermittel und die Bestimmung des Gehalts an der Berechnungsmethode zugrundeliegenden analytischen Bestandteilen erfolgt in Übereinstimmung mit den gemeinschaftlichen Probenahmeverfahren bzw. Analysemethoden für die amtliche Untersuchung von Futtermitteln.
Anzuwenden sind:
|
— |
für die Bestimmung des Rohfettgehalts: Verfahren B der Methode zur Bestimmung des Gehalts an Rohölen und -fetten, wie in Anhang III Teil H beschrieben; |
|
— |
für die Bestimmung des Stärkegehalts: die polarimetrische Methode wie in Anhang III Teil L beschrieben. |
ANHANG VIII
ANALYSEMETHODEN ZUR UNTERSUCHUNG VON FUTTERMITTELN AUF DAS VORHANDENSEIN NICHT MEHR ZUGELASSENER ZUSATZSTOFFE
Wichtiger hinweis:Zum Nachweis des Vorhandenseins nicht mehr zugelassener Zusatzstoffe in Futtermitteln können empfindlichere Analysemethoden als die in diesem Anhang beschriebenen verwendet werden.Die in diesem Anhang aufgeführten Analysemethoden werden für Bestätigungszwecke herangezogen.
A. BESTIMMUNG DES METHYLBENZOQUATGEHALTS
7-Benzyloxy-6-butyl-3-methoxycarbonyl-4-chinolon
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Methylbenzoquatgehalts von Futtermitteln. Die Bestimmungsgrenze beträgt 1 mg/kg.
2. Prinzip
Methylbenzoquat wird mit methanolischer Methansulfonsäure-Lösung aus der Probe extrahiert. Der Extrakt wird mit Dichlormethan, mittels Ionenaustauschchromatografie und anschließend erneut mit Dichlormethan gereinigt. Der Methylbenzoquatgehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
|
3.1. |
Dichlormethan. |
|
3.2. |
Methanol, HPLC-Qualität. |
|
3.3. |
Mobile Phase für die HPLC Gemisch aus Methanol (3.2) und Wasser (HPLC-Qualität) 75 + 25 (V+V). Die Lösung wird durch einen 0,22-μm-Filter (4.5) filtriert und entgast (z. B. durch 10-minütige Ultraschallbehandlung). |
|
3.4. |
Methansulfonsäure-Lösung, c = 2 % 20,0 ml Methansulfonsäure werden mit Methanol (3.2) auf 1 000 ml verdünnt. |
|
3.5. |
Salzsäure, c = 10 % 100 ml Salzsäure (ρ201,18 g/ml) werden mit Wasser auf 1 000 ml verdünnt. |
|
3.6. |
Kationenaustauschharz Amberlit CG-120 (Na), 100 bis 200 Mesh Dieses Harz wird vor der Verwendung vorbehandelt. Von dem Harz werden 100 g mit 500 ml verdünnter Salzsäure (3.5) aufgeschlämmt und auf einer Heizplatte unter ständigem Rühren zum Sieden gebracht. Es wird abkühlen gelassen, und die Säure wird dekantiert. Anschließend wird durch einen Papierfilter unter Vakuum filtriert. Das Harz wird 2-mal mit je 500 ml Wasser und danach mit 250 ml Methanol (3.2) gewaschen. Anschließend wird das Harz nochmals mit 250 ml Methanol gespült und der Filterkuchen trocken gesaugt. Das getrocknete Harz wird in einer verschlossenen Flasche aufbewahrt. |
|
3.7. |
Standardsubstanz: Methylbenzoquat (7-Benzyloxy-6-butyl-3-methoxycarbonyl-4-chinolon), rein |
|
3.7.1. |
Von der Standardsubstanz (3.7) werden 50 mg auf 0,1 mg genau eingewogen, in einem 100-ml-Messkolben in methanolischer Methansulfonsäure (3.4) gelöst, zur Marke aufgefüllt und gemischt. |
|
3.7.2. |
Von der Methylbenzoquat-Standard-Stammlösung (3.7.1) werden 5,0 ml in einen 50-ml-Messkolben überführt. Es wird mit Methanol (3.2) zur Marke aufgefüllt und gemischt. |
|
3.7.3. |
Von der Methylbenzoquat-Standardlösung (3.7.2) werden 1,0, 2,0, 3,0, 4,0 bzw. 5,0 ml jeweils in einen 25-ml-Messkolben überführt. Mit der mobilen Phase (3.3) wird zur Marke aufgefüllt und durchmischt. Diese Lösungen enthalten Methylbenzoquat in Konzentrationen von 2,0, 4,0, 6,0, 8,0 bzw. 10,0 μg/ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden. |
4. Geräte
|
4.1. |
Laborschüttler. |
|
4.2. |
Rotationsfilmverdampfer. |
|
4.3. |
Glassäule (250 × 15 mm) mit Absperrhahn und Reservoir mit einem Fassungsvermögen von ca. 200 ml. |
|
4.4. |
HPLC-Einrichtung mit UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor |
|
4.4.1. |
HPLC-Trennsäule, 300 × 4 mm, C18, 10 μm Korngröße, oder vergleichbare Säule. |
|
4.5. |
Membranfilter, 0,22 μm Porengröße. |
|
4.6. |
Membranfilter, 0,45 μm Porengröße. |
5. Verfahren
5.1. Allgemeines
|
5.1.1. |
Zur Prüfung, dass weder Methylbenzoquat noch Störsubstanzen vorhanden sind, wird eine Blindprobe untersucht. |
|
5.1.2. |
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Methylbenzoquat angereichert wurde. Die zugesetzte Menge an Methylbenzoquat sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 15 mg/kg werden 20 g der Blindprobe mit 600 μl Standard-Stammlösung (3.7.1) versetzt. Es wird gemischt und 10 min stehen gelassen, bevor mit der Extraktion (5.2) fortgefahren wird. Für den Zweck dieser Methode muss die Blindprobe ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Methylbenzoquat darf nicht nachweisbar sein. |
5.2. Extraktion
Von der vorbereiteten Probe werden 20 g auf 0,01 g genau in einen 250-ml-Erlenmeyerkolben eingewogen. Nach Zugabe von 100,0 ml Methansulfonsäure-Lösung (3.4) wird mechanisch (4.1) 30 min geschüttelt. Die Lösung wird durch ein Filterpapier filtriert und das Filtrat für die Flüssig-Flüssig-Trennung (5.3) aufbewahrt.
5.3. Flüssig-Flüssig-Trennung
Von dem durch 5.2 erhaltenen Filtrat werden 25,0 ml in einen 500-ml-Scheidetrichter überführt, der 100 ml Salzsäure (3.5) enthält. Nach Zugabe von 100 ml Dichlormethan (3.1) wird 1 min geschüttelt, die Phasentrennung abgewartet und die untere Phase (Dichlormethan) in einen 500-ml-Rundkolben ablaufen gelassen. Die Extraktion der wässrigen Phase wird 2-mal mit je 40 ml Dichlormethan wiederholt und die Extrakte werden zum ersten Extrakt im Rundkolben gegeben. Das Dichlormethanextrakt wird am Rotationsverdampfer (4.2) bei 40 oC und vermindertem Druck bis zur Trockne eingeengt. Der Rückstand wird in 20 bis 25 ml Methanol (3.2) gelöst und der Kolben verschlossen. Der gesamte Extrakt wird für die Ionenaustauschchromatografie (5.4) aufbewahrt.
5.4. Ionenaustauschromatografie
5.4.1.
In das untere Ende der Glassäule (4.3) wird ein Glaswattebausch eingebracht. Von dem behandelten Kationenaustauschharz (3.6) werden 5,0 g mit 50 ml Salzsäure (3.5) aufgeschlämmt, in die Glassäule gegeben und absetzen gelassen. Nachdem der Säureüberschuss bis knapp zur Harzoberfläche abgelaufen ist, wird die Säule so lange mit Wasser gewaschen, bis die Waschflüssigkeit neutral gegen Lackmus ist. Dann werden 50 ml Methanol (3.2) auf die Säule gegeben und bis zur Harzoberfläche ablaufen gelassen.
5.4.2.
Der Extrakt (5.3) wird mittels einer Pipette vorsichtig auf die Säule gegeben. Der Rundkolben wird 2-mal mit je 5 bis 10 ml Methanol (3.2) gespült. Die anfallenden Spüllösungen werden ebenfalls auf die Säule gegeben. Nachdem der Extrakt bis zur Harzoberfläche abgelaufen ist, wird die Säule mit 50 ml Methanol gewaschen, wobei die Flussrate höchstens 5 ml/min betragen darf. Die Waschflüssigkeit wird verworfen. Das Methylbenzoquat wird mit 150 ml methanolischer Methansulfonsäure-Lösung (3.4) von der Säule eluiert und das Eluat in einem 250-ml-Erlenmeyerkolben aufgefangen.
5.5. Flüssig-Flüssig-Trennung
Das Eluat (5.4.2) wird in einen 1-l-Scheidetrichter überführt. Der Erlenmeyerkolben wird mit 5 bis 10 ml Methanol (3.2) gespült und die anfallende Spülflüssigkeit ebenfalls in den Scheidetrichter gegeben. Dann werden 300 ml Salzsäure-Lösung (3.5) und 130 ml Dichlormethan (3.1) zugefügt. Es wird 1 min geschüttelt und die Phasentrennung abgewartet. Die untere Phase (Dichlormethan) wird in einen 500-ml-Rundkolben abgelassen. Die Extraktion der wässrigen Phase wird 2-mal mit je 70 ml Dichlormethan wiederholt, und diese Extrakte werden zum ersten Extrakt im Rundkolben gegeben.
Der Dichlormethanextrakt wird am Rotationsverdampfer (4.2) bei 40 oC und vermindertem Druck bis zur Trockne eingeengt. Der Rückstand im Rundkolben wird in ca. 5 ml Methanol (3.2) gelöst und die Lösung quantitativ in einen 10-ml-Messkolben überführt. Der Rundkolben wird 2-mal mit je 1 bis 2 ml Methanol gespült, und die Spüllösungen werden in den Messkolben überführt. Es wird mit Methanol zur Marke aufgefüllt und durchmischt. Ein aliquoter Teil wird durch einen Membranfilter (4.6) filtriert. Die Lösung wird für die HPLC-Bestimmung (5.6) aufbewahrt.
5.6. HPLC-Bestimmung
5.6.1.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
|
— |
HPLC-Trennsäule (4.4.1), |
|
— |
mobile Phase für die HPLC: Methanol-Wasser-Gemisch (3.3), |
|
— |
Durchflussrate: 1 bis 1,5 ml/min, |
|
— |
Detektionswellenlänge: 265 nm, |
|
— |
Einspritzvolumen: 20 bis 50 μl. |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.7.3), die 4 μg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.6.2.
Jede Kalibrierlösung (3.7.3) wird mehrmals eingespritzt, und die Peakhöhen (-flächen) für die einzelnen Konzentrationen werden gemessen. Es wird eine Kalibrationskurve gezeichnet, indem die mittleren Peakhöhen oder -flächen auf der Ordinate und die dazugehörigen Konzentrationen in μg/ml auf der Abszisse aufgetragen werden.
5.6.3.
Der Probenextrakt (5.5) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird, und die mittlere Peakhöhe (-fläche) der Methylbenzoquatpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Methylbenzoquatpeaks der Probenlösung wird anhand der Kalibrationskurve (5.6.2) die Konzentration der Probenlösung in μg/ml bestimmt.
Der Methylbenzoquatgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:

wobei:
|
c |
= |
Methylbenzoquatkonzentration der Probenlösung in μg/ml. |
|
m |
= |
Probeneinwaage in g. |
7. Überprüfung der Ergebnisse
7.1. Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung und der Kalibrierlösung (3.7.3), die 10 μg/ml enthält, verglichen werden.
7.1.1.
Ein Probenextrakt wird mit einer geeigneten Menge Methylbenzoquat-Standardlösung (3.7.2) versetzt. Die Menge des zugesetzten Methylbenzoquats muss dem erwarteten Methylbenzoquatgehalt des Probenextrakts entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Methylbenzoquatpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2.
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
|
a) |
Die Wellenlängen bei maximaler Absorption des Proben- und des Standardspektrums an der Peakspitze des Chromatogramms müssen innerhalb eines Bereichs übereinstimmen, der durch das Auflösungsvermögen des Detektionssystems bestimmt wird. Für die Diodenarray-Detektion beträgt dieser Bereich in der Regel ± 2 nm. |
|
b) |
Zwischen 220 und 350 nm dürfen sich das Proben- und das Standardspektrum an den Peakspitzen des Chromatogramms in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den beiden Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Standardanalyten beträgt. |
|
c) |
Zwischen 220 und 350 nm dürfen sich die Spektren des Probenextrakts im Anstieg, an der Spitze und im Abstieg des Probenpeaks in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Spektrums am Peakmaximum beträgt. |
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf bei Methylbenzoquatgehalten von 4-20 mg/kg 10 % des höheren Wertes nicht überschreiten.
7.3. Wiederfindungsrate
Für eine angereicherte Blindprobe muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
Es wurden 5 Proben von 10 Laboratorien untersucht. Jede Probe wurde doppelt analysiert.
|
|
Blindprobe |
Mehl 1 |
Pellets 1 |
Mehl 2 |
Pellets 2 |
|
Mittelwert [mg/kg] |
NN |
4,50 |
4,50 |
8,90 |
8,70 |
|
sr [mg/kg] |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
VKr [ %] |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
sR [mg/kg] |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
VKR [ %] |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Wiederfindung [ %] |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
NN |
= |
Nicht nachweisbar |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
B. BESTIMMUNG DES OLAQUINDOXGEHALTS
N-(2-Hydroxyethyl)-3-methyl-2-chinoxalin-carbamid-1,4-dioxid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Olaquindoxgehalts von Futtermitteln. Die Bestimmungsgrenze beträgt 5 mg/kg.
2. Prinzip
Die Probe wird mit einem Methanol-Wasser-Gemisch extrahiert. Der Gehalt an Olaquindox wird durch Umkehrphasen-Hochleistungsflüssigkeitschromatografie (RP-HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
|
3.1. |
Methanol. |
|
3.2. |
Methanol, HPLC-Qualität. |
|
3.3. |
Wasser, HPLC-Qualität. |
|
3.4. |
Mobile Phase für die HPLC: Wasser (3.3)-Methanol (3.2)-Gemisch, z. B. 900 + 100 (V+V). |
|
3.5. |
Standardsubstanz: Olaquindox, rein, N-(2-Hydroxyethyl)-3-methyl-2-chinoxalin-carbamid-1,4-dioxid, E 851 |
|
3.5.1. |
Von Olaquindox (3.5) werden 50 mg auf 0,1 mg genau in einen 200-ml-Messkolben eingewogen, und es werden 190 ml Wasser zugefügt. Dann wird der Kolben 20 min in ein Ultraschallbad (4.1) gestellt. Nach der Ultraschallbehandlung wird die Lösung auf Raumtemperatur gebracht, es wird mit Wasser zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt und im Kühlschrank aufbewahrt. Die Lösung muss monatlich frisch hergestellt werden. |
|
3.5.2. |
Von der Standard-Stammlösung (3.5.1) werden 10,0 ml in einen 100-ml-Messkolben überführt. Es wird mit der mobilen Phase (3.4) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt und im Kühlschrank aufbewahrt. Die Lösung muss täglich frisch hergestellt werden. |
|
3.5.3. |
Von der Standardlösung (3.5.2) werden 1,0, 2,0, 5,0, 10,0, 15,0 bzw. 20,0 ml jeweils in einen 50-ml-Messkolben überführt. Mit der mobilen Phase (3.4) wird zur Marke aufgefüllt und durchmischt. Der Kolben wird mit Aluminiumfolie umhüllt. Diese Kalibrierlösungen enthalten jeweils 0,5, 1,0, 2,5, 5,0, 7,5 bzw. 10,0 μg Olaquindox je ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden. |
4. Geräte
|
4.1. |
Ultraschallbad. |
|
4.2. |
Mechanisches Schüttelgerät. |
|
4.3. |
HPLC-Einrichtung mit UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor |
|
4.3.1. |
HPLC-Trennsäule, 250 × 4 mm, C18, 10 μm Korngröße, oder vergleichbare Säule. |
|
4.4. |
Membranfilter, 0,45 μm Porengröße. |
5. Verfahren
|
Anmerkung |
: |
Olaquindox ist lichtempfindlich. Alle Analysenschritte sind deshalb bei gedämpftem Licht oder unter Verwendung von Braunglasgeräten durchzuführen. |
5.1. Allgemeines
|
5.1.1. |
Zur Prüfung, dass weder Olaquindox noch Störsubstanzen vorhanden sind, wird eine Blindprobe untersucht. |
|
5.1.2. |
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Olaquindox angereichert wurde. Die zugesetzte Menge an Olaquindox sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 50 mg/kg werden 10,0 ml der Standard-Stammlösung (3.5.1) in einen 250-ml-Erlenmeyerkolben überführt und im Stickstoffstrom auf ca. 0,5 ml eingeengt. Dann werden 50 g der Blindprobe zugegeben. Es wird gründlich gemischt und 10 min stehen gelassen, bevor mit der Extraktion (5.2) fortgefahren wird.
|
5.2. Extraktion
Von der Probe werden 50 g auf 0,01 g genau in einen 1 000-ml-Erlenmeyerkolben eingewogen und mit 100 ml Methanol (3.1) versetzt. Der Kolben wird 5 min in das Ultraschallbad (4.1) gestellt. Es werden 410 ml Wasser zugegeben, und die Probenlösung wird weitere 15 min mit Ultraschall behandelt. Danach wird der Kolben aus dem Ultraschallbad genommen, und es wird 30 min auf dem Schüttelgerät (4.2) geschüttelt. Die Probenlösung wird durch einen Faltenfilter filtriert. Von dem Filtrat werden 10,0 ml in einen 20-ml-Messkolben überführt, es wird mit Wasser zur Marke aufgefüllt und gemischt. Ein aliquoter Teil wird durch einen Membranfilter (4.4) filtriert (siehe Bemerkung 9). Anschließend wird die HPLC-Bestimmung (5.3) durchgeführt.
5.3. HPLC-Bestimmung
5.3.1.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
|
Analysensäule (4.3.1) |
|
|
Mobile Phase (3.4): |
Wasser (3.3)-Methanol (3.2)- Gemisch 900 + 100 (V+V) |
|
Durchflussrate: |
1,5 bis 2 ml/min |
|
Detektionswellenlänge: |
380 nm |
|
Einspritzvolumen: |
20 bis 100 μl |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.5.3), die 2,5 μg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.3.2.
Jede Kalibrierlösung (3.5.3) wird mehrmals eingespritzt, und die Peakhöhen (-flächen) für die einzelnen Konzentrationen werden gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) auf der Ordinate und die dazugehörigen Konzentrationen in μg/ml auf der Abszisse aufgetragen werden.
5.3.3.
Der Probenextrakt (5.2) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird. Die mittlere Peakhöhe (-fläche) der Olaquindoxpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Olaquindoxpeaks der Probenlösung wird anhand der Kalibrationskurve (5.3.2) die Konzentration der Probenlösung in μg/ml bestimmt.
Der Olaquindoxgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:

wobei:
|
c |
= |
Olaquindoxkonzentration des Probenextrakts (5.2) in μg/ml. |
|
m |
= |
Probeneinwaage in g. |
7. Überprüfung der Ergebnisse
7.1. Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung (5.2) und der Kalibrierlösung (3.5.3), die 5,0 μg/ml enthält, verglichen werden.
7.1.1.
Ein Probenextrakt (5.2) wird mit einer geeigneten Menge Kalibrierlösung (3.5.3) versetzt. Die zugesetzte Olaquindoxmenge muss dem erwarteten Olaquindoxgehalt des Probenextrakts entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Olaquindoxpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks des nicht angereicherten Probenextrakts abweichen.
7.1.2.
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
|
a) |
Die Wellenlängen bei maximaler Absorption des Proben- und des Standardspektrums an der Peakspitze des Chromatogramms müssen innerhalb eines Bereichs übereinstimmen, der durch das Auflösungsvermögen des Detektionssystems bestimmt wird. Für die Diodenarray-Detektion beträgt dieser Bereich in der Regel ± 2 nm. |
|
b) |
Zwischen 220 und 400 nm dürfen sich das Proben- und das Standardspektrum an den Peakspitzen des Chromatogramms in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den beiden Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Standardanalyten beträgt. |
|
c) |
Zwischen 220 und 400 nm dürfen sich die Spektren des Probenextrakts im Anstieg, an der Spitze und im Abstieg des Probenpeaks in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Spektrums am Peakmaximum beträgt. |
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf bei einem Olaquindoxgehalt zwischen 10 und 200 mg/kg 15 % des höheren Wertes nicht überschreiten.
7.3. Wiederfindungsrate
Für eine angereicherte Blindprobe muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
Es wurde ein EG-Ringversuch durchgeführt, bei dem 4 Ferkelfuttermittel einschließlich eines Blindfuttermittels von bis zu 13 Laboratorien untersucht wurden. Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst.
|
|
Probe 1 |
Probe 2 |
Probe 3 |
Probe 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
Mittelwert [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
sR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
VKr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
VKR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
Sollgehalt |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Wiederfindung [ %] |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
Anzahl der Laboratorien |
|
n |
= |
Anzahl der Einzelwerte |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
9. Bemerkung
Obwohl die Methode für Futtermittel mit Olaquindoxgehalten von mehr als 100 mg/kg nicht validiert wurde, können durch Verringerung der Einwaage und/oder Verdünnung des Extrakts (5.2) auf den Konzentrationsbereich der Kalibrationskurve (5.3.2) zufriedenstellende Ergebnisse erzielt werden.
C. BESTIMMUNG DES AMPROLIUMGEHALTS
1-[(4-Amino-2-propylpyrimidin-5-yl)methyl]-2-methyl-pyridiniumchlorid-hydrochlorid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Amproliumgehalts von Futtermitteln und Vormischungen. Die Nachweisgrenze beträgt 1 mg/kg, die Bestimmungsgrenze 5 mg/kg.
2. Prinzip
Die Probe wird mit einem Methanol-Wasser-Gemisch extrahiert. Nach Verdünnung mit der mobilen Phase und Membranfiltration wird der Amproliumgehalt mittels Hochleistungsflüssigkeitschromatografie (HPLC) über ein Kationenaustauschermaterial unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
|
3.1. |
Methanol. |
|
3.2. |
Acetonitril, HPLC-Qualität. |
|
3.3. |
Wasser, HPLC-Qualität. |
|
3.4. |
Natriumdihydrogenphosphatlösung, c = 0,1 mol/l Von dem Natriumdihydrogenphosphatmonohydrat werden 13,80 g in einem 1 000-ml-Messkolben in Wasser (3.3) gelöst. Es wird mit Wasser (3.3) zur Marke aufgefüllt und gemischt. |
|
3.5. |
Natriumperchloratlösung, c = 1,6 mol/l Von dem Natriumperchloratmonohydrat werden 224,74 g in einem 1 000-ml-Messkolben in Wasser (3.3) gelöst. Es wird mit Wasser (3.3) zur Marke aufgefüllt und gemischt. |
|
3.6. |
Mobile Phase für die HPLC (siehe Bemerkung 9.1) Mischung aus Acetonitril (3.2), Natriumdihydrogenphosphatlösung (3.4) und Natriumperchloratlösung (3.5), 450 + 450 + 100 (V+V+V). Vor der Verwendung wird die Lösung durch einen 0,22-μm-Membranfilter (4.3) filtriert und entgast (z. B. mindestens 15 min im Ultraschallbad (4.4)). |
|
3.7. |
Standardsubstanz: Amprolium, rein, 1-[(4-Amino-2-propylpyrimidin-5-yl)methyl]-2-methyl-pyridiniumchlorid-hydrochlorid, E 750 (siehe 9.2) |
|
3.7.1. |
Von Amprolium (3.7) werden 50 mg auf 0,1 mg genau in einen 100-ml-Messkolben eingewogen, in 80 ml Methanol (3.l) gelöst und 10 min in ein Ultraschallbad (4.4) gestellt. Nach der Ultraschallbehandlung wird die Lösung auf Raumtemperatur gebracht; es wird mit Wasser zur Marke aufgefüllt und gemischt. Bei einer Temperatur von ≤ 4 oC ist diese Lösung 1 Monat lang haltbar. |
|
3.7.2. |
Von der Standard-Stammlösung (3.7.1) werden 5,0 ml in einen 50-ml-Messkolben pipettiert; es wird mit der Extraktionslösung (3.8) zur Marke aufgefüllt und gemischt. Bei einer Temperatur von ≤ 4 oC ist diese Lösung 1 Monat lang haltbar. |
|
3.7.3. |
Von der Amprolium-Standardlösung (3.7.2) werden 0,5, 1,0 bzw. 2,0 ml jeweils in einen 50-ml-Messkolben überführt. Mit der mobilen Phase (3.6) wird zur Marke aufgefüllt und durchmischt. Diese Lösungen enthalten jeweils 0,5, 1,0 bzw. 2,0 μg Amprolium je ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden. |
|
3.8. |
Extraktionslösung Methanol (3.1)-Wasser-Mischung 2 + 1 (V+V). |
4. Geräte
|
4.1. |
HPLC-Einrichtung mit Injektionssystem für Einspritzvolumina von 100 μl |
|
4.1.1. |
HPLC-Trennsäule für die Kationenaustauschchromatografie, 125 × 4 mm, z. B. Nucleosil 10 SA, 5 oder 10 μm Korngröße, oder vergleichbare Säule. |
|
4.1.2. |
UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor. |
|
4.2. |
Membranfilter, PTFE-Material, 0,45 μm Porengröße. |
|
4.3. |
Membranfilter, 0,22 μm Porengröße. |
|
4.4. |
Ultraschallbad. |
|
4.5. |
Mechanisches Schüttelgerät oder Magnetrührer. |
5. Verfahren
5.1. Allgemeines
5.1.1.
Für die Durchführung des Wiederfindungstests (5.1.2) muss zur Prüfung, dass weder Amprolium noch Störsubstanzen vorhanden sind, eine Blindprobe untersucht werden. Die Blindprobe muss ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Amprolium oder Störsubstanzen dürfen nicht nachweisbar sein.
5.1.2.
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Amprolium angereichert wurde. Die zugesetzte Menge an Amprolium sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 100 mg/kg werden 10,0 ml der Standard-Stammlösung (3.7.1) in einen 250-ml-Erlenmeyerkolben überführt und auf ca. 0,5 ml eingeengt. Dann werden 50 g der Blindprobe zugegeben. Es wird gründlich gemischt, 10 min stehen gelassen und nochmals mehrfach gemischt, bevor mit der Extraktion (5.2) fortgefahren wird.
Ist eine der zu untersuchenden Probe ähnliche Blindprobe nicht verfügbar (siehe 5.1.1), so kann ein Wiederfindungstest mithilfe des Additionsverfahrens durchgeführt werden. In diesem Fall wird die zu untersuchende Probe mit einer Amproliummenge angereichert, die der bereits in der Probe vorhandenen Menge entspricht. Diese Probe wird zusammen mit der nicht angereicherten Probe untersucht, und die Wiederfindungsrate kann durch Subtraktion ermittelt werden.
5.2. Extraktion
5.2.1.
Je nach Amproliumgehalt werden 5 bis 40 g der Probe auf 0,01 g genau in einen 500-ml-Erlenmeyerkolben eingewogen und mit 200 ml Extraktionslösung (3.8) versetzt. Der Kolben wird 15 min in ein Ultraschallbad (4.4) gestellt und anschließend 60 min auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer (4.5) gerührt. Ein aliquoter Teil des Extrakts wird mit der mobilen Phase (3.6) auf einen Amproliumgehalt von 0,5 bis 2 μg/ml verdünnt und gemischt (siehe Bemerkung 9.3). Von dieser verdünnten Lösung werden 5 bis 10 ml durch einen Membranfilter (4.2) filtriert. Anschließend wird die HPLC-Bestimmung (5.3) durchgeführt.
5.2.2.
Je nach Amproliumgehalt werden 1 bis 4 g der Vormischung auf 0,001 g genau in einen 500-ml-Erlenmeyerkolben eingewogen und mit 200 ml Extraktionslösung (3.8) versetzt. Der Kolben wird 15 min in ein Ultraschallbad (4.4) gestellt und anschließend 1 h auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer gerührt (4.5). Ein aliquoter Teil des Extrakts wird mit der mobilen Phase (3.6) auf einen Amproliumgehalt von 0,5 bis 2 μg/ml verdünnt und gemischt. Von dieser verdünnten Lösung werden 5 bis 10 ml durch einen Membranfilter (4.2) filtriert. Anschließend wird die HPLC-Bestimmung (5.3) durchgeführt.
5.3. HPLC-Bestimmung
5.3.1.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren
|
Ergebnissen führen: |
|
|
HPLC-Trennsäule (4.1.1): |
125 × 4 mm, Kationenaustausch Nucleosil 10 SA, 5 oder 10 μm Korngröße, oder vergleichbare Säule |
|
Mobile Phase (3.6): |
Mischung aus Acetonitril (3.2), Natriumdihydrogenphosphat-Lösung (3.4) und Natriumperchlorat-Lösung (3.5), 450 + 450 + 100 (V+V+V) |
|
Durchflussrate: |
0,7 bis 1 ml/min |
|
Detektionswellenlänge: |
264 nm |
|
Einspritzvolumen: |
100 μl |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.7.3), die 1,0 μg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.3.2.
Jede Kalibrierlösung (3.7.3) wird mehrmals eingespritzt, und die mittleren Peakhöhen (-flächen) werden für die einzelnen Konzentrationen gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) auf der Ordinate und die dazugehörigen Konzentrationen in μg/ml auf der Abszisse aufgetragen werden.
5.3.3.
Der Probenextrakt (5.2) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird. Die mittlere Peakhöhe (-fläche) der Amproliumpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Amproliumpeaks der Probenlösung wird anhand der Kalibrationskurve (5.3.2) die Konzentration der Probenlösung in μg/ml bestimmt.
Der Amproliumgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
 [mg/kg]
[mg/kg]
wobei:
|
V |
= |
Volumen der Extraktionslösung (3.8) in ml gemäß 5.2 (d. h. 200 ml), |
|
c |
= |
Amproliumkonzentration der Probenlösung (5.2) in μg/ml, |
|
f |
= |
Verdünnungsfaktor gemäß 5.2, |
|
m |
= |
Probeneinwaage in g. |
7. Überprüfung der Ergebnisse
7.1. Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung (5.2) und der Kalibrierlösung (3.7.3), die 2,0 μg/ml enthält, verglichen werden.
7.1.1.
Eine Probenlösung (5.2) wird mit einer geeigneten Menge Kalibrierlösung (3.7.3) angereichert. Die zugesetzte Amproliummenge muss dem Amproliumgehalt des Probenextrakts entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Amproliumpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2.
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
|
a) |
Die Wellenlängen bei maximaler Absorption des Proben- und des Standardspektrums an der Peakspitze des Chromatogramms müssen innerhalb eines Bereichs übereinstimmen, der durch das Auflösungsvermögen des Detektionssystems bestimmt wird. Für die Diodenarray-Detektion beträgt dieser Bereich in der Regel ± 2 nm. |
|
b) |
Zwischen 210 und 320 nm dürfen sich das Proben- und das Standardspektrum an den Peakspitzen des Chromatogramms in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den beiden Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Standardanalyten beträgt. |
|
c) |
Zwischen 210 und 320 nm dürfen sich die Spektren des Probenextrakts im Anstieg, an der Spitze und im Abstieg des Probenpeaks in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Spektrums am Peakmaximum beträgt. |
Wird eines dieser Kriterien nicht erfüllt, so gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
|
— |
15 % des höheren Werts bei einem Amproliumgehalt zwischen 25 und 500 mg/kg, |
|
— |
75 mg/kg bei einem Amproliumgehalt zwischen 500 und 1 000 mg/kg, |
|
— |
7,5 % des höheren Werts bei einem Amproliumgehalt von über 1 000 mg/kg. |
7.3. Wiederfindungsrate
Bei einer angereicherten (Blind-)Probe muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
Bei einem Ringversuch wurden 3 Geflügelfuttermittel (Proben 1 bis 3), 1 Mineralfuttermittel (Probe 4) und 1 Vormischung (Probe 5) untersucht. Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst.
|
|
Probe 1 (Blindprobe) |
Probe 2 |
Probe 3 |
Probe 4 |
Probe 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
Mittelwert [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
VKr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
VKR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Sollgehalt [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
Anzahl der Laboratorien |
|
n |
= |
Anzahl der Einzelwerte |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
9. Bemerkungen
|
9.1. |
Enthält die Probe Thiamin, so erscheint der Thiaminpeak in der Chromatografie kurz vor dem Amproliumpeak. Bei Anwendung dieser Methode müssen Amprolium und Thiamin getrennt werden. Sind Amprolium und Thiamin nicht durch die bei dieser Methode verwendete Säule (4.1.1) getrennt, sollten bis zu 50 % des Acetonitrilanteils der mobilen Phase (3.6) durch Methanol ersetzt werden. |
|
9.2. |
Gemäß dem britischen Arzneibuch zeigt das Spektrum einer Amproliumlösung (c = 0,02 mol/l) in Salzsäure (c = 0,1 mol/l) Maxima bei 246 nm und 262 nm. Die Absorption beträgt 0,84 bei 246 nm bzw. 0,80 bei 262 nm. |
|
9.3. |
Der Extrakt muss immer mit der mobilen Phase verdünnt werden, da sich sonst die Retentionszeit des Amproliumpeaks durch Veränderungen der Ionenstärke beträchtlich verschieben kann. |
D. BESTIMMUNG DES CARBADOXGEHALTS
Methyl 3-(2-chinoxalinylmethylen)carbazat N 1 ,N 4 -dioxid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Carbadoxgehalts von Futtermitteln, Vormischungen und Zubereitungen. Die Nachweisgrenze beträgt 1 mg/kg, die Bestimmungsgrenze 5 mg/kg.
2. Prinzip
Die Probe wird mit Wasser äquilibriert und anschließend mit Methanol-Acetonitril extrahiert. Bei Futtermitteln wird ein aliquoter Teil des filtrierten Extrakts auf einer Aluminiumoxid-Säule gereinigt. Bei Vormischungen und Zubereitungen wird ein aliquoter Teil des filtrierten Extrakts mit einem Gemisch aus Wasser, Methanol und Acetonitril auf eine geeignete Konzentration verdünnt. Der Carbadoxgehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
|
3.1. |
Methanol. |
|
3.2. |
Acetonitril, HPLC-Qualität. |
|
3.3. |
Essigsäure, w = 100 %. |
|
3.4. |
Aluminiumoxid: neutral, Aktivitätsstufe I. |
|
3.5. |
Methanol-Acetonitril 1 + 1 (V+V) 500 ml Methanol (3.1) werden mit 500 ml Acetonitril (3.2) gemischt. |
|
3.6. |
Essigsäure, σ = 10 % 10 ml Essigsäure (3.3) werden auf 100 ml in Wasser gelöst. |
|
3.7. |
Natriumacetat. |
|
3.8. |
Wasser, HPLC-Qualität. |
|
3.9. |
Acetat-Pufferlösung, c = 0,01 mol/l, pH = 6,0 0,82 g Natriumacetat (3.7) werden in 700 ml Wasser (3.8) gelöst, und der pH-Wert wird mit Essigsäure (3.6) auf 6,0 eingestellt. Die Lösung wird in einen 1 000-ml-Messkolben überführt. Es wird mit Wasser (3.8) zur Marke aufgefüllt und gemischt. |
|
3.10. |
Mobile Phase für die HPLC 825 ml der Acetat-Pufferlösung (3.9) werden mit 175 ml Acetonitril (3.2) vermischt. Die Lösung wird durch einen 0,22-μm-Filter (4.5) filtriert und entgast (z. B. durch 10-minütige Ultraschallbehandlung); |
|
3.11. |
Standardsubstanz Carbadox, rein: Methyl 3-(2-chinoxalinylmethylen)carbazat N1,N4-dioxid, E 850 |
|
3.11.1. |
Von der Carbadox-Standardsubstanz (3.11) werden 25 mg auf 0,1 mg genau in einen 250-ml-Messkolben eingewogen und im Ultraschallbad (4.7) in Methanol-Acetonitril (3.5) gelöst. Nach der Ultraschallbehandlung wird die Lösung auf Raumtemperatur gebracht, zur Marke mit Methanol-Acetonitril (3.5) aufgefüllt und gemischt. Der Kolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 oC ist diese Lösung 1 Monat lang haltbar. |
|
3.11.2. |
Von der Standard-Stammlösung (3.11.1) werden 2,0, 5,0, 10,0 bzw. 20,0 ml jeweils in einen 100-ml-Messkolben überführt. Es werden jeweils 30 ml Wasser hinzugefügt; es wird mit Methanol-Acetonitril (3.5) zur Marke aufgefüllt und gemischt. Die Kolben werden mit Aluminiumfolie umhüllt. Diese Lösungen enthalten jeweils 2,0, 5,0, 10,0 bzw. 20,0 μg/ml Carbadox. Kalibrierlösungen müssen vor Gebrauch frisch hergestellt werden.
|
|
3.12. |
Wasser-[Methanol-Acetonitril (3.5)]-Gemisch, z. B. 300 + 700 (V+V). 300 ml Wasser werden mit 700 ml des Methanol-Acetonitril-Gemisches (3.5) vermischt. |
4. Geräte
|
4.1. |
Laborschüttler oder Magnetrührer. |
|
4.2. |
Glasfaser-Filterpapier, z. B. Whatman GF/A oder gleichwertig. |
|
4.3. |
Glassäule (Länge 300 bis 400 mm, Innendurchmesser etwa 10 mm) mit Sinterglasfritte und Auslaufhahn
|
|
4.4. |
HPLC-Einrichtung mit Injektionssystem für Einspritzvolumina von 20 μl |
|
4.4.1. |
HPLC-Trennsäule, 300 × 4 mm, C18, 10 μm Korngröße, oder vergleichbare Säule. |
|
4.4.2. |
UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor mit einem Messbereich von 225 bis 400 nm. |
|
4.5. |
Membranfilter, 0,22 μm Porengröße. |
|
4.6. |
Membranfilter, 0,45 μm Porengröße. |
|
4.7. |
Ultraschallbad. |
5. Verfahren
|
Anmerkung |
: |
Carbadox ist lichtempfindlich. Alle Analysenschritte sind daher bei gedämpftem Licht oder unter Verwendung von Braunglasgeräten durchzuführen. Alternativ können die Glaswaren mit Aluminiumfolie umwickelt werden. |
5.1. Allgemeines
5.1.1.
Für die Durchführung des Wiederfindungstests (5.1.2) muss zur Prüfung, dass weder Carbadox noch Störsubstanzen vorhanden sind, eine Blindprobe untersucht werden. Die Blindprobe muss ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Carbadox oder Störsubstanzen dürfen nicht nachweisbar sein.
5.1.2.
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe (5.1.1) untersucht wird, die mit Carbadox angereichert wurde. Die zugesetzte Menge an Carbadox sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 50 mg/kg werden 5,0 ml der Standard-Stammlösung (3.11.1) in einen 200-ml-Erlenmeyerkolben gegeben. Die Lösung wird in einem Stickstoffstrom auf ca. 0,5 ml eingeengt. Dann werden 10 g der Blindprobe zugegeben. Es wird gemischt und 10 min stehen gelassen, bevor mit dem Extraktionsschritt (5.2) fortgefahren wird.
Ist eine der zu untersuchenden Probe ähnliche Blindprobe nicht verfügbar (siehe 5.1.1), so kann ein Wiederfindungstest mithilfe des Standard-Additionsverfahrens durchgeführt werden. In diesem Fall wird die zu untersuchende Probe mit einer Carbadoxmenge angereichert, die der bereits in der Probe vorhandenen Menge entspricht. Diese Probe wird zusammen mit der nicht angereicherten Probe untersucht, und die Wiederfindungsrate kann durch Subtraktion ermittelt werden.
5.2. Extraktion
5.2.1.
Von der Probe werden 10 g auf 0,01 g genau in einen 200-ml-Erlenmeyerkolben eingewogen. Nach Zugabe von 15,0 ml Wasser wird gemischt und 5 min stehen gelassen. Dann werden 35,0 ml Methanol-Acetonitril (3.5) hinzugefügt, der Kolben wird verschlossen und 30 min auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer (4.1) gerührt. Die Lösung wird über Glasfaser-Filterpapier (4.2) filtriert. Diese Lösung wird für die Reinigung (5.3) aufbewahrt.
5.2.2.
Von der nicht gemahlenen Probe wird 1 g auf 0,001 g genau in einen 200-ml-Erlenmeyerkolben eingewogen. Nach Zugabe von 15,0 ml Wasser wird gemischt und 5 min stehen gelassen. Dann werden 35,0 ml Methanol-Acetonitril (3.5) hinzugefügt, der Kolben wird verschlossen und 30 min auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer (4.1) gerührt. Die Lösung wird über Glasfaser-Filterpapier (4.2) filtriert.
Ein aliquoter Teil des Filtrats wird in einen 50-ml-Messkolben pipettiert. Es werden 15,0 ml Wasser hinzugefügt; es wird zur Marke mit Methanol-Acetonitril (3.5) aufgefüllt und gemischt. Die Carbadox-Konzentration der fertigen Lösung muss bei etwa 10 μg/ml liegen. Ein aliquoter Teil wird durch einen 0,45-μm-Membranfilter (4.6) filtriert.
Anschließend wird die HPLC-Bestimmung (5.4) durchgeführt.
5.2.3.
Von der nicht gemahlenen Probe werden 0,2 g auf 0,001 g genau in einen 250-ml-Erlenmeyerkolben eingewogen. Es werden 45,0 ml Wasser hinzugefügt, gemischt und 5 min stehen gelassen. Dann werden 105,0 ml Methanol-Acetonitril (3.5) hinzugefügt. Der Kolben wird verschlossen, geschüttelt, 15 min in ein Ultraschallbad (4.7) gestellt und anschließend 15 min auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer gerührt (4.1). Die Lösung wird über Glasfaser-Filterpapier (4.2) filtriert.
Ein aliquoter Teil des Filtrats wird mit dem Wasser-Methanol-Acetonitril-Gemisch (3.12) verdünnt, bis eine endgültige Carbadoxkonzentration von 10 bis 15 μg/ml erreicht ist. (Verdünnungsfaktor 10 für eine 10 %ige Zubereitung). Ein aliquoter Teil wird durch einen 0,45-μm-Membranfilter (4.6) filtriert.
Anschließend wird die HPLC-Bestimmung (5.4) durchgeführt.
5.3. Reinigung
5.3.1.
Von Aluminiumoxid (3.4) werden 4 g abgewogen und in die Glassäule (4.3) eingebracht.
5.3.2.
Von dem filtrierten Extrakt (5.2.1) werden 15 ml auf die Aluminiumoxid-Säule gegeben, und die ersten 2 ml des Eluats werden verworfen. Die folgenden 5 ml werden aufgefangen und ein aliquoter Teil davon durch einen 0,45-μm-Membranfilter (4.6) filtriert.
Anschließend wird die HPLC-Bestimmung (5.4) durchgeführt.
5.4. HPLC-Bestimmung
5.4.1.
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren
|
Ergebnissen führen: |
|
|
HPLC-Trennsäule (4.4.1): |
300 × 4 mm, C18, 10 μm Korngröße, oder vergleichbare Säule |
|
Mobile Phase (3.10): |
Gemisch aus Acetat-Pufferlösung (3.9) und Acetonitril (3.2), 825 + 175 (V+V) |
|
Durchflussrate: |
1,5 bis 2 ml/min |
|
Detektionswellenlänge: |
365 nm |
|
Einspritzvolumen: |
20 μl |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.11.2), die 5,0 μg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erhalten werden.
5.4.2.
Jede Kalibrierlösung (3.11.2) wird mehrmals eingespritzt und die Peakhöhen (-flächen) werden für die einzelnen Konzentrationen gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) auf der Ordinate und die dazugehörigen Konzentrationen in g/ml auf der Abszisse aufgetragen werden.
5.4.3.
Der Probenextrakt [(5.3.2) für Futtermittel, (5.2.2) für Vormischungen und (5.2.3) für Zubereitungen] wird mehrmals eingespritzt und die mittlere Peakhöhe (-fläche) der Carbadoxpeaks ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Carbadoxpeaks der Probenlösung wird anhand der Kalibrationskurve (5.4.2) die Carbadoxkonzentration der Probenlösung in μg/ml bestimmt.
6.1. Futtermittel
Der Carbadoxgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
 [mg/kg]
[mg/kg]
wobei:
|
c |
= |
Carbadoxkonzentration der Probenlösung (5.3.2) in μg/ml, |
|
V1 |
= |
Extraktionsvolumen in ml (d. h. 50), |
|
m |
= |
Probeneinwaage in g. |
6.2. Vormischungen und Zubereitungen
Der Carbadoxgehalt w (mg/kg) der Probe wird nach folgender Formel berechnet:
 [mg/kg]
[mg/kg]
wobei:
|
c |
= |
Carbadoxkonzentration der Probenlösung (5.2.2 oder 5.2.3) in μg/ml, |
|
V2 |
= |
Extraktionsvolumen in ml (d. h. 50 für Vormischungen; 150 für Zubereitungen), |
|
f |
= |
Verdünnungsfaktor gemäß 5.2.2 (Vormischungen) oder 5.2.3 (Zubereitungen), |
|
m |
= |
Probeneinwaage in g. |
7. Überprüfung der Ergebnisse
7.1. Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren des Probenextrakts und der Kalibrierlösung (3.11.2), die 10,0 μg/ml enthält, verglichen werden.
7.1.1.
Ein Probenextrakt wird mit einer geeigneten Menge Kalibrierlösung (3.11.2) angereichert. Die zugesetzte Carbadoxmenge muss in etwa dem vermuteten Carbadoxgehalt des Probenextrakts entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Carbadoxpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2.
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
|
a) |
Die Wellenlängen bei maximaler Absorption des Proben- und des Standardspektrums an der Peakspitze des Chromatogramms müssen innerhalb eines Bereichs übereinstimmen, der durch das Auflösungsvermögen des Detektionssystems bestimmt wird. Für die Diodenarray-Detektion beträgt dieser Bereich in der Regel ± 2 nm. |
|
b) |
Zwischen 225 und 400 nm dürfen sich das Proben- und das Standardspektrum an den Peakspitzen des Chromatogramms in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung zwischen den beiden Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Standardanalyten beträgt. |
|
c) |
Zwischen 225 und 400 nm dürfen sich die Spektren des Probenextrakts im Anstieg, an der Spitze und im Abstieg des Probenpeaks in den Bereichen zwischen 10 und 100 % relativer Absorption nicht unterscheiden. Dieses Kriterium ist erfüllt, wenn die gleichen Maxima vorliegen und die Abweichung der Spektren an keinem Beobachtungspunkt mehr als 15 % der Absorption des Spektrums am Peakmaximum beträgt. |
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf bei einem Carbadoxgehalt von 10 mg/kg und mehr 15 % des höheren Wertes nicht überschreiten.
7.3. Wiederfindungsrate
Bei einer angereicherten (Blind-)Probe muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
Bei einem Ringversuch wurden 6 Futtermittel, 4 Vormischungen und 3 Zubereitungen von 8 Laboratorien untersucht. Jede Probe wurde doppelt analysiert. (Nähere Informationen zu diesem Ringversuch sind dem Journal of the AOAC, Band 71, 1988, S. 484-490, zu entnehmen.) Die Ergebnisse (mit Ausnahme von Ausreißern) sind in der nachstehenden Tabelle zusammengefasst:
Tabelle 1
Ergebnisse des Ringversuchs für Futtermittel
|
|
Probe 1 |
Probe 2 |
Probe 3 |
Probe 4 |
Probe 5 |
Probe 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Mittelwert [mg/kg] |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
sr [mg/kg] |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
VKr [ %] |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
sR [mg/kg] |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
VKR [ %] |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Sollgehalt [mg/kg] |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Tabelle 2
Ergebnisse des Ringversuchs für Vormischungen und Zubereitungen
|
|
Vormischungen |
Zubereitungen |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Mittelwert [mg/kg] |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
sr [mg/kg] |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
VKr [ %] |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
sR [mg/kg] |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
VKR [ %] |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Sollgehalt [mg/kg] |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
Anzahl der Laboratorien |
|
n |
= |
Anzahl der Einzelwerte |
|
sr |
= |
Standardabweichung der Wiederholbarkeit |
|
VKr |
= |
Variationskoeffizient der Wiederholbarkeit |
|
sR |
= |
Standardabweichung der Vergleichbarkeit |
|
VKR |
= |
Variationskoeffizient der Vergleichbarkeit |
ANHANG IX
ENTSPRECHUNGSTABELLEN GEMÄSS ARTIKEL 6
1. Richtlinie 71/250/EWG
|
Richtlinie 71/250/EWG |
Vorliegende Verordnung |
|
Artikel 1 Unterabsatz 1 |
Artikel 3 |
|
Artikel 1 Unterabsatz 2 |
Artikel 2 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang Teil 1 |
Anhang II |
|
Anhang Teil 2 |
— |
|
Anhang Teil 3 |
— |
|
Anhang Teil 4 |
Anhang III Teil O |
|
Anhang Teil 5 |
Anhang III Teil M |
|
Anhang Teil 6 |
Anhang III Teil N |
|
Anhang Teil 7 |
Anhang III Teil Q |
|
Anhang Teil 9 |
Anhang III Teil K |
|
Anhang Teil 10 |
— |
|
Anhang Teil 11 |
— |
|
Anhang Teil 12 |
Anhang III Teil J |
|
Anhang Teil 14 |
Anhang III Teil D |
|
Anhang Teil 16 |
— |
2. Richtlinie 71/393/EWG
|
Richtlinie 71/393/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang Teil I |
Anhang III Teil A |
|
Anhang Teil II |
Anhang III Teil E |
|
Anhang Teil III |
Anhang III Teil P |
|
Anhang Teil IV |
Anhang III Teil H |
3. Richtlinie 72/199/EWG
|
Richtlinie 72/199/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Anhang I Teil 1 |
Anhang III Teil L |
|
Anhang I Teil 2 |
Anhang III Teil C |
|
Anhang I Teil 2 |
— |
|
Anhang I Teil 3 |
— |
|
Anhang I Teil 3 |
Anhang V Teil A |
|
Anhang II |
— |
4. Richtlinie 73/46/EWG
|
Richtlinie 73/46/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Anhang I Teil 1 |
Anhang III Teil B |
|
Anhang I Teil 2 |
— |
|
Anhang I Teil 3 |
Anhang III Teil I |
5. Richtlinie 76/371/EWG
|
Richtlinie 76/371/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang |
Anhang I |
6. Richtlinie 76/372/EWG
|
Richtlinie 76/372/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
— |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang |
— |
7. Richtlinie 78/633/EWG
|
Richtlinie 78/633/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang Teil 1 |
— |
|
Anhang Teil 2 |
— |
|
Anhang Teil 3 |
Anhang IV Teil C |
8. Richtlinie 81/715/EWG
|
Richtlinie 81/715/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
— |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang |
— |
9. Richtlinie 84/425/EWG
|
Richtlinie 84/425/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
— |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang |
— |
10. Richtlinie 86/174/EWG
|
Richtlinie 86/174/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 4 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang |
Anhang VII |
11. Richtlinie 93/70/EWG
|
Richtlinie 93/70/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang |
Anhang IV Teil D |
12. Richtlinie 93/117/EG
|
Richtlinie 93/117/EG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 und 5 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Anhang Teil 1 |
Anhang IV Teil E |
|
Anhang Teil 2 |
Anhang VIII Teil A |
13. Richtlinie 98/64/EG
|
Richtlinie 98/64/EG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 und 5 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Anhang Teil A |
Anhang III Teil F |
|
Anhang Teil C |
Anhang VIII Teil B |
14. Richtlinie 1999/27/EG
|
Richtlinie 1999/27/EG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 und 5 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Artikel 5 |
— |
|
Artikel 6 |
— |
|
Artikel 7 |
— |
|
Anhang Teil A |
Anhang VIII Teil C |
|
Anhang Teil B |
Anhang IV Teil F |
|
Anhang Teil C |
Anhang VIII Teil D |
15. Richtlinie 1999/76/EG
|
Richtlinie 1999/76/EG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Anhang |
Anhang IV Teil G |
16. Richtlinie 2000/45/EG
|
Richtlinie 2000/45/EG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Anhang Teil A |
Anhang IV Teil A |
|
Anhang Teil B |
Anhang IV Teil B |
|
Anhang Teil C |
Anhang III Teil G |
17. Richtlinie 2002/70/EG
|
Richtlinie 2002/70/EG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
Artikel 2 und 3 |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Artikel 5 |
— |
|
Anhang I |
Anhang I und Anhang V Teil B(I) |
|
Anhang II |
Anhang II und Anhang V Teil B(II) |
18. Richtlinie 2003/126/EG
|
Richtlinie 2003/126/EG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Artikel 5 |
— |
|
Artikel 6 |
— |
|
Anhang |
Anhang VI |






