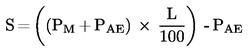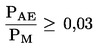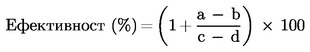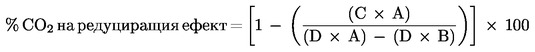Официален вестник
на Европейския съюз
-

Издание на български език
13. Индустриална политика и вътрешен пазар
TOM 039
|
Официален вестник на Европейския съюз |
- |
|
|
Издание на български език |
13. Индустриална политика и вътрешен пазар TOM 039 |
|
|
13/ 039 |
BG |
Официален вестник на Европейския съюз |
1 |
|
/ |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
Увод
В съответствие с член 58 от Акта за присъединяване на Република България и Румъния и промените в учредителните договори на Европейския съюз (ОВ L 157, 21.6.2005 г., стр. 203) текстове на актовете на институциите и на Европейската централна банка, приети преди присъединяването, изготвени от Съвета, от Комисията или от Европейската централна банка, на български и румънски език са автентични, считано от датата на присъединяване, при същите условия, както текстовете, съставени на другите официални езици на Общностите. В този член се предвижда също, че текстовете се публикуват в Официален вестник на Европейския съюз, ако текстовете на настоящите езици са публикувани в него.
В съответствие с въпросния член настоящото специално издание на Официален вестник на Европейския съюз се публикува на български език и съдържа текстовете на обвързващите актове с общо приложение. То обхваща актове, приети от 1952 г. до 31 декември 2006 г.
Текстовете, които се публикуват, са разпределени в 20 глави според класификацията, която се прилага в Указателя на действащото законодателство на Общността, както следва:
|
01. |
Общи, финансови и институционални въпроси |
|
02. |
Митнически съюз и свободно движение на стоки |
|
03. |
Земеделие |
|
04. |
Рибарство |
|
05. |
Свободно движение на работници и социална политика |
|
06. |
Право на установяване и свободно предоставяне на услуги |
|
07. |
Транспортна политика |
|
08. |
Политика на конкуренция |
|
09. |
Данъчна политика |
|
10. |
Икономическа и парична политика и свободно движение на капитали |
|
11. |
Външни отношения |
|
12. |
Енергетика |
|
13. |
Индустриална политика и вътрешен пазар |
|
14. |
Регионална политика и координация на структурните инструменти |
|
15. |
Околна среда, потребители и здравеопазване |
|
16. |
Наука, информация и култура |
|
17. |
Право относно предприятията |
|
18. |
Обща външна политика и политика на сигурност |
|
19. |
Пространство на свобода, сигурност и правосъдие |
|
20. |
Европа на гражданите |
Указателят, който се публикува два пъти годишно на официалните езици на Европейския съюз, ще бъде публикуван по-късно и на български език и ще съдържа позовавания на настоящото специално издание. По този начин указателят може да бъде използван и като индекс за настоящото специално издание.
Актовете, публикувани в настоящото специално издание, се публикуват, с малки изключения, във формата, в която са били публикувани на досегашните езици в Официален вестник. Следователно при използването на настоящото специално издание трябва да се вземат предвид последващите изменения, промени или дерогации, приети от институциите или Европейската централна банка или предвидени в Акта за присъединяване.
По изключение, в определени случаи, когато обемни технически приложения на актове се заменят по-късно от други приложения, се прави позоваване само на последния заменящ акт. Такъв е случаят основно в определени актове, които съдържат списъци на митнически кодове (глава 02), актове относно превоза на опасни вещества и опаковането и етикетирането на тези вещества (глави 07 и 13) и определени протоколи и приложения към Споразумението за ЕИП.
Също така „Правилникът за длъжностните лица“ по изключение се публикува като консолидиран текст, който съдържа всички изменения до края на 2005 г. Измененията, направени след това, се публикуват в първоначалния им вид.
В специалните издания са използвани две системи на номериране:
|
i) |
първоначалните номера на страниците заедно с датата на публикуване на Официален вестник в изданието му на италиански, немски, нидерландски и френски език, както в изданията му на английски и датски език след 1 януари 1973 г., както в изданието на гръцки език след 1 януари 1981 г., както в изданията на испански и португалски език след 1 януари 1986 г., както в изданията на фински и шведски език след 1 януари 1995 г. и в изданията на естонски, латвийски, литовски, малтийски, полски, словашки, словенски, унгарски и чешки език след 1 май 2004 г. Непоследователността при номерирането на страниците се дължи на факта, че не всички актове, публикувани тогава, са включени в настоящото специално издание. Първоначалните номера на страниците се използват при посочването на актовете, когато се прави позоваване на Официален вестник; |
|
ii) |
номерата на страниците в специалните издания, които са последователни и не следва да се използват при цитиране на актове. |
До месец юни 1967 г. номерирането на страниците в Официален вестник всяка година започва отначало. От тази дата всеки брой започва със страница първа.
От 1 януари 1968 г. Официален вестник се разделя на две части:
|
— |
законодателство (L), |
|
— |
съобщения и информации (С). |
На 1 февруари 2003 г. предишното официално наименование Официален вестник на Европейските общности бе променено вследствие на Договора от Ница и сега е Официален вестник на Европейския съюз.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
3 |
32002R2056
|
L 317/1 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕГЛАМЕНТ (ЕО) № 2056/2002 НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 5 ноември 2002 година
за изменение на Регламент (ЕО, Евратом) № 58/97 на Съвета относно структурната бизнес статистика
(текст от значение за ЕИП)
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 285 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Икономическия и социален комитет (2),
като взеха предвид становището на Европейската централна банка (3),
в съответствие с процедурата, предвидена в член 251 от Договора (4),
като имат предвид, че:
|
(1) |
Регламент (ЕО, Евратом) № 58/97 (5) установява обща рамка за събирането, изготвянето, предаването и оценката на статистически данни на Общността за структурата, дейността, конкурентоспособността и икономическите резултати на предприятията в Общността. |
|
(2) |
Развитието на паричната, икономическата и социалната интеграция на Общността изискват разширяване на общата рамка за кредитните институции, пенсионните фондове, другите финансови посредници и спомагателната финансова и застрахователна дейност. |
|
(3) |
Функционирането и развитието на вътрешния пазар увеличиха необходимостта от наличието на информация за неговата ефективност, особено що се отнася до секторите на кредитните институции, пенсионните фондове, другите финансови посредници и спомагателната финансова и застрахователна дейност. |
|
(4) |
Либерализацията на международната търговия с финансови услуги поражда необходимостта от статистически данни за предприятията в сектора на финансовите услуги в подкрепа на търговските преговори. |
|
(5) |
Изготвянето на на националните и регионалните отчети съгласно Регламент (ЕО) № 2223/96 на Съвета от 25 юни 1996 г. относно Европейската система на национални и регионални отчети в Общността (6) изисква сравнима, пълна и надеждна бизнес статистика за предприятията в областта на финансовите услуги. |
|
(6) |
Въвеждането на единната валута ще има решаващо влияние върху структурата на сектора на финансовите услуги и върху презграничните капиталови потоци, като подчертава значението на информацията относно конкурентноспособността, вътрешния пазар и интернационализацията. |
|
(7) |
За гарантиране на доброто водене на политиките, осъществявани от компетентните органи във връзка с финансовия надзор на кредитните предприятия и стабилността на финансовата система, уместно е да разполага с повече информация за кредитните институции и свързаните с тях услуги. |
|
(8) |
Развиващият се сектор на пенсионните фондове може да допринесе за стимулиране на капиталовите пазари чрез все по-широко възползване от либерализацията на инвестиционните правила. |
|
(9) |
Решение № 2179/98/ЕО на Европейския парламент и на Съвета от 24 септември 1998 г. относно преразглеждане на програмата на Европейската общност за политически действия във връзка с околната среда и устойчивото развитие „Към устойчивост“ (7), потвърди необходимостта от надеждни и сравними данни, статистическа информация и показатели, които са ключово средство за оценка на разходите за спазване на правилата в областта на околната среда. |
|
(10) |
Проведени са консултации със Статистическия програмен комитет, създаден с Решение 89/382/ЕИО, Евратом (8), Съвещателния комитет по банково дело, създаден с Директива 77/780/ЕИО (9), Комитета за статистическите данни в паричната и финансовата област и в областта на платежните баланси, създаден с Решение 91/115/ЕИО (10), и Комитета по застраховане, създаден с Директива 91/675/ЕИО (11), |
ПРИЕХА НАСТОЯЩИЯ РЕГЛАМЕНТ:
Член 1
Регламент (ЕО, Евратом) № 58/97 се изменя, както следва:
|
1. |
В член 5 се добавят следните тирета:
|
|
2. |
Добавя се текстът на приложения 6 и 7, както са определени в приложението към настоящия регламент. |
Член 2
Приложение 1 към Регламент (ЕО, Евратом) № 58/97 се изменя, както следва:
|
1. |
В раздел 5 се добавя следното изречение: „Независимо от това първата референтна година, за която следва да бъдат съставяни статистики за категориите дейност, включени в група 65.2 и подраздел 67 на NACE REV. 1, се определя в съответствие с процедурата, установена в член 13 от настоящия регламент.“; |
|
2. |
Раздел 8 се заменя със следния текст: „Раздел 8 Предаване на резултатите 1. Резултатите се предават в срок 18 месеца от края на календарната година на референтния период, с изключение на дейността от категория 65.11 на NACE REV. 1 и дейностите по NACE REV. 1, включени в приложения 5, 6 и 7. За дейността от категория 65.11 на NACE REV. 1 срокът за предаване е 10 месеца. За дейностите, включени в приложения 5, 6 и 7, срокът за предаване е определен в тези приложения. Независимо от това, срокът за предаване на резултатите за категориите дейност, включени в група 65.2 и подраздел 67 на NACE REV. 1, се определя съгласно процедурата, установена в член 13 от настоящия регламент. 2. С изключение на подраздели 65 и 66 от NACE REV. 1, предварителните национални резултати или прогнози се предават в срок 10 месеца от края на календарната година на референтния период по отношение на статистическите данни за предприятия по характеристиките, изброени по-долу:
На тези предварителни резултати или прогнози се прави разбивка по трицифрено равнище (група) на NACE REV. 1, с изключение на раздели H, I и K от NACE REV. 1, за които на тях им е направена разбивка според групите, установени в раздел 9. По отношение на подраздел 67 на NACE REV. 1 предаването на предварителни резултати или прогнози се определя в съответствие с процедурата, установена в член 13 от настоящия регламент.“ |
|
3. |
В раздел 9, раздел J се заменя със следния текст: „РАЗДЕЛ J Финансово посредничество За да бъде направено възможно изготвянето на статистически данни за Общността, държавите-членки предават националните резултати, като им правят разбивка по категории на NACE REV. 1.“ |
|
4. |
В раздел 10, параграф 1, първото изречение се заменя със следния текст: „Държавите-членки предоставят на Комисията доклад относно определянето, структурата и наличието на информация за статистическите единици, отнесени към категориите по раздели M до О на NACE REV. 1.“ |
Член 3
Приложение 2 към Регламент (ЕО, Евратом) № 58/97 се заменя със следния текст:
|
1. |
В раздел 4, парагарф 3, следните характеристики се въвеждат след изменяемия показател 21 11 00 (инвестиции в оборудване и заводи за контрол на замърсяването, и специални приспособления, които предотвратяват замърсяването — предимно оборудване за краищата на тръби): „21 12 0 —инвестиции в оборудване и заводи, свързани с по-чисти технологии („интегрирана технология“) (*).“ |
|
2. |
Бележката под линия в раздел 4, параграф 3 се заменя със следния текст:
|
|
3. |
В раздел 4, параграф 4, следните характеристики се добавят след изменяем показател 20 31 0 (Покупка на електроенергия (по стойност)): „21 14 0 —всичко текущи разходи за опазване на околната среда (*).“ |
|
4. |
В раздел 4, параграф 4, се добавя следната бележка под линия:
|
|
5. |
В раздел 5 се добавят следните параграфи: 3. Първата референтна година, за която следва да бъдат съставени статистически данни по показатели 21 12 0 и 21 14 0, е календарна 2001 година. 4. Статистическите данни по показател 21 12 0 се съставят всяка година. Статистическите данни по показател 21 14 0 се съставят на всеки три години.“. |
|
6. |
В раздел 7, параграф 6 се заменя със следния текст: 6. Резултатите за характеристики 21 11 0, 21 12 0 и 21 14 0 се разбиват до двуцифрено равнище (подраздел) на NACE REV. 1.“ |
|
7. |
В раздел 7 се добавя следният параграф: 7. Резултатите за характеристики 21 11 0, 21 12 0 и 21 14 0 се разбиват на следните екологични области: опазване на въздуха и климата, управление на отпадните води, управление на отпадъците и други дейности за опазване на околната среда. Резултатите за екологичните области се подразделят до двуцифрено равнище (подраздели) на NACE REV. 1.“ |
|
8. |
В раздел 9 се добавя следният показател: „21 11 0 —инвестиции в оборудване и заводи за контрол върху замърсяването и специални приспособления, които предпазват от замърсяване (предимно оборудване на краищата на тръби).“За характеристики 21 11 0, 21 12 0 и 21 14 0 се добавя следната бележка: „Само специфична разбивка в екологичните области биоразнообразие и ландшафт, почви и подпочвени води.“ |
|
9. |
В раздел 10 се добавя следното изречение: „За съставянето на статистически данни по характеристики 21 12 0 и 21 14 0 този преходен период може да бъде удължен най-много с още четири години, в съответствие с процедурата, установена в член 13 от настоящия регламент.“ |
Член 4
Настоящият регламент влиза в сила на двадесетия ден след публикуването му в Официален вестник на Европейските общности.
Настоящият регламент е задължителен в своята цялост и се прилага пряко във всички държави-членки.
Съставено в Брюксел на 5 ноември 2002 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
T. PEDERSEN
(1) ОВ С 154 Е, 29.5.2001 г., стр. 129 и
ОВ С 332 Е, 27.11.2001 г., стр. 340.
(2) ОВ С 260, 17.9.2001 г., стр. 54.
(3) ОВ С 131, 3.5.2001 г., стр. 5.
(4) Становище на Европейския парламент от 13 юни 2001 г. (ОВ С 53 Е, 28.2.2002 г., стр. 213), Обща позиция на Съвета от 20 юни 2002 г. (ОВ С 228, 25.9.2002 г., стр. 1) и Решение на Европейския парламент от 24 септември 2002 г. (все още непубликувано в Официален вестник).
(5) ОВ L 14, 17.1.1997 г., стр. 1. Регламент, последно изменен с Регламент (ЕО, Евратом) № 410/98 (ОВ L 52, 21.2.1998 г., стр. 1).
(6) ОВ L 310, 30.11.1996 г., стр. 1. Регламент, последно изменен с Регламент (ЕО) № 359/2002 на Европейския парламент и на Съвета (ОВ L 58, 28.2.2002 г., стр. 1).
(7) ОВ L 275, 10.10.1998 г., стр. 1.
(8) ОВ L 181, 28.6.1989 г., стр. 47.
(9) ОВ L 322, 17.12.1977 г., стр. 30. Директива, последно изменена с Директива 98/33/ЕО на Европейския парламент и на Съвета (ОВ L 204, 21.7.1998 г., стр. 29).
(10) ОВ L 59, 6.3.1991 г., стр. 9. Решение, последно изменено с Решение 96/174/ЕО (ОВ L 51, 1.3.1996 г., стр. 48).
(11) ОВ L 374, 31.12.1991 г., стр. 32.
ПРИЛОЖЕНИЕ
ПРИЛОЖЕНИЕ 6
ПОДРОБЕН МОДУЛ ЗА СТРУКТУРНИТЕ СТАТИСТИЧЕСКИ ДАННИ ЗА КРЕДИТНИТЕ ИНСТИТУЦИИ
Раздел 1
Цел
Целта на това приложение е да установи обща рамка за събирането, изготвянето, предаването и оценката на статистически данни на Общността относно структурата, дейността, конкурентноспособността и икономическите резултати на сектора на кредитните институции. Настоящият модул съдържа подробен списък на показатели, по които следва да се събират статистически данни, с цел подобряване на знанието за национално, общностно и международно развитие на сектора на кредитните институции.
Раздел 2
Обхват
Статистическите данни, които следва да бъдат съставяни, се отнасят до областите, посочени в точки i), ii) и iii) от член 2 от този регламент, и по-специално до:
|
1. |
подробния анализ на структурата, дейността, конкурентноспособността и икономическите резултати на кредитните институции; |
|
2. |
развитието и подразделянето въз основа на обща стопанска дейност, стопанска дейност по продукти, международна дейност, заетост, капитал и резерви, други активи и пасиви. |
Раздел 3
Обхват
|
1. |
Статистическите данни се съставят за дейността на кредитните институции, спадащи към категории 65.12 и 65.22 на NACE REV. 1. |
|
2. |
Статистическите данни се съставят за дейността на всички кредитни институции, посочени в член 2, параграф 1, буква а) и член 2, параграф 2 от Директива 86/635/ЕИО на Съвета от 8 декември 1986 г. относно годишните счетоводни отчети и консолидираните счетоводни отчети на банки и други финансови институции (1) (с изключение на централните банки). |
|
3. |
Клоновете на кредитните институции, посочени в член 24 от Директива 2000/12/ЕО на Европейския парламент и на Съвета от 20 март 2000 г. относно предприемането и осъществяването на дейност от кредитните институции (2), чиято дейност попада в обхвата на NACE REV. 1, категории 65.12 и 65.22, се приравняват към кредитните институции, посочени в параграф 2. |
Раздел 4
Характеристики
Характеристиките са изброени по-долу. Показателите, посочени в курсив, също са включени и в списъците на общия модул на приложение 1. Когато характеристиките са извлечени пряко от годишните счетоводни отчети, счетоводните години, които приключват през референтна година, се приравняват към тази референтна година.
Списъкът включва:
|
i) |
характеристики, изброени в член 4 от Директива 86/635/ЕИО: актив на баланса — статия 4; пасив на баланса — позициия 2 а) + 2 б) в агрегиран вид, позиции 7 + 8 + 9 + 10 + 11 + 12 + 13 + 14 в агрегиран вид; |
|
ii) |
характеристики, изброени в член 27 от Директива 86/635/ЕИО: позиция 2, позиции 3а) + 3б) + 3в) в агрегиран вид, позиция 4, позиция 5, позиция 6, позиция 7, позиции 8а) + 8б) в агрегиран вид, позиция 8б), позиция 10, позиции 11 + 12 в агрегиран вид, позиции 9 + 13 + 14 в агрегиран вид, позиции 15 + 16 в агрегиран вид, позиция 19, позиции 15 + 20 + 22 в агрегиран вид, позиция 23; |
|
iii) |
следните допълнителни показатели:
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
iv) |
характеристики, за които се съставят статистически данни всяка година:
|
Раздел 5
Първа референтна година
Първата референтна година, за която следва да бъдат съставени статистически данни за характеристиките, изброени в раздел 4, е календарната 2001 година.
Раздел 6
Представяне на резултатите
|
1. |
Резултатите се подразделят на следните отделни категории в NACE REV. 1: 65.12 и 65.22. |
|
2. |
Резултатите от регионалните статистически данни се подразделят до четирицифрово равнище (категории) на NACE REV. 1 и до равнище 1 от номенклатурата на териториалните единици (NUTS). |
Раздел 7
Предаване на резултатите
Срокът за предаване на резултатите се определя в съответствие с процедурата, установена в член 13 от настоящия регламент. Той не може да бъде по-дълъг от 10 месеца от края на референтната година.
Раздел 8
Комитет за статистическите данни във валутната и финансовата област, и в областта на платежните баланси
Комисията информира Комитета за статистическите данни във валутната и финансовата област, и в областта на платежните баланси за въвеждането на този модул и за всички мерки за приспособяване към икономическото и техническото развитие, относно събирането и статистическата обработка на данните, обработката и предаването на резултатите.
Раздел 9
Пилотни проучвания
|
1. |
За дейността, обхваната от настоящото приложение, Комисията определя следните пилотни проучвания, които се извършват от държавите-членки:
|
|
2. |
Пилотните проучвания се провеждат, за да се прецени дали получаването на данни е уместно и възможно, като предимствата от наличието на данни се сравняват с разходите за събирането им и като се вземе предвид съответната тежест за предприятията. |
Раздел 10
Преходен период
За целите на този подробен модул, преходният период няма да превишава три години от началото на първата референтна година за изготвянето на статистически данни, пососчена в раздел 5.
ПРИЛОЖЕНИЕ 7
ПОДРОБЕН МОДУЛ ЗА СТРУКТУРНИТЕ СТАТИСТИЧЕСКИ ДАННИ ЗА ПЕНСИОННИТЕ ФОНДОВЕ
Раздел 1
Цел
Целта на това приложение е да установи обща рамка за събирането, изготвянето, предаването и оценката на статистически данни на Общността относно структурата, дейността, конкурентноспособността и икономическите резултати в сектора на пенсионните фондове. Настоящият модул съдържа подробен списък на характеристиките, по които следва да се събират статистически данни, с цел по-добро запознаване с развитието на сектора на пенсионните фондове на национално, общностно и международно ниво.
Раздел 2
Области
Статистическите данни, които следва да бъдат съставяни, се отнасят за областите, посочени в точки i), ii) и iii) от член 2 от настоящия регламент и по-специално за:
|
1. |
подробния анализ на структурата, дейността, конкурентноспособността и икономическите резултати на пенсионните фондове; |
|
2. |
развитието и разпределението въз основа на обща дейност, характеристика на членовете на пенсионните фондове, международни дейности, заетост, инвестиции и пасив. |
Раздел 3
Обхват
|
1. |
Статистическите данни се съставят за всички дейности, попадащи в рамките на обхвата на категория 66.02 от NACE REV. 1. Тази категория се отнася за дейността на автономните пенсионни фондове. |
|
2. |
Следва да бъдат събирани и статистически данни за предприятията с неавтономни пенсионни фондове, които се изготвят като допълнителна дейност. |
Раздел 4
Характеристики
|
1. |
Списъкът на характеристики, установени по-долу, когато е необходимо, съдържа означение за вида на статистическите единици, за които се съставят статистически данни. Характеристиките, посочени в курсив, са включени и в списъците на общия модул на приложение 1. За характеристики, извлечени директно от годишните счетоводни отчети, счетоводните години, които приключват през референтната година, се приравняват към посочената референтна година. |
|
2. |
Демографски характеристики и характеристики на предприятия, за които се изготвят годишни статистически данни (само предприятия с автономни пенсионни фондове):
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
3. |
Характеристики на предприятията, за които се съставят годишни статистически данни (само за предприятията с неавтономни пенсионни фондове):
|
Раздел 5
Първа референтна година
Първата референтна година, за която се изготвят статистически данни за характеристиките, изброени в раздел 4, е календарната 2002 година.
Раздел 6
Изготвяне на резултатите
|
1. |
Резултатите за характеристиките, посочени в раздел 4, параграф 2, се разбиват до четирицифрено равнище (категория) на NACE REV. 1. |
|
2. |
Резултатите по показателите, изброени в раздел 4, точка 3, се подразделят до раздели на NACE REV. 1. |
Раздел 7
Предаване на резултатите
Резултатите се предават в срок 12 месеца от края на референтната година.
Раздел 8
Комитет по застрахователно дело
Комисията информира Комитета по застраховане за въвеждането на този модул и за всички мерки за адаптиране към икономическото и техническото развитие, относно събирането и статистическата обработка на данните, обработката и предаването на резултатите.
Раздел 9
Пилотни проучвания
За дейността, предмет на настоящото приложение, Комисията определя следните пилотни проучвания, които трябва да бъдат проведени от държавите-членки:
|
1. |
По-подробни данни за презграничната дейност на пенсионните фондове:
|
|
2. |
Допълнителна информация за неавтономните пенсионни фондове:
|
|
3. |
Информация за деривативните инструменти и извънбалансовите статии. Пилотните проучвания се провеждат с цел да се прецени дали получаването на данни е уместно и възможно, като предимствата от наличието на данни се отчитат в сравнение с разходите за събирането им и се взима предвид съответната тежест върху предприятията. |
Раздел 10
Преходен период
За целите на този подробен модул, преходният период няма да надвишава три години от началото на първата референтна година за изготвянето на статистически данни, посочени в раздел 5. Този период може да бъде удължен най-много с още три години по реда, в съответствие с процедурата, установена в член 13 от настоящия регламент.
(1) ОВ L 372, 31.12.1986 г., стр. 1. Директива, последно изменена с Директива 2001/65/ЕО на Европейския парламент и на Съвета (ОВ L 283, 27.10.2001 г., стр. 28).
(2) ОВ L 126, 26.5.2000 г., стр. 1. Директива, последно изменена с Директива 2000/28/ЕО (ОВ L 275, 27.10.2000 г., стр. 37).
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
14 |
32002R2245
|
L 341/28 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕГЛАМЕНТ (ЕО) № 2245/2002 НА КОМИСИЯТА
от 21 октомври 2002 година
за прилагане на Регламент (ЕО) № 6/2002 на Съвета за промишления дизайн на Общността
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Регламент (ЕО) № 6/2002 на Съвета от 12 декември 2001 г. за промишления дизайн на Общността (1), и по-специално член 107, параграф 3 от него,
като има предвид, че:
|
(1) |
Регламент (ЕО) № 6/2002 на Съвета създава система, даваща възможност за дизайн, който е в сила в цялата Общност, да се получи на базата на заявка до Службата за хармонизиране на вътрешния пазар (търговски марки и дизайни), (наричана по-долу „Служба“). |
|
(2) |
За тази цел в Регламент (ЕО) № 6/2002 на Съвета се съдържат необходимите разпоредби относно процедура, която да се следва при регистрирането на промишления дизайн на Общността, както и относно разпореждане с регистрирания промишлен дизайн на Общността относно обжалване на решения на Службата и процедура по заличаване на регистрацията на промишления дизайн на Общността. |
|
(3) |
Настоящият регламент определя необходимите мерки за изпълнение разпоредбите на Регламент (ЕО) № 6/2002. |
|
(4) |
Настоящият регламент следва да обезпечава безпроблемната и ефикасна дейност при процедурите по промишлените дизайни пред Службата. |
|
(5) |
Мерките, предвидени в настоящия регламент, са в съответствие със становището на Комитета, учреден съгласно член 109 от Регламент (ЕО) № 6/2002, |
ПРИЕ НАСТОЯЩИЯ РЕГЛАМЕНТ:
ГЛАВА I
ПРОЦЕДУРА ЗА ПОДАВАНЕ НА ЗАЯВЛЕНИЕ
Член 1
Съдържание на заявката
1. Заявката за регистриран промишлен дизайн на Общността следва да съдържа:
|
а) |
искане за регистриране на дизайн като регистриран промишлен дизайн на Общността; |
|
б) |
име, адрес и националност на заявителя и държава, в която е местожителството на заявителя или в която се намира неговото седалище, или в която се е установил. Имената на физическите лица се посочват по реда „фамилия и име/на“. Наименованията на юридическите лица се посочват по тяхното официално изписване, което може да се подаде във формата на обичайно използвано съкращение; по-нататък се посочва държавата, от чийто закон се ръководят дадените лица. Могат да се посочат и телефонните номера, както и номерата на факсовете, както и всеки друг начин за комуникация, например адрес на електронна поща. По принцип, за един заявител се посочва само един адрес; там, където са посочени няколко адреса, се взема под внимание единствено първия поред, с изключение на случаите, в които заявителят посочва един от адресите като служебен. Ако Службата е дала на заявителя номер на заявката, е достатъчно да се посочи този номер заедно с името или наименованието на заявителя; |
|
в) |
представянето на промишлен дизайн в съответствие с член 4 от настоящия регламент или, при условие че дадено заявление се отнася до двумерен дизайн и съдържа искане за отлагане на публикуване в съответствие с член 50 от Регламент (ЕО) № 6/2002 на даден промишлен дизайн съгласно член 5 от настоящия регламент; |
|
г) |
обозначаване в съответствие с член 3, параграф 3 на продукти, в които се планира дизайнът да бъде вложен или спрямо които се планира промишленият дизайн да бъде приложен; |
|
д) |
в случай че заявителят е посочил представител, се посочва името на този представител и служебния адрес на този представител в съответствие с буква б); в случай че представителят има повече от един служебен адрес или ако представителите са двама или повече с различни служебни адреси, заявителят следва да посочи кой от адресите следва да се използва за работен адрес; там, където не се направи такова обозначение, за работен адрес се приема единствено първият поред служебен адрес. В случай, че заявителите са повече от един, в заявката за общ представител може да се посочи един заявител или представител. В случай че даден представител е получил от Службата идентификационен номер, достатъчно е да се посочи този номер заедно с името на представителя.; |
|
е) |
ако е приложимо, се подава декларация, в която се предявява искане за приоритет на предишно заявление съгласно член 42 от Регламент (ЕО) № 6/2002 и се посочва датата, на която е подадена предишната заявка, както и страната, в която или за която е била подадена; |
|
ж) |
ако е приложимо, се подава декларация, в която се предявява искане за приоритет на излагане на продукти съгласно член 44 от Регламент (ЕО) № 6/2002 и се посочва наименованието на изложението и датата, на която се е състояло първото излагане на продукти, в които е вложен промишленият дизайн или спрямо които дизайнът е бил приложен; |
|
з) |
посочване на езика, на който е била подадена заявката, и на втори език съгласно член 98, параграф 2 от Регламент (ЕО) № 6/2002; |
|
и) |
подписът на заявителя или неговия/нейния представител в съответствие с член 65. |
2. В заявката може да се съдържа следното:
|
а) |
по едно описание на промишлен дизайн, което да не превишава 100 думи, и в което да e налице излагане на промишления дизайн или модел; обяснението да се отнася само до онези качества, които се проявяват във възпроизвеждането на промишления дизайн или модел; не бива да съдържа твърдения за предполагаемата новост, нито за индивидуалния характер на дизайна, нито за техническата му стойност; |
|
б) |
искане за отсрочване на публикуването на регистрация в съответствие с член 50, параграф 1 от Регламент (ЕО) № 6/2002; |
|
в) |
посочване по „класификацията от Локарно“ на продуктите, които се съдържат в заявката, тоест на класа или класовете и подкласа или подкласовете, към които са включени в съответствие с приложението към Спогодбата, с което се учредява международната класификация за промишлен дизайн, подписана в Локарно на 8 октомври 1968 г. (по-долу наричана „Спогодба от Локарно“), визирано в член 3 и с при спазване на член 2, параграф 2; |
|
г) |
посочване на автора или авторския екип на промишления дизайн на Общността или становище, подписано от заявителя относно това, че автора или авторския екип на промишления дизайн на Общността звено се отказват от правото да бъдат посочвани съгласно член 36, параграф 3, буква д) от Регламент (ЕО) № 6/2002. |
Член 2
Множествена заявка
1. Заявката може да бъде множествена заявка, когато се иска регистрирането на няколко промишлени дизайна.
2. Когато няколко промишлени дизайна, с изключение на декорация, са включени в една множествена заявка, заявката следва да се раздели, ако продуктите, в които се планира да бъдат вложени промишлените дизайни или спрямо които се възнамерява дизайните да бъдат приложени, са включени в повече от един клас по Класификацията от Локарно.
3. За всеки дизайн, който се съдържа в множествената заявка, заявителят изготвя представяне на промишления дизайн в съответствие с член 4 и посочва продукта, в който промишленият дизайн е предназначен да бъде вложен или за който е предназначен да се приложи.
4. Заявителят номерира промишлените дизайни, съдържащи се в множествената заявка последователно, като за целта използва арабски цифри.
Член 3
Класификация и обозначаване на продуктите
1. Продуктите се класифицират в съответствие с член 1 от Спогодбата от Локарно, така изменена и в сила към датата на регистриране на промишления дизайн.
2. Класификацията на продуктите е единствено за административни цели.
3. Обозначаването на продуктите се формулира по такъв начин, че да се посочи ясно естеството и да се даде възможност за класифициране само в един клас от Класификацията от Локарно, като за предпочитане се използват условията, посочени в списъка продукти, включен там.
4. Продуктите се групират според класовете в Класификацията от Локарно, като пред всяка група се посочва номерът на класа, към който е разпределена тази група продукти, и се представя по реда на класовете и подкласовете съгласно тази класификация.
Член 4
Представяне на промишления дизайн
1. Представянето на промишления дизайн следва да се състои от графична или фотографска репродукция на промишления дизайн с възможност за черно-бяло или цветно представяне. Същото следва да отговаря на следните изисквания:
|
а) |
освен в случаите, в които заявката е подадена чрез електронни средства съгласно член 67, представянето се подава на отделни листове хартия, предвидени за тази цел във формуляра, който е на разположение в Службата съгласно член 68; |
|
б) |
в случая с отделни листове хартия промишленият дизайн се възпроизвежда на непрозрачна бяла хартия, като или се залепва, или се отпечатва директно върху нея. Подава се само едно копие, като листата хартия не бива да са сгънати, нито хванати с телбод; |
|
в) |
размерът на отделния лист следва да е DIN A4 (29,7 cm х 21 сm), а повърхността, която ще се използва за възпроизвеждане не трябва да е по-голяма от 26,2 х 17 cm. От лявата страна на листа се оставя поле най-малко 2,5 cm; от горната страна на листа се посочва броят на ракурсите съгласно параграф 2, а в случай на множествена заявка, поредният номер на промишления дизайн; там не се допуска изписването на обяснителен текст, надписи, нито символи, освен обозначението „горе“ или имената и адресът на заявителя; |
|
г) |
когато заявката е подадена чрез електронни средства, графичното или фотографското възпроизвеждане на дизайна се оформя според формата за данни, определен от председателя на Службата; начинът за определяне на различните дизайни, които се съдържат в множествената заявка, както и различните ракурси, се определят от председателя на Службата; |
|
д) |
промишленият дизайн се възпроизвежда на неутрален фон и не се ретушира с мастило, нито с течен коректор. Той следва да бъде изпълнен с качество, което да позволява всички подробности от материята, по която се търси закрила, да се открояват ясно и да позволява същите да могат да се намаляват или увеличават до размер не повече от 8 × 16 cm на ракурс за включване в Регистъра на промишлените дизайни, предвиден в член 72 от Регламент (ЕО) № 6/2002, по-долу наричан „Регистър“, и за директно публикуване в Бюлетин на промишлените дизайни на Общността, посочен в член 73 от споменатия регламент. |
2. Представянето може да съдържа не повече от седем различни ракурса на промишления дизайн. Всяка графична или фотографска репродукция може да съдържа само един ракурс. Заявителят номерира всеки ракурс, като използва арабски цифри. Номерът следва да се състои от отделни числа, разделени с точка, като числото вляво от точката посочва номера на промишления дизайн, а числото вдясно от точката посочва номера на ракурса.
В случаите, в които се предвидени повече от седем ракурса, Службата може да не зачете някои от допълнителните ракурси при регистрирането и публикуването. Службата взима ракурсите в последователността, в която са номерирани от заявителя.
3. Когато заявление се отнася до промишлен дизайн, който се състои от повтарящ се мотив за повърхност, в представянето на промишления дизайн следва да се покаже пълния мотив и достатъчна част от повърхността.
Относно ограниченията за размерите се прилагат разпоредбите на параграф 1, буква в).
4. Когато заявката се отнася до промишлен дизайн, който се състои от типографско лице на знак или лице на буква, представянето на промишления дизайн следва да се състои от поредица от всички малки и големи букви от азбуката, от всички арабски цифри, придружени от текст, покриващ пет реда с това лице на буква, като буквите и цифрите следва да са в размер с височина на знака 16.
Член 5
Мостри
1. Когато заявката се отнася до двумерен промишлен дизайн и съдържа искане за отлагане на публикуването в съответствие с член 50, параграф 1 от Регламент (ЕО) № 6/2002, представянето на промишления дизайн може да се замести с мостра, залепена на лист хартия.
Заявките, към които е представена мостра, се изпращат в единичен пощенски колет или плик или се доставят направо в службата по регистрирането на промишлените дизайни.
Заявката и мострата се представят едновременно.
2. Размерът на мострите не трябва да превишава 26,2 cm х 17 cm, теглото не трябва да превишава 50 g, а дебелината — 3 mm. Мострата трябва да дава възможност за съхраняване и разгъване заедно с документите, чиито размери са предвидени в член 4, параграф 1, буква в).
3. Нетрайните и опасните за съхранение мостри не се депозират.
Мострата се депозира в пет копия; в случай на множествена заявка, за всеки промишлен дизайн се депозират по пет копия.
4. Когато заявка се отнася до промишлен дизайн, който се състои от повтарящ се мотив за повърхност, в представянето на промишления дизайн трябва да се покаже пълния мотив и достатъчна част по дължина и по широчина. Прилагат се ограниченията, предвидени в параграф 2.
Член 6
Такси за подаване на заявките
1. В момента на подаване на заявката в Службата се заплащат следните такси:
|
а) |
такса за регистрация; |
|
б) |
такса за публикуване или такса за отлагане в случай, че е било поискано отлагане на публикуването; |
|
в) |
допълнителна такса за регистрация за всеки допълнителен промишлен дизайн, включен в множествена заявка; |
|
г) |
допълнителна такса за публикация за всеки допълнителен промишлен дизайн или модел, включен в множествена заявка, или допълнителна такса за отлагане за всеки допълнителен промишлен дизайн, включен в множествена заявка в случай, че е било поискано отлагане. |
2. Когато в заявката е включено искане за отлагане на публикуването на регистрирането, се заплаща такса за публикуване и допълнителна такса за публикуване за всеки допълнителен промишлен дизайн, включен в множествена заявка, в срока, определен в член 15, параграф 4.
Член 7
Регистриране на заявката
1. Службата поставя върху документите, от които е съставена заявката, датата на получаване и регистрационния номер на заявката.
Всеки промишлен дизайн, който се съдържа в множествена заявка, се номерира от Службата в съответствие с определената от председателя система.
Службата незабавно издава на заявителя разписка, в която се посочва регистрационният номер, представянето, описание или друго определение на промишления дизайн, естеството и броят документи, както и датата на тяхното получаване.
В случай на множествена заявка в разписката, издадена от службата, се уточнява първият промишлен дизайн и броят депозирани промишлени дизайни.
2. В случай, че заявката е подадена в централната служба за индустриална собственост на държава-членка или в Службата за промишлен дизайн на Бенелюкс в съответствие с член 35 от Регламент (ЕО) № 6/2002, службата, в която е извършена регистрацията, следва да номерира всяка страница на заявката с арабски цифри. Службата, в която е извършена регистрацията, следва да обозначи документите, съставящи заявката, като посочва датата на приемане на заявката и броя на страниците, преди да изпрати заявката на службата.
Службата, в която е подадена заявката, незабавно издава на заявителя разписка, в която се посочва регистрационният номер, представянето, описание или друго определение на промишления дизайн, естеството и броят документи, както и датата на тяхното получаване.
3. В случай, че Службата получи заявка, изпратена от централната служба за промишлена интелектуална собственост на държава-членка или в Службата за промишлен дизайн на Бенелюкс, тя следва да обозначи датата на приемане на заявката и регистрационния номер и да издаде незабавно на заявителя разписка, в съответствие с параграф 1, трета и четвърта алинеи, като посочи датата на получаване в службата.
Член 8
Претендиране на право на приоритет
1. Когато в заявката се претендира право на приоритет на едно или повече предишни заявки съгласно член 42 от Регламент (ЕО) № 6/2002, заявителят посочва регистрационния номер на предишната заявка и подава копие от същото в срок от три месеца, считано от датата на подаването на заявлението съгласно член 38 от посочения регламент. Председателят на Службата определя доказателствата, които заявителят трябва да представи.
2. Ако след подаването на заявката заявителят желае да предяви претенция за приоритет за едно или повече предишни заявки съгласно член 42 от Регламент (ЕО) № 6/2002, той представя в срок от един месец, считано от датата на подаване на заявката, декларация за приоритет, като посочва датата и страната, в която или за която е направена предишната заявка.
Заявителят представя в Службата означението и доказателствата, посочени в параграф 1, в срок от три месеца, считано от получаването на декларацията за приоритет.
Член 9
Приоритет на изложение
1. Ако в заявката се претендира приоритет на изложение съгласно член 44 от Регламент (ЕО) № 6/2002, заявителят следва, наред със заявката или най-късно до три месеца, считано от датата на подаване на заявката, да подаде удостоверение, издадено при изложението от орган, отговарящ за закрилата на индустриалната собственост на изложението.
В това удостоверение се декларира, че промишленият дизайн е вложен или приложен спрямо продукта и че е изложен на изложението, като се посочва датата на откриване на изложението и, когато първото излагане на показ на продукта не съвпада с датата на откриване на изложението, се посочва датата на това първо излагане на показ. Удостоверението се придружава от надлежно заверен от този орган знак за действителното изложение на продукта.
2. Ако заявителят желае да претендира приоритет за изложение след регистрирането на заявката, в едномесечен срок, считано от датата на подаване на заявката, се представя декларацията за приоритет, в която се посочва наименованието на изложението и датата на това първо изложение на продукта, в който е вложен или спрямо който е приложен промишленият дизайн. Посочванията и доказателствата, посочени в параграф 1, се представят в Службата в тримесечен срок от получаването на декларацията за приоритет.
Член 10
Разглеждане на изискванията за дата на подаване и на формалните изисквания
1. Службата нотифицира заявителя, че дата за подаване на заявка не може да се даде, ако заявката не съдържа:
|
а) |
искане за регистрация на промишлен дизайн на Общността; |
|
б) |
информация, установяваща самоличността на заявителя; |
|
в) |
представяне на промишления дизайн съгласно член 4, параграф 1, букви г) и д) или, в зависимост от случая, на мостра. |
2. Ако нередностите, посочени в параграф 1, са отстранени в двумесечен срок, считано от получаването на известието, датата на подаването на заявка се определя спрямо датата, на която са отстранени нередностите.
Ако нередностите не са отстранени преди да изтече крайният срок, заявката не се разглежда като заявка за промишлен дизайн на Общността. Всички внесени такси се възстановяват.
3. Службата приканва заявителя да отстрани установените нередности в срок, определен от нея, ако, въпреки че е била определена дата за подаване на заявка, при разглеждането се установява, че:
|
а) |
не са спазени изискванията, изброени в членове 1, 2, 4 и 5, или другите формални изисквания за заявката, предвидени в Регламент (ЕО) № 6/2002, или в настоящия регламент; |
|
б) |
пълната сума от таксите, платими съгласно член 6, параграф 1, във връзка с Регламент (ЕО) № 2246/2002 на Комисията (2), не е получена от Службата; |
|
в) |
когато се претендира приоритет съгласно членове 8 и 9, в самата заявка или в едномесечен срок, считано от датата на регистрирането, не са съобразени другите изисквания, предвидени в тези членове; |
|
г) |
в случая на множествена заявка продуктите, в които промишлените дизайни се предназначени да се вложат или спрямо които са предназначени да се приложат, се включват към повече от един клас по Класификацията от Локарно. |
В частност, Службата следва да покани заявителя да плати дължимите такси в двумесечен срок, считано от датата на известието, заедно с таксите за закъсняло плащане, предвидени в член 107, параграф 2, букви а) до г) от Регламент (ЕО) № 6/2002 и съгласно условията, установени в Регламент (ЕО) № 2246/2002.
В случай на нередностите, посочени в първа алинея, буква г), Службата приканва заявителя да раздели множествената заявка с цел да обезпечи съответствието с изискванията на член 2, параграф 2. Тя приканва също заявителя да плати общата сума от такси за всички заявки, която се получава в резултат от разделянето на множествената заявка, в рамките на срок, който тя посочва.
След като заявителят се съобрази с искането да раздели заявката в рамките на посочения срок, датата на регистрирането на останалите заявки или заявления е датата на подаване, дадена на първоначално подадената множествена заявка.
4. Ако нередностите, определени в параграф 3, букви а) и г), не са отстранени преди да изтече определеният срок, Службата отхвърля заявката.
5. Ако платимите съгласно член 6, параграф 1, букви а) и б) такси не са внесени преди да изтече определеният срок, Службата отхвърля заявката.
6. Ако допълнителните такси, платими съгласно член 6, параграф 1, букви в) или г) такси за множествени заявки, не са внесени или не са внесени в пълен размер преди да изтече определеният срок, Службата отхвърля заявката по отношение на всички допълнителни промишлени дизайни, които не се покриват от внесената сума.
Поради липсата на критерии за определяне кои промишлени дизайни се планира да бъдат покрити, Службата подрежда промишлените дизайни по реда на номерата, в който същите са представени в съответствие с член 2, параграф 4. Службата отхвърля заявката по отношение на съдържащите се в нея промишлени дизайни, за които не са внесени допълнителни такси или не са внесени в пълен размер.
7. Ако нередностите, посочени в параграф 3, буква в), не са отстранени преди да изтече определеният срок, правото на приоритет на заявката се губи.
8. Ако някоя от нередностите, посочени в параграф 3 не е отстранена преди изтичането на посочения срок и ако тази нередност засяга само някои от промишлените дизайни, които се съдържат в множествена заявка, Службата отхвърля заявката или отказва правото на приоритет само по отношение на тези промишлени дизайни.
Член 11
Разглеждане на основанията за отказ за регистриране
1. Ако в съответствие с член 47 от Регламент (ЕО) № 6/2002 Службата прецени в течение на разглеждането съгласно член 10 от настоящия регламент, че промишленият дизайн, за който се търси закрила, не съответства на определението за промишлен дизайн, дадено в член 3, буква а) от Регламент (ЕО) № 6/2002, или че промишленият дизайн противоречи на обществения ред или на добрите нрави, Службата информира заявителя, че промишленият дизайн не подлежи на регистриране, като посочва мотивите за отказа за регистрация.
2. Службата определя срок, в рамките на който заявителят може да представи своите възражения, да оттегли своята заявка или да внесе в нея поправки, като измени представянето на промишления дизайн, при условие че се запази идентичността на промишления дизайн.
3. Ако заявителят не успее да се съобрази с мотивите за отказа за регистриране в рамките на определения срок, Службата отхвърля заявката. В случай че тези мотиви се отнасят само за отделни промишлени дизайни, съдържащи се в множествена заявка, Службата отхвърля заявката само по отношение на тези промишлени дизайни.
Член 12
Оттегляне или промени в заявката
1. Заявителят може по всяко време да оттегли заявката за промишлен дизайн на Общността или, в случая на множествена заявка, да оттегли някои от дизайните, които се съдържат в заявката.
2. По искане на заявителя могат да се поправят само името, адресът на заявителя, грешки в надписите или при копирането, както и явни грешки и при условие че тези поправки не променят представянето на промишления дизайн.
3. Заявление за поправки в заявката, съгласно параграф 2, включва:
|
а) |
регистрационният номер на заявката; |
|
б) |
името и адресът на заявителя в съответствие с член 1, параграф 1, буква б); |
|
в) |
ако заявителят е посочил свой представител, името и служебният адрес на представителя в съответствие с член 1, параграф 1, буква д); |
|
г) |
обозначаване на елемента от заявката, в който трябва да се нанесе поправката, и на същия елемент в поправения си вариант. |
4. Ако изискванията за поправка в заявката не са изпълнени, Службата уведомява заявителя за нередностите. В случай че нередностите не са отстранен в рамките на определения от Службата срок, Службата отхвърля заявлението за поправки.
5. За поправка на един и същи елемент в две и повече заявки, подадени от един и същи заявител, може да се подаде само едно заявление за поправка.
6. Параграфи 2 до 5 се прилагат mutatis mutandis по отношение на заявления за поправка на името или служебния адрес на представител, посочен от заявителя.
ГЛАВА II
ПРОЦЕДУРА ПО РЕГИСТРИРАНЕ
Член 13
Регистриране на промишлен дизайн
1. Ако заявката отговаря на изискванията, посочени в член 48 от Регламент (ЕО) № 6/2002, промишленият дизайн, който се съдържа в тази заявка, и посочванията и сведенията, предвидени в член 69, параграф 2 от настоящия регламент, се вписват в Регистъра.
2. Ако заявката съдържа искане за отлагане на публикуването съгласно член 50 от Регламент (ЕО) № 6/2002, този факт и датата на изтичане на периода на отлагането се отразяват.
3. Сумите, платими съгласно член 6, параграф 1, не се възстановяват дори и в случай, че промишленият дизайн, за който се подава заявката, не е регистриран.
Член 14
Публикуване на регистрацията
1. Регистрацията на промишления дизайн се публикува в Бюлетин на промишлените дизайни на Общността.
2. При условията на параграф 3 публикуването на регистрацията съдържа:
|
а) |
името и адреса на притежателя на промишлен дизайн на Общността (по-долу наричан „притежател“); |
|
б) |
в зависимост от случая, името и служебния адрес на посочения от притежателя представител, различен от представителя, който спада към член 77, параграф 3, първа алинея от Регламент (ЕО) № 6/2002; в случай че на един и същ служебен адрес се намира повече от един представител, се публикуват единствено името и служебният адрес на първия поред представител, като след името се изписват думите „и др.“; в случай, че са налице двама или повече представители с различни служебни адреси, се публикува единствено служебният адрес съгласно член 1, параграф 1, буква д) от настоящия регламент; а когато е посочена асоциация от представители съгласно член 62, параграф 9, се публикуват само наименованието и адресът на асоциацията; |
|
в) |
представянето на промишления дизайн съгласно член 4; когато представянето е цветно, публикацията също е цветна; |
|
г) |
в зависимост от случая, посочване, че за подновяването е подадено описание в съответствие с член 1, параграф 2, буква а); |
|
д) |
означение на продуктите, в които се възнамерява да бъде вложен или спрямо които се планира да се прилага промишленият дизайн, предшестван от броя съответни класове и подкласове по Класификацията от Локарно, и групирани по съответния начин; |
|
е) |
ако е приложимо, името на автора или авторския екип на промишления дизайн; |
|
ж) |
датата на регистриране и регистрационния номер, както и, в случая на множествена заявка, регистрационния номер на всеки промишлен дизайн; |
|
з) |
ако е приложимо, подробности по искането за приоритет съгласно член 42 от Регламент (ЕО) № 6/2002; |
|
и) |
в зависимост от случая, данни от претендиране за приоритет на изложение съгласно член 44 от Регламент (ЕО) № 6/2002; |
|
й) |
датата и регистрационния номер и датата на публикуване на регистрацията; |
|
к) |
езика, на който е извършено регистрирането, както и втория език, посочен от заявителя съгласно член 98, параграф 2 от Регламент (ЕО) № 6/2002. |
3. В случай, че заявление съдържа искане за отлагане на публикуването съгласно член 50 от Регламент (ЕО) № 6/2002 на Съвета, в Бюлетин на промишлените дизайни на Общността се публикува бележка за отлагането на публикуването заедно с името на притежателя, името на представителя, ако има такъв, датата на подаване и регистрирането, както и регистрационният номер на заявката. Не се публикуват нито представянето на промишления дизайн, нито каквито и да било подробности, които да разкриват външния му вид.
Член 15
Отлагане на публикуването
1. Когато в заявката се съдържа искане за отлагане на публикуването, съгласно член 50 от Регламент (ЕО) № 6/2002, притежателят е длъжен, заедно с искането или най-късно три месеца преди да изтече тридесетмесечният срок на отлагане, да изпълни следното:
|
а) |
да заплати таксата за публикуване, посочена в член 6, параграф 1, буква б); |
|
б) |
в случай на множествена заявка да заплати допълнителните такси за публикуване, посочени в член 6, параграф 1, буква г); |
|
в) |
в случаите, когато представяне на промишления дизайн е заменено с мостра съгласно член 5, да депозира представянето на промишления дизайн в съответствие с член 4. Това се отнася за всички промишлени дизайни, които се съдържат в множествена заявка, за които се изисква публикуване; |
|
г) |
в случай на множествена заявка да посочи ясно кой от дизайните, съдържащи се в него, трябва да бъде публикуван и кои от дизайните трябва да бъдат оставени, или в случай, че срокът на отлагане не е изтекъл, за кои промишлени дизайни отлагането трябва да се продължи. |
Когато притежателят подава искане за публикуване преди изтичането на тридесетмесечния срок на отлагане, същият е длъжен поне три месеца преди исканата дата на публикуване да изпълни изискванията, предвидени в първа алинея, букви а) до г).
2. Ако притежателят не изпълни изискванията, предвидени в параграф 1, букви в) или г), Службата го приканва да отстрани нередностите в рамките на определения срок, който в никакъв случай не може да продължава след изтичането на тридесетмесечния срок на отлагане.
3. Ако притежателят не отстрани нередностите, посочени в параграф 2, в рамките на определения срок:
|
а) |
се приема, че регистрираният промишлен дизайн на Общността не е произвел от самото начало ефектите, изброени в Регламент (ЕО) № 6/2002; |
|
б) |
ако притежателят е поискал по-ранна дата на публикуване съгласно предвиденото в параграф 1, втора алинея, приема се, че искането не е регистрирано. |
4. Ако притежателят не успее да внесе таксите, споменати в параграф 1, букви а) или б), Службата го приканва да внесе тези такси заедно с таксите, дължими за забавено плащане, предвидени в член 107, параграф 2, букви б) или г) от Регламент (ЕО) № 6/2002, и съгласно условията, посочени в Регламент (ЕО) № 2246/2002 на Комисията, в рамките на определен срок, който в никакъв случай не може да изтича след тридесетмесечния срок на отлагане.
Ако не е извършено никакво плащане в рамките на този срок, Службата уведомява притежателя, че се приема, че регистрираният промишлен дизайн на Общността не е произвел от самото начало ефектите, изброени в Регламент (ЕО) № 6/2002.
Ако по отношение на множествена заявка е извършено плащане в рамките на посочения срок, но това плащане не е достатъчно да покрие всички суми, платими съгласно параграф 1, букви а) и б), а също и дължимата такса за забавено плащане, приема се, че всички промишлени дизайни, по отношение на които не са внесени такси, не са произвели от самото начало ефектите, изброени в Регламент (ЕО) № 6/2002.
Освен ако не е ясно за кои промишлени дизайни се предполага, че ще бъдат покрити от плащането и при липсата на други критерии за определяне кои промишлени дизайни са обхванати от плащането, Службата взима промишлените дизайни по реда на техните номера на представяне в съответствие с член 2, параграф 4.
Всички промишлени дизайни, за които не е внесена допълнителна такса за публикуване или не е внесена в пълен размер, заедно с дължимата такса за забавено плащане, се приемат, че от самото начало не са имали последиците, изброени в Регламент (ЕО) № 6/2002.
Член 16
Публикуване след срока на отлагане
1. Ако притежателят е изпълнил изискванията, посочени в член 15, службата, след изтичане на срока за отлагане или в случай на искане за по-ранна дата на публикуване, доколкото това е технически възможно:
|
а) |
публикува регистрирания промишлен дизайн на Общността в Бюлетин на промишлените дизайни на Общността с поясненията, изложени в член 14, параграф 2, заедно с посочване на факта, че в заявката се е съдържало искане за отлагане на публикуването съгласно член 50 от Регламент (ЕО) № 6/2002, както и, в зависимост от случая, че е регистрирана мостра в съответствие с член 5 от настоящия регламент; |
|
б) |
отваря за публична инспекция всяко досие, свързано с промишления дизайн; |
|
в) |
отваря за публична инспекция всички вписвания в Регистъра, включително всички вписвания, изключени от инспекция съгласно член 73. |
2. Ако се прилага член 15, параграф 4, действията, споменати в параграф 1 от настоящия член, не се извършват по отношение на онези промишлени дизайни, съдържащи се в множествена заявка, за които се счита, че от самото начало не са произвели ефектите, изброени в Регламент (ЕО) № 6/2002.
Член 17
Удостоверение за регистрация
1. След публикуването Службата издава на притежателя удостоверение за регистрация, което съдържа данните от Регистъра съгласно член 69, параграф 2, и декларация, че тези данни действително са вписани в Регистъра.
2. Притежателят може да изиска заверени или незаверени копия от удостоверението за регистрация да му се изпратят срещу заплащане на такса.
Член 18
Поддържане на промишления дизайн в изменен вид
1. Ако в съответствие с член 25, параграф 6 от Регламент (ЕО) № 6/2002 регистриран промишлен дизайн на Общността се поддържа в изменен вид, промишленият дизайн на Общността в изменения си вид се вписва в Регистъра и се публикува в Бюлетин на промишлените дизайни на Общността.
2. Поддържането на промишлен дизайн в изменен вид може да включва частичен отказ от права, който да не надхвърля 100 думи, от страна на притежателя или вписване в Регистъра на промишлените дизайни на Общността на съдебно решение или решение на службата, което постановява частична недействителност на правото върху промишления дизайн.
Член 19
Промяна в името или адреса на притежателя или на неговия регистриран представител
1. Промяна в името или адреса на притежателя, която не е последица от прехвърляне на регистриран промишлен дизайн, се вписва в Регистъра по искане на притежателя.
2. Искането за промяна на името или адреса на притежателя следва да съдържа следното:
|
а) |
регистрационния номер на промишления дизайн; |
|
б) |
името и адреса на притежателя, както са вписани в Регистъра. Ако притежателят е получил идентификационен номер от Службата, достатъчно е да се посочи този номер заедно с името на притежателя; |
|
в) |
означение на променените име и адрес на притежателя в съответствие с член 1, параграф 1, буква б); |
|
г) |
ако притежателят е посочил представител, името и служебния адрес на представителя в съответствие с член 1, параграф 1, буква д). |
3. За искането, посочено в параграф 2, не се заплаща такса.
4. При промяна на името или адреса по отношение на две или повече заявления на един и същи притежател може да се подаде само едно заявление.
5. Ако изискванията, посочени в параграфи 1 и 2, не са изпълнени, Службата съобщава на заявителя за нередностите.
Ако нередностите не са отстранени в рамките на посочения от Службата срок, Службата отхвърля молбата.
6. Параграфи 1—5 се прилагат mutatis mutandis за името или адреса на регистрирания представител.
7. Параграфи 1—6 се прилагат mutatis mutandis за заявленията за промишления дизайн на Общността. Промените се вписват в досиетата, свързани със заявленията за промишления дизайн на Общността, съхранявани в службата.
Член 20
Поправка на грешки и неточности в Регистъра и в публикацията на регистрацията
Когато регистрацията на промишлен дизайн съдържа грешка или неточност, за които е виновна службата, Службата коригира грешката или неточността служебно или по искане на притежателя.
Ако заявлението е подадено от притежателя, се прилага член 19 mutatis mutandis . Такса за това заявление не се заплаща.
Службата публикува направените поправки съгласно настоящия член.
ГЛАВА III
ПОДНОВЯВАНЕ НА РЕГИСТРАЦИЯТА
Член 21
Нотифициране за изтичане на регистрацията
Най-малко шест месеца преди изтичане на регистрацията, Службата информира притежателя и всяко лице, което притежава права, вписани в Регистъра, включително лицензия по отношение на промишлен дизайн на Общността, че регистрацията приближава момента на изтичане. Липсата на нотификация не засяга изтичането на регистрацията.
Член 22
Подновяване на регистрацията
1. Заявката за подновяване на регистрацията следва да съдържа:
|
а) |
ако заявката е подадена от притежателя, се посочват неговото име и адрес в съответствие с член 1, параграф 1, буква б); |
|
б) |
ако заявката е подадена от лице, изрично упълномощено от притежателя за това, се посочват името и адресът на същото лице и доказателство, че лицето е упълномощено да подаде заявката; |
|
в) |
ако заявителят е назначил представител, името и служебния адрес на представителя в съответствие с член 1, параграф 1, буква д); |
|
г) |
регистрационният номер; |
|
д) |
в зависимост от случая, означение, че за подновяването е подадена заявка, включващо всички промишлени дизайни, покрити от множествена заявка или, ако заявлението за подновяване не обхваща всички тези промишлени дизайни, се посочват онези промишлени дизайни, за които се подава заявка за подновяване. |
2. Таксите, дължими съгласно член 13 от Регламент (ЕО) № 6/2002 за подновяване на регистрацията, са следните:
|
а) |
такса за подновяване, която, в случаите, когато от множествена заявка са обхванати няколко промишлени дизайна, следва да е пропорционална на броя промишлени дизайни, обхванати от подновяването; |
|
б) |
в зависимост от случая, допълнителната такса за забавено плащане на таксата за подновяване или закъсняло подаване на заявката за подновяване, съгласно член 13 от Регламент (ЕО) № 6/2002. |
3. Когато заявка за подновяване е подадена в сроковете, посочени в член 13, параграф 3 от Регламент (ЕО) № 6/2002, но не са изпълнени другите условия за подновяването, предвидени в същия член 13, както и в настоящия регламент, Службата инфоримира заявителя за тези нередности.
Ако заявката е подадена от лице, изрично упълномощено от притежателя за това, притежателят на промишления дизайн получава копие от нотификацията.
4. Когато не е подадена заявка за подновяване или е подадена след изтичането на срока, предвиден съгласно член 13, параграф 3, второто изречение от Регламент (ЕО) № 6/2002, или в случай че таксите не са внесени или са внесени след изтичане на съответния срок, определен от службата, Службата констатира, че регистрацията е изтекла и нотифицира за това притежателя, и, в зависимост от случая, заявителя на подновяването и лицето, вписано в Регистъра като лице с права върху промишления дизайн.
В случай на множествена регистрация, когато внесените такси са недостатъчни да покрият всички промишлени дизайни, за които е подадена заявка за подновяване, това определяне се прави само след като Службата установи за кои дизайни се предполага, че ще бъдат обхванати от внесената сума.
Поради липсата на други критерии за определяне за кои промишлени дизайни следва да се покрият, Службата подрежда промишлените дизайни по реда на номерата, в който те са представени в съответствие с член 2, параграф 4.
Службата констатира, че регистрацията е изтекла по отношение на всички промишлени дизайни, за които таксите за подновяване не са внесени или не са внесени в пълен размер.
5. Ако направената съгласно параграф 4 констатация е окончателна, Службата заличава промишления дизайн от Регистъра считано от деня, следващ този, в който изтича съществуващата регистрация.
6. Ако таксите за подновяване, предвидени съгласно параграф 2, са внесени, но регистрацията не е подновена, внесените такси се възстановяват.
ГЛАВА IV
ПРЕХВЪРЛЯНЕ, ЛИЦЕНЗИИ И ДРУГИ ПРАВА, ПРОМЕНИ
Член 23
Прехвърляне
1. Заявка за регистриране на прехвърляне съгласно Регламент (ЕО) № 6/2002 следва да съдържа:
|
а) |
регистрационния номер на промишления дизайн на Общността; |
|
б) |
подробни данни за новия притежател съгласно член 1, параграф 1, буква б); |
|
в) |
когато не всички промишлени дизайни, обхванати от множествена заявка, са включени в прехвърлянето, подробни данни за регистрираните промишлени дизайни, за които се отнася прехвърлянето; |
|
г) |
документи, надлежно установяващи прехвърлянето; |
2. Заявката може да съдържа, в зависимост от случая, името и служебния адрес на представителя на новия притежател в съответствие с член 1, параграф 1, буква д).
3. Заявката се счита за подадено само след внасянето на предвидената такса. В случай, че таксата не е внесена или не е внесена в пълен размер, Службата нотифицира заявителя по съответния начин.
4. Следното представлява достатъчно доказателство за прехвърляне съгласно параграф 1, буква г):
|
а) |
заявката за регистриране на прехвърлянето е подписана от регистрирания притежател или негов представител и от правоприемника или от негов представител; или |
|
б) |
заявката, ако е подадена от правоприемника, се придружава от декларация, подписана от регистрирания притежател или негов представител, че той е съгласен с регистрирането на правоприемника; или |
|
в) |
заявката се придружава от формуляр или документ за извършено прехвърляне, подписан от регистрирания притежател или негов представител, както и от правоприемника му или негов представител. |
5. Когато не са спазени условията, приложими по отношение на регистрацията на прехвърляне, Службата нотифицира заявителя за нередностите.
В случай, че нередностите не бъдат отстранени в рамките на срока, определен от службата, последната отхвърля заявката за регистриране на прехвърлянето.
6. За два или повече регистрирани вида промишлен дизайн на Общността може да се подаде само едно заявление за регистрация на прехвърляне, при условие че регистрираният притежател и правоприемникът са едни и същи във всеки от случаите.
7. Параграфи от 1 до 6 се прилагат, с необходимите изменения, за прехвърлянето на заявки за регистрирани промишлени дизайни на Общността. Прехвърлянето се вписва в поддържаните от Службата досиета, които се отнасят за заявката за промишлен дизайн на Общността.
Член 24
Регистриране на лицензии и други права
1. Член 23, параграф 1, букви а), б) и в) и член 23, параграфи 2, 3, 5 и 6 се прилагат mutatis mutandis за регистрацията на предоставянето или прехвърлянето на лицензия, за регистрацията на учредяване или прехвърляне на вещно право върху регистриран промишлен дизайн на Общността, както и за регистрацията на мерките на принудително изпълнение. Въпреки това, когато регистриран промишлен дизайн на Общността е предмет на дело за обявяване в несъстоятелност, искът на компетентния национален орган за вписване в Регистъра в този смисъл, не е основание за внасяне на такса.
В случай на множествена регистрация всеки регистриран промишлен дизайн на Общността може, отделно от останалите, да бъде предмет на лицензия, на вещно право, на принудително изпълнение или на производство за обявяване в несъстоятелност.
2. Когато регистриран промишлен дизайн на Общността е получил лицензия за част от Общността или за ограничен срок, в заявлението за регистрация на лицензията следва да се посочи тази част от Общността или срокът, за който е предоставена лицензията.
3. Когато условията, приложими за регистрирането на лицензии или други права, предвидени в членове 29, 30 или 32 от Регламент (ЕО) № 6/2002, в параграф 1 от настоящия член и в други приложими членове от настоящия регламент, не са изпълнени, Службата нотифицира заявителя за нередностите.
Ако нередностите не са отстранени в срока, определен от службата, последната отхвърля заявката за регистрация.
4. Параграфи 1, 2 и 3 се прилагат mutatis mutandis за лицензите и другите права, засягащи заявките за регистрирани промишлении дизайни на Общността. Лицензиите, вещните права и мерките на принудително изпълнение се вписват в поддържаните от Службата досиета, които се отнасят за заявката за промишлен дизайн на Общността.
5. Искането за неизключителна лицензия съгласно член 16, параграф 2 от Регламент (ЕО) № 6/2002 се подава в рамките на тримесечен срок, считано от датата на вписване на новия притежател в Регистъра.
Член 25
Специални разпоредби за регистриране на лицензия
1. Лицензия по отношение на регистриран промишлен дизайн на Общността се вписва в Регистъра като изключителна лицензия, ако притежателят на промишления дизайн или на лицензията поиска това.
2. Лицензия по отношение на регистриран промишлен дизайн на Общността се вписва в Регистъра като сублицензия, когато е предоставена от притежателя на лицензията, чиято лицензия е вписана в Регистъра.
3. Лицензия по отношение на регистриран промишлен дизайн на Общността се вписва в Регистъра като териториално ограничена лицензия, ако е предоставена за част от Общността.
4. Лицензия по отношение на регистриран промишлен дизайн на Общността се вписва в Регистъра като временна лицензия, ако е предоставена за ограничен срок.
Член 26
Заличаване или промяна в регистрацията на лицензии и други права
1. Регистрацията, извършена съгласно член 24, се заличава по искане на едно от заинтересованите лица.
2. Искането трябва да съдържа:
|
а) |
регистрационен номер на регистрирания промишлен дизайн на Общността или, в случай на множествена заявка, номерът на всеки промишлен дизайн; и |
|
б) |
уточнения относно правото, чиято регистрация трябва да бъде заличена. |
3. Молбата за заличаване регистрацията на лицензия или на друго право се счита за подадена само след внасянето на изискваната такса.
Ако таксата не е внесена или не е внесена в пълен размер, Службата нотифицира заявителя по съответния начин. Искането от страна на компетентна национална власт за заличаване на регистрация, когато регистриран промишлен дизайн на Общността е предмет на дело за обявяване в неплатежоспособност, не е основание за плащането на такса.
4. Заявката се придружава от документи, показващи, че регистрираното право е отпаднало, или декларация на лицензията или на притежателя на друго право в смисъл, че същият е съгласен за заличаване на регистрацията.
5. Когато изискванията за заличаване на регистрацията не са изпълнени, Службата нотифицира заявителя за нередностите. Ако нередностите не бъдат отстранени в рамките на срока, определен от службата, същата отхвърля заявката за заличаване на регистрацията.
6. Параграфи 1, 2, 4 и 5 се прилагат mutatis mutandis за заявката за промяна на регистрацията, извършена съгласно член 24.
7. Параграфи от 1 до 6 се прилагат mutatis mutandis за вписванията, извършени съгласно член 24, параграф 4.
ГЛАВА V
ОТКАЗ И НЕДЕЙСТВИТЕЛНОСТ
Член 27
Отказ
1. Декларацията за отказ съгласно член 51 от Регламент (ЕО) № 6/2002 трябва да съдържа:
|
а) |
регистрационния номер на регистрирания промишлен дизайн на Общността; |
|
б) |
името и адреса на притежателя в съответствие с член 1, параграф 1, буква б); |
|
в) |
ако е посочен представител, името и адреса на представителя в съответствие с член 1, параграф 1, буква д); |
|
г) |
ако е обявен отказ само за някои промишлени дизайни, част от множествена регистрация, се посочват промишлените дизайни, за които е обявен отказът или промишлените дизайни, за които регистрацията трябва да остане; |
|
д) |
ако, съгласно член 51, параграф 3 от Регламент (ЕО) № 6/2002, относно регистрирания промишлен дизайн на Общността е заявен частичен отказ, представяне на променения промишлен дизайн в съответствие с член 4 от настоящия регламент. |
2. Ако в Регистъра е вписано право на трето лице върху регистриран промишлен дизайн на Общността, достатъчно доказателство за неговото съгласие за отказа е декларация за съгласие за отказа, подписана от притежателя на това право или от негов представител.
Ако е регистрирана лицензия, отказът от промишления дизайн се регистрира три месеца след датата, на която притежателят представи пред Службата документи, че е информирал лицензополучателя за своето намерение за отказ от промишления дизайн. Ако притежателят представи пред Службата доказателства, че преди изтичането на този период лицензополучателят е дал своето съгласие, отказът се регистрира незабавно.
3. Ако правото върху регистриран промишлен дизайн на Общността е предмет на иск пред съда съгласно член 15 от Регламент (ЕО) № 6/2002, декларация за отказ, подписана от лицето, което претендира това право, или от негов представител, се счита за достатъчно доказателство за неговото съгласие за отказа.
4. Ако не са изпълнени изискванията, приложими за отказа, Службата съобщава нередностите на заявителя . Ако недостатъците не бъдат отстранени в рамките на срока, определен от Службата, Службата отхвърля вписването на отказа в Регистъра.
Член 28
Искане за обявяване на недействителност
1. Искането, подадено в Службата за обявяване за недействителност съгласно член 52 от Регламент (ЕО) № 6/2002 съдържа:
|
а) |
по отношение на регистриран промишлен дизайн на Общността, за който се иска обявяване на недействителност:
|
|
б) |
по отношение на основанията, посочени в искането:
|
|
в) |
по отношение на заявителя:
|
2. За искането се внася такса, предвидена в член 52, параграф 2 от Регламент (ЕО) № 6/2002.
3. Службата информира притежателя относно регистрирането на искането за обявяване на недействителност.
Член 29
Езици, които се използват при производство за недействителност
1. Искането за обявяване на недействителност се подава на езика, на който се води производството съгласно член 98, параграф 4 от Регламент (ЕО) № 6/2002.
2. Когато езикът на производството не е езика, използван при подаване на заявката, а притежателят е попълнил своите съображения на езика, използван при подаването на заявката, Службата се разпорежда за превод на тези съображения на езика на производството.
3. В срок от три години, считано от датата, определена в съответствие с член 111, параграф 2 от Регламент (ЕО) № 6/2002, Комисията представя на Комитета, посочен в член 109 от Регламент (ЕО) № 6/2002, доклад за прилагането на параграф 2 от настоящия член и, ако това е подходящо, предложения за определяне на лимит за разноските, които прави Службата в това отношение, както е предвидено в член 98, параграф 4, четвърта алинея от Регламент (ЕО) № 6/2002.
4. Комисията може да реши да представи доклада и евентуалните предложения, упоменати в параграф 3, на по-ранна дата, а Комитетът ги обсъжда като приоритетен въпрос, ако разпоредбите в параграф 2 водят до несъответстващи разноски.
5. Когато доказателствата в подкрепа на искането не са представени на езика на производството за недействителност, заявителят прилага превод на тези доказателства на този език в срок от два месеца от представянето на това доказателство.
6. Когато лицето, искащо обявяване на недействителност, или притежателят информира Службата, в рамките на два месеца от получаването на съобщението, посочено в член 31, параграф 1 от настоящия регламент, че те са се споразумели да използват различен език в съответствие с член 98, параграф 5 от Регламент (ЕО) № 6/2002, лицето, искащо обявяване на недействителност, е длъжно, ако искането не е подадено на този език, да представи превод на искането на този език в срок от един месец от горепосочената дата.
Член 30
Отхвърляне на искането за обявяване на недействителност поради недопустимост
1. Ако Службата намери, че искането за обявяване на недействителност не отговаря на член 52 от Регламент (ЕО) № 6/2002, на член 28, параграф 1 от същия регламент или на която и да било друга разпоредба от Регламент (ЕО) № 6/2002 или от настоящия регламент, Службата информира заявителя по съответния начин и го приканва да отстрани нередностите в рамките на посочен от нея срок.
В случай че недостатъците не бъдат отстранени в рамките на посочения срок, Службата отхвърля искането поради недопустимост.
2. Когато Службата констатира, че изискваните такси не са внесени, същата информира заявителя по съответния начин и го информира също, че искането ще се счита за неподадено, ако изискваните такси не бъдат внесени в рамките на посочения срок.
В случай че изискваните такси бъдат внесени след изтичането на посочения срок, те се връщат на заявителя.
3. Всяко решение за отхвърляне на искане за обявяване на недействителност съгласно параграф 1 се съобщава на заявителя.
Когато в съответствие с параграф 2 искането се счита за неподадено, на заявителя се съобщава по съответния начин.
Член 31
Разглеждане на искането за обявяване на недействителност
1. В случай, че Службата не отхвърли искане за обявяване на недействителност в съответствие с член 30, същата съобщава това решение на притежателя и изисква от него да представи своите обяснения в рамките на срок, определен от Службата.
2. В случай, че притежателят не представи никакви обяснения, Службата може да основе своето решение за недействителността върху предишни доказателства.
3. За всички обяснения, представени от притежателя, се съобщава на заявителя, към когото Службата може да се обърне за отговор в рамките на срок, какъвто определи Службата.
4. Всички съобщения съгласно член 53, точка 2 от Регламент (ЕО) № 6/2002, както и всички представени в тази връзка възражения, се изпращат на заинтересованите страни.
5. Службата може да покани страните да разрешат на спора по взаимно съгласие.
Член 32
Множество искания за обявяване на недействителност
1. Когато са подадени няколко искания за обявяване на недействителност за един и същи регистриран промишлен дизайн на Общността, Службата може да ги разгледа в едно общо производство.
Службата може впоследствие да реши да не ги разглежда по този начин.
2. Ако в хода на предварителното разглеждане на едно или повече искания се установи, че регистрираният промишлен дизайн на Общността е недействителен, Службата може да прекрати другите производства за недействителност.
Службата информира другите заявители за всяко релевантно решение, произнесено в хода на това производство.
3. След като решение, с което се обявява недействителността на промишлен дизайн, стане окончателно, исканията, по отношение на които е прекратено производството в съответствие с параграф 2, се считат за прекратени и заинтересованите заявители съответно се информират за това. Прекратяването на производствата поради този факт представлява липса на основание за произнасяне за целите на член 70, параграф 4 от Регламент (ЕО) № 6/2002.
4. Службата възстановява 50 % от таксата за недействителност, посочена в член 52, параграф 2 от Регламент (ЕО) № 6/2002, която се внася от всеки заявител, чието заявление се счита за прекратено в съответствие с параграфи 1, 2 и 3 от настоящия член.
Член 33
Участие в производството на предполагаем нарушител
Когато, съгласно член 54 от Регламент (ЕО) № 6/2002, предполагаем нарушител, поиска да участва в производството, той се подчинява на съответните разпоредби на членове 28, 29 и 30 от настоящия регламент, и по-специално трябва да представи мотивирано становище и да внесе таксата, посочена в член 52, параграф 2 от Регламент (ЕО) № 6/2002.
ГЛАВА VI
ОБЖАЛВАНЕ
Член 34
Съдържание на жалбата
1. Жалбата трябва да съдържа:
|
а) |
името и адреса на лицето, което подава жалба в съответствие с член 1, параграф 1, буква б); |
|
б) |
ако лицето, което подава жалбата, е назначило представител, името и служебния адрес на представителя в съответствие с член 1, параграф 1, буква д); |
|
в) |
изявление, което посочва атакуваното решение, и степента, в която иска решението да бъде изменено или отменено. |
2. Жалбата се подава на езика на производството, в което е било взето атакуваното решение.
Член 35
Отхвърляне на жалбата поради недопустимост
1. Ако жалбата не отговаря на изискванията на членове 55, 56 и 57 от Регламент (ЕО) № 6/2002 и член 34, параграф 1, буква в) и член 34, параграф 2 от настоящия регламент, отделението по жалбите я отхвърля поради недопустимост, освен ако всички недостатъци бъдат отстранени преди изтичането на съответния срок, определен в член 57 от Регламент (ЕО) № 6/2002.
2. Ако отделението по жалбите установи, че жалбата не отговаря на други разпоредби от Регламент (ЕО) № 6/2002 или на други разпоредби от настоящия регламент и, в частност, на член 34, параграф 1, букви а) и б), Комисията съответно информира лицето, което е подало жалбата, и изисква от него да отстрани установените нередности в рамките на срок, който същата определя. Ако нередностите не бъдат отстранени в определения срок, отделението по жалбите отхвърля жалбата поради недопустимост.
3. Ако таксата за жалбата е внесена след изтичането на срока за подаване на жалбата съгласно член 57 от Регламент (ЕО) № 6/2002, жалбата се счита, че не е била подадена, а таксата за жалбата се възстановява на лицето, което я е подало.
Член 36
Разглеждане на жалбите
1. Освен ако не е предвидено друго, разпоредбите относно производството пред инстанцията, постановила решението, срещу което е подадена жалбата, се прилагат mutatis mutandis за апелативното производство.
2. Решението на отделението по жалбите трябва да съдържа:
|
а) |
изявление, че е произнесено от отделението; |
|
б) |
датата, на която е взето решението; |
|
в) |
имената на председателя и другите членове на отделението по жалбите, взели участие в приемането на решението; |
|
г) |
името на компетентен служител от регистратурата; |
|
д) |
имената на страните и на техните представители; |
|
е) |
списък на въпросите, по които предстои да се вземе решение; |
|
ж) |
резюме на фактите; |
|
з) |
мотиви на решението; |
|
и) |
разпореждане на отделението по жалбите, включително в зависимост от случая, решението за поделяне на разноските. |
3. Решението се подписва от председателя и другите членове на отделението по жалбите, както и от служителя от регистратурата на отделението по жалбите.
Член 37
Възстановяване на таксата за жалбата
Възстановяването на таксите за жалбата се определя в случай на междинно преразглеждане или когато отделението по жалбите прецени, че жалбата е допустима, ако това възстановяване е справедливо поради съществено процесуално нарушение. В случай на междинно преразглеждане, възстановяването се определя от инстанцията, чието решение се атакува, а в останалите случаи, от отделението по жалбите.
ГЛАВА VII
РЕШЕНИЯ И СЪОБЩЕНИЯ НА СЛУЖБАТА
Член 38
Форма на решенията
1. Решенията на Службата са в писмена форма и в тях се посочват мотивите, на които те се основават.
Когато производството пред Службата е устно, решенията могат да се издават също в устна форма. Впоследствие на страните се връчва решението в писмен вид.
2. Решенията на Службата, които подлежат на обжалване, се придружават от писмено съобщение, в което се посочва, че актът на обжалване задължително се подава в Службата в писмен вид в рамките на двумесечен срок, считано от датата на съобщаване на решението, подлежащо на обжалване. Съобщенията също следва да насочват вниманието на страните към разпоредбите на членове 55, 56 и 57 от Регламент (ЕО) № 6/2002.
Страните не могат да се позовават на липсата на съобщение за възможността за производството за обжалване.
Член 39
Поправка на грешки в решенията
В решенията на Службата могат да се поправят само езикови грешки, грешки при транскрибиране и явни грешки. Същите се поправят от инстанцията, която е взела решението, служебно или по искане на една от заинтересуваните страни.
Член 40
Констатиране на загуба на права
1. В случай, че Службата открие, че загубата на какъвто и да било вид право произтича от Регламент (ЕО) № 6/2002 или от настоящия регламент, без да е било взето каквото и да било решение, Службата съобщава това на заинтересованото лице в съответствие с член 66 от Регламент (ЕО) № 6/2002 и обръща вниманието на това лице към средствата за обжалване, посочени в параграф 2 от настоящия член.
2. В случай, че заинтересованото лице счете, че констатацията на Службата не е основателна, лицето може, в срок от два месеца от съобщението, посочено в параграф 1, да поиска решение по въпроса от Службата.
Такова решение се взема само ако Службата не е съгласна с лицето, което внася искането; в противен случай Службата изменя констатацията си и информира лицето, внесло искането за вземане на решение.
Член 41
Подпис, име, печат
1. Във всяко решение, съобщение или известие от Службата се посочва отделът или поделението на службата, както и името или имената на отговорния служител или служители. Те се подписват от служителя или служителите или вместо подпис на тях се поставя отпечатан или разпечатан щемпел на службата.
2. Председателят на Службата може да разреши за идентифицирането на отдела или поделението на Службата, както и на отговорния служител или служители, да се използват други средства, освен печата на Службата, когато решенията, съобщенията и известията се изпращат по факс или по друго съобщително средство.
ГЛАВА VIII
УСТНИ ПРОИЗВОДСТВА И СЪБИРАНЕ НА ДОКАЗАТЕЛСТВА
Член 42
Призовки за устни производства
1. Страните се призовават за устното производство, предвидено в член 64 от Регламент (ЕО) № 6/2002, като тяхното внимание са насочва към параграф 3 от настоящия член. В призовката се посочва срок за явяване след един месец най-малко, освен ако страните не се съгласят на по-кратък срок.
2. При призоваването Службата обръща внимание върху точките, които, по нейно мнение, следва да бъдат обсъдени с цел да бъде взето решение.
3. Ако редовно призована страна не се яви в устното производство пред Службата като призована, производството може да продължи без тази страна.
Член 43
Разследване от страна на Службата
1. Ако Службата прецени, че е необходимо да се изслушат устните показания на страните, на свидетели и експерти или да се извърши проверка, тя взима решение за тази цел, като обявява средствата, с които възнамерява да получи доказателства, свързаните със случая факти, които предстои да се докажат, както и датата, часа и мястото на разглеждането или проверката по случая.
Ако страна поиска устни показания от свидетелите и експертите, с решение на Службата се определя срока, в рамките на който страната, подала искането, трябва да доведе до знанието на Службата имената и адресите на свидетелите и експертите, които страната иска да бъдат изслушани.
2. Срокът за явяване, даден в призовката на страна, свидетел или експерт за даване на показания, трябва да бъде най-малко един месец, освен ако те не се съгласят на по-кратък срок.
Призовката трябва да съдържа:
|
а) |
извлечение от решението, посочено в параграф 1, първа алинея, в което се посочват в частност датата, часът и мястото на нареденото разглеждане и изложение на фактите, по отношение на които предстои да бъдат изслушани страните, свидетелите и експертите; |
|
б) |
имената на страните по делото и посочване на правата, на които свидетелите или експертите могат да се позовават, съгласно член 45, параграфи 2 до 5. |
Член 44
Назначаване на експерти
1. Службата решава в каква форма трябва да се представи докладът, съставен от експерта, който тя посочи.
2. Условията по отношение на експерта включват следното:
|
а) |
точно описание на задачата на експерта; |
|
б) |
срок за представяне доклада на експерта; |
|
в) |
имената на страните по делото; |
|
г) |
посочване на правата, на които експертът може да се позовава, съгласно член 45, параграфи 2, 3 и 4. |
3. На страните се представя копие от всеки писмен документ.
4. Страните могат да искат отвод на експерт на основание на некомпетентност или на същите основания, на каквито може да се иска отвод на разследващо лице или на член на отдела или на отделението по жалбите съгласно член 132, параграфи 1 и 3 от Регламент (ЕО) № 40/94 (3). Съответният отдел на Службата се произнася по възражението.
Член 45
Разноски по вземането на показания
1. Вземането на показанията от Службата може да зависи от внасяне на депозит в Службата от страната, която е внесла искане за вземане на показания, като сумата на депозита се определя въз основа на оценка на разноските.
2. Свидетелите и експертите, които са призовани от Службата, и се явяват пред нея, имат право на възстановяване на пътни и дневни разноски в разумни граници. Службата може да им даде аванс за тези разноски. Първото изречение се прилага също и за свидетелите и експертите, които се явяват пред Службата без да са получили призовка от нея и които са дали устни показания като свидетели или като експерти.
3. Свидетелите, които имат право на възстановяване на разноски съгласно параграф 2, имат право също и на подходящо обезщетение за пропуснати приходи, като на експертите се дава правото да получават хонорари за техните услуги. Тези плащания се правят в полза на свидетелите и експертите, след като те са приключили със задълженията или задачите си в случаите, когато такива свидетели или експерти са призовани от Службата по нейна собствена инициатива.
4. Сумите и авансите за разноски, които предстои да се изплатят съгласно параграфи 1, 2 и 3, се определят от председателя на Службата и се публикуват в Официален вестник на Службата.
Сумите се изчисляват на същата база, на каквато се изчисляват и обезщетенията и възнагражденията, които служебните лица получават, а именно по степени А 4 до А 8, както е определено в Правилника за длъжностните лица на Европейските общности и в приложение VII към този Правилник.
5. Окончателното задължение по дължими или изплатени суми съгласно параграфи 1—4 е на:
|
а) |
Службата, когато Службата, по собствена инициатива, е сметнала за необходимо да изслуша устните показания на свидетели или експерти; или |
|
б) |
засегнатата страна, когато тази страна е внесла искане за вземане на устни показания от свидетели или експерти, с изключение на решението за пропорционално разпределяне и определяне на разноските съгласно членове 70 и 71 от Регламент (ЕО) № 6/2002 и член 79 от настоящия регламент. |
Страната, посочена в буква б) от първа алинея, възстановява на Службата всички надлежно изплатени авансово суми.
Член 46
Протокол от устни производства и от вземане на доказателства
1. При провеждане на устни производства, както и при събиране на доказателства се съставя протокол, съдържащ по-важното от заседанието и при взетите устни показания, съответните изказвания, направени от страните, свидетелствата на страните, свидетелите и експертите и резултатите от всяка проверка.
2. Протоколът от показанията на свидетел, експерт или страна се прочита на глас или се предава на лицето, така че лицето да може да го прегледа. В протокола се отбелязва, че тази формалност е спазена и че лицето, което е дало показания, е одобрило протокола. Когато не е получено одобрението на лицето, следва да се отбележат неговите възражения.
3. Протоколът се подписва от заетото лице, който го е съставило, и от заетото лице, който е провело устното заседание или е събрало доказателства.
4. На страните се осигурява по едно копие от протокола.
5. По искане на страните Службата им осигурява транскрипция на записите от устните разисквания, написан на машина текст от същите или друга машинно-четивна форма.
Издаването на транскрипция на тези записи се заплаща, като плащането покрива разноските, направени от Службата за изработването на такава транскрипция. Сумата, която следва да се заплати, се определя от председателя на Службата.
ГЛАВА IХ
НОТИФИЦИРАНЕ
Член 47
Общи разпоредби относно нотифицирането
1. При провеждане на производство в Службата всички нотификации, които предстои да се направят от Службата, приема формата на оригинален документ, на негово копие, заверено или подпечатано от Службата с щемпел или на компютърна разпечатка с такъв щемпел. Копията на документи, издавани от самите страни, не изискват такава заверка.
2. Нотификацията се прави:
|
а) |
по пощата в съответствие с член 48; |
|
б) |
чрез връчване в съответствие с член 49; |
|
в) |
чрез поставяне в пощенска кутия в Службата в съответствие с член 50; |
|
г) |
по факс или чрез други технически средства в съответствие с член 51; |
|
д) |
чрез публично нотифициране в съответствие с член 52. |
Член 48
Нотифициране по пощата
1. Решенията, свързани със срокове като жалби, призовки и други документи, определени от председателя на Службата, се отправят във вид на препоръчани писма с обратна разписка.
Решенията и съобщенията с други срокове се изпращат като препоръчани писма, освен ако председателят на Службата не определи друго.
Всички други съобщения се изпращат като обикновена поща.
2. Нотификациите за адресати, които нямат нито домашен адрес, нито основен служебен адрес, нито са се установили в Общността, и които не са посочили представител в съответствие с член 77, параграф 2 от Регламент (ЕО) № 6/2002, се отправят като обикновена поща, като документът, за който се изисква нотифициране, се адресира до последния известен на Службата адрес на адресата.
Нотифицирането се счита за извършено след като писмото се отправи по пощата.
3. Когато нотифицирането се осъществява с препоръчано писмо със или без обратна разписка, се счита за доставено на адресата на десетия ден след изпращането му по пощата, освен ако писмото не е успяло да достигне до адресата или е достигнало до него на по-късна дата.
В случай на спор, Службата е тази, която установява дали писмото е достигнало до мястото на предназначението си, както и датата, на която писмото е достигнало до адресата, според случая.
4. Нотифицирането с препоръчано писмо със или без обратна разписка се счита за осъществено дори и адресатът да откаже да приеме писмото.
5. До размера, който не се покрива от параграфи 1—4, се прилага законът на държавата, на чиято територия е направено нотифицирането.
Член 49
Нотифициране чрез връчване
Нотифицирането може да се осъществи на територията на Службата чрез връчване на документа на адресата, който е длъжен при връчването да потвърди получаването му.
Член 50
Нотифициране чрез депозиране в пощенска кутия в Службата
Нотифицирането на адресати, за които са предвидени пощенски кутии в Службата, може да се осъществи чрез поставяне на документа в пощенската кутия. В досиетата се внася писмено известие за поставянето. На документа следва да се означи датата на поставянето. Нотифицирането се счита за осъществено на петия ден след поставянето на документа в пощенската кутия в Службата.
Член 51
Нотифициране по факс и чрез други технически средства
1. Нотифицирането по факс се осъществява, като се изпраща или оригиналът, или копие от документа, за който предстои да се изпрати нотифициране, както е предвидено в член 47, параграф 1. Детайлите по изпращането се определят от председателят на Службата.
2. Условията и редът на нотифициране чрез други технически средства се определят от председателя на Службата.
Член 52
Публично нотифициране
1. В случай, че адресът на адресата не може да бъде установен или ако нотифицирането съгласно член 48, параграф 1 се е оказало невъзможно след втори опит от страна на службата, нотифицирането се осъществява чрез публично разпространено известие.
Това известие се публикува най-малко в Бюлетин на промишлените дизайни на Общността.
2. Председателят на Службата определя как публичното известие следва да се разпространи и определя началото на срока с продължителност от един месец, след изтичането на който за документа се счита, че е бил нотифициран.
Член 53
Нотифициране на представителите
1. В случай, че е посочен представител или когато заявителят, който е посочен на първо място в обща заявка, се счита за общ представител съгласно член 61, параграф 1, нотификациите се адресират до посочения или общия представител.
2. В случай, че са посочени няколко представители за една-единствена заинтересувана страна, е достатъчно да се изпрати нотифициране само до един от тях, освен ако не е указан специфичен служебен адрес съгласно член 1, параграф 1, буква д).
3. В случай, че няколко заинтересувани страни са посочили един общ представител, е достатъчно да се изпрати нотифициране до общия представител за един-единствен документ.
Член 54
Нередности при нотифициране
Когато даден документ е достигнал до адресата и, в случай че Службата не е в състояние да докаже, че същият е надлежно нотифициран или ако не са спазени разпоредбите, свързани с нотифицирането му, за документа се счита, че е изпратено известие на датата, посочено от Службата за дата на получаване.
Член 55
Нотифициране за документи, в случай че са налице няколко страни
За документите, които се издават от страните и съдържат съществени предложения, както и за декларация за изтегляне на съществено предложение, се изпраща известие до другите страни, за да са в течение относно това. Нотифицирането може да се отмени там, където документът не съдържа нови искания и въпросът е готов за вземане на решение.
ГЛАВА Х
СРОКОВЕ
Член 56
Изчисляване на сроковете
1. Сроковете се определят в години, месеци, седмици или дни.
2. Срокът започва да тече, считано от деня, следващ датата, на която е настъпило съответното събитие, като събитието представлява или процесуално действие или изтичане на друг срок. Когато процесуалното действие е нотифициране, съответното събитие представлява получаването на предизвестения документ, освен ако не е предвидено друго.
3. Когато срокът се изразява като една година или известен брой години, същият изтича през съответната следваща година, през месеца със същото име, на деня със същото число, каквото е числото и на месеца и деня, на който е настъпило съответното събитие. Когато съответният месец няма такова число, срокът изтича на последния ден от този месец.
4. Когато срокът се изразява като един месец или известен брой месеци, същият изтича на съответния следващ месец, на деня със същото число, каквото е числото на деня, на който е настъпило съответното събитие. Когато денят, на който е настъпило съответното събитие, е бил последният ден на даден месец, или когато съответният следващ месец няма такова число, срокът изтича на последния ден от този месец.
5. Когато срокът се изразява като една седмица или известен брой седмици, същият изтича на съответната следваща седмица, на деня със същото наименование, каквото е наименованието на деня, на който е настъпило съответното събитие.
Член 57
Продължаване на сроковете
1. Когато в Регламент (ЕО) № 6/2002 или в настоящия регламент се предвижда срока да се определя от Службата, този срок трябва, тогава когато заинтересованата страна е на адрес или основното място на дейност на същата страна, или седалището на същата страна е в рамките на Общността, да бъде не по-малко от един месец, или, тогава когато тези условия не са изпълнени, не по-малко от два месеца и не повече от шест месеца.
Когато се окаже подходящо с оглед на условията, Службата може да продължи определения срок, в случай че такова продължение бъде поискано от засегнатата страна въз основа на искане, представено преди изтичането на първоначалния срок от време.
2. Когато са налице две или повече страни, Службата може да разпореди продължаване на даден срок от време по споразумение на другите страни.
Член 58
Изтичане на срокове в особени случаи
1. В случай, че даден срок от време изтича на деня, на който Службата не е отворена за получаване на документи или на който, поради причини, различни от посочените в параграф 2, обикновената поща не се доставя по местоположението на Службата, срокът от време се продължава до първия следващ ден, на който Службата е отворена за получаване на документи и на който се доставя обикновена поща.
Дните, в които Службата не е отворена за получаване на документи, се определят от председателя на Службата преди началото на всяка календарна година.
2. В случай че даден срок от време изтича на ден, на който е налице общо прекъсване или последващо объркване на пощенските пратки в държава-членка, или между държава-членка и Службата, срокът се продължава до първия ден, следващ края на периода на прекъсването или объркването, за страни, чийто адрес или регистрирано място на дейност са разположени в заинтересованата държава или които са посочили представители с място на дейност в същата държава.
В случай че заинтересованата държава-членка е държавата, в която е разположена Службата, първата алинея се прилага за всички страни.
Срокът, посочен в първа алинея, се определя от председателя на Службата.
3. Параграфи 1 и 2 се прилагат mutatis mutandis за сроковете, предвидени от Регламент (ЕО) № 6/2002 или от настоящия регламент, в случай че предстои да се осъществяват транзакции с компетентната власт по смисъла на член 35, параграф 1, букви б) и в) от Регламент (ЕО) № 6/2002.
4. В случай че извънредно обстоятелство, като например природно бедствие или стачка, прекъсне или внесе объркване в нормалното функциониране на Службата, в резултат на което съобщителните връзки от Службата до страните, отнасящи се до изтичането на даден срок от време, бъдат забавени, онези актове, които следва да бъдат изпълнени в рамките на такъв срок, могат все пак да бъдат изпълнени и да са действителни в рамките на един месец от нотифицирането за закъсняващата съобщителна връзка.
Датата на началото и на края на всяко такова прекъсване или объркване се определя от председателя на Службата.
ГЛАВА ХI
СПИРАНЕ НА ПРОИЗВОДСТВОТО И ОТКАЗ ЗА ЗАПОЧВАНЕ НА ПРИНУДИТЕЛНО ПРОИЗВОСТВО ПО ВЪЗСТАНОВЯВАНЕ НА ДЪЛЖИМИ СУМИ
Член 59
Спиране на производство
1. Производството пред Службата се спира в следните случаи:
|
а) |
в случай на смърт или недееспособност на заявителя или на притежателя на регистриран промишлен дизайн на Общността, или на лицето, упълномощено по националното право да действа от негово име; |
|
б) |
в случай, че заявителят или притежателят на регистриран промишлен дизайн на Общността, в резултат на съдебно производство срещу негова собственост, е възпрепятстван поради юридически съображения да продължи производства пред Службата; |
|
в) |
в случай на смърт или недееспособност на представител на заявителя или на притежателя на регистриран промишлен дизайн на Общността или възпрепятстване на същия поради юридически причини в резултат на съдебно производство срещу негова собственост да продължи производството пред Службата. |
Доколкото посочените в първа алинея, буква а) събития не засягат упълномощаването на представителя, посочен съгласно член 78 от Регламент (ЕО) № 6/2002, производството се прекъсват само след заявление, внесено от такъв представител.
2. Когато в случаите, посочени в параграф 1, първа алинея, буква а) и б), Службата е била информирана за самоличността на лицето, упълномощено да продължи производството пред службата, Службата съобщава на това лице и на всички заинтересувани трети лица, че производството се възобновява, считано от датата, която се определя от Службата.
3. В случая, посочен в параграф 1, буква в), производството се възобновява, тогава когато Службата бъде информирана за посочването на нов представител на заявителя или когато Службата е информирала другите страни относно съобщението за посочването на нов представител на притежателя на промишления дизайн.
В случай, че три месеца след началото на прекъсването на производството Службата не е информирана за посочването на нов представител, същата съобщава този факт на заявителя или на притежателя на регистрирания промишлен дизайн на Общността:
|
а) |
когато член 77, параграф 2 от Регламент (ЕО) № 6/2002 е приложим, че заявката за промишлен дизайн на Общността се счита за изтеглена, ако информацията не се представи в рамките на два месеца след нотифицирането за това съобщение; или |
|
б) |
когато член 77, параграф 2 от Регламент (ЕО) № 6/2002 не е приложим, че производството със заявителя или притежателя ще бъде възобновено, считано от датата на нотифицирането за това съобщение. |
4. Сроковете, различни от срока за внасяне на такси за подновяване, в сила по отношение на заявителя или притежателя на регистриран промишлен дизайн на Общността от датата на прекъсване на производството, започва отново, считано от деня, на който е възобновено производството.
Член 60
Отказ за започване на принудително производство по възстановяване на дължими суми
Председателят на Службата може да откаже производство по принудително възстановяване на всяка дължима сума там, където сумата, която следва да се възстанови, е минимална или където такова възстановяване е твърде несигурно.
ГЛАВА ХII
ПРЕДСТАВИТЕЛСТВО
Член 61
Посочване на общ представител
1. В случай, че е налице повече от един заявител, а в заявката за регистриран промишлен дизайн на Общността не се посочва име на общ представител, заявителят, чието име фигурира в заявката като първо, се счита, че е общ представител.
При все това, в случай че един от заявителите има задължението да посочи професионален представител, за такъв представител ще се счита общият представител, освен ако заявителят, чието име фигурира в заявката като първо, също е посочил професионален представител.
Първата и втората алинеи се прилагат mutatis mutandis по отношение на трети страни, които действат заедно при внасяне на заяв ление за декларация за недействителност, както и по отношение на съвместни притежатели на регистриран промишлен дизайн на Общността.
2. В случай, че в течение на производството се осъществи прехвърляне в полза на повече от едно лице, а тези лица не са посочили общ представител, се прилага параграф 1.
В случай, че такова заявление не е възможно, Службата изисква от такива лица да посочат общ представител в рамките на два месеца. Ако на това искане не бъде отговорено, общ представител се посочва от Службата.
Член 62
Пълномощни
1. Практикуващите юристи и професионалните представители, регистрирани в списъците, водени в Службата съгласно член 78, параграф 1, букви б) или в) от Регламент (ЕО) № 6/2002, могат да подадат в Службата подписано пълномощно за включване в досиетата.
Такова пълномощно се подава, в случай че Службата изрично го изисква или, когато са налице няколко страни по производството, в които представителят действа пред Службата, това се изисква изрично от една от страните.
2. Служителите, действащи от името на физически или юридически лица по силата на член 77, параграф 3 от Регламент (ЕО) № 6/2002, подават в Службата подписано пълномощно за включване в досиетата.
3. Пълномощното може да бъде вписано на всеки от официалните езици на Общността. Същото може да покрива едно или повече заявления или регистрирани промишлени дизайни на Общността, или може да бъде във формата на генерално пълномощно, по силата на което на представителя се дава възможност да действа по отношение на всички производства пред Службата, по които лицето, което е издало пълномощното, е страна.
4. Когато, съгласно параграфи 1 или 2, следва да се регистрира пълномощно, Службата определя точно срок от време, в рамките на който такова пълномощно следва да се регистрира. В случай, че пълномощното не бъде регистрирано в срок, производството продължава с представеното лице. За всички процесуални действия, различни от регистриране на пълномощно, които се предприемат от представителя, се счита, че не са били предприети, ако представеното лице не ги одобри. Заявката по смисъла на член 77, параграф 2 от Регламент (ЕО) № 6/2002 остава незасегнато.
5. Параграфи 1, 2 и 3 се прилагат към документ, с който се оттегля пълномощно.
6. Всеки представител, който е спрял да бъде упълномощен, продължава да бъде разглеждан като представител, докато за прекратяването на неговото пълномощно бъде съобщено на Службата.
7. Освен ако не са налице някакви разпоредби за противното, съдържащи се в дадено пълномощно, същото не се прекратява пред Службата след смъртта на лицето, което го е издало.
8. Когато една и съща страна е посочила няколко представители, те могат независимо от каквито и да било разпоредби за противното, съдържащи се в техните пълномощни, да действат както колективно, така и индивидуално.
9. Пълномощното на асоциация на представители се счита за пълномощно на всеки представител, който може да докаже, че практикува в рамките на тази асоциация.
Член 63
Представителство
Всяко нотифициране или друго съобщение, адресирано от Службата до надлежно упълномощен представител, следва да има същата сила каквато би имало, в случай че е адресирано до представеното лице.
Всяко съобщение, адресирано до Службата от надлежно упълномощен представител, следва да има същата сила каквато би имало, в случай че би изхождало от представеното лице.
Член 64
Изменения на специалния списък на професионалните представители по въпросите за промишлените дизайни
1. Вписването на професионален представител в специалния списък по въпросите за промишлените дизайни, така както е изяснено в член 78, параграф 4 от Регламент (ЕО) № 6/2002, се заличава по искане на професионалния представител.
2. Вписването на професионален представител се заличава автоматично:
|
а) |
в случай на смърт или юридическа неспособност на професионалния представител; |
|
б) |
в случаите, в които професионалният представител е престанал да бъде гражданин на държава-членка, освен ако председателят на Службата е разпоредил освобождаване по силата на член 78, параграф 6, буква а) от Регламент (ЕО) № 6/2002; |
|
в) |
в случаите, в които мястото на търговска дейност или местоработата на професионалния представител е престанало да бъде на територията на Общността; |
|
г) |
в случаите, в които професионалният представител е престанал да притежава правата и пълномощията, споменати в член 78, параграф 4, буква в), първото изречение от Регламент (ЕО) № 6/2002. |
3. Вписването на професионален представител се отменя от самата Служба в случаите, в които неговите пълномощия да представя физически или юридически лица пред Службата за промишлени дизайни на Бенелюкс или централната служба за индустриална собственост на държава-членка, така както е посочено в член 78, параграф 4, буква в), първото изречение от Регламент (ЕО) № 6/2002, са били отменени.
4. Лице, чието вписване е било заличено след внасяне на искане съгласно член 78, параграф 5 от Регламент (ЕО) № 6/2002, се възстановява в списъка на професионалните представители, ако условията за заличаването са престанали да съществуват.
5. Службата за промишлени дизайни на Бенелюкс и централните служби за индустриална собственост на заинтересуваните държави-членки, там където те са осведомени за това, незабавно да информират Службата за всякакви събития, свързани с изложеното в параграфи 2 и 3.
6. Измененията и допълненията в специалния списък на професионалните представители по въпросите на промишлените дизайни се публикуват в Официален вестник на Службата.
ГЛАВА ХIII
ПИСМЕНИ СЪОБЩЕНИЯ И ФОРМУЛЯРИ
Член 65
Съобщения в писмен вид или чрез други средства
1. С изключение на изложеното в параграф 2, заявленията за регистрация на промишлен дизайн на Общността, както и всяко друго заявление или декларация, предвидени съгласно Регламент (ЕО) № 6/2002, наред с всички останали съобщения, адресирани до Службата, се представят, както следва:
|
а) |
чрез представяне на подписан оригинал на въпросния документ на Службата по пощата, с лично доставяне или по всякакъв друг начин; приложенията към представените документи не е необходимо да бъдат подписани; |
|
б) |
чрез изпращане на подписан оригинал по факса в съответствие с член 66; или |
|
в) |
чрез изпращане съдържанието на съобщението с електронни средства в съответствие с член 67. |
2. Когато заявителят се ползва от възможността, предвидена от член 36, параграф 1, буква в) от Регламент (ЕО) № 6/2002 за регистриране на мостра от промишления дизайн, заявката и мострата се представят в Службата в една пощенска пратка във формата, предписана в параграф 1, буква а) от същия член. В случай, че заявката и мострата или мострите, в случай на серийно заявление, не бъдат представени в една пощенска пратка, Службата няма да даде дата за вписване, докато не се получи и последният артикул по силата на член 10, параграф 1 от настоящия регламент.
Член 66
Съобщение по факс
1. Когато заявление за регистрация на промишлен дизайн на Общността се представя по факс, а в заявката се съдържа репродукция на промишления дизайн съгласно член 4, параграф 1, която не отговаря на изискванията на този член, изискваната репродукция, подходяща за регистрация и публикация, се представя в Службата в съответствие с член 65, параграф 1, буква а).
Когато репродукцията се получава в Службата в рамките на срок от един месец, считано от датата на получаване на факса, заявката се счита за получено в Службата на датата, на която е получен факсът.
Когато репродукцията е получена в Службата след изтичането на този срок от време, заявката се счита, че е било получено от Службата на датата, на която е получена репродукцията.
2. Когато съобщение, получено по факса, е непълно или нечетливо, или когато Службата има основателни съмнения по отношение на точността на предаването, Службата информира изпращача по съответния начин и го подканя, в рамките на срок от време, който се определя от Службата, да изпрати отново оригинала по факс или да представи оригинала в съответствие с член 65, параграф 1, буква а).
В случаите, в които на такова искане е отговорено в рамките на определения срок от време, датата на получаване на повторно изпратения факс или на оригинала се счита за дата на получаване на първоначалното съобщение, при положение че, когато недостатъкът засяга даването на дата за регистриране на заявление за вписване на промишлен дизайн на Общността, се прилагат разпоредбите относно датата за регистриране.
В случаите, в които на такова искане не е отговорено в рамките на определения срок от време, съобщението се счита, че не е получено.
3. Всяко съобщение, изпратено до Службата по факс, се счита за надлежно подписано, ако изображението на подписа личи на разпечатката, получена на факса.
4. Председателят на Службата може да обуслови допълнителни изисквания за съобщителна връзка по факс, като например, каква техника да се използва, технически подробности по съобщителната връзка, както и методи за определяне на изпращача.
Член 67
Съобщителна връзка чрез електронни средства
1. Заявленията за регистриране на промишлен дизайн на Общността могат да се представят чрез електронни средства, включително представянето на промишления дизайн, и независимо от член 65, параграф 2 в случая с регистрирането на мостра.
Условията се определят от председателя на Службата.
2. Председателят на Службата определя изискванията за съобщение чрез електронни средства, като например, каква техника да се използва, технически подробности по съобщителната връзка, както и методи за определяне на изпращача.
3. Когато съобщението се изпраща чрез електронни средства, член 66, параграф 2 се прилага mutatis mutandis.
4. Когато съобщението е изпратено на Службата посредством електронно средство, означението на името на изпращача ще се счита за равносилно на подпис.
Член 68
Формуляри
1. Службата предоставя безплатно формуляри за целите на:
|
а) |
подаване на заявка за регистриран промишлен дизайн на Общността; |
|
б) |
внасяне нa заявка за коригиране на регистриран промишлен дизайн на Общността; |
|
в) |
внасяне нa заявка за регистриране на прехвърляне и формуляр за прехвърляне, както и документ за прехвърляне, посочен в член 23, параграф 4; |
|
г) |
внасяне нa заявка за регистриране на лицензия; |
|
д) |
внасяне нa заявка за подновяване на регистрация на регистриран промишлен дизайн на Общността; |
|
е) |
внасяне на заявка за обявяване на недействителност на регистриран промишлен дизайн на Общността; |
|
ж) |
внасяне нa заявление за връщане в първоначално състояние (restitutio in integrum); |
|
з) |
внасяне на жалба; |
|
и) |
упълномощаване на представител във вид на индивидуално пълномощно и във вид на генерално пълномощно. |
2. Службата може да предоставя и други формуляри за безплатно ползване.
3. Службата предоставя формулярите, изброени в параграфи 1 и 2, на всички официални езици на Общността.
4. Службата разполага формулярите в Службата за промишлени дизайни на Бенелюкс или в централните служби за промишлена интелектуална собственост на държавите-членки безплатно.
5. Службата предоставя и формуляри в машинно-четивна форма.
6. Страните в производството пред Службата следва да използват формулярите, предвидени от Службата, или копия от тези формуляри, или формуляри със същото съдържание и формат като на тези формуляри, например формулярите, създадени посредством електронна обработка на данните.
7. Формулярите могат да бъдат попълнени по такъв начин, че да позволяват автоматично влагане на съдържанието в компютър, например посредством разпознаване на символите или чрез сканиране.
ГЛАВА ХIV
ИНФОРМАЦИЯ ЗА ОБЩЕСТВЕНОСТТА
Член 69
Регистър за промишления дизайн на Общността
1. Регистърът може да се поддържа под формата на електронна база данни.
2. В Регистъра следва да се съдържат следните вписвания:
|
а) |
датата на регистриране на заявката; |
|
б) |
регистрационният номер на заявката и регистрационния номер на всеки отделен промишлен дизайн, включен в множествена заявка; |
|
в) |
датата на публикуване на регистрацията; |
|
г) |
името, адресът и националността на заявителя и държавата на местожителството му или където се намира неговото седалище; |
|
д) |
името и служебният адрес на представителя, различен от служител, действащ като представител в съответствие с член 77, параграф 3, първа алинея от Регламент (ЕО) № 6/2002; когато е налице повече от един представител, се вписва само името и адресът на първия поред представител, като след името е изписано „и др.“; а там, където е посочена асоциация от представители, се публикува само наименованието и адресът на асоциацията; |
|
е) |
представянето на промишления дизайн; |
|
ж) |
посочване на продуктите по техните названия, предшествано от номерата на класовете и подкласовете по Класификацията от Локарно и групирани по съответния подходящ начин; |
|
з) |
данни от внесени обжалвания за приоритет по силата на член 42 от Регламент (ЕО) № 6/2002; |
|
и) |
данни от претендиране за приоритет на изложение съгласно член 44 от Регламент (ЕО) № 6/2002; |
|
й) |
там, където е приложимо, се цитира проектантът или проектантското звено съгласно член 18 от Регламент (ЕО) № 6/2002, или становище относно това, че проектантът или проектантското звено се отказва/т от правото да бъдат цитирани; |
|
к) |
езикът, на който е регистрирана заявката, и вторият език, който заявителят е посочил в своето заявление съгласно член 98, параграф 2 от Регламент (ЕО) № 6/2002; |
|
л) |
датата на регистрация на промишления дизайн в Регистъра и регистрационният номер; |
|
м) |
посочване на всякакви искания за отлагане на публикация по силата на член 50, параграф 3 от Регламент (ЕО) № 6/2002, като се посочва датата на изтичане на периода на отлагането; |
|
н) |
посочване, че по силата на член 5 е била регистрирана мостра; |
|
о) |
посочване, че по силата на член 1, параграф 2, буква а) е било регистрирано описание. |
3. Допълнително към вписванията, изложени в параграф 2, в Регистъра следва да се съдържат следните вписвания, като за всяко се отбелязва датата на регистриране на такова вписване:
|
а) |
промени в името, адреса или националността на притежателя или на държавата, в която се намира местожителството или седалището на същия; |
|
б) |
промени в името и служебният адрес на представителя, различен от представителя, включен в член 77, параграф 3, първа алинея от Регламент (ЕО) № 6/2002; |
|
в) |
когато е посочен нов представител, името и служебният адрес на такъв представител; |
|
г) |
посочване, че множествена заявка или регистрация е разделена на отделни заявки или регистрации по силата на член 37, параграф 4 от Регламент (ЕО) № 6/2002; |
|
д) |
известието за всички изменения в даден промишлен дизайн съгласно член 25, параграф 6 от Регламент (ЕО) № 6/2002, включително, ако е приложимо, позоваване на направен отказ от права или съдебно решение или решение на Службата, в което се декларира частична недействителност на правото върху промишления дизайн, както и корекции на грешки и неточности съгласно член 20 от настоящия регламент; |
|
е) |
посочване, че производството по предоставяне на права е започнало по силата на член 15, параграф 1 от Регламент (ЕО) № 6/2002, в който се засяга производството по правата; |
|
ж) |
окончателно решение или друго прекратяване на производство съгласно член 15, параграф 4, буква б) от Регламент (ЕО) № 6/2002, относно производството по правата; |
|
з) |
промяна в собствеността съгласно член 15, параграф 4, буква в) от Регламент (ЕО) № 6/2002; |
|
и) |
прехвърляния съгласно член 28 от Регламент (ЕО) № 6/2002; |
|
й) |
създаване или прехвърляне на вещно право съгласно член 29 от Регламент (ЕО) № 6/2002 и естеството на вещното право; |
|
к) |
вземания за изпълнение по силата на член 30 от Регламент (ЕО) № 6/2002 и производство по несъстоятелност по силата на член 31 от настоящия регламент; |
|
л) |
даване или прехвърляне на лицензия съгласно член 16, параграф 2 или член 32 от Регламент (ЕО) № 6/2002 и, когато е приложимо, типа лицензия съгласно член 25 от настоящия регламент; |
|
м) |
подновяване на регистрация, съгласно член 13 от Регламент (ЕО) № 6/2002, и датата, от която е в сила; |
|
н) |
запис от решение за изтичане на регистрация; |
|
о) |
декларация за пълен или частичен отказ от притежателя съгласно член 51, параграфи 1 и 3 от Регламент (ЕО) № 6/2002; |
|
п) |
датата на подаване на заявката или на подаване на насрещно искане за обявяване на недействителност в съответствие със, съответно, член 52 или член 86, параграф 2 от Регламент (ЕО) № 6/2002; |
|
р) |
датата на подаване на заявката или подаване на насрещно искане за обявяване на недействителност или всякакво друго прекратяване на производство в съответствие със, съответно, член 53 или член 86, параграф 4 от Регламент (ЕО) № 6/2002; |
|
с) |
посочване съгласно член 50, параграф 4 от Регламент (ЕО) № 6/2002, че регистрираният промишлен дизайн на Общността се счита, че не е бил действителен от самото начало съгласно посоченото в този регламент; |
|
т) |
отменяне на представител, вписан съгласно параграф 2, буква д); |
|
у) |
изменяне или заличаване в Регистъра на положенията, упоменати в букви й), л) и м). |
4. Председателят на Службата може да определя, че въпроси, различни от онези, които са споменати в параграфи 2 и 3, следва да се вписват в Регистъра.
5. Притежателят се нотифицира при всяка промяна в Регистъра.
6. С изключение на член 73, Службата, при поискване, предоставя заверени или незаверени извлечения от Регистъра срещу заплащане на такса.
ГЛАВА ХV
БЮЛЕТИН И БАЗА ДАННИ НА ПРОМИШЛЕНИТЕ ДИЗАЙНИ НА ОБЩНОСТТА
Член 70
Бюлетин на промишлените дизайни на Общността
1. Службата определя честотата на публикуване на Бюлетин на промишлените дизайни на Общността и начина, по който ще се осъществява такова публикуване.
2. Без да се засягаат разпоредбите на член 50, параграф 2 от Регламент (ЕО) № 6/2002 и с изключение на членове 14 и 16 от настоящия регламент, отнасящи се до отлагане на публикация, в Бюлетин на промишлените дизайни на Общността следва да се съдържат публикации за регистрации и публикации за вписвания, направени в Регистъра, както и други подробности, свързани с регистрациите на промишлени дизайни, публикуването на които е предвидено от Регламент (ЕО) № 6/2002 или от настоящия регламент.
3. Когато подробности, публикуването на които е предписано в Регламент (ЕО) № 6/2002 или в настоящия регламент, са публикувани в Бюлетина за промишлените дизайни на Общността, датата на изданието, показана на Бюлетина, се счита за дата на публикуването на подробностите.
4. Информацията, чието публикуване е предвидено в членове 14 и 16, там, където е подходящо, следва да се публикува на всички официални езици на Общността.
Член 71
База данни
1. Службата поддържа електронна база данни с подробностите от заявления за регистрация на промишления дизайн на Общността и вписвания в Регистъра. Службата може, въпреки ограниченията, предписани от член 50, параграфи 2 и 3 от Регламент (ЕО) № 6/2002, да предостави съдържанието на тази база данни за директен достъп или на CD-ROM, или във всякаква друга машинно-четивна форма.
2. Председателят на Службата определя условията за достъп до базата данни и начина, по който съдържанието на тази база данни може да бъде предоставено в машинно-четивна форма, включително таксите за такива действия.
ГЛАВА ХVI
ПРОВЕРКА НА ДОСИЕТА И СЪХРАНЯВАНЕ НА ДОСИЕТА
Член 72
Части от досието, изключени от проверка
Частите от досието, които се изключват от проверка съгласно член 74, параграф 4 от Регламент (ЕО) № 6/2002, са както следва:
|
а) |
документи, свързани с изключване или възражение съгласно член 132 от Регламент (ЕО) № 40/94, като за разпоредбите от този член се счита, че за тази цел се прилагат като се внесат съответните изменения в регистрирания промишлен дизайн на Общността и в заявленията относно същите; |
|
б) |
проекторешения и мнения, както и всички останали вътрешни документи, използвани за изготвянето на решения и мнения; |
|
в) |
части от досието, относно които засегнатата страна е показала специален интерес да се съхраняват поверителни преди да бъде внесена заявката за проверка на досиетата, освен ако проверката на такава част от досието е оправдана от първостепенни законни интереси на страната, предизвикала проверката. |
Член 73
Проверка на Регистъра на промишлените дизайни на Общността
Когато регистрацията е предмет на отлагане на публикация съгласно член 50, параграф 1 от Регламент (ЕО) № 6/2002:
|
а) |
достъпът до Регистъра за лица, различни от притежателя, се ограничава до името на притежателя, името на всеки представител, датата на внасяне и регистриране, картотечния номер на заявката и посочването, че публикацията е отложена, освен в случаите, в които е внесено искане от страна на притежателя или негов представител; |
|
б) |
заверените или незаверените извадки от Регистъра съдържат единствено името на титуляра, името на който и да е представител, датата на представяне и регистриране, номер на преписката на заявлението и обозначението за отлагането на публикацията, с изключение на случаите, в които заявката е подадена от притежателя или представителя му. |
Член 74
Процедури за проверка на досиета
1. Проверката на досиета на регистрирани промишлени дизайни на Общността е или на оригиналния документ, или на копия на същия, или на техническите средства за съхраняване, в случай че досиетата се съхраняват по този начин.
Искането за проверка на досиетата не се счита за внесено, докато не бъде заплатена изискваната такса.
Начините за проверка се определят от председателя на Службата.
2. Когато проверката на досиетата е свързана със заявление за регистриран промишлен дизайн на Общността или с регистриран промишлен дизайн на Общността, който е предмет на отлагане на публикация, която, бидейки предмет на такова отлагане, е била отказана преди или при изтичането на този период от време или която, по силата на член 50, параграф 4 от Регламент (ЕО) № 6/2002, се счита, че от началото не е имала действието, посочено в този регламент, в искането следва да се съдържа посочване и доказателство за това, че:
|
а) |
заявителят или притежателят на промишлен дизайн на Общността е дал съгласие за проверката; или |
|
б) |
лицето, поискало проверката, е доказало правен интерес от проверката на досието, в частност там, където заявителят или притежателят на промишлен дизайн на Общността е заявил, че след като промишленият дизайн е регистриран, той ще се позове на правата по него срещу лицето, поискало проверката. |
3. Проверката на досиетата се осъществява на територията на Службата.
4. При поискване проверка на досиетата може да се осъществи посредством издаване на копия на документи от досието. За такива копия се заплаща такса.
5. При поискване Службата издава заверени или незаверени копия от заявката за регистриран промишлен дизайн на Общността или от онези документи от досието, от които могат да се направят копия, съгласно параграф 4, и след заплащане на такса.
Член 75
Съобщаване на информация, съдържаща се в досиетата
С изключение на ограниченията, предвидени в член 74 от Регламент (ЕО) № 6/2002 и членове 72 и 73 от настоящия регламент, Службата, при поискване, може да съобщи информация от всяко досие на промишлен дизайн на Общността, за което е внесена заявка или за регистриран промишлен дизайн на Общността, за която се заплаща такса.
При все това Службата може да поиска от заявителя да провери досието на място, ако счете, че това е целесъобразно с оглед на количеството информация, която да бъде предоставена.
Член 76
Съхранение на досиетата
1. Службата съхранява досиетата, свързани със заявленията за промишления дизайн на Общността и с регистрирани промишлени дизайни на Общността, за срок от време с продължителност поне пет години, считано от края на годината, през която:
|
а) |
заявката е отхвърлена или оттеглена; |
|
б) |
регистрацията на регистриран промишлен дизайн на Общността изтича окончателно; |
|
в) |
регистриран е пълен отказ от регистриран промишлен дизайн на Общността съгласно член 51 от Регламент (ЕО) № 6/2002; |
|
г) |
регистрираният промишлен дизайн на Общността е окончателно отстранен от Регистъра; |
|
д) |
регистрираният промишлен дизайн на Общността се счита, че не е имал действието, посочено в Регламент (ЕО) № 6/2002, съгласно член 50, параграф 4 от същия. |
2. Председателят на Службата определя начина, по който се съхраняват досиетата.
ГЛАВА ХVII
АДМИНИСТРАТИВНО СЪТРУДНИЧЕСТВО
Член 77
Обмяна на информация и съобщения между Службата и компетентните органи в държавите-членки
1. Службата и централните служби за индустриална собственост на държавите-членки и на Службата за промишлени дизайни на Бенелюкс, при поискване, си съобщават съответната актуална информация за регистрирането на заявления за регистрирани промишлени дизайни на Общността, промишлени дизайни от Бенелюкс или промишлени дизайни от съответните държави, както и информация относно производството, свързано с такива заявления и промишлени дизайни, регистрирани в резултат от това. Такива съобщения не следва да са предмет на ограниченията, предвидени в член 74 от Регламент (ЕО) № 6/2002.
2. Съобщенията между Службата и съдилищата или компетентните органи на държавите-членки, които възникват от приложението на Регламент (ЕО) № 6/2002 или на настоящия регламент, се осъществяват директно помежду им.
Такова съобщение може да се осъществява също и посредством централните служби за индустриална собственост на държавите-членки или Службата за промишлен дизайн на Бенелюкс.
3. Разноските по отношение на съобщенията съгласно параграфи 1 и 2 са за сметка на институцията, осъществяваща съобщенията, които са освободени от такси.
Член 78
Проверка на досиетата от или чрез съдилищата или властите на държавите-членки
1. Проверката на досиетата, свързани с промишления дизайн на Общността, за които са внесени искания или на регистрирани промишлени дизайни на Общността от страна на съдилища или компетентни власти на държавите-членки, при положение че е поискана в такава форма, е проверка на оригинални документи или на копия от същите. Член 74 не се прилага.
2. Съдилищата или прокуратурите на държавите-членки могат, в течение на производството пред тях, да отварят досиета или копия на същите, изпратени от Службата, за проверка от трети страни. Такава проверка е предмет на член 74 от Регламент (ЕО) № 6/2002.
3. Службата не взима никаква такса за проверки съгласно параграфи 1 и 2.
4. По време на изпращане на досиетата или на копия от същите до съдилищата или прокуратурите на държавите-членки Службата посочва ограниченията, на които проверката на досиета, свързани с промишления дизайн на Общността, за които са внесени заявления, или на регистрирани промишлени дизайни на Общността, е предмет по силата на член 74 от Регламент (ЕО) № 6/2002 и член 72 от настоящия регламент.
ГЛАВА ХVIII
РАЗНОСКИ
Член 79
Пропорционално разпределение и определяне на разноските
1. Пропорционалното разпределение на разноските, съгласно член 70, параграфи 1 и 2 от Регламент (ЕО) № 6/2002, се определя в решение по заявление за обявяване на недействителност на регистриран промишлен дизайн на Общността или в решение по апелативна жалба.
2. Пропорционалното разпределение на разноските съгласно член 70, параграфи 3 и 4 от Регламент (ЕО) № 6/2002 се определя в решение по разноските, което се приема от отдела по недействителност или от апелативната комисия.
3. Към искането за определяне на разноските, съгласно член 70, параграф 6, първото изречение от Регламент (ЕО) № 6/20021, се прилага сметка за разноските заедно с подкрепящи доказателства.
Искането е допустимо само ако решението, по отношение на което се изисква определяне на разноските, е окончателно. Разноските могат да бъдат определени, след като се установи тяхната правдоподобност.
4. Искането, съгласно член 70, параграф 6, второто изречение от Регламент (ЕО) № 6/20021, за преразглеждане на решение за вписване на определени разноски, в което се излагат основанията, на които се основава, следва да се регистрира в Службата в рамките на един месец, считано от датата на нотифицирането за одобряването на разноските.
Същото не се счита за вписано, докато не се плати таксата за преразглеждане сумата на разноските.
5. Отдела за обявяване на недействителност или апелативната комисия, според случая, взимат решения по искането, посочено в параграф 4, без устно производство.
6. Таксите, които се поемат от загубилата страна по силата на член 70, параграф 1 от Регламент (ЕО) № 6/2002, се ограничават до таксите, разноските за които са направени от другата страна за заявката за обявяване на недействителност, и/или по обжалването.
7. Съществените за производството и действително направените от спечелилата страна разноски се поемат от загубилата страна в съответствие с член 70, параграф 1 от Регламент (ЕО) № 6/2002 на основата на следните максимални стойности:
|
а) |
пътни разноски на едната страна за отиване и връщане между местожителството или мястото на стопанска дейност и мястото, където се провеждат заседанията или където се взимат показанията, както следва:
|
|
б) |
дневни за едната страна, равни на дневната издръжка за служебни лица, имащи степен от А 4 до А 8, съгласно изложеното в член 13 от приложение VII към Правилник за длъжностните лица на Европейските общности; |
|
в) |
пътни разноски на представителите по смисъла на член 78, параграф 1 от Регламент (ЕО) № 6/2002, както и на свидетелите и експертите, наред с разноските, предвидени в буква а); |
|
г) |
дневни на представителите по смисъла на член 78, параграф 1 от Регламент (ЕО) № 6/2002, както и на свидетелите и експертите, наред с разноските, предвидени в буква б); |
|
д) |
разноски, възникнали при взимането на показания под формата на разпитване на свидетели, мнения на експерти или проверка — до 300 EUR на производство; |
|
е) |
представителни разноски, по смисъла на член 78, параграф 1 от Регламент (ЕО) № 6/2002:
|
|
ж) |
там, където спечелилата страна е представена от повече от един представител, по смисъла на член 78, параграф 1 от Регламент (ЕО) № 6/2002, загубилата страна поема разноските, посочени в букви в), г) и e) само за едно такова лице; |
|
з) |
загубилата страна не е задължена да възмездява на успешната страна никакви разноски, разноски и такси освен онези, които са посочени в точките от а) до ж). |
Когато взимането на свидетелски показания в някое от производствата, засегнати в първа алинея, буква е), включва разпитването на свидетели, мнения на експерти или проверка, се дава допълнителна сума за представителни разноски до 600 EUR на производство.
ГЛАВА ХIХ
ЕЗИЦИ
Член 80
Заявления и декларации
Без да се засяга предвиденото в член 98, параграф 4 от Регламент (ЕО) № 6/2002:
|
а) |
всяко заявление или декларация, свързани със заявка за регистриран промишлен дизайн на Общността, могат да се вписват на езика, който е използван при регистриране на заявката или на втория език, посочен от заявителя в неговото заявление; |
|
б) |
всяко заявление или декларация, различно от искане за обявяване на недействителност по силата на член 52 от Регламент (ЕО) № 6/2002, или декларация за отказ по силата на член 51 от същия регламент, свързано с регистриран промишлен дизайн на Общността, може да бъде вписано на един от езиците на Службата; |
|
в) |
тогава, когато е използван някой от формулярите, предоставен от Службата по силата на член 68, такива формуляри могат да се използват на всеки от официалните езици на Общността, при условие че формулярът е попълнен на един от езиците на Службата, доколкото се отнася до текстовите елементи. |
Член 81
Писмено производство
1. Без да се засяга член 98, параграфи 3 и 5 от Регламент (ЕО) № 6/2002, и освен ако в настоящия регламент не е предвидено друго, при писменото производство пред Службата, дадена страна може да използва всеки от езиците на Службата.
В случай, че избраният език не е езикът на производството, страната представя превод на този език в рамките на едномесечен срок считано от датата на представяне на оригиналния документ.
Когато заявителят за регистриран промишлен дизайн на Общността и единствена страна по производство пред Службата, а езикът, използван за вписване на заявката за регистриран промишлен дизайн на Общността, не е един от езиците на Службата, може да се впише и превод на втория език, посочен от заявителя в неговото заявление.
2. Освен ако в настоящия регламент не е предвидено друго, документите, които ще се използват в производството през Службата, могат да бъдат регистрирани на всеки официален език на Общността.
Когато езикът на такива документи не е езикът на производството, Службата може да поиска да се представи превод в рамките на период, който се определя от Службата, на такъв език, или по избор на страната по производството, на който и да е език на Службата.
Член 82
Устно производство
1. Всяка страна по устно производство пред Службата може, вместо езика на производството, да използва един от другите официални езици на Общността, при условие че същата страна се разпореди за устен превод на езика на производството.
Когато устното производство се провежда в рамките на производство, в което се разглежда заявление за регистрация на промишлен дизайн, заявителят може да използва или езика на заявката, или втория език, посочен от заявителя.
2. При устно производство относно заявление за регистрация на промишлен дизайн, персоналът на Службата може да използва езика, на който е написано заявлението, или вторият език, посочен от заявителя.
Във всички останали производства персоналът на Службата може, вместо езика на производството, да използва един от другите официални езици на Общността, при условие че страната или страните по производството се съгласи/съгласят на такова използване.
3. С оглед на взимане на свидетелски показания, при изслушването на която и да е от страните, свидетел или експерт, който не може да се изразява адекватно на езика на производството, може да използва който и да е от официалните езици на Общността.
Когато взимането на показания е решено след искане на страна по производството, при изслушването на страните, свидетелите или експертите, които се изразяват на езици, различни от езика на производството, могат да бъдат изслушани само при условие че страната, която е внесла искането, се е разпоредила за превод на този език.
При производство, засягащо заявление за регистрация на промишлен дизайн, вместо езика на заявката може да се използва вторият език, посочен от заявителя.
При всички производства със само една страна, по искане на съответната страна, Службата може да разреши отклонение от разпоредбите на настоящия параграф.
4. В случай, че страните и Службата се споразумеят по такъв въпрос, на устното производство може да се използва всеки от официалните езици на Общността.
5. Ако е необходимо, Службата може да се разпореди за своя сметка относно превод на езика на производството, или, там, където е подходящо, на другите си езици, освен ако такъв превод не е отговорност на някоя от страните по производството.
6. Изказванията на персонала на Службата, на страни по производството и на свидетели и експерти, направени на един от езиците на Службата по време на устна процедура, се вписват в протокола на езика, който е използван. Изказванията, направени на всеки друг език, се вписват на езика на производството.
Корекциите в заявленията за или в регистрацията на промишлен дизайн на Общността се вписват в протокола на езика на производството.
Член 83
Заверка на преводите
1. Ако превод на какъвто и да е документ трябва да се регистрира, Службата може да поиска регистрирането, в рамките на срок, който се посочва от нея, на удостоверение, че преводът съответства на оригиналния текст.
Когато удостоверението се отнася до превода на предишно заявление по силата на член 42 от Регламент (ЕО) № 6/2002, такъв срок от време следва да е не по-малко от три месеца след датата на вписване на заявката.
Когато удостоверението не е регистрирано в рамките на този срок от време, документът са счита, че не е получен.
2. Председателят на Службата може да определи начина, по който се заверяват преводите.
Член 84
Законна автентичност на преводите
При липса на доказателства за противното, Службата може да приеме, че даден превод съответства на съответния оригинален текст.
ГЛАВА ХХ
РЕЦИПРОЧНОСТ, ПРЕХОДЕН ПЕРИОД И ВЛИЗАНЕ В СИЛА
Член 85
Публикуване във връзка с реципрочността
1. Ако е необходимо, председателят на Службата поисква от Комисията да запита дали държава, която не е страна по Парижката конвенция за защита на индустриалната собственост или по Споразумение за създаване на Световната търговска организация, предоставя реципрочен режим по смисъла на член 41, параграф 5 от Регламент (ЕО) № 6/2002.
2. В случай, че Комисията определи, че се предоставя реципрочен режим в съответствие с параграф 1, Службата публикува комюнике в този смисъл в Официален вестник на Европейските общности.
3. Член 41, параграф 5 от Регламент (ЕО) № 6/2002 се прилага от датата на публикуването в Официален вестник на Европейските общности на съобщението, споменато в параграф 2, освен ако в съобщението не се посочва друга дата, от която да е приложимо.
Член 41, параграф 5 от Регламент (ЕО) № 6/2002 престава да бъде приложим, считано от датата на публикуването в Официален вестник на Европейските общности на съобщението на Комисията, в което да се посочва, че вече не се прилага реципрочен режим, освен ако в съобщението не се посочва друга дата, от която да е приложимо.
4. Съобщенията, посочени в параграфи 2 и 3, следва да бъдат публикувани също и в Официалния вестник на Службата.
Член 86
Преходен период
1. Всяко заявление за регистрация на промишлен дизайн на Общността, регистрирано не повече от три месеца преди датата, определена съгласно член 111, параграф 2 от Регламент (ЕО) № 6/2002, се означава от Службата с датата на регистрацията, определена съгласно тази разпоредба и с действителната дата на получаване на заявката.
2. С оглед на заявката приоритетният период от шест месеца, предвиден от членове 41 и 44 от Регламент (ЕО) № 6/2002, се изчислява считано от датата, определена съгласно член 111, параграф 2 от същия регламент.
3. Службата може да издаде разписка на заявителя преди датата, определена съгласно член 111, параграф 2 от Регламент (ЕО) № 6/2002.
4. Службата може да проучи заявката преди датата, определена съгласно член 111, параграф 2 от Регламент (ЕО) № 6/2002, и да се свърже със заявителя с оглед отстраняването на всякакви недостатъци преди тази дата.
Всякакви решения с оглед на такива заявления могат да се вземат само след тази дата.
5. Когато датата на получаване на заявление за регистриране на промишлен дизайн на Общността от Службата, в централната служба за промишлена интелектуална собственост на държава-членка или в Службата за промишлен дизайн на Бенелюкс, е преди започването на тримесечния период, посочен в член 111, параграф 2 от Регламент (ЕО) № 6/2002, заявката се счита, че не е била регистрирана.
Заявителят следва да бъде уведомен по съответния начин, а заявката следва да му се изпрати обратно.
Член 87
Влизане в сила
Настоящият регламент влиза в сила на седмия ден след публикуването му в Официален вестник на Европейските общности.
Настоящият регламент е задължителен в своята цялост и се прилага пряко във всички държави-членки.
Съставено в Брюксел на 21 октомври 2002 година.
За Комисията
Frederik BOLKESTEIN
Член на Комисията
(1) ОВ L 3, 5.1.2002 г., стр. 1.
(2) ОВ L 341, 17.12.2002 г., стр. 54.
(3) ОВ L 11, 14.1.1994 г., стр. 1.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
40 |
32002R2246
|
L 341/54 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕГЛАМЕНТ (ЕО) № 2246/2002 НА КОМИСИЯТА
от 16 декември 2002 година
относно таксите, платими на Службата за хармонизиране на вътрешния пазар (търговски марки, дизайни и модели) във връзка с регистрацията на промишлен дизайн на Общността
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Регламент (ЕО) № 6/2002 на Съвета от 12 декември 2001 г. за промишления дизайн на Общността (1), и по-специално член 107 от него,
като има предвид, че:
|
(1) |
В светлината на член 139 от Регламент (ЕО) № 40/94 на Съвета от 20 декември 1993 г. относно търговската марка на Общността (2), изменен с Регламент № 3288/94 (3), който по силата на член 97 от Регламент (ЕО) № 6/2002 е приложим също към настоящия регламент, размерите на таксите трябва да се определят на равнище, което гарантира, че съответните постъпления са по принцип достатъчни за балансиране бюджета на Службата. |
|
(2) |
Регламент (ЕО) № 2245/2002 на Комисията от 21 октомври 2002 г. относно прилагането на Регламент (ЕО) № 6/2002 на Съвета за промишления дизайн на Общността (4) урежда също условията, при които предвидените в Регламент № 6/2002 такси трябва да се заплащат на Службата. |
|
(3) |
За да се осигури необходимата гъвкавост, председателят на Службата трябва да е упълномощен да определя, при определени условия, таксите, които могат да се плащат на Службата, във връзка с услугите, които тя може да предоставя, таксите за достъп до базите данни на Службата и предоставяне съдържанието на тези бази данни в машинно-четивна форма и да определя тарифите за продажба на изданията на Службата. |
|
(4) |
С оглед да се улесни плащането на таксите, председателят на Службата трябва да е овластен да разрешава други начини на плащане, освен тези, които настоящият регламент изрично предвижда. |
|
(5) |
Мерките, предвидени в настоящия регламент, са в съответствие със становището на Комитета, създаден съгласно член 109 от Регламент (ЕО) № 6/2002, |
ПРИЕ НАСТОЯЩИЯ РЕГЛАМЕНТ:
Член 1
Предмет
Настоящият регламент определя размерите и начините на плащане на:
|
а) |
таксите, платими на Служба за хармонизация във вътрешния пазар (търговски марки и промишлен дизайн) (по-долу наричана „Службата“) съгласно Регламент (ЕО) № 6/2002 и Регламент (ЕО) № 2245/2002; |
|
б) |
тарифите, определяни от председателя на Службата съгласно член 3, параграфи 1 и 2. |
Член 2
Такси, предвидени в Регламент (ЕО) № 6/2002 и в Регламент (ЕО) № 2245/2002
Таксите, предвидени в Регламент (ЕО) № 6/2002 и в Регламент (ЕО) № 2245/2002, са дадени в приложението.
Член 3
Тарифи, определяни от председателя
1. Председателят определя сумата, платима за всяка друга услуга, предоставяна от Службата, освен тези, предвидени в приложението.
2. Председателят определя сумата, платима за Бюлетина на дизайните на Общността, както и за всяко друго издание на Службата.
3. Размерите на тарифите се определят в евро.
4. Размерите на тарифите, определени от председателя в съответствие с параграфи 1 и 2, се публикуват в Официален вестник на Службата.
Член 4
Дата на плащане на таксите и тарифите
1. Таксите и тарифите, чиято дата на плащане не е уточнена в Регламент (ЕО) № 6/2002 или в Регламент № 2245/2002, се дължат, считано от датата на подаване на заявлението за услугата, за която се дължи такса.
2. Председателят може да реши предоставянето на услуги, посочено в параграф 1, да не подлежи на предварително плащане на съответните такси и тарифи.
Член 5
Плащане на такси и тарифи
1. Таксите и тарифите, платими на Службата, трябва да се плащат в евро:
|
а) |
чрез плащане или превод по банкова сметка на Службата; |
|
б) |
чрез издаване или изпращане на чекове, платими на Службата; |
|
в) |
в брой. |
2. Председателят може да определя начините на плащания, различни от тези, уточнени в параграф 1, в частност чрез депозити по открити текущи сметки на Службата. Тези начини се публикуват в Официален вестник на Службата.
Член 6
Данни за плащането
1. Всяко плащане посочва името на лицето, което извършва плащането, и необходимите данни, за да може Службата да установи незабавно предмета на плащането. В частност, трябва да се предоставят следните данни:
|
а) |
когато се плаща такса за регистрация, предмета на плащането, а именно „такса за регистрация“, и в зависимост от случая посочването, дадено от заявителя в заявлението за регистрация на дизайн; |
|
б) |
когато се плаща такса за публикация, предмета на плащането, а именно „такса за публикация“, и в зависимост от случая посочването, дадено от заявителя в заявлението за регистрация на дизайн на общността; |
|
в) |
когато се плаща такса за публикация в съответствие с член 50, параграф 4 от Регламент (ЕО) № 6/2002, предмета на плащането, а именно „такса за публикация“ и регистрационния номер; |
|
г) |
когато се плаща такса за отлагане на публикация, предмета на плащането, а именно „такса за отлагане“ и в зависимост от случая позоваването, дадено от заявителя в заявката за регистрация на дизайн на Общността; |
|
д) |
когато се плаща такса за обявяване в несъстоятелност, регистрационния номер и името на притежателя на регистрирания дизайн на Общността, срещу когото е насочено заявлението, и предмета на плащането, а именно „такса за обявяване в несъстоятелност“. |
2. Ако предметът на плащането не може незабавно да се определи, Службата изисква от лицето, което е извършило плащането, да я нотифицира писмено за това в определен от нея срок. Ако лицето не се съобрази с искането в надлежен срок, плащането се смята за неосъществено. Платената сума се възстановява.
Член 7
Дата, на която плащането се смята за извършено
1. Датата, на която всяко плащане на Службата се смята извършено, е следната:
|
а) |
в случаите, посочени в член 5, параграф 1, буква а), датата, на която сумата на плащането е действително вписана по кредита на банковата сметка на Службата; |
|
б) |
в случая, посочен в член 5, параграф 1, буква б), датата на получаване на чека от Службата, при условие че чекът е осребрен; |
|
в) |
в случите, посочени в член 5, параграф 1, буква в), датата на получаване на сумата на плащането в брой. |
2. Когато председателят разрешава в съответствие с разпоредбите на член 5, параграф 2, други начини на плащане на таксите, освен тези, предвидени в член 5, параграф 1, той определя също датата, на която това плащане се смята извършено.
3. Когато в съответствие с параграф 1 и 2 плащането на таксата се смята извършено само след изтичането на определения срок, срокът се смята спазен, ако на Службата се представи доказателство, че лицето, което е извършило плащането:
|
а) |
в държава-членка в срока, в който плащането е трябвало да бъде извършено:
|
|
б) |
е надплатило сума, равна на 10 % от съответната такса или такси, но не по-голяма от 200 евро. |
никаква допълнителна такса не се дължи, ако едно от условията, посочени в буква а) е било изпълнено най-късно десет дни преди изтичането на срока на плащане.
4. Службата може да поиска от лицето, което е извършило плащането, да представи доказателство за датата, на която едно от условията, изброени в параграф 3, буква а), е изпълнено, и в зависимост от случая да плати допълнителната такса, посочена в параграф 3, буква б), в срок, който тя определя. Ако лицето не отговори на заявлението на Службата или ако доказателството е недостатъчно или наложената допълнителна такса не е платена навреме, срокът на плащането се смята неспазен.
Член 8
Непълно плащане
1. Срокът на плащане, по принцип, се смята спазен само ако цялата сума на таксата е платена в предвидения срок. Когато таксата не е платена изцяло, платената сума се възстановява след изтичането на срока.
2. Въпреки това Службата може, доколкото това е възможно в оставащия за изтичане срок, да даде възможност на лицето, което извършва плащането, да плати липсващата сума или, ако това е оправдано, да се откаже от малки неплатени суми, без да се засягат правата на лицето, извършващо плащането.
Член 9
Възстановяване на незначителни суми
1. Когато е платена извънредно голяма сума за покриване на такса, надплатеното не се възстановява, ако е незначително, и ако съответната страна изрично не е поискала възстановяване.
Председателят определя какво представлява незначителна сума.
2. Решенията на председателя, взети по силата на параграф 1, се публикуват в Официален вестник на Службата.
Член 10
Влизане в сила
Настоящият регламент влиза в сила от седмия ден след публикуването му в Официален вестник на Европейските общности.
Настоящият регламент е задължителен в своята цялост и се прилага пряко във всички държави-членки.
Съставено в Брюксел на 16 декември 2002 година.
За Комисията
Frederik BOLKESTEIN
Член на Комисията
(1) ОВ L 3, 5.1.2002 г., стр. 1.
(2) ОВ L 11, 14.1.1994 г. стр. 1.
(3) ОВ L 349, 31.12.1994 г., стр. 83.
(4) ОВ L 341, 17.12.2002 г., стр. 26.
ПРИЛОЖЕНИЕ
|
(в евро) |
|||||||||||||
|
230 |
||||||||||||
|
|
||||||||||||
|
115 |
||||||||||||
|
50 |
||||||||||||
|
120 |
||||||||||||
|
|
||||||||||||
|
60 |
||||||||||||
|
30 |
||||||||||||
|
40 |
||||||||||||
|
|
||||||||||||
|
20 |
||||||||||||
|
10 |
||||||||||||
|
60 |
||||||||||||
|
30 |
||||||||||||
|
10 |
||||||||||||
|
25 % от допълнителната такса |
||||||||||||
|
|
||||||||||||
|
90 |
||||||||||||
|
120 |
||||||||||||
|
150 |
||||||||||||
|
180 |
||||||||||||
|
25 % от таксата за подновяване |
||||||||||||
|
350 |
||||||||||||
|
800 |
||||||||||||
|
200 |
||||||||||||
|
200 за образец с таван 1 000, когато са представени множество искания в същото заявление за регистрация на прехвърлянето или едновременно |
||||||||||||
|
200 за образец с таван 1 000, когато са представени множество искания в същото заявление за регистрация на прехвърлянето или едновременно |
||||||||||||
|
200 за образец с таван 1 000, когато са представени множество искания в същото заявление за регистрация на лиценз или на друго право или едновременно |
||||||||||||
|
200 за образец с таван 1 000, когато са представени множество искания в същата заявление за регистрация на лиценз или на друго право или едновременно |
||||||||||||
|
|
||||||||||||
|
10 |
||||||||||||
|
30 |
||||||||||||
|
30 |
||||||||||||
|
|
||||||||||||
|
10 |
||||||||||||
|
30 |
||||||||||||
|
плюс за всяка следваща страница след десетата |
1 |
||||||||||||
|
10 |
||||||||||||
|
плюс за всяка следваща страница след десетата |
1 |
||||||||||||
|
100 |
||||||||||||
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
46 |
32003L0002
|
L 004/9 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/2/ЕО НА КОМИСИЯТА
от 6 януари 2003 година
относно ограниченията за пускането на пазара и употребата на арсен (десето адаптиране към техническия прогрес на Директива 76/769/ЕИО на Съвета)
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 76/769/ЕИО на Съвета от 27 юли 1976 г. за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки относно ограниченията за пускането на пазара и употребата на някои опасни вещества и препарати (1), последно изменена с Директива 2002/62/ЕО на Комисията (2), и по-специално член 2а от нея, въведен с Директива 89/678/ЕИО на Съвета (3),
като има предвид, че:
|
(1) |
Директива 89/677/ЕИО на Съвета (4) относно осмо изменение на Директива 76/769/ЕИО, налага някои ограничения върху продажбата и употребата на арсен. |
|
(2) |
В рамките на ревизия на законодателството на Общността, отнасящо се до употребата на съединенията на арсена при консервацията на дървесината, проведена след присъединяването на Австрия, Финландия и Швеция към Европейския съюз през 1995 г., бяха извършени оценка на риска и анализ на предимствата и недостатъците, произтичащи от налагането на по-нататъшни ограничения върху съдържанието на арсен в някои консерванти за дървесина (5). |
|
(3) |
Извършената оценка на риска бе предоставена за паралелно разглеждане (6) на Научния комитет по токсичност, екотоксичност и околна среда (НКТЕОС), който стигна до заключението, че основните рискове са идентифицирани правилно. Въпросните рискове визират рисковете за човешкото здраве по причина на изхвърлянето на отпадна дървесина, която е била обработена с консерванти, съдържащи мед, хром и арсен (МХА), и по-специално рисковете за здравето на децата по причина на употребата на обработена с МХА дървесина при изработката на спортно-развлекателни съоръжения. Бе установено, също така, и наличие на риск за водната среда при някои морски води. |
|
(4) |
Освен това, НКТЕОС изрази становище, че в светлината на острия недостиг на информация за обработената с арсен и намираща се в депата за отпадъци дървесина, е препоръчително да се упражнява предпазливост чрез ограничаване на въпросния вид обработка до случаите, в които същата се явява абсолютно необходима. |
|
(5) |
Продължавайки анализа на въздействията на арсена върху човешкото здраве (7), НКТЕОС посочи, че веществото има както генотоксично, така и добре установено канцерогенно действие, и че е целесъобразно да се приеме, че не съществува пределна граница по отношение на неговия канцерогенен ефект. |
|
(6) |
Обработената с МХА отпадна дървесина се класифицира като опасен отпадък по смисъла на Решение 2000/532/ЕО на Комисията от 3 май 2000 г., заменящо Решение 94/3/ЕО, за въвеждане на списък на отпадъци в съответствие с член 1, буква а) от Директива 75/442/ЕИО на Съвета относно отпадъците, и Решение 94/904/ЕО на Съвета за въвеждане на списък на опасни отпадъци в съответствие с член 1, параграф 4 от Директива 91/689/ЕО на Съвета относно опасните отпадъци (8), последно изменена с Решение 2001/573/ЕО на Съвета (9). |
|
(7) |
Директива 98/8/ЕО на Европейския парламент и на Съвета от 16 февруари 1998 г. относно пускането на пазара на биоцидни препарати (10) постановява разпоредби за хармонизиране на разрешителния режим при биоцидите на общностно равнище, а Регламент (ЕО) № 1896/2000 на Комисията от 7 септември 2000 г. за първата фаза от програмата, посочена в член 16, параграф 2 от Директива 98/8/ЕО на Европейския парламент и на Съвета, относно биоцидните препарати (11), поставя изискване за приоритетно третиране на консервантите за дървесина при реализирането на програмата за ревизия на законодателството, постановена с Директива 98/8/ЕО. Арсенът бе идентифициран и нотифициран в качеството му на „активно“ вещество в рамките на срока, определен с Регламент (ЕО) № 1896/2000. Пълна документация, съдържаща оценка на арсена като едно от „съществуващите“ вещества ще бъде представена до 28 март 2004 г. |
|
(8) |
За целите на оценката на риска и ръководейки се от принципа на „предпазливостта“ до хармонизирането на визираните в Директива 98/8/ЕО правила или изработването на решение по смисъла на член 6, параграф 3 от Регламент (ЕО) № 1896/2000, е необходимо да се извърши адаптиране към техническия прогрес на разпоредбите относно ограниченията върху арсена от Директива 76/769/ЕО. Въпросната директива не третира вече обработената с МХА дървесина. |
|
(9) |
Настоящата директива трябва да се прилага по начин, който не засяга законодателството на Общността, постановяващо минимални изисквания за защитата на работниците, по-специално Директива 89/391/ЕИО на Съвета от 12 юни 1989 г. за въвеждане на мерки за насърчаване подобряването на безопасността и здравето на работниците на работното място (12), Директива 90/394/ЕИО на Съвета от 28 юни 1990 г. за защита на работниците от рискове, свързани с експозиция на канцерогени по време на работа (Шеста специална директива по смисъла на член 16, параграф 1 от Директива 89/391/ЕИО) (13), последно изменена с Директива 1999/38/ЕО (14) и с Директива 98/24/ЕО на Съвета от 7 април 1998 г. за опазване на здравето и безопасността на работниците от рисковете, свързани с химични агенти на работното място (Четиринадесета специална директива по смисъла на член 16, параграф 1 от Директива 89/391/ЕИО) (15). |
|
(10) |
Предвидените в настоящата директива мерки съответстват на становището на Комитета за адаптиране към техническия прогрес на директивите за премахване на техническите пречки пред търговията с опасни вещества и препарати, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Приложение I към Директива 76/769/ЕИО се изменя в съответствие с приложението към настоящата директива.
Член 2
Държавите-членки приемат и публикуват разпоредбите, необходими за да се съобразят с настоящата директива, най-късно до 30 юни 2003 г. Те незабавно информират Комисията за това. Те прилагат тези разпоредби най-късно от 30 юни 2004 г.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 3
Настоящата директива влиза в сила на двадесетия ден след датата на публикуването ѝ в Официален вестник на Европейските общности.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 6 януари 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 262, 27.9.1976 г., стр. 201.
(2) ОВ L 183, 12.7.2002 г., стр. 58.
(3) ОВ L 398, 30.12.1989 г., стр. 24.
(4) ОВ L 398, 30.12.1989 г., стр. 19.
(5) Оценка на рисковете за здравето и околната среда по причина на арсена от състава на консервантите за дървесина и ефекта от налагането на по-нататъшни ограничения върху продажбите и употребата на същия, 1998 г.
(6) htpp://europa.eu.int/comm/food/fs/sc/sct/out18_en.html.
(7) htpp://europa.eu.int/comm/food/fs/sc/sct/out106_en.html.
(8) ОВ L 226, 6.9.2000 г., стр. 3.
(9) ОВ L 203, 28.7.2001 г., стр. 18.
(10) ОВ L 123, 24.4.1998 г., стр. 1.
(11) ОВ L 228, 8.9.2000 г., стр. 6.
(12) ОВ L 183, 29.6.1989 г., стр. 1.
(13) ОВ L 196, 26.7.1990 г., стр. 1.
(14) ОВ L 138, 1.6.1999 г., стр. 66.
(15) ОВ L 131, 5.5.1998 г., стр. 11.
ПРИЛОЖЕНИЕ
Точка 20 от приложение I към Директива 1976/769/ЕИО се заменя със следния текст:
|
„20. Съединения на арсена |
|
|||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
Без да се засяга прилагането на други разпоредби на Общността относно класифицирането, опаковането и етикетирането на опасни вещества и препарати, всеки пускан на пазара обработен дървен материал се етикетира индивидуално с обозначението „Само за професионални и промишлени инсталации и предназначения, съдържа арсен“. Освен това, всеки предлаган на пазара дървен материал на пакети трябва да бъде снабден с етикет с надпис „Работата с този продукт изисква употреба на защитни ръкавици. При рязане или други видове дървообработка трябва да се употребяват противопрахова маска и защитни очила. Отпадъците от този продукт трябва да бъдат третирани като опасни и обслужвани от лицензирана за съответната дейност фирма“. |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
49 |
32003L0003
|
L 004/12 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/3/ЕО НА КОМИСИЯТА
от 6 януари 2003 година
относно ограниченията за пускането на пазара и употребата на „синьо багрило“ (дванадесето адаптиране към техническия прогрес на Директива 76/769/ЕИО на Съвета)
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 76/769/ЕИО на Съвета от 27 юли 1976 г. за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки относно ограничения за пускането на пазара и употребата на опасни вещества и препарати (1), последно изменена с Директива 2003/2/ЕО на Комисията от 6 януари 2003 г. относно ограниченията за пускането на пазара и употребата на арсен (десето адаптиране към техническия прогрес на Директива 76/769/ЕИО на Съвета (2), и по-специално член 2а от нея, въведен с Директива 89/678/ЕИО (3) на Съвета,
като има предвид, че:
|
(1) |
Съгласно Директива 76/769/ЕИО, изменена с Директива 2002/61/ЕО на Европейския парламент и Съвета (4), някои от описаните азобагрила не трябва да се влагат в някои текстилни и кожени изделия; от своя страна, въпросните текстилни и кожени изделия не могат да се пускат на пазара, ако не удовлетворяват изискванията от цитираната директива. |
|
(2) |
Рисковете за човешкото здраве и околната среда от „синьото багрило, индекс № 611-070-00-2“ бяха оценени в съответствие с Директива 93/67/ЕИО на Комисията от 20 юли 1993, утвърждаваща принципи за оценка на рисковете за човешкото здраве и околната среда от веществата, нотифицирани в съответствие с Директива 67/548/ЕИО на Съвета (5); оценката на рисковете очерта необходимост от намаляване на рисковете от синьото багрило за околната среда, тъй като въпросното синьо багрило е силно токсично за водната среда, не се разгражда лесно и може да попада в околната среда с отпадъчните води. |
|
(3) |
За целите на опазването на околната среда трябва да се забрани пускането на пазара и употребата на синьото багрило за багрене на текстилни и кожени изделия. Синьото багрило, следователно, трябва да се добави към списъка на веществата от приложение I към Директива 76/769/ЕИО. |
|
(4) |
Ограниченията върху пускането на пазара и употребата на синьото багрило, постановени с настоящата директива, са съобразени с произтичащото от текущото състояние на познанието и технологиите наличие на подходящи заместители. |
|
(5) |
Настоящата директива трябва да се прилага по начин, който не засяга законодателството на Общността, постановяващо минимални изисквания за защитата на работниците и по-специално Директива 89/391/ЕИО на Съвета от 12 юни 1989 г. за въвеждане на мерки за насърчаване подобряването на безопасността и здравето на работниците на работното място (6) и Директива 90/394/ЕИО на Съвета от 28 юни 1990 г. за защита на работниците от рискове, свързани с експозиция на канцерогени по време на работа (шеста специална директива по смисъла на член 16, параграф 1 от Директива 89/391/ЕИО (7), последно изменена с Директива 1999/38/ЕО (8). |
|
(6) |
Предвидените в настоящата директива мерки съответстват на становището на Комитета за адаптиране към техническия прогрес на директивите за премахване на техническите пречки пред търговията с опасни вещества и препарати, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Приложение I към Директива 76/769/ЕИО се адаптира към техническия прогрес в съответствие с приложението към настоящата директива.
Член 2
Държавите-членки въвеждат в сила законовите, подзаконовите и административните разпоредби, необходими, за да се съобразят с настоящата директива не по-късно 31 декември 2003. Те незабавно информират Комисията за това. Те прилагат тези разпоредби считано от 30 юни 2004.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваването на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 3
Настоящата директива влиза в сила на двадесетия ден след публикуването ѝ в Официален вестник на Европейските общности.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 6 януари 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 262, 27.9.1976 г., стр. 201.
(2) ОВ L 4, 9.1.2001 г., стр. 13.
(3) ОВ L 398, 30.12.1989 г., стр. 24.
(4) ОВ L 243, 11.9.2002 г., стр. 15.
(5) ОВ L 227, 8.9.1993 г., стр. 9.
(6) ОВ L 183, 29.6.1989 г., стр. 1.
(7) ОВ L 196, 26.7.1990 г., стр. 1.
(8) ОВ L 138, 1.6.1999 г., стр. 66.
ПРИЛОЖЕНИЕ
|
— |
В приложение I към Директива 76/769/ЕИО, точка 43 се заменя със следната точка: |
|
„43. Азооцветители |
1. Азобагрилата, които, чрез редукционно разцепване на една или повече азогрупи, могат да отделят в готовите изделия или обагрените техни части един или повече от описаните в допълнението ароматни амини в концентрации, достатъчни за тяхното откриване, т.е. над 30 ppm, чрез метода за изпитване, утвърден в съответствие с член 2а от настоящата директива, не могат да се влагат в текстилни и кожени изделия, които могат да влизат в непосредствен и продължителен контакт с кожата или устната кухина на човека, например:
2. Освен това, текстилните и кожени изделия по точка 1 не могат да се пускат на пазара, ако не удовлетворяват изискванията, посочени в същата точка. Чрез отмяна, въпросната разпоредба няма да се прилага до 1 януари 2005 г. по отношение на текстилни изделия, произведени от вторично преработени влакна, ако амините се отделят от остатъци, произхождащи от предходно багрене на въпросните влакна, и ако посочените в тук приложения списък амини се отделят в концентрации, по-ниски от 70 ppm. 3. Азобагрилата, посочени в „Списъка на азобагрилата“, приложен към Допълнението в съответствие с настоящата директива, не могат да се пускат на пазара или използват за багрене на текстилни и кожени изделия, като вещества или като съставки на препарати, в концентрации, по-високи от 0,1 % по маса. 4. Не по-късно от 11 септември 2005 г., Комисията ще преразгледа разпоредбите за азобагрилата в светлината на новопоявилите се научни факти.“ |
|
— |
Към допълнениетосе добавя следната точка: |
„Точка 43 Азобагрила
Списък на ароматни амини
|
|
CAS номер |
Индекс номер |
ЕС номер |
Вещества |
|
1 |
92-67-1 |
612-072-00-6 |
202-177-1 |
бифенил-4-иламин 4-аминобифенил ксениламин |
|
2 |
92-87-5 |
612-042-00-2 |
202-199-1 |
бензидин |
|
3 |
95-69-2 |
|
202-441-6 |
4-хлор-o-толуидин |
|
4 |
91-59-8 |
612-022-00-3 |
202-080-4 |
2-нафтиламин |
|
5 |
97-56-3 |
611-006-00-3 |
202-591-2 |
o-аминоазотолуол 4-амино-3,2′-диметилазобензол 4-o-толиазо-o-толуидин |
|
6 |
99-55-8 |
|
202-765-8 |
5-нитро-o-толуидин |
|
7 |
106-47-8 |
612-137-00-9 |
203-401-0 |
4-хлоранилнн |
|
8 |
615-05-4 |
|
210-406-1 |
4-метокси-m-фенилендиамин |
|
9 |
101-77-9 |
612-051-00-1 |
202-974-4 |
4,4′-метилендианилин 4,4′-диаминодифенилметан |
|
10 |
91-94-1 |
612-068-00-4 |
202-109-0 |
3,3′-дихлорбензидин 3,3′-дихлорбифенил-4,4′-илендиамин |
|
11 |
119-90-4 |
612-036-00-X |
204-355-4 |
3,3′-диметоксибензидин o-дианизидин |
|
12 |
119-93-7 |
612-041-00-7 |
204-358-0 |
3,3′-диметилбензидин 4,4′-би-o-толуидин |
|
13 |
838-88-0 |
612-085-00-7 |
212-658-8 |
4,4′-метиленди-o-толуидин |
|
14 |
120-71-8 |
|
204-419-1 |
6-метокси-m-толуидин p-крезидин |
|
15 |
101-14-4 |
612-078-00-9 |
202-918-9 |
4,4′-метилен-бис-(2-хлоранилин) 2,2′-дихлор-4,4′-метилен дианилин |
|
16 |
101-80-4 |
|
202-977-0 |
4,4′-оксидианилин |
|
17 |
139-65-1 |
|
205-370-9 |
4,4′-тиодианилин |
|
18 |
95-53-4 |
612-091-00-X |
202-429-0 |
o-толуидин 2-метиланилин |
|
19 |
95-80-7 |
612-099-00-3 |
202-453-1 |
4-метил-m-фенилендиамин |
|
20 |
137-17-7 |
|
205-282-0 |
2,4,5-триметиланилин |
|
21 |
90-04-0 |
612-035-00-4 |
201-963-1 |
o-анизидин 2-метоскианилин |
|
22 |
60-09-3 |
611-008-00-4 |
200-453-6 |
4-амино азобензол |
Списък на азобагрила
|
|
CAS номер |
Индекс номер |
ЕС номер |
Вещества |
||||||
|
1 |
Не е предоставен Компонент 1:
Компонент 2:
|
611-070-00-2 |
405-665-4 |
Смес от: динатрий (6-(4-анизидин)-3-сулфонат-2-(3,5-динитро-2-оксидофенилазо)-1-нафтолат)(1-(5-хлор-2-оксидофенилазо)-2-нафтолат)хромат(1-) и тринатрий бис(6-(4-анизидин)-3-сулфонат-2-(3,5-динитро-2-оксидофенилазо)-1-нафтолат) хромат(1-)“ |
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
53 |
32003L0001
|
L 005/14 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/1/ЕО НА КОМИСИЯТА
от 6 януари 2003 година
относно адаптиране към техническия прогрес на приложение II към Директива 76/768/ЕИО на Съвета относно сближаването на законодателствата на държавите-членки, свързани с козметичните продукти
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 76/768/ЕИО на Съвета от 27 юли 1976 г. относно сближаването на законодателствата на държавите-членки, свързани с козметичните продукти (1), последно изменена с Директива 2002/34/ЕО на Комисията (2), и по-специално член 8, параграф 2 от нея,
след консултации с Научния комитет по козметични продукти и нехранителни продукти, предназначени за потребители,
като има предвид, че:
|
(1) |
Към момента пореден номер 419 от приложение II към Директива 76/768/ЕО, съдържащ списък на вещества, които не трябва да се съдържат в козметичните продукти, се привежда в съответствие с Решение 97/534/ЕО на Комисията от 30 юли 1997 г. за забрана на употребата на вещества, които представляват риск по отношение на трансмисивните спонгиформни енцефалопатии (3). Цитираното решение бе отменено с Решение 2000/418/ЕО на Комисията от 29 юни 2000 г. относно контрола върху употребата на вещества, които представляват риск по отношение на трансмисивните спонгиформни енцефалопатии, и за изменение на Решение 94/474/ЕО (4). Предвид становището на Научния комитет за козметични продукти и нехранителни продукти, предназначени за потребители (SCCNFP), е целесъобразно пореден номер 419 от приложение II към Директива 76/768/ЕО да бъде приведен в съответствие с Регламент (ЕО) № 999/2001 на Европейския парламент и на Съвета от 22 май 2001 г. относно определяне на правила за превенция, контрол и ликвидиране на някои трансмисивни спонгиформни енцефалопатии (5), последно изменен с Регламент (ЕО) № 270/2002 на Комисията (6). |
|
(2) |
В пореден номер 419 от приложение II към Директива 76/768/ЕО следва да бъде включено позоваване на специфичните рискови материали, посочени в приложение V към Регламент (ЕО) № 999/2001. |
|
(3) |
От член 22, параграф 1 от Регламент (ЕО) № 999/2001 обаче следва, че до датата на приемане на решение, след което влизат в сила член 8 от посочения регламент и приложение V към него, остават в сила разпоредбите на част А на приложение XI към същия регламент. Следователно, пореден номер 419 от приложение II към Директива 76/768/ЕИО следва да се позовава и на част А на приложение XI към Регламент (ЕО) № 999/2001. |
|
(4) |
Директива 76/768/ЕИО следва да бъде подходящо изменена, |
|
(5) |
Предвид специфичния характер на рисковите материали, държавите-членки трябва да могат да предприемат предвидените в настоящата директива мерки, без да изчакват посочената крайна дата. |
|
(6) |
Мерките, предвидени в настоящата директива, са в съответствие със становището на Комитета по адаптиране към техническия прогрес на директивите за премахването на техническите пречки в търговията в сектора на козметичните продукти, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Приложение II към Директива 76/768/ЕИО се изменя в съответствие с приложението към настоящата директива.
Член 2
1. Държавите-членки предприемат всички необходими мерки за да гарантират, че козметични продукти, които не съответстват на изискванията от настоящата директива, не се пускат на пазара от производители от Общността или от регистрирани в Общността вносители, не по-късно от 15 април 2003 г.
2. Държавите-членки предприемат всички необходими мерки за да гарантират, че посочените в параграф 1 продукти няма да бъдат продавани или предоставяни на крайни потребители най-късно след 15 април 2003 г.
Член 3
Държавите-членки въвеждат в сила законови, подзаконови и административни разпоредби, необходими, за да се съобразят с настоящата директива преди 15 април 2003 г. Те незабавно информират Комисията за това.
Когато държавите-членки приемат такива разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 4
Настоящата директива влиза в сила на третия ден след публикуването ѝ в Официален вестник на Европейските общности.
Член 5
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 6 януари 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 262, 27.9.1976 г., стр. 169.
(2) ОВ L 102, 18.4.2002 г., стр. 19.
(3) ОВ L 216, 8.8.1997 г., стр. 95.
(4) ОВ L 158, 30.6.2000 г., стр. 76.
(5) ОВ L 147, 31.5.2001 г., стр. 1.
(6) ОВ L 45, 15.2.2002 г., стр. 4.
ПРИЛОЖЕНИЕ
В пореден номер 419 от приложение II към Директива 76/768/ЕИО на Съвета изреченията:
|
„а) |
черепът, включително мозъкът и очите, сливиците и гръбначният мозък от:
|
|
б) |
далаците от овце и кози и получените от тях продукти.“ |
се заменят с изреченията:
„След посочената в член 22, параграф 1 от Регламент (ЕО) № 999/2001 на Европейския парламент и на Съвета (1) дата, специфичните рискови материали, посочени в приложение V към цитирания регламент и получените от тях продукти.
До цитираната дата специфичните рискови материали, посочени в част A на приложение XI към Регламент (ЕО) № 999/2001, и получените от тях продукти.
(1) ОВ L 147, 31.5.2001 г., стр. 1.“
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
56 |
32003D0043
|
L 013/35 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕШЕНИЕ НА КОМИСИЯТА
от 17 януари 2003 година
относно определяне на класове на огнеустойчивост за някои строителни продукти
(нотифицирано под № С(2002) 4807)
(текст от значение за ЕИП)
(2003/43/EО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 89/106/ЕИО на Съвета от 21 декември 1988 г. относно сближаването на законовите, подзаконовите и административните разпоредби на държавите-членки по отношение на строителните продукти (1), изменена с Директива 93/68/ЕИО (2), и по-специално член 20, параграф 2 от нея,
като има предвид, че:
|
(1) |
Директива 89/106/ЕИО предвижда, че за да се отчитат различните нива на защита при строежите на национално, регионално и местно равнище, може да се наложи в тълкувателните документи да се въведат класове на огнеустойчивост, отговарящи на експлоатационните характеристики на продуктите по отношение на всяко съществено изискване. Тези документи са публикувани в рамките на „Съобщение на Комисията във връзка с тълкувателните документи на Директива 89/106/ЕИО на Съвета“ (3). |
|
(2) |
Във връзка със същественото изискване за безопасност при пожар, в тълкувателен документ № 2 се дава списък на взаимносвързани мерки, които като цяло определят различен подход при разработването на стратегията за пожаробезопасност в държавите-членки. |
|
(3) |
В тълкувателен документ № 2 една от тези мерки се определя като ограничаване възникването и разпространението на пожар и дим на дадено място чрез ограничаване на възможностите на строителните продукти да допринасят за пълното развитие на пожар. |
|
(4) |
Нивото на това ограничение може да се изрази единствено чрез различните нива на огнеустойчивост на продуктите при тяхното крайно приложение. |
|
(5) |
Под формата на хармонизирано решение беше приета система от класове в Решение 2000/147/ЕО на Комисията от 8 февруари 2000 г., в съответствие с Директива 89/106/ЕИО на Съвета във връзка с класификацията на строителните продукти по отношение на тяхната огнеустойчивост (4). |
|
(6) |
В случая с някои дървени плоскости е необходимо да се използва класификацията, въведена с Решение 2000/147/ЕО. |
|
(7) |
Границата на огнеустойчивост на много строителни продукти и/или материали, в рамките на класификацията, предвидена в Решение 2000/147/ЕО, е добре установена и достатъчно добре известна на регулаторните органи по пожаробезопасност в държавите-членки, така че те не изискват изпитване на тази експлоатационна характеристика. |
|
(8) |
Мерките, предвидени в настоящото решение, са в съответствие със становището на Постоянния комитет по строителство, |
ПРИЕ НАСТОЯЩОТО РЕШЕНИЕ:
Член 1
Строителните продукти и/или материали, които удовлетворяват всичките изисквания на експлоатационната характеристика „огнеустойчивост“ без необходимост от допълнително изпитване, са посочени в приложението.
Член 2
Специфичните класове, които следва да се прилагат за различни строителни продукти и/или материали, в рамките на класификацията по огнеустойчивост, приета с Решение 2000/147/ЕО, са посочени в приложението към настоящото решение.
Член 3
Продуктите се разглеждат във връзка с тяхното крайно приложение, когато е целесъобразно.
Член 4
Адресати на настоящото решение са държавите-членки.
Съставено в Брюксел на 17 януари 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 40, 11.2.1989 г., стр. 12.
(2) ОВ L 220, 30.8.1993 г., стр. 1.
(3) ОВ С 62, 28.2.1994 г., стр. 1.
(4) ОВ L 50, 23.2.2000, стр. 14.
ПРИЛОЖЕНИЕ
В таблиците, дадени в настоящото приложение, са изброени строителни продукти и/или материали, които удовлетворяват всичките изисквания за експлоатационната характеристика „огнеустойчивост“ без необходимост от изпитване.
Таблица 1
Класове на огнеустойчивост за дървесни плоскости (1)
|
Дървени плоскости (2) |
EN номер за качество на продукта |
Минимална плътност (kg/m3) |
Минимална дебелина (mm) |
Клас (3) (с изкл. на настилки) |
Клас (4) Настилки |
|
Плочи от дървени частици |
EN 312 |
600 |
9 |
D-s2, d0 |
DFL-s1 |
|
Дървесновлакнести плоскости, твърди |
EN 622-2 |
900 |
6 |
D-s2, d0 |
DFL-s1 |
|
Дървесновлакнести плоскости, със средна твърдост |
EN 622-3 |
600 |
9 |
D-s2, d0 |
DFL-s1 |
|
400 |
9 |
E, pass |
EFL |
||
|
Дървесновлакнести плоскости, меки |
EN 622-4 |
250 |
9 |
E, pass |
EFL |
|
MDF плоскости (със средна плътност) (5) |
EN 622-5 |
600 |
9 |
D-s2, d0 |
DFL-s1 |
|
Плочи от дървени частици, слепени с цимент (6) |
EN 634-2 |
1 000 |
10 |
B-s1, d0 |
BFL-s1 |
|
OSB плоскости (7) |
EN 300 |
600 |
9 |
D-s2, d0 |
DFL-s1 |
|
Шперплат |
EN 636 |
400 |
9 |
D-s2, d0 |
DFL-s1 |
|
Плоскости от масивно дърво |
EN 13353 |
400 |
12 |
D-s2, d0 |
DFL-s1 |
(1) EN 13986
(2) Дървесни плоскости, монтирани без въздушна междина, директно срещу клас А1 или А2-s1, d0 продукти с минимална плътност 10 kg/cm3 или минимум клас D-s2, d0 продукти с минимална плътност 400 kg/m3.
(3) Клас, предвиден в Таблица 1 от приложението към Решение 2000/147/ЕО.
(4) Клас предвиден в Таблица 2 от приложението към Решение 2000/147/ЕО.
(5) Дървесно-влакнести плоскости, произвеждани по сух метод.
(6) Циментно съдържание минимум 75 % в масови единици.
(7) Плоскости с ориентирани частици.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
59 |
32003L0012
|
L 028/43 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/12/ЕО НА КОМИСИЯТА
от 3 февруари 2003 година
относно рекласифициране на гръдните имплантанти в рамките на Директива 93/42/ЕИО относно медицинските изделия
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 93/42/ЕИО на Съвета от 14 юни 1993 г. относно медицинските изделия (1), последно изменена с Директива 2001/104/ЕО на Европейския парламент и на Съвета (2), и по-специално член 13, параграф 1, буква б) от нея,
като взе предвид молбата, представена от Франция и Обединеното кралство,
като има предвид, че:
|
(1) |
Въз основа на критериите за класификация, изложени в приложение IX към Директива 93/42/ЕИО, гръдните имплантанти са по принцип медицински изделия от клас IIб. |
|
(2) |
Франция и Обединеното кралство поискаха класифициране на гръдните имплантанти като медицински изделия от клас III чрез дерогация от разпоредбите на приложение IX към Директива 93/42/ЕИО. |
|
(3) |
С цел осигуряване на възможно най-високо ниво на безопасност за гръдните имплантанти, нотифицираните органи трябва да проведат, в рамките на системата за пълно осигуряване на качеството, проверка на проектната документация на изделието в съответствие с точка 4 от приложение II към Директива 93/42/ЕИО. В съответствие с това е необходимо да се пристъпи към рекласифициране на гръдните имплантанти като медицински изделия от клас III. |
|
(4) |
Необходимо е да се определи режимът, приложим по отношение на гръдните импланти, пуснати на пазара преди 1 септември 2003 г. в съответствие с член 11, параграф 3, буква а) или член 11, параграф 3, буква б), iii) от Директива 93/42/ЕИО. |
|
(5) |
Мерките, предвидени в настоящата директива, са в съответствие със становището на Комитета по медицинските изделия, учреден съгласно член 6, параграф 2 от Директива 90/385/ЕИО на Съвета от 20 юни 1990 г. за сближаване на законодателствата на държавите-членки относно активните имплантируеми медицински изделия (3), последно изменена с Директива 93/68/ЕИО (4). |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Чрез дерогация от правилата, определени в приложение IX към Директива 93/42/ЕИО, гръдните имплантанти се рекласифицират като медицински изделия с принадлежност към клас III.
Член 2
1. Гръдните импланти, пуснати на пазара преди 1 септември 2003 г. в съответствие с член 11, параграф 3, буква а) или член 11, параграф 3, буква б), iii) от Директива 93/42/ЕИО, са обект на процедура за преоценка на съответствието в качеството им на медицински изделия от клас III преди 1 март 2004 г.
2. Чрез дерогация от член 11, параграф 11 от Директива 93/42/ЕИО, срокът на действие на решенията по отношение на гръдните импланти, взети от нотифицираните органи преди 1 септември 2003 г. в съответствие с член 11, параграф 3, буква а) от Директива 93/42/ЕИО, не може да бъде удължаван.
Член 3
1. Държавите-членки приемат и публикуват не по-късно от 1 август 2003 г. необходимите законови, подзаконови и административни разпоредби, за да се съобразят с настоящата директива. Те незабавно информират Комисията за това.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Държавите-членки прилагат тези мерки от 1 септември 2003 г.
2. Държавите-членки съобщават на Комисията текстовете на разпоредбите от националното законодателство, които те приемат в областта, уредена с настоящата директива.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 3 февруари 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 169, 12.7.1993 г., стр. 1.
(2) ОВ L 6, 10.1.2002 г., стр. 50.
(3) ОВ L 189, 20.7.1990 г., стр. 17.
(4) ОВ L 229, 30.8.1993 г., стр. 1.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
61 |
32002L0088
|
L 035/28 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2002/88/ЕО НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 9 декември 2002 година
за изменение на Директива 97/68/ЕО за сближаване на законодателствата на държавите-членки в областта на мерките срещу емисиите от замърсяващи газове и частици, изпускани от двигателите с вътрешно горене, използвани в мобилните устройства, които не са предназначени за движение по път
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 95 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Икономическия и социален комитет (2),
след консултация с Комитета на регионите,
като се произнесоха съгласно процедурата, предвидена в член 251 на Договора (3),
като взеха предвид, че:
|
(1) |
Програмата „Auto oil II“ има за цел да направи преглед на стратегиите, които се оказват ефикасни при съблюдаване на целите на Общността по отношение на качеството на въздуха. От доклада на Комисията относно резултатите от програмата „Auto oil II“ става ясно, че е необходимо да се вземат нови мерки, по-специално за намиране на специфични решения на проблемите, свързани с озона и емисиите на частици. Извършените наскоро проучвания, относно определянето на пределни стойности на газовите емисии за всяка държава показват, че е необходимо да се вземат допълнителни мерки за изпълнение на включените в европейското законодателство цели, свързани с качеството на въздуха. |
|
(2) |
Постепенно са приети стриктни норми по отношение на газовите емисии, изпускани от пътните превозни средства. Решено е също така тези норми да бъдат направени по-строги. Следователно делът на замърсяващи емисии, изпускани от мобилните устройства, които не са предназначени за движение по път, в бъдеще ще нарасне значително. |
|
(3) |
Директива 97/68/ЕО (4) въвежда пределни емисионни стойности, прилагани към замърсяващите газове и частици, отделяни от двигатели с вътрешно горене, предназначени за оборудване на мобилните устройства, непредназначени за движение по път. |
|
(4) |
Въпреки, че Директива 97/68/ЕО се прилага първоначално само към някои двигатели с компресионно запалване, петото съображение от горепосочената директива предвижда в бъдеще разширяване на нейното приложно поле, като по-специално то обхваща и бензиновите двигатели. |
|
(5) |
Газовите емисии, изпускани от малолитражни двигатели с принудително запалване (бензинови двигатели), с които са оборудвани различни типове машини, допринасят значително за задълбочаването на вече идентифицираните проблеми, свързани с качеството на въздуха, които съществуват към настоящия момент или ще се появят в бъдеще, и по-специално, които са свързани с образуването на озона. |
|
(6) |
В САЩ относно газовите емисии, изпускани от малолитражни двигатели с принудително запалване, се прилагат строги стандарти по отношение опазването на околната среда, което показва, че съществува възможност за чувствително намаление на техните вредни емисии. |
|
(7) |
Поради липсата на законодателство на Общността в тази област, е възможно на пазара да бъдат пускани двигатели, които са проектирани по остарели технологии по отношение на защитата на околната среда и които възпрепятстват осъществяването на целите за качество на въздуха в Общността, или дава възможност за прилагане в тази област на национални законодателни инструменти, които биха могли да създават пречки пред търговския обмен. |
|
(8) |
Директива 97/68/ЕО е тясно хармонизирана със съответното американско законодателство, и продължаването на това хармонизиране ще осигури предимства както за промишлеността, така и за околната среда. |
|
(9) |
Необходим е период за подготовка на европейската промишленост, и по-специално на производителите, които все още не развиват дейност на световния пазар, с цел те да бъдат в състояние да спазват нормите по отношение на газовите емисии. |
|
(10) |
Използва се двуетапен подход както в Директива 97/68/ЕО относно двигателите с компресионно запалване, така и в американската нормативна уредба относно двигателите с принудително запалване. Въпреки че би било възможно приемането на едноетапен подход в законодателството на Общността, това би довело до удължаване с четири до пет години положението на липса на нормативна уредба в тази област. |
|
(11) |
С цел да се разполага с необходимата гъвкавост за постигане на уеднаквяване на изискванията в световен план, се предвижда възможност за въвеждане на изключения, които да се прилагат в съответствие с процедурата на Комитета. |
|
(12) |
Необходимо е да се приемат съответните мерки, необходими за прилагане на настоящата директива в съответствие с решение 1999/468/ЕО на Съвета от 28 юни 1999 г., в което се определят начините на упражняване на дадените на Комисията изпълнителни правомощия (5). |
|
(13) |
Имайки предвид гореизложеното, необходимо е Директива 97/68/ЕО да бъде изменена, |
ПРИЕХА НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Директива 97/68/ЕО се изменя, както следва:
|
1. |
Към член 2:
|
|
2. |
Член 4 се изменя както следва:
|
|
3. |
В член 7 параграф 2 се заменя със следния текст: „2. Държавите-членки приемат като съответстващи на настоящата директива, изброените в приложение XII типови одобрения, както и съответните знаци за одобрение, ако има такива.“ |
|
4. |
Член 9 се изменя, както следва:
|
|
5. |
Добавя се следният член: „Член 9а График — Двигатели с принудително запалване За целите на настоящата директива двигателите с принудително запалване се разпределят в следните класове. Главен клас S: малолитражни двигатели с нетна мощност ≤ 19 kW Главен клас S се разделя на две категории: Н: двигатели, предназначени за преносими устройства N: двигатели, предназначени за непреносими устройства
След 11 август 2004 г. държавите-членки нямат право да отказват да предоставят типово одобрение на даден тип или фамилия двигатели с принудително запалване или да отказват издаването на посочения в приложение VII документ, нито да налагат други изисквания за типово одобрение, свързани с изпусканите замърсяващи емисии от мобилните устройства, непредназначени за движение по път, оборудвани с двигател, ако този двигател отговаря на изискванията на настоящата директива относно изпусканите замърсяващи газови емисии. Държавите-членки отказват извършване на типово одобрение на даден тип двигател или фамилия двигатели и издаването на посочените в приложение VII документи и отказват да предоставят всяко друго типово одобрение на мобилните устройства, непредназначени за движение по път, оборудвани с двигател след 11 август 2004 г., ако въпросният/те двигател/и не отговаря/т на изискванията на настоящата директива и ако неговите/техните замърсяващи газови емисии не отговарят на посочените пределни стойности, указани в таблицата в приложение I, точка 4.2.2.1. Държавите-членки отказват да предоставят типово одобрение на даден тип двигател или фамилия двигатели и издаването на посочените в приложение VII документи, и отказват да предоставят всяко друго типово одобрение на мобилните устройства, непредназначени за движение по път, които са оборудвани с двигател:
ако тези двигатели не отговарят на изискванията на настоящата директива и ако изпусканите от тях замърсяващи газови емисии не съответстват на посочените пределни стойности в таблицата в приложение I, точка 4.2.2.2. Шест месеца след посочените дати в параграф 3 и 4 за съответните категории двигатели, с изключение на машините и двигателите, предназначени за износ в трети страни, държавите-членки разрешават пускането на пазара на двигатели, независимо дали те са вече монтирани или не на машини, единствено, ако те отговарят на изискванията на настоящата директива. Държавите-членки разрешават обозначаването и специалната маркировка на типове или фамилии двигатели, които отговарят на допустимите стойности, указани в таблицата в приложение I, точка 4.2.2.2, преди датите, посочени в точка 4 на настоящия член, за да укажат, че съответното устройство отговаря на изискваните допустими стойности преди предвидените срокове. Следните машини се освобождават от спазване на сроковете за прилагане на максималните стойности на емисиите, определени за етап II, за период от три години, след влизането в сила на тези максимални стойности. През тези три години се прилагат определени за етап I максимални стойности на газовите емисиите за: — преносими моторни резачки: преносими устройства, предназначени за рязане на дърва с верижен трион, които изискват да се държат с две ръце, и са с работен обем на цилиндъра над 45 cm3, съгласно стандарт EN ISO 11681-1, — машини, оборудвани с дръжка в горния край (като например пробивни машини и преносими режещи машини, предназначени за използване при поддръжка на дървета): преносими устройства, оборудвани с дръжка в горния край, предназначени за пробиване на отвори или рязане на дърво с верижен трион (съгласно стандарт ISO 11681-2), — преносима машина за разчистване на храсти с двигател с вътрешно горене (храсторез): преносимо оборудване с ротативна метална или пластмасова работна повърхност, предназначено за премахване на плевели, храсти, малки дръвчета и друга подобна растителност. То трябва да бъде проектирано в съответствие със стандарт ISO 11806, за да работи в различни положения, например хоризонтално или в обърнато положение и да има работен обем на двигателя над 40 cm3, — преносими резачки за подрязване на жив плет: преносими устройства, проектирани за рязане на жив плет и храсти с помощта на една или няколко режещи повърхности, движещи се възвратно-постъпателно съгласно стандарт EN 774, — преносими циркулярни триони, работещи с двигател с вътрешно горене: преносими устройства, проектирани за рязане на твърди материали, като камък, асфалт, бетон или стомана с помощта на ротативна метална режеща повърхност и оборудвани с двигатели с работен обем надвишаващ 50 cm3, съгласно стандарт EN 1454, — непреносими двигатели от клас SN:3 с хоризонтална ос: единствено за непреносимите двигатели от клас SN:3 с хоризонтална ос на задвижване и произвеждащи енергия, равна или по-малка от 2,5 kW, използвани главно за определени промишлени цели, включително в мотокултиватори, косачки с цилиндрови двигатели, разрохквачи за зелени площи и генератори. Въпреки това, за всяка от категориите държавите-членки могат да отсрочат до две години посочените в параграфи 3, 4 и 5 дати по отношение на двигателите, чиито дати на производство предхождат тези дати.“ |
|
6. |
Член 10 се изменя, както следва:
|
|
7. |
Членове 14 и 15 се заменят със следните членове: „Член 14 Привеждане в съответствие с техническия прогрес Промените, които са необходими за привеждане в съответствие с техническия прогрес на приложенията към настоящата директива, с изключение на изискванията, посочени в приложение I, точка 1, точки от 2.1 до 2.8; и точка 4, се приемат от Комисията съобразно процедурата по член 15, параграф 2. Член 14а Процедура за дерогации Комисията разглежда евентуалните технически трудности, които могат да възникнат при спазването на изискванията, фиксирани за етап II при някои видове експлоатация на двигателите, по-специално при мобилните устройства, оборудвани с двигатели от класове SH:2 и SH:3. Ако прегледът на Комисията покаже, че по технически причини някои мобилни устройства, а по-специално оборудваните с преносими двигатели с професионално предназначение и работещи в различни положения, не могат да спазят тези срокове, тя представя към 31 декември 2003 г. доклад придружен със съответни предложения, които предвиждат за тези устройства удължаване на срока, посочен в член 9а, параграф 7, и/или други режими на отменяне на прилагането за срок от максимум пет години, освен ако са налице извънредни обстоятелства съгласно процедурата, предвидена в член 15, параграф 2. Член 15 Комитет за привеждане в съответствие с техническия прогрес 1. Комисията се подпомага в своята работа от Комитета за привеждане в съответствие с техническия прогрес на директивите относно отстраняването на пречките пред търговския обмен в областта на моторните превозни средства (наречен по-долу „комитет“). 2. В случай, че се прави позоваване на настоящия параграф, се прилагат членове 5 и 7 на Решение 1999/468/ЕО (6), като се спазват разпоредбите на член 8 от него. Периодът, предвиден в член 5, параграф 6 от Решение No 1999/468/ЕО се определя на три месеца. 3. Комитетът приема свой вътрешен правилник. |
|
8. |
Следният списък на приложенията се добавя преди текста на самите приложения. „Списък на приложенията
|
|
9. |
Приложенията се променят съобразно приложението към настоящата директива. |
Член 2
1. Държавите-членки въвеждат в действие необходимите законови, нормативни и административни разпоредби, за да се съобразят с настоящата директива, преди 11 август 2004 г. Те незабавно уведомяват Комисията за това.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваването на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
2. Държавите-членки уведомяват Комисията за текста на разпоредбите от националното законодателство, които те приемат в областта, регулирана от настоящата директива.
Член 3
Най-късно до 11 август 2004 г. Комисията представя на Европейския парламент и на Съвета доклад, а при необходимост и предложение, относно себестойността и потенциалните предимства, както и относно възможността за техническото изпълнение на намаляването на емисиите от:
|
а) |
замърсяващи частици, изпускани от малолитражните двигатели с принудително запалване, като обръщат особено внимание на двутактовите двигатели. Този доклад държи сметка за следните елементи:
|
|
б) |
превозни средства за отдих и развлечение, по-специално моторни шейни и картове, които не са посочени в момента; |
|
в) |
от отработени газове и частици, изпускани от малолитражните двигатели с компресионно запалване, чиято мощност е по-ниска от 18 kW; |
|
г) |
от отработени газове и частици, изпускани от двигатели на локомотиви с компресионно запалване. Необходимо е да се изработи изпитвателен цикъл за измерването на този тип замърсяващи емисии. |
Член 4
Настоящата директива влиза в сила в деня на публикуването ѝ в Официален вестник на Европейските общности.
Член 5
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 9 декември 2002 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
H. C. SCHMIDT
(1) ОВ С 180 Е, 26.6.2001 г., стр. 31.
(2) ОВ C 260, 17.9.2001 г., стр. 1.
(3) Становище на Европейския парламент от 2 октомври 2001 г. (ОВ С 87 Е, 11.4.2002 г., стр. 18), Обща позиция на Съвета от 25 март 2002 г. (ОВ С 145 Е, 18.6.2002 г., стр. 17) и Решение на Европейския парламент от 2 юли 2002 г. (все още непубликувано в Официален вестник).
(4) ОВ L 59, 27.2.1998 г., стр. 1. Директива, изменена с Директива 2001/63/ЕО на Комисията (ОВ L 227, 23.8.2001 г., стр. 41).
(5) ОВ L 184, 17.7.1999 г., стр. 23.
(6) ОВ L 184, 17.7.1999 г., стр. 23.“
ПРИЛОЖЕНИЕ
|
1. |
Приложение I се изменя, както следва:
|
|
2. |
Приложение II се изменя, както следва:
|
|
3. |
Приложение III се изменя, както следва:
|
|
4. |
Добавя се следното приложение. „ПРИЛОЖЕНИЕ IV ПРОЦЕДУРА НА ИЗПИТВАНЕ ЗА ДВИГАТЕЛИ С ПРИНУДИТЕЛНО ЗАПАЛВАНЕ 1. ВЪВЕДЕНИЕ 1.1. Настоящото приложение описва метода, който следва да се прилага за измерване на замърсяващите газовите емисии на двигатели, които се подлагат на изпитване. 1.2. Изпитването се провежда с двигател, който е монтиран върху изпитвателен стенд и с свързан с динамометър. 2. УСЛОВИЯ ЗА ПРОВЕЖДАНЕ НА ИЗПИТВАНЕТО 2.1. Условия за тестово изпитване на двигателя Измерват се абсолютната температура (Ta) на входящия въздух в двигателя, изразена в Kelvin и атмосферното налягане в суха среда (ps), измерено в kPa, а параметърът а се определя по следния метод:
2.1.1. Валидност на изпитването За да бъде призната валидността на изпитването, параметърът f a трябва да бъде:
2.1.2. Двигатели с охлаждане на въздуха на турбозахранването Температурата на работното тяло на охлаждането и на въздуха на турбозахранването трябва да бъдат записани. 2.2. Система за всмукване на въздух в двигателя Двигателят, който се подлага на изпитване, трябва да бъде снабден със система за всмукване на въздух, фиксирана на ± 10 % от горната граница, посочена от производителя, при наличие на нов въздушен филтър и работа на двигателя при нормални условия, както те са посочени от производителя, така че да се получи максимален дебит на въздух. За малолитражните двигатели с принудително запалване (с работен обем на цилиндъра < 1 000 cm3), трябва да се използва система, която е представителна за инсталирания двигател. 2.3. Система за отвеждане на отработените газове на двигателя Двигателят, който се подлага на изпитване, трябва да бъде снабден със система за отвеждане на отработените газове, работеща с противоналягане и регулирана на ± 10 % от горната граница, посочена от производителя за двигателя, при положение, че той работи при условия, позволяващи постигане на обявената максимална мощност при съответния режим. За малолитражните двигатели с принудително запалване (с работен обем на цилиндъра < 1 000 cm3), трябва да се използва система, която е представителна за инсталирания двигател. 2.4. Система на охлаждане Системата за охлаждане на двигателя трябва да бъде в състояние да поддържа двигателя при нормалните работни температури, предписани от производителя. Тази разпоредба се прилага към устройствата, които трябва да бъдат демонтирани, за да може да се измери мощността, например в случай, когато трябва да се демонтира вентилатора или нагнетяващия вентилатор (на охлаждането) на двигателя, за да се открие достъп да коляновия вал. 2.5. Смазочно масло За даден двигател и за определена експлоатация се използва смазочно масло, което отговаря на спецификациите на производителя на двигателя. Производителите трябва да използват смазочни моторни масла, които са представителни за наличните в търговската мрежа смазочни масла. Характеристиките на смазочното масло, използвано за изпитването, се вписват в приложение VII, допълнение 2, точка 1.2, за двигателите с принудително запалване, и се представят с резултатите от изпитването. 2.6. Регулируеми карбуратори Двигателите, които са снабдени с карбуратори с ограничено регулиране, трябва да преминат на изпитване при двете крайни положения на регулировката. 2.7. Гориво за тестовото изпитване Необходимо е да се използва еталонното гориво, посочено в приложение V. За двигателите с принудително запалване в приложение VII, допълнение 2, точка 1.1.1 са посочени октановото число и обемната маса на еталонното гориво, използвано за изпитването. При двутактовите двигатели съотношението на сместа гориво/масло трябва да отговаря на предписаното съотношение от производителя. Процентното съдържание на масло в сместа гориво/масло, която захранва двутактовия двигател, и така получената обемна маса гориво, са указани в приложение VII, допълнение 2, точка 1.1.4 за двигателите с принудително запалване. 2.8. Определяне на регулировките на динамометъра Измерването на емисиите се основава на некоригираната спирачна мощност. Допълнителните устройства, които служат само за работата на самото оборудване и които могат да бъдат монтирани на двигателя, могат да бъдат демонтирани за изпитването. Ако тези допълнителни устройства не се демонтират, погълнатата от тях мощност трябва да се определи, за да се изчислят регулировките на динамометъра, освен когато тези допълнителни устройства представляват неразделна част от двигателя (например вентилаторите за охлаждане на двигателите с въздушно охлаждане). При двигателите, които позволяват извършване на подобно коригиране, регулировките на входящото разреждане и на противоналягането в тръбата за отвеждане на отработените газове се настройват в горните граници, посочени от производителя, в съответствие с точки 2.2. и 2.3. Максималните стойности на въртящия момент в указаните изпитвателни режими се определят експериментално, за да може да се изчислят стойностите на въртящия момент за различните режими на изпитването. За двигатели, които не са създадени да работят в един диапазон от режими според крива на въртящ момент при пълно натоварване, максималният въртящ момент в изпитвателните режими се обявява от производителя. Регулировката на двигателя за всеки от режимите на изпитване се изчислява с помощта на следната формула:
където:
При съотношение:
стойността на РАЕ може да се провери от техническия орган, отговарящ за типовото одобрение. 3. ПРОВЕЖДАНЕ НА ИЗПИТВАНЕТО 3.1. Монтиране на измервателното оборудване Апаратурата и сондите за вземане на проби трябва да се инсталират в съответствие с предписанията. Когато се използва система за разреждане на отработените газове към главния кръг, системата трябва да бъде свързана към края на тръбата за отвеждане на отработените газове. 3.2. Задействане на системата за разреждане на газовете и на двигателя Системата за разреждане на газовете и двигателят трябва да бъдат задействани и подгряти, докато всички температури и налягания се стабилизират при пълно натоварване и при номинален режим (точка 3.5.2). 3.3. Регулиране на коефициента на разреждане Общият коефициент на разреждане не трябва да е по-малък от 4. При системите, които измерват концентрациите на CO2 или на NOx, съдържанието на CO2 или на NOx в първичния въздух трябва да бъде измерено в началото и в края на всяко изпитване. Отклонението между концентрациите на CO2 или на NOx в първичния въздух преди или след изпитването, не трябва да надвишава 100 милионни части (ppm) или 5 милионни части (рpm) за всяка от тях. Когато се използва система за анализ на разредените отработени газове, фоновите концентрации, които се вземат предвид, се определят чрез вземане на проба от първичния въздух в торбичка през цялото времетраене на изпитването. Измерването без прекъсване на фоновата концентрация (без торбичка за проби) може да се извърши най-малко три пъти: в началото, в края и към средата на цикъла, след което се изчисляват получените средни аритметични стойности. Ако производителят поиска, може да не се проведе измерване на фоновата концентрация. 3.4. Проверка на анализаторите Анализаторите на емисиите се нулират и еталонират. 3.5. Изпитвателен цикъл Спецификация с) на оборудването, посочено в приложение I, точка 1 а) iii). Прилагат се следните изпитвателни цикли за работа на изпитвания двигател на динамометричния стенд, в зависимост от съответното оборудване:
3.5.1.1. Режими на изпитване и тегловни коефициенти
3.5.1.2. Избор на подходящия изпитвателен цикъл Ако е известно каква е основната употреба на даден модел двигател, изпитвателният цикъл може да се избере въз основа на дадените примери в точка 3.5.1.3. Ако съществува колебание по отношение на основната употреба, подходящият изпитвателен цикъл се избира според спецификацията на двигателя. 3.5.1.3. Примери (неизчерпателен списък) Типични примери в зависимост от циклите: Цикъл D:
Цикъл G1:
Цикъл G2:
Цикъл G3:
3.5.2. Привеждане на двигателя до работна температура Двигателят и системата трябва да бъдат приведени до работна температура при максималните стойности на режима и на въртящия момент, за да се стабилизират параметрите на двигателя съгласно препоръките на производителя. Забележка: Фазата на предварително подгряване трябва също да позволи отстраняване влиянието на отлагания в системата за отвеждане на отработените газове, останали от предишно изпитване. Необходимо е да се предвиди също и фаза на стабилизиране между моментите на извършване на изпитвания, с цел да се сведе до минимум влиянието, които те биха могли да окажат един на друг. 3.5.3. Протичане на изпитванията Изпитвателните цикли G1, G2 или G3 се провеждат при спазване на възходящия ред на режимите, така както е посочено по-горе за съответния цикъл. Времето за вземане на проби за всеки режим трябва да бъде с продължителност минимум 180 секунди. Концентрациите на отработените емисии се измерват и записват през последните 120 секунди от съответното време за вземане на пробата. Продължителността на режима трябва да бъде достатъчно дълга за всяка точка на измерване, за да може двигателят да се стабилизира при работната си температура преди началото на вземане на пробата. Времетраенето на режима на работа трябва да се запише и представи в доклада за изпитването.
3.5.4. Реагиране на анализаторите Данните, които постъпват от анализаторите, се регистрират с помощта на записващо лентово устройство или се измерват с помощта на еквивалентна система за обработване на данни, а отработените газове трябва да преминават през анализаторите най-малко през последните 180 секунди на всеки цикъл. Ако торбичките за вземане на проби са използвани за измерване на разтворените CO и CO2 (виж допълнение 1, точка 1.4.4), е необходимо да се вземе мостра през последните 180 секунди на всеки изпитвателен режим, след това тя да се анализира, а резултатите от анализа да се регистрират. 3.5.5. Параметри на двигателя Режимът и натоварването на двигателя, температурата на входящия въздух и разходът на гориво трябва да се измерват за всеки изпитвателен режим след стабилизирането на двигателя. Всички други данни, които са необходими за изчисляването, трябва да бъдат записани (виж допълнение 3, точки 1.1 и 1.2). 3.6. Повторна проверка на анализаторите След изпитването за измерване на емисиите, за извършване на повторна проверка на анализаторите се използва един и същ газ за нулиране и за регулиране на чувствителността. Изпитването се счита за редовно, ако разликата в получените резултати между двете измервания е по-малка от 2 %. Допълнение 1 1. МЕТОДИ ЗА ИЗМЕРВАНЕ И ЗА ВЗЕМАНЕ НА ПРОБИ Съставните газове, изпускани от изпитвания двигател, трябва да се измерват по описаните в приложение VI методи. Тези методи определят системите за анализ, които се препоръчват за измерването на газовите емисии (точка 1.1). 1.1. Спецификация на динамометъра Използва се динамометричен стенд за двигатели, чиито характеристики са достатъчни за изпълнение на предписания в приложение IV, точка 3.5.1 изпитвателен цикъл. Измервателните уреди на въртящия момент и на скоростта трябва да позволяват измерване на спирачната мощност в посочените граници. Възможно е да се наложи извършването на допълнителни изчисления. Точността на тези измервателни инструменти трябва да бъде такава, че да не допуска надвишаване на максимално допустимите отклонения, посочени в точка 1.3. 1.2. Разход на гориво и общо количество на разредения дебит Разходомерите, които се използват за определяне на разхода на гориво, който се взема под внимание при изчисляване на емисиите (допълнение 3) трябва да притежават определената в точка 1.3 точност на измерване. Ако се използва система за разреждане към главния кръг, общият дебит на разредените отработени газове (GTOTW) трябва да бъде измерен с помощта на система PDP или CFV — приложение VI, точка 1.2.1.2. Точността на измерване трябва да съответства на разпоредбите на приложение III, допълнение 2, точка 2.2. 1.3. Прецизност на измерване Еталонирането на всички измервателни инструменти трябва да се извършва съгласно националните (международните) стандарти и да съответства на изискванията на таблици 2 и 3. Таблица 2 – Допустима неточност на инструментите за измерване на параметрите на двигателя
Таблица 3 — Допустима неточност при показанията на инструментите за измерване на останалите основни параметри
1.4. Определяне на газовите компоненти 1.4.1. Общи изисквания относно анализаторите Анализаторите трябва да притежават измервателен диапазон, който да съответства на точността, изисквана за измерване на концентрациите на компонентите на отработените газове (точка 1.4.1.1). Препоръчително е анализаторите да се използват по такъв начин, че измерваната концентрация да бъде в рамките от 15 % до 100 % от пълната скала. Концентрации по-ниски от 15 % от пълната скала също се приемат за допустими, ако стойността на пълната скала е 155 ppm (или ppm C) или по-ниска, или ако се използват системи за отчитане на показанията (компютри, устройства за записване на данни), които показват с достатъчна точност и разделителна способност стойности, по-ниски от 15 % от пълната скала. В този случай трябва да се извършат допълнителни еталонирания, за да се гарантира точността на кривите на еталониране (допълнение 2, точка 1.5.5.2 от настоящото приложение). Оборудването също така трябва да има степен на електромагнитна съвместимост (CEM), която да е в състояние да намали до минимум допълнителните грешки. 1.4.1.1. Точност на измерване Анализаторът не трябва да се отклонява от номиналната точка на еталониране с повече от ± 2 % от отчетената стойност върху цялата измервателна скала с изключение на нулево положение, когато отклонението не трябва да бъде повече от ± 0,3 % от пълната скала. Точността се определя в съответствие с изискванията за еталониране, посочени в точка 1.3. 1.4.1.2. Повторяемост Резултатите при повторяемостта трябва да са такива, че 2,5 пъти типовото отклонение при 10 последователни измервания на даден газ, използван за еталониране или за регулиране на чувствителността, не трябва да се отклонява с повече от ± 1 % от концентрацията при пълната скала за всеки използван диапазон над 100 милионни части (ppm) (или ppmC) или ± 2 % от всеки използван диапазон под 100 милионни части (ppm) (или ppmC). 1.4.1.3. Фонов шум Реакцията между два съседни пика на анализатора при газове, използвани за нулиране и еталониране или за регулиране на чувствителността, по време на който и да е 10-секунден период, не трябва да надвишава 2 % от пълната скала при всички използвани диапазони. 1.4.1.4. Отклонение от нулата Нулевото показание се определя като средната реакция, включително фоновия шум, на газ, използван за нулиране, по време на интервал от 30 секунди. Отклонението от нулата по време на период от един час трябва да бъде по-ниско от 2 % от пълната скала при използвания най-нисък диапазон. 1.4.1.5. Отклонение от скалата Показанието на горната точка на скалата се определя като средното показание, включително фоновия шум, отбелязано от газ, използван за регулиране на чувствителността по време на интервал от 30 секунди. Отклонението на показанието от горната точка на скалата за период от един час трябва да бъде по-ниско от 2 % от пълната скала при използвания най-нисък диапазон. 1.4.2. Изсушаване на газовете Отработените газове могат да бъдат измервани при наличие и отсъствие на кондензируеми фракции. Всяко евентуално използвано устройство за премахване на тези фракции трябва да има минимално влияние върху концентрацията на измерваните газове. Химическите изсушители не могат да бъдат използвани в качеството си на метод за елиминиране на водата от мострата. 1.4.3. Анализатори Точки от 1.4.3.1 до 1.4.3.5 от настоящото допълнение описват принципите на измерване, които трябва да се прилагат. приложение VI дава детайлизирано описание на измервателните системи. Газовете, които ще се измерват, се анализират с помощта на описаните по-долу устройства. Използването на линеаризационни контури се разрешава при нелинейни анализатори. 1.4.3.1. Анализ на въглеродния оксид (CO) Анализаторът за въглероден оксид трябва да бъде от недисперсивен тип с поглъщане в инфрачервения спектър (Non-Dispersive InfraRed или NDIR). 1.4.3.2. Анализ на въглеродния диоксид (CO2) Анализаторът за въглероден диоксид трябва да бъде от недисперсивен тип с поглъщане в инфрачервения спектър (Non-Dispersive InfraRed или NDIR). 1.4.3.3. Анализ на кислорода (O2) Анализаторите на кислород трябва да бъдат от тип, работещ с парамагнитен детектор (PMD), със сонда с циркониев диоксид (ZRDO) или с електрохимическа клетка (ECS). Забележка: Анализаторите със сонда с циркониев диоксид не се препоръчват, когато концентрациите на въглеводороди (HC) и на въглероден оксид (CO) са високи, както е в случая с двигателите с принудително запалване, които работят с бедна горивна смес. Уредите с електрохимическа клетка трябва да имат компенсатор на интерференцията на въглеродния диоксид (CO2) и на азотните оксиди (NОx). 1.4.3.4. Анализ на въглеводородите (HC) Когато се вземат директно проби на газове, анализаторът на въглеводородите трябва да бъде от вида загрят детектор на йонизиране на пламък (Heated Flame Ionisation Detector или HFID) и да бъде оборудван с нагорещени детектор, клапани, тръбопроводи и т. н., така че газовете да се поддържат при температура от 463 К ± 10 K (190 °С ± 10 °C). Когато се вземат проби на газове с разреждане, анализаторът на въглеводородите трябва да бъде от типа загрят детектор на йонизиране на пламък (Heated Flame Ionisation Detector или HFID) или детектор на йонизиране на пламък (FID). 1.4.3.5. Анализ на азотните оксиди (NOx) Анализаторът на азотните оксиди трябва да бъде от вида детектор с химическа луминесценция (ChemiLuminescent Detector или CLD) или загрят детектор с химическа луминесценция (Heated ChemiLuminescent Detector или HCLD), оборудван с NO2/NO конвертор, ако измерването се извършва при условия на отсъствие на кондензируеми фракции. Ако измерването се извършва при условия на наличие на кондензируеми фракции, трябва да се използва устройство HCLD, оборудван с конвертор, който се поддържа при температура, надвишаваща 328 K (55 °C), при положение че резултатът от проверката за редуциращото въздействие на водата е задоволителен (виж приложение III, допълнение 2, точка 1.9.2.2). При детекторите с химическа луминесценция CLD и загрятите детектори с химическа луминесценция HCLD стената на участъка, през който преминават пробите, трябва да се поддържа при температура от 328 K до 473 K (55 °C до 200 °C) до конвертора за измерване при отсъствие на кондензируеми фракции и до анализатора за измерване при наличие на кондензируеми фракции. 1.4.4. Вземане на проби от газовите емисии Ако върху състава на отработените газове се въздейства с независимо каква система за вторично обработване на отработените газове, пробата от отработени газове трябва да бъде взета след тази система. Сондата за вземане на проби от отработените газове трябва да се постави в точка, намираща се откъм страната на високото налягане на изпускателното гърне, но същевременно колкото се може по-далече от изпускателния отвор. За да осигури пълно смесване на отработените газове на двигателя преди вземането на пробата, може, без да бъде задължително, да се постави смесителна камера между изхода на гърнето за отработените газове и сондата за вземане на проби. Смесителната камера трябва да притежава вътрешен обем, който да не бъде по-малък от десетократния работен обем на двигателя, преминаващ изпитванията, и размерите ѝ трябва да бъдат приблизително едни и същи на височина, ширина и дълбочина, подобно на куб. Размерът на смесителната камера трябва да бъде колкото е възможно най-малък, и тя трябва да бъде свързана в точка, разположена възможно най-близко до двигателя. Линията на излизане на отработените газове от смесителната камера на гърнето за отработените газове трябва да продължава най-малко 610 мм след местоположението на сондата за вземане на проби и трябва да бъде с достатъчен диаметър, за да се намали максимално противоналягането. Температурата на вътрешната стена на смесителната камера трябва да се поддържа над точката на образуване на роса от отработените газове; препоръчва се минимална температура от 338 K (65 °C). Всички компоненти могат по избор да бъдат измервани или директно в тунела за разреждане, или чрез вземане на проби в торбичка за вземане на проби с последващо измерване на концентрацията в торбичката. Допълнение 2 1. ЕТАЛОНИРАНЕ НА ИНСТРУМЕНТИТЕ ЗА АНАЛИЗ 1.1. Въведение Всеки анализатор трябва да бъде еталониран толкова често, колкото е необходимо, за да отговаря на изискванията за точност, наложени от настоящата директива. Тази точка описва метода за еталониране, който се прилага спрямо анализаторите, описани в допълнение 1, точка 1.4.3. 1.2. Газ за еталониране Необходимо е да се спазва времетраенето на съхранение на всички еталониращи газове. Указаната от производителя крайна дата на използване на еталониращите газове трябва да бъде вписана в протокола. 1.2.1. Чисти газове Изискваната чистота за газовете се определя от указаните по-долу гранични стойности на примесите. Могат да се използват следните газове:
1.2.2. Газ за еталониране и за регулиране на чувствителността Използват се газови смеси със следния химически състав:
Забележка: Допустими са и други комбинации от газове, ако съставящите ги газове не реагират едни с други. Ефективната концентрация на даден газ за еталониране и за регулиране на чувствителността трябва да съответства на номиналната стойност с толеранс от ± 2 %. Всички концентрации на еталониращите газове трябва да бъдат указани в обемно съотношение (обемни проценти или милионни (ppm) обемни части). Газовете, използвани за еталониране и за регулиране на чувствителността, могат също така да се получат с помощта на точен смесител-дозатор (газов сепаратор) чрез разреждане с пречистен N2 или с пречистен синтетичен въздух. Точността на смесителя трябва да позволява определянето на концентрацията на разтворените еталониращи газове в рамките на ± 1,5 %. Тази точност налага да се познават първичните газове, използвани за смесването, с точност най-малко от ± 1 %, което позволява сравняване с националните или международни еталонни газове. Контролирането трябва да се извършва между 15 и 50 % от пълната скала за всяко еталониране, при което се използва смесител-дозатор. Като вариант, смесителят-дозатор може да се провери с инструмент с естествен линеен измервател, например чрез използване на газ NO с детектор тип CLD. Регулирането на скалата на измервателния инструмент трябва да се извърши с газ за регулиране на чувствителността, който е свързан директно към инструмента. Смесителят-дозатор трябва да се провери при използваните регулировки и номиналната стойност трябва да се сравни с отчитаната от инструмента концентрация. Получената разлика във всяка една точка трябва да не се различава с повече от ± 0,5 % от номиналната стойност. 1.2.3. Контрол на интерференцията с кислорода Газовете за контрол на интерференцията с кислорода трябва да съдържат пропан с 350 ppm C ± 75 ppm C въглеводороди. Стойността на концентрацията трябва да се определи при допустимия толеранс на еталониращите газове чрез хроматографски анализ на целите въглеводороди плюс примесите или чрез динамично смесване-дозиране. Азотът трябва да бъде доминиращият разтворител заедно с добавката от кислород. Изискваната дозировка при изпитването на бензинови двигатели е както следва:
1.3. Режим на използване на анализаторите и на системата за вземане на проби Режимът на работа на анализаторите трябва да отговаря на инструкциите за задействане и за работа, дадени от производителя на уреда. Минималните изисквания, указани в точки от 1.4 до 1.9, трябва също така да бъдат спазвани. За лабораторните инструменти като хроматографите GC и HPLC (за течна хроматография под високо налягане), се прилагат единствено разпоредбите на точка 1.5.4. 1.4. Изпитване за херметичност Трябва да се извърши едно изпитване за херметичност на системата. За тази цел сондата се откача от системата за отвеждане на отработените газове и краят ѝ се запушва. Помпата на анализатора се пуска в движение. След период на начално стабилизиране, всички разходомери трябва да показват нула. В противен случай трябва да се проверят тръбопроводите за вземане на проби и аномалията да бъде отстранена. Максимално допустимият процент на oтечка в частта, в която се създава вакуум, е от порядъка на 0,5 % от работния дебит за проверяваната част на системата. Дебитите на анализатора и на дериватната система могат да се използват за определяне на дебитите по време на работа. Като вариант, системата може да се изпразни посредством разреждане (вакуум) от най-малко 20 kPa (80 kPa в абсолютно налягане). След период на първоначално стабилизиране, повишаването на налягането δр (в kPa/min) в системата не трябва да надвишава:
където:
Друг метод се състои във въвеждането на постепенна промяна на концентрацията на входа на тръбопровода за вземане на проба, като се извършва превключване между газа за нулиране и газа за регулиране на чувствителността. Ако след достатъчен интервал от време измерената стойност показва концентрация, която е по-ниска от въведената първоначално, това означава, че има проблеми с еталонирането или с oтечки в системата. 1.5. Процедура по еталониране 1.5.1. Еталониране на цялото устройство Цялото устройство трябва да се еталонира и кривите на еталониране се проверяват чрез сравнение с еталонни газове. Трябва да се използват същите дебити на газа, както по време на вземането на проби от отработените газове. 1.5.2. Време за привеждане до работна температура Времето за привеждане до работна температура трябва да е съобразено с препоръките на производителя. Ако липсват указания, се препоръчва да се съблюдава минимално време от два часа за привеждане на анализаторите до работна температура. 1.5.3. Анализатори NDIR и HFID При необходимост анализаторът NDIR трябва да бъде регулиран и горивният пламък на анализатора HFID трябва да бъде оптимизиран (точка 1.9.1). 1.5.4. Хроматографи GC и HPCL Двата инструмента трябва да бъдат еталонирани според методите на лабораторните практики и в съответствие с препоръките на производителя. 1.5.5. Изработване на кривите за еталониране 1.5.5.1. Общи положения
1.5.5.2. Други методи Ако може да се докаже, че друго оборудване (например компютър, електронен превключвател на диапазони) могат да постигнат еквивалентна степен на точност, те също могат да бъдат използвани. 1.6. Проверка на еталонирането Всички нормално използвани диапазони трябва да бъдат проверени преди всеки анализ съгласно описаната по-долу процедура.
1.7. Еталониране на анализатора на трасиращия газ за определяне на дебита на отработените газове Анализаторът, който се използва за измерване на концентрациите на трасиращия газ трябва да се еталонира с помощта на еталонния газ. Кривата на еталониране се начертава, като се свържат най-малко 10 точки на еталониране, с изключение на нулата, които са разположени по такъв начин, че половината от точките да се намират между 4 % и 20 % от пълната скала на анализатора, а останалите — между 20 % и 100 % от пълната скала. Кривата на еталониране се изчислява с помощта на метода на най-малките квадрати. Кривата на еталониране не трябва да се отклонява от номиналната стойност на всяка точка на еталониране с повече от ± 1 % от пълната скала в диапазона от 20 % до 100 % от пълната скала. Тя не трябва също така да се отклонява с повече от ± 2 % от номиналната стойност на точките на еталониране в диапазона от 4 % до 20 % от пълната скала. Нулевата позиция и скалата на анализатора трябва да бъдат регулирани преди изпитването с помощта на газ за нулиране и на газ за регулиране на чувствителността, който има номинална стойност над 80 % от пълната скала на анализатора. 1.8. Изпитване за ефективността на конвертора за NOx Ефективността на конвертора, използван за превръщане на NO2 в NO, се тества по начина, указан в точки от 1.8.1 до 1.8.8 (фигура 1 от приложение III, допълнение 2). 1.8.1. Изпитвателна инсталация С изпитвателната инсталация, показана на фигура 1 от приложение III, и описаната по-долу процедура, може да се провери ефективността на конверторите с помощта на озонатор. 1.8.2. Еталониране Детекторите CLD и HCLD се еталонират съгласно спецификациите на производителя в най-често използвания диапазон с помощта на газ за нулиране и на газ за регулиране на чувствителността (последният трябва да има съдържание на NO, което отговаря на около 80 % от измервателния диапазон и концентрацията на NO2 на газовата смес трябва да бъде по-ниска от 5 % от концентрацията на NO). Анализаторът на NOx трябва да бъде поставен в режим NO, така че газът за регулиране на чувствителността да не преминава през конвертора. Отчетената концентрация трябва да се запише. 1.8.3. Изчисления Ефективността на конвертора за NOx се изчислява както следва:
където:
1.8.4. Добавяне на кислород Добавя се непрекъснато кислород или въздух за нулиране към газовия поток посредством Т-образна връзка, докато отчетената концентрация стане по-ниска с около 20 % от концентрацията за еталониране, указана в точка 1.8.2 (анализаторът е поставен в режим NO). Отчетената стойност на концентрацията (с) трябва се запише. Озонаторът остава дезактивиран по време на цялата процедура. 1.8.5. Активиране на озонатора След това озонаторът се активира, за да създаде достатъчен обем от озон, за да намали концентрацията на NO до около 20 % (минимум 10 %) от концентрацията за еталониране, указана в точка 1.8.2. Отчетената стойност на концентрацията (г) се записва (анализаторът е в режим NO). 1.8.6. Режим на анализиране на NOx След това анализаторът на NO се превключва в режим NOx, така че газовата смес (съставена от NO, от NO2, от O2 и от N2) да преминава през конвертора. Отчетената стойност на концентрацията (а) трябва да бъде записана (анализаторът е регулиран в режим NOx). 1.8.7. Дезактивиране на озонатора След това озонаторът се дезактивира. Газовата смес, описана в точка 1.8.6, преминава през конвертора и достига до детектора. Отчетената стойност на концентрацията (б) се записва (анализаторът е в режим NOx). 1.8.8. Режим на анализиране на NO След превключване в режим NO, и след като озонаторът е дезактивиран, се спира също така притокът на кислород или на синтетичен въздух. Отчетената от анализатора стойност на NOx не трябва да се различава с повече от ± 5 % от стойността, измерена съгласно точка 1.8.2 (анализаторът е в режим NO). 1.8.9. Интервал на провеждане на изпитванията Ефективността на конвертора трябва да се проверява ежемесечно. 1.8.10. Изисквана ефективност Ефективността на конвертора не трябва да бъде по-ниска от 90 %, но се препоръчва най-настоятелно тя да надвишава 95 %. Забележка: Ако анализаторът е регулиран в най-често използвания диапазон и озонаторът не позволява да се постигне намаляването от 80 % до 20 % съгласно точка 1.8.5, е необходимо да се използва най-високият диапазон, който е в състояние да отчете това намаляване. 1.9. Регулировка на FID 1.9.1. Оптимизиране на реагирането на детектора Детекторът HFID трябва да се регулира според указанията на производителя на апаратурата. За да се оптимизира реагирането на детектора в най-често използвания измервателен диапазон, трябва да се използва газ за регулиране на чувствителността, съдържащ пропан и въздух. След регулиране дебита на горивото и на въздуха според препоръките на производителя, в анализатора се вкарва газ за регулиране на чувствителността 350 ± 75 ppm C. Реагирането на определен дебит на горивото се определя въз основа на разликата между реакцията към газа за регулиране на чувствителността и към газа за нулиране. Дебитът на горивото се регулира постъпково над и под предписаната от производителя стойност. Записва се реагирането към газа за регулиране на чувствителността и към газа за нулиране при тези дебити на горивото. Начертава се кривата, която отразява разликата между реагирането към газа за регулиране на чувствителността и към газа за нулиране, и дебитът на горивото се регулира според по-високата стойност на кривата. Тази процедура представлява началното регулиране на дебита и може да се наложи впоследствие извършване на оптимизиране в зависимост от стойностите на коефициента на реагиране към въглеводородите и на резултатите от контрола на интерференцията с кислорода, в съответствие с точки 1.9.2 и 1.9.3. Ако интерференцията с кислорода или коефициентите на реагиране към въглеводородите не отговарят на следните изисквания, дебитът на въздуха трябва да се нагоди постъпково над и под указаните от производителя стойности; процедурите от точки 1.9.2 и 1.9.3 трябва да се повторят за всяка стойност на дебита. 1.9.2. Коефициенти на реагиране към въглеводородите Анализаторът се еталонира, като се използва пропан с въздух с или с пречистен синтетичен въздух, съгласно точка 1.5. Коефициентите на реагиране трябва да се определят при пускането в експлоатация на анализатор и впоследствие след извършване на процедури по цялостен преглед и поддръжка. Коефициентът на реагиране (Rе) на определен тип въглеводороди представлява отношението на стойността Cl, отчетена от FID, към газовата концентрация в бутилката, която се изразява в ppm Cl. Концентрацията на изпитвания газ трябва да бъде достатъчна, за да предизвика реакция, равна на около 80 % от пълната скала. Концентрацията трябва да се знае с точност от ± 2 % по отношение на определен гравиметричен еталон, изразен в обемни части. Освен това газовата бутилка трябва да бъде предварително поддържана в продължение на 24 часа при температура от 298 K (25 °C) ± 5 К. Изпитвателните газове, които трябва да се използват и различните диапазони, препоръчвани за определяне на коефициента на реагиране, са следните:
Тези стойности се отнасят към коефициента на реагиране (Rf) със стойност 1,00 за пропана и за пречистения синтетичен въздух. 1.9.3. Контрол на интерференцията с кислорода Контролът на интерференцията с кислорода се извършва при пускането в експлоатация на анализатор и по-късно, след процедури по извършване на цялостен преглед и поддръжка. Избира се диапазон, в който газовете за контрол на интерференцията с кислорода ще попаднат в частта над 50 % от скалата. Изпитването се извършва с пещ, регулирана на желаната температура. Газовете за определяне на интерференцията с кислорода са определени в точка 1.2.3.
1.10. Ефекти на интерференция с анализаторите на CO, на CO2, на NOx, и на O2 Газовете, които са различни от анализирания газ, могат да повлияят на отчитаните стойности по няколко начина. В инструментите NDIR и PMD се наблюдава положителна интерференция, когато газът, който е причина за интерференцията, предизвиква същия ефект като измервания газ, но в по-ниска степен от него. Отрицателна интерференция се наблюдава от една страна в инструментите NDIR, когато газът, който е причина за интерференцията, разширява диапазона на абсорбция на измервания газ, и от друга страна, в инструментите CLD, когато газът, който е причина за интерференцията, предизвиква затихване на излъчването. Контролиранията на интерференцията, упоменати в точки 1.10.1 и 1.10.2 се извършват преди пускането в експлоатация на анализатор и впоследствие след основни технически обслужвания, и при всички случаи, най-малко един път в годината. 1.10.1. Контрол на интерференцията на анализатора на CO Водата и CO2 могат да повлияят на работата на анализатора на CO. Поради това газ за регулиране на чувствителността към CO2, с концентрация от 80 % до 100 % от пълната скала на максималния диапазон, използван по време на изпитванията, се пречиства чрез преминаване през вода при температура, равна на температурата на околната среда, като показанията на анализатора се записват. Стойността на тези показания не трябва да надвишава 1 % от пълната скала за диапазоните, които са равни или надвишават 300 ppm, нито да надвишава 3 ppm за диапазоните, които са по-ниски от 300 ppm. 1.10.2. Контрол на редуциращия ефект при интерференция на анализатора на NOx Двата газа, имащи отношение към анализаторите CLD (и HCLD) са CO2 и водната пара. Степените на редуциращия ефект на тези газове са пропорционални на техните концентрации и това налага да се прибягва до изпитвания за определяне на редуциращия ефект при очакваните максимални концентрации по време на изпитванията. 1.10.2.1. Контрол на редуциращия ефект при интерференция на анализатора на CO2 През анализатора NDIR се пропуска газ за регулиране на чувствителността към CO2 с концентрация от 80 % до 100 % от пълната скала на максималния диапазон, който се използва при изпитването, и се записва измерената стойност на CO2 (A). След това газът се разрежда до около 50 % с газ за регулиране на чувствителността към NO и се пропуска през NDIR и през (H)CLD, след което се записват измерените стойности на CO2 и на NO (съответно В и С). Пропускането на CO2 се прекъсва, за да може единствено газът за регулиране на чувствителността към NO да преминава през анализатора (H)CLD, след което се записва измерената стойност за NO (D). Редуциращият ефект от интерференцията, който не трябва да бъде по-висок от 3 % от пълната скала, се определя както следва:
където:
Могат да се прилагат и други еквивалентни методи за разреждане и количествено определяне на стойностите на газа за регулиране на чувствителността към CO2 и към NO, като например динамичния метод чрез смесване/чрез дозиране. 1.10.2.2. Контрол на редуциращия ефект при интерференция с водата Това контролиране се прилага единствено при измерванията на концентрацията на газовете, в които има наличие на кондензируеми фракции. При изчисляването на редуциращия ефект от интерференцията с водата трябва да се взема предвид разреждането на газа за регулиране на чувствителността към NO във водната пара, както и съпоставянето на концентрацията на водната пара в сместа по отношение на очакваната по време на изпитването. Газ за регулиране на чувствителността към NO с концентрация от 80 % до 100 % от пълната скала на нормално използвания диапазон, се пропуска през анализатора (H)CLD и измерената стойност за NO се записва като D. След това той трябва да се пречисти чрез преминаване през вода при температура, равна на температурата на околната среда, и се пропуска през анализатора (H)CLD, като измерената стойност за NO се записва като C. Температурата на водата се определя и записва като стойност F. Насищащото налягане на парата в сместа, което съответства на температурата (F) на водата от устройството за промиване на газа, се определя и записва като стойност G. Концентрацията на водната пара (в %) в сместа се изчислява както следва:
и се записва като H. Очакваната концентрация на разредения (с водна пара) газ за регулиране на чувствителността към NO се изчислява както следва:
и се записва като De. Редуциращият ефект от интерференцията с водата не трябва да бъде по-висок от 3 % и се определя както следва:
където:
Забележка: Необходимо е газът за регулиране на чувствителността към NO да съдържа минимална концентрация на NO2 за целите на този контрол, тъй като не е взета под внимание абсорбцията на NO2 във водата при изчисляването на редуциращия ефект от интерференцията. 1.10.3. Интерференция на анализатора на O2 Реагирането на анализатора PDM, дължащо се на газове, които са различни от кислорода, е сравнително слабо. Еквивалентното съдържание в кислород на общите съставки на отработените газове е представено в таблица 1: Таблица1 – Еквивалентно съдържание в кислород
Ако е нужно да се направят измервания с висока степен на точност, трябва да се коригира получената концентрация на кислород с помощта на следната формула:
1.11. Периодичност на еталонирането Анализаторите се еталонират съгласно точка 1.5 не по-рядко от веднъж на всеки 3 месеца или след всяка поправка или промяна на системата, която е в състояние да повлияе на еталонирането. Допълнение 3 1. ОЦЕНКА И ИЗЧИСЛЕНИЕ НА ДАННИТЕ 1.1. Оценка на газовите емисии За да се оценят газовите емисии, се вземат средните стойности, отчетени от графичното регистриращо устройство през най-малко последните 120 секунди на всеки режим и се определят средните концентрации (conс) на HC, на CO, на NOx.и на CO2, постигнати по време на всеки режим въз основа на записаните средни стойности от диаграмите и съответните данни от еталонирането. Може да се използва друг тип записване, ако той гарантира получаването на еквивалентни данни. Средната фонова концентрация (concd) може да се определи въз основа на регистрираните стойности на първичния въздух, който се съдържа в торбичката, или въз основа на стойностите на фоновата концентрация, записани без прекъсване (без вземане на проба в торбичка) и съответните данни от еталонирането. 1.2. Изчисляване на газовите емисии Окончателните резултати от изпитванията се получават чрез извършване на следните операции. 1.2.1. Коригиране за преминаване от сухо към влажно състояние Измерената концентрация, ако не е била определена във влажна среда, трябва да се преобразува в стойност, измерена при влажни условия:
За брутните отработени газове:
където α представлява съотношението водород/въглерод на горивото. Концентрацията на H2 в отработените газове се изчислява по формулата:
Коефициентът Kw2 се изчислява както следва:
където Нa = абсолютна влажност на входящия въздух, измерена в g вода на kg сух въздух За разредените отработени газове: Ако измерването на СО2 е направено при влажни условия, се прилага следното уравнение:
Ако измерването на СО2 е направено при сухи условия, се прилага следното уравнение:
където α представлява съотношението водород/въглерод на горивото. Коефициентът Kw1 се изчислява с помощта на следното уравнение:
където:
За първичния въздух:
Коефициентът Kw1 се изчислява с помощта на следното уравнение:
където:
За входящия въздух (ако е различен от първичния въздух):
Коефициентът Kw2 се изчислява с помощта на следното уравнение:
където Ha = абсолютна влажност на входящия въздух, измерена в g вода на kg сух въздух. 1.2.2. Корекция в зависимост от влажността при NOx Тъй като емисиите от NOx зависят от околните атмосферни условия, концентрацията на NOx трябва да бъде умножена по коефициента KH,която взема под внимание влажността:
където Ha = абсолютна влажност на входящия въздух, измерена в g вода на kg сух въздух. 1.2.3. Изчисляване на тегловните дебити на емисиите Тегловните дебити на емисиите, Gasmass [g/h], за всеки режим се изчисляват както следва:
1.2.4. Изчисление на специфичните емисии Специфичната емисия (g/kWh) се изчислява за всеки съставен елемент:
където Pi = PM, i + PAE, i Когато спомагателните устройства, като например вентилатор или нагнетател на охлаждането не се демонтират за изпитването, мощността, която те поглъщат, се прибавя към резултатите, освен ако тези спомагателни устройства представляват съставна част от двигателя. Мощността на вентилатора или нагнетателя на охлаждането се определя при режимите, използвани при изпитването, или чрез извършване на изчисление според стандартните характеристики, или чрез извършване на практически изпитвания (приложение VII, допълнение 3). Тегловните коефициенти и номера на n използваните режими за горните изчисления са посочени в приложение IV, точка 3.5.1.1. 2. ПРИМЕРИ 2.1. Данни, отчетени за брутните отработени газове на четиритактов двигател с принудително запалване Що се отнася до експерименталните данни (таблица 3), първо се извършват изчисления за режим № 1, после се преминава към другите режими на изпитване, като се прилага същата процедура. Таблица 3 — Експериментални данни за четиритактов двигател с принудително запалване
2.1.1. Коефициент на конвертиране на стойността при сухи условия/стойността при влажни условия kw За да се конвертират измерванията на СО при сухи условия и на CO2 при влажни условия, трябва да се изчисли коефициентът на конвертиране на стойността при сухи условия/стойността при влажни условия kw:
където:
и
Таблица 4 — Стойности при влажни условия на CO и на CO2, в зависимост от режимите на изпитване
2.1.2. Емисии на НC
където:
Таблица 5 — Газови емисии на HC [g/h] в зависимост режимите на изпитване
2.1.3. Емисии на NOx Най-напред трябва да се изчисли коефициентът за корекция според влажността KH за емисиите от NOx:
Таблица 6 — Коефициент за корекция според влажността KH на емисиите от NOx в зависимост от режимите на изпитване
След това се изчислява NOxmass [g/h]:
Таблица 7 — Газови емисии на NOx [g/h] в зависимост от режимите на изпитване
2.1.4. Емисии на CO
Таблица 8 — Газови емисии на CO [g/h] в зависимост от режимите на изпитване
2.1.5. Емисии на CO2
Таблица 9 — Газови емисии на CO2 [g/h] в зависимост от режимите на изпитване
2.1.6. Специфични емисии Специфичната емисия (g/kWh) трябва да се изчислява за всеки съставен елемент поотделно:
Таблица 10 — Газови емисии [g/h] и тегловни коефициенти в зависимост от режимите на изпитване
2.2. Данни, отчетени за брутните отработени газове на двутактов двигател с принудително запалване Що се отнася до експерименталните данни (таблица 11), първо се извършват изчисленията за режим № 1, после се преминава към другите режими на изпитване, като се прилага същата процедура. Таблица 11 — Експериментални данни за двутактов двигател с принудително запалване
2.2.1. Коефициент на корекция на стойност при сухи условия/стойност при влажни условия kw За да се конвертират измерванията на СО при сухи условия и на CO2 при влажни условия, трябва да се изчисли коефициентът на корекция на стойност при сухи условия/стойност при влажни условия kw:
където:
Таблица 12 — Стойности на CO и на CO2 при влажни условия, в зависимост от режимите на изпитване
2.2.2. Емисии на НС
където:
Таблица 13 — Газови емисии на HC [g/h] в зависимост от режимите на изпитване
2.2.3. Емисии на NOx Коефициентът KH за корекция на емисиите от NOx е равен на 1 за двутактовите двигатели:
Таблица 14 — Газови емисии на NOx [g/h] в зависимост от режимите на изпитване
2.2.4. Емисии на CO
Таблица 15 — Газови емисии на CO [g/h] в зависимост от режимите на изпитване
2.2.5. Емисии на CO2
Таблица 16 — Газови емисии на CO2 [g/h] в зависимост от режимите на изпитване
2.2.6. Специфични емисии Специфичните емисии (g/kWh) трябва да се изчисляват за всеки съставен елемент поотделно:
Таблица 17 — Газови емисии [g/h] и тегловни коефициенти за два режима на изпитване
2.3. Данни, отчетени за брутните отработени газове на четиритактов двигател с принудително запалване Що се отнася до експерименталните данни (таблица 18), първо се извършват изчисленията за режим № 1, после се преминава към другите режими на изпитване, като се прилага същата процедура. Таблица 18 — Експериментални данни за четиритактов двигател с принудително запалване
2.3.1. Коефициент на корекция на стойността при сухи условия/стойността при влажни условия kw За да се конвертират измерванията на СО при сухи условия и на CO2 при влажни условия, трябва да се изчисли коефициентът на корекция на стойността при сухи условия/стойността при влажни условия kw. За разредените отработени газове:
където:
Таблица 19 — Стойности на CO и на CO2 при влажни условия за разредените отработени газове в зависимост от режимите на изпитване
За първичния въздух:
където коефициентът kw1 е същият като вече изчисленият коефициент за разтворените отработени газове. kw,d = 1 - 0,007 = 0,993
Таблица 20 — Стойности на CO и на CO2 за влажни условия за първичния въздух в зависимост от режимите на изпитване
2.3.2. Емисии на НС
където:
Таблица 21 — Газови емисии на HC [g/h] в зависимост от режимите на изпитване
2.3.3. Емисии на NOx Коефициентът KH за корекция на емисиите от NOx трябва да се изчисли по следния начин:
Таблица 22 — Коефициент за корекция според влажността KH на емисиите от NOx в зависимост от режимите на изпитване
където:
Таблица 23 — Газови емисии на NOx [g/h] в зависимост от режимите на изпитване
2.3.4. Емисии на CO
където:
Таблица 24 — Газови емисии на CO [g/h] в зависимост от режимите на изпитване
2.3.5. Eмисии на CO2
където:
Таблица 25 — Газови емисии на CO2 [g/h] в зависимост от режимите на изпитване
2.3.6. Специфични емисии Специфичните емисии (g/kWh) трябва да се изчисляват за всеки съставен елемент поотделно както следва:
Таблица 26 — Газови емисии [g/h] и тегловни коефициенти в зависимост от режимите на изпитване
Допълнение 4 1. СПАЗВАНЕ НА НОРМИТЕ, ОПРЕДЕЛЕНИ ЗА ЕМИСИИТЕ Настоящото допълнение се прилага единствено за двигателите с принудително запалване, като то започва от етап II. 1.1. Нормите за емисиите на отработените газове, изпускани от двигателите по време на изпитванията от етап II, които са определени в приложение I, допълнение 4.2, се прилагат за емисиите на двигателите, когато те се намират в период на устойчивост на характеристиките на емисиите (ПУХЕ) (PDCE), като този период е определен съгласно настоящото допълнение. 1.2. За всички двигатели, които преминават през изпитване през етап II, ако когато се подложени на изпитванията според предвидените в настоящата директива съответни процедури, всички изпитвани двигатели, които представляват една фамилия двигатели, изпускат емисии, които, след като са коригирани чрез умножаване на коефициента на влошаване (DF) (КВ), предвиден в настоящото допълнение, са по-ниски или равни на всяка от емисионните норми за етап II [пределно ниво за емисии по фамилии (FEL) при необходимост] за даден клас двигатели, за тази фамилия се признава, че отговаря на нормите за емисии за този клас двигатели. Ако даден изпитван двигател, които представлява фамилия двигатели, изпуска емисии, които след коригиране чрез умножаване на коефициента на влошаване (DF) (КВ), предвиден в настоящото допълнение, надвишават всяка от емисионните норми (FEL, при необходимост) за даден клас двигатели, се приема, че тази фамилия не отговаря на нормите за емисии за този клас двигатели. Производителят на малки серии двигатели може да приеме опционално коефициентите на влошаване, фигуриращи в таблици 1 или 2 на настоящата точка за HC + NOx и CO, или да изчисли коефициентите на влошаване за тези две категории замърсители, като следва процедурата, описана в точка 1.3.1. За технологиите, които не са взети под внимание в таблици 1 и 2 на настоящата точка, производителят трябва да използва процедурата, описана в точка 1.4 на настоящото допълнение. Таблица 1: преносими двигатели — емисии на HC + NOx и на CO — коефициенти на влошаване, определени предварително, за да бъдат взети под внимание от производителите на малки серии двигатели
Таблица 2: непреносими двигатели — емисии на HC + NOx и на CO — коефициенти на влошаване, определени предварително, за да бъдат взети под внимание от производителите на малки серии двигатели
1.3.1. Формула за изчисляване на коефициентите на влошаване за двигателите с устройство за вторична преработка на газовете:
където:
Производителите според случая избират един предварително определен коефициент на влошаване DF, или го изчисляват за всеки регламентиран замърсител, за всички фамилии двигатели, изпитвани през етап II. Тези коефициенти на влошаване DF се използват при изпитванията за типово одобрение и при изпитванията на поточните производствени линии. За двигателите, при които не се използват предварително определени коефициенти на влошаване (DF), фигуриращи в таблици 1 или 2 на настоящата точка, DF се определят по следния начин: 1.4.1.1. Най-малко върху един изпитван двигател, представляващ избраната конфигурация, и за който се смята, че има най-голяма възможност да надвиши нормите за емисии на HC + NOx (или при необходимост на FEL), и който е произведен, за да бъде представителен за произведените двигатели, се прилага цялата процедура за изпитване относно емисиите, описана в настоящата директива, след съответния брой часове на работа, необходим за стабилизиране на емисиите. 1.4.1.2. Ако се подлагат на изпитване няколко двигателя, се отчита средноаритметичната стойност на резултатите и тя се закръгля до същия брой числа след запетаята, като фигуриращите в прилаганата норма, но с едно допълнително значещо число. 1.4.1.3. Тези изпитвания относно емисиите се повтарят след остаряването на двигателя. Процедурата по стареенето трябва да бъде предвидена, за да позволи на производителя да предвиди правилно влошаването на работните характеристики на емисиите, което се очаква в периода на устойчивост на параметрите на двигателя, като в същото време държи сметка за типа на износване и за други фактори на влошаване, които са очаквани при типичните условия на експлоатация, които биха могли да се отразят на качествените показатели на емисиите. Ако се подлагат на изпитване няколко двигателя, се отчита средноаритметичната стойност на резултатите и тя се закръгля до същия брой числа след запетаята, като фигуриращите в прилаганата норма, но с едно допълнително значещо число. 1.4.1.4. В края на периода на устойчивост записаните емисии (при необходимост – средните стойности на емисиите) се делят за всеки регламентиран замърсител на стабилизираните емисии (при необходимост — средните стойности на емисиите) и резултатът се закръгля до две значещи цифри. Числото, което се получава като резултат от тази операция, представлява коефициента на влошаване КВ (DF), освен ако то е под 1,00, тогава се приема, че КВ (DF) е 1,0. 1.4.1.5. По избор на производителя могат да бъдат програмирани допълнителни точки на изпитване между точката на изпитване на стабилизираните емисии и изпитванията, които се извършват в края на периода на устойчивост на характеристиките на емисиите. Ако се програмират междинни изпитвания, точките на изпитване трябва да бъдат равномерно разпределени в периода на устойчивост на характеристиките на емисиите (ПУХЕ) (PDCE) (около два часа), а една от тези точки на изпитване трябва да се намира в средата на този период (ПУХЕ) (PDCE) (около два часа). За всеки замърсител HC + NOx и CO се начертава права линия между точките с данни, като се има предвид, че началните изпитвания се провеждат в час нула, и като се прилага метода на най-малките квадрати. Коефициентът на влошаване се изчислява като се разделят записаните емисии в края на периода на устойчивост на емисиите, записани в час нула. 1.4.1.6. Коефициентите на влошаване могат да включат други фамилии, различни от тези на чиято база те са били изчислени, при условие че производителят докаже убедително на компетентния национален орган по одобрението и преди типовото одобрение, че логично може да се очаква въпросните фамилии двигатели да бъдат с аналогични характеристики по отношение на влошаването на характеристиките на емисиите, в зависимост от модела и използваната технология. По-долу следва неизчерпателен списък на групирания в зависимост от модела и използваната технология:
2. ПЕРИОДИ НА УСТОЙЧИВОСТ НА ХАРАКТЕРИСТИКИТЕ НА ЕМИСИИТЕ ЗА ДВИГАТЕЛИТЕ ВЪВ ФАЗА II Производителят обявява категорията на устойчивост на характеристиките на газовете (ПУХЕ) (PDCE), приложима за всяка фамилия двигатели по време на типовото одобрение. Това е категорията, която се приближава най-много до предвидената продължителност на полезен живот на оборудването, върху което двигателят трябва да бъде монтиран, според производителя на двигателя. Производителят съхранява съответните данни, които обосновават избора на категорията на (ПУХЕ) (PDCE) за всяка фамилия двигатели. Тези данни са съобщават при поискване на компетентния техническия орган, отговарящ за типовото одобрение. 2.1.1. За преносимите двигатели: производителят избира дадена категория на (ПУХЕ) (PDCE) в таблица 1. Таблица 1: категории на устойчивост на характеристиките на емисиите за преносимите двигатели (в часове)
2.1.2. За непреносимите двигатели: производителят избира дадена категория на (ПУХЕ) (PDCE) в таблица 2. Таблица 2: категории на устойчивост на характеристиките на емисиите за непреносимите двигатели (в часове)
2.1.3. Производителят трябва да докаже убедително пред компетентния техническия орган, отговарящ за типовото одобрение, че обявената продължителност на полезен живот е реална. Данните, които служат за обосноваване от страна на производителя на избора на категория на устойчивост на характеристиките на емисиите (ПУХЕ) (PDCE) за дадена фамилия двигатели, може да включва следното (без този списък да се смята за изчерпателен):
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
5. |
Настоящото приложение IV се преномерира в „приложение V“ и се изменя, както следва:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
6. |
Приложение V се преномерира в приложение VI. |
|
7. |
Приложение VI се преномерира в приложение VII и се изменя, както следва.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
8. |
Приложение от VII до Х се преномерират в приложения от VIII до ХI. |
|
9. |
Добавя се следното приложение: „ПРИЛОЖЕНИЕ XII ПРИЗНАВАНЕ НА ДРУГИ НАЧИНИ ЗА ТИПОВО ОДОБРЕНИЕ Сертификатите за типово одобрение, описани по-долу, и при необходимост съответните маркировки за одобрение, се признават за еквивалентни на извършено одобрение по смисъла на настоящата директива за двигателите от категории A, B и C, така като те са определени в член 9, точка 2: 1.1. сертификатите за одобрение, издадени съгласно Директива 2000/25/ЕО; 1.2. сертификатите за одобрение, издадени съгласно Директива 88/77/ЕИО, които отговарят на предписанията, предвидени за етапи A или B по смисъла на член 2 и приложение I, точка 6.2.1 от Директива 88/77/ЕИО, чието изменение е извършено с Директива 91/542/ЕИО, или от серията изменения и поправки I/2 на Регламент 49.02 на Икономическата комисия за Европа на ООН; 1.3. сертификатите за одобрение, издадени съгласно Регламент № 96 на Икономическата комисия за Европа на Обединените нации. За двигателите от категории D, E, F и G (етап II), така както те са определени в член 9, параграф 3, описаните по-долу сертификати за типово одобрение и при необходимост съответните маркировки за одобрение се признават за еквивалентни на извършено одобрение по смисъла на настоящата директива: 2.1. сертификатите за одобрение (етап II), издадени съгласно Директива 2000/25/ЕО; 2.2. сертификатите за одобрение, издадени съгласно Директива 88/77/ЕИО, чието изменение и допълнение е извършено от Директива 99/96/EО, които съответстват на един от етапите A, B1, B2 или C, предвидени в член 2 и в точка 6.2.1 на приложение I; 2.3. серията от изменения и допълнения на Регламент № 49.03 на Икономическата комисия за Европа на Обединените нации; 2.4. сертификатите за одобрение (етап B), издадени съгласно Регламент № 96 на Икономическата комисия за Европа на Обединените нации, параграф 5.2.1, в серията изменения 01 на този регламент. |
(1) Виж приложение 4, допълнение 4, включително коефициентите за влошаване.
(2) Идентично на цикъл C1 от проектостандарт ISO 8178-4.“,
(3) Идентично на цикъл D2 от стандарт ISO 8178-4: 1996(E).
(4) Фигура 2 от стандарт ISO 8528-1: 1993(E) предлага по-добра илюстрация на определението за мощност при базова експлоатация.“.
(5) Идентично на цикъл D2 от стандарт ISO 8168-4: 1996(E).
(6) Степените на натоварване представляват стойностите на въртящия момент в проценти, съответстващи на мощността при базова експлоатация, определена като максималната мощност, налична за определен период на променлив режим на експлоатация, чиято продължителност може да бъде неопределен брой часове годишно, между технически обслужвания с обявена периодичност и при обявени условия на околната среда, при положение, че поддръжката се извършва в съответствие с предписанията на производителя. Фигура 2 от стандарт ISO 8528-1: 1993(E) илюстрира по-добре определението за мощност при базова експлоатация.
(7) За етап I се разрешава използване на стойностите 0,90 и 0,10 вместо съответно 0,85 и 0,15.
(8) Изчисленията на емисиите на отработените газове, които са описани в настоящата директива, в някои случай се основават на различни методи на измерване и/или изчисление. Поради малкия марж на стойностите на допустимия толеранс при изчисляване на емисиите на отработени газове, стойностите, които се приемат за някои параметри, използвани в съответните уравнения, трябва да бъдат по-ниски от допустимия толеранс, посочен в стандарт ISO 3046-3.
(9) При емисиите от NOx концентрацията трябва да бъде умножена по коефициента за корекция според влажността KH (коефициент за корекция според влажността при NOx).
(10) Стандартът ISO 8178-1 дава по-пълна формула за молекулярната маса на горивото [формула 50 от глава 13.5.1 б)]. Формулата взема под внимание не само съотношението водород/въглерод и съотношението кислород/въглерод, но също така и други възможни съставни елементи на горивото, като сяра и азот. Като се има предвид обаче, че двигателите с принудително запалване, посочени в директивата, се подлагат на изпитвания с бензин (посочен като референтно гориво в приложение V), който съдържа обикновено само въглерод и водород, се прибягва до използване на опростената формула.
(11) Условията за първичния въздух са същите както за входящия въздух.
(12) Не трябва да надвишава 10 % от измерената мощност по време на изпитванията.“
(13) Некоригирана мощност на двигателя, измерена съгласно разпоредбите на приложение I, точка 2.4.“
(14) В случай когато има няколко базови двигателя, тези информации трябва да бъдат посочени за всеки един от тях.
(15) Не трябва да надвишава 10 % от измерената мощност по време на изпитванията.
(16) Некоригирана мощност на двигателя, измерена съгласно разпоредбите в приложение I, точка 2.4.
(17) Пълната всмукателна система, предвидена за разглежданото приложение, трябва да се използва:
|
|
ако има опасност от значително влияние върху мощността на двигателя, |
|
|
при двигатели с принудително запалване с атмосферно засмукване, |
|
|
ако производителят го поиска. |
В останалите случаи може да се използва еквивалентна система, и трябва да се провери, че входящото налягане не се различава с повече от 100 Ра от най-високата пределна стойност, определена от производителя при работа с чист въздушен филтър.
(18) Пълната система за отвеждане на отработените газове трябва да се инсталира както е предвидено за разглежданото приложение:
|
|
ако има опасност от значително влияние върху мощността на двигателя, |
|
|
при двигатели с принудително запалване с атмосферно засмукване, |
|
|
ако производителят го поиска. |
В останалите случаи може да се монтира еквивалентна система, при условие че измереното атмосферно налягане не се отклонява с повече от 1 000 Ра от най-високата пределна стойност, определена от производителя.
(19) Ако съществува вградено към двигателя устройство за забавяне на отвеждането на отработените газове, клапата на забавящото устройство се фиксира в напълно отворено положение.
(20) При необходимост захранващото налягане на горивото може да бъде регулирано, с цел да се възпроизведе съществуващото налягане в разглежданото приложение (по-специално когато се използва система с връщане на част от горивото).
(21) Клапата за всмукване на въздуха е клапата за управление на пневматичния регулатор на инжекционната помпа. Регулаторът или инжекционната система могат да съдържат други устройства, които да са в състояние да повлияват върху качеството на впръскваното гориво.
(22) Циркулацията на охладителната течност трябва да се осъществява единствено от водната помпа на двигателя. Охлаждането на течността може да се осъществява чрез външен кръг, по такъв начин, че загубата на налягане в този кръг и входящото налягане на водната помпа да бъдат почти на 100 процента равни на тези на охладителната система на двигателя.
(23) Термостатът може да бъде фиксиран в напълно отворено положение.
(24) Ако нагнетателят или вентилаторът на охлаждането не са свалени за изпитването, мощността, която поглъщат, се прибавя към резултатите, освен в случай, когато вентилаторите на охлаждането на двигателите с въздушно охлаждане са монтирани директно върху коляновия вал. Мощността на вентилатора или на нагнетателя се определя при режимите, използвани при изпитването, или чрез извършване на изчисление въз основа на стандартните характеристики, или чрез извършването на практически изпитвания.
(25) Минимална мощност на генератора: генераторът трябва да доставя само необходимата електрическа мощност за захранването на необходимите спомагателните устройства за работата на двигателя. Ако е необходимо свързване към акумулатор, той трябва да бъде в добро техническо състояние и да е напълно зареден.
(26) Двигателите с турбозахранване с междинно охлаждане трябва да преминават изпитване с устройствата за охлаждане на турбозахранването, независимо дали те работят с въздух или течност. Ако производителят предпочита, въздушният охладител може да бъде заместен от инсталация, монтирана на изпитвателния стенд. Във всички случаи измерването на мощността при всеки режим трябва да се извършва при спадането на максималното налягане и спадането на минималната температура на въздуха на турбозахранването, засмукван в охладителя на изпитвателния стенд, така както те са специфицирани от производителя.
(27) Те могат да включват например системи за преработка на отработените газове, каталитичен конвертор, термичен реактор, вторично впръскване на въздух и противоизпарителна система за горивото.
(28) Енергията, която е необходима за електрическо стартиране или друга система за стартиране, трябва да бъде доставена от стенда.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
115 |
32002L0095
|
L 037/19 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2002/95/ЕО НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 27 януари 2003 година
относно ограничението за употребата на определени опасни вещества в електрическото и електронното оборудване
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 95 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Икономическия и социален комитет (2),
като взеха предвид становището на Комитета на регионите (3),
в съответствие с процедурата по член 251 от Договора, и в светлината на съвместния проект, одобрен от Помирителния комитет на 8 ноември 2002 г. (4),
като имат предвид, че:
|
(1) |
Несъответствията в законодателството или административните мерки, приети от държавите-членки, по отношение на ограниченията в употребата на някои опасни вещества в електрическото и електронното оборудване, могат да създадат пречки за търговията и да изменят условията на конкуренцията и могат поради това да окажат директно влияние върху установяването и функционирането на вътрешния пазар. Необходимо е да се сближат законодателствата на Държавите-членки в тази област и да се спомогне за защитата на здравето на хората и стабилно възстановяване на околната среда, премахване на отпадъците от електрическото и електронно оборудване. |
|
(2) |
Европейският съвет на заседанието си в Ница на 7, 8 и 9 декември 2000 г. подписа Резолюция на Съвета от 4 декември 2000 г. относно предохранителен принцип. |
|
(3) |
Докладът на Комисията от 30 юли 1996 г. за преразглеждане на стратегията на Общността за управление на отпадъците, акцентира на нуждата да се намали съдържанието на отпадъчни опасни вещества и посочи потенциалните ползи от общи правила на Общността за ограничаване присъствието на такива вещества в продуктите и процесите на производство. |
|
(4) |
Резолюцията на Съвета от 25 януари 1988 г. за програма за действие на Общността за борба със замърсяването на околната среда с кадмий (5) подтикна Комисията да разработи незабавно специални мерки за такава програма. Здравето на човека също е защитено с цялостна стратегия, която, по- специално, ограничава употребата на кадмий и стимулира изследванията на заместители, които трябва да бъдат направени поради тази причина. Резолюцията акцентира, че употребата на кадмий трябва да бъде ограничена до случаите, при които няма подходящи или по-безопасни алтернативи. |
|
(5) |
Наличните данни показват, че тези мерки за събиране, третиране, рециклиране и премахване на отпадъчно електрическо и електронно оборудване (ОЕЕО), както е постановено в Директива 2002/96/ЕО от 27 януари 2003 г. на Европейския парламент и на Съвета относно отпадъчно електрическо и електронно оборудване (6) са необходими за намаляването на проблемите за управление на отпадъците, свързани с въпросните тежки метали и въпросните вещества, които забавят горенето. Въпреки тези мерки, значителна част от ОЕЕО ще продължат да бъдат откривани в настоящи линии за изхвърляне. Дори ако ОЕЕО е било събрано отделно и подложено на процеси за рециклиране, неговото съдържание на живак, кадмий, олово и хром VI, PBB и PBDE има вероятност да представляват риск за здравето или околната среда. |
|
(6) |
Като се вземат под внимание техническата и икономическата приложимост, най-ефективният начин за значително намаляване на рисковете за здравето и околната среда, свързани с тези вещества, за които може да се достигне избрано ниво на защита в Общността, е заместването на тези вещества в електрическото и електронното оборудване с безопасни или по-безопасни материали. Ограничаването на употребата на тези опасни вещества е вероятно да увеличи възможностите за икономическа печалба от рециклиране на ОЕЕО и намаляване на отрицателното влияние върху здравето на работниците в заводите за рециклиране. |
|
(7) |
Веществата обхванати от настоящата директива са добре проучени научно и оценени и са били обект на различни мерки на ниво Общност и на национално ниво. |
|
(8) |
Предвидените в настоящата директива мерки вземат под внимание съществуването на международни указания и препоръки и са базирани на оценка от налична научна и техническа информация. Мерките са необходими за постигане на избраното ниво на защита за здравето на човека, здравето на животните и околната среда като се имат предвид рисковете, които липсата на тези мерки вероятно биха предизвикали в Общността. Мерките трябва да бъдат под контрол и, ако е необходимо, да бъдат коригирани, като се вземе под внимание наличната научна и техническа информация. |
|
(9) |
Директивата трябва да се прилага без да влиза в противоречие със законодателството на Общността за изискванията за безопасността и здравето и специалното законодателство на Общността за управление на отпадъците, по-специално Директива 91/157/ЕИО на Съвета от 18 март 1991 г. относно батериите и акумулаторите, съдържащи някои опасни вещества (7). |
|
(10) |
Техническото развитие на електрическото и електронното оборудване без тежки метали, полибромдифенили (PBDE) и полибромирани бифенили (PBB) трябва да бъдат взети под внимание. След като научните данни са в наличност и като се вземе под внимание предохранителния принцип забраната на други опасни вещества и тяхното заместване с по-благоприятни за околната среда алтернативи, които гарантират поне същото ниво на защита за потребителите, те трябва да бъдат проучени. |
|
(11) |
Изключения от изискванията за заместване трябва да бъдат допуснати, ако заместването не е възможно от научна или техническа гледна точка или ако негативното влияние върху околната среда и здравето, причинени от заместителя има вероятност да превиши ползите от това заместване. Заместването на опасни вещества в електрическо и електронно оборудване, трябва също да бъде направено по начин съвместим със здравето и безопасността на потребителите на електрическо и електронно оборудване (ЕЕО). |
|
(12) |
Трябва да има резервни части в наличност, ако повторната употреба, ремонтиране и удължаване на живота на продукта е от полза. |
|
(13) |
Адаптирането към научния и техническия прогрес на изключенията от изискванията относно постепенното изваждане от употреба и забрана на опасните вещества трябва да се прилага от Комисията в съответствие с процедурата на комитета. |
|
(14) |
Необходимите мерки за прилагане на настоящата директива трябва да бъдат приети в съответствие с Решение 1999/468/ЕО на Съвета от 28 юни 1999 г. относно установяването на процедурите за упражняване на изпълнителните правомощия, предоставени на Комисията (8), |
ПРИЕХА НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Цели
Целта на настоящата директива е да сближи законодателството на държавите-членки относно ограниченията за употреба на опасни вещества в електрическо и електронно оборудване и да спомогне за защитата на здравето на човека и стабилно възстановяване на околната среда, както и премахване на електрическото и електронно оборудване.
Член 2
Обхват
1. Без да засяга член 6 настоящата директива се прилага за електрическо и електронно оборудване, което попада в категории 1, 2, 3, 4, 5, 6, 7 и 10, постановени в приложение IА към Директива 2002/96/ЕО (ОЕЕО) и за електрически крушки и осветителни тела в домашни условия.
2. Настоящата директива се прилага без да засяга законодателството на Общността относно изисквания за безопасност и здраве, както и специалното законодателство на Общността за управление на отпадъците.
3. Настоящата директива няма да се прилага за резервни части за поправка или за повторна употреба на електрическо и електронно оборудване, които са пуснати на пазара преди 1 юли 2006 г.
Член 3
Определения
За целите на настоящата Директива, ще се прилагат следните определения:
|
а) |
„електрическо и електронно оборудване“ или „ЕЕО“ означава оборудване, което е зависимо от електрически ток или електромагнитни полета, за да функционира правилно и оборудване за генериране, предаване и измерване на такива електричество или полета, което попада в категориите, определени в приложение IА на Директива 2002/96/ЕО (ОЕЕО) и е създадено за употреба с електрическо напрежение, което не превишава 1 000 волта за променлив ток и 1 500 волта за прав ток; |
|
б) |
„производител“ означава всяко лице, което независимо от техниката на продажба, която използват, включително средствата за комуникация от разстояние съгласно Директива 97/7/ЕО на Европейския парламент и на Съвета от 20 май 1997 г. за защита на потребителите по отношение на договорите от разстояние (9):
|
Ако някой изключително предоставя финансиране според или съгласно някакво финансово споразумение няма да бъде смятан за „производител“, освен ако той действа като производител по смисъла на подточки i) до iii).
Член 4
Защита
1. Държавите-членки трябва да гарантират, че от 1 юли 2006 г., ново електрическо и електронно оборудване, което е пуснато на пазара, не съдържа олово, живак, кадмий, шест валентен хром, полибромирани бифенили (PBB), полибромдифенили (PBDE). Националните мерки за ограничаване и забрана на употребата на тези вещества в електрическо и електронно оборудване, които са приети в съответствие с законодателството на Общността преди приемането на настоящата директива, може да бъдат запазени до 1 юли 2006 г.
2. Параграф 1 няма да се прилага за заявките, които са изброени в приложението.
3. На базата на предложение на Комисията, Европейският парламент и Съветът ще решат веднага след като получат научните данни и в съответствие с принципите на политиката за химикалите, както е постановено в Шестата програма на Общността за действие за околната среда, забраната на други опасни вещества и тяхното заместване с по-безвредни за околната среда алтернативи, които гарантират най-малко същото равнище на защита на потребителите.
Член 5
Адаптиране към научния и техническия прогрес
1. Всички изменения, които са необходими, за да се адаптира приложението към научния и техническия прогрес за следните цели, ще бъдат приети в съответствие с процедурата, посочена в член 7, параграф 2:
|
а) |
установяване, ако е необходимо, на максимални стойности на концентрация, до които присъствието на посочени в член 4, параграф 1 вещества в специалните материали и компоненти на електрическо и електронно оборудване трябва да бъдат толерирани; |
|
б) |
изключване на материали и компоненти от електрическо и електронно оборудване от член 4, параграф 1, ако тяхното отстраняване или заменяне посредством промени в строежа или материали и компоненти, които не изискват никакви материали или вещества, посочени в него, е технически или научно неосъществимо или където отрицателното влияние върху околната среда, здравето и/или безопасността на потребителите, причинени от замяната, има вероятност да надвишат ползите за околната среда, здравето и/или безопасността на потребителите; |
|
в) |
преразглеждането на всяко изключение в приложението най-малко на четири години или четири години след като артикулът е добавен към списъка, с цел разглеждане на изхвърлянето на материали и компоненти от електрическо и електронно оборудване от приложението, ако тяхното отстраняване или замяна посредством промени в строежа или материали и компоненти, които не изискват никакви материали или вещества, посочени в член 4, параграф 1, е технически или научно възможно в случай, че отрицателно влияе върху околната среда, здравето и/или безопасността на потребителите, причинени от замяната има вероятност да надвишат ползите за околната среда, здравето и/или безопасността на потребителите. |
2. Преди приложението да бъде изменено съгласно параграф 1, Комисията, inter alia, ще консултира производителите на електрическо и електронно оборудване, собственици на заводи за рециклиране и за обработка, природозащитни организации, сдружения на работници и потребители. Коментарите ще бъдат препратени на комитета по член 7, параграф 1. Комисията трябва да предостави доклад за информацията, която получава.
Член 6
Преглед
Преди 13 февруари 2005 г., Комисията преразглеждат мерките, които са предвидени в настоящата директива, за да вземе под внимание, където е необходимо, новите научни данни.
По-специално до тази дата Комисията представя предложения за включване в обхвата на настоящата директива на оборудването, което попада в категории 8 и 9, постановените в приложение IА към Директива 2002/96/ЕО (ОЕЕО).
Комисията също проучва нуждата за адаптиране на списъка с вещества в член 4, параграф 1 на основа на научни факти и като се вземе под внимание предохранителния принцип, и, ако е подходящо — настоящите предложения на Европейския парламент и на Съвета за такива адаптации.
Особено внимание трябва да бъде обърнато на контрола върху влиянието върху околната среда и здравето на човека на опасни вещества и материали използвани в електрическо и електронно оборудване. Комисията ще изпита приложението на замяната на такива вещества и материали и ще представя предложения на Европейския парламент и на Съвета, за разширяване на обсега на член 4, както е подходящо.
Член 7
Комитет
1. Комисията се подпомага от Комитета, създаден с член 18 от Директива 75/442/ЕИО на Съвета (10).
2. Когото се прави позоваване на настоящия параграф, членове 5 и 7 от Решение 1999/468/ЕО се прилагат, като се взима под внимание член 8 от него.
Периодът предвиден в член 5, параграф 6 от Решение 1999/468/ЕО, е определен на три месеца.
3. Комитетът приема процедурен правилник.
Член 8
Санкции
Държавите-членки определят санкции, които са приложими за нарушенията на националните разпоредби, които са приети в съответствие с настоящата директива. Предвидените санкции трябва да бъдат ефективни, съразмерни и разубеждаващи.
Член 9
Транспониране
1. Държавите-членки въвеждат в сила законовите, подзаконовите и административните разпоредби, необходими за да се съобразят с настоящата директива, преди 13 август 2004 г. Те незабавно информират Комисията за това.
Когато държавите-членки приемат тези мерки, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
2. Държавите-членки съобщават на Комисията текста на всички законови, подзаконови и административни разпоредби, приета в областта, уредена с настоящата директива.
Член 10
Влизане в сила
Настоящата директива влиза в сила в деня на публикуването ѝ в Официален вестник на Европейските общности.
Член 11
Адресати
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 27 януари 2003 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
G. DRYS
(1) ОВ C 365 E, 19.12.2000 г., стр. 195 и
ОВ C 240 E, 28.8.2001 г., стр. 303.
(2) ОВ C 116, 20.4.2001 г., стр. 38.
(3) ОВ C 148, 18.5.2001 г., стр. 1.
(4) Становище на Европейския парламент от 15 май 2001 г. (ОВ C 34 E, 7.2.2002 г., стр. 109), Обща позиция на Съвета от 4 декември 2001 г. (ОВ C 90 E, 16.4.2002 г., стр. 12.) и Решение на Европейския парламент от 10 април 2002 г. (все още непубликувано в Официален вестник) и Решение на Европейския парламент от 18 декември 2002 г. и Решение на Съвета от 16 декември 2002 г.
(5) ОВ C 30, 4.2.1988 г., стр. 1.
(6) ОВ L 37, 13.2.2003 г., стр. 24.
(7) ОВ L 78, 26.3.1991 г., стр. 38. Директива, изменена с Директива 98/101/ЕО на Комисията (ОВ L 1, 5.1.1999г., стр. 1).
(8) ОВ L 184, 17.7.1999 г., стр. 23.
(9) ОВ L 144, 4.6.1997 г., стр. 19. Директива, изменена с Директива 2002 /65/ЕО (L 271, 9.10.2002 г., стр. 16).
(10) ОВ L 194, 25.7.1975 г., стр. 39.
ПРИЛОЖЕНИЕ
Заявки за олово, живак, кадмий и шествалентен хром, които са изключени от изискванията на член 4, параграф 1
1. Живак в компактни флуоресцентни лампи не превишава 5 mg на лампа.
2. Живак в прави флуоресцентни лампи за общи цели не превишава:
|
10 mg, |
||
|
5 mg, |
||
|
8 mg. |
3. Живак в прави флуоресцентни лампи за специални цели.
4. Живак в други лампи, не споменати специално в настоящото приложение.
5. Олово в стъклена електронно-лъчева тръба, електронни компоненти и флуоресцентни тръби.
6. Оловото като съставен елемент в стомана, съдържаща до 0,35 % олово от теглото, алуминий, съдържащ до 0,4 % олово от теглото и като сплав от мед, съдържаща до 4 % олово от теглото.
|
— |
Олово в мек припой с висока температура на топене (т.е. оловно-калаена припойна сплав, съдържаща повече от 85 % олово), |
|
— |
Олово в мек припой за сървъри, запаметяващи устройства и системи (изключението е дадено до 2010 г.), |
|
— |
Олово в мек припой за оборудване на инфраструктурни мрежи за включване, сигнализиране, трансмисия, както и за поддръжка на мрежи за телекомуникацията, |
|
— |
Олово в електронни керамични части (напр. пиезоелектронни средства). |
8. Кадмиево покритие освен приложенията, забранени в съответствие с Директива 91/338/ЕИО (1) за изменение на Директива 76/769/ ЕИО (2) относно ограниченията за пускането на пазара и употребата на някои опасни вещества и препарати.
9. Шествалентен хром като антикорозионно вещество за охладителни системи от въглеродна стомана в абсорбционни хладилни уредби.
10. В рамките на процедурата по член 7, параграф 2, Комисията оценява заявките за:
|
— |
декабромдифенил етер, |
|
— |
живак в прави флуоресцентни лампи за специални цели, |
|
— |
олово в мек припой за сървъри, запаметяващи устройства и системи за оборудване на инфраструктурни мрежи за включване, сигнализиране, трансмисия, както и за поддръжка на мрежи за телекомуникацията (с оглед на определянето на специално ограничение на времето за това изключение), и |
|
— |
електрически крушки, |
за приоритет, за да се установи възможно най-скоро дали тези артикули съответно подлежат на изменение.
(1) ОВ L 186,12.7.1991 г., стр. 59.
(2) ОВ L 262, 27.9.1976 г., стр. 201.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
120 |
32003L0014
|
L 041/37 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/14/ЕО НА КОМИСИЯТА
от 10 февруари 2003 година
за изменение на Директива 91/321/ЕИО относно храните за кърмачета и преходните храни
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 89/398/ЕИО на Съвета от 3 май 1989 г. за сближаване на законодателствата на държавите-членки относно храни, предназначени за специфична хранителна употреба (1), последно изменена с Директива 1999/41/ЕО на Европейския парламент и Съвета (2), и по-конкретно член 4, параграф 1 от нея,
като взе предвид становището на Научния комитет по храните,
като има предвид, че:
|
(1) |
Член 6 на Директива 91/321/ЕИО на Комисията (3) последно изменена с Директива 1999/50/ЕО (4), в който се посочва, че храните за кърмачета и преходните храни не съдържат никакво вещество в количество, което може да изложи на опасност здравето на кърмачетата и малките деца. |
|
(2) |
Въз основа на становищата, предоставени от Научния комитет по храните на 19 септември 1997 г. и 4 юни 1998 г., Директива 91/321/ЕИО определя общо максимално ниво от 0,01 mg/kg остатъци от всеки пестицид в храни за кърмачета и преходни храни. |
|
(3) |
При малък брой пестициди или метаболити на пестициди дори и максималното ниво на остатъци от 0,01 mg/kg би могло в най-лошия случай да доведе до превишаване на допустимата дневна доза при кърмачета и малки деца. Такъв е случаят с пестицидите или метаболити на пестициди, чиято допустима дневна доза е по-ниска от 0,0005 mg/kg телесно тегло. |
|
(4) |
Директива 91/321/ЕИО въвежда принципа на забрана за използването на тези пестициди в производството на селскостопански продукти, предназначени за храни за кърмачета и преходни храни. Въпросните пестициди трябва да се изброят в приложение ІХ към Директива 91/321/ЕИО. Тази забрана, обаче не гарантира непременно, че в продуктите не съдържат такива пестициди, тъй като някои от пестицидите замърсяват околната среда и във въпросните продукти може да се установи наличието на остатъци от тях. |
|
(5) |
Здравето на кърмачетата и малките деца може да се защити по-добре чрез прилагането на допълнителни изисквания, чието спазване може да се контролира чрез анализ, независимо от произхода на продукта. |
|
(6) |
Повечето от пестицидите, чиито допустими дневни дози са по-ниски от 0,0005 mg/kg телесно тегло, са вече забранени в Общността или ще бъдат забранени до месец юли 2003 г. Забранените пестициди не трябва да се откриват в храните за кърмачета и преходните храни чрез аналитични методи. Въпреки това,. някои пестициди се разграждат бавно и все още замърсяват околната среда. Те може да присъстват в храните за кърмачета и преходните храни дори и да не са били използвани. С оглед упражняване на контрол, към проблема трябва да се прилага хармонизиран подход. |
|
(7) |
В очакване на решенията на Комисията, които да определят дали разрешените пестициди отговарят изискванията за безопасност, посочени в член 5 на Директива 91/414/ЕИО на Съвета от 15 юли 1991 г. относно пускането на пазара на продукти за растителна защита (5), последно изменена с Директива 2003/5/ЕО на Комисията (6), използването на разрешените пестициди трябва да остане разрешено, доколкото техните остатъци не превишават максималните нива, определени в настоящата директива. Тези количества трябва да са на нива, които гарантират, че и в най-лошия случай стойностите на допустимите дневни дози няма да бъдат превишени при поемането им от кърмачета и малки деца. |
|
(8) |
Директива 91/321/ЕИО следва да бъде съответно изменена. |
|
(9) |
Предвидените в настоящата директива мерки са в съответствие със становището на Постоянния комитет по хранителната верига и здравето на животните, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Директива 91/321/ЕИО се изменя както следва:
|
1. |
Член 6 се изменя както следва:
|
|
2. |
Приложение ІХ се заменя с приложение І към настоящата директива. |
|
3. |
Приложение ІІ към настоящата директива се добавя като приложение Х. |
Член 2
1. Държавите-членки разрешават търговията с продукти, които съответстват на изискванията на член 6, параграф 3 от Директива 91/321/ЕИО, най-късно до 6 март 2004 г.
2. Държавите-членки забраняват търговията с продукти, които не отговарят на изискванията на член 6, параграф 3 от Директива 91/321/ЕИО, най-късно до 6 март 2005 г.
Член 3
Държавите-членки въвеждат в сила законовите, подзаконовите и административни разпоредби, необходими, за да съобразят с настоящата директива, най-късно до 6 март 2004 г. Те уведомяват незабавно Комисията за това.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 4
Настоящата директива влиза в сила на двадесетия ден след публикуването ѝ в Официален вестник на Европейския съюз.
Член 5
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 10 февруари 2003 година.
За Комисията
David BYRNE
Член на Комисията
(1) ОВ L 186, 30.6.1989 г., стр. 27.
(2) ОВ L 172, 8.7.1999 г., стр. 38.
(3) ОВ L 175, 4.7.1991 г., стр. 35.
(4) ОВ L 139, 2.6.1999 г., стр. 29.
(5) ОВ L 230, 19.8.1991 г., стр. 1.
(6) ОВ L 8, 14.1.2003 г., стр. 7.
ПРИЛОЖЕНИЕ І
„ПРИЛОЖЕНИЕ IХ
Пестициди, чиято употреба е забранена при селскостопански продукти, които са предназначени за производството на храни за кърмачета и преходни храни
Таблица 1
Химическо наименование на веществото (определение на остатъчното количество)
|
|
Дисулфотон (комбинация от дисулфотон, дисулфотон-сулфоксид и дисулфотон сулфон, изразена като дисулфотон) |
|
|
Фенсулфотион (комбинация от фенсулфотион, неговия кислороден аналог и техните сулфони, изразена като фенсулфотион) |
|
|
Фентин, изразен като трифенилтин катион |
|
|
Халоксифоп (комбинация от халоксифоп, неговите соли и естери, включително техните съединения, изразена като халоксифоп) |
|
|
Хептахлор и транс-хептахлор епоксид, изразени като хептахлор |
|
|
Хексахлорбензол |
|
|
Нитрофен |
|
|
Ометоат |
|
|
Тербуфос (комбинация от тербуфос, неговия сулфоксид и сулфон, изразена като тербуфос) |
Таблица 2
Химическо название на съединението
|
|
Алдрин и диелдрин, изразени като диелдрин |
|
|
Ендрин“ |
ПРИЛОЖЕНИЕ ІІ
„ПРИЛОЖЕНИЕ Х
Специфични максимални нива на остатъчни вещества от пестициди или метаболити на пестициди в храни за кърмачета и преходни храни
|
Химическо название на веществото |
Максимално ниво на остатък (mg/kg) |
|
Кадусафос |
0,006 |
|
Деметон-S-метил/деметон-S-метил сулфон/оксидеметонметил (поотделно или в комбинация, изразена като деметон-S-метил) |
0,006 |
|
Етопрофос |
0,008 |
|
Фипронил (комбинация от фипронил и фипронил-десулфинил, изразена като фипронил) |
0,004 |
|
Пропинеб/пропиленетиурея (комбинация от пропинеб и пропиленетиурея) |
0,006“ |
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
124 |
32003L0011
|
L 042/45 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/11/ЕО НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 6 февруари 2003 година
за изменение за двадесет и четвърти път на Директива 76/769/ЕИО на Съвета относно ограниченията за пускането на пазара и употребата на някои опасни вещества и препарати (пентабромодифенил етер, октабромодифенил етер)
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 95 от него,
като взеха предвид предложенията на Комисията (1),
като взеха предвид становището на Икономическия и социален комитет (2),
в съответствие с процедурата, установена в член 251 от Договора (3), в светлината на съвместния текст, одобрен от Помирителния комитет на 8 ноември 2002 г.,
като имат предвид, че:
|
(1) |
Съгласно член 14 от Договора трябва да се създаде пространство без вътрешни граници, в което да бъде осигурено свободното движение на стоки, хора, услуги и капитали. |
|
(2) |
Рисковете за околната среда от пентабромодифенил етер (пентаБДЕ) и октабромодифенил етер (октаБДЕ) бяха оценени в съответствие с Регламент (ЕИО) № 793/93 на Съвета от 23 март 1993 г. относно оценката и контрола на рисковете от съществуващите вещества (4). Оценките на риска за пентаБДЕ и октаБДЕ очертаха необходимост от ограничаване на рисковете, които тези вещества създават за околната среда. В своите становища от 4 февруари 2000 г. и 31 октомври 2002 г. Научният комитет по токсичност, екотоксичност и околна среда (НКТЕОС) потвърди заключенията от оценките за пентаБДЕ и октаБДЕ за необходимостта от ограничаване на рисковете за опазване на околната среда. Освен това в свое становище от 19 юни 2000 г. НКТЕОС потвърди загрижеността относно експозицията на кърмачетата на въздействието на пентаБДЕ, както и опасенията, че увеличеното съдържание на пентаБДЕ в майчиното мляко може да се дължи на все още неидентифицирана употреба на въпросния химикал. |
|
(3) |
В рамките на Регламент (ЕИО) № 793/93 Комисията прие Препоръки относно стратегия за ограничаване на риска при пентаБДЕ (5) и октаБДЕ (6), предвиждаща ограничения върху продажбата и употребата на последните за контрол на рисковете за околната среда. Освен това Комисията препоръча мерките да бъдат съобразени с нарастващата загриженост относно експозицията на кърмачетата чрез майчиното мляко. |
|
(4) |
За целите на опазването на здравето и околната среда пускането на пазара и употребата на пентаБДЕ и октаБДЕ и пускането на пазара на изделия, които съдържат едното или двете цитирани вещества, трябва да се забранят. |
|
(5) |
Присъствието на пентаБДЕ и октаБДЕ в концентрации, по-високи от 0,1 %, може да се открива с помощта на стандартни аналитични методи, например ГХ-МС (газово хроматографска масова спектрометрия). |
|
(6) |
Оценка на риска при декаБДЕ бе приключена през август 2002 г. и демонстрира редица несигурности относно възможните ефекти на това вещество върху околната среда. Общността трябва без всякакво закъснение да предприеме мерки и да приеме стратегия за ограничаване на риска. Комисията очаква резултати от разработването на стратегия за риска не по-късно от 30 юни 2003 г. След това Комисията ще анализира резултатите незабавно и ще предложи подходящи и строги мерки в отговор на идентифицираните рискове. Европейският парламент и Съветът трябва да разгледат предложението без всякакво закъснение. Одобрените от Общността ограничения по отношение на пускането на пазара и употребата на декаБДЕ трябва да влязат в сила без по-нататъшно закъснение, освен ако в резултат на допълнително тестване, възможност за каквото горецитираната оценка на риска предвижда, текущите несигурности не бъдат разрешени чрез формиране на заключение, което да елиминира възникналите опасения. |
|
(7) |
Настоящата директива не засяга законодателството на Общността, постановяващо минимални изисквания за защитата на работниците, съдържащи се в Директива 89/391/ЕИО на Съвета от 12 юни 1989 г. за въвеждане на мерки за насърчаване подобряването на безопасността и здравето на работниците на работното място (7) и базираните на нея специални директиви, и по-конкретно Директива 90/394/ЕИО на Съвета от 28 юни 1990 г. за защита на работниците от рискове, свързани с експозиция на канцерогени по време на работа (Шестата специална директива по смисъла на член 16, параграф 1 от Директива 89/391/ЕИО (8), и Директива 98/24/ЕО на Съвета от 7 април 1998 г. за опазване на здравето и безопасността на работниците от рискове, свързани с химични агенти на работното място (Четиринадесета специална директива по смисъла на член 16, параграф 1 от Директива 89/391/ЕИО) (9), |
ПРИЕХА НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Приложение I към Директива 76/769/ЕИО се изменя в съответствие с приложението към настоящата директива.
Член 2
Държавите-членки приемат и публикуват законовите, подзаконовите и административните разпоредби, необходими, за да се съобразят с настоящата директива не по-късно от 15 февруари 2004 г. Те незабавно информират Комисията за това.
Те прилагат тези мерки считано от 15 август 2004 г.
Когато държавите-членки приемат тези мерки, в тях се съдържа позоваването на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 3
Настоящата директива влиза в сила в деня на публикуването ѝ в Официален вестник на Европейския съюз.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 6 февруари 2003 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
P. EFTHYMIOU
(1) ОВ С 154 Е, 29.5.2001 г., стр. 112 и
ОВ С 25, 29.1.2002 г., стр. 472.
(2) ОВ С 193, 10.7.2001 г., стр. 27.
(3) Становище на Европейския парламент от 6 септември 2001 г. (ОВ С 72 Е, 21.3.2002 г., стр. 235), Обща позиция на Съвета от 6 декември 2001 г. (ОВ С 110 Е, 7.5.2002 г., стр. 23) и Решение на Европейския парламент от 10 април 2002 г. (все още непубликувано в Официален вестник).
(4) ОВ L 84, 5.4.1993 г., стр. 1.
(5) ОВ L 69, 10.3.2001 г., стр. 30.
(6) ОВ L 249, 17.9.2002 г., стр. 27.
(7) ОВ L 183, 29.6.1989 г., стр. 1.
(8) ОВ L 196, 26.7.1990 г., стр. 1. Директива, последно изменена с Директива 1999/38/ЕО (ОВ L 138, 1.6.1999 г., стр. 66).
(9) ОВ L 131, 5.5.1998 г., стр. 11.
ПРИЛОЖЕНИЕ
В приложение I към Директива 76/769/ЕИО се добавя следната точка 44:
|
|
В приложение I към Директива 76/769/ЕИО се добавя следната точка 45:
|
|
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
127 |
32003L0016
|
L 046/24 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/16/ЕО НА КОМИСИЯТА
от 19 февруари 2003 година
относно адаптиране към техническия прогрес на приложение III към Директива 76/768/ЕИО на Съвета относно сближаването на законодателствата на държавите-членки, свързани с козметичните продукти
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 76/768/ЕИО на Съвета от 27 юли 1976 г. относно сближаването на законодателствата на държавите-членки, свързани с козметични продукти (1), последно изменена с Директива 2003/1/ЕО на Комисията (2), и по-специално член 8, параграф 2 от нея,
след консултации с Научния комитет за козметични продукти и нехранителни продукти, предназначени за потребителите (SCCNFP),
като има предвид, че:
|
(1) |
Научният комитет за козметични продукти и нехранителни продукти, предназначени за потребителите (SCCNFP), счита, че мускусният ксилен може да се употребява безопасно в козметичните продукти, с изключение на продуктите за хигиена на устната кухина, при което теоретично максималната приета дневна доза е около 10 µg/kg. |
|
(2) |
Научният комитет за козметични продукти и нехранителни продукти, предназначени за потребителите (SCCNFP) счита, че мускусният кетон може да се употребява безопасно в козметичните продукти, с изключение на продуктите за хигиена на устната кухина, при което теоретично максималната приета дневна доза е около 14 µg/kg. |
|
(3) |
До приключване на оценката на риска по отношение на тези две вещества в съответствие с Регламент (ЕИО) № 793/93 на Съвета от 23 март 1993 г. за оценката и контрола на рисковете от съществуващите вещества (3), тези две вещества са включени временно — до 28 февруари 2003 г. — в част 2 на приложение III към Директива 76/768/ЕИО. |
|
(4) |
Оценката на риска в съответствие с изискванията на гореспоменатия регламент още не е приключила. Затова периодът за включване на мускусния ксилен и мускусния кетон в част 2 на приложение III към Директива 76/768/ЕИО следва да бъде продължен. |
|
(5) |
Мерките, предвидени в настоящата директива, са в съответствие със становището на Комитета по адаптиране към техническия прогрес на директивите за премахването на техническите пречки в търговията в сектора на козметичните продукти, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Датата „28.2.2003 г.“ се заменя с датата „30.9.2004 г.“, що се отнася до референтни номера 61 и 62 в част 2, колона ж) на приложение III към Директива 76/768/ЕИО.
Член 2
Държавите-членки въвеждат в сила необходимите законови, подзаконови и административни разпоредби, за да се съобразят с настоящата директива преди 28 февруари 2003 г. Те незабавно информират Комисията за това.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 3
Настоящата директива влиза в сила на третия ден след публикуването ѝ в Официален вестник на Европейския съюз.
Член 5
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 19 февруари 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 262, 27.9.1976 г., стр. 169.
(2) ОВ L 5, 10.1.2003 г., стр. 14.
(3) ОВ L 84, 5.4.1993 г., стр. 1.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
128 |
32003L0015
|
L 066/26 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/15/ЕO НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 27 февруари 2003 година
за изменение на Директива 76/768/ЕИО на Съвета относно сближаването на законодателствата на държавите-членки, свързани с козметични продукти
(текст от значение за ЕИП)
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 95 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Европейския икономически и социален комитет (2),
в съответствие с процедурата, установена в член 251 от Договора в светлината на общия текст, одобрен от Помирителния комитет на 3 декември 2002 г. (3),
като имат предвид, че
|
(1) |
Директива 76/768/ЕИО на Съвета (4) хармонизира по изчерпателен начин националните законодателства, свързани с козметичните продукти и има за своя главна цел защитата на общественото здраве. За да бъде постигната тази цел, продължава да бъде належащо да се провеждат определени токсикологични тестове, за да се направи оценка на безопасността на козметичните продукти. |
|
(2) |
Протоколът относно закрилата и хуманното отношение към животните, приложен с Договора от Амстердам към Договора за създаване на Европейската общност, предвижда Общността и държавите-членки да се съобразяват напълно с изискванията за хуманното отношение към животните при осъществяването на политиките на Общността, по-специално по отношение на вътрешния пазар. |
|
(3) |
Директива 86/609/ЕИО на Съвета от 24 ноември 2003 г. относно сближаването на законовите, подзаконови и административни разпоредби на държавите-членки по отношение на закрилата на животните, използвани за експериментални и други научни нужди (5) установи общи правила за използването на животни за експериментални цели в рамките на Общността и условията, при които трябва да се провеждат такива експерименти на територията на държавите-членки. По-специално, член 7 от тази Директива изисква експериментите с животни да бъдат заменени с алтернативни методи, когато такива методи съществуват и са приемливи от научна гледна точка. За да се улесни разработването и използването на алтернативни методи в козметичния сектор, при които не се използват живи животни, с Директива 93/35/ЕИО на Съвета от 14 юни 1993 г. относно изменение за шести път на Директива 76/768/ЕИО относно сближаването на законодателствата на държавите-членки в областта на козметичните продукти (6), бяха въведени специфични разпоредби. Въпреки това тези разпоредби се отнасят само до алтернативни методи, при които не се използват животни и не отчитат алтернативните методи, разработени с цел да се намали броя на животни, използвани при експериментите, или да се намалят техните страдания. Следователно, за да се постигне оптимална защита на животните, използвани за тестване на козметични продукти, докато се въведе забраната върху опитите върху животни за козметични продукти и търговията в Общността с козметични продукти, които са тествани върху животни, тези разпоредби следва да бъдат изменени, за да се осигури систематичното използване на алтернативни методи, които намаляват броя на използваните животни или намаляват причинените страдания в случаите, при които все още няма алтернативни изцяло заместващи методи, както е предвидено в параграфи 2 и 3 от член 7 от Директива 86/609/ЕИО, когато тези методи предлагат на потребителите ниво на защита, еквивалентно на нивото на защита, което те биха получили при използването на конвенционални методи, които те са предназначени да заместят. |
|
(4) |
В съответствие с Директива 86/609/ЕИО и с Директива 93/35/ЕИО, от съществено значение е, да бъде преследвана целта да се премахнат опитите върху животни за изпитване на козметични продукти и забраната да се провеждат такива експерименти да стане ефективна на територията на държавите-членки. За да може тази забрана да бъде изцяло спазвана, може да се окаже необходимо Комисията да постави на обсъждане нови предложения за изменения на Директива 86/609/ЕИО. |
|
(5) |
Понастоящем само алтернативните методи, които са научно утвърдени от Европейския център за валидиране на алтернативни методи (ECVAM) или от Организацията за икономическо сътрудничество и развитие (ОИСР) и които са приложими към целия химически сектор, се приемат систематично на ниво на Общността. Въпреки това, безопасността на козметичните продукти и техните съставки може да бъде осигурена чрез използването на алтернативни методи, за които не е задължително да се отнасят до всички приложения на химичните съставки. Следователно, използването на такива методи от цялата козметична индустрия следва да бъде насърчено и да бъде осигурено тяхното приемане на ниво на Общността, когато такива методи предлагат еквивалентно ниво на защита на потребителите. |
|
(6) |
Безопасността на готовите козметични продукти вече може да бъде осигурена въз основа на познанията върху безопасността на съставките, които те съдържат. Следователно към Директива 76/768/ЕИО могат да бъдат добавени разпоредби, забраняващи тестването върху животни на готови козметични продукти. Комисията следва да определи насоки, които да улеснят прилагането, по-специално от малки и средни предприятия, на методи, които не включват използването на животни за оценка на безопасността на готови козметични продукти. |
|
(7) |
Постепенно ще стане възможно да се осигури безопасността на съставките, използвани при производството на козметични продукти, чрез прилагането на алтернативни методи, без използването на животни, одобрени на ниво на Общността или научно утвърдени от ECVAM, като надлежно се следи развитието на валидирането в рамките на ОИСР. След консултации с Научния комитет по козметичните продукти и нехранителните продукти, предназначени за потребителите (SCCNFP) по отношение на приложимостта на утвърдените алтернативни методи в областта на козметичните продукти, Комисията следва незабавно да публикува утвърдените или одобрени методи, които са признати за приложими спрямо такива съставки. За да се достигне максимално възможна степен на защита на животните, трябва да се постави краен срок за въвеждането на окончателна забрана. |
|
(8) |
Комисията следва да състави графици на крайните срокове за забраната на търговията с козметични продукти, окончателният състав, отделните съставки на които или комбинации от съставките на които са били тествани върху животни, и за забраната на всеки тест, който понастоящем се осъществява с използване на животни, като крайният срок е най-много шест години от датата на влизане в сила на настоящата директива. Като се има предвид обаче фактът, че все още няма алтернативи за тестове, касаещи токсичността на многократните дози, репродуктивната токсичност и токсикокинетиката, подходящо е максималният краен срок на забраната за търговия с козметични продукти, за които се използват такива тестове, да бъде 10 години от датата на влизане в сила на настоящата директива. Въз основа на годишните доклади Комисията следва да бъде упълномощена да адаптира графиците в рамките на посочените по-горе максимални времеви граници. |
|
(9) |
По-добра координация на ресурсите на ниво на Общността ще допринесе за разширяване на научните знания, които са крайно необходими за разработването на алтернативни методи. За тази цел е от съществено значение Общността да продължи и увеличи своите усилия и да предприеме необходимите мерки за насърчаването на проучванията и разработки на нови алтернативни методи без използването на животни, по-специално в рамките на Шестата рамкова програма, приета с Решение № 1513/ЕО/2002 на Европейския парламент и на Съвета (7). |
|
(10) |
Признаването от страни, които не са членки на Общността, на алтернативни методи, разработени в Общността, следва да бъде насърчавано. За да се постигне тази цел, Комисията и държавите-членки следва да предприемат всички необходими мерки, за да улеснят приемането на такива методи от ОИСР. Комисията следва да положи всички усилия, в рамките на споразуменията за сътрудничество на Европейската общност, за да постигне признаване на резултатите от тестовете за безопасност, провеждани в Общността с използване на алтернативни методи, така че износът на козметични продукти, при които са били използвани такива методи, да не бъде възпрепятстван и да предотврати или избегне възможността, страните, които не са членки на Общността, да изискват повтарянето на такива тестове с използването на животни. |
|
(11) |
Следва да се предвиди възможност да се изисква върху козметичния продукт да бъде отбелязано, че не са извършвани опити с животни във връзка с неговото разработване. Комисията, като се консултира с държавите-членки, следва да разработи насоки, за да гарантира прилагането на общи критерии при използването на обозначенията и за да осигури тяхното единно интерпретиране по начин, който не въвежда потребителя в заблуждение. При разработването на такива насоки Комисията също така трябва да вземе предвид вижданията на много малки и средни предприятия, които представляват мнозинството производители, които не използват опити върху животни, съответните неправителствени организации и необходимостта потребителите да са в състояние на направят практически разграничения между продуктите въз основа на критерии, свързани с тестването върху животни. |
|
(12) |
В своето становище от 25 септември 2001 г. SCCNFP твърди, че субстанциите, класифицирани съгласно Директива 67/548/ЕИО на Съвета от 27 юни 1967 г. относно сближаването на законовите, подзаконови и административни разпоредби по отношение на класифицирането, пакетирането и етикетирането на опасни вещества (8) като канцерогенни (с изключение на веществата, които са канцерогенни само при вдишване), мутагенни или токсични за репродуктивния процес от категория 1 или 2, и вещества с подобен потенциал, не трябва да бъдат добавяни съзнателно към козметичните продукти, и че тези вещества, класифицирани в съответствие с Директива 67/548/ЕИО като канцерогенни, мутагенни или токсични за репродуктивния процес от категория 3, и вещества с подобен потенциал, не трябва да бъдат добавяни преднамерено към козметичните продукти, освен ако може да бъде доказано, че техните нива не представляват заплаха за здравето на потребителя. |
|
(13) |
Като се имат предвид рисковете, които могат да представляват за човешкото здраве веществата, класифицирани като канцерогенни, мутагенни или токсични за репродуктивния процес, категории 1, 2 и 3, в съответствие с Директива 67/548/ЕИО, тяхното използване в козметични продукти следва да бъде забранено. Вещество, класифицирано в категория 3, може да бъде използвано в козметиката, ако веществото е било оценено от SCCNFP и е било установено, че неговото използване в козметични продукти е допустимо. |
|
(14) |
За да се подобри информацията, предоставяна на потребителя, козметичните продукти следва да носят по-прецизни указания относно периода на техния срок на годност. |
|
(15) |
Някои субстанции са идентифицирани като важен причинител на алергични реакции при контакт при потребители, които са чувствителни към миризми. Следователно, за да може тези потребители да бъдат адекватно информирани, е необходимо да бъдат изменени разпоредбите на Директива 76/768/ЕИО, като се постави изискването наличието на тези субстанции да бъде споменато в списъка на съставките. Тази информация ще подобри диагностицирането на контактните алергии при такива потребители и ще им позволи да избягват употребата на козметични продукти, към които те имат непоносимост. |
|
(16) |
Редица субстанции са идентифицирани от SCCNFP като възможни причинители на алергични реакции и ще бъде необходимо да се ограничи тяхната употреба и/или да им се наложат определени условия. |
|
(17) |
Мерките, необходими за прилагането на настоящата директива, следва да бъдат приети в съответствие с Решение 1999/468/ЕО на Съвета от 28 юни 1999 г. относно установяване на процедурите за упражняване на изпълнителните пълномощия, предоставени на Комисията (9). |
|
(18) |
Разпоредбите на Директива 93/35/ЕИО, забраняващи търговията с козметични продукти, съдържащи съставки или комбинации от съставки, тествани върху животни, следва да бъдат заменени с разпоредбите на настоящата директива. С оглед постигане на правна сигурност е уместно прилагането на член 1, параграф 1 от настоящата директива да започне от 1 юли 2002 г., като изцяло се спази принципа на легитимните очаквания, |
ПРИЕХА НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Директива 76/768/ЕИО се изменя, както следва:
|
1. |
член 4, параграф 1, i) се заличава; |
|
2. |
добавят се следните членове: „Член 4а 1. Без да се засягат общите задължения, произтичащи от член 2, държавите-членки забраняват:
Не по-късно от 11 септември 2004 г. Комисията, в съответствие с член 10, параграф 2 и след консултация с Научния комитет по козметичните продукти и нехранителните продукти, предназначени за потребителите (SCCNFP), ще определи съдържанието на приложение IХ. 2. Комисията, след консултация с SCCNFP и с Европейския център за валидиране на алтернативни методи (ECVAM) и следeйки надлежно развитието на валидирането в рамките на ОИСР, определя графици за изпълнение на разпоредбите на букви а), б) и г) от параграф 1, включително крайни срокове за прекратяване на различните тестове. Графиците стават достъпни за обществеността не по-късно от 11 септември 2004 г. и се изпращат на Европейския парламент и на Съвета. Периодът за привеждане в действие се ограничава до не повече от шест години след влизането в сила на Директива 2003/15/ЕО по отношение на букви а), б) и г) от параграф 1. 2.1. По отношение на тестовете, касаещи токсичността на многократните дози, репродуктивната токсичност и токсикокинетиката, за които все още не съществуват алтернативни методи в процес на разработка, периодът за привеждане в действие на букви а) и б) се ограничава най-много до 10 години след влизането в сила на Директива 2003/15/ЕО. 2.2. Комисията проучва възможните технически трудности, свързани със спазването на забраната във връзка с опитите, по-специално тези, касаещи токсичността на многократните дози, репродуктивната токсичност и токсикокинетиката, за които не съществуват алтернативни методи в процес на разработка. Годишните доклади, представени съобразно член 9, включват информация относно междинните и окончателните резултати на тези проучвания. Въз основа на тези годишни доклади графиците, изработени в съответствие с параграф 2, могат да бъдат адаптирани в рамките на максималния срок от шест години, посочен в параграф 2, или 10 години, посочен в параграф 2.1 и след консултация с образуванията, посочени в параграф 2. 2.3. Комисията проучва напредъка и спазването на крайните срокове и възможните технически трудности за спазване на забраната. Годишните доклади, представяни съгласно член 9, съдържат информацията относно междинните и крайните резултати на тези проучвания. Ако в резултат на тези проучвания се стигне до извода, най-късно две години преди края на максималния период, указан в параграф 2.1, че по технически причини един или повече тестове, предвидени в параграф 2.1, няма да бъдат разработени и утвърдени преди изтичането на периода, посочен в параграф 2.1, Комисията информира Европейския парламент и Съвета и поставя на обсъждане законодателно предложение в съответствие с член 251 от Договора. 2.4. При изключителни обстоятелства, когато възникнат сериозни опасения относно безопасността на съществуващи козметични съставки, държава-членка може да поиска Комисията да предостави дерогация от параграф 1. Искането съдържа оценка на ситуацията и посочва необходимите мерки. На тази основа Комисията може, след консултация с SCCNFP и след като вземе мотивирано решение, да разреши дерогацията в съответствие с процедурата по член 10, параграф 2. Това разрешение определя условията, при които е направена дерогацията, като специфични цели, продължителност и отчитане на резултатите. Дерогация се предоставя само ако:
Решението за разрешаване, свързаните с него условия и достигнатите окончателни резултати се включват в годишния доклад, представян от Комисията в съответствие с член 9. 3. По смисъла на настоящия член:
Член 4б Забранява се използването на козметични продукти или вещества, класифицирани като канцерогенни, мутагенни и токсични за репродуктивния процес от категория 1, 2 и 3 съгласно приложение I към Директива 67/548/ЕИО. За тази цел Комисията предприема необходимите мерки съгласно процедурата, посочена в член 10, параграф 2. Вещество, класифицирано в категория 3, може да бъде използвано в козметиката, ако е оценено от SCCNFP със заключение, че неговото използване в козметични продукти е допустимо. |
|
3. |
член 6, параграф 1, буква в) се заменя със следното:
|
|
4. |
член 6, параграф, буква ж) се заменя със следното:
|
|
5. |
последното изречение на член 6, параграф 3 се заличава и се добавя следната алинея: „Освен това, производителят или лицето, което отговаря за пускането на продукта на пазара на Общността, могат да съобщят на потребителите, на опаковката на продукта или във всеки документ, бележка, етикет, лента или карта, съпровождащи продукта, факта, че не са провеждани опити върху животни, само ако производителят и неговите доставчици не са провеждали и не са възлагали провеждането на каквито и да са тестове върху животни на крайния продукт, или на негов прототип, или на която и да е от неговите съставки, и не са използвали каквито и да са съставки, които са били тествани върху животни от друго лице с цел разработването на нови козметични продукти. Насоките се приемат в съответствие с процедурата, посочена в член 10, параграф 2 и се публикуват в Официален вестник на Европейския съюз. Европейският парламент получава копия от проектите на мерките, предоставени на Комитета.“ |
|
6. |
текстът на член 7а, параграф 1, буква г) се заменя със следното:
|
|
7. |
към член 7а, параграф 1 се добавя следната буква:
|
|
8. |
в член 8, параграф 2 и член 8а, параграф 3 заглавието „Научен комитет по козметология“ се заменя с „Научен комитет по козметичните продукти и нехранителните продукти, предназначени за потребителите“; |
|
9. |
членове 9 и 10 се заменят със следния текст: „Член 9 Всяка година Комисията представя на Европейския парламент и на Съвета доклад относно:
Член 10 1. Комисията се подпомага от Постоянния комитет по козметичните продукти. 2. Когато се прави позоваване на настоящия параграф, се прилагат членове 5 и 7 от Решение 1999/468/ЕО, като се спазват разпоредбите на член 8 от настоящото решение. Периодът, определен с член 5, параграф 6 от Решение 1999/468/ЕО, се определя на три месеца. 3. Комитетът приема свой процедурен правилник. |
|
10. |
към приложение III, част I се добавя следния текст:
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
11. |
Добавя се приложение VIIIа, което се състои от символ, представящ отворено бурканче с крем. Комисията, в съответствие с процедурата, посочена в член 10, параграф 2, определя този символ най-късно до 11 септември 2003 г. |
Член 2
За прилагането на член 1, точка 3 по отношение на член 6, параграф 1, буква в), трета алинея от Директива 76/768/ЕИО, а също така за прилагането на член 1, точка 4 по отношение на член 6, параграф 1, буква ж), трета алинея от Директива 76/768/ЕИО:
Държавите-членки предприемат всички необходими мерки, за да гарантират, че от 11 март 2005 г. нито производителите, нито вносителите, установени в Общността, ще пускат на пазара козметични продукти, които не отговарят на изискванията на настоящата директива.
Член 3
1. Държавите-членки въвеждат в сила необходимите законови, подзаконови и административни разпоредби, за да се съобразят с настоящата директива преди 11 септември 2004 г. Те незабавно информират Комисията за това.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
2. Държавите-членки съобщават на Комисията текста на разпоредбите от националното си законодателство в областта, регулирана от настоящата директива.
Член 4
Настоящата директива влиза в сила от датата на нейното публикуване в Официален вестник на Европейския съюз.
Чрез дерогация от член 3, член 1, точка 1 се прилага от 1 юли 2002 г.
Член 5
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 27 февруари 2003 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
M. CHRISOCHOÏDIS
(1) ОВ С 311 Е, 31.10.2000 г., стр. 134 и ОВ С 51 Е, 26.2.2002 г., стр. 385.
(2) ОВ С 367, 20.12.2000 г., стр. 1.
(3) Становище на Европейския парламент от 3 април 2001 г. (ОВ С 21 Е, 24.1.2002 г., стр. 24), Обща позиция на Съвета от 14 февруари 2002 г. (ОВ С 113 Е, 14.5.2002, стр. 109) и Решение на Европейския парламент от 11 юни 2002 г. (все още непубликувано в Официален вестник). Решение на Европейския парламент от 15 януари 2003 г. и Решение на Съвета от 27 февруари 2003 г.
(4) ОВ L 262, 27.7.1976 г., стр. 169. Директива, последно изменена с Директива 2002/34/ЕО на Комисията (ОВ L 102, 18.4.2002 г., стр. 19).
(5) ОВ L 358, 18.12.1986 г., стр. 1.
(6) ОВ L 151, 23.6.1993 г., стр. 32.
(7) ОВ L 232, 29.8.2002 г., стр. 1.
(8) ОВ 196, 16.8.1967 г., стр. 1. Директива, последно изменена с Директива 2001/59/ЕО на Комисията (ОВ L 225, 21.8.2001 г., стр. 1).
(9) ОВ L 184, 17.7.1999 г., стр. 23.
(10) ОВ 196, 16.8.1967 г., стр. 1. Директива, последно изменена с Директива 2001/59/ЕО на Комисията (ОВ L 225 г., 21.8.2001 г., стр. 1).“
(11) ОВ L 140, 23.6.1995 г., стр. 26.“
(12) ОВ L 358, 18.12.1986 г., стр. 1.“
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
138 |
32003R0494
|
L 073/6 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕГЛАМЕНТ (ЕО) № 494/2003 НА КОМИСИЯТА
от 18 март 2003 година
за изменение на Регламент (ЕО) № 297/95 на Съвета относно дължимите такси на Европейската агенция за оценка на лекарствените продукти
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Регламент (ЕО) № 297/95 на Съвета от 10 февруари 1995 г. относно дължимите такси на Европейската агенция за оценка на лекарствените продукти (1), изменен с Регламент (ЕО) № 2743/98 (2), и по-специално член 12 от него,
като има предвид, че:
|
(1) |
Въз основа на член 57, параграф 1 от Регламент (ЕИО) № 2309/93 на Съвета от 22 юли 1993 г. за установяване на общностни процедури за разрешаване и контрол на лекарствените продукти за хуманна употреба и за ветеринарна употреба и за създаване на Европейска агенция за оценка на лекарствени продукти (3), последно изменен с Регламент (ЕО) № 649/98 на Комисията (4), приходите на агенцията се състоят от вноските и таксите, заплащани от предприятия за получаване и поддържане на разрешително за продажба в Общността и за други услуги, предоставяни от агенцията. |
|
(2) |
Агенцията получава по-голямата част от своите приходи от такси. |
|
(3) |
От началото на реформата в системата за такси от 1998 г., агенцията отбелязва относително намаляване на приходите от такси, поради инфлация, докато основните условия на пазара са довели до повишаване на нейните разходи. |
|
(4) |
Данните от Статистическата служба на Европейските общности (Евростат) подкрепят необходимостта от увеличаване на всички такси с 10 % с цел да се достигне същото ниво на покупателна способност, както при таксите, установени през 1998 г. |
|
(5) |
С оглед на трудните икономически условия, пред които е изправена агенцията и които влияят на нейните приходи и поставят в риск поддържането на нейната ресурсна инфраструктура и следователно нейните възможности да изпълнява своите задължения, е необходимо допълнителна промяна на всички такси освен на годишната такса с 6 %. |
|
(6) |
С оглед на нарастващата важност на проучвателните дейности, извършвани след издаването на разрешенията в работата на агенцията и за да се подобри капацитетът на Общността за идентифициране и управление на рисковете, произтичащи от използването по-специално на иновационни лекарства, годишната такса следва да се коригира с 16 %. |
|
(7) |
Общите принципи и цялостната структура на таксите ще бъде преразгледана в контекста на пълното отразяване на системата за таксите, по-специално въз основа на изменение на вече приетия Регламент (ЕИО) № 2309/93 на Съвета. Тези нови суми следователно ще се прилагат за ограничен период от време. |
|
(8) |
Мерките, предвидени в настоящия регламент, са в съответствие със становището на Постоянните комитети, |
ПРИЕ НАСТОЯЩИЯ РЕГЛАМЕНТ:
Член 1
Регламент (ЕО) № 297/95 се изменя, както следва:
|
1. |
Член 3 се изменя както следва:
|
|
2. |
Член 4 се изменя, както следва:
|
|
3. |
Член 5 се изменя, както следва:
|
|
4. |
Член 6 се изменя, както следва:
|
|
5. |
Член 7 се изменя, както следва:
|
|
6. |
Член 8 се изменя, както следва:
|
Член 2
Настоящият регламент влиза в сила в деня след публикуването му в Официален вестник на Европейския съюз.
Настоящият регламент е задължителен в своята цялост и се прилага пряко във всички държави-членки.
Съставено в Брюксел на 18 март 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 35, 15.2.1995 г., стр. 1.
(2) ОВ L 345, 19.12.1998 г., стр. 3.
(3) ОВ L 214, 24.8.1993 г., стр. 1.
(4) ОВ L 88, 24.3.1998 г., стр. 7.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
140 |
32003D0189
|
L 074/26 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕШЕНИЕ НА КОМИСИЯТА
от 18 март 2003 година
относно публикуването на справочни данни за стандарт EN 613:2000 „Независими конвекторни отоплителни уреди с газово захранване“ в съответствие с Директива 90/396/ЕИО на Съвета
(нотифицирано под № С(2003) 710)
(текст от значение за ЕИП)
(2003/189/ЕО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 90/396/ЕИО на Съвета от 29 юни 1990 г. относно сближаването на законодателството на държавите-членки относно уреди, които работят с газови горива (1), изменена с Директива 93/68/ЕИО (2), и по-специално член 6, параграф 1 от нея,
като взе предвид становището на Постоянния комитет, създаден съгласно член 5 от Директива 98/34/ЕО на Европейския парламент и на Съвета от 22 юни 1998 г. относно определяне на процедура за предоставяне на информация в областта на техническите стандарти и правила относно Услуги на Информационното Общество (3), изменена с Директива 98/48/ЕО (4),
като има предвид, че:
|
(1) |
Според член 2 от Директива 90/396/ЕИО газови уреди могат да бъдат пускани на пазара и привеждани в действие само ако при нормалната им употреба не се прави компромис с безопасността на лица, домашни животни и собственост. |
|
(2) |
Според член 5 от Директива 90/396/ЕИО се предполага, че газовите уреди отговарят на съществените изисквания, посочени в член 3 от съответната директива, ако те отговарят на приложимите към тях национални стандарти, които транспонират хармонизираните стандарти, чиито референтни номера са публикувани в Официален вестник на Европейския съюз. |
|
(3) |
От държавите-членки се изисква да публикуват референтните номера на националните стандарти, транспониращи хармонизираните стандарти, чиито референтни номера са публикувани в Официален вестник на Европейския съюз. |
|
(4) |
Обединеното кралство повдигна формално възражение относно хармонизиран стандарт EN 613:2000 „Независими конвекторни отоплителни уреди с газово захранване“, приет от Европейският комитет по стандартизация (CEN) на 13 юли 2000 г., чийто референтен номер е публикуван в Официален вестник на Европейските общности на 18 юли 2001 (5), въз основа на това, че той не задоволява напълно съществените изисквания на Директива 90/396/ЕИО, и особено онези по точки 2.1 и 3.2.2 от приложение 1 към нея, тъй като изискванията за дизайн на декоративни камини с предно стъкло не са достатъчни да осигурят високо ниво на безопасност. Обединеното кралство е особено загрижено, че с тези продукти би могло да възникне опасна ситуация в случай на натрупване на газ, тъй като акумулираният неизгорен газ би могъл да се възпламени и да причини сериозни щети. |
|
(5) |
Въз основа на информацията, получена в рамките на консултиране с националните органи, с Европейският комитет по стандартизация и с комисията, създадена с Директива 98/34/ЕО, не бе намерено доказателство, което да обосновава риска от акумулиране на газ или от газова експлозия. Следователно не беше демонстрирано, че хармонизираният стандарт EN 613:2000 не отговаря на основните изисквания на Директива 90/396/ЕИО, |
ПРИЕ НАСТОЯЩОТО РЕЩЕНИЕ:
Член 1
Позоваването на стандарт ЕN 613:2000 „Независими конвекторни отоплителни уреди с газово захранване“, приет от Европейския комитет по стандартизация (CEN) на 13 юли 2000 г. и публикуван за пръв път в Официален вестник на Еврпейските общности на 18 юли 2001 г., няма да бъде заличено от списъка на стандартите, публикувани в Официален вестник на Европейския съюз. Стандартът следователно ще продължи да съдържа презумпцията за съответствие със съответните разпоредби на Директива 90/396/ЕИО.
Член 2
Адресати на настоящото решение са държавите-членки.
Съставено в Брюксел на 18 март 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 196, 26.7.1990 г., стр. 15.
(2) ОВ L 220, 30.8.1993 г., стр. 1.
(3) ОВ L 204, 21.7.1998 г., стр. 37.
(4) ОВ L 217, 5.8.1998 г., стр. 18.
(5) ОВ C 202, 18.7.2001 г., стр. 5.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
142 |
32003D0190
|
L 074/28 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕШЕНИЕ НА КОМИСИЯТА
от 18 март 2003 година
относно публикуването на справочни данни за стандарт EN 512:1998 „Спецификации за уреди с втечнен нефтен газ — преносими уреди с втечнен нефтен газ под налягане“, точка 5.7.2.1, в съответствие с Директива 90/396/ЕИО на Съвета
(нотифицирано под № С(2003) 711)
(текст от значение за ЕИО)
(2003/190/ЕО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 90/396/ЕИО на Съвета от 29 юни 1990 г. относно сближаването на законодателствата на държавите-членки относно уреди, които работят с газови горива (1), изменена с Директива 93/68/ЕИО (2), и по-специално член 6, параграф 1 от нея,
като взе предвид становището на Постоянния комитет, създаден в съответствие с член 5 от Директива 98/34/ЕО на Европейския парламент и на Съвета от 22 юни 1998 г., относно определяне на процедура за предоставяне на информация в областта на техническите стандарти и регламентация, както и правила относно услуги на информационното общество (3), изменена с Директива 98/48/ЕО (4),
като има предвид, че:
|
(1) |
Член 2 от Директива 90/396/ЕИО предвижда, че газови уреди могат да бъдат пускани на пазара и употребявани само ако при нормална употреба не излагат на опасност сигурността на лица, домашни животни и имущество. |
|
(2) |
Съгласно член 5 от Директива 90/396/ЕИО се смята, че газовите уреди се съобразяват с основните изисквания, посочени в член 3 от съответната директива, ако те съответстват на националните стандарти, приложими към тях, които транспонират хармонизираните стандарти, чиито референтни номера са публикувани в Официален вестник на Европейския съюз. |
|
(3) |
От държавите-членки се изисква да публикуват референтните номера на националните стандарти, които транспонират хармонизираните стандарти, чиито референтни номера са публикувани в Официален вестник на Европейския съюз. |
|
(4) |
Нидерландия е повдигнала формално възражение относно точка 5.7.2.1 „уреди с прикрепяне на продупчваеми патрони“ от стандарт EN 521:1998 „Спецификации за уреди с втечнен нефтен газ — преносими уреди с втечнен нефтен газ под налягане“, приет от Европейският комитет по стандартизация (СЕN) на 21 май 1997 г., чийто референтен номер е публикуван в Официален вестник на Европейските общности на 25 юли 1998 г. (5), въз основа на това, че тя не задоволява напълно основните изисквания на Директива 90/396/ЕИО. |
|
(5) |
Според нидерландските власти преносими (къмпингови) газови уреди, захранвани в втечнен нефтен газ (LPG), при което горелката се захранва с LPG от продупчваем патрон, може — ако патронът се подмени по начин, който не съответства на инструкциите за употреба — да стане небезопасен, като така има вероятност да доведе до теч на газ и сериозни изгаряния. Нидерландските власти смятат, че такова поведение трябва да се предвиди, тъй като тези уреди се използват най-често в специфични условия, като например къмпинги и палатки за кратки периоди от годината, и не всички ползватели четат системно указанията за употреба, или пък няма достатъчно светлина за четене, така че наистина съществува риск ползвателят да не подмени патрона както трябва. |
|
(6) |
Съгласно Директива 90/396/ЕИО всички уреди трябва да бъдат придружавани от предназначени за ползвателя указания за употреба и поддръжка, които да съдържат цялата информация, необходима за безопасна употреба. От това следва, че предпоставка за безопасното функциониране на уредите, спазващи директивата, е това ползвателите да следват тези указания. |
|
(7) |
Въпреки че проблемът, повдигнат от нидерландските власти, произтича основно от поведението на ползвателите, CEN въпреки това трябва, в светлината на развитието на поведението на ползвателите и вземайки предвид специфичните условия, при които е вероятно да бъдат използвани този вид уреди, да разгледа възможността за подобряване на присъщото ниво на безопасност във връзка със смяната на газовите патрони. |
|
(8) |
За тази цел Комисията ще изиска от CEN да представи в срок от две години ревизиран вариант на EN 521:1998. След изпълнението на този мандат и в зависимост от резултатите би могло да се предвидят възможни нови решения относно настоящия вариант на стандарта. |
|
(9) |
Що се отнася до искането за оттегляне на презумпцията за съответствие, трябва да се вземе предвид фактът, че според получената информация газови патрони са в употреба повече от 30 години, като близо 50 милиона се продават всяка година в цялата Общност, и че броят инцидентите, които се случват, е изключително малък в сравнение с броя на употребяваните единици. |
|
(10) |
По-нататък трябва да се има предвид, че преносимите (къмпингови) газови уреди неизбежно представляват завишен риск при всички обстоятелства поради характеристиките на използваната техника (открит пламък, гореща повърхност и т.н.), така че рисковете, посочени във формалното възражение, съществуват при всички случаи, когато поведението на потребителите не съответства на указанията за употреба. |
|
(11) |
Трябва следователно да се заключи, въз основа на EN 521:1998, както и въз основа на подадената информация от Нидерландия, от другите национални власти, от CEN и от индустрията, и след консултации с Експертната група по газови уреди и с комитета, създаден с Директива 98/34/ЕО, че не би било в съответствие с принципа на пропорционалността и следователно не би било оправдано да се оттегли презумпцията на съответствие на точка 5.7.2.1 от EN 521:1998, |
ПРИЕ НАСТОЯЩОТО РЕШЕНИЕ:
Член 1
Справочните данни за стандарт EN 512:1998 „Спецификации за уреди с втечнен нефтен газ — преносими уреди с втечнен нефтен газ под налягане“, приет от Европейският комитет по стандартизация (CEN) на 21 май 1997 г. и публикуван за първи път в Официален вестник на Европейските общности на 25 юли 1998 г., не се оттегля от списъка на стандартите, публикувани в Официалния вестник на Европейския съюз. Стандартът следователно продължава да се ползва от презумпцията за съответствие със съответните разпоредби на Директива 90/396/ЕИО.
Член 2
Адресати на настоящото решение са държавите-членки.
Съставено в Брюксел на 18 март 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 196, 26.7.1990 г., стр. 15.
(2) ОВ L 220, 30.8.1993 г., стр. 1.
(3) ОВ L 204, 21.7.1998 г., стр. 37.
(4) ОВ L 217, 5.8.1998 г., стр. 18.
(5) ОВ C 233, 25.7.1998 г., стр. 16.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
144 |
32003L0017
|
L 076/10 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/17/ЕО НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 3 март 2003 година
за изменение на Директива 98/70/ЕО относно качеството на бензина и дизеловите горива
(текст от значение за ЕИП)
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 95 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Европейския икономически и социален комитет (2),
като взеха предвид становището на Комитета на регионите,
в съответствие с процедурата, предвидена в член 251 от Договора (3) и в светлината на съвместния проект, одобрен от Помирителната комисия на 20 януари 2003 г.,
като имат предвид, че:
|
(1) |
Директива 98/70/ЕО (4) определя екологични спецификации за горива на пазара. |
|
(2) |
Член 95 от Договора предвижда, че предложенията на Комисията, които имат за предмет установяването и функционирането на вътрешния пазар и относно inter alia опазване на здравето и околната среда, ще вземе за основа висока степен на защита, и че Европейският парламент и Съветът също ще се стремят да постигнат тази цел. |
|
(3) |
Предвижда се изменение на Директива 98/70/ЕО, за да се изпълнят условията на Общността за стандарти за качество на въздуха и свързаните с това цели и за да се включат допълнителни спецификации, за да се допълнят тези задължителни спецификации, които вече са посочени в приложение III и приложение IV на директивата. |
|
(4) |
Намаляване на съдържанието на сяра в състава на бензина и дизеловите горива се посочва като средство за постигането на тези цели. |
|
(5) |
Отрицателното действие на сярата в бензина и дизеловите горива върху ефективността технологиите за каталитична обработка на отработени газове е добре установено за пътни превозни средства все повече за непътни мобилни съоръжения. |
|
(6) |
Пътните превозни средства са все по-зависими от устройствата за каталитична обработка на отработени газове, за да бъдат в рамките на ограниченията за емисиите, посочени в Директива 70/220/ЕИО на Съвета от 20 март 1970 г. за сближаване на законодателството на държавите-членки относно мерките, които трябва да се приемат за контрол върху замърсяването на въздуха от газовете на двигателите с принудително запалване, с които са оборудвани моторните превозни средства (5) и Директива 88/77/ЕИО на Съвета от 3 декември 1987 г. за сближаване на законодателствата на държавите-членки относно мерките, които следва да се предприемат срещу емисията на газови замърсители от дизеловите двигатели, предназначени за употреба в превозни средства (6). Съответно намаляването на съдържанието на сяра в бензина и дизеловите горива вероятно ще окаже по-голямо въздействие върху отработените емисии, отколкото промените в други параметри на горивото. |
|
(7) |
Въвеждането на горива с максимално съдържание на сяра 10 mg/kg ще подобри ефективността на горивото, постижимо с новосъздадени технологии за превозни средства и трябва да бъдат разгледани, в случай че става въпрос за непътни мобилни съоръжения и трябва да доведат до значително намаляване на конвенционални замърсители на въздуха в емисиите, когато се използват при съществуващите превозни средства. Тези ползи ще компенсират увеличените емисии на СО2, свързвани с производството на бензин и дизелови горива с по-ниско съдържание на сяра. |
|
(8) |
Поради това е целесъобразно да се определят мерки, които гарантират въвеждането и наличността на горива с максимално съдържание на сяра 10 mg/kg. По отношение на това данъчните стимули доказаха, че са ефективни при насърчаването в ранните етапи на въвеждане на високо качествени горива според националните нужди и приоритети и за скъсяване на преходния период, когато две различни качества се предлагат на пазара. Използването на данъчни мерки, на подходящо национално ниво или ниво на Общността трябва да бъде насърчавано и окуражавано. |
|
(9) |
Широко разпространената наличност на горива с максимално съдържание на сяра 10 mg/kg ще осигури основа за производителите на автомобили да отбележат значителен, допълнителен напредък към подобряване на ефективността на горивата на нови превозни средства. Потенциалният принос на горива с максимално съдържание на сяра 10 mg/kg за постигане на целта на Общността от 120 g/km за средна емисия на СО2 на нов автопарк ще бъде оценено, когато настоящите екологични задължения на производителите на автомобили бъдат преразгледани през 2003 г. |
|
(10) |
Необходимо е да се гарантират, че достатъчни количества от бензин и дизелово гориво с максимално съдържание на сяра 10 mg/kg се осигуряват от 1 януари 2005 г. на подходящо балансирана географска база, за да се позволи свободното движение на нови коли, които използват тези горива, като същевременно се гарантира, че намаляването на емисиите на СО2 от новите превозни средства превъзхожда тези допълнителни емисии, свързани с производството на тези горива. |
|
(11) |
Пълното проникване на бензин и дизелово гориво с максимално съдържание на сяра 10 mg/kg следва да бъде предвидено от 1 януари 2009 г., за да се осигури на промишлеността за производство на горива да има достатъчно време, за да направи необходимите инвестиции, за да приведе в съответствие производствените си планове. В допълнение, пълното въвеждане на бензин и дизелово гориво с максимално съдържание на сяра 10 mg/kg от 1 януари 2009 г. ще намали емисиите на конвенционални замърсители от съществуващия парк от превозни средства, водещ до подобрения в качеството на въздуха, като се гарантира, че няма цялостно нарастване на емисиите на парникови газове. В този контекст ще бъде необходимо да се потвърди тази дата, в случай на дизелови горива, не по-късно от 31 декември 2005 г. |
|
(12) |
За да се опази здравето на човека и/или околната среда в конкретни населени места или в специални области, чувствителни от екологична гледна точка и от гледна точка на околната среда със специфични проблеми на замърсяването, на държавите–членки трябва да се позволи, при спазване на процедурата, установена с настоящата директива, да изискат с тези горива да бъде търгувано само ако те отговарят на по-строги екологични спецификации, свързани със замърсители, които будят тревога, различни от тези, установени в настоящата директива. Тази процедура е изключение от информационната процедура, посочена в Директива 98/34/ЕО на Европейския парламент и на Съвета от 22 юни 1998 г., за установяване на процедура за предоставянето на информация в областта на техническите стандарти и регламенти и на правила за обществени информационни услуги (7). |
|
(13) |
Емисиите от двигателите, монтирани на непътни мобилни съоръжения и трактори за селското и горското стопанство, трябва да са в съответствие с границите, определени в Директива 97/68/ЕО на Европейския парламент и на Съвета от 16 декември 1997 г. за сближаването на законодателствата на държавите-членки относно мерките за ограничаване на емисиите на газообразни и прахообразни замърсители от двигатели с вътрешно горене, инсталирани в извънпътна подвижна техника (8) и в Директива 2000/25/ЕО на Европейския парламент и на Съвета от 22 май 2000 г. относно вземане на действия срещу емисиите от газообразни и прахообразни замърсяващи околната среда вещества от двигателите, предназначени за задвижване на селскостопански и горски трактори (9). Постигането на тези граници на емисиите ще стане все по зависимо от качеството на бензина, използван от тези двигатели и за това е важно да бъде включено определение за такова гориво в Директива 98/70/ЕО. |
|
(14) |
Целесъобразно е да се предвиди единна система за мониторинга на качеството на горивата или националните системи, които гарантират също толкова достоверни резултати за системите за отчитане, за да се прецени съответствието със задължителните екологични спецификации за качеството на горивото. |
|
(15) |
Следва да се определели процедура за актуализиране на методите за измерване, използвани за да се осигури съответствие със задължителните екологични спецификации за качеството на горивото. |
|
(16) |
Необходимите мерки за прилагането на Директива 98/70/ЕО следва да се приемат в съответствие с Решение 1999/468/ЕО на Съвета от 28 юни 1999 г. относно определяне на процедурите за упражняване изпълнителните правомощията на Комисията (10). |
|
(17) |
Трябва да се предвиди преразглеждане на разпоредбите на Директива 98/70/ЕО, за да се вземе под внимание новото законодателство на Общността за качеството на въздуха и свързаните с това екологични цели, като например необходимостта да се насърчават алтернативните горива, включително биогоривата, разработването на нови технологии за намаляване на замърсяването и въздействието на металните добавки и други свързани с това въпроси за тяхното изпълнение и да се потвърди или да се съобщи датата за пълното въвеждане на дизеловите горива с максимално съдържание на сяра 10 mg/kg, за да се гарантира, че няма общо нарастване на емисиите на парникови газове. |
|
(18) |
Трябва да се предприеме изчерпателно преразглеждане на алтернативните горива, включително биогоривата, включително обсъждане на необходимостта от специално законодателство. |
|
(19) |
Държавите–членки трябва да въвеждат разпоредби за санкциите, които се прилагат при нарушаване на разпоредбите на Директива 98/70/ЕО и да гарантират, че те се прилагат. |
|
(20) |
Директива 98/70/ЕО следва съответно да се измени, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Директива 98/70/ЕО се изменя, както следва:
|
1. |
Член 2 се заменя със следното: „Член 2 Определения По смисъла на настоящата директива:
За държавите-членки с арктически или сурови зимни условия максималната точка за дестилация от 65 % е 250 °С за дизелово гориво и бензин могат да бъдат заменени с максималната точка на дестилация от 10 % (об/об) при 180 °С. |
|
2. |
Следните алинеи се добавят към член 3, параграф 2:
|
|
3. |
В член 4:
|
|
4. |
В член 6:
|
|
5. |
Член 8 се заменя със следното „Член 8 Контрол на спазването на предписанията и докладване 1. Държавите–членки контролират спазването на изискванията на членове 3 и 4, по отношение на бензина и дизеловото гориво на базата на аналитични методи, посочени в Европейските стандарти EN 228: 1999 и EN 590: 1999 съответно. 2. Държавите–членки установяват система за мониторинг на качеството на горивото в съответствие с изискванията на съответният Европейски стандарт. Използването на система за мониторинг на качеството на алтернативното гориво може да бъде позволена, в случай че такава система гарантира също толкова достоверни резултати. 3. Всяка година до 30 юни държавата–членка представя доклад за данните за качеството на националното гориво за предходната календарна година. Първият доклад се представя до 30 юни 2002 г. От 1 януари 2004 г. форматът на този доклад ще отговаря на описания в съответния европейски стандарт. В допълнение държавите–членки докладват за целия обем бензин и дизелово гориво, продадени на тяхна територия и за обема на безоловния бензин и дизеловото гориво, продадени с гориво с максимално съдържание на сяра 10 mg/kg. Освен това, държавите–членки докладват всяка година за наличността на подходящо балансирана географска основа за бензин и дизелово гориво с максимално съдържание на сяра 10 mg/kg, които са продадени на нейната територия. 4. Комисията гарантира, че информацията, предадена съгласно параграф 3 се разпространява бързо чрез подходящи средства. Комисията публикува годишно и за първи път на 31 декември 2003 г. доклад за действителното качество на горивото в различните държави–членки и географския обхват на горивата с максимално съдържание на сяра 10 mg/kg, с цел да представи общ преглед на данните за качеството на горивата в различните държави–членки.“ |
|
6. |
Член 9 се заменя със следното: „Член 9 Процес на преразглеждане 1. До 31 декември 2005 г. най-късно Комисията трябва да преразгледа спецификациите за горивото от приложения III и IV с изключение на съдържанието на сяра и да предложи изменения и допълнения, ако е целесъобразно, като се придържа към настоящите и бъдещите изисквания на законодателството на Общността в областта на емисиите от превозните средства и качеството въздуха и свързаните с това цели. По- специално Комисията разглежда:
2. Когато се разглежда предложението за следващия етап от стандартите за емисии от дизелови двигатели в нетранспортни превозни средства, Комисията трябва да утвърди паралелно с това изискваното качество на горивото. Като прави това Комисията взема под внимание значението на емисиите от този сектор, общите ползи за здравето и околната среда, последствията в държавите–членки по отношение на разпространение на горива и цените и ползите от по-ограниченото ниво на сяра, отколкото в момента се изисква за гориво за дизелови двигатели в нетранспортни приложения и след това да уеднакви съответните изисквания за качество на горивото за нетранспортни приложения с транспортния сектор до определена дата, която понастоящем се очаква да бъде 1 януари 2009 г., за да бъде потвърдено или изменено и допълнено от Комисията в нейния преглед за 2005 година. 3. В допълнение на разпоредбите на параграф 1 Комисията може, наред с другото, да представи:
|
|
7. |
Добавя се следният член: „Член 9а Санкции Държавите–членки определят санкциите, приложими за нарушаване на националните разпоредби, приети съгласно настоящата директива. Определените глоби трябва да бъдат ефективни, съразмерни и разубеждаващи.“ |
|
8. |
Първата алинея на член 10 се заменя със следното: „1. Методите за измерване, които следва да се прилагат във връзка с параметрите, определени в приложения I и III, са тези аналитични методи, определени в Европейския стандарт EN 228:1999. Методите за измерване, които следва да се прилагат във връзка с параметрите, определени в приложения II и IV, са тези аналитични методи, определени в Европейския стандарт EN 590:1999. Държавите-членки могат да приемат аналитичните методи, определени в стандартите за замяна EN 228:1999 или EN 590:1999, както е подходящо, ако може да бъде показано, че те имат поне същото ниво на прецизност, както аналитичните методи, които заместват. В случай че привеждането в съответствие на разрешените аналитични методи към техническия прогрес е необходимо, измененията и допълненията да бъдат приети от Комисията в съответствие с процедурата по член 11, параграф 2.“; |
|
9. |
Член 11 се заменя със следното: „Член 11 Процедура на Комитета 1. Комисията се подпомага от Комитета, учреден с член 12 от Директива 96/62/ЕО (19). 2. Когато се прави позоваване на настоящия параграф, член 5 и 7 Директива 1999/468/ЕО на Съвета от 28 юни 1999 г., за определяне на процедурите за упражняване на правомощията за прилагане, предоставени на Комисията (20) се прилагат, като се вземе под внимание, разпоредбите от член 8 от нея. Срокът по член 5, параграф 6 от Решение 1999/468/ЕО се определя на три месеца. 3. Комитетът приема свой процедурен правилник. |
|
10. |
Приложения от I до IV се заменят с текста в приложението към настоящата директива. |
Член 2
Държавите членки приемат и публикуват законовите, подзаконовите и административните разпоредби, необходими, за да се съобразят с настоящата директива, до 30 юни 2003 г. Те незабавно информират Комисията за това.
Държавите–членки прилагат тези мерки от 1 януари 2004 г.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 3
Директивата влиза в сила в деня на публикуването ѝ в Официален вестник на Европейския съюз.
Член 4
Адресати на настоящата директивата са държавите–членки.
Съставено в Брюксел на 3 март 2003 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
A.-A. TSOCHATZOPOULOS
(1) ОВ C 213 E, 31.7.2001 г., стр. 255.
(2) ОВ C 36, 8.2.2002 г., стр. 115.
(3) Становище на Европейския парламент от 29 ноември 2001 г. (ОВ C 153 E, 27.6.2002 г., стр. 253). Обща позиция на Съвета от 15 април 2002 г.(ОВ C 145 E, 18.6.2002 г., стр. 71) и Решение на Европейския парламент от 26 септември 2002 г. (все още непубликувано в Официалния вестник). Решение на Европейския парламент от 30 януари 2003 г. и Решение на Съвета от 6 февруари 2003 г..
(4) ОВ L 350, 28.12.1998, стр. 58; Директива, последно изменена с Директива 2000/71/ЕО на Комисията (ОВ L 287, 14.11.2000 г., стр. 46).
(5) ОВ L 76, 6.4.1970, стр. 1. Директива, последно изменена с Директива 2001/100/ЕО на Европейския парламент и на Съвета (ОВ L 16, 18.1.2001 г., стр. 32).
(6) ОВ L 36, 9.2.1988 г., стр. 33 Директива, последно изменена с Директива 2001/27/ЕО на Комисията (ОВ L 107, 18.4.2001 г., стр. 10).
(7) ОВ L 204, 21.7.1988 г., стр. 37 Директива, изменена с Директива 98/48/ЕО (ОВ L 217, 15.8.1998 г., стр. 18).
(8) ОВ L 59, 27.2.1998 г., стр. 1 Директива, последно изменена с Директива 2001/63/ЕО на Комисията (ОВ L 227, 23.8.2001 г., стр. 41).
(9) ОВ L 173, 12.7.2000 г., стр. 1.
(10) ОВ L 184, 17.7.1999 г., стр. 23.
(11) Изброяването на CN кодовете като специспецифицирани в ОМT, изменена с Регламент (ЕО) № 2031/2001 на Комисията (ОВ L 279, 23.10.2001 г., стр. 1).
(12) ОВ L 59, 27.2.1998 г., стр. 1; Директива, изменена с Директива 2001/63/ЕО на Комисията (ОВ L 227, 23.8.2001 г., стр. 41).
(13) ОВ L 173, 12.7.2000 г., стр. 1“;
(14) ОВ L 163, 29.6.1999 г., стр. 41; Директива, изменена с Решение 2001/744/ЕО на Комисията (ОВ L 278, 23.10.2001 г., стр. 35).
(15) ОВ L 100, 20.4.2000 г., стр. 57.
(16) ОВ L 100, 20.4.2000 г., стр. 55.
(17) ОВ L 40, 13.2.1999 г., стр. 49.
(18) ОВ L 44, 16.2.2000 г., стр. 1.“;
(19) ОВ L 296, 21.11.1996 г., стр. 55.
(20) ОВ L 184, 17.7.1999 г., стр. 23.“;
ПРИЛОЖЕНИЕ
ПРИЛОЖЕНИЕ I
ЕКОЛОГИЧНИ СПЕЦИФИКАЦИИ ЗА ГОРИВАТА, КОИТО СЕ ПРЕДЛАГАТ НА ПАЗАРА И СА ПРЕДНАЗНАЧЕНИ ЗА ПРЕВОЗНИ СРЕДСТВА, СНАБДЕНИ С ДВИГАТЕЛИ С ПРИНУДИТЕЛНО ЗАПАЛВАНЕ
Тип: Бензин
|
Параметър (1) |
Единица |
Граници (2) |
|||
|
минимум |
максимум |
||||
|
Теоретично октаново число |
|
95 (3) |
— |
||
|
Октаново число по моторен метод |
|
85 |
— |
||
|
Парно налягане за летния период (4) |
кРа |
— |
60,0 (5) |
||
|
Дестилация |
|
|
|
||
|
% обем/обем |
46,0 |
— |
||
|
% обем/обем |
75,0 |
— |
||
|
Въглеводороден анализ |
|
|
|
||
|
% обем/обем |
— |
18,0 (6) |
||
|
% обем/обем |
— |
42,0 |
||
|
% обем/обем |
— |
1,0 |
||
|
Съдържание на кислород |
% маса/маса |
— |
2,7 |
||
|
Кислородни съединения |
|
|
|
||
|
% обем/обем |
— |
3 |
||
|
% обем/обем |
— |
5 |
||
|
% обем/обем |
— |
10 |
||
|
% обем/обем |
— |
7 |
||
|
% обем/обем |
— |
10 |
||
|
% обем/обем |
— |
15 |
||
|
% обем/обем |
— |
10 |
||
|
Съдържание на сяра |
mg/kg |
— |
150 |
||
|
Съдържание на олово |
g/l |
— |
0,005 |
||
ПРИЛОЖЕНИЕ II
ЕКОЛОГИЧНА СПЕЦИФИКАЦИИ ЗА ГОРИВАТА, КОИТО СЕ ПРЕДЛАГАТ НА ПАЗАРА И СА ПРЕДНАЗНАЧЕНИ ЗА ПРЕВОЗНИ СРЕДСТВА С ДИЗЕЛОВИ ДВИГАТЕЛИ
Тип: Дизелово гориво
|
Параметър (8) |
Единица |
Ограничения (9) |
|||
|
минимум |
максимум |
||||
|
Октаново число |
|
51,0 |
— |
||
|
Плътност при 15 °С |
Kg/m3 |
— |
845 |
||
|
Дестилация |
|
|
|
||
|
°С |
— |
360 |
||
|
Полициклични ароматни въглеводороди |
% маса/маса |
— |
11 |
||
|
Съдържание на сяра |
mg/kg |
— |
350 |
||
ПРИЛОЖЕНИЕ III
ЕКОЛОГИЧНИ СПЕЦИФИКАЦИИ ЗА ГОРИВА, КОИТО СА ПРЕДЛАГАНИ НА ПАЗАРА И СА ПРЕДНАЗНАЧЕНИ ЗА ПРЕВОЗНИ СРЕДСТВА, КОИТО СА СНАБДЕНИ С ДВИГАТЕЛИ С ПРИНУДИТЕЛНО ЗАПАЛВАНЕ
Тип: Бензин
|
Параметър (10) |
Единица |
Ограничения (11) |
|||
|
минимум |
максимум |
||||
|
Теоретично октаново число |
|
95 (12) |
— |
||
|
Октаново число по моторен метод |
|
85 |
— |
||
|
Налягане при изпаряването, за летния период (13) |
кРа |
— |
60,0 (14) |
||
|
Дестилация |
|
|
|
||
|
% обем/обем |
46,0 |
— |
||
|
% обем/обем |
75,0 |
— |
||
|
Въглеводороден анализ |
|
|
|
||
|
% обем/обем |
— |
18,0 |
||
|
% обем/обем |
— |
35,0 |
||
|
% обем/обем |
— |
1,0 |
||
|
Съдържание на кислород |
% маса/маса |
— |
2,7 |
||
|
Кислородни съединения |
|
|
|
||
|
% обем/обем |
— |
3 |
||
|
% обем/обем |
— |
5 |
||
|
% обем/обем |
— |
10 |
||
|
% обем/обем |
— |
7 |
||
|
% обем/обем |
— |
10 |
||
|
% обем/обем |
— |
15 |
||
|
% обем/обем |
— |
10 |
||
|
Съдържание на сяра |
mg/kg |
— |
50 |
||
|
mg/kg |
— |
10 (16) |
|||
|
Съдържание на олово |
g/l |
— |
0,005 |
||
ПРИЛОЖЕНИЕ IV
ЕКОЛОГИЧНИ СПЕЦИФИКАЦИИ ЗА ГОРИВА, КОИТО СЕ ПРЕДЛАГАТ НА ПАЗАРА ЗА ПРЕВОЗНИ СРЕДСТВА С ДИЗЕЛОВИ ДВИГАТЕЛИ
Тип: Дизелово гориво
|
Параметър (17) |
Единица |
Ограничения (18) |
|||
|
минимум |
максимум |
||||
|
Октаново число |
|
51,0 |
— |
||
|
Плътност при 15 °С |
kg/m3 |
— |
845 |
||
|
Дестилация |
|
|
|
||
|
°С |
— |
360 |
||
|
Полициклични ароматни въглеводороди |
% маса/маса |
— |
11 |
||
|
Съдържание на сяра |
mg/kg |
— |
50 |
||
|
mg/kg |
— |
10 (19) |
|||
(1) Методите за изследване са тези, които са определени в EN 228:1999. Държавите–членки могат да приемат аналитични методи, определени в заменения стандарт EN 228:1999, ако може да се докаже, че той дава резултати поне толкова точни и прецизни, колкото аналитичните методи, които заменя.
(2) Стойностите, посочени в спецификацията са „истинските стойности“. При установяването на техни гранични стойности, условията на ISO 4259 „Петролни продукти – Определяне и приложение на прецизните данни във връзка с;методи за изпитване“, които са прилагани и при определяне на минимални стойности, минималната разлика от 2R над нулата е взета под внимание (R – възпроизводимост). Резултатите от отделните изследвания се тълкуват въз основа на критериите, описани в ISO 4259 (публикувано в 1995 г.).
(3) Безоловният бензин, обикновено качество може да бъде продаван с минимално октаново число по моторен метод (ОЧМ) 81 и минимално теоретично октаново число (ТОЧ) от 91.
(4) Летният период започва не по-късно от 1 май и продължава не до по-късно от 30 септември. За държавите–членки с арктически или сурови зимни условия, летния период започва не по-късно от 1 юни и завършва не преди 31 август.
(5) За държавите –членки с арктически или сурови зимни условия налягането при изпаряване не трябва да надвишава 70 кРа през летния период.
(6) Безоловният бензин, обикновено качество може да се продава с максимално олефиново съдържание 21 % обем/обем.
(7) Други моно-алкохоли и етери с точка на кипене, не по-висока от тази определена в EN 228:1999.
(8) Методите за изследване са тези, определени в EN 590:1999. Държавите –членки могат да приемат аналитични методи, определени в заменения стандарт EN 590:1999, ако може да се докаже, че той дава резултати поне толкова точни и прецизни, колкото аналитичните методи, които заменя.
(9) Стойностите, посочени в спецификацията са „истинските стойности“. При установяването на техни гранични стойности, са прилагани условията на ISO 4259 „Петролни продукти – Определяне и приложение на прецизните данни във връзка с методите за изпитване“, и при определянето на минимална стойност, е взета под внимание минимална разлика от 2R над нулата (R – възпроизводимост). Резултатите от отделните измервания се тълкуват въз основа на критериите, описани в ISO 4259 (публикувано 1995).
(10) Методите за изследване са тези, определени в EN 228:1999. Държавите –членки могат да приемат аналитични методи, определени в заменения стандарт EN 228:1999, ако може да бъде показано, че той дава резултати поне толкова точни и прецизни, колкото аналитичните методи, които заменя.
(11) Стойностите, посочени в спецификацията са „истинските стойности“. При установяването на техните гранични стойности, са прилагани условията на ISO 4259 „Петролни продукти – Определяне и приложение на прецизните данни във връзка с методите за изпитване“, и при определянето на минимална стойност е взета под внимание минимална разлика от 2R над нулата (R – възпроизводимост). Резултатите от отделните измервания се тълкуват въз основа на критериите, описани в ISO 4259 (публикувано 1995).
(12) Държавите – членки могат да решат да продължат да разрешават търговията с безоловния бензин, обикновено качество с минимално октаново число по моторен метод (ОЧМ) от 81 и минимално теоретично октаново число (ТОЧ) от 91.
(13) Летният период започва не по-късно от 1 май и продължава не до по-късно от 30 септември. За държавите–членки с арктически или сурови зимни условия, летният период започва не по-късно от 1 юни и завършва не преди 31 август.
(14) За държавите–членки с арктически или сурови зимни условия налягането при изпаряване не трябва да надвишава 70 кРа през летния период.
(15) Други моно-алкохоли и етери с точка на кипене, не по-висока от тази определена в EN 228:1999.
(16) В съответствие с член 3, параграф 2, не по-късно от 1 януари 2005 безоловният бензин, със съдържание на сяра 10 mg/kg трябва да се продават и да е разпространен на подходящо балансирана географска основа на територията на държавата-членка. До 1 януари 2009 цялото количество безоловен бензин, продаван на територията на държавата-членка трябва да има максимално съдържание на сяра 10 mg/kg.
(17) Методите за изследване са тези, определени в EN 590:1999. Държавите –членки могат да приемат аналитични методи, определени в заменения стандарт EN 590:1999, ако може да бъде доказано, че той дава резултати поне толкова точни и прецизни, колкото аналитичните методи, които заменя.
(18) Стойностите, посочени в спецификацията са „истинските стойности“. При установяването на техни гранични стойности, са прилагани условията на ISO 4259 „Петролни продукти – Определяне и приложение на прецизните данни във връзка с методите за изпитване“, и при определянето на минимална стойност, е взета под внимание минимална разлика от 2R над нулата (R – възпроизводимост). Резултатите от отделните измервания се тълкуват въз основа на критериите, описани в ISO 4259 (публикувано 1995).
(19) В съответствие с член 4, параграф 1, не по-късно от 1 януари 2005 г. дизеловото гориво, със съдържание на сяра от 10 mg/kg трябва да се продава и да се разпространява на подходящо балансирана географски основа на територията на държавата-членка. В допълнение и обект на справката в член 9, параграф 1 от 1 януари 2009 г. цялото количество дизелово гориво, продавано на територията на държавата-членка трябва да има максимално съдържание на сяра от 10 mg/kg.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
154 |
32003L0019
|
L 079/6 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/19/ЕО НА КОМИСИЯТА
от 21 март 2003 година
за изменение с цел привеждане в съответствие с техническия прогрес на Директива 97/27/ЕО на Европейския парламент и на Съвета относно масата и размерите на някои категории моторни превозни средства и на техните ремаркета
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 70/156/ЕИО на Съвета от 6 февруари 1970 г. за сближаване на законодателството на държавите-членки относно типовото одобрение на моторните превозни средства и на техните ремаркета (1), последно изменена с Директива 2001/116/ЕО на Комисията (2), и по-специално член 13, параграф 2 от нея,
като има предвид, че:
|
(1) |
Директива 97/27/ЕО на Европейския парламент и на Съвета от 22 юли 1997 г. относно масата и размерите на някои категории моторни превозни средства и на техните ремаркета, която изменя Директива 70/156/ЕИО (3), изменена с Директива 2001/85/ЕО (4), е една от специалните директиви в рамките на процедурата за типово одобрение на ЕО, установена с Директива 70/156/ЕИО. Разпоредбите на Директива 70/156/ЕИО относно системите, компонентите и обособените технически възли на превозните средства се прилагат съответно за Директива 97/27/ЕО. |
|
(2) |
В светлината на придобития опит при прилагането на Директива 97/27/ЕО е необходимо да се изменят и да се уточнят някои разпоредби в нея с оглед единно тълкуване във всички държави-членки. |
|
(3) |
Директива 96/53/ЕО на Съвета от 25 юли 1996 г. за определяне по отношение на някои пътни превозни средства, движещи се в Общността, на максимално допустимите размери в националния и международния трафик и максимално допустимото тегло в международния трафик (5), изменена с Директива 2002/7/ЕО на Европейския парламент и на Съвета (6), увеличава допустимите размери на някои моторни превозни средства, и в частност максималната дължина на автобусите. За да е възможно типовото одобряване на ЕО на превозните средства, чиято дължина достига допустимия максимум, е необходимо да се изменят съответно изискванията на Директива 97/27/ЕО. |
|
(4) |
Мерките, предвидени в настоящата директива, са в съответствие със становището на Комитета за привеждане в съответствие с техническия прогрес, създаден с Директива 70/156/ЕИО, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Приложения I—IV към Директива 97/27/ЕО се изменят в съответствие с приложението към настоящата директива.
Член 2
1. Считано от 1 октомври 2003 г., що се отнася до превозните средства, които отговарят на изискванията на Директива 97/27/ЕО, изменена с настоящата директива, държавите-членки не могат по причини, свързани с масата и размерите:
|
а) |
да отказват типово одобрение на ЕО или национално типово одобрение на тип моторно превозно средство от категориите М2, М3, N или О; |
|
б) |
да отказват да определят на тип моторно превозно средство от категориите М2, М3, N или О допустими максимални маси при регистриране/експлоатация съгласно приложение IV (когато това се изисква); |
|
в) |
да забраняват регистрацията, продажбата или въвеждането в експлоатация на тези превозни средства. |
2. Считано от 1 октомври 2004 г., държавите-членки не издават типово одобрение на ЕО и могат да отказват национално типово одобрение на тип превозно средство от категориите М2, М3, N или О на основания, свързани с масата и размерите, ако не са спазени изискванията на Директива 97/27/ЕО, последно изменена с настоящата директива.
Член 3
Настоящата директива не обезсилва одобренията, издадени в приложение на Директива 97/27/ЕО, и не пречи на разширяването на тези одобрения в съответствие с разпоредбите на директивата, по силата на която те са били издадени.
Член 4
До 9 март 2005 г. Обединеното кралство и Португалия могат на тяхна територия да отказват национално типово одобрение на тип превозно средство или да отказват или забраняват продажбата, регистрацията, въвеждането в експлоатация или употребата на превозно средство, или да считат за невалидно по смисъла на член 7, параграф 1 от Директива 70/156/ЕИО неговото удостоверение за съответствие, ако споменатото превозно средство не отговаря на критериите за управляемост, уточнени в член 8, буква а) от Директива 96/53/ЕО.
Член 5
1. Държавите-членки въвеждат в сила необходимите законови, подзаконови и административни разпоредби, за да се съобразят с настоящата директива, преди 30 септември 2003 г. Те незабавно уведомяват Комисията за това.
Когато държавите-членки приемат тези разпоредби, последните съдържат позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на това позоваване се определят от държавите-членки.
2. Държавите-членки уведомяват Комисията за текста на основните разпоредби от националното законодателство в областта, регулирана с настоящата директива.
Член 6
Настоящата директива влиза в сила от двадесетия ден след публикуването ѝ в Официален вестник на Европейския съюз.
Член 7
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 21 март 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 42, 23.2.1970 г., стр. 1.
(2) ОВ L 18, 21.1.2002 г., стр. 1.
(3) ОВ L 233, 25.8.1997 г., стр. 1.
(4) ОВ L 42, 13.2.2002 г., стр. 1.
(5) ОВ L 235, 17.9.1996 г., стр. 59.
(6) ОВ L 67, 9.3.2002 г., стр. 47.
ПРИЛОЖЕНИЕ
ПРИЛОЖЕНИЯ I—IV КЪМ ДИРЕКТИВА 97/27/ЕО СЕ ИЗМЕНЯТ, КАКТО СЛЕДВА:
А. Приложение I се изменя, както следва:
|
1. |
След заглавието на раздел 2, се добавя следният текст: „Определенията, дадени в приложение I (включително бележките под линия), и в приложение II към Директива 70/156/ЕИО, се прилагат за настоящата директива.“ |
|
2. |
Раздел 2.4.1 се изменя, както следва:
|
|
3. |
Раздел 2.4.2 се изменя, както следва:
|
|
4. |
Второто тире на раздел 2.4.3 се заменя, както следва:
|
|
5. |
Второто тире на раздел 2.4.4 се заменя, както следва:
|
|
6. |
Раздел 2.5 се заменя, както следва: 2.5. „маса на превозното средство в движение“: масата, определена в приложение I, раздел 2.6 към Директива 70/156/ЕИО.“ |
|
7. |
В раздел 2.6, второто изречение се заменя, както следва: „Определянето на категорията на превозното средство се извършва съгласно приложение II към Директива 70/156/ЕИО.“ |
|
8. |
Раздели 2.7, 2.8 и 2.9 се заменят, както следва: 2.7. „Технически допустима максимална маса върху оста (m)“: масата, съответстваща на допустимия максимален вертикален статичен товар, предаван на пътя посредством оста, фиксирана в зависимост от конструкцията на превозното средство, и на оста, и обявена от производителя на превозното средство. В случая на превозни средства от категория N1, технически допустимата максимална маса върху задната или задните оси не може да бъде надвишавана с повече от 15 %, а технически допустимата максимална маса в натоварено положение не може да бъде надвишавана с повече от 10 % или със 100 kg, като се взема предвид по-ниската от тези две стойности; тази разпоредба се прилага само в случай на превозно средство, което тегли ремарке, при положение че експлоатационната скорост е ограничена на 80 km/h или по-малко. Производителят на превозното средство уточнява това ограничение на скоростта или други евентуални експлоатационни условия в ръководството на потребителя. 2.8. „Технически допустимата максимална маса върху група от оси (μ)“: масата, съответстваща на допустимия максимален вертикален статичен товар, предаван на пътя посредством група от оси, фиксирана в зависимост от конструкцията на превозното средство, и на групата от оси, и обявена от производителя на превозното средство. 2.9. „Теглена маса“: максималният товар, предаван на пътя посредством оста или осите на тегленото или теглените превозни средства.“ |
|
9. |
Раздел 2.11 се заменя, както следва: 2.11. „Технически допустима максимална маса в точката на прикачване на моторно превозно средство“: масата, съответстваща на допустимия максимален вертикален статичен товар върху точката на прикачване в зависимост от конструкцията на моторното превозно средство и/или на теглително-прикачното устройство и която е обявена от производителя. По определение тази маса не включва масата на теглително-прикачното устройство на моторното превозно средство.“ |
|
10. |
Раздел 2.13 се заменя, както следва: 2.13. „Технически допустима максимална маса в натоварено състояние на подвижния състав (МС)“: пълната максимална маса на подвижния състав, включващ моторното превозно средство и неговото ремарке или ремаркета, която е обявена от производителя. В случаите на подвижен състав с полуремарке или ремарке с централно разположена ос, се използва технически допустимата максимална маса на осите на ремаркето, а не технически допустимото максимално тегло в натоварено положение М.“ |
|
11. |
Раздел 2.19 се заменя, както следва: 2.19. „Тип превозно средство“: превозните средства, които не се различават по някои основни аспекти, такива като:
За целите на настоящата точка аспектите, свързани с конструкцията или устройството, като например междуосието, конструкцията на осите, окачването, управлението, гумите и промените, свързани с устройството за коригиране на спирачното усилие върху осите, прибавянето или премахването на редукционни клапани, свързани с конфигурациите за използване на превозното средство като влекач на полуремаркета и като камион, и елементите, свързани с шасито (например двигател, резервоари за гориво, трансмисия и др.) не се смятат като основни точки.“ |
|
12. |
Раздел 7.2 се заменя, както следва: 7.2. Измерване на размерите Габаритните дължина, ширина и височина се измерват в съответствие с разпоредбите на раздел 2.4 върху превозното средство или средства в движение, споменати в раздел 3.3. Ако те се отклоняват с повече от 1 % от размерите, които производителят е обявил за съответните технически конфигурации в рамките на този тип превозно средство, измерените размери се използват за целите на следващите изисквания и техническата служба тогава, ако е необходимо, да извърши допълнителни измервания върху други превозни средства, освен посочените в раздел 3.3. Пределните стойности, посочени в приложение I към Директива 96/53/ЕО, не могат обаче да бъдат надвишавани.“ |
|
13. |
Раздели 7.4.2.5 и 7.4.2.5.1 се заменят, както следва: Когато превозното средство е натоварено с масата си M според едно от допустимите положения, описани в раздел 7.4.2.5.1 или 7.4.2.5.2, масата, съответстваща на товара, приложен върху оста „i“, не може да надвишава масата Mi върху тази ос, а масата, съответстваща на товара, приложен върху отделната ос или групата оси „j“, не може да надвишава масата μj. 7.4.2.5.1. Равномерното разпределяне на масата означава, че превозното средство в движение с маса от 75 kg, поставена на всяко пътническо място, е натоварено до масата М, като полезният товар е разпределен равномерно в пространството, предназначено за превоз на товари.“ |
|
14. |
Раздели 7.4.2.5.1.1 и 7.4.2.5.1.2 се заличават; |
|
15. |
Раздел 7.4.2.5.2 се заменя, както следва: 7.4.2.5.2. В случай на крайно разпределение на масата (нееднороден товар) производителят трябва да посочи крайните възможни и допустими положения на центъра на тежестта на полезния товар и/или на каросерията, и/или на вътрешните оборудвания или обзавеждания (например от 0,50 m до 1,30 m преди първата задна ос), когато превозното средство в движение с маса от 75 kg, поставена на всяко пътническо място, е натоварено до масата си М.“ |
|
16. |
Раздели от 7.4.2.5.2.1 до 7.4.2.5.3.2 се заличават; |
|
17. |
Раздел 7.4.3.2 се заменя, както следва: 7.4.3.2. Масата на превозното средство в движение, плюс масата Q, умножена по броя на седналите и правостоящите пътници, плюс масите WP, B и BX, определени в раздел 7.4.3.3.1, плюс технически допустимата максимална маса в точката на прикачване, ако от страна на производителя е монтирано теглително-прикачно устройство, не могат да надвишават масата М.“ |
|
18. |
Раздел 7.4.3.3.1 се заменя, както следва: 7.4.3.3.1. Превозното средство в движение се натоварва със: маса, съответстваща на броя Р на седящите пътници, маси Q; маса, съответстваща на броя SР на правостоящи пътници, маси Q, които са равномерно разпределени на площта, предназначена за превоз на правостоящи пътници S1; според случая, масата WP се разпределя равномерно на всяко място, предназначено за инвалидна количка; маса, равна на В (kg), равномерно разпределена в багажното отделение; маса, равна на BX (kg), равномерно разпределена на пространството на покрива, предвидено за носене на багажи, където:
|
|
19. |
Добавят се следните раздели от 7.4.3.3.2 до 7.4.3.3.2.3: В случая на превозно средство, което има променлив брой седящи места и е с площ, запазена за правостоящи пътници (S1), и/или е оборудвано за превоз на инвалидни колички, изискванията на раздели 7.4.3.2 и 7.4.3.3 трябва да се проверят за всяко от следните положения, според случая. 7.4.3.3.2.1. Заемане на всички предвидени седящи места, след това и на оставащата площ за правостоящи пътници (най-много до заявения от производителя максимален брой пътници, ако той бъде достигнат) и ако остава още пространство, заемане на всички места, които евентуално могат да се използват за инвалидни колички. 7.4.3.3.2.2. Заемане на цялото пространство, предвидено за правостоящи пътници (най-много до заявения от производителя максимален брой правостоящи пътници), след това и на всички оставащи седящи места, и ако остава още площ, заемане на всички места, които евентуално могат да се използват за инвалидни колички. 7.4.3.3.2.3. Заемане на всички места, предвидени за инвалидни колички, след това и на оставащата площ за правостоящи пътници (най-много до заявения от производителя максимален брой правостоящи пътници, ако той бъде достигнат) и на оставащите свободни места.“ |
|
20. |
Раздел 7.4.3.4 се заменя, както следва: 7.4.3.4. Когато превозното средство е в движение или е натоварено по описания в раздел 7.4.3.3.1 начин, масата, съответстваща на товара върху предната ос или върху групата предни оси, не може да бъде по-ниска от процента на масата на превозното средство в движение или от технически допустимото максимално тегло в натоварено положение „М“, определено от следната таблица:
|
||||||||||||||||||
|
21. |
Добавя се следният раздел 7.4.3.5: 7.4.3.5. Когато одобряването на превозно средство трябва да се извърши за няколко отделни класа, раздели 7.4.3.2 и 7.4.3.3 се прилагат за всеки клас.“ |
|
22. |
Заглавието на раздел 7.4.4 се заменя, както следва: 7.4.4. Изисквания за теглените каравани“. |
|
23. |
Второто изречение на раздел 7.6.1 се заменя, както следва: „За моторните превозни средства и полуремаркетата, оборудвани с повдигачи на оси (виж раздел 2.14), това изискване е валидно също в случая, когато повдигащите се оси са във вдигнато положение или когато облекчаващите натоварването оси са в разтоварено положение. То не се прилага за устройствата за подпомагане на потеглянето, като например повдигащите се оси, отговарящи на изискванията на приложение IV, раздел 3.5.“ |
|
24. |
Раздели 7.6.2, 7.6.3 и 7.6.4 се заменят, както следва: 7.6.2. Допълнителни изисквания за превозните средства от категория N: Превозното средство е неподвижно и управляващите му колела са ориентирани така че ако то започне да се движи, предният му край би трябвало да опише кръг с радиус от 12,50 m, като посредством маркиране на линия на земята се установява вертикална равнина, допирателна към тази страна на превозното средство, която е насочена към външната страна на кръга. Когато превозното средство се движи напред, следвайки кръга с радиус от 12,50 m, никой от неговите елементи не може да излиза извън вертикалната равнина с повече от 0,80 m (виж фигура Б). За превозните средства, оборудвани с повдигач на оста, това изискване е валидно също в случая, когато оста или осите са във вдигнато положение (по смисъла на раздел 2.14). За превозните средства от категория N с повдигащи се оси, когато те са във вдигнато положение, или с облекчаващи натоварването оси, когато те са в разтоварено положение, цифрата 0,80 m се заменя с 1,00 m. 7.6.3. Допълнителни изисквания за превозните средства от категории М2 или М3 Докато превозното средство е неподвижно, посредством маркиране на линия на земята се установява вертикална равнина, допирателна към тази страна на превозното средство, която е насочена към външната страна на кръга. В случая на съчленено превозно средство двете негови съчленени части се подравняват спрямо тази вертикална равнина. Когато превозното средство навлиза, движейки се по права линия, в кръга, описан в точка 7.6.1, никой от неговите елементи не може да излиза извън тази вертикална равнина с повече от 0,60 m (виж фигури В и Г). 7.6.4. Предписанията на раздели 7.6.1—7.6.3 могат също да се проверят по искане на производителя с помощта на съответни еквивалентни изчисления или чрез геометрична демонстрация. Ако по искане на производителя превозно средство от категория N без управляваща задна ос се проверява в зависимост от неговите геометрични характеристики, то се счита като отговарящо на изискванията на раздел 7.6.2 по-горе, ако разстоянието между задните колела и най-задната му част не представлява повече от 60 % от междуосието на превозното средство.“ |
|
25. |
Фигура В от раздел 7.6.3 се заменя със следната фигура: Фигура В
|
|
26. |
В раздел 7.6.3, се добавя следната фигура Г след фигура В: Фигура Г
|
|
27. |
Добавя се следният раздел 7.6.5: 7.6.5. В случая на неокомплектовани превозни средства производителят обявява максимално допустимите размери, за които превозното средство трябва да бъде проверено дали отговаря на изискванията на раздели от 7.6.1 до 7.6.3.“ |
|
28. |
Раздели 7.8.1 и 7.8.2 се заменят, както следва: 7.8.1. Максималната технически допустима маса върху точката на прикачване на моторно превозно средство, предназначено да тегли ремарке с централно разположена ос и чиято максимална технически допустима теглителна маса надвишава 3,5 тона, трябва да бъде най-малкото равностойна на 10 % от неговата технически допустима максимална теглителна маса или да бъде равна на 1 000 kg, като се взема предвид по-ниската от тези две стойности. 7.8.2. Технически допустимата максимална маса върху точката на прикачване на моторно превозно средство, предназначено да тегли ремарке с централно разположена ос и чиято технически допустима максимална теглителна маса не надвишава 3,5 тона, трябва да бъде най-малкото равностойна на 4 % от неговата технически допустима максимална теглителна маса или да бъде равна на 25 kg, като се взема предвид по-високата от тези две стойности.“ |
|
29. |
Раздел 7.10 се заменя, както следва: 7.10. Съотношение мощност на двигателя/максимална маса Моторните превозни средства трябва да имат отдавана мощност на двигателя от най-малко 5 киловата на тон от технически допустимото максимално тегло в натоварено положение на подвижния състав. В случая на пътни влекачи отдаваната мощност на двигателя трябва да бъде най-малко 2,2 киловата на тон. Отдаваната мощност на двигателя се измерва в съответствие с разпоредбите на Директива 80/1269/ЕИО на Съвета (3). |
Б. Приложение II се изменя, както следва:
|
1. |
Раздел 0.2 се заменя, както следва: 0.2. Тип.“ |
|
2. |
Раздел 13 се заменя, както следва:
|
В. Приложение III се изменя, както следва:
В добавката се добавя следният раздел 1.24.3:
1.24.3. Брой на местата, предвидени за инвалидни колички в превозните средства от категория М2 или М3:“
Г. Приложение IV се изменя, както следва:
|
1. |
Раздел 1.3.3 се заличава; |
|
2. |
Раздел 2.2.1 се изменя, както следва:
|
|
3. |
В раздел 2.2.1 последният ред от първия параграф, който гласи „осигурява изпълнението на всички съответни технически разпоредби на Директива 96/53/ЕО“ се заличава. |
|
4. |
Второто изречение на раздел 3.2 се заменя, както следва: „За тази цел повдигащата се или облекчаващата натоварването ос трябва да се спусне до нивото на земята или да се приведе автоматично в работно положение, ако оста или осите, които са най-близо до предната група оси или до предната ос на моторното превозно средство, са подложени на допустимото максимално натоварване при регистрация/при експлоатация.“ |
|
5. |
Раздел 3.3 се заличава. |
|
6. |
Четвъртото тире на раздел 3.5.1 се заменя, както следва:
|
(1) Включително 3 kg ръчен багаж.“
(2) Тази цифра се свежда до 20 % при триосните превозни средства от класове II и III, които имат две управляващи оси.“
(3) ОВ L 375, 31.12.1980 г., с. 46.“
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
162 |
32003D0224
|
L 083/70 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕШЕНИЕ НА КОМИСИЯТА
от 21 март 2003 година
относно публикуването на справочни данни за стандарт EN 1495:1997 „Повдигателни площадки — мачтови повдигателни работни площадки“ в съответствие с Директива 98/37/ЕО на Европейския парламент и на Съвета
(нотифицирано под № C(2003) 831)
(текст от значение за ЕИП)
(2003/224/ЕО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 98/37/ЕО на Европейския парламент и на Съвета от 22 юни 1998 г. относно сближаването на законодателствата на държавите-членки по отношение на машините (1), изменена с Директива 98/79/ЕО (2), и по-специално член 6, параграф 1 от нея,
като взе предвид становището на постоянния комитет, създаден в съответствие с член 5 от Директива 98/34/ЕО на Европейския парламент и на Съвета от 22 юни 1998 г. относно определяне на процедура за предоставяне на информация в областта на техническите стандарти и регламентация (3), изменена с Директива 98/48/ЕО (4),
като има предвид, че:
|
(1) |
Съгласно член 2 от Директива 98/37/ЕО машините могат да се пускат на пазара или в действие само ако не застрашават безопасността на хората, домашните животни или имущество, когато са монтирани и поддържани правилно, както и ако се използват съобразно предназначението им. |
|
(2) |
Когато национален стандарт, транспониращ хармонизиран стандарт, справочните данни на който са публикувани в Официален вестник на Европейските общности, обхваща едно или повече съществени изисквания, се предполага, че машината, изградена в съответствие с този стандарт, отговаря на приложимите съществени изисквания. |
|
(3) |
Държавите-членки са длъжни да публикуват справочните данни на националния стандарт, транспониращ хармонизираните стандарти, публикувани в Официален вестник на Европейския съюз. |
|
(4) |
Съгласно член 6, параграф 1 от Директива 98/37/ЕО Нидерландия повдигна официално възражение, според което стандарт EN 1495:1997, приет от Европейския комитет по стандартизация (CEN) на 21 април 1997 г., справочните данни на който са публикувани в Официален вестник на Европейските общности (5) на 13 март 1998 г., не отговаря напълно на съществените изисквания за безопасни и здравословни условия. |
|
(5) |
Комисията признава, че използването на въпросната машина може да се окаже опасно, тъй като стандарт EN 1495:1997 не отговаря на съществените изисквания за безопасни и здравословни условия, свързани с проектирането и конструирането на машини и предпазни елементи, посочени в приложение I към Директива 98/37/ЕО, и по-специално изискванията на член 1.5.15 „Рискове от падане“, 1.7.4 „Инструкции“ и 6.3 „Рискове от падане на лица от носещото устройство“. Относно параграф 5.3.2.4, последната алинея на 7.1.2.12, и по-специално таблица 8 и фигура 9 от стандарт EN 1495:1997, Комисията приема, че мерките, предвидени при проектиране и конструиране на площадката, не гарантират във висока степен безопасността при всички възможни приложения на продукта. |
|
(6) |
В интерес на безопасността и правната сигурност публикуването на справочните данни за този стандарт трябва да е придружено от подходящо предупреждение и държавите-членки трябва да добавят подобно предупреждение в своите национални стандарти, транспониращи стандарт EN 1495:1997. |
|
(7) |
Справочните данни за стандарт EN 1495:1997 трябва да съответно да бъдат публикувани отново, |
ПРИЕ НАСТОЯЩОТО РЕШЕНИЕ:
Член 1
Справочните данни за стандарт EN 1495:1997 се заменят от текста, посочен в приложението.
Член 2
Когато съгласно член 5, параграф 2 от Директива 98/37/ЕО държавите-членки публикуват справочните данни за националния стандарт, транспониращ хармонизирания стандарт EN 1495:1997, те добавят към публикацията предупреждение, подобно на това в справочните данни за стандарт EN 1495:1997, както е посочено в приложението.
Член 3
Адресати на настоящото решение са държавите-членки.
Съставено в Брюксел на 21 март 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 207, 23.7.1998 г., стр. 1.
(2) ОВ L 331, 7.12.1998 г., стр. 1.
(3) ОВ L 204, 21.7.1998 г., стр. 37.
(4) ОВ L 217, 5.8.1998 г., стр. 18.
(5) ОВ C 78, 13.3.1998 г., стр. 2.
ПРИЛОЖЕНИЕ
Публикуване на наименованията и справочните данни на европейски хармонизирани стандарти по Директива 98/37/ЕО на Европейския парламент и на Съвета
|
ЕОС (1) |
Данни |
Наименование на хармонизираните стандарти |
|
CEN |
EN 1495:1997 |
Повдигателни площадки — мачтови повдигателни работни площадки |
|
Предупреждение: Тази публикация не се отнася до точка 5.3.2.4, точка 7.1.2.12, последна алинея, таблица 8 и фигура 9 от стандарт EN 1495:1997, за които не дава презумпция за съответствие с разпоредбите на Директива 98/37/ЕО. |
||
Забележка:
Информация, свързана със стандартите, може да се получи или от европейските организации по стандартизация, или от националните органи по стандартизация, списъкът на които е приложен към Директива 98/34/ЕО на Европейския парламент и на Съвета (2).
Публикуването на справочните данни в Официален вестник на Европейския съюз не означава, че стандартите са достъпни на всички езици на Общността.
Комисията гарантира актуализирането на този списък.
Други хармонизирани стандарти относно машини са публикувани в предишни издания на Официален вестник на Европейския съюз. Пълен и актуализиран списък може да бъде намерен на сървъра на Европейския съюз в Интернет:
|
|
http://europa.eu.int/comm/enterprise/newapproach/standardization/harmstds/reflist/machines.html. |
(1) ЕОС (Европейска организация по стандартизация)
|
— |
CEN (Европейски комитет по стандартизация): rue de Stassart 36, B-1050 Bruxelles, тел.: (32-2) 550 08 11, факс: (32-2) 550 08 19. |
|
— |
CENELEC (Европейски комитет по електротехническа стандартизация): rue de Stassart 35, B-1050 Bruxelles, тел.: (32-2) 519 68 71, факс: (32-2) 519 69 19. |
|
— |
ETSI (Европейски институт за далекосъобщителни стандарти) телекомуникационни стандарти): 650, route des Lucioles, F-06921 Sophia Antipolis Cedex, тел.: (33-4) 92 94 42 00, факс: (33-4) 93 65 47 16. |
(2) ОВ L 204, 21.7.1998 г., стр. 37.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
165 |
32003D0312
|
L 114/50 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕШЕНИЕ НА КОМИСИЯТА
от 9 април 2003 година
за публикуването на справочни данни за стандартите, свързани с топлоизолационни продукти за сгради, геотекстил, неподвижни пожарогасителни инсталации и гипсови блокове в съответствие с Директива 89/106/ЕИО на Съвета
(нотифицирано под номер C(2003) 1161)
(текст от значение за ЕИП)
(2003/312/ЕО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 89/106/ЕИО на Съвета от 21 декември 1988 г. за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки по отношение на строителните продукти (1), изменена с Директива 93/68/ЕИО (2), и по-специално член 5, параграф 1 от нея,
като взе предвид становището на Постоянния комитет, създаден в съответствие с член 5 от Директива 98/34/ЕО на Европейския парламент и на Съвета от 22 юни 1998 г. за определяне на процедура за предоставяне на информация в областта на техническите стандарти и правила (3), изменена с Директива 98/48/ЕО (4),
като има предвид, че:
|
(1) |
Член 2 от Директива 89/106/ЕИО предвижда държавите-членки да предприемат всички необходими мерки, за да гарантират пускането на пазара на строителни продукти, само ако те ще се използват в строежи, които отговарят на съществените изисквания, посочени в член 3 от споменатата директива. |
|
(2) |
Съгласно член 4, параграф 2 от Директива 89/106/ЕИО се предполага, че строителните продукти са подходящи за употреба и позволяват на строежите, в които се използват, да отговарят на съществените изисквания, посочени в член 3 от споменатата директива, ако съответстват на приложимите национални стандарти, транспониращи хармонизираните стандарти, чиито справочни данни са публикувани в Официален вестник на Европейските общности. |
|
(3) |
Държавите-членки са длъжни да публикуват референтните номера на националния стандарт, транспониращ хармонизираните стандарти, чиито референтни номера са публикувани в Официален вестник на Европейските общности. |
|
(4) |
Съгласно член 5, параграф 1 от Директива 89/106/ЕИО Германия повдигна официално възражение относно някои хармонизирани стандарти на основание, че тези стандарти не позволяват строежите, в които ще се използват продуктите, напълно да отговарят на съществените изисквания на Директива 89/106/ЕИО. Възражението обхваща десет хармонизирани стандарта за топлоизолационни продукти за сгради, приети от Европейския комитет по стандартизация (CEN) на 23 май 2001 г., чиито референтни номера са публикувани в Официален вестник на Европейските общности (ОВЕО) на 15 декември 2001 г. (5); девет стандарта за геотекстил, приети от CEN на 13 декември 2000 г., чиито референтни номера са публикувани в ОВЕО на 26 юни 2001 г. (6); десет стандарта за неподвижни пожарогасителни инсталации, приети от CEN на 13 декември 2000 г., 21 март 2001 г. и 11 април 2001 г., чиито референтни номера са публикувани в ОВЕО на 18 юли 2001 г. (7), 15 декември 2001 г. и 14 февруари 2002 г. (8); и хармонизирания стандарт EN12859: 2001 „Гипсови блокове – определения, изисквания и методи за изпитване“, приет от CEN на 13 юни 2001 г., чийто референтен номер е публикуван в ОВЕО на 15 декември 2001 г. |
|
(5) |
Информацията, получена по време на консултациите със CEN и националните органи в комитета, създаден съгласно Директива 89/106/ЕИО, и в комитета, създаден съгласно Директива 98/34/ЕО, не съдържа доказателства за риска, приведен като аргумент от Германия. |
|
(6) |
Впоследствие не се доказа, че 30 оспорени хармонизирани стандарта не позволяват на строежите, в които се използват продуктите, да отговорят на съществените изисквания на Директива 89/106/ЕИО. |
|
(7) |
Следователно не е необходимо да се оттеглят справочните данни към тези стандарти, |
ПРИЕ НАСТОЯЩОТО РЕШЕНИЕ:
Член 1
Справочните данни на десетте стандарта за топлоизолационни продукти за сгради, посочени в таблица 1 от приложението, приети от Европейския комитет по стандартизация (CEN) на 23 май 2001 г. и публикувани за първи път в Официален вестник на Европейските общности на 15 декември 2001 г., не се оттеглят от списъка на стандартите, публикуван в Официален вестник на Европейските общности.
Член 2
Справочните данни на деветте стандарта за геотекстил, посочени в таблица 2 от приложението, приети от CEN на 13 декември 2000 г. и публикувани за първи път в Официален вестник на Европейските общности на 26 юни 2001 г., не се оттеглят от списъка на стандартите, публикуван в Официален вестник на Европейските общности.
Член 3
Справочните данни на десетте стандарта за неподвижни пожарогасителни инсталации, посочени в таблица 3 от приложението, приети от CEN на 13 декември 2000 г., 21 март 2001 г. и 11 април 2001 г. и публикувани за първи път в Официален вестник на Европейските общности на 18 юли 2001 г., 15 декември 2001 г. и 14 февруари 2002 г., не се оттеглят от списъка на стандартите, публикуван в Официален вестник на Европейските общности.
Член 4
Справочните данни на стандарт ЕN12859: 2001 „Гипсови блокове – определения, изисквания и методи за изпитване“, приет от CEN на 13 юни 2001 г. и публикуван за първи път в Официален вестник на Европейските общности на 15 декември 2001 г., не се оттеглят от списъка на стандартите, публикуван в Официален вестник на Европейските общности.
Член 5
Адресати на настоящото решение са държавите-членки.
Съставено в Брюксел на 9 април 2003 година.
За Комисията
David BYRNE
Член на Комисията
(1) ОВ L 40, 11.2.1989 г., стр. 12.
(2) ОВ L 220, 30.8.1993 г., стр. 1.
(3) ОВ L 204, 21.7.1998 г., стр. 37.
(4) ОВ L 217, 5.8.1998 г., стр. 18.
(5) ОВ C 358, 15.12.2001 г., стр. 9.
(6) ОВ C 180, 26.6.2001 г., стр. 8.
(7) ОВ С 202, 18.7.2001 г., стр. 9.
(8) ОВ С 40, 14.2.2002 г., стр. 3.
ПРИЛОЖЕНИЕ
ТАБЛИЦА 1
Публикуване на наименованията и справочните данни на хармонизираните стандарти за топлоизолационни продукти за сгради, посочени в член 1
|
ЕОС |
Референтен номер |
Наименование на хармонизирания стандарт |
Година на ратификация |
Препратка в ОВ |
|
CEN |
ЕN 13162:2001 |
Топлоизолационни продукти за сгради – продукти от минерална вата (MW), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13163:2001 |
Топлоизолационни продукти за сгради – продукти от експандиран полистирен (EPS), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13164:2001 |
Топлоизолационни продукти за сгради – продукти от екструдиран полистирен (XPS), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13165:2001 |
Топлоизолационни продукти за сгради – продукти от твърд пенополиуретан (PUR), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13166:2001 |
Топлоизолационни продукти за сгради – продукти от твърд пенофенопласт (PF), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13167:2001 |
Топлоизолационни продукти за сгради – продукти от пеностъкло (CG), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13168:2001 |
Топлоизолационни продукти за сгради – продукти от дървесна вата (WW), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13169:2001 |
Топлоизолационни продукти за сгради – продукти от експандиран перлит (EPB), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13170:2001 |
Топлоизолационни продукти за сгради – продукти от експандиран корк (ICB), произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
|
CEN |
ЕN 13171:2001 |
Топлоизолационни продукти за сгради – продукти от дървесни влакна, произведени в заводски условия – изисквания |
2001 |
C 358 15.12.2001 г. |
ТАБЛИЦА 2
Публикуване на наименованията и справочните данни на хармонизираните стандарти за геотекстилни продукти, посочени в член 2
|
ЕОС |
Референтен номер |
Наименование на хармонизирания стандарт |
Година на ратификация |
Препратка в ОВ |
|
CEN |
ЕN 13249:2000 |
Геотекстил/подобни на геотекстил продукти – характеристики, изисквани при използването им в строителството на пътища и други транспортни площи |
2000 |
C 180 26.6.2001 г. |
|
CEN |
ЕN 13251:2000 |
Геотекстил/подобни на геотекстил продукти – характеристики, изисквани за използването им в земни съоръжения, фундаменти и подпорни съоръжения |
2000 |
C 180 26.6.2001 г. |
|
CEN |
ЕN 13252:2000 |
Геотекстил/подобни на геотекстил продукти – характеристики, изисквани при използването им в дренажни системи |
2000 |
C 180 26.6.2001 г. |
|
CEN |
ЕN 13253:2000 |
Геотекстил/подобни на геотекстил продукти – характеристики, изисквани при използването им в съоръжения за ерозионен контрол |
2000 |
C 180 26.6.2001 г. |
|
CEN |
ЕN 13254:2000 |
Геотекстил/подобни на геотекстил продукти – характеристики, изисквани при използването им в строителството на резервоари и язовирни стени |
2000 |
C 180 26.6.2001 г. |
|
CEN |
ЕN 13255:2000 |
Геотекстил/подобни на геотекстил продукти – характеристики, изисквани при използването им в строителството на канали |
2000 |
C 180 26.6.2001 г. |
|
CEN |
ЕN 13256:2000 |
Геотекстил/подобни на геотекстил продукти – характеристики, изисквани при използването им в строителството на тунели и подземни съоръжения |
2000 |
C 180 26.6.2001 г. |
|
CEN |
ЕN 13257:2000 |
Геотекстил/подобни на геотекстил продукти – изисквани характеристики за използване в депа за твърди отпадъци |
2000 |
C 180 26.6.2001 г. |
|
CEN |
ЕN 13265:2000 |
Геотекстил/подобни на геотекстил продукти – изисквани характеристики за използване в обекти за задържане на течни отпадъци |
2000 |
C 180 26.6.2001 г. |
ТАБЛИЦА 3
Публикуване на наименованията и справочните данни на хармонизираните стандарти за неподвижни пожарогасителни инсталации, посочени в член 3
|
ЕОС |
Референтен номер |
Наименование на хармонизирания стандарт |
Година на ратификация |
Препратка в ОВ |
|
CEN |
ЕN 671-1:2001 |
Стационарни противопожарни системи – системи с маркуч – част 1: Макари с полутвърд маркуч |
2001 |
C 202 18.7.2001 г. |
|
CEN |
EN 671-2:2001 |
Стационарни противопожарни системи – системи с маркуч – част 1: Системи с плосък маркуч |
2001 |
C 202 18.7.2001 г. |
|
CEN |
EN 12094-5:2000 |
Неподвижни пожарогасителни инсталации – съставни елементи на инсталациите за гасене с газообразни средства: Изисквания и методи за изпитване на разпределителни вентили за високо и ниско налягане и техните активатори при системи с CO2 |
2000 |
C 202 18.7.2001 г. |
|
CEN |
EN 12094-6:2000 |
Неподвижни пожарогасителни инсталации – съставни елементи на инсталациите за гасене с газообразни средства: Изисквания и методи за изпитване на неелектрически блокиращи устройства при системи CO2 |
2000 |
C 202 18.7.2001 г. |
|
CEN |
EN 12094-7:2000 |
Неподвижни пожарогасителни инсталации – съставни елементи на инсталациите за гасене с газообразни средства: Изисквания и методи за изпитване на струйници при системи с CО2 |
2000 |
C 202 18.7.2001 г. |
|
CEN |
EN 12416-1:2001 |
Стационарни пожарогасителни системи – системи с прах – част 1: Изисквания и методи за изпитване на съставните части |
2001 |
C 40 14.2.2002 г. |
|
CEN |
EN 12094-13: 2001 |
Неподвижни пожарогасителни инсталации – съставни елементи на инсталациите за гасене с газообразни средства – част 13: Изисквания и методи за изпитване на контролни вентили и възвратни вентили |
2001 |
C 202 18.7.2001 г. |
|
CEN |
EN 12259-2: 1999/A1:2001 |
Стационарни противопожарни системи – съставни части на спринклери и системи за разпръскване на вода – мокри контролно-сигнални устройства |
2001 |
C 358 15.12.2001 г. |
|
CEN |
EN 12259-3: 2000/A1:2001 |
Стационарни противопожарни системи – съставни части на спринклери и системи за разпръскване на вода – сухи контролно-сигнални устройства |
2001 |
C 358 15.12.2001 г. |
|
CEN |
EN 12259-4: 2000/prA1 |
Стационарни противопожарни системи – съставни части на спринклери и системи за разпръскване на вода – хидравлични звукови сигнализатори |
2001 |
C 358 15.12.2001 г. |
|
CEN |
EN 12859:2001 |
Гипсови блокове – определения, изисквания и методи за изпитване |
2001 |
C 358 15.12.2001 г. |
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
170 |
32003L0030
|
L 123/42 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/30/ЕО НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 8 май 2003 година
относно насърчаването на използването на биогорива и други възобновяеми горива за транспорт
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 175, параграф 1 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Европейския икономически и социален комитет (2),
като взеха предвид становището на Комитета за регионите (3),
в съответствие с процедурата, предвидена в член 251 от Договора (4)
като имат предвид, че:
|
(1) |
Срещата на Европейския съюз в Гьотенбург на 15 и 16 юни 2001 г. постигна съгласие за стратегия на Общността за устойчиво развитие, която се състои в серия от мерки, които включват разработването на биогорива. |
|
(2) |
Естествените ресурси и тяхното благоразумно и рационално използване, както е посочено в член 174, параграф 1 на Договора, включват нефт, природен газ и твърди горива, които са основни източници на енергия, но също така и основни източници на емисии на въглероден диоксид. |
|
(3) |
Въпреки това съществува широк спектър от биомаси, които биха могли да бъдат използвани за производството на биогорива, получени от земеделски и горски продукти, както и от остатъци и отпадъци от гори и от горска индустрия и производство на храни от селскостопанска продукция. |
|
(4) |
Транспортният сектор представлява повече от 30 % от крайното потребление на енергия в Общността и нараства, тенденция, която е свързана с паралелно нарастване на емисиите на въглероден диоксид, и това нарастване ще е още по-голямо в процентно изражение в страните кандидатки след тяхното членство в Европейския съюз. |
|
(5) |
Бялата книга на Комисията „Европейска транспортна политика за 2010 г.: време за решения“, прогнозира, че емисиите на СО2 от транспорта ще се увеличат с 50 % между 1990 г. и 2010 г. до около 1 113 милиона тона, като основната отговорност за това е на автомобилния транспорт, който произвежда 84 % от емисиите на СО2, свързани с транспорта. От екологична гледна точка, Бялата книга следователно апелира зависимостта от нефта (в настоящия момент 98 %) в транспортния сектор да се намали, като се използват алтернативни горива като биогоривата. |
|
(6) |
По-широкото използване на биогоривата за транспорта е част от пакет мерки, необходими, за прилагане на Протокола от Киото, както и на всеки пакет мерки, които имат за цел изпълнението на допълнителни ангажименти в тази насока. |
|
(7) |
Нарасналата употреба на биогорива в транспорта, без да се отхвърлят други възможни алтернативни горива, включително автомобилни ТНГ и ЦНГ, е един от инструментите, чрез които Общността може да намали използването на внасяните енергия и влияние от пазара на горивата за транспорта и оттук сигурност на енергийните доставки в средносрочен и дългосрочен план. |
|
(8) |
В резултат на технологичния напредък, повечето транспортни средства в движение в Европейския съюз могат да използват ниско биогоривна смес без проблеми. Последните технологични достижения направиха възможно използването на по-висок процент биогориво в смес. Някои страни вече използват биогоривни смеси, които съдържат 10 % биогориво и повече. |
|
(9) |
Затворените транспортни паркове предлагат потенциал за използване на високи концентрации от биогорива. В някои градове затворените транспортни паркове вече работят с чисто биогориво и в някои случаи това е спомогнало да се подобри качеството на въздуха в градските райони. Поради това държавите-членки биха могли да насърчават използването на биогорива в градския транспорт. |
|
(10) |
Насърчаването на употребата на биогорива в транспорта, представлява стъпка напред към по-широкото приложение на биомасите, което ще даде възможност биогоривото да се развива по-мащабно в бъдеще, като не се изключват други възможности и по-специално възможността да се използва водород. |
|
(11) |
Изследователската политика, провеждана от държавите-членки, е свързана с увеличаване на употребата на биогорива трябва да включи водородния сектор в значителна степен и да насърчава тази възможност, като се вземе под внимание съответната рамкова програма на Общността. |
|
(12) |
Чистото растително масло от маслодайни растения, което е получено чрез пресоване, извличане или подобни процедури, нерафинирано или рафинирано, но химически непроменено, също може да се използва за биогориво в специални случаи, когато неговата употреба е съвместима с типа на съответния двигател и съответстващите изисквания за емисиите. |
|
(13) |
Нови видове горива трябва да са в съответствие с признатите технически стандарти, ако трябва да бъдат възприети в по-голяма степен от потребителите и производителите на превозни средства и по този начин да проникнат на пазара. Техническите стандарти също формират основата за изискванията, които касаят емисиите и мониторинга на емисиите. Могат да се срещнат трудности при гарантирането на съответствието на новите типове горива с настоящите технически стандарти, които до голяма степен са били развити за конвенционалните изкопаеми горива. Комисията и органите за стандартизиране трябва да наблюдават и контролират развитието, приспособяването и активното разработване на стандарти, по-специално по отношение на летливостта, така че новите типове гориво да бъдат въведени, като същевременно отговарят на същите изисквания за околната среда. |
|
(14) |
Биоетанолът и биодизелът, когато се използват за превозни средства в чиста форма или като смеси трябва да отговарят на стандартите за качество, които са определени с цел да гарантират оптималното действие на двигателя. Отбелязано е, че в случаи на биодизел за дизелови двигатели, където преработвателният процес е естерификация, могат да се приложат стандарт prEN 14214 на Европейския комитет по стандартизация (ЕКС) за метилови естери на мастни киселини и (МЕМК). Съответно ЕКС трябва да утвърди подходящи стандарти за други транспортни биогоривни продукти в Европейския съюз. |
|
(15) |
Насърчаването на използването на биогорива, като се следват устойчивите практики в земеделието и лесовъдството, посочени в правилата, които регламентират общата селскостопанска политика, може да създаде нови възможности за устойчиво селско развитие в рамките на една обща селскостопанска политика, която е ориентирана в по-голяма степен към пазара, по-конкретно към Европейския пазар, и към уважаване на процъфтяващия живот в селото и мултифункционалното земеделие и може да създаде нов пазар за иновационни земеделски продукти на настоящите и бъдещите държави-членки. |
|
(16) |
В резолюцията си от 8 юни 1998 (5) Съветът одобри Стратегия и план за действие на Комисията и за възобновяеми енергийни ресурси и изискваните специални мерки за биогоривата. |
|
(17) |
Зелената книга на Комисията „Към европейска стратегия за сигурност на енергийните доставки“ определя целта за 20 % заместване на конвенционалните горива с алтернативни горива за пътно транспортния сектор до 2020 г. |
|
(18) |
Алтернативните горива ще могат да пробият на пазара, само ако се предлагат широко и са конкурентни. |
|
(19) |
В Резолюцията от 18 юни 1998 г. (6) Европейският парламент апелира за увеличаване на пазарния дял на биогоривата до 2 % за пет години чрез пакет мерки, които включват освобождаване от данъци, финансова помощ за преработвателната промишленост и утвърждаването на задължителен процент на биогорива за петролните компании. |
|
(20) |
Оптималният метод за увеличаване на дяла на биогоривата на националните пазари и пазарите на Общността зависи от наличността на ресурсите и суровините, от националната политика и политиката на Общността за насърчаване на биогоривата и на данъчните разпоредби и на целесъобразното включване на всички заинтересовани лица/страните. |
|
(21) |
Националните политики за насърчаване на употребата на биогоривата не трябва да водят до забрани за свободното движение на горива, което отговаря на хармонизираните спецификации за околната среда, които са определени в законодателството на Общността. |
|
(22) |
Насърчаването на производството и употребата на биогорива може да допринесе за намаляването на зависимостта от вноса на енергия и емисиите на парникови газове. Освен това, биогоривата в чиста форма или като смеси могат по принцип да бъдат използвани в съществуващи превозни средства и да използват настоящата разпределителна система за гориво на моторните превозни средства. Смесването на биогориво със изкопаеми горива може да улесни намаляването на потенциалните разходи в дистрибутивната система в Общността. |
|
(23) |
Тъй като целта на предложеното действие, а именно въвеждането на общи принципи, които предвиждат минимален процент биогорива да се търгуват и разпространяват, не може да се постигне в достатъчна степен в държавите-членки поради мащаба на действието и поради това може да се постигне в по-голяма степен на равнище на Общността, Общността може да приеме мерки в съответствие с принципа на субсидиарност, както е определен в член 5 на Договора. В съответствие с принципа за пропорционалност, както е посочено в настоящия член, настоящата директива не надхвърля необходимото за постигането на целта. |
|
(24) |
Следва да се насърчават проучванията и технологичното развитие в областта на устойчивостта на биогоривата. |
|
(25) |
Увеличаване на използването на биогоривата трябва да бъде съпроводено от подробен анализ на въздействието върху околната среда, икономиката и обществото, за да се реши дали е препоръчително да се увеличи дела на биогоривата спрямо конвенционалните горива. |
|
(26) |
Трябва да се предвиди възможността за бързо привеждане в съответствие на списъка с биогорива, процента на възобновяеми компоненти и графика за въвеждане на биогорива на пазара на горива за транспорта към техническия прогрес и към резултатите от оценката на въздействието върху околната среда на първата фаза от въвеждането. |
|
(27) |
Трябва да бъдат въведени мерки за бързо разработване на стандартите за биогоривата, за да се използват в автомобилния сектор, както като чисти биогорива, така и като компоненти от смеси в конвенционални горива. Въпреки че биологично разграждаща се част от отпадъците е потенциално полезен източник за производство на биогориво, в стандарта за качество трябва да се вземе под внимание възможното наличие на замърсители в отпадъците, с цел да се избегне специфичните компоненти да увредят превозното средство или да предизвикат влошаване на емисиите. |
|
(28) |
Мерките, които имат за цел да се насърчи използването на биогорива, трябва да бъдат съвместими с целите, свързани със сигурността на доставките и опазването на околната среда, както и с целите и мерките на различните държави-членки в свързани области. За тази цел държавите-членки могат да разгледат финансово ефективни начини за популяризиране на възможностите за използване на биогоривата. |
|
(29) |
Необходимите мерки за прилагането на настоящата директива следва да бъдат приети съгласно Решение 1999/468/ ЕО на Съвета от 28.6.1999 г. относно установяването на процедурите за упражняване на изпълнителните правомощия, предоставени на Комисията (7), |
ПРИЕХА НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Настоящата директива цели насърчаването на употребата на биогорива или други възобновяеми горива, които да заменят дизела или бензина за транспортни цели във всяка държава-членка, с оглед допринасяне за постигането на цели като изпълняване на задълженията в областта на промяната на климата, екологичната безопасност на доставките и насърчаването на възобновяемите енергийни източници.
Член 2
1. За целите на настоящата директива се прилагат следните определения:
|
а) |
„биогорива“ означава течно или газообразно гориво за транспорт, което е произведено от биомаса; |
|
б) |
„биомаса“ означава биоразградима част от продукти, отпадъци или остатъци от земеделието (включително растителни и животински вещества), лесовъдство и свързани промишлености, както и биоразграждаща се част от промишлени и градски отпадъци; |
|
в) |
„други възобновяеми горива“ означава възобновяеми горива, различни от биогоривата, които произхождат от възобновяеми енергийни източници, както са определени в Директива 2001/77/ЕО (8) и използвани за транспортни цели; |
|
г) |
„енергийно съдържание“ означава ниска калоричност на гориво. |
2. Най-малко продуктите изброени по-долу трябва да се разглеждат като биогорива:
|
а) |
„биоетанол“: етанол, произведен от биомаса и/ или биологично разграждаща се част от отпадък, която се използва като биогориво; |
|
б) |
„биодизел“: метил-естер, произведен от растително или животинско масло с качество на дизел, което се използва като биогориво; |
|
в) |
„биогаз“: горивен газ, произведен от биомаса и/или от биологично разграждаща се част от отпадък, която може да бъде пречистена до продукт с качества на природен газ, който се използва като биогориво или дървесен газ; |
|
г) |
„биометанол“: метанол, произведен от биомаса, който се използва като биогориво; |
|
д) |
„биодиметилетер“: диметилетер, произведен от биомаса, който се използва като биогориво; |
|
е) |
„био-ЕТБЕ (етил-тeртио-бутил-етер)“: ЕТБЕ, които е произведен на базата на биоетанол. Процентът от обема на био-ЕТБЕ, изчислен като биологично гориво е 47 %; |
|
ж) |
„био-метил-тетрио-бутил-етер“: гориво, което е произведено на базата на биометанол. Процентът от обема на био-МТБЕ, изчислен като биологично гориво, е 36 %; |
|
з) |
„синтетични биогорива“: синтетични въглеводороди или смеси от синтетични въглеводороди, които са произведени от биомаса; |
|
и) |
„биоводород“: водород, който е произведен от биомаса и/или биологична разграждаща се част от отпадък, който се използва като биогориво; |
|
й) |
„чисто растително масло“: масло, което е произведено от маслодайни култури чрез пресоване, извличане или сходни процедури, нерафинирано или рафинирано, но химически непроменено, когато е съвместимо с типа двигател и съответните изисквания за емисии. |
Член 3
|
1. |
|
2. Биогоривата могат да бъдат направени достъпни във всяка една от следните форми:
|
а) |
като чисти биогорива или с висока концентрация в разновидности на минералното масло в съответствие със специфичните стандарти за качество за транспортни цели; |
|
б) |
като биогорива, смесени в разновидностите на минералните масла в съответствие с подходящите европейски норми, описани в техническата спецификация за транспортни горива (ЕN 228 и ЕN 590); |
|
в) |
като течности, получени от биогорива като ЕТБЕ (етил-тeртио-бутил-етер), като процента на биогориво е, както е определено в член 2, параграф 2. |
3. Държавите-членки трябва да контролират действието при използването на биогорива в смеси с дизел над 5 % от неадаптираните превозни средства и трябва, където е подходящо, да се предприемат мерки за осигуряване на съгласието със съответното законодателство на Общността за стандарти за емисии.
4. В измерванията, които правят, държавите-членки трябва да вземат под внимание цялостния климат и равновесието в околната среда на различни видове биогорива и други възобновяеми горива и могат да определят като приоритет насърчаването на тези горива, които показват много добър финансово ефективен баланс за околната среда, като също така се вземат под внимание конкурентноспособността и безопасността на доставките.
5. Държавите-членки трябва да гарантират, че е предоставена информация на обществеността за наличността на биогоривата и други възобновяеми горива. За процентите от биогоривата, смесени в деривати на минералното масло, които превишават граничната стойност от 5 % за метил-естери на мастни киселини (МЕМК) или 5 % за биоетанола, трябва да бъде наложено специално етикетиране в търговските обекти.
Член 4
1. Държавите-членки докладват на Комисията преди 1 юли всяка година за:
|
— |
предприетите мерки за насърчаване употребата на биогорива и други възобновяеми горива, които да заменят дизела или бензина за транспортни цели, |
|
— |
националните ресурси, които са отделени за производството от биомаса за енергийни цели, различни от транспорта, и |
|
— |
общите продажби на гориво за транспорт и дяловете на биогоривата, чисти или смесени, и други възобновяеми горива, които са пуснати на пазара през изминалата година. Когато е необходимо, държавите-членки трябва да докладват за всички изключителни условия при доставката на нерафинирани масла или нефтопродукти, които са оказали влияние на търговията с биогорива и други възобновяеми горива. |
В първия си доклад, след влизането в сила на настоящата директива, държавите-членки трябва да посочат нивото на техните национални примерни цели за първата фаза. В доклада за 2006 година държавите-членки трябва да отбележат техните национални примерни цели за втората фаза.
В тези доклади, диференциацията на националните цели, сравнени с референтни стойности по член 3, параграф 1, буква б), трябва да бъде обоснована и може да се основава на следните елементи:
|
а) |
обективни фактори като ограничен национален потенциал за производство на биогорива от биомаса; |
|
б) |
количеството ресурси, които са отделени за производството на биомаса за енергийни нужди, различни от транспорт и специфичните технически или климатични характеристики на националния пазар за транспортни горива; |
|
в) |
национална политики за отделяне на сходни ресурси за производството на други транспортни горива, които се основават на възобновяеми енергийни източници и в съответствие с целите на настоящата директива. |
2. До 31 декември 2006 г. най-късно и на всеки две години след това Комисията изготвя доклад за оценка до Европейския парламент и Съвета за извършения напредък в употребата на биогорива и други възобновяеми горива в държавите-членки.
Този доклад трябва да обхваща най-малко следното:
|
а) |
финансовата ефективност на предприетите от държавите-членки мерки с цел да насърчават употребата на биогорива и други възобновяеми горива; |
|
б) |
икономическите аспекти и въздействието върху околната среда на увеличаващия се дял на биогоривата и други възобновяеми горива; |
|
в) |
перспектива за жизнения цикъл на биогоривата и други възобновяеми горива с оглед на определяне на възможните мерки за бъдещо насърчаване на тези горива, които са безвредни за климата и околната среда и които имат потенциал да се превърнат в конкурентни и икономически ефективни; |
|
г) |
устойчивост на културите, които се използват за производство на биогорива, по-специално използване на земята, степента на интензивност на култивиране, редуване на културите и използване на пестициди; |
|
д) |
оценка на използването на биогорива и други възобновяеми горива по отношение на техните различни ефекти върху промяната на климата и тяхното въздействие върху намаляването на емисиите на CO2; |
|
е) |
преглед на други дългосрочни възможности по отношение на мерките за ефективност на енергията в транспорта. |
Въз основа на този доклад, когато е целесъобразно, Комисията представя предложения за привеждане в съответствие на системата от цели на Европейския парламент и на Съвета, както е посочено в член 3, параграф 1. Ако този доклад заключи, че няма вероятност примерните цели да бъдат постигнати поради причини, които са неоправдани и/или нямат връзка с нови научни доказателства, тези предложения ще бъдат насочени към националните цели, включително възможните задължителни цели, в подходяща форма.
Член 5
Списъкът в член 2, параграф 2 може да бъде приведен в съответствие с техническия прогрес в съответствие с процедурата, посочена в член 6, параграф 2. При привеждането в съответствие на този списък въздействието върху околната среда на биогоривата трябва да бъде взето под внимание.
Член 6
1. Комисията се подпомага от комитет.
2. Когато се прави справка с настоящия параграф, членове 5 и 7 от Решение 1999/468/ЕО ще се прилага, като се имат предвид разпоредбите от член 8 от него.
Срокът по член 5, параграф 6 на Решение 1999/468/ЕО се определя на 3 месеца.
3. Комитетът приема процедурен правилник.
Член 7
1. Държавите-членки въвеждат в сила необходимите законови, подзаконови и административни разпоредби, за да се съобразят с настоящата директива най-късно до 31.12.2004 г. Те незабавно уведомяват Комисията за това.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
2. Държавите-членки съобщават на Комисията разпоредбите от националното законодателство, които приемат в областта, която е обхваната от настоящата директива.
Член 8
Директивата влиза в сила в деня на публикуването ѝ в Официален вестник на Европейския съюз.
Член 9
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 8 май 2003 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
M. CHRISOCHOÏDIS
(1) ОВ C 103 E, 30.4.2002 г., стр. 205 и
ОВ C 331 E, 31.12.2002 г., стр. 291.
(2) ОВ C 149, 21.6.2002 г., стр. 7.
(3) ОВ C 278, 14.11.2002 г., стр. 29.
(4) Становище на Европейския парламент от 4 юли 2002 (все още непубликувано в Официален вестник), Обща позиция на Съвета от 18 ноември 2002 г. (ОВ C 32 E, 11.2.2003 г., стр. 1) и Решение на Европейския парламент от 12 март 2003 г. (все още непубликувано в Официален вестник).
(5) ОВ C 198, 24.6.1998 г., стр. 1.
(6) ОВ C 210, 6.7.1998 г., стр. 251.
(7) ОВ L 184, 17.7.1999 г., стр. 23.
(8) Директива 2001/77/ЕО на Европейския парламент и на Съвета от 27.9.2001 г. относно насърчаване на производството и потреблението на електроенергия от възобновяеми енергийни източници на вътрешния електроенергиен пазар (ОВ L 283, 27.10.2001 г., стр. 33).
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
175 |
32003L0040
|
L 126/34 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/40/ЕО НА КОМИСИЯТА
от 16 май 2003 година
за установяване на списъка, границите на концентрация и изискванията към етикетирането за съставките на натуралните минерални води и условията за употреба на обогатен с озон въздух за обработката на натурални минерални води и на изворни води
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 80/777/ЕИО на Съвета (1) от 15 юли 1980 г. относно сближаването на законодателствата на държавите-членки относно експлоатацията на и търговията с натурални минерални води, последно изменена с Директива 96/70/ЕО на Европейския парламент и Съвета (2), и по-специално член 11, параграф 1 от нея,
като има предвид, че:
|
(1) |
Поради техния хидрогеоложки произход някои натурални минерални води в естествено състояние могат да съдържат съставки, които ако превишават определена концентрация, могат да представляват риск за общественото здраве. Следователно се счита, че е необходимо да се установят граници на концентрацията на тези съставки в натуралните минерални води. |
|
(2) |
Член 11 от Директива 80/777/ЕИО предвижда възможността да се приемат хармонизирани граници на концентрацията на съставките в натуралните минерални води след консултация с Научния комитет по храните, както и изисквания спрямо етикетировката, за да се указва, когато това се налага, наличието на определени съставки с високи концентрации. |
|
(3) |
Научният комитет по храните даде становище (3) относно арсена, бария, флуорида, бора и мангана, както и за други съставки в натуралните минерални води, като потвърди границите, препоръчани от Световната здравна организация (СЗО) за питейна вода. |
|
(4) |
Ревизираният Кодекс на стандартите за „натуралните минерални води“ (4) съдържа, за целите на здравеопазването, списък от съставки и максимални граници за тези съставки. Той е приет въз основа на най-новите международни научни данни и осигурява достатъчна защита на общественото здраве. |
|
(5) |
Общоприето е, че приемането на ниски дози флуорид може да има благоприятно въздействие върху зъбите. За разлика от това, приемането на големи дози флуорид над допустимите граници може да има вреден ефект върху общественото здраве. Ето защо е необходимо да се установи хармонизирана максимална граница за флуорид в натуралните минерални води, която да осигури достатъчна защита за населението като цяло. |
|
(6) |
Световната здравна организация препоръча ориентировъчна стойност от 1,5 mg/l за флуорид в питейна вода, която беше валидирана за натуралните минерални води от Научния комитет по храните в гореспоменатото становище. За да се защитят кърмачетата и малките деца, които са предразположени най-много към риска от флуороза, когато съдържанието на флуорид в натурална минерална вода надвишава тази ориентировъчна стойност, е необходимо върху етикета да се направи също обозначение за този факт и то да може да бъде лесно видяно от потребителя. |
|
(7) |
Научният комитет по храните посочи ориентировъчна стойност за бора в натуралните минерални води, която се основава на препоръките на СЗО (5) от 1996 г. Въпреки това, СЗО и други международно признати научни организации извършиха оттогава нови оценки относно влиянието на бора върху общественото здраве и препоръчаха по-високи стойности. Следователно, следва да се извърши консултация с Европейския орган за безопасност на храните относно бора в натуралните минерални води, така че наличните нови научни оценки да бъдат взети предвид, и на този етап не следва да се определя максимална граница за бора. |
|
(8) |
Научният комитет по храните също така посочи в споменатото по-горе становище приемливото ниво за бария, мангана и арсена в натуралните минерални води. За другите нежелани съставки в натуралните минерални води, които могат да имат вреден ефект върху общественото здраве, ревизираният Кодекс на стандартите определя максимални граници, които осигуряват достатъчна защита на общественото здраве. Въпреки това, границата за нитратите се счита за твърде ниска, предвид наличните данни и следва да бъде изравнена с границата, предвидена за питейна вода (6). |
|
(9) |
Установената в Кодекса на стандартите максимална граница за нитрати позволява да се осигури достатъчна защита на общественото здраве и следва да служи като отправна стойност при международната търговия и търговията в рамките на Общността с натурални минерални води. Все пак, в рамките на процедурата на официалното признаване на източници на натурални минерални води, упомената в член 1 от горепосочената директива, компетентните органи на държавите-членки трябва да са в състояние да се позоват на по-ниска ориентировъчна стойност за нитратите в натуралните минерални води, каптирани на тяхна територия. |
|
(10) |
Натуралните минерални води, чието съдържание на определени съставки надхвърля максималните граници за тези съставки, с оглед на целите на общественото здраве, се подлагат на обработка, при която въпросните съставки се отделят. За да се позволи на засегнатите производители да направят инвестициите, необходими за спазването на тези нови стандарти, следва да бъдат дадени достатъчно дълги срокове преди влизането в сила на новите стандарти за такива съставки, по-специално по отношение на флуорида и никела, за които на ниво Общност няма оценена или разрешена обработка за отделяне. |
|
(11) |
За целите на официалния контрол на тези съставки е необходимо да се установят граници на колебание около границите на максимална концентрация, които да съответстват на неточностите при измерванията. |
|
(12) |
Член 4, параграф 1, буква б) от изменената Директива 80/777/ЕИО предвижда отделянето на желязо, манган, сяра и арсен от определени натурални минерални води да става чрез използването на обработка с обогатен с озон въздух, като тази обработка подлежи на оценка от Научния комитет по храните и на приемане на условията за използване на тази обработка от Постоянния комитет по хранителната верига и здравето на животните. |
|
(13) |
Научният комитет по храните предостави становище (7) относно тази обработка, което предвижда задължения както за определени методи, така и за определени резултати. Въпреки това, за да се вземат предвид както развитието на техниките за обработка с обогатен с озон въздух, така и различията на обработката, които зависят от физико-химичния състав на обработваната вода, се счита за целесъобразно да се въведат задължения само за получаваните резултати. |
|
(14) |
Освен това, обработката с обогатен с озон въздух не следва да променя състава по отношение на характеризиращите съставки по смисъла на член 7, параграф 2, буква а) от Директива 80/777/ЕИО, или да има дезинфекционно действие по смисъла на член 4, параграф 3, или да причинява образуването на остатъчни вещества от обработката, които могат да имат вредно въздействие върху общественото здраве. |
|
(15) |
Съгласно член 7, параграф 2, буква в) на горепосочената директива, натуралните минерални води, обработени с обогатен с озон въздух, трябва да съдържат на етикетите си достатъчно информация за потребителите относно извършената обработка. |
|
(16) |
В съответствие с разпоредбите на Директива 80/777/ЕИО, член 9, параграф 4, буква а), четвърто тире, разпоредбите относно обработките, предвидени в член 4 от директивата, и по-специално обработката с обогатен с озон въздух, са приложими за изворните води. |
|
(17) |
Мерките, предвидени в настоящата директива, са в съответствие със становището на Постоянния комитет по хранителната верига и здравето на животните, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Настоящата директива установява списъка от съставки в натуралните минерални води, които могат да представляват риск за общественото здраве, границите на допустимите нива на тези съставки, крайните срокове за прилагане на тези граници и изискванията към етикетирането за някои съставки. Тези съставки трябва да се съдържат във водата по естествени причини, а не поради замърсяване при източника.
Тя също така определя условията за употреба на обогатен с озон въздух за отделяне на съединения на желязо, манган, сяра и арсен от натурални минерални води или изворни води и изискванията към етикетирането за води, които са били подложени на такава обработка.
Член 2
1. Не по-късно от 1 януари 2006 г., натуралните минерални води следва да отговарят, по време на пакетирането им, на изискванията за максимални граници на концентрация, посочени в приложение I за съставките, изброени в това приложение.
2. Независимо от това, в случая на флуориди и никел, крайният срок, посочен по-горе, се удължава до 1 януари 2008 г.
3. Чрез дерогация от параграф 1, по време на процедурата на официално признаване на натурални минерални води, каптирани на тяхна територия, компетентните власти на държавите-членки могат да вземат предвид по-ниска референтна стойност за нитратите и нитритите, при условие че същата референтна стойност се прилага за всички заявления, отправени към тях.
Член 3
За целите на официалния контрол, държавите-членки спазват спецификациите, изброени в приложение II, за анализиране на съставките, изброени в приложение I.
Член 4
1. Етикетът на натурални минерални води с концентрация на флуорид над 1,5 mg/l съдържа думите „съдържа повече от 1,5 mg/l флуорид: не е подходяща за редовна консумация от кърмачета и деца под 7-годишна възраст“.
2. Информацията върху етикета, посочена в параграф 1 на настоящия член, се помества в непосредствена близост до търговското наименование и се изписва с ясно видими знаци.
3. Етикетите на натурални минерални води, които съдържат информация съгласно параграф 1 на настоящия член, посочват действителното съдържание на флуорид в съотношение към физико-химичния състав от гледна точка на съществените съставки, както е посочено в член 7, параграф 2, буква а) от Директива 80/777/ЕИО.
Член 5
1. Без да се засягат разпоредбите на член 4, параграф 1, буква б) от Директива 80/777/ЕИО, прилагането на обработка на натурални минерални води с обогатен с озон въздух трябва да бъде предварително нотифицирано на компетентните власти, които да гарантират, че:
|
а) |
използването на тази обработка е оправдано от състава на водата по отношение на съединенията на желязо, манган, сяра и арсен; |
|
б) |
операторът е взел всички необходими мерки, за да гарантира, че обработката е ефективна и безопасна, и за да позволи тя да бъде проверена от компетентните власти. |
2. Обработката на натуралните минерални води с обогатен с озон въздух трябва да отговаря на следните условия:
|
а) |
физико-химичният състав на натуралните минерални води по отношение на съществените съставки не се променя в резултат от обработката; |
|
б) |
преди обработката натуралната минерална вода трябва да съответства на микробиологичните критерии, определени в член 5, параграфи 1 и 2 от Директива 80/777/ЕИО; |
|
в) |
обработката не води до образуването на остатъчни вещества с концентрация, която надвишава максималните граници, определени в приложение III, или на остатъчни вещества, които биха могли да представляват риск за общественото здраве. |
Член 6
Съгласно член 7, параграф 2, буква в) от Директива 80/777/ЕИО, етикетировката на натуралните минерални води, които са били обработени с обогатен с озон въздух, съдържа, в близост до аналитичния състав на характеризиращите съставки, думите „вода, подложена на окисляване чрез обработка с обогатен с озон въздух“.
Член 7
Без да се засягат разпоредбите на член 9, параграф 4, буква б) от Директива 80/777/ЕИО, разпоредбите на членове 5 и 6 от настоящата директива се прилагат и за изворните води.
Член 8
1. Държавите-членки предприемат необходимите мерки, за да позволят търговията с продукти, които отговарят на изискванията на настоящата директива, не по-късно от 1 януари 2004 г.
2. Без да се засягат крайните срокове, установени в член 2, параграфи 1 и 2, държавите-членки забраняват търговията с продукти, които не отговарят на изискванията на настоящата директива, считано от 1 юли 2004 г. Независимо от това, продукти, които са опаковани и етикетирани преди 1 юли 2004 г., могат да бъдат продавани, докато бъдат изчерпани складовите наличности.
Член 9
Държавите-членки въвеждат в сила законовите, подзаконовите и административните разпоредби, необходими за да се съобразят с настоящата директива, най-късно до 31 декември 2003 г. Те незабавно информират Комисията за това.
В разпоредбите, приети съгласно настоящия параграф, се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 10
Настоящата директива влиза в сила на двадесетия ден след датата на публикуването ѝ в Официален вестник на Европейския съюз.
Член 11
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 16 май 2003 година.
За Комисията
David BYRNE
Член на Комисията
(1) ОВ L 229, 30.8.1980 г., стр. 1.
(2) ОВ L 299, 23.11.1996 г., стр. 26.
(3) Становище от 13 декември 1996 г. относно арсена, бария, флуорида, бора и мангана в натуралните минерални води.
(4) Кодекс на стандартите 108-1981, Rev 1-1997, ревизиран по време на седмата сесия на CCNMW (октомври 2000 г.).
(5) СЗО (1996): указания относно качеството на питейната вода, второ издание, том 2.
(6) Директива 98/83/ЕО на Съвета (ОВ L 330, 5.12.1998 г., стр. 32).
(7) Становище на Научния комитет по храните от 7 юни 1996 г. относно употребата на озон за отделяне на нестабилни елементи като желязо, манган и арсен от натурални минерални води.
ПРИЛОЖЕНИЕ I
Естествено съдържащи се съставки в натуралните минерални води и максимални граници, които, ако са надвишени, могат да представляват риск за общественото здраве
|
Съставки |
Максимални граници (mg/l) |
|
Антимон |
0,0050 |
|
Арсен |
0,010 (общо) |
|
Барий |
1,0 |
|
Бор |
За протокола (1) |
|
Кадмий |
0,003 |
|
Хром |
0,050 |
|
Мед |
1,0 |
|
Цианид |
0,070 |
|
Флуориди |
5,0 |
|
Олово |
0,010 |
|
Манган |
0,50 |
|
Живак |
0,0010 |
|
Никел |
0,020 |
|
Нитрати |
50 |
|
Нитрити |
0,1 |
|
Селен |
0,010 |
(1) Максималната граница за бора ще бъде определена по целесъобразност след становище на Европейския орган за безопасност на храните и по предложение на Комисията до 1 януари 2006 г.
ПРИЛОЖЕНИЕ II
Характеристики на изпълнението (1) за анализиране на съставките в приложение I
|
Съставки |
Точност на параметричната стойност в % (бележка 1) |
Прецизност на параметричната стойност (бележка 2) |
Граница на откриване в % от параметричната стойност (бележка 3) |
бележки |
||||
|
Антимон |
25 |
25 |
25 |
|
||||
|
Арсен |
10 |
10 |
10 |
|
||||
|
Барий |
25 |
25 |
25 |
|
||||
|
Бор |
|
|
|
Виж приложение I |
||||
|
Кадмий |
10 |
10 |
10 |
|
||||
|
Хром |
10 |
10 |
10 |
|
||||
|
Мед |
10 |
10 |
10 |
|
||||
|
Цианиди |
10 |
10 |
10 |
Бележка 4 |
||||
|
Флуориди |
10 |
10 |
10 |
|
||||
|
Олово |
10 |
10 |
10 |
|
||||
|
Манган |
10 |
10 |
10 |
|
||||
|
Живак |
20 |
10 |
20 |
|
||||
|
Никел |
10 |
10 |
10 |
|
||||
|
Нитрати |
10 |
10 |
10 |
|
||||
|
Нитрити |
10 |
10 |
10 |
|
||||
|
Селен |
10 |
10 |
10 |
|
||||
|
Бележка 1: точност е системната грешка и представлява разликата между средната стойност на голям брой повторени измервания и точната стойност. Бележка 2: прецизност е случайната грешка и се изразява най-общо като стандартното отклонение от средната стойност (в рамките на партида и между партиди) на представителни резултати. Приемливата точност е равна на удвоеното относително стандартно отклонение. Бележка 3: граница на откриване е:
Бележка 4: Методът следва да дава възможност да се определи общото количество цианид във всичките му форми. |
||||||||
(1) Аналитичните методи за измерване на концентрациите на съставките, изброени в приложение 1, трябва да са в състояние най-малко да измерват концентрации, равни на параметричната стойност със специфична точност, прецизност и граница на откриване. Независимо от това каква е чувствителността на използвания метод за анализ, резултатът се изразява, като се използват поне толкова десетични позиции, колкото има максималната граница, установена в приложение I.
Бележка 1: точност е системната грешка и представлява разликата между средната стойност на голям брой повторени измервания и точната стойност.
Бележка 2: прецизност е случайната грешка и се изразява най-общо като стандартното отклонение от средната стойност (в рамките на партида и между партиди) на представителни резултати. Приемливата точност е равна на удвоеното относително стандартно отклонение.
Бележка 3: граница на откриване е:
|
— |
или три пъти относителното стандартно отклонение в рамките на партида от естествен образец, съдържащ ниска концентрация на параметъра, |
|
— |
или пет пъти относителното стандартно отклонение в рамките на партида от автентичен образец. |
Бележка 4: Методът следва да дава възможност да се определи общото количество цианид във всичките му форми.
ПРИЛОЖЕНИЕ III
Максимални граници за остатъци от обработката на натурални минерални води и изворни води с обогатен с озон въздух
|
Остатъчни вещества от обработката |
Максимална граница (1) (μg/l) |
|
Разтворен озон |
50 |
|
Бромати |
3 |
|
Бромоформ |
1 |
(1) Съответствието с максималните граници се контролира от компетентните органи в държавите-членки по време на бутилиране или друга форма на пакетиране, предназначена за крайния потребител.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
181 |
32003D0424
|
L 144/9 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕШЕНИЕ НА КОМИСИЯТА
от 6 юни 2003 година
за изменение на Решение 96/603/ЕО за съставяне на списък с продукти, принадлежащи към класове А „Без принос за пожар“, предвиден в Решение 94/611/ЕО относно прилагането на член 20 от Директива 89/106/ЕИО на Съвета относно строителните продукти
(нотифицирано под № C(2003) 1673)
(текст от значение за ЕИП)
(2003/424/ЕО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 89/106/ЕИО на Съвета от 21 декември 1988 г. за сближаване на законовите, подзаконови и административни разпоредби на държавите-членки по отношение на строителните продукти (1), изменена с Директива 93/68/ЕИО (2),
като взе предвид Решение 2000/147/ЕО на Комисията от 8 февруари 2000 г., привеждащо в изпълнение Директива 89/106/ЕИО на Съвета относно класификацията според устойчивостта на пожар на строителни продукти (3), и по-специално член 1, параграф 1 от нея,
като има предвид, че:
|
(1) |
Решение 96/603/ЕО на Комисията (4), изменено с Решение 2000/605/ЕО (5), състави списък от продукти, принадлежащи към класове А „Без принос за пожар“, предвиден в таблици 1 и 2 от приложението към Решение 94/611/ЕО на Комисията (6), което описва европейската система за класификация за изразяване на устойчивостта на строителните продукти при пожар. |
|
(2) |
Решение 94/611/ЕО бе заменено с Решение 2000/147/ЕО. |
|
(3) |
Бележките към „Хоросан с неорганични спойващи агенти“ в таблицата от приложението към Решение 96/603/ЕО трябва да се приведат в съответствие с техническия напредък. |
|
(4) |
Решение 96/603/ЕО следва да бъде изменено съответно. |
|
(5) |
Мерките, предвидени в настоящото решение, са в съответствие със становището на Постоянния комитет по строителство, |
ПРИЕ НАСТОЯЩОТО РЕШЕНИЕ:
Член 1
В таблицата, посочена в приложението към Решение 96/603/ЕО, „Хоросан с неорганични спойващи агенти“, бележките „хоросан за мазилки/гипс и хоросан за подови дъски на основата на един или повече неорганични агенти, напр. цимент, вар, цимент за зидане и гипс“ се заменят от „хоросан за мазилки/гипс, хоросан за подови дъски и хоросан за зидане на основата на един или повече неорганични агенти, напр. цимент, вар, цимент за зидане и гипс“.
Член 2
Адресати на настоящото решение са държавите-членки.
Съставено в Брюксел на 6 юни 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 40, 11.2.1989 г., стр. 12.
(2) ОВ L 220, 30.8.1993 г., стр. 1.
(3) ОВ L 50, 23.2.2000 г., стр. 14.
(4) ОВ L 267, 19.10.1996 г., стр. 23.
(5) ОВ L 258, 12.10.2000 г., стр. 36.
(6) ОВ L 241, 19.9.1994 г., стр. 25.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
182 |
32003L0034
|
L 156/14 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/34/ЕО НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 26 май 2003 година
относно двадесет и трето изменение на Директива 76/769/ЕИО на Съвета относно ограниченията за пускането на пазара и употребата на някои опасни вещества и препарати (вещества, класифицирани като канцерогени, мутагени или вещества, токсични за репродукцията — CMR)
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 95 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Европейския икономически и социален комитет (2),
в съответствие с процедурата, установена в член 251 от Договора (3), в светлината на общия текст, одобрен от Помирителния комитет на 17 март 2003 г.,
като имат предвид, че:
|
(1) |
Съгласно член 14 от Договора трябва да се създаде пространство без вътрешни граници, в която да бъде осигурено свободното движение на стоки, хора, услуги и капитали. |
|
(2) |
На 29 март 1996 г. Европейският парламент и Съветът приеха Решение № 646/96/ЕО, с което бе приета програма от мерки за ограничаване на раковите заболявания в рамките за действие в сферата на общественото здравеопазване (1996—2000) (4). |
|
(3) |
За подобряване на здравната защита и безопасността на потребителите, веществата, класифицирани като канцерогенни, мутагенни или токсични за репродукцията, и препаратите, които ги съдържат, не трябва да бъдат пускани на пазара за употреба от широкия потребител. Комисията трябва да внася възможно най-бързо предложения за забрана на употребата на продукти, които съдържат такива вещества в случаите когато съществуват научни доказателства, че веществата се отделят от въпросните продукти, като влизат в контакт с широкия потребител и го излагат на риск. |
|
(4) |
Директива 94/60/ЕО на Европейския парламент и на Съвета от 20 декември 1994 г. относно четиринадесето изменение на Директива 76/769/ЕИО (5) установява, под формата на допълнение относно точки 29, 30 и 31 от приложение I към Директива 76/769/ЕИО (6), списък на вещества, класифицирани като канцерогенни, мутагенни или токсични за репродукцията от категории 1 или 2. Подобни вещества и препаратите, които ги съдържат, не трябва да бъдат пускани на пазара за употреба от широкия потребител. |
|
(5) |
Директива 94/60/ЕО предвижда, че Комисията ще представя на Европейския парламент и на Съвета предложения за разширяване на въпросния списък не по-късно от шест месеца след всяко публикуване на адаптиране към техническия прогрес на приложение I към Директива 67/548/ЕИО на Съвета от 27 юни 1967 г. за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки относно класифицирането, опаковането и етикетирането на опасни вещества (7), които съдържат вещества, класифицирани като канцерогенни, мутагенни или токсични за репродукцията от категории 1 или 2. |
|
(6) |
Директива 98/98/ЕО на Комисията от 15 декември 1998 г. относно двадесет и пето адаптиране към технически прогрес на Директива 67/548/ЕИО на Съвета (8), която адаптира, по-специално, приложение I към въпросната директива, съдържа 20 нови вещества, класифицирани като канцерогенни, мутагенни или токсични за репродукцията от категории 1 или 2, а Директива 2000/32/ЕО на Комисията от 19 май 2000 г. относно двадесет и шесто адаптиране към технически прогрес на Директива 67/548/ЕИО на Съвета (9), която адаптира, по-специално, приложение I към въпросната директива, съдържа две нови вещества, класифицирани като канцерогенни, мутагенни или токсични за репродукцията от категории 1 или 2. Тези вещества трябва да се добавят в точки 29, 30 и 31 от допълнението към приложение I към Директива 76/769/ЕИО. |
|
(7) |
Бяха взети под внимание рисковете и предимствата от новите класифицирани в съответствие горепосоченото вещества. |
|
(8) |
Настоящата директива се прилага без да засяга законодателството на Общността, постановяващо минимални изисквания за защитата на работниците, съдържащи се в Директива 89/391/ЕИО на Съвета от 12 юни 1989 г. за въвеждане на мерки за насърчаване подобряването на безопасността и здравето на работниците на работното място (10) и произтичащите от нея специални директиви, и по-конкретно Директива 90/394/ЕИО на Съвета от 28 юни 1990 г. за защита на работниците от рискове, свързани с експозиция на канцерогени по време на работа (11), |
ПРИЕХА НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Изброените в настоящото приложение вещества трябва да се добавят към веществата, изброени в точки 29, 30 и 31 от допълнението към приложение I към Директива 76/769/ЕИО.
Член 2
1. Държавите-членки приемат и публикуват не по-късно от 15 юли 2004 г. законовите, подзаконовите и административните разпоредби, необходими, за да се съобразят с настоящата директива. Те незабавно информират Комисията за това.
Те прилагат тези мерки от 15 януари 2005 г.
2. Когато държавите-членки приемат тези мерки, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 3
Настоящата директива влиза в сила на двадесетия ден след датата на публикуването ѝ в Официален вестник на Европейския съюз.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 26 май 2003 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
G. DRYS
(1) ОВ С 213 Е, 31.7.2001 г., стр. 263.
(2) ОВ С 311, 7.11.2001 г., стр. 7.
(3) Становище на Европейския парламент от 5 февруари 2002 г. (ОВ С 284 Е, 21.11.2002 г., стр. 88), Обща позиция на Съвета от 3 юни 2002 г. (ОВ С 197 Е, 20.8.2002 г., стр. 1) и Решение на Европейския парламент от 10 октомври 2002 г. (все още непубликувано в Официален вестник). Решение на Европейския парламент от 27 март 2003 г. и Решение на Съвета от 8 април 2003 г.
(4) ОВ L 95, 16.4.1996 г., стр. 9. Решение, отменено с Решение № 1786/2002/ЕО на Европейския парламент и на Съвета (ОВ L 271, 9.10.2002 г., стр. 1).
(5) ОВ L 365, 31.12.1994 г., стр. 1.
(6) ОВ L 262, 27.9.1976 г., стр. 201. Директива, последно изменена с Директива 2003/11/ЕО на Европейския парламент и на Съвета (ОВ L 42, 15.2.2003 г., стр. 45).
(7) ОВ 196, 16.8.1967 г., стр. 1. Директива, последно изменена с Директива 2001/59/ЕО на Комисията (ОВ L 225, 21.8.2001 г., стр. 1).
(8) ОВ L 355, 30.12.1998 г., стр. 1. Директива, изменена с Решение 2000/368/ЕО на Комисията (ОВ L 136, 8.6.2000 г., стр. 108).
(9) ОВ L 136, 8.6.2000 г., стр. 1.
(10) ОВ L 183, 29.6.1989 г., стр. 1.
(11) ОВ L 196, 26.7.1990 г., стр. 1. Директива, последно изменена с Директива 1999/38/ЕО (ОВ L 138, 1.6.1999 г., стр. 66).
ПРИЛОЖЕНИЕ
Точка 29 — Канцерогени: категория 2
|
Вещества |
Индекс номер |
ЕО номер |
CAS номер |
|
Кобалтов двухлорид |
027-004-00-5 |
231-589-4 |
7646-79-9 |
|
Кобалтов сулфат |
027-005-00-0 |
233-334-2 |
10124-43-3 |
|
Кадмиев флуорид |
048-006-00-2 |
232-222-0 |
7790-79-6 |
|
Хризол |
601-048-00-0 |
205-923-4 |
218-01-9 |
|
Бензо[е]пирен |
601-049-00-6 |
205-892-7 |
192-97-2 |
|
2,2'-Биоксиран; 1,2:3,4-диепоксибутан |
603-060-00-1 |
215-979-1 |
1464-53-5 |
|
2,3-Епоксипропан-1-ол; глицид |
603-063-00-8 |
209-128-3 |
556-52-5 |
|
2,4-Динитротолуол [1]; динитротолуол [2]; динитротолуол, за технически цели |
609-007-00-9 |
204-450-0 [1] |
121-14-2 [1] |
|
246-836-1 [2] |
25321-14-6 [2] |
||
|
2,6-Динитротолуол |
609-049-00-8 |
210-106-0 |
606-20-2 |
|
Хидразин-три-нитрометан |
609-053-00-X |
414-850-9 |
— |
|
Азобензол |
611-001-00-6 |
203-102-5 |
103-33-3 |
|
о-дианизидин-съдържащи азобагрила; 4,4'-диарилазо-3,3'-диметоксибифенилови багрила с изключение на цитираните на други места в приложение I към Директива 67/548/ЕИО |
611-029-00-9 |
— |
— |
|
о-толидин-съдържащи багрила; 4,4'-диарилазо-3,3'-диметилбифенилови багрила с изключение на цитираните на други места в приложение I към Директива 67/548/ЕИО |
611-030-00-4 |
— |
— |
|
1,4,5,8-тетрааминоантрахинон; C.I. дисперсно синьо 1 |
611-032-00-5 |
219-603-7 |
2475-45-8 |
Точка 30 — Мутагени: категория 2
|
Вещества |
Индекс номер |
ЕО номер |
CAS номер |
|
Кадмиев флуорид |
048-006-00-2 |
232-222-0 |
7790-79-6 |
|
Кадмиев хлорид |
048-008-00-3 |
233-296-7 |
10108-64-2 |
|
2,2'-Биоксиран; 1,2:3,4-диепоксибутан |
603-060-00-1 |
215-979-1 |
1464-53-5 |
Точка 31 — Токсични за репродукцията: категория 2
|
Вещества |
Индекс номер |
ЕО номер |
CAS номер |
|
Кадмиев флуорид |
048-006-00-2 |
232-222-0 |
7790-79-6 |
|
Кадмиев хлорид |
048-008-00-3 |
233-296-7 |
10108-64-2 |
|
2,3-Епокиспропан-1-ол; глицид |
603-063-00-8 |
209-128-3 |
556-52-5 |
|
2-Метоксипропанол |
603-106-00-0 |
216-455-5 |
1589-47-5 |
|
4,4'-изобутилетилидендифенол; 2,2-бис (4'-хидроксифенил)-4-метилпентан |
604-024-00-8 |
401-720-1 |
6807-17-6 |
|
2-Метоксипропилов ацетат |
607-251-00-0 |
274-724-2 |
70657-70-4 |
|
Тридеморф (ISO); 2,6-диметил-4-тридецилморфолин |
613-020-00-5 |
246-347-3 |
24602-86-6 |
|
Циклохексимид |
613-140-00-8 |
200-636-0 |
66-81-9 |
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
185 |
32003L0036
|
L 156/26 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/36/ЕО НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 26 май 2003 година
относно двадесет и пето изменение на Директива 76/769/ЕИО на Съвета за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки относно ограниченията за пускането на пазара и употребата на някои опасни вещества и препарати (вещества, класифицирани като канцерогени, мутагени или вещества, токсични за репродукцията — c/m/r)
(текст от значение за ЕИП)
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 95 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Европейския икономически и социален комитет (2),
в съответствие с процедурата, установена в член 251 от Договора (3),
като имат предвид, че:
|
(1) |
Директива 76/769/ЕИО на Съвета (4), постановява ограничения върху продажбата и употребата на някои опасни вещества и препарати, |
|
(2) |
Предвидените в тази директива мерки се вместват в рамките на плана за действие от Решение № 646/96/ЕО на Европейския парламент и Съвета от 29 март 1996, приемащо план от мерки за ограничаване на раковите заболявания в рамките за действие в сферата на общественото здравеопазване (1996—2000 г.) (5). |
|
(3) |
За подобряване на здравната защита и безопасността на потребителите, веществата, класифицирани като канцерогенни, мутагенни или токсични за репродукцията, и препаратите, които ги съдържат, не трябва да бъдат пускани на пазара за употреба от широкия потребител. |
|
(4) |
Директива 94/60/ЕО на Европейския парламент и Съвета от 20 декември 1994 относно четиринадесето изменение на Директива 76/769/ЕИО (6), установява под формата на допълнение относно точки 29, 30 и 31 от приложение I към Директива 76/769/ЕИО списък на вещества, класифицирани като канцерогенни, мутагенни или токсични за репродукцията от категории 1 и 2. Подобни вещества и препаратите, които ги съдържат, не трябва да бъдат пускани на пазара за употреба от широкия потребител. |
|
(5) |
Директива 94/60/ЕО предвижда, че въпросният списък ще бъде разширяван наскоро след всяко публикуване на адаптиране към техническия прогрес на приложение I към Директива 67/548/ЕИО на Съвета от 27 юни 1967 за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки относно класифицирането, опаковането и етикетирането на опасни вещества (7), което изброява вещества, класифицирани като канцерогенни, мутагенни или токсични за репродукцията от категории 1 и 2. |
|
(6) |
Директива 2001/59/ЕО на Комисията (8) относно двадесет и осмо адаптиране към технически прогрес на Директива 67/548/ЕИО на Съвета, и по-специално приложение I към цитираната директива, изброява две нови вещества, класифицирани като канцерогени от категория 1, 19 нови вещества, класифицирани като канцерогени от категория 2, пет нови вещества, класифицирани като мутагени от категория 2, едно ново вещество, класифицирано като токсично за репродукцията от категория 1, и 16 нови вещества, класифицирани като токсични за репродукцията от категория 2. |
|
(7) |
Тези вещества трябва да се добавят към списъка от допълнението към приложение I към Директива 76/769/ЕИО. |
|
(8) |
Бяха взети под внимание рисковете и предимствата от новите веществата, класифицирани в съответствие с Директива 2001/59/ЕО като канцерогени, мутагени или токсични за репродукцията. |
|
(9) |
Настоящата директива се прилага без да засяга законодателството на Общността, постановяващо минимални изисквания за защитата на работниците, съдържащи се в Директива 89/391/ЕИО на Съвета от 12 юни 1989 г. за въвеждане на мерки за насърчаване подобряването на безопасността и здравето на работниците на работното място (9), и произтичащите от нея специални директиви, и по-специално Директива 90/394/ЕИО на Съвета от 28 юни 1990 г. за защита на работниците от рискове, свързани с експозиция на канцерогени по време на работа (10), |
ПРИЕХА НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Изброените в приложението към настоящата директива вещества се добавят към веществата, изброени в допълнението относно точки 29, 30 и 31, към приложение I към Директива 76/769/ЕИО. Веществата, изброени в точка 1, буква в) от приложението към настоящата директива, се заличат от списък 2 на точка 29 от приложение I към Директива 76/769/ЕИО.
Член 2
1. Държавите-членки приемат и публикуват законовите, подзаконовите и административни разпоредби, необходими, за да се съобразят с настоящата директива до 25 юни 2004. Те незабавно информират Комисията за това.
Те прилагат тези разпоредби считано от 25 декември 2004 г.
2. Когато държавите-членки приемат тези мерки, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 3
Настоящата директива влиза в сила в деня на публикуването ѝ в Официален вестник на Европейския съюз.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 26 май 2003 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
G. DRYS
(1) ОВ C 126 Е, 28.5.2002 г., стр. 398.
(2) ОВ C 221, 17.9.2002 г., стр. 8.
(3) Становище на Европейския парламент от 11 юни 2002 г. (все още непубликувано в Официален вестник), Обща позиция на Съвета от 21 януари 2003 г. (ОВ C 64 Е, 18.3.2003 г., стр. 6) и Решение на Европейския парламент от 10 април 2003 г. (все още непубликувано в Официален вестник).
(4) ОВ L 262, 27.9.1976 г., стр. 201. Директива, последно изменена с Директива 2002/62/ЕО на Комисията (ОВ L 183, 12.7.2002 г., стр. 58).
(5) ОВ L 95, 16.4.1996 г., стр. 9. Решение, отменено на 31 декември 2002 г. с Решение№ 1786/2002/ЕО на Европейския парламент и Съвета (ОВ L 271, 9.10.2002 г., стр. 1).
(6) ОВ L 365, 31.12.1994 г., стр. 1.
(7) ОВ L 196, 16.8.1967 г., стр. 1. Директива, последно изменена с Директива 2001/59/ЕО на Комисията (ОВ L 225, 21.8.2001 г., стр. 1).
(8) ОВ L 225, 21.8.2001 г., стр. 1.
(9) ОВ L 183, 29.6.1989 г., стр. 1.
(10) ОВ L 196, 26.7.1990 г., стр. 1. Директива, последно изменена с Директива 1999/38/ЕО на Съвета (ОВ L 138, 1.6.1999 г., стр. 66).
ПРИЛОЖЕНИЕ
В допълнението към приложение I към Директива 76/769/ЕИО се изменя както следва:
|
1. |
списъците от „Точка 29 — Канцерогени“ се изменят, както следва:
|
|
2. |
в списъка за категория 2 от „Точка 30 — Мутагени“ се добавят следните вещества:
|
|
3. |
списъците от „Точка 31 — токсични за репродукцията“ се изменят както следва:
|
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
190 |
32003R1084
|
L 159/1 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕГЛАМЕНТ (ЕО) № 1084/2003 НА КОМИСИЯТА
от 3 юни 2003 година
относно проучване на измененията в условията на разрешенията за търговия с лекарствени продукти за хуманна употреба и ветеринарни лекарствени продукти, предоставяни от компетентен орган на държава-членка
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 2001/83/ЕО на Европейския парламент и на Съвета от 6 ноември 2001 г. за утвърждаване на кодекс на Общността относно лекарствени продукти за хуманна употреба (1), и по-специално член 35, параграф 1 от него,
като взе предвид Директива 2001/82/ЕО на Европейския парламент и на Съвета от 6 ноември 2001 г. за утвърждаване на кодекс на Общността относно лекарствени продукти за ветеринарна употреба (2),и по-специално член 39, параграф 1 от нея,
като има предвид, че
|
(1) |
В светлината на практическия опит в прилагането на Регламент (ЕО) № 541/95 на Комисията от 10 март 1995 г. за проверка на промените на условията на разрешително за търговия, издадено от компетентната власт на държава-членка (3), изменен с Регламент (ЕО) № 1146/98 (4), е целесъобразно да се опрости процедурата за изменение на условията за издаване на разрешение за търговия. |
|
(2) |
Някои от процедурите, определени в Регламент (ЕО) № 541/95 следва съответно да бъдат преразгледани, без да се стига до отклонения от общите принципи, на които се основават тези процедури. |
|
(3) |
Вследствие на приемането на Директива 2001/82/ЕО и Директива 2001/83/ЕО, с които бе кодифицирано законодателството на Общността в областта на лекарствените продукти за ветеринарна употреба и на лекарствените продукти за хуманната употреба, следва да се актуализират позоваванията на разпоредбите на това законодателство. |
|
(4) |
Настоящият регламент следва да продължи да прилага също и проучванията на заявленията за изменения в условията на разрешение за търговия, които се издават в съответствие с Директива 87/22/ЕИО на Съвета (5), отменена с Директива 93/41/ЕИО на Съвета (6). |
|
(5) |
Целесъобразно е да се предвиди опростена и бърза процедура за нотифициране с цел даване на възможност за въвеждане на някои незначителни изменения, които не влияят на одобреното качество, безвредността и ефикасността на продукта, без предварителна оценка от страна на съответната държава-членка. Все още обаче следва да се изисква оценка за другите видове незначителни изменения в представената документация от съответната държава-членка. |
|
(6) |
В случаите, в които се поддържа процедурата по оценяването, съответната държава-членка следва да оценява документацията от името на всички заинтересовани държави-членки с цел избягване на дублирането на работата. |
|
(7) |
Различните видове незначителни изменения следва да се класифицират съгласно условията, които следва да се изпълнят с цел определяне на процедура, която да се следва; особено необходимо е да се даде точна дефиниция на вида незначително изменение, за който не е необходимо предварително оценяване. |
|
(8) |
Необходимо е да се изясни дефиницията на термина „разширяване обхвата“ на разрешението за търговия, въпреки че все пак следва да съществува възможност за представяне на отделно, пълно заявление за разрешение за търговия за лекарствен продукт, който вече е получавал разрешение, но под друго наименование и с различно обобщение на характеристиките на продукта. |
|
(9) |
Целесъобразно е да се позволи на националните власти на съответните държави-членки да съкращават периода за даване на оценка в спешни случаи или да го удължават в случай на значителни изменения, водещи до важни промени. |
|
(10) |
Следва да се изясни графикът за процедурата, която да се следва, когато компетентният орган налага по спешност ограничителни мерки за постигане на безвредност. |
|
(11) |
Следва да се въведе по-подробно изясняване с оглед проверката на обобщението на характеристиките на продукта, етикетирането и листовката с упътванията в опаковката; въпреки това определените в настоящия регламент процедури не следва да се прилагат по отношение на промени в етикетирането или по отношение на листовката с упътванията, които не водят до промени в съдържанието на обобщението на характеристиките на продукта. |
|
(12) |
С цел постигане на по-голяма яснота е целесъобразно да се замени Регламент (ЕО) № 541/95. |
|
(13) |
Мерките, предвидени в настоящия регламент са в съответствие със становището на Постоянния комитет по лекарствени продукти за хуманна употреба и Постоянния комитет по лекарствени продукти за ветеринарна употреба, |
ПРИЕ НАСТОЯЩИЯ РЕГЛАМЕНТ:
Член 1
Предмет
Настоящият регламент определя процедурата за проучване на нотификациите за и заявленията за изменения на условията за разрешение за търговия с лекарствени продукти, които са разгледани в рамките на прилагането на Директива 87/22/ЕИО, на лекарствени продукти, които се ползват от процедурите на взаимно признаване, определени в членове 17 и 18 и член 28, параграф 4 от Директива 2001/83/ЕО или членове 21, 22 и член 32, параграф 4 от Директива 2001/82/ЕО, както и лекарствени продукти, за които има позоваване на процедурите, определени в членове 32, 33 и 34 от Директива 2001/83/ЕО или членове 36, 37 и 38 от Директива 2001/82/ЕО.
Член 2
Обхват
Настоящият регламент не се прилага за:
|
а) |
разширяване обхвата на разрешения за търговия, които отговарят на условията, определени в приложение II към настоящия регламент; |
|
б) |
прехвърляния на разрешения за търговия на нов титуляр; |
|
в) |
промени в максималната граница на остатъчните вещества, както това е определено в член 1, параграф 1, буква б) от Регламент (ЕИО) № 2377/90 на Съвета (7). |
Разширяването на обхвата, посочено в буква а) от първия параграф, се проучва в съответствие с процедурата, посочена в член 17 от Директива 2001/83/ЕО и член 21 от Директива 2001/82/ЕО.
Член 3
Дефиниции
По смисъла на настоящия регламент се прилагат следните дефиниции:
|
1. |
„Изменение в условията за разрешение за търговия“ означава:
|
|
2. |
„Незначително изменение“ от тип IА или тип IБ означава изменение в списъка в приложение I, което отговаря на изложените там условия. |
|
3. |
„Значително изменение“ от тип II означава изменение, което не може да се счита за незначително изменение или разширяване на обхвата на разрешението за търговия. |
|
4. |
„Референтна държава-членка“ означава държавата-членка, която по отношение на даден лекарствен продукт, е представила доклад-оценка, който е послужил за основа на процедурите, посочени в член 1 или алтернативно държавата-членка, избрана в това отношение от титуляра на разрешението за търговия с оглед прилагането на настоящия регламент. |
|
5. |
„Спешно ограничение, свързано с безвредността“ означава временна промяна в информация за продукта, засягаща по-специално един или повече от следните въпроси в обобщението на характеристиките на продукта, показанията, дозировката, противопоказанията, предупрежденията, биологичните видове, за които е предназначен продуктът, и периодите на изтегляне поради новопоявила се информация, имаща отношение към безопасната употреба на лекарствения продукт. |
Член 4
Процедура по нотификация при незначителни изменения от тип IА
1. С оглед на незначителните изменения от тип IА, титулярът на разрешението за търговия (наричан по-долу „титуляря“) се задължава да представи едновременно на компетентните власти на държавите-членки, в които лекарственият продукт е получил разрешение, нотификация, придружена от:
|
а) |
всички необходими документи, включително онези, които са били изменени в резултат от изменението; |
|
б) |
списък от заинтересованите държави-членки и посочване на референтната държава-членка за разглеждания лекарствен продукт; |
|
в) |
съответните такси, предвидени съгласно приложимите национални правила в съответните държави-членки. |
2. Нотификацията се отнася само за едно изменение от тип IА. Когато трябва да се направят няколко изменения в условията от тип IА по едно разрешение за търговия, се прави отделна нотификация по отношение на всяко желано изменение от тип IА, като всяка такава нотификация също така съдържа позоваване на другите нотификации.
3. Чрез дерогация от параграф 2, когато изменение от тип IА в разрешение за търговия води до поредно изменение от тип IА, една нотификация може да обхване всички поредни изменения. Тази нотификация съдържа описание на отношението между тези поредни изменения от тип IА.
4. Когато дадено изменение налага произтичащо от него преразглеждане на обобщението на характеристиките на продукта, на етикетирането и листовката/брошурата в опаковката, това се счита за част от изменението.
5. Ако нотификацията отговаря на изискванията, посочени в параграфи от 1 до 4, компетентният орган на референтната държава-членка в рамките на 14 дни след получаването на нотификацията признава валидността на тази нотификация и информира съответно останалите съответни компетентни власти и титуляра.
Всяка съответна компетентна власт при необходимост актуализира разрешението за търговия, издадено в съответствие с член 6 от Директива 2001/83/ЕО или член 5 от Директива 2001/82/ЕО.
Член 5
Процедура за нотифициране при незначителни изменения от тип IБ
1. По отношение на незначителни изменения от тип IБ титулярът на разрешение за търговия (наричан по-долу „титулярът“) представя едновременно на компетентните власти на държавите-членки, в които лекарствения продукт е получил разрешение, нотификация, придружена от:
|
а) |
всички необходими документи, включително онези, които са били изменени в резултат от изменението; |
|
б) |
списък на съответните държави-членки и посочване на референтна държава-членка за лекарствения продукт, предмет на разглеждане; |
|
в) |
съответните такси, предвидени съгласно приложимите национални правила в съответните държави-членки. |
2. Нотификацията се отнася само за едно изменение от тип IБ. Когато се налага да се направят няколко изменения тип IБ в условията на едно разрешение за търговия, се подава отделна нотификация за всяко желано изменение от тип IБ, като всяка такава нотификация съдържа също така позоваване на другите нотификации.
3. Чрез дерогация от параграф 2, когато изменение от тип IБ в разрешението за търговия води до поредно изменение от тип IА или тип IБ, една нотификация може да обхване всички поредни изменения. Тази нотификация съдържа описание на отношението между тези поредни изменения от тип I.
4. Когато дадено изменение налага произтичащо от него преразглеждане на обобщението на характеристиките на продукта, на етикетирането и листовката/брошурата в опаковката, това се счита за част от изменението.
5. Ако нотификацията отговаря на изискванията, посочени в параграфи от 1 до 4, компетентният орган на референтната държава-членка признава получаването на валидна нотификация и инициира процедурата, посочена в параграфи от 6 до 11.
6. Ако в 30-дневен срок от датата на потвърждението на получаването на валидна нотификация компетентната власт на референтната държава-членка не е изпратил на титуляра своето становище, предвидено съгласно параграф 8, нотифицираното изменение се счита за прието от всички компетентни власти на съответните държави-членки.
Компетентният орган на референтната държава-членка информира за това останалите компетентни власти на съответните държави-членки.
7. Всяки съответна компетентна власт актуализира, когато е необходимо, разрешението за търговия, предоставено в съответствие с член 6 от Директива 2001/83/ЕО или член 5 от Директива 2001/82/ЕО.
8. Когато компетентната власт на референтната държава-членка е на мнение, че нотификацията не може да бъде приета, в рамките на периода, посочен в параграф 6, той информира титуляра, който е изпратил нотификацията, като излага аргументите, на които се основава неговото становище.
9. В 30-дневен срок от получаването на становището, посочено в параграф 8, титулярът може да измени нотификацията с цел надлежно съобразяване с аргументите, изложени в становището. В такъв случай разпоредбите на параграфи 6 и 7 се прилагат и към изменената нотификация.
10. Ако титулярът не измени нотификацията, нотификацията се счита за отхвърлена. Компетентният орган на референтната държава-членка незабавно информира съответно титуляра и съответните компетентни органи.
11. В срок от 10 дни от изпращането на информацията, посочена в параграф 10, компетентните органи на заинтересованите държави-членки или титулярът могат да отнесат въпроса до Агенцията за прилагане на член 35, параграф 2 от Директива 2001/83/ЕО или член 39, параграф 2 от Директива 2001/82/ЕО.
Член 6
Процедура по одобряване на значителни изменения от тип II
1. По отношение на значителните изменения от тип II, титулярът представя едновременно на компетентните власти на държавите-членки, в които лекарствения продукт е получил разрешение, заявление, придружено от:
|
а) |
съответните данни и подкрепящи документи, посочени в членове 8 до 12 от Директива 2001/83/ЕО или членове 12 до 15 от Директива 2001/82/ЕО; |
|
б) |
данни в подкрепа на изменението, за което е внесена заявка; |
|
в) |
всички изменени в резултат на заявлението документи; |
|
г) |
допълнение или актуализирана версия на съществуващите експертни доклади/обзори/обобщенията, в които е отчетено изменението, за което е внесено заявление; |
|
д) |
списък на държави-членки, засегнати от заявката за значително изменение от тип II и посочване на референтната държава-членка за разглеждания лекарствен продукт; |
|
е) |
съответните такси, предвидени в приложимите национални правила в заинетересованите държави-членки. |
2. Една заявка се отнася само за едно значително изменение от тип II. Когато се налага да се направят няколко значителни изменения от тип II в едно разрешение за търговия, за всяко желано изменение се подава отделна заявка; във всяка такава заявка се съдържа позоваване на останалите заявки.
3. Чрез дерогация от параграф 2, когато изменение от тип II води до произтичащи от него изменения, такива изменения могат да бъдат обхванати от една заявка. Тази заявка съдържа описание на отношението между тези поредни изменения.
4. Когато дадено изменение налага произтичащо от него преразглеждане на обобщението на характеристиките на продукта, на етикетирането и листовката/брошурата в опаковката, това се счита за част от изменението.
5. Ако заявката отговаря на изискванията, посочени в параграфи от 1 до 4, компетентните органи на съответните държави-членки незабавно нотифицират компетентната власт на референтната държава-членка относно получаването на валидна заявка.
6. Компетентната власт на референтната държава-членка нотифицира другите компетентни власти на заинтересованите държави-членки и инициира процедурата, определена в членове от 7 до 13.
7. В срок от 60 дни след началото на процедурата компетентната власт на референтната държава-членка изготвя доклад-оценка и проекторешение, които се адресират до останалите заинтересовани компетентни власти.
Този период може да бъде съкратен с оглед спешния характер на въпроса, особено при проблеми, свързани с безвредността.
Този период може да бъде удължен до 90 дни за изменения, засягащи промени в или добавяне на терапевтичните показания.
Този период се удължава до 90 дни за изменения, засягащи промяна в или добавяне на целеви биологични видове, които не се използват за производството на храни.
8. В рамките на периодите, определени в параграф 7, компетентната власт на референтната държава-членка може да изиска титулярът да представи допълнителна информация в рамките на период, определен от този компетентната власт. Процедурата се преустановява до момента, в който бъде предоставена допълнителната информация. В този случай периодите, определени в параграф 7 могат да бъдат удължени за допълнителен период, който се определя от компетентната власт на референтната държава-членка.
Компетентната власт на референтната държава-членка информира другите заинтересовани компетентни власти.
9. В срок от 30 дни след получаването на проекторешението и на доклада-оценка, останалите компетентни власти на засегнатите държави-членки признават проекторешението и информират за това компетентната власт на референтната държава-членка.
Компетентният орган на референтната държава-членка приключва процедурата и информира съответно останалите компетентни власти на засегнатите държави-членки и титуляра.
10. Всякя засегната компетентна власт при необходимост изменя съответното разрешение за търговия, издадено съгласно член 6 от Директива 2001/83/ЕО или член 5 от Директива 2001/82/ЕО в съответствие с проекторешението, посочено в параграф 9.
11. Решенията, засягащи измененията, свързани с проблемите на безвредността, се изпълняват в сроковете, договорени между компетентната власт на референтната държава-членка и титуляра след консултации с останалите компетентни власт на заинтересованите държави-членки.
12. Ако в рамките на периода, определен в параграф 9, не е възможно да се постигне взаимно признаване на проекторешението на компетентния орган на референтната държава-членка от страна на един или повече компетентни органи, се прилага процедурата, посочена в член 35, параграф 2 от Директива 2001/83/ЕО или член 39, параграф 2 от Директива 2001/82/ЕО.
13. В срок от 10 дни от приключване на процедурата, посочена в параграф 8, и в случаите, в които компетентните власти на предвидените в заявлението държави-членки считат, че изменението не може да бъде прието, титулярът може да изпрати въпроса до агенцията за целите на прилагането на член 35, параграф 2 от Директива 2001/83/ЕО или на член 39, параграф 2 от Директива 2001/82/ЕО.
Член 7
Ваксини против човешки грип
1. С оглед на измененията в условията на разрешенията за търговия с ваксини против човешки грип, се прилага процедурата, определена в параграфи 2 до 5.
2. В срок от 30 дни след датата на започване на процедурата, компетентният орган на референтната държава-членка изготвя доклад-оценка въз основа на документите относно качеството, посочени в модул 3 на приложение I към Директива 2001/83/ЕО и проекторешението, който се адресира до останалите заинтересовани компетентните власти.
3. В рамките на посочения в параграф 2 период компетентният орган на референтната държава-членка може да поиска титулярът да представи допълнителна информация. Компетентният орган информира останалите компетентни власти на заинтересованите държави-членки.
4. В рамките на 12 дни от получаването на проекторешението и доклада-оценка останалите компетентни власти на съответните държави-членки признават проекторешението и информират за това компетентната власт на референтната държава-членка.
5. Клиничните данни и когато е целесъобразно, данните, засягащи устойчивостта на лекарствения продукт, се адресират от титуляра до компетентния орган на референтната държава-членка, както и до останалите компетентни органи на съответните държави-членки най-късно 12 дни след края на срока, посочен в параграф 4.
Компетената власт на референтната държава-членка дава оценка на тези данни и предлага окончателно проекторешение в 7-дневен срок от получаването на данните. Останалите засегнати компетентни органи признават окончателното проекторешение и в 7-дневен срок от получаването на окончателното проекторешение приемат решение в съответствие с окончателното проекторешение.
6. Ако в хода на посочената в параграфи от 2 до 5 процедура компетентната власт повдигне въпрос относно общественото здраве, за който той смята, че представлява пречка за взаимното признаване на решение, което трябва да бъде взето, се прилага процедурата, посочена в член 35, параграф 2 от Директива 2001/83/ЕО.
Член 8
Ситуация на пандемия по отношение на човешките заболявания
В случай на пандемична ситуация по отношение на вируса на човешкия грип, надлежно призната от Световната здравна организация или от Общността в рамките на Решение № 2119/98/ЕО на Европейския парламент и на Съвета (8), компетентните органи могат по изключение и временно да решат изменението на условията за разрешението за търговия за ваксини против човешки грип да бъде прието след получаване на заявлението и преди приключване на процедурата, определена в член 7. По време на тази процедура обаче могат да бъдат представени клинични данни относно безвредността и ефикасността.
В случай на пандемична ситуация по отношение на човешки заболявания, различни от вируса на човешкия грип, първият параграф и член 7 могат да бъдат прилагани mutatis mutandis.
Член 9
Спешни ограничителни мерки за постигане на безвредност
1. Ако титулярът, в случай на поява на риск за общественото здраве или здравето на животните, предприеме ограничителни мерки за постигане на безвредност, същият незабавно уведомява компетентните органи за това. Ако компетентните власти не повдигнат никакви възражения в рамките на 24 часа след получаването на такава информация, спешните ограничителните мерки за постигане на безвредност се считат за приети.
Спешните ограничителни мерки се изпълняват в рамките на срока, съгласуван с компетентните органи.
Съответното заявление за изменение, отразяващо спешните ограничителни мерки за постигане на безвредност, се подава незабавно и при всички случаи не по-късно от 15 дни след въвеждането на спешните ограничителни мерки за постигане на безвредност, до компетентните органи за прилагане на процедурите, посочени в член 6.
2. Когато компетентните власти наложат на титуляра спешни ограничителни мерки за постигане на безвредност, титулярът се задължава да представи заявление за изменение, като отчете спешните ограничителни мерки, свързани с безвредността, наложени от компетентните власти.
Спешната ограничителна мярка се изпълнява в рамките на срока, съгласуван с компетентните власти.
Съответното заявление за изменение, отразяващо спешната ограничителна мярка, включително необходимата документация в подкрепа на промяната, се представя незабавно и във всички случаи не по-късно от 15 дни след въвеждането на спешната ограничителна мярка на компетентните органи, отговарящи за прилагането на процедурите, посочени в член 6.
Настоящият параграф не нарушава член 36 от Директива 2001/83/ЕО и член 40 от Директива 2001/82/ЕО.
Член 10
Отмяна
Регламент (ЕО) № 541/95 се отменя.
Позоваванията на отменения регламент се считат за позовавания на настоящия регламент.
Член 11
Настоящият регламент влиза в сила на двадесетия ден след деня на публикуването му в Официален вестник на Европейския съюз.
Прилага се от 1 октомври 2003 година.
Настоящият регламент е задължителен в своята цялост и се прилага пряко във всички държави-членки.
Съставено в Брюксел на 3 юни 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 311, 28.11.2001 г., стр. 67.
(2) ОВ L 311, 28.11.2001 г., стр. 1.
(3) ОВ L 55, 11.3.1995 г., стр. 7.
(4) ОВ L 159, 3.6.1998 г., стр. 31.
(5) ОВ L 15, 17.1.1987 г., стр. 38.
(6) ОВ L 214, 24.8.1993 г., стр. 40.
(7) ОВ L 224, 18.8.1990 г., стр. 1.
(8) ОВ L 268, 3.10.1998 г., стр. 1.
ПРИЛОЖЕНИЕ I
СПИСЪК И УСЛОВИЯ ЗА НЕЗНАЧИТЕЛНИ ИЗМЕНЕНИЯ (ТИП IА И IБ) В РАЗРЕШЕНИЯТА ЗА ТЪРГОВИЯ, ПОСОЧЕНИ В ЧЛЕНОВЕ 3 ДО 5
Встъпителни бележки
Наименованията на измененията са номерирани, а подкатегориите са въведени с букви и цифри с по-дребен шрифт. Условията, необходими за да следва дадено изменение или процедура от тип IА или от тип IБ, са изведени за всяка подкатегория и са изброени под всяко изменение.
За да се обхванат и всякакви други промени, е необходимо да се подадат заявки за всякакви произтичащи или паралелни изменения, които могат да бъдат свързани с промяната, за която се подава заявка, като същевременно се дава ясно описание на връзката между тези изменения.
За нотификации, включващи сертификат за годност от Европейската фармакопея и в случаите, когато изменението засяга подадената за сертификата документация, документацията, която се изисква за тази промяна, следва да се представи на Европейската дирекция по качеството на лекарствата (EDQM). Ако сертификатът е подложен на редакция след оценяването на тази промяна, задължително се осъвременява съответното разрешение за търговия. В много случаи това става чрез нотификация тип IА.
Биологичният лекарствен продукт е продукт, чието активно вещество представлява биологично вещество. Биологично вещество е вещество, което се произвежда или извлича от биологичен източник и за което е необходима комбинация от физико-химично и биологично изпитване, като производственият процес и контролът върху него са необходими за характеризирането му и определянето на качеството му.
В резултат от това за биологични лекарствени продукти се считат следните продукти: имунологични лекарствени продукти и лекарствени продукти, произведени от човешка кръв и кръвна плазма, както това е определено съответно в член 1, параграф 4 и член 1, параграф 10 от Директива 2001/83/ЕО; имунологичните лекарствени продукти с ветеринарна употреба, както това е определено в член 1, параграф 7 от Директива 2001/82/ЕО; лекарствените продукти, влизащи в обхвата на част А от приложението към Регламент (ЕИО) № 2309/93 на Съвета (1); лекарствените продукти за усъвършенствана терапия — както това е определено в част IV от приложение I към Директива 2001/83/ЕО.
Може да се направи промяна в производствения процес на непротеинов компонент поради последващо въвеждане на биотехнологична стъпка в съответствие с разпоредбите относно изменения от тип I, № 15 или № 21, както е по-подходящо. Конкретното изменение не нарушава останалите изменения, изброени в настоящото приложение, което може да се прилага в този конкретен контекст. Въвеждането на протеинов компонент, получен вследствие на прилагане на биотехнологичен процес от списъка в част А от Приложението към Регламент (ЕИО) № 2309/93 на Съвета, в лекарствен продукт попада в обхвата на посочения регламент. То отговаря на приложимото законодателство на Общността относно специфични групи продукти (2).
Не е необходимо нотифициране на компетентните власти относно актуализирана монография от Европейската фармакопея или националната фармакопея на държава-членка в случай, че документацията се приведе в съответствие с актуализираната монография в рамките на 6-месечен срок от публикуването ѝ и в документацията на получилия разрешение лекарствен продукт се направи препратка към „актуалното издание“.
За целите на настоящия документ „процедура на изпитване“ има същия смисъл, както „процедура за анализ на данните“, като „ограничения“ има същия смисъл като „критерии за приемане“.
Комисията, след консултация с държавите-членки, агенцията и заинтересованите страни съставя и публикува подробно ръководство относно документацията, която следва да бъде представена.
|
Наименование на изменението/условия, които следва да се изпълнят |
Тип |
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия: Титулярът на разрешението за търговия остава едно и също правно образувание |
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия: Не се предизвиква объркване с наименованията на съществуващи лекарствени продукти или с непатентовано международно търговско название (INN) |
|
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия: Няма промяна в активното вещество |
|
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия: Предприятието за производство на активното вещество остава същото |
|
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия: Предприятието за производство на готовия продукт остава същото |
|
|||||||||||||||
|
|
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия: Промяна след издаване или изменение на АТС код от СЗО |
|
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия: Промяна след издаване или изменение на АТС Vet код. |
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия 1, 2 (виж по-долу) |
IА |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3, 5 |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 5 |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5 |
IБ |
||||||||||||||
|
Условия: 1, 2, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 2, 3, 4 (виж по-долу) |
IА |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IА |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия: Няма |
|
|||||||||||||||
|
IВ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 4 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5 |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 2, 4, 5 |
IВ |
||||||||||||||
|
Условия: 2, 4 |
IВ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 5 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 2, 3, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 4 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 4 (виж по-долу) |
IА |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: няма |
IБ |
||||||||||||||
|
Условия: няма |
IА |
||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 5 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 1, 2, 3 |
IБ |
||||||||||||||
|
Условия: 2, 3, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3 |
IБ |
||||||||||||||
|
Условия: 1, 2, 3 |
IА |
||||||||||||||
|
Условия: 1, 2, 3 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: няма |
IА |
||||||||||||||
|
Условия: няма |
IБ |
||||||||||||||
|
|
|||||||||||||||
|
Условия: (виж по-долу) |
IБ |
||||||||||||||
|
Условия: (виж по-долу) |
IА |
||||||||||||||
|
Условия: Пускането на ексципиента и готовия продукт и спецификациите по срока за съхранение в опакован вид се запазват. |
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 |
IА |
||||||||||||||
|
Условия: 1, 2 |
IА |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
Условия 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия: Промяната не засяга някоя основна част от материала на опаковката, която влияе върху доставката, употребата, безвредността и устойчивостта на готовия продукт |
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 4 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IА |
||||||||||||||
|
Условия: 1, 3, 4 |
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 4, 5 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5, 6 |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5, 6, 7 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3, 4, 7 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4, 7 |
IА |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3, 4, 5, 6, 7 |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5, 6, 7 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 3, 4 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2, 3 |
IА |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 4, 5 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия: 2, 3, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IА |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IА |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия: 1, 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 3 |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IА |
||||||||||||||
|
Условия: 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
(1) ОВ L 214, 24.8.1993 г., стр. 1.
(2) За храни и съставки на храни в съответствие с Регламент (ЕО) № 258/97 на Европейския парламент и Съвета (ОВ L 43, 14.2.1997 г., стр. 1), оцветители за употреба в хранително-вкусовата промишленост в рамките на Директива 94/36/ЕО на Съвета (ОВ L 237, 10.9.1994 г., стр. 13), хранителни добавки в рамките на Директива 88/388/ЕИО на Съвета (ОВ L 184, 15.7.1988 г., стр. 61), разтворители на екстракти в рамките на Директива 88/344/ЕИО на Съвета (ОВ L 157, 24.6.1988 г., стр. 28), последно изменена с Директива 92/115/ЕИО на Съвета (ОВ L 409, 31.12.1992 г., стр. 31), както и за храни и хранителни съставки, получени посредством биотехнологична стъпка, въведена в производството/продукцията, не се изисква нотификация при изменение в условията на разрешението за търговия.
ПРИЛОЖЕНИЕ II
ПРОМЕНИ В РАЗРЕШЕНИЯТА ЗА ТЪРГОВИЯ, ВОДЕЩИ ДО РАЗШИРЯВАНЕ НА ОБХВАТА НА ЗАЯВЛЕНИЕТО СЪГЛАСНО ЧЛЕН 2
Промените, изброени по-долу, ще се считат за „разширяване обхвата“ на заявлението, както е посочено в член 2.
Разширяване обхвата на съществуващо разрешение за търговия или изменение в него следва да се даде от компетентните власти.
Наименованието на лекарствения продукт за „разширяването на обхвата“ е същото, каквото е за съществуващо разрешение за търговия за даден лекарствен продукт.
Комисията, след консултации с държавите-членки, агенцията и заинтересованите страни, ще състави и публикува подробно ръководство за документацията, която следва да се представи.
Промени, изискващи разширяване обхвата на заявлението
|
1. |
Промени в активното/ите вещество/а:
|
|
2. |
Промени в концентрацията, фармацевтичната форма и начина на приемане:
|
|
3. |
Други промени, специфични за лекарствените продукти с ветеринарна употреба:
|
(1) При парентерално приемане е необходимо да се прави разлика между вътрешно артериално, интравенозно, вътрешно мускулно, подкожно и други начини. При прилагане върху птици респираторният, пероралният и очният път (небулизация) за ваксиниране се считат за еквивалентни начини на приемаие.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
213 |
32003R1085
|
L 159/24 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕГЛАМЕНТ (ЕО) № 1085/2003 НА КОМИСИЯТА
от 3 юни 2003 година
относно проучване на измененията в условията на разрешителните за търговия с лекарствени продукти за хуманна употреба и ветеринарни лекарствени продукти, попадащи в обхвата на Регламент (ЕИО) № 2309/93 на Съвета
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Регламент (ЕИО) № 2309/93 на Съвета от 22 юли 1993 г. за установяване на общностни процедури за разрешаването и надзора на лекарствените продукти за хуманна употреба и ветеринарна употреба и за създаване на Европейска агенция за оценка на лекарствени продукти (1), изменен с Регламент (ЕО) № 649/98 (2) на Комисията, и по-специално член 15, параграф 4 и член 37, параграф 4 от него,
като има предвид, че:
|
(1) |
В светлината на придобития практически опит при прилагането на Регламент (ЕО) № 542/95 на Комисията от 10 март 1995 г. за проверка на промените на условията на разрешително за търговия, попадащо в обхвата на Регламент (ЕИО) № 2309/93 на Съвета (3), изменен с Регламент (ЕО) № 1069/98 (4), е подхозящо да се опрости процедурата за промяна на условията на разрешителните за търговия. |
|
(2) |
Предвид техническото адаптиране на приложение I към Директива 2001/83/ЕО на Европейския парламент и Съвета от 6 ноември 2001 г. относно утвърждаване на кодекс на Общността относно лекарствени продукти за хуманна употреба (5), е подходящо във въпросния кодекс да се въведат разпоредби относно промените в документацията за плазма и документацията за ваксинни антигени. |
|
(3) |
Някои от процедурите, установени с Регламент (ЕО) № 542/95, следва съответно да се адаптират, без това да води до отстъпление от общите принципи, на които се основават тези процедури. |
|
(4) |
Подходящо е да се предвиди процедура за опростена и бърза нотификация, която да позволява внасянето на някои незначителни изменения, които не засягат вече утвърденото качество, безвредност или ефикасност на продукта, без предварително оценяване от страна на Европейската агенция за оценка на лекарствените продукти (наричана по-нататък „Агенцията“). Независимо от това изискването за оценка на представената документация от страна на агенцията се запазва по отношение на други типове незначителни изменения. |
|
(5) |
Различните типове незначителни изменения следва да се категоризират с цел определяне на процедурата за изпълнение; по-специално е необходимо да се даде точна дефиниция на типовете незначителни изменения, за които не се изисква предварителна оценка. |
|
(6) |
Необходимо е да се изясни дефиницията на термина „разширяване обхвата“ на разрешителното за търговия, въпреки че все пак следва да съществува възможност за представяне на отделно, пълно заявление за разрешително за търговия за лекарствен продукт, който вече е получавал разрешение, но под друго наименование и с различно обобщение на характеристиките на продукта. |
|
(7) |
Подходящо е да се позволи на Агенцията да съкращава периода за даване на оценка в спешни случаи или да го удължава в случай на значителни изменения, водещи до важни промени. |
|
(8) |
Необходимо е да се опростят административните процедури за внасяне на незначителни изменения при актуализирането на разрешителните за търговия чрез предоставяне на Комисията на възможност за обединяване на тези актуализации на шест месеца в рамките на едно общо решение. |
|
(9) |
Следва да се изясни графикът за процедурата, която да се следва, когато Комисията налага по спешност ограничителни мерки за постигане на безвредност. |
|
(10) |
Следва да се въведе по-подробно изясняване с оглед проверката на обобщението на характеристиките на продукта, етикетирането и листовката с упътванията в опаковката; въпреки това определените в настоящия регламент процедури не следва да се прилагат по отношение на промени в етикетирането или по отношение на листовката с упътванията, които не водят до промени в обобщението на характеристиките на продукта. |
|
(11) |
С цел постигане на по-голяма яснота е подходящо да се замени Регламент (ЕО) № 542/95. |
|
(12) |
Мерките, предвидени в настоящия регламент, са в съответствие със становището на Постоянния комитет по лекарствени продукти за хуманна употреба и Постоянния комитет по лекарствени продукти за ветеринарна употреба, |
ПРИЕ НАСТОЯЩИЯ РЕГЛАМЕНТ:
Член 1
Предмет
1. Настоящият регламент установява процедура за разглеждане на заявленията за изменения на условията на разрешителните за търговия, издадени в съответствие с Регламент (ЕИО) № 2309/93.
2. Настоящият регламент се отнася и за разглеждането на заявления за изменения на условията на документацията за кръвна плазма и документацията за ваксинни антигени, дефинирани в приложение I към Директива 2001/83/ЕО.
Член 2
Обхват
Настоящият регламент не се отнася за случаите на:
|
а) |
разширяване на обхвата на разрешителни за търговия, които отговарят на условията, определени в приложение II към настоящия регламент; |
|
б) |
прехвърлянето на разрешителното за търговия на друг титуляр; |
|
в) |
промени на максималните стойности на остатъчните вещества, определени в член 1, параграф 1, буква б) от Регламент (ЕИО) № 2377/90 на Съвета (6). |
Разширяването на обхвата, посочено в буква а) от първия параграф, се оценява в съответствие с процедурите, определени в членове 6—10 и членове 28—32 от Регламент (ЕИО) № 2309/93, съответно за лекарствени продукти за хуманна употреба и лекарствени препарати за ветеринарна употреба.
Член 3
Дефиниции
По смисъла на настоящия регламент се прилагат следните определения:
|
1. |
„изменение в условията на разрешително за търговия“ означава изменение в съдържанието на документите, посочени в член 6, параграфи 1 и 2 или член 28, параграфи 1 и 2 от Регламент (ЕИО) № 2309/93, във вида им към момента на приемането на решението за разрешително на търговия в съответствие с член 10 или член 32 от същия регламент, или след одобряването на съответни предшестващи изменения; |
|
2. |
„незначително изменение“ от тип IA или IБ означава описано в приложение I изменение, което отговаря на условията, определени в същото приложение; |
|
3. |
„значително изменение“ от тип II означава изменение, което не може да се разглежда като незначително изменение или разширяване на обхвата на разрешителното за търговия; |
|
4. |
„спешни ограничителни мерки за постигане на безвредност“ означава междинна промяна, дължаща се на новопоявила се информация, която има отношение към безвредната употреба на лекарствения продукт, на информацията за продукта, отнасяща се конкретно до една или няколко от следните позиции от обобщението на характеристиките на продукта: показанията, дозировката, противопоказанията, предупрежденията, видовете, за които се прилага, и сроковете за изтегляне. |
Член 4
Процедура по нотификация за незначителни изменения от тип IA
1. Титулярът на разрешителното за търговия (наричан по-нататък „титулярът“) изпраща на Агенцията нотификация за внасяне на незначителни изменения от тип IA, придружена от:
|
а) |
всички необходими документи, включително онези, които са били изменени в резултат на изменението; |
|
б) |
съответната такса, предвидена в Регламент (ЕО) № 297/95 на Съвета (7). |
2. Нотификацията се отнася само за едно изменение от тип IA. Когато се налага внасяне на няколко изменения от тип IA в условията на едно разрешително за търговия, за всяко желано изменение от тип IA се прави отделна нотификация; всяка подобна нотификация съдържа също така позоваване на останалите нотификации.
3. Чрез дерогация от параграф 2, когато изменение от тип IA в разрешително за търговия води до произтичащи от него изменения от тип IA, такива изменения могат да бъдат обхванати от една нотификация. Тази нотификация съдържа описание на връзката между тези поредни изменения от тип IA.
4. Когато дадено изменение налага произтичащо от него преразглеждане на обобщението на характеристиките на продукта, на етикетирането и листовката/брошурата в опаковката, това се счита за част от изменението.
5. Ако нотификацията отговаря на изискванията, предвидени в параграфи 1—4, агенцията потвърждава в срок до 14 дни след получаването на нотификацията валидността на съответната нотификация и информира титуляра за това.
Агенцията разпространява по целесъобразност изменените документи съгласно член 3, параграф 1.
Когато това е необходимо и въз основа на изготвено от Агенцията предложение, Комисията актуализира на всеки шест месеца разрешителнотото за търговия, предоставено съгласно член 10 или член 32 от Регламент (ЕИО) № 2309/93.
Комисията нотифицира титуляра за актуализирането на разрешителното за търговия.
Регистърът на Общността за лекарствените продукти, предвиден в членове 12 и 34 от Регламент (ЕИО) № 2309/93, се актуализира в съответствие с конкретната необходимост.
Член 5
Процедура за нотифициране на незначителни изменения от тип IБ
1. По отношение на незначителни изменения от тип IБ титулярът представя на агенцията нотификация, придружена от:
|
а) |
всички необходими документи, които показват, че условията, установени в приложение I за исканото изменение, са удовлетворени, включително всички документи с измененията, нанесени в резултат от заявлението; |
|
б) |
съответната такса, предвидена в Регламент (ЕО) № 297/95. |
2. Нотификацията се отнася само до едно изменение от тип IБ. Когато се налага да се направят няколко изменения от типа IБ в условията в едно-единствено разрешително за търговия, за всяко от желаните изменения от тип IБ се подава отделна нотификация; всяка такава нотификация съдържа също така препратка към останалите нотификации.
3. Чрез дерогация от параграф 2, когато изменение от тип IБ в разрешителното за търговия води до произтичащи от него изменения от тип IA или IБ, тези изменения могат да бъдат обхванати от нотификация за тип IБ. Това заявление съдържа описание на отношението между тези поредни изменения от тип I.
4. Когато дадено изменение налага произтичащо от него преразглеждане на обобщението на характеристиките на продукта, на етикетирането и листовката/брошурата в опаковката, това се счита за част от изменението.
5. Ако нотификацията отговаря на изискванията, предвидени в параграфи 1—4, агенцията потвърждава получаването на валидна нотификация и започва процедурата, определена в параграфи 6 —10.
6. Ако в 30-дневен срок от датата на потвърждението за получаване на валидна нотификация агенцията не изпрати на титуляра своето становище съгласно параграф 8, изменението, за която е подадено заявление, се счита за одобрено.
Агенцията информира титуляра за това.
Агенцията разпространява, когато е подходящо изменените документи, посочени в член 3, параграф 1.
7. При необходимост и въз основа на изготвено от агенцията предложение Комисията актуализира на всеки шест месеца разрешителното за търговия, предоставено съгласно член 10 или член 32 от Регламент (ЕИО) № 2309/93.
Комисията нотифицира титуляра за актуализирането на разрешителното за търговия.
Регистърът на Общността за лекарствените продукти, предвиден в членове 12 и 34 от Регламент (ЕИО) № 2309/93, се актуализира в съответствие с конкретната необходимост.
8. Ако становището на агенцията е, че нотификацията не може да бъде приета, агенцията информира за това представилия нотификацията титуляр в предвидения в параграф 6 срок, излагайки мотивите, на които се основава нейното становище.
9. В срок от 30 дни след получаването на становището, посочено в параграф 8, титулярът може да промени съдържанието на нотификацията, като съответно взема под внимание изложените в становището мотиви. В този случай отношение на изменената нотификация се прилагат разпоредбите на параграфи 6 и 7.
10. Ако титулярът не измени нотификацията, последната се счита за отхвърлена. Агенцията информира титуляра за това.
Член 6
Процедура за одобряване на съществени изменения от тип II
1. Титулярът подава до агенцията заявление за внасяне на съществени изменения от тип II, придружено от:
|
а) |
предвидените за целта данни и подкрепящи документи, посочени в член 3, параграф 1; |
|
б) |
подкрепящите данни, отнасящи се до изменението, за което се подава заявлението; |
|
в) |
всички документи, изменени в резултат на заявлението; |
|
г) |
допълнение към или актуализация на съществуващи експертни доклади/обзори/обобщения, с цел да се отчетат измененията, за които се подава заявлението; |
|
д) |
съответната такса, предвидена в Регламент (ЕО) № 297/95. |
2. Всяко заявление се отнася до само едно изменение от тип II. Когато се налага да се направят няколко изменения от тип II в условията на едно разрешително за търговия, за всяко желано изменение се подава отделно заявление; във всяко такова заявление се съдържа препратка към останалите заявления.
3. Чрез дерогация от параграф 2, когато изменение от тип II води до произтичащи от него изменения, всички такива изменения могат да бъдат обхванати от едно общо заявление. Това заявление съдържа описание на отношението между тези поредни изменения.
4. Когато дадено изменение налага произтичащо от него преразглеждане на обобщението на характеристиките на продукта, на етикетирането и листовката/брошурата в опаковката, това се счита за част от изменението.
5. Ако заявлението отговаря на изискванията, определени в параграфи от 1—4, Агенцията потвърждава получаването на валидно заявление и инициира процедурата, определена в параграфи от 6—11.
6. Компетентният комитет на агенцията представя своето становище в срок до 60 дни след започването на процедурата.
Този срок може да бъде съкратен в спешни случаи от съображения за безвредност.
Същият срок може да бъде удължен до 90 дни при изменения, свързани промените или допълнения към терапевтичните показания.
Срокът може да бъде удължен до 90 дни при изменения, свързани с внасянето на промяна или допълнения към видовете, за които е предназначен продуктът, които не се използват за производство на храни.
7. До изтичането на сроковете, определени в параграф 6, компетентният комитет може да отправи към титуляра искане за допълваща информация в определен от Комитета срок. Действието на процедурата се преустановява до момента, в който бъде осигурена допълващата информация. В този случай сроковете, определени в параграф 6, могат да бъдат удължени с допълнителен период, който се определя от Комитета.
8. Когато компетентният комитет представи свое становище, Агенцията нотифицира незабавно титуляра и Комисията и по целесъобразност изпраща на Комисията измененията, които трябва да бъдат внесени в условията на разрешителното за търговия, придружени от документите, предвидени в член 9, параграф 3 и член 31, параграф 3 от Регламент (ЕИО) № 2309/93.
9. Член 9, параграфи 1 и 2 или член 31, параграфи 1 и 2 от Регламент (ЕИО) № 2309/93 се прилагат за случаите, в които е представено становище от компетентния комитет.
10. При необходимост и въз основа на изготвено от Агенцията предложение, Комисията изменя разрешението за търговия, предоставено съгласно член 10 или член 32 от Регламент (ЕИО) № 2309/93.
Решенията, отнасящи се до измененията, свързани с проблемите на безвредността, се изпълняват в срокове, съгласувани между Комисията и титуляра.
Комисията нотифицира титуляра за измененото разрешително за търговия.
11. Регистърът на Общността за лекарствените продукти, предвиден в членове 12 и 34 от Регламент (ЕИО) № 2309/93, се актуализира в съответствие с конкретната необходимост.
Член 7
Ваксини против човешки грип
1. С оглед на измененията в условията на разрешенията за търговия с ваксини против човешки грип се прилага процедурата, определена в параграфи 2—6.
2. В срок до 45 дни след датата на получаване на валидно заявление Агенцията представя становище, основано върху доклад за извършена оценка по документите за качество, посочени в модул 3 от приложение I към Директива 2001/83/ЕО.
3. До изтичането на срока, определен в параграф 2, Агенцията може да изиска от титуляра допълваща информация.
4. Агенцията изпраща незабавно своето становище на Комисията.
Комисията приема решение за актуализиране на разрешителното за търговия, предоставено съгласно член 10 от Регламент (ЕИО) № 2309/93.
Решението се изпълнява, в случай че окончателно становище на Агенцията по параграф 5 е положително.
Комисията нотифицира титуляра за актуализирането на разрешителното за търговия.
5. Титулярът изпраща на Агенцията клиничните данни и по целесъобразност данните за стабилността на лекарствения продукт най-късно 12 дни след изтичането на срока, предвиден в параграф 2.
Агенцията оценява тези данни и представя своето окончателно становище в срок от 10 дни след получаването на данните, посочени в първата алинея. Агенцията изпраща окончателното становище до Комисията и на титуляра на разрешителното за търговия в рамките на следващите три дни.
6. Регистърът на Общността за лекарствените продукти, предвиден в член 12 от Регламент (ЕИО) № 2309/93, се актуализира в съответствие с конкретната необходимост.
Член 8
Пандемична ситуция, причинена от човешки вируси
При наличие на пандемична ситуция, причинена от човешки грипен вирус, надлежно потвърдена от Световната здравна организация или от Общността в рамките на Решение 2119/98/ЕО на Европейския парламент и Съвета (8), Комисията може да разгледа въпроса за внасянето, по изключение и временно, на изменения в разрешителните за търговия с ваксините против човешки грип, които трябва да се приемат след получаване на заявления за такива и преди края на процедурата, предвидена в член 7. Независимо от това в хода на въпросната процедура могат да бъдат представени пълни клинични данни, отнасящи се до безвредността и ефикасността.
В случай на пандемична ситуция, причинена от човешки вируси, различни от човешкия грипен вирус, първият параграф и член 7 могат да се прилагат mutatis mutandis.
Член 9
Спешни ограничителни мерки за постигане на безвредност
1. Ако в случай на поява на риск за общественото здраве или здравето на животните титулярът предприеме ограничителни мерки за постигане на безвредност, той/тя незабавно уведомява Агенцията за това. Ако агенцията не предяви възражения в рамките на 24 часа след получаването на такава информация, спешните ограничителни мерки за постигане на безвредност се считат за приети.
Спешните ограничителни мерки се изпълняват в рамките на срока, съгласуван с агенцията.
Съответното заявление за изменение, отразяващо спешните ограничителни мерки за постигане на безвредност, се подава незабавно и при всички случаи не по-късно от 15 дни след въвеждането на спешните ограничителни мерки за постигане на безвредност, до агенцията за прилагане на процедурите, посочени в член 6.
2. Когато Комисията наложи на титуляра спешни ограничителни мерки за постигане на безвредност, титулярът се задължава да представи заявление за изменение, като отчете спешните ограничителни мерки, свързани с безвредността, наложени от Комисията.
Спешните ограничителни мерки се прилагат в рамките на срока, съгласуван с агенцията.
За прилагането на процедурите, предвидени в член 6, заявлението за внасяне на изменения, отразяващо спешните ограничителни мерки за постигане на безопасност, включително съответната информация в подкрепа на изменението, се подава до агенцията веднага и при всички случаи не по-късно от 15 дни след въвеждането на спешните ограничителни мерки за постигане на безвредност.
Първата и втората алинея не нарушават членове 18 и 40 от Регламент (ЕИО) № 2309/93.
Член 10
Отмяна
Регламент (ЕО) № 542/95 се отменя.
Позоваванията на отменения регламент се считат за позовавания на настоящия регламент.
Член 11
Настоящият регламент влиза в сила на двадесетия ден след датата на публикуването му в Официален вестник на Европейския съюз.
Прилага се от 1 октомври 2003 година. По отношение обаче на проучването на заявленията за изменения в условията на документациите за кръвна плазма и документациите за ваксинни антигени, настоящият регламент се прилага от датата на влизане в сила на Директивата на Комисията за изменение на приложение I към Директива 2001/83/ЕО.
Настоящият регламент е задължителен в своята цялост и и се прилага пряко във всички държави-членки.
Съставено в Брюксел на 3 юни 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 214, 24.8.1993 г., стр. 1.
(2) ОВ L 88, 24.3.1998 г., стр. 7.
(3) ОВ L 55, 11.3.1995 г., стр. 15.
(4) ОВ L 153, 27.5.1998 г., стр. 11.
(5) ОВ L 311, 28.11.2001 г., стр. 67.
(6) ОВ L 224, 18.8.1990 г., стр. 1.
(7) ОВ L 35, 15.2.1995 г., стр. 1.
(8) ОВ L 268, 3.10.1998 г., стр. 1.
ПРИЛОЖЕНИЕ I
СПИСЪК И УСЛОВИЯ ЗА ВНАСЯНЕ НА НЕЗНАЧИТЕЛНИ ИЗМЕНЕНИЯ (ТИП IA И IБ) В РАЗРЕШИТЕЛНИТЕ ЗА ТЪРГОВИЯ, ПОСОЧЕНИ В ЧЛЕНОВЕ 3—5
Встъпителни бележки
Наименованията на измененията са номерирани, а подкатегориите са въведени с букви и цифри с по-дребен шрифт. Условията, необходими дадено изменение да следва или процедура от тип IА, или от тип IБ, са изведени за всяка подкатегория и са изброени под всяко изменение.
За да се обхванат и всякакви други промени, е необходимо да се подадат заявления за всякакви произтичащи или паралелни изменения, които могат да бъдат свързани с промяната, за която се подава заявлението, като същевременно се дава ясно описание на връзката между тези изменения.
За нотификации, включващи сертификат за пригодност от Европейската фармакопея и в случаите, когато изменението засяга подадената за сертификата документация, документацията, която се изисква за тази промяна, следва да се представи на Европейската дирекция по качеството на лекарствата (EDQM). Ако сертификатът е подложен на редакция след оценяването на тази промяна, задължително се осъвременява съответното разрешително за търговия. В много случаи това става чрез нотификация тип IА.
Биологичният лекарствен продукт е продукт, чието активно вещество представлява биологично вещество. Биологично вещество е вещество, което се произвежда или извлича от биологичен източник и за което е необходима комбинация от физико-химично и биологично изпитване, като производственият процес и контролът върху него са необходими за характеризирането му и определянето на качеството му.
В резултат от това за биологични лекарствени продукти се считат следните продукти: имунологични лекарствени продукти и лекарствени продукти, произведени от човешка кръв и кръвна плазма, както това е определено съответно в член 1, параграфи 4 и 10 от Директива 2001/83/ЕО; имунологичните лекарствени продукти с ветеринарна употреба, както това е определено в член 1, параграф 7 от Директива 2001/82/ЕО; лекарствените продукти, попадащи в обхвата на част А от приложението към Регламент (ЕИО) № 2309/93; лекарствените продукти за усъвършенствана терапия, както това е определено в част IV от приложение I към Директива 2001/83/ЕО.
Може да се направи промяна в производствения процес на непротеинов компонент поради последващо въвеждане на биотехнологична стъпка, в съответствие с разпоредбите относно изменения от тип I, № 15 или № 21, както е по-подходящо. Конкретното изменение не нарушава останалите изменения, изброени в настоящото приложение, което може да се прилага в този конкретен контекст. Въвеждането на протеинов компонент, получен вследствие на прилагане на биотехнологичен процес от списъка в част А от приложението към Регламент (ЕИО) № 2309/93 на Съвета в лекарствен продукт попада в обхвата на посочения регламент. То е съобразено с приложимото законодателство на Общността относно специфични групи продукти (1).
Не е необходимо нотифициране на компетентните органи относно актуализирана монография от Европейската фармакопея или националната фармакопея на държава-членка, в случай че документацията се приведе в съответствие с актуализираната монография в рамките на 6-месечен срок от публикуването ѝ и в документацията на получилия разрешение лекарствен продукт се направи препратка към „актуалното издание“.
За целите на настоящия документ „процедура на изпитване“ има същия смисъл, както „процедура за анализ на данните“, като „ограничения“ има същия смисъл като „критерии за приемане“.
Комисията, след консултация с държавите-членки, агенцията и заинтересованите страни, съставя и публикува подробно ръководство относно документацията, която следва да бъде представена.
|
Наименование на изменението/условия, които следва да се изпълнят |
Тип |
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Титулярът на разрешението за търговия остава едно и също юридическо лице. |
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Няма промяна в активното вещество. |
|
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Предприятието за производство на активното вещество остава същото. |
|
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Предприятието за производство на готовия продукт остава същото. |
|
|||||||||||||||
|
|
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Промяна след издаване или изменение на АТС код от СЗО |
|
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Промяна след издаване или изменение на АТС Vet код |
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1,2 (виж по-долу) |
IA |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3, 5 |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 5 |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5 |
IБ |
||||||||||||||
|
Условия: 1, 2, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 2, 3, 4 (виж по-долу) |
IA |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IA |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Няма |
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 4 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5 |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 2, 4, 5 |
IБ |
||||||||||||||
|
Условия: 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 5 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 4 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 4 (виж по-долу) |
IA |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: няма |
IБ |
||||||||||||||
|
Условия: няма |
IA |
||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 5 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 1, 2, 3 |
IБ |
||||||||||||||
|
Условия: 2, 3, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3 |
IБ |
||||||||||||||
|
Условия: 1, 2, 3 |
IA |
||||||||||||||
|
Условия: 1, 2, 3 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: няма |
IA |
||||||||||||||
|
Условия: няма |
IБ |
||||||||||||||
|
|
|||||||||||||||
|
Условия: (виж по-долу) |
IБ |
||||||||||||||
|
Условия: (виж по-долу) |
IA |
||||||||||||||
|
Условия: Пускането на ексципиента и готовия продукт и спецификациите по срока за съхранение в опакован вид се запазват. |
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 |
IA |
||||||||||||||
|
Условия: 1, 2 |
IA |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Промяната не засяга някоя основна част от материала на опаковката, която влияе върху доставката, употребата, безвредността и устойчивостта на готовия продукт. |
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 4 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IA |
||||||||||||||
|
Условия: 1, 3, 4 |
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1 (виж по-долу) |
IA |
||||||||||||||
|
словия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 4, 5 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5, 6 |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5, 6, 7 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3, 4, 7 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4, 7 |
IA |
||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3, 4, 5, 6, 7 |
IБ |
||||||||||||||
|
Условия: 1, 2, 3, 4, 5, 6, 7 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 3, 4 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2, 3 |
IA |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3, 4, 5 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 1, 2, 3, 4 |
IБ |
||||||||||||||
|
Условия: 2, 3, 4, 5 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IA |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия: 1, 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Условия: 1, 2 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 3 |
IБ |
||||||||||||||
|
Условия: 1, 2 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3 |
IБ |
|||||||||||||||
|
Условия: 2, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
|
|||||||||||||||
|
Условия: 1, 2, 3 (виж по-долу) |
IA |
||||||||||||||
|
Условия: 2, 3, 4 |
IБ |
||||||||||||||
|
Условия:
|
|
|||||||||||||||
|
IБ |
|||||||||||||||
|
Условия: Изменението се отнася единствено до въвеждането на промени в обобщението на характеристиките на продукта, етикетите и опаковъчните листовки/вложки за вземане под внимание на научното становище, предоставено в контекста на позоваването в съответствие с членове 31 и 32 от Директива 2001/83/ЕО или членове 35 и 36 от Директива 2001/82/ЕО. |
|
|||||||||||||||
|
|
|||||||||||||||
|
IA |
|||||||||||||||
|
IA |
|||||||||||||||
|
IA |
|||||||||||||||
|
Условия: Оставащата/те информация/и за продуктите трябва да осигурява/т достатъчно инструкции за дозиране и продължителност на приложението в обобщението на характеристиките на продукта. |
|
|||||||||||||||
(1) Към храни и съставки на храни, отговарящи на Регламент (ЕО) № 258/97 на Европейския парламент и Съвета (ОВ L 43, 14.2.1997 г., стр. 1), оцветители за употреба в хранително-вкусовата промишленост в обхвата на Директива 94/36/ЕО на Съвета (ОВ L 237, 10.9.1994 г., стр. 13), хранителни добавки в рамките на Директива 88/388/ЕИО на Съвета (ОВ L 184, 15.7.1988 г., стр. 61), разтворители на екстракти по смисъла на Директива 88/344/ЕИО на Съвета (ОВ L 157, 24.6.1988 г., стр. 28), последно изменена с Директива 92/115/ЕИО (ОВ L 409, 31.12.1992 г., стр. 31), както и към храни и хранителни съставки, получени посредством биотехнологична стъпка, въведена в производството/продукцията, не се изисква нотификация при изменение в условията на разрешението за търговия.
ПРИЛОЖЕНИЕ II
ПРОМЕНИ В РАЗРЕШИТЕЛНОТО ЗА ТЪРГОВИЯ, НАЛАГАЩИ НЕОБХОДИМОСТ ОТ РАЗШИРЯВАНЕ ОБХВАТА НА ЗАЯВЛЕНИЕТО ПО СМИСЪЛА НА ЧЛЕН 2
Описаните по-долу промени ще се считат за „разширяване на обхвата“ на заявлението по смисъла на член 2.
Разширяването на обхвата или промяната на съществуващо разрешително за търговия ще се разрешава от Общността.
Наименованието на лекарствения продукт за „разширяване на обхвата“ ще бъде същото като наименованието на лекарствения продукт в съществуващото разрешително за търговия.
В рамките на консултации с държавите-членки, агенцията и заинтересованите страни Комисията ще изготви и публикува подробни ръководства относно подлежащата на представяне документация.
Промени, за които се подава заявление за разширяване на обхвата
|
1. |
Промени по отношение на активното/ите вещество/а
|
|
2. |
Промeни в концентрацията, фармацевтичната форма и начина на приемане:
|
|
3. |
Други промени, които са специфични за ветеринерните лекарствени продукти, които се приемат от животни за получаване на храна
|
(1) При парентерално приемане е необходимо да се прави разлика между вътрешно артериално, интравенозно, вътрешно мускулно, подкожно и други начини. При прилагане върху птици респираторният, пероралният и очният път (небулизация) за ваксиниране се считат за еквивалентни начини на приемане.
|
13/ 39 |
BG |
Официален вестник на Европейския съюз |
235 |
32003L0063
|
L 159/46 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2003/63/EО НА КОМИСИЯТА
от 25 юни 2003 година
за изменение на Директива 2001/83/EО на Европейския парламент и на Съвета за въвеждане на кодекс на Общността относно лекарствените продукти за хуманна употреба
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 2001/83/EО на Европейския парламент и на Съвета от 6 ноември 2001 г. за въвеждане на кодекс на Общността по отношение на лекарствените продукти за хуманна употреба (1), последно изменена с Директива 2002/98/EО (2), и по-специално член 120 от нея,
като има предвид, че:
|
(1) |
Всеки лекарствен продукт за хуманна употреба, за който се планира да бъде пуснат на пазара на Европейската общност, трябва да получи разрешение за търговия, издадено от компетентна власт. Предвид получаването на разрешение за търговия, трябва да бъде представена документация за кандидастване, съдържаща данни и документи, свързани с резултатите от проведените върху дадения лекарствен продукт изпитвания и проби. |
|
(2) |
Е необходимо да се адаптират подробните научни и технически изисквания от приложение I към Директива 2001/83/ЕО, като се вземе предвид научно-техническият прогрес и по-специално обширният комплекс от нови изисквания, произтичащи от наскоро приетото законодателство. Представянето и съдържанието на документацията за кандидатстване за разрешение за търговия следва да бъдат усъвършенствани с цел улесняване на оценката и по-добро използване на определени части от документацията, които са общи за някои лекарствени продукти. |
|
(3) |
В рамките на Международната конференция по хармонизация (IСН) през 2000 г. се стигна до съгласие да се предвиди хармонизиран формат и терминология за Общия технически документ, посредством който биха могли да се постигнат хомогенна организация и представяне на документацията за кандидатстване за разрешение за търговия с лекарствени продукти за хуманна употреба. Следователно, следва незабавно да се въведат стандартизирани изисквания за заявлението за разрешение за търговия в изпълнение на Общия технически документ. |
|
(4) |
Стандартизираните изисквания за документацията за заявление за разрешение за търговия (в хармонизиран формат) следва да бъдат приложими към всякакъв тип лекарствени продукти за хуманна употреба, независимо от процедурата за даване на разрешение за търговия. Някои лекарствени продукти обаче се характеризират с такива специфични особености, че не могат да отговорят на всички изисквания. С оглед на такива особени положения следва да се предвиди представяне на опростена документация. |
|
(5) |
За безвредността на биологичните лекарствени продукти се разчита на стриктния контрол над техните изходни материали. Изискванията за годност на лицата донори и изследването на дарения изходен материал за лекарствени продукти, които се изработват въз основа на кръвната плазма, са установени в Директива 2002/98/ЕО, която определя стандарти за качество и безвредност при вземането, изследването, обработката, съхранението и разпространението на човешка кръв и кръвни съставки и изменя Директива 2001/83/ЕО. Член 109 от Директива 2001/83/ЕО е изменен. Лекарствените продукти, които се изработват въз основа на кръвната плазма, сами по себе си представляват биологични лекарствени продукти, чието производство се основава на внимателното боравене с човешката плазма като изходен материал. За да се отчете фактът, че един и същи плазмен материал в повечето случаи се използва за няколко лекарствени продукта и че в резултат на това съществена част от документацията относно разрешаването на търговията може да се окаже една и съща за голям брой други документации за съвършено различни лекарствени продукти, които се извличат от кръвната плазма, е подходящо да се установи нова система с оглед опростяването на процедурите за одобрение и последващи изменения в лекарствените продукти на основата на кръвна плазма. За тази цел следва да се въведе концепцията за основна документация за плазма (ОДП), особено с цел да се даде възможност за консолидиране на опита на национално равнище и с помощта на координирането от страна на ЕМЕА – за изработването на отделна оценка. ОДП следва да служи като самостоятелен документ, който да е отделна част от документацията за разрешение за търговия и с помощта на който да се постигне хармонизиран контрол върху съответната информация, отнасяща се до изходния материал, използван за производството на лекарствени продукти на базата на кръвна плазма. Системата на ОДП следва да се състои от двустепенно оценяване: първо, оценка на ОДП, осъществена на общностно равнище, резултатът от която, тоест сертификатът за съответствие със законодателството на Общността за всяка ОДП, следва да се вземе предвид от всички национални компетентни власти, като се вземат мерки да не се допуска каквато и да било последваща преоценка; второ, оценка на готовия лекарствен продукт на базата на кръвната плазма, съдържаща изменената част от ОДП (двете основни части на съдържанието, произход на плазмата и качество-безвредност на плазмата). Всичко това следва да остане задача на компетентните власти, които са дали разрешението за търговия с лекарствения продукт на основата на кръвната плазма. |
|
(6) |
В случай че става дума за ваксини за хуманна употреба, един и същи антиген може да се окаже общ за няколко лекарствени продукта (ваксини), при което всякакво изменение в този антиген, ipso facto, да даде отражение върху няколко ваксини, разрешени с различни процедури. С цел да се опростят съществуващите процедури за оценка на такива ваксини както за предоставяне на първоначално разрешение за търговия, така и за последващи изменения, нанесени в него вследствие на изменения в производствения процес и изпитването на отделните антигени, влизащи в състава на комбинирани ваксини, следва да се въведе нова система за основна документация за ваксинни антигени (ОДВА). Тази ОДВА ще даде възможност за консолидиране на опита на национално равнище и с помощта на координирането от страна на ЕМЕА – за изработването на отделна оценка на необходимия ваксинен антиген. ОДВА следва да служи като самостоятелен документ, който да е отделна част от документацията за разрешение за търговия и да дава цялата необходима информация от биологичния и химичния характер, свързана със специфичен антиген, който представлява едно от активните вещества в една или няколко комбинирани ваксини. |
|
(7) |
Системата от ОДВА следва да се състои от двустепенно оценяване: първо, оценка на ОДВА, осъществена на общностно равнище, резултатът от която, тоест сертификатът за съответствие със законодателството на Общността за всяка ОДВА, трябва да се отчете от всички национални компетентни власти, като се вземат мерки да не се допуска каквато и да било последваща преоценка; второ, оценка на готовия лекарствен продукт (комбинирана ваксина), съдържаща изменения антиген, което е задача на компетентните власти, които са дали разрешението за търговия с комбинираната ваксина. |
|
(8) |
Растителните лекарствени продукти съществено се различават от конвенционалните лекарствени продукти, тъй като поради присъщите си свойства се асоциират с твърде специфичното понятие за растителни вещества и растителни препарати. Поради това е подходящо да се определят специфични изисквания по отношение на тези продукти с оглед на стандартизираните изисквания за разрешение за търговия с тях. |
|
(9) |
Лечението на редица придобити и наследствени патологични функционални нарушения у хората изисква възприемането на новаторски подходи, основани на развитието на биотехнологични методи. Тези методи включват използването на лекарствени продукти за усъвършенствана терапия, основани на процеси, съсредоточени върху различни биомолекули, произведени чрез генно пренасяне (лекарствени продукти за генна терапия) и манипулирани или обработени клетки (лекарствени продукти за клетъчна терапия) като активни вещества. |
|
(10) |
Доколкото те постигат същественото си действие по метаболитен, физиологичен и имунологичен начин за възстановяване, коригиране или изменение на физиологичните функции при хората, тези нововъведени комплексни терапевтични продукти представляват нова категория биологични лекарствени продукти по смисъла на членове 1 и 2 от Директива 2001/83/ЕО. Общите принципи, които вече се прилагат спрямо тези продукти, следва да бъдат точно определени от научна и техническа гледна точка, като следва да се определят специфичните изисквания с оглед на стандартизирането на изискванията за издаване на разрешение за търговия. |
|
(11) |
Директива 2001/83/ЕО следва да бъде съответно изменена. |
|
(12) |
Мерките, предвидени в настоящата директива, са в съответствие със становището на Постоянния комитет за лекарствени продукти за хуманна употреба, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Директива 2001/83/ЕО се изменя, както следва:
|
а) |
в член 22, втори параграф изразът „част 4, буква Ж“ се заменя със следното: „Част II, точка 6“; |
|
б) |
приложение I се заменя с текста в приложението към настоящата директива. |
Член 2
Държавите-членки въвеждат в сила необходимите законови, подзаконови и административни разпоредби, за да се съобразят с настоящата директива, най-късно до 31 октомври 2003 г. Те незабавно информират Комисията за това.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Настоящата директива се прилага от 1 юли 2003 г.
Член 3
Настоящата директива влиза в сила на третия ден след публикуването ѝ в Официален вестник на Европейския съюз.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 25 юни 2003 година.
За Комисията
Erkki LIIKANEN
Член на Комисията
(1) ОВ L 311, 28.11.2001 г., стр. 67.
(2) ОВ L 33, 8.2.2003 г., стр. 30.
ПРИЛОЖЕНИЕ
Приложение I към Директива 2001/83/ЕО се заменя със следното:
„ПРИЛОЖЕНИЕ I
АНАЛИТИЧНИ, ФАРМАКОТОКСИКОЛОГИЧНИ И КЛИНИЧНИ НОРМИ И ПРОТОКОЛИ ЗА ИЗПИТВАНИЯТА НА ЛЕКАРСТВЕНИТЕ ПРОДУКТИ
СЪДЪРЖАНИЕ
Увод и общи принципи
|
Част I: |
Изисквания за стандартизирана документация за разрешение за търговия |
|
1. |
Модул 1: Административна информация |
|
1.1. |
Съдържание |
|
1.2. |
Формуляр за заявление |
|
1.3. |
Обобщение на характеристиките на продукта, етикетиране и листовка с упътвания в опаковката |
|
1.3.1. |
Обобщение на характеристиките на продукта |
|
1.3.2. |
Етикетиране и листовка с упътвания в опаковката |
|
1.3.3. |
Макети и образци |
|
1.3.4. |
Обобщения на характеристиките на продукти, вече одобрени от държавите-членки |
|
1.4. |
Информация за експертите |
|
1.5. |
Специфични изисквания към различните типове заявления |
|
1.6. |
Оценка на риска за околната среда |
|
2. |
Модул 2: Обобщения |
|
2.1. |
Общо съдържание |
|
2.2. |
Увод |
|
2.3. |
Цялостно обобщение по качеството |
|
2.4. |
Обзор на неклиничните характеристики |
|
2.5. |
Обзор на клиничните характеристики |
|
2.6. |
Обобщение на неклиничните характеристики |
|
2.7. |
Обобщение на клиничните характеристики |
|
3. |
Модул 3: Химична, фармацевтична и биологична информация относно лекарствените продукти, съдържащи химични и/или биологични активни вещества |
|
3.1. |
Формат и представяне |
|
3.2. |
Съдържание: основни принципи и изисквания |
|
3.2.1. |
Активно/и вещество/а |
|
3.2.1.1. |
Обща информация и информация относно изходните материали и суровините |
|
3.2.1.2. |
Процес на производство на активното/ите вещество/а |
|
3.2.1.3. |
Характеристика на активното/ите вещество/а |
|
3.2.1.4. |
Контрол върху активното/ите вещество/а |
|
3.2.1.5. |
Референтни стандарти или материали |
|
3.2.1.6. |
Система от съдове и запушване на съдовете за съхраняване на активното вещество |
|
3.2.1.7. |
Устойчивост на активното/ите вещество/а |
|
3.2.2. |
Готов лекарствен продукт |
|
3.2.2.1. |
Описание и състав на готовия лекарствен продукт |
|
3.2.2.2. |
Фармацевтична разработка |
|
3.2.2.3. |
Процес на производство на готовия лекарствен продукт |
|
3.2.2.4. |
Контрол върху ексципиентите |
|
3.2.2.5. |
Контрол върху готовия лекарствен продукт |
|
3.2.2.6. |
Референтни стандарти и материали |
|
3.2.2.7. |
Система съдове и запушване на съдовете за съхраняване на готовия лекарствен продукт |
|
3.2.2.8. |
Устойчивост на готовия лекарствен продукт |
|
4. |
Модул 4: Неклинични доклади |
|
4.1. |
Формат и представяне |
|
4.2. |
Съдържание: основни принципи и изисквания |
|
4.2.1. |
Фармакология |
|
4.2.2. |
Фармакокинетика |
|
4.2.3. |
Токсикология |
|
5. |
Модул 5: Доклади от клиничните изследвания |
|
5.1. |
Формат и представяне |
|
5.2. |
Съдържание: основни принципи и изисквания |
|
5.2.1. |
Доклади от биофармацевтичните изследвания |
|
5.2.2. |
Доклади от изследванията, свързани с фармакокинетиката с използване на човешки биологични материали |
|
5.2.3. |
Доклади от хуманните фармакокинетични изследвания |
|
5.2.4. |
Доклади от хуманните фармакодинамични изследвания |
|
5.2.5. |
Доклади от изследванията на ефикасността и безвредността |
|
5.2.5.1. |
Доклади от контролираните клинични изследвания, отнасящи се до представеното показание |
|
5.2.5.2. |
Доклади от изследванията върху докладите от неконтролираните клинични изследвания на анализите на данните от повече от едно изследване и други доклади от клинични изследвания |
|
5.2.6. |
Доклади от опита, придобит след получаване на разрешение за търговия |
|
5.2.7. |
Формуляри за доклади по отделни случаи и списъци с индивидуални пациенти |
|
Част II: |
Специфична документация за разрешение за търговия и изисквания към нея |
|
1. |
Лекарствени продукти с утвърдено лечебно приложение |
|
2. |
Сходни по същество лекарствени продукти |
|
3. |
Допълнителни данни, изисквани в специфични ситуации |
|
4. |
Сходни биологични лекарствени продукти |
|
5. |
Лекарствени продукти с установени комбинации |
|
6. |
Документация за заявления при изключителни обстоятелства |
|
7. |
Комбинирани заявления за разрешения за търговия |
|
Част III: |
Специални лекарствени продукти |
|
1. |
Биологични лекарствени продукти |
|
1.1. |
Лекарствени продукти на базата на кръвната плазма |
|
1.2. |
Ваксини |
|
2. |
Радиофармацевтици и прекурсори |
|
2.1. |
Радиофармацевтици |
|
2.2. |
Радиофармацевтични прекурсори за целите на радиоактивната маркировка |
|
3. |
Хомеопатични лекарствени продукти |
|
4. |
Растителни лекарствени продукти |
|
5. |
Лекарствени продукти сираци |
|
Част IV: |
Лекарствени продукти за усъвършенствана терапия |
|
1. |
Лекарствени продукти за генна терапия (хуманни и ксеногенни) |
|
1.1. |
Разнообразие на лекарствените продукти за генна терапия |
|
1.2. |
Специфични изисквания във връзка с модул 3 |
|
2. |
Лекарствени продукти за терапия със соматични клетки (хуманни и ксеногенни) |
|
3. |
Специфични изисквания към лекарствените продукти за генна терапия и терапия със соматични клетки (хуманни и ксеногенни) във връзка с модули 4 и 5 |
|
3.1. |
Модул 4 |
|
3.2. |
Модул 5 |
|
3.2.1. |
Хуманна фармакология и изследвания върху ефикасността ѝ |
|
3.2.2. |
Безвредност |
|
4. |
Специално становище относно лекарствените продукти за ксенотрансплантация |
Увод и общи принципи
(1) Данните и документите, придружаващи заявлението за разрешение за търговия съгласно член 8 и член 10, параграф 1 се представят в съответствие с изискванията, формулирани в настоящото приложение и следват ръководствата, публикувани от Комисията в Правилника за лекарствените продукти в Европейската общност, том 2 Б, Препоръка към заявителите, Лекарствени продукти за хуманна употреба, Представяне и съдържание на документацията, Общ технически документ (ОТД).
(2) Данните и документите се представят в пет модула: в модул 1 се представят конкретните административни данни за Европейската общност; в модул 2 се представят обобщенията по качеството, неклиничните и клиничните обобщения; в модул 3 се представя химична, фармацевтична и биологична информация; в модул 4 се представят неклиничните доклади и в модул 5 се представят клиничните доклади. Това представяне се изпълнява на общ за всички региони на IСН (1) (Европейската общност, Съединените американски щати, Япония). Тези пет модула се представят в строго съответствие с формата, съдържанието и системата за номериране, подробно представени в том 2 Б от Бележката към заявителите, посочена по-горе.
(3) Представянето на ОТД на Европейската общност е приложимо за всички видове заявления за разрешение за търговия независимо от приложимата процедура (тоест централизирана, на взаимно признаване или национална) и от това, дали те се основават на пълно или съкратено заявление. Представянето е приложимо също и за всички типове продукти, включително новите химични образувания (НХО), радиофармацевтиците, производните от кръвната плазма, ваксините, растителните лекарствени продукти и т.н.
(4) При съставянето на документацията за кандидатстване за разрешение за търговия заявителите вземат предвид също и научните ръководства по отношение на качеството, безвредността и ефикасността на лекарствените продукти за хуманна употреба, както са приети от Комитета за патентованите лекарствени продукти (СРМР) и публикувани от Европейската агенция за оценка на лекарствата (ЕМЕА), както и останалите фармацевтични ръководства на Общността, публикувани от Комисията в различни томове на Правилника за лекарствените продукти в Европейската общност.
(5) По отношение на частта от документацията, която се отнася до качествата (химични, фармацевтични и биологични), са приложими всички монографии, включително общите монографии и общите глави от Европейската фармакопея.
(6) Производственият процес следва да съответства на изискванията на Директива 91/356/ЕИО на Комисията за установяване на принципите и ръководствата на добрата производствена практика (ДПП) за лекарствените продукти за хуманна употреба (2) и на принципите и ръководствата на ДПП, публикувани от Комисията в Правилника за лекарствените продукти в Европейската общност, том 4.
(7) Цялата информация, отнасяща се до оценката на въпросните лекарствени продукти, се включва в заявлението, независимо дали е благоприятна или неблагоприятна за продукта. По-специално се представят всички подробности относно всякакви незавършени или прекратени фармакотоксикологични или клинични изпитвания и опити, проведени във връзка с лекарствения продукт, и/или завършени опити, отнасящи се до терапевтичните показания, които не са включени в заявлението.
(8) Всички клинични опити, проведени на територията на Европейската общност, следва да съответстват на изискванията на Директива 2001/20/ЕО на Европейския парламент и на Съвета за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки относно прилагането на добрата клинична практика при провеждането на клинични изпитвания на лекарствени продукти за хуманна употреба (3). За да бъдат взети предвид при оценяването на дадено заявление, клиничните изследвания, проведени извън границите на Европейската общност и имащи отношение към лекарствените продукти, предназначени за използване в Европейската общност, се проектират, изпълняват и докладват, като се отчитат добрата клинична практика и етичните принципи въз основа на принципите, които съответстват на заложените в разпоредбите на Директива 2001/20/ЕО. Те се провеждат в съответствие с етичните принципи, отразени например в Декларацията от Хелзинки.
(9) Неклиничните (фармакотоксикологичните) изследвания се провеждат в съответствие с разпоредбите относно добрата лабораторна практика, установени в Директива 87/18/ЕИО на Съвета за хармонизиране на законовите, подзаконовите и административните разпоредби, отнасящи се до прилагането на принципите на добрата лабораторна практика и проверката на тяхното прилагане при анализите на химични вещества (4) и Директива 88/320/ЕИО на Съвета относно инспекцията и проверките на добрата лабораторна практика (ДЛП) (5).
(10) Държавите-членки обезпечават провеждането на всички изпитвания върху животни в съответствие с Директива 86/609/ЕИО на Съвета от 24 ноември 1986 г. за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки по отношение на защитата на животните, използвани за експериментални и други научни цели.
(11) За целите на мониторинга на оценката риск—полза на компетентните власти се представя всяка нова информация извън първоначалното заявление, както и цялата информация, свързана с фармацевтичната бдителност. След издаването на разрешение за търговия всяко изменение на данните в документацията се представя на компетентните власти съгласно изискванията на Регламент (ЕО) № 1084/2003 на Комисията (6) и Регламент (ЕО) № 1085/2003 на Комисията (7) или ако е релевантно към въпроса, в съответствие с националните разпоредби, както и с изискванията на том 9 от публикацията на Комисията Правила за лекарствените продукти в Европейската общност.
Настоящото приложение е разделено на четири различни части:
|
— |
В част I са описани форматът на заявлението, обобщението на характеристиките на продукта, етикетирането и листовката с упътвания в опаковката, както и изискванията за стандартните заявления (модули от 1 до 5). |
|
— |
В част II е уредена дерогацията за „Специалните заявления“, тоест утвърдени приложения на лекарствените продукти, сходни по същество продукти, установени комбинации, сходни биологични продукти, извънредни обстоятелства, както и смесени заявления (с библиографска част и част, посветена на собствените изследвания). |
|
— |
В част III са разгледани „Особените изисквания към заявлението“ по отношение на биологичните лекарствени продукти (основна документация за плазмата, основна документация за ваксинни антигени), радиофармацевтици, хомеопатични лекарствени продукти, растителни лекарствени продукти и лекарства сираци. |
|
— |
В част IV са разгледани „Лекарствените продукти за усъвършенствана терапия“ и специфичните изисквания към лекарствените продукти за генна терапия (с използване на човешката автологична, алогенна или ксеногенна система) и лекарствените продукти за клетъчна терапия с човешки и животински произход, както и лекарствените продукти за ксеногенна трансплантация. |
ЧАСТ I
ИЗИСКВАНИЯ ЗА СТАНДАРТИЗИРАНА ДОКУМЕНТАЦИЯ ЗА РАЗРЕШЕНИЕ ЗА ТЪРГОВИЯ
1. МОДУЛ 1: АДМИНИСТРАТИВНА ИНФОРМАЦИЯ
1.1. Съдържание
Представя се пълното съдържание на модули от 1 до 5 от документацията, представена за разрешение за търговия.
1.2. Формуляр за заявление
Лекарственият продукт, който представлява предмет на заявлението, се идентифицира по собственото си наименование и наименованието на активното/ите вещество/а, заедно с фармацевтичната си форма, начина на приложение, концентрацията и окончателния вид, включително опаковката.
Подават се името и адресът на заявителя заедно с името и адреса на производителите, както и обектите, участващи на различните етапи от производството (включително производителя на крайния продукт и производителите на активното/ите вещество/а, а където е приложимо – и името и адресът на вносителя).
Заявителят определя какъв тип заявление ще подава и посочва какви образци са включени, ако има и такива.
Към административните данни се прилагат копия от разрешението за производство, както е определено в член 40, заедно със списък на страните, в които е получено такова разрешение, копия от всички обобщения на характеристиките на продукта в съответствие с член 11, както е одобрено от държавите-членки, както и списък на страните, в които е представено заявление за разрешение.
Както е подчертано във формуляра за заявление, заявителите представят, inter alia, подробности за лекарствените продукти, които са предмет на заявлението, правната основа за заявлението, предложените за разрешение за търговия титуляр и производител/и, информация относно статута на лекарствата сираци, научна консултация и програма за педиатрична разработка.
1.3. Обобщение на характеристиките на продукта, етикетиране и листовка с упътвания в опаковката
1.3.1. Обобщение на характеристиките на продукта
Заявителят представя обобщение на характеристиките на продукта в съответствие с член 11.
1.3.2. Етикетиране и листовка с упътвания в опаковката
Представя се предложение за текста за етикет за първичната и външната опаковка, заедно с предложение за листовка с упътвания в опаковката. Те отговарят на всички задължителни условия, изброени в дял V относно етикетите на лекарствените продукти за хуманна употреба (член 63) и листовка с упътвания в опаковката (член 59).
1.3.3. Макети и образци
Заявителят представя образец и/или макети на първичната и външната опаковка и за листовката с упътвания в опаковката на дадения лекарствен продукт.
1.3.4. Обобщения на характеристиките на продуктите, вече одобрени от държавите-членки
Към административните данни се прилагат копия от всички обобщения на характеристиките на продукта, както е определено в членове 11 и 21, одобрени от държавите-членки, където е приложимо, заедно със списък на страните, в които е представено заявление за разрешение.
1.4. Информация за експертите
В съответствие с член 12, параграф 2 експертите трябва да представят подробни доклади от своите наблюдения върху документите и данните, които представляват документацията за разрешение за търговия, и в частност модули 3, 4 и 5 (химична, фармацевтична и биологична документация и съответно неклинична и клинична документация). От експертите се изисква да разгледат критичните точки, свързани с качеството на лекарствения продукт и проведените върху животни и хора изследвания, като съберат и представят всички данни, имащи значение за оценката.
За изпълнението на тези изисквания се представя цялостно обобщение за качеството, неклиничен обзор (данни от проведени върху животни изследвания), както и клиничен обзор, който се включва в модул 2 на документацията за заявлението за разрешение за търговия. В модул 1 се представя декларация, подписана от експертите, заедно с кратка автобиографична справка за тяхното образование, обучение и стаж по професията. Експертите имат подходяща техническа или професионална квалификация. Декларира се и професионалната връзка между експерта и заявителя.
1.5. Специфични изисквания към различните типове заявления
Специфичните изисквания към различните типове заявления са разгледани в част II от настоящото приложение.
1.6. Оценка на риска за околната среда
Когато е приложимо, заявленията за разрешения за търговия включват преглед на оценката на риска, в която се дава преценка относно възможните рискове за околната среда вследствие на използването и/или изхвърлянето на лекарствения продукт, както и предложения за подходящи разпоредби за етикетирането. Разглежда се рискът за околната среда, свързан с пускането на лекарствени продукти, съдържащи или състоящи се от генетично модифицирани организми (ГМО) по смисъла на член 2 от Директива 2001/18/ЕО на Европейския парламент и на Съвета от 12 март 2001 г. относно съзнателното освобождаване в околната среда на генетично модифицирани организми и за отмяна на Директива 90/220/ЕИО на Съвета (8).
Информацията, отнасяща се до риска за околната среда, се представя като допълнение към модул 1.
Информацията се представя съгласно разпоредбите на Директива 2001/18/ЕО, като се вземат предвид документите с ръководства, публикувани от Комисията във връзка с изпълнението на посочената директива.
Информацията се състои от:
|
— |
уводна част; |
|
— |
копие от писменото/ите съгласие/я за съзнателно освобождаване на ГМО в околната среда с научноизследователски и развойни цели съгласно част Б от Директива 2001/18/ЕО; |
|
— |
информацията, която се изисква съгласно приложения от II до IV към Директива 2001/18/ЕО, включително методи за откриване и идентифициране, както и уникалния код на ГМО, плюс всякаква допълнителна информация по отношение на ГМО или продукт, имащ значение за оценката на риска за околната среда. |
|
— |
доклад за оценката на риска за околната среда (ОРОС), изготвен на базата на информацията, посочена в приложения III и IV към Директива 2001/18/ЕО и в съответствие с приложение II към Директива 2001/18/ЕО; |
|
— |
като се вземе предвид горепосочената информация, както и ОРОС, заключение, което предлага подходяща стратегия за управление на риска, която, доколкото е свързана с ГМО и въпросния продукт, включва план за наблюдение след издаването на разрешение за търговия и определянето на всякакви специални подробности, които е необходимо да се включат в обобщението на характеристиките на продукта, в етикетирането и листовката с упътвания в опаковката; |
|
— |
подходящи мерки за информиране на обществеността. |
Включват се също подпис и печат на автора, сведения за образованието, обучението и стажа му по професията, както и декларация за отношенията между автора и заявителя.
2. МОДУЛ 2: ОБОБЩЕНИЯ
Този модул има за цел да направи обобщение на химичните, фармацевтичните и биологичните данни, неклиничните данни и клиничните данни, представени в модули 3, 4 и 5 от документацията за разрешение за търговия, както и да представи докладите/прегледите, описани в член 12 от настоящата директива.
Разглеждат се и се анализират критичните точки. Представят се обобщенията с фактите, заедно с табличните формати. В тези доклади се включват препратки към табличните формати или към информацията, която се съдържа в главната документация, представена в модул 3 (химична, фармацевтична и биологична документация), модул 4 (неклинична документация) и модул 5 (клинична документация).
Информацията, която се съдържа в модул 2, се представя в съответствие с формата, съдържанието и системата за номериране, посочени в том 2 от Бележката към заявителите. Обзорите и обобщенията отговарят на основните принципи и изисквания, установени по-долу в настоящия документ:
2.1. Общо съдържание
Модул 2 включва съдържание на научната документация, представена в модули от 2 до 5.
2.2. Увод
Представя се информация за фармакологичния клас, начина на действие и предлаганото клинично приложение на продукта, за който се иска разрешение за търговия.
2.3. Цялостно обобщение за качеството
В цялостното обобщение за качеството се представя преглед на информацията, свързана с химичните, фармацевтичните и биологичните данни.
Набляга се на ключовите критични параметри и въпроси, свързани с аспектите на качеството, а в случаите, в които не са били следвани съответните ръководства, се представя и съответната аргументация. Този документ следва обхвата и профила на съответните подробни данни, представени в модул 3.
2.4. Обзор на неклиничните характеристики
Изисква се пълна и критична оценка на неклиничната оценка на лекарствения продукт при животните/ин витро. Включват се и обсъждане и аргументация на стратегията за изпитванията и за отклоненията от съответните ръководства.
С изключение на биологичните лекарствени продукти се включва оценка на примесите и продуктите, които се получават при разпадането, заедно с тяхното потенциално фармакологично и токсикологично въздействие. Обсъждат се естествените последици от всякакви разлики в хиралността, химичната форма и вида примеси между съединението, което е използвано при неклиничните изследвания, и продукта, който ще бъде търгуван.
Относно биологичните лекарствени продукти се прави оценка на сравнимостта на материала, използван в неклиничните изследвания, и лекарствения продукт, предназначен за търговия.
Всички нови ексципиенти са предмет на специална оценка по отношение на безвредността.
Характеристиките на лекарствения продукт се определят така, както е посочено в неклиничните изследвания, и се обсъждат последиците от изводите по отношение на безвредността на лекарствения продукт за предвиденото клинично приложение в хуманната медицина.
2.5. Обзор на клиничните характеристики
Клиничният преглед има за цел да представи критичен анализ на клиничните данни, включени в клиничното обобщение в модул 5. Представя се подходът към клиничната разработка на лекарствения продукт, с включен проект за критично изследване, свързаните с него решения, както и реализирането на изследванията.
Представя се кратък преглед на клиничните заключения, като се включват и важните ограничения, както и оценка за ползите и рисковете, която да се основава на изводите от клиничните изследвания. Изисква се и излагане на интерпретация относно начина, по който заключенията по ефикасността и безвредността подкрепят предложената доза и търсените показания, както и оценка на начина, по който обобщението на характеристиките на продукта и останалите подходи ще оптимизират ползите и ще избегнат рисковете.
Разясняват се въпросите относно ефикасността и безвредността, които се срещат в разработката и остават неразрешени.
2.6. Обобщение на неклиничните характеристики
Резултатите от фармакологичните, фармакокинетичните и токсикологичните изследвания, проведени върху животни/ин витро, се представят като фактология в писмени и таблични обобщения, които се излагат в следния ред:
|
— |
Увод |
|
— |
Писмено фармакологично обобщение |
|
— |
Таблично фармакологичното обобщение |
|
— |
Писмено фармакокинетично обобщение |
|
— |
Таблично фармакокинетично обобщение |
|
— |
Писмено токсикологично обобщение |
|
— |
Таблично токсикологично обобщение |
2.7. Обобщение на клиничните характеристики
Представя се подробно фактологично обобщение на клиничната информация относно лекарствения продукт, включена в модул 5. Обобщението включва резултатите от биофармацевтичните изследвания, от клиничните фармакологични изследвания, както и от изследванията по клиничната ефикасност и безвредност. Изисква се и кратък обзор на отделните изследвания.
Резюмираната клинична информация се представя, като се спазва следният ред:
|
— |
Обобщение по биофармацевтиката и свързаните с нея аналитични методи |
|
— |
Обобщение на клиничните фармакологични изследвания |
|
— |
Обобщение по клиничната ефикасност |
|
— |
Обобщение по клиничната безвредност |
|
— |
Кратък обзор на отделните изследвания |
3. МОДУЛ 3: ХИМИЧНА, ФАРМАЦЕВТИЧНА И БИОЛОГИЧНА ИНФОРМАЦИЯ ОТНОСНО ЛЕКАРСТВЕНИТЕ ПРОДУКТИ, СЪДЪРЖАЩИ ХИМИЧНИ И/ИЛИ БИОЛОГИЧНИ АКТИВНИ ВЕЩЕСТВА
3.1. Формат и представяне
Общият профил на модул 3 е следният:
|
— |
Съдържание |
|
— |
Изложение на данните
|
|
— |
Библиографска справка |
3.2. Съдържание: основни принципи и изисквания
(1) Химичните, фармацевтичните и биологичните данни, които се представят, включват по отношение на активното/ите вещество/а и по отношение на готовия лекарствен продукт всички необходими сведения относно: разработването, производствения процес, характеристиките и свойствата, операциите и изискванията по контрола на качеството и устойчивостта, както и описание на състава и представяне на готовия лекарствения продукт.
(2) Представят се два основни комплекта документация – относно активното/ите вещество/а и съответно относно готовия лекарствен продукт.
(3) Освен това в настоящия модул се представя подробна информация относно изходните материали и суровините, използвани по време на операциите по производството на активното/ите вещество/а и на ексципиентите, включени във формулата на готовия лекарствен продукт.
(4) Всички процедури и методи, използвани за производството и контрола на активното вещество и готовия лекарствен продукт, се описват с достатъчно подробности, така че да се даде възможност те да бъдат повторени по време на контролните изпитвания, провеждани по искане на компетентните власти. Всички изпитателни процедури отговарят на състоянието на научния прогрес към момента на провеждането им и подлежат на утвърждаване. Представят се и резултатите от изследвания по утвърждаването. При изпитателните процедури, включени в Европейската фармакопея, това описание се замества с подходяща подробна библиографска справка с препратки към монографията/ите и общите глави.
(5) Монографиите от Европейската фармакопея са приложими към всички вещества, препарати и фармацевтични форми, които са включени в нея. По отношение на останалите вещества всяка държава-членка може да изисква да се спазва собствената ѝ фармакопея.
Въпреки това там, където даден материал в Европейската фармакопея или във фармакопеята на държава-членка е изготвен по метод, при който е възможно да се оставят примесите, които не се контролират в монографията по фармакопеята, тези примеси и максимално допустимите им равнища трябва да се декларират и трябва да се опише подходяща процедура за изпитване. В случаите, когато дадена спецификация, която е включена в монография от Европейската фармакопея или във фармакопеята на съответната държава-членка, може да се окаже недостатъчна за осугуряването на качеството на веществото, компетентните власти могат да поискат от титуляра на разрешението за търговия по-подходящи спецификации. Компетентните власти уведомяват властите, от чиято компетентност е въпросната фармакопея. Титулярът на разрешението за търговия представя данни по евентуално недостатъчните сведения, както и допълнително представените спецификации на органите, от чиято компетентност е въпросната фармакопея.
Когато в Европейската фармакопея са включени аналитични процедури, това описание се замества във всеки съответен раздел с подходяща подробна библиографска справка с препратки към монографията/ите и общата/ите глава/и.
(6) В случаите, когато изходните материали, суровините, активното/ите вещество/а или ексципиентите не са описани нито в Европейската фармакопея, нито във фармакопеята на държава-членка, може да се приеме съответствие с монография от фармакопеята на трета страна. В такива случаи заявителят представя копие от монографията, придружено от потвърждение относно процедурите за анализ на данните, съдържащи се в монографията, както и от превод, където това е необходимо.
(7) Когато изходните материали, суровините, активното/ите вещество/а или инертните пълнители представлява предмет на монография от Европейската фармакопея, заявителят може да кандидатства за сертификат за годност, който, след като бъде издаден от Европейската дирекция по качеството на медикаментите, се представя в съответния раздел на настоящия модул. За тези сертификати за годност съгласно монография от Европейската фармакопея се счита, че заместват съответните данни от съответните секции, описани в настоящия модул. Производителят представя на заявителя писмено уверение, че производственият процес не е бил подлаган на изменения от момента на издаването на сертификата за годност от Европейската дирекция по качеството на медикаментите.
(8) С цел активното вещество да се дефинира по-добре, производителят или заявителят може да организира следното:
|
i) |
подробно описание на производствения процес, |
|
ii) |
качествен контрол по време на производство, и |
|
iii) |
утвърждаване на процеса, |
което да бъде подадено от производителя на активното вещество като отделен документ направо до компетентните власти във вид на основна документация на активното вещество.
В такъв случай обаче производителят снабдява заявителя с всички данни, които могат да се окажат необходими на последния при поемане на отговорност за лекарствения продукт. Производителят потвърждава писмено на заявителя, че гарантира съгласуваността на качеството на отделните партиди и че няма да променя производствения процес или спецификациите, без да уведоми заявителя за това свое действие. Документите и данните в подкрепа на заявлението за такава промяна се представят на компетентните власти; тези документи и данни се представят също и на заявителя, когато те засягат откритата част от основната документация на активното вещество.
(9) Специалните мерки, засягащи предотвратяването на пренасяне на спонгиформни енцефалопатии (материали с произход от преживно животно): на всяка стъпка от производствения процес заявителят трябва да демонстрира съответствието на използваните материали с Ръководството относно минимизиране на риска от пренасяне на спонгиформната енцефалопатия по животните чрез лекарствени продукти и нейните актуализирани версии, публикувани от Комисията в Официален вестник на Европейския съюз. Демонстрирането на съответствие с гореспоменатото ръководство може да се извърши или като се представи (за предпочитане) сертификат за годност по отношение на съответната монография от Европейската фармакопея, издаден от Европейската дирекция по качеството на медикаментите, или като се предоставят научни данни, които да аргументират такова съответствие.
(10) По отношение на случайно привнесените вещества се представя информация, в която да се дава оценка на риска с оглед на потенциалното замърсяване със случайно привнесени вещества, независимо дали същите са с вирусен или невирусен характер така, както е формулирано в съответните ръководства, както и в съответната обща монография и обща глава от Европейската фармакопея.
(11) Цялата специална апаратура и съоръжения, които могат да се ползват на който и да е етап от производствения процес и операциите по контрол над производството на лекарствения продукт, се описват подробно и по подходящ начин.
(12) Там, където е приложимо и необходимо, върху медицинската апаратура се поставя маркировка „СЕ“ съгласно изискванията на законодателството на Общността.
Специално внимание се отделя на следните избрани елементи:
3.2.1. Активно/и вещество/а
3.2.1.1. Обща информация и информация относно изходните материали и суровините
|
a) |
Информацията относно номенклатурата на активното вещество се представя, като се включат препоръчителните Международно непатентовано наименование (INN), наименование по Европейската фармакопея, ако е приложимо, химическо/и наименование/я. Представят се също и структурната формула, включително относителната и абсолютната стереохимична формула, молекулната формула и относителната молекулна маса. Ако е подходящо, за биотехнологичните лекарствени продукти се представя схематичната аминокиселинна последователност и относителната молекулярна маса. Представя се списък на физикохимичните и съответно другите свойства на активното вещество, включително биологичната активност при биологичните лекарствени продукти. |
|
б) |
За целите на настоящото приложение под „изходен материал“ се разбира всяко вещество, от което се произвежда или извлича активно вещество. За биологичните лекарствени продукти под „изходен материал“ се разбира всяко вещество с биологичен произход, като например микроорганизми, органи и тъкани както с растителен, така и с животински произход, клетки или течности (включително кръв и плазма) с хуманен или животински произход, както и биотехнологични клетъчни конструкции (клетъчни субстрати, рекомбинирани или нерекомбинирани, включително първични клетки Биологичният лекарствен продукт е продукт, чието активно вещество е биологично вещество. Биологичното вещество е вещество, което се произвежда или извлича от биологичен източник и за чиято характеристика и за определянето на чието качество се провежда комбинация от физико—химико—биологични изпитвания, заедно с производствения процес и контрола върху него. За биологични лекарствени продукти се считат следните: имунологични лекарствени продукти и лекарствени продукти – производни от човешката кръв и човешката кръвна плазма, определени в член 1, параграфи 4 и 10; лекарствените продукти, обхванати от част А от приложението към Регламент (ЕИО) № 2309/93; лекарствените продукти за усъвършенствана терапия, както са определени в част IV от настоящото приложение. Всички останали вещества, които се използват за производство или извличане на активно/и вещество/а, но от които това активно вещество не е пряко производно, като например реагентите, средите за клетъчните култури, серум от телешки ембриони, адитиви и буфери, които се използват в хроматографията и т.н., са известни като суровини. |
3.2.1.2. Процес на производство на активното/ите вещество/а
|
а) |
Описанието на производствения процес на активното/ите вещество/а представлява ангажиментът, който се поема от заявителя по отношение на производството. За да се опишат адекватно производственият процес и управлението на този процес, следва да се представи подходяща информация съгласно ръководствата, публикувани от агенцията. |
|
б) |
Всички материали, необходими за целите на производството на активно/и вещество/а, се представят във вид на списък, като се определя мястото на всеки материал в производствения процес. Представя се информация относно качеството и контрола върху тези материали. Представя се и информация, показваща, че материалите отговарят на стандартите, съответстващи на тяхното планирано приложение. Представя се списък на суровините, както и документация относно качеството и контрола на тези материали. Посочват се също и името, адресът и отговорността на всеки производител, включително подизпълнителите, както и всички производствени мощности, включени в производствения процес и изпитванията. |
|
в) |
Относно биологичните лекарствени продукти се прилагат следните допълнителни изисквания: Описват се и се документират произходът и историята на изходните материали. Относно специалните мерки за предотвратяване пренасянето на спонгиформни енцефалопатии по животните, заявителят следва да покаже, че активното/ите вещество/а отговаря на Препоръката относно минимизиране на риска от пренасяне на спонгиформната енцефалопатия по животните чрез лекарствени продукти и нейните актуализирани версии, публикувани от Комисията в Официален вестник на Европейския съюз. Когато се използват клетъчни банки, за клетъчните характеристики се показва, че не са претърпели изменения при преходното ниво, използвано за производството и по-нататък. Семенните материали, клетъчните банки, контейнерите с плазма или серум и там, където е възможно, материалите, на които те са производни, се подлагат на изпитване за случайно привнесени вещества. Ако присъствието на потенциално патогенни случайно привнесени вещества е неизбежно, съответният материал се използва само след като претърпи допълнителна обработка, която да гарантира елиминирането и/или дезактивирането им, което подлежи на потвърждаване. Когато това е възможно, производството на ваксини се основава на система от серии семена и на установени клетъчни банки. Що се отнася до бактерийните и вирусните ваксини, характеристиките на инфектиращия агент се демонстрират върху семената. Освен това устойчивостта на характеристиките на отслабване при живите ваксини се демонстрират върху семената; ако това доказателство не се окаже достатъчно, характеристиките на отслабване се демонстрират също и на етап производство. За лекарствените продукти, производни от човешка кръв или кръвна плазма, в съответствие с разпоредбите, формулирани в част III от настоящото приложение, се описват и се документират произходът и критериите за събиране, транспортиране и съхраняване на изходния материал. Описват се производствените мощности и съоръжения. |
|
г) |
Изпитванията и критериите за приемане на всеки важен етап, сведенията относно качеството и контрола върху междинните вещества, както и процесът на даване на потвърждение и/или оценка се осъществяват по подходящ начин. |
|
д) |
Ако присъствието на потенциално патогенни случайно привнесени вещества е неизбежно, съответният материал се използва само след като претърпи допълнителна обработка, която да гарантира елиминирането и/или дезактивирането им, за което се получава и потвърждение в раздела, отнасящ се до оценката на вирусната безвредност. |
|
е) |
Представя се описание и обсъждане на по-важните изменения по отношение на производствения процес при разработката и/или на обекта, в който се произвежда активното/ите вещество/а. |
3.2.1.3. Характеристика на активното/ите вещество/а
Представят се данни, разясняващи структурата и останалите характеристики на активното/ите вещество/а.
Представя се потвърждение по отношение на структурата на активното/ите вещество/а въз основа на физикохимични и/или имунологично-химични и/или биологични методи, както и информация относно примесите.
3.2.1.4. Контрол върху активното/ите вещество/а
Представя се подробна информация по спецификациите, използвани за рутинен контрол на активното/ите вещество/а, аргументация в полза на избора на тези спецификации, методи на анализ и тяхното потвърждение.
Представят се резултатите от осъществените проверки върху отделни партиди, произведени по време на разработването.
3.2.1.5. Референтни стандарти или материали
Референтните препарати и стандарти се определят и описват подробно. Когато е необходимо, се използват справочни химични и биологични материали от Европейската фармакопея.
3.2.1.6. Система от съдове и запушване на съдовете за съхраняване на активното/ите вещество/а
Представя се описание на системата от съдове и запушване на съдовете за съхраняване на активното/ите вещество/а.
3.2.1.7. Устойчивост на активното/ите вещество/а
|
а) |
Резюмират се видовете проведени изследвания, използваните протоколи и резултатите от изследванията. |
|
б) |
В подходящ формат се представят подробните резултати от изследванията по устойчивостта, включително сведенията от процедурите за анализ на данните, използвани за генериране на данни, както и утвърждаването на тези процедури. |
|
в) |
Представя се протокол по устойчивостта след издаване на разрешението, както и ангажимент по отношение на устойчивостта. |
3.2.2. Готов лекарствен продукт
3.2.2.1. Описание и състав на готовия лекарствен продукт
Представят се описание на готовия лекарствен продукт и неговия състав. Сведенията включват описание на фармацевтичната форма и състава на всички съставки на готовия лекарствен продукт, тяхното количество на база единица, както и функциите на съставките на:
|
— |
активното/ите вещество/а |
|
— |
съставката/ите на ексципиентите, независимо от тяхното естество и използвано количество, включително оцветителите, консервантите, спомагателните вещества, стабилизиращите вещества, сгъстителите, емулгаторите, овкусителите и ароматизиращите вещества и др., |
|
— |
съставките, предназначени за поглъщане или прилагане по друг начин на пациента, от външния слой покритие на лекарствените продукти (твърди капсули, меки капсули, капсули с анално приложение, филм—таблети и др.), |
|
— |
тези подробности се придружават от всякакви необходими данни, засягащи вида съд и където това е необходимо, начина на запушване на тези съдове, заедно с подробности за механизмите, с които лекарственият продукт ще се използва или прилага и които ще придружават самия лекарствен продукт. |
„Обичайната терминология“, която трябва да се използва за описание на съставките на лекарствените продукти, независимо от приложението на останалите разпоредби на член 8, параграф 3, буква в), означава:
|
— |
по отношение на веществата, включени в Европейската фармакопея, или ако не са включени, във фармакопеята на някоя от държавите-членки, основното наименование в заглавието на въпросната монография с препратка към посочената фармакопея, |
|
— |
по отношение на останалите вещества, международните непатентовани наименования (INN), препоръчани от Международната здравна организация, или ако не е възможно – точното научно обозначение; веществата, които нямат международни нетърговски имена (INN) или точно научно обозначение, се описват в справка, в която се включва начинът на тяхното приготвяне, от какво са приготвени, какви добавки са включени и ако е необходимо – други важни подробности, |
|
— |
по отношение на оцветителите – обозначение с код „Е“, дадено им в Директива 78/25/ЕИО на Съвета от 12 декември 1977 г. за сближаване на правилата на държавите-членки по отношение на оцветителите, разрешени за използване в лекарствените продукти (9) и/или Директива 94/36/ЕО на Европейския парламент и на Съвета от 30 юни 1994 г. относно цветовете за използване в хранителни продукти (10). |
С цел представяне на „количествения състав“ на активното/ите вещество/а на готовите лекарствени продукти е необходимо, в зависимост от фармацевтичната форма, за всяко активно вещество да се посочат: масата или броят единици биологична активност, или единици за доза, или за единица маса, или за единица обем.
Активнотите вещества, представени под формата на съединения или производни, се обозначава/т количествено чрез общата му/им маса и ако е необходимо или важно – чрез масата активно вещество или вещества на молекулата.
За лекарствените продукти, съдържащи активно вещество, което е предмет на заявление за разрешение за търговия на територията на която и да е от държавите-членки за първи път, количествената справка за активното вещество, което представлява сол или хидрат, е изразена систематизирано чрез масата на активното вещество или вещества на молекулата. Всички лекарствени продукти, получили впоследствие разрешение в държавите-членки, разполагат със справка, в която количественият състав за едно и също активно вещество е отразен по един и същи начин.
Единиците за биологична активност се използват за вещества, които не могат да се дефинират молекулярно. Когато дадена международна единица за биологична активност е определена от Международната здравна организация, се използва именно тази единица. Когато дадена международна единица за биологична активност не е определена, единиците за биологична активност се изразяват по такъв начин, че да представят недвусмислена информация относно активността на веществата посредством използване, където е приложимо, на единиците от Европейската фармакопея.
3.2.2.2. Фармацевтична разработка
Тази глава се посвещава на информацията относно развойните изследвания, които се провеждат с цел да се установи, че формата на дозировката, формулата, производственият процес, системата за запушване на съдовете, микробиологичните свойства и указанията за приложение са подходящи за планираното приложение, указано в документацията на заявлението за разрешение за търговия.
Изследванията, описани в настоящата глава, се различават от обичайните контролни изпитвания, които се провеждат съгласно спецификациите. Определят се и се описват най-важните параметри на формулата и характерните свойства на процеса, които е възможно да повлияят на серийната възпроизводимост, действието на лекарствения продукт и качеството на лекарствения продукт. Към допълнителните подкрепящи данни, когато това е подходящо, се дават препратки към съответните глави от модул 4 (Доклади от неклиничните изследвания) и модул 5 (Доклади от клиничните изследвания) от документацията на заявлението за разрешение за търговия.
|
а) |
Документира се съвместимостта на активното вещество с инертните пълнители, както и ключовите физикохимични характеристики на активното вещество, които могат да повлияят на действието на готовия лекарствен продукт или на съвместимостта на различните активни вещества в случаите на комбинираните продукти. |
|
б) |
Документират се ексципиентите и по-точно по отношение на съответните им функции и концентрация. |
|
в) |
Представя се описание на разработването на готовия продукт, като се взема предвид предложеният начин на приемане и приложение. |
|
г) |
Дава се потвърждение за всякакви излишъци във формулата/ите. |
|
д) |
Що се отнася до физикохимичните и биологичните свойства, разглежда се и се документира всеки параметър, който има отношение към действието на готовия продукт. |
|
е) |
Разглежда се изборът на производствен процес и усъвършенстването му, както и разликите между различните производствени процеси, които се използват за производството на базисните клинични серии, както и процесът, който се използва за производството на готовия лекарствен продукт. |
|
ж) |
Документират се годността на съдовете и начинът им на запушване за целите на съхранението, транспортирането и използването на готовия лекарствен продукт. Може да се наложи да се разгледа възможното взаимодействие между лекарствения продукт и съда за съхранението му. |
|
з) |
Микробиологичните свойства на дозиращата форма по отношение на нестерилните и стерилните продукти следва да отговарят на предписанията на Европейската фармакопея и се документират в съответствие с нея. |
|
и) |
С цел да се представи подходяща подкрепяща информация за етикетирането, се документира съвместимостта на готовия лекарствен продукт с разредителя или дозиращото/ите устройство/а. |
3.2.2.3. Процес на производство на готовия лекарствен продукт
|
a) |
Описанието на метода на производство, придружаващо заявлението за разрешение за търговия съгласно член 8, параграф 3, буква г), се изготвя по такъв начин, че да се даде подходящ кратък преглед на естеството на използваните операции. За тази цел се включва най-малко следното:
Посочват се името, адресът и отговорността на всеки производител, включително подизпълнителите, както и всички производствени мощности, включени в производствения процес и изпитванията. |
|
б) |
Включват се данни за контролните изпитвания на продукта, които могат да се проведат на междинен етап от производствения процес с оглед обезпечаването на последователността на производствения процес. Тези изпитвания са от основно значение за проверката на съответствието на лекарствения продукт с формулата, когато по изключение даден кандидат предложи аналитичен метод за изпитване на готовия лекарствен продукт, който не включва количествен анализ на всички активни вещества (или на всички ексципиенти, които подлежат на същите изисквания като активните вещества). Същото се отнася за случаите, в които контролът по качеството на готовия лекарствен продукт зависи от контролните изпитвания на самия производствен процес и особено когато лекарственият продукт по същество е дефиниран чрез метода си на приготвяне. |
|
в) |
Представят се описанието, документацията и резултатите от валидиращите изследвания на най-важните етапи или най-важните количествени анализи, използвани в производствения процес. |
3.2.2.4. Контрол върху ексципиентите
|
а) |
Изброяват се всички материали, необходими за производството на ексципиенти, като се посочва мястото на всеки един в производствения процес. Представя се информация относно качеството и контрола на тези материали. Представя се информация, която да показва, че материалите отговарят на нормите, приети по отношение на тяхното приложение. Оцветителите при всички случаи отговарят на изискванията на Директиви 78/25/ЕИО и/или 94/36/ЕО. Освен това оцветителите отговарят на изискванията на Директива 95/45/ЕО на Съвета, както е изменена. |
|
б) |
За всеки ексципиент се представят подробни спецификации. Процедурите за анализ на данните се описват и се утвърждават надлежно. |
|
в) |
Специално внимание се отделя на ексципиентите с хуманенен или животински произход. По отношение на специалните мерки за предотвратяване пренасянето на спонгиформни енцефалопатии с животински произход заявителят представя аргументация също и за ексципиентите, от която да следва, че лекарственият продукт е произведен в съответствие с Ръководството относно минимизиране на риска от пренасяне на спонгиформната енцефалопатия по животните чрез лекарствени продукти и нейните актуализирани версии, публикувани от Комисията в Официален вестник на Европейския съюз. Аргументация в полза на съответствието с горепосоченото указание може да се постигне или чрез представяне на сертификат за годност съгласно съответната монография относно пренасянето на спонгиформни енцефалопатии от Европейската фармакопея (за предпочитане), или чрез представяне на научни данни в потвърждение на такова съответствие. |
|
г) |
Нововъведени ексципиенти
|
3.2.2.5. Контрол върху готовия лекарствен продукт
За контрола върху готовия лекарствен продукт се представя серия от готовия лекарствен продукт, която включва всички единици от дадена фармацевтична форма, които са изработени от едно и също изходно количество материал и са преминали през едни и същи серии производство и/или процедури по стерилизиране, или в случаите с непрекъснатия производствен цикъл – всички единици са произведени в един и същи отрязък от време.
Освен ако няма подходящо основание за друго, максимално допустимото отклонение в съдържанието на активното вещество на готовия лекарствен продукт не превишава ± 5 % в момента на излизане от цеха за производство.
Представя се подробна информация по спецификациите, аргументация относно избора (пускане и трайност в неотворен вид), методите за анализ и тяхното потвърждение.
3.2.2.6. Референтни стандарти или материали
Референтните препарати и нормите, които се използват за изпитване на готовия лекарствен продукт, се определят и описват подробно, ако не са предварително представени в раздела, отнасящ се до активното вещество.
3.2.2.7. Система съдове и затваряне на съдовете за съхраняване на готовия лекарствен продукт
Представя се описание на системата съдове и затваряне на съдовете за съхраняване на готовия лекарствен продукт, включително идентичността на всеки материал за изработване на непосредствената опаковка. Спецификациите включват описание и идентификация. Там където е подходящо, се включват методи, невключени във фармакопеята (с потвърждение).
Относно нефункционалните външни опаковъчни материали се представя само кратко описание. Относно функционалните външни опаковъчни материали се представя и допълнително описание.
3.2.2.8. Устойчивост на готовия лекарствен продукт
|
а) |
Обобщават се видовете проведени изследвания, използваните протоколи и резултатите от изследванията; |
|
б) |
В подходящ формат се представят подробните резултати от изследванията по устойчивостта, включително сведения относно процедурите за анализ на данните, използвани при генерирането на данни и потвърждаването на тези процедури; относно ваксините – когато е необходимо, се представят сведения за кумулативната устойчивост; |
|
в) |
Представя се протокол за устойчивостта след разрешението и ангажименти по отношение на устойчивостта. |
4. МОДУЛ 4: НЕКЛИНИЧНИ ДОКЛАДИ
4.1. Формат и представяне
Общото съдържание на модул 4 е, както следва:
|
— |
Съдържание |
|
— |
Доклади от изследванията
|
|
— |
Библиография |
4.2. Съдържание: основни принципи и изисквания
Специално внимание се отделя на следните избрани елементи:
|
(1) |
Фармакологичните и токсикологичните изпитвания трябва да покажат:
|
|
(2) |
За биологичните лекарствени продукти, например имунологичните лекарствени продукти и лекарствените продукти, производни от човешката кръв и кръвна плазма, изискванията на настоящия модул може да трябва да бъдат приспособени за отделните продукти; следователно заявителят представя аргументация на програмата за изпитванията, която ще се провежда. При установяването на програмата за изпитванията следва да се вземе предвид следното: всички изпитвания, при които се изисква многократно прилагане на лекарствения продукт, се проектират с оглед на възможното индуциране на антитела и възможното взаимодействие с тях; изследване на репродуктивната функция, на ембрионалната токсичност/ токсичността при плода, на мутагенния потенциал и на канцерогенния потенциал. Когато се появят съмнения относно съставките извън активното/ите вещество/а, изследването може да се замени с утвърждаване на тяхното премахване. |
|
(3) |
Токсикологията и фармакокинетиката на ексципиентите, използвани за първи път в областта на фармацевтиката, се подлагат на проучвания. |
|
(4) |
Когато съществува вероятност от значително разграждане по време на престой на склад на лекарствения продукт, се взема под внимание и токсикологията на продуктите от разграждането. |
4.2.1. Фармакология
Фармакологичното изследване следва две различни линии на подход:
|
— |
първо, действията, имащи отношение към предложеното терапевтично приложение, се подлагат на адекватно проучване и се описват. Когато е възможно, се прилага признат и утвърден количествен анализ както ин виво, така и ин витро. Новите експериментални методи се описват толкова подробно, че да се даде възможност за тяхното многократно повторение. Резултатите се представят количествено, като се използват например криви на ефекта при различните дози, времеви криви и т.н. Когато е възможно, се правят сравнения с данните, свързани с дадено вещество или вещества с подобно терапевтично действие. |
|
— |
Второ, заявителят проучва потенциалните нежелателни фармакодинамични ефекти върху физиологичните функции. Тези проучвания се провеждат с предварително разкриване на терапевтичната гама. Експерименталните методи се описват подробно, освен ако не представляват отработени стандартни процедури, така че да се даде възможност за тяхното многократно повторение, като изследователят се задължава да установи тяхната валидност. Изследва се всяко съмнение за изменение на реакции, възникнали в резултат от многократното прилагане на веществото. |
Относно фармакодинамичното взаимодействие на лекарствения продукт се провеждат изпитвания на комбинации на активни вещества, които се налагат или във връзка с предпоставки от фармакологичен характер, или във връзка с показания за терапевтичното действие на тези вещества. В първия случай фармакодинамичното изследване показва взаимодействията, които биха могли да придадат на комбинация значение за терапевтичното приложение. Във втория случай, когато за комбинацията се търси научна аргументация за терапевтично експериментиране, с изследването се определя дали очакваните от комбинацията ефекти могат да се демонстрират върху животни, както и дали е възможно поне да се изследва значението на всякакви съпътстващи ефекти.
4.2.2. Фармакокинетика
Фармакокинетиката означава изучаване съдбата на активното вещество, на неговите метаболити в организма, и покрива изучаването на абсорбцията, разпределението, обмяната на веществата (биотрансформацията им) и отделянето на тези вещества.
Изучаването на тези различни фази може да се провежда главно посредством физични, химични и евентуално биологични методи, както и посредством наблюдение на действителната фармакодинамична дейност на самото вещество.
Информацията относно разпределението и елиминирането е необходима във всички случаи, когато такива данни се оказват незаменими за определянето на дозировката при хора, както и по отношение на химико-терапевтичните вещества (антибиотици и т.н.) и веществата, чието приложение зависи от техните нефармакодинамични въздействия (например многобройни диагностични агенти и др.).
Изследвания ин витро също могат да се провеждат, като се извлича полза от предимството при използването на човешки материал за сравнение с животински материал (например свързването на белтъчините, обмяната на веществата, взаимодействието между различните лекарства).
Необходимо е фармакокинетично проучване на всички фармакологично активни вещества. При новите комбинации между известни вещества, които са били изследвани в съответствие с предписанията на настоящата директива, може да не се изискват фармакокинетични изследвания, ако изпитванията на токсичността и терапевтичните експерименти оправдават тяхното пропускане.
Фармакокинетичната програма се разработва така, че да позволява сравнение и екстраполиране между човешкия и животинския материал.
4.2.3. Токсикология
|
a) |
Токсичност при единична доза Изпитването на токсичността на единична доза означава количествено и качествено проучване на токсичните реакции, които е възможно да възникнат вследствие на единично приложение на активното/ите вещество/а, съдържащо/и се в лекарствения продукт, в пропорциите и физикохимичното състояние, в което то/те се съдържа/т в действителния продукт. Изпитванията на токсичността на единична доза задължително се провеждат съгласно съответните ръководства, публикувани от агенцията. |
|
б) |
Токсичност на многократна доза Изпитванията на токсичността на многократна доза имат за цел да разкрият всички физиологични и/или анатомо-патологични изменения, които възникват при многократното приложение на активното вещество или комбинацията от активни вещества при изследване, както и да се определи взаимовръзката между тези изменения и дозирането. Като цяло е желателно да се изпълнят две изпитвания – едното краткосрочно, с продължителност до четири седмици, а другото – дългосрочно. Продължителността на последното зависи от условията на клиничното приложение. Целта му е да покаже възможните странични ефекти, на които следва да се обърне внимание по време на клиничните изследвания. Продължителността се определя в съответните ръководства, публикувани от агенцията. |
|
в) |
Генетична токсичност Целта на изследването на мутагенния и кластогенния потенциал е да се разкрият възможните изменения вследствие на въздействието на дадено вещество върху генетичния материал на хора или клетки. Мутагенните вещества могат да представляват опасност за здравето, при положение че излагането на мутаген носи в себе си риска от индуциране на бактериална мутация с възможни наследствени нарушения, както и риска от соматични мутации, включително такива, които водят до рак. Тези изследвания са задължителни за всички нови вещества. |
|
г) |
Канцерогенност Обикновено се изисква провеждането на изпитвания за разкриване на канцерогенни ефекти:
|
|
д) |
Репродуктивна токсичност и токсичност на развитието Посредством подходящи изпитвания се провежда изследване на възможното разстройство на мъжката или женската репродуктивна функция, както и на вредните въздействия върху потомството. Тези изпитвания включват изследвания относно въздействието върху мъжката или женската репродуктивна функция, изследвания върху токсичните и тератогенните ефекти на всички стадии на развитие от зачатието до половата зрялост, като се включват и латентните ефекти, когато лекарственият продукт във фаза проучване е приложен на женски индивид по време на бременност. В случай на пропускане на тези изследвания се представя подходяща аргументация. В зависимост от посоченото приложение на лекарствения продукт е възможно да се изискват и допълнителни изследвания по отношение на развитието на плода по време на приложението на лекарствения продукт. Обикновено се провеждат изследвания върху токсичността при зародиша и плода на два вида бозайници, единият от които следва да не е гризач. Провеждат се перинатални и постнатални изследвания върху поне един вид бозайник. Ако за метаболизма на лекарствения продукт при даден биологичен вид е известно, че е подобен на метаболизма при човека, желателно е този вид също да бъде включен. Желателно е също и единият от видовете да е същият, като при провеждането на изследванията на токсичността при многократно прилагане на дозата. Състоянието на научните познания към момента, когато се представят изследванията, също се взема под внимание, когато се определя проектът за изследванията. |
|
е) |
Местна поносимост Целта на изследванията на местната поносимост е да установи с положителност дали лекарствените продукти (включително активнотите вещества и ексципиентите) се понасят от местата в организма, в които те могат да встъпят в контакт с лекарствения продукт след неговото клинично прилагане. Стратегията на изпитванията е такава, че всички механични ефекти от приложението на лекарствения продукт, както и чисто физикохимичното му действие, могат да се отличават от токсикологичните и фармакодинамичните. Изпитването на местната поносимост към даден препарат се извършва, след като се премине през предварителна подготовка за приложение спрямо хора, като при лечението на контролната/ите група/и се използва носител и/или ексципиенти. Когато е необходимо, се включват вещества за положителен контрол/референтни вещества. Разработването на изпитвания на местната поносимост (избор на биологични видове, продължителност, честота и път на прилагане, дозировка) зависи от начина, по който ще бъде изследван проблемът и от предложените условия за прилагане в клиничната практика. Там, където е подходящо, се изпълнява реверсивност на локалните нарушения. Изследванията върху животни могат да се заменят с утвърдени изпитвания ин витро, при положение че резултатите от изпитванията са със сравнимо качество и полза за целите на оценката на безвредността. Относно химикалите, които се прилагат кожно (например дермално, анално, вагинално), се дава оценка на потенциала на осезанието в поне една от изпитните системи, които са на разположение в момента (количествен анализ при морското свинче или количествен анализ на локалните лимфни възли). |
5. МОДУЛ 5: ДОКЛАДИ ОТ КЛИНИЧНИТЕ ИЗСЛЕДВАНИЯ
5.1. Формат и представяне
Общото съдържание на модул 5 е, както следва:
|
— |
Съдържание за докладите от клиничните изследвания |
|
— |
Таблично изброяване на всички клинични изследвания |
|
— |
Доклади от клиничните изследвания
|
|
— |
Библиография |
5.2. Съдържание: основни принципи и изисквания
Следва да се обърне специално внимание на следните избрани елементи:
|
а) |
Клиничните подробности, представени съгласно член 8, параграф 3, i) и член 10, параграф 1, трябва да дават възможност за съставянето на достатъчно обосновано и научно валидно становище дали лекарственият продукт отговаря на критериите за предоставяне на разрешение за търговия. Вследствие на това съществено изискване е резултатите от всички клинични изследвания да бъдат съобщени – били те благоприятни или неблагоприятни. |
|
б) |
Клиничните изследвания трябва винаги да се предшестват от подходящи фармакологични и токсикологични изпитвания, които се провеждат върху животни съгласно изискванията в модул 4 от настоящото приложение. Изследователят трябва да се запознае със заключенията от фармакологичните и токсикологичните изследвания, следователно заявителят трябва да му предостави най-малко брошура, в която да са включени всички важни сведения, станали известни преди началото на клиничното изпитване, включително химичните, фармацевтичните и биологичните данни, токсикологичните, фармакокинетичните и фармакодинамичните данни при животните и резултатите от по-ранните клинични опити, снабдени с подходящи данни в подкрепа на естеството, мащаба и продължителността на предложения опит; пълните фармакологични и токсикологични доклади се представят при поискване. Относно материалите с хуманен или животински произход се използват всички налични средства с цел обезпечаване предотвратяването на предаване на заразни агенти преди началото на опита. |
|
в) |
Титулярите на разрешение за търговия следва да разполагат с основните документи за клинични опити (включително формуляри за доклади по случаите), различни от медицинските документации на субекта, които да се съхраняват от притежателите на данните:
Медицинските документации на субекта следва да се задържат съгласно приложимото законодателство и в съответствие с максималния период от време, разрешен от болничното заведение, институция или частно здравно заведение. Документите обаче се съхраняват за по-дълъг период от време, ако това се изисква от приложимите регулационни разпоредби или от споразумение със спонсора. Спонсорът или друг притежател на данните задържа всички останали документи, имащи отношение към опита, докато продуктът получи разрешение. Тази документация включва: протокола, включително обосновката, целите, статистическия проект и методологията на опита, заедно с условията, при които се провежда опитът, както и подробности от продукта на изследването, референтния лекарствен продукт и/или използваното плацебо; стандартните оперативни процедури; всички писмени становища от протокола, заедно с процедурите; брошурата на изследователя; доклад от всяко изследване на субект; окончателния доклад; сертификатите от проверки, ако има такива. Окончателният доклад се задържа от спонсора или от следващия притежател за срок от пет години след изтичането на срока на разрешението за лекарствения продукт. Освен изпитванията, които се провеждат в рамките на Европейската общност, титулярът на разрешение за търговия предприема всякакви други действия с цел архивиране на документацията съгласно разпоредбите на Директива 2001/20/ЕО и в изпълнение на подробните ръководства. Всички промени в собствеността върху данните се документират. Всички данни и документи се представят при поискване от съответните органи. |
|
г) |
Подробностите от всеки клиничен опит трябва да съдържат достатъчно данни, които да дават възможност за даване на обективна оценка:
|
|
д) |
Подробностите по клиничните изследвания, споменати по-горе, се предоставят на съответните компетентни власти. При сключването на договора си с компетентните власти обаче изследователят може да пропусне част от тази информация. Пълната информация се предоставя само при поискване. В своите заключения по експерименталния материал изследователят изразява мнение относно безвредността на продукта при нормални условия на приложение, поносимостта на продукта, ефикасността му, както и всякакви други полезни сведения, свързани с показанията и противопоказанията, дозировката и средната продължителност на лечението, както и всякакви специални мерки, които да се вземат по време на лечението и клиничните симптоми при предозиране. При докладване на резултатите от изследвания, провеждани в няколко изследователски центъра, главният изследовател изразява в своите заключения становище относно безвредността и ефикасността на изследвания лекарствен продукт от името на всички изследователски центрове. |
|
е) |
Клиничните наблюдения за всяко изследване се обобщават, като се включва следното:
|
|
ж) |
Освен това изследователят винаги посочва своите наблюдения върху:
|
|
з) |
Подробностите, засягащи нова комбинация от лекарствени вещества, трябва да са идентични на тези, които се изискват за новите лекарствени продукти, като същевременно се привеждат основания в полза на безвредността и ефикасността на комбинацията. |
|
и) |
Пълното или частичното пропускане на данни трябва да се обясни. Ако по време на опитите се получат някакви неочаквани резултати, трябва да се проведат и подложат на анализ по-нататъшни предклинични токсикологични и фармакологични изследвания. |
|
й) |
Ако лекарственият продукт е предназначен за дългосрочно прилагане, се посочват подробности относно всички разновидности фармакологично действие, което се проявява след многократна употреба, както и установяване на дозировката при дългосрочна употреба. |
5.2.1. Доклади от биофармацевтичните изследвания
Представят се докладите за биологичната годност, сравнителната биологична годност, докладите за биологичната еквивалентност, докладите от корелационните изследвания ин витро и ин виво, както и биоаналитичните и аналитичните методи.
Освен това там, където е необходимо, се прави оценка на биологичната пригодност, с цел да се покаже биологичната еквивалентност на лекарствените продукти, за които става дума в член 10, параграф 1, буква а).
5.2.2. Доклади по изследванията, свързани с фармакокинетиката с използване на човешки биологични материали
По смисъла на настоящото приложение, под „хуманни биологични материали“ се разбират всякакви белтъчини, клетки, тъкани и други сродни материали, извлечени от човешки източници, които материали се използват ин витро и ин виво за оценка на фармакокинетичните свойства на лекарствените вещества.
Във връзка с това се представят доклади от изследванията по свързване на белтъчините в плазмата, хепатитния метаболизъм, както и изследванията върху взаимодействията на активното/ите вещество/а и изследвания по отношение на други видове човешки материали.
5.2.3. Доклади от хуманните фармакокинетични изследвания
|
a) |
На изследване подлежат следните характеристики:
Описват се клинично значимите белези, включително реалните последици на кинетичните данни по отношение на режима на дозировката, особено при пациентите от рисковите групи, както и разликите между човека и животинските видове, използвани в предклиничните изследвания. Освен стандартните многократни фармакокинетични изследвания на образците, фармакокинетичните анализи на населението, основани върху случайно взети отделни проби по време на клиничните изследвания, също могат да предизвикат въпроси относно приноса на външните и вътрешните фактори към разнообразието на връзката доза—фармакокинетични реакции. Представят се докладите по фармакокинетичните изследвания и изследванията на първоначалната поносимост при здрави индивиди и при пациенти, докладите по фармакокинетичните изследвания за оценка на ефектите на външните и вътрешните фактори, както и докладите от фармакокинетичните изследвания на населението. |
|
б) |
Ако е прието лекарственият продукт да се прилага едновременно с други лекарствени продукти, се посочват данни за проведените изпитвания на съвместното приложение, които да покажат възможно изменение на фармакологичното действие. Фармакокинетичните взаимодействия между активното вещество и други лекарствени продукти или вещества също се подлагат на изследване. |
5.2.4. Доклади от хуманните фармакодинамични изследвания
|
a) |
Фармакодинамичното действие се представя в корелация с ефикасността, като тук се включва следното:
Описва се фармакодинамичното действие, несвързано с ефикасността. Демонстрирането на фармакодинамичните ефекти върху хора само по себе си не е достатъчно за аргументиране на изводите по отношение на някой отделно взет потенциален терапевтичен ефект. |
|
б) |
Ако е прието лекарственият продукт да се прилага едновременно с други лекарствени продукти, се дават подробности относно проведените изпитвания на съвместното приложение, които да покажат възможно изменение на фармакологичното действие. Фармакодинамичните взаимодействия между активното вещество и други лекарствени продукти или вещества също се подлагат на изследване. |
5.2.5. Доклади от изследванията на ефикасността и безвредността
5.2.5.1. Доклади от контролираните клинични изследвания, отнасящи се до представеното показание
Като цяло клиничните изследвания се извършват по възможност във вид на „контролирани клинични изследвания“, рандомизират се по съответния начин спрямо плацебото и спрямо утвърден лекарствен продукт с доказана терапевтична стойност; по отношение на какъвто и да било друг проект се дава аргументация. Лечението на контролните групи варира при отделните случаи, а също така зависи от решенията, които се вземат с оглед етиката и терапевтичната област; по такъв начин в някои случаи може се окаже по-подходящо да се сравни ефикасността на даден нов лекарствен продукт с тази на вече утвърден лекарствен продукт с доказана терапевтична стойност, отколкото да се сравнява с ефекта на плацебото.
|
(1) |
Доколкото това е възможно и особено при изследвания, при които не е възможно ефектът на продукта да се измери по обективен начин, се предприемат стъпки, които да предотвратят отклоненията, особено посредством методите на рандомизирането и затъмняването. |
|
(2) |
Протоколът от изследването трябва да съдържа подробно описание на статистическите методи, които се използват, броя пациенти и причините за избора (включват се изчисления относно мощността на изследването), нивото на важност, както и описание на статистическата единица. Документират се взетите за предотвратяването на отклонението мерки, особено методите на рандомизирането. Включването на голям брой субекти в даден опит не се разглежда като адекватно заместване на добре проведен и контролиран опит. |
Данните относно безвредността се разглеждат, като се вземат предвид ръководствата, публикувани от Комисията, като се обръща специално внимание на фактите, появили се в резултат от промяната в дозировката, както и в резултат от появата на необходимост от употреба на съпътстващо лекарство, някои сериозни странични реакции, явления, от които произтича оттегляне, както и смъртни случаи. Идентифицират се всички пациенти или групи пациенти, принадлежащи към контингента с повишен риск, като следва да се обърне специално внимание на потенциално уязвимите пациенти, чийто брой може да е незначителен, например деца, бременни жени, възрастни хора с крехко здраве, хора с отбелязани аномалии при обмяната на веществата или в отделителната система и т.н. Описват се последиците от оценката на безвредността по отношение на възможната употреба на лекарствения продукт.
5.2.5.2. Доклади от изследванията върху докладите от неконтролираните клинични изследвания на анализите на данните от повече от едно изследване и други доклади от клинични изследвания
Представят се такива доклади.
5.2.6. Доклади от опита, придобит след получаване на разрешение за търговия
Ако лекарственият продукт вече е получил разрешение в трети страни, при възможност се представя информация относно странични реакции спрямо съответния лекарствен продукт и лекарствените продукти, които съдържат същото активно/и вещество/а във връзка с дозировките.
5.2.7. Формуляри за доклади по отделни случаи и списъци с индивидуални пациенти
Когато в съответствие със съответните ръководства, публикувани от агенцията, се предават формулярите за докладите по случаите и списъците с отделни пациенти, те се подреждат и представят в същия ред, в който са предадени и съответно заведени докладите по клиничните изследвания.
ЧАСТ II
СПЕЦИФИЧНА ДОКУМЕНТАЦИЯ ЗА РАЗРЕШЕНИЕ ЗА ТЪРГОВИЯ И ИЗИСКВАНИЯ КЪМ НЕЯ
Някои лекарствени продукти притежават специфични свойства, които са от такъв характер, че е необходимо да се променят всички изисквания относно документацията за разрешение за търговия, формулирани в част I от настоящото приложение. С оглед на такива специфични ситуации заявителите се придържат към подходяща и изменена форма на представяне на документацията.
1. ЛЕКАРСТВЕНИ ПРОДУКТИ С УТВЪРДЕНО ЛЕЧЕБНО ПРИЛОЖЕНИЕ
За лекарствените продукти, чието/ито активно/и вещество/а има „утвърдено лечебно приложение“ по смисъла на член 10, параграф 1, буква а), ii), с призната ефикасност и приемливо равнище на безвредност, се прилагат следните специфични правила.
Заявителят представя модули 1, 2 и 3 така, както е описано в част I от настоящото приложение.
В модули 4 и 5 подробната научна библиография разглежда неклиничните и клиничните характеристики.
С цел да се демонстрира утвърдената употреба на лекарствения продукт, се прилагат следните специфични правила:
|
a) |
Факторите, които се вземат предвид с цел установяването на утвърдено лечебно приложение на съставки на лекарствени продукти са, както следва:
Следователно е възможно да са необходими различни периоди от време за установяването на утвърденото лечебно приложение на различните вещества. Във всеки случай обаче периодът от време, който се изисква за установяването на утвърденото лечебно приложение на дадена съставка на даден лекарствен продукт, не може да бъде по-малък от десет години от първата системна и документирана употреба на даден лекарствен продукт в рамките на Общността. |
|
б) |
В документацията, представена от заявителя, следва да бъдат обхванати всички аспекти при оценяването на безвредността и/или ефикасността, както и да бъде включен или да има позоваване на преглед на литературата по въпроса, като се вземат предвид изследванията, проведени преди пускането в търговската мрежа и след това, както и всичко, публикувано в научните издания по отношение на опита под формата на епидемиологични изследвания и особено на сравнителни епидемиологични изследвания. Следва да се представи цялата документация, както благоприятната, така и неблагоприятната. Относно разпоредбите за „утвърдено лечебно приложение“ от особена важност е да се изясни, че „библиографската справка“, в която има препратки към други източници на доказателства (изследвания, проведени след пускане на даден лекарствен продукт на пазара, епидемиологични изследвания и т.н.), а не само данните, свързани с изпитванията и опитите, могат да служат като валидно доказателство за безвредността и ефикасността на даден продукт, ако в заявлението по задоволителен начин се обяснява и аргументира използването на тези източници на информация. |
|
в) |
Специално внимание трябва да се обърне на всякакви липсващи сведения, като трябва да се представи аргументация защо може да се подкрепи приемливо ниво на безвредността и/или ефикасността въпреки липсата на някои изследвания. |
|
г) |
В неклиничните и/или клиничните прегледи трябва да се обясни значението на всички представени данни, имащи отношение към даден продукт, който се различава от продукта, предвиден за пускане на пазара. |
|
д) |
Опитът, придобит след пускане на пазара на други продукти, които съдържат същите съставки, е от особено значение, поради което заявителите следва да наблегнат особено върху този въпрос. |
2. СХОДНИ ПО СЪЩЕСТВО ЛЕКАРСТВЕНИ ПРОДУКТИ
|
а) |
Заявленията на базата на член 10, параграф 1, буква а), i) (сходни по същество лекарствени продукти) съдържат данните, описани в модули 1, 2 и 3 от част I на настоящото приложение, при условие че заявителят е получил съгласието на титуляра на първоначалното разрешение за търговия за препращане към съдържанието на неговите модули 4 и 5. |
|
б) |
Заявленията на базата на член 10, параграф 1, буква а), iii) (сходни по същество лекарствени продукти) съдържат данните, описани в модули 1, 2 и 3 от част I на настоящото приложение заедно с данните, показващи биологичната годност и биоеквивалентността с оригиналния лекарствен продукт, при условие че последният не е биологичен лекарствен продукт (виж част II, 4 — Сходни лекарствени продукти). |
За тези продукти неклиничните/клиничните обзори/обобщения се фокусират върху следните елементи:
|
— |
основанията да се твърди сходство по същество; |
|
— |
обобщение на примесите в сериите на активното/ите вещество/а, както и на готовия продукт (и където е приложимо, на продуктите от разграждането, получаващи се по време на съхранението), предложени за използване в продукта, с който ще се търгува, заедно с оценка на тези примеси; |
|
— |
оценка на изследванията на биоеквивалентността или обосновка изследванията да не се провеждат, като се вземат предвид ръководствата относно „Изследване на биологичната годност и биоеквивалентността“; |
|
— |
актуализиран списък на публикуваната литература, свързана с веществото и настоящото приложение. Приема се анотация на статиите в списанията „peer review“ за тази цел; |
|
— |
всяко твърдение в обобщението на характеристиките на продукта, което не е известно или за което не може да се направи заключение от свойствата на лекарствения продукт и/или неговата терапевтична група, следва да се разгледа в неклиничните/клиничните обзори/обобщения и да се докаже чрез публикуваната литература и/или допълнителни изследвания; |
|
— |
ако е приложимо, при твърдение за сходство по същество заявителят следва да представи допълнителни данни, с цел да се докаже еквивалентността на безвредното и ефективно действие на различни соли, естери или производни на разрешено активно вещество. |
3. ДОПЪЛНИТЕЛНИ ДАННИ, КОИТО СЕ ИЗИСКВАТ В СПЕЦИФИЧНИ СИТУАЦИИ
Когато активното вещество на сходен по същество лекарствен продукт съдържа същата терапевтична част като оригиналния разрешен продукт, свързана с различна сол/естер комплекс/производно се представя доказателство, че няма промяна във фармакокинетиката на частта, фармакодинамиката и/или токсичността, която би могла да промени профила на безвредност/ефикасност. Ако случаят не е такъв, тази асоциация се смята за ново активно вещество.
Ако лекарственият продукт е предвиден за различна терапевтична употреба или се представя в различна фармацевтична форма, или се прилага по различен начин, или в различна концентрация, или с различна дозировка, се представят резултати от подходящи токсикологични и фармакологични тестове и/или клинични изпитания.
4. СХОДНИ БИОЛОГИЧНИ ЛЕКАРСТВЕНИ ПРОДУКТИ
Възможно е разпоредбите на член 10, параграф 1, буква а), iii) да се окажат недостатъчни по отношение на биологичните лекарствени продукти. Ако сведенията, които се изискват при сходните по същество продукти (генерични продукти), не позволяват да се покаже сходното естество на два биологични лекарствени продукта, се представят допълнителни данни, особено по токсикологичния и клиничния профил.
Когато даден биологичен лекарствен продукт, както е определен в част I, параграф 3.2 от настоящото приложение, който се отнася до даден оригинален лекарствен продукт, за който е издадено разрешение за търговия в рамките на Общността, се представя за получаване на разрешение за търговия от независим кандидат след изтичането на периода за запазване на данните, се прилага следният подход.
|
— |
Подлежащата на представяне информация не се изчерпва с модули 1, 2 и 3 (фармацевтични, биологични и химични данни), подкрепени с данните за биоеквивалентността и биологичната годност. Видът и количеството допълнителни данни (тоест токсикологичните и другите неклинични и подходящи клинични данни) се определят на базата на всеки отделен случай в съответствие със съответните научни предписания. |
|
— |
Поради разнообразието на биологичните лекарствени продукти от компетентните власти се изискват определени изследвания, предвидени в модули 4 и 5, като се вземат предвид специфичните характеристики на всеки отделен лекарствен продукт. |
Общите принципи, които трябва да се приложат, са насочени към публикуваното от агенцията ръководство, отчитащо характеристиките на дадения лекарствен продукт. В случай че лекарствен продукт, за който първоначално е издадено разрешение за търговия, има повече от едно показание, следва да се даде оправдание на ефикасността и безвредността на лекарствения продукт, за който се твърди, че е сходен, или ако е необходимо, показанията, за чието наличие се претендира, се демонстрират поотделно.
5. ЛЕКАРСТВЕНИ ПРОДУКТИ С УСТАНОВЕНИ КОМБИНАЦИИ
Заявленията, основани на член 10, параграф 1, буква б), се отнасят единствено за лекарствени продукти, произведени от поне две активни вещества, за които не е получено предварително разрешение като за лекарствен продукт с установена комбинация.
В такива заявления се представя пълна документация (модули от 1 до 5) на лекарствен продукт с установена комбинация. Когато е приложимо, се представя информация относно производствените предприятия, външните агенти и оценка на безвредността.
6. ДОКУМЕНТАЦИЯ ЗА ЗАЯВЛЕНИЯ ПРИ ИЗКЛЮЧИТЕЛНИ ОБСТОЯТЕЛСТВА
Когато съгласно член 22 заявителят демонстрира, че не е в състояние да представи всеобхватни данни относно ефикасността и безвредността при нормални условия на употреба поради това, че:
|
— |
показанията, за които е предназначена употребата на въпросния продукт, са толкова рядко срещани, че заявителят по обясними причини не може да представи пълен доказателствен материал, или |
|
— |
състоянието на научните познания към момента е такова, че не могат да се представят всеобхватни сведения, или |
|
— |
би било против общоприетите принципи на лекарската етика да се събират такива сведения, |
разрешението за търговия може да се даде, при условие че се спазват някои специфични задължения.
Тези задължения могат да включват следното:
|
— |
заявителят изпълнява определена програма от изследвания в рамките на периода от време, определен от компетентния орган, като резултатите от тези изследвания формират основата за преоценка на съотношението полза—риск, |
|
— |
въпросният лекарствен продукт може да се предоставя единствено по лекарско предписание, като в определени случаи може да се употребява само под стриктен лекарски контрол, евентуално в болнично заведение, а при радиофармацевтиците – само под контрола на оправомощено лице, |
|
— |
листовката с упътвания в опаковката, както и всяка медицинска информация, привличат вниманието на общопрактикуващия лекар към факта, че съществуващите данни за въпросния лекарствен продукт все още не съответстват на изискванията в определени специфични отношения. |
7. КОМБИНИРАНИ ЗАЯВЛЕНИЯ ЗА РАЗРЕШЕНИЕ ЗА ТЪРГОВИЯ
Под комбинирани заявления за разрешения за търговия следва да се разбира документацията за заявления за разрешение за търговия, когато модули 4 и/или 5 се състоят от комбинация от доклади по ограничени неклинични и/или клинични изследвания, осъществени от заявителя, като са включени и библиографски справки. Всички останали модули отговарят на структурата, описана в част I от настоящото приложение. Компетентният орган приема предложения формат, представен от заявителя на базата на всеки случай поотделно.
ЧАСТ III
СПЕЦИАЛНИ ЛЕКАРСТВЕНИ ПРОДУКТИ
В настоящата част са изложени специалните изисквания по отношение на естеството на определени лекарствени продукти.
1. БИОЛОГИЧНИ ЛЕКАРСТВЕНИ ПРОДУКТИ
1.1. Лекарствени продукти на базата на кръвна плазма
По отношение на лекарствените продукти, извлечени от човешка кръв или кръвна плазма, и чрез дерогация от разпоредбите на модул 3, изискванията за документацията, посочени в „Информация, свързана с изходните материали и суровините“, „изходните материали, приготвени от човешка кръв/плазма“ могат да се заменят с основна документация за плазмата в съответствие с настоящата част.
a) Принципи
По смисъла на настоящото приложение:
|
— |
„основна документация за плазмата“ означава отделна документация, която е отделена от документацията за разрешение за търговия, в която се дава цялата необходима подробна информация относно характеристиките на цялата човешка кръвна плазма, използвана като изходен материал и/или като суровина за производството на субфракции/междинни фракции, съставки на инертните материали и активното/ите вещество/а, които представляват част от лекарствените продукти или медицинските устройства, разгледани в Директива 2000/70/ЕО на Европейския парламент и на Съвета от 16 ноември 2000 г. за изменение на Директива 93/42/ЕО на Съвета относно медицинските устройства, които включват устойчиви производни от човешка кръв или човешка кръвна плазма (11). |
|
— |
Всеки център или предприятие, в което се осъществява разделяне на фракции/обработка на човешка кръвна плазма, подготвя и осъвременява пакет подробна информация относно ОДП. |
|
— |
Основната документация за плазмата се представя от заявителя за разрешение за търговия или титуляра на разрешение за търговия на агенцията или на компетентния орган. Когато заявителят за разрешение за търговия или титулярът на разрешение за търговия не съвпада с титуляра на основната документация за плазмата, ОДП става достъпна за заявителя или титуляра на разрешение за търговия, за да се предаде на компетентните власти. Във всеки случай заявителят или титулярът на разрешение за търговия поема отговорността за лекарствения продукт. |
|
— |
Компетентната власт, която оценява разрешението за търговия изчаква агенцията да издаде сертификат, преди да се произнесе по заявлението. |
|
— |
Всяка документация за разрешение за търговия, в която се съдържа съставка—производна от кръвна плазма, се позовава на основната документация за плазма, съответстваща на плазмата, използвана за изходен материал/суровина. |
б) Съдържание
В съответствие с разпоредбите на член 109, изменен с Директива 2002/98/ЕО, който се отнася до изискванията към донорите и изпитването на даренията, основната документация за плазмата съдържа информация за плазмата, използвана за изходен материал/суровина, и по-специално:
|
(1) |
Произход на плазмата
|
|
(2) |
Качество и безвредност на плазмата
|
|
(3) |
Съществуващата система между производителя на лекарствения продукт, произведен от кръвна плазма и/или институцията, която се занимава с кръвните фракции/обработка на кръвта, от една страна, и центровете и институтите за изпитване, от друга страна, която определя условията за тяхното взаимодействие и съгласуваните помежду им спецификации. Освен това основната документация за плазмата включва списък на лекарствените продукти, за които е валидна основната документация за плазмата; дали на лекарствените продукти е издадено разрешение за търговия или те се намират в процес на издаване на такова разрешение за търговия, включително лекарствените продукти, посочени в член 2 от Директива 2001/20/ЕО на Европейския парламент и на Съвета, отнасяща се до прилагането на добрата клинична практика при провеждането на клинични опити върху лекарствените продукти за хуманна употреба. |
в) Оценяване и сертифициране
|
— |
При лекарствените продукти, които все още не са получили разрешение, заявителят за разрешение за търговия представя пълна документация на компетентния орган, придружена от отделна основна документация за плазмата в случаите, когато все още не съществува такова. |
|
— |
Основната документация за плазмата е предмет на научно и техническо оценяване, което се осъществява от агенцията. Ако оценката за основната документация за плазмата е положителна, се издава сертификат съгласно законодателството на Общността, който се придружава от доклад по оценката. Издаденият сертификат важи на територията на цялата Общност. |
|
— |
Основната документация за плазмата се актуализира и подлежи на ежегодно подновяване. |
|
— |
Измененията, които впоследствие се въвеждат по отношение на условията за основната документация за плазмата, трябва да следват процедурата по оценяването, установена в Регламент (ЕО) № 542/95 на Комисията от 10 март 1995 г. (12) относно проучването на измененията в разрешението за търговия, включени в обхвата на Регламент (ЕИО) № 2309/93 на Съвета от 22 юли 1993 г. за установяване на общностни процедури за предоставяне на разрешение и за надзор на лекарствените продукти за хуманна и ветеринарна употреба и относно създаване на Европейска агенция за оценка на лекарствените продукти (13). Условията за оценка на тези промени са установени в Регламент (ЕО) № 1085/2003 на Комисията. |
|
— |
Като втора стъпка от разпоредбите на първо, второ, трето и четвърто тире компетентните власти, които ще издават или вече са издали разрешение за търговия, вземат предвид сертифицирането, подновяването на сертификата или измененията в основната документация за плазмата на съответния лекарствен продукт. |
|
— |
Чрез дерогация от разпоредбите на второ тире от настоящата точка (оценяване и сертифициране), когато дадена основна документация за плазмата съответства само на лекарствените продукти, произведени от кръв/кръвна плазма, чието разрешение за търговия е ограничено в рамките само на една от държавите-членки, националната компетентна власт на тази държава-членка извършва научно-техническо оценяване на съответната основна документация за плазмата. |
1.2. Ваксини
Относно ваксините за хуманна употреба, чрез дерогация от разпоредбите на модул 3 относно „Активните вещества“ и в случай, че са основани върху системата основна документация за плазмата на ваксинния антиген, се прилагат следните изисквания.
Документацията за разрешение за търговия на дадена ваксина, различна от ваксината за човешкия грип, задължително включва основната документация на ваксинния антиген, който представлява активното/ите вещество/а на тази ваксина.
a) Принципи
По смисъла на настоящото приложение:
|
— |
„Основна документация на ваксинния антиген“ означава отделна част от документацията на заявлението за разрешение за търговия за ваксина, която съдържа цялата необходима информация от биологично, фармацевтично и химично естество и се отнася до всяко от активните вещества, което представлява част от този лекарствен продукт. Отделната част може да бъде обща за една или повече моновалентни и/или комбинирани ваксини, представени от един и същи кандидат или титуляр на разрешение за търговия. |
|
— |
Дадена ваксина може да съдържа един или повече отделни ваксинни антигени. Активните вещества са толкова на брой, колкото и ваксинните антигени, присъстващи в дадена ваксина. |
|
— |
Комбинираната ваксина съдържа най-малко два различни ваксинни антигена, които служат за предотвратяване на едно или няколко инфекциозни заболявания. |
|
— |
Моновалентната ваксина е ваксина, която съдържа един ваксинен антиген, който служи за предотвратяване само на едно инфекциозно заболяване. |
б) Съдържание
Основната документация на ваксинния антиген съдържа следната извлечена от съответната част (Активно вещество) на модул 3 информация относно „Данни за качеството“ така, както е очертано в част I от настоящото приложение:
|
|
Активно вещество
|
в) Оценяване и сертифициране
|
— |
За нови ваксини, които съдържат нов ваксинен антиген, заявителят представя на компетентната власт пълната документация за разрешение за търговия, в която да са включени всички основни документации за ваксинните антигени, съответстващи на всеки ваксинен антиген, който представлява част от новата ваксина, там, където не съществува такава основна документация на ваксинния антиген за даден отделен ваксинен антиген. Научно-техническата оценка за всяка основна документация на ваксинния антиген се извършва от агенцията. Ако оценката е положителна, се издава сертификат за съответствие с европейското законодателство за всяка основна документация на ваксинния антиген, който се придружава от доклад по оценката. Сертификатът важи на територията на цялата Общност. |
|
— |
Разпоредбите на първо тире се прилагат за всяка ваксина, която се състои от нова комбинация от ваксинни антигени, независимо дали един или повече от тези ваксинни антигени са част от ваксини, които вече са получили разрешение в Общността. |
|
— |
Измененията в съдържанието на дадена основна документация на ваксинния антиген за ваксина, получила разрешение в рамките на Общността, са предмет на научно-техническа оценка, която се осъществява от агенцията съгласно процедурата, установена в Регламент (ЕО) № 1085/2003 на Комисията. Ако оценката е положителна, агенцията издава сертификат за съответствие със законодателството на Общността за всяка основна документация на ваксинния антиген. Сертификатът се прилага на територията на цялата Общност. |
|
— |
Чрез дерогация от разпоредбите на първо, второ и трето тире от настоящата буква (Оценяване и сертифициране), там, където дадена основна документация на ваксинния антиген на ваксина съответства само на ваксина, която е предмет на разрешение за търговия, което не е било/няма да бъде издадено в съответствие с процедурите на Общността, и при положение че разрешената ваксина съдържа антигени, които не са преминали оценяване по процедурата на Общността, научно-техническата оценка на въпросната основна документация на ваксинния антиген и последващите изменения по нея се извършват от националния компетентна власт, който е издал разрешението за търговия. |
|
— |
Като втора стъпка от разпоредбите на първо, второ, трето и четвърто тире компетентният орган, който е издал или ще издаде разрешение за търговия, взема предвид сертифицирането, повторното сертифициране или изменението на основната документация на ваксинния антиген на въпросния/те лекарствен/и продукт/и. |
2. РАДИОФАРМАЦЕВТИЦИ И ПРЕКУРСОРИ
2.1. Радиофармацевтици
За целите на настоящата глава в заявленията въз основа на член 6, параграф 2 и член 9 се представя пълна документация, в която са включени следните специфични подробности:
Модул 3
|
a) |
В контекста на радиофармацевтичния кит, който следва да бъде обозначен с етикет като радиоактивен, след като бъде доставен от производителя, активното вещество е онази част от формулата, за която се смята, че носи или че свързва радионуклида. Описанието на метода на производство на радиофармацевтичните китове съдържа данни за производството на кита и данни за препоръчителната довършителна обработка за производството на радиоактивния лекарствен продукт. Необходимите спецификации на радионуклида се описват там, където е подходящо, в съответствие с общата монография или с конкретните монографии от Европейската фармакопея. Освен това се описва и структурата на радиоактивното съединение. За радионуклидите се разглеждат включените ядрени реакции. В генератора за активни вещества се считат както първичните радионуклиди, така и вторичните радионуклиди. |
|
б) |
Представят се данни за естеството на радионуклидите, идентичността на изотопа, възможните примеси, носителя, употребата и специфичната активност. |
|
в) |
Изходните материали включват материалите — цели на облъчването. |
|
г) |
Представят се съображения относно химичната/радиоактивната чистота и нейната връзка с биологичното разпространение. |
|
д) |
Описват се радионуклидната чистота и специфичната активност. |
|
е) |
За генераторите се изискват данни от изпитването за първични и вторични радионуклиди. Относно елуатите на генераторите се представят изпитванията за първични и вторични радионуклиди и други съставни части на системата за генериране. |
|
ж) |
Изискването да се изрази съдържанието на активното/ите вещество/а чрез масата на активните вещества важи само за радиофармацевтичните китове. По отношение на радионуклидите – радиоактивността се изразява в бекерели на дадена дата и при необходимост се дава времето според съответния часови пояс. Отбелязва се и видът радиация. |
|
з) |
При китовете, в спецификациите на готовия продукт се включват и изпитванията върху действието на продуктите след въвеждането на радиоактивните изотопи. Включват се подходящите проверки на радиохимичната и радионуклидната чистота на радиоактивното белязано съединение. Всеки материал, който е важен за въвеждането на радиоактивните изотопи, се определя и се подлага на количествен анализ. |
|
и) |
Представя се информация за устойчивостта на радионуклидните генератори, радионуклидните китове и белязаните с радиоактивни изотопи продукти. Документира се устойчивостта по време на употреба на радиофармацевтици във флакони за многократна употреба. |
Модул 4
Оценява се, че токсичността може да се свърже с дозата радиация. В диагнозата това е последица от използването на радиофармацевтици; при терапията това е търсено свойство. Следователно оценката на безвредността и ефикасността на радиофармацевтиците разглежда изискванията за лекарствените продукти и аспектите на радиационната дозиметрия. Излагането на даден орган или тъкан на радиация се документира. Изчислява се поетата по определен начин радиационна доза, като се използва конкретна международно призната система.
Модул 5
Когато е приложимо, се представят резултатите от клиничните изследвания, аргументацията за които иначе е представена в клиничните прегледи.
2.2. Радиофармацевтични прекурсори за целите на радиоактивното маркиране
В специфичния случай с радиофармацевтичния прекурсор, предназначен единствено за целите на маркирането с радиоактивни изотопи, първостепенната цел е да се представи информация, която да е насочена към възможните последици от недостатъчно ефикасното маркиране с радиоактивни изотопи или разпадане ин виво на белязаната с радиоактивни изотопи химична връзка, тоест въпроси, свързани с въздействието на свободния радионуклид върху пациента. Освен това е необходимо да се представи съответната информация във връзка с професионалните рискове, тоест въздействието на излагането на облъчване върху болничния персонал и върху околната среда.
По-специално там, където е приложимо, се представя следната информация:
Модул 3
Разпоредбите на модул 3 важат за регистрирането на радиофармацевтичните прекурсори, както е определено по-горе (букви от а) до и), когато е приложимо.
Модул 4
По отношение на токсичността на единичната доза и на многократно прилаганата доза се представят резултатите от проведените изследвания в съответствие с разпоредбите, отнасящи се до добрата лабораторна практика, предвидени в Директиви 87/18/ЕИО и 88/320/ЕИО на Съвета, освен ако не е оправдано по друг начин.
Изследванията върху мутагенността на радиоизотопите не се считат за полезни в този случай.
Представят се сведения относно химичната токсичност и отлагането на съответния „студен“ нуклид.
Модул 5
Клиничната информация, генерирана въз основа на клиничните изследвания с използване на самия прекурсор, не се счита за имаща отношение в конкретния случай на радиофармацевтичния прекурсор, който е предназначен единствено за целите на маркирането с радиоизотопи.
Въпреки това се представя информация, от която да личи клиничната полза от радиофармацевтичния прекурсор тогава, когато същият е прикрепен към съответните молекули носители.
3. ХОМЕОПАТИЧНИ ЛЕКАРСТВЕНИ ПРОДУКТИ
В този раздел са изложени конкретните разпоредби, отнасящи се до прилагането на модули 3 и 4 по отношение на хомеопатичните лекарствени продукти, както са описани в член 1, параграф 5.
Модул 3
Разпоредбите на модул 3 се прилагат към представените документи в съответствие с член 15 в опростената процедура за регистрация на хомеопатични лекарствени продукти, посочена в член 14, параграф 1, както и към документите за издаване на разрешение на други хомеопатични лекарствени продукти, посочени в член 16, параграф 1, като се вземат предвид следните изменения.
|
a) |
Терминология Латинското название на хомеопатичния продукт, описано в документацията за разрешение за търговия, трябва да съответства на латинското наименование в Европейската фармакопея или при липса на такова, в официалната фармакопея на държава-членка. Когато е подходящо, се представя и традиционното наименование, използвано в държавата-членка. |
|
б) |
Контрол върху изходните материали Подробностите и документите по изходните материали, тоест всички използвани материали, включително суровините и междинните материали до крайното разтваряне, които се включват в готовия лекарствен продукт, са снабдени с допълнителни данни относно хомеопатичния продукт, които да се приложат към материалите в заявлението. Общите изисквания по отношение на качеството се прилагат към всички изходни материали и суровини, включително суровините и междинните материали до крайното разтваряне, които задължително се включват в готовия лекарствен продукт. Ако това е възможно, в случай на присъствие на токсични компоненти, се изисква техният количествен анализ, при условие че качеството не може да се проконтролира при крайното разреждане, когато те следва да се включат поради високата степен на разреждане. Всяка фаза на производствения процес от изходните материали до крайното разреждане до получаване на готовия лекарствен продукт се описва подробно. В случай че в производствения процес съществува фаза разреждане, всички стъпки, включени във фазата разреждане, се извършват в съответствие с методите за производство на хомеопатични продукти, изложени в съответните монографии от Европейската фармакопея или в случай на липса на такива, в официалната фармакопея на държава-членка. |
|
в) |
Контролни изпитвания на готовия лекарствен продукт Към готовите хомеопатични лекарствени продукти се прилагат общите изисквания за качество, като заявителят представя надлежна аргументация, в случай на необходимост от отклонения от тези изисквания. Провеждат се определяне и количествен анализ на всички съставки със значение от гледна точка на токсикологията. Ако могат да се представят аргументи, че за дадени съставки със значение от гледна точка на токсикологията не е възможно да се проведе определяне или количествен анализ, например поради степента на разреждане в готовия лекарствен продукт, качеството се демонстрира посредством пълно удостоверяване на производствения процес и процеса на разреждане. |
|
г) |
Изпитвания за устойчивост Трябва да се демонстрира устойчивостта на готовия лекарствен продукт. Данните за устойчивостта от запасите от хомеопатични продукти обикновено се пренасят върху разтворите/праховете, получени от тях. Ако не може да се проведе определяне или количествен анализ на активното вещество, например поради степента на разреждане в готовия лекарствен продукт, могат да се вземат под внимание данните за устойчивостта от готовата фармацевтична форма. |
Модул 4
Разпоредбите на модул 4 се прилагат при опростената регистрация на хомеопатичните лекарствени продукти, посочени в член 14, параграф 1, заедно със следните спецификации.
Следва да се представи обосновка за всякаква липсваща информация, например трябва да се представи обосновка защо може да се подкрепи демонстриране на приемливо равнище на безвредност, въпреки липсата на някои изследвания.
4. РАСТИТЕЛНИ ЛЕКАРСТВЕНИ ПРОДУКТИ
Към заявленията за растителни лекарствени продукти се прилага пълна документация, в която се включват задължително следните конкретни подробности.
Модул 3
Разпоредбите, засягащи модул 3, включително съответствието с монография/и от Европейската фармакопея, с прилагат и към разрешението на растителните лекарствени продукти. Взема се под внимание състоянието на науката към момента, в който се подава заявлението.
Следва да се вземат под внимание следните специфични за растителните лекарствени продукти аспекти:
(1) Растителни вещества и растителни препарати
За целите на настоящото приложение термините „растителни вещества и растителни препарати“ следва да се считат за еквивалентни на понятията „растителни лекарства и растителни лекарствени препарати“ съгласно формулировката в Европейската фармакопея.
По отношение на номенклатурата на растителното вещество се представят научното описание на дадено растение (род, вид, разновидност и автор), както и хемотипа (когато е приложимо), частите на растенията, определението на растителния препарат, съотношението на растителното вещество към растителния препарат, другите наименования (синонимите, посочени в други фармакопеи), както и лабораторният шифър.
По отношение на номенклатурата на растителния препарат се представят научното описание на растението (род, вид, разновидност и автор), както и хемотипът (когато е приложимо), частите на растенията, определението на растителния препарат, съотношението на растителното вещество към растителния препарат, разтворителя на екстракта, останалите наименования (синоними, упоменати в други фармакопеи) и лабораторния код.
За да се документира разделът за структурата на растителното/ите вещество/а и растителния/те препарат/и, там, където е приложимо, се представят физичната форма, описание на съставките с познато терапевтично действие или маркери (молекулярна формула, относителна молекулярна маса, структурна формула, включително относителна и абсолютна стереохимия, молекулярната формула и относителната молекулярна маса), както и други съставки.
За да се документира разделът за производителя на растителното вещество, там, където е подходящо, се представят името, адресът и отговорността на всеки доставчик, включително подизпълнителите, всеки производствен обект или производствено съоръжение, включен/о в производствения процес/събирането и изпитването на растителното вещество.
За да се документира разделът за производителя на растителния препарат, там където е подходящо, се представят името, адресът и отговорността на всеки доставчик, включително подизпълнителите, всеки производствен обект или производствено съоръжение, включен/о в производствения процес и изпитването на растителния препарат.
Що се отнася до описанието на производствения процес и управлението на производствения процес по отношение на растителното вещество се представя информация, с която да се опише по подходящ начин производството на растенията и събирането на растенията, включително географския източник на лекарственото растение, както и условията за отглеждане, събиране, сушене и съхранение.
Що се отнася до описанието на производствения процес и управлението на производствения процес във връзка с растителния препарат се представя информация, с която да се опише по подходящ начин производството на растителния препарат, включително описание на обработката, разтворителите, реагентите, етапите на пречистването и стандартизацията.
Що се отнася до разработването на производствения процес, там, където е подходящо, се представя кратко обобщение, в което се описва разработването на растителното/те вещество/а и растителния/те препарат/и, като се вземе под внимание предложеният начин на приложение и употреба. Там, където е приложимо, се дискутират резултатите, в които се сравнява фитохимичният състав на растителното/ите вещество/а и растителния/те препарат/и, използвани в подкрепящите библиографски данни и там, където е приложимо, растителното/ите вещество/а и растителния/те препарат/и, които влизат в съдържанието на растителния лекарствен продукт като активно/и вещество/а.
По отношение на изясняване на структурата и на останалите характеристики на растителното вещество се представят сведения относно ботаническата, макроскопската, микроскопската и фитохимичната характеристика, както и ако е необходимо, относно биологичната активност на билковото вещество.
По отношение на изясняване на структурата и на останалите характеристики на растителния препарат се представя информация относно фито- и физикохимичната характеристика и ако е необходимо, относно биологичната активност на растителния препарат.
Представят се спецификации за растителното/ите вещество/а и растителния/те препарат/и.
Представят се процедурите за провеждането на анализите, използвани за изпитване на растителното/ите вещество/а и растителния/те препарат/и там, където е приложимо.
По отношение на утвърждаването на процедурите за провеждането на анализите, когато е приложимо, се представя информация за процедурите за провеждането на анализите, включително експериментални данни относно процедурите за провеждането на анализите, използвани за изпитване на растителното/ите вещество/а и растителния/те препарат/и.
По отношение на серийните анализи, когато е приложимо, се представя описание на серийните анализи на растителното/ите вещество/а и растителния/те препарат/и, включително тези на веществата по фармакопеята.
Когато е приложимо, се представя аргументация за спецификациите на растителното/ите вещество/а и растителния/те препарат/и.
Представя се информация за референтните стандарти и материали, използвани при изпитванията на растителното/ите вещество/а и растителния/те препарат/и, там, където е приложимо.
Там, където растителното вещество и растителният препарат са предмет на монография, заявителят може да кандидатства за сертификат за годност, който се издава от Европейската дирекция по качество на лекарствата.
(2) Растителни лекарствени продукти
По отношение на разработването на формулата се представя кратко обобщение на растителния лекарствен продукт, като се вземе предвид предложеният начин на приложение и употреба. Когато е подходящо, се обсъждат резултатите, в които се сравняват фитохимичният състав на продуктите, използвани в подкрепа на библиографските данни, и растителният лекарствен продукт, за който се кандидатства.
5. ЛЕКАРСТВЕНИ ПРОДУКТИ СИРАЦИ
|
— |
В случая с лекарствен продукт сирак по смисъла на Регламент (ЕО) № 141/2000 могат да се прилагат общите разпоредби от част II-6 (Извънредни обстоятелства). В такъв случай заявителят се аргументира в клиничното и неклиничното обобщение относно причините, поради които не е възможно да се представи пълна информация, и представя аргументация относно баланса риск—полза при въпросното лекарство сирак. |
|
— |
Когато кандидат за разрешение за търговия за лекарство сирак се позове на разпоредбите на член 10, параграф 1, буква а), ii) и част II-1 от настоящото приложение (Лекарствени продукти с утвърдено лечебно приложение), системната документирана употреба на въпросното вещество може да се позове — чрез дерогация — на употребата на това вещество съгласно разпоредбите на член 5 от настоящата директива. |
ЧАСТ IV
ЛЕКАРСТВЕНИ ПРОДУКТИ ЗА УСЪВЪРШЕНСТВАНА ТЕРАПИЯ
Лекарствените продукти за усъвършенствана терапия се основават на производствени процеси, чиято главна характеристика представляват различни биологични молекули, произведени в резултат от генно пренасяне и/или биологично усъвършенствани терапевтични модифицирани клетки като активни вещества или част от активните вещества.
За такива лекарствени продукти представянето на документацията за разрешение за търговия изпълнява изискванията за формата, описани в част I от настоящото приложение.
Прилагат се модули от 1 до 5. По отношение на съзнателното освобождаване в околната среда на генетично модифицирани организми се обръща внимание на продължителното присъствие на генетично модифицираните организми в организма на реципиента, както и възможното размножаване и/или модификация на генетично модифицираните организми при пускане на същите в околната среда. Информацията, която засяга риска за околната среда, се представя в приложението към модул 1.
1. ЛЕКАРСТВЕНИ ПРОДУКТИ ЗА ГЕННА ТЕРАПИЯ (ХУМАННИ И КСЕНОГЕННИ)
За целите на настоящото приложение „лекарствен продукт за генна терапия“ означава продукт, получен в резултат на комплекс от производствени процеси с цел пренасяне, които се изпълняват или ин виво, или екс виво на даден профилактичен, диагностичен или терапевтичен ген (тоест част от нуклеиновата киселина) към човешки/животински клетки и последващия израз на такова пренасяне ин виво. Пренасянето на гени включва система от изрази, съдържаща се в системата за предаване, позната под наименованието „вектор“, която може да бъде от вирусен и невирусен произход. Векторът също може да бъде включен в човешка или животинска клетка.
1.1. Разнообразие от лекарствени продукти за генна терапия
a) Лекарствени продукти за генна терапия на основата на алогенни или ксеногенни клетки
Векторът е предварително подготвен и се съхранява преди пренасянето му в клетките на приемника.
Клетките са получени предварително и могат да се обработват като клетъчна банка (събрани в банка клетки или банка, организирана на базата на намирането на първични клетки) с ограничена жизнеспособност.
Генетично модифицираните от вектора клетки представляват активно вещество.
С цел получаване на готов продукт могат да се предприемат допълнителни стъпки. По същество такъв лекарствен продукт е предназначен за употреба от определен брой пациенти.
б) Лекарствени продукти за генна терапия с използване на автологични човешки клетки
Активното вещество представлява серия от предварително изготвени вектори, които се съхраняват преди да бъдат пренесени в автологичните клетки.
С цел получаване на готов продукт могат да се предприемат допълнителни стъпки.
Тези продукти се изготвят от клетки, получени от отделен пациент. След получаването им клетките се променят генетично, като се използва предварително подготвен вектор, съдържащ подходящ ген, който е бил подготвен предварително и представлява активното вещество. Препаратът се инжектира отново на пациента, като по дефиниция такъв препарат е предназначен само за конкретен пациент. Целият производствен процес от извеждането на клетките от пациента до повторното им въвеждане в пациента се счита за една интервенция.
в) Прилагане на предварително подготвени вектори с вграден генетичен материал (профилактичен, диагностичен или терапевтичен)
Активното вещество представлява серия предварително подготвени вектори.
С цел получаване на готов продукт могат да се предприемат допълнителни стъпки. Този тип лекарствен продукт е предназначен за употреба от определен брой пациенти.
Пренасянето на генетичния материал може да се извърши посредством директно инжектиране на предварително подготвения вектор на реципиентите.
1.2. Специфични изисквания по отношение на модул 3
Лекарствените продукти за генна терапия включват:
|
— |
чиста нуклеинова киселина |
|
— |
сложна нуклеинова киселина или невирусни вектори |
|
— |
вирусни вектори |
|
— |
генетично модифицирани клетки |
Що се отнася до останалите лекарствени продукти, могат да се определят трите главни елемента на производствения процес, тоест:
|
— |
изходни материали: материали, от които се произвежда активното вещество, например генът обект, изразни плазмиди, клетъчни банки и запаси от вируси или невирусен вектор; |
|
— |
активно вещество: рекомбиниращ вектор, вирус, чисти или сложни плазмиди, вирусопроизвеждащи клетки, генетично променени при лабораторни условия клетки; |
|
— |
готов лекарствен продукт: активно вещество, формулирано крайната си непосредствена опаковка за планирано лечебно приложение. В зависимост от вида лекарствен продукт за генна терапия е възможно начинът на приложение и условията за употреба да изискват третиране на живо на клетките на пациента (виж точка 1.1, буква б). |
Специално внимание се отделя на следните въпроси:
|
a) |
Предоставя се информация относно главните характеристики на лекарствения продукт за генна терапия, включително относно неговия израз в целевата клетъчна популация. Представя се информация относно източника, строежа, характеристиката и утвърждаването на кодираната генна поредица, включително нейната цялост и устойчивост. Освен терапевтичния ген се представят пълната поредица други гени, регулиращите елементи и гръбнакът на вектора. |
|
б) |
Представя се информация относно характеристиката на вектора, използван за пренасянето и предаването на гена. Тук се включва неговата физико-химична характеристика и/или биологичната/имунологичната характеристика. За лекарствени продукти, в които се използват микроорганизми, например бактерии или вируси, с цел улесняването на пренасянето на гените (биологично генно пренасяне), се представят данни относно патогенезата на родителската верига и относно нейния тропизъм по отношение на конкретни тъкани и видове клетки, както и относно цикличната зависимост на клетките от тяхното взаимодействие. За лекарствени продукти, при които се използват небиологични средства с цел улесняването на пренасянето на гените, се представят физико-химичните свойства на съставките поотделно и в комбинация. |
|
в) |
Принципите за организирането на клетъчни банки или семенни запаси и характеризирането на семената се прилагат по подходящ начин и върху лекарствените продукти за генна терапия. |
|
г) |
Представя се източникът на клетките, които приемат рекомбинанта. Документират се характеристиките на човешкия източник, като например възраст, пол, резултати от микробиологични и вирусни изследвания, критерии за изключване, както и страна на произход. Относно клетките от животински произход се подава подробна информация във връзка със следните въпроси:
Документира се описанието на методологията на извличане на клетките, включително място, вид тъкан, процес на работа, транспортиране, съхранение и начини за проследяване, както и контрол по време на процеса на извличане на клетките. |
|
д) |
Оценката на опасността от вируси, както и начините за проследяване на продуктите от донора до готовия лекарствен продукт, представляват съществена част от документацията, която следва да се предаде. Например присъствието на вирус, свързан с разпространението в запасите на вирусни вектори, които не отговарят за разпространението, трябва да бъде изключено. |
2. ЛЕКАРСТВЕНИ ПРОДУКТИ ЗА ТЕРАПИЯ СЪС СОМАТИЧНИ КЛЕТКИ (ХУМАННИ И КСЕНОГЕННИ)
По смисъла на настоящото приложение „лекарствени продукти за терапия със соматични клетки“ означава употреба в хуманната медицина на автологични (произхождащи от самия пациент), алогенни (произхождащи от друг човек) или ксеногенни (произхождащи от животни) живи соматични клетки, чиито биологични характеристики са съществено променени в резултат на манипулациите с тях с цел получаване на терапевтичен, диагностичен или профилактичен ефект с метаболитни, фармакологични или имунологични средства. Тези манипулации включват разширяването или активизирането на популациите автологични клетки екс виво (например приемна имунотерапия), използването на алогенни и ксеногенни клетки, асоциирани с медицински механизми, използвани екс виво или ин виво (например микрокапсули, вътрешни матрични скелета, биологически разрушими или не).
Специални изисквания към лекарствените продукти за клетъчна терапия по отношение на модул 3
Лекарствените продукти за терапия със соматични клетки включват:
|
— |
Клетки, манипулирани така, че да се изменят техните имунологични, метаболитни и други функционални свойства в количествен и качествен аспект; |
|
— |
Сортирани, селекционирани и манипулирани и впоследствие подложени на производствен процес с цел получаване на готов лекарствен продукт; |
|
— |
Манипулирани и комбинирани с неклетъчни компоненти клетки (например биологични или инертни матрици или медицински механизми), прилагащи принципа на планираното действие в готовия лекарствен продукт; |
|
— |
Деривати от автологични клетки, преобразувани ин витро при специални условия за работа с културата; |
|
— |
Генетично променени или манипулирани по друг начин клетки с цел изразяване на неочаквани предварително хомологични или нехомологични функционални свойства. |
Целият производствен процес – от извличането на клетките от пациента (автологична ситуация) до повторното инжектиране в пациента се счита за единична интервенция.
Що се отнася до останалите лекарствени продукти, могат да се посочат три елемента на производствения процес:
|
— |
изходни материали: материали, от които се произвежда активното вещество, тоест органи, тъкани, телесни течности или клетки; |
|
— |
активното вещество: манипулирани клетки, клетъчни лизати, размножаващи се чрез пролиферация клетки и клетки, използвани в конюнкция с инертни матрици и медицински механизми; |
|
— |
готови лекарствени продукти: активното вещество, формулирано в крайната си непосредствена опаковка за дадена планирана медицинска употреба. |
|
a) |
Общи сведения за активното/ите вещество/а Активните вещества при лекарствените продукти за клетъчна терапия се състоят от клетки, които в резултат от обработка в лабораторни условия показват профилактични, диагностични или терапевтични свойства, различаващи се от изходните физиологични или биологични свойства. В настоящия раздел се описват видът клетки и културата, с която се работи. Документират се тъканите, органите и биологичните вещества, от които идват клетките, както и автологичният, алогенният или ксеногенният характер на материала от донора, както и географският му произход. Извличането на клетките, вземането на проби от клетки и съхраняването им, преди да се пристъпи към по-нататъшна обработка, също се документират. Представя се видът проведена манипулация и физиологичната функция на клетките, използвани като активно вещество. |
|
б) |
Информация относно изходните материали за активното/ите вещество/а |
1. Човешки соматични клетки
Лекарствените продукти за терапия със соматични клетки с използване на клетки от човешки произход се произвеждат от определен брой (комплект) жизнеспособни клетки, които се извличат при производствения процес, започващ или на ниво органи или тъкани, извлечени от човешки организъм, или на ниво ясно определена система клетъчна банка, в която даден комплект клетки разчита на определени непрекъснати клетъчни линии. За целите на тази глава „активни вещества“ означава семенният комплект човешки клетки, а „готов лекарствен продукт“ означава семенният комплект човешки клетки, формулиран за дадена планирана употреба в хуманитарната медицина.
Изходните материали, както и всяка стъпка от производствения процес, се документират в пълна степен, като се включват и аспектите, засягащи вирусната безвредност.
(1) Органи, тъкани, телесни течности и клетки от човешки произход
Документират се характеристиките на човешкия източник, като например възраст, пол, микробиологичен статус, критерии за изключване и страна на произход.
Документира се описанието на вземането на проби, включително мястото, типа, процеса на работа, комплектуването, транспортирането, съхранението и начините за проследяване, както и контрола върху вземането на проби.
(2) Системи клетъчни банки
Съответните изисквания, описани в част I, се прилагат и за подготовката и качествения контрол над системите клетъчни банки. Тук са обхванати главно алогенните или ксеногенните клетки.
(3) Помощни материали или помощни медицински механизми
Представя се информация относно използването на всякакви суровини (например цитокини, фактори на растежа, среди на култури) или относно възможни помощни продукти и медицински механизми, например механизми за сортиране на клетките, биологично съвместими полимери, матрици, влакна, вериги в смисъл на биологичната съвместимост, функционалност, както и риска от инфекциозни агенти.
2. Животински соматични клетки (ксеногенни)
Представя се подробна информация, свързана със следните въпроси:
|
— |
Произход на животните |
|
— |
Животновъдство и грижа за животните |
|
— |
Трансгенни животни (методи на създаване, характеристики на трансгенните клетки, характер на въведения ген) |
|
— |
Предпазни мерки и наблюдение на инфекциите сред животните донори |
|
— |
Изпитвания за инфекциозни агенти, включително вертикално разпространяващи се микроорганизми (също и ендогенни ретровируси) |
|
— |
Материална база |
|
— |
Системи за съхранение на клетки |
|
— |
Контрол на изходните материали и суровини. |
a) Сведения за производствения процес на активното/ите вещество/а и на готовия лекарствен продукт
Документират се различните стъпки от производствения процес, като например разпадането на органите и тъканите, подбора на търсената клетъчна популация, клетъчна култура в лабораторни условия, преобразуване на клетките от физикохимични агенти или пренасяне на гени.
б) Характеризиране на активното/ите вещество/а
С оглед планираната употреба се представя цялата необходима информация по характеризирането на търсената клетъчна популация в смисъл на идентичност (видове по произход, генетика на клетката, морфологичен анализ), чистота (случайно привнесени микробни агенти и клетъчни примеси), действие (дефинирана биологична активност) и пригодност (кариология и изпитвания за канцерогенност).
в) Фармацевтична разработка на готовия лекарствен продукт
Освен конкретния начин на употреба (венозна инфузия, местна инжекция, трансплантация по хирургичен път) се представя също и информация относно евентуалното използване на помощни медицински механизми (биологично съвместими полимери, матрици, влакна, вериги) в смисъл на биологичната съвместимост и дълготрайност.
г) Проследяване
Представя се подробно направена графика, която да дава възможност за проследяване на продуктите от донора до готовия лекарствен продукт.
3. СПЕЦИФИЧНИ ИЗИСКВАНИЯ КЪМ ЛЕКАРСТВЕНИТЕ ПРОДУКТИ ЗА ГЕННА ТЕРАПИЯ И ТЕРАПИЯ СЪС СОМАТИЧНИ КЛЕТКИ (ЧОВЕШКА И КСЕНОГЕННА) ВЪВ ВРЪЗКА С МОДУЛИ 4 И 5
3.1. Модул 4
За лекарствените продукти за генна терапия и терапия със соматични клетки е признато, че конвенционалните изисквания, изложени в модул 4, относно неклиничните изпитвания на лекарствените продукти, може невинаги да са подходящи поради уникалните и разнообразни структурни и биологични свойства на въпросните продукти, включително високата степен на специфика при даден вид, спецификата на даден субект, имунологичните бариери и различията в плеотропичните реакции.
Обосновката в подкрепа на неклиничната разработка и използваните критерии при избор на подходящи видове и модели се описват по съответния начин в модул 2.
Може да се окаже необходимо да бъдат идентифицирани и разработени нови модели с цел подпомагане на екстраполацията на конкретните открития по функционалните крайни точки и токсичността при активността на продуктите в човешкия организъм в естествени условия. Представя се научна аргументация за използването на тези животински модели на заболявания в подкрепа на концепцията за безвредността, както и за да се даде доказателство за концепцията за ефикасността.
3.2. Модул 5
Ефикасността на лекарствените продукти за усъвършенствана терапия задължително се демонстрира по начина, по който е описана в модул 5. За някои продукти и за някои терапевтични показания обаче е възможно да не могат да бъдат извършени всички необходими конвенционални клинични изпитвания. Всички отклонения от съществуващите ръководства се обосновават в модул 2.
Клиничната разработка на лекарствените продукти за усъвършенствана терапия със сигурност се отличава с някои специфични белези поради комплексността и лабилното естество на активните вещества. За целта се изискват допълнителни решения поради аспекти, свързани с жизнеспособността, начина на размножаване, миграцията и диференциацията на клетките (терапия на соматични клетки), поради особените клинични обстоятелства, при които се използват продуктите или поради особения начин на действие посредством негенен израз (соматична генна терапия).
В заявлението за разрешение за търговия с лекарствени продукти за усъвършенствана терапия следва да се обърне внимание на особените рискове, които се асоциират с подобни продукти и възникват във връзка с потенциалното заразяване с инфекциозни агенти. Особено следва да се наблегне на ранните стадии от развитието, от една страна, включително избора на донори в случая с лекарствените продукти за клетъчна терапия, и на терапевтичната намеса като цяло, включително правилното боравене с продукта и неговата правилна употреба – от друга.
Освен това в модул 5 от заявлението следва да се съдържат представляващи интерес данни относно мерките за проучване, контрол и наблюдение на функциите и развитието на живите клетки в организма на реципиента с цел предотвратяване на предаването на инфекциозни агенти на реципиента и свеждането до минимум на всякакъв потенциален риск за общественото здраве.
3.2.1. Хуманна фармакология и изследвания на ефикасността
Изследванията по хуманна фармакология следва да осигуряват информация относно очаквания начин на действие, очакваната ефикасност на базата на аргументирани крайни точки, биологично разпространение, подходяща доза, график на приемането, както и методи на прилагане или променливост в употребата, желателни за целите на изследванията за ефикасност.
Конвенционалните фармакокинетични изследвания може да не се отнасят до някои продукти на усъвършенстваната терапия. Понякога изследванията при здрави доброволци не са осъществими, като съществува вероятност от поява на трудности при установяването на дозата и кинетиката и при определянето на клиничния риск. Въпреки това е необходимо да се проучат разпространението и поведението в естествени условия на генния продукт и продължителността на желаното генно изражение. Прилагат се подходящи изпитвания и ако е необходимо, се разработват такива изпитвания за проследяването на клетъчния продукт или клетъчното изражение на желания ген в човешкия организъм, както и за наблюдение върху функциите на клетките, на които е приложен продуктът или които са били трансфектирани.
Оценката на ефикасността и безвредността на даден лекарствен продукт за усъвършенствана терапия трябва да включва подробно описание и оценка на терапевтичната процедура като цяло, включително специални начини на приложение (като например трансфекция на клетки ин виво, манипулации ин витроo или използване на техники за интервенция), както и изпитване на възможните свързани режими (включително имунопотискащо, антивирусно, цитотоксично лечение).
Цялата процедура трябва да бъде изпитана в клинични опити, след което се описва в информацията за продукта.
3.2.2. Безвредност
Вземат се предвид въпросите на безвредността, произтичащи от имунната реакция към лекарствените продукти или към изразените протеини, имунното отхвърляне, потискането на имунната система, както и разрушаването на имуноизолационните механизми.
Някои лекарствени продукти за генна терапия и за терапия на соматични клетки (например терапевтични продукти за ксеногенна клетъчна терапия) може да съдържат частици, отговарящи за размножаването и/или инфекциозни агенти. Възможно е да се наложи наблюдение над пациента по отношение на възможното развитие на инфекции и/или патологични усложнения във връзка с тях по време на периода преди даването на разрешението и/или през периода след получаването на разрешението; освен това може да се наложи това наблюдение да бъде разширено в смисъл на прекратяване на контактите на пациента, включително със здравните работници.
Рискът от заразяване с потенциално преносими агенти не може да бъде напълно елиминиран по време на употреба на някои лекарствени продукти за генна терапия и за терапия на соматични клетки. Рискът обаче може да бъде сведен до минимум посредством подходящи мерки, както е описано в модул 3.
Мерките, включени в производствения процес, трябва да се изпълняват заедно с прилагане на методи на изпитване, контрол на качеството и подходящи методи за наблюдение, които следва да се опишат в модул 5.
Може да се наложи ограничаване – временно или постоянно – на употребата на някои лекарствени продукти за усъвършенствана терапия на соматични клетки до институции, които разполагат с опита и базата, с които да обезпечат спазването на конкретните мерки за безопасност на пациентите. Подобен подход може да бъде подходящ за определени генно-терапевтични медицински продукти, които се свързват с потенциален риск от инфекциозни причинители със способност за повторно заразяване.
Следва да се вземат под внимание и дългосрочните аспекти на наблюдението върху развитието на късни прояви на усложнения, които се отразяват в подадените документи там, където е необходимо.
Когато е подходящо, заявителят следва да представи подробен план за управление на риска, който да обхваща клиничните и лабораторните данни за пациента, текущите епидемиологични данни, както и ако е важно, данните от архивите на образци от тъкани от донора и реципиента. Тази система е необходима, за да се осигури възможност за проследяване на лекарствения продукт и бързо реагиране на подозрителни модели на странични реакции.
4. СПЕЦИАЛНО СТАНОВИЩЕ ОТНОСНО ЛЕКАРСТВЕНИТЕ ПРОДУКТИ ЗА КСЕНОТРАНСПЛАНТАЦИЯ
По смисъла на настоящото приложение под „ксенотрансплантация“ се разбира всяка процедура, в която е включено трансплантиране, имплантиране или инфузия в човешки реципиент или на живи тъкани, или на органи, извлечени от животни, или на течности, клетки, тъкани или органи от човешки организъм, които са били подложени на контакт екс виво с живи нечовешки животински клетки, тъкани или органи.
Специално се набляга на изходните материали.
Във връзка с това се представя подробна информация относно следните въпроси съгласно конкретните ръководства:
|
— |
Животни донори |
|
— |
Животновъдство и грижа за животните |
|
— |
Генетично модифицирани животни (методи на създаване, характеристики на трансгенните клетки, характер на въведения или извлечения ген) |
|
— |
Предпазни мерки и наблюдение на инфекциите сред животните донори |
|
— |
Изпитвания за инфекциозни агенти |
|
— |
Материална база |
|
— |
Контрол на изходните материали и суровини |
|
— |
Проследяване. |
(1) Международна конференция по хармонизация на техническите изисквания при регистриране на лекарствени продукти за хуманна употреба.
(2) ОВ L 193, 17.7.1991 г., стр. 30.
(3) ОВ L 121, 1.5.2001 г., стр. 34.
(4) ОВ L 15, 17.1.1987 г., стр. 29.
(5) ОВ L 145, 11.6.1988 г., стр. 35.
(6) ОВ L 159, 27.6.2003 г., стр. 1.
(7) ОВ L 159, 27.6.2003 г., стр. 24.
(8) ОВ L 106, 17.4.2001 г., стр. 1.
(9) ОВ L 11, 14.1.1978 г., стр. 18.
(10) ОВ L 237, 10.9.1994 г., стр. 13.
(11) ОВ L 313, 13.12.2000 г., стр. 22.
(12) ОВ L 55, 11.3.1995 г., стр. 15.
(13) ОВ L 214, 24.8.1993 г., стр. 1.“