ISSN 1725-2520
doi:10.3000/17252520.L_2009.220.dan
Den Europæiske Unions
Tidende
L 220

Dansk udgave
Retsforskrifter
52. årgang
24. august 2009
|
ISSN 1725-2520 doi:10.3000/17252520.L_2009.220.dan |
||
|
Den Europæiske Unions Tidende |
L 220 |
|
|
|
||
|
Dansk udgave |
Retsforskrifter |
52. årgang |
|
Indhold |
|
I Retsakter vedtaget i henhold til traktaterne om oprettelse af Det Europæiske Fællesskab/Euratom, hvis offentliggørelse er obligatorisk |
Side |
|
|
|
FORORDNINGER |
|
|
|
* |
Kommissionens forordning (EF) nr. 761/2009 af 23. juli 2009 om tilpasning til den tekniske udvikling af forordning (EF) nr. 440/2008 om fastlæggelse af forsøgsmetoder i henhold til Europa-Parlamentets og Rådets forordning (EF) nr. 1907/2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH) ( 1 ) |
|
|
|
|
|
(1) EØS-relevant tekst |
|
DA |
De akter, hvis titel er trykt med magre typer, er løbende retsakter inden for rammerne af landbrugspolitikken og har normalt en begrænset gyldighedsperiode. Titlen på alle øvrige akter er trykt med fede typer efter en asterisk. |
I Retsakter vedtaget i henhold til traktaterne om oprettelse af Det Europæiske Fællesskab/Euratom, hvis offentliggørelse er obligatorisk
FORORDNINGER
|
24.8.2009 |
DA |
Den Europæiske Unions Tidende |
L 220/1 |
KOMMISSIONENS FORORDNING (EF) Nr. 761/2009
af 23. juli 2009
om tilpasning til den tekniske udvikling af forordning (EF) nr. 440/2008 om fastlæggelse af forsøgsmetoder i henhold til Europa-Parlamentets og Rådets forordning (EF) nr. 1907/2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH)
(EØS-relevant tekst)
KOMMISSIONEN FOR DE EUROPÆISKE FÆLLESSKABER HAR —
under henvisning til traktaten om oprettelse af Det Europæiske Fællesskab,
under henvisning til Europa-Parlamentets og Rådets forordning (EF) nr. 1907/2006 af 18. december 2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH), om oprettelse af et europæisk kemikalieagentur og om ændring af direktiv 1999/45/EF og ophævelse af Rådets forordning (EØF) nr. 793/93 og Kommissionens forordning (EF) nr. 1488/94 samt Rådets direktiv 76/769/EØF og Kommissionens direktiv 91/155/EØF, 93/67/EØF, 93/105/EF og 2000/21/EF (1), særlig artikel 13, stk. 3, og
ud fra følgende betragtninger:
|
(1) |
Kommissionens forordning (EF) nr. 440/2008 (2) indeholder de testmetoder, der skal anvendes til at bestemme stoffers fysisk-kemiske egenskaber, toksicitet og økotoksicitet i forbindelse med forordning (EF) nr. 1907/2006. |
|
(2) |
Der er behov for ajourføring af forordning (EF) nr. 440/2008 derved, at nogle testmetoder ændres, og der indsættes flere nye testmetoder, som OECD har vedtaget. De interesserede parter er blevet hørt om dette forslag. Ændringerne udgør en tilpasning af de pågældende metoder til den videnskabelige og tekniske udvikling. |
|
(3) |
Bestemmelserne om damptryk, som indfører den nye effusionsmetode, bør revideres. |
|
(4) |
Det er påkrævet at tilføje en ny metode til måling af fibres længdevægtede geometriske middeldiameter. |
|
(5) |
Det er hensigtsmæssigt at ajourføre forordning (EF) nr. 440/2008 ved, at der med høj prioritet indsættes en ny in vitro-testmetode for hudirritation, således at antallet af dyr, der anvendes til dyreforsøg, begrænses til et minimum i overensstemmelse med Rådets direktiv 86/609/EØF af 24. november 1986 om indbyrdes tilnærmelse af medlemsstaternes love og administrative bestemmelser om beskyttelse af dyr, der anvendes til forsøg og andre videnskabelige formål (3). Selvom OECD stadig drøfter et udkast til in vitro-testmetode for hudirritation, bør metode B 46 i dette særlige tilfælde indføjes i denne forordning. Metode B 46 bør ajourføres hurtigst muligt, efter at der er opnået enighed i OECD, eller hvis der fremkommer yderligere oplysninger, der berettiger en sådan ændring. |
|
(6) |
Væksthæmningstesten for alger bør revideres, idet der indsættes yderligere arter, og således at den opfylder behovene for vurdering af kemikaliers farlighed og klassificering. |
|
(7) |
Der er behov for at tilføje en ny metode til måling af aerob mineralisering i overfladevand, som består i en test med simuleret bionedbrydning, og en ny metode til vurdering af toksiciteten over for slægten Lemna, som er en væksthæmningstest. |
|
(8) |
Forordning (EF) nr. 440/2008 bør derfor ændres i overensstemmelse hermed. |
|
(9) |
Foranstaltningerne i denne forordning er i overensstemmelse med udtalelse fra det udvalg, der er nedsat ved artikel 133 i forordning (EF) nr. 1907/2006 — |
UDSTEDT FØLGENDE FORORDNING:
Artikel 1
Bilaget til forordning (EF) nr. 440/2008 ændres således:
|
1) |
Del A ændres således:
|
|
2) |
Del B ændres således: Teksten i bilag III til denne forordning indsættes som kapitel B.46. |
|
3) |
Del C ændres således:
|
Artikel 2
Denne forordning træder i kraft på tredjedagen efter offentliggørelsen i Den Europæiske Unions Tidende.
Denne forordning er bindende i alle enkeltheder og gælder umiddelbart i hver medlemsstat.
Udfærdiget i Bruxelles, den 23. juli 2009.
På Kommissionens vegne
Stavros DIMAS
Medlem af Kommissionen
(1) EUT L 396 af 30.12.2006, s. 1.
(2) EUT L 142 af 31.5.2008, s. 1.
(3) EFT L 358 af 18.12.1986, s. 1.
BILAG I
|
A.4. |
DAMPTRYK |
1. METODE
Metoden er ækvivalent med OECD TG 104 (2004).
1.1. INDLEDNING
Denne reviderede version af metode A.4 (1) omfatter én supplerende metode, effusionsmetoden: isoterm termogravimetri, som er udviklet til stoffer med meget lavt tryk (ned til 10–10 Pa). Da der er et særligt behov for metoder til måling af damptrykket af stoffer med lavt damptryk, er de øvrige procedurer i denne metode revurderet med hensyn til andre anvendelsesområder.
Ved termodynamisk ligevægt er damptrykket af et rent stof alene en funktion af temperaturen. Grundprincipperne findes beskrevet andetsteds (2)(3).
Ingen enkelt måleteknik kan anvendes på hele trykområdet fra mindre end 10–10 til 105 Pa. I denne metode til damptrykmåling indgår otte metoder, som kan anvendes i forskellige damptrykintervaller. I tabel 1 sammenlignes de forskellige metoders anvendelighed og måleområde. Metoderne kan kun bruges på forbindelser, der ikke dekomponeres under de pågældende testbetingelser. I tilfælde, hvor de eksperimentelle metoder af tekniske grunde ikke kan anvendes, kan et tilnærmet damptryk beregnes, og i tillægget beskrives en anbefalet beregningsmetode.
1.2. DEFINITIONER OG ENHEDER
Ved damptrykket af et stof forstås mætningstrykket over stoffet i fast eller flydende tilstand.
Som trykenhed bør anvendes SI-enheden, der er pascal (Pa). Nedenfor er angivet andre, tidligere anvendte enheder og de tilhørende omregningsfaktorer:
|
1 Torr |
= |
1 mm Hg |
= |
1,333 × 102 Pa |
|
1 atmosfære |
= |
1,013 × 105 Pa |
|
|
|
1 bar |
= |
105 Pa |
|
|
SI-enheden for temperatur er kelvin (K). Omregning af grader celsius til kelvin sker med formlen:
T = t + 273,15
hvor T er kelvin-temperaturen eller den termodynamiske temperatur, og t er temperaturen i grader celsius.
Tabel 1
|
Målemetode |
Stof |
Anslået repeterbarhed |
Anslået Reproducerbarhed |
Anbefalet område |
|
|
Faststof |
Væske |
||||
|
Den dynamiske metode |
Letsmelteligt |
Ja |
indtil 25 % 1 til 5 % |
indtil 25 % 1 til 5 % |
103 Pa til 2 × 103 Pa 2 × 103 Pa til 105 Pa |
|
Den statiske metode |
Ja |
Ja |
5 til 10 % |
5 til 10 % |
10 Pa til 105 Pa 10–2 Pa til 105 Pa (1) |
|
Isoteniskop-metoden |
Ja |
Ja |
5 til 10 % |
5 til 10 % |
102 Pa til 105 Pa |
|
Effusionsmetoden: damptrykbalance |
Ja |
Ja |
5 til 20 % |
indtil 50 % |
10–3 til 1 Pa |
|
Effusionsmetoden: Knudsen-celle |
Ja |
Ja |
10 til 30 % |
— |
10–10 til 1 P |
|
Effusionsmetoden: isotermisk termogravimetri |
Ja |
Ja |
5 til 30 % |
indtil 50 % |
10–10 til 1 Pa |
|
Gasmætningsmetoden |
Ja |
Ja |
10 til 30 % |
indtil 50 % |
10–10 til 103 Pa |
|
Metoden med roterende kugle |
Ja |
Ja |
10 til 20 % |
— |
10–4 til 0,5 Pa |
1.3. TESTENS PRINCIP
Sædvanligvis bestemmes damptrykket ved forskellige temperaturer. Inden for et begrænset temperaturområde er logaritmen til damptrykket af et rent stof en lineær funktion af den reciprokke termodynamiske temperatur ifølge den forenklede Clausius-Clapeyron-ligning:
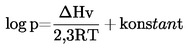
hvor:
|
p |
= |
damptrykket i pascal |
|
ΔHv |
= |
fordampningsvarmen i J mol–1 |
|
R |
= |
den universelle gaskonstant, 8,314 J mol–1 K–1 |
|
T |
= |
temperaturen i K |
1.4. REFERENCESTOFFER
Referencestoffer behøver ikke anvendes. De bruges hovedsagelig til lejlighedsvis kontrol af en metodes præstationer og til at sammenligne resultaterne fra forskellige metoder.
1.5. BESKRIVELSE AF METODEN
1.5.1. Den dynamiske metode (Cottrells metode)
1.5.1.1. Princip
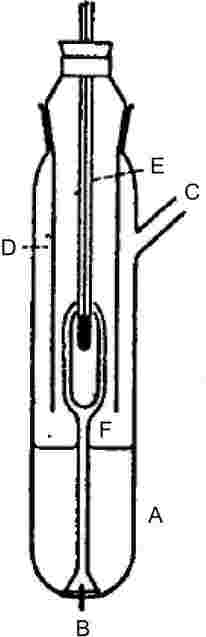
Damptrykket bestemmes ved måling af stoffets kogepunktstemperatur ved forskellige givne tryk mellem ca. 103 og 105 Pa. Metoden anbefales også til kogepunktsbestemmelse. Til det formål er metoden brugbar op til 600 K. Væskers kogepunktstemperatur er ca. 0,1 °C højere i en dybde af 3-4 cm end ved overfladen på grund af det hydrostatiske tryk fra væskesøjlen. I Cottrells metode (4) er termometeret placeret i dampen over væskeoverfladen, og den kogende væske bringes til konstant at pumpe sig selv ud over termometerets kugle. Kuglen er dækket af et tyndt lag væske, der er i ligevægt med dampen ved atmosfæretryk. Termometeret viser derfor det sande kogepunkt uden fejl pga. overhedning eller hydrostatisk tryk. Den oprindelige pumpe, som Cottrell anvendte, ses i figur 1. Rør A indeholder den kogende væske. En indlejret platintråd B i bunden gør kogningen mere ensartet. Siderøret C fører til en kondensator, og kappen D forhindrer det kolde kondensat i at nå termometeret E. Når væsken i A koger, bliver bobler og væske opfanget af tragten og via de to arme af pumpen F ledt ud over termometerets kugle.
|
Figur 1
|
Figur 2
|
Cottrell-pumpe (4)
|
A: |
Termoelement |
|
B: |
Vakuum-stødpudevolumen |
|
C: |
Trykmåler |
|
D: |
Vakuum |
|
E: |
Målepunkt |
|
F: |
Varmeelement, ca. 150 W |
1.5.1.2. Apparatur
I figur 2 er vist et meget nøjagtigt apparat, der arbejder efter Cottrells princip. Det består af et rør med en kogesektion i underdelen, en svaler i midten og en afgang med flange i overdelen. Cottrell-pumpen er anbragt i kogesektionen, der opvarmes af en elektrisk varmepatron. Temperaturen måles af et termoelement med kappe eller af et modstandstermometer, der er ført gennem flangen foroven. Afgangen er tilsluttet trykreguleringssystemet. Dette består af en vakuumpumpe, et stødpudevolumen, en trykregulator for tilførsel af nitrogen til trykregulering og et manometer.
1.5.1.3. Fremgangsmåde
Stoffet anbringes i kogesektionen. Faste stoffer, der ikke er pulverformige, kan volde problemer, som dog undertiden kan løses ved opvarmning af kølekappen. Apparatet forsegles ved flangen, og stoffet afgasses. Skumdannende stoffer kan ikke måles med denne metode.
Derefter sættes det laveste ønskede tryk, og der tændes for opvarmningen. Samtidig tilsluttes temperaturføleren til en skriver.
Ligevægt er nået, når der registreres konstant kogetemperatur ved konstant tryk. Særlig omhu må udvises for at undgå stødkogning. Desuden skal kondensationen i svaleren være fuldstændig. Ved damptryksbestemmelse for letsmeltelige faste stoffer skal man passe på, at svaleren ikke bliver tilstoppet.
Efter at dette ligevægtspunkt er nået, sættes et højere tryk. Processen fortsættes på denne måde, indtil 105 Pa er nået (i alt 5 til 10 målepunkter). Som kontrol gentages ligevægtspunkterne ved faldende værdier af trykket.
1.5.2. Den statiske metode
1.5.2.1. Princip
Ved den statiske metode (5) bestemmes damptrykket i termodynamisk ligevægt ved en fastlagt temperatur. Metoden er velegnet til enkeltstoffer samt væsker og faste stoffer bestående af flere komponenter i området fra 10–1 til 105 Pa samt, med tilstrækkelig omhu, i området 1 til 10 Pa.
1.5.2.2. Apparatur
Apparatet består af et termostatbad (præcision ±0,2 K), en prøvebeholder tilsluttet en vakuumledning, et manometer og et system til trykregulering. Prøvekammeret (figur 3a) er tilsluttet vakuumledningen gennem en ventil og et differensmanometer (et U-rør indeholdende en egnet manometervæske), der fungerer som nulindikator. I differensmanometeret kan anvendes kviksølv, siliconeolie og phthalater, afhængigt af trykområdet og teststoffets kemiske egenskaber. Kviksølv bør dog om muligt undgås af hensyn til miljøet. Teststoffet må ikke være nævneværdigt opløseligt i væsken i U-røret eller kunne reagere med denne. I stedet for U-røret kan anvendes en trykmåler (figur 3b). I manometeret kan kviksølv anvendes i området fra normalt tryk ned til 102 Pa, mens siliconeolier og phthalater er egnede ved tryk under 102 Pa ned til 10 Pa. Der findes andre trykmålere, som kan anvendes ved tryk under 102 Pa, og opvarmede membrankapacitetsmanometre kan endda bruges ved tryk under 10–1 Pa. Temperaturen måles enten på ydervæggen af den beholder, som indeholder prøven, eller i selve beholderen.
1.5.2.3. Fremgangsmåde
I det i figur 3a beskrevne apparatur fyldes U-røret med den valgte væske, som skal afgasses ved forhøjet temperatur, før der aflæses. Teststoffet anbringes i apparatet og afgasses ved reduceret temperatur. For prøver med flere komponenter skal temperaturen være tilstrækkelig lav til at sikre, at materialets sammensætning ikke ændres. Ligevægt kan nås hurtigere ved omrøring. Prøven kan køles med flydende nitrogen eller tøris, men der må udvises forsigtighed, så kondensation af luft eller pumpevæske undgås. Mens ventilen over prøvebeholderen er åben, tilsluttes sugningen i nogle minutter for at fjerne luften. Om nødvendigt gentages afgasningen nogle gange.
|
Figur 3a
|
Figur 3b
|
Når prøven opvarmes med lukket ventil, stiger damptrykket. Derved ændres væskeligevægten i U-røret. For at kompensere herfor lukkes der nitrogen eller luft ind i apparatet, indtil differenstrykindikatoren igen viser nul. Det nødvendige tryk hertil kan aflæses på manometeret eller på et instrument med større præcision. Dette tryk svarer til stoffets damptryk ved måletemperaturen. På det i figur 3b viste apparatet aflæses damptrykket direkte.
Damptrykket bestemmes med passende små temperaturintervaller (ca. 5 til 10 målepunkter i alt) op til den ønskede maksimumtemperatur.
Lavtemperaturaflæsningerne skal gentages som kontrol. Hvis værdierne fra gentagelserne ikke falder sammen med den kurve, der er opnået ved stigende temperatur, kan dette have følgende årsager:
|
i) |
prøven indeholder stadig luft (f.eks. ved højviskøse materialer) eller flygtige stoffer, der frigives ved opvarmning |
|
ii) |
stoffet undergår en kemisk reaktion i det undersøgte temperaturområde (f.eks. dekomposition eller polymerisering). |
1.5.3. Isoteniskop-metoden
1.5.3.1. Princip
Isoteniskopet (6) bygger på den statiske metodes princip. I metoden anbringes en prøve i en kugle, der holdes ved konstant temperatur og er tilsluttet til et manometer og en vakuumpumpe. Urenheder, der er flygtigere end stoffet, fjernes ved afgasning ved reduceret tryk. Prøvens damptryk ved de valgte temperaturer afbalanceres ved et kendt tryk af inert gas. Isoteniskopet er oprindeligt udviklet til måling af damptrykket af visse flydende kulbrinter, men egner sig også til undersøgelse af faste stoffer. Metoden egner sig normalt ikke til systemer med flere komponenter. Resultaterne er kun behæftet med små fejl for prøver indeholdende ikke-flygtige urenheder. Det anbefalede område er 102 til 105 Pa.
1.5.3.2. Apparatur
Et eksempel på en måleanordning er vist i figur 4. En komplet beskrivelse kan findes i ASTM D 2879-86 (6).
1.5.3.3. Fremgangsmåde
Når teststoffet er en væske, fungerer den selv som manometervæske i differensmanometeret. Isoteniskopet påfyldes så meget af væsken, at manometerets kugle og korte gren er fyldt. Isoteniskopet tilslutte et vakuumsystem og udpumpes, hvorefter det fyldes med nitrogen. Udpumpningen og gennemskylningen af systemet gentages to gange for at fjerne tilbageværende oxygen. Det påfyldte isoteniskop anbringes vandret, så prøven fordeler sig i et tyndt lag i prøvekuglen og manometeret. Trykket i systemet nedsættes til 133 Pa, og prøven opvarmes forsigtigt, til den netop koger (fjernelse af opløste gasser). Isoteniskopet anbringes derefter, så prøven løber tilbage i kuglen og fylder manometerets korte gren. Trykket holdes på 133 Pa. Prøvekuglens udtrukne spids opvarmes med en svag flamme, indtil den afgivne damp fra teststoffet har udvidet sig så meget, at en del af prøven fra den øverste del af kuglen og manometergrenen fortrænges ind i manometeret og derved danner et dampfyldt, nitrogenfrit rum. Isoteniskopet anbringes derefter i et termostatbad, og nitrogentrykket indstilles, til det er lig prøvens. I ligevægt er nitrogentrykket lig med teststoffets damptryk.
Figur 4

Til faste stoffer anvendes som manometervæske siliconeolie eller phthalater, afhængigt af tryk- og temperaturområdet. Den afgassede manometervæske fyldes i en udposning på isoteniskopets lange gren. Det faste stof, der skal undersøges, anbringes i prøvekuglen og afgasses ved en forhøjet temperatur. Derefter holdes isoteniskopet skråt, så manometervæsken kan løbe ind i U-røret.
1.5.4. Effusionsmetoden: damptrykvægt (7)
1.5.4.1. Princip
Teststoffet opvarmes i en lille ovn og anbringes i en udpumpet glasklokke. Ovnen er dækket af et låg med små huller med kendt diameter. Den damp, som fra stoffet afgives gennem et af hullerne, ledes op mod vægtskålen på en meget fintmærkende vægt, der ligeledes er placeret i den udpumpede glasklokke. I nogle udformninger er vægtskålen omgivet af en køleboks, der leder varmen bort til omgivelserne ved varmeledning, og den køles ved stråling, så den udslippende damp kondenserer på den. Dampstrålens impuls påfører en kraft på vægten. Damptrykket kan beregnes på to måder: direkte af kraften på vægtskålen og af fordampningshastigheden ved hjælp af Hertz-Knudsen-ligningen (2):

hvor:
|
G |
= |
fordampningshastighed (kg s–1 m–2) |
|
M |
= |
molær masse (g mol–1) |
|
T |
= |
temperatur (K) |
|
R |
= |
den universelle gaskonstant (J mol–1 K–1) |
|
P |
= |
damptryk (Pa) |
Det anbefalede område er 10–3 til 1 Pa.
1.5.4.2. Apparatur
Det generelle princip i apparatet er vist i figur 5.
Figur 5

|
A: |
Bundplade |
F: |
Køleboks og kølestang |
|
B: |
Drejespoleinstrument |
G: |
Muffelovn |
|
C: |
Glasklokke |
H: |
Dewarkar med flydende nitrogen |
|
D: |
Vægt med vægtskål |
I: |
Måling af prøvens temperatur |
|
E: |
Anordning til måling af vakuum |
J: |
Teststof |
1.5.5. Effusionsmetoden: Knudsen-celle
1.5.5.1. Princip
Metoden bygger på bestemmelse af den masse af dampformigt teststof, der pr. tidsenhed strømmer ud af en Knudsen-celle (8) gennem en mikroskopisk åbning under ekstremt vakuum. Massen af udstrømmede damp kan enten bestemmes af cellens massetab eller ved at lade dampen kondensere ved lav temperatur og bestemme den fordampede stofmængde ved kromatografi. Damptrykket beregnes ved at anvende Hertz-Knudsen-relationen (se punkt 1.5.4.1) med korrektionsfaktorer, der afhænger af apparaturets parametre (9). Det anbefalede område er 10–10 til 1 Pa (10)(11)(12)(13)(14).
1.5.5.2. Apparatur
Apparatets generelle princip er illustreret i figur 6.
Figur 6
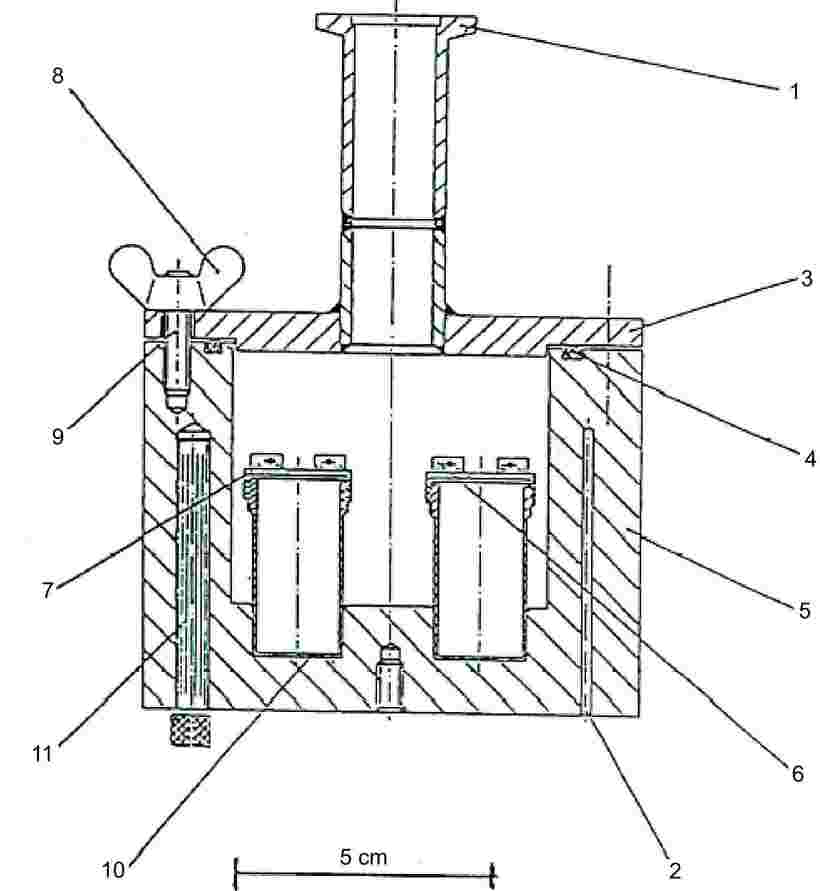
|
1: |
Vakuumtilslutning |
7: |
Skruelåg |
|
2: |
Huller fra platinmodstandstermometer eller temperaturmåling og -kontrol |
8: |
Vingemøtrikker |
|
3: |
Låg til vakuumbeholder |
9: |
Bolte |
|
4: |
O-ring |
10: |
Effusionsceller af rustfrit stål |
|
5: |
Vakuumbeholder af aluminium |
11: |
Varmepatron |
|
6: |
Anordning til isætning og udtagning af effusionsceller |
|
|
1.5.6. Effusionsmetoden: isotermisk termogravimetri
1.5.6.1. Princip
Metoden bygger på bestemmelse af stigningen i teststoffets fordampningshastighed ved forhøjet temperatur og atmosfæretryk ved hjælp af termogravimetri (10)(15)(16)(17)(18)(19)(20). Fordampningshastighederne vT bestemmes ved, at den valgte forbindelse eksponeres for en atmosfære af langsomt strømmende inert gas, mens vægttabet overvåges ved fastlagte isotermiske temperaturer T i kelvin i passende tidsrum. Damptrykkene pT beregnes af vT-værdierne ved udnyttelse af den lineære sammenhæng mellem logaritmen til damptrykket og logaritmen til fordampningshastigheden. Om nødvendigt kan der ekstrapoleres til temperaturerne 20 og 25 °C ved regressionsanalyse af log pT som funktion af 1/T. Metoden er egnet til stoffer med damptryk helt ned til 10–10 Pa (10–12 mbar) og med en renhed så tæt som muligt på to 100 % for at undgå fejlfortolkning af målte vægttab.
1.5.6.2. Apparatur
Det generelle princip i testopstillingen er vist i figur 7.
Figur 7
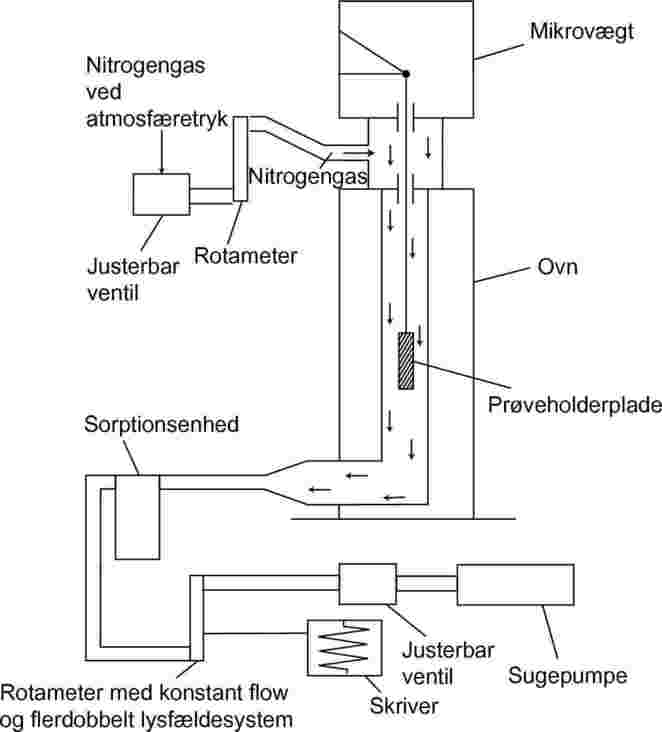
Prøveholderpladen er ophængt i en mikrovægt i et kammer med kontrolleret temperatur, hvor der strømmer tør nitrogengas forbi den, som fjerner de fordampede molekyler af teststof. Efter at have forladt kammeret bliver gasstrømmen renset i en sorptionsenhed.
1.5.6.3. Fremgangsmåde
Teststoffet påføres som et homogent lag på en matteret glasplade. For faste stoffer vædes pladen ensartet med en opløsning af stoffet i et egnet opløsningsmiddel og tørres i inert atmosfære. Målingen sker ved, at den overtrukne plade hænges op i den termogravimetriske analysator, hvorefter dens vægttab registreres kontinuerligt som funktion af tiden.
Fordampningshastigheden vT ved en nærmere angivet temperatur beregnes af prøvepladens vægttab Dm ved hjælp af udtrykket
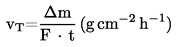
hvor F er teststoflagets overfladeareal, sædvanligvis prøvepladens overfladeareal, og t er tiden svarende til vægttabet Δm.
Damptrykket pT beregnes af funktionen for fordampningshastigheden vT:
log pT = C + D log vT
hvor C og D er konstanter, der er specifikke for den anvendte testopstilling og afhænger af målekammerets diameter og af gassens strømningshastighed. Disse konstanter bestemmes én gang for alle ved måling på et sæt forbindelser med kendt damptryk og regression af log pT mod log vT (11)(21)(22).
Sammenhængen mellem damptrykket pT og temperaturen T i kelvin er givet ved
log pT = A + B 1/T
hvor A og B er konstanter, der fås ved regression af log pT mod 1/T. Heraf kan damptrykket ved en vilkårlig temperatur beregnes ved ekstrapolation.
1.5.7. Gasmætningsmetoden (23)
1.5.7.1. Princip
En inert gas ledes ved rumtemperatur og med kendt flowhastighed gennem eller hen over en prøve af teststoffet, tilstrækkelig langsomt til at sikre mætning. Det er afgørende, at gassen når til mætning. Det transporterede stof opfanges, sædvanligvis med hjælp af et sorptionsmiddel, og dets mængde bestemmes. I stedet for opsamling af dampen og efterfølgende analyse kan man benytte in-line analyse, f.eks. ved gaskromatrografi, til kvantitativ bestemmelse af den transporterede mængde materiale. Damptrykket beregnes, idet det forudsættes, at idealgasloven gælder, og at det totale tryk af en gasblanding er lig summen af gassernes partialtryk. Teststoffets partialtryk, dvs. damptrykket, beregnes af det kendte totale gasvolumen og vægten af det transporterede materiale.
Gasmætningsmetoden er anvendelig på faste og flydende stoffer. Den kan anvendes på damptryk ned til 10–10 Pa (10)(11)(12)(13)(14). Metoden er mest pålidelig ved damptryk under 103 Pa. Over 103 Pa bliver damptrykket sædvanligvis overvurderet, antagelig som følge af aerosoldannelse. Da damptrykket måles ved stuetemperatur, undgår man at skulle ekstrapolere fra høje temperaturer, hvilket ofte kan medføre alvorlige fejl.
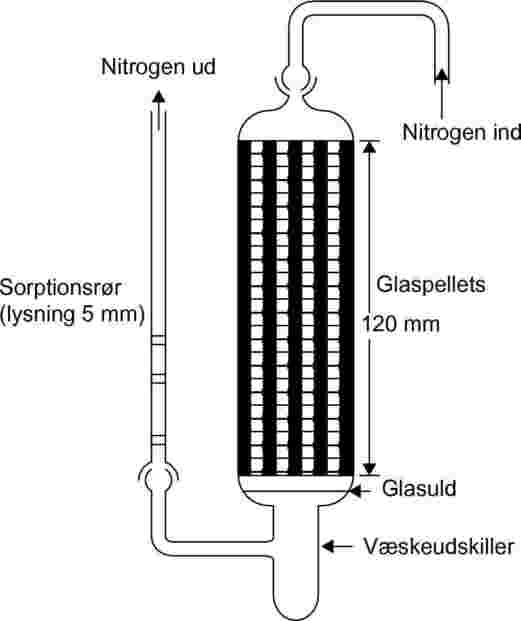
1.5.7.2. Apparatur
Metoden kræver brug af et termostateret hus. Skitsen i figur 8 viser et hus, der med tre holdere til faste og tre til flydende prøver giver mulighed for tredobbelte analyser af enten et fast eller et flydende teststof. Temperaturen styres med en nøjagtighed på ±0,5 °C eller bedre.
Figur 8
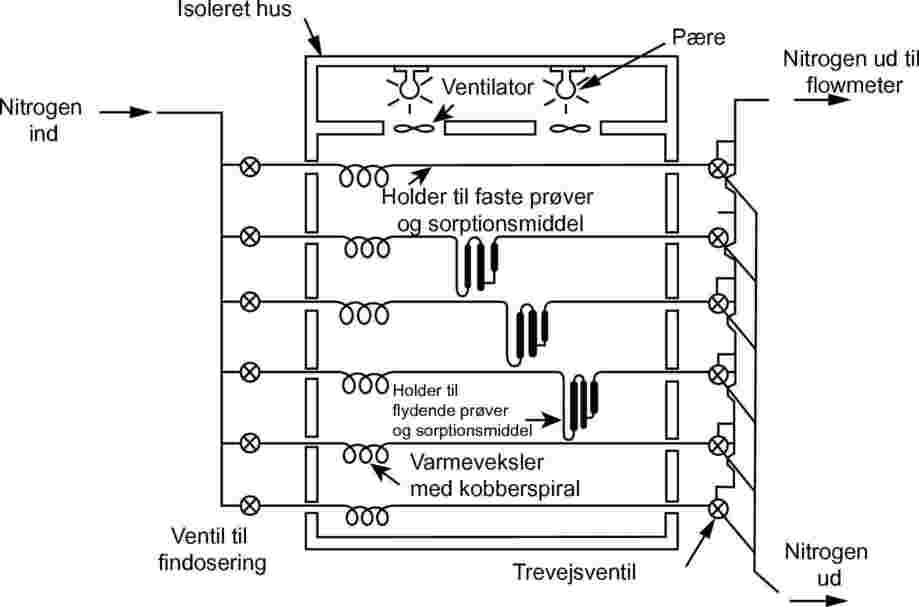
Som bæregas anvendes sædvanligvis nitrogen, men brug af en anden gas kan undertiden være nødvendig (24). Bæregassen skal være tør. Gasstrømmen opdeles i 6 strømme, der styres ved hjælp af nåleventiler (med en åbning på ca. 0,79 mm) og ledes ind i huset gennem et kobberrør med en lysning på 3,8 mm. Efter temperaturudligning ledes gassen gennem prøven og sorptionsfælden og forlader huset.
Faste prøver anbringes med glasuldspropper på hver side i et glasrør med 5 mm lysning (se figur 9). Figur 10 viser en prøveholder til væskeformige prøver og sorptionssystemet. Den mest reproducerbare metode til måling af væskers damptryk er at påføre væsken på glaspellets eller på et inert sorptionsmiddel såsom silica og pakke holderen med disse pellets. Bæregassen kan i stedet ledes gennem en grov glasfritte og gennemboble en kolonne med flydende teststof.
|
Figur 9
|
Figur 10
|
Sorptionssystemet indeholder en forreste sorptionssektion og en backup-sorptionssektion. Ved meget lave damptryk et det kun små mængder, der indfanges af sorptionsmidlet, og adsorption til glasulden og glasrøret mellem prøven og sorptionsmidlet kan være et alvorligt problem.
En anden effektiv metode til opsamling af det fordampede materiale er dampfælder kølet med tøris. De giver ikke modtryk i mætningskolonnen, og det opfangede materiale kan let fjernes fuldstændigt.
1.5.7.3. Fremgangsmåde
Flowhastigheden af den udstrømmende bæregas måles ved rumtemperatur. Flowhastigheden kontrolleres ofte under testen for at sikre nøjagtig bestemmelse af det totale volumen bæregas. Hertil bør fortrinsvis anvendes kontinuerlig overvågning med et masseflowmeter. For at gasfasen skal blive mættet, kan det være nødvendigt med en ret lang kontakttid og dermed en lav flowhastighed af gassen (25).
Ved testens slutning skal den forreste sorptionssektion og backup-sorptionssektionen analyseres hver for sig. Stoffet på hver sektion desorberes ved tilsætning af et opløsningsmiddel. De resulterende opløsninger analyseres kvantitativt til bestemmelse af den desorberede vægt fra hver sektion. Valg af analysemetode (herunder valg af sorptionsmiddel og opløsningsmiddel til desorption) træffes ud fra teststoffets beskaffenhed. Effektiviteten af desorptionen bestemmes ved, at en kendt mængde teststof injiceres på sorptionsmidlet, hvorefter det desorberes, og den genfundne mængde bestemmes. Det er vigtigt at bestemme effektiviteten af desorptionen ved en koncentration svarende til prøvens og under samme betingelser som i testen.
For at sikre, at bæregassen er mættet med teststof, anvendes tre forskellige flowhastigheder af gassen. Hvis det beregnede damptryk viser sig at være uafhængigt af flowhastigheden, kan gassen antages at være mættet.
Damptrykket beregnes af udtrykket:

hvor:
|
P |
= |
damptryk (Pa) |
|
W |
= |
masse af det fordampede teststof (g) |
|
V |
= |
volumen af den mættede gas (m3) |
|
R |
= |
den universelle gaskonstant 8,314 (J mol–1 K–1) |
|
T |
= |
temperatur (K) |
|
M |
= |
teststoffets molmasse (g mol–1) |
De målte volumener skal korrigeres for tryk- og temperaturforskelle mellem flowmeteret og mætningskolonnen.
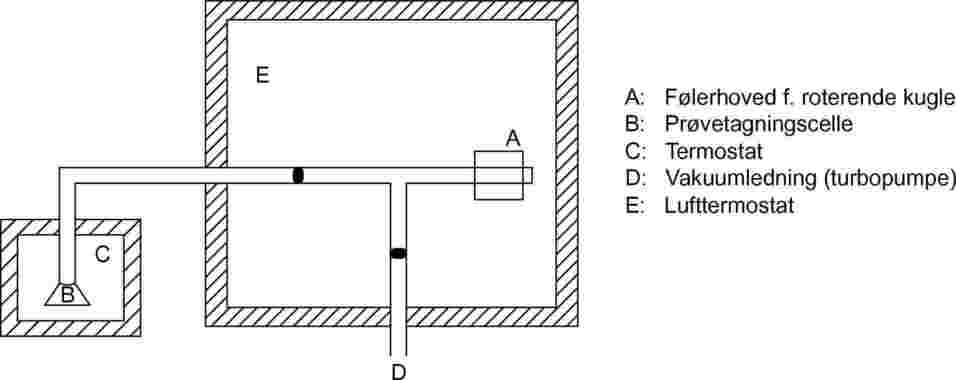
1.5.8. Roterende kugle
1.5.8.1. Princip
I metoden anvendes et viskosimeter, hvis måleelement er en lille stålkugle, som holdes svævende i et magnetisk felt og sættes i rotation af roterende felter (26)(27)(28). Kuglens rotationshastighed måles med detektorspoler. Når kuglen har nået en given rotationshastighed, sædvanligvis ca. 400 o./min., standses energitilførslen til kuglen, som derefter bremses af gasfriktionen. Faldet i rotationshastigheden registreres som funktion af tiden. Damptrykket afledes af den trykafhængige nedbremsning af stålkuglen. Det anbefalede område er 10–4 til 0,5 Pa.
1.5.8.2. Apparatur
Testopstillingen er vist skematisk i figur 11. Målehovedet er placeret i et termostateret hus, der er reguleret inden for 0,1 °C. Prøveholderen er placeret i et separat hus, der ligeledes er reguleret inden for 0,1 °C. Alle de øvrige dele af testopstillingen holdes ved højere temperatur for at undgå kondensation. Hele apparatet er tilsluttet et højvakuum-system.
Figur 11
2. DATA OG RAPPORTERING
2.1. DATA
Damptrykket skal bestemmes ved mindst to temperaturer med en af ovenstående metoder. I området 0 til 50 °C bør fortrinsvis anvendes tre eller flere temperaturer for at eftervise damptrykkurvens linearitet. Ved effusionsmetoden (Knudsen-celle og isotermisk termogravimetri) og ved gasmætningsmetoden anbefales 120 til 150 °C som temperaturområde for målingen i stedet for 0 til 50 °C.
2.2. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
— |
den anvendte metode |
|
— |
nøjagtig specifikation af stoffet (identitet og urenheder) og eventuelle indledende rensningstrin |
|
— |
mindst to værdier af damptryk og temperatur (helst tre eller flere) — i området fra 0 til 50 °C (eller 120 til 150 °C) |
|
— |
mindst en af temperaturerne bør være 25 °C eller derunder, hvis dette er teknisk muligt med den valgte metode |
|
— |
alle originale data |
|
— |
en kurve over log p som funktion af 1/T |
|
— |
et beregnet damptryk ved 20 eller 25 °C. |
Hvis der iagttages en omdannelse (tilstandsændring, dekomposition), skal følgende oplysninger noteres:
|
— |
ændringens art |
|
— |
den temperatur, ved hvilken ændringen finder sted ved atmosfæretryk |
|
— |
damptryk ved 10 og 20 °C under omdannelsestemperaturen og 10 og 20 °C over denne temperatur (bortset fra omdannelse fra fast stof til gas). |
Alle oplysninger og bemærkninger, der er relevante for fortolkningen af resultaterne, skal rapporteres, specielt hvad angår urenheder og stoffets fysiske tilstand.
3. LITTERATUR
|
(1) |
De Europæiske Fællesskabers Tidende L 383 A, 26-47 (1992). |
|
(2) |
Ambrose, D. (1975). Experimental Thermodynamics, Vol. II, Le Neindre, B., and Vodar, B., Eds., Butterworths, London. |
|
(3) |
Weissberger R., ed. (1959). Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Vol. I, Part I. Chapter IX, Interscience Publ., New York. |
|
(4) |
Glasstone, S. (1946). Textbook of Physical Chemistry, 2nd ed., Van Nostrand Company, New York. |
|
(5) |
NF T 20-048 AFNOR (September 1985). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within a range from 10–1 to 105 Pa — Static method. |
|
(6) |
ASTM D 2879-86, Standard test method for vapour pressure — temperature relationship and initial decomposition temperature of liquids by isoteniscope. |
|
(7) |
NF T 20-047 AFNOR (September 1985). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within range from 10–3 to 1 Pa — Vapour pressure balance method. |
|
(8) |
Knudsen, M. (1909). Ann. Phys. Lpz., 29, 1979; (1911), 34, 593. |
|
(9) |
Ambrose, D., Lawrenson, I.J., Sprake, C.H.S. (1975). J. Chem. Thermodynamics 7, 1173. |
|
(10) |
Schmuckler, M.E., Barefoot, A.C., Kleier, D.A., Cobranchi, D.P. (2000), Vapor pressures of sulfonylurea herbicides; Pest Management Science 56, 521-532. |
|
(11) |
Tomlin, C.D.S. (ed.), The Pesticide Manual, Twelfth Edition (2000). |
|
(12) |
Friedrich, K., Stammbach, K., Gas chromatographic determination of small vapour pressures determination of the vapour pressures of some triazine herbicides. J. Chromatog. 16 (1964), 22-28. |
|
(13) |
Grayson, B.T., Fosbraey, L.A., Pesticide Science 16 (1982), 269-278. |
|
(14) |
Rordorf, B.F., Prediction of vapor pressures, boiling points and enthalpies of fusion for twenty-nine halogenated dibenzo-p-dioxins, Thermochimia Acta 112 Issue 1 (1987), 117-122. |
|
(15) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection; Pesticide Science 4 (1973) 137-147. |
|
(16) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection II. Application to Formulated Products; Pesticide Science 5 (1974) 393-400. |
|
(17) |
Gückel, W., Kaestel, R., Lewerenz, J., Synnatschke, G., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection. Part III: The Temperature Relationship between Vapour Pressure and Evaporation Rate; Pesticide Science 13 (1982) 161-168. |
|
(18) |
Gückel, W., Kaestel, R., Kroehl, T., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part IV: An Improved Thermogravimetric Determination Based on Evaporation Rate; Pesticide Science 45 (1995) 27-31. |
|
(19) |
Kroehl, T., Kaestel, R., Koenig, W., Ziegler, H., Koehle, H., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part V: Thermogravimetry Combined with Solid Phase MicroExtraction (SPME); Pesticide Science, 53 (1998) 300-310. |
|
(20) |
Tesconi, M., Yalkowsky, S.H., A Novel Thermogravimetric Method for Estimating the Saturated Vapor Pressure of Low-Volatility Compounds; Journal of Pharmaceutical Science 87(12) (1998) 1512-20. |
|
(21) |
Lide, D.R. (ed.), CRC Handbook of Chemistry and Physics, 81th ed.(2000), Vapour Pressure in the Range -25 °C to 150 °C. |
|
(22) |
Meister, R.T. (ed.), Farm Chemicals Handbook, Vol. 88 (2002). |
|
(23) |
40 CFR, 796. (1993). pp 148-153, Office of the Federal Register, Washington DC. |
|
(24) |
Rordorf B.F. (1985). Thermochimica Acta 85, 435. |
|
(25) |
Westcott et al. (1981). Environ. Sci. Technol. 15, 1375. |
|
(26) |
Messer G., Röhl, P., Grosse G., and Jitschin W. (1987). J. Vac. Sci. Technol. (A), 5(4), 2440. |
|
(27) |
Comsa G., Fremerey J.K., and Lindenau, B. (1980). J. Vac. Sci. Technol. 17(2), 642. |
|
(28) |
Fremerey, J.K. (1985). J. Vac. Sci. Technol. (A), 3(3), 1715. |
(1) Ved brug af kapacitansmanometer.
Tillæg
Metode til tilnærmet beregning
INDLEDNING
Tilnærmede værdier af damptrykket kan anvendes:
|
— |
til at afgøre, hvilken testmetode der er velegnet |
|
— |
til at give et skøn eller en grænseværdi i tilfælde, hvor den eksperimentelle metode af tekniske grunde ikke kan anvendes. |
METODE TIL TILNÆRMET BEREGNING
Damptrykket af faste stoffer og væsker kan beregnes tilnærmet med den modificerede Watson-korrelation (a). De eneste eksperimentelle data, der kræves, er det normale kogepunkt. Metoden kan anvendes i hele trykområdet fra 105 Pa til 10–5 Pa.
Detaljerede oplysninger om metoden findes i »Handbook of Chemical Property Estimation Methods« (b). Se også OECD Environmental Monograph No.67 (c).
BEREGNINGSMETODE
Damptrykket beregnes af følgende udtryk:

hvor:
|
T |
= |
den betragtede temperatur |
|
Tb |
= |
normalkogepunktet |
|
Pvp |
= |
damptrykket ved temperaturen T |
|
ΔHvb |
= |
fordampningsvarmen |
|
ΔZb |
= |
kompressibilitetsfaktoren (anslået værdi 0,97) |
|
m |
= |
en empirisk faktor, der afhænger af den fysiske tilstand ved den betragtede temperatur |
Endvidere gælder:
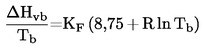
hvor KF er en empirisk faktor, der tager hensyn til stoffets polaritet. Faktoren KF er for en række forbindelser af forskellig art angivet i reference (b).
I mange tilfælde indeholder de foreliggende data en angivelse af kogepunktet ved reduceret tryk. I så fald beregnes damptrykket af følgende udtryk:

hvor T1 er kogepunktet ved det reducerede tryk P1.
RAPPORT
Hvis den tilnærmede beregningsmetode anvendes, skal beregningen være fuldstændig dokumenteret i rapporten.
REFERENCER
|
(a) |
Watson, K.M. (1943). Ind. Eng. Chem, 35, 398. |
|
(b) |
Lyman, W.J., Reehl, W.F., Rosenblatt, D.H. (1982). Handbook of Chemical Property Estimation Methods, McGraw-Hill. |
|
(c) |
OECD Environmental Monograph No.67. Application of Structure-Activity Relationships to the Estimation of Properties Important in Exposure Assessment (1993). |
BILAG II
|
A.22. |
FIBRES LÆNGDEVÆGTEDE GEOMETRISKE MIDDELDIAMETER |
1. METODE
1.1. INDLEDNING
Denne metode beskriver en procedure til måling af den længdevægtede geometriske middeldiameter (Length Weighted Geometric Mean Diameter — LWGMD) af syntetiske mineralfibre i løs vægt (Man Made Mineral Fibres — MMMF). Idet LWGMD af populationen med 95 % sandsynlighed vil ligge mellem 95 % konfidensgrænserne (LWGMD ± to standardfejl) af prøven, vil den fremkomne værdi (testværdien) være den laveste 95 % konfidensgrænse af prøven (dvs. LWGMD — 2 standardfejl). Metoden er baseret på et opdateret udkast (juni 1994) til HSE's industriprocedure, som blev fastlagt på et møde mellem ECFIA og HSE i Chester den 26. september 1993, og som er udviklet for og med udgangspunkt i et yderligere forsøg blandt forskellige laboratorier (1, 2). Denne målemetode kan anvendes til at bestemme fiberdiameteren af bulk-materialer eller produkter, der indeholder MMMF'er, herunder ildfaste keramiske fibre (RCF), Man-Made Vitreous Fibres (MMVF — fællesbetegnelse for syntetiske fibre indeholdende siliciumdioxid og en række andre metaloxider) og endelig krystallinske og polykrystallinske fibre.
Længdevægtning er en metode, der skal kompensere for den virkning af diameterfordelingen, der skyldes brud på lange fibre, når materialer udsættes for prøveudtagning eller håndtering. Der anvendes geometrisk statistik (geometrisk middelværdi) til at måle størrelsesfordelingen af MMMF-diametre, idet disse diametre almindeligvis har størrelsesfordelinger, der tilnærmer sig log-normal.
Måling af både længde og diameter er ensformigt og tidskrævende, men hvis man kun måler de fibre, der rører en uendelig tynd linje på et SEM-synsfelt, vil sandsynligheden for at vælge en given fiber være proportional med dens længde. Da der her tages hensyn til længden i beregninger om længdevægtning, er der kun behov for diameteren, hvorefter LWGMD-2SE kan beregnes som beskrevet.
1.2. DEFINITIONER
Partikel: et objekt med et længde-breddeforhold på mindre end 3:1.
Fiber: et objekt med et længde-breddeforhold (formatforhold) på mindst 3:1.
1.3. ANVENDELSESOMRÅDE OG BEGRÆNSNINGER
Metoden er udviklet med fokus på diameterfordelinger med mediandiametre fra 0,5 μm til 6 μm. Større diametre kan måles ved hjælp af mindre SEM-forstørrelser, men metoden bliver gradvist mere begrænset med finere fiberfordelinger, og derfor anbefales en TEM-måling (transmission electron microscope), hvis mediandiameteren er under 0,5 μm.
1.4. Principper for testmetoden
Der udtages en række repræsentative kerneprøver fra fibertæppet eller fra de løse bulkfibre. Bulkfibrene afkortes i en knusningsproces, hvorefter der spredes en repræsentativ delprøve i vand. Der udtages måleprøver, der filtreres gennem et polykarbonatfilter med en porestørrelse på 0,2 μm, hvorefter de forberedes til undersøgelse i scanning-elektronmikroskop (SEM-teknologi). Fiberdiametrene måles ved en skærmforstørrelse på x 10 000 eller højere (1) ved hjælp af en linjeskæringspunktmetode for at give et objektivt estimat af mediandiameteren. Det laveste 95 % konfidensinterval (baseret på en en-sidet test) beregnes for at give et estimat af den laveste værdi af materialets geometriske middelfiberdiameter.
1.5. Beskrivelse af testmetoden
1.5.1. Sikkerhedsforanstaltninger
Medarbejdernes eksponering for luftbårne fibre bør minimeres, og der bør anvendes aftræksskab eller handskekasse ved håndtering af tørre fibre. Der bør gennemføres periodisk overvågning af medarbejdernes eksponering med henblik på at vurdere effektiviteten af de anvendte kontrolmetoder. Ved arbejde med MMMF'er skal der bæres engangshandsker for at reducere hudirritation og for at forebygge krydskontaminering.
1.5.2. Apparatur og udstyr
|
— |
presse og matricer (der kan producere 10 MPa) |
|
— |
polykarbonat-kapillærfilter med en porestørrelse på 0,2 μm (25 mm diameter) |
|
— |
membranfilter i ester-cellulose med en porestørrelse på 5 μm til brug som backing-filter |
|
— |
apparatur til glasfiltrering (eller engangsfiltreringssystemer), der passer til filtre med en diameter på 25 mm (f.eks. Millipore glas-mikroanalysesæt, type nr. XX10 025 00) |
|
— |
frisk destilleret vand, der er filtreret gennem et filter med en porestørrelse på 0,2 μm for at fjerne mikroorganismer |
|
— |
sputter-coater med guld eller guld/palladium target |
|
— |
scanning elektronmikroskop med en opløsning ned til 10 nm og med forstørrelse x 10 000 |
|
— |
diverse: spatler, skalpelblade type 24, pincetter, SEM-rør, carbon-lim eller carbon-klæbebånd, silver-dag |
|
— |
ultralydsonde eller bordmonteret ultralydsbad |
|
— |
kerneprøvebor eller propbor til udtagelse af kerneprøver af MMMF-tæppe. |
1.5.3. Testprocedure
1.5.3.1. Prøveudtagning
Til tæpper og måtter anvendes der et 25 mm kerneprøvebor eller propbor til udtagning af stikprøver af tværsnittet. De bør være jævnt fordelt over hele bredden af et kort stykke tæppe eller udtages fra tilfældige områder, hvis der er lange tæppelængder til rådighed. Man kan anvende det samme udstyr til udtagning af tilfældige stikprøver fra løse fibre. Hvis det er muligt, skal der udtages seks stikprøver for at afspejle lokale variationer i bulkmaterialet.
De seks kerneprøver knuses med et pressestempel på 50 mm diameter ved 10 MPa. Materialet blandes med spatel og presses igen ved 10 MPa. Endelig fjernes materialet fra pressen og opbevares i en lukket glasflaske.
1.5.3.2. Behandling af stikprøver
Hvis det er nødvendigt, kan det organiske bindemiddel fjernes ved at anbringe fibrene i en ovn ved 450 °C i ca. en time.
Ved hjælp af Cone-and-Quarter-teknikken opdeles stikprøven (dette skal udføres i et støvskab).
Med en spatel tilsættes en lille mængde (< 0,5 g) af stikprøven til 100 ml frisk destilleret vand, der er filtreret gennem et 0,2 μm membranfilter (alternative former for ultra-rent vand kan også bruges, hvis de har vist sig tilfredsstillende). Prøven dispergeres omhyggeligt ved hjælp af en ultralyd-sonde med en effekt på 100 W, der er indstillet, så der opstår kavitation. (Hvis der ikke kan tilvejebringes en sonde, kan følgende metode anvendes: Ryst og vend i 30 sekunder; derefter ultralydsbehandling i bordmonteret ultralydsbad i fem minutter, og endelig skal der igen rystes og vendes i 30 sekunder).
Umiddelbart efter dispersion af fibrene udtages der et antal måleprøver (f.eks. tre måleprøver på 3, 6 og 10 ml) med en pipette med stor åbning (kapacitet: 2-5 ml).
Hver delprøve vacuumfiltreres gennem et 0,2 μm polykarbonatfilter med et MEC backing-filter med en porestørrelse på 5 µm og med en 25 mm glasfiltertragt med cylindrisk beholder. Der hældes ca. 5 ml filtreret destilleret vand i tragten, og delprøven tilføres derefter langsomt vandet med pipette, idet pipettespidsen holdes under væskeoverfladen. Pipette og beholder skal gennemskylles omhyggeligt efter afpipetteringen, idet tynde fibre har en tendens til at samles på overfladen.
Filtret fjernes forsigtigt og tages af fra backing-filtret, inden det sættes til tørre i en beholder.
Med en vippende bevægelse udskæres et kvart eller et halvt filterudsnit af det filtrerede bundfald ved hjælp af et skalpelblad type 24. Udskæringen anbringes forsigtigt på en SEM-stub ved hjælp af en klæbende carbon-tab eller carbon-lim. Der skal påsættes Silver-dag mindst tre steder for at optimere den elektriske kontakt på filterkanterne og på stubben. Når den påførte lim/silver-dag er tør, sputtercoates ca. 50 nm guld eller guld/palladium på overfladen af bundfaldet.
1.5.3.3. Kalibrering og betjening af SEM
1.5.3.3.1. Kalibrering
SEM-kalibrering bør kontrolleres mindst en gang om ugen (og helst hver dag) ved hjælp af et Certified Calibration Grid. Kalibreringen bør kontrolleres i forhold til en certificeret standard. Hvis den målte værdi (SEM) ikke ligger inden for ±2 % af den certificerede værdi, skal SEM-kalibreringen gentages eller justeres.
SEM skal kunne arbejde med mindst en synlig minimumdiameter på 0,2 µm ved hjælp af en reel stikprøvematrix ved en forstørrelse på x 2 000.
1.5.3.3.2. Betjening
SEM skal arbejde ved 10 000 forstørrelse (2) og indstilles til at give en god opløsning med et acceptabelt billede ved langsomme scan-rates, f.eks. 5 sekunder pr. frame. Selv om betjeningen af forskellige SEM'er kan variere noget, så gælder det generelt, at man for at opnå den bedste synlighed og opløsning ved materialer med relativt lav atomvægt bør anvende stigende spændinger på 5-10 keV og indstille apparaturet til lille punktstørrelse og kort arbejdsafstand. Da man arbejder lineært gennem materialet, skal man vælge en 0o hældning for at minimere genfokusering eller, hvis SEM'en er udstyret med et eucentrisk trin, skal denne arbejdsafstand anvendes. Der kan vælges en lavere forstørrelse, hvis materialet ikke indeholder små (diameter) fibre, og fiberdiameteren er stor (> 5 µm).
1.5.3.4. Størrelsesvurdering
1.5.3.4.1. Undersøgelse ved lille forstørrelse til vurdering af stikprøven
Først skal stikprøven undersøges ved lille forstørrelse for at afdække eventuel sammenklumpning af store fibre og for at vurdere fibertætheden. Hvis der konstateres for stor sammenklumpning, anbefales det, at der forberedes en ny prøve.
Af hensyn til den statistiske nøjagtighed er det nødvendigt at måle et minimumsantal fibre. Ligeledes er høj fibertæthed at foretrække, da undersøgelse af tomme felter er tidskrævende og uden værdi for analysen. Hvis filtret derimod er overbelastet, er det svært at måle alle målbare fibre, og desuden risikerer man ikke at få målt de små fibre, da de let ligger skjult af de større fibre.
En systematisk fejl i retning af en for høj værdi af LWGMD kan optræde som følge af fibertætheder på over 150 fibre pr. millimeter lineært snit. På den anden side vil lave fiberkoncentrationer forøge analysetiden, og derfor kan det ofte bedre betale sig at forberede en prøve med en tæthed, der ligger nær optimal tæthed, end at blive ved med at tælle på filtre med lave koncentrationer. Den optimale fibertæthed vil give et gennemsnit på ca. et eller to tælbare fibre pr. felt ved 5 000 forstørrelse. Den optimale tæthed vil imidlertid afhænge af størrelsen (diameteren) af fibrene. Derfor skal analysemedarbejderen støtte sig til en ekspertvurdering for at kunne afgøre, om fibertætheden er nær ved det optimale eller ej.
1.5.3.4.2. Længdevægtning af fiberdiametre
Det er kun de fibre, der rører (eller ligger på tværs af) en (uendelig) tynd linje afsat på SEM'ens skærm, der tælles. Derfor er der afsat en horisontal (eller vertikal) linje tværs gennem skærmens centrum.
Alternativt er der afsat et enkelt punkt i skærmens centrum, hvorefter der foretages en kontinuerlig scanning i en retning på tværs af filtret. Enhver fiber, der har et formatforhold større end 3:1, og som berører eller går gennem dette punkt, bliver underkastet måling af diameter, hvorefter resultatet registreres.
1.5.3.4.3. Størrelsesvurdering af fibre
Det anbefales, at der måles mindst 300 fibre. Hver fiber måles kun én gang ved skæringspunktet med den linje eller det punkt, der er afsat på billedet (eller tæt ved skæringspunktet, hvis fiberkanterne er skjult). Hvis man støder på fibre med uensartede tværsnit, skal man foretage en måling, der repræsenterer fiberens gennemsnitsdiameter. Man skal være meget omhyggelig med at udpege kanten og måle den korteste afstand mellem fiberens kanter. Størrelsesvurderingen kan ske online eller offline på lagrede billeder eller fotos. Det anbefales at bruge systemer til halvautomatisk billedmåling, der henter data direkte ind i et regneark, da de sparer tid og udelukker afskriftsfejl. Endelig automatiserer de beregningerne.
Enderne af lange fibre skal undersøges ved lille forstørrelse for at sikre, at de ikke krummer tilbage i målefeltet og derved måles flere gange.
2. DATA
2.1. BEHANDLING AF RESULTATERNE
Fiberdiametre følger almindeligvis ikke en normalfordeling. Hvis man gennemfører en log-transformation, er det dog muligt at opnå en fordeling, der ligger nær ved normalfordelingen.
Den aritmetiske middelværdi (mean lnD) og standardafvigelsen (SDlnD) af den naturlige logaritme (lnD) til n fiberdiametre (D) beregnes.
|
|
(1) |
|
|
(2) |
Standardafvigelsen divideres med kvadratroden af antallet af målinger (n) for at nå frem til standardfejlen (SElnD).
|
|
(3) |
To gange standardfejlen trækkes fra middelværdien, og eksponentialfunktionen af denne værdi (middelværdi minus to standardfejl) beregnes for at få den geometriske middelværdi minus to geometriske standardfejl.
|
|
(4) |
3. RAPPORTERING
TESTRAPPORT
Testrapporten skal mindst indeholde følgende oplysninger:
|
— |
værdien af LWGMD-2SE |
|
— |
enhver afvigelse og især de afvigelser, der vil kunne påvirke præcisionen eller nøjagtigheden af resultaterne med tilhørende behørigt begrundede tilfælde. |
4. REFERENCER
|
1. |
B. Tylee SOP MF 240. Health and Safety Executive. Februar 1999. |
|
2. |
G. Burdett and G. Revell. Development of a standard method to measure the length-weigthed geometric mean fibre diameter: Results of the Second inter-laboratory exchange. IR/L/MF/94/07. Project R42.75 HPD. Health and Safety Executive. Research and Laboratory Services Division. 1994. |
(1) Denne forstørrelsesværdi gælder for 3 µm fibre. For 6 µm fibre vil en forstørrelse på × 5 000 være mere passende.
(2) For 3 μm fibres vedkommende, se forrige note.
BILAG III
|
B.46. |
IN VITRO-HUDIRRITATION: TEST MED REKONSTRUERET HUMAN EPIDERMIS-MODEL |
1. METODE
1.1. INDLEDNING
Hudirritation defineres som fremkomsten af reversible hudskader efter påføring af teststof i op til 4 timer (som defineret i FN's globale harmoniserede system for klassificering og mærkning af kemikalier (GHS)) (1). Denne testmetode omfatter en in vitro-procedure, som afhængigt af informationsbehovet kan give mulighed for at bestemme stoffers hudirritation og udgøre en selvstændig erstatningstest i en teststrategi med en »weight of evidence«-tilgang (2).
Vurderingen af hudirritation har typisk omfattet brug af forsøgsdyr (se metode B.4) (3). Af hensyn til dyrevelfærden giver metode B.4 mulighed for at bestemme hudætsning/hudirritation ved hjælp af en sekventiel teststrategi, der består af validerede in vitro- og ex vivo-metoder, hvorved det undgås, at dyr udsættes for smerter og lidelser. Der er tre validerede in vitro-testmetoder og -testretningslinjer, B.40, B.40a og TG 435 (4, 5, 6), som er relevante for ætsningsdelen af den sekventielle teststrategi i B.4.
Denne testmetode bygger på modeller af rekonstrueret human epidermis, som på grund af deres overordnede udformning (brug af humant afledte epidermiskeratinocytter som cellekilde, repræsentativt væv og cellearkitektur) ret nøje efterligner de biokemiske og fysiologiske egenskaber i de øvre dele af menneskers hud, dvs. epidermis. Ved den procedure, der er beskrevet i denne testmetode, kan det afgøres, om lokalirriterende stoffer tilhører kategori 2 ifølge FN's GHS fareidentifikation. Testmetoden rummer også et sæt ydeevnestandarder til brug for vurdering af lignende og modificerede testmetoder, der er baseret på rekonstrueret human epidermis (7).
To in vitro-testmetoder, hvor der benyttes rekonstrueret human epidermis-modeller, som forhandles under betegnelserne EpiSkin™ og EpiDerm™, har været underkastet prævaliderings-, optimerings- og valideringundersøgelser (8, 9, 10, 11, 12, 13, 14, 15, 16, 17). Disse referencer byggede på R 38. Visse aspekter af omberegningen med henblik på GHS behandles i reference 25. Metoder med samme ydeevne som EpiSkin™ (valideret referencemetode 1) anbefales som selvstændige erstatningsmetoder for in vivo-test med kaniner til klassificering af lokalirriterende stoffer i GHS kategori 2. I forbindelse med klassificering af lokalirriterende stoffer i GHS kategori 2 anbefales metoder med samme ydeevne som EpiDerm™ (valideret referencemetode 2) kun som screeningmetode eller som en del af en sekventiel teststrategi med en »weight of evidence«-tilgang. Inden en foreslået metode til in vitro-undersøgelse af hudirritation med en rekonstrueret human epidermis-model kan benyttes i forbindelse med lovgivningen, skal dens pålidelighed og relevans (nøjagtighed) og grænserne for dens påtænkte anvendelse bestemmes, så det er sikkert, at den på de punkter er sammenlignelig med den validerede referencemetode 1, og det skal ske i overensstemmelse med ydeevnestandarderne i testmetoden (tillægget).
To andre in vitro-testmetoder med rekonstrueret human epidermis er valideret ifølge kravene i denne testmetode med omtrent samme resultat som den validerede referencemetode 1 (18). Det er den modificerede EpiDerm™-testmetode (modificeret referencemetode 2) og SkinEthic RHE™-testmetoden (analog til metode 1).
1.2. DEFINITIONER
I denne testmetode anvendes følgende definitioner:
Nøjagtighed: Overensstemmelsen mellem testmetodens resultater og accepterede referenceværdier. Den er et mål for testmetodens ydeevne og et af aspekterne af relevans. Udtrykket benyttes ofte som synonym for »konkordans«, forstået som den andel korrekte udfald, testmetoden fører til.
Batchkontrolstof: Referencestof, som med det pågældende væv giver et udfald for cellernes levedygtighed midt i måleområdet.
Cellelevedygtighed: En parameter, der måler den samlede aktivitet i en cellepopulation, f.eks. udtrykt ved de cellulære mitokondriedehydrogenasers evne til at reducere vitalfarvestoffet MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid, thiazolylblåt], og som, afhængigt af det valgte endpoint og testens udformning, korrelerer med det totale antal levende celler og/eller deres levedygtighed.
ET50 : Den eksponeringstid, der nedsætter cellernes levedygtighed med 50 % ved påføring af et markørstof i en nærmere angivet fast koncentration; se også IC50.
Falsk negativ-rate: Andelen af alle positive stoffer, som metoden ukorrekt har udpeget som negative. Den er en indikator for testmetodens ydeevne.
Falsk positiv-rate: Andelen af alle negative (ikke-aktive) stoffer, der ukorrekt er udpeget som positive. Den er en indikator for testmetodens ydeevne.
Uendelig dosis: Den mængde teststof, som er påført på huden ud over den mængde, der er nødvendig for helt at dække hudoverfladen på ensartet måde.
GHS (det globale harmoniserede system for klassificering og mærkning af kemikalier): Et system til klassificering af stoffer og blandinger efter standardiserede typer og niveauer af fysisk fare, sundhedsfare og miljøfare med tilhørende kommunikationselementer såsom piktogrammer, signalord, risikosætninger, sikkerhedssætninger og sikkerhedsdatablade, således at oplysninger om stoffernes og blandingernes skadelige virkninger kan videreformidles med henblik på at beskytte mennesker (f.eks. arbejdsgivere, arbejdstagere, transportører, forbrugere og redningspersonel) og miljø (1) og gennemført i EU ved forordning (EF) nr. 1272/2008.
IC50 : Den koncentration, ved hvilken et markørstof nedsætter vævets levedygtighed med 50 % (IC50) efter en fast eksponeringstid; se også ET50.
Ydeevnestandarder: Standarder, som med udgangspunkt i en valideret referencemetode giver mulighed for at vurdere, om en foreslået testmetode, som er mekanistisk og funktionelt tilsvarende, er sammenlignelig. De indeholder bl.a. I) de centrale dele af testmetoden, II) en minimumsliste med referencestoffer, som er udvalgt blandt de stoffer, der er benyttet til at påvise, at den validerede referencemetodes ydeevne er acceptabel, III) de sammenlignelige niveauer af nøjagtighed og pålidelighed, som er fastsat efter, hvad der er opnået med den validerede referencemetode, og som det skal påvises, at den foreslåede testmetode kan yde, ved vurdering ud fra minimumslisten med referencestoffer.
Pålidelighed: Mål for, i hvilken grad en testmetode kan udføres reproducerbart i og mellem laboratorier over en længere periode, når den udføres efter samme protokol. Den bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier.
Følsomhed: Andelen af alle positive/aktive stoffer, der klassificeres korrekt ved testen. Den er et mål for nøjagtigheden af en testmetode, der fører til kategoriske resultater, og har stor betydning for vurdering af en testmetodes relevans.
Specificitet: Andelen af alle negative/ikke-aktive stoffer, der klassificeres korrekt ved testen. Den er et mål for nøjagtigheden af en testmetode, der fører til kategoriske resultater, og har stor betydning for vurdering af en testmetodes relevans.
Hudirritation: Fremkaldelse af reversibel skade på huden ved påføring af teststoffet i indtil 4 timer. Hudirritation er en lokalt fremkaldt ikke-immunogen reaktion, som indtræder kort tid efter stimuleringen (24). Den karakteriseres især ved, at den reversible proces indebærer inflammatoriske reaktioner og de fleste af de karakteristiske kliniske tegn på irritation (rødme, ødem, kløe og smerte), der er knyttet til en inflammatorisk proces.
1.3. ANVENDELSESOMRÅDE OG BEGRÆNSNINGER
Det er en begrænsning for de test med rekonstrueret human epidermis, som henhører under denne testmetode, at de kun muliggør klassificering af stoffer som hudirriterende i kategori 2 ifølge FN's GHS. Da de ikke giver mulighed for klassificering af stoffer i den valgfrie kategori 3 ifølge FN's GHS, vil alle andre stoffer ikke blive klassificeret (ingen kategori). Afhængigt af behovet for lovgivning og om der i fremtiden bliver indføjet nye endpoints, foretaget forbedringer eller udviklet nye analoge metoder, kan der blive behov for revision af denne testmetode.
Denne testmetode muliggør fareidentifikation af lokalirriterende stoffer med én komponent (19), men den giver ikke fyldestgørende oplysninger om hudætsning. Gasser og aerosoler kan ikke testes; blandinger er endnu ikke vurderet ved en valideringsundersøgelse.
1.4. TESTMETODENS PRINCIP
Teststoffet påføres på en tredimensional rekonstrueret human epidermis-model, som består af normale humant afledte epidermiskeratinocytter, som ved dyrkning har dannet en stærkt differentieret flerlagsmodel af den humane epidermis. Den består af velordnede basallag, stratum spinosum og stratum granulosum og et flerlaget stratum corneum, hvori intercellulære lamellære lipidlag er til stede i samme mønstre, som man finder in vivo.
Princippet bag testen med rekonstrueret human epidermis-model bygger på den forudsætning, at lokalirriterende stoffer kan trænge gennem stratum corneum ved diffusion og er cytotoksiske for cellerne i de underliggende lag. Cellernes levedygtighed måles ved, at dehydrogenase omdanner vitalfarvestoffet MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid, thiazolylblåt; Einecs-nr. 206-069-5, CAS-nr. 298-93-1)] til et blåt formazansalt, der måles kvantitativt efter ekstraktion fra vævene (20). Lokalirriterende stoffer identificeres ved deres evne til at nedsætte cellernes levedygtighed til en bestemt tærskelværdi eller derunder (f.eks. ≤ 50 % for lokalirriterende stoffer i kategori 2 ifølge FN's GHS). Stoffer, der fører til en levedygtighed for cellerne, der ligger over den fastlagte tærskel, klassificeres ikke (f.eks. > 50 % ingen kategori).
Modelsystemer med rekonstrueret human epidermis kan benyttes til test af faste stoffer, væsker, halvfaste stoffer og vokser. Væsker kan være vandige eller ikke-vandige, og faste stoffer kan være opløselige eller uopløselige i vand. Faste stoffer bør testes som fint pulver, når det overhovedet er muligt. Da der ved valideringen af testsystemerne med rekonstrueret human epidermis-model er undersøgt 58 omhyggeligt udvalgte stoffer, der repræsenterer et bredt spektrum af kemiske stofgrupper, forventes metoderne at kunne anvendes generelt til alle kemiske stofgrupper (16). Valideringen omfatter 13 lokalirriterende stoffer i GHS-kategori 2. Bemærk, at ikkeætsende syrer, baser, salte og andre uorganiske stoffer ikke var omfattet af valideringen, og at visse grupper af organiske stoffer, der vides at være lokalirriterende, såsom hydroperoxider, phenoler og overfladeaktive stoffer heller ikke eller kun i begrænset omfang var med i valideringen.
1.5. GODTGØRELSE AF KOMPETENCE
Inden et laboratorium påbegynder rutinemæssig brug af en valideret metode, der følger denne testmetode, bør de godtgøre deres tekniske kompetence ved hjælp af de ti anbefalede stoffer i tabel 1. I denne testmetode betragtes kemikalier i den valgfrie kategori 3 i FN's GHS som ikke-klassificerede. Inden nye lignende (analoge) testmetoder, der udvikles under denne testmetode, og som strukturelt og funktionelt ligner de validerede referencemetoder, og modificeringer af validerede metoder tages i brug af de kontrollerende myndigheder til testning, bør det ved hjælp af ydeevnestandarderne i tillægget til denne testmetode godtgøres, at den nye testmetode har tilsvarende pålidelighed og nøjagtighed.
Tabel 1
Kompetencestoffer, der udgør en delmængde af referencestofferne i tillægget
|
Stof |
CAS-nummer |
In vivo-vurdering |
Fysisk form |
GHS-kategori |
|
naphthaleneddikesyre |
86-87-3 |
0 |
S |
Ingen kat. |
|
isopropanol |
67-63-0 |
0,3 |
L |
Ingen kat. |
|
methylstearat |
112-61-8 |
1 |
S |
Ingen kat. |
|
heptylbutyrat |
5870-93-9 |
1,7 |
L |
Valgfri kat. 3 |
|
hexylsalicylat |
6259-76-3 |
2 |
L |
Valgfri kat. 3 |
|
cyclamenaldehyd |
103-95-7 |
2,3 |
L |
Kat. 2 |
|
1-bromhexan |
111-25-1 |
2,7 |
L |
Kat. 2 |
|
butylmethacrylat |
97-88-1 |
3 |
L |
Kat. 2 |
|
1-methyl-3-phenyl-1-piperazin |
5271-27-2 |
3,3 |
S |
Kat. 2 |
|
heptanal |
111-71-7 |
4 |
L |
Kat. 2 |
1.6. BESKRIVELSE AF METODEN
Nedenfor følger en beskrivelse af komponenter og procedurer i en test til vurdering af hudirritation med rekonstrueret human epidermis-model. En rekonstrueret human epidermis-model kan konstrueres, fremstilles eller købes i handelen (f.eks. EpiSkin™, EpiDerm™ og SkinEthic RHE™). Der er standardiserede testprotokoller for EpiSkin™, EpiDerm™ og SkinEthic RHE™ til rådighed på http://ecvam.jrc.ec.europa.eu (21, 22, 23). Test bør udføres på følgende måde:
1.6.1. Komponenterne i rekonstrueret human epidermis-modellen
1.6.1.1. Almindelige modelbetingelser
Der skal anvendes normale humane keratinocytter til at konstruere epithelet. Der skal være flere lag af levedygtige epithelceller (basallag, stratum spinosum, stratum granulosum) til stede under et fungerende stratum corneum. Stratum corneum skal være flerlaget og indeholde den fedtprofil, der gør den til en effektiv barriere, der kan modstå hurtig gennemtrængning af cytotoksiske markørstoffer, f.eks. natriumdodecylsulfat (SDS) eller Triton X-100. Barrierefunktionen kan bedømmes enten ved at bestemme den koncentration, hvor et markørstof nedsætter vævets levedygtighed med 50 % (IC50) efter en fast eksponeringstid, eller ved at bestemme den eksponeringstid, der er nødvendig for at nedsætte levedygtigheden med 50 % (ET50), når markørstoffet påføres i en nærmere fastsat konstant koncentration. Modellens fysiske udformning skal forhindre, at stof passerer uden om stratum corneum til det levedygtige væv, hvilket ville forringe modelleringen af hudens eksponering. Hudmodellen skal være fri for kontamination med bakterier, virus, mykoplasma og svampe.
1.6.1.2. Funktionsmæssige modelbetingelser
1.6.1.2.1. Levedygtighed
MTT foretrækkes til kvantificering af levedygtigheden (20). Absorbansen (optical density — OD) af den farvestofmængde, der kan ekstraheres (ved opløsning) fra vævet fra behandlingen med negativ kontrol (NC), skal være mindst 20 gange absorbansen af det rene ekstraktionsmiddel. Det skal godtgøres, at vævet fra behandling med negativ kontrol er en stabil kultur (leverer enslydende levedygtighedsmålinger) i hele testeksponeringsperioden.
1.6.1.2.2. Barrierefunktion
Stratum corneum og dets lipidsammensætning skal være tilstrækkelig til at modstå hurtig gennemtrængning af cytotoksiske markørstoffer, f.eks. SDS eller Triton X-100, bestemt ved IC50 eller ET50.
1.6.1.2.3. Morfologi
Rekonstrueret hud/epidermis skal af behørigt kvalificeret personale undersøges histologisk for human hud/epidermis-lignende struktur (herunder flerlaget stratum corneum).
1.6.1.2.4. Reproducerbarhed
De resultater, metoden giver med en bestemt model, skal udvise tidsmæssig reproducerbarhed, helst godtgjort ved hjælp af et egnet batchkontrolstof (se tillægget).
1.6.1.2.5. Kvalitetskontroller (QC) i modellen
Hver batch af den anvendte epidermismodel skal overholde veldefinerede produktionsgodkendelseskriterier, hvoraf levedygtighed (punkt 1.6.1.2.1) og barrierefunktion (punkt 1.6.1.2.2) er de vigtigste. Der skal af leverandøren af hudmodellen (eller forsøgslederen, hvis modellen er internt fremstillet) fastsættes en interval for acceptable værdier af IC50 eller ET50 (øvre og nedre grænse). Vævenes barriereegenskaber kontrolleres af laboratoriet efter modtagelsen. Kun resultater, der er fremkommet med kvalificerede væv, kan accepteres til at give en pålidelig vurdering af lokalirriterende virkninger. Nedenfor er acceptintervallerne for de validerede referencemetoder angivet som eksempler.
Tabel 2
Eksempler på batchgodkendelseskriterier til kvalitetskontrol
|
|
Nedre acceptgrænse |
Middelværdi i acceptinterval |
Øvre acceptgrænse |
|
Valideret referencemetode 1 (behandling i 18 h med SDS) |
IC50 = 1,0 mg/ml |
IC50 = 2,32 mg/ml |
IC50 = 3,0 mg/ml |
|
Valideret referencemetode 2 (1 % Triton X-100) |
ET50 = 4,8 hr |
ET50 = 6,7 hr |
ET50 = 8,7 hr |
1.6.1.3. Påføring af test- og kontrolstoffer
Der anvendes et passende antal vævsgentagelser til hver behandling og til kontroller (mindst tredobbeltbestemmelse pr. analyseserie). For både flydende og faste stoffer gælder det, at der skal påføres tilstrækkelig meget teststof til, at hudoverfladen dækkes med et jævnt lag, samtidig med at uendelig dosis undgås (se 1.2 Definitioner), dvs. mindst 25 μL/cm2 eller 25 mg/cm2. I tilfælde af faste stoffer fugtes epidermisoverfladen med deioniseret eller destilleret vand inden påføringen, så der sikres god hudkontakt. Faste stoffer bør testes som fint pulver, når det overhovedet er muligt. Efter eksponeringsperioden vaskes teststoffet omhyggeligt af hudoverfladen med vandig buffer eller 0,9 % NaCl. Afhængigt af, hvilken rekonstrueret human epidermis-model der anvendes, kan eksponeringsperioden variere mellem 15 og 60 minutter og inkubationstemperaturen mellem 20 og 37 °C. Der er nærmere oplysninger i de tre metoders standardprocedurer (21, 22, 23).
Der skal i hver undersøgelse anvendes samtidig negativ kontrol og positive kontroller (PC), så det godtgøres, at levedygtigheden (NC), barrierefunktionen og vævets deraf følgende følsomhed (PC) ligger inden for et forud fastsat acceptinterval. Som positivt kontrolstof foreslås 5 % vandig SDS. Som negativt kontrolstof foreslås vand eller phosphatbufferet saltopløsning (PBS).
1.6.1.4. Måling af cellelevedygtighed
Det vigtigste i testproceduren er, at levedygtigheden ikke måles umiddelbart efter eksponeringen med teststofferne, men først efter en tilstrækkelig lang inkubationsperiode efter behandlingen i frisk substrat af de renskyllede væv fra behandlingen. En sådan periode betyder, dels at huden kan komme sig over svagt irriterende virkninger, dels at eventuelle cytotoksiske virkninger træder tydeligt frem. En inkubationsperiode efter behandlingen på 42 timer viste sig i testoptimeringfasen (9, 10, 11, 12, 13) at være optimal, og der blev derfor benyttet en sådan ved valideringen af referencetestmetoderne.
Måling af omdannelse af MTT er en valideret kvantitativ metode, som skal benyttes til måling af cellelevedygtigheden. Den er forenelig med brug i et tredimensionalt væv. Hudlappen anbringes i en MTT-opløsning af passende koncentration (f.eks. 0,3-1 mg/mL) i tre timer. Det udfældede blå formazan ekstraheres fra vævet med et opløsningsmiddel (f.eks. isopropanol eller sur isopropanol), og formazankoncentrationen beregnes ud fra måling af absorbansen i et højst ±30 nm bredt bånd ved 570 nm.
Teststoffets optiske egenskaber eller dets kemiske reaktion med MTT kan interferere med målingen og føre til et forkert skøn over levedygtigheden (da teststoffet kan standse, mindske eller forårsage farvedannelsen). Det kan forekomme, hvis et specifikt teststof ikke bliver fjernet fuldstændigt fra huden ved skylning, eller hvis det er trængt ned i epidermis. Hvis teststoffet reagerer direkte med MTT, hvis det selv er farvet, eller hvis det bliver farvet under behandlingen af vævet, skal der anvendes yderligere kontroller til at spore og korrigere for teststoffernes interferens med levedygtighedsmålingen. En direkte test for MTT-reduktion er nærmere beskrevet i testmetodeprotokollen for de validerede referencemetoder (21, 22, 23). Ikke-specifik farvning (NSC) som følge af interferens må ikke være større end 30 % af den negative kontrol (med henblik på korrektion). Er NSC > 30 %, anses teststoffet for uforeneligt med testen.
1.6.1.5. Acceptkriterier for målingen
For hver måling med gyldige batcher (se punkt 1.6.1.2.5) skal væv, der er behandlet med negativ kontrol, give absorbansresultater, der afspejler kvaliteten af de væv, der har været underkastet alle forsendelses- og modtagelsestrin og alle processer i irritationsprotokollen. Absorbansværdierne af kontrollerne må ikke ligge lavere end forud fastsatte nedre grænser. På tilsvarende måde skal væv, der er behandlet med positiv kontrol, dvs. 5 % vandig SDS, afspejle vævenes følsomhed og deres evne til at reagere på et lokalirriterende stof under de aktuelle forsøgsvilkår (f.eks. levedygtighed ≤ 40 % for valideret referencemetode 1 og ≤ 20 % for valideret referencemetode 2). Der skal defineres passende mål for den tilknyttede variation mellem multiple bestemmelser på samme væv (f.eks. skal en eventuel standardafvigelse være ≤ 18 %).
2. DATA
2.1. DATA
For hver behandling skal data fra de enkelte multipelbestemmelser (f.eks. absorbansværdier og beregnet cellelevedygtighedsprocent for hvert teststof samt klassificeringen) fremlægges i tabelform, herunder data fra eventuelle gentagelser af forsøget. Desuden skal gennemsnit og standardafvigelse angives for hvert forsøg. For hvert testet stof oplyses der om eventuelt iagttagne vekselvirkninger med MTT-reagenset og farvede teststoffer.
2.2. FORTOLKNING AF RESULTATER
De absorbansværdier, som måles ved hver undersøgelse, anvendes til beregning af den procentuelle levedygtighed i forhold til den negative kontrol, som vilkårligt sættes til 100 %. Den tærskelværdi for cellelevedygtighedsprocent, som adskiller lokalirriterende stoffer fra ikke-klassificerede stoffer, og de statistiske metoder, der benyttes til at vurdere resultaterne og udpege lokalirriterende stoffer, skal klart defineres, dokumenteres og bevises at være hensigtsmæssige. Tærskelværdierne for vurdering som lokalirriterende ifølge de validerede referencemetoder er som følger:
Teststoffet betragtes som værende hudirriterende i kategori 2 ifølge FN's GHS:
|
i) |
hvis vævslevedygtigheden efter eksponering og inkubation efter behandling er mindre end eller lig med (≤) 50 %. |
Teststoffet anses for ikke at tilhøre nogen kategori:
|
ii) |
hvis vævslevedygtigheden efter eksponering og inkubation efter behandling er større end (>) 50 %. |
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
Test- og kontrolstoffer:
|
— |
kemiske navne, f.eks. IUPAC-navn eller CAS-navn og -nummer, hvis de kendes |
|
— |
stoffets renhed og sammensætning (i vægtprocent) |
|
— |
fysiske-kemiske egenskaber, som er relevante for undersøgelsen (f.eks. fysisk tilstand, stabilitet og flygtighed, pH og opløselighed i vand, hvis kendt) |
|
— |
eventuel behandling af test/kontrolstoffer inden testen (f.eks. opvarmning, formaling) |
|
— |
opbevaringsforhold. |
Begrundelse for valg af hudmodel og forsøgsprotokol.
Testbetingelser:
|
— |
anvendt cellesystem |
|
— |
kalibreringsinformationer for udstyr og båndpas, som er anvendt til måling af cellelevedygtighed (f.eks. spektrofotometer) |
|
— |
komplet dokumentation for den anvendte hudmodel, herunder om dens ydeevne. Der skal bl.a. gives oplysning om
|
|
— |
detaljer om den anvendte testprocedure |
|
— |
anvendte testdoser, eksponeringens varighed og længden af inkubationsperioden efter behandlingen |
|
— |
beskrivelse af eventuelle ændringer i testproceduren |
|
— |
reference til historiske data for modellen. Der skal bl.a. gives oplysning om
|
|
— |
beskrivelse af de anvendte evalueringskriterier, herunder begrundelse for valg af prædiktionsmodellens afskæringspunkt(er). |
Resultater:
|
— |
angivelse i tabelform af data fra hver enkelt testprøve |
|
— |
beskrivelse af andre observerede virkninger. |
Diskussion af resultater.
Konklusioner.
4. LITTERATUR
|
(1) |
United Nations (UN) (2007). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Second revised edition, UN New York and Geneva, 2007. Se følgende websted: http://www.unece.org/trans/danger/publi/ghs/ghs_rev02/02files_e.html. |
|
(2) |
REACH: Guidance on Information Requirements and Chemical Safety Assessment. Se følgende websted: http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1232447649. |
|
(3) |
Testmetode B4. AKUT TOKSICITET: HUDIRRITATION/ÆTSNING. |
|
(4) |
Testmetode B.40. IN VITRO-HUDÆTSNING: MÅLING AF TRANSKUTAN ELEKTRISK MODSTAND (TER). |
|
(5) |
Testmetode B.40.A. IN VITRO-HUDÆTSNING: TEST MED HUMAN HUDMODEL. |
|
(6) |
OECD (2006). Test Guideline 435. OECD Guideline for the Testing of Chemicals. In Vitro Membrane Barrier Test Method. Adopted July 19, 2006. Se følgende websted: http://www.oecd.org/document/22/0,2340,en_2649_34377_1916054_1_1_1_1,00.html. |
|
(7) |
ECVAM (2009) Performance Standards for applying human skin models to in vitro skin irritation. Se under »Download Study Documents« på følgende websted: http://ecvam.jrc.ec.europa.eu. |
|
(8) |
Fentem, J.H., Briggs, D., Chesné, C., Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Roguet, R., van de Sandt, J.J.M. & Botham, P. (2001). A prevalidation study on in vitro tests for acute skin irritation. Results and evaluation by the Management Team. Toxicology in Vitro 15, 57-93. |
|
(9) |
Portes, P., Grandidier, M.H., Cohen, C. & Roguet, R.(2002). Refinement of the EPISKIN protocol for the assessment of acute skin irritation of chemicals: follow-up to the ECVAM prevalidation study. Toxicology in Vitro 16, 765-770. |
|
(10) |
Kandárová, H., Liebsch, M., Genschow, E., Gerner, I., Traue, D., Slawik, B. & Spielmann, H. (2004). Optimisation of the EpiDerm test protocol for the upcoming ECVAM validation study on in vitro skin irritation tests. ALTEX 21, 107-114. |
|
(11) |
Kandárová, H., Liebsch, M., Gerner, I., Schmidt, E., Genschow, E., Traue, D. & Spielmann H. (2005) The EpiDerm Test Protocol fort the Upcoming ECVAM Validation Study on In Vitro Skin Irritation Tests — An Assessment of the Performance of the Optimised Test. ATLA 33, 351-367. |
|
(12) |
Cotovio, J., Grandidier, M.-H., Portes, P., Roguet, R. & G. Rubinsteen. (2005). The In Vitro Acute Skin Irritation of Chemicals: Optimisation of the EPISKIN Prediction Model within the Framework of the ECVAM Validation Process. ATLA 33, 329-249. |
|
(13) |
Zuang, V., Balls, M., Botham, P.A., Coquette, A., Corsini, E., Curren, R.D., Elliot, G.R., Fentem, J.H., Heylings, J.R., Liebsch, M., Medina, J., Roguet, R., van De Sandt, J.J.M., Wiemann, C. & Worth, A.(2002). Follow-up to the ECVAM prevalidation study on in vitro tests for acute skin irritation. ECVAM Skin Irritation Task Force Report 2. ATLA 30,109-129. |
|
(14) |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007) The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559-601. |
|
(15) |
Hoffmann, S. (2006). ECVAM Skin Irritation Validation Study Phase II: Analysis of the Primary Endpoint MTT and the Secondary Endpoint IL1-α. 135 pp. + annexes. Se under »Download Study Documents« på følgende websted: http://ecvam.jrc.ec.europa.eu. |
|
(16) |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603-619. |
|
(17) |
J. Cotovio, M.-H. Grandidier, D. Lelièvre, R. Roguet, E. Tinois-Tessonneaud, J. Leclaire (2007). In vitro acute skin irritancy of chemicals using the validated EPISKIN model in a tiered strategy — Results and performances with 184 cosmetic ingredients, AATEX, Special Issue-proceedings from WC6. Vol.14, 351-358. |
|
(18) |
Erklæring fra ESAC om ajourførte EpiDerm-forsøg og tilsvarende SkinEthic-forsøg. 5. november 2008. |
|
(19) |
EC (2006). Europa-Parlamentets og Rådets forordning (EF) nr. 1907/2006 af 18. december 2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH), om oprettelse af et europæisk kemikalieagentur og om ændring af direktiv 1999/45/EF og ophævelse af Rådets forordning (EØF) nr. 793/93 og Kommissionens forordning (EF) nr. 1488/94 samt Rådets direktiv 76/769/EØF og Kommissionens direktiv 91/155/EØF, 93/67/EØF, 93/105/EF og 2000/21/EF. Den Europæiske Unions Tidende L 396 af 30.12.2006, s. 1. OPOCE, Luxembourg. |
|
(20) |
Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55-63. |
|
(21) |
EpiSkin™ SOP, Version 1.6 (January 2005). Validation of the EpiSkin Skin Irritation Test — 42 Hours Assay For The Prediction of Acute Skin Irritation of Chemicals. Se under »Download Study Documents« på følgende websted: http://ecvam.jrc.ec.europa.eu. |
|
(22) |
EpiDerm™ SOP, Version 5.0 (October 2004). Draft Standard Operating Procedure. In Vitro Skin Irritation Test: Human Skin Model. Model: EpiDerm™- 200. Se under »Download Study Documents« på følgende websted: http://ecvam.jrc.ec.europa.eu. |
|
(23) |
SkinEthic RHE™ SOP. Se senere under »Download Study Documents« på følgende websted: http://ecvam.jrc.ec.europa.eu. |
|
(24) |
Harvell, J.D., Lammintausta, K. Maibach H.I. (1995) Irritant contact dermatitis IN: Guin J.D. Practical Contact Dermatitis Mc Graw-Hill New York, pp 7-18. |
|
(25) |
Griesinger C, Barroso J & Zuang V: ECVAM background document on the recent adaptations of the ECVAM performance standards for in vitro skin irritation testing in the context of the drafting process of an EU test method and an OECD draft test guideline. Ispra, 13. november 2008. |
Tillæg
Vurdering af ydeevnen ved de foreslåede metoder til in vitro-undersøgelse af hudirritation med en rekonstrueret human epidermis-model
INDLEDNING
De procedurer, der foreslås anvendt i forbindelse med denne testmetode, skal vurderes med hensyn til pålidelighed og nøjagtighed ved hjælp af stoffer, der repræsenterer hele Draize-pointskalaen for irritation. Når den foreslåede procedure bedømmes ved hjælp af de 20 anbefalede referencestoffer (tabel 2), skal den have samme niveau af pålidelighed og nøjagtighed som den validerede referencemetode 1 (tabel 3) (1). Der redegøres nærmere for nøjagtigheds- og pålidelighedskrav i punkt II og III nedenfor. Der indgår ikke-klassificerede og klassificerede (i kategori 2 ifølge FN's GHS) stoffer, som repræsenterer relevante kemiske stofgrupper, således at den foreslåede testmetode kan sammenlignes med den validerede referencemetode 1 med hensyn til pålidelighed og ydeevne (følsomhed, specificitet, falsk negativ-rate, falsk postiv-rate og nøjagtighed). Inden testmetoden tages i brug til test af nye stoffer, skal det fastslås, om den er pålidelig, og om den kan identificere irriterende stoffer i kategori 2 ifølge FN's GHS korrekt.
YDEEVNESTANDARDER
Ydeevnestandarderne omfatter følgende tre elementer: I) de centrale dele af testmetoden, II) referencestoffer, III) fastlagte værdier for nøjagtighed og pålidelighed (2). De foreliggende ydeevnestandarder bygger på de ydeevnestandarder, der er fastlagt efter afslutning af ECVAM-valideringsundersøgelsen af hudirritation (3).
I) De centrale dele af testmetoden
Almindelige modelbetingelser
Der skal anvendes normale humane keratinocytter til at konstruere epithelet. Der skal være flere lag af levedygtige epithelceller (basallag, stratum spinosum, stratum granulosum) til stede under et fungerende stratum corneum. Stratum corneum skal være flerlaget og indeholde den fedtprofil, der gør den til en effektiv barriere, der kan modstå hurtig gennemtrængning af cytotoksiske markørstoffer, f.eks. SDS eller Triton X-100. Barrierefunktionen kan bedømmes enten ved at bestemme den koncentration, hvor et markørstof nedsætter vævets levedygtighed med 50 % (IC50) efter en fast eksponeringstid, eller ved at bestemme den eksponeringstid, der er nødvendig for at nedsætte levedygtigheden med 50 % (ET50), når markørstoffet påføres i en nærmere fastsat konstant koncentration. Modellens fysiske udformning skal forhindre, at stof passerer uden om stratum corneum til det levedygtige væv, hvilket ville forringe modelleringen af hudens eksponering. Hudmodellen skal være fri for kontamination med bakterier, virus, mykoplasma og svampe.
Funktionsmæssige modelbetingelser
Levedygtighed
MTT foretrækkes til kvantificering af levedygtigheden (4). Absorbansen (optical density — OD) af den farvestofmængde, der kan ekstraheres (ved opløsning) fra vævet fra behandlingen med negativ kontrol (NC), skal være mindst 20 gange absorbansen af det rene ekstraktionsmiddel. Det skal godtgøres, at vævet fra behandling med negativ kontrol er en stabil kultur (leverer enslydende levedygtighedsmålinger) i hele testeksponeringsperioden.
Barrierefunktion
Stratum corneum og dets lipidsammensætning skal være tilstrækkelig til at modstå hurtig gennemtrængning af cytotoksiske markørstoffer, f.eks. SDS eller Triton X-100, bestemt ved IC50 eller ET50.
Morfologi
Rekonstrueret hud/epidermis skal af behørigt kvalificeret personale undersøges histologisk for human hud/epidermis-lignende struktur (herunder flerlaget stratum corneum).
Reproducerbarhed
De resultater, metoden giver med en bestemt model, skal udvise tidsmæssig reproducerbarhed, helst godtgjort ved hjælp af et egnet batchkontrolstof (se definitionerne i punkt 1.2).
Kvalitetskontroller (QC) i modellen
Hver batch af den anvendte epidermismodel skal overholde veldefinerede produktionsgodkendelseskriterier, hvoraf levedygtighed og barrierefunktion er de vigtigste. Der skal af leverandøren af hudmodellen (eller forsøgslederen, hvis modellen er internt fremstillet) fastsættes en interval for acceptable værdier af IC50 eller ET50 (øvre og nedre grænse). Vævenes barriereegenskaber kontrolleres af laboratoriet efter modtagelsen. Kun resultater, der er fremkommet med kvalificerede væv, kan accepteres til at give en pålidelig vurdering af lokalirriterende virkninger. Nedenfor er acceptintervallerne for de validerede referencemetoder angivet som eksempler.
Tabel 1
Eksempler på batchgodkendelseskriterier til kvalitetskontrol
|
|
Nedre acceptgrænse |
Middelværdi i acceptinterval |
Øvre acceptgrænse |
|
Valideret referencemetode 1 (behandling i 18 h med SDS) |
IC50 = 1,0 mg/ml |
IC50 = 2,32 mg/ml |
IC50 = 3,0 mg/ml |
|
Valideret referencemetode 2 (1 % Triton X-100) |
ET50 = 4,8 hr |
ET50 = 6,7 hr |
ET50 = 8,7 hr |
II) Referencestoffer
Referencestoffer benyttes til at fastslå, om en foreslået ny in vitro-testmetode med rekonstrueret human epidermis, som er bevist at have tilstrækkelig strukturel og funktionel lighed med de validerede referencemetoder, eller som består i en mindre modfikation af en valideret referencemetode, har samme ydeevne som den validerede referencemetode 1 (1). Blandt de 20 referencestoffer på listen i tabel 2 er der stoffer i forskellige interessante kemiske stofgrupper og stoffer i kategori 2 ifølge FN's GHS. På listen er der ti stoffer i kategori 2 ifølge FN's GHS, tre stoffer i den valgfrie kategori 3 ifølge FN's GHS og syv ikke-klassificerede stoffer. I denne testmetode betragtes stoffer i den valgfrie kategori 3 som ikke-klassificerede. Disse referencestoffer er det mindste antal stoffer, der skal indgå i vurderingen af nøjagtighed og pålidelighed af en foreslået testmetode for hudirritation med rekonstrueret human epidermis. Hvis et stof på listen ikke kan fås, kan der benyttes andre stoffer, som der foreligger tilstrækkelige in vivo-referencedata om. Hvis det ønskes for at udbygge vurderingen af den foreslåede testmetodes nøjagtighed, kan der tilføjes flere stoffer, som repræsenterer andre kemiske stofgrupper, og som der foreligger tilstrækkelige in vivo-referencedata om, til minimumslisten over referencestoffer.
Tabel 2
Referencestoffer til bestemmelse af nøjagtigheds- og pålidelighedsværdier for hudirritationsmodeller med rekonstrueret human epidermis
|
Stof (1) |
CAS-nummer |
Einecs-nummer |
Fysisk form |
In vivo-vurdering |
GHS in vitro-kategori |
GHS in vivo-kategori |
|
1-brom-4-chlorbutan |
6940-78-9 |
230-089-3 |
L |
0 |
Kat. 2 |
Ingen kat. |
|
diethylphthalat |
84-66-2 |
201-550-6 |
L |
0 |
Ingen kat. |
Ingen kat. |
|
naphthaleneddikesyre |
86-87-3 |
201-705-8 |
S |
0 |
Ingen kat. |
Ingen kat. |
|
allylphenoxyacetat |
7493-74-5 |
231-335-2 |
L |
0,3 |
Ingen kat. |
Ingen kat. |
|
isopropanol |
67-63-0 |
200-661-7 |
L |
0,3 |
Ingen kat. |
Ingen kat. |
|
4-(methylthio)-benzaldehyd |
3446-89-7 |
222-365-7 |
L |
1 |
Kat. 2 |
Ingen kat. |
|
methylstearat |
112-61-8 |
203-990-4 |
S |
1 |
Ingen kat. |
Ingen kat. |
|
heptylbutyrat |
5870-93-9 |
227-526-5 |
L |
1,7 |
Ingen kat. |
Valgfri kat. 3 |
|
hexylsalicylat |
6259-76-3 |
228-408-6 |
L |
2 |
Ingen kat. |
Valgfri kat. 3 |
|
tri-isobutylphosphat |
126-71-6 |
204-798-3 |
L |
2 |
Kat. 2 |
Valgfri kat. 3 |
|
1-decanol |
112-30-1 |
203-956-9 |
L |
2,3 |
Kat. 2 |
Kat. 2 |
|
cyclamenaldehyd |
103-95-7 |
203-161-7 |
L |
2,3 |
Kat. 2 |
Kat. 2 |
|
1-bromhexan |
111-25-1 |
203-850-2 |
L |
2,7 |
Kat. 2 |
Kat. 2 |
|
2-chlormethyl-3,5-dimethyl-4-methoxypyridinhydrochlorid |
86604-75-3 |
434-680-9 |
S |
2,7 |
Kat. 2 |
Kat. 2 |
|
a-terpineol |
98-55-5 |
202-680-6 |
L |
2,7 |
Kat. 2 |
Kat. 2 |
|
di-n-propyldisulfid |
629-19-6 |
211-079-8 |
L |
3 |
Ingen kat. |
Kat. 2 |
|
butylmethacrylat |
97-88-1 |
202-615-1 |
L |
3 |
Kat. 2 |
Kat. 2 |
|
benzenthiol, 5-(1,1-dimethylethyl)-2-methyl |
7340-90-1 |
438-520-9 |
L |
3,3 |
Kat. 2 |
Kat. 2 |
|
1-methyl-3-phenyl-1-piperazin |
5271-27-2 |
431-180-2 |
S |
3,3 |
Kat. 2 |
Kat. 2 |
|
heptanal |
111-71-7 |
203-898-4 |
L |
4 |
Kat. 2 |
Kat. 2 |
Stofferne i tabel 2 er et repræsentativt udsnit af de 58 stoffer, der indgik i den internationale ECVAM-valideringsundersøgelse af hudirritation (1). De er udvalgt ud fra følgende kriterier:
|
— |
de fås i handelen |
|
— |
de repræsenterer hele Draize-pointskalaen (fra ikke-irriterende til stærkt irriterende) |
|
— |
de har en veldefineret kemisk struktur |
|
— |
de er repræsentative for den validerede metodes reproducerbarhed og prædiktionsevne som fastslået ved ECVAM-valideringsundersøgelsen |
|
— |
de er repræsentative for de typer kemisk funktion, der indgår i valideringsprocessen |
|
— |
de har ikke nogen ekstremt toksisk profil (f.eks. kræftfremkaldende eller reproduktionstoksiske) og indebærer heller ikke urimeligt høje bortskaffelsesomkostninger. |
III) Definerede nøjagtigheds- og pålidelighedsværdier
De foreslåede metoder skal være sammenlignelige med den validerede referencemetode 1 (tabel 3) med hensyn til ydeevne (følsomhed, specificitet, falsk negativ-rate, falsk postiv-rate og nøjagtighed), dvs. at følsomheden skal være 80 % eller derover, specificiteten skal være 70 % eller derover, og nøjagtigheden skal være 75 % eller derover. Ydeevnen skal beregnes for alle de klassificeringer, som de forskellige deltagende laboratorier når frem til med de 20 stoffer. Hvert laboratorium skal fastsætte klassificeringen af de enkelte stoffer ud fra gennemsnittet af levedygtigheden over de forskellige udførte analyseserier (mindst tre gyldige serier).
Tabel 3
Ydeevne for den validerede referencemetode 1 (2)
|
Testmetode |
Antal stoffer |
Følsomhed |
Specificitet |
Falsk negativ-rate |
Falsk positiv-rate |
Nøjagtighed |
|
Valideret referencemetode 1 (3) |
58 |
87,2 % (4) |
71,1 % (5) |
12,8 % |
29,9 % |
74,7 % |
|
Valideret referencemetode 1 (3) |
20 |
90 % |
73,3 % |
10 % |
26,7 % |
81,7 % |
Den foreslåede testmetodes pålidelighed skal være sammenlignelig med de validerede referencemetoders.
Reproducerbarhed inden for laboratoriet
En vurdering af variationen inden for laboratoriet skal vise, at klassificeringerne (kategori 2/ingen kategori) ved forskellige uafhængige analyseserier med de 20 referencestoffer i ét enkelt laboratorium er overensstemmende i 90 % af tilfældene eller derover.
Reproducerbarhed mellem laboratorier
En vurdering af reproducerbarheden mellem laboratorier er ikke afgørende, hvis de foreslåede testmetoder kun skal benyttes i ét laboratorium. For metoder, der ønskes overført mellem laboratorier, skal klassificeringerne (kategori 2/ingen kategori) ved forskellige uafhængige analyseserier med de 20 referencestoffer, der foretages i helst mindst tre laboratorier, være overensstemmende i 80 % af tilfældene eller derover.
REFERENCER
|
1. |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007) The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559-601. |
|
2. |
OECD (2005) Guidance Document No. 34 on the validation and international acceptance of new or updated test methods for hazard assessment. OECD, Paris. |
|
3. |
ECVAM (2007) Performance Standards for applying human skin models to in vitro skin irritation. Se under »Download Study Documents« på følgende websted: http://ecvam.jrc.ec.europa.eu. Konsulteret den 27.10.2008. |
|
4. |
Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55-63. |
|
5. |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603-619. |
(1) Disse 20 referencestoffer er et repræsentativt udsnit af de 58 stoffer, som oprindelig blev brugt til at validere referencemetode 1 (EpiSkin™). Der foreligger en fuldstændig liste over teststofferne og kriterierne for udvælgelsen (5).
(2) Tabel 3 viser ydeevnen for den validerede referencemetode 1, for så vidt angår dens evne til korrekt at identificere lokalirriterende stoffer (kategori 2 ifølge FN's GHS) og ikke-klassificerede stoffer (såvel ingen kategori som valgfri kategori 3) for henholdsvis 58 og 20 referencestoffer (tabel 2).
(3) EpiSkin™.
(4) Baseret på 13 lokalirriterende stoffer i GHS-kategori 2.
(5) Baseret på 45 lokalirriterende stoffer i GHS-kategori 3 eller ikke kategoriseret i GHS.
BILAG IV
|
C.3. |
FERSKVANDSALGER OG CYANOBAKTERIER, VÆKSTHÆMNINGSTEST |
1. METODE
Metoden er ækvivalent med OECD TG 201 (2006) (1).
1.1. INDLEDNING
Testmetoderne bliver med mellemrum gennemgået og ajourført på baggrund af den videnskabelige udvikling. Der er nødvendigt at revidere testmetode C.3, således at den omfatter flere arter og opfylder kravene til risikovurdering og klassificering af kemiske stoffer. Revisionen er foretaget på grundlag af de omfattende praktiske erfaringer med testen, den videnskabelige udvikling inden for algetoksicitetsundersøgelser samt de kontrollerende myndigheders udstrakte anvendelse af testen, siden denne oprindeligt blev indført.
1.2. DEFINITIONER
I denne testmetode anvendes følgende definitioner og forkortelser:
Biomasse, den tørre vægt af levende materiale, som er til stede i en population, i et givet volumen, f.eks. mg alger/liter testopløsning. »Biomasse« defineres sædvanligvis som masse, men skal i denne test forstås som masse pr. volumenenhed. I denne test anvendes typisk målinger af surrogater for biomasse som f.eks. celletal, fluorescens mv., hvorfor betegnelsen »biomasse« ligeledes dækker disse surrogatmåleværdier.
Variationskoefficient, et dimensionsløst mål for variabiliteten af en parameter, defineret som forholdet mellem standardafvigelsen og middelværdien. Den kan også angives i procent. Middelværdien af variationskoefficienten for den gennemsnitlige specifikke væksthastighed i replikate kontrolkulturer beregnes som følger:
|
1. |
Variationskoefficienten beregnes som % af den gennemsnitlige specifikke væksthastighed ved hjælp af de trinvist beregnede daglige vækstrater for den pågældende replikat. |
|
2. |
Der beregnes et gennemsnit af alle værdier beregnet i punkt 1. Deraf fås den gennemsnitlige variationskoefficient for den beregnede trinvise daglige specifikke vækstrate i replikate kontrolkulturer. |
ECx, den koncentration af teststof opløst i testsubstratet, der medfører en væksthæmning på x % (f.eks. 50 %) af testorganismen inden for en angiven eksponeringsperiode (som angives udtrykkeligt, hvis den er forskellig fra den fulde eller normale testperiode). For entydigt at skelne mellem EC-værdier afledt af vækstrate (»rate«) eller af udbytte (»yield«) benyttes de respektive symboler »ErC« og »EyC«.
Næringssubstrat, det komplette, syntetiske næringssubstrat, hvori algerne vokser, når de eksponeres for teststoffet. Teststoffet vil normalt være opløst i testsubstratet.
Væksthastighed (gennemsnitlig specifik væksthastighed), den logaritmiske stigning i biomasse i eksponeringsperioden.
Laveste koncentration med observeret effekt (Lowest Observed Effect Concentration, LOEC), den laveste testede koncentration, ved hvilken stoffet har vist statistisk signifikant (p < 0,05) vækstnedsættende virkning i forhold til kontrolprøverne inden for en given eksponeringsperiode. Alle testkoncentrationer over LOEC skal dog have en skadelig virkning mindst svarende til den, der iagttages ved LOEC. Er disse to betingelser ikke opfyldt, skal der fyldestgørende redegøres for, hvordan den pågældende LOEC (og dermed NOEC) er valgt.
Nuleffekt-koncentration (No Observed Effect Concentration (NOEC)), testkoncentrationen umiddelbart under LOEC.
Responsvariabel, en variabel, som anvendes til vurdering af den toksiske virkning og er afledt af vilkårlige målte parametre, der beskriver biomassen ved andre beregningsmetoder. I nærværende metode er vækstrater og udbytte responsvariabler, der er afledt ved måling af biomasse direkte eller af et af de nævnte surrogater.
Specifik væksthastighed, en responsvariabel, der er defineret som forholdet mellem ændringen i den naturlige logaritme til en observationsparameter (i nærværende testmetode, biomasse) og det tilsvarende tidsrum.
Udbytte, værdien af en målevariabel ved eksponeringsperiodens slutning minus værdien af variablen ved eksponeringsperiodens start, dvs. stigningen i biomasse under testen.
1.3. TESTMETODENS ANVENDELIGHED
Metoden er bedst anvendelig på vandopløselige stoffer, som under forsøgsbetingelserne forventes at forblive i vandet. Til testning af stoffer, der er flygtige, stærkt adsorberende, farvede eller tungtopløselige i vand, eller som kan ændre tilgængeligheden af næringsstoffer eller mineraler i testsubstratet, kan visse ændringer af den beskrevne fremgangsmåde være nødvendige (f.eks. lukket system eller konditionering af testbeholderne). Anvisninger for nogle sådanne ændringer er givet i (2) (3) (4).
1.4. TESTENS PRINCIP
Testens formål er at bestemme teststoffets påvirkning af væksten af ferskvandsmikroalger og/eller cyanobakterier. Testorganismer i eksponentiel vækst eksponeres for teststoffet i batchkulturer i et tidsrum af normalt 72 timer. Til trods for testens forholdsvis korte varighed giver den mulighed for at vurdere virkningerne på flere generationer.
Systemets respons er væksthæmningen i en række algekulturer (testenheder), der eksponeres for forskellige koncentrationer af et teststof. Responsen udtrykkes som funktion af eksponeringskoncentrationen og sammenholdes med gennemsnitsvæksten i replikate, ikke eksponerede kulturer. For at systemets respons på toksiske virkninger kan komme fuldt til udtryk (optimal følsomhed), lader man kulturerne vokse uhæmmet eksponentielt med tilstrækkelige næringsstoffer og kontinuerlig belysning tilstrækkelig længe til, at hæmningen af den specifikke væksthastighed kan måles.
Vækst og væksthæmning bestemmes kvantitativt ved måling af algebiomassen som funktion af tiden. Algebiomasse defineres som tør vægt pr. volumenenhed, f.eks. mg alger/liter testopløsning. Tør vægt er imidlertid vanskelig at måle, hvorfor der anvendes surrogatparametre. Af sådanne surrogater er celletal det mest anvendte. Andre surrogatparametre er cellevolumen, fluorescens, optisk tæthed mv. Omregningsfaktoren mellem den målte surrogatparameter og biomassen skal være kendt.
Endepunktet for testen er væksthæmning, udtrykt som logaritmisk stigning i biomasse (gennemsnitlig specifik væksthastighed) i eksponeringsperioden. Ud fra de gennemsnitlige specifikke væksthastigheder, der er registreret i en række testopløsninger, bestemmes den koncentration, der frembringer en bestemt hæmning x % (f.eks. 50 %) af væksthastigheden, og denne udtrykkes som ErCx (f.eks. ErC50).
Når denne metode anvendes i forbindelse med EU-lovgivningen, skal resultaterne beregnes på grundlag af den gennemsnitlige specifikke væksthastighed af de grunde, der er anført i punkt 2.2. I metoden anvendes udbytte som en supplerende responsvariabel, der kan være nødvendig for at efterkomme myndighedsbestemmelser i visse stater. Udbyttet defineres som biomassen ved eksponeringsperiodens slutning minus biomassen ved eksponeringsperiodens start. Ud fra det udbytte, der er registreret i en række testopløsninger, bestemmes den koncentration, der frembringer en bestemt hæmning x % (f.eks. 50 %) i udbyttet, og denne angives som EyCx (f.eks. EyC50).
Derudover kan den laveste koncentration med observeret effekt (LOEC) og nuleffekt-koncentrationen (NOEC) bestemmes statistisk.
1.5. OPLYSNINGER OM TESTSTOFFET
Med henblik på fastsættelse af testbetingelser vil det være nyttigt at kende teststoffets strukturformel, renhed, lysstabilitet, stabilitet under testbetingelserne, lysabsorptionsegenskaber, pKa og resultaterne af omdannelsesundersøgelser, herunder bionedbrydeligheden i vand.
Teststoffets vandopløselighed, octanol/vand-fordelingskoefficient (Pow) og damptryk skal være kendt, og til kvantitativ bestemmelse af stoffet i testopløsningerne skal der foreligge en pålidelig analysemetode, hvis genfindingsevne og detektionsgrænse rapporteres.
1.6. REFERENCESTOF
Til kontrol af testmetoden kan testes et eller flere referencestoffer, f.eks. 3,5-dichlorphenol, som er anvendt i den internationale ringtest (4). Kaliumdichromat kan ligeledes anvendes som referencestof for grønalger. Det er ønskeligt, at der testes et referencestof mindst to gange årligt.
1.7. TESTENS VALIDITET
Til afgørelse af, om en test er valid, anvendes følgende præstationskriterier:
|
— |
Biomassen i kontrolkulturerne skal være steget eksponentielt med mindst en faktor 16 i løbet af testperioden på 72 timer. Dette svarer til en specifik væksthastighed på 0,92 dag–1. For de oftest anvendte arter er væksthastigheden sædvanligvis betydeligt højere (se tillæg 1). Dette kriterium er ikke nødvendigvis opfyldt for arter, der er mere langsomtvoksende end de i tillæg 1 anførte. I så fald må testperioden forlænges, så der opnås en vækst til mindst det 16-dobbelte i kontrolkulturerne, og så væksten er eksponentiel i hele testperioden. For at opretholde uhæmmet eksponentiel vækst under testen kan testperioden afkortes til ikke under 48 h, når blot den minimale multiplikationsfaktor på 16 nås. |
|
— |
Den gennemsnitlige variationskoefficient af den trinvise specifikke væksthastighed (ved 72-timers test, dag 0-1, 1-2 og 2-3) i kontrolkulturerne (se punkt 1.2 under »variationskoefficient«) må ikke være over 35 %. Vedrørende beregning af trinvis specifik væksthastighed: se andet afsnit i punkt 2.2.1. Kriteriet gælder for gennemsnitsværdien af variationskoefficienterne for replikate kontrolkulturer. |
|
— |
Variationskoefficienten for de gennemsnitlige specifikke væksthastigheder i hele testperioden i replikate kontrolkulturer må ikke være over 7 % i test med Pseudokirchneriella subcapitata og Desmodesmus subspicatus. For andre, mindre hyppigt benyttede testede arter må værdien ikke være over 10 %. |
1.8. BESKRIVELSE AF METODEN
1.8.1. Apparatur
Testbeholdere og andet apparatur, der kommer i berøring med testopløsningerne, skal være udført helt i glas eller andet kemisk inaktivt materiale. Det pågældende udstyr skal være omhyggelig vasket for at sikre, at ingen organiske eller uorganiske kontaminanter kan forstyrre algevæksten eller testopløsningernes sammensætning.
Som testbeholdere anvendes sædvanligvis glaskolber af en størrelse, der giver tilstrækkeligt kulturvolumen til målingerne under testen og tilstrækkelig masseoverførsel af CO2 fra atmosfæren (se andet afsnit i punkt 1.8.9). Bemærk, at der skal være tilstrækkeligt væskevolumen til de analytiske bestemmelser (se femte afsnit i punkt 1.8.11).
Derudover er der brug for følgende udstyr eller en del deraf:
|
— |
dyrkningsapparatur: Det anbefales at benytte et kabinet eller kammer med mulighed for fastholdelse af den valgte inkubationstemperatur inden for ±2 °C. |
|
— |
instrumenter til lysmåling: Man må være opmærksom på, at den anvendte metode til lysintensitetsmåling, specielt den anvendte type detektor (kollektor), vil påvirke måleværdien. Til målingerne skal fortrinsvis benyttes en sfærisk detektor (4 π-detektor) (som reagerer på direkte og reflekteret lys fra alle vinkler over og under måleplanet) eller en 2 π-detektor (som reagerer på lys fra alle vinkler over måleplanet). |
|
— |
apparatur til bestemmelse af algebiomasse: Celletal, der er den oftest anvendte surrogatparameter for algebiomasse, kan bestemmes med en elektronisk partikeltæller, et mikroskop med tællekammer eller et flowcytometer. Andre biomassesurrogater kan måles med flowcytometer, fluorimeter, spektrofotometer eller kolorimeter. Det er hensigtsmæssigt at beregne en omregningsfaktor fra celletal til tør vægt. Ved spektrofotometrisk bestemmelse af lave biomassekoncentrationer kan det være nødvendigt at bruge kuvetter med en lysvej på mindst 4 cm for at få brugbare målinger. |
1.8.2. Testorganismer
Der kan anvendes forskellige arter af ikke-fastsiddende mikroalger og cyanobakterier. De i tillæg 1 opførte stammer har vist sig velegnede ved brug af fremgangsmåden i denne testmetode.
Benyttes andre arter, skal stamme og/eller oprindelse angives. Det skal være godtgjort, at der kan opretholdes eksponentiel vækst af de valgte testalger i hele testperioden under de givne betingelser.
1.8.3. Næringssubstrat
To alternative næringssubstrater, OECD-substratet og AAP-substratet, anbefales. Substraternes sammensætning er angivet i tillæg 2. Bemærk, at de to mediers initiale pH-værdi og bufferkapacitet (som regulerer pH-stigningen) er forskellige. Testningen kan derfor give forskelligt resultat afhængigt af substratet, specielt ved test af ioniserende stoffer.
Til visse formål kan det være nødvendigt at modificere substratet, f.eks. ved testning af metaller og chelaterende midler, eller når testning finder sted ved afvigende pH-værdier. Modificerede substrater skal beskrives i detaljer og deres anvendelse begrundes (3) (4).
1.8.4. Initial biomassekoncentration
Den initiale biomasse i testkulturerne skal være ens i alle testkulturer og tilstrækkelig lav til at muliggøre eksponentiel vækst i hele inkubationsperioden uden risiko for, at næringsstoffer opbruges. Den initiale biomasse må ikke udgøre mere end 0,5 mg/l som tør vægt. Følgende initiale cellekoncentrationer anbefales:
|
Pseudokirchneriella subcapitata |
5 × 103-104 |
celler/ml |
|
Desmodesmus subspicatus |
2-5 × 103 |
celler/ml |
|
Navicula pelliculosa |
104 |
celler/ml |
|
Anabaena flos-aquae |
104 |
celler/ml |
|
Synechococcus leopoliensis |
5 × 104-105 |
celler/ml |
1.8.5. Teststofkoncentrationer
Der kan udføres indledende test til at fastlægge det koncentrationsområde, hvor der kan forventes effekt. Til den endelige test skal vælges mindst fem koncentrationer, der er ordnet i en geometrisk serie med en faktor på højst 3,2. For teststoffer med flad koncentrations-responskurve kan en højere faktor være berettiget. Koncentrationsserien skal fortrinsvis dække det område, der medfører 5-75 % hæmning af algevæksthastigheden.
1.8.6. Replikater og kontroller
Testens design skal indeholde tre replikater ved hver testkoncentration. Hvis der ikke kræves bestemmelse af NOEC, kan testens design ændres, så der anvendes flere forskellige koncentrationer og færre replikater for hver koncentration. Der skal være mindst tre replikater af kontrolprøverne, og ideelt dobbelt så mange som antallet af replikater for hver testkoncentration.
Der kan fremstilles et separat sæt testopløsninger til analytisk bestemmelse af teststofkoncentrationerne (se fjerde og sjette afsnit i punkt 1.8.11).
Anvendes et opløsningsmiddel til at bringe teststoffet i opløsning, skal testen omfatte ekstra kontroller indeholdende opløsningsmidlet i samme koncentration som i testkulturerne.
1.8.7. Fremstilling af podekultur
For at lade de testede alger tilpasse sig til testbetingelserne og sikre, at algerne er i den eksponentielle vækstfase, når de benyttes til podning af testopløsningerne, fremstilles en podekultur i testsubstratet 2-4 dage, før testen begynder. Mængden af algebiomasse skal tilpasses, så der bliver eksponentiel vækst i podekulturen, indtil testen begynder. Podekulturen skal inkuberes under samme betingelser som testkulturerne. Stigningen i biomasse i podekulturen skal måles for at sikre, at væksten er inden for normalområdet for den pågældende teststamme under dyrkningsbetingelserne. Et eksempel på fremgangsmåden ved dyrkning af alger er beskrevet i tillæg 3. For at undgå synkrone celledelinger under testen kan et ekstra formeringstrin af podekulturen være nødvendigt.
1.8.8. Fremstilling af testopløsninger
Alle testopløsninger skal indeholde samme koncentration af næringssubstrat og initial biomasse af testalger. Testopløsningerne af de valgte koncentrationer fremstilles sædvanligvis ved, at en stamopløsning af teststoffet opblandes med vækstmedium og podekultur. Stamopløsningerne fremstilles normalt ved opløsning af teststoffet i testsubstrat.
Opløsningsmidler, f.eks. acetone, t-butylalkohol og dimethylformamid, kan bruges som bæremedier, når tungtopløselige stoffer skal tilsættes testsubstratet (2) (3). Opløsningsmidlets koncentration må ikke være over 100 µl/l, og det skal tilsættes i samme koncentration til alle kulturer (også kontrollerne) i testserien.
1.8.9. Inkubation
Testbeholderne lukkes med luftgennemtrængelige propper. Testbeholderne omrystes og anbringes i dyrkningsapparatet. Under testen skal algerne holdes opslæmmet og CO2-overførslen lettes. Kontinuerlig omrystning eller omrøring er derfor nødvendig. Kulturerne skal være reguleret til en temperatur i området 21 til 24 °C, og den fastsatte temperatur skal holdes inden for ±2 °C. Til andre arter end de i tillæg 1 anførte, f.eks. tropiske arter, kan højere temperaturer være hensigtsmæssigt, forudsat at validitetskriterierne kan opfyldes. Det anbefales at placere kolberne i tilfældig orden og dagligt bytte rundt på dem i inkubatoren.
pH af kontrolsubstratet må højst stige med 1,5 enheder under testen. For metaller og forbindelser, der kun er delvis ioniseret ved pH-værdier omkring testens pH, kan det være nødvendigt at begrænse pH-afvigelsen for at få reproducerbare og veldefinerede resultater.. Begrænsning af afvigelsen til < 0,5 pH-enhed er teknisk mulig og kan opnås ved at sikre tilstrækkelig CO2-masseoverførsel fra den omgivende luft til testopløsningen, f.eks. ved at øge omrystningshastigheden. En anden mulighed er at nedsætte CO2-behovet ved enten at reducere den initiale biomasse eller testens varighed.
Den overflade, som kulturerne inkuberes på, skal have kontinuerlig, ensartet fluorescerende belysning, f.eks. af typen »cool-white« eller »daylight«. Forskellige stammer af alger og cyanobakterier har forskelligt lysbehov. Der skal vælges en passende lysintensitet i forhold til den anvendte organisme. Til de anbefalede arter af grønalger skal lysintensiteten i testopløsningernes niveau vælges i området 60-120 µE·m–2·s–1, målt i det fotosyntetisk effektive bølgelængdeområde 400-700 nm med en egnet detektor. Visse arter, navnlig Anabaena flos-aquae, vokser godt ved lavere lysintensitet og kan tage skade af højere intensitet. Til sådanne arter bør vælges en gennemsnitlig lysintensitet i området 40-60 µE·m–2·s–1. (For lysmåleinstrumenter kalibreret i lux vil den anbefalede lysintensitet på 60-120 µE·m–2·s–1 med cool-white-lys tilnærmelsesvis svare til området 4 440-8 880 lux.) Lysintensiteten må ikke afvige mere end ±15 % fra den gennemsnitlige lysintensitet i hele inkubationsområdet.
1.8.10. Testens varighed
Testens varighed er normalt 72 timer. Kortere eller længere varighed kan dog anvendes, forudsat at alle validitetskriterierne i punkt 1.7 kan opfyldes.
1.8.11. Målinger og analytiske bestemmelser
Algebiomassen i hver kolbe bestemmes mindst én gang dagligt i hele testperioden. Hvis målingerne foretages på små volumener, der er udtaget af testopløsningen med pipette, bør disse ikke erstattes.
Måling af biomasse foretages enten ved manuel celletælling i mikroskop eller med en elektronisk partikeltæller (i celletal og/eller biovolumen). Andre teknikker kan anvendes, f.eks. flowcytometri, in vitro eller in vivo chlorophyl-fluorescens (6) (7) eller optisk densitet, forudsat at der kan påvises tilfredsstillende korrelation med biomasse i hele det biomasseområde, der forekommer under testen.
Opløsningernes pH skal måles ved testens start og slutning.
Hvis der foreligger en analysemetode til bestemmelse af teststoffet i det anvendte koncentrationsområde, skal testopløsningerne analyseres til efterprøvning af intialkoncentrationerne og af, at eksponeringskoncentrationerne overholdes under testen.
Hvis eksponeringskoncentrationerne under testen forventes at afvige mindre end 20 % fra de nominelle værdier, kan det være tilstrækkeligt at bestemme en høj og en lav teststofkoncentration og en koncentration omkring den forventede EC50 ved testens begyndelse og slutning. Når koncentrationen ikke kan forventes at holde sig inden for 80-120 % af den nominelle værdi, anbefales analytisk bestemmelse af alle testkoncentrationer ved testens begyndelse og slutning. Til flygtige, ustabile eller stærkt adsorberende stoffer anbefales det at udtage supplerende prøver til analyse med 24 timers mellemrum i hele eksponeringsperioden for bedre at bestemme tabet af teststof. For sådanne stoffer vil ekstra replikater være nødvendige. I alle tilfælde behøver teststofkoncentrationen kun bestemmes på én replikat testbeholder ved hver testkoncentration (eller på indholdet af de poolede beholdere for hver replikat).
Testmedier, der er fremstillet specielt til analyse af eksponeringskoncentrationerne under testen, skal behandles på samme måde som dem, der anvendes til testning, dvs. podes med alger og inkuberes ved identiske betingelser. Hvis koncentrationen af opløst teststof kræves analyseret, kan det være nødvendigt at separere algerne fra substratet. Separationen skal fortrinsvis ske ved centrifugering med lav g-kraft, tilstrækkelig til at få algerne til at bundfælde.
Hvis det kan konstateres, at prøvestofkoncentrationen har været holdt tilfredsstillende inden for ±20 % af den nominelle eller målte begyndelseskoncentration under hele testen, kan analysen af resultaterne baseres på nominelle eller målte startværdier. Er afvigelsen fra den nominelle eller målte initialkoncentration større end ±20 %, skal analysen af resultaterne baseres på den geometriske middelkoncentration under eksponeringen eller på modeller, der beskriver faldet i teststofkoncentrationen (3) (8).
Algevæksthæmning er et mere dynamisk testsystem end de fleste andre korttidstest for akvatisk toksicitet. De faktiske eksponeringskoncentrationer kan derfor være vanskelige at fastlægge, navnlig for adsorberende stoffer, der testes ved lav koncentration. I sådanne tilfælde er stoffets forsvinden fra opløsningen ved adsorption til den voksende algebiomasse ikke ensbetydende med, at det forsvinder fra testsystemet. Ved analysen af testresultatet skal det kontrolleres, om et fald i teststofkoncentrationen under testen ledsages af et fald i væksthæmningen. Hvis det er tilfældet, kan det overvejes at benytte en egnet model, der beskriver faldet i teststofkoncentrationen (8). Hvis ikke, kan det være hensigtsmæssigt at basere analysen af resultaterne på de initiale (nominelle eller målte) koncentrationer.
1.8.12. Andre observationer
Der skal foretages mikroskopisk undersøgelse for at verificere, at podekulturen fremtræder normal og sund, og konstatere, om algernes udseende er unormalt ved testens slutning (som en mulig følge af eksponeringen for teststoffet).
1.8.13. Grænsetest
Under visse omstændigheder, f.eks. når en indledende test viser, at teststoffet er uden toksiske virkninger i koncentrationer på op til 100 mg·l–1, dog ikke over stoffets opløselighed i testsubstratet, kan der udføres en grænsetest, hvor reaktionen i en kontrolgruppe sammenholdes med reaktionen i én behandlingsgruppe (100 mg·l–1 eller en koncentration lig med opløseligheden). Det anbefales kraftigt, at dette understøttes ved analyse af eksponeringskoncentrationen. Alle de beskrevne testbetingelser og validitetskriterier gælder for grænsetesten, bortset fra, at antallet af behandlingsreplikater skal være mindst seks. Responsvariablerne i kontrol- og behandlingsgruppen kan analyseres med en statistisk test til sammenligning af middelværdier, f.eks. Students t-test. Er variansen i de to grupper uens, skal der anvendes en t-test justeret for forskel i varians.
1.8.14. Modifikation i tilfælde af stærkt farvede stoffer
Belysningen (lysintensiteten) skal ligge i den højeste ende af det interval, der forskrives i testmetoden, dvs. 120 µE·m–2·s–1 eller højere.
Lysvejen forkortes ved at reducere testopløsningernes volumen (til intervallet 5-25 ml).
Der sørges for tilstrækkelig omrøring (f.eks. ved moderat rystning), for at algerne kan blive udsat for stærk belysning ved kulturoverfladen.
2. DATA
2.1. OPTEGNING AF VÆKSTKURVER
Biomassen i testbeholderne kan angives i enheder svarende til den til målingen anvendte surrogatparameter (f.eks. celletal eller fluorescens).
Der opstilles en tabel over den beregnede biomassekoncentration i testkulturer og kontroller sammen med teststofkoncentrationer og måletidspunkter, målt med en mindste opløsning på hele timer, og derudfra optegnes vækstkurverne. Både logaritmisk og lineær afbildning kan være nyttig på dette første trin, men logaritmisk afbildning er obligatorisk og giver sædvanligvis en bedre fremstilling af variationerne i vækstmønsteret i løbet af testperioden. Bemærk, at eksponentiel vækst frembringer en ret linje, når den afbildes på en logaritmisk skala, og at linjens hældning angiver den specifikke væksthastighed.
Ud fra kurverne undersøges det, om kontrolkulturerne vokser eksponentielt med den forventede hastighed igennem hele testen. Alle datapunkter og kurvernes udseende kontrolleres kritisk, og rådata og de anvendte metoder gås efter for eventuelle fejl. Navnlig kontrolleres alle datapunkter, der synes at afvige med en systematisk fejl. Det er indlysende, at hvis metodefejl kan konstateres og/eller må anses for stærkt sandsynlige, må det pågældende punkt markeres som afvigende og udelades af den efterfølgende statistiske analyse. (En algekoncentration på nul i den ene af tre replikate testbeholdere kan tyde på, at beholderen enten ikke er blevet podet korrekt eller ikke har været tilstrækkelig rengjort.) Begrundelsen for at afvise et datapunkt som afvigende skal klart angives i testrapporten. Som begrundelse godtages kun (sjældne) metodefejl, ikke blot ringe præcision. Til identifikation af afvigende værdier har statistiske metoder kun begrænset værdi ved problemer af denne art og kan ikke erstatte en faglig vurdering. Afvigende værdier (mærket som sådanne) skal helst bibeholdes blandt de datapunkter, der vises ved efterfølgende fremstilling af data i grafer eller tabeller.
2.2. RESPONSVARIABLER
Testens formål er at bestemme teststoffets virkninger på væksten af alger. I nærværende testmetode beskrives to responsvariabler, da medlemsstaterne har forskellige præferencer og myndighedskrav. For at testresultaterne skal kunne godtages i alle medlemsstater, skal virkningerne vurderes ved hjælp af begge de nedenfor beskrevne responsvariabler a) og b).
|
a) |
gennemsnitlig specifik væksthastighed: Denne responsvariabel beregnes på grundlag af den logaritmiske stigning i biomasse i løbet af testperioden, angivet pr. døgn. |
|
b) |
udbytte: Denne responsvariabel er biomassen ved testens slutning minus biomassen ved testens start. |
Når denne metode anvendes i forbindelse med EU-lovgivningen, skal resultaterne beregnes på grundlag af den gennemsnitlige specifikke væksthastighed, af de grunde, der er anført nedenfor. Det skal bemærkes, at toksicitetsværdier beregnet ved hjælp af disse to responsvariabler ikke er sammenlignelige, og denne forskel må haves for øje ved anvendelse af testens resultater. ECx-værdier baseret på gennemsnitlig specifik væksthastighed (ErCx) vil sædvanligvis være højere end resultater baseret på udbytte (EyCx), når testbetingelserne i denne metode er overholdt, hvilket følger af det matematiske grundlag for de to beregningsmåder. Dette må ikke opfattes som en forskel i følsomhed mellem de to responsvariabler, men kun, at der er tale om to matematisk forskellige størrelser. Begrebet gennemsnitlig specifik væksthastighed bygger på det generelle eksponentielle vækstmønster for alger i ikke-begrænsede kulturer, mens toksicitet vurderes på grundlag af virkningerne på væksthastigheden og ikke afhænger af den absolutte størrelse af den specifikke væksthastighed af kontrollen, koncentrations-responskurvens hældning eller testens varighed. Resultater, der er baseret på responsvariablen udbytte, afhænger derimod af alle disse øvrige variabler. EyCx afhænger af den specifikke væksthastighed af de algearter, der anvendes i hver test, og af den maksimale specifikke væksthastighed, som kan variere mellem de forskellige arter og endda algestammer. Denne responsvariabel bør ikke anvendes til at sammenligne følsomheden over for giftstoffer hos forskellige algearter eller -stammer. Skønt det videnskabeligt foretrukne grundlag til vurdering af toksicitet er den gennemsnitlige specifikke væksthastighed, indeholder nærværende testmetode toksicitetsvurderinger baseret på udbytte for at imødekomme de aktuelle myndighedskrav i visse stater.
2.2.1. Gennemsnitlig specifik væksthastighed
Den gennemsnitlige specifikke væksthastighed for en given periode beregnes som den logaritmiske stigning i biomasse ved hjælp af ligningen for hver enkelt testbeholder med kontrol- og behandlingsprøver:
 , (dag–1)
, (dag–1)
hvor:
|
µ i-j |
: |
er den gennemsnitlige specifikke væksthastighed fra tiden i til j |
|
X i |
: |
er biomassen til tiden i |
|
X j |
: |
er biomassen til tiden j. |
For hver behandlings- og kontrolgruppe beregnes en gennemsnitlig væksthastighed samt estimater for variansen.
Den gennemsnitlige specifikke væksthastighed i hele testperioden (sædvanligvis dag 0-3) beregnes ved at bruge den nominelt podede biomasse som startværdi i stedet for en målt startværdi, da der herved sædvanligvis opnås størst præcision. Hvis udstyret til biomassemåling tillader tilstrækkelig nøjagtig bestemmelse af den lille mængde podet biomasse (f.eks. med flowcytometer), kan den målte initiale biomassekoncentration anvendes. Desuden foretages trinvis beregning af den specifikke væksthastighed for hver dag af testen (dag 0-1, 1-2 og 2-3), og det undersøges, om væksthastigheden i kontrollen forbliver konstant (se validitetskriterierne i punkt 1.7). Er den specifikke væksthastighed væsentligt lavere på dag ét end den totale gennemsnitlige specifikke væksthastighed, kan dette tyde på, at der er tale om en lag-fase. Mens en lag-fase i kontrolkulturerne kan minimeres og i praksis elimineres ved korrekt formering af forkulturen, kan en lag-fase i eksponerede kulturer tyde på, at der er tale om restitution efter indledende toksisk stress, eller at eksponeringen efter den initiale eksponering er mindsket pga. tab af teststof (herunder sorption på algebiomassen). Trinvis beregning af væksthastigheden kan derfor anvendes til at vurdere teststoffets virkninger i eksponeringsperioden. Væsentlige forskelle mellem den trinvist beregnede væksthastighed og den gennemsnitlige væksthastighed tyder på en afvigelse fra konstant eksponentiel vækst, og nøje granskning af vækstkurverne er da nødvendig.
Den procentuelle hæmning af væksthastigheden beregnes for hver behandlingsreplikat af udtrykket:

hvor:
|
%Ir |
: |
procent hæmning af den gennemsnitlige specifikke væksthastighed |
|
µC |
: |
middelværdi af den gennemsnitlige specifikke væksthastighed (µ) i kontrolgruppen |
|
µT |
: |
gennemsnitlig specifik væksthastighed for behandlingsreplikaten. |
Når der anvendes opløsningsmiddel til fremstilling af testopløsningerne, skal hæmningen i procent beregnes ved hjælp af opløsningsmiddelkontroller i stedet for kontroller uden opløsningsmiddel.
2.2.2. Udbytte
Udbyttet beregnes som biomassen ved testens slutning minus biomassen ved testens start for hver enkelt beholder med kontrol- eller behandlingsprøve. For hver testkoncentration og kontrol beregnes en gennemsnitsværdi af udbyttet samt estimater for variansen. Udbyttehæmningen i procent (%Iy) kan beregnes for hver behandlingsreplikat som følger:

hvor:
|
% Iy |
: |
udbyttehæmning i procent |
|
YC |
: |
gennemsnitligt udbytte i kontrolgruppen |
|
YT |
: |
størrelsen af udbyttet for behandlingsreplikaten. |
2.3. OPTEGNING AF KONCENTRATIONS-RESPONSKURVE
Hæmningen i procent afsættes som funktion af logaritmen til teststofkoncentrationen, og grafen undersøges nøje, idet der ses bort fra alle datapunkter, der er udpeget som afvigende i første fase. Der lægges en jævn linje gennem datapunkterne, enten på fri hånd eller med computer-interpolation, for at få et første indtryk af koncentrations-responssammenhængen, hvorefter der fortsættes med en mere dybtgående metode, helst en computerbaseret statistisk metode. Afhængigt af den påtænkte anvendelse af data, dataenes kvalitet (præcision) og mængde samt adgangen til værktøjer til dataanalyse kan det blive besluttet (undertiden med god grund) at standse dataanalysen på dette trin og blot aflæse nøgletallene EC50 og EC10 (og/eller EC20) på frihåndskurven (se også afsnittet nedenfor om stimulerende virkninger). Af gyldige grunde til ikke at benytte en statistisk metode kan nævnes:
|
— |
Data er ikke egnede til at give mere pålidelige resultater ved hjælp af computermetoder end ved et fagligt skøn — nogle computerprogrammer kan i sådanne situationer endda være ude af stand til at finde en pålidelig løsning (f.eks. hvis iterationen ikke konvergerer). |
|
— |
Stimuleret vækstrespons kan ikke behandles fyldestgørende med de forhåndenværende computerprogrammer (se nedenfor). |
2.4. STATISTISKE METODER
Målet er at formulere en kvantitativ koncentrations-responssammenhæng ved hjælp af regressionsanalyse. Der kan anvendes vægtet lineær regression efter forudgående lineariserende transformation af responsdataene — til f.eks. probit-, logit- eller Weibull-enheder (9), men det må foretrækkes at benytte ikke-lineære regressionsmetoder, da de bedre håndterer de uundgåelige uregelmæssigheder i data og afvigelser fra jævne fordelinger. I området tæt på ingen hæmning eller total hæmning kan sådanne uregelmæssigheder blive forstørret af transformationen og derved forstyrre analysen (9). Det må bemærkes, at standardanalysemetoder, hvor der bruges probit-, logit- eller Weibull-transformerede variabler, er bestemt til brug på quantale data (f.eks. død eller overlevelse) og må modificeres for at kunne håndtere vækst- eller biomassedata. Særlige metoder til bestemmelse af ECx-værdier ud fra kontinuerte data findes i henvisning (10) (11) (12). Brugen af ikke-lineær regressionsanalyse er nærmere beskrevet i tillæg 4.
For hver responsvariabel, der skal analyseres, anvendes koncentrations-responsrelationen til at beregne punktestimater for ECx-værdier. Når det er muligt, skal der bestemmes 95 % konfidensgrænser for hvert estimat. Responsdataenes tilpasningsgrad til regressionsmodellen vurderes enten grafisk eller statistisk. Regressionsanalysen skal foretages ud fra responsværdierne for de enkelte replikater, ikke gruppegennemsnit. Hvis ikke-lineær kurvetilpasning er vanskelig eller umulig på grund af for stor spredning i data, kan problemet omgås ved udførelse af regressionen på gruppegennemsnittet som en praktisk måde at mindske indflydelsen fra formodet afvigende værdier på. Når denne mulighed udnyttes, skal det i testrapporten anføres, at der her er afveget fra den normale procedure, fordi kurvetilpasning med de enkelte replikater ikke har givet et godt resultat.
Hvis de tilgængelige regressionsmodeller eller -metoder er uegnede til dataene, kan estimater og konfidensgrænser for EC50 desuden fås ved lineær interpolation med bootstrapping (13).
For at beregne et estimat for LOEC og dermed NOEC og for teststoffets virkninger på væksthastigheden må middelværdierne for behandlingerne sammenholdes ved hjælp af variansanalyse. Gennemsnittet for hver koncentration sammenholdes derefter med gennemsnittet for kontrollerne ved hjælp af en egnet multisammenlignings- eller trend test-metode. Dunnetts eller Williams’ test kan eventuelt bruges (14) (15) (16) (17) (18). Det skal vurderes, om variansanalysens forudsætning om varianshomogenitet er opfyldt. Denne vurdering kan foretages grafisk eller ved en formel test (18). Hertil kan Levenes eller Bartletts test anvendes Manglende opfyldelse af forudsætningen om varianshomogenitet kan undertiden afhjælpes ved logaritmisk transformation af dataene. Hvis variansen er ekstremt heterogen og ikke kan korrigeres ved transformation, må analyse med step-down Jonkheere trend test overvejes. Der er supplerende vejledning for bestemmelse af NOEC i (12).
Den senere tids videnskabelige udvikling har ført til en anbefaling om, at NOEC-begrebet afskaffes og erstattes med regressionsbaserede punktestimater for ECx. Der er ikke fastlagt en passende værdi af x til denne algetest. Et område på 10 til 20 % synes at være passende (afhængigt af den valgte responsvariabel), og det må foretrækkes at rapportere både EC10 og EC20.
2.5. VÆKSTSTIMULATION
Ved lave koncentrationer ses undertiden vækststimulation (negativ hæmning). Dette kan enten skyldes hormese (»toksisk stimulation«), eller at der er tilsat vækststimulerende faktorer sammen med testmaterialet til det anvendte minimalsubstrat. Bemærk, at tilsætning af uorganiske næringsstoffer ikke skulle have nogen direkte virkning, da testsubstratet skal indeholde overskud af næringsstoffer under hele testen. Sædvanligvis kan der ses bort fra lavdosisstimulation i EC50-beregninger, medmindre den er ekstrem. Er den ekstrem, eller skal der beregnes en ECx-værdi for en lav x-værdi, kan særlige procedurer dog være nødvendige. Om muligt bør det undgås at lade stimulerende responsværdier udgå af dataanalysen, og kan den forhåndenværende kurvetilpasningssoftware ikke acceptere en lav grad af stimulation, kan der anvendes lineær interpolation med bootstrapping. Hvis stimulationen er ekstrem, kan det overvejes at anvende en hormese-model (19).
2.6. IKKE-TOKSISK VÆKSTHÆMNING
Lysabsorberende teststoffer kan nedsætte væksthastigheden, fordi de ved at skygge mindsker den tilgængelige lysmængde. Sådanne fysiske virkninger må adskilles fra toksiske virkninger ved ændring af testbetingelserne og skal rapporteres særskilt. Vejledning findes i (2) (3).
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal indeholde følgende:
Teststof:
|
— |
fysisk tilstand og relevante fysisk-kemiske egenskaber, herunder vandopløselighed |
|
— |
kemiske identifikationsdata, herunder renhed. |
Testede arter:
|
— |
stamme, leverandør eller kilde og anvendte dyrkningsbetingelser. |
Testbetingelser:
|
— |
startdato og varighed for testen |
|
— |
beskrivelse af testens design: testbeholdere, kulturvolumen, biomassetæthed ved testens start |
|
— |
substratets sammensætning |
|
— |
testkoncentrationer og replikater (f.eks. antal replikater, antal testkoncentrationer og den anvendte geometriske progression) |
|
— |
beskrivelse af fremstillingen af testopløsningerne, herunder brug af opløsningsmiddel osv. |
|
— |
dyrkningsapparatur |
|
— |
lysintensitet og -kvalitet (kilde, homogenitet) |
|
— |
temperatur |
|
— |
testede koncentrationer: de nominelle testkoncentrationer og alle resultater af bestemmelser af teststofkoncentrationen i testbeholderne. Metodens genfindingsgrad og kvantificeringsgrænse i testmatrixen skal rapporteres |
|
— |
alle fravigelser af nærværende testmetode |
|
— |
metode til bestemmelse af biomasse og påvisning af korrelation mellem den målte parameter og den tørre vægt. |
Resultater:
|
— |
pH-værdi ved testens start og slutning for alle behandlinger |
|
— |
biomasse for hver kolbe ved hvert målepunkt samt metode til måling af biomasse |
|
— |
vækstkurver (biomasse som funktion af tiden) |
|
— |
beregnede responsvariabler for hver behandlingsreplikat, med gennemsnitsværdier og variationskoefficient for replikaterne |
|
— |
grafisk fremstilling af sammenhængen mellem koncentration og virkning |
|
— |
estimater for toksiciteten for responsvariablerne, f.eks. EC50, EC10, EC20 med tilhørende konfidensintervaller. LOEC og NOEC (hvis disse er beregnet) og de statistiske metoder, der anvendt til at bestemme dem |
|
— |
hvis variansanalyse er anvendt, størrelsen af den virkning, det er muligt at påvise (f.eks. mindste signifikante forskel) |
|
— |
enhver vækststimulation, der er fundet ved nogen behandling |
|
— |
eventuelle andre observerede effekter, f.eks. morfologiske ændringer af algerne |
|
— |
diskussion af resultaterne, herunder den eventuelle betydning af fravigelser fra nærværende testmetode. |
4. LITTERATUR
|
(1) |
OECD TG 201 (2006) Freshwater Alga and Cyanobacteria, Growth Inhibition Test. |
|
(2) |
ISO 1998 Water quality — Guidance for algal growth inhibition tests with poorly soluble materials, volatile compounds, metals and waster water. ISO/DIS 14442. |
|
(3) |
OECD 2000: Guidance Document on Aquatic Toxicity Testing of Difficult Substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment, no. 23. |
|
(4) |
ISO 1998: Water quality — Sampling — Part 16: General Guidance for Biotesting. ISO 5667-16. |
|
(5) |
ISO 1993: Water quality — Algal growth inhibition test. ISO 8692. |
|
(6) |
Mayer, P., Cuhel, R. and Nyholm, N. (1997). A simple in vitro fluorescence method for biomass measurements in algal growth inhibition tests. Water Research 31: 2525-2531. |
|
(7) |
Slovacey, R.E. and Hanna, P.J. In vivo fluorescence determinations of phytoplancton chlorophyll, Limnology & Oceanography 22,5 (1977), pp.919-925. |
|
(8) |
Simpson, S.L., Roland, M.G.E., Stauber, J.L. and Batley, G.E. (2003) Effect of declining toxicant concentrations on algal bioassay endpoints. Environ. Toxicol. Chem 22, 2073-2079. |
|
(9) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713-718. |
|
(10) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157-167. |
|
(11) |
Bruce, R.D., and Versteeg, D.J. (1992). A statistical procedure for modelling continuous toxicity data. Env. Toxicol. Chem. 11:1485-1494. |
|
(12) |
OECD. (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data. |
|
(13) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05-88. USEPA, Duluth, MN. |
|
(14) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc. 50: 1096-1121. |
|
(15) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics 20: 482-491. |
|
(16) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27: 103-117. |
|
(17) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics 28: 510-531. |
|
(18) |
Draper, N.R. and Smith, H. (1981). Applied Regression Analysis, second edition. Wiley, New York. |
|
(19) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93-96. |
Tillæg 1
Stammer, der er påvist at være velegnede til testen
Grønalger
|
— |
Pseudokirchneriella subcapitata (tidligere benævnt Selenastrum capricornutum), ATCC 22662, CCAP 278/4, 61.81 SAG |
|
— |
Desmodesmus subspicatus (tidligere benævnt Scenedesmus subspicatus) 86.81 SAG |
Diatoméer
|
— |
Navicula pelliculosa, UTEX 664 |
Cyanobakterier
|
— |
Anabaena flos-aquae, UTEX 1444, ATCC 29413, CCAP 1403/13A |
|
— |
Synechococcus leopoliensis, UTEX 625, CCAP 1405/1 |
Kilder til stammer
De anbefalede stammer fås som monokulturer fra følgende samlinger (i alfabetisk rækkefølge):
|
|
|
|
|
|
|
|
|
|
|
|
Udseende og karakteristika af anbefalede arter
|
|
P. subcapitata |
D. subspicatus |
N. pelliculosa |
A. flos-aquae |
S. leopoliensis |
|
Udseende |
Krumme, skrueformede enkeltceller |
Ovale, hovedsagelig enkeltceller |
Stave |
Kæder af ovale celler |
Stave |
|
Størrelse (L × W), µm |
8-14 × 2-3 |
7-15 × 3-12 |
7,1 × 3,7 |
4,5 × 3 |
6 × 1 |
|
Cellevolumen (µm3/celle) |
40-60 (1) |
60-80 (1) |
40-50 (1) |
30-40 (1) |
2,5 (2) |
|
Tør vægt af celle (mg/celle) |
2-3 × 10–8 |
3-4 × 10–8 |
3-4 × 10–8 |
1-2 × 10–8 |
2-3 × 10–9 |
|
Væksthastighed (3) (dag–1) |
1,5-1,7 |
1,2-1,5 |
1,4 |
1,1-1,4 |
2,0-2,4 |
Særlige anbefalinger for dyrkning og håndtering af de anbefalede testarter
Pseudokirchneriella subcapitata og Desmodesmus subspicatus
Disse grønalgearter er sædvanligvis lette at holde i forskellige næringssubstrater. Oplysninger om egnede substrater kan fås fra kultursamlingerne. Cellerne er normalt solitære, og måling af celletætheden kan let foretages med elektronisk partikeltæller eller i mikroskop.
Anabaena flos-aquae
Stamkulturer kan holdes med forskellige næringssubstrater. Det er især vigtigt at undgå, at batchkulturen kommer ud af den logaritmiske vækstfase, når den fornyes, da restitution på dette punkt er vanskelig.
Anabaena flos-aquae udvikler aggregater af sammenfiltrede kæder af celler. Størrelsen af aggregaterne kan være forskellig, afhængigt af dyrkningsbetingelserne. Det kan være nødvendigt at bryde sådanne aggregater op, før biomassen kan bestemmes ved tælling i mikroskop eller med en elektronisk partikeltæller.
For at nedsætte variabiliteten af tællingerne kan kæderne brydes op ved sonikering af delprøver. Hvis sonikeringen fortsættes længere end nødvendigt for at bryde kæderne til kortere længder, kan cellerne blive ødelagt. Intensitet og varighed af sonikeringen skal være ens for alle behandlinger.
For at medvirke til at kompensere for variabiliteten skal der tælles et tilstrækkeligt antal felter på hæmocytometeret (mindst 400 celler). Derved fås mere pålidelige tæthedsbestemmelser i mikroskop.
Til bestemmelse af det totale cellevolumen af Anabaena kan en elektronisk partikeltæller anvendes efter forudgående forsigtig sonikering for at bryde cellekæderne. Sonikeringsenergien skal tilpasses, så bristning af celler undgås.
Ved hjælp af hvirvelomrører eller en tilsvarende velegnet metode skal det sikres, at den anvendte algesuspension til podning af testbeholderne er godt opblandet og homogen.
Testbeholderne anbringes på et roterende eller frem- og tilbagegående rystebord ved ca. 150 o./min. I stedet kan der anvendes intermitterende omrystning for at mindske Anabaenas tilbøjelighed til sammenklumpning. I tilfælde af sammenklumpning skal der drages omsorg for, at prøverne til biomassebestemmelse bliver repræsentative. Energisk omrystning kan være nødvendig før prøvetagning for at skille algeklumperne ad.
Synechococcus leopoliensis
Stamkulturer kan holdes med forskellige næringssubstrater. Oplysninger om egnede substrater kan fås fra kultursamlingerne.
Synechococcus leopoliensis vokser som solitære, stavformede celler. Cellerne er meget små, hvilket komplicerer tælling i mikroskop til biomassebestemmelse. Elektroniske partikeltællere, der er udstyret til tælling af partikler ned til ca. 1 µm, er gode. In vitro fluorometrisk måling kan ligeledes anvendes.
Navicula pelliculosa
Stamkulturer kan holdes med forskellige næringssubstrater. Oplysninger om egnede substrater kan fås fra kultursamlingerne. Bemærk, at substratet skal indeholde silikat.
Navicula pelliculosa kan danne aggregater under visse vækstbetingelser. På grund af dannelse af lipider vil algecellerne undertiden have tendens til at akkumulere i overfladefilmen. Under sådanne omstændigheder må der ved udtagning af delprøver til biomassebestemmelse træffes særlige foranstaltninger for at sikre, at prøverne er repræsentative. Det kan være nødvendigt med kraftig omrystning, f.eks. med en hvirvelomrører.
(1) Målt med elektronisk partikeltæller.
(2) Beregnet af størrelsen.
(3) Hyppigste observerede væksthastighed i OECD-substrat ved en lysintensitet på ca. 70 µE·m–2·s–1 og 21 °C.
Tillæg 2
Næringssubstrater
Et af nedenstående to næringssubstrater kan anvendes:
OECD-substrat: Det oprindelige substrat fra OECD TG 201, også i henhold til ISO 8692
US. EPA-substrat AAP, ligeledes i henhold til ASTM.
Til fremstilling af sådanne substrater skal anvendes kemikalier af reagens- eller analysekvalitet og deioniseret vand.
Sammensætning af AAP-substratet (US. EPA) og OECD TG 201-substratet
|
Komponent |
EPA |
OECD |
||
|
|
mg/l |
mM |
mg/l |
mM |
|
NaHCO3 |
15,0 |
0,179 |
50,0 |
0,595 |
|
NaNO3 |
25,5 |
0,300 |
|
|
|
NH4Cl |
|
|
15,0 |
0,280 |
|
MgCl2·6(H2O) |
12,16 |
0,0598 |
12,0 |
0,0590 |
|
CaCl2·2(H2O) |
4,41 |
0,0300 |
18,0 |
0,122 |
|
MgSO4·7(H2O) |
14,6 |
0,0592 |
15,0 |
0,0609 |
|
K2HPO4 |
1,044 |
0,00599 |
|
|
|
KH2PO4 |
|
|
1,60 |
0,00919 |
|
FeCl3·6(H2O) |
0,160 |
0,000591 |
0,0640 |
0,000237 |
|
Na2EDTA·2(H2O) |
0,300 |
0,000806 |
0,100 |
0,000269 (1) |
|
H3BO3 |
0,186 |
0,00300 |
0,185 |
0,00299 |
|
MnCl2·4(H2O) |
0,415 |
0,00201 |
0,415 |
0,00210 |
|
ZnCl2 |
0,00327 |
0,000024 |
0,00300 |
0,0000220 |
|
CoCl2·6(H2O) |
0,00143 |
0,000006 |
0,00150 |
0,00000630 |
|
Na2MoO4·2(H2O) |
0,00726 |
0,000030 |
0,00700 |
0,0000289 |
|
CuCl2.2(H2O) |
0,000012 |
0,00000007 |
0,00001 |
0,00000006 |
|
pH |
7,5 |
8,1 |
||
I testen med diatoméen Navicula pelliculosa skal begge substrater suppleres med Na2SiO3·9H2O til en koncentration på 1,4 mg Si/l.
Substratets pH opnås ved ligevægt mellem substratets carbonatsystem og partialtrykket af CO2 i atmosfærisk luft. En omtrentlig sammenhæng mellem pH ved 25 °C og den molære bicarbonatkoncentration er givet ved:
pHeq = 11,30 + log [HCO3]
Ved 15 mg NaHCO3/l er pHeq = 7,5 (U.S. EPA-substrat), og ved 50 mg NaHCO3/l er pHeq = 8,1 (OECD-substrat).
Testsubstraternes sammmensætning af grundstoffer
|
Grundstof |
EPA |
OECD |
|
|
mg/l |
mg/l |
|
C |
2,144 |
7,148 |
|
N |
4,202 |
3,927 |
|
P |
0,186 |
0,285 |
|
K |
0,469 |
0,459 |
|
Na |
11,044 |
13,704 |
|
Ca |
1,202 |
4,905 |
|
Mg |
2,909 |
2,913 |
|
Fe |
0,033 |
0,017 |
|
Mn |
0,115 |
0,115 |
Fremstilling af OECD-substrat
|
Næringsstof |
Koncentration i stamopløsning |
|
Stamopløsning 1: makronæringsstoffer |
|
|
NH4Cl MgCl2·6H2O CaCl2·2H2O MgSO4·7H2O KH2PO4 |
1,5 g·l–1 1,2 g·l–1 1,8 g·l–1 1,5 g·l–1 0,16 g·l–1 |
|
Stamopløsning 2: jern |
|
|
FeCl3·6H2O Na2EDTA·2H2O |
64 mg·l–1 100 mg·l–1 |
|
Stamopløsning 3: sporstoffer |
|
|
H3BO3 MnCl2·4H2O ZnCl2 CoCl2·6H2O CuCl2·2H2O Na2MoO4·2H2O |
185 mg·l–1 415 mg·l–1 3 mg·l–1 1,5 mg·l–1 0,01 mg·l–1 7 mg·l–1 |
|
Stamopløsning 4: bicarbonat |
|
|
NaHCO3 |
50 g·l–1 |
|
Na2SiO3·9H20 |
|
Stamopløsningen steriliseres ved membranfiltrering (middelporediameter 0,2 µm) eller autoklavering (120 °C, 15 min.). Opløsningerne opbevares i mørke ved 4 °C.
Stamopløsning 2 og 4 må ikke autoklaveres, men skal steriliseres ved membranfiltrering.
Næringssubstrater fremstilles ved tilsætning af et passende volumen stamopløsning 1-4 til vand:
Til 500 ml steriliseret vand tilsættes der:
|
— |
10 ml stamopløsning 1 |
|
— |
1 ml stamopløsning 2 |
|
— |
1 ml stamopløsning 3 |
|
— |
1 ml stamopløsning 4. |
Der tilsættes steriliseret vand til 1 000 ml.
Giv substratet tid til at ækvilibrere med atmosfærens CO2, om nødvendigt ved nogle timers gennembobling med sterilfiltreret luft.
Fremstilling af AAP-substrat
|
A1.1. |
1 ml af hver stamopløsning i A1.2.1-A1.2.7 tilsættes til ca. 900 ml deioniseret eller destilleret vand og fortyndes derefter til 1 liter. |
|
A1.2. |
Stamopløsninger af makronæringsstoffer fremstilles ved opløsning af nedenstående i 500 ml deioniseret eller destilleret vand. Reagens A1.2.1, A1.2.2, A1.2.3 og A1.2.4 kan kombineres til én stamopløsning. |
|
A1.2.1. |
NaNO3 -12,750 g |
|
A1.2.2. |
MgCl2·6H2O -6,082 g |
|
A1.2.3. |
MgCl2·2H2O -2,205 g |
|
A1.2.4. |
Stamopløsning af mikronæringsstoffer — (se A1.3) |
|
A1.2.5. |
MgSO4·7H2O -7,350 g |
|
A1.2.6. |
K2HPO4 -0,522 g |
|
A1.2.7. |
NaHCO3 -7,500 g |
|
A1.2.8. |
Na2SiO3·9H2O — se bemærkning A1.1. BEMÆRKNING A1.1: Anvendes kun til diatomé-testarter. Kan tilsættes direkte (202,4 mg) eller som stamopløsning til en endelige Si-koncentration i substratet på 20 mg/l. |
|
A1.3. |
Stamopløsningen af mikronæringsstoffer fremstilles ved opløsning af nedenstående i 500 ml deioniseret eller destilleret vand: |
|
A1.3.1. |
H3BO3 -92,760 mg |
|
A1.3.2. |
MnCl2·4H2O -207,690 mg |
|
A1.3.3. |
ZnCl2 -1,635 mg |
|
A1.3.4. |
FeCl3·6H2O -79,880 mg |
|
A1.3.5. |
CoCl2· 6H2O -0,714 mg |
|
A1.3.6. |
Na2MoO4· 2H2O -3,630 mg |
|
A1.3.7. |
CuCl2· 2H2O -0,006 mg |
|
A1.3.8. |
Na2EDTA·2H2O -150,000 mg [dinatrium-(ethylendinitrilo-)tetraacetat] |
|
A1.3.9. |
Na2SeO4·5H2O -0,005 mg, se bemærkning A1.2. BEMÆRKNING A1.2: Bruges kun i substrat til stamkulturer af diatoméarter. |
|
A1.4. |
pH justeres til 7,5 ± 0,1 med 0,1 N eller 1,0 N NaOH eller HCl. |
|
A1.5. |
Substratet filtreres over i en steril beholder, enten gennem et 0,22 μm membranfilter, hvis der skal bruges partikeltæller, eller et 0,45 μm filter, hvis der ikke skal bruges partikeltæller. |
|
A1.6. |
Substratet opbevares i mørke ved ca. 4 °C, til det skal bruges. |
(1) Det molære forhold mellem EDTA og jern er lidt over 1. Derved undgås udfældning af jern, samtidig med at chelatering af tungmetalioner minimeres.
Tillæg 3
Eksempel på fremgangsmåde ved dyrkning af alger
Almindelige bemærkninger
Formålet med dyrkningen efter følgende metode er at fremstille algekulturer til toksicitetstest.
Det skal ved hjælp af egnede metoder sikres, at algekulturerne ikke er inficeret med bakterier. Axeniske kulturer kan være ønskeligt, men der skal fremstilles og anvendes monokulturer af alger.
Alle operationer skal udføres med steril teknik for at undgå kontaminering med bakterier og andre alger.
Udstyr og materialer
Se under testmetode: Apparatur.
Metoder til fremstilling af algekulturer
Fremstilling af næringssubstrater (medier):
Alle næringssalte i substratet fremstilles som koncentrerede stamopløsninger, der opbevares mørkt og koldt. Disse opløsninger steriliseres ved filtrering eller autoklavering.
Substratet fremstilles ved, at den korrekte mængde stamopløsning sættes til destilleret vand, idet der udvises omhu for at undgå kontaminering. Til faste substrater tilsættes 0,8 procent agar.
Stamkultur:
Stamkulturerne er små algekulturer, der regelmæssigt overføres til friskt substrat og anvendes som udgangsmateriale ved testen. Hvis kulturerne ikke jævnligt bruges, udstryges de på skråagarrør. Herfra overføres de til friskt substrat mindst hver anden måned.
Stamkulturerne dyrkes i koniske kolber indeholdende det pågældende substrat (volumen ca. 100 ml). Når algerne inkuberes ved 20 °C med kontinuerlig belysning, skal de overføres en gang om ugen.
Ved overførslen føres en del af den »gamle« kultur med sterile pipetter til en kolbe med friskt substrat, så initialkoncentrationen for hurtigtvoksende arter er ca. 100 gange mindre end i den gamle kultur.
Væksthastigheden for en art kan bestemmes af vækstkurven. Kendes denne, kan man beregne, ved hvilken tæthed kulturen skal overføres til nyt substrat. Dette skal ske, før kulturen når dødsfasen.
Forkultur:
Forkulturen har til formål at producere en mængde alger, der er egnede til podning af testkulturer. Forkulturen inkuberes under testbetingelserne og anvendes, mens den endnu er i eksponentiel vækst, normalt efter en inkubationsperiode på 2 til 4 dage. Når algekulturer indeholder deforme eller abnorme celler, skal de kasseres.
Tillæg 4
Dataanalyse ved ikke-lineær regression
Generelle betragtninger
Responsen i algetest og andre test af mikrobiel vækst — vækst af biomasse — er i sig selv en kontinuert eller metrisk variabel — en proceshastighed, når væksthastigheden anvendes, og integralet af væksthastigheden med hensyn til tiden, når biomasse vælges. Begge henføres til den tilsvarende gennemsnitsrespons for replikate, ikke-eksponerede kontroller, der udviser maksimal respons ved de anvendte betingelser — med lys og temperatur som de primære bestemmende faktorer i algetesten. Systemet er distribueret eller homogent, og biomassen kan betragtes som en helhed uden hensyn til enkeltceller. Variansfordelingen for den type respons, der ses i et sådant system, afhænger udelukkende af eksperimentelle faktorer (dette beskrives typisk ved, at fejlen er log-normalfordelt eller normalfordelt). Dette er modsat typiske bioassay-responser med kvantale data, hvor det dominerende bidrag til variansen ofte antages at være de enkelte organismers tolerance (der typisk er binomialfordelt). Responsen fra kontrollerne er her nulniveau eller baggrundsniveau.
I det ukomplicerede tilfælde aftager den normaliserede eller relative respons, r, monotont fra 1 (ingen hæmning) til 0 (100 procents hæmning). Bemærk, at der til alle responser er knyttet en fejl, og at tilsyneladende negative hæmningsværdier ved beregningen kan tilskrives tilfældige fejl alene.
Regressionsanalyse
Modeller
Med regressionsanalyse tilstræbes det at beskrive koncentrations-responskurven som en matematisk regressionsfunktion Y = f (C) eller oftere F (Z), hvor Z = log C. Med den omvendte funktion C = f–1 (Y) kan der beregnes ECx-værdier, herunder EC50, EC10 og EC20, med tilhørende 95 % konfidensgrænser. Flere forskellige enkle matematiske funktioner har vist sig velegnede til at beskrive den koncentrations-responssammenhæng, der optræder i væksthæmningstest med alger. Af sådanne funktioner kan nævnes den logistiske ligning, den asymmetriske Weibull-ligning og log-normalfordelingsfunktionen, der alle er S-formede kurver, der asymptotisk går mod én for C → 0, og mod nul for C → uendeligt.
Anvendelse af modeller med kontinuerte tærskelfunktioner (f.eks. »the Kooijman model for inhibition of population growth« Kooijman et al. 1996) er for nylig blevet foreslået som et alternativ til asymptotiske modeller. I denne model antages, at der ikke er nogen virkninger ved koncentrationer under en bestemt tærskel, EC0+, som estimeres ved at ekstrapolere responskoncentrationsforholdet til skæring med koncentrationsaksen ved hjælp af en simpel kontinuert funktion, der ikke er differentiabel i udgangspunktet.
Bemærk, at analysen kan bestå i en simpel minimering af summer af residualkvadrater (når variansen forudsættes konstant) eller af vægtede kvadrater, når der kompenseres for variansheterogenitet.
Fremgangsmåde
Fremgangsmåden kan sammenfattes således: Der vælges en passende funktion, Y = f (C), som tilpasses til dataene ved ikke-lineær regression. Det må foretrækkes at anvende målinger fra hver enkelt kolbe frem for gennemsnitsværdier for replikaterne, da man derved får mest mulig information fra dataene. Hvis variansen derimod er høj, tyder praktisk erfaring på, at der ved brug af gennemsnitsværdier af replikaterne fås en mere robust matematisk beregning, der er mindre påvirkelig af systematiske fejl i data end bibeholdelse af de enkelte datapunkter.
Den tilpassede kurve og de målte data afsættes, og det undersøges, om tilpasningen af kurven er tilfredsstillende. Analyse af residualer kan være et særdeles nyttigt redskab til dette formål. Hvis den valgte funktion for koncentrations-responssammenhængen ikke beskriver hele kurven eller en vigtig del af denne som f.eks. responsen i koncentrationslavpunktet, vælges en anden mulighed for kurvetilpasning — f.eks. en ikke-symmetrisk kurve som Weibul-funktionen i stedet for en symmetrisk. Negativ hæmning kan være et problem ved brug af f.eks. log-normalfordelingsfunktionen, så der også her er brug for en alternativ regressionsfunktion. Det kan ikke anbefales at tildele en nulværdi eller en lille positiv værdi til sådanne negative værdier, da det forvrænger fejlfordelingen. Det kan være hensigtsmæssigt at foretage separate kurvetilpasninger, f.eks. på den del af kurven, hvor hæmningen er lille, for at beregne et estimat for EClow x-værdierne. Af den tilpassede ligning beregnes (ved »omvendt beregning«, C = f–1 (Y)) karakteristiske punktestimater ECx, og som minimum rapporteres EC50-værdien og et eller to estimater for EClow x. De praktiske erfaringer fra testningerne har vist, at algetesten sædvanligvis er tilstrækkelig præcis til at give et rimelig nøjagtigt estimat ved 10 % hæmning, hvis der er tilstrækkeligt med datapunkter — medmindre der ved lave koncentrationer optræder stimulation som konfunderende faktor. Nøjagtigheden af et estimat for EC20 er ofte betydeligt bedre end af en EC10-værdi, da EC20 sædvanligvis ligger på den tilnærmelsesvis lineære del af den centrale del af koncentrations-responskurven. Undertiden kan EC10 være vanskelig at fortolke på grund af vækststimulation. Selv om EC10 således normalt fås med tilstrækkelig nøjagtighed, anbefales det således, at man desuden altid rapporterer EC20-værdien.
Vægtningsfaktorer
Den eksperimentelle varians er sædvanligvis ikke konstant, men indeholder typisk en proportional komponent, hvorfor rutinemæssig brug af vægtet regression vil være en fordel. Vægtningsfaktorerne til en sådan analyse antages normalt at være omvendt proportionale med variansen:
Wi = 1/Var(ri)
Mange regressionsprogrammer kan bruges til vægtet regressionsanalyse med vægtningsfaktorer angivet i en tabel. For nemheds skyld bør vægtningsfaktorerne normaliseres ved multiplikation med n/Σ wi (hvor n er antallet af datapunkter), således at deres sum bliver lig én.
Normalisering af respons
Normalisering med gennemsnitsresponsen i kontrollerne medfører visse principielle problemer foruden en ret kompliceret variansstruktur. Når responsværdierne divideres med kontrollernes gennemsnitsrespons for at få hæmningen i procent, indføres derved en ekstra fejl, der skyldes fejlen på middelværdien for kontrollerne. Medmindre denne fejl er ubetydelig, skal vægtningsfaktorerne i regressionen og konfidensgrænserne korrigeres for kovarians med kontrollen (17). Bemærk, at det er vigtigt med en høj præcision af estimatet for middelværdien af responsen i kontrollerne, for at den samlede varians af den relative repons kan blive minimeret. Denne varians kan angives således:
(indeks i henviser til koncentrationsniveau i og indeks 0 til kontrollerne)
Yi = Relativ respons = ri/r0 = 1 — I = f (Ci)
med variansen:
Var (Yi) = Var (ri/r0) ≈ (∂Yi/∂ ri)2·Var(ri) + (∂ Yi/∂ r0)2·Var (r0)
og, eftersom
(∂ Yi/∂ ri) = 1/r0, og (∂ Yi/∂ r0) = ri/r0 2
med normalfordelte data og replikaterne mi og m0:
Var(ri) = σ2/mi
bliver den totale varians af den relative respons Yi:
Var(Yi) = σ2/(r0 2 mi) + ri 2·σ2/r0 4 m0
Fejlen på middelværdien for kontrollerne er omvendt proportional med kvadratroden af det antal replikater af kontrollerne, som gennemsnittet er beregnet af, og undertiden kan det være berettiget at medtage historiske data for på denne måde at mindske fejlen stærkt. En alternativ metode er at undlade normalisering af dataene og i stedet tilpasse de absolutte responsværdier, også responsdata for kontrollerne, men herved indføres responsværdien for kontrollerne som en ekstra parameter, der skal indgå i tilpasningen ved ikke-lineær regression. Med en sædvanlig to-parametres regressionsligning kræver denne metode tilpasning af tre parametre og dermed flere datapunkter end ikke-lineær regression på data, der er normaliseret med en forud fastlagt respons for kontrollerne.
Omvendte konfidensintervaller
Beregning af konfidensintervaller i ikke-lineær regression ved hjælp af omvendt estimation er ret kompliceret og hører ikke til standardvalgene i almindelige statistikprogrampakker. Tilnærmede konfidensgrænser kan beregnes med standardprogrammer til ikke-lineær regression med re-parameterisering (Bruce og Versteeg, 1992), hvorved den matematiske ligning omskrives med de ønskede punktestimater, f.eks. med EC10 og EC50 som de parametre, der skal estimeres. (Lad funktionen være I = f (α, β, koncentration), og benyt definitionsrelationerne f (α, β, EC10) = 0,1 og f (α, β, EC50) = 0,5 til at erstatte funktionen f (α, β, koncentration) med den ækvivalente funktion g (EC10, EC50, koncentration.)
En mere direkte beregning (Andersen et al, 1998) foretages ved at bibeholde den oprindelige ligning og bruge Taylor-udvikling omkring middelværdierne af ri og r0.
På det seneste har »boot strap«-metoder vundet udbredelse. I sådanne metoder estimeres en empirisk variansfordeling ved hjælp af de målte data sammen med hyppig resampling, der styres af en generator af tilfældige tal.
Referencer
Kooijman, S.A.L.M.; Hanstveit, A.O.; Nyholm, N. (1996): No-effect concentrations in algal growth inhibition tests. Water Research, 30, 1625-1632.
Bruce, R.D. and Versteeg, D.J.(1992) A Statistical Procedure for Modelling Continuous Ecotoxicity Data. Env. Toxicol. Chem.11, 1485-1494.
Andersen, J.S., Holst, H., Spliid, H., Andersen, H., Baun, A. & Nyholm, N. (1998): Continuous ecotoxicological data evaluated relative to a control response. Journal of Agricultural, Biological and Environmental Statistics, 3, 405-420.
BILAG V
|
C.25. |
AEROB MINERALISERING I OVERFLADEVAND — SIMULERET BIONEDBRYDNINGSFORSØG |
1. METODE
Metoden er ækvivalent med OECD TG 309 (2004) (1).
1.1. INDLEDNING
Formålet med forsøget er at bestemme det tidsmæssige forløb af bionedbrydningen af et prøvestof med lav koncentration i aerobt naturligt vand og at kvantificere iagttagelserne i form af kinetiske hastighedsudtryk. Dette simuleringsforsøg er et shake-flask batchforsøg i laboratoriet og har til formål at bestemme hastigheden af aerob bionedbrydning af organiske stoffer i prøver af naturligt vand (ferskvand, brakvand eller søvand). Det bygger på ISO/DIS 14592-1 (2) og indeholder desuden elementer fra prøvemetode C.23 og C.24 (3)(4). Langvarige forsøg kan valgfrit udføres som semikontinuerte forsøg i stedet for batchforsøg for at undgå ældning af mikromiljøet i forsøget. Hovedformålet med simuleringsforsøget er at bestemme mineraliseringen af prøvestoffet i overfladevand, og mineraliseringen danner grundlaget for de udtryk, der formulerer nedbrydningskinetikken. Et sekundært, valgfrit formål med forsøget er imidlertid at skaffe oplysninger om den primære nedbrydning og dannelsen af de vigtigste omdannelsesprodukter. Identifikation af omdannelsesprodukter og, om muligt, kvantitativ bestemmelse af disse er særligt vigtig for stoffer, der mineraliseres meget langsomt (f.eks. med en halveringstid for total residual–14C på over 60 dage). Pga. begrænsningerne i analyseteknikken bør der normalt anvendes højere prøvestofkoncentration (f.eks. > 100 μg/l) til identifikation og kvantificering af vigtige omdannelsesprodukter.
I dette forsøg forstås ved »lav koncentration« en koncentration, der er så lav (f.eks. mindre end 1 μg/l til 100 μg/l), at den i forsøget iagttagne bionedbrydningskinetik med sikkerhed afspejler den, der forventes at gælde i miljøet. I forhold til den samlede masse bionedbrydeligt kulstofsubstrat, der er til rådighed i det i forsøget anvendte naturlige vand, vil prøvestoffet, der er til stede i lav koncentration, kun være et sekundært substrat. Dette forudsætter, at den forventede bionedbrydningskinetik er af første orden (»non-growth« kinetik), og at prøvestoffet kan nedbrydes ved »co-metabolisme«. Ved første ordens kinetik er nedbrydningshastigheden (mg/l/dag) proportional med substratkoncentrationen, der aftager med tiden. Ved ægte første ordens kinetik er den specifikke nedbrydningshastighedskonstant, k, uafhængig af tiden og af koncentrationen. Det vil sige, at k ikke ændrer sig nævneværdigt i samme forsøg og ikke ændrer sig med den tilsatte koncentration mellem forskellige forsøg. Ved den specifikke nedbrydningshastighedskonstant forstås den relative koncentrationsændring pr. tidsenhed: k. Ved første ordens kinetik er nedbrydningshastigheden (mg/l/dag) proportional med substratkoncentrationen, der aftager med tiden. Ved ægte første ordens kinetik er den specifikke nedbrydningshastighedskonstant, k, uafhængig af tiden og af koncentrationen. Det vil sige, at k ikke ændrer sig nævneværdigt i samme forsøg og ikke ændrer sig med den tilsatte koncentration mellem forskellige forsøg. Ved den specifikke nedbrydningshastighedskonstant forstås den relative koncentrationsændring pr. tidsenhed: k = (1/C) · (dC/dt). Skønt der under de foreskrevne betingelser normalt forventes første ordens kinetik, kan der være omstændigheder, hvor andre kinetiske love giver bedre overensstemmelse. Afvigelser fra første ordens kinetik ses f.eks., hvis det er massetransportfænomener, f.eks. diffusionshastigheden, snarere end den biologiske reaktionshastighed, der er hastighedsbegrænsende for den biologiske omdannelse. Data kan imidlertid næsten altid beskrives ved pseudo første ordens kinetik, når det accepteres, at hastighedskonstanten er koncentrationsafhængig.
Inden forsøget bør der foreligge oplysninger om biologisk nedbrydning af prøvestoffet ved højere koncentrationer (f.eks. fra rutinemæssige screeningstest) og oplysninger om abiotisk nedbrydning, omdannelsesprodukter og relevante fysisk-kemiske egenskaber, hvilket vil lette planlægning af forsøget og fortolkning af resultaterne. Ved at anvende 14C-mærket prøvestof og bestemme fasefordelingen af 14C ved forsøgets afslutning kan den endelige bionedbrydelighed bestemmes. Er prøvestoffet umærket, kan den endelige bionedbrydning kun bestemmes, hvis der testes en højere koncentration, og alle vigtige omdannelsesprodukter kendes.
1.2. DEFINITIONER
Primær bionedbrydning: Den strukturændring (omdannelse) af et kemisk stof, der frembringes af mikroorganismer og medfører, at stoffet mister sin kemiske identitet.
Funktionel bionedbrydning: Den strukturændring (omdannelse) af et kemisk stof, som frembringes af mikroorganismer og medfører tab af en bestemt egenskab.
Endelig aerob bionedbrydning: Den af mikroorganismer i tilstedeværelse af oxygen frembragte nedbrydning af et kemisk stof til carbondioxid, vand og uorganiske salte af eventuelle andre grundstoffer, der er til stede (mineralisering) og produktion af ny biomasse og organiske mikrobielle biosynteseprodukter.
Mineralisering: Den af mikroorganismer i tilstedeværelse af oxygen frembragte nedbrydning af et kemisk stof eller organisk materiale til carbondioxid, vand og uorganiske salte af eventuelle andre grundstoffer, der er til stede.
Lag-fase: Den tid, der går fra et forsøg begynder, indtil de nedbrydende mikroorganismer har tilpasset sig, og nedbrydningsgraden for et kemisk stof eller organisk materiale har nået et detekterbart omfang (f.eks. 10 % af den teoretiske maksimale bionedbrydning, evt. mindre, afhængigt af måleteknikkens nøjagtighed).
Maksimal bionedbrydningsgrad: Den grad af bionedbrydning af et kemisk stof eller organisk materiale, angivet i procent, der finder sted i et forsøg, og over hvilken der ikke sker yderligere bionedbrydning under forsøget.
Primært substrat: En række naturlige kulstof- og energikilder, der tjener til vækst og opretholdelse af den mikrobielle biomasse.
Sekundært substrat: En substratkomponent, der er til stede i så lav koncentration, at dens nedbrydning kun tilfører ubetydelige mængder kulstof og energi til de kompetente mikroorganismer i forhold til den mængde kulstof og energi, der tilføres ved nedbrydning af hovedsubstratets komponenter (de primære substrater).
Nedbrydningshastighedskonstant: En første ordens eller pseudo første ordens hastighedskonstant k (d–1), der angiver nedbrydningsprocessers hastighed. For et batch-forsøg beregnes k af den første del af nedbrydningskurven efter slutningen af lag-fasen.
Halveringstid, t1/2 (d): En betegnelse til karakterisering af hastigheden af en første ordens reaktion. Halveringstiden er tidsrummet svarende til en formindskelse af koncentrationen med en faktor 2. Sammenhængen mellem halveringstiden og hastighedskonstanten er givet ved udtrykket t1/2 = ln2/k.
Nedbrydningshalveringstid, DT50 (d): En betegnelse, der anvendes til kvantitativ angivelse af resultatet af bionedbrydningsforsøg. Det er det tidsrum, inklusive lag-fasen, der er nødvendigt for at nå 50 % bionedbrydning.
Detektionsgrænse (limit of detection (LOD)) og kvantificeringsgrænse (limit of quantification (LOQ)): Detektionsgrænsen er den koncentration af et stof, under hvilken stoffets identitet ikke kan skelnes fra analysebetingede artefakter. Kvantificeringsgrænsen er den koncentration af et stof, under hvilken koncentrationen ikke kan bestemmes med acceptabel nøjagtighed.
Opløst organisk kulstof (dissolved organic carbon (DOC)): Den del af det organiske kulstofindhold i en prøve af vand, som ikke kan fjernes ved den foreskrevne faseadskillelse som f.eks. ved centrifugering ved 40 000 ms–2 i 15 min. eller ved membranfiltrering med porestørrelse 0,2 μm-0,45 μm i diameter.
Total organisk 14C-aktivitet (total organic 14C activity (TOA)): Den totale 14C-aktivitet, der er knyttet til organisk kulstof.
Opløst organisk 14C-aktivitet (dissolved organic 14C activity (DOA)): Den totale 14C-aktivitet, der er knyttet til opløst organisk kulstof.
Partikelbåren organisk 14C-aktivitet (particulate organic 14C activity (POA)): Den totale 14C-aktivitet, der er knyttet til partikelformigt organisk kulstof.
1.3. PRØVEMETODENS ANVENDELIGHED
Denne simuleringsmetode er anvendelig til ikke-flygtige eller svagt flygtige organiske stoffer, der testes ved lave koncentrationer. Når der bruges kolber med åben forbindelse til atmosfæren (f.eks. med vatprop), kan stoffer med Henrys lov konstant mindre end omkring 1 Pa·m3/mol (ca. 10–5 atm·m3/mol) i praksis regnes for ikke-flygtige. Ved at anvende lukkede kolber med headspace kan man teste svagt flygtige stoffer (med Henrys lov konstant < 100 Pa·m3/mol eller < 10–3 atm·m3/mol) uden tab fra prøvesystemet. Tab af 14C-mærkede stoffer kan forekomme, hvis der ikke er truffet passende forholdsregler, når CO2 fjernes. I sådanne tilfælde kan det være nødvendigt at opfange CO2 i en intern absorber med alkali eller i et eksternt CO2-absorbersystem (direkte 14CO2-bestemmelse, jf. tillæg 3). Til fastlæggelse af bionedbrydningens kinetik skal prøvestofkoncentrationen være lavere end stoffets opløselighed i vand. Det skal dog bemærkes, at man i litteraturen kan finde væsentligt højere værdier for vandopløseligheden end prøvestoffets opløselighed i naturlige vande. Om ønsket kan opløseligheden af prøvestoffer med ringe vandopløselighed bestemmes ved brug af de naturlige vande, som forsøget omfatter.
Metoden kan dels anvendes til simulering af bionedbrydning i overfladevand fri for grove partikler (»pelagisk forsøg«), dels i uklart vand som f.eks. nær grænsefladen mellem vand og sediment (»forsøg med opslæmmet sediment«).
1.4. FORSØGETS PRINCIP
Forsøget udføres som batchforsøg med inkubering af prøvestoffet enten med overfladevand alene (»pelagisk forsøg«) eller overfladevand modificeret med en opslæmmet mængde faststof/sediment på 0,01 til 1 g tør vægt pr. liter (»forsøg med opslæmmet sediment«) til simulering af en vandmasse med opslæmmede faststoffer eller genopslæmmet sediment. I de fleste overfladevande vil koncentrationen af opslæmmet faststof/sediment typisk ligge i den nedre ende af dette interval. Prøvekolberne inkuberes i mørke ved rumtemperatur under aerobe forhold og med omrystning. Prøvestoffet skal anvendes i mindst to forskellige koncentrationer til fastlæggelse af nedbrydningskinetikken. De anvendte koncentrationer skal indbyrdes afvige med en faktor 5 til 10 og skal være repræsentative for det forventede koncentrationsområde i miljøet. Den maksimale prøvestofkoncentration må ikke være over 100 µg/l, dog må en maksimal prøvekoncentration på 10 µg/l eller derunder foretrækkes for at sikre, at bionedbrydningen følger første ordens kinetik. Den laveste koncentration må ikke være over 10 µg/l, men en laveste prøvekoncentration på 1-2 μg/l eller under 1 μg/l må foretrækkes. En så lav koncentration kan normalt analyseres tilfredsstillende ved hjælp af gængse 14C-mærkede stoffer. Analyseteknikkens begrænsninger gør det ofte umuligt at måle prøvestoffets koncentration med den krævede nøjagtighed, hvis prøvestoffet tilsættes i en koncentration ≤ 100 µg/l (jf. andet afsnit i punkt 1.7.2). Til identifikation og kvantitativ bestemmelse af de vigtigste omdannelsesprodukter kan anvendes højere prøvestofkoncentration (> 100 µg/l, undertiden > 1 mg/l), hvis man ikke råder over en specifik analysemetode med lav detektionsgrænse. Anvendes en høj prøvestofkoncentration, kan resultaterne muligvis ikke anvendes til at beregne første ordens nedbrydningskonstant og halveringstid, da nedbrydningen antagelig ikke vil følge første ordens kinetik.
Nedbrydningen følges med passende tidsintervaller ved måling enten af residual–14C eller, hvis en specifik kemisk analyse anvendes, residualkoncentration af prøvestoffet. 14C-mærkning af den mest stabile del af molekylet giver mulighed for bestemmelse af den totale mineralisering, mens 14C-mærkning af en mindre stabil del af molekylet i lighed med en specifik analysemetode kun gør det muligt at bestemme den primære bionedbrydning. Den mest stabile del omfatter dog ikke nødvendigvis molekylets relevante funktionelle enhed (dvs. den enhed, der kan være knyttet til en specifik egenskab som toksicitet, bioakkumulering osv.). I så fald kan det være hensigtsmæssigt at 14C-mærke prøvestoffet i den funktionelle gruppe for at følge eliminationen af den specifikke egenskab.
1.5. OPLYSNINGER OM PRØVESTOFFET
Både radioaktivt mærket og umærket prøvestof kan anvendes i forsøget. 14C-mærkning anbefales, og mærkning bør normalt finde sted i molekylets mest stabile del(e) (jf. også punkt 1.4). I stoffer indeholdende mere end én aromatisk ring skal helst et eller flere kulstofatomer i hver ring være 14C-mærket. I bindinger, der let brydes, skal desuden et eller flere kulstofatomer på begge sider af bindingen helst være 14C-mærket. Prøvestoffets kemiske og/eller radiokemiske renhed skal være > 95 %. For radioaktivt mærkede stoffer skal den specifikke aktivitet helst være ca. 50 μCi/mg (1,85 MBq) eller derover for at lette 14C-målingerne i forsøg med lav initialkoncentration. Følgende oplysninger om prøvestoffet skal foreligge:
|
— |
vandopløselighed [metode A.6] |
|
— |
opløselighed i organiske(e) opløsningsmiddel (-midler) (stoffer, der tilføres med opløsningsmiddel eller er tungtopløselige i vand) |
|
— |
for stoffer, som kan protoniseres eller deprotoniseres, dissociationskonstant (pKa) [OECD TG 112] (5) |
|
— |
damptryk [metode A.4] og Henrys konstant |
|
— |
kemisk stabilitet i vand og i mørke (hydrolyse) [metode C.7]. |
Når tungtopløselige stoffer testes i søvand, kan det desuden være nyttigt at kende udsaltningskonstanten (eller »Setschenows konstant«) Ks, der er defineret ved udtrykket: log (S/S') = Ks Cm, where S and S' er stoffets opløselighed i henholdsvis ferskvand og søvand, og Cm er den molære saltkoncentration.
Udføres forsøget som »forsøg med opslæmmet sediment«, skal der foreligge følgende oplysninger:
|
— |
fordelingskoefficient n-octanol/vand [metode A.8] |
|
— |
adsorptionskoefficient [metode C.18]. |
Desuden kan følgende oplysninger være nyttige:
|
— |
stoffets koncentration i miljøet, hvis den kendes eller er beregnet |
|
— |
prøvestoffets toksicitet over for mikroorganismer [metode C.11] |
|
— |
umiddelbar og/eller naturlig bionedbrydelighed [metode C.4 A-F, C.12, C.9, OECD TG 302 (5)] |
|
— |
aerob eller anaerob bionedbrydelighed i omdannelsesforsøg i jord og sediment/vand [metode C.23 og C.24]. |
1.6. REFERENCESTOF
Som referencestof anvendes et stof, der normalt er let nedbrydeligt under aerobe forhold (f.eks. anilin eller natriumbenzoat). Anilin og natriumbenzoat nedbrydes sædvanligvis på mindre end to uger. Formålet med referencestofferne er at sikre, at prøvevandets mikrobielle aktivitet er inden for visse grænser, dvs. at vandet indeholder en aktiv mikrobiel population.
1.7. KVALITETSKRITERIER
1.7.1. Genfinding
Umiddelbart efter tilsætning af prøvestoffet skal hver begyndelseskoncentration verificeres ved måling af 14C-aktiviteten eller, for umærkede stoffer, ved kemisk analyse, idet der mindst anvendes dobbelte prøver. Dette vil give oplysninger om analysemetodens anvendelighed og repeterbarhed og om, hvor homogent prøvestoffet er fordelt. Ved den efterfølgende dataanalyse anvendes normalt den målte initiale 14C-aktivitet eller prøvestofkoncentration i stedet for den nominelle koncentration, så der tages højde for sorptionstab og doseringsfejl. For 14C-mærket prøvestof fås genfindingsgraden ved forsøgets slutning af en massebalance (jf. sidste afsnit i punkt 1.8.9.4). Ideelt skal massebalancen for radioaktivt mærket stof ligge mellem 90 % og 110 %, mens der for umærkede prøvestoffer bør opnås en analysenøjagtighed svarende til en genfinding af initialmængden på mellem 70 % og 110 %. De nævnte områder må opfattes som mål, ikke som acceptkriterier for forsøget. Hvis det ønskes, kan analysenøjagtigheden for prøvestoffet bestemmes ved en lavere koncentration end initialkoncentrationen samt for de vigtigste omdannelsesprodukter.
1.7.2. Analysemetodens repeterbarhed og følsomhed
Repeterbarheden af analysemetoden (herunder udvindingsgraden for den indledende ekstraktion) ved kvantitativ bestemmelse af prøvestof og, hvis relevant, omdannelsesprodukter kontrolleres ved fem replikate analyser på hver enkelt ekstrakt af overfladevand.
Analysemetodens detektionsgrænse for prøvestoffet og for omdannelsesprodukterne skal så vidt muligt udgøre mindst 1 % af den initiale mængde, der tilføres prøvesystemet. Kvantificeringsgrænsen skal være højst 10 % af den tilsatte koncentration. Til kemisk analyse af mange organiske stoffer og omdannelsesprodukter heraf må prøvestoffet ofte tilsættes i forholdsvis høj koncentration, dvs. > 100 μg/l.
1.8. BESKRIVELSE AF PRØVEMETODEN
1.8.1. Udstyr
Forsøget kan udføres i koniske eller cylindriske kolber af passende kapacitet (f.eks. 0,5 eller 1,0 liter), lukket med silicone- eller gummiprop, eller i serumflasker med CO2-tæt låg (f.eks. med butylgummi-septum). En anden mulighed er at udføre forsøget med flere kolber og høste hele kolbens indhold, mindst som dobbeltprøver, ved hvert prøvetagningstidspunkt (jf. sidste afsnit i punkt 1.8.9.1). For ikke-flygtige prøvestoffer, der ikke er radioaktivt mærkede, kræves ikke gastætte propper eller låg. Velegnede er løse vatpropper, der forhindrer kontaminering fra luften (jf. andet afsnit i punkt 1.8.9.1). Svagt flygtige kemiske stoffer bør testes i et system af biometertypen med let omrøring af vandoverfladen. For at sikre mod bakteriel kontaminering kan beholderne valgfrit steriliseres ved opvarmning eller autoklavering inden brug. Desuden anvendes følgende standardlaboratorieudstyr:
|
— |
rystebord eller magnetomrørere til konstant omrøring af prøvekolberne |
|
— |
centrifuge |
|
— |
pH-meter |
|
— |
turbidimeter til nephelometrisk turbiditetsmåling |
|
— |
ovn eller mikroovn til bestemmelse af tør vægt |
|
— |
apparatur til membranfiltrering |
|
— |
autoklave eller ovn til varmesterilisering af glasapparatur |
|
— |
udstyr til håndtering af 14C-mærkede stoffer |
|
— |
udstyr til kvantitativ bestemmelse af 14C-aktivitet i prøver fra CO2-opsamlingsvæsker og, om nødvendigt, fra sedimentprøver |
|
— |
analyseudstyr til bestemmelse af prøvestof (og referencestof), hvis der anvendes specifik kemisk analyse (f.eks. gaskromatograf eller højtryksvæskekromatograf). |
1.8.2. Stamopløsninger af prøvestoffet
Til fremstilling af stamopløsningerne af prøve- og referencestof skal anvendes deioniseret vand (jf. første afsnit i punkt 1.8.7). Det deioniserede vand skal være frit for stoffer, der kan være toksiske for mikroorganismer, og indholdet af opløst organisk kulstof (dissolved organic carbon (DOC)) må ikke være over 1 mg/l (6).
1.8.3. Indsamling og transport af overfladevand
Prøvetagningsstedet, hvor overfladevandet indsamles, skal vælges i henhold til formålet med forsøget i den givne situation. Ved valg af prøvetagningssted må der tages hensyn til oplysninger om eventuel tidligere udledning fra landbrug, industri eller husholdninger. Hvis det akvatiske miljø vides at have været forurenet med prøvestoffet eller andre stoffer med analog struktur inden for de foregående fire år, bør det ikke anvendes til indsamling af vandprøver, medmindre undersøgeren udtrykkelig har ønsket at undersøge nedbrydningshastigheden på tidligere eksponerede steder. Vandets pH og temperatur skal måles på indsamlingsstedet. Desuden skal indsamlingsdybden og vandets udseende (f.eks. farve og turbiditet) noteres (jf. punkt 3). Oxygenkoncentrationen og/eller redoxpotentialet i vand og i sedimentets overfladelag skal måles for at påvise, at der er aerobe betingelser, medmindre dette er åbenbart, bedømt ud fra prøvens udseende og tidligere erfaringer med det pågældende sted. Overfladevandet skal transporteres i en omhyggeligt rengjort beholder. Under transporten må prøvens temperatur ikke være nævneværdigt højere end forsøgstemperaturen. Ved længere transporttid end 2-3 timer anbefales afkøling til 4 °C. Vandprøven må ikke fryses.
1.8.4. Opbevaring og klargøring af overfladevand
Forsøget bør fortrinsvis igangsættes inden for ét døgn efter prøveindsamlingen. Er opbevaring af vandet nødvendig, bør dette begrænses til kortest mulig tid og må i intet tilfælde overstige fire uger. Vandprøven skal opbevares ved 4 °C med beluftning, indtil den skal bruges. Før brug skal grove partikler fjernes, f.eks. ved filtrering med nylonfilter med maskevidde ca. 100 μm, med et groft papirfilter eller ved bundfældning.
1.8.5. Klargøring af vand modificeret med sediment (valgfrit)
Anvendes opslæmmet sediment, tilsættes sedimentet til kolberne indeholdende naturligt vand (som er filtreret for at fjerne grove partikler som beskrevet i punkt 1.8.4), så der dannes en opslæmning med en faststofkoncentration mellem 0,01 og 1 g/l. Overfladesedimentet skal være fra samme indsamlingssted som vandprøven. Afhængigt af det pågældende akvatiske miljø kan overfladesedimentet enten karakteriseres som sediment med højt organisk kulstofindhold (2,5-7,5 %) og fin tekstur eller med lavt organisk kulstofindhold (0,5-2,5 %) og grov tekstur (3). Overfladesedimentet kan klargøres således: Ved hjælp af et transparent plastrør udtages adskillige sedimentkerner, de øverste aerobe lag skæres af (fra overfladen til en dybde af maks. 5 mm) straks efter prøvetagningen, hvorefter de hældes sammen. Den resulterende sedimentprøve transporteres i en beholder med stort luft-headspace for at opretholde aerobe betingelser (prøven afkøles til 4 °C, hvis transporttiden er over 2-3 timer). Sedimentprøven opslæmmes i prøvevandet i forholdet 1:10 og opbevares ved 4 °C med beluftning, indtil den skal bruges. Opbevaringstiden for sedimentet bør begrænses mest muligt og må i intet tilfælde overstige fire uger.
1.8.6. Semikontinuert metode (valgfri)
Det kan være nødvendigt med langvarig inkubering (flere måneder), hvis der optræder en lang lag-fase, før nævneværdig nedbrydning af prøvestoffet kan måles. Vides dette i forvejen fra tidligere forsøg med stoffet, kan forsøget indledes med den semikontinuerte metode, hvor en del af prøvevandet eller -opslæmningen udskiftes med mellemrum (jf. tillæg 2). I stedet kan det normale batchforsøg ændres til et semikontinuert forsøg, hvis der ikke er sket nogen nedbrydning af prøvestoffet efter ca. 60 dages testning med batchmetoden (jf. andet afsnit i punkt 1.8.8.3).
1.8.7. Tilsætning af prøvestoffet (eller referencestoffet)
For stoffer med stor vandopløselighed (> 1 mg/l) og lav flygtighed (Henrys konstant < 1 Pa·m3/mol eller < 10–5 atm·m3/mol) kan der fremstilles en stamopløsning i deioniseret vand (jf. punkt 1.8.2); en passende mængde stamopløsning tilsættes prøvebeholderne, så den ønskede koncentration opnås. Volumenet af den tilsatte stamopløsning bør holdes så lavt, som det er praktisk muligt (< 10 % af volumenet af den endelige opløsning, om muligt). En anden mulighed er at bruge et større volumen prøvevand til at opløse prøvestoffet, hvilket kan betragtes som et alternativ til brug af organiske opløsningsmidler.
Hvis det ikke kan undgås, må stamopløsninger af ikke-flygtige stoffer med ringe vandopløselighed fremstilles ved hjælp af et flygtigt organisk opløsningsmiddel, men opløsningsmidlet må ikke tilsættes systemet i en mængde på over 1 % v/v og må ikke have negativ indvirkning på den mikrobielle aktivitet. Opløsningsmidlet må ikke påvirke prøvestoffets stabilitet i vand. Opløsningsmidlet skal fjernes, så der kun er en yderst ringe mængde tilbage, som ikke nævneværdigt øger DOC-indholdet i prøvevandet eller -suspensionen. Dette skal efterprøves ved stofspecifik analyse eller om muligt ved analyse for DOC (6). Det skal tilstræbes at begrænse den overførte mængde opløsningsmiddel til det absolut nødvendige og sikre, at den tilsatte mængde prøvestof er opløselig i det endelige volumen prøvevand. Tilsætning af prøvestoffet til prøvebeholderne kan ske ved hjælp af andre teknikker som beskrevet i (7) og (8). Når der bruges et organisk opløsningsmiddel til tilsætning af prøvestoffet, skal der køres opløsningsmiddelkontrolprøver, som indeholder prøvevandet (uden tilsætning) samt prøvevand tilsat referencestof, og som behandles på samme måde som aktive prøvebeholdere modificeret med prøvestof i stabiliserende opløsningsmiddel. Formålet med opløsningsmiddelkontrolprøverne er at undersøge eventuel negativ indvirkning af opløsningsmidlet på den mikrobielle population, således som dette kan konstateres ved nedbrydningen af referencestoffet.
1.8.8. Forsøgsbetingelser
1.8.8.1. Forsøgstemperatur
Inkubering skal enten ske i mørke (hvilket bør foretrækkes) eller i diffust lys ved kontrolleret temperatur (± 2 °C), som kan være felttemperaturen eller en standardtemperatur på 20-25 °C. Felttemperaturen kan enten være prøvens faktiske temperatur på prøvetagningstidspunktet eller en gennemsnitlig felttemperatur på prøvetagningsstedet.
1.8.8.2. Omrøring
Der skal sørges for konstant omrystning eller omrøring, så partikler og mikroorganismer holdes opslæmmet. Omrøringen fremmer desuden overførslen af oxygen fra prøvebeholderens headspace til væsken, så aerobe betingelser kan opretholdes. Kolberne anbringes i rystebord (ca. 100 omdrejninger/min.) eller forsynes med magnetomrører. Omrystning eller omrøring skal være kontinuerlig. Omrystning eller omrøring skal dog være så svag som muligt og samtidig opretholde en homogen opslæmning.
1.8.8.3. Forsøgets varighed
Forsøget bør normalt ikke strække sig over mere end 60 døgn, medmindre den semikontinuerte metode med periodisk udskiftning af prøvesuspensionen anvendes (jf. punkt 1.8.6 og tillæg 2). Er nedbrydningen af prøvestoffet begyndt i løbet af de første 60 dage, kan batchforsøget dog forlænges til maksimalt 90 dage. Nedbrydningen overvåges med passende tidsintervaller ved bestemmelse af 14C-residualaktivitet eller den udviklede 14CO2 (jf. punkt 1.8.9.4) og/eller ved kemisk analyse (punkt 1.8.9.5). Inkubationstiden skal være tilstrækkeligt lang til, at nedbrydningsprocessen kan bedømmes. Nedbrydningsgraden skal helst være over 50 %. For langsomt nedbrydelige stoffer skal nedbrydningsgraden være tilstrækkelig (normalt mere end 20 % nedbrydning) til, at nedbrydningshastighedskonstanten kan bestemmes.
Testsystemets pH og oxygenkoncentration skal måles periodisk, medmindre erfaringer fra tilsvarende tidligere forsøg med vand- og sedimentprøver indsamlet på samme sted gør sådanne målinger unødvendige. Under visse betingelser kan metaboliseringen af det primære substrat ved langt større koncentration i vand eller sediment muligvis medføre CO2-udvikling og oxygenforbrug i et omfang, der ændrer forsøgsbetingelserne væsentligt.
1.8.9. Fremgangsmåde
1.8.9.1. Klargøring af kolber til pelagisk forsøg
Overfør en passende mængde af prøvevandet til prøvekolberne, højst en tredjedel af kolbens volumen og mindst ca. 100 ml. Også hvis der anvendes flere kolber (for at kunne høste en hel kolbe ved hvert prøvetagningstidspunkt), vil den mest hensigtsmæssige mængde prøvevand være ca. 100 ml, da små prøvevolumener kan påvirke lag-fasens varighed. Prøvestoffet tilsættes fra en stamopløsning som beskrevet i punkt 1.8.2 og 1.8.7. Der skal anvendes mindst to forskellige prøvestofkoncentrationer, der afviger indbyrdes med en faktor 5 til 10, for at nedbrydningskinetikken kan fastlægges og nedbrydningshastighedskonstanten beregnes. Begge de valgte koncentrationer skal være under 100 µg/l og helst i området < 1-10 µg/l.
Kolberne lukkes med propper eller låg, der er uigennemtrængelige for luft og CO2. Til ikke–14C-mærkede, ikke-flygtige prøvestoffer kan anvendes løse vatpropper, der beskytter mod kontaminering fra luften (jf. punkt 1.8.1), forudsat at alle vigtige nedbrydningsprodukter vides at være ikke-flygtige, og at der anvendes indirekte CO2-bestemmelse (se tillæg 3).
Kolberne inkuberes ved den valgte temperatur (se punkt 1.8.8.1). Prøver til kemisk analyse eller 14C-måling udtages ved forsøgets begyndelse (dvs. før bionedbrydningen begynder, se punkt 1.7.1) og derefter med passende tidsintervaller i løbet af forsøget. Prøverne kan udtages som delprøver (f.eks. 5 ml aliquoter) fra hver replikat, eller ved at høste en hel kolbe på hvert prøvetagningstidspunkt. Mineraliseringen af prøvestoffet kan bestemmes enten indirekte eller direkte (se tillæg 3). Til pålidelig bestemmelse af hastighedskonstanten kræves sædvanligvis mindst fem prøvetagningspunkter i nedbrydningsfasen (dvs. efter at lag-fasen er slut), medmindre det for hurtigt nedbrydelige stoffer kan godtgøres, at tre prøvetagningspunkter er tilstrækkeligt. For ikke hurtigt nedbrydelige stoffer kan der let foretages flere målinger i nedbrydningsfasen, og der skal i så fald anvendes flere datapunkter til bestemmelse af k. En fast tidsplan for prøvetagningen kan ikke angives, da bionedbrydningshastigheden varierer. Ved langsom nedbrydning anbefales dog ugentlig prøveudtagning. Er prøvestoffet hurtigt nedbrydeligt, udtages prøver dagligt de første tre dage, derefter hver anden eller hver tredje dag. Under visse omstændigheder, således ved stoffer, der hydrolyseres meget hurtigt, kan det være nødvendigt at tage prøver en gang i timen. Inden forsøget gennemføres, anbefales det at udføre et indledende forsøg til bestemmelse af passende prøvetagningsintervaller. Hvis der skal være prøver til rådighed til nærmere specifik analyse, tilrådes det at tage ekstra prøver og derefter vælge dem, der skal analyseres, ved forsøgets slutning i henhold til en bagudrettet strategi, dvs. de sidste prøver analyseres først (se vejledningen i andet afsnit i punkt 1.8.9.5 vedrørende prøvernes stabilitet ved opbevaring).
1.8.9.2. Antal kolber og prøver
Kør et tilstrækkeligt antal prøvekolber til, at der er:
|
— |
prøvekolber: mindst to kolber for hver prøvestofkoncentration (helst mindst tre), dog flere kolber for hver koncentration hvis der høstes hele prøvekolber (betegnet FT) ved hvert prøvetagningstidspunkt |
|
— |
prøvekolber til beregning af massebalance: mindst to kolber (betegnet FM) for hver prøvekoncentration |
|
— |
blindkontrol, intet prøvestof: mindst én blindprøvekolbe (betegnet FB), kun indeholdende prøvevand |
|
— |
referencekontrol: to kolber (betegnet FC) med referencestof (f.eks. anilin eller natriumbenzoat i en koncentration af 10 µg/l). Formålet med referencekontrollen er at bekræfte et minimum af mikrobiel aktivitet. Hvis det er hensigtsmæssigt, kan der anvendes et radioaktivt mærket referencestof, også når nedbrydningen af prøvestoffet overvåges ved kemiske analyser |
|
— |
steril kontrol: en eller to kolber (betegnet FS) indeholdende steriliseret prøvevand, til undersøgelse af eventuel abiotisk nedbrydning eller anden ikke-biologisk bortfjernelse af prøvestoffet. Den biologiske aktivitet kan enten standses ved autoklavering (121 °C; 20 min.) af prøvevandet, ved tilsætning af et giftstof (f.eks. natriumazid (NaN3) i en koncentration af 10-20 g/l, kviksølvchlorid (HgCl2) i en koncentration af 100 mg/l eller formalin i en koncentration af 100 mg/l) eller ved gammabestråling. Anvendes HgCl2, skal dette bortskaffes som giftigt affald. For vand med sediment tilsat i store mængder er det ikke let at opnå sterile forhold; i så fald anbefales gentagne autoklaveringer (f.eks. tre gange). Der må tages højde for, at autoklaveringen kan ændre sedimentets sorptionsegenskaber |
|
— |
opløsningsmiddelkontroller indeholdende prøvevand og prøvevand med referencestof: dobbelt sæt kolber behandlet med samme mængde opløsningsmiddel og efter samme fremgangsmåde som ved tilsætning af prøvestoffet. Formålet hermed er at afsløre eventuelle skadelige virkninger af opløsningsmidlet ved bestemmelse af nedbrydningen af referencestoffet. |
Ved forsøgets tilrettelæggelse må forsøgslederen veje betydningen af et øget antal forsøg op mod et øget antal prøvetagningstidspunkter. Det nøjagtige antal kolber, der er nødvendigt, afhænger af den anvendte metode til bestemmelse af nedbrydningen (se tredje afsnit i punkt 1.8.9.1, punkt 1.8.9.4 og tillæg 3).
Der skal udtages to delprøver (f.eks. på 5 ml aliquoter) fra hver prøvekolbe på hvert prøvetagningstidspunkt. Anvendes mange kolber for at kunne høste hele kolber, skal der høstes mindst to kolber ved hvert prøvetagningstidspunkt (se første afsnit i punkt 1.8.9.1).
1.8.9.3. Klargøring af kolber til forsøg med opslæmmet sediment (valgfrit)
Der tilsættes det nødvendige volumen prøvevand og, i givet fald, sediment, til prøvekarrene (se punkt 1.8.5). Klargøring af kolber til forsøg med opslæmmet sediment foretages som ved det pelagiske forsøg (se punkt 1.8.9.1 og 1.8.9.2). Der bør fortrinsvis anvendes serumflasker eller kolber af tilsvarende form. De lukkede kolber anbringes vandret på rystebord. Åbne kolber til ikke–14C-mærkede, ikke-flygtige stoffer skal selvfølgelig anbringes stående; i så fald anbefales brug af magnetomrører og magneter overtrukket med glas. Om nødvendigt beluftes kolberne for at opretholde aerobe betingelser.
1.8.9.4. Radiokemiske bestemmelser
Den udviklede 14CO2 bestemmes indirekte og direkte (se tillæg 3). 14CO2 bestemmes indirekte ved forskellen mellem den initiale 14C-aktivitet i prøvevand eller -opslæmning og den totale residualaktivitet på prøvetagningstidspunktet, målt efter forudgående forsuring af prøven til pH 2-3 og fjernelse af CO2. Derved er uorganisk kulstof fjernet, og den målte resterende aktivitet kommer fra organisk materiale. Indirekte 14CO2-betemmelse bør ikke benyttes, hvis der dannes vigtige flygtige omdannelsesprodukter ved omdannelse af prøvestoffet (se tillæg 3). Om muligt bør 14CO2-udviklingen måles direkte (se tillæg 3) i mindst én prøvekolbe ved hvert prøvetagningstidspunkt. Derved kan både massebalance og bionedbrydning kontrolleres, men dette kan kun lade sig gøre ved forsøg i lukkede kolber.
Måles den udviklede 14CO2 direkte under forsøget, skal der ved forsøgets start opsættes ekstra kolber hertil. Direkte 14CO2-bestemmelse anbefales, hvis der dannes vigtige omdannelsesprodukter ved prøvestoffets omdannelse. Ved hvert målepunkt skal de ekstra flasker forsures til pH 2-3, og 14CO2 opsamles i en intern eller ekstern absorber (se tillæg 3).
I stedet kan koncentrationen af 14C-mærket prøvestof og vigtige omdannelsesprodukter bestemmes ved radiokromatografi (f.eks. tyndtlagskromatografi, RAD-TLC) eller ved HPLC med radiokemisk detektion.
Det kan frit vælges, om man vil bestemme fasefordelingen af den resterende radioaktivitet (se tillæg 1) og resterende prøvestof og omdannelsesprodukter.
Ved forsøgets slutning bestemmes massebalancen ved direkte 14CO2-måling med separate prøvekolber, fra hvilke der ikke er udtaget nogen prøver under forsøget (se tillæg 3).
1.8.9.5. Specifik kemisk analyse
Er en følsom specifik analysemetode til rådighed, kan den primære bionedbrydning bestemmes ved måling af den totale residualkoncentration af prøvestoffet i stedet for ved brug af radioaktiv mærkning. Anvendes et radioaktivt mærket prøvestof (til måling af total mineralisering), kan der ved sideløbende specifikke kemiske analyser fås værdifulde supplerende oplysninger og efterprøvning af metoden. Specifik kemisk analyse kan desuden anvendes til bestemmelse af omdannelsesprodukter, der dannes ved nedbrydning af prøvestoffet; dette anbefales for stoffer, der mineraliseres med en halveringstid på over 60 dage. For hvert prøvetagningstidspunkt måles og rapporteres koncentrationen af prøvestof og omdannelsesprodukter (som koncentration og som procent af tilført mængde). Generelt kræves et omdannelsesprodukt identificeret, hvis det på noget prøvetagningstidspunkt forefindes i en koncentration ≥ 10 % af den tilførte koncentration, medmindre andet med rimelighed kan begrundes. Ligeledes må det overvejes at identificere omdannelsesprodukter, hvis koncentration stiger konstant under forsøget, også selv om koncentrationen ikke overstiger den nævnte grænse, da dette kan være et tegn på, at de persisterer. Hvis prøvestoffet kan tænkes at være genstand for hurtig abiotisk omdannelses (f.eks. hydrolyse), må det overvejes at analysere for omdannelsesprodukterne i sterile kontroller. Nødvendigheden af kvantificering og identifikation af omdannelsesprodukter må vurderes konkret i det enkelte tilfælde, og begrundelser gives i rapporten. Ekstraktion med organisk opløsningsmiddel skal anvendes i henhold til anvisningerne i den pågældende analysemetode.
Alle prøver skal opbevares ved 2 til 4 °C i lufttæt emballage, hvis analysen foretages inden for 24 timer (hvilket må foretrækkes). Ved længere opbevaring skal prøverne fryses ved en temperatur under –18 °C eller konserveres kemisk. Forsuring kan ikke anbefales som konserveringsmetode, da forsurede prøver kan være ustabile. Hvis prøverne ikke analyseres inden for 24 timer, men skal opbevares længere, skal der udføres et forsøg, der godtgør stabiliteten af de betragtede stoffer ved opbevaring under –18 °C eller i konserveret tilstand. Omfatter analysemetoden enten ekstraktion med opløsningsmiddel eller fastfase-ekstraktion (SPE), skal ekstraktionen finde sted straks efter prøvetagning eller efter højst 24 timers opbevaring af prøven i nedkølet tilstand.
Afhængigt af analysemetodens følsomhed kan større prøvevolumener end angivet i punkt 1.8.1 være nødvendige. Forsøget kan let udføres med et prøvevolumen på én liter i kolber med et volumen på 2-3 liter, hvorved der kan udtages prøver på ca. 100 ml.
2. DATA OG RAPPORTERING
2.1. BEHANDLING AF RESULTATER
2.1.1. Grafisk fremstilling af data
Alle prøvetagningstidspunkter afrundes til hele timer (medmindre stoffet nedbrydes væsentligt inden for minutter til timer), men ikke til hele døgn. Der optegnes en kurve over prøvestoffets beregnede residualaktivitet (for 14C-mærkede prøvestoffer) eller residualkoncentration (for umærkede stoffer) som funktion af tiden i både lineær og semilogaritmisk afbildning (se figur 1a og 1b). Har nedbrydning fundet sted, sammenholdes resultaterne for kolber FT med resultaterne for kolber FS. Hvis der er mindre end 10 % forskel mellem gennemsnitsresultaterne for kolberne med prøvestof (FT) og de sterile kolber (FS), kan den iagttagne nedbrydning antages at være overvejende abiotisk. Hvis nedbrydningen i kolberne FS er mindre, kan disse tal benyttes til at korrigere tallene for kolberne FT (ved fratrækning) for at beregne omfanget af bionedbrydningen. Når der foretages valgfrie analyser for vigtige nedbrydningsprodukter, skal der forelægges grafiske kurver over deres dannelse og aftagen foruden en kurve over faldet i prøvestofindholdet.
Lag-fasens varighed tL beregnes af nedbrydningskurven (semilogaritmisk afbildning) ved at ekstrapolere kurvens lineære del til et punkt svarende til en nedbrydning lig nul eller i stedet ved at bestemme tiden svarende til en nedbrydning på ca. 10 % (se figur 1a og 1b). Af den semilogaritmiske afbildning bestemmes første ordens hastighedskonstanten k og middelfejlen på denne ved lineær regression på kurven over ln (14C-residualaktivitet eller prøvestofkoncentration) som funktion af tiden. Navnlig for 14C-målinger må der kun anvendes data hørende til kurvens første lineære del, der følger efter lag-fasens slutning, og der må gives præference til nogle få udvalgte, repræsentative data frem for et større antal mere usikre data. Usikkerheden indbefatter her de naturlige fejl i den anbefalede direkte brug af målt 14C residualaktivitet (se nedenfor). Hvis nedbrydningen følger et bifasisk mønster, kan det undertiden være relevant at beregne to forskellige hastighedskonstanter. Hertil defineres to forskellige faser af nedbrydningskurven. Hastighedskonstanten k og halveringstiden t½ = ln2/k beregnes for hver enkelt af de replikate kolber, når der er udtaget delprøver fra samme kolbe, eller ved hjælp af gennemsnitsværdier, når der høstes hele kolber på hvert prøvetagningstidspunkt (se sidste afsnit i punkt 1.8.9.2). Ved førstnævnte fremgangsmåde skal hastighedskonstant og halveringstid rapporteres for hver enkelt replikat kolbe og som gennemsnitsværdi med middelfejl. Er der anvendt høje koncentrationer af prøvestoffet, kan nedbrydningskurven afvige betydeligt fra den rette linje (i semilogaritmisk afbildning), og der gælder ikke nødvendigvis første ordens kinetik. Det har derfor ingen mening at fastlægge en halveringstid. For et begrænset dataområde kan man dog anvende pseudo første ordens kinetik og beregne halveringstiden DT50 for nedbrydningen (den tid, der medgår til 50 % nedbrydning). Man må dog være opmærksom på, at nedbrydningens tidsforløb uden for det valgte dataområde ikke kan forudsiges ved hjælp af DT50, der blot er en deskriptor for et givet datasæt. Der findes let tilgængelige analyseredskaber, som letter statistiske beregninger og kurvefitting, og brug af sådan software anbefales.
Foretages der specifikke kemiske analyser, beregnes hastighedskonstanter og halveringstider for primær nedbrydning som beskrevet ovenfor for total mineralisering. Hvis den primære nedbrydning udgør den hastighedsbegrænsende proces, kan der undertiden anvendes datapunkter fra hele nedbrydningsforløbet. Dette skyldes, at målingerne er direkte, i modsætning til målingerne af 14C-aktivitet.
Anvendes 14C-mærkede stoffer, skal der opstilles en massebalance i procent af den tilsatte startkoncentration, i det mindste ved forsøgets slutning.
2.1.2. Residualaktivitet
Ved bionedbrydning af den 14C-mærkede del af et organisk stof omdannes størstedelen af 14C til 14CO2, mens en anden del udnyttes til vækst af biomasse og/eller syntese af ekstracellulære metabolitter. »Fuldstændig« biologisk nedbrydning af et stof medfører derfor ikke 100 % omdannelse af kulstofindholdet til 14CO2. Den 14C, der indbygges i produkter dannet ved biosyntese, frigives efterfølgende langsomt som 14CO2 ved »sekundær mineralisering«. Kurver, der viser residualaktiviteten af organisk 14C (målt efter fjernelse af CO2) eller den producerede 14CO2 som funktion af tiden, har derfor en »hale«, efter at nedbrydningen er løbet til ende. Dette komplicerer den kinetiske fortolkning af data, og til dette formål bør kun den initiale del af kurven (efter lag-fasens afslutning, og inden ca. 50 % nedbrydning er nået) normalt anvendes til beregning af en hastighedskonstant for nedbrydningen. Hvis prøvestoffet nedbrydes, er den totale organiske 14C-residualaktivitet altid større end den 14C-aktivitet, der skyldes tilbageværende intakt prøvestof. Hvis prøvestoffet nedbrydes ved en første ordens reaktion, og en konstant brøkdel α mineraliseres til CO2, vil den initiale hældning af 14C-depletionskurven (total organisk 14C som funktion af tiden) være α gange hældningen af den tilsvarende kurve over koncentrationen af prøvestoffet (eller, mere præcist, den del af prøvestoffet, der er mærket med 14C). Hvis målingerne af den totale organiske 14C-aktivitet bruges uden korrektion, vil hastighedskonstanten for nedbrydningen derfor være lavt regnet. I litteraturen er beskrevet metoder til bestemmelse af prøvestofkoncentrationen ud fra den målte radiokemiske aktivitet ved brug af nogle forenklede forudsætninger (2)(9)(10)(11). Sådanne metoder kan lettest anvendes på hurtigt nedbrydelige stoffer.
2.2. FORTOLKNING AF RESULTATER
Hvis k findes at være uafhængig af den tilsatte koncentration (dvs. den beregnede k-værdi er omtrent ens ved forskellige prøvestofkoncentrationer), kan første ordens hastighedskonstanten antages at være repræsentativ for de anvendte prøvebetingelser, dvs. prøvestoffet, vandprøven og forsøgstemperaturen. I hvilket omfang resultaterne kan generaliseres eller ekstrapoleres til andre systemer, kræver det ekspertviden at vurdere. Anvendes en høj prøvestofkoncentration, så nedbrydningen ikke følger første ordens kinetik, kan data ikke benyttes til direkte beregning af en første ordens hastighedskonstant eller en tilsvarende halveringstid. Dog vil data fra forsøg med høj prøvestofkoncentration stadig være nyttige til beregning af den totale mineraliseringsgrad og/eller detektion og kvantificering af omdannelsesprodukter.
Hvis man kender tabsprocenten fra andre tabsprocesser end bionedbrydning (f.eks. hydrolyse eller fordampning), kan de trækkes fra de nettotab, der iagttages under forsøget, hvorved man kan få en tilnærmet bestemmelse af bionedbrydningshastigheden. Data for hydrolyse vil kunne fås fra f.eks. den sterile kontrol eller fra sideløbende forsøg med højere prøvestofkoncentration.
Indirekte og direkte bestemmelse af 14CO2 (punkt 1.8.9.4 og tillæg 3) kan kun bruges til at bestemme graden af prøvestoffets mineralisering til CO2. Koncentrationen af 14C-mærket prøvestof og dannelsen af vigtige omdannelsesprodukter (tredje afsnit i punkt 1.8.9.4) kan analyseres ved radiokromatografi (RAD-TLC) eller HPLC. For at halveringstiden kan bestemmes direkte, må der ikke være vigtige omdannelsesprodukter (defineret som ≥ 10 % af den tilsatte mængde prøvestof) til stede. Hvis der er vigtige omdannelsesprodukter, som her defineret, til stede, er en detaljeret vurdering af data nødvendig. En sådan vurdering kan bestå i yderligere undersøgelser og/eller identifikation af omdannelsesprodukterne (se første afsnit i punkt 1.8.9.5), medmindre disses skæbne med rimelig sikkerhed kan vurderes ud fra tidligere erfaringer (f.eks. oplysninger om nedbrydningsvej). Da den andel af prøvestoffets kulstofindhold, der omdannes til CO2, er forskellig (hovedsagelig afhængig af koncentrationen af prøvestoffet og øvrige tilgængelige substrater, prøvebetingelserne og det mikrobielle miljø), giver forsøget ikke umiddelbar mulighed for beregning af den endelige bionedbrydning som i et DOC die-away forsøg, men resultatet svarer til det, der fås af et respirometrisk forsøg. Mineraliseringsgraden vil således højst være lig med minimumsniveauet af fuldstændig bionedbrydning. For at få et mere fyldestgørende billede af den fuldstændige bionedbrydning (mineralisering og inkorporation i biomasse) bør analysen af fasefordelingen af 14C foretages ved forsøgets slutning (se tillæg 1).14C-indholdet i den samlede partikelmængde vil dels bestå af 14C indbygget i bakteriel biomasse, dels 14C sorberet til organiske partikler.
2.3. FORSØGETS VALIDITET
Hvis referencestoffet ikke nedbrydes inden for det forventede tidsrum (for anilin og natriumbenzoat sædvanligvis mindre end to uger), er forsøgets validitet tvivlsom og må nærmere efterprøves, eller forsøget må i stedet gentages med en ny vandprøve. I en ISO ringtest af metoden med deltagelse af syv laboratorier beliggende forskellige steder i Europa lå hastighedskonstanten for nedbrydning af anilin efter tilpasning mellem 0,3 og 1,7 dag–1 med et gennemsnit på 0,8 d–1 ved 20 oC og en middelfejl på ±0,4 d–1 (t½ = 0,9 dag). Typiske lag-tider var 1 til 7 dage. De undersøgte vande rapporteredes at have en bakteriel biomasse svarende til 103-104 kolonidannende enheder (CFU) pr. ml. Nedbrydningshastigheden i næringsstofrige mellemeuropæiske vande var større end i oligotrofe nordiske vande, hvilket kan skyldes forskelle i trofisk status eller tidligere eksponering for kemiske stoffer.
Den totale genfinding (ved massebalance) ved forsøgets slutning bør være mellem 90 % og 110 % for radioaktivt mærkede stoffer, mens den initiale genfinding ved forsøgets begyndelse bør være mellem 70 % og 110 % for umærkede stoffer. De angivne områder må imidlertid kun opfattes som mål og bør ikke anvendes som acceptkriterier for testen.
3. FORSØGSRAPPORT
Undersøgelsens art, dvs. pelagisk eller med opslæmmet sediment, skal klart angives i forsøgsrapporten, som desuden skal indeholde i det mindste følgende oplysninger:
For prøvestof og referencestof(fer):
|
— |
generisk navn, kemisk navn, (anbefalet IUPAC og/eller CAS navn), CAS-nummer, strukturformel (med angivelse af placeringen af 14C, når radioaktivt mærket stof anvendes) og relevante fysisk-kemiske egenskaber (se punkt 1.5 og 1.6) |
|
— |
kemisk navn, CAS-nummer, strukturformel (med angivelse af placeringen af 14C, når radioaktivt mærket stof anvendes) og relevante fysisk-kemiske egenskaber for stoffer anvendt som standarder til identifikation og kvantificering af omdannelsesprodukter |
|
— |
renhed (urenheder) af prøve- og referencestof |
|
— |
for mærkede stoffer, radiokemisk renhed og specifik aktivitet (når det er relevant). |
For overfladevand:
Der skal som minimum gives følgende oplysninger om den udtagne vandprøve:
|
— |
beliggenhed og beskrivelse af prøvetagningsstedet, herunder så vidt muligt oplysninger om tidligere forurening |
|
— |
dato og klokkeslæt for indsamling af prøven |
|
— |
næringsstofindhold (total N, ammonium, nitrit, nitrat, total P, opløst orthophosphat) |
|
— |
prøvetagningsdybde |
|
— |
prøvens udseende (f.eks. farve og turbiditet) |
|
— |
DOC og TOC |
|
— |
BOD |
|
— |
temperatur og pH på prøvetagningsstedet og -tidspunktet |
|
— |
oxygen- eller redoxpotentiale (kun nødvendigt, hvis der ikke tydeligt ses at være aerobe betingelser) |
|
— |
saltindhold eller ledeevne (for søvand og brakvand) |
|
— |
opslæmmet faststof (for uklare prøver) |
|
— |
eventuelle andre relevante oplysninger om prøvetagningsstedet på prøvetagningstidspunktet (f.eks. aktuelle eller historiske oplysninger om strømhastigheden i vandløb eller havstrømme, nærliggende større udledninger og udledningernes art, vejrforholdene i tiden forud for prøvetagningen) |
og, valgfrit:
|
— |
mikrobiel biomasse (f.eks. direkte tælling med acridinorange-farvning, eller kolonidannende enheder) |
|
— |
uorganisk kulstof |
|
— |
koncentration af chlorophyll-a som specifik bestemmelse af algebiomasse. |
Hvis forsøget udføres med opslæmmet sediment, skal der desuden gives følgende oplysninger om sedimentet:
|
— |
prøvetagningsdybde for sedimentet |
|
— |
sedimentets fremtræden (som f.eks. farvet, mudret, siltholdig eller sandholdig) |
|
— |
tekstur (f.eks. angivet som % groft sand, fint sand, silt og ler) |
|
— |
tør vægt af opslæmmet faststof i g/l, TOC-koncentration eller vægttab ved forbrænding som mål for indholdet af organisk stof |
|
— |
pH |
|
— |
oxygen- eller redoxpotentiale (kun nødvendigt, hvis der ikke tydeligt ses at være aerobe betingelser). |
Forsøgsbetingelser:
|
— |
forsinkelse mellem indsamling og anvendelse i laboratorieforsøget, opbevaring og forbehandling af prøven, dato for undersøgelserne |
|
— |
mængde prøvestof tilført, koncentration under forsøget og referencestof |
|
— |
tilsætningsmetode for prøvestoffet, herunder brug af eventuelt opløsningsmiddel |
|
— |
anvendt volumen overfladevand og sediment (hvis anvendt), og volumen udtaget til analyse ved hvert prøvetagningstidspunkt |
|
— |
beskrivelse af det anvendte prøvesystem. |
Hvis forsøget ikke udføres i mørke, oplysninger om betingelserne »diffus belysning«:
|
— |
oplysninger om den eller de anvendte metoder til sterilisering af kontrolprøver (f.eks. autoklaveringstemperatur og -tid samt antal autoklaveringer) |
|
— |
inkubationstemperatur |
|
— |
oplysninger om analyseteknik og metode(r) anvendt til radiokemiske målinger samt til kontrol af massebalance og målinger af fasefordeling (hvis sådanne er udført) |
|
— |
antal gentagelser. |
Resultater:
|
— |
genfindingsprocenter (se punkt 1.7.1) |
|
— |
reproducerbarhed og følsomhed af de anvendte analysemetoder, herunder detektionsgrænse (LOD) og kvantificeringsgrænse (LOQ) (se punkt 1.7.2) |
|
— |
alle målte data (herunder prøvetagningstidspunkter) og beregnede værdier i tabelform og nedbrydningskurver; for hver prøvekoncentration og for hver replikat kolbe angives den lineære korrelationskoefficient for kurvehældningen i logaritmisk afbildning, den beregnede lag-fase og en første ordens eller pseudo første ordens hastighedskonstant (hvis dette er muligt) samt den tilsvarende nedbrydningshalveringstid (t50) |
|
— |
relevante værdier angives som gennemsnit af de værdier, der er iagttaget i de enkelte replikater, f.eks. lag-fasens varighed, nedbrydningshastighedskonstanten og nedbrydningshalveringstiden (t50) |
|
— |
systemet rubriceres enten som ikke tilpasset eller tilpasset, bedømt ud fra nedbrydningskurvens udseende og ud fra den mulige indvirkning af prøvekoncentrationen |
|
— |
resultaterne af den endelige massebalancekontrol og resultaterne af eventuelle fasefordelingsmålinger |
|
— |
den andel af 14C, der er mineraliseret, og, når specifikke analyser anvendes, den endelige primære nedbrydningsgrad |
|
— |
identifikation, molær koncentration og procentdel tilsat stof og, når det er relevant, vigtige omdannelsesprodukter (se første afsnit i punkt 1.8.9.5) |
|
— |
formodet omdannelsesvej, når det er relevant |
|
— |
diskussion af resultater. |
4. LITTERATUR
|
1. |
OECD TG 309 (2004) Aerobic Mineralisation in surface water — Simulation Biodegradation Test. |
|
2. |
ISO/DIS 14592-1 (1999) Water quality — Evaluation of the aerobic biodegradability of organic compounds at low concentrations — Part 1: Shake flask batch test with surface water or surface water/sediment suspensions. |
|
3. |
Prøvemetode C.23. Aerob og anaerob omdannelse i jord. |
|
4. |
Prøvemetode C.24. Aerob og anaerob omdannelse i akvatiske sedimentsystemer. |
|
5. |
OECD (1993). Guidelines for the Testing of Chemicals. OECD, Paris. |
|
6. |
ISO 8245 (1999). Water quality — Guidelines on the determination of total organic carbon (TOC) and dissolved organic carbon (DOC). |
|
7. |
ISO 10634 (1995). Water quality — Guidance for the preparation and treatment of poorly water-soluble organic compounds for the subsequent evaluation of their biodegradability in an aqueous medium. |
|
8. |
OECD draft (2000). Guidance Document on aquatic toxicity testing of difficult substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment. No 22. |
|
9. |
Simkins, S. and Alexander, M. (1984). Models for mineralization kinetics with the variables of substrate concentration and population density. Appl. Environ. Microbiol.47, 394-401. |
|
10. |
Ingerslev, F. and N. Nyholm. (2000). Shake-flask test for determination of biodegradation rates of 14C-labeled chemicals at low concentrations in surface water systems. Ecotoxicol. Environ. Saf. 45, 274-283. |
|
11. |
ISO/CD 14592-1 (1999). Ring test report: Water Quality — Evaluation of the aerobic biodegradability of organic compounds at low concentrations part 1 — report of 1998/1999 ring-test. Shake flask batch test with surface water or surface water/sediment suspensions. |
Tillæg 1
Fasefordeling af 14C
Til kontrol af metoden skal rutinemålingerne af total organisk 14C-residualaktivitet (TOA) suppleres med massebalancemålinger, hvor den udviklede 14CO2 bestemmes direkte efter opsamling i absorber (se tillæg 3). Positiv 14CO2-dannelse er i sig selv et direkte tegn på bionedbrydning i modsætning til abiotisk nedbrydning eller andre tabsmekanismer som fordampning og sorption. Andre nyttige oplysninger til karakterisering af bionedbrydningsegenskaberne kan fås af målinger af fordelingen af TOA på opløst tilstand (opløst organisk 14C-aktivitet, DOA) og fast partikelform (POA) efter separation af partiklerne ved membranfiltrering eller centrifugering. POA består af prøvestof, der er sorberet på den mikrobielle biomasse og på andre partikler samt af kulstof fra prøvestoffet, der er anvendt til syntese af nyt cellemateriale og derved indbygget i den partikelformige biomassefraktion. Dannelsen af opløst organisk 14C kan bestemmes som DOA, når bionedbrydningen er slut (svarende til plateauet af kurven over nedbrydningen som funktion af tiden).
Fasefordelingen af residual–14C i udvalgte prøver bestemmes ved filtrering af prøven på et 0,22 μm eller 0,45 μm membranfilter af et materiale, der ikke adsorberer nævneværdige mængder prøvestof (polycarbonatfiltre kan være egnede). Er sorptionen af prøvestoffet på filteret for stor til at kunne ignoreres (dette skal verificeres inden forsøget), kan højhastighedscentrifugering (2 000 g; 10 min.) anvendes i stedet for filtrering.
Derefter behandles filtratet eller centrifugatet som beskrevet i tillæg 3 for ufiltrerede prøver. Membranfiltratet opløses i en egnet scintillationsvæske og tælles på sædvanlig måde, idet korrektion for quenching normalt kun foretages ved hjælp af kanalforholdet for ekstern standard eller behandling af prøverne med et blegemiddel. Er der anvendt centrifugering, skal den pellet, der er dannet af partikelfraktionen, genopslæmmes i 1-2 ml destilleret vand og overføres til et scintillationsglas. Derefter vaskes to gange med 1 ml destilleret vand, og vaskevandet overføres til scintillationsglasset. Opslæmningen kan om nødvendigt indlejres i en gel til væskescintillationstælling.
Tillæg 2
Semikontinuert metode
Vanskelige stoffer kan kræve inkubation i indtil flere måneder for at opnå tilstrækkelig nedbrydning. Forsøget bør normalt ikke vare over 60 dage, medmindre den normale vandprøves egenskaber opretholdes ved udskiftning af prøveopslæmningen. Er nedbrydningen af prøvestoffet begyndt inden for de første 60 dage, kan forsøget dog forlænges til maksimalt 90 dage, uden at forsøgsopslæmningen udskiftes.
Ved langvarig inkubation kan mangfoldigheden af det mikrobielle samfund blive nedsat som følge af forskellige tabsmekanismer og muligvis opbrugning af essentielle næringsstoffer og primære kulstofsubstrater i vandprøven. Derfor anbefales det at anvende en semikontinuert metode for at få tilfredsstillende bestemmelse af nedbrydningshastigheden for langsomt nedbrydelige stoffer. Den semikontinuerte metode bør anvendes fra forsøgets start, hvis tidligere erfaring viser, at der kræves en inkubationsperiode på tre måneder for at opnå 20 % nedbrydning af stoffet. En anden mulighed er, at det normale batchforsøg ændres til et semikontinuert forsøg, hvis der ikke er sket nogen nedbrydning af prøvestoffet, efter at forsøget har kørt i ca. 60 dage med batchmetoden. Den semikontinuerte udførelse af forsøget kan standses og forsøget fortsættes som et batchforsøg, når der er konstateret en væsentlig nedbrydning (f.eks. > 20 %).
I det semikontinuerte forsøg udskiftes ca. en tredjedel af forsøgsopslæmningens volumen hver anden uge med friskindsamlet vand, som er tilsat prøvestof svarende til initialkoncentrationen. Anvendes den valgfrie metode med opslæmmet sediment, skal udskiftningsvandet desuden tilsættes sediment svarende til initialkoncentration (mellem 0,01 og 1 g/l). Udføres forsøget med opslæmmet sediment, er det vigtigt, at systemet holdes fuldstændig opslæmmet, også mens vandet skiftes, og at der er samme opholdstid for faste stoffer og for vand, da man ellers ikke opnår den tilsigtede overensstemmelse med et homogent vandigt system uden fastliggende faser. Af disse grunde bør initialkoncentrationen af opslæmmet sediment helst være i den nedre del af det foreskrevne interval, når den semikontinuerte metode anvendes.
Den angivne tilsætning af prøvestof forudsætter, at den delvise udskiftning af prøveopslæmningen ikke medfører overskridelse af den initiale prøvestofkoncentration, for at man skal undgå den tilpasning, der ofte ses ved høje prøvestofkoncentrationer. Da metoden både indebærer re-inokulering og kompensation for opbrugte næringsstoffer og primære substrater, genetableres den oprindelige mikrobielle mangfoldighed, og forsøget kan i princippet forlænges ubegrænset. Når den semikontinuerte metode anvendes, at det vigtigt at bemærke, at residualkoncentrationen af prøvestoffet skal korrigeres for de mængder prøvestof, der tilsættes og fjernes ved hver udskiftningsgang. For stoffer, der kun udviser ringe sorption, kan man frit vælge, om man vil bruge totalkoncentrationen eller den opløste koncentration af prøvestoffet. Sorptionen er uden betydning (< 5 %) ved de foreskrevne betingelser (0,1-1 g faststof/l) for stoffer med log Kow < 3 (gælder for neutrale, lipofile forbindelser). Dette illustreres af følgende beregningseksempel. 0,1 g faststof /l svarer til ca. 10 mg kulstof pr. liter (ved en vægtbrøk kulstof, fC = 0,01). Idet det forudsættes, at
Log Kow (for prøvestoffet) = 3
Koc = 0,42 × Kow
Fordelingskoefficient, Kd = fC × Koc
Vil den opløste andel af totalkoncentrationen (C-water (Cw)/C-total (Ct) være:
Cw/Ct = 1/(1 + Kd × SS) = 1(1 + Koc × fC × SS) = 1/(1 + 0,42 × 103 × 0,01 × 0,1 × 10–3) = 0,999
Tillæg 3
Bestemmelse af 14CO2
Indirekte 14CO2-bestemmelse
Til rutinemæssige målinger er indirekte bestemmelse normalt den mindst tidskrævende og mest nøjagtige metode, når prøvestoffet er ikke-flygtigt og ikke omdannes til flygtige omdannelsesprodukter. De ufiltrerede prøver overføres simpelthen til scintillationsglas, f.eks. i en mængde af 5 ml. En passende initial aktivitet i prøverne er 5 000 dpm-10 000 dpm (80-170 Bq), og en mindste initial aktivitet er ca. 1 000 dpm. CO2-indholdet fjernes efter forsuring til pH 2-3 med 1-2 dråber koncentreret H3PO4 eller HCl. CO2 -fjernelse kan ske ved gennembobling med luft i ½-1 time. I stedet kan glassene omrystes kraftigt i 1-2 timer (f.eks. på en mikropladeryster) eller med svagere omrystning natten over. Effektiviteten af CO2-fjernelsen skal kontrolleres (ved forlængelse af beluftnings- eller omrystningsperioden). Der tilsættes derefter en scintillationsvæske egnet til tælling af vandige prøver, prøven homogeniseres i en omrører, og radioaktiviteten måles med væskescintillationstælling, idet man fratrækker den baggrundsaktivitet, der er fundet i blindprøverne (FB). Medmindre prøvevandet er stærkt farvet eller har et højt partikelindhold, vil prøverne normalt udvise ensartet quenching, og det vil være tilstrækkeligt at korrigere for quenching ved hjælp af en ekstern standard. Hvis prøvevandet er stærkt farvet, kan det være nødvendigt at tilsætte en intern standard til at korrigere for quenching. Er partikelkoncentrationen høj, kan det være umuligt at få en homogen opløsning eller gel, eller prøverne vil udvise store forskelle i quenching. I så fald kan man benytte den tællemetode, der er beskrevet nedenfor for prøveslam. Udføres forsøget med opslæmmet sediment, kan 14CO2 måles indirekte ved udtagning af en homogen 10 ml prøve af prøvevand/prøveopslæmning og adskillelse af faserne ved centrifugering med passende hastighed (f.eks. 40 000 m/s2 i 15 min.). Den vandige fase behandles derefter som ovenfor beskrevet. 14C-aktiviteten i partikelfasen (POA) bestemmes ved genopslæmning af sedimentet i et lille volumen destilleret vand, overførsel til scintillationsglas og tilsætning af scintillationsvæske, så der dannes en gel (særlige scintillationsvæsker fås hertil). Alt efter partiklernes beskaffenhed (f.eks. indholdet af organisk stof) kan det være en fordel at behandle prøven natten over med et vævsopløsende middel og derefter homogenisere den i en omrører, før scintillationsvæsken tilsættes. I stedet kan POA bestemmes ved forbrænding i overskud af oxygen ved hjælp af en oxidiser. Ved tællingen skal der altid medtages interne standarder, og det kan være nødvendigt at korrigere hver enkelt prøve for quenching ved tilsætning af intern standard.
Direkte 14CO2-bestemmelse
Hvis den udviklede 14CO2 måles direkte, skal det ske ved at opsætte ekstra kolber ved forsøgets begyndelse og høste prøvekolberne i hvert målepunkt ved forsuring af kolberne til pH 2-3 og indsamling af 14CO2 i en intern (anbragt i hver prøvekolbe ved forsøgets begyndelse) eller ekstern absorber. Som absorptionsmiddel kan enten anvendes alkali (f.eks. 1 N NaOH-opløsning eller en NaOH-pellet), ethanolamin eller en ethanolamin-baseret absorber, eller andre gængse absorbere. Til direkte måling af 14CO2 skal kolberne lukkes med septa f.eks. af butylgummi.
Figur 1a
Eksempel på aritmetisk kurve over data (residualaktiviteten som funktion af tiden)
Figur 1b
Eksempel på semilogaritmisk kurve over data (ln til residualaktiviteten som funktion af tiden)
BILAG VI
|
C.26. |
LEMNA SP. VÆKSTHÆMNINGSTEST |
1. METODE
Metoden er ækvivalent med OECD TG 221 (2006) (1). Der er opnået bred tilslutning blandt EU's myndigheder til, at Lemna-testen er et velegnet alternativ til en algetest for stærkt farvede stoffer (2)(3).
1.1. INDLEDNING
Formålet med nærværende testmetode er at vurdere stoffers toksicitet over for akvatiske ferskvandsplanter af slægten Lemna (andemad). Den er baseret på eksisterende retningslinjer (4)(5)(6)(7)(8)(9), men der er foretaget ændringer, som afspejler den senere tids forskning og drøftelser vedrørende en række vigtige punkter. Den foreslåede metode er blevet valideret ved en international ringtest (10).
I denne testmetode beskrives toksicitetstestning med Lemna gibba og Lemna minor, der begge har været genstand for omfattende undersøgelser og er omhandlet i ovennævnte standarder. Taksonomien for Lemna spp. er vanskelig, idet den kompliceres af, at der findes en bred vifte af fænotyper. Genetisk variation i responsen på giftstoffer hos Lemna kan forekomme, men de aktuelt foreliggende oplysninger om denne kilde til variation er utilstrækkelige som grundlag for at anbefale en bestemt klon til brug i nærværende testmetode. Det skal bemærkes, at testen ikke udføres axenisk, men nogle af testprocessens trin er foranstaltninger, der skal holde kontamineringen med andre organismer nede på et minimum.
Der gives en nærmere beskrivelse af testning med udskiftning af substratet (semistatisk forsøg og gennemstrømningsforsøg) og uden udskiftning (statisk). Afhængigt af målsætningen for testen og myndighedernes krav anbefales det at overveje brug af den semistatiske metode og gennemstrømningsmetoden f.eks. til stoffer, der hurtigt forlader opløsningen ved fordampning, fotonedbrydning, udfældning eller bionedbrydning. Nærmere anvisninger er givet i (11).
1.2. DEFINITIONER
Følgende definitioner og forkortelser anvendes i denne testmetode:
Biomasse: den tørre vægt af det levende materiale, som er til stede i en population. I denne test måles typisk surrogater for biomasse, f.eks. bladantal eller bladareal, og betegnelsen »biomasse« benyttes således også for sådanne surrogater.
Chlorose: gulning af bladvæv.
Klon: en organisme eller celle, der er opstået af en enkelt celle ved vegetativ formering. Individer fra samme klon er derfor genetisk identiske.
Koloni: en samling af moder- og datterblade (sædvanligvis 2 til 4), der sidder fast på hinanden. Betegnes undertiden en plante.
ECx : den koncentration af teststof opløst i testsubstratet, der medfører en væksthæmning på x % (f.eks. 50 %) af Lemna i løbet af en nærmere bestemt eksponeringsperiode (som udtrykkeligt skal angives, hvis den er forskellig fra den fulde eller normale testperiode). For klart at skelne mellem EC-værdier afledt af henholdsvis væksthastighed og udbytte anvendes betegnelsen »ErC« for væksthastighed (rate) og »EyC« for udbytte (yield), efterfulgt af den anvendte målevariabel, f.eks. »ErC (bladantal)«.
Gennemstrømningstest: en test, hvor testopløsningen udskiftes kontinuerligt.
Blad: en individuel/enkelt »bladlignende« struktur hos andemadplanten. Det er den mindste enhed, dvs. det mindste individ, der kan formere sig.
Opsvulmethed: blade, der fremtræder udbulede eller opsvulmede.
Vækst: en stigning i en målevariabel i løbet af testperioden, f.eks. bladantal, tør vægt, våd vægt eller bladareal.
Væksthastighed (gennemsnitlig specifik væksthastighed): den logaritmiske biomasseforøgelse i eksponeringsperioden.
Laveste koncentration med observeret effekt (Lowest Observed Effect Concentration, LOEC): den laveste testede koncentration, ved hvilken stoffet har vist statistisk signifikant (p < 0,05) vækstnedsættende virkning i forhold til kontrolprøverne inden for en given eksponeringsperiode. Alle testkoncentrationer over LOEC skal dog have en skadelig virkning mindst svarende til den, der iagttages ved LOEC. Kan disse to betingelser ikke opfyldes, skal der fyldestgørende redegøres for, hvordan den pågældende LOEC (og dermed NOEC) er valgt.
Målevariabler: enhver type variabel, der måles for at udtrykke endepunktet for en test ved hjælp af en eller flere forskellige responsvariabler. I denne metode er bladantal, bladareal, frisk vægt og tør vægt målevariabler.
Monokultur: en kultur med én planteart.
Nekrose: dødt (dvs. hvidt eller gennemblødt) bladvæv.
Nuleffekt-koncentration (No Observed Effect Concentration, NOEC): testkoncentrationen umiddelbart under LOEC.
Fænotype: de observerbare kendetegn for en organisme, som er bestemt af samspillet mellem dens gener og dens omgivelser.
Responsvariabler: variabler, som anvendes til vurdering af den toksiske virkning og er afledt af målte parametre, der beskriver biomassen ved forskellige beregningsmetoder. I nærværende metode er væksthastighed og udbytte responsvariabler, der er afledt af målevariabler som bladantal, bladareal, frisk vægt eller tør vægt.
Semistatisk test (udskiftningstest): en test, hvor testopløsningen udskiftes med bestemte intervaller i løbet af testen.
Statisk test: en testmetode, hvor testopløsningen ikke udskiftes under testen.
Endepunkt for en test: den generelle faktor, som teststoffet fremkalder en ændring af i forhold til kontrollen som mål for testen. I denne testmetode er endepunktet væksthæmning, som kan udtrykkes ved forskellige responsvariabler, der er baseret på en eller flere målevariabler.
Testsubstrat: det komplette, syntetiske næringssubstrat, hvorpå testplanterne vokser, når de eksponeres for teststoffet. Normalt er teststoffet opløst i testsubstratet.
Udbytte: værdien af en målevariabel for biomasse ved eksponeringsperiodens slutning minus værdien af samme variabel ved eksponeringsperiodens start.
1.3. TESTENS PRINCIP
Eksponentielt voksende plantekulturer af slægten Lemna dyrkes som monokultur ved forskellige koncentrationer af teststoffet i syv dage. Testens formål er at kvantificere stofrelaterede virkninger på den vegetative vækst i dette tidsrum, baseret på vurdering af udvalgte målevariabler. Bladantallet er den primære målevariabel. Der måles desuden mindst én anden målevariabel, (totalt bladareal, tør vægt eller frisk vægt), da nogle stoffer kan påvirke andre målevariabler langt mere end bladantallet. De stofrelaterede virkninger kvantificeres ved, at væksten i testopløsningerne sammenholdes med væksten i kontrolprøverne, hvorudfra man bestemmer den koncentration, som frembringer en bestemt hæmning x % (f.eks. 50 %) af væksthastigheden, og som betegnes ECx (f.eks. EC50).
Testens endepunkt er hæmning af væksten, der er udtrykt som den logaritmiske stigning i målevariablen (den gennemsnitlige specifikke væksthastighed) i eksponeringsperioden. Ud fra de gennemsnitlige specifikke væksthastigheder, der er registreret i en række testopløsninger, bestemmes den koncentration, der frembringer en bestemt hæmning x % (f.eks. 50 %) af væksthastigheden, og denne betegnes ErCx (f.eks. ErC50).
I denne metode anvendes udbytte som en supplerende responsvariabel, der kan være nødvendig for at efterkomme myndighedsbestemmelserne i visse stater. Udbyttet defineres som værdien af målevariablerne ved eksponeringsperiodens slutning minus værdien af variablerne ved eksponeringsperiodens start. Ud fra det udbytte, der er registreret i en række testopløsninger, bestemmes den koncentration, der frembringer en bestemt hæmning x % (f.eks. 50 %) af udbyttet, og denne betegnes EyCx (f.eks. EyC50).
Derudover kan den laveste koncentration med observeret effekt (LOEC) og nuleffekt-koncentrationen (NOEC) bestemmes statistisk.
1.4. OPLYSNINGER OM TESTSTOFFET
Der skal foreligge en tilstrækkeligt følsom analysemetode til kvantitativ bestemmelse af stoffet i testsubstratet.
Af oplysninger om teststoffet, der kan være nyttige ved fastsættelse af testbetingelserne, kan nævnes strukturformel, renhed, vandopløselighed, stabilitet i vand, lysstabilitet, pKa, Kow, damptryk og bionedbrydelighed. Vandopløseligheden og damptrykket kan bruges til at beregne Henrys konstant, som angiver, om der kan forventes væsentligt tab af teststof i løbet af testperioden. Derved fås en indikation af, om der skal træffes særlige skridt til at kontrollere sådanne tab. Er oplysningerne om teststoffets opløselighed og stabilitet usikre, anbefales det at vurdere disse ved testbetingelserne, dvs. med samme næringssubstrat, temperatur og belysning som i testen.
Når regulering af testsubstratets pH er særlig vigtig, f.eks. ved testning af metaller eller hydrolytisk ustabile stoffer, anbefales det at tilsætte en buffer til næringssubstratet (se første afsnit i punkt 1.7.4). I (11) er der givet vejledning for testning af stoffer, hvis fysisk-kemisk egenskaber vanskeliggør testning.
1.5. REFERENCESTOF
Til kontrol af testmetoden kan der testes et eller flere referencestoffer, f.eks. 3,5-dichlorphenol, som er anvendt i den internationale ringtest (10). Det tilrådes, at testning af et referencestof finder sted mindst to gange årligt eller, hvis testningsfrekvensen er lavere, sideløbende med toksicitetsbestemmelsen af et teststof.
1.6. TESTENS VALIDITET
For at testen skal være valid, skal fordoblingstiden for bladantallet i kontrollen være mindre end 2,5 dage (60 h), tilnærmelsesvis svarende til en syvdobling på syv dage og en gennemsnitlig specifik væksthastighed på 0,275 d–1. Med de testsubstrater og testbetingelser, der er beskrevet i denne testmetode, kan dette kriterium opfyldes ved at gennemføre testen under statiske betingelser (8). Kriteriet forventes desuden at kunne opfyldes under semistatiske forhold og med gennemstrømning. Beregningen af fordoblingstiden er vist i punkt 2.1.
1.7. BESKRIVELSE AF METODEN
1.7.1. Apparatur
Alt udstyr, der kommer i berøring med testsubstraterne, skal være udført helt i glas eller andet kemisk inaktivt materiale. Glasapparatur, der anvendes til dyrkning og testning, skal være renset for kemiske kontaminanter, der kan udvaskes i testsubstratet, og skal være sterilt. Testbeholderne skal være tilstrækkeligt vide til, at blade fra forskellige kolonier i kontrolbeholderne kan vokse uden at overlappe hinanden ved testens slutning. Rødderne må gerne røre bunden af testbeholderen, men en mindste dybde på 20 mm og et mindste rumfang på 100 ml i hver testbeholder tilrådes. Valget af testbeholder er ikke afgørende, når blot disse krav er opfyldt. Bægerglas, krystallisationsskåle eller petriskåle af glas med passende mål har alle vist sig velegnede. Testbeholderne skal have låg, der nedsætter fordampning og utilsigtet kontaminering til et minimum og samtidig tillader det nødvendige luftskifte. Testbeholderne, og især disses låg, må ikke skygge eller ændre lysets spektrale sammensætning.
Kulturerne og testbeholderne må ikke opbevares sammen. Dette opnås bedst ved, at klimakamre, inkubatorer eller lokaler er separate. Belysning og temperatur skal kunne reguleres og holdes på et konstant niveau (se punkt 1.7.8).
1.7.2. Testorganisme
Som testorganisme anvendes i denne test enten Lemna gibba eller Lemna minor. Tillæg 1 indeholder en kort beskrivelse af de arter af andemad, der er blevet anvendt til toksicitetstestning. Plantemateriale kan fås fra en kultursamling, fra et andet laboratorium eller i felten. Er planterne indsamlet i felten, skal de opbevares i mindst otte uger i kultur i samme substrat, som benyttes til testningen, før de anvendes. Indsamlingssteder for startkulturer i felten skal være fri for åbenbare forureningskilder. Hvis de leveres fra et andet laboratorium eller fra en kultursamling, skal de opbevares på tilsvarende måde i mindst tre uger. Kilden til plantematerialet og den testede art og klon (hvis denne kendes) skal altid rapporteres.
Der skal anvendes monokulturer, som er fri for synlig kontaminering med andre organismer såsom alger og protozoer. Sunde planter af L. minor består af kolonier på mellem to og fem blade, mens sunde kolonier af L. gibba kan bestå af op til syv blade.
De testede planters kvalitet og ensartethed har stor indflydelse på testresultatet, hvorfor de bør udvælges nøje. Der skal anvendes unge, hurtigtvoksende planter uden synlige forandringer eller misfarvning (chlorose). Kulturer af god kvalitet er kendetegnet af en høj andel af kolonier med mindst to blade. Et stort antal enkeltblade er tegn på miljøbelastning som f.eks. begrænset adgang til næringsstoffer, og plantemateriale fra sådanne kulturer bør ikke anvendes til testning.
1.7.3. Dyrkning
For at undgå at skulle vedligeholde kulturerne så ofte (f.eks. når Lemna-test ikke påtænkes anvendt i en periode) kan kulturerne opbevares ved reduceret belysning og temperatur (4-10 °C). En nærmere beskrivelse af dyrkningen er givet i tillæg 2. Hvis der er tydelige tegn på kontaminering med alger eller andre organismer, udtages en delprøve af Lemna-blade, som overfladesteriliseres og derefter overføres til friskt substrat (se tillæg 2). I så fald kasseres resten af den kontaminerede kultur.
Mindst syv dage før testning overføres et tilstrækkeligt antal kolonier aseptisk til friskt, sterilt substrat og dyrkes i 7-10 dage under testbetingelserne.
1.7.4. Testsubstrat
Der anbefales forskellige substrater til Lemna minor og Lemna gibba som beskrevet nedenfor. Det må nøje overvejes, om der skal anvendes pH-buffer i testsubstratet (MOPS (4-morpholinpropan sulfonsyre, CAS-nr.: 1132-61-2, Einecs-nr.: 214-478-5) i L. minor-substrat og NaHCO3 i L. gibba-substrat), hvis det mistænkes for at kunne reagere med teststoffet og påvirke tegnene på dets toksicitet. Steinberg-substrat (12) er ligeledes acceptabelt, når blot validitetskriterierne er opfyldt.
Til dyrkning af og testning med L. minor anbefales en modificeret version af Lemna-næringssubstratet efter svensk standard (SIS). Sammensætningen af dette substrat er givet i tillæg 3.
Til dyrkning af og testning med L. gibba anbefales næringssubstratet 20X AAP, som er beskrevet i tillæg 3.
Steinberg-substratet, som beskrives i tillæg 3, er også velegnet til L. minor og kan desuden bruges til L. gibba, når blot validitetskriterierne er opfyldt.
1.7.5. Testopløsninger
Testopløsninger fremstilles sædvanligvis ved fortynding af en stamopløsning. Stamopløsningerne af teststoffet fremstilles normalt ved opløsning af stoffet i næringssubstrat.
Den højeste testede koncentration af teststoffet skal sædvanligvis ikke være større end stoffets vandopløselighed under testbetingelserne. Det skal dog bemærkes, at Lemna spp. flyder ovenpå og kan blive eksponeret for stoffer, som samler sig i vand-luft-grænsefladen (f.eks. stoffer, der er tungtopløselige i vand, hydrofobe eller overfladeaktive). Under sådanne omstændigheder vil eksponeringen skyldes andet materiale end det opløste materiale, og alt efter teststoffets egenskaber kan testkoncentrationerne være større end opløseligheden i vand. For teststoffer med lav vandopløselighed kan det være nødvendigt at fremstille en koncentreret stamopløsning eller -dispersion af stoffet ved hjælp af et organisk opløsnings- eller dispergeringsmiddel for at lette tilsætning af nøjagtige mængder teststof til testsubstratet og medvirke til, at det dispergeres eller opløses. Brug af sådanne midler bør på enhver måde søges undgået. Brug af sådanne hjælpeopløsnings- og dispergeringsmidler må ikke medføre fytotoksisk virkning. Som eksempler på almindeligt anvendte opløsningsmidler, der er uden fytotoksisk virkning i koncentrationer op til 100 µl·l–1, kan nævnes acetone og dimethylformamid. Hvis der anvendes et opløsnings- eller dispergeringsmiddel, skal dets slutkoncentration rapporteres og skal holdes så lav som muligt (≤ 100 µl·l–1), og der skal være samme koncentration af opløsnings- eller dispergeringsmiddel i alle behandlings- og kontrolgrupper. Nærmere anvisninger kan findes i (11).
1.7.6. Behandlings- og kontrolgrupper
Det vil lette valget af passende teststofkoncentrationer, hvis man på forhånd kender til teststoffets toksicitet over for Lemna, f.eks. fra et indledende forsøg. I den endelige toksicitetstest skal der normalt være mindst fem teststofkoncentrationer, der danner en geometrisk række. Kvotienten mellem testkoncentrationerne skal helst ikke være over 3,2, men kan dog være større, når koncentrations-responskurven er flad. Anvendes færre end fem koncentrationer, skal dette begrundes. Der skal anvendes mindst tre replikater for hver testkoncentration.
Ved fastsættelse af området for testkoncentrationerne (i det indledende forsøg og/eller i den endelige toksicitetstest) skal følgende tages i betragtning:
|
— |
Ved bestemmelse af en ECx-værdi skal denne ligge i det område, testkoncentrationerne dækker, for at den skal blive bestemt med et tilstrækkeligt konfidensniveau. Ved beregning af en EC50-værdi skal den højeste testkoncentration således være større end EC50-værdien. Ligger EC50-værdien uden for testkoncentrationernes område, bliver de tilhørende konfidensintervaller store, så det kan blive umuligt at foretage en pålidelig vurdering af modellens statistiske fit. |
|
— |
Hvis målet er at bestemme en LOEC/NOEC-værdi, skal den laveste testkoncentration være så lav, at væksten ikke er væsentligt lavere end i kontrolprøven. Desuden skal den højeste testkoncentration være så høj, at væksten er væsentligt mindre end i kontrolprøven. Er det ikke tilfældet, må testen gentages med et andet koncentrationsområde (medmindre den højeste koncentration svarer til opløselighedsgrænsen eller til den maksimalt krævede grænsekoncentration, f.eks. 100 mg·l–1). |
Hver test skal omfatte kontroller med samme næringssubstrat, samme antal blade og kolonier og samme miljøbetingelser og procedurer som testbeholderne, men uden teststof. Hvis der anvendes et opløsnings- eller dispergeringsmiddel, skal der indgå en ekstra kontrolbehandling med samme koncentration af opløsnings/dispergeringsmiddel som i testbeholderne med teststof. Antallet af replikate kontrolprøver (og, i givet fald, prøver med opløsningsmiddel) skal mindst være lig antallet af beholdere for hver testkoncentration, og ideelt skal der være dobbelt så mange.
Hvis der ikke kræves bestemmelse af NOEC, kan testens design ændres, så der er flere forskellige koncentrationer og færre replikater for hver koncentration. Der skal dog være mindst tre replikater af kontrolprøverne.
1.7.7. Eksponering
Kolonier bestående af 2 til 4 synlige blade overføres fra podekulturen i tilfældig orden til testbeholderne under aseptiske betingelser. Hver testbeholder skal indeholde i alt 9 til 12 blade. Der skal være samme antal blade og kolonier i hver testbeholder. Erfaringerne med brugen af metoden og oplysninger fra ringtest har vist, at tre replikater pr. behandling med 9 til 12 blade som startværdi i hver replikat er tilstrækkeligt til at påvise vækstforskelle mellem behandlingerne svarende til en hæmning på 4 til 7 %, beregnet som væksthastighed (10 til 15 %, beregnet som udbytte) (10).
Testbeholdernes placering i inkubatoren skal være randomiseret for at minimere indflydelsen af rumlige forskelle i lysintensitet eller temperatur. Desuden skal testen enten have blokdesign, eller også skal testbeholderne omplaceres tilfældigt ved hver observation (eller hyppigere).
Hvis en indledende stabilitetstest viser, at teststofkoncentrationen ikke kan opretholdes (dvs. den målte koncentration kommer ned under 80 % af den målte initialkoncentration) gennem hele testperioden (7 dage), anbefales et semistatisk testregime. Dette gennemføres ved, at kolonierne eksponeres for friskfremstillede test- og kontrolopløsninger mindst to gange i løbet af testen (f.eks. på dag 3 og 5). Hyppigheden af eksponeringen for friskt substrat afhænger af teststoffets stabilitet. Meget ustabile eller flygtige stoffer kan kræve hyppigere eksponering, for at koncentrationerne kan holdes tilnærmelsesvis konstante. I visse tilfælde kan det være nødvendigt at bruge en gennemstrømningsmetode (11)(13).
Eksponeringsscenariet ved påføring på bladene (spray) er ikke omhandlet i denne metode. Der henvises i stedet til (14).
1.7.8. Inkubationsbetingelser
Belysningen skal være konstant og komme fra lysstofrør med »warm« eller »cool white« lys med en styrke, der vælges i området 85-135 μE·m–2·s–1, målt i det fotosyntetisk aktive spektrum (400-700 nm) i punkter med samme afstand fra lyskilden som bladene af Lemna (svarende til 6 500-10 000 lux). Lysstyrken over testområdet må ikke afvige mere end ± 15 % fra den valgte lysstyrke. Den anvendte metode til lysdetektion og -måling, specielt den anvendte type sensor, vil påvirke måleværdien. Sfæriske sensorer (som reagerer på lys fra alle vinkler over og under måleplanet) og cosinus-sensorer (som reagerer på lys fra alle vinkler over måleplanet) må foretrækkes frem for retningsbestemte sensorer og vil give højere aflæsninger fra en lyskilde bestående af mange punkter som den her beskrevne.
Temperaturen i testbeholderne skal være 24 ± 2 °C. Kontrolsubstratets pH må højst stige med 1,5 enheder under testen. Dog vil en afvigelse på mere end 1,5 enhed ikke gøre testen ugyldig, når det kan vises, at validitetskriterierne er opfyldt. I særlige tilfælde, f.eks. testning af ustabile stoffer eller metaller, må der udvises ekstra opmærksomhed over for pH-afvigelse. Nærmere vejledning er givet i (11).
1.7.9. Varighed
Testen afsluttes 7 dage efter, at planterne er overført til testbeholderne.
1.7.10. Målinger og analytiske bestemmelser
Ved testens begyndelse tælles og registreres antallet af blade i testkarrene, idet der sørges for, at fremstående, klart synlige blade medtages. Antal blade med normalt og unormalt udseende bestemmes ved testens begyndelse, mindst hvert tredje døgn i løbet af eksponeringsperioden (dvs. mindst to gange i løbet af testperioden på syv dage) samt ved testens slutning. Ændringer i planternes udvikling, f.eks. i bladstørrelse, udseende, tegn på nekrose, chlorose eller opsvulmethed, opbrydning af kolonierne eller tab af disses flydeevne, samt i røddernes længde og udseende, skal registreres. Særlige kendetegn ved testsubstratet (f.eks. ikke-opløst materiale i testbeholderen eller algevækst på denne) skal ligeledes registreres.
Foruden at bladantallet registreres under testen, vurderes teststoffets virkninger på en (eller flere) af følgende målevariabler:
|
i) |
totalt bladareal |
|
ii) |
tør vægt |
|
iii) |
frisk vægt. |
Bestemmelse af det totale bladareal har den fordel, at det kan bestemmes for hver testbeholder og kontrolbeholder både ved testens start, under testen og ved dens slutning. Den tørre og friske vægt bestemmes ved testens start på en prøve af podekulturen, der er repræsentativ for den, der anvendes til at begynde testen, samt ved testens slutning med plantemateriale fra hver test- og kontrolbeholder. Hvis der ikke måles bladareal, er tør vægt at foretrække frem for frisk vægt.
Bestemmelse af totalt bladareal, tør vægt og frisk vægt kan foretages således:
|
i) |
Totalt bladareal: Det totale bladareal af alle kolonier kan bestemmes ved billedanalyse: Der kan afbildes en silhuet af testbeholder og planter med et videokamera (idet beholderen anbringes på en lyskasse), og det resulterende billede digitaliseres. Derefter kan det totale bladareal i testbeholderen bestemmes ved kalibrering med flade former med kendt areal. Der skal sørges for at udelukke interferens fra testbeholderens rand. En alternativ, men mere omstændelig metode er at fotokopiere testbeholdere og planter, klippe den resulterende silhuet af kolonierne ud og bestemme arealet med en bladareal-analysator eller ved hjælp af millimeterpapir. Også andre teknikker kan anvendes (f.eks. vejning af papiret svarende til silhuetten og sammenholdelse med et enhedsareal). |
|
ii) |
Tør vægt: Alle kolonier indsamles fra hver testbeholder og skylles med destilleret eller deioniseret vand. De duppes fri for overskydende vand og tørres derefter ved 60 °C, til vægten er konstant. Eventuelle rodstumper skal medtages. Den tørre vægt skal angives med en nøjagtighed mindst svarende til 0,1 mg. |
|
iii) |
Frisk vægt: Alle kolonier overføres til på forhånd vejede rør af polystyren (eller andet inert materiale), hvis afrundede bund er forsynet med et lille hul (1 mm). Rørene centrifugeres derefter ved 3 000 o./min. i 10 minutter ved rumtemperatur. Rørene, der indeholder de nu tørrede kolonier, vejes igen, og den friske vægt beregnes ved fratrækning af vægten af de tomme rør. |
1.7.10.1. Hyppighed af målinger og analytiske bestemmelser
Hvis testen udføres statisk, skal pH for hver behandling måles ved testens start og slutning. Udføres testen semistatisk, foretages pH-måling på hver portion »frisk« testopløsning før hver udskiftning foruden på de tilsvarende »brugte« opløsninger.
Lysintensiteten skal måles i vækstkammeret, inkubatoren eller lokalet i punkter med samme afstand til lyskilden som bladene af Lemna. Målingerne foretages mindst én gang i løbet af testen. Mindst én gang dagligt måles temperaturen af substratet i en surrogatbeholder, der opbevares ved samme betingelser i klimakammeret, inkubatoren eller lokalet.
Under testen bestemmes teststofkoncentrationerne med passende intervaller. Mindstekravet i statiske test er bestemmelse af koncentrationerne ved testens start og slutning.
I semistatiske test, hvor teststofkoncentrationen ikke forventes at forblive inden for ± 20 % af den nominelle koncentration, er det nødvendigt at analysere alle frisk fremstillede testopløsninger, og de samme opløsninger analyseres ved hver udskiftning (se tredje afsnit i punkt 1.7.7). For de test, hvor den målte startkoncentration af teststoffet ikke er inden for ± 20 % af den nominelle værdi, men hvor der kan fremlægges tilstrækkelig dokumentation for, at startkoncentrationerne er repeterbare og stabile (dvs. inden for området 80-120 % af startkoncentrationen), kan det godtages, at der kun foretages kemisk bestemmelse af den højeste og laveste testkoncentration. I alle tilfælde behøver teststofkoncentrationen før udskiftning kun bestemmes på én replikat testbeholder ved hver testkoncentration (eller på indholdet af de poolede beholdere for hver replikat).
For gennemstrømningstest vil det være hensigtsmæssigt med en prøvetagningsplan svarende til den, der er beskrevet for semistatiske test, herunder analyse ved testens start, midtpunkt og slutning, men måling af »brugt« testopløsning er ikke hensigtsmæssig i dette tilfælde. I sådanne test skal flowhastigheden af fortyndingsmiddel og teststofopløsning eller teststof-stamopløsning kontrolleres dagligt.
Hvis det kan konstateres, at prøvestofkoncentrationen har været holdt tilfredsstillende inden for ± 20 % af den nominelle eller målte startkoncentration under hele testen, kan analysen af resultaterne baseres på nominelle eller målte startværdier. Er afvigelsen fra den nominelle eller målte startkoncentration større end ± 20 %, skal analysen af resultaterne baseres på den geometriske middelkoncentration under eksponeringen eller på modeller, der beskriver faldet i teststofkoncentrationen (11).
1.7.11. Grænsetest
Under visse omstændigheder, f.eks. når en indledende test viser, at teststoffet er uden toksiske virkninger i koncentrationer på op til 100 mg·l–1, dog ikke over stoffets opløselighed i testsubstratet, kan der udføres en grænsetest, hvor reaktionen i en kontrolgruppe sammenholdes med reaktionen i én behandlingsgruppe (100 mg·l–1 eller en koncentration lig med opløselighedsgrænsen). Det anbefales kraftigt, at dette understøttes ved analyse af eksponeringskoncentrationen. Alle de beskrevne testbetingelser og validitetskriterier finder anvendelse på en grænsetest bortset fra, at antallet af behandlingsreplikater skal fordobles. Væksten i kontrol- og behandlingsgruppen kan analyseres med en statistisk test til sammenligning af middelværdier, f.eks. Students t-test.
2. DATA OG RAPPORTERING
2.1. FORDOBLINGSTID
Til bestemmelse af fordoblingstiden (Td) for bladantallet og forsøgets overensstemmelse med dette validitetskriterium (punkt 1.6) anvendes følgende formel på de data, der er indsamlet for kontrolbeholderne:
Td = ln 2/μ
hvor μ er den gennemsnitlige specifikke væksthastighed, der bestemmes som beskrevet i første og andet afsnit i punkt 2.2.1.
2.2. RESPONSVARIABLER
Testens formål er at bestemme teststoffets virkninger på den vegetative vækst af Lemna. I nærværende testmetode beskrives to responsvariabler, da præferencer og myndighedskrav er forskellige i medlemsstaterne. For at testresultaterne skal kunne godtages i alle medlemsstater, skal virkningerne vurderes ved hjælp af begge de nedenfor beskrevne responsvariabler a) og b).
|
a) |
Gennemsnitlig specifik væksthastighed: Denne responsvariabel beregnes på grundlag af den logaritmiske ændring i bladantallet samt den logaritmiske ændring i en anden måleparameter (totalt bladareal, tør vægt eller frisk vægt) med tiden (udtrykt pr. dag) i kontrollerne og i hver behandlingsgruppe. Den kaldes undertiden relativ væksthastighed (15). |
|
b) |
Udbytte: Denne responsvariabel beregnes på grundlag af ændringen i bladantallet og desuden ændringen i en anden måleparameter (totalt bladareal, tør vægt eller frisk vægt) i kontrollerne og i hver behandlingsgruppe indtil testens slutning. |
Det skal bemærkes, at toksicitetsværdier beregnet ved hjælp af disse to responsvariabler ikke er sammenlignelige, og at forskellen mellem dem må holdes for øje ved anvendelse af testens resultater. ECx-værdier baseret på gennemsnitlig specifik væksthastighed (ErCx) vil sædvanligvis være højere end resultater baseret på udbytte (EyCx), når testbetingelserne i denne metode er overholdt, hvilket følger af det matematiske grundlag for de to beregningsmåder. Dette må ikke opfattes som en forskel i følsomhed mellem de to responsvariabler, men kun, at der er tale om to matematisk forskellige størrelser. Begrebet gennemsnitlig specifik væksthastighed bygger på det generelle eksponentielle vækstmønster for andemad i ikke-begrænsede kulturer, idet toksiciteten vurderes på grundlag af virkningerne på væksthastigheden og ikke afhænger af den absolutte størrelse af den specifikke væksthastighed af kontrollen, koncentrations-responskurvens hældning eller testens varighed. Resultater, der er baseret på responsvariablen udbytte, afhænger derimod af alle disse øvrige variabler. EyCx afhænger af den specifikke væksthastighed af de arter af andemad, der anvendes i hver test, og af den maksimale specifikke væksthastighed, som kan variere mellem de forskellige arter og endda kloner. Denne responsvariabel bør ikke anvendes til at sammenligne følsomheden over for giftstoffer mellem forskellige arter af andemad, endsige forskellige kloner. Skønt det videnskabeligt foretrukne grundlag til vurdering af toksicitet er den gennemsnitlige specifikke væksthastighed, indeholder nærværende testmetode også toksicitetsvurderinger baseret på udbytte for at imødekomme de aktuelle myndighedskrav i visse stater.
Beregningen af toksicitet skal baseres på bladantal og på mindst én anden målevariabel (totalt bladareal, tør vægt eller frisk vægt), da visse stoffer kan påvirke andre målevariabler langt mere end bladantallet. Denne virkning ville ikke blive opdaget ved beregning af bladantallet alene.
Bladantallet og eventuelle andre registrerede målevariabler, dvs. totalt bladareal, tør vægt eller frisk vægt, samles i en tabel sammen med teststofkoncentrationerne for hvert målepunkt. Den efterfølgende dataanalyse, f.eks. beregning af estimater for LOEC, NOEC eller ECx, skal baseres på værdierne for de enkelte replikater og ikke beregnede gennemsnit for hver behandlingsgruppe.
2.2.1. Gennemsnitlig specifik væksthastighed
Den gennemsnitlige specifikke væksthastighed for en given periode beregnes som den logaritmiske stigning i vækstvariablerne — bladantal og en anden målevariabel (totalt bladareal, tør vægt eller frisk vægt) — ved hjælp af nedenstående udtryk for hver replikat af kontrol og behandlingsprøve:

hvor:
|
— |
: |
μi-j |
: |
er den gennemsnitlige specifikke væksthastighed i tidsrummet fra i til j |
|
— |
: |
Ni |
: |
er værdien af målevariablen i test- eller kontrolbeholderen til tiden i |
|
— |
: |
Nj |
: |
er værdien af målevariablen i test- eller kontrolbeholderen til tiden j |
|
— |
: |
t |
: |
er tidsrummet fra i til j |
For hver behandlings- og kontrolgruppe beregnes en gennemsnitlig væksthastighed samt estimater for variansen.
Den gennemsnitlige specifikke væksthastighed skal beregnes for hele testperioden (tiden »i« i ovenstående formel er testens starttid, og tiden »j« er testens sluttid). For hver testkoncentration og kontrol beregnes en gennemsnitsværdi af den specifikke væksthastighed samt estimater for variansen. Derudover foretages en trinvis beregning af væksthastigheden for at vurdere teststoffets virkninger i eksponeringsperioden (f.eks. ved betragtning af logaritmisk transformerede vækstkurver). Væsentlige forskelle mellem den trinvist beregnede væksthastighed og den gennemsnitlige væksthastighed tyder på en afvigelse fra konstant eksponentiel vækst, og nøje granskning af vækstkurverne er da nødvendig. I så fald kan et forsigtigt skøn opstilles ved at sammenholde de specifikke væksthastigheder fra behandlede kulturer i det tidsrum, hvor hæmningen er maksimal, med de tilsvarende værdier for kontrolprøver i samme tidsrum.
Hæmningen i procent af væksthastigheden (Ir) kan derefter beregnes for hver testkoncentration (behandlingsgruppe) af følgende udtryk:

hvor:
|
— |
: |
% Ir |
: |
er hæmningen i procent af den gennemsnitlige specifikke væksthastighed |
|
— |
: |
μC |
: |
er gennemsnitsværdien af μ i kontrolgruppen |
|
— |
: |
μT |
: |
er gennemsnitsværdien af μ i behandlingsgruppen |
2.2.2. Udbytte
Virkningerne på udbyttet bestemmes på grundlag af to målevariabler, bladantallet og en anden målevariabel (totalt bladareal, tør vægt eller frisk vægt) i hver testbeholder ved testens start og slutning. For tør vægt og frisk vægt bestemmes start-biomassen på grundlag af en prøve af blade, der tages fra samme batch som den, der er anvendt til at pode testbeholderne (se andet afsnit i punkt 1.7.3). For hver testkoncentration og kontrol beregnes en gennemsnitsværdi af udbyttet samt estimater for variansen. Udbyttehæmningen i procent (% Iy) kan beregnes for hver behandlingsgruppe som følger:
hvor:
|
— |
: |
% Iy |
: |
er udbyttereduktionen i procent |
|
— |
: |
bC |
: |
er slut-biomasse minus start-biomasse for kontrolgruppen |
|
— |
: |
bT |
: |
er slut-biomasse minus start-biomasse for behandlingsgruppen |
2.2.3. Optegning af koncentrations-responskurver
Der afsættes koncentrations-responskurver over sammenhængen mellem den gennemsnitlige hæmningsprocent for responsvariablerne (Ir eller Iy, beregnet som vist i sidste afsnit i punkt 2.2.1 eller i punkt 2.2.2) og logaritmen til teststofkoncentrationen.
2.2.4. Beregning af ECx
Estimaterne for ECx (f.eks. EC50) baseres på gennemsnitsværdien dels af specifik væksthastighed (ErCx), dels af udbytte (EyCx), der hver især er baseret på bladantal og endnu en målevariabel (totalt bladareal, tør vægt eller frisk vægt). Dette skyldes, at der findes teststoffer med forskellig virkning på bladantallet og på de øvrige målevariabler. Som parametre for toksicitet er der derfor fire ECx-værdier for hvert beregnet hæmningsniveau x: ErCx (bladantal), ErCx (totalt bladareal, tør vægt eller frisk vægt), EyCx (bladantal) og EyCx (totalt bladareal, tør vægt eller frisk vægt).
2.3. STATISTISKE METODER
Målet er at formulere den kvantitative koncentrations-responssammenhæng ved hjælp af regressionsanalyse. Der kan anvendes vægtet lineær regression efter forudgående lineariserende transformation af responsdataene — til f.eks. probit-, logit- eller Weibull-enheder (16), men det må foretrækkes at benytte ikke-lineære regressionsmetoder, da de bedre håndterer de uundgåelige uregelmæssigheder i data og afvigelser fra jævne fordelinger. I området tæt på enten ingen hæmning eller total hæmning kan sådanne uregelmæssigheder blive forstørret ved transformation og derved gribe forstyrrende ind i analysen (16). Det må bemærkes, at standardanalysemetoder, hvor der bruges probit-, logit- eller Weibull-transformerede variabler, er bestemt til brug på binære data (f.eks. død eller overlevelse) og må modificeres for at kunne benyttes til data for væksthastighed eller udbytte. Særlige metoder til bestemmelse af ECx-værdier ud fra kontinuerte data findes i henvisning (17)(18) og (19).
For hver responsvariabel, der skal analyseres, anvendes koncentrations-responsrelationen til at beregne punktestimater for ECx-værdier. Når det er muligt, skal der bestemmes 95 % konfidensgrænser for hvert estimat. Responsdataenes tilpasningsgrad til regressionsmodellen vurderes enten grafisk eller statistisk. Regressionsanalysen skal foretages på responsværdierne for de enkelte replikater, ikke på gruppegennemsnit.
Hvis de tilgængelige regressionsmodeller eller -metoder er uegnede til dataene, kan estimater og konfidensgrænser for EC50 desuden fås ved lineær interpolation med bootstrapping (20).
Til beregning af et estimat for LOEC og dermed NOEC må middelværdierne for behandlingerne sammenholdes ved hjælp af variansanalyse. Gennemsnittet for hver koncentration sammenholdes derefter med gennemsnittet for kontrollerne ved hjælp af en egnet multisammenlignings- eller trend test-metode. Dunnetts eller Williams’ test kan være nyttig (21)(22)(23)(24). Det må vurderes, om variansanalysens forudsætning om varianshomogenitet er opfyldt. Denne vurdering kan foretages grafisk eller ved en formel test (25). Hertil kan Levenes eller Bartletts test anvendes. Manglende opfyldelse af forudsætningen om varianshomogenitet kan undertiden afhjælpes ved logaritmisk transformation af dataene. Hvis variansen er ekstremt heterogen og ikke kan korrigeres ved transformation, må analyse med step-down Jonkheere trend test overvejes. Supplerende vejledning for bestemmelse af NOEC er givet i (19).
Den senere tids videnskabelige udvikling har ført til en anbefaling om, at NOEC-begrebet afskaffes og erstattes med regressionsbaserede punktestimater ECx. Der er ikke fastlagt en passende værdi af x til denne Lemna-test. Et område på 10 til 20 % synes imidlertid at være passende (afhængigt af den valgte responsvariabel), og det må foretrækkes at rapportere både EC10 og EC20.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal indeholde følgende:
Teststoffet:
|
— |
fysisk tilstand og fysisk-kemiske egenskaber, herunder vandopløselighed |
|
— |
kemiske identifikationsdata (f.eks. CAS-nr.), herunder renhed. |
Testede arter:
|
— |
videnskabeligt navn, klon (hvis den kendes) og kilde. |
Testbetingelser:
|
— |
den anvendte testmetode (statisk, semistatisk eller gennemstrømningstest) |
|
— |
startdato og varighed for testen |
|
— |
testsubstrat |
|
— |
beskrivelse af testens design: testbeholdere og låg, opløsningsvolumener, antal kolonier og blade pr. testbeholder ved testens start |
|
— |
testkoncentrationer (nominelle eller målte) og antal replikater for hver koncentration |
|
— |
metoder til fremstilling af stam- og testopløsninger, herunder eventuelle opløsnings- og dispergeringsmidler |
|
— |
testtemperaturen |
|
— |
lyskilde, lysintensitet og -homogenitet |
|
— |
pH-værdierne af test- og kontrolsubstraterne |
|
— |
teststofkoncentrationer og analysemetoder med fyldestgørende oplysninger om kvalitetsvurdering (valideringsundersøgelser, standardafvigelser eller konfidensgrænser for analyserne) |
|
— |
metoder til bestemmelse af bladantal og andre målevariabler, f.eks. tør vægt, frisk vægt og bladareal |
|
— |
alle fravigelser af nærværende testmetode. |
Resultater:
|
— |
rådata: antal blade og andre målevariabler i hver test- og kontrolbeholder ved hver observation og hver analyse |
|
— |
gennemsnit og standardafvigelse for hver målevariabel |
|
— |
vækstkurver for hver koncentration (brug af logaritmisk transformerede målevariabler anbefales, se andet afsnit i punkt 2.2.1) |
|
— |
fordoblingstid/væksthastighed i kontrolprøven, baseret på bladantal |
|
— |
beregnede responsvariabler for hver behandlingsreplikat, med gennemsnitsværdier og variationskoefficient for replikaterne |
|
— |
grafisk fremstilling af sammenhængen mellem koncentration og virkning |
|
— |
estimater for toksicitetsendepunkter for responsvariablerne, f.eks. EC50, EC10, EC20, med tilhørende konfidensintervaller; LOEC og/eller NOEC (hvis disse er beregnet) og de statistiske metoder, der anvendt til at beregne dem |
|
— |
hvis variansanalyse er anvendt, størrelsen af den virkning, det er muligt at påvise (f.eks. mindste signifikante forskel) |
|
— |
enhver vækststimulation, der er fundet ved nogen behandling |
|
— |
ethvert visuelt tegn på fytotoksicitet samt observationer af testopløsningerne |
|
— |
diskussion af resultaterne, herunder den eventuelle betydning af fravigelser fra nærværende testmetode. |
4. LITTERATUR
|
(1) |
OECD TG 221 (2006) Lemna Sp. Growth Inhibition Test. |
|
(2) |
Der redegøres nærmere for brugen af Lemna-undersøgelser for farvede stoffer i afsnit 13.5.3 i EU's Manual of Decisions fra juli 2006, se: http://ecb.jrc.ec.europa.eu/new-chemicals |
|
(3) |
Guidance on information requirements and chemical safety assessment — Chapter R.7b: Endpoint specific guidance; Table 7.8.3 Summary of difficult substance testing issues, available at http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1234958685#A |
|
(4) |
ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415-91 (Reapproved 1998). pp. 733-742. In, Annual Book of ASTM Standards, Vol. 11.05 Biological Effects and Environmental Fate; Biotechnology; Pesticides, ASTM, West Conshohocken, PA. |
|
(5) |
USEPA — United States Environmental Protection Agency. (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., »Public draft«. EPA 712-C-96-156. 8pp. |
|
(6) |
AFNOR — Association Française de Normalisation. (1996). XP T 90-337: Détermination de l’inhibition de la croissance de Lemna minor. 10pp. |
|
(7) |
SSI — Swedish Standards Institute. (1995). Water quality — Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15pp. (på svensk). |
|
(8) |
Environment Canada. (1999). Biological Test Method: Test for Measuring the Inhibition of Growth Using the Freshwater Macrophyte, Lemna minor. EPS 1/RM/37-120 pp. |
|
(9) |
Environment Canada. (1993) Proposed Guidelines for Registration of Chemical Pesticides: Non-Target Plant Testing and Evaluation. Canadian Wildlife Service, Technical Report Series No. 145. |
|
(10) |
Sims I., Whitehouse P., and Lacey R. (1999) The OECD Lemna Growth Inhibition Test. Development and Ring-testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc — Environment Agency. |
|
(11) |
OECD (2000). Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publications, Series on Testing and Assessment No.23. |
|
(12) |
ISO DIS 20079. Water Quality — Determination of the Toxic Effect of Water Constituents and Waste Water to Duckweed (Lemna minor) — Duckweed Growth Inhibition Test. |
|
(13) |
Walbridge C. T. (1977). A flow-through testing procedure with duckweed (Lemna minor L.). Environmental Research Laboratory — Duluth, Minnesota 55804. US EPA Report No. EPA-600/3-77 108. September 1977. |
|
(14) |
Lockhart W. L., Billeck B. N. and Baron C. L. (1989). Bioassays with a floating plant (Lemna minor) for effects of sprayed and dissolved glyphosate. Hydrobiologia, 118/119, 353-359. |
|
(15) |
Huebert, D.B. and Shay J.M. (1993) Considerations in the assessment of toxicity using duckweeds. Environmental Toxicology and Chemistry, 12, 481-483. |
|
(16) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713-718. |
|
(17) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157-167. |
|
(18) |
Bruce R.D. and Versteeg D.J. (1992) A statistical procedure for modelling continuous toxicity data. Environmental Toxicology and Chemistry, 11, 1485-1494. |
|
(19) |
OECD. (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data. |
|
(20) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05-88. USEPA, Duluth, MN. |
|
(21) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc., 50, 1096-1121. |
|
(22) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics, 20, 482-491. |
|
(23) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics, 27: 103-117. |
|
(24) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics, 28: 510-531. |
|
(25) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93-96. |
Tillæg 1
Beskrivelse af Lemna spp.
Vandplanten med trivialnavnet andemad, Lemna spp., tilhører familien Lemnaceae, der omfatter en række arter, som er udbredt over hele verden og består af fire slægter. Deres respektive udseende og taksonomi er beskrevet udtømmende (1)(2). Lemna gibba og L. minor er repræsentative arter fra tempererede områder og finder almindelig anvendelse i toksicitetstest. Begge arter har flydende eller nedsænket skiveformet stilk (blad) og en meget tynd rod, der udgår fra midten af undersiden på hvert blad. Lemna spp. danner sjældent blomster, og planterne formerer sig vegetativt ved at skyde nye blade (3). De unge planter er blegere og har kortere rødder end de ældre planter og består af to-tre blade af forskellig størrelse. Lemnas enkle struktur, ukønnede formering og korte generationstid gør planten meget velegnet til laboratorietest (4)(5).
På grund af artsbetingede forskelle i følsomhed er sammenligning af følsomhed kun gyldig inden for samme art.
Eksempler på arter af Lemna, der er blevet anvendt til testning: Henvisninger vedrørende de forskellige arter
Lemna aequinoctialis: Eklund, B. (1996). The use of the red alga Ceramium strictum and the duckweed Lemna aequinoctialis in aquatic ecotoxicological bioassays. Licentiate in Philosophy Thesis 1996:2. Dep. of Systems Ecology, Stockholm University.
Lemna major: Clark, N. A. (1925). The rate of reproduction of Lemna major as a function of intensity and duration of light. J. phys. Chem., 29: 935-941.
Lemna minor: United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., »Public draft«. EPA 712-C-96-156. 8pp.
Association Française de Normalisation (AFNOR). (1996). XP T 90-337: Détermination de l’inhibition de la croissance de Lemna minor. 10pp.
Swedish Standards Institute (SIS). (1995). Water quality — Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15pp. (på svensk).
Lemna gibba: ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415-91 (Reapproved 1998). pp. 733-742.
United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., »Public draft«. EPA 712-C-96-156. 8pp.
Lemna paucicostata: Nasu, Y., Kugimoto, M. (1981). Lemna (duckweed) as an indicator of water pollution. I. The sensitivity of Lemna paucicostata to heavy metals. Arch. Environ. Contam. Toxicol., 10:1959-1969.
Lemna perpusilla: Clark, J. R. et al. (1981). Accumulation and depuration of metals by duckweed (Lemna perpusilla). Ecotoxicol. Environ. Saf., 5:87-96.
Lemna trisulca: Huebert, D. B., Shay, J. M. (1993). Considerations in the assessment of toxicity using duckweeds. Environ. Toxicol. and Chem., 12:481-483.
Lemna valdiviana: Hutchinson, T.C., Czyrska, H. (1975). Heavy metal toxicity and synergism to floating aquatic weeds. Verh.-Int. Ver. Limnol., 19:2102-2111.
Kilder til Lemna-arter
|
University of Toronto Culture Collection of Algae and Cyanobacteria |
|
Department of Botany, University of Toronto |
|
Toronto, Ontario, Canada, M5S 3 B2 |
|
Tlf. +1 4169783641 |
|
Fax +1 4169785878 |
|
E-mail: jacreman@botany.utoronto.ca |
|
http://www.botany.utoronto.ca/utcc |
|
North Carolina State University |
|
Forestry Dept |
|
Duckweed Culture Collection |
|
Campus Box 8002 |
|
Raleigh, NC 27695-8002 |
|
USA |
|
Tlf. +1 9195157572 |
|
astomp@unity.ncsu.edu |
|
Institute of Applied Environmental Research (ITM) Stockholm University |
|
SE-106 91 STOCKHOLM |
|
SVERIGE |
|
Tlf. +46 86747240 |
|
Fax: +46 86747636 |
|
Federal Environmental Agency (UBA) |
|
FG III 3.4 |
|
Schichauweg 58 |
|
12307 Berlin |
|
TYSKLAND |
|
E-mail: lemna@uba.de |
|
http://www.umweltbundesamt.de/contact.htm |
Litteratur
|
(1) |
Hillman, W.S. (1961). The Lemnaceae or duckweeds: A review of the descriptive and experimental literature. The Botanical Review, 27:221-287. |
|
(2) |
Landolt, E. (1986). Biosystematic investigations in the family of duckweed (Lemnaceae). Vol. 2. Geobotanischen Inst. ETH, Stiftung Rubel, Zürich, Switzerland. |
|
(3) |
Björndahl, G. (1982). Growth performance, nutrient uptake and human utilization of duckweeds (Lemnaceae family). ISBN 82-991150-0-0. The Agricultural Research Council of Norway, University of Oslo. |
|
(4) |
Wang, W. (1986). Toxicity tests of aquatic pollutants by using common duckweed. Environmental Pollution, Ser B, 11:1-14. |
|
(5) |
Wang, W. (1990). Literature review on duckweed toxicity testing. Environmental Research, 52:7-22. |
Tillæg 2
Vedligeholdelse af stamkultur
Ved lavere temperatur (4-10 °C) kan stamkulturer vedligeholdes i længere tid uden at skulle gendannes. Næringssubstratet til Lemna kan være det samme som det, der anvendes ved testning, men andre næringsrige substrater kan anvendes til stamkulturer.
Med jævne mellemrum udtages et antal unge, lysegrønne planter og overføres aseptisk til nye dyrkningsbeholdere med friskt substrat. Under de her foreslåede køligere forhold kan dyrkning af subkulturer ske med op til tre måneders mellemrum.
Der skal anvendes kemisk rene (syrevaskede) og sterile dyrkningsbeholdere af glas samt aseptisk håndtering. Hvis en stamkultur bliver kontamineret med. f.eks. alger eller svampe, må der foretages indgreb til at eliminere kontaminanten. For algers og de fleste andre kontaminanters vedkommende kan dette opnås ved overfladesterilisation. Der udtages en prøve af det kontaminerede plantemateriale, og rødderne klippes bort. Materialet omrystes derefter kraftigt med rent vand, efterfulgt af nedsænkning i en 0,5 % (v/v) opløsning af natriumhypochlorit i mellem 30 sekunder og 5 minutter. Plantematerialet skylles derefter med sterilt vand og overføres i et par portioner til dyrkningsbeholdere indeholdende friskt næringssubstrat. Mange af bladene vil dø som følge af denne behandling, specielt hvis der anvendes længere eksponeringsperioder, men nogle af de overlevende vil sædvanligvis være fri for kontaminering. Disse kan derefter bruges til podning af nye kulturer.
Tillæg 3
Substrater
Der anbefales forskellige næringssubstrater til L. minor og L. gibba. Til L. minor anbefales et modificeret substrat efter Swedish Standard (SIS), mens substratet 20X AAP anbefales til L. gibba. Nedenfor er sammensætningen af de to substrater angivet. Til fremstilling af sådanne substrater skal anvendes kemikalier af reagens- eller analysekvalitet og deioniseret vand.
Lemna-næringssubstrat efter Swedish Standard (SIS)
|
— |
Stamopløsning I-V steriliseres ved autoklavering (120 °C, 15 min.) eller ved membranfiltrering (porediameter ca. 0,2 µm). |
|
— |
Stamopløsning VI (og valgfrit VII) steriliseres kun ved membranfiltrering og må ikke autoklaveres. |
|
— |
Sterile stamopløsninger skal opbevares køligt og mørkt. Stamopløsning I-V skal kasseres efter seks måneder, mens holdbarheden for stamopløsning VI (og valgfrit VII) er én måned.
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
— |
Til fremstilling af én liter SIS-substrat tilsættes følgende til 900 ml deioniseret vand:
Bemærkning: Til visse teststoffer kan der yderligere være brug for en stamopløsning VII (MOPS-buffer) (se sidste afsnit i punkt 1.4). |
|
— |
pH justeres til 6,5 ± 0,2 med enten 0,1 M eller 1 M HCl eller NaOH, og der fyldes op til én liter med deioniseret vand. |
20X AAP-næringssubstrat
Stamopløsninger fremstilles i sterilt, destilleret eller deioniseret vand.
Sterile stamopløsninger skal opbevares køligt og mørkt. Under disse betingelser vil stamopløsningerne have en holdbarhed på mindst 6-8 uger.
Der fremstilles fem næringssubstrat-stamopløsninger (A1, A2, A3, B og C) til 20X AAP-substrat, idet der anvendes kemikalier af reagenskvalitet. Næringssubstratet fremstilles ved, at der tilsættes 20 ml af hver næringssubstrat-stamopløsning til ca. 850 ml deioniseret vand. pH justeres til 7,5 ± 0,1 med enten 0,1 M eller 1 M HCl eller NaOH, og der fyldes op til én liter med deioniseret vand. Substratet filtreres gennem et membranfilter med en porediameter på ca. 0,2 μm over i en steril beholder.
Det næringssubstrat, der skal bruges til testningen, skal fremstilles 1-2 dage før brug, for at pH kan stabiliseres. Næringssubstratets pH skal kontrolleres før brug og om nødvendigt justeres igen med 0,1 M eller 1 M NaOH eller HCl som beskrevet ovenfor.
|
Stamopløsning nr. |
Stof |
Koncentration i stamopløsningen (g·l–1) (1) |
Koncentration i færdigt substrat (mg·l–1) (1) |
Færdigt substrat |
|
|
Grundstof |
Koncentration (mg·l–1) (1) |
||||
|
A1 |
NaNO3 MgCl2·6H2O CaCl2·2H2O |
26 12 4,4 |
510 240 90 |
Na, N Mg Ca |
190, 84 58,08 24,04 |
|
A2 |
MgSO4·7H2O |
15 |
290 |
S |
38,22 |
|
A3 |
K2HPO4·3H2O |
1,4 |
30 |
K, P |
9,4, 3,7 |
|
B |
H3BO3 MnCl2·4H2O FeCl3·6H2O Na2EDTA·2H2O ZnCl2 CoCl2·6H2O Na2MoO4·2H2O CuCl2·2H2O |
0,19 0,42 0,16 0,30 3,3 mg·l–1 1,4 mg·l–1 7,3 mg·l–1 0,012 mg·l–1 |
3,7 8,3 3,2 6,0 66 μg·l–1 29 μg·l–1 145 μg·l–1 0,24 μg·l–1 |
B Mn Fe — Zn Co Mo Cu |
0,65 2,3 0,66 — 31 μg·l–1 7,1 μg·l–1 58 μg·l–1 0,080 μg·l–1 |
|
C |
NaHCO3 |
15 |
300 |
Na, C |
220, 43 |
Fodnote: Den teoretisk mest passende endelige bicarbonatkoncentration (som vil overflødiggøre nævneværdig pH-justering) er 15 mg/l, ikke 300 mg/l. Den traditionelle brug af 20X AAP-substratet og ringtesten af metoden er imidlertid baseret på 300 mg/l. (I. Sims, P. Whitehouse og R. Lacey. (1999) The OECD Lemna Growth Inhibition Test. Development and Ring-testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc — Environment Agency.
STEINBERG-substrat (efter ISO 20079)
Koncentrationer og stamopløsninger
|
— |
Det modificerede Steinberg-substrat anvendes i ISO 20079 til Lemna minor alene (da der deri kun tillades brug af Lemna minor), men også med Lemna gibba er der opnået gode testresultater. |
|
— |
Til fremstilling af substratet skal der anvendes kemikalier af reagens- eller analysekvalitet og deioniseret vand. |
|
— |
Næringssubstratet fremstilles af stamopløsninger eller af det 10-dobbelt koncentrerede substrat, som har den maksimale substratkoncentration uden udfældning. |
Tabel 1
pH-stabiliseret STEINBERG-substrat (modificeret efter Altenburger)
|
Stof |
Næringssubstrat |
||
|
Makronæringsstoffer |
molvægt |
mg/l |
mmol/l |
|
KNO3 |
101,12 |
350,00 |
3,46 |
|
Ca(NO3)2 · 4H2O |
236,15 |
295,00 |
1,25 |
|
KH2PO4 |
136,09 |
90,00 |
0,66 |
|
K2HPO4 |
174,18 |
12,60 |
0,072 |
|
MgSO4 · 7H2O |
246,37 |
100,00 |
0,41 |
|
Mikronæringsstoffer |
molvægt |
µg/l |
µmol/l |
|
H3BO3 |
61,83 |
120,00 |
1,94 |
|
ZnSO4 · 7H2O |
287,43 |
180,00 |
0,63 |
|
Na2MoO4 · 2H2O |
241,92 |
44,00 |
0,18 |
|
MnCl2 · 4H2O |
197,84 |
180,00 |
0,91 |
|
FeCl3 · 6H2O |
270,21 |
760,00 |
2,81 |
|
EDTA-dinatrium-dihydrat |
372,24 |
1 500,00 |
4,03 |
Tabel 2
Stamopløsninger (makronæringsstoffer)
|
g/l |
||
|
Stamopløsning 1: |
|||
|
KNO3 |
17,50 |
||
|
KH2PO4 |
4,5 |
||
|
K2HPO4 |
0,63 |
||
|
Stamopløsning 2: |
|||
|
MgSO4 · 7H2O |
5,00 |
||
|
Stamopløsning 3: |
|||
|
Ca(NO3)2 · 4H2O |
14,75 |
||
Tabel 3
Stamopløsninger (mikronæringsstoffer)
|
mg/l |
||
|
Stamopløsning 4: |
|||
|
H3BO3 |
120,0 |
||
|
Stamopløsning 5: |
|||
|
ZnSO4 · 7H2O |
180,0 |
||
|
Stamopløsning 6: |
|||
|
Na2MoO4 · 2H2O |
44,0 |
||
|
Stamopløsning 7: |
|||
|
MnCl2 · 4H2O |
180,0 |
||
|
Stamopløsning 8: |
|||
|
FeCl3 · 6H2O |
760,00 |
||
|
EDTA-dinatrium-dihydrat |
1 500,00 |
||
|
— |
Stamopløsning 2 og 3 og 4 til 7 kan pooles hver for sig (idet de nødvendige koncentrationer tages i betragtning). |
|
— |
For at få opnå længere holdbarhed autoklaveres stamopløsningerne ved 121 °C i 20 min., eller sterilfiltreres (0,2 µm) som alternativ. Brug af sterilfiltrering (0,2 µm) tilrådes kraftigt til stamopløsning 8. |
Fremstilling af den endelige koncentration af STEINBERG-substratet (modificeret)
|
— |
Der tilsættes 20 ml stamopløsning 1, 2 og 3 (se tabel 2) til ca. 900 ml deioniseret vand for at undgå udfældning. |
|
— |
Der tilsættes 1,0 ml stamopløsning 4, 5, 6, 7 og 8 (se tabel 3). |
|
— |
pH skal være 5,5 ± 0,2 (justeres ved tilsætning af mindst mulig NaOH-opløsning eller HCl). |
|
— |
Der fyldes op med vand til 1 000 ml. |
|
— |
Hvis stamopløsningerne er steriliseret, og det anvendte vand er af den korrekte kvalitet, kræves ingen yderligere sterilisering. Hvis steriliseringen finder sted på det endelige substrat, skal stamopløsning 8 tilsættes efter autoklavering (ved 121 °C i 20 min.). |
Fremstilling af det 10-dobbelt koncentrerede STEINBERG-substrat (modificeret) til midlertidig opbevaring
|
— |
Der tilsættes 20 ml stamopløsning 1, 2 og 3 (se tabel 2) til ca. 30 ml vand for at undgå udfældning. |
|
— |
Der tilsættes 1,0 ml stamopløsning 4, 5, 6, 7 og 8 (se tabel 3). Der fyldes op med vand til 100 ml. |
|
— |
Hvis stamopløsningerne er steriliseret, og det anvendte vand er af den korrekte kvalitet, kræves ingen yderligere sterilisering. Hvis steriliseringen finder sted på det endelige substrat, skal stamopløsning 8 tilsættes efter autoklavering (ved 121 °C i 20 min.). |
|
— |
pH af substratet (i den endelige koncentration) skal være 5,5 ± 0,2. |
(1) Medmindre noteret.