BILAG II
VIDENSKABELIGE KRAV TIL RISIKOVURDERING AF GENETISK MODIFICEREDE FØDEVARER OG FODERSTOFFER
I. INDLEDNING
1. DEFINITIONER
I dette bilag forstås ved:
1. »fareidentifikation«: identifikation af biologiske, kemiske og fysiske agenser, der kan forårsage sundhedsskadelige virkninger, og som kan være til stede i en bestemt fødevare/et bestemt foderstof eller i en gruppe af fødevarer/foderstoffer
2. »farekarakterisering«: kvalitativ og/eller kvantitativ vurdering af arten af de sundhedsskadelige virkninger, der er forbundet med biologiske, kemiske eller fysiske agenser, som kan være til stede i fødevarer og foderstoffer
3. »risikokarakterisering«: et kvalitativt og/eller kvantitativt skøn, under hensyntagen til de relevante usikkerheder, over sandsynligheden for og sværhedsgraden af kendte eller potentielle sundhedsskadelige virkninger i en given population, baseret på farekarakterisering og vurdering af eksponeringen.
2. SÆRLIGE ASPEKTER
2.1. Indsættelse af markørgener og andre nukleinsyresekvenser, som ikke er nødvendige for at opnå den ønskede egenskab
For at lette risikovurderingen skal ansøgeren bestræbe sig på at minimere forekomsten af indsatte nukleinsyresekvenser, der ikke er nødvendige for at opnå den ønskede egenskab.
Ved genetisk modifikation af planter og andre organismer anvendes der ofte markørgener for at gøre det lettere at udvælge og identificere genetisk modificerede celler, som indeholder interessegenet, der er indsat i værtsorganismens genom, blandt det store flertal af ikke-transformerede celler. Ansøgeren skal udvælge sådanne markørgener med omhu, og artikel 4, stk. 2, i direktiv 2001/18/EF skal overholdes. På denne baggrund skal ansøgeren derfor sigte mod at udvikle GMO'er uden brug af antibiotikaresistens-markørgener.
2.2. Risikovurdering af genetisk modificerede fødevarer og foderstoffer, der indeholder stablede transformationsbegivenheder (stacked events)
Til risikovurdering af genetisk modificerede fødevarer og foderstoffer, der indeholder stablede transformationsbegivenheder, som er opnået ved konventionel krydsning af genetisk modificerede planter indeholdende en eller flere transformationsbegivenheder, skal ansøgeren fremlægge en risikovurdering af hver enkelt transformationsbegivenhed eller, jf. artikel 3, stk. 6, i denne forordning, henvise til ansøgning(er), der allerede er indgivet. Risikovurderingen af genetisk modificerede fødevarer og foderstoffer, der indeholder stablede transformationsbegivenheder, skal også omfatte en vurdering af følgende aspekter:
|
a) |
transformationsbegivenhedernes stabilitet |
|
b) |
transformationsbegivenhedernes ekspression |
|
c) |
potentielle synergistiske eller antagonistiske virkninger af kombinationen af de pågældende transformationsbegivenheder — vurderes i overensstemmelse med afsnit 1.4 (Toksikologi), 1.5 (Allergenicitet) og 1.6 (Ernæringsmæssig vurdering). |
For genetisk modificerede fødevarer og foderstoffer, der indeholder, består af eller er fremstillet af genetisk modificerede planter, hvis dyrkning er forbundet med produktion af genetisk modificeret materiale indeholdende flere underkombinationer af transformationsbegivenheder (segregationsafgrøder), skal ansøgningen omfatte alle underkombinationer, som endnu ikke er tilladt, uafhængigt af deres oprindelse. I sådanne tilfælde skal ansøgeren fremlægge en videnskabelig begrundelse for, at det ikke er nødvendigt at fremlægge forsøgsdata vedrørende de pågældende underkombinationer, eller, i mangel af en sådan videnskabelig begrundelse, fremlægge de relevante forsøgsdata.
For genetisk modificerede fødevarer og foderstoffer, der indeholder, består af eller er fremstillet af genetisk modificerede planter, hvis dyrkning ikke indebærer produktion af genetisk modificeret materiale indeholdende flere forskellige underkombinationer af transformationsbegivenheder (ikke-segregationsafgrøder), skal ansøgningen kun omfatte den kombination, der skal markedsføres.
Bestemmelserne i dette afsnit finder tilsvarende anvendelse på transformationsbegivenheder, der er kombineret med andre metoder, såsom co- og retransformation.
II. VIDENSKABELIGE KRAV
1. FAREIDENTIFIKATION OG -KARAKTERISERING
1.1. Oplysninger om recipientplanter eller (hvis det er relevant) forældreplanter
|
1.1.1. |
Ansøgeren skal fremlægge omfattende oplysninger om recipient- eller (i givet fald) forældreplanterne, så det er muligt at:
|
|
1.1.2. |
Til de formål, der er omhandlet i punkt 1.1.1, skal ansøgeren fremlægge følgende oplysninger:
|
1.2. Molekylær karakterisering
1.2.1. Oplysninger om den genetiske modifikation
Ansøgeren skal fremlægge fyldestgørende oplysninger om den genetiske modifikation, så det er muligt at:
|
a) |
identificere den/de nukleinsyre(r), der ønskes indsat, og relevante vektorsekvenser, som potentielt kan videregives til recipientplanten |
|
b) |
karakterisere det den/de nukleinsyre(r), der faktisk er indsat i planten. |
1.2.1.1. Beskrivelse af de metoder, der er anvendt til den genetiske modifikation
Ansøgeren skal fremlægge oplysninger om følgende:
|
a) |
gentransformationsmetoden, herunder relevante referencer |
|
b) |
recipientplantematerialet |
|
c) |
Agrobacterium-arten og –stammen og andre mikrober, hvis en sådan/sådanne er anvendt i gentransformationsprocessen |
|
d) |
hjælperplasmiderne, hvis sådanne plasmider er anvendt i gentransformationsprocessen |
|
e) |
kilden til bærer-nukleinsyren/rne, hvis en sådan/sådanne er anvendt i den genetiske transformationsproces. |
1.2.1.2. Den anvendte vektors art og oprindelse
Ansøgeren skal fremlægge følgende oplysninger:
|
a) |
et fysisk kort over de funktionelle elementer og andre plasmid/vektorkomponenter sammen med de relevante oplysninger, der er nødvendige for at kunne fortolke de molekylære analyser (f.eks. restriktionssteder, primernes position ved polymerasekædereaktion (PCR) og probernes placering i Southern-analyser). Den region, der skal indsættes, skal angives tydeligt |
|
b) |
et skema, hvori hver enkelt bestanddel af plasmidet/vektoren (herunder den region, der skal indsættes) samt dens størrelse, oprindelse og tilsigtede funktion er angivet. |
1.2.1.3. Kilden til den/de til transformationen anvendte nukleinsyre(r) samt størrelse og tilsigtet funktion af hver bestanddel af den region, der skal indsættes
Ansøgeren skal fremlægge oplysninger om donororganismen eller donorganismerne og om den eller de nukleinsyresekvenser, der skal indsættes, for at afgøre, hvorvidt arten af donororganismen/donororganismerne eller nukleinsekvensen/nukleinsekvenserne potentielt kan være forbundet med en sikkerhedsrisiko.
Oplysningerne om funktionen af den eller de nukleinsyreregioner, der skal indsættes, skal omfatte følgende elementer:
|
a) |
hele sekvensen af den eller de nukleinsyre(r), der skal indsættes, herunder oplysninger om enhver aktivt foretaget ændring af den eller de pågældende sekvenser i donororganismen/donormekanismerne |
|
b) |
langvarig sikker anvendelse af det eller de genprodukter, der fremkommer af de regioner, der skal indsættes |
|
c) |
oplysninger om det mulige forhold mellem genprodukterne og kendte toksiner, næringshæmmende stoffer og allergener. |
Oplysningerne om hvert enkelt donororganisme skal omfatte:
|
— |
den taksonomiske klassifikation |
|
— |
erfaringerne med anvendelsen for så vidt angår fødevare- og fodersikkerheden. |
1.2.2. Oplysninger om den genetisk modificerede plante
1.2.2.1. Overordnet beskrivelse af de egenskaber og karakteristika, der er blevet indført eller ændret
Oplysningerne i dette punkt kan begrænses til en generel beskrivelse af den eller de indførte egenskaber og de deraf følgende ændringer i plantens fænotype og metabolisme.
Når den indførte egenskab f.eks. er herbicidtolerance, skal ansøgeren fremlægge oplysninger om aktivstoffets virkningsmekanisme og dets metabolisme i planten.
1.2.2.2. Oplysninger om faktisk indsatte/deleterede sekvenser
Ansøgeren skal fremlægge følgende oplysninger:
|
a) |
Størrelse og antal kopier af alle påviselige inserter, både komplette og delvise; dette bestemmes sædvanligvis ved Southern-analyse. Probe/restriktionsenzym-kombinationer, der anvendes til dette formål, skal sikre fuldstændig dækning af de sekvenser, der vil kunne indsættes i den genetisk modificerede plante, såsom dele af plasmidet/vektoren eller bærer-nukleinsyre(r) eller fremmed(e) nukleinsyre(r), der bliver tilbage i den genetisk modificerede plante. Southern-analysen skal dække hele det/de pågældende transgene lokus/loki samt flankerende sekvenser og omfatte alle relevante kontroller. Til bestemmelse af antallet af kopier af insertet kan der også anvendes supplerende metoder (f.eks. realtids-PCR). |
|
b) |
Det indsatte genetiske materiales opbygning og sekvens ved hvert insertionssted i et standardiseret elektronisk format med henblik på at identificere ændringer i de indsatte sekvenser sammenlignet med den sekvens, der skal indsættes. |
|
c) |
I tilfælde af deletion(er), om muligt den eller de deleterede regioners størrelse og funktion. |
|
d) |
Insertets/inserternes subcellulære placering (i kerne, kloroplaster eller mitokondrier eller bevaret i en ikke-integreret) samt metoder til bestemmelse af det/dem. |
|
e) |
Sekvensoplysninger i et standardiseret elektronisk format for de flankerende regioner 5' og 3' på hvert insertionssted til påvisning af afbrydelser af kendte gener. Der udføres bioinformatikanalyser under anvendelse af ajourførte databaser til søgning efter ligheder både artsinternt og mellem forskellige arter. Når det drejer sig om genetiske modificerede planter, der indeholder stablede transformationsbegivenheder, skal der foretages en vurdering af sikkerheden med hensyn til potentiel interaktion mellem eventuelle utilsigtede modifikationer på hvert enkelt insertionssted. |
|
f) |
Åbne læserammer (i det følgende benævnt »ORF'er« (Open Reading Frames)) og defineret som enhver nukleotidsekvens, der omfatter en streng af codoner, som ikke er afbrudt af en stopcodon i samme læseramme), der fremkommer som et resultat af den genetiske modifikation, enten ved forbindelsespunkterne med genomisk DNA eller som følge af interne omgrupperinger af insertet/inserterne. ORF'erne analyseres mellem stop-codoner, uden begrænsninger med hensyn til deres længde. Der udføres bioinformatikanalyser for at undersøge mulige ligheder med kendte toksiner eller allergener, idet der anvendes ajourførte databaser. Databasernes beskrives, og det oplyses, hvilken version der er anvendt. Afhængigt af de indsamlede oplysninger kan yderligere analyser (f.eks. en transkriptionsanalyse) være påkrævede til fuldstændiggørelse af risikovurderingen. |
1.2.2.3. Oplysninger om insertets/inserternes ekspression
Ansøgeren skal fremlægge oplysninger, der gør det muligt at:
|
— |
påvise, hvorvidt den indsatte/modificerede sekvens resulterer i de tilsigtede ændringer på protein-, RNA- og/eller metabolitniveau |
|
— |
karakterisere eventuel utilsigtet ekspression af nye ORF'er, som ved undersøgelserne i henhold til punkt 1.2.2.2, litra f), er identificeret som en potentiel sikkerhedsrisiko. |
Ansøgeren skal til disse formål fremlægge oplysninger om følgende:
|
a) |
Metode(r), der er anvendt til ekspressionsanalyse, ledsaget af ydeevnekarakteristikaene. |
|
b) |
Oplysninger om insertets udviklingsmæssige ekspression i løbet af plantens livscyklus. Kravet om oplysninger om udviklingsmæssig ekspression skal overvejes i hvert enkelt tilfælde under hensyntagen til den anvendte promoter, den eller de tilsigtede virkninger af modifikationen og ansøgningens anvendelsesområde. |
|
c) |
Dele af planten, hvori insertet/de modificerede sekvenser udtrykkes. |
|
d) |
Eventuel utilsigtet ekspression af nye ORF'er, som ved undersøgelserne i henhold til punkt 1.2.2.2, litra f), er identificeret som en potentiel sikkerhedsrisiko. |
|
e) |
Data om ekspression af protein, herunder rådata, skal være tilvejebragt ved markforsøg og relatere sig til de betingelser, afgrøden dyrkes under. Der skal altid fremlægges data om ekspressionsniveauer fra de dele af planten, der anvendes til fødevare- og foderbrug. Der skal desuden fremlægges oplysninger om ekspressionen af målgener i andre dele af planten, når der er anvendt vævsspecifikke promotorer, og når det er relevant for sikkerhedsvurderingen. For så vidt angår ekspression af protein skal der som minimum tilvejebringes data fra tre vækstlokaliteter eller fra én lokalitet over tre sæsoner. Permutation (ombytning) af lokaliteter og sæsoner kan accepteres, forudsat at minimumskravet er opfyldt. Hvis insertets art begrunder det (f.eks. ved inaktivering (silencing), eller hvor biokemiske stier aktivt er blevet ændret), analyseres specifikke RNA'er eller metabolitter. For så vidt angår inaktivering (silencing) ved hjælp af RNAi-ekspression skal der søges efter potentielle ikke-målgener ved in silico-analyse for at vurdere, om den genetiske modifikation vil kunne påvirke ekspressionen af andre gener, der giver anledning til sikkerhedsmæssige betænkeligheder. |
|
f) |
For så vidt angår stabling (stacking) af transformationsbegivenheder ved konventionel krydsning skal der fremlægges ekspressionsdata, så der kan foretages en vurdering af de potentielle interaktioner mellem begivenheder, som evt. er forbundet med yderligere sikkerhedsmæssige betænkeligheder med hensyn til ekspressionen af protein og egenskaber i forhold til én enkelt transformationsbegivenhed. Sammenligningen skal foretages ved anvendelse af data fra planter, der er dyrket i de samme markforsøg. I individuelle tilfælde, hvor der er sikkerhedsmæssige betænkeligheder, kan der være behov for yderligere oplysninger. |
1.2.2.4. Insertets genetiske stabilitet og den genetisk modificerede plantes fænotypiske stabilitet
Ansøgeren skal fremlægge oplysninger, der gør det muligt at:
|
a) |
påvise den genetiske stabilitet ved den/de pågældende transgene locus/loci samt den fænotypiske stabilitet og arvemønsteret/arvemønstrene for den eller de indførte egenskaber |
|
b) |
hvis der er tale om stablede transformationsbegivenheder, godtgøre, at hver af de begivenheder, der er kombineret i planten, har de samme molekylære egenskaber og karakteristika som i planterne med én enkelt transformationsbegivenhed. |
Ansøgeren skal i denne forbindelse fremlægge data, der dokumenterer stabilitet over flere (normalt fem) generationer eller vækstcyklusser, for planter indeholdende én enkelt transformationsbegivenhed. Det er tilstrækkeligt med data fra den første og den sidste vækstcyklus. Kilden til det materiale, der er anvendt til analysen, skal angives. De tilvejebragte data skal analyseres ved brug af passende statistiske metoder.
For så vidt angår stablede transformationsbegivenheder skal der foretages en sammenligning mellem de oprindelige begivenheder og de stablede transformationsbegivenheder, idet der skal anvendes plantemateriale, som er repræsentativt for det, der er beregnet til kommerciel produktion. Ansøgeren skal fremlægge en behørig begrundelse for valget af de anvendte plantematerialer. Sammenligningen skal omfatte en sammenligning af sekvenserne af inserterne og de flankerende regioner fra henholdsvis GM-planter, der indeholder én enkelt transformationsbegivenhed, og planter indeholdende stablede transformationsbegivenheder.
Ansøgeren skal vurdere den genetiske stabilitet ved transformationsbegivenheden/transformationsbegivenhederne efter passende molekylære metoder, jf. punkt 1.2.2.2.
1.2.2.5. Potentiel risiko i tilknytning til horisontal genoverførsel
Ansøgeren skal vurdere sandsynligheden for horisontal genoverførsel fra produktet til mennesker, dyr og mikroorganismer og potentiel risiko i tilknytning hertil, når intakt(e) og funktionsdygtig(e) nukleinsyre(r) er blevet tilbage i den/det genetisk modificerede fødevare/foderstof.
1.2.3. Konklusioner af den molekylære karakterisering
Den molekylære karakterisering skal give oplysninger om insertets/inserternes struktur og ekspression og om stabiliteten ved den eller de tilsigtede egenskaber. Dette gælder også i situationer, hvor transformationsbegivenheder er stablet ved konventionelle avlsmetoder.
Det skal specifikt angives, hvorvidt den molekylære karakterisering af den eller de genetiske modifikationer giver anledning til sikkerhedsmæssige overvejelser for så vidt angår afbrydelse af endogene gener eller regulatoriske sekvenser.
Den molekylære karakterisering skal også sigte mod at klarlægge, hvorvidt den eller de genetiske modifikationer rejser spørgsmål for så vidt angår sandsynligheden for, at der vil blive produceret andre proteiner/stoffer end dem, der er påtænkt, især nye toksiner eller allergener.
Potentielle utilsigtede ændringer som omhandlet i dette punkt skal behandles i den eller de relevante supplerende dele af sikkerhedsvurderingen.
1.3. Sammenlignende analyse
Den sammenlignende analyse af sammensætningen samt de agronomiske og fænotypiske karakteristika skal sammen med den molekylære karakterisering danne udgangspunkt for tilrettelæggelsen og gennemførelsen af risikovurderingen af en ny/et nyt genetisk modificeret fødevare/foderstof.
Målet skal være at identificere følgende ligheder og forskelle:
|
a) |
ligheder og forskelle mellem den genetisk modificerede plante og dens konventionelle modstykke med hensyn til sammensætning, agronomisk ydeevne og fænotypiske karakteristika (tilsigtede og utilsigtede ændringer) |
|
b) |
ligheder og forskelle mellem den/det genetisk modificerede fødevare/foderstof og dens/dets konventionelle modstykke med hensyn til sammensætningen. |
Hvis der ikke kan findes et egnet konventionelt modstykke, kan der ikke foretages en sammenlignende sikkerhedsvurdering, og følgelig gennemføres der i sådanne tilfælde en sikkerheds- og ernæringsvurdering af den genetisk modificerede fødevare eller det genetisk modificerede foderstof som den, der foretages af nye fødevarer omfattet af Europa-Parlamentets og Rådets forordning (EF) nr. 258/97 (1), som ikke har konventionelle modstykker (f.eks. hvis den genetisk modificerede fødevarer eller det genetisk modificerede foderstof ikke er nært beslægtet med en fødevare eller et foderstof, for hvilken/hvilket der foreligger oplysninger om langvarig sikker anvendelse, eller hvis en eller flere særlige egenskaber er indført med den hensigt at bibringe komplekse ændringer i sammensætningen af den genetisk modificerede fødevare eller det genetisk modificerede foderstof).
1.3.1. Valg af konventionelt modstykke og yderligere komparatorer
Hvis der er tale om vegetativt formerede afgrøder, skal det konventionelle modstykke i princippet være den nær-isogene sort, der er anvendt til at skabe den transgene linje.
For afgrøder, der formerer sig ved kønnet formering, skal det konventionelle modstykke have en genetisk baggrund, der svarer til den genetisk modificerede plantes. Når den genetisk modificerede plante er blevet udviklet ved tilbagekrydsning, skal der vælges et konventionelt modstykke med en genetisk baggrund, der minder mest muligt om den genetisk modificerede plantes.
Ansøgeren kan desuden inkludere en komparator med en genetisk baggrund, der ligger tættere på den genetisk modificerede plantes end det konventionelle modstykkes (f.eks. en negativ segregant).
For så vidt angår herbicidtolerante genetisk modificerede planter sammenlignes tre testmaterialer med henblik på at vurdere, hvorvidt den forventede landbrugspraksis påvirker ekspressionen af de undersøgte endpoints, nemlig: den genetisk modificerede plante, som eksponeres for det påtænkte herbicid, det konventionelle modstykke, behandlet efter konventionelle herbicidforvaltningsmetoder, og den genetisk modificerede plante, behandlet efter den samme konventionelle herbicidforvaltningsmetode.
For så vidt angår stablede transformationsbegivenheder er det ikke altid muligt at anvende et konventionelt modstykke med en genetisk baggrund, der ligger lige så tæt på den genetisk modificerede plante som et konventionelt modstykke, der normalt anvendes til én enkelt transformationsbegivenhed. Under sådanne omstændigheder skal ansøgeren fremlægge behørig begrundelse for valget af det konventionelle modstykke med en vurdering af dets begrænsninger med hensyn til risikovurdering. Desuden kan som yderligere komparatorer medtages genetisk modificerede linjer med kun én forældreplante eller genetisk modificerede linjer, der indeholder en underkombination af de stablede transformationsbegivenheder, for hvilke der er indgivet en ansøgning, eller negative segreganter frembragt fra disse genetisk modificerede linjer. Ansøgeren skal fremlægge detaljerede oplysninger, der begrunder valget af yderligere komparatorer.
Ansøgeren skal altid fremlægge oplysninger om avlsprogrammet (herkomst) vedrørende den genetisk modificerede plante, det konventionelle modstykke og, hvis det er relevant, yderligere komparatorer sammen med en behørig begrundelse for valget heraf. Beskrivelsen af langvarig sikker anvendelse af det konventionelle modstykke skal være behørigt understøttet af både kvalitative og kvantitative data.
EFSA's videnskabelige udtalelse om retningslinjer for udvælgelse af komparatorer til risikovurdering af genetisk modificerede planter og afledte fødevarer og foderstoffer (»Guidance on selection of comparators for the risk assessment of genetically modified plants and derived food and feed«) (2) indeholder mere detaljerede retningslinjer for anvendelsen af bestemmelserne i dette punkt.
1.3.2. Forsøgsplan og statistisk analyse af data fra markforsøg til sammenlignende analyse
1.3.2.1. Beskrivelse af protokollerne for forsøgsplanen
a) Principper for forsøgsplanen
Der skal udføres markforsøg til frembringelse af materiale til den sammenlignende analyse med henblik på at fastlægge, hvorvidt den genetisk modificerede plante og/eller de genetisk modificerede fødevarer/foderstoffer er forskellig(e) fra de konventionelle modstykker og/eller ækvivalent(e) med ikke genetisk modificerede referencesorter med kendt langvarig sikker anvendelse.
Den sammenlignende analyse skal for hvert endpoint omfatte følgende to metoder:
|
i) |
test for forskel til efterprøvning af, hvorvidt den genetisk modificerede plante er forskellig fra sit konventionelle modstykke og derfor vil kunne betragtes som en fare, afhængigt af, hvori den konstaterede forskel består, samt af eksponeringens omfang og art |
|
ii) |
test for ækvivalens til efterprøvning af, hvorvidt den genetisk modificerede plante er ækvivalent med ikke genetisk modificerede referencesorter, når der ses bort fra den eller de indførte egenskaber. |
Ved testning for forskel er nulhypotesen, at der ikke er forskel på GMO'en og dens konventionelle modstykke, mod den alternative hypotese, at der faktisk er forskel.
Hvis der anvendes supplerende komparator(er) til risikovurderingen, skal der foretages en test for forskelle mellem den genetisk modificerede plante og hver af de supplerende komparatorer efter kravene i punkt 1.3.2.2 med henblik på test for forskellen mellem den genetisk modificerede plante og dens konventionelle modstykke.
Ved testning for ækvivalens er nulhypotesen, at forskellen mellem GMO'en og de anvendte referencesorter mindst er lig med en nærmere bestemt mindstestørrelse (jf. punkt 1.3.2.2), mod den alternative hypotese, at der ikke er nogen forskel eller en mindre forskel end det fastsatte minimum mellem GMO'en og de anvendte referencesorter.
Nulhypotesen skal forkastes, for at det kan konkluderes, at GMO'en og de anvendte referencesorter entydigt er ækvivalente for så vidt angår det undersøgte endpoint. Ækvivalensgrænserne, der anvendes ved ækvivalenstestningen, skal være behørigt repræsentative for den naturlige variation, der kan forventes for referencesorter med kendt langvarig sikker anvendelse.
b) Specifikke protokoller for forsøgsplanen
Den naturlige variation kan have flere årsager: Variation inden for en given sort skyldes miljømæssige faktorer, og variation mellem forskellige sorter opstår som følge af en kombination af genetiske og miljømæssige faktorer. For at kunne identificere og vurdere forskelle, der kun kan tilskrives genotyper, er det meget vigtigt at kontrollere miljøvariabiliteten. Forsøgsplanen for markforsøgene skal derfor omfatte ikke genetisk modificerede referencesorter, idet der skal anvendes tilstrækkeligt mange til at sikre et tilfredsstillende skøn over variabiliteten, som er en forudsætning for at kunne fastsætte ækvivalensgrænserne. Alle testmaterialer bestående af genetisk modificerede planter, konventionelle modstykker, referencesorter og, hvis det er relevant, yderligere komparatorer skal randomiseres i plots inden for et enkelt felt på hver lokalitet, sædvanligvis i et fuldstændig randomiseret eller randomiseret blokforsøg. De forskellige lokaliteter, der vælges til markforsøg, skal afspejle de forskellige klimatiske og agronomiske betingelser, som den pågældende afgrøde skal dyrkes under; valget skal begrundes udtrykkeligt. Valget af ikke genetisk modificerede referencesorter skal være passende for de valgte lokaliteter og skal begrundes udtrykkeligt. Hvis lokaliterne omfatter en begrænset vifte af vækstbetingelser, skal ansøgeren gentage markforsøgene over mere end ét år.
Testmaterialerne i form af genetisk modificerede planter, konventionelt modstykke og, hvis det et relevant, yderligere komparatorer skal være identisk for alle forsøgsgentagelser på hver enkelt lokalitet. Desuden skal der, medmindre der foreligger en udtrykkelig begrundelse for, at dette ikke er nødvendigt, på hver lokalitet være mindst tre passende ikke genetisk modificerede referencesorter af afgrøden med kendt langvarig sikker anvendelse, som samtidig skal være identiske mellem replikater. Replikationen på hvert lokalitet er antallet af resultater for hvert testmateriale; replikationen bør aldrig være mindre end fire på en hvilken som helst lokalitet. Hvis der kun foreligger to relevante referencesorter på en given lokalitet, skal replikationen dog være seks på den pågældende lokalitet; hvis der kun foreligger én relevant referencesort, skal replikationen være otte.
Hvert markforsøg skal gentages på mindst otte lokaliteter, der er udvalgt som repræsentative for de forskellige sandsynlige recipientmiljøer, hvor planten vil skulle dyrkes. Markforsøgene kan gennemføres inden for et enkelt år eller spredes over flere år. De ikke genetisk modificerede referencesorter kan variere fra lokalitet til lokalitet, og der skal anvendes mindst seks forskellige referencesorter i hele sættet af markforsøg.
Når den genetisk modificerede plante testes sammen med andre genetisk modificerede planter af samme afgrødeart (såsom Zea mays), kan materialet til den sammenlignende vurdering af disse forskellige genetisk modificerede planter produceres samtidig på samme lokalitet og inden for rammerne af det samme markforsøg, ved at de forskellige genetisk modificerede planter og de(n) relevante komparator(er) placeres i den samme randomiserede blok. Dette kræver, at følgende to betingelser er overholdt til punkt og prikke:
|
i) |
Det konventionelle modstykke og, hvis det er relevant, yderligere komparatorer skal altid være til stede sammen med den genetisk modificerede plante inden for samme blok |
|
ii) |
Alle de forskellige genetisk modificerede planter og deres komparator(er) samt alle ikke genetisk modificerede referencesorter, der anvendes til at teste ækvivalens med de pågældende genetisk modificerede planter, skal være fuldstændig randomiseret inden for hver enkelt blok. |
Hvis antallet af plots pr. blok, der er nødvendige til et sådant markforsøg, overstiger 16, kan der anvendes et delvist balanceret ufuldstændigt blokforsøg med det formål at reducere antallet af plots pr. blok, ved at nogle af de genetisk modificerede planter og deres komparator(er) fjernes fra hver enkelt blok. Dette kræver, at følgende to betingelser er overholdt til punkt og prikke:
|
i) |
Konventionelle modstykker skal altid være til stede sammen med den genetisk modificerede plante, den hører sammen med, inden for samme blok. |
|
ii) |
Alle ikke genetisk modificerede referencesorter skal være til stede i hver af de ufuldstændige blokke og være fuldstændig randomiserede med planterne og deres komparatorer(er). |
Markforsøgene skal beskrives tilstrækkelig udførligt, således at der oplyses om vigtige parametre såsom forvaltningen/behandlingen af området forud for såningen, sådatoen, jordbundstype, brug af herbicider, klimatiske forhold og andre dyrknings/miljøforhold under væksten og på høsttidspunktet samt betingelserne under oplagringen af det høstede materiale.
EFSA's udtalelse om statistiske betragtninger vedrørende sikkerhedsvurdering af GMO'er (»Statistical considerations for the safety evaluation of GMOs«) (3) indeholder mere detaljerede retningslinjer for anvendelsen af bestemmelserne i dette punkt.
1.3.2.2. Statistisk analyse
Analyser af data skal fremlægges i et tydeligt format, idet der skal anvendes standardiserede videnskabelige enheder. De rådata og den programmeringskode, der er anvendt til den statistiske analyse, skal fremlægges i en redigerbar form.
Det kan være nødvendigt med datatransformation for at sikre normalfordeling og for at opnå en passende skala, hvori den statistiske effekt er additiv. For mange endpointresponsvariablers vedkommende burde logaritmisk transformation være hensigtsmæssig. I sådanne tilfælde fortolkes enhver forskel mellem det genetisk modificerede materiale og andet testmateriale som en kvotient på den naturlige skala. Giver logaritmisk transformation imidlertid ikke tilfredsstillende resultater, skal man overveje den naturlige eller en anden skala.
Den samlede variabilitet for hvert endpoint, der er observeret i markforsøgene, estimeres og fordeles efter passende statistiske modeller for at udlede to konfidensintervaller og fastsætte en nedre og en øvre ækvivalensgrænse på basis af den variabilitet, der er observeret referencesorterne imellem. Det ene konfidensinterval anvendes til testen for forskelle; det andet interval og ækvivalensgrænserne anvendes til ækvivalenstesten.
Der anvendes en blandet lineær statistisk model til beregning af konfidensintervallerne for begge test (dvs. testen for forskelle og ækvivalenstesten), mens der gøres brug af en lidt anderledes model til beregning af de ækvivalensgrænser, der skal anvendes i ækvivalenstesten.
Der angives en indikatorvariabel (ucentreret i den blandede model) med I, således at I = 1 for et feltplot med ikke genetisk modificerede referencesorter, og ellers er I = 0. De stokastiske faktorer for model 1 bør altså være, men ikke nødvendigvis udelukkende, dem, der repræsenterer variationen: i) mellem testmaterialerne (et sæt, som omfatter den genmodificerede plante, dens konventionelle modstykke, hver af de ikke genetisk modificerede referencesorter og eventuelle supplerende komparatorer), ii) i interaktionen mellem testmaterialerne og I, iii) mellem lokaliteter og iv) mellem blokke i lokaliteterne. Model 2 skal svare til model 1, dog således at den stokastiske faktor, der repræsenterer interaktionen mellem testmaterialerne og I, er udeladt.
Den faste faktor for begge modeller bør have lige så mange niveauer, som der er testmaterialer, og repræsentere kontrasterne mellem middelværdierne for testmaterialerne. Testmaterialerne er, jf. ovenfor, den genetisk modificerede plante, dens konventionelle modstykke, sættet af ikke genetisk modificerede referencesorter og eventuelle yderligere testmaterialer. Sættet af ikke genetisk modificerede referencesorter betragtes som et enkelt niveau af den faste faktor. For så vidt angår testen for forskelle er komponenten af den pågældende faste faktor kontrasten på 1 frihedsgrad mellem den genetisk modificerede plante og dens konventionelle modstykke. For så vidt angår ækvivalenstesten er komponenten af den pågældende faste faktor kontrasten på 1 frihedsgrad mellem den genetisk modificerede plante og sættet af ikke genetisk modificerede referencesorter.
Både testen for forskelle og ækvivalenstesten skal gennemføres under anvendelse af sammenhængen mellem hypoteseprøvningen og konfidensgrænserne. Ækvivalenstestning skal gennemføres efter en metode med to ensidede test (TOST-metoden (two one-sided tests)), hvor nulhypotesen om ikke-ækvivalens forkastes, når begge konfidensgrænser falder inden for ækvivalensgrænserne. Valget af en konfidensgrad på 90 % modsvarer det sædvanlige niveau på 95 % for statistisk testning af ækvivalens.
Resultaterne af testen for forskelle og ækvivalenstesten skal angives for alle endpoints samtidig i ét eller nogle få diagrammer.
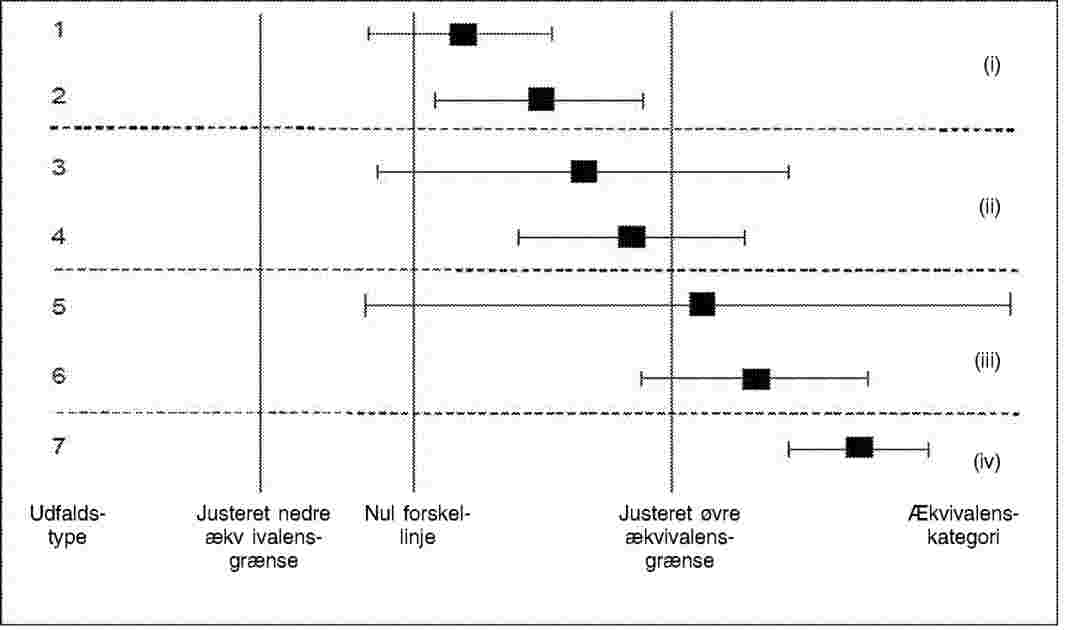
Der skal i diagrammet/diagrammerne være en linje, der angiver nul forskel mellem det genetisk modificerede materiale og dets konventionelle modstykke samt — for hvert endpoint — de justerede ækvivalensgrænser (nedre og øvre), middelforskellen mellem det genetisk modificerede materiale og dets konventionelle modstykke og konfidensintervallet for denne forskel (jf. de mulige udfald for et enkelt endpoint er vist i diagrammet i figur 1).
Når der ud over det konventionelle modstykke også anvendes andet testmateriale som komparator, vises middelforskellen mellem det genetisk modificerede materiale og den pågældende komparator, dens konfidensgrænser og dens justerede ækvivalensgrænser i diagrammet/diagrammerne for hver supplerende komparator, idet denne henføres til samme (nul)basislinje, som er defineret af det konventionelle modstykke. Nul forskel-linjen på den logaritmiske skala svarer til en enhedsmultiplikationsfaktor på den naturlige skala. Den vandrette akse skal være mærket med værdier, der nærmere angiver ændringen på den naturlige skala. Ved logaritmisk transformation vil ændringer på 2x og ½x være fordelt med lige stor indbyrdes afstand på hver side af nul forskel-linjen.
Om end en vis andel af falske signifikante forskelle er at forvente, skal ansøgeren rapportere om og diskutere alle signifikante forskelle, der er observeret mellem den genetisk modificerede afgrøde, dens konventionelle modstykke og, hvis det er relevant, andet testmateriale, med fokus på deres biologiske betydning (jf. afsnit 3 om risikokarakterisering).
Der skal ved rapporteringen gives udførlige oplysninger for hvert enkelt analyseret endpoint, med angivelse af:
|
a) |
de antagelser, der ligger til grund for analysen |
|
b) |
udførlig beskrivelse af de valgte blandede modeller, herunder faste og stokastiske effekter |
|
c) |
resultaterne af alle test af vekselvirkninger mellem testmaterialerne og lokaliteterne |
|
d) |
faste effekter samt den relevante estimerede residualvarians, der sammenlignes med, og varianskomponenter for de stokastiske faktorer |
|
e) |
estimerede frihedsgrader |
|
f) |
eventuelle yderligere relevante statistiske oplysninger. |
Der skal redegøres for de sandsynlige virkninger af andre vækstbetingelser, der ikke er undersøgt i markforsøget.
Figur 1: Forenklet udgave af et diagram over sammenlignende vurdering, som viser de syv mulige udfaldstyper for hvert enkelt endpoint. Efter justering af ækvivalensgrænserne kan en enkelt konfidensgrænse (for forskellen) bidrage visuelt til at vurdere udfaldet af begge test (testen for forskelle og ækvivalenstesten). I det illustrerede eksempel er det kun den øvre justerede ækvivalensgrænse, der vurderes. Følgende er vist: middelværdien for den genetisk modificerede afgrøde i en passende målestok (kassen), konfidensgrænserne (whiskers) for forskellen mellem den genetisk modificerede afgrøde og dens konventionelle modstykke (stregen viser konfidensintervallet), en lodret linje, som angiver nul forskel (til test for forskel), og lodrette linjer, som angiver de justerede ækvivalensgrænser (til test for ækvivalens). Ved udfaldstype 1, 3 og 5 kan nulhypotesen om ingen forskel ikke forkastes; ved udfaldstype 2, 4, 6 og 7 er den genetisk modificerede afgrøde forskellig fra sit konventionelle modstykke. Til ækvivalensfortolkningen er fire kategorier (i)-iv)) angivet: I kategori i) forkastes nulhypotesen om ikke-ækvivalens til fordel for ækvivalens; i kategori ii), iii) og iv) kan ikke-ækvivalens ikke afvises.
|
A. |
For så vidt angår test for forskel skal alle udfald fra diagrammet kategoriseres som angivet herunder, og de relevante konklusioner drages.
|
|
B. |
For så vidt angår test for ækvivalens skal alle udfald fra diagrammet kategoriseres som angivet herunder, og de relevante konklusioner drages.
I tilfælde af betydelig forskel og/eller manglende ækvivalens for et givent endpoint skal der foretages yderligere statistiske analyser for at vurdere, hvorvidt der er vekselvirkninger mellem et eller flere af testmaterialerne og den pågældende lokalitet, eventuelt ved hjælp af en simpel standard-ANOVA-test. Uanset hvilken metode der anvendes, skal de relevante oplysninger gives for hvert enkelt endpoint, med angivelse af: a) de antagelser, der ligger til grund for analysen, og, hvis det er relevant: b) frihedsgrader, c) den estimerede residualvarians for hver enkelt variationskilde samt varianskomponenter og d) eventuelle yderligere relevante statistiske oplysninger. Disse supplerende analyser har til formål at lette fortolkningen af observerede væsentlige forskelle og at undersøge potentielle vekselvirkninger mellem testmateriale og andre faktorer. EFSA's udtalelse om statistiske betragtninger vedrørende sikkerhedsvurdering af GMO'er (»Statistical considerations for the safety evaluation of GMOs« (4)) indeholder mere detaljerede retningslinjer for anvendelsen af bestemmelserne i dette punkt. |
1.3.3. Udvælgelse af materiale og forbindelser til analyse
Det er af afgørende betydning at analysere plantematerialets sammensætning, når man sammenligner den/det genetisk modificerede fødevare/foderstof med dens/dets konventionelle modstykke. Materialet, der skal anvendes til den sammenlignende vurdering, vælges under hensyntagen til anvendelserne af den genetisk modificerede plante og arten af den genetiske modifikation. For så vidt angår herbicidtolerante genetisk modificerede planter skal der anvendes tre testmaterialer: den genetisk modificerede plante, som eksponeres for det påtænkte herbicid, det konventionelle modstykke, behandlet efter konventionelle herbicidforvaltningsmetoder, og den genetisk modificerede plante, behandlet efter de samme konventionelle herbicidforvaltningsmetoder. Medmindre andet er behørigt begrundet, skal de relevante analyser udføres på de uforarbejdede landbrugsråvarer, idet det normalt primært er via disse, materialet kommer ind i fødevare- og foderproduktions- og forarbejdningskæden. Om fornødent gennemføres der yderligere analyser af forarbejdede produkter (såsom fødevarer og foderstoffer, fødevareingredienser, fodermidler, fødevare- og fodertilsætningsstoffer eller fødevarearomaer) i de tilfælde, hvor det er relevant (jf. også punkt 1.3.6). Prøveudtagning, analyse og forberedelse af testmateriale skal gennemføres i overensstemmelse med relevante kvalitetsstandarder.
1.3.4. Sammenlignende analyse af sammensætningen
Ud over analysen af indholdet af de nye udtrykte proteiner (jf. punkt 1.2.2.3) skal analysen af sammensætningen omfatte et passende antal forbindelser. Ansøgeren skal altid som minimum fremlægge en proximatanalyse (af bl.a. vandindhold og samlet askeindhold) og en analyse af de vigtigste makro- og mikronæringsstoffer, næringshæmmende forbindelser, naturlige toksiner og allerede identificerede allergener samt andre sekundære plantemetabolitter, der er karakteristiske for bestemte afgrødeplantearter, jf. konsensusdokumenterne fra Organisationen for Økonomisk Samarbejde og Udvikling (OECD) om betragtninger vedrørende sammensætningen af nye plantesorter (OECD's konsensusdokumenter) (5). Til analyse udvælges de vitaminer og mineraler, der er til stede i ernæringsmæssigt signifikante mængder, og/eller som bidrager ernæringsmæssigt signifikant til kosten i de mængder, som planten indtages i. Hvilke analyser der specifikt kræves, afhænger af arten af den undersøgte planteart, men analyserne skal under alle omstændigheder omfatte en detaljeret vurdering i overensstemmelse med, hvilke virkninger der tilsigtes med den genetiske modifikation, næringsværdien og plantens anvendelse. Ansøgeren skal være særlig opmærksom på de vigtigste næringsstoffer såsom proteiner, kulhydrater, lipider/fedtstoffer, fibre, vitaminer og mineraler. F.eks. skal der inkluderes en fedtsyreprofil for olierige planter (vigtigste individuelle mættede, enkeltumættede og flerumættede fedtsyrer) og en aminosyreprofil (individuelle proteinaminosyrer og de vigtigste ikke-proteinaminosyrer) for planter, der anvendes som en vigtig proteinkilde. For vegetative plantedele, der anvendes til foderbrug, er en analyse af bestanddelene i planternes cellevæg desuden påkrævet.
Ansøgeren skal ligeledes fremlægge en analyse af vigtige toksiner, der er naturligt til stede i recipientplanten og kan have negative virkninger for menneskers/dyrs sundhed, afhængigt af giftighed og mængde. Koncentrationerne af sådanne forbindelser skal vurderes efter planteart og den foreslåede anvendelse af fødevaren/foderstoffet. Også næringshæmmende forbindelser såsom fordøjelsesenzym-inhibitorer og allerede identificerede allergener skal undersøges.
Karakteristikaene ved den indførte egenskab vil kunne udløse yderligere analyse af specifikke forbindelser, herunder metabolitter fra potentielt ændrede stofskifteveje. Ansøgeren skal, hvis det er relevant, overveje at inkludere andre forbindelser end de vigtigste næringsstoffer, de vigtigste toksiner, næringshæmmende stoffer og allergener identificeret ved OECD's konsensusdokumenter og begrunde valget af de pågældende forbindelser.
1.3.5. Sammenlignende analyse af agronomiske og fænotypiske karakteristika
Ansøgeren skal fremlægge en sammenligning mellem den genetisk modificerede plante og dens konventionelle modstykke. Denne sammenligning skal gøre det muligt for ansøgeren at identificere utilsigtede virkninger som følge af den genetiske modifikation og skal også omfatte plantens biologi og agronomiske egenskaber, herunder fælles avlsparametre (såsom ydeevne, plantemorfologi, blomstringstidspunkt, graddøgn til modning, varigheden af pollenets levedygtighed, forsvar over for plantepatogener og skadelige insekter og følsomhed over for abiotisk stress). Protokollerne for disse markforsøg skal overholde de specifikationer, der er fastsat i punkt 1.3.2.
Hvis transformationsbegivenheder er stablet ved konventionel krydsning, kan de agronomiske og fænotypiske karakteristika også være ændret. Mulige forskelle i fænotypiske karakteristika og agronomiske egenskaber som følge af stablede transformationsbegivenheder skal vurderes i markforsøg. Hvis det er relevant, skal ansøgeren fremlægge yderligere oplysninger om agronomiske egenskaber ved de stablede begivenheder fra yderligere markforsøg.
1.3.6. Virkninger af forarbejdning
Ansøgeren skal vurdere, hvorvidt det er sandsynligt, at de anvendte forarbejdnings- og/eller konserveringsteknikker vil ændre genetisk modificerede slutprodukters karakteristika i forhold til deres respektive konventionelle modstykker. Ansøgeren skal fremlægge en tilstrækkelig detaljeret beskrivelse af de forskellige forarbejdningsteknikker med særligt fokus på de trin i processen, der kan resultere i betydelige ændringer i produktets indhold, kvalitet eller renhed.
Genetisk modifikation kan specifikt vedrøre stofskiftevejene, hvilket kan resultere i ændrede koncentrationer af ikke-proteinstoffer eller i nye metabolitter (såsom i ernæringsmæssigt forbedrede fødevarer). Vurderingen af forarbejdede produkter kan foretages sammen med vurderingen af den genetisk modificerede plante med hensyn til sikkerheden ved den genetiske modifikation, eller et forarbejdet produkt kan vurderes separat. Ansøgeren skal fremlægge en videnskabelig begrundelse for risikovurderingen af de pågældende produkter. Ansøgeren tager i hvert enkelt tilfælde stilling til, om der er behov for at fremlægge yderligere forsøgsdata.
Afhængigt af produktet skal der, hvis det er relevant, fremlægges oplysninger om sammensætning, mængden af uønskede stoffer, næringsværdi og metabolisme samt om den påtænkte anvendelse.
Afhængigt af arten af det eller de nye udtrykte proteiner skal der, hvis det er relevant, foretages en vurdering af, i hvilket omfang forarbejdningstrinnene medfører koncentration eller eliminering, denaturering og/eller nedbrydning af det eller de pågældende proteiner i det færdige produkt.
1.3.7. Konklusion
Af konklusionen af den sammenlignende analyse skal følgende klart fremgå:
|
a) |
hvorvidt de agronomiske og fænotypiske karakteristika ved den genetisk modificerede plante, under hensyntagen til naturlig variation, er forskellige fra det konventionelle modstykkes karakteristika og/eller ækvivalente med referencesorternes, når der ses bort fra den eller de indførte egenskaber |
|
b) |
hvorvidt den/det genetisk modificerede fødevares/foderstofs karakteristika med hensyn til sammensætningen, under hensyntagen til naturlig variation, er forskellige fra det konventionelle modstykkes karakteristika og/eller ækvivalente med referencesorternes, når der ses bort fra den eller de indførte egenskaber |
|
c) |
karakteristika, med hensyn til hvilke den genetisk modificerede plante eller den/det genetisk modificerede fødevare/foderstof, under hensyntagen til naturlig variation, er forskellig(t) fra det konventionelle modstykke og/eller ikke ækvivalent med referencesorterne, og som det er nødvendigt at undersøge yderligere |
|
d) |
hvorvidt der i tilfælde, hvor transformationsbegivenheder er kombineret ved konventionel krydsning, er indikationer på vekselvirkninger mellem de kombinerede transformationsbegivenheder. |
1.4. Toksikologi
De toksikologiske virkninger af ændringer af hele genetisk modificerede fødevarer og foderstoffer foranlediget af den genetiske modifikation, f.eks. introduktion af nye gener, geninaktivering (gene silencing) eller overekspression af et endogent gen, skal vurderes.
Den toksikologiske vurdering skal:
|
a) |
påvise, at den eller de tilsigtede virkninger af den genetiske modifikation ikke har negative virkninger for menneskers og dyrs sundhed |
|
b) |
dokumentere, at utilsigtede virkninger af den eller de genetiske modifikationer, som på basis af de forudgående sammenlignende molekylær-, sammensætnings- eller fænotypeanalyser er påvist eller antages at have fundet sted, ikke har nogen negative virkninger for menneskers eller dyrs sundhed |
|
c) |
identificere potentielle negative virkninger af nye bestanddele og fastslå det højeste dosisniveau, der ikke resulterer i negative virkninger. Der kan udledes et acceptabelt daglig indtag (ADI) af individuelle forbindelser hos mennesker fra oplysninger tilvejebragt ved et passende dyreforsøg, idet der anvendes usikkerheds- eller sikkerhedsfaktorer, hvormed der tages hensyn til forskelle mellem de undersøgte dyrearter og mennesker samt individuelle forskelle forskellige personer imellem |
|
d) |
identificere potentielle negative virkninger på hele genetisk modificerede fødevarer/foderstoffer eller afklaring af usikkerhedselementer, ved hjælp af et 90-dages fodringsforsøg. |
Ansøgeren skal tage stilling til arten af de toksikologiske undersøgelser, der skal gennemføres på nye bestanddele og hele genetisk modificerede fødevarer/foderstoffer, på basis af resultatet af den molekylære og sammenlignende analyse, der er omhandlet i afsnit 1.2 og 1.3, nemlig de konstaterede forskelle mellem det genetisk modificerede produkt og dets konventionelle modstykke, herunder såvel tilsigtede som utilsigtede ændringer. Ansøgeren skal også vurdere resultaterne af de gennemførte toksikologiske undersøgelser med henblik på at vurdere behovet for at foretage yderligere undersøgelser af nye bestanddele eller hele genetisk modificerede fødevarer/foderstoffer, jf. afsnit 1.4.4.2 og 1.4.4.3.
Ansøgeren skal tage hensyn til tilstedeværelse af nye udtrykte proteiner, eventuel tilstedeværelse af andre nye bestanddele og/eller eventuelle ændringer i mængden af naturlige bestanddele ud over normal variation. De specifikke informationskrav og teststrategier er beskrevet i punkt 1.4.1-1.4.4.
Med hensyn til ansøgninger, hvis anvendelsesområde omfatter eller er begrænset til genetisk modificerede fødevarer og foderstoffer, der er fremstillet af genetisk modificerede planter, skal der fremlægges toksikologiske undersøgelser af de forarbejdede produkter, undtagen hvis ansøgeren fremlægger en risikovurdering af den genetisk modificerede plante (eller relevante dele heraf), der dokumenterer sikkerheden ved denne, og der ikke er indikationer på, at de forarbejdede genetisk modificerede fødevarer og foderstoffer vil være forskellige fra deres respektive konventionelle modstykker. Ansøgeren skal begrunde dette på behørig vis.
Toksikologiske undersøgelser, der har til formål at vurdere risikoen for menneskers og/eller dyrs sundhed, skal supplere hinanden. De fleste undersøgelser, der kræves til vurdering af sikkerheden ved genetisk modificerede fødevarer, kan også anvendes til vurdering af genetisk modificerede foderstoffer.
Ud over eksponeringen af forbrugere og dyr via indtagelse af fødevarer og foderstoffer skal ansøgeren rapportere om eventuelle negative virkninger, som mennesker kan blive udsat for, fordi de eksponeres for genetisk modificerede fødevarer og fodermidler i forbindelse med deres erhvervsvirksomhed, f.eks. landbrugsarbejde eller forarbejdning af frø. Der skal gennemføres relevante undersøgelser med henblik på at beskrive sådanne indikationer på potentielle negative virkninger yderligere.
Ansøgeren skal til toksicitetstest anvende internationalt anerkendte protokoller og testmetoder (jf. afsnit 1.7, tabel 1 og 2). Tilpasning af disse protokoller eller anvendelse af metoder, der adskiller sig fra disse protokoller, skal begrundes i ansøgningen.
1.4.1. Undersøgelse af nye udtrykte proteiner
Ansøgeren skal fremlægge en vurdering af alle nye udtrykte proteiner. Det besluttes i hvert enkelt tilfælde, hvilke undersøgelser der skal anvendes til klarlægning af et nyt udtrykt proteins potentielle toksicitet, afhængigt af den tilgængelige viden om, hvor proteinet stammer fra, om proteinets funktion eller aktivitet samt om dets historie for så vidt angår menneskers eller dyrs indtag af proteinet. Hvad angår proteiner, der udtrykkes i den genetisk modificerede plante, er specifik toksicitetstestning som omhandlet i dette afsnit ikke påkrævet i tilfælde, hvor der foreligger behørig dokumentation for langvarig sikker anvendelse til konsum og/eller som foder for både planten og de nye udtrykte proteiner. I sådanne tilfælde skal ansøgeren fremlægge de nødvendige oplysninger vedrørende langvarig sikker anvendelse af proteinerne.
Hvor specifik testning er påkrævet, skal det undersøgte protein være ækvivalent med det nye udtrykte protein, som det udtrykkes i den genetisk modificerede plante. Hvis der, fordi der ikke er en tilstrækkelig mængde testmateriale fra planten til rådighed, anvendes et protein, der produceres af mikroorganismer, skal det påvises, at der strukturelt, biokemisk og funktionelt er ækvivalens mellem den mikrobielle substitut og det nye udtrykte planteprotein. Der er til påvisningen af ækvivalens især behov for at sammenligne molekylvægt, aminosyresekvens, posttranslationel modifikation, immunologisk reaktivitet og, for så vidt angår enzymer, enzymaktivitet. I tilfælde af forskelle mellem det i planten udtrykte protein og dets mikrobielle substitut vurderes betydningen af disse forskelle for sikkerhedsundersøgelserne.
For at dokumentere, at nye udtrykte proteiner er sikre, skal ansøgeren fremlægge følgende:
|
a) |
En molekylær og biokemisk karakterisering af det nye udtrykte protein, herunder fastlæggelse af primær struktur, molekylvægt (f.eks. ved hjælp af massespektrometri), undersøgelser af posttranslationelle modifikationer og en beskrivelse af dets funktion. Når der er tale om nye udtrykte enzymer, skal der ligeledes fremlægges oplysninger om enzymaktiviteten, herunder temperatur og pH-værdi for optimal aktivitet, substratspecificitet og eventuelle reaktionsprodukter. Også de potentielle vekselvirkninger med andre plantebestanddele skal vurderes. |
|
b) |
En opdateret søgning efter homologi til proteiner, der vides at forårsage negative virkninger, f.eks. toksiske proteiner. En søgning efter homologi til proteiner med en normal metabolisk eller strukturel funktion kan også give værdifulde oplysninger. Det oplyses, hvilke databaser og metoder der er anvendt til søgningen. |
|
c) |
En beskrivelse af proteinets stabilitet under de relevante betingelser, der gør sig gældende under forarbejdning og oplagring, og af den forventede behandling af fødevaren/foderstoffet. Virkningerne af ændringer i temperatur og pH-værdi skal undersøges, og potentielle modifikationer af proteinerne (f.eks. denaturering) og/eller produktion af stabile proteinfragmenter fremkommet ved sådanne behandlinger skal karakteriseres. |
|
d) |
Data om resistens hos det nye udtrykte protein over for proteaser (såsom pepsin), f.eks. fra in vitro-undersøgelser under anvendelse af passende, standardiserede test. Stabile nedbrydningsprodukter skal karakteriseres og vurderes med hensyn til deres potentielle sundhedsskadelige virkninger i relation til deres biologiske aktivitet. |
|
e) |
En 28-dages undersøgelse af det nye udtrykte proteins orale toksicitet hos gnavere ved gentagen dosering. Afhængigt af resultatet af den 28-dages toksicitetsundersøgelse skal der, hvis det er relevant, fremlægges yderligere målrettede undersøgelser, herunder en analyse af immunotoksicitetet. |
Testning for akut toksicitet af de nye udtrykte proteiner i genetisk modificerede planter har kun begrænset merværdi for risikovurderingen af menneskers og dyrs gentagne indtagelse af genetisk modificerede fødevarer/foderstoffer og skal ikke fremlægges som en del af de undersøgelser, der foretages i henhold til dette punkt.
Ansøgeren skal gennemføre undersøgelser med kombineret indgift af proteiner, når den genetiske modifikation resulterer i ekspression af to eller flere proteiner i den genetisk modificerede plante, og når der på basis af videnskabelig viden påvises en mulighed for synergistiske eller antagonistiske vekselvirkninger, der udgør en potentiel sikkerhedsrisiko.
1.4.2. Undersøgelse af andre nye bestanddele end proteiner
Ansøgeren skal fremlægge en risikovurdering af andre nye identificerede bestanddele end proteiner. Denne skal, alt efter omstændighederne i hvert enkelt tilfælde, omfatte en vurdering af proteinernes giftighed og af behovet for toksikologisk testning samt bestemmelse af proteinernes koncentration i den/det genetisk modificerede fødevare/foderstof. For at fastslå sikkerheden ved nye bestanddele, for hvilke der ikke foreligger dokumentation for langvarig sikker anvendelse i fødevarer og foderstoffer, skal ansøgeren fremlægge oplysninger svarende til dem, der er beskrevet i retningslinjerne for fremlæggelse af oplysninger i forbindelse med vurdering af fødevaretilsætningsstoffer (»Guidance for submissions for food additive evaluations by the EFSA Panel on Food Additives and Nutrient Sources added to Food«) (6) af 16. august 2012 og i Kommissionens forordning (EF) nr. 429/2008 af 25. april 2008 om gennemførelsesbestemmelser til Europa-Parlamentets og Rådets forordning (EF) nr. 1831/2003 for så vidt angår udarbejdelse og indgivelse af ansøgninger samt vurdering og godkendelse af fodertilsætningsstoffer (7). Der skal blandt andet fremlægges oplysninger om et sæt nøgleundersøgelser af f.eks. metabolisme/toksikokinetik, subkronisk toksicitet, genotoksicitet, kronisk toksicitet, karcinogenicitet samt reproduktions- og udviklingstoksicitet, ledsaget af alle andre relevante undersøgelser. Afsnit 1.7, tabel 1, indeholder specifikke retningslinjer for dyreforsøg. Afsnit 1.7, tabel 2, indeholder forsøgsprotokoller for genotoksicitetsundersøgelser.
1.4.3. Oplysninger om ændrede mængder af bestanddele af fødevaren/foderstoffet
Dette punkt finder kun anvendelse, hvis den tilsigtede eller utilsigtede virkning af den genetiske modifikation ville resultere i, at mængden af bestanddele af fødevarer og foderstoffer ændres mere, end hvad der kan tilskrives den naturlige variation.
For at påvise sikkerheden ved den ændrede mængde af naturlige fødevare- og foderstofbestanddele, som f.eks. makro- og mikronæringsstoffer, næringshæmmende stoffer og naturlige toksiner samt andre sekundære plantemetabolitter, skal ansøgeren forelægge en detaljeret risikovurdering, som bygger på kendskabet til disse bestanddeles fysiologiske funktion og/eller toksiske egenskaber.
Resultatet af denne risikovurdering afgør, om og i hvilket omfang ansøgeren skal fremlægge yderligere toksikologiske undersøgelser af bestemte fødevare- og foderstofbestanddele som supplement til det 90-dages fodringsforsøg med gnavere med hele den/det genetisk modificerede fødevare/foderstof.
1.4.4. Undersøgelse af hele den/det genetisk modificerede fødevare/foderstof
Ansøgeren skal primært basere sin risikovurdering af den/det genetisk modificerede fødevare/foderstof på molekylær karakterisering, sammenlignende agronomisk og fænotypisk analyse og en omfattende analyse af sammensætningen samt på den toksikologiske vurdering af de identificerede tilsigtede og utilsigtede virkninger, herunder et 90-dages fodringsforsøg med gnavere med hele den/det genetisk modificerede fødevare/foderstof, jf. punkt 1.4.4.1. Under de omstændigheder, der er beskrevet i punkt 1.4.4.2 og 1.4.4.3, skal der gennemføres yderligere toksikologiske undersøgelser af hele den/det genetisk modificerede fødevare/foderstof.
1.4.4.1. 90-dages fodringsforsøg med gnavere med hele genetisk modificerede fødevarer/foderstoffer
Ansøgeren skal inkludere et 90-dages fodringsforsøg, hvor gnavere fodres med hele fødevarer og foderstoffer, til vurdering af fødevarer og foderstoffer, der indeholder, består af eller er fremstillet af genetisk modificerede planter med én enkelt transformationsbegivenhed eller med stablede begivenheder, som ikke er opnået ved konventionel krydsning af genetisk modificerede planter indeholdende én enkelt transformationsbegivenhed.
For så vidt angår stablede transformationsbegivenheder opnået ved konventionel krydsning af genetisk modificerede planter, der indeholder en eller flere transformationsbegivenheder, skal der inkluderes et 90-dages fodringsforsøg, hvor gnavere fodres med hele fødevarer og foderstoffer, med hver af de genetisk modificerede planter med én enkelt transformationsbegivenhed, som er anvendt. Der inkluderes yderligere et 90-dages fodringsforsøg, hvor gnavere fodres med hele fødevarer og foderstoffer af den genetisk modificerede plante med stablede transformationsbegivenheder, såfremt der konstateres indikationer på potentielle negative virkninger i forbindelse med vurderingen af i) inserternes stabilitet, ii) inserternes ekspression og iii) potentielle synergistiske eller antagonistiske virkninger af kombinationen af transformationsbegivenhederne.
Udformningen af toksicitetsundersøgelsen med genetisk modificerede fødevarer og foderstoffer bør udføres i henhold til »subkronisk oral toksicitetstest — et 90-dages forsøg over gentagne orale dosers toksicitet på gnavere« (se tabel 1) efter en tilpasset protokol. I princippet skal der anvendes mindst to testdoser og en negativ kontrol. Den højeste dosis skal være den maksimalt opnåelige, uden at den forårsager ernæringsmæssig ubalance; den laveste dosis skal indeholde den/det testede fødevare og/eller foderstof i en mængde, der altid ligger over det forventede indtag hos mennesker/måldyr. De analyserede genetisk modificerede fødevarer og foderstoffer bør være relevante for det produkt, der indtages. For så vidt angår herbicidtolerante genetisk modificerede planter skal det testede materiale komme fra den genetisk modificerede plante eksponeret for det påtænkte herbicid. Når det er muligt, skal oplysninger om naturlig variation af testparametre udledes af historiske data snarere end ved inddragelse i forsøgene af referencesorter bestående af kommercielt tilgængelige fødevarer og foderstoffer fremstillet af ikke genetisk modificerede planter med kendt langvarig sikker anvendelse. Den statistiske analyse skal fokusere på påvisning af eventuelle forskelle mellem testmaterialet og kontrolprøven. Der bør anvendes en teststyrkeanalyse til at estimere, hvilken stikprøvestørrelse der skal til for at påvise en forud fastsat biologisk relevant effektstørrelse med en bestemt styrke og et bestemt signifikansniveau. Der findes nærmere retningslinjer for, hvordan undersøgelsen skal gennemføres, i EFSA's retningslinjer for gennemførelse af 90-dages undersøgelser af hele fødevarers/foderstoffers toksicitet hos gnavere ved gentagen dosering (»Guidance on conducting repeated-dose 90-day oral toxicity study in rodents on whole food/feed«) (8).
1.4.4.2. Dyreforsøg til testning for reproduktions- og udviklingstoksicitet
Når oplysninger, der kræves i henhold til punkt 1.4.1, 1.4.2 og 1.4.3, vedrørende den/det genetisk modificerede fødevare/foderstof peger i retning af potentiel reproduktionstoksicitet, udviklingstoksicitet eller kronisk toksicitet, eller hvis 90-dages-fodringsforsøget med gnavere giver indikationer på negative virkninger (såsom funktionelle og/eller histologiske forandringer i neurologiske, endokrine, reproduktive eller immunologiske væv/organer), gennemføres der passende testning. Protokollerne for testning for reproduktionstoksicitet, udviklingstoksicitet og kronisk toksicitet (jf. afsnit 1.7, tabel 1) kan tilpasses til undersøgelse af hele den/det genetisk modificerede fødevare/foderstof.
Da 90-dages-fodringsforsøget med gnavere udelukkende har til formål at påvise virkninger for reproduktionsorganernes vægt og histopatologi hos voksne individer og ikke gør det muligt at påvise andre virkninger på reproduktion eller udvikling, skal hele fødevaren/foderstoffet underkastes testning ud over et 90-dages fodringsforsøg med gnavere, hvis der er konstateret farer for så vidt angår sådanne virkninger.
1.4.4.3. Andre dyreforsøg til klarlægning af genetisk modificerede fødevarers og foderstoffers sikkerhed og karakteristika (jf. også punkt 1.6.1 og 1.6.2)
Der skal forelægges fodringsforsøg af arter i målgruppen, når oplysninger, der kræves i henhold til punkt 1.4.1, 1.4.2 og 1.4.3, vedrørende den/det genetisk modificerede fødevare/foderstof eller 90-dages-fodringsforsøget med gnavere giver formodning om negative virkninger. Forsøgene skal fokusere på sikkerheden ved nye bestanddele (nye udtrykte proteiner og andre nye bestanddele), på identifikation og karakterisering af utilsigtede virkninger og på de ernæringsmæssige virkninger af alle tilsigtede, væsentlige ændringer i den genetisk modificerede plantes sammensætning (jf. også afsnit 1.6).
Forsøg af denne type begrænses til plantematerialer, der egner sig til at indgå i dyrenes kost, og som ernæringsmæssigt kan matches med et egnet kontrolfoder.
1.4.4.4. Fortolkning af betydningen af dyreforsøg
Relevante virkninger observeret i dyreforsøg skal vurderes af eksperter med henblik på at identificere potentielle konsekvenser for menneskers og dyrs sundhed og for at vurdere deres betydning for sikkerheden ved den/det genetisk modificerede fødevare/foderstof. Denne vurdering kan understøttes af supplerende oplysninger og betragtninger. Der bør lægges vægt på det forhold, at visse virkninger kan være specifikke for forsøgsdyret, men ikke for mennesker, som følge af forskelle arterne imellem.
Ansøgeren skal især tage stilling til dosis/virkningsforholdene for parametre, der er blevet ændret (dvs. øgede doser/tilsvarende stigninger i ændringerne), eftersom disse er en stærk indikation på, at virkning af den testede forbindelse. Hvis en forskel kun kan konstateres ved den højeste anvendte dosis, skal det overvejes at inkludere andre faktorer for at fastslå, hvorvidt der er en sammenhæng med behandlingen. Oplysninger om baggrundsvariabiliteten for en given parameter kan tilvejebringes af ansøgeren på grundlag af data for andre dyr af samme art/stamme, der er testet i samme eller andre forsøg, eller fra internationalt harmoniserede databaser.
I forsøg, hvor der anvendes dyr af begge køn, kan forandringer, der kun finder sted i dyr af det ene køn, stadig være relevante indikatorer for en virkning, afhængigt af den parameter, der ændres, og den mekanisme, hvorved forandringen kan være blevet forårsaget. F.eks. kan dyr af det ene køn være mere — eller ligefrem specifikt — udsat for ændringer forårsaget af en given bestanddel end dyr af det modsatte køn, hvilket f.eks. gælder for endokrine virkninger.
Ansøgeren skal også identificere eventuelle indbyrdes sammenhænge mellem observerede ændringer i individuelle parametre, som kan forstærke indikationerne på, at en virkning har fundet sted. F.eks. vil leverskader, som kan observeres i selve leveren som forandringer i histopatologi, makroskopisk patologi og organvægt, også tydeligt kunne ses af de ændrede mængder af visse forbindelser, der stammer fra leveren, såsom enzymer eller bilirubin (i serum).
Med hensyn til den mulige årsag til en observeret virkning skal sandsynligheden for en årsagssammenhæng tages i betragtning, ikke kun for testforbindelsen, men også for andre faktorer, som også kan have påvirket resultaterne (såsom faldende kropsvægt som følge af et reduceret indtag af mindre velsmagende kost). Data, der understøtter en hypotese om en årsagssammenhæng mellem testforbindelsen og virkninger i forsøgsdyr, kan bl.a. omfatte prognosedata om sandsynlige virkninger fra in vitro- og in silico-forsøg samt dosis/virkningsforhold observeret i dyreforsøget.
1.4.5. Konklusion af den toksikologiske vurdering
I konklusionen af den toksikologiske vurdering skal det angives, hvorvidt:
|
a) |
potentielle negative virkninger, der er påvist i andre dele af sikkerhedsvurderingen, er blevet bekræftet eller afvist |
|
b) |
de tilgængelige oplysninger om det eller de nye udtrykte proteiner og andre nye bestanddele, der er fremkommet ved den genetiske modifikation, giver indikationer på potentielle negative virkninger, navnlig hvorvidt og ved hvilke dosisniveauer de negative virkninger er påvist i specifikke undersøgelser |
|
c) |
oplysningerne om naturlige bestanddele, der er til stede i andre mængder, end det er tilfældet i det konventionelle modstykke, giver indikationer på potentielle negative virkninger, navnlig hvorvidt og ved hvilke dosisniveauer de negative virkninger er påvist i specifikke undersøgelser |
|
d) |
der er påvist negative virkninger i undersøgelser af hele den/det genetisk modificerede fødevare/foderstof, og ved hvilke dosisniveauer. |
Ansøgeren skal vurdere resultatet af den toksikologiske vurdering på grundlag af det forventede indtag af den/det genetisk modificerede fødevare/foderstof (jf. afsnit 2).
1.5. Allergenicitet
Fødevareallergi er en negativ reaktion på fødevarer og er et alvorligt folkesundhedsproblem. Fødevareallergi adskiller sig fra toksiske reaktioner og intolerans. Allergi er en patologisk afvigelse i immunresponset på et bestemt stof, som kun berører nogle mennesker, hos hvem en kombination af ændringer i miljøet og genetiske anlæg har resulteret i allergisk sensibilisering.
Hos personer med allergi kan ganske små mængder af en fødevare, som langt størstedelen af befolkningen kan tåle, undertiden fremkalde både alvorlige symptomer og dødsfald. Det er ikke allergenet som sådan, men derimod den allergiske persons afvigende reaktion over for allergenet, der forårsager den sundhedsskadelige virkning.
Fødevareallergi kan forårsages af forskellige immunmekanismer. IgE-medieret fødevareallergi er dog den almindeligste form for fødevareallergi; det er denne form, der forårsager de alvorligste reaktioner, og den eneste form, der forårsager livstruende reaktioner. Det er IgE-medieret fødevareallergi, der har været fokus på i forbindelse med risikovurdering af GMO'ers allergenicitet. Det er væsentligt at nævne, at fødevareallergi omfatter to separate faser: først sensibilisering, hvor der ikke opstår symptomer, men hvor immunsystemets evne til at reagere øges drastisk, og senere udløsning (provokation) med kliniske manifestationer.
Når allergenet eller allergenerne, dvs. den sensibiliserende fødevare eller fødevarebestanddel, indtages, nedbrydes det/de til en vis grad af fordøjelsesenzymer, absorberes af mavetarmsystemets slimhinder (små mængder endda af mundens slimhinde) og bearbejdes i immunsystemets specialiserede celler, hvorefter det bringes sammen med de reagerende immunceller, som frembringer et immunrespons. Sensibilisering kan også forekomme, hvis fødevareallergenet kommer i kontakt med huden eller indåndes.
Størstedelen af de bestanddele, der giver fødevarer og pollen allergifremkaldende egenskaber, er proteiner. Visse proteolyseprodukter, nemlig peptidfragmenter, kan bevare en del af det native proteins allergenicitet og kan derfor også betragtes som allergener.
Den specifikke allergirisiko ved GMO'er er forbundet med: i) eksponering for nye udtrykte proteiner, som kan være til stede i spiselige dele af planterne eller i pollen; dette punkt vedrører transgenets biologiske herkomst; og ii) ændringer i allergeniciteten af hele planter og produkter fremstillet heraf, f.eks. som følge af overekspression af naturlige endogene allergener som en utilsigtet virkning af den genetiske modifikation; dette afsnit vedrører selve recipientplantens biologi.
EFSA's videnskabelige udtalelse af 30. juni 2010 om vurdering af allergeniciteten af genetisk modificerede planter og mikroorganismer samt afledte fødevarer og foderstoffer (»Scientific Opinion on the assessment of allergenicity of GM plants and microorganisms and derived food and feed«) (9) indeholder mere detaljerede retningslinjer for anvendelsen af bestemmelserne i dette punkt.
1.5.1. Vurdering af det nye udtrykte proteins allergenicitet
Allergenicitet ikke er en iboende, fuldt forudsigelig egenskab hos et givent protein, men derimod en biologisk aktivitet, som forudsætter en vekselvirkning med personer, der er disponeret for allergi i kraft af deres arveanlæg. Allergenicitet afhænger derfor af den genetiske diversitet og variabilitet hos atopikere. Allergiske reaktioners hyppighed, sværhedsgrad og specificitet afhænger samtidig af geografiske og miljømæssige faktorer. Grundet denne mangel på fuldstændig forudsigelighed er det nødvendigt at tage stilling til flere forskellige aspekter i allergenicitetsvurderingen for at tilvejebringe et samlet evidensgrundlag, der begrænser usikkerheden vedrørende det eller de pågældende proteiner til et minimum.
Når man undersøger de strukturelle karakteristika og de biologiske og fysisk-kemiske egenskaber ved et nyt udtrykt protein, er det meget vigtigt, at det undersøgte protein er ækvivalent med det nye udtrykte protein i den genetisk modificerede plante med hensyn til struktur og aktivitet. Undersøgelser udført med oprensede målproteiner fremstillet ved ekspression i organismer såsom Escherichia coli kan accepteres, forudsat at den mikrobielle substituts egenskaber er identiske med egenskaberne hos det protein, der udtrykkes i planten, således at der tages hensyn til alle posttranslationelle modifikationer, som specifikt forekommer i planten.
Ansøgeren skal kontrollere, hvorvidt kilden til transgenet er allergen. Når det indsatte genetiske materiale stammer fra hvede, rug, byg, havre eller dermed beslægtede kornsorter, skal ansøgeren tillige undersøge de nye udtrykte proteiner for deres mulige rolle i forbindelse med udløsning af cøliaki eller andre enteropatier, der ikke er IgE-medieret. Hvor transformationsbegivenheder er blevet stablet, skal ansøgeren fremlægge en vurdering af enhver sandsynlighed for øget allergenicitet hos mennesker og dyr i hvert enkelt tilfælde. Disse potentielle virkninger kan forårsages af additive, synergistiske eller antagonistiske virkninger af genprodukterne.
Ansøgeren skal følge en integreret, sag til sag-tilgang — dvs. en bevisvægtet fremgangsmåde — ved vurderingen af nye udtrykte proteiners potentielle allergenicitet. Denne fremgangsmåde skal omfatte følgende:
|
a) |
En sammenligning af aminosyrers sekvenshomologi i henholdsvis det nye udtrykte protein og kendte allergener Der skal i hvert enkelt tilfælde foretages en søgning efter sekvenshomologier og/eller strukturelle ligheder mellem det udtrykte protein og kendte allergener for at påvise eventuel IgE-krydsreaktivitet mellem det nye udtrykte protein og kendte allergener. Ansøgeren skal sikre, at databasernes kvalitet og dækning ikke findes bedre. Det alignmentbaserede kriterium med en sekvensidentitet på 35 % i forhold til et kendt allergen over et vindue af mindst 80 aminosyrer betragtes som mindstekravet. Alle sekvensalignmentparametre, der er anvendt i analysen, skal oplyses, herunder beregningen af procentuel identitet (PID). Beregningen af PID foretages på et vindue af 80 aminosyrer med gaps, således at indsatte gaps betragtes som manglende overensstemmelse. I visse tilfælde kan der, ved vurdering af korte peptidfragmenter såsom ORF'er, foretages en søgning efter sekvenser af identiske eller kemisk til hinanden svarende aminosyrerester. Denne søgning må dog, på grund af sin meget begrænsede følsomhed/specificitet, ikke rutinemæssigt anvendes til påvisning af potentielle lineære IgE-bindende epitoper. |
|
b) |
Specifik serumscreening Når der er indikationer på sekvenshomologi eller strukturelle ligheder, findes der en vigtig procedure til vurdering af sandsynligheden for, at eksponering for de nye udtrykte proteiner vil fremkalde en allergisk reaktion hos personer, der allerede er sensibiliseret over for krydsreaktive proteiner, på basis af in vitro-test, i hvilke det måles, i hvilket omfang specifikt IgE fra serum fra allergiske patienter er i stand til at binde testproteinet eller -proteinerne. IgE-responset hos mennesker varierer fra person til person med hensyn til specificitet og affinitet. Navnlig IgE-antistoffernes specificitet til de forskellige allergener i en given fødevare/kilde og/eller til de forskellige epitoper, der er til stede på et givent protein, kan variere fra allergiker til allergiker. For at optimere testens følsomhed skal der anvendes individuelle sera fra velkarakteriserede personer med allergi. Ansøgeren skal foretage specifik serumscreening i følgende tilfælde:
Specifik serumscreening udføres med individuelle sera fra personer med en dokumenteret og velkarakteriseret allergi over for kilden eller det potentielt krydsreagerende allergen ved hjælp af relevante immunokemiske test. IgE-bindingsassays (f.eks. radio- eller enzym-allergosorbent-assay (RAST eller EAST), enzymbundet immunosorbent-assay (ELISA) og elektroforese, efterfulgt af immunblotting med specifikke IgE-holdige sera) er egnede metoder. |
|
c) |
Pepsinresistens og in vitro-fordøjelighedstest Allergene proteiner har længe været anset for at være stabile over for proteasenedbrydning. Selv om det er fastslået, at der ikke eksisterer en absolut sammenhæng, er proteiners resistens over for pepsinnedbrydning et supplerende kriterium, som der skal tages hensyn til i den bevisvægtede tilgang til vurdering af allergenicitet. Pepsinresistenstesten udføres normalt under ret standardiserede forhold, med lav pH og store andele pepsin i forhold til protein. Det er almindeligt anerkendt, at pepsinresistenstesten ikke afspejler de fysiologiske betingelser for nedbrydningen. Fordøjeligheden af de nye udtrykte proteiner i specifikke befolkningsgrupper, f.eks. hos børn og hos personer med fordøjelsesproblemer, kan vurderes ved hjælp af in vitro-fordøjelighedstest under andre betingelser. Eftersom det protein, der er kodet af de nye indsatte gener, vil være til stede i produktet som en kompleks matrix, skal der i yderligere in vitro-fordøjelighedstest tages hensyn til virkningerne af eventuelle vekselvirkninger mellem proteinet og andre bestanddele af matricen samt virkningerne af forarbejdningen. Afhængigt af resultatet af in vitro-fordøjelighedstesten skal der foretages en sammenligning af de intakte, de varmedenaturerede og de pepsinfordøjede proteiner til IgE-binding, da ændret fordøjelighed kan påvirke det nye udtrykte proteins allergenicitet. |
|
d) |
Yderligere undersøgelser Selv om der ikke hidtil er valideret yderligere test, såsom cellebaserede assays eller in vivo-test på dyremodeller, til lovgivningsmæssige formål, kan disse test give nyttig supplerende information, f.eks. om potentialet hos det nye udtrykte protein til at forårsage de novo-sensibilisering. |
1.5.2. Vurdering af allergeniciteten af den/det genetisk modificerede fødevare/foderstof
Når recipientplanten vides at være allergen, skal ansøgeren vurdere enhver potentiel ændring i allergeniciteten af den/det genetisk modificerede fødevare/foderstof ved at sammenligne allergenets repertoire med dets konventionelle modstykkes repertoire. I den forbindelse skal især potentiel overekspression af naturlige endogene allergener i den genetisk modificerede plante undersøges.
Ansøgeren skal følge en sag til sag-tilgang ved sådanne undersøgelser alt efter de tilgængelige oplysninger om recipientplantens allergifremkaldende potentiale. Der anvendes generelt analysemetoder såsom proteomik under anvendelse af sera fra allergiske personer som prober. Sera fra klinisk velkarakteriserede personer med allergi, som er referencemateriale i IgE-bindingsundersøgelser, vil i nogle tilfælde kun være til rådighed i begrænset antal og mængde. For at begrænse brugen af humane sera til et minimum kan der tilvejebringes vigtige foreløbige oplysninger om sandsynligheden for en utilsigtet ændring af den genetisk modificerede plantes samlede allergenicitet ved at anvende sera fra dyr, der er sensibiliseret i forsøg under veldefinerede betingelser, og ved at medtage relevante påviste endogene allergener i den sammenlignende analyse af sammensætningen.
Ansøgeren skal desuden fremlægge oplysninger om forekomsten af allergi hos personer, der arbejder med eller kommer i kontakt med eller i nærheden af dyrkning af genetisk modificerede planter, i det omfang sådanne oplysninger foreligger.
1.5.3. Adjuvansevne
Adjuvanter er stoffer, som, når de indgives sammen med et antigen, forbedrer immunresponset over for dette antigen og derfor også kan forstærke det allergiske respons. I tilfælde, hvor kendte funktionelle aspekter ved det nye udtrykte protein eller strukturel lighed med kendte stærke adjuvanter kan give formodning om mulig adjuvansaktivitet, skal ansøgeren vurdere de pågældende proteiners mulige rolle som adjuvanter. For så vidt angår allergener kan vekselvirkninger med andre bestanddele af fødevarematricen og/eller forarbejdning ændre en adjuvants struktur og biotilgængelighed og således ændre dens biologiske aktivitet.
1.5.4. Konklusion af allergenicitetsvurderingen
I konklusionen af allergenicitetsvurderingen skal det angives:
|
a) |
hvorvidt det er sandsynligt, at det eller de nye proteiner er allergene |
|
b) |
hvorvidt det er sandsynligt, at den genetisk modificerede fødevare/det genetiske modificerede foderstof er mere allergent end dens/dets konventionelle modstykke. |
Når der er en sandsynlighed for øget allergenicitet som følge af den genetiske modifikation, skal den/det genetisk modificerede fødevare/foderstof karakteriseres yderligere på baggrund af det forventede indtag (jf. afsnit 2). Ansøgeren skal foreslå passende betingelser for markedsføring (f.eks. overvågning efter markedsføringen og mærkning).
1.6. Ernæringsmæssig vurdering
1.6.1. Mål for den ernæringsmæssige vurdering
Ansøgeren skal fremlægge en ernæringsmæssig vurdering for at dokumentere, at:
|
a) |
markedsføringen af den/det genetisk modificerede fødevare/foderstof ikke er ernæringsmæssigt ufordelagtig for dyr eller mennesker. Denne vurdering skal bl.a. beskrive betydningen i ernæringsmæssig henseende af nye udtrykte proteiner, andre nye bestanddele og ændringer i mængden af fødevare- og foderstofbestanddele samt potentielle ændringer i forbrugernes eller dyrets kost samlet set |
|
b) |
utilsigtede virkninger af den genetiske modifikation, som på basis af de forudgående molekylær-, sammensætnings- eller fænotypeanalyser, der er foretaget i overensstemmelse med afsnit 1.2 og 1.3, er påvist eller kan antages at have fundet sted, ikke har influeret negativt på næringsværdien af den/det genetisk modificerede fødevare/foderstof. |
For så vidt angår stablede transformationsbegivenheder, der er kombineret ved konventionel krydsning, skal ansøgeren fremlægge en vurdering af de potentielle ændringer i næringsværdien, som vil kunne forårsages af synergistiske eller antagonistiske virkninger af genprodukterne, herunder ændringer i sammensætningen. Dette kan være særlig relevant, hvis den kombinerede ekspression af de nye indsatte gener har uventede virkninger på biokemiske synteseveje.
1.6.2. Aspekter, der skal tages hensyn til med henblik på ernæringsmæssig vurdering af genetisk modificerede fødevarer og foderstoffer
Den ernæringsmæssige vurdering af genetisk modificerede fødevarer og foderstoffer skal omfatte:
|
a) |
de genetisk modificerede fødevarers/foderstoffers sammensætning med hensyn til indholdet af næringsstoffer og næringshæmmende stoffer (jf. undersøgelserne af sammensætningen som beskrevet i afsnit 1.3) |
|
b) |
biotilgængeligheden og den biologiske effektivitet af næringsstoffer i fødevaren/foderstoffet under hensyntagen til de potentielle påvirkninger fra transport, oplagring og forventet behandling af fødevaren og foderstoffet |
|
c) |
det forventede indtag af fødevaren/foderstoffet (jf. afsnit 2) og virkningerne heraf i ernæringsmæssig henseende. |
Har den sammenlignende analyse afdækket karakteristika ved den/det genetisk modificerede fødevares/foderstofs sammensætning, som er forskellige fra det konventionelle modstykke og/eller ikke er ækvivalente med referencesorternes karakteristika, skal den ernæringsmæssige betydning heraf vurderes på grundlag af den aktuelle videnskabelige viden. Hvis denne vurdering lader konkludere, at der er ernæringsmæssig ækvivalens mellem den/det genetisk modificerede fødevare/foderstof og dens/dets konventionelle modstykke, er der ikke behov for yderligere undersøgelser. Hvis vurderingen af de oplysninger, der er tilvejebragt med den sammenlignende analyse, derimod ikke muliggør en konklusion om den ernæringsmæssige ækvivalens, skal der gennemføres yderligere ernæringsmæssige undersøgelser. Der skal gennemføres sammenlignende vækstundersøgelser med unge individer af hurtigt voksende dyrearter (f.eks. slagtekyllinger som dyremodel for ikke-drøvtyggere, lam for drøvtyggere eller andre hurtigt voksende arter).
1.6.3. Ernæringsmæssige undersøgelser af genetisk modificerede fødevarer
Ansøgeren skal træffe afgørelse om behovet for og udformningen af ernæringsmæssige undersøgelser på grundlag af den eller de indførte egenskaber, resultatet af den sammenlignende analyse og 90-dages-fodringsforsøget, hvis det foreligger. Der kan tilvejebringes supplerende oplysninger om næringsværdien fra sammenlignende undersøgelser af vækstforøgelsen hos andre dyrearter, f.eks. slagtekyllinger, til ernæringsmæssig vurdering af genetisk modificerede foderstoffer. Ved gennemførelse af ernæringsmæssige undersøgelser skal kontrolfoderet omfatte det konventionelle modstykke og eventuelt supplerende komparatorer. For så vidt angår herbicidtolerante genetisk modificerede planter skal det testede materiale komme fra den genetisk modificerede plante eksponeret for det påtænkte herbicid.
Genetisk modificerede fødevarer, der er modificeret med henblik på at give forbrugeren sundhedsmæssige fordele i forhold til konventionelle fødevarer, vil kunne gavne bestemte befolkningsgrupper eller delpopulationer og samtidig udgøre en risiko for andre. I tilfælde, hvor det er nødvendigt at fastlægge en ændret biotilgængelighed, som potentielt vil kunne være problematisk for visse delpopulationer, skal indholdet af næringsstoffet i fødevaren bestemmes under hensyntagen til alle de forskellige former, forbindelsen kan forekomme i. Metoderne til undersøgelse af biotilgængeligheden vælges i hvert enkelt tilfælde alt efter næringsstoffet eller andre bestanddele, fødevaren, der indeholder disse bestanddele, samt sundheden, ernæringstilstanden og kostvanerne i den eller de befolkningsgrupper, der forventes at ville indtage fødevaren.
1.6.4. Ernæringsmæssige undersøgelser af genetisk modificerede foderstoffer
Ansøgeren skal træffe afgørelse om behovet for og udformningen af yderligere ernæringsmæssige undersøgelser på grundlag af den eller de indførte egenskaber, resultatet af den sammenlignende analyse og i givet fald 90-dages-fodringsforsøget. Der kan tilvejebringes supplerende oplysninger om næringsværdien fra sammenlignende undersøgelser af vækstforøgelsen hos andre dyrearter, f.eks. slagtekyllinger, til ernæringsmæssig vurdering af genetisk modificerede foderstoffer. Ved gennemførelse af ernæringsmæssige undersøgelser skal kontrolfoderet omfatte det konventionelle modstykke og eventuelt supplerende komparatorer.
Når der er tale om genetisk modificerede foderstoffer med forbedrede ernæringsmæssige karakteristika, skal der udføres fodringsforsøg af målarter af dyr bestemt til fødevareproduktion for at vurdere indvirkningen på foderet. Hvad angår genetisk modificerede planter, der er modificeret for at forbedre indholdet og biotilgængeligheden af næringsstoffer, skal der udføres forsøg af målarter af dyr bestemt til fødevareproduktion med henblik på at bestemme biotilgængeligheden af individuelle næringsstoffer i den genetisk modificerede plante i forhold til dens konventionelle modstykke. For genetisk modificerede planter, der specifikt er modificeret med henblik på at forbedre dyrenes ydelse via øget næringsstoftæthed (f.eks. øget indhold af olie) eller opnå en højere koncentration af et bestemt næringsstof (f.eks. en essentiel aminosyre eller et vitamin), skal der sammensættes et passende kontrolfoder af plantens konventionelle modstykke ved hertil at tilsætte det pågældende næringsstof i en mængde, der svarer til den, som er opnået med ændringerne i den genetisk modificerede plante. For så vidt angår sideprodukter (såsom mel af oliefrø), hvorfra den ingrediens, som den genetiske modifikation vedrører, er blevet ekstraheret, kan disse sammenlignes med sideprodukter fremstillet af det konventionelle modstykke.
Fodringsforsøg med målarter skal omfatte vækst- og/eller slutfedningsperioden indtil slagtetidspunktet for kyllinger, svin og fedekvæg, størstedelen af laktationscyklen for malkekøer eller æglægningscyklen for æglæggende høner eller vagtler. For foderstoffer, der udelukkende er beregnet til akvakultur, gennemføres vækstundersøgelser med akvatiske arter såsom karper, havkat, laksefisk eller typiske planteædere.
Når det er relevant, skal der fremlægges forsøg med forskellig udformning, der dokumenterer, at den ernæringsmæssigt forbedrede genetisk modificerede plante har den forventede næringsværdi. Den nøjagtige forsøgsplan og det statistiske system, der anvendes til fodringsforsøg med dyr bestemt til fødevareproduktion med henblik på at undersøge næringsværdien af genetisk modificerede foderstoffer, der er modificeret for at forbedre ernæringsmæssige karakteristika, fastlægges i overensstemmelse med, hvilken dyreart der er tale om, og hvilken type planteegenskaber der undersøges, samt omfanget af de forventede virkninger. Forsøgskosten skal sammensættes på en sådan måde, at de vigtigste målte endpoints påvirkes af en forskel i mængden og/eller tilgængeligheden af det pågældende næringsstof. Endpointmålingerne fastlægges i overensstemmelse med, hvilken målart der anvendes i forsøget, men skal omfatte foderindtag, kropsvægt, dyrenes ydeevne og biotilgængeligheden af næringsstoffer.
Rapporten om fodringsforsøg (10) fra arbejdsgruppen under EFSA's GMO-panel indeholder mere detaljerede retningslinjer for anvendelsen af bestemmelserne i dette punkt.
1.6.5. Konklusion af den ernæringsmæssige vurdering
I konklusionen af den ernæringsmæssige vurdering skal det angives, hvorvidt den/det genetisk modificerede fødevare/foderstof, under hensyntagen til naturlig variation, er ækvivalent med dens/dets konventionelle modstykke med hensyn til næringsværdi.
Ansøgeren skal vurdere resultatet af den ernæringsmæssige vurdering på baggrund af det forventede indtag af den/det genetisk modificerede fødevare/foderstof (jf. afsnit 2).
1.7. Standardiserede retningslinjer for toksicitetstest
Ansøgeren skal til toksicitetstest anvende internationalt anerkendte retningslinjer og testmetoder som beskrevet i Kommissionens forordning (EF) nr. 440/2008 af 30. maj 2008 om fastlæggelse af forsøgsmetoder i henhold til Europa-Parlamentets og Rådets forordning (EF) nr. 1907/2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH) (11) (jf. tabel 1 og 2). En ikke-udtømmende liste over validerede testmetoder, som skal anvendes i det fornødne omfang, eventuelt i en tilpasset form, til toksikologisk undersøgelse af GMO'er, findes i tabel 1 og 2.
Testmetodernes ydeevne afhænger af arten af de genetisk modificerede fødevarer og foderstoffer, arten af den genetiske modifikation og derved fremkomne tilsigtede og utilsigtede ændringer, den påtænkte anvendelse og eksponering/indtag samt af den foreliggende viden. Visse af disse test er udviklet til vurdering af risici på arbejdspladsen (jf. afsnit 1.4 og 1.5).
Tabel 1
Ikke-udtømmende liste over validerede testmetoder for kemikalier, jf. forordning (EF) nr. 440/2008, som kan anvendes i en eventuelt tilpasset form til toksikologisk undersøgelse af GMO'er
|
Betegnelse |
Henvisning til metoden i del B i bilaget til forordning (EF) nr. 440/2008 |
|
AKUT TOKSICITET (DERMAL) |
B.3. |
|
HUDSENSIBILISERING |
B.6. |
|
TOKSICITET VED GENTAGEN DOSERING (28 DAGE, ORAL) |
B.7. |
|
TOKSICITET VED GENTAGEN DOSERING (28 DAGE, DERMAL) |
B.9. |
|
SUBKRONISK ORAL TOKSICITETSTEST — ET 90-DAGES FORSØG OVER GENTAGNE ORALE DOSERS TOKSICITET PÅ GNAVERE |
B.26. |
|
UNDERSØGELSE AF KRONISK TOKSICITET |
B.30. |
|
KARCINOGENICITETSUNDERSØGELSE |
B32. |
|
KOMBINERET UNDERSØGELSE AF KARCINOGENICITET/KRONISK TOKSICITET |
B.33. |
|
REPRODUKTIONSTOKSICITETSUNDERSØGELSE I EN GENERATION |
B.34. |
|
REPRODUKTIONSTOKSICITETSUNDERSØGELSE I TO GENERATIONER |
B.35. |
|
TOKSIKOKINETIK |
B.36. |
|
NEUROTOKSICITETSUNDERSØGELSE I GNAVERE |
B.43. |
Tabel 2
Genotoksicitetsundersøgelser, jf. forordning (EF) nr. 440/2008
|
Betegnelse |
Henvisning til metoden i del B i bilaget til forordning (EF) nr. 440/2008 |
|
MUTAGENICITET — IN VIVO-TEST FOR KROMOSOMABERRATIONER I KNOGLEMARV HOS PATTEDYR |
B.11. |
|
MUTAGENICITET — IN VIVO-TEST FOR MIKROKERNER I ERYTHROCYTTER HOS PATTEDYR |
B.12. |
|
MUTAGENICITET: TILBAGEMUTATIONSTEST MED BAKTERIER |
B.13/14. |
|
MUTAGENICITETSTESTNING OG SCREENING FOR KARCINOGENICITET — GENMUTATION — SACCHAROMYCES CEREVlSIAE |
B.15. |
|
MITOTISK REKOMBINATION — SACCHAROMYCES CEREVISIAE |
B.16. |
|
DNA-SKADE OG DNA-REPARATION — IKKE PLANLAGT DNA-SYNTESE — PATTEDYRCELLER IN VITRO |
B.18. |
|
MUTAGENICITET — IN VITRO-TEST FOR GENMUTATION I PATTEDYRCELLER |
B.17. |
|
SØSTERKROMATIDOMBYTNING (SCE) IN VITRO |
B.19. |
|
CELLETRANSFORMATIONSTEST — PATTEDYRCELLER IN VITRO |
B.21. |
|
TEST FOR KROMOSOMABERRATIONER I SPERMATOGONIER HOS PATTEDYR |
B.23. |
2. VURDERING AF EKSPONERINGEN — FORVENTET INDTAG/OMFANG AF ANVENDELSEN
Et skøn over det forventede indtag skal indgå som et væsentligt element i risikovurderingen af genetisk modificerede fødevarer og foderstoffer og er også påkrævet ved den ernæringsmæssige vurdering. Ansøgeren skal fremlægge oplysninger om den tilsigtede funktion, rolle i kosten og det forventede omfang af anvendelsen i EU af de genetisk modificerede fødevarer og foderstoffer. Desuden angives det forventede koncentrationsområde for aktivt modificerede nyproducerede proteiner eller eksisterende planteproteiner i de(n) genetisk modificerede fødevare(r) og foderstof(fer), der skal markedsføres.
På grundlag af repræsentative data om forbruget af produkter fra de respektive konventionelle planter skal ansøgeren estimere det forventede gennemsnitlige og maksimale indtag af de genetisk modificerede fødevarer og foderstoffer. Der kan anvendes sandsynlighedsbaserede metoder til at fastlægge intervaller af sandsynlige værdier frem for enkeltværdier eller punktestimater. Ansøgeren skal identificere og tage hensyn til særlige grupper af EU-populationen med en forventet højere eksponering og skal tage hensyn til sådanne højere eksponeringsniveauer i risikovurderingen. Gøres der brug af antagelser i eksponeringsvurderingen, skal disse beskrives. Vurderingen baseres på de nyeste metoder og passende forbrugsdata. Som supplerende input til vurderingen af indtaget kan der anvendes oplysninger om import- og produktionsmængder.
Ansøgeren skal ved egnede metoder bestemme koncentrationerne af de nye udtrykte proteiner, andre nye bestanddele samt endogene fødevare- og foderstofbestanddele, hvis koncentrationer er blevet ændret i de dele af den genetisk modificerede plante, der er bestemt til fødevare- eller foderbrug, som et resultat af den genetiske modifikation (f.eks. som følge af ændrede stofskifteveje). Det forventede indtag af disse bestanddele skal estimeres under hensyntagen til påvirkningerne fra forarbejdning, oplagring og forventet behandling af den/det pågældende fødevare/foderstof, f.eks. potentiel akkumulering eller reduktion. I tilfælde, hvor den genetiske modifikation har resulteret i en ændret mængde af en naturlig bestanddel, eller hvis en ny bestanddel forekommer naturligt i andre fødevarer og foderstoffer, vurderes den forventede ændring i det samlede indtag af denne bestanddel med udgangspunkt i såvel realistiske scenarier som worst case-scenarier.
Ansøgeren skal fremlægge oplysninger om menneskers/dyrs kendte eller forventede indtag af tilsvarende genetisk modificerede fødevarer og foderstoffer og om andre kilder til eksponering for de henholdsvis nye og naturlige bestanddele, herunder eksponeringsmængder og -hyppighed og andre faktorer, der har indflydelse på eksponeringen.
3. RISIKOKARAKTERISERING
3.1. Indledning
Ansøgeren skal basere sin risikokarakterisering af genetisk modificerede planter og fødevarer/foderstoffer på data tilvejebragt ved fareidentifikation og farekarakterisering samt på oplysninger om eksponering/indtag. Ansøgeren skal sikre, at risikokarakteriseringen er dækkende, ved at tage hensyn til al tilgængelig dokumentation fra en række analyser, herunder molekylæranalyse, analyser af fænotypiske og agronomiske karakteristika, analyser af sammensætning og toksicitet og testning for allergenicitet. Ansøgeren skal tage hensyn til indikationer, der udledes af risikokarakteriseringen, som kan nødvendiggøre særlige aktiviteter vedrørende overvågning efter markedsføringen af genetisk modificerede fødevarer eller foderstoffer.
Ansøgeren skal i forbindelse med risikokarakteriseringen dokumentere, at identifikationen og karakteriseringen af farer er tilendebragt. Ansøgeren skal tage stilling til og redegøre for kvaliteten af eksisterende data og oplysninger. Det skal af denne redegørelse klart fremgå, hvorledes der er taget hensyn til disse oplysninger ved fastlæggelsen af den endelige risikokarakterisering.
Ansøgeren skal fremlægge skøn over de usikkerheder, der er forbundet med hver enkelt undersøgelse samt med risikovurderingens forskellige faser. Ansøgeren skal kvantificere dem i så høj grad som muligt. Der sondres mellem usikkerheder, der afspejler naturlig variation i biologiske parametre (herunder udsving i følsomheden hos bestemte populationer), og mulige responsvariationer arterne imellem.
Alt efter spørgsmålet, der skal behandles, og de tilgængelige oplysninger skal ansøgeren foretage en kvalitativ og om muligt en kvantitativ risikokarakterisering. Betingelserne for den estimerede risiko og de relevante usikkerheder skal afgrænses så præcist som muligt.
3.2. Aspekter, som der skal tages stilling til i forbindelse med risikokarakteriseringen
Afhængigt af arten af den genetiske modifikation skal ansøgeren, hvis det er relevant, foretage en integreret risikovurdering af genetisk modificerede planter i overensstemmelse med afsnit 3.1. Denne risikovurdering skal gennemføres med udgangspunkt i det enkelte tilfælde afhængigt af den modificerede plante og arten af den genetiske modifikation, praksis med hensyn til dyrkningen af den genetisk modificerede plante og anvendelserne af den/det genetisk modificerede fødevare/foderstof. Ansøgeren skal tage hensyn til de forskellige aspekter, der er behandlet i forbindelse med trinnene fareidentifikation, farekarakterisering og eksponering. Ansøgeren foretager en samlet evaluering af resultaterne af disse betragtninger i forbindelse med risikokarakteriseringen. Listen over emner i dette afsnit er ikke udtømmende.
3.2.1. Molekylær karakterisering
Det er vigtigt at vurdere donor og recipientplantens karakteristika og tidligere anvendelse, når det skal afgøres, hvorvidt der er behov for specifikke analyser, såsom forekomst af specifikke toksiner eller allergener i den ikke-modificerede recipientplante, som utilsigtet vil kunne øges som følge af den genetiske modifikation.
Ansøgeren skal fremlægge og diskutere transformationsprotokoller og strategier for molekylær karakterisering samt de anvendte metoders specificitet og følsomhed i forhold til den tilsigtede samt muligvis utilsigtet indsættelse og ekspression af gensekvenser.
Hvis der ved analysen af sekvenser er identificeret en potentiel fare, skal ansøgeren påvise, hvordan fremgangsmåder som bioinformatikanalyse, analyser af sammensætning og agronomiske analyser samt eventuelt fodringsforsøg med hele den/det genetisk modificerede fødevare/foderstof bidrager til sikkerhedsvurderingen. Værdien af de opnåede resultater skal evalueres i lyset af den tilgængelige viden om, hvordan genomdatabaserne over den pågældende afgrødeart eller beslægtede arter er opbygget og fungerer.
For så vidt angår genetisk modificerede planter indeholdende stablede transformationsbegivenheder skal der foretages en evaluering af de yderligere risici, der muligvis opstår som følge af kombinationseffekten af de stakkede gener.
3.2.2. Sammenlignende analyse
Det første mål for den sammenlignende analyse er at identificere eventuelle forskelle mellem den genetisk modificerede plante og dens konventionelle modstykke og, hvis det er relevant, yderligere komparatorer. Det andet mål for den sammenlignende analyse er at identificere eventuel manglende ækvivalens mellem den genetisk modificerede plante og dens referencesorter. Forskellene og/eller den manglende ækvivalens bør vurderes med hensyn til deres mulige virkning for fødevare- og foderstofsikkerheden og ernæringsmæssige egenskaber, idet der tages hensyn til naturlig variation. Den estimerede risiko og dermed forbundne usikkerhed bør være så præcis som mulig og tages i betragtning.
Ansøgeren skal godtgøre, at den sammenlignende analyse af den genetisk modificerede plante og dens konventionelle modstykke med hensyn til deres karakteristika for så vidt angår agronomi, morfologi og sammensætning er foretaget i overensstemmelse med kravene i denne forordning. Valget af konventionelt modstykke og, hvis det er relevant, yderligere komparatorer skal begrundes.
3.2.3. Fødevare- og fodersikkerheden i forbindelse med indtagelse
Ansøgeren skal vurdere de tilvejebragte data for at estimere mulige kortsigtede og langsigtede risici for menneskers eller dyrs sundhed ved indtagelse af genetisk modificerede fødevarer eller foder med hensyn til ekspression af nye proteiner/metabolitter samt væsentlige ændringer i mængden af de oprindelige planteproteiner/metabolitter i genetisk modificerede fødevarer/foderstoffer. Denne evaluering skal omfatte en grundig analyse af hver enkelt undersøgelses og den samlede informationsmængdes relevans og begrænsninger.
Ansøgeren skal undersøge intervallet af observerede niveauer for forbindelser, der vides at forekomme i det konventionelle modstykke og i referencesorter. Variabiliteten kan skyldes forskelle, der er genotypeafhængige, miljøafhængige eller forårsaget af genotype x's vekselvirkninger med miljøet. Der kan desuden tages hensyn til intervallet af observerede niveauer i en bred vifte af fødevarer og foderstoffer, der er repræsentative for menneskers og dyrs ernæring, for så vidt som det afspejler de mængder af den pågældende forbindelse, som forbrugerne vil kunne blive eksponeret for.
Hvis det konstateres, at individuelle bestanddele og/eller hele den/det genetisk modificerede fødevare/foderstof fremkalder negative virkninger i specifikke undersøgelser, skal der fremlægges oplysninger om dosis/virkningsforhold, tærskelværdier, forsinket indtræden af negative virkninger, risici for bestemte befolkningsgrupper og brug af usikkerhedsfaktorer ved ekstrapolering af data fra dyr til mennesker.
Ansøgeren skal tage stilling til data om karakteristika ved de nye forbindelser, der forekommer i den genetisk modificerede plante, herunder potentielle biologiske virkninger i mennesker og dyr. Hvis de pågældende forbindelser har kendte sundhedsskadelige virkninger, og der i lovgivningen specifikt er fastsat maksimalgrænseværdier for forekomst af disse forbindelser i planten eller produkter heraf, skal der tages hensyn til disse grænseværdier. Ellers tages der hensyn til referenceværdierne for acceptabelt eller tolerabelt indtag, såsom det acceptable daglige indtag (ADI) eller den øvre grænse for sikkert indtag (UL), i forhold til det forventede indtag. Indtages forbindelsen allerede via fødevarer uden negative virkninger, betragtes forbrugernes indtag via en almindelig kost som sikkert.
Ansøgeren skal vurdere oplysningerne om virkningerne af forarbejdning af de nye forbindelser. Potentiel akkumulering/depletion i fødevarer og foderstoffer, der indgår i menneskers eller dyrs ernæring, skal tages i betragtning. Ansøgeren skal også vurdere betydningen af forskelle som følge af kemiske reaktioner, der vides at forekomme under forarbejdningsbetingelser.
I tilfælde, hvor der er foretaget mere komplekse genetiske modifikationer, f.eks. ved overførsel af flere gener i én enkelt genkonstruktion, retransformation af allerede eksisterende genetisk modificerede linjer og stabling af transformationsbegivenheder ved konventionel krydsning af genetisk modificerede forældreplanter, skal ansøgeren fremlægge og diskutere strategier for vurdering af eventuelle risici i tilknytning til potentielle vekselvirkninger mellem de nye udtrykte proteiner, nye metabolitter og oprindelige plantebestanddele. Ved vurderingen skal der tages hensyn til alle tilgængelige oplysninger, herunder de nye udtrykte proteiners virkningsmekanisme, den genetisk modificerede plantes molekylære karakteristika og karakteristika med hensyn til sammensætning/agronomi og resultaterne af toksicitetsundersøgelser og fodringsforsøg.
Ansøgeren skal vurdere de oplysninger, der er tilvejebragt til vurdering af det allergifremkaldende potentiale hos nye udtrykte proteiner i genetisk modificerede planter, for så vidt angår indførelse af nye allergene proteiner i fødevare- og foderplanter og potentiel fremkaldelse af allergiske reaktioner hos følsomme individer, samt oplysninger, der dokumenterer, at den genetiske modifikation ikke forårsager utilsigtede ændringer i endogene allergene proteiners karakteristika og/eller ekpressionsniveauer i den genetisk modificerede fødevare. Især skal valget af testmodeller begrundes med hensyn til specificitet, forudsigelighed og valideringsstatus.
Med hensyn til skøn over indtaget af genetisk modificerede fødevarer skal ansøgeren vurdere de anvendte metoder for så vidt angår usikkerhederne i forbindelse med forudberegningen af langtidsindtaget. Der skal sættes særlig fokus på genetisk modificerede planter, der har til formål at ændre fødevarens og foderets ernæringsmæssige karakteristika. Behovet for overvågning efter markedsføringen af disse genetisk modificerede produkter skal diskuteres som en mekanisme, der gør det muligt at påvise ændringer i mønstret for det samlede indtag af den modificerede fødevare i praksis og at fastslå, i hvilket omfang disse har fundet sted, samt hvorvidt produktet har fremkaldt kendte (bi)virkninger eller uventede bivirkninger. Hvis overvågning efter markedsføringen vurderes at være påkrævet, skal der fremlægges oplysninger om pålideligheden, følsomheden og specificiteten af de metoder, der foreslås anvendt.
3.3. Resultatet af risikokarakteriseringen
Ansøgeren skal i overensstemmelse med artikel 4 og 16 i forordning (EF) nr. 1829/2003 sikre, at den endelige risikokarakterisering klart dokumenterer følgende:
|
a) |
Den/det genetisk modificerede fødevare/foderstof har ingen negative virkninger for menneskers eller dyrs sundhed. |
|
b) |
Den genetisk modificerede fødevare afviger ikke fra den fødevare, som den er bestemt til at erstatte, i et sådant omfang, at den ved normal indtagelse vil være ernæringsmæssigt ufordelagtig for forbrugeren. |
|
c) |
Den genetisk modificerede fødevare vildleder ikke forbrugeren. |
|
d) |
Det genetisk modificerede foderstof skader ikke og vildleder ikke forbrugeren ved at ændre de særlige kendetegn for animalske produkter. |
|
e) |
Det genetisk modificerede foderstof afviger ikke fra det foderstof, som det er bestemt til at erstatte, i et sådant omfang, at det ved normal indtagelse vil være ernæringsmæssigt ufordelagtigt for dyr eller mennesker. |
Ansøgeren skal klart angive, hvilke antagelser der er anvendt i risikovurderingen for at forudsige sandsynligheden for og sværhedsgraden af negative virkninger i en given population, samt arten og omfanget af de usikkerheder, der er forbundet med fastlæggelsen af disse risici.
Ansøgeren skal også udførligt begrunde, hvorfor der er eller ikke er fremlagt et forslag til mærkning, jf. artikel 13, stk. 2, litra a), og stk. 3, og artikel 25, stk. 2, litra c), og stk. 3, i forordning (EF) nr. 1829/2003.
(1) EFT L 43 af 14.2.1997, s. 1.
(2) EFSA Journal 2011; 9(5):2149.
(3) EFSA Journal 2010; 8(1):1250.
(4) EFSA Journal 2010; 8(1):1250.
(5) http://www.oecd.org/document/15/0,3746,en_2649_34385_46726799_1_1_1_1,00.html.
(6) http://www.efsa.europa.eu/en/efsajournal/pub/2760.htm.
(7) EUT L 133 af 22.5.2008, s. 1.
(8) EFSA Journal 2011; 9(12):2438.
(9) EFSA Journal 2010; 8(7):1700.
(10) EFSA, 2008-rapport fra arbejdsgruppen under EFSA's GMO-panel om fodringsforsøg, 2008. Safety and nutritional assessment of GM plants and derived food and feed. The role of animal feeding trials. Food and Chemical Toxicology 46 (2008), s. 2-70.