2009R0152 — DA — 17.07.2014 — 004.002
Dette dokument er et dokumentationsredskab, og institutionerne påtager sig intet ansvar herfor
|
KOMMISSIONENS FORORDNING (EF) NR152/2009 af 27. januar 2009 om prøveudtagnings- og analysemetoder til offentlig kontrol af foder (EUT L 054 af 26.2.2009, s. 1) |
Ændret ved:
|
|
|
Tidende |
||
|
No |
page |
date |
||
|
KOMMISSIONENS FORORDNING (EU) Nr. 278/2012 af 28. marts 2012 |
L 91 |
8 |
29.3.2012 |
|
|
KOMMISSIONENS FORORDNING (EU) Nr. 51/2013 af 16. januar 2013 |
L 20 |
33 |
23.1.2013 |
|
|
L 197 |
1 |
20.7.2013 |
||
|
L 188 |
1 |
27.6.2014 |
||
Berigtiget ved:
KOMMISSIONENS FORORDNING (EF) NR152/2009
af 27. januar 2009
om prøveudtagnings- og analysemetoder til offentlig kontrol af foder
(EØS-relevant tekst)
KOMMISSIONEN FOR DE EUROPÆISKE FÆLLESSKABER HAR —
under henvisning til traktaten om oprettelse af Det Europæiske Fællesskab,
under henvisning til Europa-Parlamentets og Rådets forordning (EF) nr. 882/2004 af 29. april 2004 om offentlig kontrol med henblik på verifikation af, at foderstof- og fødevarelovgivningen samt dyresundheds- og dyrevelfærdsbestemmelserne overholdes ( 1 ), særlig artikel 11, stk. 4, litra a), b) og c), og
ud fra følgende betragtninger:|
(1) |
Følgende retsakter blev vedtaget til gennemførelse af direktiv 70/373/EØF og gælder fortsat i henhold til artikel 61, stk. 2, i forordning (EF) nr. 882/2004: — Kommissionens første direktiv 71/250/EØF af 15. juni 1971 om fastsættelse af fællesskabsanalysemetoder til den officielle kontrol af foderstoffer ( 2 ) — Kommissionens andet direktiv 71/393/EØF af 18. november 1971 om fastsættelse af fællesskabsanalysemetoder til officiel kontrol af foderstoffer ( 3 ) — Kommissionens tredje direktiv 72/199/EØF af 27. april 1972 om fastsættelse af fællesskabsanalysemetoder til officiel kontrol af foderstoffer ( 4 ) — Kommissionens fjerde direktiv 73/46/EØF af 5. december 1972 om fastsættelse af fællesskabsanalysemetoder til officiel kontrol af foderstoffer ( 5 ) — Kommissionens første direktiv 76/371/EØF af 1. marts 1976 om fastsættelse af fællesskabsmåder for udtagning af prøver til den officielle kontrol med foderstoffer ( 6 ) — Kommissionens syvende direktiv 76/372/EØF af 1. marts 1976 om fastsættelse af fællesskabsanalysemetoder til den officielle kontrol med foderstoffer ( 7 ) — Kommissionens ottende direktiv 78/633/EØF af 15. juni 1978 om fastsættelse af fællesskabsanalysemetoder til den officielle kontrol med foderstoffer ( 8 ) — Kommissionens niende direktiv 81/715/EØF af 31. juli 1981 om fastsættelse af fællesskabsanalysemetoder til den officielle kontrol med foderstoffer ( 9 ) — Kommissionens tiende direktiv 84/425/EØF af 25. juli 1984 om fastsættelse af fællesskabsanalysemetoder til den officielle kontrol med foderstoffer ( 10 ) — Kommissionens direktiv 86/174/EØF af 9. april 1986 om fastsættelse af metoden til beregning af næringsværdien i foderblanding til fjerkræ ( 11 ) — Kommissionens ellevte direktiv 93/70/EØF af 28. juli 1993 om fastsættelse af fællesskabsanalysemetoder til den officielle kontrol med foderstoffer ( 12 ) — Kommissionens tolvte direktiv 93/117/EF af 17. december 1993 om fastsættelse af fællesskabsanalysemetoder til den officielle kontrol med foderstoffer ( 13 ) — Kommissionens direktiv 98/64/EF af 3. september 1998 om fastsættelse af fællesskabsanalysemetoder til bestemmelse af aminosyrer, råfedt og olaquindox i foderstoffer og om ændring af direktiv 71/393/EØF ( 14 ) — Kommissionens direktiv 1999/27/EF af 20. april 1999 om fastsættelse af fællesskabsanalysemetoder til bestemmelse af amprolium, diclazuril og carbadox i foderstoffer og om ændring af direktiv 71/250/EØF og 73/46/EØF og ophævelse af direktiv 74/203/EØF ( 15 ) — Kommissionens direktiv 1999/76/EF af 23. juli 1999 om fastsættelse af fællesskabsanalysemetoder til bestemmelse af lasalocid natrium i foderstoffer ( 16 ) — Kommissionens direktiv 2000/45/EF af 6. juli 2000 om fællesskabsmetoder til bestemmelse af vitamin A, vitamin E og tryptophan i foderstoffer ( 17 ) — Kommissionens direktiv 2002/70/EF af 26. juli 2002 om krav til bestemmelse af indholdet af dioxin og dioxinlignende PCB i foderstoffer ( 18 ) — Kommissionens direktiv 2003/126/EF af 23. december 2003 om analysemetoder til bestemmelse af animalske bestanddele med henblik på officiel kontrol af foderstoffer ( 19 ). |
|
(2) |
Idet direktiv 70/373/EØF blev afløst af forordning (EF) nr. 882/2004/2004, bør gennemførelsesretsakterne til samme direktiv afløses af én enkelt forordning. Samtidig bør metoderne tilpasses den udvikling, der er sket på det videnskabelige og teknologiske område. Metoder, der ikke længere udfylder deres formål på tilfredsstillende vis, bør udgå. Det er hensigten, at prøveudtagningsbestemmelserne på et tidspunkt skal opdateres for at tage hensyn til den seneste udvikling inden for metoderne til produktion, opbevaring, transport og markedsføring af foder, men det vil for nuværende være mest hensigtsmæssigt at fastholde de eksisterende prøveudtagningsbestemmelser. |
|
(3) |
Direktiv 71/250/EØF, 71/393/EØF, 72/199/EØF, 73/46/EØF, 76/371/EØF, 76/372/EØF, 78/633/EØF, 81/715/EØF, 84/425/EØF, 86/174/EØF, 93/70/EØF, 93/117/EF, 98/64/EF, 1999/27/EF, 1999/76/EF, 2000/45/EF, 2002/70/EF og 2003/126/EF bør derfor ophæves. |
|
(4) |
Foranstaltningerne i denne forordning er i overensstemmelse med udtalelse fra Den Stående Komité for Fødevarekæden og Dyresundhed — |
UDSTEDT FØLGENDE FORORDNING:
Artikel 1
Udtagning af prøver som led i den offentlige kontrol af foder, navnlig for så vidt angår bestemmelse af bestanddele, herunder materiale, der indeholder eller består af eller er fremstillet af genetisk modificerede organismer, fodertilsætningsstoffer som defineret i Europa-Parlamentets og Rådets forordning (EF) nr. 1831/2003 ( 20 ), uønskede stoffer som defineret i Europa-Parlamentets og Rådets direktiv 2002/32/EF ( 21 ), foretages efter de metoder, der er anført i bilag I.
Den prøveudtagningsmetode, der er anført i bilag I, finder anvendelse på kontrol af foder, for så vidt angår bestemmelse af pesticidrester som defineret i Europa-Parlamentets og Rådets forordning (EF) nr. 396/2005 ( 22 ) og kontrol af overensstemmelse med forordning (EU) nr. 619/2011.
Artikel 2
Klargøring af prøver til analyse og angivelse af resultater foretages efter de metoder, der er beskrevet i bilag II.
Artikel 3
Analyser som led i den offentlige kontrol af foder foretages efter de metoder, der er beskrevet i bilag III (Analysemetoder til kontrol af sammensætningen af fodermidler og foderblandinger), bilag IV (Analysemetoder til kontrol af indholdet af godkendte tilsætningsstoffer i foder), bilag V (Analysemetoder til kontrol for uønskede stoffer i foder) og bilag VI (Analysemetoder til bestemmelse af animalske bestanddele som led i den offentlige kontrol af foder).
Artikel 4
Næringsværdien af foderblandinger til fjerkræ beregnes som angivet i bilag VII.
Artikel 5
De i bilag VIII beskrevne analysemetoder til kontrol for ulovlig forekomst af fodertilsætningsstoffer, der ikke længere er tilladt, anvendes til verifikationsformål.
Artikel 6
Direktiv 71/250/EØF, 71/393/EØF, 72/199/EØF, 73/46/EØF, 76/371/EØF, 76/372/EØF, 78/633/EØF, 81/715/EØF, 84/425/EØF, 86/174/EØF, 93/70/EØF, 93/117/EF, 98/64/EF, 1999/27/EF, 1999/76/EF, 2000/45/EF, 2002/70/EF og 2003/126/EF ophæves.
Henvisninger til de ophævede direktiver gælder som henvisninger til nærværende forordning og læses efter sammenligningstabellerne i bilag IX.
Artikel 7
Denne forordning træder i kraft på tyvendedagen efter offentliggørelsen i Den Europæiske Unions Tidende.
Den anvendes fra den 26. august 2009
Denne forordning er bindende i alle enkeltheder og gælder umiddelbart i hver medlemsstat.
BILAG I
PRØVEUDTAGNINGSMETODER
1. FORMÅL OG ANVENDELSESOMRÅDE
Prøver, der er bestemt til offentlig kontrol af foder, udtages i overensstemmelse med de nedenfor anførte metoder. De således fremkomne prøver skal betragtes som repræsentative for de portioner, der prøvetages af.
Formålet med repræsentativ prøveudtagning er at udtage en brøkdel af et parti på en sådan måde, at fastlæggelsen af de særlige karakteristika ved denne brøkdel repræsenterer gennemsnitsværdien af partiets karakteristika. Der udtages prøver af partiet ved gentagne gange at udtage enkeltprøver på forskellige steder i partiet. Disse enkeltprøver kombineres ved sammenblanding, så de udgør en samleprøve, hvorfra der klargøres repræsentative slutprøver ved hjælp af repræsentativ prøveneddeling.
Hvis det ved en visuel kontrol viser sig, at en del af det foder, der skal udtages prøver af, er af en anden kvalitet end resten af foderet i samme parti, adskilles de pågældende dele fra resten af foderet og behandles som et særskilt delparti. Hvis det ikke er muligt at opdele foderet i særskilte delpartier, udtages prøver som fra et parti. I sådanne tilfælde nævnes dette i prøveudtagningsrapporten.
Såfremt det konstateres, at foder, der udtages prøver af i overensstemmelse med bestemmelserne i denne forordning, og som ikke opfylder EU's krav, udgør en del af et parti foder af samme klasse eller betegnelse, antages det, at alt foder i det pågældende parti er berørt, medmindre en nærmere gennemgang ikke giver belæg for, at den resterende del af partiet ikke skulle opfylde EU's krav.
2. DEFINITIONER
|
— |
Parti (lot eller batch) : En identificerbar mængde foder, hvorom det er fastslået, at det har fælles karakteristika såsom oprindelse, type, emballagetype, pakkevirksomhed, afsender eller mærkning og i tilfælde af en produktionsproces en produktionsenhed fra ét anlæg, hvor der anvendes ensartede produktionsparametre, eller et antal af sådanne enheder, når de er fremstillet fortløbende og oplagres sammen. |
|
— |
Portion der prøvetages af : Et parti eller en identificerbar del af partiet eller delpartiet. |
|
— |
Plomberet prøve : En prøve, der er plomberet på en måde, som forhindrer enhver adgang til prøven uden at bryde eller fjerne plomben. |
|
— |
Enkeltprøve : En mængde, som er udtaget på et enkelt sted i den portion, der prøvetages af. |
|
— |
Samleprøve : En samling af enkeltprøverne, udtaget fra den portion, der prøvetages af. |
|
— |
Reduceret prøve : En del af en samleprøve, fremkommet ved repræsentativ reduktion af denne. |
|
— |
Slutprøve : En del af den reducerede prøve eller af den homogeniserede samleprøve. |
|
— |
Laboratorieprøve : Prøve bestemt til laboratorieundersøgelse (som modtaget af laboratoriet), der kan være en slutprøve, en reduceret prøve eller en samleprøve. |
3. ALMINDELIGE BESTEMMELSER
— Prøveudtagningspersonale: Prøverne skal udtages af personer, som er autoriseret hertil af den kompetente myndighed.
— Prøven plomberes på en måde, som forhindrer enhver adgang til prøven uden at bryde eller fjerne plomben. Plombens etiket skal være klart identificerbar og synlig. Alternativt kan prøven anbringes i en beholder, der kan lukkes på en sådan måde, at den ikke kan åbnes uden at medføre uoprettelig skade på beholderen, så man undgår genanvendelse af beholderen.
— Identifikation af prøven: Prøven skal være mærket på en uudslettelig måde og skal identificeres på en sådan måde, at der er en utvetydig forbindelse til prøveudtagningsrapporten.
— Fra hver enkelt samleprøve udtages mindst to slutprøver: mindst én til offentlig kontrol (håndhævelsesformål) og en til lederen af foderstofvirksomheden (kontraprøve). Endelig kan der udtages én slutprøve til referenceformål. Hvis hele samleprøven homogeniseres, udtages slutprøverne af den homogeniserede samleprøve, medmindre denne fremgangsmåde er i modstrid med medlemsstaternes forskrifter for så vidt angår rettighederne for lederen af en foderstofvirksomhed.
4. APPARATUR
|
4.1. |
Det apparatur, der anvendes til prøveudtagning, skal være udført i materialer, der ikke kan forurene de produkter, der skal udtages prøver af. Apparatur, der er bestemt til at blive anvendt flere gange, skal være let at rengøre for at undgå krydsforurening. |
|
4.2. |
Anbefalet apparatur til udtagning af prøver af foder i fast form.
4.2.1. Manuel prøveudtagning 4.2.1.1. Skovl med flad bund og lodrette sider
4.2.2. Mekanisk prøveudtagning Passende mekanisk apparatur kan anvendes til udtagning af prøver af foder i bevægelse. Passende betyder, at der mindst udtages prøver af hele flowets tværsnit. Prøveudtagning af foder i bevægelse (med høj flowhastighed) kan udføres med automatisk prøvetagningsudstyr. 4.2.3. Prøvedeler Til neddeling af prøver på en repræsentativ måde anvendes apparatur, der deler prøverne i tilnærmelsesvis lige store dele, hvis det er muligt og hensigtsmæssigt. |
5. KVANTITATIVE KRAV FOR SÅ VIDT ANGÅR ANTAL ENKELTPRØVER
— De kvantitative krav i punkt 5.1 og 5.2, for så vidt angår antallet af enkeltprøver, gælder for portioner, der prøvetages af, med en vægt på op til 500 tons, og hvoraf der kan udtages prøver på en repræsentativ måde. Den anførte fremgangsmåde for prøveudtagning gælder også for mængder, som er større end den foreskrevne maksimale størrelse af portioner, der prøvetages af, hvis der ses bort fra det højeste antal enkeltprøver, som er angivet i nedenstående tabeller, og antallet af enkeltprøver fastlægges ved hjælp af den kvadratrodsformel, der er fastsat i den relevante del af fremgangsmåden (jf. punkt 5.3), og den mindste størrelse af samleprøven øges proportionelt hermed. Dette forhindrer ikke, at et stort parti kan opdeles i mindre delpartier, og at der udtages prøver af hvert delparti i overensstemmelse med fremgangsmåden i punkt 5.1 og 5.2.
— Størrelsen af portionen, der prøvetages af, skal være således, at der kan udtages enkeltprøver af alle dens bestanddele.
— I tilfælde af meget store partier eller delpartier (> 500 tons) og partier, som transporteres eller oplagres på en sådan måde, at der ikke kan udtages prøver i overensstemmelse med den fremgangsmåde, der er fastsat i punkt 5.1 og 5.2, anvendes den i punkt 5.3 fastsatte fremgangsmåde.
— Hvis lederen af foderstofvirksomheden i henhold til lovgivningen har pligt til at overholde bestemmelserne i denne forordning som led i et obligatorisk overvågningssystem, kan lederen af foderstofvirksomheden afvige fra de kvantitative krav, som er fastsat i dette afsnit, for at tage hensyn til operationelle karakteristika, forudsat at lederen af foderstofvirksomheden over for den kompetente myndighed har godtgjort ækvivalensen af fremgangsmåden for prøveudtagning med hensyn til repræsentativitet og efter godkendelse fra den kompetente myndighed.
— I særlige tilfælde, hvis det ikke er muligt at anvende den fastsatte prøveudtagningsmetode for så vidt angår de kvantitative krav på grund af uacceptabel forretningsmæssig beskadigelse af partiet (som følge af emballeringsform, transportmiddel, opbevaringsforhold osv.), kan der anvendes en alternativ prøveudtagningsmetode, under forudsætning af at den er så repræsentativ som muligt og beskrives og dokumenteres fuldt ud.
5.1. Kvantitative krav for så vidt angår enkeltprøver i forbindelse med kontrol af stoffer og produkter, der er homogent fordelt i foderet
5.1.1. Uemballeret foder i fast form
|
Størrelsen af den portion, der prøvetages af |
Mindste antal enkeltprøver |
|
≤ 2,5 tons |
7 |
|
> 2,5 tons |
Kvadratroden af 20 gange det antal tons, som den portion, der prøvetages af, vejer (1), op til 40 enkeltprøver |
|
(1) Er den fremkomne størrelse ikke et helt tal, afrundes til nærmeste højere hele tal. |
|
5.1.2. Uemballeret foder i flydende form
|
Størrelsen af den portion, der prøvetages af |
Mindste antal enkeltprøver |
|
≤ 2,5 tons eller ≤ 2 500 liter |
4 (1) |
|
> 2,5 tons eller > 2 500 liter |
7 (1) |
|
(1) Hvis det ikke er muligt at gøre væsken homogen, øges antallet af enkeltprøver. |
|
5.1.3. Emballeret foder
Foder (i fast og flydende form) kan være emballeret i sække, poser, dåser, tønder mv., der i tabellen er omhandlet som enheder. Af store enheder (≥ 500 kg eller liter) udtages prøver i overensstemmelse med bestemmelserne for uemballeret foder (jf. punkt 5.1.1 og 5.1.2).
|
Størrelsen af den portion, der prøvetages af |
Mindste antal enheder, hvoraf (mindst) én enkeltprøve skal udtages (1) |
|
1 til 20 enheder |
1 enhed (1) |
|
21 til 150 enheder |
3 enheder (2) |
|
151 til 400 enheder |
5 enheder (3) |
|
> 400 enheder |
►C1 ¼ af kvadratroden af antallet af enheder, som udgør den portion, der prøvetages (1), op til 40 enheder ◄ |
|
(1) I tilfælde af at åbningen af en enhed kan påvirke analysen (f.eks. letfordærveligt vådt foder), udgøres en enkeltprøve af den uåbnede enhed. (2) For enheder, hvis indhold ikke overstiger 1 kg eller 1 liter, udgøres en enkeltprøve af indholdet af én original enhed. (3) Er den fremkomne størrelse ikke et helt tal, afrundes til nærmeste højere hele tal. |
|
5.1.4. Foderblokke og mineralske sliksten
For hver 25 enheder, der prøvetages, udtages mindst én blok eller sliksten, dog højst 4 blokke eller sliksten.
For blokke eller sliksten, der ikke vejer over 1 kg stykket, udgøres en enkeltprøve af indholdet af én blok eller én sliksten.
5.1.5. Grovfoder/foderplanter
|
Størrelsen af den portion, der prøvetages af |
Mindste antal enkeltprøver (1) |
|
≤ 5 tons |
5 |
|
> 5 tons |
Kvadratroden af 5 gange det antal tons, som den portion, der prøvetages af, udgør (2), op til 40 enkeltprøver |
|
(1) Det erkendes, at det i visse situationer (f.eks. ved ensilage) ikke er muligt at udtage de krævede enkeltprøver uden at forårsage uacceptabel beskadigelse af partiet. En alternativ prøveudtagningsmetode kan anvendes i sådanne situationer, og inden ikrafttrædelsen af denne forordning vil der blive udarbejdet en vejledning om prøveudtagning af sådanne partier. (2) Er den fremkomne størrelse ikke et helt tal, afrundes til nærmeste højere hele tal. |
|
5.2. Kvantitative krav for så vidt angår enkeltprøver i forbindelse med kontrol af bestanddele eller stoffer, der sædvanligvis er uhomogent fordelt i foderet
Disse kvantitative krav for så vidt angår enkeltprøver skal anvendes i følgende situationer:
— kontrol med forekomst af aflatoksin, meldrøje, andre mykotoksiner og skadelige botaniske urenheder i fodermidler
— kontrol af krydsforurening fra en bestanddel, herunder genmodificeret materiale eller stof, der sædvanligvis er uhomogent fordelt i fodermidler.
Hvis kontrolmyndigheden har stærk mistanke om, at der forekommer en uhomogen fordeling, og i tilfælde af krydsforurening fra en bestanddel eller et stof i en foderblanding, kan de kvantitative krav i nedenstående tabel anvendes.
|
Størrelsen af den portion, der prøvetages af |
Mindste antal enkeltprøver |
|
< 80 tons |
Jf. de kvantitative krav i punkt 5.1. Antal enkeltprøver, som skal udtages, ganges med 2,5. |
|
≥ 80 tons |
100 |
5.3. Kvantitative krav for så vidt angår enkeltprøverne i tilfælde af meget store partier
I tilfælde af store portioner, der prøvetages af (> 500 tons), er antallet af enkeltprøver, der skal udtages = 40 enkeltprøver + kvadratroden af antal tons i forbindelse med kontrol af stoffer og produkter, der er homogent fordelt i foderet, eller 100 enkeltprøver + kvadratroden af antal tons i forbindelse med kontrol af bestanddele eller stoffer, der sædvanligvis er uhomogent fordelt i fodermidler.
6. KVANTITATIVE KRAV FOR SÅ VIDT ANGÅR SAMLEPRØVER
|
Der skal være én samleprøve for hver portion der prøvetages af. |
||
|
|
Fodertype |
|
|
6.1. |
Uemballeret foder |
4 kg |
|
6.2. |
Emballeret foder: |
4 kg (3) |
|
6.3. |
Flydende eller halvflydende foder: |
4 liter |
|
6.4. |
Foderblokke eller mineralske sliksten: |
|
|
6.4.1. |
med en stykvægt på over 1 kg |
4 kg |
|
6.4.2. |
med en stykvægt på højst 1 kg |
vægten af 4 originale blokke eller sliksten |
|
6.5. |
Grovfoder/foderplanter |
4 kg (4) |
|
(1) Hvis det foder, der udtages prøver af, er af stor værdi, kan der udtages en mindre mængde samleprøve, under forudsætning af at dette beskrives og dokumenteres i prøveudtagningsrapporten. (2) I henhold til bestemmelserne i Kommissionens forordning (EU) nr. 619/2011 af 24. juni 2011 om prøveudtagnings- og analysemetoder til offentlig kontrol af foder for så vidt angår forekomst af genetisk modificeret materiale, som er under godkendelse, eller for hvilket godkendelsen er udløbet (EUT L 166 af 25.6.2011, s. 9) skal samleprøven til kontrol af forekomst af genetisk modificeret materiale indeholde mindst 35 000 frø/korn. ►C1 Dette betyder, at for majs skal størrelsen af samleprøven være mindst 10,5 kg og for sojabønner 7 kg. ◄ For andre frø og korn, såsom byg, hirse, havre, ris, rug, hvede og rapsfrø, svarer størrelsen af samleprøven på 4 kg til mere end 35 000 frø/korn. (3) I tilfælde af emballeret foder er det heller ikke altid muligt at opnå en størrelse på 4 kg for samleprøven afhængigt af størrelsen af de enkelte enheder. (4) For så vidt angår grovfoder eller foderplanter med lav massefylde (f.eks. hø, halm) skal samleprøven være på mindst 1 kg. |
||
7. KVANTITATIVE KRAV FOR SÅ VIDT ANGÅR SLUTPRØVER
Slutprøver
Der kræves analyse af mindst én slutprøve. Mængden af slutprøven, der skal analyseres, må ikke være mindre end:
|
Foder i fast form |
|
|
Flydende eller halvflydende foder |
500 ml (1) |
|
(1) I henhold til bestemmelserne i forordning (EU) nr. 619/2011 skal slutprøver til kontrol af forekomst af genetisk modificeret materiale indeholde mindst 10 000 frø/korn. ►C1 Dette betyder, at for majs skal størrelsen af slutprøven være mindst 3 000 g og for sojabønner 2 000 g. ◄ For andre frø og korn, såsom byg, hirse, havre, ris, rug, hvede og rapsfrø, svarer størrelsen af slutprøven på 500 g til mere end 10 000 frø/korn. (2) Hvis størrelsen af samleprøven er betydeligt mindre end 4 kg eller liter (jf. fodnoterne i afsnit 6), kan der ligeledes udtages en mindre mængde slutprøve, under forudsætning af at dette beskrives og dokumenteres i prøveudtagningsrapporten. (3) I tilfælde af prøveudtagning af bælgfrugter, korn og nødder til bestemmelse af indholdet af pesticidrester skal minimumsstørrelsen af slutprøven være 1 kg i overensstemmelse med bestemmelserne i Kommissionens direktiv 2002/63/EF (EFT L 187 af 16.7.2002, s. 30). |
|
8. PRØVEUDTAGNINGSMETODE FOR MEGET STORE PARTIER ELLER PARTIER, SOM OPBEVARES ELLER TRANSPORTERES PÅ EN MÅDE, SÅ PRØVEUDTAGNING I HELE PARTIET IKKE ER MULIG
8.1. Generelle principper
Hvis transport eller opbevaring af et parti forhindrer udtagning af enkeltprøver i hele partiet, bør prøveudtagning af sådanne partier foretages, når partiet er i flow.
I tilfælde af store lagerbygninger, der er bestemt til oplagring af foder, bør virksomhederne opfordres til at installere udstyr i lagerbygningerne, som (automatisk) foretager prøveudtagning i hele det oplagrede parti.
Ved anvendelse af de fremgangsmåder for prøveudtagning, som er omhandlet i dette afsnit 8, underrettes lederen af foderstofvirksomheden eller dennes repræsentant om den pågældende fremgangsmåde for prøveudtagning. Hvis lederen af foderstofvirksomheden eller dennes repræsentant anfægter denne fremgangsmåde for prøveudtagning, skal lederen af foderstofvirksomheden eller dennes repræsentant give den kompetente myndighed mulighed for at udtage prøver af hele det pågældende parti for vedkommendes regning.
8.2. Store partier, som transporteres med skib
8.2.1. Dynamisk prøveudtagning af store partier, som transporteres med skib
Prøveudtagning af store partier i skibe foretages bedst, mens produktet er i flow (dynamisk prøveudtagning).
Prøveudtagningen skal foretages pr. lastrum (enhed, der fysisk kan adskilles). Lastrummene tømmes imidlertid ikke hver for sig, så den oprindelige fysiske adskillelse eksisterer ikke længere efter overførslen til lagerfaciliteter. Prøveudtagning kan derfor foretages enten på baggrund af den oprindelige fysiske adskillelse eller på baggrund af opdelingen efter overførslen til lagerfaciliteterne.
Losningen af et skib kan vare flere dage. Normalt gennemføres prøveudtagningen med regelmæssige mellemrum under hele losningens varighed. Det er dog ikke altid muligt eller hensigtsmæssigt, at en officiel inspektør er til stede med henblik på prøveudtagning i løbet af hele lossearbejdet. Det er derfor tilladt at gennemføre prøveudtagning på en del af hele partiet. Antallet af enkeltprøver bestemmes under hensyntagen til størrelsen af den portion, der prøvetages af.
Hvis der er udtaget prøver af en del af et parti foder af samme klasse eller betegnelse, og det konstateres, at denne del af partiet ikke opfylder EU's krav, antages det, at alt foder i det pågældende parti er berørt, medmindre en nærmere gennemgang ikke giver belæg for, at den resterende del af partiet ikke skulle opfylde EU’s krav.
Selv om den officielle prøve udtages automatisk, skal der være en inspektør til stede. Såfremt den automatiske prøveudtagning gennemføres med forudindstillede parametre, som ikke kan ændres under prøveudtagningen, og enkeltprøverne indsamles i en plomberet beholder, som forhindrer eventuelt svig, er inspektørens tilstedeværelse kun nødvendig ved påbegyndelsen af prøveudtagningen, hver gang prøvebeholderen skal udskiftes og ved afslutningen af prøveudtagningen.
8.2.2. Prøveudtagning af partier, som transporteres med skib ved statisk prøveudtagning
Hvis prøveudtagningen udføres på en statisk måde, anvendes den samme fremgangsmåde som den, der gælder for lagerfaciliteter (siloer), som er tilgængelige ovenfra (jf. punkt 8.4.1).
Prøveudtagningen gennemføres på den tilgængelige del (ovenfra) af partiet/lastrummet. Antallet af enkeltprøver bestemmes under hensyntagen til størrelsen af den portion, der prøvetages af. Hvis der er udtaget prøver af en del af et parti foder af samme klasse eller betegnelse, og det konstateres, at denne del af partiet ikke opfylder EU's krav, antages det, at alt foder i det pågældende parti er berørt, medmindre en nærmere gennemgang ikke giver belæg for, at den resterende del af partiet ikke skulle opfylde EU’s krav.
8.3. Prøveudtagning af store partier, der er oplagret i lagerbygninger
Prøveudtagningen gennemføres på den tilgængelige del af partiet. Antallet af enkeltprøver bestemmes under hensyntagen til størrelsen af den portion der prøvetages af. Hvis der er udtaget prøver af en del af et parti foder af samme klasse eller betegnelse, og det konstateres, at denne del af partiet ikke opfylder EU's krav, antages det, at alt foder i det pågældende parti er berørt, medmindre en nærmere gennemgang ikke giver belæg for, at den resterende del af partiet ikke skulle opfylde EU’s krav.
8.4. Udtagning af prøver fra lagerfaciliteter (siloer)
8.4.1. Udtagning af prøver fra siloer, som er (let) tilgængelige ovenfra
Prøveudtagningen gennemføres på den tilgængelige del af partiet. Antallet af enkeltprøver bestemmes under hensyntagen til størrelsen af den portion, der prøvetages af. Hvis der er udtaget prøver af en del af et parti foder af samme klasse eller betegnelse, og det konstateres, at denne del af partiet ikke opfylder EU's krav, antages det, at alt foder i det pågældende parti er berørt, medmindre en nærmere gennemgang ikke giver belæg for, at den resterende del af partiet ikke skulle opfylde EU’s krav.
8.4.2. Udtagning af prøver fra siloer, som ikke er tilgængelige ovenfra (lukkede siloer)
8.4.2.1.
Der kan ikke udtages prøver af foder, der er oplagret i sådanne siloer, på en statisk måde. Hvis der skal udtages prøver af foder i en sådan silo, og det ikke er muligt at flytte partiet, skal der derfor træffes aftale med virksomhedslederen om, at vedkommende meddeler inspektøren, hvornår den pågældende silo vil blive tømt, så der kan udtages prøver, mens foderet er i flow.
8.4.2.2.
Fremgangsmåden for prøveudtagning indebærer overførsel til en beholder af en størrelse på 50-100 kg, hvorfra prøven udtages. Størrelsen af samleprøven skal svare til hele partiet, og antallet af enkeltprøver skal beregnes på baggrund af mængden i den silo, hvorfra der overføres foder til en beholder med henblik på prøveudtagning. Hvis der er udtaget prøver af en del af et parti foder af samme klasse eller betegnelse, og det konstateres, at denne del af partiet ikke opfylder EU's krav, antages det, at alt foder i det pågældende parti er berørt, medmindre en nærmere gennemgang ikke giver belæg for, at den resterende del af partiet ikke skulle opfylde EU’s krav.
8.5. Udtagning af prøver af uemballeret foder i store lukkede beholdere
Der kan som regel kun udtages prøver af sådanne partier, når beholderne aflæsses. Det er i visse tilfælde ikke muligt at aflæsse beholderne ved indførsel eller kontrol, og prøveudtagningen bør derfor gennemføres, når beholderne aflæsses.
9. FREMGANGSMÅDE VED UDTAGNING, KLARGØRING OG EMBALLERING AF PRØVERNE
9.1. Generelt
Prøverne udtages og klargøres så hurtigt som muligt under hensyntagen til de forholdsregler, som er nødvendige for at undgå, at produktet forandres eller forurenes. Instrumenter og også overflader og beholdere, som er beregnet til at rumme prøverne, skal være rene og tørre.
9.2. Enkeltprøver
Enkeltprøver skal udtages tilfældigt fra hele den portion, der prøvetages af. Deres størrelse skal være tilnærmelsesvis ens.
Enkeltprøvens størrelse skal være på mindst 100 g eller 25 g i tilfælde af grovfoder eller foderplanter med lav massefylde.
I tilfælde af at der i henhold til den fremgangsmåde for prøveudtagning, der er fastsat i afsnit 8, skal udtages færre end 40 enkeltprøver, fastlægges størrelsen af enkeltprøverne på baggrund af den størrelse, der kræves for samleprøver (jf. afsnit 6).
I tilfælde af prøveudtagning af prøver af mindre partier af emballeret foder, hvor der i henhold til de kvantitative krav skal udtages et begrænset antal enkeltprøver, skal en enkeltprøve udgøres af indholdet af én original enhed, hvis indhold ikke overstiger 1 kg eller 1 liter.
I tilfælde af prøveudtagning af emballeret foder, der består af små enheder (f.eks. < 250 g), afhænger antallet af enkeltprøver af enhedernes størrelse.
9.2.1. Uemballeret foder
Hvor det er passende, kan prøveudtagningen foretages, mens partiet er i bevægelse (pålæsning eller aflæsning).
9.2.2. Emballeret foder
Efter at det i afsnit 5 fastsatte antal enheder til prøveudtagning er udvalgt, udtages en del af indholdet af hver enhed ved hjælp af spyd eller skovl. Om nødvendigt skal prøverne udtages, efter at enhederne er tømt hver for sig.
9.2.3. Flydende eller halvflydende foder, som er homogent eller kan gøres homogent
Efter at det i afsnit 5 fastsatte antal enheder til prøveudtagning er udvalgt, homogeniseres indholdet om nødvendigt, og der udtages en mængde fra hver enhed.
Enkeltprøverne kan eventuelt udtages under aftapning af partiet.
9.2.4. Flydende eller halvflydende foder, som ikke kan gøres homogent
Efter at det i afsnit 5 fastsatte antal enheder til prøveudtagning er udvalgt, udtages prøver fra forskellige niveauer.
Enkeltprøver kan ligeledes udtages, når partiet aftappes, men de første fraktioner kasseres.
Under alle omstændigheder må den samlede udtagne mængde ikke være mindre end 10 liter.
9.2.5. Foderblokke og mineralske sliksten
Efter at det i afsnit 5 fastsatte antal blokke eller sliksten til prøveudtagning er udvalgt, kan der udtages en del af hver blok eller sliksten. I tilfælde af mistanke om en uhomogen blok eller sliksten, kan hele blokken eller slikstenen udtages som prøve.
For blokke eller sliksten, der ikke vejer over 1 kg stykket, udgøres en enkeltprøve af indholdet af én blok eller én sliksten.
9.3. Klargøring af samleprøver
Enkeltprøverne sammenblandes, så de udgør en enkelt samleprøve.
9.4. Klargøring af slutprøver
Samleprøven blandes omhyggeligt ( 23 ).
— Hver prøve anbringes i en egnet beholder. Alle nødvendige forholdsregler tages for at undgå, at prøven forurenes eller forfalskes, eller at sammensætningen ændres under transport eller opbevaring.
— Ved kontrol af bestanddele eller stoffer, der er homogent fordelt i foderet, kan samleprøven reduceres på en repræsentativ måde til mindst 2,0 kg eller 2,0 liter (reduceret prøve) ( 24 ) helst ved anvendelse af en mekanisk eller en automatisk prøvedeler. Ved kontrol af forekomst af pesticidrester i bælgfrugter, korn og trænødder er minimumsstørrelsen af den reducerede prøve 3 kg. Hvis fodertypen umuliggør anvendelse af en prøvedeler, eller hvis der ikke er en prøvedeler til rådighed, kan prøven reduceres ved firdeling (kvartning). Fra de reducerede prøver klargøres slutprøverne (til kontrol-, kontraprøve- og referenceformål) af tilnærmelsesvis samme størrelse og under overholdelse af de kvantitative krav i afsnit 7. Ved kontrol af bestanddele, herunder genetisk modificeret materiale eller stoffer, der sædvanligvis er uhomogent fordelt i fodermidler, skal samleprøven være:
—
— fuldstændigt homogeniseret og derefter opdelt i slutprøver eller
— reduceret til mindst 2 kg eller 2 liter ( 25 ) ved anvendelse af en mekanisk eller automatisk prøvedeler. Kun i tilfælde, hvor fodertypen forhindrer anvendelse af en prøvedeler, kan prøven om nødvendigt reduceres ved firdeling. Med henblik på kontrol af forekomst af genetisk modificeret materiale i henhold til forordning (EU) nr. 619/2011 skal den reducerede prøve indeholde mindst 35 000 frø/korn for at opnå de slutprøver, der kræves til håndhævelses-, kontraprøve- og referenceformål på mindst 10 000 frø/korn (jf. fodnote (**) i afsnit 6 og fodnote (*) i afsnit 7).
9.5. Emballering af prøver
Beholdere eller pakninger plomberes og forsynes med etiketter på en sådan måde, at de ikke kan åbnes, uden at plomben beskadiges. Hele etiketten skal anbringes inden for plomben.
9.6. Forsendelse af prøver til laboratoriet
Prøven sendes så hurtigt som muligt til det udpegede analyselaboratorium sammen med de for analysen nødvendige oplysninger.
10. REGISTRERING AF PRØVEUDTAGNING
Hver prøve skal registreres, således at portionen, der prøvetages af, og dens størrelse utvetydigt kan identificeres.
Desuden skal enhver afvigelse fra den fremgangsmåde for prøveudtagning, der er fastsat i denne forordning, registreres.
De registrerede oplysninger stilles til rådighed for det officielle kontrollaboratorium samt for lederen af foderstofvirksomheden og/eller det laboratorium, som er udpeget af foderstofvirksomheden.
BILAG II
ALMINDELIGE BESTEMMELSER VEDRØRENDE ANALYSEMETODER FOR FODER
A. KLARGØRING AF PRØVER TIL ANALYSE
1. Formål
De nedenfor beskrevne fremgangsmåder omhandler klargøring til analyse af prøver, der efter udtagning sendes til kontrollaboratorierne i overensstemmelse med bilag I.
Laboratorieprøverne klargøres således, at de i henhold til analysemetoderne afvejede mængder er homogene og repræsentative for slutprøverne.
2. Forholdsregler
Fremgangsmåden for klargøring af prøven afhænger af, hvilke analysemetoder der skal anvendes, og hvilke bestanddele eller stoffer der skal kontrolleres. Det er derfor yderst vigtigt at sikre, at den fremgangsmåde for klargøring af prøven, der følges, passer til den anvendte analysemetode og til de bestanddele eller stoffer, der skal kontrolleres.
Alle nødvendige arbejdsgange udføres, så det så vidt muligt undgås at forurene prøven og ændre dens sammensætning.
Formaling, blanding og sigtning gennemføres så hurtigt som muligt, og således at prøven udsættes mindst muligt for luft og lys. Der anvendes ikke kværne og andre formalingsapparater med væsentlig tilbøjelighed til at frembringe varme i prøven.
Særligt varmefølsomt foder bør formales manuelt. Det sikres tillige, at apparaturet i sig selv ikke forurener.
Kan klargøringen ikke foretages uden væsentlige ændringer af prøvens vandindhold, bestemmes vandindholdet før og efter klargøringen efter den metode, der er fastsat i del A i bilag III.
3. Fremgangsmåde
3.1. Generel fremgangsmåde
Testdelprøven udtages af slutprøven. Neddeling efter keglemetoden og firdeling (kvartning) kan ikke anbefales, da disse metoder giver risiko for uhomogene delprøver.
3.1.1.
— Den sigtede slutprøve blandes og opsamles i en egnet, ren og tør beholder med lufttæt lukning. Slutprøven blandes igen for at sikre fuldstændig homogenisering umiddelbart før afvejning til analyse (testdelprøve).
3.1.2.
— Medmindre andet er angivet for analysemetoden, tørres slutprøven, så den får et vandindhold på 8-12 %, i overensstemmelse med den fremgangmåde for fortørring, der er anført i punkt 4.3 om metoden til bestemmelse af vandindhold i del A i bilag III. Der fortsættes som beskrevet i punkt 3.1.1.
3.1.3.
— Slutprøven opsamles i en ren, tør og egnet beholder med lufttæt lukning. Slutprøven blandes grundigt for at sikre fuldstændig homogenisering umiddelbart før afvejning til analyse (testdelprøve).
3.1.4.
— Slutprøver, der ikke kan klargøres i overensstemmelse med en af de ovenfor beskrevne fremgangsmåder, behandles på anden måde, så der skabes sikkerhed for, at de mængder, der afvejes til analyse (testdelprøve), er homogene og repræsentative for slutprøverne.
3.2. Særlig fremgangsmåde i tilfælde af undersøgelse ved visuel eller ved mikroskopisk kontrol eller i tilfælde, hvor samleprøven er homogeniseret
— I tilfælde af en undersøgelse ved visuel kontrol (uden anvendelse af mikroskop) anvendes hele laboratorieprøven til undersøgelsen.
— I tilfælde af en mikroskopisk undersøgelse kan laboratoriet reducere samleprøven eller reducere den reducerede prøve yderligere. Slutprøver til kontraprøve- og eventuelt referenceformål udtages efter en fremgangsmåde svarende til den fremgangsmåde, der følges for slutprøver til kontrolformål.
— Hvis hele samleprøven er homogeniseret, udtages slutprøverne fra den homogeniserede samleprøve.
4. Opbevaring af prøverne
Prøverne opbevares ved en temperatur, der ikke ændrer deres sammensætning. Prøver, der er bestemt til analyse af vitaminer eller særligt lysfølsomme stoffer, opbevares under sådanne betingelser, at prøven ikke påvirkes negativt af lys.
B. KRAV TIL REAGENSER OG APPARATUR, DER ANVENDES I FORBINDELSE MED ANALYSEMETODERNE
1. Medmindre andet er angivet for analysemetoderne, skal alle reagenser være af analysekvalitet (p.a.). Ved udførelse af en sporstofanalyse kontrolleres reagensernes renhed ved en blindprøve. Resultatet af blindprøven er afgørende for, om der kræves yderligere oprensning af reagenserne.
2. Til alle former for fremstilling af opløsninger, fortynding, skylning eller vaskning, der er angivet i forbindelse med analysemetoderne, uden at opløsnings- eller fortyndingsmidlets art er omtalt, anvendes vand. Som generel regel gælder det, at der anvendes demineraliseret eller destilleret vand. I særlige tilfælde, som angivet i de beskrevne analysemetoder, er det nødvendigt at underkaste vandet særlige oprensningsprocesser.
3. Standardapparatur, der normalt findes på kontrollaboratorier, er ikke omtalt i forbindelse med analysemetoderne; derimod omtales specialinstrumenter og -udstyr samt instrumenter og udstyr, hvis brug der stilles særlige krav til. Instrumenter og apparatur skal være rene, især ved bestemmelse af meget små stofmængder.
C. ANVENDELSE AF ANALYSEMETODERNE OG ANGIVELSE AF RESULTATER
1. Ekstraktionsprocedure
Der er flere metoder, der omfatter en specifik ekstraktionsprocedure. Som hovedregel kan der anvendes andre ekstraktionsprocedurer end den i metoden angivne, under forudsætning af at den anvendte ekstraktionsprocedure bevisligt frembyder en ekstraktionseffektivitet for den analyserede matrix, der svarer til effektiviteten af den i metoden omtalte fremgangsmåde.
2. Oprensningsprocedure
Der er flere metoder, der omfatter en specifik oprensningsprocedure. Som hovedregel kan der anvendes andre oprensningsprocedurer end den i metoden angivne, under forudsætning af at den anvendte oprensningsprocedure bevisligt giver analyseresultater for den analyserede matrix, der svarer til de resultater, som opnås med den i metoden omtalte fremgangsmåde.
3. Antal bestemmelser
I forbindelse med analyse af uønskede stoffer gælder det, at hvis resultatet af den første bestemmelse er væsentligt (> 50 %) lavere end den specifikation, der skal kontrolleres, er yderligere bestemmelser ikke påkrævet, såfremt der anvendes tilfredsstillende kvalitetsprocedurer. I andre tilfælde er det nødvendigt at foretage en ekstra analyse (anden bestemmelse) for at udelukke muligheden for intern krydsforurening eller utilsigtet sammenblanding af prøver. Gennemsnittet af de to bestemmelser, hvor der tages hensyn til måleusikkerheden, anvendes til at verificere, om kravene er opfyldt.
Ved kontrol af det angivne indhold af et stof eller en ingrediens gælder det, at hvis resultatet af den første bestemmelse bekræfter det angivne indhold, dvs. hvis analyseresultatet ligger inden for det acceptable variationsområde for det angivne indhold, er yderligere bestemmelser ikke påkrævet, såfremt der anvendes tilfredsstillende kvalitetsprocedurer. I andre tilfælde er det nødvendigt at foretage en ekstra analyse (anden bestemmelse) for at udelukke muligheden for intern krydsforurening eller utilsigtet sammenblanding af prøver. Gennemsnittet af de to bestemmelser, hvor der tages hensyn til måleusikkerheden, anvendes til at verificere, om kravene er opfyldt.
I visse tilfælde er det acceptable variationsområde fastsat i lovgivningen, som f.eks. i Europa-Parlamentets og Rådets forordning (EF) nr. 767/2009 af 13. juli 2009 om markedsføring og anvendelse af foder, om ændring af Europa-Parlamentets og Rådets forordning (EF) nr. 1831/2003, og om ophævelse af Rådets direktiv 79/373/EØF, Kommissionens direktiv 80/511/EØF, Rådets direktiv 82/471/EØF, 83/228/EØF, 93/74/EØF, 93/113/EF og 96/25/EF og Kommissionens beslutning 2004/217/EF ( 26 ).
4. Afrapportering af anvendt analysemetode
Analyserapporten skal angive den anvendte analysemetode.
5. Afrapportering af analyseresultater
Analyseresultatet angives som anført i analysemetoden med et tilstrækkeligt antal betydende cifre og korrigeres om nødvendigt til samme vandindhold, som slutprøven havde før prøveklargøringen.
6. Måleusikkerhed og genfindingsprocent i forbindelse med analyse af uønskede stoffer
For så vidt angår uønskede stoffer som omhandlet i direktiv 2002/32/EF bør et produkt til foderbrug anses for ikke at overholde det fastsatte maksimumsindhold, hvis analyseresultatet beregnet ved et vandindhold på 12 % vurderes at overstige maksimumsindholdet under hensyntagen til den ekspanderede måleusikkerhed og korrektion for genfinding. Det vurderes, om maksimumsindholdet er overholdt, ved at vurdere analyseresultatet korrigeret for genfinding og fratrukket ekspanderet måleusikkerhed. Dette gælder kun i tilfælde, hvor analysemetoden tillader vurdering af måleusikkerheden og korrektion for genfinding (hvilket f.eks. ikke er tilfældet ved mikroskopering).
Analyseresultatet skal afrapporteres som følger (hvis den anvendte analysemetode tillader vurdering af måleusikkerhed og genfindingsprocent):
a) ved korrektion for genfinding den angivne genfindingsprocent. Ved en genfindingsprocent på 90-110 % er korrektion for genfinding ikke påkrævet
b) som »x +/– U«, hvor x er analyseresultatet og U er den ekspanderede måleusikkerhed, idet der anvendes en dækningsfaktor på 2, hvilket giver et konfidensniveau på ca. 95 %.
Analyseresultatet kan dog, hvis resultatet af analysen er væsentligt (> 50 %) lavere end den specifikation, der skal kontrolleres, hvis der anvendes tilfredsstillende kvalitetsprocedurer, og hvis analysen udelukkende har til formål at kontrollere, at gældende lovgivning er overholdt, indberettes uden korrektion for genfinding, ligesom det i sådanne tilfælde kan undlades at indberette genfindingsprocent og måleusikkerhed.
BILAG III
ANALYSEMETODER TIL KONTROL AF SAMMENSÆTNINGEN AF FODERMIDLER OG FODERBLANDINGER
A. BESTEMMELSE AF VANDINDHOLD
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme vandindholdet i foder. For så vidt angår foder indeholdende flygtige stoffer, såsom organiske syrer, skal det bemærkes, at der sammen med vandindholdet også bestemmes betydelige mængder flygtige stoffer.
Metoden er ikke beregnet på analyse af mejeriprodukter, der anvendes som fodermidler, analyse af mineralske stoffer og blandinger, der overvejende består af mineralske stoffer, analyse af animalsk og vegetabilsk fedt eller analyse af olieholdige frø og frugter.
2. Princip
Prøven tørres under nærmere angivne betingelser, som er afhængige af foderets art. Vægttabet bestemmes ved vejning. For fast foder med højt vandindhold er yderligere fortørring nødvendig.
3. Apparatur
3.1. Formalingsapparat udført i et materiale, der ikke er fugtabsorberende, og som er let at gøre rent, muliggør en hurtig og ensartet formaling uden mærkbar varmeudvikling, så vidt muligt forhindrer kontakt med den omgivende luft og opfylder kravene i punkt 4.1.1 og 4.1.2 (f. eks. mikrohammermøller, mikroformalingsapparater med vandkøling, demontable keglemøller, langsomtløbende keglemøller og tandskivemøller)
3.2. Analysevægt med 1 mg aflæsningsnøjagtighed
3.3. Tørre vejekar af korrosionsbestandigt metal eller glas med lufttæt lukning; nyttearealet skal være stort nok til, at prøven kan fordeles med ca. 0,3 g/cm2
3.4. Elektrisk opvarmet, temperaturreguleret tørreskab (± 2 oC), som sikrer hurtig regulering af temperaturen og med god ventilation ( 27 ).
3.5. Elektrisk opvarmet justerbart vakuumtørreskab med oliepumpe, som enten er forsynet med en anordning til tilførsel af tørret varmluft eller med et tørremiddel (f.eks. calciumoxid)
3.6. Ekssikkator med tyk, perforeret plade af metal eller porcelæn indeholdende et virksomt tørringsmiddel.
4. Fremgangsmåde
|
NB |
De under dette punkt beskrevne arbejdsgange skal udføres straks efter åbningen af de pakninger, som indeholder prøverne. Analyserne skal foretages mindst to gange. |
4.1. Klargøring
4.1.1.
Der udtages mindst 50 g af prøven, som — om nødvendigt — formales eller neddeles på fornøden vis for at undgå varierende fugtighedsgrader (se punkt 6).
4.1.2.
Der udtages mindst 50 g af prøven. Den formales, så kornstørrelsen ved sigtning med masker på 0,5 mm tillader passage af mindst 50 %, og at der ved sigtning med runde masker på 1 mm bliver højst 10 % tilbage.
4.1.3.
Der udtages ca. 25 g af prøven, afvejet med 10 mg nøjagtighed, som blandes med en tilsvarende mængde vandfrit sand, afvejet med 10 mg nøjagtighed, indtil der fremkommer en homogen vare.
4.2. Tørring
4.2.1.
Et vejekar med låg (3.3) vejes med 1 mg nøjagtighed. Ca. 5 g af prøven afvejes med 1 mg nøjagtighed i det tarerede kar og fordeles jævnt. Karret stilles efter aftagning af låget ind i et til 103 oC opvarmet tørreskab. For at temperaturen i tørreskabet ikke skal falde for stærkt, skal karret stilles hurtigt ind i skabet. Der tørres i 4 timer, idet tørringstiden regnes fra det tidspunkt, hvor temperaturen i tørreskabet atter er kommet op på 103 oC. Efter åbning af tørreskabet lukkes karret med låget, tages ud af skabet, henstilles til afkøling i 30-45 minutter i ekssikkatoren (3.6) og vejes derefter med 1 mg nøjagtighed.
For prøver af foder overvejende bestående af fedt foretages yderligere en tørring på 30 minutter i tørreskabet ved 130 oC. Forskellen mellem de to vejeresultater må ikke overstige 0,1 % vandindhold.
4.2.2.
Et vejekar med låg (3.3) vejes med 0,5 mg nøjagtighed. Ca. 5 g af den formalede prøve afvejes med 1 mg nøjagtighed i det tarerede kar og fordeles jævnt. Karret stilles efter aftagning af låget ind i et til 130 oC opvarmet tørreskab. For at temperaturen i tørreskabet ikke skal falde for stærkt, skal karret stilles hurtigt ind i skabet. Der tørres i 2 timer, idet tørringstiden regnes fra det tidspunkt, hvor temperaturen i tørreskabet atter er kommet op på 130 oC. Efter åbning af tørreskabet lukkes karret med låget, tages ud af skabet, henstilles til afkøling i 30–45 minutter i ekssikkatoren (3.6) og vejes derefter med 1 mg nøjagtighed.
4.2.3. Foderblandinger indeholdende mere end 4 % saccharose eller lactose: fodermidler såsom johannesbrødskrå, hydrolyserede kornprodukter, maltspirer, sukkerroesnitter, fiskepressevand og sukker, samt foderblandinger med mere end 25 % mineralske salte inkl. krystalvand
Et vejekar med låg (3.3) vejes med 0,5 mg nøjagtighed. Ca. 5 g af prøven afvejes med 1 mg nøjagtighed i det tarerede kar og fordeles jævnt. Karret stilles efter aftagning af låget ind i et til 80-85 oC opvarmet vakuumtørreskab (3.5). For at temperaturen i tørreskabet ikke skal falde for stærkt, skal karret stilles hurtigt ind i skabet.
Trykket indstilles til 100 torr, og prøven tørres i 4 timer ved dette tryk enten under tilførsel af varm, tør luft eller ved hjælp af et tørringsmiddel (ca. 300 g til 20 prøver). I sidstnævnte tilfælde afbrydes forbindelsen til vakuumpumpen, så snart det foreskrevne tryk er nået. Tørringstiden regnes fra det tidspunkt, hvor temperaturen i tørreskabet atter er kommet op på 80–85 oC. Efter tørringstidens udløb bringes tørreskabet forsigtigt op på atmosfærisk tryk. Efter åbning af tørreskabet lukkes karret straks med låget, tages ud af skabet, henstilles til afkøling i 30–45 minutter i ekssikkatoren (3.6) og vejes derefter med 1 mg nøjagtighed. Der tørres i yderligere 30 minutter under vakuum i tørreskabet ved 80–85 oC, og derefter vejes på ny. Forskellen mellem de to vejeresultater må ikke overstige 0,1 % vandindhold.
4.3. Fortørring
4.3.1.
Fast foder med højt vandindhold, og hvis formaling er vanskelig, skal fortørres som følger:
50 g af den ikke-formalede prøve (om nødvendigt foretages en grov neddeling af presset eller klumpet foder) afvejes med 10 mg nøjagtighed i en egnet beholder (f. eks. en aluminiumskål på 20 × 12 cm med en kant på 0,5 cm). Der tørres i tørreskab ved 60-70 oC, indtil vandindholdet er reduceret til mellem 8 og 12 %. Beholderen tages ud af tørreskabet og henstilles utildækket til afkøling i laboratoriet i 1 time, hvorefter der vejes med 10 mg nøjagtighed. Alt efter foderets art foretages, som beskrevet i punkt 4.1.1, derefter straks en formaling, og der tørres som beskrevet i punkt 4.2.1 eller 4.2.3.
4.3.2.
Korn med et vandindhold på over 17 % skal fortørres som følger:
50 g af de ikke-formalede korn afvejes med 10 mg nøjagtighed i en egnet beholder (f. eks. en aluminiumskål på 20 × 12 cm med en kant på 0,5 cm) og tørres i tørreskab i 5-7 minutter ved 130 oC, hvorefter prøven tages ud af tørreskabet. Prøven henstilles utildækket til afkøling i laboratoriet i 2 timer, hvorefter der vejes med 10 mg nøjagtighed. Straks efter formales som beskrevet i punkt 4.1.2 og tørres som beskrevet i punkt 4.2.2.
5. Beregning af resultater
Prøvens vandindhold (X) i procent beregnes efter følgende formler:
5.1. Tørring uden fortørring
![]()
hvor:
|
m |
= |
prøvens begyndelsesvægt i gram |
|
m0 |
= |
den tørre prøves vægt i gram. |
5.2. Tørring med fortørring
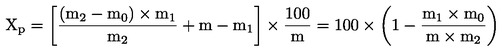
hvor:
|
m |
= |
prøvens begyndelsesvægt i gram |
|
m1 |
= |
prøvens vægt i gram efter fortørring |
|
m2 |
= |
prøvens vægt i gram efter formaling |
|
m0 |
= |
den tørre prøves vægt i gram. |
5.3. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige 0,2 % vandindhold i absolut værdi.
6. Bemærkning
Hvis det viser sig at være nødvendigt med formaling og der i den forbindelse må påregnes en ændring af materialets vandindhold, skal analyseresultaterne for foderets bestanddele korrigeres i overensstemmelse med originalprøvens vandindhold.
B. BESTEMMELSE AF VANDINDHOLDET I ANIMALSK OG VEGETABILSK FEDT
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme animalske og vegetabilske fedtstoffers og oliers vandindhold (vand og flygtige stoffer).
2. Princip
Prøven tørres ved 103 °C, indtil vægten ikke mere formindskes (vægttabet mellem to på hinanden følgende vejninger må højst være på 1 mg). Vægttabet bestemmes ved vejning.
3. Apparatur
3.1. Fladbundet beholder af korrosionsbestandigt materiale, diameter: 8-9 cm, højde: ca. 3 cm
3.2. Termometer med forstærket kugle og udvidelsesrum i den øverste ende, inddelt i grader fra ca. 80 °C til mindst 110 °C, længde: ca. 10 cm
3.3. Sandbad eller elektrisk varmeplade
3.4. Ekssikkator indeholdende et effektivt tørremiddel
3.5. Analysevægt.
4. Fremgangsmåde
Ca. 20 g af den homogeniserede prøve afvejes med 1 mg nøjagtighed i den tørre, tarerede beholder (3.1), hvori termometret (3.2) er anbragt. Prøven opvarmes på sandbadet eller varmepladen (3.3) under stadig omrøring med termometret, så den når en temperatur på 90 °C i løbet af ca. 7 minutter.
Varmen dæmpes efter den hyppighed, med hvilken boblerne stiger op fra beholderens bund. Temperaturen må ikke overstige 105 °C. Bliv ved med at røre og skrabe bunden, indtil der ikke dannes flere bobler.
For at sikre, at fugtigheden helt er fjernet, gentages opvarmningen til 103 °C ± 2 ° flere gange, med afkøling til 93 °C mellem de på hinanden følgende opvarmninger. Derefter henstilles til afkøling til rumtemperatur i ekssikkatoren (3.4) og vejes. Denne proces gentages, indtil vægttabet mellem to afvejninger ikke overstiger 2 mg.
|
NB |
Hvis prøvens vægt øges efter gentagne opvarmninger, er dette tegn på oxidering af fedtstoffet. I så fald beregnes resultatet af den afvejning, der er blevet foretaget, umiddelbart inden vægtforøgelsen begyndte. |
5. Beregning af resultater
Prøvens vandindhold (X) i procent beregnes efter følgende formel:
![]()
hvor:
|
m |
= |
prøvens vægt i gram |
|
m1 |
= |
beholderens og indholdets vægt i gram inden opvarmning |
|
m2 |
= |
beholderens og indholdets vægt i gram efter opvarmning. |
Resultater på under 0,05 % opføres som »mindre end 0,05 %«.
Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige 0,05 % vandindhold i absolut værdi.
C. BESTEMMELSE AF INDHOLDET AF RÅPROTEIN
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af råprotein i foder på basis af nitrogenindholdet (Kjeldahls metode).
2. Princip
Prøven destrueres med svovlsyre under tilstedeværelse af en katalysator. Syreopløsningen gøres alkalisk med en natriumhydroxidopløsning. Ammoniakken destilleres og opsamles i en nøjagtigt afmålt mængde svovlsyre, idet den overskydende mængde svovlsyre titreres med en standardnatriumhydroxidopløsning.
Alternativt destilleres den frigjorte ammoniak i et overskud af borsyreopløsning, hvorefter der titreres med en saltsyre- eller svovlsyreopløsning.
3. Reagenser
3.1. Kaliumsulfat.
3.2. Katalysator: kobber(II)oxid (CuO) eller kobber(II)sulfatpentahydrat (CuSO4 · 5H2O).
3.3. Granuleret zink.
3.4. Svovlsyre, ρ20 = 1,84 g/ml.
3.5. Svovlsyre, standardopløsning, c(H2SO4) = 0,25 mol/liter.
3.6. Svovlsyre, standardopløsning, c(H2SO4) = 0,10 mol/liter.
3.7. Svovlsyre, standardopløsning, c(H2SO4) = 0,05 mol/liter.
3.8. Indikator af methylrødt; 300 mg methylrødt opløses i 100 ml ethanol, σ = 95-96 % (v/v).
3.9. Natriumhydroxidopløsning (teknisk kvalitet kan bruges), β = 40 g/100 ml (m/v: 40 %).
3.10. Natriumhydroxid, standardopløsning, c(NaOH) = 0,25 mol/liter.
3.11. Natriumhydroxid, standardopløsning, c(NaOH) = 0,10 mol/liter.
3.12. Granuleret pimpsten, vasket i saltsyre og udglødet.
3.13. Acetanilid (smeltepunkt = 114 oC, N-indhold = 10,36 %).
3.14. Saccharose (nitrogenfri).
3.15. Borsyre (H3BO3).
3.16. Indikatoropløsning af methylrødt: 100 mg methylrødt opløses i 100 ml ethanol eller methanol.
3.17. Opløsning af bromcresolgrønt: 100 mg bromcresolgrønt opløses i 100 ml ethanol eller methanol.
3.18. Borsyreopløsning (10–40 g/liter, afhængigt af det anvendte apparatur)
Anvendes kolorimetrisk endpointbestemmelse, skal borsyreopløsningerne tilføres methylrødt- og bromcresolgrøntindikatorer. Fremstilles 1 liter borsyreopløsning, tilsættes der, inden tilpasning af volumen, 7 ml indikatoropløsning af methylrødt (3.16) og 10 ml opløsning af bromcresolgrønt (3.17).
Borsyreopløsningens pH-værdi vil kunne variere fra batch til batch afhængigt af vandet, der bruges. Det vil ofte være nødvendigt at justere med en lille mængde alkali for at opnå en positiv blindprøve.
|
NB |
En god justering opnås normalt ved at tilsætte ca. 3–4 ml NaOH (3.11) til 1 liter borsyreopløsning på 10 g/liter. Opløsningen opbevares ved rumtemperatur og beskyttes under opbevaringen mod lys og ammoniakdampe. |
3.19. Standardsaltsyreopløsning, c(HCl) = 0,10 mol/liter.
|
NB |
Der kan anvendes opløsninger i andre koncentrationer (3.5, 3.6, 3.7, 3.10, 3.11 og 3.19), forudsat at der korrigeres herfor i beregningerne. Koncentrationen skal altid angives med fire decimaler. |
4. Apparatur
Apparat egnet til destruktion, destillation og titrering efter Kjeldahls metode.
5. Fremgangsmåde
5.1. Destruktion
1 g af prøven afvejes med 0,001 g nøjagtighed og overføres til destruktionskolben. Der tilsættes 15 g kaliumsulfat (3.1), en passende mængde katalysator (3.2) (0,3 –0,4 g kobber(II)oxid eller 0,9 –1,2 g kobber(II)sulfatpentahydrat), 25 ml svovlsyre (3.4) og eventuelt nogle få korn pimpsten (3.12) og blandes.
Kolben opvarmes, forsigtigt i begyndelsen, og omrystes om nødvendigt af og til forsigtigt, indtil prøven er forkullet og skummet forsvundet. Derefter opvarmes kraftigere, indtil væsken koger jævnt. Opvarmningen er tilstrækkelig, hvis den kogende syre fortætter sig på kolbens væg. Det må forhindres, at siderne bliver overophedet, og at organiske partikler klæber til dem.
Når opløsningen bliver klar og lysegrøn, fortsættes kogningen i yderligere to timer. Derefter henstilles kolben til afkøling.
5.2. Destillation
Der tilsættes forsigtigt tilstrækkeligt vand til, at sulfaterne opløses fuldstændigt. Kolben henstår til afkøling, hvorefter der om nødvendigt tilsættes nogle få zinkkorn (3.3). Der fortsættes som beskrevet i punkt 5.2.1 eller 5.2.2.
5.2.1.
I destillationsapparatets opsamlingskolbe anbringes en nøjagtigt afmålt mængde på 25 ml svovlsyre (3.5 eller 3.7) afhængigt af det formodede nitrogenindhold. Der tilsættes nogle få dråber indikator af methylrødt (3.8).
Destruktionskolben forbindes med destillationsapparatets svaler, og svalerens nederste del nedsænkes i væsken i opsamlingskolben til en dybde af mindst 1 cm (se bemærkning 8.3). 100 ml natriumhydroxidopløsning (3.9) overføres forsigtigt til destruktionskolben, uden at der mistes ammoniak (se bemærkning 8.1). Kolben opvarmes, indtil ammoniakken er destilleret over.
5.2.2.
Foretages titreringen af destillatets ammoniakindhold manuelt, følges den nedenfor beskrevne fremgangsmåde. Er destillationsenheden fuldt automatiseret, så også titreringen af destillatets ammoniakindhold sker automatisk, følges brugsanvisningen til destillationsenheden.
En opsamlingskolbe med 25-30 ml af borsyreopløsningen (3.18) anbringes under svalerens afløbsåbning, således at tilførselsrøret befinder sig under overfladen i den overskydende borsyreopløsning. Destillationsenheden indstilles til at give 50 ml natriumhydroxidopløsning (3.9). Destillationsenheden betjenes i overensstemmelse med brugsanvisningen, og den ammoniak, der er frigivet ved tilførslen af natriumhydroxidopløsningen, afdampes. Destillatet opsamles i forlaget med borsyre. Destillatmængden (dampdestillationstiden) afhænger af mængden af nitrogen i prøven. Brugsanvisningen følges.
|
NB |
I en halvautomatisk destillationsenhed sker tilførslen af overskydende natriumhydroxid samt dampdestillationen automatisk. |
5.3. Titrering
Der fortsættes som beskrevet i punkt 5.3.1 eller 5.3.2.
5.3.1.
Den overskydende svovlsyre i opsamlingskolben titreres med natriumhydroxidopløsning (3.10 eller 3.11) afhængigt af koncentrationen af den anvendte svovlsyre, indtil ækvivalenspunktet er nået.
5.3.2.
Med en burette titreres opsamlingskolbens indhold med standardsaltsyreopløsningen (3.19) eller med standardsvovlsyreopløsningen (3.6), og den anvendte titrantmængde aflæses.
Anvendes kolorimetrisk endpointbestemmelse, er ækvivalenspunktet nået ved det første tegn på pinkfarvning af indholdet. Buretteaflæsningen anslås med 0,05 ml nøjagtighed. En oplyst magnetisk omrørerplade eller en fotometrisk detektor vil kunne lette visualiseringen af ækvivalenspunktet.
Processen kan gøres automatisk ved anvendelse af en dampdestillationsenhed med automatisk titrering.
Brugsanvisningen til den enkelte destillationsenhed eller destillationsenhed/titrator følges.
|
NB |
Anvendes et automatisk titreringssystem, begynder titreringen umiddelbart efter, at destillationen er begyndt, og borsyreopløsningen på 1 % (3.18) anvendes. Gøres der brug af en fuldautomatisk destillationsenhed, kan den automatiske titrering af ammoniakken også foretages med endpointbestemmelse, idet der så anvendes et potentiometrisk pH-system. I dette tilfælde anvendes en automatisk titrator med pH-meter. pH-metret skal være korrekt kalibreret i området pH 4-pH 7 i overensstemmelse med almindelige laboratorie-pH-kalibreringsprocedurer. pH-titreringsækvivalenspunktet nås ved en pH-værdi på 4,6 , som er det sted på titreringskurven, hvor hældningen er stejlest. |
5.4. Blindprøve
For at sikre, at reagenserne er nitrogenfrie, foretages der en blindprøve (destruktion, destillation og titrering) ved brug af 1 g saccharose (3.14) i stedet for prøven.
6. Beregning af resultater
Beregningerne foretages som beskrevet i punkt 6.1 eller 6.2.
6.1. Titreringsberegning, jf. 5.3.1
Indholdet af råprotein, udtrykt i vægtprocent, beregnes efter følgende formel:
![]()
hvor:
|
Vo |
= |
mængde (ml) NaOH (3.10 eller 3.11) forbrugt i blindprøven |
|
V1 |
= |
mængde (ml) NaOH (3.10 eller 3.11) forbrugt ved titreringen af prøven |
|
c |
= |
koncentration (mol/liter) af natriumhydroxid (3.10 eller 3.11) |
|
m |
= |
prøvens vægt i gram. |
6.2. Titreringsberegning, jf. 5.3.2
6.2.1.
Indholdet af råprotein, udtrykt i vægtprocent, beregnes efter følgende formel:
![]()
hvor:
|
m |
= |
testportionens vægt i gram |
|
c |
= |
koncentration (mol/liter) af standardsaltsyreopløsningen (3.19) |
|
V0 |
= |
mængde (ml) saltsyre anvendt i blindprøven |
|
V1 |
= |
mængde (ml) saltsyre anvendt i testportionen. |
6.2.2.
Indholdet af råprotein, udtrykt i vægtprocent, beregnes efter følgende formel:
![]()
hvor:
|
m |
= |
testportionens vægt i gram |
|
c |
= |
koncentration (mol/liter) af standardsvovlsyreopløsningen (3.6) |
|
V0 |
= |
mængde (ml) svovlsyre (3.6) anvendt i blindprøven |
|
V1 |
= |
mængde (ml) svovlsyre (3.6) anvendt i testportionen. |
7. Kontrol af metoden
7.1. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige:
— 0,2 % i absolut værdi for råproteinindhold på under 20 %
— 1,0 % i relativ værdi (af det højeste resultat) for råproteinindhold på 20–40 %
— 0,4 % i absolut værdi for råproteinindhold på over 40 %.
7.2. Nøjagtighed
Analysen (destruktion, destillation og titrering) foretages på 1,5 –2,0 g acetanilid (3.13) under tilstedeværelse af 1 g saccharose (3.14). 1 g acetanilid kræver 14,80 ml svovlsyre (3.5). Genfindingsprocenten skal være mindst 99 %.
8. Bemærkninger
8.1. Apparaturet kan være manuelt, halvautomatisk eller automatisk. Hvis apparaturet kræver overførsel mellem destruktions- og destillationstrinet, skal sådan overførsel kunne ske uden tab. Hvis destillationsapparatets kolbe ikke har skilletragt, tilsættes natriumhydroxiden, lige inden kolben forbindes med svaleren, idet væsken hældes langsomt ned langs siden.
8.2. Hvis destruktionsproduktet størkner, gentages bestemmelsen med brug af en større mængde svovlsyre (3.4) end angivet ovenfor.
8.3. For produkter med lavt nitrogenindhold kan mængden af svovlsyre (3.7), der skal bruges i opsamlingskolben, om nødvendigt mindskes til 10 eller 15 ml og fyldes op med vand til 25 ml.
8.4. Til rutineanalyser kan der anvendes alternative analysemetoder til bestemmelse af råprotein, men Kjeldahls metode som beskrevet her i del C er referencemetoden. Det skal for hver enkelt matrix godtgøres, at de resultater, der opnås med den alternative metode (f.eks. DUMAS), svarer til dem, der opnås med referencemetoden. Eftersom resultaterne, der opnås med en alternativ metode, kan afvige en smule fra de resultater, der opnås med referencemetoden, selv efter ækvivalenskontrol, er det nødvendigt i analyserapporten at angive, hvilken analysemetode der er anvendt til bestemmelse af råprotein.
D. BESTEMMELSE AF URINSTOF
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af urinstof i foder.
2. Princip
Prøven opslæmmes i vand under tilsætning af et klaringsmiddel. Suspensionen filtreres. Filtratets indhold af urinstof bestemmes, efter tilsætning af 4-dimethylaminobenzaldehyd (4-DMAB), ved måling af ekstinktionen ved en bølgelængde på 420 nm.
3. Reagenser
3.1. 4-Dimethylaminobenzaldehydopløsning: 1,6 g 4-DMAB opløses i 100 ml 96 % ethanol, og der tilsættes 10 ml saltsyre (ρ20 = 1,19 g/ml). Dette reagens kan højst holde sig i to uger.
3.2. Carrez-reagens I: 21,9 g zinkacetat, Zn(CH3COO)2 · 2H2O, og 3 g iseddike opløses i vand. Der fyldes op med vand til 100 ml.
3.3. Carrez-reagens II: 10,6 g kaliumferrocyanid, K4Fe(CN)6 · 3H2O, opløses i vand. Der fyldes op med vand til 100 ml.
3.4. Aktivt kul, der ikke absorberer urinstoffet (skal kontrolleres).
3.5. Urinstof, 0,1 % opløsning (w/v).
4. Apparatur
4.1. Mekanisk rysteapparat: ca. 35–40 omdrejninger i minuttet
4.2. Reagensglas: 160 × 16 mm, med slibprop
4.3. Spektrofotometer.
5. Fremgangsmåde
5.1. Analyse af prøven
2 g af prøven afvejes med 1 mg nøjagtighed og kommes sammen med 1 g aktivt kul (3.4) i en 500 ml målekolbe. Der tilsættes 400 ml vand og 5 ml Carrez-reagens I (3.2) og blandes i ca. 30 sekunder, hvorefter der tilsættes 5 ml Carrez-reagens II (3.3). Væsken blandes i 30 minutter i rysteapparatet. Der fyldes op med vand til mærket, omrystes og filtreres.
5 ml af de klare og farveløse filtrater udtages med pipette og hældes i reagensglassene med slibprop, hvorefter der tilsættes 5 ml 4-DMAB-opløsning (3.1) og blandes. Glassene anbringes i vandbad ved 20 oC (+/- 4 oC). Efter 15 minutter måles ekstinktionen i prøveopløsningen i spektrofotometret ved 420 nm ved sammenligning med reagensernes blindprøveopløsning.
5.2. Kalibreringskurve
Der udtages volumener på 1, 2, 4, 5 og 10 ml af urinstofopløsningen (3.5), disse hældes i 100 ml målekolber, og der fyldes op med vand til mærket. Der udtages 5 ml af hver opløsning, hver af dem tilsættes 5 ml 4-DMAB-opløsning (3.1), der homogeniseres, og ekstinktionen måles, som angivet ovenfor, ved sammenligning med en kontrolopløsning, som indeholder 5 ml 4-DMAB og 5 ml vand uden urinstof. Kalibreringskurven tegnes.
6. Beregning af resultater
Bestem mængden af urinstof i prøven ved benyttelse af kalibreringskurven.
Resultatet angives i procent af prøven.
7. Bemærkninger
7.1. Hvis indholdet af urinstof er større end 3 %, reduceres analyseprøven til 1 gram, eller den oprindelige opløsning fortyndes så meget, at der ikke er mere end 50 mg urinstof i 500 ml.
7.2. Hvis indholdet af urinstof er ringe, forøges prøvens volumen i det omfang, filtratet vedbliver at være klart og farveløst.
7.3. Hvis prøven indeholder simple kvælstofforbindelser, som for eksempel aminosyrer, foretages målingen af ekstinktionen ved 435 nm.
E. BESTEMMELSE AF FLYGTIGE KVÆLSTOFHOLDIGE BASER
I. BESTEMMELSE VED MIKRODIFFUSION
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af flygtige kvælstofholdige baser, udtrykt som ammoniak, i foder.
2. Princip
Prøven ekstraheres med vand, og opløsningen klares og filtreres. De flygtige kvælstofholdige baser udskilles efter tilsætning af kaliumcarbonatopløsning ved mikrodiffusion, opfanges i en borsyreopløsning og titreres med svovlsyre.
3. Reagenser
3.1. Trichloreddikesyre, 20 % opløsning (w/v).
3.2. Indikator: 33 mg bromcresolgrønt og 65 mg methylrødt opløses i 100 ml ethanol 95–96 % (v/v).
3.3. Borsyreopløsning: 10 g borsyre opløses i en 1 liter målekolbe indeholdende 200 ml ethanol 95–96 % (v/v) og 700 ml vand. 10 ml af indikatoren (3.2) tilsættes. Opløsningen blandes og bringes om nødvendigt under tilsætning af natriumhydroxidopløsning til at antage en lyserød farve. 1 ml af denne opløsning kan binde op til 300 μg NH3.
3.4. Mættet kaliumcarbonatopløsning: 100 g kaliumcarbonat opløses i 100 ml kogende vand. Efter afkøling filtreres opløsningen.
3.5. Svovlsyre, 0,01 mol/liter.
4. Apparatur
4.1. Mekanisk rysteapparat: ca. 35–40 omdrejninger i minuttet
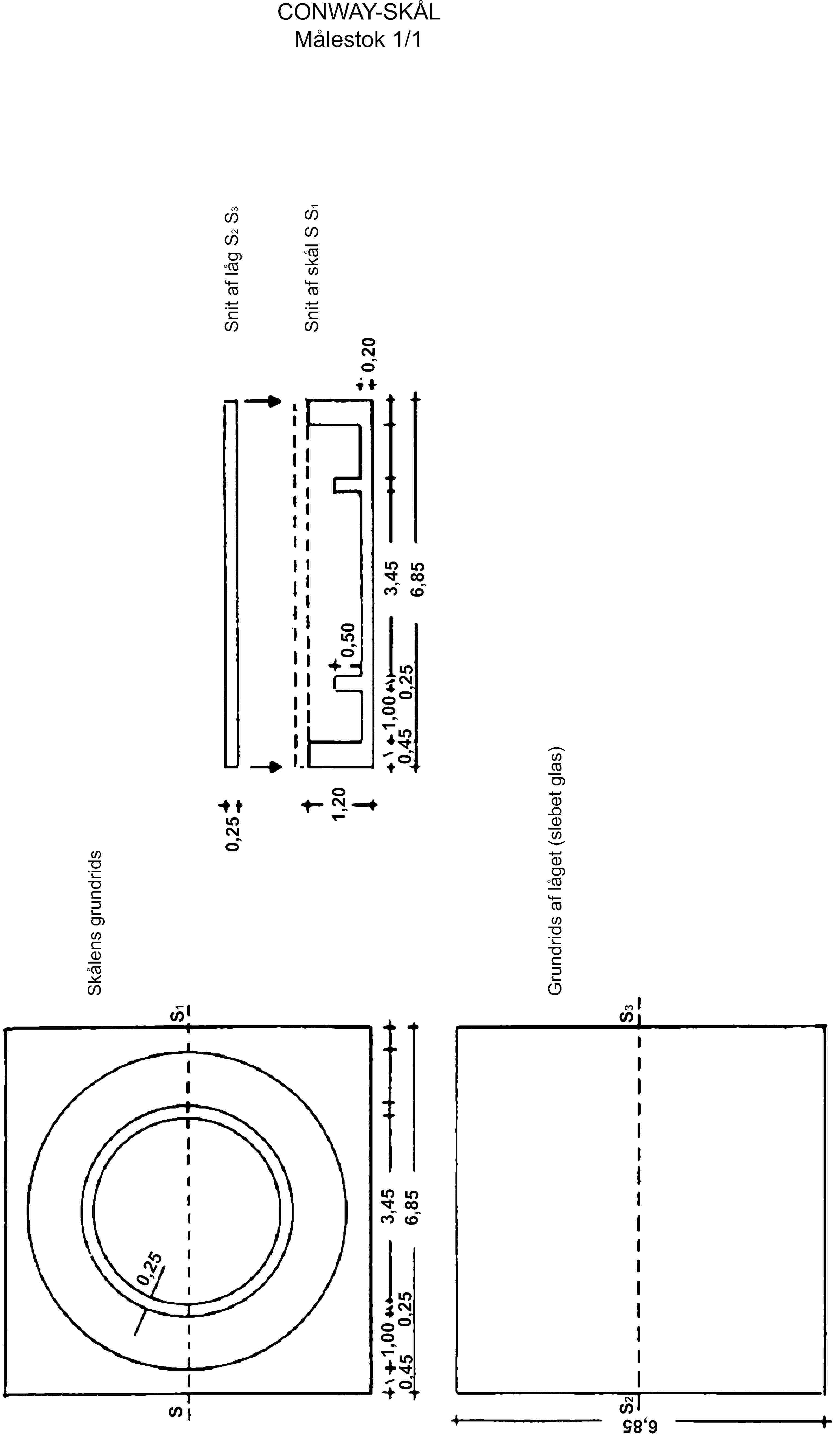
4.2. Conwayskåle (se skitse) af glas eller plastic
4.3. Mikroburetter inddelt i 1/100 ml.
5. Fremgangsmåde
10 g af prøven afvejes med 1 mg nøjagtighed, kommes sammen med 100 ml vand i en 200 ml målekolbe og blandes eller omrystes i 30 minutter i rysteapparatet. Der tilsættes 50 ml trichloreddikesyreopløsning (3.1), fyldes op med vand til mærket, omrystes kraftigt og filtreres gennem et foldefilter.
1 ml borsyreopløsning (3.3) afpipetteres i Conwayskålens midte, hvorefter 1 ml af prøvefiltratet overføres til skålens ring. Skålen tildækkes delvist med det let indfedtede låg. 1 ml af den mættede kaliumcarbonatopløsning (3.4) kommes hurtigt ned i ringen, og skålen lukkes, så den er lufttæt. Skålen drejes forsigtigt i vandret stilling, så de to reagenser blandes. Der inkuberes i mindst 4 timer ved rumtemperatur eller i 1 time ved 40 oC.
De i borsyreopløsningen indeholdte flygtige baser titreres derefter med svovlsyre (3.5) ved anvendelse af en mikroburette (4.3).
På samme måde udføres en blindprøve under udeladelse af analyseprøven.
6. Beregning af resultater
1 ml 0,01 mol/liter svovlsyre svarer til 0,34 mg ammoniak.
Resultatet angives i procent af prøven.
Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige:
— 10 % i relativ værdi ved indhold på mindre end 1,0 % ammoniak
— 0,1 % i absolut værdi ved indhold på 1,0 % ammoniak og derover.
7. Bemærkning
Har prøven et indhold på mere end 0,6 % ammoniak, fortyndes det oprindelige filtrat.
II. BESTEMMELSE VED DESTILLATION
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af flygtige kvælstofholdige baser, udtrykt som ammoniak, i fiskemel, som praktisk talt ikke indeholder urinstof. Metoden anvendes kun ved indhold på mindre end 0,25 % ammoniak.
2. Princip
Prøven ekstraheres med vand, og opløsningen klares og filtreres. De flygtige kvælstofholdige baser udskilles efter tilsætning af magnesiumoxid ved kogetemperatur og opfanges i en bestemt mængde svovlsyre, idet den overskydende mængde svovlsyre tilbagetitreres med en natriumhydroxidopløsning.
3. Reagenser
3.1. Trichloreddikesyre, 20 % opløsning (w/v)
3.2. Magnesiumoxid
3.3. Skumdæmpende emulsion (f.eks. silikone)
3.4. Svovlsyre, 0,05 mol/liter
3.5. Natriumhydroxidopløsning, 0,1 mol/liter
3.6. Methylrødtopløsning, 0,3 % i 95–96 % (v/v) ethanol.
4. Apparatur
4.1. Mekanisk rysteapparat: ca. 35–40 omdrejninger i minuttet
4.2. Destillationsapparatur af Kjeldahl-typen.
5. Fremgangsmåde
10 g af prøven afvejes med 1 mg nøjagtighed og kommes sammen med 100 ml vand i en 200 ml målekolbe og blandes eller omrystes i 30 minutter i rysteapparatet. Der tilsættes 50 ml trichloreddikesyreopløsning (3.1), fyldes op med vand til mærket, omrystes kraftigt og filtreres gennem et foldefilter.
Afhængigt af det formodede indhold af flygtige kvælstofholdige baser udtages en mængde af det klare filtrat (normalt 100 ml). Det fortyndes til 200 ml, og der tilsættes 2 g magnesiumoxid (3.2) samt nogle få dråber skumdæmpende emulsion (3.3). Opløsningen skal reagere alkalisk over for lakmuspapir; i modsat fald tilsættes yderligere magnesiumoxid (3.2). Der fortsættes som beskrevet i punkt 5.2 og 5.3 for analysemetoden til bestemmelse af indholdet af råprotein (del C i dette bilag).
På samme måde udføres en blindprøve under udeladelse af analyseprøven.
6. Beregning af resultater
1 ml 0,05 mol/liter svovlsyre svarer til 1,7 mg ammoniak.
Resultatet angives i procent af prøven.
Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige 10 % ammoniak i relativ værdi.
F. BESTEMMELSE AF AMINOSYRER (BORTSET FRA TRYPTOPHAN)
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af frie (syntetiske og naturlige) og totale (peptidbundne og frie) aminosyrer i foder ved brug af en aminosyreanalysator. Metoden kan anvendes til følgende aminosyrer: cyst(e)in, methionin, lysin, threonin, alanin, arginin, asparaginsyre, glutaminsyre, glycin, histidin, isoleucin, leucin, phenylalanin, prolin, serin, tyrosin og valin.
Metoden skelner ikke mellem salte af aminosyrer og kan ikke differentiere mellem D- og L-former af aminosyrer. Den er ikke anvendelig til bestemmelse af tryptophan- eller hydroxyanaloger af aminosyrer.
2. Princip
2.1. Frie aminosyrer
Frie aminosyrer ekstraheres med fortyndet saltsyre. Kvælstofholdige makromolekyler, der ekstraheres samtidig, udfældes med sulfosalicylsyre og fjernes ved filtrering. Den filtrerede opløsnings pH-værdi justeres til 2,20 . Aminosyrerne adskilles ved ionbytningskromatografi og bestemmes ved reaktion med ninhydrin ved fotometrisk påvisning ved 570 nm.
2.2. Totale aminosyrer
Den valgte metode afhænger af de aminosyrer, der skal undersøges. Cyst(e)in og methionin skal oxideres til henholdsvis cysteinsyre og methioninsulfon inden hydrolyse. Tyrosin skal bestemmes i hydrolysater af uoxiderede prøver. Alle de øvrige aminosyrer, der er nævnt i punkt 1, kan bestemmes i enten den oxiderede eller den uoxiderede prøve.
Oxidering sker ved 0 oC med en phenolholdig permyresyre. Overskydende oxideringsreagens destrueres med natriumdisulfit. Den oxiderede eller uoxiderede prøve hydrolyseres med saltsyre (3.20) i 23 timer. Hydrolysatets pH-værdi justeres til 2,20 . Aminosyrerne adskilles ved ionbytningskromatografi og bestemmes ved reaktion med ninhydrin ved fotometrisk påvisning ved 570 nm (440 nm for prolin).
3. Reagenser
Der skal anvendes dobbeltdestilleret vand eller vand af tilsvarende kvalitet (ledningsevne: < 10 μS).
3.1. Hydrogenperoxid, w (w/v) = 30 %.
3.2. Myresyre, w (w/v) = 98–100 %.
3.3. Phenol.
3.4. Natriumdisulfit.
3.5. Natriumhydroxid.
3.6. 5-Sulfosalicylsyre-dihydrat.
3.7. Saltsyre, massefylde: ca. 1,18 g/ml.
3.8. Tri-natriumcitrat-dihydrat.
3.9. 2,2 '-Thiodiethanol (thiodiglycol).
3.10. Natriumchlorid.
3.11. Ninhydrin.
3.12. Petroleumsether, kogepunktsinterval: 40–60 oC.
3.13. Norleucin eller anden forbindelse, der er egnet til brug som intern standard.
3.14. Nitrogengas (< 10 ppm oxygen).
3.15. 1-Octanol.
3.16. Aminosyrer
3.16.1. Standardstoffer som anført i punkt 1. Rene forbindelser, der ikke indeholder krystalvand. Tørres under vakuum over P205 eller H2SO4 i 1 uge inden brug.
3.16.2. Cysteinsyre.
3.16.3. Methioninsulfon.
3.17. Natriumhydroxidopløsning, c = 7,5 mol/liter:
300 g NaOH (3.5) opløses i vand, og der fyldes op til 1 liter.
3.18. Natriumhydroxidopløsning, c = 1 mol/liter:
40 g NaOH (3.5) opløses i vand, og der fyldes op til 1 liter.
3.19. Phenolholdig myresyreopløsning:
889 g myresyre (3.2) blandes med 111 g vand, og der tilsættes 4,73 g phenol (3.3).
3.20. Hydrolyseblanding, c = 6 mol HCl/liter indeholdende 1 g phenol/liter:
Der tilsættes 1 g phenol (3.3) til 492 ml HCl (3.7) og fyldes op med vand til 1 liter.
3.21. Ekstraktionsopløsning, c = 0,1 mol HCl/liter indeholdende 2 % thiodiglycol: 8,2 ml HCl (3.7) opløses i ca. 900 ml vand, hvorefter der tilsættes 20 ml thiodiglycol (3.9) og fyldes op med vand til 1 liter (3.7 og 3.9 må ikke blandes direkte).
3.22. 5-Sulfosalicylsyreopløsning, ß = 6 %:
60 g 5-sulfosalicylsyre (3.6) opløses i vand, og der fyldes op med vand til 1 liter.
3.23. Oxideringsreagens (permyresyre-phenol):
0,5 ml hydrogenperoxid (3.1) blandes med 4,5 ml phenolholdig myresyreopløsning (3.19) i et lille bægerglas. Inkuberes ved 20–30 oC i 1 time for at danne permyresyre. Herefter afkøles opløsningen i isbad (15 minutter), inden den tilsættes prøven.
Advarsel: Undgå kontakt med huden, og bær beskyttelsesbeklædning.
3.24. Citratbuffer, c = 0,2 mol Na+/liter, pH 2,20 :
19,61 g natriumcitrat (3.8), 5 ml thiodiglycol (3.9), 1 g phenol (3.3) og 16,50 ml HCl (3.7) opløses i ca. 800 ml vand. pH justeres til 2,20 . Der fyldes op med vand til 1 liter.
3.25. Elueringsbuffere fremstillet ifølge betingelserne for den benyttede analysator (4.9).
3.26. Ninhydrinreagens fremstillet ifølge betingelserne for den benyttede analysator (4.9).
3.27. Standardopløsninger af aminosyrer. Opløsningerne opbevares ved < 5 oC.
3.27.1. Standardstamopløsning af aminosyrer (3.16.1)
c = 2,5 μmol/ml af hver i saltsyre.
Kan fås i handelen.
3.27.2. Standardstamopløsning af cysteinsyre og methioninsulfon, c = 1,25 μmol/ml
0,2115 g cysteinsyre (3.16.2) og 0,2265 g methioninsulfon (3.16.3) opløses i citratbuffer (3.24) i en 1 liter målekolbe, og der fyldes op til mærket med citratbuffer. Opløsningen opbevares ved < 5 oC i højst 12 måneder. Denne opløsning benyttes ikke, hvis standardstamopløsningen (3.27.1) indeholder cysteinsyre og methioninsulfon.
3.27.3. Standardstamopløsning af den interne standard, f.eks. norleucin, c = 20 μmol/ml
0,6560 g norleucin (3.13) opløses i citratbuffer (3.24) i en målekolbe, og der fyldes op med citratbuffer til 250 ml. Opløsningen opbevares ved < 5 oC i højst 6 måneder.
3.27.4. Kalibreringsopløsning af standardaminosyrer til brug med hydrolysater, c = 5 nmol/50 μl cysteinsyre og methioninsulfon og c = 10 nmol/50 μl af de øvrige aminosyrer. 2,2 g natriumchlorid (3.10) opløses i et 100 ml bægerglas med 30 ml citratbuffer (3.24). Der tilsættes 4,00 ml standardstamopløsning af aminosyrer (3.27.1), 4,00 ml standardstamopløsning af cysteinsyre og methioninsulfon (3.27.2) og, hvis benyttet, 0,50 ml standardstamopløsning af intern standard (3.27.3). pH justeres til 2,20 med natriumhydroxid (3.18).
Opløsningen overføres kvantitativt til en 50 ml målekolbe, hvorefter der fyldes op til mærket med citratbuffer (3.24) og blandes.
Opløsningen opbevares ved < 5 oC i højst 3 måneder.
Se også bemærkning 9.1.
3.27.5. Kalibreringsopløsning af standardaminosyrer til brug med hydrolysater fremstillet under punkt 5.3.3.1 og til brug med ekstrakter (5.2). Kalibreringsopløsningen fremstilles efter punkt 3.27.4, men der tilsættes ikke natriumchlorid.
Opløsningen opbevares ved < 5 oC i højst 3 måneder.
4. Apparatur
4.1. 100 eller 250 ml rundbundet kolbe med tilbagesvaler.
4.2. 100 ml borsilikatglasflaske med skruelåg forsynet med gummi/teflonpakning (f.eks. Duran, Schott) til brug i tørreskab.
4.3. Tørreskab med motordreven ventilation og temperaturindstilling med en nøjagtighed på ± 2 oC.
4.4. pH-meter (aflæsning med tre decimaler).
4.5. Membranfilter (0,22 μm).
4.6. Centrifuge.
4.7. Rotationsvakuumfordamper.
4.8. Mekanisk rysteapparat eller magnetomrører.
4.9. Aminosyreanalysator eller HPLC-udstyr med ionbyttekolonne, anordning til ninhydrin-postkolonne-derivatisering og fotometrisk detektor.
Kolonnen pakkes med sulfoneret polystyrenresin, der kan adskille aminosyrerne fra hinanden og fra andre ninhydrinpositive stoffer. Pumperne til buffer- og ninhydrinopløsningen skal i den tid, der omfatter såvel standardkalibreringskørslen som prøveanalysen, have en flowstabilitet på ±0,5 %.
Nogle aminosyreanalysatorer kan benyttes, hvor hydrolysatet har en natriumkoncentration på c = 0,8 mol/liter og indeholder den resterende myresyre fra oxideringstrinnet. Andre analysatorer giver ikke tilfredsstillende adskillelse af visse aminosyrer, hvis hydrolysatet indeholder overskydende myresyre og/eller høje natriumionkoncentrationer. I så fald reduceres syrevolumen ved inddampning til ca. 5 ml efter hydrolysen og inden pH-justeringen. Inddampningen foretages under vakuum ved højst 40 oC.
5. Fremgangsmåde
5.1. Klargøring af prøven
Prøven formales, så den kan passere gennem en 0,5 mm sigte. Meget fugtige prøver skal inden formalingen enten lufttørres ved højst 50 oC eller frysetørres. Prøver med et højt fedtindhold ekstraheres med petroleumsether (3.12) inden formaling.
5.2. Bestemmelse af frie aminosyrer i foder og forblandinger
En passende mængde (1–5 g) af den klargjorte prøve (5.1) afvejes med en nøjagtighed på 0,2 mg i en konisk kolbe, og der tilsættes 100,0 ml ekstraktionsopløsning (3.21). Blandingen rystes i 60 minutter på mekanisk rysteapparat eller magnetomrører (4.8). Efter henstand og bundfældning afpipetteres 10,0 ml af supernatanten i et 100 ml bægerglas.
Der tilsættes 5,0 ml sulfosalicylsyreopløsning (3.22) under omrøring, og der omrøres fortsat med magnetomrører i 5 minutter. Supernatanten filtreres eller centrifugeres for at fjerne eventuelt bundfald. 10,0 ml af den klarede opløsning kommes i et 100 ml bægerglas, og pH justeres til 2,20 med natriumhydroxidopløsning (3.18). Opløsningen overføres med citratbuffer (3.24) til en målekolbe af passende størrelse, og der fyldes op til mærket med citratbufferopløsningen (3.24).
Hvis der bruges en intern standard, tilsættes 1,00 ml intern standard (3.27.3) for hver 100 ml slutopløsning, og der fyldes op til mærket med bufferopløsningen (3.24).
Derefter foretages kromatografi som beskrevet i punkt 5.4.
Hvis ekstraktet ikke kromatograferes samme dag, skal det opbevares ved < 5 oC.
5.3. Bestemmelse af totale aminosyrer
5.3.1.
0,1 –1 g af den klargjorte prøve (5.1) afvejes med en nøjagtighed på 0,2 mg i:
— en 100 ml rundbundet kolbe (4.1) til åben hydrolyse (5.3.2.3), eller
— en 250 ml rundbundet kolbe (4.1), hvis der kræves en lav natriumkoncentration (5.3.3.1), eller
— en 100 ml flaske med skruelåg (4.2) til lukket hydrolyse (5.3.2.4).
Den afvejede prøveportion skal have et kvælstofindhold på ca. 10 mg og et vandindhold på højst 100 mg.
Kolben/flasken anbringes i isbad og nedkøles til 0 oC, hvorefter der tilsættes 5 ml oxideringsblanding (3.23) og omrøres ved hjælp af en glasspatel med bøjet spids. Kolben/flasken, der indeholder spatelen, lukkes med lufttæt film. Isbadet med den lukkede beholder anbringes i et køleskab ved 0 oC og henstår i 16 timer. Efter 16 timer tages det ud af køleskabet, og det overskydende oxideringsreagens destrueres ved tilsætning af 0,84 g natriumdisulfit (3.4).
Der fortsættes som beskrevet i punkt 5.3.2.1.
5.3.2.
5.3.2.1.
Den oxiderede prøve (5.3.1) tilsættes 25 ml hydrolyseblanding (3.20). Alle prøverester, der måtte sidde fast på beholderen og spatelen, skal omhyggeligt vaskes ned.
Alt efter den benyttede hydrolysemetode fortsættes som beskrevet i punkt 5.3.2.3 eller 5.3.2.4.
5.3.2.2.
0,1 –1 g af den klargjorte prøve (5.1) afvejes med en nøjagtighed på 0,2 mg i en 100 ml eller 250 ml rundbundet kolbe (4.1) eller en 100 ml flaske med skruelåg (4.2). Den afvejede prøveportion skal have et kvælstofindhold på ca. 10 mg. Der tilsættes forsigtigt 25 ml hydrolyseblanding (3.20), som blandes med prøven. Der fortsættes som beskrevet i punkt 5.3.2.3 eller 5.3.2.4.
5.3.2.3.
Der tilsættes tre glasperler til blandingen i kolben (klargjort som beskrevet i punkt 5.3.2.1 eller 5.3.2.2). Den opvarmes til kogning og holdes under konstant kogning med tilbagesvaler i 23 timer. Efter afslutning af hydrolysen skylles tilbagesvaleren med 5 ml citratbuffer (3.24). Kolben aftages og afkøles i isbad.
Der fortsættes som beskrevet i punkt 5.3.3.
5.3.2.4.
Flasken med blandingen klargjort som beskrevet i punkt 5.3.2.1 eller 5.3.2.2 anbringes i et tørreskab (4.3) ved 110 oC. For at forhindre trykopbygning (på grund af udvikling af gasser) og undgå eksplosion lægges skruelåget i den første time løst på flasken. Flasken må ikke lukkes med skruelåget. Efter en times forløb lukkes flasken med skruelåget og henstår i tørreskabet (4.3) i 23 timer. Efter afslutning af hydrolysen tages flasken ud af tørreskabet, skruelåget åbnes forsigtigt, og kolben anbringes i isbad, hvor den henstår til afkøling.
Alt efter metoden for justering af pH-værdien (5.3.3) overføres kolbens indhold kvantitativt med citratbuffer (3.24) til et 250 ml bægerglas eller til en 250 ml rundbundet kolbe.
Der fortsættes som beskrevet i punkt 5.3.3.
5.3.3.
Alt efter aminosyreanalysatorens (4.9) natriumtolerance foretages justeringen af pH-værdien som beskrevet i punkt 5.3.3.1 eller 5.3.3.2.
5.3.3.1.
Det er tilrådeligt at bruge en intern standardstamopløsning (3.27.3), hvis der benyttes aminosyreanalysatorer, der kræver en lav natriumkoncentration (hvor syreindholdet skal reduceres).
I så fald tilsættes 2,00 ml af den interne standardstamopløsning (3.27.3) til hydrolysatet før inddampningen.
Der tilsættes 2 dråber 1-octanol (3.15) til det efter punkt 5.3.2.3 eller 5.3.2.4 frembragte hydrolysat.
Volumen reduceres ved hjælp af rotationsfordamperen (4.7) til 5–10 ml under vakuum ved 40 oC. Hvis volumen ved et uheld reduceres til under 5 ml, kasseres hydrolysatet, og analysen gentages.
pH justeres til 2,20 med natriumhydroxidopløsning (3.18), og der fortsættes som beskrevet i punkt 5.3.4.
5.3.3.2. :
Det efter 5.3.2.3 eller 5.3.2.4 frembragte hydrolysat neutraliseres delvis ved under omrøring at tilsætte 17 ml natriumhydroxidopløsning (3.17), samtidig med at temperaturen holdes under 40 oC.
pH justeres til 2,20 med natriumhydroxidopløsning (3.17 og 3.18) ved rumtemperatur. Der fortsættes som beskrevet i punkt 5.3.4.
5.3.4.
Hydrolysatet (5.3.3.1 eller 5.3.3.2) justeret til en pH-værdi på 2,20 overføres med citratbuffer (3.24) kvantitativt til en 200 ml målekolbe, og der fyldes op til mærket med buffer (3.24).
Hvis en intern standard anvendes, og denne ikke allerede er tilsat, tilsættes der 2,00 ml intern standard (3.27.3) og fyldes op til mærket med citratbuffer (3.24). Der blandes omhyggeligt.
Derefter foretages kromatografi som beskrevet i punkt 5.4.
Hvis prøveopløsningen ikke kromatograferes samme dag, skal den opbevares ved < 5 oC.
5.4. Kromatografi
Inden kromatografi opvarmes ekstraktet (5.2) eller hydrolysatet (5.3.4) til rumtemperatur. Blandingen rystes, og en passende mængde filtreres gennem et 0,22 μm membranfilter (4.5). Der foretages ionbytningskromatografi af den fremstillede klarede opløsning ved brug af en aminosyreanalysator (4.9).
Indsprøjtningen kan foretages manuelt eller automatisk. Hovedsagen er, at der altid tilsættes samme mængde opløsning ± 0,5 % til kolonnen til analyse af standardopløsninger og prøveopløsninger, undtagen i tilfælde, hvor der bruges en intern standard. Forholdet mellem natrium og aminosyre i standard- og prøveopløsningerne skal være så ens som muligt.
Kalibreringshyppigheden afhænger normalt af stabiliteten af ninhydrinreagenset og det anvendte analysatorsystem. Standard- eller prøveopløsningen fortyndes med citratbuffer (3.24), således at der for standarden fremkommer et topareal på 30–200 % af arealet af toppene for aminosyrerne i prøveopløsningen.
Kromatografien af aminosyrerne vil variere lidt afhængigt af den anvendte type analysator og kolonnematerialet. Systemet skal kunne adskille aminosyrerne fuldstændigt fra hinanden og fra øvrige ninhydrinpositive stoffer. Kromatografisystemet skal i sit arbejdsområde give lineært respons på ændringer i mængderne af aminosyrer, der tilsættes kolonnen.
Hvis der analyseres en ækvimolær opløsning (af de aminosyrer, der skal bestemmes), skal der ved kromatografien opnås nedenstående forhold mellem dal og tophøjde. Den ækvimolære opløsning skal indeholde mindst 30 % af den største mængde af hver aminosyre, der kan måles præcist med aminosyreanalysatoren (4.9).
Ved adskillelsen af threonin og serin må forholdet mellem dal og tophøjde for den laveste af de to sammenhængende aminosyretoppe ikke være større end 2:10. (Hvis der kun bestemmes cyst(e)in, methionin, threonin og lysin, vil utilstrækkelig adskillelse fra nabotoppe indvirke uheldigt på bestemmelsen.) For alle øvrige aminosyrer skal adskillelsen være bedre end 1:10.
Systemet skal sikre, at lysin adskilles fra artefakter fra andre lysinlignende stoffer og ornithin.
6. Beregning af resultater
Arealet af toppene i prøve- og standardopløsningerne måles for hver enkelt aminosyre, og mængden (X) beregnes i gram aminosyre pr. kg prøve:
![]()
Ved anvendelse af en intern standard multipliceres med:
![]()
|
A |
= |
topareal for hydrolysat eller ekstrakt |
|
B |
= |
topareal for standardkalibreringsopløsning |
|
C |
= |
topareal for intern standard i hydrolysat eller ekstrakt |
|
D |
= |
topareal for intern standard, standardkalibreringsopløsning |
|
M |
= |
molarvægt af den aminosyre, der skal bestemmes |
|
c |
= |
standardopløsningens koncentration i μmol/ml |
|
m |
= |
prøvens vægt i gram (korrigeret til oprindelig vægt hvis tørret eller affedtet) |
|
V |
= |
ml totalhydrolysat (5.3.4) eller ml beregnet total fortyndingsvolumen af ekstraktet (6.1). |
Såvel cystin som cystein bestemmes som cysteinsyre i hydrolysater af den oxiderede prøve, men beregnes som cystin (C6H12N2O4S2, M 240,30 g/mol) ved anvendelse af M 120,15 g/mol (= 0,5 × 240,30 g/mol).
Methionin bestemmes som methioninsulfon i hydrolysater af den oxiderede prøve, men beregnes som methionin ved anvendelse af M af methionin (149,21 g/mol).
Tilsat fri methionin bestemmes efter ekstraktion som methionin, hvorved der benyttes samme M til beregningen.
6.1. Totale fortyndingsvolumen af ekstrakter (F) til bestemmelse af frie aminosyrer (5.2) beregnes som følger:
![]()
|
V |
= |
slutekstraktets volumen. |
7. Evaluering af metoden
Metoden er blevet afprøvet ved en international ringanalyse i 1990 med anvendelse af fire forskellige typer foder (svinefoderblanding, slagtefjerkræblanding, proteinkoncentrat og en forblanding). Resultaterne af middel- og standardafvigelse efter fjernelse af outliers er vist i tabellerne under dette punkt:
Middelværdi i g/kg
|
Referencemateriale |
Aminosyre |
|||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
|
|
Svinefoderblanding |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|
Slagtefjerkræblanding |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|
Proteinkoncentrat |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|
Forblanding |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|
n = antal deltagende laboratorier. |
||||
7.1. Repeterbarhed
Repeterbarheden for de undersøgte aminosyrer udtrykt som »standardafvigelse inden for laboratorier« for de ovennævnte prøver er gengivet i nedenstående tabel.
Standardafvigelse inden for laboratorier (Sr) i g/kg
|
Referencemateriale |
Aminosyre |
|||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
|
|
Svinefoderblanding |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|
Slagtefjerkræblanding |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|
Proteinkoncentrat |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|
Forblanding |
1,30 n= 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|
n = antal deltagende laboratorier. |
||||
Variationskoefficient ( %) for standardafvigelsen inden for laboratorier (Sr)
|
Referencemateriale |
Aminosyre |
|||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
|
|
Svinefoderblanding |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|
Slagtefjerkræblanding |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|
Proteinkoncentrat |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|
Forblanding |
2,2 n= 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|
n = antal deltagende laboratorier. |
||||
7.2 Reproducerbarhed
Resultaterne af standardafvigelse mellem laboratorier for ovennævnte undersøgelser er vist i nedenstående skema.
Standardafvigelse mellem laboratorier (SR) i g/kg
|
Referencemateriale |
Aminosyre |
|||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
|
|
Svinefoderblanding |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|
Slagtefjerkræblanding |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|
Proteinkoncentrat |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|
Forblanding |
2,49 n= 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|
n = antal deltagende laboratorier. |
||||
Variationskoefficient ( %) for standardafvigelsen mellem laboratorier (SR)
|
Referencemateriale |
Aminosyre |
|||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
|
|
Svinefoderblanding |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|
Slagtefjerkræblanding |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|
Proteinkoncentrat |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|
Forblanding |
4,3 n= 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|
n = antal deltagende laboratorier. |
||||
8. Anvendelse af referencematerialer
Det undersøges, om metoden er brugt korrekt, ved at foretage gentagne målinger af certificerede referencematerialer, når sådanne forefindes. Det anbefales at kalibrere med certificeret aminosyrekalibreringsopløsning.
9. Bemærkninger
9.1. På grund af forskelle mellem aminosyreanalysatorer skal slutkoncentrationerne af kalibreringsopløsningerne af standardaminosyrerne (se punkt 3.27.4 og 3.27.5) og hydrolysatet (se punkt 5.3.4) betragtes som en retningslinje.
Apparatets lineære responsskala skal kontrolleres for alle aminosyrer.
Standardopløsningen fortyndes med citratbuffer for at opnå toparealer midt på skalaen.
9.2. Hvis der anvendes HPLC-udstyr til at analysere hydrolysaterne, skal kørselsbetingelserne optimeres i henhold til fabrikantens anbefaling.
9.3. Hvis metoden anvendes på foder, der indeholder mere end 1 % chlorid (koncentrat, mineralfoder, fodertilskud), vil der kunne ske en undervurdering af methionin, og der skal foretages en særlig behandling.
G. BESTEMMELSE AF TRYPTOPHAN
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme det samlede indhold af tryptophan (totaltryptophan) og frit tryptophan i foder. Der differentieres ikke mellem D- og L-former.
2. Princip
Til bestemmelse af totaltryptophan hydrolyseres prøven i basisk miljø i en mættet bariumhydroxidopløsning og holdes opvarmet til 110 oC i 20 timer. Efter hydrolysen tilsættes der intern standard.
Til bestemmelse af frit tryptophan ekstraheres prøven i svagt sur væske under tilstedeværelse af intern standard.
Tryptophanet og den interne standard i hydrolysatet eller i ekstraktet bestemmes ved HPLC med fluorescensdetektor.
3. Reagenser
3.1. Der benyttes dobbeltdestilleret vand eller vand af tilsvarende kvalitet (ledningsevne: < 10 μS/cm).
3.2. Standardstof: tryptophan (renhedsgrad/indhold ≥ 99 %), tørret i vakuum over phosphorpentoxid.
3.3. Internt standardstof: α-methyltryptophan (renhedsgrad/indhold ≥ 99 %), tørret i vakuum over phosphorpentoxid.
3.4. Bariumhydroxidoctahydrat (man skal passe på ikke at udsætte Ba(OH)2.8H2O for meget for luft, da der ellers dannes BaCO3, som kan interferere med bestemmelsen) (se bemærkning 9.3).
3.5. Natriumhydroxid.
3.6. Orthophosphorsyre, w (w/w) = 85 %.
3.7. Saltsyre, ρ20 = 1,19 g/ml.
3.8. Methanol, svarende til HPLC-kvalitet.
3.9. Petroleumsether, kogepunktsinterval: 40–60 oC.
3.10. Natriumhydroxidopløsning, c = 1 mol/liter: 40,0 g NaOH (3.5) opløses i vand, og der fyldes op med vand (3.1) til 1 liter.
3.11. Saltsyre, c = 6 mol/liter:
492 ml saltsyre (3.7) fortyndes til 1 liter med vand.
3.12. Saltsyre, c = 1 mol/liter:
82 ml saltsyre (3.7) fortyndes til 1 liter med vand.
3.13. Saltsyre, c = 0,1 mol/liter:
8,2 ml saltsyre (3.7) fortyndes til 1 liter med vand.
3.14. Orthophosphorsyre, c = 0,5 mol/liter:
34 ml orthophosphorsyre (3.6) fortyndes til 1 liter med vand (3.1).
3.15. Koncentreret opløsning af tryptophan (3.2), c = 2,50 μmol/ml:
I en 500 ml målekolbe opløses 0,2553 g tryptophan (3.2) i saltsyre (3.13), og der fyldes op til mærket med saltsyre (3.13). Opløsningen opbevares ved - 18 oC i højst 4 uger.
3.16. Koncentreret opløsning af intern standard, c = 2,5 μmol/ml:
I en 500 ml målekolbe opløses 0,2728 g α-methyltryptophan (3.3) i saltsyre (3.13), og der fyldes op til mærket med saltsyre (3.13). Opløsningen opbevares ved - 18 oC i højst 4 uger.
3.17. Standardkalibreringsopløsning af tryptophan og intern standard:
2,00 ml koncentreret opløsning af tryptophan (3.15) og 2,00 ml koncentreret opløsning af intern standard (α-methyltryptophan) (3.16) fortyndes med vand (3.1) og methanol (3.8) til omtrent samme volumen og omtrent samme methanolkoncentration (10–30 %) som det færdige hydrolysat.
Opløsningen skal fremstilles umiddelbart før brugen.
Der sørges for beskyttelse mod direkte sollys under fremstillingen.
3.18. Eddikesyre.
3.19. 1,1,1-Trichlor-2-methyl-2-propanol.
3.20. Ethanolamin, w (w/w) > 98 %.
3.21. Opløsning af 1 g 1,1,1-trichlor-2-methyl-2-propanol (3.19) i 100 ml methanol (3.8).
3.22. Mobil fase til HPLC: 3,00 g eddikesyre (3.18) + 900 ml vand (3.1) +50,0 ml af opløsningen (3.21) af 1,1,1-trichlor-2-methyl-2-propanol (3.19) i methanol (3.8) (1 g pr. 100 ml). pH justeres til 5,00 med ethanolamin (3.20). Der fyldes op med vand (3.1) til 1 000 ml.
4. Apparatur
4.1. HPLC-udstyr med spektrofluorometrisk detektor
4.2. HPLC-kolonne, 125 mm × 4 mm, C18, 3 μm pakkemateriale, eller tilsvarende
4.3. pH-meter
4.4. Polypropylenflaske, 125 ml, med vid hals og skruelåg
4.5. Membranfilter, 0,45 μm
4.6. Autoklav, 110 (± 2) oC, 1,4 (±0,1 ) bar
4.7. Mekanisk rysteapparat eller magnetomrører
4.8. Vortex-blander.
5. Fremgangsmåde
5.1. Klargøring af prøver
Prøven formales, så den kan passere gennem en 0,5 mm sigte. Meget fugtige prøver skal inden formalingen enten lufttørres ved højst 50 oC eller frysetørres. Prøver med et højt fedtindhold ekstraheres med petroleumsether (3.9) inden formaling.
5.2. Bestemmelse af frit tryptophan (ekstrakt)
En passende mængde (1–5 g) af den klargjorte prøve (5.1) afvejes med en nøjagtighed på 1 mg i en konisk kolbe. Der tilsættes 100,0 ml saltsyre (3.13) og 5,00 ml koncentreret opløsning af intern standard (3.16). Der omrystes eller blandes i 60 minutter på mekanisk rysteapparat eller magnetomrører (4.7). Efter henstand og bundfældning afpipetteres 10,0 ml af supernatanten i et bægerglas. Der tilsættes 5 ml orthophosphorsyre (3.14). pH justeres til 3 med natriumhydroxid (3.10). Der tilsættes så meget methanol (3.8), at slutkoncentrationen af methanol bliver 10–30 %. Opløsningen overføres til en målekolbe af passende størrelse og fortyndes med vand til et volumen, der passer til kromatografien (ca. samme volumen som standardkalibreringsopløsningen (3.17)).
Nogle få ml af opløsningen filtreres gennem et 0,45 μm membranfilter (4.5), inden den indsprøjtes på HPLC-kolonnen. Derefter foretages kromatografi som beskrevet i punkt 5.4.
Standardopløsninger og ekstrakter beskyttes mod direkte sollys. Hvis ekstrakterne ikke kan analyseres samme dag, kan de opbevares ved 5 oC i højst 3 dage.
5.3. Bestemmelse af totaltryptophan (hydrolysat)
Med en nøjagtighed på 0,2 mg afvejes 0,1 –1 g af den klargjorte prøve (5.1) i polypropylenflasken (4.4). Den afvejede prøveportion skal have et kvælstofindhold på ca. 10 mg. Der tilsættes 8,4 g bariumhydroxidoctahydrat (3.4) og 10 ml vand. Der blandes på vortex-blander (4.8) eller magnetomrører (4.7). Den teflonbelagte magnetpind efterlades i blandingen. Flaskens vægge skylles med 4 ml vand. Skruelåget skrues løst på flasken, der sættes ind i autoklaven (4.6) med kogende vand i 30–60 minutter. Autoklaven lukkes, og der autoklaveres ved 110 (± 2) oC i 20 timer.
Temperaturen i autoklaven sænkes til lige under 100 oC, inden den åbnes. For at undgå udkrystallisering af Ba(OH)2 · 8H2O tilsættes der til den varme blanding 30 ml stuevarmt vand. Der omrystes eller omrøres forsigtigt og tilsættes 2,00 ml koncentreret intern standardopløsning (α-methyltryptophan) (3.16). Flasken afkøles i vand/isbad i 15 minutter.
Derefter tilsættes 5 ml orthophosphorsyre (3.14). Medens flasken stadig står i kølebadet, neutraliseres der med saltsyre (3.11) under omrøring, og pH justeres til 3,0 med saltsyre (3.12). Der tilsættes så meget methanol, at slutkoncentrationen af methanol bliver 10–30 %. Opløsningen overføres til en målekolbe af passende størrelse og fortyndes med vand til et volumen, der passer til kromatografien (f.eks. 100 ml). Der må ikke ske udfældning under methanoltilsætningen.
Nogle få ml af opløsningen filtreres gennem et 0,45 μm membranfilter (4.5), inden den indsprøjtes på HPLC-kolonnen. Derefter foretages kromatografi som beskrevet i punkt 5.4.
Standardopløsninger og hydrolysater beskyttes mod direkte sollys. Hvis hydrolysaterne ikke kan analyseres samme dag, kan de opbevares ved 5 oC i højst 3 dage.
5.4. HPLC-bestemmelse
Nedenstående betingelser for isokratisk eluering er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater (se også bemærkning 9.1 og 9.2).
|
HPLC-kolonne (4.2): |
125 mm × 4 mm, C18, 3 pakkemateriale, μm eller tilsvarende |
|
Kolonnetemperatur: |
Rumtemperatur |
|
Mobil fase (3.22): |
3,00 g eddikesyre (3.18) + 900 ml vand (3.1) +50,0 ml af opløsningen (3.21) af 1,1 ,1-trichlor- 2-methyl-2-propanol (3.19) i methanol (3.8) (1 g pr. 100 ml). pH justeres til 5,00 med ethanolamin (3.20). Der fyldes op med vand (3.1) til 1 000 ml |
|
Flow: |
1 ml/minuttet |
|
Total gennemløbstid: |
ca. 34 minutter |
|
Detektionsbølgelængde: |
excitation: 280 nm, emission: 356 nm |
|
Injektionsvolumen: |
20 μl. |
6. Beregning af resultater
Mængden af tryptophan (X) beregnes i gram pr. 100 g prøve:
![]()
|
A |
= |
topareal for intern standard, standardkalibreringsopløsning (3.17) |
|
B |
= |
topareal for tryptophan, ekstrakt (5.2) eller hydrolysat (5.3) |
|
V1 |
= |
volumen i ml (2 ml) af den mængde koncentreret tryptophanopløsning (3.15), der er tilsat til kalibreringsopløsningen (3.17) |
|
c |
= |
koncentration i μmol/ml (= 2,50 ) af den koncentrerede tryptophanopløsning (3.15), der er tilsat til kalibreringsopløsningen (3.17) |
|
V2 |
= |
volumen i ml af den mængde koncentreret opløsning af intern standard (3.16), der er tilsat til ekstraktet (5.2) (= 5,00 ml) eller til hydrolysatet (5.3) (= 2,00 ml) |
|
C |
= |
topareal for intern standard, ekstrakt (5.2) eller hydrolysat (5.3) |
|
D |
= |
topareal for tryptophan, standardkalibreringsopløsning (3.17) |
|
V3 |
= |
volumen i ml (= 2,00 ml) af den mængde koncentreret opløsning af intern standard (3.16), der er tilsat til standardkalibreringsopløsningen (3.17) |
|
m |
= |
prøvens vægt i gram (korrigeret til oprindelig vægt, hvis den er tørret og/eller affedtet) |
|
M |
= |
tryptophans molarvægt (= 204,23 g/mol). |
7. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige 10 % i relativ værdi (af det højeste resultat).
8. Resultater af ringanalyse
Der er gennemført en EF-ringanalyse (4. ringanalyse), hvor 3 prøver blev analyseret i op til 12 laboratorier med henblik på certificering af hydrolysemetoden. Der blev foretaget 5-dobbelt bestemmelse på hver prøve. Resultaterne er vist i nedenstående tabel:
|
|
Prøve 1 Svinefoder |
Prøve 2 Svinefoder suppleret med L-tryptophan |
Prøve 3 Foderkoncentrat til svin |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Middelværdi [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [%] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [%] |
6,3 |
6,0 |
2,2 |
|
L |
= |
antal laboratorier, der har indleveret resultater |
|
n |
= |
antal enkeltværdier efter fjernelse af outliers (Cochran-Dixon outlier-test) |
|
sr |
= |
standardafvigelse for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
r |
= |
repeterbarhed |
|
R |
= |
reproducerbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed, % |
|
CVR |
= |
variationskoefficient for reproducerbarhed, %. |
Der er også gennemført en EF-ringanalyse (3. ringanalyse), hvor 2 prøver blev analyseret i op til 13 laboratorier med henblik på certificering af metoden til ekstraktion af frit tryptophan. Der blev foretaget 5-dobbelt bestemmelse på hver prøve. Resultaterne er vist i nedenstående tabel:
|
|
Prøve 4 Blanding af soja og hvede |
Prøve 5 Blanding af soja og hvede (= prøve 4) med tilsætning af tryptophan (0,457 g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Middelværdi [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [%] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [%] |
4,71 |
5,11 |
|
L |
= |
antal laboratorier, der har indleveret resultater |
|
n |
= |
antal enkeltværdier efter fjernelse af outliers (Cochran-Dixon outlier-test) |
|
sr |
= |
standardafvigelse for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
r |
= |
repeterbarhed |
|
R |
= |
reproducerbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed, % |
|
CVR |
= |
variationskoefficient for reproducerbarhed, %. |
Der er gennemført endnu en EF-ringanalyse, hvor 4 prøver blev analyseret i op til 7 laboratorier med henblik på certificering af tryptophan med hensyn til hydrolyse. Resultaterne er vist herunder. Der blev foretaget 5-dobbelt bestemmelse på hver prøve.
|
|
Prøve 1 Svinefoderblanding (CRM 117) |
Prøve 2 Fiskemel med lavt fedtindhold (CRM 118) |
Prøve 3 Sojamel (CRM 119) |
Prøve 4 Skummetmælkspulver (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Middelværdi [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
Sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [%] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [%] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
antal laboratorier, der har indleveret resultater |
|
n |
= |
antal enkeltværdier efter fjernelse af outliers (Cochran-Dixon outlier-test) |
|
sr |
= |
standardafvigelse for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
r |
= |
repeterbarhed |
|
R |
= |
reproducerbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed, % |
|
CVR |
= |
variationskoefficient for reproducerbarhed, %. |
9. Bemærkninger
9.1. Nedenstående særlige kromatograferingsbetingelser kan give bedre adskillelse af tryptophan og α-methyltryptophan.
Isokratisk eluering efterfulgt af gradientrensning af kolonnen som følger:
|
HPLC-kolonne: |
125 mm × 4 mm, C18, 5 μm pakkemateriale, eller tilsvarende |
||
|
Kolonnetemperatur: |
32 oC |
||
|
Mobil fase: |
A: 0,01 mol/liter KH2PO4/methanol, 95 + 5 (v + v) B: methanol |
||
|
Gradientprogram: |
0 minutter |
100 % A |
0 % B |
|
|
15 minutter |
100 % A |
0 % B |
|
|
17 minutter |
60 % A |
40 % B |
|
|
19 minutter |
60 % A |
40 % B |
|
|
21 minutter |
100 % A |
0 % B |
|
|
33 minutter |
100 % A |
0 % B |
|
Flow: |
1,2 ml/minuttet |
||
|
Total gennemløbstid: |
ca. 33 minutter. |
||
9.2. Kromatograferingen varierer, alt efter hvilken type HPLC og kolonnemateriale der benyttes. Der skal vælges et system, der giver adskillelse til basislinjen mellem tryptophan og den interne standard. Det er desuden vigtigt, at nedbrydningsprodukter adskilles helt fra tryptophan og den interne standard. Der køres hydrolysater uden intern standard for at kontrollere, at der ikke ligger urenheder på basislinjen under den interne standard. Det er vigtigt, at gennemløbstiden er lang nok til, at alle nedbrydningsprodukter elueres, da toppe med lang elueringstid ellers kan interferere i efterfølgende kromatograferinger.
Kromatografisystemet skal i sit arbejdsområde give lineært respons. Det lineære respons kontrolleres med konstant (dvs. den normale) koncentration af intern standard og varierende koncentrationer af tryptophan. Det er vigtigt, at både toppen for tryptophan og toppen for intern standard ligger inden for HPLC/fluorescenssystemets lineære område. Hvis blot en af toppene er enten for lille eller for stor, gentages analysen med en anden prøvestørrelse og/eller et andet slutvolumen.
9.3. Bariumhydroxid
Bariumhydroxid bliver med tiden vanskeligere at opløse. Det medfører, at opløsningen til HPLC-bestemmelse bliver uklar, hvilket kan betyde, at resultaterne for tryptophan bliver for lave.
H. BESTEMMELSE AF RÅFEDT
1. Formål og anvendelsesområde
Denne metode anvendes til bestemmelse af råfedtindholdet i foder. Den er ikke beregnet til analyse af olieholdige frø og frugter.
Anvendelsen af de to fremgangsmåder, der er beskrevet nedenfor, afhænger af arten og sammensætningen af foderet og af grunden til, at analysen gennemføres.
1.1. Metode A — Råfedt, som kan ekstraheres direkte
Denne metode anvendes til vegetabilske fodermidler med undtagelse af dem, der er medtaget under metode B.
1.2. Metode B — Råfedt i alt
Denne metode anvendes til animalske fodermidler samt til alle foderblandinger. Den skal anvendes til alle materialer, hvorfra råfedt ikke kan ekstraheres fuldstændigt uden forudgående hydrolyse, såsom gluten, gær, kartoffelproteiner og produkter, der underkastes processer som ekstrudering, omdannelse til flager og opvarmning.
1.3. Fortolkning af resultater
I samtlige tilfælde, hvor der opnås et højere resultat ved anvendelse af metode B end ved metode A, skal de resultater, der er opnået ved metode B, accepteres som den sande værdi.
2. Princip
2.1. Metode A
Prøven ekstraheres med petroleumsether. Opløsningsmidlet afdampes, og remanensen tørres og vejes.
2.2. Metode B
Prøven behandles med saltsyre under opvarmning. Blandingen afkøles og filtreres. Den vaskede og tørrede remanens behandles herefter som beskrevet under metode A.
3. Reagenser
3.1. Petroleumsether, kogepunktsinterval: 40–60 oC. Bromtallet skal være under 1, og inddampningsresten under 2 mg/100 ml
3.2. Natriumsulfat, vandfrit
3.3. Saltsyre, c = 3 mol/liter
3.4. Filtreringsmiddel, f.eks. kiselgur, Hyflo-supercel.
4. Apparatur
4.1. Ekstraktionsapparat. Dersom apparatet er udstyret med et hævertrør (Soxhlet-apparat), indstilles varmekilden, så der sker ca. 10 tømninger i timen; dersom apparatet ikke er udstyret med hævertrør, skal tilbageløbet være på ca. 10 ml i minuttet.
4.2. Ekstraktionshætter skal være fri for stof, der er opløseligt i petroleumsether, og skal have en porøsitet i overensstemmelse med kravene under 4.1.
4.3. Tørreskab, enten et vakuumtørreskab indstillet til 75 oC ± 3 oC eller et tørreskab med luftcirkulation indstillet til 100 oC ± 3 oC.
5. Fremgangsmåde
5.1. Metode A (se punkt 8.1)
5 g af prøven afvejes med en nøjagtighed på 1 mg, overføres til en ekstraktionshætte (4.2) og tildækkes med en affedtet vatprop.
Hætten anbringes i et ekstraktionsapparat (4.1), og der ekstraheres i 6 timer med petroleumsether (3.1). Petroleumsetherekstraktet opsamles i en tør, tareret kolbe indeholdende stykker af pimpsten ( 28 ).
Opløsningsmidlet afdampes. Remanensen tørres derpå i kolben i 1 1/2 time i tørreskab (4.3). Efter afkøling i ekssikkator vejes remanensen. Der tørres i yderligere 30 minutter for at sikre, at fedtets vægt forbliver konstant (vægttabet mellem to på hinanden følgende vejninger må højst være på 1 mg).
5.2. Metode B
2,5 g af prøven afvejes med en nøjagtighed på 1 mg (se punkt 8.2) og overføres til et 400 ml bægerglas eller en 300 ml konisk kolbe, hvorefter der tilsættes 100 ml saltsyre (3.3) og stykker af pimpsten. Bægerglasset tildækkes med et urglas, eller hvis der anvendes en konisk kolbe, forsynes denne med en tilbagesvaler. Blandingen bringes til let kogning over en svag flamme eller på varmeplade i ca. 1 time. Materialet skal forhindres i at sætte sig fast på beholderens sider.
Beholderen afkøles, og der tilsættes så meget filtreringsmiddel (3.4), at der ikke opstår noget fedttab ved filtreringen. Der filtreres gennem et fugtigt, fedtfrit dobbelt filtrerpapir. Remanensen vaskes med koldt vand, indtil filtratet er neutralt. Det kontrolleres, at filtratet ikke indeholder fedt. Tilstedeværelse af fedt viser, at der inden hydrolysen skal foretages en ekstraktion af prøven med petroleumsether ved anvendelse af metode A.
Det dobbelte filtrerpapir med remanensen lægges på et urglas og tørres i tørreskab (4.3) ved 100 oC ± 3 oC i 1 ½ time.
Det dobbelte filtrerpapir med den tørre remanens anbringes i ekstraktionshætte (4.2) og tildækkes med en affedtet vatprop. Hætten anbringes i et ekstraktionsapparat (4.1), hvorefter der fortsættes som beskrevet i punkt 5.1, andet og tredje afsnit.
6. Angivelse af resultater
Remanensens vægt angives i procent af prøven.
7. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført af den samme person på den samme prøve må ikke overstige:
— 0,2 % i absolut værdi for råfedtindhold på under 5 %
— 4,0 % i relativ værdi (af det højeste resultat) for indhold på 5–10 %
— 0,4 % i absolut værdi for indhold på over 10 %.
8. Bemærkninger
8.1. For produkter med højt fedtindhold, som er vanskelige at formale, eller som ikke egner sig til udtagning af en reduceret homogen prøve, anvendes følgende fremgangsmåde:
20 g af prøven afvejes med en nøjagtighed på 1 mg og blandes med mindst 10 g vandfrit natriumsulfat (3.2). Derpå ekstraheres med petroleumsether (3.1) som angivet under punkt 5.1. Det herved opsamlede ekstrakt tilsættes petroleumsether (3.1) ad 500 ml, og blandingen rystes. Af opløsningen udtages 50 ml, som hældes i en lille, tør, tareret kolbe indeholdende stykker af pimpsten. Opløsningsmidlet afdampes; derpå tørres og fortsættes som beskrevet i punkt 5.1, sidste afsnit.
Ekstraktionsresten i hætten befries for opløsningsmidlet og formales til en kornstørrelse på 1 mm, hvorefter den hældes tilbage i ekstraktionshætten (der tilsættes ikke natriumsulfat); derpå fortsættes som beskrevet i punkt 5.1, andet og tredje afsnit.
Fedtindholdet beregnes som en procentdel af prøven ved anvendelse af nedenstående formel:
(10m1 + m2) × 5
hvor:
|
m1 |
= |
vægt i gram af remanensen efter den første ekstraktion (den alikvote del af ekstraktet) |
|
m2 |
= |
vægt i gram af remanensen efter den anden ekstraktion. |
8.2. For produkter med lavt fedtindhold kan der anvendes en afvejning på 5 g.
8.3. Foder med højt vandindhold til selskabsdyr vil det kunne være nødvendigt at blande med vandfrit natriumsulfat forud for hydrolyse og ekstraktion som ved metode B.
8.4. I punkt 5.2 vil det kunne være mere effektivt at anvende varmt vand i stedet for koldt vand til vaskning af remanensen efter filtrering.
8.5. Det kan for visse typer foder være nødvendigt at forlænge tørringstiden på 1 ½ time. Overdreven tørring undgås, da en sådan kan føre til lave resultater. En mikrobølgeovn kan også anvendes.
8.6. Præekstraktion ved metode A forud for hydrolyse og reekstraktion ved metode B anbefales, hvis råfedtindholdet er højere end 15 %. Dette afhænger til en vis grad af arten af foderet og af arten af fedtet i foderet.
I. BESTEMMELSE AF TRÆSTOF
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af fedtfri organiske stoffer i foder, som er uopløselige i fortyndede syre- og baseopløsninger, og som normalt betegnes som træstof.
2. Princip
Prøven, der om nødvendigt er affedtet, behandles i kogende opløsninger af først svovlsyre og derefter kaliumhydroxid i bestemte koncentrationer. Remanensen adskilles ved filtrering i en glasfilterdigel, hvorefter det vaskes, tørres, vejes og foraskes ved 475–500 oC. Vægttabet ved foraskningen svarer til træstofindholdet i prøven.
3. Reagenser
3.1. Svovlsyre, c = 0,13 mol/liter
3.2. Skumdæmpende middel (f.eks. n-octanol)
3.3. Filtreringshjælpemiddel (Celite 545 el. lign.) opvarmet til 500 oC i 4 timer (8.6)
3.4. Acetone
3.5. Petroleumsether, kogepunktsinterval: 40–60 oC
3.6. Saltsyre, c = 0,5 mol/liter
3.7. Kaliumhydroxidopløsning, c = 0,23 mol/liter.
4. Apparatur
4.1. Varmeenhed til kogning med svovlsyre og kaliumhydroxid med holder til filterdiglen (4.2) og afgangsrør med hane til væskeafløb samt vakuumpumpe, eventuelt med trykluft. Enheden forvarmes før daglig brug med kogende vand i 5 minutter.
4.2. Glasfilterdigel med filterplade af sintret glas, porestørrelse: 40–90 μm. Inden den første gang tages i anvendelse, opvarmes den i nogle få minutter til 500 oC og afkøles (8.6).
4.3. Kogecylinder på mindst 270 ml med tilbagesvaler.
4.4. Tørreskab med termostat.
4.5. Muffelovn med termostat.
4.6. Ekstraktionsapparat med holder til filterdiglen (4.2) og afgangsrør med hane til væskeafløb samt vakuumpumpe.
4.7. Forbindelsesringe til samling af varmeenhed (4.1), filterdigel (4.2) og kogecylinder (4.3) og til samling af koldekstraktionsapparat (4.6) og filterdigel.
5. Fremgangsmåde
I glasfilterdiglen (4.2) afvejes med 1 mg nøjagtighed 1 g af den klargjorte prøve (se bemærkning 8.1, 8.2 og 8.3), og der tilsættes 1 g filtreringshjælpemiddel (3.3).
Varmeenheden (4.1) og filterdiglen (4.2) samles. Kogecylinderen (4.3) forbindes til diglen. 150 ml svovlsyre (3.1), der er opvarmet til kogepunktet, overføres til kogecylinderen, og om nødvendigt tilsættes nogle få dråber skumdæmpende middel (3.2).
Væsken opvarmes til kogepunktet i løbet af 5 ± 2 minutter og koges livligt i nøjagtig 30 minutter.
Afgangshanen (4.1) åbnes, og svovlsyren filtreres under vakuum gennem filterdiglen. Remanensen på filterdiglen vaskes under vakuum tre gange med 30 ml kogende vand hver gang. Filtret med remanensen suges tørt efter hver vask.
Afgangshanen lukkes, og 150 ml kogende kaliumhydroxidopløsning (3.7) overføres til kogecylinderen. Der tilsættes nogle få dråber skumdæmpende middel (3.2). Væsken opvarmes til kogepunktet i løbet af 5 ± 2 minutter og koges livligt i nøjagtig 30 minutter. Remanensen filtreres og vaskes på samme måde som efter behandling med svovlsyre.
Efter den sidste vask suges bundfaldet tørt, og diglen med indhold forbindes til koldekstraktionsapparatet (4.6). Remanensen i diglen vaskes under vakuum tre gange med 25 ml acetone (3.4) hver gang og suges tørt efter hver vask.
Filterdiglen med indhold tørres i tørreskabet ved 130 oC indtil konstant vægt. Efter hver tørring afkøles diglen i en ekssikkator og vejes straks. Herefter anbringes den i muffelovnen, og indholdet foraskes ved 475–500 oC i mindst 30 minutter. Dette gentages indtil konstant vægt (vægttabet mellem to på hinanden følgende vejninger må højst være på 2 mg).
Efter hver foraskning afkøles diglen først i ovnen og derefter i ekssikkatoren, inden den vejes.
Der foretages en blindprøve uden prøve. Vægttabet ved foraskningen må ikke overstige 4 mg.
6. Beregning af resultater
Træstofindholdet som procent af prøven beregnes efter følgende formel:
![]()
hvor:
|
m |
= |
prøvens vægt i gram |
|
m0 |
= |
vægttabet i gram efter foraskning under bestemmelsen |
|
m1 |
= |
vægttabet i gram ved foraskning af remanensen af blindprøven. |
7. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige:
— 0,6 % i absolut værdi for træstofindhold på under 10 %
— 6 % i relativ værdi (af det højeste resultat) for træstofindhold på 10 % og derover.
8. Bemærkninger
8.1. Foder, der indeholder over 10 % råfedt, skal affedtes med petroleumsether (3.5) inden analysen. Filterdiglen (4.2) med indhold forbindes med koldekstraktionsapparatet (4.6) og vaskes under vakuum tre gange med 30 ml petroleumsether hver gang. Prøven suges tør, og diglen med indhold forbindes til varmeenheden (4.1). Derefter fortsættes som beskrevet i punkt 5.
8.2. Foder, der indeholder fedt, som ikke kan ekstraheres direkte med petroleumsether (3.5), skal affedtes som beskrevet i punkt 8.1 og efter kogning med syre affedtes endnu en gang. Efter kogning med syre og efterfølgende vask forbindes diglen med indhold til koldekstraktionsapparatet (4.6) og vaskes tre gange med 30 ml acetone efterfulgt af tre gange vask med 30 ml petroleumsether. Filtret suges tørt, og analysen fortsættes som beskrevet i punkt 5 med behandlingen med kaliumhydroxid.
8.3. Hvis foderet indeholder over 5 % carbonater, udtrykt som calciumcarbonat, forbindes diglen (4.2) med den afvejede prøve til varmeenheden (4.1). Prøven vaskes tre gange med 30 ml saltsyre (3.6). Efter hver tilsætning skal prøven henstå i ca. 1 minut, inden den filtreres. Der vaskes én gang med 30 ml vand, hvorefter der fortsættes som beskrevet i punkt 5.
8.4. Hvis der anvendes apparatur i form af et stativ (flere digler forbundet med samme varmeenhed), må der aldrig foretages to enkeltbestemmelser af samme analyseprøve i samme prøverække.
8.5. Hvis det efter kogning viser sig vanskeligt at filtrere syre- og baseopløsningerne, føres der trykluft gennem varmeenhedens afgangsrør, hvorefter filtreringen fortsættes.
8.6. Udglødningstemperaturen må ikke være over 500 oC, hvilket skal sikre, at diglerne af sintret glas holder længst muligt. Der drages omsorg for at undgå store temperaturudsving under opvarmning og afkøling.
J. BESTEMMELSE AF SUKKER
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af reducerende sukker og totalsukker efter invertering, udtrykt som glucose eller, ved omregning med faktoren 0,95 , som saccharose. Metoden finder anvendelse på foderblandinger. Der er fastsat særlige metoder for andre typer foder. Om nødvendigt bestemmes lactosen særskilt, idet der tages hensyn hertil ved beregningen af resultaterne.
2. Princip
Sukkeret opløses i fortyndet ethanol; opløsningen klares ved hjælp af Carrez-reagens I og II. Efter afdampning af ethanol foretages bestemmelserne før og efter inverteringen efter Luff-Schoorl-metoden.
3. Reagenser
3.1. Ethanolopløsning, 40 % (v/v), massefylde: 0,948 g/ml ved 20 oC, neutraliseret til phenolphthalein.
3.2. Carrez-reagens I: 21,9 g zinkacetat, Zn(CH3COO)2·2H2O, og 3 g iseddike opløses i vand. Der fyldes op med vand til 100 ml.
3.3. Carrez-reagens II: 10,6 g kaliumferrocyanid, K4Fe(CN)6 · 3H2O, opløses i vand. Der fyldes op med vand til 100 ml.
3.4. Methylorange, 0,1 % opløsning (w/v).
3.5. Saltsyre, 4 mol/liter.
3.6. Saltsyre, 0,1 mol/liter.
3.7. Natriumhydroxidopløsning, 0,1 mol/liter.
3.8. Luff-Schoorl-reagens:
Under forsigtig omrøring hældes citronsyreopløsningen (3.8.2) over i natriumcarbonatopløsningen (3.8.3). Kobbersulfatopløsningen (3.8.1) tilsættes, og der fyldes op med vand til 1 liter. Det henstår natten over og filtreres.
Koncentrationen af det således opnåede reagens kontrolleres (Cu 0,05 mol/liter; Na2CO3 1 mol/liter) (se punkt 5.4, sidste afsnit). Opløsningens pH-værdi skal være ca. 9,4 .
3.8.1. Kobbersulfatopløsning: 25 g kobbersulfat, CuSO4 · 5H2O, frit for jern, opløses i 100 ml vand.
3.8.2. Citronsyreopløsning: 50 g citronsyre, C6H8O7.H2O, opløses i 50 ml vand.
3.8.3. Natriumcarbonatopløsning: 143,8 g vandfrit natriumcarbonat opløses i ca. 300 ml varmt vand. Opløsningen henstilles til afkøling.
3.9. Natriumthiosulfatopløsning, 0,1 mol/liter.
3.10. Stivelsesopløsning: 5 g opløselig stivelse tilsættes 30 ml vand og opblandes i 1 liter kogende vand. Der koges i 3 minutter, henstilles til afkøling og tilsættes eventuelt 10 mg kviksølviodid som konserveringsmiddel.
3.11. Svovlsyre, 3 mol/liter.
3.12. Kaliumiodid, 30 % opløsning (w/v).
3.13. Granuleret pimpsten udkogt i saltsyre, vasket med vand og tørret.
3.14. 3-methylbutan-l-ol.
4. Apparatur
Mekanisk rysteapparat: ca. 35–40 omdrejninger i minuttet.
5. Fremgangsmåde
5.1. Ekstraktion af prøven
2,5 g af prøven afvejes med 1 mg nøjagtighed og hældes i en 250 ml målekolbe. Der tilsættes 200 ml ethanol (3.1) og blandes i 1 time i rysteapparatet. Der tilsættes 5 ml Carrez-reagens I (3.2) og omrøres i ca. 30 sekunder. Derefter tilsættes 5 ml Carrez-reagens II (3.3) og omrøres på ny i 1 minut. Der fyldes op til mærket med ethanol (3.1), hvorefter der homogeniseres og filtreres. 200 ml af filtratet udtages og inddampes til ca. halvdelen af volumenet, hvorved størstedelen af ethanolen fordamper. Fordampningsresten overføres kvantitativt ved hjælp af varmt vand til en 200 ml målekolbe og afkøles, der fyldes op til mærket med vand og homogeniseres, og om nødvendigt filtreres. Denne opløsning benyttes til bestemmelse af reducerende sukker samt, efter invertering, af totalsukker.
5.2. Bestemmelse af reducerende sukker
Med pipette udtages højst 25 ml af opløsningen indeholdende under 60 mg reducerende sukker, udtrykt som glucose. Om nødvendigt fyldes op med destilleret vand til 25 ml, og indholdet af reducerende sukker bestemmes efter Luff-Schoorl-metoden. Resultatet angives i procent glucose.
5.3. Bestemmelse af totalsukker efter invertering
Med pipette udtages 50 ml opløsning, som overføres til en 100 ml målekolbe. Der tilsættes nogle få dråber methylorangeopløsning (3.4) og derefter, forsigtigt og under stadig omrøring, saltsyre (3.5), indtil der sker et tydeligt omslag til rødt. Der tilsættes 15 ml saltsyre (3.6), og kolben anbringes på vandbad i stærk kogning og efterlades der i 30 minutter. Der afkøles hurtigt til ca. 20 oC og tilsættes 15 ml natriumhydroxidopløsning (3.7). Der fyldes op med vand til 100 ml og homogeniseres. Der udtages en mængde, der ikke overstiger 25 ml, og som indeholder mindre end 60 mg reducerende sukker, udtrykt som glucose. Om nødvendigt fyldes op med destilleret vand til 25 ml, og indholdet af reducerende sukker bestemmes efter Luff-Schoorl-metoden. Resultatet angives i procent glucose eller, ved multiplikation med faktoren 0,95 , saccharose.
5.4. Titrering efter Luff-Schoorl-metoden
Med pipette udtages 25 ml af Luff-Schoorl-reagenset (3.8), som overføres til en 300 ml erlenmeyerkolbe; der tilsættes nøjagtig 25 ml af den klarede sukkeropløsning. Der tilsættes 2 korn pimpsten (3.13) og opvarmes under omrystning med hånden over en fri flamme af middelhøjde, og væsken bringes i kog på ca. 2 minutter. Erlenmeyerkolben anbringes umiddelbart derefter på en metaltrådsdug, som er forsynet med et asbesttrådnet med et hul på ca. 6 cm i diameter, hvorunder man i forvejen har tændt en flamme. Denne skal være afpasset således, at alene erlenmeyerkolbens bund opvarmes. Kolben forbindes med en tilbagesvaler. Væsken koges i nøjagtig 10 minutter og afkøles straks derefter ved hjælp af koldt vand, hvorpå der efter ca. 5 minutters forløb titreres som følger:
Der tilsættes 10 ml kaliumiodidopløsning (3.12) og straks derefter (forsigtigt på grund af risikoen for dannelse af skum i stor mængde) 25 ml svovlsyre (3.11). Der titreres så med natriumthiosulfatopløsning (3.9), indtil der viser sig en matgul farve, hvorefter der tilsættes stivelse (3.10) som indikator, og titreringen færdiggøres.
Den samme titrering foretages med en nøjagtigt afmålt blanding af 25 ml Luff-Schoorl-reagens (3.8) og 25 ml vand, efter at der er tilsat 10 ml kaliumiodidopløsning (3.12) og 25 ml svovlsyre (3.11), dog uden at bringe den i kog.
6. Beregning af resultater
Ved hjælp af tabellen beregnes den mængde glucose i mg, der svarer til forskellen mellem resultaterne af de to titreringer, udtrykt i mg natriumthiosulfat 0,1 mol/liter. Resultatet angives i procent af prøven.
7. Specielle fremgangsmåder
7.1. For så vidt angår foder med et højt melasseindhold og andet lidet homogent foder afvejes 20 g, som anbringes i en 1 liter målekolbe med 500 ml vand. Blandes i 1 time i rysteapparatet og klares ved hjælp af Carrez-reagens I (3.2) og II (3.3), som beskrevet i punkt 5.1, idet der dog benyttes en fire gange større mængde af hvert reagens. Der fyldes op til mærket med 80 % ethanol (v/v).
Derefter homogeniseres og filtreres. Ethanolen afdampes som beskrevet i punkt 5.1. Findes der ikke dekstrineret stivelse, fyldes der op med destilleret vand.
7.2. For så vidt angår melasse og fodermidler med et højt indhold af sukker, men praktisk talt ingen stivelse (johannesbrød, tørrede sukkerroesnitter osv.) afvejes 5 g i en 250 ml målekolbe, hvorefter der tilsættes 200 ml destilleret vand og blandes i 1 time eller mere, om nødvendigt, i rysteapparatet. Blandingen klares ved hjælp af Carrez-reagens I (3.2) og II (3.3), som beskrevet i punkt 5.1. Der fyldes op til mærket med koldt vand, homogeniseres og filtreres. Til bestemmelse af totalsukker benyttes fremgangsmåden, der er beskrevet under punkt 5.3.
8. Bemærkninger
8.1. Det anbefales at tilsætte ca. 1 ml 3-methylbutan-l-ol (3.14) (uden hensyntagen til volumenet) før kogning med Luff-Schoorl-reagenset for at undgå skumdannelse.
8.2. Forskellen mellem indholdet af totalsukker efter invertering, udtrykt som glucose, og indholdet af reducerende sukker, udtrykt som glucose, giver, multipliceret med 0,95 , procentindholdet af saccharose.
8.3. Til bestemmelse af indholdet af reducerende sukker, med undtagelse af lactose, kan der anvendes to metoder:
8.3.1. For at foretage en omtrentlig beregning multipliceres med 0,675 det ved en separat bestemmelse konstaterede indhold af lactose, og det fundne resultat trækkes fra indholdet af reducerende sukker.
8.3.2. For at foretage en præcis beregning af det reducerende sukker, med undtagelse af lactosen, er det nødvendigt at lægge den samme analyseprøve til grund ved de to definitive bestemmelser. Den ene analyse udføres på en del af opløsningen, der opnås efter punkt 5.1, den anden på en del af opløsningen, der opnås ved bestemmelsen af lactose efter den hertil fastlagte metode (efter gæring af de andre sukkerarter og klaring).
I begge tilfælde bestemmes det tilstedeværende indhold af sukker efter Luff-Schoorl-metoden og beregnes i mg glucose. De to værdier trækkes fra hinanden, og forskellen angives i procent af prøven.
Eksempel
De to udtagne volumener svarer for hver bestemmelse til en analyseprøve på 250 mg.
I det første tilfælde forbruges der 17 ml natriumthiosulfatopløsning 0,1 mol/liter, hvad der svarer til 44,2 mg glucose; i det andet tilfælde forbruges der 11 ml, hvad der svarer til 27,6 mg glucose.
Forskellen andrager 16,6 mg glucose.
Indholdet af reducerende sukker (uden lactose) beregnet som glucose er altså:
![]()
Tabel over værdierne for 25 ml Luff-Schoorl-reagens
ml Na2S2O30,1 mol/liter, 2 minutters opvarmning, 10 minutters kogning
|
Na2S2O3 0,1 mol/liter |
Glucose, fructose, inverteret sukker C6H12O6 |
Lactose C12H22O11 |
Maltose C12H22O11 |
Na2S2O3 0,1 mol/liter |
|||
|
ml |
mg |
forskel |
mg |
forskel |
mg |
forskel |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. BESTEMMELSE AF LACTOSE
1. Formål og anvendelsesområde
Metoden gør det muligt at bestemme indholdet af lactose i foder, der indeholder mere end 0,5 % heraf.
2. Princip
Sukkeret opløses i vand. Opløsningen underkastes en gæring med Saccharomyces cerevisiae, som ikke angriber lactosen. Efter klaring og filtrering bestemmes filtratets lactoseindhold efter Luff-Schoorl-metoden.
3. Reagenser
3.1. Saccharomyces cerevisiae-opslæmning: 25 g frisk gær opslæmmes i 100 ml vand. Suspensionen kan holde sig i højst 1 uge i køleskab.
3.2. Carrez-reagens I: 21,9 g zinkacetat, Zn (CH3COO)2.2H2O, og 3 g iseddike opløses i vand. Der fyldes op med vand til 100 ml.
3.3. Carrez-reagens II: 10,6 g kaliumferrocyanid, K4Fe(CN)6.3H2O, opløses i vand. Der fyldes op med vand til 100 ml.
3.4. Luff-Schoorl-reagens:
Under forsigtig omrøring hældes citronsyreopløsningen (3.4.2) over i natriumcarbonatopløsningen (3.4.3). Kobbersulfatopløsningen (3.4.1) tilsættes, og der fyldes op med vand til 1 liter. Det henstår natten over og filtreres. Koncentrationen af det således opnåede reagens kontrolleres (Cu 0,05 mol/liter; Na2CO3 1 mol/liter). Opløsningens pH-værdi skal være ca. 9,4 .
3.4.1. Kobbersulfatopløsning: 25 g kobbersulfat, CuSO4.5H2O, frit for jern, opløses i 100 ml vand.
3.4.2. Citronsyreopløsning: 50 g citronsyre, C6H8O7.H2O, opløses i 50 ml vand.
3.4.3. Natriumcarbonatopløsning: 143,8 g vandfrit natriumcarbonat opløses i ca. 300 ml varmt vand. Opløsningen henstilles til afkøling.
3.5. Granuleret pimpsten udkogt i saltsyre, vasket med vand og tørret.
3.6. Kaliumiodid, 30 % opløsning (w/v).
3.7. Svovlsyre, 3 mol/liter.
3.8. Natriumthiosulfatopløsning, 0,1 mol/liter.
3.9. Stivelsesopløsning: 5 g opløselig stivelse tilsættes 30 ml vand og opblandes i 1 liter kogende vand. Der koges i 3 minutter, henstilles til afkøling og tilsættes eventuelt 10 mg kviksølviodid som konserveringsmiddel.
4. Apparatur
Vandbad forsynet med termostat, indstillet til 38–40 oC.
5. Fremgangsmåde
1 g af prøven afvejes med 1 mg nøjagtighed, og denne portion anbringes i en 100 ml målekolbe. Der tilsættes 25–30 ml vand. Kolben opvarmes i 30 minutter i kogende vandbad, hvorefter der afkøles til ca. 35 oC. Der tilsættes 5 ml gærsuspension (3.1) og homogeniseres. Kolben henstår i 2 timer i vandbad ved en temperatur på 38–40 oC. Afkøles derpå til ca. 20 oC.
Der tilsættes 2,5 ml Carrez-reagens I (3.2), og blandingen rystes i 30 sekunder; dernæst tilsættes 2,5 ml Carrez-reagens II (3.3), og der rystes på ny i 30 sekunder. Der fyldes op med vand til 100 ml, blandes og filtreres. Med pipette udtages en mængde filtrat, der ikke overstiger 25 ml, og som så vidt muligt indeholder 40–80 mg lactose, og dette overføres til en 300 ml erlenmeyerkolbe. Om nødvendigt fyldes op med vand til 25 ml.
På samme måde udføres en blindprøve med 5 ml gærsuspension (3.1). Lactoseindholdet bestemmes efter Luff-Schoorl-metoden som følger: Der tilsættes nøjagtig 25 ml Luff-Schoorl-reagens (3.4) og 2 korn pimpsten (3.5). Der opvarmes under omrystning med hånden over en fri flamme af middelhøjde, og væsken bringes i kog på ca. 2 minutter. Erlenmeyerkolben anbringes umiddelbart derefter på en metaltrådsdug, som er forsynet med et asbesttrådnet med et hul på ca. 6 cm i diameter, hvorunder man i forvejen har tændt en flamme. Denne skal være afpasset således, at alene erlenmeyerkolbens bund opvarmes. Kolben forbindes med en tilbagesvaler. Væsken koges i nøjagtig 10 minutter. Derpå afkøles der straks ved hjælp af koldt vand, og efter ca. 5 minutters forløb titreres der som følger:
Der tilsættes 10 ml kaliumiodidopløsning (3.6) og straks derefter (forsigtigt på grund af risikoen for dannelse af skum i stor mængde) 25 ml svovlsyre (3.7). Der titreres så med natriumthiosulfatopløsning (3.8), indtil der viser sig en matgul farve, hvorefter der tilsættes stivelse (3.9) som indikator, og titreringen færdiggøres.
Den samme titrering foretages med en nøjagtigt afmålt blanding af 25 ml Luff-Schoorl-reagens (3.4) og 25 ml vand, efter at der er tilsat 10 ml kaliumiodidopløsning (3.6) og 25 ml svovlsyre (3.7), dog uden at bringe den i kog.
6. Beregning af resultater
Ved hjælp af nedenstående tabel beregnes den mængde lactose i mg, der svarer til forskellen mellem resultaterne af de to titreringer, udtrykt i ml natriumthiosulfatopløsning 0,1 mol/liter.
Resultatet for vandfri lactose angives i procent af prøven.
7. Bemærkning
Til produkter, der indeholder mere end 40 % gæringsdygtigt sukker, skal der benyttes mere end 5 ml gærsuspension (3.1).
Tabel over værdierne for 25 ml Luff-Schoorl-reagens
ml Na2S2O30,1 mol/liter, 2 minutters opvarmning, 10 minutters kogning
|
Na2S2O3 0,1 mol/liter |
Glucose, fructose, inverteret sukker C6H12O6 |
Lactose C12H22O11 |
Maltose C12H22O11 |
Na2S2O3 0,1 mol/liter |
|||
|
ml |
mg |
forskel |
mg |
forskel |
mg |
forskel |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. BESTEMMELSE AF STIVELSE
POLARIMETRISK METODE
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af stivelse og nedbrydningsprodukter heraf med høj molekylvægt i foder til kontrol af, om reglerne vedrørende angivelse af energiværdien (jf. bilag VII) og direktiv 96/25/EF ( 29 ) er overholdt.
2. Princip
Metoden er baseret på dobbeltbestemmelse. Ved den første bestemmelse behandles prøven med fortyndet saltsyre. Efter klaring og filtrering måles opløsningens optiske drejning polarimetrisk.
Ved den anden bestemmelse ekstraheres prøven med 40 % ethanol. Efter behandling af filtratet med saltsyre klares og filtreres, og den optiske drejning måles under samme betingelser som ved den første bestemmelse.
Forskellen mellem de to målinger, multipliceret med en kendt faktor, giver prøvens stivelsesindhold.
3. Reagenser
3.1. Saltsyre, 25 % opløsning (w/w), massefylde: 1,126 g/ml.
3.2. Saltsyre, 1,13 % opløsning (w/v)
Koncentrationen skal kontrolleres ved titrering med 0,1 mol/liter natriumhydroxidopløsning under tilstedeværelse af 0,1 % (w/v) methylrødt i 94 % (v/v) ethanol. Til neutralisering af 10 ml kræves der 30,94 ml NaOH 0,1 mol/liter.
3.3. Carrez-reagens I: 21,9 g zinkacetat, Zn (CH3COO)2.2H2O, og 3 g iseddike opløses i vand. Der fyldes op med vand til 100 ml.
3.4. Carrez-reagens II: 10,6 g kaliumferrocyanid, K4Fe(CN)6.3H2O, opløses i vand. Der fyldes op med vand til 100 ml.
3.5. Ethanol, 40 % opløsning (v/v), massefylde: 0,948 g/ml ved 20 oC.
4. Apparatur
4.1. 250 ml erlenmeyerkolbe med standardslibhals og tilbagesvaler
4.2. Polarimeter eller saccharimeter.
5. Fremgangsmåde
5.1. Klargøring af prøven
Prøven formales, så den kan passere fuldstændigt gennem en sigte med 0,5 mm runde masker.
5.2. Bestemmelse af den totale optiske drejning (P eller S) (se bemærkning 7.1)
2,5 g af den formalede prøve afvejes med 1 mg nøjagtighed i en 100 ml målekolbe, og der tilsættes 25 ml saltsyre (3.2). Kolben rystes, indtil prøvematerialet er jævnt fordelt, og der tilsættes yderligere 25 ml saltsyre (3.2). Derefter nedsænkes kolben i et kogende vandbad. I de første 3 minutter rystes kolben kraftigt og med regelmæssige mellemrum for at forhindre klumpdannelse. Vandmængden i vandbadet skal være tilstrækkelig til fortsat at holde vandet i kog, når kolben nedsænkes. Under omrystningen må kolben ikke tages op af vandbadet. Efter nøjagtig 15 minutter tages kolben op af vandbadet, hvorefter der tilsættes 30 ml koldt vand og straks afkøles til 20 oC.
Der tilsættes 5 ml Carrez-reagens I (3.3) og omrystes i ca. 30 sekunder. Derpå tilsættes 5 ml Carrez-reagens II (3.4), og der omrystes atter i ca. 30 sekunder. Der fyldes op med vand til mærket, blandes og filtreres: Hvis filtratet ikke er fuldstændig klart, hvilket sjældent forekommer, skal bestemmelsen gentages med en større mængde Carrez-reagens I og II, f.eks. 10 ml.
Opløsningens optiske drejning måles i et 200 mm rør med et polarimeter eller et saccharimeter.
5.3. Bestemmelse af den optiske drejning (P' eller S') for de i 40 % ethanol opløselige stoffer
5 g af prøven afvejes med 1 mg nøjagtighed i en 100 ml målekolbe, og der tilsættes ca. 80 ml ethanol (3.5) (se bemærkning 7.2). Kolben henstår i 1 time ved rumtemperatur. I dette tidsrum rystes kolben kraftigt seks gange, så prøvematerialet blandes omhyggeligt med ethanolen. Der fyldes op med ethanol (3.5) til mærket, blandes og filtreres.
50 ml af filtratet (svarende til 2,5 g af prøven) afpipetteres i en 250 ml erlenmeyerkolbe, og der tilsættes 2,1 ml saltsyre (3.1). Kolben rystes kraftigt, sluttes til en tilbagesvaler og nedsænkes i et bad med kogende vand. Efter nøjagtig 15 minutter tages erlenmeyerkolben op af vandbadet, og indholdet skylles over i en 100 ml målekolbe med en lille mængde koldt vand og afkøles til 20 oC.
Derpå klares med Carrez-reagens I (3.3) og II (3.4), fyldes op med vand til mærket, blandes og filtreres, og den optiske drejning måles som beskrevet i punkt 5.2, andet og tredje afsnit.
6. Beregning af resultater
Indholdet af stivelse ( %) beregnes som følger:
6.1. Polarimetriske målinger
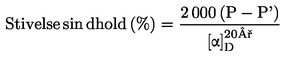
|
P |
= |
total optisk drejning i cirkelgrader |
|
P' |
= |
optisk drejning i cirkelgrader af de i 40 % (v/v) ethanol opløselige stoffer |
|
|
= |
den rene stivelses specifikke optiske drejning. Til denne faktor D anvendes følgende generelt accepterede værdier:
|
6.2. Saccharimetriske målinger
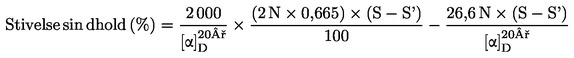
|
S |
= |
total optisk drejning i saccharimetergrader |
|
S' |
= |
optisk drejning i saccharimetergrader af de i 40 % (v/v) ethanol opløselige stoffer |
|
N |
= |
saccharosens vægt i gram i 100 ml vand ved en vejlængde på 200 mm, som giver en optisk drejning på 100 saccharimetergrader. Denne vægt varierer alt efter saccharimetertype: 16,29 g for franske saccharimetre 26,00 g for tyske saccharimetre 20,00 g for blandede saccharimetre. |
|
|
= |
den rene stivelses specifikke optiske drejning (se punkt 6.1). |
6.3. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ved indhold på under 40 % stivelse ikke overstige 0,4 % i absolut værdi og ved indhold på 40 % og derover ikke overstige 1 % i relativ værdi.
7. Bemærkninger
7.1. Hvis prøven indeholder over 6 % carbonater, beregnet som calciumcarbonat, skal de destrueres med den nøjagtige mængde fortyndet svovlsyre inden bestemmelsen af den totale optiske drejning.
7.2. For produkter med højt lactoseindhold, f.eks. mælkeserum i pulverform eller skummetmælkspulver, anvendes efter tilsætning af 80 ml ethanol (3.5) følgende fremgangsmåde: Kolben tilsluttes en tilbagesvaler og nedsænkes i et vandbad på 50 oC i 30 minutter. Efter afkøling fortsættes som beskrevet i punkt 5.3.
7.3. Ved tilstedeværelse i foder i større mængde kan følgende fodermidler interferere ved bestemmelse af stivelsesindholdet efter den polarimetriske metode, som derved kan give forkerte resultater:
— (sukker)roeprodukter, såsom ekstraherede (sukker)roesnitter, (sukker)roemelasse, ekstraherede (sukker)roesnitter tilsat melasse, (sukker)roevinasse, (roe)sukker
— citruskvas
— hørfrø, hørfrøkage, hørfrøskrå
— rapsfrø, rapskage, rapsskrå, rapsskaller
— solsikkefrø, solsikkeskrå, solsikkeskrå, delvis afskallet
— kokoskage, kokosskrå
— kartoffelkvas
— tørret gær
— produkter med stort inulinindhold (f.eks. snitter og skrå af jordskokker)
— grever.
M. BESTEMMELSE AF RÅASKE
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af råaske i foder.
2. Princip
Prøven foraskes ved 550 oC; remanensen vejes.
3. Reagenser
Ammoniumnitrat, 20 % opløsning (w/v).
4. Apparatur
4.1. Varmeplade
4.2. Elektrisk muffelovn med termostat
4.3. Foraskningsdigler af kvarts, porcelæn eller platin, rektangulære (ca. 60 × 40 × 25 mm) eller runde (diameter: 60–75 mm, højde: 20–40 mm).
5. Fremgangsmåde
Ca. 5 g af prøven afvejes med 1 mg nøjagtighed (2,5 g for produkter, der har tendens til at svulme op) i en foraskningsdigel, der i forvejen er opvarmet til 550 oC, afkølet og tareret. Diglen anbringes på varmepladen og opvarmes gradvist, indtil stoffet forkuller. Der foraskes som beskrevet i punkt 5.1 eller 5.2.
5.1. Diglen stilles ind i den kalibrerede muffelovn, der er indstillet på 550 oC. Den henstår ved denne temperatur, indtil der fås hvid, lysegrå eller rødlig aske, der tilsyneladende er fri for kulpartikler. Diglen anbringes i en ekssikkator, henstilles til afkøling og vejes med det samme.
5.2. Diglen stilles ind i den kalibrerede muffelovn, der er indstillet på 550 oC. Der foraskes i 3 timer. Diglen anbringes i en ekssikkator, henstilles til afkøling og vejes med det samme. Der foraskes i yderligere 30 minutter for at sikre, at askens vægt forbliver konstant (vægttabet mellem to på hinanden følgende vejninger må højst være på 1 mg).
6. Beregning af resultater
Vægten af remanensen beregnes, idet taraen trækkes fra.
Resultatet angives i procent af prøven.
7. Bemærkninger
7.1. Asken af de stoffer, der er vanskelige at foraske, skal underkastes en første foraskning på mindst 3 timer, afkøles og tilsættes nogle få dråber 20 % ammoniumnitratopløsning eller vand (dog forsigtigt for at undgå, at asken spredes eller danner klumper). Foraskningen fortsættes efter tørring i tørreskab. Gentag processen, indtil foraskningen er fuldstændig.
7.2. For stoffer, der ikke kan behandles efter den i punkt 7.1. beskrevne metode, benyttes følgende fremgangsmåde: Efter 3 timers foraskning bringes asken ved hjælp af varmt vand over på et lille askefrit filter. Filtret med indhold foraskes i den oprindelige digel. Filtratet anbringes i den afkølede digel, inddampes til tørhed, foraskes og vejes.
7.3. Når det drejer sig om fedtstoffer, afvejes med størst mulig nøjagtighed en prøve på 25 g i en digel af passende størrelse. Prøven forkulles ved at antænde stoffet ved hjælp af en lunte af askefrit filtrerpapir. Fugtes efter forbrændingen med den mindste mængde vand, der er nødvendig. Tørres og foraskes som beskrevet i punkt 5.
N. BESTEMMELSE AF ASKE UOPLØSELIG I SALTSYRE
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet i foder af mineralske bestanddele, som er uopløselige i saltsyre. Der er fastsat to fremgangsmåder alt afhængigt af prøvens art.
1.1. Fremgangsmåde A: finder anvendelse på organiske fodermidler og på de fleste typer foderblandinger.
1.2. Fremgangsmåde B: finder anvendelse på mineralstoffer og mineralstofblandinger såvel som på foderblandinger, hvis indhold af aske uopløselig i saltsyre, bestemt efter fremgangsmåde A, er større end 1 %.
2. Princip
2.1. Fremgangsmåde A: Prøven foraskes, asken koges med saltsyre, og den uopløselige remanens frafiltreres og vejes.
2.2. Fremgangsmåde B: Prøven behandles med saltsyre. Opløsningen filtreres, remanensen foraskes, og den fundne aske behandles som beskrevet under fremgangsmåde A.
3. Reagenser
3.1. Saltsyre, 3 mol/liter
3.2. Trichloreddikesyre, 20 % opløsning (w/v)
3.3. Trichloreddikesyre, 1 % opløsning (w/v).
4. Apparatur
4.1. Varmeplade
4.2. Elektrisk muffelovn med termostat
4.3. Foraskningsdigler af kvarts, porcelæn eller platin, rektangulære (ca. 60 × 40 × 25 mm) eller runde (diameter: 60–75 mm, højde: 20–40 mm).
5. Fremgangsmåde
5.1. Fremgangsmåde A
Prøven foraskes efter den fremgangsmåde, der er beskrevet for bestemmelse af råaske. Man kan også benytte den aske, der opnås ved denne bestemmelse.
Asken anbringes i et 250–400 ml bægerglas med 75 ml saltsyre (3.1). Væsken bringes forsigtigt til kogning og koges sagte i 15 minutter. Den varme opløsning filtreres gennem et askefrit filtrerpapir, og remanensen vaskes med varmt vand, indtil den sure reaktion forsvinder. Filtret med remanensen tørres, og der foraskes i en tareret digel ved en temperatur på mindst 550 oC og højst 700 oC. Afkøles i en ekssikkator og vejes.
5.2. Fremgangsmåde B
5 g af prøven afvejes med 1 mg nøjagtighed i et 250–400 ml bægerglas. Der tilsættes 25 ml vand og derefter 25 ml saltsyre (3.1), blandes og ventes, indtil opbrusningen ophører. Der tilsættes yderligere 50 ml saltsyre (3.1). Det afventes, at en eventuel luftudvikling er overstået, hvorefter bægerglasset anbringes på kogende vandbad og efterlades dér i 30 minutter eller mere, om nødvendigt, så eventuelt forekommende stivelse hydrolyseres fuldstændigt. Der varmfiltreres gennem et askefrit filter, og filtret vaskes med 50 ml varmt vand (se punkt 7, »Bemærkning«). Filtret med remanensen anbringes i en foraskningsdigel, og der tørres og foraskes ved en temperatur på mindst 550 oC og højst 700 oC. Asken anbringes i et 250–400 ml bægerglas med 75 ml saltsyre (3.1). Derefter fortsættes som beskrevet i punkt 5, andet afsnit.
6. Beregning af resultater
Vægten af remanensen beregnes, idet taraen trækkes fra. Resultatet angives i procent af prøven.
7. Bemærkning
Hvis filtreringen viser sig vanskelig, gentages bestemmelsen, idet de 50 ml saltsyre (3.1) erstattes med 50 ml trichloreddikesyreopløsning på 20 % (3.2), og idet filtret vaskes med en varm trichloreddikesyreopløsning på 1 % (3.3).
O. BESTEMMELSE AF CARBONATER
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af carbonater, der normalt udtrykkes som calciumcarbonat, i de fleste foderstoffer.
I visse tilfælde (f.eks. ferrocarbonat) må der dog anvendes en særlig metode.
2. Princip
Carbonaterne spaltes med saltsyre; den frigjorte kuldioxid opsamles i et måleglas, og dens volumen sammenlignes med det volumen, der under de samme betingelser frigøres af en bekendt mængde calciumcarbonat.
3. Reagenser
3.1. Saltsyre, massefylde: 1,10 g/ml
3.2. Calciumcarbonat
3.3. Svovlsyre ca. 0,05 mol/liter, farvet med methylrødt.
4. Apparatur
Scheibler-Dietrich-apparat (se skitse) eller tilsvarende apparat.
5. Fremgangsmåde
Alt efter prøvens carbonatindhold afvejes en prøve som angivet nedenfor:
— 0,5 g for produkter, der indeholder 50–100 % carbonater, udtrykt som calciumcarbonat
— 1 g for produkter, der indeholder 40–50 % carbonater, udtrykt som calciumcarbonat
— 2–3 g for øvrige produkter.
Prøven anbringes i apparatets specialflaske (4), der er forsynet med et lille rør af brudsikkert materiale, som indeholder 10 ml saltsyre (3.1), og flasken forbindes med apparatet. Tregangshanen (5) drejes, således at røret (1) står i forbindelse med luften udenfor. Ved hjælp af det bevægelige rør (2), som er fyldt med farvet svovlsyre (3.3) og forbundet med det gradinddelte rør (1), bringes væskens niveau til nulpunktet på skalaen. Hanen (5) drejes, således at rørene (1) og (3) kommer i forbindelse med hinanden, og det kontrolleres, at væskeniveauet er på nulpunktet.
Saltsyren (3.1) lades langsomt flyde hen over prøven, idet flasken (4) hældes. Trykket udlignes ved at sænke røret (2). Flasken (4) rystes, indtil kuldioxidudviklingen er helt ophørt.
Trykket genoprettes ved at føre væsken tilbage til det samme niveau i rørene (1) og (2). Der aflæses efter få minutters forløb, når volumenet er blevet konstant.
Under de samme betingelser udføres et sammenlignende forsøg med 0,5 g calciumcarbonat (3.2).
6. Beregning af resultater
Indholdet af carbonater, udtrykt som calciumcarbonat, beregnes efter følgende formel:
![]()
hvor:
|
X |
= |
% (w/w) carbonater i prøven, udtrykt som calciumcarbonat |
|
V |
= |
ml CO2 frigjort af prøven |
|
V1 |
= |
ml CO2 frigjort af 0,5 g CaCO3 |
|
m |
= |
prøvens vægt i gram. |
7. Bemærkninger
7.1. Når prøven vejer mere end 2 g, fyldes der forinden 15 ml destilleret vand i flasken (4), og indholdet blandes, før forsøget påbegyndes. Der benyttes samme vandvolumen til det sammenlignende forsøg.
7.2. Anvendes et apparat med et andet volumen end Scheibler-Dietrich-apparatet, må man afpasse analyseprøven, sammenligningsmængden og resultatberegningen herefter.
P. BESTEMMELSE AF TOTALPHOSPHOR
FOTOMETRISK METODE
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af totalphosphor i foder. Metoden er specielt egnet til undersøgelse af foder med et ringe phosphorindhold. I bestemte tilfælde (phosphorrige produkter) kan der anvendes en gravimetrisk metode.
2. Princip
Prøven destrueres enten ved forbrænding (når der er tale om organisk foder) eller ved syreoplukning (mineralske stoffer og flydende foder) og opløses i syre. Opløsningen behandles med vanadatmolybdatreagenset. Ekstinktionen af den gulfarvede opløsning måles ved 430 nm i spektrofotometret.
3. Reagenser
3.1. Calciumcarbonat.
3.2. Saltsyre, ρ20 = 1,10 g/ml (ca. 6 mol/liter).
3.3. Salpetersyre, ρ20 = 1,045 g/ml.
3.4. Salpetersyre, ρ20 = 1,38 –1,42 g/ml.
3.5. Svovlsyre, ρ20 = 1,84 g/ml.
3.6. Vanadatmolybdatreagens: 200 ml ammoniumheptamolybdatopløsning (3.6.1) blandes med 200 ml ammoniummonovandatopløsning (3.6.2) og 134 ml salpetersyre (3.4) i en 1 liter målekolbe, hvorefter der fyldes op med vand til mærket.
3.6.1. Ammoniumheptamolybdatopløsning: 100 g ammoniumheptamolybdat, (NH4) 6Mo7O24.4H2O, opløses i varmt vand. Der tilsættes 10 ml ammoniakvand (massefylde: 0,91 g/ml) og fyldes op med vand til 1 liter.
3.6.2. Ammoniummonovanadatopløsning: 2,35 g ammoniummonovanadat, NH4VO3, opløses i 400 ml varmt vand. 20 ml fortyndet salpetersyre (7 ml HNO3 (3.4) + 13 ml H2O) tilsættes langsomt under stadig omrøring, og der fyldes op med vand til 1 liter.
3.7. Standardphosphoropløsning på 1 mg pr. ml: 4,387 g kaliumdihydrogenphosphat, KH2PO4, opløses i vand, og der fyldes op med vand til 1 liter.
4. Apparatur
4.1. Foraskningsdigler af kvarts, porcelæn eller platin
4.2. Elektrisk muffelovn med termostat indstillet på 550 oC
4.3. 250 ml Kjeldahlkolbe
4.4. Målekolber og mikromålepipetter
4.5. Spektrofotometer
4.6. Reagensglas, ca. 16 mm diameter med NS 14,5 ; rumindhold: 25–30 ml.
5. Fremgangsmåde
5.1. Fremstilling af opløsningen
Alt efter prøvens art fremstilles opløsningen som beskrevet i punkt 5.1.1 eller 5.1.2.
5.1.1.
1 g eller mere af prøven afvejes med 1 mg nøjagtighed og hældes i en Kjeldahlkolbe. Der tilsættes 20 ml svovlsyre (3.5), hvorefter der omrystes for at gennemvæde materialet med syren og for at forhindre, at materialet sætter sig fast på kolbens inderside. Derpå opvarmes blandingen og holdes i kog i 10 minutter. Efter en let afkøling tilsættes der 2 ml salpetersyre (3.4); der opvarmes svagt og afkøles atter noget. Herefter tilsættes på ny lidt salpetersyre (3.4) og opvarmes til kogetemperatur, og dette gentages, indtil opløsningen er farveløs. Derpå afkøles der; der tilsættes lidt vand, og væsken overføres kvantitativt til en 500 ml målekolbe ved skylning af Kjeldahlkolben med varmt vand. Efter afkøling fyldes der op med vand til mærket, homogeniseres og filtreres.
5.1.2.
Ca. 2,5 g af prøven afvejes med 1 mg nøjagtighed i en foraskningsdigel og blandes omhyggeligt med 1 g calciumcarbonat (3.1). I muffelovnen foraskes der ved 550 oC, indtil asken er hvid eller grå (små mængder kul er ikke et problem). Asken overføres til et 250 ml bægerglas. Der tilsættes 20 ml vand og saltsyre (3.2), indtil opbrusningen ophører. Der tilsættes yderligere 10 ml saltsyre (3.2). Derpå anbringes bægerglasset på sandbad, og der inddampes til fuldstændig tørhed for at gøre kiselsyren uopløselig. Remanensen opløses i 10 ml salpetersyre (3.3) og opvarmes til kogetemperatur i 5 minutter på sandbad eller varmeplade, idet remanensen dog ikke skal tørre helt. Væsken overføres til en 500 ml målekolbe, idet bægerglasset gentagne gange skylles med varmt vand. Efter afkøling fyldes der op med vand til mærket, homogeniseres og filtreres.
5.2. Udvikling af farvningen og måling af ekstinktionen
En alikvot del af det efter 5.1.1 eller 5.1.2 udvundne filtrat fortyndes for at få en koncentration af phosphor på højst 40 μg/ml. 10 ml af denne opløsning overføres til et reagensglas (4.6), og der tilsættes 10 ml vanadatmolybdatreagens (3.6). Blandingen homogeniseres og henstår i mindst 10 minutter ved 20 oC. Derpå måles ekstinktionen i spektrofotometret ved 430 nm i forhold til en opløsning af 10 ml vand og 10 ml vanadatmolybdatreagens (3.6).
5.3. Kalibreringskurve
Af standardopløsningen (3.7) fremstilles opløsninger, der indeholder henholdsvis 5, 10, 20, 30 og 40 μg phosphor pr. ml. Til hver 10 ml af disse opløsninger tilsættes 10 ml vanadatmolybdatreagens (3.6). Der homogeniseres, og blandingen henstår i mindst 10 minutter ved 20 oC. Derpå måles ekstinktionen under de ovenfor i punkt 5.2 beskrevne betingelser. Kalibreringskurven tegnes, idet ekstinktionsværdierne indtegnes op ad ordinaten, og den tilsvarende phosphormængde ud ad abscissen. Kurven er lineær for phosphorkoncentrationer på 0–40 μg/ml.
6. Beregning af resultater
Indholdet af phosphor i prøven bestemmes ved benyttelse af kalibreringskurven.
Resultatet angives i procent af prøven.
Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige:
— 3 % i relativ værdi (af det højeste resultat) for phosphorindhold på mindre end 5 %
— 0,15 % i absolut værdi for phosphorindhold på 5 % og derover.
Q. BESTEMMELSE AF CHLOR AF CHLORIDER
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af chlor af vandopløselige chlorider, der normalt udtrykkes som natriumchlorid. Den kan anvendes på alt foder.
2. Princip
Chloriderne opløses i vand. Dersom produktet indeholder organisk stof, foretages en klaring. Opløsningen gøres let sur ved tilsætning af salpetersyre, og chloriderne bundfældes i form af sølvchlorid ved hjælp af en sølvnitratopløsning. Overskuddet af sølvnitrat titreres med en ammoniumthiocyanatopløsning efter Volhards metode.
3. Reagenser
3.1. Ammoniumthiocyanatopløsning, 0,1 mol/liter.
3.2. Sølvnitratopløsning, 0,1 mol/liter.
3.3. Mættet ammoniumferrisulfatopløsning (NH4)Fe(SO4)2.
3.4. Salpetersyre, massefylde: 1,38 g/ml.
3.5. Diethylether.
3.6. Acetone.
3.7. Carrez-reagens I: 21,9 g zinkacetat, Zn (CH3COO)2·2H2O, og 3 g iseddike opløses i vand. Der fyldes op med vand til 100 ml.
3.8. Carrez-reagens II: 10,6 g kaliumferrocyanid, K4Fe(CN)6 · 3H2O, opløses i vand. Der fyldes op med vand til 100 ml.
3.9. Aktivt kul, chloridfrit og ikke-chloridadsorberende.
4. Apparatur
Mekanisk rysteapparat: ca. 35–40 omdrejninger i minuttet.
5. Fremgangsmåde
5.1. Fremstilling af opløsningen
Alt efter prøvens art fremstilles en opløsning som beskrevet i punkt 5.1.1., 5.1.2 eller 5.1.3.
Der udføres til sammenligning en blindprøve uden analyseprøven.
5.1.1.
Der afvejes med 1 mg nøjagtighed en analyseprøve på ikke mere end 10 g, der ikke indeholder mere end 3 g chlor i form af chlorider, og denne anbringes i en 500 ml målekolbe med 400 ml vand på ca. 20 oC. Væsken blandes i 30 minutter i rysteapparatet, og der fyldes op til mærket, homogeniseres og filtreres.
5.1.2.
Ca. 5 g af prøven afvejes med 1 mg nøjagtighed og anbringes med 1 g aktivt kul i en 500 ml målekolbe. Der tilsættes 400 ml vand på ca. 20 oC og 5 ml Carrez-reagens I (3.7), omrøres i 30 sekunder og tilsættes derefter 5 ml Carrez-reagens II (3.8). Væsken blandes i 30 minutter i rysteapparatet, og der fyldes op til mærket, homogeniseres og filtreres.
5.1.3.
Opløsningen fremstilles som beskrevet i punkt 5.1.2, men filtreres ikke. Opløsningen dekanteres (om nødvendigt centrifugeres), med pipette udtages 100 ml af supernatanten, og dette overføres til en 200 ml målekolbe. Der blandes med acetone (3.6) og fyldes op til mærket med denne opløsningsvæske, hvorefter der homogeniseres og filtreres.
5.2. Titrering
Med pipette overføres til en erlenmeyerkolbe 25–100 ml af filtratet (alt efter det formodede chlorindhold), der er opnået efter punkt 5.1.1, 5.1.2 eller 5.1.3. Den alikvote mængde må ikke indeholde mere end 150 mg klor (Cl). Om nødvendigt fortyndes med vand op til mindst 50 ml, hvorefter der tilsættes 5 ml salpetersyre (3.4), 20 ml mættet ammoniumferrisulfatopløsning (3.3) og 2 dråber ammoniumthiocyanatopløsning (3.1), som påfyldes ved hjælp af en burette, der er fyldt til nulpunktstregen. Dernæst påfyldes ved hjælp af en burette sølvnitratopløsningen (3.2), således at der opnås et overskud på 5 ml. Der tilsættes 5 ml diethylether (3.5) og omrystes kraftigt for at sikre fuldstændig udfældning. Overskuddet af sølvnitrat titreres med ammoniumthiocyanatopløsning (3.1), indtil omslaget til den brunrøde farve har holdt sig i 1 minut.
6. Beregning af resultater
Mængden af chlor (X), udtrykt i % natriumchlorid, beregnes efter følgende formel:
![]()
hvor:
|
V1 |
= |
tilsatte ml sølvnitratopløsning 0,1 mol/liter |
|
V2 |
= |
ml ammoniumthiocyanatopløsning 0,1 mol/liter, forbrugt ved titreringen |
|
m |
= |
prøvens vægt. |
Hvis blindprøven viser et forbrug af sølvnitratopløsning 0,1 mol/liter, trækkes denne værdi fra volumenet (V1 - V2).
7. Bemærkninger
7.1. Titreringen kan også foregå ved potentiometri.
7.2. For meget fedtholdige produkter foretages der en forudgående affedtning med diethylether eller petroleumsether.
7.3. For så vidt angår fiskemel kan titreringen foretages efter Mohrs metode.
BILAG IV
ANALYSEMETODER TIL KONTROL AF INDHOLDET AF GODKENDTE TILSÆTNINGSSTOFFER I FODER
A. BESTEMMELSE AF VITAMIN A
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af vitamin A (retinol) i foder og forblandinger. Ved vitamin A forstås all-trans-retinylalkohol og dens cis-isomerer, som bestemmes ved denne metode. Indholdet af vitamin A udtrykkes i internationale enheder (IU) pr. kg. En IU svarer til aktiviteten af 0,300 μg all-trans-vitamin A-alkohol eller 0,344 μg all-trans-vitamin A-acetat eller 0,550 μg all-trans-vitamin A-palmitat.
Bestemmelsesgrænsen er 2 000 IU vitamin A pr. kg.
2. Princip
Prøven hydrolyseres med kaliumhydroxid opløst i ethanol, og vitamin A ekstraheres med petroleumsether. Opløsningsmidlet fjernes ved inddampning, og remanensen opløses i methanol og fortyndes om nødvendigt til den ønskede koncentration. Vitamin A-indholdet bestemmes ved omvendt-fase-HPLC med anvendelse af UV- eller fluorescensdetektor. Kromatograferingsparametrene vælges således, at all-trans-vitamin A-alkohol og dens cis-isomerer ikke adskilles.
3. Reagenser
3.1. Ethanol, σ = 96 %.
3.2. Petroleumsether, kogepunktsinterval: 40–60 oC.
3.3. Methanol.
3.4. Kaliumhydroxidopløsning, c = 50 g/100 ml.
3.5. Natriumascorbatopløsning, c = 10 g/100 ml (se bemærkning 7.7).
3.6. Natriumsulfid, Na2S· x H2O (x = 7–9).
3.6.1. Natriumsulfidopløsning, c = 0,5 mol/liter i glycerol, β = 120 g/liter (for x = 9) (se bemærkning 7.8).
3.7. Phenolphthaleinopløsning, c = 2 g/100 ml i ethanol (3.1).
3.8. 2-Propanol.
3.9. Mobil fase til HPLC: blanding af methanol (3,3 ) og vand, f.eks. 980 + 20 (v + v). Det nøjagtige forhold bestemmes af den anvendte kolonnes egenskaber.
3.10. Nitrogen, oxygenfrit.
3.11. All-trans-vitamin A-acetat, ekstra rent, med certificeret aktivitet, f.eks. 2,80 × 106 IU/g.
3.11.1. Stamopløsning af all-trans-vitamin A-acetat: Der afvejes med 0,1 mg nøjagtighed 50 mg vitamin A-acetat (3.11) i en 100 ml målekolbe. Efter opløsning i 2-propanol (3.8) fyldes der op til mærket med samme opløsningsmiddel. Denne opløsning har en nominel koncentration på 1 400 IU vitamin A pr. ml. Det nøjagtige indhold bestemmes efter punkt 5.6.3.1.
3.12. All-trans-vitamin A-palmitat, ekstra rent, med certificeret aktivitet, f.eks. 1,80 × 106 IU/g.
3.12.1. Stamopløsning af all-trans-vitamin A-palmitat: Der afvejes med 0,1 mg nøjagtighed 80 mg vitamin A-palmitat (3.12) i en 100 ml målekolbe. Efter opløsning i 2-propanol (3.8) fyldes der op til mærket med samme opløsningsmiddel. Denne opløsning har en nominel koncentration på 1 400 IU vitamin A pr. ml. Det nøjagtige indhold bestemmes efter punkt 5.6.3.2.
3.13. 2,6-Di-tert-butyl-4-methylphenol (BHT) (se bemærkning 7.5).
4. Apparatur
4.1. Rotationsfordamper
4.2. Udstyr af brunt glas
4.2.1. Fladbundede eller koniske kolber, 500 ml, med slib
4.2.2. Målekolber med slibprop og tynd hals, 10, 25, 100 og 500 ml
4.2.3. Skilletragte, koniske, 1 000 ml, med slibprop
4.2.4. Pærekolber, 250 ml, med slib
4.3. Allihn-svaler, kappelængde 300 mm, med slibhals, med adapter til gastilførsel
4.4. Foldefiltrerpapir til faseseparation, diameter: 185 mm (f.eks. Schleicher & Schüll 597 HY 1/2)
4.5. HPLC-udstyr med injektionssystem
4.5.1. HPLC-kolonne, 250 mm × 4 mm, C18, 5 eller 10 μm pakkemateriale, eller tilsvarende (præstationskriterium: kun én top for alle retinolisomerer under HPLC-betingelserne)
4.5.2. UV- eller fluorescensdetektor, med variabel bølgelængde
4.6. Spektrofotometer med 10 mm kvartskuvetter
4.7. Vandbad med magnetomrører
4.8. Ekstraktionsapparat (se figur 1) bestående af følgende:
4.8.1. Cylinderglas på 1 liter med slibhals og -prop
4.8.2. Slibindsats med sidearm og justerbart rør, der er ført igennem midten. Det justerbare rør skal være U-formet i den nedre ende og være tilspidset i den anden ende, således at det øverste væskelag i glasset kan overføres til en skilletragt.
5. Fremgangsmåde
|
NB |
Vitamin A er følsomt over for lys (UV) og oxidering. Alt arbejde udføres afskærmet mod lys (udstyr af brunt glas eller glasudstyr omviklet med aluminiumfolie) og oxygen (skylning med nitrogen). Under ekstraktionen udskiftes luften over væsken med nitrogen (overtryk udlignes ved jævnligt at lette på proppen). |
5.1. Klargøring af prøven
Prøven formales, så den kan passere gennem en sigte med 1 mm maskevidde, men varmeudvikling skal undgås. Formalingen foretages umiddelbart før afvejning og forsæbning, da der ellers kan gå vitamin A tabt.
5.2. Forsæbning
Afhængigt af vitamin A-indholdet afvejes der med en nøjagtighed på 1 mg 2–25 g af prøven i en 500 ml fladbundet eller konisk kolbe (4.2.1). Der tilsættes under forsigtig blanding i rækkefølge 130 ml ethanol (3.1), ca. 100 mg BHT (3.13), 2 ml natriumascorbatopløsning (3.5) og 2 ml natriumsulfidopløsning (3.6). Der sættes en svaler på kolben, som anbringes i vandbad med magnetomrører (4.7). Der opvarmes til kogning og koges med tilbagesvaling i 5 minutter. Dernæst tilsættes der 25 ml kaliumhydroxidopløsning (3.4) gennem svaleren (4.3), og der koges med tilbagesvaling i endnu 25 minutter under omrøring og under langsom nitrogengennemstrømning. Svaleren skylles med ca. 20 ml vand, og kolbens indhold afkøles til rumtemperatur.
5.3. Ekstraktion
Forsæbningsopløsningen overføres kvantitativt til en 1 000 ml skilletragt (4.2.3) eller ekstraktionsapparatet (4.8) ved skylning med i alt 250 ml vand. Derefter skylles forsæbningskolben først med 25 ml ethanol (3.1) og dernæst med 100 ml petroleumsether (3.2), idet skyllevæskerne overføres til skilletragten eller ekstraktionsapparatet. Forholdet mellem vand og ethanol i den samlede væskemængde skal være ca. 2:1. Der omrystes kraftigt i 2 minutter, og blandingen henstår til adskillelse i 2 minutter.
5.3.1.
Når væsken er skilt i to lag (se bemærkning 7.3), overføres petroleumsetherlaget til en anden skilletragt (4.2.3). Ekstraktionen gentages to gange med 100 ml petroleumsether (3.2) og derefter to gange med 50 ml petroleumsether (3.2).
De samlede ekstrakter i skilletragten vaskes to gange med 100 ml vand hver gang (ved forsigtig omdrejning, så der ikke dannes emulsion) og derefter flere gange ved rystning med 100 ml vand, indtil vandet ikke farves ved tilsætning af phenolphthaleinopløsning (3.7) (fire gange er normalt tilstrækkeligt). Det vaskede ekstrakt filtreres over i en 500 ml målekolbe (4.2.2) gennem et tørt foldefilter til faseseparation (4.4), således at eventuelt opslæmmet vand fjernes. Skilletragten og filtret skylles med 50 ml petroleumsether (3.2), og der fyldes op til mærket med petroleumsether (3.2) og blandes omhyggeligt.
5.3.2.
Når væsken er skilt i to lag (se bemærkning 7.3), udskiftes cylinderglassets (4.8.1) prop med slibindsatsen (4.8.2), og den nederste, U-formede del af det justerbare rør anbringes, så dets munding er lige over skillefladen. Ved at sætte nitrogentryk på sidearmen overføres det lette lag af petroleumsether til en 1 000 ml skilletragt (4.2.3). Der tilsættes 100 ml petroleumsether (3.2) til cylinderglasset, og det tilproppes og rystes omhyggeligt. Når væsken er skilt i to lag, overføres det lette lag til skilletragten som før. Ekstraktionen gentages med endnu 100 ml petroleumsether (3.2) og dernæst med 2 portioner 50 ml petroleumsether (3.2), og alle lagene af petroleumsether overføres til skilletragten.
De samlede petroleumsetherekstrakter i skilletragten vaskes som beskrevet i punkt 5.3.1, og der fortsættes som beskrevet der.
5.4. Fremstilling af prøveopløsning til HPLC-analyse
En alikvot mængde af petroleumsetherekstraktet (fra 5.3.1 eller 5.3.2) afpipetteres i en 250 ml pærekolbe (4.2.4). Der inddampes til næsten tørhed på rotationsfordamper (4.1) under reduceret tryk med en vandbadstemperatur på højst 40 oC. Trykket udlignes til atmosfæretryk, ved at der ledes nitrogen (3.10) ind i kolben, og kolben fjernes fra rotationsfordamperen. Resterende opløsningsmiddel fjernes under en nitrogenstrøm (3.10), og remanensen opløses straks i en kendt mængde (10–100 ml) methanol (3.3) (vitamin A-koncentrationen skal ligge i intervallet 5–30 IU/ml).
5.5. HPLC-bestemmelse
Separationen af vitamin A finder sted på en C18-kolonne med omvendt fase (4.5.1), og koncentrationen måles med en UV-detektor (325 nm) eller en fluorescensdetektor (excitation: 325 nm, emission: 475 nm) (4.5.2).
Der indsprøjtes en alikvot mængde (f.eks. 20 μl) af methanolopløsningen fra punkt 5.4, og der elueres med den mobile fase (3.9). Middeltophøjden (-arealet) for flere indsprøjtninger af samme prøveopløsning og middeltophøjderne (-arealerne) for flere indsprøjtninger af kalibreringsopløsningerne (5.6.2) beregnes.
Nedenstående betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater.
|
HPLC-kolonne (4.5.1): |
250 mm × 4 mm, C18, 5 eller 10 μm pakkemateriale, eller tilsvarende |
|
Mobil fase (3.9): |
blanding af methanol (3.3) og vand, f.eks. 980 + 20 (v + v) |
|
Flow: |
1–2 ml/minuttet |
|
Detektor (4.5.2): |
UV-detektor (325 nm) eller fluorescensdetektor (excitation: 325 nm/emission: 475 nm). |
5.6. Kalibrering
5.6.1.
20 ml stamopløsning af vitamin A-acetat (3.11.1) eller vitamin A-palmitat (3.12.1) afpipetteres i en 500 ml fladbundet eller konisk kolbe (4.2.1), og der hydrolyseres som beskrevet i punkt 5.2, dog uden tilsætning af BHT. Derefter ekstraheres der med petroleumsether (3.2) som beskrevet i punkt 5.3 og fyldes op til 500 ml med petroleumsether (3.2). 100 ml af denne opløsning inddampes til næsten tørhed på rotationsfordamper (se punkt 5.4), resterende opløsningsmiddel fjernes under en nitrogenstrøm (3.10), og remanensen opløses i 10,0 ml methanol (3.3). Opløsningens nominelle vitamin A-koncentration er 560 IU pr. ml. Det nøjagtige indhold bestemmes efter punkt 5.6.3.3. Arbejdsstandardopløsningen skal fremstilles umiddelbart før brugen.
Der afpipetteres 2,0 ml af denne arbejdsstandardopløsning i en 20 ml målekolbe, hvorefter der fyldes op til mærket med methanol (3.3) og blandes. Denne fortyndede arbejdsstandardopløsning har en nominel vitamin A-koncentration på 56 IU pr. ml.
5.6.2.
Henholdsvis 1,0 , 2,0 , 5,0 og 10,0 ml af den fortyndede arbejdsstandardopløsning overføres til hver sin 20 ml målekolbe, hvorefter der fyldes op til mærket med methanol (3.3) og blandes. Opløsningernes nominelle vitamin A-koncentration er 2,8 , 5,6 , 14,0 og 28,0 IU pr. ml.
Der indsprøjtes 20 μl af hver kalibreringsopløsning flere gange, og middeltophøjderne (-arealerne) bestemmes. Kalibreringskurven tegnes ved hjælp af middeltophøjderne (-arealerne) under hensyntagen til resultaterne af UV-kontrollen (5.6.3.3).
5.6.3.
5.6.3.1.
Der afpipetteres 2,0 ml stamopløsning af vitamin A-acetat (3.11.1) i en 50 ml målekolbe (4.2.2), hvorefter der fyldes op til mærket med 2-propanol (3.8). Opløsningens nominelle vitamin A-koncentration er 56 IU pr. ml. Der afpipetteres 3,0 ml af denne fortyndede opløsning af vitamin A-acetat i en 25 ml målekolbe, hvorefter der fyldes op til mærket med 2-propanol (3.8). Opløsningens nominelle vitamin A-koncentration er 6,72 IU pr. ml. Opløsningens UV-spektrum i området 300–400 nm måles mod 2-propanol (3.8) i spektrofotometret (4.6). Ekstinktionsmaksimum skal ligge mellem 325 nm og 327 nm.
Indholdet af vitamin A beregnes efter følgende formel:
IU vitamin A/ml = E326 × 19,0
(
![]()
for vitamin A-acetat = 1 530 ved 326 nm i 2-propanol)
5.6.3.2.
Der afpipetteres 2,0 ml stamopløsning af vitamin A-palmitat (3.12.1) i en 50 ml målekolbe (4.2.2), hvorefter der fyldes op til mærket med 2-propanol (3.8). Opløsningens nominelle vitamin A-koncentration er 56 IU pr. ml. Der afpipetteres 3,0 ml af denne fortyndede opløsning af vitamin A-palmitat i en 25 ml målekolbe, hvorefter der fyldes op til mærket med 2-propanol (3.8). Opløsningens nominelle vitamin A-koncentration er 6,72 IU pr. ml. Opløsningens UV-spektrum i området 300–400 nm måles mod 2-propanol (3.8) i spektrofotometret (4.6). Ekstinktionsmaksimum skal ligge mellem 325 nm og 327 nm.
Indholdet af vitamin A beregnes efter følgende formel:
IU vitamin A/ml = E326 × 19,0
(
![]()
for vitamin A-palmitat = 957 ved 326 nm i 2-propanol)
5.6.3.3.
Der afpipetteres 3,0 ml af den ufortyndede arbejdsstandardopløsning af vitamin A som fremstillet under punkt 5.6.1 i en 50 ml målekolbe (4.2.2), hvorefter der fyldes op til mærket med 2-propanol (3.8). Der afpipetteres 5,0 ml af denne opløsning i en 25 ml målekolbe, hvorefter der fyldes op til mærket med 2-propanol (3.8). Opløsningens nominelle vitamin A-koncentration er 6,72 IU pr. ml. Opløsningens UV-spektrum i området 300–400 nm måles mod 2-propanol (3.8) i spektrofotometret (4.6). Ekstinktionsmaksimum skal ligge mellem 325 nm og 327 nm.
Indholdet af vitamin A beregnes efter følgende formel:
IU vitamin A/ml = E325 × 18,3
(
![]()
for vitamin A-alkohol = 1 821 ved 325 nm i 2-propanol)
6. Beregning af resultater
Ud fra middelhøjden (-arealet) af prøveopløsningens vitamin A-toppe bestemmes koncentrationen i prøveopløsningen i IU/ml ved benyttelse af kalibreringskurven (5.6.2).
Indholdet w af vitamin A i IU pr. kg prøve beregnes efter følgende formel:
![]()
[UI/kg]
hvor:
|
c |
= |
vitamin A-koncentrationen i prøveopløsningen (5.4) i IU/ml |
|
V1 |
= |
volumen af prøveopløsning (5.4) i ml |
|
V2 |
= |
volumen af den i 5.4 udtagne alikvot i ml |
|
m |
= |
testportionens vægt i gram. |
7. Bemærkninger
7.1. For prøver med lav vitamin A-koncentration kan man med fordel samle petroleumsetherekstrakterne fra to forsæbningsportioner (afvejet mængde: 25 g) i én prøveopløsning til HPLC-bestemmelse.
7.2. Den prøve, der tages ud til analyse, må ikke indeholde mere end 2 g fedtstof.
7.3. Hvis faserne ikke skiller, kan der tilsættes ca. 10 ml ethanol (3.1) for at bryde emulsionen.
7.4. Med torskeleverolie og andre rene fedtstoffer skal forsæbningstiden øges til 45–60 minutter.
7.5. Der kan anvendes hydroquinon i stedet for BHT.
7.6. Ved brug af en normal-fase-kolonne er det muligt at adskille retinolisomererne. I så fald er det dog ved beregningerne nødvendigt at lægge højderne (arealerne) af samtlige cis- og trans-isomerers toppe sammen.
7.7. Der kan anvendes ca. 150 mg ascorbinsyre i stedet for natriumascorbatopløsning.
7.8. Der kan anvendes ca. 50 mg EDTA i stedet for natriumsulfidopløsning.
7.9. Ved analyse af vitamin A i mælkeerstatninger skal især følgende tages i betragtning:
— Ved forsæbning (5.2): Det kan på grund af fedtmængden i prøven være nødvendigt at øge mængden af kaliumhydroxidopløsning (3.4).
— Ved ekstraktion (5.3): Det kan på grund af dannelse af emulsioner være nødvendigt at justere forholdet på 2:1 mellem vand og ethanol.
Det kontrolleres med en genfindingstest på yderligere en testportion, at den anvendte analysemetode giver pålidelige resultater for denne matrix (mælkeerstatning). Er genfindingsprocenten lavere end 80 %, korrigeres analyseresultatet for genfinding.
8. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige 15 % i relativ værdi (af det højeste resultat).
9. Resultater af ringanalyse ( 30 )
|
|
Forblanding |
Forblandet foder |
Mineralkoncentrat |
Proteinfoder |
Foder til smågrise |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
Middelværdi [IU/kg] |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
Sr [IU/kg] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [IU/kg] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
SR [IU/kg] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [IU/kg] |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
CVR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enkeltværdier |
|
Sr |
= |
standardafvigelse for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
r |
= |
repeterbarhed |
|
R |
= |
reproducerbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed |
B. BESTEMMELSE AF VITAMIN E
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af vitamin E i foder og forblandinger. Indholdet af vitamin E udtrykkes i mg DL-α -tocopherolacetat pr. kg. 1 mg DL-α-tocopherolacetat svarer til 0,91 mg DL-α-tocopherol (vitamin E).
Bestemmelsesgrænsen er 2 mg vitamin E pr. kg. Denne bestemmelsesgrænse er kun realistisk ved brug af fluorescensdetektor. Ved brug af UV-detektor er bestemmelsesgrænsen 10 mg/kg.
2. Princip
Prøven hydrolyseres med kaliumhydroxid opløst i ethanol, og vitamin E ekstraheres med petroleumsether. Opløsningsmidlet fjernes ved inddampning, og remanensen opløses i methanol og fortyndes om nødvendigt til den ønskede koncentration. Vitamin E-indholdet bestemmes ved omvendt-fase-HPLC med anvendelse af UV- eller fluorescensdetektor.
3. Reagenser
3.1. Ethanol, σ = 96 %.
3.2. Petroleumsether, kogepunktsinterval: 40–60 oC.
3.3. Methanol.
3.4. Kaliumhydroxidopløsning, c = 50 g/100 ml.
3.5. Natriumascorbatopløsning, c = 10 g/100 ml (se bemærkning 7.7).
3.6. Natriumsulfid, Na2S· x H2O (x = 7–9).
3.6.1. Natriumsulfidopløsning, c = 0,5 mol/liter i glycerol, β = 120 g/liter (for x = 9) (se bemærkning 7.8).
3.7. Phenolphthaleinopløsning, c = 2 g/100 ml i ethanol (3.1).
3.8. Mobil fase til HPLC: blanding af methanol (3.3) og vand, f.eks. 980 + 20 (v + v). Det nøjagtige forhold bestemmes af den anvendte kolonnes egenskaber.
3.9. Nitrogen, oxygenfrit.
3.10. DL-α-tocopherolacetat, ekstra rent, med certificeret aktivitet.
3.10.1. Stamopløsning af DL-α-tocopherolacetat: Der afvejes med 0,1 mg nøjagtighed 100 mg DL-α-tocopherolacetat (3.10) i en 100 ml målekolbe. Efter opløsning i ethanol (3.1) fyldes der op til mærket med samme opløsningsmiddel. 1 ml af denne opløsning indeholder 1 mg DL-α-tocopherolacetat (UV-kontrol, se punkt 5.6.1.3; stabilisering, se bemærkning 7.4.).
3.11. DL-α-tocopherol, ekstra rent, med certificeret aktivitet
3.11.1. Stamopløsning af DL-α-tocopherol: Der afvejes med 0,1 mg nøjagtighed 100 mg DL-α-tocopherol (3.11) i en 100 ml målekolbe. Efter opløsning i ethanol (3.1) fyldes der op til mærket med samme opløsningsmiddel. 1 ml af denne opløsning indeholder 1 mg DL-α-tocopherol (UV-kontrol, se punkt 5.6.2.3; stabilisering, se bemærkning 7.4).
3.12. 2,6-Di-tert-butyl-4-methylphenol (BHT) (se bemærkning 7.5).
4. Apparatur
4.1. Rotationsfordamper
4.2. Udstyr af brunt glas
4.2.1. Fladbundede eller koniske kolber, 500 ml, med slib
4.2.2. Målekolber med slibprop og tynd hals, 10, 25, 100 og 500 ml
4.2.3. Skilletragte, koniske, 1 000 ml, med slibprop
4.2.4. Pærekolber, 250 ml, med slib
4.3. Allihn-svaler, kappelængde 300 mm, med slibhals, med adapter til gastilførsel
4.4. Foldefiltrerpapir til faseseparation, diameter: 185 mm (f.eks. Schleicher & Schüll 597 HY 1/2)
4.5. HPLC-udstyr med injektionssystem
4.5.1. HPLC-kolonne, 250 mm × 4 mm, C18, 5 eller 10 μm pakkemateriale, eller tilsvarende
4.5.2. UV- eller fluorescensdetektor, med variabel bølgelængde
4.6. Spektrofotometer med 10 mm kvartskuvetter
4.7. Vandbad med magnetomrører
4.8. Ekstraktionsapparat (se figur 1) bestående af følgende:
4.8.1. Cylinderglas på 1 liter med slibhals og -prop
4.8.2. Slibindsats med sidearm og justerbart rør, der er ført igennem midten. Det justerbare rør skal være U-formet i den nedre ende og være tilspidset i den anden ende, således at det øverste væskelag i glasset kan overføres til en skilletragt.
5. Fremgangsmåde
|
NB |
Vitamin E er følsomt over for lys (UV) og oxidering. Alt arbejde udføres afskærmet mod lys (udstyr af brunt glas eller glasudstyr omviklet med aluminiumfolie) og oxygen (skylning med nitrogen). Under ekstraktionen udskiftes luften over væsken med nitrogen (overtryk udlignes ved jævnligt at lette på proppen). |
5.1. Klargøring af prøven
Prøven formales, så den kan passere gennem en sigte med 1 mm maskevidde, men varmeudvikling skal undgås. Formalingen foretages umiddelbart før afvejning og forsæbning, da der ellers kan gå vitamin E tabt.
5.2. Forsæbning
Afhængigt af vitamin E-indholdet afvejes der med en nøjagtighed på 0,01 g 2–25 g af prøven i en 500 ml fladbundet eller konisk kolbe (4.2.1). Der tilsættes under forsigtig blanding i rækkefølge 130 ml ethanol (3.1), ca. 100 mg BHT (3.12), 2 ml natriumascorbatopløsning (3.5) og 2 ml natriumsulfidopløsning (3.6). Der sættes en svaler på kolben, som anbringes i vandbad med magnetomrører (4.7). Der opvarmes til kogning og koges med tilbagesvaling i 5 minutter. Dernæst tilsættes der 25 ml kaliumhydroxidopløsning (3.4) gennem svaleren (4.3), og der koges med tilbagesvaling i endnu 25 minutter under omrøring og under langsom nitrogengennemstrømning. Svaleren skylles med ca. 20 ml vand, og kolbens indhold afkøles til rumtemperatur.
5.3. Ekstraktion
Forsæbningsopløsningen overføres kvantitativt til en 1 000 ml skilletragt (4.2.3) eller ekstraktionsapparatet (4.8) ved skylning med i alt 250 ml vand. Derefter skylles forsæbningskolben først med 25 ml ethanol (3.1) og dernæst med 100 ml petroleumsether (3.2), idet skyllevæskerne overføres til skilletragten eller ekstraktionsapparatet. Forholdet mellem vand og ethanol i den samlede væskemængde skal være ca. 2:1. Der omrystes kraftigt i 2 minutter, og blandingen henstår til adskillelse i 2 minutter.
5.3.1.
Når væsken er skilt i to lag (se bemærkning 7.3), overføres petroleumsetherlaget til en anden skilletragt (4.2.3). Ekstraktionen gentages to gange med 100 ml petroleumsether (3.2) og derefter to gange med 50 ml petroleumsether (3.2).
De samlede ekstrakter i skilletragten vaskes to gange med 100 ml vand hver gang (ved forsigtig omdrejning, så der ikke dannes emulsion) og derefter flere gange ved rystning med 100 ml vand, indtil vandet ikke farves ved tilsætning af phenolphthaleinopløsning (3.7) (fire gange er normalt tilstrækkeligt). Det vaskede ekstrakt filtreres over i en 500 ml målekolbe (4.2.2) gennem et tørt foldefilter til faseseparation (4.4), således at eventuelt opslæmmet vand fjernes. Skilletragten og filtret skylles med 50 ml petroleumsether (3.2), og der fyldes op til mærket med petroleumsether (3.2) og blandes omhyggeligt.
5.3.2.
Når væsken er skilt i to lag (se bemærkning 7.3), udskiftes cylinderglassets (4.8.1) prop med slibindsatsen (4.8.2), og den nederste, U-formede del af det justerbare rør anbringes, så dets munding er lige over skillefladen. Ved at sætte nitrogentryk på sidearmen overføres det lette lag af petroleumsether til en 1 000 ml skilletragt (4.2.3). Der tilsættes 100 ml petroleumsether (3.2) til cylinderglasset, og det tilproppes og rystes omhyggeligt. Når væsken er skilt i to lag, overføres det lette lag til skilletragten som før. Ekstraktionen gentages med endnu 100 ml petroleumsether (3.2) og dernæst med 2 portioner 50 ml petroleumsether (3.2), og alle lagene af petroleumsether overføres til skilletragten.
De samlede petroleumsetherekstrakter i skilletragten vaskes som beskrevet i punkt 5.3.1, og der fortsættes som beskrevet der.
5.4. Fremstilling af prøveopløsning til HPLC-analyse
En alikvot mængde af petroleumsetherekstraktet (fra 5.3.1 eller 5.3.2) afpipetteres i en 250 ml pærekolbe (4.2.4). Der inddampes til næsten tørhed på rotationsfordamper (4.1) under reduceret tryk med en vandbadstemperatur på højst 40 oC. Trykket udlignes til atmosfæretryk, ved at der ledes nitrogen (3.9) ind i kolben, og kolben fjernes fra rotationsfordamperen. Resterende opløsningsmiddel fjernes under en nitrogenstrøm (3.9), og remanensen opløses straks i en kendt mængde (10–100 ml) methanol (3.3) (koncentrationen af DL-α-tocopherol skal ligge i intervallet 5–30 μg/ml).
5.5. HPLC-bestemmelse
Separationen af vitamin E finder sted på en C18-kolonne med omvendt fase (4.5.1), og koncentrationen måles med en UV-detektor (292 nm) eller en fluorescensdetektor (excitation: 295 nm, emission: 330 nm) (4.5.2).
Der indsprøjtes en alikvot mængde (f.eks. 20 μl) af methanolopløsningen fra punkt 5.4, og der elueres med den mobile fase (3.8). Middeltophøjderne (-arealerne) for flere indsprøjtninger af samme prøveopløsning og middeltophøjderne (-arealerne) for flere indsprøjtninger af kalibreringsopløsningerne (5.6.2) beregnes.
Nedenstående betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater.
|
HPLC-kolonne (4.5.1): |
250 mm × 4 mm, C18, 5 eller 10 μm pakkemateriale, eller tilsvarende |
|
Mobil fase (3.8): |
blanding af methanol (3.3) og vand, f.eks. 980 + 20 (v + v) |
|
Flow: |
1–2 ml/minuttet |
|
Detektor (4.5.2): |
fluorescensdetektor (excitation: 295 nm/emission: 330 nm) eller UV detektor (292 nm) |
5.6. Kalibrering (DL-α-tocopherolacetat eller DL-α-tocopherol)
5.6.1.
5.6.1.1.
25 ml stamopløsning af DL-α-tocopherolacetat (3.10.1) afpipetteres i en 500 ml fladbundet eller konisk kolbe (4.2.1), og der hydrolyseres som beskrevet i punkt 5.2. Derefter ekstraheres der med petroleumsether (3.2) som beskrevet i punkt 5.3 og fortyndes til 500 ml med petroleumsether. 25 ml af denne opløsning inddampes til næsten tørhed på rotationsfordamper (se punkt 5.4), resterende opløsningsmiddel fjernes under en nitrogenstrøm (3.9), og remanensen opløses i 25,0 ml methanol (3.3). Opløsningens nominelle koncentration er 45,5 μg DL-α-tocopherol pr. ml svarende til 50 μg DL-α-tocopherolacetat pr. ml. Arbejdsstandardopløsningen skal fremstilles umiddelbart før brugen.
5.6.1.2.
Henholdsvis 1,0 , 2,0 , 4,0 og 10,0 ml af arbejdsstandardopløsningen overføres til hver sin 20 ml målekolbe, hvorefter der fyldes op til mærket med methanol (3.3) og blandes. Opløsningernes nominelle koncentration er 2,5 , 5,0 , 10,0 og 25,0 μg DL-α-tocopherolacetat pr. ml, dvs. 2,28 , 4,55 , 9,10 og 22,8 μg DL-α-tocopherol pr. ml.
Der indsprøjtes 20 μl af hver kalibreringsopløsning flere gange, og middeltophøjderne (-arealerne) bestemmes. Kalibreringskurven tegnes ved hjælp af middeltophøjderne (-arealerne).
5.6.1.3.
5,0 ml af stamopløsningen af DL-α-tocopherolacetat (3.10.1) fortyndes til 25,0 ml med ethanol, og opløsningens UV-spektrum i området 250–320 nm måles mod ethanol (3.1) i spektrofotometret (4.6).
Absorptionsmaksimum skal ligge ved 284 nm:
![]()
43,6 ved 284 nm i ethanol
Ved denne fortynding skal der opnås en ekstinktionsværdi på 0,84 –0,88 .
5.6.2.
5.6.2.1.
Der afpipetteres 2 ml stamopløsning af DL-α-tocopherol (3.11.1) i en 50 ml målekolbe, hvorefter der fyldes op til mærket med methanol (3.3). Opløsningens nominelle koncentration er 40 μg DL-α-tocopherol pr. ml svarende til 44,0 μg DL-α-tocopherolacetat pr. ml. Arbejdsstandardopløsningen skal fremstilles umiddelbart før brugen.
5.6.2.2.
Henholdsvis 1,0 , 2,0 , 4,0 og 10,0 ml af arbejdsstandardopløsningen overføres til hver sin 20 ml målekolbe, hvorefter der fyldes op til mærket med methanol (3.3) og blandes. Opløsningernes nominelle koncentration er 2,0 , 4,0 , 8,0 og 20,0 μg DL-α-tocopherol pr. ml, dvs. 2,20 , 4,40 , 8,79 og 22,0 μg DL-α-tocopherolacetat pr. ml.
Der indsprøjtes 20 μl af hver kalibreringsopløsning flere gange, og middeltophøjderne (-arealerne) bestemmes. Kalibreringskurven tegnes ved hjælp af middeltophøjderne (-arealerne).
5.6.2.3.
2,0 ml af stamopløsningen af DL-α-tocopherol (3.11.1) fortyndes til 25,0 ml med ethanol, og opløsningens UV-spektrum i området 250-320 nm måles mod ethanol (3.1) i spektrofotometret (4.6). Absorptionsmaksimum skal ligge ved 292 nm:
![]()
= 75,8 ved 292 nm i ethanol
Ved denne fortynding skal der opnås en ekstinktionsværdi på 0,6 .
6. Beregning af resultater
Ud fra middelhøjden (-arealet) af prøveopløsningens vitamin E-toppe bestemmes koncentrationen i prøveopløsningen i μg/ml (beregnet som α-tocopherolacetat) ved benyttelse af kalibreringskurven (5.6.1.2 eller 5.6.2.2).
Indholdet w af vitamin E i mg/kg for prøven beregnes efter følgende formel:
![]()
[mg/kg]
hvor:
|
c |
= |
vitamin E-koncentration (som α-tocopherolacetat) i prøveopløsningen (5.4) i μg/ml |
|
V1 |
= |
volumen af prøveopløsning (5.4) i ml |
|
V2 |
= |
volumen af den i 5.4 udtagne alikvot i ml |
|
m |
= |
testportionens vægt i gram. |
7. Bemærkninger
7.1. For prøver med lav vitamin E-koncentration kan man med fordel samle petroleumsetherekstrakterne fra to forsæbningsportioner (afvejet mængde: 25 g) i én prøveopløsning til HPLC-bestemmelse.
7.2. Den prøve, der tages ud til analyse, må ikke indeholde mere end 2 g fedtstof.
7.3. Hvis faserne ikke skiller, kan der tilsættes ca. 10 ml ethanol (3.1) for at bryde emulsionen.
7.4. Efter spektrofotometrisk måling af opløsningen af DL-α-tocopherolacetat eller DL-α-tocopherol ifølge henholdsvis 5.6.1.3 og 5.6.2.3 tilsættes der ca. 10 mg BHT (3.12) til opløsningen (3.10.1 eller 3.10.2), hvorefter den opbevares i køleskab (højst 4 uger).
7.5. Der kan anvendes hydroquinon i stedet for BHT.
7.6. Ved brug af en normal-fase-kolonne er det muligt at adskille α-, β-, γ- og δ-tocopherol.
7.7. Der kan anvendes ca. 150 mg ascorbinsyre i stedet for natriumascorbatopløsning.
7.8. Der kan anvendes ca. 50 mg EDTA i stedet for natriumsulfidopløsning.
7.9. Vitamin E-acetat hydrolyserer meget hurtigt i basisk miljø og er derfor yderst følsomt over for oxidering, navnlig under tilstedeværelse af mikromineraler såsom jern eller kobber. Bestemmelse af vitamin E i forblandinger i mængder på over 5 000 mg/kg vil kunne resultere i nedbrydning af vitamin E. HPLC-bestemmelse med enzymatisk behandling af vitamin E-indholdet, hvor alkalisk forsæbning udelades, er derfor at anbefale i verifikationsøjemed.
8. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige 15 % i relativ værdi (af det højeste resultat).
9. Resultater af ringanalyse ( 31 )
|
|
Forblanding |
Forblandet foder |
Mineralkoncentrat |
Proteinfoder |
Foder til smågrise |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
Middelværdi [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [%] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
sR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
SR [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [%] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enkeltværdier |
|
sr |
= |
standardafvigelse for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
r |
= |
repeterbarhed |
|
R |
= |
reproducerbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed |
C. BESTEMMELSE AF MIKROMINERALERNE JERN, KOBBER, MANGAN OG ZINK
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme mikromineralerne jern, kobber, mangan og zink i foder. Bestemmelsesgrænserne er:
— jern (Fe): 20 mg/kg
— kobber (Cu): 10 mg/kg
— mangan (Mn): 20 mg/kg
— zink (Zn): 20 mg/kg.
2. Princip
Prøven opløses i saltsyre, efter at eventuelt organisk stof er destrueret. Jern, kobber, mangan og zink bestemmes efter en passende fortynding ved anvendelse af atomabsorptionsspektrofotometri.
3. Reagenser
Indledning
Ved fremstillingen af reagenser og analyseopløsninger anvendes vand, som ikke indeholder de kationer, der skal bestemmes. Vandet renses enten ved en dobbelt destillation af vandet i et borsilikat- eller kvartsdestillationsapparat eller ved dobbelt behandling på ionbytter.
Reagenserne skal mindst være af analysekvalitet. Fravær af de mineraler, som skal bestemmes, kontrolleres ved blindbestemmelse. Om nødvendigt oprenses reagenserne yderligere.
I stedet for stamopløsninger, som beskrevet nedenfor, kan kommercielle stamopløsninger anvendes, på betingelse af at de kontrolleres før brug.
3.1. Saltsyre, massefylde: 1,19 g/ml.
3.2. Saltsyre, 6 mol/liter.
3.3. Saltsyre, 0,5 mol/liter.
3.4. Flussyre 38–40 % (v/v) med et indhold af jern (Fe) på mindre end 1 mg/liter og med en inddampningsrest på mindre end 10 mg (som sulfat)/liter.
3.5. Svovlsyre, massefylde: 1,84 g/ml.
3.6. Hydrogenperoxid, ca. 100 vol. oxygen (30 vægtprocent).
3.7. Standardjernopløsning (1 000 μg Fe/ml) fremstilles som følger (fås en tilsvarende opløsning i handelen, kan denne anvendes): 1 g jerntråd opløses i 200 ml 6 mol/liter saltsyre (3.2), hvorefter der tilsættes 16 ml hydrogenperoxid (3.6) og fyldes op med vand til 1 liter.
3.7.1. Arbejdsstandardjernopløsning (100 μg Fe/ml) fremstilles ved at fortynde 1 del standardopløsning (3.7) med 9 dele vand.
3.8. Standardkobberopløsning (1 000 μg Cu/ml) fremstilles som følger (fås en tilsvarende opløsning i handelen, kan denne anvendes):
— 1 g kobber i pulverform opløses i 25 ml 6 mol/liter saltsyre (3.2), hvorefter der tilsættes 5 ml hydrogenperoxid (3.6) og fyldes op med vand til 1 liter.
3.8.1. Arbejdsstandardkobberopløsning (10 μg Cu/ml) fremstilles ved at fortynde 1 del stamopløsning (3.8) med 9 dele vand og derefter fortynde 1 del af den således opnåede opløsning med 9 dele vand.
3.9. Standardmanganopløsning (1 000 μg Mn/ml) fremstilles som følger (fås en tilsvarende opløsning i handelen, kan denne anvendes):
— 1 g mangan i pulverform opløses i 25 ml 6 mol/liter saltsyre (3.2), og der fyldes op til 1 liter med vand.
3.9.1. Arbejdsstandardmanganopløsning (10 μg Mn/ml) fremstilles ved at fortynde 1 del stamopløsning (3.9) med 9 dele vand og derefter fortynde 1 del af den således opnåede opløsning med 9 dele vand.
3.10. Standardzinkopløsning (1 000 μg Zn/ml) fremstilles som følger (fås en tilsvarende opløsning i handelen, kan denne anvendes):
— 1 g zink i bånd eller bladform opløses i 25 ml 6 mol/liter saltsyre (3.2), og der fyldes op med vand til 1 liter.
3.10.1. Arbejdsstandardzinkopløsning (10 μg Zn/ml) fremstilles ved at fortynde 1 del stamopløsning (3.10) med 9 dele vand og derefter fortynde 1 del af den således opnåede opløsning med 9 dele vand.
3.11. Lanthanchloridopløsning: 12 g lanthanoxid opløses i 150 ml vand, hvorefter der tilsættes 100 ml 6 mol/liter saltsyre (3.2) og fyldes op med vand til 1 liter.
4. Apparatur
4.1. Muffelovn med temperaturregulering og helst pyrometer.
4.2. Glasudstyr skal være af modstandsdygtigt borsilikat, og det anbefales, at det pågældende apparatur udelukkende anvendes til bestemmelse af mikromineraler.
4.3. Atomabsorptionsspektrofotometer, som opfylder de relevante metodekrav med hensyn til følsomhed og nøjagtighed i det krævede område.
5. Fremgangsmåde ( 32 )
5.1. Prøver indeholdende organisk stof
5.1.1. ( 33 )
5.1.1.1. 5–10 g af prøven afvejes med 0,2 mg nøjagtighed i en kvarts- eller platindigel (se bemærkning b)), prøven tørres i tørreskab ved 105 oC, og diglen anbringes i en kold muffelovn (4.1). Ovnen lukkes (se bemærkning c)), og temperaturen øges gradvis til 450–475 oC i løbet af ca. 90 minutter. Denne temperatur holdes i 4–16 timer (f.eks. natten over) for at fjerne kulstofholdigt materiale, hvorefter ovnen åbnes til afkøling (se bemærkning d)).
Asken fugtes med vand og overføres til et 250 ml bægerglas. Diglen skylles med i alt ca. 5 ml saltsyre (3.1), som overføres langsomt og omhyggeligt til bægerglasset (en voldsom reaktion kan forekomme på grund af dannelse af CO2). Der tilsættes saltsyre (3.1) dråbevis under omrøring, indtil enhver opbrusning er ophørt. Der inddampes til tørhed under lejlighedsvis omrøring med en glasspatel.
Der tilsættes først 15 ml 6 mol/liter saltsyre (3.2) og dernæst 120 ml vand til remanensen. Der omrøres med glasspatelen, som skal forblive i bægerglasset, og dette tildækkes med et urglas. Indholdet bringes til svag kogning og holdes på kogepunktet, indtil der tilsyneladende ikke opløses mere aske. Der filtreres gennem et askefrit filtrerpapir over i en 250 ml målekolbe. Bægerglasset vaskes, og der filtreres med 5 ml varm 6 mol/liter saltsyre (3.2) samt to gange med kogende vand. Målekolben fyldes op til mærket med vand (HCl-koncentration: ca. 0,5 mol/liter).
5.1.1.2. Hvis remanensen på filtret er sort (kulstof), sættes den tilbage i ovnen, og der foraskes igen ved 450–475 oC. Denne foraskning, som kun kræver nogle få timer (ca. 3–5 timer), er gennemført, når asken er hvid eller næsten hvid. Remanensen opløses i ca. 2 ml saltsyre (3.1) og inddampes til tørhed, hvorefter der tilsættes 5 ml 6 mol/liter saltsyre (3.2). Der opvarmes, opløsningen filtreres over i målekolben, og der fyldes op til mærket med vand (ca. 0,5 mol/liter HCl-koncentration).
Bemærkninger:
a) Ved bestemmelse af mikromineraler er det vigtigt at være opmærksom på risikoen for forurening især med zink, kobber og jern. Derfor skal det udstyr, som anvendes til klargøring af prøverne, være fri for disse metaller.
For at mindske den almindelige forureningsrisiko arbejdes i en støvfri atmosfære med pinligt rent udstyr og omhyggeligt afvaskede glasvarer. Bestemmelsen af zink er særligt følsom over for mange typer forurening, f.eks. fra glas, reagenser, støv osv.
b) Vægten af den prøve, der skal foraskes, beregnes ud fra det omtrentlige forventede indhold af mikromineraler i foderet i relation til det anvendte spektrofotometers følsomhed. For visse typer foder, hvis mikromineralindhold er lavt, kan det være nødvendigt at starte med en prøve på 10–20 g og begrænse den endelige opløsning til kun 100 ml.
c) Foraskning skal udføres i en lukket ovn uden gennemblæsning med luft eller oxygen.
d) Pyrometrets temperaturudvisning må ikke overstige 475 oC.
5.1.2.
5.1.2.1.
For hvert af de grundstoffer, som skal bestemmes, skal der ud fra arbejdsstandardopløsningerne 3.7.1, 3.8.1, 3.9.1 og 3.10.1 fremstilles en række kalibreringsopløsninger, hver med en HCl-koncentration på ca. 0,5 mol/liter og (for så vidt angår jern, mangan og zink) en lanthanchloridkoncentration svarende til 0,1 % La (w/v).
De valgte koncentrationer for mikromineraler skal ligge inden for det anvendte spektrofotometers følsomhedsområde. Som eksempler er i tabellerne nedenfor angivet sammensætningen af typiske rækker af kalibreringsopløsninger. Afhængigt af spektrofotometret og dets følsomhed kan det dog være nødvendigt at vælge andre koncentrationer
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml arbejdsstandardopløsning (3.7.1) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lanthanchloridopløsning (3.11) — der fyldes op med vand til 100 ml |
|||||||
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml arbejdsstandardopløsning (3.8.1) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml arbejdsstandardopløsning (3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lanthanchloridopløsning (3.11) — der fyldes op med vand til 100 ml |
|||||||
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml arbejdsstandardopløsning (3.10.1) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lanthanchloridopløsning (3.11) — der fyldes op med vand til 100 ml |
|||||||
5.1.2.2.
Til bestemmelse af kobber kan den opløsning, som er fremstillet efter punkt 5.1.1, normalt anvendes direkte. Hvis det er nødvendigt for at bringe dens koncentration ind i kalibreringsopløsningernes område, kan en alikvot mængde afpipetteres i en 100 ml målekolbe, hvorefter der fyldes op til mærket med 0,5 mol/liter saltsyre (3.3).
Til bestemmelse af jern, mangan og zink afpipetteres en alikvot mængde af den efter punkt 5.1.1 fremstillede opløsning i en 100 ml målekolbe, der tilsættes 10 ml lanthanchloridopløsning (3.11), og der fyldes op til mærket med 0,5 mol/liter saltsyre (3.3). (se også punkt 8, »Bemærkning«).
5.1.2.3.
Blindbestemmelsen skal indeholde alle trinene under fremgangsmåden, blot skal prøvematerialet udelades. Kalibreringsopløsning »0« må ikke anvendes som blind.
5.1.2.4.
Atomabsorptionen for kalibreringsopløsningerne og den opløsning, der skal analyseres, måles ved anvendelse af en oxiderende luftacetylenflamme ved følgende bølgelængder:
Fe: 248,3 nm
Cu: 324,8 nm
Mn: 279,5 nm
Zn: 213,8 nm
Hver af målingerne gentages fire gange.
5.2. Mineralfoder
Hvis prøven ikke indeholder organisk materiale, er forudgående foraskning unødvendig. Fremgangsmåden i punkt 5.1.1.1 følges, idet der startes fra andet afsnit. Fordampning i tilstedeværelse af flussyre kan udelades.
6. Beregning af resultater
Ved anvendelse af en kalibreringskurve beregnes koncentrationen af mikromineralet i den opløsning, som skal analyseres, og resultatet angives i mg mikromineral pr. kg prøve (ppm).
7. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført af den samme person på den samme prøve må ikke overstige:
— 5 mg/kg i absolut værdi for indhold af det pågældende mikromineral på op til 50 mg/kg
— 10 % af det højeste resultat for indhold af det pågældende mikromineral på 50–100 mg/kg
— 10 mg/kg i absolut værdi for indhold af det pågældende mikromineral på 100–200 mg/kg;
— 5 % af det højeste resultat for indhold af det pågældende mikromineral på over 200 mg/kg.
8. Bemærkning
Tilstedeværelse af store mængder phosphater kan interferere på bestemmelsen af jern, mangan og zink. En sådan interferens skal korrigeres gennem tilsætning af lanthanchloridopløsning (3.11). Hvis imidlertid vægtforholdet Ca + Mg/P er > 2 i prøven, kan tilsætning af lanthanchlorid (3.11) til den opløsning, der skal analyseres, og til kalibreringsopløsningerne udelades.
D. BESTEMMELSE AF HALOFUGINON
DL-trans-7-brom-6-chlor-3-[3-(3-hydroxy-2-piperidyl)acetonyl]-quinazolin-4(3H)-on-hydrobromid
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af halofuginon i foder. Bestemmelsesgrænsen er 1 mg/kg.
2. Princip
Efter behandling med varmt vand ekstraheres halofuginon som fri base med ethylacetat og separeres derefter som hydrochlorid over i en vandig syreopløsning. Ekstraktet oprenses ved ionbytningskromatografi. Indholdet af halofuginon bestemmes ved omvendt fase-HPLC med anvendelse af UV-detektor.
3. Reagenser
3.1. Acetonitril, svarende til HPLC-kvalitet.
3.2. Amberlit XAD-2-resin.
3.3. Ammoniumacetat.
3.4. Ethylacetat.
3.5. Iseddike.
3.6. Halofuginonstandardstof (DL-trans-7-brom-6-chlor-3-[3-hydroxy-2-piperidyl)acetonyl]-quinazolin-4-(3H)-on-hydrobromid, E 764).
3.6.1.
50 mg halofuginon (3.6) afvejes med 0,1 mg nøjagtighed i en 500 ml målekolbe og opløses i ammoniumacetatbufferopløsning (3.18), hvorefter der fyldes op med bufferopløsning til mærket og blandes. Denne opløsning er stabil i tre uger ved 5 oC ved opbevaring i mørke.
3.6.2.
Henholdsvis 1,0 , 2,0 , 3,0 , 4,0 og 6,0 ml af standardstamopløsningen (3.6.1) overføres til hver sin 100 ml målekolbe. Der fyldes op til mærket med mobil fase (3.21) og blandes. Disse opløsninger har en koncentration på henholdsvis 1,0 , 2,0 , 3,0 , 4,0 og 6,0 μg/ml halofuginon. Disse opløsninger skal fremstilles umiddelbart før brugen.
3.7. Saltsyre, ρ20 = ca. 1,16 g/ml.
3.8. Methanol.
3.9. Sølvnitrat.
3.10. Natriumascorbat.
3.11. Natriumcarbonat.
3.12. Natriumchlorid.
3.13. EDTA (dinatriumethylendiamintetraacetat).
3.14. Vand, svarende til HPLC-kvalitet.
3.15. Natriumcarbonatopløsning, c = 10 g/100 ml.
3.16. Saltmættet natriumcarbonatopløsning, c = 5 g/100 ml
50 g natriumcarbonat (3.11) opløses i vand og fortyndes til 1 liter, hvorefter der tilsættes natriumchlorid (3.12), indtil opløsningen er mættet.
3.17. Saltsyre, ca. 0,1 mol/liter
10 ml HCI (3.7) fortyndes med vand til 1 liter.
3.18. Ammoniumacetatbufferopløsning, ca. 0,25 mol/liter
19,3 g ammoniumacetat (3.3) og 30 ml eddikesyre (3.5) opløses i vand (3.14) og fortyndes til 1 liter.
3.19. Amberlit XAD-2-resin-materiale
En passende mængde amberlit (3.2) vaskes med vand, til alle chloridioner er fjernet, således som det viser sig ved kontrol med sølvnitrat (3.20) på den kasserede, vandige fase. Resinet vaskes derefter med 50 ml methanol (3.8), methanolen kasseres, og resinet opbevares under frisk methanol.
3.20. Sølvnitratopløsning, ca. 0,1 mol/liter
0,17 g sølvnitrat (3.9) opløses i 10 ml vand.
3.21. Mobil fase til HPLC
500 ml acetonitril (3.1) blandes med 300 ml ammoniumacetatbufferopløsning (3.18) og 1 200 ml vand (3.14). pH justeres til 4,3 med eddikesyre (3.5). Der filtreres gennem et 0,22 μm filter (4.8), og opløsningen afgasses (f.eks. ved ultralydsbehandling i 10 minutter). Denne opløsning er stabil i en måned, hvis den opbevares mørkt i en lukket beholder.
4. Apparatur
4.1. Ultralydsbad
4.2. Rotationsfordamper
4.3. Centrifuge
4.4. HPLC-udstyr med ultraviolet detektor med variabel bølgelængde eller med diodearraydetektor
4.4.1. HPLC-kolonne, 300 mm × 4 mm, C18, 10 μm pakkemateriale, eller tilsvarende
4.5. Glaskolonne (300 mm × 10 mm) med filter af sintret glas og stophane
4.6. Glasfiberfiltre, diameter: 150 mm
4.7. Membranfiltre, 0,45 μm
4.8. Membranfiltre, 0,22 μm.
5. Fremgangsmåde
|
NB |
Halofuginon er som fri base ustabilt i alkaliske opløsninger og ethylacetatopløsninger. Må ikke henstå i ethylacetat i over 30 minutter. |
5.1. Generelt
5.1.1. Der analyseres en blindprøve for at kontrollere, at hverken halofuginon eller interfererende stoffer er til stede.
5.1.2. Der udføres en genfindingstest ved at analysere blindprøven, som er blevet spiket ved tilsætning af en mængde halofuginon svarende til den, der er til stede i prøven. For at nå op på et niveau på 3 mg/kg tilsættes 300 μl af standardstamopløsningen (3.6.1) til 10 g blindprøve. Der blandes og ventes i 10 minutter, inden der fortsættes med ekstraktion (5.2).
|
NB |
Til denne metode anvendes en blindprøve af samme type som analyseprøven, og som ved analyse viser sig ikke at indeholde halofuginon. |
5.2. Ekstraktion
10 g af den klargjorte prøve afvejes med en nøjagtighed på 0,1 g i et 200 ml centrifugeglas, hvorefter der tilsættes 0,5 g natriumascorbat (3.10), 0,5 g EDTA (3.13) og 20 ml vand og blandes. Centrifugeglasset anbringes i vandbad (80 oC) i 5 minutter. Efter afkøling til rumtemperatur tilsættes 20 ml natriumcarbonatopløsning (3.15) og blandes. Der tilsættes straks 100 ml ethylacetat (3.4), og glasset rystes kraftigt i hånden i 15 sekunder. Derefter anbringes glasset i 3 minutter i ultralydsbadet (4.1), og proppen løsnes. Der centrifugeres i 2 minutter, og ethylacetatfasen hældes gennem et glasfiberfilter (4.6) over i en 500 ml skilletragt. Ekstraktionen af prøven gentages med en ny portion på 100 ml ethylacetat. De samlede ekstrakter vaskes i 1 minut med 50 ml saltmættet natriumcarbonatopløsning (3.16), og den vandige fase kasseres.
Den organiske fase ekstraheres i 1 minut med 50 ml saltsyre (3.17). Den nederste syrefase aftappes i en 250 ml skilletragt. Den organiske fase ekstraheres igen i 1,5 minutter med yderligere 50 ml saltsyre og blandes med det første ekstrakt. De samlede syreekstrakter vaskes ved omrystning i ca. 10 sekunder med 10 ml ethylacetat (3.4).
Den vandige fase overføres kvantitativt til en 250 ml rundbundet kolbe, og den organiske fase kasseres. Al den resterende ethylacetat inddampes fra syreopløsningen ved brug af en rotationsfordamper (4.2). Vandbadets temperatur må ikke være over 40 oC. Under et vakuum på ca. 25 mbar vil al den resterende ethylacetat blive fjernet i løbet af 5 minutter ved 38 oC.
5.3. Oprensning
5.3.1.
Der klargøres en XAD-2-kolonne for hvert prøveekstrakt. 10 g behandlet amberlit (3.19) overføres med methanol (3.8) til en glaskolonne (4.5). Der anbringes en lille tot glasuld på toppen af resinkolonnen. Methanolen aftappes fra kolonnen, og resinet vaskes med 100 ml vand, idet der standses, når væsken når toppen af resinkolonnen. Kolonnen henstår for at komme i ligevægt i 10 minutter inden brug. Man må aldrig lade kolonnen løbe tør.
5.3.2.
Ekstraktet (5.2) overføres kvantitativt til den klargjorte amberlitkolonne (5.3.1), og der elueres; eluatet kasseres. Elueringshastigheden må ikke være over 20 ml/minuttet. Den rundbundede kolbe skylles med 20 ml saltsyre (3.17), som derefter benyttes til at rense resinkolonnen. Eventuelt resterende syreopløsning fjernes med luft. Skyllevæskerne kasseres. 100 ml methanol (3.8) tilsættes til kolonnen, og 5–10 ml elueres; eluatet opsamles i en 250 ml rundbundet kolbe. Den resterende methanol henstår i 10 minutter for at komme i ligevægt med resinkolonnen, og elueringen fortsættes ved en hastighed på ikke over 20 ml/minuttet; eluatet opsamles i samme rundbundede kolbe. Methanolen inddampes på rotationsfordamperen (4.2); vandbadets temperatur må ikke være på over 40 oC. Remanensen overføres kvantitativt til en 10 ml målekolbe ved brug af den mobile fase (3.21). Der fyldes op til mærket med mobil fase og blandes. En alikvot mængde filtreres gennem et membranfilter (4.7). Denne opløsning gemmes til HPLC-bestemmelsen (5.4).
5.4. HPLC-bestemmelse
5.4.1.
Nedenstående betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater.
HPLC-kolonne (4.4.1)
Mobil fase til HPLC (3.21)
Flow: 1,5 –2 ml/minuttet
Detektionsbølgelængde: 243 nm
Injektionsvolumen: 40–100 μl.
Kromatografisystemets stabilitet kontrolleres, ved at der flere gange indsprøjtes kalibreringsopløsning (3.6.2) med 3,0 μg/ml, indtil der opnås konstante tophøjder (/-arealer) og retentionstider.
5.4.2.
Hver kalibreringsopløsning (3.6.2) indsprøjtes flere gange, og højden af toppene (arealerne) bestemmes for hver koncentration. Der tegnes en kalibreringskurve med kalibreringsopløsningernes middeltophøjder eller -arealer som ordinat og de tilsvarende koncentrationer i μg/ml som abscisse.
5.4.3.
Prøveekstraktet (5.3.2) indsprøjtes flere gange, idet der benyttes samme mængde som til kalibreringsopløsningerne, og middeltophøjden (-arealet) af halofuginontoppene bestemmes.
6. Beregning af resultater
Ud fra middelhøjden (-arealet) af prøveopløsningens halofuginontoppe bestemmes koncentrationen i prøveopløsningen i μg/ml ved benyttelse af kalibreringskurven (5.4.2).
Prøvens indhold af halofuginon w (mg/kg) beregnes efter følgende formel:
![]()
hvor:
|
c |
= |
prøveopløsningens halofuginonkoncentration i μg/ml |
|
m |
= |
testportionens vægt i gram. |
7. Validering af resultater
7.1. Identitet
Analyttens identitet kan bekræftes ved ko-kromatografi eller ved at benytte en diodearraydetektor, hvormed spektrene for prøveekstraktet og kalibreringsopløsningen (3.6.2) indeholdende 6,0 μg/ml sammenlignes.
7.1.1.
Et prøveekstrakt spikes ved tilsætning af en passende mængde kalibreringsopløsning (3.6.2). Den tilsatte mængde halofuginon skal svare til den skønnede mængde halofuginon, der er fundet i prøveekstraktet.
Kun højden af halufuginontoppen må være blevet øget, efter at der er taget hensyn til både den tilsatte mængde og fortyndingen af ekstraktet. Toppens bredde skal i den halve højde ligge inden for ± 10 % af den oprindelige bredde.
7.1.2.
Resultaterne vurderes i overensstemmelse med følgende kriterier:
a) Den maksimale absorptionsbølgelængde for prøvens og standardens spektre, målt ved toppens spids på kromatogrammet, skal være den samme inden for en margin, der afhænger af detektionssystemets opløsningsevne. For diodearraydetektion ligger denne typisk inden for ± 2 nm.
b) Mellem 225 og 300 nm må prøvens og standardens spektre registreret ved toppens spids på kromatogrammet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af standardanalyttens absorbans.
c) Mellem 225 og 300 nm må spektrene ved toppens forside, spids og bagside frembragt med prøveekstraktet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem spektrene i intet observeret punkt overstiger 15 % af spektrets absorbans i spidsen.
Hvis et af disse kriterier ikke er opfyldt, er analyttens tilstedeværelse ikke bekræftet.
7.2. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må højst være på 0,5 mg/kg for halofuginonindhold på op til 3 mg/kg.
7.3. Genfinding
For den spikede blindprøve skal genfindingen være mindst 80 %.
8. Resultater af ringanalyse
Der er gennemført en ringanalyse ( 34 ) hvor 3 prøver blev analyseret af 8 laboratorier.
Resultater
|
|
Prøve A (blind) ved modtagelse |
Prøve B (mel) |
Prøve C (piller) |
||
|
|
|
ved modtagelse |
efter 2 måneder |
ved modtagelse |
efter 2 måneder |
|
Middelværdi [mg/kg] |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [ %] |
— |
16 |
18 |
14 |
17 |
|
Genf. [ %] |
|
86 |
74 |
88 |
75 |
|
ND |
= |
ikke påvist |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed ( %) |
|
Genf. |
= |
genfinding ( %). |
E. BESTEMMELSE AF ROBENIDIN
1,3-Bis[(4-chlorbenzyliden)amino]guanidinhydrochlorid
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af robenidin i foder. Bestemmelsesgrænsen er 5 mg/kg.
2. Princip
Prøven ekstraheres med sur methanol. Ekstraktet tørres, og en alikvot mængde oprenses på en aluminiumoxidkolonne. Robenidinet elueres fra kolonnen med methanol og koncentreres, og rumfanget øges i passende grad ved tilsætning af mobil fase. Indholdet af robenidin bestemmes ved omvendt-fase-HPLC med anvendelse af UV-detektor.
3. Reagenser
3.1. Methanol.
3.2. Sur methanol
4,0 ml saltsyre (ρ20 = 1,18 g/ml) overføres til en 500 ml målekolbe, hvorefter der fyldes op til mærket med methanol (3.1) og blandes. Denne opløsning skal fremstilles umiddelbart før brugen.
3.3. Acetonitril, svarende til HPLC-kvalitet
3.4. Molekylsi
Type 3A, 8–12 mesh (1,6 –2,5 mm kugler, krystallinsk aluminiumsilikat, porediameter: 0,3 mm).
3.5. Aluminiumoxid, sur, aktivitetskvalitet I til søjlekromatografi
100 g aluminiumoxid overføres til en egnet beholder, og der tilsættes 2,0 ml vand. Beholderen tilproppes og rystes i ca. 20 minutter. Væsken opbevares i en godt tilproppet beholder.
3.6. Kaliumdihydrogenphosphatopløsning, c = 0,025 mol/liter
3,40 g kaliumdihydrogenphosphat opløses i vand (HPLC-kvalitet) i en 1 000 ml målekolbe, hvorefter der fyldes op til mærket og blandes.
3.7. Dinatriumhydrogenphosphatopløsning, c = 0,025 mol/liter
3,55 g vandfrit (eller 4,45 g dihydrat eller 8,95 g dodecahydrat) dinatriumhydrogenphosphat opløses i vand (svarende til HPLC-kvalitet) i en 1 000 ml målekolbe, hvorefter der fyldes op til mærket og blandes.
3.8. Mobil fase til HPLC
Følgende reagenser blandes:
650 ml acetonitril (3.3)
250 ml vand (svarende til HPLC-kvalitet)
50 ml kaliumdihydrogenphosphatopløsning (3.6)
50 ml dinatriumhydrogenphosphatopløsning (3.7).
Der filtreres gennem et 0,22 μm filter (4.6), og opløsningen afgasses (f.eks. ved ultralydsbehandling i 10 minutter).
3.9. Standardstof
Rent robenidin: 1,3 -bis[(4-chlorbenzyliden)amino)] guanidinhydrochlorid.
3.9.1.
30 mg robenidinstandardstof (3.9) afvejes med en nøjagtighed på 0,1 mg og opløses i sur methanol (3.2) i en 100 ml målekolbe, hvorefter der fyldes op til mærket med samme opløsningsmiddel og blandes. Kolben omvikles med aluminiumfolie og opbevares i mørke.
3.9.2.
10,0 ml af standardstamopløsningen (3.9.1) overføres til en 250 ml målekolbe, hvorefter der fyldes til mærket med mobil fase (3.8) og blandes. Kolben omvikles med aluminiumfolie og opbevares i mørke.
3.9.3.
Henholdsvis 5,0 , 10,0 , 15,0 , 20,0 og 25,0 ml af standardmellemopløsningen (3.9.2) overføres til hver sin 50 ml målekolbe. Der fyldes op til mærket med mobil fase (3.8) og blandes. Disse opløsninger svarer til henholdsvis 1,2 , 2,4 , 3,6 , 4,8 og 6,0 μg/ml robenidin. Disse opløsninger skal fremstilles umiddelbart før brugen.
3.10. Vand, svarende til HPLC-kvalitet.
4. Apparatur
4.1. Glaskolonne
Fremstillet af brunt glas og forsynet med stophane og med en kapacitet på ca. 150 ml, indvendig diameter: 10–15 mm, længde: 250 mm.
4.2. Mekanisk rysteapparat eller magnetomrører
4.3. Rotationsfordamper
4.4. HPLC-udstyr med ultraviolet detektor med variabel bølgelængde eller med diodearraydetektor, som arbejder i området fra 250 til 400 nm
4.4.1. HPLC-kolonne: 300 mm × 4 mm, C18, 10 μm pakkemateriale, eller tilsvarende
4.5. Glasfiberfiltrerpapir (Whatman GF/A eller tilsvarende)
4.6. Membranfiltre, 0,22 μm
4.7. Membranfiltre, 0,45 μm.
5. Fremgangsmåde
|
NB |
Robenidin er lysfølsomt. Der anvendes udstyr af brunt glas i alle arbejdsgange. |
5.1. Generelt
5.1.1. Der analyseres en blindprøve for at kontrollere, at hverken robenidin eller interfererende stoffer er til stede.
5.1.2. Der udføres en genfindingstest ved at analysere blindprøven (5.1.1), som er blevet spiket ved tilsætning af en mængde robenidin svarende til den, der er til stede i prøven. For at nå op på et niveau på 60 mg/kg overføres 3,0 ml af standardstamopløsningen (3.9.1) til en 250 ml konisk kolbe. Opløsningen inddampes til ca. 0,5 ml i en nitrogenstrøm. Der tilsættes 15 g af blindprøven, blandes og ventes i 10 minutter, inden der fortsættes med ekstraktion (5.2).
|
NB |
Til denne metode anvendes en blindprøve af samme type som analyseprøven, og som ved analyse viser sig ikke at indeholde robenidin. |
5.2. Ekstraktion
Ca. 15 g af den klargjorte prøve afvejes med en nøjagtighed på 0,01 g, overføres til en 250 ml konisk kolbe og tilsættes 100,0 ml sur methanol (3.2), hvorefter kolben tilproppes og rystes i 1 time på rysteapparatet (4.2). Opløsningen filtreres gennem et glasfiberfiltrerpapir (4.5), og hele filtratet opsamles i en 150 ml konisk kolbe. Der tilsættes 7,5 mg molekylsi (3.4), tilproppes og omrystes i 5 minutter. Derefter filtreres omgående gennem et glasfiberfiltrerpapir. Denne opløsning anvendes til oprensningsprocessen (5.3).
5.3. Oprensning
5.3.1.
En lille prop af glasuld anbringes i den nedre ende af en glaskolonne (4.1) og skubbes helt ned med en glasstang. 11,0 g af den klargjorte aluminiumoxid (3.5) afvejes og overføres til kolonnen. Eksponeringen for luft begrænses mest muligt på dette stadium. Der bankes let på kolonnens nederste ende for at få aluminiumoxiden til at sætte sig.
5.3.2.
Med pipette overføres 5,0 ml af det under (5.2) fremstillede prøveekstrakt til kolonnen. Man holder spidsen af pipetten nær kolonnevæggen og lader opløsningen absorbere af aluminiumoxiden. Robenidinet elueres fra kolonnen under anvendelse af 100 ml methanol (3.1) med en gennemstrømningshastighed på 2–3 ml/minuttet, og eluatet opsamles i en 250 ml rundbundet kolbe. Methanolopløsningen inddampes til tørhed under reduceret tryk ved 40 oC ved hjælp af en rotationsfordamper (4.3). Remanensen opløses i 3–4 ml mobil fase (3.8) og overføres kvantitativt til en 10 ml målekolbe. Kolben skylles med flere portioner på 1–2 ml mobil fase, som overføres til målekolben. Der fyldes op til mærket med samme opløsningsmiddel og blandes. En alikvot mængde filtreres gennem et 0,45 μm membranfilter (4.7). Denne opløsning gemmes til HPLC-bestemmelsen (5.4).
5.4. HPLC-bestemmelse
5.4.1.
Følgende betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater:
HPLC-kolonne (4.4.1)
Mobil fase til HPLC (3.8)
Flow: 1,5 –2 ml/minuttet
Detektorbølgelængde: 317 nm
Injektionsvolumen: 20–50 μl.
Kromatografisystemets stabilitet kontrolleres, ved at der flere gange indsprøjtes kalibreringsopløsning (3.9.3) med 3,6 μg/ml, indtil der opnås konstante tophøjder og retentionstider.
5.4.2.
Hver kalibreringsopløsning (3.9.3) indsprøjtes flere gange, og højden af toppene (arealerne) bestemmes for hver koncentration. Der tegnes en kalibreringskurve med kalibreringsopløsningernes middeltophøjder eller -arealer som ordinat og de tilsvarende koncentrationer i μg/ml som abscisse.
5.4.3.
Prøveekstraktet (5.3.2) indsprøjtes flere gange, idet der benyttes samme mængde som til kalibreringsopløsningerne, og middeltophøjden (-arealet) af robenidintoppene bestemmes.
6. Beregning af resultater
Ud fra middelhøjden (-arealet) af prøveopløsningens robenidintoppe bestemmes koncentrationen i prøveopløsningen i μg/ml ved benyttelse af kalibreringskurven (5.4.2).
Prøvens indhold af robenidin w (mg/kg) beregnes efter ved følgende formel:
![]()
hvor:
|
c |
= |
prøveopløsningens robenidinkoncentration i μg/ml |
|
m |
= |
testportionens vægt i gram. |
7. Validering af resultater
7.1. Identitet
Analyttens identitet kan bekræftes ved ko-kromatografi eller ved at benytte en diodearraydetektor, hvormed spektrene for prøveekstraktet og kalibreringsopløsningen (3.9.3) indeholdende 6 μg/ml sammenlignes.
7.1.1.
Et prøveekstrakt spikes ved tilsætning af en passende mængde kalibreringsopløsning (3.9.3). Den tilsatte mængde robenidin skal svare til den skønnede mængde robenidin, der er fundet i prøveekstraktet.
Kun højden af robenidintoppen må være blevet øget, efter at der er taget hensyn til både den tilsatte mængde og fortyndingen af ekstraktet. Toppens bredde skal i den halve højde ligge inden for ca. 10 % af den oprindelige bredde.
7.1.2.
Resultaterne vurderes i overensstemmelse med følgende kriterier:
a) Den maksimale absorptionsbølgelængde for prøvens og standardens spektre, målt ved toppens spids på kromatogrammet, skal være den samme inden for en margin, der afhænger af detektionssystemets opløsningsevne. Ved diodearraydetektion er den typisk ca. 2 nm.
b) Mellem 250 og 400 nm må prøvens og standardens spektre registreret ved toppens spids på kromatogrammet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af standardanalyttens absorbans.
c) Mellem 250 og 400 nm må spektrene ved toppens forside, spids og bagside frembragt med prøveekstraktet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem spektrene i intet observeret punkt overstiger 15 % af spektrets absorbans i spidsen.
Hvis et af disse kriterier ikke er opfyldt, er analyttens tilstedeværelse ikke bekræftet.
7.2. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige 10 % af det højeste resultat for robenidinindhold på over 15 mg/kg.
7.3. Genfinding
For den spikede blindprøve skal genfindingen være mindst 85 %.
8. Resultater af ringanalyse
Der er gennemført en EF-ringanalyse, hvor 4 prøver af fjerkræ- og kaninfoder i form af mel eller piller blev analyseret af 12 laboratorier. Der blev foretaget dobbeltanalyse af hver prøve. Resultaterne er vist i nedenstående skema:
|
|
Fjerkræ |
Kaniner |
||
|
|
Mel |
Piller |
Mel |
Piller |
|
Middelværdi [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [%] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [%] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Genfinding [%] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
standardafvigelse for repeterbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed, % |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed, %. |
F. BESTEMMELSE AF DICLAZURIL
2,6-Chlor-alpha-(4-chlorphenyl)-4-[4,5-dihydro-3,5-dioxo-1,2,4-triazin-2(3H)-yl]-benzenacetonitril
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af diclazuril i foder og forblandinger. Detektionsgrænsen er 0,1 mg/kg, og bestemmelsesgrænsen er 0,5 mg/kg.
2. Princip
Efter tilsætning af en intern standard ekstraheres prøven med sur methanol. For foders vedkommende oprenses en alikvot del af ekstraktet på en C18-fastfase-ekstraktionspatron. Diclazuril elueres fra patronen med en blanding af sur methanol og vand. Efter inddampning opløses remanensen i DMF/vand. For forblandingers vedkommende inddampes ekstraktet, og remanensen opløses i DMF/vand. Indholdet af diclazuril bestemmes ved ternær gradient-HPLC i omvendt fase med anvendelse af UV-detektor.
3. Reagenser
3.1. Vand, svarende til HPLC-kvalitet.
3.2. Ammoniumacetat.
3.3. Tetrabutylammoniumhydrogensulfat (TBHS).
3.4. Acetonitril, svarende til HPLC-kvalitet.
3.5. Methanol, svarende til HPLC-kvalitet.
3.6. N, N-Dimethylformamid (DMF).
3.7. Saltsyre, ρ20 = 1,19 g/ml.
3.8. Standardstof: diclazuril II-24: 2,6-chlor-alpha-(4-chlorphenyl)-4-[4,5-dihydro-3,5-dioxo-1,2,4-triazin-2(3H)-yl)-benzenacetonitril med garanteret renhed, E 771.
3.8.1.
Med en nøjagtighed på 0,1 mg afvejes 25 mg diclazurilstandardstof (3.8) i en 50 ml målekolbe. Der opløses i DMF (3.6), fyldes op til mærket med DMF (3.6) og blandes. Kolben omvikles med aluminiumfolie, eller der anvendes en brun kolbe, og opløsningen opbevares i køleskab. Opløsningen er stabil i en måned ved en temperatur på ≤ 4 oC.
3.8.2.
Der overføres 5,00 ml af standardstamopløsningen (3.8.1) til en 50 ml målekolbe, fyldes op til mærket med DMF (3.6) og blandes. Kolben omvikles med aluminiumfolie, eller der anvendes en brun kolbe, og opløsningen opbevares i køleskab. Opløsningen er stabil i en måned ved en temperatur på ≤ 4 oC.
3.9. Internt standardstof: 2,6-dichlor-α-(4-chlorphenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3H)-yl)-α-methylbenzenacetonitril.
3.9.1.
Med en nøjagtighed på 0,1 mg afvejes 25 mg internt standardstof (3.9) i en 50 ml målekolbe. Der opløses i DMF (3.6), fyldes op til mærket med DMF (3.6) og blandes. Kolben omvikles med aluminiumfolie, eller der anvendes en brun kolbe, og opløsningen opbevares i køleskab. Opløsningen er stabil i en måned ved en temperatur på ≤ 4 oC.
3.9.2.
Der overføres 5,00 ml af den interne standardstamopløsning (3.9.1) til en 50 ml målekolbe, fyldes op til mærket med DMF (3.6) og blandes. Kolben omvikles med aluminiumfolie, eller der anvendes en brun kolbe, og opløsningen opbevares i køleskab. Opløsningen er stabil i en måned ved en temperatur på ≤ 4 oC.
3.9.3.
(p = nominelt indhold af diclazuril i forblandingen i mg/kg)
Med en nøjagtighed på 0,1 mg afvejes p/10 mg af det interne standardstof i en 100 ml målekolbe. Der opløses i DMF (3.6) i et ultralydsbad (4.6), fyldes op til mærket med DMF og blandes. Kolben omvikles med aluminiumfolie, eller der anvendes en brun kolbe, og kolben opbevares i køleskab. Opløsningen er stabil i en måned ved en temperatur på ≤ 4 oC.
3.10. Kalibreringsopløsning, 2 μg/ml
Der afpipetteres 2,00 ml diclazurilstandardopløsning (3.8.2) og 2,00 ml intern standardopløsning (3.9.2) i en 50 ml målekolbe. Der tilsættes 16 ml DMF (3.6), fyldes op til mærket med vand og blandes. Opløsningen skal fremstilles umiddelbart før brugen.
3.11. C18-fastfase-ekstraktionspatron, f.eks. Bond Elut, størrelse: 1 cm3, sorbentmasse: 100 mg.
3.12. Ekstraktionsopløsningsmiddel: sur methanol
Der afpipetteres 5,0 ml saltsyre (3.7) i 1 000 ml methanol (3.5) og blandes.
3.13. Mobil fase til HPLC
3.13.1. Eluent A: ammoniumacetat-tetrabutylammoniumhydrogensulfat-opløsning
Der opløses 5 g ammoniumacetat (3.2) og 3,4 g TBHS (3.3) i 1 000 ml vand (3.1) og blandes.
3.13.2. Eluent B: acetonitril (3.4).
3.13.3. Eluent C: methanol (3.5).
4. Apparatur
4.1. Mekanisk rysteapparat
4.2. Udstyr til ternær gradient-HPLC
4.2.1. HPLC-kolonne, Hypersil ODS, 3 μm pakkemateriale, 100 mm x 4,6 mm, eller tilsvarende
4.2.2. UV-detektor med variabel bølgelængde eller diodearraydetektor
4.3. Rotationsfordamper
4.4. Membranfilter, 0,45 μm
4.5. Vakuummanifold
4.6. Ultralydsbad.
5. Fremgangsmåde
5.1. Generelt
5.1.1.
Der analyseres en blindprøve for at kontrollere, at hverken diclazuril eller interfererende stoffer er til stede. Blindprøven skal være af samme type som den prøve, der skal undersøges, og ved analyse må diclazuril eller interfererende stoffer ikke påvises.
5.1.2.
Der udføres en genfindingstest ved at analysere blindprøven, som er blevet spiket ved tilsætning af en mængde diclazuril svarende til den, der er til stede i prøven. For at nå op på et niveau på 1 mg/kg tilsættes der 0,1 ml af standardstamopløsningen (3.8.1) til 50 g af en blindprøve, der blandes omhyggeligt, og efter henstand i 10 minutter blandes igen flere gange, før man går videre (5.2).
Hvis der ikke er en blindprøve af samme type som analyseprøven til rådighed (se punkt 5.1.1), kan der alternativt udføres en genfindingstest ved hjælp af standardtilsætningsmetoden. I dette tilfælde spikes den prøve, der skal analyseres, ved tilsætning af en mængde diclazuril svarende til den, der allerede er til stede i prøven. Denne prøve analyseres sammen med den ikke-spikede prøve, og genfindingen kan beregnes ved subtraktion.
5.2. Ekstraktion
5.2.1.
Med en nøjagtighed på 0,01 g afvejes der ca. 50 g prøve. Det overføres til en 500 ml konisk kolbe, der tilsættes 1,00 ml intern standardopløsning (3.9.2) og 200 ml ekstraktionsopløsningsmiddel (3.12), og kolben tilproppes. Blandingen rystes natten over på rysteapparatet (4.1). Henstilles til bundfældning i 10 minutter. En 20 ml alikvot af supernatanten overføres til en egnet glasbeholder og fortyndes med 20 ml vand. Denne opløsning overføres til en ekstraktionspatron (3.11) og ledes igennem ved hjælp af vakuum (4.5). Patronen vaskes med 25 ml af en blanding af ekstraktionsopløsningsmiddel (3.12) og vand, 65 + 35 (v + v). De opsamlede fraktioner kasseres, og forbindelserne elueres med 25 ml af en blanding af ekstraktionsopløsningsmiddel (3.12) og vand, 80 + 20 (v + v). Denne fraktion inddampes, indtil den netop når tørhed, ved hjælp af rotationsfordamperen (4.3) ved 60 oC. Remanensen opløses i 1,0 ml DMF (3.6), og der tilsættes 1,5 ml vand (3.1) og blandes. Der filtreres gennem et membranfilter (4.4). Der fortsættes med HPLC-bestemmelse (5.3).
5.2.2.
Med en nøjagtighed på 0,001 g afvejes der ca. 1 g prøve. Den overføres til en 500 ml konisk kolbe, der tilsættes 1,00 ml intern standardopløsning (3.9.3) og 200 ml ekstraktionsopløsningsmiddel (3.12), og kolben tilproppes. Blandingen rystes natten over på rysteapparatet (4.1). Henstilles til bundfældning i 10 minutter. En alikvot på 10 000 /p ml (p = nominelt indhold af diclazuril i forblandingen i mg/kg) af supernatanten overføres til en rundbundet kolbe af passende størrelse. Der inddampes, indtil den netop når tørhed, under reduceret tryk ved hjælp af rotationsfordamperen (4.3) ved 60 oC. Remanensen opløses i 10,0 ml DMF (3.6), og der tilsættes 15,0 ml vand (3.1) og blandes. Der fortsættes med HPLC-bestemmelse (5.3).
5.3. HPLC-bestemmelse
5.3.1.
Nedenstående betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater.
|
HPLC-kolonne (4.2.1): |
Hypersil ODS, 100 mm × 4,6 mm, 3 μm pakkemateriale, eller tilsvarende |
|
|
Mobil fase: |
Eluent A (3.13.1): |
Vandig opløsning af ammoniumacetat og tetrabutylammoniumhydrogen-sulfat |
|
|
Eluent B (3.13.2): |
acetonitril |
|
|
Eluent C (3.13.3): |
methanol |
|
Elueringsmåde: |
— lineær gradient — startbetingelser: A + B + C = 60 + 20 + 20 (V + V + V) — efter 10 minutters gradienteluering — i 30 minutter til: A + B + C = 45 + 20 + 35 (V + V + V). Der skylles med B i 10 minutter |
|
|
Flow: |
1,5 — 2 ml/minuttet |
|
|
Injektionsvolumen: |
20 μl |
|
|
Detektorbølgelængde: |
280 nm. |
|
Kromatografisystemets stabilitet kontrolleres, ved at der flere gange indsprøjtes kalibreringsopløsning (3.10) med 2,0 μg/ml, indtil der opnås konstante tophøjder og retentionstider.
5.3.2.
Der indsprøjtes 20 μl af kalibreringsopløsningen (3.10) flere gange, og middeltophøjden (-arealet) af toppene for diclazuril og intern standard bestemmes.
5.3.3.
Der indsprøjtes 20 μl af prøveopløsningen (5.2.1. eller 5.2.2) flere gange, og middeltophøjden (-arealet) af toppene for diclazuril og intern standard bestemmes.
6. Beregning af resultater
6.1. Foder
Diclazurilindholdet w (mg/kg) i prøven beregnes efter følgende formel:
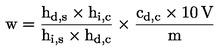
[mg/kg]
hvor:
|
hd, s |
= |
tophøjde (-areal) for diclazuril i prøveopløsningen (5.2.1) |
|
hi, s |
= |
tophøjde (-areal) for den interne standard i prøveopløsningen (5.2.1) |
|
hd, c |
= |
tophøjde (-areal) for diclazuril i kalibreringsopløsningen (3.10) |
|
hi, c |
= |
tophøjde (-areal) for den interne standard i kalibreringsopløsningen (3.10) |
|
cd, c |
= |
kalibreringsopløsningens diclazurilkoncentration i μg/ml (3.10) |
|
m |
= |
testportionens vægt i gram |
|
V |
= |
prøveekstraktets volumen ifølge 5.2.1 (dvs. 2,5 ml). |
6.2. Forblandinger
Diclazurilindholdet w (mg/kg) i prøven beregnes efter følgende formel:
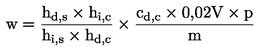
[mg/kg]
hvor:
|
hd, c |
= |
tophøjde (-areal) for diclazuril i kalibreringsopløsningen (3.10) |
|
hi, c |
= |
tophøjde (-areal) for den interne standard i kalibreringsopløsningen (3.10) |
|
hd, s |
= |
tophøjde (-areal) for diclazuril i prøveopløsningen (5.2.2) |
|
hi, s |
= |
tophøjde (-areal) for den interne standard i prøveopløsningen (5.2.2) |
|
cd, c |
= |
kalibreringsopløsningens diclazurilkoncentration i μg/ml (3.10) |
|
m |
= |
testportionens vægt i gram |
|
V |
= |
prøveekstraktets volumen ifølge 5.2.2 (dvs. 25 ml) |
|
p |
= |
nominelt indhold af diclazuril i forblandingen i mg/kg. |
7. Validering af resultater
7.1. Identitet
Analyttens identitet kan bekræftes ved ko-kromatografi eller ved at benytte en diodearraydetektor, hvormed spektrene for prøveekstraktet (5.2.1 eller 5.2.2) og kalibreringsopløsningen (3.10) sammenlignes.
7.1.1.
Et prøveekstrakt (5.2.1 eller 5.2.2) spikes ved tilsætning af en passende mængde kalibreringsopløsning (3.10). Den tilsatte mængde diclazuril skal svare til den mængde diclazuril, der er fundet i prøveekstraktet.
Kun højden af diclazuriltoppen og intern standard-toppen må være blevet øget, efter at der er taget hensyn til både den tilsatte mængde og fortyndingen af ekstraktet. Toppens bredde skal i den halve højde ligge inden for ± 10 % af den oprindelige bredde af diclazuriltoppen eller intern standard-toppen for det ikke-spikede prøveekstrakt.
7.1.2.
Resultaterne vurderes i overensstemmelse med følgende kriterier:
a) Den maksimale absorptionsbølgelængde for prøvens og standardens spektre, målt ved toppens spids på kromatogrammet, skal være den samme inden for en margin, der afhænger af detektionssystemets opløsningsevne. For diodearraydetektion ligger denne typisk inden for ± 2 nm.
b) Mellem 230 og 320 nm må prøvens og standardens spektre registreret ved toppens spids på kromatogrammet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af standardanalyttens absorbans.
c) Mellem 230 og 320 nm må spektrene ved toppens forside, spids og bagside frembragt med prøveekstraktet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af absorbansen for spektret i toppens spids.
Hvis et af disse kriterier ikke er opfyldt, er analyttens tilstedeværelse ikke bekræftet.
7.2. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige:
— 30 % i relativ værdi (af det højeste resultat) for diclazurilindhold på 0,5 –2,5 mg/kg
— 0,75 mg/kg for diclazurilindhold på 2,5 –5 mg/kg
— 15 % i relativ værdi (af det højeste resultat) for diclazurilindhold på mere end 5 mg/kg.
7.3. Genfinding
For en spiket (blind)prøve skal genfindingen være mindst 80 %.
8. Resultater af ringanalyse
Der er gennemført en ringanalyse, hvor 5 prøver blev analyseret af 11 laboratorier. Disse prøver bestod af to forblandinger; den ene var blandet med en organisk matrix (O 100), og den anden med en uorganisk matrix (A 100). Det teoretiske indhold var 100 mg diclazuril pr. kg. De tre blandede kyllingefoderstoffer var fremstillet af 3 forskellige producenter (NL) (Ll/Z1/K1). Det teoretiske indhold var 1 mg diclazuril pr. kg. Laboratorierne blev instrueret om at analysere hver af prøverne én gang eller som dobbeltbestemmelse (Journal of AOAC International, bind 77, nr. 6, 1994, s. 1359–1361, indeholder nærmere oplysninger om denne ringprøve). Resultaterne er vist i nedenstående tabel:
|
|
Prøve 1 A 100 |
Prøve 2 O 100 |
Prøve 3 L1 |
Prøve 4 Z1 |
Prøve 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Middelværdi |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr ( %) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR ( %) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Nominelt indhold (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enkeltværdier |
|
Sr |
= |
standardafvigelse for repeterbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed. |
9. Bemærkninger
Diclazurilresponset skal tidligere være påvist at være lineær i de koncentrationsområder, hvori der måles.
G. BESTEMMELSE AF LASALOCIDNATRIUM
Natriumsalt af en monocarboxylsyrepolyether, der dannes af Streptomyces lasaliensis
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af lasalocidnatrium i foder og forblandinger. Detektionsgrænsen er 5 mg/kg, og bestemmelsesgrænsen er 10 mg/kg.
2. Princip
Lasalocidnatrium ekstraheres af prøven i sur methanol og bestemmes ved omvendt fase-HPLC med anvendelse af spektrofluorometrisk detektor.
3. Reagenser
3.1. Kaliumdihydrogenphosphat (KH2PO4).
3.2. Orthophosphorsyre, w (w/w) = 85 %.
3.3. Orthophosphorsyreopløsning, c = 20 %
23,5 ml ortophosphorsyre (3.2) fortyndes med vand til 100 ml.
3.4. 6-Methyl-2-heptylamin(1,5-dimethylhexylamin), w (w/w) = 99 %.
3.5. Methanol, svarende til HPLC-kvalitet.
3.6. Saltsyre, massefylde: 1,19 g/ml.
3.7. Phosphatbufferopløsning, c = 0,01 mol/liter
1,36 g KH2PO4 (3.1) opløses i 500 ml vand (3.11), og der tilsættes 3,5 ml orthophosphorsyre (3.2) og 10,0 ml 6-methyl-2-heptylamin (3.4). pH-værdien justeres til 4,0 med orthophosphorsyreopløsning (3.3), og der fortyndes til 1 000 ml med vand (3.11).
3.8. Sur methanol
Der overføres 5,0 ml saltsyre (3.6) til en 1 000 ml målekolbe, fyldes op til mærket med methanol (3.5) og blandes. Opløsningen skal fremstilles umiddelbart før brugen.
3.9. Mobil fase til HPLC, phosphatbuffermethanolopløsning 5 + 95, (V + V)
5 ml phosphatbufferopløsning (3.7) blandes med 95 ml methanol (3.5).
3.10. Lasalocidnatriumstandardstof med garanteret renhed, C34H53O8Na (natriumsalt af en monocarboxylsyrepolyether, der dannes af Streptomyces lasaliensis), E 763.
3.10.1.
50 mg lasalocidnatrium (3.10) afvejes med en nøjagtighed på 0,1 mg i en 100 ml målekolbe og opløses i sur methanol (3.8), og der fyldes op til mærket med samme opløsningsmiddel og blandes. Denne opløsning skal fremstilles umiddelbart før brugen.
3.10.2.
Der afpipetteres 10,0 ml standardstamopløsning (3.10.1) i en 100 ml målekolbe, fyldes op til mærket med sur methanol (3.8) og blandes. Opløsningen skal fremstilles umiddelbart før brugen.
3.10.3.
Henholdsvis 1,0 , 2,0 , 4,0 , 5,0 og 10,0 ml af standardmellemopløsningen (3.10.2) overføres til hver sin 50 ml målekolbe. Der fyldes op til mærket med sur methanol (3.8) og blandes. Disse opløsninger svarer til henholdsvis 1,0 , 2,0 , 4,0 , 5,0 og 10,0 μg lasalocidnatrium pr. ml. Disse opløsninger skal fremstilles umiddelbart før brugen.
3.11. Vand, svarende til HPLC-kvalitet.
4. Apparatur
4.1. Ultralydsbad (eller vandbad med rysteanordning) med temperaturkontrol
4.2. Membranfiltre, 0,45 μm
4.3. HPLC-udstyr med injektionssystem, egnet til injektionsvolumener på 20 μl
4.3.1. HPLC-kolonne, 125 mm x 4 mm, omvendt-fase-C18, 5 μm pakkemateriale, eller tilsvarende
4.3.2. Spektrofluorometer med variabel indstilling af excitations- og emissionsbølgelængden.
5. Fremgangsmåde
5.1. Generelt
5.1.1.
Til gennemførelse af genfindingstesten (5.1.2) analyseres en blindprøve for at kontrollere, at hverken lasalocidnatrium eller interfererende stoffer er til stede. Blindprøven skal være af samme type som den prøve, der skal undersøges, og ved analyse må lasalocidnatrium eller interfererende stoffer ikke påvises.
5.1.2.
Der udføres en genfindingstest ved at analysere blindprøven, som er blevet spiket ved tilsætning af en mængde lasalocidnatrium svarende til den, der er til stede i prøven. For at nå op på et niveau på 100 mg/kg overføres der 10,0 ml standardstamopløsning (3.10.1) til en 250 ml konisk kolbe, og opløsningen inddampes til ca. 0,5 ml. Der tilsættes 50 g af blindprøven og blandes omhyggeligt, og efter henstand i 10 minutter blandes der på ny flere gange, inden der fortsættes med ekstraktion (5.2).
Hvis der ikke er en blindprøve af samme type som analyseprøven til rådighed (se punkt 5.1.1), kan der alternativt udføres en genfindingstest ved hjælp af standardtilsætningsmetoden. I dette tilfælde spikes den prøve, der skal analyseres, ved tilsætning af en mængde lasalocidnatrium svarende til den, der allerede er til stede i prøven. Denne prøve analyseres sammen med den ikke-spikede prøve, og genfindingen beregnes ved subtraktion.
5.2. Ekstraktion
5.2.1.
Med en nøjagtighed på 0,01 g afvejes 5–10 g af prøven i en 250 ml konisk kolbe med prop. Der tilsættes 100,0 ml sur methanol (3.8) med pipette. Proppen sættes løst på, og der omrystes forsigtigt, så prøven opslæmmes. Kolben anbringes i ultralydsbad (4.1) ved ca. 40 oC i 20 minutter, hvorefter den fjernes og afkøles til rumtemperatur. Kolben henstår i ca. 1 time, indtil opslæmningen har bundfældet sig, hvorpå en alikvot mængde filtreres gennem et 0,45 μm membranfilter (4.2) ned i en egnet beholder. Der fortsættes med HPLC-bestemmelse (5.3).
5.2.2.
Med en nøjagtighed på 0,001 g afvejes ca. 2 g af den ikke-formalede forblanding i en 250 ml målekolbe. Der tilsættes 100,0 ml sur methanol (3.8) og omrystes forsigtigt, så prøven opslæmmes. Kolben med indhold anbringes i ultralydsbad (4.1) ved ca. 40 oC i 20 minutter, hvorefter den fjernes og afkøles til rumtemperatur. Der fortyndes op til mærket med sur methanol (3.8) og blandes omhyggeligt. Kolben henstår i 1 time, indtil opslæmningen har bundfældet sig, hvorpå en alikvot mængde filtreres gennem et 0,45 μm membranfilter (4.2). En passende mængde af det klare filtrat fortyndes med sur methanol (3.8), så der fremkommer en endelig prøveopløsning, som indeholder ca. 4 μg/ml lasalocidnatrium. Der fortsættes med HPLC-bestemmelse (5.3).
5.3. HPLC-bestemmelse
5.3.1.
Nedenstående betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater.
|
HPLC-kolonne (4.3.1): |
125 mm × 4 mm, omvendt-fase-C18, 5 μm pakkemateriale, eller tilsvarende |
|
Mobil fase (3.9): |
Blanding af phosphatbufferopløsning (3.7) og methanol (3.5), 5 + 95 (v + v) |
|
Flow : |
1,2 ml/minuttet |
|
Detektionsbølgelængde: |
|
|
Excitation: |
310 nm |
|
Emission: |
419 nm |
|
Injektionsvolumen: |
20 μl. |
Kromatografisystemets stabilitet kontrolleres, ved at der flere gange indsprøjtes kalibreringsopløsning (3.10.3) med 4,0 μg/ml, indtil der opnås konstante tophøjder (/-arealer) og retentionstider.
5.3.2.
Hver kalibreringsopløsning (3.10.3) indsprøjtes flere gange, og middeltophøjderne (-arealerne) bestemmes for hver koncentration. Der tegnes en kalibreringskurve med middeltophøjderne (-arealerne) som ordinat og de tilsvarende koncentrationer i μg/ml som abscisse.
5.3.3.
Prøveekstrakterne fra 5.2.1 eller 5.2.2 indsprøjtes flere gange, idet der benyttes samme mængde som til kalibreringsopløsningerne, og middeltophøjderne (-arealerne) for lasalocidnatriumtoppene bestemmes.
6. Beregning af resultater
Ud fra middeltophøjden (-arealet), der er frembragt ved indsprøjtning af prøveopløsningen (5.3.3), bestemmes koncentrationen af lasalocidnatrium (μg/ml) ved benyttelse af kalibreringskurven.
6.1. Foder
Indholdet af lasalocidnatrium w (mg/kg) i prøven beregnes efter følgende formel:
![]()
[mg/kg]
hvor:
|
c |
= |
lasalocidnatriumkoncentrationen i prøveopløsningen (5.2.1) i μg/ml |
|
V1 |
= |
prøveekstraktets volumen ifølge 5.2.1 i ml (dvs. 100) |
|
m |
= |
testportionens vægt i gram. |
6.2. Forblandinger
Indholdet af lasalocidnatrium w (mg/kg) i prøven beregnes efter følgende formel:
![]()
[mg/kg]
hvor:
|
c |
= |
lasalocidnatriumkoncentrationen i prøveopløsningen (5.2.2) i μg/ml |
|
V2 |
= |
prøveekstraktets volumen ifølge 5.2.2 i ml (dvs. 250) |
|
f |
= |
fortyndingsfaktor ifølge 5.2.2 |
|
m |
= |
testportionens vægt i gram. |
7. Validering af resultater
7.1. Identitet
Metoder baseret på spektrofluorometri er mindre udsatte for interferens end metoder, hvor der anvendes UV-detektion. Analyttens identitet kan bekræftes ved ko-kromatografi.
7.1.1.
Et prøveekstrakt (5.2.1 eller 5.2.2) spikes ved tilsætning af en passende mængde kalibreringsopløsning (3.10.3). Den tilsatte mængde lasalocidnatrium skal svare til den mængde lasalocidnatrium, der er fundet i prøveekstraktet. Kun højden af lasalocidnatriumtoppen må være blevet øget, efter at der er taget hensyn til både den tilsatte mængde lasalocidnatrium og fortyndingen af ekstraktet. Toppens bredde skal i den halve højde ligge inden for ± 10 % af bredden af den oprindelige top for det ikke-spikede prøveekstrakt.
7.2. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige:
— 15 % i relativ værdi (af det højeste resultat) for lasalocidnatriumindhold på 30–100 mg/kg
— 15 mg/kg for lasalocidnatriumindhold på 100–200 mg/kg
— 7,5 % i relativ værdi (af det højeste resultat) for lasalocidnatriumindhold på over 200 mg/kg.
7.3. Genfinding
For den spikede foder(blind)prøve skal genfindingen være mindst 80 %. For de spikede forblandingprøver skal genfindingen være mindst 90 %.
8. Resultater af ringanalyse
Der er gennemført en ringanalyse ( 35 ), hvor 2 forblandinger (prøve 1 og 2) og 5 foderstoffer (prøve 3–7) blev analyseret af 12 laboratorier. Der blev foretaget dobbeltanalyse af hver prøve. Resultaterne er vist i nedenstående tabel:
|
|
Prøve 1 Forblanding (kyllinger) |
Prøve 2 Forblanding (kalkuner) |
Prøve 3 Kalkunpellets |
Prøve 4 Kyllingegranulat |
Prøve 5 Kalkunfoder |
Prøve 6 Kyllingefoder A |
Prøve 7 Kyllingefoder B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Middelværdi [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
Sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
SR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Nominelt indhold [mg/kg] |
5 000 (1) |
16 000 (1) |
80 (1) |
105 (1) |
120 (1) |
50 (2) |
35 (2) |
|
(1) Indhold oplyst af fabrikanten. (2) Foder tilberedt på laboratoriet. |
|||||||
|
L |
= |
antal laboratorier |
|
n |
= |
antal enkeltværdier |
|
Sr |
= |
standardafvigelse for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed, % |
|
CVR |
= |
variationskoefficient for reproducerbarhed, %. |
BILAG V
ANALYSEMETODER TIL KONTROL FOR UØNSKEDE STOFFER I FODER
A. BESTEMMELSE AF FRI GOSSYPOL OG TOTALGOSSYPOL
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af fri gossypol, totalgossypol og kemisk nært beslægtede substanser i frø, mel og kager af bomuldsfrø samt i foderblandinger indeholdende disse fodermidler, hvis indholdet af fri gossypol, totalgossypol og kemisk nært beslægtede stoffer er på over 20 mg/kg.
2. Princip
Gossypol ekstraheres under tilstedeværelse af 3-amino-1-propanol, enten med en blanding af propan-2-ol og hexan til bestemmelse af fri gossypol eller med dimethylformamid til bestemmelse af totalgossypol, og omsættes med anilin til gossypol-dianilin, hvis ekstinktion måles ved 440 nm.
3. Reagenser
3.1. Propan-2-ol-hexan-blanding: 60 volumendele propan-2-ol blandes med 40 volumendele n-hexan.
3.2. Opløsningsmiddel A: I en 1 liter målekolbe fyldes der ca. 500 ml propan-2-ol-hexan-blanding (3.1), 2 ml 3-amino-1-propanol, 8 ml iseddike og 50 ml vand, og der fyldes op med propan-2-ol-hexan-blanding (3.1) til mærket. Dette reagens er stabilt i 1 uge.
3.3. Opløsningsmiddel B: I en 100 ml målekolbe afpipetteres 2 ml 3-amino-1-propanol og 10 ml iseddike, hvorefter der afkøles til rumtemperatur og fyldes op med N, N-dimethylformamid til mærket. Dette reagens er stabilt i 1 uge.
3.4. Anilin: Såfremt ekstinktionen i blindprøven er større end 0,022 , destilleres anilinen over zinkstøv, idet de første og sidste 10 % af destillatet kasseres. I køleskab er dette reagens ved opbevaring i vel tillukket, brunt glas holdbart i flere måneder.
3.5. Standardgossypolopløsning A: I en 250 ml målekolbe opløses 27,9 mg gossypolacetat med opløsningsmiddel A (3.2), og der fyldes op til mærket med dette. 50 ml af denne opløsning afpipetteres i en 250 ml målekolbe og der fyldes op til mærket med opløsningsmiddel A. Gossypolkoncentrationen i denne opløsning er 0,02 mg/ml. Før brugen henstår denne opløsning i 1 time ved rumtemperatur.
3.6. Standardgossypolopløsning B: I en 50 ml målekolbe opløses 27,9 mg gossypolacetat i opløsningsmiddel B (3.3), og der fyldes op til mærket med dette. Gossypolkoncentrationen i denne opløsning er 0,5 mg/ml.
Standardgossypolopløsning A og B er stabile i 24 timer, såfremt de beskyttes mod lys.
4. Apparatur
4.1. Mekanisk rysteapparat: ca. 35 omdrejninger i minuttet
4.2. Spektrofotometer.
5. Fremgangsmåde
5.1. Afvejning af prøven
Størrelsen af den afvejede stofmængde afhænger af det formodede indhold af gossypol i prøven. Det er fordelagtigt at arbejde med en lille afvejning og med en relativt stor alikvot del af filtratet for at få en tilstrækkelig mængde gossypol til en nøjagtig fotometrisk måling. Til bestemmelse af fri gossypol i frø, mel og kager af bomuldsfrø må afvejningen ikke være på mere end 1 g, medens den for foderblandinger kan være på op til 5 g. En alikvot del af filtratet på 10 ml er i de fleste tilfælde passende; den skal indeholde 50–100 μg gossypol. Til bestemmelse af totalgossypol skal afvejningen ligge på 0,5 –5 g, således at der i en alikvot del af filtratet på 2 ml vil være et indhold på 40–200 μg gossypol.
Analyser udføres ved en rumtemperatur på ca. 20 oC.
5.2. Bestemmelse af fri gossypol
Den afvejede stofmængde anbringes i en 250 ml kolbe med slibhals, hvis bund er dækket med knust glas. 50 ml af opløsningsmiddel A (3.2) tilsættes med pipette, kolben lukkes, og der blandes i 1 time i rysteapparat. Derpå filtreres gennem et tørt filter, og filtratet opsamles i en lille kolbe med slibhals. Under filtreringen tildækkes tragten med et urglas.
Lige store alikvote dele af filtratet indeholdende 50–100 μg gossypol afpipetteres i to 25 ml målekolber (A og B). Eventuelt fyldes op med opløsningsmiddel A (3.2) til 10 ml. Derpå suppleres indholdet af kolben A med propan-2-ol-hexan-blandingen (3.1) til mærket. Denne opløsning anvendes som referenceopløsning ved målingen af prøveopløsningen.
10 ml opløsningsmiddel A (3.2) afpipetteres i to andre 25 ml målekolber (C og D). Indholdet i kolben (C) suppleres med propan-2-ol-hexan-blandingen (3.1) til mærket. Denne opløsning anvendes som referenceopløsning ved målingen af blindprøveopløsningen.
Hver af målekolberne (D) og (B) tilsættes 2 ml anilin (3.4). Der opvarmes i 30 minutter på kogende vandbad til udvikling af farvningen. Derefter afkøles til rumtemperatur, fortyndes med propan-2-ol-hexan-blandingen (3.1) til mærket og homogeniseres, hvorefter det hele henstår i 1 time.
Derpå måles i spektrofotometret ved 440 nm i glaskuvetter på 1 cm tykkelse blindprøveopløsningens (D) ekstinktion i forhold til referenceopløsningen (C) og prøveopløsningens (B) ekstinktion i forhold til referenceopløsningen (A).
Ekstinktionen af blindprøveopløsningen trækkes fra ekstinktionen af prøveopløsningen (= korrigeret ekstinktion). På basis af den fundne værdi beregnes indholdet af fri gossypol som beskrevet i punkt 6.
5.3. Bestemmelse af totalgossypol
En afvejet stofmængde indeholdende 1–5 mg gossypol kommes i en 50 ml målekolbe; derpå tilsættes 10 ml af opløsningsmidlet B (3.3). Samtidig klargøres en blindprøve, idet 10 ml af opløsningsmidlet B (3.3) kommes i en anden 50 ml målekolbe. Begge målekolber opvarmes i 30 minutter på kogende vandbad, og der afkøles til rumtemperatur. Kolbernes indhold fortyndes med propan-2-ol-hexan-blandingen (3.1) til mærket, homogeniseres og henstår i 10–15 minutter. Derpå filtreres, og filtratet opsamles i kolber med slibhals.
Der afpipetteres 2 ml af prøvefiltratet i hver af to 25 ml målekolber og 2 ml af filtratet fra blindprøven i hver af to andre 25 ml målekolber. En kolbe i hver række fyldes op med propan-2-ol-hexan-blandingen (3.1) til 25 ml. Disse opløsninger anvendes som referenceopløsninger.
Hver af de to andre målekolber tilsættes 2 ml anilin (3.4). Der opvarmes i 30 minutter på kogende vandbad til udvikling af farvningen. Derefter afkøles til rumtemperatur, fortyndes med propan-2-ol-hexan-blandingen (3.1) til 25 ml og homogeniseres, hvorefter det hele henstår i 1 time.
Ekstinktionen måles som beskrevet i punkt 5.2 for fri gossypol. På basis af den fundne værdi beregnes indholdet af totalgossypol som beskrevet i punkt 6.
6. Beregning af resultater
Beregningen af resultaterne kan foretages på basis af den specifikke ekstinktion (6.1) eller ved benyttelse af en kalibreringskurve (6.2).
6.1. På basis af den specifikke ekstinktion
Den specifikke ekstinktion er under de angivne betingelser som følger:
|
Fri gossypol: |
|
|
Totalgossypol: |
|
Indholdet af fri gossypol eller totalgossypol i prøven beregnes efter følgende formel:
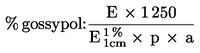
hvor:
|
E |
= |
korrigeret ekstinktion, som bestemt under punkt 5.2 |
|
p |
= |
afvejet stofmængde i gram |
|
a |
= |
alikvot del af filtratet i ml. |
6.2. Ved hjælp af en kalibreringskurve
6.2.1.
Der klargøres 2 rækker med hver fem 25 ml målekolber. I begge rækker afpipetteres 2,0 , 4,0 , 6,0 , 8,0 og 10,0 ml af standardgossypolopløsningen A (3.5), og rumfanget suppleres til 10 ml med opløsningsmidlet A (3.2). I hver række stilles en 25 ml målekolbe, som kun indeholder 10 ml af opløsningsmidlet A (3.2) (blindprøve).
Kolberne i den første række, inklusive blindprøven, fyldes op med propan-2-ol-hexan-blandingen (3.1) til 25 ml (referencerækken).
Hver af kolberne i den anden række, inklusive blindprøven, tilsættes 2 ml anilin (3.4). Der opvarmes i 30 minutter på kogende vandbad til udvikling af farvningen. Derefter afkøles til rumtemperatur, fortyndes med propan-2-ol-hexan-blandingen (3.1) til mærket og homogeniseres, hvorefter det hele henstår i 1 time (standardrækken).
Under de i 5.2 beskrevne betingelser måles ekstinktionen af standardrækkeopløsningerne ved sammenligning med de tilsvarende opløsninger i referencerækken. Kalibreringskurven tegnes, idet ekstinktionsværdierne og gossypolmængderne (i μg) indtegnes over for hinanden.
6.2.2.
Der klargøres seks 50 ml målekolber. I den første afpipetteres 10 ml af opløsningsmidlet B (3.3), og i de øvrige henholdsvis 2,0 , 4,0 , 6,0 , 8,0 og 10,0 ml af gossypolstandardopløsningen B (3.6). Indholdet i hver af kolberne suppleres med opløsningsmidlet B (3.3) til 10 ml, opvarmes i 30 minutter i kogende vandbad, afkøles til rumtemperatur, fortyndes med propan-2-ol-hexan-blandingen (3.1) til mærket og homogeniseres.
I to rækker med seks 25 ml målekolber afpipetteres i hver 2,0 ml af denne opløsning. Kolberne i den første række fyldes op med propan-2-ol-hexan-blandingen (3.1) til 25 ml (referencerækken).
I hver af kolberne i den anden række kommes 2 ml anilin (3.4). Der opvarmes i 30 minutter i kogende vandbad. Derefter afkøles til rumtemperatur, fortyndes med propan-2-ol-hexan-blandingen (3.1) til mærket og homogeniseres, hvorefter det hele henstår i 1 time (standardrækken).
Under de i 5.2 beskrevne betingelser måles ekstinktionen af standardrækkeopløsningerne ved sammenligning med de tilsvarende opløsninger i referencerækken. Kalibreringskurven tegnes, idet ekstinktionsværdierne og gossypolmængderne (i μg) indtegnes over for hinanden.
6.3. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige:
— 15 % i relativ værdi (af det højeste resultat) for gossypolindhold på mindre end 500 ppm
— 75 ppm i absolut værdi for gossypolindhold på 500–750 ppm
— 10 % i relativ værdi (af det højeste resultat) for gossypolindhold på over 750 ppm.
B. BESTEMMELSE AF INDHOLDET AF DIOXINER (PCDD'er/PCDF'er) OG PCB'er
KAPITEL I
Prøveudtagningsmetoder og fortolkning af analyseresultater
1. Formål og anvendelsesområde
Prøver til offentlig kontrol af indholdet af polychlorerede dibenzo-p-dioxiner (PCDD'er), polychlorerede dibenzofuraner (PCDF'er), dioxinlignende polychlorerede biphenyler (PCB'er) ( 36 ) og ikke-dioxinlignende PCB'er i foder udtages som beskrevet i bilag I. De kvantitative krav i forbindelse med kontrol af stoffer og produkter, der er homogent fordelt i foderet, jf. punkt 5.1 i bilag I, skal overholdes. De derved fremkomne samleprøver betragtes som repræsentative for de partier eller delpartier, de er udtaget fra. På grundlag af det indhold, der er konstateret i laboratorieprøverne, fastslås det, om de i direktiv 2002/32/EF fastsatte grænseværdier er overholdt.
Ved anvendelsen af denne del (del B) gælder definitionerne i bilag I til Kommissionens beslutning 2002/657/EF ( 37 ).
Desuden forstås i denne del (del B) ved:
»screeningsmetoder«: metoder til udvælgelse af stikprøver med indhold af PCDD'er/PCDF'er og dioxinlignende PCB'er, der overskrider grænseværdierne eller indgrebstærsklerne. Sådanne metoder skal give mulighed for omkostningseffektivitet og høj produktivitet med hensyn til antallet af screenede prøver og derved forbedre mulighederne for at opdage nye hændelser med høj eksponering og sundhedsrisici for forbrugerne. Screeningsmetoder skal baseres på bioanalytiske metoder eller GC-MS-metoder. Resultater fra prøver, der overstiger afskæringsværdien, skal verificeres ved en fuldstændig fornyet analyse af den oprindelige prøve efter en verifikationsmetode til kontrol af, at grænseværdien er overholdt
»verifikationsmetoder«: metoder, der giver fuldstændig eller supplerende information, så PCDD'er/PCDF'er og dioxinlignende PCB'er kan identificeres og kvantificeres entydigt på grænseværdi- eller om nødvendigt indgrebstærskelniveauet. De pågældende metoder omfatter anvendelse af gaskromatografi/højtopløsende massespektrometri (GC-HRMS) eller gaskromatografi/tandemmassespektrometri (GC-MS/MS).
2. Partiets eller delpartiets overholdelse af grænseværdien
2.1. For så vidt angår ikke-dioxinlignende PCB'er
Partiet overholder grænseværdien, hvis analyseresultatet ikke overskrider den grænseværdi for ikke-dioxinlignende PCB'er, der er fastsat i direktiv 2002/32/EF, idet der tages hensyn til måleusikkerheden.
Partiet overholder ikke grænseværdien, hvis analyseresultatets øvre koncentration ( 38 ) — bekræftet ved endnu en analyse ( 39 ) — overskrider grænseværdien, der er fastsat i direktiv 2002/32/EF, idet der tages hensyn til måleusikkerheden. Middelværdien af de to bestemmelser, under hensyntagen til måleusikkerheden, anvendes til at verificere, om kravene er opfyldt.
Der tages hensyn til måleusikkerheden på en af følgende måder:
— ved at beregne den ekspanderede usikkerhed, idet der anvendes en dækningsfaktor på 2, hvilket giver et konfidensniveau på ca. 95 %. Et parti eller delparti opfylder ikke kravene (dvs. er ikke-overensstemmende), hvis den målte værdi minus U ligger over grænseværdien
— ved at fastlægge beslutningsgrænsen (CCα) i overensstemmelse med punkt 3.1.2.5 i bilag I til beslutning 2002/657/EF. Et parti eller delparti er ikke-overensstemmende, hvis den målte værdi er lig med eller højere end CCα.
Første, andet og tredje afsnit gælder for analyseresultater af prøver udtaget ved offentlig kontrol. For kontraprøveanalyser eller analyser til referenceformål gælder de nationale regler.
2.2. For så vidt angår PCDD'er/PCDF'er og dioxinlignende PCB'er
Partiet overholder grænseværdierne, hvis resultatet fra en enkelt analyse
— udført efter en screeningsmetode med en andel af falsk overensstemmende resultater på under 5 % viser, at indholdet ikke overskrider den pågældende grænseværdi for PCDD'er/PCDF'er eller for summen af PCDD'er/PCDF'er og dioxinlignende PCB'er, der er fastsat i direktiv 2002/32/EF
— udført efter en verifikationsmetode ikke overskrider den pågældende grænseværdi for PCDD'er/PCDF'er eller for summen af PCDD'er/PCDF'er og dioxinlignende PCB'er, der er fastsat i direktiv 2002/32/EF, idet der tages hensyn til måleusikkerheden.
For screeningsassays fastlægges en afskæringsværdi, som lægges til grund for beslutninger om, hvorvidt de relevante grænseværdier, der er fastsat for enten PCDD'er/PCDF'er eller for summen af PCDD'er/PCDF'er og dioxinlignende PCB'er, er overholdt i prøverne.
Partiet overholder ikke grænseværdien, hvis analyseresultatets øvre koncentration ( 40 ) — tilvejebragt efter en verifikationsmetode og bekræftet ved endnu en analyse — overskrider grænseværdien, der er fastsat i direktiv 2002/32/EF, idet der tages hensyn til måleusikkerheden ( 41 ). Middelværdien af de to bestemmelser, under hensyntagen til måleusikkerheden, anvendes til at verificere, om kravene er opfyldt.
Der tages hensyn til måleusikkerheden på en af følgende måder:
— ved at beregne den ekspanderede usikkerhed, idet der anvendes en dækningsfaktor på 2, hvilket giver et konfidensniveau på ca. 95 %. Et parti eller delparti er ikke-overensstemmende, hvis den målte værdi minus U ligger over grænseværdien. Hvis PCDD'er/PCDF'er og dioxinlignende PCB'er bestemmes separat, anvendes summen af den anslåede ekspanderede usikkerhed på de separate analyseresultater vedrørende PCDD'er/PCDF'er og dioxinlignende PCB'er på summen af PCDD'er/PCDF'er og dioxinlignende PCB'er
— ved at fastlægge beslutningsgrænsen (CCα) i overensstemmelse med punkt 3.1.2.5 i bilag I til beslutning 2002/657/EF. Et parti eller delparti er ikke-overensstemmende, hvis den målte værdi er lig med eller højere end CCα.
Første til fjerde afsnit gælder for analyseresultater af prøver udtaget ved offentlig kontrol. For kontraprøveanalyser eller analyser til referenceformål gælder de nationale regler.
3. Resultater over de indgrebstærskler, der er fastsat i bilag ii til direktiv 2002/32/EF
Indgrebstærskler er et redskab til udvælgelse af prøver i tilfælde, hvor det er nødvendigt at identificere en forureningskilde og træffe foranstaltninger, der reducerer eller fjerner den. Med screeningsmetoder fastlægges der relevante afskæringsværdier til udvælgelse af de pågældende prøver. Er det nødvendigt med en betydelig indsats for at identificere en kilde og reducere eller fjerne forureningen, kan det være hensigtsmæssigt at bekræfte overskridelsen af indgrebstærsklerne ved endnu en analyse under anvendelse af en verifikationsmetode og under hensyntagen til måleusikkerheden ( 42 ).
KAPITEL II
Klargøring af prøver og krav til analysemetoder, der anvendes ved offentlig kontrol af indholdet af dioxiner (pcdd'er/pcdf'er) og dioxinlignende pcb'er i foder
1. Anvendelsesområde
Kravene i dette kapitel anvendes, når foder analyseres med henblik på offentlig kontrol af indholdet af 2,3,7,8-substituerede polychlorerede dibenzo-p-dioxiner og polychlorerede dibenzofuraner (PCDD'er/PCDF'er) og dioxinlignende polychlorerede biphenyler (dioxinlignende PCB'er) og til andre forskriftsmæssige formål.
Indholdet af PCDD'er/PCDF'er og dioxinlignende PCB'er i foder kan overvåges under anvendelse af to forskellige typer analysemetoder:
a) Screeningsmetoder
Formålet med screeningsmetoder er at udvælge stikprøver med indhold af PCDD'er/PCDF'er og dioxinlignende PCB'er, der overskrider grænseværdierne eller indgrebstærsklerne. Screeningsmetoder skal give mulighed for omkostningseffektivitet og høj produktivitet med hensyn til antallet af screenede prøver og derved forbedre mulighederne for at opdage nye hændelser med høj eksponering og sundhedsrisici for forbrugerne. De bør anvendes med henblik på at undgå falsk overensstemmende resultater. Metoderne kan omfatte bioanalytiske analysemetoder og GC-MS-metoder.
Med screeningsmetoder sammenholdes analyseresultatet med en afskæringsværdi, som giver et ja/nej-svar på, om grænseværdien eller indgrebstærsklen er overskredet. Koncentrationen af PCDD'er/PCDF'er og summen af PCDD'er/PCDF'er og dioxinlignende PCB'er i prøver, der mistænkes for at være ikke-overensstemmende i forhold til grænseværdien, skal bestemmes/bekræftes efter en verifikationsmetode.
Screeningsmetoder kan desuden give en indikation af indholdet af PCDD'er/PCDF'er og dioxinlignende PCB'er i prøven. Anvendes der bioanalytiske screeningsmetoder, udtrykkes resultatet som bioanalytiske ækvivalenter (BEQ), mens det udtrykkes i toksicitetsækvivalenter (TEQ), hvis der anvendes fysisk-kemiske GC-MS-metoder. De numerisk angivne resultater fra screeningsmetoder er egnede til at påvise overensstemmelse eller mistænkt ikke-overensstemmelse eller overskridelse af indgrebstærskler og giver en indikation af koncentrationsintervallet i tilfælde af opfølgning efter verifikationsmetoder. De egner sig ikke til formål som at vurdere baggrundsniveauer, foretage skøn over indtag, følge udviklingen i niveauer over tid eller revurdere indgrebstærskler og grænseværdier.
b) Verifikationsmetoder
Verifikationsmetoder gør det muligt entydigt at identificere og kvantificere PCDD'er/PCDF'er og dioxinlignende PCB'er i en prøve og giver fuldstændig information på kongenerniveau. Disse metoder muliggør således kontrol af grænseværdier og indgrebstærskler, herunder bekræftelse af resultater fra screeningsmetoder. Resultaterne kan tillige anvendes til andre formål såsom at bestemme lave baggrundsniveauer i forbindelse med overvågningen af foder, følge udviklingen i niveauer over tid, vurdere eksponeringen og opbygge en database til brug for en eventuel revurdering af indgrebstærskler og grænseværdier. De er også vigtige redskaber til at klarlægge kongenermønstre med henblik på at identificere kilden til en eventuel forurening. De pågældende metoder omfatter anvendelse af GC-HRMS. Også GC-MS/MS kan anvendes til at bekræfte overholdelse eller manglende overholdelse af grænseværdien.
2. Baggrund
Til beregning af koncentrationer af toksicitetsækvivalenter (TEQ) ganges koncentrationerne af de enkelte stoffer i en given prøve med deres respektive toksicitetsækvivalensfaktor (TEF) (jf. fodnote (42) i kapitel I), og summen heraf giver den samlede koncentration af dioxinlignende forbindelser udtrykt i TEQ.
I denne del (del B) forstås ved den accepterede specifikke bestemmelsesgrænse for en enkeltkongener det laveste indhold af analyt, der kan påvises med rimelig statistisk sikkerhed, og som opfylder identifikationskriterierne som beskrevet i internationalt anerkendte standarder, f.eks. i standard EN 16215:2012 (Foderstoffer — Bestemmelse af dioxiner og dioxinlignende PCB-forbindelser samt indikator-PCB-forbindelser ved GC/HRMS) og/eller EPA-metode 1613 og 1668, som ændret.
Bestemmelsesgrænsen for en enkeltkongener kan identificeres som
a) koncentrationen af en analyt i prøveekstraktet, som giver et instrumentrespons for de to forskellige ioner, der skal undersøges, med et signal-støj-forhold på 3:1 for det svageste rådatasignal, eller
b) såfremt beregningen af signal-støj-forholdet af tekniske grunde ikke giver pålidelige resultater, punktet med laveste koncentration på en kalibreringskurve, som giver en acceptabel (≤ 30 %) og ensartet (som minimum målt i begyndelsen og i slutningen af en analyserække af prøver) afvigelse i forhold til den gennemsnitlige relative responsfaktor, beregnet for alle punkter på kalibreringskurven i hver prøverække. LOQ beregnes ud fra punktet med laveste koncentration under hensyntagen til genfindingen af interne standarder og prøvemængde.
Bioanalytiske screeningsmetoder vil ikke give resultater på kongenerniveau, men giver udelukkende en indikation ( 43 ) af TEQ-niveauet, udtrykt i bioanalytiske ækvivalenter (BEQ), hvorved der tages hensyn til det forhold, at det ikke nødvendigvis er alle forbindelser i et prøveekstrakt, som giver respons i testen, der opfylder samtlige TEQ-princippets forudsætninger.
Screenings- og verifikationsmetoder kan kun anvendes til kontrol af en bestemt matrix, hvis metoderne er tilstrækkeligt følsomme til på pålidelig vis at påvise forekomst på indgrebstærskel- eller grænseværdiniveauet.
3. Kvalitetssikringskrav
|
3.1. |
Der skal træffes foranstaltninger til at undgå krydskontaminering i alle faser af prøveudtagnings- og analyseproceduren. |
|
3.2. |
Prøverne opbevares og transporteres i beholdere af glas, aluminium, polypropylen eller polyethylen, som egner sig til opbevaring, uden at mængden af PCDD'er/PCDF'er og dioxinlignende PCB'er i prøverne påvirkes. Spor af papirstøv fjernes fra prøvebeholderen. |
|
3.3. |
Opbevaring og transport af foderprøven skal foregå, så prøvens integritet bevares. |
|
3.4. |
Alle laboratorieprøver findeles og blandes grundigt, hvis det er relevant, efter en metode, for hvilken det er godtgjort, at den resulterer i fuldstændig homogenisering (f.eks. så prøven kan sigtes gennem en 1 mm-sigte). Prøverne tørres før formalingen, hvis vandindholdet er for højt. |
|
3.5. |
Det kontrolleres, at reagenser, glasudstyr og andet udstyr ikke påvirker TEQ- eller BEQ-baserede resultater. |
|
3.6. |
Der foretages en blindprøveanalyse ved at gennemføre hele analyseproceduren, blot med udeladelse af prøven. |
|
3.7. |
For så vidt angår bioanalytiske metoder kontrolleres det, at alt glasudstyr og alle opløsningsmidler, der anvendes til analyser, er fri for forbindelser, der interfererer med påvisningen af målforbindelser i måleområdet. Glasudstyr skylles med opløsningsmidler eller opvarmes til temperaturer, der er tilstrækkeligt høje til at fjerne spor af PCDD'er/PCDF'er, dioxinlignende forbindelser og interfererende forbindelser fra udstyrets overflader. |
|
3.8. |
Den prøvemængde, der anvendes til ekstraktionen, skal være tilstrækkelig til, at kravene vedrørende et tilstrækkelig lavt måleområde, som dækker grænseværdi- eller indgrebstærskelkoncentrationerne, er opfyldt. |
|
3.9. |
De specifikke procedurer for klargøring af prøver, som anvendes for de pågældende produkter, skal være i overensstemmelse med internationalt anerkendte retningslinjer. |
4. Krav til laboratorier
|
4.1. |
I henhold til forordning (EF) nr. 882/2004 skal laboratorier være akkrediteret af et anerkendt organ, der fungerer i overensstemmelse med ISO-vejledning 58, så det sikres, at de anvender analysekvalitetssikring. Laboratorier akkrediteres ifølge EN ISO/IEC 17025-standarden. |
|
4.2. |
Laboratoriets præstationer dokumenteres ved løbende, vellykket deltagelse i laboratoriesammenligninger vedrørende bestemmelse af PCDD'er/PCDF'er og dioxinlignende PCB'er i de relevante fodermatrixer og koncentrationsområder. |
|
4.3. |
Laboratorier, der anvender screeningsmetoder til rutinemæssig kontrol af prøver, skal etablere et tæt samarbejde med laboratorier, der anvender verifikationsmetoden — både i forbindelse med kvalitetskontrol og til bekræftelse af analyseresultater for mistænkte prøver. |
5. Grundlæggende krav til procedurer for analyse af dioxiner (PCDD'er/PCDF'er) og dioxinlignende PCB'er
5.1. Lavt måleområde og bestemmelsesgrænser
For PCDD'er/PCDF'er skal de påviselige mængder befinde sig i det øvre femtogram-interval (10–15 g) på grund af visse af disse forbindelsers ekstremt høje toksicitet. For de fleste PCB-kongenere er det tilstrækkeligt med en bestemmelsesgrænse i nanogram-intervallet (10–9 g). Til måling af de mere toksiske dioxinlignende PCB-kongenere (især non-ortho-substituerede kongenere) skal den nedre del af måleområdet ligge i den nedre del af pikogram-intervallet (10–12 g). For alle andre PCB-kongenere er det tilstrækkeligt med en bestemmelsesgrænse i nanogram-intervallet (10–9 g).
5.2. Høj selektivitet (specificitet)
|
5.2.1. |
Der skal kunne skelnes mellem PCDD'er, PCDF'er og dioxinlignende PCB'er og en lang række andre, potentielt interfererende forbindelser i prøveekstraktet, som forekommer i koncentrationer, der er op til mange gange højere end de relevante analytters. For GC-MS-metoders vedkommende skal der kunne skelnes mellem forskellige kongenere, f.eks. mellem toksiske kongenere (såsom de 17 2,3,7,8-substituerede PCDD'er/PCDF'er og 12 dioxinlignende PCB'er) og andre kongenere. |
|
5.2.2. |
Bioanalytiske metoder skal kunne påvise målforbindelserne som summen af PCDD'er/PCDF'er og/eller dioxinlignende PCB'er. Ved oprensningen af prøver skal det tilstræbes at fjerne forbindelser, der giver falsk ikke-overensstemmende resultater, og forbindelser, der kan svække responset og give falsk overensstemmende resultater. |
5.3. Høj nøjagtighed (korrekthed og præcision, tilsyneladende bioassay-genfinding)
|
5.3.1. |
For så vidt angår GC-MS-metoder skal bestemmelsen give et pålideligt estimat over den korrekte koncentration i en prøve. Høj nøjagtighed er nødvendig for at undgå, at et resultat af en analyse afvises på grundlag af lav pålidelighed af det bestemte TEQ-niveau. Nøjagtighed udtrykkes som korrekthed (forskellen mellem den målte middelværdi for en analyt i et certificeret materiale og dens certificerede værdi, udtrykt i procent af den certificerede værdi) og præcision (RSDR relativ standardafvigelse beregnet ud fra resultater, der er fremkommet under reproducerbarhedsbetingelser). |
|
5.3.2. |
For bioanalytiske metoder bestemmes den tilsyneladende bioassay-genfinding. Ved tilsyneladende bioassay-genfinding forstås BEQ-niveauet beregnet ud fra TCDD- eller PCB 126-kalibreringskurven, korrigeret for blindprøven og efterfølgende divideret med TEQ-niveauet som bestemt efter verifikationsmetoden. Formålet er at korrigere for faktorer såsom tabet af PCDD'er/PCDF'er og dioxinlignende forbindelser i ekstraktions– og oprensningsfasen, medekstraherede forbindelser, som forstærker eller svækker responset (henholdsvis agonistisk og antagonistisk virkning), kvaliteten af kurvetilpasningen eller forskelle mellem værdierne for henholdsvis toksicitetsækvivalensfaktoren (TEF) og den relative styrke (REP). Den tilsyneladende bioassay-genfinding beregnes ud fra passende referenceprøver med repræsentative kongenermønstre omkring det relevante niveau. |
5.4. Validering i nærheden af grænseværdien og kvalitetskontrol generelt
|
5.4.1. |
Laboratorierne skal dokumentere en metodes ydeevne i nærheden af grænseværdien, f.eks. 0,5, 1 og 2 gange grænseværdien med en acceptabel variationskoefficient for gentagne analyser, som led i valideringsproceduren og i forbindelse med rutinemæssige analyser. |
|
5.4.2. |
Som interne kvalitetskontrolforanstaltninger gennemføres der regelmæssigt blindprøvekontrol og spikingforsøg eller analyse af kontrolprøver (om muligt med certificeret referencemateriale). Kvalitetskontrolkort for blindprøvekontroller, spikingforsøg eller analyser af kontrolprøver registreres og kontrolleres for at sikre, at den analytiske ydeevne er i overensstemmelse med gældende krav. |
5.5. Bestemmelsesgrænse
|
5.5.1. |
For bioanalytiske screeningsmetoders vedkommende er fastlæggelse af bestemmelsesgrænsen (LOQ) ikke et ufravigeligt krav, men det skal godtgøres, at metoden gør det muligt at skelne blindprøveværdien fra afskæringsværdien. I forbindelse med fastlæggelsen af et BEQ-niveau fastsættes et rapporteringsniveau med henblik på håndtering af prøver, som giver et respons under dette niveau. Det skal dokumenteres, at rapporteringsniveauet adskiller sig — som minimum med en faktor tre — fra procedureblindprøver med et respons under måleområdet. Det skal derfor beregnes ud fra prøver med et indhold af målforbindelserne omkring det krævede minimumsniveau og ikke ud fra et bestemt signal-støj-forhold eller en assay-blindprøve. |
|
5.5.2. |
LOQ for en verifikationsmetode skal være på ca. en femtedel af grænseværdien. |
5.6. Analysekriterier
For at sikre pålidelige resultater af verifikations- eller screeningsmetoder skal følgende kriterier være opfyldt i nærheden af grænseværdien eller indgrebstærsklen for henholdsvis TEQ- eller BEQ-værdien, bestemt enten som samlet TEQ (som summen af PCDD'er/PCDF'er og dioxinlignende PCB'er) eller separat for henholdsvis PCDD'er/PCDF'er og dioxinlignende PCB'er:
|
|
Screening efter bioanalytiske eller fysisk-kemiske metoder |
Verifikationsmetoder |
|
Falsk overensstemmende-andel (1) |
< 5 % |
|
|
Korrekthed |
|
– 20 % til + 20 % |
|
Repeterbarhed (RSDr) |
< 20 % |
|
|
Intern reproducerbarhed (RSDR) |
< 25 % |
< 15 % |
|
(1) I forhold til grænseværdierne. |
||
5.7. Specifikke krav til screeningsmetoder
|
5.7.1. |
Både GC-MS-metoder og bioanalytiske metoder kan anvendes til screening. For GC-MS-metoder skal kravene i punkt 6 være opfyldt. Der er fastsat specifikke krav til cellebaserede bioanalytiske metoder i punkt 7. |
|
5.7.2. |
Laboratorier, der anvender screeningsmetoder til rutinemæssig kontrol af prøver, skal etablere et tæt samarbejde med laboratorier, der anvender verifikationsmetoden. |
|
5.7.3. |
Screeningsmetodens ydeevne skal efterprøves i forbindelse med rutinemæssige analyser, ved analysekvalitetskontrol og ved løbende metodevalidering. Der skal opereres med et fast program for kontrol af overensstemmende resultater. |
|
5.7.4. |
Kontrol af eventuel hæmning af celleresponset og cytotoxicitet 20 % af prøveekstrakterne måles i forbindelse med rutinemæssig screening med og uden 2,3,7,8-TCDD tilsat i overensstemmelse med grænseværdien eller indgrebstærsklen med henblik på at kontrollere, om responset måske hæmmes af interfererende stoffer i prøveekstraktet. Den målte koncentration af den spikede prøve sammenholdes med summen af koncentrationen af det ikke-spikede ekstrakt og spikingkoncentrationen. Hvis denne målte koncentration er over 25 % mindre den beregnede (sum-)koncentration, er dette en indikation af mulig signalhæmning, og den pågældende prøve underkastes en GC-HRMS-verifikationsanalyse. Resultaterne registreres i kvalitetskontrolkort. |
|
5.7.5. |
Kvalitetskontrol af overensstemmende prøver Ca. 2-10 % af de overensstemmende prøver, afhængigt af prøvematrix og laboratoriets erfaring, bekræftes ved hjælp af GC-HRMS. |
|
5.7.6. |
Bestemmelse af andelen af falsk overensstemmende resultater fra kvalitetskontroldata Andelen af falsk overensstemmende resultater fra screening af prøver, der ligger under og over grænseværdien eller indgrebstærsklen, bestemmes. Den faktiske andel af falsk overensstemmende resultater skal ligge under 5 %. Når der foreligger mindst 20 bekræftede resultater pr. matrix/matrixgruppe fra kvalitetskontrollen af overensstemmende prøver, drages konklusionerne vedrørende andelen af falsk overensstemmende resultater på grundlag af denne database. Resultaterne fra prøver, der er analyseret i ringtest eller i forbindelse med forureningshændelser og dækker et koncentrationsområde på op til f.eks. 2 gange grænseværdien, kan også tælles med i de mindst 20 resultater, der skal lægges til grund for evalueringen af andelen af falsk overensstemmende resultater. Prøverne skal dække de mest almindelige kongenermønstre og repræsentere forskellige kilder. Selv om screeningsassays fortrinsvis skal have til formål at påvise prøver, for hvilke indgrebstærsklen er overskredet, er kriteriet for bestemmelse af andelen af falsk overensstemmende resultater grænseværdien, under hensyntagen til måleusikkerheden ved verifikationsmetoden. |
|
5.7.7. |
Potentielt ikke-overensstemmende prøver fra screening skal altid verificeres ved en fuldstændig fornyet analyse af den oprindelige prøve efter en verifikationsanalysemetode. Disse prøver kan også anvendes til at evaluere andelen af falsk ikke-overensstemmende resultater. For screeningsmetoder er andelen af falsk ikke-overensstemmende resultater andelen af resultater, for hvilke det ved hjælp af en verifikationsanalyse er bekræftet, at de opfylder kravene (dvs. er overensstemmende), efter at prøven i en tidligere screening er erklæret for potentielt ikke-overensstemmende. Vurderingen af screeningsmetodens hensigtsmæssighed baseres på sammenholdelse af antallet af falsk ikke-overensstemmende prøver med det samlede antal kontrollerede prøver. Denne andel skal være tilstrækkeligt lille til, at screening med fordel kan anvendes. |
|
5.7.8. |
Bioanalytiske metoder skal, i det mindste under valideringsbetingelser, give en brugbar indikation af TEQ-niveauet, beregnet og udtrykt som BEQ. Også for bioanalytiske metoder, der gennemføres under repeterbarhedsbetingelser, er den interne RSDr typisk lavere end reproducerbarheden RSDR. |
6. Specifikke krav, som GC-MS-metoder skal opfylde med henblik på screening eller verifikation
6.1. Acceptabel difference mellem WHO-TEQ-resultaters øvre og nedre koncentration
Differencen mellem den øvre og den nedre koncentration må ikke overstige 20 % i forbindelse med bekræftelse af overskridelse af grænseværdien eller i tilfælde af behov for indgrebstærskler.
6.2. Genfindingskontrol
|
6.2.1. |
For at validere analyseproceduren tilsættes 13C-mærkede 2,3,7,8-chlorsubstituerede PCDD'er/PCDF'er og 13C-mærkede dioxinlignende PCB'er som intern standard helt fra begyndelsen af analysen, f.eks. inden ekstraktion. Der tilsættes mindst én kongener for hver af de tetra- til octa-chlorerede homologe grupper af PCDD'er/PCDF'er og mindst én kongener for hver af de homologe grupper af dioxinlignende PCB'er (alternativt tilsættes mindst én kongener for hver massespektrometrisk udvalgt ionregistreringsfunktion, der anvendes til overvågning af PCDD'er/PCDF'er og dioxinlignende PCB'er). For verifikationsmetoders vedkommende anvendes alle 17 13C-mærkede 2,3,7,8-substituerede PCDD'er/PCDF'er og alle 12 13C-mærkede dioxinlignende PCB'er som intern standard. |
|
6.2.2. |
Der bestemmes også relative responsfaktorer for de kongenere, som der ikke tilsættes en 13C-mærket analog for, ved hjælp af relevante kalibreringsopløsninger. |
|
6.2.3. |
For vegetabilsk foder og animalsk foder, der indeholder under 10 % fedt, er det obligatorisk at tilsætte interne standarder inden ekstraktionen. For animalsk foder, der indeholder over 10 % fedt, tilsættes de interne standarder enten før eller efter fedtekstraktionen. Der foretages en hensigtsmæssig validering af ekstraktionseffektiviteten, afhængigt af det trin, hvor de interne standarder tilsættes, og af, om resultaterne er produktbaserede eller fedtbaserede. |
|
6.2.4. |
Inden GC-MS-analyse tilsættes en eller to genfindingsstandarder (surrogat). |
|
6.2.5. |
Det er nødvendigt med genfindingskontrol. Niveauet for genfinding af de enkelte interne standarder ved verifikationsmetoder skal ligge på mellem 60 og 120 %. Lavere eller højere genfinding for enkeltkongenere, navnlig for visse hepta- og octa-chlorerede dibenzo-p-dioxiner og dibenzofuraner, kan accepteres, på betingelse af at deres bidrag til TEQ-værdien ikke udgør mere end 10 % af den samlede TEQ-værdi (baseret på summen af PCDD'er/PCDF'er og dioxinlignende PCB'er). Niveauet for genfinding ved GC-MS-screeningsmetoder skal ligge på mellem 30 og 140 %. |
6.3. Fjernelse af interfererende stoffer
— Der gennemføres separation af PCDD'er/PCDF'er fra interfererende chlorerede forbindelser som f.eks. ikke-dioxinlignende PCB'er og chlorerede diphenylethere ved hjælp af egnede kromatografiske metoder (helst med florisil-, alumina- og/eller carbonkolonne).
— Gaskromatografisk separation af isomerer skal være på under < 25 % målt mellem toppene for 1,2,3,4,7,8-HxCDF og 1,2,3,6,7,8-HxCDF.
6.4. Kalibrering med standardkurve
Kalibreringskurven skal dække det relevante grænseværdi- eller indgrebstærskelniveau.
6.5. Specifikke kriterier vedrørende verifikationsmetoder
— For GC-HRMS:
—
Ved HRMS skal opløsningsevnen typisk være mindst 10 000 for hele masseintervallet ved en dalværdi på 10 %.
Opfyldelse af yderligere identifikations- og bekræftelseskriterier som beskrevet i internationalt anerkendte standarder, f.eks. i standard EN 16215:2012 (Foderstoffer — Bestemmelse af dioxiner og dioxinlignende PCB-forbindelser samt indikator-PCB-forbindelser ved GC/HRMS) og/eller EPA-metode 1613 og 1668, som ændret.
— For GC-MS/MS:
—
Overvågning af mindst 2 specifikke prækursor-ioner, hver med et specifikt tilsvarende transitionsprodukt-ion for alle mærkede og ikke mærkede analytter inden for analysens anvendelsesområde.
Tilladt maksimumstolerance for relative ionintensiteter på ± 15 % for udvalgte transitionsprodukt-ioner sammenholdt med beregnede eller målte værdier (gennemsnit fra kalibreringsstandarder) ved anvendelse af identiske MS/MS-betingelser, især kollisionsenergi og kollisionsgastryk, for hver transition af en analyt.
Opløsningen for hver quadrupol skal enten kunne sidestilles med eller være bedre end enhedsmasseopløsningen (enhedsmasseopløsning: opløsning, der er tilstrækkelig til at adskille en masseenhed i to toppe) for at minimere eventuelle interferenser på den pågældende analyt.
Opfyldelse af de yderligere kriterier som beskrevet i internationalt anerkendte standarder, f.eks. i standard EN 16215:2012 (Foderstoffer — Bestemmelse af dioxiner og dioxinlignende PCB-forbindelser samt indikator-PCB-forbindelser ved GC/HRMS) og/eller EPA-metode 1613 og 1668, som ændret, undtagen pligten til at anvende GC-HRMS.
7. Specifikke krav til bioanalytiske metoder
Bioanalytiske metoder er metoder baseret på anvendelse af biologiske principper, såsom cellebaserede assays, receptorassays eller immunoassays. Dette punkt (punkt 7) indeholder krav til bioanalytiske metoder generelt.
Med en screeningsmetode klassificeres en prøve principielt som overensstemmende eller som mistænkt for at være ikke-overensstemmende. I det øjemed sammenholdes det beregnede BEQ-niveau med afskæringsværdien (jf. punkt 7.3). Prøver, der giver resultater under afskæringsværdien, erklæres for overensstemmende, mens prøver, der ligger på eller over afskæringsværdien, erklæres for mistænkt for at være ikke-overensstemmende og nødvendiggør analyse efter en verifikationsmetode. I praksis kan et BEQ-niveau på 2/3 af grænseværdien være afskæringsværdi, såfremt der sikres en andel af falsk overensstemmende resultater på under 5 % og en acceptabel andel af falsk ikke-overensstemmende resultater. Med særskilte grænseværdier for henholdsvis PCDD'er/PCDF'er og summen af PCDD'er/PCDF'er og dioxinlignende PCB'er er passende bioassay-afskæringsværdier for PCDD'er/PCDF'er en forudsætning for at kunne kontrollere prøvernes overensstemmelse uden fraktionering. Til kontrol af prøver, for hvilke indgrebstærsklerne er overskredet, vil en passende procentdel af den enkelte indgrebstærskel være en egnet afskæringsværdi.
Der kan desuden for visse bioanalytiske metoders vedkommende fastsættes et vejledende niveau udtrykt i BEQ for prøver i måleområdet, der ligger over rapporteringsgrænsen (jf. punkt 7.1.1 og 7.1.6).
7.1. Evaluering af testresponset
7.1.1. Generelle krav
— Ved beregning af koncentrationerne ud fra en TCDD-kalibreringskurve vil værdierne i den nederste og den øverste del af kurven vise en stor spredning (en høj variationskoefficient (CV)). Måleområdet er det område, hvor CV er på under 15 %. Den nedre del af måleområdet (rapporteringsniveauet) sættes højere — som minimum med en faktor tre — end procedureblindprøverne. Den øvre del af måleområdet er normalt repræsenteret ved EC70-værdien (70 % af den højeste effektive koncentration), men er lavere, hvis CV ligger over 15 % inden for dette interval. Måleområdet fastsættes i forbindelse med validering. Afskæringsværdierne (jf. punkt 7.3) skal ligge klart inden for måleområdet.
— Standardopløsninger og prøveekstrakter testes mindst to gange. Ved dobbeltbestemmelse skal en standardopløsning eller et kontrolekstrakt, der testes i 4-6 huller fordelt over hele pladen, give et respons eller en koncentration (kun muligt i måleområdet) baseret på en CV på < 15 %.
7.1.2. Kalibrering
7.1.2.1. Kalibrering med standardkurve
— Indholdet i prøver anslås ved at sammenholde testresponset med en kalibreringskurve for TCDD (eller PCB 126 eller en standardblanding af PCDD/PCDF/dioxinlignende PCB) med henblik på at beregne BEQ-niveauet i ekstraktet og efterfølgende i prøven.
— Kalibreringskurver skal indbefatte 8-12 koncentrationer (som minimum dobbeltbestemmelser), idet der skal være tilstrækkeligt med koncentrationer i den nederste del af kurven (måleområdet). Der lægges særlig vægt på kvaliteten af kurvetilpasningen i måleområdet. R2-værdien er som sådan af begrænset eller slet ingen værdi som grundlag for en goodness-of-fit-vurdering ved ikke-lineær regression. Der opnås et bedre fit ved at minimere forskellen mellem beregnede og observerede niveauer i kurvens måleområde, f.eks. ved at minimere summen af kvadrerede residualer.
— Det anslåede indhold i prøveekstraktet korrigeres efterfølgende for det BEQ-niveau, der er beregnet for en matrix/opløsningsmiddel-blindprøve (for at tage urenheder fra de anvendte opløsningsmidler og kemikalier i betragtning), og den tilsyneladende genfinding (som beregnes ud fra BEQ-niveauet i passende referenceprøver med repræsentative kongenermønstre omkring grænseværdien eller indgrebstærsklen). Til korrektion for genfinding skal den tilsyneladende genfinding ligge inden for det krævede interval (jf. punkt 7.1.4). Referenceprøver, der anvendes til korrektion for genfinding, skal opfylde kravene i punkt 7.2.
7.1.2.2. Kalibrering med referenceprøver
Alternativt kan der anvendes en kalibreringskurve fremstillet på grundlag af mindst fire referenceprøver (jf. punkt 7.2.4: en matrixblindprøve plus tre referenceprøver på 0,5, 1 og 2 gange grænseværdien eller indgrebstærsklen), hvorved behovet for at korrigere for blindprøve og genfinding bortfalder. I dette tilfælde kan det testrespons, som svarer til 2/3 af grænseværdien (jf. punkt 7.3), beregnes direkte ud fra disse prøver og anvendes som afskæringsværdi. Til kontrol af prøver, for hvilke indgrebstærsklerne er overskredet, vil en passende procentdel af disse indgrebstærskler være en egnet afskæringsværdi.
7.1.3. Separat bestemmelse af henholdsvis PCDD'er/PCDF'er og dioxinlignende PCB'er
Ekstrakter kan opdeles i fraktioner indeholdende henholdsvis PCDD'er/PCDF'er og dioxinlignende PCB'er, således at TEQ-niveauet (i BEQ) kan angives særskilt for henholdsvis PCDD'er/PCDF'er og dioxinlignende PCB'er. Der anvendes fortrinsvis en PCB 126-standardkalibreringskurve til evaluering af resultaterne for den fraktion, der indeholder dioxinlignende PCB'er.
7.1.4. Tilsyneladende bioassay-genfinding
Den tilsyneladende bioassay-genfinding beregnes ud fra passende referenceprøver med repræsentative kongenermønstre omkring grænseværdien eller indgrebstærsklen og udtrykkes som procent af BEQ-niveauet i forhold til TEQ-niveauet. Forskellene mellem TEF- og REP-faktorer for dioxinlignende PCB'er kan, afhængigt af, hvilken type assay og TEF'er ( 44 ) der anvendes, resultere i lave tilsyneladende genfindingsprocenter for dioxinlignende PCB'er i forhold til PCDD'er/PCDF'er. Hvis PCDD'er/PCDF'er og dioxinlignende PCB'er bestemmes separat, er den tilsyneladende bioassay-genfinding derfor 20-60 % for dioxinlignende PCB'er og 50-130 % for PCDD'er/PCDF'er (disse intervaller gælder for TCDD-kalibreringskurven). Eftersom dioxinlignende PCB'ers bidrag til summen af PCDD'er/PCDF'er og dioxinlignende PCB'er kan variere afhængigt af forskellige matrixer og prøver, afspejler den tilsyneladende bioassay-genfinding for summen af PCDD'er/PCDF'er og dioxinlignende PCB'er disse intervaller og skal være på 30-130 %. Enhver indvirkning af væsentligt ændrede TEF-værdier på EU-lovgivningen for PCDD'er/PCDF'er og dioxinlignende PCB'er nødvendiggør en ændring af disse intervaller.
7.1.5. Kontrol af genfinding efter oprensning
Tabet af forbindelser under oprensningen kontrolleres i forbindelse med validering. En blindprøve tilsat en blanding af de forskellige kongenere underkastes oprensning (mindst n = 3), og genfinding og variabilitet kontrolleres efter en verifikationsmetode. Genfindingen skal være på mellem 60 og 120 %, især for kongenere, der bidrager med over 10 % af TEQ-niveauet i forskellige blandinger.
7.1.6. Rapporteringsgrænse
Ved indberetning af BEQ-niveauer fastlægges der en rapporteringsgrænse ud fra relevante matrixprøver med typiske kongenermønstre, men ikke ud fra standardernes kalibreringskurve, idet præcisionen i den nederste del af kurven er lav. Der skal tages hensyn til virkningerne af ekstraktion og oprensning. Rapporteringsgrænsen sættes væsentligt højere (som minimum med en faktor tre) end procedureblindprøverne.
7.2. Anvendelse af referenceprøver
|
7.2.1. |
Referenceprøver skal repræsentere prøvematrix, kongenermønstre og koncentrationsområder for PCDD'er/PCDF'er og dioxinlignende PCB'er omkring grænseværdien eller indgrebstærsklen. |
|
7.2.2. |
Hver analyserække skal omfatte en matrixblindprøve eller, hvor dette ikke er muligt, en procedureblindprøve samt en referenceprøve ved grænseværdien eller indgrebstærsklen. Disse prøver ekstraheres og analyseres samtidig under identiske betingelser. Referenceprøven skal udvise en klart kraftigere reaktion end blindprøven, så der er sikkerhed for testens egnethed. Prøverne kan anvendes til korrektion for blindprøve og genfinding. |
|
7.2.3. |
Referenceprøver, der udvælges til korrektion for genfinding, skal være repræsentative for de klargjorte prøver, hvilket betyder, at bestemte kongenermønstre ikke må føre til, at niveauerne vurderes for lavt. |
|
7.2.4. |
Der kan inkluderes ekstra referenceprøver på f.eks. 0,5 og 2 gange grænseværdien eller indgrebstærsklen for at vise analysemetodens ydeevne inden for det interval, der er relevant for kontrollen af grænseværdien eller indgrebstærsklen. Kombineret kan disse prøver anvendes til beregning af BEQ-niveauerne i de klargjorte prøver (jf. punkt 7.1.2.2). |
7.3. Fastlæggelse af afskæringsværdier
Relationen mellem bioanalytiske resultater i BEQ og resultater fra verifikationsmetoden i TEQ fastlægges, f.eks. ved hjælp af matrixtilpassede kalibreringsforsøg med referenceprøver tilsat 0, 0,5, 1 og 2 gange grænseværdien, med seks gentagelser på hvert niveau (n = 24). Korrektionsfaktorer (blindprøve og genfinding) kan beregnes skønsmæssigt ud fra dette forhold, men skal efterprøves i overensstemmelse med punkt 7.2.2.
Der fastlægges afskæringsværdier for at kunne afgøre, om en prøve overholder de fastsatte grænseværdier, eller for, hvor det er relevant, at kunne kontrollere indgrebstærsklerne i forhold til de pågældende grænseværdier eller indgrebstærskler, der er fastsat for enten PCDD'er/PCDF'er og dioxinlignende PCB'er hver for sig eller for summen af PCDD'er/PCDF'er og dioxinlignende PCB'er. De repræsenteres ved det nedre endepunkt i fordelingen af bioanalytiske resultater (korrigeret for blindprøve og genfinding), som svarer til verifikationsmetodens beslutningsgrænse, med et konfidensniveau på 95 %, hvilket indebærer en falsk overensstemmende-andel på < 5 %, og en RSDR på < 25 %. Verifikationsmetodens beslutningsgrænse er grænseværdien, under hensyntagen til måleusikkerheden.
Afskæringsværdien (i BEQ) beregnes som beskrevet i enten punkt 7.3.1, punkt 7.3.2 eller punkt 7.3.3 (jf. figur 1).
|
7.3.1. |
Anvendelse af det nedre bånd i forventningsintervallet på 95 % ved verifikationsmetodens beslutningsgrænse:
hvor:
|
|
7.3.2. |
Beregning ud fra bioanalytiske resultater (korrigeret for blindprøve og genfinding) fra flere analyser af prøver (n ≥ 6) forurenet i koncentrationer ved verifikationsmetodens beslutningsgrænse som det nedre endepunkt i fordelingen af data ved den tilsvarende BEQ-middelværdi: Afskæringsværdi = BEQDL — 1,64 × SDR hvor:
|
|
7.3.3. |
Beregning som middelværdi af bioanalytiske resultater (i BEQ, korrigeret for blindprøve og genfinding) fra flere analyser af prøver (n ≥ 6) forurenet i koncentrationer på 2/3 af grænseværdien eller indgrebstærsklen, baseret på den observation, at dette niveau ligger på omkring den afskæringsværdi, der er fastlagt i henhold til punkt 7.3.1 eller punkt 7.3.2: 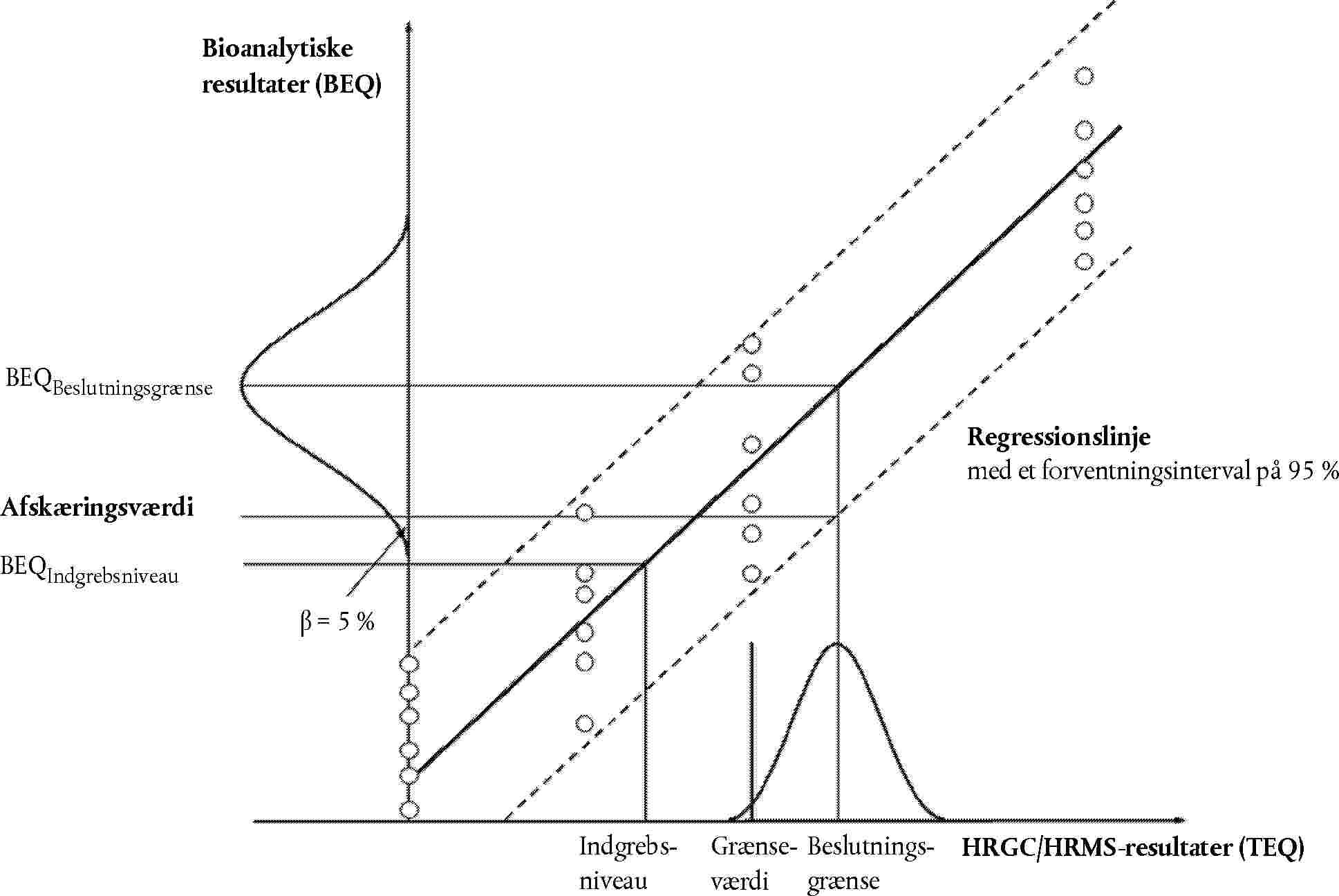 Figur 1.
Figur 1.
Beregning af afskæringsværdier med et konfidensniveau på 95 %, hvilket indebærer en falsk overensstemmende-andel på < 5 %, og en RSDR på < 25 %: 1. fra det nedre bånd i forventningsintervallet på 95 % ved verifikationsmetodens beslutningsgrænse, 2. fra flere analyser af prøver (n ≥ 6) forurenet i koncentrationer ved verifikationsmetodens beslutningsgrænse som det nedre endepunkt i fordelingen af data (i figuren repræsenteret ved en klokkelignende kurve) ved den tilsvarende BEQ-middelværdi. |
|
7.3.4. |
Begrænsninger for afskæringsværdier BEQ-baserede afskæringsværdier beregnet ud fra RSDR, som er tilvejebragt i forbindelse med validering med et begrænset antal prøver med forskellige matrix/kongenermønstre, kan være højere end de TEQ-baserede grænseværdier eller indgrebstærskler, fordi der opnås en højere præcision end den, der kan opnås i rutinemæssige analyser, når et ukendt spektrum af mulige kongenermønstre skal kontrolleres. I sådanne tilfælde beregnes afskæringsværdierne med udgangspunkt i en RSDR på 25 % eller — om muligt — to tredjedele af grænseværdien eller indgrebstærsklen. |
7.4. Karakteristika for metodens ydeevne
|
7.4.1. |
Da interne standarder ikke kan anvendes i bioanalytiske metoder, skal der gennemføres repeterbarhedsanalyser for bioanalytiske metoder for at tilvejebringe oplysninger om standardafvigelsen inden for og mellem de enkelte analyserækker. Repeterbarheden skal være på under 20 %, mens den interne reproducerbarhed skal ligge på under 25 %. Til grund lægges de beregnede niveauer i BEQ efter korrektion for blindprøve og genfinding. |
|
7.4.2. |
Det dokumenteres som led i valideringsprocessen, at testen kan skelne mellem en blindprøve og et indhold på afskæringsværdien, således at det er muligt at identificere prøver over den tilsvarende afskæringsværdi (jf. punkt 7.1.2). |
|
7.4.3. |
Målforbindelser, mulige interferenser og højeste tolererede værdier for blindprøver fastlægges. |
|
7.4.4. |
Standardafvigelsen i responset eller i den koncentration, der beregnes ud fra responset (kun muligt i måleområdet), ved tredobbelt bestemmelse af et prøveekstrakt må ikke være på over 15 %. |
|
7.4.5. |
De ukorrigerede resultater af referenceprøven/prøverne udtrykt i BEQ (blindprøve samt ved grænseværdien eller indgrebstærsklen) bruges til at evaluere den bioanalytiske metodes ydeevne over et konstant tidsrum. |
|
7.4.6. |
Kvalitetskontrolkort for procedureblindprøver og for hver enkelt type referenceprøve registreres og kontrolleres med henblik på at sikre, at den analytiske ydeevne er i overensstemmelse med gældende krav, især for procedureblindprøverne for så vidt angår den påkrævede minimumsdifference i forhold til den nedre del af måleområdet og for referenceprøverne med hensyn til den interne reproducerbarhed. Procedureblindprøver holdes under kontrol for at undgå falsk overensstemmende resultater ved fratrækning af værdierne. |
|
7.4.7. |
Resultaterne for mistænkte prøver fra verifikationsmetoder samt 2-10 % af de overensstemmende prøver (mindst 20 prøver pr. matrix) indsamles og lægges til grund for en evaluering af screeningsmetodens ydeevne og relationen mellem BEQ og TEQ. Denne database kan anvendes til revurdering af de afskæringsværdier, der finder anvendelse på rutinemæssige prøver til de validerede matrixer. |
|
7.4.8. |
En metodes gode ydeevne kan også dokumenteres ved deltagelse i ringtest. Resultaterne fra prøver, der er analyseret i ringtest og dækker et koncentrationsområde på op til f.eks. 2 gange grænseværdien, kan inkluderes i evalueringen af andelen af falsk overensstemmende resultater, såfremt laboratoriet kan dokumentere gode præstationer. Prøverne skal dække de mest almindelige kongenermønstre og repræsentere forskellige kilder. |
|
7.4.9. |
I forbindelse med hændelser kan afskæringsværdierne revurderes på baggrund af de specifikke matrix- og kongenermønstre, der observeres under den pågældende hændelse. |
8. Indberetning af resultater
8.1. Verifikationsmetoder
|
8.1.1. |
Så vidt den anvendte analyseprocedure tillader det, skal analyseresultaterne omfatte værdierne for de enkelte PCDD/PCDF- og dioxinlignende PCB-kongenere og angives som nedre koncentrationer, øvre koncentrationer og middelkoncentrationer med henblik på at sikre, at indberetningen af resultater omfatter så mange oplysninger som muligt, så resultaterne kan fortolkes i overensstemmelse med specifikke krav. |
|
8.1.2. |
Rapporten skal omfatte oplysninger om den metode, der er anvendt til ekstraktion af PCDD'er/PCDF'er og dioxinlignende PCB'er. |
|
8.1.3. |
Tallene for genfinding af de enkelte interne standarder stilles til rådighed, hvis genfindingen ligger uden for det interval, der er angivet i punkt 6.2.5, hvis grænseværdien er overskredet (i dette tilfælde genfindingen for en af to analyser), og i øvrige tilfælde efter anmodning. |
|
8.1.4. |
Da der skal tages hensyn til måleusikkerheden, når det afgøres, om en prøve er overensstemmende, skal denne parameter være tilgængelig. Analyseresultaterne indberettes derfor som x +/- U, hvor x er analyseresultatet og U er den ekspanderede måleusikkerhed, idet der anvendes en dækningsfaktor på 2, hvilket giver et konfidensniveau på ca. 95 %. Hvis PCDD'er/PCDF'er og dioxinlignende PCB'er bestemmes separat, anvendes summen af den anslåede ekspanderede usikkerhed på de separate analyseresultater vedrørende PCDD'er/PCDF'er og dioxinlignende PCB'er på summen af PCDD'er/PCDF'er og dioxinlignende PCB'er |
|
8.1.5. |
Hvis der tages hensyn til måleusikkerheden ved at anvende CCα (se beskrivelsen i denne del (del B), kapitel I, punkt 2.2), indberettes denne parameter. |
|
8.1.6. |
Resultaterne angives i samme enheder og med (mindst) samme antal betydende cifre som de grænseværdier, der er fastsat i direktiv 2002/32/EF. |
8.2. Bioanalytiske screeningsmetoder
|
8.2.1. |
Resultatet af screeningen udtrykkes som værende »overensstemmende« eller som »mistænkt for at være ikke-overensstemmende« (»mistænkt«). |
|
8.2.2. |
Derudover kan et resultat for PCDD'er/PCDF'er og/eller dioxinlignende PCB'er udtrykt i BEQ — ikke TEQ — oplyses. |
|
8.2.3. |
Prøver, der giver et respons under rapporteringsgrænsen, udtrykkes som værende »under rapporteringsgrænsen«. |
|
8.2.4. |
Rapporten skal for hver enkelt type prøvematrix give oplysninger om grænseværdien eller indgrebstærsklen, som vurderingen er baseret på. |
|
8.2.5. |
Rapporten skal indeholde oplysninger om, hvilken type test der er anvendt, samt om det grundlæggende prøvningsprincip og den anvendte kalibreringsmåde. |
|
8.2.6. |
Rapporten skal omfatte oplysninger om den metode, der er anvendt til ekstraktion af PCDD'er/PCDF'er og dioxinlignende PCB'er. |
|
8.2.7. |
I tilfælde, hvor prøver mistænkes for at være ikke-overensstemmende, skal rapporten indeholde en bemærkning om den foranstaltning, der skal træffes. Koncentrationen af PCDD'er/PCDF'er og summen af PCDD'er/PCDF'er og dioxinlignende PCB'er i prøver med forhøjet indhold skal bestemmes/bekræftes efter en verifikationsmetode |
KAPITEL III
Klargøring af prøver og krav til analysemetoder, der anvendes ved offentlig kontrol af indholdet af ikke-dioxinlignende PCB'er (PCB 28, 52, 101, 138, 153 OG 180)
1. Anvendelsesområde
Kravene i dette kapitel anvendes, når foder analyseres med henblik på offentlig kontrol af indholdet af ikke-dioxinlignende polychlorerede biphenyler (ikke-dioxinlignende PCB'er) og til andre forskriftsmæssige formål.
2. Påvisningsmetoder, der kan anvendes
Gaskromatografi/elektronindfangningsdetektion (GC-ECD), GC-LRMS, GC-MS/MS, GC-HRMS eller tilsvarende metoder.
3. Identifikation og bekræftelse af de relevante analytter
|
3.1. |
Relativ retentionstid i forhold til interne standarder eller referencestandarder (tilladt afvigelse: +/- 0,25 %). |
|
3.2. |
Gaskromatografisk separation af alle seks indikator-PCB'er (PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 og PCB 180) fra interfererende stoffer, navnlig fra co-eluerende PCB'er, især hvis niveauet i de pågældende prøver ligger i nærheden af de ved lov fastsatte grænser, og en overskridelse skal bekræftes. [Kongenere, der erfaringsmæssigt ofte co-eluerer, er f.eks.. PCB 28/31, PCB 52/69 og PCB 138/163/164. For så vidt angår GC-MS skal der også tages hensyn til mulige interferenser fra fragmenter af højere chlorerede kongenere.] |
|
3.3. |
Krav til GC-MS-metoder Overvågning af mindst: a) to specifikke ioner ved HRMS b) to specifikke ioner med en m/z-værdi på > 200 eller tre specifikke ioner med en m/z-værdi på > 100 ved LRMS c) 1 prækursor-ion og 2 produkt-ioner ved MS-MS. Tilladte maksimumstolerancer for isotopforholdet for udvalgte massefragmenter: Relativ afvigelse for isotopforholdet for udvalgte massefragmenter fra teoretisk isotopforhold eller kalibreringsstandard for målionen (den hyppigst forekommende af de målte ioner) og kvalifikatorionen/ionerne:
|
||||||||||||||||||
|
3.4. |
Krav til GC-ECD-metoder Resultater, der overskrider tolerancegrænsen, bekræftes med to GC-kolonner med stationære faser med forskellig polaritet. |
4. Dokumentation for metodens ydeevne
Metodens ydeevne valideres i nærheden af grænseværdien (0,5 til 2 gange grænseværdien) med en acceptabel variationskoefficient for gentagne analyser (jf. kravene til intern reproducerbarhed (intermediær præcision) i punkt 9).
5. Bestemmelsesgrænse
Blindprøverne må ikke repræsentere over 30 % af den forureningsgrad, som svarer til grænseværdien ( 45 ).
6. Kvalitetskontrol
Regelmæssig blindprøvekontrol, analyse af spikede prøver, kvalitetskontrolprøver og deltagelse i laboratoriesammenligninger vedrørende de relevante matrixer.
7. Genfindingskontrol
|
7.1. |
Der anvendes egnede interne standarder med fysisk-kemiske egenskaber, der er sammenlignelige med de relevante analytters. |
|
7.2. |
Tilsætning af interne standarder: Tilsætning til produkter (inden ekstraktion og oprensning). |
|
7.3. |
Krav til metoder, hvor alle seks isotopmærkede indikator-PCB-kongenere anvendes: a) Resultater korrigeres for genfinding af interne standarder. b) Genfindingsprocenten for isotopmærkede interne standarder skal være på mellem 50 og 120 %. c) En lavere eller højere genfindingsprocent er acceptabel for enkeltkongenere, der bidrager til summen af de seks indikator-PCB'er med under 10 %. |
|
7.4. |
Krav til metoder, hvor ikke alle seks isotopmærkede interne standarder eller andre interne standarder anvendes: a) Genfindingen af de(n) interne standard(er) kontrolleres for hver enkelt prøve. b) Genfindingen af interne standarder skal være på mellem 60 og 120 %. c) Resultater korrigeres for genfinding af interne standarder. |
|
7.5. |
Genfindingen af umærkede kongenere kontrolleres ved hjælp af spikede prøver eller kvalitetskontrolprøver med koncentrationer i nærheden af grænseværdien. Genfindingen af disse kongenere anses for acceptabel, hvis genfindingsprocenten er på mellem 70 og 120 %. |
8. Krav til laboratorier
I henhold til forordning (EF) nr. 882/2004 skal laboratorier være akkrediteret af et anerkendt organ, der fungerer i overensstemmelse med ISO-vejledning 58, så det sikres, at de anvender analysekvalitetssikring. Laboratorier akkrediteres ifølge EN ISO/IEC 17025-standarden.
9. Karakteristika for metodens ydeevne: kriterier vedrørende summen af de seks indikator-PCB'er ved grænseværdien:
|
Korrekthed |
– 30 til + 30 % |
|
Intermediær præcision (RSD %) |
≤ 20 % |
|
Forskel mellem øvre og nedre koncentration (beregnet) |
≤ 20 % |
10. Indberetning af resultater
|
10.1. |
Så vidt den anvendte analyseprocedure tillader det, skal analyseresultaterne omfatte værdierne for de enkelte PCB-kongenere og registreres som nedre koncentrationer, øvre koncentrationer og middelkoncentrationer, med henblik på at registreringen af resultater omfatter så mange oplysninger som muligt, så resultaterne kan fortolkes i overensstemmelse med specifikke krav. |
|
10.2. |
Rapporten skal omfatte oplysninger om den metode, der er anvendt til ekstraktion af PCB'er og fedtstoffer. |
|
10.3. |
Tallene for genfinding af de enkelte interne standarder stilles til rådighed, hvis genfindingen ligger uden for det interval, der er angivet i punkt 7, hvis grænseværdien er overskredet, og i øvrige tilfælde efter anmodning. |
|
10.4. |
Da der skal tages hensyn til måleusikkerheden, når det afgøres, om en prøve er overensstemmende, skal denne parameter ligeledes være tilgængelig. Analyseresultaterne indberettes derfor som x +/- U, hvor x er analyseresultatet og U er den ekspanderede måleusikkerhed, idet der anvendes en dækningsfaktor på 2, hvilket giver et konfidensniveau på ca. 95 %. |
|
10.5. |
Hvis der tages hensyn til måleusikkerheden ved at anvende CCα (se beskrivelsen i kapitel I, punkt 2.1), indberettes denne parameter. |
|
10.6. |
Resultaterne angives i samme enheder og med (mindst) samme antal betydende cifre som de grænseværdier, der er fastsat i direktiv 2002/32/EF. |
BILAG VI
ANALYSEMETODER TIL BESTEMMELSE AF ANIMALSKE BESTANDDELE SOM LED I DEN OFFENTLIGE KONTROL AF FODER
1. FORMÅL OG ANVENDELSESOMRÅDE
Bestemmelse af animalske bestanddele i foder skal udføres ved lysmikroskopi eller PCR (polymerasekædereaktion) i overensstemmelse med de bestemmelser, der er fastsat i dette bilag.
Disse to metoder gør det muligt at påvise tilstedeværelsen af animalske bestanddele i fodermidler og foderblandinger. De gør det imidlertid ikke muligt at beregne mængden af sådanne bestanddele i fodermidler og foderblandinger. Begge metoder har en påvisningsgrænse under 0,1 % (w/w).
PCR-metoden gør det muligt at identificere, hvilken taksonomisk gruppe animalske bestanddele, der er til stede i fodermidler og foderblandinger, tilhører.
Disse metoder skal finde anvendelse i forbindelse med kontrollen med anvendelsen af forbuddene i artikel 7, stk. 1, i og bilag IV til forordning (EF) nr. 999/2001 og i artikel 11, stk. 1, i forordning (EF) nr. 1069/2009.
Afhængigt af hvilken type foder der afprøves, kan disse metoder anvendes inden for en enkelt operationel protokol, enten alene eller tilsammen i overensstemmelse med de standardprocedurer (SOP), som er fastsat af EU-referencelaboratoriet for animalske proteiner i foderstoffer (EURL-AP) og offentliggjort på dets websted ( 46 ).
2. METODER
2.1. Lysmikroskopi
2.1.1. Princip
Animalske bestanddele, der kan være til stede i fodermidler og foderblandinger, som er sendt til analyse, identificeres ud fra typiske kendetegn, der kan identificeres ved mikroskopi (bl.a. muskelfibre og andre kødpartikler, brusk, knogler, horn, hår, børster, blod, fjer, æggeskaller, fiskeben og skæl).
2.1.2. Reagenser og udstyr
2.1.2.1. Reagenser
2.1.2.1.1. Koncentreringsmiddel
2.1.2.1.1.1. Tetrachlorethylen (massefylde 1,62)
2.1.2.1.2. Farvningsreagens
|
2.1.2.1.2.1. |
Alizarinrødtopløsning (2,5 ml 1M saltsyre fortyndes i 100 ml vand, og der tilsættes 200 mg alizarinrødt til opløsningen) |
2.1.2.1.3. Monteringsmedier
2.1.2.1.3.1. Lud (NaOH 2,5 % w/v eller KOH 2,5 % w/v)
2.1.2.1.3.2. Glycerol (ufortyndet, viskositet: 1 490 cP)
2.1.2.1.3.3. Norland ® Optical Adhesive 65 (viskositet: 1 200 cP) eller en harpiks med tilsvarende egenskaber med henblik på præparering af permanente objektglas
2.1.2.1.4. Monteringsmedier med farvende egenskaber
|
2.1.2.1.4.1. |
Lugolopløsning (2 g kaliumiodid opløses i 100 ml vand, og der tilsættes 1 g jod under hyppig omrystning) |
|
2.1.2.1.4.2. |
Cystinreagens (2 g blyacetat og 10 g NaOH i 100 ml vand) |
|
2.1.2.1.4.3. |
Fehlings væske (klargjort før brug af lige dele (1/1) af to stamopløsninger A og B. Opløsning A: 6,9 g kobber(II)sulfatpentahydrat opløses i 100 ml vand. Opløsning B: 34,6 g kaliumnatriumtartrattetrahydrat og 12 g NaOH opløses i 100 ml vand) |
|
2.1.2.1.4.4. |
Tetramethylbenzidin/hydrogenperoxid (1 g 3,3«,5,5’tetramethylbenzidin (TMB) opløses i 100 ml iseddike og 150 ml vand. Før brug blandes 4 dele af denne TMB-opløsning med 1 del 3 %-hydrogenperoxid) |
2.1.2.1.5. Skyllevæsker
2.1.2.1.5.1. Ethanol ≥ 96 % (teknisk kvalitet)
2.1.2.1.5.2. Acetone (teknisk kvalitet)
2.1.2.1.6. Blegemiddel
2.1.2.1.6.1. Natriumhypochloritopløsning, som fås i handelen (9-14 % aktivt chlor).
2.1.2.2. Udstyr
2.1.2.2.1. Analysevægt med 0,001 g aflæsningsnøjagtighed
2.1.2.2.2. Formalingsudstyr (mølle eller morter)
2.1.2.2.3. Sigter med kvadratiske masker med en maskevidde på 0,25 mm og 1 mm
|
2.1.2.2.4. |
Konisk glasskilletragt med et rumindhold på 250 ml med teflon eller slebet stophane ved keglens bund. Stophanens åbning skal have en diameter på ≥ 4 mm. Alternativt kan et glasbæger med konisk bund anvendes, forudsat at laboratoriet har dokumenteret, at påvisningsniveauet svarer til det, der opnås ved at benytte den koniske glasskilletragt. 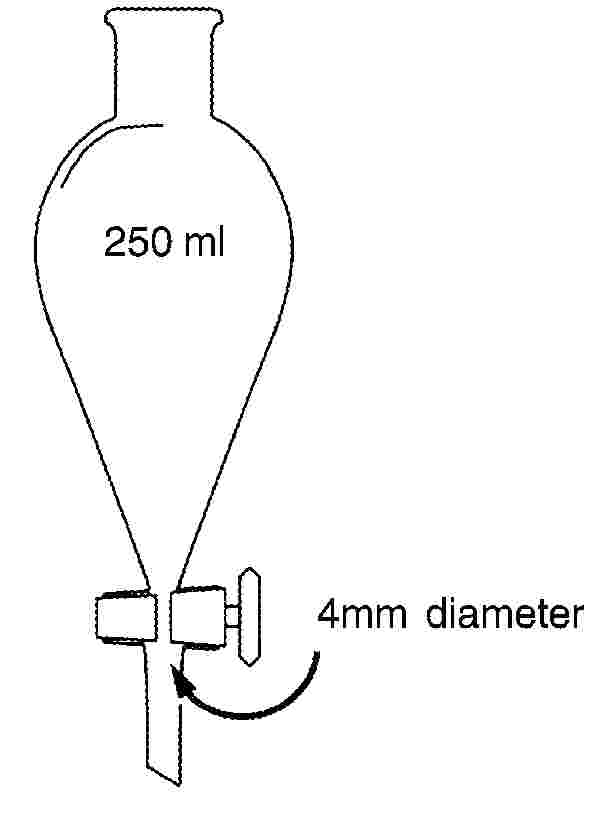
|
|
2.1.2.2.5. |
Stereomikroskop med et endeligt forstørrelsesinterval på mindst 6,5 × til 40 × |
|
2.1.2.2.6. |
Sammensat mikroskop med et endeligt forstørrelsesinterval på mindst 100 × til 400 × med transmitteret lys/lysfelt. Alternativt kan polariseret lys og differentiel interferenskontrast anvendes |
|
2.1.2.2.7. |
Standardlaboratorieglasudstyr |
|
2.1.2.2.8. |
Udstyr til klargøring af objektglas: klassiske objektglas, konkave objektglas, dækglas (20 × 20 mm), pincetter, tynd spatel |
2.1.3. Udtagning og forberedelse af prøver
2.1.3.1. Prøveudtagning
Der skal anvendes en repræsentativ prøve, som er udtaget i overensstemmelse med bestemmelserne i bilag I.
2.1.3.2. Forholdsregler
For at undgå krydskontaminering på laboratoriet skal alt genanvendeligt udstyr rengøres omhyggeligt før brugen. Skilletragtens dele adskilles inden rengøring. Skilletragtens dele og glasudstyr forvaskes manuelt og vaskes derefter i en vaskemaskine. Sigter rengøres ved brug af en børste med stive syntetiske børster. Det anbefales at foretage en afsluttende rengøring af sigter med acetone og komprimeret luft efter sigtning af fedtholdigt materiale, som f.eks. fiskemel.
2.1.3.3. Forberedelse af andre prøver end fedt eller olie
|
2.1.3.3.1. |
Tørring af prøver: Prøver med et vandindhold på over 14 % skal tørres inden håndteringen. |
|
2.1.3.3.2. |
Sigtning af prøver på forhånd: Det anbefales at sigte (1 mm-sigte) foder i pilleform og kerner på forhånd og derefter forberede og analysere begge fraktionerne som separate prøver. |
|
2.1.3.3.3. |
Delprøveudtagning og formaling: Der udtages en delprøve på mindst 50 g af prøven, og den formales efterfølgende. |
|
2.1.3.3.4. |
Ekstraktion og forberedelse af sedimentet: En portion på 10 g (med en nøjagtighed på 0,01 g) af den formalede delprøve overføres til skilletragten eller glasbægeret med konisk bund, og der tilsættes 50 ml tetrachlorethylen. Den portion, der overføres til tragten, udgør højst 3 g, når det drejer sig om fiskemel eller andre rene animalske produkter, mineralske ingredienser eller forblandinger, som genererer mere end 10 % sediment. Blandingen omrystes kraftigt i mindst 30 sekunder, og der tilsættes forsigtigt yderligere mindst 50 ml tetrachlorethylen, idet den indvendige side af tragten vaskes for at fjerne eventuelle partikler, der har sat sig fast. Den resulterende blanding henstår i mindst 5 minutter, før sedimentet skilles fra ved åbning af hanen. Hvis der anvendes et glasbæger med konisk bund, omrøres blandingen kraftigt i mindst 15 sekunder, og eventuelle partikler, der har sat sig fast på siden af glasbægeret, vaskes omhyggeligt ned langs indersiden med mindst 10 ml ren tetrachlorethylen. Blandingen henstår i 3 minutter og omrøres igen i 15 sekunder, og eventuelle partikler, der har sat sig fast på siden af glasbægeret, vaskes omhyggeligt ned langs indersiden med mindst 10 ml ren tetrachlorethylen. Den resulterende blanding henstår i mindst 5 minutter, hvorefter den flydende fraktion fjernes og kasseres ved omhyggelig dekantering, idet der udvises omhu for ikke at miste noget af sedimentet. Sedimentet tørres og vejes med en nøjagtighed på 0,001 g. Hvis mere end 5 % af sedimentet består af partikler på over 0,50 mm, sigtes det (0,25 mm-sigte), og begge fraktionerne undersøges. |
|
2.1.3.3.5. |
Ekstraktion og forberedelse af flotatet: Efter at sedimentet er skilt fra med den ovenfor beskrevne metode, bør der være to faser tilbage i skilletragten: en flydende, der består af tetrachlorethylen, og en fast, der er dannet af floteringsmateriale (flydelaget). Denne faste fase er flotatet, og det skal skilles fra ved at hælde tetrachlorethylenet helt fra tragten ved at åbne hanen. Ved at skilletragten vendes, overføres flotatet til en stor petriskål, og det lufttørres i et stinkskab. Hvis mere end 5 % af flotatet består af partikler på over 0,50 mm, sigtes det (0,25 mm-sigte), og begge fraktionerne undersøges. |
|
2.1.3.3.6. |
Forberedelse af råmateriale: En portion på mindst 5 g af den formalede delprøve forberedes. Hvis mere end 5 % af råmaterialet består af partikler på over 0,50 mm, sigtes det (0,25 mm-sigte), og begge fraktionerne undersøges. |
2.1.3.4. Forberedelse af prøver, der består af fedt eller olie
Følgende protokol følges for forberedelse af prøver, der består af fedt eller olie:
— Fedt i fast form opvarmes i en ovn, indtil det er blevet flydende.
— Med pipette overføres 40 ml fedt eller olie fra bunden af prøven til et centrifugerør.
— Der centrifugeres i 10 minutter ved 4 000 omdrejninger i minuttet.
— Hvis fedtet efter centrifugering er fast, opvarmes det i en ovn, indtil det er blevet flydende.
— Der centrifugeres endnu en gang ved 4 000 omdrejninger i 5 minutter.
— Med en lille ske eller en spatel overføres halvdelen af de dekanterede urenheder til objektglas til undersøgelse. Det anbefales at bruge glycerol som monteringsmedium.
— De resterende urenheder anvendes til at forberede sedimentet som beskrevet i punkt 2.1.3.3.
2.1.3.5. Brug af farvningsreagenser
For at lette korrekt identifikation af de animalske bestanddele kan laboranten anvende farvningsreagenser under prøveforberedelsen i overensstemmelse med retningslinjerne, der er udstedt af EURL-AP og offentliggjort på dets websted.
Hvis der anvendes alizarinrødtopløsning til at farve sedimentet, anvendes følgende protokol:
— Det tørrede sediment hældes i et reagensglas og skylles to gange med ca. 5 ml ethanol (der anvendes vortex-apparat i 30 sekunder, opløsningsmidlet skal henstå i ca. 1 minut 30 sekunder, så det bundfælder sig, før det hældes fra).
— Sedimentet bleges ved tilsætning af mindst 1 ml natriumhypochloritopløsning. Lad reaktionen fortsætte i 10 minutter. Røret fyldes med vand, sedimentet henstår i 2-3 minutter, og vandet og de opslæmmede partikler hældes forsigtigt fra.
— Sedimentet renses yderligere to gange med ca. 10 ml vand (der anvendes vortex-apparat i 30 sekunder, lad bundfælde, og hæld hver gang vandet fra).
— Der tilsættes 2-10 dråber alizarinrødtopløsning, og blandingen vortexes. Reaktionen skal have lov at finde sted i 30 sekunder, og det farvede sediment renses to gange med ca. 5 ml ethanol efterfulgt af én skylning med acetone (der anvendes hver gang vortex-apparat i 30 sekunder, opløsningsmidlet skal henstå ca. 1 minut, før det hældes fra).
— Det farvede sediment tørres.
2.1.4. Mikroskopisk undersøgelse
2.1.4.1. Præparering af objektglas
Der forberedes præparater af sedimentet og, afhængigt af laborantens valg, af enten flotatet eller råmaterialet. Hvis der har været anvendt sigtning ved prøveforberedelsen, forberedes begge fraktionerne (både den fine og den grove fraktion). De testportioner af fraktioner, der udstryges på objektglas, skal være repræsentative for hele fraktionen.
Der præpareres et tilstrækkeligt antal objektglas for at sikre, at en fuldstændig undersøgelsesprotokol, jf. punkt 2.1.4.2, kan gennemføres.
Objektglas monteres med passende monteringsmedium i overensstemmelse med de standardprocedurer, der er fastsat af EURL-AP og offentliggjort på dets websted. Objektglassene skal være dækket med dækglas.
2.1.4.2. Observationsprotokoller vedrørende påvisning af animalske partikler i foderblandinger og fodermidler
De præparerede objektglas skal observeres i overensstemmelse med de observationsprotokoller, der er fastsat i diagram 1 for foderblandinger og fodermidler, dog ikke rent fiskemel, eller i diagram 2 for rent fiskemel.
Mikroskopiobservationerne udføres med sammensat mikroskop på sedimentet og, afhængigt af laborantens valg, på enten flotatet eller råmaterialet. Stereomikroskop kan benyttes som supplement til sammensat mikroskop for de grove fraktioner. Hvert objektglas skal screenes i sin helhed ved forskellige forstørrelser.
Minimumsantallet af objektglas, der skal observeres på hvert enkelt trin af observationsprotokollen, skal overholdes strengt, medmindre det ikke med hele fraktionsmaterialet er muligt at nå op på det fastsatte antal objektglas. Højst 6 objektglas pr. bestemmelse skal observeres.
For at lette identifikationen af partiklernes type og oprindelse kan laboranten bruge støtteværktøjer såsom beslutningsstøttesystemer, billedbiblioteker og referenceprøver.
2.1.4.3. Antal bestemmelser
Hvis der efter en første bestemmelse, der er gennemført i overensstemmelse med den relevante observationsprotokol, jf. diagram 1 eller diagram 2, ikke er påvist nogen animalske partikler af en given type (dvs. landdyr eller fisk), er det ikke nødvendigt med en yderligere bestemmelse, og resultatet af analysen indberettes med anvendelse af den terminologi, der er fastsat i punkt 2.1.5.1.
Hvis der efter en første bestemmelse, der er gennemført i overensstemmelse med den relevante observationsprotokol, jf. diagram 1 eller diagram 2, er påvist 1-5 animalske partikler af en given type (dvs. landdyr eller fisk), foretages der en yderligere bestemmelse på en ny delprøve på 50 g. Hvis der efter denne anden bestemmelse er påvist 0-5 animalske partikler af den samme type, skal resultatet af analysen indberettes med anvendelse af den terminologi, der er fastsat i punkt 2.1.5.2, ellers foretages der en tredje bestemmelse på en ny delprøve på 50 g. Hvis summen af partikler af en given type, der er påvist ved de to bestemmelser, efter den første og anden bestemmelse imidlertid er større end 15, er det ikke nødvendigt med en yderligere bestemmelse, og resultatet af analysen indberettes direkte med anvendelse af den terminologi, der er fastsat i punkt 2.1.5.3. Hvis summen af animalske partikler af en given type, der er påvist ved de tre bestemmelser, efter den tredje bestemmelse er større end 15, indberettes resultatet af analysen med anvendelse af den terminologi, der er fastsat i punkt 2.1.5.3. Ellers indberettes resultatet af analysen med anvendelse af den terminologi, der er fastsat i punkt 2.1.5.2.
Hvis der efter en første bestemmelse, der er gennemført i overensstemmelse med den relevante observationsprotokol, jf. diagram 1 eller diagram 2, er påvist mere end 5 animalske partikler af en given type (dvs. landdyr eller fisk), indberettes resultatet af analysen med anvendelse af den terminologi, der er fastsat i punkt 2.1.5.3.
2.1.5. Angivelse af resultaterne
Ved indberetningen af resultaterne skal laboratoriet angive, hvilken type materiale analysen er blevet udført på (sediment, flotat eller råmateriale), og hvor mange bestemmelser der er blevet foretaget.
Laboratorierapporten skal mindst indeholde oplysninger om tilstedeværelsen af bestanddele fra landdyr og fra fisk.
De forskellige tilfælde afrapporteres på følgende måde.
|
2.1.5.1. |
Hvis der ikke er blevet påvist nogen animalske partikler af en given type: — Der har ved lysmikroskopi ikke kunnet påvises partikler fra landdyr i den forelagte prøve. — Der har ved lysmikroskopi ikke kunnet påvises partikler fra fisk i den forelagte prøve. |
|
2.1.5.2. |
Hvis der er blevet påvist i gennemsnit 1-5 animalske partikler af en given type: — Der har ved lysmikroskopi i gennemsnit pr. bestemmelse kunnet påvises højst 5 partikler fra landdyr i den forelagte prøve. Partiklerne blev identificeret som … [knogler, brusk, muskelvæv, hår, horn …]. Da dette lave indhold ligger under påvisningsgrænsen for mikroskopimetoden, kan risiko for et falsk positivt resultat ikke udelukkes eller, hvis det er relevant: — Der har ved lysmikroskopi i gennemsnit pr. bestemmelse kunnet påvises højst 5 partikler fra fisk i den forelagte prøve. Partiklerne blev identificeret som … [fiskeben, fiskeskæl, brusk, muskelvæv, øresten, gælle …]. Da dette lave indhold ligger under påvisningsgrænsen for mikroskopimetoden, kan risiko for et falsk positivt resultat ikke udelukkes. Hvis der er foretaget sigtning på forhånd, skal det oplyses i laboratorierapporten, hvilken fraktion (sigtet fraktion, pelleteret fraktion eller kerner) de animalske partikler er blevet påvist i, for så vidt som påvisning af animalske partikler udelukkende i den sigtede fraktion kan være tegn på en forurening fra omgivelserne. |
|
2.1.5.3. |
Hvis der er blevet påvist i gennemsnit mere end 5 animalske partikler af en given type: — Der har ved lysmikroskopi i gennemsnit pr. bestemmelse kunnet påvises mere end 5 partikler fra landdyr i den forelagte prøve. Partiklerne blev identificeret som … [knogler, brusk, muskelvæv, hår, horn …] eller, hvis det er relevant: — Der har ved lysmikroskopi i gennemsnit pr. bestemmelse kunnet påvises mere end 5 partikler fra fisk i den forelagte prøve. Partiklerne blev identificeret som … [fiskeben, fiskeskæl, brusk, muskelvæv, øresten, gælle …]. Hvis der er foretage sigtning på forhånd, skal det oplyses i laboratorierapporten, hvilken fraktion (sigtet fraktion, pelleteret fraktion eller kerner) de animalske partikler er blevet påvist i, for så vidt som påvisning af animalske partikler udelukkende i den sigtede fraktion kan være tegn på en forurening fra omgivelserne. |
2.2. PCR
2.2.1. Princip
Deoxyribonukleinsyre (dna)-fragmenter af animalsk oprindelse, som kan forekomme i fodermidler og foderblandinger påvises med en genetisk amplifikationsmetode ved PCR, der er målrettet mod artsspecifikke dna-sekvenser.
PCR-metoden kræver, at der først foretages en dna-ekstraktion. Derefter amplificeres det fremkomne dna-ekstrakt for at påvise de dyrearter, assayet er målrettet mod.
2.2.2. Reagenser og udstyr
2.2.2.1. Reagenser
2.2.2.1.1. Reagenser til dna-ekstraktion
Der må kun anvendes reagenser, der er godkendt af EURL-AP og offentliggjort på dets websted.
2.2.2.1.2. Reagenser til genetisk amplifikation
2.2.2.1.2.1. Primere og sonder
Der må kun anvendes primere og sonder med oligonukleotidsekvenser, der er valideret af EURL-AP ( 47 ).
2.2.2.1.2.2. Mastermix
Der må kun anvendes mastermix-opløsninger, som ikke indeholder reagenser, der kan føre til falske resultater på grund af tilstedeværelsen af animalsk dna ( 48 ).
2.2.2.1.2.3. Dekontamineringsreagenser
2.2.2.1.2.3.1. Saltsyreopløsning (0,1N)
2.2.2.1.2.3.2. Blegemiddel (natriumhypochloritopløsning (0,15 % aktivt chlor))
2.2.2.1.2.3.3. Ikke-ætsende reagenser til dekontaminering af bekosteligt udstyr såsom analysevægte (f.eks. DNA EraseTM fra MP Biomedicals).
2.2.2.2. Udstyr
2.2.2.2.1. Analysevægt med 0,001 g aflæsningsnøjagtighed
2.2.2.2.2. Formalingsudstyr
2.2.2.2.3. Thermocycler, der muliggør tidstro PCR
2.2.2.2.4. Mikrocentrifuge til mikrocentrifugerør
2.2.2.2.5. Mikropipettesæt, som gør det muligt, at der afpipetteres fra 1-1 000 μl
|
2.2.2.2.6. |
Molekylærbiologisk standardudstyr af plast: mikrocentrifugerør, plastspidser med filter til mikropipetter, plader til thermocycler |
|
2.2.2.2.7. |
Frysere til at opbevare prøver og reagenser. |
2.2.3. Udtagning og forberedelse af prøver
2.2.3.1. Prøveudtagning
Der skal anvendes en repræsentativ prøve, som er udtaget i overensstemmelse med bestemmelserne i bilag I.
2.2.3.2. Forberedelse af prøver
Forberedelsen af laboratorieprøver indtil dna-ekstraktion skal opfylde kravene i bilag II. Der udtages en delprøve på mindst 50 g af prøven, og den formales efterfølgende.
Prøveforberedelsen skal foregå i et andet lokale end dem, der er beregnet til dna-ekstraktion og til genetisk amplifikation-reaktioner som beskrevet i ISO 24276.
Der forberedes to testportioner på mindst 100 mg hver.
2.2.4. Dna-ekstraktion
Dna-ekstraktionen udføres på hver testportion, der er forberedt ved hjælp af den standardprocedure, der er fastsat af EURL-AP og offentliggjort på dets websted.
Der forberedes to ekstraktionskontroller for hver ekstraktionsserie som beskrevet i ISO 24276:
— en ekstraktionsblindkontrol
— en positiv dna-ekstraktionskontrol.
2.2.5. Genetisk amplifikation
Den genetiske amplifikation foretages ved anvendelse af de metoder, som er valideret for de enkelte arter, der kræver identificering. Disse metoder er fastsat i de standardprocedurer, der er fastsat af EURL-AP og offentliggjort på dets websted. Hvert dna-ekstrakt analyseres i mindst to forskellige fortyndinger for at evaluere hæmning.
Der forberedes to amplifikationskontroller pr. målart som beskrevet i ISO 24276:
— Der anvendes en positiv dna-målkontrol for alle plader eller serier af PCR-assays.
— Der anvendes en amplifikationsreagenskontrol (også kaldet no template control (NTC)) for alle plader eller serier af PCR-assays.
2.2.6. Fortolkning og angivelse af resultater
Ved indberetningen af resultaterne skal laboratoriet som minimum angive vægten af de anvendte testportioner, den anvendte ekstraktionsmetode, antallet af foretagne bestemmelser og metodens påvisningsgrænse.
Resultaterne fortolkes og indberettes ikke, hvis den positive dna-ekstraktionskontrol og de positive dna-målkontroller ikke giver positive resultater for det mål, assayet vedrører, og amplifikationensreagenskontrollen samtidig er negativ.
Hvis resultaterne af de to testportioner ikke er overensstemmende, gentages som minimum den genetiske amplifikation. Hvis laboratoriet har mistanke om, at dna-ekstrakterne kan være årsag til uoverensstemmelsen, foretages der en ny dna-ekstraktion og efterfølgende genetisk amplifikation, inden resultaterne fortolkes.
Den endelige angivelse af resultater skal være baseret på integration og fortolkning af resultaterne af de to testportioner i overensstemmelse med de standardprocedurer, der er fastsat af EURL-AP og offentliggjort på dets websted.
2.2.6.1. Negativt resultat
Et negativt resultat afrapporteres på følgende måde:
Der blev ikke påvist noget dna fra X i den forelagte prøve (X står for den dyreart eller gruppe af dyrearter, assayet er målrettet mod).
2.2.6.2. Positivt resultat
Et positivt resultat afrapporteres på følgende måde:
Der blev påvist dna fra X i den forelagte prøve (X står for den dyreart eller gruppe af dyrearter, assayet er målrettet mod).
BILAG VII
METODE TIL BEREGNING AF NÆRINGSVÆRDIEN AF FODER TIL FJERKRÆ
1. Beregningsmetode og angivelse af næringsværdi
Næringsværdien i foderblandinger til fjerkræ beregnes efter nedenstående formel ud fra procentsatserne for visse bestanddele i foderet. Denne værdi udtrykkes i megajoule (MJ) omsættelig energi (ME), korrigeret for nitrogen, pr. kg foderblanding:
MJ/kg ME = 0,1551 × % råprotein + 0,3431 × % råfedt + 0,1669 × % stivelse + 0,1301 × % totalsukker (udtrykt som saccharose).
2. Tolerancer for de angivne værdier
Hvis der i forbindelse med den offentlige kontrol konstateres en afvigelse (øget eller reduceret næringsværdi af foderet) mellem kontrollens resultat og den angivne næringsværdi, kan en mindstetolerance på 0,4 MJ/kg ME tillades.
3. Angivelse af resultater
Det resultat, der opnås ved ovenstående formel, angives med en decimal.
4. Prøveudtagning og analyse
Udtagningen af prøver af foderblandingen og bestemmelsen af indholdet af bestanddele som angivet i beregningsmetoden foretages i overensstemmelse med Fællesskabets prøveudtagnings- og analysemetoder som led i den offentlige kontrol med foder.
Følgende metoder anvendes:
— Til bestemmelse af indholdet af råfedt: metode B under den i del H i bilag III beskrevne metode til bestemmelse af råfedt
— Til bestemmelse af indholdet af stivelse: den polarimetriske metode som beskrevet i del L i bilag III.
BILAG VIII
ANALYSEMETODER TIL KONTROL FOR ULOVLIG FOREKOMST AF FODERTILSÆTNINGSSTOFFER, DER IKKE LÆNGERE ER TILLADT
Vigtigt:
Der kan anvendes mere følsomme analysemetoder til påvisning af ulovlig forekomst i foder af tilsætningsstoffer, der ikke længere er tilladt, end dem, der er beskrevet i dette bilag.
De i dette bilag beskrevne analysemetoder anvendes til verifikationsformål.
A. BESTEMMELSE AF METHYLBENZOQUAT
7-Benzyloxy-6-Butyl-3-Methoxycarbonyl-4-Quinolon
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af methylbenzoquat i foder. Bestemmelsesgrænsen er 1 mg/kg.
2. Princip
Methylbenzoquat ekstraheres fra prøven med en opløsning af methansulfonsyre i methanol. Ekstraktet oprenses med dichlormethan, ved ionbytningskromatografi og derefter igen med dichlormethan. Methylbenzoquatindholdet bestemmes ved omvendt fase-HPLC med anvendelse af UV-detektor.
3. Reagenser
3.1. Dichlormethan.
3.2. Methanol, svarende til HPLC-kvalitet.
3.3. Mobil fase til HPLC
Blanding af methanol (3.2) og vand (svarende til HPLC-kvalitet), 75 + 25 (v + v)
Der filtreres gennem et 0,22 μm filter (4.5), og opløsningen afgasses (f.eks. ved ultralydsbehandling i 10 minutter).
3.4. Methansulfonsyreopløsning, c = 2 %
20,0 ml methansulfonsyre fortyndes til 1 000 ml med methanol (3.2).
3.5. Saltsyreopløsning, c = 10 %
100 ml saltsyre (ρ20 = 1,18 g/ml) fortyndes til 1 000 ml med vand.
3.6. Kationbytterresin Amberlite CG-120 (Na), 100–200 mesh
Resinet forbehandles inden brug. 100 g resin opslæmmes med 500 ml saltsyreopløsning (3.5) og opvarmes på varmeplade til kogning under stadig omrøring. Opslæmningen henstår til afkøling, og syren dekanteres fra. Derefter filtreres gennem et filtrerpapir under vakuum. Resinet vaskes to gange med vand i portioner på 500 ml og derefter med 250 ml methanol (3.2). Resinet skylles med yderligere 250 ml methanol og tørres, ved at der sendes luft gennem filterkagen. Det tørrede resin opbevares i en tilproppet flaske.
3.7. Standardstof: ren methylbenzoquat (7-benzyloxy-6-butyl-3-methoxycarbonyl-4-quinolon)
3.7.1.
Med en nøjagtighed på 0,1 mg afvejes 50 mg standard (3.7), som opløses i methansulfonsyreopløsning (3.4) i en 100 ml målekolbe, hvorefter der fyldes op til mærket og blandes.
3.7.2.
5,0 ml af methylbenzoquatstandardstamopløsningen (3.7.1) overføres til en 50 ml målekolbe, hvorefter der fyldes op til mærket med methanol (3.2) og blandes.
3.7.3.
Henholdsvis 1,0 , 2,0 , 3,0 , 4,0 og 5,0 ml af methylbenzoquatstandardmellemopløsningen (3.7.2) overføres til hver sin 25 ml målekolbe. Der fyldes op til mærket med mobil fase (3.3) og blandes. Disse opløsninger har en koncentration på henholdsvis 2,0 , 4,0 , 6,0 , 8,0 og 10,0 μg/ml methylbenzoquat. Disse opløsninger skal fremstilles umiddelbart før brugen.
4. Apparatur
4.1. Mekanisk rysteapparat
4.2. Rotationsfordamper
4.3. Glaskolonne (250 mm × 15 mm), forsynet med stophane og med en kapacitet på ca. 200 ml
4.4. HPLC-udstyr med ultraviolet detektor med variabel bølgelængde eller med diodearraydetektor
4.4.1. HPLC-kolonne: 300 mm × 4 mm, C18, 10 μm pakkemateriale, eller tilsvarende
4.5. Membranfiltre, 0,22 μm
4.6. Membranfiltre, 0,45 μm.
5. Fremgangsmåde
5.1. Generelt
5.1.1. Der analyseres en blindprøve for at kontrollere, at hverken methylbenzoquat eller interfererende stoffer er til stede.
5.1.2. Der udføres en genfindingstest ved at analysere blindprøven, som er blevet spiket ved tilsætning af en mængde methylbenzoquat svarende til den, der er til stede i prøven. For at nå op på et niveau på 15 mg/kg tilsættes 600 μl af standardstamopløsningen (3.7.1) til 20 g blindprøve. Der blandes og ventes i 10 minutter, inden der fortsættes med ekstraktion (5.2).
|
NB |
: |
Til denne metode anvendes en blindprøve af samme type som analyseprøven, og som ved analyse viser sig ikke at indeholde methylbenzoquat. |
5.2. Ekstraktion
Ca. 20 g af den klargjorte prøve afvejes med en nøjagtighed på 0,01 g og overføres til en 250 ml konisk kolbe. Der tilsættes 100,0 ml methanolsulfonsyreopløsning (3.4) og omrystes mekanisk (4.1) i 30 minutter. Opløsningen filtreres gennem filtrerpapir, og filtratet gemmes til væske-væskefordelingen (5.3).
5.3. Væske-væskefordeling
25,0 ml af det under (5.2) opnåede filtrat overføres til en 500 ml skilletragt indeholdende 100 ml saltsyreopløsning (3.5). Der tilsættes 100 ml dichlormethan (3.1) til tragten og omrystes i 1 minut. Efter at de to lag har fået tid til at skilles, aftappes det nederste lag (dichlormethan) i en 500 ml rundbundet kolbe. Ekstraktionen af den vandige fase gentages med yderligere to portioner dichlormethan på hver 40 ml, og disse ekstrakter kombineres med det første ekstrakt i den rundbundede kolbe. Dichlormethanekstraktet inddampes til tørhed på rotationsfordamperen (4.2) ved 40 oC under reduceret tryk. Remanensen opløses i 20–25 ml methanol (3.2), hvorefter kolben tilproppes, og hele ekstraktet gemmes til ionbytningskromatografi (5.4).
5.4. Ionbytningskromatografi
5.4.1.
En prop af glasuld anbringes i den nedre ende af en glaskolonne (4.3). Der klargøres en opslæmning på 5,0 g af det behandlede kationbytterresin (3.6) med 50 ml saltsyre (3.5), som hældes i glaskolonnen, hvorefter man lader den få tid til at sætte sig. Den overskydende syre lades løbe ud, indtil den står lige over resinoverfladen, og kolonnen vaskes med vand, indtil vaskevandet er neutralt over for lakmuspapir. 50 ml methanol (3.2) overføres til kolonnen og lades sive ned til resinoverfladen.
5.4.2.
Det under (5.3) opnåede ekstrakt overføres forsigtigt til kolonnen med pipette. Den rundbundede kolbe skylles med to portioner på 5-10 ml methanol (3.2), og disse skyllevæsker overføres til kolonnen. Ekstraktet lades løbe ned til resinoverfladen, og kolonnen vaskes med 50 ml methanol, idet det sikres, at gennemstrømningshastigheden ikke overstiger 5 ml pr. minut. Den udstrømmende væske kasseres. Methylbenzoquatet elueres fra kolonnen under anvendelse af 150 ml methansulfonsyreopløsning (3.4), og eluatet fra kolonnen opsamles i en 250 ml konisk kolbe.
5.5. Væske-væskefordeling
Det under (5.4.2) opnåede eluat overføres til en 1 liter skilletragt. Den koniske kolbe skylles med 5–10 ml methanol (3.2), og skyllevæskerne kombineres med indholdet i skilletragten. Der tilsættes 300 ml saltsyreopløsning (3.5) og 130 ml dichlormethan (3.1) og omrystes i 1 minut. Efter at de to faser har fået tid til at skilles, aftappes det nederste (dichlormethan) lag i en 500 ml rundbundet kolbe. Ekstraktionen af den vandige fase gentages med yderligere to portioner dichlormethan på hver 70 ml, og disse ekstrakter kombineres med det første ekstrakt i den rundbundede kolbe.
Dichlormethanekstraktet inddampes til tørhed på rotationsfordamperen (4.2) ved 40 oC under reduceret tryk. Remanensen opløses i kolben med ca. 5 ml methanol (3.2), og denne opløsning overføres kvantitativt til en 10 ml målekolbe. Den rundbundede kolbe skylles med yderligere to portioner methanol på hver 1–2 ml, som overføres til målekolben. Der fyldes op til mærket med methanol og blandes. En alikvot mængde filtreres gennem et membranfilter (4.6). Denne opløsning gemmes til HPLC-bestemmelsen (5.6).
5.6. HPLC-bestemmelse
5.6.1.
Følgende betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater.
— HPLC-kolonne (4.4.1)
— Mobil fase til HPLC: blanding af methanol og vand (3.3)
— Flow: 1–1,5 ml/minuttet
— Detektionsbølgelængde: 265 nm
— Injektionsvolumen: 20–50 μl.
Kromatografisystemets stabilitet kontrolleres, ved at der flere gange indsprøjtes kalibreringsopløsning (3.7.3) med 4 μg/ml, indtil der opnås konstante tophøjder eller -arealer og retentionstider.
5.6.2.
Hver kalibreringsopløsning (3.7.3) indsprøjtes flere gange, og højden af toppene (arealerne) bestemmes for hver koncentration. Der tegnes en kalibreringskurve med kalibreringsopløsningernes middeltophøjder eller -arealer som ordinat og de tilsvarende koncentrationer i μg/ml som abscisse.
5.6.3.
Prøveekstraktet (5.5) indsprøjtes flere gange, idet der benyttes samme mængde som til kalibreringsopløsningerne, og middeltophøjden (-arealet) af methylbenzoquattoppene bestemmes.
6. Beregning af resultater
Ud fra middelhøjden (-arealet) af prøveopløsningens methylbenzoquattoppe bestemmes koncentrationen i prøveopløsningen i μg/ml ved benyttelse af kalibreringskurven (5.6.2).
Prøvens indhold af methylbenzoquat w (mg/kg) beregnes efter følgende formel:
![]()
hvor:
|
c |
= |
prøveopløsningens methylbenzoquatkoncentration i g/ml |
|
m |
= |
testportionens vægt i gram. |
7. Validering af resultater
7.1. Identitet
Analyttens identitet kan bekræftes ved ko-kromatografi eller ved at benytte en diodearraydetektor, hvormed spektrene for prøveekstraktet og kalibreringsopløsningen (3.7.3) indeholdende 10 μg/ml sammenlignes.
7.1.1.
Et prøveekstrakt spikes ved tilsætning af en passende mængde standardmellemopløsning (3.7.2). Den tilsatte mængde methylbenzoquat skal svare til den skønnede mængde methylbenzoquat i prøveekstraktet.
Kun højden af methylbenzoquattoppen må være blevet øget, efter at der er taget hensyn til både den tilsatte mængde og fortyndingen af ekstraktet. Toppens bredde skal i den halve højde ligge inden for ca. 10 % af den oprindelige bredde.
7.1.2.
Resultaterne vurderes i overensstemmelse med følgende kriterier:
a) Den maksimale absorptionsbølgelængde for prøvens og standardens spektre, målt ved toppens spids på kromatogrammet, skal være den samme inden for en margin, der afhænger af detektionssystemets opløsningsevne. Ved diodearraydetektion er den typisk ca. 2 nm.
b) Mellem 220 og 350 nm må prøvens og standardens spektre registreret ved toppens spids på kromatogrammet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af standardanalyttens absorbans.
c) Mellem 220 og 350 nm må spektrene ved toppens forside, spids og bagside frembragt med prøveekstraktet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem spektrene i intet observeret punkt overstiger 15 % af spektrets absorbans i spidsen.
Hvis et af disse kriterier ikke er opfyldt, er analyttens tilstedeværelse ikke bekræftet.
7.2. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige: 10 % i relativ værdi (af det højeste resultat) for methylbenzoquatindhold på 4–20 mg/kg.
7.3. Genfinding
For den spikede blindprøve skal genfindingen være mindst 90 %.
8. Resultater af ringanalyse
5 prøver blev analyseret af 10 laboratorier. Der blev foretaget dobbeltanalyse af hver prøve.
|
|
Blind |
Mel 1 |
Piller 1 |
Mel 2 |
Piller 2 |
|
Middelværdi [mg/kg] |
IP |
4,50 |
4,50 |
8,90 |
8,70 |
|
Sr [mg/kg] |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [ %] |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
SR [mg/kg] |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [ %] |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Genfinding [ %] |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
IP |
= |
Ikke påvist |
|
Sr |
= |
standardafvigelse for repeterbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed, % |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed, %. |
B. BESTEMMELSE AF OLAQUINDOX
2-[N-2'-(hydroxyethyl)carbamoyl]-3-methylquinoxalin-N 1 ,N 4 -dioxid
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af olaquindox i foder. Bestemmelsesgrænsen er 5 mg/kg.
2. Princip
Prøven ekstraheres med en blanding af vand og methanol. Indholdet af olaquindox bestemmes ved omvendt fase-HPLC med anvendelse af UV detektor.
3. Reagenser
3.1. Methanol.
3.2. Methanol, svarende til HPLC-kvalitet.
3.3. Vand, svarende til HPLC-kvalitet.
3.4. Mobil fase til HPLC
Blanding af vand (3.3) og methanol (3.2), 900 + 100 (V + Vv).
3.5. Standardstof: rent olaquindox 2-[N-2'-(hydroxyethyl)carbamoyl]-3-methylquinoxalin-N1,N4-dioxid, E 851.
3.5.1.
Med en nøjagtighed på 0,1 mg afvejes 50 mg olaquindox (3.5) i en 200 ml målekolbe, og der tilsættes ca. 190 ml vand. Derefter anbringes kolben i 20 minutter i et ultralydsbad (4.1). Efter ultralydsbehandlingen bringes opløsningen til rumtemperatur, hvorefter der fyldes op til mærket med vand og blandes. Kolben omvikles med aluminiumfolie og opbevares i køleskab. Fremstilles hver måned.
3.5.2.
10,0 ml af standardstamopløsningen (3.5.1) overføres til en 100 ml målekolbe, hvorefter der fyldes op til mærket med den mobile fase (3.4) og blandes. Kolben omvikles med aluminiumfolie og opbevares i køleskab. Fremstilles dagligt.
3.5.3.
Henholdsvis 1,0 , 2,0 , 5,0 , 10,0 , 15,0 og 20,0 ml af standardmellemopløsningen (3.5.2) overføres til hver sin 50 ml målekolbe. Der fyldes op til mærket med mobil fase (3.4) og blandes. Kolberne omvikles med aluminiumfolie. Disse opløsninger svarer til henholdsvis 0,5 , 1,0 , 2,5 , 5,0 , 7,5 og 10,0 μg olaquindox pr. ml.
Fremstilles dagligt.
4. Apparatur
4.1. Ultralydsbad
4.2. Mekanisk rysteapparat
4.3. HPLC-udstyr med ultraviolet detektor med variabel bølgelængde eller med diodearraydetektor
4.3.1. HPLC-kolonne, 250 mm × 4 mm, C18, 10 μm pakkemateriale, eller tilsvarende
4.4. Membranfiltre, 0,45 μm.
5. Fremgangsmåde
|
NB |
Olaquindox er lysfølsomt. Alle procedurer udføres i dæmpet belysning eller under anvendelse af udstyr af brunt glas. |
5.1. Generelt
5.1.1. Der analyseres en blindprøve for at kontrollere, at hverken olaquindox eller interfererende stoffer er til stede.
5.1.2. Der udføres en genfindingstest ved at analysere blindprøven, som er blevet spiket ved tilsætning af en mængde olaquindox svarende til den, der er til stede i prøven. For at nå op på et niveau på 50 mg/kg overføres 10,0 ml af standardstamopløsningen (3.5.1) til en 250 ml konisk kolbe, hvorefter opløsningen inddampes til ca. 0,5 ml. Der tilsættes 50 g af blindprøven og blandes omhyggeligt, og efter henstand i 10 minutter blandes der på ny flere gange, inden der fortsættes med ekstraktion (5.2).
|
NB |
Til denne metode anvendes en blindprøve af samme type som analyseprøven, og som ved analyse viser sig ikke at indeholde olaquindox. |
5.2. Ekstraktion
Med en nøjagtighed på 0,01 g afvejes der ca. 50 g af prøven, som overføres til en 1 000 ml konisk kolbe, hvorefter der tilsættes 100 ml methanol (3.1), og kolben anbringes i 5 minutter i ultralydsbad (4.1). Der tilsættes 410 ml vand, og kolben anbringes i ultralydsbad i yderligere 15 minutter. Derefter fjernes kolben fra ultralydsbadet og rystes i 30 minutter på rysteapparatet (4.2), og indholdet filtreres gennem et foldet filter. 10,0 ml af filtratet overføres til en 20 ml målekolbe, hvorefter der fyldes op til mærket med vand og blandes. En alikvot mængde filtreres gennem et membranfilter (4.4) (se bemærkning, punkt 9). Der fortsættes med HPLC-bestemmelse (5.3).
5.3. HPLC-bestemmelse
5.3.1.
Nedenstående betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater.
|
Analysekolonne (4.3.1) |
|
|
Mobil fase (3.4): |
Blanding af vand (3.3) og methanol (3.2), 900 + 100 (V + V) |
|
Flow: |
1,5 –2 ml/minuttet |
|
Detektionsbølgelængde: |
380 nm |
|
Injektionsvolumen: |
20–100 μl. |
Kromatografisystemets stabilitet kontrolleres, ved at der flere gange indsprøjtes kalibreringsopløsning (3.5.3) med 2,5 μg/ml, indtil der opnås konstante tophøjder og retentionstider.
5.3.2.
Hver kalibreringsopløsning (3.5.3) indsprøjtes flere gange, og middeltophøjderne (-arealerne) bestemmes for hver koncentration. Der tegnes en kalibreringskurve med kalibreringsopløsningernes middeltophøjder (-arealer) som ordinat og de tilsvarende koncentrationer i μg/ml som abscisse.
5.3.3.
Prøveekstraktet (5.2) indsprøjtes flere gange, idet der benyttes samme mængde som til kalibreringsopløsningerne, og middeltophøjden (-arealet) af olaquindoxtoppene bestemmes.
6. Beregning af resultater
Ud fra middelhøjden (-arealet) af olaquindoxtoppene i prøveopløsningen bestemmes koncentrationen i prøveopløsningen i μg/ml ved benyttelse af kalibreringskurven (5.3.2).
Olaquindoxindholdet w i mg/kg for prøven beregnes efter følgende formel:
![]()
hvor:
|
c |
= |
olaquindoxkoncentrationen i prøveekstraktet (5.2) i μg/ml |
|
m |
= |
testportionens vægt i gram (5.2). |
7. Validering af resultater
7.1. Identitet
Analyttens identitet kan bekræftes ved ko-kromatografi eller ved at benytte en diodearraydetektor, hvormed spektrene for prøveekstraktet (5.2) og kalibreringsopløsningen (3.5.3) indeholdende 5,0 μg/ml sammenlignes.
7.1.1.
Et prøveekstrakt (5.2) spikes ved tilsætning af en passende mængde kalibreringsopløsning (3.5.3). Den tilsatte mængde olaquindox skal svare til den mængde olaquindox, der er fundet i prøveekstraktet.
Kun højden af olaquindoxtoppen må være blevet øget, efter at der er taget hensyn til både den tilsatte mængde og fortyndingen af ekstraktet. Toppens bredde skal i den halve højde ligge inden for ± 10 % af den oprindelige bredde af olaquindoxtoppen for det ikke-spikede prøveekstrakt.
7.1.2.
Resultaterne vurderes i overensstemmelse med følgende kriterier:
a) Den maksimale absorptionsbølgelængde for prøvens og standardens spektre, målt ved toppens spids på kromatogrammet, skal være den samme inden for en margin, der afhænger af detektionssystemets opløsningsevne. For diodearraydetektion ligger denne typisk inden for ± 2 nm.
b) Mellem 220 og 400 nm må prøvens og standardens spektre registreret ved toppens spids på kromatogrammet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af standardanalyttens absorbans.
c) Mellem 220 og 400 nm må spektrene ved toppens forside, spids og bagside frembragt med prøveekstraktet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af absorbansen for spektret i toppens spids.
Hvis et af disse kriterier ikke er opfyldt, er analyttens tilstedeværelse ikke bekræftet.
7.2. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige 15 % i relativ værdi (af det højeste resultat) for olaquindoxindhold på 10–200 mg/kg.
7.3. Genfinding
For den spikede blindprøve skal genfindingen være mindst 90 %.
8. Resultater af ringanalyse
Der er gennemført en EF-ringanalyse, hvor 4 prøver af foder til smågrise, herunder en blindprøve, blev analyseret i op til 13 laboratorier. Resultaterne er vist herunder:
|
|
Prøve 1 |
Prøve 2 |
Prøve 3 |
Prøve 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
Middelværdi [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
Nominelt indhold |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Genfinding ( %) |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enkeltværdier |
|
Sr |
= |
standardafvigelse for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed. |
9. Bemærkning
Selv om metoden ikke er valideret for foder med et olaquindoxindhold på over 100 mg/kg, er det muligt at opnå tilfredsstillende resultater ved at reducere prøvemængden og/eller fortynde ekstraktet (5.2) for at opnå en koncentration inden for kalibreringskurven (5.3.2).
C. BESTEMMELSE AF AMPROLIUM
1-[(4-amino-2-propyl-5-pyrimidinyl)methyl]-2-picoliniumchloridhydrochlorid
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af amprolium i foder og forblandinger. Detektionsgrænsen er 1 mg/kg, og bestemmelsesgrænsen er 5 mg/kg.
2. Princip
Prøven ekstraheres med en methanol-vand-blanding. Efter fortynding med den mobile fase og membranfiltrering bestemmes indholdet af amprolium ved kationbytter-HPLC med anvendelse af UV-detektor.
3. Reagenser
3.1. Methanol.
3.2. Acetonitril, svarende til HPLC-kvalitet.
3.3. Vand, svarende til HPLC-kvalitet.
3.4. Natriumdihydrogenphosphatopløsning, c = 0,1 mol/liter
13,80 g natriumdihydrogenphosphatmonohydrat opløses i vand (3.3) i en 1 000 ml målekolbe; der fyldes op til mærket med vand (3.3) og blandes.
3.5. Natriumperchloratopløsning, c =1,6 mol/liter
224,74 g natriumperchloratmonohydrat opløses i vand (3.3) i en 1 000 ml målekolbe; der fyldes op til mærket med vand (3.3) og blandes.
3.6. Mobil fase til HPLC (se bemærkning 9.1)
Blanding af acetonitril (3.2), natriumdihydrogenphosphatopløsning (3.4) og natriumperchloratopløsning (3.5), 450 + 450 + 100 (v + v + v). Før anvendelse filtreres der gennem et 0,22 μm membranfilter (4.3), og opløsningen afgasses (f.eks. i ultralydsbad (4.4) i mindst 15 minutter).
3.7. Standardstof: rent amprolium, 1-[(4-amino-2-propylpyrimidin-5-yl)methyl]-2-methylpyridiniumchloridhydrochlorid, E 750 (se punkt 9.2).
3.7.1.
50 mg amprolium (3.7) afvejes med en nøjagtighed på 0,1 mg i en 100 ml målekolbe og opløses i 80 ml methanol (3.1), og kolben anbringes i 10 minutter i ultralydsbad (4.4). Efter ultralydsbehandlingen bringes opløsningen til rumtemperatur, hvorefter der fyldes op til mærket med vand og blandes. Opløsningen er stabil i en måned ved en temperatur på ≤ 4 oC.
3.7.2.
5,0 ml af standardstamopløsningen (3.7.1) afpipetteres i en 50 ml målekolbe, og der fyldes op til mærket med ekstraktionsopløsningsmidlet (3.8) og blandes. Opløsningen er stabil i en måned ved en temperatur på ≤ 4 oC.
3.7.3.
Henholdsvis 0,5 , 1,0 og 2,0 ml af standardmellemopløsningen (3.7.2) overføres til hver sin 50 ml målekolbe. Der fyldes op til mærket med mobil fase (3.6) og blandes. Disse opløsninger svarer til henholdsvis 0,5 , 1,0 og 2,0 μg amprolium pr. ml. Disse opløsninger skal fremstilles umiddelbart før brugen.
3.8. Ekstraktionsopløsningsmiddel
Blanding af methanol (3.1) og vand, 2 + 1 (v + v).
4. Apparatur
4.1. HPLC-udstyr med injektionssystem, egnet til injektionsvolumener på 100 μl
4.1.1. HPLC-kolonne, 125 mm x 4 mm, kationbytter Nucleosil 10 SA, 5 eller 10 μm pakkemateriale, eller tilsvarende
4.1.2. UV-detektor med variabel bølgelængde eller diodearraydetektor
4.2. Membranfilter, PTFE-, 0,45 μm
4.3. Membranfilter, 0,22 μm
4.4. Ultralydsbad
4.5. Mekanisk rysteapparat eller magnetomrører.
5. Fremgangsmåde
5.1. Generelt
5.1.1.
Til gennemførelse af genfindingstesten (5.l.2) analyseres en blindprøve for at kontrollere, at hverken amprolium eller interfererende stoffer er til stede. Blindprøven skal være af samme type som den prøve, der skal undersøges, og ved analyse må amprolium eller interfererende stoffer ikke påvises.
5.1.2.
Der udføres en genfindingstest ved at analysere blindprøven, som er blevet spiket ved tilsætning af en mængde amprolium svarende til den, der er til stede i prøven. For at nå op på et niveau på 100 mg/kg overføres der 10,0 ml standardstamopløsning (3.7.1) til en 250 ml konisk kolbe, og opløsningen inddampes til ca. 0,5 ml. Der tilsættes 50 g af blindprøven og blandes omhyggeligt, og efter henstand i 10 minutter blandes der på ny flere gange, inden der fortsættes med ekstraktion (5.2).
Hvis der ikke er en blindprøve af samme type som analyseprøven til rådighed (se punkt 5.1.1), kan der alternativt udføres en genfindingstest ved hjælp af standardtilsætningsmetoden. I dette tilfælde spikes den prøve, der skal analyseres, ved tilsætning af en mængde amprolium svarende til den, der allerede er til stede i prøven. Denne prøve analyseres sammen med den ikke-spikede prøve, og genfindingen kan beregnes ved subtraktion.
5.2. Ekstraktion
5.2.1.
Med en nøjagtighed på 0,01 g afvejes der 5–40 g af prøven, afhængigt af amproliumindholdet, i en 500 ml konisk kolbe, og der tilsættes 200 ml ekstraktionsopløsningsmiddel (3.8). Kolben anbringes i ultralydsbad (4.4) og henstår i 15 minutter. Kolben fjernes fra ultralydsbadet og rystes i 1 time på rysteapparatet eller omrøres på magnetomrøreren (4.5). En alikvot mængde af ekstraktet fortyndes med den mobile fase (3.6) til et amproliumindhold på 0,5 –2 μg/ml og blandes (se bemærkning 9.3). 5–10 ml af denne fortyndede opløsning filtreres på et membranfilter (4.2). Der fortsættes med HPLC-bestemmelse (5.3).
5.2.2.
Med en nøjagtighed på 0,001 g afvejes 1–4 g af forblandingen, afhængigt af amproliumindholdet, i en 500 ml konisk kolbe, og der tilsættes 200 ml ekstraktionsopløsningsmiddel (3.8). Kolben anbringes i ultralydsbad (4.4) og henstår i 15 minutter. Kolben fjernes fra ultralydsbadet og rystes i 1 time på rysteapparatet eller omrøres på magnetomrøreren (4.5). En alikvot mængde af ekstraktet fortyndes med den mobile fase (3.6) til et amproliumindhold på 0,5 –2 μg/ml og blandes. 5–10 ml af denne fortyndede opløsning filtreres på et membranfilter (4.2). Der fortsættes med HPLC-bestemmelse (5.3).
5.3. HPLC-bestemmelse
5.3.1.
Nedenstående betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater.
|
|
|
|
HPLC-kolonne (4.1.1): |
125 mm × 4 mm, kationbytter Nucleosil 10 SA, 5 eller 10 μm pakkemateriale, eller tilsvarende |
|
Mobil fase (3.6): |
Blanding af acetonitril (3.2), natriumdihydrogenphosphatopløsning (3.4) og natriumperchloratopløsning (3.5), 450 + 450 + 100 (v + v + v). |
|
Flow : |
0,7 –1 ml/minuttet |
|
Detektionsbølgelængde: |
264 nm |
|
Injektionsvolumen: |
100 μl. |
Kromatografisystemets stabilitet kontrolleres, ved at der flere gange indsprøjtes kalibreringsopløsning (3.7.3) med 1,0 μg/ml, indtil der opnås konstante tophøjder og retentionstider.
5.3.2.
Hver kalibreringsopløsning (3.7.3) indsprøjtes flere gange, og middeltophøjderne (-arealerne) bestemmes for hver koncentration. Der tegnes en kalibreringskurve med kalibreringsopløsningernes middeltophøjder (-arealer) som ordinat og de tilsvarende koncentrationer i μg/ml som abscisse.
5.3.3.
Prøveekstraktet (5.2) indsprøjtes flere gange, idet der benyttes samme mængde som til kalibreringsopløsningerne, og middeltophøjden (-arealet) af amproliumtoppene bestemmes.
6. Beregning af resultater
Ud fra middelhøjden (-arealet) af prøveopløsningens amproliumtoppe bestemmes koncentrationen i prøveopløsningen i μg/ml ved benyttelse af kalibreringskurven (5.3.2).
Amproliumindholdet w i mg/kg for prøven er givet ved følgende formel:
![]()
[mg/kg]
hvor:
|
V |
= |
volumen ekstraktionsopløsningsmiddel (3.8) i ml ifølge 5.2 (dvs. 200 ml) |
|
c |
= |
amproliumkoncentrationen i prøveekstraktet (5.2) i μg/ml |
|
f |
= |
fortyndingsfaktor ifølge 5.2 |
|
m |
= |
testportionens vægt i gram. |
7. Validering af resultater
7.1. Identitet
Analyttens identitet kan bekræftes ved ko-kromatografi eller ved at benytte en diodearraydetektor, hvormed spektrene for prøveekstraktet (5.2) og kalibreringsopløsningen (3.7.3) indeholdende 2,0 μg/ml sammenlignes.
7.1.1.
Et prøveekstrakt (5.2) spikes ved tilsætning af en passende mængde kalibreringsopløsning (3.7.3). Den tilsatte mængde amprolium skal svare til den mængde amprolium, der er fundet i prøveekstraktet.
Kun højden af amproliumtoppen må være blevet øget, efter at der er taget hensyn til både den tilsatte mængde og fortyndingen af ekstraktet. Toppens bredde skal i den halve højde ligge inden for ± 10 % af den oprindelige bredde af amproliumtoppen for det ikke-spikede prøveekstrakt.
7.1.2.
Resultaterne vurderes i overensstemmelse med følgende kriterier:
a) Den maksimale absorptionsbølgelængde for prøvens og standardens spektre, målt ved toppens spids på kromatogrammet, skal være den samme inden for en margin, der afhænger af detektionssystemets opløsningsevne. For diodearraydetektion ligger denne typisk inden for ± 2 nm.
b) Mellem 210 og 320 nm må prøvens og standardens spektre registreret ved toppens spids på kromatogrammet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af standardanalyttens absorbans.
c) Mellem 210 og 320 nm må spektrene ved toppens forside, spids og bagside frembragt med prøveekstraktet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af absorbansen for spektret i toppens spids.
Hvis et af disse kriterier ikke er opfyldt, er analyttens tilstedeværelse ikke bekræftet.
7.2. Repeterbarhed
Forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve må ikke overstige:
— 15 % i relativ værdi (af det højeste resultat) for amproliumindhold på 25–500 mg/kg
— 75 mg/kg for amproliumindhold på 500–1 000 mg/kg
— 7,5 % i relativ værdi (af det højeste resultat) for amproliumindhold på mere end 1 000 mg/kg.
7.3. Genfinding
For en spiket (blind)prøve skal genfindingen være mindst 90 %.
8. Resultater af ringanalyse
Der er gennemført en ringanalyse, hvor 3 kyllingefoderstoffer (prøve 1–3), 1 mineralfoder (prøve 4) og 1 forblanding (prøve 5) blev analyseret. Resultaterne er vist i nedenstående tabel:
|
|
Prøve 1 (blindprøve) |
Prøve 2 |
Prøve 3 |
Prøve 4 |
Prøve 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
Middelværdi [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
Sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
SR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Nominelt indhold [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enkeltværdier |
|
Sr |
= |
standardafvigelse for repeterbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed. |
9. Bemærkninger
9.1. Hvis prøven indeholder thiamin, forekommer thiamintoppen i kromatogrammet kort før amproliumtoppen. Ved at følge denne metode skal amprolium og thiamin adskilles. Hvis amprolium og thiamin ikke adskilles med den kolonne (4.1.1), der anvendes i denne metode, erstattes op til 50 % af acetonitrildelen af den mobile fase (3.6) med methanol.
9.2. Ifølge British Pharmacopoeia viser spektret for en amproliumopløsning (c = 0,02 mol/liter) i saltsyre (c = 0,1 mol/liter) maksima ved 246 nm og 262 nm. Absorbansen skal være 0,84 ved 246 nm og 0,80 ved 262 nm.
9.3. Ekstraktet skal altid fortyndes med den mobile fase, da retentionstiden for amproliumtoppen ellers kan forskydes væsentligt på grund af ændringer i ionstyrken.
D. BESTEMMELSE AF CARBADOX
Methyl-3-(2-quinoxalinylmethylen)carbazat-N 1 ,N 4 -dioxid
1. Formål og anvendelsesområde
Denne metode gør det muligt at bestemme indholdet af carbadox i foder, forblandinger og tilberedninger. Detektionsgrænsen er 1 mg/kg, og bestemmelsesgrænsen er 5 mg/kg.
2. Princip
Prøven bringes i ligevægt med vand og ekstraheres med methanol-acetonitril. For foders vedkommende oprenses en alikvot mængde af det filtrerede ekstrakt på en aluminiumoxidkolonne. For forblandingers og tilberedningers vedkommende fortyndes en alikvot mængde af det filtrerede ekstrakt til en passende koncentration med vand, methanol og acetonitril. Indholdet af carbadox bestemmes ved omvendt fase-HPLC med anvendelse af UV-detektor.
3. Reagenser
3.1. Methanol.
3.2. Acetonitril, svarende til HPLC-kvalitet.
3.3. Eddikesyre, w = 100 %.
3.4. Aluminiumoxid: neutral, aktivitetskvalitet I.
3.5. Methanol-acetonitril, 1 + 1 (v + v)
500 ml methanol (3.1) blandes med 500 ml acetonitril (3.2).
3.6. Eddikesyre, σ = 10 %
10 ml eddikesyre (3.3) fortyndes til 100 ml med vand.
3.7. Natriumacetat.
3.8. Vand, svarende til HPLC-kvalitet.
3.9. Acetatbufferopløsning, c = 0,01 mol/liter, pH = 6,0
0,82 g natriumacetat (3.7) opløses i 700 ml vand (3.8), og pH justeres til 6,0 med eddikesyre (3.6). Opløsningen overføres til en 1 000 ml målekolbe, og der fyldes op til mærket med vand (3.8) og blandes.
3.10. Mobil fase til HPLC
825 ml acetatbufferopløsning (3.9) blandes med 175 ml acetonitril (3.2).
Der filtreres gennem et 0,22 μm filter (4.5), og opløsningen afgasses (f.eks. ved ultralydsbehandling i 10 minutter).
3.11. Standardstof
Rent carbadox: Methyl-3-(2-quinoxalinylmethylen)carbazat-N1,N4-dioxid, E 850
3.11.1.
25 mg carbadoxstandardstof (3.11) afvejes med en nøjagtighed på 0,1 mg i en 250 ml målekolbe og opløses i methanol-acetonitril (3.5) ved ultralydsbehandling (4.7). Efter ultralydsbehandlingen bringes opløsningen til rumtemperatur, hvorefter der fyldes op til mærket med methanol-acetonitril (3.5) og blandes. Kolben omvikles med aluminiumfolie, eller der anvendes brunt glasudstyr, og opløsningen opbevares i køleskab. Opløsningen er stabil i en måned ved en temperatur på ≤ 4 oC.
3.11.2.
Henholdsvis 2,0 , 5,0 , 10,0 og 20,0 ml af standardstamopløsningen (3.11.1) overføres til hver sin 100 ml målekolbe. Der tilsættes 30 ml vand, fyldes op til mærket med methanol-acetonitril (3.5) og blandes. Kolberne omvikles med aluminiumfolie. Disse opløsninger svarer til henholdsvis 2,0 , 5,0 , 10,0 og 20,0 μg/ml carbadox.
Kalibreringsopløsninger skal fremstilles umiddelbart før brugen.
|
NB |
Til bestemmelse af carbadox i foder, som indeholder mindre end 10 mg/kg, skal der fremstilles kalibreringsopløsninger med en koncentration på under 2,0 μg/ml. |
3.12. Blanding af vand og methanol-acetonitril (3.5), 300 + 700 (v + v)
300 ml vand blandes med 700 ml af methanol-acetonitril-blandingen (3.5).
4. Apparatur
4.1. Mekanisk rysteapparat eller magnetomrører.
4.2. Glasfiberfiltrerpapir (Whatman GF/A eller tilsvarende).
4.3. Glaskolonne (længde 300–400 mm, indvendig diameter ca. 10 mm) med sintret glasfritte og aftapningsventil.
|
NB |
Der kan også anvendes en glaskolonne med stophane eller en glaskolonne med en tilspidset ende; i dette tilfælde anbringes en lille prop af glasuld i den nedre ende og skubbes helt ned med en glasstav. |
4.4. HPLC-udstyr med injektionssystem, egnet til injektionsvolumener på 20 μl.
4.4.1. HPLC-kolonne, 300 mm × 4 mm, C18, 10 μm pakkemateriale, eller tilsvarende.
4.4.2. UV-detektor med variabel bølgelængde eller diodearraydetektor, som arbejder i området fra 225 til 400 nm.
4.5. Membranfilter, 0,22 μm.
4.6. Membranfilter, 0,45 μm.
4.7. Ultralydsbad.
5. Fremgangsmåde
|
NB |
Carbadox er lysfølsomt. Alle procedurer udføres i dæmpet belysning eller under anvendelse af brunt glasudstyr eller glasudstyr omviklet med aluminiumfolie. |
5.1. Generelt
5.1.1.
Til gennemførelse af genfindingstesten (5.1.2) analyseres en blindprøve for at kontrollere, at hverken carbadox eller interfererende stoffer er til stede. Blindprøven skal være af samme type som den prøve, der skal undersøges, og ved analyse må carbadox eller interfererende stoffer ikke påvises.
5.1.2.
Der udføres en genfindingstest ved at analysere blindprøven (5.1.1), som er blevet spiket ved tilsætning af en mængde carbadox svarende til den, der er til stede i prøven. For at nå op på et niveau på 50 mg/kg overføres 5,0 ml af standardstamopløsningen (3.11.1) til en 200 ml konisk kolbe. Opløsningen inddampes til ca. 0,5 ml i en nitrogenstrøm. Der tilsættes 10 g af blindprøven, blandes og ventes i 10 minutter, inden der fortsættes med ekstraktion (5.2).
Hvis der ikke er en blindprøve af samme type som analyseprøven til rådighed (se punkt 5.1.1), kan der alternativt udføres en genfindingstest ved hjælp af standardtilsætningsmetoden. I dette tilfælde spikes prøven ved tilsætning af en mængde carbadox svarende til den, der allerede er til stede i prøven. Denne prøve analyseres sammen med den ikke-spikede prøve, og genfindingen kan beregnes ved subtraktion.
5.2. Ekstraktion
5.2.1.
Med en nøjagtighed på 0,01 g afvejes 10 g af prøven, som overføres til en 200 ml konisk kolbe. Der tilsættes 15,0 ml vand og blandes, og blandingen henstår i 5 minutter for at komme i ligevægt. Der tilsættes 35,0 ml methanol-acetonitril (3.5), tilproppes og omrystes i 30 minutter på rysteapparatet eller omrøres på magnetomrøreren (4.1). Opløsningen filtreres gennem et glasfiberfiltrerpapir (4.2). Denne opløsning anvendes til oprensningsprocessen (5.3).
5.2.2.
Med en nøjagtighed på 0,001 g afvejes 1 g af den ikke-formalede prøve, som overføres til en 200 ml konisk kolbe. Der tilsættes 15,0 ml vand og blandes, og blandingen henstår i 5 minutter for at komme i ligevægt. Der tilsættes 35,0 ml methanol-acetonitril (3.5), tilproppes og omrystes i 30 minutter på rysteapparatet eller omrøres på magnetomrøreren (4.1). Opløsningen filtreres gennem et glasfiberfiltrerpapir (4.2).
En alikvot mængde af filtratet afpipetteres i en 50 ml målekolbe. Der tilsættes 15,0 ml vand, fyldes op til mærket med methanol-acetonitril (3.5) og blandes. Carbadoxkoncentrationen i den endelige opløsning skal være cirka 10 μg/ml. En alikvot mængde filtreres gennem et 0,45 μm filter (4.6).
Der fortsættes med HPLC-bestemmelse (5.4).
5.2.3.
Med en nøjagtighed på 0,001 g afvejes 0,2 g af den ikke-formalede prøve, som overføres til en 250 ml konisk kolbe. Der tilsættes45,0 ml vand og blandes, og blandingen henstår i 5 minutter for at komme i ligevægt. Der tilsættes 105,0 ml methanol-acetonitril (3.5), tilproppes og homogeniseres. Prøven lydbehandles (4.7) i 15 minutter efterfulgt af omrystning eller omrøring i 15 minutter (4.1). Opløsningen filtreres gennem et glasfiberfiltrerpapir (4.2).
En alikvot mængde af filtratet fortyndes med blandingen af vand og methanol-acetonitril (3.12) til en endelig carbadoxkoncentration på 10-15 μg/ml (for en 10 % tilberedning er fortyndingsfaktoren 10). En alikvot mængde filtreres gennem et 0,45 μm filter (4.6).
Der fortsættes med HPLC-bestemmelse (5.4).
5.3. Oprensning
5.3.1.
Der afvejes 4 g aluminiumoxid (3.4), som overføres til glaskolonnen (4.3).
5.3.2.
Der sættes 15 ml af den filtrerede ekstrakt (5.2.1) på aluminiumoxidsøjlen, og de første 2 ml eluat kasseres. De næste 5 ml opsamles, og en alikvot mængde filtreres gennem et 0,45 μm filter (4.6).
Der fortsættes med HPLC-bestemmelse (5.4).
5.4. HPLC-bestemmelse
5.4.1.
Følgende betingelser er vejledende; andre betingelser kan benyttes, forudsat at de giver tilsvarende resultater:
|
|
|
|
HPLC-kolonne (4.1.1): |
300 mm × 4 mm, C18, 10 μmpakkemateriale, eller tilsvarende |
|
Mobil fase (3.10): |
Blanding af acetatbufferopløsning (3.9) og acetonitril (3.2), 825 + 175 (v + v) |
|
Flow: |
1,5 –2 ml/minuttet |
|
Detektionsbølgelængde: |
365 nm |
|
Injektionsvolumen: |
20 μl. |
Kromatografisystemets stabilitet kontrolleres, ved at der flere gange indsprøjtes kalibreringsopløsning (3.11.2) med 5,0 μg/ml, indtil der opnås konstante tophøjder (-arealer) og retentionstider.
5.4.2.
Hver kalibreringsopløsning (3.11.2) indsprøjtes flere gange, og tophøjderne (-arealerne) bestemmes for hver koncentration. Der tegnes en kalibreringskurve med kalibreringsopløsningernes middeltophøjder eller -arealer som ordinat og de tilsvarende koncentrationer i μg/ml som abscisse.
5.4.3.
Prøveekstraktet ((5.3.2) for foder, (5.2.2) for forblandinger og (5.2.3) for tilberedninger) indsprøjtes flere gange, og middeltophøjden (-arealet) for carbadoxtoppene bestemmes.
6. Beregning af resultater
Ud fra middelhøjden (-arealet) af prøveopløsningens carbadoxtoppe bestemmes carbadoxkoncentrationen i prøveopløsningen i μg/ml ved benyttelse af kalibreringskurven (5.4.2).
6.1. Foder
Indholdet af carbadox w (mg/kg) i prøven beregnes efter følgende formel:
![]()
[mg/kg]
hvor:
|
c |
= |
carbadoxkoncentrationen i prøveekstraktet (5.3.2) i μg/ml |
|
V1 |
= |
ekstraktionsvolumen i ml (dvs. 50) |
|
m |
= |
testportionens vægt i gram. |
6.2. Forblandinger og tilberedninger
Indholdet af carbadox w (mg/kg) i prøven beregnes efter følgende formel:
![]()
[mg/kg]
hvor:
|
c |
= |
carbadoxkoncentrationen i prøveekstraktet (5.2.2 eller 5.2.3) i μg/ml |
|
V2 |
= |
ekstraktionsvolumen i ml (dvs. 50 for forblandinger; 150 for tilberedninger) |
|
f |
= |
fortyndingsfaktor ifølge 5.2.2 (forblandinger) eller 5.2.3 (tilberedninger) |
|
m |
= |
testportionens vægt i gram. |
7. Validering af resultater
7.1. Identitet
Analyttens identitet kan bekræftes ved ko-kromatografi eller ved at benytte en diodearraydetektor, hvormed spektrene for prøveekstraktet og kalibreringsopløsningen (3.11.2) indeholdende 10,0 μg/ml sammenlignes.
7.1.1.
Et prøveekstrakt spikes ved tilsætning af en passende mængde kalibreringsopløsning (3.11.2). Den tilsatte mængde carbadox skal svare til den skønnede mængde carbadox, der er fundet i prøveekstraktet.
Kun højden af carbadoxtoppen må være blevet øget, efter at der er taget hensyn til både den tilsatte mængde og fortyndingen af ekstraktet. Toppens bredde skal i den halve højde ligge inden for ca. 10 % af den oprindelige bredde.
7.1.2.
Resultaterne vurderes i overensstemmelse med følgende kriterier:
a) Den maksimale absorptionsbølgelængde for prøvens og standardens spektre, målt ved toppens spids på kromatogrammet, skal være den samme inden for en margin, der afhænger af detektionssystemets opløsningsevne. For diodearraydetektion ligger denne typisk inden for ± 2 nm.
b) Mellem 225 og 400 nm må prøvens og standardens spektre registreret ved toppens spids på kromatogrammet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10–100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem de to spektre i intet observeret punkt overstiger 15 % af standardanalyttens absorbans.
c) Mellem 225 og 400 nm må spektrene ved toppens forside, spids og bagside frembragt med prøveekstraktet ikke være forskellige fra hinanden i de dele af spektret, der ligger inden for 10-100 % relativ absorbans. Dette kriterium er opfyldt, når de samme maksima er til stede, og afvigelsen mellem spektrene i intet observeret punkt overstiger 15 % af spektrets absorbans i spidsen.
Hvis et af disse kriterier ikke er opfyldt, er analyttens tilstedeværelse ikke bekræftet.
7.2. Repeterbarhed
For indhold på 10 mg/kg og derover må forskellen mellem resultaterne af to parallelle bestemmelser udført på samme prøve ikke overstige 15 % i relativ værdi (af det højeste resultat).
7.3. Genfinding
For en spiket (blind)prøve skal genfindingen være mindst 90 %.
8. Resultater af ringanalyse
Der er gennemført en ringanalyse, hvor 6 foderstoffer, 4 forblandinger og 3 tilberedninger blev analyseret af 8 laboratorier. Der blev foretaget dobbeltanalyse af hver prøve (Journal of the AOAC, bind 71, 1988, s. 484–490, indeholder nærmere oplysninger om denne ringanalyse). Resultaterne (ekskl. outliers) er vist nedenfor:
Skema 1
Resultater fra ringanalysen af foder
|
|
Prøve 1 |
Prøve 2 |
Prøve 3 |
Prøve 4 |
Prøve 5 |
Prøve 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Middelværdi (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr ( %) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR ( %) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Nominelt indhold (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Skema 2
Resultater fra ringanalysen af forblandinger og tilberedninger
|
|
Forblandinger |
Tilberedninger |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Middelværdi (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr ( %) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR ( %) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Nominelt indhold (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enkeltværdier |
|
Sr |
= |
standardafvigelse for repeterbarhed |
|
CVr |
= |
variationskoefficient for repeterbarhed |
|
SR |
= |
standardafvigelse for reproducerbarhed |
|
CVR |
= |
variationskoefficient for reproducerbarhed. |
BILAG IX
SAMMENLIGNINGSTABELLER, JF. ARTIKEL 6
1. Direktiv 71/250/EØF
|
Direktiv 71/250/EØF |
Denne forordning |
|
Artikel 1, stk. 1 |
Artikel 3 |
|
Artikel 1, stk. 2 |
Artikel 2 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget, del 1 |
Bilag II |
|
Bilaget, del 2 |
— |
|
Bilaget, del 3 |
— |
|
Bilaget, del 4 |
Bilag III, del O |
|
Bilaget, del 5 |
Bilag III, del M |
|
Bilaget, del 6 |
Bilag III, del N |
|
Bilaget, del 7 |
Bilag III, del Q |
|
Bilaget, del 9 |
Bilag III, del K |
|
Bilaget, del 10 |
— |
|
Bilaget, del 11 |
— |
|
Bilaget, del 12 |
Bilag III, del J |
|
Bilaget, del 14 |
Bilag III, del D |
|
Bilaget, del 16 |
— |
2. Direktiv 71/393/EØF
|
Direktiv 71/393/EØF |
Denne forordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget, del I |
Bilag III, del A |
|
Bilaget, del II |
Bilag III, del E |
|
Bilaget, del III |
Bilag III, del P |
|
Bilaget, del IV |
Bilag III, del H |
3. Direktiv 72/199/EØF
|
Direktiv 72/199/EØF |
Denne forordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Bilag I, del 1 |
Bilag III, del L |
|
Bilag I, del 2 |
Bilag III, del C |
|
Bilag I, del 3 |
— |
|
Bilag I, del 4 |
— |
|
Bilag I, del 5 |
Bilag V, del A |
|
Bilag II |
— |
4. Direktiv 73/46/EØF
|
Direktiv 73/46/EØF |
Denne forordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Bilag I, del 1 |
Bilag III, del B |
|
Bilag I, del 2 |
— |
|
Bilag I, del 3 |
Bilag III, del I |
5. Direktiv 76/371/EØF
|
Direktiv 76/371/EØF |
Denne forordning |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget |
Bilag I |
6. Direktiv 76/372/EØF
|
Direktiv 76/372/EØF |
Denne forordning |
|
Artikel 1 |
— |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget |
— |
7. Direktiv 78/633/EØF
|
Direktiv 78/633/EØF |
Denne forordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget, del 1 |
— |
|
Bilaget, del 2 |
— |
|
Bilaget, del 3 |
Bilag IV, del C |
8. Direktiv 81/715/EØF
|
Direktiv 81/715/EØF |
Denne forordning |
|
Artikel 1 |
— |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget |
— |
9. Direktiv 84/425/EØF
|
Direktiv 84/425/EØF |
Denne forordning |
|
Artikel 1 |
— |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget |
— |
10. Direktiv 86/174/EØF
|
Direktiv 86/174/EØF |
Denne forordning |
|
Artikel 1 |
Artikel 4 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget |
Bilag VII |
11. Direktiv 93/70/EØF
|
Direktiv 93/70/EØF |
Denne forordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget |
Bilag IV, del D |
12. Direktiv 93/117/EF
|
Direktiv 93/117/EF |
Denne forordning |
|
Artikel 1 |
Artikel 3 og 5 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Bilaget, del 1 |
Bilag IV, del E |
|
Bilaget, del 2 |
Bilag VIII, del A |
13. Direktiv 98/64/EF
|
Direktiv 98/64/EF |
Denne forordning |
|
Artikel 1 |
Artikel 3 og 5 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Bilaget, del A |
Bilag III, del F |
|
Bilaget, del C |
Bilag VIII, del B |
14. Direktiv 1999/27/EF
|
Direktiv 1999/27/EF |
Denne forordning |
|
Artikel 1 |
Artikel 3 og 5 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Artikel 5 |
— |
|
Artikel 6 |
— |
|
Artikel 7 |
— |
|
Bilaget, del A |
Bilag VIII, del C |
|
Bilaget, del B |
Bilag IV, del F |
|
Bilaget, del C |
Bilag VIII, del D |
15. Direktiv 1999/76/EF
|
Direktiv 1999/76/EF |
Denne forordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Bilaget |
Bilag IV, del G |
16. Direktiv 2000/45/EF
|
Direktiv 2000/45/EF |
Denne forordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Bilaget, del A |
Bilag IV, del A |
|
Bilaget, del B |
Bilag IV, del B |
|
Bilaget, del C |
Bilag III, del G |
17. Direktiv 2002/70/EF
|
Direktiv 2002/70/EF |
Denne forordning |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
Artikel 2 og 3 |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Artikel 5 |
— |
|
Bilag I |
Bilag I og bilag V, del B, afsnit I |
|
Bilag II |
Bilag II og bilag V, del B, afsnit II |
18. Direktiv 2003/126/EF
|
Direktiv 2003/126/EF |
Denne forordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
— |
|
Artikel 3 |
— |
|
Artikel 4 |
— |
|
Artikel 5 |
— |
|
Artikel 6 |
— |
|
Bilaget |
Bilag VI |
( 1 ) EUT L 165 af 30.4.2004, s. 1. Berigtiget i EUT L 191 af 28.5.2004, s. 1.
( 2 ) EFT L 155 af 12.7.1971, s. 13.
( 3 ) EFT L 279 af 20.12.1971, s. 7.
( 4 ) EFT L 123 af 29.5.1972, s. 6.
( 5 ) EFT L 83 af 30.3.1973, s. 21.
( 6 ) EFT L 102 af 15.4.1976, s. 1.
( 7 ) EFT L 102 af 15.4.1976, s. 8.
( 8 ) EFT L 206 af 29.7.1978, s. 43.
( 9 ) EFT L 257 af 10.9.1981, s. 38.
( 10 ) EFT L 238 af 6.9.1984, s. 34.
( 11 ) EFT L 130 af 16.5.1986, s. 53.
( 12 ) EFT L 234 af 17.9.1993, s. 17.
( 13 ) EFT L 329 af 30.12.1993, s. 54.
( 14 ) EFT L 257 af 19.9.1998, s. 14.
( 15 ) EFT L 118 af 6.5.1999, s. 36.
( 16 ) EFT L 207 af 6.8.1999, s. 13.
( 17 ) EFT L 174 af 13.7.2000, s. 32.
( 18 ) EFT L 209 af 6.8.2002, s. 15.
( 19 ) EUT L 339 af 24.12.2003, s. 78.
( 20 ) EUT L 268 af 18.10.2003, s. 29.
( 21 ) EFT L 140 af 30.5.2002, s. 10.
( 22 ) EUT L 70 af 16.3.2005, s. 1.
( 23 ) Eventuelle klumper findeles (om nødvendigt ved at udskille dem og derpå føre dem tilbage til samleprøven).
( 24 ) Undtagen i tilfælde af grovfoder eller foderplanter med lav massefylde.
( 25 ) Undtagen i tilfælde af grovfoder eller foderplanter med lav massefylde.
( 26 ) EUT L 229 af 1.9.2009, s. 1.
( 27 ) Til tørring af korn, mel, gryn og grutning skal tørreskabet have en sådan varmekapacitet, at det, når det er indstillet på en temperatur på 131 oC, igen når op på denne temperatur på mindre end 45 minutter, efter at det maksimale antal prøver, som skal tørres samtidig, er sat ind i skabet. Ventilationen skal være af en sådan beskaffenhed, at hvis samtlige prøver af blød hvede, som skabet kan rumme, tørres samtidig i to timer, må resultaterne sammenlignet med de efter 4 timers tørring opnåede resultater højst afvige med 0,15 %.
( 28 ) Såfremt fedtet senere skal undersøges kvalitativt, erstattes pimpstensstykkerne af glaskugler.
( 29 ) EFT L 125 af 23.5.1996, s. 35.
( 30 ) Udført af arbejdsgruppen for foder under Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
( 31 ) Udført af arbejdsgruppen for foder under Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
( 32 ) Der kan også anvendes andre destruktionsmetoder, forudsat at de bevisligt giver tilsvarende resultater (som f.eks. mikrobølgedestruktion).
( 33 ) Grøntfoder (frisk eller tørret) indeholder ofte store mængder vegetabilsk silicium, som kan binde mikromineraler og derfor må fjernes. Til prøver af denne type foder skal derfor følgende ændrede fremgangsmåde anvendes. Det under punkt 5.1.1.1 beskrevne udføres indtil filtreringen. Filtrerpapiret indeholdende den uopløselige remanens vaskes 2 gange med kogende vand og anbringes i en kvarts- eller platindigel. Der foraskes i muffelovn (4.1) ved en temperatur på under 550 °C, indtil alt kulstofholdigt materiale er forsvundet fuldstændig. Efter afkøling tilsættes nogle få dråber vand og derefter 10-15 ml flussyre (3.4), og der inddampes til tørhed ved ca. 150 °C. Hvis der stadig er silicium i inddampningsresten, genopløses denne i nogle få ml flussyre (3.4) og inddampes til tørhed. Der tilsættes 5 dråber svovlsyre (3.5) og opvarmes, indtil der ikke mere afgives hvide dampe. Efter tilsætning af 5 ml 6 mol/liter saltsyre (3.2) og ca. 30 ml vand opvarmes opløsningen; derefter filtreres denne ned i en 250 ml målekolbe, og der fyldes op med vand til mærket (HCl-koncentration ca. 0,5 mol/l). Bestemmelsen fortsættes derefter fra punkt 5.1.2.
( 34 ) The Analyst 108, 1983, s. 1252-1256.
( 35 ) Analyst, 1995, 120, s. 2175–2180.
( 36 ) Skema over TEF (= toksicitetsækvivalensfaktorer) for dioxiner, furaner og dioxinlignende PCB'er:
WHO-TEF til vurdering af risikoen for mennesker baseret på konklusionerne fra Verdenssundhedsorganisationens (WHO) ekspertmøde i Genève i juni 2005 om det internationale program for sikkerhed i forbindelse med kemikalier (IPCS) (Martin van den Berg et al., The 2005 World Health Organization Re-evaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-like Compounds. Toxicological Sciences 93(2), 223-241 (2006)).
|
Kongener |
TEF-værdi |
Kongener |
TEF-værdi |
|
Dibenzo-p-dioxiner (»PCDD'er«) og dibenzo-p-furaner (»PCDF'er«) |
»Dioxinlignende« PCB'er: non-ortho-PCB'er + mono-ortho-PCB'er |
||
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
Non-ortho-PCB'er |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0003 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,03 |
|
OCDD |
0,0003 |
|
|
|
|
|
Mono-ortho-PCB'er |
|
|
2,3,7,8-TCDF |
0,1 |
PCB 105 |
0,00003 |
|
1,2,3,7,8-PeCDF |
0,03 |
PCB 114 |
0,00003 |
|
2,3,4,7,8-PeCDF |
0,3 |
PCB 118 |
0,00003 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 123 |
0,00003 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 156 |
0,00003 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 157 |
0,00003 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00003 |
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
PCB 189 |
0,00003 |
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0003 |
|
|
Anvendte forkortelser: »T« = tetra; »Pe« = penta; »Hx« = hexa; »Hp« = hepta; »O« = octa; »CDD« = chlordibenzodioxin; »CDF« = chlordibenzofuran; »CB« = chlorbiphenyl.
( 37 ) Kommissionens beslutning 2002/657/EF af 14. august 2002 om gennemførelsesbestemmelser til Rådets direktiv 96/23/EF for så vidt angår analysemetoders ydeevne og fortolkning af resultater (EFT L 221 af 17.8.2002, s. 8).
( 38 ) »Øvre koncentration« betyder, at hver ikke-bestemt kongeners bidrag anses for at være lig med bestemmelsesgrænsen. »Nedre koncentration« betyder, at hver ikke-bestemt kongeners bidrag anses for at være nul. »Middelkoncentration« betyder, at hver ikke-bestemt kongeners bidrag anses for at være lig med halvdelen af bestemmelsesgrænsen.
( 39 ) Generelt finder kravene vedrørende dobbeltanalyse i bilag II, kapitel C, punkt 3, anvendelse. For verifikationsmetoder, hvor der anvendes 13C-mærket intern standard til de relevante analytter, er dobbeltanalyse dog kun et krav, hvis resultatet af den første bestemmelse efter sådanne metoder ikke er overensstemmende. En sådan ekstra analyse er nødvendig for at udelukke muligheden for intern krydskontaminering eller utilsigtet sammenblanding af prøver. Hvis analysen gennemføres i forbindelse med en hændelse med forurening, kan bekræftelse ved en ekstra analyse undlades, såfremt de prøver, der er udtaget til analyse, ved hjælp af sporbarhed kan relateres til forureningshændelsen, og det konstaterede indhold ligger væsentligt over grænseværdien.
( 40 ) »Øvre koncentration« betyder, at hver ikke-bestemt kongeners bidrag til toksicitetsækvivalenten (TEQ) anses for at være lig med bestemmelsesgrænsen. »Nedre koncentration« betyder, at hver ikke-bestemt kongeners bidrag til TEQ anses for at være nul. »Middelkoncentration« betyder, at hver ikke-bestemt kongeners bidrag til TEQ anses for at være lig med halvdelen af bestemmelsesgrænsen.
( 41 ) Generelt finder kravene vedrørende dobbeltanalyse i bilag II, kapitel C, punkt 3, anvendelse. For verifikationsmetoder, hvor der anvendes 13C-mærket intern standard til de relevante analytter, er dobbeltanalyse dog kun et krav, hvis resultatet af den første bestemmelse efter sådanne metoder ikke er overensstemmende. En sådan ekstra analyse er nødvendig for at udelukke muligheden for intern krydskontaminering eller utilsigtet sammenblanding af prøver. Hvis analysen gennemføres i forbindelse med en hændelse med forurening, kan bekræftelse ved en ekstra analyse undlades, såfremt de prøver, der er udtaget til analyse, ved hjælp af sporbarhed kan relateres til forureningshændelsen, og det konstaterede indhold ligger væsentligt over grænseværdien.
( 42 ) Forklarende bemærkninger og krav vedrørende dobbeltanalyse til kontrol af indgrebstærskler: jf. fodnote (42) (afsnit om grænseværdier).
( 43 ) Bioanalytiske metoder er ikke specifikt udformet til kongenerne i TEF-skemaet. Der kan i prøveekstraktet forekomme andre strukturelt beslægtede AhR-aktive forbindelser, som bidrager til den samlede prøvereaktion. Resultaterne af bioanalytiske metoder kan derfor ikke være et estimat, men snarere en indikation af TEQ-indholdet i prøven.
( 44 ) Gældende krav er baseret på TEF'erne, som er offentliggjort i: M. Van den Berg et al., Toxicol Sci 93 (2), 223-241 (2006).
( 45 ) Det må på det kraftigste anbefales, at bidraget fra reagensblindprøven til indholdet af et forurenende stof i en prøve er lavere. Det er laboratoriets ansvar at kontrollere variationen i blindprøveniveauet, især hvis blindprøveindholdet fratrækkes.
( 46 ) http://eurl.craw.eu/
( 47 ) Listen over disse primere og sonder for hver dyreart, assayet er målrettet mod, findes på EURL-AP's websted.
( 48 ) Der findes eksempler på mastermix, der er funktionelle, på EURL-AP's websted.