|
(8)
|
I del B indsættes følgende kapitler:
"B.63 SCREENINGSTEST FOR REPRODUKTIONS-/UDVIKLINGSTOKSICITET
INDLEDNING
|
1.
|
Denne forsøgsmetode svarer til OECD-testvejledning (Test Guideline (TG)) 421 (2016). OECD-vejledningerne om testning af kemikalier bliver revideret regelmæssigt på baggrund af den videnskabelige udvikling. Den oprindelige screeningstestvejledning 421 blev vedtaget i 1995 med udgangspunkt i en protokol for en indledende screeningstest for reproduktionstoksicitet (»Preliminary Reproduction Toxicity Screening Test«), der blev drøftet på to ekspertmøder i henholdsvis London i 1990 (1) og Tokyo i 1992 (2).
|
|
2.
|
Denne forsøgsmetode er blevet opdateret med endepunkter for hormonforstyrrende stoffer som en opfølgning på de højt prioriterede aktiviteter, som OECD indledte i 1998 for at revidere eksisterende testvejledninger og udvikle nye testvejledninger for screening og testning af potentielt hormonforstyrrende stoffer (3). OECD TG 407 (undersøgelse af toksicitet ved gentagen dosering (28 dage, oral) i gnavere, kapitel B.7 i dette bilag) blev f.eks. i 2008 forbedret med parametre, der kan påvise testkemikaliers endokrine virkemåde. Formålet med at opdatere testvejledning 421 var at indlemme endepunkter for hormonforstyrrende stoffer i testvejledningerne om screening, hvor eksponeringsperioderne omfatter følsomme udviklingsmæssige perioder (prænatale eller tidligt postnatale perioder).
|
|
3.
|
De valgte supplerende endepunkter for hormonforstyrrende stoffer, som også er at finde i testvejledning 443 (udvidet reproduktionstoksicitetsundersøgelse i én generation, kapitel B.56 i dette bilag), blev indlemmet i testvejledning 421 på baggrund af en feasibilityundersøgelse, hvor der blev set på de videnskabelige og tekniske spørgsmål i forbindelse med deres indlemmelse samt eventuelle tilpasninger af testens udformning som følge heraf (4).
|
|
4.
|
Denne forsøgsmetode er beregnet til at generere begrænsede oplysninger om et testkemikalies virkninger på han- og hundyrs reproduktionsevne, herunder kønskirtlernes funktion, parringsadfærd, befrugtning, conceptus’ udvikling og fødsel. Den er ikke et alternativ til og erstatter ikke de eksisterende forsøgsmetoder B.31, B.34, B.35 og B.56.
|
INDLEDENDE OVERVEJELSER
|
5.
|
Denne screeningsforsøgsmetode kan anvendes til at indsamle indledende oplysninger om eventuelle virkninger på reproduktion og/eller udvikling, enten tidligt i vurderingen af kemikaliers toksikologiske egenskaber eller i forbindelse med problematiske kemikalier. Den kan også anvendes som led i en række indledende screeningstest af eksisterende kemikalier, om hvilke der kun foreligger få eller ingen toksikologiske oplysninger, som en forundersøgelse til bestemmelse af dosisinterval ved mere omfattende reproduktions-/udviklingsrelaterede undersøgelser, eller når det ellers er relevant. Ved gennemførelse af undersøgelsen følges de vejledende principper og overvejelser, der er anført i OECD’s vejledende dokument nr. 19 om anerkendelse, vurdering og anvendelse af kliniske tegn som humane endepunkter for forsøgsdyr, der bruges i sikkerhedsevalueringer (5).
|
|
6.
|
Denne forsøgsmetode giver ikke udtømmende oplysninger om alle aspekter af reproduktion og udvikling. Den giver navnlig kun begrænsede muligheder for at påvise postnatale tegn på prænatal eksponering eller virkninger, der induceres under postnatal eksponering. Bl.a. som følge af det relativt lave antal dyr i doseringsgrupperne, endepunkternes selektivitet og undersøgelsens korte varighed kan denne metode ikke bruges som endelig dokumentation for, at kemikalier ikke har nogen virkning. Når der ikke foreligger data fra andre reproduktions-/udviklingstoksicitetstest, kan positive resultater desuden anvendes til indledende farevurdering og som udgangspunkt for beslutninger om nødvendigheden af og tidsplanen for yderligere testning.
|
|
7.
|
De resultater, der opnås ved hjælp af de endokrinrelaterede parametre, bør ses i sammenhæng med »OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals« (6). Inden for denne begrebsramme er den forbedrede OECD TG 421 omfattet af niveau 4 som et in vivo-assay, der giver data om skadelige virkninger på endokrinrelevante endepunkter. Et endokrint signal alene vil dog ikke nødvendigvis blive opfattet som tilstrækkelig dokumentation for, at testkemikaliet er et hormonforstyrrende stof.
|
|
8.
|
I forsøgsmetoden indgives testkemikaliet oralt. Det kan være nødvendigt at ændre forsøgsmetoden, hvis der anvendes andre eksponeringsveje.
|
|
9.
|
Før forsøgsmetoden bruges på en blanding for at generere data i lovgivningsøjemed, bør det overvejes, om den vil give resultater, som er egnede til formålet, og i givet fald hvorfor. Sådanne overvejelser er ikke nødvendige, når der foreligger et myndighedskrav om test af blandingen.
|
|
10.
|
De anvendte definitioner findes i tillæg 1.
|
PRINCIP FOR TESTEN
|
11.
|
Testkemikaliet indgives i graduerede doser til flere grupper hanner og hunner. Hannerne doseres i mindst fire uger og frem til og med dagen før planlagt aflivning (dette omfatter mindst to uger før parring, under parringsperioden og ca. to uger efter parring). I lyset af hannernes begrænsede doseringsperiode inden parring er deres fertilitet ikke nødvendigvis nogen særlig følsom indikator for testikeltoksicitet. Derfor er det særdeles vigtigt, at der foretages en detaljeret histologisk undersøgelse af testiklerne. Kombinationen af en doseringsperiode på to uger inden parring og efterfølgende parrings-/fertilitetsobservationer og en samlet doseringsperiode på mindst fire uger efterfulgt af en detaljeret histopatologisk undersøgelse af hannernes kønskirtler anses for at være tilstrækkeligt til at påvise størstedelen af virkningerne på hannernes fertilitet og spermatogenese.
|
|
12.
|
Hunnerne doseres gennem hele undersøgelsen. Dette omfatter to uger inden parring (med det formål at dække mindst to hele brunstcyklusser), det variable tidsrum frem til befrugtning, drægtighedsperioden og mindst 13 dage efter fødslen frem til og med dagen før planlagt aflivning.
|
|
13.
|
Undersøgelsens varighed efter tilvænning til forholdene og vurdering af brunstcyklussen før dosering afhænger af de enkelte hunner og er på ca. 63 dage [mindst 14 dage før parring, (op til) 14 dages parring, 22 dages drægtighed og 13 dages laktation].
|
|
14.
|
I eksponeringsperioden observeres dyrene hver dag omhyggeligt for tegn på toksicitet. Dyr, der dør eller aflives i løbet af forsøgsperioden, obduceres, og de overlevende dyr aflives og obduceres ved forsøgets afslutning.
|
BESKRIVELSE AF METODEN
Valg af dyreart
|
15.
|
Denne forsøgsmetode er beregnet til rotter. Hvis de parametre, der er anført i denne forsøgsmetode, undersøges hos andre gnaverarter, gives en detaljeret begrundelse herfor. I det internationale valideringsprogram for påvisning af hormonforstyrrende stoffer i OECD TG 407 (som svarer til kapitel B.7 i dette bilag) blev der kun anvendt rotter. Der bør ikke anvendes stammer med ringe frugtbarhed eller kendt høj forekomst af udviklingsdefekter. Der bør anvendes sunde jomfruelige dyr, som ikke tidligere har været brugt i forsøg. Forsøgsdyrene beskrives med hensyn til art, stamme, køn, vægt og alder. Ved undersøgelsens begyndelse bør vægtvariationen mellem de anvendte dyr være mindst mulig og ikke over 20 % af gennemsnitsvægten for hvert køn. Hvis undersøgelsen gennemføres som en forundersøgelse til en længerevarende undersøgelse eller en undersøgelse af en hel generation, bør der så vidt muligt anvendes dyr fra samme stamme og af samme oprindelse i begge undersøgelser.
|
Miljø og fodring
|
16.
|
Alle procedurer skal overholde lokale standarder for pasning af forsøgsdyr. Temperaturen i forsøgslokalet skal være 22 °C (± 3 °C). Den relative luftfugtighed skal være mindst 30 % og helst ikke over 70 %, hvilket dog ikke gælder under rengøring af lokalet, men 50-60 % bør tilstræbes. Belysningen skal være kunstig og bestå af 12 timers lys og 12 timers mørke. For så vidt angår fodring, kan der anvendes konventionelt laboratoriefoder med ubegrænset adgang til vand. Valget af foder kan påvirkes af behovet for at sikre en passende iblanding af et testkemikalie, når dette indgives ad denne vej.
|
|
17.
|
Dyrene bør anbringes i bure med små grupper af samme køn. Dyrene kan anbringes enkeltvis, hvis dette er sagligt begrundet. Hvis flere dyr anbringes i samme bur, bør der højst være fem dyr pr. bur. Parring bør finde sted i bure, der er egnede til formålet. Drægtige hunner bør anbringes alene i et bur og have adgang til redemateriale. Lakterende hunner anbringes alene i et bur sammen med deres afkom.
|
|
18.
|
Foderet bør analyseres regelmæssigt for forurenende stoffer. En prøve af foderet bør opbevares, indtil rapporten er udarbejdet.
|
Klargøring af dyrene
|
19.
|
Sunde unge voksne dyr udvælges tilfældigt til kontrol- og behandlingsgrupperne. Burene anbringes på en sådan måde, at de mulige følger af burenes placering minimeres. Dyrene identificeres entydigt og holdes i deres bure i mindst fem dage, inden undersøgelsen påbegyndes, så de kan vænne sig til laboratorieforholdene.
|
Fremstilling af doser
|
20.
|
Det anbefales, at testkemikaliet indgives oralt, medmindre andre indgivelsesveje er bedre egnede. Ved oral indgivelse indgives testkemikaliet normalt med sonde. Alternativt kan testkemikalier indgives via foder eller drikkevand.
|
|
21.
|
Hvor det er nødvendigt, opløses eller suspenderes kemikaliet i et egnet bærestof. Det anbefales, at man først vurderer muligheden for at benytte en vandig opløsning/suspension, derefter en opløsning/emulsion i olie (f.eks. majsolie) og først derefter eventuel opløsning i andre bærestoffer. For andre bærestoffer end vand skal bærestoffets toksiske egenskaber være kendt. Testkemikaliets stabilitet og homogenitet i bærestoffet skal bestemmes.
|
FREMGANGSMÅDE
Dyrenes antal og køn
|
22.
|
Det anbefales, at hver gruppe startes op med mindst 10 hanner og 12-13 hunner. Hunnernes brunstcyklus vurderes før eksponering, og dyr, der ikke udviser typiske fire-fem-dages cyklusser, medtages ikke i undersøgelsen. Derfor anbefales det at starte ud med ekstra hunner for at nå op på 10 hunner pr. gruppe. Medmindre der er tale om markante toksiske virkninger, forventes det, at dette vil give mindst otte drægtige hunner pr. gruppe, hvilket normalt er det lavest accepterede antal drægtige hunner pr. gruppe. Formålet hermed er at sikre, at der produceres tilstrækkelig meget afkom til, at der kan foretages en meningsfuld vurdering af testkemikaliets potentielle virkning på fertilitet, drægtighed, moderdyrenes adfærd og dieadfærd samt vækst og udvikling i F1-generationen fra befrugtning og frem til dag 13 efter fødslen.
|
Dosering
|
23.
|
Der bør anvendes generelt mindst tre testgrupper og en kontrolgruppe. Dosisniveauerne kan baseres på oplysninger fra test af akut toksicitet eller på resultater fra undersøgelser med gentagen dosering. Bortset fra behandlingen med testkemikaliet bør dyr i kontrolgruppen behandles på samme måde som testgruppens dyr. Hvis der anvendes et bærestof til indgivelse af testkemikaliet, bør kontrolgruppen have den samme mængde bærestof som den gruppe, der modtager den største mængde bærestof.
|
|
24.
|
Dosisniveauerne bør udvælges under hensyntagen til eventuelle foreliggende data om toksicitet og (toksiko)kinetik. Der bør endvidere tages hensyn til, at der kan være forskel på følsomheden hos drægtige og ikke-drægtige dyr. Det højeste dosisniveau bør vælges med henblik på at fremkalde toksiske virkninger, men ikke død eller alvorlig lidelse. Derefter bør en aftagende række dosisniveauer vælges, som giver mulighed for at påvise et eventuelt dosis/effekt-forhold, og som laveste dosis et niveau, hvor ingen skadelige virkninger observeres (NOAEL). Ved fastlæggelse af den aftagende række dosisniveauer svarer de optimale intervaller ofte til en faktor på to til fire, og i mange tilfælde må tilføjelse af en fjerde behandlingsgruppe foretrækkes frem for meget store intervaller (f.eks. større end en faktor 10) mellem doseringerne.
|
|
25.
|
Hvis der observeres generel toksicitet (f.eks. reduceret kropsvægt, virkninger på lever, hjerte, lunger eller nyrer osv.) eller andre forandringer, der eventuelt ikke er toksiske virkninger (f.eks. reduceret indtagelse af føde eller forstørret lever), bør de observerede virkninger på de endokrinfølsomme endepunkter fortolkes med forsigtighed.
|
Grænsetest
|
26.
|
Såfremt et oralt enkeltdosisniveau på mindst 1 000 mg/kg kropsvægt/dag eller en ækvivalent procentdel tilsat foderet eller drikkevandet med de til dette forsøg beskrevne metoder ikke frembringer observerbare toksiske virkninger, og hvis der ikke på grundlag af data fra strukturelt beslægtede stoffer kan ventes toksiske virkninger, kan en fuldstændig undersøgelse med anvendelse af flere dosisniveauer betragtes som unødvendig. Grænsetesten gælder, medmindre data om human eksponering viser, at der er behov for anvendelse af et højere oralt dosisniveau. For andre indgivelsesmåder – f.eks. inhalation eller påføring på huden – kan testkemikaliets fysisk-kemiske egenskaber ofte være bestemmende for den maksimalt opnåelige koncentration.
|
Indgivelse af doser
|
27.
|
Dyrene modtager testkemikaliet hver dag syv dage om ugen. Hvis testkemikaliet indgives med sonde, bør det gøres i en enkelt dosis med anvendelse af en gastrisk sonde eller et passende intubationsrør. Hvor stor en væskemængde, der kan indgives på én gang, afhænger af forsøgsdyrets størrelse. Denne mængde må højst være 1 ml/100 g kropsvægt, bortset fra vandige opløsninger, hvor der kan anvendes 2 ml/100 g kropsvægt. Hvis der ikke er tale om irriterende eller ætsende testkemikalier, hvis virkninger sædvanligvis forstærkes ved højere koncentrationer, bør variabiliteten af testmængden minimeres ved justering af koncentrationen, således at der sikres en konstant mængde ved alle dosisniveauer.
|
|
28.
|
Når testkemikalier indgives via foder eller drikkevand, er det vigtigt at sikre, at de pågældende mængder af testkemikaliet ikke griber forstyrrende ind i den normale ernærings- eller vandbalance. Når testkemikaliet indgives via foderet, kan det enten gives ved konstant koncentration i foderet (ppm) eller ved et konstant dosisniveau i forhold til dyrenes kropsvægt. Det anvendte alternativ bør anføres. Ved indgivelse med sonde bør dosis gives på nogenlunde samme tidspunkt hver dag og justeres mindst en gang om ugen, så dosisniveauet holdes konstant i forhold til kropsvægten.
|
Forsøgsplan
|
29.
|
Doseringen af begge køn påbegyndes senest to uger før parring, efter at dyrene har haft mindst fem dage til at vænne sig til laboratorieforholdene, og hunnerne er blevet screenet for en normal brunstcyklus (i en tougersperiode forud for behandling). Undersøgelsen planlægges således, at vurderingen af brunstcyklus påbegyndes hurtigt efter, at dyrene er blevet fuldt kønsmodne. Dette kan variere en smule for de forskellige rottestammer i forskellige laboratorier og er f.eks. for Sprague Dawley-rotter, når dyrene er 10 uger gamle, og for Wistar-rotter, når de er omkring 12 uger gamle. Moderdyr og deres afkom aflives dag 13 efter fødslen eller umiddelbart derefter. Dagen for fødslen (dvs. hvor fødslen afsluttes) defineres som dag 0 efter fødslen. Hunner, der ikke udviser nogen tegn på kopulation, aflives 24-26 dage efter den sidste dag i parringsperioden. Doseringen fortsættes hos begge køn i parringsperioden. Hannerne bør stadig doseres efter parringsperioden, og mindst indtil den samlede doseringsperiode på mindst 28 dage er afsluttet. Derefter aflives de, eller doseringen fortsættes med henblik på eventuel fornyet parring, hvis dette findes hensigtsmæssigt.
|
|
30.
|
Moderdyrene doseres hver dag gennem hele drægtighedsperioden og mindst frem til og med dag 13 efter fødslen eller dagen før aflivning. I undersøgelser, hvor testkemikaliet indgives ved inhalation eller påføring på huden, bør doseringen fortsættes mindst frem til og med dag 19 i drægtighedsperioden, og doseringen bør genoptages hurtigst muligt og senest PND 4.
|
|
31.
|
Der findes et diagram over forsøgsplanen i tillæg 2.
|
Parringsmetode
|
32.
|
I denne undersøgelse bør der normalt anvendes 1:1-parring (én han og én hun). Dette princip kan fraviges, hvis hannen skulle dø. Hunnen bør anbringes sammen med den samme han, indtil der observeres tegn på kopulation, eller indtil der er gået to uger. Hunnerne undersøges hver morgen for tilstedeværelse af sperma eller vaginalprop. Dag 0 i drægtighedsperioden defineres som den dag, hvor tegn på parring bekræftes (der observeres vaginalprop eller sperma). Lykkes parring ikke, kan fornyet parring af hunnerne med afprøvede hanner fra samme gruppe overvejes.
|
Kuldstørrelse
|
33.
|
På dag 4 efter fødslen standardiseres hvert kuld, ved at overskydende unger fjernes ved vilkårlig udvælgelse, så hvert kuld i videst muligt omfang kommer til at bestå af fire eller fem unger pr. køn, alt efter den normale kuldstørrelse i den anvendte rottestamme. Der bør tages blodprøver fra to af de overskydende unger, som samles og bruges til bestemmelse af T4-niveauer i serum. Selektiv fjernelse af unger, f.eks. baseret på kropsvægt eller anogenital afstand (AGD), er ikke hensigtsmæssig. Når antallet af han- eller hununger gør det umuligt at få fire eller fem af hvert køn i hvert kuld, er en delvis standardisering (f.eks. seks hanner og fire hunner) acceptabel. Ingen unger fjernes, når et kuld er på mindre end målet for fjernelse (otte eller 10 unger/kuld). Hvis der kun er én unge over målet for fjernelse, vil der kun blive fjernet én unge, som vil blive brugt til indsamling af blod til eventuel vurdering af T4 i serum.
|
|
34.
|
Hvis ikke kuldstørrelsen standardiseres, aflives to unger pr. kuld dag 4 efter fødslen, og der tages blodprøver til måling af thyreoideahormonkoncentrationer i serum. Hvis det er muligt, skal disse to unger pr. kuld være hununger, så hanungerne bevares til vurdering af bibeholdelsen af brystvorter, medmindre fjernelsen af disse to unger betyder, at der ikke er flere hunner tilbage til afsluttende vurdering. Der fjernes ingen unger, når kuldet er på mindre end otte eller 10 unger/kuld (alt efter den normale kuldstørrelse i den anvendte rottestamme). Hvis der kun er én overskydende unge i forhold til den normale kuldstørrelse, vil der kun blive fjernet én unge, som vil blive brugt til indsamling af blod til eventuel vurdering af T4 i serum.
|
Observationer i levende dyr
Kliniske observationer
|
35.
|
Gennem hele testperioden bør der foretages generelle kliniske observationer mindst én gang om dagen og hyppigere, hvis der observeres tegn på toksicitet. Observationerne bør fortrinsvis finde sted på samme tidspunkt(er) hver dag og under hensyntagen til det tidspunkt, hvor de forventede virkninger efter dosering topper. Relevante adfærdsændringer, tegn på vanskelig eller forlænget fødsel og alle toksicitetstegn, herunder mortalitet, bør registreres. Disse registreringer bør omfatte tidspunktet for toksicitetstegnenes indtræden, grad og varighed.
|
Kropsvægt og foder-/vandindtagelse
|
36.
|
Han- og hundyr bør vejes den dag, doseringen indledes, og derefter mindst en gang om ugen og ved forsøgets afslutning. I drægtighedsperioden bør hunnerne vejes på dag 0, 7, 14 og 20 og senest 24 timer efter fødslen (dag 0 eller 1 efter fødslen) samt mindst på dag 4 og 13 efter fødslen. Disse observationer bør rapporteres individuelt for hvert voksent dyr.
|
|
37.
|
Før parring og i drægtigheds- og laktationsperioden bør foderindtaget måles mindst en gang om ugen. Måling af foderindtaget i parringsperioden er valgfrit. Hvis testkemikaliet indgives via drikkevandet, bør vandindtagelsen i disse perioder også måles.
|
Brunstcyklusser
|
38.
|
Brunstcyklusserne bør overvåges, inden behandlingen indledes, for at udvælge de hunner til undersøgelsen, der har en regelmæssig cyklus (jf. punkt 22). Vaginalt smear bør også overvåges dagligt fra behandlingens indledning, og indtil der findes tegn på parring. Hvis der er problemer med akutte stressvirkninger, der kan ændre brunstcyklussen, når doseringen indledes, kan laboratorierne eksponere forsøgsdyrene i to uger og derefter indsamle vaginalt smear hver dag i mindst to uger før parring for at overvåge brunstcyklussen og fortsætte overvågningen i parringsperioden, indtil der findes tegn på parring. Udtagning af celleprøver fra vagina/cervix skal ske forsigtigt for at undgå at forstyrre slimhinden, hvilket ellers kunne inducere pseudodrægtighed (7) (8).
|
Parametre for afkom
|
39.
|
Drægtighedsperioden bør registreres og beregnes fra drægtighedsdag 0. Hvert kuld undersøges hurtigst muligt efter fødslen til bestemmelse af ungernes antal og køn, dødfødte, levendefødte, små uudviklede unger (unger, der er signifikant mindre end ungerne i kontrolgruppen) og tilstedeværelse af alvorlige abnormiteter.
|
|
40.
|
De levende unger tælles, og deres køn bestemmes, og kuldet vejes senest 24 timer efter fødslen (dag 0 eller 1 efter fødslen) samt mindst på dag 4 og 13 efter fødslen. Ud over observationerne i punkt 35 bør al unormal adfærd hos afkommet registreres.
|
|
41.
|
Ungernes AGD bør måles den samme dag efter fødslen mellem PND 0 og PND 4. Ungernes kropsvægt bør registreres den dag, AGD måles, og AGD bør normaliseres til et mål for ungernes størrelse, helst kubikroden af kropsvægten (9). Antallet af brystvorter/areolae hos hanungerne tælles på PND 12 eller 13 som anbefalet i OECD GD 151 (10).
|
Klinisk biokemi
|
42.
|
Der tages blodprøver fra et bestemt sted i henhold til nedenstående:
|
—
|
fra mindst to unger pr. kuld på dag 4 efter fødslen, hvis antallet af unger gør det muligt (jf. punkt 33-34)
|
|
—
|
fra alle moderdyr og mindst to unger pr. kuld ved forsøgets afslutning på dag 13 og
|
|
—
|
fra alle voksne hanner ved forsøgets afslutning.
|
|
Alle blodprøver opbevares under egnede forhold. Blodprøver fra dag 13-unger og voksne hanner undersøges for niveauet af thyreoideahormoner (T4) i serum. Der kan eventuelt foretages yderligere vurdering af T4 i blodprøver fra moderdyr og dag 4-unger. Andre hormoner kan eventuelt også måles. Blod fra unger til thyreoideahormonanalyse kan samles pr. kuld. Thyreoideahormoner (T4 og TSH) bør fortrinsvis måles som en samlet værdi.
|
43.
|
Følgende faktorer kan påvirke variabiliteten og de absolutte koncentrationer i hormonanalyserne:
|
—
|
aflivningstidspunkt på grund af de forskellige hormonkoncentrationer i løbet af døgnet
|
|
—
|
aflivningsmetode, der har til formål at undgå unødig stress af dyrene, som kan påvirke hormonkoncentrationerne
|
|
—
|
analysesæt til hormonanalyser, der kan have forskellige standardkurver.
|
|
|
44.
|
Plasmaprøver, der specifikt er beregnet til hormonanalyse, tages på nogenlunde samme tidspunkt på dagen. De numeriske værdier, der fås ved analyse af hormonkoncentrationer, varierer ved anvendelse af forskellige analysesæt på markedet.
|
Patologi
Makroskopisk undersøgelse
|
45.
|
På aflivningstidspunktet eller ved død under forsøget undersøges de voksne dyr makroskopisk for eventuelle abnormiteter eller patologiske forandringer. Særlig opmærksomhed bør rettes mod kønsorganerne. Antallet af implantationssteder bør registreres. Det vaginale smear bør undersøges om morgenen på obduktionsdagen for at bestemme stadiet i brunstcyklussen og muliggøre korrelation med den histopatologiske undersøgelse af ovarierne.
|
|
46.
|
På alle voksne handyr renses testikler og bitestikler samt prostata og vesiculae seminales og prostata anterior som én samlet enhed for alt vedhængende væv, og de vejes hurtigst muligt (våd vægt) efter dissektion, så udtørring undgås. Derudover kan eventuelle organvejninger omfatte levator ani plus bulbocavernosus-muskelkomplekset, Cowpers kirtler og glans penis i hanner samt parrede ovarier (våd vægt) og uterus (herunder cervix) i hunner. Disse valgfrie vejninger bør foretages hurtigst muligt efter dissektion.
|
|
47.
|
Døde unger og unger, der aflives på dag 13 efter fødslen eller hurtigt derefter, bør som minimum undersøges omhyggeligt på kroppens ydre for større abnormiteter. Særlig opmærksomhed bør udvises over for de eksterne kønsorganer, som bør undersøges for tegn på forandringer i udviklingen. På dag 13 præserveres skjoldbruskkirtlen fra en han- og en hununge pr. kuld.
|
|
48.
|
Ovarier, testikler, sekundære kønsorganer (uterus og cervix, bitestikler, prostata, vesiculae seminales og prostata anterior), skjoldbruskkirtel og alle organer, der udviser makroskopiske læsioner, fra alle voksne dyr præserveres. Fiksering med formalin anbefales ikke til rutinemæssig undersøgelse af testikler og bitestikler. Anvendelse af Bouins væske eller modificeret Davidson-fikseringsmiddel er en acceptabel metode til præservering af dette væv (11). Tunica albuginea kan punkteres forsigtigt og overfladisk i begge ender af organet med en kanyle, der muliggør hurtig penetration af fikseringsmidlet.
|
Histopatologisk undersøgelse
|
49.
|
Der bør foretages en detaljeret histologisk undersøgelse af ovarier, testikler og bitestikler (hvor der lægges særlig vægt på spermatogenesens faser og histopatologien af den interstitielle cellestruktur i testiklerne) fra dyrene i højdosisgruppen og kontrolgruppen. De øvrige præserverede organer, herunder skjoldbruskkirtler fra unger og voksne dyr, kan undersøges om nødvendigt. Skjoldbruskkirtlens vægt kan bestemmes efter fiksering. Rensningen for vedhængende væv bør også foretages meget omhyggeligt og først efter fiksering for at undgå beskadigelse af vævet. Beskadiget væv kan kompromittere den histopatologiske analyse. Undersøgelserne skal udvides til dyrene i de andre doseringsgrupper, hvis der observeres ændringer i højdosisgruppen. I vejledningen om histopatologi (11) findes der flere oplysninger om dissektion, fiksering, udtagning af snit og histopatologisk undersøgelse af endokrine væv.
|
DATA OG RAPPORTERING
Data
|
50.
|
Der bør fremlægges data om hvert enkelt dyr. Derudover bør alle data sammenfattes i tabelform med angivelse for hver forsøgsgruppe af antal dyr ved forsøgets begyndelse, antal dyr, som er fundet døde under forsøget eller aflivet af humane grunde, tidspunkt for eventuel død eller human aflivning, antal fertile dyr, antal drægtige hundyr, antal dyr, som viser tegn på toksisk virkning, beskrivelse af iagttagne toksicitetstegn, herunder begyndelsestidspunkt, varighed og sværhed af alle de toksiske virkninger, arten af de histopatologiske forandringer og alle relevante data om kuldene. Tillæg 3 indeholder en sammenfattende rapport i tabelform, der har vist sig at være særdeles velegnet til vurdering af reproduktions-/udviklingsmæssige virkninger.
|
|
51.
|
Som følge af undersøgelsens begrænsede omfang er de statistiske analyser i form af test for »signifikans« af begrænset værdi for mange endepunkter, især de reproduktionsrelaterede endepunkter. Hvis der anvendes statistiske analyser, bør den valgte metode være egnet til fordeling af de undersøgte variabler og vælges, inden undersøgelsen indledes. Statistiske analyser af AGD og bibeholdelse af brystvorter (»nipple retention«) bør baseres på data fra de enkelte unger under hensyntagen til virkninger på de enkelte kuld. Når det er hensigtsmæssigt, er det kuldene, der er analyseenheden. Statistiske analyser af ungernes kropsvægt bør baseres på data fra de enkelte unger under hensyntagen til kuldstørrelsen. På grund af den lille gruppestørrelse kan anvendelsen af historiske kontroldata (om f.eks. kuldstørrelse), når sådanne foreligger, også være en hjælp i forbindelse med fortolkningen af undersøgelsesresultaterne.
|
Vurdering af resultater
|
52.
|
Resultaterne af denne toksicitetsundersøgelse bør vurderes med hensyn til de observerede virkninger samt obduktionsfund og mikroskopiske fund. Vurderingen skal omfatte sammenhæng mellem dyrenes eksponering for testkemikaliet og tilstedeværelse eller fravær, forekomst og sværhedsgrad af abnormiteter, herunder makroskopiske læsioner, identificerede målorganer, manglende fertilitet, unormale kliniske fund, påvirkning af reproduktions- og ynglepræstationer, ændringer af kropsvægt, indvirkning på mortaliteten og eventuelle andre toksiske virkninger.
|
|
53.
|
På grund af hannernes korte behandlingsperiode bør histopatologien for testikler og bitestikler ses i sammenhæng med fertilitetsdataene ved vurdering af den reproduktionsmæssige virkning på hannerne. Anvendelsen af historiske kontroldata om reproduktion/udvikling (f.eks. med hensyn til kuldstørrelse, AGD, bibeholdelse af brystvorter (»nipple retention«), T4-niveauer i serum) kan også, når sådanne foreligger, være en hjælp i forbindelse med fortolkningen af undersøgelsesresultaterne.
|
|
54.
|
Med henblik på kvalitetskontrol foreslås det, at historiske kontroldata indsamles, og at der beregnes variationskoefficienter for numeriske data, især for parametre med relation til påvisning af hormonforstyrrende virkninger. Disse data kan bruges i forbindelse med sammenligninger, når de faktiske undersøgelser evalueres.
|
Testrapport
|
55.
|
Testrapporten bør indeholde følgende oplysninger:
|
|
Testkemikalie:
|
—
|
oprindelse, batchnummer, sidste anvendelsesdato, hvis dette foreligger
|
|
—
|
testkemikaliets stabilitet, hvis kendt.
|
|
|
|
Stof med kun én bestanddel:
|
—
|
fysisk udseende, vandopløselighed og yderligere relevante fysisk-kemiske egenskaber
|
|
—
|
kemisk identifikation, f.eks. IUPAC-navn eller CAS-navn, CAS-nummer, SMILES- eller InChI-kode, strukturformel, renhed, kemisk identitet af urenheder, i det omfang det er relevant og praktisk muligt, osv.
|
|
|
|
Stof med flere bestanddele, UVCB-stoffer og blandinger:
|
—
|
kendetegnet så vidt muligt ved kemisk identitet (jf. ovenfor), kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber.
|
|
|
|
Eventuelt bærestof:
|
—
|
begrundelse for valg af bærestof, hvis dette ikke er vand.
|
|
|
|
Forsøgsdyr:
|
—
|
den anvendte art/stamme
|
|
—
|
dyrenes antal, alder og køn
|
|
—
|
oprindelse, miljøbetingelser, foder mv.
|
|
—
|
de enkelte dyrs vægt ved testens begyndelse
|
|
—
|
begrundelse for andre arter end rotte.
|
|
|
|
Testbetingelser:
|
—
|
begrundelse for valg af dosisniveau
|
|
—
|
oplysninger om formulering af testkemikalie eller iblanding heraf i foderet, opnået koncentration, foderblandingens stabilitet og homogenitet
|
|
—
|
oplysninger om indgivelse af testkemikaliet
|
|
—
|
eventuel omregning af testkemikaliets koncentration (ppm) i foder/drikkevand til faktisk dosis (mg pr. kg kropsvægt pr. dag)
|
|
—
|
oplysninger om foder- og vandkvalitet
|
|
—
|
detaljeret beskrivelse af randomiseringsproceduren til eventuel udvælgelse af unger til fjernelse.
|
|
|
|
Resultater:
|
—
|
kropsvægt og ændringer i kropsvægt
|
|
—
|
foderindtagelse og indtagelse af vand, hvis disse data foreligger
|
|
—
|
oplysninger om toksisk reaktion inddelt efter køn og dosis, herunder fertilitet, drægtighed og andre tegn på toksicitet
|
|
—
|
drægtighedsperiodens længde
|
|
—
|
toksiske eller andre virkninger på reproduktion, afkom, postnatal vækst osv.
|
|
—
|
art, sværhedsgrad og varighed af kliniske symptomer (hvad enten disse er reversible eller ej)
|
|
—
|
antal voksne hunner med normal eller anormal brunstcyklus og cyklusvarighed
|
|
—
|
antal levendefødte og postimplantationstab
|
|
—
|
data om ungernes kropsvægt
|
|
—
|
AGD for alle unger (og kropsvægt på dagen for AGD-måling)
|
|
—
|
bibeholdelse af brystvorter (»Nipple retention«) hos hanunger
|
|
—
|
thyreoideahormonniveauer, dag 13-unger og voksne hanner (og eventuelt moderdyr og dag 4-unger)
|
|
—
|
antal unger med makroskopisk synlige abnormiteter, makroskopisk vurdering af eksterne genitalia, antal små uudviklede unger
|
|
—
|
dødstidspunkt under undersøgelsen, eller hvorvidt dyrene overlevede til undersøgelsens afslutning
|
|
—
|
antal implantationer, kuldstørrelse og kuldenes vægt på registreringstidspunktet
|
|
—
|
kropsvægt ved aflivning og organvægt for forældredyrene
|
|
—
|
detaljeret beskrivelse af alle histopatologiske fund
|
|
—
|
absorptionsdata (hvis de foreligger)
|
|
—
|
når det er hensigtsmæssigt, statistisk behandling af resultaterne.
|
|
Diskussion af resultater
Konklusioner
|
Fortolkning af resultater
|
56.
|
Ved denne undersøgelse vurderes den reproduktions-/udviklingsmæssige toksicitet i forbindelse med gentagen dosering (jf. punkt 5 og 6). Den kan vise, om det er nødvendigt at foretage yderligere undersøgelser, og udstikker retningslinjer for udformningen af efterfølgende undersøgelser. Med hensyn til fortolkningen af de reproduktions- og udviklingsrelaterede resultater henvises til OECD’s vejledende dokument 43 (12). OECD’s vejledende dokument nr. 106 om histologisk vurdering af endokrine og reproduktionsrelaterede test i gnavere (11) indeholder oplysninger om præpareringen og vurderingen af (endokrine) organer og vaginalt smear, der kan være nyttige i forbindelse med denne testvejledning.
|
LITTERATUR
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic and Cooperation and Development, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Available Upon Request at Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Available Upon Request at Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. og Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. og Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. og Reynolds V.L. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organisation for Economic Cooperation and Development, Paris.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No106), Organisation for Economic Cooperation and Development, Paris.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
Tillæg 1
DEFINITIONER (SE OGSÅ OECD GD 150 (6))
Androgenicitet: et kemikalies evne til at fungere som et naturligt androgent hormon (f.eks. testosteron) i en pattedyrsorganisme.
Antiandrogenicitet: et kemikalies evne til at undertrykke virkemåden for et naturligt androgent hormon (f.eks. testosteron) i en pattedyrsorganisme.
Antiøstrogenicitet: et kemikalies evne til at undertrykke virkemåden for et naturligt østrogent hormon (f.eks. østradiol 17ß) i en pattedyrsorganisme.
Antithyreoid aktivitet: et kemikalies evne til at undertrykke virkemåden for et naturligt thyreoideahormon (f.eks. T3) i en pattedyrsorganisme.
Kemikalie: et stof eller en blanding.
Udviklingstoksicitet: manifestering af reproduktionstoksicitet, som repræsenterer prænatale, perinatale, postnatale, strukturelle eller funktionsmæssige forstyrrelser i afkommet.
Dosering: et generelt udtryk, som omfatter dosis samt doseringsfrekvens og -varighed.
Dosis: mængden af indgivet testkemikalie. Dosis angives som vægt af testkemikalie pr. enhed af forsøgsdyrets vægt pr. dag (f.eks. mg/kg kropsvægt/dag) eller som en konstant koncentration i foderet.
Synlig toksicitet: et generelt udtryk, som angiver klare tegn på toksicitet efter indgivelse af testkemikaliet. Disse tegn bør være tilstrækkelige til farevurdering og bør være af en sådan karakter, at en forøgelse af den indgivne dosis må forventes at resultere i udvikling af svære toksiske virkninger og sandsynligvis død.
Fertilitetsforringelse: forstyrrelse af hanners eller hunners reproduktionsfunktioner eller -evne.
Toksisk virkning på moderdyret: skadelig virkning på drægtige hunner, der opstår enten specifikt (direkte virkning) eller ikke-specifikt (indirekte virkning).
NOAEL: forkortelse for "no-observed-adverse-effect level" (niveau, hvor ingen skadelige virkninger observeres). Dette er det højeste dosisniveau, hvor der ikke observeres behandlingsrelaterede skadelige virkninger.
Østrogenicitet: et kemikalies evne til at fungere som et naturligt østrogent hormon (f.eks. østradiol 17ß) i en pattedyrsorganisme.
Reproduktionstoksicitet: fremkaldelse af skadelige virkninger på afkommet og/eller forringelse af hanners og hunners reproduktionsfunktioner eller -evne.
Testkemikalie: et stof eller en blanding, der testes med denne forsøgsmetode.
Thyreoid aktivitet: et kemikalies evne til at fungere som et naturligt thyreoideahormon (f.eks. T3) i en pattedyrsorganisme.
Validering: en videnskabelig proces til karakterisering af en forsøgsmetodes operationelle krav og begrænsninger og påvisning af dens pålidelighed og relevans til et bestemt formål.
Tillæg 2
DIAGRAM OVER FORSØGSPLANEN, SOM VISER DEN MAKSIMALE VARIGHED AF UNDERSØGELSEN BASERET PÅ EN FULD 14-DAGES PARRINGSPERIODE

Tillæg 3
SAMMENFATTENDE RAPPORT I TABELFORM OVER REPRODUKTIONS-/UDVIKLINGSMÆSSIGE VIRKNINGER
|
OBSERVATIONER
|
VÆRDIER
|
|
|
|
Dosering (enheder)
|
0 (kontrol)
|
…
|
…
|
…
|
…
|
|
Startede par (N)
|
|
|
|
|
|
|
Brunstcyklus (som minimum gennemsnitlig varighed og frekvens for uregelmæssige cyklusser)
|
|
|
|
|
|
|
Hunner, som viser tegn på kopulation (N)
|
|
|
|
|
|
|
Hunner, som bliver drægtige (N)
|
|
|
|
|
|
|
Undfangelse, dag 1-5 (N)
|
|
|
|
|
|
|
Undfangelse, dag 6-… (21) (N)
|
|
|
|
|
|
|
Drægtighed ≤ 21 dage (N)
|
|
|
|
|
|
|
Drægtighed = 22 dage (N)
|
|
|
|
|
|
|
Drægtighed 23 ≥ dage (N)
|
|
|
|
|
|
|
Moderdyr med levende nyfødte unger (N)
|
|
|
|
|
|
|
Moderdyr med levende unger på dag 4 efter fødslen (N)
|
|
|
|
|
|
|
Implantater/moderdyr (gennemsnit)
|
|
|
|
|
|
|
Levende unger/moderdyr ved fødslen (gennemsnit)
|
|
|
|
|
|
|
Levende unger/moderdyr på dag 4 (gennemsnit)
|
|
|
|
|
|
|
Kønsfordeling (han/hun) ved fødslen (gennemsnit)
|
|
|
|
|
|
|
Kønsfordeling (han/hun) på dag 4 (gennemsnit)
|
|
|
|
|
|
|
Kuldenes vægt ved fødslen (gennemsnit)
|
|
|
|
|
|
|
Kuldenes vægt på dag 4 (gennemsnit)
|
|
|
|
|
|
|
Ungernes vægt ved fødslen (gennemsnit)
|
|
|
|
|
|
|
Ungernes vægt på tidspunktet for AGD-måling (gennemsnit hanner, gennemsnit hunner)
|
|
|
|
|
|
|
Ungernes AGD på den samme dag efter fødslen, fødsel – dag 4 (gennemsnit hanner, gennemsnit hunner, PND-angivelse)
|
|
|
|
|
|
|
Ungernes vægt på dag 4 (gennemsnit)
|
|
|
|
|
|
|
Hanungernes bibeholdelse af brystvorter ("nipple retention") på dag 13 (gennemsnit)
|
|
|
|
|
|
|
Ungernes vægt på dag 13 (gennemsnit)
|
|
|
|
|
|
|
|
|
UNGER MED ABNORMITETER
|
|
Moderdyr med 0
|
|
|
|
|
|
|
Moderdyr med 1
|
|
|
|
|
|
|
Moderdyr med ≥ 2
|
|
|
|
|
|
|
|
|
TAB AF AFKOM
|
|
|
|
Prænatalt/efter implantation (implantationer minus levendefødte)
|
|
Hunner med 0
|
|
|
|
|
|
|
Hunner med 1
|
|
|
|
|
|
|
Hunner med 2
|
|
|
|
|
|
|
Hunner med ≥ 3
|
|
|
|
|
|
|
|
|
Postnatalt (levendefødte minus levende på dag 13 efter fødslen)
|
|
Hunner med 0
|
|
|
|
|
|
|
Hunner med 1
|
|
|
|
|
|
|
Hunner med 2
|
|
|
|
|
|
|
Hunner med ≥ 3
|
|
|
|
|
|
B.64 KOMBINERET SCREENINGSTEST FOR TOKSICITET VED GENTAGEN DOSERING OG REPRODUKTIONS-/UDVIKLINGSTOKSICITET
INDLEDNING
|
1.
|
Denne forsøgsmetode svarer til OECD-testvejledning (Test Guideline (TG)) 422 (2016). OECD-vejledningerne om testning af kemikalier bliver revideret regelmæssigt på baggrund af den videnskabelige udvikling. Den oprindelige screeningstestvejledning 422 blev vedtaget i 1996 med udgangspunkt i en protokol for en kombineret screeningstest for gentagen dosering og reproduktion/udvikling (»Combined Repeat Dose and Reproductive/Developmental Screening Test«), der blev drøftet på to ekspertmøder i henholdsvis London i 1990 (1) og Tokyo i 1992 (2).
|
|
2.
|
Denne forsøgsmetode kombinerer screening for reproduktions-/udviklingstoksicitet, som er baseret på de erfaringer, medlemsstaterne har gjort ved anvendelse af den oprindelige metode på eksisterende kemikalier, der fremstilles i store mængder, i forsøg med positive kontrolstoffer (3) (4) med screening for toksicitet ved gentagen dosering i overensstemmelse med OECD-testvejledning 407 (toksicitet ved gentagen dosering (28 dage, oral) hos gnavere, der svarer til kapitel B.7 i dette bilag).
|
|
3.
|
Denne forsøgsmetode er blevet opdateret med endepunkter for hormonforstyrrende stoffer som en opfølgning på de højt prioriterede aktiviteter, som OECD indledte i 1998 for at revidere eksisterende testvejledninger og udvikle nye testvejledninger for screening og testning af potentielt hormonforstyrrende stoffer (5). I denne forbindelse blev testvejledning 407 (der svarer til kapitel B.7 i dette bilag) i 2008 forbedret med parametre, der er egnede til påvisning af testkemikaliers endokrine virkemåde. Formålet med at opdatere testvejledning 422 var at indlemme endepunkter for hormonforstyrrende stoffer i testvejledningerne om screening, hvor eksponeringsperioderne omfatter følsomme udviklingsmæssige perioder (prænatale eller tidligt postnatale perioder).
|
|
4.
|
De valgte supplerende endepunkter for hormonforstyrrende stoffer, som også er at finde i testvejledning 443 (udvidet reproduktionstoksicitetsundersøgelse i én generation, som svarer til kapitel B.56 i dette bilag), blev indlemmet i testvejledning 422 på baggrund af en feasibilityundersøgelse, hvor der blev set på de videnskabelige og tekniske spørgsmål i forbindelse med deres indlemmelse samt eventuelle tilpasninger af testens udformning som følge heraf (6).
|
|
5.
|
Denne forsøgsmetode er beregnet til at generere begrænsede oplysninger om et testkemikalies virkninger på han- og hundyrs reproduktionsevne, herunder kønskirtlernes funktion, parringsadfærd, befrugtning, conceptus’ udvikling og fødsel. Den er ikke et alternativ til og erstatter ikke de eksisterende forsøgsmetoder B.31, B.34, B.35 og B.56.
|
INDLEDENDE OVERVEJELSER
|
6.
|
Efter udførelse af en akut test med henblik på indledende vurdering af et testkemikalies toksicitet, kan der udføres en oral toksicitetstest ved anvendelse af gentagen dosering til bestemmelse og vurdering af testkemikaliets toksiske egenskaber. Denne undersøgelse skaffer viden om mulige sundhedsfarer, som kan opstå ved gentagen eksponering i en relativt begrænset periode. Metoden omfatter den grundlæggende undersøgelse for toksicitet ved gentagen dosering, der kan bruges til kemikalier, for hvilke en 90-dages undersøgelse ikke er berettiget (f.eks. når produktionsmængden ikke overstiger visse grænser), eller som en forundersøgelse forud for en langtidsundersøgelse. Ved gennemførelse af undersøgelsen følges de vejledende principper og overvejelser, der er anført i OECD’s vejledende dokument nr. 19 om anerkendelse, vurdering og anvendelse af kliniske tegn som humane endepunkter for forsøgsdyr, der bruges i sikkerhedsevalueringer (7).
|
|
7.
|
Forsøgsmetoden omfatter desuden en screeningstest for reproduktions-/udviklingstoksicitet og kan derfor også anvendes til indhentning af indledende oplysninger om eventuelle virkninger på han- og hundyrs reproduktionsevne, herunder kønskirtlernes funktion, parringsadfærd, befrugtning, conceptus’ udvikling og fødsel, enten tidligt i vurderingen af testkemikaliers toksikologiske egenskaber eller i forbindelse med problematiske kemikalier. Denne forsøgsmetode giver ikke udtømmende oplysninger om alle aspekter af reproduktion og udvikling. Den giver navnlig kun begrænsede muligheder for at påvise postnatale tegn på prænatal eksponering eller virkninger, der induceres under postnatal eksponering. Bl.a. som følge af endepunkternes selektivitet og undersøgelsens korte varighed kan denne metode ikke bruges som endelig dokumentation for, at kemikalier ikke har nogen reproduktions-/udviklingsmæssig virkning. Når der ikke foreligger data fra andre reproduktions-/udviklingstoksicitetstest, kan positive resultater desuden anvendes til indledende farevurdering og som udgangspunkt for beslutninger om nødvendigheden af og tidsplanen for yderligere testning.
|
|
8.
|
De resultater, der opnås ved hjælp af de endokrinrelaterede parametre, bør ses i sammenhæng med »OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals« (8). Inden for denne begrebsramme er den forbedrede OECD TG 422 omfattet af niveau 4 som et in vivo-assay, der giver data om skadelige virkninger på endokrinrelevante endepunkter. Et endokrint signal alene vil dog ikke nødvendigvis blive opfattet som tilstrækkelig dokumentation for, at testkemikaliet er et hormonforstyrrende stof.
|
|
9.
|
Med denne metode lægges der også vægt på neurologiske virkninger som et specifikt endepunkt, og dyrene skal underkastes omhyggelige kliniske observationer med henblik på indhentning af flest mulige oplysninger. Metoden tager sigte på at identificere kemikalier med neurotoksisk potentiale, som i givet fald bør undersøges nærmere. Herudover kan metoden også vise, om der er grundlæggende tegn på immunologiske virkninger.
|
|
10.
|
Når der ikke foreligger data fra andre systemiske toksicitetstest, reproduktions-/udviklingstoksicitetstest, neurotoksicitetstest og/eller immunotoksicitetstest, kan positive resultater anvendes til indledende farevurdering og som udgangspunkt for beslutninger om nødvendigheden af og tidsplanen for yderligere testning. Testen kan være særdeles egnet i forbindelse med OECD’s Screening Information Data Set (SIDS) ved vurdering af eksisterende kemikalier, for hvilke der kun foreligger få eller ingen toksikologiske oplysninger, og kan bruges som et alternativ til at udføre to separate test for henholdsvis toksicitet ved gentagen dosering (OCD TG 407, der svarer til kapitel B.7 i dette bilag) og reproduktions-/udviklingstoksicitet (OECD TG 421, der svarer til kapitel B.63 i dette bilag). Den kan endvidere anvendes som en forundersøgelse til bestemmelse af dosisinterval ved mere omfattende reproduktions-/udviklingsrelaterede undersøgelser, eller når det ellers findes relevant.
|
|
11.
|
Generelt formodes det, at der er forskel på følsomheden hos drægtige og ikke-drægtige dyr. Det kan derfor være mere kompliceret at bestemme dosisniveauer i denne kombinerede test, som er egnede til både at evaluere generel systemisk toksicitet og specifik reproduktions-/udviklingstoksicitet, end når testene foretages enkeltvis. Derudover kan det være vanskeligere at fortolke testresultaterne med hensyn til generel systemisk toksicitet, end når der foretages en separat undersøgelse med gentagen dosering, især når serum- og histopatologiparametrene ikke samtidig vurderes i undersøgelsen. På grund af disse tekniske vanskeligheder kræver det omfattende erfaring med toksicitetstestning at foretage denne kombinerede screeningstest. Ud over det mindre antal anvendte dyr kan den kombinerede test på den anden side også være bedre til at skelne mellem de direkte reproduktions-/udviklingsmæssige virkninger og dem, der er sekundære i forhold til andre (systemiske) virkninger.
|
|
12.
|
I denne test er doseringsperioden længere end de sædvanlige 28 dage for en undersøgelse med gentagen dosering. Til gengæld bruges der færre dyr af hvert køn pr. gruppe i forhold til, når der gennemføres en undersøgelse med gentagen dosering på de sædvanlige 28 dage ud over en screeningstest for reproduktions-/udviklingstoksicitet.
|
|
13.
|
I forsøgsmetoden indgives testkemikaliet oralt. Det kan være nødvendigt at ændre forsøgsmetoden, hvis der anvendes andre eksponeringsveje.
|
|
14.
|
Før forsøgsmetoden bruges på en blanding for at generere data i lovgivningsøjemed, bør det overvejes, om den vil give resultater, som er egnede til formålet, og i givet fald hvorfor. Sådanne overvejelser er ikke nødvendige, når der foreligger et myndighedskrav om test af blandingen.
|
|
15.
|
De anvendte definitioner findes i tillæg 1.
|
PRINCIP FOR TESTEN
|
16.
|
Testkemikaliet indgives i graduerede doser til flere grupper hanner og hunner. Hannerne doseres i mindst fire uger frem til og med dagen før planlagt aflivning (dette omfatter mindst to uger før parring, under parringsperioden og ca. to uger efter parring). I lyset af hannernes begrænsede doseringsperiode inden parring er deres fertilitet ikke nødvendigvis nogen særlig følsom indikator for testikeltoksicitet. Derfor er det særdeles vigtigt, at der foretages endetaljeret histologisk undersøgelse af testiklerne. Kombinationen af en doseringsperiode på to uger inden parring og efterfølgende parrings-/fertilitetsobservationer og en samlet doseringsperiode på mindst fire uger efterfulgt af en detaljeret histopatologisk undersøgelse af hannernes kønskirtler anses for at være tilstrækkeligt til at påvise størstedelen af virkningerne på hannernes fertilitet og spermatogenese.
|
|
17.
|
Hunnerne doseres gennem hele undersøgelsen. Dette omfatter to uger inden parring (med det formål at dække mindst to hele brunstcyklusser), det variable tidsrum frem til befrugtning, drægtighedsperioden og mindst 13 dage efter fødslen frem til og med dagen før planlagt aflivning.
|
|
18.
|
Undersøgelsens varighed efter tilvænning til forholdene og vurdering af brunstcyklussen før dosering afhænger af de enkelte hunner og er på ca. 63 dage [mindst 14 dage før parring, (op til) 14 dages parring, 22 dages drægtighed og 13 dages laktation].
|
|
19.
|
I eksponeringsperioden observeres dyrene hver dag omhyggeligt for tegn på toksicitet. Dyr, der dør eller aflives under forsøget, obduceres, og de overlevende dyr aflives og obduceres ved forsøgets afslutning.
|
BESKRIVELSE AF METODEN
Valg af dyreart
|
20.
|
Denne forsøgsmetode er beregnet til rotter. Hvis de parametre, der er anført i denne TG 422, undersøges hos andre gnaverarter, gives en detaljeret begrundelse herfor. I det internationale valideringsprogram for påvisning af hormonforstyrrende stoffer vedrørende TG 407 blev der kun anvendt rotter. Der bør ikke anvendes stammer med ringe frugtbarhed eller kendt høj forekomst af udviklingsdefekter. Der bør anvendes sunde jomfruelige dyr, som ikke tidligere har været brugt i forsøg. Forsøgsdyrene beskrives med hensyn til art, stamme, køn, vægt og alder. Ved undersøgelsens start bør variationen i dyrenes vægt være minimal og ikke overstige ± 20 % af gennemsnitsvægten for hvert køn. Hvis undersøgelsen gennemføres som en forundersøgelse til en længerevarende undersøgelse eller en undersøgelse af en hel generation, bør der så vidt muligt anvendes dyr fra samme stamme og af samme oprindelse i begge undersøgelser.
|
Miljø og fodring
|
21.
|
Alle procedurer skal overholde lokale standarder for pasning af forsøgsdyr. Temperaturen i forsøgslokalet skal være 22 °C (± 3 °C). Den relative luftfugtighed skal være mindst 30 % og helst ikke over 70 %, hvilket dog ikke gælder under rengøring af lokalet. Belysningen skal være kunstig og bestå af 12 timers lys og 12 timers mørke. For så vidt angår fodring, kan der anvendes konventionelt laboratoriefoder med ubegrænset adgang til vand. Valget af foder kan påvirkes af behovet for at sikre en passende iblanding af et testkemikalie, når dette indgives ad denne vej.
|
|
22.
|
Dyrene bør anbringes i bure med små grupper af samme køn. Dyrene kan anbringes enkeltvis, hvis dette er sagligt begrundet. Hvis flere dyr anbringes i samme bur, bør der højst være fem dyr pr. bur. Parring bør finde sted i bure, der er egnede til formålet. Drægtige hunner bør anbringes alene i et bur og have adgang til redemateriale. Lakterende hunner anbringes alene i et bur sammen med deres afkom.
|
|
23.
|
Foderet bør analyseres regelmæssigt for forurenende stoffer. En prøve af foderet bør opbevares, indtil rapporten er udarbejdet.
|
Klargøring af dyrene
|
24.
|
Sunde unge voksne dyr fordeles vilkårligt i forsøgsgrupper og bure. Burene anbringes på en sådan måde, at de mulige følger af burenes placering minimeres. Dyrene identificeres entydigt og holdes i deres bure i mindst fem dage, inden undersøgelsen påbegyndes, så de kan vænne sig til laboratorieforholdene.
|
Fremstilling af doser
|
25.
|
Det anbefales, at testkemikaliet indgives oralt, medmindre andre indgivelsesveje er bedre egnede. Ved oral indgivelse indgives testkemikaliet normalt med sonde. Alternativt kan testkemikalier dog også indgives via foder eller drikkevand.
|
|
26.
|
Hvor det er nødvendigt, opløses eller suspenderes kemikaliet i et egnet bærestof. Det anbefales, at man først vurderer muligheden for at benytte en vandig opløsning/suspension, derefter en opløsning/suspension i olie (f.eks. majsolie), og først derefter eventuel opløsning i andre bærestoffer. For ikke-vandige bærestoffer skal bærestoffets toksiske egenskaber være kendt. Testkemikaliets stabilitet og homogenitet i bærestoffet skal bestemmes.
|
FREMGANGSMÅDE
Dyrenes antal og køn
|
27.
|
Det anbefales, at hver gruppe startes op med mindst 10 hanner og 12-13 hunner. Hunnernes brunstcyklus vurderes før eksponering, og dyr, der ikke udviser typiske fire-fem-dages cyklusser, medtages ikke i undersøgelsen. Derfor anbefales det at starte ud med ekstra hunner for at nå op på 10 hunner pr. gruppe. Medmindre der er tale om markante toksiske virkninger, forventes det, at dette vil give mindst otte drægtige hunner pr. gruppe, hvilket normalt er det lavest accepterede antal drægtige hunner pr. gruppe. Formålet hermed er at sikre, at der produceres tilstrækkelig meget afkom til, at der kan foretages en meningsfuld vurdering af testkemikaliets potentielle virkning på fertilitet, drægtighed, moderdyrenes adfærd og dieadfærd samt vækst og udvikling i F1-generationen fra befrugtning og frem til dag 13 efter fødslen. Hvis der påregnes aflivning af dyr under undersøgelsen, bør det oprindelige antal dyr forøges med det antal dyr, der påregnes aflivet i løbet af undersøgelsen. Det kan overvejes at udsætte en yderligere satellitgruppe på fem dyr af hvert køn i kontrolgruppen og i gruppen med den højeste dosis, således at man kan observere evt. reversibilitet, persistens eller forsinket indtræden af systemisk toksicitet i mindst 14 dage efter behandlingen. Dyrene i satellitgruppen parres ikke og vil derfor ikke blive anvendt til vurdering af reproduktions-/udviklingstoksicitet.
|
Dosering
|
28.
|
Der bør anvendes generelt mindst tre testgrupper og en kontrolgruppe. Foreligger der ikke egnede data om generel toksicitet, kan der udføres en forundersøgelse til bestemmelse af dosisinterval (dyr af samme stamme og herkomst) til bestemmelse af de doser, der skal anvendes. Bortset fra behandlingen med testkemikaliet bør dyr i kontrolgruppen behandles på samme måde som testgruppens dyr. Hvis der anvendes et bærestof til indgivelse af testkemikaliet, bør kontrolgruppen have den samme mængde bærestof som den gruppe, der modtager den største mængde bærestof.
|
|
29.
|
Dosisniveauerne bør udvælges under hensyntagen til eventuelle foreliggende data om toksicitet og (toksiko)kinetik. Der bør endvidere tages hensyn til, at der kan være forskel på følsomheden hos drægtige og ikke-drægtige dyr. Det højeste dosisniveau bør vælges med henblik på at fremkalde toksiske virkninger, men ikke død eller åbenlys lidelse. Derefter vælges en aftagende række dosisniveauer, som giver mulighed for at påvise et eventuelt dosis/effekt-forhold og ingen skadelige virkninger ved det laveste dosisniveau. De optimale intervaller svarer ofte til en faktor på to til fire, og i mange tilfælde må tilføjelse af en fjerde behandlingsgruppe foretrækkes frem for meget store intervaller (f.eks. større end en faktor 10) mellem dosisniveauerne.
|
|
30.
|
Hvis der observeres generel toksicitet (f.eks. reduceret kropsvægt, virkninger på lever, hjerte, lunger eller nyrer osv.) eller andre forandringer, der eventuelt ikke er toksiske virkninger (f.eks. reduceret indtagelse af føde eller forstørret lever), bør de observerede virkninger på de endokrinfølsomme endepunkter fortolkes med forsigtighed.
|
Grænsetest
|
31.
|
Såfremt et oralt enkeltdosisniveau på mindst 1 000 mg/kg kropsvægt/dag eller en ækvivalent procentdel tilsat foderet eller drikkevandet (baseret på kropsvægten) med de til dette forsøg beskrevne metoder ikke frembringer observerbare toksiske virkninger, og hvis der ikke på grundlag af data fra strukturelt beslægtede stoffer kan ventes toksiske virkninger, kan en fuldstændig undersøgelse med anvendelse af flere dosisniveauer betragtes som unødvendig. Grænsetesten gælder, medmindre data om human eksponering viser, at der er behov for anvendelse af et højere dosisniveau. For andre indgivelsesmåder – f.eks. inhalation eller påføring på huden – kan testkemikaliets fysisk-kemiske egenskaber ofte være bestemmende for den maksimalt opnåelige eksponering.
|
Indgivelse af doser
|
32.
|
Dyrene modtager testkemikaliet hver dag syv dage om ugen. Hvis testkemikaliet indgives med sonde, bør det gøres i en enkelt dosis med anvendelse af en gastrisk sonde eller et passende intubationsrør. Hvor stor en væskemængde, der kan indgives på én gang, afhænger af forsøgsdyrets størrelse. Denne mængde må højst være 1 ml/100 g kropsvægt, bortset fra vandige opløsninger, hvor der kan anvendes 2 ml/100 g kropsvægt. Hvis der ikke er tale omirriterende eller ætsende testkemikalier, hvis virkninger sædvanligvis forstærkes ved højere koncentrationer, bør variabiliteten af testmængden minimeres ved justering af koncentrationen, således at der sikres en konstant mængde ved alle dosisniveauer.
|
|
33.
|
Når testkemikaliet indgives via foder eller drikkevand, er det vigtigt at sikre, at de pågældende mængder af testkemikaliet ikke griber forstyrrende ind i den normale ernærings- eller vandbalance. Når testkemikaliet indgives via foderet, kan det enten gives ved konstant koncentration i foderet (ppm) eller ved et konstant dosisniveau i forhold til dyrenes kropsvægt. Det anvendte alternativ bør anføres. Ved indgivelse med sonde bør dosis gives på nogenlunde samme tidspunkt hver dag og justeres mindst en gang om ugen, så dosisniveauet holdes konstant i forhold til kropsvægten. Hvis den kombinerede undersøgelse anvendes som forløber for en længerevarende eller fuldstændig reproduktionstoksicitetsundersøgelse, skal der i begge undersøgelser gives samme fodersammensætning.
|
Forsøgsplan
|
34.
|
Doseringen af begge køn påbegyndes to uger før parring, efter at dyrene har haft mindst fem dage til at vænne sig til laboratorieforholdene, og hunnerne er blevet screenet for en normal brunstcyklus (i en tougersperiode forud for behandling). Undersøgelsen planlægges således, at vurderingen af brunstcyklus påbegyndes hurtigt efter, at dyrene er blevet fuldt kønsmodne. Dette kan variere en smule for de forskellige rottestammer i forskellige laboratorier og er f.eks. for Sprague Dawley-rotter, når dyrene er 10 uger gamle, og for Wistar-rotter, når de er omkring 12 uger gamle. Moderdyr og deres afkom aflives dag 13 efter fødslen eller umiddelbart derefter. For at moderdyrene skal kunne faste natten over forud for blodprøvetagning (hvis dette foretrækkes), skal moderdyrene og deres afkom ikke nødvendigvis aflives samme dag. Dagen for fødslen (dvs. hvor fødslen afsluttes) defineres som dag 0 efter fødslen. Hunner, der ikke udviser nogen tegn på kopulation, aflives 24-26 dage efter den sidste dag i parringsperioden. Doseringen fortsættes hos begge køn i parringsperioden. Hannerne bør stadig doseres efter parringsperioden, og mindst indtil den samlede doseringsperiode på mindst 28 dage er afsluttet. Derefter aflives de, eller doseringen fortsættes med henblik på eventuel fornyet parring, hvis dette findes hensigtsmæssigt.
|
|
35.
|
Moderdyrene doseres hver dag gennem hele drægtighedsperioden og mindst frem til og med dag 13 efter fødslen eller dagen før aflivning. I undersøgelser, hvor testkemikaliet indgives ved inhalation eller påføring på huden, bør doseringen fortsættes mindst frem til og med dag 19 i drægtighedsperioden, og doseringen bør genoptages hurtigst muligt og senest den fjerde dag efter fødslen (PND 4).
|
|
36.
|
Eventuelle dyr i en satellitgruppe, der anvendes til opfølgningsobservationer, parres ikke. De holdes mindst i yderligere 14 dage efter den planlagte aflivning af moderdyrene uden indgivelse af testkemikalie, således at det kan registreres, om der er forsinkede, vedvarende eller reversible toksiske virkninger.
|
|
37.
|
Der findes et diagram over forsøgsplanen i tillæg 2.
|
Brunstcyklusser
|
38.
|
Brunstcyklusserne bør overvåges, inden behandlingen indledes, for at udvælge de hunner til undersøgelsen, der har en regelmæssig cyklus (jf. punkt 27). Vaginalt smear bør også overvåges dagligt fra behandlingens indledning, og indtil der findes tegn på parring. Hvis der er problemer med akutte stressvirkninger, der kan ændre brunstcyklussen, når doseringen indledes, kan laboratorierne eksponere forsøgsdyrene i to uger og derefter indsamle vaginalt smear hver dag for at overvåge brunstcyklussen i mindst to uger før parring og fortsætte overvågningen i parringsperioden, indtil der findes tegn på parring. Udtagning af celleprøver fra vagina/cervix skal ske forsigtigt for at undgå at forstyrre slimhinden, hvilket ellers kunne inducere pseudodrægtighed (8) (9).
|
Parringsmetode
|
39.
|
I denne undersøgelse bør der normalt anvendes 1:1-parring (én han og én hun). Dette princip kan fraviges, hvis hannen skulle dø. Hunnen bør anbringes sammen med den samme han, indtil der observeres tegn på kopulation, eller indtil der er gået to uger. Hunnerne undersøges hver morgen for tilstedeværelse af sperma eller vaginalprop. Dag 0 i drægtighedsperioden defineres som den dag, hvor tegn på parring bekræftes (der observeres vaginalprop eller sperma). Lykkes parring ikke, kan fornyet parring af hunnerne med afprøvede hanner fra samme gruppe overvejes.
|
Kuldstørrelse
|
40.
|
På dag 4 efter fødslen standardiseres hvert kuld, ved at overskydende unger fjernes ved vilkårlig udvælgelse, så hvert kuld i videst muligt omfang kommer til at bestå af fire eller fem unger pr. køn, alt efter den normale kuldstørrelse i den anvendte rottestamme. Der tages blodprøver fra to af de overskydende unger, som samles og bruges til bestemmelse af T4-niveauer i serum. Selektiv fjernelse af unger, f.eks. baseret på kropsvægt eller anogenital afstand (AGD), er ikke hensigtsmæssig. Når antallet af han- eller hununger gør det umuligt at få fire eller fem af hvert køn i hvert kuld, er en delvis standardisering (f.eks. seks hanner og fire hunner) acceptabel. Ingen unger fjernes, når et kuld er på mindre end målet for fjernelse (otte eller 10 unger/kuld). Hvis der kun er én unge over målet for fjernelse, vil der kun blive fjernet én unge, som vil blive brugt til indsamling af blod til eventuel vurdering af T4 i serum.
|
|
41.
|
Hvis ikke kuldstørrelsen standardiseres, aflives to unger pr. kuld dag 4 efter fødslen, og der tages blodprøver til måling af thyreoideahormonkoncentrationer i serum. Hvis det er muligt, skal disse to unger pr. kuld være hununger, så hanungerne bevares til vurdering af bibeholdelsen af brystvorter, medmindre fjernelsen af disse to unger betyder, at der ikke er flere hunner tilbage til afsluttende vurdering. Der fjernes ingen unger, når kuldet er på mindre end otte eller 10 unger/kuld (alt efter den normale kuldstørrelse i den anvendte rottestamme). Hvis der kun er én overskydende unge i forhold til den normale kuldstørrelse, vil der kun blive fjernet én unge, som vil blive brugt til indsamling af blod til eventuel vurdering af T4 i serum.
|
Observationer
|
42.
|
Der bør foretages generelle kliniske observationer mindst én gang om dagen, fortrinsvis på samme tidspunkt(er) hver dag og under hensyntagen til det tidspunkt, hvor de forventede virkninger efter dosering topper. Dyrenes helbredstilstand bør registreres. Dyrene observeres for morbiditet og mortalitet mindst to gange om dagen.
|
|
43.
|
Én gang før den første eksponering (med henblik på individbaserede sammenligninger) og mindst én gang om ugen derefter foretages der indgående kliniske observationer af alle forældredyr. Med henblik på disse observationer tages dyrene ud af deres egne bure og anbringes i et standardindelukke. Observationerne udføres om muligt på samme tidspunkt hver dag. De bør registreres omhyggeligt, fortrinsvis ved anvendelse af et scoringssystem, der udtrykkeligt er defineret af testlaboratoriet. Det er vigtigt at sikre, at testbetingelserne varierer minimalt, og at observationerne om muligt foretages af observatører, der ikke har kendskab til behandlingen. Observationerne bør bl.a. rettes mod ændringer i hud, pels, øjne, slimhinder, forekomst af sekretion og ekskretion samt autonom aktivitet (f.eks. tåreflåd, piloerektion, pupilstørrelse, usædvanligt respirationsmønster). Derudover registreres forandringer i gang, holdning og respons på håndtering såvel som forekomst af kloniske eller toniske bevægelser, stereotypi (f.eks. overdreven soignering, repetitive cirkelbevægelser), vanskelig eller langvarig fødsel eller mærkelig adfærd (f.eks. selvmutilation, baglæns gang) (10).
|
|
44.
|
Én gang i løbet af undersøgelsen vurderes reaktiviteten over for sensoriske stimuli af forskellig art (f.eks. auditive, visuelle og proprioceptive stimuli) (8) (9) (11), gribestyrke (12) og motorisk aktivitet (13) hos fem hanner og fem hunner, der udvælges vilkårligt fra hver gruppe. Nærmere oplysninger om de fremgangsmåder, der kan benyttes, findes i de respektive referencer. Andre alternative fremgangsmåder end de refererede kan imidlertid også anvendes. Hos hannerne bør disse funktionelle observationer foretages i slutningen af doseringsperioden, kort tid inden den planlagte aflivning, men inden der tages blodprøve til hæmatologi eller klinisk kemi (jf. punkt 53-56, også fodnote 1). Hunnerne bør være i fysiologisk samme tilstand under disse funktionelle test og skal helst testes én gang i løbet af den sidste laktationsuge (f.eks. LD 6-13), kort tid inden den planlagte aflivning. Moderdyr og unger holdes adskilt i så kort tid som muligt.
|
|
45.
|
Funktionelle observationer, der foretages én gang i slutningen af undersøgelsen, kan udelades, hvis undersøgelsen gennemføres som en forundersøgelse til en efterfølgende undersøgelse af subkronisk toksicitet (90 dage) eller en længerevarende undersøgelse. I så fald bør de funktionelle observationer medtages i den opfølgende undersøgelse. På den anden side kan data vedrørende funktionelle observationer fra denne undersøgelse med gentagen dosering lette valget af dosisniveauer for en efterfølgende undersøgelse af subkronisk toksicitet eller en længerevarende undersøgelse.
|
|
46.
|
I undtagelsestilfælde kan funktionelle observationer ligeledes udelades for grupper, der viser så tydelige tegn på toksicitet, at det i signifikant grad ville indvirke på resultaterne af den funktionelle test.
|
|
47.
|
Drægtighedsperioden bør registreres og beregnes fra drægtighedsdag 0. Hvert kuld undersøges hurtigst muligt efter fødslen til bestemmelse af ungernes antal og køn, dødfødte, levendefødte, små uudviklede unger (unger, der er signifikant mindre end ungerne i kontrolgruppen) og tilstedeværelse af alvorlige abnormiteter.
|
|
48.
|
De levende unger tælles, og deres køn bestemmes, og kuldet vejes senest 24 timer efter fødslen (dag 0 eller 1 efter fødslen) samt mindst på dag 4 og dag 13 efter fødslen. Ud over observationerne på forældredyr (jf. punkt 43 og 44) skal al unormal adfærd hos afkommet også registreres.
|
|
49.
|
Ungernes AGD bør måles den samme dag efter fødslen mellem PND 0 og PND 4. Ungernes kropsvægt registreres den dag, AGD måles, og AGD normaliseres til et mål for ungens størrelse, helst kubikroden af kropsvægten (14). Antallet af brystvorter/areolae hos hanungerne tælles på PND 12 eller 13 som anbefalet i OECD GD 151 (15).
|
Kropsvægt og foder-/vandindtagelse
|
50.
|
Han- og hundyr bør vejes den dag, doseringen indledes, og derefter mindst en gang om ugen og ved forsøgets afslutning. I drægtighedsperioden vejes hunnerne på dag 0, 7, 14 og 20 og senest 24 timer efter fødslen (dag 0 eller 1 efter fødslen) samt mindst på dag 4 og dag 13 efter fødslen. Disse observationer bør rapporteres individuelt for hvert voksent dyr.
|
|
51.
|
Før parring og i drægtigheds- og laktationsperioden bør foderindtaget måles mindst en gang om ugen. Måling af foderindtaget i parringsperioden er valgfrit. Hvis testkemikaliet indgives via drikkevandet, skal vandindtagelsen i disse perioder også måles.
|
Hæmatologi
|
52.
|
Én gang i løbet af undersøgelsen foretages følgende hæmatologiske undersøgelser på fem hanner og fem hunner, der udvælges vilkårligt fra hver gruppe: hæmatokrit, hæmoglobinkoncentration, erythrocyttælling, retikulocyttælling, total og differential leucocyttælling, blodpladetælling og måling af blodets koaguleringsevne/-tid. Andre analyser, som bør udføres, hvis testkemikaliet eller dets putative metabolitter har eller mistænkes for at have oxiderende egenskaber, omfatter methæmoglobinkoncentration og Heinz-legemer.
|
|
53.
|
Der bør tages blodprøver fra et bestemt, angivet sted. Hunnerne bør være i fysiologisk samme tilstand ved blodprøvetagning. For at undgå praktiske problemer som følge af de forskellige tidspunkter for drægtighedsperiodernes indledning kan blodprøver fra hunnerne tages ved udløbet af perioden før parring i stedet for umiddelbart inden eller i forbindelse med dyrenes aflivning. Blodprøver fra hannerne bør fortrinsvis tages umiddelbart inden eller i forbindelse med dyrenes aflivning. Som et alternativ er det også muligt at tage blodprøver fra hannerne ved udløbet af perioden før parring, hvis der tages blodprøver fra hunnerne på dette tidspunkt.
|
|
54.
|
Alle blodprøver opbevares under egnede forhold.
|
Klinisk biokemi
|
55.
|
Klinisk-biokemiske bestemmelser med henblik på undersøgelse af vigtige toksiske virkninger i væv, og navnlig i nyrer og lever, foretages med blodprøver, der udtages fra de udvalgte fem hanner og fem hunner fra hver gruppe. Det anbefales, at dyrene faster natten over forud for blodprøvetagning (22). Undersøgelser af plasma eller serum bør omfatte natrium, kalium, glukose, total kolesterol, urinstof, kreatinin, total protein og albumin, mindst to enzymer, hvis forekomst kan være tegn på hepatocellulære effekter (f.eks. alanin aminotransferase, aspartat aminotransferase og sorbitol dehydrogenase) og galdesyrer. Målinger af yderligere enzymer (fra lever eller andre organer) og bilirubin kan under visse omstændigheder give nyttige oplysninger.
|
|
56.
|
Der tages blodprøver fra et bestemt sted i henhold til nedenstående:
|
—
|
fra mindst to unger pr. kuld på dag 4 efter fødslen, hvis antallet af unger gør det muligt (jf. punkt 40-41)
|
|
—
|
fra alle moderdyr og mindst to unger pr. kuld ved forsøgets afslutning på dag 13 og
|
|
—
|
fra alle voksne hanner ved forsøgets afslutning.
|
Alle blodprøver opbevares under egnede forhold. Blodprøver fra dag 13-unger og voksne hanner undersøges for niveauet af thyreoideahormoner (T4) i serum. Der kan eventuelt foretages yderligere vurdering af T4 i blodprøver fra moderdyr og dag 4-unger. Andre hormoner kan eventuelt også måles. Blod fra unger til thyreoideahormonanalyse kan samles pr. kuld. Thyreoideahormoner (T4 og TSH) bør fortrinsvis måles som en samlet værdi.
|
|
57.
|
Følgende urinanalyser kan eventuelt udføres på fem vilkårligt udvalgte hanner fra hver gruppe i undersøgelsens sidste uge til bestemmelse af udseende, volumen, osmolalitet eller relativ densitet, pH, protein, glukose og blod eller blodceller.
|
|
58.
|
Endvidere kan det overvejes at gennemføre undersøgelser af serummarkører for generelle vævsskader. Andre bestemmelser, der bør foretages, hvis testkemikaliets kendte egenskaber kan eller forventes at kunne indvirke på tilsvarende metaboliske funktioner, omfatter kalcium, phosphat, triglycerider og glukose ved faste, visse hormoner, methæmoglobin og cholinesterase. Disse forhold undersøges individuelt.
|
|
59.
|
Følgende faktorer kan påvirke variabiliteten og de absolutte koncentrationer i hormonanalyserne:
|
—
|
aflivningstidspunkt på grund af de forskellige hormonkoncentrationer i løbet af døgnet
|
|
—
|
aflivningsmetode, der har til formål at undgå unødig stress af dyrene, som kan påvirke hormonkoncentrationerne
|
|
—
|
analysesæt til hormonanalyser, der kan have forskellige standardkurver.
|
|
|
60.
|
Plasmaprøver, der specifikt er beregnet til hormonanalyse, tages på nogenlunde samme tidspunkt på dagen. De numeriske værdier, der fås ved analyse af hormonkoncentrationer, varierer ved anvendelse af forskellige analysesæt på markedet.
|
|
61.
|
Hvis ikke der foreligger passende historiske data, bør det overvejes at bestemme hæmatologiske og klinisk-biokemiske variabler, inden eksponering påbegyndes, eller så vidt muligt hos en gruppe dyr, der ikke er med i forsøgsgrupperne. Dataene for hunner skal komme fra lakterende dyr.
|
PATOLOGI
Makroskopisk undersøgelse
|
62.
|
Alle voksne forsøgsdyr underkastes en fuldstændig og tilbundsgående makroskopisk undersøgelse, der omfatter omhyggelig undersøgelse af kroppens ydre, alle åbninger samt af kraniehule, brysthule og bughule og indholdet heraf. Særlig opmærksomhed bør rettes mod kønsorganerne. Antallet af implantationssteder bør registreres. Det vaginale smear bør undersøges på obduktionsdagen for at bestemme stadiet i brunstcyklussen og muliggøre korrelation med den histopatologiske undersøgelse af hunnernes kønsorganer.
|
|
63.
|
På alle voksne handyr renses testikler og bitestikler samt prostata og vesiculae seminales og prostata anterior som én samlet enhed for alt vedhængende væv, og de vejes hurtigst muligt (våd vægt) efter dissektion, så udtørring undgås. Derudover kan eventuelle organvejninger omfatte levator ani plus bulbocavernosus-muskelkomplekset, Cowpers kirtler og glans penis i hanner samt parrede ovarier (våd vægt) og uterus (herunder cervix) i hunner. Disse valgfrie vejninger bør foretages hurtigst muligt efter dissektion. Alle de voksne dyrs ovarier, testikler, bitestikler og sekundære kønsorganer samt alle organer, der udviser makroskopiske læsioner, præserveres.
|
|
64.
|
Skjoldbruskkirtlerne fra alle voksne hanner og hunner samt fra én dag 13-hanunge og én dag 13-hununge fra hvert kuld præserveres i det bedst egnede fikseringsmiddel med henblik på den påtænkte efterfølgende histopatologiske undersøgelse. Skjoldbruskkirtlens vægt kan bestemmes efter fiksering. Rensningen for vedhængende væv bør også foretages meget omhyggeligt og først efter fiksering for at undgå beskadigelse af vævet. Beskadiget væv kan kompromittere den histopatologiske analyse. Der tages blodprøver fra et bestemt angivet sted umiddelbart inden eller i forbindelse med dyrenes aflivning. Prøverne opbevares under egnede forhold (jf. punkt 56).
|
|
65.
|
Derudover befries lever, nyrer, binyrer, thymus, milt, hjerne og hjerte fra mindst fem voksne hanner og hunner, som udvælges vilkårligt fra hver gruppe (undtaget er de dyr, der er døende og/eller aflives, inden undersøgelsen er afsluttet), for alt vedhængende væv, og de vejes hurtigst muligt (våd vægt) efter dissektion, så udtørring undgås. Følgende væv bør præserveres i det bedst egnede fikseringsmiddel både med hensyn til vævstype og den påtænkte efterfølgende histopatologiske undersøgelse: alle makroskopiske læsioner, hjerne (repræsentative regioner, herunder cerebrum, cerebellum og pons), rygmarv, øjne, mavesæk, tyktarm og tyndtarm (herunder Peyerpletter), lever, nyrer, binyrer, milt, hjerte, thymus, luftrør og lunger (præserveret ved oppumpning med fikseringsmiddel og efterfølgende immersion), kønskirtler (testikler og ovarier), sekundære kønsorganer (uterus og cervix, bitestikler, prostataog vesiculae seminales plus prostata anterior), vagina, urinblære, lymfeknuder (ud over den nærmeste drænende lymfeknude bør en anden lymfeknude udtages på baggrund af laboratoriets erfaring (16)), perifer nerve (ischias- eller tibianerven), om muligt tæt på musklen, skeletmuskel og knogle med knoglemarv (knoglemarvssnit eller som alternativ et nyligt præpareret knoglemarvsaspirat). Det anbefales, at testikler fikseres ved immersion i Bouins væske eller modificeret Davidsons fikseringsmiddel (16) (17) (18). Fiksering med formalin anbefales ikke til disse væv. Tunica albuginea kan punkteres forsigtigt og overfladisk i begge ender af organet med en kanyle, der muliggør hurtig penetration af fikseringsmidlet. Kliniske og andre fund kan foranledige undersøgelse af andet væv. Også særlige organer, der sandsynligvis kan være mål for testkemikaliets kendte virkninger, bør præserveres.
|
|
66.
|
Følgende væv kan give værdifulde indikationer på endokrinrelaterede virkninger: kønskirtler (testikler og ovarier), sekundære kønsorganer (uterus med cervix, bitestikler, vesiculae seminales med prostata anterior, dorsolateral og ventral prostata), vagina, hypofyse, brystkirtler fra handyr og binyrer. Forandringer i brystkirtler fra handyr er ikke tilstrækkeligt dokumenteret, men denne parameter kan være meget følsom over for stoffer med østrogen virkemåde. Observation af organer/væv, der ikke er anført i punkt 65, er valgfrit.
|
|
67.
|
Døde unger og unger, der aflives på dag 13 efter fødslen eller hurtigt derefter, bør som minimum undersøges omhyggeligt på kroppens ydre for større abnormiteter. Særlig opmærksomhed bør udvises over for de eksterne kønsorganer, som bør undersøges for tegn på forandringer i udviklingen.
|
Histopatologisk undersøgelse
|
68.
|
Der bør foretages en fuldstændig histopatologisk undersøgelse af de præserverede organer og væv fra de udvalgte dyr i kontrolgruppen og højdosisgruppen (idet der lægges særlig vægt på spermatogenesens faser i hannernes kønskirtler og histopatologien i testiklernes interstitielle cellestruktur). Skjoldbruskkirtlerne fra ungerne og de resterende voksne dyr kan undersøges om nødvendigt. Disse undersøgelser bør omfatte dyr fra andre dosisgrupper, hvis der i højdosisgruppen observeres eksponeringsrelaterede ændringer. I vejledningen om histopatologi (10) findes der flere oplysninger om dissektion, fiksering, udtagning af snit og histopatologisk undersøgelse af endokrine væv.
|
|
69.
|
Alle makroskopiske læsioner skal undersøges. Til fastsættelse af NOAEL’er bør målorganer fra andre dosisgrupper undersøges, navnlig fra grupper, der menes at udvise et NOAEL.
|
|
70.
|
Hvis der anvendes en satellitgruppe, bør der udføres en histopatologisk undersøgelse af væv og organer, der udviser toksisk virkning i behandlingsgrupperne.
|
DATA OG RAPPORTERING
Data
|
71.
|
Der bør fremlægges data om hvert enkelt dyr. Derudover bør alle data sammenfattes i tabelform med angivelse for hver forsøgsgruppe af antal dyr ved forsøgets begyndelse, antal dyr, som er fundet døde under testen eller aflivet af humane grunde, tidspunkt for eventuel død eller aflivning, antal fertile dyr, antal drægtige hundyr, antal dyr, som viser tegn på toksisk virkning, beskrivelse af iagttagne toksicitetstegn, herunder begyndelsestidspunkt, varighed og sværhed af alle de toksiske virkninger, arten af de histopatologiske forandringer og alle relevante data om kuldene. Tillæg 3 indeholder en sammenfattende rapport i tabelform, der har vist sig at være særdeles velegnet til vurdering af reproduktions-/udviklingsmæssige virkninger.
|
|
72.
|
Når det er muligt, bedømmes resultaterne ved en egnet og almindeligt anerkendt statistisk metode. Sammenligninger af virkningen i et dosisinterval gør det muligt at undgå at bruge flere t-test. De statistiske metoder udvælges i forbindelse med undersøgelsens tilrettelæggelse. Statistiske analyser af AGD og bibeholdelse af brystvorter (»nipple retention«) bør baseres på data fra de enkelte unger under hensyntagen til virkninger på de enkelte kuld. Når det er hensigtsmæssigt, er det kuldene, der er analyseenheden. Statistiske analyser af ungernes kropsvægt bør baseres på data fra de enkelte unger under hensyntagen til kuldstørrelsen. Som følge af undersøgelsens begrænsede omfang er de statistiske analyser i form af test for »signifikans« af begrænset værdi for mange endepunkter, især de reproduktionsrelaterede endepunkter. Nogle af de hyppigst anvendte metoder, især de parameterbaserede test for central tendens, er uegnede. Hvis der anvendes statistiske analyser, bør den valgte metode være egnet til fordeling af de undersøgte variabler og vælges, inden undersøgelsen indledes.
|
Vurdering af resultater
|
73.
|
Resultaterne af denne toksicitetsundersøgelse bør vurderes med hensyn til de observerede virkninger samt obduktionsfund og mikroskopiske fund. Vurderingen skal omfatte sammenhæng mellem dyrenes eksponering for testkemikaliet og tilstedeværelse eller fravær, forekomst og sværhedsgrad af abnormiteter, herunder makroskopiske læsioner, identificerede målorganer, manglende fertilitet, unormale kliniske fund, påvirkning af reproduktions- og ynglepræstationer, ændringer af kropsvægt, indvirkning på mortaliteten og eventuelle andre toksiske virkninger.
|
|
74.
|
På grund af hannernes korte behandlingsperiode bør histopatologien for testikler og bitestikler ses i sammenhæng med fertilitetsdataene ved vurdering af reproduktionsvirkningen på hannerne. Anvendelsen af historiske kontroldata om reproduktion/udvikling (f.eks. med hensyn til kuldstørrelse, AGD, bibeholdelse af brystvorter (»nipple retention«), T4-niveauer i serum) kan også, når sådanne foreligger, være en hjælp i forbindelse med fortolkningen af undersøgelsesresultaterne.
|
|
75.
|
Med henblik på kvalitetskontrol foreslås det, at historiske kontroldata indsamles, og at der beregnes variationskoefficienter for numeriske data, især for parametre med relation til påvisning af hormonforstyrrende virkninger. Disse data kan bruges i forbindelse med sammenligninger, når de faktiske undersøgelser evalueres.
|
Testrapport
|
76.
|
Testrapporten bør indeholde følgende oplysninger:
|
|
Testkemikalie:
|
—
|
oprindelse, batchnummer, sidste anvendelsesdato, hvis dette foreligger
|
|
—
|
testkemikaliets stabilitet, hvis kendt.
|
|
|
|
Stof med kun én bestanddel:
|
—
|
fysisk udseende, vandopløselighed og yderligere relevante fysisk-kemiske egenskaber
|
|
—
|
kemisk identifikation, f.eks. IUPAC-navn eller CAS-navn, CAS-nummer, SMILES- eller InChI-kode, strukturformel, renhed, kemisk identitet af urenheder, i det omfang det er relevant og praktisk muligt, osv.
|
|
|
|
Stof med flere bestanddele, UVCB-stoffer og blandinger:
|
—
|
kendetegnet så vidt muligt ved kemisk identitet (jf. ovenfor), kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber
|
. |
|
|
Eventuelt bærestof:
|
—
|
begrundelse for valg af bærestof, hvis dette ikke er vand.
|
|
|
|
Forsøgsdyr:
|
—
|
den anvendte art/stamme
|
|
—
|
dyrenes antal, alder og køn
|
|
—
|
oprindelse, miljøbetingelser, foder mv.
|
|
—
|
de enkelte dyrs vægt ved testens begyndelse
|
|
—
|
begrundelse for andre arter end rotte.
|
|
|
|
Testbetingelser:
|
—
|
begrundelse for valg af dosisniveau
|
|
—
|
oplysninger om formulering af testkemikalie eller iblanding heraf i foderet, opnået koncentration, foderblandingens stabilitet og homogenitet
|
|
—
|
oplysninger om indgivelse af testkemikaliet
|
|
—
|
eventuel omregning af testkemikaliets koncentration (ppm) i foder/drikkevand til faktisk dosis (mg pr. kg kropsvægt pr. dag)
|
|
—
|
oplysninger om foder- og vandkvalitet
|
|
—
|
detaljeret beskrivelse af randomiseringsproceduren til eventuel udvælgelse af unger til fjernelse.
|
|
|
|
Resultater:
|
—
|
kropsvægt og ændringer i kropsvægt
|
|
—
|
foderindtagelse og indtagelse af vand, om muligt
|
|
—
|
oplysninger om toksisk reaktion inddelt efter køn og dosis, herunder fertilitet, drægtighed og andre tegn på toksicitet
|
|
—
|
drægtighedsperiodens længde
|
|
—
|
toksisk virkning eller andre virkninger på reproduktion, afkom, postnatal vækst osv.
|
|
—
|
art, sværhedsgrad og varighed af kliniske symptomer (hvad enten disse er reversible eller ej)
|
|
—
|
vurdering af sanseevne, gribestyrke og motorisk aktivitet
|
|
—
|
hæmatologiske målinger med relevante udgangsværdier
|
|
—
|
klinisk-biokemiske målinger med relevante udgangsværdier
|
|
—
|
antal voksne hunner med normal eller anormal brunstcyklus og cyklusvarighed
|
|
—
|
antal levendefødte og postimplantationstab
|
|
—
|
antal unger med makroskopisk synlige abnormiteter - makroskopisk vurdering af eksterne genitalia, antal små uudviklede unger
|
|
—
|
dødstidspunkt under undersøgelsen, eller hvorvidt dyrene overlevede til undersøgelsens afslutning
|
|
—
|
antal implantationer, kuldstørrelse og kuldenes vægt på registreringstidspunktet
|
|
—
|
data om ungernes kropsvægt
|
|
—
|
AGD for alle unger (og kropsvægt på dagen for AGD-måling)
|
|
—
|
bibeholdelse af brystvorter (»Nipple retention«) hos hanunger
|
|
—
|
thyreoideahormonniveauer, dag 13-unger og voksne hanner (og eventuelt moderdyr og dag 4-unger)
|
|
—
|
kropsvægt ved aflivning og organvægt for forældredyrene
|
|
—
|
detaljeret beskrivelse af alle histopatologiske fund
|
|
—
|
absorptionsdata (hvis de foreligger)
|
|
—
|
når det er hensigtsmæssigt, statistisk behandling af resultaterne.
|
|
|
Fortolkning af resultater
|
77.
|
Ved denne undersøgelse vurderes den reproduktions-/udviklingsmæssige toksicitet i forbindelse med gentagen dosering. Da der lægges vægt på endepunkter for både generel toksicitet og reproduktions-/udviklingstoksicitet, vil resultaterne af undersøgelsen gøre det muligt at skelne mellem reproduktions-/udviklingsmæssige virkninger, som opstår uden generel toksicitet, og dem, der kun kommer til udtryk på niveauer, som også er toksiske for forældredyrene (jf. punkt 7-11). Den kan vise, om det er nødvendigt at foretage yderligere undersøgelser, og udstikke retningslinjer for udformningen af efterfølgende undersøgelser. Med hensyn til fortolkningen af de reproduktions- og udviklingsrelaterede resultater henvises til OECD’s vejledende dokument 43 (19). OECD’s vejledende dokument 106 om histologisk vurdering af endokrine og reproduktionsrelaterede test i gnavere (16) indeholder oplysninger om præpareringen og vurderingen af (endokrine) organer og vaginalt smear, der kan være nyttige i forbindelse med denne forsøgsmetode.
|
LITTERATUR
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Available upon request at Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. og Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. og Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998, Available upon request at Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M. og Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. og Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. og Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. og Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp og V.L. Reynolds. (1999). »Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights«, Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Organisation for Economic Cooperation and Development, Paris.
|
|
(16)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 106) Organisation for Economic Cooperation and Development, Paris.
|
|
(17)
|
Hess RA og Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE og Heindel JJ (Eds.). Academic Press: San Diego, CA, s. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organisation for Economic Cooperation and Development, Paris.
|
Tillæg 1
DEFINITIONER (SE OGSÅ (20) OECD GD 150)
Androgenicitet et kemikalies evne til at fungere som et naturligt androgent hormon (f.eks. testosteron) i en pattedyrsorganisme.
Antiandrogenicitet et kemikalies evne til at undertrykke virkemåden for et naturligt androgent hormon (f.eks. testosteron) i en pattedyrsorganisme.
Antiøstrogenicitet et kemikalies evne til at undertrykke virkemåden for et naturligt østrogent hormon (f.eks. østradiol 17ß) i en pattedyrsorganisme.
Antithyreoid aktivitet et kemikalies evne til at undertrykke virkemåden for et naturligt thyreoideahormon (f.eks. T3) i en pattedyrsorganisme.
Kemikalie: et stof eller en blanding.
Udviklingstoksicitet manifestering af reproduktionstoksicitet, som repræsenterer prænatale, perinatale, postnatale, strukturelle eller funktionsmæssige forstyrrelser i afkommet.
Dosis: mængden af indgivet testkemikalie. Dosis angives som vægt af testkemikalie pr. enhed af forsøgsdyrets vægt pr. dag (f.eks. mg/kg kropsvægt/dag) eller som en konstant koncentration i foderet.
Dosering: et generelt udtryk, som omfatter dosis samt doseringsfrekvens og -varighed.
Synlig toksicitet et generelt udtryk, som angiver klare tegn på toksicitet efter indgivelse af testkemikaliet. Disse tegn bør være tilstrækkelige til farevurdering og bør være af en sådan karakter, at en forøgelse af den indgivne dosis må forventes at resultere i udvikling af svære toksiske virkninger og sandsynligvis død.
Fertilitetsforringelse forstyrrelse af hanners eller hunners reproduktionsfunktioner eller -evne.
Toksisk virkning på moderdyret skadelig virkning på drægtige hunner, der opstår enten specifikt (direkte virkning) eller ikke-specifikt (indirekte virkning), og som skyldes deres drægtighed.
NOAEL forkortelse for "no-observed-adverse-effect level" (niveau, hvor ingen skadelige virkninger observeres). Dette er det højeste dosisniveau, hvor der ikke observeres behandlingsrelaterede skadelige virkninger.
Østrogenicitet et kemikalies evne til at fungere som et naturligt østrogent hormon (f.eks. østradiol 17ß) i en pattedyrsorganisme.
Reproduktionstoksicitet fremkaldelse af skadelige virkninger på afkommet og/eller forringelse af hanners og hunners reproduktionsfunktioner eller -evne.
Testkemikalie: et stof eller en blanding, der testes med denne forsøgsmetode.
Thyreoid aktivitet et kemikalies evne til at fungere som et naturligt thyreoideahormon (f.eks. T3) i en pattedyrsorganisme.
Validering en videnskabelig proces til karakterisering af en forsøgsmetodes operationelle krav og begrænsninger og påvisning af dens pålidelighed og relevans til et bestemt formål.
Tillæg 2
DIAGRAM OVER FORSØGSPLANEN, SOM VISER DEN MAKSIMALE VARIGHED AF UNDERSØGELSEN BASERET PÅ EN FULD 14-DAGES PARRINGSPERIODE

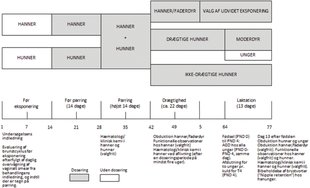
Tillæg 3
SAMMENFATTENDE RAPPORT I TABELFORM OVER REPRODUKTIONS-/UDVIKLINGSMÆSSIGE VIRKNINGER
|
OBSERVATIONER
|
VÆRDIER
|
|
Dosering (enheder)
|
0 (kontrol)
|
…
|
…
|
…
|
…
|
|
Startede par (N)
|
|
|
|
|
|
|
Brunstcyklus (som minimum gennemsnitlig varighed og frekvens for uregelmæssige cyklusser)
|
|
|
|
|
|
|
Hunner, som viser tegn på kopulation (N)
|
|
|
|
|
|
|
Hunner, som bliver drægtige (N)
|
|
|
|
|
|
|
Undfangelse, dag 1-5 (N)
|
|
|
|
|
|
|
Undfangelse, dag 6-… (23) (N)
|
|
|
|
|
|
|
Drægtighed ≤ 21 dage (N)
|
|
|
|
|
|
|
Drægtighed = 22 dage (N)
|
|
|
|
|
|
|
Drægtighed ≥ 23 dage (N)
|
|
|
|
|
|
|
Moderdyr med levende nyfødte unger (N)
|
|
|
|
|
|
|
Moderdyr med levende unger på dag 4 efter fødslen (N)
|
|
|
|
|
|
|
Implantater/moderdyr (gennemsnit)
|
|
|
|
|
|
|
Levende unger/moderdyr ved fødslen (gennemsnit)
|
|
|
|
|
|
|
Levende unger/moderdyr på dag 4 (gennemsnit)
|
|
|
|
|
|
|
Kønsfordeling (han/hun) ved fødslen (gennemsnit)
|
|
|
|
|
|
|
Kønsfordeling (han/hun) på dag 4 (gennemsnit)
|
|
|
|
|
|
|
Kuldenes vægt ved fødslen (gennemsnit)
|
|
|
|
|
|
|
Kuldenes vægt på dag 4 (gennemsnit)
|
|
|
|
|
|
|
Ungernes vægt ved fødslen (gennemsnit)
|
|
|
|
|
|
|
Ungernes vægt på tidspunktet for AGD-måling (gennemsnit hanner, gennemsnit hunner)
|
|
|
|
|
|
|
Ungernes AGD på den samme dag efter fødslen, fødsel - dag 4 (gennemsnit hanner, gennemsnit hunner, PND-angivelse)
|
|
|
|
|
|
|
Ungernes vægt på dag 4 (gennemsnit)
|
|
|
|
|
|
|
Ungernes vægt på dag 13 (gennemsnit)
|
|
|
|
|
|
|
Hanungernes bibeholdelse af brystvorter ("nipple retention") på dag 13 (gennemsnit)
|
|
|
|
|
|
|
UNGER MED ABNORMITETER
|
|
Moderdyr med 0
|
|
|
|
|
|
|
Moderdyr med 1
|
|
|
|
|
|
|
Moderdyr med ≥ 2
|
|
|
|
|
|
|
TAB AF AFKOM
|
|
Prænatalt (implantationer minus levendefødte)
|
|
Hunner med 0
|
|
|
|
|
|
|
Hunner med 1
|
|
|
|
|
|
|
Hunner med 2
|
|
|
|
|
|
|
Hunner med ≥ 3
|
|
|
|
|
|
|
Postnatalt (levendefødte minus levende på dag 13 efter fødslen)
|
|
Hunner med 0
|
|
|
|
|
|
|
Hunner med 1
|
|
|
|
|
|
|
Hunner med 2
|
|
|
|
|
|
|
Hunner med ≥ 3
|
|
|
|
|
|
B.65
IN VITRO-MEMBRANBARRIEREFORSØGSMETODE FOR HUDÆTSNING
INDLEDNING
|
1.
|
Denne forsøgsmetode svarer til OECD-testvejledning (Test Guideline (TG)) 435 (2015). Hudætsning defineres som fremkomsten af irreversibel ødelæggelse af huden, der viser sig som en synlig nekrose gennem epidermis til dermis efter påføring af et testkemikalie som defineret i De Forenede Nationers (FN’s) globale harmoniserede system for klassificering og mærkning af kemikalier (GHS) (1) og Den Europæiske Unions (EU’s) forordning 1272/2008 om klassificering, mærkning og emballering af stoffer og blandinger (CLP) (24). Denne forsøgsmetode, som svarer til den opdaterede OECD-testvejledning 435, er en in vitro-membranbarriereforsøgsmetode, der kan anvendes til identifikation af ætsende kemikalier. I denne forsøgsmetode anvendes en kunstig membran, der reagerer på ætsende kemikalier på samme måde som dyrehud in situ.
|
|
2.
|
Hudætsning er traditionelt blevet vurderet ved påføring af et testkemikalie på huden på levende dyr og vurdering af omfanget af vævsbeskadigelsen efter et bestemt tidsrum (2). Ud over denne forsøgsmetode er der blevet vedtaget flere andre in vitro-forsøgsmetoder som alternativer (3) (4) til den traditionelle in vivo-procedure med kaninhud (kapitel B.4 i dette bilag, som svarer til OECD TG 404), der anvendes til identifikation af ætsende kemikalier (2). Den trindelte UN GHS-test- og evalueringsstrategi for vurdering og klassificering af hudætsning og OECD’s vejledende dokument om integrerede tilgange til testning og vurdering (IATA) vedrørende hudirritation/-ætsning anbefaler, at der anvendes validerede og accepterede in vitro-forsøgsmetoder under modul 3 og 4 (1) (5). IATA beskriver flere moduler, som grupperer informationskilder og analyseværktøjer, og vejleder i, i) hvordan eksisterende forsøgsdata og ikke-forsøgsdata integreres i og anvendes til vurdering af kemikaliers hudirritations-/hudætsningspotentiale, og ii) foreslår en tilgang, når der er behov for yderligere testning, herunder når der findes negative resultater (5). I denne modulopdelte tilgang kan positive resultater fra in vitro-forsøgsmetoder anvendes til at klassificere et kemikalie som ætsende uden dyreforsøg, hvorved anvendelsen af dyr reduceres, og det undgås at påføre dyrene de smerter og lidelser, de kan blive udsat for, når de anvendes til dette formål.
|
|
3.
|
Der er foretaget valideringsundersøgelser af den in vitro-membranbarrieremodel, der findes i handelen under betegnelsen Corrositex® (6) (7) (8), som påviser en overordnet nøjagtighed for forudsigelse af hudætsning på 79 % (128/163), en følsomhed på 85 % (76/89) og en specificitet på 70 % (52/74) for en database med 163 stoffer og blandinger (7). På baggrund af denne anerkendte validitet er den validerede referencemetode (VRM) blevet anbefalet til at indgå som en del af en trindelt teststrategi til vurdering af kemikaliers potentielle hudætsningsfare (5) (7). Inden en in vitro-membranbarrieremodel til vurdering af hudætsning kan benyttes i lovgivningsøjemed, skal dens pålidelighed og relevans (nøjagtighed) og grænserne for dens påtænkte anvendelse bestemmes for at sikre, at den er sammenlignelig med VRM’erne (9), i overensstemmelse med de foruddefinerede ydeevnestandarder (PS) (10). OECD-aftalen om gensidig anerkendelse af data vil først være garanteret, når en foreslået ny eller opdateret metode i henhold til ydeevnestandarderne er blevet gennemgået og medtaget i den tilsvarende OECD-testvejledning. For øjeblikket er kun en in vitro-metode omfattet af OECD-testvejledning 435 og denne forsøgsmetode, nemlig den Corrositex®-model, der findes i handelen.
|
|
4.
|
Andre forsøgsmetoder til vurdering af hudætsning er baseret på anvendelse af rekonstitueret human hud (OECD TG 431) (3) og isoleret rottehud (OECD TG 430) (4). Denne testvejledning muliggør endvidere underopdeling af ætsende kemikalier i de tre UN GHS-underkategorier for ætsende virkning og de tre UN-transportemballagegrupper for ætsningsfare. Denne testvejledning blev oprindeligt vedtaget i 2006 og opdateret i 2015 for at referere til det vejledende IATA-dokument og opdatere listen over kompetencestoffer.
|
DEFINITIONER
|
5.
|
De anvendte definitioner er forklaret i tillægget.
|
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER
|
6.
|
Den test, der beskrives i denne forsøgsmetode, muliggør identifikation af ætsende testkemikalier og underopdeling af ætsende testkemikalier i henhold til UN GHS/CLP (tabel 1). Derudover kan forsøgsmetoden anvendes til at træffe beslutninger om specifikke kemikalieklassers ætsende og ikke-ætsende virkning, f.eks. organiske og uorganiske syrer, syrederivater (25) og baser til visse transportrelaterede formål (7) (11) (12). Denne forsøgsmetode beskriver engenerisk procedure, der svarer til den validerede referencemetode (7). Forsøgsmetoden giver ikke fyldestgørende oplysninger om hudirritation, men det skal bemærkes, at TM B.46 (der svarer til OECD TG 439) specifikt vedrører in vitro-test af hudirritation (13). For fuldt ud at kunne vurdere de lokale virkninger på huden efter en enkelt dermal eksponering bør OECD’s vejledende dokument om integrerede tilgange til testning og vurdering (IATA) følges (5).
Tabel 1
UN GHS-kategori og -underkategorier for hudætsning (26)
|
Kategori for hudætsning (kategori 1) (for myndigheder, der ikke anvender underkategorier)
|
Underkategorier for hudætsning (26) (for myndigheder, der anvender underkategorier, herunder CLP-forordningen)
|
Ætsende i ≥ 1 af 3 dyr
|
|
Eksponering
|
Observation
|
|
Ætsende
|
Underkategori for ætsning 1A
|
≤ 3 minutter
|
≤ 1 time
|
|
Underkategori for ætsning 1B
|
> 3 minutter/≤ 1 time
|
≤ 14 dage
|
|
Underkategori for ætsning 1C
|
> 1 time/≤ 4 timer
|
≤ 14 dage
|
|
|
7.
|
En af den validerede referencemetodes begrænsninger (7) er, at mange ikke-ætsende kemikalier og nogle ætsende kemikalier muligvis ikke er kvalificeret til testning ud fra resultaterne af den indledende kompatibilitetstest (jf. punkt 13). Vandige kemikalier med en pH på mellem 4,5 og 8,5 er ofte ikke kvalificeret til testning. Ikke desto mindre var 85 % af de kemikalier, der blev testet i dette pH-interval, ikke ætsende ved dyreforsøg (7). In vitro-membranbarrieremetoden kan anvendes til testning af faste stoffer (vandopløselige eller uopløselige i vand), væsker (vandige eller ikke-vandige) og emulsioner. Testkemikalier, der ikke forårsager en påviselig ændring under kompatibilitetstesten (dvs. farveændring i kemikaliedetektionssystemet (CDS) i den validerede referenceforsøgsmetode), kan imidlertid ikke testes med membranbarrieremetoden og bør testes ved hjælp af andre forsøgsmetoder.
|
PRINCIP FOR TESTEN
|
8.
|
Testsystemet består af to dele: en syntetisk makromolekylær biobarriere og et kemikaliedetektionssystem (CDS). Ved hjælp af CDS-membranbarrieren påviser denne forsøgsmetode skader forårsaget af ætsende testkemikalier efter påføring af et testkemikalie på overfladen af den syntetiske makromolekylære membranbarriere (7), idet disse skader formodes forårsaget af de samme ætsende mekanismer som dem, der opererer på levende hud.
|
|
9.
|
Membranbarrierens penetration (eller gennembrud) kan måles ved en række procedurer eller CDS, herunder ændring af farven på en pH-indikatorfarve eller af en anden af indikatoropløsningens egenskaber under barrieren.
|
|
10.
|
Det bør bestemmes, at membranbarrieren er gyldig, dvs. relevant og pålidelig, til det påtænkte formål. Dette indebærer sikring af, at forskellige kemiske produkter er homogene med hensyn til deres barriereegenskaber, f.eks. at de er i stand til at opretholde en barriere mod ikke-ætsende kemikalier, og at de kan kategorisere kemikaliernes ætsende egenskaber i henhold til de forskellige UN GHS-underkategorier for ætsning (1). Den tildelte klassificering baseres på den tid, det tager for et kemikalie at trænge igennem membranbarrieren og ind til indikatoropløsningen.
|
PÅVISNING AF KOMPETENCE
|
11.
|
Inden laboratorierne påbegynder rutinemæssig brug af in vitro-membranbarrieremetoden, som er i overensstemmelse med denne forsøgsmetode, bør de påvise deres tekniske kompetence ved korrekt identifikation af de 12 anbefalede kompetencestoffer i tabel 2. I situationer, hvor et stof på listen ikke kan fås, eller hvor det er berettiget, kan et andet stof anvendes, hvis der foreligger tilstrækkelige in vivo- og in vitro-referencedata herfor (f.eks. fra listen over referencekemikalier (10)), forudsat at der benyttes de samme udvælgelseskriterier som beskrevet i tabel 1.
Tabel 2
Kompetencestoffer
(27)
|
Stof (28)
|
CAS RN
|
Kemisk gruppe
|
UN GHS-underkategori in vivo (29)
|
UN GHS-underkategori in vitro (29)
|
|
Bortrifluoriddihydrat
|
13319-75-0
|
Uorganiske syrer
|
1A
|
1A
|
|
Salpetersyre
|
7697-37-2
|
Uorganiske syrer
|
1A
|
1A
|
|
Phosphorpentachlorid
|
10026-13-8
|
Prækursorer for uorganiske syrer
|
1A
|
1A
|
|
Valerylchlorid
|
638-29-9
|
Syrechlorider
|
1B
|
1B
|
|
Natriumhydroxid
|
1310-73-2
|
Uorganiske baser
|
1B
|
1B
|
|
1-(2-aminoethyl)piperazin
|
140-31-8
|
Alifatiske aminer
|
1B
|
1B
|
|
Benzensulfonylchlorid
|
98-09-9
|
Syrechlorider
|
1C
|
1C
|
|
N,N-dimethylbenzylamin
|
103-83-3
|
Aniliner
|
1C
|
1C
|
|
Tetraethylenpentamin
|
112-57-2
|
Alifatiske aminer
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Phenoler
|
NC
|
NC
|
|
Nonylacrylat
|
2664-55-3
|
Acrylater/methacrylater
|
NC
|
NC
|
|
Natriumbicarbonat
|
144-55-8
|
Uorganiske salte
|
NC
|
NC
|
|
FREMGANGSMÅDE
|
12.
|
Nedenstående punkter beskriver de komponenter og procedurer, som en forsøgsmetode til vurdering af ætsende virkning med en kunstig membranbarriere består af (7) (15), baseret på den aktuelle VRM, dvs. Corrositex®, som findes i handelen. Membranbarrieren og kompatibilitets-/indikatoropløsningen og kategoriseringsopløsningen kan konstrueres, fremstilles eller købes i handelen, som det er tilfældet med VRM’en Corrositex®. Der findes et eksempel på en forsøgsmetodeprotokol for den validerede referenceforsøgsmetode (7). Testen bør foretages ved stuetemperatur (17-25 °C), og komponenterne skal opfylde nedenstående betingelser.
|
Testkemikalie-kompatibilitetstest
|
13.
|
Inden membranbarrieretesten foretages, udføres en kompatibilitetstest for at fastslå, om CDS kan detektere testkemikaliet. Hvis ikke CDS kan detektere testkemikaliet, er membranbarrieretesten ikke egnet til at vurdere det pågældende testkemikalies potentielle ætsende virkning, og der anvendes en anden forsøgsmetode. CDS og eksponeringsforholdene i kompatibilitetstesten bør afspejle eksponeringen i den efterfølgende membranbarrieretest.
|
Tidsbestemt kategoritest for testkemikaliet
|
14.
|
Hvis det passer til forsøgsmetoden, underkastes et testkemikalie, der er kvalificeret i henhold til kompatibilitetstesten, en tidsbestemt kategoritest, dvs. en screeningstest, for at skelne mellem svage og stærke syrer eller baser. I den validerede referenceforsøgsmetode anvendes f.eks. en tidsbestemt kategoriseringstest til at vise, hvilken af to tidsrammer der bør anvendes, alt efter om der påvises en signifikant sur eller basisk reserve. Der bør anvendes to forskellige tidsrammer for gennembrud til bestemmelse af den ætsende virkning og UN GHS-underkategorien for hudætsning på baggrund af testkemikaliets sure eller basiske reserve.
|
MEMBRANBARRIEREFORSØGSMETODENS KOMPONENTER
Membranbarriere
|
15.
|
Membranbarrieren består af to komponenter: en proteinholdig makromolekylær vandig gel og en permeabel støttemembran. Den proteinholdige gel bør være uigennemtrængelig for væsker og faste stoffer, men kan ætses og gøres permeabel. Den fuldt konstruerede membranbarriere bør opbevares under forud fastsatte betingelser, som har vist sig at forhindre, at gelen ødelægges, f.eks. ved udtørring, bakterievækst, flytning, revnedannelse, der ville forringe dens ydeevne. Den acceptable opbevaringsperiode skal fastsættes, og membranbarrierepræparaterne må ikke anvendes efter udløbet af denne periode.
|
|
16.
|
Den permeable støttemembran yder den proteinholdige gel mekanisk støtte under gelatineringsprocessen, og mens den eksponeres for testkemikaliet. Støttemembranen skal forhindre, at gelen synker sammen eller flytter sig, og alle testkemikalier skal kunne trænge igennem den.
|
|
17.
|
Den proteinholdige gel, der består af protein, f.eks. keratin, collagen eller blandinger af proteiner, som danner en gelmatrice, er målet for testkemikaliet. Det proteinholdige materiale anbringes på støttemembranens overflade og gelatinerer, inden membranbarrieren placeres over indikatoropløsningen. Den proteinholdige gel skal have samme tykkelse og tæthed overalt og være uden luftbobler eller andre defekter, der risikerer at påvirke dens funktionelle integritet.
|
Kemikaliedetektionssystem (CDS)
|
18.
|
Indikatoropløsningen, der er den samme opløsning som den, der bruges i kompatibilitetstesten, skal reagere på testkemikaliets tilstedeværelse. Der skal bruges en pH-indikatorfarve eller -farvekombination, f.eks. cresolrød og methylorange, der skifter farve som reaktion på testkemikaliets tilstedeværelse. Målesystemet kan være visuelt eller elektronisk.
|
|
19.
|
Relevansen og pålideligheden af de detektionssystemer, der udvikles til detektion af testkemikaliets passering gennem barrieremembranen, skal vurderes for at påvise den række kemikalier, der kan detekteres, og de kvantitative begrænsninger for detektionen.
|
TESTENS YDEEVNE
Samling af forsøgsmetodens komponenter
|
20.
|
Membranbarrieren anbringes i et hætteglas (eller rør), som indeholder indikatoropløsningen, således at hele støttemembranen er i kontakt med indikatoropløsningen, og der ikke er luftbobler til stede. Der bør udvises forsigtighed for at bevare barrierens integritet.
|
Påføring af testkemikaliet
|
21.
|
En passende mængde testkemikalie, f.eks. 500 μl, hvis der er tale om en væske, eller 500 mg fint pulver, hvis der er tale om et fast stof (7), påføres forsigtigt membranbarrierens opadvendte overflade og fordeles jævnt. Der forberedes et passende antal replikater, f.eks. fire (7), for hvert testkemikalie og deres tilhørende kontroller (jf. punkt 23-25). Tidspunktet for testkemikaliets påføring på membranbarrieren registreres. For at sikre, at korte ætsningstider bliver registreret nøjagtigt, anføres tidspunkterne for testkemikaliets indføring i de forskellige hætteglas separat.
|
Måling af membranbarrierepenetration
|
22.
|
Hvert hætteglas overvåges nøje, og tidspunktet for den første ændring af indikatoropløsningen, dvs. barrierepenetration, registreres, og tiden mellem påføring på og penetration af membranbarrieren bestemmes.
|
Kontroller
|
23.
|
I test, hvor der bruges et bærestof eller opløsningsmiddel sammen med testkemikaliet, bør bærestoffet eller opløsningsmidlet være kompatibelt med membranbarrieresystemet, dvs. det må ikke ændre membranbarrieresystemets integritet, og det må ikke ændre testkemikaliets ætsende virkning. Når det er relevant, bør der testes kontroller med opløsningsmiddel (eller bærestof) sideløbende med testkemikaliet for at påvise opløsningsmidlets kompatibilitet med membranbarrieresystemet.
|
|
24.
|
En positiv (ætsende) kontrol med mellemhøj ætsende aktivitet, f.eks. 110 ± 15 mg natriumhydroxid (UN GHS-underkategori 1B) (7), bør testes sideløbende med testkemikaliet for at vurdere, om testsystemet fungerer, som det skal. Det kan være hensigtsmæssigt at teste en anden positiv kontrol fra samme kemiske gruppe som testkemikaliet med henblik på at evaluere et ætsende testkemikalies relative ætsende potentiale. Der bør vælges positive kontroller med mellemkraftig ætsende virkning (f.eks. UN GHS-underkategori 1B) med henblik på at påvise ændringer i penetrationstiden, der kan være uacceptabelt meget længere eller kortere end den fastsatte referenceværdi og dermed viser, at testsystemet ikke fungerer korrekt. I denne forbindelse er stærkt ætsende (UN GHS-underkategori 1A) og ikke-ætsende kemikalier af begrænset relevans. Et ætsende kemikalie fra UN GHS-underkategori 1B ville påvise en for kort eller lang gennembrudstid. Et svagt ætsende kemikalie (UN GHS-underkategori 1C) kan eventuelt benyttes som positiv kontrol til at måle forsøgsmetodens evne til konsekvent at skelne mellem svagt ætsende og ikke-ætsende kemikalier. Uanset den anvendte tilgang skal der opstilles et acceptabelt responsinterval for positive kontroller med udgangspunkt i det historiske interval for de anvendte positive kontrollers gennembrudstider, som f.eks. gennemsnit ± 2-3 standardafvigelser. I hver undersøgelse skal den præcise gennembrudstid for den positive kontrol bestemmes, så afvigelser, der ligger uden for det acceptable interval, kan påvises.
|
|
25.
|
Der bør endvidere testes en negativ (ikke-ætsende) kontrol, f.eks. 10 % citronsyre, 6 % propionsyre (7), sideløbende med testkemikaliet som en anden kvalitetskontrolforanstaltning til påvisning af membranbarrierens funktionelle integritet.
|
Acceptkriterier for undersøgelsen
|
26.
|
I henhold til de opstillede tidsparametre for de enkelte UN GHS-underkategorier for ætsende virkning anvendes den tid (i minutter), der går mellem et testkemikalies påføring på membranbarrieren og barrierepenetrationen, til at forudsige testkemikaliets ætsende virkning. For at en undersøgelse kan opfattes som acceptabel, skal den sideløbende positive kontrol give den forventede penetrationsresponstid (f.eks. 8-16 minutters gennembrudstid for natriumhydroxid, hvis dette stof anvendes som positiv kontrol), den sideløbende negative kontrol skal være ikke-ætsende, og en eventuel kontrol med opløsningsmiddel skal hverken være ætsende eller ændre testkemikaliets ætsende potentiale. Inden laboratorierne påbegynder rutinemæssig brug af en metode, som er i overensstemmelse med denne forsøgsmetode, skal de påvise deres tekniske kompetence ved hjælp af de 12 anbefalede stoffer i tabel 2. Inden nye analoge metoder, der udvikles under denne forsøgsmetode, og som strukturelt og funktionelt ligner den validerede referencemetode (14), tages i brug til testning i lovgivningsøjemed, bør de foruddefinerede ydeevnestandarder bruges til at påvise den nye metodes pålidelighed og nøjagtighed.
|
Fortolkning af resultater og testkemikaliernes klassificering efter ætsende virkning
|
27.
|
Den tid (i minutter), der går mellem testkemikaliets påføring på membranbarrieren og barrierepenetrationen, bruges til at klassificere testkemikaliet i UN GHS-underkategorierne for ætsende virkning (1) og eventuelt i UN-emballagegrupperne (16). De tidsmæssige afskæringsværdier for de tre underkategorier for ætsende virkning opstilles for hver enkelt foreslået forsøgsmetode. De endelige beslutninger om afskæringstider bør tage hensyn til behovet for at minimere underklassificering af ætsningsfare (dvs. falske negative). I denne testvejledning bør afskæringstiderne for Corrositex®, som beskrevet i tabel 3, anvendes, da denne er den eneste forsøgsmetode, der for øjeblikket er omfattet af denne testvejledning (7).
Tabel 3
Corrositex®-forudsigelsesmodel
|
Gennemsnitlig gennembrudstid (min.)
|
UN GHS-forudsigelse (32)
|
|
Kategori 1-testkemikalier (30) (bestemt ved metodens kategoriseringstest)
|
Kategori 2-testkemikalier (31) (bestemt ved metodens kategoriseringstest)
|
|
0-3 min.
|
0-3 min.
|
Ætsende valgfri underkategori 1A
|
|
> 3 til 60 min.
|
> 3 til 30 min.
|
Ætsende valgfri underkategori 1B
|
|
> 60 til 240 min.
|
> 30 til 60 min.
|
Ætsende valgfri underkategori 1C
|
|
> 240 min.
|
> 60 min.
|
Ikke-ætsende
|
|
DATA OG RAPPORTERING
Data
|
28.
|
Den tid (i minutter), der går mellem påføring og barrierepenetration for henholdsvis testkemikaliet og den eller de positive kontroller, rapporteres i tabelform som individuelle replikatdata sammen med gennemsnit ± standardafvigelsen for hvert forsøg.
|
Testrapport
|
29.
|
Testrapporten bør indeholde følgende oplysninger:
|
|
Testkemikalie og kontrolstoffer:
|
—
|
Stof med kun én bestanddel: kemisk identifikation, f.eks. IUPAC-navn eller CAS-navn, CAS-nummer, SMILES- eller InChI-kode, strukturformel, renhed, kemisk identitet af urenheder, i det omfang det er relevant og praktisk muligt, osv.
|
|
—
|
Stof med flere bestanddele, UVCB-stoffer og blandinger: kendetegnet så vidt muligt ved kemisk identitet (jf. ovenfor), kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber
|
|
—
|
fysisk udseende, vandopløselighed og yderligere relevante fysisk-kemiske egenskaber
|
|
—
|
oprindelse, evt. batchnummer
|
|
—
|
behandling af testkemikaliet/kontrolstoffet inden testning, hvis det er relevant (f.eks. opvarmning, formaling)
|
|
—
|
testkemikaliets stabilitet, sidste anvendelsesdato eller dato for ny analyse, hvis dette er kendt
|
|
|
|
Bærestof:
|
—
|
identifikation, koncentration (hvis relevant), anvendt mængde
|
|
—
|
begrundelse for valg af bærestof.
|
|
|
|
Anvendt in vitro-membranbarrieremodel og -protokol, herunder påvist nøjagtighed og pålidelighed
|
|
|
Testbetingelser:
|
—
|
beskrivelse af anvendt apparatur og anvendte præpareringsprocedurer
|
|
—
|
den anvendte in vitro-membranbarrieres oprindelse og sammensætning
|
|
—
|
indikatoropløsningens sammensætning og egenskaber
|
|
—
|
testkemikalie- og kontrolstofmængde
|
|
—
|
beskrivelse af og begrundelse for den tidsbestemte kategoriseringstest
|
|
—
|
observationstidspunkter
|
|
—
|
beskrivelse af de benyttede vurderings- og klassificeringskriterier
|
|
—
|
påvisning af kompetence til at anvende forsøgsmetoden inden rutinemæssig brug ved at teste kompetencekemikalierne.
|
|
|
|
Resultater:
|
—
|
opstilling i tabelform af individuelle rådata fra individuelle test og kontroller for hvert replikat
|
|
—
|
beskrivelse af andre observerede virkninger
|
|
—
|
den afledte klassificering med henvisning til den anvendte forudsigelsesmodel og/eller de anvendte beslutningskriterier.
|
|
Diskussion af resultaterne
Konklusioner
|
LITTERATUR
|
(1)
|
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth Revised Edition, UN New York and Geneva, 2013. Tilgængelig på: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Kapitel B.4 i dette bilag, Akut hudirritation/-ætsning.
|
|
(3)
|
Kapitel B.40.A i dette bilag, In vitro-hudætsning: forsøgsmetode med rekonstrueret human epidermis (RhE).
|
|
(4)
|
Kapitel B.40 i dette bilag, In vitro-hudætsning: Transkutan elektrisk modstand (TER).
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. og Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495).
|
|
(8)
|
Gordon V.C., Harvell J.D. og Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (No 34).
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (September 20, 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Kapitel B.46 i dette bilag, In vitro-hudirritation: forsøgsmetode med rekonstrueret human epidermis. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Tilgængelig på: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Tilgængelig på: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
United Nations (UN) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Tilgængelig på: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Tillæg
DEFINITIONER
Nøjagtighed
: overensstemmelsen mellem forsøgsmetodens resultater og accepterede referenceværdier. Den er et mål for forsøgsmetodens ydeevne og et af aspekterne af relevans. Udtrykket benyttes ofte som synonym for "konkordans", forstået som den andel af korrekte udfald, forsøgsmetoden fører til (9).
Kemikalie
: et stof eller en blanding.
Kemikaliedetektionssystem (CDS)
: et visuelt eller elektronisk målesystem med en indikatoropløsning, der reagerer på et testkemikalies tilstedeværelse, f.eks. ved ændring af en pH-indikatorfarve eller -farvekombination. Systemet reagerer på testkemikaliets tilstedeværelse med en farveændring eller med en anden form for kemisk eller elektrokemisk reaktion.
Konkordans
: et mål for ydeevnen af en forsøgsmetode, der fører til kategoriske resultater, og et af aspekterne af relevans. Udtrykket benyttes af og til som synonym for nøjagtighed og defineres som andelen af alle kemikalier, der klassificeres korrekt som positive eller negative. Konkordansen afhænger i høj grad af forekomsten af positiver i de undersøgte typer testkemikalier (9).
GHS (det globale harmoniserede system for klassificering og mærkning af kemikalier)
: et system til klassificering af kemikalier (stoffer og blandinger) efter standardiserede typer og niveauer af fysisk fare, sundhedsfare og miljøfare med tilhørende kommunikationselementer såsom piktogrammer, signalord, faresætninger, sikkerhedssætninger og sikkerhedsdatablade, således at oplysninger om kemikaliernes skadelige virkninger kan videreformidles med henblik på at beskytte mennesker (f.eks. arbejdsgivere, arbejdstagere, transportører, forbrugere og redningspersonel) og miljøet (1).
IATA
: integreret tilgang til testning og vurdering.
Blanding
: blanding eller opløsning, der er sammensat af to eller flere stoffer.
Stof med kun én bestanddel
: et stof, der defineres af sin mængdemæssige sammensætning, hvori en hovedbestanddel udgør mindst 80 % (w/w).
Stof med flere bestanddele
: et stof, der defineres af sin mængdemæssige sammensætning, hvori flere end én hovedbestanddel er til stede i en koncentration på ≥ 10 % (w/w) og < 80 % (w/w). Et stof, der består af flere forskellige bestanddele, er resultatet af en fremstillingsproces. Forskellen mellem en blanding og et stof med flere bestanddele er, at blandingen er fremstillet ved blanding af to eller flere stoffer uden kemisk reaktion. Et stof, der består af flere forskellige bestanddele, er resultatet af en kemisk reaktion.
NC
: Ikke-ætsende.
Ydeevnestandarder
: standarder, der med udgangspunkt i en valideret forsøgsmetode danner grundlag for at vurdere, om en foreslået forsøgsmetode, som er mekanistisk og funktionelt tilsvarende, er sammenlignelig. Disse standarder omfatter i) de centrale dele af forsøgsmetoden, ii) en minimumsliste med referencekemikalier, som er udvalgt blandt de kemikalier, der er benyttet til at påvise, at den validerede forsøgsmetodes ydeevne er acceptabel, og iii) de tilsvarende niveauer af pålidelighed og nøjagtighed, som er fastsat efter, hvad der er opnået med den validerede forsøgsmetode, og som det skal påvises, at den foreslåede forsøgsmetode kan yde ved vurdering ud fra minimumslisten med referencekemikalier (9).
Relevans
: beskrivelse af sammenhængen mellem forsøgsmetoden og den pågældende virkning og dens meningsfuldhed og brugbarhed til et bestemt formål. Det angives, i hvilket omfang forsøgsmetoden måler eller forudsiger den pågældende biologiske virkning korrekt. Forsøgsmetodens nøjagtighed (konkordans) indgår i vurderingen af dens relevans (9).
Pålidelighed
: mål for, i hvilken grad en forsøgsmetode kan reproduceres i og mellem laboratorier over en længere periode, når den udføres efter samme protokol. Den bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier (9).
Følsomhed
: andelen af alle positive/aktive kemikalier, der klassificeres korrekt ved forsøgsmetoden. Den er et mål for nøjagtigheden af en forsøgsmetode, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en forsøgsmetodes relevans (9).
In vivo-hudætsning
: fremkaldelse af irreversibel skade på huden, dvs. synlig nekrose gennem epidermis og ned i dermis efter påføring af et testkemikalie i indtil fire timer. Typiske ætsningsreaktioner er sår, blødning, blodige skorper, efter 14 dages observation affarvning som følge af blegning af huden, hele områder med alopeci samt ardannelse. Tvivlsomme læsioner bør overvejes vurderet ved histopatologisk undersøgelse.
Specificitet
: andelen af alle negative/inaktive kemikalier, der klassificeres korrekt ved forsøgsmetoden. Den er et mål for nøjagtigheden af en forsøgsmetode, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en forsøgsmetodes relevans (9).
Stof
: et grundstof og forbindelser heraf, naturlige eller industrielt fremstillede, indeholdende sådanne tilsætningsstoffer, som er nødvendige til bevarelse af stoffets stabilitet, og sådanne urenheder, som følger af fremstillingsprocessen, bortset fra opløsningsmidler, som kan udskilles, uden at det påvirker stoffets stabilitet eller ændrer dets sammensætning.
Testkemikalie
: alle stoffer eller blandinger, der testes ved hjælp af denne forsøgsmetode.
UVCB
: stoffer med ukendt eller variabel sammensætning, komplekse reaktionsprodukter eller biologiske materialer.
B.66 STABILT TRANSFEKTEREDE IN VITRO-TRANSAKTIVERINGSASSAYS TIL PÅVISNING AF ØSTROGENRECEPTORAGONISTER OG -ANTAGONISTER
GENEREL INDLEDNING
OECD’s ydeevnebaserede testvejledning
|
1.
|
Denne forsøgsmetode svarer til OECD-testvejledning (Test Guideline (TG)) 455 (2016). TG 455 er en ydeevnebaseret testvejledning (PBTG), der beskriver metoden for stabilt transfekterede in vitro-transaktiveringsassays til påvisning af østrogenreceptoragonister og -antagonister (ER TA-assays). Den omfatter flere mekanistisk og funktionelt tilsvarende forsøgsmetoder til identifikation af østrogenreceptoragonister og -antagonister (dvs. ERα og/eller ERβ) og ventes at lette udviklingen af nye lignende eller modificerede forsøgsmetoder i overensstemmelse med de principper for validering, som er fastsat i OECD’s vejledende dokument om validering og international accept af nye eller opdaterede forsøgsmetoder til farevurdering (1). De fuldt validerede referenceforsøgsmetoder (tillæg 2 og tillæg 3), som danner grundlag for denne PBTG, er:
|
—
|
det stabilt transfekterede TA-assay (STTA) (2), som bruger (h) ERα-HeLa-9903-cellelinjen, og
|
|
—
|
VM7Luc ER TA-assayet (3), som anvender VM7Luc4E2-cellelinjen (33), der overvejende angiver hERα med en vis medvirken fra hERβ(4) (5).
|
Der findes ydeevnestandarder (PS) (6) (7), som bør anvendes til udvikling og validering af lignende assays vedrørende den samme fareegenskab. De gør det muligt rettidigt at ændre PBTG 455, så nye lignende assays kan blive tilføjet den opdaterede PBTG. Lignende assays vil dog først blive tilføjet efter OECD’s kontrol og validering af, at ydeevnestandarderne er opfyldt. De assays, der er omfattet af TG 455, kan uden forskel anvendes til at opfylde OECD-medlemslandenes krav til testresultaterne om østrogenreceptortransaktivering, samtidig med at de drager fordel af OECD-aftalen om gensidig anerkendelse af data.
|
Baggrund og principper for de assays, der er omfattet af denne forsøgsmetode
|
2.
|
OECD iværksatte i 1998 en højt prioriteret aktivitet med henblik på revision af eksisterende og udvikling af nye testvejledninger vedrørende screening og testning af potentielle hormonforstyrrende kemikalier. OECD’s begrebsramme for testning og vurdering af potentielle hormonforstyrrende kemikalier blev revideret i 2012. Den oprindelige og reviderede begrebsramme er vedlagt som bilag til OECD’s vejledende dokument om standardiserede testvejledninger til vurdering af kemikaliers hormonforstyrrende virkning (8). Begrebsrammen omfatter fem niveauer, idet hvert niveau svarer til et niveau for biologisk kompleksitet. De ER-transaktiveringsassays (ER TA-assays), som beskrives i denne forsøgsmetode, tilhører niveau 2, der omfatter »in vitro-assays, der giver data om en eller flere udvalgte endokrine mekanismer/veje«. Denne forsøgsmetode er til in vitro-transaktiveringsassays (TA-assays), som er beregnet til at påvise østrogenreceptoragonister og -antagonister.
|
|
3.
|
Østrogeners interaktion med ER’er kan påvirke transkriptionen af østrogenkontrollerede gener, hvilket kan føre til induktion eller inhibering af cellulære processer, herunder processer, som er nødvendige for celleproliferation, normal fosterudvikling og reproduktiv funktion (9) (10) (11). Forstyrrelse af normale østrogensystemer kan potentielt udløse skadelige virkninger på normal udvikling (ontogenese), reproduktiv sundhed og det reproduktive systems integritet.
|
|
4.
|
In vitro-transaktiveringsassays er baseret på stoffernes direkte eller indirekte interaktion med en specifik receptor, som regulerer transkriptionen af et reportergenprodukt. Sådanne assays er i stor udstrækning blevet anvendt til at vurdere genekspression, der reguleres af specifikke kernereceptorer som f.eks. ER’er (12) (13) (14) (15) (16). De er blevet foreslået til påvisning af østrogentransaktivering, som reguleres af ER’en (17) (18) (19). Der findes mindst to større undertyper af kerne-ER’er, α og β, som er kodet af forskellige gener. De respektive proteiner har forskellige biologiske funktioner og forskellig vævsfordeling og ligandaffinitet (20) (21) (22) (23) (24) (25) (26). Kerne-ERα medierer den klassiske østrogenrespons (27) (28) (29) (30), og derfor er de fleste af de modeller, der for øjeblikket udvikles til at måle ER-aktivering eller -inhibering, ERα-specifikke. Disse assays anvendes til påvisning af kemikalier, deraktiverer (eller inhiberer) ER’en efter ligandbinding, hvorefter receptor-ligand-komplekset binder sig til specifikke DNA-responselementer og transaktiverer et reportergen, hvilket resulterer i et markørproteins øgede celleekspression. Der kan anvendes forskellige reporterresponser i disse assays. I luciferasebaserede systemer omdanner luciferaseenzymet luciferinmediet til et bioluminescensprodukt, som måles kvantitativt med et luminometer. Andre eksempler på almindelige reportere er fluorescerende protein og LacZ-genet, som koder β-galactosidase, et enzym, der kan omdanne det farveløse medium X-gal (5-brom-4-chlor-indolyl-galactopyranosid) til et blåt produkt, der kan kvantificeres med et spektrofotometer. Disse reportere kan vurderes hurtigt og billigt med analysesæt, der kan fås i handelen.
|
|
5.
|
Valideringsundersøgelser af STTA- og VM7Luc TA-assays har påvist deres relevans og pålidelighed i overensstemmelse med deres formål (3) (4) (5) (30). Ydeevnestandarder for luminescensbaserede ER TA-assays ved anvendelse af brystcellelinjer er omfattet af ICCVAM-evalueringsrapporten Test Method Evaluation Report on the LUMI-CELL® ER (VM7Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals (3). Disse ydeevnestandarder er blevet ændret, så de kan anvendes på både STTA- og VM7Luc TA-assays (2).
|
|
6.
|
Definitioner og forkortelser brugt i denne forsøgsmetode er beskrevet i tillæg 1.
|
Anvendelsesområde og begrænsninger i forbindelse med TA-assays
|
7.
|
Disse assays foreslås til screenings- og prioriteringsformål, men kan også give mekanistiske oplysninger, som kan anvendes i en weight-of-the-evidence-analyse. De analyserer den TA, som kemikaliets binding til ER’ene inducerer i et in vitro-system. Resultaterne bør således ikke direkte ekstrapoleres til den komplekse signalering og regulering af det intakte hormonsystem in vivo.
|
|
8.
|
TA’en, som medieres af ER’ene, betragtes som en af de centrale mekanismer ved hormonforstyrrelse (ED), skønt der findes andre mekanismer, gennem hvilke ED kan forekomme, bl.a. i) interaktioner med andre receptorer og enzymsystemer i hormonsystemet, ii) hormonsyntese, iii) metabolisk aktivering og/eller inaktivering af hormoner, iv) fordeling af hormoner til målvæv og v) clearance af hormoner fra kroppen. Ingen af de assays, der er omfattet af denne forsøgsmetode, behandler disse virkemåder.
|
|
9.
|
Denne forsøgsmetode ser på kemikaliers evne til at aktivere (dvs. handle som agonister) og undertrykke (dvs. handle som antagonister) ER-afhængig transkription. Nogle kemikalier kan udvise både celletypeafhængig agonist- og antagonistaktivitet og er kendt som selektive østrogenreceptormodulatorer (SERM’er). Kemikalier, der er negative i disse assays, kan vurderes i en ER-bindingsassay, før det konkluderes, at kemikaliet ikke binder sig til receptoren. Desuden vil disse assays formentlig kun oplyse om forældremolekylets aktivitet, da in vitro-cellesystemerne har begrænset metaboliserende kapacitet. I betragtning af at der kun blev anvendt enkeltstoffer under valideringen, er anvendeligheden på forsøgsblandinger ikke blevet behandlet. Forsøgsmetoden kan ikke desto mindre teoretisk set benyttes til testning af stoffer med flere bestanddele, UVCB’er og blandinger. Før forsøgsmetoden anvendes på et stof med flere bestanddele, et UVCB eller en blanding til generering af data i lovgivningsøjemed, bør det overvejes, om den vil give resultater, som er egnede til formålet, og i givet fald hvorfor. Sådanne overvejelser er ikke nødvendige, når der foreligger et myndighedskrav om test af blandingen.
|
|
10.
|
Til orientering viser tabel 1 agonist-testresultaterne for de 34 stoffer, som blev testet i de to fuldt validerede referenceforsøgsmetoder, der beskrives i denne forsøgsmetode. Af disse stoffer er 26 klassificeret som definitive ER-agonister og otte som negative baseret på offentliggjorte rapporter, herunder in vitro-assays for ER-binding og TA og/eller det uterotrofiske assay (2) (3) (18) (31) (32) (33) (34). Tabel 2 viser antagonist-testresultaterne for de 15 stoffer, som blev testet i de to fuldt validerede referenceforsøgsmetoder, der beskrives i denne forsøgsmetode. Af disse stoffer er fire klassificeret som definitive/formodede ER-antagonister og 10 som negative baseret på offentliggjorte rapporter, herunder in vitro-assays for ER-binding og TA (2) (3) (18) (31). Med henvisning til dataene i tabel 1 og 2 var der 100 % overensstemmelse mellem de to referenceforsøgsmetoder med hensyn til klassificeringen af samtlige stoffer med undtagelse af ét stof (mifepriston) i antagonistassayet, og samtlige stoffer blev klassificeret korrekt som ER-agonister/-antagonister eller negative. Der findes yderligere oplysninger om denne kemikaliegruppe samt om supplerende kemikalier, som er testet i STTA- og VM7Luc ER TA-assays i løbet af valideringsundersøgelserne, i ydeevnestandarderne for ER TA (6) (7), tillæg 2 (tabel 1, 2 og 3).
|
Tabel 1
Oversigt over resultaterne af STTA- og VM7Luc ER TA-assays for stoffer testet i begge agonistassays og klassificeret som ER-agonister (POS) eller negative (NEG)
|
|
Stof
|
CAS RN
|
STTA-assay (35)
|
VM7Luc ER TA-assay (36)
|
Datakilde til klassificering (38)
|
|
ER TA- aktivitet
|
PC10-værdi (M)
|
PC50-værdi (2) (M)
|
ER TA-aktivitet
|
EC50-værdi (2), (37) (M)
|
Andre ER TA’er (34)
|
ER-binding
|
Uterotrofisk
|
|
1
|
17ß-østradiol (1)
|
50-28-2
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
5,63 × 10-12
|
POS(227/227)
|
POS
|
POS
|
|
2
|
17α-østradiol (1)
|
57-91-0
|
POS
|
7,24 × 10-11
|
6,44 × 10-10
|
POS
|
1,40 × 10-9
|
POS(11/11)
|
POS
|
POS
|
|
3
|
17α-ethinyløstradiol (1)
|
57-63-6
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
7,31 × 10-12
|
POS(22/22)
|
POS
|
POS
|
|
4
|
17β-trenbolon
|
10161-33-8
|
POS
|
1,78 × 10-8
|
2,73 × 10-7
|
POS
|
4,20 × 10-8
|
POS(2/2)
|
NT
|
NT
|
|
5
|
19-nortestosteron (1)
|
434-22-0
|
POS
|
9,64 × 10-9
|
2,71 × 10-7
|
POS
|
1,80 × 10-6
|
POS(4/4)
|
POS
|
POS
|
|
6
|
4-cumylphenol (1)
|
599-64-4
|
POS
|
1,49 × 10-7
|
1,60 × 10-6
|
POS
|
3,20 × 10-7
|
POS(5/5)
|
POS
|
NT
|
|
7
|
4-tert-octylphenol (1)
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
POS
|
3,19 × 10-8
|
POS(21/24)
|
POS
|
POS
|
|
8
|
Apigenin (1)
|
520-36-5
|
POS
|
1,31 × 10-7
|
5,71 × 10-7
|
POS
|
1,60 × 10-6
|
POS(26/26)
|
POS
|
NT
|
|
9
|
Atrazin (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(30/30)
|
NEG
|
NT
|
|
10
|
Bisphenol A (1)
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
POS
|
5,33 × 10-7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisphenol B (1)
|
77-40-7
|
POS
|
2,36 × 10-8
|
2,11 × 10-7
|
POS
|
1,95 × 10-7
|
POS(6/6)
|
POS
|
POS
|
|
12
|
Butylbenzylphthalat (1)
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
POS
|
1,98 × 10-6
|
POS(12/14)
|
POS
|
NEG
|
|
13
|
Corticosteron (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(6/6)
|
NEG
|
NT
|
|
14
|
Coumestrol (1)
|
479-13-0
|
POS
|
1,23 × 10-9
|
2,00 × 10-8
|
POS
|
1,32 × 10-7
|
POS(30/30)
|
POS
|
NT
|
|
15
|
Daidzein (1)
|
486-66-8
|
POS
|
1,76 × 10-8
|
1,51 × 10-7
|
POS
|
7,95 × 10-7
|
POS(39/39)
|
POS
|
POS
|
|
16
|
Diethylstilbestrol (1)
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POS
|
3,34 × 10-11
|
POS(42/42)
|
POS
|
NT
|
|
17
|
Di-n-butylphthalat
|
84-74-2
|
POS
|
4,09 × 10-6
|
|
POS
|
4,09 × 10-6
|
POS(6/11)
|
POS
|
NEG
|
|
18
|
Ethylparaben
|
120-47-8
|
POS
|
5,00 × 10-6
|
(ingen PC50)
|
POS
|
2,48 × 10-5
|
POS
|
|
NT
|
|
19
|
Østron (1)
|
53-16-7
|
POS
|
3,02 × 10-11
|
5,88 × 10-10
|
POS
|
2,34 × 10-10
|
POS(26/28)
|
POS
|
POS
|
|
20
|
Genistein (1)
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
POS
|
2,71 × 10-7
|
POS(100/102)
|
POS
|
POS
|
|
21
|
Haloperidol
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kaempferol (1)
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
POS
|
3,99 × 10-6
|
POS(23/23)
|
POS
|
NT
|
|
23
|
Chlordecon (1)
|
143-50-0
|
POS
|
7,11 × 10-7
|
7,68 × 10-6
|
POS
|
4,91 × 10-7
|
POS(14/18)
|
POS
|
NT
|
|
24
|
Ketoconazol
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(2/2)
|
NEG
|
NT
|
|
25
|
Linuron (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(8/8)
|
NEG
|
NT
|
|
26
|
meso-hexestrol (1)
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POS
|
1,65 × 10-11
|
POS(4/4)
|
POS
|
NT
|
|
27
|
Methyltestosteron (1)
|
58-18-4
|
POS
|
1,73 × 10-7
|
4,11 × 10-6
|
POS
|
2,68 × 10-6
|
POS(5/6)
|
POS
|
NT
|
|
28
|
Morin
|
480-16-0
|
POS
|
5,43 × 10-7
|
4,16 × 10-6
|
POS
|
2,37 × 10-6
|
POS(2/2)
|
POS
|
NT
|
|
29
|
Norethynodrel (1)
|
68-23-5
|
POS
|
1,11 × 10-11
|
1,50 × 10-9
|
POS
|
9,39 × 10-10
|
POS(5/5)
|
POS
|
NT
|
|
30
|
p,p’-Methoxychlor (1)
|
72-43-5
|
POS
|
1,23 × 10-6
|
(ingen PC50)b (2)
|
POS
|
1,92 × 10-6
|
POS(24/27)
|
POS
|
POS
|
|
31
|
Phenobarbital (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(2/2)
|
NEG
|
NT
|
|
32
|
Reserpin
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(4/4)
|
NEG
|
NT
|
|
33
|
Spironolacton (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(4/4)
|
NEG
|
NT
|
|
34
|
Testosteron
|
58-22-0
|
POS
|
2,82 × 10-8
|
9,78 × 10-6
|
POS
|
1,75 × 10-5
|
POS(5/10)
|
POS
|
NT
|
|
Forkortelser: CAS RN = Registernummer i Chemical Abstracts Service; M = molær; EC50 = halvdelen af den maksimale effektive koncentration af et testkemikalie; NEG = negativ; POS = positiv; NT = ikke testet; PC10 (og PC50) = koncentrationen af et teststof, ved hvilken responsen er 10 % (eller 50 % for PC50) af den respons, der fremkaldes af den positive kontrol (E2, 1nM), på hver plade.
|
Tabel 2
Sammenligning af resultater af STTA- og VM7Luc ER TA-assays for stoffer testet i begge antagonistassays og klassificeret som ER-antagonister (POS) eller negative (NEG)
|
|
Stof (3)
|
CAS RN
|
ER STTA-assay (40)
|
VM7Luc ER TA-assay (41)
|
ER STTA-kandidat-virkninger (43)
|
ICCVAM (44) konsensusklassifikation
|
MeSH (45) kemisk gruppe
|
Produktgruppe (46)
|
|
ER TA-aktivitet
|
IC50-værdi (39) (M)
|
ER TA-aktivitet
|
IC50-værdi (39), (42) (M)
|
|
1
|
4-hydroxytamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
POS
|
2,08 × 10-7
|
moderat POS
|
POS
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt
|
|
2
|
Dibenzo[a.h]anthracen
|
53-70-3
|
POS
|
Ingen IC50
|
POS
|
Ingen IC50
|
POS
|
PP
|
Polycyklisk forbindelse
|
Laboratoriekemikalie, naturprodukt
|
|
3
|
Mifepriston
|
84371-65-3
|
POS
|
5,61 × 10-6
|
NEG
|
—
|
let POS
|
NEG
|
Steroid
|
Farmaceutisk produkt
|
|
4
|
Raloxifen HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
POS
|
1,19 × 10-9
|
moderat POS
|
POS
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt
|
|
5
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
POS
|
8,17 × 10-7
|
POS
|
POS
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt
|
|
6
|
17β-østradiol
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
7
|
Apigenin
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Heterocyklisk forbindelse
|
Farvestof, naturprodukt, farmaceutisk mellemprodukt
|
|
8
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Heterocyklisk forbindelse
|
Herbicid
|
|
9
|
Di-n-butylphthalat
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Ester, phthalsyre
|
Kosmetisk ingrediens, industrikemikalie, blødgøringsmiddel
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
ikke testet
|
PN
|
Heterocyklisk forbindelse, pyrimidin
|
Fungicid
|
|
11
|
Flavon
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Flavonoid, heterocyklisk forbindelse
|
Naturprodukt, farmaceutisk produkt
|
|
12
|
Flutamid
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Amid
|
Farmaceutisk produkt, veterinær
|
|
13
|
Genistein
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Flavonoid, heterocyklisk forbindelse
|
Naturprodukt, farmaceutisk produkt
|
|
14
|
p-n-nonylphenol
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
ikke testet
|
NEG
|
Phenol
|
Kemisk mellemprodukt
|
|
15
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Kulbrinte (cyklisk)
|
Naturprodukt
|
|
Forkortelser: CAS RN = Registernummer i Chemical Abstracts Service; M = molær; IC50 = halvdelen af den maksimale inhiberende koncentration af et testkemikalie; NEG = negativ; PN = formodet negativ; POS = positiv; PP = formodet positiv.
|
ER TA-ASSAYETS KOMPONENTER
De centrale dele af assayets komponenter
|
11.
|
Denne forsøgsmetode gælder for assays, hvor der anvendes en stabilt transfekteret eller endogen ERα-receptor og en stabilt transfekteret reportergenkonstruktion ved kontrol af et eller flere østrogenresponselementer. Andre receptorer såsom ERβ kan dog være til stede. Dette er de centrale dele af assayets komponenter.
|
Kontroller
|
12.
|
Grundlaget for de foreslåede sideløbende referencestandarder for de forskellige agonist- og antagonistassays beskrives. Eventuelle sideløbende kontroller (negative, opløsningsmiddel og positive) er et tegn på, at assayet virker under testbetingelserne og danner grundlag for sammenligninger mellem forsøgene. De indgår normalt i acceptkriterierne for et givet forsøg (1).
|
Standardprocedurer for kvalitetskontrol
|
13.
|
Standardprocedurer for kvalitetskontrol udføres som beskrevet for hvert assay for at sikre, at cellelinjen forbliver stabil gennem mange passeringer, forbliver mykoplasmafri (dvs. fri for kontaminering med bakterier) og bevarer evnen til at give de forventede ER-medierede responser over tid. Cellelinjerne kontrolleres desuden for korrekt identitet samt for andre kontaminanter (f.eks. svamp, gær og vira).
|
Påvisning af laboratoriets kompetence
|
14.
|
Inden laboratorierne tester ukendte kemikalier med et assay under denne forsøgsmetode, skal de påvise deres kompetence med anvendelsen af det pågældende assay. Til påvisning af denne kompetence skal laboratorierne teste de 14 kompetencestoffer, der er anført i tabel 3 (agonistassayet), og de 10 kompetencestoffer, der er anført i tabel 4 (antagonistassayet). Denne kompetencetest vil endvidere bekræfte testsystemets responsivitet. Listen over kompetencestoffer indeholder en del af de referencestoffer, der er anført i ydeevnestandarderne for ER TA-assays (6). Disse stoffer findes i handelen, repræsenterer kemiske grupper, der sædvanligvis er forbundet med ER-agonist- eller -antagonistaktivitet, udviser et passende styrkeinterval, som forventes for ER-agonister/-antagonister (dvs. stærk til svag) og omfatter negative. Testen af kompetencestofferne bør gentages mindst to gange på forskellige dage. Kompetence påvises ved korrekt klassificering (positiv/negativ) af hvert kompetencestof. Kompetencetesten gentages af de enkelte teknikere, når de lærer et nyt assay. Alt efter celletype kan nogle af disse kompetencestoffer opføre sig som SERM’er og udvise aktivitet som både agonister og antagonister. Kompetencestofferne klassificeres imidlertid i tabel 3 og 4 efter deres kendte primære aktivitet, som skal anvendes til kompetencevurdering.
|
|
15.
|
Til påvisning af laboratoriernes ydeevne og med henblik på kvalitetskontrol udarbejder de historiske databaser over agonister og antagonister med oplysninger om referencestandarder (f.eks. 17β-østradiol og tamoxifen), positive og negative kontrolkemikalier og kontrol med opløsningsmiddel (f.eks. DMSO). Indledningsvis genereres databasen ud fra mindst 10 uafhængige agonistanalyseserier (f.eks. 17β-østradiol) og 10 uafhængige antagonistanalyseserier (f.eks. tamoxifen). Laboratoriet tilføjer resultaterne fra fremtidige analyser af disse referencestandarder og kontroller med opløsningsmiddel for at øge databasen og sikre bioassayets konsekvens og ydeevne over tid.
|
Tabel 3
Liste over (14) kompetencestoffer til agonistassay
(54)
|
Nr. (53)
|
Stof
|
CAS RN
|
Forventet respons (47)
|
STTA-assay
|
VM7Luc ER TA-assay
|
MeSH-kemikaliegruppe (51)
|
Produktgruppe (52)
|
|
PC10-værdi (M) (48)
|
PC50-værdi (M) (48)
|
Testkoncentrationsinterval (M)
|
VM7Luc EC50-værdi (M) (49)
|
Højeste konc. ved bestemmelse af dosisinterval (M) (50)
|
|
14
|
Diethylstilbestrol
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt, veterinær
|
|
12
|
17α-østradiol
|
57-91-0
|
POS
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
15
|
meso-hexestrol
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Kulbrinte (cyklisk), phenol
|
Farmaceutisk produkt, veterinær
|
|
11
|
4-tert-octylphenol
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Phenol
|
Kemisk mellemprodukt
|
|
9
|
Genistein
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoid, heterocyklisk forbindelse
|
Naturprodukt, farmaceutisk produkt
|
|
6
|
Bisphenol A
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Phenol
|
Kemisk mellemprodukt
|
|
2
|
Kaempferol
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoid, heterocyklisk forbindelse
|
Naturprodukt
|
|
3
|
Butylbenzylphthalat
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Carboxylsyre, ester, phthalsyre
|
Blødgøringsmiddel, industrikemikalie
|
|
4
|
p,p’-methoxychlor
|
72-43-5
|
POS
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Kulbrinte (halogeneret)
|
Pesticid, veterinær
|
|
1
|
Ethylparaben
|
120-47-8
|
POS
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Carboxylsyre, phenol
|
Farmaceutisk produkt, konserveringsmiddel
|
|
17
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Heterocyklisk forbindelse
|
Herbicid
|
|
20
|
Spironolacton
|
52-01-7
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Lacton, steroid
|
Farmaceutisk produkt
|
|
21
|
Ketoconazol
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Heterocyklisk forbindelse
|
Farmaceutisk produkt
|
|
22
|
Reserpin
|
50-55-5
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Heterocyklisk forbindelse, indol
|
Farmaceutisk produkt, veterinær
|
|
Forkortelser: CAS RN = Registernummer i Chemical Abstracts Service; EC50 = halvdelen af den maksimale effektive koncentration af et testkemikalie; NEG = negativ; POS = positiv; PC10 (og PC50) = koncentrationen af et teststof, ved hvilken responsen er 10 % (eller 50 % for PC50) af den respons, der fremkaldes af den positive kontrol (E2, 1nM), på hver plade.
|
Tabel 4
Liste over (10) kompetencestoffer til antagonistassay
|
|
Stof (4)
|
CAS RN
|
ER STTA-assay (55)
|
VM7Luc ER TA-assay (56)
|
ER STTA (55) -kandidatvirkninger
|
ICCVAM (59) konsensusklassifikation
|
MeSH (60) kemisk gruppe
|
Produktgruppe (61)
|
|
ER TA-aktivitet
|
IC50 (M)
|
Testkoncentrationsinterval (M)
|
ER TA-aktivitet
|
IC50
(57) (M)
|
Højeste konc. ved bestemmelse af dosisinterval (M) (58)
|
|
1
|
4-hydroxytamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
10-12 – 10-7
|
POS
|
2,08 × 10-7
|
2,58 × 10-4
|
moderat POS
|
POS
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt
|
|
2
|
Raloxifen HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
10-12 – 10-7
|
POS
|
1,19 × 10-9
|
1,96 × 10-4
|
moderat POS
|
POS
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt
|
|
3
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
10-10 – 10-5
|
POS
|
8,17 × 10-7
|
2,69 × 10-4
|
POS
|
POS
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt
|
|
4
|
17β-østradiol
|
50-28-2
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,67 × 10-3
|
skal være negativ (*4)
|
PN
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
5
|
Apigenin
|
520-36-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Heterocyklisk forbindelse
|
Farvestof, naturprodukt, farmaceutisk mellemprodukt
|
|
6
|
Di-n-butylphthalat
|
84-74-2
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Ester, phthalsyre
|
Kosmetisk ingrediens, industrikemikalie, blødgøringsmiddel
|
|
7
|
Flavon
|
525-82-6
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,50 × 10-4
|
skal være negativ (*4)
|
PN
|
Flavonoid, heterocyklisk forbindelse
|
Naturprodukt, farmaceutisk produkt
|
|
8
|
Genistein
|
446-72-0
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
skal være negativ (*4)
|
NEG
|
Flavonoid, heterocyklisk forbindelse
|
Naturprodukt, farmaceutisk produkt
|
|
9
|
p-n-nonylphenol
|
104-40-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
4,54 × 10-4
|
ikke testet
|
NEG
|
Phenol
|
Kemisk mellemprodukt
|
|
10
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,38 × 10-4
|
skal være negativ (*4)
|
NEG
|
Kulbrinte (cyklisk)
|
Naturprodukt
|
|
Forkortelser: CAS RN = Registernummer i Chemical Abstracts Service; M = molær; IC50 = halvdelen af den maksimale inhiberende koncentration af et testkemikalie; NEG = negativ; PN = formodet negativ; POS = positiv.
|
Acceptkriterier for analyseserien
|
16.
|
Accept eller forkastelse af en analyseserie er baseret på vurdering af de resultater, som er opnået for de referencestandarder og kontroller, der er anvendt til hvert enkelt forsøg. Referencestandardernes værdier for PC50 (EC50) eller IC50 skal opfylde acceptkriterierne som fastsat i det valgte assay (jf. tillæg 2 for STTA, jf. tillæg 3 for VM7Luc ER TA), og alle positive/negative kontroller skal klassificeres korrekt for hvert accepteret forsøg. Evnen til at gennemføre assayet konsekvent skal påvises ved udvikling og vedligeholdelse af en historisk database for referencestandarderne og kontrollerne (jf. punkt 15). Standardafvigelser (SD) eller variationskoefficienter (CV) for gennemsnit af referencestandardernes kurvefittingsparametre fra flere forsøg kan anvendes som et mål for reproducerbarhed inden for laboratoriet. Endvidere skal følgende principper for acceptkriterier overholdes:
|
—
|
Dataene skal være tilstrækkelige til kvantitativ vurdering af ER-aktivering (ved agonistassay) eller -undertrykkelse (ved antagonistassay) (dvs. effektivitet og styrke).
|
|
—
|
Den gennemsnitlige reporteraktivitet for referencekoncentrationen af referenceøstrogen skal mindst være det minimum, der er fastsat i assayet, i forhold til aktiviteten i kontrollen med bærestof (opløsningsmiddel) for at sikre en passende følsomhed. For STTA- og VM7Luc ER TA-assays er dette fire gange aktiviteten i den gennemsnitlige kontrol med bærestof på hver plade.
|
|
—
|
De testede koncentrationer skal forblive inden for testkemikaliernes opløselighedsinterval og må ikke påvise cytotoksisk effekt.
|
|
Analyse af data
|
17.
|
Den fastlagte procedure for datafortolkning for hvert assay skal anvendes til klassificering af en positiv og negativ respons.
|
|
18.
|
Hvis acceptkriterierne opfyldes (punkt 16), er det tegn på, at assayet fungerer korrekt, men det er ingen garanti for, at alle analyseserier vil producere nøjagtige data. Kan resultaterne af den første analyseserie gentages, er det den bedste garanti for, at der blev produceret nøjagtige data. Hvis to analyseserier giver reproducerbare resultater (hvis f.eks. resultaterne af begge analyseserier viser, at et testkemikalie er positivt), er det ikke nødvendigt at gennemføre en tredje analyseserie.
|
|
19.
|
Hvis to analyseserier ikke giver reproducerbare resultater (hvis f.eks. et testkemikalie er positivt i én analyseserie og negativt i den anden analyseserie), eller hvis der stilles krav om større sikkerhed omkring resultatet af assayet, bør der gennemføres mindst tre uafhængige analyseserier. I dette tilfælde baseres klassificeringen på de to overensstemmende resultater af de tre.
|
Generelle kriterier for datafortolkning
|
20.
|
Der findes for øjeblikket ikke nogen almindeligt anerkendt metode til fortolkning af ER TA-data. Både kvalitative (f.eks. positiv/negativ) og/eller kvantitative (f.eks. EC50, PC50, IC50) vurderinger af ER-medieret aktivitet bør være baseret på empiriske data og solid videnskabelig vurdering. Når det er muligt, skal de positive resultater karakteriseres ved både virkningens omfang sammenlignet med kontrollen med bærestof (opløsningsmiddel) eller referenceøstrogenet og den koncentration, hvorved virkningen opstår (f.eks. EC50, PC50, RPCMax, IC50 osv.).
|
Testrapport
|
21.
|
Testrapporten bør indeholde følgende oplysninger:
|
|
Assay:
|
—
|
kontrol/referencestandard/testkemikalie
|
|
—
|
oprindelse, batchnummer, frist for anvendelse, hvis dette foreligger
|
|
—
|
testkemikaliets stabilitet, hvis denne er kendt
|
|
—
|
testkemikaliets opløselighed og stabilitet i opløsningsmidlet, hvis dette er kendt
|
|
—
|
måling af pH-værdi, osmolalitet og bundfald i det dyrkningsmedium, som testkemikaliet blev tilsat, hvor det er relevant.
|
|
|
|
Stof med kun én bestanddel:
|
—
|
fysisk udseende, vandopløselighed og yderligere relevante fysisk-kemiske egenskaber
|
|
—
|
kemisk identifikation, f.eks. IUPAC-navn eller CAS-navn, CAS-nummer, SMILES- eller InChI-kode, strukturformel, renhed, kemisk identitet af urenheder, i det omfang det er relevant og praktisk muligt, osv.
|
|
|
|
Stof med flere bestanddele, UVCB-stoffer og blandinger:
|
—
|
kendetegnet så vidt muligt ved kemisk identitet (jf. ovenfor), kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber.
|
|
|
|
Opløsningsmiddel/bærestof:
|
—
|
karakterisering (art, leverandør og batch)
|
|
—
|
Begrundelse for valg af opløsningsmiddel/bærestof
|
|
—
|
testkemikaliets opløselighed og stabilitet i opløsningsmidlet/bærestoffet, hvis dette er kendt
|
|
|
|
Celler:
|
—
|
celletype og -oprindelse:
|
—
|
udtrykkes ER endogent? Hvis nej, hvilke(n) receptor(er) blev transfekteret?
|
|
—
|
anvendt(e) reporterkonstruktion(er) (herunder oprindelsesarter)
|
|
—
|
eventuel udvælgelsesmetode til vedligeholdelse af stabil transfektion
|
|
—
|
er transfektionsmetoden relevant for stabile linjer?
|
|
|
—
|
antal cellegenerationer (fra optøning)
|
|
—
|
cellernes generationsantal ved optøning
|
|
—
|
metoder til opretholdelse af cellekulturer.
|
|
|
|
Testbetingelser:
|
—
|
opløselighedsbegrænsninger
|
|
—
|
Beskrivelse af de anvendte metoder til vurdering af levedygtighed
|
|
—
|
mediesammensætning og CO2-koncentration
|
|
—
|
testkemikaliekoncentrationer
|
|
—
|
mængde tilsat bærestof og testkemikalie
|
|
—
|
inkubationstemperatur og -fugtighed
|
|
—
|
celletæthed ved behandlingsstart og undervejs i behandlingen
|
|
—
|
positive og negative referencestandarder
|
|
—
|
reporterreagenser (produktnavn, leverandør og batch)
|
|
—
|
kriterier for bedømmelse af analyseserier som positive, negative eller usikre.
|
|
|
|
Testgodkendelse:
|
—
|
foldinduktioner for hver assayplade og angivelse af, hvorvidt de opfylder assayets minimumskrav baseret på historiske kontroller
|
|
—
|
faktiske værdier for acceptkriterier, f.eks. log10EC50, log10PC50, logIC50 og Hills kurve-værdier, for sideløbende positive kontroller/referencestandarder.
|
|
|
|
Resultater:
|
—
|
rådata og normaliserede data
|
|
—
|
maksimalt foldinduktionsniveau
|
|
—
|
den laveste effektive koncentration (LEC), hvis den foreligger
|
|
—
|
RPCMax-, PCMax-, PC50-, IC50- og/eller EC50-værdier, hvis disse er relevante
|
|
—
|
koncentration/respons-forhold, når det er muligt
|
|
—
|
eventuelle statistiske analyser sammen med en måling af fejl og konfidens (f.eks. middelfejlen på middeltallet (SEM), standardafvigelse, variationskoefficient eller 95 % CI) og en beskrivelse af, hvordan disse værdier er opnået.
|
|
|
|
Diskussion af resultaterne
|
|
LITTERATUR
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34.), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): s. 5367-73.
|
|
(5)
|
Rogers J.M. og Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): s. 67-82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173.), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174.), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150.), Organisation for Economic Cooperation and Development, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: s. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): s. 1073-89.
|
|
(11)
|
Younes M. og Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): s. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): s. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): s. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti-Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): s. 91-98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals, 28(1): s. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol, 71(10): s. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): s. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERαIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): s. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism, 82(12): s. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): s. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): s. 105-12.
|
|
(25)
|
Deroo B.J. og Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): s. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): s. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. og Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor-Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): s. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): s. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: s. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3): s. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose-Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. og Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Tillæg 1
DEFINITIONER OG FORKORTELSER
Acceptkriterier
: minimumsstandarder for udførelse af forsøgskontroller og referencestandarder. Alle acceptkriterier skal være opfyldt, for at et forsøg kan betragtes som gyldigt.
Nøjagtighed (konkordans)
: overensstemmelsen mellem forsøgsmetodens resultater og accepterede referenceværdier. Den er et mål for forsøgsmetodens ydeevne og et af aspekterne af relevans. Udtrykket benyttes ofte som synonym for "konkordans" forstået som den andel af korrekte udfald, forsøgsmetoden fører til (1).
Agonist
: et stof, der giver en respons, f.eks. transkription, når det binder til en bestemt receptor.
Antagonist
: en type receptorligand eller kemikalie, som ikke selv fremkalder en biologisk respons ved binding til en receptor, men blokerer eller dæmper agonist-medierede responser.
Antiøstrogenaktivitet
: et kemikalies evne til at undertrykke virkningen af 17β-østradiol medieret gennem østrogenreceptorer.
Cellemorfologi
: form og udseende af celler, der dyrkes i encellede lag på en vævsdyrkningsplade med én brønd. Døende celler udviser ofte en abnorm cellemorfologi.
CF
: OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupters (OECD's konceptuelle ramme for test og vurdering af hormonforstyrrende kemikalier).
Trækul-/dextranbehandling
: behandling af serum, der benyttes i cellekultur. Behandling med trækul/dextran (ofte omtalt som "stripping") fjerner endogene hormoner og hormonbindende proteiner.
Kemikalie
: et stof eller en blanding
Cytotoksisk effekt
: skadelige virkninger i cellestruktur eller -funktion, som i sidste ende forårsager celledød og kan afspejles i en reduktion i antallet af celler i brønden ved afslutningen af eksponeringsperioden eller en reduktion i kapaciteten til måling af cellefunktionen ved sammenligning med den sideløbende bærestofkontrol.
CV
: Coefficient of variation (variationskoefficient)
DCC-FBS
: Dextran-coated charcoal treated fetal bovine serum (føtalt kalveserum behandlet med dextranovertrukket trækul).
DMEM
: Dulbecco's Modification of Eagle's Medium
DMSO
: Dimethylsulfoxid
E2
: 17β-østradiol
EC50
: halvdelen af den maksimale effektive koncentration af et testkemikalie
ED
: hormonforstyrrende virkning
hERα
: human østrogenreceptor alfa
hERß
: human østrogenreceptor beta
EFM
: østrogenfrit medium. Dulbecco's Modification of Eagle's Medium (DMEM) suppleret med 4,5 % aktivt kul-/dextranbehandlet FBS, 1.9 % L-glutamin og 0,9 % pen-Strep.
ER
: østrogenreceptor
ERE
: østrogenresponselement
Østrogenaktivitet
: et kemikalies evne til at efterligne 17β-østradiol i dets evne til at binde sig til og aktivere østrogenreceptorer. hERα-medieret østrogenaktivitet kan spores med denne forsøgsmetode.
ERTA
: transaktivering af østrogenreceptorer
FBS
: føtalt kalveserum
HeLa
: en udødelig human cervikal cellelinje
HeLa9903
: en underklon af en HeLa-celle, hvori hERααog et luciferasereportergen er blevet stabilt transfekteret
IC
50
: halvdelen af den maksimale effektive koncentration af et inhiberende testkemikalie
ICCVAM
: US Interagency Coordinating Committee on the Validation of Alternative Methods.
Reproducerbarhed mellem laboratorier
: mål for, i hvilken grad forskellige kvalificerede laboratorier kan opnå de samme resultater kvalitativt og kvantitativt, når de anvender samme protokol og tester de samme stoffer. Reproducerbarheden mellem laboratorier bestemmes forud for og under valideringsprocessen og angiver, i hvilken grad et assay kan overføres mellem laboratorier med et vellykket resultat. Kaldes også ekstern reproducerbarhed (1).
Reproducerbarhed inden for laboratorier
: bestemmelse af, i hvilken grad kvalificeret personale på det samme laboratorium kan reproducere resultater ved at anvende en specifik protokol på forskellige tidspunkter. Kaldes også "intern reproducerbarhed" (1).
LEC
: den laveste effektive koncentration er testkemikaliets laveste koncentration, som fremkalder en respons (dvs. den laveste testkemikaliekoncentration, ved hvilken foldinduktionen er statistisk forskellig fra den sideløbende bærestofkontrol)
Me-too test
: populært udtryk for et assay, som strukturelt og funktionelt ligner en valideret og anerkendt referenceforsøgsmetode. Benyttes som synonym for tilsvarende forsøgsmetode
MT
: metallothionein
MMTV
: mamatumorvirus hos mus
OHT
: 4-hydroxytamoxifen
PBTG
: ydeevnebaseret forsøgsretningslinje
PC (positiv kontrol)
: et stærkt aktivt stof, fortrinsvis 17ß-østradiol, der er inkluderet i alle test for at bidrage til, at assayet fungerer tilfredsstillende
PC
10
: den koncentration af et testkemikalie, hvor den målte aktivitet i et agonist-assay er 10 % af den maksimale aktivitet, der er fremkaldt af PC (E2 ved 1 nM for STTA-assayet) i hver plade.
PC
50
: den koncentration af et testkemikalie, hvor den målte aktivitet i et agonist-assay er 50 % af den maksimale aktivitet, der er fremkaldt af PC (E2 ved den referencekoncentration, der er specificeret i forsøgsmetoden) i hver plade.
PC
Max
: den koncentration af et testkemikalie, som fremkalder RPCMax
Ydeevnestandarder
: standarder, der med udgangspunkt i et valideret assay danner grundlag for at vurdere, om et foreslået assay, som er mekanistisk og funktionelt tilsvarende, er sammenligneligt. De indeholder (1) de centrale dele af assayet, (2) en minimumsliste over referencekemikalier, som er udvalgt blandt de kemikalier, der anvendes til at påvise, at den validerede forsøgsmetodes ydeevne er acceptabel, og (3) de sammenlignelige niveauer af nøjagtighed og pålidelighed, som er fastsat efter, hvad der er opnået med den validerede forsøgsmetode, og som det skal påvises, at det foreslåede assay kan yde ved vurdering ud fra minimumslisten over referencekemikalier (1).
Kompetencestoffer
: en delmængde af referencestoffer, der indgår i ydeevnestandarderne, og som kan anvendes af laboratorier til at påvise teknisk kompetence med en standardiseret forsøgsmetode. Udvælgelseskriterier for disse stoffer omfatter typisk, at de repræsenterer responsområdet, fås i handelen, og at der foreligger referencedata af høj kvalitet.
Kompetence
: dokumenteret evne til at gennemføre et assay korrekt, før ukendte stoffer testes.
Referenceøstrogen (positiv kontrol, PC)
: 17β-østradiol (E2, CAS 50-28-2).
Referencestandard
: et referencestof, der anvendes til at påvise tilstrækkeligheden af et assay. 17β-østradiol er referencestandarden for STTA og VM7Luc ER TA-assays.
Referenceforsøgsmetoder
: de assays, hvorpå PBTG 455 er baseret.
Relevans
: beskrivelse af sammenhængen mellem et assay og den pågældende virkning og dens meningsfuldhed og brugbarhed til et bestemt formål. Det angives, i hvilket omfang assayet måler eller forudsiger den relevante biologiske virkning korrekt. Assayets nøjagtighed (konkordans) indgår i vurderingen af dets relevans (1).
Pålidelighed
: mål for, i hvilken grad et assay kan reproduceres i og mellem laboratorier over en længere periode, når det udføres efter samme protokol. Det bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier.
RLU
: relative lysenheder
RNA
: ribonukleinsyre
RPC
Max
: maksimalt responsniveau fremkaldt af et testkemikalie, udtrykt som en procentdel af den respons, der fremkaldes af 1 nM E2 på den samme plade
RPMI
: RPMI 1 640-medium suppleret med 0,9 % Pen-Strep og 8,0 % føtalt kalveserum (FBS)
Analyseserie
: et individuelt forsøg, der vurderer den kemiske påvirkning af assayets biologiske udfald. Hver enkelt analyseserie er et fuldstændigt forsøg udført på replikatbrønde med celler udpladet fra en fælles cellepool på samme tid.
Uafhængig analyseserie
: et særskilt uafhængigt forsøg, der vurderer den kemiske påvirkning af assayets biologiske udfald ved hjælp af celler fra en anden pool og frisk fortyndede kemikalier, udført på forskellige dage eller på den samme dag af andet personale.
Standardafvigelse
: standardafvigelse
Følsomhed
: andelen af alle positive/aktive stoffer, der klassificeres korrekt ved assayet. Den er et mål for nøjagtigheden af et assay, der fører til kategoriske resultater, og har stor betydning for vurdering af et assays relevans (1).
Specificitet
: andelen af alle negative/inaktive stoffer, som klassificeres korrekt ved testen. Den er et mål for nøjagtigheden af et assay, der fører til kategoriske resultater, og har stor betydning for vurdering af et assays relevans (1).
Stabil transfektion
: når DNA transfekteres ind i dyrkede celler på en sådan måde, at det integreres stabilt i cellernes genom og resulterer i en stabil ekspression af transfekterede gener. Kloner af stabilt transfekterede celler udvælges af stabile markører (f.eks. modstand mod G418)
STTA-assay
: stabilt transfekteret transaktiveringsassay, ERα-transkriptionsaktiveringsassay, der anvender HeLa 9 903-cellelinje
Undersøgelse
: hele rækken af forsøgsarbejdsopgaver, der udføres for at vurdere et enkelt specifikt stof ved hjælp af et bestemt assay. En undersøgelse omfatter alle skridt, herunder opløsningstest af teststoffet i testmediet, forprøve til bestemmelse af dosisintervaller, alle nødvendige omfattende analyseserier, dataanalyser, kvalitetssikring, vurderinger af den cytotoksiske effekt osv. Gennemførelse af en undersøgelse gør det muligt at klassificere testkemikaliets aktivitet på det toksicitetsmål (dvs. aktiv, inaktiv eller usikker), der vurderes med det anvendte assay, og foretage et skøn over styrke i forhold til det positive referencekemikalie.
Stof
: i henhold til REACH (62) defineres et stof som et grundstof og forbindelser heraf, naturligt eller industrielt fremstillet, indeholdende sådanne tilsætningsstoffer, som er nødvendige til bevarelse af stoffets stabilitet, og sådanne urenheder, som følger af fremstillingsprocessen, bortset fra opløsningsmidler, der kan udskilles, uden at det påvirker stoffets stabilitet eller ændrer dets sammensætning. Der benyttes en lignende definition i forbindelse med UN GHS (1).
TA (Transaktivering): igangsættelse af mRNA-syntese som reaktion på et bestemt kemisk signal såsom binding af et østrogen til østrogenreceptoren
Assay
: i forbindelse med denne forsøgsmetode er et assay en af de metoder, der accepteres som gyldig med hensyn til opfyldelse af de skitserede ydeevnekriterier. Assayets komponenter omfatter f.eks. den specifikke cellelinje med tilhørende vækstbetingelser, særlige medier, hvori testen udføres, pladeopstillingsbetingelser, opstilling og opløsninger af testkemikalier sammen med alle andre nødvendige kvalitetskontrolforanstaltninger og tilhørende dataevalueringsskridt.
Testkemikalie
: alle stoffer eller blandinger, der testes ved hjælp af denne forsøgsmetode
Transskription
: mRNA-syntese
UVCB
: kemiske stoffer med ukendt eller variabel sammensætning, komplekse reaktionsprodukter eller biologiske materialer
Valideret forsøgsmetode
: et assay, for hvilket der er gennemført valideringsundersøgelser for at fastslå relevansen (herunder nøjagtigheden) og pålideligheden til et bestemt formål. Det er vigtigt at være opmærksom på, at en valideret forsøgsmetode måske ikke har en tilstrækkelig ydeevne med hensyn til nøjagtighed og pålidelighed til at være acceptabel til det planlagte formål (1).
Validering
: den proces, hvorved pålideligheden og relevansen af en bestemt tilgang, metode, proces eller vurdering eller et bestemt assay fastslås til et defineret formål (1).
VC (bærestofkontrol)
: Det opløsningsmiddel, der anvendes til at opløse test- og kontrolkemikalier, testes alene som bærestof uden opløst kemikalie.
VM7
: en udødeliggjort adenocarcinomcelle, som endogent udtrykker østrogenreceptoren
VM7Luc4E2
: VM7Luc4E2-cellelinjen blev afledt af VM7 udødeliggjorte humant afledte adenocarcinomceller, som endogent udtrykker begge former for østrogenreceptorer (ERα og ERβ) og er blevet stabilt transfekteret med plasmidet pGudLuc7.ERE. Dette plasmid indeholder fire kopier af et syntetisk oligonukleotid, som indeholder østrogenresponselementet umiddelbart før promotoren for brysttumorvirus hos mus (MMTV) og luciferasegenet fra ildfluer.
Svagt positiv kontrol
: et svagt aktivt stof, der vælges fra listen over referencekemikalier, og som indgår i alle forsøg for at bidrage til, at assayet fungerer tilfredsstillende.
Tillæg 2
TRANSAKTIVERINGSASSAY MED STABILT TRANSFEKTERET HUMAN ØSTROGENRECEPTOR-α TIL PÅVISNING AF ØSTROGENAGONIST- OG ØSTROGENANTAGONISTAKTIVITET I KEMIKALIER, DER ANVENDER HERα-HELA-9903-CELLELINJEN
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER (JF. OGSÅ GENEREL INDLEDNING)
|
1.
|
Dette transaktiveringsassay (TA) anvender hERα-HeLa-9903-cellelinjen til at påvise østrogenagonistaktivitet medieret gennem human østrogenreceptor alfa (hERα). Den valideringsundersøgelse af det stabilt transfekterede transaktiveringsassay (STTA), som blev foretaget af det japanske Chemicals Evaluation and Research Institute (CERI), der anvender hERα-HeLa-9903-cellelinjen til at påvise østrogenagonist- og østrogenantagonistaktivitet medieret gennem human østrogenreceptor alfa (hERα), viste assayets relevans og pålidelighed i overensstemmelse med dets formål (1).
|
|
2.
|
Dette assay er specifikt udformet til at påvise hERα-medieret TA ved at måle kemiluminescens som effektparameter. Der findes imidlertid rapporter om ikke-receptormedierede luminescenssignaler ved højere koncentrationer af fytoøstrogen end 1 μM på grund af overaktivering af luciferasereportergenet (2) (3). Mens dosis/respons-kurven tyder på, at der sker en egentlig aktivering af ER-systemet ved lavere koncentrationer, skal den luciferaseekspression, som opnås ved høje koncentrationer af fytoøstrogener eller lignende kemiske stoffer, der mistænkes for at forårsage fytoøstrogenlignende overaktivering af luciferasereportergenet, undersøges nøje i stabilt transfekterede ER TA-assaysystemer (tillæg 1).
|
|
3.
|
Afsnittene "GENEREL INDLEDNING" og "ER TA-ASSAYETS KOMPONENTER" bør læses, før dette assay benyttes til lovmæssige formål. Definitioner og forkortelser, der benyttes i denne testvejledning, er beskrevet i tillæg 2.1.
|
PRINCIP FOR ASSAYET (SE OGSÅ GENEREL INDLEDNING)
|
4.
|
Assayet bruges til at signalere binding af østrogenreceptoren til en ligand. Efter ligandbindingen forflytter receptorligandkomplekset sig til kernen, hvor det binder specifikke DNA-responselementer og transaktiverer et ildflue-luciferasereportergen, hvilket resulterer i øget cellulær ekspression af luciferaseenzym. Luciferin er et medium, der omdannes af luciferaseenzymet til et bioluminescensprodukt, som måles kvantitativt med et luminometer. Luciferaseaktivitet kan vurderes hurtigt og billigt med forskellige typer testkit, der kan fås i handelen.
|
|
5.
|
Testsystemet anvender hERα-HeLa-9903-cellelinjen, der er afledt af en human cervikal tumor med to stabilt indsatte konstruktioner: (i) hERα-ekspressionskonstruktion (med indkodning af den humane receptor i fuld længde) og (ii) en ildflueluciferasereporter-konstruktion med fem tandem-gentagelser af et vitellogenint østrogenresponselement (ERE) drevet af et metallothioneinpromotor (MT) TATA-element fra mus. MT TATA-genkonstruktionen i mus har vist sig at have den bedste ydeevne og anvendes derfor normalt. Denne hERα-HeLa-9903-cellelinje kan derfor måle et testkemikalies evne til at fremkalde hERα-medieret transaktivering af luciferasegenekspression.
|
|
6.
|
Ved anvendelse af ER-agonistassays er datafortolkningen baseret på, om det maksimale responsniveau fremkaldt af et testkemikalie er lig med eller overstiger en agonistrespons, der svarer til 10 % af det, der fremkaldes af en maksimalt inducerende (1 nM) koncentration af den positive kontrol (PC) 17β-østradiol (E2) (dvs. PC10). Ved anvendelse af et ER-antagonistassay er datafortolkningen baseret på, om responsen viser en mindst 30 % aktivitetsreduktion fra den respons, der induceres af spike-in-kontrollen (25 pM af E2) uden cytotoksisk effekt. Dataanalyse og -fortolkning diskuteres nærmere i punkt 34-48.
|
FREMGANGSMÅDE
Cellelinjer
|
7.
|
Den stabilt transfekterede hERα-HeLa-9903-cellelinje bør anvendes til assayet. Cellelinjen kan fås fra den japanske Collection of Research Bioresources (JCRB) Cell Bank (63) ved underskrivelse af en materialeoverførselsaftale (MTA).
|
|
8.
|
Der bør kun anvendes mykoplasmafrie celler i test. RT-PCR (realtidspolymerasekædereaktion) er den foretrukne metode til en sensitiv påvisning af mykoplasmainfektion (4) (5) (6).
|
Cellelinjens stabilitet
|
9.
|
For at overvåge cellelinjens stabilitet bør E2, 17α-østradiol, 17α-methyltestosteron og corticosteron anvendes som referencestandarder for agonistassayet, og der bør mindst én gang, hver gang assayet udføres, måles en fuldstændig koncentration/respons-kurve i testkoncentrationsområdet, som er angivet i tabel 1, og resultaterne bør være i overensstemmelse med resultaterne i tabel 1.
|
|
10.
|
Ved anvendelse af et antagonistassay bør der samtidig med hver analyseserie måles fuldstændige koncentrationskurver for to referencestandarder, tamoxifen og flutamid. En korrekt kvalitativ klassificering som positiv eller negativ for de to kemikalier bør overvåges.
|
Celledyrknings- og udpladningsbetingelser
|
11.
|
Celler bør opbevares i Eagle’s Minimum Essential Medium (EMEM) uden fenolrød, suppleret med 60 mg/l antibiotisk kanamycin og 10 % dextranovertrukket trækulsbehandlet føtalt kalveserum (DCC-FBS), i en CO2-inkubator (5 % CO2) ved 37 ± 1oC. Ved en konfluens på 75-90 % kan celler dyrkes i subkultur ved 10 ml 0,4 × 105 – 1 × 105 celler/ml pr. 100 mm celledyrkningsskål. Celler bør suspenderes med 10 % FBS-EMEM (der er det samme som EMEM med DCC-FBS) og dernæst udplades i brønde på en mikroplade med en tæthed på 1 × 104 celler/(100 μl pr. brønd). Herefter bør cellerne præinkuberes i en 5 % CO2-inkubator ved 37 ± 1oC i 3 timer før den kemiske eksponering. Plastudstyr bør være fri for østrogenaktivitet.
|
|
12.
|
For at opretholde responsens integritet bør cellerne dyrkes til mere end én generation fra den nedfrosne beholdning i det konditionerede dyrkningsmedium og bør ikke dyrkes til mere end 40 generationer. For hERα-HeLa-9903-cellelinjen vil dette være under tre måneder. Cellernes ydeevne kan dog reduceres, hvis de dyrkes under uhensigtsmæssige dyrkningsbetingelser.
|
|
13.
|
DCC-FBS kan fremstilles som beskrevet i tillæg 2.2 eller fås i handelen.
|
Acceptkriterier
Positive og negative referencestandarder for ER-agonistassay
|
14.
|
Før og i løbet af undersøgelsen kontrolleres testsystemets responsivitet ved hjælp af passende koncentrationer af et stærkt østrogen: E2, et svagt østrogen (17α-østradiol), en meget svag agonist (17α-methyltestosteron) og et negativt stof (corticosteron). Acceptable områdeværdier fra valideringsundersøgelsen (1) er anført i tabel 1. Disse fire sammenfaldende referencestandarder bør inkluderes i hvert forsøg, og resultaterne bør falde inden for de angivne acceptable grænser. Hvis dette ikke er tilfældet, bør årsagen til, at acceptkriterierne ikke er opfyldt, bestemmes (f.eks. cellehåndtering samt kvalitet og koncentration af serum og antibiotika), og assayet gentages. Når acceptkriterierne eropfyldt, er det væsentligt at sikre en minimumsvariation mellem EC50-, PC50- og PC10-værdier og en konsekvent anvendelse af materialer til celledyrkning. De fire sideløbende referencestandarder, som bør inkluderes i hvert forsøg (der udføres under samme betingelser, herunder materialer, cellegenerationsniveau og teknikere), kan sikre assayets følsomhed, fordi PC10-værdier af de tre positive referencestandarder bør ligge inden for det acceptable område, og det samme bør PC50- og EC50-værdier, hvor de kan beregnes (se tabel 1).
|
Tabel 1
Acceptable områdeværdier for de fire referencestandarder for ER-agonistassays
|
Navn
|
logPC50
|
logPC10
|
logEC50
|
Hills kurve
|
Måleområde
|
|
17β-østradiol (E2) CAS-nr.: 50-28-2
|
-11,4~-10,1
|
< -11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-østradiol CAS-nr.: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Corticosteron CAS-nr.: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-methyltestosteron CAS-nr.: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5M
|
Positive og negative referencestandarder for ER-antagonistassay
|
15.
|
Før og i løbet af undersøgelsen bør testsystemets responsivitet kontrolleres ved hjælp af passende koncentrationer af et positivt stof (tamoxifen) og et negativt stof (flutamid). Acceptable områdeværdier fra valideringsundersøgelsen (1) er anført i tabel 2. Disse to sideløbende referencestandarder inkluderes i hvert forsøg, og resultaterne bedømmes korrekt som vist i kriterierne. Hvis dette ikke er tilfældet, bestemmes årsagerne til, at kriterierne ikke er opfyldt (f.eks. cellehåndtering samt kvalitet og koncentration af serum og antibiotika), og assayet gentages. Endvidere bør IC50-værdier for et positivt stof (tamoxifen) beregnes, og resultaterne bør falde inden for de givne acceptable grænser. Når acceptkriterierne er opfyldt, er det væsentligt at sikre minimumsvariation mellem IC50-værdier og konsekvent anvendelse af materialer til celledyrkning. De to sammenfaldende referencestandarder, som bør inkluderes i hvert forsøg (der udføres under samme betingelser, herunder materialer, cellegenerationsniveau og teknikere), kan sikre assayets følsomhed (se tabel 2).
|
Tabel 2
Kriterier og acceptable områdeværdier for de to referencestandarder for ER-antagonistassay
|
Navn
|
Kriterier
|
LogIC50
|
Måleområde
|
|
Tamoxifen CAS-nr.: 10540-29-1
|
Positiv: IC50 bør beregnes
|
-5,942~-7,596
|
10-10~10-5M
|
|
Flutamid CAS-nr.: 13311-84-7
|
Negativ: IC30 bør ikke beregnes
|
—
|
10-10~10-5M
|
Positiv kontrol og bærestofkontrol
|
16.
|
Den positive kontrol (PC) for ER-agonistassay (1 nM af E2) og for ER-antagonistassay (10 μM TAM) bør testes mindst in triplo på hver plade. Det bærestof, som anvendes til at opløse et testkemikalie, bør testes som en bærestofkontrol (VC) mindst in triplo på hver plade. Hvis der ved den positive kontrol (PC) anvendes et andet bærestof end testkemikaliet, bør der ud over denne bærestofkontrol (VC) testes en anden VC mindst in triplo på den samme plade med PC.
|
Kvalitetskriterier for ER-agonistassay
|
17.
|
Den gennemsnitlige luciferaseaktivitet for den positive kontrol (1 nM E2) bør være mindst 4 fold den gennemsnitlige VC på hver plade. Dette kriterium fastlægges på grundlag af pålideligheden af værdierne for effektparametrene fra valideringsundersøgelsen (historisk mellem 4 og 30 fold).
|
|
18.
|
Med hensyn til kvalitetskontrollen af assayet bør foldinduktionen svarende til PC10-værdien af den sideløbende PC (1 nM E2) være større end 1+2 standardafvigelser af foldinduktionsværdien (=1) af den sideløbende VC. Med henblik på prioritering kan PC10-værdien være nyttig til at forenkle den nødvendige dataanalyse sammenlignet med en statistisk analyse. Selv om en statistisk analyse giver oplysninger om signifikans, er en sådan analyse ikke en kvantitativ parameter med hensyn til koncentrationsbaseret potentiale og er således mindre nyttig til prioriteringsformål.
|
Kvalitetskriterier for ER-antagonistassay
|
19.
|
Den gennemsnitlige luciferaseaktivitet af spike-in-kontrollen (25 nM E2) bør være mindst 4 fold den gennemsnitlige VC på hver plade. Dette kriterium fastlægges på grundlag af pålideligheden af værdierne for effektparametrene fra valideringsundersøgelsen.
|
|
20.
|
Med hensyn til kvalitetskontrollen af assayet bør den relative transkriptionsaktivering (RTA) af 1 nM E2 være større end 100 %, RTA af 1 μM 4-hydroxytamoxifen (OHT) bør ligge under 40,6 %, og RTA af 100 μM digitonin (Dig) bør være under 0 %.
Påvisning af laboratoriets kompetence (jf. punkt 14 og tabel 3 og 4 i "ER TA-ASSAYETS KOMPONENTER" under denne forsøgsmetode).
|
Bærestof
|
21.
|
Dimethylsulfoxid (DMSO) eller et passende opløsningsmiddel med samme koncentration, som anvendes til de forskellige positive og negative kontroller og testkemikalier, bør anvendes som den sideløbende VC. Testkemikalier bør opløses i et opløsningsmiddel, som opløser det pågældende testkemikalie og kan blandes med cellemediet. Vand, ethanol (95-100 % renhed) og DMSO er egnede bærestoffer. Hvis der anvendes DMSO, bør niveauet ikke overstige 0,1 % (v/v). For ethvert bærestof bør det påvises, at den maksimalt anvendte mængde ikke er cytotoksisk og ikke indvirker på assayets ydeevne.
|
Præparering af testkemikalier
|
22.
|
Generelt bør testkemikalier opløses i DMSO eller et andet egnet opløsningsmiddel og fortyndes serielt med det samme opløsningsmiddel i et fælles forhold på 1:10 for at fremstille opløsninger til fortynding med mediet.
|
Opløselighed og cytotoksisk effekt: Overvejelser vedrørende bestemmelse af dosisinterval
|
23.
|
Der bør udføres en forprøve for at bestemme et passende koncentrationsområde for det kemikalie, der skal testes, og fastslå, om der kan være nogen problemer forbundet med testkemikaliets opløselighed og cytotoksiske effekt. Indledningsvis testes kemikalier op til den maksimale koncentration på 1 μl/ml, 1 mg/ml eller 1 mM, (den laveste af de tre). Baseret på omfanget af den cytotoksiske effekt eller manglende opløselighed observeret i forprøven bør den første endelige analyseserie teste kemikaliet ved log-seriefortyndinger med start ved den maksimale acceptable koncentration (f.eks. 1 mM, 100 μM, 10 μM osv.), og idet der noteres tilstedeværelse af uklarheder eller cytotoksisk effekt. Koncentrationer i den anden og, om nødvendigt, i den tredje analyseserie bør justeres, hvis det er relevant, for bedre at karakterisere koncentration/respons-kurven og undgå koncentrationer, der er uopløselige eller fremkalder for høj cytotoksisk effekt.
|
|
24.
|
For ER-agonister og -antagonister kan tilstedeværelsen af stigende cytotoksicitetsniveauer i væsentlig grad ændre eller fjerne den typiske sigmoidale respons og bør tages i betragtning ved fortolkningen af data. Der bør anvendes forsøgsmetoder vedrørende cytotoksisk effekt, som kan give oplysninger om 80 % levedygtighed i celler, idet der benyttes et passende assay baseret på laboratoriets erfaringer.
|
|
25.
|
Skulle resultaterne af cytotoksicitetstesten vise, at koncentrationen af testkemikaliet har reduceret antallet af celler med 20 % eller derover, bør denne koncentration betragtes som cytotoksisk, og der bør i vurderingen ses bort fra koncentrationer på eller over den cytotoksiske koncentration.
|
Eksponering for kemikaliet og organisering af assaypladen
|
26.
|
Proceduren for kemiske fortyndinger (trin 1 og 2) og celleeksponering (trin 3) kan gennemføres på følgende måde:
|
Trin 1: Hvert testkemikalie fortyndes serielt i DMSO eller et passende opløsningsmiddel og tilsættes til brøndene på en mikrotiterplade for at opnå endelige seriekoncentrationer som fastlagt ved forprøven til bestemmelse af dosisinterval (typisk i en serie på f.eks. 1 mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM og 10 pM (10-3-10-11 M)) til test in triplo.
Trin 2: Kemikaliefortynding: Fortynd først 1,5 μl af testkemikaliet i opløsningsmidlet til en mængde på 500 μl medium.
Trin 3: Eksponering af cellerne for kemikaliet: Tilsæt 50 μl af fortyndingen med mediet (præpareret under trin 2) til en assaybrønd indeholdende 104 celler/100 μl/brønd.
Den anbefalede endelige mængde medium, der kræves til hver brønd, er 150 μl. Testprøver og referencestandarder kan tilordnes som vist i tabel 3 og tabel 4.
Tabel 3
Eksempel på pladekoncentrationsfordeling af referencestandarderne på assaypladen i ER-agonistassayet
|
Række
|
17α-methyltestosteron
|
Corticosteron
|
17α-østradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
konc. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
konc. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
konc. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
konc. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
konc. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
konc. 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
konc. 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Bærestofkontrol (0,1 % DMSO), PC: Positiv kontrol (1 nM E2)
|
|
27.
|
Referencestandarderne (E2, 17α-østradiol, 17α-methyltestosteron og corticosteron) testes i hver analyseserie (tabel 3). PC-brønde behandlet med 1 nM af E2, som kan frembringe en E2-induktion, og VC-brønde behandlet med DMSO (eller et passende opløsningsmiddel) alene bør inkluderes på hver testassayplade (tabel 4). Hvis der anvendes celler fra forskellige kilder (f.eks. forskelligt generationsnummer, forskellig batch osv.) i det samme forsøg, bør referencestandarderne testes for hver cellekilde.
|
Tabel 4
Eksempel på pladekoncentrationsfordeling for test- og pladekontrolkemikalier på assaypladen i ER-agonistassayet
|
Række
|
Testkemikalie 1
|
Testkemikalie 2
|
Testkemikalie 3
|
Testkemikalie 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
konc. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
konc. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
konc. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
konc. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
konc. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
konc. 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
konc. 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Bærestofkontrol (0,1 % DMSO), PC: Positiv kontrol (1 nM E2)
|
Tabel 5
Eksempel på pladekoncentrationsfordeling af referencestandarder på assaypladen i ER-antagonistassayet
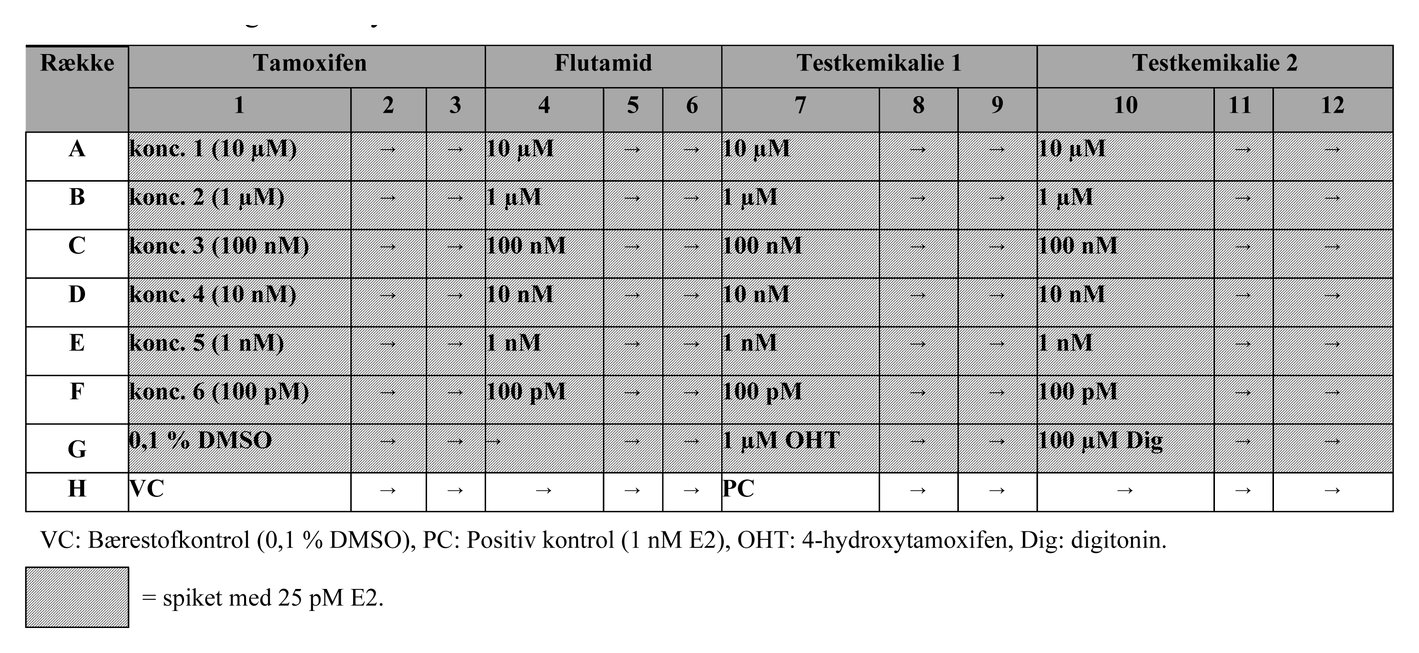
|
28.
|
For at vurdere kemikaliers antagonistaktivitet bør assaybrønde placeret i rækker fra A til G spikes med 25 pM E2. Referencestandarderne (tamoxifen og flutamid) testes i hver analyseserie. I hver testassayplade bør der inkluderes PC-brønde behandlet med 1 nM E2, der kan benyttes til kvalitetskontrol af hERα-HeLa-9903-cellelinje, VC-brønde behandlet med DMSO (eller et egnet opløsningsmiddel), 0,1 % DMSO-brønde behandlet med DMSO-tilsætning til den spikede E2 svarende til "spike-in-kontrol", brønde behandlet med den endelige koncentration 1 μM OHT og brønde behandlet med 100 μM Dig (tabel 5). Den efterfølgende assayplade bør følge det samme pladelayout uden referencestandardbrønde (tabel 6). Hvis der anvendes celler fra forskellige kilder (f.eks. forskelligt generationsnummer, forskellig batch osv.) i det samme forsøg, bør referencestandarderne testes for hver cellekilde.
|
Tabel 6
Eksempel på pladekoncentrationsfordeling for test- og pladekontrolkemikalier på assaypladen i ER-antagonistassayet

|
29.
|
De manglende randeffekter bør bekræftes, hvis det er relevant, og hvis der er mistanke om randeffekter, bør pladelayoutet ændres for at undgå sådanne effekter. F.eks. kan der anvendes et pladelayout, hvor der ses bort fra randbrønde.
|
|
30.
|
Efter tilsætning af kemikalierne bør assaypladerne inkuberes i en 5 % CO2-inkubator ved 37 ± 1oC i 20-24 timer for at fremkalde reportergenprodukter.
|
|
31.
|
Der skal tages særlige hensyn ved meget flygtige forbindelser. I sådanne tilfælde kan de tilstødende kontrolbrønde generere falsk positive resultater, og dette bør tages i betragtning i lyset af forventede og historiske kontrolværdier. I de få tilfælde, hvor der kan være bekymring for flygtighed, anbefales det at anvende pladeforseglere, som kan bidrage til effektivt at isolere individuelle brønde under testningen.
|
|
32.
|
Der bør udføres gentagne endelige test for det samme kemikalie på forskellige dage for at sikre uafhængighed.
|
Luciferaseassay
|
33.
|
Til assayet kan der anvendes et kommercielt luciferaseassayreagens [f.eks. Steady-Glo® Luciferase Assay System (Promega, E2510 eller tilsvarende)] eller et standard luciferaseassaysystem (f.eks. Promega, E1500 eller tilsvarende), så længe acceptkriterierne opfyldes. Assayreagenser bør vælges på grundlag af det anvendte luminometers følsomhed. Når der bruges et standardsystem til luciferaseassays, bør der anvendes et cellekulturlysisreagens (f.eks. Promega, E1531 eller tilsvarende), før mediet tilsættes. Luciferasereagenset bør anvendes efter producentens vejledning.
|
ANALYSE AF DATA
ER-agonistassay
|
34.
|
For ved anvendelse af et ER-agonistassay at opnå den relative transkriptionsaktivitet for PC (1 nM af E2) kan luminescenssignalerne fra den samme plade analyseres i overensstemmelse med følgende trin (andre tilsvarende matematiske processer kan også accepteres):
|
Trin 1. Beregn middelværdien for VC.
Trin 2. Træk middelværdien af VC fra hver brøndværdi for at normalisere dataene.
Trin 3. Beregn gennemsnittet for den normaliserede PC.
Trin 4. Divider den normaliserede værdi af hver brønd på pladen med middelværdien af den normaliserede PC (PC=100 %).
Den endelige værdi for hver brønd er den relative transkriptionsaktivitet for den pågældende brønd sammenlignet med PC-responsen.
Trin 5. Beregn middelværdien af den relative transaktionsaktivitet for hver af testkemikaliets koncentrationsgrupper. Der er to dimensioner for responsen: den gennemsnitlige transkriptionsaktivitet (respons) og den koncentration, hvorved responsen opstår (se næste afsnit).
Overvejelser vedrørende EC50-, PC50 - og PC10-induktion
|
35.
|
Den fuldstændige koncentration/respons-kurve er nødvendig til beregning af EC50, men dette er måske ikke altid muligt eller praktisk gennemførligt på grund af begrænsninger i testkoncentrationsområdet (f.eks. på grund af cytotoksicitets- eller opløselighedsproblemer). Da EC50 og det maksimale induktionsniveau (svarende til den højeste værdi af Hill-ligningen) er informative parametre, bør disse parametre angives, hvor det er muligt. Til beregning af EC50 og det maksimale induktionsniveau bør der anvendes egnet statistisk software (f. eks. det statistiske software Graphpad Prism). Hvis Hills logistiske ligning skal anvendes på dataene om koncentrationsrespons, bør EC50 beregnes efter følgende ligning (7):
Y=bund + (top-bund) / (1+10 exp ((log EC50 -X) × Hills kurve)), hvor:
X er logaritmen af koncentration, og
Y er responsen, og Y starter ved bunden og går til toppen i en sigmoidal kurve. Bunden er fastsat til nul i Hills logistiske ligning.
|
|
36.
|
For hvert testkemikalie angives følgende:
RPCMax, som er det maksimale responsniveau fremkaldt af et testkemikalie, udtrykt som en procentdel af den respons, som fremkaldes af 1 nM E2 på den samme plade, samt PCMax (koncentration, der er forbundet med RPCMax), og
for positive kemikalier, de koncentrationer, der fremkalder PC10 og, hvis det er relevant, PC50.
|
|
37.
|
PCx-værdien kan beregnes ved at interpolere mellem to punkter i X-Y-koordinatsystemet, ét umiddelbart over og ét umiddelbart under en PCx-værdi. Når de datapunkter, der ligger umiddelbart over og under PCx-værdien, har koordinaterne henholdsvis (a,b) og (c,d), kan PCx-værdien beregnes ved hjælp af følgende ligning:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
Beskrivelser af PC-værdierne er anført i figur 1 nedenfor.
Figur 1
Eksempel på, hvordan man udleder PC-værdier. PC (1 nM E2) tilføres på hver assayplade.
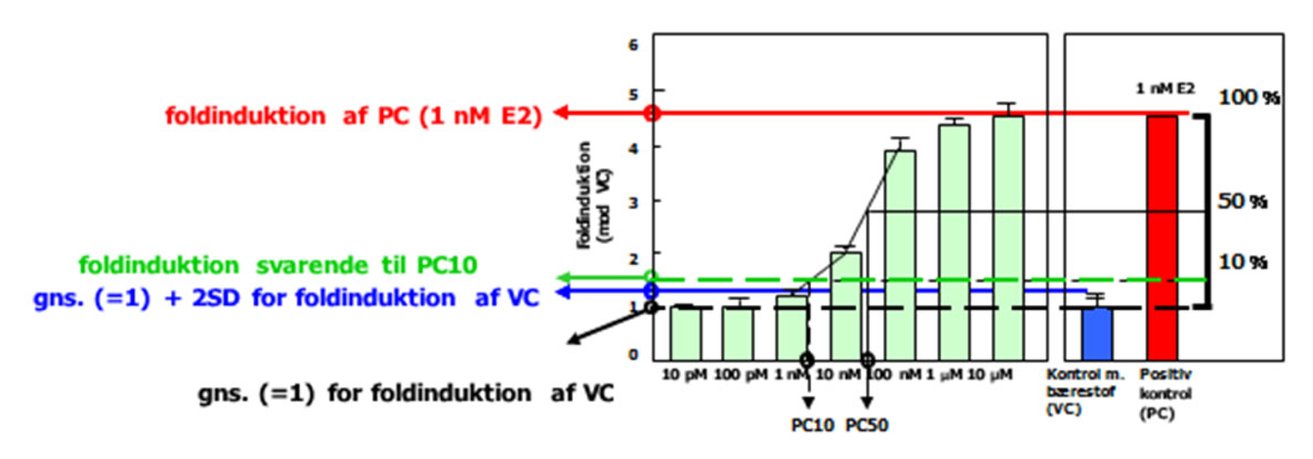
|
ER-antagonistassay
|
39.
|
For ved anvendelse af et ER-antagonistassay at opnå den relative transkriptionsaktivitet (RTA) i forhold til den spikede kontrol (25 pM E2) kan luminescenssignalerne fra den samme plade analyseres i overensstemmelse med følgende trin (andre tilsvarende matematiske processer kan også accepteres):
Trin 1. Beregn middelværdien for VC.
Trin 2. Træk middelværdien af VC fra hver brøndværdi for at normalisere dataene. Trin 3. Beregn middelværdien for den normaliserede spikede kontrol.
Trin 4. Divider den normaliserede værdi af hver brønd på pladen med middelværdien af den normaliserede spikede kontrol (spiket kontrol=100 %).
Den endelige værdi af hver brønd er den relative transkriptionsaktivitet for den pågældende brønd sammenlignet med responsen i den spikede kontrol.
Trin 5. Beregn middelværdien af den relative transaktionsaktivitet for hver behandling.
|
Overvejelser vedrørende IC30- og IC50-induktion
|
40.
|
For positive kemikalier angives de koncentrationer, der fremkalder IC30 og, hvis det er relevant, IC50.
|
|
41.
|
ICx-værdien kan beregnes ved at interpolere mellem to punkter i X-Y-koordinatsystemet, ét umiddelbart over og ét umiddelbart under en ICx-værdi. Når de datapunkter, der ligger umiddelbart over og under ICx-værdien, har koordinaterne henholdsvis (c,d) og (a,b), kan ICx-værdien beregnes ved hjælp af følgende ligning:
lin ICx = a-(b-(100-x)) (a-c) /(b-d)
Figur 2
Eksempel på udledning af IC-værdier. Den spikede kontrol (25 pM E2) tilføres på hver assayplade.

RTA: relativ transkriptionsaktivitet
|
|
42.
|
Resultaterne bør baseres på to (eller tre) uafhængige analyseserier. Hvis to analyseserier giver sammenlignelige og derfor reproducerbare resultater, er det ikke nødvendigt at gennemføre en tredje analyseserie. For at resultaterne kan godkendes, skal de:
|
—
|
opfylde acceptkriterierne (se Acceptkriterier, punkt 14-20)
|
|
Kriterier for datafortolkning
Tabel 7
Positive og negative beslutningskriterier i et ER-agonistassay
|
Positive
|
Hvis den opnåede RPCMax er lig med eller overstiger 10 % af responsen for den positive kontrol i mindst to af to eller to af tre analyseserier.
|
|
Negative
|
Hvis den opnåede RPCMax ikke kommer op på mindst 10 % af responsen for den positive kontrol i to af to eller to af tre analyseserier.
|
Tabel 8
Positive og negative beslutningskriterier i et ER-antagonistassay
|
Positive
|
Hvis IC30 beregnes i mindst to af to eller to af tre analyseserier.
|
|
Negative
|
Hvis IC30 ikke beregnes i to af to eller to af tre analyseserier.
|
|
43.
|
Kriterierne for datafortolkning fremgår af tabel 7 og 8. Positive resultater vil blive karakteriseret ved både effektens størrelsesorden og den koncentration, hvor effekten opstår. Når resultater udtrykkes som en koncentration, hvor 50 % (PC50) eller 10 % (PC10) af PC-værdierne nås for agonistassayet, og 50 % (IC50) eller 30 % (IC30) af den spikede kontrolværdi hæmmes for antagonistassayet, opfyldes begge disse mål. Et testkemikalie bestemmes til at være positivt, hvis den maksimale respons, der fremkaldes af testkemikaliet (RPCMax), er lig med eller overstiger 10 % af responsen fra PC i mindst to af to eller to af tre analyseserier, mens et testkemikalie betragtes som negativt, hvis RPCMax ikke kommer op på mindst 10 % af responsen fra den positive kontrol i to af to eller to af tre analyseserier.
|
|
44.
|
Beregningerne af PC10, PC50 og PCMax i et ER-agonistassay og IC30 og IC50 i et ER-antagonistassay kan foretages ved hjælp af et regneark, der er tilgængeligt sammen med testvejledningen på OECD’s offentlige websted (64).
|
|
45.
|
Det bør være tilstrækkeligt at opnå PC10- eller PC50- og IC30- eller IC50-værdier mindst to gange. Skulle den deraf følgende basislinje for data i det samme koncentrationsområde udvise en variation med en uacceptabelt høj variationskoefficient (CV; %), kan dataene dog ikke anses for pålidelige, og årsagen til den høje variation bør afdækkes. CV af rådata fra tre forsøg (dvs. data om luminescensintensitet) for de datapunkter, som anvendes til beregning af PC10, bør ligge under 20 %.
|
|
46.
|
Hvis acceptkriterierne opfyldes, er det tegn på, at assaysystemet fungerer korrekt, men det er ingen garanti for, at en given analyseserie vil producere nøjagtige data. Kan resultaterne af den første analyseserie gentages, er det den bedste garanti for, at der blev produceret nøjagtige data.
|
|
47.
|
Ved et ER-agonistassay, hvor der er behov for flere oplysninger end dem, der skal bruges til screening og prioritering til denne testvejledning for positive testkemikalier, især for PC10-PC49-kemikalier samt kemikalier, der er mistænkt for at overstimulere luciferase, kan det bekræftes, at den observerede luciferaseaktivitet udelukkende er en ERα-specifik respons, der anvender en ERα-antagonist (se tillæg 2.1).
|
TESTRAPPORT
|
48.
|
Jf. punkt 20 i "ER TA-ASSAYETS KOMPONENTER".
|
LITTERATUR
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environmental Health and Safety Publications, Series on Testing and Assessment, (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1459-1469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4252-4263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769-71.
|
|
(6)
|
Dussurget O. og Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. og Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Tillæg 2.1
FALSK POSITIVE RESULTATER: VURDERING AF IKKE-RECEPTORMEDIEREDE LUMINESCENSSIGNALER
|
1.
|
Falsk positive fund i ER-agonistassayet kan være genereret af ikke-ER-medieret aktivering af luciferasegenet eller direkte aktivering af genproduktet eller urelateret fluorescens. Sådanne virkninger angives ved en ufuldstændig eller usædvanlig dosis/respons-kurve. Hvis der er mistanke om sådanne virkninger, bør det undersøges, hvordan en ER-antagonist (f.eks. 4-hydroxytamoxifen (OHT) ved en ikke-toksisk koncentration) påvirker responsen. Den rene antagonist ICI 1827 80 er måske ikke egnet til dette formål, da en tilstrækkelig koncentration af ICI 1827 80 kan sænke VC-værdien, og dette vil påvirke dataanalysen.
|
|
2.
|
For at sikre validiteten af denne tilgang skal følgende testes på den samme plade:
|
—
|
det ukendte kemikalies agonistiske aktivitet med/uden 10 μM OHT
|
|
—
|
1 nM E2 (in triplo) som agonist-PC
|
|
—
|
1 nM E2 + OHT (in triplo)
|
|
Kriterier for datafortolkning
Bemærk: Alle brønde bør behandles med den samme koncentration af bærestoffet.
|
—
|
Såfremt det ukendte kemikalies agonistaktivitet IKKE påvirkes af behandlingen med ER-antagonist, klassificeres det som "negativt".
|
|
—
|
Såfremt det ukendte kemikalies agonistaktivitet hæmmes fuldstændigt, anvendes beslutningskriterierne.
|
|
—
|
Såfremt agonistaktiviteten ved den laveste koncentration svarer til eller overstiger PC10-responsen, er det ukendte kemikalie hæmmet i en grad, der svarer til eller overstiger PC10-responsen. Forskellen i respons mellem de brønde, der er behandlet, og de brønde, der ikke er behandlet med ER-antagonist, beregnes, og denne forskel anses for at være den korrekte respons og bør anvendes til beregning af de rette parametre, som gør det muligt at træffe en klassificeringsafgørelse.
|
Dataanalyse
Kontrollér standarden for ydeevne.
Kontrollér CV mellem brønde, som er behandlet under de samme betingelser.
|
1.
|
Beregn gennemsnittet af VC.
|
|
2.
|
Fratræk gennemsnittet af VC fra værdien af hver brønd, som ikke er behandlet med OHT.
|
|
3.
|
Beregn gennemsnittet af OHT.
|
|
4.
|
Fratræk gennemsnittet af VC fra værdien af hver brønd, som er behandlet med OHT.
|
|
5.
|
Beregn gennemsnittet af PC.
|
|
6.
|
Beregn den relative transkriptionsaktivitet for alle øvrige brønde i forhold til PC.
|
Tillæg 2.2
FREMSTILLING AF SERUM BEHANDLET MED DEXTRANOVERTRUKKET TRÆKUL (DCC)
|
1.
|
Behandlingen af serum med dextranovertrukket trækul (DCC) er en almindelig metode til at fjerne østrogenforbindelser fra serum, som tilsættes cellemedium for at udelukke den afvigende respons, som er knyttet til resterende østrogen i serum. 500 ml føtalt kalveserum (FBS) kan behandles efter denne fremgangsmåde.
|
Komponenter
|
2.
|
Der kræves følgende materialer og udstyr:
|
Materialer
|
magnesiumchloridhexahydrat (MgCl2 6H2O)
|
|
1 M HEPES-bufferopløsning (pH 7,4)
|
|
ultrarent vand filtreret gennem et filtersystem
|
Udstyr
autoklaveret glasbeholder (størrelsen bør eventuelt tilpasses) almindelig laboratoriecentrifuge (hvor temperaturen kan sættes til 4 oC)
Fremgangsmåde
|
3.
|
Den følgende fremgangsmåde er tilpasset til anvendelse af 50 ml centrifugerør:
[Dag 1] Fremstil dextranovertrukket kulsuspension med 1 l ultrarent vand indeholdende 1,5 mM MgCl2, 0,25 M saccharose, 2,5 g trækul, 0,25 g dextran og 5 mM HEPES, og omrør ved 4 oC natten over.
[Dag 2] Dispensér suspensionen i 50 ml centrifugerør, og centrifugér ved 10 000 o/min. ved 4 oC i 10 minutter. Fjern supernatanten, og opbevar halvdelen af kulsedimentet ved 4 oC til anvendelse på dag 3. Suspendér den anden halvdel af kullet med FBS, der er blevet optøet forsigtigt for at undgå bundfald og varmeinaktiveret ved 56 oC i 30 minutter, og overfør herefter til en autoklaveret glasbeholder som f.eks. en Erlenmeyerkolbe. Omrør denne suspension forsigtigt ved 4 oC natten over.
[Dag 3] Dispensér suspensionen med FBS i centrifugerør, og centrifugér med 10 000 o/min. ved 4 oC i 10 minutter. Indsaml FBS, og overfør til det nye kulsediment, der blev fremstillet og opbevaret på dag 2. Suspendér kulsedimentet, og omrør denne suspension forsigtigt i en autoklaveret glasbeholder ved 4 oC natten over.
[Dag 4] Dispensér suspensionen til centrifugering ved 10 000 o/min. ved 4 oC i 10 minutter, og sterilisér supernatanten ved filtrering gennem et 0,2 μm sterilt filter. Denne DCC-behandlede FBS bør opbevares ved -20 oC og kan bruges i op til et år.
|
Tillæg 3
VM7LUC-ØSTROGENRECEPTOR-TRANSAKTIVERINGSASSAY TIL PÅVISNING AF ØSTROGENRECEPTORAGONISTER OG -ANTAGONISTER.
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER (JF. OGSÅ GENEREL INDLEDNING)
|
1.
|
Dette assay anvender VM7Luc4E2-cellelinjen (65). Det er blevet valideret af National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) og Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) (1). VM7Luc-cellelinjer angiver overvejende endogen ERα og en mindre mængde endogen ERβ (2) (3) (4).
|
|
2.
|
Dette assay kan finde anvendelse på en lang række stoffer, forudsat at de kan opløses i dimethylsulfoxid (DMSO, CASRN 67-68-5), ikke reagerer med DMSO eller celledyrkningsmediet og ikke er cytotoksiske ved de testede koncentrationer. Hvis anvendelse af DMSO ikke er mulig, kan der anvendes et andet bærestof som ethanol eller vand (jf. punkt 12). Den påviste ydeevne for VM7Luc ER TA-(ant)agonistassayet tyder på, at de data, der genereres med dette assay, kan give oplysninger om ER-medierede virkningsmekanismer og kan tages i betragtning til prioritering af stoffer til yderligere test.
|
|
3.
|
Dette assay er specifikt udformet til at påvise hERα- og hERß-medieret TA ved at måle kemiluminescens som effektparameter. Anvendelsen af kemiluminescens i bioassays er udbredt, fordi luminescens har et højt forhold mellem signal og baggrund (10). Aktiviteten som følge af ildflueluciferase i cellebaserede assays kan imidlertid forveksles med stoffer, der hæmmer luciferaseenzymet og forårsage både tilsyneladende hæmning og øget luminescens på grund af proteinstabilisering (10). I nogle luciferasebaserede ER-reportergenassays er der endvidere rapporteret om ikke-receptormedierede luminescenssignaler ved højere fytoøstrogenkoncentrationer end 1 μM på grund af overaktivering af luciferasereportergenet (9) (11). Mens dosis/respons-kurven tyder på, at der sker en egentlig aktivering af ER-systemet ved lavere koncentrationer, skal den luciferaseekspression, som er opnået ved høje koncentrationer af fytoøstrogener eller lignende kemiske forbindelser, der mistænkes for at forårsage fytoøstrogenlignende overaktivering af luciferasereportergenet, undersøges nøje i stabilt transfekterede ER TA-assaysystemer (see Appendix 2).
|
|
4.
|
Afsnittene "GENEREL INDLEDNING" og "ER TA-ASSAYETS KOMPONENTER" bør læses, før dette assay benyttes til lovmæssige formål. Definitioner og forkortelser brugt i denne forsøgsmetode er beskrevet i tillæg 1.
|
PRINCIP FOR ASSAYET (SE OGSÅ GENEREL INDLEDNING)
|
5.
|
Assayet benyttes til at angive ER-ligandbinding efterfulgt af kromosomtranslokation af receptorligandkomplekset til kernen. I kernen binder receptorligandkomplekset til specifikke DNA-responselementer og transaktiverer reportergenet (luc), hvorved der fremkommer luciferase og den efterfølgende udsendelse af lys, som kan kvantificeres ved hjælp af et luminometer. Luciferaseaktivitet kan vurderes hurtigt og billigt med forskellige typer testkit, der kan fås i handelen. VM7Luc ER TA anvender en ER-responsiv human brystadenocarcinomcellelinje, VM7, som er blevet stabilt transfekteret med en ildflue-luc-reporterkonstruktion under kontrol af fire østrogenresponselementer anbragt ovenfor mamatumorviruspromoteren i mus (MMTV) for at opspore stoffer med in vitro-ER-agonistaktivitet eller -antagonistaktivitet. Denne MMTV-promoter udviser kun mindre krydsreaktivitet med andre steroide og ikke-steroide hormoner (8). Kriterier for datafortolkning er nærmere beskrevet i punkt 41. Kort beskrevet identificeres en positiv respons ved en koncentration/respons-kurve, som indeholder mindst tre punkter med ikke-overlappende fejltærskler (gennemsnit ± standardafvigelse) samt en ændring i amplitude (normaliseret relativ lysenhed [RLU]) på mindst 20 % af den maksimale værdi for referencestandarden (17β-østradiol [E2, CASRN 50-28-2] for agonistassayet, raloxifen HCl [Ral, CASRN 84 449-90-1]/E2 for antagonistassayet).
|
FREMGANGSMÅDE
Cellelinje
|
6.
|
Den stabilt transfekterede VM7Luc4E2-cellelinje bør anvendes til assayet. Cellelinjen er i øjeblikket kun tilgængelig med en teknisk licensaftale fra University of California, Davis, Californien, USA (66), og fra Xenobiotic Detection Systems Inc., Durham, North Carolina, USA (67).
|
Cellelinjens stabilitet
|
7.
|
For at opretholde cellelinjens stabilitet og integritet bør cellerne dyrkes i mere end én generation fra den nedfrosne beholdning i celleopbevaringsmedium (jf. punkt 9). Celler bør ikke dyrkes i mere end 30 generationer. For VM7Luc4E2-cellelinjen vil 30 generationer være ca. tre måneder.
|
Celledyrknings- og udpladningsbetingelser
|
8.
|
De procedurer, der er anført i Guidance on Good Cell Culture Practice (5) (6), bør følges for at sikre kvaliteten af alle materialer og metoder med henblik på at bevare integriteten, validiteten og reproducerbarheden af det arbejde, der udføres.
|
|
9.
|
VM7Luc4E2-celler opbevares i RPMI 1 640-medium suppleret med 0,9 % Pen-Strep og 8,0 % føtalt kalveserum (FBS) i en dedikeret vævsdyrkningsinkubator ved 37 ± 1 °C, 90 % ± 5 % fugtighed og 5,0 % ± 1 % CO2/luft.
|
|
10.
|
Når VM7Luc4E2-cellerne når op på ~80 % konfluens, bliver de dyrket i subkultur og konditioneres til et østrogenfrit miljø i 48 timer, før cellerne udplades i plader med 96 brønde til eksponering for testkemikalier og analyse af østrogenafhængig induktion af luciferaseaktivitet. Det østrogenfrie medium (EFM) Dulbecco’s Modification of Eagle’s Medium (DMEM) uden fenolrød suppleret med 4,5 % kul-/dextranbehandlet FBS, 1,9 % L-glutamin og 0,9 % Pen-Strep. Alt plastikudstyr skal være fri for østrogenaktivitet [se detaljeret protokol (7)].
|
Acceptkriterier
|
11.
|
Godkendelse eller forkastelse af en test er baseret på vurdering af referencestandard og kontrolresultater fra hvert enkelt forsøg, der udføres på en plade med 96 brønde. Hver referencestandard testes i mange koncentrationer, og der er mange prøver af hver reference- og kontrolkoncentration. Resultaterne sammenlignes med kvalitetskontrollerfor de parametre, der er udledt af historiske agonist- og antagonistdatabaser, og som hvert laboratorium har genereret under påvisningen af laboratoriets kompetence. De historiske databaser ajourføres løbende med referencestandard og kontrolværdier. Ændringer i udstyr eller laboratoriebetingelser kan gøre det nødvendigt at generere ajourførte historiske databaser.
|
Agonisttest
Test til bestemmelse af dosisinterval
|
•
|
Induktion: Pladeinduktion bør måles ved at dividere den gennemsnitligt højeste E2-referencestandard for relativ lysenhedsværdi (RLU) med den gennemsnitlige RLU-værdi for DMSO-kontrollen. Der opnås normalt fem fold induktion, men med henblik på godkendelse bør induktionen være større end eller lig med fire fold.
|
|
•
|
DMSO-kontrolresultater: RLU-værdierne for kontrolkulturen med opløsningsmiddel bør ligge inden for 2,5 gange standardafvigelsen for den historiske kontrolkultur med opløsningsmiddel for den gennemsnitlige RLU-værdi.
|
|
•
|
Forsøg, der ikke opfylder nogen af acceptkriterierne, bør kasseres og gentages.
|
Omfattende test
Den omfatter acceptkriterier fra testen til bestemmelse af agonist-dosisinterval og følgende:
|
•
|
Referencestandardresultater: Referencestandarden for koncentration/respons-kurven for ER2 bør være sigmoidal i form og have mindst tre værdier inden for den lineære del af koncentration/respons-kurven.
|
|
•
|
Positive kontrolresultater: RLU-værdierne for methoxychlorkontrollen bør være større end gennemsnittet for DMSO plus tre gange standardafvigelsen fra gennemsnittet for DMSO.
|
|
•
|
Forsøg, der ikke opfylder et eneste acceptkriterium, bør kasseres og gentages.
|
Antagonisttest
Test til bestemmelse af dosisinterval
|
•
|
Reduktion: Pladereduktion måles ved at dividere den gennemsnitligt højeste RLU-værdi for Ral/E2-referencestandarden med den gennemsnitlige RLU-værdi for DMSO-kontrollen. Der opnås normalt fem fold reduktion, men med henblik på godkendelse bør reduktionen være større end eller lig med tre fold.
|
|
•
|
E2-kontrolresultater: RLU-værdierne for E2-kontrollen bør ligge inden for 2,5 gange standardafvigelsen for den historiske gennemsnitlige RLU-værdi for E2-kontrollen.
|
|
•
|
DMSO-kontrolresultater: RLU-værdierne for DMSO-kontrollen bør ligge inden for 2,5 gange standardafvigelsen for den historiske gennemsnitlige RLU-værdi for kontrolkulturen med opløsningsmiddel.
|
|
•
|
Forsøg, der ikke opfylder et eneste acceptkriterium, kasseres og gentages.
|
Omfattende test
Den omfatter acceptkriterier fra testen til bestemmelse af antagonist-dosisinterval og følgende:
|
•
|
Referencestandardresultater: Koncentration/respons-kurven for referencestandarden for Ral/E2 bør være sigmoidal i form og have mindst tre værdier inden for den lineære del af koncentration/respons-kurven.
|
|
•
|
Positive kontrolresultater: RLU-værdier for tamoxifen/E2-kontrollen bør ligge under den gennemsnitlige E2-kontrol minus tre gange standardafvigelsen fra gennemsnittet for E2-kontrollen.
|
|
•
|
Forsøg, der ikke opfylder et eneste acceptkriterium, kasseres og gentages.
|
Referencestandarder, positive kontroller og bærestofkontroller
Bærestofkontrol (agonist- og antagonistassays)
|
12.
|
Det bærestof, der anvendes til at opløse testkemikalierne, bør testes som en bærestofkontrol. Det bærestof, der blev anvendt under valideringen af VM7Luc ER TA-assayet, var 1 % (v/v) dimethylsulfoxid (DMSO, CASRN 67-68-5) (jf. punkt 24). Hvis der anvendes et andet bærestof end DMSO, testes alle referencestandarder, kontroller og testkemikalier i det samme bærestof, hvis det er relevant.
|
Referencestandard (bestemmelse af agonist-dosisinterval)
|
13.
|
Referencestandarden er E2 (CASRN 50-28-2). Til testen til bestemmelse af dosisinterval består referencestandarden af en seriel fortynding af fire koncentrationer af E2 (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 og 2,87 x 10-12 M), idet hver koncentration testes i to brønde.
|
Referencestandard (agonist omfattende)
|
14.
|
E2 til omfattende testning består af en seriel 1:2-fortynding bestående af 11 koncentrationer (fra 3,67 x 10-10 til 3,59 x 10-13 M) E2 i to brønde.
|
Referencestandard (bestemmelse af antagonist-dosisinterval)
|
15.
|
Referencestandarden er en kombination af Ral (CASRN 84 449-90-1) og E2 (CASRN 50-28-2). Ral/E2 til testen til bestemmelse af dosisinterval består af en seriel fortynding af tre koncentrationer af Ral (3,06 x 10-9, 7,67 x 10-10 og 1,92 x 10-10 M) plus en fast koncentration (9,18 × 10-11 M) af E2 i to brønde.
|
Referencestandard (antagonist omfattende)
|
16.
|
Ral/E2 til omfattende testning består af en seriel 1:2-fortynding af Ral (fra 2,45x 10-8 til 9,57 x 10-11 M) plus en fast koncentration (9,18 × 10-11 M) E2 bestående af ni koncentrationer af Ral/E2 i to brønde.
|
Svagt positiv kontrol (agonist)
|
17.
|
Den svagt positive kontrol er 9,06 x 10-6 M p,p’-methoxychlor (methoxychlor; CASRN 72-43-5) i EFM.
|
Svagt positiv kontrol (antagonist)
|
18.
|
Den svagt positive kontrol består af tamoxifen (CASRN 10 540-29-1) 3,36 x 10-6 M med 9,18 × 10-11 M E2 i EFM.
|
E2-kontrol (antagonistassay alene)
|
19.
|
E2-kontrol er 9,18 × 10-11 M E2 i EFM og anvendt som en negativ grundkontrol.
|
Foldinduktion (agonist)
|
20.
|
Induktionen af luciferaseaktivitet i referencestandarden (E2) måles ved at dividere den gennemsnitlige højeste E2-referencestandardværdi for RLU med den gennemsnitlige DMSO-kontrolværdi for RLU, og resultatet bør være større end fire fold.
|
Foldreduktion (antagonist)
|
21.
|
Den gennemsnitlige luciferaseaktivitet for referencestandarden (Ral/E2) måles ved at dividere den gennemsnitlige højeste RLU-værdi for Ral/E2-referencestandarden med den gennemsnitlige RLU-værdi for DMSO-kontrollen, som bør være større end tre fold.
Påvisning af laboratoriets kompetence (jf. punkt 14 og tabel 3 og 4 i "ER TA-ASSAYETS KOMPONENTER" for denne forsøgsmetode)
|
Bærestof
|
22.
|
Testkemikalier bør opløses i et opløsningsmiddel, som opløser testkemikaliet og kan blandes med cellemediet. Vand, ethanol (95-100 % renhed) og DMSO er egnede bærestoffer. Hvis der anvendes DMSO, bør niveauet ikke overstige 1 % (v/v). For ethvert bærestof bør det påvises, at den maksimalt anvendte mængde ikke er cytotoksisk og ikke indvirker på assayets ydeevne. Referencestandarder og kontroller opløses i 100 % opløsningsmiddel og fortyndes herefter ned til passende koncentrationer i EFM.
|
Præparering af testkemikalier
|
23.
|
Testkemikalier opløses i 100 % DMSO (eller et passende opløsningsmiddel) og fortyndes herefter ned til passende koncentrationer i EFM. Lad alle testkemikalier ækvilibrere til stuetemperatur, før de opløses og fortyndes. Testkemikalieopløsninger fremstilles frisk til hvert forsøg. Opløsningerne bør ikke have udtalt bundfald eller uklarhed. Lagre af referencestandarder og kontroller kan fremstilles i større mængder; dog bør den endelige referencestandard, kontrolfortyndinger og testkemikalier fremstilles frisk til hvert forsøg og anvendes inden for 24 timer efter fremstillingen.
|
Opløselighed og cytotoksisk effekt: Overvejelser vedrørende bestemmelse af dosisinterval
|
24.
|
Test til bestemmelse af dosisinterval består af syv seriefortyndinger i forholdet 1:10 (dobbeltbestemmelse). Indledningsvis testes testkemikalierne op til højeste koncentration på 1 mg/ml (~1 mM) til agonisttest og 20 μg/ml (~10 μM) til antagonisttest. Forsøg til bestemmelse af dosisinterval benyttes til at fastlægge følgende:
|
|
—
|
testkemikaliets startkoncentrationer, der anvendes under omfattende testning
|
|
—
|
testkemikaliefortyndinger (1:2 eller 1:5), der anvendes under omfattende testning
|
|
25.
|
En vurdering af cellernes levedygtighed/cytotoksiske effekt er inkluderet i protokollerne for agonist- og antagonistassays (7) og er indarbejdet i test til bestemmelse af dosisinterval og omfattende test. Den cytotoksicitetsmetode, som blev benyttet til at vurdere cellernes levedygtighed under valideringen af VM7Luc ER TA (1), var en skaleret kvalitativ, visuel observationsmetode; der kan dog benyttes en kvantitativ metode til bestemmelse af den cytotoksiske effekt (jf. protokollen (7)). Data fra testkemikaliekoncentrationer, der forårsager over 20 % reduktion i levedygtighed, kan ikke anvendes.
|
Eksponering for testkemikaliet og organisering af assaypladen
|
26.
|
Celler tælles og udplades i vævsdyrkningsplader med 96 brønde (2 x 105 celler pr. brønd) i EFM og inkuberes i 24 timer, så cellerne kan binde sig til pladen. EFM fjernes og erstattes med test- og referencekemikalier i EFM og inkuberes i 19-24 timer. Der skal tages særlige hensyn til meget flygtige stoffer, da tilstødende kontrolbrønde kan generere falsk positive resultater. I sådanne tilfælde anbefales "pladeforseglere", der kan bidrage til effektivt at isolere individuelle brønde under testningen.
|
Test til bestemmelse af dosisinterval
|
27.
|
Ved bestemmelse af dosisinterval benyttes alle brønde i en plade med 96 brønde til at teste op til seks testkemikalier som syv seriefortyndinger i forholdet 1:10 (dobbeltbestemmelse) (jf. figur 1 og 2).
|
|
—
|
Ved test til bestemmelse af agonist-dosisinterval anvendes fire koncentrationer af E2 (dobbeltbestemmelse) som referencestandard og fire replikatbrønde til DMSO-kontrollen.
|
|
—
|
Ved test til bestemmelse af antagonist-dosisinterval anvendes tre koncentrationer af Ral/E2 med 9,18 × 10-11 M E2 (dobbeltbestemmelse) som referencestandard med tre replikatbrønde til E2- og DMSO-kontrollen.
|
Figur 1
Layout for en testplade med 96 brønde til bestemmelse af agonist-dosisinterval

Forkortelser: E2-1 til E2-4 = koncentrationer af E2-referencestandard (fra høj til lav); TC1-1 til TC1-7 = koncentrationer (fra høj til lav) af testkemikalie 1 (TC1); TC2-1 til TC2-7 = koncentrationer (fra høj til lav) af testkemikalie 2 (TC2); TC3-1 til TC3-7 = koncentrationer (fra høj til lav) af testkemikalie 3 (TC3); TC4-1 til TC4-7 = koncentrationer (fra høj til lav) af testkemikalie 4 (TC4); TC5-1 til TC5-7 = koncentrationer (fra høj til lav) af testkemikalie 5 (TC5); TC6-1 til TC6-7 = koncentrationer (fra høj til lav) af testkemikalie 6 (TC6); VC = bærestofkontrol (DMSO [1 % (v/v) EFM.]).
Figur 2
Layout for en testplade med 96 brønde til bestemmelse af antagonist-dosisinterval

Forkortelser: E2 = E2-kontrol; Ral-1 til Ral-3 = koncentrationer af Raloxifen/E2-referencestandard (fra høj til lav); TC1-1 til TC1-7 = koncentrationer (fra høj til lav) af testkemikalie 1 (TC1); TC2-1 til TC2-7 = koncentrationer (fra høj til lav) af testkemikalie 2 (TC2); TC3-1 til TC3-7 = koncentrationer (fra høj til lav) af testkemikalie 3 (TC3); TC4-1 til TC4-7 = koncentrationer (fra høj til lav) af testkemikalie 4 (TC4); TC5-1 til TC5-7 = koncentrationer (fra høj til lav) af testkemikalie 5 (TC5); TC6-1 til TC6-7 = koncentrationer (fra høj til lav) af testkemikalie 6 (TC6); VC = bærestofkontrol (DMSO [1 % (v/v) EFM.]).
Bemærk: Alle testkemikalier testes med 9,18 × 10-11 M E2.
|
28.
|
Den anbefalede endelige mængde medium, der kræves til hver brønd, er 200 μl. Brug kun testplader, hvor cellerne i alle brønde giver en levedygtighed på 80 % og derover.
|
|
29.
|
Bestemmelse af startkoncentrationer til omfattende
agonist
-testning er indgående beskrevet i agonistprotokollen (7). Kort forklaret anvendes følgende kriterier:
|
|
—
|
Hvis ingen punkter på testkemikaliets koncentrationskurve ligger over middel plus tre gange DMSO-kontrollens standardafvigelse, udføres der omfattende testning under anvendelse af 11 seriefortyndinger i forholdet 1:2 begyndende ved den maksimale opløselige koncentration.
|
|
—
|
Hvis ingen punkter på testkemikaliets koncentrationskurve ligger over middel plus tre gange DMSO-kontrollens standardafvigelse, bør den startkoncentration, der skal anvendes i 11-punktsplanen for fortynding ved omfattende testning, være en log højere end den koncentration, som giver den højeste justerede RLU-værdi i bestemmelsen af dosisinterval. 11-punktsplanen for fortynding er baseret på fortyndinger i forholdet 1:2 eller 1:5 efter følgende kriterier:
|
Der bør anvendes 11 seriefortyndinger i forholdet 1:2, hvis det deraf følgende koncentrationsområde vil omfatte hele responsområdet baseret på den koncentration/respons-kurve, der genereres i testen til bestemmelse af dosisinterval. Ellers bruges en 1:5-fortynding.
|
|
|
—
|
Hvis et testkemikalie udviser en bifasisk koncentration/respons-kurve i testen til bestemmelse af dosisinterval, opløses begge faser også i omfattende testning.
|
|
30.
|
Bestemmelse af startkoncentrationer til omfattende
antagonist
-testning er indgående beskrevet i antagonistprotokollen (7). Kort forklaret anvendes følgende kriterier:
|
|
—
|
Hvis ingen punkter på testkemikaliets koncentrationskurve ligger under middel minus tre gange standardafvigelsen for E2-kontrollen, udføres der en omfattende testning under anvendelse af 11 seriefortyndinger i forholdet 1:2 begyndende ved den maksimale opløselige koncentration.
|
|
—
|
Hvis ingen punkter på testkemikaliets koncentrationskurve ligger under middel minus tre gange E2-kontrollens standardafvigelse, bør den startkoncentration, der skal anvendes i 11-punktsplanen for fortynding ved omfattende testning, være en af følgende:
|
—
|
den koncentration, der giver den laveste justerede RLU-værdi ved bestemmelsen af dosisinterval
|
|
—
|
den maksimale opløselige koncentration (jf. antagonistprotokollen (7), figur 14-2)
|
|
—
|
den laveste cytotoksiske koncentration (jf. antagonistprotokollen (7), figur 14-3 for et beslægtet eksempel).
|
|
|
—
|
11-punktsplanen for fortynding er baseret på enten en seriefortynding i forholdet 1:2 eller 1:5 efter følgende kriterier:
|
Der bør anvendes 11 seriefortyndinger i forholdet 1:2, hvis det deraf følgende koncentrationsområde vil omfatte hele responsområdet baseret på den koncentration/respons-kurve, der genereres i testen til bestemmelse af dosisinterval. Ellers anvendes en 1:5-fortynding.
|
|
Omfattende test
|
31.
|
Omfattende testning består af 11 seriefortyndinger (seriefortyndinger i forholdet 1:2 eller 1:5 baseret på startkoncentrationer for omfattende testkriterier), idet hver koncentration testes i tredobbelte brønde på pladen med 96 brønde (jf. figur 3 og 4).
|
|
—
|
Ved omfattende agonist-testning anvendes 11 koncentrationer af E2 (dobbeltbestemmelse) som referencestandard. Fire replikatbrønde til DMSO-kontrol og fire replikatbrønde til methoxychlor-kontrol (9,06 x 10-6 M) medtages på hver plade.
|
|
—
|
Ved omfattende antagonist-testning anvendes ni koncentrationer af Ral/E2 med 9,18 × 10-11 M E2 (dobbeltbestemmelse) som referencestandard med fire replikatbrønde til E2 9,18 x 10-11 M kontrol, fire replikatbrønde til DMSO-kontrol og fire replikatbrønde til tamoxifen 3,36 x 10-6 M.
|
Figur 3
Layout for en testplade med 96 brønde til omfattende agonisttest
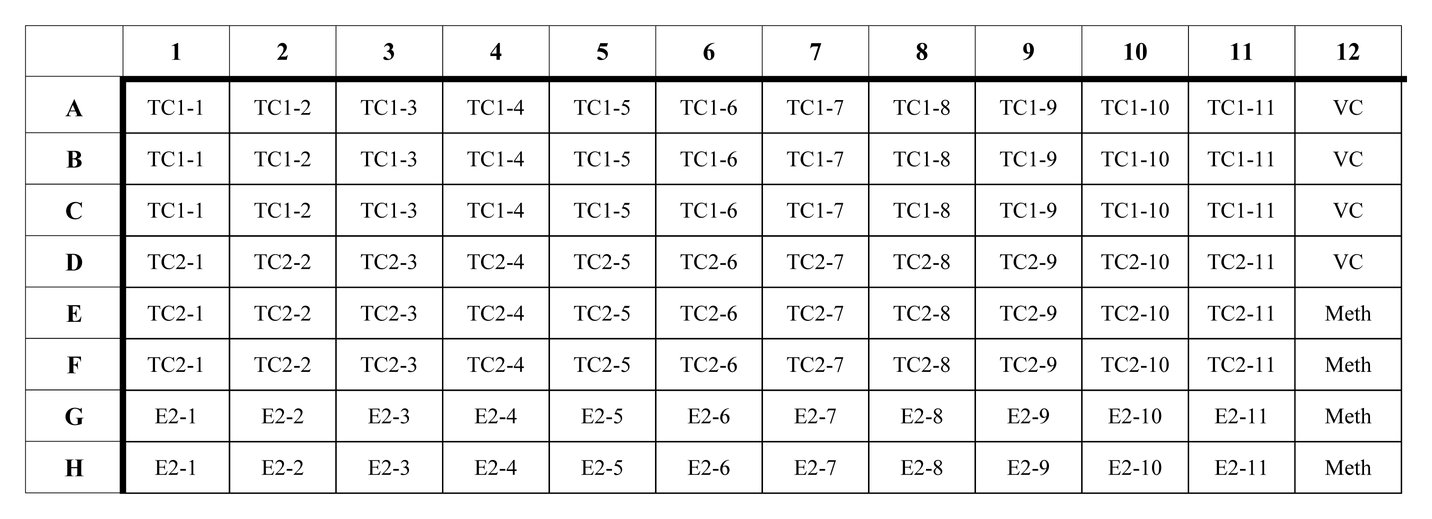
Forkortelser: TC1-1 til TC1-11 = koncentrationer (fra høj til lav) af testkemikalie 1; TC2-1 til TC2-11 = koncentrationer (fra høj til lav) af testkemikalie 2; E2-1 til E2-11 = koncentrationer af E2-referencestandard (fra høj til lav); Meth = p,p’ methoxychlor svagt positiv kontrol; VC = bærestofkontrol (DMSO, 1 % (v/v) EFM).
Figur 4
Layout for en plade med 96 brønde til omfattende agonisttest

Forkortelser: E2 = E2-kontrol; Ral-1 til Ral-9 = koncentrationer af raloxifen/E2-referencestandard (fra høj til lav); Tam = tamoxifen/E2 svagt positiv kontrol; TC1-1 til TC1-11 = koncentrationer (fra høj til lav) af testkemikalie 1 (TC1); TC2-1 til TC2-11 = koncentrationer (fra høj til lav) af testkemikalie 2 (TC2); VC = bærestofkontrol (DMSO [1 % (v/v) EFM.]).
Bemærk: Som anført indeholder alle reference- og testbrønde en fast koncentration af E2 (9,18 x 10-11 M)
|
32.
|
Gentagne omfattende test for det samme kemikalie udføres på forskellige dage for at sikre uafhængighed. Der bør foretages mindst to omfattende test. Hvis testresultaterne modsiger hinanden (f.eks. én test er positiv, den anden er negativ), eller hvis én af testene er utilstrækkelig, udføres der en tredje supplerende test.
|
Måling af luminescens
|
33.
|
Luminescens måles i området 300-650 nm ved hjælp af et injektionsluminometer og med software, som kontrollere injektionsvolumen og måleinterval (7). Lysemission fra hver brønd angives som RLU pr. brønd.
|
ANALYSE AF DATA
Bestemmelse af EC50 /IC50
|
34.
|
EC50-værdien (halvdelen af den maksimale effektive koncentration af et testkemikalie [agonister]) og IC50-værdien (halvdelen af den maksimale inhiberende koncentration af et testkemikalie [antagonister]) bestemmes ud fra koncentration/respons-data. For testkemikalier, der er positive ved en eller flere koncentrationer, beregnes den koncentration af testkemikaliet, som forårsager en respons på halvdelen af det maksimale (IC50 eller EC50) ved hjælp af en Hill-funktionsanalyse eller et passende alternativ. Hill-funktionen er en logistisk matematisk model med fire parametre, som knytter testkemikaliekoncentrationen til responsen (typisk efter en sigmoidal kurve) ved hjælp af nedenstående ligning:
|
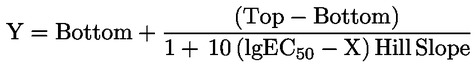
hvor:
Y= respons (dvs RLU’er)
X= logaritmen af koncentrationen
Bund= minimumrespons
Top= maksimumrespons
lg EC50 (eller lg IC50)= logaritmen af X som responsen midtvejs mellem top og bund
Hills kurve= kurvens stejlhed.
Modellen beregner den bedste tilpasning for parametrene top, bund, Hills kurve samt IC50 og EC50. Til beregning af EC50 og IC50-værdier bør der anvendes passende statistisk software (f.eks. Graphpad PrismR).
Bestemmelse af afvigende værdier (outliers)
|
35.
|
En god statistisk bedømmelse kan lettes ved at medtage (men ikke nøjes med) Q-testen (jf. agonist- og antagonistprotokollerne (7) til bestemmelse af "ubrugelige" brønde, som vil blive udelukket fra dataanalysen.
|
|
36.
|
For E2-referencestandardreplikater (prøvestørrelse på to) betragtes enhver justeret RLU-værdi til et replikat ved en given koncentration af E2 som en afvigende værdi, hvis dens værdi ligger mere end 20 % over eller under den justerede RLU-værdi for den pågældende koncentration i den historiske database.
|
Indsamling og justering af luminometerdata til testen til bestemmelse af dosisinterval
|
37.
|
Rådata fra luminometeret overføres til en regnearkskabelon designet til assayet. Det bestemmes, om der er afvigende datapunkter, som skal fjernes. (Jf. Kriterier for accept af testen med hensyn til parametre, der bestemmes i analyserne.) Følgende beregninger bør foretages:
|
Agonist
|
Trin 1
|
Beregn middelværdien for DMSO-bærestofkontrollen (VC).
|
|
Trin 2
|
Træk middelværdien af DMSO VC fra hver brøndværdi for at normalisere dataene.
|
|
Trin 3
|
Beregn den gennemsnitlige foldinduktion for referencestandarden (E2).
|
|
Trin 4
|
Beregn den gennemsnitlige EC50-værdi for testkemikalierne.
|
Antagonist
|
Trin 1
|
Beregn middelværdien for DMSO VC.
|
|
Trin 2
|
Træk middelværdien af DMSO VC fra hver brøndværdi for at normalisere dataene.
|
|
Trin 3
|
Beregn den gennemsnitlige foldreduktion for referencestandarden (Ral/E2).
|
|
Trin 4
|
Beregn middelværdien for E2-referencestandarden.
|
|
Trin 5
|
Beregn den gennemsnitlige IC50-værdi for testkemikalierne.
|
Indsamling og justering af luminometerdata til omfattende test
|
38.
|
Rådata fra luminometeret overføres til en regnearkskabelon designet til assayet. Det bestemmes, om der er afvigende datapunkter, som skal fjernes. (Jf. Kriterier for accept af testen med hensyn til parametre, der bestemmes i analyserne.) Følgende beregninger foretages:
|
Agonist
|
Trin 1
|
Beregn middelværdien for DMSO VC.
|
|
Trin 2
|
Træk middelværdien af DMSO VC fra hver brøndværdi for at normalisere dataene.
|
|
Trin 3
|
Beregn den gennemsnitlige foldinduktion for referencestandarden (E2).
|
|
Trin 4
|
Beregn den gennemsnitlige EC50-værdi for E2 og testkemikalierne.
|
|
Trin 5
|
Beregn den gennemsnitlige justerede RLU-værdi for methoxychlor.
|
Antagonist
|
Trin 1
|
Beregn middelværdien for DMSO VC.
|
|
Trin 2
|
Træk middelværdien af DMSO VC fra hver brøndværdi for at normalisere dataene.
|
|
Trin 3
|
Beregn den gennemsnitlige foldinduktion for referencestandarden (Ral/E2).
|
|
Trin 4
|
Beregn den gennemsnitlige IC50-værdi for Ral/E2 og testkemikalierne.
|
|
Trin 5
|
Beregn den gennemsnitlige justerede RLU-værdi for tamoxifen.
|
|
Trin 6
|
Beregn middelværdien for E2-referencestandarden.
|
Kriterier for datafortolkning
|
39.
|
VM7Luc ER TA er tænkt som en del af en "weight of evidence"-analyse, der skal bidrage til at prioritere stoffer til ED-testning in vivo. En del af denne prioriteringsprocedure er klassificeringen af testkemikaliet som positivt eller negativt i forbindelse med enten ER-agonist- eller ER-antagonistaktivitet. De positive og negative beslutningskriterier, der anvendes i VM7Luc ER TA-valideringsundersøgelsen, er beskrevet i tabel 1.
|
Tabel 1
Positive og negative beslutningskriterier
|
|
|
AGONISTAKTIVITET
|
|
Positive
|
|
—
|
Alle testkemikalier, der klassificeres som positive for ER-agonistaktivitet, bør have en koncentration/respons-kurve bestående af en basislinje efterfulgt af en positiv hældning, inden den ender i et plateau eller en top. I nogle tilfælde kan der kun defineres to af disse karakteristika (basislinje–hældning eller hældning–top).
|
|
—
|
Den linje, der definerer den positive hældning bør indeholde mindst tre punkter med ikke-overlappende fejlsøjler (gennemsnit ± standardafvigelse). Der ses bort fra de punkter, der danner basislinjen, men den lineære del af kurven kan omfatte toppen eller første punkt i plateauet.
|
|
—
|
En positiv klassificering kræver en responsamplitude, forskellen mellem basislinje og top, på mindst 20 % af den maksimale værdi for referencestandarden E2 (dvs. 2 000 RLU’er eller derover, når den maksimale responsværdi for referencestandarderne [E2] er justeret til 10 000 RLU’er).
|
|
—
|
Om muligt bør der beregnes en EC50-værdi for hvert positivt testkemikalie.
|
|
|
Negative
|
Den gennemsnitlige justerede RLU for en given koncentration ligger på eller under den gennemsnitlige RLU-værdi for DMSO-kontrollen plus tre gange standardafvigelsen for DMSO RLU.
|
|
Utilstrækkelige
|
Data, der ikke kan fortolkes som valide til at vise enten tilstedeværelse eller fravær af aktivitet på grund af større kvalitative eller kvantitative begrænsninger, anses for at være utilstrækkelige og kan ikke bruges til at bestemme, om testkemikaliet er positivt eller negativt. Kemikalierne bør testes igen.
|
|
|
|
ANTAGONISTAKTIVITET
|
|
Positive
|
|
—
|
Ud fra testkemikaliedata genereres en koncentration/respons-kurve bestående af en basislinje, som efterfølges af en negativ hældning.
|
|
—
|
Den linje, der definerer den negative hældning, bør indeholde mindst tre punkter med ikke-overlappende fejlsøjler. Der ses bort fra de punkter, der danner basislinjen, men den lineære del af kurven kan omfatte det første punkt i plateauet.
|
|
—
|
Der bør mindst være en 20 % reduktion i aktiviteten i forhold til den maksimale værdi for referencestandarden Ral/E2 (dvs. 8 000 RLU eller derunder, når den maksimale responsværdi for referencestandarden [Ral/E2] justeres til 10 000 RLU’er).
|
|
—
|
De højeste ikke-cytotoksiske koncentrationer af testkemikaliet bør være under eller lig med 1x10-5 M.
|
|
—
|
Om muligt bør der beregnes en IC50-værdi for hvert positivt testkemikalie.
|
|
|
Negative
|
Alle datapunkter ligger over EC80-værdi (80 % af E2-responsen eller 8 000 RLU’er) ved koncentrationer under 1,0 x 10-5 M.
|
|
Utilstrækkelige
|
Data, der ikke kan fortolkes som valide til at vise enten tilstedeværelse eller fravær af aktivitet på grund af større kvalitative eller kvantitative begrænsninger, anses for at være utilstrækkelige og kan ikke bruges til at bestemme, om testkemikaliet er positivt eller negativt. Kemikaliet bør testes igen.
|
|
40.
|
Positive resultater vil om muligt blive karakteriseret ved både effektens størrelsesorden og den koncentration, hvor effekten opstår. Eksempler på positive, negative og utilstrækkelige data er vist i figur 5 og 6.
|
Figur 5
Agonisteksempler: positive, negative og utilstrækkelige data

Den stiplede linje viser 20 % af E2-responsen, 2 000 justerede og normaliserede RLU’er.
Figur 6
Antagonisteksempler: positive, negative og utilstrækkelige data
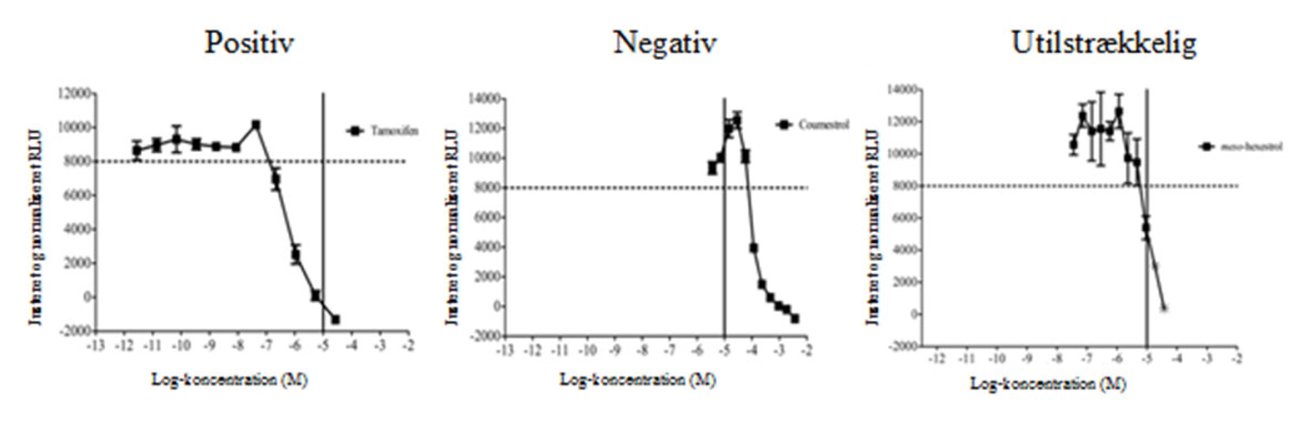
Den stiplede linje viser 80 % af Ral/E2-responsen, 8 000 justerede og normaliserede RLU’er.
Den ubrudte linje viser 1,00 x 10-5 M. For at blive anset for positiv bør en respons ligge under 8 000 RLU-linjen og ved koncentrationer under 1,00 x 10-5 M.
De koncentrationer, der er markeret med asterisk (*) på meso-hexestrol-grafen, viser levedygtighedsscorer på "2" eller derover.
Testresultater for meso-hexestrol anses for at være utilstrækkelige data, fordi den eneste respons under 8 000 RLU opstår ved 1,00 x 10-5 M.
|
41.
|
Beregningerne af EC50 og IC50 kan foretages ved hjælp af en Hill-funktion med fire parametre (se agonistprotokollen og antagonistprotokollen for yderligere oplysninger (7)). Hvis acceptkriterierne opfyldes, viser det, at systemet fungerer korrekt, men det sikrer ikke, at enhver analyseserie vil producere nøjagtige data. Kan resultaterne af den første analyseserie gentages, er det den bedste garanti for, at der blev produceret nøjagtige data (jf. punkt 19 i "ER TA-ASSAYETS KOMPONENTER").
|
TESTRAPPORT
|
42.
|
Jf. punkt 20 i "ER TA-ASSAYETS KOMPONENTER".
|
LITTERATUR
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5 367-5 373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5 941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): s. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: s. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7 850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1 459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10):4 252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. og Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Tillæg 4
TRANSAKTIVERINGSASSAY MED STABILT TRANSFEKTERET HUMAN ØSTROGENRECEPTOR-Α TIL PÅVISNING AF ØSTROGENAGONIST- OG ØSTROGENANTAGONISTAKTIVITET I KEMIKALIER, DER ANVENDER ERΑ CALUX-CELLELINJEN
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER (JF. OGSÅ GENEREL INDLEDNING)
|
1.
|
ERα CALUX-transaktiveringsassayet anvender den humane U2OS-cellelinje til at påvise østrogenagonist- og østrogenantagonistaktivitet medieret gennem human østrogenreceptor alfa (hERα). Valideringsundersøgelsen af bioassayet af det stabilt transfekterede ERα CALUX udført af BioDetection Systems BV (Amsterdam, Nederlandene) påviste relevansen og pålideligheden af assayet i overensstemmelse med dets formål (1). ERα CALUX-cellelinjen udtrykker stabilt transfekteret human ERα alene (2) (3).
|
|
2.
|
Dette assay er specifikt udformet til at påvise hERα-medieret transaktivering ved at måle bioluminescens som effektparameter. Anvendelsen af bioluminescens benyttes almindeligvis i bioassays på grund af det høje forhold mellem signal og støj (4).
|
|
3.
|
Det er rapporteret, at højere koncentrationer af fytoøstrogen end 1 μM har overaktiveret luciferasereportergenet, hvilket har medført ikke-receptormedieret luminescens (5) (6) (7). Derfor skal højere koncentrationer af fytoøstrogener eller andre lignende forbindelser, som kan overaktivere luciferaseekspression, undersøges nøje i stabilt transfekterede ER-transaktiveringsassays (jf. tillæg 2).
|
|
4.
|
Afsnittene "GENEREL INDLEDNING" og "ER TA-ASSAYETS KOMPONENTER" bør læses, før dette assay benyttes til lovmæssige formål. Definitioner og forkortelser brugt i denne forsøgsmetode er beskrevet i tillæg 1.
|
PRINCIP FOR ASSAYET (SE OGSÅ GENEREL INDLEDNING)
|
5.
|
Bioassayet benyttes til at vurdere ER-ligandbinding og efterfølgende kromosomtranslokation af receptorligandkomplekset til kernen. I kernen binder receptorligandkomplekset specifikke DNA-responselementer og transaktiverer et ildflue-luciferasereportergen, hvilket resulterer i øget cellulær ekspression af luciferaseenzymet. Efter tilsætningen af luciferasesubstratet luciferin omdannes luciferin til et bioluminescerende produkt. Det frembragte lys kan let påvises og kvantificeres ved hjælp af et luminometer.
|
|
6.
|
Testsystemet anvender stabilt transfekterede ERα CALUX-celler. ERα CALUX-celler stammer fra den humane osteoblastiske osteosarkom-U2OS-cellelinje. Humane U2OS-celler blev stabilt transfekteret med 3xHRE-TATA-Luc og pSG5-neo-hERα under anvendelse af metoden til samtidig udfældning af calciumfosfat. U2OS-cellelinjen blev valgt som den cellelinje med den største modtagelighed for østrogen (og andre steroide hormoner) baseret på den observation, at U2OS-cellelinjen kun udviste ringe eller ingen endogen receptoraktivitet. Manglen på endogene receptorer blev alene vurderet ved hjælp af luciferase-reporterplasmider, som ikke viste nogen aktivitet ved tilsætning af receptorligander. Endvidere støttede denne cellelinje stærke hormonmedierede reaktioner, når der flygtigt indførtes beslægtede receptorer (2) (3) (8).
|
|
7.
|
Test af kemikalier for østrogen- eller antiøstrogenaktivitet ved hjælp af ERα CALUX-cellelinjen omfatter en prescreening-analyseserie og omfattende analyseserier. Under prescreening-analyseserien bestemmes opløselighed, cytotoksisk effekt og et justeret koncentrationsområde for testkemikalier med henblik på omfattende testning. Under de omfattende analyseserier testes de justerede koncentrationsområder for testkemikalier i ERα CALUX-bioassays efterfulgt af klassificering af testkemikalierne for agonisme eller antagonisme.
|
|
8.
|
Kriterier for datafortolkning er nærmere beskrevet i punkt 59. Kort fortalt anses et testkemikalie for at være positivt for agonisme, såfremt mindst to på hinanden følgende koncentrationer af testkemikaliet viser en respons, som er lig med eller højere end 10 % af den maksimale respons for referencestandarden 17β-østradiol (PC10). Et testkemikalie anses for at være positivt for antagonisme, såfremt mindst to på hinanden følgende koncentrationer af testkemikaliet viser en respons, som er lig med eller lavere end 80 % af den maksimale respons for referencestandarden tamoxifen (PC80).
|
FREMGANGSMÅDE
Cellelinjer
|
9.
|
Den stabilt transfekterede U2OS ERα CALUX-cellelinje bør anvendes til assayet. Cellelinjen kan fås fra BioDetection Systems BV, Amsterdam, Nederlandene med en teknisk licensaftale.
|
|
10.
|
Der bør kun anvendes mykoplasmafrie cellekulturer. De cellebatcher, der anvendes, bør enten være certificeret negative for mykoplasmakontaminering, eller der bør foretages en mykoplasmatest før brug. RT-PCR (realtidspolymerasekædereaktion) bør anvendes til sensitiv påvisning af mykoplasmainfektion (9).
|
Cellelinjens stabilitet
|
11.
|
For at bevare CALUX-cellernes stabilitet og integritet bør cellerne opbevares i flydende nitrogen (-80 °C). Efter optøning af cellerne med henblik på start af en ny kultur bør cellerne dyrkes i subkultur mindst to gange, før de anvendes til at vurdere kemikaliernes østrogene agonist- og antagonistaktivitet. Celler bør ikke dyrkes i subkultur i mere end 30 generationer.
|
|
12.
|
For at overvåge cellelinjens stabilitet over tid bør det agonistiske og antagonistiske testsystems reaktionsevne kontrolleres ved at vurdere EC50- eller IC50-referencestandarden. Endvidere bør den relative induktion af den positive (PC) og negative kontrol (NC) overvåges. Resultaterne bør være i overensstemmelse med acceptkriterierne for det agonistiske (tabel 3C) eller antagonistiske ERα CALUX-bioassay (tabel 4C). Referencestandarder, positive og negative kontroller er anført i tabel 1 og tabel 2 for den henholdsvis agonistiske og antagonistiske måde.
|
Celledyrknings- og udpladningsbetingelser
|
13.
|
U2OS-celler dyrkes i dyrkningsmedium (DMEM/F12 (1:1) med fenolrød som pH-indikator suppleret med føtalt kalveserum (7,5 %), ikke-essentielle aminosyrer (1 %), 10 enheder/ml penicillin, streptomycin og geneticin (G-418) som selektionsmarkør). Cellerne anbringes i en CO2-inkubator (5 % CO2) ved 37 °C og 100 % fugtighed. Når cellerne når en konfluens på 85-95 %, bør de enten dyrkes i subkultur eller forberedes til udsåning på mikrotiterplader med 96 brønde. I sidstnævnte tilfælde bør cellerne resuspenderes med 1x105 celler/ml i østrogenfrit assaymedium (DMEM/F12 (1:1) medium uden fenolrød, suppleret med føtalt kalveserum behandlet med dextranovertrukket trækul (5 % (v/v)), ikke-essentielle aminosyrer (1 % (v/v)), 10 enheder/ml penicillin og streptomycin) og udplades i mikrotiterplader med 96 brønde (100 μl homogeniseret cellesuspension). Celler bør præinkuberes i en CO2-inkubator (5 % CO2, 370C, 100 % fugtighed) i 24 timer forud for eksponeringen. Plastudstyr skal være østrogenfrit.
|
Acceptkriterier
|
14.
|
Agonist- og antagonistaktiviteter i testkemikaliet/-kemikalierne testes i testserier. En testserie består af maksimalt seks mikrotiterplader. Hver testserie indeholder mindst én fuld serie af opløsninger af en referencestandard, en positiv kontrol, en negativ kontrol og kontrolkulturer med opløsningsmiddel. Figur 1 og 2 viser pladeopstillingen for agonist- og antagonist-testserier.
|
|
15.
|
Der bør foretages tredobbelt analyse af hver fortynding af referencestandarder, testkemikalier, alle kontrolkulturer med opløsningsmiddel samt positive og negative kontroller. Hver af de tredobbelte analyser skal opfylde kravene i tabel 3A og tabel 4A.
|
|
16.
|
En fuldstændig serie af fortyndinger af referencestandarden (17β-østradiol for agonisme, tamoxifen for antagonisme) måles på den første plade i hver testserie. For at kunne sammenligne analyseresultaterne for de resterende fem mikrotiterplader med den første mikrotiterplade, som indeholder den fuldstændige koncentration/respons-kurve for referencestandarden, skal alle plader indeholde tre kontroller: kontrolkultur med opløsningsmiddel, den højeste koncentration af den testede referencestandard og referencestandardens omtrentlige EC50- (agonisme) eller IC50- (antagonisme) koncentration. Forholdet mellem de gennemsnitlige kontroller på den første plade og de resterende fem plader bør opfylde kravene i tabel 3C (agonisme) eller tabel 4C (antagonisme).
|
|
17.
|
For hver af mikrotiterpladerne inden for en testserie beregnes z-faktoren (10). z-faktoren beregnes ved hjælp af reaktionerne ved den højeste og den laveste koncentration af referencestandarden. En mikrotiterplade anses for gyldig, såfremt den opfylder kravene i tabel 3C (agonisme) eller tabel 4C (antagonisme).
|
|
18.
|
Referencestandarden bør påvise en sigmoidal dosis/respons-kurve. EC50 eller IC50 udledt af responsen fra serien af fortyndinger af referencestandarden skal opfylde kravene i tabel 3C (agonisme) eller tabel 4C (antagonisme).
|
|
19.
|
Hver testserie bør indeholde en positiv og en negativ kontrol. Den beregnede relative induktion af både den positive og negative kontrol bør opfylde kravene i tabel 3C (agonisme) eller tabel 4C (antagonisme).
|
|
20.
|
Under alle målinger bør induktionsfaktoren for den højeste koncentration af referencestandarden måles ved at dividere den gennemsnitlige højeste relative lysenhedsrespons (RLU) for referencestandarden 17β-østradiol med den gennemsnitlige RLU-respons for referencekontrolkulturen med opløsningsmiddel. Denne induktionsfaktor skal opfylde mindstekravene til foldinduktionen som anført i tabel 3C (agonisme) eller tabel 4C (antagonisme).
|
|
21.
|
Kun mikrotiterplader, der opfylder alle de ovennævnte acceptkriterier, betragtes som gyldige og kan anvendes til at vurdere responsen af testkemikalierne.
|
|
22.
|
Acceptkriterierne finder anvendelse på både prescreening-analyseserie og omfattende analyseserier.
|
Tabel 1
Koncentrationer af referencestandard, positiv kontrol (PC) og negativ kontrol (NC) for det agonistiske CALUX-bioassay
|
|
Stof
|
CAS RN
|
Måleområde (M)
|
|
Referencestandard
|
17β-østradiol
|
50-28-2
|
1*10-13 - 1*10-10
|
|
Positiv kontrol (PC)
|
17α-methyltestosteron
|
58-18-4
|
3*10-6
|
|
Negativ kontrol (NC)
|
corticosteron
|
50-22-6
|
1*10-8
|
Tabel 2
Koncentrationer af referencestandard, positiv kontrol (PC) og negativ kontrol (NC) for det antagonistiske CALUX-bioassay
|
|
Stof
|
CAS RN
|
Måleområde (M)
|
|
Referencestandard
|
tamoxifen
|
10540-29-1
|
3*10-9 - 1*10-5
|
|
Positiv kontrol (PC)
|
4-hydroxytamoxifen
|
68047-06-3
|
1*10-9
|
|
Negativ kontrol (NC)
|
resveratrol
|
501-36-0
|
1*10-5
|
Tabel 3
Acceptkriterier for det agonistiske ERα CALUX-bioassay
|
A – individuelle prøver på en plade
|
Kriterium
|
|
1
|
Maksimal procentvis standardafvigelse for tredobbelte brønde (til NC, PC, hver fortynding af testkemikaliet og referencestandarden, undtagen C0)
|
< 15 %
|
|
2
|
Maksimal procentvis standardafvigelse for tredobbelte brønde (til kontrolkultur med opløsningsmiddel for referencestandard og testkemikalie (C0, SC))
|
< 30 %
|
|
3
|
Maksimal LDH-udsivning som et mål for cytotoksisk effekt.
|
< 120 %
|
|
B - inden for en enkelt mikrotiterplade
|
|
|
4
|
Forholdet mellem kontrolkulturen med opløsningsmiddel for referencestandarden (C0, plade 1) og kontrolkulturen med opløsningsmiddel for testkemikaliet (SC, plade 2 til x)
|
0,5 til 2,0
|
|
5
|
Forholdet mellem den omtrentlige EC50 og højeste referencestandardkoncentrationer på plade 1 og den omtrentlige EC50 og højeste referencestandardkoncentrationer på plade 2 til x (C4, C8)
|
0,70 til 1,30
|
|
6
|
Z-faktor for hver plade
|
> 0,6
|
|
C - inden for en enkelt analyseserie (alle plader inden for én serie)
|
|
|
7
|
Sigmoidal kurve for referencestandarden
|
Ja (17ß-østradiol)
|
|
8
|
EC50-niveau for referencestandard 17ß-østradiol
|
4*10-12 – 4*10-11 M
|
|
9
|
Minimumsfoldinduktion for den højeste koncentration af 17ß-østradiol i forhold til kontrolkulturen med opløsningsmiddel for referencestandarden.
|
5
|
|
10
|
Relativ induktion (%) PC
|
> 30 %
|
|
11
|
Relativ induktion (%) NC
|
< 10 %
|
Omtr.: omtrentlig, PC: positiv kontrol, NC: negativ kontrol, SC: kontrolkultur med opløsningsmiddel for testkemikalie, C0: kontrolkultur med opløsningsmiddel for referencestandard, Standardafvigelse: standardafvigelse, LDH: laktatdehydrogenase.
Tabel 4
Acceptkriterier for det antagonistiske ERα CALUX-bioassay
|
A – individuelle prøver på en plade
|
Kriterium
|
|
1
|
Maksimal procentvis standardafvigelse for tredobbelte brønde (til NC, PC, hver fortynding af testkemikaliet og referencestandarden, kontrolkultur med opløsningsmiddel (C0))
|
< 15 %
|
|
2
|
Maximal procentvis standardafvigelse for tredobbelte brønde (til bærestofkontrol (VC) og højeste referencestandardkoncentration (C8))
|
< 30 %
|
|
3
|
Maksimal LDH-udsivning som et mål for cytotoksisk effekt.
|
< 120 %
|
|
B - inden for en enkelt mikrotiterplade
|
|
|
4
|
Forholdet mellem kontrolkulturen med opløsningsmiddel for referencestandarden (C0, plade 1) og kontrolkulturen med opløsningsmiddel for testkemikaliet (SC, plade 2 til x)
|
0,70 til 1,30
|
|
5
|
Forholdet mellem den omtrentlige IC50-referencestandardkoncentrationer på plade 1 og de omtr. IC50-referencestandardkoncentrationer på plade 2 til x (C4)
|
0,70 til 1,30
|
|
6
|
Forholdet mellem de højeste referencestandardkoncentrationer på plade 1 og de højeste referencestandardkoncentrationer på plade 2 til x (C8)
|
0,50 til 2,0
|
|
7
|
Z-faktor for hver plade
|
> 0,6
|
|
C - inden for en enkelt analyseserie (alle plader inden for én serie)
|
|
|
8
|
Sigmoidal kurve for referencestandarden
|
Ja (tamoxifen)
|
|
9
|
IC50-niveau for referencestandard (tamoxifen)
|
1*10-8 - 1*10-7 M
|
|
10
|
Minimumsfoldinduktion for kontrolkultur med opløsningsmiddel for referencestandarden i forhold til den højeste koncentration af tamoxifen
|
2,5
|
|
11
|
Relativ induktion (%) PC
|
< 70 %
|
|
12
|
Relativ induktion (%) NC
|
> 85 %
|
Omtr.: omtrentlig, PC: positiv kontrol, NC: negativ kontrol, VC: bærestofkontrol (kontrolkultur med opløsningsmiddel uden fast koncentration af agonistreferencestandard), SC: kontrolkultur med opløsningsmiddel for testkemikalie, C0: kontrolkultur med opløsningsmiddel for referencestandard, Standardafvigelse: standardafvigelse, LDH: laktatdehydrogenase.
Kontrolkultur med opløsningsmiddel/bærestof, referencestandarder, positive kontroller, negative kontroller
|
23.
|
For både prescreening-analyseserien og de omfattende analyseserier bør der anvendes den samme kontrolkultur med opløsningsmiddel/bærestof og de samme referencestandarder, positive kontroller og negative kontroller. Endvidere bør koncentrationen for referencestandarder, positive kontroller og negative kontroller være de samme.
|
Kontrolkultur med opløsningsmiddel
|
24.
|
Det opløsningsmiddel, der anvendes til at opløse testkemikalierne, bør testes som en kontrolkultur med opløsningsmiddel. Dimethylsulfoxid (DMSO, 1 % (v/v), CASRN 67-68-5) blev anvendt som bærestof under valideringen af ERα CALUX-bioassayet. Hvis der anvendes et andet opløsningsmiddel end DMSO, testes alle referencestandarder, kontroller og testkemikalier i det samme bærestof. Bemærk, at kontrolkulturen med opløsningsmiddel til antagonistiske undersøgelser indeholder en fast koncentration af agonistreferencestandarden 17β-østradiol (omtrentlig EC50-koncentration). Til test af det opløsningsmiddel, der skal anvendes til antagonistiske undersøgelser, fremstilles og testes en bærestofkontrol.
|
Bærestofkontrol (antagonisme)
|
25.
|
Til test for antagonisme suppleres assaymediet med en fast koncentration af agonistreferencestandarden 17β-østradiol (omtrentlig EC50-koncentration). Til test af det opløsningsmiddel, som anvendes til at opløse testkemikalierne for antagonisme, fremstilles et assaymedium uden en fast koncentration af agonistreferencestandarden 17β-østradiol. Denne kontrol anføres som bærestofkontrol. Dimethylsulfoxid (DMSO, 1 % (v/v), CASRN 67-68-5) blev anvendt som bærestof under valideringen af ERα CALUX-bioassayet. Hvis der anvendes et andet opløsningsmiddel end DMSO, testes alle referencestandarder, kontroller og testkemikalier i det samme bærestof.
|
Referencestandarder
|
26.
|
Den agonistiske referencestandard er 17β-østradiol (tabel 1). Referencestandarderne omfatter en række fortyndinger med otte koncentrationer af 17β-østradiol (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11, 1*10-10 M).
|
|
27.
|
Den antagonistiske referencestandard er tamoxifen (tabel 2). Referencestandarderne omfatter en række fortyndinger med otte koncentrationer af tamoxifen (3*10-9, 1*10-8, 3*10-8, 1*10-7, 3*10-7, 1*10-6, 3*10-6, 1*10-5 M). Hver af koncentrationerne af den antagonistiske referencestandard inkuberes sammen med en fast koncentration af den agonistiske referencestandard 17β-østradiol (3*10-12 M).
|
Positiv kontrol
|
28.
|
Den positive kontrol for agonistiske undersøgelser er 17α-methyltestosteron (tabel 1).
|
|
29.
|
Den positive kontrol for antagonistiske undersøgelser er 4-hydroxytamoxifen (tabel 2). Den antagonistiske positive kontrol inkuberes sammen med en fast koncentration af den agonistiske referencestandard 17β-østradiol (3*10-12 M).
|
Negativ kontrol
|
30.
|
Den negative kontrol for agonistiske undersøgelser er corticosteron (tabel 1).
|
|
31.
|
Den negative kontrol for antagonistiske undersøgelser er resveratrol (tabel 2). Den negative kontrol inkuberes sammen med en fast koncentration af den agonistiske referencestandard 17β-østradiol (3*10-12 M).
|
Påvisning af laboratoriets kompetence (jf. punkt 14 og tabel 3 og 4 i "ER TA-ASSAYETS KOMPONENTER" for denne forsøgsmetode)
Bærestof
|
32.
|
Det opløsningsmiddel, der anvendes til at opløse testkemikalier, skal opløse testkemikaliet fuldstændigt og skal kunne blandes med cellemediet. DMSO, vand og ethanol (95-100 % renhed) er egnede opløsningsmidler. Såfremt DMSO anvendes som opløsningsmiddel bør den maksimale koncentration af DMSO under inkubationen ikke overstige 1 % (v/v). Før brug testes opløsningsmidlet for manglende cytotoksisk effekt og interferens med assayets ydeevne.
|
Fremstilling af referencestandarder, positive kontroller, negative kontroller og testkemikalier
|
33.
|
Referencestandarder, positive kontroller, negative kontroller og testkemikalier opløses i 100 % DMSO (eller et egnet opløsningsmiddel). Egnede (serie-) fortyndinger fremstilles herefter i det samme opløsningsmiddel. Lad alle stoffer ækvilibrere til stuetemperatur, før de opløses. Frisk fremstillede stamopløsninger af referencestandarder, positive kontroller, negative kontroller og testkemikalier bør ikke have udtalt bundfald eller uklarhed. Referencestandard- og kontrollagre kan fremstilles i større mængder. Stamopløsninger af testkemikalier fremstilles frisk før hvert forsøg. Endelige fortyndinger af referencestandarder, positive kontroller, negative kontroller og testkemikalier fremstilles frisk til hvert forsøg og anvendes inden for 24 timer efter fremstillingen.
|
Opløselighed, cytotoksisk effekt og bestemmelse af dosisinterval
|
34.
|
Under prescreening-analyseserien bestemmes testkemikaliernes opløselighed i det valgte opløsningsmiddel. Der fremstilles en maksimum-stamkoncentration på 0,1 M. Såfremt denne koncentration giver opløselighedsproblemer, fremstilles der lavere stamopløsninger, indtil testkemikalierne er fuldt opløst. Under prescreening-analyseserien testes seriefortyndinger af testkemikaliet i forholdet 1:10. Den maksimale assaykoncentration til agonist- og antagonisttestning er 1 mM. Efter prescreeningen udledes et passende justeret koncentrationsområde til testkemikalier, som testes under de omfattende analyseserier. De opløsninger, der anvendes til omfattende testning, bør være 1x, 3x, 10x, 30x, 100x, 300x, 1000x og 3000x.
|
|
35.
|
Testning for cytotoksisk effekt er inkluderet i protokollen for agonist- og antagonistassays (11). Testning for cytotoksisk effekt er indarbejdet i både prescreening-analyseserien og i omfattende analyseserier. Den metode, som blev benyttet til at vurdere cytotoksisk effekt under valideringen af ERα CALUX-bioassayet, var udsivningstesten for laktatdehydrogenase (LDH) kombineret med kvalitativ visuel cellekontrol (jf. tillæg 4.1) efter eksponering for testkemikalier. Andre kvantitative metoder kan dog benyttes til bestemmelse af cytotoksisk effekt (f.eks. tetrazolium-baseret kolorimetrisk (MTT) assay eller cytotoksicitets-CALUX bioassay). Generelt betragtes testkemikaliekoncentrationer, der udviser mere end 20 % reduktion i cellernes levedygtighed, som cytotoksiske og kan derfor ikke anvendes til datavurdering. Med hensyn til LDH-udsivningsassayet betragtes koncentrationen af testkemikaliet som cytotoksisk, når den procentvise LDH-udsivning er højere end 120 %.
|
Eksponering for testkemikalie og organisering af assaypladen
|
36.
|
Efter trypsinering af en konfluerende kolbe med dyrkede celler genopslæmmes cellerne med 1x105 celler/ml i østrogenfrit assaymedium. 100 μl genopslæmmede celler udplades i de inderste brønde på en mikrotiterplade med 96 brønde. De yderste brønde fyldes med 200 μl phosphatbufret fysiologisk saltopløsning (PBS) (jf. figur 1 og 2). De udpladede celler præinkuberes i 24 timer i en CO2-inkubator (5 % CO2, 37 °C, 100 % fugtighed).
|
|
37.
|
Efter præinkubation kontrolleres pladerne for visuel cytotoksisk effekt (jf. tillæg 4.1), kontaminering og konfluens. Kun plader, som ikke udviser visuel cytotoksisk effekt og kontaminering og har en konfluens på minimum 85 %, anvendes til test. Mediet fra de inderste brønde fjernes omhyggeligt og erstattes af 200 μl østrogenfrit assaymedium, som indeholder egnede fortyndingsserier af referencestandarder, testkemikalier, positive kontroller, negativekontroller og kontrolkulturer med opløsningsmiddel (tabel 5: agonistundersøgelser, Tabel 6: antagonistundersøgelser). Alle referencestandarder, testkemikalier, positive kontroller, negative kontroller og kontrolkulturer med opløsningsmiddel testes in triplo. I figur 1 angives pladelayoutet til agonisttestning. I figur 2 angives pladelayoutet til antagonisttestning. Pladelayoutet til prescreening-testning og omfattende testning er identisk. Til antagonisttestning indeholder alle inderste brønde undtagen brøndene til bærestofkontrol (VC) også en fast koncentration af agonistreferencestandard 17β-østradiol (3*10-12 M). Bemærk, at referencestandarderne C8 og C4 tilsættes til hver TC-plade.
|
|
38.
|
Efter eksponering af cellerne for alle kemikalier inkuberes mikrotiterpladerne med 96 brønde i endnu 24 timer i en CO2-inkubator (5 % CO2, 37 °C, 100 % fugtighed).
|
Figur 1
Pladelayout for mikrotiterplader med 96 brønde til prescreening og vurdering af agonisteffekt.

C0 = referencestandard-opløsningsmiddel
C(1-8) = fortyndingsserier (1-8, lave til høje koncentrationer) af referencestandarden
PC = positiv kontrol
NC = negativ kontrol
TCx-(1-8) = fortyndinger (1-8, lave til høje koncentrationer) af testkemikalie til prescreening-analyseserie og vurdering af agonistisk effekt af testkemikalie x
SC = kontrolkultur med opløsningsmiddel for testkemikaliet (optimalt det samme opløsningsmiddel som i C0, men eventuelt fra en anden batch).
Grå felter: = Yderste brønde fyldt op med 200 μl PBS.
Figur 2
Pladelayout for mikrotiterplader med 96 brønde til antagonistisk prescreening og vurdering af antagonistisk effekt.

C0 = referencestandard-opløsningsmiddel
C(1-8) = fortyndingsserier (1-8, lave til høje koncentrationer) af referencestandarden
NC = negativ kontrol
PC = positiv kontrol
TCx-(1-8) = fortyndinger (1-8, lave til høje koncentrationer) af testkemikalie til prescreening-analyseserie og vurdering af agonistisk effekt af testkemikalie x
SC = kontrolkultur med opløsningsmiddel for testkemikaliet (optimalt det samme opløsningsmiddel som i C0, men eventuelt fra en anden batch).
VC = bærestofkontrol (kontrolkultur med opløsningsmiddel uden fast koncentration af agonistreferencestandard 17β-østradiol).
Grå felter: = Yderste brønde fyldt op med 200 μl PBS.
Bemærk: Alle inderste brønde undtagen brøndene til bærestofkontrol (VC) indeholder også en fast koncentration af agonistreferencestandard 17β-østradiol (3,0*10-12 M).
Måling af luminescens
|
39.
|
Måling af luminescens er nærmere beskrevet i agonist- og antagonistassayprotokollen (10). Dyrkningsmediet fra brøndene bør fjernes, og cellerne lyseres efterfulgt af 24 timers inkubation for at åbne cellemembranen, så luciferaseaktiviteten kan måles.
|
|
40.
|
Til måling af luminiscensen kræves et luminometer udstyret med to injektorer. Luciferasereaktionen startes med injektion af substratet luciferin. Reaktionen standes ved tilsætning af 0,2 M NaOH. Reaktionen standses for at forhindre, at der overføres luminescens fra én brønd til en anden.
|
|
41.
|
Lys, der udsendes fra hver brønd, angives som relative lysenheder (RLU’er) pr. brønd.
|
Prescreening-analyseserie
|
42.
|
Resultaterne af prescreening-analysen bruges til at bestemme et justeret koncentrationsområde for testkemikalier med henblik på omfattende testning. Vurderingen af resultaterne af prescreening-analysen og bestemmelsen af det justerede koncentrationsområde for testkemikalier med henblik på omfattende testning beskrives indgående i agonist- og antagonistassayprotokollen (10). Her anføres et kort resumé af procedurerne for bestemmelse af koncentrationsområdet for testkemikalier til agonist- og antagonisttestning. Jf. retningslinjer for udformning af seriefortynding i tabel 5 og 6.
|
Valg af koncentrationer til vurdering af agonistiske virkninger
|
43.
|
Under prescreening-analyseserien testes testkemikalierne ved hjælp af serier af fortyndinger som anført i tabel 5 (agonisme) og 6 (antagonisme). Alle koncentrationer testes i tredobbelte brønde i overensstemmelse med pladelayoutet i figur 1 (agonisme) eller 2 (antagonisme).
|
|
44.
|
Kun analyseresultater, der opfylder acceptkriterierne (tabel 3), betragtes som gyldige og kan anvendes til at vurdere responsen af testkemikalierne. Såfremt en eller flere mikrotiterplader i analyseserien ikke opfylder acceptkriterierne, analyseres de pågældende mikrotiterplader igen. I tilfælde af at den første plade med den fuldstændige serie af fortyndinger af referencestandarden ikke opfylder acceptkriterierne, skal hele testserien (seks plader) analyseres igen.
|
|
45.
|
De indledende koncentrationsområder for testkemikalier bør justeres, og prescreening-analyseserien gentages, såfremt:
|
—
|
der observeres cytotoksisk effekt. Prescreening-proceduren gentages med lavere ikke-cytotoksiske koncentrationer af testkemikaliet
|
|
—
|
prescreeningen af testkemikaliet ikke udviser en fuld dosis/respons-kurve, fordi de testede koncentrationer genererer maksimal induktion. Prescreening-analyseserien gentages under anvendelse af lavere koncentrationer af testkemikaliet.
|
|
|
46.
|
Når der observeres et gyldigt dosis/effekt-forhold, vælges den (laveste) koncentration, hvor der observeres maksimal induktion uden cytotoksisk effekt. Den højeste koncentration af det testkemikalie, som skal testes i omfattende analyseserier, bør være tre gange denne valgte koncentration.
|
|
47.
|
En fuldstændig justeret fortyndingsserie med testkemikaliet fremstilles med fortyndingstrin som anført i tabel 5, idet der startes med den højeste koncentration som fastsat ovenfor.
|
|
48.
|
Et testkemikalie, som ikke fremkalder nogen agonistisk virkning, testes i omfattende analyseserier, der starter med den højeste ikke-cytotoksiske koncentration, som identificeres under prescreeningen.
|
Valg af koncentrationer til vurdering af antagonistiske virkninger
|
49.
|
Kun analyseresultater, der opfylder acceptkriterierne (tabel 4), betragtes som gyldige og kan anvendes til at vurdere testkemikaliernes respons. Såfremt en eller flere mikrotiterplader i analyseserien ikke opfylder acceptkriterierne, analyseres de pågældende mikrotiterplader igen. I tilfælde af at den første plade med den fuldstændige serie af fortyndinger af referencestandarden ikke opfylder acceptkriterierne, skal hele testserien (seks plader) analyseres igen.
|
|
50.
|
De indledende koncentrationsområder for testkemikalier bør justeres, og prescreening-analyseserien gentages, såfremt:
|
—
|
der observeres cytotoksisk effekt. Prescreening-proceduren gentages med lavere ikke-cytotoksiske koncentrationer af testkemikaliet
|
|
—
|
prescreeningen af testkemikaliet ikke udviser en fuld dosis/respons-kurve, fordi de testede koncentrationer genererer maksimal inhibering. Prescreeningen gentages under anvendelse af lavere koncentrationer af testkemikaliet.
|
|
|
51.
|
Når der konstateres et gyldigt dosis/effekt-forhold, vælges den (laveste) koncentration, hvor der observeres maksimal inhibering uden cytotoksisk effekt. Den højeste koncentration af det testkemikalie, som skal testes i omfattende analyseserier, bør være tre gange denne valgte koncentration.
|
|
52.
|
En fuldstændig justeret fortyndingsserie af testkemikaliet fremstilles med fortyndingstrin som anført i tabel 6, idet der startes med den højeste koncentration som bestemt ovenfor.
|
|
53.
|
Testkemikalier, som ikke fremkalder nogen antagonistiske virkninger, testes i omfattende analyseserier, der starter med den højeste ikke-cytotoksiske koncentration, som er testet under prescreeningen.
|
Omfattende analyseserier
|
54.
|
Efter valg af de justerede koncentrationsområder testes testkemikalierne grundigt ved hjælp af fortyndingsserierne i tabel 5 (agonisme) og 6 (antagonisme). Alle koncentrationer testes i tredobbelte brønde i overensstemmelse med pladelayoutet i figur 1 (agonisme) eller 2 (antagonisme).
|
|
55.
|
Kun analyseresultater, der opfylder acceptkriterierne (tabel 3 og 4), betragtes som gyldige og kan anvendes til at vurdere testkemikaliernes respons. Såfremt en eller flere mikrotiterplader i analyseserien ikke opfylder acceptkriterierne, analyseres de pågældende mikrotiterplader igen. I tilfælde af at den første plade med den fuldstændige serie af fortyndinger af referencestandarden ikke opfylder acceptkriterierne, skal hele testserien (seks plader) analyseres igen.
|
Tabel 5
Koncentration og fortyndinger af referencestandarder, kontroller og testkemikalier, som anvendes til agonisttestning
|
Reference-17β-østradiol
|
TCx – prescreening-analyseserie
|
TCx – omfattende analyseserie
|
Kontroller
|
|
konc. (M)
|
fortynding
|
fortynding
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10000000 x
|
TCx-1
|
3000 x
|
PC
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1000000 x
|
TCx-2
|
1000 x
|
NC
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx - testkemikalie x
PC - positiv kontrol (17α-methyltestosteron)
NC - negativ kontrol (corticosteron)
C0 - kontrolkultur med opløsningsmiddel efter referencestandard
SC - kontrolkultur med opløsningsmiddel for testkemikalie
|
Tabel 6
Koncentration og fortyndinger af referencestandarder, kontroller og testkemikalier, som anvendes til antagonisttestning
|
Referencetamoxifen
|
TCx – prescreening-analyseserie
|
TCx – omfattende analyseserie
|
Kontroller
|
|
konc. (M)
|
fortynding
|
fortynding
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10000000 x
|
TCx-1
|
3000 x
|
PC
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1000000 x
|
TCx-2
|
1000 x
|
NC
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Suppleret agonist
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
konc. (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-østradiol
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx - testkemikalie x
PC - positiv kontrol (4-hydroxytamoxifen)
NC - negativ kontrol (resveratrol)
C0 - kontrolkultur med opløsningsmiddel efter referencestandard
SC - kontrolkultur med opløsningsmiddel for testkemikalie
VC - bærestofkontrol (indeholder ikke fast koncentration af agonistreferencestandarden 17β-østradiol (3,0*10-12 M).
|
Indsamling af data og dataanalyse
|
56.
|
Efter prescreening og omfattende analyseserier bestemmes EC10, EC50, PC10, PC50 og maksimal induktion (TCxmaks.) for et testkemikalie til agonistisk testning. Til antagonistisk testning beregnes IC20, IC50, PC80, PC50 og minimumsinduktion (TCxmin.). I figur 3 (agonisme) og 4 (antagonisme) er disse parametre gengivet grafisk. De nødvendige parametre beregnes på grundlag af den relative induktion for hvert testkemikalie (i forhold til referencestandardens maksimale induktion (=100 %). Ikke-lineær regression (variabel hældning, 4 parametre) anvendes til vurdering af data i overensstemmelse med følgende ligning:
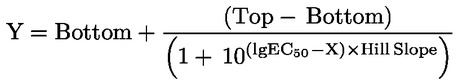
hvor:
X = Logaritmen af dosis eller koncentration
Y = Respons (relativ induktion (%))
Top = Maksimumsinduktion (%)
Bund = minimumsinduktion (%)
LogEC50 = Logaritmen af koncentrationen, hvor der observeres 50 % af den maksimale respons.
Hills kurve = hældningsfaktor eller Hills kurve
|
|
57.
|
Rådata fra luminometeret angivet som relative lysenheder (RLU’er) overføres til det regneark til dataanalyse, som er udformet til prescreening og omfattende analyseserier. Rådata bør opfylde acceptkriterierne i tabel 3A og 3B (agonisme) eller 4A og 4B (antagonisme). Såfremt rådataene opfylder acceptkriterierne, udføres følgende beregningstrin for at bestemme de nødvendige parametre:
|
Agonisme
|
—
|
Træk den gennemsnitlige RLU for kontrollen af referencestandarden med opløsningsmiddel fra hver af referencestandardernes råanalysedata.
|
|
—
|
Træk den gennemsnitlige RLU for kontrolkulturen med opløsningsmiddel for testkemikaliet fra hver af testkemikaliernes råanalysedata.
|
|
—
|
Beregn den relative induktion for hver koncentration af referencestandarden. Sæt induktionen for den højeste koncentration af referencestandarden til 100 %.
|
|
—
|
Beregn den relative induktion for hver koncentration af testkemikaliet sammenlignet med den højeste koncentration af referencestandarden til 100 %.
|
|
—
|
Vurder analyseresultaterne efter ikke-lineær regression (variabel hældning, 4 parametre).
|
|
—
|
Bestem EC50 og EC10 for referencestandarden.
|
|
—
|
Bestem EC50 og EC10 for testkemikalierne.
|
|
—
|
Bestem den relative maksimumsinduktion for testkemikaliet (TCmaks.).
|
|
—
|
Bestem PC10 og PC50 for testkemikalierne.
|
For testkemikalier kan der ikke altid opnås en fuld dosis/respons-kurve, f.eks. på grund af cytotoksicitets- eller opløselighedsproblemer. Derfor kan EC50, EC10 og PC50 ikke bestemmes. I så fald kan kun PC10 og TCmaks. bestemmes.
Antagonisme
|
—
|
Træk den gennemsnitlige RLU for den højeste referencestandardkoncentration fra hver af referencestandardernes råanalysedata.
|
|
—
|
Træk den gennemsnitlige RLU for den højeste referencestandardkoncentration fra hver af testkemikaliernes råanalysedata.
|
|
—
|
Beregn den relative induktion for hver koncentration af referencestandarden. Sæt induktionen for den laveste koncentration af referencestandarden til 100 %.
|
|
—
|
Beregn den relative induktion for hver koncentration af testkemikaliet sammenlignet med den laveste koncentration af referencestandarden til 100 %.
|
|
—
|
Vurder analyseresultaterne efter ikke-lineær regression (variabel hældning, 4 parametre).
|
|
—
|
Bestem IC50 og IC20 for referencestandarden.
|
|
—
|
Bestem IC50 og IC20 for testkemikalierne.
|
|
—
|
Bestem den relative minimumsinduktion for testkemikaliet (TCmin.).
|
|
—
|
Bestem PC80 og PC50 for testkemikalierne.
|
Figur 3
Oversigt over parametre bestemt i agonistassayet
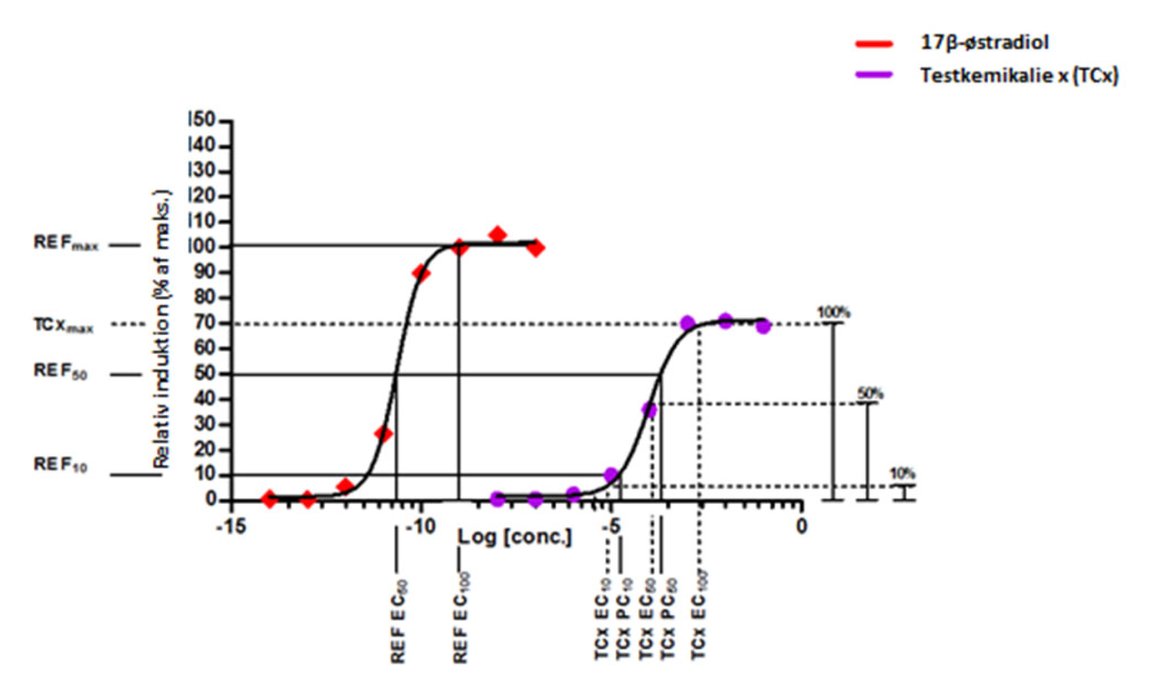
EC10 = koncentration af et stof, hvor der observeres 10 % af dets maksimale respons
EC50 = koncentration af et stof, hvor der observeres 50 % af dets maksimale respons
PC10 = koncentration af et testkemikalie, hvis respons er lig med EC10 af referencestandarden
PC50 = koncentration af et testkemikalie, hvis respons er lig med EC50 af referencestandarden.
TCxmaks. = maksimal relativ induktion af testkemikalie.
Figur 4
Oversigt over parametre bestemt i antagonistassayet
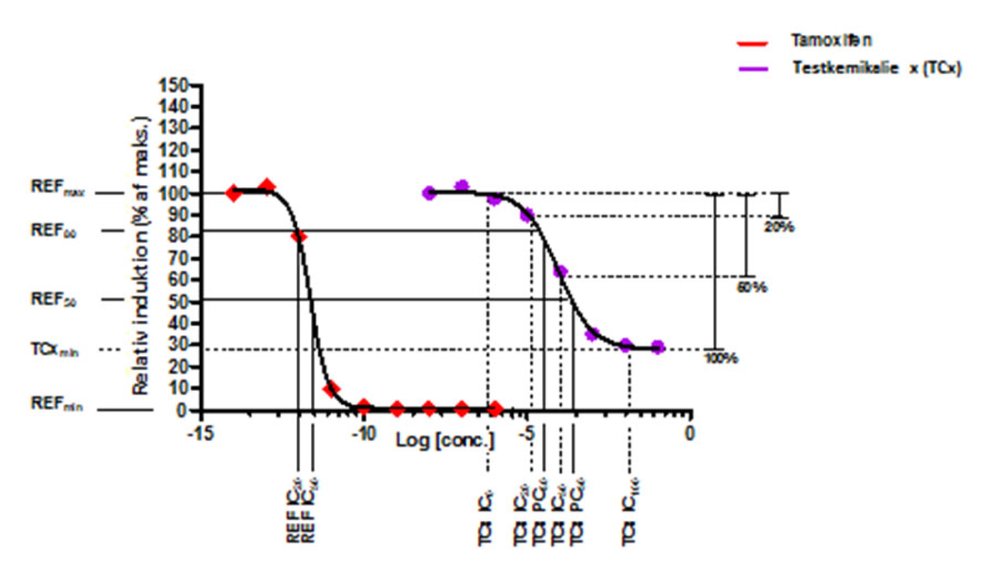
IC20 = koncentration af et stof, hvor der observeres 80 % af dets maksimale respons (20 % inhibering).
IC50 = koncentration af et stof, hvor der observeres 50 % af dets maksimale respons (50 % inhibering).
PC80 = koncentration af et testkemikalie, hvis respons er lig med IC20 af referencestandarden
PC50 = koncentration af et testkemikalie, hvis respons er lig med IC50 af referencestandarden
TCxmin. = relativ minimumsinduktion af testkemikalie.
For testkemikalier kan der ikke altid opnås en fuld dosis/respons-kurve, f.eks. på grund af cytotoksicitets- eller opløselighedsproblemer. Derfor kan IC50, IC20 og PC50 ikke bestemmes. I så fald kan kun PC20 og TCmin. bestemmes.
|
58.
|
Resultaterne bør baseres på to (eller tre) uafhængige analyseserier. Hvis to analyseserier giver sammenlignelige og derfor reproducerbare resultater, er det ikke nødvendigt at gennemføre en tredje analyseserie. For at resultaterne kan godkendes, skal de:
|
—
|
opfylde acceptkriterierne (se Acceptkriterier, punkt 14-22)
|
|
Kriterier for datafortolkning
|
59.
|
For at fortolke data og afgøre, om et testkemikalie anses for positivt eller negativt, anvendes følgende kriterier:
|
For hver omfattende analyseserie anses et testkemikalie for positivt, såfremt:
|
1
|
TCmaks. er lig med eller overstiger 10 % af referencestandardens maksimale respons (REF10).
|
|
2
|
Mindst to på hinanden følgende koncentrationer af testkemikaliet er lig med eller overstiger REF10.
|
|
|
For hver omfattende analyseserie anses et testkemikalie for negativt, såfremt:
|
1
|
TCmaks ikke overstiger 10 % af referencestandardens maksimale respons (REF10).
|
|
2
|
Under to koncentrationer af testkemikaliet er lig med eller overstiger REF10.
|
|
|
For hver omfattende analyseserie anses et testkemikalie for positivt, såfremt:
|
1
|
TCmin. er lig med eller lavere end 80 % af referencestandardens maksimale respons (REF80 = 20 % inhibering).
|
|
2
|
Mindst to på hinanden følgende koncentrationer af testkemikaliet er lig med eller lavere end REF80
|
|
|
For hver omfattende analyseserie anses et testkemikalie for negativt, såfremt:
|
1
|
TCmin. overstiger 80 % af referencestandardens maksimale respons (REF80 = 20 % inhibering).
|
|
2
|
Under to koncentrationer af testkemikaliet er lig med eller lavere end REF80.
|
|
|
|
60.
|
For at karakterisere styrken af et testkemikalies positive respons rapporteres virkningens omfang (agonisme: TCmaks., antagonisme: TCmin.) og den koncentration, hvor virkningen opstår (agonisme: EC10, EC50, PC10, PC50, antagonisme: IC20, IC50, PC80, PC50).
|
TESTRAPPORT
|
61.
|
Jf. punkt 20 i "ER TA-ASSAYETS KOMPONENTER".
|
LITTERATUR
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organisationen for Økonomisk Samarbejde og Udvikling, Paris.
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, og van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J og Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V og Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B og Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, og van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173-187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M og Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, og Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Scr., 4, 67-73.
|
|
(11)
|
Besselink H, Middelhof I, og Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, Nederlandene.
|
Tillæg 4.1
VISUEL KONTROL AF CELLELEVEDYGTIGHED
B.67 IN VITRO-TEST FOR GENMUTATION I PATTEDYRCELLER VED ANVENDELSE AF THYMIDIN-KINASEGENET
INDLEDNING
|
1.
|
Denne forsøgsmetode svarer til OECD’s testvejledning (Test Guideline) 490 (2016). Forsøgsmetoderne gennemgås regelmæssigt og revideres på baggrund af den videnskabelige udvikling, myndighedskrav og hensyn til dyrevelfærd. Muselymfomassayet (MLA) og TK6-testen ved anvendelse af thymidin-kinase-locus (TK) var oprindelig indeholdt i forsøgsmetode B.17. Efterfølgende har MLA-ekspertarbejdsgruppen under den internationale workshop for genotoksicitetstest (IWGT) udviklet internationalt harmoniserede anbefalinger vedrørende assayacceptkriterier og datafortolkning for MLA (1)(2)(3)(4)(5), og disse anbefalinger er indarbejdet i denne nye forsøgsmetode, B.67. Denne forsøgsmetode er skrevet til MLA, og fordi den også anvender TK locus, til TK6-testen. Mens MLA har fundet udbredt anvendelse til lovmæssige formål, anvendes TK6 langt mindre hyppigt. Det skal bemærkes, at de to cellelinjer til trods for ligheden mellem effektparametrene ikke er substituerbare, og at der i lovmæssige programmer kan være angivet en præference for den ene i forhold til den anden med henblik på en særlig lovmæssig anvendelse. F.eks. påviste valideringen af MLA, at den er hensigtsmæssig til påvisning ikke blot af genmutation, men også et testkemikalies evne til at fremkalde strukturel kromosombeskadigelse. Denne forsøgsmetode er en del af en række forsøgsmetoder vedrørende genetisk toksikologi. OECD har udarbejdet et dokument, som indeholder kortfattede oplysninger om testning for genetisk toksikologi, og en oversigt over de seneste ændringer af OECD’s testvejledning om genetisk toksicitet (6).
|
|
2.
|
Formålet med in vitro-testen for genmutation i pattedyrceller er at påvise genmutationer, som induceres af kemikalier. De cellelinjer, der anvendes i disse test, måler fremadmutationer i reportergener, specifikt det endogene thymidin-kinasegen (TK for humane celler og TK for gnaverceller, under ét benævnt TK i denne forsøgsmetode). Denne forsøgsmetode er beregnet til brug med to cellelinjer: L5178Y TK+/--3.7.2C muselymfomcellelinje (generelt benævnt L5178Y) og TK6 human lymphoblastoid cellelinje (generelt benævnt TK6). Skønt de to cellelinjer varierer på grund af deres oprindelse, cellevækst, p53-status, osv., kan der foretages TK-genmutationstest på samme måde i begge celletyper som beskrevet i denne forsøgsmetode.
|
|
3.
|
Thymidin-kinasegenets autosomale og heterozygotiske karakter gør det muligt at påvise levedygtige kolonier, hvis celler mangler i enzymet thymidin-kinase efter mutation fra TK+/-
til TK-/-
. Denne mangel kan skyldes genetiske hændelser, der påvirker TK-genet, herunder både genmutationer (punktmutationer, frameshift-mutationer, små deletioner osv.) og kromosomale hændelser (store deletioner, genomlejringer og mitotisk rekombination). Sidstnævnte hændelser angives som tab af heterozygoti, som er en almindelig genetisk forandring af anti-onkogener i human tumordannelse. Teoretisk kan tab af hele det TK-genbærende kromosom som følge af tensvækkelse og/eller mitotisk non-disjunction påvises i MLA. En kombination af cytogenetisk og molekylær analyse viser nemlig klart, at nogle MLA TK-mutanter er resultatet af non-disjunction. "Weight of evidence"-analysen viser imidlertid, at TK-genmutationstest ikke pålideligt kan påvise aneugener ved anvendelse af standardcytotoksicitetskriterier (som beskrevet i denne forsøgsmetode), og derfor er det ikke hensigtsmæssigt at anvende disse test til at påvise aneugener (7)(8)(9).
|
|
4.
|
I TK-genmutationstest genereres der to tydelige fænotypiske klasser af TK-mutanter: de normalt voksende mutanter, som vokser med samme hastighed som heterozygotiske TK-celler, og langsomt voksende mutanter, der vokser med længere fordoblingstider. De normalt voksende og langsomt voksende mutanter anerkendes som mutanter i både store og små kolonier i MLA og som mutanter i en tidligt fremkommen koloni og mutanter i en sent fremkommen koloni i TK6. Den molekylære og cytogenetiske karakter af MLA-mutanter i både store og små kolonier er blevet udforsket i detaljer (8)(10)(11)(12)(13). Den molekylære og cytogenetiske karakter af tidligt fremkomne og sent fremkomne TK6-mutanter er også blevet grundigt undersøgt (14)(15)(16)(17). Langsomt voksende mutanter for begge celletyper har lidt genetiske skader, der formentlig indebærer vækstregulerende gen(er) i nærheden af TK-locus, hvilket resulterer i længere fordoblingstider og dannelse af sent fremkomne eller små kolonier (18). Induktion af langsomt voksende mutanter er sat i forbindelse med kemikalier, som fremkalder større strukturelle ændringer på kromosomniveau. Celler, hvis skade ikke indebærer formodede vækstregulerende gener i nærheden af TK-locus, vokser med omtrent samme hastighed som modercellerne og bliver normalt voksende mutanter. Induktion af primært normalt voksende mutanter er sat i forbindelse med kemikalier, der primært fungerer som punktmutagener. Det er derfor væsentligt at tælle både langsomt voksende og normalt voksende mutanter med henblik på at genvinde alle mutanterne og skabe indsigt i, hvilke(n) type(r) skade (mutagener mod klastogener), testkemikaliet fremkalder (10)(12)(18)(19).
|
|
5.
|
Forsøgsmetoden er organiseret, så den giver generelle oplysninger, som gælder for både MLA og TK6 samt specialiseret vejledning til individuelle test.
|
|
6.
|
De anvendte definitioner er forklaret i tillæg 1.
|
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER
|
7.
|
Normalt kræver en in vitro-test anvendelse af en kilde til eksogen metabolisk aktivering. Det eksogene metaboliske aktiveringssystem efterligner ikke in vivo-betingelserne fuldstændig.
|
|
8.
|
Man bør omhyggeligt undgå betingelser, der kan føre til et artefakt positivt resultat (dvs. eventuel interaktion med testsystemet), der ikke skyldes interaktion mellem testkemikaliet og cellens genetiske materiale, men omfatter ændringer i pH eller osmolalitet, interaktion med mediets komponenter (20) (21) eller for højt cytotoksicitetsniveau (22)(23)(24). Et cytotoksicitetsniveau, der er højere end det højst anbefalede cytotoksicitetsniveau, jf. punkt 28, anses for at være for højt til MLA og TK6. Endvidere skal det bemærkes, at testkemikalier, som er thymidinanaloge eller har en adfærd som thymidinanaloge kan øge mutantfrekevensen gennem selektiv vækst af de spontane baggrundsmutanter under cellebehandling og kræver supplerende forsøgsmetoder for at sikre en tilstrækkelig vurdering (25).
|
|
9.
|
For fremstillede nanomaterialer kan der være behov for særlige tilpasninger af denne forsøgsmetode, men disse er ikke beskrevet i forsøgsmetoden.
|
|
10.
|
Før forsøgsmetoden anvendes til at teste en blanding for at generere data til lovmæssige formål, bør det overvejes, om den vil give resultater, som er egnede til formålet, og i givet fald hvorfor. Sådanne overvejelser er ikke nødvendige, når der foreligger et myndighedskrav om test af blandingen.
|
|
11.
|
Mutantceller, som mangler i thymidin-kinase-enzymaktiviteten på grund af en TK
+/-- til TK
-/--mutation, er resistente over for de cytostatiske virkninger af den pyrimidinanaloge trifluorthymidin (TFT). Celler, som indeholder TK, er følsomme over for TFT, som inhiberer cellemetabolismen og standser den videre celledeling. Det betyder, at mutantceller kan formere sig under tilstedeværelse af TFT og danne synlige kolonier, mens celler, der indeholder TK-enzym, ikke kan.
|
PRINCIP FOR TESTEN
|
12.
|
Celler i suspensionskultur eksponeres for testkemikaliet både med og uden eksogen metabolisk aktivering (se punkt 19) i et passende tidsrum (jf. punkt 33), hvorefter der ved dyrkning i subkultur bestemmes cytotoksisk effekt og gives mulighed for fænotypeekspression, inden mutanterne udvælges. Cytotoksisk effekt bestemmes ved relativ total vækst (RTG – jf. punkt 25) for MLA og ved relativ overlevelse (RS – jf. punkt 26) for TK6. De behandlede kulturer bibeholdes så lang tid i dyrkningsmediet, afhængigt af hver celletype (jf. punkt 37), at fænotypeekspressionen af inducerede mutationer bliver næsten optimal. Efter fænotypeekspressionen bestemmes mutantfrekevensen ved udsåning af et kendt antal celler i et dyrkningsmedium med det selektive stof til påvisning af mutantkolonier og i et medium uden selektivt stof til bestemmelse af kloningseffektivitet (levedygtigheden). Efter en passende inkuberingstid tælles kolonierne. Mutantfrekvensen beregnes ud fra antallet af mutantkolonier korrigeret for kloningseffektiviteten på tidspunktet for mutantudvælgelsen.
|
BESKRIVELSE AF METODEN
Forberedelser
Celler
|
13.
|
For MLA: Da MLA blev udviklet og karakteriseret ved hjælp af underlinje TK
+/- -3.7.2C af L5178Y-celler, skal denne specifikke underlinje anvendes til MLA. L5178Y-cellelinjen blev udledt af et methylcholanthren-induceret thymisk lymfom fra en DBA-2-mus (26). Clive behandlede sammen med sine medarbejdere L5178Y-celler (af Clivebetegnet som TK+/+-3) med ethylmethansulfonat og isolerede en TK-/--klon (betegnet som TK-/--3,7) ved hjælp af bromdeoxyuridin som det selektive stof. Fra TK-/- klonen blev der isoleret og karakteriseret en spontan TK+/--klon (benævnt TK+/--3.7.2.) og en underklon (benævnt TK+/--3.7.2C) til brug i MLA (27). Karyotypen for cellelinjen er blevet offentliggjort (28)(29)(30)(31). Det normale kromosomtal er 40. Der er et metacentrisk kromosom (t12,13), som bør tælles som ét kromosom. Musens TK-locus ligger i blomsterenden af kromosom 11. L5178Y TK
+/- -3.7.2C-cellelinjen har mutationer i begge p53-alleler og producerer mutant-p53 protein (32) (33). Testens evne til at påvise omfattende skade skyldes formentlig TK+/--3.7.2C-cellelinjens p53-status (17).
|
|
14.
|
For TK6: TK6 er en human lymfoblastcellelinje. Modercellelinjen er en Epstein-Barr-virustransformeret cellelinje, WI-L2, som oprindelig blev udledt af en femårig dreng med arvelig spherocytose. Den første isolerede klon, HH4, blev mutageniseret med ICR191, og der blev genereret en TK heterozygotisk cellelinje, TK6, (34). TK6-celler er næsten diploide, og den repræsentative karyotype er 47, XY, 13+, t(14, 20), t(3, 21) (35). Det humane TK-locus ligger på den lange arm af kromosom 17. TK6 er en p53-kompetent cellelinje, fordi den har en vildtype p53-sekvens i begge alleler og kun udtrykker p53-protein af vildtypen (36).
|
|
15.
|
For både MLA og TK6 anbefales det, at forsøgslaboratoriet – når det første gang etablerer eller erstatter en stamkultur – sikrer sig, at der ikke er nogen mykoplasma-kontaminering, karyotyperer cellerne eller maler de kromosomer, som huser TK-locus, og kontrollerer populationens fordoblingstider. Den normale celledelingstid for celler, der anvendes i laboratoriet, fastslås og bør være i overensstemmelse med de offentliggjorte celleegenskaber (16)(19)(37). Denne stamkultur opbevares ved -150 °C eller derunder og anvendes til at fremstille alle arbejdscellekulturer.
|
|
16.
|
Enten forud for etablering af en lang række kryopræserverede arbejdskulturer eller lige før anvendelsen i et forsøg kan det være nødvendigt at rense kulturen for allerede eksisterende mutantceller [med mindre mutantfrekvensen (MF) i kontrolkulturen med opløsningsmiddel allerede ligger inden for det acceptable område – jf. tabel 2 med hensyn til MLA]. Dette opnås ved hjælp af methotrexat (aminopterin) til at fravælge celler, der mangler TK, og tilsætte thymidin, hypoxanthin og glycin (L5178Y) eller 2’-deoxycytidin (TK6) til kulturen for at sikre optimal vækst af de TK-kompetente celler (19)(38)(39), og (40) for TK6. Der kan findes generel rådgivning om god praksis for vedligeholdelse af cellekulturer samt specifik rådgivning for L5178Y- og TK6-celler i (19)(31)(37)(39)(41). For laboratorier, som har brug for stamcellekulturer til indledning af enten MLA eller TK6 eller ønsker nye stamcellekulturer, findes der et register over velkarakteriserede celler (37).
|
Medier og dyrkningsbetingelser
|
17.
|
Til vedligeholdelse af kulturerne bør der til begge test anvendes egnede dyrkningsmedier og inkubationsbetingelser (f.eks. dyrkningsskåle, fugtig atmosfære på 5 % CO2, inkubationstemperatur på 37 °C). Cellekulturer bør altid vedligeholdes under betingelser, der sikrer, at de vokser i logfase. Det er især vigtigt at vælge medier og dyrkningsbetingelser, som sikrer optimal cellevækst i ekspressionsperioden og under kloning for både mutantceller og ikke-mutantceller. For MLA og TK6 er det også vigtigt, at dyrkningsbetingelserne sikrer optimal vækst af både den store koloni/tidlige fremkomst og den lille koloni/sene fremkomst af TK-mutanter. Nærmere oplysninger om dyrkning, bl.a. behovet for at opvarme inaktivt hesteserum korrekt, hvis der anvendes RPMI-medium under mutantudvælgelsen, kan findes i (19)(31)(38)(39)(40)(42).
|
Fremstilling af kulturer
|
18.
|
Celler opformeres fra stamkulturer, udsås i dyrkningsmedium med en sådan tæthed, at suspensionskulturer vil fortsætte med at vokse eksponentielt i behandlings- og ekspressionsperioderne.
|
Metabolisk aktivering
|
19.
|
Der bør anvendes eksogene metaboliserende systemer, når der gøres brug af L5178Y- og TK6-celler, fordi de har utilstrækkelige endogene metaboliske egenskaber. Det mest almindeligt anvendte system, der anbefales som standard, medmindre der er begrundelse for andet, er en cofaktorsuppleret postmitochondriefraktion (S9), som fremstilles af levere fra gnavere (normalt rotter), der er behandlet med enzyminducerende stoffer såsom Aroclor 1254 (43)(44)(45) eller en blanding af phenobarbital og β-naphthoflavon (46)(47)(48)(49)(50)(51). Sidstnævnte blanding er ikke istrid med Stockholmkonventionen om persistente organiske miljøgifte (52) og har vist sig at være lige så effektiv som Aroclor 1254 til induktion af oxidaser med blandet funktion (45)(46)(47)(48)(49). S9-fraktionen anvendes normalt i en koncentration på 1-2 %, men kan øges til 10 % (v/v) i det færdige testmedium. Valget af type og koncentration af eksogent metabolisk aktiveringssystem eller anvendte metaboliske inducerende stoffer kan påvirkes af gruppen af testkemikalier.
|
Præparering af testkemikaliet
|
20.
|
Testkemikalier i fast form bør præpareres i passende opløsningsmidler og eventuelt fortyndes inden behandling af cellerne (jf. punkt 21). Testkemikalier i væskeform kan enten tilsættes direkte til testsystemet og/eller fortyndes inden behandlingen af testsystemet. Gasser og flygtige testkemikalier bør testes ved passende modifikation af standardprotokollerne, f.eks. behandling i forseglede dyrkningsskåle (53)(54)(55). Testkemikaliet bør fremstilles lige inden behandlingen, medmindre det via stabilitetsdata påvises, at opbevaring er acceptabelt.
|
TESTBETINGELSER
Opløsningsmidler
|
21.
|
Der vælges et opløsningsmiddel, der optimerer testkemikaliets opløselighed uden at påvirke testforløbet negativt, f.eks. i form af en ændring af cellevækst, påvirkning af testkemikaliets integritet, reaktion med dyrkningsskåle eller blokering af metabolismeaktiveringssystemet. Det anbefales så vidt muligt først at overveje at anvende et vandigt opløsningsmiddel (eller dyrkningsmedium). Veletablerede opløsningsmidler er vand eller dimethylsulfoxid. Organiske opløsningsmidler bør generelt ikke overstige 1 % (v/v), og vandige opløsningsmidler (fysiologisk saltopløsning eller vand) bør ikke overstige 10 % (v/v) i det færdige behandlingsmedium. Hvis der anvendes andet end ikke-veletablerede opløsningsmidler (f.eks. ethanol eller acetone), bør disse underbygges af data om deres kompatibilitet med testkemikalierne, testsystemet og fraværet af genetisk toksicitet ved den anvendte koncentration. Uden disse understøttende data er det vigtigt at medtage ubehandlede kontroller (jf. tillæg 1, Definitioner) for at påvise, at det valgte opløsningsmiddel ikke forårsager skadevirkninger eller mutagene virkninger.
|
MÅLING AF CYTOTOKSISK EFFEKT OG VALG AF DOSISKONCENTRATIONER
|
22.
|
Ved valg af højeste koncentration af testkemikaliet bør koncentrationer, der kan frembringe artefakt positiv respons, undgås, f.eks. koncentrationer, der frembringer for høj cytotoksisk effekt (jf. punkt 28), udfældning i dyrkningsmediet (jf. punkt 29) eller væsentlige ændringer i pH eller osmolalitet (jf. punkt 8). Hvis testkemikaliet frembringer en væsentlig ændring i mediets pH på tilsætningstidspunktet, kan pH-værdien justeres ved bufring af det endelige behandlingsmedium for at undgå artefakt positive resultater og for at opretholde passende dyrkningsbetingelser.
|
|
23.
|
Valget af koncentration baseres på cytotoksisk effekt og andre overvejelser (jf. punkt 27-30). Evaluering af cytotoksisk effekt i en indledende test kan være nyttig for at sikre en bedre definition af de koncentrationer, der skal anvendes i hovedforsøget, men en indledende test er ikke påkrævet. Selv om der foretages en indledende vurdering af den cytotoksiske effekt, skal denne stadig måles i hovedforsøget for hver enkelt kultur. Hvis der udføres et forsøg til bestemmelse af dosisinterval, bør det dække en bred vifte af koncentrationer og kan enten afsluttes på dag 1 efter behandling eller gennemføres under den to dage lange ekspression og til mutantudvælgelsen (hvis det skulle vise sig, at de anvendte koncentrationer er egnede).
|
|
24.
|
Cytotoksisk effekt bør bestemmes for hver enkelt testkultur og kontrolkultur, idet metoder for MLA (2) og TK6 (15) defineres ved internationalt aftalt praksis.
|
|
25.
|
For både agar- og mikrobrøndversionerne af MLA: Cytotoksisk effekt vurderes ved hjælp af relativ total vækst (RTG), som oprindelig blev defineret af Clive og Spector i 1975 (2). Dette mål omfatter den relative suspensionsvækst (RSG: testkultur over for kontrolkultur med opløsningsmiddel) under cellebehandlingen, ekspressionstiden og den relative kloningseffektivitet (RCE: testkultur over for kontrolkultur med opløsningsmiddel) på det tidspunkt, hvor der vælges mutanter (2). Det skal bemærkes, at RSG omfatter ethvert celletab, som opstår i testkulturen under behandling (jf. formlen i tillæg 2).
|
|
26.
|
For TK6: Cytotoksisk effekt bør evalueres ved anvendelse af relativ overlevelse (RS), dvs. cellernes kloningseffektivitet, når de udplades umiddelbart efter behandling, justeret for ethvert celletab under behandlingen baseret på celletal sammenholdt med den negative kontrol (med en overlevelsesprocent på 100) (jf. formlen i tillæg 2).
|
|
27.
|
Mindst fire testkoncentrationer (bortset fra opløsningsmiddel og positive kontroller), der opfylder acceptkriterierne (egnet cytotoksisk effekt, antal celler mv.), bør vurderes. Selv om det anbefales at benytte dobbeltbestemmelse, kan der ved hver af de testede koncentrationer også anvendes replikate eller enkelte behandlede kulturer. De resultater, der opnås for replikate kulturer ved en given koncentration, bør rapporteres separat, men kan samles med henblik på dataanalyse (55). For testkemikalier, der udviser ringe eller ingen cytotoksisk effekt, vil koncentrationsintervaller på ca. 2 til 3 fold normalt være egnede. Hvis der forekommer cytotoksisk effekt, bør de valgte koncentrationer dække et cytotoksicitetsområde fra det, der frembringer cytotoksisk effekt som beskrevet i punkt 28, til koncentrationer, hvor der er moderat og ringe eller ingen cytotoksisk effekt. Mange testkemikalier udviser kraftige koncentration/respons-kurver, og for at dække hele cytotoksicitetsområdet eller undersøge koncentration/respons-forholdet nærmere kan det være nødvendigt at bruge mere samlede koncentrationer og mere end fire koncentrationer, navnlig i situationer, hvor der er behov for at gentage forsøget (jf. punkt 70). Det kan være særligt vigtigt at bruge mere end fire koncentrationer, når der anvendes enkeltkulturer.
|
|
28.
|
Hvis den maksimale koncentration er baseret på cytotoksisk effekt, bør den højeste koncentration sigte mod at nå mellem 20 og 10 % RTG for MLA og mellem 20 og 10 % RS for TK6 (punkt 67).
|
|
29.
|
For tungtopløselige testkemikalier, der ikke er cytotoksiske ved lavere koncentrationer end den laveste uopløselige koncentration, bør den højeste analyserede koncentration producere uklarheder eller bundfald, som er synligt med det blotte øje eller i et inverteret mikroskop efter afslutningen af behandlingen med testkemikaliet. Selv om der forekommer cytotoksisk effekt over den laveste uopløselige koncentration, tilrådes det at gennemføre testen ved kun én koncentration, der frembringer uklarhed eller synligt bundfald, fordi artefakte virkninger kan skyldes bundfaldet. Da der ved MLA og TK6 anvendes suspensionskulturer, bør det omhyggeligt sikres, at bundfaldet ikke griber forstyrrende ind i udførelsen af testen. Det kan også være nyttigt at bestemme opløseligheden i dyrkningsmediet inden forsøget.
|
|
30.
|
Hvis der ikke observeres bundfald eller begrænsende cytotoksisk effekt, bør testkoncentrationen højst være 10 mM, 2 mg/ml eller 2 μl/ml (den laveste af de tre) (57) (58). Hvis testkemikaliets sammensætning ikke er defineret, f.eks. et stof af ukendt eller variabel sammensætning, komplekse reaktionsprodukter eller biologiske materialer (dvs. kemiske stoffer af ukendt eller variabel sammensætning (UVCB’er)), miljømæssige udtræk mv., kan der være behov for at øge den højeste koncentration (f.eks. 5 mg/ml) ved manglende tilstrækkelig cytotoksisk effekt for at øge koncentrationen af hver af komponenterne. Det bør dog bemærkes, at disse krav kan variere for humanlægemidler (59).
|
Kontroller
|
31.
|
Der bør under alle forsøgsbetingelser indgå samtidige negative kontroller (jf. punkt 21), hvor behandlingsmediet kun består af opløsningsmiddel, og som behandles på samme måde som de øvrige kulturer.
|
|
32.
|
Der skal foretages samtidige positive kontroller for at påvise laboratoriets evne til at identificere mutagener i henhold til testprotokollen, påvise effektiviteten af det eksogene metabolismeaktiveringssystem, hvor det er relevant, og i tilstrækkelig grad påvise både små/sent fremkomne og store/tidligt fremkomne TK-mutanter. Eksempler på positive kontroller er anført i tabel 1 nedenfor. Alternative positive kontrolstoffer kan anvendes, hvis der er belæg for det. Eftersom in vitro-test af pattedyrceller for genetisk toksicitet er tilstrækkeligt standardiserede til kortvarige behandlinger (3-4 timer), der udføres samtidig med og uden metabolisk aktivering af samme behandlingsvarighed, kan brugen af positive kontroller begrænses til en mutagenkrævende metabolisk aktivering. I dette tilfælde vil denne ene positive kontrolrespons påvise såvel aktiviteten i metabolismeaktiveringssystemet som testsystemets reaktionsevne. Anvendes der langtidsbehandling (dvs. 24 timer uden postmitochondrisk fraktion), bør den dog have sin egen positive kontrol, da behandlingens varighed er forskellig fra testen med metabolisk aktivering. Hver positiv kontrol bør anvendes ved en eller flere koncentrationer, der forventes at give reproducerbare og påviselige forøgelser i forhold til baggrundsværdierne, for at påvise testsystemets følsomhed, og responsen bør ikke bringes i fare på grund af en cytotoksisk effekt, der overstiger grænserne i denne TM (jf. punkt 28).
|
Tabel 1
Anbefalede referencestoffer til vurdering af laboratoriets kompetence og udvælgelse af positive kontroller
|
Kategori
|
Stof
|
CAS RN
|
|
1. Aktive mutagener uden metabolisk aktivering
|
|
Methylmethansulfonat
|
66-27-3
|
|
Mitomycin C
|
50-07-7
|
|
4-nitroquinolin-N-oxid
|
56-57-5
|
|
2. Mutagener, som kræver metabolisk aktivering
|
|
Benzo(a)pyren
|
50-32-8
|
|
Cyclophosphamidmonohydrat
|
50-18-0 (6055-19-2)
|
|
7,12-dimethylbenzanthracen
|
57-97-6
|
|
3-methylcholanthren
|
56-49-5
|
FREMGANGSMÅDE
Behandling med testkemikalie
|
33.
|
Celler under formering behandles med testkemikaliet med og uden tilstedeværelse af et metabolismeaktiveringssystem. Eksponeringen skal vare et passende stykke tid (normalt er 3-4 timer passende). Det bør dog bemærkes, at disse krav kan variere for humanlægemidler (59). I tilfælde, hvor kortvarig behandling med MLA giver negative resultater, og der foreligger oplysninger, som tyder på, at der er behov for længere behandling (f.eks. nukleosidanaloger, tungt opløselige kemikalier (5)(59)), bør det overvejes at udføre testen med længere behandling, dvs. 24 timer uden postmitochondrisk fraktion.
|
|
34.
|
Det antal celler, der mindst skal anvendes til hver testkultur (kontrol og behandling) i hver enkelt testfase, bør baseres på den spontane mutantfrekevens. En generel vejledning er at behandle og opformere et tilstrækkeligt antal celler i hver forsøgskultur for at bevare mindst 10, men ideelt set 100, spontane mutanter i alle faser af testen (behandling, fænotypeekspression og mutantudvælgelse (56).
|
|
35.
|
For MLA ligger den anbefalede acceptable spontane mutantfrekvens mellem 35-140 × 10-6 (agarversion) og 50-170 × 10-6 (mikrobrøndversion) (jf. tabel 2). For at have mindst 10 og ideelt set 100 spontane mutanter, der overlever behandling for hver testkultur, er det nødvendigt at behandle mindst 6 × 106 celler. Behandling af dette antal celler og bibeholdelse af et tilstrækkeligt antal celler under ekspression og kloning til mutantudvægelse sikrer et tilstrækkeligt antal spontane mutanter (10 eller derover) i alle faser af forsøget, selv for kulturer, som behandles ved koncentrationer, der resulterer i 90 % cytotoksisk effekt (som målt ved en RTG på 10 %) (19)(38)(39).
|
|
36.
|
For TK6 ligger den spontane mutantfrekvens generelt mellem 2 og 10 × 10-6. For at have mindst 10 spontane mutanter, der overlever behandling for hver kultur, er det nødvendigt at behandle mindst 20 × 106 celler. Behandling af dette antal celler giver et tilstrækkeligt antal spontane mutanter (10 eller derover), selv for kulturer behandlet ved koncentrationer, der forårsager 90 % cytotoksisk effekt under behandling (10 % RS). Endvidere skal der dyrkes et tilstrækkeligt antal celler i ekspressionsperioden, som udplades til mutantudvælgelse (60).
|
Fænotypeekspressionsperiode og måling af cytotoksisk effekt og mutantfrekvens
|
37.
|
Ved afslutningen af behandlingsperioden dyrkes celler i en fastsat periode for at give mulighed for en næsten optimal fænotypeekspression af nyligt inducerede mutanter, som er specifikke for hver enkelt cellelinje. For MLA varer fænotypeekspressionsperioden 2 dage. For TK6 varer fænotypeekspressionsperioden 3-4 dage. Hvis der anvendes en 24-timers behandling, begynder ekspressionsperioden efter behandlingens afslutning.
|
|
38.
|
I fænotypeekspressionsperioden optælles cellerne dagligt. For MLA benyttes de daglige celletællinger til at beregne den daglige suspensionsvækst (SG). Efter den to dage lange ekspressionsperiode suspenderes cellerne i dyrkningsmedium med og uden selektivt stof med henblik på henholdsvis bestemmelse af antallet af mutanter (selektionsplader) og kloningseffektivitet (levedygtighedsplader). For MLA findes der to lige acceptable metoder til udvælgelse af mutanter til kloning: ved den ene anvendes agar, og ved den anden flydende vækstmedium på plader med 96 brønde (19)(38)(39). Kloning i TK6 udføres ved hjælp af flydende medier og plader med 96 brønde (16).
|
|
39.
|
Trifluorthymidin (TFT) er det eneste selektive stof, der anbefales til TK-mutanter (61).
|
|
40.
|
For MLA tælles agarplader og mikrobrøndplader efter 10-12 dages inkubation. For TK6 registreres kolonier på mikrobrøndplader efter 10-14 dage for tidligt fremkomne mutanter. For at nyttiggøre de langsomt voksende (sent fremkomne) TK6-mutanter er det nødvendigt at fodre cellerne igen med dyrkningsmedium og TFT efter optælling af de tidligt fremkomne mutanter og herefter inkubere pladerne i yderligere 7-10 dage (62). Se punkt 42 og 44 for en diskussion af optællingen af langsomt og normalt voksende TK-mutanter.
|
|
41.
|
De korrekte beregninger for de to test, herunder de to metoder (agar og mikrobrønd) til MLA, findes i tillæg 2. Til agarmetoden for MLA tælles kolonier, og antallet af mutantkolonier justeres ved kloningseffektiviteten for at beregne en MF. Til mikrobrøndversionen af MLA og TK6 bestemmes kloningseffektiviteten både for selektions- og kloningseffektivitetspladerne i overensstemmelse med Poisson-fordelingen (63). MF beregnes på grundlag af disse to kloningseffektiviteter.
|
Karakterisering af mutantkolonier
|
42.
|
Hvis testkemikaliet for MLA er positivt (jf. punkt 62-63), udføres kolonikarakterisering ved bestemmelse af kolonistørrelse eller vækst i mindst én af testkulturerne (normalt den højeste acceptable positive koncentration) og i de negative og positive kontrolkulturer. Er testkemikaliet negativt (jf. punkt 64) udføres karakteriseringen af mutantkolonien i de negative og positive kontrolkulturer. Ved mikrobrøndmetoden for MLA defineres mutanter fra små kolonier som dem, der dækker under 25 % af brøndens diameter, og mutanter fra store kolonier som dem, der dækker over 25 % af brøndens diameter. Ved agarmetoden benyttes en automatisk kolonitæller til at optælle mutantkolonier og bestemme kolonistørrelse. Metoder til bestemmelse af kolonistørrelse er nærmere beskrevet i litteraturen (19)(38)(40). Karakterisering af kolonier på den negative og positive kontrol er nødvendig for at påvise, at undersøgelserne er foretaget korrekt.
|
|
43.
|
Testkemikaliet kan ikke bestemmes til at være negativt, hvis mutanterne fra både de store og små kolonier ikke er tilstrækkeligt påvist i den positive kontrol. Karakterisering af kolonier kan give generelle oplysninger om testkemikaliets evne til at forårsage punktmutationer og/eller kromosomale hændelser (punkt 4).
|
|
44.
|
TK6: Normalt voksende og langsomt voksende mutanter differentieres ved en forskel i inkubationstid (jf. punkt 40). For TK6 registreres både de tidligt og sent fremkomne mutanter for alle kulturerne, herunder de negative og positive kontroller. Kolonikarakterisering af den negative og positive kontrol er nødvendig for at påvise, at undersøgelserne foretages korrekt. Testkemikaliet kan ikke bestemmes til at være negativt, hvis både de tidligt fremkomne og sent fremkomne mutanter ikke er tilstrækkeligt påvist i den positive kontrolkultur. Karakterisering af kolonier kan give generelle oplysninger om testkemikaliets evne til at forårsage punktmutationer og/eller kromosomale hændelser (punkt 4).
|
Laboratoriets kompetencer
|
45.
|
For at påvise tilstrækkelig erfaring med testen, før den tages i brug til rutinetest, bør laboratoriet have udført en række forsøg med positive referencekemikalier, som virker via forskellige mekanismer (mindst et aktivt med og et aktivt uden metabolisk aktivering udvalgt blandt kemikalierne i tabel 1), og forskellige negative kontroller (herunder ubehandlede kulturer og forskellige opløsningsmidler/bærestoffer). Disse positive og negative kontrolresponser bør være i overensstemmelse med litteraturen. Dette krav gælder ikke for laboratorier, der har erfaring, dvs. som har en historisk database til rådighed som defineret i punkt 47-50. For MLA bør de værdier, som er opnået for både positive og negative kontroller, være i overensstemmelse med IWGT’s anbefalinger (jf. tabel 2).
|
|
46.
|
Et udvalg af positive kontrolstoffer (jf. tabel 1) bør undersøges med korte og lange behandlinger (hvis der benyttes lange behandlinger) uden metabolisk aktivering og også med kort behandling med metabolisk aktivering med henblik på at påvise evnen til at spore mutagene kemikalier, bestemme effektiviteten af metabolismeaktiveringssystemet og påvise cellevækstbetingelsernes egnethed under behandling, fænotypeekspression og mutantudvælgelse samt bedømmelsesprocedurernes egnethed. Der bør vælges en række koncentrationer af de udvalgte stoffer, som giver en reproducerbar og koncentrationsafhængig stigning over baggrunden for at påvise testsystemets følsomhed og dynamiske rækkevidde.
|
Historiske kontroldata
|
47.
|
Laboratoriet bør fastlægge:
|
—
|
et historisk positivt kontrolområde og en historisk positiv kontrolfordeling
|
|
—
|
et historisk negativt (ubehandlet, opløsningsmiddel) kontrolområde og en historisk negativ kontrolfordeling.
|
|
|
48.
|
Når der første gang indhentes data til en historisk negativ kontrolfordeling, bør de samtidige negative kontroller stemme overens med offentliggjorte negative kontroldata. Efterhånden som der tilføjes flere forsøgsdata til kontrolfordelingen, bør de samtidige negative kontroller ideelt set ligge inden for kontrolgrænsen på 95 % af denne fordeling (64)(65).
|
|
49.
|
Laboratoriets database over historiske negative kontroller bør i første omgang opbygges med mindst 10 forsøg, men fortrinsvis bestå af mindst 20 forsøg udført under sammenlignelige forsøgsbetingelser. Laboratorierne bør anvende kvalitetskontrolmetoder som f.eks. kontrolkort (f.eks. C-diagrammer eller X-søjlediagrammer (65)) til at fastslå, hvor variable deres positive og negative kontroldata er, samt vise, at metodologien er "under kontrol" i deres laboratorium (66). Yderligere oplysninger og anbefalinger om, hvordan de historiske data kan opbygges og bruges, kan findes i litteraturen (64).
|
|
50.
|
Negative kontroldata bør bestå af mutantfrekvenser fra enkeltkulturer eller fortrinsvis fra replikate kulturer som beskrevet i punkt 27. Sideløbende negative kontroller bør ideelt set ligge inden for kontrolgrænsen på 95 % af fordelingen af laboratoriets historiske negative kontroldatabase. Såfremt data fra negative kontroller falder uden for kontrolgrænsen på 95 %, kan de eventuelt optages i den historiske kontrolfordeling, så længe disse data ikke er ekstreme afvigende værdier, der er dokumentation for, at testsystemet er "under kontrol" (jf. punkt 49), og for, at der ikke er begået tekniske eller menneskelige fejl.
|
|
51.
|
Der bør tages hensyn til eventuelle ændringer af forsøgsprotokollen med hensyn til, om dataene stemmer overens med laboratoriets eksisterende historiske kontroldatabaser. Ved større uoverensstemmelser bør der etableres en ny historisk kontroldatabase.
|
DATA OG RAPPORTERING
Præsentation af resultaterne
|
52.
|
Præsentation af data for både MLA og TK6 bør for både behandlede kulturer og kontrolkulturer omfatte de nødvendige data til beregning af cytotoksisk effekt (henholdsvis RTG og RS) og mutantfrekevenser, som beskrevet nedenfor.
|
|
53.
|
For MLA bør der anføres data for hver enkelt kultur for RSG, RTG, kloningseffektivitet på tidspunktet for mutantudvælgelsen og antallet af mutantkolonier (for agarversionen) eller antal tomme brønde (for mikrobrøndversionen). MF opgives som antallet af mutantceller pr. én million overlevende celler. Hvis reaktionen er positiv, bør MF’er i små og store kolonier (og eller procentdelen af den samlede MF) angives for mindst én koncentration af testkemikaliet (normalt den højeste positive koncentration) og de negative og positive kontroller. I tilfælde af en negativ reaktion bør MF’er i små og store kolonier angives for den negative og den positive kontrol.
|
|
54.
|
For TK6 anføres data for hver enkelt kultur for RS, kloningseffektivitet på tidspunktet for mutantudvælgelse og antallet af tomme brønde for tidligt fremkomne og sent fremkomne mutanter. MF udtrykkes som antal mutantceller pr. antal overlevende celler og bør omfatte den samlede MF samt MF (og/eller procentdel af den samlede MF) af de tidligt og sent fremkomne mutanter.
|
Acceptkriterier
|
55.
|
For både MLA og TK6 bør følgende kriterier opfyldes, før de samlede resultater for et specifikt testkemikalie bestemmes:
|
—
|
To forsøgsbetingelser (kort behandling med og uden metabolisk aktivering – jf. punkt 33) blev gennemført, medmindre en af dem gav positive resultater.
|
|
—
|
Antallet af analyserbare celler og koncentrationer er tilstrækkeligt (jf. punkt 27, 34-36).
|
|
—
|
Kriterierne for valg af højeste koncentration er i overensstemmelse med kriterierne i punkt 28-30.
|
|
Acceptkriterier for negative og positive kontroller
|
56.
|
Expert MLA Workgroup’s analyse af en omfattende mængde MLA-data førte til international enighed om specifikke acceptkriterier for MLA (1)(2)(3)(4)(5). Derfor giver denne forsøgsmetode specifikke anbefalinger om bestemmelse af, om negative og positive kontroller kan accepteres, og om vurdering af resultater for individuelle stoffer i MLA. TK6 har en langt mindre database og er ikke blevet vurderet af en arbejdsgruppe.
|
|
57.
|
For MLA bør hvert forsøg vurderes med hensyn til, om den ubehandlede kontrolkultur/kontrolkultur med opløsningsmiddel opfylder IWGT MLA Workgroup’s acceptkriterier ((4) og tabel 2, nedenfor) for: (1) MF (bemærk, at IWGT’s acceptable MF’er er forskellige for agar- og mikrobrøndversionerne af MLA), (2) kloningseffektivitet (CE) på tidspunktet for mutantudvælgelse og (3) suspensionsvækst (SG) for kontrolkulturen med opløsningsmiddel (jf. formlen i tillæg 2).
Tabel 2
Acceptkriterier for MLA
|
Parameter
|
Metode med blød agar
|
Mikrobrøndmetode
|
|
Mutantfrekvens
|
35-140 × 10-6
|
50-170 × 10-6
|
|
Kloningseffektivitet
|
65-120 %
|
65-120 %
|
|
Suspensionsvækst:
|
8-32 fold (3-4 timers behandling) 32-180 fold (24 timers behandling, hvis den gennemføres)
|
8-32 fold (3-4 timers behandling) 32-180 fold (24 timers behandling, hvis den gennemføres)
|
|
|
58.
|
Ved MLA bør hver test også vurderes med hensyn til, om de positive kontroller opfylder mindst ét af følgende to acceptkriterier, der er udviklet af IWGT-arbejdsgruppen:
|
—
|
Den positive kontrol bør påvise en absolut stigning i den samlede MF, dvs. en stigning over den spontane baggrunds-MF (en induceret MF (IMF)) på mindst 300 × 10-6. Mindst 40 % af IMF bør afspejles i MF for den lille koloni.
|
|
—
|
Den positive kontrol har en stigning i den lille MF-koloni på mindst 150 × 10-6 over det, der observeres i den samtidige ubehandlede kontrolkultur/kontrolkulturen med opløsningsmiddel (en lille IMF-kultur på 150 × 10-6).
|
|
|
59.
|
Ved TK6 vil en test kunne godtages, hvis den samtidige negative kontrol betragtes som acceptabel som supplement til laboratoriets historiske negative kontroldatabase som beskrevet i punkt 48-49. Endvidere bør de sideløbende positive kontroller (jf. punkt 32) fremkalde responser, der er forenelige med responserne i den historiske positive kontroldatabase, og medføre en statistisk signifikant stigning sammenlignet med den sideløbende negative kontrol.
|
|
60.
|
For begge test bør den øvre cytotoksicitetsgrænse, som er observeret i den positive kontrolkultur, være den samme som for forsøgskulturerne. Dvs. at RTG/RS ikke bør ligge under 10 %. Det er tilstrækkeligt at anvende en enkelt koncentration (eller en af koncentrationerne fra de positive kontrolkulturer, hvis der anvendes mere end én koncentration) til at påvise, at acceptkriterierne for den positive kontrol er blevet opfyldt. Yderligere skal MF for den positive kontrol ligge inden for det acceptable område, der er fastsat for laboratoriet.
|
Vurdering og fortolkning af resultater
|
61.
|
For MLA er der udført et betydeligt arbejde om biologisk relevans og kriterier for en positiv respons af Mouse Lymphoma Expert Workgroup under IWGT (4). Derfor giver denne forsøgsmetode specifikke anbefalinger til fortolkning af testkemikalieresultaterne fra MLA (jf. punkt 62-64). TK6 har en langt mindre database og er ikke blevet vurderet af en arbejdsgruppe. Derfor formuleres anbefalingerne til fortolkning af data for TK6 i mere generelle vendinger (jf. punkt 65-66). Supplerende anbefalinger gælder for begge test (jf. punkt 67-71).
|
MLA
|
62.
|
En metode til at definere positive og negative responser anbefales for at sikre, at den øgede MF er biologisk relevant. I stedet for statistisk analyse, som normalt benyttes til andre test, bygger den på anvendelse af forud induceret mutantfrekvens (dvs. øget MF over sideløbende kontrol), der betegnes den samlede vurderingsfaktor (GEF), som er baseret på analyse af data om fordelingen af den negative kontrol-MF fra deltagende laboratorier (4). For agarversionen af MLA er GEF 90 × 10-6, og for mikrobrøndversionen af MLA er GEF 126 × 10-6.
|
|
63.
|
Forudsat at alle acceptkriterier er opfyldt, anses et testkemikalie for at være klart positivt, hvis stigningen i MF over den sideløbende baggrund under en af de undersøgte forsøgsbetingelser (jf. punkt 33) overstiger GEF, og stigningen er koncentrationsafhængig (f.eks. under anvendelse af en trendtest). Testkemikaliet anses herefter for at kunne fremkalde mutation i dette testsystem.
|
|
64.
|
Forudsat at alle acceptkriterier er opfyldt, anses et testkemikalie for at være klart negativt, hvis der under alle de undersøgte forsøgsbetingelser (jf. punkt 33) ikke er nogen koncentrationsafhængig respons, eller, hvis der er en stigning i MF, den ikke overstiger GEF. Testkemikaliet anses herefter for ikke at kunne fremkalde mutationer i dette testsystem.
|
TK6
|
65.
|
Såfremt alle acceptkriterier er opfyldt, anses et testkemikalie for at være klart positivt, hvis følgende er opfyldt under en af de undersøgte forsøgsbetingelser (jf. punkt 33):
|
—
|
mindst én af testkoncentrationerne udviser en statistisk signifikant stigning sammenlignet med den sideløbende negative kontrol
|
|
—
|
stigningen er koncentrationsafhængig, når den evalueres med en egnet trendtest (jf. punkt 33)
|
|
—
|
et eller flere af resultaterne ligger uden for fordelingen af de historiske negative kontroldata (f.eks. den Poisson-baserede kontrolgrænse på 95 %, jf. punkt 48).
|
Når alle disse kriterier er opfyldt, anses testkemikaliet for at kunne fremkalde mutation i dette testsystem. Anbefalinger til de mest hensigtsmæssige statistiske metoder kan findes i litteraturen (66)(67).
|
|
66.
|
Såfremt alle acceptkriterier er opfyldt, anses et testkemikalie for at være klart negativt, hvis følgende er opfyldt under alle de undersøgte forsøgsbetingelser (jf. punkt 33):
|
—
|
ingen af testkoncentrationerne udviser en statistisk signifikant stigning sammenlignet med den sideløbende negative kontrol
|
|
—
|
der ikke indtræder en koncentrationsafhængig stigning ved evaluering med en egnet trendtest
|
|
—
|
alle resultaterne ligger inden for fordelingen af de historiske negative kontroldata (f.eks. den Poisson-baserede kontrolgrænse på 95 %). jf. punkt 48).
|
Testkemikaliet anses herefter for ikke at kunne fremkalde mutationer i dette testsystem.
|
For både MLA og TK6:
|
67.
|
Hvis den maksimale koncentration er baseret på cytotoksisk effekt, bør den højeste koncentration sigte mod at nå mellem 20 og 10 % RTG/RS. Der er enighed om, at der bør udvises omhu ved fortolkning af positive resultater, hvor der kun er fundet mellem 20 og 10 % RTG/RS, og et resultat vil ikke blive anset for at være positivt, såfremt stigningen i MF først opstod ved eller under 10 % RTG/RS (hvis det vurderes) (2)(59).
|
|
68.
|
Der er nogle omstændigheder, hvor supplerende oplysninger kan være en hjælp til bestemmelsen af, at et testkemikalie ikke er et mutagent stof, når der ikke er nogen kultur, som viser en RTG-værdi mellem 10 og 20 % RTG/RS. Disse situationer er beskrevet i det følgende: (1) Der er ingen evidens for mutagenicitet (f.eks. ingen dosisrespons, ingen muantfrekevenser over dem, der ses i den sideløbende negative kontrol eller historiske baggrundsområder osv.) i en række datapunkter inden for 100 % til 20 % RTG/RS, og der er mindst ét datapunkt mellem 20 og 25 % RTG/RS. (2) Der er ingen mutagenicitet (f.eks. ingen dosisrespons, ingen mutantfrekevenser over dem, der ses i den sideløbende negative kontrol eller historiske baggrundsområder osv.) i en række datapunkter mellem 100 % til 25 % RTG/RS, og der er også et negativt datapunkt lidt over 10 % RTG/RS. I begge disse situationer kan det konkluderes, at testkemikaliet er negativt.
|
|
69.
|
Der er ikke krav om verifikation af en klart positiv eller negativ respons.
|
|
70.
|
Hvis responsen hverken er klart negativ eller klart positiv som beskrevet ovenfor og/eller for at bidrage til at fastslå den biologiske relevans af et resultat, bør dataene vurderes af eksperter, og/eller der bør foretages yderligere undersøgelser. Det kan være nyttigt at gennemføre et nyt forsøg, eventuelt med ændrede forsøgsbetingelser (f.eks. koncentrationsinterval for at øge sandsynligheden for at nå datapunkter inden for 10-20 % RTG/RS-området, ved hjælp af andre metabolismeaktiveringsbetingelser (dvs. S9-koncentration eller S9-oprindelse) og behandlingens varighed).
|
|
71.
|
I sjældne tilfælde giver dataene selv efter yderligere undersøgelser ikke mulighed for at konkludere, om resultaterne er positive eller negative. Konklusionen er derfor, at testkemikalieresponsen er usikker (fortolkes på den måde, at der er lige stor sandsynlighed for, at den er positiv eller negativ).
|
TESTRAPPORT
|
72.
|
Testrapporten bør indeholde følgende oplysninger:
|
|
Testkemikalie:
|
—
|
oprindelse, batchnummer, sidste anvendelsesdato, hvis dette foreligger
|
|
—
|
testkemikaliets stabilitet, hvis denne er kendt
|
|
—
|
testkemikaliets opløselighed og stabilitet i opløsningsmidlet, hvis dette er kendt
|
|
—
|
måling af pH-værdi, osmolalitet og bundfald i det dyrkningsmedium, som testkemikaliet blev tilsat, hvor det er relevant.
|
|
|
Stof med kun én bestanddel:
|
—
|
fysisk udseende, vandopløselighed og yderligere relevante fysisk-kemiske egenskaber
|
|
—
|
kemisk identifikation, f.eks. IUPAC-navn eller CAS-navn, CAS-nummer, SMILES- eller InChI-kode, strukturformel, renhed, kemisk identitet af urenheder, i det omfang det er relevant og praktisk muligt, osv.
|
|
|
|
Stof med flere bestanddele, UVCB-stoffer og blandinger:
|
—
|
kendetegnet så vidt muligt ved kemisk identitet (jf. ovenfor), kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber.
|
|
|
|
|
Opløsningsmiddel:
|
—
|
begrundelse for valg af opløsningsmiddel
|
|
—
|
procentdel af opløsningsmiddel i det endelige dyrkningsmedium.
|
|
|
|
Celler:
|
|
For laboratoriets stamkulturer:
|
—
|
celletype og oprindelse samt historie i testlaboratoriet
|
|
—
|
karyotypiske karakteristika og/eller normalt kromosomtal
|
|
—
|
metoder til opretholdelse af cellekulturer
|
|
—
|
cellernes fordoblingstid.
|
|
|
|
|
Testbetingelser:
|
—
|
begrundelse for valg af koncentrationer og antal cellekulturer, herunder f.eks. data om cytotoksisk effekt og opløselighedsbegrænsninger
|
|
—
|
mediesammensætning, CO2-koncentration, fugtighedsniveau
|
|
—
|
testkemikaliets koncentration udtrykt som endelig koncentration i dyrkningsmediet (f.eks. μg eller mg/ml eller mM af dyrkningsmediet)
|
|
—
|
koncentration (og/eller volumen) af opløsningsmiddel og tilsat testkemikalie i dyrkningsmediet
|
|
—
|
celletæthed under behandlingen
|
|
—
|
metabolismeaktiveringssystemets type og sammensætning (kilde til S9, metode til fremstilling af den postmitochondriske fraktionsblanding (S9), koncentration eller rumfang af S9-blandingen og S9 i det endelige dyrkningsmedium, kvalitetskontrol af S9)
|
|
—
|
positive og negative kontrolstoffer, endelige koncentrationer for hver behandlingsbetingelse
|
|
—
|
ekspressionsperiodens længde (herunder antal udsåede celler, subkulturer og eventuel feeding)
|
|
—
|
det selektive stofs identitet og koncentration
|
|
—
|
for MLA anføres den anvendte version (agar eller mikrobrønd)
|
|
—
|
kriterier for accept af testene
|
|
—
|
metoder til tælling af levedygtige celler og mutantceller
|
|
—
|
metoder til måling af cytotoksisk effekt
|
|
—
|
eventuelle supplerende oplysninger vedrørende cytotoksisk effekt og anvendt metode
|
|
—
|
inkubationstidens varighed efter udpladning
|
|
—
|
definition af kolonier, hvor størrelse og type lægges til grund (herunder kriterier for, hvilke kolonier der er "små" og "store")
|
|
—
|
kriterier for bedømmelse af undersøgelsen som positiv, negativ eller usikker
|
|
—
|
anvendte metoder til bestemmelse af pH, osmolalitet, hvis den udføres, og bundfald, hvis det er relevant.
|
|
|
|
Resultater:
|
—
|
antal behandlede celler og antal celler dyrket i subkultur for hver kultur
|
|
—
|
toksicitetsparametre (RTG for MLA og RS for TK6)
|
|
—
|
tegn på bundfald og tidspunkt for bestemmelse
|
|
—
|
antal celler udpladet i selektivt og ikke-selektivt dyrkningsmedium
|
|
—
|
antal kolonier i det ikke-selektive dyrkningsmedium og antal resistente kolonier i det selektive medium samt den tilhørende mutantfrekvens
|
|
—
|
bestemmelse af størrelse for de negative og positive kontroller, og hvis testkemikaliet er positivt, mindst én koncentration og deraf afhængig mutantfrekevens
|
|
—
|
koncentration/respons-sammenhængen, når det er muligt
|
|
—
|
sideløbende negative (opløsningsmiddel) og positive kontroller (koncentrationer og opløsningsmidler)
|
|
—
|
data for tidligere negative (opløsningsmiddel) og positive kontroller (koncentrationer og opløsningsmidler) med angivelse af intervaller, gennemsnit og standardafvigelser antal test, der danner grundlag for de historiske kontroller
|
|
—
|
statistiske analyser (for de enkelte kulturer og eventuelt for samlede replikater) og eventuelle P-værdier og for MLA, GEF-vurderingen.
|
|
|
|
Diskussion af resultaterne
|
|
LITTERATUR
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. og Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. og Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. og Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. og Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. og Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. og O’Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. og Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. og Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., og Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. og Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. og Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. og Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. og Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. og Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. og Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. og Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. og Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. og Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan og M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. og Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. og Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. og Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. og Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. og Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. og Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. og Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. og Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. og Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. og Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. og DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. og Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. og Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411-416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. og Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuskript under udarbejdelse).
|
|
(38)
|
Lloyd M. og Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. og Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan Z og Caldwell(Eds) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. og Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. og Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. og Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. og Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. og Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. og De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. og Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. og Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. og Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, s. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. og Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. og McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, s. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. og Brooks, A.L. (1983). Evaluation of an exposure system using cells grown on collagen gels for detecting highly volatile mutagens in the CHO/HGPRT mutation assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. og Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), Cambridge University Press, s. 66-101.
|
|
(57)
|
Morita T., Honma M. og Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. og Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Tilgængelig på: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. og Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. og Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. og Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. og Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. og Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley og Sons, New York 2nd Edition.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 199.), Organisation for Economic Cooperation and Development, Paris.
|
|
(67)
|
Fleiss J.L., Levin B. og Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Tillæg 1
DEFINITIONER
Aneugen
: alle kemikalier eller processer, der ved interaktion med komponenterne i den mitotiske og meiotiske celledelingscyklus forårsager aneuploidi i celler eller organismer.
Aneuploidi
: enhver afvigelse fra det normale diploide (eller haploide) kromosomtal af et eller flere kromosomer, men ikke af et eller flere komplette kromosomsæt (polyploidi).
Basesubstitutionsmutagener
: kemikalier, der forårsager substitution af basepar i DNA.
Kemikalie
: et stof eller en blanding
Kloningseffektivitet
: procentdel af celler, som udplades med lav tæthed og kan vokse til en koloni, der kan tælles.
Klastogen
: alle kemikalier eller processer, der forårsager strukturelle kromosomaberrationer i populationer af celler eller organismer.
Cytotoksisk effekt
: for de assays, som er omfattet af denne forsøgsmetode, identificeres cytotoksisk effekt som en reduktion i relativ total vækst (RTG) eller relativ overlevelse (RS) for henholdsvis MLA og TK6.
Fremadmutation
: en genmutation fra forældretypen til mutanten, som medfører ændring i eller tab af det kodede proteins enzymaktivitet eller funktion.
Frameshift-mutagener
: kemikalier, der fremkalder addition eller deletion af et eller flere basepar i DNA-molekylet.
Genotoksisk
: et generelt udtryk, der omfatter alle former for DNA- eller kromosombeskadigelse, herunder DNA-brud, addukter, omlejringer, mutationer, kromosomaberrationer og aneuploidi. Det er ikke alle former for genotoksiske virkninger, der forårsager mutationer eller stabil kromosomskade.
Mitotisk rekombination
: under mitosen, rekombination mellem homologe kromatider, der muligvis medfører induktion af brud på DNA-dobbeltstrenge eller tab af heterozygoti.
Mutagen
: forårsager arvelige ændringer af DNA-baseparsekvens(er) i gener eller i kromosomstrukturen (kromosomaberrationer).
Mutantfrekvens (MF)
: antallet af observerede mutantceller divideret med antallet af levedygtige celler.
Fænotypeekspressionsperiode
: det tidsrum efter behandling, hvor den genetiske ændring repareres i genomet, og alle eksisterende genprodukter forsvinder, således at det fænotypiske karakteristikum ændres.
Relativ overlevelse (RS)
: RS bruges til at måle behandlingsrelateret cytotoksisk effekt i TK6. Det er cellernes relative kloningseffektivitet (CE), når de udplades umiddelbart efter cellebehandlingen, justeret for ethvert celletab under behandlingen sammenholdt med kloningseffektiviteten i den negative kontrol.
Relativ suspensionsvækst (RSG)
: For MLA: testkulturens relative totale to dage lange suspensionsvækst sammenlignet med den samlede to dage lange suspensionsvækst for den negative kontrolkultur/kontrolkulturen med opløsningsmiddel (Clive og Spector, 1975). RSG bør omfatte testkulturens relative vækst sammenlignet med den negative kontrolkultur/kontrolkulturen med opløsningsmiddel i behandlingsperioden.
Relativ total vækst (RTG)
: RTG bruges til at måle behandlingsrelateret cytotoksisk effekt i MLA. Den er et mål for testkulturernes relative (i forhold til bærestofkontrollen) vækst under behandlingen, den to dage lange ekspression og udvælgelsen af mutanter til kloning i testforløbet. RSG for hver testkultur ganges med testkulturens relative kloningseffektivitet på tidspunktet for mutantudvælgelsen og angivet i forhold til kloningseffektiviteten for den negative kontrolkultur/kontrolkulturen med opløsningsmiddel (Clive og Spector, 1975).
S9-leverfraktioner
: supernatant fra leverhomogenat efter 9 000 g centrifugering, dvs. ekstrakt fra rå lever.
S9-blanding
: blanding med postmitochondrisk fraktion (S9) af lever og andre medvirkende faktorer, som er nødvendige for at skabe metabolisk enzymaktivitet.
Suspensionsvækst (SG)
: foldstigning i antallet af celler i løbet af behandlings- og ekspressionsfaserne af MLA. SG beregnes ved at gange foldstigningen på dag 1 med foldstigningen på dag 2 for den korte (3-4-timers) behandling. Benyttes en 24-timers behandling, er SG foldstigningen i løbet af 24-timers behandlingen ganget med foldstigningerne på ekspressionsdag 1 og 2.
Kontrolkultur med opløsningsmiddel
: generelt begreb, der angiver de kontrolkulturer, som kun modtager det opløsningsmiddel, der anvendes til at opløse testkemikaliet.
Testkemikalie
: alle stoffer eller blandinger, der testes ved hjælp af denne forsøgsmetode
Ubehandlet kontrol
: kulturer, der ikke modtager behandling (dvs. hverken testkemikalie eller opløsningsmiddel), men behandles på samme måde som de kulturer, der modtager testkemikaliet.
Tillæg 2
FORMLER
Cytotoksisk effekt
For begge versioner (agar og mikrobrønd) af MLA
Cytotoksisk effekt defineres som den relative totale vækst (RTG), der omfatter den relative suspensionsvækst (RSG) i den to dage lange suspensionsperiode og den relative kloningseffektivitet (RCE), der opnås på tidspunktet for udvælgelsen af mutanter. RTG, RSG og RCE angives alle som en procentdel.
Beregning af RSG: Suspensionsvækst 1 (SG1) er vækstraten mellem dag 0 og dag 1 (cellekoncentration på dag 1/cellekoncentration på dag 0), og suspensionsvækst 2 (SG2) er vækstraten mellem dag 1 og dag 2 (cellekoncentration på dag 2/cellekoncentration på dag 1). RSG er den samlede SG (SG1 × SG2) for den behandlede kultur sammenlignet med den ubehandlede kultur/kontrolkulturen med opløsningsmiddel. Det vil sige, at RSG = (SG1(test) × SG2(test)] / [SG1(kontrol) × SG2(kontrol)). SG1 beregnes ud fra den initiale cellekoncentration, som blev anvendt ved påbegyndelsen af cellebehandlingen. Dette gælder for en eventuel differentierende cytotoksisk effekt, der opstår i testkulturen/testkulturerne under cellebehandlingen.
RCE er testkulturens relative kloningseffektivitet sammenlignet med den relative kloningseffektivitet for den ubehandlede kultur/kontrolkulturen med opløsningsmiddel, der opnås på tidspunktet for udvælgelsen af mutanter.
Relativ total vækst (RTG):
RTG = RSG × RCE
TK6
Relativ overlevelse (RS):
Cytotoksisk effekt vurderes ved anvendelse af den relative overlevelse, dvs. cellernes kloningseffektivitet (CE), når de udplades umiddelbart efter behandling, justeret for ethvert celletab under behandlingen sammenholdt med kloningseffektiviteten i de negative kontroller (tildeles en overlevelsesprocent på 100 %). Justeringen for celletab under behandling kan beregnes som følger:
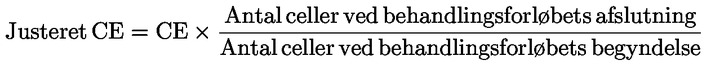
RS for en kultur, der er behandlet med et testkemikalie, beregnes som:

Mutantfrekvens for både MLA og TK6
Mutantfrekvens (MF) er mutantkoloniers kloningsseffektivitet i selektivt dyrkningsmedium (CEM) justeret for kloningsseffektiviteten i ikke-selektivt medium på mutantudvælgelsestidspunktet (CEV). Dvs. MF=CEM/CEV. Beregningen af disse to former for klondannelseseffektivitet er beskrevet nedenfor for kloningsmetoder for agar og mikrobrønd.
MLA-agarversion: I den version af MLA, der anvender blød agar, fås antallet af kolonier på mutantselektionspladen (CM) og antallet af kolonier på pladen uden selektion eller kloningseffektivitet (levedygtig tælling) (CV) ved direkte at tælle klonerne. Når der er udpladet 600 celler med henblik på kloningseffektivitet (CE) til plader med mutantudvælgelse (CEM) og plader uden udvælgelse eller kloningseffektivitet (levedygtig tælling) (CEV), og der anvendes 3 × 106-celler til udvælgelse af mutanter, er
CEM = CM / (3 × 106) = (CM / 3) × 10-6
CEV = CV / 600
MLA- og TK6-mikrobrøndversion:
I mikrobrøndversionen af MLA bestemmes CM og CV som produktet af det samlede antal mikrobrønde (TW) og det sandsynlige antal kolonier pr. brønd (P) på plader med mikrobrønde.
CM = PM × TWM
CV = PV × TWV
Ud fra nul-betingelsen i Poisson-fordelingen (Furth et al., 1981), angives P ved
P = - ln (EW / TW)
hvor EW er tomme brønde, og TW er totale brønde. Derfor gælder følgende:
CEM = CM / TM = (PM × TWM) / TM
CEV = CV / TV = (PV × TWV) / TV
For mikrobrøndversionen af MLA beregnes mutantfrekevenser for små og store kolonier på samme måde ved anvendelse af det relevante antal tomme brønde for små og store kolonier.
For TK6 er mutantfrekvenser for små og store kolonier baseret på tidligt og sent fremkomne mutanter.
B.68 FORSØGSMETODE MED KORTVARIG EKSPONERING IN VITRO TIL IDENTIFIKATION AF i) KEMIKALIER, DER FREMKALDER ALVORLIGE ØJENSKADER, OG ii) KEMIKALIER, DER IKKE KRÆVER KLASSIFICERING FOR ØJENIRRITATION ELLER ALVORLIGE ØJENSKADER
INDLEDNING
|
1.
|
Denne forsøgsmetode svarer til OECD’s testvejledning (Test Guideline (TG)) 491 (2017). Forsøgsmetoden med kortvarig eksponering (STE) er en in vitro-metode, som kan anvendes under visse omstændigheder og med særlige begrænsninger til fareklassificering og mærkning af kemikalier (stoffer og blandinger), der fremkalder alvorlige øjenskader samt kemikalier, der ikke kræver klassificering hverken for alvorlige øjenskader eller for øjenirritation som defineret i De Forenede Nationers (FN) globalt harmoniserede system for klassificering og mærkning af kemikalier (GHS) (kategori 1) og Den Europæiske Unions (EU) forordning nr. 1272/2008 om klassificering, mærkning og emballering af stoffer og blandinger (CLP) (68).
|
|
2.
|
I mange år er kemikaliers potentielle risiko for øjenskade primært blevet vurderet ved anvendelse af en in vivo-kaninøjentest (TM B.5 (8), som svarer til OECD TG 405). Det er generelt accepteret, at der i den nærmeste fremtid ikke vil være en enkelt alternativ in vitro-test, der fuldt ud vil kunne erstatte in vivo-kaninøjentesten til forudsigelse af hele spektret af alvorlige øjenskade-/øjenirritationsreaktioner for forskellige kemikaliegrupper. Strategiske kombinationer af alternative forsøgsmetoder i en (trinvis) teststrategi kan dog muligvis fuldt ud erstatte kaninøjentesten (2). Top-down-tilgangen er beregnet til testning af kemikalier, som baseret på eksisterende information kan forventes at have et højt irritationspotentiale eller fremkalde alvorlige øjenskader. Omvendt er bottom-up-tilgangen beregnet til testning af kemikalier, der baseret på eksisterende information ikke ventes at forårsage tilstrækkelig øjenirritation til, at der kræves en klassificering. Selv om STE-forsøgsmetoden ikke betragtes som værende en fuldstændig erstatning for in vivo-kaninøjentesten, er den egnet til brug som del af en trindelt teststrategi for forskriftsmæssig klassificering og mærkning såsom top-down/bottom-up-tilgangen til uden yderligere testning at identificere (i) kemikalier, der fremkalder alvorlige øjenskader (UN GHS/CLP-kategori 1), og (ii) kemikalier (bortset fra meget flygtige stoffer og alle andre faste kemikalier end overfladeaktive stoffer), som ikke kræver klassificering for øjenirritation eller alvorlige øjenskader (UN GHS/CLP – uden for kategori) (1)(2). Et kemikalie, der hverken forventes at forårsage alvorlige øjenskader (UN GHS/CLP-kategori 1) eller UN GHS/CLP – uden for kategori (hverken fremkalder alvorlige øjenskader eller øjenirritation) ved STE-forsøgsmetoden, vil dog skulle testes yderligere for at fastslå en endelig klassificering. Desuden bør de relevante kompetente myndigheder høres, før STE anvendes i en bottom-up-tilgang i henhold til andre klassifikationsordninger end UN GHS/CLP. Valget af den mest egnede forsøgsmetode og anvendelsen af denne forsøgsmetode bør ses i forbindelse med OECD’s vejledende dokument om integrerede tilgange til testning og vurdering for alvorlige øjenskader og øjenirritation (14).
|
|
3.
|
Formålet med denne forsøgsmetode er at beskrive de procedurer, som anvendes til at vurdere et testkemikalies potentielle risiko for at forårsage øjenskader baseret på dets evne til at fremkalde en cytotoksisk effekt ved forsøgsmetoden med kortvarig eksponering. Kemikaliers cytotoksiske effekt på cornea-epithelceller er en vigtig virkemåde, der fører til cornea-epithelskade og øjenirritation. Cellernes levedygtighed ved STE-forsøgsmetoden vurderes ved kvantitativ måling efter ekstraktion fra celler af blåt formazansalt produceret af de levende celler ved enzymomdannelse af vitalfarvestoffet MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid), også kaldet thiazolylblåt tetrazolium-bromid (3). Den opnåede cellelevedygtighed sammenholdes med kontrolkulturen med opløsningsmiddel (relativ levedygtighed) og bruges til at anslå testkemikaliets potentielle risiko for øjet. Et testkemikalie klassificeres som UN GHS/CLP-kategori 1, når både 5 %- og 0,05 %-koncentrationerne medfører en cellelevedygtighed under eller lig med (≤) 70 %. Omvendt forudses et kemikalie at kunne klassificeres som UN GHS/CLP – uden for kategori 1, når både 5 %- og 0,05 %-koncentrationerne medfører en cellelevedygtighed over (>) 70 %.
|
|
4.
|
Betegnelsen "testkemikalie" anvendes i denne forsøgsmetode om det, der testes, og vedrører ikke anvendeligheden af STE-forsøgsmetoden til testning af stoffer og/eller blandinger. Definitioner findes i tillægget.
|
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER
|
5.
|
Denne forsøgsmetode er baseret på en protokol udviklet af Kao Corporation (4), som var genstand for to forskellige valideringsundersøgelser: en, der blev foretaget af valideringsudvalget under det japanske selskab for alternativertil dyreforsøg (Validation Committee of the Japanese Society for Alternative to Animal Experiments (JSAAE)) (5), og et andet, der blev foretaget af det japanske center for validering af alternative metoder (Japanese Center for the Validation of Alternative Methods (JaCVAM)) (6). Der blev foretaget et peer review af NICEATM/ICCVAM på grundlag af valideringsundersøgelsesrapporter og background review-dokumenter om forsøgsmetoden (7).
|
|
6.
|
Når data opnået med STE-forsøgsmetoden på 125 kemikalier (bl.a. både stoffer og blandinger) blev anvendt til at identificere kemikalier (stoffer og blandinger), der medfører alvorlige øjenskader (UN GHS/CLP-kategori 1) (1), udviste de en generel nøjagtighed på 83 % (104/125), en falsk positiv-rate på 1 % (1/86) og en falsk negativ-rate på 51 % (20/39) sammenlignet med in vivo-kaninøjentesten (7). Falsk negativ-raten er ikke kritisk i den aktuelle sammenhæng, eftersom alle testkemikalier, der medfører en cellelevedygtighed på ≤ 70 % ved en 5 % koncentration og > 70 % ved en 0,05 % koncentration efterfølgende vil blive testet med andre tilstrækkeligt validerede in vitro-forsøgsmetoder eller, som en sidste mulighed, i in vivo-kaninøjentesten alt efter de forskriftsmæssige krav og i overensstemmelse med en sekventiel testningsstrategi med den "weight of evidence"-analyse, som i øjeblikket anbefales (1) (8). Der blev hovedsagelig testet stoffer med kun én bestanddel, skønt der også findes en begrænset mængde data om test af blandinger. Forsøgsmetoden kan ikke desto mindre teknisk set benyttes til test af stoffer med forskellige bestanddele og i forskellige blandinger. Før forsøgsmetoden anvendes til at teste en blanding for at generere data til lovmæssige formål, bør det imidlertid overvejes, om den vil give resultater, som er egnede til formålet, og i givet fald hvorfor. Sådanne overvejelser er ikke nødvendige, når der foreligger et myndighedskrav om test af blandingen. STE-forsøgsmetoden viste ingen andre særlige mangler, da den blev benyttet til at identificere testkemikalier som UN GHS/CLP-kategori 1. Undersøgeren kan overveje at anvende denne forsøgsmetode på testkemikalier, hvorved en cellelevedygtighed på ≤ 70 ved både 5 % og 0,05 % koncentration bør accepteres som indikation på alvorlig øjenskade, der bør klassificeres som UN GHS/CLP-kategori 1 uden yderligere test.
|
|
7.
|
Når data opnået med STE-forsøgsmetoden på 130 kemikalier (bl.a. både stoffer og blandinger) anvendes til at identificere kemikalier (stoffer og blandinger), som ikke kræver klassificering for øjenirritation og alvorlige øjenskader (dvs. UN GHS/CLP – uden for kategori), udviste de en generel nøjagtighed på 85 % (110/130), en falsk negativ-rate på 12 % (9/73) og en falsk positiv-rate på 19 % (11/57) sammenlignet med in vivo-kaninøjentesten (7). Hvis meget letflygtige stoffer og andre faste stoffer end overfladeaktive stoffer udelukkes fra datasættet, forbedres den generelle nøjagtighed til 90 % (92/102), falsk negativ-raten til 2 % (1/54) og falsk positiv-raten til 19 % (9/48) (7). Som følge heraf er de potentielle mangler ved STE-forsøgsmetoden, når den benyttes til at identificere testkemikalier, som ikke kræver klassificering for øjenirritation og alvorlige øjenskader (UN GHS/CLP – uden for kategori), en høj falsk negativ-rate for i) meget flygtige stoffer med et damptryk over 6 kPa og ii) andre faste kemikalier (stoffer og blandinger) end overfladeaktive stoffer og blandinger, som udelukkende består af overfladeaktive stoffer. Sådanne kemikalier er udelukket fra STE-forsøgsmetodens anvendelsesområde (7).
|
|
8.
|
Bortset fra de kemikalier, der er nævnt i punkt 6 og 7, indeholder det datasæt, som blev genereret af STE-forsøgsmetoden, også interne data om 40 blandinger, som sammenholdt med in vivo-Draize-øjentesten udviste en nøjagtighed på 88 % (35/40), en falsk positiv-rate på 50 % (5/10) og en falsk negativ-rate på 0 % (0/30) for forudsigelse af blandinger, der ikke kræver klassificering i henhold til UN GHS/CLP-klassificeringssystemer (9). STE-forsøgsmetoden kan derfor benyttes til at identificere blandinger som UN GHS/CLP – uden for kategori ved en bottom-up-tilgang med undtagelse af andre faste blandinger end dem, der udelukkende består af overfladeaktive stoffer som en udvidelse af dens begrænsning til faste stoffer. Endvidere bør blandinger, der indeholder stoffer med et højere damptryk end 6kPa, vurderes omhyggeligt for at undgå undervurdering i de potentielle forudsigelser og bør begrundes i hvert enkelt tilfælde.
|
|
9.
|
STE-forsøgsmetoden kan ikke benyttes til identifikation af testkemikalier som UN GHS/CLP-kategori 2 eller UN GHS-kategori 2A (øjenirritation) eller 2B (mild øjenirritation) på grund af det betydelige antal kemikalier i UN GHS/CLP-kategori 1, der er undervurderet som kategori 2, 2A eller 2B og kemikalier i UN GHS/CLP – uden for kategori, der er overvurderet som kategori 2, 2A eller 2B (7). Til dette formål kan der være behov for at gennemføre yderligere testning med en anden egnet metode.
|
|
10.
|
STE-forsøgsmetoden er egnet til testkemikalier, der er opløst eller har været ensartet suspenderet i mindst fem minutter i fysiologisk saltopløsning, 5 % dimethylsulfoxid (DMSO) i fysiologisk saltopløsning eller mineralolie. STE-forsøgsmetoden er ikke egnet til testkemikalier, der er uopløselige eller ikke kan suspenderes ensartet i mindst fem minutter i fysiologisk saltopløsning, 5 % DMSO i fysiologisk saltopløsning eller mineralolie. Anvendelsen af mineralolie i STE-forsøgsmetoden er mulig på grund af den kortvarige eksponering. Derfor er STE-forsøgsmetoden egnet til at forudsige vanduopløselige testkemikaliers (dvs. f.eks. langkædede fedtalkoholer eller ketonstoffer) potentielle risiko for at forårsage øjenskader, forudsat at de kan blandes i mindst ét af de tre ovennævnte opløsningsmidler (4).
|
|
11.
|
Betegnelsen "testkemikalie" anvendes i denne forsøgsmetode om det, der testes (69), og vedrører ikke STE-forsøgsmetodens anvendelighed til testning af stoffer og/eller blandinger.
|
PRINCIP FOR TESTEN
|
12.
|
STE-forsøgsmetoden er et cytotoksicitetsbaseret in vitro-assay, som udføres på et sammenflydende enkeltlag af cornea-celler fra kanin fra Statens Seruminstitut (SIRC) dyrket på en polycarbonat-mikroplade med 96 brønde (4). Efter fem minutters eksponering for testkemikaliet måles den cytotoksiske effekt kvantitativt som den relative levedygtighed af SIRC-celler ved anvendelse af MTT-assayet (4). Nedsat cellelevedygtighed anvendes til at forudsige potentielle skadelige virkninger, som medfører øjenskade.
|
|
13.
|
Det er rapporteret, at 80 % af en opløsning dryppet i et kaninøje udskilles gennem saccus conjunctivalis inden for 3-4 minutter, mens en højere opløsning end 80 % dryppet i det menneskelige øje udskilles inden for 1-2 minutter (10). Ved STE-forsøgsmetoden forsøges det at tilnærme disse eksponeringstider ved anvendelse af cytotoksisk effekt som en effektparameter for at vurdere omfanget af skade på SIRC-celler efter en fem minutter lang eksponering for testkemikaliet.
|
PÅVISNING AF KOMPETENCE
|
14.
|
Inden laboratorier påbegynder rutinemæssig brug af STE-metoden, som beskrives i denne forsøgsmetode, bør de påvise deres tekniske kompetence ved korrekt identifikation af de 11 anbefalede stoffer i tabel 1. Disse stoffer blev udvalgt til at repræsentere det fuldstændige responsområde for alvorlige øjenskader eller øjenirritation på grundlag af resultaterne af in vivo-kaninøjentest (TG 405) og UN GHS/CLP-klassificeringssystemet (1). Andre udvælgelseskriterier var, at stofferne skulle være kommercielt tilgængelige, at in vivo-referencedata af høj kvalitet skulle kunne fås i handelen, og at in vitro-data af høj kvalitet fra STE-forsøgsmetoden skulle være tilgængelige (3). I situationer, hvor et stof på listen ikke kan fås, eller hvor det er begrundet, kan et andet stof anvendes, hvis der foreligger tilstrækkelige in vivo- og in vitro-referencedata herfor, forudsat at der benyttes de samme kriterier som beskrevet her.
Tabel 1
Listen over kompetencestoffer
|
Stof
|
CAS RN
|
Kemikaliegruppe (70)
|
Fysisk form
|
In vivo UN GHS/CLP-kategori (71)
|
Opløsningsmiddel i STE-test
|
STE UN GHS/CLP-kat.
|
|
Benzalkoniumchlorid (10 %, vandig)
|
8001-54-5
|
Oniumforbindelse
|
Flydende
|
Kategori 1
|
Fysiologisk saltopløsning
|
Kategori 1
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Ether
|
Flydende
|
Kategori 1
|
Fysiologisk saltopløsning
|
Kategori 1
|
|
Acid Red 92
|
18472-87-2
|
Heterocyklisk forbindelse, bromforbindelse, chlorforbindelse
|
Fast
|
Kategori 1
|
Fysiologisk saltopløsning
|
Kategori 1
|
|
Natriumhydroxid
|
1310-73-2
|
Alkali, uorganisk kemikalie
|
Fast
|
Kategori 1 (72)
|
Fysiologisk saltopløsning
|
Kategori 1
|
|
Butyrolacton
|
96-48-0
|
Lacton, heterocyklisk forbindelse
|
Flydende
|
Kategori 2A (kategori 2 i CLP)
|
Fysiologisk saltopløsning
|
Der kan ikke fremsættes nogen forudsigelser
|
|
1-octanol
|
111-87-5
|
Alkohol
|
Flydende
|
Kategori 2A/B (73) (kategori 2 i CLP)
|
Mineralolie
|
Der kan ikke fremsættes nogen forudsigelser
|
|
Cyclopentanol
|
96-41-3
|
Alkohol, kulbrinte, cyklisk
|
Flydende
|
Kategori 2A/B (74) (kategori 2 i CLP)
|
Fysiologisk saltopløsning
|
Der kan ikke fremsættes nogen forudsigelser
|
|
2-ethoxyethylacetat
|
111-15-9
|
Alkohol, Ether
|
Flydende
|
Uden for kategori
|
Fysiologisk saltopløsning
|
Uden for kategori
|
|
Dodecan
|
112-40-3
|
Kulbrinte, cyklisk
|
Flydende
|
Uden for kategori
|
Mineralolie
|
Uden for kategori
|
|
Methylisobutylketon
|
108-10-1
|
Keton
|
Flydende
|
Uden for kategori
|
Mineralolie
|
Uden for kategori
|
|
1,1-dimethylguanidinsulfat
|
598-65-2
|
Amidin, svovlforbindelse
|
Fast
|
Uden for kategori
|
Fysiologisk saltopløsning
|
Uden for kategori
|
|
Forkortelser: CAS RN = Registernummer i Chemical Abstracts Service
|
|
FREMGANGSMÅDE
Præparering af cellemonolaget
|
15.
|
Cornea-cellelinjen fra kanin, SIRC, bør anvendes til at udføre STE-forsøgsmetoden. Det anbefales, at SIRC-celler anskaffes fra en velkvalificeret cellebank som f.eks. American Type Culture Collection CCL60.
|
|
16.
|
SIRC-celler dyrkes ved 37 °C under 5 % CO2 og fugtig atmosfære i en dyrkningsbeholder med et dyrkningsmedium bestående af Eagle’s minimum essential medium (MEM) suppleret med 10 % føtalt kalveserum (FBS), 2 mM L-glutamin, 50-100 enheder/ml penicillin og 50-100 μg/ml streptomycin. Celler, der er flydt sammen i dyrkningsflasken, bør adskilles ved hjælp af en opløsning af trypsin-ethylendiamintetraeddikesyre med eller uden brug af en celleskraber. Celler opformeres (f.eks. 2-3 generationer) i en dyrkningsflaske, før de anvendes til rutinetest og bør højst dyrkes i 25 generationer fra optøning.
|
|
17.
|
Celler, der er klar til brug i STE-forsøget, præpareres herefter ved passende tæthed og udsås på plader med 96 brønde. Den anbefalede celleudsåningstæthed er 6,0 × 103 celler pr. brønd, når cellerne anvendes fire dage efter udsåning, eller 3,0 × 103 celler pr. brønd, når cellerne anvendes fem dage efter udsåning ved en dyrkningsvolumen på 200 μl. Celler, der anvendes til STE-forsøget og udsås i et dyrkningsmedium med en passende tæthed, vil nå en konfluens på over 80 % på forsøgstidspunktet, dvs. 4-5 dage efter udsåningen.
|
Påføring af testkemikalier og kontrolstoffer
|
18.
|
Førstevalget af opløsningsmiddel til opløsning eller suspension af testkemikalier er fysiologisk saltopløsning. Hvis testkemikaliet udviser lav opløselighed eller ikke kan opløses eller suspenderes ensartet i mindst fem minutter i fysiologisk saltopløsning, anvendes 5 % DMSO (CAS#67-68-5) i fysiologisk saltopløsning som et sekundært valg af opløsningsmiddel. Til testkemikalier, der ikke kan opløses eller suspenderes ensartet i mindst fem minutter i enten en fysiologisk saltopløsning eller 5 % DMSO i fysiologisk saltopløsning, anvendes mineralolie (CAS#8042-47-5) som et tredjevalg af opløsningsmiddel.
|
|
19.
|
Testkemikalier opløses eller suspenderes ensartet i det valgte opløsningsmiddel ved en koncentration på 5 % (w/w) og yderligere fortyndet med en 10-fold seriefortynding til 0,5 % og 0,05 % koncentration. Hvert testkemikalie skal testes ved koncentrationer på både 5 % og 0,05 %. Celler dyrket på en plade med 96 brønde eksponeres for 200 μl/brønd med enten en 5 %- eller en 0,05 %-koncentration af testkemikalieopløsningen (eller -suspensionen) i fem minutter ved stuetemperatur. Testkemikalier (stoffer med kun én bestanddel eller stoffer med flere bestanddele eller blandinger) anses for at være ublandede stoffer og fortyndes eller suspenderes i overensstemmelse med metoden uanset deres renhed.
|
|
20.
|
Dyrkningsmediet, som er beskrevet i punkt 16, anvendes som mediekontrol på hver plade for hver repetition. Cellerne skal desuden eksponeres for prøver af kontrolkultur med opløsningsmiddel på hver plade for hver repetition. De opløsningsmidler, der er anført i punkt 18, har ingen negative virkninger på SIRC-cellers levedygtighed.
|
|
21.
|
Ved STE-forsøgsmetoden skal 0,01 % natriumlaurylsulfat (SLS) i fysiologisk saltopløsning anvendes som positiv kontrol på hver plade for hver repetition. For at beregne cellernes levedygtighed i den positive kontrol skal hver plade for hver repetition også omfatte en kontrol med fysiologisk saltopløsning.
|
|
22.
|
En blindprøve er nødvendig for at bestemme kompensationen for optisk tæthed og bør udføres på brønde, som kun indeholder phosphatbufret fysiologisk saltopløsning, men ingen calcium og magnesium (PBS-) eller celler.
|
|
23.
|
Hver prøve (testkemikalie ved 5 % og 0,05 %, mediekontrol, kontrolkultur med opløsningsmiddel og positiv kontrol) bør testes tre gange i hver repetition ved at eksponere cellerne for 200 μl af det rette test- eller kontrolkemikalie i fem minutter ved stuetemperatur.
|
|
24.
|
Referencestoffer er nyttige i forbindelse med vurdering af øjenirritationspotentialet for ukendte kemikalier fra en særlig kemisk stofgruppe eller produktgruppe eller vurdering af det relative irritationspotentiale for et øjenirriterende stof inden for et specifikt område af irritationsresponser.
|
Måling af cellelevedygtighed
|
25.
|
Efter eksponering vaskes cellerne to gange med 200 μl PBS, og 200 μl MTT-opløsning (0,5 mg MTT/ml dyrkningsmedium) tilsættes. Efter en to timer lang reaktionstid i en inkubator (37 °C, 5 % CO2) dekanteres MTT-opløsningen, MTT-formazan ekstraheres ved at tilsætte 200 μl 0,04 N hydrochloridsyre-isopropanol i 60 minutter i mørke ved stuetemperatur, og absorbansen af MTT-formazanopløsningen måles ved 570 nm med en pladelæser. Der opstår kun interferens mellem testkemikalier og MTT-assayet (med farvestoffer eller direkte MTT-opløsninger), hvis en betydelig mængde testkemikalie tilbageholdes i testsystemet som følge af rensning efter eksponering, hvilket er tilfældet med rekonstrueret human cornea i 3D eller rekonstrueret human epidermisvæv i 3D, men er ikke relevant for 2D-cellekulturer, som anvendes til STE-forsøgsmetoden.
|
Fortolkning af resultater og forudsigelsesmodel
|
26.
|
De værdier for optisk tæthed (OD), der er opnået for hvert testkemikalie, benyttes herefter til at beregne cellelevedygtighed i forhold til kontrolturen med opløsningsmiddel, der er fastsat til 100 %. Den relative cellelevedygtighed angives som en procentdel og opnås ved at dividere testkemikaliets OD med OD for kontrolkulturen med opløsningsmiddel efter fratrækning af blindprøvens OD fra begge værdier.
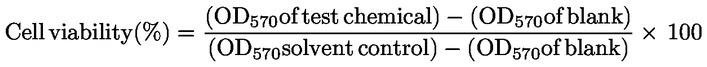
Tilsvarende angives den relative cellelevedygtighed for hver kontrolkultur med opløsningsmiddel som en procentdel og fås ved at dividere OD for hver kontrolkultur med opløsningsmiddel med OD for mediekontrollen efter fratrækning af blindprøvens OD fra begge værdier.
|
|
27.
|
Der bør udføres tre uafhængige repetitioner, som hver indeholder tre replikatbrønde (dvs. n=9). Det aritmetiske gennemsnit af de tre brønde for hvert testkemikalie og hver kontrolkultur med opløsningsmiddel i hver uafhængig repetition bruges til at beregne det aritmetiske gennemsnit for relativ cellelevetid. Det endelige aritmetiske gennemsnit for cellelevedygtighed beregnes ud fra de tre uafhængige repetitioner.
|
|
28.
|
Cut-off-værdierne for cellelevedygtighed til identificering af testkemikalier, som medfører alvorlige øjenskader (UN GHS/CLP-kategori 1), og testkemikalier, der ikke kræver klassificering for øjenirritation eller alvorlige øjenskader (UN GHS/CLP – uden for kategori) er angivet nedenfor.
|
Tabel 2
Forudsigelsesmodel for STE-forsøgsmetoden
|
Cellernes levedygtighed
|
UN GHS/CLP-klassificering
|
Anvendelse
|
|
Ved 5 %
|
Ved 0,05 %
|
|
> 70 %
|
> 70 %
|
Uden for kategori
|
Stoffer og blandinger med undtagelse af: i) meget flygtige stoffer med et damptryk over 6 kPa (75) og ii) andre faste kemikalier (stoffer og blandinger) end overfladeaktive stoffer og blandinger, som kun består af overfladeaktive stoffer
|
|
≤ 70 %
|
> 70 %
|
Der kan ikke fremsættes nogen forudsigelser
|
Ikke relevant
|
|
≤ 70 %
|
≤ 70 %
|
Kategori 1
|
Stoffer og blandinger (76)
|
Acceptkriterier
|
29.
|
Testresultaterne anses for at være acceptable, når alle de følgende kriterier er opfyldt:
|
a)
|
Mediekontrollens optiske tæthed (eksponeret for dyrkningsmediet) bør være 0,3 eller højere efter fratrækning af blindprøvens optiske tæthed.
|
|
b)
|
Levedygtigheden for kontrolkulturen med opløsningsmiddel bør være 80 % eller derover i forhold til mediekontrollen. Såfremt der anvendes mange kontrolkulturer med opløsningsmiddel i hver repetition, bør hver af disse kontroller vise en større cellelevedygtighed end 80 % for at kvalificere de testede testkemikalier med de pågældende opløsningsmidler.
|
|
c)
|
Den cellelevedygtighed, der opnås med den positive kontrol (0,01 % SLS), bør ligge inden for standardafvigelserne for det historiske gennemsnit. De øvre og nedre acceptgrænser for den positive kontrol bør opdateres hyppigt, dvs. hver tredje måned eller hver gang, der udføres en acceptabel test i laboratorier, hvor der sjældent udføres test (dvs. sjældnere end én gang om måneden). Såfremt laboratoriet ikke gennemfører et tilstrækkeligt antal forsøg for at fastlægge en statistisk robust, positiv kontrolfordeling, kan det accepteres at benytte de øvre og nedre acceptgrænser, som fastsættes af metodeudvikleren, dvs. mellem 21,1 % og 62,3 %, i overensstemmelse med laboratoriets tidligere data, mens en intern fordeling opbygges under de første rutinetest.
|
|
d)
|
Standardafvigelsen for den endelige cellelevedygtighed, som udledes af tre uafhængige repetitioner, bør ligge under 15 % for både 5 %- og 0,05 %-koncentrationer af testkemikaliet.
Hvis et eller flere af disse kriterier ikke opfyldes, bør der ses bort fra resultaterne, og tre andre uafhængige repetitioner udføres.
|
|
DATA OG RAPPORTERING
Data
|
30.
|
Data rapporteres for hver enkelt brønd (f.eks. værdier for cellernes levedygtighed) for hver repetition og samlet gennemsnit, standardafvigelse og klassificering.
|
Testrapport
|
31.
|
Testrapporten bør indeholde følgende oplysninger:
|
|
Testkemikalie og kontrolstoffer
|
—
|
Stof med kun én bestanddel: kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-registreringsnummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
—
|
stof med flere bestanddele, UVCB-stoffer og blandinger: kendetegnet så vidt muligt ved f.eks. kemisk identitet (jf. ovenfor), renhed, kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber (jf. ovenfor) i det omfang, det er muligt
|
|
—
|
fysisk tilstand, flygtighed, pH, LogP, molekylvægt, kemisk gruppe og yderligere fysisk-kemiske egenskaber, som er relevante for undersøgelsen, i det omfang det er muligt
|
|
—
|
renhed, kemisk identitet af urenheder i det omfang, det er relevant og praktisk muligt, osv.
|
|
—
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
—
|
opbevaring og stabilitet i det omfang, det er muligt.
|
|
|
|
Betingelser for forsøgsmetoden og procedurer
|
—
|
navn og adresse på sponsoren, testfaciliteten og undersøgelseslederen
|
|
—
|
beskrivelse af den anvendte forsøgsmetode
|
|
—
|
anvendte cellelinjer, deres oprindelse, antal generationer og graden af konfluens for de celler, der anvendes til test
|
|
—
|
detaljer om den anvendte testprocedure
|
|
—
|
antal anvendte gentagelser og replikater
|
|
—
|
koncentrationer af testkemikaliet (hvis en anden end de anbefalede)
|
|
—
|
begrundelse for valg af opløsningsmiddel for de enkelte testkemikalier
|
|
—
|
varigheden af eksponeringen for testkemikaliet (hvis en anden end den anbefalede)
|
|
—
|
beskrivelse af eventuelle ændringer i testproceduren
|
|
—
|
beskrivelse af de anvendte vurderings- og beslutningskriterier
|
|
—
|
henvisning til det tidligere positive kontrolgennemsnit og standardafvigelse:
|
|
—
|
påvisning af laboratoriets kompetence til at anvende forsøgsmetoden (f.eks. ved test af kompetencestoffer) eller til at påvise reproducerbare resultater af forsøgsmetoden over tid.
|
|
|
|
Resultater
|
—
|
for hvert testkemikalie og kontrolstof og hver testet koncentration bør de enkelte OD-værdier for hver replikatbrønd opstilles i tabelform, og følgende angives: det aritmetiske gennemsnit af OD-værdier for hver uafhængig repetition, den procentvise cellelevedygtighed for hver uafhængig repetition og det endelige aritmetiske gennemsnit for cellelevedygtighed og standardafvigelse i procent over tre repetitioner
|
|
—
|
resultater for dyrkningsmedium, opløsningsmiddel og positiv kontrol, der påviser egnede acceptkriterier for undersøgelsen
|
|
—
|
beskrivelse af andre observerede virkninger
|
|
—
|
den samlede afledte klassificering med henvisning til den anvendte forudsigelsesmodel og/eller de anvendte beslutningskriterier.
|
|
|
|
Diskussion af resultaterne
|
|
LITTERATUR
|
(1)
|
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York and Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Tilgængelig på: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, s. 1 855-1 869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Tilgængelig på: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Kapitel B.5 i dette bilag, Akut øjenirritation/-ætsning.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS og Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1 648-1 653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Bruxelles, Belgien.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442–449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Organisation for Economic Cooperation and Development, Paris.
|
Tillæg
DEFINITIONER
Nøjagtighed
: overensstemmelsen mellem forsøgsmetodens resultater og accepterede referenceværdier. Den er et mål for forsøgsmetodens ydeevne og et af aspekterne af relevans. Udtrykket benyttes ofte som synonym for konkordans, forstået som den andel af korrekte udfald, forsøgsmetoden giver (13).
Referencestof
: et stof, der anvendes som standard til sammenligning med et testkemikalie. Et referencestof skal have følgende egenskaber: (i) en eller flere ensartede og pålidelige kilder, (ii) strukturel og funktionel lighed med den stofgruppe, der testes, (iii) kendte fysisk/kemiske egenskaber, (iv) støttedata om kendte egenskaber og (v) kendt styrke i det ønskede responsområde.
Bottom-up-tilgang
: en trinvis tilgang til et testkemikalie, som mistænkes for ikke at kræve klassificering for øjenirritation eller alvorlige øjenskader, og som starter med bestemmelse af kemikalier, der ikke kræver klassificering (negativt resultat) i forhold til andre kemikalier (positivt resultat).
Kemikalie
: et stof eller en blanding.
Øjenirritation
: frembringelse af forandring i øjet, som opstår efter påføring af et testkemikalie på øjets anteriore overflade, og som er fuldt reversibelt inden for 21 dage efter påføring. Synonymt med "Reversible virkninger på øjet" og UN GHS/CLP-kategori 2.
Falsk negativ-rate
: den andel af alle positive kemikalier, som ved en forsøgsmetode fejlagtigt identificeres som negative. Det er en indikator for forsøgsmetodens ydeevne.
Falsk positiv-rate
: den andel af alle negative kemikalier, som ved en forsøgsmetode fejlagtigt identificeres som positive. Det er en indikator for forsøgsmetodens ydeevne.
Fare
: den iboende egenskab ved et middel eller en situation, som kan forårsage skadelige virkninger, når en organisme, et system eller en (sub)population eksponeres for det pågældende middel.
Mediekontrol
: et ubehandlet replikat, som indeholder alle et testsystems komponenter. Prøven behandles sammen med prøver behandlet med testkemikaliet og andre kontroller for at afgøre, hvorvidt opløsningsmidlet interagerer med testsystemet.
Blanding
: blanding eller opløsning, der består af to eller flere stoffer.
Stof med kun én bestanddel
: et stof, der defineres af sin mængdemæssige sammensætning, hvori en hovedbestanddel udgør mindst 80 % (w/w).
MTT
: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid thiazolylblåt tetrazolium-bromid.
Stof med flere bestanddele
: et stof, der defineres af sin mængdemæssige sammensætning, hvori flere end én hovedbestanddel er til stede i en koncentration på ≥ 10 % (w/w) og < 80 % (w/w). Et stof, der består af flere forskellige bestanddele, er resultatet af en fremstillingsproces. Forskellen mellem en blanding og et stof med forskellige bestanddele er, at blandingen er fremstillet ved at blande to eller flere stoffer uden kemisk reaktion. Et stof, der består af flere forskellige bestanddele, er resultatet af en kemisk reaktion.
OD
: optisk tæthed.
Positiv kontrol
: et replikat, som indeholder alle komponenterne fra et testsystem og er behandlet med et stof, som vides at inducere en positiv respons. For at sikre, at det er muligt at vurdere variabiliteten i den positive kontrolrespons over tid, bør den positive respons ikke være overdrevent stor.
Relevans
: beskrivelse af sammenhængen mellem testen og den pågældende virkning og dens meningsfuldhed og brugbarhed til et bestemt formål. Det angives, i hvilket omfang testen måler eller forudsiger den pågældende biologiske virkning korrekt. Forsøgsmetodens nøjagtighed (konkordans) indgår i vurderingen af dens relevans (10).
Pålidelighed
: mål for, i hvilken grad en forsøgsmetode kan reproduceres i og mellem laboratorier over en længere periode, når den udføres efter samme protokol. Den bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier og repeterbarhed inden for laboratorier (13).
Følsomhed
: andel af alle positive/aktive kemikalier, der klassificeres korrekt ved testen. Det er et mål for nøjagtigheden af en forsøgsmetode, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en forsøgsmetodes relevans (10).
Alvorlig øjenskade
: frembringelse af vævsbeskadigelse i øjet eller alvorlig fysisk synsnedsættelse, som opstår efter påføring af et testkemikalie på øjets anteriore overflade, og som ikke er fuldt reversibel inden for 21 dage efter påføring. Synonymt med "irreversible virkninger på øjet" og UN GHS/CLP-kategori 1.
Opløsningsmiddel-/bærestofkontrol
: en ubehandlet prøve, som indeholder alle komponenterne fra et testsystem, herunder det opløsningsmiddel eller bærestof, som behandles sammen med de testkemikaliebehandlede prøver og andre kontroller for at fastlægge basisresponsen for de prøver, som er behandlet med testkemikaliet opløst i det samme opløsningsmiddel eller bærestof. Når denne prøve testes med en samtidig mediekontrol, viser den også, hvorvidt opløsningsmidlet eller bærestoffet interagerer med testsystemet.
Specificitet
: andel af alle negative/inaktive kemikalier, der klassificeres korrekt ved testen. Det er et mål for nøjagtigheden af en forsøgsmetode, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en forsøgsmetodes relevans (13).
Stof
: et grundstof og forbindelser heraf, naturlige eller industrielt fremstillet, indeholdende sådanne tilsætningsstoffer, som er nødvendige til bevarelse af stoffets stabilitet, og sådanne urenheder, som følger af fremstillingsprocessen, bortset fra opløsningsmidler, der kan udskilles, uden at det påvirker stoffets stabilitet eller ændrer dets sammensætning.
Overfladeaktivt stof
: også kaldet for afspændingsmiddel. Dette kemikalie, f.eks. et vaske- og rengøringsmiddel, der kan reducere overfladespændingen i en væske og dermed give den mulighed for at skumme eller trænge gennem faste stoffer, kaldes også for befugtningsmiddel.
Testkemikalie
: ethvert stof eller enhver blanding, der testes ved hjælp af denne forsøgsmetode.
Trindelt testningsstrategi
: ved anvendelse af den trinvise teststrategi gennemgås alle eksisterende oplysninger om et testkemikalie i en bestemt rækkefølge ved anvendelse af en "weight of evidence"-proces på hvert trin for at fastslå, om der foreligger tilstrækkelige oplysninger til at træffe en fareklassificeringsafgørelse, inden der fortsættes til næste trin. Hvis et testkemikalies irritationspotentiale kan underbygges på grundlag af eksisterende oplysninger, er der ikke behov for at foretage yderligere test. Hvis et testkemikalies irritationspotentiale ikke kan underbygges på grundlag af eksisterende oplysninger, foretages en trindelt sekventiel dyreforsøgsprocedure, indtil der kan foretages en utvetydig klassificering.
Top-down-tilgang
: trinvis tilgang, som anvendes for et testkemikalie, der mistænkes for at forårsage alvorlig øjenskade, og som starter med bestemmelse af kemikalier, som forårsager alvorlig øjenskade (positivt resultat), i forhold til andre kemikalier (negativt resultat).
De Forenede Nationers globalt harmoniserede system for klassificering og mærkning af kemikalier (United Nations Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: Et system til klassificering af kemikalier (stoffer og blandinger) efter standardiserede typer og niveauer af fysisk fare, sundhedsfare og miljøfare med tilhørende kommunikationselementer såsom piktogrammer, signalord, faresætninger, sikkerhedssætninger og sikkerhedsdatablade, således at oplysninger om kemikaliernes skadelige virkninger kan videreformidles med henblik på at beskytte mennesker (f.eks. arbejdsgivere, arbejdstagere, transportører, forbrugere og redningspersonel) og miljø (1).
UN GHS/CLP-kategori 1
: se "Alvorlige øjenskader".
UN GHS/CLP-kategori 2
: se "Øjenirritation".
UN GHS/CLP – uden for kategori
: Kemikalier, der ikke er klassificeret som øjenirriterende stoffer i UN GHS/CLP-kategori 1 eller 2 (eller UN GHS-kategori 2A 2Beller 2B).
UVCB
: stoffer med ukendt eller variabel sammensætning, komplekse reaktionsprodukter eller biologiske materialer.
B.69 FORSØGSMETODE MED REKONSTRUERET HUMANT CORNEA-LIGNENDE EPITHEL (RhCE) TIL PÅVISNING AF KEMIKALIER, SOM IKKE KRÆVER KLASSIFICERING OG MÆRKNING FOR ØJENIRRITATION ELLER ALVORLIGE ØJENSKADER
INDLEDNING
|
1.
|
Denne forsøgsmetode svarer til OECD’s testvejledning (Test Guideline (TG)) 492 (2017). Alvorlige øjenskader defineres som frembringelse af vævsbeskadigelse i øjet eller alvorlig fysisk synsnedsættelse, som opstår efter påføring af et testkemikalie på øjets anteriore overflade, og som ikke er fuldt reversibel inden for 21 dage efter påføring som defineret i De Forenede Nationers globale harmoniserede system for klassificering og mærkning af kemikalier (UN GHS) (1) og Den Europæiske Unions forordning (EU) 1272/2008 om klassificering, mærkning og emballering af stoffer og blandinger (CLP) (77). Øjenirritation defineres også i overensstemmelse med UN GHS og CLP som frembringelse af forandringer i øjet, der opstår efter påføring af et testkemikalie på øjets anteriore overflade og er fuldt reversible inden for 21 dage efter påføring. Testkemikalier, der medfører alvorlige øjenskader, er klassificeret som UN GHS/CLP-kategori 1, mens kemikalier, der medfører øjenirritation, er klassificeret som UN GHS/CLP-kategori 2. Testkemikalier, der ikke klassificeres for øjenirritation eller alvorlig øjenskade, defineres på samme måde som de kemikalier, der ikke opfylder kravene med hensyn til klassificering som UN GHS/CLP-kategori 1 eller 2 (2A eller 2B), dvs. de betegnes som UN GHS/CLP – uden for kategori.
|
|
2.
|
Vurderingen af alvorlige øjenskader/øjenirritation har typisk omfattet brug af forsøgsdyr (TM B.5) (2). Valget af den mest egnede forsøgsmetode og anvendelsen af denne forsøgsmetode skal ses i sammenhæng med OECD’s vejledende dokument om integrerede tilgange til testning og vurdering for alvorlige øjenskader og øjenirritation (39).
|
|
3.
|
Denne forsøgsmetode beskriver en in vitro-procedure, som gør det muligt at identificere kemikalier (stoffer og blandinger), der ikke kræver klassificering og mærkning for øjenirritation eller alvorlige øjenskader i overensstemmelse med UN GHS/CLP. Den gør brug af rekonstrueret humant cornea-lignende epithel (RhCE), som nøje efterligner det humane cornea-epithels histologiske, morfologiske, biokemiske og fysiologiske egenskaber. Fire andre in vitro-forsøgsmetoder er blevet valideret, betragtet som videnskabeligt gyldige og vedtaget som TM B.47 (3), B.48 (4), B.61 (5) og B.68 (6), hvis effektparameter for menneskers sundhed er alvorlige øjenskader/øjenirritation.
|
|
4.
|
To validerede test, der anvender RhCE-modeller, der kan fås i handelen, er inkluderet i denne forsøgsmetode. Der er foretaget valideringsundersøgelser til vurdering af øjenirritation/alvorlige øjenskader (7)(8)(9)(10)(11)(12)(13) under anvendelse af EpiOcular™ Eye Irritation Test (EIT) og SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). Som testsystem gør hver af disse test brug af RhCE-vævsmodeller, der kan fås i handelen, og som i den følgende tekst omtales som de validerede referencemetoder – henholdsvis VRM 1 og VRM 2. På grundlag af disse valideringsundersøgelser og det uafhængige peer review heraf (9)(12) blev det konkluderet, at EpiOcular™ EIT og SkinEthic™ HCE EIT kan identificere kemikalier korrekt (både stoffer og blandinger), som ikke kræver klassificering og mærkning for øjenirritation eller alvorlige øjenskader i overensstemmelse med UN GHS, og testene blev anbefalet som videnskabeligt gyldige til det pågældende formål (13).
|
|
5.
|
Det er i øjeblikket generelt accepteret, at der i den nærmeste fremtid ikke vil være en enkelt in vitro-forsøgsmetode, der fuldt ud vil kunne erstatte in vivo-Draize-øjentesten(2)(14) til forudsigelse af hele spektret af alvorlige øjenskader/øjenirritation for forskellige kemikaliegrupper. Strategiske kombinationer af flere alternative forsøgsmetoder i (trinvise) teststrategier såsom bottom-up/top-down-tilgangen kan dog muligvis fuldt ud erstatte Draize-øjentesten (15). Bottom-up-tilgangen (15) er beregnet til at blive anvendt, når et kemikalie på grundlag af eksisterende oplysninger ikke forventes at forårsage tilstrækkelig øjenirritation til at kræve en klassificering, mens top-down-tilgangen (15) er beregnet til at blive anvendt, når et kemikalie på grundlag af eksisterende oplysninger forventes at forårsage alvorlige øjenskader. EpiOcular™ EIT og SkinEthic™ HCE EIT anbefales til at identificere kemikalier, der ikke kræver klassificering for øjenirritation eller alvorlige øjenskader ifølge UN GHS/CLP (uden for kategori), uden yderligere testning inden for en teststrategi som bottom-up/top-down-tilgangen i henhold til Scott et al., f.eks. som et indledende skridt i bottom-up-tilgangen eller som et af de sidste skridt i top-down-tilgangen (15). EpiOcular™ EIT og SkinEthic™ HCE EIT er dog ikke beregnet til at kunne sondre mellem UN GHS/CLP-kategori 1 (alvorligeøjenskader) og UN GHS/CLP-kategori 2 (øjenirritation). Denne opdeling vil skulle foretages på et andet trin i teststrategien (15). Et testkemikalie, der med EpiOcular™ EIT eller SkinEthic™ HCE EIT identificeres som et kemikalie, der kræver klassificering for øjenirritation/alvorlige øjenskader, vil således kræve yderligere testning (in vitro og/eller in vivo), for at der kan nås en endelig konklusion (UN GHS/CLP – uden for kategori, kategori 2 eller kategori 1) under anvendelse af f.eks. TM B.47, B.48, B.61 eller B.68.
|
|
6.
|
Formålet med denne forsøgsmetode er at beskrive den procedure, som anvendes til at vurdere et testkemikalies potentielle risiko for at forårsage øjenskader baseret på dets evne til at fremkalde en cytotoksisk effekt i et RhCE-væv som målt ved MTT-assayet (16) (jf. punkt 21). RhCE-vævets levedygtighed efter eksponering for et testkemikalie bestemmes ved sammenligning med væv, der behandles med det negative kontrolstof (levedygtighed i %) og anvendes herefter til at forudsige testkemikaliets risiko for at forårsage øjenskade.
|
|
7.
|
Der foreligger ydeevnestandarder (17) for at lette valideringen af nye eller ændrede in vitro-RhCE-baserede test, der ligner EpiOcular™ EIT og SkinEthic™ HCE EIT, i overensstemmelse med OECD’s vejledende dokument nr. 34 (18) og give mulighed for rettidig ændring af OECD TG 492, så de kan blive opført heri. Gensidig godkendelse af data i henhold til OECD-aftalen vil kun være garanteret for test, der er valideret i henhold til ydeevnestandarderne, hvis disse test er gennemgået og medtaget i OECD’s tilhørende testvejledning.
|
DEFINITIONER
|
8.
|
Definitioner findes i tillæg 1.
|
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER
|
9.
|
Denne forsøgsmetode er baseret på tredimensionale RhCE-vævsmodeller, som fås i handelen. De er fremstillet ved anvendelse af enten primære humane epidermis-keratinocytter (dvs. EpiOcular™ OCL-200) eller humane udødeliggjorte cornea-epithelceller (dvs. SkinEthic™ HCE/S). EpiOcular™ OCL-200- og SkinEthic™ HCE/S-RhCE-vævsmodeller ligner den tredimensionale opbygning af in vivo-cornea-epithel og fremstilles ved hjælp af celler fra relevante arter (19)(20). Endvidere måler testene direkte cytotoksisk effekt som følge af kemikaliets penetration gennem cornea og forårsagelse af celle- og vævsskade. Den cytotoksiske reaktion bestemmer herefter det samlede resultat af alvorlige øjenskader/øjenirritation in vivo. Cellebeskadigelse kan opstå ved flere virkemåder (jf. punkt 20), men cytotoksisk effekt spiller en vigtig, hvis ikke primær, mekanistisk rolle for bestemmelse af den samlede reaktion på et kemikalie i form af alvorlige øjenskader/øjenirritation, som giver sig udtryk in vivo, hovedsagelig ved cornea-uklarhed, iritis, conjunctiva-rødme og/eller conjunctiva-chemosis, uanset hvilke fysisk-kemiske processer, der ligger til grund for vævsbeskadigelsen.
|
|
10.
|
En lang række af kemikalier, som dækker en bred vifte af kemiske stoffer, kemikaliegrupper, molekylvægte, LogP’er, kemiske strukturer osv., er blevet testet i den valideringsundersøgelse, som ligger til grund for denne forsøgsmetode. EpiOcular™ EIT-valideringsdatabasen indeholdt 113 kemikalier i alt, der dækker 95 forskellige organiske funktionelle grupper ifølge en analyse af OECD’s QSAR-værktøjskasse (8). De fleste af disse kemikalier var stoffer med kun én bestanddel, men flere stoffer med flere bestanddele (bl.a. 3 homopolymerer, 5 copolymerer og 10 kvasi-polymerer) blev også medtaget i undersøgelsen. Med hensyn til fysisk form og UN GHS/CLP-kategorier fordelte de 113 testede kemikalier sig som følger: 13 væsker i kategori 1, 15 faste stoffer i kategori 1, 6 væsker i kategori 2A, 10 faste stoffer i kategori 2A, 7 væsker i kategori 2B, 7 faste stoffer i kategori 2B, 27 væsker uden for kategori og 28 faste stoffer uden for kategori (8). SkinEthic™ HCE EIT-valideringsdatabasen indeholdt 200 kemikalier i alt, der dækker 165 forskellige organiske funktionelle grupper (8)(10)(11). De fleste af disse kemikalier var stoffer med kun én bestanddel, men flere stoffer med flere bestanddele (bl.a. 10 polymerer) blev også medtaget i undersøgelsen. Med hensyn til fysisk form og UN GHS/CLP-kategorier fordelte de 200 testede kemikalier sig som følger: 27 væsker i kategori 1, 24 faste stoffer i kategori 1, 19 væsker i kategori 2A, 10 faste stoffer i kategori 2A, 9 væsker i kategori 2B, 8 faste stoffer i kategori 2B, 50 væsker uden for kategori og 53 faste stoffer uden for kategori (10)(11).
|
|
11.
|
Denne forsøgsmetode kan benyttes til test af stoffer og blandinger og faste stoffer, væsker, halvfaste stoffer og vokser. Væskerne kan være vandige eller ikke-vandige, og de faste stoffer kan være vandopløselige eller uopløselige i vand. Faste stoffer skal males til et fint pulver, før de påføres, når det overhovedet er muligt. Der kræves ingen anden forbehandling af teststoffet. Gasser og aerosoler er ikke vurderet ved en valideringsundersøgelse. Selv om det er tænkeligt, at disse stoffer kan undersøges ved hjælp af RhCE-teknologi, muliggør den nuværende forsøgsmetode ikke undersøgelse af gasser og aerosoler.
|
|
12.
|
Testkemikalier, der absorberer lys i det samme område som MTT-formazan (naturligt eller efter behandling), og testkemikalier, som direkte kan interferere med målinger af vævets levedygtighed og reducere vitalfarvestoffet MTT (til MTT-formazan), kan kræve anvendelse af tilpassede kontroller med henblik på korrektioner. Hvilken type af tilpassede kontroller, der kan være nødvendige, varierer alt efter den type interferens, testkemikaliet fremkalder, og den procedure, der benyttes til at kvantificere MTT-formazan (jf. punkt 36-42).
|
|
13.
|
Resultater, der er fremkommet ved undersøgelser til prævalidering (21)(22) og fuld validering (8)(10)(11), har vist, at både EpiOcular™ EIT og SkinEthic™ HCE EIT kan overføres til laboratorier, der betragtes som uerfarne med gennemførelsen af assays, og også kan reproduceres i og mellem laboratorier. På grundlag af disse undersøgelser er niveauet for reproducerbarhed med hensyn til konkordans i de forudsigelser, der kan forventes af EpiOcular™ EIT vedrørende data om 113 kemikalier, i størrelsesordenen 95 % i laboratorier og 93 % mellem laboratorier. Niveauet for reproducerbarhed med hensyn til konkordans i de forudsigelser, der kan forventes af SkinEthic™ HCE EIT vedrørende data om 120 kemikalier, er i størrelsesordenen 92 % i laboratorier og 95 % mellem laboratorier.
|
|
14.
|
EpiOcular™ EIT kan anvendes til at identificere kemikalier, der ikke kræver klassificering for øjenirritation eller alvorlige øjenskader i overensstemmelse med UN GHS- og CLP-klassificeringssystemet. Ud fra de data, der fremgår af valideringsundersøgelsen (8), har EpiOcular™ EIT en samlet nøjagtighed på 80 % (baseret på 112 kemikalier), en følsomhed på 96 % (baseret på 57 kemikalier), en falsk negativ-rate på 4 % (baseret på 57 kemikalier), en specificitet på 63 % (baseret på 55 kemikalier) og en falsk positiv-rate på 37 % (baseret på 55 kemikalier), sammenholdt med referencedata fra in vivo-kaninøjentest (TM B.5) (2)(14), der er klassificeret i overensstemmelse med UN GHS- og CLP-klassificeringssystemet. En undersøgelse, hvor 97 flydende agrokemiske formuleringer blev testet med EpiOcular™ EIT, viste lignende resultater af forsøgsmetoden for denne type blandinger som dem, der blev opnået i valideringsundersøgelsen (23). De 97 formuleringer fordelte sig som følger: 21 i kategori 1, 19 i kategori 2A, 14 i kategori 2B og 43 uden for kategori, klassificeret efter UN GHS-klassificeringssystemet på grundlag af referencedata fra in vivo-kaninøjentesten (TM B.5) (2)(14). Der blev opnået en samlet nøjagtighed på 82 % (baseret på 97 formuleringer), en følsomhed på 91 % (baseret på 54 formuleringer), en falsk negativ-rate på 9 % (baseret på 54 formuleringer), en specificitet på 72 % (baseret på 43 formuleringer) og en falsk positiv-rate på 28 % (baseret på 43 formuleringer) (23).
|
|
15.
|
SkinEthic™ HCE EIT kan anvendes til at identificere kemikalier, som ikke kræver klassificering for øjenirritation eller alvorlige øjenskader efter UN GHS- og CLP-klassificeringssystemet. Ud fra de data, der fremgår af valideringsundersøgelsen (10)(11), har SkinEthic™ HCE EIT en samlet nøjagtighed på 84 % (baseret på 200 kemikalier), en følsomhed på 95 % (baseret på 97 kemikalier), en falsk negativ-rate på 5 % (baseret på 97 kemikalier), en specificitet på 72 % (baseret på 103 kemikalier) og en falsk positiv-rate på 28 % (baseret på 103 kemikalier), sammenholdt med referencedata fra in vivo-kaninøjentest (TM B.5) (2)(14), der er klassificeret i overensstemmelse med UN GHS- og CLP-klassificeringssystemet.
|
|
16.
|
De falsk negativ-rater, som blev opnået med begge RhCE-test for enten stoffer eller blandinger, falder inden for den 12 % samlede sandsynlighed for, at kemikalier identificeres som enten UN GHS/CLP-kategori 2 eller UN GHS/CLP – uden for kategori ved in vivo-Draize-øjentesten i gentagne test. Dette skyldes metodens iboende variabilitet indenfor testen (24). De falsk positive-rater, som blev opnået med begge RhCE-forsøgsmetoder med stoffer eller blandinger, er ikke kritiske i forbindelse med denne forsøgsmetode, eftersom alle testkemikalier, der fremkalder en vævslevedygtighed svarende til eller lavere end de fastsatte tærskelværdier (jf. punkt 44), vil kræve yderligere test med andre in vitro-forsøgsmetoder eller som sidste mulighed hos kaniner alt efter de forskriftsmæssige krav ved anvendelse af en sekventiel teststrategi i en »weight-of-evidence«-analyse. Disse forsøgsmetoder kan anvendes til alle typer af kemikalier, hvor der bør accepteres et negativt resultat, som ikke klassificerer et kemikalie for øjenirritation og alvorlige øjenskader (UN GHS/CLP – uden for kategori). De relevante kompetente myndigheder bør høres, før EpiOcular™ EIT og SkinEthic™ HCE EIT anvendes i henhold til andre klassifikationsordninger end UN GHS/CLP.
|
|
17.
|
En begrænsning af denne forsøgsmetode er, at den ikke gør det muligt at sondre mellem øjenirritation/reversible virkninger for øjet (kategori 2) og alvorlige øjenskader/irreversible virkninger for øjet (kategori 1) som defineret i UN GHS og CLP, og heller ikke mellem øjenirriterende stoffer (valgfri kategori 2A) og milde øjenirriterende stoffer (valgfri kategori 2B), som defineret i UN GHS (1). Til disse formål kræves der yderligere testning med andre in vitro-forsøgsmetoder.
|
|
18.
|
Betegnelsen »testkemikalie« anvendes i denne forsøgsmetode om det, der testes (78), og vedrører ikke RhCE-forsøgsmetodens anvendelighed til testning af stoffer og/eller blandinger.
|
PRINCIP FOR TESTEN
|
19.
|
Testkemikaliet påføres lokalt på mindst to tredimensionale RhCE-vævsmodeller, og vævets levedygtighed måles efter eksponering og en inkubationsperiode efter behandling. RhCE-væv rekonstrueres fra primære humane epidermis-keratinocytter eller humane udødeliggjorte cornea-epithelceller, som er blevet dyrket i flere dage, så de danner et lagdelt, stærkt differentieret pladeepithel, som morfologisk ligner det, der findes i den menneskelige cornea. EpiOcular™ RhCE-vævsmodellen består af mindst tre levedygtige cellelag og en ikke-keratiniseret overflade, der udviser en cornea-lignende struktur svarende til den, som findes in vivo. SkinEthic™ HCE RhCE-vævsmodellen består af mindst fire levedygtige cellelag, herunder søjleformede basalceller, midlertidige vingeceller og overfladiske pladeceller svarende til det normale humane cornea-epithel (20)(26).
|
|
20.
|
Kemikalieinducerede alvorlige øjenskader/øjenirritation, der in vivo hovedsagelig giver sig udslag i cornea-uklarhed, iritis, conjunctiva-rødme og/eller conjunctiva-chemosis, er en følge af en kaskade af begivenheder, der begynder med kemikaliets penetration gennem cornea og/eller conjunctiva og fremkaldelse af skader på cellerne. Cellebeskadigelse kan opstå ved flere virkemåder, herunder: lysis af cellemembranen (f.eks. ved overfladeaktive stoffer, organiske opløsningsmidler), koagulering af makromolekyler (især proteiner) (f.eks. ved overfladeaktive stoffer, organiske opløsningsmidler, baser og syrer), forsæbning af lipider (f.eks. ved baser) samt alkylering og andre kovalente interaktioner med makromolekyler (f.eks. ved blegemidler, peroxider og alkylatorer) (15)(27)(28). Det er dog blevet påvist, at cytotoksisk effekt spiller en vigtig, om ikke primær, mekanistisk rolle for bestemmelse af den samlede reaktion på et kemikalie i form af alvorlige øjenskader/øjenirritation, uanset hvilke fysisk-kemiske processer, der ligger til grund for vævsbeskadigelsen (29)(30). Endvidere bestemmes et kemikalies risiko for at forårsage alvorlige øjenskader/øjenirritation primært af omfanget af den indledende skade (31), der hænger sammen med omfanget af celledød (29) og med omfanget af de efterfølgende reaktioner og senere resultater (32). Således påvirker let irriterende stoffer normalt kun corneas overfladeepithel, de mildt og moderat irriterende stoffer primært epithelet og stroma i overfladen, mens stærkt øjenirriterende stoffer beskadiger epithelet, dyb stroma og til tider corneas endothel (30)(33). Målingen af RhCE-vævsmodellens levedygtighed efter lokal eksponering for et testkemikalie for at identificere kemikalier, som ikke kræver klassificering for alvorlige øjenskader/øjenirritation (UN GHS/CLP – uden for kategori), er baseret på den antagelse, at alle kemikalier, der fremkalder alvorlige øjenskader eller øjenirritation, vil inducere cytotoksisk effekt i cornea-epithelet og/eller conjunctiva.
|
|
21.
|
RhCE-vævs levedygtighed måles typisk ved enzymomdannelse af vitalfarvestoffet MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid, thiazolylblåt tetrazolium-bromid, CAS-nummer 298-93-1) forårsaget af de levedygtige celler i vævet til et blåt MTT-formazansalt, der måles kvantitativt efter ekstraktion fra vævene (16). Kemikalier, som ikke kræver klassificering og mærkning efter UN GHS/CLP (uden for kategori), identificeres som kemikalier, der ikke nedsætter vævets levedygtighed under en fastsat tærskel (dvs. vævslevedygtighed > 60 % i EpiOcular™ EIT og SkinEthic™ HCE EITL (79) eller > 50 %, i SkinEthic™ HCE EITS (80)) (jf. punkt 44).
|
PÅVISNING AF KOMPETENCE
|
22.
|
Inden laboratorier påbegynder rutinemæssig brug af RhCE-test til lovmæssige formål, bør de påvise deres tekniske kompetence ved korrekt at forudsige de 15 kompetencekemikalier, der er anført i tabel 1. Disse kemikalier blev udvalgt blandt de kemikalier, som anvendes i valideringsundersøgelserne af VRM’er (8)(10)(11). De udvalgte kemikalier omfatter så vidt muligt kemikalier, der: (i) dækker forskellige fysiske former, ii) dækker det fulde spektrum af in vivo-reaktioner i form af alvorlige øjenskader/øjenirritation baseret på resultater af høj kvalitet opnået ved in vivo-kaninøjentesten som reference (TM B.5) (2)(14) og UN GHS-klassificeringssystemet (dvs. kategori 1, 2A, 2B eller uden for kategori) (1) og CLP-klassificeringssystemet (dvs. kategori 1, 2 eller uden for kategori), (iii) dækker de forskellige in vivo-klassificeringsfaktorer (24)(25), (iv) er repræsentative for de kemikaliegrupper, der blev anvendt i valideringsundersøgelsen (8)(10)(11), (v) dækker et godt og bredt udsnit af organiske funktionelle grupper (8)(10)(11), vi) har veldefinerede kemiske strukturer (8)(10)(11), (vii) er farvede og/eller direkte MTT-opløsninger, (viii) gav reproducerbare resultater ved RhCE-forsøgsmetoderne under valideringer af dem, (ix) blev korrekt forudset ved RhCE-forsøgsmetoderne under valideringsundersøgelsen af dem, (x) dækker det fulde spektrum af in vitro-reaktioner baseret på data af høj kvalitet om RhCE-forsøgsmetoder (0-100 % levedygtighed), xi) findes i handelen og xii) ikke er forbundet med uoverkommelige anskaffelses- og/eller bortskaffelsesomkostninger. Hvis et stof på listen ikke kan fås eller af andre gyldige grunde ikke kan anvendes, kan der anvendes et andet kemikalie, som opfylder ovennævnte kriterier, f.eks. blandt de kemikalier, der blev anvendt ved valideringen af VRM. Sådanne afvigelser bør dog begrundes.
Tabel 1
Liste over kompetencekemikalier
|
Kemisk betegnelse
|
CAS RN
|
Organisk funktionel gruppe (81)
|
Fysisk form
|
VRM1-levedygtighed (%) (82)
|
VRM2-levedygtighed (%) (83)
|
VRM-forudsigelse
|
MTT-reduktionsmiddel
|
Farveinterf.
|
|
In vivo-kategori 1 (84)
|
|
|
Methylthioglycolat
|
2365-48-2
|
Carboxylsyre-ester, thioalkohol
|
L
|
10,9 ± 6,4
|
5,5 ± 7,4
|
Der kan ikke fremsættes nogen forudsigelser
|
Y (stærk)
|
N
|
|
Hydroxyethylacrylat
|
818-61-1
|
Acrylat, alkohol
|
L
|
7,5 ± 4,7 (85)
|
1,6 ± 1,0
|
Der kan ikke fremsættes nogen forudsigelser
|
N
|
N
|
|
2,5-dimethyl-2,5-hexanediol
|
110-03-2
|
alkohol
|
S
|
2,3 ± 0,2
|
0,2 ± 0,1
|
Der kan ikke fremsættes nogen forudsigelser
|
N
|
N
|
|
Natriumoxalat
|
62-76-0
|
Oxocarboxylsyre
|
S
|
29,0 ± 1,2
|
5,3 ± 4,1
|
Der kan ikke fremsættes nogen forudsigelser
|
N
|
N
|
|
In vivo-kategori 2A (84)
|
|
|
2,4,11,13-tetraazatetradecan-diimidamid, N,N″-bis(4-chlorphenyl)-3,12-diimino-, di-D-gluconat (20 %, vandig) (86)
|
18472-51-0
|
Aromatisk heterocyklisk halid, arylhalogenid, dihydroxylgruppe, guanidin
|
L
|
4,0 ± 1,1
|
1,3 ± 0,6
|
Der kan ikke fremsættes nogen forudsigelser
|
N
|
Y (svag)
|
|
Natriumbenzoat
|
532-32-1
|
Aryl, carboxylsyre
|
S
|
3,5 ± 2,6
|
0,6 ± 0,1
|
Der kan ikke fremsættes nogen forudsigelser
|
N
|
N
|
|
In vivo-kategori 2B (84)
|
|
|
Diethyltoluamid
|
134-62-3
|
Benzamid
|
L
|
15,6 ± 6,3
|
2,8 ± 0,9
|
Der kan ikke fremsættes nogen forudsigelser
|
N
|
N
|
|
2,2-dimethyl-3-methylenbicyclo [2.2.1] heptan
|
79-92-5
|
Forgrenet alkan med tertiært kulstof, alken, bicycloheptan, forbundne kulstofringe, cycloalkan
|
S
|
4,7 ± 1,5
|
15,8 ± 1,1
|
Der kan ikke fremsættes nogen forudsigelser
|
N
|
N
|
|
In vivo Uden for kategori (84)
|
|
|
1-ethyl-3-methylimidazolethylsulfat
|
342573-75-5
|
Alkoxy, ammoniumsalt, aryl, imidazol, sulfat
|
L
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Uden for kat.
|
N
|
N
|
|
Dicaprylylether
|
629-82-3
|
Alkoxy, Ether
|
L
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Uden for kat.
|
N
|
N
|
|
Piperonylbutoxid
|
51-03-6
|
Alkoxy, benzodioxol, benzyl, Ether
|
L
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Uden for kat.
|
N
|
N
|
|
Polyethylenglycol (PEG-40)-hydrogeneret ricinusolie
|
61788-85-0
|
Acylal, alkohol, allyl, Ether
|
Viskøs
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Uden for kat.
|
N
|
N
|
|
1-(4-chlorphenyl)-3-(3,4-dichlorphenyl)-urinstof
|
101-20-2
|
Aromatisk heterocyklisk halid, arylhalogenid, urinstofderivater
|
S
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Uden for kat.
|
N
|
N
|
|
2,2′-methylen-bis-(6-(2H-benzotriazol-2-yl)-4-(1,1,3,3-tetramethylbutyl)-phenol)
|
103597-45-1
|
Forgrenet alkan med kvartært kulstof, smeltede carbocykliske forbindelser, aromatiske, smeltede mættede heterocykliske forbindelser, prækursorer quinoide forbindelser, tert-butyl
|
S
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Uden for kat.
|
N
|
N
|
|
Kalium-tetrafluorborat
|
14075-53-7
|
Uorganisk salt
|
S
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Uden for kat.
|
N
|
N
|
|
Forkortelser:
CAS RN = Registernummer i Chemical Abstracts Service UN GHS = De Forenede Nationers globalt harmoniserede system for klassificering og mærkning af kemikalier (1), VRM1 = valideret referencemetode, EpiOcular™ EIT, VRM2 = valideret referencemetode, SkinEthic™ HCE EIT, Farveinterf. = farveinterferens med måling af standardabsorbans (optisk densitet (OD)) af MTT-formazan.
|
|
|
23.
|
Som led i påvisningen af kompetence anbefales det, at brugere kontrollerer vævenes barriereegenskaber efter modtagelse som beskrevet af producenten af RhCE-vævsmodellen (jf. punkt 25, 27 og 30). Dette er navnlig vigtigt, hvis vævene sendes over lange afstande/er længe undervejs. Når en test er helt fastlagt, og kompetencen med hensyn til brugen heraf erhvervet og påvist, vil denne rutinemæssige kontrol ikke være nødvendig. Når en test benyttes rutinemæssigt, anbefales det dog fortsat at vurdere barriereegenskaberne med regelmæssige mellemrum.
|
FREMGANGSMÅDE
|
24.
|
De test, der i øjeblikket er dækket af denne forsøgsmetode, er de videnskabeligt validerede EpiOcular™ EIT og SkinEthic™ HCE EIT (9)(12)(13), der omtales som de validerede referencemetoder (henholdsvis VRM1 og VRM2). Standardprocedurer for RhCE-forsøgsmetoderne er tilgængelige og bør anvendes ved gennemførelse og brug af forsøgsmetoderne i et laboratorium (34)(35). Følgende punkter og tillæg 2 indeholder en beskrivelse af de vigtigste komponenter og procedurer i RhCE-testene.
|
KOMPONENTERNE I RHCE-FORSØGSMETODEN
Almindelige betingelser
|
25.
|
Relevante humant afledte celler bør anvendes til at rekonstruere cornea-lignende tredimensionalt epithelvæv, som bør bestå af gradvist lagdelte, men ikke forhornede celler. RhCE-vævsmodellen præpareres i indsatser med en porøs syntetisk membran, som næringsstoffer kan passere igennem til cellerne. Der bør være flere lag levedygtige, ikke-keratiniserede epithelceller til stede i det rekonstruerede cornea-lignende epithel. RhCE-vævsmodellens epitheloverflade skal have direkte luftkontakt, så der kan ske direkte lokal eksponering for testkemikalierne på en måde, der ligner den måde, hvorpå cornea-epithelet vil være eksponeret in vivo. RhCE-vævsmodellen skal danne en tilstrækkeligt robust funktionel barriere til at kunne modstå hurtig gennemtrængning af cytotoksiske referencestoffer, f.eks. Triton X-100 eller natriumdodecylsulphat (SDS). Barrierefunktionen påvises og kan vurderes ved at bestemme enten den eksponeringstid, der er nødvendig for at nedsætte vævets levedygtighed med 50 % (ET50) efter påføring af et referencestof ved en fastsat koncentration (f.eks. 100 μl 0,3 % (v/v) Triton X-100) eller den koncentration, hvorved et referencestof nedsætter vævenes levedygtighed med 50 % (IC50) efter en fastsat eksponeringstid (f.eks. 30 minutters behandling med 50 μl SDS) (jf. punkt 30). RhCE-vævsmodellens fysiske udformning skal forhindre, at testkemikaliet passerer uden om kanten af det levedygtige væv, hvilket kan forringe modelleringen af eksponeringen af cornea. De humant afledte celler, der anvendes til at opbygge RhCE-vævsmodellen, skal være fri for kontamination med bakterier, virus, mykoplasma og svampe. Leverandøren skal kontrollere vævsmodellens sterilitet for fravær af kontaminering med svampe og bakterier.
|
Funktionsmæssige betingelser
Levedygtighed
|
26.
|
MTT-assayet foretrækkes til kvantificering af vævslevedygtigheden (16). Levedygtige celler i RhCE-vævsmodellen reducerer vitalfarvestoffet MTT til blåt MTT-formazanbundfald, som herefter ekstraheres fra vævet ved anvendelseaf isopropanol (eller et lignende opløsningsmiddel). Det ekstraherede MTT-formazan kan kvantificeres ved anvendelse af enten måling af en standardabsorbans (optisk densitet (OD)) eller en HPLC/UPLC-spektrofotometriprocedure (36). OD af ekstraktionsmidlet alene bør være tilstrækkeligt lille, dvs. OD < 0,1. Brugere af RhCE-vævsmodellen skal sikre, at hver batch af den anvendte RhCE-vævsmodel overholder de fastlagte kriterier for den negative kontrol. Intervaller for de acceptable OD-værdier til negativ kontrol for VRM’er er anført i tabel 2. En bruger af HPLC/UPLC-spektrofotometri bør som acceptkriterie for den negative kontrol anvende de OD-intervaller til negativ kontrol, der er anført i tabel 2. Det skal i testrapporten godtgøres, at vævet fra behandling med det negative kontrolstof er en stabil kultur (giver enslydende målinger af vævslevedygtighed) i hele testeksponeringsperioden. En lignende procedure bør følges af vævsproducenten som led i kvalitetskontrollen ved godkendelse af en vævsbatch, men i dette tilfælde kan andre godkendelseskriterier end dem, der er anført i tabel 2, finde anvendelse. Udvikleren/leverandøren af RhCE-vævsmodellen skal fastsætte et interval (øvre og nedre grænse) for acceptable OD-værdier for negativ kontrol (i betingelserne for kvalitetskontrol af forsøgsmetoden).
|
Tabel 2
Intervaller for acceptable OD-værdier for negativ kontrol (for testbrugere)
|
Test
|
Nedre acceptgrænse
|
Øvre acceptgrænse
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (for protokoller for både væsker og faste stoffer)
|
> 0,8 (87)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (for protokoller for både væsker og faste stoffer)
|
> 1,0
|
≤ 2,5
|
Barrierefunktion
|
27.
|
RhCE-vævsmodellen skal være tilstrækkeligt tyk og robust til at kunne modstå hurtig gennemtrængning af cytotoksiske referencestoffer, som anslået af f.eks. ET50 (Triton X-100) eller IC50 (SDS) (tabel 3). Udvikleren/leverandøren af den anvendte RhCE-vævsmodel skal påvise barrierefunktionen for hver enkelt batch af RhCE-vævsmodellen ved levering af vævene til slutbrugeren (jf. punkt 30).
|
Morfologi
|
28.
|
En histologisk undersøgelse af RhCE-vævsmodellen skal påvise en human cornea-lignende epithelstruktur (bl.a. mindst tre lag levedygtige epithelceller og en ikke-keratiniseret overflade). For VRM’er har udvikleren/leverandøren fastlagt passende morfologi, og den behøver derfor ikke påvises igen af brugeren af en forsøgsmetode for hver enkelt anvendt vævsbatch.
|
Reproducerbarhed
|
29.
|
Resultaterne af forsøgsmetoden med positive og negative kontroller skal vise tidsmæssig reproducerbarhed.
|
Kvalitetskontrol
|
30.
|
RhCE-vævsmodellen bør kun anvendes, hvis udvikleren/leverandøren påviser, at hver batch af den anvendte RhCE-vævsmodel overholder veldefinerede produktionsgodkendelseskriterier, hvoraf levedygtighed (punkt 26) og barrierefunktion (punkt 27) er de vigtigste. Udvikleren/leverandøren af RhCE-vævsmodellen skal fastsætte et interval for acceptable værdier (øvre og nedre grænser) for barrierefunktionerne som målt ved ET50 eller IC50 (jf. punkt 25 og 26). De ET50- og IC50-godkendelsesintervaller, som udvikleren/leverandøren af RhCE-vævsmodeller (anvendt i VRM’er) som kriterie for batchfrigivelse, er angivet i tabel 3. Udvikleren/leverandøren af RhCE-vævsmodellen skal fremlægge data, der dokumenterer overensstemmelse med alle produktionsgodkendelseskriterier, for brugere af forsøgsmetoden, således at disse kan medtage de pågældende oplysninger i testrapporten. Kun resultater, som er opnået med væv, der opfylder alle disse kriterier for produktionsgodkendelse, kan accepteres til at give en pålidelig vurdering af kemikalier, som ikke kræver klassificering og mærkning for øjenirritation eller alvorlige øjenskader i overensstemmelse med UN GHS/CLP.
|
Tabel 3
Kriterium for batchgodkendelse ved kvalitetskontrol
|
Test
|
Nedre acceptgrænse
|
Øvre acceptgrænse
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (100 μl 0,3 % (v/v) Triton X-100)
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (30 minutters behandling med 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Påføring af testkemikalie og kontrolstoffer
|
31.
|
Der anvendes mindst to vævsgentagelser for hvert testkemikalie og hvert kontrolstof i hver analyseserie. Der anvendes to forskellige behandlingsprotokoller, en for flydende testkemikalier og en for faste testkemikalier (34)(35). For begge metoder og protokoller fugtes vævsmodellens overflade med kalcium- og magnesiumfrit Dulbeccos phosphatbufret fysiologisk saltopløsning (Ca2+/Mg2+-frit DPBS) før påføring af testkemikalier for at efterligne fugtbetingelserne i det menneskelige øje. Vævsbehandlingen indledes med eksponering for testkemikalie(r) og kontrolstoffer. For begge behandlingsprotokoller i de to VRM’er påføres en tilstrækkelig mængde testkemikalie eller kontrolstof til, at epitheloverfladen dækkes med et jævnt lag, og der samtidig undgås en ubegrænset dosis (jf. punkt 32 og 33) (tillæg 2).
|
|
32.
|
Testkemikalier, der kan pipetteres ved 37 °C eller lavere temperaturer (om nødvendigt ved hjælp af en pipette med positiv fortrængning), behandles som væsker i VRM’erne, og ellers som faste stoffer (jf. punkt 33). I VRM’erne fordeles testkemikaliet jævnt over vævsoverfladen (dvs. en påføring på minimum 60 μl/cm2) (jf. tillæg 2, (33)(34)). De kapillarvirkninger (virkninger på overfladespændingen), som kan opstå på grund af de lave volumener, der påføres indsatsen (på vævets overflade), bør så vidt muligt undgås for at sikre korrekt dosering af vævet. Væv, der behandles med flydende testkemikalier, inkuberes i 30 minutter under standarddyrkningsbetingelser (37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH). Ved afslutningen af eksponeringsperioden fjernes det flydende testkemikalie og kontrolstofferne forsigtigt fra vævsoverfladen ved skylning med rigelig Ca2+/Mg2+-frit DPBS ved stuetemperatur. Dette skylletrin følges af nedsænkning i frisk dyrkningsmedium ved stuetemperatur (for at fjerne testkemikalie, der eventuelt er absorberet i vævet) i en forud fastsat periode, som varierer alt efter den anvendte VRM. Kun for VMR1 anvendes en inkubation efter eksponering i frisk dyrkningsmedium under standarddyrkningsbetingelser før udførelsen af MTT-assayet (jf. tillæg 2, (34)(35)).
|
|
33.
|
Testkemikalier, som ikke kan pipetteres ved temperaturer op til 37 °C, behandles som faste stoffer i VRM’er. Mængden af testkemikalie skal være tilstrækkelig til at dække hele vævets overflade, dvs. at der bør påføres mindst 60 mg/cm2 (tillæg 2). Når det er muligt, bør faste stoffer testes som fint pulver. Væv, der behandles med faste testkemikalier, inkuberes i en forud fastsat periode (afhængigt af den anvendte VRM) under standarddyrkningsbetingelser (jf. tillæg 2, (34)(35)). Ved afslutningen af eksponeringsperioden fjernes det faste testkemikalie og kontrolstofferne forsigtigt fra vævsoverfladen ved skylning med rigelig Ca2+/Mg2+-frit DPBS ved stuetemperatur. Dette skylletrin følges af en nedsænkning i frisk dyrkningsmedium ved stuetemperatur efter eksponering (for at fjerne testkemikalie, der eventuelt er absorberet i vævet) i en forud fastsat periode, som varierer alt efter den anvendte VRM, og en inkubation i frisk medium under standarddyrkningsbetingelser, før MTT-assayet udføres (jf. tillæg 2 (34)(35)).
|
|
34.
|
Sideløbende negative og positive kontroller bør medtages i hver analyseserie for at påvise, at vævenes levedygtighed (bestemt med den negative kontrol) og følsomhed (bestemt med den positive kontrol) ligger inden for de acceptintervaller, som defineres på grundlag af tidligere data. Den sideløbende negative kontrol giver også referencen (100 % vævslevedygtighed) til at beregne den relative procentvise levedygtighed for de væv, der behandles med testkemikaliet (% levedygtighedstest). Det anbefalede positive kontrolstof, der skal anvendes med VRM’erne, er ren methylacetat (CAS-nr. 79-20-9, fås i handelen fra f.eks. Sigma-Aldrich, Cat# 45997, væske). De anbefalede negative kontrolstoffer, der skal anvendes med VRM1 og VRM2, er ultrarent henholdsvis H2O- og Ca2+/Mg2+-frit DPBS. Det var disse kontrolstoffer, der blev anvendt i valideringsundersøgelserne af VRM’erne, og det er dem, de fleste tidligere data foreligger for. Anvendelsen af egnede alternative positive eller negative kontrolstoffer skal være tilstrækkeligt videnskabeligt begrundet. Negative og positive kontroller bør testes med de(n) samme protokol(ler) som dem, der benyttes for de testkemikalier, der er medtaget i analyseserien (dvs. for væsker og/eller faste stoffer). Denne anvendelse bør følges af eksponering for behandlingen, skylning, nedsænkning efter eksponering og eventuelt inkubation efter eksponering som beskrevet for kontroller, som gennemføres sideløbende med flydende testkemikalier (jf. punkt 32) eller for kontroller, der gennemføres sideløbende med faste testkemikalier (jf. punkt 33), før MTT-assayet udføres (jf. punkt 35) (34)(35)). En enkelt række negative og positive kontroller er tilstrækkelig for alle testkemikalier med samme fysiske form (væsker eller faste stoffer), der medtages i den samme analyseserie.
|
Måling af vævslevedygtighed
|
35.
|
MTT-assayet er en standardiseret kvantitativ metode (16), som skal benyttes til måling af vævslevedygtighed i forbindelse med denne forsøgsmetode. Den er forenelig med brug i en tredimensional vævsmodel. MTT-assayet udføres straks efter inkubationstiden efter eksponering. I VRM’er anbringes RhCE-vævsmodelprøven i 0,3 ml MTT-opløsning til 1 mg/ml i 180 ± 15 minutter under standarddyrkningsbetingelser. Vitalfarvestoffet MTT reduceres til blåt MTT-formazanbundfald af de levedygtige celler i RhCE-vævsmodellen. Det udfældede blå MTT-formazan ekstraheres herefter fra vævet ved hjælp af en passende mængde isopropanol (eller et lignende opløsningsmiddel) (34) (35). Væv, der testes med flydende testkemikalier, bør ekstraheres både fra vævenes top og bund, mens væv, der testes med faste testkemikalier og farvede væsker, kun ekstraheres fra vævets bund (for at minimere potentiel kontaminering af isopropanol-ekstraktionsopløsningen med testkemikalie, som måske er forblevet på vævet. Væv, der testes med flydende testkemikalier, som ikke med det samme vaskes af, kan også ekstraheres fra vævets bund alene. De negative og positive kontrolstoffer, der testes samtidig, bør behandles på samme måde som det testede kemikalie. Det ekstraherede MTT-formazan kan kvantificeres enten ved en måling af standardabsorbans (OD) ved anvendelse af et højst ± 30 nm bredt filterbånd ved 570 nm eller ved anvendelse af en HPLC/UPLC-spektrofotometriprocedure (jf. punkt 42) (11)(36).
|
|
36.
|
Testkemikaliets optiske egenskaber eller dets kemiske reaktion med MTT kan interferere med målingen af MTT-formazan og føre til et forkert skøn over vævslevedygtigheden. Testkemikalier kan interferere med målingen af MTT-formazan ved direkte reduktion af MTT til blåt MTT-formazan og/eller ved farveinterferens, hvis testkemikaliet naturligt eller på grund af behandlingsprocedurer absorberer i det samme absorbansinterval som MTT-formazan (dvs. omkring 570 nm). Der bør udføres forudgående kontroller før testning for at identificere potentielle direkte MTT-reduktionsmidler og/eller kemikalier, som fremkalder farveinterferens, og der bør udføres supplerende kontroller for at spore og korrigere for potentiel interferens fra sådanne testkemikalier (jf. punkt 37-41). Dette er især vigtigt, når et bestemt testkemikalie ikke er helt fjernet fra RhCE-vævsmodellen ved skylning, eller når det gennemtrænger det cornea-lignende epithel og derfor er til stede i RhCE-vævsmodellerne, når MTT-assayet er udført. For testkemikalier, der absorberer lys i det samme interval som MTT-formazan (naturligt eller efter behandling), som ikke er kompatibelt med måling af standardabsorbans (OD) af MTT-formazan på grund af for stærk interferens, dvs. stærk absorption ved 570 ± 30 nm, kan der benyttes en HPLC/UPLC-spektrofotometriprocedure til at måle MTT-formazan (jf. punkt 41 og 42) (11)(36). Det er nærmere beskrevet i SOP’erne for VRM’erne, hvordan man sporer og korrigerer for direkte MTT-reduktion og farvestoffernes interferens (34)(35). I henholdsvis tillæg III og IV findes der også illustrative flowdiagrammer over, hvordan man identificerer og håndterer direkte MTT-opløsninger og/eller farveinterfererende kemikalier for VRM1 og VRM2.
|
|
37.
|
For at identificere den potentielle interferens fra testkemikalier, der absorberer lys i det samme interval som MTT-formazan (naturligt eller efter behandling), og afgøre, om der er behov for yderligere kontrol, tilsættes testkemikaliet til vand og/eller isopropanol og inkuberes i en passende tid ved stuetemperatur (jf. tillæg 2, (34) (35)). Hvis testkemikaliet i vand og/eller isopropanol absorberer tilstrækkeligt lys i intervallet 570 ± 20 nm til VRM1 (jf. tillæg 3), eller hvis en farvet opløsning opnås ved blanding af testkemikaliet med vand til VRM2 (jf. tillæg 4), antages testkemikaliet at interferere med målingen af standardabsorbansen (OD) for MTT-formazan, og der bør foretages yderligere farvestofkontrol, eller alternativt bør der anvendes en HPLC/UPLC-spektrofotometriprocedure, og i så fald er disse kontroller ikke nødvendige (jf. punkt 41 og 42 og tillæg III og IV) (34)(35). Når der foretages en måling af standardabsorbansen (OD), påføres hvert interfererende testkemikalie på mindst to levedygtige vævsgentagelser, som gennemgår hele testproceduren, men inkuberes med dyrkningsmedium i stedet for MTT-opløsning under MTT-inkubationstrinnet for at generere en kontrol af en ikke-specifik farve i levende væv (NSCliving) (34)(35). NSCliving-kontrollen skal udføres sideløbende med testningen af det farvede testkemikalie, og i tilfælde af flere test skal der foretages en uafhængig NSCliving-kontrol for hver test, der udføres (i hver analyseserie) på grund af den iboende biologiske variabilitet i levende væv. En korrekt vævslevedygtighed beregnes som: den procentvise vævslevedygtighed opnået med levende væv eksponeret for det interfererende testkemikalie og inkuberet med MTT-opløsning (% levedygtighedstest) minus den procentvise ikke-specifik farve opnået med levende væv eksponeret for det interfererende testkemikalie og inkuberet med medium uden MTT kørt samtidig med den test, der korrigeres (% NSCliving), dvs. korrekt vævslevedygtighed = [% levedygtighedstest] - [% NSCliving].
|
|
38.
|
For at identificere direkte MTT-reduktionsmidler bør hvert testkemikalie tilsættes til en frisklavet MTT-opløsning. En passende mængde testkemikalie tilsættes til en MTT-opløsning, og blandingen inkuberes i ca. tre timer under standarddyrkningsbetingelser (jf. tillæg III og IV) (34)(35). Hvis MTT-blandingen, der indeholder testkemikaliet (eller suspension til uopløselige testkemikalier), bliver blå/violet, antages testkemikaliet direkte at reducere MTT, og der bør foretages en yderligere funktionskontrol på ikke-levedygtige RhCE-vævsmodeller uafhængigt af anvendelsen af målingen af standardabsorbans (OD) eller en HPLC/UPLC-spektrofotometriprocedure. Ved denne supplerende funktionskontrol anvendes dødt væv, hvor der kun er metabolisk residualaktivitet, men absorberer og tilbageholder testkemikaliet på samme måde som levedygtigt væv. Dødt væv under VRM1 præpareres ved eksponering for lav temperatur (»freeze-killed«). Dødt væv under VRM2 præpareres ved langvarig inkubation (f.eks. i mindst 24 ± 1 timer) i vand efterfulgt af opbevaring ved lave temperatur (»water-killed«). Hvert MTT-reducerende testkemikalie påføres på mindst to døde vævsgentagelser, som gennemgår hele testproceduren for at generere en ikke-specifik MTT-reduktionskontrol (NSMTT) (34)(35). En enkelt NSMTT-kontrol er tilstrækkelig pr. testkemikalie, uanset hvor mange uafhængige test/analyseserier der udføres. En korrekt vævslevedygtighed beregnes som: den procentvise vævslevedygtighed, der opnås med levende væv, som eksponeres for MTT-reduktionsmidlet (% levedygtighedstest) minus den procentvise ikke-specifikke MTT-reduktion, der opnås med det døde væv, som udsættes for det samme MTT-reduktionsmiddel, beregnet i forhold til den negative kontrol, der foretages sideløbende med den test, der korrigeres (% NSMTT), dvs. den faktiske vævslevedygtighed = [% levedygtighedstest] - [% NSMTT].
|
|
39.
|
Testkemikalier, der identificeres som kemikalier, der fremkalder både farveinterferens (jf. punkt 37) og direkte MTT-reduktion (jf. punkt 38), vil også kræve et tredje sæt kontroller, når målingen af standardabsorbansen (OD) udføres, ud over NSMTT- og NSCliving-kontrollerne, der er beskrevet ovenfor. Dette er sædvanligvis tilfældet med mørke testkemikalier, der absorberer lys i intervallet 570 ± 30 nm (f.eks. blå, violette, sorte), fordi deres egen farve forstyrrer vurderingen af deres evne til direkte at reducere MTT, som beskrevet i punkt 38. Dette nødvendiggør anvendelsen af NSMTT-kontroller som standard sammen med NSCliving-kontroller. Testkemikalier, for hvilke der udføres både NSMTT- og NSCliving-kontroller, kan blive absorberet og tilbageholdt af både levende og dødt væv. Derfor kan NSMTT-kontrollen i dette tilfælde ikke blot korrigere for testkemikaliets potentielle direkte MTT-reduktion, men også for den farveinterferens, der opstår, når testkemikaliet absorberes og tilbageholdes af dødt væv. Dette kan medføre dobbeltkorrektion for farveinterferens, da NSCliving-kontrollen allerede korrigerer for farveinterferens, som opstår, når testkemikaliet absorberes og tilbageholdes af levende væv. For at undgå eventuel dobbeltkorrektionfor farveinterferens skal der gennemføres en tredje kontrol for ikke-specifik farve i dødt væv (NSCkilled) (jf. tillæg III og IV) (34)(35). I denne yderligere kontrol påføres testkemikaliet på mindst to døde vævsprøver, som gennemgår hele testproceduren, men inkuberes med dyrkningsmedium i stedet for MTT-opløsning under MTT-inkubationstrinnet. En enkelt NSCkilled-kontrol er tilstrækkelig pr. testkemikalie, uanset hvor mange uafhængige test/analyseserier der er udført, men bør udføres sideløbende med NSMTT-kontrollen og med den samme vævsbatch. En korrekt vævslevedygtighed beregnes som: den procentvise vævslevedygtighed opnået med levende væv eksponeret for testkemikaliet (% levedygtighedstest) minus % NSMTT minus % NSCliving
plus den procentvise ikke-specifikke farve opnået med dødt væv eksponeret for det interfererende testkemikalie og inkuberet med dyrkningsmedium uden MTT beregnet i forhold til den negative kontrol, som gennemføres samtidig med den test, der korrigeres (% NSCkilled), dvs. korrekt vævslevedygtighed = [% levedygtighedstest] - [% NSMTT] - [% NSCliving] + [% NSCkilled].
|
|
40.
|
Det er vigtigt at bemærke, at en ikke-specifik MTT-reduktion og ikke-specifik farveinterferens kan øge vævsekstraktets OD (når der udføres målinger af standardabsorbans) over spektrofotometrets linearitetsinterval, og at ikke-specifik MTT-reduktion også kan øge vævsekstraktets topareal for MTT-formazan (ved udførelse af HPLC/UPLC-målinger ved hjælp af spektrofotometri) over spektrofotometrets linearitetsinterval. På dette grundlag er det vigtigt for hvert laboratorium at bestemme linearitetsintervallet for deres spektrofotometers OD/topareal med f.eks. MTT-formazan (CAS # 57360-69-7), som fås i handelen fra f.eks. Sigma-Aldrich (Cat# M2003), før der indledes testning af testkemikalier til lovmæssige formål.
|
|
41.
|
Måling af standardabsorbans (OD) ved hjælp af et spektrofotometer er hensigtsmæssig til at vurdere direkte MTT-opløsningsmidler og farveinterfererende testkemikalier, når den observerede interferens med måling af MTT-formazan ikke er for stærk (dvs. OD-værdierne af vævsekstrakter opnået med testkemikaliet uden nogen korrektion for direkte MTT-reduktion og/eller farveinterferens ligger inden for spektrofotometrets lineære interval). Ikke desto mindre bør resultater for testkemikalier, som producerer % NSMTT og/eller % NSCliving ≥ 60 % (VRM1 og VRM2 for væskeprotokol) eller 50 % (VRM2 for faste stoffers protokol), af den negative kontrol tages med forsigtighed, da dette er den fastlagte tærskelværdi, som anvendes i VRM’erne for at skelne mellem klassificerede og ikke-klassificerede kemikalier (jf. punkt 44). Standardabsorbans (OD) kan dog ikke måles, når interferensen med målingen af MTT-formazan er for stærk (dvs. fører til ukorrigerede OD-værdier for testvævsekstrakten, som falder uden for spektrofotometrets lineære interval). Farvede testkemikalier eller testkemikalier, som bliver farvede i kontakt med vand eller isopropanol, der interfererer for stærkt med målingen af standardabsorbans (OD) af MTT-formazan, kan stadig vurderes ved anvendelse af HPLC/UPLC-spektrofotometri (jf. tillæg III og IV). Dette skyldes, at HPLC/UPLC-systemet giver mulighed for at separere MTT-formazan fra kemikaliet, før det kvantificeres (36). Af denne grund kræves NSCliving- eller NSCkilled-kontroller aldrig, når der anvendes HPLC/UPLC-spektrofotometri uafhængigt af det testede kemikalie. NSMTT-kontroller bør ikke desto mindre benyttes, hvis testkemikaliet mistænkes for direkte at reducere MTT (efter proceduren i punkt 38). NSMTT-kontroller bør også benyttes med testkemikalier, der har en farve (deres egen eller en farve, der kommer frem i vand), som forhindrer vurderingen af deres evne til direkte at reducere MTT som beskrevet i punkt 38. Ved benyttelse af HPLC/UPLC-spektrofotometri til at måle MTT-formazan beregnes den procentvise vævslevedygtighed som en procentdel af toparealet for MTT-formazan, der opnås med levende væv, som eksponeres for testkemikaliet i forhold til toparealet for MTT formazan opnået med den sideløbende negative kontrol. For testkemikalier, der direkte kan reducere MTT, beregnes korrekt vævslevedygtighed som: % levedygtighedstest
minus % NSMTT som beskrevet i den sidste sætning i punkt 38. Endelig bør det bemærkes, at direkte MTT-opløsninger eller direkte MTT-opløsningsmidler, der også er farveinterfererende, og som tilbageholdes i vævet efter behandling og reducerer MTT så stærkt, at de fører til OD-værdier (ved anvendelse måling af standard-OD) eller toparealer (ved anvendelse af UPLC/HPLC-spektrofotometri) for de testede vævsekstrakter, som falder uden for spektrofotometrets linearitetsinterval, ikke kan vurderes med RhCE-forsøgsmetoder, skønt disse kun forventes at opstå i meget sjældne situationer.
|
|
42.
|
HPLC/UPLC-spektrofotometri kan anvendes med alle typer af testkemikalier (farvede, ikke-farvede, MTT-opløsninger og opløsninger uden MTT) til måling af MTT-formazan (11)(36). På grund af de mange forskellige HPLC/UPLC-spektrofotometri-systemer er det ikke muligt for den enkelte bruger at skabe nøjagtig de samme betingelser. Kvalificeringen af HPLC/UPLC-spektrofotometrisystemet bør som sådan påvises, før det anvendes til at kvantificere MTT-formazan fra vævsekstrakter ved at opfylde acceptkriterierne til en række parametre for standardkvalificering baseret på dem, der er beskrevet i retningslinjerne for industrien om bioanalytisk metodevalidering fra den amerikanske Food and Drug Administration (FDA) (36)(38). Disse centrale parametre og deres acceptkriterier er vist i tillæg 5. Når acceptkriterierne i tillæg 5 er blevet opfyldt, anses HPLC/UPLC-spektrofotometrisystemet for at være kvalificeret og klar til at måle MTT-formazan under de forsøgsbetingelser, som er beskrevet i denne forsøgsmetode.
|
Acceptkriterier
|
43.
|
For hver analyseserie, hvor der anvendes RhCE-vævsbatcher, som opfylder kvalitetskontrollen (jf. punkt 30), skal væv behandlet med det negative kontrolstof give OD-resultater, der afspejler kvaliteten af de væv, som har gennemgået alle forsendelses- og modtagelsestrin og alle protokolprocesser, og må ikke ligge uden for de tidligere fastsatte grænser som beskrevet i tabel 2 (jf. punkt 26). På tilsvarende måde skal væv, der behandles med det positive kontrolstof, dvs. methylacetat, udvise en gennemsnitlig vævslevedygtighed på < 50 % i forhold til den negative kontrol ved VRM1 med protokollen for enten flydende eller faste stoffer og ≤ 30 % (protokollen for væsker) eller ≤ 20 % (protokollen for faste stoffer) i forhold til den negative kontrol ved VRM2 og således afspejle vævenes evne til at reagere på et lokalirriterende testkemikalie under forsøgsmetodens vilkår (34)(35). Variabiliteten mellem vævsprøver af testkemikalier og kontrolstoffer skal ligge inden for de accepterede grænser (dvs. at forskellen i levedygtighed mellem to vævsprøver skal ligge under 20 %, eller standardafvigelsen mellem tre vævsprøver må ikke overstige 18 %). Hvis enten den negative kontrol eller positive kontrol i en analyseserie ligger uden for de accepterede intervaller, anses analyseserien for »ikke-kvalificeret« og bør gentages. Hvis variabiliteten mellem vævsprøver for et testkemikalie ligger uden for de accepterede intervaller, skal testen anses for at være »ikke-kvalificeret«, og testkemikaliet skal testes igen.
|
Fortolkning af resultater og forudsigelsesmodel
|
44.
|
OD-værdier/toparealer opnået med vævsekstraktprøven for hvert testkemikalie anvendes til at beregne den gennemsnitlige vævslevedygtighed i procent (gennemsnit mellem vævsprøver) normaliseret til den negative kontrol, som fastsættes til 100 %. Tærskelværdien for den procentvise vævslevedygtighed for identifikation af testkemikalier, der ikke kræver klassificering for øjenirritation eller alvorlige øjenskader (UN GHS/CLP – uden for kategori), er anført i tabel 4. Resultaterne skal således fortolkes som følger:
|
—
|
Testkemikaliet identificeres således, at det ikke kræver klassificering og mærkning ifølge UN GHS/CLP (uden for kategori), hvis den gennemsnitlige procentvise vævslevedygtighed efter eksponering og inkubation efter eksponering ligger over (>) tærskelværdien af den fastsatte procentvise vævslevedygtighed som vist i tabel 4. I så fald kræves der ingen yderligere testning efter andre forsøgsmetoder.
|
|
—
|
Såfremt den gennemsnitlige procentvise vævslevedygtighed efter eksponering og inkubation efter eksponering ligger under eller er lig med (≤) tærskelværdien for den fastsatte procentvise vævslevedygtighed, kan der ikke fremsættes nogen forudsigelse, som det fremgår af tabel 4. I så fald vil yderligere testning efter andre forsøgsmetoder være påkrævet, fordi RhCE-forsøgsmetoderne viser et vist antal falsk positive resultater (jf. punkt 14-15) og ikke kan ende mellem UN GHS/CLP-kategori 1 og 2 (jf. punkt 17).
|
Tabel 4
Forudsigelsesmodeller ifølge UN GHS- og CLP-klassificering
|
VRM
|
Uden for kategori
|
Der kan ikke fremsættes nogen forudsigelser
|
|
VRM 1 - EpiOcular™ EIT (for begge protokoller)
|
Gennemsnitlig vævslevedygtighed > 60 %
|
Gennemsnitlig vævslevedygtighed ≤ 60 %
|
|
VRM 2 - SkinEthic™ HCE EIT (for protokollen for væsker)
|
Gennemsnitlig vævslevedygtighed > 60 %
|
Gennemsnitlig vævslevedygtighed ≤ 60 %
|
|
VRM2 - SkinEthic™ HCE EIT (for protokollen for faste stoffer)
|
Gennemsnitlig vævslevedygtighed > 50 %
|
Gennemsnitlig vævslevedygtighed ≤ 50 %
|
|
|
45.
|
En enkelt test bestående af mindst to vævsprøver bør være tilstrækkelig for et testkemikalie, når resultatet er entydigt. I tilfælde af tvetydige resultater såsom diskordante gentagne målinger, og/eller hvis den gennemsnitlige procentvise vævslevedygtighed er 60 ± 5 % (VRM1 og VRM2 for protokollen for væsker) eller 50 ± 5 % (VRM2 for protokollen for faste stoffer), skal det dog overvejes at foretage en anden test samt en tredje, hvis resultaterne af de første to test er diskordante.
|
|
46.
|
Forskellige tærskelværdier for procentvis vævslevedygtighed, der adskiller klassificerede og ikke-klassificerede testkemikalier, kan overvejes for specifikke typer blandinger, hvis det er passende og begrundet, med henblik på at øge forsøgsmetodens samlede resultater for de pågældende typer blandinger (jf. punkt 14). Referencekemikalier kan være nyttige ved vurdering af risikoen for alvorlige øjenskader/øjenirritation ved ukendte testkemikalier eller en ukendt produktgruppe eller ved vurdering af et klassificeret kemikalies potentielle relative øjenætsende toksicitet inden for et specifikt område af irritationsresponser.
|
DATA OG RAPPORTERING
Data
|
47.
|
Data fra individuelle vævsgentagelser i en analyseserie (f.eks. OD-værdier/toparealer for MTT-formazan og beregnede data for procentvis vævslevedygtighed for testkemikaliet og kontrolstoffer samt forudsigelse fra den endelige RhCE-forsøgsmetode) rapporteres i tabelform for hvert testkemikalie, herunder om nødvendigt data fra gentagne test. Endvidere rapporteres den gennemsnitlige procentvise vævslevedygtighed og forskel i levedygtighed mellem to vævsgentagelser (hvis n = 2 vævsgentagelser) eller standardafvigelse (hvis n ≥ 3 vævsgentagelser) for hvert testkemikalie og kontrolstof. Alle observerede interferenser for et testkemikalie med målingen af MTT-formazan gennem direkte MTT-reduktion og/eller farvet interferens rapporteres for hvert testet kemikalie.
|
Testrapport
|
48.
|
Testrapporten skal indeholde følgende oplysninger:
Testkemikalie
|
|
Stof med kun én bestanddel
|
—
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-registreringsnummer/-numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
—
|
fysisk tilstand, flygtighed, pH, LogP, molekylvægt, kemisk gruppe og yderligere fysisk-kemiske egenskaber, som er relevante for undersøgelsen, i det omfang det er muligt
|
|
—
|
renhed, urenheders kemiske identitet, i det omfang det er relevant og praktisk muligt, osv.
|
|
—
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
—
|
opbevaring og stabilitet i det omfang, det er muligt.
|
|
|
|
Stof med flere bestanddele, UVCB-stoffer og blandinger
|
—
|
kendetegnet så vidt muligt ved f.eks. kemisk identitet (jf. ovenfor), renhed, kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber (jf. ovenfor) i det omfang, det er muligt
|
|
—
|
fysisk tilstand og yderligere fysisk-kemiske egenskaber, som er relevante for undersøgelsen, i det omfang det er muligt
|
|
—
|
renhed, urenheders kemiske identitet, i det omfang det er relevant og praktisk muligt, osv.
|
|
—
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
—
|
opbevaring og stabilitet i det omfang, det er muligt.
|
|
Positive og negative kontrolstoffer
|
—
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-registreringsnummer/-numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
—
|
fysisk tilstand, flygtighed, molekylvægt, kemisk gruppe og yderligere fysisk-kemiske egenskaber, som er relevante for undersøgelsen, i det omfang det er muligt
|
|
—
|
renhed, urenheders kemiske identitet, i det omfang det er relevant og praktisk muligt, osv.
|
|
—
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
—
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
—
|
begrundelse for eventuel anvendelse af en anden negativ kontrol end ultrarent H2O eller Ca2+/Mg2+-frit DPBS
|
|
—
|
begrundelse for eventuel anvendelse af en anden positiv kontrol end rent methylacetat
|
|
—
|
henvisning til tidligere positive og negative kontrolresultater, der udviser passende acceptkriterier for analyseserien.
|
Oplysninger om sponsor og testfacilitet
|
—
|
navn og adresse på sponsoren, testfaciliteten og undersøgelseslederen.
|
|
—
|
RhCE-vævsmodel og anvendt protokol (eventuelt med begrundelse for valg)
|
Betingelser for forsøgsmetoden
|
—
|
den anvendte RhCE-vævsmodel, herunder batchnummer
|
|
—
|
bølgelængde og båndpas (hvis relevant) anvendt til kvantificering af MTT-formazan og måleudstyrets linearitetsinterval (f.eks. spektrofotometer)
|
|
—
|
beskrivelse af den metode, der blev anvendt til kvantificering af MTT-formazan
|
|
—
|
eventuelt beskrivelse af det anvendte HPLC/UPLC-spektrofotometrisystem
|
|
—
|
komplet dokumentation for den anvendte RhCE-vævsmodel, herunder om dens ydeevne. Der skal bl.a. gives oplysning om:
|
i)
|
kvalitetskontrol vedrørende levedygtighed (leverandør)
|
|
ii)
|
levedygtighed under betingelserne for forsøgsmetoden (bruger)
|
|
iii)
|
kvalitetskontrol vedrørende barrierefunktion
|
|
iv)
|
morfologi (hvis tilgængelig)
|
|
v)
|
reproducerbarhed og forudsigelsesevne
|
|
vi)
|
anden kvalitetskontrol af RhCE-vævsmodellen, hvis tilgængelig
|
|
|
—
|
reference til historiske data for RhCE-modellen. Der skal bl.a. gives oplysning om: om kvalitetskontroldataene kan accepteres, sammenlignet med data fra tidligere batcher
|
|
—
|
erklæring om, at testfaciliteten har dokumenteret kompetence i anvendelsen af forsøgsmetoden før rutinemæssig anvendelse ved testning af kompetencekemikalier
|
Acceptkriterier for analyseserier og test
|
—
|
positive og negative kontrolgennemsnitsværdier og acceptintervaller på grundlag af historiske data
|
|
—
|
acceptabel variabilitet mellem vævsprøver til positiv og negativ kontrol
|
|
—
|
acceptabel variabilitet mellem vævsprøver til testkemikaliet.
|
Testprocedure
|
—
|
detaljer om den anvendte testprocedure
|
|
—
|
doser af det anvendte testkemikalie og anvendte kontrolstoffer
|
|
—
|
varighed og temperatur for eksponering, nedsænkning efter eksponering og inkubationstid efter eksponering (hvor det er relevant)
|
|
—
|
beskrivelse af eventuelle ændringer i forsøgsmetoden
|
|
—
|
eventuel angivelse af kontroller anvendt til direkte MTT-reduktionsmidler og/eller farvede testkemikalier
|
|
—
|
antal vævsprøver anvendt pr. testkemikalie og kontrol (positiv kontrol, negativ kontrol, NSMTT, NSCliving og NSCkilled, hvis relevant)
|
Resultater
|
—
|
angivelse i tabelform af data fra hvert enkelt testkemikalie og hvert enkelt kontrolstof til hver analyseserie (herunder gentagne forsøg, hvis det er relevant) og hver gentagen måling, herunder OD-værdier eller topareal for MTT-formazan, vævslevedygtighedsprocent, gennemsnitlig vævslevedygtighedsprocent, vævsprøveforskelle eller standardafvigelse og endelig forudsigelse
|
|
—
|
eventuelle resultater af kontroller anvendt for direkte MTT-reduktionsmidler og/eller farvede testkemikalier, herunder OD-værdi eller topareal for MTT-formazan, % NSMTT, % NSCliving, % NSCkilled, forskelle mellem vævsprøver eller standardafvigelse, endelig korrekt vævslevedygtighedsprocent og endelig forudsigelse
|
|
—
|
resultater opnået med testkemikaliet/-kemikalierne og kontrolstofferne i forhold til de fastlagte acceptkriterier for analyseserier og test
|
|
—
|
beskrivelse af andre observerede virkninger, f.eks. farvning af væv på grund af et farvet testkemikalie.
|
Diskussion af resultaterne
Konklusion
|
LITTERATUR
|
(1)
|
UN (2015). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York and Geneva: United Nations. Tilgængelig på: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Kapitel B.5 i dette bilag, Akut øjenirritation/-ætsning.
|
|
(3)
|
Kapitel B.47 i dette bilag, Forsøgsmetode til påvisning af oksecornea-uklarhed og -permeabilitet til identifikation af i) kemikalier, der medfører alvorlig øjenskade, og ii) kemikalier, der ikke kræver klassificering for øjenirritation eller alvorlig øjenskade.
|
|
(4)
|
Kapitel B.48 i dette bilag, Isoleret kyllingeøje-forsøgsmetode til identifikation af i) kemikalier, der fremkalder alvorlig øjenskade og ii) kemikalier, der ikke kræver klassificering.
|
|
(5)
|
Kapitel B.61 i dette bilag, Fluoresceinudsivnings-forsøgsmetode til identifikation af øjenætsende stoffer og stærkt øjenirriterende stoffer.
|
|
(6)
|
Kapitel B.68 i dette bilag, Forsøgsmetode med kortvarig eksponering in vitro til identifikation af i) kemikalier, der fremkalder alvorlige øjenskader, og ii) kemikalier, der ikke kræver klassificering for øjenirritation eller alvorlige øjenskader.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2010, 261-266.
|
|
(8)
|
EC EURL ECVAM (2014). EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Tilgængelig på: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28173 DA doi: 10.2787/043697. Tilgængelig på: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28175 EN; doi: 10.2787/390390. Tilgængelig på: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuskript under udarbejdelse).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OECD (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisation for Economic Cooperation and Development, Paris.
|
|
(18)
|
OECD (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, s. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1 476-1 488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, s. 749-815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, s. 665-697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922-936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115-130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2 610–2 625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41-54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Tilgængelig på: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Tilgængelig på: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. May 2001. Tilgængelig på: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications, Organisation for Economic Cooperation and Development, Paris.
|
Tillæg 1
DEFINITIONER
Nøjagtighed
: overensstemmelsen mellem forsøgsmetodens resultater og accepterede referenceværdier. Den er et mål for forsøgsmetodens ydeevne og et af aspekterne af "relevans". Udtrykket benyttes ofte som synonym for "konkordans", forstået som den andel af korrekte udfald, forsøgsmetoden fører til (18).
Referencekemikalie
: et kemikalie, der anvendes som standard til sammenligning med et testkemikalie. Et referencekemikalie skal have følgende egenskaber: i) en eller flere ensartede og pålidelige kilder til identifikation og karakterisering heraf, ii) strukturel og funktionel lighed og/eller kemisk lighed eller lighed med hensyn til produktgruppe med de(t) kemikalie(r), der testes, iii) kendte fysisk-kemiske egenskaber, iv) støttedata om kendte virkninger og v) kendt styrke i det ønskede responsområde.
Bottom-up-tilgang
: trinvis tilgang til et testkemikalie mistænkt for ikke at kræve klassificering og mærkning for øjenirritation eller alvorlige øjenskader, der starter med bestemmelse af kemikalier, som ikke kræver klassifikation og mærkning (negativt resultat) fra andre kemikalier (positivt resultat).
Kemikalie
: et stof eller en blanding.
Konkordans
: se "Nøjagtighed".
Cornea
: den transparente del af øjeæblets forside, som dækker øjets iris og pupil, og hvorigennem lys kommer ind i øjets indre.
CV
: variationskoefficient
Dev.
: afvigelse.
EIT
: øjenirritationstest.
EURL ECVAM
: EU-referencelaboratorium for alternativer til dyreforsøg.
Øjenirritation
: frembringelse af forandringer i øjet, som opstår efter påføring af et testkemikalie på øjets anteriore overflade, og som er fuldt reversible inden for 21 dage efter påføring. Synonymt med "Reversible virkninger på øjet" og "UN GHS/CLP-kategori 2".
ET50
: den eksponeringstid, der er nødvendig for at nedsætte vævets levedygtighed med 50 % ved påføring af et referencekemikalie i en nærmere angivet fast koncentration.
Falsk negativ-rate
: den andel af alle positive stoffer som ved en forsøgsmetode fejlagtigt identificeres som negativ. Det er en indikator for forsøgsmetodens ydeevne.
Falsk positiv-rate
: Den andel af alle negative stoffer, som ved en forsøgsmetode fejlagtigt identificeres som positiv. Det er en indikator for forsøgsmetodens ydeevne.
Fare
: den iboende egenskab ved et middel eller en situation, som kan forårsage skadelige virkninger, når en organisme, et system eller en (sub)population eksponeres for det pågældende middel.
HCE
: SkinEthic™ Human Corneal Epithelium (humant cornea-epithel)
HPLC
: High Performance Liquid Chromatography (højtryksvæskekromatografi)
IC50
: koncentration, hvor et referencekemikalie nedsætter vævets levedygtighed med 50 % efter en fast eksponeringstid (f.eks. 30 minutters behandling med SDS).
Ubegrænset dosis
: den mængde testkemikalie, som er påført på RhCE-vævsmodellen ud over den mængde, der er nødvendig for helt at dække epithelets overflade på en ensartet måde.
Irreversible virkninger på øjet
: se "Alvorlige øjenskader".
LLOQ
: Lower Limit of Quantification (laveste kvantitative bestemmelsesgrænse)
LogP
: logaritmen af oktanol/vand-fordelingskoefficient
Blanding
: blanding eller opløsning, der består af to eller flere stoffer.
Stof med kun én bestanddel
: et stof, der defineres af sin mængdemæssige sammensætning, hvori en hovedbestanddel udgør mindst 80 % (w/w).
Stof med flere bestanddele
: et stof, der defineres af sin mængdemæssige sammensætning, hvori flere end én hovedbestanddel er til stede i en koncentration på ≥ 10 % (w/w) og < 80 % (w/w). Et stof, der består af flere forskellige bestanddele, er resultatet af en fremstillingsproces. Forskellen mellem en blanding og et stof med forskellige bestanddele er, at blandingen er fremstillet ved at blande to eller flere stoffer uden kemisk reaktion. Et stof, der består af flere forskellige bestanddele, er resultatet af en kemisk reaktion.
MTT
: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid thiazolylblåt tetrazolium-bromid.
Negativ kontrol
: en prøve, som indeholder alle komponenterne fra et forsøgssystem og er behandlet med et stof, som ikke vides at inducere en positiv respons i testsystemet. Prøven behandles sammen med prøver behandlet med testkemikaliet og andre kontroller og anvendes til at bestemme 100 % vævslevedygtighed.
Ikke klassificeret
: kemikalier, der ikke er klassificeret for øjenirritation (UN GHS/CLP-kategori 2, UN GHS-kategori 2A eller 2B) eller alvorlige øjenskader (UN GHS/CLP-kategori 1). Synonymt med "UN GHS/CLP – uden for kategori".
NSCkilled
: ikke-specifik farve i dødt væv
NSCliving
: ikke-specifik farve i levende væv
NSMTT
: ikke-specifik MTT-reduktion.
OD
: optisk tæthed.
Ydeevnestandarder
: standarder, som med udgangspunkt i en valideret forsøgsmetode, der blev betragtet som videnskabeligt valid, danner grundlag for at vurdere, om en foreslået forsøgsmetode, som er mekanistisk og funktionelt tilsvarende, er sammenlignelig. De indeholder bl.a.: i) de centrale dele af forsøgsmetoden, ii) en minimumsliste med referencekemikalier udvalgt blandt de kemikalier, der benyttes til at påvise, at den validerede forsøgsmetodes ydeevne er acceptabel, og (iii) de sammenlignelige niveauer af nøjagtighed og pålidelighed, som er fastsat efter, hvad der er opnået med den validerede forsøgsmetode, og som det skal påvises, at den foreslåede forsøgsmetode kan yde, ved vurdering ud fra minimumslisten med referencekemikalier (18).
Positiv kontrol
: en prøve, som indeholder alle komponenterne fra et forsøgssystem og er behandlet med et stof, som vides at inducere en positiv respons i testsystemet. Prøven behandles sammen med prøver behandlet med testkemikaliet og andre kontroller. For at sikre, at det er muligt at vurdere variabiliteten i den positive kontrolrespons over tid, bør den positive respons ikke være overdrevent stor.
Relevans
: beskrivelse af sammenhængen mellem testen og den pågældende virkning og dens meningsfuldhed og brugbarhed til et bestemt formål. Det angives, i hvilket omfang testen måler eller forudsiger den pågældende biologiske virkning korrekt. Forsøgsmetodens nøjagtighed (konkordans) indgår i vurderingen af dens relevans (18).
Pålidelighed
: mål for, i hvilken grad en forsøgsmetode kan reproduceres i og mellem laboratorier over en længere periode, når den udføres efter samme protokol. Den bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier og repeterbarhed inden for laboratorier (18).
Erstatningstest
: test, der erstatter en test, som bruges rutinemæssigt og er godkendt til fareidentifikation og/eller risikovurdering, og som vurderes at sikre en tilsvarende eller bedre beskyttelse af menneskers og dyrs sundhed eller miljøet alt afhængigt af situationen i forhold til den godkendte test, i alle testsituationer og for alle kemikalier (18).
Reproducerbarhed
: overensstemmelse mellem resultater af gentagne test af samme testkemikalie med samme testprotokol (jf. "Pålidelighed") (18).
Reversible virkninger på øjet
: se "Øjenirritation".
RhCE
: rekonstrueret humant cornea-lignende epithel
Analyseserie
: en analyseserie består af et eller flere testkemikalier, der testes samtidig med en negativ kontrol og en positiv kontrol.
Standardafvigelse
: standardafvigelse
Følsomhed
: andelen af alle positive/aktive testkemikalier, der klassificeres korrekt ved testen. Det er et mål for nøjagtigheden af en forsøgsmetode, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en forsøgsmetodes relevans (18).
Alvorlig øjenskade
: frembringelse af vævsbeskadigelse i øjet eller alvorlig fysisk synsnedsættelse, som opstår efter påføring af et testkemikalie på øjets anteriore overflade, og som ikke er fuldt reversibel inden for 21 dage efter påføring. Synonymt med "Irreversible virkninger på øjet" og "UN GHS/CLP-kategori 1".
Standardprocedurer (SOP)
: formelle skriftlige procedurer med detaljeret beskrivelse af, hvordan rutinemæssig og testspecifik laboratoriepraksis bør udføres. De er et krav i henhold til GLP.
Specificitet
: andelen af alle negative/inaktive testkemikalier, der klassificeres korrekt ved testen. Det er et mål for nøjagtigheden af en forsøgsmetode, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en forsøgsmetodes relevans (18).
Stof
: et grundstof og forbindelser heraf, i naturlig tilstand eller industrielt fremstillet, herunder sådanne tilsætningsstoffer, som er nødvendige til bevarelse af stoffets stabilitet, og sådanne urenheder, som følger af fremstillingsprocessen, bortset fra opløsningsmidler, der kan udskilles, uden at det påvirker stoffets stabilitet eller ændrer dets sammensætning.
Test
: et enkelt testkemikalie, der sideløbende testes i mindst to vævsprøver, som defineret i den tilsvarende standardprocedure.
Vævslevedygtighed
: parameter, der måler den samlede aktivitet i en cellepopulation i et rekonstrueret væv som deres evne til at reducere vitalfarvestoffet MTT, og som, afhængigt af den effektparameter, der måles, og udformningen af testen, korrelerer med det samlede antal levende celler og/eller deres levedygtighed.
Top-down-tilgang
: trinvis tilgang, der anvendes for et kemikalie, som mistænkes for at forårsage alvorlige øjenskader, der starter med bestemmelse af kemikalier, som forårsager alvorlige øjenskader (positivt resultat), i forhold til andre kemikalier (negativt resultat).
Testkemikalie
: alle stoffer eller blandinger, der testes ved hjælp af denne forsøgsmetode
Trindelt testningsstrategi
: trindelt teststrategi, hvor der anvendes sekventielle forsøgsmetoder. Alle eksisterende oplysninger om et testkemikalie gennemgås i en bestemt rækkefølge ved anvendelse af en "weight of evidence"-proces på hvert trin for at fastslå, om der foreligger tilstrækkelige oplysninger til at træffe en fareklassificeringsafgørelse, inden der fortsættes til strategiens næste trin. Hvis et testkemikalies farepotentiale/styrke kan underbygges på et givet trin baseret på eksisterende oplysninger, er der ikke behov for at foretage yderligere test (18).
ULOQ
: Upper Limit of Quantification (øverste kvantitative bestemmelsesgrænse)
De Forenede Nationers globalt harmoniserede system for klassificering og mærkning af kemikalier (United Nations Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: et system til klassificering af kemikalier (stoffer og blandinger) efter standardiserede typer og niveauer af fysisk fare, sundhedsfare og miljøfare med tilhørende kommunikationselementer såsom piktogrammer, signalord, faresætninger, sikkerhedssætninger og sikkerhedsdatablade, således at oplysninger om kemikaliernes skadelige virkninger kan videreformidles med henblik på at beskytte mennesker (f.eks. arbejdsgivere, arbejdstagere, transportører, forbrugere og redningspersonel) og miljøet (1).
UN GHS/CLP-kategori 1
: se "Alvorlige øjenskader".
UN GHS/CLP-kategori 2
: se "Øjenirritation".
UN GHS/ CLP – uden for kategori
: kemikalier, der ikke opfylder kravene med hensyn til klassificering som UN GHS/CLP-kategori 1 eller 2 (eller UN GHS-kategori 2A eller 2B). Synonymt med "Ikke-klassificeret".
UPLC
: Ultra-High Performance Liquid Chromatography (ultra-højtryksvæskekromatografi)
UVCB
: stoffer med ukendt eller variabel sammensætning, komplekse reaktionsprodukter eller biologiske materialer.
Gyldig forsøgsmetode
: en forsøgsmetode, der anses for at være tilstrækkeligt relevant og pålidelig til et bestemt formål, og som er baseret på videnskabeligt forsvarlige principper. En forsøgsmetode har aldrig absolut gyldighed, men er kun gyldig i forbindelse med et bestemt formål (18).
Valideret forsøgsmetode
: en forsøgsmetode, for hvilken der er gennemført valideringsundersøgelser for at fastslå relevansen (herunder nøjagtigheden) og pålideligheden til et bestemt formål. Det er vigtigt at være opmærksom på, at en valideret forsøgsmetode måske ikke har tilstrækkelig ydeevne med hensyn til nøjagtighed og pålidelighed til at være acceptabel til det planlagte formål (18).
VRM
: Valideret referencemetode
VRM1
: EpiOcular™ EIT omtales som valideret referencemetode 1.
VRM2
: SkinEthic™ HCE EIT omtales som valideret referencemetode 2.
"Weight of evidence"-analyse
: En proces, hvor styrkerne og svaghederne ved forskellige oplysninger afvejes for at nå frem til og underbygge en konklusion om et testkemikalies farepotentiale.
Tillæg 2
Vigtigste Komponenter i Rhce-Test Valideret til Identifikation af Kemikalier, som Ikke Kræver Klassificering og Mærkning for Øjenirritation Eller Alvorlige Øjenskader
|
Testkomponenter
|
EpiOcular™ EIT
(VRM 1)
|
SkinEthic™ HCE EIT
(VRM 2)
|
|
Protokoller
|
Væsker
(kan pipetteres ved 37 ± 1oC eller lavere temperaturer i 15 min.)
|
Faste stoffer
(kan ikke pipetteres)
|
Væsker og viskøse stoffer
(kan pipetteres)
|
Faste stoffer
(kan ikke pipetteres)
|
|
Modelareal
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Antal vævsprøver
|
Mindst 2
|
Mindst 2
|
Mindst 2
|
Mindst 2
|
|
Forhåndskontrol for farveinterferens
|
50 μl + 1 ml H2O i 60 min. ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH (ikke-farvede testkemikalier) eller 50 μl + 2 ml isopropanol blandet i 2-3 timer ved stuetemp. (farvede testkemikalier)
|
 Hvis testkemikaliets OD ved 570 ± 20 nm efter fratrækning af OD for isopropanol eller vand er > 0,08 (som svarer til ca. 5 % af den gennemsnitlige OD for den negative kontrol), bør der foretages tilpassede "living" kontroller. Hvis testkemikaliets OD ved 570 ± 20 nm efter fratrækning af OD for isopropanol eller vand er > 0,08 (som svarer til ca. 5 % af den gennemsnitlige OD for den negative kontrol), bør der foretages tilpassede "living" kontroller.
|
|
50 mg + 1 ml H2O i 60 min. ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
(ikke-farvede testkemikalier) og/eller
50 mg + 2 ml isopropanol blandet i 2-3 timer ved stuetemp. (farvede og ikke-farvede testkemikalier)
|
 Hvis testkemikaliets OD ved 570 ± 20 nm efter fratrækning af OD for isopropanol eller vand er > 0,08 (som svarer til ca. 5 % af den gennemsnitlige OD af den negative kontrol), bør der foretages tilpassede "living" kontroller. Hvis testkemikaliets OD ved 570 ± 20 nm efter fratrækning af OD for isopropanol eller vand er > 0,08 (som svarer til ca. 5 % af den gennemsnitlige OD af den negative kontrol), bør der foretages tilpassede "living" kontroller.
|
|
10 μl + 90 μl H2O blandet i 30 ± 2 min. ved stuetemp. (RT, 18-28oC)
|
 Hvis testkemikaliet er farvet, bør der gennemføres tilpassede "living"-kontroller Hvis testkemikaliet er farvet, bør der gennemføres tilpassede "living"-kontroller
|
|
10 mg + 90 μl H2O blandet i 30 ± 2 min. ved stuetemp.
|
 Hvis testkemikaliet er farvet, bør der gennemføres tilpassede "living"-kontroller Hvis testkemikaliet er farvet, bør der gennemføres tilpassede "living"-kontroller
|
|
|
Forhåndskontrol for direkte MTT-reduktion
|
50 μl + 1 ml MTT 1 mg/ml opløsning i 180 ± 15 min. ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
 Hvis opløsningen bliver blå/violet, bør der gennemføres tilpassede "freeze-killed"-kontroller Hvis opløsningen bliver blå/violet, bør der gennemføres tilpassede "freeze-killed"-kontroller
(50 μl sterilt afioniseret vand i MTT-opløsning bruges som negativ kontrol)
|
|
50 mg + 1 ml MTT 1 mg/ml opløsning i 180 ± 15 min. ved 37 ± 2oC, 5 ± 1 % CO2, ≥95 % RH
|
 Hvis opløsningen bliver blå/violet, bør der gennemføres tilpassede "freeze-killed"-kontroller Hvis opløsningen bliver blå/violet, bør der gennemføres tilpassede "freeze-killed"-kontroller
(50 μl sterilt afioniseret vand i MTT-opløsning bruges som negativ kontrol)
|
|
30 μl + 300 μl MTT 1 mg/ml opløsning i 180 ± 15 min. ved 37 ± 2oC, 5 ± 1, 5 ± 1 % CO2, ≥ 95 % RH
|
 Hvis opløsningen bliver blå/violet, bør der gennemføres tilpassede "water-killed"-kontroller Hvis opløsningen bliver blå/violet, bør der gennemføres tilpassede "water-killed"-kontroller
(30 μl sterilt afioniseret vand i MTT-opløsning bruges som negativ kontrol)
|
|
30 mg + 300 μl MTT 1 mg/ml opløsning i 180 ± 15 min. ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
 Hvis opløsningen bliver blå/violet, bør der gennemføres tilpassede "water-killed"-kontroller Hvis opløsningen bliver blå/violet, bør der gennemføres tilpassede "water-killed"-kontroller
(30 μl sterilt afioniseret vand i MTT-opløsning bruges som negativ kontrol)
|
|
|
Forbehandling
|
20 μl Ca2+/Mg2+-frit DPBS
i 30±2 min. ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH, beskyttet mod lys.
|
20 μl Ca2+/Mg2+ -frit DPBS
i 30±2 min. ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH, beskyttet mod lys.
|
-
|
-
|
|
Behandlingsdoser og påføring
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) ved hjælp af et kalibreret værktøj (f.eks. en strøget skefuld, der er kalibreret, så den rummer 50 mg natriumchlorid).
|
10 μl Ca2+/Mg2+-frit DPBS + 30 ± 2 μl (60 μl/cm2)
Til viskøse stoffer benyttes et nylonnet
|
30 μl Ca2+/Mg2+-frit DPBS + 30 ± 2 mg (60 mg/cm2)
|
|
Eksponeringstid og temperatur
|
30 min. (± 2 min.)
i dyrkningsmedium
ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
6 timer (± 0,25 timer)
i dyrkningsmedium
ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
30 min. (± 2 min.)
i dyrkningsmedium
ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
4 timer (± 0,1 timer)
i dyrkningsmedium
ved 37 ± 2oC, 5 ± 1 % CO2, ≥95 % RH
|
|
Skylning ved stuetemperatur
|
3 gange i 100 ml Ca2+/Mg2+-frit DPBS
|
3 gange i 100 ml Ca2+/Mg2+-frit DPBS
|
20 ml Ca2+/Mg2+-frit DPBS
|
25 ml Ca2+/Mg2+-frit DPBS
|
|
Nedsænkning efter eksponering
|
12 min. (± 2 min.) ved stuetemp. i dyrkningsmedium
|
25 min. (± 2 min.) ved stuetemp. i dyrkningsmedium
|
30 min (± 2 min.) ved 37oC, 5 % CO2, 95 % RH i dyrkningsmedium
|
30 min. (± 2 min.) ved stuetemp. i dyrkningsmedium
|
|
Inkubation efter eksponering
|
120 min. (± 15 min.) i dyrkningsmedium ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
18 timer (± 0,25 min.) i dyrkningsmedium ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
Ingen
|
18 timer (± 0,5 timer) i dyrkningsmedium ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Negativ kontrol
|
50 μl H2O
Testes samtidig
|
50 μl H2O
Testes samtidig
|
30 ± 2 μl Ca2+/Mg2+-frit DPBS
Testes samtidig
|
30 ± 2 μl Ca2+/Mg2+-frit DPBS
Testes samtidig
|
|
Positiv kontrol
|
50 μl methylacetat
Testes samtidig
|
50 μl methylacetat
Testes samtidig
|
30 ± 2 μl methylacetat
Testes samtidig
|
30 ± 2 μl methylacetat
Testes samtidig
|
|
MTT-opløsning
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
MTT-inkubationstid og temperatur
|
180 min. (± 15 min.) ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min. (± 15 min.) ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min. (± 15 min.) ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min. (± 15 min.) ved 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Ekstraktionsopløsningsmiddel
|
2 ml isopropanol
(ekstraktion fra top og bund af indsatsen ved at gennembore vævet)
|
2 ml isopropanol
(ekstraktion fra bund af indsatsen ved at gennembore vævet)
|
1,5 ml isopropanol
(ekstraktion fra top og bund af indsatsen)
|
1,5 ml isopropanol
(ekstraktion fra bund af indsatsen)
|
|
Ekstraktionstid og -temperatur
|
2-3 timer under omrystning (~120 o/min.) ved stuetemp. eller natten over ved 4-10oC
|
2-3 timer under omrystning (~120 o/min.) ved stuetemp. eller natten over ved 4-10oC
|
4 timer under omrystning (~120 o/min.) ved stuetemp. eller mindst natten over uden omrystning ved 4-10oC
|
Mindst 2 timer under omrystning (~120 o/min.) ved stuetemp.
|
|
OD-aflæsning
|
570 nm (550-590 nm)
uden referencefilter
|
570 nm (550-590 nm)
uden referencefilter
|
570 nm (540-600 nm)
uden referencefilter
|
570 nm (540-600 nm)
uden referencefilter
|
|
Kvalitetskontrol af væv
|
Behandling med 100 μl 0,3 % (v/v) Triton X-100
12,2 min. ≤ ET50 ≤ 37,5 min.
|
Behandling med 100 μl 0,3 % (v/v) Triton X-100
12,2 min. ≤ ET50 ≤ 37,5 min.
|
behandling i 30 min. med SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
behandling i 30 min. med SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Acceptkriterier
|
|
1.
|
Den gennemsnitlige OD for vævsprøver behandlet med negativt kontrolstof bør være > 0,8 og < 2,5.
|
|
2.
|
Den gennemsnitlige levedygtighed for vævsprøver eksponeret i 30 min. for positivt kontrolstof, udtrykt som % af den negative kontrol, bør være < 50 %.
|
|
3.
|
Forskellen i levedygtighed mellem to vævsprøver bør være under 20 %.
|
|
|
1.
|
Den gennemsnitlige OD for vævsprøver behandlet med negativt kontrolstof bør være > 0,8 og < 2,5.
|
|
2.
|
Den gennemsnitlige levedygtighed for vævsprøver eksponeret i 6 timer for positivt kontrolstof, udtrykt som % af den negative kontrol, bør være < 50 %
|
|
3.
|
Forskellen i levedygtighed mellem to vævsprøver bør være under 20 %.
|
|
|
1.
|
Den gennemsnitlige OD for vævsprøver behandlet med negativt kontrolstof bør være > 1,0 og ≤ 2,5.
|
|
2.
|
Den gennemsnitlige levedygtighed for vævsprøver eksponeret i 30 min. for positivt kontrolstof, udtrykt som % af den negative kontrol, bør være ≤ 30 %.
|
|
3.
|
Forskellen i levedygtighed mellem to vævsprøver bør være under 20 %.
|
|
|
1.
|
Den gennemsnitlige OD for vævsprøver behandlet med negativt kontrolstof bør være > 1,0 og ≤ 2,5.
|
|
2.
|
Den gennemsnitlige levedygtighed for vævsprøver eksponeret i 4 timer for positivt kontrolstof, udtrykt som % af den negative kontrol, bør være ≤ 20 %.
|
|
3.
|
Forskellen i levedygtighed mellem to vævsprøver bør være under 20 %.
|
|
Tillæg 3
ILLUSTRATIVT FLOWDIAGRAM OVER RETNINGSLINJER FOR, HVORDAN MAN IDENTIFICERER OG HÅNDTERER DIREKTE MTT-REDUKTIONSMIDLER OG/ELLER FARVEINTERFERERENDE KEMIKALIER BASERET PÅ VRM1 SOP

Tillæg 4
ILLUSTRATIVT FLOWDIAGRAM OVER RETNINGSLINJER FOR, HVORDAN MAN IDENTIFICERER OG HÅNDTERER DIREKTE MTT-REDUKTIONSMIDLER OG/ELLER FARVEINTERFERERENDE KEMIKALIER BASERET PÅ VRM2 SOP

Tillæg 5
NØGLEPARAMETRE OG ACCEPTKRITERIER FOR KVALIFICERING AF ET HPLC/UPLC-SPEKTROFOTOMETRISYSTEM TIL MÅLING AF MTT-FORMAZAN EKSTRAHERET FRA RHCE-VÆVSMODELLER
|
Parameter
|
Protokol fra FDA-vejledning (36)(38)
|
Acceptkriterier
|
|
Selektivitet
|
Analyse af isopropanol, "living blank" (isopropanolekstrakt fra modeller af levende RhCE-væv uden behandling), "dead blank" (isopropanolekstrakt fra modeller af dødt RhCE-væv uden behandling) og et farvestof (f.eks. methylenblåt)
|
Arealinterferens ≤ 20 % af ArealLLOQ
(88)
|
|
Præcision
|
Kvalitetskontroller (dvs. MTT-formazan ved 1,6 μg/ml, 16 μg/ml og 160 μg/ml ) i isopropanol (n=5)
|
CV ≤ 15 % eller ≤ 20 % for LLOQ
|
|
Nøjagtighed
|
Kvalitetskontroller i isopropanol (n=5)
|
%Dev ≤ 15 % eller ≤ 20 % for LLOQ
|
|
Matrixvirkning
|
Kvalitetskontrol i "living blank" (n=5)
|
85 % ≤ matrixvirkning i % ≤ 115 %
|
|
Overførsel
|
Analyse af isopropanol efter en ULOQ (89) standard
|
Arealinterferens ≤ 20 % af ArealLLOQ
|
|
Reproducerbarhed (inden for en dag)
|
Tre uafhængige kalibreringskurver (baseret på seks på hinanden følgende 1/3-opløsninger af MTT-formazan i isopropanol begyndende ved ULOQ, dvs. 200 μg/ml).
Kvalitetskontroller i isopropanol (n=5)
|
Kalibreringskurver: %Dev ≤ 15 % eller ≤ 20 % for LLOQ
Kvalitetskontroller: %Dev ≤ 15 % og CV ≤ 15 %
|
|
Reproducerbarhed (mellem flere dage)
|
Dag 1: en kalibreringskurve og kvalitetskontroller i isopropanol (n=3)
Dag 2: en kalibreringskurve og kvalitetskontroller i isopropanol (n=3)
Dag 3: en kalibreringskurve og kvalitetskontroller i isopropanol (n=3)
|
|
MTT-formazans kortsigtede stabilitet i RhCE-vævsekstrakt
|
Kvalitetskontroller i "living blank" (n=3) analyseret på præpareringsdagen og efter 24 timers opbevaring ved stuetemperatur
|
%Dev ≤ 15 %
|
|
MTT-formazans langsigtede stabilitet i RhCE-vævsekstrakt (hvis påkrævet)
|
Kvalitetskontroller i "living blank" (n=3) analyseret på præpareringsdagen og efter flere dages opbevaring ved -20 °C
|
%Dev ≤ 15 %
|
B.70
IN VITRO-ASSAYS MED HUMAN REKOMBINANT ØSTROGENRECEPTOR (hrER) TIL PÅVISNING AF KEMIKALIER MED ER-AFFINITET
GENEREL INDLEDNING
OECD’s ydeevnebaserede forsøgsretningslinje
|
1.
|
Denne forsøgsmetode svarer til OECD Test Guideline (TG) 493 (2015). TG 493 er en ydeevnebaseret forsøgsretningslinje (PBTG) med beskrivelse af metoden for in vitro assays med human rekombinant østrogenreceptor til påvisning af stoffer med østrogenreceptor-affinitet (hrER-affinitetassays). Den omfatter to mekanistisk og funktionelt lignende assays til identifikation af østrogenreceptorbindere (dvs. ERα) og ventes at lette udvikingen af nye lignende eller modificerede assays i overensstemmelse med de principper for validering, som er fastsat i OECD’s Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment (1). De fuldt validerede referenceforsøgsmetoder (tillæg 2 og tillæg 3), som danner grundlag for denne PBTG, er:
|
—
|
Freyberger-Wilson (FW) In Vitro Estrogen Receptor (ER) Binding Assay Using a Full Length Human Recombinant ERα (2) og
|
|
—
|
The Chemical Evaluation and Research Institute’s (CERI) In Vitro Estrogen Receptor Binding Assay Using a Human Recombinant Ligand Binding Domain Protein (2).
|
Der findes ydeevnestandarder (PS) (3) for at lette udvikling og validering af lignende forsøgsmetoder til den samme fareegenskab og give mulighed for rettidig ændring af PBTG 493, så nye lignende assays kan føjes til en opdateret PBTG. Lignende testassays vil dog først blive tilføjet efter OECD’s kontrol af og samtykke til, at ydeevnestandarderne er opfyldt. De assays, der er omfattet af TG 493, kan uden forskel anvendes til at opfylde OECD-medlemslandenes krav til forsøgsresultaterne om østrogenreceptor-affinitet, samtidig med at de drager fordel af OECD’s gensidige anerkendelse af data.
|
Baggrund og principper for de assays, der indgår i denne forsøgsmetode
|
2.
|
OECD iværksatte i 1998 en højt prioriteret aktivitet om revision af eksisterende og udvikling af nye forsøgsretningslinjer til screening og test af potentielle hormonforstyrrende kemikalier. OECD’s begrebsramme for testning og vurdering af potentielle hormonforstyrrende kemikalier blev revideret i 2012. Den oprindelige og reviderede begrebsramme er vedlagt som bilag til det vejledende dokument, Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (4). Begrebsrammen omfatter fem niveauer, idet hvert niveau svarer til forskellige niveauer af biologisk kompleksitet. De ER-affinitetsassays, som beskrives i denne forsøgsmetode, er niveau 2, der omfatter »in vitro-assays, der giver data om en eller flere udvalgte endokrine mekanismer/veje«. Denne forsøgsmetode er til in vitro-receptoraffinitetsassays udformet til at identificere ligander for den humane østrogenreceptor alfa (ERα).
|
|
3.
|
Relevansen af in vitro-ER-affinitets-assayet for biologiske funktioner er tydeligt påvist. ER-affinitetsassays er udformet til at identificere kemikalier, som har potentiale til at forstyrre østrogenhormonvejen, og er i udstrakt grad blevet anvendt i den seneste snes år til at karakterisere ER-vævsfordeling samt til at identificere ER-agonister/-antagonister. Disse assays afspejler ligand-receptor-interaktionen, som er det indledende skridt på østrogensignaleringsvejen og er væsentlig for reproduktionsfunktionen hos alle hvirveldyr.
|
|
4.
|
Østrogeners interaktion med ER’er kan påvirke transkription af østrogenkontrollerede gener og inducere ikke-genomiske virkninger, der kan føre til induktion eller inhibering af cellulære processer, herunder processer, som er nødvendige for celleproliferation, normal fosterudvikling og reproduktiv funktion (5)(6)(7). Forstyrrelse af normale østrogensystemer kan have potentiale til at udløse skadelige virkninger på normal udvikling (ontogenese), reproduktiv sundhed og det reproduktive systems integritet. Mangelfuld ER-signalering kan medføre virkninger som øget risiko for hormonbetinget cancer, forringet forplantningsevne og ændringer i fostervækst og -udvikling (8).
|
|
5.
|
In vitro-affinitetsassays er baseret på direkte interaktion mellem et stof med et specifikt bindingssted for receptorliganden, som regulerer gentranskriptionen. Den vigtigste komponent for bindingsassayet vedrørende human rekombinant østrogenreceptor alfa (hrERα) måler en radioaktivt mærket ligands evne ([3H]17β-østradiol) til at binde sig til ER under tilstedeværelse af stigende koncentrationer af et testkemikalie (dvs. konkurrent). Testkemikalier, som har en høj ER-affinitet, konkurrerer med den radioaktivt mærkede ligand ved en lavere koncentration sammenlignet med kemikalier med lavere affinitet for receptoren. Dette assay består af to vigtige komponenter: et mætningsbindingsforsøg for at karakterisere parametrene for interaktion mellem receptor og ligand og dokumentere ER-specificitet efterfulgt af et forsøg med kompetitiv binding, der karakteriserer konkurrencen mellem et testkemikalie og en radioaktivt mærket ligand for binding til ER.
|
|
6.
|
CERI’s valideringsstudier og FW-bindingsassays har påvist deres relevans og pålidelighed i overensstemmelse med deres formål (2).
|
|
7.
|
Definitioner og forkortelser brugt i denne forsøgsmetode er beskrevet i tillæg 1.
|
Anvendelsesområde og begrænsninger i forbindelse med receptorbindingsassays
|
8.
|
Disse assays foreslås til screenings- og prioriteringsformål, men kan også give oplysninger til en molekylær initiering (MIE), som kan anvendes i en »weight of evidence«-analyse. De analyserer kemikaliets binding til ERα-ligandens bindingsområde i et in vitro-system. Således bør resultaterne ikke direkte ekstrapoleres til den komplekse signalering og regulering af det intakte hormonsystem in vivo.
|
|
9.
|
Binding af den naturlige ligand, 17β-østradiol, er det indledende skridt i en række molekylære begivenheder, der aktiverer transkriptionen af målgener og i sidste ende kulminerer med en fysiologisk forandring (9). Binding til ERα-ligandbindingsområdet betragtes som en af de centrale mekanismer ved ER-medieret hormonforstyrrelse (ED), skønt der findes andre mekanismer, gennem hvilke ED kan forekomme, bl.a. (i) interaktioner med andre ERα-steder end ligandbindingslommen, (ii) interaktioner med andre receptorer af relevans for østrogensignalering, ER-β og G-proteinkoblet østrogenreceptor, andre receptorer og enzymsystemer i hormonsystemet, (iii) hormonsyntese, (iv) metabolisk aktivering og/eller inaktivering af hormoner, (v) fordeling af hormoner til målvæv og (vi) udskilning af hormoner fra kroppen. Ingen af de assays, der er omfattet af denne forsøgsmetode, behandler disse virkemåder.
|
|
10.
|
Denne forsøgsmetode behandler stoffers evne til at binde sig til humant ERα og skelner ikke mellem ERα-agonister og -antagonister. Disse assays behandler heller ikke yderligere downstream-begivenheder såsom gentranskription eller fysiologiske forandringer. I betragtning af at alene enkeltstoffer med kun én bestanddel blev anvendt under valideringen, er anvendeligheden på forsøgsblandinger ikke blevet behandlet. Assays kan dog teoretisk set benyttes til test af stoffer med forskellige bestanddele og blandinger. Før forsøgsmetoden anvendes til at teste en blanding for at generere data til lovmæssige formål, bør det overvejes, om den vil give resultater, som er egnede til formålet, og i givet fald hvorfor. Sådanne overvejelser er ikke nødvendige, når der foreligger et myndighedskrav om test af blandingen.
|
|
11.
|
De cellefrie receptorsystemer har ingen iboende metabolisk kapacitet, og de blev ikke valideret i kombination med metaboliske enzymsystemer. Det kunne dog være muligt at indarbejde metabolisk aktivitet i forsøgsdesignet, men dette ville kræve en yderligere valideringsindsats.
|
|
12.
|
Kemikalier, der kan denaturere proteinet (dvs. receptorprotein), såsom overfladeaktivt stof eller kemikalier, der kan ændre assaybufferens pH-værdi, kan ikke testes eller kan kun testes ved koncentrationer uden sådanne interaktioner. Ellers er det koncentrationsområde, som kan testes i assays for et testkemikalie, begrænset ved sin opløselighed i assaybufferen.
|
|
13.
|
Til orientering viser tabel 1 testresultaterne for de 24 stoffer, som blev testet i begge de fuldt validerede assays, der beskrives i denne forsøgsmetode. Af disse stoffer er 17 klassificeret som ER-bindere og 6 som ikke-bindere baseret på offentliggjorte rapporter, herunder in vitro-assays for ER-transkriptionsaktivering og/eller det uterotrofiske assay (9)(10)(11)(12)(13)(14)(15). I henhold til de data, der er opsummeret i tabel 1, var der næsten 100 % overensstemmelse mellem de to assays om klassificeringer af alle stoffer op til 10-4M, og hvert stof blev korrekt klassificeret som en ER-binder eller ikke-binder. Supplerende information om denne gruppe af stoffer samt supplerende stoffer, som er testet i ER-bindingsassays i løbet af valideringsundersøgelserne, er anført i ydeevnestandarderne for hrER-bindingsassayet (3), tillæg 2 (tabel 1, 2 og 3).
Tabel 1
Klassificering af stoffer som ER-bindere eller ikke-bindere ved testning i FW- og CERI-hrER-bindingsassays med sammenligning med forventet respons
|
|
Stof
|
CAS RN
|
Forventet respons
|
FW-assay
|
CERI-assay
|
MESH-kemikalie: Gruppe
|
Produktgruppe
|
|
|
Koncentrations-område (M)
|
Klassificering
|
Koncentrations-område (M)
|
Klassificering
|
|
1
|
17β-østradiol
|
50-28-2
|
Binder
|
1×10-11 – 1×10-6
|
Binder
|
1×10-11 – 1×10-6
|
Binder
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
2
|
Norethynodrel
|
68-23-5
|
Binder
|
3×10-9 – 30×10-4
|
Binder
|
3×10-9 – 30×10-4
|
Binder
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
3
|
Norethindron
|
68-22-4
|
Binder
|
3×10-9 – 30×10-4
|
Binder
|
3×10-9 – 30×10-4
|
Binder
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
4
|
Di-n-butylphthalat
|
84-74-2
|
Ikke-binder (*5)
|
1×10-10 – 1×10-4
|
Ikke-binder (*6)
(1)
|
1×10-10 – 1×10-4
|
Ikke-binder (*6)
(1)
|
Kulbrinte (cyklisk), ester
|
Blødgører, kemisk mellemprodukt
|
|
5
|
DES
|
56-53-1
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kulbrinte (cyklisk), phenol
|
Farmaceutisk produkt, veterinær
|
|
6
|
17α-ethynyløstradiol
|
57-63-6
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
7
|
Meso-hexestrol
|
84-16-2
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kulbrinte (cyklisk), phenol
|
Farmaceutisk produkt, veterinær
|
|
8
|
Genistein
|
446-72-0
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kulbrinte (heterocyklisk), flavonoid
|
Naturprodukt
|
|
9
|
Equol
|
531-95-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Phytoøstrogenmetabolit
|
Naturprodukt
|
|
10
|
Butylparaben (n butyl-4-hydroxybenzoat)
|
94-26-8
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Paraben
|
Konserveringsmiddel
|
|
11
|
Nonylphenol (blanding)
|
84852-15-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Alkylphenol
|
Mellemprodukt
|
|
12
|
o,p’-DDT
|
789-02-6
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Organiske chlorforbindelser
|
Insekticid
|
|
13
|
Corticosteron
|
50-22-6
|
Ikke-binder (*5)
|
1×10-10 – 1×10-4
|
Ikke-binder
|
1×10-10 – 1×10-4
|
Ikke-binder
|
Steroid
|
Naturprodukt
|
|
14
|
Zearalenon
|
17924-92-4
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kulbrinte (heterocyklisk), lacton
|
Naturprodukt
|
|
15
|
Tamoxifen
|
10540-29-1
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt, veterinær
|
|
16
|
5α-dihydrotestosteron
|
521-18-6
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Steroid, ikke-phenolisk
|
Naturprodukt
|
|
17
|
Bisphenol A
|
80-05-7
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Phenol
|
Kemisk mellemprodukt
|
|
18
|
4-n-heptylphenol
|
1987-50-4
|
Binder
|
1×10-10 – 1×10-3
|
Usikker (5)
|
1×10-10 – 1×10-3
|
Binder
|
Alkylphenol
|
Mellemprodukt
|
|
19
|
Kepon (chlordecon)
|
143-50-0
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Kulbrinter (halogenerede)
|
Pesticid
|
|
20
|
Benz(a)anthracen
|
56-55-3
|
Ikke-binder
|
1×10-10 – 1×10-3
|
Ikke-binder (6)
|
1×10-10 – 1×10-3
|
Ikke-binder (6)
|
Aromatiske kulbrinter
|
Mellemprodukt
|
|
21
|
Enterolacton
|
78473-71-9
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
1×10-10 – 1×10-3
|
Binder
|
Phytoøstrogen
|
Naturprodukt
|
|
22
|
Progesteron
|
57-83-0
|
Ikke-binder (*5)
|
1×10-10 – 1×10-4
|
Ikke-binder
|
1×10-10 – 1×10-4
|
Ikke-binder
|
Steroid
|
Naturprodukt
|
|
23
|
Octyltriethoxysilan
|
2943-75-1
|
Ikke-binder
|
1×10-10 – 1×10-3
|
Ikke-binder
|
1×10-10 – 1×10-3
|
Ikke-binder
|
Silan
|
Overflademodifikator
|
|
24
|
Atrazin
|
1912-24-9
|
Ikke-binder (*5)
|
1×10-10 – 1×10-4
|
Ikke-binder
|
1×10-10 – 1×10-4
|
Ikke-binder
|
heterocyklisk forbindelse
|
Herbicid
|
|
KOMPONENTER I hrER-BINDINGSASSAY
De centrale dele af assayets komponenter
|
14.
|
Denne forsøgsmetode gælder for assays, der anvender en ER-receptor og en passende stærk ligand til receptoren, der kan bruges som markør/sporstof for assayet og kan fortrænges med stigende koncentrationer af et testkemikalie. Bindingsassays indeholder følgende to hovedkomponenter: 1) mætningsbinding og 2) kompetitiv binding. Mætningsbindingsassayet benyttes til at bekræfte receptorpræparaternes specificitet og aktivitet, mens det kompetitive bindingsforsøg benyttes til at vurdere et testkemikalies evne til at binde sig til hrER.
|
Kontroller
|
15.
|
Grundlaget for forslaget til sideløbende referenceøstrogen og kontroller bør beskrives. Sideløbende kontroller (opløsningsmiddel (bærestof), positiv (ER-binder, stærk og svag affinitet), negativ (ikke-binder)), hvor det er relevant, viser, at assayet virker under testbetingelser og danner grundlag for sammenligninger af forsøg. De indgår normalt i acceptkriterierne for et givet forsøg (1). Fuldstændige koncentrationskurver for referenceøstrogen og kontroller (dvs. svag binder og ikke-binder) anvendes på én plade under hver analyseserie. Alle andre plader skal indeholde: 1) en høj (næsten fuld fortrængning af radioaktivt mærket ligand) og middelhøj (næsten IC50) koncentration af E2 og svag binder, hver især in triplo 2) en kontrolkultur med opløsningsmiddel og ikke-specifik binding, hver især in triplo.
|
Procedurer for standardkvalitetskontrol
|
16.
|
Procedurer for standardkvalitetskontrol udføres som beskrevet for hvert assay for at sikre aktive receptorer og korrekte kemiske koncentrationer, og at tolerancegrænser forbliver stabile gennem mange replikationer og bevarer evnen til at give de forventede ER-bindingsresponser over tid.
|
Påvisning af laboratoriets kompetence
|
17.
|
Inden ukendte kemikalier testes med et af de assays, der indgår i denne forsøgsmetode, skal det enkelte laboratorium påvise kompetence til at bruge assayet ved at udføre mætningsassays til at bekræfte ER-præparatets specificitet og aktivitet og kompetitive bindingsassays med referenceøstrogen og kontroller (svag binder og ikke-binder). Laboratoriet skal oprette en historisk database med resultater for referenceøstrogenet og de kontroller, der er genereret fra 3-5 uafhængige forsøg gennemført på forskellige dage. Disse forsøg skal danne grundlaget for referenceøstrogenet og de historiske kontroller for laboratoriet og vil blive anvendt til en delvis vurdering af, om assayet kan godkendes til fremtidige analyseserier.
|
|
18.
|
Testsystemets responsivitet vil også blive bekræftet ved at teste de kompetencestoffer, som er anført i tabel 2. Listen over kompetencestoffer er en del af de referencestoffer, der er anført i Performance Standards for the ER binding assays (3). Disse stoffer kan fås i handelen, repræsenterer kemikaliegrupper, der i almindelighed er forbundet med ER-bindingsaktivitet, og udviser et passende styrkeområde, som forventes for ER-bindere (dvs. stærk til svag) og ikke-bindere (dvs. negative). For hvert kompetencestof skal de testede koncentrationer dække det område, der er anført i tabel 2. Der udføres mindst tre forsøg for hvert stof, og resultaterne skal være i overensstemmelse med den forventede kemiske aktivitet. Hvert forsøg gennemføres uafhængigt (dvs. med friske receptorfortyndinger, kemikalier og reagens) med tre replikater for hver koncentration. Kompetence påvises ved korrekt klassificering (positiv/negativ) af hvert kompetencestof. Kompetencetest udføres af hver enkelt tekniker, når han/hun lærer et assay.
Tabel 2
Liste over kontroller og kompetencestoffer for kompetitive hrER-bindingsassays
(90)
|
Nr.
|
Stof
|
CAS RN (91)
|
Forventet respons (92)
(93)
|
Testkoncentrationsområde (M)
|
MeSH-kemikaliegruppe (94)
|
Produktgruppe (95)
|
|
Kontroller (referenceøstrogen, svag binder, ikke-binder)
|
|
1
|
17ß-østradiol (E2)
|
50-28-2
|
Binder
|
1×10-11 – 1×10-6
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
2
|
Norethynodrel (eller) Norethindron
|
68-23-5 (eller) 68-22-4
|
Binder
|
3×10-9 – 30×10-6
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
3
|
Octyltriethoxysilan
|
2943-75-1
|
Ikke-binder
|
1×10-10 – 1×10-3
|
Silan
|
Overflademodifikator
|
|
Kompetencestoffer (95)
|
|
4
|
Diethylstilbestrol
|
56-53-1
|
Binder
|
1×10-11 – 1×10-6
|
Kulbrinte (cyklisk), phenol
|
Farmaceutisk produkt, veterinær
|
|
5
|
17α-ethynyløstradiol
|
57-63-6
|
Binder
|
1×10-11 – 1×10-6
|
Steroid
|
Farmaceutisk produkt, veterinær
|
|
6
|
Meso-hexestrol
|
84-16-2
|
Binder
|
1×10-11 – 1×10-6
|
Kulbrinte (cyklisk), phenol
|
Farmaceutisk produkt, veterinær
|
|
7
|
Tamoxifen
|
10540-29-1
|
Binder
|
1×10-11 – 1×10-6
|
Kulbrinte (cyklisk)
|
Farmaceutisk produkt, veterinær
|
|
8
|
Genistein
|
446-72-0
|
Binder
|
1×10-10 – 1×10-3
|
Heterocyklisk forbindelse, flavonoid
|
Naturprodukt
|
|
9
|
Bisphenol A
|
80-05-7
|
Binder
|
1×10-10 – 1×10-3
|
Phenol
|
Kemisk mellemprodukt
|
|
10
|
Zearalonon
|
17924-92-4
|
Binder
|
1×10-11 – 1×10-3
|
Heterocyklisk forbindelse, lacton
|
Naturprodukt
|
|
11
|
Butylparaben
|
94-26-8
|
Binder
|
1×10-11 – 1×10-3
|
Carboxylsyre, phenol
|
Konserveringsmiddel
|
|
12
|
Atrazin
|
1912-24-9
|
Ikke-binder
|
1×10-11 – 1×10-6
|
heterocyklisk forbindelse
|
Herbicid
|
|
13
|
Di-n-butylphthalat (DBP) (96)
|
84-74-2
|
Ikke-binder (97)
|
1×10-10 – 1×10-4
|
Kulbrinte (cyklisk), ester
|
Blødgører, kemisk mellemprodukt
|
|
14
|
Corticosteron
|
50-22-6
|
Ikke-binder
|
1×10-11 – 1×10-4
|
Steroid
|
Naturprodukt
|
|
Opløselighedstest og bestemmelse af dosisinterval for testkemikalier
|
19.
|
Der udføres en foreløbig test for at bestemme opløselighedsgrænsen for hvert testkemikalie og for at afdække det passende koncentrationsområde, der skal anvendes, når testen udføres. Opløselighedsgrænsen for hvert testkemikalie skal indledningsvis bestemmes i opløsningsmidlet og yderligere bekræftes under assaybetingelser. De endelige koncentrationer, der testes i assayet, bør ikke overstige 1 mM. Test af områdebestemmelsen består af en kontrolkultur med opløsningsmiddel sammen med otte log-seriefortyndinger, der begynder ved den maksimalt acceptable koncentration (f.eks. 1 mM eller lavere, baseret på opløselighedsgrænsen), og idet der noteres tilstedeværelse af uklarhed eller bundfald. Koncentrationer i det andet og tredje forsøg bør tilpasses, hvis det er relevant, for bedre at karakterisere koncentrationsresponskurven.
|
Acceptkriterier for testanalyseserien
|
20.
|
Godkendelse eller forkastelse af en analyseserie er baseret på vurdering af de resultater, der er opnået for referenceøstrogen og den kontrol, der er anvendt til hvert enkelt forsøg. For det første skal de fuldstændige koncentrationskurver for referencekontrollerne fra hvert forsøg for plade 1 opfylde resultatmålingsmetoderne med parametrene for kurvetilpasningen (f.eks. IC50 og Hills kurve) baseret på de resultater, der er rapporteret for de respektive protokoller for CERI- og FW-assayene (tillæg 2 og 3) og de historiske kontroldata fra det laboratorium, der udfører testen. Alle kontroller (referenceøstrogen, svag binder og ikke-binder) skal være klassificeret korrekt for hvert forsøg. For det andet skal kontrollerne på alle efterfølgende plader vurderes for overensstemmelse med plade 1. Der skal benyttes et tilstrækkeligt koncentrationsområde for testkemikaliet for klart at definere toppen af den kompetitive bindingskurve. Variabilitet mellem replikater ved hver koncentration af testkemikaliet samt mellem de tre uafhængige analyseserier skal være rimelige og videnskabeligt forsvarlige. Evnen til at gennemføre assayet konsistent skal påvises ved udvikling og vedligeholdelse af en historisk database for referenceøstrogenet og kontrollerne. Standardafvigelser eller variationskoefficienter (CV) for gennemsnit af kurver for referenceøstrogen og svage bindere, der fungerer som kontrol, og som lever op til parametrene fra flere forsøg, kan anvendes som et mål for reproducerbarhed i laboratorier. Der bør anlægges en faglig vurdering ved gennemgang af pladekontrolresultaterne fra hver analyseserie samt for hvert testkemikalie.
Endvidere skal følgende principper for acceptkriterier overholdes:
|
—
|
Der skal være tilstrækkelige data til en kvantitativ vurdering af ER-binding.
|
|
—
|
De testede koncentrationer skal forblive inden for testkemikaliets opløselighedsområde.
|
|
Analyse af data
|
21.
|
Den definerede fremgangsmåde ved analyse af data om mætning og kompetitiv binding skal overholde de centrale principper for karakterisering af interaktioner mellem receptor og ligand. Mæthedsbindingsdata analyseres normalt under anvendelse af en ikke-lineær regressionsmodel, der tager højde for samlet og ikke-specifik binding. En korrektion for ligandnedbrydning (f.eks. Swillens, 1995 (19)) kan være nødvendig ved bestemmelse af Bmax og Kd. Data fra kompetitive bindingsassays omdannes normalt (f.eks. procentvis specifik binding og testkemikaliekoncentration (log M)). Skøn over log (IC50) for hvert testkemikalie bør bestemmes ved anvendelse af passende ikke-lineært kurvetilpasningssoftware, så det passer til en Hill-ligning med fire parametre. Efter en indledende analyse bør der foretages en gennemgang af kurvetilpasningsparametrene og en visuel gennemgang af, hvor godt bindingsdataene passer til den genererede kompetitive bindingskurve. I nogle tilfælde kan det være nødvendigt at foretage en supplerende analyse for at få den bedste kurvetilpasning (f.eks. begrænsning af top og/eller bund af kurven, anvendelse af 10 %-reglen, jf. tillæg 4 og henvisning 2 (afsnit III.A.2).
|
|
22.
|
Hvis acceptkriterierne opfyldes (punkt 20), er det tegn på, at assaysystemet fungerer korrekt, men det er ingen garanti for, at enhver test vil producere nøjagtige data. Kan de korrekte resultater af den første test gentages, er det bedste tegn på, at der blev produceret nøjagtige data.
|
Generelle kriterier for datafortolkning
|
23.
|
Der er i øjeblikket ingen universelt anerkendt metode for fortolkning af ER-bindingsdata. Både kvalitative (f.eks. binder/ikke-binder) og/eller kvantitative (f.eks. log IC50, relativ affinitet (RBA) osv.) vurderinger af hrER-medieret aktivitet bør være baseret på empiriske data og en solid videnskabelig vurdering.
|
Testrapport
|
24.
|
Testrapporten bør indeholde følgende oplysninger:
|
|
Kontrol/reference/testkemikalie
|
—
|
oprindelse, batchnummer, frist for anvendelse, hvis dette foreligger
|
|
—
|
testkemikaliets stabilitet, hvis denne er kendt
|
|
—
|
testkemikaliets opløselighed og stabilitet i opløsningsmidlet, hvis dette er kendt
|
|
—
|
måling af pH-værdi, osmolalitet og bundfald i det dyrkningsmedium, som testkemikaliet blev tilsat, hvor det er relevant.
|
|
|
|
Stof med kun én bestanddel:
|
—
|
fysisk udseende, vandopløselighed og yderligere relevante fysisk-kemiske egenskaber
|
|
—
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn, CAS-nummer, SMILES eller InChI-kode, strukturformel, renhed, kemisk identitet af urenheder i det omfang, det er relevant og praktisk muligt osv.
|
|
|
|
Stof med flere bestanddele, UVCB-stoffer og blandinger:
|
—
|
så vidt muligt kendetegnet ved kemisk identitet (jf. ovenfor), kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber
|
|
|
|
Opløsningsmiddel/bærestof:
|
—
|
karakterisering (art, leverandør og batch)
|
|
—
|
begrundelse for valg af opløsningsmiddel/bærestof
|
|
—
|
testkemikaliets opløselighed og stabilitet i opløsningsmidlet/bærestoffet, hvis dette er kendt
|
|
|
|
Receptorer:
|
—
|
receptorkilde (leverandør, katalognr., parti, receptorart, aktiv receptorkoncentration fra leverandøren, certificering fra leverandøren)
|
|
—
|
karakterisering af receptorer (herunder mætningsbindingsresultater): Kd, Bmax
|
|
—
|
opbevaring af receptorer
|
|
—
|
radioaktivt mærket ligand:
|
|
—
|
leverandør, katalognr., parti, specifik aktivitet
|
|
|
|
Testbetingelser:
|
—
|
begrænsninger for opløselighed under assaybetingelser
|
|
—
|
sammensætning af bindingsbuffer
|
|
—
|
sporstofkoncentration (dvs. radioaktivt mærket ligand)
|
|
—
|
testkemikaliekoncentrationer
|
|
—
|
procent bærestof i det endelige assay
|
|
—
|
inkubationstemperatur og -periode
|
|
—
|
metode for bunden/fri separation
|
|
—
|
positive og negative kontrol-/referencestoffer
|
|
—
|
kriterier for bedømmelse af test som positive, negative eller usikre
|
|
|
|
Testgodkendelse:
|
—
|
faktiske IC50-værdier og værdier for Hills kurve for sideløbende positive kontrol-/referencestoffer
|
|
|
|
Resultater:
|
—
|
primære og bundne/frie data
|
|
—
|
bekræftelseskontrol af denaturering, hvis det er relevant
|
|
—
|
den laveste effektive koncentration (LEC), hvis den foreligger
|
|
—
|
RBA og/eller IC50-værdier, hvis det er relevant
|
|
—
|
koncentration/respons-sammenhængen, når det er muligt
|
|
—
|
eventuelle statistiske analyser sammen med en måling af fejl og konfidens (f.eks. SEM (middelfejlen på middeltallet), standardafvigelse, CV eller 95 % CI) og en beskrivelse af, hvordan disse værdier blev opnået
|
|
|
|
Diskussion af resultater:
|
—
|
anvendelse af 10 %-reglen
|
|
Konklusion
|
LITTERATUR
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: s. 20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): s. 1073-89.
|
|
(7)
|
Younes M. og Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): s. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. s. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Tillæg 1
DEFINITIONER OG FORKORTELSER
10 %-regel
: mulighed for at se bort fra datapunkter i analyser, hvor gennemsnittet af replikater for det procentvise specifikt bundne [3H]17β-østradiol ligger på 10 % eller mere over den, som blev observeret for gennemsnitsværdien ved en lavere koncentration (jf. tillæg 4).
Acceptkriterier
: minimumsstandarder for udførelse af forsøgskontroller og referencestandarder. Alle acceptkriterier skal være opfyldt, for at et forsøg kan betragtes som gyldigt.
Nøjagtighed (konkordans)
: overensstemmelsen mellem forsøgsmetodens resultater og accepterede referenceværdier. Den er et mål for forsøgsmetodens ydeevne og et af aspekterne af relevans. Udtrykket benyttes ofte som synonym for "konkordans", forstået som den andel af korrekte udfald, som assayet fører til (1).
CF
: OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupters (OECD's konceptuelle ramme for test og vurdering af hormonforstyrrende kemikalier).
Kemikalie
: et stof eller en blanding
CV
: Coefficient of variation (variationskoefficient)
E2
: 17β-østradiol
ED
: hormonforstyrrende virkning
hERα
: human østrogenreceptor alfa
ER
: østrogenreceptor
Østrogenaktivitet
: et kemikalies evne til at efterligne 17β-østradiol med hensyn til dettes evne til at binde sig til østrogenreceptorer Binding til hERα kan påvises med denne forsøgsmetode.
IC
50
: halvdelen af den maksimale effektive koncentration af et inhiberende testkemikalie
ICCVAM
: US Interagency Coordinating Committee on the Validation of Alternative Methods.
Reproducerbarhed mellem laboratorier
: mål for, i hvilken grad forskellige kvalificerede laboratorier kan opnå de samme resultater kvalitativt og kvantitativt, når de anvender samme protokol og tester de samme stoffer. Reproducerbarheden mellem laboratorier bestemmes forud for og under valideringsprocessen og angiver, i hvilken grad et assay kan overføres mellem laboratorier med et vellykket resultat. Kaldes også ekstern reproducerbarhed (1).
Reproducerbarhed inden for laboratorier
: bestemmelse af, i hvilken grad kvalificeret personale på det samme laboratorium kan reproducere resultater ved at anvende en specifik protokol på forskellige tidspunkter. Kaldes også "intern reproducerbarhed" (1).
LEC
: laveste effektive koncentration er den laveste koncentration af testkemikaliet, som giver en respons (dvs. den laveste testkemikaliekoncentration, ved hvilken foldinduktionen er statistisk forskellig fra den sideløbende bærestofkontrol).
Me-too test
: populært udtryk for et assay, som strukturelt og funktionelt ligner en valideret og anerkendt referenceforsøgsmetode. Benyttes som synonym for lignende forsøgsmetoder
PBTG
: ydeevnebaseret forsøgsretningslinje
Ydeevnestandarder
: standarder, der med udgangspunkt i en valideret forsøgsmetode danner grundlag for at vurdere, om et foreslået assay, som er mekanistisk og funktionelt tilsvarende, er sammenligneligt. De indeholder (1) de centrale dele af assayet, (2) en minimumsliste over referencekemikalier, som er udvalgt blandt de kemikalier, der anvendes til at påvise, at den validerede forsøgsmetodes ydeevne er acceptabel, og (3) de sammenlignelige niveauer af nøjagtighed og pålidelighed, som er fastsat efter, hvad der er opnået med den validerede forsøgsmetode, og som det skal påvises, at det foreslåede assay kan yde ved vurdering ud fra minimumslisten over referencekemikalier (1).
Kompetencestoffer
: en delmængde af referencestoffer, der indgår i ydeevnestandarderne, og som laboratorier kan anvende til at påvise teknisk kompetence med et standardiseret assay. Udvælgelseskriterier for disse stoffer omfatter typisk, at de repræsenterer responsområdet, fås i handelen, og at der foreligger referencedata af høj kvalitet.
Kompetence
: dokumenteret evne til at gennemføre et assay korrekt, før ukendte stoffer testes.
Referenceøstrogen
: 17ß-østradiol (E2, CAS 50-28-2)
Referenceforsøgsmetoder
: de assays, hvorpå PBTG 493 er baseret
RBA
: Relativ affinitet. RBA af et stof beregnes som en procentdel af logaritmen (IC50) til stoffet i forhold til logaritmen (IC50) af 17β-østradiol.
Relevans
: beskrivelse af sammenhængen mellem et assay og den pågældende virkning og dens meningsfuldhed og brugbarhed til et bestemt formål. Det angives, i hvilket omfang assayet måler eller forudsiger den relevante biologiske virkning korrekt. Assayets nøjagtighed (konkordans) indgår i vurderingen af dets relevans (1).
Pålidelighed
: mål for, i hvilken grad et assay kan reproduceres i og mellem laboratorier over en længere periode, når det udføres efter samme protokol. Det bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier.
Standardafvigelse
: standardafvigelse
Testkemikalie
: alle stoffer eller blandinger, der testes ved hjælp af denne forsøgsmetode
Valideret forsøgsmetode
: et assay, for hvilket der er gennemført valideringsundersøgelser for at fastslå relevansen (herunder nøjagtigheden) og pålideligheden til et bestemt formål. Det er vigtigt at være opmærksom på, at en valideret forsøgsmetode måske ikke har en tilstrækkelig ydeevne med hensyn til nøjagtighed og pålidelighed til at være acceptabel til det planlagte formål (1).
Validering
: den proces, hvorved pålideligheden og relevansen af en bestemt tilgang, metode, proces eller vurdering eller et bestemt assay fastslås til et defineret formål (1).
Tillæg 2
FREYBERGER-WILSONS IN VITRO-ASSAYS VEDRØRENDE ØSTROGENRECEPTORMÆTNING (ERΑ) OG KOMPETITIV BINDING VED ANVENDELSE AF REKOMBINANT ERΑ I FULD LÆNGDE
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER (JF. OGSÅ GENEREL INDLEDNING)
|
1.
|
Dette in vitro-assay vedrørende østrogenreceptormætning (ERα) og kompetitiv binding anvender human receptor ERα(hrERα) i fuld længde, som produceres i og isoleres fra baculovirusinficerede insektceller. Protokollen, som blev udviklet af Freyberger og Wilson, gennemgik en international valideringsundersøgelse i flere laboratorier (2), der har påvist dens relevans og pålidelighed til assayets tilsigtede formål.
|
|
2.
|
Dette assay er en screeningsprocedure til påvisning af stoffer, som kan binde sig til hrERα i fuld længde. Det benyttes til at bestemme et testkemikalies evne til at konkurrere med 17β-østradiol om binding til hrERα. Kvantitative assayresultater kan omfatte IC50 (et mål for den koncentration af testkemikaliet, der er nødvendig for at fortrænge halvdelen af [3H]-17β-østradiol fra hrERα), og testkemikaliernes relative affiniteter for hrERα sammenlignet med 17β-østradiol. Til kemikaliescreeningsformål kan acceptable kvalitative assayresultater omfatte klassificeringer af testkemikalier som enten hrERα-bindere, ikke-bindere eller usikre baseret på kriterier beskrevet for bindingskurver.
|
|
3.
|
Assayet anvender en radioaktivt mærket ligand, som kræver, at laboratoriet har licens til radioaktive materialer. Alle procedurer med radioisotoper og farlige kemikalier skal følge forskrifter og procedurer som beskrevet i national lovgivning.
|
|
4.
|
Afsnittene "GENEREL INDLEDNING" og "hrER-BINDINGSASSAYETS KOMPONENTER" skal læses, før dette assay benyttes til lovmæssige formål. Definitioner og forkortelser, der benyttes i denne testvejledning, er beskrevet i tillæg 1.
|
PRINCIPPER FOR ASSAYET (JF. OGSÅ GENEREL INDLEDNING)
|
5.
|
hrERα-bindingsassayet måler en radioaktivt mærket ligands ([3H]17β-østradiol) evne til at binde sig til ER under tilstedeværelse af stigende koncentrationer af et testkemikalie (dvs. konkurrent). Testkemikalier, som har en høj ER-affinitet, konkurrerer med den radioaktivt mærkede ligand ved en lavere koncentration sammenlignet med kemikalier med lavere affinitet for receptoren.
|
|
6.
|
Dette assay består af to vigtige komponenter: et mætningsbindingsforsøg for at karakterisere parametrene for interaktion mellem receptor og ligand efterfulgt af et forsøg med kompetitiv binding, der karakteriserer konkurrencen mellem et testkemikalie og en radioaktivt mærket ligand for binding til ER.
|
|
7.
|
Formålet med mætningsbindingsforsøget er at karakterisere en særlig batch af receptorer for affinitet og antal i forberedelsen af det kompetitive bindingsforsøg. Mætningsbindingsforsøget måler, under ligevægtsbetingelser, en fast østrogenreceptorkoncentrations affinitet for dens naturlige ligand (repræsenteret ved dissociationskonstanten, Kd) og koncentrationen af aktive receptorsteder (Bmax).
|
|
8.
|
Det kompetitive bindingsforsøg måler et stofs affinitet til at konkurrere med [3H]17β-østradiol om binding til ER. Affiniteten kvantificeres af koncentrationen af testkemikaliet, som under ligevægtsbetingelser, inhiberer 50 % af den specifikke binding af [3H]17β-østradiol (kaldet den "inhiberende koncentration på 50 %" eller IC50). Denne kan også vurderes ved hjælp af den relative affinitet (RBA, i forhold til IC50 for østradiol målt separat i den samme analyseserie). Det kompetitive bindingsforsøg måler bindingen af [3H]17β-østradiol ved en fast koncentration under tilstedeværelse af en bred vifte (otte størrelsesordener) af testkemikaliekoncentrationer. Dataene tilpasses herefter, hvor det er muligt, så de danner Hill-ligningen (Hill, 1910), der beskriver en kompetitiv binders fortrængning af den radioaktive ligand ét sted. Omfanget af fortrængningen af det radioaktivt mærkede østradiol ved ligevægt anvendes til at karakterisere testkemikaliet som en binder, ikke-binder eller som et kemikalie, der genererer en usikker respons.
|
FREMGANGSMÅDE
Påvisning af acceptabel ydeevne for hrERα-protein
|
9.
|
Før der rutinemæssigt udføres mætningsassays og kompetitive bindingsassays, skal det påvises, at hver ny batch hrERα fungerer korrekt i det laboratorium, hvor det vil blive anvendt. Ydeevnen påvises gennem en totrinsproces. Det drejer sig om følgende trin:
|
—
|
Der udføres et [3H]-17β-østradiol-bindingsassay for at påvise hrERα-specificitet og mætning. Ikke-lineær regressionsanalyse af disse data (f.eks. BioSoft, McPherson, 1985; Motulsky, 1995) og det efterfølgende Scatchard-plot bør dokumentere hrERα-affiniteten for [3H]-17β-østradiol (Kd) og antallet af receptorer (Bmax) for hver batch hrERα.
|
|
—
|
Der udføres et kompetitivt bindingsassay ved anvendelse af kontrolstofferne (referenceøstrogen (17β-østradiol)), en svag binder (f.eks. norethynodrel eller norethindron) og en ikke-binder (octyltriethoxysilan, OTES). Hvert laboratorium bør oprette en historisk database for at dokumentere konsistensen af IC50 og andre relevante værdier for referenceøstrogenet og den svage binder blandt forsøg og forskellige batcher af hrERα. Parametrene for de kompetitive bindingskurver for kontrolstofferne bør ligge inden for de grænser for konfidensintervallet på 95 % (jf. tabel 1), der blev udviklet ved hjælp af data fra laboratorier, som deltog i valideringsundersøgelsen for dette assay (2).
|
|
Tabel 1
Ydeevnekriterier udviklet for referenceøstrogen og svage bindere, FW-hrER bindingsassay
|
Stof
|
Parameter
|
Gennemsnit (7)
|
Standardafvigelse (n)
|
95 %-konfidensintervaller (8)
|
|
Nedre grænse
|
Øvre grænse
|
|
17β-østradiol
|
Top (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Bund (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Hills kurve
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
LogIC50 (M)
|
-8,92 (9)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Norethynodrel
|
Top (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Bund (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Hills kurve
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
Log IC50(M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Norethindron (9)
|
Top (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Bund (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Hills kurve
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
LogIC50(M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Påvisning af laboratoriets kompetence
|
10.
|
Jf. punkt 17 og 18 og tabel 2 i afsnittet "hrER-BINDINGSASSAYETS KOMPONENTER" i denne forsøgsmetode. Hvert assay (mætning og kompetitiv binding) bør bestå af tre uafhængige analyseserier (dvs. friske fortyndinger af receptor, kemikalier og reagenser) på forskellige dage, og hver analyseserie bør indeholde tre replikater.
|
Bestemmelse af receptor(hrERα)koncentration
|
11.
|
Den aktive receptorkoncentration varierer lidt efter batch og opbevaringsforhold. Af denne grund bør den koncentration af aktiv receptor, der modtages fra leverandøren, bestemmes. Dette vil give den rette koncentration af aktiv receptor på tidspunktet for analyseserien.
|
|
12.
|
Under forhold svarende til kompetitiv binding (dvs. 1 nM [3H]-østradiol) bør nominelle koncentrationer på 0,25, 0,5, 0,75 og 1 nM receptor inkuberes i fravær af (total binding) og under tilstedeværelse (ikke-specifik binding) af 1 μM umærket østradiol. Specifik binding beregnet som forskellen mellem total og ikke-specifik binding afsættes i forhold til den nominelle receptorkoncentration. Den receptorkoncentration, som giver specifikke bindingsværdier svarende til 20 % af de tilsatte radioaktivt mærkede stoffer, hænger sammen med den tilsvarende nominelle receptorkoncentration, og denne receptorkoncentration anvendes til mætningsforsøg og kompetitive bindingsforsøg. En slutkoncentration for hrER på 0,5 nM vil ofte opfylde denne betingelse.
|
|
13.
|
Hvis 20 %-kriteriet gentagne gange ikke kan opfyldes, kontrolleres forsøgs-setuppet for potentielle fejl. Hvis 20 %-kriteriet ikke opfyldes, kan det tyde på, at der er meget lidt aktiv receptor i rekombinantbatchen, og det bør overvejes at anvende en anden receptorbatch.
|
Mætningsassay
|
14.
|
Otte stigende koncentrationer af [3H]17β-østradiol vurderes in triplo under følgende tre betingelser (jf. tabel 2):
|
—
|
i fravær af umærket 17β-østradiol og under tilstedeværelse af ER Dette er bestemmelsen af den totale binding ved måling af radioaktiviteten i de brønde, som kun har [3H]17β-østradiol.
|
|
—
|
under tilstedeværelse af en 1000-fold uforholdsmæssig koncentration af umærket 17β-østradiol i forhold til mærket 17β-østradiol og tilstedeværelse af ER. Formålet med denne betingelse er at mætte de aktive bindingssteder med umærket 17β-østradiol og, ved at måle radioaktiviteten i brøndene, bestemme den ikke-specifikke binding. Eventuelt tilbageværende varmt østradiol, som kan binde sig til receptoren, anses for at kunne binde sig til et ikke-specifikt sted, da koldt østradiol bør have en så høj koncentration, at det binder sig til alle tilgængelige specifikke steder på receptoren
|
|
—
|
i fravær af umærket 17β-østradiol og af ER (bestemmelse af samlet radioaktivitet).
|
|
Fremstilling af [3H]-17β-østradiol og umærkede 17β-østradiol-opløsninger
|
15.
|
Fortyndinger af [3H]-17β-østradiol fremstilles ved tilsætning af assaybuffer til en 12 nM stamopløsning af [3H]-17β-østradiol for at opnå koncentrationer, der indledningsvis ligger fra 0,12 nM til 12 nM. Ved at tilsætte 40 μl af disse opløsninger til de respektive assaybrønde på en mikrotiterplade med 96 brønde (i en endelig mængde på 160 μl) fås den endelige assaykoncentration fra 0,03 til 3,0 nM. Fremstilling af assaybuffer, [3H]-17β-østradiolstamopløsning og fortyndinger samt bestemmelse af koncentrationerne er beskrevet i detaljer i FW-protokollen (2).
|
|
16.
|
Fortyndinger af 17β-østradiol-opløsninger i ethanol fremstilles ved at tilsætte assaybuffer for at få otte stigende koncentrationer, der indledningsvis ligger fra 0,06 μM til 6 μM. Ved at tilsætte 80 μl af disse opløsninger til de respektive assaybrønde på en mikrotiterplade med 96 brønde (i en endelig mængde på 160 μl) fremkommer de endelige assaykoncentrationer, der ligger fra 0,03 μM til 3 μM. Den endelige koncentration af umærket 17β-østradiol i brøndene i det enkelte ikke-specifikke bindingsassay bør være 1000 fold den mærkede [3H]-17β-østradiolkoncentration. Fremstilling af fortyndinger af umærket 17β-østradiol er beskrevet i detaljer i FW-protokollen (2).
|
|
17.
|
Den nominelle receptorkoncentration, som giver en specifik binding på 20 ± 5 %, bør anvendes (jf. punkt 12-13). hrERα-opløsningen fremstilles umiddelbart inden anvendelsen.
|
|
18.
|
Mikrotiterpladerne med 96 brønde forberedes som illustreret i tabel 2 med 3 replikater pr. koncentration. Et eksempel på pladekoncentration og volumenfordeling af [3H]-17β-østradiol, umærket 17β-østradiol, buffer og receptor er anført i tillæg 2.2.
|
Tabel 2
Layout for en mikrotiterplade til mætningsbindingsassay
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Total binding (opløsningsmiddel)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E
2
+ ER
+ 0,10 μM E
2
|
Ikke-specifik binding
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E
2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
[3H] E2: [3H]-17β-østradiol
ER: østrogenreceptor
E2: Eumærket 17β-østradiol
|
|
19.
|
Assaymikrotiterplader inkuberes ved 2-8 °C i 16-20 timer og anbringes på en rotator i inkubationsperioden.
|
Måling af [3H]-17β-østradiol bundet til hrERα
|
20.
|
[3H]-17β-østradiol bundet til hrERα separeres fra fri [3H]-17β-østradiol ved at tilsætte 80 μl kold DCC-suspension til hver brønd, ryste mikrotiterpladerne i 10 minutter og centrifugere i 10 minutter ved omkring 2500 o/min. For at minimere dissociation af bundet [3H]-17β-østradiol fra hrERα under denne proces er det særdeles vigtigt, at buffere og assaybrønde holdes mellem 2 og 8 °C, og at hvert skridt udføres hurtigt. En rysteanordning til mikrotiterplader er nødvendig for at behandle plader effektivt og hurtigt.
|
|
21.
|
50 μl supernatant, som indeholder hrERα-bundet [3H]-17β-østradiol, bør herefter tages ekstremt forsigtigt for at undgå enhver kontaminering af brøndene ved kontakt med DCC og anbringes på en anden mikrotiterplade.
|
|
22.
|
200 μl scintillationsvæske, der kan omdanne kinetisk energi fra nukleare emissioner til lysenergi, tilsættes herefter til hver brønd (A1-B12 og D1 til E12). Brønd G1-H12 (identificeret som dpms i alt) udgør seriefortyndinger af [3H]-17β-østradiol (40 μl), som bør hældes direkte i scintillationsvæsken i brøndene på målepladen som anført i tabel 3, dvs. at disse brønde kun indeholder 200 μl af scintillationsvæsken og den rette fortynding af [3H]-17β-østradiol. Disse mål viser, hvor meget [3H]-17β-østradiol i dpms der blev tilsat til hvert sæt brønde til den totale binding og ikke-specifikke binding.
|
Tabel 3
Layout for en mikrotiterplade til mætningsbindingsassay, måling af radioaktivitet
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3 H] E2 + ER
|
Total binding (opløsningsmiddel)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E
2
+ ER
+ 0,10 μM E
2
|
Ikke-specifik binding
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E
2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H] E
2 (dpms i alt)
|
0,06 nM [3H] E
2
|
0,08 nM [3H] E
2
|
0,10 nM [3H] E
2
|
dpms i alt (*7)
|
|
H
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2: [[3H]-17β-østradiol
ER: østrogenreceptor
E2: umærket 17β-østradiol
dpms: disintegrationer pr. minut
|
|
23.
|
Målingen indledes efter mindst 2 timer, og tælletiden bør være 40 minutter pr. brønd. Der bør anvendes en scintillationstæller til en mikrotiterplade til bestemmelse af dpm/brønd med korrektion for quenching. Er der ikke adgang til en scintillationstæller til mikrotiterplader, kan prøverne måles i en konventionel tæller. Under disse forhold kan det overvejes at nedsætte tælletiden.
|
Kompetitivt bindingsassay
|
24.
|
Det kompetitive bindingsassay måler bindingen af en enkelt koncentration af [3H]-17β-østradiol under tilstedeværelse af stigende koncentrationer af et testkemikalie. Der bør anvendes tre sideløbende replikater ved hver koncentration inden for én analyseserie. Endvidere bør der udføres tre ikke-sideløbende analyseserier for hvert kemikalie, der testes. Assayet bør opsættes med en eller flere mikrotiterplader med 96 brønde.
|
Kontroller
|
25.
|
Ved udførelsen af assayet medtages samtidig opløsningsmiddel og kontroller (dvs. referenceøstrogen, svag binder og ikke-binder) i hvert forsøg. Fuldstændige koncentrationskurver for referenceøstrogen og kontroller (dvs. svag binder og ikke-binder) anvendes på én plade under hver analyseserie. Alle øvrige plader bør hver indeholde i) en høj- (maksimal fortrængning) og middel- (ca. IC50) koncentration af E2 og svag binder in triplo, ii) kontrolkultur med opløsningsmiddel og ikke-specifik binding, hver især mindst in triplo. Procedurer for fremstilling af assaybuffer, kontroller, [3H]-17β-østradiol, hrERα og testkemikalieopløsninger er beskrevet i reference 2 (bilag K, jf. FW-assayprotokol).
|
Kontrolkultur med opløsningsmiddel
|
26.
|
Kontrolkulturen med opløsningsmiddel viser, at opløsningsmidlet ikke interagerer med testsystemet og måler også total binding (TB). Ethanol er det foretrukne opløsningsmiddel. Såfremt den højeste koncentration af testkemikaliet ikke er opløseligt i ethanol, kan DMSO anvendes. Koncentrationen af ethanol eller DMSO, hvis det anvendes, i de endelige assaybrønde er 1,5 % og må ikke overstige 2 %.
|
Bufferkontrol
|
27.
|
Bufferkontrollen (BC) bør hverken indeholde opløsningsmiddel eller testkemikalie, men alle assayets øvrige komponenter. Resultaterne af bufferkontrollen sammenlignes med opløsningsmiddelkontrollen for at kontrollere, at det anvendte opløsningsmiddel ikke påvirker assaysystemet.
|
Stærk binder (referenceøstrogen)
|
28.
|
17β-østradiol (CAS 50-28-2) er den endogene ligand og binder sig med høj affinitet til ER, undertype alfa. En standardkurve, som anvender umærket 17β-østradiol, fremstilles til hvert hrERα kompetitivt bindingsassay, så der kan foretages en variabilitetsvurdering ved gennemførelse af assayet over længere tid i det samme laboratorium. Otte opløsninger af umærket 17β-østradiol fremstilles i ethanol med koncentrationer i assaybrønde i området 100 nM-10 pM (-7[logM] til -11[logM]), med følgende intervaller: (-7[logM], -8[logM], -8,5[logM], -9[logM], - 9,5[logM], -10[logM], -11[logM]). Den højeste koncentration af umærket 17β-østradiol (1 μM) tjener også som ikke-specifik bindingsindikator. Denne koncentration er angivet ved mærket "NSB" i tabel 4, selv om den også indgår i standardkurven.
|
Svag binder
|
29.
|
En svag binder (norethynodrel (CAS 68-23-5) eller norethindron (CAS 68-22-4)) bør medtages for at påvise hvert forsøgs følsomhed og for at give mulighed for en vurdering af variabiliteten, når assayet udføres over længere tid. Otte opløsninger af den svage binder fremstilles i ethanol med koncentrationer i assaybrøndene i området fra 3 nM til 30 μM (-8,5[logM] til -4,5[logM]) med følgende intervaller: -4,5[logM], -5[logM], -5,5[logM], -6[logM], -6,5[logM], -7[logM], -7,5[logM], -8,5[logM].
|
Ikke-binder
|
30.
|
Octyltriethoxysilan (OTES, CAS 2943-75-1) bør anvendes som den negative kontrol (ikke-binder). Det giver sikkerhed for, at det med det udførte assay kan påvises, når testkemikalierne ikke binder sig til hrERα. Otte opløsninger af ikke-binderen fremstilles i ethanol med koncentrationer i assaybrøndene i området fra 0,1 nM til 1000 μM (-10[logM] til -3[logM]) i log-stigninger. Di-n-butylphthalat (DBP) kan bruges som en alternativ ikke-binder som kontrol. Det er påvist, at dens maksimale opløselighed er -4[logM].
|
hrERα-koncentration
|
31.
|
Den mængde receptor, som giver en specifik binding på 20 ± 5 % af 1 nM radioaktivt mærket ligand (jf. punkt 12-13 i tillæg 2), bør anvendes. hrERα-opløsningen fremstilles umiddelbart inden anvendelsen.
|
[3H]-17β-østradiol
|
32.
|
Koncentrationen af [3H]-17β-østradiol i assaybrøndene skal være 1,0 nM.
|
Testkemikalier
|
33.
|
I første omgang er det nødvendigt at foretage en opløselighedstest for at fastslå opløselighedsgrænsen for hvert testkemikalie og for at afdække det passende koncentrationsområde, der skal anvendes, når testprotokollen udføres. Opløselighedsgrænsen for hvert testkemikalie skal indledningsvis bestemmes i opløsningsmidlet og yderligere bekræftes under assaybetingelser. Den endelige koncentration, som testes i assayet, bør ikke overstige 1 mM. Områdebestemmelsen består af en kontrolkultur med opløsningsmiddel sammen med otte log-seriefortyndinger, der begynder ved den maksimalt acceptable koncentration (f.eks. 1 mM eller lavere, baseret på opløselighedsgrænsen), og idet der noteres tilstedeværelse af uklarhed eller bundfald (jf. også punkt 35). Testkemikaliet testes ved anvendelse af otte logaritmiske kurver beregnet efter koncentrationsintervaller som defineret ved den foregående test til bestemmelse af dosisinterval. Koncentrationer i det andet og tredje forsøg bør tilpasses, hvis det er relevant, for bedre at karakterisere koncentrationsresponskurven.
|
|
34.
|
Fortyndinger af testkemikaliet fremstilles i et passende opløsningsmiddel (jf. punkt 26 i tillæg 2). Hvis den højeste koncentration af testkemikaliet ikke kan opløses i hverken ethanol eller DMSO, og tilsætning af mere opløsningsmiddel ville medføre, at opløsningsmiddelkoncentrationen i det endelige rør blev større end den acceptable grænse, kan den højeste koncentration reduceres til den næste lavere koncentration. I så fald kan en supplerende koncentration tilsættes i den lave ende af koncentrationsserien. Andre koncentrationer i serien bør forblive uændrede.
|
|
35.
|
Testkemikalieopløsningerne bør overvåges nøje, når de tilsættes til assaybrønden, da testkemikaliet kan danne bundfald ved tilsætning til assaybrønden. Data for alle brønde, der indeholder bundfald, bør udelukkes fra kurvetilpasningen, og årsagen til udelukkelsen af data noteres.
|
|
36.
|
Hvis der allerede findes oplysninger fra andre kilder, som angiver en log (IC50) for et testkemikalie, kan det være hensigtsmæssigt at sætte en geometrisk afstand mellem fortyndingerne (dvs. 0,5 logenheder omkring den forventede log(IC50)). Det endelige resultat bør afspejle en tilstrækkelig spredning af koncentrationer på begge sider af log(IC50), herunder "top" og "bund", således at bindingskurven kan karakteriseres tilstrækkeligt.
|
Organisering af assaypladen
|
37.
|
Mærkede mikrotiterplader bør forberedes under hensyntagen til seksdobbelte inkubationer med koder for kontrolkultur med opløsningsmiddel, den højeste koncentration af referenceøstrogenet, der også fungerer som indikator for ikke-specifik binding (NSB) og bufferkontrol, og under hensyntagen til tredobbelte inkubationer med koder for hver af de otte koncentrationer af den ikke-bindende kontrol (octyltriethoxysilan), de syv lavere koncentrationer for referenceøstrogenet, de otte koncentrationsdosisniveauer for den svage binder og de otte koncentrationer af hvert testkemikalie. Et eksempel på layout for pladediagrammet til de fulde koncentrationskurver for referenceøstrogen og kontrol er anført nedenfor i tabel 4. Supplerende mikrotiterplader bruges til testkemikalier og bør omfatte pladekontroller, dvs.: 1) en høj- (maksimal fortrængning) og middel- (ca. IC50) koncentration af E2 og svag binder in triplo, 2) kontrolkultur med opløsningsmiddel og ikke-specifik binding, hver især seksdobbelt (tabel 5). Et eksempel på arbejdsark til layout for en mikrotiterplade til et kompetitivt assay, der anvender tre ukendte testkemikalier, er anført i tillæg 2.3. De koncentrationer, som er angivet i tabel 4 og 5, er assayets endelige koncentrationer. Den maksimale koncentration for E2 bør være 1×10-7 M, og for den svage binder bør den højeste anvendte koncentration for den svage binder på plade 1 anvendes. IC50-koncentrationen skal bestemmes af laboratoriet på grundlag af deres historiske kontroldatabase. Det forventes, at denne værdi vil ligne den, der blev observeret i valideringsundersøgelserne (jf. tabel 1).
|
Tabel 4
Layout for mikrotiterpladen til et kompetitivt bindingsassay, fulde koncentrationskurver for referenceøstrogen og kontroller (plade 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (kun opløsningsmiddel)
|
TB (kun opløsningsmiddel)
|
NSB
|
NSB
|
|
B
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
C
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Blindprøve (*8)
|
|
D
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
E
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
F
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
H
|
Blindprøve (til varm) (*9)
|
Blindprøve (til varm) (*9)
|
Bufferkontrol
|
Bufferkontrol
|
|
I dette eksempel er den svage binder norethinodrel (NE)
|
Tabel 5
Layout til mikrotiterpladen til et kompetitivt bindingsassay, fulde koncentrationskurver til testkemikalier og pladekontroller
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (kun opløsningsmiddel)
|
TB (kun opløsningsmiddel)
|
NSB
|
NSB
|
|
B
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
C
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
D
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
E
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
F
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
H
|
NE (IC50)
|
NE (1×10-4,5)
|
E2 (IC50)
|
E2 (1×10-7)
|
|
I dette eksempel er den svage binder norethinodrel (NE)
|
Gennemførelse af kompetitivt bindingsassay
|
38.
|
Som vist i tabel 6 tilsættes 80 μl kontrolkultur med opløsningsmiddel, bufferkontrol, referenceøstrogen, svag binder, ikke-binder og testkemikalier fremstillet i assaybuffer til brøndene. Herefter tilsættes 40 μl af en 4 nM [3H]-17β-østradiolopløsning til hver enkelt brønd. Efter en let rotation i 10-15 minutter ved 2-8 °C tilsættes 40 μl hrERα-opløsning. Assaymikrotiterpladerne inkuberes ved 2-8 °C i 16-20 timer og anbringes på en rotator i inkubationsperioden.
|
Tabel 6
Mængde af assaykomponenter til kompetitivt bindingsassay med hrER, mikrotiterplader
|
Mængde (μl)
|
Indholdsstof
|
|
80
|
Umærket 17β-østradiol, norethynodrel, OTES, testkemikalier, opløsningsmiddel eller buffer
|
|
40
|
4 nM [3H]-17β-østradiolopløsning
|
|
40
|
hrERα-opløsning, koncentration som bestemt
|
|
160
|
Samlet mængde i hver assaybrønd
|
|
39.
|
Kvantificeringen af [3H]-17β-østradiol bundet til hrERα efter separation af [3H]-17β-østradiol bundet til hrERα fra fri [3H]-17β-østradiol ved at tilsætte 80 μl kold DCC-suspension til hver brønd bør herefter udføres som beskrevet i punkt 20-23 for mætningsbindingsassayet.
|
|
40.
|
Brønd H1-6 (identificeret som blindprøve (til varm) i tabel 4) repræsenterer dpms i [3H]-mærket østradiol i 40 μl. Delprøven på 40 μl hældes direkte i scintillationsvæsken i brønd H1-H6.
|
Acceptkriterier
Mætningsbindingsassay
|
41.
|
Den specifikke bindingskurve bør nå et plateau i takt med anvendelsen af stigende koncentrationer af [3H]-17β-østradiol, hvilket viser mætningen af hrERα med ligand.
|
|
42.
|
Den specifikke binding ved 1 nM [3H]-17β-østradiol bør ligge inden for det acceptable område på 15-25 % af den gennemsnitlige målte samlede radioaktivitet, der tilsættes på tværs af analyseserier. Lejlighedsvise lette udsving uden for dette område er acceptable, men hvis analyseserier konsekvent ligger uden for dette område, eller en særlig analyseserie ligger væsentligt uden for dette område, bør proteinkoncentrationen justeres, og mætningsassayet gentages.
|
|
43.
|
Dataene bør generere et lineært Scatchard-plot.
|
|
44.
|
Den ikke-specifikke binding bør ikke være uforholdsmæssigt stor. Værdien for den ikke-specifikke binding bør typisk være < 35 % af den totale binding. Forholdet kan dog undertiden overstige denne grænse ved måling af en meget lav dpm for den laveste testede koncentration af radioaktivt mærket 17β-østradiol.
|
Kompetitivt bindingsassay
|
45.
|
Stigende koncentrationer af umærket 17β-østradiol bør fortrænge [3H]-17β-østradiol fra receptoren på en måde, der stemmer overens med en kompetitiv binding ét sted.
|
|
46.
|
IC50-værdien for referenceøstrogenet (dvs. 17β-østradiol) bør omtrent svare til den molære koncentration af [3H]-17β-østradiol plus den Kd, der er bestemt ud fra mætningsbindingsassayet.
|
|
47.
|
Den samlede specifikke binding bør til stadighed ligge inden for det acceptable område på 20 ± 5 %, når den gennemsnitlige målte koncentration af den samlede radioaktivitet, som tilsættes til hver brønd, var 1 nM på tværs af analyseserier. Lejlighedsvise lette udsving uden for dette område er acceptable, men hvis analyseserier konsekvent ligger uden for dette område, eller en særlig analyseserie ligger væsentligt uden for området, bør proteinkoncentrationen justeres.
|
|
48.
|
Opløsningsmidlet bør ikke ændre assayets følsomhed eller reproducerbarhed. Resultaterne af kontrolkulturen med opløsningsmiddel (TB-brønde) sammenlignes med bufferkontrollen for at kontrollere, at det anvendte opløsningsmiddel ikke påvirker assaysystemet. Resultaterne af TB og bufferkontrol bør være sammenlignelige, hvis opløsningsmidlet ikke påvirker assayet.
|
|
49.
|
Ikke-binderen bør ikke fortrænge mere end 25 % af [3H]-17β-østradiol fra hrERα ved test op til 10-3 M (OTES) eller 10-4 M (DBP).
|
|
50.
|
Der blev udviklet ydeevnekriterier for referenceøstrogen og to svage bindere (f.eks. norethynodrel, norethindron) ved hjælp af data fra valideringsundersøgelsen af FW hrER-bindingsassayet (bilag N i reference 2). Der er anført 95 %-konfidensintervaller for den gennemsnitlige (n) +/- standardafvigelse for alle kontrolanalyseserier på tværs af de laboratorier, der deltog i valideringsundersøgelsen. Der blev beregnet 95 %-konfidensintervaller for de kurvetilpassede parametre (dvs. top, bund, Hills kurve, logIC50) for referenceøstrogen og svage bindere og for log10RBA af de svage bindere i forhold til referenceøstrogen, og de angives som ydeevnekriterier for positive kontroller. Tabel 1 viser forventede intervaller for kurvetilpassede parametre, der kan benyttes som ydeevnekriterier. I praksis kan området for IC50 variere lidt baseret på Kd af receptorpræparatet og ligandkoncentrationen.
|
|
51.
|
Der blev ikke udviklet ydeevnekriterier for kurvetilpassede parametre til testkemikalier på grund af den brede vifte af eksisterende potentielle testkemikalier og variation i potentielle affiniteter og udfald (f.eks. fuld kurve, delkurve, ingen kurvetilpasning). Der bør dog anlægges en faglig vurdering ved gennemgang af resultater fra hver analyseserie for et testkemikalie. Der skal benyttes et tilstrækkeligt koncentrationsområde for testkemikaliet for klart at definere toppen (f.eks. 90-100 % af bindingen) af den kompetitive kurve. Variabiliteten mellem replikater ved hver koncentration af testkemikaliet samt mellem de tre ikke-sideløbende analyseserier skal være rimelig og videnskabeligt forsvarlig. Kontroller fra hver analyseserie for et testkemikalie skal tilnærmes resultatmålinger rapporteret for dette FW-assay og skal stemme overens med tidligere kontroldata fra de respektive laboratorier.
|
ANALYSE AF DATA
Mætningsbindingsassay
|
52.
|
Der måles både samlet og ikke-specifik binding. På grundlag af disse værdier beregnes specifik binding af stigende koncentrationer af [3H]-17β-østradiol under ligevægtsbetingelser ved fratrækning af ikke-specifik binding fra samlet binding. En graf over specifik binding i forhold til [3H]-17β-østradiolkoncentration bør nå et plateau for maksimal specifik binding som tegn på mætning af hrERα med [3H]-17β-østradiol. Endvidere bør analyse af data dokumentere bindingen af [3H]-17β-østradiol til et enkelt bindingssted med høj affinitet. Ikke-specifik, samlet og specifik binding bør vises på en mætningsbindingskurve. Ved yderligere analyse af disse data bør der benyttes en ikke-lineær regressionsanalyse (f.eks. BioSoft, McPherson, 1985; Motulsky, 1995) med en endelig visning af data som et Scatchard-plot.
|
|
53.
|
Dataanalysen skal bestemme Bmax og Kd på grundlag af de totale bindingsdata alene ud fra den antagelse, at ikke-specifik binding er lineær, medmindre der gives en begrundelse for at benytte en anden metode. Der bør desuden benyttes en robust regression ved bestemmelse af den bedste tilpasning, medmindre der gives en begrundelse. Den valgte metode til robust regression skal anføres. Der bør altid korrigeres for liganddepletering (f.eks. ved hjælp af Swillens metode, 1995) ved bestemmelse af Bmax og Kd ud fra mætningsbindingsdata.
|
Kompetitivt bindingsassay
|
54.
|
Den kompetitive bindingskurve plottes som specifik [3H]-17β-østradiolbinding mod konkurrentens koncentration (log10-enheder). Den koncentration af testkemikaliet, der inhiberer 50 % af den maksimale specifikke [3H]-17β-østradiolbinding, er IC50-værdien.
|
|
55.
|
Skøn over log(IC50)-værdier for de positive kontroller (f.eks. referenceøstrogen og svag binder) bestemmes ved anvendelse af en passende ikke-lineær kurvetilpasningssoftware, som passer til en Hill-ligning med fire parametre (f.eks. BioSoft, McPherson, 1985; Motulsky, 1995). Top, bund, hældning og log(IC50) skal normalt være frie ved tilpasning af disse kurver. Der bør benyttes robust regression ved bestemmelse af den bedste tilpasning, medmindre der gives en begrundelse. Der bør ikke korrigeres for liganddepletion. Efter den indledende analyse skal hver bindingskurve revideres for at sikre, at den passer til modellen. Den relative affinitet (RBA) for den svage binder beregnes som en procentdel af log (IC50) for den svage binder i forhold til log (IC50) for 17β-østradiol. Resultaterne fra de positive kontroller og ikke-binderkontrollen bør vurderes ved hjælp af målinger af assayets ydeevne i punkt 45-50 i dette tillæg 2.
|
|
56.
|
Data for alle testkemikalier analyseres gennem en trinvis tilgang for at sikre, at dataene analyseres indgående, og at hver kompetitiv bindingskurve klassificeres korrekt. Det anbefales, at hver analyseserie for et testkemikalie indledningsvis gennemgår en standardiseret dataanalyse, som er identisk med den, der benyttes for referenceøstrogen og kontroller for svag binder (jf. punkt 55 ovenfor). Når analysen er færdig, foretages en teknisk gennemgang af kurvetilpasningsparametrene samt en visuel gennemgang af, hvor godt dataene passer til den genererede kompetitive bindingskurve for hver analyseserie. Under denne tekniske gennemgang er observationer af et koncentrationsafhængigt fald i den procentvise specifikt bundne [3H]-17β-østradiol, lave variabilitet mellem de tekniske replikater ved hver kemisk koncentration og konsistens i tilpassede parametre mellem de tre analyseserier et godt tegn på, at assayet og dataanalyserne blev gennemført korrekt.
|
Fortolkning af data
|
57.
|
Forudsat at alle acceptkriterier er opfyldt, anses et testkemikalie for at være en binder for hrERα, hvis der kan tilpasses en bindingskurve, og det laveste punkt på responskurven inden for dataområdet er under 50 % (figur 1).
|
|
58.
|
Såfremt alle acceptkriterier er opfyldt, anses et testkemikalie for at være en ikke-binder for hrERα, hvis:
|
—
|
der kan tilpasses en bindingskurve, og det laveste punkt på den tilpassede responskurve inden for dataområdet ligger over 75 %, eller
|
|
—
|
der ikke kan tilpasses en bindingskurve, og den laveste ikke-udjævnede, gennemsnitlige, procentvise binding blandt koncentrationsgrupperne i dataene ligger over 75 %.
|
|
|
59.
|
Testkemikalierne anses for usikre, hvis ingen af de ovennævnte betingelser er opfyldt (f.eks. det laveste punkt på den tilpassede responskurve ligger mellem 76 og 51 %).
|
Tabel 7
Kriterier for klassificering på grundlag af et testkemikalies kompetitive bindingskurve.
|
Klassificering
|
Kriterier
|
|
Bindera
|
En bindingskurve kan tilpasses.
Det laveste punkt på responskurven inden for dataområdet er mindre end 50 %.
|
|
Ikke-binderb
|
Hvis en bindingskurve kan tilpasses,
er det laveste punkt på den tilpassede responskurve inden for dataområdet over 75 %.
Hvis en bindingskurve ikke kan tilpasses,
er den laveste ikke-udjævnede, gennemsnitlige, procentvise binding blandt koncentrationsgrupperne i dataene over 75 %.
|
|
Usikkerc
|
Enhver testbar analyseserie, som hverken er en binder eller en ikke-binder s
(f.eks. Det laveste punkt på den tilpassede responskurve er mellem 76 % og 51 %).
|
Figur 1
Eksempler på klassificering af testkemikalier ved anvendelse af kompetitiv bindingskurve
|
60.
|
Flere analyseserier for et testkemikalie, der gennemføres i et laboratorium, kombineres ved at tildele hver analyseserie en numerisk værdi og beregne gennemsnittet på tværs af analyseserierne som vist i tabel 8. Resultater for de kombinerede analyseserier i hvert laboratorium sammenlignes med den forventede klassificering for hvert testkemikalie.
|
Tabel 8
Metode til klassificering af testkemikalier ved anvendelse af flere analyseserier i et laboratorium
|
Tildeling af værdi til hver analyseserie:
|
|
|
Klassificering
|
Numerisk værdi
|
|
Binder
|
2
|
|
Usikker
|
1
|
|
Ikke-binder
|
0
|
|
Klassificering af gennemsnitlig numerisk værdi på tværs af analyseserier:
|
|
|
Klassificering
|
Numerisk værdi
|
|
Binder
|
Gennemsnit ≥ 1,5
|
|
Usikker
|
0,5 ≤ Gennemsnit < 1,5
|
|
Ikke-binder
|
Gennemsnit < 0,5
|
TESTRAPPORT
|
61.
|
Jf. punkt 24 i "hrER-BINDINGSASSAYETS KOMPONENTER" i denne forsøgsmetode.
|
Tillæg 2.1
TERMINOLOGI
[3H]E2
: 17β-østradiol radioaktivt mærket med tritium
DCC
: dextranovertrukket trækul
E2
: umærket 17β-østradiol (inert)
Assaybuffer
: 10 mM tris, 10 mg bovint serumalbumin/ml, 2 mM DTT, 10 % glycerol, 0,2 mM leupeptin, pH 7,5
hrERα
: human rekombinant østrogenreceptor alfa
Replikat
: en af flere brønde, der har samme indhold ved de samme koncentrationer, og som analyseres sideløbende inden for én analyseserie. I denne protokol testes hver koncentration af testkemikaliet in triplo, dvs. at der er tre replikater, som analyseres samtidig ved hver koncentration af testkemikaliet.
Analyseserie
: et fuldstændigt sæt assaybrønde på mikrotiterplade, der kører sideløbende, og som giver alle nødvendige oplysninger til at karakterisere bindingen af et testkemikalie til hrERα (nemlig samlet [3H]-17β-østradiol tilsat til assaybrønden, maksimal binding af [3H]-17β-østradiol til hrERα, ikke-specifik binding og total binding ved forskellige koncentrationer af testkemikaliet). En analyseserie kan bestå af så lidt som én assaybrønd (dvs. replikat) pr. koncentration, men eftersom denne protokol kræver tredobbelt analyse, består én analyseserie af tre assaybrønde pr. koncentration. Endvidere kræver denne protokol tre uafhængige (dvs. ikke-sideløbende) analyseserier pr. kemikalie.
Tillæg 2.2
TYPISK [3H]-17Β-ØSTRADIOLMÆTNINGSASSAY MED TRE REPLIKATBRØNDE
|
Typisk [3H]-17β-østradiolmætningsassay med tre replikatbrønde
|
|
Placering
|
Replikat
|
Kode for brøndtype
|
Varm E2 Begyndelseskoncentration (nM)
|
Varm E2 Volumen (μl)
|
Varm E2 Slutkoncentration (nM)
|
Kold E2 Begyndelseskoncentration (μM)
|
Kold E2 Volumen (μl)
|
Kold E2 Slutkoncentration (μM)
|
Buffervolumen (μl)
|
Receptorvolumen (μl)
|
Samlet volumen i brøndene
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Varm
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Varm
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Varm
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Varm
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Varm
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Varm
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Varm
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Varm
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Varm
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Varm
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Varm
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Varm
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Varm
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Varm
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Varm
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Varm
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Varm
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Varm
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Varm
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Varm
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Varm
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Varm
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Varm
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Varm
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Bemærk, at de "varme" brønde er tomme under inkubation. De 40 μl tilsættes kun til scintillationstælling.
|
Tillæg 2.3
KOMPETITIVT BINDINGSASSAY: BRØNDLAYOUT
|
Plade
|
Placering
|
Replikat
|
Brøndtype
|
Brøndkode
|
Koncentrationskode
|
Konkurrent Begyndelseskoncentration (M)
|
hrER-stamkultur (μl)
|
Buffervolumen (μl)
|
Sporstof (varm E2) Volumen (μl)
|
Volumen fra fortyndingsplade (μl)
|
Slutvolumen (μl)
|
Konkurrent Slutkoncentration (M)
|
|
S
|
A1
|
1
|
total binding
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
total binding
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
total binding
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
total binding
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
total binding
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
total binding
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A8
|
2
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A9
|
3
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A10
|
1
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A11
|
2
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A12
|
3
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
B1
|
1
|
kold E2
|
S
|
S1
|
2.00E-07
|
40
|
—
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
B2
|
2
|
kold E2
|
S
|
S1
|
2.00E-07
|
40
|
—
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
B3
|
3
|
kold E2
|
S
|
S1
|
2.00E-07
|
40
|
—
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
B4
|
1
|
kold E2
|
S
|
S2
|
2.00E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
B5
|
2
|
kold E2
|
S
|
S2
|
2.00E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
B6
|
3
|
kold E2
|
S
|
S2
|
2.00E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
B7
|
1
|
kold E2
|
S
|
S3
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
B8
|
2
|
kold E2
|
S
|
S3
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
B9
|
3
|
kold E2
|
S
|
S3
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
B10
|
1
|
kold E2
|
S
|
S4
|
2.00E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
B11
|
2
|
kold E2
|
S
|
S4
|
2.00E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
B12
|
3
|
kold E2
|
S
|
S4
|
2.00E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
C1
|
1
|
kold E2
|
S
|
S5
|
6.00E-10
|
40
|
—
|
40
|
80
|
160
|
3.0E-10
|
|
S
|
C2
|
2
|
kold E2
|
S
|
S5
|
6.00E-10
|
40
|
—
|
40
|
80
|
160
|
3.0E-10
|
|
S
|
C3
|
3
|
kold E2
|
S
|
S5
|
6.00E-10
|
40
|
—
|
40
|
80
|
160
|
3.0E-10
|
|
S
|
C4
|
1
|
kold E2
|
S
|
S6
|
2.00E-10
|
40
|
—
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C5
|
2
|
kold E2
|
S
|
S6
|
2.00E-10
|
40
|
—
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C6
|
3
|
kold E2
|
S
|
S6
|
2.00E-10
|
40
|
—
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C7
|
1
|
kold E2
|
S
|
S7
|
2.00E-11
|
40
|
—
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C8
|
2
|
kold E2
|
S
|
S7
|
2.00E-11
|
40
|
—
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C9
|
3
|
kold E2
|
S
|
S7
|
2.00E-11
|
40
|
—
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C10
|
1
|
blindprøve
|
blindprøve
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
blindprøve
|
blindprøve
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
blindprøve
|
blindprøve
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
norethynodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
—
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
D2
|
1
|
norethynodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
—
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
D3
|
1
|
norethynodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
—
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
D4
|
1
|
norethynodrel
|
NE
|
WP2
|
2.00E-05
|
40
|
—
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D5
|
1
|
norethynodrel
|
NE
|
WP2
|
2.00E-05
|
40
|
—
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D6
|
1
|
norethynodrel
|
NE
|
WP2
|
2.00E-05
|
40
|
—
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D7
|
1
|
norethynodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
—
|
40
|
80
|
160
|
3.0E-06
|
|
S
|
D8
|
1
|
norethynodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
—
|
40
|
80
|
160
|
3.0E-06
|
|
S
|
D9
|
1
|
norethynodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
—
|
40
|
80
|
160
|
3.0E-06
|
|
S
|
D10
|
1
|
norethynodrel
|
NE
|
WP4
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
D11
|
1
|
norethynodrel
|
NE
|
WP4
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
D12
|
1
|
norethynodrel
|
NE
|
WP4
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
E1
|
1
|
norethynodrel
|
NE
|
WP
|
6.00E-07
|
40
|
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
E2
|
2
|
norethynodrel
|
NE
|
WP
|
6.00E-07
|
40
|
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
E3
|
3
|
norethynodrel
|
NE
|
WP
|
6.00E-07
|
40
|
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
E4
|
1
|
norethynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E5
|
2
|
norethynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E6
|
3
|
norethynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E7
|
1
|
norethynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
—
|
40
|
80
|
160
|
3.0E-08
|
|
S
|
E8
|
2
|
norethynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
—
|
40
|
80
|
160
|
3.0E-08
|
|
S
|
E9
|
3
|
norethynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
—
|
40
|
80
|
160
|
3.0E-08
|
|
S
|
E10
|
1
|
norethynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
E11
|
2
|
norethynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
E12
|
3
|
norethynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2.00E-03
|
40
|
—
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2.00E-03
|
40
|
—
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2.00E-03
|
40
|
—
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2.00E-04
|
40
|
—
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2.00E-04
|
40
|
—
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2.00E-04
|
40
|
—
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2.00E-05
|
40
|
—
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2.00E-05
|
40
|
—
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2.00E-05
|
40
|
—
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2.00E-07
|
40
|
—
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2.00E-07
|
40
|
—
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2.00E-07
|
40
|
—
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2.00E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2.00E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2.00E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2.00E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2.00E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2.00E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2.00E-10
|
40
|
—
|
40
|
—
|
160
|
1.0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2.00E-10
|
40
|
—
|
40
|
—
|
160
|
1.0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2.00E-10
|
40
|
—
|
40
|
—
|
160
|
1.0E-10
|
|
S
|
H1
|
1
|
varm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
varm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
varm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
varm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
varm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
varm
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
bufferkontrol
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
bufferkontrol
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
bufferkontrol
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
bufferkontrol
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
bufferkontrol
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
bufferkontrol
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Bemærk, at de "varme" brønde er tomme under inkubation. De 40 μl tilsættes kun til scintillationstælling.
|
Kompetitivt bindingsassay: brøndlayout
|
|
Plade
|
Placering
|
Replikat
|
Brøndtype
|
Brøndkode
|
Koncentrations-kode
|
Konkurrent Begyndelseskoncentration (M)
|
hrER-stamkultur (μl)
|
Buffervolumen (μl)
|
Sporstof (varm E2) Volumen (μl)
|
Volumen fra fortyndingsplade (μl)
|
Slutvolumen (μl)
|
Konkurrent Slutkoncentration (M)
|
|
P1
|
A1
|
1
|
total binding
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
total binding
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
total binding
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
total binding
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
total binding
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
total binding
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A8
|
2
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A9
|
3
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A10
|
1
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A11
|
2
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A12
|
3
|
kold E2 (høj)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B1
|
1
|
Testkemikalie 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B2
|
2
|
Testkemikalie 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B3
|
3
|
Testkemikalie 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B4
|
1
|
Testkemikalie 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B5
|
2
|
Testkemikalie 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B6
|
3
|
Testkemikalie 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B7
|
1
|
Testkemikalie 1
|
TC1
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B8
|
2
|
Testkemikalie 1
|
TC1
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B9
|
3
|
Testkemikalie 1
|
TC1
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B10
|
1
|
Testkemikalie 1
|
TC1
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B11
|
2
|
Testkemikalie 1
|
TC1
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B12
|
3
|
Testkemikalie 1
|
TC1
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
C1
|
1
|
Testkemikalie 1
|
TC1
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C2
|
2
|
Testkemikalie 1
|
TC1
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C3
|
3
|
Testkemikalie 1
|
TC1
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C4
|
1
|
Testkemikalie 1
|
TC1
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C5
|
2
|
Testkemikalie 1
|
TC1
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C6
|
3
|
Testkemikalie 1
|
TC1
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C7
|
1
|
Testkemikalie 1
|
TC1
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C8
|
2
|
Testkemikalie 1
|
TC1
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C9
|
3
|
Testkemikalie 1
|
TC1
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C10
|
1
|
Testkemikalie 1
|
TC1
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
C11
|
2
|
Testkemikalie 1
|
TC1
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
C12
|
3
|
Testkemikalie 1
|
TC1
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
D1
|
1
|
Testkemikalie 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D2
|
2
|
Testkemikalie 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D3
|
3
|
Testkemikalie 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D4
|
1
|
Testkemikalie 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D5
|
2
|
Testkemikalie 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D6
|
3
|
Testkemikalie 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D7
|
1
|
Testkemikalie 2
|
TC2
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D8
|
2
|
Testkemikalie 2
|
TC2
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D9
|
3
|
Testkemikalie 2
|
TC2
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D10
|
1
|
Testkemikalie 2
|
TC2
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
D11
|
2
|
Testkemikalie 2
|
TC2
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
D12
|
3
|
Testkemikalie 2
|
TC2
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
E1
|
1
|
Testkemikalie 2
|
TC2
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E2
|
2
|
Testkemikalie 2
|
TC2
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E3
|
3
|
Testkemikalie 2
|
TC2
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E4
|
1
|
Testkemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E5
|
2
|
Testkemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E6
|
3
|
Testkemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E7
|
1
|
Testkemikalie 2
|
TC2
|
7
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E8
|
2
|
Testkemikalie 2
|
TC2
|
7
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E9
|
3
|
Testkemikalie 2
|
TC2
|
7
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E10
|
1
|
Testkemikalie 2
|
TC2
|
8
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
E11
|
2
|
Testkemikalie 2
|
TC2
|
8
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
E12
|
3
|
Testkemikalie 2
|
TC2
|
8
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
F1
|
1
|
Testkemikalie 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F2
|
2
|
Testkemikalie 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F3
|
3
|
Testkemikalie 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F4
|
1
|
Testkemikalie 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F5
|
2
|
Testkemikalie 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F6
|
3
|
Testkemikalie 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F7
|
1
|
Testkemikalie 3
|
TC3
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F8
|
2
|
Testkemikalie 3
|
TC3
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F9
|
3
|
Testkemikalie 3
|
TC3
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F10
|
1
|
Testkemikalie 3
|
TC3
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
F11
|
2
|
Testkemikalie 3
|
TC3
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
F12
|
3
|
Testkemikalie 3
|
TC3
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
G1
|
1
|
Testkemikalie 3
|
TC3
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G2
|
2
|
Testkemikalie 3
|
TC3
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G3
|
3
|
Testkemikalie 3
|
TC3
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G4
|
1
|
Testkemikalie 3
|
TC3
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G5
|
2
|
Testkemikalie 3
|
TC3
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G6
|
3
|
Testkemikalie 3
|
TC3
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G7
|
1
|
Testkemikalie 3
|
TC3
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G8
|
2
|
Testkemikalie 3
|
TC3
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G9
|
3
|
Testkemikalie 3
|
TC3
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G10
|
1
|
Testkemikalie 3
|
TC3
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
G11
|
2
|
Testkemikalie 3
|
TC3
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
G12
|
3
|
Testkemikalie 3
|
TC3
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
H1
|
1
|
norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
norethynodrel
|
NE
|
|
1.00E-4.5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
norethynodrel
|
NE
|
|
1.00E-4.5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
norethynodrel
|
NE
|
|
1.00E-4.5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
kold E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
kold E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
kold E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
kold E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
kold E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
kold E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
Tillæg 3
CHEMICAL EVALUATION AND RESEARCH INSTITUTE'S (CERI) IN VITRO-ASSAY VEDRØRENDE ØSTROGENRECEPTORBINDING VED ANVENDELSE AF ET LIGANDBINDINGSDOMÆNE AF HUMANT REKOMBINANT ERΑ-PROTEIN
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER (JF. OGSÅ GENEREL INDLEDNING)
|
1.
|
Dette in vitro-assay vedrørende østrogenreceptor(ERα)-mætning og kompetitiv binding anvender et ligandbindingsdomæne (LBD) af humant ERα (hrERα). Denne proteinmodel blev produceret af Chemicals Evaluation Research Institute (CERI), Japan og findes som et glutathion-S-transferase (GST) fusionsprotein og udtrykkes i E. coli. CERI-protokollen gennemgik en international valideringsundersøgelse (2) i flere laboratorier, der har påvist dens relevans og pålidelighed i forbindelse med assayets tilsigtede formål.
|
|
2.
|
Assayet er en screeningsprocedure til påvisning af stoffer, der kan binde sig til hrERα. Det bruges til at bestemme et testkemikalies evne til at konkurrere med 17β-østradiol om binding til hrERα-LBD. Kvantitative assayresultater kan omfatte IC50 (et mål for den koncentration af testkemikaliet, der er nødvendig for at fortrænge halvdelen af [3H]-17β-østradiol fra hrERα), og testkemikaliernes relative affiniteter for hrERα sammenlignet med 17β-østradiol. Til kemikaliescreeningsformål kan acceptable kvalitative assayresultater omfatte klassificeringer af testkemikalier som enten hrERα-bindere, ikke-bindere eller usikre baseret på kriterier beskrevet for bindingskurver.
|
|
3.
|
Assayet anvender en radioaktivt mærket ligand, som kræver, at laboratoriet har licens til radioaktive materialer. Alle procedurer med radioisotoper og farlige kemikalier skal følge forskrifter og procedurer som beskrevet i national lovgivning.
|
|
4.
|
Afsnittene "GENEREL INDLEDNING" og "hrER-BINDINGSASSAYETS KOMPONENTER" skal læses, før dette assay benyttes til lovmæssige formål. Definitioner og forkortelser, der benyttes i denne testvejledning, er beskrevet i tillæg 1.
|
PRINCIPPER FOR ASSAYET (JF. OGSÅ GENEREL INDLEDNING)
|
5.
|
hrERα-bindingsassayet måler en radioaktivt mærket ligands ([3H]17β-østradiol) evne til at binde sig til ER under tilstedeværelse af stigende koncentrationer af et testkemikalie (dvs. konkurrent). Testkemikalier, som har en høj ER-affinitet, konkurrerer med den radioaktivt mærkede ligand ved en lavere koncentration sammenlignet med kemikalier med lavere affinitet for receptoren.
|
|
6.
|
Dette assay består af to vigtige komponenter: et mætningsbindingsforsøg for at karakterisere parametrene for interaktion mellem receptor og ligand efterfulgt af et forsøg med kompetitiv binding, der karakteriserer konkurrencen mellem et testkemikalie og en radioaktivt mærket ligand for binding til ER.
|
|
7.
|
Formålet med mætningsbindingsforsøget er at karakterisere en særlig batch af receptorer for affinitet og antal i forberedelsen af det kompetitive bindingsforsøg. Mætningsbindingsforsøget måler, under ligevægtsbetingelser, en fast østrogenreceptorkoncentrations affinitet for dens naturlige ligand (repræsenteret ved dissociationskonstanten, Kd) og koncentrationen af aktive receptorsteder (Bmax).
|
|
8.
|
Det kompetitive bindingsforsøg måler et stofs affinitet til at konkurrere med [3H]17β-østradiol med hensyn til binding til ER. Affiniteten kvantificeres af koncentrationen af testkemikaliet, som under ligevægtsbetingelser inhiberer 50 % af den specifikke binding af [3H]17β-østradiol (kaldet den "inhiberende koncentration på 50 %" eller IC50). Denne kan også vurderes ved hjælp af den relative affinitet (RBA, i forhold til IC50 af østradiol målt separat i den samme analyseserie). Det kompetitive bindingsforsøg måler bindingen af [3H]17β-østradiol ved en fast koncentration under tilstedeværelse af en bred vifte (otte størrelsesordener) af testkemikaliekoncentrationer. Dataene tilpasses herefter, hvor det er muligt, så de danner Hill-ligningen (Hill, 1910), der beskriver en kompetitiv binders fortrængning af den radioaktive ligand ét sted. Omfanget af fortrængningen af det radioaktivt mærkede østradiol ved ligevægt anvendes til at karakterisere testkemikaliet som en binder, ikke-binder eller som et kemikalie, der genererer en usikker respons.
|
FREMGANGSMÅDE
Påvisning af acceptabel ydeevne for hrERα-protein
|
9.
|
Før der rutinemæssigt udføres mætningsassays og kompetitive bindingsassays, skal det påvises, at hver ny batch hrERα fungerer korrekt i det laboratorium, hvor det vil blive anvendt. Ydeevnen påvises gennem en totrinsproces. Det drejer sig om følgende trin:
|
—
|
Der udføres et [3H]-17β-østradiol-mætnings- og bindingsassay for at påvise hrERα-specificitet og mætning. Ikke-lineær regressionsanalyse af disse data (f.eks. BioSoft, McPherson, 1985; Motulsky, 1995) og det efterfølgende Scatchard-plot bør dokumentere hrERα-affiniteten for [3H]-17β-østradiol (Kd) og antallet af receptorer (Bmax) for en bestemt batch hrERα.
|
|
—
|
Der udføres et kompetitivt bindingsassay ved anvendelse af kontrolstofferne (referenceøstrogen (17β-østradiol), en svag binder (f.eks. norethynodrel eller norethindron) og en ikke-binder (octyltriethoxysilan, OTES). Hvert laboratorium bør oprette en historisk database for at dokumentere konsistensen af IC50 og de relevante værdier for referenceøstrogenet og den svage binder blandt forsøg og forskellige batcher af hrERα. Parametrene for de kompetitive bindingskurver for kontrolstofferne bør desuden ligge inden for de grænser for konfidensintervallet på 95 % (jf. tabel 1), der blev udviklet ved hjælp af data fra laboratorier, som deltog i valideringsundersøgelsen for dette assay (2).
|
Tabel 1
Ydeevnekriterier udviklet for referenceøstrogen og svage bindere, CERI's hrER-bindingsassay
|
Stof
|
Parameter
|
Gennemsnit (10)
|
Standard-afvigelse (n)
|
95 %-konfidensintervaller (11)
|
|
Nedre grænse
|
Øvre grænse
|
|
17β-østradiol
|
Top
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Bund
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Hills kurve
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
LogIC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Norethynodrel
|
Top
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Bund
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Hills kurve
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
LogIC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Norethindron (12)
|
Top
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Bund
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Hills kurve
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
LogIC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Påvisning af laboratoriets kompetence
|
10.
|
Jf. punkt 17 og 18 og tabel 2 i afsnittet "hrER-BINDINGSASSAYETS KOMPONENTER" i denne forsøgsmetode. Hvert assay (mætning og kompetitiv binding) bør bestå af tre uafhængige analyseserier (dvs. friske fortyndinger af receptor, kemikalier og reagenser) på forskellige dage, og hver analyseserie bør indeholde tre replikater.
|
Bestemmelse af receptor(hrERα)koncentration
|
11.
|
Den aktive receptorkoncentration varierer lidt efter batch og opbevaringsforhold. Af denne grund bør den koncentration af aktiv receptor, der modtages fra leverandøren, bestemmes. Dette vil give den rette koncentration af aktiv receptor på tidspunktet for analyseserien.
|
|
12.
|
Under forhold svarende til kompetitiv binding (dvs. 0,5 nM [3H]-østradiol) bør nominelle koncentrationer på 0,1, 0,2, 0,4 og 0,6 nM receptor inkuberes i fravær af (total binding) og under tilstedeværelse (ikke-specifik binding) af 1 μM umærket østradiol. Specifik binding beregnet som forskellen mellem total og ikke-specifik binding afsættes i forhold til den nominelle receptorkoncentration. Den receptorkoncentration, som giver specifikke bindingsværdier svarende til 40 % af de tilsatte radioaktivt mærkede stoffer, hænger sammen med den tilsvarende receptorkoncentration, og denne receptorkoncentration anvendes til mætningsforsøg og kompetitive bindingsforsøg. En slutkoncentration for hrER på 0,2 nM vil ofte opfylde denne betingelse.
|
|
13.
|
Hvis 40 %-kriteriet gentagne gange ikke kan opfyldes, kontrolleres forsøgs-setuppet for potentielle fejl. Hvis 40 %-kriteriet ikke opfyldes, kan det tyde på, at der er meget lidt aktiv receptor i rekombinantbatchen, og det bør overvejes at anvende en anden receptorbatch.
|
Mætningsassay
|
14.
|
Otte stigende koncentrationer af [3H]17β-østradiol vurderes in triplo under følgende tre betingelser (jf. tabel 2):
|
a.
|
i fravær af umærket 17β-østradiol og under tilstedeværelse af ER Dette er den kvantitative analyse af den totale binding ved måling af radioaktiviteten i de brønde, som kun har [3H]17β-østradiol
|
|
b.
|
under tilstedeværelse af en 2000-fold uforholdsmæssig koncentration af umærket 17β-østradiol i forhold til mærket 17β-østradiol og tilstedeværelse af ER. Formålet med denne betingelse er at mætte de aktive bindingssteder med umærket 17β-østradiol og, ved at måle radioaktiviteten i brøndene, bestemme den ikke-specifikke binding. Eventuelt tilbageværende varmt østradiol, som kan binde sig til receptoren, anses for at kunne binde sig til et ikke-specifikt sted, da koldt østradiol bør have en så høj koncentration, at det binder sig til alle tilgængelige specifikke steder på receptoren
|
|
c.
|
i fravær af umærket 17β-østradiol og af ER (bestemmelse af samlet radioaktivitet).
|
Fremstilling af [3H]-17β-østradiol, umærkede 17β-østradiol-opløsninger og hrERα
|
15.
|
En 40 nM-opløsning af [3H]-17β-østradiol fremstilles af en 1 μM stamopløsning af [3H]-17β-østradiol i DMSO ved at tilsætte DMSO (for at fremstille 200 nM) og assaybuffer ved stuetemperatur (for at fremstille 40 nM). Ved hjælp af denne 40 nM-opløsning fremstilles serierne af [3H]-17β-østradiolfortyndinger i et område fra 0,313 nM til 40 nM med assaybuffer ved stuetemperatur (som vist i kolonne 12 i tabel 2). Assayets slutkoncentrationer i området fra 0,0313 til 4,0 nM fås ved at tilsætte 10 μl af disse opløsninger til de respektive assaybrønde på en mikrotiterplade med 96 brønde (jf. tabel 2 og 3). Fremstilling af assaybuffer, beregning af den originale stamopløsning af [3H]-17β-østradiol baseret på dens specifikke aktivitet, fremstilling af fortyndinger og bestemmelse af koncentrationerne er beskrevet i detaljer i CERI-protokollen (2).
|
|
16.
|
Fortyndinger af opløsninger af umærket 17β-østradiol fremstilles af en stamopløsning af 1 nM 17β-østradiol ved tilsætning af assaybuffer for at få otte stigende koncentrationer, der indledningsvis ligger fra 0,625 μM til 80 μM. Assayets slutkoncentrationer i området fra 0,0625 til 8 nM fås ved at tilsætte 10 μl af disse opløsninger til de respektive assaybrønde på en mikrotiterplade med 96 brønde til måling af ikke-specifik binding (jf. tabel 2 og 3). Fremstilling af fortyndinger af umærket 17β-østradiol er beskrevet i detaljer i CERI-protokollen (2).
|
|
17.
|
Den receptorkoncentration, som giver 40 ± 10 % specifik binding, bør anvendes (jf. punkt 12-13). hrERα-opløsningen bør fremstilles med iskold assaybuffer umiddelbart før brug, dvs. efter at alle brønde til total binding, ikke-specifik binding og varm ligand alene er blevet fremstillet.
|
|
18.
|
Mikrotiterpladerne med 96 brønde forberedes som illustreret i tabel 2 med 3 replikater pr. [3H]-17β-østradiolkoncentration. Volumenfordelingen af [3H]-17β-østradiol, umærket 17β-østradiol, buffer og receptor er anført i tabel 3.
Tabel 2
Layout for en mikrotiterplade til mætningsbindingsassay
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
Til måling af TB
|
Til måling af NSB
|
Til bestemmelse af varm ligand alene
|
|
Umærkede E2-fortyndinger til pladekolonne 4-6.
|
[3H]E2-fortyndinger til pladekolonne 1-9.
|
|
A
|
0,0313 nM [3H] E2+ ER
|
0,0313 nM [3H] E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H] E2+ ER
|
0,0625 nM [3H] E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H] E2+ ER
|
0,125 nM [3H] E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H] E2+ ER
|
0,250 nM [3H] E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [ H] E2+ ER
|
0,50 nM [3H] E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H] E2+ ER
|
1,00 nM [3H] E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H] E2+ ER
|
2,00 nM [3H] E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H] E2+ ER
|
4,00 nM [3H] E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
-
|
total binding
|
|
NSB
|
-
|
ikke-specifik binding
|
|
[3H] E2
|
-
|
[3H]17β-østradiol
|
|
E2
|
-
|
umærket 17β-østradiol
|
|
Tabel 3
Reagensmængder til mætningsmikrotiterpladen
|
Kolonnenummer
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Fremstillingstrin
|
TB-brønde
|
NSB-brønde
|
Varm ligand alene
|
|
Komponentvolumen til reaktionsbrønde og tilsætningsrækkefølge
|
Buffer
|
60 μl
|
50 μl
|
90 μl
|
|
umærket E2 fra kolonne 11 i tabel 2
|
–
|
10 μl
|
–
|
|
[3H]E2 fra kolonne 12 i tabel 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
–
|
|
Samlet reaktionsvolumen
|
100 μl
|
100 μl
|
100 μl
|
|
Inkubation
|
REAKTION EFTER 2 TIMERS INKUBATION
|
Kvantificering af radioaktivitet lige efter fremstilling. Ingen inkubation
|
|
Behandling med 0,4 % DCC
|
Ja
|
Ja
|
Nej
|
|
Volumen af 0,4 % DCC
|
100 μl
|
100 μl
|
–
|
|
Filtrering
|
Ja
|
Ja
|
Nej
|
|
MÅLING AF DPMS
|
|
Kvantificeringsvolumen tilsat til scintillationscocktail
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Assaymikrotiterplader til bestemmelse af total binding og ikke-specifik binding inkuberes ved stuetemperatur (22-28 °C) i to timer.
|
|
Måling af [3H]-17β-østradiol bundet til hrERα
|
20.
|
Efter inkubationsperioden på to timer bør [3H]-17β-østradiol bundet til hrERα separeres fra fri [3H]-17β-østradiol ved tilsætning af 100 μl iskold 0,4 % DCC-suspension til brøndene. Pladerne anbringes herefter på is i 10 minutter, og reaktionsblandingen og DCC-blandingen filtreres ved overførsel til et mikrotiterpladefilter for at fjerne DCC. 100 μl af filtratet tilsættes herefter til scintillationsvæsken i LSC-glas til bestemmelse af disintegration pr. minut (dpms) pr. glas ved væskescintillationstælling.
|
|
21.
|
Er der ikke adgang til et mikropladefilter, kan DCC fjernes ved centrifugering. 50 μl supernatant, der indeholder hrERα-bundet [3H]-17β-østradiol, tages herefter ekstremt forsigtigt for at undgå enhver kontaminering af brøndene ved kontakt med DCC og anvendes til scintillationstælling.
|
|
22.
|
Betingelsen om den varme ligand alene anvendes til bestemmelse af disintegration pr. minut (dpm) af [3H]-17β-østradiol tilsat til assaybrøndene. Radioaktiviteten kvantificeres lige efter fremstillingen. Disse brønde bør ikke inkuberes og bør ikke behandles med DCC-suspension, men deres indhold bør hældes direkte i scintillationsvæsken. Disse mål viser, hvor meget [3H]-17β-østradiol i dpms der blev tilsat til hvert sæt brønde til den totale binding og ikke-specifikke binding.
|
Kompetitivt bindingsassay
|
23.
|
Det kompetitive bindingsassay måler bindingen af en enkelt koncentration af [3H]-17β-østradiol under tilstedeværelse af stigende koncentrationer af et testkemikalie. Der bør anvendes tre sideløbende replikater ved hver koncentration inden for én analyseserie. Endvidere bør der udføres tre ikke-sideløbende analyseserier for hvert kemikalie, der testes. Assayet bør opsættes med en eller flere mikrotiterplader med 96 brønde.
|
Kontroller
|
24.
|
Ved udførelsen af assayet medtages samtidig opløsningsmiddel og kontroller (dvs. referenceøstrogen, svag binder og ikke-binder) i hvert forsøg. Fuldstændige koncentrationskurver for referenceøstrogen og kontroller (dvs. svag binder og ikke-binder) anvendes på én plade under hver analyseserie. Alle andre plader bør indeholde i) en høj- (maksimal fortrængning dvs. næsten fuld fortrængning af radioaktivt mærket ligand) og medium- (næsten IC50) koncentration med E2 og svag binder in triplo ii) kontrolkultur med opløsningsmiddel og ikke-specifik binding, hver især in triplo. Procedurer for fremstilling af assaybuffer, [3H]-17β-østradiol, hrERα og testkemikalieopløsninger er beskrevet i detaljer i CERI-protokollen (2).
|
Kontrolkultur med opløsningsmiddel
|
25.
|
Kontrolkulturen med opløsningsmiddel viser, at opløsningsmidlet ikke interagerer med testsystemet og måler også total binding (TB). DMSO er det foretrukne opløsningsmiddel. Såfremt den højeste koncentration af testkemikaliet ikke er opløseligt i DMSO, kan ethanol anvendes. DMSO-koncentrationen i slutassaybrøndene bør ligge på 2,05 % og kan øges op til 2,5 % i tilfælde af testkemikaliets manglende opløselighed. DMSO-koncentrationer over 2,5 % bør ikke anvendes, fordi højere koncentrationer af opløsningsmidlet interfererer med assayet. For testkemikalier, som ikke er opløselige i DMSO, men er opløselige i ethanol, kan der højst anvendes 2 % ethanol i assayet uden interferens.
|
Bufferkontrol
|
26.
|
Bufferkontrollen (BC) bør hverken indeholde opløsningsmiddel eller testkemikalie, men alle assayets øvrige komponenter. Resultaterne af bufferkontrollen sammenlignes med opløsningsmiddelkontrollen for at kontrollere, at det anvendte opløsningsmiddel ikke påvirker assaysystemet.
|
Stærk binder (referenceøstrogen)
|
27.
|
17β-østradiol (CAS 50-28-2) er den endogene ligand og binder sig med høj affinitet til ER, undertype alfa. En standardkurve, som anvender umærket 17β-østradiol, fremstilles til hvert hrERα kompetitivt bindingsassay, så der kan foretages en variabilitetsvurdering ved gennemførelse af assayet over længere tid i det samme laboratorium. Otte opløsninger af umærket 17β-østradiol fremstilles i DMSO og assaybuffer med slutkoncentrationer i assaybrøndene, så de kan bruges til standardkurven med følgende intervaller: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. Den højeste koncentration af umærket 17β-østradiol (1 μM) bør fungere som indikator for ikke-specifik binding. Denne koncentration er angivet ved mærket "NSB" i tabel 4, selv om den også indgår i standardkurven.
|
Svag binder
|
28.
|
En svag binder (norethynodrel (CAS 68-23-5) eller alternativt norethindron (CAS 68-22-4)) bør medtages for at påvise følsomheden af hvert forsøg og for at give mulighed for en vurdering af variabiliteten, når assayet udføres over længere tid. Otte opløsninger af den svage binder fremstilles i DMSO og assaybuffer med slutkoncentrationer i assaybrøndene som følger: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 og 10–9 M.
|
Ikke-binder
|
29.
|
Octyltriethoxysilan (OTES, CAS 2943-75-1) bør anvendes som den negative kontrol (ikke-binder). Det giver sikkerhed for, at der med det udførte assay påvises testkemikalier, som ikke binder sig til hrERα. Otte opløsninger af ikke-binder fremstilles i DMSO og assaybuffer, med slutkoncentrationer i assaybrøndene som følger: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Di-n-butylphthalat (DBP, CAS 84-72-2) kan anvendes som en alternativ ikke-binder, men kun testet op til 10–4 M. Det er påvist, at den maksimale opløselighed af DBP i assayet er 10–4M.
|
hrERα-koncentration
|
30.
|
Den mængde receptor, som giver en specifik binding på 40 ± 10 %, bør anvendes (jf. punkt 12-13 i tillæg 3). hrERα-opløsningen bør fremstilles ved fortynding af den funktionelle hrERα i iskold assaybuffer, umiddelbart inden brug.
|
[3H]-17β-østradiol
|
31.
|
Slutkoncentrationen af [3H]-17β-østradiol i assaybrøndene skal være 0,5 nM.
|
Testkemikalier
|
32.
|
I første omgang er det nødvendigt at foretage en opløselighedstest for at fastslå opløselighedsgrænsen for hvert testkemikalie og for at afdække det passende koncentrationsområde, der skal anvendes, når testprotokollen udføres. Opløselighedsgrænsen for hvert testkemikalie skal indledningsvis bestemmes i opløsningsmidlet og herefter yderligere bekræftes under assaybetingelser. Den slutkoncentration, der testes i assayet, bør ikke overstige 1 mM. Områdebestemmelsen omfatter en kontrolkultur med opløsningsmiddel sammen med mindst otte log-seriefortyndinger, der begynder ved den maksimale acceptable koncentration (f.eks. 1 mM eller lavere, baseret på opløselighedsgrænsen), og idet der noteres tilstedeværelse af uklarhed eller bundfald (jf. også punkt 35 i tillæg 3). Når koncentrationsområdet for testningen er fastlagt, testes et testkemikalie ved anvendelse af otte log-koncentrationer med passende intervaller som defineret i den foregående test til bestemmelse af dosisinterval. Koncentrationer, som testes i det andet og tredje forsøg, bør yderligere tilpasses, hvis det er relevant, for om nødvendigt bedre at kunne karakterisere koncentration/respons-kurven.
|
|
33.
|
Fortyndinger af testkemikaliet fremstilles i det relevante opløsningsmiddel (jf. punkt 25 i tillæg 3). Hvis den højeste koncentration af testkemikaliet hverken kan opløses i DMSO eller ethanol, og tilsætning af mere opløsningsmiddel vil medføre, at opløsningsmiddelkoncentrationen i slutrøret bliver større end den acceptable grænse, kan den højeste koncentration reduceres til den næste lavere koncentration. I så fald kan en supplerende koncentration tilsættes i den lave ende af koncentrationsserien. Andre koncentrationer i serien bør forblive uændrede.
|
|
34.
|
Testkemikalieopløsningerne bør overvåges nøje, når de tilsættes til assaybrønden, da testkemikaliet kan danne bundfald ved tilsætning til assaybrønden. Data for alle brønde, der indeholder bundfald, bør udelukkes fra kurvetilpasningen, og årsagen til udelukkelsen af data noteres.
|
|
35.
|
Hvis der allerede findes oplysninger fra andre kilder, som angiver en log (IC50) for et testkemikalie, kan det være hensigtsmæssigt at sætte en tættere geometrisk afstand mellem fortyndingerne omkring den forventede log(IC50) (dvs. 0,5 log-enheder). Slutresultaterne bør vise nok tilstrækkelig spredning af koncentrationer på begge side af log(IC50), herunder "top" og "bund", således at bindingskurven kan karakteriseres tilstrækkeligt.
|
Organisering af assaypladen
|
36.
|
Mærkede mikrotiterplader bør forberedes ved anvendelse af seksdobbelte inkubationer for kontrolkulturen med opløsningsmiddel, den højeste koncentration af referenceøstrogen (E2), der også fungerer som indikator for ikke-specifik binding (NSB), bufferkontrol, og tredobbelte inkubationer for hver af de otte koncentrationer af den ikke-bindende kontrol (octyltriethoxysilan), de syv lavere koncentrationer af referenceøstrogen (E2), de otte koncentrationer af den svage binder (norethynodrel eller norethindron) og de otte koncentrationer af hvert testkemikalie (TC). Et layouteksempel på pladelayoutdiagrammet til de fulde koncentrationskurver for referenceøstrogen og kontroller er anført nedenfor i tabel 4. Supplerende mikrotiterplader bruges til testkemikaliet og bør omfatte pladekontroller (dvs. i) en høj- (maksimal fortrængning) og middel- (ca. IC50) koncentration af E2 og svag binder in triplo, ii) kontrolkultur med opløsningsmiddel (som total binding) og ikke-specifik binding, hver især seksdobbelt (tabel 5). Et eksempel på et arbejdsark for layout til en mikrotiterplade til et kompetitivt assay med tre ukendte testkemikalier er anført i tillæg 3.3. De koncentrationer, der er anført på arbejdsarket og i tabel 4 og 5, henviser til de slutkoncentrationer, som er anvendt i hver assaybrønd. Den maksimale koncentration for E2 bør være 1×10–7 M, og for den svage binder bør den højeste anvendte koncentration for den svage binder på plade 1 anvendes. IC50-koncentrationen skal bestemmes af laboratoriet på grundlag af deres historiske kontroldatabase. Forventningen er, at denne værdi vil ligne den, der blev observeret i valideringsundersøgelserne (jf. tabel 1).
|
Tabel 4
Layout for mikrotiterpladen til et kompetitivt bindingsassay (98)
(99), fulde koncentrationskurver for referenceøstrogen og kontroller (plade 1).
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Bufferkontrol og positiv kontrol (E2)
|
Svagt positiv (norethynodrel)
|
Negativ kontrol (OTES)
|
TB og NSB
|
|
A
|
Blindprøve (*14)
|
1×10–9 M
|
1×10–10 M
|
TB (kontrolkultur med opløsningsmiddel) (2,05 % DMSO)
|
|
B
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
C
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
E
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Bufferkontrol
|
|
F
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
G
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Blindprøve (til varm) (*15)
|
|
H
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
Tabel 5
Layout til mikrotiterpladen til et kompetitivt bindingsassay, supplerende plader til testkemikalier (TC) og pladekontroller.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Testkemikalie 1 (TC-1)
|
Testkemikalie 2(TC-2)
|
Testkemikalie 3(TC-3)
|
Kontroller
|
|
A
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10–7M)
|
|
B
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10
-4,5
M)
|
|
D
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (10-6 M E2)
|
|
F
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
G
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
TB (kontrolkultur med opløsningsmiddel)
|
|
H
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
I dette eksempel er den svage binder norethinodrel (NE)
|
Gennemførelse af kompetitivt bindingsassay
|
37.
|
Mens brønde til total binding og blindprøver (til varm) undtages som vist i tabel 6, sættes 50 μl assaybuffer i hver brønd og blandes med 10 μl af kontrolkulturen med opløsningsmiddel, referenceøstrogen (E2), svag binder, ikke-binder og testkemikalier, henholdsvis 10 μl af en 5 nM [3H]-17β-østradiolopløsning. Herefter blev der tilsat 30 μl iskold receptoropløsning til hver plade og blandet forsigtigt. hrERα-opløsningen bør være det sidste reagens, der tilsættes. Assaymikrotiterplader bør inkuberes ved stuetemperatur (22-28 °C) i to timer.
Tabel 6
Mængde af assaykomponenter til kompetitivt bindingsassay med hrER, mikrotiterplader
|
Fremstillingstrin
|
Andre end TB-brønde
|
TB-brønde
|
Blindprøve (til varm)
|
|
Komponentvolumen til reaktionsbrønde og tilsætningsrækkefølge
|
Assaybuffer ved stuetemperatur
|
50 μl
|
60 μl
|
90 μl
|
|
Umærket E2, svag binder, ikke-binder, opløsningsmiddel og testkemikalier (*16)
|
10 μl
|
–
|
–
|
|
[3H]-17β-østradiol, der giver en slutkoncentration på 0,5 nM (dvs. 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα-koncentration som fastlagt (jf. punkt 12-13)
|
30 μl
|
30 μl
|
–
|
|
Samlet mængde i hver assaybrønd
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Kvantificeringen af [3H]-17β-østradiol bundet til hrERα efter separation af [3H]-17β-østradiol bundet til hrERα fra fri [3H]-17β-østradiol ved tilsætning af 100 μl iskold DCC-suspension til hver brønd bør herefter udføres som beskrevet i punkt 21-23 i tillæg 3 for mætningsbindingsassayet.
|
|
39.
|
Brønd G10-12 og H10-12 (identificeret som blindprøve (til varm) i tabel 4) repræsenterer dpms i [3H]-mærket østradiol i 10 μl. Delprøven på 10 μl hældes direkte i scintillationsvæsken.
|
Acceptkriterier
Mætningsbindingsassay
|
40.
|
Den specifikke bindingskurve bør nå et plateau i takt med anvendelsen af stigende koncentrationer af [3H]-17β-østradiol, hvilket viser mætningen af hrERα med ligand.
|
|
41.
|
Den specifikke binding ved 0,5 nM [3H]-17β-østradiol bør ligge inden for det acceptable område på 30-50 % af den gennemsnitlige målte samlede radioaktivitet, der tilsættes på tværs af analyseserier. Lejlighedsvise lette udsving uden for dette område er acceptable, men hvis analyseserier konsekvent ligger uden for dette område, eller en særlig analyseserie ligger væsentligt uden for dette område, bør proteinkoncentrationen justeres, og mætningsassayet gentages.
|
|
42.
|
Dataene bør generere et lineært Scatchard-plot.
|
|
43.
|
Den ikke-specifikke binding bør ikke være uforholdsmæssigt stor. Værdien for den ikke-specifikke binding bør typisk være < 35 % af den totale binding. Forholdet kan dog undertiden overstige denne grænse ved måling af en meget lav dpm for den laveste testede koncentration af radioaktivt mærket 17β-østradiol.
|
Kompetitivt bindingsassay
|
44.
|
Stigende koncentrationer af umærket 17β-østradiol bør fortrænge [3H]-17β-østradiol fra receptoren på en måde, der stemmer overens med en kompetitiv binding ét sted.
|
|
45.
|
IC50-værdien for referenceøstrogenet (dvs. 17β-østradiol) bør omtrent svare til den molære koncentration af [3H]-17β-østradiol plus den Kd, der er bestemt ud fra mætningsbindingsassayet.
|
|
46.
|
Den totale specifikke binding bør til stadighed ligge inden for det acceptable område på 40 ± 10 %, når den gennemsnitlige målte koncentration af den samlede radioaktivitet tilsat til hver brønd var 0,5 nM på tværs af analyseserier. Lejlighedsvise lette udsving uden for dette område er acceptable, men hvis analyseserier konsekvent ligger uden for dette område, eller en særlig analyseserie ligger væsentligt uden for området, bør proteinkoncentrationen justeres.
|
|
47.
|
Opløsningsmidlet bør ikke ændre assayets følsomhed eller reproducerbarhed. Resultaterne af kontrolkulturen med opløsningsmiddel (TB-brønde) sammenlignes med bufferkontrollen for at kontrollere, at det anvendte opløsningsmiddel ikke påvirker assaysystemet. Resultaterne af TB og bufferkontrol bør være sammenlignelige, hvis opløsningsmidlet ikke påvirker assayet.
|
|
48.
|
Ikke-binderen bør ikke fortrænge mere end 25 % af [3H]-17β-østradiol fra hrERα ved test op til 10–3 M (OTES) eller 10–4 M (DBP).
|
|
49.
|
Der blev udviklet ydeevnekriterier for referenceøstrogen og to svage bindere (f.eks. norethynodrel, norethindron) ved hjælp af data fra valideringsundersøgelsen for CERI's hrER-bindingsassay (bilag N i reference 2). Der er anført 95 %-konfidensintervaller for den gennemsnitlige ± standardafvigelse (n) af alle kontrolanalyseserier på tværs af fire laboratorier, som deltog i valideringsundersøgelsen. Der blev beregnet 95 %-konfidensintervaller for de kurvetilpassede parametre (dvs. top, bund, Hills kurve og logIC50) for referenceøstrogen og svage bindere og for log10RBA af de svage bindere i forhold til referenceøstrogen. Tabel 1 viser forventede intervaller for kurvetilpassede parametre, der kan benyttes som ydeevnekriterier. I praksis kan området for IC50 variere lidt baseret på den eksperimentelt udledte Kd af receptorpræparatet og den ligandkoncentration, som blev anvendt til assayet.
|
|
50.
|
Der blev ikke udviklet ydeevnekriterier for kurvetilpassede parametre for testkemikalier på grund af den brede vifte af eksisterende potentielle testkemikalier og variation i potentielle affiniteter og udfald (f.eks. fuld kurve, delkurve, ingen kurvetilpasning). Der bør dog anlægges en faglig vurdering ved gennemgang af resultater fra hver analyseserie for et testkemikalie. Der skal benyttes et tilstrækkeligt koncentrationsområde for testkemikaliet for klart at definere toppen (f.eks. 90-100 % af bindingen) af den kompetitive kurve. Variabiliteten mellem replikater ved hver koncentration af testkemikaliet samt mellem de tre ikke-sideløbende analyseserier skal være rimelig og videnskabeligt forsvarlig. Kontroller fra hver analyseserie for et testkemikalie skal tilnærmes resultatmålinger rapporteret for dette CERI-assay og skal stemme overens med tidligere kontroldata fra de respektive laboratorier.
|
ANALYSE AF DATA
Mætningsbindingsassay
|
51.
|
Der måles både samlet og ikke-specifik binding. På grundlag af disse værdier beregnes specifik binding af stigende koncentrationer af [3H]-17β-østradiol under ligevægtsbetingelser ved fratrækning af ikke-specifik binding fra samlet binding. En graf over specifik binding i forhold til [3H]-17β-østradiolkoncentration bør nå et plateau for maksimal specifik binding som tegn på mætning af hrERα med [3H]-17β-østradiol. Endvidere bør analyse af data dokumentere bindingen af [3H]-17β-østradiol til et enkelt bindingssted med høj affinitet. Ikke-specifik, samlet og specifik binding bør vises på en mætningsbindingskurve. Ved yderligere analyse af disse data bør der benyttes en ikke-lineær regressionsanalyse (f.eks. BioSoft, McPherson, 1985; Motulsky, 1995) med en endelig visning af data som et Scatchard-plot.
|
|
52.
|
Dataanalysen skal bestemme Bmax og Kd på grundlag af de totale bindingsdata alene ud fra den antagelse, at ikke-specifik binding er lineær, medmindre der gives en begrundelse for at benytte en anden metode. Der bør desuden benyttes en robust regression ved bestemmelse af den bedste tilpasning, medmindre der gives en begrundelse. Den valgte metode til robust regression skal anføres. Der bør altid korrigeres for liganddepletering (f.eks. ved hjælp af Swillens metode, 1995) ved bestemmelse af Bmax og Kd ud fra mætningsbindingsdata.
|
Kompetitivt bindingsassay
|
53.
|
Den kompetitive bindingskurve plottes som specifik [3H]-17β-østradiolbinding mod konkurrentens koncentration (log10-enheder). Den koncentration af testkemikaliet, der inhiberer 50 % af den maksimale specifikke [3H]-17β-østradiolbinding, er IC50-værdien.
|
|
54.
|
Skøn over log(IC50)-værdier for de positive kontroller (f.eks. referenceøstrogen og svag binder) bestemmes ved anvendelse af en passende ikke-lineær kurvetilpasningssoftware, som passer til en Hill-ligning med fire parametre (f.eks. BioSoft, McPherson, 1985; Motulsky, 1995). Top, bund, hældning og log(IC50) skal normalt være frie ved tilpasning af disse kurver. Der bør benyttes robust regression ved bestemmelse af den bedste tilpasning, medmindre der gives en begrundelse. Der bør ikke korrigeres for liganddepletion. Efter den indledende analyse skal hver bindingskurve revideres for at sikre, at den passer til modellen. Den relative affinitet (RBA) for den svage binder beregnes som en procentdel af log (IC50) for den svage binder i forhold til log (IC50) for 17β-østradiol. Resultaterne fra de positive kontroller og ikke-binderkontrollen bør vurderes ved hjælp af målinger af assayets ydeevne i punkt 44-49 i dette tillæg 3.
|
|
55.
|
Data for alle testkemikalier analyseres gennem en trinvis tilgang for at sikre, at dataene analyseres indgående, og at hver kompetitiv bindingskurve klassificeres korrekt. Det anbefales, at hver analyseserie for et testkemikalie indledningsvis gennemgår en standardiseret dataanalyse, som er identisk med den, der benyttes for referenceøstrogen og kontroller for svag binder (jf. punkt 54 i dette tillæg 3). Når analysen er færdig, foretages en teknisk gennemgang af kurvetilpasningsparametrene samt en visuel gennemgang af, hvor godt dataene passer til den genererede kompetitive bindingskurve for hver analyseserie. Under denne tekniske gennemgang er observationer af et koncentrationsafhængigt fald i den procentvise specifikt bundne [3H]-17β-østradiol, lav variabilitet mellem de tekniske replikater ved hver testkemikaliekoncentration og konsistens i tilpassede parametre mellem de tre analyseserier et godt tegn på, at assayet og dataanalyserne blev behørigt gennemført.
|
Fortolkning af data
|
56.
|
Forudsat at alle acceptkriterier er opfyldt, anses et testkemikalie for at være en binder for hrERα, hvis der kan tilpasses en bindingskurve, og det laveste punkt på responskurven inden for dataområdet er under 50 % (figur 1).
|
|
57.
|
Såfremt alle acceptkriterier er opfyldt, anses et testkemikalie for at være en ikke-binder for hrERα, hvis:
|
—
|
der kan tilpasses en bindingskurve, og det laveste punkt på den tilpassede responskurve inden for dataområdet ligger over 75 %, eller
|
|
—
|
der ikke kan tilpasses en bindingskurve, og den laveste ikke-udjævnede, gennemsnitlige, procentvise binding blandt koncentrationsgrupperne i dataene ligger over 75 %.
|
|
|
58.
|
Testkemikalierne anses for usikre, hvis ingen af de ovennævnte betingelser er opfyldt (f.eks. det laveste punkt på den tilpassede responskurve ligger mellem 76 og 51 %).
|
Tabel 7
Kriterier for klassificering på grundlag af et testkemikalies kompetitive bindingskurve
|
Klassificering
|
Kriterier
|
|
Bindera
|
En bindingskurve kan tilpasses.
Det laveste punkt på responskurven inden for dataområdet er mindre end 50 %.
|
|
Ikke-binderb
|
Hvis en bindingskurve kan tilpasses,
er det laveste punkt på den tilpassede responskurve inden for dataområdet over 75 %.
Hvis en bindingskurve ikke kan tilpasses,
er den laveste ikke-udjævnede, gennemsnitlige, procentvise binding blandt koncentrationsgrupperne i dataene over 75 %.
|
|
Usikkerc
|
Enhver testbar analyseserie, som hverken er en binder eller en ikke-binder
(f.eks. Det laveste punkt på den tilpassede responskurve er mellem 76 % og 51 %).
|
Figur 1
Eksempler på klassificering af testkemikalier ved anvendelse af kompetitiv bindingskurve.
|
59.
|
Flere analyseserier for et testkemikalie, der gennemføres i et laboratorium, kombineres ved at tildele hver analyseserie en numerisk værdi og beregne gennemsnittet på tværs af analyseserierne som vist i tabel 8. Resultater for de kombinerede analyseserier i hvert laboratorium sammenlignes med den forventede klassificering for hvert testkemikalie.
|
Tabel 8
Metode til klassificering af testkemikalier ved anvendelse af flere analyseserier i et laboratorium
|
Tildeling af værdi til hver analyseserie:
|
|
Klassificering
|
Numerisk værdi
|
|
Binder
|
2
|
|
Usikker
|
1
|
|
Ikke-binder
|
0
|
|
Klassificering af gennemsnitlig numerisk værdi på tværs af analyseserier:
|
|
Klassificering
|
Numerisk værdi
|
|
Binder
|
Gennemsnit ≥ 1,5
|
|
Usikker
|
0,5 ≤ Gennemsnit < 1,5
|
|
Ikke-binder
|
Gennemsnit < 0,5
|
TESTRAPPORT
|
60.
|
Jf. punkt 24 i "hrER-BINDINGSASSAYETS KOMPONENTER" i denne forsøgsmetode.
|
Tillæg 3.1
TERMINOLOGI
[3H]E2
: 17β-østradiol radioaktivt mærket med tritium
DCC
: dextranovertrukket trækul
E2
: umærket 17β-østradiol (inert)
Assaybuffer
: 10 mM Tris-HCl, pH 7,4, indeholdende 1 mM EDTA, 1 mM EGTA, 1 mM NaVO3, 10 % glycerol, 0,2 mM leupeptin, 1 mM dithiothreitol og 10 mg/ml bovint serumalbumin
hrERα
: human rekombinant østrogenreceptor alfa (ligandbindingsområde)
Replikat
: en af flere brønde, der har samme indhold ved de samme koncentrationer, og som analyseres sideløbende inden for én analyseserie. I denne protokol testes hver koncentration af testkemikaliet in triplo, dvs. at der er tre replikater, som analyseres samtidig ved hver koncentration af testkemikaliet.
Analyseserie
: et fuldstændigt sæt assaybrønde på mikrotiterplade, der kører sideløbende, og som giver alle nødvendige oplysninger til at karakterisere bindingen af et testkemikalie til hrERα (nemlig samlet [3H]-17β-østradiol tilsat til assaybrønden, maksimal binding af [3H]-17β-østradiol til hrERα, ikke-specifik binding og total binding ved forskellige koncentrationer af testkemikaliet). En analyseserie kan bestå af så lidt som én assaybrønd (dvs. replikat) pr. koncentration, men eftersom denne protokol kræver tredobbelt analyse, består én analyseserie af tre assaybrønde pr. koncentration. Endvidere kræver denne protokol tre uafhængige (dvs. ikke-sideløbende) analyseserier pr. kemikalie.
Tillæg 3.2
KOMPETITIVT BINDINGSASSAY: BRØNDLAYOUT
|
Plade
|
Placering
|
Replikat
|
Brøndtype
|
Brøndkode
|
Koncentrations-kode
|
Konkurrent Begyndelses koncentration (M)
|
hrER-stamkultur (μl)
|
Buffervolumen (μl)
|
Sporstof (varm E2) Volumen (μl)
|
Volumen fra fortyndingsplade (μl)
|
Slutvolumen (μl)
|
Konkurrent Slutkon centration (M)
|
|
S
|
A1
|
1
|
Blindprøve
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Blindprøve
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Blindprøve
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
kold E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
B2
|
2
|
kold E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
B3
|
3
|
kold E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
C1
|
1
|
kold E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
C2
|
2
|
kold E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
C3
|
3
|
kold E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
D1
|
1
|
kold E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
D2
|
2
|
kold E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
D3
|
3
|
kold E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
E1
|
1
|
kold E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
E2
|
2
|
kold E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
E3
|
3
|
kold E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
F1
|
1
|
kold E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
F2
|
2
|
kold E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
F3
|
3
|
kold E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
G1
|
1
|
kold E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
G2
|
2
|
kold E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
G3
|
3
|
kold E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
H1
|
1
|
kold E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
H2
|
2
|
kold E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
H3
|
3
|
kold E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
A4
|
1
|
norethynodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
A5
|
2
|
norethynodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
A6
|
3
|
norethynodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B4
|
1
|
norethynodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
B5
|
2
|
norethynodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
B6
|
3
|
norethynodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C4
|
1
|
norethynodrel
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
C5
|
2
|
norethynodrel
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
C6
|
3
|
norethynodrel
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
D4
|
1
|
norethynodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D5
|
2
|
norethynodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D6
|
3
|
norethynodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
E4
|
1
|
norethynodrel
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
E5
|
2
|
norethynodrel
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
E6
|
3
|
norethynodrel
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
F4
|
1
|
norethynodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F5
|
2
|
norethynodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F6
|
3
|
norethynodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
G4
|
1
|
norethynodrel
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
G5
|
2
|
norethynodrel
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
G6
|
3
|
norethynodrel
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
H4
|
1
|
norethynodrel
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
H5
|
2
|
norethynodrel
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
H6
|
3
|
norethynodrel
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
A10
|
1
|
total binding
|
TB
|
TB1
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
A11
|
2
|
total binding
|
TB
|
TB2
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
A12
|
3
|
total binding
|
TB
|
TB3
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
B10
|
4
|
total binding
|
TB
|
TB4
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
B11
|
5
|
total binding
|
TB
|
TB5
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
B12
|
6
|
total binding
|
TB
|
TB6
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
C10
|
1
|
kold E2 (høj)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
C11
|
2
|
kold E2 (høj)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
C12
|
3
|
kold E2 (høj)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D10
|
4
|
kold E2 (høj)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D11
|
5
|
kold E2 (høj)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D12
|
6
|
kold E2 (høj)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E10
|
1
|
Bufferkontrol
|
BC
|
BC1
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
E11
|
2
|
Bufferkontrol
|
BC
|
BC2
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
E12
|
3
|
Bufferkontrol
|
BC
|
BC3
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
F10
|
4
|
Bufferkontrol
|
BC
|
BC4
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
F11
|
5
|
Bufferkontrol
|
BC
|
BC5
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
F12
|
6
|
Bufferkontrol
|
BC
|
BC6
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
G10 (*17)
|
1
|
Blindprøve (til varm)
|
Varm
|
H1
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
G11 (*17)
|
2
|
Blindprøve (til varm)
|
Varm
|
H2
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
G12 (*17)
|
3
|
Blindprøve (til varm)
|
Varm
|
H3
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
H10 (*17)
|
4
|
Blindprøve (til varm)
|
Varm
|
H4
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
H11 (*17)
|
5
|
Blindprøve (til varm)
|
Varm
|
H5
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
H12
|
6
|
Blindprøve (til varm)
|
Varm
|
H6
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
P1
|
A1
|
1
|
Ukendt 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A2
|
2
|
Ukendt 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A3
|
3
|
Ukendt 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B1
|
1
|
Ukendt 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B2
|
2
|
Ukendt 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B3
|
3
|
Ukendt 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C1
|
1
|
Ukendt 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C2
|
2
|
Ukendt 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C3
|
3
|
Ukendt 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D1
|
1
|
Ukendt 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D2
|
2
|
Ukendt 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D3
|
3
|
Ukendt 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E1
|
1
|
Ukendt 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E2
|
2
|
Ukendt 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E3
|
3
|
Ukendt 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F1
|
1
|
Ukendt 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F2
|
2
|
Ukendt 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F3
|
3
|
Ukendt 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G1
|
1
|
Ukendt 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G2
|
2
|
Ukendt 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G3
|
3
|
Ukendt 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H1
|
1
|
Ukendt 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H2
|
2
|
Ukendt 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H3
|
3
|
Ukendt 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A4
|
1
|
Ukendt 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A5
|
2
|
Ukendt 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A6
|
3
|
Ukendt 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B4
|
1
|
Ukendt 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B5
|
2
|
Ukendt 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B6
|
3
|
Ukendt 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C4
|
1
|
Ukendt 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C5
|
2
|
Ukendt 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C6
|
3
|
Ukendt 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D4
|
1
|
Ukendt 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D5
|
2
|
Ukendt 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D6
|
3
|
Ukendt 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E4
|
1
|
Ukendt 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E5
|
2
|
Ukendt 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E6
|
3
|
Ukendt 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F4
|
1
|
Ukendt 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F5
|
2
|
Ukendt 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F6
|
3
|
Ukendt 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G4
|
1
|
Ukendt 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G5
|
2
|
Ukendt 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G6
|
3
|
Ukendt 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H4
|
1
|
Ukendt 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H5
|
2
|
Ukendt 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H6
|
3
|
Ukendt 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A7
|
1
|
Ukendt 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A8
|
2
|
Ukendt 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A9
|
3
|
Ukendt 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B7
|
1
|
Ukendt 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B8
|
2
|
Ukendt 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B9
|
3
|
Ukendt 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C7
|
1
|
Ukendt 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C8
|
2
|
Ukendt 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C9
|
3
|
Ukendt 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D7
|
1
|
Ukendt 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D8
|
2
|
Ukendt 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D9
|
3
|
Ukendt 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E7
|
1
|
Ukendt 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E8
|
2
|
Ukendt 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E9
|
3
|
Ukendt 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F7
|
1
|
Ukendt 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F8
|
2
|
Ukendt 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F9
|
3
|
Ukendt 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G7
|
1
|
Ukendt 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G8
|
2
|
Ukendt 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G9
|
3
|
Ukendt 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H7
|
1
|
Ukendt 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H8
|
2
|
Ukendt 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H9
|
3
|
Ukendt 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A10
|
1
|
Kontrol E2 (maks.)
|
S
|
E2maks1
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A11
|
2
|
Kontrol E2 (maks.)
|
S
|
E2maks2
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A12
|
3
|
Kontrol E2 (maks.)
|
S
|
E2maks3
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
B10
|
1
|
Kontrol E2 (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Kontrol E2 (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Kontrol E2 (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Kontrol NE (maks.)
|
S
|
Nemax1
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4.5
|
|
P1
|
C11
|
2
|
Kontrol NE (maks.)
|
S
|
Nemax2
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4.5
|
|
P1
|
C12
|
3
|
Kontrol NE (maks.)
|
S
|
Nemax3
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4.5
|
|
P1
|
D10
|
1
|
Kontrol NE (IC50)
|
S
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Kontrol NE (IC50)
|
S
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Kontrol NE (IC50)
|
S
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
kold E2 (høj)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E11
|
2
|
kold E2 (høj)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E12
|
3
|
kold E2 (høj)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F10
|
4
|
kold E2 (høj)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F11
|
5
|
kold E2 (høj)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F12
|
6
|
kold E2 (høj)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
G10
|
1
|
total binding
|
TB
|
TB1
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
G11
|
2
|
total binding
|
TB
|
TB2
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
G12
|
3
|
total binding
|
TB
|
TB3
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
H10
|
4
|
total binding
|
TB
|
TB4
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
H11
|
5
|
total binding
|
TB
|
TB5
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
H12
|
6
|
total binding
|
TB
|
TB6
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
Tillæg 4
OVERVEJELSER OVER ANALYSEN AF DATA FRA DET KOMPETITIVE HRER-BINDINGSASSAY
|
1.
|
Det kompetitive hrERα-bindingsassay måler bindingen af en enkelt koncentration af [3H]-17β-østradiol under tilstedeværelse af stigende koncentrationer af et testkemikalie. Den kompetitive bindingskurve plottes som specifik [3H]-17β-østradiolbinding mod konkurrentens koncentration (log10-enheder). Den koncentration af testkemikaliet, der inhiberer 50 % af den maksimale specifikke [3H]-17β-østradiolbinding, er IC50.
|
Dataanalyse for referenceøstrogen og svag binder (1)
|
2.
|
Data fra kontrolanalyseserierne omdannes (dvs. procent [3H]-17β-østradiol-specifik binding og log-koncentrationen af kontrolkemikaliet) til videre analyse. Skøn over log(IC50)-værdier for de positive kontroller (f.eks. referenceøstrogen og svag binder) bestemmes ved anvendelse af en passende ikke-lineær kurvetilpasningssoftware, som passer til en Hill-ligning med fire parametre (f.eks. BioSoft, GraphPad Prism) (2). Top, bund, hældning og log( IC50) kan normalt være frie ved tilpasning af disse kurver. Der bør benyttes robust regression ved bestemmelse af den bedste tilpasning, medmindre der gives en begrundelse. Den valgte metode til robust regression skal anføres. Det var ikke nødvendigt at korrigere for liganddepletering for FW- eller CERI's hrER-assays, men kan om nødvendigt overvejes. Efter den indledende analyse skal hver bindingskurve revideres for at sikre, at den passer til modellen. Den relative affinitet (RBA) for den svage binder kan beregnes som en procentdel af log (IC50) for den svage binder i forhold til log (IC50) for 17β-østradiol. Resultater for de positive kontroller og ikke-binderkontrollen bør vurderes ved anvendelse af mål for assayets ydeevne og acceptkriterier som beskrevet i denne forsøgsmetode (punkt 20), tillæg 2 (FW-assay, punkt 41-51) og tillæg 3 (CERI-assay, punkt 41-51). Eksempler på tre analyseserier for referenceøstrogen og svag binder er vist i figur 1.
|
Figur 1
Eksempler på de kompetitive bindingskurver for referenceøstrogen og den svage binder, der fungerer som kontrol

Dataanalyse for testkemikalier
|
3.
|
Data for alle testkemikalier analyseres gennem en trinvis tilgang for at sikre, at dataene analyseres indgående, og at hver kompetitiv bindingskurve klassificeres korrekt. Hver analyseserie for et testkemikalie bør indledningsvis gennemgå en standardiseret dataanalyse, som er identisk med den, der benyttes for referenceøstrogen og kontroller for svag binder. Når analysen er færdig, foretages en teknisk gennemgang af kurvetilpasningsparametrene samt en visuel gennemgang af, hvor godt dataene passer til den genererede kompetitive bindingskurve for hver analyseserie. Under denne tekniske gennemgang er observationer af et koncentrationsafhængigt fald i den procentvise specifikt bundne [3H]-17β-østradiol, lav variabilitet mellem de tekniske replikater ved hver kemisk koncentration og konsistens i tilpassede parametre mellem de tre analyseserier et godt tegn på, at assayet og dataanalyserne blev gennemført korrekt. Der bør anlægges en faglig vurdering ved gennemgang af resultater fra hver analyseserie for et testkemikalie, og de data, der benyttes til at klassificere hvert testkemikalie som binder eller ikke-binder skal være videnskabeligt forsvarlige.
|
|
4.
|
Undertiden kan der være eksempler på data, som kræver supplerende opmærksomhed med henblik på at analysere og fortolke hrER-bindingsdataene korrekt. Tidligere undersøgelser havde vist tilfælde, hvor det kan være kompliceret at analysere og fortolke kompetitive receptorbindingsdata på grund af en stigning i den procentvise specifikke binding, ved testning af kemikalier ved de højeste koncentrationer (figur 2). Dette er et velkendt problem, som man er stødt på ved anvendelse af protokoller til en række kompetitive receptorbindingsassays (3). I disse tilfælde observeres en koncentrationsafhængig respons ved lavere koncentrationer, men i takt med at koncentrationen af testkemikaliet nærmer sig opløselighedsgrænsen, falder fortrængningen af [3H]17β-østradiol ikke længere. I disse tilfælde viser data for de højere koncentrationer, at assayets biologiske grænse er nået. F.eks. forbindes dette fænomen mange gange med kemisk uopløselighed og bundfald ved høje koncentrationer eller kan også afspejle overskridelse af det dextranovertrukne trækuls kapacitet til at fange ubundet radioaktivt mærket ligand under adskillelsesproceduren ved de højeste kemiske koncentrationer. Hvis sådanne datapunkter ignoreres, når data om kompetitiv binding tilpasses en sigmoidal kurve, kan det undertiden føre til en forkert klassificering af et testkemikalies ER-bindingspotentiale (figur 2). For at undgå dette omfatter protokollen for FW- og CERI-hrER-bindingsassays en mulighed for at se bort fra datapunkter i de analyser, hvor replikatgennemsnittet af den procentvise specifikt bundne [3H]17β-østradiol ligger 10 % eller mere over den gennemsnitsværdi, som blev observeret ved en lavere koncentration (dvs. dette omtales almindeligvis som 10 %-reglen). Denne regel kan kun benyttes én gang for en given kurve, og der skal være data tilbage for mindst seks koncentrationer, således at kurven kan klassificeres korrekt.
|
Figur 2
Eksempler, kompetitive bindingskurver med og uden anvendelse af 10 %-reglen

|
5.
|
Passende anvendelse af 10 %-reglen til at korrigere disse kurver bør nøje overvejes og forbeholdes de tilfælde, hvor der er stærk indikation for en hrER-binder. Under udførelsen af forsøg til valideringsundersøgelsen af FW-hrER-bindingsassayet blev det observeret, at 10 %-reglen undertiden havde en utilsigtet og uforudset konsekvens. Kemikalier, som ikke interagerede med receptoren (dvs. reelle ikke-bindere), udviste ofte variabilitet omkring 100 % radioligandbinding, som var større end 10 % på tværs af det testede koncentrationsområde. Hvis den laveste værdi skulle være ved en lav koncentration, kan dataene fra alle højere koncentrationer potentielt slettes fra analysen ved anvendelse af 10 %-reglen, selv om disse koncentrationer kunne være nyttige til at fastslå, at kemikaliet er en ikke-binder. Figur 3 viser eksempler, hvor anvendelsen af 10 %-reglen ikke er hensigtsmæssig
|
Figur 3
Eksempler, data om kompetitiv binding, hvor anvendelsen af 10 %-reglen ikke er hensigtsmæssig


Henvisninger
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Motulsky H. og Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, s. 187-191. www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71
IN VITRO HUDSENSIBILISERINGSASSAYS AF DEN CENTRALE BEGIVENHED AKTIVERING AF DENDRITISKE CELLER I ADVERSE OUTCOME PATHWAY (AOP) FOR HUDSENSIBILISERING
GENEREL INDLEDNING
Forsøgsmetode baseret på den centrale begivenhed aktivering af dendritiske celler
|
1.
|
Et hudsensibiliserende stof er et stof, der inducerer en allergisk reaktion ved hudkontakt som defineret ved De Forenede Nationers globalt harmoniserede system til klassificering og mærkning af kemikalier (UN GHS) (1) og Den Europæiske Unions (EU) forordning nr. 1272/2008 om klassificering, mærkning og emballering af stoffer og blandinger (CLP-forordningen) (100). Der hersker almindelig enighed om de centrale biologiske begivenheder, der ligger til grund for hudsensibilisering. Den nuværende viden om kemiske og biologiske mekanismer i forbindelse med hudsensibilisering er sammenfattet i en Adverse Outcome Pathway (AOP) under OECD’s AOP-program (2) fra den molekylære udløsende begivenhed over de mellemliggende begivenheder til den skadelige virkning, nemlig allergisk kontaktdermatitis. I dette tilfælde er den molekylære udløsende begivenhed (dvs. den første centrale begivenhed) den kovalente binding af elektrofile stoffer til nukleofile centre i hudproteiner. Den anden centrale begivenhed i denne AOP finder sted i keratinocytterne og omfatter inflammatoriske reaktioner samt ændringer i genekspression i tilknytning til specifikke cellesignalveje, f.eks. antioxidant-/elektrofilresponselement (ARE)- afhængige veje. Den tredje centrale begivenhed er aktivering af dendritiske celler, som typisk vurderes ved ekspression af bestemte celleoverflademarkører, kemokiner og cytokiner. Den fjerde centrale begivenhed er T-celleaktivering, som vurderes indirekte ved Local Lymph Node Assay (assay på lokale lymfeknuder – LLNA) i mus (3).
|
|
2.
|
Denne forsøgsmetode svarer til OECD-testvejledning (TG) 442E (2017). Heri beskrives in vitro-assays vedrørende de mekanismer, der er beskrevet under den centrale begivenhed aktivering af dendritiske celler i hudsensibiliserings-AOP’en (2). Denne forsøgsmetode omfatter test, der anvendes til at underbygge differentieringen mellem hudsensibiliserende og ikke-sensibiliserende stoffer i henhold til UN GHS og CLP.
|
|
De test, der er beskrevet i denne forsøgsmetode, er:
|
—
|
Human Cell Line Activation-test (h-CLAT)
|
|
—
|
U937 cell line activation-test (U-SENS™)
|
|
—
|
Interleukin-8 Reporter Gene Assay (IL-8 Luc assay).
|
|
|
|
3.
|
De test, der indgår i denne forsøgsmetode og den tilsvarende OECD-testvejledning, kan afvige fra den fremgangsmåde, der anvendes til generering af data, og de målte resultater, men de kan anvendes uden forskel til at opfylde landenes behov for testresultater vedrørende den centrale begivenhed aktivering af dendritiske celler i hudsensibiliserings-AOP’en, samtidig med at de drager fordel af OECD-aftalen om gensidig godkendelse af data.
|
Baggrund og principper for de test, der indgår i forsøgsmetoden baseret på den centrale begivenhed
|
4.
|
Vurderingen af hudsensibilisering har typisk omfattet brug af laboratoriedyr. Med de klassiske metoder, der anvender marsvin, Magnusson og Kligmans Guinea Pig Maximisation Test (GPMT) og Buehler-testen (TM B.6 (4)) vurderer man både induktions- og udløsningsfasen ved hudsensibilisering. Musetest, LLNA (TM B.42) (3) og dens to ikke-radioaktive ændringer, LLNA: DA (TM B.50 (5)) og LLNA: BrdU-ELISA (TM B.51 (6)) vurderer alle udelukkende induktionsresponsen og er også blevet accepteret, eftersom de indebærer en fordel i forhold til marsvineforsøg, hvad angår dyrevelfærd og en objektiv måling af latenstiden for hudsensibilisering.
|
|
5.
|
På det seneste er der blevet indført mekanistisk baserede in chemico- og in vitro-forsøgsmetoder vedrørende den første centrale begivenhed (TM B.59; direkte peptidreaktivitetsassay (DPRA) (7)) og den anden centrale begivenhed (TM B.60;ARE-Nrf2-luciferase-forsøgsmetoden (8)) i hudsensibiliserings-AOP’en som et bidrag til evalueringen af kemikaliers potentielle hudsensibiliseringsfare.
|
|
6.
|
De test, der er beskrevet i denne forsøgsmetode, måler enten ændringen i ekspressionen af celleoverflademarkør(er), der er knyttet til processen med aktivering af monocyter og dendritiske celler efter eksponering for allergifremkaldende stoffer (f.eks. CD54, CD86), eller ændringer i ekspressionen af IL-8, et cytokin knyttet til aktiveringen af dendritiske celler. Hudsensibiliserende stoffer inducerer angiveligt ekspression af cellemembranmarkører som f.eks. CD40, CD54, CD80, CD83 og CD86 og inducerer desuden proinflammatoriske cytokiner som f.eks. IL-1β og TNF-α og adskillige kemokiner, herunder IL-8 (CXCL8) og CCL3 (9) (10) (11) (12), knyttet til aktivering af dendritiske celler (2).
|
|
7.
|
Da aktivering af dendritiske celler imidlertid kun udgør en central begivenhed i hudsensibiliserings-AOP’en (2) (13), er den information, der genereres med test, som måler markører for aktivering af dendritiske celler alene, muligvis ikke tilstrækkelig til at drage konklusioner om, hvorvidt kemiske stoffer har et hudsensibiliseringspotentiale eller ej. Derfor skal de data, der genereres med de test, som er beskrevet i denne forsøgsmetode, støtte differentieringen mellem hudsensibiliserende stoffer (dvs. UN GHS-/CLP-kategori 1) og ikke-sensibiliserende stoffer, når de anvendes i en integreret tilgang til testning og vurdering (IATA) sammen med andre relevante supplerende oplysninger fra f.eks. in vitro-assays, der vedrører andre centrale begivenheder i hudsensibiliserings-AOP’en, samt andre metoder end test, herunder analogislutning fra tilsvarende kemikalier (13). Eksempler på anvendelse af data genereret med disse test i definerede tilgange, dvs. tilgange, som er standardiserede både i forhold til den række af informationskilder, der anvendes, og i forhold til den procedure, der anvendes på dataene med henblik på at udlede prognoser, er blevet offentliggjort (13) og kan anvendes som nyttige elementer i IATA.
|
|
8.
|
De test, der er beskrevet i denne forsøgsmetode, kan ikke anvendes alene, hverken til at kategorisere hudsensibiliserende stoffer i underkategorierne 1A og 1B som defineret i UN GHS-/CLP, af myndigheder, der anvender disse to valgfrie underkategorier, eller til at forudsige styrken i forbindelse med beslutninger om sikkerhedsvurdering. Afhængigt af den juridiske ramme kan de positive udfald, som disse metoder genererer, imidlertid anvendes alene til at klassificere et kemikalie i UN GHS-/CLP-kategori 1.
|
|
9.
|
Betegnelsen »testkemikalie« anvendes i denne forsøgsmetode om det, der testes (101), og vedrører ikke anvendeligheden af test til testning af stoffer med én bestanddel, stoffer med flere bestanddele og/eller blandinger. I øjeblikket foreligger der begrænsede oplysninger om anvendeligheden af test på stoffer med flere bestanddele/blandinger (14) (15). Testene kan ikke desto mindre teknisk set benyttes til stoffer med flere bestanddele og blandinger. Før denne forsøgsmetode anvendes til test af en blanding til generering af data med et lovmæssigt formål, bør det imidlertid overvejes, om den vil give resultater, som er egnede til formålet, og i givet fald hvorfor (102). Sådanne overvejelser er ikke nødvendige, når der foreligger et myndighedskrav om testning af blandingen. Ved test af stoffer med flere bestanddele eller blandinger bør der desuden tages hensyn til cytotoksiske bestanddeles eventuelle interferens med de observerede responser.
|
LITTERATUR
|
(1)
|
United Nations (UN) (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1 Tilgængelig på: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment, No. 168. Tilgængelig på: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Kapitel B.42 i dette bilag: Assay på lokale lymfeknuder.
|
|
(4)
|
Kapitel B.6 i dette bilag: Hudsensibilisering.
|
|
(5)
|
Kapitel B.50 i dette bilag: Hudsensibilisering: Assay på lokale lymfeknuder: DA.
|
|
(6)
|
Kapitel B.51 i dette bilag: Hudsensibilisering: Assay på lokale lymfeknuder: BrdU-ELISA.
|
|
(7)
|
Kapitel B.59 i dette bilag: In chemico-hudsensibilisering: direkte peptidreaktivitetsassay (DPRA).
|
|
(8)
|
Kapitel B.60 i dette bilag: In vitro-hudsensibilisering: ARE-Nrf2-luciferase-forsøgsmetoden.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL og Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H og Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y og Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Series on Testing and Assessment, No. 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Tillæg 1
IN VITRO-HUDSENSIBILISERING: HUMAN CELL LINE ACTIVATION TEST (H-CLAT)
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER
|
1.
|
h-CLAT måler ændringer i ekspressionen af celleoverflademarkører i tilknytning til processen med aktivering af monocyter og dendritiske celler (DC) (dvs. CD86 og CD54) i human monocytisk leukæmi-cellelinjen THP-1 efter eksponering for allergifremkaldende stoffer (1) (2). De målte ekspressionsniveauer for celleoverflademarkørerne CD86 og CD54 anvendes dernæst til støtte for differentieringen mellem hudsensibiliserende stoffer og ikke-sensibiliserende stoffer.
|
|
2.
|
h-CLAT er blevet evalueret i en valideringsundersøgelse koordineret af Den Europæiske Unions Referencelaboratorium for Alternativer til Dyreforsøg (EURL ECVAM), og EURL ECVAM's videnskabelige rådgivende udvalg (ESAC) foretog efterfølgende uafhængige peer reviews. På grundlag af al tilgængelig dokumentation og input fra myndigheder og interessenter anbefalede EURL ECVAM (3), at h-CLAT anvendes som en del af en IATA til støtte for differentieringen mellem hudsensibiliserende stoffer og ikke-sensibiliserende stoffer med henblik på fareklassificering og mærkning. Litteraturen indeholder eksempler på anvendelse af h-CLAT-data i kombination med andre oplysninger (4) (5) (6) (7) (8) (9) (10) (11).
|
|
3.
|
Det viste sig, at h-CLAT kunne overføres til laboratorier med erfaring inden for celledyrkningsteknikker og flowcytometrianalyse. Graden af reproducerbarhed i de forudsigelser, der kan forventes fra testen, ligger i størrelsesordenen 80 % inden for og mellem laboratorier (3) (12). De resultater, der er blevet genereret i valideringsundersøgelsen (13) og andre offentliggjorte undersøgelser (14), indikerer generelt, at nøjagtigheden af differentieringen mellem hudsensibiliserende stoffer (dvs. UN GHS-/CLP-kategori 1) og ikke-sensibiliserende stoffer i forhold til LLNA-resultaterne er 85 % (N=142) med en følsomhed på 93 % (94/101) og en specificitet på 66 % (27/41) (baseret på en ny analyse foretaget af EURL ECVAM (12), hvor alle de eksisterende data er blevet taget i betragtning, og der ikke er blevet taget hensyn til de negative resultater for kemikalier med en log Kow på mere end 3,5 som beskrevet i punkt 4). Det er mere sandsynligt, at falsk negative forudsigelser i forbindelse med h-CLAT vil vedrøre kemikalier med lav til moderat hudsensibiliserende styrke (dvs. UN GHS-/CLP - underkategori 1B) end kemikalier med en høj hudsensibiliserende styrke (dvs. UN GHS-/CLP-underkategori 1A) (4) (13) (15). Samlet set viser denne information nytteværdien af h-CLAT som bidrag til identifikation af hudsensibiliseringsfarer. De nøjagtige værdier, der her angives for h-CLAT som en selvstændig test, er imidlertid kun vejledende, da forsøgsmetoden bør betragtes i kombination med andre informationskilder i forbindelse med en IATA og i overensstemmelse med bestemmelserne i punkt 7 og 8 i den generelle indledning. I forbindelse med vurderingen af metoder til test af hudsensibilisering uden brug af dyr bør man desuden holde sig for øje, at LLNA-forsøg samt andre dyreforsøg måske ikke fuldt ud afspejler situationen hos mennesker.
|
|
4.
|
På grundlag af de data, der er tilgængelige i øjeblikket, har h-CLAT vist sig at være relevant for testkemikalier, der omfatter en række forskellige organiske funktionelle grupper, reaktionsmekanismer, hudsensibiliseringsstyrker (som fastlagt ved in vivo-undersøgelser) og fysisk-kemiske egenskaber (3) (14) (15). h-CLAT-metoden kan anvendes til at teste kemikalier, der er opløselige, eller som danner en stabil dispergering (dvs. en kolloid opløsning eller en suspension, i hvilken testkemikaliet ikke bundfældes eller separeres fra opløsningsmidlet/bærestoffet i forskellige faser) i et passende opløsningsmiddel/bærestof (jf. punkt 14). Testkemikalier med en log Kow på over 3,5 har tendens til at give falsk negative resultater (14). Der bør derfor ikke tages hensyn til negative resultater i forbindelse med testkemikalier med en log Kow på over 3,5. De positive resultater, der opnås med testkemikalier med en log Kow på over 3,5, kan imidlertid fortsat anvendes til støtte for påvisningen af testkemikaliet som et hudsensibiliserende stof. På grund af den anvendte cellelinjes begrænsede metaboliske kapacitet (16) og på grund af testbetingelserne kan pro-haptener (dvs. stoffer, der kræver enzymatisk aktivering f.eks. via P450-enzymer) og præ-haptener (dvs. stoffer, der aktiveres ved oxidering), især med en langsom oxideringsrate, også give negative resultater i h-CLAT (15). Fluorescerende testkemikalier kan vurderes ved hjælp af h-CLAT (17), men stærkt fluorescerende stoffer, der udsender den samme bølgelængde som fluoresceinisothiocyanat (FITC) eller propidiumiodid (PI), vil interferere med den flowcytometriske detektion og kan derfor ikke evalueres korrekt ved hjælp af FITC-konjugerede antistoffer eller PI. I sådanne tilfælde kan henholdsvis andre flourochrom-mærkede antistoffer eller andre cytotoksicitetsmarkører anvendes, når blot det kan påvises, at de giver resultater svarende til FITC-mærkede antistoffer (jf. punkt 24) eller PI (jf. punkt 18) f.eks. via testning af kompetencestofferne i tillæg 1-2. I lyset af ovenstående bør negative resultater fortolkes i lyset af de anførte begrænsninger og sammen med andre informationskilder inden for rammerne af IATA. I de tilfælde, hvor der foreligger beviser for, at h-CLAT-metoden ikke kan anvendes på andre specifikke kategorier af testkemikalier, bør denne metode ikke anvendes til disse specifikke kategorier.
|
|
5.
|
Som beskrevet ovenfor understøtter h-CLAT differentieringen mellem hudsensibiliserende og ikke-sensibiliserende stoffer. Den kan imidlertid også potentielt bidrage til vurderingen af sensibiliseringsstyrken (4) (5) (9), når den bruges i integrerede tilgange såsom IATA. Der er dog behov for yderligere arbejde, fortrinsvis baseret på humane data, for at fastslå, hvordan h-CLAT-resultaterne eventuelt kan anvendes til styrkevurderingen.
|
|
6.
|
Definitioner findes i tillæg 1.1.
|
PRINCIP FOR TESTEN
|
7.
|
h-CLAT-metoden er et in vitro-assay, der måler ændringerne af ekspressionen af celleoverflademarkører (dvs. CD86 og CD54) på en human monocytisk leukæmi-cellelinje, THP-1 celler, efter 24 timers eksponering for testkemikaliet. Disse overflademolekyler er typiske markører for monocytisk THP-1-aktivering og kan efterligne aktivering af dendritiske celler, der spiller en afgørende rolle i priming af T-celler. Ændringerne i ekspressionen af overflademarkører måles ved hjælp af flowcytometri efter cellefarvning med flourochrom-mærkede antistoffer. Der foretages en samtidig cytotoksicitetsmåling med henblik på at måle, om der forekommer øget ekspression af overflademarkører ved subcytotoksiske koncentrationer. Overflademarkørernes relative fluorescerende intensitet sammenlignet med opløsningsmiddel-/bærestofkontrollen beregnes ved hjælp af forudsigelsesmodellen (jf. punkt 26) til støtte for differentieringen mellem hudsensibiliserende stoffer og ikke-sensibiliserende stoffer.
|
PÅVISNING AF KOMPETENCE
|
8.
|
Før rutinemæssig brug af den test, der beskrives i dette tillæg til forsøgsmetoden B.71, bør laboratorierne godtgøre deres tekniske kompetence ved hjælp af de ti kompetencestoffer, der findes opført i tillæg 1.2. Desuden bør brugerne af testen opretholde en historisk database over data, der er genereret ved hjælp af reaktivitetskontrollerne (jf. punkt 11) og ved hjælp af de positive kontroller og opløsningsmiddel-/bærestofkontrollerne (jf. punkt 20-22), og de bør anvende disse data til at bekræfte, at reproducerbarheden af testen i deres laboratorium opretholdes over tid.
|
FREMGANGSMÅDE
|
9.
|
Denne forsøgsmetode er baseret på h-CLAT databasetjenesten om alternative metoder til dyreforsøg (DB-ALM), protokol nr. 158 (18), som repræsenterer den protokol, der blev anvendt i valideringsundersøgelsen koordineret af EURL ECVAM. Det anbefales, at denne protokol anvendes ved gennemførelsen og anvendelsen af h-CLAT-metoden i laboratoriet. Følgende er en beskrivelse af h-CLAT-metodens vigtigste komponenter og procedurer, der består af to trin: dosisbestemmelsesassay og CD86/CD54-ekspressionsmåling.
|
Præparering af celler
|
10.
|
Human monocytisk leukæmi-cellelinjen, THP-1, bør anvendes til udførelse af h-CLAT-metoden. Det anbefales, at celler (TIB-202™) anskaffes fra en velrenommeret cellebank som f.eks. American Type Culture Collection.
|
|
11.
|
THP-1 celler dyrkes i en fugtig atmosfære ved 37oC og 5 % CO2 og i RPMI-1640 medium suppleret med 10 % føtalt kalveserum (FBS), 0,05 mM 2-mercaptoethanol, 100 enheder/ml penicillin og 100 μg/ml streptomycin. Anvendelse af penicillin og streptomycin i dyrkningsmediet kan undgås. I sådanne tilfælde bør brugerne imidlertid kontrollere, at fraværet af antibiotika i dyrkningsmediet ikke indvirker på resultaterne, f.eks. ved at teste de kompetencestoffer, der findes opført i tillæg 1.2. For at minimere risikoen for kontaminering bør god celledyrkningspraksis under alle omstændigheder følges, uanset om der er antibiotika i celledyrkningsmediet eller ej. THP-1 celler podes rutinemæssigt hver 2. eller 3. dag ved en celletæthed på 0,1 til 0,2 × 106 celler/ml. De bør opretholdes ved celletætheder fra 0,1 til 1,0 × 106 celler/ml. Inden cellerne anvendes til testning, bør de valideres ved at gennemføre en reaktivitetskontrol. Reaktivitetskontrollen af cellerne bør udføres ved hjælp af de positive kontroller, 2,4-dinitrochlorbenzen (DNCB) (CAS nr. 97-00-7, ≥ 99 % renhed) og nikkelsulfat (NiSO4) (CAS nr. 10101-97-0, ≥ 99 % renhed), og den negative kontrol, mælkesyre (LA) (CAS nr. 50-21-5, ≥ 85 % renhed), to uger efter optøning. Både DNCB og NiSO4 bør give en positiv respons for såvel CD86- som CD54-celleoverflademarkører, og LA bør give en negativ respons for såvel CD86- som CD54-celleoverflademarkører. Kun celler, der bestod reaktivitetskontrollen, bør anvendes til assayet. Cellerne kan opformeres i op til to måneder efter optøning. Der bør højst være 30 opformeringer. Reaktivitetskontrollen bør foretages i henhold til de fremgangsmåder, der er beskrevet i punkt 20-24.
|
|
12.
|
Til testning podes THP-1-cellerne ved en celletæthed på enten 0,1 × 106 celler/ml eller 0,2 × 106 celler/ml, og de forkultiveres i dyrkningsflasker i henholdsvis 72 timer eller 48 timer. Det er vigtigt, at celletætheden i dyrkningsflasken lige efter forkultiveringsperioden er så stabil som muligt i hvert enkelt eksperiment (ved at anvende en af de to ovennævnte forkultiveringsmetoder), fordi celletætheden i dyrkningsflasken lige efter forkultiveringen vil kunne påvirke ekspressionen af CD86/CD54, der induceres af allergener (19). På dagen for testningen genopslæmmes de celler, der høstes fra dyrkningsflasken, i et nyt dyrkningsmedium med en celletæthed på 2 × 106 celler/ml. Derefter fordeles cellerne i en fladbundet 24-brønds plade (500 μl, 1 × 106 celler/brønd) eller en fladbundet 96-brønds plade (80 μl, 1,6 × 105 celler/brønd).
|
Dosisbestemmelsesassay
|
13.
|
Der udføres et dosisbestemmelseassay for at fastlægge CV75, som er den koncentration af testkemikaliet, der resulterer i 75 % cellelevedygtighed (CV) sammenlignet med opløsningsmiddel-/bærestofkontrollen. CV75-værdien anvendes til at fastlægge koncentrationen af testkemikalierne til CD86/CD54-ekspressionsmålingen (jf. punkt 20-24).
|
Fremstilling af testkemikalier og kontrolstoffer
|
14.
|
Testkemikalierne og kontrolstofferne fremstilles på dagen for testen. I h-CLAT-metoden opløses testkemikalierne, eller de dispergeres stabilt (jf. også punkt 4) i fysiologisk saltopløsning eller medium som første opløsningsmiddel-/bærestofmulighed eller dimethylsulfoxid (DMSO, ≥ 99 % renhed) som anden opløsningsmiddel-/bærestofmulighed, hvis testkemikaliet ikke er opløseligt eller ikke danner en stabil dispersion i de to førstnævnte opløsningsmidler/bærestoffer, til slutkoncentrationer på 100 mg/ml (i fysiologisk saltopløsning eller medium) eller 500 mg/ml (i DMSO). Andre opløsningsmidler/bærestoffer end de ovenfor beskrevne kan anvendes, hvis der er videnskabeligt belæg herfor. Der bør tages hensyn til testkemikaliets stabilitet i det endelige opløsningsmiddel/bærestof.
|
|
15.
|
Begyndende med 100 mg/ml (i fysiologisk saltopløsning eller medium) eller 500 mg/ml (i DMSO) stamopløsninger af testkemikalierne foretages følgende fortyndinger:
|
—
|
fysiologisk saltopløsning eller medium som opløsningsmiddel/bærestof: Der fremstilles otte stamopløsninger (otte koncentrationer) ved hjælp af tofoldsseriefortyndinger, idet det tilsvarende opløsningsmiddel/bærestof anvendes. Disse stamopløsninger fortyndes derefter igen 50-fold i dyrkningsmediet (arbejdsopløsninger). Hvis den højeste slutkoncentration på pladen på 1 000 μg/ml er ikke-toksisk, bør maksimumskoncentrationen fastsættes igen ved at udføre en ny cytotoksicitetstest. Slutkoncentrationen på pladen bør ikke være højere end 5 000 μg/ml for testkemikalier, der er opløst eller stabilt dispergeret i en fysiologisk saltopløsning eller et medium.
|
|
—
|
DMSO som opløsningsmiddel/bærestof: Der fremstilles otte stamopløsninger (otte koncentrationer) ved hjælp af tofoldsseriefortyndinger, idet det tilsvarende opløsningsmiddel/bærestof anvendes. Disse stamopløsninger fortyndes yderligere 250-fold i dyrkningsmediet (arbejdsopløsninger). Slutkoncentrationen på pladen bør ikke være højere end 1 000 μg/ml, selv om denne koncentration er ikke-toksisk.
|
Arbejdsopløsningerne anvendes til slut til eksponering ved at tilsætte en mængde af arbejdsopløsningen svarende til mængden af THP-1 cellesuspension i pladen (jf. også punkt 17) for at opnå en yderligere tofoldsfortynding (normalt er den endelige koncentrationsserie i pladen 7,81–1 000 μg/ml).
|
|
16.
|
Den opløsningsmiddel-/bærestofkontrol, der anvendes i h-CLAT-metoden, er dyrkningsmedium (testkemikalier, der er opløst eller stabilt dispergeret (jf. punkt 4) enten i fysiologisk saltopløsning eller medium) eller DMSO (testkemikalier, der er opløst eller stabilt dispergeret i DMSO), der er testet ved en enkelt slutkoncentration i pladen på 0,2 %. Der foretages den samme fortynding som den, der er beskrevet for arbejdsopløsninger i punkt 15.
|
Påføring af testkemikalier og kontrolstoffer
|
17.
|
Det dyrkningsmedium eller de arbejdsopløsninger, der er beskrevet i punkt 15 og 16, blandes i forholdet 1:1 (v/v) med de cellesuspensioner, der er forberedt i den fladbundede 24-brønds eller 96-brønds plade (jf. punkt 12). De behandlede plader inkuberes derefter i 24±0,5 timer ved 37oC og 5 % CO2. Der bør træffes forholdsregler for at undgå, at flygtige testkemikalier fordamper, og for at undgå krydskontaminering med testkemikalier mellem brøndene, f.eks. ved at forsegle pladerne inden inkubation med testkemikalierne (20).
|
Farvning med propidiumiodid (PI)
|
18.
|
Efter 24±0,5 timers eksponering overføres cellerne til prøveglas og indsamles ved centrifugering. Supernatanterne kasseres, og de tilbageværende celler genopslæmmes i 200 μl (96-brønds plade) eller 600 μl (24-brønds plade) phosphatbufret fysiologisk saltopløsning indeholdende 0,1 % bovint serumalbumin (farvebuffer). 200 μl af cellesuspensionen overføres til en rundbundet 96-brønds plade (ved 96 brønde) eller et mikrorør (ved 24 brønde) og vaskes to gange med 200 μl (ved 96 brønde) eller 600 μl (24 brønde) farvebuffer. Endelig genopslæmmes cellerne i farvebufferen (f.eks. 400 μl), og PI-opløsningen (f.eks. 20 μl) tilsættes (f.eks. er PI-opløsningens slutkoncentration 0,625 μg/ml). Andre cytotoksicitetsmarkører som f.eks. 7-aminoactinomycin D (7-AAD), trypanblåt eller andre kan anvendes, hvis det kan påvises, at denne alternative farvning giver resultater svarende til PI, f.eks. via testning af de kompetencestoffer, der findes opført i tillæg 1.2.
|
Cytotoksicitetsmåling ved hjælp af flowcytometri og vurdering af CV75-værdien
|
19.
|
PI-optaget analyseres ved hjælp af flowcytometri med kanal FL-3. Der høstes i alt 10 000 levende celler (PI negativ). Cellelevedygtigheden kan beregnes af cytometeranalyseprogrammet ved hjælp af nedenstående ligning. Når cellelevedygtigheden er lav, bør der høstes op til 30 000 celler inklusive døde celler. Alternativt kan der høstes data i et minut efter, at analysen er startet.

CV75 værdien (jf. punkt 13), dvs. en koncentration, der viser overlevelse af 75 % THP-1-celler (25 % cytotoksisk effekt), beregnes ved log-lineær interpolation ved hjælp af følgende ligning:
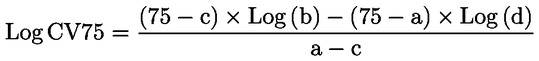
hvor:
a er minimumsværdien for cellelevedygtighed over 75 %
c er maksimumsværdien for cellelevedygtighed under 75 %
b og d er de koncentrationer, der giver henholdsvis a- og c-værdien for cellelevedygtighed.

Der kan anvendes andre fremgangsmåder til at udlede CV75-værdien, når blot det påvises, at dette ikke har nogen indvirkning på resultaterne (f.eks. via testning af kompetencestofferne).
|
CD86/CD54-ekspressionsmåling
Præparering af testkemikalier og kontrolstoffer
|
20.
|
Det egnede opløsningsmiddel/bærestof (fysiologisk saltopløsning, medium eller DMSO, jf. punkt 14) anvendes til at opløse eller dispergere testkemikalierne stabilt. Testkemikalierne opløses først i den koncentration, der svarer til 100-foldfortynding (fysiologiske saltopløsninger eller medium) eller 500-foldfortynding (DMSO) af 1,2 × CV75 fastlagt i dosisbestemmelsesassayet (jf. punkt 19). Hvis CV75 ikke kan bestemmes (dvs. hvis en tilstrækkelig cytotoksisk effekt ikke observeres i dosisbestemmelsesassayet), bør den højeste opløselige eller stabilt dispergerede koncentration af testkemikaliet fremstillet med hvert enkelt opløsningsmiddel/bærestof anvendes som startkoncentration. Bemærk, at slutkoncentrationen i pladen ikke bør overstige 5 000 μg/ml (ved fysiologisk saltopløsning eller medium) eller 1 000 μg/ml (ved DMSO). Dernæst laves der 1,2-foldsseriefortyndinger ved hjælp af det tilsvarende opløsningsmiddel/bærestof for at opnå stamopløsninger (otte koncentrationer fra 100×1,2 × CV75 til 100×0,335 × CV75 (fysiologisk saltopløsning eller medium) eller fra 500×1,2 × CV75 til 500×0,335 × CV75 (DMSO)), som testes ved hjælp af h-CLAT-metoden (se et eksempel på et doseringsskema i DB-ALM protokol nr. 158). Stamopløsningerne fortyndes yderligere 50-fold (fysiologisk saltopløsning eller medium) eller 250-fold (DMSO) i dyrkningsmediet (arbejdsopløsninger). Disse arbejdsopløsninger anvendes til slut til eksponering med en yderligere to-foldsfortyndingsfaktor i pladen. Hvis resultaterne ikke opfylder de i punkt 29 og 30 opstillede acceptkriterier for cellelevedygtighed, kan dosisbestemmelsesassayet gentages for at fastlægge en mere præcis CV75-værdi. Bemærk, at der kun kan anvendes 24-brønds plader til CD86/CD54-ekspressionsmåling.
|
|
21.
|
Opløsningsmiddel-/bærestofkontrollen fremstilles som beskrevet i punkt 16. Den positive kontrol, der anvendes i h-CLAT-metoden, er DNCB (jf. punkt 11), hvortil stamopløsningerne fremstilles i DMSO og fortyndes som beskrevet for stamopløsningerne i punkt 20. DNCB bør anvendes som den positive kontrol for CD86/CD54-ekspressionsmåling ved en enkelt slutkoncentration i pladen (typisk 4,0 μg/ml). For at opnå en 4,0 μg/ml koncentration af DNCB i pladen fremstilles en 2 mg/ml stamopløsning af DNCB i DMSO, som fortyndes yderligere 250-fold med dyrkningsmedium til en arbejdsopløsning på 8 μg/ml. Alternativt kan CV75-værdien af DNCB, der bestemmes i hver enkelt forsøgsfacilitet, anvendes som den positive kontrolkoncentration. Andre egnede positive kontroller kan anvendes, hvis der foreligger historiske data, som kan anvendes til at udlede sammenlignelige acceptkriterier for analyseserien. Bemærk, at slutkoncentrationen i pladen ikke bør overstige 5 000 μg/ml (fysiologisk saltopløsning eller medium) eller 1 000 μg/ml (DMSO). Acceptkriterierne for analyseserien er de samme som dem, der er beskrevet for testkemikaliet (jf. punkt 29), med undtagelse af det sidste acceptkriterium, da den positive kontrol testes ved en enkelt koncentration.
|
Påføring af testkemikalier og kontrolstoffer
|
22.
|
Der er behov for en test for hvert enkelt testkemikalie og kontrolstof for at opnå en forudsigelse. Hvert eksperiment består af mindst to uafhængige analyseserier for CD86/CD54-ekspressionsmåling (jf. punkt 26-28). Hver enkelt uafhængig analyseserie foretages på forskellige dage eller på samme dag, forudsat at der for hver analyseserie: a) fremstilles uafhængige friske stamopløsninger og arbejdsopløsninger af testkemikaliet og antistofopløsningerne og b) anvendes uafhængigt høstede celler (dvs. cellerne indsamles fra forskellige dyrkningsflasker). Cellerne kan imidlertid komme fra den samme generation. De testkemikalier og kontrolstoffer, der er fremstillet som arbejdsopløsninger (500 μl), blandes med 500 μl opslæmmede celler (1x106 celler) i forholdet 1:1, og cellerne inkuberes i 24±0,5 timer som beskrevet i punkt 20 og 21. Ved hver analyseserie er et enkelt replikat for hver koncentration af testkemikaliet og kontrolstoffet tilstrækkelig, fordi en forudsigelse opnås fra mindst to uafhængige analyseserier.
|
Cellefarvning og analyse
|
23.
|
Efter 24±0,5 timers eksponering overføres cellerne fra 24-brønds pladen til prøveglas, indsamles ved centrifugering og vaskes derefter to gange med 1 ml farvebuffer (om nødvendigt kan der foretages yderligere vaskninger). Efter vask blokeres cellerne med 600 μl blokerende opløsning (farvebuffer indeholdende 0,01 % (vægt/volumen) globulin (Cohn fraktion II, III, human; SIGMA, #G2388-10G eller tilsvarende)) og inkuberes ved 4oC i 15 min. Efter blokering fordeles cellerne i tre delprøver a 180 μl i en rundbundet 96-brønds plade eller et mikrorør.
|
|
24.
|
Efter centrifugering farves cellerne med 50 μl FITC-mærkede anti-CD86-, anti-CD54- eller muse-IgG1 (isotype)-antistoffer ved 4oC i 30 min. De antistoffer, der er beskrevet i h-CLAT DB-ALM protokol nr. 158 (18), bør anvendes ved at opløse 3:25 v/v (til CD86 (BD-PharMingen, #555657; Klon: Fun-1)) eller 3:50 volumen/volumen (til CD54 (DAKO, #F7143; Klon: 6.5B5) og IgG1 (DAKO, #X0927)) med farvebuffer. Disse antistofopløsningsfaktorerblev af testudviklerne fastlagt som dem, der giver det bedste signal/støjforhold. Det er testudviklernes erfaring, at antistoffernes fluorescensintensitet normalt er konsekvent mellem forskellige partier. Brugerne kan imidlertid overveje at titrere antistofferne under deres egne laboratorieforhold for at finde frem til de koncentrationer, der er bedst egnede. Andre fluorchrom-mærkede anti-CD86- og/eller anti-CD54-antistoffer kan anvendes, hvis det kan påvises, at de giver resultater svarende til FITC-konjugerede antistoffer, f.eks. via testning af de kompetencestoffer, der findes opført i tillæg 1.2. Det skal bemærkes, at ændring af klonen eller leverandøren af antistofferne som beskrevet i h-CLAT DB-ALM protokol nr. 158 (18) kan påvirke resultaterne. Efter vaskning to gange eller mere med 150 μl af farvebufferen genopslæmmes cellerne i farvebufferen (f.eks. 400 μl), og PI-opløsningen (f.eks. 20 μl for at opnå en endelig koncentration på 0,625 μg/ml) eller en anden cytotoksicitetsmarkøropløsning (jf. punkt 18) tilsættes. Ekspressionsniveauerne for CD86 og CD54 og cellelevedygtighed analyseres ved hjælp af flowcytometri.
|
DATA OG RAPPORTERING
Dataevaluering
|
25.
|
Ekspressionen af CD86 og CD54 analyseres ved hjælp af flowcytometri ved registreringskanal FL-1. På grundlag af det geometriske gennemsnit for fluorescensintensitet (MFI) beregnes den relative fluorescensintensitet (RFI) af CD86 og CD54 for positive kontrolceller (ctrl) og kemikaliebehandlede celler i henhold til nedenstående ligning:
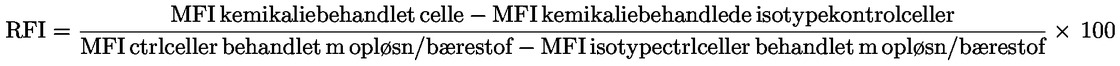
Cellelevedygtigheden fra isotypekontrolcellerne (der er farvet med muse-IgG1 (isotype)-antistoffer) beregnes ligeledes i henhold til den i punkt 19 beskrevne ligning.
|
Forudsigelsesmodel
|
26.
|
I forbindelse med CD86/CD54-ekspressionsmåling testes hvert testkemikalie i mindst to uafhængige analyseserier for at udlede en enkelt forudsigelse (POSITIV eller NEGATIV). En h-CLAT-forudsigelse anses for at være POSITIV, hvis mindst en af følgende betingelser er opfyldt i to ud af to eller mindst to ud af tre uafhængige analyseserier, ellers anses h-CLAT-forudsigelsen for at være NEGATIV (figur 1):
|
—
|
RFI for CD86 svarer til eller er højere end 150 % ved enhver testet koncentration (med en cellelevedygtighed ≥ 50 %).
|
|
—
|
RFI for CD54 svarer til eller er højere end 200 % ved enhver testet koncentration (med en cellelevedygtighed ≥ 50 %).
|
|
|
27.
|
Det fremgår af det ovenstående, at såfremt de første to analyseserier begge er positive for CD86 og/eller begge er positive for CD54, anses h-CLAT-forudsigelsen for at være POSITIV, og det er ikke nødvendigt at gennemføre en tredje analyseserie. Tilsvarende, hvis de to første analyseserier er negative for begge markører, anses h-CLAT-forudsigelsen for at være NEGATIV (med behørig hensyntagen til bestemmelserne i punkt 30), uden at det er nødvendigt at gennemføre en tredje analyseserie. Hvis de to første analyseserier imidlertid ikke er overensstemmende for mindst en af markørerne (CD54 eller CD86), er der behov for en tredje analyseserie, og den endelige forudsigelse vil blive baseret på flertallet af de tre individuelle analyseserier (dvs. to ud af tre). Det skal i denne forbindelse bemærkes, at hvis der foretages to uafhængige analyseserier, og den ene kun er positiv for CD86 (herefter benævnt P1), mens den anden kun er positiv for CD54 (herefter benævnt P2), bør der gennemføres en tredje analyseserie. Hvis denne tredje analyseserie er negativ for begge markører (herefter benævnt N), anses h-CLAT-forudsigelsen for at være NEGATIV. Hvis en tredje analyseserie på den anden side er positiv for en af markørerne (P1 eller P2) eller for begge markørerne (herefter benævnt P12), anses h-CLAT-forudsigelsen for at være POSITIV.

Figur 1: Den forudsigelsesmodel, der anvendes i h-CLAT-metoden. En h-Clat-forudsigelse bør overvejes inden for rammerne af en IATA og i overensstemmelse med bestemmelserne i punkt 7 og 8 i den generelle indledning. P1: analyseserie med kun CD86 positiv; P2: analyseserie med kun CD54 positiv; P12: analyseserie med både CD86 og CD54 positiv; N: analyseserie med hverken CD86 eller CD54 positiv.#Boksene viser de relevante kombinationer af resultater fra de tre analyseserier på grundlag af de resultater, der er opnået i de to første analyseserier vist i boksen ovenfor, men afspejler ikke den rækkefølge, i hvilken de er opnået. |
|
28.
|
For de testkemikalier, der forudsiges at være POSITIVE med h-CLAT, kan der, hvis det ønskes, fastsættes to værdier for effektiv koncentration (EC), EC150 for CD86 og EC200 for CD54, dvs. den koncentration, ved hvilken testkemikalierne gav en RFI på 150 eller 200. Disse EC-værdier kan også bidrage til vurderingen af sensibiliseringsstyrken (9), når de bruges i integrerede tilgange såsom IATA (4) (5) (6) (7) (8). De kan beregnes ved hjælp af følgende ligninger:
EC 150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
hvor
Aconc er den laveste koncentration udtrykt i μg/ml med RFI > 150 (CD86) eller 200 (CD54)
25Bconc er den højeste koncentration udtrykt i μg/ml med RFI < 150 (CD86) eller 200 (CD54)
ARFI er RFI ved den laveste koncentration med RFI > 150 (CD86) eller 200 (CD54)
BRFI er RFI ved den højeste koncentration med RFI < 150 (CD86) eller 200 (CD54).
Med henblik på mere præcist at udlede EC150- og EC200-værdierne, kan det være nødvendigt med tre uafhængige analyseserier for CD86/CD54-ekspressionsmåling. De endelige EC150- og EC200-værdier fastsættes derefter som en gennemsnitsværdi af EC-værdierne beregnet på grundlag af tre uafhængige analyseserier. Når kun to ud af tre uafhængige analyseserier opfylder kriterierne for positivitet (jf. punkt 26-27), vælges den højeste EC150- eller EC200-værdi af de to beregnede værdier.
|
Acceptkriterier
|
29.
|
Følgende acceptkriterier bør være opfyldt, når h-CLAT-metoden anvendes (22) (27):
|
—
|
Cellelevedygtigheden i mediekontrollen og opløsningsmiddel-/bærestofkontrollen bør være højere end 90 %.
|
|
—
|
I opløsningsmiddel-/bærestofkontrollen bør RFI-værdierne for både CD86 og CD54 ikke overstige de positive kriterier (CD86 RFI ≥ 150 % og CD54 RFI ≥ 200 %). RFI-værdierne i opløsningsmiddel-/bærestofkontrollen beregnes ved hjælp af den formel, der er beskrevet i punkt 25 ("MFI for kemikaliet" erstattes med "MFI for opløsningsmiddel/bærestof", og "MFI for opløsningsmiddel/bærestof" erstattes af "MFI for (medium) kontrol").
|
|
—
|
>I både mediekontrollen og opløsningsmiddel-/bærestofkontrollen bør MFI-forholdet mellem både CD86 og CD54 og isotypekontrollen være > 105 %.
|
|
—
|
I den positive kontrol (DNCB), bør RFI-værdierne for både CD86 og CD54 opfylde de positive kriterier (CD86 RFI ≥ 150 og CD54 RFI ≥ 200), og cellelevedygtigheden bør være højere end 50 %.
|
|
—
|
For testkemikaliet bør cellelevedygtigheden være højere end 50 % ved mindst fire testede koncentrationer i hver analyseserie.
|
|
|
30.
|
Negative resultater accepteres kun for testkemikalier, der fremviser en cellelevedygtighed på under 90 % ved den højeste testede koncentration (dvs. 1,2 × CV75 i henhold til den fortyndingsserie, der er beskrevet i punkt 20). Hvis cellelevedygtigheden ved 1,2 × CV75 svarer til eller er højere end 90 %, bør det negative resultat kasseres. I sådanne tilfælde anbefales det, at det forsøges at finjustere dosisbestemmelsen ved at gentage bestemmelsen af CV75. Det bemærkes, at når 5 000 μg/ml i fysiologisk saltopløsning (eller medium eller andre opløsningsmidler/bærestoffer), 1 000 μg/ml i DMSO eller den højeste opløselige koncentration anvendes som den maksimale testkoncentration af et testkemikalie, er et negativt resultat acceptabelt, selv om cellelevedygtigheden ligger på over 90 %.
|
Testrapport
|
31.
|
Testrapporten bør indeholde følgende oplysninger:
|
Testkemikalie
|
Stof med kun én bestanddel
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
fysisk udseende, log Kow, vandopløselighed, DMSO-opløselighed, molekylvægt og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt
|
|
renhed, urenheders kemiske identitet i det omfang, det er relevant og praktisk muligt osv.
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel/bærestof for de enkelte testkemikalier.
|
|
|
Stof med flere bestanddele, UVCB-stoffer og blandinger
|
så vidt muligt kendetegnet ved f.eks. kemisk identitet (jf. ovenfor), renhed, kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber (jf. ovenfor) i det omfang, det er muligt
|
|
fysisk udseende, vandopløselighed, DMSO-opløselighed og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt
|
|
molekylvægt eller tilsyneladende molekylvægt i tilfælde af blandinger/polymerer med kendt sammensætning eller andre oplysninger, der er relevante for undersøgelsens gennemførelse
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel/bærestof for de enkelte testkemikalier.
|
|
|
|
Kontrol
|
Positiv kontrol
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
fysisk udseende, log Kow, vandopløselighed, DMSO-opløselighed, molekylvægt og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt og relevant
|
|
renhed, urenheders kemiske identitet i det omfang, det er relevant og praktisk muligt osv.
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
reference til historiske positive kontrolresultater, der udviser passende acceptkriterier for analyseserien, hvis det er relevant.
|
|
|
|
Negativ kontrol og opløsningsmiddel-/bærestofkontrol
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
renhed, urenheders kemiske identitet i det omfang, det er relevant og praktisk muligt osv.
|
|
fysisk udseende, molekylvægt og yderligere relevante fysisk-kemiske egenskaber ved brug af andre opløsningsmidler/bærestoffer end dem, der er nævnt i testvejledningen, i det omfang, det er muligt
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel/bærestof for de enkelte testkemikalier.
|
|
|
Testbetingelser
|
navn og adresse på sponsoren, testfaciliteten og undersøgelseslederen
|
|
beskrivelse af den anvendte test
|
|
anvendt cellelinje, opbevaringsforhold og kilde (f.eks. det anlæg, hvorfra den er anskaffet)
|
|
den anvendte flowcytometri (f.eks. model), herunder instrumentindstillinger, globulin, antistoffer og anvendt cytotoksicitetsmarkør
|
|
den procedure, der er blevet anvendt til at påvise laboratoriets kompetence til at anvende forsøgsmetoden via testning af kompetencestoffer, og den metode, der er blevet anvendt til at påvise reproducerbare resultater over tid, f.eks. historiske kontroldata og/eller historiske reaktivitetskontroldata.
|
|
|
Acceptkriterier for testen
|
cellelevedygtigheds-, MFI- og RFI-værdier opnået ved opløsningsmiddel-/bærestofkontrol sammenlignet med acceptintervallerne
|
|
cellelevedygtigheds- og RFI-værdier opnået ved den positive kontrol sammenlignet med acceptintervallerne
|
|
cellelevedygtigheden ved alle de testede koncentrationer af det testede kemikalie.
|
|
|
Forsøgsmetode
|
antal anvendte analyseserier
|
|
koncentrationer af testkemikaliet, anvendt påførsels- og eksponeringstid (hvis en anden end den anbefalede)
|
|
beskrivelse af de anvendte evaluerings- og beslutningskriterier
|
|
beskrivelse af eventuelle ændringer i forsøgsmetoden.
|
|
|
Resultater
|
tabulering af data, herunder CV75 (hvis muligt), individuelle geometriske MFI-, RFI-, cellelevedygtighedsværdier, EC150/EC200-værdier (hvis muligt), opnået for testkemikaliet og for den positive kontrol i hver enkelt analyseserie, og en angivelse af klassificeringen af testkemikaliet i forhold til forudsigelsesmodellen
|
|
beskrivelse af eventuelle andre relevante observationer, hvis det er relevant.
|
|
|
Diskussion af resultater
|
diskussion af de resultater, der er opnået med h-CLAT-metoden
|
|
vurdering af testresultaterne inden for rammerne af en IATA, hvis der foreligger andre relevante oplysninger.
|
|
|
LITTERATUR
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767-773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Tilgængelig på: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations.
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Tilgængelig på: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing.
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report. Tilgængelig på: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations.
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation test: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), s. 23 ff. Tilgængelig på: http://ecvam-dbalm.jrc.ec.europa.eu/.
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, , Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organization for Economic Cooperation and Development, Paris, France, 2005, 96 ff.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Tilgængelig på: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(23)
|
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1 Tilgængelig på: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Tillæg 1.1
DEFINITIONER
Nøjagtighed
: Overensstemmelsen mellem forsøgsmetodens resultater og accepterede referenceværdier. Den er et mål for testens ydeevne og et af aspekterne af relevans. Udtrykket benyttes ofte som synonym for "konkordans", forstået som den andel af korrekte udfald, som en forsøgsmetode fører til (21).
AOP (Adverse Outcome Pathway)
: Sekvens af begivenheder fra den kemiske struktur af et målkemikalie eller en gruppe af ensartede kemikalier gennem den molekylære udløsende begivenhed til et in vivo-resultat af betydning (22).
Kemikalie
: Et stof eller en blanding.
CV75
: Den anslåede koncentration, der giver en cellelevedygtighed på 75 %.
EC150
: De koncentrationer, der viser RFI-værdier på 150 i CD86-ekspressionen.
EC200
: De koncentrationer, der viser RFI-værdier på 200 i CD54-ekspressionen.
Flowcytometri
: En cytometrisk teknik, i hvilken celler opslæmmet i en væske flyder en ad gangen gennem et fokus bestående af magnetiserende lys, som spredes i mønstre, der er karakteristiske for cellerne og deres bestanddele. Cellerne mærkes hyppigt med fluorescerende markører, så lyset først absorberes og derefter udsendes ved ændrede frekvenser.
Fare
: Den iboende egenskab ved et stof eller en situation, som kan have en skadelig virkning, når en organisme, et system eller en (sub)population eksponeres for det pågældende stof.
IATA (integreret tilgang til testning og vurdering)
: En struktureret tilgang, der bruges til identifikation af farer (potentiale), farebeskrivelse (styrke) og/eller sikkerhedsvurdering (potentiale/styrke og eksponering) af et kemikalie eller en gruppe af kemikalier, som på strategisk vis integrerer og vægter alle relevante data som grundlag for lovgivningsmæssige beslutninger om mulige farer og/eller risici og/eller behovet for yderligere målrettede og derfor færrest mulige test.
Mediekontrol
: Et ubehandlet replikat, som indeholder alle komponenter fra et forsøgssystem. Prøven behandles sammen med prøver behandlet med testkemikaliet og andre kontroller for at afgøre, hvorvidt opløsningsmidlet/bærestoffet interagerer med forsøgssystemet.
Blanding
: Blanding eller opløsning, der består af to eller flere stoffer.
Stof med kun én bestanddel
: Et stof, der defineres ved dets mængdemæssige sammensætning, hvori en hovedbestanddel udgør mindst 80 % (w/w).
Stof med flere bestanddele
: Et stof, der defineres ved dets mængdemæssige sammensætning, hvori mere end en hovedbestanddel er til stede i en koncentration på ≥ 10 % (w/w) og < 80 % (w/w). Et stof, der består af flere forskellige bestanddele, er resultatet af en fremstillingsproces. Forskellen mellem en blanding og et stof med forskellige bestanddele er, at blandingen er fremstillet ved blanding af to eller flere stoffer uden kemisk reaktion. Et stof, der består af flere forskellige bestanddele, er resultatet af en kemisk reaktion.
Positiv kontrol
: Et replikat, som indeholder alle komponenterne fra et forsøgssystem og er behandlet med et stof, som vides at inducere en positiv respons. For at sikre at det er muligt at vurdere variabiliteten i den positive kontrolrespons over tid, bør den positive respons ikke være overdrevent stor.
Præ-haptener
: Kemikalier, der bliver allergifremkaldende via abiotisk omdannelse.
Pro-haptener
: Kemikalier, der kræver en enzymaktivering for at udløse det hudsensibiliserende potentiale.
Relativ fluorescensintensitet (RFI)
: Relative værdier af det geometriske gennemsnit af fluorescensintensitet (MFI) i kemisk behandlede celler sammenlignet med MFI i celler behandlet med opløsningsmiddel/bærestof.
Relevans
: Beskrivelse af sammenhængen mellem testen og den pågældende virkning og dens meningsfuldhed og brugbarhed til et bestemt formål. Det angives, i hvilket omfang testen måler eller forudsiger den pågældende biologiske virkning korrekt. Testens nøjagtighed (konkordans) indgår i vurderingen af relevansen (21).
Pålidelighed
: Mål for, i hvilken grad en test kan udføres reproducerbart i og mellem laboratorier over tid, når den udføres efter samme protokol. Den bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier og repeterbarhed inden for laboratorier (21).
Analyseserie
: En analyseserie består af et eller flere testkemikalier, der testes samtidig med en opløsningsmiddel-/bærestofkontrol og med en positiv kontrol.
Følsomhed
: Andel af alle positive/aktive kemikalier, der klassificeres korrekt ved testen. Følsomheden er et mål for nøjagtigheden af en test, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en tests relevans (21).
Farvebuffer
: En phosphatbufret fysiologisk saltopløsning indeholdende 0,1 % bovint serumalbumin.
Opløsningsmiddel-/bærestofkontrol
: En ubehandlet prøve, som indeholder alle komponenterne fra et testsystem, herunder det anvendte opløsningsmiddel/bærestof. Den anvendes til at fastlægge basislinjeresponsen for de prøver, som er behandlet med testkemikaliet opløst eller stabilt dispergeret i det samme opløsningsmiddel/bærestof. Når denne prøve testes med en samtidig mediekontrol, viser den også, hvorvidt opløsningsmidlet eller bærestoffet interagerer med testsystemet.
Specificitet
: Andel af alle negative/inaktive kemikalier, der klassificeres korrekt ved testen. Det er et mål for nøjagtigheden af et forsøg, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en tests relevans (21).
Stof
: Et grundstof og forbindelser heraf, i naturlig tilstand eller industrielt fremstillet, herunder sådanne tilsætningsstoffer, som er nødvendige til bevarelse af stoffets stabilitet, og sådanne urenheder, som følger af fremstillingsprocessen, bortset fra opløsningsmidler, der kan udskilles, uden at det påvirker stoffets stabilitet eller ændrer dets sammensætning.
Testkemikalie
: Alle stoffer eller blandinger, der testes ved hjælp af denne metode.
De Forenede Nationers globalt harmoniserede system for klassificering og mærkning af kemikalier (United Nations Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: System til klassificering af kemikalier (stoffer og blandinger) efter standardiserede typer og niveauer af fysisk fare, sundhedsfare og miljøfare med tilhørende kommunikationselementer såsom piktogrammer, signalord, faresætninger, sikkerhedssætninger og sikkerhedsdatablade, således at oplysninger om kemikaliernes skadelige virkninger kan videreformidles med henblik på at beskytte mennesker (f.eks. arbejdsgivere, arbejdstagere, transportører, forbrugere og redningspersonel) og miljøet (23).
UVCB
: Stoffer med ukendt eller variabel sammensætning, komplekse reaktionsprodukter eller biologiske materialer.
Gyldig test
: En test, der anses for at være tilstrækkeligt relevant og pålideligt til et bestemt formål, og som er baseret på videnskabeligt forsvarlige principper. En test er aldrig gyldig i absolut forstand, men kun i relation til et bestemt formål (21).
Tillæg 1.2
KOMPETENCESTOFFER
Før den rutinemæssige brug af den test, der er beskrevet i dette tillæg til forsøgsmetode B.71, bør laboratorierne godtgøre deres tekniske kompetence ved at opnå den forventede h-CLAT-forudsigelse på korrekt vis for de ti kompetencestoffer, der anbefales i tabel 1, og ved at opnå de CV75-, EC150- og EC200-værdier, der falder ind under de respektive referenceintervaller for otte ud af de ti kompetencestoffer. Disse kompetencestoffer er udvalgt således, at de repræsenterer responsområdet for hudsensibiliseringsfare. Andre udvælgelseskriterier var, at kemikalierne er kommercielt tilgængelige, at der foreligger in vivo-referencedata af høj kvalitet, og at der er genereret in vitro-data af høj kvalitet med h-CLAT-forsøgsmetoden. Der foreligger ligeledes offentliggjorte referencedata for h-CLAT-metoden (3) (14).
Tabel 1
Anbefalede stoffer til godtgørelse af teknisk kompetence i tilknytning til h-CLAT-metoden
|
Kompetencestoffer
|
CAS RN
|
Fysisk form
|
In vivo-forudsigelse (103)
|
CV75-reference-område i μg/ml (104)
|
h-CLAT-resultater for CD86 (EC150-reference-interval i μg/ml) (104)
|
h-CLAT-resultater for CD54 (EC200-reference-interval i μg/ml) (104)
|
|
2,4-Dinitrochlorbenzen
|
97-00-7
|
Fast stof
|
Sensibiliserende stof (ekstrem)
|
2-12
|
Positiv (0,5-10)
|
Positiv (0,5-15)
|
|
4-Phenylenediamin
|
106-50-3
|
Fast stof
|
Sensibiliserende stof (stærk)
|
5-95
|
Positiv (<40)
|
Negativ (>1,5) (105)
|
|
Nikkelsulfat
|
10101-97-0
|
Fast stof
|
Sensibiliserende stof (moderat)
|
30-500
|
Positiv (<100)
|
Positiv (10-100)
|
|
2-Mercaptobenzothiazol
|
149-30-4
|
Fast stof
|
Sensibiliserende stof (moderat)
|
30-400
|
Negativ (>10) (105)
|
Positiv (10-140)
|
|
R(+)-Limonen
|
5989-27-5
|
Væske
|
Sensibiliserende stof (svag)
|
>20
|
Negativ (>5) (105)
|
Positiv (<250)
|
|
Imidazolidinyl-urinstof
|
39236-46-9
|
Fast stof
|
Sensibiliserende stof (svag)
|
25-100
|
Positiv (20-90)
|
Positiv (20-75)
|
|
Isopropanol
|
67-63-0
|
Væske
|
Ikke-sensibiliserende
|
>5 000
|
Negativ >5 000
|
Negativ >5 000
|
|
Glycerol
|
56-81-5
|
Væske
|
Ikke-sensibiliserende
|
>5 000
|
Negativ >5 000
|
Negativ >5 000
|
|
Mælkesyre
|
50-21-5
|
Væske
|
Ikke-sensibiliserende
|
1500-5 000
|
Negativ >5 000
|
Negativ >5 000
|
|
4-Aminobenzoesyre
|
150-13-0
|
Fast stof
|
Ikke-sensibiliserende
|
> 1 000
|
Negativ (>1 000 )
|
Negativ (>1 000 )
|
|
Forkortelser: CAS RN = Registernummer i Chemical Abstracts Service
|
Tillæg 2
IN VITRO-HUDSENSIBILISERING: U937 CELL LINE ACTIVATION TEST (U-SENS™)
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER
|
1.
|
U-SENS™ måler ændringer i ekspressionen af celleoverflademarkører i tilknytning til processen med aktivering af monocyter og dendritiske celler (DC) (dvs. CD86) i den humane histiocytære lymfomcellelinje U937 efter eksponering for allergifremkaldende stoffer (1). De målte ekspressionsniveauer for celleoverflademarkøren CD86 i cellelinjen U937 anvendes dernæst til støtte for differentieringen mellem hudsensibiliserende stoffer og ikke-sensibiliserende stoffer.
|
|
2.
|
U-SENS™ er blevet evalueret i en valideringsundersøgelse (2) koordineret af l’Oreal, og efterfølgende foretog det videnskabelige rådgivende udvalg (ESAC) under EU-referencelaboratoriet for alternativer til dyreforsøg (EURL ECVAM) uafhængige peer reviews (3). På grundlag af alle de tilgængelige beviser og input fra myndigheder og interessenter anbefalede EURL ECVAM (4), at U-SENS™ anvendes som en del af en IATA til støtte for differentieringen mellem hudsensibiliserende stoffer og ikke-sensibiliserende stoffer med henblik på fareklassificering og mærkning. OECD drøfter i øjeblikket i sit vejledende dokument om rapportering af strukturerede tilgange til dataintegration og individuelle informationskilder, der anvendes inden for IATA for hudsensibilisering, en række casestudier, der beskriver forskellige forsøgsstrategier og forudsigelsesmodeller. En af de forskelligt definerede tilgange er baseret på U-SENS™-assayet (5). Der kan også findes eksempler på anvendelsen af U-SENS™-data i kombination med anden information, herunder historiske data og eksisterende gyldige humane data (6), andre steder i litteraturen (4) (5) (7).
|
|
3.
|
Det viste sig, at U-SENS™-metoden kunne overføres til laboratorier med erfaring inden for celledyrkningsteknikker og flowcytometrianalyse. Graden af reproducerbarhed i de forudsigelser, der kan forventes fra testen, ligger i størrelsesordenen henholdsvis 90 % og 84 % inden for og mellem laboratorier (8). De resultater, der genereres i valideringsundersøgelsen (8), og andre offentliggjorte undersøgelser (1), viser generelt, at nøjagtigheden med hensyn til sondring mellem hudsensibiliserende stoffer (dvs. UN GHS-/CLP-kategori 1) og ikke-sensibiliserende stoffer er på 86 % (N=166) med en følsomhed på 91 % (118/129) og en specificitet på 65 % (24/37) sammenlignet med LLNA-resultaterne. Sammenlignet med resultater fra mennesker er nøjagtigheden med hensyn til sondring mellem hudsensibiliserende stoffer (dvs. UN GHS-/CLP-kategori 1) og ikke-sensibiliserende stoffer på 77 % (N=101) med en følsomhed på 100 % (58/58) og en specificitet på 47 % (20/43). Sammenlignet med LLNA er det mere sandsynligt, at falsk negative forudsigelser i forbindelse med U-SENS™ vil vedrøre kemikalier med lav til moderat hudsensibiliserende styrke (dvs. UN GHS-/CLP-underkategori 1B) end kemikalier med en høj hudsensibiliserende styrke (dvs. UN GHS-/CLP-underkategori 1A) (1) (8) (9). Samlet set viser disse oplysninger nytteværdien af U-SENS™-metoden som bidrag til identifikation af hudsensibiliseringsfarer. De nøjagtige værdier, der her angives for U-SENS™ som en selvstændig test, er imidlertid kun vejledende, da testen bør betragtes i kombination med andre informationskilder i forbindelse med en IATA og i overensstemmelse med bestemmelserne i punkt 7 og 8 i den generelle indledning. I forbindelse med vurderingen af metoder til test af hudsensibilisering uden brug af dyr bør man desuden holde sig for øje, at LLNA-forsøg samt andre dyreforsøg måske ikke fuldt ud afspejler situationen hos mennesker.
|
|
4.
|
På grundlag af de data, der foreligger i øjeblikket, viste U-SENS™-testen sig at kunne anvendes på testkemikalier (herunder ingredienser i kosmetik, f.eks. konserveringsmidler, overfladeaktive stoffer, aktive stoffer, farvestoffer) omfattende en lang række organiske funktionelle gruppers fysisk-kemiske egenskaber, hudsensibiliseringsstyrke (som bestemt i in vivo-undersøgelser) og det spektrum af reaktionsmekanismer, som man ved, er knyttet til hudsensibilisering (dvs. Michael acceptor, Schiff basedannelse, acyloverførselsmiddel, bimolekylær nucleofil substitutionsreaktion [SN2], eller nukleofil aromatisk substitution [SNAr]) (1) (8) (9) (10). U-SENS™-metoden kan anvendes til at teste kemikalier, der er opløselige, eller som danner en stabil dispersion (dvs. en kolloid opløsning eller en suspension, i hvilken testkemikaliet ikke bundfældes eller separeres fra opløsningsmidlet/bærestoffet i forskellige faser) i et passende opløsningsmiddel/bærestof (jf. punkt 13). Kemikalier, der i datasættet blev angivet som præ-haptener (dvs. stoffer, der aktiveres ved oxidering) eller pro-haptener (dvs. stoffer, der kræver en enzymaktivering, f.eks. via P450 enzymer),blev korrekt forudsagt af U-SENS™ (1) (10). Membranforstyrrende stoffer kan føre til falsk positive resultater som følge af en ikke-specifik stigning i CD86-ekspressionen, eftersom tre ud af syv falske positiver i tilknytning til in vivo-referenceklassificeringen var overfladeaktive stoffer (1). Positive resultater med overfladeaktive stoffer som sådan bør betragtes med forsigtighed, mens negative resultater af overfladeaktive stoffer fortsat kan anvendes til støtte for identifikationen af testkemikaliet som et ikke-sensibiliserende stof. Fluorescerende testkemikalier kan vurderes med U-SENS™ (1), men stærkt fluorescerende testkemikalier, der udsender den samme bølgelængde som fluoresceinisothiocyanat (FITC) eller propidiumiodid (PI), vil interferere med den flowcytometriske påvisning og kan derfor ikke evalueres korrekt ved hjælp af FITC-konjugerede antistoffer (potentielt falske negativer) eller PI (levedygtighed er ikke målbar). I sådanne tilfælde kan henholdsvis andre flourochrom-mærkede antistoffer eller andre cytotoksicitetsmarkører anvendes, når blot det kan påvises, at de giver resultater svarende til FITC-mærkede antistoffer eller PI (jf. punkt 18) f.eks. via testning af de kompetencestoffer, der findes opført i tillæg 2-2. I lyset af det ovenstående bør positive resultater med overfladeaktive stoffer og negative resultater med stærke fluorescerende testkemikalier fortolkes på baggrund af de fastsatte begrænsninger og sammen med andre informationskilder inden for rammerne af IATA. I de tilfælde, hvor der foreligger beviser for, at U-SENS™-metoden ikke kan anvendes på andre specifikke kategorier af testkemikalier, bør denne metode ikke anvendes til disse specifikke kategorier.
|
|
5.
|
Som beskrevet ovenfor understøtter U-SENS™ differentieringen mellem hudsensibiliserende og ikke-sensibiliserende stoffer. Den kan imidlertid også bidrage til vurderingen af sensibiliseringsstyrken, når den anvendes i integrerede tilgange såsom IATA. Der er dog behov for yderligere arbejde, fortrinsvis baseret på humane data, for at fastslå, hvordan U-SENS™-resultaterne eventuelt kan anvendes til styrkevurderingen.
|
|
6.
|
Definitioner findes i tillæg 2.1.
|
PRINCIP FOR TESTEN
|
7.
|
U-SENS™-metoden er et in vitro-assay, der måler ændringerne i ekspressionen af CD86-celleoverflademarkøren på en human histiocytær lymfomcellelinje, U937, efter 45±3 timers eksponering for testkemikaliet. CD86-overflademarkøren er en typisk markør for U937-aktivering. CD86 er kendt som et medstimulerende molekyle, der kan efterligne monocytisk aktivering, som spiller en afgørende rolle i primingen af T-celler. Ændringerne i ekspressionen af CD86-overflademarkøren måles ved hjælp af flowcytometri efter cellefarvning med fluoresceinisothiocyanat (FITC)-mærkede antistoffer. Cytotoksicitetsmålingen foretages (f.eks. ved hjælp af PI) samtidig med henblik på at vurdere, om forøgelsen af ekspressionen af CD86-celleoverflademarkøren optræder ved subcytotoksiske koncentrationer. Stimuleringsindekset (SI) for CD86-celleoverflademarkøren sammenlignet med opløsningsmiddel-/bærestofkontrollen beregnes ved hjælp af forudsigelsesmodellen (jf. punkt 19) til støtte for differentieringen mellem hudsensibiliserende stoffer og ikke-sensibiliserende stoffer.
|
PÅVISNING AF KOMPETENCE
|
8.
|
Forud for den rutinemæssige brug af den test, der beskrives i dette tillæg til forsøgsmetoden B.71, bør laboratorierne godtgøre deres tekniske kompetence ved hjælp af de ti kompetencestoffer, der findes opført i tillæg 2.2. i overensstemmelse med Good in vitro Method Practices (11). Desuden bør brugerne af testen opretholde en historisk database over data, der er genereret ved hjælp af reaktivitetskontrollerne (jf. punkt 11) og ved hjælp af de positive kontroller og opløsningsmiddel-/bærestofkontrollerne (jf. punkt 15-16), og de bør anvende disse data til at bekræfte, at reproducerbarheden af testen i deres laboratorium opretholdes over tid.
|
FREMGANGSMÅDE
|
9.
|
Dette forsøg er baseret på U-SENS™ DataBase service on ALternative Methods to animal experimentation (DB-ALM) protokol nr. 183 (12). Der bør anvendes standardprocedurer (SOP) ved gennemførelsen og anvendelsen af U-SENS™-metoden i laboratoriet. Et automatiseret system til gennemførelse af U-SENS™ kan anvendes, hvis det kan påvises, at det giver tilsvarende resultater, f.eks. via testning af de kompetencestoffer, der findes opført i tillæg 2.2. Følgende er en beskrivelse af de vigtigste komponenter og procedurer i forbindelse med U-SENS™-testen.
|
Præparering af celler
|
10.
|
Den humane histiocytære lymfomcellelinje, U937 (13), bør anvendes til udførelse af U-SENS™-testen. Celler (klon CRL1593.2) bør anskaffes fra en velrenommeret cellebank som f.eks. American Type Culture Collection.
|
|
11.
|
U937-celler dyrkes i en fugtig atmosfære ved 37 °C og 5 % CO2 og i RPMI-1 640-medium suppleret med 10 % føtalt kalveserum (FBS), 2 mM L-glutamin, 100 enheder/ml penicillin og 100 μg/ml streptomycin (komplet medium). U937-celler opformeres rutinemæssigt hver 2. eller 3. dag ved en celletæthed på henholdsvis 1,5 eller 3 × 105 celler/ml. Celletætheden bør ikke overstige 2 × 106 celler/ml, og cellelevedygtigheden målt ved trypanblå-udelukkelse bør være ≥ 90 % (bør ikke anvendes ved den første opformering efter optøning). Inden cellerne anvendes til testning, bør hver enkelt celleportion, FCS eller antistoffer valideres ved at gennemføre en reaktivitetskontrol. Reaktivitetskontrollen af cellerne bør foretages ved hjælp af den positive kontrol, picrylsulfonsyre (2,4,6-Trinitrobenzensulfonsyre: TNBS) (CASRN 2 508-19-2, ≥ 99 % renhed), og den negative kontrol, mælkesyre (LA) (CASRN 50-21-5, ≥ 85 % renhed), mindst en uge efter optøning. Ved reaktivitetskontrollen bør seks slutkoncentrationer testes for hver af de to kontroller (TNBS: 1, 12,5, 25, 50, 75, 100μg/ml og LA: 1, 10, 20, 50, 100, 200μg/ml). TNBS opløst i komplet medium bør give en positiv og koncentrationsbestemt respons for CD86 (f.eks. når en positiv koncentration, CD86 SI ≥ 150, efterfølges af en koncentration med en øget CD86 SI), og LA opløst i komplet medium bør give en negativ respons for CD86 (jf. punkt 21). Kun den celleportion, der har bestået reaktivitetskontrollen to gange, bør anvendes til assayet. Celler kan opformeres op til syv uger efter optøning. Der bør højst foretages 21 opformeringer. Reaktivitetskontrollen bør foretages i henhold til de fremgangsmåder, der er beskrevet i punkt 18-22.
|
|
12.
|
Til testning podes U937-cellerne ved en celletæthed på enten 3 × 105 celler/ml eller 6 × 105 celler/ml, og de forkultiveres i dyrkningsflasker i henholdsvis to dage eller én dag. Der kan anvendes andre forkultiveringsforhold end de ovenfor nævnte, hvis der fremlægges tilstrækkeligt videnskabeligt belæg herfor, f.eks. via en testning af de kompetencestoffer, der findes opført i tillæg 2.2. På dagen for testningen genopslæmmes de celler, der høstes fra dyrkningsflasken, i nyt dyrkningsmedium med en celletæthed på 5 × 105 celler/ml. Derefter fordeles cellerne i en fladbundet 96-brønds plade med 100 μl (endelig celletæthed på 0,5 × 105 celler/brønd).
|
Fremstilling af testkemikalier og kontrolstoffer
|
13.
|
Der foretages en vurdering af opløseligheden forud for testningen. Med henblik herpå opløses testkemikalierne, eller de dispergeres stabilt ved en koncentration på 50 mg/ml i et komplet medium som det første valg af opløsningsmiddel eller i dimethylsulfoxid (DMSO, ≥ 99 % renhed) som det andet valg af opløsningsmiddel/bærestof, hvis testkemikaliet ikke er opløseligt i det komplette mediumopløsningsmiddel/-bærestof. Når testkemikaliet skal anvendes i testen, opløses det til en slutkoncentration på 0,4 mg/ml i komplet medium, hvis kemikaliet er opløseligt i dette opløsningsmiddel/bærestof. Hvis kemikaliet kun er opløseligt i DMSO, opløses det til en koncentration på 50 mg/ml. Andre opløsningsmidler/bærestoffer end de ovenfor beskrevne kan anvendes, hvis der er videnskabeligt belæg herfor. Der bør tages hensyn til testkemikaliets stabilitet i det endelige opløsningsmiddel/bærestof.
|
|
14.
|
Testkemikalierne og kontrolstofferne fremstilles på dagen for testen. Da der ikke foretages et dosisbestemmelsesassay, bør seks slutkoncentrationer testes ved første analyseserie (1, 10, 20, 50, 100 og 200 μg/ml) i det tilsvarende opløsningsmiddel/bærestof enten i komplet medium eller i 0,4 % DMSO i mediet. Ved de efterfølgende analyseserier begyndende med 0,4 mg/ml i komplet medium eller 50 mg/ml i DMSO fremstilles opløsninger af testkemikaliet (dvs. mindst fire koncentrationer) ved hjælp af det tilsvarende opløsningsmiddel/bærestof. Arbejdsopløsningerne anvendes til slut til behandling ved at tilsætte lige store mængder af U937-cellesuspensionen (jf. punkt 11 ovenfor) til mængden af arbejdsopløsningen på pladen for at opnå en yderligere tofoldsopløsning (12). Koncentrationerne (mindst fire koncentrationer) til eventuelle yderligere analyseserier vælges på grundlag af de individuelle resultater af alle de tidligere analyseserier (8). De slutkoncentrationer, der kan anvendes, er 1, 2, 3, 4, 5, 7,5, 10, 12,5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 og 200 μg/ml. Den maksimale slutkoncentration er 200 μg/ml.Hvis der observeres en positiv CD86-værdi ved 1 μg/ml, evalueres en koncentration på 0,1 μg/ml for at finde frem til den koncentration af testkemikaliet, der ikke inducerer CD86 over den positive tærskel. EC150 (den koncentration, ved hvilken et kemikalie når den positive CD86-tærskel på 150 %, jf. punkt 19) beregnes for hver analyseserie, hvis en positiv CD86-koncentration/respons observeres. Hvor testkemikaliet inducerer en positiv CD86-respons, der ikke er koncentrationafhængig, er EC150 måske ikke relevant som beskrevet i U-SENS™ DB-ALM protokol nr. 183 (12). For hver analyseserie beregnes CV70 (den koncentration, ved hvilket kemikaliet når cytotoksicitetsgrænsen på 70 %, se punkt 19), når det er muligt (12). Med henblik på at undersøge koncentration/responsvirkningen af stigningen i CD86 bør en hvilken som helst koncentration fra de koncentrationer, der kan anvendes, vælges jævnt fordelt mellem EC150 (eller den højeste CD86-negative ikke-cytotoksiske koncentration) og CV70 (eller den højeste tilladte koncentration, dvs. 200 μg/ml). Der bør testes minimum fire koncentrationer i hver analyseserie, idet mindst to af koncentrationerne skal være de samme som i den/de tidligere analyseserie/analyseserier med henblik på sammenligning.
|
|
15.
|
Den opløsningsmiddel-/bærestofkontrol, der anvendes i U-SENS™-testen, er komplet medium (for testkemikalier, der er opløst eller stabilt dispergeret i komplet medium) (jf. punkt 4) eller 0,4 % DMSO i komplet medium (for testkemikalier, der er opløst eller stabilt dispergeret i DMSO).
|
|
16.
|
Den positive kontrol, der anvendes i U-SENS™-testen, er TNBS (jf. punkt 11), der fremstilles i komplet medium. TNBS bør anvendes som den positive kontrol ved CD86-ekspressionsmåling ved en enkelt slutkoncentration i pladen (50 μg/ml), der giver en cellelevedygtighed på > 70 %. For at opnå en 50 μg/ml koncentration af TNBS i pladen fremstilles 1 M (dvs. 293 mg/ml) stamopløsning af TNBS i komplet medium, som fortyndes yderligere 2 930-fold med komplet medium til en arbejdsopløsning på 100 μg/ml. Mælkesyre (LA, CAS 50-21-5) bør anvendes som den negative kontrol ved 200 μg/ml opløst i komplet medium (fra en stamopløsning på 0,4 mg/ml). I hver enkelt plade for hver enkelt analyseserie fremstilles tre replikater af komplet medium ubehandlet kontrol, opløsningsmiddel-/bærestofkontrol, negative og positive kontroller (12). Andre egnede positive kontroller kan anvendes, hvis der foreligger historiske data, som kan anvendes til at udlede sammenlignelige acceptkriterier for analyseserien. Acceptkriterierne for analyseserien er de samme som dem, der er beskrevet for testkemikaliet (jf. punkt 12).
|
Påføring af testkemikalier og kontrolstoffer
|
17.
|
Den opløsningsmiddel-/bærestofkontrol og de arbejdsopløsninger, der er beskrevet i punkt 14-16, blandes i forholdet 1:1 (v/v) med de cellesuspensioner, der er forberedt i den fladbundede 96-brønds plade (jf. punkt 12). De behandlede plader inkuberes derefter i 45±3 timer ved 37 °C og 5 % CO2. Forud for inkubationen forsegles pladerne med en semipermeabel membran for at undgå fordampning af flygtige testkemikalier og krydskontaminering mellem celler, der er behandlet med testkemikalier (12).
|
Cellefarvning
|
18.
|
Efter 45±3 timers eksponering overføres cellerne til en V-formet mikrotiterplade og indsamles ved centrifugering. Opløselighedsinterferens defineres som krystaller eller dråber, der observeres under mikroskop 45 ± 3 timer efter behandling (før cellefarvning). Supernatanterne kasseres, og de tilbageværende celler vaskes en gang med 100 μl af en iskold phosphatbufret fysiologisk saltopløsning (PBS) indeholdende 5 % føtalt kalveserum (farvebuffer). Efter centrifugering genopslæmmes cellerne med 100 μl af farvebufferen og farves med 5 μl (dvs. 0,25 μg) FITC-mærket anti-CD86- eller muse-IgG1 (isotype)-antistoffer ved 4 °C i 30 min. i mørke. De antistoffer, der er beskrevet i U-SENS™ DB-ALM protokol nr. 183 (12), bør anvendes (for CD86: BD-PharMingen #5556 57 Klon: Fun-1, eller Caltag/Invitrogen # MHCD8601 Klon: BU63; og for IgG1: BD-PharMingen #5557 48 eller Caltag/Invitrogen # GM4992). Det er testudviklernes erfaring, at antistoffernes fluorescensintensitet normalt er konsekvent mellem forskellige partier. Andre kloner eller leverandører af antistoffer, der har bestået reaktivitetskontrollen, kan anvendes til assayet (jf. punkt 11).Brugerne kan dog overveje at titrere antistofferne under deres egne laboratorieforhold for at finde frem til de koncentrationer, der er bedst egnede. Andre påvisningssystemer som f.eks. fluorchrom-mærkede anti-CD86-antistoffer kan anvendes, hvis det kan påvises, at de giver resultater svarende til FITC-konjugerede antistoffer, f.eks. via testning af de kompetencestoffer, der findes opført i tillæg 2.2. Efter vask med 100 μl farvebuffer to gange og en gang med 100 μl iskold PBS genopslæmmes cellerne i iskold PBS (f.eks. 125 μl for prøver, der analyseres manuelt rør for rør, eller 50 μl, når der anvendes en auto-sampler plade), og PI-opløsning tilsættes (slutkoncentration på 3 μg/ml). Andre cytotoksicitetsmarkører som f.eks. 7-Aminoactinomycin D (7-AAD), eller trypanblåt kan anvendes, hvis det kan påvises, at denne alternative farvning giver resultater svarende til PI, f.eks. via testning af de kompetencestoffer, der findes opført i tillæg 2.2.
|
Flowcytometrianalyse:
|
19.
|
Ekspressionsniveauerne for CD86 og cellelevedygtigheden analyseres ved hjælp af flowcytometri. Cellerne vises i et punktdiagram over størrelse (FSC) og granularitet (SSC) i logaritmisk skala for klart at identificere populationen i en første gate R1 og eliminere affald. Der høstes i alt 10 000 celler i gate R1 for hver enkelt brønd. Celler fra den samme gate R1 vises i et FL3- eller FL4/SSC-punktdiagram. De levedygtige celler beskrives ved at anbringe en anden gate R2, der frasorterer populationen af propidiumiodid-negative celler (kanal FL3 eller FL4). Cellelevedygtigheden kan beregnes af cytometeranalyseprogrammet ved hjælp af nedenstående ligning. Når cellelevedygtigheden er lav, kan der høstes op til 20 000 celler inklusive døde celler. Alternativt kan der høstes data i et minut efter, at analysen er startet.

Procentdelen af FL1-positive celler måles dernæst blandt disse levedygtige celler, der er gated på R2 (inden for R1). Celleoverfladeekspressionen af CD86 analyseres i et FL1/SSC punktdiagram gated på levedygtige celler (R2).
For komplet medium-/IgG1-brønde sættes analysemarkøren tæt på hovedpopulationen, så de komplette mediekontroller har IgG1, der ligger inden for målområdet på 0,6 til 0,9 %.
Farveinterferens defineres som en ændring af det FITC-mærkede IgG1-punktdiagram (IgG1 FL1 Geo Mean SI ≥ 150 %).
Stimuleringsindekset (SI) for CD86 i kontrolcellerne (ubehandlede eller i 0,4 % DMSO) og de kemisk behandlede celler beregnes i henhold til nedenstående ligning:.
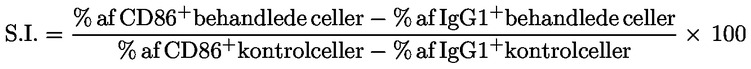
% af ubehandlede IgG1+ kontrolceller: benævnt procentdelen af FL1-positive IgG1-celler defineret med analysemarkøren (accepteret interval på ≥ 0,6 % og < 1,5 %, jf. punkt 22) blandt de ubehandlede levedygtige celler.
% af IgG1+/CD86+-kontrolceller/behandlede celler: benævnt procentdelen af FL1-positive IgG1/CD86-celler målt uden at flytte analysemarkøren blandt de levedygtige kontrolceller/behandlede celler.
|
DATA OG RAPPORTERING
Dataevaluering
|
20.
|
Følgende parametre beregnes i U-SENS™ -testen: CV70-værdien, dvs. en koncentration, der giver en U937-celleoverlevelse på 70 % (30 % cytotoksisk effekt), og EC150-værdien, dvs. den koncentration, ved hvilken testkemikalierne inducerede et CD86-stimuleringsindeks (SI) på 150 %.
CV70 beregnes ved log-lineær interpolation ved hjælp af følgende ligning:
CV70 = C1 + [(V1 - 70) / (V1 – V2) * (C2 – C1)]
hvor:
V1 er minimumsværdien for cellelevedygtighed over 70 %
V2 er maksimumsværdien for cellelevedygtighed under 70 %
C1 og C2 er de koncentrationer, der viser henholdsvis V1 og V2-værdien for cellelevedygtighed.

Der kan anvendes andre fremgangsmåder til at udlede CV70-værdien, når blot det påvises, at dette ikke har nogen indvirkning på resultaterne (f.eks. via testning af kompetencestofferne).
EC150 beregnes ved log-lineær interpolation ved hjælp af følgende ligning:
EC150 = C1 + [(150 – SI1) / (SI2 – SI1) * (C2 – C1)]
hvor:
C1 er den højeste koncentration udtrykt i μg/ml med et CD86 SI < 150 % (SI 1)
C2 er den laveste koncentration udtrykt i μg/ml med et CD86 SI < 150 % (SI 2).

EC150- og CV70-værdierne beregnes
|
—
|
For hver analyseserie: De individuelle EC150- og CV70-værdier anvendes som redskaber til at undersøge koncentration/responsvirkningen af stigningen i CD86 (jf. punkt 14).
|
|
—
|
Den overordnede CV70 bestemmes på grundlag af den gennemsnitlige levedygtighed (12).
|
|
—
|
De overordnede EC150-værdier bestemmes for det testkemikalie, der forudsiges at være POSITIVT med U-SENS™, på grundlag af de gennemsnitlige SI for CD86-værdierne (jf. punkt 21) (12).
|
|
Forudsigelsesmodel
|
21.
|
I forbindelse med CD86-ekspressionsmåling testes hvert testkemikalie i mindst fire koncentrationer og i mindst to uafhængige analyseserier (der foretages på forskellige dage) for at udlede en enkelt forudsigelse (POSITIV eller NEGATIV).
|
—
|
Den individuelle konklusion på en U-SENS™-analyseserie betragtes som NEGATIV (herefter N), hvis SI for CD86 er på under 150 % ved alle ikke-cytotoksiske koncentrationer (cellelevedygtighed ≥ 70 %), og hvis der ikke observeres nogen interferens (cytotoksisk effekt, opløselighed: jf. punkt 18 eller farve: jf. punkt 19, uanset ved hvilke ikke-cytotoksiske koncentrationer interferens påvises). I alle andre tilfælde: SI for CD86 er højere end eller lig med 150 %, og/eller interferens observeres, den individuelle konklusion på en U-SENS™-analyseserie betragtes som POSITIV (herefter P).
|
|
—
|
En U-SENS™-forudsigelse betragtes som NEGATIV, hvis mindst to uafhængige analyseserier er negative (N) (figur 1). Hvis de to første analyseserier begge er negative (N), betragtes U-SENS™-forudsigelsen som NEGATIV, og en tredje analyseserie er unødvendig.
|
|
—
|
En U-SENS™-forudsigelse betragtes som POSITIV, hvis mindst to uafhængige analyseserier er positive (P) (figur 1). Hvis de to første analyseserier begge er positive (P), betragtes U-SENS™-forudsigelsen som POSITIV, og en tredje analyseserie er unødvendig.
|
|
—
|
Da der ikke foretages et dosisbestemmelsesassay, er der en undtagelse, hvis SI for CD86 i første analyseserie er højere end eller lig med 150 % udelukkende ved den højeste ikke-cytotoksiske koncentration. Analyseserien betragtes i dette tilfælde som ikke entydig (NC), og yderligere koncentrationer (mellem den højeste ikke-cytotoksiske koncentration og den laveste cytotoksiske koncentration – jf. punkt 20) bør testes i supplerende analyseserier. Hvis en analyseserie påvises som NC, bør der foretages mindst to ekstra analyseserier, og en fjerde analyseserie, hvis forsøg 2 og 3 ikke er overensstemmende (N og/eller P) (figur 1). Opfølgende analyseserier vil blive betragtet som positive, selv hvis en af de ikke-cytotoksiske koncentrationer giver en CD86-værdi på eller over 150 %, eftersom koncentrationen er blevet tilpasset det specifikke testkemikalie. Den endelige forudsigelse baseres på flertalsresultatet af de tre eller fire individuelle analyseserier (dvs. to ud af tre eller to ud af fire) (figur 1).
|

Figur 1: Den forudsigelsesmodel, der anvendes i U-SENS™-testen. En U-SENS™-forudsigelse bør overvejes inden for rammerne af en IATA og i overensstemmelse med bestemmelserne i punkt 4 og punkt 7, 8 og 9 i den generelle indledning.
N: Analyseserie med ingen CD86 positiv eller interferens observeret.
P: Analyseserie med CD86 positiv og/eller interferens observeret.
NC: Ikke entydig. Første analyseserie med Ikke entydig, hvor CD86 udelukkende er positiv ved den højeste ikke-cytotoksiske koncentration.
#: En Ikke entydig (NC) individuel konklusion, der kun tilskrives den første analyseserie, medfører automatisk et behov for en tredje analyseserie for at opnå et flertal af positive (P) eller negative (N) konklusioner i mindst to ud af tre uafhængige analyseserier.
$: Boksene viser de relevante kombinationer af resultater fra de tre analyseserier på grundlag af de resultater, der er opnået i de to første analyseserier vist i boksen ovenfor.
o: Boksene viser de relevante kombinationer af resultater fra de fire analyseserier på grundlag af de resultater, der er opnået i de tre første analyseserier vist i boksen ovenfor.
|
Acceptkriterier
|
22.
|
Følgende acceptkriterier bør være opfyldt, når U-SENS™-testen anvendes (12):
|
—
|
Efter eksponeringsperioden på 45±3 timer skulle den gennemsnitlige levedygtighed af de tre prøver af ubehandlede U937-celler være > 90 %, og ingen afvigelse i CD86-ekspressionen observeres. Den grundlæggende CD86-ekspression af ubehandlede U937-celler skulle ligge i intervallet ≥ 2 % og ≤ 25 %.
|
|
—
|
Når DMSO anvendes som opløsningsmiddel, vurderes gyldigheden af DMSO-bærestofkontrollen ved at beregne et DMSO SI sammenlignet med ubehandlede celler, og den gennemsnitlige levedygtighed af celler i de tre prøver skulle være > 90 %. DMSO-bærestofkontrollen er gyldig, hvis middelværdien af dens tre CD86 SI var lavere end 250 % af middelværdien for de tre CD86 SI af ubehandlede U937-celler.
|
|
—
|
Analyseserierne betragtes som gyldige, hvis mindst to ud af tre IgG1-værdier for ubehandlede U937-celler lå inden for intervallet ≥ 0,6 % og < 1,5 %.
|
|
—
|
Den samtidigt gennemførte negative kontrol (mælkesyre) betragtes som gyldig, hvis mindst to ud af tre replikater var negative (CD86 SI < 150 %) og ikke-cytotoksiske (cellelevedygtighed ≥ 70 %).
|
|
—
|
Den positive kontrol (TNBS) betragtes som gyldig, hvis mindst to ud af tre replikater var positive (CD86 SI ≥ 150 %) og ikke-cytotoksiske (cellelevedygtighed ≥ 70 %).
|
|
Testrapport
|
23.
|
Testrapporten bør indeholde følgende oplysninger:
|
Testkemikalie
Stof med kun én bestanddel
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
fysisk udseende, opløselighed i komplet medium, DMSO-opløselighed, molekylvægt og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt
|
|
renhed, urenheders kemiske identitet i det omfang, det er relevant og praktisk muligt osv.
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel/bærestof for de enkelte testkemikalier.
|
Stof med flere bestanddele, UVCB-stoffer og blandinger:
|
så vidt muligt kendetegnet ved f.eks. kemisk identitet (jf. ovenfor), renhed, kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber (jf. ovenfor) i det omfang, det er muligt
|
|
fysisk udseende, opløselighed i komplet medium, DMSO-opløselighed og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt
|
|
molekylvægt eller tilsyneladende molekylvægt i tilfælde af blandinger/polymerer med kendt sammensætning eller andre oplysninger, der er relevante for undersøgelsens gennemførelse
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel/bærestof for de enkelte testkemikalier.
|
|
|
Kontrol
Positiv kontrol
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
fysisk udseende, DMSO-opløselighed, molekylvægt og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt og relevant
|
|
renhed, urenheders kemiske identitet i det omfang, det er relevant og praktisk muligt osv.
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
reference til historiske positive kontrolresultater, der udviser passende acceptkriterier for analyseserien, hvis det er relevant.
|
Negativ kontrol og opløsningsmiddel-/bærestofkontrol
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
renhed, urenheders kemiske identitet i det omfang, det er relevant og praktisk muligt osv.
|
|
fysisk udseende, molekylvægt og yderligere relevante fysisk-kemiske egenskaber ved brug af andre opløsningsmidler/bærestoffer end dem, der er nævnt i testvejledningen, i det omfang, det er muligt
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel/bærestof for de enkelte testkemikalier.
|
|
|
Testbetingelser
|
navn og adresse på sponsoren, testfaciliteten og undersøgelseslederen
|
|
beskrivelse af den anvendte test
|
|
anvendt cellelinje, opbevaringsforhold og kilde (f.eks. det anlæg, hvorfra den er anskaffet)
|
|
den anvendte flowcytometri (f.eks. model), herunder de anvendte instrumentindstillinger, antistoffer og den anvendte cytotoksicitetsmarkør
|
|
den procedure, der er blevet anvendt til at påvise laboratoriets kompetence til at anvende forsøgsmetoden via testning af kompetencestoffer, og den metode, der er blevet anvendt til at påvise reproducerbare resultater over tid, f.eks. historiske kontroldata og/eller historiske reaktivitetskontroldata.
|
|
|
Acceptkriterier for testen
|
cellelevedygtighed og CD86 SI-værdier opnået ved opløsningsmiddel-/bærestofkontrol sammenlignet med acceptintervallerne
|
|
cellelevedygtighed og SI-værdier opnået ved den positive kontrol sammenlignet med acceptintervallerne
|
|
cellelevedygtighed ved alle de testede koncentrationer af det testede kemikalie.
|
|
|
Forsøgsmetode
|
antal anvendte analyseserier
|
|
koncentrationer af testkemikaliet, anvendt påførsels- og eksponeringstid (hvis en anden end den anbefalede)
|
|
beskrivelse af de anvendte evaluerings- og beslutningskriterier
|
|
beskrivelse af eventuelle ændringer i forsøgsmetoden.
|
|
|
Resultater
|
tabulering af data, herunder CV70 (hvis muligt), SI, cellelevedygtighedsværdier, EC150-værdier (hvis muligt), opnået for testkemikaliet og for den positive kontrol i hver enkelt analyseserie, og en angivelse af klassificeringen af testkemikaliet i forhold til forudsigelsesmodellen
|
|
beskrivelse af eventuelle andre relevante observationer, hvis det er relevant.
|
|
|
Diskussion af resultaterne
|
diskussion af de resultater, der er opnået med U-SENS™-testen
|
|
vurdering af testresultaterne inden for rammerne af en IATA, hvis der foreligger andre relevante oplysninger.
|
|
Konklusioner
|
LITTERATUR
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EC EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Tilgængelig på: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations.
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN doi 10.2 787/8157 37. Tilgængelig på: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Tilgængelig på: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: [http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683-1 696.
|
|
(11)
|
OECD. (2018). Udkast til vejledende dokument: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), s. 33 ff. Tilgængelig på:[http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
United Nations (UN) (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York & Geneva: United Nations Publications. Tilgængelig på: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. Tilgængelig på: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Tillæg 2.1
DEFINITIONER
Nøjagtighed
: Overensstemmelsen mellem forsøgsmetodens resultater og accepterede referenceværdier. Den er et mål for testens ydeevne og et af aspekterne af relevans. Udtrykket benyttes ofte som synonym for »konkordans«, forstået som den andel af korrekte udfald, som en forsøgsmetode fører til (14).
AOP (Adverse Outcome Pathway)
: Sekvens af begivenheder fra den kemiske struktur af et målkemikalie eller en gruppe af ensartede kemikalier gennem den molekylære udløsende begivenhed til et in vivo-resultat af betydning (15).
CD86-koncentration/respons
: Der forekommer koncentrationsafhængighed (eller koncentration/respons), når en positiv koncentration (CD86 SI ≥ 150) efterfølges af en koncentration med en stigende CD86 SI.
Kemikalie
: Et stof eller en blanding.
CV70
: Den anslåede koncentration, der viser en cellelevedygtighed på 70 %.
Afvigelse
: En afvigelse defineres ved i) den korrigerede %CD86+-værdi af det ubehandlede kontrolreplikat 3 er mindre end 50 % af den gennemsnitlige korrigerede %CD86+-værdi for de ubehandlede kontrolreplikater 1 og 2, og ii) den korrigerede %CD86+-værdi af det negative kontrolreplikat 3 er mindre end 50 % af den gennemsnitlige korrigerede %CD86+-værdi for de negative kontrolreplikater 1 og 2.
EC150
: De anslåede koncentrationer, der giver 150 % SI for CD86-ekspression.
Flowcytometri
: En cytometrisk teknik, i hvilken celler opslæmmet i en væske flyder en ad gangen gennem et fokus bestående af magnetiserende lys, som spredes i mønstre, der er karakteristiske for cellerne og deres bestanddele. Cellerne mærkes hyppigt med fluorescerende markører, så lyset først absorberes og derefter udsendes ved ændrede frekvenser.
Fare
: Den iboende egenskab ved et stof eller en situation, som kan have en skadelig virkning, når en organisme, et system eller en (sub)population eksponeres for det pågældende stof.
IATA (integreret tilgang til testning og vurdering)
: En struktureret tilgang, der bruges til identifikation af farer (potentiale), farebeskrivelse (styrke) og/eller sikkerhedsvurdering (potentiale/styrke og eksponering) af et kemikalie eller en gruppe af kemikalier, som på strategisk vis integrerer og vægter alle relevante data som grundlag for lovgivningsmæssige beslutninger om mulige farer og/eller risici og/eller behovet for yderligere målrettede og derfor færrest mulige test.
Blanding
: Blanding eller opløsning, der består af to eller flere stoffer.
Stof med kun én bestanddel
: Et stof, der defineres ved dets mængdemæssige sammensætning, hvori en hovedbestanddel udgør mindst 80 % (w/w).
Stof med flere bestanddele
: Et stof, der defineres ved dets mængdemæssige sammensætning, hvori mere end en hovedbestanddel er til stede i en koncentration på ≥ 10 % (w/w) og < 80 % (w/w). Et stof, der består af flere forskellige bestanddele, er resultatet af en fremstillingsproces. Forskellen mellem en blanding og et stof med forskellige bestanddele er, at blandingen er fremstillet ved blanding af to eller flere stoffer uden kemisk reaktion. Et stof, der består af flere forskellige bestanddele, er resultatet af en kemisk reaktion.
Positiv kontrol
: Et replikat, som indeholder alle komponenterne fra et forsøgssystem og er behandlet med et stof, som vides at inducere en positiv respons. For at sikre at det er muligt at vurdere variabiliteten i den positive kontrolrespons over tid, bør den positive respons ikke være overdrevent stor.
Præ-haptener
: Kemikalier, der bliver allergifremkaldende via abiotisk omdannelse, f.eks. via oxidation.
Pro-haptener
: Kemikalier, der kræver en enzymaktivering for at udløse det hudsensibiliserende potentiale.
Relevans
: Beskrivelse af sammenhængen mellem testen og den pågældende virkning og dens meningsfuldhed og brugbarhed til et bestemt formål. Det angives, i hvilket omfang testen måler eller forudsiger den pågældende biologiske virkning korrekt. Testens nøjagtighed (konkordans) indgår i vurderingen af relevansen (14).
Pålidelighed
: Mål for, i hvilken grad en test kan udføres reproducerbart i og mellem laboratorier over tid, når den udføres efter samme protokol. Pålideligheden bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier og repeterbarhed inden for laboratorier (14).
Analyseserie
: En analyseserie består af et eller flere testkemikalier, der testes samtidig med en opløsningsmiddel-/bærestofkontrol og med en positiv kontrol.
Følsomhed
: Andel af alle positive/aktive kemikalier, der klassificeres korrekt ved testen. Følsomheden er et mål for nøjagtigheden af en test, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en tests relevans (14).
SI
: Stimuleringsindeks. Relative værdier for det geometriske gennemsnit af fluorescensintensitet i kemisk behandlede celler sammenlignet med celler behandlet med opløsningsmiddel.
Opløsningsmiddel-/bærestofkontrol
: En ubehandlet prøve, som indeholder alle komponenterne fra et testsystem, herunder det anvendte opløsningsmiddel/bærestof. Den anvendes til at fastlægge basislinjeresponsen for de prøver, som er behandlet med testkemikaliet opløst eller stabilt dispergeret i det samme opløsningsmiddel/bærestof. Når denne prøve testes med en samtidig mediekontrol, viser den også, hvorvidt opløsningsmidlet eller bærestoffet interagerer med testsystemet.
Specificitet
: Andel af alle negative/inaktive kemikalier, der klassificeres korrekt ved testen. Specificiteten er et mål for nøjagtigheden af en test, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en tests relevans (14).
Farvebuffer
: En phosphatbufret fysiologisk saltopløsning indeholdende 5 % føtalt kalveserum.
Stof
: Et grundstof og forbindelser heraf, i naturlig tilstand eller industrielt fremstillet, herunder sådanne tilsætningsstoffer, som er nødvendige til bevarelse af stoffets stabilitet, og sådanne urenheder, som følger af fremstillingsprocessen, bortset fra opløsningsmidler, der kan udskilles, uden at det påvirker stoffets stabilitet eller ændrer dets sammensætning.
Testkemikalie
: Alle stoffer eller blandinger, der testes ved hjælp af denne forsøgsmetode.
De Forenede Nationers globalt harmoniserede system for klassificering og mærkning af kemikalier (United Nations Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: Et system til klassificering af kemikalier (stoffer og blandinger) efter standardiserede typer og niveauer af fysisk fare, sundhedsfare og miljøfare med tilhørende kommunikationselementer såsom piktogrammer, signalord, faresætninger, sikkerhedssætninger og sikkerhedsdatablade, således at oplysninger om kemikaliernes skadelige virkninger kan videreformidles med henblik på at beskytte mennesker (f.eks. arbejdsgivere, arbejdstagere, transportører, forbrugere og redningspersonel) og miljøet (16).
UVCB
: Stoffer med ukendt eller variabel sammensætning, komplekse reaktionsprodukter eller biologiske materialer.
Gyldig test
: En test, der anses for at være tilstrækkeligt relevant og pålideligt til et bestemt formål, og som er baseret på videnskabeligt forsvarlige principper. En test er aldrig gyldig i absolut forstand, men kun i relation til et bestemt formål (14).
Tillæg 2.2
KOMPETENCESTOFFER
Forud for den rutinemæssige brug af den test, der er beskrevet i dette tillæg til forsøgsmetode B.71, bør laboratorierne godtgøre deres tekniske kompetence ved at opnå den forventede U-SENS™-forudsigelse på korrekt vis for de ti stoffer, der anbefales i tabel 1, og ved at opnå de CV70- og EC150-værdier, der falder ind under de respektive referenceintervaller for otte ud af de ti kompetencestoffer. Disse kompetencestoffer er udvalgt således, at de repræsenterer responsområdet for hudsensibiliseringsfare. Andre udvælgelseskriterier var, at kemikalierne er kommercielt tilgængelige, at der foreligger in vivo-referencedata af høj kvalitet, og at der er genereret in vitro-data af høj kvalitet med U-SENS™-forsøgsmetoden. Der foreligger ligeledes offentliggjorte referencedata for U-SENS™-forsøgsmetoden (1) (8).
Tabel 1
Anbefalede stoffer til godtgørelse af teknisk kompetence i tilknytning til U-SENS™-testen
|
Kompetencestoffer
|
CAS RN
|
Fysisk form
|
In vivo forudsigelse (106)
|
U-SENS Opløsningsmiddel/bærestof
|
U-SENS CV70-reference-interval i μg/ml (107)
|
U-SENS EC150-reference-interval i μg/ml) (107)
|
|
4-Phenylenediamin
|
106-50-3
|
Fast stof
|
Sensibiliserende stof (stærk)
|
Komplet medium (108)
|
<30
|
Positiv (≤10)
|
|
Picrylsulfonsyre
|
2508-19-2
|
Væske
|
Sensibiliserende stof (stærk)
|
Komplet medium
|
>50
|
Positiv (≤50)
|
|
Diethylmaleat
|
141-05-9
|
Væske
|
Sensibiliserende stof (moderat)
|
DMSO
|
10-100
|
Positiv (≤20)
|
|
Resorcinol
|
108-46-3
|
Fast stof
|
Sensibiliserende stof (moderat)
|
Komplet medium
|
>100
|
Positiv (≤50)
|
|
Kanelalkohol
|
104-54-1
|
Fast stof
|
Sensibiliserende stof (svag)
|
DMSO
|
>100
|
Positiv (10-100)
|
|
4-Allylanisol
|
140-67-0
|
Væske
|
Sensibiliserende stof (svag)
|
DMSO
|
>100
|
Positiv (<200)
|
|
Sakkarin
|
81-07-2
|
Fast stof
|
Ikke-sensibiliserende
|
DMSO
|
>200
|
Negativ (>200)
|
|
Glycerol
|
56-81-5
|
Væske
|
Ikke-sensibiliserende
|
Komplet medium
|
>200
|
Negativ (>200)
|
|
Mælkesyre
|
50-21-5
|
Væske
|
Ikke-sensibiliserende
|
Komplet medium
|
>200
|
Negativ (>200)
|
|
Salicylsyre
|
69-72-7
|
Fast stof
|
Ikke-sensibiliserende
|
DMSO
|
>200
|
Negativ (>200)
|
|
Forkortelser: CAS RN = Registernummer i Chemical Abstracts Service
|
Tillæg 3
IN VITRO-HUDSENSIBILISERING: IL-8 LUC-ASSAY
INDLEDENDE OVERVEJELSER OG BEGRÆNSNINGER
|
1.
|
I modsætning til assays, der analyserer ekspressionen af celleoverflademarkører, måler IL8-Luc-assay ændringerne i IL-8-ekspressionen, et cytokin knyttet til aktiveringen af dendritiske celler (DC). I IL-8 reporter-cellelinjen udledt af THP-1 (THP-G8, fastlagt på grundlag af human akut monocytisk leukæmi-cellelinjen THP-1) måles IL-8-ekspressionen efter eksponering for sensibiliserende stoffer (1). Ekspressionen af luciferase anvendes derefter til at understøtte differentieringen mellem sensibiliserende stoffer og ikke-sensibiliserende stoffer.
|
|
2.
|
IL-8 Luc-assayet er blevet evalueret i en valideringsundersøgelse (2) udført af det japanske center for validering af alternative metoder (JaCVAM), økonomi-, handels- og industriministeriet (METI), og det japanske selskab for alternativer til dyreforsøg (JSAAE) og efterfølgende underkastet en uafhængig peer review (3) under ledelse af JaCVAM og sundheds-, arbejds- og velfærdsministeriet (MHLW) med støtte fra det internationale samarbejde om alternative forsøgsmetoder (ICATM). På grundlag af al tilgængelig dokumentation og input fra myndigheder og interessenter anses IL-8 Luc-assayet for at være nyttigt som en del af IATA til støtte for differentieringen mellem sensibiliserende stoffer og ikke-sensibiliserende stoffer med henblik på fareklassificering og mærkning. Der findes i litteraturen eksempler på anvendelsen af data fra IL-8 Luc-assayet i kombination med andre oplysninger (4) (5) (6).
|
|
3.
|
Det viste sig, at IL-8 Luc-assayet kunne overføres til laboratorier med erfaring i celledyrkning og luciferasemåling. Reproducerbarheden inden for og mellem laboratorier var på henholdsvis 87,7 % og 87,5 % (2). Data genereret i valideringsundersøgelsen (2) og andre publicerede værker (1) (6) viser, at IL-8 Luc-assayet i modsætning til LLNA bedømte 118 ud af 143 kemikalier som positive eller negative og 25 kemikalier som ikke entydige, og nøjagtigheden af IL-8 Luc-assayet med hensyn til differentiering mellem hudsensibiliserende stoffer (UN GHS-/CLP-kategori 1) og ikke-sensibiliserende stoffer (UN GHS-/CLP-ingen kategori) er 86 % (101/118) med en følsomhed på 96 % (92/96) og en specificitet på 41 % (9/22). Når der ses bort fra stoffer, der ligger uden for det nedenfor beskrevne anvendelsesområde (punkt 5), bedømte IL-8 Luc-assayet 113 ud af 136 kemikalier som positive eller negative og 23 kemikalier som ikke entydige, og nøjagtigheden af IL-8 Luc-assayet er 89 % (101/113) med en følsomhed på 96 % (92/96) og en specificitet på 53 % (9/17). Ved hjælp af de i Urbisch et al. (7) nævnte humandata bedømte IL-8 Luc-assayet 76 ud af 90 kemikalier som positive eller negative og 14 kemikalier som ikke entydige, og nøjagtigheden er 80 % (61/76), følsomheden er 93 % (54/58) og specificiteten er 39 % (7/18). Når der ses bort fra stoffer, der ligger uden for anvendelsesområdet, bedømte IL-8 Luc-assayet 71 ud af 84 kemikalier som positive eller negative og 13 kemikalier som ikke entydige, og nøjagtigheden er 86 % (61/76)med en følsomhed på 93 % (54/58) og en specificitet på 54 % (7/18). Det er mere sandsynligt, at falsk negative forudsigelser i forbindelse med IL-8 Luc-assayet vil forekomme i tilknytning til kemikalier med lav til moderat hudsensibiliserende styrke (UN GHS-/CLP-underkategori 1B) end i tilknytning til kemikalier med en høj hudsensibiliserende styrke (UN GHS-/CLP-underkategori 1A) (6). Samlet set fremgår det af oplysningerne, at IL-8 Luc-assayet kan bidrage til identificeringen af hudsensibiliseringsfarer. Den angivne nøjagtighed for IL-8 Luc-assayet som en enkeltstående test er kun vejledende, da testen bør vurderes i kombination med andre informationskilder i forbindelse med en IATA og i overensstemmelse med bestemmelserne i punkt 7 og 8 i den generelle indledning. Når forsøgsmetoder uden brug af dyr vurderes i tilknytning til hudsensibilisering, bør man desuden huske på, at LLNA og andre dyreforsøg måske ikke fuldt ud afspejler situationen hos mennesker.
|
|
4.
|
På grundlag af de data, der er tilgængelige i øjeblikket, har IL-8 Luc-assayet vist sig at være relevant for testkemikalier, der omfatter en række forskellige organiske funktionelle grupper, reaktionsmekanismer, hudsensibiliseringsstyrker (som fastlagt ved in vivo-undersøgelser) og fysisk-kemiske egenskaber (2) (6).
|
|
5.
|
Selv om IL-8 Luc-assayet anvender X-VIVOTM 15 som opløsningsmiddel, bedømte det korrekt kemikalier med log Kow >3,5 og kemikalier med en vandopløselighed på omkring 100 μg/ml som beregnet ved EPI SuiteTM, og dens kapacitet til at påvise sensibiliserende stoffer med dårlig vandopløselighed er bedre end for IL-8 Luc-assayet, der anvender dimethylsulfoxid (DMSO) som opløsningsmiddel (2). De negative resultater for testkemikalier, der ikke er opløst ved 20 mg/ml, kan dog medføre falsk negative resultater på grund af den manglende evne til at opløses i X-VIVOTM 15. De negative resultater for disse kemikalier bør derfor ikke tages i betragtning. Der sås en høj andel af falske negativer for anhydrider i valideringsundersøgelsen. På grund af cellelinjens begrænsede metaboliske kapacitet (8) og testbetingelserne kan pro-haptener (stoffer, der kræver metabolisk aktivering) og præ-haptener (stoffer, der aktiveres ved oxidation) desuden give negative resultater i assayet. Selv om de negative resultater for stoffer, der mistænkes for at være præ-/pro-haptener, skal fortolkes med forsigtighed, bedømte IL-8 Luc-assayet imidlertid 11/11 præ-haptener, 6/6 pro-haptener, og 6/8 præ-/pro-haptener i IL-8 Luc-assay-datasættet korrekt (2). På grundlag af den seneste detaljerede gennemgang af tre forsøg uden brug af dyr (DPRA, KeratinoSens™ og h-CLAT) til påvisning af præ- og pro-haptener (9), og på grundlag af den omstændighed, at THP-G8-celler, der anvendes i IL-8 Luc-assayet er en cellelinje, der er udledt af THP-1, som anvendes i h-CLAT, kan IL-8 Luc-assayet også bidrage til at øge følsomheden af forsøg uden brug af dyr og til at påvise præ- og pro-haptener i kombination med andre test. De overfladeaktive stoffer, der er blevet testet indtil videre, gav (falsk) positive resultater uanset deres type (f.eks. kationaktiv, anionaktiv eller nonionaktiv). Endelig kan kemikalier, der interfererer med luciferase, maskere dets aktivitet/måling, hvilket forårsager en tydelig inhibering eller øget luminescens (10). Eksempelvis findes der rapporter om, at fytoøstrogenkoncentrationer på over 1μM forstyrrede luminescenssignalerne i andre luciferasebaserede reportergenassays på grund af overaktivering af luciferasereportergenet. Som følge heraf skal den luciferaseekspression, som er opnået ved høje koncentrationer af fytoøstrogener eller forbindelser, der mistænkes for at forårsage fytoøstrogenlignende aktivering af luciferasereportergenet, undersøges nøje (11). Det fremgår af det ovenstående, at overfladeaktive stoffer, anhydrider og kemikalier, der interfererer med luciferase, ligger uden for anvendelsesområdet for dette assay. I de tilfælde, hvor der foreligger beviser for, at IL-8 Luc-assayet ikke kan anvendes på andre specifikke kategorier af testkemikalier, bør denne metode ikke anvendes til disse specifikke kategorier.
|
|
6.
|
Som beskrevet ovenfor understøtter IL-8 Luc-assayet differentieringen mellem hudsensibiliserende og ikke-sensibiliserende stoffer. Der er behov for yderligere arbejde, fortrinsvis baseret på humane data, med henblik på at bestemme, om IL-8 Luc-resultaterne kan bidrage til styrkevurderingen, når den betragtes i kombination med andre informationskilder.
|
|
7.
|
Definitioner findes i tillæg 3.1.
|
PRINCIP FOR TESTEN
|
8.
|
IL-8 Luc-assayet anvender en human monocytisk leukæmi-cellelinje, THP-1, der er erhvervet fra American Type Culture Collection (Manassas, VA, USA). Ved hjælp af denne cellelinje udviklede afdelingen for dermatologi, Tohoku University School of Medicine, en THP-1-udledt IL-8 reporter-cellelinje, THP-G8, der indeholder henholdsvis Stable Luciferase Orange (SLO) og Stable Luciferase Red (SLR) luciferasegenerne under kontrol af IL-8- og glyceraldehyd 3-fosfat dehydrogenase (GAPDH)- aktivatorer (1). Dette giver mulighed for kvantitativ måling af luciferasegeninduktion ved hjælp af veldokumenterede lysproducerende luciferasesubstrater som en indikator for aktiviteten af IL-8 og GAPDH i celler efter eksponering for sensibiliserende kemikalier.
|
|
9.
|
Assaysystemet med to farver omfatter luciferase, der udsender orange lys (SLO; λmax = 580 nm) (12) for genekspressionen af IL-8-aktivatoren, og luciferase, der udsender rødt lys (SLR; λmax = 630 nm) (13) for genekspressionen af den interne kontrolaktivator, GAPDH. De to luciferaser udsender forskellige farver, når de reagerer med firefly d-luciferin, og deres luminescens måles samtidigt i en et-trinsreaktion ved at dele emissionen fra assayblandingen ved hjælp af et optisk filter (14) (tillæg 3.2).
|
|
10.
|
THP-G8-cellerne behandles med testkemikaliet i 16 timer, hvorefter SLO luciferaseaktiviteten (SLO-LA), der afspejles i IL-8-aktivatoraktiviteten, og SLR luciferaseaktiviteten (SLR-LA), der afspejles i GAPDH-aktivatoraktiviteten, måles. For at gøre forkortelserne letforståelige betegnes SLO-LA og SLR-LA som henholdsvis IL8LA og GAPLA. Tabel 1 indeholder en beskrivelse af de udtryk, der er knyttet til luciferaseaktiviteten i IL-8 Luc-assayet. De målte værdier anvendes til at beregne den standardiserede IL8LA-værdi (nIL8LA), som er forholdet mellem IL8LA og GAPLA; induktionen af nIL8LA (Ind-IL8LA), der er forholdet mellem det aritmetiske gennemsnit af firdobbelt-målte værdier for nIL8LA i THP-G8-celler behandlet med et testkemikalie og værdierne for nIL8LA i ubehandlede THP-G8-celler; og inhiberingen af GAPLA (Inh-GAPLA), der er forholdet mellem det aritmetiske gennemsnit af firdobbelt-målte værdier for GAPLA i THP-G8-celler behandlet med et testkemikalie og værdierne for GAPLA i ubehandlede THP-G8-celler, og anvendes som en indikator for cytotoksisk effekt.
Tabel 1
Beskrivelse af de udtryk, der er knyttet til luciferaseaktiviteten i IL-8 Luc-assayet
|
Forkortelser
|
Definition
|
|
GAPLA
|
SLR luciferaseaktivitet, der afspejler GAPDH-aktivatoraktiviteten
|
|
IL8LA
|
SLO luciferaseaktivitet, der afspejler IL-8-aktivatoraktiviteten
|
|
nIL8LA
|
IL8LA/GAPLA
|
|
Ind-IL8LA
|
nIL8LA i THP-G8-celler behandlet med kemikalier/nIL8LA i ubehandlede celler
|
|
Inh-GAPLA
|
GAPLA i THP-G8-celler behandlet med kemikalier/GAPLA i ubehandlede celler
|
|
CV05
|
Den laveste koncentration af kemikaliet, ved hvilken Inh-GAPLA bliver < 0,05.
|
|
|
11.
|
Der stilles ydeevnestandarder (15) til rådighed for at lette valideringen af ændrede in vitro-IL-8-luciferaseforsøg svarende til IL-8 Luc-assayet og for at muliggøre en rettidig ændring af OECD-testvejledning 442E, så de kan medtages. OECD's Mutual Acceptance of Data (MAD) vil kun være garanteret for forsøg, der er valideret i henhold til ydeevnestandarderne, hvis disse test er gennemgået og medtaget i OECD-testvejledning 442E (16).
|
PÅVISNING AF KOMPETENCE
|
12.
|
Forud for den rutinemæssige brug af det forsøg, der er beskrevet i dette tillæg til forsøgsmetode B.71, bør laboratorierne godtgøre deres tekniske kompetence ved hjælp af de ti kompetencestoffer, der er opført i tillæg 3.3. i overensstemmelse med Good in vitro Method Practices (17). Desuden bør brugerne af testen opretholde en historisk database over data, der er genereret ved hjælp af reaktivitetskontrollerne (jf. punkt 15) og ved hjælp af de positive kontroller og opløsningsmiddel-/bærestofkontrollerne (jf. punkt 21-24), og de bør anvende disse data til at bekræfte, at reproducerbarheden af testen i deres laboratorium opretholdes over tid.
|
FREMGANGSMÅDE
|
13.
|
Der foreligger standardprocedurer (SOP) for IL-8 Luc-assayet, og de bør anvendes ved udførelsen af testen (18). Laboratorier, der er villige til at udføre testen, kan erhverve recombinant THP-G8-cellelinjerne fra GPC Lab. Co.Ltd., Tottori, Japan, når de har underskrevet en materialeoverførselsaftale (MTA) i overensstemmelse med betingelserne i OECD-skabelonen. Følgende punkter indeholder en beskrivelse af de vigtigste komponenter og fremgangsmåder i assayet.
|
Præparering af celler
|
14.
|
THP-G8-cellelinjen fra GPC Lab. Co. Ltd., Tottori, Japan, bør anvendes til udførelse af IL-8 Luc-assayet (jf. punkt 8 og 13). Ved modtagelsen opformeres cellerne (f.eks. 2-4 opformeringer) og opbevares frosne som en homogen stamme. Celler fra denne stamme kan opformeres højst 12 gange eller i højst seks uger. Det medium, der anvendes til opformering, er RPMI-1640 dyrkningsmedium indeholdende 10 % føtalt kalveserum (FBS), antibiotika-/antimykotikumopløsning (100U/ml penicillin G, 100μg/ml streptomycin og 0,25 μg/ml amfotericin B i 0,85 % fysiologisk saltopløsning) (f.eks. GIBCO Cat#15240-062), 0,15 μg/ml Puromycin (f.eks. CAS:58-58-2) og 300 μg/ml G418 (f.eks. CAS:108321-42-2).
|
|
15.
|
Inden cellerne anvendes til testning, bør de valideres ved at gennemføre en reaktivitetskontrol. Denne kontrol bør foretages 1-2 uger eller 2-4 generationer efter optøning ved hjælp af den positive kontrol, 4-nitrobenzylbromid (4-NBB) (CAS:100-11-8, ≥ 99 % renhed) og den negative kontrol, mælkesyre (LA) (CAS:50-21-5, ≥ 85 % renhed). 4-NBB bør give en positiv respons på Ind-IL8LA (≥1.4), mens mælkesyre bør give en negativ respons på Ind-IL8LA (< 1,4). Kun celler, der består reaktivitetskontrollen, anvendes til assayet. Reaktivitetskontrollen bør foretages i henhold til de fremgangsmåder, der er beskrevet i punkt 22-24.
|
|
16.
|
Når THP-G8-cellerne skal anvendes til testning, podes de ved en tæthed på 2 til 5 × 105 celler/ml, og forkultiveres i dyrkningsflasker i 48-96 timer. På dagen for gennemførelse af testen vaskes celler høstet fra dyrkningsflasken med RPMI-1640 indeholdende 10 % FBS uden nogen antibiotika, og genopslæmmes derefter med RPMI-1640 indeholdende 10 % FBS uden nogen antibiotika ved 1 × 106 celler/ml. Derefter fordeles cellerne i en fladbundet sort 96-brønds plade (f.eks. Costar Cat#3603) med 50 μl (5 × 104 celler/brønd).
|
Fremstilling af testkemikalie og kontrolstoffer
|
17.
|
Testkemikaliet og kontrolstofferne fremstilles på dagen for testen. I forbindelse med IL-8 Luc-assayet opløses testkemikalierne i X-VIVOTM 15, et kommercielt tilgængeligt serumfrit medium (Lonza, 04-418Q), til slutkoncentrationen på 20 mg/ml. X-VIVOTM 15 tilsættes til 20 mg af testkemikaliet (uanset kemikaliets opløselighed) i et mikrocentrifugerør, og der fyldes op til et volumen på 1 ml, hvorefter der omrystes kraftigt, og der rystes i en rotor ved en maksimumshastighed på 8 o/min. i 30 min. ved rumtemperatur på omkring 20oC. Hvis kemikalier i fast form fortsat er uopløselige, ultralydsbehandles de desuden, indtil kemikaliet er opløst fuldstændigt eller er stabilt dispergeret. Når testkemikalierne er opløselige i X-VIVOTM 15, fortyndes opløsningen med en faktor 5 med X-VIVOTM 15 og anvendes som en X-VIVOTM 15 stamopløsning af testkemikaliet (4 mg/ml). Når testkemikalierne ikke er opløselige i X-VIVOTM 15, rystes blandingen igen i rotoren i mindst 30 min., hvorefter den centrifugeres ved 15000 o/min. (≈20000g) i 5 min. Den fremkaldte supernatant anvendes som en X-VIVOTM 15 stamopløsning af testkemikaliet. Hvis der benyttes andre opløsningsmidler som f.eks. DMSO, vand eller dyrkningsmediet, bør der fremlægges tilstrækkeligt videnskabeligt belæg for dette. Den detaljerede procedure for opløsning af kemikalier fremgår af tillæg 3.5. De X-VIVOTM 15-opløsninger, der er beskrevet i punkt 18-23, blandes i forholdet 1:1 (v/v) med de cellesuspensioner, der er forberedt i den fladbundede sorte 96-brønds plade (jf. punkt 16).
|
|
18.
|
Den første analyseserie er beregnet til at bestemme den cytotoksiske koncentration og undersøge kemikaliernes hudsensibiliserende potentiale. Ved hjælp af X-VIVOTM 15 foretages der seriefortyndinger af X-VIVOTM 15 stamopløsningerne af testkemikalierne med en fortyndingsfaktor to (jf. tillæg 3.5) ved hjælp af en 96-brønds blok (f.eks. Costar Cat#EW-01729-03). Derefter tilsættes 50 μl/brønd fortyndet opløsning til 50 μl af cellesuspensionen i en fladbundet sort 96-brønds plade. Når testkemikalierne er opløselige i X-VIVOTM 15, ligger slutkoncentrationerne for testkemikalierne i intervallet fra 0,002 til 2 mg/ml (tillæg 3.5). Når testkemikalierne ikke er opløselige i X-VIVOTM 15 ved 20 mg/ml, anvendes kun fortyndingsfaktorer i intervallet fra 2 til 210, selv om de faktiske slutkoncentrationer af testkemikaliet forbliver usikre og afhænger af den mættede koncentration af testkemikalierne i X-VIVOTM 15 stamopløsningen.
|
|
19.
|
I de efterfølgende analyseserier (dvs. det andet, tredje og fjerde replikat) laves X-VIVOTM 15 stamopløsningen i en koncentration, der er fire gange højere end koncentrationen for cellelevedygtighed 05 (CV05; den laveste koncentration, ved hvilken Inh-GAPLA bliver < 0,05) i det første forsøg. Hvis Inh-GAPLA ikke falder under 0,05 ved den højeste koncentration i den første analyseserie, laves X-VIVOTM 15 stamopløsningen som den højeste koncentration ved første analyseserie. Koncentrationen af CV05 beregnes ved at dividere koncentrationen af stamopløsningen i første analyseserie med fortyndingsfaktoren for CV05 (X) (fortyndingsfaktor CV05 (X); den fortyndingsfaktor, der er nødvendig for at fortynde stamopløsningen til CV05) (jf. tillæg 3.5). For testkemikalier, der ikke er opløselige i X-VIVO ved 20 mg/ml, fastsættes CV05 som koncentrationen af stamopløsningen × 1/X. Til 2.-4. analyseserie fremstilles en ny stamopløsning på 4 × CV05 (tillæg 3.5).
|
|
20.
|
Seriefortyndinger af X-VIVOTM 15 anden stamopløsning foretages med en fortyndingsfaktor på 1,5 ved hjælp af en 96-brønds blok. Derefter tilsættes 50 μl/brønd fortyndet opløsning til 50 μl af cellesuspensionen i brøndene i en fladbundet sort 96-brønds plade. Hver enkelt koncentration af hvert testkemikalie bør testes i fire brønde. Prøverne blandes derefter på en pladeryster og inkuberes i 16 timer ved 37oC og 5 % CO2, hvorefter luciferaseaktiviteten måles som beskrevet nedenfor.
|
|
21.
|
Kontrolkulturen med opløsningsmiddel er blandingen af 50 μl/brønd af X-VIVOTM 15 og 50 μl/brønd af cellesuspensionen i RPMI-1640 indeholdende 10 % FBS.
|
|
22.
|
Den anbefalede positive kontrol er 4-NBB. 20 mg af 4-NBB fremstilles i et 1,5-ml mikrocentrifugeglas, hvortil der tilsættes X-VIVOTM 15 op til 1 ml. Glasset omrystes kraftigt, og der rystes i en rotor ved en maksimumshastighed på 8 o/min. i mindst 30 min. Efter centrifugering ved 20000g i 5 min. fortyndes supernatanten med en faktor 4 med X-VIVOTM 15, og 500 μl af den fortyndede supernatant overføres til en 96-brønds blok. Den fortyndede supernatant fortyndes med X-VIVOTM 15 med faktorer på 2 og 4, og 50 μl af opløsningen tilsættes til 50 μl af THP-G8-cellesuspensionen i brøndene i den fladbundede 96-brønds plade (tillæg 3.6). Hver enkelt koncentration af den positive kontrol bør testes i fire brønde. Pladen rystes på en pladeryster og inkuberes i en CO2 inkubator i 16 timer (37oC, 5 % CO2), hvorefter luciferaseaktiviteten måles som beskrevet i punkt 29.
|
|
23.
|
Den anbefalede negative kontrol er mælkesyre LA. 20 mg af mælkesyren fremstilles i et 1,5-ml mikrocentrifugeglas, hvortil der tilsættes X-VIVOTM 15 op til 1 ml (20 mg/ml). 20 mg/ml af mælkesyreopløsningen fortyndes med en faktor 5 med X-VIVOTM 15 (4 mg/ml). 500 μl af denne 4 mg/ml mælkesyreopløsning overføres til en brønd i en 96-brønds blok. Denne opløsning fortyndes med en faktor 2 med X-VIVOTM 15 og fortyndes derefter igen med en faktor 2 for at opnå 2 mg/ml og 1 mg/ml opløsninger. 50 μl af disse tre opløsninger og bærestofkontrollen (X-VIVOTM 15) tilsættes til 50 μl af THP-G8-cellesuspensionen i brøndene i den fladbundede sorte 96-brønds plade. Hver enkelt koncentration af den negative kontrol testes i fire brønde. Pladen rystes på en pladeryster og inkuberes i en CO2 inkubator i 16 timer (37oC, 5 % CO2), hvorefter luciferaseaktiviteten måles som beskrevet i punkt 29.
|
|
24.
|
Andre egnede positive kontroller kan anvendes, hvis der foreligger historiske data, som kan anvendes til at udlede sammenlignelige acceptkriterier for analyseserien.
|
|
25.
|
Der bør træffes forholdsregler for at undgå, at flygtige testkemikalier fordamper, og for at undgå krydskontaminering med testkemikalier mellem brøndene, f.eks. ved at forsegle pladerne inden inkubation med testkemikalierne.
|
|
26.
|
De negative testkemikalier og kontrolkulturen med opløsningsmiddel kræver 2-4 analyseserier, for at der kan udledes en positiv eller negativ forudsigelse (jf. tabel 2). De enkelte analyseserier foretages på forskellige dage med en frisk X-VIVOTM 15 stamopløsning af testkemikalier og uafhængigt høstede celler. Cellerne kan stamme fra den samme opformering.
|
Målinger af luciferaseaktivitet
|
27.
|
Luminescens måles ved hjælp af et 96-brønds mikropladeluminometer med optiske filtre, f.eks. Phelios (ATTO, Tokyo, Japan), Tristan 941 (Berthold, Bad Wildbad, Germany) og ARVO-serien (PerkinElmer, Waltham, MA, USA). Luminometeret skal kalibreres for hver test for at sikre reproducerbarhed (19). Denne kalibrering kan foretages ved hjælp af rekombinante luciferaser, der udsender orange og rødt lys.
|
|
28.
|
100 μl opvarmet Tripluc® Luciferase assay reagens (Tripluc) overføres til hver enkelt brønd på pladen indeholdende cellesuspensionen behandlet med kemikaliet eller ubehandlet. Pladen rystes i 10 min. ved rumtemperatur på omkring 20oC. Pladen anbringes i luminometeret, så lucififeraseaktiviteten kan måles. Bioluminescensen måles i 3 sekunder uden (F0) og med (F1) det optiske filter. Anvendes der alternative indstillinger, f.eks. som følge af den anvendte luminometermodel, bør dette begrundes.
|
|
29.
|
Parametrene for hver enkelt koncentration beregnes ud fra de målte værdier, f.eks. IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA og den gennemsnitlige ±SD (standardafvigelse) for IL8LA, den gennemsnitlige ±SD for GAPLA, den gennemsnitlige ±SD for nIL8LA, den gennemsnitlige ±SD for Ind-IL8LA, den gennemsnitlige ±SD for Inh-GAPLA og 95 % konfidensintervallet for Ind-IL8LA. Definitionerne af de parametre, der anvendes i dette punkt, findes i henholdsvis tillæg 3.1 og 3.4.
|
|
30.
|
Forud for måling opnås farveopløsningsevnen i flerfarvede reporterassays generelt ved hjælp af detektorer (luminometer og pladelæser) udstyret med optiske filtre såsom kontrastfiltre (passage af lange eller korte bølgelængder) eller båndpasfiltre. Transmissionskoefficienterne for filtrene for hver enkelt bioluminescensignalfarve bør kalibreres forud for testning, jf. tillæg 3.2.
|
DATA OG RAPPORTERING
Dataevaluering
|
31.
|
Kriterierne for en positiv/negativ afgørelse kræver for hver analyseserie følgende:
|
—
|
En IL-8 Luc-assay-forudsigelse betragtes som positiv, hvis et testkemikalie har et Ind-IL8LA ≥ 1,4, og den nedre grænse for 95 % konfidensintervallet for Ind-IL8LA ≥ 1,0.
|
|
—
|
En IL-8 Luc-assay-forudsigelse betragtes som negativ, hvis et testkemikalie har et Ind-IL8LA < 1,4, og/eller den nedre grænse for 95 % konfidensintervallet for Ind-IL8LA < 1,0.
|
|
Forudsigelsesmodel
|
32.
|
Testkemikalier, der giver to positive resultater i 1., 2., 3. eller 4. analyseserie, identificeres som positive, mens de testkemikalier, der giver tre negative resultater i 1., 2., 3. eller 4. analyseserie, identificeres som formodet negative (tabel 2). Blandt de formodede negative kemikalier betragtes kemikalier, der er opløst i koncentrationen 20 mg/ml i X-VIVOTM 15 som negative, mens kemikalier, der ikke er opløst i koncentrationen 20 mg/ml i X-VIVOTM 15 ikke bør tages i betragtning (figur 1).
Tabel 2
Kriterier for identificering af positive og formodet negative
|
1. analyseserie
|
2. analyseserie
|
3. analyseserie
|
4. analyseserie
|
Endelig forudsigelse
|
|
Positiv
|
Positiv
|
–
|
–
|
Positiv
|
|
Negativ
|
Positiv
|
–
|
Positiv
|
|
Negativ
|
Positiv
|
Positiv
|
|
Negativ
|
Formodet negativ
|
|
Negativ
|
Positiv
|
Positiv
|
–
|
Positiv
|
|
Negativ
|
Positiv
|
Positiv
|
|
Negativ
|
Formodet negativ
|
|
Negativ
|
Positiv
|
Positiv
|
Positiv
|
|
Negativ
|
Formodet negativ
|
|
Negativ
|
–
|
Formodet negativ
|
|
Figur 1
Forudsigelsesmodel for den endelige bedømmelse
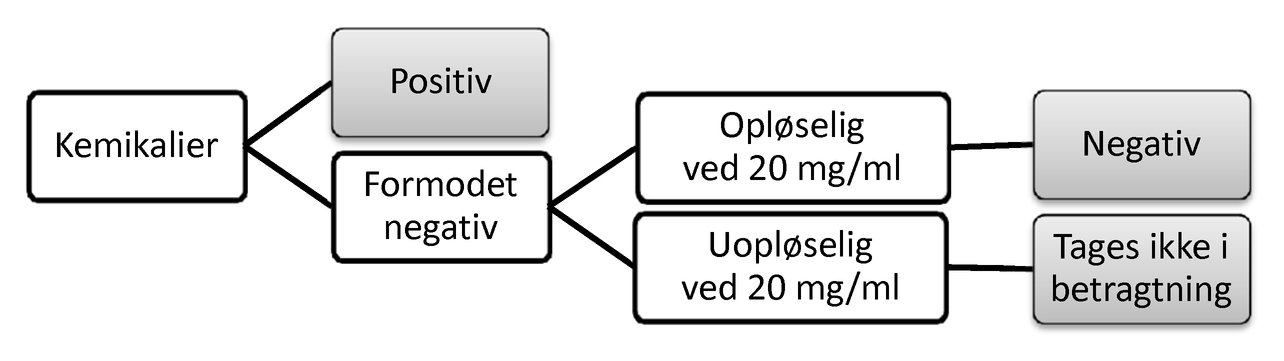
Acceptkriterier
|
33.
|
Følgende acceptkriterier bør være opfyldt, når IL-8 Luc-assayet anvendes:
|
—
|
Ind-IL8LA bør være over 5,0 ved mindst en koncentration af den positive kontrol, 4-NBB, i hver enkelt analyseserie.
|
|
—
|
Ind-IL8LA bør være under 1,4 ved enhver koncentration af den negative kontrol, mælkesyre, i hver enkelt analyseserie.
|
|
—
|
Data fra plader, for hvilke GAPLA i kontrolbrøndene med celler og Tripluc, men uden kemikalier er mindre end 5 gange værdien for en brønd, der kun indeholder testmedium (50 μl/brønd RPMI-1640 indeholdende 10 % FBS og 50 μl/brønd X-VIVOTM 15), bør forkastes.
|
|
—
|
Data fra plader, for hvilke Inh-GAPLA for alle koncentrationer af test- eller kontrolkemikalierne er mindre end 0,05, bør kasseres. I dette tilfælde bør den første test gentages, så den højeste koncentration af den gentagne test er den laveste slutkoncentration for den foregående test.
|
|
Testrapport
|
34.
|
Testrapporten bør indeholde følgende oplysninger:
|
Testkemikalier
Stof med kun én bestanddel:
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
fysisk udseende, vandopløselighed, molekylvægt og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt
|
|
renhed, urenheders kemiske identitet i det omfang, det er relevant og praktisk muligt osv.
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opløselighed i X-VIVOTM 15. For kemikalier, der er opløselige i X-VIVOTM 15, uanset om der ses bundfald eller flotation efter centrifugering
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel/bærestof for de enkelte testkemikalier, hvis X-VIVOTM 15 ikke er blevet anvendt.
|
Stof med flere bestanddele, UVCB-stoffer og blandinger:
|
så vidt muligt kendetegnet ved f.eks. kemisk identitet (jf. ovenfor), renhed, kvantitativ forekomst og bestanddelenes relevante fysisk-kemiske egenskaber (jf. ovenfor) i det omfang, det er muligt
|
|
fysisk udseende, vandopløselighed og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt
|
|
molekylvægt eller tilsyneladende molekylvægt i tilfælde af blandinger/polymerer med kendt sammensætning eller andre oplysninger, der er relevante for undersøgelsens gennemførelse
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opløselighed i X-VIVOTM 15. For kemikalier, der er opløselige i X-VIVOTM 15, uanset om der ses bundfald eller flotation efter centrifugering
|
|
opbevaringsforhold og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel/bærestof for de enkelte testkemikalier, hvis X-VIVOTM 15 ikke er blevet anvendt.
|
|
|
Kontrol
Positiv kontrol:
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre, SMILES- eller InChI-kode, strukturformel og/eller andre identifikatorer
|
|
fysisk udseende, vandopløselighed, molekylvægt og yderligere relevante fysisk-kemiske egenskaber i den udstrækning, det er muligt og relevant
|
|
renhed, kemisk identitet af urenheder i det omfang, det er relevant og praktisk muligt osv.
|
|
eventuel behandling inden testen (f.eks. opvarmning, formaling)
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
reference til historiske positive kontrolresultater, der udviser passende acceptkriterier for analyseserie, hvis det er relevant.
|
Negativ kontrol:
|
kemisk identifikation, f.eks. IUPAC- eller CAS-navn(e), CAS-nummer/numre og/eller andre identifikatorer
|
|
renhed, kemisk identitet af urenheder i det omfang, det er relevant og praktisk muligt osv.
|
|
fysisk udseende, molekylvægt og yderligere relevante fysisk-kemiske egenskaber ved brug af andre negative kontroller end dem, der er nævnt i testvejledningen, i det omfang, det er muligt
|
|
opbevaring og stabilitet i det omfang, det er muligt
|
|
begrundelse for valg af opløsningsmiddel for de enkelte testkemikalier.
|
|
|
Testbetingelser
|
navn og adresse på sponsoren, testfaciliteten og undersøgelseslederen
|
|
beskrivelse af den anvendte test
|
|
anvendt cellelinje, opbevaringsforhold og kilde (f.eks. det anlæg, hvorfra den er anskaffet)
|
|
partinummer og oprindelse af FBS, leverandørnavn, partinummer på den sorte 96-brønds fladbundede plade og partinummer på Tripluc-reagensen
|
|
opformeringsnummer og den celletæthed, der er anvendt til testning
|
|
den anvendte celletællingsmetode til opformering inden testen, og de foranstaltninger, der er truffet for at sikre en homogen cellefordeling
|
|
Det anvendte luminometer (f.eks. model), herunder instrumentindstillinger, det anvendte luciferasesubstrat og dokumentation for hensigtsmæssige luminescensmålinger baseret på den kontrol, der er beskrevet i tillæg 3.2
|
|
den fremgangsmåde, der er anvendt til at påvise laboratoriets kompetence til at anvende forsøgsmetoden (f.eks. ved testning af kompetencestoffer) eller til at påvise reproducerbare resultater af forsøgsmetoden over tid.
|
|
|
Forsøgsmetode
|
antal replikater og foretagne analyseserier
|
|
koncentrationer af testkemikaliet, tilsætningsprocedure og eksponeringstid (hvis forskellig fra det anbefalede)
|
|
beskrivelse af de anvendte evaluerings- og beslutningskriterier
|
|
beskrivelse af de benyttede undersøgelsesacceptkriterier
|
|
beskrivelse af eventuelle ændringer i forsøgsmetoden.
|
|
|
Resultater
|
målinger af IL8LA og GAPLA
|
|
beregninger af nIL8LA, Ind-IL8LA og Inh-GAPLA
|
|
95 % konfidensinterval for Ind-IL8LA
|
|
en graf, der viser dosis/respons-kurver for induktion af luciferaseaktivitet og levedygtighed
|
|
beskrivelse af eventuelle andre relevante observationer, hvis det er relevant.
|
|
|
Diskussion af resultater
|
diskussion af de resultater, der er opnået med IL-8 Luc-assayet
|
|
vurdering af resultaterne af assayet inden for rammerne af en IATA, hvis der foreligger andre relevante oplysninger.
|
|
Konklusion
|
LITTERATUR
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K og Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc-assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc-assay. Series on Testing og Assessment No 267, ENV/JM/MONO(2017)19. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H og Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y og Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N og Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation test: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A og Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J og Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M og Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, og Ohmiya Y. (1999). Cloning, sequence analysis, og expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M og Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OECD (2017). Publiceres senere - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OECD, Paris, France
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OECD, Paris, France.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Tilgængelig på: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol. Tilgængelig på: http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, og Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OECD, Paris, France.
|
|
(21)
|
De Forenede Nationer, 2015. Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1 Tilgængelig på: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Tillæg 3.1
DEFINITIONER
Nøjagtighed
: Overensstemmelsen mellem forsøgsmetodens resultater og accepterede referenceværdier. Den er et mål for testens ydeevne og et af aspekterne af relevans. Udtrykket benyttes ofte som synonym for »konkordans«, forstået som den andel af korrekte udfald, som en forsøgsmetode fører til (16).
AOP (Adverse Outcome Pathway)
: Sekvens af begivenheder fra den kemiske struktur af et målkemikalie eller en gruppe af ensartede kemikalier gennem den molekylære udløsende begivenhed til et in vivo -resultat af betydning (20).
Kemikalie
: Et stof eller en blanding.
CV05
: Cellelevedygtighed 05, dvs. den minimumskoncentration, ved hvilken kemikalier viser mindre end 0,05 af Inh-GAPLA.
FInSLO-LA
: Forkortelse, der er anvendt i valideringsrapporten og i tidligere publikationer vedrørende IL-8 Luc-assayet for at henvise til Ind-IL8LA. Jf. definitionen af Ind-IL8LA.
GAPLA
: Luciferaseaktiviteten i stabil luciferaserød (SLR) (λmaks. = 630 nm), reguleret af GAPDH-vækstfremmeren, viser cellelevedygtigheden og det levedygtige celleantal.
Fare
: Den iboende egenskab ved et stof eller en situation, som kan have en skadelig virkning, når en organisme, et system eller en (sub)population eksponeres for det pågældende stof.
IATA (integreret tilgang til testning og vurdering)
: En struktureret tilgang, der bruges til identifikation af farer (potentiale), farebeskrivelse (styrke) og/eller sikkerhedsvurdering (potentiale/styrke og eksponering) af et kemikalie eller en gruppe af kemikalier, som på strategisk vis integrerer og vægter alle relevante data som grundlag for lovgivningsmæssige beslutninger om mulige farer og/eller risici og/eller behovet for yderligere målrettede og derfor færrest mulige test.
II-SLR-LA
: Forkortelse, der er anvendt i valideringsrapporten og i tidligere publikationer vedrørende IL-8 Luc-assayet for at henvise til Inh-GAPLA. Jf. definitionen af Inh-GAPLA.
IL-8 (Interleukin-8)
: et cytokin udledt af endotele celler, fibroblaster, keratinocyter, makrofager og monocyter, der forårsager kemotaksi af neutrofiler og T-cellelymfocytter.
IL8LA
: Luciferaseaktiviteten i stabil luciferaseorange (SLO) (λmaks. = 580 nm), reguleret af IL-8-promoteren.
Ind-IL8LA
: Foldinduktion for nIL8LA. Denne værdi opnås ved at dividere nIL8LA for THP-G8-celler behandlet med kemikalier med nIL8LA for ikke-stimulerede THP-G8-celler og betegner kemikaliers induktion af IL-8-promoteraktivitet.
Inh-GAPLA
: Inhibering af GAPLA. Opnås ved at dividere GAPLA for THP-G8 behandlet med kemikalier med GAPLA for ubehandlede THP-G8 og betegner kemikaliers cytotoksiske effekt.
Minimumsinduktionstærskel (MIT)
: Den laveste koncentration, ved hvilken et kemikalie opfylder de positive kriterier.
Blanding
: Blanding eller opløsning, der består af to eller flere stoffer.
Stof med kun én bestanddel
: Et stof, der defineres ved dets mængdemæssige sammensætning, hvori en hovedbestanddel udgør mindst 80 % (w/w).
Stof med flere bestanddele
: Et stof, der defineres ved dets mængdemæssige sammensætning, hvori mere end en af hovedbestanddelene er til stede i en koncentration på ≥ 10 % (w/w) og < 80 % (w/w). Et stof, der består af flere forskellige bestanddele, er resultatet af en fremstillingsproces. Forskellen mellem en blanding og et stof med forskellige bestanddele er, at blandingen er fremstillet ved blanding af to eller flere stoffer uden kemisk reaktion. Et stof, der består af flere forskellige bestanddele, er resultatet af en kemisk reaktion.
nIL8LA
: SLO luciferaseaktiviteten, der afspejler IL-8-promoteraktiviteten (IL8LA) standardiseret ved hjælp af SLR luciferaseaktiviteten, der afspejler GAPDH-promoteraktiviteten (GAPLA). Denne værdi betegner IL-8-promoteraktiviteten efter hensyntagen til cellelevedygtigheden eller celleantallet.
nSLO-LA
: Forkortelse, der er anvendt i valideringsrapporten og i tidligere publikationer vedrørende IL-8 Luc-assayet for at henvise til nIL8LA. Jf. nIL8LA for en definition.
Positiv kontrol
: Et replikat, som indeholder alle komponenterne fra et forsøgssystem og er behandlet med et stof, som vides at inducere en positiv respons. For at sikre at det er muligt at vurdere variabiliteten i den positive kontrolrespons over tid, bør den positive respons ikke være overdrevent stor.
Præ-haptener
: Kemikalier, der bliver sensibiliserende stoffer via abiotisk omdannelse.
Pro-haptener
: Kemikalier, der kræver en enzymaktivering for at udløse det hudsensibiliserende potentiale.
Relevans
: Beskrivelse af sammenhængen mellem testen og den pågældende virkning og dens meningsfuldhed og brugbarhed til et bestemt formål. Det angives, i hvilket omfang testen måler eller forudsiger den pågældende biologiske virkning korrekt. Testens nøjagtighed (konkordans) indgår i vurderingen af relevansen (16).
Pålidelighed
: Mål for, i hvilken grad en test kan udføres reproducerbart i og mellem laboratorier over tid, når den udføres efter samme protokol. Pålideligheden bedømmes ved beregning af reproducerbarhed inden for og mellem laboratorier og repeterbarhed inden for laboratorier (16).
Analyseserie
: En analyseserie består af et eller flere testkemikalier, der testes samtidig med en opløsningsmiddel-/bærestofkontrol og med en positiv kontrol.
Følsomhed
: Andel af alle positive/aktive kemikalier, der klassificeres korrekt ved testen. Følsomheden er et mål for nøjagtigheden af en test, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en tests relevans (16).
SLO-LA
: Forkortelse, der er anvendt i valideringsrapporten og i tidligere publikationer vedrørende IL-8 Luc-assayet for at henvise til IL8LA. Jf. definitionen af IL8LA.
SLR-LA
: Forkortelse, der er anvendt i valideringsrapporten og i tidligere publikationer vedrørende IL-8 Luc-assayet for at henvise til GAPLA. Jf. definitionen af GAPLA.
Opløsningsmiddel-/bærestofkontrol
: En ubehandlet prøve, som indeholder alle komponenterne fra et testsystem, herunder det anvendte opløsningsmiddel/bærestof. Den anvendes til at fastlægge basislinjeresponsen for de prøver, som er behandlet med testkemikaliet opløst eller stabilt dispergeret i det samme opløsningsmiddel/bærestof. Når denne prøve testes med en samtidig mediekontrol, viser den også, hvorvidt opløsningsmidlet eller bærestoffet interagerer med testsystemet.
Specificitet
: Andel af alle negative/inaktive kemikalier, der klassificeres korrekt ved testen. Specificiteten er et mål for nøjagtigheden af en test, som giver kategoriske resultater, og er et vigtigt kriterium ved vurderingen af en tests relevans (16).
Stof
: Kemiske stoffer og forbindelser heraf, naturlige eller industrielt fremstillede, indeholdende sådanne tilsætningsstoffer, som er nødvendige til bevarelse af stoffets stabilitet, og sådanne urenheder, som følger af fremstillingsprocessen, bortset fra opløsningsmidler, som kan udskilles, uden at det påvirker stoffets stabilitet eller ændrer dets sammensætning.
Afspændingsmiddel
: Også kaldet for overfladeaktivt stof. Dette stof, f.eks. et vaske- og rengøringsmiddel, kan reducere overfladespændingen i en væske og dermed give den mulighed for at skumme eller trænge ind i faste stoffer. Det kaldes også for befugtningsmiddel. (TG437)
Testkemikalie
: Alle stoffer eller blandinger, der testes ved hjælp af denne metode.
THP-G8
: En IL-8-reportercellelinje, der anvendes i IL-8 Luc-assayet. Den humane makrofaglignende cellelinje THP-1 blev transfekteret med henholdsvis SLO og SLR luciferasegener under kontrollen af IL-8- og GAPDH-promotere.
De Forenede Nationers globalt harmoniserede system for klassificering og mærkning af kemikalier (United Nations Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: System til klassificering af kemikalier (stoffer og blandinger) efter standardiserede typer og niveauer af fysisk fare, sundhedsfare og miljøfare med tilhørende kommunikationselementer såsom piktogrammer, signalord, faresætninger, sikkerhedssætninger og sikkerhedsdatablade, således at oplysninger om kemikaliernes skadelige virkninger kan videreformidles med henblik på at beskytte mennesker (f.eks. arbejdsgivere, arbejdstagere, transportører, forbrugere og redningspersonel) og miljøet (21).
UVCB
: Stoffer med ukendt eller variabel sammensætning, komplekse reaktionsprodukter eller biologiske materialer.
Gyldig forsøgsmetode
: En test, der anses for at være tilstrækkeligt relevant og pålideligt til et bestemt formål, og som er baseret på videnskabeligt forsvarlige principper. En test er aldrig gyldig i absolut forstand, men kun i relation til et bestemt formål.
Tillæg 3.2
PRINCIP FOR MÅLING AF LUCIFERASEAKTIVITETEN OG BESTEMMELSE AF OPTISKE FILTRES TRANSMISSIONSKOEFFICIENTER FOR SLO OG SLR
MultiReporter Assay System – Tripluc – kan anvendes sammen med et luminometer af mikropladetypen med et flerfarvepåvisningssystem, der kan forsynes med et optisk filter (f.eks. Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). Det optiske filter, der anvendes til måling, er 600–620 nm filter, der tillader passage af lange eller korte bølgelængder, eller 600–700 nm båndpasfiltre.
Måling af tofarvede luciferaser med et optisk filter
Dette er et eksempel på brug af Phelios AB-2350 (ATTO). Dette luminometer er udstyret med et 600 nm filter, der tillader passage af lange bølgelængder (R60 HOYA Co.), 600 nm LP (filter 1) til opsplitning af SLO (λmaks. = 580 nm) og SLR (λmaks. = 630 nm) luminescens.
Med henblik på at bestemme transmissionskoefficienterne for 600 nm LP måles først ved hjælp af rensede SLO og SLR luciferaseenzymer i) SLO- og SLR-bioluminescensintensiteten uden filter (F0), ii) SLO og SLR bioluminescensintensiteten, der passerede igennem 600 nm LP (filter 1), og derefter iii) beregnes transmissionskoefficienterne for 600 nm LP til SLO og SLR som anført nedenfor.
|
Transmissionskoefficienter
|
Forkortelse
|
Definition
|
|
SLO
|
Filter 1 Transmissions-koefficienter
|
=κOR60
|
Filterets transmissions-koefficient for SLO
|
|
SLR
|
Filter 1 Transmissions-koefficienter
|
κRR60
|
Filterets transmissions-koefficient for SLR
|
Når intensiteten af SLO og SLR i prøven defineres som henholdsvis O og R, beskrives i) intensiteten af lys uden filter (alle optiske) F0 og ii) intensiteten af lys, der udsendes via 600 nm LP (filter 1) F1, som nedenfor.
F0=O+R
F1=κOR60 x O + κRR60 x R
Disse formler kan omformuleres således:

Ved hjælp af de beregnede transmissionskoefficienter (κOR60 og κRR60) og de målte F0 og F1, kan du beregne O- og R-værdien som følger:

Materialer og metoder til bestemmelse af transmissionskoefficient
|
1)
|
Reagenser
Enkelte rensede luciferaseenzymer:
|
Lyofiliseret renset SLO enzym
|
|
Lyofiliseret renset SLR enzym
|
|
(som i forbindelse med valideringsarbejdet blev anskaffet fra GPC Lab. Co. Ltd., Tottori, Japan med cellelinje THP-G8)
|
Assay reagens:
|
Tripluc® Luciferase assay reagens (f.eks. fra TOYOBO Cat#MRA-301)
|
Medium: til luciferase assay (30 ml, opbevaret ved 2-8 °C)
|
|
Reagens
|
Konc.
|
Slutkonc. i medium
|
Krævet mængde
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
2)
|
Fremstilling af enzymopløsning
Opløs lyofiliseret renset luciferaseenzym i et reagensglas ved at tilsætte 200 μl 10 ~ 100 mM Tris/HCl eller Hepes/HCl (pH 7.5 ~ 8.0) suppleret med 10 % (w/v) glycerol, del enzymopløsningen i 10 μl delprøver i 1,5 ml engangsrør, og opbevar dem i en fryser ved -80 °C. Den nedfrosne enzymopløsning kan anvendes i op til seks måneder. Ved brug tilsættes 1 ml af mediet til luciferase assay (RPMI-1640 med 10 % FBS) til hvert enkelt rør indeholdende enzymopløsninger (fortyndet enzymopløsning), og de opbevares på is for at forhindre deaktivering.
|
|
3)
|
Måling af bioluminescens
Optø Tripluc® Luciferase assay reagens (Tripluc), og opbevar det ved rumtemperatur enten i vandbad eller ved rumtemperatur. Start luminometeret, 30 min. inden målingen påbegyndes, så fotomultiplikatoren kan stabiliseres. Overfør 100 μl af den fortyndede enzymopløsning til en sort 96-brønds plade (fladbundet) (SLO referenceprøven til #B1, #B2, #B3, SLR referenceprøven til #D1, #D2, #D3). Overfør dernæst 100 μl opvarmet Tripluc til hver enkelt brønd på pladen indeholdende den fortyndede enzymopløsning ved hjælp af en pipetman. Omryst pladen i 10 min. ved rumtemperatur (omkring 25 °C) ved hjælp af en pladeryster. Opstår der bobler i opløsningerne i brøndene, fjernes de. Anbring pladen i luminometeret, og mål luciferaseaktiviteten. Bioluminescensen måles i 3 sekunder uden (F0) og med (F1) det optiske filter.
Transmissionskoefficienten for det optiske filter blev beregnet som følger:
Transmissionskoefficient (SLO (κOR60)) = (#B1 af F1+ #B2 af F1+ #B3 af F1) / (#B1 af F0+ #B2 af F0+ #B3 of F0)
Transmissionskoefficient (SLR (κRR60)) = (#D1 af F1+ #D2 af F1+ #D3 af F1) / (#D1 af F0+ #D2 af F0+ #D3 of F0)
Beregnede transmissionskoefficienter anvendes til alle målinger, der foretages ved hjælp af det samme luminometer.
|
Kvalitetskontrol af udstyr
De fremgangsmåder, der er beskrevet i IL-8 Luc-protokollen, bør anvendes (18).
Tillæg 3.3
KOMPETENCESTOFFER
Forud for den rutinemæssige brug af de forsøg, der er beskrevet i dette tillæg til forsøgsmetode B.71, bør laboratorierne godtgøre deres tekniske kompetence ved at opnå den forventede IL-8 Luc-assay-forudsigelse for de 9 stoffer, der anbefales i tabel 1, og ved at opnå værdier, der falder inden for de respektive referenceintervaller for mindst otte ud af 9 kompetencestoffer (udvalgt til at repræsentere reaktionsintervallet for hudsensibiliseringsfare). Andre udvælgelseskriterier var, at kemikalierne er kommercielt tilgængelige, og at der foreligger in vivo-referencedata af høj kvalitet samt in vitro-data af høj kvalitet, der er genereret med IL-8 Luc-assayet. Der foreligger ligeledes offentliggjorte referencedata for IL-8 Luc-assayet (6) (1).
Tabel 1
Anbefalede stoffer til godtgørelse af teknisk kompetence i tilknytning til IL-8 Luc-assayet
|
Kompetencestoffer
|
CAS nr.
|
Fysisk tilstand
|
Opløselighed i X-VIVO15 ved 20 mg/ml
|
In vivo-forudsigelse (109)
|
IL-8 Luc-forudsigelse (110)
|
Referenceinterval (μg/ml) (111)
|
|
|
CV05
(112)
|
IL-8 Luc MIT (113)
|
|
2,4-Dinitrochlorbenzen
|
97-00-7
|
Fast stof
|
Uopløselig
|
Sensibiliserende stof (Ekstrem)
|
Positiv
|
2,3-3,9
|
0,5-2,3
|
|
Formaldehyd
|
50-00-0
|
Væske
|
Opløseligt
|
Sensibiliserende stof (Stærk)
|
Positiv
|
9-30
|
4-9
|
|
2-Mercaptobenzothiazol
|
149-30-4
|
Fast stof
|
Uopløselig
|
Sensibiliserende stof (Moderat)
|
Positiv
|
250-290
|
60-250
|
|
Ethylendiamin
|
107-15-3
|
Væske
|
Opløseligt
|
Sensibiliserende stof (Moderat)
|
Positiv
|
500-700
|
0,1-0,4
|
|
Ethylenglycoldimethacrylat
|
97-90-5
|
Væske
|
Uopløselig
|
Sensibiliserende stof (Svag)
|
Positiv
|
>2000
|
0,04-0,1
|
|
4-Allylanisol (Estragol)
|
140-67-0
|
Væske
|
Uopløselig
|
Sensibiliserende stof (Svag)
|
Positiv
|
>2000
|
0,01-0,07
|
|
Streptomycinsulfat
|
3810-74-0
|
Fast stof
|
Opløseligt
|
Ikke-sensibiliserende
|
Negativ
|
>2000
|
>2000
|
|
Glycerol
|
56-81-5
|
Væske
|
Opløseligt
|
Ikke-sensibiliserende
|
Negativ
|
>2000
|
>2000
|
|
Isopropanol
|
67-63-0
|
Væske
|
Opløseligt
|
Ikke-sensibiliserende
|
Negativ
|
>2000
|
>2000
|
|
Forkortelser: CAS-nr. = Registernummer i Chemical Abstracts Service.
|
Tillæg 3.4
INDEKSER OG BEDØMMELSESKRITERIER
nIL8LA (nSLO-LA)
j-ende repetition (j = 1-4) af i-ende koncentration (i = 0-11) måles for henholdsvis IL8LA (SLO-LA) og GAPLA (SLR-LA). Den standardiserede IL8LA-værdi betegnes som nIL8LA (nSLO-LA) og defineres som:
nIL8LAij = IL8LAij/GAPLAij
Dette er den grundlæggende måleenhed i dette assay.
Ind-IL8LA (FInSLO-LA)
Foldstigningen for den gennemsnitlige nIL8LA (nSLO-LA) for repetition i den i-ende koncentration sammenlignet med 0-koncentrationen, Ind-IL8LA, er det primære mål for dette assay. Dette forhold kan sættes på følgende formel:

Det førende laboratorium har foreslået, at en værdi på 1,4 svarer til et positivt resultat for det testede kemikalie. Denne værdi er baseret på undersøgelsen af det førende laboratoriums historiske data. Dataforvaltningsteamet anvendte dernæst denne værdi i alle faser af valideringsundersøgelsen. Det primære resultat, Ind-IL8LA, er forholdet mellem to aritmetiske gennemsnit, som det fremgår af ligningen.
95 % konfidensinterval (95 % CI)
95 % konfidensintervallet (95 % CI) baseret på forholdet kan anses for at vise nøjagtigheden af dette primære resultat. Den nedre grænse for 95 % CI ≥ 1 indikerer, at nIL8LA ved den i-ende koncentration er betydeligt højere end ved kontrolkultur med opløsningsmiddel. 95 % CI kan fastsættes på flere måder. Vi anvendte den metode, der er kendt som Fiellers teorem, i denne undersøgelse. Dette teorem for 95 % konfidensinterval udregnes ved hjælp af følgende formel:
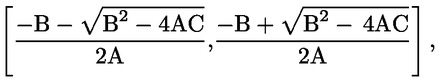
hvor




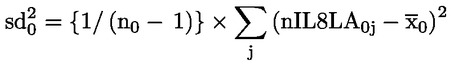
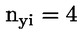

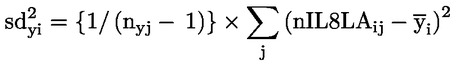
er 97,5 percentilen af den centrale t-fordeling med ν af frihedsgraden, hvor

Inh-GAPLA (II-SLR-LA)
Inh-GAPLA er forholdet mellem den gennemsnitlige GAPLA (SLR-LA)-værdi for repetitionen af den i-ende koncentration sammenlignet med den gennemsnitlige GAPLA (SLR-LA)-værdi for kontrolkulturen med opløsningsmiddel og kan sættes på følgende formel:
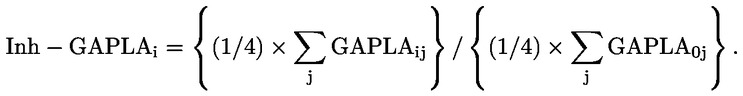
Da GAPLA er denominatoren for nIL8LA, forårsager ekstremt små værdier en stor variation i nIL8LA. Derfor kan Ind-IL8LA-værdier med ekstremt små værdier for Inh-GAPLA (under 0,05) anses for at være meget upræcise. [[G1]]
Tillæg 3.5
SKEMA OVER METODERNE TIL OPLØSNING AF KEMIKALIER TIL IL-8 LUC-ASSAYET
|
a)
|
For kemikalier, der er opløselige i X-VIVOTM 15 ved 20 mg/ml

|
|
b)
|
For kemikalier, der ikke er opløselige i X-VIVOTM 15 ved 20 mg/ml
|
Tillæg 3.6
SKEMA OVER METODEN TIL OPLØSNING AF 4-NBB TIL DEN POSITIVE KONTROL I FORBINDELSE MED IL-8 LUC-ASSAYET

" |